|
8)
|
W części B dodaje się rozdziały w brzmieniu:
„B.63 BADANIE KLASYFIKACYJNE TOKSYCZNOŚCI REPRODUKCYJNEJ I ROZWOJOWEJ
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna wytycznej OECD dotyczącej badań (TG) nr 421 (2016). Okresowo dokonuje się przeglądu wytycznych OECD dotyczących badania substancji chemicznych pod kątem postępu naukowego. Pierwotna wytyczna dotycząca badań klasyfikacyjnych 421 została przyjęta w 1995 r. na podstawie protokołu „Wstępne badanie klasyfikacyjne toksyczności reprodukcyjnej (Preliminary Reproduction Toxicity Screening Test)” omówionego na dwóch posiedzeniach ekspertów: w Londynie w 1990 r. (1) i w Tokio w 1992 r. (2).
|
|
2.
|
Niniejsza metoda badawcza została zaktualizowana o właściwe punkty końcowe badania substancji zaburzającej funkcjonowanie układu hormonalnego w ramach działań następczych do działania o wysokim priorytecie podjętego przez OECD w 1998 r., mającego na celu zmianę istniejących wytycznych i opracowanie nowych wytycznych dotyczących badań klasyfikacyjnych i badania potencjalnych substancji zaburzających funkcjonowanie układu hormonalnego (3). Na przykład wytyczną dotyczącą badań nr OECD 407 (28-dniowe badanie toksyczności doustnej wywołanej powtarzanym dawkowaniem u gryzoni, rozdział B.7 niniejszego załącznika) wzbogacono w 2008 r. o parametry odpowiednie do wykrywania aktywności endokrynologicznej badanych substancji chemicznych. Celem aktualizacji wytycznej dotyczącej badań nr 421 było uwzględnienie niektórych właściwych punktów końcowych substancji zaburzającej funkcjonowanie układu hormonalnego w badaniach klasyfikacyjnych TG, gdzie okresy narażenia obejmują niektóre wrażliwe okresy w rozwoju (okresy przed lub wcześnie po urodzeniu).
|
|
3.
|
Wybrane dodatkowe właściwe punkty końcowe badania substancji zaburzającej funkcjonowanie układu hormonalnego, stanowiące również część wytycznej dotyczącej badań 443 (Rozszerzone badanie toksyczności reprodukcyjnej w jednym pokoleniu, rozdział B.56 niniejszego załącznika), zostały włączone do wytycznej TG 421 na podstawie studium wykonalności dotyczącego kwestii naukowych i technicznych związanych z ich włączeniem, jak również możliwych dostosowań projektu badań potrzebnych do ich włączenia (4).
|
|
4.
|
Niniejsza metoda badawcza ma na celu wygenerowanie ograniczonej ilości informacji dotyczących wpływu badanej substancji chemicznej na efektywność reprodukcyjną samców i samic, taką jak funkcja gonad, zachowanie kopulacyjne, zapłodnienie, rozwój jaja płodowego i poród. Nie jest ona metodą alternatywną dla istniejących metod badań B.31, B.34, B.35 lub B.56, ani ich nie zastępuje.
|
ZAŁOŻENIA WSTĘPNE
|
5.
|
Niniejsza metoda badania klasyfikacyjnego może być stosowana w celu dostarczenia wstępnych informacji na temat możliwego wpływu na reprodukcję lub rozwój, albo na wczesnym etapie oceny właściwości toksykologicznych substancji chemicznych lub na przedmiotowe substancje chemiczne. Może być również stosowana jako część zestawu wstępnych badań klasyfikacyjnych dla istniejących substancji chemicznych, w przypadku których dostępne są niewiele informacji toksykologicznych lub nie ma ich wcale, jako badanie ustalające zakres dawek dla bardziej ekstensywnych badań nad rozrodczością/rozwojem, lub w przypadku gdy jest to uznane za istotne w inny sposób. Przy prowadzeniu badania należy przestrzegać zasad i ustaleń określonych w dokumencie zawierającym wytyczne OECD nr 19 „Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations” (5) (Wytyczne dotyczące uznawania, oceny i wykorzystywania objawów klinicznych jako punktów humanitarnego zakończenia w odniesieniu do zwierząt doświadczalnych wykorzystywanych w ocenach bezpieczeństwa).
|
|
6.
|
Niniejsza metoda badawcza nie zapewnia pełnych informacji na temat wszystkich aspektów reprodukcji i rozwoju. W szczególności oferuje jedynie ograniczone środki wykrywania poporodowych przejawów narażenia prenatalnego lub skutków, które mogą być wywołane podczas narażenia postnatalnego. Ze względu (między innymi) na stosunkowo małą liczbę zwierząt w grupach dawkowania, selektywność punktów końcowych oraz krótki czas trwania badania, metoda ta nie dostarczy dowodów na definitywne stwierdzenie braku skutków. Ponadto wobec braku danych z innych badań toksyczności reprodukcyjnej/rozwojowej, wyniki dodatnie są przydatne do wstępnej oceny zagrożeń i przyczyniają się do podejmowania decyzji dotyczących konieczności i terminów dodatkowych badań.
|
|
7.
|
Wyniki uzyskane za pomocą parametrów związanych z układem hormonalnym należy rozpatrywać w kontekście dokumentu „OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals” („Ramy koncepcyjne OECD dotyczące testowania i oceny substancji chemicznych zaburzających funkcjonowanie układu hormonalnego”) (6). W niniejszych ramach koncepcyjnych rozszerzona wytyczna dotycząca badań OECD nr 421 zawarta jest na poziomie 4 jako test in vivo dostarczający danych na temat niekorzystnego wpływu na właściwe punkty końcowe w badaniu wpływu na układ hormonalny. Sygnał endokrynny może jednak nie być uważany za wystarczający dowód na to, że badana substancja chemiczna jest substancją zaburzającą funkcjonowanie układu hormonalnego.
|
|
8.
|
Niniejsza metoda badawcza zakłada doustną drogę podawania badanej substancji chemicznej. W przypadku stosowania innych dróg narażenia, może być konieczne dokonanie modyfikacji.
|
|
9.
|
Przed zastosowaniem przedmiotowej metody badawczej z użyciem mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy zastanowić się, czy zastosowanie tej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak, to dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny.
|
|
10.
|
Stosowane definicje znajdują się w dodatku 1.
|
ZASADA BADANIA
|
11.
|
Badana substancja chemiczna podawana jest w stopniowanych dawkach kilku grupom samców i samic. Samce powinny otrzymywać dawki substancji przez co najmniej cztery tygodnie do dnia przed planowanym uśmierceniem włącznie (obejmuje to co najmniej dwa tygodnie przed kryciem, w okresie krycia i około dwóch tygodni po kryciu). Ze względu na ograniczone dawkowanie przed kryciem u samców, płodność może nie być szczególnie czułym wskaźnikiem toksyczności w odniesieniu do jąder. W związku z tym niezbędne jest szczegółowe badanie histologiczne jąder. Uważa się, że połączenie dwutygodniowego okresu dawkowania w okresie przed kojarzeniem i późniejszych obserwacji krycia/płodności z ogólnym okresem dawkowania wynoszącym co najmniej cztery tygodnie, po którym następuje szczegółowa histopatologia gonad samców, jest wystarczające do wykrycia większości wpływu na płodność i spermatogenezę samców.
|
|
12.
|
Samice powinny otrzymywać dawki przez cały czas trwania badania. Obejmuje to dwa tygodnie przed kryciem (w celu objęcia co najmniej dwóch pełnych cykli estrogenowych), zmienny czas do zapłodnienia, czas trwania ciąży i co najmniej trzynaście dni po porodzie, do dnia planowanego uśmiercenia włącznie.
|
|
13.
|
Czas trwania badania, po aklimatyzacji i ocenie cyklu estrogenowego przed dawkowaniem, zależy od efektywności samic i wynosi około 63 dni, [co najmniej 14 dni okresu przed kojarzeniem, (do) 14 dni krycia, 22 dni ciąży, 13 dni laktacji].
|
|
14.
|
Podczas okresu dawkowania każdego dnia zwierzęta są dokładnie obserwowane w celu wykrycia oznak toksyczności. Zwierzęta, które zginą lub zostaną uśmiercone w okresie badania, są poddawane autopsji, a zwierzęta, które przeżyją, są po zakończeniu badania uśmiercane i poddawane autopsji.
|
OPIS METODY
Wybór gatunku zwierząt
|
15.
|
Niniejsza metoda badawcza jest przeznaczona do stosowania z wykorzystaniem szczurów. W przypadku gdy parametry określone w niniejszej metodzie badawczej badane są z wykorzystaniem innego gatunku gryzoni, należy podać uzasadnienie. Szczur był jedynym gatunkiem wykorzystywanym w międzynarodowym programie walidacyjnym dotyczącym substancji zaburzających funkcjonowanie układu hormonalnego w wytycznej dotyczącej badań OECD nr 407 (odpowiadającej rozdziałowi B.7 niniejszego załącznika). Nie należy stosować szczepów o niskiej płodności lub powszechnie znanej wysokiej częstości występowania wad rozwojowych. Należy wykorzystać zdrowe zwierzęta dziewicze, które nie były wcześniej poddawane procedurom badawczym. Badane zwierzęta należy scharakteryzować z uwzględnieniem ich gatunku, szczepu, płci, masy i wieku. W momencie rozpoczęcia badania różnice w masie ciała wykorzystywanych zwierząt powinny być minimalne i nie powinny przekraczać 20 % średniej masy ciała u każdej z płci. W przypadku gdy badanie przeprowadza się jako badanie wstępne w stosunku do badania długoterminowego lub pełnopokoleniowego, zaleca się, aby w obu badaniach wykorzystywać zwierzęta tego samego szczepu i źródła.
|
Warunki utrzymywania i karmienia
|
16.
|
Wszystkie procedury powinny być zgodne z lokalnymi normami utrzymywania zwierząt laboratoryjnych. Temperatura w pomieszczeniu, w którym przetrzymuje się zwierzęta doświadczalne, powinna wynosić 22 °C (± 3 °). Wilgotność względna, poza okresem sprzątania pomieszczenia, powinna wynosić co najmniej 30 % i raczej poniżej 70 %, należy starać się ją utrzymać na poziomie 50–60 %. Oświetlenie powinno być sztuczne w cyklu 12 godzin z dostępem światła i 12 godzin bez dostępu światła. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Na wybór paszy może mieć wpływ potrzeba zapewnienia odpowiedniej domieszki badanej substancji chemicznej w przypadku podawania jej w ramach opisywanej metody.
|
|
17.
|
Zwierzęta należy przetrzymywać razem, w małych grupach składających się z osobników tej samej płci; można je przetrzymywać pojedynczo, jeżeli ma to naukowe uzasadnienie. W przypadku przetrzymywania w grupach, w jednej klatce należy umieszczać nie więcej niż pięć zwierząt. Procedury krycia powinny być przeprowadzane w klatkach nadających się do tego celu. Ciężarne samice powinny być przetrzymywane w osobnych klatkach i mieć zapewnione materiały do budowy gniazda. Samice w okresie ciąży będą przebywać w indywidualnych klatkach ze swoim potomstwem.
|
|
18.
|
Paszę należy regularnie analizować pod kątem obecności zanieczyszczeń. Próbkę paszy należy zachować do momentu ukończenia sprawozdania.
|
Przygotowanie zwierząt
|
19.
|
Zdrowe młode dorosłe osobniki zwierząt są losowo przydzielane do grup kontrolnych oraz grup poddawanych działaniu substancji. Klatki należy rozmieścić w taki sposób, aby zminimalizować potencjalny wpływ ich układu. Zwierzęta są identyfikowane jednoznacznie i przetrzymywane w klatkach przez co najmniej pięć dni przed rozpoczęciem badania, żeby umożliwić aklimatyzację do warunków laboratoryjnych.
|
Przygotowanie dawek
|
20.
|
Zaleca się, aby badana substancja chemiczna była podawana doustnie, chyba że za bardziej odpowiednią uzna się inną drogę podawania. W przypadku wybrania doustnej drogi podawania, badaną substancję chemiczną zazwyczaj podaje się za pomocą sondy; można jednak również zastosować metodę alternatywną polegającą na podaniu badanych substancji chemicznych w paszy lub w wodzie do picia.
|
|
21.
|
W razie potrzeby badaną substancję chemiczną rozpuszcza się lub przygotowuje jako zawiesinę w odpowiednim nośniku. Zaleca się, w miarę możliwości, najpierw rozważyć zastosowanie roztworu wodnego/zawiesiny, następnie roztworu/emulsji w oleju (np. z zastosowaniem oleju kukurydzianego), a potem możliwego roztworu w innych nośnikach. W przypadku nośników innych niż woda właściwości toksyczne tego nośnika powinny być znane. Należy określić stabilność i jednorodność badanej substancji chemicznej w nośniku.
|
PROCEDURA
Liczba i płeć zwierząt
|
22.
|
Zaleca się, aby w każdej grupie na początku znajdowało się co najmniej 10 samców i 12–13 samic. Ocena samic będzie prowadzona przed narażeniem pod kątem cykliczności estrogenowej, a do badania nie zostaną włączone zwierzęta, które nie wykażą typowych 4–5 dniowych cykli; w związku z tym zaleca się zastosowanie dodatkowych samic w celu uzyskania 10 samic w każdej grupie. Poza przypadkiem, w którym występują wyraźne efekty toksyczne, oczekuje się, że zapewni to co najmniej 8 ciężarnych samic na grupę, co zazwyczaj stanowi najniższą dopuszczalną liczbę ciężarnych samic na grupę. Celem jest wyprodukowanie odpowiedniej liczby ciąż i potomstwa w celu zapewnienia znaczącej oceny wpływu badanej substancji chemicznej na płodność, ciążę, zachowanie macierzyńskie i zwierząt ssących oraz wzrost i rozwój potomstwa F1 w okresie od chwili poczęcia do 13 dnia po porodzie.
|
Dawkowanie
|
23.
|
Zasadniczo należy wykorzystać co najmniej trzy grupy badane i jedną grupę kontrolną. Poziomy dawek mogą opierać się na informacjach z badań dotyczących ostrej toksyczności lub na wynikach badań wielokrotnej dawki. Z wyjątkiem podawania badanej substancji chemicznej zwierzęta w grupie kontrolnej należy traktować w identyczny sposób jak zwierzęta w grupie badanej. Jeżeli przy podawaniu badanej substancji chemicznej jest stosowany nośnik, grupa kontrolna powinna otrzymać największą użytą objętość nośnika.
|
|
24.
|
Poziomy dawki powinny być wybierane, biorąc pod uwagę jakiekolwiek istniejące dostępne dane o toksyczności i dane toksykokinetczne. Należy także wziąć pod uwagę, że mogą istnieć różnice w czułości między zwierzętami w ciąży a tymi niebędącymi w ciąży. Należy wybrać taki najwyższy poziom dawkowania, który powoduje oznaki toksyczności, ale nie powoduje śmierci lub silnego cierpienia. Następnie należy dobierać poziomy dawkowania w sekwencji malejącej w taki sposób, aby ukazać wszystkie reakcje powiązane z dawkowaniem i brak obserwowanych szkodliwych zmian przy najmniejszym poziomie dawkowania (NOAEL). Często do ustalenia malejących poziomów dawkowania optymalne jest zastosowanie poziomów dawkowania różniących się od dwu- do czterokrotnie, a zamiast stosować bardzo odległe (np. różniące się więcej niż 10-krotnie) poziomy dawkowania, często lepiej jest dodać czwartą grupę badaną.
|
|
25.
|
Jeśli obserwowana jest ogólna toksyczność (np. spadek masy ciała, wpływ na wątrobę, serce, płuca lub nerki itp.) bądź inne zmiany, które nie muszą być efektami toksycznymi (np. zmniejszone pobieranie pokarmu, powiększenie wątroby), należy zachować ostrożność interpretując obserwowane skutki w punktach końcowych wpływu na układ hormonalny.
|
Badanie graniczne
|
26.
|
Jeżeli badanie z podawaniem doustnym przy jednym poziomie dawki równym co najmniej 1 000 mg/kg masy ciała/dzień lub, w przypadku podawania w paszy lub w wodzie do picia, równoważny procent w paszy lub wodzie do picia, stosując procedury opisane dla tego badania, nie wywołuje widocznych objawów zatrucia i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych, dotyczących substancji strukturalnie pokrewnych, wtedy pełne badanie przy użyciu szeregu poziomów dawki może nie być konieczne. Ten test graniczny stosuje się z wyjątkiem przypadków, w których ekspozycja ludzi wskazuje na konieczność użycia wyższego poziomu dawki doustnej. W odniesieniu do innych dróg podawania, na przykład wziewnej lub stosowania na skórę, fizyczne własności chemiczne badanych substancji chemicznych mogą często wyznaczać najwyższe możliwe do osiągnięcia stężenie.
|
Podawanie dawek
|
27.
|
Zwierzętom podaje się badaną substancję chemiczną codziennie, przez siedem dni w tygodniu. Gdy badana substancja chemiczna jest podawana przez sondę, należy ją podawać zwierzętom w pojedynczej dawce przy użyciu sondy żołądkowej lub odpowiedniej kaniuli intubacyjnej. Maksymalna objętość płynu, jaką można podać jednorazowo, zależy od wielkości badanego zwierzęcia. Objętość ta nie powinna przekraczać 1 ml/100 g masy ciała, oprócz roztworów wodnych, w przypadku których można użyć 2 ml/100 g masy ciała. Poza drażniącymi lub żrącymi badanymi substancjami chemicznymi, które zwykle dają zaostrzone objawy przy wyższych stężeniach, zmienność objętości w badaniu powinna być zminimalizowana przez dostosowanie stężenia, aby zapewnić stałą objętość przy wszystkich poziomach dawki.
|
|
28.
|
W przypadku substancji chemicznych podawanych z paszą lub wodą do picia należy dopilnować, aby ilości podawanej badanej substancji chemicznej nie zakłócały zwykłego odżywiania lub gospodarki wodnej organizmu. Przy podawaniu badanej substancji chemicznej wraz z pokarmem można zastosować stałe stężenie w pokarmie (ppm) lub też stały poziom dawki w zależności do masy ciała zwierzęcia; należy podać zastosowany wariant. W przypadku badanej substancji chemicznej podawanej przez sondę, dawka powinna być podawana każdego dnia o podobnej porze i w razie konieczności modyfikowana co najmniej raz na tydzień, aby utrzymać stały poziom dawki w zależności od masy ciała zwierzęcia.
|
Harmonogram doświadczalny
|
29.
|
Podawanie dawek substancji obu płciom powinno rozpocząć się co najmniej 2 tygodnie przed kryciem, po zaaklimatyzowaniu się osobników przez co najmniej pięć dni i poddaniu samic badaniu przesiewowemu pod kątem normalnych cykli estrogenowych (w dwa tygodnie przed okresem poddawania działaniu substancji). Badanie powinno być zaprogramowane w taki sposób, żeby ocena cyklu estrogenowego rozpoczynała się zaraz po osiągnięciu przez zwierzęta pełnej dojrzałości płciowej. Pod tym względem mogą występować nieznaczne różnice u różnych szczepów szczurów wykorzystywanych w różnych laboratoriach, np. szczury szczepu Sprague Dawley osiągają dojrzałość płciową po 10 tygodniach życia, a szczepu Wistar – po około 12 tygodniach. Matki posiadające potomstwo należy uśmiercić 13. dnia po porodzie lub wkrótce po tym dniu. Dzień narodzin (tj. dzień zakończenia porodu) określa się jako dzień 0 po porodzie. Samice, u których nie zaobserwowano oznak kopulacji, uśmierca się po 24–26 dniach od ostatniego dnia okresu krycia. Dawkowanie należy kontynuować u obu płci przez okres krycia. Dawkowanie u samców należy nadal stosować po okresie krycia co najmniej do zakończenia minimalnego łącznego okresu dawkowania wynoszącego 28 dni. Następnie samce są uśmiercane lub ewentualnie zatrzymywane i kontynuuje się dawkowanie w celu ewentualnego drugiego krycia, jeśli uzna się to za stosowne.
|
|
30.
|
Codziennie dawkowanie u rodzicielskich samic powinno dalej trwać przez cały okres ciąży, co najmniej do 13. dnia po porodzie włącznie, lub do dnia poprzedzającego uśmiercenie. W przypadku badań, w których badaną substancję chemiczną podaje się drogą wziewną lub skórną, dawkowanie powinno trwać co najmniej do 19. dnia ciąży włącznie i powinno być ono wznowione najszybciej jak to możliwe – nie później niż w czwartym dniu po urodzeniu.
|
|
31.
|
Diagram przedstawiający harmonogram doświadczenia znajduje się w dodatku 2.
|
Procedura krycia
|
32.
|
Podczas tego badania należy zazwyczaj wykorzystywać krycia 1:1 (jeden samiec na jedną samicę). W przypadku wystąpienia sporadycznych zgonów samców mogą pojawić się wyjątki. Samicę należy umieścić razem z tym samym samcem na okres trwający do momentu odnotowania kopulacji lub dwa tygodnie. Każdego dnia rano samice należy poddać badaniu na obecność plemników lub czopa pochwowego. Dzień 0 ciąży określa się jako dzień, w którym potwierdzono wystąpienie krycia (wykrycie czopa pochwowego lub plemników). Jeśli kojarzenie w parze nie uda się, można rozważyć krycie samic samcami o dowiedzionej płodności.
|
Liczebność miotu
|
33.
|
Czwartego dnia po porodzie można skorygować liczebność każdego miotu, eliminując dodatkowe, losowo wybrane młode w miocie, by otrzymać, na ile to możliwe, cztery lub pięć samic i cztery lub pięć samców na miot, w zależności od typowej liczebności miotów w szczepie wykorzystanych szczurów. Próbki krwi należy pobrać od dwóch młodych nieprzydzielonych do żadnej kohorty, zebrać i wykorzystać do określenia poziomów surowicy T4. Wybiórcza eliminacja młodych, np. na podstawie masy ciała lub odległości anogenitalnej jest nieodpowiednia. Ilekroć liczba młodych samców lub samic uniemożliwia uzyskanie czterech lub pięciu osobników każdej płci na miot, dopuszczalne jest częściowe dostosowanie (na przykład sześć samców i cztery samice). Żadne młode nie zostanie wyeliminowane, gdy liczebność miotu spadnie poniżej celu eliminacji (8 lub 10 młodych/miot) Jeżeli dostępne jest tylko jedno młode powyżej celu eliminacji, tylko jedno młode zostanie wyeliminowane i wykorzystane do pobrania krwi do ewentualnej oceny surowicy T4.
|
|
34.
|
Jeżeli liczebność miotu nie została skorygowana, dwa młode na miot uśmierca się 4. dnia po urodzeniu, a próbki krwi pobiera się do pomiaru stężeń hormonów tarczycy w surowicy. Jeżeli to możliwe, dwa młode na miot powinny stanowić młode samice, aby zachować młode samce na potrzeby oceny zatrzymywania płynu w brodawkach sutkowych, oprócz przypadku, w którym eliminacja tych młodych powoduje, że nie pozostaną żadne samice do oceny końcowej przy uśmiercaniu. Żadne młode nie zostanie wyeliminowane, gdy rozmiar miotu spadnie poniżej 8 lub 10 młodych/miot (w zależności od typowego rozmiaru miotu w szczepie wykorzystanych szczurów). Jeżeli dostępne jest tylko jedno młode powyżej typowej liczebności miotu, tylko jedno młode zostanie wyeliminowane i wykorzystane do pobrania krwi do ewentualnej oceny surowicy T4.
|
Obserwacje w trakcie życia
Obserwacje kliniczne
|
35.
|
W trakcie okresu badania ogólne obserwacje kliniczne należy przeprowadzać co najmniej raz dziennie, a w przypadku zaobserwowania oznak toksyczności należy zwiększyć ich częstotliwość. Najlepiej prowadzić je każdego dnia o tej samej porze, biorąc pod uwagę szczytowy okres przewidywanych objawów po dawkowaniu. Należy odnotować zmiany w zachowaniu mające związek z badaniami, objawy trudnego lub długotrwałego porodu oraz wszelkie objawy toksyczności, włączając śmiertelność. Zapisy te powinny zawierać czas pojawienia się, stopień ciężkości i czas trwania objawów toksyczności.
|
Masa ciała i spożycie pokarmu/wody
|
36.
|
Samce i samice należy zważyć w pierwszym dniu dawkowania, a następnie co najmniej raz tygodniowo oraz w momencie zakończenia dawkowania. Samice w okresie ciąży należy ważyć w dniach 0, 7, 14 i 20 oraz w ciągu 24 godzin od porodu (w dniu 0 lub 1 po porodzie), a także co najmniej 4 i 13 dni po porodzie. Obserwacje te należy umieścić w sprawozdaniu oddzielnie w odniesieniu do każdego dorosłego zwierzęcia.
|
|
37.
|
Przed kryciem, w okresie ciąży oraz laktacji spożycie pokarmu należy mierzyć co najmniej raz w tygodniu. W okresie krycia pomiar spożycia pokarmu nie jest obowiązkowy. Gdy badana substancja chemiczna podawana jest za pośrednictwem wody do picia, w trakcie tych okresów należy także mierzyć spożycie wody.
|
Cykle estrogenowe
|
38.
|
Monitorowanie cykli estrogenowych powinno odbywać się przed rozpoczęciem poddawania działaniu substancji, aby do badania wybrać samice o regularnej cykliczności (zob. pkt 22). Należy także codziennie, od rozpoczęcia poddawania działaniu substancji do potwierdzenia krycia, monitorować rozmazy śluzówki pochwy. Jeżeli istnieje obawa związana z wystąpieniem dotkliwych skutków stresu mogących zmienić cykle estrogenowe wraz z rozpoczęciem dawkowania, laboratoria mogą przez dwa tygodnie narażać zwierzęta doświadczalne, a następnie przez co najmniej dwa tygodnie codziennie pobierać rozmazy śluzówki pochwy by monitorować cykl estrogenowy w trakcie okresu poprzedzającego krycie, ciągle monitorując go w okresie krycia, aż do momentu jego zaistnienia. Przy pozyskiwaniu komórek pochwy / szyjki macicy należy zachować ostrożność, aby nie dopuścić do zaburzeń błony śluzowej, które mogłyby wywoływać ciążę rzekomą (7)(8).
|
Parametry potomstwa
|
39.
|
Począwszy od dnia 0 ciąży należy odnotowywać i liczyć czas jej trwania. Każdy miot należy zbadać tak szybko po urodzeniu jak to możliwe, w celu ustalenia liczby i płci młodych, płodów urodzonych martwo, płodów urodzonych żywo, słabowitych młodych w miocie (młodych o znacznie mniejszych rozmiarach niż odpowiadające młode kontrolne) oraz obecności poważnych nieprawidłowości.
|
|
40.
|
Należy policzyć i ustalić płeć żywych młodych, a następnie zważyć mioty w ciągu 24 godzin od porodu (w dniu 0 lub 1. dniu po porodzie) oraz co najmniej 4. i 13. dnia po porodzie. W dodatku do obserwacji opisanych w pkt 35 należy odnotować wszelkie nieprawidłowe zachowania potomstwa.
|
|
41.
|
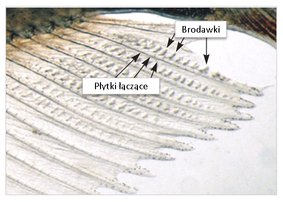
Odległość anogenitalną każdego młodego należy mierzyć w tym samym dniu po urodzeniu – między 0. a 4. dniem po urodzeniu. W dniu pomiaru odległości anogenitalnej należy dokonać pomiaru masy ciała młodego, a odległość anogenitalną należy znormalizować w odniesieniu do pomiaru wielkości młodego, najlepiej jako pierwiastek kwadratowy masy ciała (9). Liczbę brodawek sutkowych / otoczek brodawek u młodych samców należy mierzyć, według zaleceń zawartych w wytycznej OECD nr 151, w 12. lub 13. dnia po urodzeniu (10).
|
Biochemia kliniczna
|
42.
|
Próbki krwi z określonego miejsca pobiera się według następującego planu:
|
—
|
od co najmniej dwóch młodych na miot 4. dnia po urodzeniu jeżeli liczba młodych na to pozwala (zob. pkt 33–34);
|
|
—
|
od wszystkich matek oraz co najmniej dwóch młodych z każdego miotu przy zakończeniu badania 13. dnia; oraz
|
|
—
|
od wszystkich dorosłych samców w dniu zakończenia badań.
|
|
Wszystkie próbki krwi przechowuje się w odpowiednich warunkach. Próbki krwi pobrane od 13. dniowych młodych oraz dorosłych samców ocenia się pod kątem poziomów surowicy hormonów tarczycy (T4). Jeżeli uzna się za stosowne, przeprowadzana jest dalsza ocena T4 w próbkach krwi od matek oraz 4. dniowych młodych. Można opcjonalnie przeprowadzić pomiar innych hormonów. Krew młodych z każdego miotu można połączyć do celów analizy hormonów tarczycy. Hormony tarczycy (T4 i TSH) zaleca się mierzyć jako „całkowite”.
|
43.
|
Na zmienność i wartości bezwzględne stężeń przy oznaczaniu hormonów mogą mieć wpływ następujące czynniki:
|
—
|
czas uśmiercenia zwierzęcia w związku z dobową zmiennością stężeń hormonów;
|
|
—
|
metoda uśmiercenia zastosowana w celu uniknięcia zbędnego stresu dla zwierząt, mogącego wpłynąć na stężenia hormonów;
|
|
—
|
zestawy do oznaczania hormonów, których krzywe standardowe mogą się różnić.
|
|
|
44.
|
Próbki osocza przeznaczone konkretnie do oznaczenia poziomu hormonów należy pobierać w porównywalnej porze dnia. Wartości liczbowe uzyskane w toku analizy stężeń hormonów różnią się w zależności od zastosowanego dostępnego w handlu zestawu testowego.
|
Patologia
Pełne rozpoznanie histopatologiczne
|
45.
|
W momencie uśmiercenia lub śmierci w okresie trwania badania dorosłe zwierzęta należy zbadać makroskopowo pod kątem nieprawidłowości lub zmian patologicznych. Szczególną uwagę należy zwrócić na organy układu rozrodczego. Należy odnotować liczbę miejsc zagnieżdżenia. Rozmazy śluzówki pochwy należy zbadać z rana w dniu autopsji, aby określić stadium cyklu estrogenowego oraz umożliwić korelację z histopatologią jajników.
|
|
46.
|
Jądra i najądrza, a także gruczoł krokowy i kanaliki nasienne z całymi gruczołami koagulującymi wszystkich dorosłych samców należy, w stosownym przypadku, wyczyścić z wszelkich przylegających tkanek, a ich mokrą masę, a ich mokrą masę należy pobrać jak najszybciej po dysekcji, aby zapobiec jej wyschnięciu. Ponadto opcjonalne masy narządów mogą w przypadku samców obejmować zespół mięśni dźwigacza odbytu i mięśni opuszkowo-jamistych, gruczoły Cowpera i żołądź, a w przypadku samic parę jajników (mokra masa) i macicę (wraz z szyjką); jeżeli są uwzględnione, masy te należy określić jak najszybciej po dysekcji.
|
|
47.
|
Martwe młode i młode zabite 13. dnia po porodzie lub wkrótce potem należy co najmniej przebadać zewnętrznie z zachowaniem ostrożności pod kątem poważnych nieprawidłowości. Szczególną uwagę należy zwrócić na zewnętrzne narządy rozrodcze, które należy zbadać pod kątem zmian rozwojowych. W 13. dniu należy zachować tarczycę jednego samca i jednej młodej samicy na miot.
|
|
48.
|
Należy zachować jajniki, jądra, płciowe narządy dodatkowe (macicę wraz z szyjką, najądrza, gruczoł krokowy, kanaliki nasienne i gruczoły koagulujące), tarczycę i wszystkie narządy wykazujące makroskopowe zmiany patologiczne wszystkich dorosłych zwierząt. Do rutynowych badań jąder i najądrzy nie zaleca się utrwalania w formalinie. Akceptowalną metodą w przypadku tych tkanek jest wykorzystanie utrwalacza Bouina lub zmodyfikowanego utrwalacza Davidsona (11). Błonę białawą trzeba delikatnie i płytko przekłuć igłą na obu biegunach narządu w celu umożliwienia szybkiego wniknięcia płynu utrwalającego.
|
Histopatologia
|
49.
|
Należy wykonać szczegółowe badanie histologiczne jajników, jąder i najądrzy (ze szczególnym naciskiem na stadia spermatogenezy i histopatologię śródmiąższowej struktury komórek jąder) zwierząt znajdujących się w grupie otrzymującej najwyższą dawkę oraz zwierząt znajdujących się w grupie kontrolnej. Można w razie potrzeby zbadać pozostałe zachowane organy, w tym tarczyce pochodzące od młodych i dorosłych zwierząt. Masę tarczycy można określić po utrwaleniu. Należy zachować dużą ostrożność przy okrawaniu i wykonywać je dopiero po utrwaleniu, w celu uniknięcia uszkodzenia tkanek. Mogłoby ono zakłócić analizę histopatologiczną. Badania powinny być rozciągnięte na inne grupy badane, gdy w grupie otrzymującej najwyższe dawki obserwuje się zmiany. W wytycznych dotyczących histopatologii (11) podano dodatkowe informacje dotyczące wycinania, utrwalania, dzielenia i histopatologii tkanek układu hormonalnego.
|
DANE I SPRAWOZDAWCZOŚĆ
Dane
|
50.
|
Należy podać dane każdego zwierzęcia. Ponadto wszystkie dane należy podsumować w formie tabeli wykazującej, w odniesieniu do każdej grupy badanej i każdego pokolenia, liczbę zwierząt na początku badania, liczbę zwierząt, które znaleziono martwe podczas badania lub zwierząt uśmierconych ze względów humanitarnych, czas śmierci lub humanitarnego uśmiercenia, liczbę zwierząt płodnych, liczbę ciężarnych samic, liczbę zwierząt wykazujących zaobserwowane oznaki toksyczności, z podaniem czasu ich pojawienia się, w tym czasu trwania, ciężkości wszelkich skutków toksyczności, rodzajów zmian histopatologicznych oraz wszelkich odnośnych danych miotów. W dodatku 3 przedstawiono podsumowanie w formie tabeli, które okazało się być bardzo przydatne przy ocenianiu efektu reprodukcyjnego/rozwojowego.
|
|
51.
|
Ze względu na ograniczone wymiary badania, analizy statystyczne w postaci testów na „istotność” mają ograniczoną wartość dla wielu punktów końcowych, zwłaszcza reprodukcyjnych punktów końcowych. Jeżeli wykorzystywane są analizy statystyczne, wybrana metoda powinna być odpowiednia dla rozkładu badanej zmiennej i powinna być wybrana przed rozpoczęciem badania. Analiza statystyczna odległości anogenitalnej i zatrzymywania płynu w brodawkach powinna być oparta na danych dotyczących poszczególnych młodych z uwzględnieniem wpływu na miot. W stosownych przypadkach za jednostkę analityczną uznaje się miot. Analiza statystyczna masy ciała młodych powinna być oparta na danych dotyczących poszczególnych młodych z uwzględnieniem liczebności miotu. Ze względu na małą wielkość grupy, wykorzystanie historycznych danych dotyczących kontroli (np. dotyczących wielkości miotu), w przypadku gdy są one dostępne, może być również przydatne jako pomoc w interpretacji badania.
|
Ocena wyników
|
52.
|
Wyniki tego badania toksyczności powinny być oceniane w kategoriach zaobserwowanych skutków, wyników autopsji i badań mikroskopowych. Ocena obejmuje zależność pomiędzy dawką badanej substancji chemicznej a obecnością lub brakiem, częstością i ciężkością nieprawidłowości, w tym poważnych zmian patologicznych, wskazanymi organami docelowymi, bezpłodnością, nieprawidłowościami klinicznymi, zmienionymi funkcjami rozrodczymi i funkcjonowaniem miotów, zmianami masy ciała, wpływem na śmiertelność i wszelkimi innymi skutkami toksycznymi.
|
|
53.
|
Ze względu na krótki okres poddania działaniu substancji w odniesieniu do samców, przy ocenie wpływu na rozrodczość u samców należy uwzględnić histopatologię jąder i najądrzy oraz dane dotyczące płodności. Wykorzystanie historycznych danych dotyczących kontroli odnoszących się do reprodukcji/rozwoju (np. dotyczących wielkości miotu, odległości anogenitalnej, zatrzymywania płynu w brodawkach, poziomu T4 w surowicy), w przypadku gdy są one dostępne, może być również przydatne jako pomoc w interpretacji badania.
|
|
54.
|
Proponuje się, aby do celów kontroli jakości zbierać historyczne dane kontrolne i obliczać współczynniki zmienności dla danych liczbowych, szczególnie w odniesieniu do parametrów związanych z wykrywaniem substancji zakłócających funkcjonowanie układu hormonalnego. Dane te można wykorzystać do celów porównawczych przy ocenie bieżących badań.
|
Sprawozdanie z badania
|
55.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Badana substancja chemiczna:
|
—
|
źródło, numer partii, termin przydatności, jeżeli są znane;
|
|
—
|
stabilność badanej substancji chemicznej, jeżeli jest znana.
|
|
|
|
substancja jednoskładnikowa:
|
—
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń, stosownie do przypadku i jeśli jest to praktycznie wykonalne, itp.
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszaniny:
|
—
|
opisane w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników.
|
|
|
|
Nośnik (w stosownych przypadkach):
|
—
|
uzasadnienie wyboru nośnika, jeśli jest inny niż woda.
|
|
|
|
Zwierzęta doświadczalne:
|
—
|
wykorzystany gatunek/szczep;
|
|
—
|
liczba, wiek i płeć zwierząt;
|
|
—
|
źródło pochodzenia, warunki utrzymywania, pasza itp.;
|
|
—
|
masa ciała poszczególnych zwierząt na początku badania;
|
|
—
|
uzasadnienie wyboru gatunku, jeśli jest inny niż szczur.
|
|
|
|
Warunki badania:
|
—
|
uzasadnienie wyboru poziomu dawkowania;
|
|
—
|
szczegóły dotyczące postaci użytkowej badanej substancji chemicznej/przygotowania pokarmu, osiągnięte stężenie, stabilność i jednorodność preparatu;
|
|
—
|
szczegółowe informacje dotyczące podawania badanej substancji chemicznej;
|
|
—
|
wskaźnik konwersji stężenia badanej substancji chemicznej w pokarmie / wodzie pitnej (ppm) do dawki rzeczywistej (mg/kg masy ciała/dzień), jeśli ma to zastosowanie;
|
|
—
|
szczegółowe informacje dotyczące jakości pokarmu i wody;
|
|
—
|
szczegółowy opis procedury randomizacji stosowanej do doboru młodych do eliminacji w przypadku, gdy się jej dokonuje.
|
|
|
|
Wyniki:
|
—
|
masa ciała / zmiany masy ciała,
|
|
—
|
spożycie pokarmu, spożycie wody (jeżeli dane są dostępne);
|
|
—
|
dane dotyczące efektu toksycznego ze względu na płeć i dawkę, włączając płodność, ciążę i wszelkie inne objawy toksyczności;
|
|
—
|
skutki toksyczne lub inne wywierane na reprodukcję, potomstwo, wzrost pourodzeniowy itp.;
|
|
—
|
rodzaj, nasilenie i czas trwania obserwowanych objawów klinicznych (tak odwracalnych, jak i nieodwracalnych);
|
|
—
|
liczba dorosłych samic, u których występuje normalny lub nienormalny cykl estrogenowy i czas trwania cyklu;
|
|
—
|
liczba młodych urodzonych żywo i strat poimplantacyjnych,
|
|
—
|
dane dotyczące masy ciała młodego;
|
|
—
|
odległość anogenitalna u wszystkich młodych (oraz masa ciała na dzień pomiaru odległości anogenitalnej);
|
|
—
|
zatrzymywanie płynu w brodawkach u młodych płci męskiej;
|
|
—
|
poziom hormonów tarczycy, młode w 13. dniu żucia i dorosłe samce (oraz matki i młode w 4. dniu życia, jeśli dokonano pomiaru)
|
|
—
|
liczba młodych z wyraźnie widocznymi nieprawidłowościami, ocena wzrokowa zewnętrznych narządów płciowych, liczba słabowitych młodych w miocie;
|
|
—
|
czas zgonu podczas badania lub informacja o tym, czy zwierzęta przetrwały do czasu zakończenia badania;
|
|
—
|
liczba zagnieżdżeń, liczebność miotu i masa poszczególnych młodych w miocie w momencie rejestracji;
|
|
—
|
masa ciała w chwili uśmiercenia oraz masa narządów u zwierząt rodzicielskich;
|
|
—
|
szczegółowy opis wyników badania histopatologicznego;
|
|
—
|
dane dotyczące absorpcji (jeżeli są dostępne);
|
|
—
|
w stosownych przypadkach statystyczne opracowanie wyników.
|
|
Omówienie wyników.
Wnioski
|
Interpretacja wyników
|
56.
|
Badanie dostarczy ocen toksyczności reprodukcyjnej/rozwojowej związanej z zastosowaniem powtarzanego dawkowania (zob. pkt 5 i 6). Może ono stanowić wskazówkę co do potrzeby prowadzenia dalszych badań i stanowić wskazówkę przy projektowaniu kolejnych badań. Przy interpretowaniu wyników dotyczących rozrodczości i rozwoju pomocny może być dokument zawierający wytyczne OECD nr 43 (12). Wytyczne OECD nr 106 w sprawie oceny histologicznej badań endokrynologicznych i reprodukcyjnych u gryzoni („OECD Guidance Document No 106 on Histologic Evaluation of Endocrine and Reproductive Tests in Rodents”) (11) zawierają informacje na temat przygotowania i oceny narządów (endokrynologicznych) i rozmazów śluzówki pochwy, które mogą być pomocne w odniesieniu do tej wytycznej dotyczącej badań.
|
BIBLIOGRAFIA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokio, 27–29 października 1992 Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10–11 marca 1998 r. Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. [W:] Environment, Health and Safety Publications, Series on Testing and Assessment (nr 217). Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. [w:] seria dotycząca badań i oceny nr 19. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 150. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. i Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies. „Birth Defects Research” część B nr 80 (2), s. 84–97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, w: C.R. Auston i R.V. Short, (red.), Reproduction in Mammals, t. 1 Germ Cells and Fertilization. Cambridge, Nowy Jork.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. i Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights. „Reproductive Toxicology” nr 13, s. 383–390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study, [W:] Environment, Health and Safety Publications, Series on Testing and Assessment (nr 151). Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 106. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. [W:] Environment, Health and Safety Publications, Series on Testing and Assessment (nr 43). Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
Dodatek 1
DEFINICJE (ZOB. RÓWNIEŻ WYTYCZNA OECD NR 150 (6))
Działanie androgenowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu androgenowego (np. testosteronu) w organizmach ssaków.
Działanie antyandrogenowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu androgenowego (np. testosteronu) w organizmach ssaków.
Działanie antyestrogenowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu estrogenowego (np. 17ß-estradiolu) w organizmach ssaków.
Działanie antytyroidowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu tarczycy (np. T3) w organizmach ssaków.
Substancja chemiczna oznacza substancję lub mieszaninę.
Toksyczność rozwojowa: objawy toksyczności reprodukcyjnej, reprezentującej zaburzenia przedporodowe, okołoporodowe, poporodowe, strukturalne lub czynnościowe u potomstwa.
Dawkowanie jest terminem ogólnym obejmującym dawkę, częstotliwość i czas trwania dawkowania.
Dawka jest to ilość podawanej badanej substancji chemicznej. Dawkę wyraża się jako masę badanej substancji chemicznej na jednostkę masy ciała zwierzęcia doświadczalnego na dzień (np. mg/kg masy ciała/dzień) lub jako stałe stężenie w diecie.
Wyraźna toksyczność jest to określenie ogólne opisujące wyraźne oznaki toksyczności występujące po podaniu badanej substancji chemicznej. Powinny one być wystarczające do oceny ryzyka i mieć taki charakter, że można oczekiwać, iż zwiększenie podawanej dawki spowoduje pojawienie się silnych oznak toksyczności i prawdopodobną śmiertelność.
Upośledzenie płodności oznacza zaburzenie funkcji reprodukcyjnych lub zdolności reprodukcyjnych samców lub samic.
Toksyczność matczyna: niekorzystny skutek dla ciężarnych samic, występujący swoiście (skutek bezpośredni) lub nieswoiście (skutek pośredni).
NOAEL jest to skrót oznaczający poziom dawkowania, przy którym nie obserwuje się szkodliwych zmian. Jest to najwyższy poziom dawkowania, przy którym nie obserwuje się szkodliwych efektów związanych z podawaniem substancji na skutek jej podawania.
Działanie estrogenowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu estrogenowego (np. 17ß-estradiolu) w organizmach ssaków.
Toksyczność reprodukcyjna oznacza szkodliwy wpływ na potomstwo lub upośledzenie funkcji lub zdolności reprodukcyjnych u samców i samic.
Badana substancja chemiczna oznacza każdą substancję lub mieszaninę badaną za pomocą niniejszej metody badawczej.
Działanie tyroidowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu tarczycy (np. T3) w organizmach ssaków.
Walidacja jest to proces naukowy mający na celu scharakteryzowanie wymogów operacyjnych i ograniczeń metody badawczej i wykazanie jej wiarygodności oraz odpowiedniości do danego celu.
Dodatek 2
SCHEMAT EKSPERYMENTALNEGO PLANU WSKAZUJĄCY MAKSYMALNY CZAS TRWANIA BADANIA, OPARTY NA PEŁNYM 14-DNIOWYM OKRESIE KRYCIA

Dodatek 3
TABELARYCZNE SPRAWOZDANIE PODSUMOWUJĄCE WPŁYW NA REPRODUKCJĘ/ROZWÓJ
|
OBSERWACJE
|
WARTOŚCI
|
|
|
|
Dawkowanie (jednostki)
|
0 (kontrola)
|
…
|
…
|
…
|
…
|
|
Zapoczątkowane pary (N)
|
|
|
|
|
|
|
Cykl estrogenowy (co najmniej średnia długość i częstość występowania nieregularnych cykli)
|
|
|
|
|
|
|
Samice wykazujące oznaki kopulacji (N)
|
|
|
|
|
|
|
Samice zachodzące w ciążę (N)
|
|
|
|
|
|
|
Dni poczęcia 1–5 (N)
|
|
|
|
|
|
|
Dni poczęcia 6–… (21) (N)
|
|
|
|
|
|
|
Ciąża ≤ 21 dni (N)
|
|
|
|
|
|
|
Ciąża = 22 dni (N)
|
|
|
|
|
|
|
Ciąża ≥ 23 dni (N)
|
|
|
|
|
|
|
Matki z urodzonymi żywymi młodymi (N)
|
|
|
|
|
|
|
Matki z żywymi młodymi w 4. dniu po porodzie (N)
|
|
|
|
|
|
|
Implanty/matka (średnia)
|
|
|
|
|
|
|
Żywe młode/matkę przy porodzie (średnia)
|
|
|
|
|
|
|
Żywe młode/matka w 4 dniu (średnia)
|
|
|
|
|
|
|
Proporcja płci (samiec/samica) przy porodzie (średnia)
|
|
|
|
|
|
|
Proporcja płci (samiec/samica) w 4 dniu (średnia)
|
|
|
|
|
|
|
Masa miotu przy porodzie (średnia)
|
|
|
|
|
|
|
Masa miotu w 4 dniu (średnia)
|
|
|
|
|
|
|
Masa ciała młodego przy porodzie (średnia)
|
|
|
|
|
|
|
Masa ciała młodego przy pomiarze odległości anogenitalnej (średnia dla samców, średnia dla samic)
|
|
|
|
|
|
|
Odległość anogenitalna młodego w tym samym dniu po urodzeniu, urodzenie – 4. dzień (średnia dla samców, średnia dla samic, zaznaczyć dzień po urodzeniu)
|
|
|
|
|
|
|
Masa ciała młodego w 4 dniu (średnia)
|
|
|
|
|
|
|
Zatrzymywanie płynu w brodawkach u młodych płci męskiej w 13 dniu (średnia)
|
|
|
|
|
|
|
Masa ciała młodego w 13 dniu (średnia)
|
|
|
|
|
|
|
|
|
MŁODE Z ZABURZENIAMI
|
|
Matki z 0
|
|
|
|
|
|
|
Matki z 1
|
|
|
|
|
|
|
Matki z ≥ 2
|
|
|
|
|
|
|
|
|
UTRATA POTOMSTWA
|
|
|
|
Przedurodzeniowe/postimplantacyjne (implantacje minus żywe urodzenia)
|
|
Samice z 0
|
|
|
|
|
|
|
Samice z 1
|
|
|
|
|
|
|
Samice z 2
|
|
|
|
|
|
|
Samice z ≥ 3
|
|
|
|
|
|
|
|
|
Pourodzeniowe (żywe urodzenia minus żywe 13. dnia po urodzeniu)
|
|
Samice z 0
|
|
|
|
|
|
|
Samice z 1
|
|
|
|
|
|
|
Samice z 2
|
|
|
|
|
|
|
Samice z ≥ 3
|
|
|
|
|
|
B.64 POŁĄCZONE BADANIE Z UŻYCIEM DAWKI POWTARZANEJ I BADANIE KLASYFIKACYJNE TOKSYCZNOŚCI REPRODUKCYJNEJ/ROZWOJOWEJ
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej dotyczącej badań OECD nr 422 (2016). Okresowo dokonuje się przeglądu wytycznych OECD dotyczących badania substancji chemicznych pod kątem postępu naukowego. Pierwotna wytyczna dotycząca badań klasyfikacyjnych nr 422 została przyjęta w 1996 r. na podstawie protokołu „Połączone badanie z użyciem dawki powtarzanej i badanie klasyfikacyjne reprodukcyjne/rozwojowe (Combined Repeat Dose and Reproductive/Developmental Screening Test)” omówionego na dwóch posiedzeniach ekspertów: w Londynie w 1990 r. (1) i w Tokio w 1992 r. (2).
|
|
2.
|
Niniejsza metoda badawcza łączy część dotyczącą badań klasyfikacyjnych toksyczności reprodukcyjnej/rozwojowej, która opiera się na doświadczeniu zdobytym w państwach członkowskich w związku ze stosowaniem pierwotnej metody w odniesieniu do istniejących substancji chemicznych produkowanych w dużych objętościach oraz w badaniach eksploracyjnych z substancjami służącymi do kontroli dodatniej (3) (4), oraz część dotyczącą badania toksyczności z użyciem dawki powtarzanej, zgodnie z wytyczną dotyczącą badań OECD nr 407 (28-dniowe badanie toksyczności doustnej wywołanej powtarzanym dawkowaniem u gryzoni, odpowiadająca rozdziałowi B.7 niniejszego załącznika).
|
|
3.
|
Niniejsza metoda badawcza została zaktualizowana o właściwe punkty końcowe dotyczące substancji zaburzającej funkcjonowanie układu hormonalnego w ramach działań następczych do działania o wysokim priorytecie podjętego przez OECD w 1998 r., mającego na celu zmianę istniejących wytycznych i opracowanie nowych wytycznych dotyczących badań klasyfikacyjnych i badania potencjalnych substancji zaburzających funkcjonowanie układu hormonalnego (5). W tym kontekście wytyczną dotyczącą badań nr 407 (opowiadającą rozdziałowi B.7 niniejszego załącznika) wzbogacono w 2008 r. o parametry odpowiednie do wykrywania aktywności endokrynologicznej badanych substancji chemicznych. Celem aktualizacji wytycznej dotyczącej badań nr 422 było uwzględnienie niektórych właściwych punktów końcowych substancji zaburzającej funkcjonowanie układu hormonalnego w badaniach klasyfikacyjnych TG, gdzie okresy narażenia obejmują niektóre wrażliwe okresy w rozwoju (okresy przed lub wcześnie po urodzeniu).
|
|
4.
|
Wybrane dodatkowe właściwe punkty końcowe dotyczące substancji zaburzającej funkcjonowanie układu hormonalnego, stanowiące również część wytycznej dotyczącej badań nr 443 (Rozszerzone badanie toksyczności reprodukcyjnej w jednym pokoleniu, odpowiadająca rozdziałowi B.56 niniejszego załącznika), zostały włączone do wytycznej dotyczącej badań nr 422 na podstawie studium wykonalności dotyczącego kwestii naukowych i technicznych związanych z ich włączeniem, jak również możliwych dostosowań projektu badań potrzebnych do ich włączenia (6).
|
|
5.
|
Niniejsza metoda badawcza ma na celu wygenerowanie ograniczonej ilości informacji dotyczących wpływu badanej substancji chemicznej na efektywność reprodukcyjną samców i samic, taką jak funkcja gonad, zachowanie kopulacyjne, zapłodnienie, rozwój jaja płodowego i poród. Nie jest ona metodą alternatywną dla istniejących metod badań B.31, B.34, B.35 lub B.56, ani ich nie zastępuje.
|
ZAŁOŻENIA WSTĘPNE
|
6.
|
Przy szacowaniu i ocenie właściwości toksycznych badanej substancji chemicznej oznaczenie toksyczności doustnej przy zastosowaniu powtarzanego dawkowania można prowadzić po uzyskaniu początkowej informacji o toksyczności na podstawie badań toksyczności ostrej. Badanie to dostarcza informacji na temat możliwych zagrożeń dla zdrowia, które mogą wynikać z powtarzającego się narażenia przez stosunkowo ograniczony czas. Metoda obejmuje podstawowe badanie toksyczności wywołanej powtarzanym dawkowaniem, które można przeprowadzić w przypadku substancji chemicznych, dla których nie ma obowiązku wykonania badania 90-dniowego (np. wtedy, gdy wielkość produkcji nie przekracza pewnych limitów) lub jako badanie wstępne do badania długoterminowego. Przy prowadzeniu badania należy przestrzegać zasad i ustaleń określonych w dokumencie zawierającym wytyczne OECD nr 19 „Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations” (7) (Wytyczne dotyczące uznawania, oceny i wykorzystywania objawów klinicznych jako punktów humanitarnego zakończenia w odniesieniu do zwierząt doświadczalnych wykorzystywanych w ocenach bezpieczeństwa).
|
|
7.
|
Ponadto obejmuje ono badanie klasyfikacyjne toksyczności reprodukcyjnej/rozwojowej, a zatem może być również wykorzystywane w celu dostarczenia wstępnych informacji na temat możliwych skutków dla sprawności reprodukcyjnej samców i samic, takich jak funkcja gonad, zachowanie kopulacyjne, zapłodnienie, rozwój jaja płodowego i poród, albo na wczesnym etapie oceny właściwości toksykologicznych badanych substancji chemicznych lub na przedmiotowe badane substancje chemiczne. Niniejsza metoda badawcza nie zapewnia pełnych informacji na temat wszystkich aspektów reprodukcji i rozwoju. W szczególności oferuje jedynie ograniczone środki wykrywania poporodowych przejawów narażenia prenatalnego lub skutków, które mogą być wywołane podczas narażenia postnatalnego. Ze względu (między innymi) na selektywność punktów końcowych oraz krótki czas trwania badania, metoda ta nie dostarczy dowodów na definitywne stwierdzenie braku skutków reprodukcyjnych/rozwojowych. Ponadto wobec braku danych z innych badań toksyczności reprodukcyjnej/rozwojowej, wyniki dodatnie są przydatne do wstępnej oceny zagrożeń i przyczyniają się do podejmowania decyzji dotyczących konieczności i terminów dodatkowych badań.
|
|
8.
|
Wyniki uzyskane za pomocą parametrów związanych z układem hormonalnym należy rozpatrywać w kontekście dokumentu „OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals” („Ramy koncepcyjne OECD dotyczące testowania i oceny substancji chemicznych zaburzających funkcjonowanie układu hormonalnego”) (8). W niniejszych ramach koncepcyjnych rozszerzona wytyczna dotyczącej badań OECD nr 422 zawarta jest na poziomie 4 jako test in vivo dostarczający danych na temat niekorzystnego wpływu na właściwe punkty końcowe dotyczące układu hormonalnego. Sygnał endokrynny może jednak nie być uważany za wystarczający dowód na to, że badana substancja chemiczna jest substancją zaburzającą funkcjonowanie układu hormonalnego.
|
|
9.
|
Metoda badawcza kładzie również nacisk na objawy neurologiczne jako określony punkt końcowy, i podkreślana jest konieczność uważnych obserwacji klinicznych zwierząt, w celu otrzymania możliwie największej ilości informacji. Metoda ta powinna identyfikować chemikalia o potencjale neurotoksycznym, które mogą również dawać podstawy do dalszego dogłębnego badania tego aspektu. Ponadto metoda ta może również w podstawowym zakresie może wskazywać na skutki dla układu immunologicznego.
|
|
10.
|
Wobec braku danych z innych badań toksyczności ogólnoustrojowej, toksyczności reprodukcyjnej/rozwojowej, neurotoksyczności lub immunotoksyczności, wyniki dodatnie są przydatne do wstępnej oceny zagrożeń i przyczyniają się do podejmowania decyzji dotyczących konieczności i terminów dodatkowych badań. Badanie może być szczególnie użyteczne jako część Screening Information Data Set (SIDS) OECD do oceny istniejących substancji chemicznych, w odniesieniu do których istnieje niewiele lub nie ma dostępnych informacji toksykologicznych i może służyć jako metoda alternatywna dla przeprowadzenia dwóch oddzielnych badań, odpowiednio toksyczności z użyciem dawki powtarzanej (wytyczna dotycząca badań OCD nr 407, odpowiadająca rozdziałowi B.7 niniejszego załącznika) i toksyczności reprodukcyjnej/rozwojowej (wytyczna dotycząca badań OECD nr 421, odpowiadająca rozdziałowi B.63 niniejszego załącznika). Może być również stosowana jako badanie ustalające zakres dawek dla bardziej ekstensywnych badań nad rozrodczością/rozwojem, lub w przypadku gdy jest to uznane za istotne w inny sposób.
|
|
11.
|
Zakłada się zasadniczo, że istnieją różnice w czułości między zwierzętami ciężarnymi a tymi niebędącymi w ciąży. W związku z tym określenie w ramach tego badania łączonego poziomów dawek, które wystarczają do oceny zarówno ogólnej toksyczności ogólnoustrojowej i specyficznej toksyczności reprodukcyjnej i rozwojowej, może być bardziej skomplikowane niż w przypadku przeprowadzenia indywidualnych badań odrębnie. Interpretacja wyników badania w odniesieniu do ogólnej toksyczności ogólnoustrojowej może być ponadto trudniejsze niż przeprowadzanie odrębnego badania dawki powtórzonej, w szczególności kiedy parametry dotyczące surowicy i histopatologii nie są poddane ocenie w tym samym punkcie czasu badania. Ze względu na te złożoności techniczne przeprowadzenie tego łączonego badania klasyfikacyjnego wymaga znacznego doświadczenia w badaniu toksyczności. Z drugiej strony badanie łączne może, poza wykorzystaniem mniejszej liczby osób, stanowić lepszy sposób odróżnienia bezpośrednich skutków dotyczących reprodukcji i rozwoju od tych, które są drugorzędne względem innych skutków (ogólnoustrojowych).
|
|
12.
|
Okres dawkowania jest dłuższy w tym badaniu niż w standardowym 28-dniowym badaniu dawki powtórzonej. Wykorzystuje się w nim jednak mniej zwierząt obojga płci w poszczególnych grupach w porównaniu z sytuacją, w której przeprowadza się standardowe 28-dniowe badanie dawki powtórzonej poza badaniem klasyfikacyjnym toksyczności reprodukcyjnej i rozwojowej.
|
|
13.
|
Niniejsza metoda badawcza zakłada doustną drogę podawania badanej substancji chemicznej. W przypadku stosowania innych dróg narażenia, może być konieczne dokonanie modyfikacji.
|
|
14.
|
Przed zastosowaniem przedmiotowej metody badawczej z użyciem mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy zastanowić się, czy zastosowanie tej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak, to dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny.
|
|
15.
|
Stosowane definicje znajdują się w dodatku 1.
|
ZASADA BADANIA
|
16.
|
Badana substancja chemiczna podawana jest w stopniowanych dawkach kilku grupom samców i samic. Samce powinny otrzymywać dawki przez co najmniej cztery tygodnie do dnia przed planowanym uśmierceniem włącznie (obejmuje to co najmniej dwa tygodnie przed kryciem, w okresie krycia i około dwóch tygodni po kryciu). Ze względu na ograniczone dawkowanie przed kryciem u samców, płodność nie może być szczególnie czułym wskaźnikiem toksyczności w odniesieniu do jąder. W związku z tym niezbędne jest szczegółowe badanie histologiczne jąder. Uważa się, że połączenie dwutygodniowego okresu dawkowania w okresie przed kojarzeniem i późniejszych obserwacji krycia/płodności z ogólnym okresem dawkowania wynoszącym co najmniej cztery tygodnie, po którym następuje szczegółowa histopatologia gonad samców, jest wystarczające do wykrycia większości wpływu na płodność i spermatogenezę samców.
|
|
17.
|
Samice powinny otrzymywać dawki przez cały czas trwania badania. Obejmuje to dwa tygodnie przed kryciem (w celu objęcia co najmniej dwóch pełnych cykli estrogenowych), zmienny czas do zapłodnienia, czas trwania ciąży i co najmniej trzynaście dni po porodzie, do dnia planowanego uśmiercenia włącznie.
|
|
18.
|
Czas trwania badania, po aklimatyzacji i ocenie cyklu estrogenowego przed dawkowaniem, zależy od efektywności samic i wynosi około 63 dni, [co najmniej 14 dni okresu przed kojarzeniem, (do) 14 dni krycia, 22 dni ciąży, 13 dni laktacji].
|
|
19.
|
Podczas okresu dawkowania każdego dnia zwierzęta są dokładnie obserwowane w celu wykrycia oznak toksyczności. Zwierzęta, które zginą lub zostaną uśmiercone w okresie badania, są poddawane autopsji, a zwierzęta, które przeżyją, są po zakończeniu badania uśmiercane i poddawane autopsji.
|
OPIS METODY
Wybór gatunku zwierząt
|
20.
|
Niniejsza metoda badawcza jest przeznaczona do stosowania z wykorzystaniem szczurów. W przypadku gdy parametry określone w TG 422 badane są z wykorzystaniem innego gatunku gryzoni, należy podać uzasadnienie. Szczur był jedynym gatunkiem wykorzystywanym w międzynarodowym programie walidacyjnym dotyczącym substancji zaburzających funkcjonowanie układu hormonalnego w oparciu o TG 407. Nie należy stosować szczepów o niskiej płodności lub powszechnie znanej wysokiej częstości występowania wad rozwojowych. Należy wykorzystać zdrowe zwierzęta dziewicze, które nie były wcześniej poddawane procedurom badawczym. Badane zwierzęta należy scharakteryzować z uwzględnieniem ich gatunku, szczepu, płci, masy i wieku. W momencie rozpoczęcia badania różnice w masie ciała wykorzystywanych zwierząt powinny być minimalne i nie powinny przekraczać ± 20 % średniej masy ciała u każdej z płci. W przypadku gdy badanie przeprowadza się jako badanie wstępne w stosunku do badania długoterminowego lub pełnopokoleniowego, zaleca się, aby w obu badaniach wykorzystywać zwierzęta tego samego szczepu i źródła.
|
Warunki utrzymywania i karmienia
|
21.
|
Wszystkie procedury powinny być zgodne z lokalnymi normami utrzymywania zwierząt laboratoryjnych. Temperatura w pomieszczeniu, w którym przetrzymuje się zwierzęta doświadczalne, powinna wynosić 22 °C (± 3 °). Wilgotność względna, poza okresem sprzątania pomieszczenia, powinna wynosić co najmniej 30 % i najlepiej byłoby, gdyby utrzymywała się na poziomie 70 %. Oświetlenie powinno być sztuczne w cyklu 12 godzin z dostępem światła i 12 godzin bez dostępu światła. Do żywienia można stosować konwencjonalne pasze laboratoryjne z nieograniczonym dostępem do wody pitnej. Na wybór paszy może mieć wpływ potrzeba zapewnienia odpowiedniej domieszki badanej substancji chemicznej w przypadku podawania jej w ramach opisywanej metody.
|
|
22.
|
Zwierzęta należy przetrzymywać razem, w małych grupach składających się z osobników tej samej płci; można je przetrzymywać pojedynczo, jeżeli ma to naukowe uzasadnienie. W przypadku przetrzymywania w grupach, w jednej klatce należy umieszczać nie więcej niż pięć zwierząt. Procedury krycia powinny być przeprowadzane w klatkach nadających się do tego celu. Ciężarne samice powinny być przetrzymywane w osobnych klatkach i mieć zapewnione materiały do budowy gniazda. Samice w okresie ciąży będą przebywać w indywidualnych klatkach ze swoim potomstwem.
|
|
23.
|
Paszę należy regularnie analizować pod kątem obecności zanieczyszczeń. Próbkę paszy należy zachować do momentu ukończenia sprawozdania.
|
Przygotowanie zwierząt
|
24.
|
Zdrowe młode dorosłe osobniki zwierząt wybiera się losowo i przypisuje do grup poddawanych działaniu substancji i do klatek. Klatki należy rozmieścić w taki sposób, aby zminimalizować potencjalny wpływ ich układu. Zwierzęta są identyfikowane jednoznacznie i przetrzymywane w klatkach przez co najmniej pięć dni przed rozpoczęciem badania, żeby umożliwić aklimatyzację do warunków laboratoryjnych.
|
Przygotowanie dawek
|
25.
|
Zaleca się, aby badana substancja chemiczna była podawana doustnie, chyba że za bardziej odpowiednią uzna się inną drogę podawania. W przypadku wybrania doustnej drogi podawania, badaną substancję chemiczną zazwyczaj podaje się za pomocą sondy; można jednak również zastosować metodę alternatywną polegającą na podaniu badanych substancji chemicznych w paszy lub w wodzie do picia.
|
|
26.
|
W razie potrzeby badaną substancję chemiczną rozpuszcza się lub przygotowuje jako zawiesinę w odpowiednim nośniku. Zaleca się, w miarę możliwości, najpierw rozważyć zastosowanie roztworu wodnego/zawiesiny, następnie roztworu/zawiesiny w oleju (np. z zastosowaniem oleju kukurydzianego), a potem możliwego roztworu w innych nośnikach. W przypadku zastosowania nośników niewodnych właściwości toksyczne danego nośnika powinny być znane. Należy określić stabilność i jednorodność badanej substancji chemicznej w nośniku.
|
PROCEDURA
Liczba i płeć zwierząt
|
27.
|
Zaleca się, aby w każdej grupie na początku znajdowało się co najmniej 10 samców i 12–13 samic. Ocena samic będzie prowadzona przed narażeniem pod kątem cykliczności estrogenowej, a do badania nie zostaną włączone zwierzęta, które nie wykażą typowych 4–5 dniowych cykli; w związku z tym zaleca się zastosowanie dodatkowych samic w celu uzyskania 10 samic w każdej grupie. Poza przypadkiem, w którym występują wyraźne efekty toksyczne, oczekuje się, że zapewni to co najmniej 8 ciężarnych samic na grupę, co zazwyczaj stanowi najniższą dopuszczalną liczbę ciężarnych samic na grupę. Celem jest wyprodukowanie odpowiedniej liczby ciąż i potomstwa w celu zapewnienia znaczącej oceny wpływu badanej substancji chemicznej na płodność, ciążę, zachowanie macierzyńskie i zwierząt ssących oraz wzrost i rozwój potomstwa F1 w okresie od chwili poczęcia do 13 dnia po porodzie. Jeśli planuje się uśmiercanie w trakcie badania, liczbę należy zwiększyć o liczbę zwierząt przeznaczonych do uśmiercenia przed zakończeniem badań. Należy rozważyć włączenie dodatkowej satelitarnej grupy pięciu zwierząt obojga płci do grupy kontrolnej i grupy otrzymującej najwyższe dawki w celu obserwacji odwracalności, trwałości lub opóźnionego występowania skutków toksycznych przez co najmniej 14 dni po zakończeniu podawania substancji. Zwierzęta wchodzące w skład grup satelitarnych nie będą kryte, a zatem nie zostaną wykorzystane w ocenie toksyczności reprodukcyjnej/rozwojowej.
|
Dawkowanie
|
28.
|
Zasadniczo należy wykorzystać co najmniej trzy grupy badane i jedną grupę kontrolną. Jeżeli nie ma dostępu do odpowiednich danych dotyczących ogólnej toksyczności, można wykonać badanie ustalające zakres (z wykorzystaniem zwierząt tego samego szczepu, pochodzących z tego samego źródła) w celu określenia dawek, które mają być zastosowane. Z wyjątkiem podawania badanej substancji chemicznej zwierzęta w grupie kontrolnej należy traktować w identyczny sposób jak zwierzęta w grupie badanej. Jeżeli przy podawaniu badanej substancji chemicznej jest stosowany nośnik, grupa kontrolna powinna otrzymać największą użytą objętość nośnika.
|
|
29.
|
Poziomy dawki powinny być wybierane, biorąc pod uwagę jakiekolwiek istniejące dostępne dane o toksyczności i dane toksykokinetczne. Należy także wziąć pod uwagę, że mogą istnieć różnice w czułości między zwierzętami w ciąży a tymi niebędącymi w ciąży. Należy wybrać taki najwyższy poziom dawkowania, który powoduje oznaki toksyczności, ale nie zgon lub widoczne cierpienie. Następnie należy dobierać poziomy dawkowania w sekwencji malejącej w taki sposób, aby ukazać wszystkie reakcje powiązane z dawkowaniem i brak niekorzystnych zmian przy najmniejszym poziomie dawkowania. Często optymalne jest zastosowanie poziomów dawkowania różniących się od dwu- do czterokrotnie, a zamiast stosować bardzo odległe (np. różniące się więcej niż 10-krotnie) poziomy dawkowania, często lepiej jest dodać czwartą grupę badaną.
|
|
30.
|
Jeśli obserwowana jest ogólna toksyczność (np. spadek masy ciała, wpływ na wątrobę, serce, płuca lub nerki itp.) bądź inne zmiany, które nie muszą być efektami toksycznymi (np. zmniejszone pobieranie pokarmu, powiększenie wątroby), należy zachować ostrożność interpretując obserwowane skutki w punktach końcowych wpływu na układ hormonalny.
|
Badanie graniczne
|
31.
|
Jeżeli badanie z podawaniem doustnym przy jednym poziomie dawki równym co najmniej 1 000 mg/kg masy ciała/dzień lub, w przypadku podawania w pożywieniu, równoważnej procentowej zawartości w pożywieniu lub wodzie pitnej (na podstawie ustalonej masy ciała), z zastosowaniem procedur opisanych dla tego badania, nie wywołuje widocznych efektów toksycznych i jeżeli nie należałoby spodziewać się toksyczności na podstawie danych dotyczących substancji strukturalnie pokrewnych, wówczas pełne badanie przy użyciu kilku poziomów dawki może nie być konieczne. Można zastosować badanie graniczne z wyjątkiem sytuacji, gdy narażenie ludzi wskazuje potrzebę zastosowania wyższego poziomu dawki. W odniesieniu do innych dróg podawania, na przykład wziewnej lub stosowania na skórę, fizyczne własności chemiczne badanych substancji chemicznych mogą często decydować o najwyższym możliwym do osiągnięcia narażeniu.
|
Podawanie dawek
|
32.
|
Zwierzętom podaje się badaną substancję chemiczną codziennie, przez siedem dni w tygodniu. Gdy badana substancja chemiczna jest podawana przez sondę, należy ją podawać zwierzętom w pojedynczej dawce przy użyciu sondy żołądkowej lub odpowiedniej kaniuli intubacyjnej. Maksymalna objętość płynu, jaką można podać jednorazowo, zależy od wielkości badanego zwierzęcia. Objętość ta nie powinna przekraczać 1 ml/100 g masy ciała, oprócz roztworów wodnych, w przypadku których można użyć 2 ml/100 g masy ciała. Poza drażniącymi lub żrącymi badanymi substancjami chemicznymi, które zwykle dają zaostrzone objawy przy wyższych stężeniach, zmienność objętości w badaniu powinna być zminimalizowana przez dostosowanie stężenia, aby zapewnić stałą objętość przy wszystkich poziomach dawki.
|
|
33.
|
W przypadku substancji chemicznych podawanych w paszy lub w wodzie do picia należy dopilnować, aby ilości podawanej badanej substancji chemicznej nie zakłócały zwykłego odżywiania lub gospodarki wodnej organizmu. Przy podawaniu badanej substancji chemicznej wraz z pokarmem można zastosować stałe stężenie w pokarmie (ppm) lub też stały poziom dawki w zależności od masy ciała zwierzęcia; należy podać zastosowany wariant. W przypadku badanej substancji chemicznej podawanej przez sondę, dawka powinna być podawana każdego dnia o podobnej porze i w razie konieczności modyfikowana co najmniej raz na tydzień, aby utrzymać stały poziom dawki w zależności od masy ciała zwierzęcia. W przypadku wykorzystania badania łączonego jako badania wstępnego przed badaniem długoterminowym lub pełnym badaniem toksyczności reprodukcyjnej, należy w obu badaniach stosować podobną dietę.
|
Harmonogram doświadczalny
|
34.
|
Podawanie dawek osobnikom obu płci należy rozpocząć co najmniej dwa tygodnie przed kryciem, co najmniej pięć dni po aklimatyzacji i po poddaniu samic badaniu przesiewowemu pod kątem normalnych cykli estrogenowych (przez dwa tygodnie przed okresem poddawania działaniu substancji). Badanie powinno być zaprogramowane w taki sposób, żeby ocena cyklu estrogenowego rozpoczynała się zaraz po osiągnięciu przez zwierzęta pełnej dojrzałości płciowej. Pod tym względem mogą występować nieznaczne różnice u różnych szczepów szczurów wykorzystywanych w różnych laboratoriach, np. szczury szczepu Sprague Dawley osiągają dojrzałość płciową po 10 tygodniach życia, a szczepu Wistar – po około 12 tygodniach. Matki posiadające potomstwo należy uśmiercić 13. dnia po porodzie lub wkrótce po tym dniu. Aby umożliwić wstrzymanie podawania zwierzętom pokarmu matkom przed pobraniem krwi (jeżeli preferuje się taki wariant), matki i ich potomstwo nie muszą być uśmiercone tego samego dnia. Dzień narodzin (tj. dzień zakończenia porodu) określa się jako dzień 0 po porodzie. Samice, u których nie zaobserwowano oznak kopulacji, uśmierca się po 24–26 dniach od ostatniego dnia okresu krycia. Dawkowanie należy kontynuować u obu płci przez okres krycia. Dawkowanie u samców należy nadal stosować po okresie krycia co najmniej do zakończenia minimalnego łącznego okresu dawkowania wynoszącego 28 dni. Następnie samce są uśmiercane lub ewentualnie zatrzymywane i kontynuuje się dawkowanie w celu ewentualnego drugiego krycia, jeśli uzna się to za stosowne.
|
|
35.
|
Codziennie dawkowanie u rodzicielskich samic powinno dalej trwać przez cały okres ciąży, co najmniej do 13. dnia po porodzie włącznie, lub do dnia poprzedzającego uśmiercenie. W przypadku badań, w których badaną substancję chemiczną podaje się drogą wziewną lub skórną, dawkowanie powinno trwać co najmniej do 19. dnia ciąży włącznie i powinno zostać wznowione najszybciej jak to możliwe, nie później niż w czwartym dniu po urodzeniu.
|
|
36.
|
Jeżeli uwzględniono zwierzęta z grupy satelitarnej przeznaczone do obserwacji uzupełniających, nie wykorzystuje się ich do krycia. Należy je trzymać przez co najmniej 14 kolejnych dni po pierwszym planowanym uśmierceniu matek bez podawania substancji w celu wykrycia opóźnionego pojawienia się, utrzymywania lub ustąpienia efektów toksyczności.
|
|
37.
|
Diagram przedstawiający harmonogram doświadczenia znajduje się w dodatku 2.
|
Cykle estrogenowe
|
38.
|
Monitorowanie cykli estrogenowych powinno odbywać się przed rozpoczęciem poddawania działaniu substancji, aby do badania wybrać samice o regularnej cykliczności (zob. pkt 27). Należy także codziennie, od rozpoczęcia poddawania działaniu substancji do potwierdzenia krycia, monitorować rozmazy śluzówki pochwy. Jeżeli istnieje obawa o wystąpienie dotkliwych skutków stresu mogących zmienić cykle estrogenowe wraz z rozpoczęciem dawkowania, laboratoria mogą przez dwa tygodnie narażać zwierzęta doświadczalne, a następnie przez co najmniej dwa tygodnie codziennie pobierać rozmazy śluzówki pochwy, by monitorować cykl estrogenowy w okresie poprzedzającym krycie i ciągle monitorować go w okresie krycia, aż do momentu zaistnienia oznak krycia. Przy pozyskiwaniu komórek pochwy/szyjki macicy należy zachować ostrożność, aby nie dopuścić do uszkodzenia błony śluzowej, które mogłoby wywoływać ciążę rzekomą (8)(9).
|
Procedura krycia
|
39.
|
Podczas tego badania należy zazwyczaj wykorzystywać krycia 1:1 (jeden samiec na jedną samicę). W przypadku wystąpienia sporadycznych zgonów samców mogą pojawić się wyjątki. Samicę należy umieścić razem z tym samym samcem na okres trwający do momentu odnotowania kopulacji lub dwa tygodnie. Każdego dnia rano samice należy poddać badaniu na obecność plemników lub czopa pochwowego. Dzień 0 ciąży określa się jako dzień, w którym potwierdzono wystąpienie krycia (wykrycie czopa pochwowego lub plemników). Jeśli kojarzenie w danej parze nie uda się, można rozważyć krycie samic samcami o dowiedzionej płodności.
|
Liczebność miotu
|
40.
|
Czwartego dnia po porodzie można skorygować liczebność każdego miotu, eliminując dodatkowe, losowo wybrane młode w miocie, by otrzymać, na ile to możliwe, cztery lub pięć samic i cztery lub pięć samców na miot, w zależności od typowej liczebności miotów w szczepie wykorzystanych szczurów. Próbki krwi należy pobrać od dwóch młodych nieprzydzielonych do żadnej kohorty, zebrać je i wykorzystać do określenia poziomów surowicy T4. Selektywna eliminacja młodych, np. oparta na masie ciała lub odległości anogenitalnej, jest nieodpowiednia. Ilekroć liczba młodych samców lub samic uniemożliwia uzyskanie czterech lub pięciu osobników każdej płci na miot, dopuszczalne jest częściowe dostosowanie (na przykład sześć samców i cztery samice). Żadne młode nie zostanie wyeliminowane, gdy liczebność miotu spadnie poniżej celu eliminacji (8 lub 10 młodych/miot) Jeżeli dostępne jest tylko jedno młode powyżej celu eliminacji, tylko jedno młode zostanie wyeliminowane i wykorzystane do pobrania krwi do ewentualnej oceny surowicy T4.
|
|
41.
|
Jeżeli liczebność miotu nie została skorygowana, dwa młode na miot uśmierca się 4. dnia po urodzeniu, a próbki krwi pobiera się do pomiaru stężeń hormonów tarczycy w surowicy. Jeżeli to możliwe, dwa młode na miot powinny stanowić młode samice, aby zachować młode samce na potrzeby ocen zatrzymywania płynu w brodawkach sutkowych, oprócz przypadku, w którym eliminacja tych młodych powoduje, że nie pozostaną żadne samice do oceny końcowej przy uśmiercaniu. Żadne młode nie zostanie wyeliminowane, gdy rozmiar miotu spadnie poniżej 8 lub 10 młodych/miot (w zależności od typowego rozmiaru miotu w szczepie wykorzystanych szczurów). Jeżeli dostępne jest tylko jedno młode powyżej typowej liczebności miotu, tylko jedno młode zostanie wyeliminowane i wykorzystane do pobrania krwi do ewentualnej oceny surowicy T4.
|
Obserwacje
|
42.
|
Ogólne obserwacje kliniczne powinny być przeprowadzane co najmniej raz dziennie, najlepiej każdego dnia o tej samej porze, biorąc pod uwagę szczytowy okres przewidywanych objawów po dawkowaniu. Należy rejestrować stan zdrowia zwierząt. Co najmniej dwa razy dziennie wszystkie zwierzęta poddaje się obserwacjom pod kątem zachorowalności i śmiertelności.
|
|
43.
|
Dla wszystkich rodzicielskich zwierząt należy przeprowadzać szczegółowe obserwacje kliniczne: jeden raz przed pierwszym narażeniem (w celu umożliwienia porównania badanych zwierząt), a następnie co najmniej raz w tygodniu. Obserwacji należy dokonywać poza klatką zwierzęcia, najlepiej w warunkach standardowych i każdego dnia o tej samej porze. Obserwacje należy starannie odnotowywać najlepiej za pomocą systemów punktacji wyraźnie określonych przez laboratorium badawcze. Należy dołożyć starań w celu dopilnowania, aby odchylenia w warunkach badania były minimalne, preferowane jest prowadzenie obserwacji przez osoby, które nie wiedzą, jaka substancja jest podawana. Odnotowane objawy powinny obejmować między innymi zmiany w skórze, sierści, oczach, błonach śluzowych, wystąpienie wydzielin i wydalin oraz aktywność autonomicznej części układu nerwowego (np. łzawienie, piloreksja, rozmiar źrenicy, zmieniony rytm oddychania). Należy odnotować także zmiany w sposobie chodzenia, postawie i reakcji na dotyk, jak również obecność ruchów klonicznych lub tonicznych, zachowań stereotypowych (np. nadmierne czyszczenie się, powtarzające się krążenie w kółko), trudny lub długotrwały poród oraz dziwne zachowania (np. samookaleczanie się, chodzenie do tyłu) (10).
|
|
44.
|
Raz w ciągu badania należy przeprowadzić ocenę reaktywności na bodźce różnego typu (np. słuchowe, wzrokowe i czucia głębokiego) (8)(9)(11), ocenę siły uchwytu (12) oraz ocenę aktywności motorycznej (13) na pięciu samcach i pięciu samicach losowo wybranych z każdej z grup. Dalsze szczegółowe informacje na temat procedur, jakie można zastosować, podano w odpowiednich odniesieniach. Można jednak także zastosować również procedury inne niż te podane w odniesieniach. W przypadku samców danych obserwacji funkcjonalnych należy dokonać pod koniec ich okresu dawkowania, krótko przed planowanym uśmierceniem ale przed pobieraniem próbek krwi do analizy pod kątem hematologii lub chemii klinicznej (zob. pkt 53–56, w tym przypis 1). W trakcie tych badań funkcjonalnych samice powinny być w fizjologicznie podobnym stanie i najlepiej badać je raz w trakcie ostatniego tygodnia laktacji (np. LD 6–13), na krótko przed planowanym uśmierceniem. W miarę możliwości należy zminimalizować czas, w którym matki i młode przebywają osobno.
|
|
45.
|
Obserwacje funkcjonalne przeprowadzane tylko raz odnośnie do końca badań mogą być pominięte, gdy badanie prowadzone jest jako badanie wstępne do późniejszego badania podchronicznego (90-dniowego) lub badania długofalowego. W takim przypadku obserwacje funkcjonalne należy zamieścić w tym badaniu uzupełniającym. Z drugiej strony, dostępność danych dotyczących obserwacji czynnościowych z tego badania z powtarzanym dawkowaniem może zwiększyć możliwości doboru poziomów dawkowania do celów późniejszego badania toksyczności podprzewlekłej lub badania długofalowego.
|
|
46.
|
Wyjątkowo można pominąć obserwacje czynnościowe również w odniesieniu do grup, które oprócz tego przejawiają oznaki toksyczności nasilone na tyle, że mogłyby znacznie zakłócić wykonanie badania funkcjonalnego.
|
|
47.
|
Począwszy od dnia 0 ciąży należy odnotowywać i liczyć czas jej trwania. Każdy miot należy zbadać tak szybko po urodzeniu jak to możliwe, w celu ustalenia liczby i płci młodych, płodów urodzonych martwo, płodów urodzonych żywo, słabowitych młodych w miocie (młodych o znacznie mniejszych rozmiarach niż odpowiadające młode kontrolne) oraz obecności poważnych nieprawidłowości.
|
|
48.
|
Należy zliczyć i ustalić płeć żywych młodych, a mioty należy zważyć w ciągu 24 godzin od porodu (w dniu 0 lub 1. dnia po porodzie) oraz co najmniej 4. i 13. dnia po porodzie. W dodatku do obserwacji dotyczących zwierząt rodzicielskich (zob. pkt 43 i 44), należy odnotować wszelkie nieprawidłowe zachowania potomstwa.
|
|
49.
|
Odległość anogenitalną każdego młodego należy mierzyć w tym samym dniu po urodzeniu – między 0. a 4. dniem po urodzeniu. W dniu pomiaru odległości anogenitalnej należy dokonać pomiaru masy ciała młodego, a odległość anogenitalną należy znormalizować w odniesieniu do pomiaru wielkości młodego, najlepiej jako pierwiastek kwadratowy masy ciała (14). Liczbę brodawek sutkowych / otoczek brodawek u młodych samców należy mierzyć, według zaleceń zawartych w wytycznej OECD nr 151, 12. lub 13. dnia po urodzeniu (15).
|
Masa ciała i spożycie pokarmu/wody
|
50.
|
Samce i samice należy zważyć w pierwszym dniu dawkowania, a następnie co najmniej raz tygodniowo oraz w momencie zakończenia dawkowania. Samice w okresie ciąży należy ważyć w dniach 0, 7, 14 i 20 oraz w ciągu 24 godzin od porodu (w dniu 0 lub 1. dniu po porodzie), a także co najmniej 4. i 13. dnia po porodzie. Obserwacje te należy umieścić w sprawozdaniu oddzielnie w odniesieniu do każdego dorosłego zwierzęcia.
|
|
51.
|
Przed kryciem, w okresie ciąży oraz laktacji spożycie pokarmu należy mierzyć co najmniej raz w tygodniu. W okresie krycia pomiar spożycia pokarmu nie jest obowiązkowy. W tych okresach należy także mierzyć spożycie wody, jeżeli badana substancja chemiczna podawana jest za pośrednictwem tego nośnika.
|
Hematologia
|
52.
|
W okresie badania na pięciu samcach i pięciu samicach losowo wybranych z każdej z grup należy jednorazowo przeprowadzić następujące analizy hematologiczne: hematokryt, stężenia hemoglobiny, liczba erytrocytów, liczba retikulocytów, całkowita i różnicowa liczba leukocytów, liczba płytek krwi i pomiar czasu krzepnięcia krwi. Jeśli badana substancja chemiczna lub jej potencjalne metabolity mają właściwości utleniające lub podejrzewa się, że mogą mieć takie właściwości, należy również przeprowadzić inne oznaczenia, w tym stężenia methemoglobiny i ciałek Heinza.
|
|
53.
|
Próbki krwi należy pobierać z wyznaczonego miejsca. Podczas pobierania próbek samice powinny być w fizjologicznie podobnym stanie. Aby uniknąć utrudnień praktycznych związanych ze zmiennością na początku ciąży, pobrania próbek krwi samic można dokonać na końcu okresu poprzedzającego krycie, ponieważ jest to metoda alternatywna do pobierania próbek tuż przed, lub w ramach procedury eutanazji zwierząt. Najlepiej, by próbki krwi samców pobierać tuż przed eutanazją zwierząt lub jako element tej procedury. Ewentualnie pobieranie próbek krwi od samców można także przeprowadzić pod koniec okresu poprzedzającego krycie, w przypadku gdy ten punkt czasowy był preferowany u samic.
|
|
54.
|
Próbki krwi należy przechowywać we właściwych warunkach.
|
Biochemia kliniczna
|
55.
|
Oznaczenia biochemii klinicznej w celu zbadania głównych efektów toksycznych w tkankach, a w szczególności wpływu na nerki i wątrobę, powinny być wykonywane na próbkach krwi pobranych od wybranych pięciu samców i pięciu samic z każdej grupy. Zalecane jest wstrzymanie podawania pożywienia zwierzętom przez noc poprzedzającą pobranie krwi (22). Badania osocza i surowicy powinny obejmować sód, potas, glukozę, cholesterol całkowity, mocznik, kreatyninę, białko całkowite i albuminę, co najmniej dwa enzymy wskazujące na skutki komórkowe w wątrobie (takie jak aminotransferaza alaninowa, aminotransferaza asparaginowa i dehydrogenaza sorbitolu) oraz kwasy żółciowe. W pewnych okolicznościach przydatnych informacji mogą dostarczyć pomiary dodatkowych enzymów (pochodzenia wątrobowego lub innego) oraz bilirubiny.
|
|
56.
|
Próbki krwi z określonego miejsca pobiera się według następującego planu:
|
—
|
od co najmniej dwóch młodych na miot 4. dnia po urodzeniu, jeżeli liczba młodych na to pozwala (zob. pkt 40–41);
|
|
—
|
od wszystkich matek oraz co najmniej dwóch młodych z każdego miotu przy zakończeniu badania 13. dnia; oraz
|
|
—
|
od wszystkich dorosłych samców w dniu zakończenia badania.
|
Wszystkie próbki krwi przechowuje się w odpowiednich warunkach. Próbki krwi pobrane od 13-dniowych młodych oraz dorosłych samców ocenia się pod kątem poziomów surowicy hormonów tarczycy (T4). Jeżeli uzna się za stosowne, przeprowadzana jest dalsza ocena T4 w próbkach krwi od matek oraz 4-dniowych młodych. Jeżeli uzna się to za stosowne, można przeprowadzić opcjonalny pomiar innych hormonów. Krew młodych z każdego miotu można połączyć do celów analizy hormonów tarczycy. Hormony tarczycy (T4 i TSH) zaleca się mierzyć jako „całkowite”.
|
|
57.
|
Fakultatywnie u pięciu przypadkowo wybranych samców w każdej grupie można przeprowadzić następujące oznaczenia analityczne w moczu w ciągu ostatniego tygodnia badań; przejrzystość, objętość, osmolalność lub ciężar właściwy, pH, białka, glukoza i komórki krwi.
|
|
58.
|
Dodatkowo należy rozważyć zastosowanie do badań surowicy znaczników ogólnego uszkodzenia tkanek. Inne oznaczenia, które powinny być przeprowadzone, jeżeli znane właściwości badanej substancji chemicznej mogą lub podejrzewa się, że mogą wpływać na pokrewne profile metaboliczne, obejmują wapń, fosforan, triglicerydy i glukozę po poście, hormony specyficzne, metahemoglobinę i cholinoesterazę. Kwestie te należy identyfikować indywidualnie dla każdego przypadku.
|
|
59.
|
Na zmienność i wartości bezwzględne stężeń przy oznaczaniu hormonów mogą mieć wpływ następujące czynniki:
|
—
|
czas uśmiercenia zwierzęcia w związku z dobową zmiennością stężeń hormonów;
|
|
—
|
metoda uśmiercenia zastosowana w celu uniknięcia zbędnego stresu dla zwierząt, mogącego wpłynąć na stężenia hormonów;
|
|
—
|
zestawy do oznaczania hormonów, których krzywe standardowe mogą się różnić.
|
|
|
60.
|
Próbki osocza przeznaczone konkretnie do oznaczenia poziomu hormonów należy pobierać w porównywalnej porze dnia. Wartości liczbowe uzyskane w toku analizy stężeń hormonów różnią się w zależności od zastosowanego dostępnego w handlu zestawu testowego.
|
|
61.
|
Jeżeli historyczne dane odniesienia są niewystarczające, należy rozważyć oznaczenie zmiennych hematologicznych i biochemii klinicznej przed rozpoczęciem podawania dawek lub, lepiej, w grupie zwierząt niebędących zwierzętami doświadczalnymi. W przypadku samic dane muszą dotyczyć zwierząt w okresie laktacji.
|
PATOLOGIA
Pełne rozpoznanie histopatologiczne
|
62.
|
Wszystkie dorosłe zwierzęta w badaniu powinny podlegać szczegółowemu pełnemu rozpoznaniu histopatologicznemu, które obejmuje staranne badanie zewnętrznej powierzchni ciała, wszystkich otworów ciała, jamy czaszkowej, klatki piersiowej i jamy brzusznej oraz ich zawartości. Szczególną uwagę należy zwrócić na organy układu rozrodczego. Należy odnotować liczbę miejsc zagnieżdżenia. Rozmazy śluzówki pochwy należy zbadać w dniu autopsji, aby określić stadium cyklu estrogenowego oraz umożliwić korelację z histopatologią organów rozrodczych samicy.
|
|
63.
|
Jądra i najądrza, a także gruczoł krokowy i kanaliki nasienne z całymi gruczołami koagulującymi wszystkich dorosłych samców należy, w stosownym przypadku, wyczyścić z wszelkich przylegających tkanek, a ich mokrą masę należy pobrać jak najszybciej po dysekcji, aby zapobiec jej wyschnięciu. Ponadto opcjonalne masy narządów mogą w przypadku samców obejmować zespół mięśni dźwigacza odbytu i mięśni opuszkowo-jamistych, gruczoły Cowpera i żołądź, a w przypadku samic parę jajników (mokra masa) i macicę (wraz z szyjką); jeżeli są uwzględnione, masy te należy określić jak najszybciej po dysekcji. Należy zachować jajniki, jądra, najądrza, płciowe narządy dodatkowe i jakiekolwiek narządy wykazujące makroskopowe zmiany patologiczne wszystkich dorosłych zwierząt.
|
|
64.
|
Gruczoły tarczowe wszystkich dorosłych samców i samic oraz jednego młodego, 13-dniowego samca i samicy powinny być zakonserwowane w najwłaściwszym środku utrwalającym dla zamierzonych kolejnych badań histopatologicznych. Masę tarczycy można określić po utrwaleniu. Należy zachować dużą ostrożność przy okrawaniu i wykonywać je dopiero po utrwaleniu, w celu uniknięcia uszkodzenia tkanek. Mogłoby ono zakłócić analizę histopatologiczną. Próbki krwi należy pobierać z wyznaczonego miejsca tuż przed eutanazją zwierząt lub jako element jej procedury i przechowywać w odpowiednich warunkach (zob. pkt 56).
|
|
65.
|
Ponadto wątroby, nerki, nadnercza, grasice, śledziony, mózgi i serca co najmniej pięciu dorosłych, losowo wybranych z każdej grupy samców i samic (poza tymi, które znaleziono w agonii lub poddano eutanazji przed zakończeniem badania) należy wyczyścić z wszelkich przylegających tkanek, a ich mokrą masę, aby uniknąć wyschnięcia, należy pobrać możliwie najszybciej po dysekcji. Następujące tkanki powinny być zakonserwowane w najwłaściwszym środku utrwalającym dla obu typów tkanek i zamierzonych kolejnych badań histopatologicznych: wszystkie poważne zmiany patologiczne, mózg (reprezentatywne obszary, w tym kresomózgowie, móżdżek i most), rdzeń kręgowy, oko, żołądek, jelito cienkie i grube (w tym kępki Peyera), wątrobę, nerki, nadnercza, śledzionę, serce, grasicę, tchawicę i płuca (zakonserwowane przez napełnienie środkiem utrwalającym i następnie zanurzenie), gonady (jądra i jajniki), płciowe narządy dodatkowe (macicę i szyjkę macicy, najądrza, prostatę, pęcherzyki nasienne wraz z gruczołami koagulującymi), pochwę, pęcherz moczowy, węzły chłonne (poza najbliższym węzłem chłonnym należy pobrać inny węzeł w zależności od doświadczenia laboratorium (16)), nerw obwodowy (kulszowy lub piszczelowy), najlepiej w bliskiej odległości od mięśnia, mięsień szkieletowy i kość szkieletową wraz ze szpikiem kostnym (wycinek lub ewentualnie świeżo pobrany szpik kostny). Zaleca się utrwalenie jąder poprzez zanurzenie w utrwalaczu Bouina lub zmodyfikowanym utrwalaczu Davidsona (16)(17)(18); nie zaleca się utrwalania tych tkanek w formalinie. Błonę białawą trzeba delikatnie i płytko przekłuć igłą na obu biegunach narządu w celu umożliwienia szybkiego wniknięcia płynu utrwalającego. Wyniki badań klinicznych i inne ustalenia mogą wskazać na konieczność zbadania dodatkowych tkanek. Organy uznane za prawdopodobne organy docelowe, w oparciu o znane właściwości substancji badanej, także należy zachować.
|
|
66.
|
Ważnych wskazówek co do efektów związanych z układem hormonalnym mogą dostarczyć badania następujących tkanek: gonad (jajników i jąder), płciowych narządów dodatkowych (macicy wraz z szyjką, najądrzy, pęcherzyków nasiennych z gruczołami koagulacyjnymi, części grzbietowo-bocznej i brzusznej prostaty), pochwy, przysadki mózgowej, męskich gruczołów mlekowych i gruczołu nadnerczowego. Zmiany w męskich gruczołach mlekowych nie są zbyt dobrze udokumentowane, ale parametr ten może być bardzo czuły na substancje o działaniu estrogenowym. Obserwacja organów/tkanek niewykazanych w pkt 65 jest opcjonalna.
|
|
67.
|
Martwe młode i młode zabite 13. dnia po porodzie lub wkrótce potem należy co najmniej przebadać zewnętrznie z zachowaniem ostrożności pod kątem poważnych nieprawidłowości. Szczególną uwagę należy zwrócić na zewnętrzne narządy rozrodcze, które należy zbadać pod kątem zmian rozwojowych.
|
Histopatologia
|
68.
|
Należy wykonać szczegółową histopatologię zachowanych organów i tkanek zwierząt wytypowanych w grupach kontrolnych i grupach otrzymujących wysoką dawkę (ze szczególnym naciskiem na stadia spermatogenezy w gonadach samców i histopatologię śródmiąższowej struktury komórek jąder). Gruczoły tarczowe pochodzące od młodych i pozostałych dorosłych zwierząt w razie potrzeby mogą być badane. Badania te powinny być rozciągnięte na inne grupy badane, jeśli w grupie otrzymującej najwyższe dawki obserwuje się zmiany związane z działaniem substancji. W wytycznych dotyczących histopatologii (10) podano dodatkowe informacje dotyczące wycinania, utrwalania, dzielenia i histopatologii tkanek układu hormonalnego.
|
|
69.
|
Należy przebadać wszystkie uszkodzenia. Aby ułatwić objaśnienie poziomów dawkowania, przy których nie obserwuje się szkodliwych zmian, należy zbadać organy docelowe w innych grupach otrzymujących dawkę, a w szczególności w grupach, które te poziomy rzekomo wykazują.
|
|
70.
|
Jeśli wykorzystuje się grupę satelitarną, należy przeprowadzić badanie histopatologiczne tych tkanek i organów, w których – w grupach otrzymujących substancję badaną – stwierdzono skutki podania substancji.
|
DANE I SPRAWOZDAWCZOŚĆ
Dane
|
71.
|
Należy podać dane każdego zwierzęcia. Ponadto wszystkie dane należy podsumować w formie tabeli wykazującej, w odniesieniu do każdej grupy badanej i każdego pokolenia, liczbę zwierząt na początku badania, liczbę zwierząt, które znaleziono martwe podczas badania lub zwierząt uśmierconych ze względów humanitarnych, czas śmierci lub uśmiercenia, liczbę zwierząt płodnych, liczbę ciężarnych samic, liczbę zwierząt wykazujących zaobserwowane oznaki toksyczności, z podaniem czasu ich pojawienia się, w tym czasu trwania, ciężkości wszelkich skutków toksyczności, rodzajów zmian histopatologicznych oraz wszelkich odnośnych danych miotów. W dodatku 3 przedstawiono podsumowanie w formie tabeli, które okazało się być bardzo przydatne przy ocenianiu efektów reprodukcyjnych/rozwojowych.
|
|
72.
|
O ile to możliwe, wyniki liczbowe należy poddawać ocenie za pomocą właściwych i ogólnie przyjętych metod statystycznych. Przy porównywaniu skutków dla różnych dawek należy unikać stosowania wielokrotnego porównywania testem t-Studenta. Metody statystyczne należy wybrać podczas planowania badania. Analiza statystyczna odległości anogenitalnej i zatrzymywania płynu w brodawkach powinna być oparta na danych dotyczących poszczególnych młodych z uwzględnieniem wpływu na miot. W stosownych przypadkach za jednostkę analityczną uznaje się miot. Analiza statystyczna masy ciała młodych powinna być oparta na danych dotyczących poszczególnych młodych z uwzględnieniem liczebności miotu. Ze względu na ograniczone wymiary badania, analizy statystyczne w postaci testów na „istotność” mają ograniczoną wartość dla wielu punktów końcowych, zwłaszcza reprodukcyjnych punktów końcowych. Niewłaściwe są niektóre z najszerzej stosowanych metod, zwłaszcza badania parametryczne dotyczące średnich. Jeżeli wykorzystywane są analizy statystyczne, wybrana metoda powinna być odpowiednia dla rozkładu badanej zmiennej i powinna być wybrana przed rozpoczęciem badania.
|
Ocena wyników
|
73.
|
Wyniki tego badania toksyczności powinny być oceniane w kategoriach zaobserwowanych skutków, wyników autopsji i badań mikroskopowych. Ocena obejmuje zależność pomiędzy dawką badanej substancji chemicznej a obecnością lub brakiem, częstością i ciężkością nieprawidłowości, w tym poważnych zmian patologicznych, wskazanymi organami docelowymi, bezpłodnością, nieprawidłowościami klinicznymi, zmienionymi funkcjami rozrodczymi i funkcjonowaniem miotów, zmianami masy ciała, wpływem na śmiertelność i wszelkimi innymi skutkami toksycznymi.
|
|
74.
|
Ze względu na krótki okres poddania działaniu substancji w odniesieniu do samców, przy ocenie wpływu na rozrodczość u samców należy uwzględnić histopatologię jąder i najądrzy oraz dane dotyczące płodności. Wykorzystanie historycznych danych dotyczących kontroli odnoszących się do reprodukcji/rozwoju (np. dotyczących wielkości miotu, odległości anogenitalnej, zatrzymywania płynu w brodawkach, poziomu T4 w surowicy), w przypadku gdy są one dostępne, może być również przydatne jako pomoc w interpretacji badania.
|
|
75.
|
Proponuje się, aby do celów kontroli jakości zbierać historyczne dane kontrolne i obliczać współczynniki zmienności dla danych liczbowych, szczególnie w odniesieniu do parametrów związanych z wykrywaniem substancji zakłócających funkcjonowanie układu hormonalnego. Dane te można wykorzystać do celów porównawczych przy ocenie bieżących badań.
|
Sprawozdanie z badania
|
76.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Badana substancja chemiczna:
|
—
|
źródło, numer partii, termin przydatności, jeżeli są znane;
|
|
—
|
stabilność badanej substancji chemicznej, jeżeli jest znana.
|
|
|
|
substancja jednoskładnikowa:
|
—
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń, stosownie do przypadku i jeśli jest to praktycznie wykonalne, itp.
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszaniny:
|
—
|
opisane w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników.
|
|
|
|
Nośnik (w stosownych przypadkach):
|
—
|
uzasadnienie wyboru nośnika, jeśli jest inny niż woda.
|
|
|
|
Zwierzęta doświadczalne:
|
—
|
wykorzystany gatunek/szczep;
|
|
—
|
liczba, wiek i płeć zwierząt;
|
|
—
|
źródło pochodzenia, warunki utrzymywania, pasza itp.;
|
|
—
|
masa ciała poszczególnych zwierząt na początku badania;
|
|
—
|
uzasadnienie wyboru gatunku, jeśli jest inny niż szczur.
|
|
|
|
Warunki badania:
|
—
|
uzasadnienie wyboru poziomu dawkowania;
|
|
—
|
szczegóły dotyczące przygotowania formy użytkowej badanej substancji chemicznej / przygotowania paszy, uzyskanego stężenia, trwałości i jednorodności preparatu,
|
|
—
|
szczegółowe informacje dotyczące podawania badanej substancji chemicznej;
|
|
—
|
wskaźnik konwersji stężenia badanej substancji chemicznej w pokarmie / wodzie pitnej (ppm) do dawki rzeczywistej (mg/kg masy ciała/dzień), jeśli ma to zastosowanie;
|
|
—
|
szczegółowe informacje dotyczące jakości pokarmu i wody;
|
|
—
|
szczegółowy opis procedury randomizacji stosowanej do doboru młodych do eliminacji w przypadku, gdy się jej dokonuje.
|
|
|
|
Wyniki:
|
—
|
masa ciała / zmiany masy ciała,
|
|
—
|
dane dotyczące spożycia pokarmu oraz wody, w stosownych przypadkach;
|
|
—
|
dane dotyczące efektu toksycznego ze względu na płeć i dawkę, włączając płodność, ciążę i wszelkie inne objawy
|
|
—
|
długość trwania ciąży; wpływ toksyczności lub innych skutków na reprodukcję, potomstwo i rozwój poporodowy itp.;
|
|
—
|
rodzaj, nasilenie i czas trwania obserwowanych objawów klinicznych (tak odwracalnych, jak i nieodwracalnych);
|
|
—
|
oceny aktywności sensorycznej, siły uchwytu i aktywności motorycznej;
|
|
—
|
badania hematologiczne wraz z odpowiednimi wartościami odniesienia;
|
|
—
|
badania biochemii klinicznej wraz z odpowiednimi wartościami odniesienia;
|
|
—
|
liczba dorosłych samic, u których występuje normalny lub nienormalny cykl estrogenowy i czas trwania cyklu;
|
|
—
|
liczba młodych urodzonych żywo i strat poimplantacyjnych;
|
|
—
|
liczba młodych z wyraźnie widocznymi nieprawidłowościami; ocena wzrokowa zewnętrznych narządów płciowych, liczba słabowitych młodych w miocie;
|
|
—
|
czas zgonu podczas badania lub informacja o tym, czy zwierzęta przetrwały do czasu zakończenia badania;
|
|
—
|
liczba zagnieżdżeń, liczebność miotu i masa poszczególnych młodych w miocie w momencie rejestracji;
|
|
—
|
dane dotyczące masy ciała młodego;
|
|
—
|
odległość anogenitalna u wszystkich młodych (oraz masa ciała na dzień pomiaru odległości anogenitalnej);
|
|
—
|
zatrzymywanie płynu w brodawkach u młodych płci męskiej;
|
|
—
|
poziom hormonów tarczycy, młode w 13. dniu życia i dorosłe samce (oraz matki i młode w 4. dniu życia, jeśli dokonano pomiaru)
|
|
—
|
masa ciała w chwili uśmiercenia oraz masa narządów u zwierząt rodzicielskich;
|
|
—
|
szczegółowy opis wyników badania histopatologicznego;
|
|
—
|
dane dotyczące absorpcji (jeżeli są dostępne);
|
|
—
|
w stosownych przypadkach statystyczne opracowanie wyników.
|
|
|
Interpretacja wyników
|
77.
|
Badanie dostarczy ocen toksyczności reprodukcyjnej/rozwojowej związanej z zastosowaniem powtarzanego dawkowania. W szczególności, ponieważ szczególną uwagę przywiązuje się do punktów końcowych toksyczności ogólnej i toksyczności reprodukcyjnej/rozwojowej, wyniki badania pozwolą na odróżnienie skutków reprodukcyjnych/rozwojowych występujących przy braku ogólnej toksyczności od występujących jedynie na poziomach, które są toksyczne również dla zwierząt rodzicielskich (zob. pkt 7–11). Może ono stanowić wskazówkę co do potrzeby prowadzenia dalszych badań i może dostarczyć wytycznych przy opracowywaniu kolejnych badań. Przy interpretowaniu wyników dotyczących rozrodczości i rozwoju pomocny może być dokument zawierający wytyczne OECD nr 43 (19). Wytyczne OECD nr 106 w sprawie oceny histologicznej badań endokrynologicznych i reprodukcyjnych u gryzoni (OECD Guidance Document 106 on Histologic Evaluation of Endocrine and Reproductive Tests in Rodents) (16) zawierają informacje na temat przygotowania i oceny narządów (endokrynologicznych) i rozmazów śluzówki pochwy, które mogą być pomocne w niniejszej metodzie badawczej.
|
BIBLIOGRAFIA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokio, 27–29 października 1992. Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu.
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. i Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD),„J. Toxicol, Sci.” nr 19, s. 141–149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. i Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide.„Fundamental and Applied Toxicol.” nr 18, s. 89–95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, Dokument dostępny na wniosek w Organizacji Współpracy Gospodarczej i Rozwoju w Paryżu, 10–11 marca 1998.
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. [W:] Environment, Health and Safety Publications, Series on Testing and Assessment (nr 217). Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 19. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M. i Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, „Birth Defects Research” część B, nr 80 (2), 84–97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, w: C.R. Auston i R.V. Short, (red.), Reproduction in Mammals: I. Germ Cells and Fertilization. Cambridge, Nowy Jork.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals, [W:] Environmental Health Criteria Document nr 60.
|
|
(11)
|
Moser V.C., McDaniel K.M. i Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz, „Toxicol. AppliedPharmacol.” nr 108, s. 267–283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. i Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice, „Neurobehav. Toxicol.” nr 1, s. 233–236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments, „Neurotoxicol. Teratol.” nr 13, s. 599–609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp i V.L. Reynolds. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, „Reproductive Toxicology” nr 13, s. 383–390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study, [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 151. Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(16)
|
OECD (2009) Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents, [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 106. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(17)
|
Hess RA i Moore BJ. (1993). Histological Methods for the Evaluation of the Testis [W:] „Methods in Reproductive Toxicology”, R.E. Chapin i J.J. Heindel (red.), Academic Press: San Diego, Kalifornia, s. 52–85.
|
|
(18)
|
J.R. Latendresse, A.R. Warbrittion, H. Jonassen, D.M. Creasy (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid, „Toxicol. Pathol.” nr 30, s. 524–533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. [W:] Environment, Health and Safety Publications, Series on Testing and Assessment (nr 43). Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption nr 150, Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
Dodatek 1
DEFINICJE (ZOB. RÓWNIEŻ WYTYCZNA OECD NR 150 (20))
Działanie androgenowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu androgenowego (np. testosteronu) w organizmach ssaków.
Działanie antyandrogenowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu androgenowego (np. testosteronu) w organizmach ssaków.
Działanie antyestrogenowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu estrogenowego (np. 17ß-estradiolu) w organizmach ssaków.
Działanie antytyroidowe jest to zdolność substancji chemicznej do hamowania działania naturalnego hormonu tarczycy (np. T3) w organizmach ssaków.
Substancja chemiczna oznacza substancję lub mieszaninę.
Toksyczność rozwojowa objawy toksyczności reprodukcyjnej, reprezentującej zaburzenia przedporodowe, okołoporodowe, poporodowe, strukturalne lub czynnościowe u potomstwa.
Dawka jest to ilość podawanej badanej substancji chemicznej. Dawkę wyraża się jako masę badanej substancji chemicznej na jednostkę masy ciała zwierzęcia doświadczalnego na dzień (np. mg/kg masy ciała/dzień) lub jako stałe stężenie w diecie.
Dawkowanie jest terminem ogólnym obejmującym dawkę, częstotliwość i czas trwania dawkowania.
Wyraźna toksyczność jest to określenie ogólne opisujące wyraźne oznaki toksyczności występujące po podaniu badanej substancji chemicznej. Powinny one być wystarczające do oceny ryzyka i mieć taki charakter, że można oczekiwać, iż zwiększenie podawanej dawki spowoduje pojawienie się silnych oznak toksyczności i prawdopodobną śmiertelność.
Upośledzenie płodności oznacza zaburzenie funkcji reprodukcyjnych lub zdolności reprodukcyjnych samców lub samic.
Toksyczność matczyna niekorzystny skutek dla ciężarnych samic, występujący swoiście (skutek bezpośredni) lub nieswoiście (skutek pośredni) i związany ze stanem ciężarnym.
NOAEL jest to skrót oznaczający poziom dawkowania, przy którym nie obserwuje się szkodliwych zmian. Jest to najwyższy poziom dawkowania, przy którym nie obserwuje się szkodliwych efektów związanych z podawaniem substancji na skutek jej podawania.
Działanie estrogenowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu estrogenowego (np. 17ß-estradiolu) w organizmach ssaków.
Toksyczność reprodukcyjna oznacza szkodliwy wpływ na potomstwo lub upośledzenie funkcji lub zdolności reprodukcyjnych u samców i samic.
Badana substancja chemiczna oznacza każdą substancję lub mieszaninę badaną za pomocą niniejszej metody badawczej.
Działanie tyroidowe jest to zdolność substancji chemicznej do działania przypominającego działanie naturalnego hormonu tarczycy (np. T3) w organizmach ssaków.
Walidacja jest to proces naukowy mający na celu scharakteryzowanie wymogów operacyjnych i ograniczeń metody badawczej i wykazanie jej wiarygodności oraz odpowiedniości do danego celu.
Dodatek 2
SCHEMAT HARMONOGRAMU DOŚWIADCZALNEGO WSKAZUJĄCY MAKSYMALNY CZAS TRWANIA BADANIA, OPARTY NA PEŁNYM 14-DNIOWYM OKRESIE KRYCIA


Dodatek 3
TABELARYCZNE SPRAWOZDANIE PODSUMOWUJĄCE WPŁYW NA REPRODUKCJĘ/ROZWÓJ
|
OBSERWACJE
|
WARTOŚCI
|
|
Dawkowanie (jednostki)
|
0 (kontrola)
|
…
|
…
|
…
|
…
|
|
Zapoczątkowane pary (N)
|
|
|
|
|
|
|
Cykl estrogenowy (co najmniej średnia długość i częstość występowania nieregularnych cykli)
|
|
|
|
|
|
|
Samice wykazujące oznaki kopulacji (N)
|
|
|
|
|
|
|
Samice zachodzące w ciążę (N)
|
|
|
|
|
|
|
Dni poczęcia 1–5 (N)
|
|
|
|
|
|
|
Dni poczęcia 6–… (23) (N)
|
|
|
|
|
|
|
Ciąża ≤ 21 dni (N)
|
|
|
|
|
|
|
Ciąża = 22 dni (N)
|
|
|
|
|
|
|
Ciąża ≥ 23 dni (N)
|
|
|
|
|
|
|
Matki z urodzonymi żywymi młodymi (N)
|
|
|
|
|
|
|
Matki z żywymi młodymi w 4. dniu po porodzie (N)
|
|
|
|
|
|
|
Implanty/matka (średnia)
|
|
|
|
|
|
|
Żywe młode/matka przy porodzie (średnia)
|
|
|
|
|
|
|
Żywe młode/matka w 4. dniu (średnia)
|
|
|
|
|
|
|
Proporcja płci (samiec/samica) przy porodzie (średnia)
|
|
|
|
|
|
|
Proporcja płci (samiec/samica) w 4 dniu (średnia)
|
|
|
|
|
|
|
Masa miotu przy porodzie (średnia)
|
|
|
|
|
|
|
Masa miotu w 4 dniu (średnia)
|
|
|
|
|
|
|
Masa ciała młodego przy porodzie (średnia)
|
|
|
|
|
|
|
Masa ciała młodego przy pomiarze odległości anogenitalnej (średnia dla samców, średnia dla samic)
|
|
|
|
|
|
|
Odległość anogenitalna młodego w tym samym dniu po urodzeniu, urodzenie – 4. dzień (średnia dla samców, średnia dla samic, zaznaczyć dzień po urodzeniu)
|
|
|
|
|
|
|
Masa ciała młodego w 4. dniu (średnia)
|
|
|
|
|
|
|
Masa ciała młodego w 13. dniu (średnia)
|
|
|
|
|
|
|
Zatrzymywanie płynu w brodawkach u młodych płci męskiej w 13 dniu (średnia)
|
|
|
|
|
|
|
MŁODE Z ZABURZENIAMI
|
|
Matki z 0
|
|
|
|
|
|
|
Matki z 1
|
|
|
|
|
|
|
Matki z ≥ 2
|
|
|
|
|
|
|
UTRATA POTOMSTWA
|
|
Przed urodzeniem (implantacje minus żywe urodzenia)
|
|
Samice z 0
|
|
|
|
|
|
|
Samice z 1
|
|
|
|
|
|
|
Samice z 2
|
|
|
|
|
|
|
Samice z ≥ 3
|
|
|
|
|
|
|
Pourodzeniu (żywe urodzenia minus żywe 13. dnia po urodzeniu)
|
|
Samice z 0
|
|
|
|
|
|
|
Samice z 1
|
|
|
|
|
|
|
Samice z 2
|
|
|
|
|
|
|
Samice z ≥ 3
|
|
|
|
|
|
B.65 METODA BADAWCZA BARIERY MEMBRANOWEJ IN VITRO W PRZYPADKU DZIAŁANIA ŻRĄCEGO NA SKÓRĘ
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej OECD nr 435 (2015). Działanie żrące na skórę oznacza powstawanie nieodwracalnych uszkodzeń skóry, które przejawiają się jako widoczna martwica naskórka i skóry właściwej, w wyniku kontaktu z badaną substancję chemiczną zgodnie z definicją Globalnie Zharmonizowanego Systemu Klasyfikacji i Oznakowania Chemikaliów (GHS) Organizacji Narodów Zjednoczonych (ONZ) (1) i z rozporządzeniem Unii Europejskiej (UE) nr 1272/2008 w sprawie klasyfikacji, oznakowania i pakowania substancji i mieszanin (CLP) (24). Niniejsza metoda badawcza, równoważna zaktualizowanej metodzie opisanej w dotyczącej badań wytycznej OECD nr 435 zapewnia metodę badawczą bariery membranowej in vitro, którą można zastosować, aby zidentyfikować żrące substancje chemiczne. W ramach metody badawczej wykorzystuje się sztuczną membranę zaprojektowaną, aby reagowała na żrące substancje chemiczne w sposób podobny do skóry zwierząt in situ.
|
|
2.
|
Tradycyjnie ocenę działania żrącego na skórę przeprowadzano, nakładając badaną substancję chemiczną na skórę żywych zwierząt i oceniając stopień uszkodzenia tanki po upłynięciu ustalonego okresu (2). Oprócz obecnie stosowanej metody, jako metody alternatywne dla standardowej procedury in vivo dotyczącej skóry królików (rozdział B.4 niniejszego załącznika, równoważnej metodzie opisanej w dotyczącej badań wytycznej OECD nr 404) stosowanej do identyfikowania żrących substancji chemicznych (2) przyjęto szereg innych metod badawczych in vitro (3)(4). W ramach zintegrowanej strategii GHS ONZ dotyczącej badania i analizowania w zakresie oceny i klasyfikacji działania żrącego na skórę oraz wytycznych OECD w sprawie zintegrowanego podejścia do badań i oceny (IATA) podrażnienia skóry / działania żrącego na skórę zaleca się stosowanie zweryfikowanych i zaakceptowanych metod badawczych in vitro w ramach modułu 3 i 4 (1)(5). W ramach IATA opisano szereg modułów grupujących źródła informacji i narzędzia służące do analizy oraz (i) przedstawiono wytyczne dotyczące sposobu integracji i wykorzystania istniejących danych pochodzących z badań i metod niebadawczych do oceny potencjałów substancji chemicznych do podrażnienia i działania żrącego na skórę, a także (ii) zaproponowano podejście, w przypadku gdy potrzebne są dalsze badania, w tym jeśli stwierdzono wyniki ujemne (5). W ramach tego podejścia modułowego można wykorzystać wyniki dodatnie z metod badawczych in vitro, aby zaklasyfikować substancję chemiczną jako żrącą bez konieczności przeprowadzania badań na zwierzętach, tym samym ograniczając i doskonaląc wykorzystanie zwierząt oraz zapobiegając bólowi i stresowi, który mógłby wystąpić, jeśli wykorzystywano by zwierzęta w tym celu.
|
|
3.
|
Zakończono badania walidacyjne dotyczące modelu bariery membranowej in vitro dostępne na rynku jako Corrositex® (6)(7)(8), wykazując ogólną dokładność w przewidywaniu działania żrącego na skórę wynoszącą 79 % (128/163), wrażliwości – 85 % (76/89) i specyficzności – 70 % (52/74) w odniesieniu do bazy danych składającej się ze 163 substancji i mieszanin (7). Na podstawie jej potwierdzonej zasadności tę zwalidowaną metodę referencyjną (VRM) zalecono w celu zastosowania jako część zintegrowanej strategii badań dotyczącej oceniania potencjału substancji chemicznych w zakresie zagrożenia działaniem żrącym na skórę (5)(7). Zanim będzie można wykorzystać do celów regulacyjnych model bariery membranowej in vitro w przypadku działania żrącego na skórę, należy określić jej wiarygodność, przydatność (dokładność) oraz ograniczenia dotyczące jej proponowanego wykorzystania, aby zapewnić jej podobieństwo do VRM (9) zgodnie z wcześniej określonymi standardami wykonywania badań (PS) (10). Wzajemne uznawanie danych OECD będzie zagwarantowane wyłącznie po poddaniu przeglądowi każdej nowej lub zaktualizowanej metody zgodnej ze standardem wykonywania badań i włączeniu jej do równoważnych wytycznych OECD dotyczących badań. Obecnie wytyczne OECD dotyczące badań nr 435 obejmują wyłącznie jedną metodę in vitro i niniejszą metodę badawczą, dostępny na rynku model Corrositex®.
|
|
4.
|
Inne metody badawcze dotyczące badania działania żrącego na skórę opierają się na wykorzystaniu zregenerowanej ludzkiej skóry (wytyczne OECD dotyczące badań nr 431) (3) oraz wyizolowanej skóry szczura (wytyczne OECD dotyczące badań nr 430) (4). Te wytyczne dotyczące badań przewidują również podzielenie żrących substancji chemicznych na trzy podkategorie GHS ONZ w odniesieniu do działania żrącego oraz trzy grupy opakowaniowe ONZ w celu transportu w związku z zagrożeniem działaniem żrącym. Te wytyczne dotyczące badań przyjęto pierwotnie w 2006 r. i zaktualizowano w 2015 r., aby odnieść się do wytycznych IATA i zaktualizować wykaz substancji służących do wykazania biegłości.
|
DEFINICJE
|
5.
|
Stosowane definicje znajdują się w dodatku.
|
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
6.
|
Badanie opisane w niniejszej metodzie badawczej umożliwia zidentyfikowanie żrących badanych substancji chemicznych oraz pozwala na podzielenie żrących badanych substancji chemicznych na podkategorie zgodnie z GHS ONZ / rozporządzeniem CLP (tabela 1). Ponadto taką metodę badawczą można wykorzystać, aby podjąć decyzje dotyczące działania żrącego i braku działania żrącego szczególnych klas substancji chemicznych, np. kwasów organicznych i nieorganicznych, pochodnych kwasów (25) oraz podstaw określonych celów badania transportu (7)(11)(12). Niniejsza metoda badawcza opisuje ogólną procedurę podobną do zwalidowanej referencyjnej metody badawczej (7). Chociaż niniejsza metoda badawcza nie dostarcza wystarczających informacji na temat podrażnienia skóry, należy zauważyć, że metoda badawcza B.46 (równoważna metodzie opisanej w dotyczącej badań wytycznej OECD nr 439) dotyczy konkretnie badania in vitro, powodującego skutki dla zdrowia w postaci podrażnienia skóry (13). Aby dokonać pełnej oceny miejscowego działania na skórę po pojedynczym narażeniu przez skórę, należy zapoznać się z wytycznymi OECD dotyczącymi zintegrowanych podejść do badań i oceny (5).
Tabela 1
Kategoria i podkategorie działania żrącego na skórę GHS ONZ (26)
|
Kategoria działania żrącego (kategoria 1) (dla organów niestosujących podkategorii)
|
Podkategorie potencjalnego działania żrącego (26) (dla organów stosujących podkategorie, w tym rozporządzenie CLP)
|
Działanie żrące w przypadku ≥ 1 z 3 zwierząt
|
|
Narażanie
|
Uwagi
|
|
Żrące
|
Działanie żrące podkategorii 1A
|
≤ 3 minuty
|
≤ 1 godzina
|
|
Działanie żrące podkategorii 1B
|
> 3 minuty / ≤ 1 godzina
|
≤ 14 dni
|
|
Działanie żrące podkategorii 1C
|
> 1 godzina / ≤ 4 godziny
|
≤ 14 dni
|
|
|
7.
|
Ograniczenie zwalidowanej metody referencyjnej (7) polega na możliwości niezakwalifikowania do badania wielu nieżrących substancji chemicznych i niektórych żrących substancji chemicznych na podstawie wyników wstępnego badania zgodności (zob. pkt 13). Uwodnione substancje chemiczne o pH mieszącym się w zakresie 4,5–8,5 często nie kwalifikują się do objęcia badaniem; jednak podczas badań na zwierzętach okazało się, że 85 % zbadanych substancji chemicznych o pH mieszczącym się w tym zakresie nie ma właściwości żrących (7). Metodę badawczą bariery membranowej in vitro można stosować w celu zbadania substancji stałych (rozpuszczalnych lub nierozpuszczalnych w wodzie), cieczy (wodnych lub niewodnych) i emulsji. Badanych substancji chemicznych niepowodujących wykrywalnej zmiany w badaniu zgodności (tj. zmiana barwy w systemie wykrywania substancji chemicznych zwalidowanej referencyjnej metody badawczej) nie można poddać badaniu metodą bariery membranowej, ale należy je badać przy użyciu innych metod badawczych.
|
ZASADA BADANIA
|
8.
|
Układ badawczy składa się z dwóch elementów: syntetycznej makrocząsteczkowej bariery biologicznej i systemu wykrywania substancji chemicznych; niniejsza metoda badawcza pozwala wykrywać za pośrednictwem systemu wykrywania substancji chemicznych uszkodzenie bariery membranowej spowodowane przez badane żrące substancje chemiczne w następstwie zastosowania badanej substancji chemicznej na powierzchni syntetycznej makrocząsteczkowej bariery membranowej (7), przypuszczalnie na podstawie takiego samego mechanizmu lub takich samych mechanizmów działania żrącego na żywą skórę.
|
|
9.
|
Penetrację bariery membranowej (lub jej przebicie) można zmierzyć wykorzystując w tym celu szereg procedur lub systemów wykrywania substancji chemicznych, w tym zmiany barwy barwnika we wskaźniku pH lub innej właściwości roztworu wskaźnika umieszczonego pod barierą.
|
|
10.
|
Należy stwierdzić, czy bariera membranowa jest ważna, tj. właściwa i wiarygodna pod względem zamierzonego zastosowania. Działanie to obejmuje zapewnienie, by poszczególne preparaty były spójne pod względem właściwości bariery, np. zdolności do zachowania bariery w przypadku nieżrących substancji chemicznych, zdolności do kategoryzacji właściwości żrących substancji chemicznych w poszczególnych podkategoriach działania żrącego GHS ONZ (1). Przypisana klasyfikacja opiera się na czasie potrzebnym, aby substancja chemiczna przeniknęła przez barierę membranową do roztworu wskaźnika.
|
WYKAZANIE BIEGŁOŚCI
|
11.
|
Przed przystąpieniem do rutynowego stosowania metody badawczej bariery membranowej in vitro zgodnej z niniejszą metodą badawczą laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo klasyfikując 12 substancji służących do wykazania biegłości, które przedstawiono w tabeli 2. W przypadku gdy wykazana substancja nie jest dostępna lub gdy jest to uzasadnione, można użyć innej substancji, w odniesieniu do której dostępne są odpowiednie dane porównawcze z badań in vivo i in vitro (np. z wykazu referencyjnych substancji chemicznych (10)), pod warunkiem zastosowania tych samych kryteriów wyboru, które opisano w tabeli 1.
Tabela 2
Substancje służące do wykazania biegłości
(27)
|
Substancja (28)
|
Numer CAS
|
Klasa chemiczna
|
Podkategoria in vivo GHS ONZ (29)
|
Podkategoria in vitro GHS ONZ (29)
|
|
Trójfluorek boru – dwuwodny
|
13319-75-0
|
Kwasy nieorganiczne
|
1A
|
1A
|
|
Kwas azotowy
|
7697-37-2
|
Kwasy nieorganiczne
|
1A
|
1A
|
|
Pentachlorek fosforu
|
10026-13-8
|
Prekursory kwasów nieorganicznych
|
1A
|
1A
|
|
Chlorek walerylu
|
638-29-9
|
Chlorki kwasowe
|
1B
|
1B
|
|
Wodorotlenek sodu
|
1 310 -73-2
|
Zasady nieorganiczne
|
1B
|
1B
|
|
1-(2-Aminoetylo)piperazyna
|
140-31-8
|
Aminy alifatyczne
|
1B
|
1B
|
|
Chlorek benzenosulfonylu
|
98-09-9
|
Chlorki kwasowe
|
1C
|
1C
|
|
N,N-Dimetylobenzyloamina
|
103-83-3
|
Anilina
|
1C
|
1C
|
|
Tetraetylenopentamina
|
112-57-2
|
Aminy alifatyczne
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenole
|
NŻ
|
NŻ
|
|
Akrylan nonylu
|
2664-55-3
|
Akrylany/metakrylany
|
NŻ
|
NŻ
|
|
Wodorowęglan sodu
|
144-55-8
|
Sole nieorganiczne
|
NŻ
|
NŻ
|
|
PROCEDURA
|
12.
|
W poniższych punktach opisano elementy i procedury metody badawczej sztucznej bariery membranowej służące ocenie działania żrącego (7)(15), oparte na obecnej zwalidowanej metodzie referencyjnej, tj. dostępnym na rynku badaniu Corrositex®. Barierę membranową oraz wskaźnik zgodności i roztwory wykorzystywane do kategoryzacji można stworzyć, przygotować lub nabyć, tak jak w przypadku zwalidowanej metody referencyjnej Corrositex®. Dostępny jest protokół próbnej metody badawczej dotyczący zwalidowanej referencyjnej metody badawczej (7). Badanie należy przeprowadzać w temperaturze otoczenia (17–25 °C), a elementy powinny spełniać następujące warunki.
|
Badanie zgodności badanej substancji chemicznej
|
13.
|
Przez przystąpieniem do badania bariery membranowej przeprowadza się badanie zgodności w celu określenia, czy system wykrywania substancji chemicznych wykrywa badaną substancję chemiczną. Jeżeli system wykrywania substancji chemicznych nie wykrywa badanej substancji chemicznej, metoda badawcza bariery membranowej nie jest odpowiednia, aby ocenić potencjalne działanie żrące tej szczególnej badanej substancji chemicznej i należy zastosować inną metodę badawczą. System wykrywania substancji chemicznych i warunki narażenia na działanie substancji powinny odzwierciedlać narażenie podczas następującego badania bariery membranowej.
|
Badanie kategorii skali czasu badanej substancji chemicznej
|
14.
|
W przypadkach stosownych dla metody badawczej badaną substancję chemiczną zakwalifikowaną na podstawie badania zgodności należy poddać badaniu kategorii skali czasu, tj. badaniu klasyfikacyjnemu pozwalającemu rozróżnić kwasy słabe i mocne lub zasady. Na przykład w zwalidowanej referencyjnej metodzie badawczej badanie kategoryzacji skali czasu wykorzystuje się, aby wskazać, którą z dwóch skal czasu należy wykorzystać w zależności od wykrycia znacznej rezerwy kwasowej lub alkalicznej. Dwie różne skale czasu wytrzymałości należy stosować w celu określenia działania żrącego i podkategorii działania żrącego na skórę GHS ONZ, w zależności od rezerwy kwasowej lub alkalicznej badanej substancji chemicznej.
|
ELEMENTY METODY BADAWCZEJ BARIERY MEMBRANOWEJ
Bariera membranowa
|
15.
|
Bariera membranowa składa się z dwóch elementów: białkowego makrocząsteczkowego wodnistego żelu i przepuszczalnej, podtrzymującej membrany. Białkowy żel nie powinien przepuszczać cieczy i substancji stałych, ale może ulec zniszczeniu i stać się przepuszczalny. W pełni zbudowaną barierę membranową należy przechowywać w określonych wcześniej warunkach, które zapobiegają pogorszeniu się jakości żelu, tj. jego wyschnięciu, wzrostowi mikroorganizmów, przesunięciu, krakingowi, które miałyby niekorzystny wpływ na jego działanie. Należy określić dopuszczalny okres przechowywania i nie używać preparatów bariery membranowej po upłynięciu tego okresu.
|
|
16.
|
Przepuszczalna podtrzymująca membrana zapewnia mechaniczne podparcie dla białkowego żelu w trakcie procesu formowania żelu i narażenia na badaną substancję chemiczną. Podtrzymująca membrana powinna zapobiegać opadaniu lub przesuwaniu się żelu oraz powinna łatwo przepuszczać wszystkie badane substancje chemiczne.
|
|
17.
|
Białkowy żel, składający się z białka, np. keratyny, kolagenu lub mieszanin białek, tworzący macierz żelową, odgrywa rolę celu badanej substancji chemicznej. Materiał białkowy umieszcza się na powierzchni podtrzymującej membrany i umożliwia się wytworzenie z niego żelu przed umieszczeniem bariery membranowej nad roztworem wskaźnika. Żel białkowy powinien mieć taką samą objętość i gęstość na całej powierzchni oraz nie powinny występować w nim pęcherzyki powietrza lub wady, które mogłyby wpłynąć na jego integralność funkcjonalną.
|
System wykrywania substancji chemicznych
|
18.
|
Roztwór wskaźnika będący takim samym roztworem, jaki wykorzystano do badania zgodności, powinien reagować na obecność badanej substancji chemicznej. Jako barwnik we wskaźniku pH lub mieszaninę barwników należy stosować np. czerwień krezolową i oranż metylowy, które zmienią swoje zabarwienie w reakcji na obecność badanej substancji chemicznej. System pomiarowy może być wizualny lub elektroniczny.
|
|
19.
|
Systemy wykrywania, które opracowano w celu wykrywania przechodzenia badanej substancji chemicznej przez barierę membranową, należy oceniać pod względem ich istotności i wiarygodności w celu wykazania zakresu substancji chemicznych, które można wykryć, i ilościowych granic wykrywalności.
|
WYKONANIE BADANIA
Zestaw elementów metody badawczej
|
20.
|
Bariera membranowa umieszczona jest we fiolce (lub w rurce) zawierającej roztwór wskaźnika, w taki sposób, by cała powierzchnia podtrzymującej membrany miała kontakt z roztworem wskaźnika oraz by nie powstawały pęcherzyki powietrza. Należy dopilnować, by bariera zachowała integralność.
|
Podawanie badanej substancji chemicznej
|
21.
|
Stosowną ilość badanej substancji chemicznej, np. 500 μl cieczy lub 500 mg drobno sproszkowanej substancji stałej (7) nakłada się ostrożnie na górną powierzchnię bariery membranowej i równo po niej rozprowadza. Dla każdej badanej substancji chemicznej i jej odpowiednich kontroli przygotowuje się odpowiednią liczbę kontrprób, np. cztery (7) (zob. pkt 23–25). Odnotowuje się czas stosowania badanej substancji chemicznej na barierę membranową. Aby zapewnić właściwe odnotowanie krótkich okresów działania żrącego, okresy stosowania badanej substancji chemicznej we fiolkach z kontrpróbą bada się z przesunięciem.
|
Pomiar przenikania przez barierę membranową
|
22.
|
Należy odpowiednio monitorować każdą fiolkę oraz odnotować czas pierwszej zmiany roztworu wskaźnika, tj. przeniknięcia przez barierę, a także określać upływ czasu między zastosowaniem i przeniknięciem bariery membranowej.
|
Kontrole
|
23.
|
Podczas badań, które obejmują stosowanie nośnika lub rozpuszczalnika wraz z badaną substancją chemiczną, nośnik lub rozpuszczalnik powinien być zgodny z układem bariery membrany, tj. nie powinien zmieniać integralności układu bariery membranowej i nie powinien wpływać na działanie żrące badanej substancji chemicznej. W stosownych przypadkach grupę kontrolną rozpuszczalnika (lub nośnika) należy zbadać jednocześnie z badaną substancję chemiczną, aby wykazać zgodność rozpuszczalnika z układem bariery membranowej.
|
|
24.
|
Kontrolę dodatnią (żrącą) o średnim działaniu żrącym, np. 110 ± 15 mg wodorotlenek sodu (podkategoria działania żrącego GHS ONZ 1B) (7), należy badać jednocześnie z badaną substancją chemiczną, aby ocenić, czy sposób działania układu badawczego jest akceptowalny. Druga kontrola dodatnia, która należy do takiej samej klasy chemicznej, jak badana substancja chemiczna, może być przydatna, aby ocenić względne potencjał działania żrącego żrącej badanej substancji chemicznej. Należy wybierać kontrole dodatnie o średnim działaniu żrącym (np. należące do podkategorii 1B GHS ONZ), aby wykryć zmiany w czasie przenikania, które mogą być niedopuszczalnie dłuższe lub krótsze niż ustanowiona wartość odniesienia, wskazując tym samym, że układ badawczy nie funkcjonuje prawidłowo. Do tego celu substancje chemiczne o wysokim działaniu żrącym (podkategoria 1A GHS ONZ) lub nieżrące substancje chemiczne mają ograniczoną użyteczność. Żrąca substancja chemiczna należąca do podkategorii 1B GHS ONZ umożliwiłaby wykrywanie zbyt szybkiego lub zbyt wolnego czasu wytrzymałości. Substancję chemiczną żrącą w niewielkim stopniu (podkategoria 1C GHS ONZ) można wykorzystać jako kontrolę dodatnią w celu zmierzenia zdolności metody badawczej do ciągłego rozróżniania substancji chemicznych żrących w niewielkim stopniu od nieżrących substancji chemicznych. Niezależnie od zastosowanego podejścia należy opracować dopuszczalny zakres reakcji w kontroli dodatniej na podstawie wcześniejszego zakresu czasu wytrzymałości dla zastosowanych kontroli dodatnich, takich jak średnia ± 2-3 odchyleń standardowych. Podczas każdego badania należy określić dokładny czas wytrzymałości w odniesieniu do kontroli dodatniej, aby możliwe było wykrycie odchyleń od dopuszczalnego zakresu.
|
|
25.
|
Kontrolę ujemną (nieżrącą), np. 10 % kwas cytrynowy, 6 % kwas propanowy (7), również należy zbadać jednocześnie z badaną substancją chemiczną, jako kolejny środek kontroli jakości w celu wykazania integralności funkcjonalnej bariery membranowej.
|
Kryteria dopuszczalności badania
|
26.
|
Zgodnie z ustanowionymi parametrami czasu dla każdej z podkategorii działań żrących GHS ONZ, czas (w minutach), jaki upłynął od zastosowanie badanej substancji chemicznej na barierę membranową a przeniknięciem bariery, wykorzystuje się w celu przewidzenia działania żrącego badanej substancji chemicznej. Aby badanie można było uznać za dopuszczalne, równoczesna kontrola dodatnia powinna wykazywać oczekiwany czas reakcji przenikania (np. czas wytrzymałości 8–16 minut w przypadku wykorzystania wodorotlenku sodu jako kontroli dodatniej), równoczesna kontrola ujemna nie powinna mieć działania żrącego, a w stosownych przypadkach równoczesna kontrola z rozpuszczalnikiem nie powinna być żrąca, ani nie powinna wpływać na potencjał działania żrącego badanej substancji chemicznej. Przed przystąpieniem do rutynowego stosowania metody zgodnej z niniejszą metodą badawczą, laboratoria powinny wykazać swoją biegłość techniczną przy użyciu 12 substancji zalecanych w tabeli 2. W przypadku nowych metod zwanych „me-too”, opracowanych zgodnie z niniejszą metodą badawczą, które są podobne do zwalidowanej metody referencyjnej pod względem strukturalnym i funkcjonalnym (14), należy zastosować wcześniej określone standardy wykonywania badań, aby wykazać wiarygodność i dokładność nowej metody przed zastosowaniem jej podczas badań regulacyjnych (10).
|
Interpretacja wyników i klasyfikacja badanych substancji chemicznych pod względem działania żrącego
|
27.
|
Czas (w minutach), jaki upłynął od zastosowania badanej substancji chemicznej na barierę membranową a przeniknięciem bariery, wykorzystuje się, aby zaklasyfikować badaną substancję chemiczną do podkategorii działania żrącego GHS ONZ (1) oraz, w stosownych przypadkach, do grupy opakowaniowej ONZ (16). Wartości czasu odcięcia dla każdej z trzech podkategorii działania żrącego ustala się dla każdej proponowanej metody badawczej. Ostateczne decyzje dotyczące czasu odcięcia powinny uwzględniać potrzebę ograniczenia zaniżania klasyfikacji zagrożenia związanego z działaniem żrącym (tj. wyników fałszywie ujemnych). Zgodnie z obecną wytyczną dotyczącą badań należy wykorzystać czas odcięcia Corrositex® opisany w tabeli 3, ponieważ model ten stanowi obecnie jedyną metodę badawczą zgodną z wytyczną dotyczącą badań (7).
Tabela 3
Model prognozowania Corrositex®
|
Średni czas wytrzymałości (min.)
|
Prognoza GHS ONZ (32)
|
|
Badane substancje chemiczne kategorii 1 (30) (określone na podstawie badania kategoryzacji w ramach metody)
|
Badane substancje chemiczne kategorii 2 (31) (określone na podstawie badania kategoryzacji w ramach metody)
|
|
0–3 min.
|
0–3 min.
|
Żrąca podkategoria działania żrącego 1A
|
|
> 3–60 min.
|
> 3–30 min.
|
Żrąca podkategoria działania żrącego 1B
|
|
> 60–240 min.
|
> 30–60 min.
|
Żrąca podkategoria działania żrącego 1C
|
|
> 240 min.
|
> 60 min.
|
Substancja nieżrąca
|
|
DANE I SPRAWOZDAWCZOŚĆ
Dane
|
28.
|
Czas (w minutach), jaki upłynął od zastosowania substancji do przeniknięcia bariery w odniesieniu do badanej substancji chemicznej i kontroli dodatnich, należy zgłosić w postaci tabeli jako poszczególne dane dla powtórzeń, a także średnie ± odchylenie standardowe dla każdej próby.
|
Sprawozdanie z badania
|
29.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Badana substancja chemiczna oraz substancje kontrolne:
|
—
|
Substancja jednoskładnikowa: dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń, stosownie do przypadku i jeśli jest to praktycznie wykonalne itp.;
|
|
—
|
substancja wieloskładnikowa, UVCB i mieszanina: opisana w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników;
|
|
—
|
Wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
źródło, numer partii (jeżeli dostępny);
|
|
—
|
w stosownych przypadkach obróbka badanej substancji chemicznej/substancji kontrolnej przed badaniem (np. ogrzanie, rozdrobnienie);
|
|
—
|
stabilność badanej substancji chemicznej, termin przydatności lub data ponownej analizy, jeżeli są znane;
|
|
—
|
warunki przechowywania.
|
|
|
|
Nośnik:
|
—
|
identyfikacja, stężenie (jeżeli dotyczy) użyta objętość;
|
|
—
|
uzasadnienie wyboru nośnika.
|
|
|
|
Wykorzystany model bariery membranowej in vitro i protokół, w tym wykazana dokładność i wiarygodność
|
|
|
Warunki badania:
|
—
|
Opis wykorzystanej aparatury i zastosowanych procedur przygotowania;
|
|
—
|
Pochodzenie i skład wykorzystanej bariery membranowej in vitro;
|
|
—
|
Skład i właściwości roztworu wskaźnika;
|
|
—
|
Ilości badanej substancji chemicznej i substancji kontrolnych;
|
|
—
|
Opis i uzasadnienie badania kategoryzacji skali czasu;
|
|
—
|
Opis stosowanych kryteriów oceny i klasyfikacji;
|
|
—
|
Wykazanie efektywności w wykonywaniu metody badawczej przed rutynowym użyciem poprzez badanie substancji chemicznych przeznaczonych do oceny biegłości.
|
|
|
|
Wyniki:
|
—
|
Tabelaryczne zestawienie poszczególnych danych surowych dotyczących poszczególnych badań i próbek kontrolnych dla każdej kontrpróby;
|
|
—
|
opis innych zaobserwowanych skutków;
|
|
—
|
ustalona na tej podstawie klasyfikacja w odniesieniu do modelu prognozowania lub zastosowanych kryteriów podejmowania decyzji.
|
|
Omówienie wyników
Wnioski
|
BIBLIOGRAFIA
|
(1)
|
Organizacja Narodów Zjednoczonych (ONZ), Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS), wydanie piąte zmienione, ONZ Nowy Jork i Genewa 2013. Dostępne na stronie internetowej: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(2)
|
Rozdział B.4 niniejszego załącznika, Ostre działanie drażniące na skórę.
|
|
(3)
|
Rozdział B.40bis niniejszego załącznika, Badanie działania żrącego na skórę metodą In Vitro: metoda badawcza z użyciem zrekonstruowanego ludzkiego naskórka.
|
|
(4)
|
Rozdział B.40 niniejszego załącznika, Badanie działania żrącego na skórę in vitro: Test przezskórnej oporności elektrycznej (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 203, Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. i Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and evaluation by the Management Team.„Toxicology In Vitro” nr 12, s. 483–524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. [W:] Publikacja NIH nr 99-4495, NIEHS.
|
|
(8)
|
Gordon V.C., Harvell J.D. i Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. [W:] In vitro Skin Toxicology: Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, s. 37–45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 34.
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX〈 Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy,„ATLA” nr 29, s. 96–97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision), (20 września 2002 r.). Waszyngton: U.S. DOT.
|
|
(13)
|
Rozdział B.46 niniejszego załącznika, Badanie podrażnień skóry in vitro: Metoda badania z użyciem zrekonstruowanego ludzkiego naskórka. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. [W:] Publikacja NIH nr 04-4510, NIEHS. Dostępne na stronie internetowej: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Dostępne na stronie internetowej: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Organizacja Narodów Zjednoczonych (ONZ), UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), ONZ, 2013. Dostępne na stronie internetowej: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf
|
Dodatek
DEFINICJE
Dokładność
: stopień zgodności wyników zastosowanej metody badawczej z przyjętymi wartościami odniesienia. Jest to miara efektywności metody badawczej i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem »zgodność« na określenie odsetka prawidłowych wyników uzyskiwanych przy użyciu metody badawczej (9).
Substancja chemiczna
: substancja lub mieszanina.
System wykrywania substancji chemicznych
: Wizualny lub elektroniczny system pomiarowy zawierający roztwór wskaźnika, który reaguje na obecność badanej substancji chemicznej, np. przez zmianę barwy barwnika we wskaźniku pH lub mieszanin barwników, które zmienią swoje zabarwienie w reakcji na obecność badanej substancji chemicznej, lub przez inny rodzaj reakcji chemicznych lub elektromechanicznych.
Zgodność
: jest to miara efektywności metody badawczej w odniesieniu do metod badawczych, które dają wyniki kategoryczne i jest ona jednym z aspektów istotności. To określenie jest czasami stosowane wymiennie z określeniem „dokładność”; definiuje się jako odsetek wszystkich badanych substancji chemicznych, które są prawidłowo sklasyfikowane jako dodatnie lub ujemne. Zgodność jest w dużym stopniu uzależniona od przewagi dodatnich wyników w rodzajach badanej substancji chemicznej (9).
GHS (Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów)
: system obejmujący klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz odpowiednie elementy komunikacyjne, takie jak: piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki, mające informować na temat szkodliwego działania tych substancji chemicznych w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) oraz środowiska (1).
IATA
: zintegrowane podejście do badań i oceny.
Mieszanina
: mieszanina lub roztwór składający się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
Substancja wieloskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥ 10 % (w/w) i < 80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się w wyniku zmieszania co najmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
NŻ
: substancja niemająca działania żrącego.
Standardy wykonywania badań (PS)
: normy oparte na zwalidowanej metodzie badania, zapewniające podstawę do oceny porównywalności proponowanej metody badawczej, która jest podobna do metody referencyjnej pod względem mechanistycznym i funkcjonalnym. Uwzględnia się (i) istotne elementy metody badania; (ii) podstawowy wykaz chemicznych substancji odniesienia wybranych spośród substancji chemicznych wykorzystywanych w celu wykazania dopuszczalnej efektywności zwalidowanej metody badania; oraz (iii) podobny poziom dokładności i wiarygodności, na podstawie wyników uzyskanych dla zwalidowanej metody badawczej, który proponowana metoda badania powinna osiągnąć przy ocenie z wykorzystaniem podstawowego wykazu chemicznych substancji odniesienia (9).
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono istotne i użyteczne dla konkretnego celu. Jest to zakres, w jakim metoda badawcza prawidłowo mierzy lub przewiduje biologiczny oczekiwany skutek. Istotność obejmuje uwzględnienie dokładności (zgodności) metody badania (9).
Wiarygodność
: miary zakresu, w jakim metoda badawcza może być przeprowadzana w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jej przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją poprzez obliczenie odtwarzalności wewnątrz- i międzylaboratoryjnej (9).
Czułość
: odsetek wszystkich substancji chemicznych dających wynik dodatni / aktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą danej metody badawczej. Jest to miara dokładności metody badania, która daje wyniki kategoryczne, stanowiąca parametr istotny dla oceny istotności metody badania (9).
Działanie żrące na skórę in vivo
: powstanie nieodwracalnego uszkodzenia skóry; tj. widocznej martwicy przebiegającej przez naskórek w głąb skóry, spowodowanej naniesieniem badanej substancji chemicznej na okres nieprzekraczający czterech godzin. Reakcjami na działanie żrące są wrzody, krwotoki, krwawiące strupy oraz – po zakończeniu 14-dniowej obserwacji – odbarwienia wynikłe ze zblednięcia skóry, obszary jednolitej alopecji, a także blizny. Celem oceny wątpliwych uszkodzeń można przeprowadzić badanie histopatologiczne.
Swoistość
: odsetek wszystkich substancji chemicznych dających wynik ujemny / nieaktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą danej metody badawczej. Jest to miara dokładności metody badania, która daje wyniki kategoryczne, stanowiąca parametr istotny dla oceny istotności metody badania (9).
Substancja
: pierwiastek chemiczny i jego związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania trwałości produktu i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem wszelkich rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
B.66 STABILNIE TRANSFEKOWANA TRANSAKTYWACJA W BADANIACH IN VITRO DO WYKRYWANIA AGONISTÓW i ANTAGONISTÓW RECEPTORA ESTROGENOWEGO
WPROWADZENIE OGÓLNE
Wytyczna OECD dotycząca badań w oparciu o wyniki
|
1.
|
Niniejsza metoda badawcza jest równoważna wytycznej OECD dotyczącej badań (TG) nr 455 (2016). TG 455 jest wytyczną dotyczącą badań w oparciu o wyniki, która opisuje metodykę stabilnie transfekowanej transaktywacji w testach in vitro do wykrywania agonistów i antagonistów receptora estrogenowego (badania aktywacji transkrypcji receptora estrogenowego – ER TA). Obejmuje ona szereg metod badań podobnych pod względem mechanistycznym i funkcjonalnym mających na celu określenie agonistów lub antagonistów receptora estrogenowego (tj. ERα, lub ERβ), które powinny ułatwiać opracowanie nowych – podobnych lub zmienionych – metod badawczych zgodnie z zasadami walidacji określonymi w wytycznych OECD Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1). W pełni zwalidowane referencyjne metody badań (dodatek 2 i dodatek 3), które stanowią podstawę tej wytycznej dotyczącej badań w oparciu o wyniki, to:
|
—
|
test stabilnie transfekowanej transaktywacji (STTA) (2) wykorzystaniem linii komórkowej (h) ERα-HeLa-9903; oraz
|
|
—
|
test VM7Luc ER TA (3) przy użyciu linii komórkowej VM7Luc4E2 (33), która wyraża przede wszystkim hERα z niewielkim udziałem hERβ(4)(5).
|
W celu opracowania i walidacji podobnych testów dotyczących tego samego punktu końcowego zagrożenia dostępne są standardy wykonywania badań (6) (7), które należy stosować. Umożliwiają one terminową zmianę wytycznej dotyczącej badań w oparciu o wyniki nr 455, aby do zaktualizowanej wytycznej dotyczącej badań w oparciu o wyniki można było dodać nowe podobne testy; podobne testy zostaną jednak dodane dopiero po przeglądzie przeprowadzonym przez OECD i po uzgodnieniu przez tę organizację, że standardy wykonywania badań zostały spełnione. Testy ujęte w wytycznej dotyczącej badań nr 455 można stosować w sposób nieograniczony w celu spełnienia wymogów państw członkowskich OECD w zakresie wyników badań dotyczących transaktywacji receptora estrogenowego przy jednoczesnym korzystaniu z wzajemnego uznawania danych OECD.
|
Kontekst i zasady dotyczące testów zawartych w niniejszej metodzie badawczej
|
2.
|
W 1998 r. OECD podjęła działania o wysokim priorytecie, mające na celu zmianę istniejących wytycznych i opracowanie nowych wytycznych dotyczących badań na potrzeby badań klasyfikacyjnych i badań substancji potencjalnie zaburzających funkcjonowanie układu hormonalnego. W 2012 r. zmieniono ramy koncepcyjne OECD dotyczące badania i oceny substancji potencjalnie zaburzających funkcjonowanie układu hormonalnego. Oryginalne i zmienione ramy koncepcyjne uwzględniono jako załączniki do wytycznych OECD Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (8). Ramy koncepcyjne składają się z pięciu poziomów, a każdy z nich odpowiada innemu poziomowi złożoności biologicznej. Transaktywacja receptora estrogenowego opisana w niniejszej metodzie badawczej należy do poziomu 2, który obejmuje „testy in vitro dostarczające danych na temat wybranych mechanizmów/ścieżek układy hormonalnego”. Niniejsza metoda badawcza dotyczy testów transaktywacji in vitro stworzonych, aby określić agonistów i antagonistów receptora estrogenowego.
|
|
3.
|
Interakcja estrogenów z receptorami estrogenowymi może wpłynąć na transkrypcję genów regulowanych przez estrogen, co może prowadzić do indukcji lub inhibicji procesów komórkowych, w tym tych koniecznych do proliferacji komórek, prawidłowego rozwoju płodu i funkcji reprodukcyjnej (9)(10)(11). Zakłócenie prawidłowego układu estrogenowego może potencjalnie wywołać niekorzystny wpływ na prawidłowy rozwój (ontogenezę), zdrowie reprodukcyjne i integralność układu rozrodczego.
|
|
4.
|
Badania aktywacji transkrypcji in vitro opierają się na bezpośredniej lub pośredniej interakcji między substancjami zawierającymi szczególny receptor, który reguluje transkrypcję produktu genu reporterowego. Takie testy wykorzystywano w dużym stopniu w celu oceny ekspresji genów regulowanej przez szczególne receptory jądrowe, takie jak receptory estrogenowe (12)(13)(14)(15)(16). Proponowano je w celu wykrywania transaktywacji estrogenu regulowanej przez receptor estrogenowy (17)(18)(19). Istnieją co najmniej dwa główne podtypy jądrowych receptorów estrogenowych, α i β, które są zakodowane przez odrębne geny. Odpowiednie białka mają różne biologiczne funkcje, a także różny rozkład w tkankach i powinowactwo wiązania ligandów (20)(21)(22)(23)(24)(25)(26). Jądrowy ERα pośredniczy w klasycznej reakcji estrogennej (27)(28)(29)(30) i z związku z tym większość opracowywanych obecnie modeli mających na celu zmierzenie aktywacji lub inhibicji ER dotyczy ERα. Badania wykonuje się w celu określenia substancji chemicznych, które aktywują (lub hamują) receptor estrogenowy w następstwie wiązania ligandów, po którym kompleks receptor-ligand wiąże się z określonymi elementami reakcji DNA oraz transaktywuje gen reporterowy, powodując zwiększoną ekspresję komórkową białka markerowego. Podczas testów można wykorzystać różne reakcje genów reporterowych. W układach opartych na lucyferazie enzym lucyferaza przekształca substrat lucyferyny w produkt bioluminescencyjny, który można zmierzyć ilościowo przy użyciu luminometru. Inne przykłady częstych genów reporterowych obejmują białko fluorescencji i gen LacZ, który koduje β-galaktozydazę, enzym przetwarzający bezbarwny substrat X-gal (5- bromo-4-chloro-indolilo-galaktopiranozyd) w niebieski produkt, który można określić ilościowo przy użyciu spektrofotometru. Te geny reporterowe można ocenić w szybki i niedrogi sposób, wykorzystując dostępne na rynku zestawy badawcze.
|
|
5.
|
Badania walidacyjne dotyczące badań aktywacji transkrypcji STTA i VM7Luc wykazały istotność i wiarygodność tych metod w odniesieniu do ich przeznaczenia (3)(4)(5)(30). Standardy wykonywania badań dotyczące testów ER TA opartych na luminescencji przy użyciu linii komórkowych raka piersi zawarto w dokumencie ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Te standardy wykonywania badań zmieniono, aby miały zastosowanie zarówno do badań aktywacji transkrypcji STTA jak i VM7Luc (2).
|
|
6.
|
Definicje i skróty stosowane w niniejszej metodzie badawczej opisano w dodatku 1.
|
Zakres badań aktywacji transkrypcji i związane z nimi ograniczenia
|
7.
|
Testy te proponuje się do celów badań przesiewowych i ustalania substancji priorytetowych, ale mogą one dostarczać również informacji mechanistycznych, które można wykorzystać w ramach podejścia opartego na wadze dowodów. Testy te dotyczą transaktywacji wywołanej przez substancję chemiczną wiążącą się z receptorami estrogenowymi w układzie in vitro. W związku z tym wyników nie należy ekstrapolować bezpośrednio do złożonej sygnalizacji i regulacji nieuszkodzonego układu hormonalnego in vivo.
|
|
8.
|
Transaktywację, w której pośredniczą ER, uznaje się za jeden z kluczowych mechanizmów zaburzania funkcjonowania układu hormonalnego, chociaż istnieją także inne mechanizmy, które mogą wywołać zaburzenia funkcjonowania układu hormonalnego, między innymi (i) interakcje z innymi receptorami i układami enzymatycznymi w układzie hormonalnym, (ii) synteza hormonów, (iii) aktywacja metaboliczna lub dezaktywacja hormonów, (iv) dystrybucja hormonów do tkanek docelowych i (v) usunięcie hormonów z ciała. Żaden z testów wchodzących w skład niniejszej metody badawczej nie dotyczy tych sposobów działania.
|
|
9.
|
Niniejsza metoda badawcza dotyczy zdolności substancji chemicznych do aktywowania (tj. działania jako agoniści), a także do hamowania (tj. działania jako antagoniści) transkrypcji zależnej od ER. Niektóre substancje chemiczne, które w zależności od rodzaju komórek wykazują zarówno działanie agonistyczne, jak i antagonistyczne, określa się jako selektywne modulatory receptora estrogenowego (SERM). Substancje chemiczne, które w tych testach dają wynik ujemny, można ocenić za pomocą testu wiązania ER przed stwierdzeniem, że dana substancja chemiczna nie wiąże się z receptorem. Ponadto testy mogą zapewnić informacje wyłącznie dotyczące działanie pierwotnej molekuły, biorąc pod uwagę ograniczone zdolności metaboliczne układów komórek in vitro. Biorąc pod uwagę, że podczas walidacji wykorzystano jedynie pojedyncze substancje, nie zbadano zastosowania do mieszanin stosowanych w badaniu. Teoretycznie metoda badawcza może jednak mieć zastosowanie do badania zarówno substancji wieloskładnikowych, substancji o nieznanym lub zmiennym składzie, złożonych produktów reakcji lub materiały biologicznych, jak i mieszanin. Przed zastosowaniem przedmiotowej metody badawczej w odniesieniu do substancji wieloskładnikowej, UVCB lub mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy zastanowić się, czy zastosowanie niniejszej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak – dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny.
|
|
10.
|
Do celów informacyjnych w tabeli 1 przedstawiono wyniki testu agonisty 34 substancji, które zbadano za pomocą obu w pełni zwalidowanych referencyjnych metod badań opisanych w niniejszej metodzie badawczej. 26 z tych substancji sklasyfikowano jako jednoznacznych agonistów ER a 8 jako ujemne na podstawie opublikowanych sprawozdań, w tym testów in vitro wiązania i transaktywacji lub testu wzrostu macicy (2)(3)(18)(31)(32)(33)(34). W tabeli 2 przedstawiono wyniki testu antagonistycznego 15 substancji, które zbadano za pomocą obu w pełni zwalidowanych referencyjnych metod badań opisanych w niniejszej metodzie badawczej. Cztery z tych substancji sklasyfikowano jako jednoznacznych/przypuszczalnych antagonistów ER a 10 jako ujemne na podstawie opublikowanych sprawozdań, w tym testów in vitro wiązania i transaktywacji ER (2)(3)(18)(31). Jeżeli chodzi o dane podsumowane w tabeli 1 i tabeli 2, wyniki obu referencyjnych metod badawczych dotyczące klasyfikacji wszystkich substancji, poza jedną (mifepryston), w odniesieniu do testu antagonisty były stuprocentowo zgodne, a wszystkie substancje sklasyfikowano prawidłowo jako agonistów/antagonistów ER lub jako substancje ujemne. Dodatkowe informacje dotyczące tej grupy substancji chemicznych, a także dodatkowe substancje chemiczne zbadane za pomocą testów STTA i VM7Luc ER TA podczas badań walidacyjnych zawarto w dodatku 2 (tabela 1, 2 i 3) do standardów wykonywania badań ER TA (6)(7).
|
Tabela 1
Przegląd wyników testów STTA i VM7Luc ER TA dotyczących substancji zbadanych za pomocą obu testów agonistycznych, które sklasyfikowano jako agonistów ER dodatnich (DOD.) lub ujemnych (UJEM.)
|
|
Substancja
|
Numer CAS
|
Test STTA (35)
|
Test VM7Luc ER TA (36)
|
Źródło danych do celów klasyfikacji (38)
|
|
Działanie ER TA
|
wartość PC10 (M)
|
wartość PC50
(2) (M)
|
Działanie ER TA
|
Wartość EC50
(2), (37) (M)
|
Inne testy ER TA (34)
|
Wiązanie ER
|
Wzrost macicy
|
|
1
|
17β-estradiol (1)
|
50-28-2
|
DOD.
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
DOD.
|
5,63 × 10-12
|
DOD. (227/227)
|
DOD.
|
DOD.
|
|
2
|
17α-estradiol (1)
|
57-91-0
|
DOD.
|
7,24 × 10-11
|
6,44 × 10-10
|
DOD.
|
1,40 × 10-9
|
DOD. (11/11)
|
DOD.
|
DOD.
|
|
3
|
Etynyloestradiol 17-α (1)
|
57-63-6
|
DOD.
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
DOD.
|
7,31 × 10-12
|
DOD. (22/22)
|
DOD.
|
DOD.
|
|
4
|
17β-trenbolon
|
10161-33-8
|
DOD.
|
1,78 × 10-8
|
2,73 × 10-7
|
DOD.
|
4,20 × 10-8
|
DOD. (2/2)
|
NB
|
NB
|
|
5
|
19-nortestosterone (1)
|
434-22-0
|
DOD.
|
9,64 × 10-9
|
2,71 × 10-7
|
DOD.
|
1,80 × 10-6
|
DOD. (4/4)
|
DOD.
|
DOD.
|
|
6
|
4-kumylfenol (1)
|
599-64-4
|
DOD.
|
1,49 × 10-7
|
1,60 × 10-6
|
DOD.
|
3,20 × 10-7
|
DOD. (5/5)
|
DOD.
|
NB
|
|
7
|
4-tert-Oktylofenol (1)
|
140-66-9
|
DOD.
|
1,85 × 10-9
|
7,37 × 10-8
|
DOD.
|
3,19 × 10-8
|
DOD. (21/24)
|
DOD.
|
DOD.
|
|
8
|
Apigenin (1)
|
520-36-5
|
DOD.
|
1,31 × 10-7
|
5,71 × 10-7
|
DOD.
|
1,60 × 10-6
|
DOD. (26/26)
|
DOD.
|
NB
|
|
9
|
Atrazyn (1)
|
1912-24-9
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (30/30)
|
UJEM.
|
NB
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
DOD.
|
2,02 × 10-8
|
2,94 × 10-7
|
DOD.
|
5,33 × 10-7
|
DOD. (65/65)
|
DOD.
|
DOD.
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
DOD.
|
2,36 × 10-8
|
2,11 × 10-7
|
DOD.
|
1,95 × 10-7
|
DOD. (6/6)
|
DOD.
|
DOD.
|
|
12
|
Ftalan benzylu butylu (1)
|
85-68-7
|
DOD.
|
1,14 × 10-6
|
4,11 × 10-6
|
DOD.
|
1,98 × 10-6
|
DOD. (12/14)
|
DOD.
|
UJEM.
|
|
13
|
Kortykosteron (1)
|
50-22-6
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (6/6)
|
UJEM.
|
NB
|
|
14
|
Kumestrol (1)
|
479-13-0
|
DOD.
|
1,23 × 10-9
|
2,00 × 10-8
|
DOD.
|
1,32 × 10-7
|
DOD. (30/30)
|
DOD.
|
NB
|
|
15
|
Daidzein (1)
|
486-66-8
|
DOD.
|
1,76 × 10-8
|
1,51 × 10-7
|
DOD.
|
7,95 × 10-7
|
DOD. (39/39)
|
DOD.
|
DOD.
|
|
16
|
Dietylostylbestrol (1)
|
56-53-1
|
DOD.
|
< 1,00 × 10-11
|
2,04 × 10-11
|
DOD.
|
3,34 × 10-11
|
DOD. (42/42)
|
DOD.
|
NB
|
|
17
|
Ftalan di-n-butylu
|
84-74-2
|
DOD.
|
4,09 × 10-6
|
|
DOD.
|
4,09 × 10-6
|
DOD. (6/11)
|
DOD.
|
UJEM.
|
|
18
|
Etyloparaben
|
120-47-8
|
DOD.
|
5,00 × 10-6
|
(PC50 nie występuje)
|
DOD.
|
2,48 × 10-5
|
DOD.
|
|
NB
|
|
19
|
Estron (1)
|
53-16-7
|
DOD.
|
3,02 × 10-11
|
5,88 × 10-10
|
DOD.
|
2,34 × 10-10
|
DOD. (26/28)
|
DOD.
|
DOD.
|
|
20
|
Genistein (1)
|
446-72-0
|
DOD.
|
2,24 × 10-9
|
2,45 × 10-8
|
DOD.
|
2,71 × 10-7
|
DOD. (100/102)
|
DOD.
|
DOD.
|
|
21
|
Haloperydol
|
52-86-8
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (2/2)
|
UJEM.
|
NB
|
|
22
|
Kemferol (1)
|
520-18-3
|
DOD.
|
1,36 × 10-7
|
1,21 × 10-6
|
DOD.
|
3,99 × 10-6
|
DOD. (23/23)
|
DOD.
|
NB
|
|
23
|
Kepone (1)
|
143-50-0
|
DOD.
|
7,11 × 10-7
|
7,68 × 10-6
|
DOD.
|
4,91 × 10-7
|
DOD. (14/18)
|
DOD.
|
NB
|
|
24
|
Ketokonazol
|
65277-42-1
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (2/2)
|
UJEM.
|
NB
|
|
25
|
Linuron (1)
|
330-55-2
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (8/8)
|
UJEM.
|
NB
|
|
26
|
mezoheksestrol (1)
|
84-16-2
|
DOD.
|
< 1,00 × 10-11
|
2,75 × 10-11
|
DOD.
|
1,65 × 10-11
|
DOD. (4/4)
|
DOD.
|
NB
|
|
27
|
Metylotestosteron (1)
|
58-18-4
|
DOD.
|
1,73 × 10-7
|
4,11 × 10-6
|
DOD.
|
2,68 × 10-6
|
DOD. (5/6)
|
DOD.
|
NB
|
|
28
|
Moryna
|
480-16-0
|
DOD.
|
5,43 × 10-7
|
4,16 × 10-6
|
DOD.
|
2,37 × 10-6
|
DOD. (2/2)
|
DOD.
|
NB
|
|
29
|
Noretynodrel (1)
|
68-23-5
|
DOD.
|
1,11 × 10-11
|
1,50 × 10-9
|
DOD.
|
9,39 × 10-10
|
DOD. (5/5)
|
DOD.
|
NB
|
|
30
|
p,p’-Metoksychlor (1)
|
72-43-5
|
DOD.
|
1,23 × 10-6
|
(PC50 nie występuje) (2)
|
DOD.
|
1,92 × 10-6
|
DOD. (24/27)
|
DOD.
|
DOD.
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (2/2)
|
UJEM.
|
NB
|
|
32
|
Rezerpina
|
50-55-5
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (4/4)
|
UJEM.
|
NB
|
|
33
|
Spironolakton (1)
|
52-01-7
|
UJEM.
|
—
|
—
|
UJEM.
|
—
|
UJEM. (4/4)
|
UJEM.
|
NB
|
|
34
|
Testosteron
|
58-22-0
|
DOD.
|
2,82 × 10-8
|
9,78 × 10-6
|
DOD.
|
1,75 × 10-5
|
DOD. (5/10)
|
DOD.
|
NB
|
|
Skróty: Numer CAS = numer w rejestrze Chemical Abstracts Service; M = molowy; EC50 = połowa maksymalnego stężenia efektywnego badanej substancji; UJEM. = ujemny; DOD. = dodatni; NB = nie badano; PC10 (and PC50) = stężenie badanej substancji, przy którym reakcja stanowi 10 % (lub 50 % w przypadku PC50) reakcji kontroli dodatniej (E2, 1nM) na każdej płytce.
|
Tabela 2
Porównanie wyników testów STTA i VM7Luc ER TA dotyczących substancji zbadanych za pomocą obu testów antagonisty, które sklasyfikowano jako antagonistów ER dodatnich (DOD.) lub ujemnych (UJEM.)
|
|
Substancja (3)
|
Numer CAS
|
Test STTA ER (39)
|
Test VM7Luc ER TA (40)
|
Potencjalne wyniki ER STTA (42)
|
Uzgodniona klasyfikacja ICCVAM (43)
|
Klasa chemiczna MeSH (44)
|
Klasa produktowa (45)
|
|
Działanie ER TA
|
wartość IC50
(4) (M)
|
Działanie ER TA
|
wartość IC50
(4), (41) (M)
|
|
1
|
4-hydroksy-tamoksyfen
|
68047-06-3
|
DOD.
|
3,97 × 10-9
|
DOD.
|
2,08 × 10-7
|
umiarkowany DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
2
|
Dibenzo[a,h]antracen
|
53-70-3
|
DOD.
|
IC50 nie występuje
|
DOD.
|
IC50 nie występuje
|
DOD.
|
ZD
|
Związek policykliczny
|
Laboratoryjna substancja chemiczna, produkt naturalny
|
|
3
|
Mifepryston
|
84371-65-3
|
DOD.
|
5,61 × 10-6
|
UJEM.
|
—
|
umiarkowany DOD
|
UJEM.
|
Steryd
|
Produkt leczniczy
|
|
4
|
Chlorowodorek raloksyfenu
|
82640-04-8
|
DOD.
|
7,86 × 10-10
|
DOD.
|
1,19 × 10-9
|
umiarkowany DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
5
|
Tamoksyfen
|
10540-29-1
|
DOD.
|
4,91 × 10-7
|
DOD.
|
8,17 × 10-7
|
DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
6
|
17β-estradiol
|
50-28-2
|
UJEM.
|
—
|
UJEM.
|
—
|
ZU
|
ZU
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
7
|
Apigenin
|
520-36-5
|
UJEM.
|
—
|
UJEM.
|
—
|
UJEM.
|
UJEM.
|
Związek heterocykliczny
|
Barwnik, produkt naturalny, półprodukt farmaceutyczny
|
|
8
|
Atrazyna
|
1912-24-9
|
UJEM.
|
—
|
UJEM.
|
—
|
UJEM.
|
ZU
|
Związek heterocykliczny
|
Herbicyd
|
|
9
|
Ftalan di-n-butylu
|
84-74-2
|
UJEM.
|
—
|
UJEM.
|
—
|
UJEM.
|
UJEM.
|
Ester, Kwas ftalowy
|
Składnik kosmetyczny, przemysłowa substancja chemiczna, plastyfikator
|
|
10
|
Fenarymol
|
60168-88-9
|
UJEM.
|
—
|
UJEM.
|
—
|
nie badano
|
ZU
|
Związek heterocykliczny, pirymidyna
|
Środek grzybobójczy
|
|
11
|
Flawon
|
525-82-6
|
UJEM.
|
—
|
UJEM.
|
—
|
ZU
|
ZU
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny, produkt leczniczy
|
|
12
|
Flutamid
|
13311-84-7
|
UJEM.
|
—
|
UJEM.
|
—
|
UJEM.
|
ZU
|
Amidy
|
Produkt leczniczy, środek weterynaryjny
|
|
13
|
Genisteina
|
446-72-0
|
UJEM.
|
—
|
UJEM.
|
—
|
ZU
|
UJEM.
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny, produkt leczniczy
|
|
14
|
p-n-nonylofenol
|
104-40-5
|
UJEM.
|
—
|
UJEM.
|
—
|
nie badano
|
UJEM.
|
Fenol
|
Półprodukt
|
|
15
|
Resweratrol
|
501-36-0
|
UJEM.
|
—
|
UJEM.
|
—
|
ZU
|
UJEM.
|
Węglowodór (cykliczny)
|
Produkt naturalny
|
|
Skróty: Numer CAS = numer w rejestrze Chemical Abstracts Service; M = molowy; IC50 = połowa maksymalnego stężenia hamującego badanej substancji; UJEM. = ujemny; ZU = zakładany ujemne; DOD. = dodatni; ZD = zakładany dodatni.
|
ELEMENTY TESTU ER TA
Istotne elementy testu
|
11.
|
Niniejsza metoda badawcza ma zastosowanie do testów, podczas których wykorzystuje się stabilnie transfekowany lub endogenny receptor estrogenowy alfa oraz stabilnie transfekowany konstrukt genu reporterowego podlegający kontroli co najmniej jednego elementu reakcji na estrogen; mogą jednak występować inne receptory, takie jak ERβ. Są to istotne elementy testu.
|
Kontrole
|
12.
|
Należy opisać podstawę proponowanych równoległych wzorców referencyjnych dla każdego testu agonisty i antagonisty. Jednoczesne kontrole (ujemne, z rozpuszczalnikiem i dodatnie) w stosownych przypadkach służą jako wskazówka, że test działa w warunkach badania, a także stanowią podstawę porównań między doświadczeniami; kontrole te wchodzą zwykle w skład kryteriów dopuszczalności danego doświadczenia (1).
|
Standardowe procedury kontroli jakości
|
13.
|
Standardowe procedury kontroli jakości należy przeprowadzać jak opisano dla każdego testu w celu zapewnienia, by każda linia komórkowa utrzymywała stabilność przez wiele pasaży, by pozostawała niezanieczyszczona mykoplazmą (tj. niezanieczyszczona skażeniem bakteryjnym) oraz by zachowała zdolność do zapewnienia oczekiwanych reakcji, w których pośredniczy ER, na przestrzeni czasu. Należy przeprowadzić dalszą kontrolę linii komórkowych, aby sprawdzić ich właściwą tożsamość, a także inne zanieczyszczenia (np. grzyby, drożdże i wirusy).
|
Wykazanie biegłości laboratorium
|
14.
|
Przed przeprowadzeniem badania nieznanych substancji chemicznych przy użyciu któregokolwiek z testów przeprowadzanych w ramach niniejszej metody badawczej każde laboratorium powinno wykazać biegłość w stosowaniu testu. Aby wykazać biegłość, każde laboratorium powinno zbadać 14 substancji służących do wykazania biegłości wymienionych w tabeli 3 w odniesieniu do testu agonisty i 10 substancji służących do wykazania biegłości wymienionych w tabeli 4 w odniesieniu do testu antagonisty. To badanie biegłości potwierdzi również reaktywność układu badawczego. Wykaz substancji służących do wykazania biegłości stanowi podzbiór substancji odniesienia przedstawionych w standardach wykonywania badań dotyczących testów ER TA (6). Substancje te są dostępne na rynku, należą do klas substancji chemicznych łączących się często z aktywnością agonistów lub antagonistów ER, wykazują odpowiedni zakres siły działania oczekiwanej w przypadku agonistów/antagonistów ER (tj. wysoki lub słaby) oraz obejmują substancje ujemne. Badanie substancji służących do wykazania biegłości należy powtórzyć co najmniej dwukrotnie, w innych dniach. Biegłość wykazuje się w drodze poprawnej klasyfikacji (dodatnia/ujemna) każdej substancji służącej do wykazania biegłości. Podczas nauki przeprowadzania testów każdy technik powinien powtórzyć badanie biegłości. W zależności od rodzaju komórki, niektóre z tych substancji służących do wykazania biegłości mogą zachowywać się jak SERM i wykazywać działanie zarówno agonistyczne, jak i antagonistyczne. Substancje służące do wykazania biegłości są jednak sklasyfikowane w tabelach 3 i 4 według ich znanego i dominującego działania, które należy wykorzystać w celu oceny biegłości.
|
|
15.
|
Aby wykazać efektywność oraz do celów kontroli jakości każde laboratorium powinno stworzyć bazy danych historycznych dotyczących działania agonistycznego i antagonistycznego wraz z wzorcami referencyjnymi (np. 17β-estradiol i tamoksyfen), substancjami chemicznymi służącymi do kontroli dodatniej i ujemnej i danymi dotyczącymi kontroli z rozpuszczalnikiem (np. DMSO). Na początek bazę danych należy wygenerować z co najmniej 10 niezależnych prób dotyczących agonistów (np. 17β-estradiol) i 10 niezależnych serii badawczych antagonistów (np. tamoksyfen). Wyniki przyszłych analiz tych wzorców referencyjnych i kontroli z rozpuszczalnikiem należy dodawać do bazy danych, aby ją powiększyć, co pozwoli z czasem zapewnić spójność i efektywność testu biologicznego przeprowadzanego przez laboratorium.
|
Tabela 3
Wykaz (14) substancje służące do wykazania biegłości w odniesieniu do testu agonisty
(46)
|
Nr (53)
|
Substancja
|
Numer CAS
|
Przewidywana reakcja (47)
|
Test STTA
|
Test VM7Luc ER TA
|
Klasa chemiczna MeSH (51)
|
Klasa produktowa (52)
|
|
wartość PC10 (M) (48)
|
wartość PC50 (M) (48)
|
Zakres badanych stężeń (M)
|
Wartość EC50 VM7Luc (M) (49)
|
Najwyższe stężenie na potrzeby ustalenia zakresu dawkowania (M) (50)
|
|
14
|
Dietylostylbestrol
|
56-53-1
|
DOD.
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Węglowodór (cykliczny)
|
Produkt leczniczy, środek weterynaryjny
|
|
12
|
17α-estradiol
|
57-91-0
|
DOD.
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
15
|
mezoheksestrol
|
84-16-2
|
DOD.
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Węglowodór (cykliczny), fenol
|
Produkt leczniczy, środek weterynaryjny
|
|
11
|
4-tert-oktylofenol
|
140-66-9
|
DOD.
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenol
|
Półprodukt
|
|
9
|
Genisteina
|
446-72-0
|
DOD.
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny, produkt leczniczy
|
|
6
|
Bisfenol A
|
80-05-7
|
DOD.
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenol
|
Półprodukt
|
|
2
|
Kemferol
|
520-18-3
|
DOD.
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny
|
|
3
|
Ftalan benzylu butylu
|
85-68-7
|
DOD.
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Kwas karboksylowy, ester, kwas ftalowy
|
Plastyfikator, przemysłowa substancja chemiczna
|
|
4
|
p,p’-Metoksychlor
|
72-43-5
|
DOD.
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Węglowodór (halogenowany)
|
Pestycyd, środek weterynaryjny
|
|
1
|
Etyloparaben
|
120-47-8
|
DOD.
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Kwas karboksylowy, fenol
|
Produkt leczniczy, konserwant
|
|
17
|
Atrazyna
|
1912-24-9
|
UJEM.
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Związek heterocykliczny
|
Herbicyd
|
|
20
|
Spironolakton
|
52-01-7
|
UJEM.
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lakton, Steryd
|
Produkt leczniczy
|
|
21
|
Ketokonazol
|
65277-42-1
|
UJEM.
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Związek heterocykliczny
|
Produkt leczniczy
|
|
22
|
Rezerpina
|
50-55-5
|
UJEM.
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Związek heterocykliczny, indol
|
Produkt leczniczy, środek weterynaryjny
|
|
Skróty: Numer CAS = numer w rejestrze Chemical Abstracts Service; EC50 = połowa maksymalnego stężenia efektywnego badanej substancji; UJEM. = ujemny; DOD. = dodatni; PC10 (and PC50) = stężenie badanej substancji, przy którym reakcja stanowi 10 % (lub 50 % w przypadku PC50) reakcji kontroli dodatniej (E2, 1nM) na każdej płytce.
|
Tabela 4
Wykaz (10) substancji służących do wykazania biegłości w odniesieniu do testu antagonisty
|
|
Substancja (5)
|
Numer CAS
|
Test STTA ER (54)
|
Test VM7Luc ER TA (55)
|
Potencjalne wyniki ER STTA (54)
|
ICCVAM (58) ICCVAM 5
|
Klasa chemiczna MeSH (59)
|
Klasa produktowa (60)
|
|
Działanie ER TA
|
IC50 (M)
|
Zakres badanych stężeń (M)
|
Działanie ER TA
|
IC50
(56) (M)
|
Najwyższe stężenie na potrzeby ustalenia zakresu dawkowania (M) (57)
|
|
1
|
4-hydroksy-tamoksyfen
|
68047-06-3
|
DOD.
|
3,97 × 10-9
|
10-12 – 10-7
|
DOD.
|
2,08 × 10-7
|
2,58 × 10-4
|
umiarkowany DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
2
|
Chlorowodorek raloksyfenu
|
82640-04-8
|
DOD.
|
7,86 × 10-10
|
10-12 – 10-7
|
DOD.
|
1,19 × 10-9
|
1,96 × 10-4
|
umiarkowany DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
3
|
Tamoksyfen
|
10540-29-1
|
DOD.
|
4,91 × 10-7
|
10-10 – 10-5
|
DOD.
|
8,17 × 10-7
|
2,69 × 10-4
|
DOD.
|
DOD.
|
Węglowodór (cykliczny)
|
Produkt leczniczy
|
|
4
|
17β-estradiol
|
50-28-2
|
UJEM.
|
—
|
10-9 – 10-4
|
UJEM.
|
—
|
3,67 × 10-3
|
będą ujemne (*4)
|
ZU
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
5
|
Apigenin
|
520-36-5
|
UJEM.
|
—
|
10-9 – 10-4
|
UJEM.
|
—
|
3,70 × 10-4
|
UJEM.
|
UJEM.
|
Związek heterocykliczny
|
Barwnik, produkt naturalny, półprodukt farmaceutyczny
|
|
6
|
Ftalan di-n-butylu
|
84-74-2
|
UJEM.
|
—
|
10-8 – 10-3
|
UJEM.
|
—
|
3,59 × 10-3
|
UJEM.
|
UJEM.
|
Ester, Kwas ftalowy
|
Składnik kosmetyczny, przemysłowa substancja chemiczna, plastyfikator
|
|
7
|
Flawon
|
525-82-6
|
UJEM.
|
—
|
10-8 – 10-3
|
UJEM.
|
—
|
4,50 × 10-4
|
będą ujemne (*4)
|
ZU
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny, produkt leczniczy
|
|
8
|
Genisteina
|
446-72-0
|
UJEM.
|
—
|
10-9 – 10-4
|
UJEM.
|
—
|
3,70 × 10-4
|
będą ujemne (*4)
|
UJEM.
|
Flawonoid, związek heterocykliczny
|
Produkt naturalny, produkt leczniczy
|
|
9
|
p-n-nonylofenol
|
104-40-5
|
UJEM.
|
—
|
10-9 – 10-4
|
UJEM.
|
—
|
4,54 × 10-4
|
nie badano
|
UJEM.
|
Fenol
|
Półprodukt
|
|
10
|
Resweratrol
|
501-36-0
|
UJEM.
|
—
|
10-8 – 10-3
|
UJEM.
|
—
|
4,38 × 10-4
|
będą ujemne (*4)
|
UJEM.
|
Węglowodór (cykliczny)
|
Produkt naturalny
|
|
Skróty: Numer CAS = numer w rejestrze Chemical Abstracts Service; M = molowy; IC50 = połowa maksymalnego stężenia hamującego badanej substancji; UJEM. = ujemny; ZU = zakładany ujemne; DOD. = dodatni
|
Kryteria dopuszczalności serii badawczej
|
16.
|
Akceptacji lub odrzucenia serii badawczej dokonuje się na podstawie oceny wyników uzyskanych w odniesieniu do wzorców referencyjnych i kontroli stosowanych w przypadku każdego doświadczenia. Wartości PC50 (EC50) lub IC50 w odniesieniu do wzorców referencyjnych powinny spełniać kryteria dopuszczalności przewidziane dla wybranego testu (dla STTA zob. dodatek 2, dla VM7Luc ER TA zob. dodatek 3), a wszystkie kontrole dodatnie/ujemne powinny być prawidłowo sklasyfikowane w odniesieniu do każdego zaakceptowanego doświadczenia. Zdolność do spójnego przeprowadzania testu należy wykazać poprzez opracowanie i utrzymywanie bazy danych historycznych w odniesieniu do wzorców referencyjnych i kontroli (zob. pkt 15). Odchylenia standardowe (SD) lub współczynniki zmienności (CV) dla środków parametrów dopasowania krzywej wzorców referencyjnych z wielu doświadczeń mogą być stosowane jako miara odtwarzalności laboratoryjnej. Ponadto powinny być spełnione następujące zasady dotyczące kryteriów dopuszczalności:
|
—
|
Dane powinny być wystarczające do ilościowej oceny aktywacji receptora estrogenowego (dla testu agonisty) lub jego supresji (dla testu antagonisty) (tj. skuteczności i siły działania).
|
|
—
|
Średnia aktywność reporterowa w odniesieniu do referencyjnego stężenia estrogenów odniesienia powinna być co najmniej równa minimum podanemu w testach dotyczących kontroli z nośnikiem (z rozpuszczalnikiem) w celu zapewnienia odpowiedniej czułości. W odniesieniu do testów STTA i VM7Luc ER TA jest to czterokrotność średniej grupy kontrolnej nośnika na każdej płytce.
|
|
—
|
Badane stężenia powinny pozostać w zakresie rozpuszczalności badanych substancji chemicznych i nie powinny wykazywać cytotoksyczności.
|
|
Analiza danych
|
17.
|
Zdefiniowana procedura interpretacji danych w odniesieniu do każdego testu powinna być stosowana do klasyfikacji odpowiedzi dodatniej i ujemnej.
|
|
18.
|
Spełnienie kryteriów dopuszczalności (pkt 16) wskazuje, że test funkcjonuje prawidłowo, ale nie gwarantuje, że którakolwiek konkretna seria badawcza dostarczy dokładnych danych. Powtarzanie się wyników pierwszej serii badawczej stanowi najlepszą wskazówkę, że dostarczono dokładnych danych. Jeżeli dwie serie badawcze dają odtwarzalne wyniki (np. obie serie badawcze wskazują, że badana substancja chemiczna jest dodatnia), wykonanie trzeciej serii nie jest konieczne.
|
|
19.
|
Jeżeli dwie serie badawcze nie dają odtwarzalnych wyników (np. badana substancja chemiczna jest dodatnia w jednej serii, a ujemna w drugiej) lub jeżeli wymagana jest większa pewność co do wyniku tego testu, należy przeprowadzić co najmniej trzy niezależne serie badawcze. W tym przypadku klasyfikacji dokonuje się na podstawie dwóch zgodnych wyników z trzech.
|
Ogólne kryteria interpretacji danych
|
20.
|
Obecnie nie istnieje powszechnie uzgodniona metoda interpretacji danych pochodzących z ER TA. Zarówno jakościowe (np. dodatnie/ujemne) jak i ilościowe (np. EC50, PC50, IC50) oceny aktywności z udziałem receptora estrogenowego powinny jednak być oparte na danych empirycznych i rzetelnej ocenie naukowej. W przypadku, gdy jest to możliwe, wyniki dodatnie powinny charakteryzować się zarówno wielkością skutku w porównaniu z kontrolą z nośnikiem (z rozpuszczalnikiem) lub estrogenu odniesienia, jak i stężeniem, przy którym występuje skutek (np. EC50, PC50, RPCMax, IC50 itd.).
|
Sprawozdanie z badania
|
21.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Test:
|
—
|
kontrola/wzorzec referencyjny/badana substancja chemiczna;
|
|
—
|
źródło, numer partii, termin przydatności, jeżeli jest dostępny;
|
|
—
|
stabilność badanej substancji chemicznej, jeżeli jest znana;
|
|
—
|
rozpuszczalność i stabilność badanej substancji chemicznej w rozpuszczalniku, jeżeli są znane;
|
|
—
|
pomiar pH, osmolalności i osadu w podłożu, do którego dodano badaną substancję chemiczną, stosownie do przypadku.
|
|
|
|
Substancja jednoskładnikowa:
|
—
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń, stosownie do przypadku i jeśli jest to praktycznie wykonalne, itp.
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszaniny:
|
—
|
opisane w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników.
|
|
|
|
Rozpuszczalnik/nośnik:
|
—
|
charakterystyka (charakter, dostawca i partia);
|
|
—
|
uzasadnienie wyboru rozpuszczalnika/nośnika;
|
|
—
|
rozpuszczalność i stabilność badanej substancji chemicznej w rozpuszczalniku/nośniku, jeżeli są znane.
|
|
|
|
Komórki:
|
—
|
rodzaj i źródło komórek:
|
—
|
Czy receptor estrogenowy jest endogenicznie wyrażony? Jeśli nie, to który receptor (które receptory) poddano transfekcji?
|
|
—
|
Zastosowany (zastosowane) konstrukt reporterowy (konstrukty reporterowe) (w tym gatunki źródłowe);
|
|
—
|
Metoda wyboru w odniesieniu do utrzymania stabilnej transfekcji (w stosownych przypadkach);
|
|
—
|
Czy metoda transfekcji jest odpowiednia dla linii stabilnych?
|
|
|
—
|
liczba pasaży komórek (od rozmrożenia);
|
|
—
|
liczba pasaży komórek w momencie rozmrożenia;
|
|
—
|
metody utrzymywania kultur komórkowych.
|
|
|
|
Warunki badania:
|
—
|
granice rozpuszczalności;
|
|
—
|
opis zastosowanych metod oceny żywotności;
|
|
—
|
skład podłoża; stężenie CO2;
|
|
—
|
stężenia badanej substancji chemicznej;
|
|
—
|
objętość dodanego nośnika i badanej substancji chemicznej;
|
|
—
|
temperatura i wilgotność inkubacji;
|
|
—
|
czas poddawania działaniu substancji chemicznej;
|
|
—
|
zagęszczenie komórek na początku i w trakcie poddania działaniu substancji;
|
|
—
|
kontrole dodatnie i ujemne wzorce referencyjne;
|
|
—
|
odczynniki reporterowe (nazwa produktu, dostawca i partia);
|
|
—
|
kryteria uznania serii badawczej za dodatnią, ujemną lub niejednoznaczną.
|
|
|
|
Kontrola dopuszczalności:
|
—
|
krotności indukcji w odniesieniu do każdej płytki testowej oraz czy spełniają one minimum wymagane w ramach tego testu na podstawie historycznych kontroli;
|
|
—
|
rzeczywiste wartości w odniesieniu do kryteriów dopuszczalności, np. log10EC50, log10PC50, logIC50 oraz krzywa Hilla w odniesieniu do równoczesnych kontroli dodatnich/wzorców referencyjnych.
|
|
|
|
Wyniki:
|
—
|
dane surowe i znormalizowane;
|
|
—
|
maksymalny poziom krotności indukcji;
|
|
—
|
dane dotyczące cytotoksyczności;
|
|
—
|
jeżeli istnieje, najniższe stężenie efektywne (LEC);
|
|
—
|
W stosownych przypadkach wartości RPCMax, PCMax, PC50, IC50 lub EC50;
|
|
—
|
w stosownych przypadkach zależność stężenie-odpowiedź;
|
|
—
|
analizy statystyczne, jeśli takie istnieją, wraz z miarą błędu i wiarygodności (np. SEM, SD, CV lub 95 % CI) oraz opisem sposobu uzyskania tych wartości.
|
|
|
BIBLIOGRAFIA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 34. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 225. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis. „Cancer. Res.” nr 58(23), s. 5367–5373.
|
|
(5)
|
Rogers J.M. i Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology:„Journal of Basic and Applied Research” nr 13(1), s. 67–82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 173. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 174. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(8)
|
OECD (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. [w:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 150. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept.„Climacteric” nr 5, suplement 2, s. 20–26.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated?„Endocr. Relat. Cancer” nr 16(4): s. 1073–1089.
|
|
(11)
|
Younes M. i Honma N. (2011). Estrogen Receptor Beta. „Arch. Pathol. Lab. Med.”, 135(1): s. 63–26.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses.„Journal of Chromatography B” nr 777(1–2), s. 179–189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities. „Toxicol. Sci.” nr 89(1), s. 173–187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions. „Toxicology Letters” nr 126(2), s. 91–98.
|
|
(15)
|
R.D. Combes, (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans. „ATLA Alternatives to Laboratory Animals” nr 28(1), s. 81–118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta. „Biochem. Pharmacol.” nr 71(10), s. 1459–1469.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens. „Toxicol. Lett.” nr 102–103, s. 677–680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action. „Journal of Endocrinology” nr 163(3), s. 379–383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERα In Vivo and In Vitro.„Biochemical and Biophysical Research Communications” nr 243(1), s. 122–126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern. „Journal of Clinical Endocrinology and Metabolism” nr 82(12), s. 4258–4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements.„Molecular and Cellular Endocrinology” nr 299(2), s. 204–211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists. „Mol. Pharmacolog.” nr 54(1), s. 105–112.
|
|
(25)
|
Deroo B.J. i Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies. „Molecular Endocrinology” nr 24(9), s. 1703–1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells. „Experimental Biology and Medicine” nr 230(8), s. 558–568.
|
|
(27)
|
Anderson J.N. Clark J.H. i Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses. „Biochemical and Biophysical Research Communications” nr 48(6), s. 1460–1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA. „Journal of Steroid Biochemistry” nr 3(3), s. 515–522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus. „Recent Progress in Hormone Research” nr 24, s. 45–80.
|
|
(30)
|
E.V. Jensen et al.(1967), Estrogen-Receptor Interactions in Target Tissues. „Archives d’Anatomie Microscopique et de Morphologie Experimentale” nr 56(3), s. 547–569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Dodatek D, Substances Tested in the ER TA Assay. NIH Publication Report (Nr 03–4505).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1. „Environ. Health Persp.” nr 109, s. 785–94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose-Response Studies. „Environ. Health Persp.” nr 111, s. 1530–1549.
|
|
(34)
|
J. Kanno et al.(2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies. „Environ. Health Persp.” nr 111, s. 1550-1558.
|
|
(35)
|
Geisinger et al. (1989), Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors. „Cancer” nr 63, s. 280–288.
|
|
(36)
|
Baldwin et al. (1989), BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro. „In Vitro Cell. Dev. Biol. – Animal” nr 34, s. 649–654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant. „Mol. Endo.” nr 28, s. 2072–2081.
|
|
(38)
|
J.M. Rogers i M.S. Denison (2000), Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals. „In Vitro & Molec. Toxicol.” nr 13, s. 67–82.
|
Dodatek 1
DEFINICJE I SKRÓTY
Kryteria dopuszczalności
: minimalne normy dotyczące przeprowadzania kontroli doświadczalnych i wzorców referencyjnych. Aby doświadczenie zostało uznane za ważne, należy spełnić wszystkie kryteria dopuszczalności.
Dokładność (zgodność)
: stopień zgodności wyników testu z przyjętymi wartościami odniesienia. Jest to miara efektywności testu i jeden z aspektów jego istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na określenie odsetka prawidłowych wyników testu (1).
Agonista
: Substancja, która powoduje reakcję, np. transkrypcję, gdy wiąże się z określonym receptorem.
Antagonista
: Rodzaj receptora liganda lub substancji chemicznej, która nie wywołuje reakcji biologicznej po związaniu z receptorem, ale blokuje lub tłumi reakcje agonistów.
Działanie antyestrogenne
: oznacza zdolność substancji chemicznej do hamowania działania 17β-estradiolu przekazywanego przez receptory estrogenowe.
Morfologia komórki
: Kształt i wygląd komórek wyhodowanych w jednowarstwowej hodowli w pojedynczym dołku płytki do hodowli tkankowych. Umierające komórki często wykazują nieprawidłową morfologię komórkową.
CF
: ramy koncepcyjne OECD dotyczące testowania i oceny substancji zaburzających funkcjonowanie układu hormonalnego.
Poddanie działaniu węgla drzewnego / dekstranu
: Oczyszczanie surowicy stosowanej w hodowli komórkowej. Poddanie działaniu węgla drzewnego/dekstranu (często nazywane „odpędzaniem”) powoduje usunięcie endogennych hormonów i białek wiążących hormony.
Substancja chemiczna
: substancja lub mieszanina.
Cytotoksyczność
: Szkodliwe skutki dla struktury lub funkcji komórki, które mogą ostatecznie spowodować śmierć komórki i mogą być odzwierciedlone przez zmniejszenie liczby komórek w dołku pod koniec okresu narażenia lub zmniejszenie zdolności do pomiaru funkcji komórkowej w porównaniu z równoczesną grupą kontrolną nośnika.
CV
: Współczynnik zmienności
DCC-FBS
: Pokryta dekstranem bydlęca surowica płodowa poddana działaniu węgla drzewnego.
DMEM
: Dulbecco’s Modification of Eagle’s Medium
DMSO
: Sulfotlenek dimetylu
E2
: 17β-estradiol
EC50
: Połowa maksymalnego stężenia efektywnego badanej substancji chemicznej.
ED
: zaburzenia endokrynologiczne
hERα
: ludzki receptor estrogenowy alfa
hERß
: ludzki receptor estrogenowy beta
EFM
: podłoże bez estrogenu. Dulbecco's Modification of Eagle's Medium (DMEM) uzupełniony 4,5 % bydlęcą surowicą płodową poddaną działaniu węgla drzewnego / dekstranu, 1,9 % L-glutaminą i 0,9 % Pen-Strep.
ER
: receptor estrogenowy
ERE
: element reakcji estrogenowej
Aktywność estrogenna
: zdolność substancji chemicznej do naśladowania 17β-estradiolu w jego zdolności do wiązania się z receptorami estrogenowymi i aktywowania ich receptorów. Za pomocą niniejszej metody badawczej można wykryć aktywność estrogenną za pośrednictwem hERα.
ERTA
: transaktywacja receptora estrogenowego
FBS
: bydlęca surowica płodowa
HeLa
: nieśmiertelna ludzka linia komórek szyjki macicy
HeLa9903
: subklon komórkowy HeLa, do którego dokonano stabilnej transfekcji ludzkiego receptora estrogenowegoααi genu reporterowego lucyferazy
IC
50
: połowa maksymalnego stężenia efektywnego badanej hamującej substancji chemicznej.
ICCVAM
: Międzyagencyjny Komitet Koordynacyjny ds. Uznawania Metod Alternatywnych.
Odtwarzalność międzylaboratoryjna
: miara zakresu, w jakim różne wykwalifikowane laboratoria, przy użyciu tego samego protokołu i testując tę samą badaną substancję, mogą uzyskać jakościowo i ilościowo podobne wyniki. Odtwarzalność międzylaboratoryjna jest ustalana w ramach procesów poprzedzających walidację i obejmujących walidację i wskazuje, w jakim stopniu test można z powodzeniem przekazywać między laboratoriami: nazywana jest również odtwarzalnością międzylaboratoryjną (1).
Odtwarzalność wewnątrzlaboratoryjna
: ustalenie stopnia, w jakim wykwalifikowani pracownicy w tym samym laboratorium mogą z powodzeniem odtworzyć wyniki, stosując szczegółowy protokół w różnych momentach. Nazywana również „odtwarzalnością laboratoryjną” (1).
LEC
: najniższe stężenie efektywne to najniższe stężenie badanej substancji chemicznej, które powoduje reakcję (tj. najniższe stężenie badanej substancji chemicznej, przy którym krotność indukcji jest statystycznie różna niż równoległa grupa kontrolna nośnika).
Badanie me-too
: potoczne określenie testu, które jest strukturalnie i funkcjonalnie podobne do zwalidowanej i zaakceptowanej referencyjnej metody badania. Stosowane zamiennie z podobną metodą badań.
MT
: metalotioneina
MMTV
: wirus mysiego nowotworu gruczołu piersiowego
OHT
: 4-hydroksy-tamoksyfen
PBTG
: wytyczne dotyczące badań w oparciu o wyniki
PC (kontrola dodatnia)
: silnie aktywna substancja, najlepiej 17ß-estradiol, która jest włączona do wszystkich badań w celu zapewnienia prawidłowego funkcjonowania testu.
PC
10
: stężenie badanej substancji chemicznej, przy którym mierzona aktywność w ramach testu agonisty wynosi 10 % maksymalnej aktywności wywołanej przez PC (E2 dla 1nM w odniesieniu do analizy STTA) na każdej płytce.
PC
50
: stężenie badanej substancji chemicznej, przy którym mierzona aktywność w ramach testu agonisty wynosi 50 % maksymalnej aktywności wywołanej przez PC (E2 przy referencyjnym stężeniu określonym w metodzie badawczej) na każdej płytce.
PC
Max
: stężenie badanej substancji chemicznej wywołujące RPCMax
Standardy wykonywania badań (PS)
: normy oparte na zwalidowanym teście, zapewniające podstawę do oceny porównywalności proponowanego testu, który jest podobny do metody referencyjnej pod względem mechanistycznym i funkcjonalnym. Uwzględnia się: 1) istotne elementy testu; 2) podstawowy wykaz chemicznych substancji odniesienia, wybranych spośród substancji chemicznych wykorzystywanych w celu wykazania akceptowalnej efektywności zwalidowanej metody badania; oraz 3) podobny poziom dokładności i wiarygodności, na podstawie wyników uzyskanych dla zwalidowanej metody badania, który proponowany test powinien osiągnąć przy ocenie z wykorzystaniem podstawowego wykazu chemicznych substancji odniesienia (1).
Substancje służące do wykazania biegłości
: podzbiór substancji odniesienia zawartych w standardach wykonywania badań, które mogą być stosowane przez laboratoria w celu wykazania kompetencji technicznych za pomocą znormalizowanej metody badawczej. Kryteria kwalifikacji w przypadku tych substancji obejmują zazwyczaj takie czynniki, jak to, że reprezentują one zakres reakcji, są dostępne na rynku i że dostępne są wysokiej jakości dane porównawcze.
Biegłość
: wykazana zdolność do prawidłowego wykonania testu przed badaniem nieznanych substancji.
Estrogen odniesienia (kontrola dodatnia, PC)
: 17β-estradiol (E2, CAS 50-28-2).
Wzorzec referencyjny
: substancja odniesienia wykorzystana do wykazania prawidłowości testu. 17β-estradiol jest wzorcem referencyjnym na potrzeby testów STTA i VM7Luc ER TA.
Referencyjne metody badawcze
: Testy oparte na PBTG 455.
Istotność
: opis powiązania testu z oczekiwanym skutkiem oraz czy jest on znaczący i użyteczny z punktu widzenia określonego celu. Jest to zakres, w jakim test pozwala prawidłowo zmierzyć lub przewidzieć biologiczny skutek będący przedmiotem zainteresowania. Istotność obejmuje uwzględnienie dokładności (zgodności) testu (1).
Wiarygodność
: miara zakresu, w jakim test może zostać wykonany w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jego przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją w drodze obliczenia odtwarzalności wewnątrz- i międzylaboratoryjnej.
RLU
: względne jednostki światła.
RNA
: kwas rybonukleinowy.
PC
Max
: maksymalny poziom reakcji wywołanej przez badaną substancję chemiczną, wyrażony jako procent reakcji wywołanej przez 1 nM E2 na tej samej płytce.
RPMI
: pożywka RPMI 1640 zawierająca 0,9 % Pen-Strep i 8 % bydlęcej surowicy.
Seria badawcza
: pojedyncze doświadczenie oceniające działanie substancji chemicznej na wynik biologiczny testu. Każda seria badawcza stanowi kompletne doświadczenie wykonane w dołkach z kontrpróbą komórek pobranych ze wspólnej puli komórek w tym samym czasie.
Niezależna seria badawcza
: oddzielne, niezależne doświadczenie oceniające działanie substancji chemicznej na wynik biologiczny testu, przy użyciu komórek z innej puli, świeżo rozcieńczonych substancji chemicznych, przeprowadzane w różne dni lub w tym samym dniu przez różnych pracowników.
SD
: odchylenie standardowe.
Czułość
: odsetek wszystkich obecnych/aktywnych substancji prawidłowo sklasyfikowanych w ramach testu. Jest to miara dokładności testu, która daje wyniki kategoryczne i stanowi parametr istotny do celów oceny przydatności testu (1).
Swoistość
: odsetek wszystkich nieobecnych/nieaktywnych substancji prawidłowo sklasyfikowanych w badaniu. Jest to miara dokładności testu, która daje wyniki kategoryczne i stanowi parametr istotny do celów oceny przydatności testu (1).
Stabilna transfekcja
: Kiedy DNA jest przenoszone do hodowanych komórek w taki sposób, że jest stabilnie zintegrowane z genomem komórek, co skutkuje stabilną ekspresją genów, których transfekcji się dokonuje. Klony komórek, w odniesieniu do których dokonano stabilnej transfekcji, są wybierane przez stabilne markery (np. odporność na G418).
Test STTA
: test stabilnie transfekowanej transaktywacji, badanie aktywacji transkrypcji ERα z wykorzystaniem linii komórkowej HeLa 9903.
Badanie
: Pełen zakres prac doświadczalnych wykonanych w celu oceny pojedynczej, specyficznej substancji przy użyciu konkretnego testu. Badanie obejmuje wszystkie etapy, w tym badania rozcieńczenia badanej substancji w pożywce użytej do badań, wstępne serie badawcze zakresu dawkowania, wszystkie niezbędne kompleksowe serie badawcze, analizy danych, zapewnienia jakości, oceny cytotoksyczności itd. Zakończenie badania pozwala na klasyfikację aktywności badanej substancji chemicznej na docelowej toksyczności (tj. aktywna, nieaktywna lub niejednoznaczna), która jest oceniana za pomocą zastosowanej analizy oraz oszacowanie siły działania w stosunku do dodatniej referencyjnej substancji chemicznej.
Substancja
: w rozporządzeniu REACH (61) substancję zdefiniowano jako „pierwiastek chemiczny lub jego związki w stanie, w jakim występują w przyrodzie lub zostają uzyskane za pomocą procesu produkcyjnego, z wszelkimi dodatkami wymaganymi do zachowania ich trwałości oraz wszelkimi zanieczyszczeniami powstałymi w wyniku zastosowanego procesu, wyłączając rozpuszczalniki, które można oddzielić bez wpływu na stabilność i skład substancji”. Bardzo podobna definicja jest stosowana w kontekście Globalnie Zharmonizowanego Systemu Klasyfikacji i Oznakowania Chemikaliów (1).
TA (transaktywacja)
: rozpoczęcie syntezy mRNA w reakcji na specyficzny sygnał chemiczny, taki jak wiązanie estrogenu z receptorem estrogenowym.
Test
: w kontekście niniejszej metody badawczej, test jest jedną z metod przyjętych jako ważne dla spełnienia określonych kryteriów efektywności. Elementy testu obejmują na przykład określoną linię komórkową wraz z powiązanymi warunkami wzrostu, specyficzną pożywkę, w której przeprowadzane jest badanie, warunki ustawienia płytki, rozmieszczenie i rozcieńczenia badanych substancji chemicznych wraz z innymi wymaganymi środkami kontroli jakości oraz związane z nimi etapy oceny danych.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
Transkrypcja
: synteza mRNA.
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
Zweryfikowana metoda badawcza
: test, w odniesieniu do którego zakończono badania walidacyjne w celu określenia jej istotności (w tym dokładności) i wiarygodności w odniesieniu do konkretnego celu. Należy zauważyć, że zweryfikowana metoda badawcza może nie wykazywać dostatecznej efektywności z punktu widzenia dokładności i wiarygodności, aby można było ją uznać za dopuszczalną w odniesieniu do danego celu (1).
Walidacja
: proces, za pomocą którego ustalana jest wiarygodność i adekwatność danego podejścia, metody, testu, procesu lub oceny w określonym celu (1).
VC (grupa kontrolna nośnika)
: rozpuszczalnik używany do rozpuszczania badanych i kontrolnych substancji chemicznych jest badany wyłącznie jako nośnik bez rozpuszczonych substancji chemicznych.
VM7
: Unieśmiertelniona komórka gruczolakoraka, która endogennie wyraża receptor estrogenowy.
VM7Luc4E2
: linia komórkowa VM7Luc4E2 została wyprowadzona z unieśmiertelnionych komórek gruczolakoraka VM7, które endogennie wyrażają obie formy receptora estrogenowego (ERα i ERβ) i dokonano ich stabilnej transfekcji przy użyciu plazmidu pGudLuc7.ERE. Plazmid zawiera cztery kopie syntetycznego oligonukleotydu zawierającego element reakcji estrogenowej w obszarze promotora powyżej sekwencji kodującej genu wirusa mysiego nowotworu gruczołu piersiowego (MMTV) oraz gen lucyferazy robaczka świętojańskiego.
Słaba kontrola dodatnia
: słabo aktywna substancja wybrana z wykazu referencyjnych substancji chemicznych, która jest włączona do wszystkich badań w celu zapewnienia prawidłowego funkcjonowania testu.
Dodatek 2
TEST TRANSAKTYWACJI STABILNIE TRANSFEKOWANEGO LUDZKIEGO RECEPTORA ESTROGENOWEGO W CELU WYKRYCIA ESTROGENOWEJ DZIAŁANIA AGONISTYCZNEGO I ANTAGONISTYCZNEGO SUBSTANCJI CHEMICZNYCH Z WYKORZYSTANIEM LINII KOMÓRKOWEJ HERΑ-HELA-9903
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
1.
|
W niniejszym teście transaktywacji (TA) wykorzystuje się linię komórkową hERα-HeLa-9903 do wykrywania estrogenowej aktywności agonistycznej przekazywanej przez ludzki receptor estrogenowy alfa (hERα). Badanie walidacyjne testu stabilnie transfektowanej transaktywacji (STTA) przeprowadzone przez japoński Instytut Oceny i Badań Substancji Chemicznych (CERI), wykorzystujące linię komórkową hERα-HeLa-9903 do wykrywania estrogenowego działania agonistycznego i antagonistycznego przekazywanego przez ludzki receptor estrogenowy alfa (hERα), dowiodło adekwatności i wiarygodności testu w odniesieniu do jego zamierzonego celu (1).
|
|
2.
|
Szczególnym przeznaczeniem tego testu jest wykrywanie transaktywacji za pośrednictwem hERα poprzez pomiar chemiluminescencji jako punktu końcowego. Z powodu nadaktywacji lucyferazowego genu reporterowego zgłoszono jednak sygnały niezwiązane z receptorami pośredniczącymi w luminescencji przy stężeniach fitoestrogenu przekraczających 1 μM (2)(3). Chociaż krzywa dawka-efekt wskazuje, że rzeczywista aktywacja systemu receptora estrogenowego występuje przy niższych stężeniach, w stabilnej transfekcji systemów badania aktywacji transkrypcji receptora estrogenowego należy ostrożnie badać ekspresję lucyferazy uzyskaną przy wysokich stężeniach fitoestrogenów lub podobnych związków chemicznych, co do których przypuszcza się, że wywołują one – podobnie jak fitoestrogeny – nadaktywację lucyferazowego genu reporterowego (dodatek 1).
|
|
3.
|
Przed rozpoczęciem stosowania tego testu do celów regulacyjnych należy zapoznać się z sekcjami „WPROWADZENIE OGÓLNE” oraz „ELEMENTY BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”. Definicje i skróty stosowane w niniejszej wytycznej dotyczącej badań opisano w dodatku 2.1.
|
ZASADA WYKONANIA TESTU (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
4.
|
Test ten służy do wiązania sygnału receptora estrogenowego z ligandem. Po wiązaniu ligandu kompleks receptor-ligand przenosi się do jądra komórkowego, gdzie wiąże określone elementy odpowiedzi DNA i transaktywuje gen reporterowy lucyferazy robaczka świętojańskiego, powodując zwiększoną ekspresję komórkową enzymu lucyferazy. Lucyferyna jest substratem przekształcanym przez enzym lucyferaza przekształca w produkt bioluminescencyjny, który można zmierzyć ilościowo przy użyciu luminometru. Aktywność lucyferazy można ocenić w szybki i niedrogi sposób, wykorzystując szereg dostępnych na rynku zestawów badawczych.
|
|
5.
|
Układ badawczy wykorzystuje linię komórkową hERα-HeLa-9903, pochodzącą z ludzkiego guza szyjki macicy, z dwoma stabilnie wstawionymi konstruktami: (i) konstrukt ekspresji hERαα (kodujący ludzki receptor o pełnej długości), oraz (ii) konstrukt reportera lucyferazy ogniotrwałej, zawierający pięć tandemowych powtórzeń elementu reakcji estrogenowej (ERE) witellogeniny napędzanego przez element promotora TATA metalotioneiny (MT) myszy. Wykazano, że konstrukt genu TATA MT myszy ma najlepszą wydajność i dlatego jest powszechnie stosowany. W konsekwencji linia komórkowa hERα-HeLa-9903 może mierzyć zdolność badanej substancji chemicznej do wywoływania transaktywacji za pośrednictwem hERα ekspresji genu lucyferazy.
|
|
6.
|
W przypadku testu agonisty receptora estrogenowego interpretacji danych dokonuje się na podstawie tego, czy maksymalny poziom reakcji wywołany przez badaną substancję chemiczną jest równy lub przekracza reakcję agonisty równą 10 % reakcji wywołanej przez maksymalne indukujące (1 nM) stężenie kontroli dodatniej (PC) 17β-estradiolu (E2) (tj. PC10). W przypadku testu antagonisty receptora estrogenowego interpretacji danych dokonuje się na podstawie tego, czy reakcja wykazuje co najmniej 30-procentowa redukcja działania w wyniku reakcji wywołanej przez wzbogacenie kontroli (25 pM E2) bez cytotoksyczności. Analiza danych i interpretacja zostały szczegółowo omówione w pkt 34–48.
|
PROCEDURA
Linie komórkowe
|
7.
|
W teście należy wykorzystać stabilnie transfekowaną linię komórkową hERα-HeLa-9903. Przedmiotową linię komórkową można uzyskać od banku komórek Japońskiego Zbioru Zasobów Biologicznych do Celów Badawczych (JCRB) (62), po podpisaniu porozumienia o transferze materiału.
|
|
8.
|
Do badań należy używać wyłącznie komórek charakteryzujących się tym, że nie zawierają mykoplazmy. RT-PCR (reakcja łańcuchowa polimerazy w czasie rzeczywistym) jest metodą do wyboru w odniesieniu do czułego wykrywania zakażenia mykoplazmą (4)(5)(6).
|
Stabilność linii komórkowej
|
9.
|
W celu monitorowania stabilności linii komórkowej należy stosować E2, 17α-estradiol, 17α-metylotestosteron i kortykosteron jako wzorce referencyjne na potrzeby testu agonisty, a pełna krzywa zależności stężenie-odpowiedź w zakresie badanego stężenia podanym w tabeli 1 powinna być mierzona co najmniej raz za każdym razem, gdy wykonuje się test, a wyniki powinny być zgodne z wynikami podanymi w tabeli 1.
|
|
10.
|
W przypadku testu antagonisty, pełne krzywe stężeń dla dwóch wzorców referencyjnych, tamoksyfenu i flutamidu, powinny być mierzone jednocześnie z każdą serią badawczą. Należy monitorować prawidłową klasyfikację jakościową jako dodatnie lub ujemne w odniesieniu do tych dwóch substancji chemicznych.
|
Kultura komórkowa i warunki posiewu
|
11.
|
Komórki powinny być utrzymywane w podłożu Eagle'a bez czerwieni fenolowej, uzupełnionym 60 mg/l antybiotyku kanamycyny i 10 % pokrytej dekstranem, poddanej działaniu węgla drzewnego bydlęcej surowicy płodowej (DCC-FBS), w inkubatorze CO2 (5 % CO2) w temperaturze 37±1 °C. Po osiągnięciu 75 % – 90 % konfluencji, komórki mogą być hodowane wtórnie w 10 ml 0,4 x 105 – 1 x 105 komórek/ml w odniesieniu do 100 mm naczynia do kultury komórkowej. Komórki powinny być zawieszone z 10 % FBS-EMEM (który jest taki sam jak EMEM z DCC-FBS), a następnie umieszczone w dołkach mikropłytki z gęstością 1 x 104 komórek/(100 μl na dołek). Następnie należy dokonać wstępnej inkubacji komórek w 5 % inkubatorze CO2 w temperaturze 37 °± 1 °C przez 3 godziny przed narażeniem na działanie substancji chemicznej. Wyroby z tworzyw sztucznych powinny być wolne od aktywności estrogenowej.
|
|
12.
|
Aby zachować integralność reakcji, komórki powinny wyrastać z zamrożonego zapasu w kondycjonowanych pożywkach przez więcej niż jeden pasaż i nie powinny być hodowane dłużej niż przez 40 pasaży. W odniesieniu do linii komórkowej hERα-HeLa-9903 będzie to mniej niż trzy miesiące. Efektywność komórek może jednak zostać zmniejszona, jeżeli są one hodowane w nieodpowiednich warunkach hodowli kultur.
|
|
13.
|
DCC-FBS można przygotować w sposób opisany w dodatku 2.2 lub uzyskać ze źródeł komercyjnych.
|
Kryteria dopuszczalności
Dodatnie i ujemne wzorce referencyjne w odniesieniu do testu agonisty receptora estrogenowego
|
14.
|
Przed badaniem i w jego trakcie należy zweryfikować reaktywność układu badawczego, stosując odpowiednie stężenia silnego estrogenu: E2, słabego estrogenu (17α-estradiol), bardzo słabego agonisty (17α-metylotestosteron) i substancji ujemnej (kortykosteron). Dopuszczalne wartości zakresu uzyskane z badania walidacyjnego (1) podano w tabeli 1. Te 4 równoległe wzorce referencyjne powinny być włączone do każdego doświadczenia, a wyniki powinny mieścić się w podanych dopuszczalnych granicach. Jeżeli tak nie jest, należy określić przyczynę niespełnienia kryteriów dopuszczalności (np. obsługa komórek oraz surowica i antybiotyki w odniesieniu do jakości i stężenia) i powtórzyć test. Po spełnieniu kryteriów dopuszczalności, w celu zapewnienia minimalnej zmienności wartości EC50, PC50 i PC10, niezbędne jest konsekwentne stosowanie materiałów do hodowli komórkowej. Cztery jednoczesne wzorce referencyjne, które powinny być włączone do każdego doświadczenia (przeprowadzanego w tych samych warunkach, w tym w odniesieniu do materiałów, poziomu pasażowania komórek i techników), mogą zapewnić czułość testu, ponieważ PC10 trzech dodatnich wzorców referencyjnych powinny mieścić się w dopuszczalnym zakresie, podobnie jak PC50 i EC50, w przypadkach gdy można je obliczyć (zob. tabela 1).
|
Tabela 1
Dopuszczalne wartości zakresu czterech wzorców referencyjnych w odniesieniu do testu agonisty receptora estrogenowego
|
Nazwa
|
logPC50
|
logPC10
|
logEC50
|
Krzywa Hilla
|
Zakres badania
|
|
17β-estradiol (E2) Nr CAS: 50-28-2
|
-11,4~-10,1
|
< -11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-estradiol Nr CAS: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Kortykosteron Nr CAS: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-metylotestosteron Nr CAS: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Dodatnie i ujemne wzorce referencyjne w odniesieniu do testu antagonisty receptora estrogenowego
|
15.
|
Przed badaniem i w jego trakcie należy zweryfikować reaktywność układu badawczego, stosując odpowiednie stężenia substancji dodatniej (tamoksyfen) oraz substancji ujemnej (flutamid). Dopuszczalne wartości zakresu uzyskane z badania walidacyjnego (1) podano w tabeli 2. Te dwa równoległe wzorce referencyjne powinny być włączone do każdego doświadczenia, a wyniki powinny być oceniane jako prawidłowe, jak pokazano w kryteriach. Jeżeli tak nie jest, należy określić przyczynę niespełnienia kryteriów (np. obsługa komórek oraz surowica i antybiotyki w odniesieniu do jakości i stężenia) i powtórzyć test. Ponadto należy obliczyć wartości IC50 dla substancji dodatniej (tamoksyfen), a wyniki powinny mieścić się w podanych dopuszczalnych granicach. Po spełnieniu kryteriów dopuszczalności, w celu zapewnienia minimalnej zmienności wartości IC50, niezbędne jest konsekwentne stosowanie materiałów do hodowli komórkowej. Dwa jednoczesne wzorce referencyjne, które powinny być włączone do każdego doświadczenia (przeprowadzanego w tych samych warunkach, jeśli chodzi o materiały, poziom pasażowania komórek i techników), mogą zapewnić czułość testu (zob. tabela 2).
|
Tabela 2
Kryteria i dopuszczalne wartości zakresu dwóch wzorców referencyjnych w odniesieniu do testu antagonisty receptora estrogenowego
|
Nazwa
|
Kryteria
|
LogIC50
|
Zakres badania
|
|
Tamoksyfen Nr CAS: 10540-29-1
|
dodatnie: należy wyliczyć IC50
|
-5942~-7596
|
10-10~10-5M
|
|
Flutamid Nr CAS: 13311-84-7
|
ujemne: nie należy obliczać IC30
|
—
|
10-10~10-5M
|
Kontrole dodatnie i grupy kontrolne nośnika
|
16.
|
Kontrolę dodatnią (PC) w odniesieniu do testu agonistycznego receptora estrogenowego (1 nM E2) oraz w odniesieniu do testu antagonistycznego receptora estrogenowego (10 μM TAM) należy przeprowadzić co najmniej trzykrotnie na każdej płytce. Nośnik stosowany do rozpuszczenia badanej substancji chemicznej należy zbadać jako grupę kontrolną nośnika (VC) co najmniej trzykrotnie na każdej płytce. Oprócz tej VC, jeżeli w ramach PC używany jest inny nośnik niż badana substancja chemiczna, należy przeprowadzić inną VC co najmniej trzykrotnie na tej samej płytce z PC.
|
Kryteria jakości w odniesieniu do testu agonisty receptora estrogenowego
|
17.
|
Średnia aktywność lucyferazy w kontroli dodatniej (1 nM E2) powinna być co najmniej czterokrotnie większa od średniej VC na każdej płytce. Kryterium to ustalono na podstawie wiarygodności wartości punktów końcowych z badania walidacyjnego (historycznie od czterech do trzydziestu razy).
|
|
18.
|
W odniesieniu do kontroli jakości testu krotność indukcji odpowiadająca wartości PC10 jednoczesnej PC (1 nM E2) powinna być większa niż 1+2SD wartości krotności indukcji (= 1) jednoczesnej VC. Dla celów ustalania priorytetów, wartość PC10 może być przydatna do uproszczenia analizy danych wymaganej w porównaniu z analizą statystyczną. Mimo że analiza statystyczna dostarcza informacji na temat istotności, analiza taka nie jest parametrem ilościowym w odniesieniu do potencjału na podstawie stężeń, a zatem jest mniej przydatna do celów ustalania priorytetów.
|
Kryteria jakości w odniesieniu do testu antagonisty receptora estrogenowego
|
19.
|
Średnia aktywność lucyferazy we wzbogaceniu kontroli (25 nM E2) powinna być co najmniej czterokrotnie większa od średniej VC na każdej płytce. Kryterium to ustalono na podstawie wiarygodności wartości punktów końcowych z badania walidacyjnego.
|
|
20.
|
W odniesieniu do kontroli jakości testu względna aktywacja transkrypcyjna (RTA) 1 nM E2 powinna być większa niż 100 %, RTA 1 μM 4-kydroksytamoksyfenu (OHT) powinna być mniejsza niż 40,6 %, a RTA 100 μM digitoniny (Dig) powinna być mniejsza niż 0 %.
Wykazanie biegłości laboratorium (zob. pkt 14 oraz tabela 3 i 4 w „ELEMENTY BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO” niniejszej metody badawczej).
|
Nośnik
|
21.
|
Sulfotlenek dimetylowy (DMSO), lub odpowiedni rozpuszczalnik, o tym samym stężeniu, co w przypadku różnych kontroli dodatnich i ujemnych, a badane substancje chemiczne powinny być stosowane jako jednoczesna kontrola z nośnikiem. Badaną substancję chemiczną należy rozpuścić w nośniku, który rozpuszcza tę badaną substancję chemiczną i jest mieszalny z podłożem komórkowym. Odpowiednie nośniki stanowią woda, etanol (95–100 % czystości) i sufotlenek dimetylowy. Jeżeli stosuje się sufotlenek dimetylowy, poziom nie powinien przekroczyć 0,1 % (obj./obj.). Dla każdego nośnika należy wykazać, że maksymalna zastosowana objętość nie wykazuje właściwości cytotoksycznych i nie ma wpływu na skuteczność testu.
|
Przygotowanie badanych substancji chemicznych
|
22.
|
Ogólnie rzecz biorąc, badane substancje chemiczne powinny być rozpuszczone w sulfotlenku dimetylowym lub innym odpowiednim rozpuszczalniku i seryjnie rozcieńczane tym samym rozpuszczalnikiem w stosunku 1:10 w celu przygotowania roztworów do rozcieńczenia z podłożem.
|
Rozpuszczalność i cytotoksyczność: Uwagi dotyczące ustalania zakresu dawkowania
|
23.
|
Należy przeprowadzić badanie wstępne w celu określenia właściwego zakresu stężeń substancji chemicznych, które mają być badane, oraz w celu upewnienia się, czy badana substancja chemiczna może mieć problemy z rozpuszczalnością i cytotoksycznością. Początkowo substancje chemiczne są badane do maksymalnego stężenia 1 μl/ml, 1 mg/ml lub 1 mM, w zależności od tego, które jest najniższe. Na podstawie zakresu cytotoksyczności lub braku rozpuszczalności zaobserwowanego w badaniu wstępnym, pierwsza określona seria badawcza powinna obejmować badanie substancji chemicznej w ciągach stężeń wzrastających w postępie logarytmicznym, począwszy od maksymalnego dopuszczalnego stężenia (np. 1 mM, 100 μM, 10 μM itd.) oraz odnotowanej obecności zmętnienia, osadu lub cytotoksyczności. Stężenia w drugiej, a w razie potrzeby w trzeciej serii badawczej należy odpowiednio dostosować, aby lepiej scharakteryzować krzywą zależności stężenie-odpowiedź oraz aby uniknąć stężeń, które okazały się nierozpuszczalne lub wywołują nadmierną cytotoksyczność.
|
|
24.
|
W odniesieniu do agonistów i antagonistów receptora estrogenowego obecność rosnących poziomów cytotoksyczności może znacząco zmienić lub wyeliminować typową reakcję sigmoidalną i powinna być brana pod uwagę przy interpretacji danych. Należy stosować metody badania cytotoksyczności, które mogą dostarczyć informacji dotyczących 80 % żywotności komórek, wykorzystując odpowiednie testy oparte na doświadczeniu laboratoryjnym.
|
|
25.
|
Jeżeli wyniki badania cytotoksyczności wykażą, że stężenie badanej substancji chemicznej zmniejszyło liczbę komórek o przynajmniej 20 %, stężenie to należy uznać za cytotoksyczne, a stężenia na poziomie co najmniej stężenia cytotoksycznego powinny być wyłączone z oceny.
|
Narażenie na działanie badanej substancji chemicznej oraz struktura płytki testowej
|
26.
|
Procedurę rozcieńczeń substancji chemicznych (etapy 1 i 2) oraz narażenia komórek (etap 3) można przeprowadzić w następujący sposób:
Etap 1: Każda badana substancja chemiczna powinna być seryjnie rozcieńczona w sulfotlenku dimetylowym lub odpowiednim rozpuszczalniku i dodana do dołków mikropłytki w celu uzyskania końcowych stężeń seryjnych, jak określono we wstępnym badaniu zakresu dawkowania (zazwyczaj w seriach np. 1 mM, 100 μM, 10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM i 10 pM (10-3-10-11 M)) potrójnego badania.
Etap 2: Rozcieńczenie substancji chemicznej: Najpierw rozcieńczyć 1,5 μl badanej substancji chemicznej w rozpuszczalniku do objętości 500 μl podłoża.
Etap 3: Narażenie komórek na działanie substancji chemicznej: Dodać 50 μl rozcieńczenia z podłożem (przygotowanym w etapie 2) do dołka testowego zawierającego 104 komórek/100 μl/dołek.
Zalecana ostateczna objętość podłoża wymagana dla każdego dołka wynosi 150 μl. Próbki do badań i wzorce referencyjne mogą być przyporządkowane w sposób przedstawiony w tabeli 3 i tabeli 4.
Tabela 3
Przykładowe przyporządkowanie stężenia na płytkach do wzorców referencyjnych na płytce testowej w teście agonisty receptora estrogenowego
|
Wiersz
|
17α-metylotestosteron
|
Kortykosteron
|
17α-estradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
stężenie 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
stężenie 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
stężenie 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
stężenie 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
stężenie 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
stężenie 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
stężenie 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Grupa kontrolna nośnika (0,1 % sulfotlenek dimetylowy); PC: Kontrola dodatnia (1 nM E2)
|
|
|
27.
|
Wzorce referencyjne (E2, 17α-estradiol, 17-μετψλοτεστοστερον ι κορτψκοστερον) ναλε ψ βαδα ω κα δεφ σεριι βαδαωχζεφ (ταβελα 3). Dołki kontroli dodatniej poddane działaniu 1 nM E2, w których może nastąpić maksymalna indukcja E2 i dołki kontroli z nośnikiem poddane działaniu sulfotlenku dimetylowego (lub odpowiedniego rozpuszczalnika) należy uwzględnić na każdej płytce testowej (tabela 4). Jeżeli w tym samym doświadczeniu wykorzystywane są komórki z różnych źródeł (np. inna liczba pasaży, różne partie itd.), wzorce referencyjne powinny być badane w odniesieniu do każdego źródła komórek.
Tabela 4
Przykładowe przyporządkowanie stężenia na płytkach do badanych i pochodzących z płytki kontrolnych substancji chemicznych na płytce testowej w teście agonisty receptora estrogenowego
|
Wiersz
|
Badana substancja chemiczna nr 1
|
Badana substancja chemiczna nr 2
|
Badana substancja chemiczna nr 3
|
Badana substancja chemiczna nr 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
stężenie 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
stężenie 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
stężenie 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
stężenie 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
stężenie 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
stężenie 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
stężenie 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Grupa kontrolna nośnika (0,1 % sulfotlenek dimetylowy); PC: Kontrola dodatnia (1 nM E2)
|
Tabela 5
Przykładowe przyporządkowanie stężenia na płytkach do wzorców referencyjnych na płytce testowej w teście antagonisty receptora estrogenowego

|
|
28.
|
Aby ocenić aktywność antagonistyczną substancji chemicznych, dołki testowe znajdujące się w rzędach od A do G powinny być wzbogacone 25 pM E2. Wzorce referencyjne(tamoksyfen i flutamid) należy badać w każdej serii badawczej. Dołki kontroli dodatniej poddane działaniu 1 nM E2, które mogą być wykorzystywane jako kontrola jakości linii komórkowej hERα-HeLa-9903, dołki kontroli z nośnikiem poddane działaniu sulfotlenku dimetylowego (lub odpowiedniego rozpuszczalnika), dołki 0,1 % sulfotlenku dimetylowego poddane działaniu dodatku sulfotlenku dimetylowego do wzbogaconego E2 odpowiadającego „wzbogaceniu kontroli”, dołki poddane działaniu końcowego stężenia 1 μM OHT oraz dołki poddane działaniu 100 μM Dig należy uwzględnić na każdej płytce testowej (tabela 5). Kolejna płytka testowa powinna być wykonana według tego samego układu płytki bez dołków z wzorcami porównawczymi (tabela 6). Jeżeli w tym samym doświadczeniu wykorzystywane są komórki z różnych źródeł (np. inna liczba pasaży, różne partie itd.), wzorce referencyjne powinny być badane w odniesieniu do każdego źródła komórek.
Tabela 6
Przykładowe przyporządkowanie stężenia na płytkach do badanych i pochodzących z płytki kontrolnych substancji chemicznych na płytce testowej w teście antagonisty receptora estrogenowego

|
|
29.
|
Brak efektów styku powinien zostać potwierdzony, w stosownych przypadkach, a jeśli podejrzewa się efekty styku, należy zmienić układ płytki, aby uniknąć takich efektów. Można na przykład zastosować układ płytek z wyłączeniem dołków skrajnych.
|
|
30.
|
Po dodaniu substancji chemicznych należy dokonać inkubacji płytek testowych w 5 % inkubatorze CO2 w temperaturze 37 °± 1 °C przez 20–24 godzin w celu wywołania produktów genu reporterowego.
|
|
31.
|
Należy zwrócić szczególną uwagę na związki charakteryzujące się wysoką lotnością. W takich przypadkach położone obok dołki kontrolne mogą generować fałszywie dodatnie wyniki, co należy uwzględnić w świetle oczekiwanych i historycznych wartości kontroli. W nielicznych przypadkach, w których zmienność może budzić obawy, zaleca się zastosowanie „uszczelniaczy płytek”, które podczas badania mogą pomóc w skutecznym odizolowaniu poszczególnych dołków.
|
|
32.
|
Aby zapewnić niezależność badań ostatecznych dotyczących tej samej substancji chemicznej, ich powtórki należy przeprowadzać w różne dni.
|
Test lucyferazy
|
33.
|
W teście można stosować komercyjny odczynnik do testów lucyferazy [np. System Steady-Glo® Luciferase Assay (Promega, E2510 lub równoważny)] lub standardowy system testów na lucyferazę (np. Promega, E1500 lub równoważny), pod warunkiem spełnienia kryteriów dopuszczalności. Odczynniki testowe należy dobierać na podstawie czułości luminometru, który ma być użyty. W przypadku stosowania standardowego systemu testu lucyferazy przed dodaniem substratu należy użyć odczynnika do lizy kultury komórkowej (np. Promega, E1531 lub równoważnego). Odczynnik lucyferazy należy stosować zgodnie z instrukcjami producenta.
|
ANALIZA DANYCH
Test agonisty receptora estrogenowego
|
34.
|
W przypadku testu agonisty receptora estrogenowego w celu uzyskania względnej aktywności transkrypcyjnej do kontroli dodatniej (1 nM E2), sygnały luminescencyjne z tej samej płytki mogą być analizowane według następujących kroków (dopuszczalne są również inne równoważne procesy matematyczne):
Krok 1. Obliczyć średnią wartość VC.
Krok 2. Odjąć średnią wartość VC od wartości każdego dołka, aby znormalizować dane.
Krok 3. Obliczyć średnią wartość znormalizowanej PC.
Krok 4. Podzielić znormalizowaną wartość każdego dołka w danej płytce przez średnią wartość znormalizowanej PC (PC = 100 %).
Ostateczną wartością każdego dołka jest względna aktywność transkrypcyjna dla tego dołka w stosunku do reakcji PC.
Krok 5. Obliczyć średnią wartość względnej aktywności transkrypcyjnej dla każdej grupy stężeń badanej substancji chemicznej. Reakcja ma dwa wymiary: uśrednioną aktywność transkrypcyjną (reakcja) i stężenie, przy którym występuje reakcja (zob. następna sekcja).
|
Kwestie indukcji EC50, PC50 i PC10
|
35.
|
Pełna krzywa zależności stężenie-odpowiedź jest wymagana do obliczenia EC50, ale nie zawsze może być to osiągalne lub praktyczne ze względu na ograniczenia zakresu stężeń w badaniu (na przykład z powodu cytotoksyczności lub problemów z rozpuszczalnością). Ponieważ jednak EC50 i maksymalny poziom indukcji (odpowiadający górnej wartości równania Hilla) są parametrami informacyjnymi, parametry te powinny być w miarę możliwości zgłaszane. Aby obliczyć wartość EC50 i maksymalny poziom indukcji, należy skorzystać z odpowiedniego oprogramowania do obliczeń statystycznych (np. oprogramowanie statystyczne Graphpad Prism). Jeśli równanie logistyczne Hilla ma zastosowanie do danych związanych z zależnością stężenie-odpowiedź, EC50 należy obliczyć za pomocą następującego równania (7):
Y = najniższa wartość + (najwyższa wartość -najniższa wartość/ (1+10 wykł. ((log EC50 -X) x krzywa Hilla)) gdzie:
X oznacza logarytm stężenia; oraz
Y oznacza reakcję i Y przebiega od najniższej wartości do najwyższej w postaci krzywej sigmoidalnej. Najniższa wartość jest ustalona na zero w równaniu logistycznym Hilla.
|
|
36.
|
W odniesieniu do każdej badanej substancji chemicznej należy podać następujące informacje:
RPCMax, które oznacza maksymalny poziom reakcji wywołanej przez badaną substancję chemiczną, wyrażony jako procent reakcji wywołanej przez 1 nM E2 na tej samej płytce, a także PCMax (stężenie związane z PCMax); oraz
w przypadku dodatnich substancji chemicznych – stężenia, które indukują PC10 oraz, w stosownych przypadkach, PC50.
|
|
37.
|
Wartość PCx można obliczyć poprzez interpolację między 2 punktami na współrzędnej X-Y, jednym bezpośrednio powyżej i jednym bezpośrednio poniżej wartości PCx. Jeżeli punkty danych leżące bezpośrednio powyżej i poniżej wartości PCx mają odpowiednio współrzędne (a,b) i (c,d), wówczas wartość PCx można obliczyć przy użyciu następującego równania:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Opisy wartości PC podano na rysunku 1 poniżej.
Rysunek 1
Przykład wyprowadzania wartości PC. PC (1 nM E2) jest uwzględniony na każdej płytce testowej

|
Test antagonisty receptora estrogenowego
|
39.
|
W przypadku testu antagonisty receptora estrogenowego w celu uzyskania względnej aktywności transkrypcyjnej do wzbogacenia kontroli (25 pM E2), sygnały luminescencyjne z tej samej płytki mogą być analizowane według następujących kroków (dopuszczalne są również inne równoważne procesy matematyczne):
Krok 1. Obliczyć średnią wartość VC.
Krok 2. Odjąć średnią wartość VC od wartości każdego dołka, aby znormalizować dane. Krok 3. Obliczyć średnią wartość znormalizowanego wzbogacenia kontroli.
Krok 4. Podzielić znormalizowaną wartość każdego dołka danej płytki przez średnią wartość znormalizowanego wzmocnienia w kontroli (wzbogacenie kontroli = 100 %).
Ostateczną wartością każdego dołka jest względna aktywność transkrypcyjna dla tego dołka w stosunku do wzmocnienia reakcji kontroli.
Krok 5. Obliczyć średnią wartość względnej aktywności transkrypcyjnej dla każdego poddania działaniu substancji.
|
Parametry indukcji IC30 i IC50
|
40.
|
W przypadku dodatnich substancji chemicznych należy podać stężenia, które indukują IC30 oraz, w stosownych przypadkach, IC50.
|
|
41.
|
Wartość ICx można obliczyć poprzez interpolację między 2 punktami na współrzędnej X-Y, jednym bezpośrednio powyżej i jednym bezpośrednio poniżej wartości ICx. Jeżeli punkty danych leżące bezpośrednio powyżej i poniżej wartości ICx mają odpowiednio współrzędne (c,d) i (a,b), wówczas wartość ICx można obliczyć przy użyciu następującego równania:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Rysunek 2
Przykład wyprowadzania wartości VC. Wzbogacenie kontroli (25 pM E2) jest uwzględnione na każdej płytce testowej

RTA: względna aktywność transkrypcyjna
|
|
42.
|
Wyniki powinny opierać się na dwóch (lub trzech) niezależnych seriach badawczych. Jeżeli dwie serie badawcze dają porównywalne i tym samym odtwarzalne wyniki, nie jest konieczne wykonywanie trzeciej serii. Aby wyniki zostały dopuszczone, powinny:
|
—
|
Spełniać kryteria dopuszczalności (zob. kryteria dopuszczalności pkt 14–22);
|
|
Kryteria interpretacji danych
Tabela 7
Dodatnie i ujemne kryteria podejmowania decyzji w odniesieniu do testu agonisty receptora estrogenowego
|
Dodatnia
|
Jeżeli osiągnięto RPCMax, które jest równe lub przekracza 10 % reakcji kontroli dodatniej w co najmniej dwóch z dwóch lub dwóch z trzech serii badawczych.
|
|
Ujemna
|
Jeżeli RPCMax nie osiągnie co najmniej 10 % reakcji kontroli dodatniej w dwóch z dwóch lub dwóch z trzech serii badawczych.
|
Tabela 8
Dodatnie i ujemne kryteria podejmowania decyzji w odniesieniu do testu antagonisty receptora estrogenowego
|
Dodatnia
|
Jeżeli IC30 oblicza się w co najmniej dwóch z dwóch lub dwóch z trzech serii badawczych.
|
|
Ujemna
|
Jeżeli IC30 nie da się obliczyć w dwóch z dwóch lub dwóch z trzech serii badawczych.
|
|
43.
|
Kryteria interpretacji danych przedstawiono w tabelach 7 i 8. Wyniki dodatnie będą charakteryzować się zarówno wielkością skutku, jak i stężeniem, przy którym występuje skutek. Wyrażanie wyników jako stężenia, przy którym 50 % (PC50) lub 10 % (PC10) wartości kontroli dodatniej jest osiągane w odniesieniu do testu agonisty, a 50 % (IC50) lub 30 % (IC30) wzbogaconej wartości kontrolnej jest hamowane w odniesieniu testu antagonistycznego, osiąga oba te cele. Badana substancja chemiczna jest jednak uważana za dodatnią, jeżeli maksymalna reakcja wywołana przez badaną substancję chemiczną (RPCMax) jest równa lub przekracza 10 % reakcji kontroli dodatniej w co najmniej dwóch z dwóch lub dwóch z trzech serii badawczych, natomiast badana substancja chemiczna jest uważana za ujemną, jeżeli RPCMax nie osiągnie co najmniej 10 % reakcji kontroli dodatniej w dwóch z dwóch lub dwóch z trzech serii badawczych.
|
|
44.
|
Obliczeń PC10, PC5 i PCMaks w teście agonisty receptora estrogenowego oraz IC30 i IC50 w teście antagonisty receptora estrogenowego można dokonać, wykorzystując arkusz kalkulacyjny, który jest dostępny na publicznej stronie internetowej OECD poświęconej wytycznym dotyczącym badań (63).
|
|
45.
|
Co najmniej podwójne uzyskanie wartości PC10 lub PC50 i IC30 lub IC50 powinno wystarczyć. Jeżeli jednak wynikająca z tego linia podstawowa dotycząca danych będących w tym samym zakresie stężeń wykaże zmienność z niedopuszczalnie wysokim współczynnikiem zmienności (CV; %), dane te mogą zostać uznane za niewiarygodne i w takim wypadku należy określić źródło wysokiej zmienności. CV potrójnych danych surowych (tj. danych dotyczących intensywności luminescencji) punktów danych wykorzystywanych do obliczenia PC10 powinno wynosić mniej niż 20 %.
|
|
46.
|
Spełnienie kryteriów dopuszczalności wskazuje, że system testów funkcjonuje prawidłowo, ale nie gwarantuje, że którakolwiek konkretna seria badawcza dostarczy dokładnych danych. Powielenie wyników pierwszej serii badawczej gwarantuje największe prawdopodobieństwo uzyskania dokładnych danych.
|
|
47.
|
W przypadku testu agonisty receptora estrogenowego tam, gdzie istnieje potrzeba uzyskania dodatkowych informacji w celach rozpoznania i ustalenia priorytetów niniejszej wytycznej dotyczącej badań w odniesieniu do badanych substancji chemicznych, a zwłaszcza substancji chemicznych PC10–PC49, jak również substancji chemicznych podejrzewanych o nadmierną stymulację lucyferazy, można potwierdzić, że jej obserwowana aktywność jest wyłącznie reakcją szczególną dla receptora estrogenowego alfa, przy wykorzystaniu antagonisty receptora estrogenowego alfa (zob. dodatek 2.1).
|
SPRAWOZDANIE Z BADANIA
|
48.
|
Zob. pkt 20 dotyczący „ELEMENTÓW BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”.
|
BIBLIOGRAFIA
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 225. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta. „Biochem. Pharmacol.” nr 71, s. 1459–1469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta.„Endocrinol.” nr 139, s. 4252–4263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction.„FEMS Microbiol. Lett.” nr 78(1), s. 89–94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products.„J. Vet. Med. Sci.” nr 57(4), s. 769–771.
|
|
(6)
|
Dussurget O. i Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera.„Appl. Environ. Microbiol.” nr 60(3), s. 953–959.
|
|
(7)
|
De Lean A., Munson P.J. i Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves.„Am. J. of Physiol.” nr 235, E97–El02.
|
Dodatek 2.1
WYNIKI FAŁSZYWIE DODATNIE: OCENA SYGNAŁÓW LUMINESCENCJI NIEPRZEKAZYWANYCH ZA POMOCĄ RECEPTORA
|
1.
|
Wyniki fałszywie dodatnie w badaniu antagonisty receptora estrogenowego mogą pojawiać się na skutek aktywacji genu lucyferazy niezależnej od receptora estrogenowego lub na skutek bezpośredniej aktywacji produktu genu lub niepowiązanej fluorescencji. Wyniki takie wskazuje niekompletna lub nietypowa krzywa dawka-efekt. Jeżeli podejrzewa się wystąpienie takich wyników, należy zbadać wpływ wyników antagonisty receptora estrogenowego (np. 4-hydroksytamoksyfen (OHT) w stężeniu nietoksycznym) na reakcję. Czysty antagonista ICI 182780 może być niewłaściwy do tego celu, ponieważ wystarczające stężenie ICI 182780 może obniżyć wartość VC, a to w rezultacie wpłynie na analizę danych.
|
|
2.
|
Aby zapewnić zasadność tego podejścia, na tej samej płytce należy przeprowadzić badanie następujących elementów:
|
—
|
aktywności agonistycznej nieznanej substancji chemicznej zawierającej/niezawierającej 10 μM OHT;
|
|
—
|
1 nM E2 (trzykrotnie) jako agonisty PC;
|
|
—
|
1 nM E2 + OHT (trzykrotnie).
|
|
Kryteria interpretacji danych
Uwaga: Wszystkie dołki powinny otrzymać jednakowe stężenie nośnika.
|
—
|
Jeżeli aktywność agonistyczna nieznanej substancji chemicznej NIE zostanie naruszona w wyniku stosowania antagonisty receptora estrogenowego, klasyfikuje się ją jako „ujemną”.
|
|
—
|
Jeżeli aktywność agonistyczna nieznanej substancji chemicznej jest całkowicie zablokowana, zastosowanie mają kryteria podejmowania decyzji.
|
|
—
|
Jeżeli aktywność agonistyczna w najniższym stężeniu jest równa bądź przekracza reakcję PC10, nieznana substancja chemiczna jest blokowana w stopniu równym bądź przekraczającym reakcję PC10. Różnicę w reakcjach między dołkami poddanymi działaniu antagonisty receptora estrogenowego i dołkami niepoddanymi takiemu działaniu oblicza się i należy uznać ją za rzeczywistą reakcję oraz wykorzystać do obliczania odpowiednich parametrów umożliwiających podjęcie decyzji o klasyfikacji.
|
Analiza danych
Skontrolować standardy wykonywania badań.
Skontrolować CV między dołkami poddanymi działaniu substancji w takich samych warunkach.
|
2.
|
Odjąć średnią VC od wartości każdego dołka niepoddanego działaniu OHT.
|
|
4.
|
Odjąć średnią VC od wartości każdego dołka poddanego działaniu OHT.
|
|
6.
|
Obliczyć względną aktywność transkrypcyjną wszystkich innych dołków w stosunku do PC.
|
Dodatek 2.2
PRZYGOTOWANIE SUROWICY PODDANEJ DZIAŁANIU WĘGLA DRZEWNEGO POKRYTEGO DEKSTRANEM
|
1.
|
Poddawanie surowicy działaniu węgla drzewnego pokrytego dekstranem stanowi ogólną metodę usuwania związków estrogenowych z surowicy, którą dodaje się do podłoża komórkowego w celu wykluczenia subiektywnej reakcji związanej z pozostałościami estrogenów w surowicy. W trakcie tej procedury działaniu można poddać 500 ml bydlęcej surowicy płodowej.
|
Komponenty
|
2.
|
Wymagane będą następujące materiały i sprzęt:
|
Materiały
|
aktywowany węgiel drzewny;
|
|
sześciowodny chlorek magnezu (MgCl2 6H2O);
|
|
1 M roztworu buforowego KEPES (pH 7,4);
|
|
woda ultraczysta uzyskana za pomocą systemu filtracyjnego.
|
Sprzęt
Wysterylizowany w autoklawie szklany pojemnik (rozmiar należy odpowiednio dostosować) Standardowa wirówka laboratoryjna (której temperaturę można ustawić na 4 °C).
Procedura
|
3.
|
Niniejszą procedurę dostosowano do wykorzystania probówek wirówkowych o pojemności 50 ml:
[Dzień 1] Należy przygotować zawiesinę węgla drzewnego pokrytego dekstranem z 1 l wody ultraczystej zawierającej 1,5 mM MgCl2, 0,25 M sacharozy, 2,5 g węgla drzewnego, 0,25 g dekstranu i 5 mM HEPES i mieszać nocą w temperaturze 4 °C.
[Dzień 2] Rozmieścić zawiesinę w probówkach wirówki o pojemności 50 ml i wirować przez 10 min z prędkością 10 000 obr./min w temperaturze 4 °C. Usunąć supernatant, a połowę osadu z węgla drzewnego przechowywać w temperaturze 4 °C do wykorzystania w 3. dniu. Pozostałą część węgla drzewnego zawiesić za pomocą bydlęcej surowicy płodowej, którą należy uprzednio ostrożnie rozmrozić, by uniknąć strącenia i przez 30 min inaktywować ciepłem w temperaturze 56 °C, a następnie umieścić w wysterylizowanym w autoklawie szklanym pojemniku (np. w kolbie Erlenmeyera). Zawiesinę mieszać ostrożnie nocą w temperaturze 4 °C.
[Dzień 3] Rozmieścić zawiesinę zawierającą bydlęcą surowicę płodową w probówkach wirówki do wirowania przez 10 min z prędkością 10 000 obr./min w temperaturze 4 °C. Zebrać bydlęcą surowicę płodową i przenieść ją do nowego osadu z węgla drzewnego przygotowanego i przechowywanego od 2. dnia. Zawiesić osad z węgla drzewnego i nocą ostrożnie mieszać tę zawiesinę w wysterylizowanym w autoklawie szklanym pojemniku w temperaturze 4 °C.
[Dzień 4] Rozmieścić zawiesinę w probówkach wirówki do wirowania przez 10 min z prędkością 10 000 obr./min w temperaturze 4 °C oraz sterylizować supernatant poprzez filtrację sterylnym filtrem 0,2 μm. Ten węgiel drzewny pokryty dekstranem poddany działaniu bydlęcej surowicy płodowej należy przechowywać w temperaturze –20 °C i można go wykorzystywać przez rok.
|
Dodatek 3
TEST TRANSAKTYWACJI RECEPTORA ESTROGENOWEGO VM7LUC DOTYCZĄCE IDENTYFIKACJI AGONISTÓW I ANTAGONISTÓW RECEPTORA ESTROGENOWEGO
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
1.
|
W tym teście stosuje się linię komórkową VM7Luc4E2 (64). Badanie to zostało zatwierdzone przez Amerykański Krajowy Program Toksykologiczny Międzyagencyjnego Centrum Oceny Alternatywnych Metod Toksykologicznych oraz Międzyagencyjny Komitet Koordynacyjny ds. Uznawania Metod Alternatywnych (1). Linie komórkowe VM7Luc ulegają ekspresji głównie endogennego receptor estrogenowego alfa i niewielkiej ilości endogennego receptora estrogenowego beta (2)(3)(4).
|
|
2.
|
Test ten ma zastosowanie w odniesieniu do szerokiego zakresu substancji pod warunkiem, że można je rozpuścić w sulfotlenku dimetylu (nr CAS 67-68-5), nie wchodzą w reakcję z nim lub podłożem do hodowli komórkowej, a także nie są cytotoksyczne w badanych stężeniach. Jeżeli zastosowanie sulfotlenku dimetylowego nie jest możliwe, można zastosować inny nośnik, np. etanol lub wodę (zob. pkt 12). Wykazana efektywność badania (ant)agonisty transaktywacji receptora estrogenowego VM7Luc wskazuje, że dane wygenerowane za pomocą tego testu mogą być źródłem informacji o mechanizmach działania pośredniczących w funkcjonowaniu receptorów estrogenowych i mogą być brane pod uwagę w momencie ustalania priorytetów dla substancji w celu przeprowadzenia dalszych badań.
|
|
3.
|
Szczególnym przeznaczeniem tego testu jest wykrywanie transaktywacji zależnej od ludzkiego receptora estrogenowego alfa i ludzkiego receptora estrogenowego beta poprzez pomiar chemiluminescencji jako punktu końcowego.α Wykorzystanie chemiluminescencji w testach biologicznych jest powszechne, ponieważ luminescencję cechuje wysoki stosunek sygnału do tła. Aktywność lucyferazy robaczka świętojańskiego w testach prowadzonych na komórkach może być jednak zakłócona przez substancje, które hamują wytwarzanie enzymu lucyferazy, powodując zarówno widoczną inhibicję, jak i zwiększoną luminescencję spowodowaną stabilizacją białka. Ponadto w przypadku pewnych testów genu reporterowego receptora estrogenowego opartych na lucyferazie, z powodu nadaktywacji lucyferazowego genu reporterowego zgłoszono sygnały niezwiązane z receptorami pośredniczącymi w luminescencji przy stężeniach fitoestrogenu przekraczających 1 μM (9) (11). Chociaż krzywa dawka-efekt wskazuje, że rzeczywista aktywacja systemu receptora estrogenowego występuje przy niższych stężeniach, w stabilnej transfekcji systemów badania aktywacji transkrypcji receptora estrogenowego należy ostrożnie badać ekspresję lucyferazy uzyskaną przy wysokich stężeniach fitoestrogenów lub podobnych związków chemicznych, co do których przypuszcza się, że wywołują one – podobnie jak fitoestrogeny – nadaktywację lucyferazowego genu reporterowego.
|
|
4.
|
Przed rozpoczęciem stosowania tego testu do celów regulacyjnych należy zapoznać się z sekcjami „WPROWADZENIE OGÓLNE” oraz „ELEMENTY BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”. Definicje i skróty stosowane w niniejszej metodzie badawczej są opisane w dodatku 1.
|
ZASADA WYKONANIA TESTU (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
5.
|
Celem testu jest wskazanie wiązania ligandów, a następnie przeniesienie kompleksu receptor-ligand do jądra komórkowego. W jądrze komórkowym kompleks receptor-ligand wiąże się z określonymi elementami reakcji DNA oraz transaktywuje gen reporterowy (luc), co skutkuje wydobywaniem się lucyferazy, a w związku z tym powstaniem emisji światła, którą można określić ilościowo za pomocą luminometru. Aktywność lucyferazy można ocenić w szybki i niedrogi sposób, wykorzystując szereg dostępnych na rynku zestawów pomiarowych. Mechanizm transaktywacji receptora estrogenowego VM7Luc wykorzystuje reagującą na receptor estrogenowy ludzką linię komórkową gruczolaka piersi VM7, którą stabilnie transfekuje się za pomocą reportera luc robaczka świętojańskiego pod kontrolą czterech elementów reakcji estrogenowej umieszczonych powyżej promotora wirusa mysiego nowotworu gruczołu piersiowego, aby wykryć substancje wykazujące aktywność agonistów lub antagonistów receptora estrogenowego in vitro. Ten promotor mysiego nowotworu gruczołu piersiowego okazuje jedynie niewielką reakcję krzyżową z innymi hormonami sterydowymi i niesterydowymi (8). Kryteria dotyczące interpretacji danych szczegółowo opisano w pkt 41. Krótko mówiąc, reakcję dodatnią stwierdza się na podstawie krzywej zależności stężenie-odpowiedź, składającej się z co najmniej trzech punktów z nienakładającymi się na siebie słupkami błędu (średnim ±SD), jak również na podstawie zmiany amplitudy (ujednoliconych względnych jednostek światła) o co najmniej 20 % maksymalnej wartości dla wzorca referencyjnego (17β-estradiol [E2; nr CAS 50-28-2] w odniesieniu do testu agonisty, raloksyfen HCl [Ral; nr CAS 84449-90-1]/E2 w odniesieniu do testu antagonisty).
|
PROCEDURA
Linia komórkowa
|
6.
|
W teście należy stosować stabilnie transfekowaną linię komórkową VM7Luc4E2. Dana linia komórkowa jest dostępna jedynie na podstawie technicznej umowy licencyjnej pochodzącej z Uniwersytetu Kalifornijskiego, Davis, Kalifornia, Stany Zjednoczone (65) oraz z Xenobiotic Detection Systems Inc., Durnham, Północna Karolina, Stany Zjednoczone (66).
|
Stabilność linii komórkowej
|
7.
|
Aby zachować stabilność i integralność linii komórkowej, komórki powinny wyrastać z zamrożonego zapasu w pożywkach utrzymujących przez więcej niż jeden pasaż (zob. pkt 9). Komórek nie powinno się hodować przez więcej niż 30 pasaży. W przypadku linii komórkowej VM7Luc4E2, 30 pasaży może trwać około trzy miesiące.
|
Kultura komórkowa i warunki posiewu
|
8.
|
Należy przestrzegać procedur określonych w wytycznej dotyczącej dobrych praktyk w zakresie kultury komórkowej (5) (6), aby zagwarantować wysoką jakość wszystkich materiałów i metod, co pozwoli zachować integralność, zasadność i odtwarzalność wszelkich prowadzonych prac.
|
|
9.
|
Komórki VM7Luc4E2 zachowuje się w pożywce RPMI 1640 zawierającej 0,9 % Pen-Strep i 8,0 % bydlęcej surowicy płodowej w dedykowanym inkubatorze do hodowli tkankowej w temperaturze 37 oC ±1 oC, wilgotności 90 % ±5 % i atmosferze zawierającej 5,0 % ±1 % CO2.
|
|
10.
|
Do chwili osiągnięcia konfluencji na poziomie około 80 %, komórki VM7Luc4E2 wysiewa się na hodowle wtórne i kondycjonuje do środowiska wolnego od estrogenów przez 48 godzin przed posianiem komórek w 96-dołkowych płytkach w celu poddania ich działaniu badanych substancji chemicznych i analizy zależnej od estrogenów indukcji aktywności lucyferazy. Pożywka wolna od estrogenów zawiera Dulbecco's Modification of Eagle's Medium uzupełniony 4,5 % bydlęcą surowicą płodową pozbawioną czerwieni fenolowej i poddaną działaniu węgla drzewnego / dekstranu, 1,9 % L-glutaminą i 0,9 % Pen-Strep. Wszystkie wyroby z tworzyw sztucznych powinny być wolne od aktywności estrogenowej [zob. protokół szczegółowy (7)].
|
Kryteria dopuszczalności
|
11.
|
Akceptacji lub odrzucenia badania dokonuje się na podstawie oceny wyników kontroli i wzorca referencyjnego uzyskanych z każdego doświadczenia przeprowadzonego na 96-dołkowej płytce. Każdy wzorzec referencyjny bada się w wielu stężeniach i istnieje wiele próbek każdego stężenia referencyjnego i stężenia kontroli. Wyniki porównuje się do wyników kontroli jakości pod względem tych parametrów, które pochodziły z bazy danych historycznych agonistów i antagonistów wygenerowanych przez każde laboratorium podczas wykazywania efektywności. Bazy danych historycznych ciągle aktualizuje się o wartości wzorców referencyjnych i kontroli. Zmiany w sprzęcie lub warunkach laboratoryjnych mogą wymagać wygenerowania zaktualizowanych baz danych historycznych.
|
Badanie agonisty
Badanie ustalające zakres dawkowania
|
•
|
indukcja: Indukcję płytki należy mierzyć, dzieląc najwyższą średnią wartość względnej jednostki światła wzorca referencyjnego E2 przez średnią wartość względnej jednostki światła kontroli z sulfotlenkiem dimetylowym. Zazwyczaj osiąga się pięciokrotność indukcji, jednak dla celów dopuszczalności krotność indukcji powinna być większa bądź równa 4.
|
|
•
|
wyniki kontroli z sulfotlenkiem dimetylowym: Wartości względnej jednostki światła kontroli z rozpuszczalnikiem powinny mieścić się w granicach 2,5-krotności odchylenia standardowego historycznej średniej wartości względnej jednostki światła kontroli z rozpuszczalnikiem.
|
|
•
|
doświadczenie, które nie spełni któregokolwiek z kryteriów dopuszczalności należy odrzucić i powtórzyć.
|
Badanie kompleksowe
W jego skład wchodzą kryteria dopuszczalności pochodzące z badania ustalającego zakres dawkowania oraz następujące elementy:
|
•
|
wyniki wzorca referencyjnego: Krzywa zależności stężenie-odpowiedź wzorca referencyjnego E2 powinna mieć kształt sigmoidalny, a w liniowej części zawierać co najmniej trzy wartości.
|
|
•
|
wyniki kontroli dodatniej: Wartości względnej jednostki światła kontroli z metoksychlorem powinny być wyższe niż średnia sulfotlenku dimetylowego powiększona o trzykrotność odchylenia standardowego ze średniej sulfotlenku dimetylowego.
|
|
•
|
doświadczenie, które nie spełni któregokolwiek z kryteriów dopuszczalności należy odrzucić i powtórzyć.
|
Badanie antagonisty
Badanie ustalające zakres dawkowania
|
•
|
redukcja: redukcję płytki mierzy się, dzieląc najwyższą średnią wartość względnej jednostki światła wzorca referencyjnego Ral/E2 przez średnią wartość względnej jednostki światła kontroli z sulfotlenkiem dimetylowym. Zazwyczaj osiąga się pięciokrotność redukcji, jednak dla celów dopuszczalności krotność redukcji powinna być większa bądź równa 3.
|
|
•
|
wyniki kontroli z E2: wartości względnej jednostki światła kontroli z E2 powinny mieścić się w granicach 2,5-krotności odchylenia standardowego historycznej średniej wartości względnej jednostki światła kontroli z E2.
|
|
•
|
wyniki kontroli z sulfotlenkiem dimetylowym: wartości względnej jednostki światła kontroli z sulfotlenkiem dimetylowym powinny mieścić się w granicach 2,5-krotności odchylenia standardowego historycznej średniej wartości względnej jednostki światła kontroli z rozpuszczalnikiem.
|
|
•
|
Doświadczenie, które nie spełni któregokolwiek z kryteriów dopuszczalności, zostanie odrzucone i powtórzone.
|
Badanie kompleksowe
W jego skład wchodzą kryteria dopuszczalności pochodzące z badania ustalającego zakres dawkowania oraz następujące elementy:
|
•
|
wyniki wzorca referencyjnego: Krzywa zależności stężenie-odpowiedź wzorca referencyjnego Ral/E2 powinna mieć kształt sigmoidalny i zawierać co najmniej trzy wartości w swojej liniowej części.
|
|
•
|
wyniki kontroli dodatniej: Wartości względnej jednostki światła kontroli z tamoksyfenem/E2 powinny być niższe niż średnia kontroli z E2 pomniejszona o trzykrotność odchylenia standardowego ze średniej kontroli z E2.
|
|
•
|
Doświadczenie, które nie spełni któregokolwiek z kryteriów dopuszczalności, zostanie odrzucone i powtórzone.
|
Wzorce referencyjne, kontrole dodatnie i grupy kontrolne nośnika
Grupa kontrolna nośnika (testy agonistów i antagonistów)
|
12.
|
Nośnik stosowany do rozpuszczenia badanej substancji chemicznej należy zbadać jako grupę kontrolną nośnika. Nośnikiem stosowanym w trakcie walidacji badania aktywacji transkrypcji receptora estrogenowego VM7Luc był sufotlenek dimetylowy 1 % (obj./obj.) (nr CAS 67-68-5) (zob. pkt 24). Jeśli stosuje się nośnik inny niż sufotlenek dimetylowy, wszystkie wzorce referencyjne, grupy kontrolne i badane substancje chemiczne należy, jeżeli jest to stosowne, zbadać w tym samym nośniku.
|
Wzorzec referencyjny (agonista ustalający zakres dawkowania)
|
13.
|
Wzorcem referencyjnym jest E2 (nr CAS 50-28-2). W przypadku badań ustalających zakres dawkowania, wzorzec referencyjny składa się z serii stężeń wzrastających w postępie geometrycznym, zawierającej cztery stężenia E2 (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 i 2,87 x 10-12 M), przy czym każde z nich badane jest w zduplikowanych dołkach.
|
Wzorzec referencyjny (kompleksowy agonista)
|
14.
|
W przypadku badań kompleksowych, E2 składa się z serii stężeń wzrastających w postępie geometrycznym w stosunku 1:2, zawierającej w zduplikowanych dołkach 11 stężeń E2 (w zakresie od 3,67 x 10-10 do 3,59 x 10-13 M).
|
Wzorzec referencyjny (antagonista ustalający zakres dawkowania)
|
15.
|
Wzorcem referencyjnym jest połączenie Ral (nr CAS 84449-90-1) i E2 (nr CAS 50-28-2). W przypadku badań ustalających zakres dawkowania, Ral/E2 składa się z serii stężeń wzrastających w postępie geometrycznym zawierającej w zduplikowanych dołkach trzy stężenia Ral (3,06 x 10-9, 7,67 x 10-10 i 1,92 x 10-10M) plus stałe stężenie (9,18 × 10-11 M) E2.
|
Wzorzec referencyjny (kompleksowy antagonista)
|
16.
|
W przypadku badań kompleksowych, Ral/E2 składa się z serii stężeń wzrastających w postępie geometrycznym w stosunku 1:2 Ral (w zakresie od 2,45x 10-8 do 9,57 x 10-11M) plus stałego stężenia (9,18 × 10-11 M) E2 zawierającego w zduplikowanych dołkach dziewięć stężeń Ral/E2.
|
Słaba kontrola dodatnia (agonista)
|
17.
|
Słabą kontrolą dodatnią jest 9,06 x 10-6 M p,p′-metoksychlor (metoksychlor; nr CAS 72-43-5) w podłożu bez estrogenu.
|
Słaba kontrola dodatnia (antagonista)
|
18.
|
Słaba kontrola dodatnia składa się z tamoksyfenu (nr CAS 10540-29-1) 3,36 x 10-6 M zawierającego 9,18 × 10-11 M E2 w podłożu bez estrogenu.
|
Kontrola z E2 (wyłącznie test antagonisty)
|
19.
|
Kontrolą z E2 jest 9,18 × 10-11 M E2 w podłożu bez estrogenu oraz kontrola ujemna stosowana jako punkt odniesienia.
|
Krotność indukcji (agonista)
|
20.
|
Indukcję aktywności lucyferazy wzorca referencyjnego (E2) mierzy się, dzieląc najwyższą średnią wartość względnej jednostki światła wzorca porównawczego E2 przez średnią wartość względnej jednostki światła kontroli z sulfotlenkiem dimetylowym, a otrzymana krotność indukcji powinna być wyższa niż 4.
|
Krotność redukcji (antagonista)
|
21.
|
Średnią aktywność lucyferazy wzorca referencyjnego (Ral/E2) mierzy się, dzieląc najwyższą średnią wartość względnej jednostki światła wzorca porównawczego E2 przez średnią wartość względnej jednostki światła kontroli z sulfotlenkiem dimetylowym, a jej krotność powinna być wyższa niż 3.
Wykazanie biegłości laboratorium (zob. pkt 14 oraz tabela 3 i 4 w „ELEMENTACH BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO” niniejszej metody badawczej).
|
Nośnik
|
22.
|
Badaną substancję chemiczną należy rozpuścić w nośniku, który rozpuszcza badaną substancję chemiczną i jest mieszalny z podłożem komórkowym. Odpowiednie nośniki stanowią woda, etanol (95–100 % czystości) i sufotlenek dimetylowy. Jeżeli stosuje się sulfotlenek dimetylowy, poziom nie powinien przekroczyć 1 % (obj./obj.). Dla każdego nośnika należy wykazać, że maksymalna zastosowana objętość nie wykazuje właściwości cytotoksycznych i nie ma wpływu na efektywność testu. Wzorce referencyjne i kontrole rozpuszcza się w rozpuszczalniku 100 %, a następnie rozcieńcza do właściwych stężeń w podłożu bez estrogenu.
|
Przygotowanie badanych substancji chemicznych
|
23.
|
Badane substancje chemiczne rozpuszcza się w sulfotlenku dimetylowym 100 % (lub stosownym rozpuszczalniku), a następnie rozcieńcza do właściwych stężeń w podłożu bez estrogenu. Badane substancje chemiczne, przed rozpuszczeniem i rozcieńczeniem, należy pozostawić w spokoju aż do momentu osiągnięcia temperatury pokojowej. Do każdego doświadczenia należy przygotować świeże roztwory badanych substancji chemicznych. Roztwory nie powinny zawierać zauważalnego osadu ani nie powinny być mętne. Wzorzec referencyjny i roztwory kontrolne można przygotować w dużej ilości; ostateczny wzorzec referencyjny, rozcieńczenia kontrolne oraz badane substancje chemiczne należy jednak przygotować świeżo do każdego doświadczenia i wykorzystać w ciągu 24 godzin od przygotowania.
|
Rozpuszczalność i cytotoksyczność: Uwagi dotyczące ustalania zakresu dawkowania
|
24.
|
Badanie ustalające zakres składa się z siedmiopunktowej podwojonej serii badawczej stężeń wzrastających w postępie geometrycznym w stosunku 1:10. Badane substancje chemiczne wstępnie bada się do maksymalnego stężenia wynoszącego 1 mg/ml (~1 mM) w przypadku badania agonisty oraz do 20 μg/ml (~10 μM) w przypadku badania antagonisty. Doświadczenia ustalające zakres dawkowania wykorzystuje się do określenia:
|
|
—
|
początkowych stężeń badanych substancji chemicznych wykorzystywanych podczas badań kompleksowych;
|
|
—
|
rozcieńczeń badanych substancji chemicznych (w stosunkach 1:2 lub 1:5) wykorzystywanych podczas badań kompleksowych.
|
|
25.
|
Ocena żywotności komórek / cytotoksyczności wchodzi w zakres protokołów dotyczących badania agonisty i antagonisty (7) oraz obejmuje badanie ustalające zakres dawkowania i badanie kompleksowe. Metoda dotycząca cytotoksyczności, którą stosowano do oceny żywotności komórek podczas walidacji transaktywacji receptora estrogenowy VM7Luc (1), była skalowaną jakościowo metodą obserwacji wzrokowej; do określenia cytotoksyczności można wykorzystać jednak metodę ilościową (zob. protokół (7)). Nie można wykorzystywać danych dotyczące stężeń badanych substancji chemicznych powodujących redukcję żywotności żywotności o ponad 20 %.
|
Narażenie na działanie badanej substancji chemicznej oraz organizacja płytki testowej
|
26.
|
Komórki zlicza się i posiewa na 96-dołkowych płytkach z kulturą tkankową (2 × 105 komórek na dołek) w podłożu bez estrogenu, a następnie inkubuje przez 24 godziny, by umożliwić komórkom osadzenie się na płytce. Podłoże bez estrogenu usuwa się i zastępuje badanymi i porównawczymi substancjami chemicznymi w podłożu bez estrogenu, a następnie inkubuje przez 19–24 godzin. Należy zwrócić szczególną uwagę na substancje charakteryzujące się wysoką lotnością, ponieważ pobliskie dołki kontrolne mogą generować wyniki fałszywie dodatnie. W takich przypadkach zaleca się zastosowanie „uszczelniaczy płytek”, które podczas badania mogą pomóc w skutecznym odizolowaniu poszczególnych dołków.
|
Badania ustalające zakres dawkowania
|
27.
|
W badaniu ustalającym zakres dawkowania wykorzystuje się wszystkie dołki 96-dołkowej płytki, aby prowadzić badanie maksymalnie sześciu substancji chemicznych w postaci siedmiopunktowej podwojonej próby serii stężeń wzrastających w postępie geometrycznym w stosunku 1:10 (zob. rysunki 1 i 2).
|
|
—
|
W badaniu ustalającym zakres dawkowania agonisty jako wzorzec referencyjny wykorzystuje się cztery podwojone stężenia E2 i cztery dołki stanowiące kontrpróbę kontroli z sulfotlenkiem dimetylowym.
|
|
—
|
W badaniu ustalającym zakres dawkowania antagonisty jako wzorzec referencyjny wykorzystuje się trzy podwojone stężenia Ral/E2 z zawartością 9,18 × 10-11 M E2, z trzema dołkami stanowiącymi kontrpróbę kontroli z E2 i kontroli z sulfotlenkiem dimetylowym.
|
Rysunek 1
Badanie ustalające zakres dawkowania agonisty – rozkład na 96-dołkowej płytce

Skróty: E2-1 do E2-4 = stężenia wzorca referencyjnego E2 (od wysokiego do niskiego); TC1-1 do TC1-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 1 (TC1); TC2-1 do TC2-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 2 (TC2); TC3-1 do TC3-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 3 (TC3); TC4-1 do TC4-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 4 (TC4); TC5-1 do TC5-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 5 (TC5); TC6-1 do TC6-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 6 (TC6); VC = grupa kontrolna nośnika (sulfotlenek dimetylowy [1 % obj./obj. w podłożu bez estrogenu]).
Rysunek 2
Badanie ustalające zakres dawkowania antagonisty – rozkład na 96-dołkowej płytce

Skróty: E2 = kontrola z E2; Ral-1 do Ral-3 = stężenia wzorca referencyjnego raloksyfenu/E2 (od wysokiego do niskiego); TC1-1 do TC1-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 1 (TC1); TC2-1 do TC2-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 2 (TC2); TC3-1 do TC3-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 3 (TC3); TC4-1 do TC4-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 4 (TC4); TC5-1 do TC5-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 5 (TC5); TC6-1 do TC6-7 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 6 (TC6); VC = grupa kontrolna nośnika (sulfotlenek dimetylowy [1 % obj./obj. w podłożu bez estrogenu]).
Uwaga: Wszystkie badane substancje chemiczne bada się w obecności E2 9,18 × 10-11 M.
|
28.
|
Zalecana ostateczna objętość podłoża wymagana dla każdego dołka wynosi 200 μl. Należy stosować jedynie płytki testowe, na których komórki we wszystkich dołkach wykazują żywotność na poziomie co najmniej 80 %.
|
|
29.
|
Określanie początkowych stężeń w odniesieniu do kompleksowego badania agonisty szczegółowo opisano w protokole dotyczącym agonisty (7). Krótko mówiąc, zastosowano następujące kryteria:
|
|
—
|
Jeżeli na krzywej przedstawiającej stężenia badanej substancji chemicznej nie istnieją punkty znajdujące się powyżej średniej powiększonej o trzykrotność odchylenia standardowego kontroli z sulfotlenkiem dimetylowym, kompleksowe badanie zostanie przeprowadzone z zastosowaniem 11-punktowej serii stężeń wzrastających w postępie geometrycznym w stosunku 1:2, począwszy od maksymalnego stężenia rozpuszczalnego.
|
|
—
|
Jeżeli na krzywej przedstawiającej stężenia badanej substancji chemicznej istnieją punkty znajdujące się powyżej średniej powiększonej o trzykrotność odchylenia standardowego kontroli z sulfotlenkiem dimetylowym, początkowe stężenie, które należy zastosować w kompleksowym badaniu w odniesieniu do 11-punktowego schematu rozcieńczeń powinno być o 1 jednostkę logarytmiczną wyższe niż stężenie dające najwyższą dostosowaną wartość względnej jednostki światła w badaniu ustalającym zakres dawkowania. 11-punktowy schemat rozcieńczeń będzie, w związku z poniższymi kryteriami oparty na rozcieńczeniach w stosunkach 1:2 albo 1:5:
|
11-punktową serię stężeń wzrastających w postępie geometrycznym w stosunku 1:2 należy zastosować, gdy wynikły zakres stężeń obejmie pełen zakres reakcji opartych na krzywej stężenie-odpowiedź generowanych w badaniu ustalającym zakres dawkowania. W innym przypadku należy zastosować rozcieńczenie w stosunku 1:5.
|
|
|
—
|
Jeżeli badana substancja chemiczna podczas badania ustalającego zakres dawkowania wykazuje dwufazową krzywą stężenie-odpowiedź, obie fazy należy również wyjaśnić w badaniu kompleksowym.
|
|
30.
|
Określanie początkowych stężeń w odniesieniu do kompleksowego badania antagonisty szczegółowo opisano w protokole dotyczącym antagonisty (7). Krótko mówiąc, zastosowano następujące kryteria:
|
|
—
|
Jeżeli na krzywej dotyczącej stężenia badanej substancji chemicznej nie istnieją punkty znajdujące się poniżej średniej pomniejszonej o trzykrotność odchylenia standardowego kontroli z E2, kompleksowe badanie zostanie przeprowadzone z zastosowaniem 11-punktowej serii stężeń wzrastających w postępie geometrycznym w stosunku 1:2, począwszy od maksymalnego stężenia rozpuszczalnego.
|
|
—
|
Jeżeli na krzywej dotyczącej stężenia badanej substancji chemicznej istnieją punkty znajdujące się poniżej średniej pomniejszonej o trzykrotność odchylenia standardowego kontroli z E2, początkowe stężenie, które należy zastosować w kompleksowym badaniu w odniesieniu do 11-punktowego schematu rozcieńczeń powinno być:
|
—
|
stężeniem dającym najniższą dostosowaną wartość względnej jednostki światła w badaniu ustalającym zakres dawkowania;
|
|
—
|
najwyższym rozpuszczalnym stężeniem (zob. protokół dotyczący antagonisty (7), rysunek 14-2);
|
|
—
|
najniższym cytotoksycznym stężeniem (dla przykładu pokrewnego zob. protokół dotyczący antagonisty (7), rysunek 14-3).
|
|
|
—
|
11-punktowy schemat rozcieńczeń będzie, w związku z poniższymi kryteriami, oparty na serii albo rozcieńczeniu w stosunku 1:2 lub 1:5:
|
11-punktową serię stężeń wzrastających w postępie geometrycznym w stosunku 1:2 należy zastosować, gdy wynikły zakres stężeń obejmie pełen zakres reakcji opartych na krzywej stężenie-odpowiedź generowanych w badaniu ustalającym zakres dawkowania. W innym przypadku należy zastosować rozcieńczenie w stosunku 1:5.
|
|
Badania kompleksowe
|
31.
|
Badanie kompleksowe składa się z 11-punktowych serii stężeń wzrastających w postępie geometrycznym (seriach stężeń wzrastających w postępie geometrycznym w stosunku 1:2 albo 1:5, opartych na początkowym stężeniu na podstawie kryteriów badań kompleksowych), przy czym każde stężenie bada się w trzech dołkach na 96-dołkowej płytce (zob. rysunki 3 i 4).
|
|
—
|
W badaniu kompleksowym agonisty jako wzorzec referencyjny stosuje się 11 podwójnych stężeń E2. Na każdej płytce znajdują się cztery dołki stanowiące kontrpróbę kontroli z sulfotlenkiem dimetylowym i cztery dołki stanowiące kontrpróbę kontroli z metoksychlorem (9,06 x 10-6 M).
|
|
—
|
W badaniu kompleksowym antagonisty jako wzorzec referencyjny wykorzystuje się dziewięć podwojonych stężeń Ral/E2 z zawartością 9,18 × 10-11 M E2, przy czterech dołkach stanowiących kontrpróbę kontroli z E2 9,18 x 10-11 M, czterech dołkach stanowiących kontrpróbę kontroli z sulfotlenkiem dimetylowym oraz czterech dołkach stanowiących kontrpróbę tamoksyfenu 3,36 x 10-6M.
|
Rysunek 3
Badanie kompleksowe agonisty – rozkład na 96-dołkowej płytce

Skróty: TC1-1 do TC1-11 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 1; TC2-1 do TC2-11 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 2; E2-1 do E2-11 = stężenia wzorca referencyjnego E2 (od wysokiego do niskiego); Meth = p,p’ methoksychlor, słaba kontrola dodania; VC = grupa kontrolna nośnika z sulfotlenkiem dimetylowym (1 % obj./obj.) z podłożem bez estrogenu
Rysunek 4
Badanie kompleksowe antagonisty – rozkład na 96-dołkowej płytce

Skróty: E2 = kontrola z E2; Ral-1 do Ral-9 = stężenia wzorca referencyjnego raloksyfenu/E2 (od wysokiego do niskiego); Tam = tamoksyfen/E2 słaba kontrola dodatnia TC1-1 do TC1-11 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 1 (TC1); TC2-1 do TC2-11 = stężenia (od wysokiego do niskiego) badanej substancji chemicznej 2 (TC2); VC = grupa kontrolna nośnika (sulfotlenek dimetylowy [1 % obj./obj. w podłożu bez estrogenu]).
Uwaga: Jak wspomniano, wszystkie dołki referencyjne i badawcze zawierają stałe stężenie E2 (9,18 x 10-11M).
|
32.
|
Aby zapewnić niezależność badań kompleksowych dotyczących tej samej substancji chemicznej, ich powtórki należy przeprowadzać w różne dni. Należy przeprowadzić co najmniej dwa badania kompleksowe. Jeżeli wyniki badań są sprzeczne (np. jedno badanie jest dodatnie, a drugie ujemne), lub gdy jedno z badań jest nieodpowiednie, należy przeprowadzić trzecie, dodatkowe badanie.
|
Pomiar luminescencji
|
33.
|
Luminescencję mierzy się w zakresie od 300 do 650 nm, wykorzystując do tego luminometr iniekcyjny i oprogramowanie kontrolujące objętość iniekcji i interwał pomiaru (7). Emisję światła z każdego dołka wyraża się jako względną jednostkę światła/dołek.
|
ANALIZA DANYCH
Określenie EC50 /IC50
|
34.
|
Wartość EC50 (połowę maksymalnego stężenia efektywnego badanej substancji chemicznej [agonistów]) oraz wartość IC50 (połowę maksymalnego stężenia hamującego badanej substancji chemicznej [antagonistów]) określa się za pomocą danych dotyczących zależności stężenie-odpowiedź. W przypadku badanych substancji chemicznych, które są dodatnie w co najmniej jednym stężeniu, stężenie badanej substancji chemicznej, które wywołuje reakcję połowicznie maksymalną (IC50 lub EC50) oblicza się, przeprowadzając analizę funkcji Hill lub jej odpowiednią alternatywy. Funkcja Hill stanowi czteroparametrowy logistyczny model matematyczny, przedstawiający stosunek stężenia badanej substancji chemicznej do reakcji (zazwyczaj w postaci krzywej sigmoidalnej), który oblicza się za pomocą następującego równania:
|

gdzie:
Y= reakcja (tj. względne jednostki światła);
X= logarytm stężenia;
Dół= minimalna reakcja;
Góra= najwyższa reakcja;
lg EC50 (or lg IC50)= logarytm z X jako pośrednia reakcja między najwyższą wartością a najniższą;
Nachylenie= stromość krzywej.
W tym modelu oblicza się najlepszą możliwość dostosowania parametrów najwyższej wartości, najniższej wartości, nachylenia oraz IC50 i EC 50. Aby obliczyć wartości EC50 i IC50, należy skorzystać z odpowiedniego oprogramowania do obliczeń statystycznych (np. z oprogramowania do obliczeń statystycznych Graphpad PrismR).
Określenie wyników odstających
|
35.
|
Właściwy osąd statystyczny można by uprościć, stosując między innymi test Q (zob. protokoły dotyczące agonisty i antagonisty (7)), by określić dołki „nienadające się do wykorzystania”, które zostaną wykluczone z analizy danych).
|
|
36.
|
W przypadku kontrprób wzorca referencyjnego E2 (przy wielkości próby wynoszącej dwie kontrpróby), każdą dostosowaną wartość względnej jednostki światła w odniesieniu do kontrpróby w danym stężeniu E2 uznaje się za odstającą, jeżeli jej wartość w bazie danych historycznych jest o 20 % wyższa lub niższa od wartości względnej jednostka światła dla tego stężenia.
|
Gromadzenie i dostosowywanie danych z luminometru na potrzeby badań ustalających zakres
|
37.
|
Dane surowe z luminometra należy przenieść na szablon arkusza kalkulacyjnego zaprojektowanego na potrzeby danego testu. Należy ustalić, czy istnieją punkty danych odstających, które należy usunąć. (W odniesieniu do parametrów określonych w analizie zob. kryteria dopuszczalności badania.) Należy dokonać następujących obliczeń:
|
Agonista
|
Krok 1
|
Obliczyć średnią wartość dla grupy kontrolnej nośnika (VC) z sulfotlenkiem dimetylowym.
|
|
Krok 2
|
Odjąć średnią wartość sulfotlenku dimetylowego od wartości każdego dołka, aby znormalizować dane.
|
|
Krok 3
|
Obliczyć średnią krotność indukcji w odniesieniu do wzorca referencyjnego (E2).
|
|
Krok 4
|
Obliczyć średnią wartość EC50 w odniesieniu do badanych substancji chemicznych.
|
Antagonista
|
Krok 1
|
Obliczyć średnią wartość dla VC sulfotlenku dimetylowego.
|
|
Krok 2
|
Odjąć średnią wartość sulfotlenku dimetylowego od wartości każdego dołka, aby znormalizować dane.
|
|
Krok 3
|
Obliczyć średnią krotność redukcji w odniesieniu do wzorca referencyjnego (Ral/E2).
|
|
Krok 4
|
Obliczyć średnią wartość wzorca referencyjnego E2.
|
|
Krok 5
|
Obliczyć średnią wartość IC50 w odniesieniu do badanych substancji chemicznych.
|
Gromadzenie i dostosowywanie danych z luminometru na potrzeby badań kompleksowych
|
38.
|
Dane surowe z luminometra należy przenieść na szablon arkusza kalkulacyjnego zaprojektowanego na potrzeby danego testu. Należy ustalić, czy istnieją punkty danych odstających, które należy usunąć. (W odniesieniu do parametrów określonych w analizie zob. kryteria dopuszczalności badania.) Dokonuje się następujących obliczeń:
|
Agonista
|
Krok 1
|
Obliczyć średnią wartość dla VC sulfotlenku dimetylowego.
|
|
Krok 2
|
Odjąć średnią wartość sulfotlenku dimetylowego od wartości każdego dołka, aby znormalizować dane.
|
|
Krok 3
|
Obliczyć średnią krotność indukcji w odniesieniu do wzorca referencyjnego (E2).
|
|
Krok 4
|
Obliczyć średnią wartość EC50 w odniesieniu do E2 i badanych substancji chemicznych.
|
|
Krok 5
|
Obliczyć średnią dostosowaną wartość względnej jednostki światła dla metoksychloru.
|
Antagonista
|
Krok 1
|
Obliczyć średnią wartość dla VC sulfotlenku dimetylowego.
|
|
Krok 2
|
Odjąć średnią wartość sulfotlenku dimetylowego od wartości każdego dołka, aby znormalizować dane.
|
|
Krok 3
|
Obliczyć średnią krotność indukcji w odniesieniu do wzorca referencyjnego (Ral/E2).
|
|
Krok 4
|
Obliczyć średnią wartość IC50 w odniesieniu do Ral/E2 i badanych substancji chemicznych.
|
|
Krok 5
|
Obliczyć średnią dostosowaną wartość względnej jednostki światła dla tamoksyfenu.
|
|
Krok 6
|
Obliczyć średnią wartość wzorca referencyjnego E2.
|
Kryteria interpretacji danych
|
39.
|
Transaktywacja receptora estrogenowego VM7Luc ma stanowić część wagi dowodów, aby pomóc w ustaleniu priorytetów substancji do badań in vivo na obecność ED. Część niniejszej procedury ustalania priorytetów będzie stanowić klasyfikowanie badanej substancji chemicznej jako dodatniej lub ujemnej w odniesieniu do aktywności agonisty albo antagonisty receptora estrogenowego. W tabeli 1 przedstawiono dodatnie i ujemne kryteria podejmowania decyzji stosowane w badaniu walidacyjnym transaktywacji receptora estrogenowego VM7Luc.
|
Tabela 1
Dodatnie i ujemne kryteria podejmowania decyzji
|
|
|
AKTYWNOŚĆ AGONISTY
|
|
Dodatnia
|
|
—
|
Wszystkie badane substancje chemiczne sklasyfikowane jako dodatnie pod względem aktywności agonisty receptora estrogenowego powinny zawierać krzywą zależności stężenie-odpowiedź, składającą się z linii podstawowej, po której następuje nachylenie dodatnie, a następnie plateau lub wierzchołek. W niektórych przypadkach można określić jedynie dwie z tych cech charakterystycznych (linia podstawowa-nachylenie lub nachylenie-wierzchołek).
|
|
—
|
Linia określająca nachylenie dodatnie powinna składać się z co najmniej trzech punktów zawierających nienakładające się na siebie słupki błędu (średnia ±SD). Nie uwzględnia się punktów formujących linię podstawową, jednak liniowa część krzywej może zawierać wierzchołek lub pierwszy punkt plateau.
|
|
—
|
W przypadku dodatniej klasyfikacji wymaga się wskazania amplitudy reakcji – różnicy między linią podstawową a wierzchołkiem przy maksymalnej wartości wzorca referencyjnego (E2) wynoszącej co najmniej 20 % (tj. co najmniej 2 000 względnych jednostek światła, podczas gdy maksymalną wartość reakcji wzorców referencyjnych [E2] dostosowano do wartości 10 000 względnych jednostek światła).
|
|
—
|
Jeżeli jest to możliwe, wartość EC50 należy obliczyć w odniesieniu do każdej dodatniej substancji chemicznej.
|
|
|
Ujemna
|
Średnia dostosowana wartość względnej jednostki światła w odniesieniu do danego stężenia jest równa lub niższa od średniej wartości względnej jednostki światła kontroli z sulfotlenkiem dimetylowym plus trzykrotności odchylenia standardowego względnej jednostki światła sulfotlenku dimetylowego.
|
|
Niedostateczna
|
Dane, których nie można interpretować jako prawidłowe pod kątem obecności albo nieobecności aktywności substancji z uwagi na znaczne ograniczenia jakościowe lub ilościowe uznaje się za niedostateczne oraz niezdatne do wykorzystania przy określaniu, czy badana substancja chemiczna jest dodatnia czy ujemna. Badanie substancji chemicznych należy przeprowadzić ponownie.
|
|
|
|
AKTYWNOŚĆ ANTAGONISTY
|
|
Dodatnia
|
|
—
|
Dane dotyczące badanej substancji chemicznej kreślą krzywą zależności stężenie-odpowiedź, składającą się z linii podstawowej, po której następuje nachylenie ujemne.
|
|
—
|
Linia określająca nachylenie ujemne powinna składać się z co najmniej trzech punktów zawierających nienakładające się na siebie słupki błędu; nie wyklucza się punktów formujących linię podstawową, jednak liniowa część krzywej może zawierać pierwszy punkt plateau.
|
|
—
|
W przypadku wzorca referencyjnego (Ral/E2) powinno zaistnieć zmniejszenie aktywności o co najmniej 20 % w stosunku do wartości maksymalnej (tj. co najwyżej 8 000 względnych jednostek światła, podczas gdy maksymalną wartość reakcji wzorca referencyjnego [Ral/E2] dostosowano do wartości 10 000 względnych jednostek światła).
|
|
—
|
Najwyższe niecytotoksyczne stężenia badanej substancji chemicznej powinny być mniejsze bądź równe 1x10-5 M.
|
|
—
|
Jeżeli jest to możliwe, wartość IC50 należy obliczyć w odniesieniu do każdej dodatniej badanej substancji chemicznej.
|
|
|
Ujemna
|
Wszystkie punkty danych znajdują się powyżej wartości EC80 (80 % reakcji E2 lub 8 000 względnych jednostek światła), w stężeniach poniżej 1,0 x 10-5 M.
|
|
Niedostateczna
|
Dane, których nie można interpretować jako prawidłowe pod kątem obecności albo nieobecności aktywności substancji z uwagi na znaczne ograniczenia jakościowe lub ilościowe uznaje się za niedostateczne oraz niezdatne do wykorzystania przy określaniu, czy badana substancja chemiczna jest dodatnia czy ujemna. Badanie substancji chemicznej należy przeprowadzić ponownie.
|
|
40.
|
Wyniki dodatnie będą charakteryzować się zarówno wielkością skutku, oraz ilekroć to możliwe – stężeniem, przy którym występuje skutek. Przykłady dodatnich, ujemnych oraz niedostatecznych danych przedstawiono na rysunkach 5 i 6.
|
Rysunek 5
Przykłady agonistów: dane dodatnie, ujemne lub niedostateczne

Linią przerywaną oznaczono poziom 20 % reakcji E2, czyli 2 000 dostosowanych i znormalizowanych względnych jednostek światła.
Rysunek 6
Przykłady antagonistów: dane dodatnie, ujemne lub niedostateczne

Linią przerywaną oznaczono poziom 80 % reakcji Ral/E2, czyli 8 000 dostosowanych i znormalizowanych względnych jednostek światła.
Linią ciągłą oznaczono 1,00 × 10-5 M. W przypadku reakcji, którą zamierza się uznać za dodatnią, liczba względnych jednostek światła nie powinna przekraczać poziomu 8 000, a jej stężenia – 1,00 × 10–5 M.
Stężenia oznaczone gwiazdką na wykresie mezoheksestrolu wskazują na wyniki żywotności „2” lub większe.
Wyniki badania mezoheksestrolu uważa się za dane nieodpowiednie, ponieważ jedyna reakcja, która jest niższa niż 8 000 RLU zachodzi przy 1,00 × 10–5 M.
|
41.
|
Obliczeń EC50 i IC50 można dokonać, wykorzystując czteroparametrową funkcję Hill (w celu uzyskania bardziej szczegółowych informacji zob. protokoły dotyczące agonistów i antagonistów (7)). Spełnienie kryteriów dopuszczalności wskazuje, że system działa prawidłowo, ale nie gwarantuje, że którakolwiek konkretna seria badawcza dostarczy dokładnych danych. Powielenie wyników pierwszej serii badawczej gwarantuje największe prawdopodobieństwo uzyskania dokładnych danych (zob. pkt 19 dotyczący „ELEMENTÓW BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”).
|
SPRAWOZDANIE Z BADANIA
|
42.
|
Zob. pkt 20 dotyczący „ELEMENTÓW BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”.
|
BIBLIOGRAFIA
|
(1)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists. National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), „ALTEX” nr 23 (wyd. uzupełnione), s. 270–273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, „Alternatives to Laboratory Animals” nr 33, s. 261–287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals.„NIH Publication” nr 11–7850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals.„In Vitro Mol. Toxicol.” nr 13(1) s. 67–82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta. „Biochem. Pharmacol.” nr 71(10), s. 1459–69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology.„Chemistry and Biology” nr 17(6), s. 646–57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. „Endocrinology” nr 139(10), s. 4252–63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, „Cancer” nr 63, s. 280–288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro. „In Vitro Cell. Dev. Biol. – Animal” nr 34, s. 649–654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant. „Mol. Endo.” nr 28, s. 2072–2081.
|
|
(15)
|
Rogers, J.M. i Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, „In Vitro & Molec. Toxicol.” nr 13, s. 67–82.
|
Dodatek 4
TEST TRANSAKTYWACJI STABILNIE TRANSFEKOWANEGO LUDZKIEGO RECEPTORA ESTROGENOWEGO W CELU WYKRYCIA ESTROGENOWEGO DZIAŁANIA AGONISTYCZNEGO I ANTAGONISTYCZNEGO SUBSTANCJI CHEMICZNYCH Z WYKORZYSTANIEM LINII KOMÓRKOWEJ Z EKSPRESJĄ AKTYWOWANYCH CHEMICZNIE GENÓW LUCYFERAZY RECEPTORA ESTROGENOWEGO ALFA
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
1.
|
Do celów wykrywania estrogenowej aktywności agonistycznej i antagonistycznej przekazywanej przez ludzki receptor estrogenowy alfa (hERα), w teście transaktywacji ekspresji aktywowanych chemicznie genów lucyferazy receptora estrogenowego alfa wykorzystuje się ludzką linię komórkową U2OS. Badanie walidacyjne stabilnie transfekowanego testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa przeprowadzone przez BioDetection Systems BV (Amsterdam, Niderlandy) wykazało stosowność i wiarygodność badania w odniesieniu do jego zamierzonego celu (1). W linii komórkowej z ekspresją aktywowanych chemicznie genów lucyferazy receptora estrogenowego alfa zachodzi ekspresja jedynie stabilnie transfekowanego ludzkiego receptora estrogenowego alfa (2)(3).
|
|
2.
|
Szczególnym przeznaczeniem tego testu jest wykrywanie transaktywacji zależnej od ludzkiego receptora estrogenowego alfa poprzez pomiar bioluminescencji jako punktu końcowego. Bioluminescencja, ze względu na wysoki stosunek sygnału do szumu (4), jest powszechnie stosowana w testach biologicznych.
|
|
3.
|
Stwierdzono, że stężenia fitoestrogenu wyższe niż 1 μM powodują nadmierną aktywację genu reporterowego lucyferazy, w wyniku czego powstaje luminescencja niezależna od receptora (5)(6)(7). W związku z tym wyższe stężenia fitoestrogenów lub podobnych związków mogących powodować nadmierną aktywację ekspresji lucyferazy należy dokładnie zbadać w trakcie stabilnie transfekowanych testów transaktywacji receptora estrogenowego (zob. dodatek 2).
|
|
4.
|
Przed rozpoczęciem stosowania tego testu do celów regulacyjnych należy zapoznać się z sekcjami „WPROWADZENIE OGÓLNE” oraz „ELEMENTY BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”. Definicje i skróty stosowane w niniejszej metodzie badawczej opisano w dodatku 1.
|
ZASADA WYKONANIA TESTU (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
5.
|
Celem testu biologicznego jest ocena wiązania ligandów receptora estrogenowego, a następnie przeniesienie kompleksu receptor-ligand do jądra komórkowego. W jądrze komórkowym kompleks receptor-ligand wiąże określone elementy odpowiedzi DNA i transaktywuje gen reporterowy lucyferazy robaczka świętojańskiego, powodując zwiększoną ekspresję komórkową enzymu lucyferazy. Po dodaniu lucyferazy jako substratu lucyferyny, lucyferyna przekształca się w produkt bioluminescencyjny. Wytworzone światło można w łatwy sposób wykryć i za pomocą luminometru określić ilościowo.
|
|
6.
|
W układzie badawczym wykorzystuje się stabilnie transfekowane komórki ekspresji aktywowanych chemicznie genów lucyferazy receptora estrogenowego alfa. Komórki ekspresji aktywowanych chemicznie genów lucyferazy pochodzą z ludzkiej osteoblastycznej linii komórkowej kostniakomięsaka o nazwie U2OS. Ludzkie komórki U2OS stabilnie transfekowano z 3xHRE-TATA-Luc i pSG5-neo-hERα metodą współstrącania fosforanu wapnia. Linię komórkową U2OS wybrano ze względu na najwyższą odpowiedniość do pełnienia roli wskaźnikowej linii komórkowej reagującej na estrogeny (oraz inne hormony steroidowe), opierając się na obserwacji, że linia komórkowa U2OS wykazywała niewielką lub zerową aktywność receptora endogennego. Nieobecność receptorów endogennych oceniono, korzystając jedynie z plazmidów reporterowych lucyferazy, nie wykazujących aktywności po dodaniu ligandów receptorowych. Ponadto ta linia komórkowa, w momencie przejściowego wprowadzenia receptorów poznawczych stanowiła wsparcie dla reakcji zależnych od silnych hormonów (2)(3)(8).
|
|
7.
|
Badanie substancji chemicznych pod kątem aktywności estrogenowej lub antyestrogenowej przy zastosowaniu linii komórkowej z ekspresją aktywowanych chemicznie genów lucyferazy receptora estrogenowego alfa obejmuje wstępną serię badawczą i kompleksowe serie badawcze. We wstępnej serii badawczej określa się rozpuszczalność, cytotoksyczność oraz zawężony zakres stężeń badanych substancji chemicznych, które służą do wykonania kompleksowych serii badawczych. W kompleksowych seriach badawczych w ramach testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa przeprowadza się badanie zawężonych zakresów stężeń badanych substancji chemicznych, a następnie badane substancje chemiczne klasyfikuje się pod kątem agonizmu lub antagonizmu.
|
|
8.
|
Kryteria dotyczące interpretacji danych opisano szczegółowo w pkt 59. Krótko mówiąc, badaną substancję chemiczną uznaje się za dodatnią pod kątem agonizmu w przypadku, gdy co najmniej dwa następujące po sobie stężenia badanej substancji chemicznej wykazują reakcję, która jest wyższa bądź równa 10 % maksymalnej reakcji wzorca referencyjnego 17β-estradiolu (PC10). Badaną substancję chemiczną uznaje się za dodatnią pod kątem antagonizmu, w przypadku gdy co najmniej dwa następujące po sobie stężenia badanej substancji chemicznej wykazują reakcję, która jest niższa bądź równa 80 % maksymalnej reakcji wzorca referencyjnego tamoksyfenu (PC80).
|
PROCEDURA
Linie komórkowe
|
9.
|
W teście należy stosować stabilnie transfekowaną linię komórkową U2OS z ekspresją aktywowanych chemicznie genów lucyferazy receptora estrogenowego alfa. Linię komórkową można pozyskać z BioDetection Systems BV (Amsterdam, Niderlandy) na podstawie technicznej umowy licencyjnej.
|
|
10.
|
Należy stosować jedynie komórki wolne od mykoplazmy. Partie komórkowe wykorzystane do badań powinny albo dawać wyniki ujemne pod względem zanieczyszczenia mykoplazmą, albo przed ich zastosowaniem powinno się wykonać badanie na obecność mykoplazmy. RT-PCR (reakcję łańcuchową polimerazy w czasie rzeczywistym) należy zastosować w odniesieniu do czułego wykrywania zakażenia mykoplazmą (9).
|
Stabilność linii komórkowej
|
11.
|
Aby zachować stabilność i integralność komórek ekspresji aktywowanych chemicznie genów lucyferazy, należy przechowywać je w ciekłym azocie (w temperaturze -80 °C). Po rozmrożeniu komórek w celu rozpoczęcia nowej hodowli, komórki, zanim rozpocznie się ich stosowanie do oceny agonistycznej i antagonistycznej aktywności substancji chemicznych, należy co najmniej dwukrotnie wysiać na hodowle wtórne. Komórki nie powinny wydzielać kultury pochodnej przez więcej niż 30 pasaży.
|
|
12.
|
Aby monitorować stabilność linii komórkowej w czasie, należy zweryfikować antagonistyczną i agonistyczną responsywność układu badawczego, czego można dokonać, przeprowadzając ocenę wartości EC50 lub IC50 wzorca referencyjnego. Ponadto należy monitorować indukcję względną próbki kontroli dodatniej (PC) i próbki kontroli ujemnej (NC). Wyniki powinny być zgodne z kryteriami dopuszczalności dotyczącymi agonistycznego (tabela 3C) lub antagonistycznego (tabela 4C) testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa. W tabeli 1 (dla trybu agonistycznego) i 2 (dla trybu antagonistycznego) przedstawiono wzorce referencyjne, kontrole dodatnie i ujemne.
|
Kultura komórkowa i warunki posiewu
|
13.
|
Komórki U2OS należy hodować w substancji biogennej (pożywce DMEM/F12 (1:1) zawierającej czerwony fenol pełniący rolę wskaźnika pH, uzupełnionej bydlęcą surowicą płodową (7,5 %), nieegzogennymi aminokwasami (1 %), 10 jednostkami/ml penicyliny oraz markerem selekcyjnym w postaci streptomycyny i genotycyny (G418)). Komórki należy umieścić w inkubatorze CO2 (w atmosferze zawierającej 5 % CO2) w temperaturze 37 °C, przy 100 % wilgotności. Kiedy komórki osiągną 85–95 % konfluencji, należy wysiać je na hodowle wtórne albo przygotować do posiania w płytkach 96-dołkowych. W drugim przypadku komórki należy zawiesić ponownie w 1x105 komórek/ml w pożywce testowej wolnej od estrogenu (pożywce DMEM/F12 (1:1) niezawierającej czerwonego fenolu, uzupełnionej pokrytą dekstranem bydlęcą surowicą płodową poddaną działaniu węgla drzewnego (5 % obj./obj.), nieegzogennymi aminokwasami (1 % obj./obj.), 10 jednostkami/ml penicyliny i streptomycyny) oraz rozmieścić je w dołkach płytek 96-dołkowych (100 μl homogenizowanej zawiesiny komórki). 24 godziny przed rozpoczęciem narażania komórki należy poddać wstępnej inkubacji w inkubatorze CO2 (w atmosferze zawierającej 5 % CO2, w 37 °C, przy 100 % wilgotności). Wyroby wykonane z tworzywa sztucznego powinny być wolne od estrogenu.
|
Kryteria dopuszczalności
|
14.
|
Działania agonistyczne i antagonistyczne badanej substancji chemicznej lub badanych substancji chemicznych bada się w serii badań. Badanie składa się z maksymalnie sześciu mikropłytek. W skład każdej serii badań wchodzi co najmniej jedna seria rozcieńczeń wzorca referencyjnego, próbka kontroli dodatniej, próbka kontroli ujemnej oraz kontrole z rozpuszczalnikiem. Na rysunkach 1 i 2 przedstawiono układ płytek na potrzeby serii badań agonistycznych i antagonistycznych.
|
|
15.
|
Każde rozcieńczenie wzorców referencyjnych, badanych substancji chemicznych, wszystkich kontroli z rozpuszczalnikiem oraz kontroli dodatnich i ujemnych należy trzykrotnie przeanalizować. Każda z tych analiz powinna spełniać wymagania określone w tabeli 3A i tabeli 4A.
|
|
16.
|
Pomiar pełnej serii rozcieńczeń wzorca referencyjnego (w przypadku agonizmu – 17β-estradiolu; w przypadku antagonizmu – tamoksyfenu) przeprowadza się na pierwszej płytce podczas każdej serii badań. Aby umożliwić zestawienie wyników analizy pozostałych pięciu mikropłytek z pierwszą mikropłytką obejmującą pełną krzywą zależności stężenie-odpowiedź wzorca referencyjnego, wszystkie płytki powinny zawierać trzy próbki kontrolne: kontrolę z rozpuszczalnikiem, najwyższe stężenie badanego wzorca porównawczego oraz przybliżone stężenie EC50 (w przypadku agonizmu) lub IC50 (w przypadku antagonizmu) wzorca referencyjnego. Stosunek średnich próbek kontrolnych na pierwszej płytce do pozostałych pięciu płytek powinien spełniać warunki określone w tabeli 3C (agonizm) lub 4C (antagonizm).
|
|
17.
|
Wskaźnik „z” oblicza się dla każdej z mikropłytek objętych serią badań (10). Wskaźnik „z” należy obliczyć, korzystając z reakcji w najwyższym i najniższym stężeniu wzorca referencyjnego. Mikropłytkę uznaje się za prawidłową, gdy spełnia wymagania określone w tabeli 3C (agonizm) lub 4C (antagonizm).
|
|
18.
|
Wzorzec referencyjny powinien wykazać sygmoidalną krzywą zależności dawka-efekt. Wartości EC50 lub IC50 uzyskane z reakcji serii rozcieńczeń wzorca referencyjnego powinny spełniać wymagania określone w tabeli 3C (agonizm) lub 4C (antagonizm).
|
|
19.
|
W skład każdej serii badań powinna wchodzić próbka kontroli dodatniej i próbka kontroli ujemnej. Obliczona względna indukcja zarówno próbki kontroli dodatniej, jak i ujemnej powinna spełniać minimalne wymagania określone w tabeli 3C (agonizm) lub 4C (antagonizm).
|
|
20.
|
Podczas wszystkich obliczeń współczynnik sygnału najwyższego stężenia wzorca referencyjnego należy mierzyć, dzieląc średnią najwyższą reakcję względnej jednostki światła wzorca referencyjnego 17β-estradiolu przez średnią reakcję względnej jednostki światła kontroli z rozpuszczalnikiem referencyjnym. Ten współczynnik sygnału powinien spełniać minimalne wymagania krotności indukcji określone w tabeli 3C (agonizm) lub 4C (antagonizm).
|
|
21.
|
Wyłącznie mikropłytki, które spełniają wszystkie wymienione powyżej kryteria dopuszczalności uznaje się za poprawne i można je wykorzystać do przeprowadzenia oceny w zakresie reakcji badanych substancji chemicznych.
|
|
22.
|
Kryteria dopuszczalności mają zastosowanie zarówno do wstępnych serii badawczych, jak i do kompleksowych serii badawczych.
|
Tabela 1
Stężenia wzorca referencyjnego, kontroli dodatniej (PC) oraz kontroli ujemnej (NC) w odniesieniu do testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie agonistyczne
|
|
Substancja
|
NR CAS
|
Zakres badania (M)
|
|
Wzorzec referencyjny
|
17β-estradiol
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Kontrola dodatnia (PC)
|
17α-metylotestosteron
|
58-18-4
|
3*10-6
|
|
Kontrola ujemna (NC)
|
kortykosteron
|
50-22-6
|
1*10-8
|
Tabela 2
Stężenia wzorca referencyjnego, kontroli dodatniej (PC) oraz kontroli ujemnej (NC) w odniesieniu do testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie antagonistyczne
|
|
Substancja
|
NR CAS
|
Zakres badania (M)
|
|
Wzorzec referencyjny
|
tamoksyfen
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Kontrola dodatnia (PC)
|
4-hydroksy-tamoksyfen
|
68047-06-3
|
1*10-9
|
|
Kontrola ujemna (NC)
|
resweratrol
|
501-36-0
|
1*10-5
|
Tabela 3
Kryteria dopuszczalności agonistycznego testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa
|
A – poszczególne próbki na płytce
|
Kryterium
|
|
1
|
Maksymalne % SD trzech dołków (w przypadku NC, PC, każdego rozcieńczenia badanej substancji chemicznej i wzorca referencyjnego, z wyłączeniem C0)
|
< 15 %
|
|
2
|
Maksymalne % SD trzech dołków (w przypadku wzorca referencyjnego oraz kontroli z rozpuszczalnikiem badanej substancji chemicznej (C0, SC))
|
< 30 %
|
|
3
|
Maksymalna utrata dehydrogenazy mleczanowej jako wskaźnik cytotoksyczności.
|
< 120 %
|
|
B – w obrębie pojedynczej mikropłytki
|
|
|
4
|
Stosunek kontroli z rozpuszczalnikiem wzorca referencyjnego (C0; płytka 1) do kontroli z rozpuszczalnikiem badanej substancji chemicznej (SC; płytki od 2–x)
|
0,5–2,0
|
|
5
|
Stosunek przybliżonego EC50 i najwyższych stężeń wzorca referencyjnego na płytce 1 oraz przybliżonego EC50 i najwyższych stężeń wzorca referencyjnego na płytce 2 do x (C4, C8)
|
0,70 do 1,30
|
|
6
|
Czynnik „Z” w przypadku każdej płytki
|
> 0,6
|
|
C – w ramach pojedynczej serii analiz (wszystkie płytki w ramach jednej serii)
|
|
|
7
|
Krzywa sigmoidalna wzorca referencyjnego
|
Tak (17β-estradiol)
|
|
8
|
Wzorzec referencyjny zakresu EC50 (17ß-estradiol)
|
4*10-12 – 4*10-11 M
|
|
9
|
Minimalna krotność indukcji najwyższego stężenia 17ß-estradiolu w odniesieniu do kontroli z rozpuszczalnikiem wzorca referencyjnego.
|
5
|
|
10
|
Indukcja względna (%) PC.
|
> 30 %
|
|
11
|
Indukcja względna (%) NC
|
< 10 %
|
Przybliżony: w przybliżeniu; PC: kontrola dodatnia: NC: kontrola ujemna; SC: kontrola z rozpuszczalnikiem badanej substancji chemicznej; C0: kontrola z rozpuszczalnikiem wzorca referencyjnego; SD: odchylenie standardowe; LDH: dehydrogenaza mleczanowa
Tabela 4
Kryteria dopuszczalności antagonistycznego testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa
|
A – poszczególne próbki na płytce
|
Kryterium
|
|
1
|
Maksymalne % SD trzech dołków (w przypadku NC, PC, każdego rozcieńczenia badanej substancji chemicznej i wzorca referencyjnego, kontrola z rozpuszczalnikiem (C0))
|
< 15 %
|
|
2
|
Maksymalne % SD trzech dołków (w przypadku grupy kontrolnej nośnika (VC) i najwyższego stężenia wzorca referencyjnego (C8))
|
< 30 %
|
|
3
|
Maksymalna utrata dehydrogenazy mleczanowej jako wskaźnik cytotoksyczności.
|
< 120 %
|
|
B – w obrębie pojedynczej mikropłytki
|
|
|
4
|
Stosunek kontroli z rozpuszczalnikiem wzorca referencyjnego (C0; płytka 1) do kontroli z rozpuszczalnikiem badanej substancji chemicznej (SC; płytki od 2 do x)
|
0,70 do 1,30
|
|
5
|
Stosunek przybliżonego stężenia IC50 we wzorcu referencyjnym na płytce 1 do przybliżonego stężenia IC50 we wzorcu referencyjnym na płytkach od 2 do x (C4)
|
0,70 do 1,30
|
|
6
|
Stosunek najwyższego stężenia wzorca referencyjnego na płytce 1 do najwyższego stężenia wzorca referencyjnego na płytkach od 2 do x (C8)
|
0,50 do 2,0
|
|
7
|
Czynnik „Z” w przypadku każdej płytki
|
> 0,6
|
|
C – w ramach pojedynczej serii analiz (wszystkie płytki w ramach jednej serii)
|
|
|
8
|
Krzywa sigmoidalna wzorca referencyjnego
|
Tak (Tamoksyfen)
|
|
9
|
Wzorzec referencyjny zakresu IC50 (Tamoksyfen)
|
1*10-8 – 1*10-7 M
|
|
10
|
Minimalna krotność indukcji kontroli z rozpuszczalnikiem wzorca referencyjnego w odniesieniu do najwyższego stężenia Tamoksyfenu.
|
2,5
|
|
11
|
Indukcja względna (%) PC.
|
< 70 %
|
|
12
|
Indukcja względna (%) NC
|
> 85 %
|
Przybliżony: w przybliżeniu; PC: kontrola dodatnia: NC: kontrola ujemna; VC: grupa kontrolna nośnika (grupa kontrolna z rozpuszczalnikiem bez stałego stężenia wzorca referencyjnego agonisty); SC: kontrola z rozpuszczalnikiem badanej substancji chemicznej; C0: kontrola z rozpuszczalnikiem wzorca referencyjnego; SD: odchylenie standardowe; LDH: dehydrogenaza mleczanowa
Kontrola z rozpuszczalnikiem/nośnikiem, wzorce referencyjne, kontrole dodatnie, kontrole ujemne
|
23.
|
Zarówno w przypadku wstępnej serii badawczej, jak i kompleksowych serii badawczych należy stosować tę samą kontrolę z rozpuszczalnikiem/nośnikiem, te same wzorce referencyjne, kontrole dodatnie i kontrole ujemne. Ponadto stężenie wzorców referencyjnych, kontroli dodatnich i kontroli ujemnych powinno być takie samo.
|
Kontrola z rozpuszczalnikiem
|
24.
|
Rozpuszczalnik stosowany do rozpuszczenia badanej substancji chemicznej należy zbadać jako kontrolę z rozpuszczalnikiem. Sufotlenek dimetylowy (DMSO, 1 % (obj./obj.); nr CAS 67-68-5) zastosowano jako nośnik podczas walidacji testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa. Jeśli stosuje się rozpuszczalnik inny niż DMSO, wszystkie wzorce referencyjne, grupy kontrolne i badane substancje chemiczne należy zbadać w tym samym nośniku. Należy zauważyć, że kontrola z rozpuszczalnikiem dotycząca badań antagonistów obejmuje stałe stężenie wzorca referencyjnego agonisty – 17β-estradiolu (w przybliżeniu stężenie EC50). Aby zbadać rozpuszczalnik wykorzystany w badaniach antagonistów, należy przygotować i zbadać grupę kontrolną nośnika.
|
Grupa kontrolna nośnika (antagonizm)
|
25.
|
W celu zbadania antagonizmu, pożywkę testową uzupełnia się o stałe stężenie wzorca referencyjnego agonisty – 17β-estradiolu (w przybliżeniu stężenie EC50). Aby zbadać rozpuszczalnik zastosowany do rozpuszczenia badanej substancji chemicznej w celu uzyskania antagonizmu, należy przygotować pożywkę testową bez stałego stężenia wzorca referencyjnego agonisty – 17β-estradiolu. Ta próbka kontrolna jest oznaczona jako kontrola nośnika. Sufotlenek dimetylowy (DMSO, 1 % (obj./obj.); nr CAS 67-68-5) zastosowano jako nośnik podczas walidacji testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa. Jeśli stosuje się rozpuszczalnik inny niż DMSO, wszystkie wzorce referencyjne, grupy kontrolne i badane substancje chemiczne należy zbadać w tym samym nośniku.
|
Wzorce referencyjne
|
26.
|
Wzorzec referencyjny agonisty to 17β-estradiol (tabela 1). Wzorce referencyjne składają się z serii rozcieńczeń ośmiu stężeń 17β-estradiolu (1*10-13; 3*10-13; 1*10-12; 3*10-12; 6*10-12; 1*10-11; 3*10-11; 1*10-10 M).
|
|
27.
|
Wzorzec referencyjny antagonisty to tamoksyfen (tabela 2). Wzorce referencyjne składają się z serii rozcieńczeń ośmiu stężeń tamoksyfenu (3*10-9; 1*10-8; 3*10-8; 1*10-7; 3*10-7; 1*10-6; 3*10-6; 1*10-5 M). Każde stężenie wzorca referencyjnego antagonisty poddaje się inkubacji wspólnie ze stałym stężeniem wzorca referencyjnego agonisty – 17β-estradiolu (3*10-12 M).
|
Kontrola dodatnia
|
28.
|
Kontrola dodatnia w przypadku badań agonistów to 17α-metylotestosteron (tabela 1).
|
|
29.
|
Kontrola dodatnia w przypadku badań antagonistów to 4-hydroksy-tamoksyfen (tabela 2). Antagonistyczną kontrolę dodatnią poddaje się inkubacji wspólnie ze stałym stężeniem wzorca referencyjnego agonisty – 17β-estradiolu (3*10-12 M).
|
Kontrola ujemna
|
30.
|
Kontrola ujemna w przypadku badań agonistów to kortykosteron (tabela 1).
|
|
31.
|
Kontrola ujemna w przypadku badań antagonistów to resweratrol (tabela 2). Antagonistyczną kontrolę ujemną poddaje się inkubacji wspólnie ze stałym stężeniem wzorca referencyjnego agonisty – 17β-estradiolu (3*10-12 M).
|
Wykazanie biegłości laboratorium (zob. pkt 14 oraz tabela 3 i 4 w „ELEMENTY BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO” niniejszej metody badawczej)
Nośnik
|
32.
|
Rozpuszczalnik stosowany do rozpuszczenia badanej substancji chemicznej powinien całkowicie rozpuścić badaną substancję chemiczną i powinien być mieszalny z podłożem komórkowym. DMSO, woda i etanol (95–100 % czystości) to odpowiednie rozpuszczalniki. W przypadku gdy jako rozpuszczalnik stosuje się DMSO, maksymalne stężenie DMSO w trakcie inkubacji nie powinno przekroczyć 1 % (obj./obj.). Przed zastosowaniem należy zbadać rozpuszczalnik w kierunku braku cytotoksyczności i oddziaływania na efektywność testu.
|
Przygotowanie wzorców referencyjnych, kontroli dodatnich, kontroli ujemnych i badanych substancji chemicznych
|
33.
|
Wzorce referencyjne, kontrole dodatnie, kontrole ujemne i badane substancje chemiczne rozpuszcza się w 100 % DMSO (lub właściwym rozpuszczalniku). Następnie należy przygotować właściwe stężenia (serię stężeń wzrastających w postępie geometrycznym) w tym samym rozpuszczalniku. Przed rozpuszczeniem należy pozwolić, aby temperatura wszystkich substancji wyrównała się z temperaturą pokojową. Świeżo przygotowane roztwory podstawowe wzorców referencyjnych, kontroli dodatnich, kontroli ujemnych i badanych substancji chemicznych nie powinny zawierać zauważalnego osadu ani nie powinny być mętne. Wzorzec referencyjny i roztwory kontrolne można przygotować w dużej ilości. Przed każdym doświadczeniem należy przygotowywać świeże roztwory podstawowe badanych substancji chemicznych. Do każdego doświadczenia należy przygotować świeże ostateczne rozcieńczenia wzorców referencyjnych, kontroli dodatnich, kontroli ujemnych i badanych substancji chemicznych i wykorzystać w ciągu 24 godzin od przygotowania.
|
Rozpuszczalność, cytotoksyczność i zakres dawkowania.
|
34.
|
We wstępnej serii badawczej określa się rozpuszczalność badanej substancji chemicznej w wybranym rozpuszczalniku. Przygotowuje się maksymalne stężenie roztworu wynoszące 0,1 M. W przypadku gdy to stężenie wykazuje problemy z rozpuszczalnością, należy przygotowywać roztwory podstawowe o niższym stężeniu, do czasu aż badane substancje chemiczne zostaną w pełni rozpuszczone. We wstępnej serii badawczej bada się serię stężeń badanej substancji chemicznej wzrastających w postępie geometrycznym w stosunku 1:10. Maksymalne stężenie próby w przypadku badania agonistów lub antagonistów wynosi 1 mM. Po przeprowadzeniu próby wstępnej określa się właściwy zawężony zakres czystego stężenia badanej substancji chemicznej, które należy zbadać w kompleksowych seriach badawczych. Stężenia wykorzystywane w badaniu kompleksowym powinny wynosić 1x, 3x, 10x, 30x, 100x, 300x, 1000x i 3000x.
|
|
35.
|
Badanie cytotoksyczności zostało uwzględnione w protokole testów agonisty i antagonisty (11). Zarówno wstępna seria badawcza, jak i kompleksowe serie badawcze obejmują badanie cytotoksyczności. Metodą zastosowaną, aby ocenić cytotoksyczność podczas walidacji testu biologicznego z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na działanie receptora estrogenowego alfa było badanie utraty dehydrogenazy mleczanowej (LDH) w połączeniu z jakościową kontrolą wzrokową komórek (zob. dodatek 4.1) po narażeniu na działanie badanych substancji chemicznych. Aby określić cytotoksyczność można zastosować jednak inne jakościowe metody (np. analizę kolometryczną opartą na MTT (test MTT) lub test biologiczny z ekspresją aktywowanych chemicznie genów lucyferazy wrażliwych na cytotoksyczność). Zasadniczo stężenia badanej substancji chemicznej wykazujące ponad 20 % zmniejszenie żywotności komórek uznaje się za cytotoksyczne i w związku z tym nie można ich wykorzystać do oceny danych. Jeżeli chodzi o badanie utraty LDH, stężenie badanej substancji chemicznej uważa się za cytotoksyczne, w przypadku gdy procentowa utrata LDH jest wyższa niż 120 %.
|
Narażenie na działanie badanej substancji chemicznej oraz organizacja płytki testowej
|
36.
|
Po przeprowadzeniu trypsynizacji kolby zlewającej się hodowli komórek, komórki ponownie zawiesza się w pożywce testowej wolnej od estrogenu w ilości 1x105 komórek/ml. Sto μl ponownie zawieszonych komórek posiewa się na wewnętrznych dołkach płytki 96-dołkowej. Zewnętrzne dołki wypełnia się 200 μl roztworem chlorku sodu buforowanym fosforanami (PBS) (zob. rysunek 1 i 2). Wysiane komórki poddaje się wstępnej inkubacji przez 24 godziny w inkubatorze CO2 (w atmosferze zawierającej 5 % CO2, w 37 °C, przy 100 % wilgotności).
|
|
37.
|
Po wstępnej inkubacji płytki poddaje się kontroli w kierunku widocznej cytotoksyczności (zob. dodatek 4.1), skażenia i konfluencji. Do badania wykorzystuje się wyłącznie płytki wykazujące brak widocznej cytotoksyczności, skażenia i posiadające co najmniej 85 % konfluencję. Usuwa się dokładnie podłoże z wewnętrznych dołków i zastępuje je 200 μl pożywki testowej wolnej od estrogenu, zawierającej odpowiednie serie rozcieńczeń wzorców referencyjnych, badanych substancji chemicznych, kontroli dodatnich, kontroli ujemnych i kontroli z rozpuszczalnikiem (tabela 5: badanie agonisty; tabela 6: badanie antagonisty). Wszystkie wzorce referencyjne, badane substancje chemiczne, kontrole dodatnie, kontrole ujemne i kontrole z rozpuszczalnikiem bada się trzykrotnie. Na rysunku 1 przedstawiono rozkład na płytce służącej do badania agonisty. Na rysunku 2 przedstawiono rozkład na płytce służącej do badania antagonisty. Rozkład na płytce służącej do badania wstępnego i kompleksowego jest taki sam. W przypadku badania antagonisty wszystkie wewnętrzne dołki z wyjątkiem dołków grupy kontrolnej nośnika (VC) również zawierają stałe stężenie wzorca referencyjnego agonisty – 17β-estradiolu (3*10-12 M). Należy zauważyć, że wzorce referencyjne C8 i C4 należy dodać do każdej płytki TC.
|
|
38.
|
Po narażeniu komórek na działanie wszystkich substancji chemicznych płytki 96-dołkowe należy poddać wstępnej inkubacji przez kolejne 24 godziny w inkubatorze CO2 (w atmosferze zawierającej 5 % CO2, w 37 °C, przy 100 % wilgotności).
|
Rysunek 1
Rozkład na płytce w przypadku płytek 96-dołkowych służących do przeprowadzenia próby wstępnej i oceny wpływu agonistycznego.

C0 = rozpuszczalnik wzorca referencyjnego.
C(1-8) = serie rozcieńczeń (1–8, od niskich do wysokich stężeń) wzorca referencyjnego.
PC = kontrola dodatnia.
NC = kontrola ujemna.
TCx-(1-8) = rozcieńczenia (1–8, od niskich do wysokich stężeń) badanej substancji chemicznej wykorzystywanej do przeprowadzenia wstępnej serii badawczej i oceny wpływu agonistycznego badanej substancji chemicznej x.
SC = kontrola z rozpuszczalnikiem badanej substancji chemicznej (optymalnie ten sam rozpuszczalnik co w przypadku C0, ale w miarę możliwości z innej partii).
Komórki oznaczone kolorem szarym: = Zewnętrzne dołki, wypełnione 200 μl PBS.
Rysunek 2
Rozkład na płytce w przypadku płytek 96-dołkowych służących do przeprowadzenia antagonistycznej próby wstępnej i oceny wpływu antagonistycznego.

C0 = rozpuszczalnik wzorca referencyjnego.
C(1-8) = serie rozcieńczeń (1–8, od niskich do wysokich stężeń) wzorca referencyjnego.
NC = kontrola ujemna.
PC = kontrola dodatnia.
TCx-(1-8) = rozcieńczenia (1–8, od niskich do wysokich stężeń) badanej substancji chemicznej wykorzystywanej do przeprowadzenia wstępnej serii badawczej i oceny wpływu agonistycznego badanej substancji chemicznej x.
SC = kontrola z rozpuszczalnikiem badanej substancji chemicznej (optymalnie ten sam rozpuszczalnik co w przypadku C0, ale w miarę możliwości z innej partii).
VC = grupa kontrolna nośnika (kontrola rozpuszczalnika bez stałego stężenia wzorca referencyjnego agonisty – 17β-estradiolu).
Komórki oznaczone kolorem szarym: = Zewnętrzne dołki, wypełnione 200 μl PBS.
Uwaga: wszystkie wewnętrzne dołki z wyjątkiem dołków grupy kontrolnej nośnika (VC) również zawierają stałe stężenie wzorca referencyjnego agonisty – 17β-estradiolu (3,0*10-12 M)
Pomiar luminescencji
|
39.
|
Pomiar luminescencji szczegółowo opisano w protokole testu dotyczącego agonisty i antagonisty (10). Po 24 godzinach inkubacji należy usunąć podłoże z dołków i poddać komórki procesowi lizy, aby otworzyć błonę komórkową i umożliwić dokonanie pomiaru aktywności lucyferazy.
|
|
40.
|
Aby dokonać pomiaru luminescencji, procedura ta wymaga luminometra wyposażonego w 2 iniektory. Reakcję lucyferazy rozpoczyna się poprzez wstrzyknięcie substratu – lucyferyny. Reakcję przerywa się poprzez dodanie 0,2 M NaOH. Reakcję przerywa się, aby zapobiec przeniesieniu luminescencji z jednego dołka do drugiego.
|
|
41.
|
Światło emitowane z każdego dołka wyraża się jako względną jednostkę światła (RLU) w przeliczeniu na dołek.
|
Wstępna seria badawcza
|
42.
|
Wyniki analizy wstępnej wykorzystuje się, aby określić zawężony zakres stężenia badanych substancji chemicznych służący do przeprowadzenia badania kompleksowego. Ocenę wyników analizy wstępnej i określenie zawężonego zakresu stężenia badanej substancji chemicznej służącej do przeprowadzenia badania kompleksowego szczegółowo opisano w protokole próby dotyczącej agonisty i antagonisty (10). Poniżej podano krótkie streszczenie procedur służących do określenia zakresu stężenia badanych substancji chemicznych służących do przeprowadzenia badania agonisty i antagonisty. Zob. tabela 5 i 6, aby zapoznać się z wytycznymi dotyczącymi planu serii stężeń wzrastających w postępie geometrycznym.
|
Wybór stężeń do przeprowadzenia oceny wpływu agonistycznego
|
43.
|
We wstępnej serii badawczej należy zbadać badane substancje chemiczne, wykorzystując serię rozcieńczeń zgodnie z tabelą 5 (agonista) i 6 (antagonista). Wszystkie stężenia należy zbadać w trzech dołkach zgodnie z rozkładem na płytce wskazanym na rysunku 1 (agonista) lub 2 (antagonista).
|
|
44.
|
Wyłącznie wyniki analizy, które spełniają kryteria dopuszczalności (tabela 3) uznaje się za poprawne i można je wykorzystać do przeprowadzenia oceny w zakresie reakcji badanych substancji chemicznych. W przypadku gdy jedna mikropłytka lub większa ich liczba w serii analiz nie spełnia kryteriów dopuszczalności, odpowiednie mikropłytki należy ponownie poddać analizie. W przypadku gdy pierwsza płytka zawierająca całą serię rozcieńczeń wzorca referencyjnego nie spełni kryteriów dopuszczalności, konieczne jest ponowne poddanie analizie całej serii badań (6 płytek).
|
|
45.
|
Należy dostosować zakresy stężenia początkowego badanych substancji chemicznych i powtórzyć wstępną serię badawczą, w przypadku gdy:
|
—
|
zaobserwowano cytotoksyczność. Należy powtórzyć wstępną procedurę przy niższych niecytotoksycznych stężeniach badanej substancji chemicznej;
|
|
—
|
próba wstępna badanych substancji chemicznych nie wykazuje pełnej krzywej dawka-efekt, ponieważ badane stężenia generują maksymalną indukcję. Należy powtórzyć wstępną serię badawczej, stosując niższe stężenia badanej substancji chemicznej.
|
|
|
46.
|
Jeśli zaobserwowano poprawną zależność dawka-odpowiedź, należy wybrać (najniższe) stężenie, w przypadku którego obserwuje się maksymalną indukcję, i które nie wykazuje cytotoksyczności. Najwyższe stężenie badanej substancji chemicznej, która ma zostać zbadana w kompleksowych seriach badawczych, powinno być 3 razy wyższe niż przedmiotowe wybrane stężenie.
|
|
47.
|
Całą serię czystych rozcieńczeń badanej substancji chemicznej należy przygotować zgodnie z etapami rozcieńczania wskazanymi w tabeli 5, rozpoczynając od najwyższego stężenia, jak określono powyżej.
|
|
48.
|
Badaną substancję chemiczną, która nie wykazuje wpływu agonistycznego należy zbadać w kompleksowych seriach badawczych, rozpoczynając od najwyższego, niecytotoksycznego stężenia określonego we wstępnej serii badawczej.
|
Wybór stężeń do przeprowadzenia oceny wpływu antagonistycznego
|
49.
|
Wyłącznie wyniki analizy, które spełniają kryteria dopuszczalności (tabela 4) uznaje się za poprawne i można je wykorzystać do przeprowadzenia oceny w zakresie reakcji badanych substancji chemicznych. W przypadku gdy jedna mikropłytka lub większa ich liczba w serii analiz nie spełnia kryteriów dopuszczalności, odpowiednie mikropłytki należy ponownie poddać analizie. W przypadku gdy pierwsza płytka zawierająca całą serię rozcieńczeń wzorca referencyjnego nie spełni kryteriów dopuszczalności, konieczne jest ponowne poddanie analizie całej serii badań (6 płytek).
|
|
50.
|
Należy dostosować zakresy stężenia początkowego badanych substancji chemicznych i powtórzyć wstępną serię badawczą, w przypadku gdy:
|
—
|
zaobserwowano cytotoksyczność. Należy powtórzyć wstępną procedurę przy niższych niecytotoksycznych stężeniach badanej substancji chemicznej;
|
|
—
|
próba wstępna badanych substancji chemicznych nie wykazuje pełnej krzywej dawka-efekt, ponieważ badane stężenia generują maksymalną inhibicję. Należy powtórzyć próbę wstępną, stosując niższe stężenia badanej substancji chemicznej.
|
|
|
51.
|
Jeśli stwierdzono poprawną zależność dawka-odpowiedź, należy wybrać (najniższe) stężenie, w przypadku którego obserwuje się maksymalną inhibicję, i które nie wykazuje cytotoksyczności. Najwyższe stężenie badanej substancji chemicznej, która ma zostać zbadana w kompleksowych seriach badawczych, powinno być 3 razy wyższe niż przedmiotowe wybrane stężenie.
|
|
52.
|
Całą serię czystych rozcieńczeń badanej substancji chemicznej należy przygotować zgodnie z etapami rozcieńczania wskazanymi w tabeli 6, rozpoczynając od najwyższego stężenia, jak określono powyżej.
|
|
53.
|
Badane substancje chemiczne, które nie wykazują wpływu antagonistycznego należy zbadać w kompleksowych seriach badawczych, rozpoczynając od najwyższego, niecytotoksycznego stężenia badanego we wstępnej serii badawczej.
|
Kompleksowe serie badawcze
|
54.
|
Po dokonaniu wyboru zawężonych zakresów stężenia należy kompleksowo zbadać badane substancje chemiczne, wykorzystując serię rozcieńczeń zgodnie z tabelą 5 (agonista) i 6 (antagonista). Wszystkie stężenia należy zbadać w trzech dołkach zgodnie z rozkładem na płytce wskazanym na rysunku 1 (agonista) lub 2 (antagonista).
|
|
55.
|
Wyłącznie wyniki analizy, które spełniają kryteria dopuszczalności (tabela 3 i 4) uznaje się za poprawne i można je wykorzystać do przeprowadzenia oceny w zakresie reakcji badanych substancji chemicznych. W przypadku gdy jedna mikropłytka lub większa ich liczba w serii analiz nie spełnia kryteriów dopuszczalności, odpowiednie mikropłytki należy ponownie poddać analizie. W przypadku gdy pierwsza płytka zawierająca całą serię rozcieńczeń wzorca referencyjnego nie spełni kryteriów dopuszczalności, konieczne jest ponowne poddanie analizie całej serii badań (6 płytek).
|
Tabela 5
Stężenie i rozcieńczenia wzorców referencyjnych, kontroli i badanych substancji chemicznych stosowanych do przeprowadzenia badania agonisty
|
Wzorzec porównawczy – 17β-estradiol
|
TCx – wstępna seria badawcza
|
TCx – kompleksowa seria badawcza
|
Kontrole
|
|
stężenie (M)
|
rozcieńczenie
|
rozcieńczenie
|
stężenie (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx – badana substancja chemiczna x
PC – kontrola dodatnia (17α-metylotestosteron)
NC – kontrola ujemna (kortykosteron)
C0 – kontrola z rozpuszczalnikiem wzorca referencyjnego
SC – kontrola z rozpuszczalnikiem badanej substancji chemicznej
|
Tabela 6
Stężenie i rozcieńczenia wzorców referencyjnych, kontroli i badanych substancji chemicznych stosowanych do przeprowadzenia badania antagonisty
|
Wzorzec porównawczy – tamoksyfen
|
TCx – wstępna seria badawcza
|
TCx – kompleksowa seria badawcza
|
Kontrole
|
|
stężenie (M)
|
rozcieńczenie
|
rozcieńczenie
|
stężenie (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Wzbogacony agonista
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
stężenie (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx – badana substancja chemiczna x
PC – kontrola dodatnia (4-hydroksy-tamoksyfen)
NC – kontrola ujemna (resweratrol)
C0 – kontrola z rozpuszczalnikiem wzorca referencyjnego
SC – kontrola z rozpuszczalnikiem badanej substancji chemicznej
VC – grupa kontrolna nośnika (nie zawiera stałego stężenia wzorca referencyjnego agonisty – 17β-estradiolu (3,0*10-12 M))
|
Gromadzenie i analiza danych
|
56.
|
Po przeprowadzeniu wstępnej serii badawczej i kompleksowych serii badawczych należy określić EC10, EC50, PC10, PC50 i maksymalną indukcję (TCmax) badanej substancji chemicznej służącej do przeprowadzenia badania agonisty. Do celów przeprowadzenia badania antagonisty należy obliczyć IC20, IC50, PC80, PC50 i minimalną indukcję (TCxmin). Na rysunku 3 (agonista) i 4 (antagonista) podano graficzne przedstawienie tych parametrów. Wymagane parametry oblicza się na podstawie indukcji względnej każdej badanej substancji chemicznej (od względnej do maksymalnej indukcji wzorca referencyjnego (= 100 %)). Do celów przeprowadzenia oceny danych należy zastosować regresję nieliniową (zmienne nachylenie, 4 parametry) zgodnie z poniższym równaniem:

gdzie:
X = Logarytm dawkowania lub stężenia
Y = Reakcja (indukcja względna (%))
Najwyższa wartość = Maksymalna indukcja (%)
Najniższa wartość = Minimalna indukcja (%)
LogEC50 = Logarytm stężenia, w przypadku którego obserwuje się 50 % maksymalnej reakcji
Krzywa Hilla = Współczynnik nachylenia krzywej Hilla
|
|
57.
|
Dane surowe z luminometra, wyrażone jako względna jednostka światła (RLU) należy przenieść do arkusza kalkulacyjnego analizy danych opracowanego do celów wstępnej serii badawczej i kompleksowych serii badawczych. Dane surowe powinny spełniać kryteria dopuszczalności wskazane w tabeli 3A i 3B (agonista) lub 4A i 4B (antagonista). W przypadku gdy dane surowe spełniają kryteria dopuszczalności, przeprowadza się następujące etapy obliczeń, aby określić wymagane parametry:
|
Agonizm
|
—
|
Odjąć średnią RLU kontroli z rozpuszczalnikiem wzorca porównawczego od wszelkich surowych danych uzyskanych w wyniku analizy wzorców referencyjnych.
|
|
—
|
Odjąć średnią RLU kontroli z rozpuszczalnikiem badanej substancji chemicznej od wszelkich surowych danych uzyskanych w wyniku analizy badanych substancji chemicznych.
|
|
—
|
Obliczyć indukcję względną każdego stężenia wzorca referencyjnego. Ustalić indukcję najwyższego stężenia wzorca referencyjnego na 100 %.
|
|
—
|
Obliczyć indukcję względną każdego stężenia badanej substancji chemicznej w porównaniu z najwyższym stężeniem wzorca referencyjnego jako 100 %.
|
|
—
|
Przeprowadzić ocenę wyników analizy zgodnie z regresją nieliniową (zmienne nachylenie, 4 parametry).
|
|
—
|
Określić EC50 i EC10 wzorca referencyjnego.
|
|
—
|
Określić EC50 i EC10 badanych substancji chemicznych.
|
|
—
|
Określić maksymalną względną indukcję badanej substancji chemicznej (TCmax).
|
|
—
|
Określić PC10 i PC50 badanych substancji chemicznych.
|
W przypadku badanej substancji chemicznej możliwe jest, że pełna krzywa dawka-efekt nie zawsze zostanie uzyskana, np. ze względu na cytotoksyczność lub problemy z rozpuszczalnością. W związku z nie można określić EC50, EC10 i PC50. W takich przypadkach można określić jedynie PC10 i TCmax.
Antagonizm
|
—
|
Odjąć średnią RLU kontroli z najwyższego stężenia wzorca referencyjnego od wszelkich surowych danych uzyskanych w wyniku analizy wzorców referencyjnych.
|
|
—
|
Odjąć średnią RLU kontroli z najwyższego stężenia wzorca referencyjnego od wszelkich surowych danych uzyskanych w wyniku analizy badanych substancji chemicznych.
|
|
—
|
Obliczyć indukcję względną każdego stężenia wzorca referencyjnego. Ustalić indukcję najniższego stężenia wzorca referencyjnego na 100 %.
|
|
—
|
Obliczyć indukcję względną każdego stężenia badanej substancji chemicznej w porównaniu z najniższym stężeniem wzorca referencyjnego jako 100 %.
|
|
—
|
Przeprowadzić ocenę wyników analizy zgodnie z regresją nieliniową (zmienne nachylenie, 4 parametry).
|
|
—
|
Określić IC50 i IC20 wzorca referencyjnego.
|
|
—
|
Określić IC50 i IC20 badanych substancji chemicznych.
|
|
—
|
Określić minimalną względną indukcję badanej substancji chemicznej (TCmin).
|
|
—
|
Określić PC80 i PC50 badanych substancji chemicznych.
|
Rysunek 3
Przegląd parametrów określonych w teście agonisty
EC10 = stężenie substancji, przy którym obserwuje się 10 % jej maksymalnej reakcji.
EC50 = stężenie substancji, przy którym obserwuje się 50 % jej maksymalnej reakcji.
PC10 = stężenie badanej substancji chemicznej, przy którym jej reakcja jest równa EC10 wzorca referencyjnego.
PC50 = stężenie badanej substancji chemicznej, przy którym jej reakcja jest równa EC50 wzorca referencyjnego.
TCxmax = maksymalna indukcja względna badanej substancji chemicznej.
Rysunek 4
Przegląd parametrów określonych w teście antagonisty
IC20 = stężenie substancji, przy którym obserwuje się 80 % jej maksymalnej reakcji (20 % inhibicji).
IC50 = stężenie substancji, przy którym obserwuje się 50 % jej maksymalnej reakcji (50 % inhibicji).
PC80 = stężenie badanej substancji chemicznej, przy którym jej reakcja jest równa IC20 wzorca referencyjnego.
PC50 = stężenie badanej substancji chemicznej, przy którym jej reakcja jest równa IC50 wzorca referencyjnego.
TCxmin = minimalna indukcja względna badanej substancji chemicznej.
W przypadku badanej substancji chemicznej możliwe jest, że pełna krzywa dawka-efekt nie zawsze zostanie uzyskana, np. ze względu na cytotoksyczność lub problemy z rozpuszczalnością. W związku z nie można określić IC50, IC20 i PC50. W takich przypadkach można określić jedynie PC20 i TCmin.
|
58.
|
Wyniki powinny opierać się na dwóch (lub trzech) niezależnych seriach badawczych. Jeżeli dwie serie badawcze dają porównywalne i tym samym odtwarzalne wyniki, nie jest konieczne wykonywanie trzeciej serii. Aby wyniki zostały dopuszczone, powinny:
|
—
|
spełniać kryteria dopuszczalności (zob. kryteria dopuszczalności pkt 14–22);
|
|
Kryteria interpretacji danych
|
59.
|
Do celów interpretacji danych i podjęcia decyzji, czy badaną substancję chemiczną uznaje się za dodatnią lub ujemną stosuje się następujące kryteria:
|
W przypadku każdej kompleksowej serii badawczej badaną substancję chemiczną uznaje się za dodatnią, jeśli:
|
1
|
TCmax jest równe 10 % maksymalnej reakcji wzorca referencyjnego (REF10) lub je przekracza.
|
|
2
|
Co najmniej 2 kolejne stężenia badanej substancji chemicznej są równe REF10 lub je przekraczają.
|
|
|
W przypadku każdej kompleksowej serii badawczej badaną substancję chemiczną uznaje się za ujemną, jeśli:
|
1
|
TCmax nie przekracza 10 % maksymalnej reakcji wzorca referencyjnego (REF10).
|
|
2
|
Mniej niż 2 kolejne stężenia badanej substancji chemicznej są równe REF10 lub je przekraczają.
|
|
|
W przypadku każdej kompleksowej serii badawczej badaną substancję chemiczną uznaje się za dodatnią, jeśli:
|
1
|
TCmin jest równe lub niższe niż 80 % maksymalnej reakcji wzorca referencyjnego (REF80 = 20 % inhibicji).
|
|
2
|
Co najmniej 2 kolejne stężenia badanej substancji chemicznej są równe lub niższe niż REF80.
|
|
|
W przypadku każdej kompleksowej serii badawczej badaną substancję chemiczną uznaje się za ujemną, jeśli:
|
1
|
TCmin przekracza 80 % maksymalnej reakcji wzorca referencyjnego (REF80 = 20 % inhibicji).
|
|
2
|
Mniej niż 2 kolejne stężenia badanej substancji chemicznej są równe lub niższe niż REF80.
|
|
|
|
60.
|
Aby scharakteryzować siłę działania reakcji dodatniej badanej substancji chemicznej należy zgłosić wielkość wpływu (agonista: TCmax; antagonista: TCmin) oraz stężenie, przy którym nastąpił wpływ (agonista: EC10, EC50, PC10, PC50; antagonista: IC20, IC50, PC80, PC50).
|
SPRAWOZDANIE Z BADANIA
|
61.
|
Zob. pkt 20 dotyczący „ELEMENTÓW BADANIA AKTYWACJI TRANSKRYPCJI RECEPTORA ESTROGENOWEGO”.
|
BIBLIOGRAFIA
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 240. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. „Toxicol. Sci.” nr 83(1), s. 136–148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, i van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ, „Endocrinology” nr 142(3), s. 1156–1166.
|
|
(4)
|
Thorne N, Inglese J i Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, [W:] „Chemistry and Biology” nr 17(6), s. 646–57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V i Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta, „Biochem. Pharmacol.” nr 71, s. 1459–1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B i Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta, „Endocrinol.” nr 139, s. 4252–4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism, J. Steroid. „Biochem. Mol. Biol.” nr 122, s. 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, i van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities, „Toxicol. Sci.” nr 89(1), s. 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M i Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. „J. Vet. Med. Sci.” nr 57(4), s. 769–771.
|
|
(10)
|
Zhang J-H, Chung TDY i Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays, „J. Biomol. Scr.” nr 4, s. 67–73.
|
|
(11)
|
Besselink H, Middelhof I, i Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells, BioDetection Systems BV (BDS), Amsterdam, Niderlandy.
|
Dodatek 4.1:
KONTROLA WZROKOWA ŻYWOTNOŚCI KOMÓREK
B.67 TESTY MUTACJI GENOWYCH NA KOMÓRKACH SSAKÓW IN VITRO Z WYKORZYSTANIEM GENU KINAZY TYMIDYNOWEJ
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej OECD nr 490 (2016). Metody badawcze są okresowo poddawane przeglądowi i zmianom w związku z postępem naukowym, potrzebami regulacyjnymi i dobrostanem zwierząt. Początkowo test na mysich chłoniakach i test TK6 z wykorzystaniem locusu kinazy tymidynowej wchodziły w skład metody badawczej B.17. Następnie grupa robocza ekspertów ds. badania mysich chłoniaków międzynarodowych warsztatów z badań genotoksycznych (IWGT) opracowała zharmonizowane na szczeblu międzynarodowym zalecenia w zakresie kryteriów dopuszczalności testu oraz interpretacji danych dotyczących badania mysich chłoniaków (1)(2)(3)(4)(5) i zalecenia te włączono do niniejszej nowej metody badawczą B.67. Niniejszą metodę badawczą opisano w odniesieniu do badania mysich chłoniaków, a ponieważ wykorzystuje również locus TK, także badania TK6. Mimo że badanie mysich chłoniaków stosowano w dużym zakresie do celów regulacyjnych, TK6 stosowano znacznie rzadziej. Należy zauważyć, że pomimo podobieństwa między punktami końcowymi te dwie linie komórkowe nie są zamienne i w programach regulacyjnych można prawomocnie wyrazić preferencję w stosunku do jednej w odniesieniu do określonego zastosowania regulacyjnego. Na przykład w ramach walidacji badania mysich chłoniaków wykazano, że jest ono odpowiednie do wykrycia nie tylko mutacji genowej, ale również zdolności badanej substancji chemicznej do wywoływania strukturalnej aberracji chromosomowej. Niniejsza metoda badawcza stanowi część cyklu metod badawczych dotyczących toksykologii genetycznej. OECD opracowała dokument, który zawiera zwięzłe informacje na temat badań w zakresie toksykologii genetycznej oraz przegląd najnowszych zmian, jakie wprowadzono do wytycznych OECD dotyczących badań w zakresie toksykologii genetycznej (6).
|
|
2.
|
Celem testów mutacji genowych na komórkach ssaków in vitro jest wykrycie mutacji genowych wywołanych przez substancje chemiczne. Linie komórkowe wykorzystane w tych badaniach mierzą mutacje pierwotne w genach reporterowych, w szczególności w endogennym genie kinazy tymidynowej (TK w odniesieniu do komórek ludzkich i Tk w przypadku komórek gryzoni, które w niniejszej metodzie badawczej zbiorczo nazywa się TK). Niniejsza metoda badawcza jest przeznaczona do stosowania dwóch linii komórkowych: linii komórkowej mysiego chłoniaka L5178Y TK+/--3.7.2C (ogólnie nazywanej L5178Y) oraz linii komórkowej ludzkiej komórki limfoblastycznej TK6 (ogólnie nazywanej TK6). Chociaż te dwie linie komórkowe różnią się z powodu swojego pochodzenia, wzrostu komórki, statusu p-53 itp. badania mutacji genowej TK można przeprowadzać w podobny sposób w obu rodzajach komórek, jak opisano w niniejszej metodzie badawczej.
|
|
3.
|
Autosomalny i heterozygotyczny charakter genu kinazy tymidynowej umożliwia wykrycie żywotnych kolonii, których komórki wykazują niedobór enzymu kinazy tymidynowej w następstwie mutacji z TK+/-
do TK. Niedobór ten może wynikać ze zdarzeń genetycznych wpływających na gen TK, w tym zarówno mutacji genowych (mutacji punktowej, mutacji zmiany fazy odczytu, niewielkich delecji itp.), jak i zdarzeń chromosomowych (dużych delecji, rearanżacji chromosomów i rekombinacji mitotycznej). Zdarzenia chromosomowe wyraża się jako utratę heterozygotyczności, co stanowi powszechną zmianę genetyczną antyonkogenów w ludzkiej onkogenezie. Teoretycznie utratę całego chromosomu zawierającego gen TK wynikającą z uszkodzenia wrzeciona lub mitotycznej nondysjunkcji można wykryć w badaniu mysich chłoniaków. W rzeczywistości połączenie cytogenetycznej i molekularnej analizy wyraźnie wykazuje, że niektóre mutanty kinazy tymidynowej w badaniu mysich chłoniaków są wynikiem nondysjunkcji. Waga dowodów wykazuje jednak, że w ramach badania mutacji genowej TK nie można skutecznie wykryć aneugenów, stosując standardowe kryteria cytotoksyczności (jak opisano w niniejszej metodzie badawczej) i w związku z tym stosowanie tych badań w celu wykrycia aneugenów nie jest właściwe (7)(8)(9).
|
|
4.
|
W ramach badań mutacji genowej TK generuje się dwie różne fenotypowe klasy mutacji TK; mutanty rozwijające się zwyczajnie, rozwijające się w tym samym tempie co heterozygotyczne komórki kinazy tymidynowej oraz mutanty rozwijające się powoli, które rozwijają się przy wydłużonym czasie podwojenia. W badaniu mysich chłoniaków mutanty rozwijające się zwyczajnie i mutanty rozwijające się powoli uznaje się za mutanty dużych i małych kolonii oraz za mutanty wcześnie i późno pojawiających się kolonii w badaniu TK6. Charakter cytogenetyczny oraz charakter budowy cząsteczkowej mutantów badania mysich chłoniaków, zarówno małych, jak i dużych kolonii, został szczegółowo zbadany (8)(10)(11)(12)(13). Charakter cytogenetyczny oraz charakter budowy cząsteczkowej wcześnie i późno pojawiających się mutantów TK6 również obszernie zbadano (14)(15)(16)(17). Mutanty rozwijające się powoli w przypadku obu rodzajów komórek doznały uszkodzenia genetycznego obejmującego rzekome geny regulujące wzrost w pobliżu locusu TK, co skutkuje wydłużonym czasem podwojenia oraz utworzeniem się późno pojawiających się lub małych kolonii (18). Indukcję mutantów rozwijających się powoli przypisuje się substancjom chemicznym, które powodują duże zmiany strukturalne na poziomie chromosomów. Komórki, których uszkodzenie nie obejmuje rzekomych genów regulujących wzrost w pobliżu locusu TK rozwijają się w tempie podobnym do komórek macierzystych i stają się mutantami rozwijającymi się zwyczajnie. Indukcję mutantów głównie rozwijających się zwyczajnie przypisuje się substancjom chemicznym działającym przede wszystkim jako mutageny powodujące mutację punktową. W związku z tym istotne jest policzenie zarówno mutantów rozwijających się powoli, jak i mutantów rozwijających się zwyczajnie, aby wykryć wszystkie mutanty i dostarczyć informacji na temat rodzajów uszkodzenia (mutagenów i klastogenów) wywołanego przez badaną substancję chemiczną (10)(12)(18)(19).
|
|
5.
|
Metodę badawczą zorganizowano w taki sposób, aby dostarczyć ogólnych informacji mających zastosowanie zarówno do badania mysich chłoniaków, jak i badania TK6 oraz zapewnić specjalistyczne wytyczne dotyczące poszczególnych badań.
|
|
6.
|
Zastosowane definicje znajdują się w dodatku 1.
|
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
7.
|
Badania przeprowadzane in vitro wymagają zazwyczaj wykorzystania egzogennego źródła aktywacji metabolicznej. Egzogenny układ metabolizujący nie jest w stanie całkowicie naśladować warunków in vivo.
|
|
8.
|
Należy zachować ostrożność, aby uniknąć wystąpienia warunków, które mogłyby doprowadzić do zniekształconych wyników dodatnich (tj. do potencjalnej interakcji z układem badawczym) niespowodowanych interakcją między badaną substancją chemiczną a materiałem genetycznym komórki; do takich warunków zalicza się zmiany pH lub osmolalność, interakcje z elementami podłoża (20)(21) bądź zbyt wysokie poziomy cytotoksyczności (22)(23)(24). Cytotoksyczność przekraczającą zalecane górne poziomy cytotoksyczności ustanowione w pkt 28 uznaje się za nadmierną dla badania mysich chłoniaków i badania TK6. Ponadto należy zauważyć, że badane substancje chemiczne będące analogami tymidyny lub zachowujące się jak analogi tymidyny mogą zwiększyć częstość występowania mutacji poprzez selektywny rozwój spontanicznych mutantów w tle w trakcie poddawania komórki działaniu substancji oraz wymagają zastosowania dodatkowych metod badawczych, aby przeprowadzić właściwą ocenę (25).
|
|
9.
|
W przypadku wytworzonych nanomateriałów konieczne może być specjalne dostosowanie niniejszej metody badawczej, które jednak nie zostało tu opisane.
|
|
10.
|
Przed zastosowaniem niniejszej metody badawczej służącej zbadaniu mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy zastanowić się, czy zastosowanie niniejszej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak – dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny.
|
|
11.
|
Zmutowane komórki charakteryzujące się niedoborem aktywności enzymów kinazy tymidynowej z powodu mutacji TK
+/- do TK
-/- są odporne na działanie cytostatyczne trifluorotymidyny będącej analogiem pirymidyny. Komórki, w przypadku których odnotowuje się prawidłowy poziom enzymów TK, są wrażliwe na trifluorotymidynę, pod wpływem której dochodzi do inhibicji metabolizmu komórkowego i wstrzymania dalszego podziału komórki. Tym samym zmutowane komórki mogą dzielić się w obecności trifluorotymidyny i tworzyć widoczne kolonie, podczas gdy komórki, które zawierają enzymy TK – nie.
|
ZASADA BADANIA
|
12.
|
Komórki w zawiesinie są poddawane działaniu badanej substancji chemicznej, zarówno z udziałem lub bez udziału egzogennego źródła aktywacji (zob. pkt 19), przez odpowiedni okres (zob. pkt 33), po czym wydziela się z nich kulturę pochodną, aby oznaczyć poziom cytotoksyczności oraz wywołać ekspresję fenotypową przed wybraniem zmutowanych komórek. Cytotoksyczność określa się poprzez względny wzrost całkowity (zob. pkt 25) w przypadku badania mysich chłoniaków oraz poprzez względny wskaźnik przeżyć (zob. pkt 26) w przypadku badania TK6. Hodowle poddane działaniu substancji są utrzymywane na podłożu przez okres odpowiedni dla każdego wybranego rodzaju komórki (zob. pkt 37), aby doprowadzić do zbliżonej do optymalnej ekspresji fenotypowej wywołanych mutacji. Po uzyskaniu ekspresji fenotypowej oznacza się częstość występowania mutacji, dokonując posiewu określonej liczby komórek na podłożu zawierającym czynnik selektywny w celu wykrycia kolonii zmutowanych komórek oraz na podłożu pozbawionym takiego czynnika selektywnego, aby oznaczyć skuteczność klonowania (żywotność). Kolonie liczy się po upływie odpowiedniego okresu inkubacji. Częstość występowania mutacji oblicza się na podstawie liczby kolonii zmutowanych komórek skorygowanej o wartość wskaźnika skuteczności klonowania w momencie wyboru zmutowanych komórek.
|
OPIS METODY
Czynności przygotowawcze
Komórki
|
13.
|
W odniesieniu do badania mysich chłoniaków: Ponieważ badanie mysich chłoniaków opracowano i scharakteryzowano wykorzystując linię TK
+/- -3.7.2C należącą do linii komórkowych L5178Y, tę konkretną linię należy zastosować w odniesieniu do badania mysich chłoniaków. Linię komórkową L5178Y uzyskano z chłoniaka grasicy u myszy DBA-2 wywołanego metylocholatrenem (26). Clive i jego współpracownicy poddali komórki L5178Y (które Clive oznaczył jako TK+/+ -3) działaniu metanosulfonatu etylu oraz wyizolowali klon TK-/- (oznaczony jako TK-/- -3.7), wykorzystując 5-bromo-2-dezoksyurydynę jako czynnik selektywny. Z klona TK-/- wyizolowano i scharakteryzowano spontanicznego klona TK+/- (oznaczonego jako TK+/- -3.7.2.) i subklona (oznaczonego jako TK+/--3.7.2C) do zastosowania w badaniu mysich chłoniaków (27). Opublikowano kariotyp linii komórkowej (28)(29)(30)(31). Zmienna liczba chromosomów wynosi 40. Istnieje jeden chromosom metacentryczny (t12;13), który należy liczyć jako jeden chromosom. Locus TK myszy znajduje się na dystalnym końcu chromosomu 11. Linia komórkowa L5178Y TK
+/- -3.7.2C zawiera mutacje w obu allelach p53 i powoduje mutację białka p53 (32)(33). Status p53 linii komórkowej TK+/--3.7.2C prawdopodobnie odpowiada za zdolność badania do wykrycia uszkodzenia na dużą skalę (17).
|
|
14.
|
W odniesieniu do TK6: TK6 to linia ludzkiej komórki limfoblastycznej. Macierzysta linia komórkowa to linia komórkowa zmodyfikowana przez wirus Epsteina-Barra, WI-L2, który pierwotnie uzyskano od 5-letniego samca z dziedziczną sferozytozą. Pierwszy wyizolowany klon, HH4, poddano mutacji za pośrednictwem ICR191 i wygenerowano heterozygotyczną linię komórkową TK – TK6(34). Komórki TK6 są niemal diploidalne, a reprezentatywny kariotyp to 47, XY, 13+, t(14; 20), t(3; 21)(35). Locus ludzkiej TK znajduje się na długim ramieniu chromosomu 17. TK6 to linia komórkowa zawierająca białko p53, ponieważ zawiera sekwencję p53 typu dzikiego w obu allelach i opisuje wyłącznie białko p53 typu dzikiego (36).
|
|
15.
|
Zarówno w przypadku badania mysich chłoniaków, jak i TK6 zaleca się, aby przy pierwszym wyznaczaniu lub uzupełnianiu głównej kultury komórkowej laboratorium badawcze zapewniło brak skażenia mykoplazmą, przeprowadziło analizę chromosomów komórki lub oznaczyło kolorem chromosomy zawierające locus TK oraz sprawdziło czas podwojenia populacji. Należy ustalić normalny czas trwania cyklu komórkowego w przypadku komórek wykorzystywanych w laboratorium badawczym, który powinien być zgodny z opublikowanymi właściwościami komórek (16)(19)(37). Główną strukturę komórkową należy przechowywać w temperaturze -150 °C lub niższej i wykorzystywać do przygotowywania wszystkich roboczych kultur komórkowych.
|
|
16.
|
Przed ustaleniem dużej liczby kultur roboczych poddanych krioprezerwacji, albo tuż przed wykorzystaniem w doświadczeniu konieczne może być oczyszczenie kultury z wcześniej istniejących zmutowanych komórek [o ile częstość występowania mutacji w kontroli z rozpuszczalnikiem nie osiągnęła już dopuszczalnego zakresu – zob. tabela 2 dotycząca badania mysich chłoniaków]. Osiąga się to, stosując metotreksat (aminopterynę), aby dokonać selekcji względem komórek, w których występuje niedobór TK oraz dodając tymidynę, hipoksantynę i glicynę (L5178Y) lub 2-deoksycytydynę (TK6) do kultury w celu zapewnienia optymalnego wzrostu komórek zawierających TK (19)(38)(39) oraz (40) w odniesieniu do TK6. Ogólne porady dotyczące dobrych praktyk w zakresie utrzymania kultury komórkowej, jak również szczegółowe porady w zakresie komórek L5178Y i TK6 znajdują się w pkt (19)(31)(37)(39)(41). Dla laboratoriów wymagających głównych kultur komórkowych do zapoczątkowania badania mysich chłoniaków lub TK6, albo pozyskania nowych głównych kultur komórkowych dostępne jest repozytorium dobrze scharakteryzowanych komórek (37).
|
Podłoża i warunki hodowli kultur
|
17.
|
W przypadku obu badań w celu utrzymania kultur należy zastosować odpowiednie podłoże oraz warunki inkubacji (np. naczynia do hodowli komórkowych, wilgotne środowisko o zawartości CO2 wynoszącej 5 %, temperatura inkubacji w wysokości 37 °C). Kultury komórkowe powinny być zawsze utrzymywane w warunkach zapewniających fazę logarytmicznego wzrostu. W tym kontekście szczególne znaczenie ma dobranie podłoża i warunków hodowli kultur w taki sposób, aby zapewnić optymalny wzrost komórek w okresie ekspresji oraz klonowanie zarówno dla komórek zmutowanych, jak i dla komórek niezmutowanych. W przypadku badania mysich chłoniaków i TK6 istotne jest również, aby warunki hodowli kultur zapewniały optymalny wzrost zarówno dużych kolonii / wcześnie pojawiających się mutantów TK, jak i małych kolonii / późno pojawiających się mutantów TK. Więcej szczegółów dotyczących kultury, w tym konieczność właściwego podgrzania surowicy końskiej, jeśli podczas wyboru zmutowanych komórek stosuje się podłoże RPMI znajduje się w pkt (19)(31)(38)(39)(40)(42).
|
Przygotowanie kultur
|
18.
|
Komórki są rozmnażane z kultur wyjściowych i wysiewane na podłoże w takiej gęstości, by hodowle komórek w zawiesinie mogły nadal rosnąć wykładniczo przez okres poddawania działaniu substancji i okres ekspresji.
|
Aktywacja metaboliczna
|
19.
|
Egzogenne układy metabolizujące powinny być wykorzystywane w przypadku, gdy stosuje się komórki L5178Y i TK6, ponieważ mają one nieodpowiednią endogenną wydolność metaboliczną. Najpowszechniej stosowanym układem, który jest domyślnie zalecany, jeżeli w danym przypadku nie występują czynniki uzasadniające zastosowanie innego układu, jest frakcja postmitochondrialna suplementowanego kofaktora (S9) przygotowywana z wątrób gryzoni (najczęściej szczurów) poddawanych działaniu substancji indukujących enzymy takich jak Aroclor 1254 (43)(44)(45) lub połączenie fenobarbitalu i β-naftoflawonu (46)(47)(48)(49)(50)(51). Stosowanie tego ostatniego połączenia substancji nie narusza postanowień Konwencji sztokholmskiej w sprawie trwałych zanieczyszczeń organicznych (52), przy czym połączenie to okazało się równie skuteczne jak Aroclor 1254 w indukowaniu oksydaz wieloczynnościowych (45)(46)(47)(48)(49). Frakcja S9 jest zazwyczaj wykorzystywana w stężeniach mieszczących się w przedziale 1–2 %, ale stężenie to może zostać zwiększone do 10 % (objętościowo) w końcowym podłożu badawczym. Klasa badanych substancji chemicznych może wywierać wpływ na rodzaj i stężenie stosowanego egzogennego układu metabolizującego lub czynnika pobudzającego metabolizm.
|
Przygotowanie badanej substancji chemicznej
|
20.
|
Badane substancje chemiczne w stanie stałym należy rozpuścić w odpowiednich rozpuszczalnikach oraz – w stosownych przypadkach – rozcieńczyć przed poddaniem komórek ich działaniu (zob. pkt 21). Badane substancje chemiczne w stanie ciekłym można dodać do układu badawczego bezpośrednio lub rozcieńczyć je przed poddaniem układu badawczego ich działaniu. Badane substancje chemiczne w stanie gazowym lub lotnym należy badać, wprowadzając stosowne modyfikacje w standardowych protokołach, np. poddając układ działaniu substancji w uszczelnionych naczyniach do hodowli komórkowych (53)(54)(55). Preparaty badanej substancji chemicznej należy przygotowywać tuż przed poddaniem komórek jej działaniu, chyba że dane dotyczące stabilności wskazują, że dopuszczalne jest składowanie substancji.
|
WARUNKI BADANIA
Rozpuszczalniki
|
21.
|
Rozpuszczalnik powinien zostać dobrany w taki sposób, aby zapewnić optymalną rozpuszczalność badanej substancji chemicznej bez wywierania niepożądanego wpływu na przebieg badania, który może przejawiać się np. zmianą tempa wzrostu komórek, wpływem na integralność badanej substancji chemicznej, wejściem w reakcję z naczyniami do hodowli komórkowych lub zakłóceniem funkcjonowania układu metabolizującego. Zaleca się, aby w miarę możliwości w pierwszej kolejności rozważyć możliwość zastosowania wodnego rozpuszczalnika (lub podłoża). Do sprawdzonych rozpuszczalników zalicza się wodę lub sulfotlenek dimetylu. Zasadniczo zawartość rozpuszczalników organicznych nie powinna przekraczać 1 % (obj.) a zawartość rozpuszczalników wodnych (soli fizjologicznej lub wody) w końcowym podłożu użytym do badania nie powinna przekroczyć 10 % (obj.). W przypadku korzystania z rozpuszczalników innych niż sprawdzone (np. etanolu lub acetonu), decyzja o ich zastosowaniu powinna być uzasadniona danymi potwierdzającymi zgodność tych rozpuszczalników z badanymi substancjami chemicznymi, układem badawczym, a także brak właściwości świadczących o toksyczności genetycznej w zastosowanym stężeniu. W przypadku braku takich danych potwierdzających, należy przeprowadzić dodatkowe kontrole (zob. dodatek 1, definicje), aby wykazać, że wybrany rozpuszczalnik nie wywołuje żadnych szkodliwych ani mutagennych skutków.
|
DOKONYWANIE POMIARU CYTOTOKSYCZNOŚCI I WYBÓR BADANEGO STĘŻENIA
|
22.
|
Przy ustalaniu najwyższego stężenia badanej substancji chemicznej należy unikać stężeń, które mogą doprowadzić do uzyskania zniekształconych wyników dodatnich, np. stężeń wywołujących nadmierną cytotoksyczność (zob. pkt 28), wytrącanie substancji w podłożu (zob. pkt 29) lub znaczne zmiany w pH lub osmolalności (zob. pkt 8). Jeżeli badana substancja chemiczna wywołuje znaczną zmianę pH podłoża w momencie jej dodania, poziom pH można skorygować, buforując końcowe podłoże użyte do badania, aby uniknąć zniekształconych wyników dodatnich i utrzymać odpowiednie warunki hodowli kultur.
|
|
23.
|
Wyboru stężenia dokonuje się na podstawie poziomu cytotoksyczności oraz innych parametrów (zob. pkt 27–30). Choć dokonanie oceny cytotoksyczności na etapie badania wstępnego może przyczynić się do precyzyjniejszego określenia stężeń, które mają zostać zastosowane w ramach głównego doświadczenia, wykonanie takiego badania wstępnego nie jest wymagane. Nawet w przypadku dokonania wstępnej oceny cytotoksyczności, w ramach głównego doświadczenia należy wykonać ponownie pomiar cytotoksyczności w odniesieniu do każdej kultury. Jeśli przeprowadza się doświadczenie dotyczące zakresu dawkowania, powinno ono obejmować szeroki zakres stężeń i może zostać przerwane 1. dnia po poddaniu działaniu substancji, albo przeprowadzone za pośrednictwem ekspresji trwającej 2 dni i do momentu wybrania zmutowanych komórek (jeżeli okaże się, że zastosowane stężenia są właściwe).
|
|
24.
|
Cytotoksyczność należy określić dla każdej pojedynczej badanej kultury i kultury kontrolnej: metody dotyczące badania mysich chłoniaków (2) oraz TK6 (15) określa praktyka uzgodniona na szczeblu międzynarodowym.
|
|
25.
|
Zarówno w przypadku wersji badania mysich chłoniaków wykorzystującej agar, jak i wersji badania mysich chłoniaków wykorzystującej mikropłytki: Ocenę cytotoksyczności należy przeprowadzić, wykorzystując względny wzrost całkowity (RTG), który opracował Clive i Spector w 1975 r. (2) Miernik ten obejmuje względny wzrost zawieszania (RSG: badana kultura w stosunku do kontroli z rozpuszczalnikiem) podczas poddawania komórki działaniu substancji, czas ekspresji i względna skuteczność klonowania (RCE: badana kultura w stosunku do kontroli z rozpuszczalnikiem) w momencie wyboru mutantów (2). Należy zauważyć, że względny wzrost zawieszania obejmuje wszelką utratę komórek mającą miejsce w badanej kulturze podczas poddawania działaniu substancji (zob. dodatek 2 dotyczący wzorów).
|
|
26.
|
W odniesieniu do TK6: Ocenę cytotoksyczności należy przeprowadzić w oparciu o względny wskaźnik przeżyć (RS), tj. wskaźnik skuteczności klonowania komórek posianych natychmiast po poddaniu działaniu substancji skorygowany o wszelkie komórki utracone w trakcie działania substancji, na podstawie liczby komórek w stosunku do kontroli ujemnych (którym przypisano wskaźnik przeżycia wynoszący 100 %) (odpowiedni wzór zawarto w dodatku 2).
|
|
27.
|
Należy ocenić co najmniej cztery badane stężenia (poza kontrolami z rozpuszczalnikiem i kontrolami dodatnimi), które spełniają kryteria dopuszczalności (odpowiednia cytotoksyczność, liczba komórek itp.). Choć zaleca się korzystanie ze zduplikowanych kultur, w odniesieniu do każdego badanego stężenia dopuszcza się możliwość stosowania kontrprób albo pojedynczych kultur poddawanych działaniu substancji. Wyniki uzyskane na podstawie zduplikowanych kultur przy danym stężeniu należy zgłaszać odrębnie, ale dopuszcza się możliwość ich łączenia na potrzeby analizy danych (55). W przypadku badanych substancji chemicznych wykazujących niewielką cytotoksyczność lub niewykazujących cytotoksyczności na ogół odpowiednie będzie zastosowanie 2–3-krotnych przedziałów stężenia. W przypadku wystąpienia cytotoksyczności należy wybrać stężenia, aby obejmowały zakres cytotoksyczności rozpoczynający się od stężenia, które wykazuje cytotoksyczność, jak opisano w pkt 28, oraz uwzględniający stężenia, w których wystąpiła umiarkowana i niewielka cytotoksyczność lub nie wystąpiła żadna cytotoksyczność. W przypadku wielu badanych substancji chemicznych można zaobserwować strome krzywe ilustrujące zależność stężenie-odpowiedź, dlatego też aby uwzględnić cały zakres cytotoksyczności lub szczegółowo zbadać zależność stężenie-odpowiedź, konieczne może okazać się zastosowanie więcej niż czterech stężeń o bardziej zbliżonych wartościach – dotyczy to zwłaszcza przypadków, w których zachodzi konieczność powtórzenia doświadczenia (zob. pkt 70). Stosowanie więcej niż 4 stężeń może mieć szczególnie istotne znaczenie w przypadku korzystania z pojedynczych kultur.
|
|
28.
|
Jeżeli maksymalne stężenie określono na podstawie cytotoksyczności, należy dążyć do tego, by poziom takiego maksymalnego stężenia doprowadził do uzyskania względnego wzrostu całkowitego na poziomie od 20 do 10 % w przypadku badania mysich chłoniaków oraz względnego wskaźnika przeżyć na poziomie od 20 do 10 % w przypadku TK6 (pkt 67).
|
|
29.
|
W przypadku słabo rozpuszczalnych badanych substancji chemicznych, które nie wykazują cytotoksyczności w stężeniach niższych od najniższego stężenia nierozpuszczalnego, najwyższy poziom stężenia wykorzystanego w badaniu powinien wywołać zmętnienie lub doprowadzić do wytrącenia się osadu widocznego gołym okiem lub przez mikroskop odwrócony pod koniec poddawania działaniu badanej substancji. Nawet jeżeli cytotoksyczność występuje powyżej najniższego stężenia nierozpuszczalnego, zaleca się przeprowadzenie badania przy zastosowaniu wyłącznie jednego stężenia wywołującego zmętnienie lub skutkującego wytrącaniem się widocznego osadu, ponieważ osad może prowadzić do uzyskania zniekształconych wyników. Ponieważ w badaniu mysich chłoniaków i TK6 stosuje się hodowlę komórek w zawiesinie należy zadbać o to, aby pojawienie się osadu nie zakłóciło przebiegu badania. W tym kontekście pomocne może okazać się również oznaczenie rozpuszczalności w podłożu przed przeprowadzeniem doświadczenia.
|
|
30.
|
Jeżeli nie zaobserwowano żadnego osadu ani ograniczenia cytotoksyczności, najwyższe badane stężenie powinno odpowiadać 10 mM, 2 mg/ml lub 2 μl/ml, w zależności od tego, która z tych wartości jest najniższa (57) (58). Jeżeli badana substancja chemiczna nie ma określonego składu, np. substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne [tj. substancje chemiczne o nieznanym lub zmiennym składzie (UVCB)], wyciągi pochodzące ze środowiska itp., w przypadku braku dostatecznej cytotoksyczności zwiększenie stężenia poszczególnych składników może wiązać się z koniecznością podwyższenia najwyższego stężenia (np. 5 mg/ml). Należy jednak zwrócić uwagę na fakt, że wymagania te mogą być inne w przypadku produktów leczniczych przeznaczonych dla ludzi (59).
|
Kontrole
|
31.
|
Dla każdego warunku doświadczalnego należy włączyć jednoczesne kontrole ujemne (zob. pkt 21) obejmujące sam rozpuszczalnik w podłożu użytym do badania, w odniesieniu do którego podjęto takie same czynności jak w odniesieniu do kultur poddanych działaniu substancji.
|
|
32.
|
Jednoczesne kontrole dodatnie są niezbędne do wykazania zdolności danego laboratorium do zidentyfikowania mutagenów w warunkach określonych w stosowanym protokole badania oraz skuteczności egzogennego układu metabolizującego (w stosownych przypadkach), oraz do wykazania właściwego wykrycia zarówno małych / późno pojawiających się mutantów TK, jak i dużych / wcześnie pojawiających się mutantów TK. Przykładowe substancje służące do kontroli dodatniej przedstawiono w tabeli 1 poniżej. W uzasadnionych przypadkach dopuszcza się możliwość stosowania alternatywnych substancji służących do kontroli dodatniej. Ponieważ testy na komórkach ssaków in vitro pod kątem toksyczności genetycznej są w dostatecznym stopniu ustandaryzowane, jeżeli chodzi o krótkoterminowe (3–4 godzinny) poddanie działaniu substancji chemicznej przeprowadzane jednocześnie z udziałem lub bez udziału aktywacji metabolicznej, zastosowanie kontroli dodatniej można ograniczyć do mutagenu wymagającego aktywacji metabolicznej. W takim przypadku wspomniana pojedyncza reakcja kontroli dodatniej potwierdzi zarówno aktywność układu metabolizującego, jak i reaktywność układu badawczego. W przypadku długoterminowego poddawania działaniu substancji chemicznej (tj. 24 godziny bez S9) należy zastosować jednak oddzielną kontrolę dodatnią, ponieważ czas podawania badanej substancji chemicznej będzie się różnił od czasu w badaniu z wykorzystaniem aktywacji metabolicznej. Każdą kontrolę dodatnią należy zastosować przy co najmniej jednym stężeniu, od którego oczekuje się wytworzenia odtwarzalnego oraz wykrywalnego wzrostu wykraczającego poza poziom wyjściowy, w celu wykazania czułości układu badawczego, a reakcja nie powinna być zakłócona cytotoksycznością wykraczającą poza limity określone w niniejszej metodzie badawczej (zob. pkt 28).
|
Tabela 1
Substancje odniesienia, z których zaleca się korzystać przy przeprowadzaniu oceny biegłości laboratorium i przy dokonywaniu wyboru kontroli dodatnich
|
Kategoria
|
Substancja
|
Numer CAS
|
|
1. Mutageny aktywne bez aktywacji metabolicznej
|
|
Metanosulfonian metylu
|
66-27-3
|
|
Mitomycyna C
|
50-07-7
|
|
N-tlenek 4-nitrochinoliny
|
56-57-5
|
|
2. Mutageny wymagające aktywacji metabolicznej
|
|
Benzo[a]piren
|
50-32-8
|
|
Cyklofosfamid (jednowodny)
|
50-18-0 (6055-19-2)
|
|
7,12-antracen dimetylobenzenu
|
57-97-6
|
|
3-Metylocholantren
|
56-49-5
|
PROCEDURA
Poddanie działaniu badanej substancji chemicznej
|
33.
|
Rozmnażające się komórki są poddawane działaniu badanej substancji chemicznej z udziałem i bez udziału układu metabolizującego. Narażenie na działanie badanej substancji powinno trwać dostatecznie długo (z reguły przyjmuje się, że odpowiedni okres narażenia na działanie substancji wynosi od 3 do 4 godzin). Należy jednak zwrócić uwagę na fakt, że wymagania te mogą być inne w przypadku produktów leczniczych przeznaczonych dla ludzi (59). Jeżeli chodzi o badanie mysich chłoniaków, w przypadkach, w których krótkoterminowe poddanie działaniu substancji chemicznej daje wyniki ujemne i istnieją dane wskazujące na potrzebę dłuższego poddawania działaniu substancji [np. analogów nukleozydowych, słabo rozpuszczalnych chemikaliów, (5)(59)], należy rozważyć przeprowadzenie badania obejmującego dłuższe poddawanie działaniu substancji, tj. 24 godziny bez S9.
|
|
34.
|
Minimalną liczbę komórek wykorzystywanych w odniesieniu do każdej kultury (kontrolnej i poddawanej działaniu substancji) na poszczególnych etapach badania należy ustalić na podstawie wskaźnika spontanicznej częstości występowania mutacji. Co do zasady, zaleca się poddawanie działaniu substancji i pasażowanie takiej liczby komórek w każdej hodowli eksperymentalnej, która będzie wystarczająca do tego, aby utrzymać poziom co najmniej 10, a najlepiej 100, spontanicznie powstałych mutantów na wszystkich etapach badania (poddanie działaniu substancji, ekspresja fenotypowa, wybór mutanta) (56).
|
|
35.
|
W przypadku badania mysich chłoniaków zalecana dopuszczalna spontaniczna częstość występowania mutacji znajduje się w przedziale 35–140 × 10-6 (wersja wykorzystująca agarem) oraz 50-170 × 10-6 (wersja wykorzystująca mikropłytki) (zob. Tabela 2). Aby osiągnąć we wszystkich badanych kulturach poziom co najmniej 10, a najlepiej 100, spontanicznie powstałych mutantów, które przeżywają poddanie działaniu substancji, konieczne jest poddanie działaniu substancji co najmniej 6 × 106 komórek. Poddawanie takiej liczby komórek działaniu substancji i utrzymywanie wystarczającej liczby komórek podczas ekspresji i klonowania do celów wybrania mutanta, zapewnia wystarczającą liczbę spontanicznych mutacji (przynajmniej 10) podczas wszystkich etapów eksperymentu, również w kulturach poddanych działaniu substancji w stężeniach charakteryzujących się cytotoksycznością na poziomie 90 % (określoną za pomocą względnego wzrostu całkowitego na poziomie 10 %) (19)(38)(39).
|
|
36.
|
W przypadku badania TK6, spontaniczna częstość występowania mutacji waha się zazwyczaj w przedziale od 2 do 10 × 10-6. Aby osiągnąć we wszystkich badanych kulturach poziom co najmniej 10 spontanicznie powstałych mutantów, konieczne jest poddanie działaniu substancji co najmniej 20 × 106 komórek. Poddawanie takiej liczby komórek działaniu substancji zapewnia wystarczającą liczbę spontanicznie powstałych mutantów (przynajmniej 10) również w kulturach poddanych działaniu substancji w stężeniach charakteryzujących się cytotoksycznością na poziomie 90 % (względny wskaźnik przeżyć wynoszący 10 %). Ponadto w okresie ekspresji należy wyhodować wystarczającą liczbę komórek, a następnie posiać te komórki w celu dokonania wyboru mutantów (60).
|
Czas ekspresji fenotypowej oraz pomiar cytotoksyczności i częstości występowania mutacji
|
37.
|
Po zakończeniu okresu poddawania działaniu substancji komórki hoduje się przez określony czas, aby umożliwić niemal optymalną ekspresję fenotypową nowo wywołanych mutacji; specyficzną dla każdej linii komórkowej. W przypadku badania mysich chłoniaków czas ekspresji fenotypowej wynosi 2 dni. Jeżeli chodzi o badanie TK6, czas ekspresji fenotypowej wynosi 3–4 dni. W przypadku zastosowania 24-godzinnego poddania działaniu substancji okres ekspresji rozpoczyna się po zakończeniu poddawania działaniu substancji.
|
|
38.
|
W czasie ekspresji fenotypowej komórki zlicza się codziennie. W przypadku badania mysich chłoniaków do obliczania dziennego wzrostu zawieszania wykorzystuje się liczbę komórek w danym dniu. Po 2 dniach ekspresji fenotypowej komórki zawiesza się w podłożu z udziałem lub bez udziału czynnika selektywnego, aby odpowiednio ustalić liczbę mutantów (płytki wyboru) i określić skuteczność klonowania (płytki żywotności). Jeżeli chodzi o badanie mysich chłoniaków, istnieją dwie jednakowo dopuszczalne metody wybierania mutantów za pomocą klonowania; w jednej wykorzystuje się miękki agar, a w drugiej podłoże ciekłe na 96-dołkowych płytkach (19)(38)(39). Klonowanie w przypadku badania TK6 przeprowadza się z wykorzystaniem ciekłego podłoża i 96-dołkowych płytek (16).
|
|
39.
|
W przypadku mutacji TK jedynym zalecanym czynnikiem selektywnym jest trifluorotymidyna (TFT) (61).
|
|
40.
|
W przypadku badania mysich chłoniaków płytki agarowe i mikropłytki zlicza się po 10–12 dniach inkubacji. W przypadku badania TK6 kolonie na mikropłytkach ocenia się po 10–14 dniach pod kątem wcześnie pojawiających się mutacji. Aby uzyskać wolną rosnące (późno pojawiające się) mutacje TK6, należy po policzeniu wcześnie pojawiających się mutacji ponownie nakarmić komórki pożywką i TFT i inkubować płytki przez kolejne 7–10 dni (62). W pkt 42 i 44 przedstawiono argumenty dotyczące zliczania mutacji TK cechujących się powolnym i normalnym wzrostem.
|
|
41.
|
Odpowiednie obliczenia dotyczące dwóch badań wykorzystujące te dwie metody (za pomocą agaru i mikropłytek) w przypadku badania mysich chłoniaków znajdują się w dodatku 2. Jeżeli chodzi o metodę wykorzystującą agar do badania mysich chłoniaków, kolonie zlicza się, a liczbę kolonii zmutowanych komórek koryguje się o skuteczność klonowania w celu obliczenia częstości występowania mutacji. W wersji badania mysich chłoniaków i badania TK6 przy wykorzystaniu mikropłytek skuteczność klonowania zarówno w przypadku płytek wyboru, jak i płytek skuteczności klonowania, określa się zgodnie z rozkładem Poissona (63). Częstość występowania mutacji oblicza się na podstawie tych dwóch wartości skuteczności klonowania.
|
Charakterystyka kolonii zmutowanych komórek
|
42.
|
W przypadku badania mysich chłoniaków, jeżeli badana substancja chemiczna daje wynik dodatni (zob. pkt 62–63), charakterystykę kolonii za pomocą sortowania według rozmiarów lub wzrostu należy przeprowadzić na przynajmniej jednej z badanych kolonii (najwyższe pozytywne stężenie) oraz na dodatnich i ujemnych kontrolach. Jeżeli wynik badanej substancji chemicznej jest ujemny (zob. pkt 64), charakterystykę kolonii zmutowanych komórek należy przeprowadzić na dodatnich i ujemnych kontrolach. W przypadku metody badania mysich chłoniaków na mikropłytkach mutanty małych kolonii definiuje się jako pokrywające poniżej 25 % średnicy dołka, a mutanty dużych kolonii jako pokrywające powyżej 25 % średnicy dołka. W przypadku metody wykorzystującej agar do zliczania kolonii zmutowanych komórek i sortowania kolonii według rozmiarów używa się licznika kolonii. Informacje szczegółowe na temat podejść do sortowania kolonii według rozmiarów znajdują się w literaturze (19)(38)(40). Do wykazania, że badania przeprowadzono poprawnie, konieczna jest charakterystyka kolonii przeprowadzona na dodatnich i ujemnych kontrolach.
|
|
43.
|
Jeżeli wyniku badanej substancji chemicznej nie można uznać za ujemny, jeżeli mutanty zarówno dużych, jak i małych kolonii nie zostaną odpowiednio wykryte w dodatniej kontroli. Charakterystykę kolonii można wykorzystać do przedstawienia ogólnych informacji dotyczących zdolności badanej substancji chemicznej do wywoływania mutacji punktowych lub zdarzeń chromosomowych (pkt 4).
|
|
44.
|
TK6: Mutacje rosnące normalnie i powoli rozróżnia się za pomocą różnic w okresie inkubacji (zob. pkt 40). W przypadku badania TK6 zarówno wcześnie jak i późno pojawiające się mutacje ocenia się we wszystkich kulturach, w tym w kontrolach ujemnych i dodatnich. Do wykazania, że badania przeprowadzono poprawnie, konieczna jest charakterystyka kolonii przeprowadzona na ujemnych i dodatnich kontrolach. Wyniku badanej substancji chemicznej nie można uznać za ujemny, jeżeli zarówno wcześnie pojawiające się, jak i późno pojawiające się mutacje nie zostaną odpowiednio wykryte w dodatniej kontroli. Charakterystykę kolonii można wykorzystać do przedstawienia ogólnych informacji dotyczących zdolności badanej substancji chemicznej do wywoływania mutacji punktowych lub zdarzeń chromosomowych (pkt 4).
|
Biegłość laboratorium
|
45.
|
Aby można było wykazać, że dane laboratorium dysponuje dostatecznym doświadczeniem w zakresie przeprowadzania badań przed wykorzystaniem go do przeprowadzenia rutynowego badania, laboratorium to musiało przeprowadzić szereg doświadczeń, podczas których stosowano substancje odniesienia służące do kontroli z wykorzystaniem różnych mechanizmów (co najmniej jedna substancja aktywna i jedna substancja aktywna przy braku aktywacji metabolicznej spośród substancji wymienionych w tabeli 1) oraz różnych kontroli ujemnych (uwzględniając kultury niepoddane działaniu substancji chemicznej oraz różne rozpuszczalniki/nośniki). Reakcje otrzymane w wyniku takiej kontroli dodatniej i ujemnej powinny odpowiadać reakcjom opisanym w literaturze. Wymóg ten nie dotyczy laboratoriów, które dysponują odpowiednim doświadczeniem, tj. laboratoriów prowadzących bazę danych historycznych, o której mowa w pkt 47–50. W przypadku badania mysich chłoniaków wartości otrzymane zarówno w wyniku kontroli dodatnich i ujemnych powinny być zgodne z zaleceniami międzynarodowych warsztatów z badań genotoksycznych (International Workshops on Genotoxicity Testing) (zob. Tabela 2).
|
|
46.
|
Aby wykazać biegłość w zakresie wykrywania mutagennych substancji chemicznych, ustalania skuteczności układu metabolizującego i wykazywania odpowiedniości warunków wzrostu komórek poddawanych działaniu substancji, ekspresji fenotypowej i wyboru mutantów oraz procedur oceny punktowej należy zbadać szereg substancji chemicznych służących do kontroli dodatniej (zob. tabela 1) przy krótkoterminowym i długoterminowym poddawaniu działaniu substancji chemicznej (w przypadku stosowania długoterminowego poddawania działaniu substancji chemicznej) przy braku aktywacji metabolicznej oraz przy krótkoterminowym poddawaniu działaniu substancji chemicznej w obecności aktywacji metabolicznej. Należy dobrać zakres stężeń wybranych substancji tak, aby uzyskać powtarzalne i powiązane ze stężeniem wzrosty powyżej tła w celu wykazania czułości i zakresu oznaczania układu badawczego.
|
Dane historyczne dotyczące kontroli
|
47.
|
Laboratorium powinno ustalić:
|
—
|
zakres i rozkład historycznych wyników kontroli dodatniej,
|
|
—
|
zakres i rozkład historycznych wyników kontroli ujemnej (bez poddania działaniu substancji chemicznej, z rozpuszczalnikiem).
|
|
|
48.
|
Przy pierwszym gromadzeniu danych dotyczących rozkładu historycznych wyników kontroli ujemnej wyniki jednoczesnej kontroli ujemnej powinny być zgodne z opublikowanymi danymi dotyczącymi kontroli ujemnej. W miarę dodawania kolejnych danych doświadczalnych do rozkładu kontroli jednoczesne kontrole ujemne powinny w idealnych warunkach mieścić się w granicach kontrolnych wyznaczających przedział 95 % dla tego rozkładu (64)(65).
|
|
49.
|
Prowadzona przez dane laboratorium baza danych historycznych dotyczących kontroli ujemnych powinna początkowo zawierać dane z co najmniej 10 doświadczeń, a najlepiej z co najmniej 20 doświadczeń przeprowadzonych w porównywalnych warunkach doświadczalnych. Laboratoria powinny stosować metody kontroli jakości, takie jak karty kontrolne (np. karty C lub karty X-średnie (65)) w celu określania stopnia zmienności gromadzonych przez nie danych dotyczących kontroli dodatniej i ujemnej oraz w celu wykazania, że stosowana przez nie metodologia jest »pod kontrolą« (66). Więcej informacji szczegółowych i zaleceń dotyczących sposobu gromadzenia i wykorzystywania danych historycznych można znaleźć w literaturze (64).
|
|
50.
|
Dane dotyczące kontroli ujemnej powinny obejmować częstość występowania mutacji w pojedynczej kulturze lub, najlepiej, w zduplikowanych kulturach, jak opisano w pkt 27. Jednoczesne kontrole ujemne powinny w idealnych warunkach mieścić się w granicach kontrolnych wyznaczających przedział 95 % dla rozkładu danych zawartych w prowadzonej przez laboratorium bazie danych historycznych dotyczących kontroli ujemnej. W przypadku gdy dane dotyczące kontroli ujemnej nie mieszczą się w granicy kontrolnej wyznaczającej przedział 95 %, włączenie ich do rozkładu kontroli historycznej może być dopuszczalne, pod warunkiem że dane te nie stanowią skrajnych obserwacji nietypowych, istnieją dowody świadczące o tym, iż dany układ badawczy znajduje się „pod kontrolą” (zob. pkt 48), a także dowody wykluczające błąd techniczny czy ludzki.
|
|
51.
|
Wszelkie zmiany w protokole doświadczalnym należy rozpatrywać pod kątem ich spójności z dotychczas prowadzonymi przez laboratorium bazami danych historycznych dotyczących kontroli. Wszelkie poważne niespójności powinny skutkować utworzeniem nowej bazy danych historycznych dotyczących kontroli.
|
DANE I SPRAWOZDAWCZOŚĆ
Przedstawienie wyników
|
52.
|
Zarówno w przypadku badania mysich chłoniaków, jak i badania TK6, przedstawienie danych powinno zgodnie z poniższym opisem uwzględniać – zarówno w odniesieniu do kultur, które zostały poddane działaniu substancji oraz kultur kontrolnych – dane niezbędne do obliczenia cytotoksyczności (odpowiednio względny wzrost całkowity i względny wskaźnik przeżyć) oraz częstości występowania mutacji.
|
|
53.
|
W przypadku badania mysich chłoniaków należy dostarczyć dane dotyczące poszczególnych kultur określające względny wzrost zawieszania, względny wzrost całkowity, skuteczność klonowania w momencie wybierania mutantów oraz liczbę kolonii zmutowanych komórek (w wersji wykorzystującej agar) lub liczbę pustych dołków (w wersji wykorzystującej mikropłytki). Częstość występowania mutacji powinna być wyrażona jako liczba zmutowanych komórek na milion komórek przeżywających. Jeżeli reakcja daje wynik dodatni, należy podać częstość występowania mutacji w małych i dużych koloniach (lub procent częstości występowania mutacji) w odniesieniu do przynajmniej jednego stężenia substancji badanej (zazwyczaj najwyższe pozytywne stężenie) oraz do ujemnych i dodatnich kontroli. Jeżeli reakcja daje wynik ujemny, należy podać częstość występowania mutacji w małych i dużych koloniach w odniesieniu do ujemnych i dodatnich kontroli.
|
|
54.
|
W przypadku badania TK6 należy dostarczyć dane dotyczące poszczególnych kultur określające względny wskaźnik przeżyć, skuteczność klonowania w momencie wybierania mutantów oraz liczbę pustych dołków w odniesieniu do wcześnie i późno pojawiających się mutacji. Częstość występowania mutacji powinna być wyrażona jako liczba zmutowanych komórek na milion komórek przeżywających i powinna obejmować zarówno całkowitą częstość występowania mutacji, jak i częstość występowania wcześnie i późno pojawiających się mutacji (lub procent częstości występowania mutacji).
|
Kryteria dopuszczalności
|
55.
|
Zarówno w przypadku badania mysich chłoniaków, jak i badania TK6, przed ustaleniem ogólnych wyników dotyczących danej badanej substancji chemicznej należy spełnić następujące kryteria:
|
—
|
Przeprowadzono dwa warunki doświadczalne (krótkoterminowe poddanie działaniu substancji z udziałem i bez udziału aktywacji metabolicznej – zob. pkt 33) do momentu uzyskania w jednym z nich wyników dodatnich.
|
|
—
|
Należy poddać analizie wystarczającą liczbę komórek i stężeń (zob. pkt 27, 34–36).
|
|
—
|
Kryteria wyboru najwyższego stężenia są spójne z kryteriami opisanymi w pkt 28–30.
|
|
Kryteria dopuszczalności dotyczące ujemnych i dodatnich kontroli
|
56.
|
W wyniku przeprowadzenia przez grupę roboczą ekspertów ds. badania mysich chłoniaków w ramach międzynarodowych warsztatów z badań genotoksycznych (International Workshops on Genotoxicity Testing) analizy szerokiego zakresu danych dotyczących badania mysich chłoniaków powstało uzgodnione stanowisko na temat szczegółowych kryteriów dopuszczalności dotyczących badania mysich chłoniaków (1)(2)(3)(4)(5). W związku z tym w ramach niniejszej metody badawczej przedstawiono szczegółowe zalecenia dotyczące ustalania dopuszczalności kontroli ujemnych i dodatnich oraz oceny wyników dotyczących poszczególnych substancji w ramach badania mysich chłoniaków. Baza danych dotyczących badania TK6 jest dużo mniejsza i grupa robocza nie przeprowadziła jego oceny.
|
|
57.
|
W przypadku badania mysich chłoniaków każdy eksperyment należy poddać ocenie pod kątem sprawdzenia, czy kontrola niepoddawana działaniu substancji / z rozpuszczalnikiem spełnia kryteria dopuszczalności grupy roboczej ds. badania mysich chłoniaków międzynarodowych warsztatów z badań genotoksycznych ((4) i tabela 2 poniżej) dotyczące: (1) częstości występowania mutacji (należy zauważyć, że częstość występowania mutacji dopuszczalna przez międzynarodowe warsztaty z badań genotoksycznych jest różna w wersjach badania mysich chłoniaków z wykorzystaniem agaru i mikropłytek), (2) skuteczności klonowania w momencie wybierania mutantów oraz (3) wzrostu zawieszania (wzory znajdują się w dodatku 2).
Tabela 2
Kryteria dopuszczalności dotyczące badania mysich chłoniaków
|
Parametr
|
Metoda badania na miękkim agarze
|
Metoda badania na mikropłytkach
|
|
Częstość występowania mutacji
|
35–140 × 10-6
|
50–170 × 10-6
|
|
Skuteczność klonowania
|
65–120 %
|
65–120 %
|
|
Wzrost zawiesiny
|
8–32 -krotne (3–4 godziny poddawania działaniu substancji) 32–180 -krotne (24 godziny poddawania działaniu substancji, jeżeli będzie stosowane)
|
8–32 -krotne (3–4 godziny poddawania działaniu substancji) 32–180 -krotne (24 godziny poddawania działaniu substancji, jeżeli będzie stosowane)
|
|
|
58.
|
W przypadku badania mysich chłoniaków każde badanie należy również poddać ocenie pod kątem sprawdzenia, czy dodatnia(-e) kontrola(-e) spełniają co najmniej jedno z następujących kryteriów dopuszczalności opracowanych przez grupę roboczą międzynarodowych warsztatów z badań genotoksycznych:
|
—
|
Kontrola dodatnia powinna wskazywać bezwzględny wzrost całkowitej częstości występowania mutacji, tzn. przekroczenie poziomu spontanicznej częstości występowania mutacji w tle [indukowana częstość występowania mutacji (IMF)] o co najmniej 300 × 10-6. Co najmniej 40 % indukowanej częstości występowania mutacji powinno zostać odzwierciedlone w częstości występowania mutacji w małych koloniach.
|
|
—
|
W dodatniej kontroli odnotowano wzrost częstości występowania mutacji w małych koloniach na poziomie co najmniej 150 × 10-6 powyżej wzrostu odnotowanego w jednoczesnej kontroli niepoddawanej działaniu substancji / z rozpuszczalnikiem (indukowana częstość występowania mutacji w małych koloniach na poziomie 150 × 10-6).
|
|
|
59.
|
W przypadku TK6 badanie będzie dopuszczalne, jeżeli uznaje się, że dopuszczalne jest włączenie danych dotyczących jednoczesnej kontroli ujemnej do prowadzonej przez laboratorium bazy danych historycznych dotyczących kontroli ujemnej, jak opisano w pkt 48–49. Jednoczesne kontrole dodatnie (zob. pkt 32) powinny ponadto wywoływać reakcje zgodne z reakcjami odnotowanymi w bazie danych historycznych dotyczących kontroli dodatniej i wykazywać statystycznie istotny wzrost w porównaniu z jednoczesną kontrolą ujemną.
|
|
60.
|
W obu badaniach górna granica cytotoksyczności zaobserwowanej w kulturze kontroli dodatniej powinna być taka sama, jak w kulturach eksperymentalnych. Oznacza to, że względny wzrost całkowity / względny wskaźnik przeżyć nie powinny być niższe niż 10 %. Aby wykazać spełnienie kryteriów dopuszczalności w odniesieniu do kontroli dodatniej, wystarczy zastosować jedno stężenie (lub jedno ze stężeń kultur w ramach kontroli dodatniej, jeżeli zastosowanie więcej niż jedno stężenie). Częstość występowania mutacji w kontroli dodatniej musi się ponadto zmieścić w dopuszczalnym zakresie ustalonym dla danego laboratorium.
|
Ocena i interpretacja wyników
|
61.
|
W przypadku badania mysich chłoniaków grupa robocza ekspertów ds. mysich chłoniaków międzynarodowych warsztatów z badań genotoksycznych wykonała ogromną pracę dotyczącą biologicznego znaczenia i kryteriów reakcji dodatniej. W związku z tym w niniejszej metodzie badawczej przedstawiono szczegółowe zalecenia na temat interpretacji wyników dotyczących badanej substancji chemicznej uzyskanych w badaniu mysich chłoniaków (zob. pkt 62–64). Baza danych dotyczących badania TK6 jest dużo mniejsza i grupa robocza nie przeprowadziła jego oceny. W związku z tym zalecenia na temat interpretacji danych dotyczących TK6 przedstawiono w bardziej ogólnej formie (zob. pkt 65–66). Do obu badań mają zastosowanie dodatkowe zalecenia (zob. pkt 67–71).
|
Badanie mysich chłoniaków
|
62.
|
Aby zagwarantować, że zwiększona częstość występowania mutacji ma biologiczne znaczenie, zaleca się zastosowanie metody określania reakcji dodatnich i ujemnych. Zamiast stosowanej zazwyczaj w innych badaniach analizy statystycznej, metoda ta polega na wykorzystaniu predefiniowanej indukowanej częstości występowania mutacji (tj. wzrostu częstości występowania mutacji powyżej poziomu w ramach jednoczesnej kontroli) określanej jako globalny czynnik oceny (Global Evaluation Factor), który opiera się na analizie rozkładu danych dotyczących częstości występowania mutacji w kontroli ujemnej otrzymanych od laboratoriów uczestniczących (4). W wersji badania mysich chłoniaków wykorzystującej agar, globalny czynnik oceny wynosi 90 × 10-6, w wersji badania mysich chłoniaków wykorzystującej mikropłytki globalny czynnik oceny wynosi 126 × 10-6.
|
|
63.
|
O ile wszystkie kryteria dopuszczalności są spełnione, uznaje się, że badana substancja chemiczna daje wynik wyraźnie dodatni, jeżeli w dowolnych ze zbadanych warunków doświadczalnych (zob. pkt 33), wzrost częstości występowania mutacji powyżej jednoczesnego tła przekracza globalny czynnik oceny i jest powiązany ze stężeniem (np. z zastosowaniem testu tendencji). Badaną substancję chemiczną uznaje się następnie za zdolną do wywołania mutacji w danym układzie badawczym.
|
|
64.
|
O ile wszystkie kryteria dopuszczalności są spełnione, uznaje się, że badana substancja chemiczna daje wynik wyraźnie ujemny, jeżeli we wszystkich zbadanych warunkach doświadczalnych (zob. pkt 33) nie zachodzi reakcja powiązana ze stężeniem lub, jeżeli ma miejsce wzrost częstości występowania mutacji – nie przekracza globalnego czynnika oceny. Badaną substancję chemiczną uznaje się następnie za niezdolną do wywołania mutacji w danym układzie badawczym.
|
TK6
|
65.
|
O ile wszystkie kryteria dopuszczalności są spełnione, uznaje się, że badana substancja chemiczna daje wynik wyraźnie dodatni, jeżeli w dowolnych ze zbadanych warunków doświadczalnych (zob. pkt 33):
|
—
|
co najmniej jedno z badanych stężeń wykazuje statystycznie istotny wzrost w porównaniu z jednoczesną kontrolą ujemną,
|
|
—
|
ocena przeprowadzona z zastosowaniem odpowiedniego testu tendencji pokazuje, że wzrost jest powiązany ze stężeniem (zob. pkt 33)
|
|
—
|
którykolwiek z wyników znajduje się poza rozkładem danych historycznych dotyczących kontroli ujemnej (np. poza granicą kontrolną wyznaczającą przedział 95 % na podstawie rozkładu Poissona; zob. pkt 48).
|
W przypadku gdy wszystkie wspomniane kryteria są spełnione, uznaje się, że badana substancja chemiczna jest w stanie wywołać mutacje w danym układzie badawczym. Zalecenia dotyczące najodpowiedniejszych metod statystycznych można znaleźć w literaturze (66)(67).
|
|
66.
|
O ile wszystkie kryteria dopuszczalności są spełnione, uznaje się, że badana substancja chemiczna daje wynik wyraźnie ujemny, jeżeli we wszystkich zbadanych warunkach doświadczalnych (zob. pkt 33):
|
—
|
żadne z badanych stężeń nie wykazuje statystycznie istotnego wzrostu w porównaniu z równoczesną kontrolą ujemną,
|
|
—
|
ocena przeprowadzona z zastosowaniem odpowiedniego testu tendencji pokazuje, że wzrost nie jest powiązany ze stężeniem,
|
|
—
|
wszystkie wyniki mieszczą się w rozkładzie danych historycznych dotyczących kontroli ujemnej (np. w granicach kontrolnych wyznaczających przedział 95 % na podstawie rozkładu Poissona; zob. pkt 48).
|
Badaną substancję chemiczną uznaje się następnie za niezdolną do wywołania mutacji w danym układzie badawczym.
|
Zarówno w przypadku badania mysich chłoniaków i badania TK6:
|
67.
|
Jeżeli maksymalne stężenie określono na podstawie cytotoksyczności, należy dążyć do tego, by poziom takiego maksymalnego stężenia doprowadził do uzyskania względnego wzrostu całkowitego / względnego wskaźnika przeżyć na poziomie od 20 do 10 %. Istnieje zgoda co do tego, że należy zachować ostrożność podczas interpretowania wyników dodatnich występujących jedynie między 20 i 10 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć, a wyniku nie uznaje się za dodatni, jeżeli wzrost częstości występowania mutacji nastąpił tylko na poziomie lub poniżej 10 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć (jeżeli podlegał ocenie) (2)(59).
|
|
68.
|
W niektórych okolicznościach dodatkowe informacje mogą okazać się pomocne w ustaleniu, czy badana substancja chemiczna nie jest mutagenna, kiedy w żadnej z kultur nie odnotowuje się wartości względnego wzrostu całkowitego między 10–20 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć. Są to następujące sytuacje: (1) Nie ma objawów mutagenności (np. brak zależności dawka-odpowiedź, brak częstości występowania mutacji wyższych niż zaobserwowane w jednoczesnej kontroli ujemnej lub w historycznych zakresach tła itp.) w szeregu punktów danych w zakresie 100–20 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć, a co najmniej jeden punkt danych znajduje się w zakresie 20–25 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć. (2) Nie ma objawów mutagenności (np. brak zależności dawka-odpowiedź, brak częstości występowania mutacji wyższych niż zaobserwowane w jednoczesnej kontroli ujemnej lub w historycznych zakresach tła itp.) w szeregu punktów danych w zakresie 100–25 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć, a jeden punkt danych jest ujemny i jego wartość wynosi niewiele poniżej 10 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć. W obu tych sytuacjach można stwierdzić, że badana substancja chemiczna daje wynik ujemny.
|
|
69.
|
Weryfikacja jednoznacznie dodatniej lub ujemnej reakcji nie jest wymagana.
|
|
70.
|
Jeżeli reakcja nie jest ani wyraźnie dodatnia, ani wyraźnie ujemna, jak opisano powyżej, oraz aby ułatwić ustalenie biologicznego znaczenia wyniku, dane powinny zostać ocenione w oparciu o opinię eksperta lub wyniki dalszych badań. Przydatne może być powtórne przeprowadzenie doświadczenia, potencjalnie z zastosowaniem zmienionych warunków doświadczalnych [np. z zastosowaniem odstępów czasu między stężeniami w celu zwiększenia prawdopodobieństwa uzyskania punktów danych w zakresie 10–20 % względnego wzrostu całkowitego / względnego wskaźnika przeżyć, wykorzystując inne warunki aktywacji metabolicznej (tj. stężenie S9 lub pochodzenie S9) oraz czas poddawania działaniu substancji chemicznej].
|
|
71.
|
W rzadkich przypadkach nawet po przeprowadzeniu dalszych badań uzyskany zbiór danych uniemożliwi uznanie wyników za dodatnie lub ujemne. W takiej sytuacji należy uznać, że reakcja badanej substancji chemicznej jest niejednoznaczna (tj. może zostać zinterpretowana zarówno jako dodatnia, jak i jako ujemna).
|
SPRAWOZDANIE Z BADANIA
|
72.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Badana substancja chemiczna:
|
—
|
źródło, numer partii, termin przydatności, jeżeli jest dostępny;
|
|
—
|
stabilność badanej substancji chemicznej, jeżeli jest znana;
|
|
—
|
rozpuszczalność i stabilność badanej substancji chemicznej w rozpuszczalniku, jeżeli są znane;
|
|
—
|
pomiar pH, osmolalności i osadu w podłożu, do którego dodano badaną substancję chemiczną, stosownie do przypadku.
|
|
|
Substancja jednoskładnikowa:
|
—
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń, stosownie do przypadku i jeśli jest to praktycznie wykonalne, itp.
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszaniny:
|
—
|
opisane w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników.
|
|
|
|
|
Rozpuszczalnik:
|
—
|
uzasadnienie wyboru rozpuszczalnika;
|
|
—
|
zawartość procentowa rozpuszczalnika w końcowym podłożu.
|
|
|
|
Komórki:
|
|
W przypadku głównych kultur laboratoryjnych:
|
—
|
rodzaj i źródło komórek oraz historia w laboratorium badawczym;
|
|
—
|
cechy kariotypu lub zmienna liczba chromosomów;
|
|
—
|
metody utrzymywania kultur komórkowych;
|
|
—
|
czasy podwojenia komórek.
|
|
|
|
|
Warunki badania:
|
—
|
uzasadnienie wyboru stężeń oraz liczby kultur komórkowych; z uwzględnieniem np. danych dotyczących cytotoksyczności oraz granic rozpuszczalności;
|
|
—
|
skład podłoża, stężenie CO2, poziom wilgotności;
|
|
—
|
stężenie badanej substancji chemicznej wyrażone jako stężenie końcowe w podłożu (np. μg lub mg/ml lub mM podłoża);
|
|
—
|
stężenie (lub objętość) rozpuszczalnika i badanej substancji chemicznej dodanych do podłoża;
|
|
—
|
czas poddawania działaniu substancji chemicznej;
|
|
—
|
zagęszczenie komórek w trakcie poddawania działaniu substancji;
|
|
—
|
typ oraz skład układu metabolizującego (źródło frakcji S9, metoda przygotowania preparatu frakcji S9, stężenie lub objętość preparatu frakcji S9 i frakcji S9 w końcowym podłożu, kontrole jakości frakcji S9);
|
|
—
|
substancje służące do kontroli dodatniej i ujemnej, stężenia końcowe w poszczególnych warunkach poddawania działaniu substancji chemicznej;
|
|
—
|
długość okresu ekspresji (w tym liczba posianych komórek i podkultur oraz – w stosownych przypadkach – schematy żywienia);
|
|
—
|
nazwa czynnika selektywnego i jego stężenie;
|
|
—
|
w przypadku badania mysich chłoniaków należy wskazać zastosowaną wersję badania (agar lub mikropłytki)
|
|
—
|
kryteria dopuszczalności badań;
|
|
—
|
metody wykorzystywane do wyliczenia liczby żywotnych i zmutowanych komórek;
|
|
—
|
metody wykorzystywane do pomiaru cytotoksyczności;
|
|
—
|
wszelkie informacje uzupełniające istotne w kontekście cytotoksyczności i stosowanej metody;
|
|
—
|
czas trwania okresów inkubacji po posiewie;
|
|
—
|
definicja kolonii, których rozmiary oraz typy są brane pod uwagę (łącznie z kryteriami dotyczącymi „małych” oraz „dużych” kolonii, w stosownych przypadkach);
|
|
—
|
kryteria uznawania wyników badań za dodatnie, ujemne lub niejednoznaczne;
|
|
—
|
metody stosowane w celu ustalenia poziomu pH, osmolalności, jeżeli jest badana i strącania, w stosownych przypadkach.
|
|
|
|
Wyniki:
|
—
|
liczba komórek poddanych działaniu substancji chemicznej i liczba komórek w hodowli pochodnej z każdej kultury;
|
|
—
|
parametry toksyczności (względny wzrost całkowity w przypadku badania mysich chłoniaków i względny wskaźnik przeżyć w przypadku badania TK6);
|
|
—
|
oznaki strącania i czas oznaczenia;
|
|
—
|
liczba komórek posianych na podłożu selektywnym i nieselektywnym;
|
|
—
|
liczba kolonii na podłożu nieselektywnym i liczba odpornych kolonii na podłożu selektywnym oraz powiązane częstości występowania mutacji;
|
|
—
|
sortowanie według rozmiarów w przypadku dodatnich i ujemnych kontroli oraz jeżeli badana substancja chemiczna daje wynik dodatni, przy co najmniej jednym stężeniu, oraz powiązane częstości występowania mutacji;
|
|
—
|
w stosownych przypadkach zależność stężenie-odpowiedź;
|
|
—
|
dane dotyczące jednoczesnych kontroli ujemnych (z rozpuszczalnikiem) i dodatnich (stężenia i rozpuszczalniki);
|
|
—
|
dane dotyczące historycznych kontroli ujemnych (z rozpuszczalnikiem) i dodatnich (stężenia i rozpuszczalniki) z uwzględnieniem zakresów, średnich i odchyleń standardowych; liczba badań, na których oparto kontrole historyczne;
|
|
—
|
analizy statystyczne (dla poszczególnych kultur oraz, w stosownych przypadkach, połączonych kontrprób) oraz p-wartości, jeżeli występują; oraz w przypadku badania mysich chłoniaków ocena globalnego czynnika oceny.
|
|
|
BIBLIOGRAFIA
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. i Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, „Environ. Mol. Mutagen.” nr 35 (3), s. 185–190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. i Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana (kwiecień 2000) „Environ. Mol. Mutagen.” nr 40 (4), s. 292–299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. i Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, „Mutation Res.” nr 540, s. 127–140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. i Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, „Environ. Mol. Mutagen.” nr 47 (1), s. 1–5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. i Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment,„Mutation. Res.” nr 627 (1), s. 36–40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. [W:] Publikacje na temat środowiska, seria dotycząca badań i oceny nr 234. Paryż: OECD.
|
|
(7)
|
M.D. Fellows, T. Luker, A. Cooper i M.R. O'Donovan (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors.„Mutation, Res.” nr 746 (1), s. 21–28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. i Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction.„Mutation Res.” nr 493 (1–2), s. 101–114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. i Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, „Toxicol. Sci.” nr 109 (1), s. 96–105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., i Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. „Proc. National. Academy. Sci. USA” nr 87 (1), s. 51–55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. i Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, „Mutation Res.” nr 84 (1), s. 169–181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. i Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, „Mutation Res.” nr 147 (5), s. 237–242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. i Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells.„Mutation, Res.” nr 151 (1), s. 161–174.
|
|
(14)
|
Liber H.L., Call K.M. i Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells.„Mutation, Res.” nr 178 (1), s. 143–153.
|
|
(15)
|
Li C.Y., Yandell D.W. i Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. „Somat. Cell Mol. Genet.” nr 18 (1), s. 77–87.
|
|
(16)
|
Honma M., Hayashi M. i Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells.„Mutation. Res.” nr 374 (1), s. 89–98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. i Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity.„Mol. Carcinogen.” nr 28 (4), s. 203–14.
|
|
(18)
|
Amundson S.A. i Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants.„Mutation Res.” nr 267 (1), s. 89–95.
|
|
(19)
|
Schisler M.R., Moore M.M. i Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. [W:] Protocols in Genotoxicity Assessment A. Dhawan i M. Bajpayee (red.), Springer Protocols, Humana Press, s. 27–50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. i Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, „Mutation Res.” nr 634 (1–2), s. 177–183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. i Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. „Environ. Mol. Mutagen.” nr 49 (6), s. 439–452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations.„Environ. Mutagen.” nr 8 (6), s. 879–886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. i Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. „Mutation Res.” nr 268 (2), s. 297–305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. i Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9.„Mutation Res.” nr 257, s. 147–204.
|
|
(25)
|
Wang J., Heflich R.H. i Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay.„Mutation Res.” nr 626 (1–2), s. 185–190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro.„Ann. N.Y. Academy Sci.” nr 76: s. 673–680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. i Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System.„Mutation Res.” nr 59 (1), s. 61–108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. i Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, „Mutation Res.” nr 147 (5), s. 243–253.
|
|
(29)
|
Sawyer J.R., Moore M.M. i Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, „Mutation Res.” nr 214 (2), s. 181–193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. i Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, „Environ. Mol. Mutagen.” nr 47 (2), s. 127–131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. i Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, „Environ. Mol. Mutagen.” nr 55 (1), s. 35–42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. i DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene.„Mutation. Res.” nr 373 (2), s. 157–165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. i Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells.„Mutagen.” nr 19 (4), s. 263–268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. i Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay.„Biochem. Biophys. Res. Commun.” nr 84 (2), s. 411–416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells.„Environ. Mol. Mutagen.” nr 45 (2–3), s. 162–176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. i Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines.„Cancer. Res.” nr 55 (1), s. 12–15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Tekst w przygotowaniu).
|
|
(38)
|
Lloyd M. i Kidd D. (2012). The Mouse Lymphoma Assay. [W:] Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods. Parry i Parry (red.), Humana Press. ISBN, 978-1-61779-420-9, s. 35–54.
|
|
(39)
|
Mei N., Guo X. i Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. [W:] Optimization in Drug Discover: In Vitro Methods, Z Yan i Caldwell (red.), wydanie drugie, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. i Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts.„Mutation Res.” nr 94 (2), s. 467–485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. i Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA nr 33 (3), s. 261–287.
|
|
(42)
|
Moore M.M. i Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, „Mutation Res.” nr 104 (4–5): s. 287–294.
|
|
(43)
|
Ames B.N., McCann J. i Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test.„Mutation Res.” nr 31 (6), s. 347–364.
|
|
(44)
|
Maron D.M. i Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. „Mutation Res.” nr 113 (3–4), s. 173–215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. i De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes.„Mutation Res.” nr 37 (1), s. 83–90.
|
|
(46)
|
Matsuoka A., Hayashi M. i Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro.„Mutation Res.” nr 66 (3): s. 277–290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver.„J. Environ. Pathol. Toxicol.” nr 4 (1), s. 55–65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. i Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays.„Mutagen.” nr 7 (3), s. 175–177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. i Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, [W:] In Vitro Metabolic Activation in Mutagenesis Testing. F.J. de Serres et al. (red.). North-Holland: Elsevier, s. 85–88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations.„Mutation Res.” nr 312 (3), s. 241–261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. i Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9. „Environ. Mol. Mutagen.” nr 28 (1), s. 51–59.
|
|
(52)
|
UNEP (2001). Konwencja sztokholmska w sprawie trwałych zanieczyszczeń organicznych, Program Narodów Zjednoczonych ds. Ochrony Środowiska (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. i McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. [W:] Genotoxic Effects of Airborne Agents, R.R. Tice, D.L. Costa i K.M. Schaich (red.). Nowy Jork, Plenum, s. 91–103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. i Brooks, A.L. (1983). for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay.„Environ. Mutagen.” nr 5 (6), s. 795–801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. i Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay.„Mutation Res.” nr 652 (2), s. 122–130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation, [W:] Statistical Evaluation of Mutagenicity Test Data, D.J. Kirkland (red.), CambridgeUniversity Press, s. 66–101.
|
|
(57)
|
Morita T., Honma M. i Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity.„Mutation Res.” nr 741 (1–2), s. 32–56.
|
|
(58)
|
Brookmire L., Chen J.J. i Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay.„Environ. Mol. Mutagen.” nr 54 (1), s. 36–43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Dostępne na stronie internetowej: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. i Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells.„Environ. Mol. Mutagen.”, nr 52 (5), s. 373–384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. i Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells.„Mutation Res.” nr 85 (5), s. 363–378.
|
|
(62)
|
Liber H.L., Yandell D.W. i Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus.„Mutation Res.” nr 216 (1), s. 9–17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. i Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates.„Anal. Biochem.” nr 110 (1), s. 1–8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. i Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data.„Mutation Res.” nr 723 (2), s. 87–90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. Nowy Jork: John Wiley and Sons, wydanie drugie.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 199. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(67)
|
Fleiss J.L., Levin B. i Paik M.C. (2003). Statistical Methods for Rates and Proportions, wydanie trzecie, Nowy Jork: John Wiley & Sons.
|
Dodatek 1
DEFINICJE
Aneugen
: Każda substancja chemiczna lub proces, który poprzez reakcję z elementami mitotycznego i mejotycznego cyklu podziału komórek prowadzi do aneuploidii w komórkach lub organizmach.
Aneuploidia
: Wszelkie odchylenia od zwykłej diploidalnej (lub haploidalnej) liczby chromosomów w pojedynczym chromosomie lub w większej ich liczbie, ale nie w całym zestawie lub w całych zestawach chromosomów (poliploidalność).
Mutageny powodujące substytucję
: Substancje chemiczne, które powodują podstawienie par zasad w DNA.
Substancja chemiczna
: substancja lub mieszanina.
Skuteczność klonowania
: odsetek komórek posiewanych z niską gęstością, będących w stanie przekształcić się w zdatną do zliczenia kolonię.
Klastogen
: Każda substancja chemiczna lub proces, które powodują strukturalne aberracje chromosomowe w populacjach komórek lub organizmów.
Cytotoksyczność
: W odniesieniu do badań objętych niniejszą metodą badawczą, cytotoksyczność określa się jako obniżenie względnego wzrostu całkowitego w przypadku badania mysich chłoniaków lub względnego wskaźnika przeżyć w przypadku badania TK6.
Mutacja pierwotna
: Mutacja genu z rodzaju macierzystego do mutanta, który powoduje zmianę lub stratę aktywności enzymatycznej funkcji zakodowanego białka.
Mutageny typu zmiany fazy odczytu
: Substancje chemiczne, które powodują dodanie lub delecję pojedynczej pary zasad lub wielu par zasad w cząstce DNA.
Genotoksyczny
: Termin ogólny, obejmujący wszystkie rodzaje uszkodzeń DNA lub chromosomu, w tym przerwy, przegrupowania adduktów, mutacje, aberracje chromosomowe i aneuploidię. Nie wszystkie rodzaje skutków genotoksycznych powodują mutacje lub stałe uszkodzenia chromosomu.
Rekombinacja mitotyczna
: Zachodząca podczas mitozy rekombinacja między chromatydami homologicznymi, której możliwe skutki to indukowanie pęknięć łańcucha DNA lub utrata heterozygotyczności.
Mutagenny
: Powoduje dziedziczne zmiany w sekwencji lub sekwencjach par zasad DNA w genach lub w strukturze chromosomów (aberracje chromosomowe).
Częstość występowania mutacji (MF)
: Liczba zaobserwowanych zmutowanych komórek podzielona przez liczbę komórek zdolnych do życia.
Czas ekspresji fenotypowej
: Czas po poddaniu działaniu substancji chemicznej, w którym zmiana genetyczna utrwalana jest w genomie, a wszelkie istniejące wcześniej produkty genu są oddzielane do tego stopnia, że zmianie ulega cecha fenotypowa.
Względny wskaźnik przeżyć (RS)
: Względny wskaźnik przeżyć wykorzystuje się do mierzenia cytotoksyczności związanej z poddawaniem działaniu substancji chemicznej w badaniu TK6. Stanowi względną skuteczność klonowania komórek posianych natychmiast po poddaniu działaniu substancji, skorygowany o wszelkie komórki utracone w trakcie działania substancji w stosunku do skuteczności klonowania w kontroli ujemnej.
Względny wzrost zawieszania (RSG)
: W przypadku badania mysich chłoniaków względny, całkowity, dwudniowy wzrost zawieszania badanej kultury w porównaniu z całkowitym dwudniowym całkowitym dwudniowym wzrostem zawieszania w kontroli ujemnej / kontroli z rozpuszczalnikiem (Clive i Spector, 1975). Względny wzrost zawieszania powinien obejmować względny wzrost badanej kultury w porównaniu z kontrolą ujemną / kontrolą z rozpuszczalnikiem w trakcie okresu poddawania działaniu substancji.
Względny wzrost całkowity (RTG)
: Względny wzrost całkowity wykorzystuje się do mierzenia cytotoksyczności związanej z poddawaniem działaniu substancji chemicznej w badaniu mysich chłoniaków. Jest to miara względnego (w stosunku do grupy kontrolnej nośnika) wzrostu badanych kultur w trakcie etapów badania poddawania działaniu substancji, dwudniowej ekspresji i wybierania mutantów za pomocą klonowania. Względny wzrost zawieszania każdej badanej kultury mnoży się przez względną skuteczność klonowania badanej kultury w momencie wybierania mutantów oraz wyraża się go w odniesieniu do skuteczności klonowania w kontroli ujemnej / kontroli z rozpuszczalnikiem (Clive i Spector, 1975).
Frakcje S9 uzyskane z wątroby
: Supernatant z homogenatu wątroby po odwirowaniu przy 9 000 g, tj. surowy ekstrakt z wątroby.
Preparat frakcji S9
: Preparat złożony z frakcji S9 uzyskanej z wątroby i kofaktorów niezbędnych do aktywności metabolicznej enzymów.
Wzrost zawieszania (SG)
: Wielokrotny wzrost liczby komórek w trakcie etapów poddawania działaniu substancji i ekspresji w badaniu mysich chłoniaków. Wzrost zawieszania oblicza się poprzez pomnożenie wielokrotnego wzrostu na dzień 1 przez wielokrotny wzrost na dzień 2 w przypadku krótkoterminowego (3–4 h) poddawania działaniu substancji. W przypadku zastosowania 24-godzinnego poddania działaniu substancji wzrost zawieszania stanowi wielokrotny wzrost podczas 24-godzinnego poddawania działaniu substancji pomnożony przez wartości wielokrotnego wzrostu w 1. i 2. dniu ekspresji.
Kontrola z rozpuszczalnikiem
: ogólny termin opisujący kultury kontrolne otrzymujące wyłącznie rozpuszczalnik stosowany do rozpuszczenia badanej substancji chemicznej.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
Kontrole niepoddawane działaniu substancji
: Kontrolne niepoddawane działaniu substancji są to kultury, które nie są poddawane działaniu żadnej substancji (tj. ani badanej substancji chemicznej, ani rozpuszczalnika), lecz które są przetwarzane taki sam sposób jako hodowle otrzymujące badaną substancję chemiczną.
Dodatek 2
WZORY
Cytotoksyczność
W przypadku obu wersji badania mysich chłoniaków (wykorzystującej agar i wykorzystującej mikropłytki)
Cytotoksyczność określa się jako względny wzrost całkowity (RTG), który obejmuje względny wzrost zawieszania (RSG) podczas dwudniowego okresu ekspresji oraz względną skuteczność klonowania (RCE) uzyskaną w momencie wybierania mutantów. Względny wzrost całkowity, względny wzrost zawieszania oraz względną skuteczność klonowania wyraża się jako procent.
Obliczanie względnego wzrostu zawieszania: Pierwsza wartość wzrostu zawieszania (SG1) to szybkość wzrostu między dniem 0 a dniem 1 (stężenie komórek na dzień 1 / stężenie komórek na dzień 0), a druga wartość wzrostu zawieszenia (SG2) to szybkość wzrostu między dniem 1 a dniem 2 (stężenie komórek na dzień 2 / stężenie komórek na dzień 1). Względny wzrost zawieszania to całkowity wzrost zawieszania (SG1 x SG2) dla danej kultury poddawanej działaniu substancji w porównaniu z kontrolą niepoddawaną działaniu substancji / kontroli z rozpuszczalnikiem. Czyli: RSG = [SG1(test) x SG2(test)] / [SG1(control) x SG2(control)] SG1 należy obliczyć na podstawie wstępnego stężenia komórek zastosowanego na początku poddawania komórek działaniu substancji. Pozwala to uwzględnić wszelką różnicową cytotoksyczność, która występuje w badanych kulturach w trakcie poddawania komórek działaniu substancji chemicznej.
Względna skuteczność klonowania to względna skuteczność klonowania badanej kultury w porównaniu ze względną skutecznością klonowania w kontroli ujemnej / kontroli z rozpuszczalnikiem uzyskaną w momencie wybierania mutantów.
Względny wzrost całkowity (RTG):
RTG = RSG x RCE
TK6
Względny wskaźnik przeżyć (RS):
Ocenę cytotoksyczności przeprowadza się w oparciu o względny wskaźnik przeżyć, tj. wskaźnik skuteczności klonowania komórek posianych natychmiast po poddaniu działaniu substancji, skorygowany o wszelkie komórki utracone w trakcie działania substancji w stosunku do skuteczności klonowania w kontrolach ujemnych (którym przypisano wskaźnik przeżycia wynoszący 100 %). Dostosowanie z tytułu utraty komórek podczas poddawania działaniu substancji można obliczyć następująco:
Względny wskaźnik przeżyć kultury poddanej działaniu badanej substancji chemicznej oblicza się w następujący sposób:

Częstość występowania mutacji zarówno w badaniu mysich chłoniaków, jak i badaniu TK6
Częstość występowania mutacji (MF) to skuteczność klonowania kolonii zmutowanych komórek w podłożu selektywnym (CEm) skorygowana o skuteczność klonowania w podłożu nieselektywnym w momencie wybierania mutantów (CEV). Czyli, MF = CEM/CEV Poniżej opisano obliczenia tych dwóch wartości skuteczności klonowania w metodzie klonowania z wykorzystaniem agaru i metodzie klonowania z wykorzystaniem mikropłytek.
Wersja badania mysich chłoniaków z wykorzystaniem agaru: W wersji badania mysich chłoniaków z wykorzystaniem miękkiego agaru, liczbę kultur na płytce wyboru mutantów (CM) oraz liczbę kultur na płytce niewybranych lub skuteczności klonowania (liczba zdolnych do życia)(CV) uzyskuje się dzięki bezpośredniemu zliczeniu klonów. Jeżeli umieszczono 600 komórek na płytkach skuteczności klonowania (CE), wybierania mutantów (CEM) i płytkach niewybranych lub skuteczności klonowania (liczba zdolnych do życia)(CV), a 3 x 106 komórek wykorzystano do wybierania mutantów,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
Wersja badania mysich chłoniaków i TK6 wykorzystująca mikropłytki:
W wersji badania mysich chłoniaków wykorzystującej mikropłytki CM i CV określa się jako wynik całkowitej liczby mikropłytek (TW) i prawdopodobnej liczby kultur na płytkę (P) na mikropłytkach.
CM = PM x TWM
CV = PV x TWV
Poczynając od wyrazu zerowego rozkładu Poissona (Furth i in., 1981), P uzyskuje się z
P = - ln (EW / TW)
Gdzie EW oznacza puste dołki, a TW wszystkie dołki. W związku z tym
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
W wersji badania mysich chłoniaków na mikropłytkach częstości występowania mutacji w małych i dużych koloniach oblicza się w ten sam sposób, wykorzystując odpowiednią liczbę pustych dołków dla małych i dużych kolonii.
W przypadku badania TK6 częstości występowania mutacji w małych i dużych koloniach są oparte na wcześnie pojawiających się i późno pojawiających się mutacjach.
B.68 METODA BADAWCZA IN VITRO KRÓTKIEGO OKRESU NARAŻENIA DO CELÓW IDENTYFIKACJI i) SUBSTANCJI CHEMICZNYCH POWODUJĄCYCH POWAŻNE USZKODZENIE OCZU ORAZ ii) SUBSTANCJI CHEMICZNYCH, KTÓRE NIE WYMAGAJĄ ZAKLASYFIKOWANIA JAKO SUBSTANCJE DRAŻNIĄCE OCZY LUB POWODUJĄCE POWAŻNE USZKODZENIE OCZU.
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej OECD (TG) nr 491 (2017). Metoda badania krótkiego okresu narażenia (STE) jest metodą badawczą in vitro, którą można stosować w niektórych okolicznościach i przy określonych ograniczeniach na potrzeby klasyfikacji pod względem zagrożeń i oznakowania substancji chemicznych (substancji i mieszanin) powodujących poważne uszkodzenie oczu, jak również substancji chemicznych, które nie wymagają klasyfikacji jako powodujące poważne uszkodzenie lub podrażnienie oczu, określonych przez Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ) (1) oraz rozporządzenie Unii Europejskiej (WE) nr1272/2008 w sprawie klasyfikacji, oznakowania i pakowania substancji i mieszanin (rozporządzenie CLP) (67).
|
|
2.
|
Potencjalne działanie substancji chemicznych szkodliwe dla oczu oceniano przez wiele lat przede wszystkim za pomocą badania na oku królika in vivo (metoda badawcza B.5 (8), równoważna z wytyczną OECD TG 405). Powszechnie uznaje się, że w dającej się przewidzieć przyszłości żadne pojedyncze alternatywne badanie in vitro nie będzie w stanie w pełni zastąpić badania na oku królika in vivo na potrzeby prognozowania pełnego zakresu działania drażniącego różnych klas chemicznych. Jest jednak możliwe, że strategiczne połączenia alternatywnych metod badawczych przeprowadzonych w ramach (zintegrowanej) strategii badań będą mogły w pełni zastąpić badanie na oku królika (2). Podejście odgórne opracowano na potrzeby badania substancji chemicznych, w przypadku których można oczekiwać w oparciu o istniejącą wiedzę, że posiada wysoki potencjał działania drażniącego lub spowodowania poważnego uszkodzenia oczu. Z kolei podejście oddolne opracowano na potrzeby badania substancji chemicznych, w przypadku których można oczekiwać w oparciu o istniejącą wiedzę, że nie spowoduje podrażnienia oczu w stopniu wymagającym zaklasyfikowania. Chociaż uważa się, że metoda badawcza krótkiego okresu narażenia nie może całkowicie zastąpić badania na oku królika in vivo, jest odpowiednia do stosowania w ramach zintegrowanej strategii badań do celów regulacyjnej klasyfikacji i oznakowania, np. w podejściu odgórnym/oddolnym, do identyfikacji bez dalszych badań (i) substancji chemicznych powodujących poważne uszkodzenie oczu (kategoria 1 wg GHS ONZ / rozporządzenia CLP) oraz (ii) substancji chemicznych (z wyjątkiem wysoce lotnych substancji i wszystkich substancji chemicznych w stanie stałym, które nie są środkami powierzchniowo czynnymi), które nie wymagają klasyfikacji jako powodujące poważne uszkodzenie lub podrażnienie oczu (brak kategorii wg GHS ONZ / rozporządzenia CLP) (1)(2). Konieczne będzie jednak przeprowadzenie dodatkowego badania w odniesieniu do substancji chemicznej, w przypadku której nie przewidziano, że spowoduje poważne uszkodzenie oczu (kategoria 1 wg GHS ONZ / rozporządzenia CLP) lub której nie zaklasyfikowano do grupy substancji oznaczonej jako „brak kategorii” wg GHS ONZ / rozporządzenia CLP (nie powoduje poważnego uszkodzenia lub podrażnienia oczu) w ramach metody badawczej krótkiego okresu narażenia, w celu ustanowienia ostatecznej klasyfikacji. Ponadto przed zastosowaniem metody badawczej krótkiego okresu narażenia w ramach podejścia oddolnego zgodnie z systemami klasyfikacji innymi niż GHS ONZ / rozporządzeniem CLP należy skonsultować się z odpowiednimi organami regulacyjnymi. Na wybór najbardziej odpowiedniej metody badawczej i zastosowanie niniejszej metody badawczej należy patrzeć w kontekście wytycznych OECD w sprawie zintegrowanych podejść do badań i oceny dotyczących poważnych uszkodzeń i podrażnień oczu (14).
|
|
3.
|
Niniejsza metoda badawcza ma na celu opisanie procedur stosowanych do oceny potencjalnego działania szkodliwego dla oczu badanej substancji chemicznej na podstawie jej potencjału wywoływania cytotoksyczności w metodzie badawczej krótkiego okresu narażenia. Cytotoksyczny wpływ substancji chemicznych na komórki nabłonka rogówki stanowi istotny sposób działania wywołujący uszkodzenie nabłonka rogówki i podrażnienie oka. Żywotność komórek ocenia się w ramach metody badawczej krótkiego okresu narażenia za pomocą pomiaru ilościowego – po ekstrakcji z komórek – niebieskiej soli formazanu, wytworzonej w żywych komórkach metodą enzymatycznej konwersji barwnika przyżyciowego MTT (bromek 3-(4,5-dimetylotiazol-2-ilo)-2,5-difenylotetrazoliowy) (3). Uzyskaną w ten sposób żywotność komórek porównuje się do kontroli z rozpuszczalnikiem (względna żywotność) i wykorzystuje do oszacowania potencjalnego działania szkodliwego dla oczu badanej substancji chemicznej. Badaną substancje chemiczną klasyfikuje się jako substancję kategorii 1 wg GHZ ONZ / rozporządzenia CLP, jeżeli zarówno stężenia 5 % i 0,05 % prowadzą do żywotności komórek niższej niż lub równej (≤) 70 %. Z drugiej strony przewiduje się, że substancja chemiczna zalicza się do grupy substancji „brak kategorii” wg GHZ ONZ / rozporządzenia CLP, jeżeli zarówno stężenia 5 % i 0,05 % prowadzą do żywotności komórek wyższej niż (>) 70 %.
|
|
4.
|
Termin „badana substancja chemiczna” jest używany w niniejszej metodzie badawczej w odniesieniu do badanej substancji, i nie jest powiązany z zastosowaniem metody badawczej krótkiego okresu narażenia do badania substancji lub mieszanin. Definicje znajdują się w dodatku.
|
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
5.
|
Niniejsza metoda badawcza bazuje na protokole opracowanym przez Kao Corporation (4), który został poddany dwóm różnym badaniom walidacyjnym: jednemu, które zostało przeprowadzone przez Komitet Walidacyjny Japońskiego Towarzystwa ds. Rozwiązań Alternatywnych Wobec Eksperymentów na Zwierzętach (JSAAE) (5) i drugiemu, które zostało przeprowadzone przez Japońskie Centrum ds. Uznawania Metod Alternatywnych (JaCVAM) (6). Wzajemna ocena została przeprowadzona przez NICEATM/ICCVAM na podstawie treści sprawozdań z zastosowania metody badawczej oraz dokumentów przeglądowych poświęconych metodzie badawczej (7).
|
|
6.
|
W przypadku wykorzystania metody badawczej krótkiego okresu narażenia do identyfikacji substancji chemicznych (substancji i mieszanin) na 125 substancjach chemicznych (zarówno substancjach, jak i mieszaninach) powodujących poważne uszkodzenie oczu (kategoria 1 wg GHS ONZ / rozporządzenia CLP) metoda ta wykazuje ogólną dokładność na poziomie 83 % (104/125), odsetek wyników fałszywie dodatnich na poziomie 1 % (1/86) oraz odsetek wyników fałszywie ujemnych na poziomie 51 % (20/39) w porównaniu do danych uzyskanych w wyniku metody badań na oku królika in vivo (7). Uzyskany odsetek wyników fałszywie ujemnych nie ma decydującego znaczenia w tym kontekście, ponieważ wszystkie badane substancje chemiczne, które charakteryzują się żywotnością komórek ≤ 70 % przy stężeniu 5 % i > 70 % przy stężeniu 0,05 %, byłyby następnie badane za pomocą innych odpowiednio zwalidowanych metod badawczych in vitro, lub – w ostateczności – w ramach badania na oku królika in vivo, w zależności od obowiązujących wymogów regulacyjnych oraz zgodnie ze strategią badań sekwencyjnych oraz aktualnie zalecanymi podejściami bazującymi na analizie wagi dowodów (1)(8). Badaniu poddano głównie substancje jednoskładnikowe, chociaż istnieje również ograniczona ilość danych dotyczących badania mieszanin. Niniejsza metoda badawcza ma jednak zastosowanie techniczne w odniesieniu do badania zarówno substancji wieloskładnikowych, jak i mieszanin. Przed zastosowaniem niniejszej metody badawczej z użyciem mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy jednak zastanowić się nad tym, czy zastosowanie niniejszej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak – dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli istnieje wymóg regulacyjny dotyczący badania danej mieszaniny. Zastosowanie metody badawczej krótkiego okresu narażenia w celu zidentyfikowania badanej substancji chemicznej jako substancji kategorii 1 wg GHS ONZ / rozporządzenia CLP nie ujawniło żadnych innych konkretnych niedociągnięć. Osoby przeprowadzające badanie mogą rozważyć zastosowanie niniejszej metody badawczej w odniesieniu do badanych substancji chemicznych, wskutek czego żywotność komórek ≤ 70 % zarówno przy stężeniu 5 %, jak i przy stężeniu 0,05 %, należy zaakceptować jako wskazującą na reakcję wywołującą poważne uszkodzenie oczu, w którym to przypadku należy daną substancję zaklasyfikować do kategorii 1 wg GHS ONZ / rozporządzenia CLP bez dalszych badań.
|
|
7.
|
W przypadku wykorzystania metody badawczej krótkiego okresu narażenia do identyfikacji substancji chemicznych (substancji i mieszanin) na 130 substancjach chemicznych (zarówno substancjach, jak i mieszaninach), które nie muszą zostać sklasyfikowane jako powodujące podrażnienie i poważne uszkodzenie oczu (tj. brak kategorii wg GHS ONZ / rozporządzenia CLP), metoda ta wykazuje ogólną dokładność na poziomie 85 % (110/130), odsetek wyników fałszywie ujemnych na poziomie 12 % (9/73) oraz odsetek wyników fałszywie dodatnich na poziomie 19 % (11/57) w porównaniu do danych uzyskanych w wyniku metody badań na oku królika in vivo (7). W przypadku wykluczenia wysoce lotnych substancji i substancji stałych innych niż środki powierzchniowo czynne ze zbioru danych, ogólna dokładność wzrasta do 90 % (92/102), odsetek wyników fałszywie ujemnych do 2 % (1/54), a odsetek wyników fałszywie dodatnich do 19 % (9/48)(7). W rezultacie potencjalne niedociągnięcia związane ze stosowaniem metody badawczej krótkiego okresu narażenia w celu zidentyfikowania badanych substancji chemicznych, które nie wymagają sklasyfikowania jako substancje powodujące podrażnienie i poważne uszkodzenie oczu (brak kategorii wg GHS ONZ / rozporządzenia CLP), obejmują wysoki odsetek wyników fałszywie ujemnych w przypadku (i) wysoce lotnych substancji charakteryzujących się prężnością par powyżej 6 kPa i (ii) stałych substancji chemicznych (substancji i mieszanin) innych niż środki powierzchniowo czynne i mieszaniny, w których skład wchodzą wyłącznie środki powierzchniowo czynne. Takie substancje chemiczne są wykluczone z dziedziny zastosowania metody badawczej krótkiego okresu narażenia (7).
|
|
8.
|
Poza substancjami chemicznymi, o których mowa w pkt 6 i 7, zbiór danych wygenerowany wskutek zastosowania metody badawczej krótkiego okresu narażenia zawiera również dane z badań wewnętrznych przeprowadzonych na 40 mieszaninach, które – w porównaniu z testem działania drażniącego na oko in vivo wg Draize'a – wykazały dokładność na poziomie 88 % (35/40), odsetek wyników fałszywie dodatnich na poziomie 50 % (5/10) oraz odsetek wyników fałszywie ujemnych na poziomie 0 % (0/30), do celów potencjalnego zidentyfikowania mieszanin niewymagających sklasyfikowania w ramach systemów klasyfikacji GHS ONZ / rozporządzenia CLP (9). Dlatego też metodę badawczą krótkiego okresu narażenia można stosować w celu zidentyfikowania mieszanin jako mieszanin nieprzypisanych do żadnej kategorii wg GHS ONZ / rozporządzenia CLP w ramach podejścia oddolnego, z wyjątkiem mieszanin w postaci stałej innych niż mieszaniny składające się wyłącznie ze środków powierzchniowo czynnych z uwagi na rozszerzenie stosowanego ograniczenia na substancje stałe. Ponadto mieszaniny, w których skład wchodzą substancje o prężności par przekraczającej 6 kPa, powinny być ocenianie w taki sposób, aby zagwarantować uniknięcie potencjalnych przypadków zaniżenia prognoz, przy czym w takiej sytuacji odpowiednie uzasadnienie należy sporządzać stosownie do specyfiki danego przypadku.
|
|
9.
|
Metody badawczej krótkiego okresu narażenia nie można stosować do identyfikacji badanych substancji chemicznych jak substancji należących do kategorii 2 wg GHS ONZ / rozporządzenia CLP lub do kategorii 2A (podrażniające oczy) lub 2B (delikatnie podrażniające oczy) wg GHS ONZ z uwagi na znaczną liczbę substancji chemicznych należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP, które wskutek zaniżenia prognoz są klasyfikowane jako należące do kategorii 2, 2A lub 2B wg GHS ONZ / rozporządzenia CLP, oraz znaczną liczbę substancji chemicznych nieprzypisanych do żadnej kategorii, które z uwagi na zawyżenie prognoz są klasyfikowane jako należące do kategorii 2, 2A lub 2B (7). W tym celu konieczne może być przeprowadzenie dodatkowego badania, z wykorzystaniem innej odpowiedniej metody.
|
|
10.
|
Metoda badawcza krótkiego okresu narażenia jest odpowiednia dla badanych substancji chemicznych, które zostały rozpuszczone lub jednolicie zawieszone przez co najmniej 5 minut w soli fizjologicznej, 5 % roztworze sulfotlenku dimetylu w soli fizjologicznej lub oleju mineralnym. Metoda badawcza krótkiego okresu narażenia nie jest odpowiednia dla nierozpuszczalnych badanych substancji chemicznych ani dla badanych substancji chemicznych, które nie mogą zostać jednolicie zawieszone przez co najmniej 5 minut w soli fizjologicznej, 5 % roztworze sulfotlenku dimetylu w soli fizjologicznej lub oleju mineralnym. Stosowanie oleju mineralnego w ramach metody badawczej krótkiego okresu narażenia uznaje się za dopuszczalne z uwagi na krótki okres narażenia. Dlatego też metodę badawczą krótkiego okresu narażenia uznaje się za odpowiednią do przewidywania potencjalnego działania szkodliwego dla oczu nierozpuszczalnych w wodzie badanych substancji chemicznych (np. alkoholi tłuszczowych o długich łańcuchach lub ketonów), o ile są one mieszalne z co najmniej jednym z trzech zaproponowanych powyżej rozpuszczalników (4).
|
|
11.
|
Termin „badana substancja chemiczna” jest używany w niniejszej metodzie badawczej w odniesieniu do substancji będącej przedmiotem badania (68), i nie jest powiązany z zastosowaniem metody badawczej krótkiego okresu narażenia do badania substancji lub mieszanin.
|
ZASADA BADANIA
|
12.
|
Metoda badawcza krótkiego okresu narażenia to test in vitro bazujący na cytotoksyczności, który przeprowadza się na zlewającej się warstwie komórek rogówki królika spreparowanej przez Statens Seruminstitut (SIRC) hodowanej na 96-dołkowej poliwęglanowej mikropłytce (4). Po pięciominutowym okresie narażenia na działanie badanej substancji chemicznej przeprowadza się ilościowy pomiar cytotoksyczności rozumianej jako względna żywotność komórek SIRC w ramach badania MTT (4). Obniżona żywotność komórek jest wykorzystywana do przewidywania potencjalnych działań niepożądanych, które mogą doprowadzić do uszkodzenia oczu.
|
|
13.
|
Zgromadzone informacje wskazują, że 80 % roztworu zakroplonego do oka królika jest wydalane za pośrednictwem worka spojówkowego w okresie od trzech do czterech minut, podczas gdy ponad 80 % roztworu zakroplonego do oka człowieka jest wydalane w okresie od jednej minuty do dwóch minut (10). Metoda badawcza krótkiego okresu narażenia ma na celu zbliżenie wartości tych okresów narażenia i wykorzystanie cytotoksyczności w charakterze punktu końcowego służącego ocenieniu skali uszkodzenia komórek SIRC po pięciominutowym okresie narażenia na oddziaływanie badanej substancji chemicznej.
|
WYKAZANIE BIEGŁOŚCI
|
14.
|
Przed przystąpieniem do rutynowego stosowania metody badawczej krótkiego okresu narażenia opisanej w niniejszej metodzie badawczej laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo klasyfikując jedenaście substancji wskazanych w tabeli 1. Wspomniane substancje dobrano w taki sposób, by reprezentowały pełne spektrum reakcji na poważne uszkodzenie oczu lub podrażnienie oczu w oparciu o wyniki badań na oku królika in vivo (wytyczna TG 405) oraz system klasyfikacji GHS ONZ / rozporządzenia CLP (1). Przy wyborze substancji chemicznej kierowano się również następującymi kryteriami: dostępnością tych substancji na rynku, dostępnością wysokiej jakości danych referencyjnych z badań in vivo oraz dostępnością wysokiej jakości danych z badań in vitro uzyskanych dzięki zastosowaniu metody badawczej krótkiego okresu narażenia (3). Jeżeli figurująca w wykazie substancja jest niedostępna lub jeżeli będzie to uzasadnione, dopuszcza się możliwość zastosowania innej substancji, dla której dostępne są odpowiednie dane referencyjne z badań in vivo i in vitro, pod warunkiem zastosowania tych samych kryteriów co kryteria opisane w tym miejscu.
Tabela 1
Wykaz substancji służących do wykazania biegłości
|
Substancja
|
Numer CAS
|
Klasa chemiczna (69)
|
Stan skupienia
|
Kategoria (70) badania in vivo według klasyfikacji GHS ONZ / rozporządzenia CLP
|
Rozpuszczalnik w badaniu przeprowadzanym zgodnie z metodą badawczą krótkiego okresu narażenia
|
Kategoria badania przeprowadzanego zgodnie z metodą badawczą krótkiego okresu narażenia według klasyfikacji GHS ONZ / rozporządzenia CLP
|
|
Chlorek bezalkoniowy (10 %, roztwór wodny)
|
8 001 -54-5
|
Związek oniowy
|
Ciecz
|
Kategoria 1
|
Sól fizjologiczna
|
Kategoria 1
|
|
Triton X-100 (100 %)
|
9 002 -93-1
|
eter
|
Ciecz
|
Kategoria 1
|
Sól fizjologiczna
|
Kategoria 1
|
|
Czerwień kwasowa 92
|
18 472 -87-2
|
Związek heterocykliczny; związek bromu; związek chloru
|
Stały
|
Kategoria 1
|
Sól fizjologiczna
|
Kategoria 1
|
|
Wodorotlenek sodu
|
1 310 -73-2
|
Zasada; nieorganiczna substancja chemiczna
|
Stały
|
Kategoria 1 (71)
|
Sól fizjologiczna
|
Kategoria 1
|
|
Butyrolakton
|
96-48-0
|
Lakton; związek heterocykliczny
|
Ciecz
|
Kategoria 2A (kategoria 2 zgodnie z rozporządzeniem CLP)
|
Sól fizjologiczna
|
Nie można przewidzieć
|
|
1-oktanol
|
111-87-5
|
alkohol
|
Ciecz
|
Kategoria 2A/B (72) (kategoria 2 zgodnie z rozporządzeniem CLP)
|
Olej mineralny
|
Nie można przewidzieć
|
|
Cyklopentanol
|
96-41-3
|
Alkohol; węglowodór cykliczny
|
Ciecz
|
Kategoria 2A/B (73) (kategoria 2 zgodnie z rozporządzeniem CLP)
|
Sól fizjologiczna
|
Nie można przewidzieć
|
|
Octan 2-etoksyetylu
|
111-15-9
|
Alkohol; eter
|
Ciecz
|
Brak kategorii
|
Sól fizjologiczna
|
Brak kategorii
|
|
Dodekan
|
112-40-3
|
Węglowodór acykliczny
|
Ciecz
|
Brak kategorii
|
Olej mineralny
|
Brak kategorii
|
|
Keton metylowo-izobutylowy
|
108-10-1
|
Keton
|
Ciecz
|
Brak kategorii
|
Olej mineralny
|
Brak kategorii
|
|
Siarczan 1,1-dimetyloguanidyny
|
598-65-2
|
Amidyna; związek siarki
|
Stały
|
Brak kategorii
|
Sól fizjologiczna
|
Brak kategorii
|
|
Skróty: Nr CAS = numer w rejestrze Chemical Abstracts Service.
|
|
PROCEDURA
Przygotowanie jednowarstwowej hodowli komórek
|
15.
|
Do badania przeprowadzanego zgodnie z metodą badawczą krótkiego okresu narażenia należy wykorzystywać linię komórkową rogówki królika SIRC. Zaleca się, aby komórki SIRC były pozyskiwane z dobrze przygotowanego banku komórek, takiego jak zbiór kultur typu amerykańskiego CCL60.
|
|
16.
|
Komórki SIRC hoduje się w kolbie z kulturami zawierającej minimalne niezbędne podłoże Eagle'a uzupełnione 10 % roztworem bydlęcej surowicy płodowej, 2 mM L-glutaminy, 50–100 jednostkami/ml penicyliny i 50–100 μg/ml streptomycyny w temperaturze 37 °C, przy stężeniu CO2 poniżej 5 % i w wilgotnym środowisku. Komórki, które zlały się w kolbie z kulturami, powinny zostać odseparowane za pomocą roztworu trypsyny i kwasu wersenowego, potencjalnie przy wykorzystaniu skrobaka do komórek. Komórki rozmnaża się (np. 2–3 pasaże) w kolbie z kulturami przed ich poddaniem rutynowym badaniom, przy czym od momentu ich rozmrożenia nie powinny być one poddane więcej niż 25 pasażom.
|
|
17.
|
Komórki gotowe do ich poddania badaniu przeprowadzanemu zgodnie z metodą badawczą krótkiego okresu narażenia są następnie przygotowywane przy zachowaniu odpowiedniego zagęszczenia i posiewane na płytkach 96-dołkowych. Zalecane zagęszczenie posiewu komórek wynosi 6,0 × 103 komórek na dołek, jeżeli komórki mają zostać wykorzystane po upływie czterech dni od dnia ich posiania, lub 3,0 × 103 komórek na dołek, jeżeli komórki mają zostać wykorzystane po upływie pięciu dnia od dnia ich posiania, przy objętości kultur wynoszącej 200 μl. Komórki wykorzystywane w badaniu przeprowadzanym zgodnie z metodą badawczą krótkiego okresu narażenia posiane na podłożu przy zachowaniu odpowiedniego zagęszczenia posiewu osiągną poziom konfluencji przekraczający 80 % w momencie przeprowadzenia badania, tj. po upływie czterech lub pięciu dni od posiewu.
|
Podawanie badanej i kontrolnej substancji chemicznej
|
18.
|
Preferowanym rozpuszczalnikiem do rozcieńczania badanych substancji chemicznych lub tworzenia zawiesiny z tych substancji jest sól fizjologiczna. Jeżeli badana substancja chemiczna charakteryzuje się niską rozpuszczalnością lub nie może zostać rozpuszczona ani jednolicie zawieszona przez co najmniej pięć minut w soli fizjologicznej, jako rozpuszczalnik drugiego wyboru wykorzystuje się 5 % roztwór sulfotlenku dimetylowego (nr CAS 67-68-5) w soli fizjologicznej. Jeżeli badane substancje chemiczne nie mogą zostać rozpuszczone ani jednolicie zawieszone przez co najmniej pięć minut w soli fizjologicznej ani w 5 % roztworze sulfotlenku dimetylowego w soli fizjologicznej, jako rozpuszczalnik trzeciego wyboru wykorzystuje się olej mineralny (nr CAS 8042-47-5).
|
|
19.
|
Badane substancje chemiczne rozpuszcza się lub zawiesza się jednolicie w wybranym rozpuszczalniku w stężeniu 5 % (w/w), po czym rozcieńcza się je w ramach seryjnego procesu 10-krotnego rozcieńczania do stężenia 0,5 % i 0,05 %. Każda badana substancja chemiczna musi zostać zbadana zarówno przy stężeniu 5 %, jak i przy stężeniu 0,05 %. Komórki hodowane na płytce 96-dołkowej są poddawane działaniu 200 μl roztworu (lub zawiesiny) badanej substancji chemicznej w stężeniu 5 % albo 0,05 % na dołek przez okres pięciu minut w temperaturze pokojowej. Badane substancje chemiczne (substancje jednoskładnikowe lub substancje wieloskładnikowe lub mieszaniny) uznaje się za czyste substancje oraz substancje rozcieńczone lub zawieszone zgodnie z tą metodą, niezależnie od ich czystości.
|
|
20.
|
Podłoże opisane w pkt 16 wykorzystuje się w odniesieniu do każdej płytki i każdego powtórzenia jako podłoże na potrzeby badania. Ponadto w ramach każdego powtórzenia komórki we wszystkich dołkach poddaje się również działaniu próbek kontroli z rozpuszczalnikiem. W przypadku rozpuszczalników wymienionych w pkt 18 potwierdzono, że nie wywierają one żadnego szkodliwego wpływu na żywotność komórek SIRC.
|
|
21.
|
W ramach metody badawczej krótkiego okresu narażenia należy korzystać z 0,01 % roztworu dodecylo siarczanu sodu w soli fizjologicznej jako kontroli dodatniej w odniesieniu do każdej płytki przy każdym powtórzeniu. Aby obliczyć żywotność komórek w kontroli dodatniej, każda płytka w ramach każdego powtórzenia musi również zawierać kontrolę z rozpuszczalnikiem w postaci soli fizjologicznej.
|
|
22.
|
Przeprowadzenie próby ślepej jest konieczne w celu ustalenia poziomu korekty z tytułu gęstości optycznej; próbę taką powinno się przeprowadzić na dołkach zawierających wyłącznie sól fizjologiczną buforowaną fosforanami, bez wapniu i magnezu (PBS) ani komórek.
|
|
23.
|
Każda próbka (badana substancja chemiczna w stężeniu 5 % i 0,05 %, kontrola podłoża, kontrola z rozpuszczalnikiem i kontrola dodatnia) powinna zostać zbadana trzykrotnie w ramach każdego powtórzenia poprzez narażenie komórek na oddziaływanie 200 μl odpowiedniej badanej lub kontrolnej substancji chemicznej przez okres pięciu minut w temperaturze pokojowej.
|
|
24.
|
Substancje wzorcowe są przydatne do oceny potencjalnego działania drażniącego dla oczu nieznanych chemikaliów z określonej klasy chemicznej lub produktowej lub do oceny względnego potencjalnego działania drażniącego substancji drażniącej dla oczu w określonym zakresie reakcji w postaci podrażnienia.
|
Pomiar żywotności komórek
|
25.
|
Po narażeniu na działanie substancji komórki przemywa się dwukrotnie 200 μl PBS i dodaje się do nich 200 μl roztworu MTT (0,5 mg MTT / ml podłoża). Po upływie dwugodzinnego czasu reakcji w inkubatorze (37 °C, 5 % CO2) roztwór MTT dekantuje się po czym ekstrahuje się z niego formazan, dodając 200 μl roztworu 0,04 N kwasu chlorowodorowego – izopropanolu przez 60 minut bez dostępu światła w temperaturze pokojowej, po czym dokonuje się pomiaru absorbancji roztworu formazanu w MTT przy 570 nm za pomocą czytnika płytek. Do interferencji badanych substancji chemicznych w teście MTT (wywołanej barwnikami lub bezpośrednimi reduktorami MTT) dochodzi wyłącznie w przypadku zatrzymania znacznej ilości badanej substancji chemicznej w układzie badawczym po zakończeniu płukania po okresie narażenia – do takiej sytuacji może dojść w przypadku tkanek ludzkiej rogówki zrekonstruowanej w trójwymiarze lub zrekonstruowanego ludzkiego naskórka, ale nie w przypadku dwuwymiarowych kultur komórkowych wykorzystywanych w metodzie badawczej krótkiego okresu narażenia.
|
Interpretacja wyników i model prognozowania
|
26.
|
Wartości gęstości optycznej (OD) otrzymane dla każdej badanej substancji chemicznej są następnie wykorzystywane do obliczenia żywotności komórek w stosunku do kontroli z rozpuszczalnikiem, dla której przyjmuje się 100 %. Względną żywotność komórek wyraża się jako odsetek stanowiący iloraz wartości OD badanej substancji chemicznej i wartości OD kontroli z rozpuszczalnikiem po odjęciu wartości OD próby ślepej od obydwu tych wartości.

Podobnie względną żywotność komórek każdej kontroli z rozpuszczalnikiem wyraża się jako odsetek stanowiący iloraz wartości OD każdej kontroli z rozpuszczalnikiem i wartości OD kontroli podłoża po odjęciu wartości OD próby ślepej od obydwu tych wartości.
|
|
27.
|
Należy przeprowadzić trzy niezależne powtórzenia, każde na trzech dołkach stanowiących kontrpróbę (tj. n = 9). Średnią arytmetyczną obliczoną dla trzech dołków w ramach każdego niezależnego powtórzenia dla każdej badanej substancji chemicznej i kontroli z rozpuszczalnikiem wykorzystuje się do obliczenia średniej arytmetycznej względnej żywotności komórek. Ostateczną średnią arytmetyczną żywotności komórek oblicza się na podstawie wyników trzech niezależnych powtórzeń.
|
|
28.
|
Wartości graniczne żywotności komórek służące do identyfikowania badanych substancji chemicznych jako powodujące poważne uszkodzenie oczu (kategoria 1 wg GHS ONZ / rozporządzenia CLP) oraz badanych substancji chemicznych, które nie wymagają zaklasyfikowania do substancji powodujących podrażnienie lub poważne uszkodzenie oczu (brak kategorii wg GHS ONZ / rozporządzenia CLP), podano poniżej.
|
Tabela 2
Model prognozowania dla metody badawczej krótkiego okresu narażenia
|
Żywotność komórek
|
Klasyfikacja GHS ONZ / rozporządzenie CLP
|
Możliwość zastosowania
|
|
Przy 5 %
|
Przy 0,05 %
|
|
> 70 %
|
> 70 %
|
Brak kategorii
|
Substancje i mieszaniny, z wyjątkiem: (i) wysoce lotnych substancji charakteryzujących się prężnością par powyżej 6 kPa (74); oraz (ii) stałych chemikaliów (substancji i mieszanin) innych niż środki powierzchniowo czynne i mieszaniny składające się wyłącznie ze środków powierzchniowo czynnych
|
|
≤ 70 %
|
> 70 %
|
Nie można przewidzieć
|
Brak możliwości zastosowania
|
|
≤ 70 %
|
≤ 70 %
|
Kategoria 1
|
Substancje i mieszaniny (75)
|
Kryteria dopuszczalności
|
29.
|
Wyniki badania uznaje się za dopuszczalne w przypadku spełnienia wszystkich poniższych kryteriów:
|
a)
|
gęstość optyczna kontroli podłoża (narażonej na oddziaływanie podłoża) powinna wynosić 0,3 lub być wyższa po odjęciu gęstości optycznej próby ślepej;
|
|
b)
|
żywotność kontroli z rozpuszczalnikiem powinna odpowiadać 80 % żywotności kontroli podłoża lub być wyższa. Jeżeli w ramach każdego powtórzenia wykorzystuje się szereg kontroli z rozpuszczalnikiem, aby daną badaną substancję chemiczną poddaną badaniu z wykorzystaniem tych rozpuszczalników można było uznać za kwalifikującą się, każda z tych kontroli powinna charakteryzować się żywotnością komórek wyższą niż 80 %;
|
|
c)
|
żywotność komórek uzyskana w kontroli dodatniej (0,01 % SDS) powinna mieścić się w przedziale dwóch odchyleń standardowych średniej historycznej. Górne i dolne limity dopuszczalności kontroli dodatniej powinny być często aktualizowane, tj. co najmniej co trzy miesiące lub za każdym razem, gdy dopuszczalne badanie przeprowadza się w laboratoriach, w których rzadko przeprowadza się badania (tj. rzadziej niż raz na miesiąc). Jeżeli laboratorium nie przeprowadziło dostatecznie dużej liczby doświadczeń, aby ustalić istotny statystycznie rozkład danych z kontroli dodatniej, dopuszcza się możliwość zastosowania górnych i dolnych limitów dopuszczalności ustanowionych przez podmiot, który opracował daną metodę badawczą, tj. od 21,1 % do 62,3 % zgodnie z historycznymi danymi laboratoryjnymi znajdującymi się w posiadaniu tego podmiotu, natomiast rozkład wewnętrzny można określić w trakcie pierwszych rutynowych badań.
|
|
d)
|
Odchylenie standardowe ostatecznej żywotności komórek wyprowadzone z trzech niezależnych powtórzeń powinno być niższe niż 15 % zarówno w przypadku 5 % stężenia badanej substancji chemicznej, jak i w przypadku 0,05 % stężenia tej substancji.
Jeżeli przynajmniej jedno z powyższych kryteriów nie zostało spełnione, wyniki należy odrzucić, po czym należy przeprowadzić kolejne trzy niezależne powtórzenia.
|
|
DANE I SPRAWOZDAWCZOŚĆ
Dane
|
30.
|
Należy przedstawić dane dotyczące poszczególnych dołków (np. wartości żywotności komórek) dla każdego powtórzenia, jak również średnią wszystkich wyników, wartość SD oraz informacje na temat klasyfikacji.
|
Sprawozdanie z badania
|
31.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Badana substancja chemiczna oraz substancje kontrolne:
|
—
|
Substancja jednoskładnikowa: dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(y) w rejestrze CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
—
|
substancja wieloskładnikowa, UVCB i mieszanina: opisana w miarę możliwości np. za pomocą nazwy chemicznej (zob. powyżej), czystości, występowania ilościowego oraz istotnych właściwości fizykochemicznych składników (zob. powyżej), w zakresie, w jakim są dostępne;
|
|
—
|
stan skupienia, lotność, pH, LogP, masa cząsteczkowa, klasa chemiczna oraz dodatkowe istotne właściwości fizykochemiczne mające znaczenie dla przebiegu badania w zakresie, w jakim są dostępne;
|
|
—
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
—
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
—
|
dane na temat warunków przechowywania i stabilności w zakresie, w jakim są dostępne;
|
|
|
|
Warunki i procedury metody badawczej
|
—
|
nazwa i adres sponsora i placówki przeprowadzającej badanie oraz dane kierownika badań;
|
|
—
|
opis zastosowanej metody badawczej;
|
|
—
|
informacje na temat zastosowanej linii komórkowej, jej źródła, liczby pasaży oraz konfluencji komórek wykorzystanych w badaniu;
|
|
—
|
szczegółowe dane na temat zastosowanej procedury badawczej;
|
|
—
|
liczba zastosowanych powtórzeń i użytych kontrprób;
|
|
—
|
zastosowane stężenia badanej substancji chemicznej (jeżeli różnią się od zalecanych);
|
|
—
|
uzasadnienie wyboru rozpuszczalnika w odniesieniu do każdej badanej substancji chemicznej;
|
|
—
|
czas narażenia na działanie badanej substancji chemicznej (jeżeli różni się od zalecanego);
|
|
—
|
opis wszelkich modyfikacji procedury badawczej;
|
|
—
|
opis zastosowanych kryteriów oceny i podejmowania decyzji;
|
|
—
|
odniesienie do historycznych średnich wyników kontroli dodatniej i odchylenia standardowego (SD):
|
|
—
|
wykazanie biegłości laboratorium w zakresie stosowania metody badawczej (np. poprzez badanie substancji służących do wykazania biegłości) lub wykazanie odtwarzalnego przeprowadzenia metody badawczej w miarę upływu czasu.
|
|
|
|
Wyniki
|
—
|
dla każdej badanej substancji chemicznej i dla każdej substancji kontrolnej, a także dla każdego badanego stężenia, należy przedstawić tabelę zawierającą indywidualne wartości OD dla każdego dołka stanowiącego kontrpróbę, średnią arytmetyczną wartości OD dla każdego niezależnego powtórzenia, procentową wartość żywotności komórek dla każdego niezależnego powtórzenia oraz ostateczną średnią arytmetyczną procentowej żywotności komórek i SD dla trzech powtórzeń;
|
|
—
|
wyniki kontroli podłoża, rozpuszczalnika i kontroli dodatniej potwierdzające spełnienie odpowiednich kryteriów dopuszczalności badania;
|
|
—
|
opis innych zaobserwowanych skutków;
|
|
—
|
ustalona na tej podstawie ogólna klasyfikacja odnosząca się do zastosowanego modelu prognozowania / zastosowanych kryteriów podejmowania decyzji.
|
|
|
BIBLIOGRAFIA
|
(1)
|
Organizacja Narodów Zjednoczonych (ONZ) (2013), Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS), wydanie piąte zmienione; Nowy Jork i Genewa: publikacje Organizacji Narodów Zjednoczonych. ISBN: 978-92-1-117006-1. Dostępne na stronie internetowej: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches.„Toxicol. In Vitro” nr 24, s. 1–9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. „J. Immunol. Methods” nr 65, s. 55–63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. „Toxicol. In Vitro” nr 22, s. 760–770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. „Toxicol. In Vitro” nr 25, s. 796–809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. „Toxicol. In Vitro” nr 27, s. 1855–1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Dostępne na stronie internetowej: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Rozdział B.5 niniejszego załącznika, Działanie żrące/silnie drażniące na oczy.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS i Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. „J. Pharm. Sci” s. 1648–1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Sprawozdanie techniczne (nr 48 (2)), Bruksela, Belgia.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. „Fundam. Appl. Toxicol.” nr 18, s. 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 34. Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 263. Organizacja Współpracy Gospodarczej i Rozwoju.
|
Dodatek
DEFINICJE
Dokładność
: stopień zgodności wyników zastosowanej metody badawczej z przyjętymi wartościami odniesienia. Jest to miara efektywności metody badawczej i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na oznaczenie odsetka prawidłowych wyników uzyskiwanych przy użyciu metody badawczej (13).
Substancja wzorcowa
: substancja używana jako wzorzec do porównań z badaną substancją chemiczną. Substancja wzorcowa powinna cechować się następującymi właściwościami: (i) stałe i wiarygodne źródło(-a); (ii) podobieństwo strukturalne i funkcjonalne do badanej klasy substancji; (iii) znane właściwości fizyczne i chemiczne; (iv) dane potwierdzające występowanie znanych skutków oraz (v) znany potencjał w zakresie pożądanych reakcji.
Podejście oddolne
: stopniowe podejście stosowane w odniesieniu do badanej substancji chemicznej, w przypadku której podejrzewa się, że nie wymaga zaklasyfikowania jako substancja powodująca podrażnienie lub poważne uszkodzenie oka; takie podejście polega na odróżnieniu substancji chemicznych niewymagających zaklasyfikowania (wynik ujemny) od pozostałych substancji chemicznych (wynik dodatni).
Substancja chemiczna
: substancja lub mieszanina.
Podrażnienie oka
: zmiany w oku spowodowane zaaplikowaniem badanej substancji chemicznej na wierzchnią warstwę oka, które są w pełni odwracalne w ciągu 21 dni po zaaplikowaniu. Określenie używane zamiennie z określeniem „odwracalne skutki działania na oczy” oraz ze sformułowaniem „kategoria 2 wg GHS ONZ / rozporządzenia CLP”.
Odsetek wyników fałszywie ujemnych
: odsetek wszystkich substancji chemicznych dających wynik dodatni fałszywie zidentyfikowanych po zastosowaniu metody badawczej jako dające wynik ujemny. Jest to jeden ze wskaźników efektywności metody badawczej.
Odsetek wyników fałszywie dodatnich
: odsetek wszystkich substancji chemicznych dających wynik ujemny fałszywie zidentyfikowanych po zastosowaniu metody badawczej jako dające wynik dodatni. Jest to jeden ze wskaźników efektywności metody badawczej.
Zagrożenie
: nieodłączna właściwość czynnika lub sytuacja, która może potencjalnie doprowadzić do niekorzystnych skutków w przypadku narażenia organizmu, systemu lub (sub) populacji na taki czynnik.
Kontrola podłoża
: kontrpróba niepoddana działaniu badanej substancji chemicznej zawierająca wszystkie składniki układu badawczego. Próbka ta jest przetwarzana razem z próbkami poddanymi działaniu badanej substancji chemicznej oraz z innymi próbkami kontrolnymi w celu określenia, czy rozpuszczalnik wchodzi w reakcję z układem badawczym.
Mieszanina
: mieszanina lub roztwór, które składają się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
MTT
: bromek 3-(4,5-dimetylotiazol-2-ilo)-2,5-difenylotetrazoliowy MTT.
Substancja wieloskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥ 10 % (w/w) i < 80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się w wyniku zmieszania co najmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
OD
: gęstość optyczna.
Kontrola dodatnia
: kontrpróba zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że wywołuje reakcję dodatnią. Aby zapewnić możliwość oceny zmienności reakcji w kontroli dodatniej w czasie, siła reakcji w postaci silnego działania drażniącego nie może być zbyt duża.
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono znaczące i użyteczne z punktu widzenia określonego celu. Jest to zakres, w jakim badanie prawidłowo mierzy lub przewiduje biologiczny oczekiwany skutek. Istotność obejmuje uwzględnienie dokładności (zgodności) metody badawczej (10).
Wiarygodność
: miary zakresu, w jakim metoda badawcza może być przeprowadzana w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jej przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją poprzez obliczenie odtwarzalności międzylaboratoryjnej i wewnątrzlaboratoryjnej (13).
Czułość
: odsetek wszystkich substancji chemicznych dających wynik dodatni / aktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą badania. Jest to miara dokładności metody badawczej, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności metody badawczej (10).
Poważne uszkodzenie oczu
: uszkodzenie tkanki oka lub poważne fizyczne pogorszenie widzenia spowodowane zaaplikowaniem badanej substancji chemicznej na przednią powierzchnię oka, które nie jest w pełni odwracalne w ciągu 21 dni po zaaplikowaniu. Określenie używane zamiennie z określeniem „nieodwracalne skutki działania na oczy” oraz ze sformułowaniem „kategoria 1 wg GHS ONZ / rozporządzenia CLP”.
Grupa kontrolna z rozpuszczalnikiem/nośnikiem
: próbka niepoddana działaniu badanej substancji, zawierająca wszystkie składniki układu badawczego, w tym rozpuszczalnik lub nośnik, która jest przetwarzana razem z próbkami poddanymi działaniu badanej substancji oraz z innymi próbkami kontrolnymi w celu określenia wyjściowej reakcji dla próbek poddanych działaniu badanej substancji chemicznej rozpuszczonej w tym samym rozpuszczalniku lub nośniku. W przypadku badania z jednoczesną kontrolą podłoża próbka ta wykazuje również, czy rozpuszczalnik lub nośnik wchodzi w reakcję z układem badawczym.
Swoistość
: odsetek wszystkich substancji chemicznych dających wynik ujemny / nieaktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą badania. Jest to miara dokładności metody badawczej, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności metody badawczej (13).
Substancja
: pierwiastek chemiczny i jego związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania trwałości produktu i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem wszelkich rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Środek powierzchniowo czynny
: zwany także surfaktantem, jest to substancja chemiczna, np. detergent, która może zmniejszać napięcie powierzchniowe cieczy, umożliwiając tym samym jej pienienie lub przenikanie w ciała stałe; substancja ta jest także zwana środkiem zwilżającym.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
Wielopoziomowa strategia badań
: strategia badań sekwencyjnych, w ramach której wszystkie istniejące informacje na temat badanej substancji chemicznej są analizowane na każdym poziomie w określonym porządku z zastosowaniem procesu uwzględniającego wagę dowodów w celu określenia, czy dostępna jest wystarczająca ilość informacji do podjęcia decyzji o klasyfikacji zagrożenia, przed przejściem do następnego poziomu. Jeżeli na podstawie dostępnych informacji można przypisać badanej substancji chemicznej potencjał w zakresie wywołania podrażnienia, dodatkowe badania nie są wymagane. Jeżeli na podstawie dostępnych informacji nie można przypisać badanej substancji chemicznej potencjału w zakresie wywołania podrażnień, przeprowadza się procedurę badań sekwencyjnych na zwierzętach do momentu, w którym będzie można dokonać jednoznacznej klasyfikacji.
Podejście odgórne
: stopniowe podejście stosowane w odniesieniu do badanej substancji chemicznej, w przypadku której podejrzewa się, że powoduje poważne uszkodzenie oczu; takie podejście polega na odróżnieniu substancji chemicznych powodujących poważne uszkodzenie oczu (wynik dodatni) od pozostałych substancji chemicznych (wynik ujemny).
Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ)
: system, w ramach którego proponuje się klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz omawia się odpowiednie elementy komunikacyjne, takie jak: piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki substancji i produktów niebezpiecznych, aby przekazać informacje na temat ich szkodliwego działania w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) i środowiska (1).
Kategoria 1 wg GHS ONZ / rozporządzenia CLP
: zob. „Poważne uszkodzenie oczu”.
Kategoria 2 wg GHS ONZ / rozporządzenia CLP
: zob. „Działanie drażniące na oczy”.
Brak kategorii wg GHS ONZ / rozporządzenia CLP
: Substancje chemiczne, które nie zostały zaklasyfikowane jako należące do kategorii 1 ani 2 wg GHS ONZ / rozporządzenia CLP (lub do kategorii 2A ani 2B wg GHS ONZ).
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
B.69 METODA BADAWCZA WYKORZYSTYWANA W RAMACH BADANIA NA MODELU ZREKONSTRUOWANEGO LUDZKIEGO NABŁONKA PRZYPOMINAJĄCEGO ROGÓWKĘ (RhCE) STOSOWANA DO IDENTYFIKACJI SUBSTANCJI CHEMICZNYCH NIEWYMAGAJĄCYCH KLASYFIKACJI I OZNAKOWANIA POD WZGLĘDEM DZIAŁANIA DRAŻNIĄCEGO NA OCZY LUB POWAŻNYCH USZKODZEŃ OCZU
WPROWADZENIE
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w wytycznej OECD dotyczącej badań (TG) nr 492 (2017). Poważne uszkodzenie oczu oznacza uszkodzenie tkanki oka lub poważne fizyczne pogorszenie widzenia spowodowane zaaplikowaniem badanej substancji chemicznej na przednią powierzchnię oka, które nie jest w pełni odwracalne w ciągu 21 dni po zaaplikowaniu, zgodnie z definicją zawartą w Globalnie Zharmonizowanym Systemie Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ) (1) oraz w rozporządzeniu Unii Europejskiej (WE) nr 1272/2008 w sprawie klasyfikacji, oznakowania i pakowania substancji i mieszanin (rozporządzenie CLP) (76). Ponadto zgodnie z GHS ONZ i rozporządzeniem CLP podrażnienie oka oznacza zmiany w oku spowodowane zaaplikowaniem badanej substancji chemicznej na wierzchnią warstwę oka, które są w pełni odwracalne w ciągu 21 dni po zaaplikowaniu. Badane substancje chemiczne powodujące poważne uszkodzenie oczu klasyfikuje się jako substancje należące do kategorii 1 wg GHS ONZ i rozporządzenia CLP, natomiast substancje chemiczne powodujące podrażnienie oczu klasyfikuje się jako substancje należące do kategorii 2 wg GHS ONZ i rozporządzenia CLP. Badane substancje chemiczne niesklasyfikowane jako powodujące podrażnienie lub poważne uszkodzenie oczu, definiuje się jako te substancje, które nie spełniają wymogów zaklasyfikowania jako należące do kategorii 1 lub 2 (2A lub 2B) wg GHS ONZ i rozporządzenia CLP, tj. określa się je jako nienależące do żadnej kategorii – brak kategorii wg GHS ONZ i rozporządzenia CLP.
|
|
2.
|
Ocenę służącą ustaleniu, czy doszło do poważnego uszkodzenia oczu / podrażnienia oczu, przeprowadza się zazwyczaj na zwierzętach laboratoryjnych (metoda badawcza B.5 (2)). Na wybór najbardziej odpowiedniej metody badawczej i zastosowanie niniejszej metody badawczej należy patrzeć w kontekście wytycznych OECD w sprawie zintegrowanych podejść do badań i oceny dotyczących poważnych uszkodzeń i podrażnień oczu (39).
|
|
3.
|
Niniejsza metoda badawcza opisuje procedurę badania in vitro zapewniającą możliwość zidentyfikowania chemikaliów (substancji i mieszanin) niewymagających sklasyfikowania i oznakowania jako chemikalia powodujące podrażnienie oczu lub poważne uszkodzenie oczu zgodnie z GHS ONZ i rozporządzeniem CLP. Wykorzystuje się w niej zrekonstruowany ludzki nabłonek przypominający rogówkę (RhCE), który wiernie imituje właściwości histologiczne, morfologiczne, biochemiczne i fizjologiczne właściwości nabłonka rogówki ludzkiej. Zweryfikowano również cztery inne metody badawcze in vitro uznawane za potwierdzone naukowo, przyjmując je jako metody badawcze B.47 (3), B.48 (4), B.61 (5) i B.68 (6) odnoszące się do punktu końcowego polegającego na poważnym uszkodzeniu oczu / podrażnieniu oczu człowieka.
|
|
4.
|
W niniejszej metodzie badawczej uwzględniono dwa zweryfikowane badania przeprowadzone przy wykorzystaniu dostępnych na rynku modeli RhCE. Przeprowadzono badania walidacyjne służące do ocenienia stopnia podrażnienia oczu / poważnego uszkodzenia oczu przy wykorzystaniu badania działania drażniącego na oczy EpiOcular™ oraz badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ (7)(8)(9)(10)(11)(12)(13). W ramach każdego z tych badań w charakterze układu badawczego wykorzystywano dostępne na rynku wytwory tkankowe RhCE, które określa się poniżej jako zwalidowane metody referencyjne – odpowiednio VRM 1 i VRM 2. Na podstawie wyników wspomnianych badań walidacyjnych oraz ich niezależnej wzajemnej oceny (9)(12) stwierdzono, że badanie działania drażniącego na oczy EpiOcular™ i badanie działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ umożliwiają prawidłowe zidentyfikowanie chemikaliów (substancji i mieszanin) niewymagających klasyfikacji i oznakowania pod względem działania drażniącego na oczy lub poważnego uszkodzenia oczu zgodnie z GHS ONZ, oraz zarekomendowano uznanie tych badań za potwierdzone naukowo w tym celu (13).
|
|
5.
|
Obecnie powszechnie uznaje się, że w dającej się przewidzieć przyszłości żadna pojedyncza metoda badawcza in vitro nie będzie w stanie w pełni zastąpić testu działania drażniącego na oko in vivo wg Draize’a (2)(14) na potrzeby prognozowania pełnego zakresu działania drażniącego różnych klas chemicznych. Strategiczne połączenia szeregu alternatywnych metod badawczy w ramach (wielopoziomowych) strategii badań takich jak podejście oddolne/odgórne mogą jednak być w stanie w pełni zastąpić test działania drażniącego na oko wg Draize’a (15). Podejście oddolne (15) ma w założeniu być stosowane wówczas, gdy na podstawie istniejących informacji oczekuje się, że substancja chemiczna nie będzie wywoływała podrażnienia oczu w stopniu wymagających jej sklasyfikowania, podczas gdy podejście odgórne (15) ma być stosowane wówczas, gdy na podstawie istniejących informacji oczekuje się, że substancja chemiczna doprowadzi do poważnego uszkodzenia oczu. W celu zidentyfikowania substancji chemicznych niewymagających sklasyfikowania jako substancje wywołujące podrażnienie oczu lub poważne uszkodzenie oczu wg klasyfikacji GHS ONZ / rozporządzenia CLP (brak kategorii) bez konieczności przeprowadzenia dalszych badań zaleca się stosowanie badania działania drażniącego na oczy EpiOcular™ i badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ w ramach strategii badania zakładającej stosowanie podejścia oddolnego/odgórnego zaproponowanej przez Scotta et al., np. strategii, w której podejście oddolne stanowi pierwszy etap, lub strategii, w której podejście odgórne stanowi jeden z ostatnich etapów (15). Badanie działania drażniącego na oczy EpiOcular™ oraz badanie działania drażniącego na oczy na ludzkim nabłonku rogówki nie zapewniają jednak możliwości rozróżnienia między kategorią 1 wg GHS ONZ / rozporządzeniem CLP (poważne uszkodzenie oczu) a kategorią 2 wg GHS ONZ / rozporządzeniem CLP (działanie drażniące na oczy). Takie rozróżnienie będzie musiało zostać dokonane na innym poziomie strategii badania (15). Badana substancja chemiczna uznana za wymagającą sklasyfikowania jako powodująca podrażnienie oczu / poważne uszkodzenie oczu w ramach badania działania drażniącego na oczy EpiOcular™ lub w ramach badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ będzie zatem musiała zostać poddana dodatkowym badaniom (in vitro lub in vivo), aby można było sformułować ostateczny wniosek (brak kategorii, kategoria 2 lub kategoria 1 wg GHS ONZ / rozporządzenia CLP), na przykład przy wykorzystaniu metody badawczej B.47, B.48, B.61 lub B.68.
|
|
6.
|
Niniejsza metoda badawcza ma na celu opisanie procedur stosowanych do oceny potencjalnego działania szkodliwego dla oczu badanej substancji chemicznej na podstawie jej potencjału wywoływania cytotoksyczności w wytworze tkankowym RhCE zgodnie z pomiarem w ramach testu MTT (16) (zob. pkt 21). Żywotność wytworu tkankowego RhCE po narażeniu na działanie badanej substancji chemicznej ustala się w porównaniu z tkankami poddanymi działaniu substancji wykorzystywanej w ramach kontroli ujemnej (procentowa wartość żywotności), po czym wartość tą stosuje się przy przewidywaniu potencjalnego działania szkodliwego dla oczu badanej substancji chemicznej.
|
|
7.
|
Aby usprawnić proces walidacji nowych lub zmienionych metod przeprowadzania badań in vitro bazujących na RHCE podobnych do badania działania drażniącego na oczy EpiOcular™ i badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ i aby umożliwić terminowe modyfikowanie wytycznych OECD dotyczących badań nr 492 w celu uwzględnienia tych metod badawczych, można skorzystać z dostępnych standardów wykonywania badań (17) opracowanych zgodnie z zasadami ustanowionymi w wytycznych OECD nr 34 (18). Wzajemne uznawanie danych (MAD) zgodnie z porozumieniem OECD będzie możliwe wyłącznie w przypadku badań zweryfikowanych zgodnie ze standardami wykonywania badań, jeżeli te badania zostały poddane przeglądowi i włączone do odpowiednich wytycznych OECD dotyczących badań.
|
DEFINICJE
|
8.
|
Definicje znajdują się w dodatku 1.
|
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
9.
|
Niniejszą metodę badawczą przeprowadza się na dostępnych na rynku trójwymiarowych wytworach tkankowych RhCE wytwarzanych przy wykorzystaniu pierwotnych keratynocytów naskórka ludzkiego (tj. EpiOcular™ OCL-200) albo unieśmiertelnionych komórek nabłonkowych ludzkiej rogówki (tj. SkinEthic™ HCE/s). Struktura wytworów tkankowych RhCE EpiOcular™ OCL-200 i SkinEthic™ HCE/S jest podobna do trójwymiarowej struktury komórek nabłonkowych rogówki in vivo, a wytwory te wytwarza się przy wykorzystaniu komórek gatunków będących przedmiotem zainteresowania (19)(20). Badania zapewniają ponadto możliwość bezpośredniego pomiaru poziomu cytotoksyczności wywołanej przeniknięciem substancji chemicznej przez rogówkę, co skutkuje uszkodzeniem komórek i tkanek; ogólny wynik badania in vivo przejawiający się poważnym uszkodzeniem oczu / podrażnieniem oczu zależy od efektu cytotoksycznego, do którego dochodzi w następstwie przeniknięcia substancji chemicznej przez rogówkę. Choć do uszkodzenia komórek może dojść w następstwie szeregu działań (zob. pkt 20), cytotoksyczność odgrywa istotną – o ile nie najistotniejszą – mechanistyczną rolę w procesie ustalania, czy działanie substancji chemicznej wywołuje ogólną reakcję w postaci poważnego uszkodzenia oczu / podrażnienia oczu, która w badaniu in vivo przejawia się głównie zmętnieniem rogówki, zapaleniem tęczówki, zaczerwienieniem błony spojówkowej lub obrzękiem spojówki, niezależnie od procesów fizykochemicznych leżących u podstaw uszkodzenia tkanek.
|
|
10.
|
W ramach badania walidacyjnego leżącego u podstaw niniejszej metody badawczej zbadano szerokie spektrum substancji chemicznych obejmujące wiele różnych rodzajów chemikaliów, klas chemicznych, mas cząsteczkowych, LogP, struktur chemicznych itp. Baza danych służących do walidacji badania działania drażniącego na oczy EpiOcular™ zawierała łącznie 113 substancji chemicznych i obejmowała 95 różnych organicznych grup funkcyjnych zgodnie z wynikami analizy zestawu narzędzi OECD QSAR (8). Choć substancje jednoskładnikowe stanowiły większość tych substancji chemicznych, w badaniu uwzględniono również szereg substancji wieloskładnikowych (w tym 3 homopolimery, 5 kopolimerów i 10 quasi-polimerów). Jeżeli chodzi o stan skupienia i kategorie wg GHS ONZ / rozporządzenia CLP, 113 zbadanych substancji chemicznych sklasyfikowano w następujący sposób: 13 substancji jako ciecze należące do kategorii 1, 15 substancji jako substancje stałe należące do kategorii 1, 6 substancji jako ciecze należące do kategorii 2A, 10 substancji jako substancje stałe należące do kategorii 2A, 7 substancji jako ciecze należące do kategorii 2B, 7 substancji jako ciała stałe należące do kategorii 2B, 27 substancji jako ciecze nieprzypisane do żadnej kategorii i 28 substancji jako ciała stałe nienależące do żadnej kategorii (8). Baza danych służących do walidacji badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ zawierała łącznie 200 substancji chemicznych i obejmowała 165 różnych organicznych grup funkcyjnych (8)(10)(11). Choć substancje jednoskładnikowe stanowiły większość tych substancji chemicznych, w badaniu uwzględniono również szereg substancji wieloskładnikowych (w tym 10 polimerów). Jeżeli chodzi o stan skupienia i kategorie wg GHS ONZ / rozporządzenia CLP, 200 zbadanych substancji chemicznych sklasyfikowano w następujący sposób: 27 substancji jako ciecze należące do kategorii 1, 24 substancji jako substancje stałe należące do kategorii 1, 19 substancji jako ciecze należące do kategorii 2A, 10 substancji jako substancje stałe należące do kategorii 2A, 9 substancji jako ciecze należące do kategorii 2B, 8 substancji jako ciała stałe należące do kategorii 2B, 50 substancji jako ciecze nieprzypisane do żadnej kategorii i 53 substancje jako ciała stałe nienależące do żadnej kategorii (10)(11).
|
|
11.
|
Niniejsza metoda badawcza ma zastosowanie do substancji i mieszanin, a także do ciał stałych, cieczy, ciał półstałych i wosków. Ciecze mogą być wodne lub niewodne; substancje stałe mogą być rozpuszczalne lub nierozpuszczalne w wodzie. W miarę możliwości substancje stałe należy przed zastosowaniem zmielić na drobnoziarnisty proszek; żadna inna wstępna obróbka próbki nie jest wymagana. W ramach badania walidacyjnego nie oceniano gazów ani aerozoli. Chociaż nie można wykluczyć, że da się je badać z wykorzystaniem technologii RhCE, niniejsza metoda badawcza nie pozwala na badanie gazów i aerozoli.
|
|
12.
|
Badane substancje chemiczne pochłaniające światło w takim samym spektrum co formazan MTT (w warunkach naturalnych lub po poddaniu działaniu substancji) oraz badane substancje chemiczne zdolne do bezpośredniego obniżenia zawartości barwnika przyżyciowego MTT (do formazanu MTT) mogą zakłócać pomiary żywotności tkanek, co może wiązać się z koniecznością zastosowania przystosowanych kontroli w celu dokonania korekty. Rodzaj przystosowanych kontroli, których przeprowadzenie może okazać się konieczne, będzie różnił się w zależności od rodzaju zakłócenia wywoływanego przez badaną substancję chemiczną i od rodzaju procedury zastosowanej w celu określenia ilości formazanu MTT (zob. pkt 36–42).
|
|
13.
|
Wyniki badań przedwalidacyjnych (21)(22) i pełnych badań walidacyjnych (8)(10)(11) potwierdziły, że zarówno badanie działania drażniącego na oczy EpiOcular™, jak i badanie działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ mogą być przeprowadzane również w laboratoriach uznawanych za niedoświadczone w dziedzinie przeprowadzania tego rodzaju testów, a ich wyniki mogą zostać odtworzone w laboratoriach i między laboratoriami. Na podstawie wyników tych badań, poziom odtwarzalności zgodności prognoz, którego można oczekiwać w przypadku badania działania drażniącego na oczy EpiOcular™ w oparciu o dane dotyczące 113 substancji chemicznych mieści się w granicach 95 % wewnątrz laboratoriów i 93 % pomiędzy laboratoriami. Poziom odtwarzalności zgodności prognoz, którego można oczekiwać w przypadku badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ w oparciu o dane dotyczące 120 substancji chemicznych mieści się w granicach 92 % wewnątrz laboratoriów i 95 % pomiędzy laboratoriami.
|
|
14.
|
Badanie działania drażniącego na oczy EpiOcular™ można przeprowadzać w celu zidentyfikowania substancji chemicznych niewymagających sklasyfikowania jako wywołujące podrażnienie oczu lub poważne uszkodzenie oczu zgodnie z systemem klasyfikacji GHS ONZ / systemem klasyfikacji ustanowionym w rozporządzeniu CLP. Biorąc pod uwagę dane pozyskane w ramach badania walidacyjnego (8), badanie działania drażniącego na oczy EpiOcular™ charakteryzowało się ogólną dokładnością na poziomie 80 % (na podstawie wyników badania przeprowadzonego na 112 substancjach chemicznych), czułością na poziomie 96 % (na podstawie wyników badania przeprowadzonego na 57 substancjach chemicznych), odsetkiem wyników fałszywie ujemnych na poziomie 4 % (na podstawie wyników badania przeprowadzonego na 57 substancjach chemicznych), swoistością na poziomie 63 % (na podstawie wyników badania przeprowadzonego na 55 substancjach chemicznych) oraz odsetkiem wyników fałszywie dodatnich na poziomie 37 % (na podstawie wyników badania przeprowadzonego na 55 substancjach chemicznych) w zestawieniu z danymi z referencyjnego badania in vivo na oku królika (metoda badawcza B.5) (2)(14) sklasyfikowanego zgodnie z systemem klasyfikacji GHS ONZ / systemem klasyfikacji ustanowionym w rozporządzeniu CLP. W badaniu, w ramach którego zbadano 97 ciekłych preparatów agrochemicznych w formie użytkowej za pomocą badania działania drażniącego na oczy EpiOcular™, wyniki zastosowania tej metody badawczej w odniesieniu do tego rodzaju mieszanin były zbliżone do wyników badania walidacyjnego (23). 97 postaci użytkowych sklasyfikowano w następujący sposób: 21 postaci użytkowych do kategorii 1, 19 postaci użytkowych do kategorii 2A i 14 postaci użytkowych do kategorii 2B, przy czym 43 postacie użytkowe nie zostały sklasyfikowane do żadnej kategorii zgodnie z systemem klasyfikacji GHS ONZ w zestawieniu z danymi z badania in vivo na oku królika (metoda badawcza B.5) (2)(14). Badanie charakteryzowało się ogólną dokładnością na poziomie 82 % (na podstawie wyników badania przeprowadzonego na 97 postaciach użytkowych), czułością na poziomie 91 % (na podstawie wyników badania przeprowadzonego na 54 postaciach użytkowych), odsetkiem wyników fałszywie ujemnych na poziomie 9 % (na podstawie wyników badania przeprowadzonego na 54 postaciach użytkowych), swoistością na poziomie 72 % (na podstawie wyników badania przeprowadzonego na 43 postaciach użytkowych) oraz odsetkiem wyników fałszywie dodatnich na poziomie 28 % (na podstawie wyników badania przeprowadzonego na 43 postaciach użytkowych) (23).
|
|
15.
|
Badanie działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ można przeprowadzać w celu zidentyfikowania substancji chemicznych niewymagających sklasyfikowania jako wywołujące podrażnienie oczu lub poważne uszkodzenie oczu zgodnie z systemem klasyfikacji GHS ONZ / systemem klasyfikacji ustanowionym w rozporządzeniu CLP. Biorąc pod uwagę dane pozyskane w ramach badania walidacyjnego (10)(11), badanie działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ charakteryzowało się ogólną dokładnością na poziomie 84 % (na podstawie wyników badania przeprowadzonego na 200 substancjach chemicznych), czułością na poziomie 95 % (na podstawie wyników badania przeprowadzonego na 97 substancjach chemicznych), odsetkiem wyników fałszywie ujemnych na poziomie 5 % (na podstawie wyników badania przeprowadzonego na 97 substancjach chemicznych), swoistością na poziomie 72 % (na podstawie wyników badania przeprowadzonego na 103 substancjach chemicznych) oraz odsetkiem wyników fałszywie dodatnich na poziomie 28 % (na podstawie wyników badania przeprowadzonego na 103 substancjach chemicznych) w zestawieniu z danymi z referencyjnego badania in vivo na oku królika (metoda badawcza B.5) (2)(14) sklasyfikowanego zgodnie z systemem klasyfikacji GHS ONZ / systemem klasyfikacji ustanowionym w rozporządzeniu CLP.
|
|
16.
|
Odsetki wyników fałszywie ujemnych uzyskane w obydwu badaniach RhCE przeprowadzonych na substancjach albo mieszaninach mieszczą się w wynoszącym 12 % przedziale prawdopodobieństwa, że substancje chemiczne zostaną zidentyfikowane jako należące do kategorii 2 wg GHS ONZ / rozporządzenia CLP albo jako nienależące do żadnej kategorii wg GHS ONZ / rozporządzenia CLP w przypadku przeprowadzenia testu Draize’a na oku in vivo w ramach powtórzonych badań; jest to spowodowane zmiennością wewnętrzną badania nieodłącznie związaną ze stosowaniem tej metody (24). Odsetek wyników fałszywie dodatnich uzyskanych po zastosowaniu obydwu metod badawczych RhCE w odniesieniu do substancji albo mieszanin nie ma decydującego znaczenia w przypadku niniejszej metody badawczej, ponieważ wszystkie badane substancje chemiczne, których oddziaływanie prowadzi do osiągnięcia żywotności tkanek równej ustanowionym wartościom progowym lub niższej od tych wartości (zob. pkt 44), będą wymagały zastosowania dodatkowych metod badawczych in vitro lub – w ostateczności w przypadku królików, w zależności od obowiązujących wymogów regulacyjnych – zastosowania strategii badań sekwencyjnych w ramach podejścia bazującego na wadze dowodów. Wspomniane metody badawcze można zastosować w odniesieniu do wszystkich rodzajów substancji chemicznych, w przypadku których wynik ujemny powinien zostać uznany za niedający podstaw do sklasyfikowania substancji chemicznej za powodującą podrażnienie i poważne uszkodzenie oczu (brak kategorii wg GHS ONZ i rozporządzenia CLP). Przed przeprowadzeniem badania działania drażniącego na oczy EpiOcular™ i badania działania drażniącego na oczy na ludzkim nabłonku rogówki SkinEthic™ zgodnie z systemami klasyfikacji innymi niż GHS ONZ / system klasyfikacji ustanowiony w rozporządzeniu CLP należy skonsultować się z odpowiednimi organami regulacyjnymi.
|
|
17.
|
Ograniczenie niniejszej metody badawczej jest związane z faktem, że nie zapewnia ona możliwości dokonania rozróżnienia między podrażnieniem oczu / wywarciem odwracalnego wpływu na oczy (kategoria 2) a poważnym uszkodzeniem oczu / wywarciem nieodwracalnego wpływu na oczy (kategoria 1) wg klasyfikacji GHS ONZ / klasyfikacji ustanowionej w rozporządzeniu CLP ani między substancjami wywołującymi podrażnienie oczu (fakultatywna kategoria 2A) a substancjami wywołującymi lekkie podrażnienie oczu (fakultatywna kategoria 2B) zgodnie z klasyfikacją GHS ONZ (1). Aby dokonać takiego rozróżnienia, konieczne jest przeprowadzenie dalszych badań przy wykorzystaniu innych metod badawczych in vitro.
|
|
18.
|
Termin „badana substancja chemiczna” jest używany w niniejszej metodzie badawczej w odniesieniu do tego, co jest przedmiotem badania (77), i nie jest powiązany z zastosowaniem metody badawczej RhCE do badania substancji lub mieszanin.
|
ZASADA BADANIA
|
19.
|
Badaną substancję chemiczną nanosi się miejscowo na co najmniej dwa trójwymiarowe wytwory tkankowe RhCE, po czym dokonuje się pomiaru żywotności tkanek po okresie narażenia i okresie inkubacji tkanek przemytych. Tkanki RhCE rekonstruuje się przy wykorzystaniu pierwotnych keratynocytów naskórka ludzkiego lub unieśmiertelnionych komórek nabłonkowych ludzkiej rogówki, które były hodowane przez kilka dni w celu wytworzenia warstwowego, wysoce zróżnicowanego nabłonka płaskiego podobnego do nabłonka występującego w ludzkiej rogówce pod względem morfologicznym. Wytwór tkankowy RhCE EpiOcular™ składa się z co najmniej 3 warstw zdolnych do życia komórek oraz niezrogowaciałej powierzchni o strukturze zbliżonej do struktury rogówki, na której przeprowadza się badanie in vivo. Wytwór tkankowy RhCE imitujący ludzki nabłonek rogówki SkinEthic™ składa się z co najmniej 4 warstw zdolnych do życia komórek, uwzględniając kolumnowe komórki bazowe, przejściowe komórki boczne i sztuczne komórki płaskie zbliżone do komórek występujących w prawdziwym nabłonku rogówki (20)(26).
|
|
20.
|
Poważne uszkodzenie oczu / podrażnienie oczu wywołane oddziaływaniem substancji chemicznych, które przejawia się in vivo głównie zmętnieniem rogówki, zapaleniem tęczówki, zaczerwienieniem błony spojówkowej lub obrzękiem spojówki, stanowi rezultat szeregu zdarzeń, począwszy od przeniknięcia substancji chemicznej przez rogówkę lub błonę spojówkową, a skończywszy na uszkodzeniu komórek. Do uszkodzenia komórek może dość w wyniku szeregu działań, m.in.: lizy błony komórkowej (np. wskutek oddziaływania środków powierzchniowo czynnych, rozpuszczalników organicznych); koagulacji makrocząsteczek (głównie białek) (np. wskutek oddziaływania środków powierzchniowo czynnych, rozpuszczalników organicznych, zasad i kwasów); zmydlania lipidów (np. wskutek oddziaływania zasad); alkilowania makrocząsteczek lub innych interakcji kowalencyjnych z makrocząsteczkami (np. wskutek oddziaływania wybielaczy, nadtlenków i alkilatorów) (15)(27)(28). Wykazano jednak, że cytotoksyczność odgrywa istotną – o ile nie najistotniejszą – mechanistyczną rolę w procesie ustalania, czy działanie substancji chemicznej wywołuje ogólną reakcję w postaci poważnego uszkodzenia oczu / podrażnienia oczu niezależnie od procesów fizykochemicznych leżących u podstaw uszkodzenia tkanek (29)(30). Ponadto potencjał wywołania poważnego uszkodzenia oczu / podrażnienia oczu przez daną substancję chemiczną ustala się przede wszystkim na podstawie skali początkowych obrażeń (31), która odpowiada skali uśmiercenia komórek (29) oraz skali późniejszych reakcji i ewentualnych rezultatów (32). Dlatego też substancje wywołujące lekkie podrażnienie zasadniczo wpływają wyłącznie na nabłonek rogówki, substancje wywołujące łagodne i umiarkowane podrażnienie wpływają głównie na nabłonek i sztuczną tkankę podskórną, natomiast substancje silnie drażniące uszkadzają nabłonek, głęboko położoną tkankę podskórną i niekiedy śródbłonek rogówki (30)(33). Pomiar żywotności wytworu tkankowego RhCE po miejscowym narażeniu na działanie badanej substancji chemicznej przeprowadzany w celu zidentyfikowania substancji chemicznych niewymagających sklasyfikowania jako wywołujące poważne uszkodzenie oczu / podrażnienie oczu (brak kategorii wg klasyfikacji GHS ONZ / rozporządzenia CLP) opiera się na założeniu, że wszystkie substancje chemiczne wywołujące poważne uszkodzenie oczu lub podrażnienie oczu będą wywierały cytotoksyczny wpływ na nabłonek lub błonę spojówkową.
|
|
21.
|
Żywotność tkanek RhCE klasycznie mierzy się metodą enzymatycznej konwersji barwnika przyżyciowego MTT [bromek 3-(4,5-dimetylotiazol-2-ilo)-2,5-difenylotetrazoliowy; MTT; numer CAS 298-93-1] przez komórki żywotne tkanki w niebieską sól formazanu MTT, którą oznacza się ilościowo po ekstrakcji z tkanek (16). Substancje chemiczne, które zgodnie z GHS ONZ / rozporządzeniem CLP nie wymagają sklasyfikowania i oznakowania (brak kategorii), identyfikuje się jako substancje nieobniżające żywotności tkanek poniżej ustalonego progu (tj. żywotność tkanek > 60 % w przypadku badania działania drażniącego na oczy EpiOcular™ i badania działania drażniącego na oczy EITL SkinEthic™ (78) lub > 50 % w przypadku badania działania drażniącego na oczy EITS SkinEthic™ (79)) (zob. pkt 44).
|
WYKAZANIE BIEGŁOŚCI
|
22.
|
Przed przystąpieniem do rutynowego przeprowadzania badań RhCE do celów regulacyjnych laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo przewidując działanie piętnastu substancji chemicznych przeznaczonych do oceny biegłości wymienionych w tabeli 1. Wspomniane substancje chemiczne zostały wybrane spośród substancji chemicznych wykorzystywanych w badaniach walidacyjnych dotyczących VRM (8)(10)(11). Wybrane substancje chemiczne obejmują – w zakresie, w jakim jest to możliwe – substancje, które: (i) występują w różnych stanach skupienia; (ii) obejmują pełne spektrum reakcji in vivo w postaci poważnego uszkodzenia oczu / podrażnienia oczu stwierdzonych w oparciu o wysokiej jakości wyniki uzyskane po przeprowadzeniu porównania z wynikami badania na oku królika in vivo (metoda badawcza B.5) (2)(14) oraz poprzez odniesienie do systemu klasyfikacji GHS ONZ (tj. kategorie 1, 2A, 2B lub brak kategorii) (1) i systemem klasyfikacji ustanowionym w rozporządzeniu CLP (tj. kategorie 1, 2 lub brak kategorii); (iii) obejmują różne kryteria klasyfikacji in vivo (24)(25); (iv) są reprezentatywne dla klas chemicznych wykorzystywanych w badaniu walidacyjnym (8)(10)(11); (v) reprezentują odpowiednią i szeroką grupę organicznych grup funkcyjnych (8)(10)(11); (vi) mają dobrze określone struktury chemiczne (8)(10)(11); (vii) są zabarwione lub stanowią bezpośrednie reduktory MTT; (viii) doprowadziły do uzyskania odtwarzalnych wyników w ramach metod badawczych RhCE podczas ich walidacji; (ix) zostały prawidłowo zidentyfikowane w ramach metod badawczych RhCE w trakcie poświęconych im badań walidacyjnych; (x) obejmują pełne spektrum reakcji in vitro w oparciu o wysokiej jakości dane uzyskane w ramach metod badawczych RhCE (0–100 % żywotności); (xi) są dostępne na rynku; oraz (xii) ich pozyskiwanie lub utylizacja nie wiąże się z koniecznością ponoszenia nadmiernie wysokich kosztów. W sytuacjach, w których substancja chemiczna figurująca w wykazie jest niedostępna lub nie może zostać wykorzystana z innych uzasadnionych względów, dopuszcza się możliwość zastosowania innej substancji chemicznej spełniającej opisane powyżej kryteria, np. substancji wybranej spośród substancji chemicznych wykorzystywanych do walidacji VRM. Takie odstępstwa powinny jednak być uzasadnione.
Tabela 1
Wykaz substancji chemicznych przeznaczonych do oceny biegłości
|
Nazwa systematyczna
|
Numer CAS
|
Organiczna grupa funkcyjna (80)
|
Stan skupienia
|
Żywotność VRM1 (%) (81)
|
Żywotność VRM2 (%) (82)
|
Prognoza dotycząca VRM
|
Reduktor MTT
|
Zakłócenie barwy
|
|
Kategoria 1 in vivo
(83)
|
|
|
Tioglikolan metylu
|
2365-48-2
|
Ester kwasu karboksylowego; tioalkohol
|
C
|
10,9±6,4
|
5,5±7,4
|
Nie można przewidzieć
|
T (silny)
|
N
|
|
Akrylan hydroksyetylu
|
818-61-1
|
Akrylan; alkohol
|
C
|
7,5±4,7 (84)
|
1,6±1,0
|
Nie można przewidzieć
|
N
|
N
|
|
2,5-dimetylo-2,5-heksanodiol
|
110-03-2
|
alkohol
|
S
|
2,3±0,2
|
0,2±0,1
|
Nie można przewidzieć
|
N
|
N
|
|
Szczawian sodu
|
62-76-0
|
Kwas oksokarboksylowy
|
S
|
29,0±1,2
|
5,3±4,1
|
Nie można przewidzieć
|
N
|
N
|
|
Kategoria 2A in vivo (83)
|
|
|
N,N″-bis(4-chlorofenylo)-3,12-diimino-di-D-glukonian diimidu amidowego 2,4,11,13-tetraazatetradekanu (20 %, roztwór wodny) (85)
|
18472-51-0
|
Heterocykliczny halogenek aromatyczny; halogenek arylu; grupa dihydroksylowa; guanidyna
|
C
|
4,0±1,1
|
1,3±0,6
|
Nie można przewidzieć
|
N
|
T (słaba)
|
|
Benzoesan sodu
|
532-32-1
|
Aryl; Kwas karboksylowy
|
S
|
3,5±2,6
|
0,6±0,1
|
Nie można przewidzieć
|
N
|
N
|
|
Kategoria 2B in vivo
(83)
|
|
|
Ν,Ν-dietylo-m-toluamid
|
134-62-3
|
Benzamid
|
C
|
15,6±6,3
|
2,8±0,9
|
Nie można przewidzieć
|
N
|
N
|
|
2,2-dmetylo-3-metylenobicyclo[2.2.1]heptan
|
79-92-5
|
Alkan rozgałęziony o trzeciorzędowy węgiel; Alken; Bicykloheptan; Związki karbocykliczne z pierścieniem połączonym mostkiem; Cykloalkan
|
S
|
4,7±1,5
|
15,8±1,1
|
Nie można przewidzieć
|
N
|
N
|
|
Substancje chemiczne określane mianem „brak kategorii” wykorzystywane do badań in vivo
(83)
|
|
|
1-etylo-3-metyloimidazoliowy siarczan etylu
|
342573-75-5
|
Alkoksy; Sól amonowa; Aryl; Imidazol; Siarczan
|
C
|
79,9±6,4
|
79,4±6,2
|
Brak kategorii
|
N
|
N
|
|
Eter (di)oktylu
|
629-82-3
|
Alkoksy; eter
|
C
|
97,8±4,3
|
95,2±3,0
|
Brak kategorii
|
N
|
N
|
|
Butotlenek piperonylu
|
51-03-6
|
Alkoksy; Benzodioksol; Benzyl; eter
|
C
|
104,2±4,2
|
96,5±3,5
|
Brak kategorii
|
N
|
N
|
|
Olej rycynowy uwodorniony glikolem polietylenowym (PEG-40)
|
61788-85-0
|
Acylal; Alkohol; Allil; eter
|
Lepka konsystencja
|
77,6±5,4
|
89,1±2,9
|
Brak kategorii
|
N
|
N
|
|
1-(4-chlorofenylo)-3-(3,4-dichlorofenylo)mocznik
|
101-20-2
|
Heterocykliczny halogenek aromatyczny; Halogenek arylu; Pochodne mocznika
|
S
|
106,7±5,3
|
101,9±6,6
|
Brak kategorii
|
N
|
N
|
|
2,2′-metyleno-bis-(6-(2H-benzotriazol-2-yl)-4-(1,1,3,3-tetrametylobutyl)-fenol)
|
103597-45-1
|
Alkan rozgałęziony o czwartorzędowy węgiel; Skondensowany karbocykliczny związek aromatyczny; Skondensowane nasycone związki heterocykliczne; Związki będące prekursorami chinonowymi; związki tert-butylowe
|
S
|
102,7±13,4
|
97,7±5,6
|
Brak kategorii
|
N
|
N
|
|
Tetrofluoroboran potasu
|
14075-53-7
|
Sól nieorganiczna
|
S
|
88,6±3,3
|
92,9±5,1
|
Brak kategorii
|
N
|
N
|
|
Skróty:
Numer CAS = numer w rejestrze Chemical Abstracts Service; GHS ONZ = Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (1); VRM1 = zwalidowana metoda referencyjna, badanie działania drażniącego na oczy EpiOcular™; VRM2 = zwalidowana metoda referencyjna, badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™; Zakłócenie barwy = pomiar zakłócenia barwy za pomocą absorbancji wzorca (gęstość optyczna (OD)) formazanu MTT.
|
|
|
23.
|
W ramach wykazywania efektywności zaleca się, aby użytkownicy sprawdzili właściwości bariery cechujące tkanki po ich otrzymaniu, zgodnie ze wskazaniami producenta wytworu tkankowego RhCE (zob. pkt 25, 27 i 30). Jest to szczególnie istotne, jeżeli tkanki są wysłane na duże odległości lub w długich okresach czasu. Gdy badanie zostało z powodzeniem wprowadzone oraz nabyto biegłość w jego przeprowadzaniu i zostało to wykazane, weryfikacja taka nie będzie konieczna w ramach rutynowej procedury. W przypadku rutynowego zastosowania badania zaleca się jednak, aby w dalszym ciągu oceniać właściwości bariery w regularnych odstępach czasu.
|
PROCEDURA
|
24.
|
Niniejsza metoda badawcza obejmuje obecnie potwierdzone naukowo badanie działania drażniącego na oczy EpiOcular™ oraz badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™ (odpowiednio VRM1 i VRM2). Standardowe procedury operacyjne dotyczące metod badawczych RhCE są dostępne i należy z nich korzystać przy wdrażaniu i stosowaniu metod badawczych w warunkach laboratoryjnych (34)(35). W poniższych punktach oraz w dodatku 2 opisano najistotniejsze elementy i procedury badań RhCE.
|
SKŁADOWE METODY BADANIA RHCE
Warunki ogólne
|
25.
|
W celu odtworzenia trójwymiarowej tkanki nabłonka przypominającej rogówkę, która powinna się składać ze stopniowo nawarstwionych ale nie zrogowaciałych komórek, należy użyć odpowiednich komórek pochodzących od człowieka. Wytwór tkankowy RhCE przygotowuje się w insertach z porowatą membraną syntetyczną, przez którą składniki odżywcze mogą się przedostać do komórek. W zrekonstruowanym nabłonku przypominającym rogówkę powinno się znajdować wiele warstw żywotnych, niezrogowaciałych komórek nabłonka. Powierzchnia nabłonkowa wytworu tkankowego RhCE powinna mieć bezpośredni kontakt z powietrzem w celu umożliwienia bezpośredniego miejscowego narażenia na działanie badanej substancji w podobny sposób, jak w przypadku narażenia nabłonka rogówki w badaniu in vivo. Wytwór tkankowy RhCE powinien uformować funkcjonalną barierę, która jest na tyle wytrzymała, aby uniemożliwiała szybkie przenikanie cytotoksycznych wzorcowych substancji, np. Triton X-100 lub soli sodowej siarczanu dodecylu (SDS). Funkcję bariery należy wykazać i można ocenić poprzez ustalenie czasu narażenia na działanie potrzebnego do zmniejszenia żywotności komórek o 50 % (ET50) po zastosowaniu substancji wzorcowej w określonym, stałym stężeniu (np., 100 μl 0,3 % (obj./obj.) Triton X-100) lub stężenia, w którym substancja wzorcowa zmniejsza żywotność tkanek o 50 % (IC50) po ustalonym czasie narażenia na działanie (np. 30-minutowym poddaniu działaniu substancji z wykorzystaniem 50 μl SDS) (zob. pkt 30). Właściwości izolacyjne wytworu tkankowego RhCE powinny zapobiegać przedostawaniu się badanej substancji chemicznej przez krawędź żywej tkanki, gdyż w innym razie modelowanie narażenia rogówki mogłoby być niewłaściwe. Pochodzące od człowieka komórki, które wykorzystuje się do stworzenia wytworu tkankowego RhCE, nie powinny być skażone bakteriami, wirusami, mykoplazmą ani grzybami. Dostawca powinien sprawdzić sterylność wytworu tkankowego pod kątem braku skażenia grzybami i bakteriami.
|
Warunki funkcjonalne
Żywotność
|
26.
|
Testem stosowanym do określenia ilościowego żywotności tkanki jest test MTT (16). Komórki żywotne wytworu tkankowego RhCE redukują barwnik przyżyciowy MTT do wytrąconego niebieskiego formazanu MTT, który jest później ekstrahowany z komórki przy użyciu izopropanolu (lub podobnego rozpuszczalnika). Ekstrahowany formazan MTT można określić ilościowo, stosując pomiar absorbancji wzorca (gęstość optyczna – OD) lub procedurę spektrofotometrii HPLC/UPLC (36). OD samego rozpuszczalnika do ekstrakcji powinna być wystarczająco niska, tj. OD < 0,1. Użytkownicy wytworu tkankowego RhCE powinni zapewnić zgodność każdej partii zastosowanego wytworu tkankowego RhCE z określonymi kryteriami kontroli ujemnej. Zakresy dopuszczalności kontroli ujemnej wartości OD dotyczące VRM przedstawiono w tabeli 2. Użytkownik spektrofotometrii HPLC/UPLC powinien stosować kontrolę ujemną zakresów OD określonych w tabeli 2 jako warunku dopuszczenia kontroli ujemnej. W sprawozdaniu z badania należy udokumentować stabilność w hodowli (uzyskiwanie podobnych wyników pomiarów żywotności tkanki) tkanki poddanej działaniu substancji kontroli ujemnej w badanym okresie narażenia. Podobną procedurę powinien stosować producent tkanek w ramach zwolnienia serii na podstawie kontroli jakości, ale w tym przypadku mogą mieć zastosowanie inne kryteria dopuszczalności niż kryteria określone w tabeli 2. Zakres dopuszczalności (górna i dolna granica) kontroli ujemnej wartości OD (w warunkach metody badania kontroli jakości) powinni określić producenci/dostawcy wytworu tkankowego RhCE.
|
Tabela 2
Zakresy dopuszczalności dla kontroli ujemnej wartości OD (dla użytkowników badania)
|
Badanie
|
Dolna granica dopuszczalności
|
Górna granica dopuszczalności
|
|
Badanie działania drażniącego na oczy EpiOcular™ (OCL-200) – VRM1 (zarówno w przypadku protokołów dotyczących cieczy, jak i substancji stałych)
|
> 0,8 (86)
|
< 2,5
|
|
badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™ (HCE/S) – VRM2 (zarówno w przypadku protokołów dotyczących cieczy, jak i substancji stałych)
|
> 1,0
|
≤ 2,5
|
Funkcja bariery
|
27.
|
Wytwór tkankowy RhCE powinien być na tyle gruby i szczelny, aby powstrzymać szybkie przenikanie cytotoksycznych substancji wzorcowych, zgodnie z oszacowaniem np. za pomocą ET50 (Triton X-100) lub IC50 (SDS) (tabela 3). Producent/dostawca wytworu tkankowego RhCE powinien w momencie dostarczenia tkanek użytkownikowi końcowemu wykazać funkcję bariery każdej partii zastosowanego wytworu tkankowego RhCE (zob. pkt 30).
|
Morfologia
|
28.
|
Badanie histologiczne wytworu tkankowego RhCE powinno wykazać istnienie struktury nabłonka przypominającego rogówkę (w tym co najmniej trzech warstw żywotnych komórek nabłonkowych i niezrogowaciałej powierzchni). W przypadku VRM odpowiednią morfologię ustala producent/dostawca, a więc użytkownik metody badawczej nie musi jej ponownie wykazywać dla każdej partii tkanek.
|
Odtwarzalność
|
29.
|
Wyniki dodatniej kontroli oraz ujemnych w ramach metody badawczej powinny wykazywać odtwarzalność w czasie.
|
Kontrola jakości (QC)
|
30.
|
Wytwór tkankowy RhCE należy stosować jedynie w przypadku, gdy producent lub dostawca wykaże, że każda partia wykorzystywanego wytworu tkankowego RhCE spełnia określone kryteria zwolnienia do produkcji, spośród których najbardziej istotne są te dotyczące żywotności (pkt 26)i funkcji bariery (pkt 27). Zakres dopuszczalności (górna i dolna granica) dotyczący funkcji bariery zmierzonej za pomocą ET50 lub IC50 (zob. pkt 25 i 26) powinien określić producent/dostawca wytworu tkankowego RhCE. Zakres dopuszczalności ET50 and IC50 zastosowany przez producenta/dostawcę wytworów tkankowych RhCE (wykorzystywanych w VRM) jako kryterium zwolnienia serii na podstawie kontroli jakości przedstawiono w tabeli 3. Producent/dostawca wytworu tkankowego RhCE powinien dostarczyć dane potwierdzające zgodność ze wszystkimi kryteriami zwolnienia do produkcji użytkownikom metody badawczej, tak aby mogli oni uwzględnić te informacje w sprawozdaniu z badania. Do wiarygodnego przewidzenia, że dane substancje chemiczne nie wymagają zaklasyfikowania jako substancje drażniące oczy lub powodujące poważne uszkodzenie oczu zgodnie z GHS ONZ / rozporządzeniem CLP, można przyjąć wyłącznie wyniki uzyskane z użyciem tkanek spełniających wszystkie kryteria zwolnienia do produkcji.
|
Tabela 3
Kryterium zwolnienia serii na podstawie kontroli jakości
|
Badanie
|
Dolna granica dopuszczalności
|
Górna granica dopuszczalności
|
|
Badanie działania drażniącego na oczy EpiOcular™ (OCL-200) – VRM1 (100 μl 0,3 % (obj./obj.) Triton X-100)
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™ (HCE/S) – VRM2 (30-minutowe poddanie działaniu substancji z wykorzystaniem 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Podawanie badanej i kontrolnej substancji chemicznej
|
31.
|
W każdej serii badawczej należy użyć co najmniej dwóch kontrprób tkanek w przypadku każdej badanej i kontrolnej substancji chemicznej. Stosuje się dwa różne protokoły poddawania działaniu substancji – jeden dla badanych substancji chemicznych w stanie ciekłym, a drugi dla badanych substancji chemicznych w stanie stałym (34)(35). W przypadku obu metod i protokołów powierzchnię wytworu tkankowego należy przed podaniem badanych substancji chemicznych zwilżyć roztworem chlorku sodu buforowanym fosforanami Dulbecco wolnym od wapnia i magnezu (DPBS wolny od Ca2+/Mg2+), aby odwzorować mokre warunki w ludzkim oku. Poddawanie tkanek działaniu substancji rozpoczyna się od narażenia ich na działanie badanych i kontrolnych substancji chemicznych. W przypadku obu protokołów podawania działaniu substancji w obu VRM należy nałożyć na skórę dostateczną ilość badanej lub kontrolnej substancji chemicznej, tak aby równomiernie pokrywała powierzchnię nabłonka, unikając jednak nakładania dawki nieskończonej (zob. pkt 32 i 33) (dodatek 2).
|
|
32.
|
Badane substancje chemiczne, które można podawać pipetą w temperaturach na poziomie 37 °C lub niższych (w stosownych przypadkach wykorzystując pipetę typu D), traktuje się w VRM jako ciecze, a w innym przypadku należy je traktować jako substancje stałe (zob. pkt 33). W VRM badaną substancję chemiczną równomiernie rozprowadza się na powierzchni tkanki (tj. stosuje się co najmniej 60 μl/cm2) (zob. dodatek 2, (33)(34)). Aby zagwarantować właściwe dawkowane tkanek, należy w jak największym stopniu uniknąć wystąpienia efektów kapilarnych (efektów napięcia powierzchniowego), które mogą wyniknąć z nałożenia na insert (na powierzchnię tkanki) niewielkiej ilości substancji. Tkanki podawane działaniu badanych substancji chemicznych w stanie ciekłym inkubuje się przez 30 minut w normalnych warunkach hodowli kultur (37±2 °C, 5±1 % CO2, ≥ 95 % RH). Pod koniec okresu narażenia badaną należy ostrożnie usunąć substancję chemiczną w stanie ciekłym i substancje kontrolne z powierzchni tkanki stosując intensywne płukanie za pomocą DPBS wolnego od Ca2+/Mg2+ w temperaturze pokojowej. Po etapie płukania następuje zanurzenie po zakończeniu narażenia w świeżym podłożu w temperaturze pokojowej (w celu usunięcia badanej substancji chemicznej wchłoniętej przez tkankę) na wcześniej określony czas, zależny od zastosowanej VRM. Jedynie w przypadku VRM1 przed przeprowadzeniem testu MTT należy zastosować inkubację po zakończeniu narażenia w świeżym podłożu w normalnych warunkach hodowli kultur (zob. dodatek 2, (34) (35)).
|
|
33.
|
Badane substancje chemiczne, których nie można podawać pipetą w temperaturach na poziomie 37 °C lub niższych traktuje się w VRM jako substancje stałe. Ilość podawanej badanej substancji chemicznej powinna być wystarczająca do pokrycia całej powierzchni tkanki, tj. należy nałożyć co najmniej 60 mg/cm2 (dodatek 2). W miarę możliwości substancje stałe powinny być badane w postaci drobnoziarnistego proszku. Tkanki poddane działaniu badanych substancji chemicznych w stanie stałym inkubuje się przez wcześniej określony czas (w zależności od zastosowanej VRM) w normalnych warunkach hodowli kultur (zob. dodatek 2, (34) (35)). Pod koniec okresu narażenia badaną należy ostrożnie usunąć substancję chemiczną w stanie stałym i substancje kontrolne z powierzchni tkanki stosując intensywne płukanie za pomocą DPBS wolnego od Ca2+/Mg2+ w temperaturze pokojowej. Po etapie płukania następuje zanurzenie po zakończeniu narażenia w świeżym podłożu w temperaturze pokojowej (w celu usunięcia badanej substancji chemicznej wchłoniętej przez tkankę) na wcześniej określony czas, zależny od zastosowanej VRM, oraz zanurzenie po narażeniu w świeżym podłożu w normalnych warunkach hodowli kultur przed przeprowadzeniem testu MTT (zob. dodatek 2, (34)(35)).
|
|
34.
|
W każdej serii badawczej należy uwzględnić równoczesną kontrolę ujemną i kontrolę dodatnią, aby wykazać, że żywotność (określona w kontroli ujemnej) oraz czułość (określona w kontroli dodatniej) tkanek mieszczą się w zakresach dopuszczalności ustalonych na podstawie danych historycznych. Jednoczesna kontrola ujemna dostarcza również wartość bazową (100 % żywotności tkanek) na potrzeby obliczenia względnej procentowej żywotności tkanek poddanych działaniu badanej substancji chemicznej (%Viabilitytest). Substancją służącą do kontroli dodatniej zalecaną do stosowania w VRM jest czysty octan metylu (nr CAS 79-20-9, dostępny na rynku od np. Sigma-Aldrich, nr katalogowy 45997; ciecz). Substancjami służącymi do kontroli ujemnej zalecanymi do stosowania w VRM1 i VRM2 są odpowiednio ultraczyste H2O oraz DPBS wolny od Ca2+/Mg2+. Były to substancje kontrolne wykorzystane w badaniach walidacyjnych dotyczących VRM i istnieje na ich temat najwięcej danych historycznych. W przypadku zastosowania odpowiednich alternatywnych substancji służących do kontroli ujemnej lub dodatniej należy to odpowiednio naukowo uzasadnić. Kontrole dodatnie i ujemne należy badać za pomocą tych samych protokołów, które wykorzystano do badania substancji chemicznych w danej serii badawczej (tj. cieczy lub substancji stałych). Po takim zastosowaniu należy przeprowadzić, w stosownych przypadkach, poddanie narażeniu na działanie substancji, płukanie, zanurzenie po zakończeniu narażenia oraz inkubację po zakończeniu narażenia, jak opisano w przypadku prób kontrolnych poddawanych badaniu równolegle z badanymi substancjami chemicznych w stanie ciekłym (zob. pkt 32) lub w przypadku kontroli poddawanych badaniu równolegle z badanymi substancjami chemicznych w stanie stałym (zob. pkt 33) przed przeprowadzeniem testu MTT (zob. pkt 35) (34)(35). W przypadku wszystkich badanych substancji chemicznych o tym samym stanie skupienia (ciecze lub substancje stałe), będących częścią jednej serii badawczej, wystarczy jeden zestaw kontroli ujemnych i dodatnich.
|
Pomiary żywotności tkanek
|
35.
|
Test MTT stanowi uznaną metodą ilościową (16), którą należy wykorzystywać do pomiarów żywotności tkanek w ramach niniejszej metody badawczej. Można ją stosować do badań z użyciem trójwymiarowego wytworu tkankowego. Test MTT przeprowadza się natychmiast po okresie inkubacji po zakończeniu narażenia. W ramach VRM wytwór tkankowy RhCE umieszcza się w 0,3 ml roztworu MTT przy 1 mg/ml na 180±15 min w normalnych warunkach hodowli kultur. Komórki żywotne wytworu tkankowego RhCE redukują barwnik przyżyciowy MTT do wytrąconego niebieskiego formazanu MTT. Produkt wytrącania niebieskiego formazanu MTT ekstrahuje się następnie z tkanki przy użyciu odpowiedniej ilości izopropanolu (lub podobnego rozpuszczalnika) (34)(35). Tkanki zbadane za pomocą badanych substancji chemicznych w stanie ciekłym należy ekstrahować zarówno z góry, jak i z dołu tkanki, podczas gdy tkanki zbadane za pomocą badanych substancji chemicznych w stanie stałym oraz cieczy zabarwionych należy ekstrahować jedynie z dołu tkanki (w celu zminimalizowania ewentualnego skażenia roztworu ekstrakcyjnego izopropanolu jakąkolwiek badaną substancją chemiczną, która mogła pozostać na tkance). Tkanki zbadane za pomocą badanych substancji chemicznych w stanie ciekłym, które nie są łatwe do zmycia, również można ekstrahować jedynie z dołu tkanki. Badane równolegle substancje służące do kontroli ujemnej i dodatniej należy potraktować podobnie do badanej substancji chemicznej. Ekstrahowany formazan MTT można określić ilościowo albo przy pomocy pomiaru absorbancji wzorca (OD) przy 570 nm, wykorzystując filtr w paśmie maksimum ±30 nm, albo przy pomocy procedury spektrofotometrii HPLC/UPLC (zob. pkt 42) (11)(36).
|
|
36.
|
Właściwości optyczne badanej substancji chemicznej lub jej oddziaływanie chemiczne na MTT mogą zakłócać pomiar formazanu MTT, prowadząc do błędnego oszacowania żywotności tkanki. Badane substancje chemiczne mogą zakłócać pomiar formazanu MTT poprzez bezpośrednią redukcję MTT do niebieskiego formazanu MTT lub na skutek zakłócenia barwy, jeżeli badana substancja chemiczna jest chłonna, w sposób naturalny lub w wyniku procedur poddania działaniu badanej substancji w tym samym zakresie gęstości optycznej co formazan MTT (tj. ok 570 nm). Przed badaniem należy wykonać wstępne kontrole, aby umożliwić identyfikację potencjalnych bezpośrednich reduktorów MTT lub substancji chemicznych zakłócających barwę oraz należy wykorzystać dodatkowe próby kontrolne do wykrycia i skorygowania potencjalnej interferencji takich badanych substancji chemicznych (zob. pkt 37–41). Jest to istotne zwłaszcza w przypadku niepełnego usunięcia określonej substancji badanej z wytworu tkankowego RhCE za pomocą płukania lub gdy substancja ta przeniknie przez nabłonek przypominający rogówkę, przez co pozostaje obecna w wytworach tkankowych RhCE podczas przeprowadzania testu MTT. W przypadku badanych substancji chemicznych pochłaniających światło w takim samym zakresie, jak formazan MTT (naturalnie lub po poddaniu działaniu substancji), które nie są zgodne z pomiarem absorbancji wzorca (OD) formazanu MTT ze względu na zbyt silną interferencję, tj. silną absorbcję przy 570±30 nm, można zastosować procedurę spektrofotometrii HPLC/UPLC do pomiaru formazanu MTT (zob. pkt 41 i 42) (11)(36). Szczegółowy opis sposobu wykrywania i skorygowania redukcji MTT i zakłóceń przez barwniki jest dostępny w standardowych procedurach operacyjnych dotyczących VRM (34)(35). Przykładowe schematy zawierające wytyczne dotyczące sposobu identyfikacji i obchodzenia się z bezpośrednimi reduktorami MTT lub substancjami chemicznymi zakłócającymi barwę w VRM1 i VRM2 przedstawiono również odpowiednio w dodatkach III i IV.
|
|
37.
|
W celu identyfikacji potencjalnej interferencji wynikającej z tego, że badane substancje chemiczne pochłaniają światło w tym samym zakresie co formazan MTT (naturalnie lub po poddaniu działaniu substancji), i podjęcia decyzji o konieczności zastosowania dodatkowych kontroli badaną substancję chemiczną dodaje się do wody lub izopropanolu i inkubuje się odpowiednio długo w temperaturze pokojowej (zob. dodatek 2, (34)(35)). Jeżeli w wodzie lub izopropanolu badana substancja chemiczna pochłania wystarczającą ilość światła w zakresie 570 ± 20 nm, w przypadku VRM1 (zob. dodatek 3), lub jeżeli w wyniku zmieszania badanej substancji chemicznej z wodą uzyskuje się barwiony roztwór, w przypadku VRM2 (zob. dodatek 4), zakłada się, że badana substancja chemiczna zakłóca pomiar absorbancji wzorca (OD) formazanu MTT i należy przeprowadzić kolejne pogłębione kontrole z barwnikiem lub, alternatywnie, należy zastosować procedurę spektrofotometrii HPLC/UPLC i wtedy te kontrole nie są konieczne (zob. pkt 41 i 42 oraz dodatki III i IV)(34)(35). Podczas dokonywania pomiaru absorbancji wzorca (OD) należy nałożyć każdą zakłócającą badaną substancję chemiczną na co najmniej dwie kontrpróby żywych tkanek, które w celu uzyskania barwy nieswoistej w kontroli żywych tkanek (NSCliving) podlegają pełnej procedurze badawczej, ale podczas etapu inkubacji z MTT są inkubowane z pożywką zamiast z roztworem MTT (34)(35). Kontrolę NSCliving należy przeprowadzać równolegle z badaniem barwionej substancji chemicznej, a w przypadku wielokrotnego badania, niezależną kontrolę NSCliving należy przeprowadzić podczas każdego wykonanego badania (w każdej serii badawczej) z powodu naturalnej różnorodności biologicznej żywych tkanek. Rzeczywistą żywotność tkanek oblicza się jako: odsetek żywotności tkanek uzyskanej w wyniku narażenia żywych tkanek na zakłócanie badaną substancją chemiczną i inkubowanych w roztworze MTT (%Viabilitytest), pomniejszony o odsetek tkanek o barwie nieswoistej uzyskany w wyniku narażenia żywych tkanek na zakłócenie badaną substancją chemiczną i inkubowanych na podłożu bez MTT, w badaniu przeprowadzonym jednocześnie z korygowanym badaniem (%NSCliving), tj. rzeczywista żywotność tkanek = [%Viabilitytest] - [%NSCliving].
|
|
38.
|
W celu identyfikacji bezpośrednich reduktorów MTT, każdą badaną substancję chemiczną należy dodać do świeżo przygotowanego roztworu MTT. Do roztworu MTT dodaje się odpowiednią ilość badanej substancji chemicznej i inkubuje się tę mieszaninę przez około 3 godziny w normalnych warunkach hodowli kultur (zob. dodatki III i IV)(34)(35). Jeżeli mieszanina MTT zawierająca badaną substancję chemiczną (lub zawiesina w przypadku nierozpuszczalnej badanej substancji chemicznej) zmieni barwę na niebieską/fioletową, zakłada się, że badana substancja chemiczna bezpośrednio redukuje MTT i należy przeprowadzić pogłębioną kontrolę funkcjonalną nieżywotnych wytworów tkankowych RhCE, stosując niezależnie pomiar absorbancji wzorca (OD) lub procedurę spektrofotometrii HPLC/UPLC. Do tej dodatkowej kontroli funkcjonalnej używa się martwych tkanek, które wykazują jedynie resztkową aktywność metaboliczną, ale wchłaniają i zatrzymują badaną substancję chemiczną w podobny sposób, co tkanki żywotne. Martwe tkanki w VRM1 przygotowuje się, narażając je na działanie niskiej temperatury („uśmiercenie przez zamrożenie”). Martwe tkanki w VRM2 przygotowuje się za pomocą przedłużone inkubacji (np. co najmniej 24±1 h) w wodzie, po czym następuje przechowywanie w niskiej temperaturze („uśmiercenie wodą”). Każda substancja chemiczna redukująca MTT nanoszona jest na co najmniej dwie kontrpróby martwych tkanek, które przechodzą przez pełną procedurę badawczą, by wygenerować kontrolę nieswoistej redukcji MTT (NSMTT) (34)(35). Pojedyncza kontrola NSMTT jest wystarczająca w odniesieniu do każdej badanej substancji chemicznej, niezależnie od liczby przeprowadzonych niezależnych badań/serii badawczych. Rzeczywistą żywotność tkanek oblicza się jako: odsetek żywotności tkanek uzyskaną przy pomocy żywych tkanek narażonych na działanie reduktora MTT (%Viabilitytest) pomniejszony o odsetek nieswoistej redukcji MTT uzyskany w wyniku narażenia martwych tkanek na działanie tego samego reduktora MTT, obliczony relatywnie względem kontroli ujemnej przeprowadzonej jednocześnie z korygowanym badaniem (%NSMTT), tj. rzeczywista żywotność tkanek = [%Viabilitytest] - [%NSMTT].
|
|
39.
|
Badane substancje chemiczne, które zidentyfikowano jako powodujące zarówno zakłócenie barwy (zob. pkt 37), jak bezpośrednią redukcję MTT (zob. pkt 38), będą podczas wykonywania pomiaru absorbancji wzorca (OD) wymagały również trzeciego zestawu kontroli, oprócz kontroli NSMTT i NSCliving opisanych w poprzednich punktach. Dzieje się tak zazwyczaj w przypadku badanych substancji chemicznych o ciemnym zabarwieniu, które pochłaniają światło w zakresie 570±30 nm (np. niebieska, fioletowa, czarna), ponieważ ich barwa swoista utrudnia ocenę zdolności do bezpośredniej redukcji MTT, jak opisano w pkt 38. Stwarza to konieczność domyślnego stosowania kontroli NSMTT wraz z kontrolami NSCliving. Badane substancje chemiczne, w odniesieniu do których przeprowadza się kontrole NSMTT i NSCliving, mogą zostać wchłonięte i zatrzymane zarówno przez żywe, jak i martwe tkanki. Zatem w tym przypadku kontrola NSMTT może nie tylko korygować potencjalną bezpośrednią redukcję MTT wywołaną przez badaną substancję chemiczną, ale również zakłócenie barwy wynikającą z wchłaniania i zatrzymywania badanej substancji chemicznej przez martwe tkanki. Mogłoby to doprowadzić do podwójnej korekty zakłócenia barwy, ponieważ kontrola NSCliving koryguje już zakłócenie barwy wynikające z wchłaniania i zatrzymywania badanej substancji chemicznej przez żywe tkanki. Aby uniknąć podwójnej korekty możliwej w przypadku zakłócenia barwy, należy przeprowadzić trzecią kontrolę nieswoistego zabarwienia martwych tkanek (NSCkilled) (zob. dodatki III i IV)(34)(35). W trakcie tej dodatkowej kontroli badana substancja chemiczna nakładana jest na co najmniej dwie kontrpróby martwych kontrprób tkanek na czas narażenia, które podlegają pełnemu badaniu, ale są inkubowane z pożywką zamiast z roztworem MTT w trakcie etapu inkubacji z MTT. Pojedyncza kontrola NSCkilled jest wystarczająca dla każdej badanej substancji chemicznej, niezależnie od liczby przeprowadzonych niezależnych badań/serii badawczych, lecz powinna być przeprowadzana jednocześnie z kontrolą NSMTT z wykorzystaniem tej samej partii tkanki. Rzeczywistą żywotność tkanek oblicza się jako: odsetek żywotności tkanek uzyskanych za pomocą żywych tkanek narażonych na działanie badanej substancji chemicznej (%Viabilitytest) pomniejszony o %NSMTT, pomniejszony o%NSCliving, zwiększony o odsetek tkanek o nieswoistej barwie uzyskany z martwych tkanek narażonych na zakłócenie badaną substancją chemiczną i inkubowanych na podłożu bez MTT, obliczony w stosunku do kontroli ujemnej przeprowadzonej jednocześnie z korygowanym badaniem (%NSCkilled), tj. rzeczywista żywotność tkanek = [%Viabilitytest] - [%NSMTT] - [%NSCliving] + [%NSCkilled].
|
|
40.
|
Należy zauważyć, że nieswoista redukcja MTT i nieswoiste zakłócenia barwy mogą zwiększyć OD (podczas przeprowadzania pomiarów absorbancji wzorca) ekstraktu tkankowego powyżej zakresu liniowości spektrofotometru i że niespecyficzna redukcja MTT może również zwiększyć powierzchnię piku formazanu MTT (podczas przeprowadzania pomiaru spektrofotometrii HPLC/UPLC) ekstraktu tkankowego powyżej zakresu liniowości spektrofotometru. W związku z tym istotne jest, aby każde laboratorium przed rozpoczęciem badania badanych substancji chemicznych do celów regulacyjnych określiło liniowość zakresu OD / powierzchni piku swojego spektrofotometru za pomocą np. formazanu MTT (CAS # 57360-69-7) dostępnego na rynku np. od Sigma-Aldrich (nr katalogowy M2003).
|
|
41.
|
Pomiar absorbancji wzorca (OD) z wykorzystaniem spektrofotometru jest odpowiedni do oceny bezpośrednich reduktorów MTT oraz zakłócających barwę badanych substancji chemicznych, gdy odnotowane zakłócenie pomiaru formazanu MTT nie jest zbyt silne (np. gdy OD ekstraktów tkankowych uzyskanych za pomocą badanej substancji chemicznej bez skorygowania bezpośredniej redukcji MTT lub interferencji koloru mieszczą się w liniowym zakresie spektrofotometru). Niemniej jednak wyniki badanych substancji chemicznych wytwarzających %NSMTT lub %NSCliving ≥ 60 % (VRM1 oraz VRM2 w przypadku protokołu dotyczącego cieczy) lub 50 % (VRM2 w przypadku protokołu dotyczącego substancji stałych) kontroli ujemnej powinny być przyjmowane z ostrożnością, ponieważ stanowi to ustaloną granicę odcięcia stosowaną w VRM do odróżniania substancji chemicznych sklasyfikowanych od niesklasyfikowanych (zob. pkt 44). Nie można jednak przeprowadzić pomiaru absorbancji wzorca (OD) jeżeli zakłócenia pomiaru formazanu MTT są zbyt silne (np. jeżeli prowadzą do sytuacji, w której nieskorygowane wartości OD badanych ekstraktów tkankowych znajdą się poza liniowym zakresem spektrofotometru). Barwne badane substancje chemiczne lub badane substancje chemiczne barwiące się przy kontakcie z wodą lub izopropanolem, które zbyt silnie zakłócają pomiar absorbancji wzorca (OD) formazanu MTT, można wciąż poddać ocenie, wykorzystując spektrofotometrię HPLC/UPLC (zob. dodatki III i IV). Wynika to z tego, że system HPLC/UPLC umożliwia oddzielenie formazanu MTT od badanej substancji chemicznej przed jej oznaczeniem ilościowym (36). Z tego powodu kontrole NSCliving lub NSCkilled nie są nigdy wymagane, gdy wykorzystuje się spektrofotometrię HPLC/UPLC, niezależnie od badanej substancji chemicznej. Kontrole NSMTT należy jednak stosować, jeżeli istnieje podejrzenie, że badana substancja chemiczna bezpośrednio redukuje MTT (zgodnie z procedurą opisaną w pkt 38). Kontrole NSMTT należy również stosować w przypadku badania substancji chemicznych mających zabarwienie (swoiste lub pojawiające się, kiedy substancja znajduje się w wodzie), który utrudnia ocenę zdolności substancji do bezpośredniej redukcji MTT, jak opisano w pkt 38. W przypadku stosowania spektrofotometrii HPLC/UPLC do pomiaru formazanu MTT procent żywotności tkanek oblicza się jako procent MTT powierzchni piku formazanu uzyskanej z żywych tkanek poddanych działaniu badanej substancji chemicznej w stosunku do piku formazanu MTT uzyskanego przy równoczesnej kontroli ujemnej. W przypadku badanych substancji chemicznych zdolnych do bezpośredniej redukcji MTT, rzeczywistą żywotność tkanek oblicza się jako: %Viabilitytest
minus %NSMTT, jak opisano w ostatnim zdaniu pkt 38. Na koniec należy zauważyć, że bezpośrednich reduktorów MTT lub bezpośrednich reduktorów MTT powodujących również zakłócenie barwy, które są zatrzymywane w tkankach po poddaniu działaniu substancji i redukują MTT tak bardzo, że prowadzą do sytuacji, w której wartości OD (przy użyciu standardowego pomiaru OD) lub powierzchnie piku (przy użyciu spektrofotometrii UPLC/HPLC) badanych ekstraktów tkankowych wykraczają poza zakres liniowości spektrofotometru – nie można ocenić za pomocą metod badawczych RhCE, chociaż przewiduje się, że wystąpią tylko w bardzo rzadkich sytuacjach.
|
|
42.
|
Spektrofotometria HPLC/UPLC może być stosowana ze wszystkimi rodzajami badanych substancji chemicznych (barwnych, bezbarwnych, reduktorów MTT i substancji nieredukujących MTT) do pomiaru formazanu MTT (11)(36). Ponieważ systemy spektrofotometrii HPLC/UPLC są zróżnicowane, ustalanie przez wszystkich użytkowników dokładnie tych samych warunków dotyczących systemu jest niepraktyczne. W związku z tym należy wykazać kwalifikację systemu spektrofotometrii HPLC/UPLC przed jego zastosowaniem, aby określić ilościowo formazan MTT z ekstraktów tkankowych poprzez spełnienie kryteriów dopuszczalności zestawu standardowych parametrów kwalifikacyjnych w oparciu na kryteriach opisanych w wytycznych Agencji Żywności i Leków Stanów Zjednoczonych dotyczących przemysłu walidacji metodą bioanalityczną (36)(38). Te kluczowe parametry i kryteria ich dopuszczalności przedstawiono w dodatku 5. Po spełnieniu kryteriów dopuszczalności określonych w dodatku 5, system spektrofotometrii HPLC/UPLC uznaje się za kwalifikowany i gotowy do pomiaru formazanu MTT w warunkach doświadczalnych opisanych w niniejszej metodzie badawczej.
|
Kryteria dopuszczalności
|
43.
|
W przypadku każdej serii badawczej z wykorzystaniem partii tkanek RhCE, które przeszły kontrolę jakości (zob. pkt 30), tkanki poddane działaniu substancji przeznaczonej do celów kontroli ujemnej powinny wykazywać wartość OD odzwierciedlającą jakość tkanek, które przeszły wszystkie etapy wysyłki i odbioru oraz wszystkie procesy protokołu badania, i nie powinny mieścić się poza ustalonymi na podstawie danych historycznych granicami opisanymi w tabeli 2 (zob. pkt 26). Podobnie, tkanki poddane działaniu substancji służącej do kontroli dodatniej, tj. octan metylu, powinny wykazywać średnią żywotność tkanki < 50 % w odniesieniu do kontroli ujemnej w ramach VRM1 przy zastosowaniu albo protokołu dotyczącego cieczy, albo dotyczącego substancji stałych; oraz ≤ 30 % (protokół dotyczący cieczy) lub ≤ 20 % (protokół dotyczący substancji stałych) w odniesieniu do kontroli negatywnej w VRM2, tym samym odzwierciedlając zdolność tkanek do reakcji na drażniącą badaną substancję chemiczną w warunkach danej metody badawczej (34)(35). Zmienność między kontrpróbami tkanek w przypadku badanych substancji chemicznych i substancji kontrolnych powinna mieścić się w dopuszczalnych granicach (tj. różnica w żywotności między dwoma kontrpróbami tkanek powinna wynosić mniej niż 20 % lub odchylenie standardowe między trzema kontrpróbami tkanek nie powinno przekraczać 18 %). Jeżeli uwzględniona w serii badawczej kontrola ujemna lub dodatnia nie mieści się w dopuszczalnych zakresach, seria badawcza zostaje uznana za „niekwalifikującą się” i należy ją powtórzyć. Jeżeli zmienność między kontrpróbami tkanek w przypadku danej badanej substancji chemicznej wykracza poza dopuszczalne zakresy, badanie trzeba uznać za „niekwalifikujące się” i należy ponownie przebadać daną badaną substancję chemiczną.
|
Interpretacja wyników i model prognozowania
|
44.
|
Wartości / powierzchnie piku OD uzyskane przy wykorzystaniu kontrprób ekstraktów tkankowych dla każdej badanej substancji chemicznej należy wykorzystać do obliczenia średniej procentowej żywotności tkanek (średniej między kontrpróbami tkanek) znormalizowanej w odniesieniu do kontroli ujemnej, której żywotność ustala się na 100 %. Wartość graniczną procentowej żywotności tkanek służącą do identyfikowania badanych substancji chemicznych, które nie wymagają zaklasyfikowania do substancji powodujących podrażnienie lub poważne uszkodzenie oczu (brak kategorii wg GHS ONZ / rozporządzenia CLP), podano w tabeli 4. Wynik należy zatem interpretować w następujący sposób:
|
—
|
Badaną substancję chemiczną określa się jako niewymagającą klasyfikacji i oznakowania wg GHS ONZ i rozporządzenia CLP (brak kategorii), jeżeli średnia procentowa żywotności tkanek po narażeniu i inkubacji po narażeniu jest wyższa (>) niż ustalona wartość graniczna procentowej żywotności tkanek, jak przedstawiono w tabeli 4. W takim przypadku nie ma konieczności przeprowadzenia dalszych badań w ramach innych metod badawczych.
|
|
—
|
Jeżeli średnia procentowa żywotności tkanek po narażeniu i inkubacji po narażeniu jest niższa lub równa (≤) niż ustalona wartość graniczna procentowej żywotności tkanek, nie można przewidzieć działania, jak przedstawiono w tabeli 4. W takim przypadku niezbędne będzie przeprowadzenie dalszych badań w ramach innych metod badawczych, ponieważ metody badawcze RhCE dają szereg wyników fałszywie dodatnich (zob. pkt 14–15) i nie mogą rozróżnić między kategoriami 1 i 2 wg GHS ONZ i rozporządzenia CLP (zob. pkt 17).
|
Tabela 4
Modele prognozowania wg klasyfikacji GHS ONZ / rozporządzenia CLP
|
Kategoria VRM
|
Brak kategorii
|
Nie można przewidzieć działania
|
|
VRM1 - badanie działania drażniącego na oczy EpiOcular™ (w przypadku obu protokołów)
|
Średnia żywotność tkanek > 60 %
|
Średnia żywotność tkanek ≤ 60 %
|
|
VRM2 – badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™ (w przypadku protokołu dotyczącego cieczy)
|
Średnia żywotność tkanek > 60 %
|
Średnia żywotność tkanek ≤ 60 %
|
|
VRM2 – badanie działania drażniącego na oczy z wykorzystaniem ludzkiego nabłonka rogówki SkinEthic™ (w przypadku protokołu dotyczącego substancji stałych)
|
Średnia żywotność tkanek > 50 %
|
Średnia żywotność tkanek ≤ 50 %
|
|
|
45.
|
Jeśli wynik jest jednoznaczny, w przypadku badanej substancji chemicznej wystarczy pojedyncze badanie składające się z co najmniej dwóch kontrprób tkanek. W przypadku wyników granicznych, np. niezgodności powtarzalnych pomiarów lub średniej procentowej żywotności tkanek równej 60 ± 5 % (VRM1 i VRM2 w przypadku protokołu dotyczącego cieczy) lub 50 ± 5 % (VRM2 w przypadku protokołu dotyczącego substancji stałych), należy jednak rozważyć przeprowadzenie drugiego badania, jak również trzeciego w przypadku niezgodności między wynikami dwóch pierwszych badań.
|
|
46.
|
W stosownych i uzasadnionych przypadkach w odniesieniu do określonych rodzajów mieszanin można rozważyć zastosowanie innych wartości granicznych procentowej żywotności tkanek służących do odróżnienia badanych substancji chemicznych sklasyfikowanych i nieklasyfikowanych, aby zwiększyć ogólną skuteczność metody badawczej dla tych rodzajów mieszanin (zob. pkt 14). Wzorcowe substancje chemiczne mogą się okazać przydatne do oceny potencjału poważnego uszkodzenia oczu/podrażnienia oczu nieznanych badanych substancji chemicznych lub klasy produktowej lub do oceny względnego potencjału toksyczności sklasyfikowanej substancji chemicznej dla oczu w określonym zakresie reakcji dodatnich.
|
DANE I SPRAWOZDAWCZOŚĆ
Dane
|
47.
|
Dane z poszczególnych kontrprób tkanek w serii badawczej (np. dane dotyczące wartości OD / powierzchni piku formazanu MTT i obliczonej procentowej żywotności tkanek dla każdej badanej substancji chemicznej i kontroli, łącznie z końcową prognozą dotyczącą metody badawczej RhCE) należy przedstawić w formie tabelarycznej w odniesieniu do każdej badanej substancji chemicznej, w stosownych przypadkach uwzględniając dane z powtórzonych badań. Należy również przedstawić średnią procentową żywotność tkanek i różnicę w żywotności między dwoma kontrpróbami tkanek (jeżeli n = 2 kontrpróby tkanek) lub SD (jeżeli n ≥ 3 kontrprób tkanek) w odniesieniu do poszczególnych badanych substancji chemicznych i kontroli. Należy zgłosić, w odniesieniu do każdej badanej substancji chemicznej, wszelkie odnotowane zakłócenia w pomiarze formazanu MTT wynikające ze spowodowanej przez badaną substancję chemiczną bezpośredniej redukcji MTT lub zakłócenia barwy.
|
Sprawozdanie z badania
|
48.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
Badana substancja chemiczna
|
|
Substancja jednoskładnikowa
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(y) w rejestrze CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
—
|
stan skupienia, lotność, pH, LogP, masa cząsteczkowa, klasa chemiczna oraz dodatkowe istotne właściwości fizykochemiczne mające znaczenie dla przebiegu badania w zakresie, w jakim są dostępne;
|
|
—
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
—
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
—
|
dane na temat warunków przechowywania i stabilności w zakresie, w jakim są dostępne;
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszanina
|
—
|
maksymalnie szczegółowa charakterystyka wykonana np. za pomocą nazwy chemicznej (zob. powyżej), czystości, występowania ilościowego oraz istotnych właściwości fizykochemicznych składników (zob. powyżej), w zakresie, w jakim są dostępne;
|
|
—
|
stan skupienia oraz dodatkowe istotne właściwości fizykochemiczne mające znaczenie dla przebiegu badania w zakresie, w jakim są dostępne;
|
|
—
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
—
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
—
|
dane na temat warunków przechowywania i stabilności w zakresie, w jakim są dostępne;
|
|
Substancje służące do kontroli dodatniej i ujemnej
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(y) w rejestrze CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
—
|
stan skupienia, lotność, masa cząsteczkowa, klasa chemiczna oraz dodatkowe istotne właściwości fizykochemiczne mające znaczenie dla przebiegu badania w zakresie, w jakim są dostępne;
|
|
—
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
—
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
—
|
warunki przechowywania i stabilności w dostępnym zakresie;
|
|
—
|
uzasadnienie zastosowania innej kontroli ujemnej niż ultraczysta H2O lub DPBS wolne od Ca2+/Mg2+, w stosownych przypadkach;
|
|
—
|
uzasadnienie zastosowania innej kontroli dodatniej niż czysty octan metylu, w stosownych przypadkach;
|
|
—
|
odniesienie do wyników historycznych kontroli dodatnich i ujemnych, w których zastosowano odpowiednie kryteria dopuszczalności serii badawczej.
|
Informacje dotyczące sponsora i placówki przeprowadzającej badanie
|
—
|
nazwa i adres sponsora i placówki przeprowadzającej badanie oraz dane kierownika badań.
|
|
—
|
Zastosowany wytwór tkankowy RhCE i protokół (oraz uzasadnienie wyboru w stosownych przypadkach)
|
Warunki metody badawczej
|
—
|
zastosowany wytwór tkankowy RhCE, w tym numer partii;
|
|
—
|
długość fali i filtr środkowoprzepustowy (w stosownych przypadkach) stosowany do określenia ilościowego formazanu MTT oraz zakres liniowości urządzenia pomiarowego (np. spektrofotometru);
|
|
—
|
opis metody wykorzystanej do oceny ilościowej formazanu MTT;
|
|
—
|
opis stosowanego systemu spektrofotometrycznego HPLC/UPLC, w stosownych przypadkach;
|
|
—
|
pełne informacje podstawowe na temat stosowanego wytworu tkankowego RhCE, z uwzględnieniem jego efektywności. Powinno to między innymi obejmować:
|
i)
|
kontrolę jakości dotyczącą żywotności (dostawca);
|
|
ii)
|
żywotność w warunkach metody badawczej (użytkownik);
|
|
iii)
|
kontrolę jakości dotyczącą funkcji bariery;
|
|
iv)
|
morfologię, jeżeli jest dostępna;
|
|
v)
|
odtwarzalność i zdolność predykcyjną;
|
|
vi)
|
inne kontrole jakości (QC) dotyczące wytworu tkankowego RhCE, jeżeli są dostępne;
|
|
|
—
|
odwołanie do danych historycznych wytworu tkankowego RhCE. Powinno to między innymi obejmować: dopuszczalność danych QC z odwołaniem do danych dotyczących serii historycznych;
|
|
—
|
oświadczenie, że placówka badawcza wykazała się biegłością w stosowaniu metody badawczej przed przystąpieniem do jej rutynowego stosowania za pomocą badania substancji chemicznych przeznaczonych do oceny biegłości;
|
Kryteria dopuszczalności badania i serii badawczej
|
—
|
średnie wartości oraz zakresy dopuszczalności dodatniej i ujemnej kontroli na podstawie danych historycznych;
|
|
—
|
dopuszczalna zmienność wśród kontrprób tkanek w przypadku dodatniej i ujemnej kontroli;
|
|
—
|
dopuszczalna zmienność między kontrpróbami tkanek w przypadku badanej substancji chemicznej;
|
Procedura badania
|
—
|
informacje szczegółowe dotyczące zastosowanej procedury badawczej;
|
|
—
|
dawki podawanych badanych substancji chemicznych i substancji kontrolnych;
|
|
—
|
czas trwania i temperatura podczas okresów narażenia, zanurzenia po zakończeniu narażenia oraz inkubacji po zakończeniu narażenia (w stosownych przypadkach);
|
|
—
|
opis wszelkich modyfikacji w procedurze badawczej;
|
|
—
|
wskazanie prób kontroli używanych w odniesieniu do bezpośrednich reduktorów MTT lub badanych substancji chemicznych, które w wyniku reakcji przechodzą w barwny produkt, w stosownych przypadkach;
|
|
—
|
liczba kontrprób tkanek wykorzystanych na badaną substancję chemiczną i kontrolę (kontrola dodatnia, kontrola ujemna oraz NSMTT, NSCliving i NSCkilled, w stosownych przypadkach);
|
Wyniki
|
—
|
tabelaryczne zestawienie danych uzyskanych w badaniu poszczególnych badanych substancji chemicznych i substancji kontrolnych dla każdej serii badawczej (uwzględniając, w stosownych przypadkach, powtórzone doświadczenia) i każdego pomiaru kontrpróby, w tym wartości OD lub powierzchnia piku formazanu MTT, procentowa żywotność tkanek, średnia procentowa żywotność tkanek, różnica między kontrpróbami lub SD oraz prognoza końcowa;
|
|
—
|
w stosownych przypadkach wyniki kontroli używanych w odniesieniu do bezpośrednich reduktorów MTT lub barwnych substancji chemicznych, w tym wartości OD lub powierzchnia piku formazanu MTT, %NSMTT, %NSCliving, %NSCkilled, różnica pomiędzy kontrpróbami tkanki lub SD, ostateczna prawidłowa procentowa żywotność tkanek oraz prognoza końcowa;
|
|
—
|
wyniki uzyskane z zastosowaniem badanych substancji chemicznych i substancji kontrolnych w odniesieniu do określonych kryteriów dopuszczalności serii badawczej i badania;
|
|
—
|
opis innych zaobserwowanych skutków, np. zabarwienie tkanek przez barwioną substancja chemiczna;
|
Omówienie wyników
Wniosek
|
BIBLIOGRAFIA
|
(1)
|
ONZ (2015). Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS), ST/SG/AC.10/30/Rev.6, wydanie szóste zmienione, Nowy Jork i Genewa: Organizacja Narodów Zjednoczonych. Dostępne na stronie internetowej: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Rozdział B.5 niniejszego załącznika, Działanie żrące/silnie drażniące na oczy.
|
|
(3)
|
Rozdział B.47 niniejszego załącznika, Metoda badania zmętnienia i przepuszczalności rogówki u bydła do celów identyfikacji i) substancji chemicznych powodujących poważne uszkodzenie oczu oraz ii) substancji chemicznych, które nie wymagają zaklasyfikowania jako substancje drażniące oczy lub powodujące poważne uszkodzenie oczu.
|
|
(4)
|
Rozdział B.48 niniejszego załącznika, Metoda badania na izolowanym oku kurzym do celów identyfikacji i) substancji chemicznych powodujących poważne uszkodzenie oczu oraz ii) substancji chemicznych, które nie wymagają zaklasyfikowania.
|
|
(5)
|
Rozdział B.61 niniejszego załącznika, Metoda badania utraty fluoresceiny do celów identyfikacji substancji żrących i silnie drażniących dla oczu.
|
|
(6)
|
Rozdział B.68 niniejszego załącznika, Metoda badawcza in vitro krótkiego okresu narażenia do celów identyfikacji i) substancji chemicznych powodujących poważne uszkodzenie oczu oraz ii) substancji chemicznych, które nie wymagają zaklasyfikowania jako substancje drażniące oczy lub powodujące poważne uszkodzenie oczu.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX nr 27, Specjalne wydanie 2010, s. 261–266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Dostępne na stronie internetowej: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
Naukowy komitet doradczy EURL ECVAM (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. Opinia ESAC nr 2014-03 z dnia 17 listopada 2014 r.; EUR 28173 EN; doi: 10.2787/043697. Dostępne na stronie internetowej: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals.„Toxicol. In Vitro” nr 31, s. 43–53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing.„Toxicol. In Vitro” nr 34, s. 55–70.
|
|
(12)
|
Naukowy komitet doradczy EURL ECVAM (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). Opinia ESAC nr 2016-02 z dnia 24 czerwca 2016 r.; EUR 28175 EN; doi: 10.2787/390390. Dostępne na stronie internetowej: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Tekst w przygotowaniu).
|
|
(14)
|
J.H. Draize, G. Woodard i H.O. Calvery (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes.„Journal of Pharmacol. and Exp. Therapeutics” nr 82, s. 377–390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches.„Toxicol. In Vitro” nr 24, s. 1–9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. „Journal of Immunological Methods” Methods nr 65, s. 55–63.
|
|
(17)
|
OECD (2016). Seria dotycząca badań i oceny nr 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(18)
|
OECD (2005). Seria dotycząca badań i oceny nr 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. „Altern. Lab. Anim.” nr 39, s. 339–364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. [W:] Alternative Toxicological Methods, H. Salemi S.A. Katz (red.), CRC Press, s. 147–159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation.„Toxicol. In Vitro” nr 27, s. 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation.„Toxicol. In Vitro” nr 27, s. 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. „Altern. Lab. Anim.” nr 43, s. 1–18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. „Arch.” Toxicol. nr 88, s. 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). „Arch.” Toxicol. nr 91, s. 521–547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium.„Molecular Vision” nr 17, s. 113–126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. [W:] Advances in Modern Toxicology: Dermatoxicology F.N Marzulli i H.I. Maibach (red.), wydanie czwarte, s. 749–815. Waszyngton, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. [W:] Cassaret and Doull’s Toxicology: The Basic Science of Poisons C.D. Klaassen (red.), wydanie siódme, s. 665–697. Withby, ON, Kanada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. „Invest. Ophthalmol. Vis. Sci.” nr 39, s. 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. „Reg. Tox. Pharmacol.” nr 36, s. 106–117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests.„Toxicol. In Vitro” nr 15, s. 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. „Invest. Ophthalmol. Vis. Sci.” nr 39, s. 2610–2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test.„Cutan.”Ocul. Toxicol. nr 25, s. 41–54.
|
|
(34)
|
EpiOcular™ EIT SOP, wersja 8 (z dnia 5 marca 2013 r.). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Dostępne na stronie internetowej: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, wersja 1. (z dnia 20 lipca 2015 r.). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Dostępne na stronie internetowej: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals.„Toxicol. In Vitro” nr 29, s. 741–761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times.„Altern. Lab. Anim.” nr 43, s. 101–127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration, maj 2001. Dostępne na stronie internetowej: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. [W:] Seria dotycząca badań i oceny nr 263, publikacje na temat środowiska. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
Dodatek 1
DEFINICJE
Dokładność
: stopień zgodności wyników zastosowanej metody badawczej z przyjętymi wartościami odniesienia. Jest to miara efektywności metody badawczej i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na oznaczenie odsetka prawidłowych wyników uzyskiwanych przy użyciu metody badawczej (18).
Wzorcowa substancja chemiczna
: substancja chemiczna używana jako wzorzec do porównań z badaną substancją chemiczną. Wzorcowa substancja chemiczna powinna mieć następujące cechy: (i) stałe i wiarygodne źródło(-a) na potrzeby jej identyfikacji i charakterystyki; (ii) podobieństwo strukturalne, funkcjonalne, chemiczne lub związane z klasą produktową do badanych substancji chemicznych; (iii) znane właściwości fizyczne i chemiczne; (iv) dane potwierdzające znane skutki; oraz (v) znaną siłę działania w zakresie pożądanych reakcji.
Podejście oddolne
: stopniowe podejście stosowane w odniesieniu do badanej substancji chemicznej, w przypadku której podejrzewa się, że nie wymaga zaklasyfikowania i oznakowania jako substancja powodująca podrażnienie lub poważne uszkodzenie oka; takie podejście polega na odróżnieniu substancji chemicznych niewymagających zaklasyfikowania i oznakowania (wynik ujemny) od pozostałych substancji chemicznych (wynik dodatni).
Substancja chemiczna
: substancja lub mieszanina.
Zgodność
: zob. „dokładność”.
Rogówka
: przezroczysta warstwa w przedniej części gałki ocznej, która okrywa tęczówkę i źrenicę oraz wpuszcza światło do wnętrza oka.
CV
: Współczynnik zmienności.
Dev
: odchylenie.
EIT
: badanie działania drażniącego na oczy.
EURL ECVAM
: laboratorium referencyjne Unii Europejskiej ds. metod alternatywnych wobec testów na zwierzętach.
Podrażnienie oka
: zmiany w oku spowodowane zaaplikowaniem badanej substancji chemicznej na wierzchnią warstwę oka, które są w pełni odwracalne w ciągu 21 dni po zaaplikowaniu. Określenie używane zamiennie z określeniem „odwracalne skutki działania na oczy” oraz ze sformułowaniem „kategoria 2 wg GHS ONZ / rozporządzenia CLP”.
ET50
: czas narażenia na działanie potrzebny do zmniejszenia żywotności tkanek o 50 % po zastosowaniu wzorcowej substancji chemicznej w określonym, stałym stężeniu.
Odsetek wyników fałszywie ujemnych
: odsetek wszystkich substancji dających wynik dodatni fałszywie zidentyfikowanych po zastosowaniu metody badawczej jako dające wynik ujemny. Jest to jeden ze wskaźników efektywności metody badawczej.
Odsetek wyników fałszywie dodatnich
: odsetek wszystkich substancji dających wynik ujemny fałszywie zidentyfikowanych po zastosowaniu metody badawczej jako dające wynik dodatni. Jest to jeden ze wskaźników efektywności metody badawczej.
Zagrożenie
: nieodłączna właściwość czynnika lub sytuacja, która może potencjalnie doprowadzić do niekorzystnych skutków w przypadku narażenia organizmu, systemu lub (sub) populacji na taki czynnik.
HCE
: ludzki nabłonek rogówki SkinEthic™.
HPLC
: wysokosprawna chromatografia cieczowa.
IC50
: stężenie, w którym wzorcowa substancja chemiczna powoduje zmniejszenie żywotności tkanek o 50 % po stałym czasie narażenia na działanie (np. 30-minutowe poddanie działaniu substancji z wykorzystaniem SDS).
Dawka nieskończona
: ilość badanej substancji chemicznej nakładana na wytwór tkankowy RhCE przekraczająca ilość konieczną do całkowitego, jednolitego pokrycia powierzchni nabłonka.
Nieodwracalne skutki działania na oczy
: zob. „Poważne uszkodzenie oczu”.
LLOQ
: dolna granica oznaczalności.
LogP
: Logarytm współczynnika podziału oktanol-woda
Mieszanina
: mieszanina lub roztwór, które składają się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
Substancja wieloskładnikowa
: substancja zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥ 10 % (w/w) i < 80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się w wyniku zmieszania co najmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
MTT
: bromek 3-(4,5-dimetylotiazol-2-ilo)-2,5-difenylotetrazoliowy MTT.
Kontrola ujemna
: próbka zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że nie wywołuje reakcji dodatniej w układzie badawczym. Próbkę tę przetwarza się razem z próbkami poddanymi działaniu badanej substancji chemicznej i z innymi próbkami kontrolnymi oraz wykorzystuje się ją do określenia 100 % żywotności komórek.
Niesklasyfikowane
: substancje chemiczne, które nie zostały zaklasyfikowane jako substancje podrażniające oczy (kategoria 2 wg GHS/CLP ONZ, kategoria 2A lub 2B wg GHS ONZ) ani substancje powodujące poważne uszkodzenie oczu (kategoria 1 wg GHS/CLP ONZ). Określenie używane zamiennie ze sformułowaniem „brak kategorii wg GHS/CLP ONZ”.
NSCkilled
: barwa nieswoista martwych tkanek.
NSCliving
: barwa nieswoista żywych tkanek.
NSMTT
: nieswoista redukcja MTT.
OD
: gęstość optyczna.
Standardy wykonywania badań (PS)
: standardy oparte na zwalidowanej metodzie badawczej, którą uznano za naukowo uzasadnioną, zapewniające podstawę do oceny porównywalności proponowanej metody badania, która jest podobna do metody referencyjnej pod względem mechanistycznym i funkcjonalnym. Uwzględnia się: (i) zasadnicze elementy metody badawczej; (ii) podstawowy wykaz chemicznych substancji odniesienia wybranych spośród substancji chemicznych wykorzystywanych w celu wykazania dopuszczalnej efektywności zwalidowanej metody badania; oraz (iii) podobny poziom dokładności i wiarygodności, na podstawie wyników uzyskanych dla zwalidowanej metody badania, który proponowana metoda badania powinna osiągnąć przy ocenie z wykorzystaniem podstawowego wykazu chemicznych substancji odniesienia (18).
Kontrola dodatnia
: próbka zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że wywołuje reakcję dodatnią w układzie badawczym. Próbkę tę przetwarza się razem z próbkami poddanymi działaniu badanej substancji chemicznej i z innymi próbkami kontrolnymi. Aby zapewnić możliwość oceny zmienności reakcji w kontroli dodatniej w czasie, siła reakcji w postaci silnego działania drażniącego nie może być zbyt duża.
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono znaczące i użyteczne z punktu widzenia określonego celu. Jest to zakres, w jakim badanie prawidłowo mierzy lub przewiduje biologiczny oczekiwany skutek. Istotność obejmuje uwzględnienie dokładności (zgodności) metody badania (18).
Wiarygodność
: miary zakresu, w jakim metoda badawcza może być przeprowadzana w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jej przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją poprzez obliczenie odtwarzalności wewnątrzlaboratoryjnej i międzylaboratoryjnej (18).
Badanie zastępcze
: badanie, które ma na celu zastąpienie badania stosowanego rutynowo i zaakceptowanego do identyfikacji zagrożeń lub oceny ryzyka i w odniesieniu do którego ustalono, że w porównaniu do zaakceptowanego badania zapewnia odpowiednio równoważną lub lepszą ochronę zdrowia ludzi lub zwierząt lub środowiska we wszystkich możliwych warunkach badania i w odniesieniu do wszystkich możliwych substancji chemicznych (18).
Odtwarzalność
: zgodność wyników uzyskanych w wyniku powtarzanego badania tej samej badanej substancji chemicznej przy użyciu tego samego protokołu badania (zob. „Wiarygodność”) (18).
Odwracalne skutki działania na oczy
: zob. „Działanie drażniące na oczy”.
RhCE
: zrekonstruowany ludzki nabłonek przypominający rogówkę.
Seria badawcza
: Seria badawcza obejmuje przynajmniej jedną badaną substancję chemiczną, badaną równocześnie z kontrolą ujemną i kontrolą dodatnią.
SD
: odchylenie standardowe.
Czułość
: odsetek wszystkich dodatnich/aktywnych substancji chemicznych prawidłowo sklasyfikowanych w badaniu. Jest to miara dokładności metody badania, która daje wyniki kategoryczne, stanowiąca parametr istotny dla oceny istotności metody badania (18).
Poważne uszkodzenie oczu
: uszkodzenie tkanki oka lub poważne fizyczne pogorszenie widzenia spowodowane zaaplikowaniem badanej substancji chemicznej na przednią powierzchnię oka, które nie są w pełni odwracalne w ciągu 21 dni po zaaplikowaniu. Określenie jest używane zamiennie z określeniem „nieodwracalne skutki działania na oczy” oraz ze sformułowaniem „kategoria 1 wg GHS i CLP ONZ”.
Standardowe procedury operacyjne (SPO)
: formalne, sporządzone w formie pisemnej procedury szczegółowo opisujące sposób, w jaki należy przeprowadzać właściwe rutynowe operacje laboratoryjne oraz operacje laboratoryjne właściwe dla danego badania. Procedur tych wymaga się w ramach dobrej praktyki laboratoryjnej.
Swoistość
: odsetek wszystkich negatywnych/nieczynnych badanych chemikaliów prawidłowo sklasyfikowanych w badaniu. Jest to miara dokładności metody badania, która daje wyniki kategoryczne, stanowiąca parametr istotny dla oceny istotności metody badania (18).
Substancja
: pierwiastek chemiczny i jego związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania trwałości produktu i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem wszelkich rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Badanie
: pojedyncza badana substancja chemiczna badana jednocześnie na co najmniej dwóch kontrpróbach tkanek, co określono w odpowiadającej standardowej procedurze operacyjnej.
Żywotność tkanek
: parametr określający całkowitą aktywność populacji komórek w zrekonstruowanych tkankach jako ich zdolność do obniżenia zawartości barwnika przyżyciowego MTT, który, w zależności od pomiaru końcowego i zastosowanego schematu badania odpowiada całkowitej liczbie lub żywotności komórek.
Podejście odgórne
: stopniowe podejście stosowane w odniesieniu do substancji chemicznej, w przypadku której podejrzewa się, że powoduje poważne uszkodzenie oczu; takie podejście polega na odróżnieniu substancji chemicznych powodujących poważne uszkodzenie oczu (wynik dodatni) od pozostałych substancji chemicznych (wynik ujemny).
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
Wielopoziomowa strategia badań
: Strategia badań sekwencyjnych, w ramach której metody badawcze stosuje się w sposób sekwencyjny. Wszystkie istniejące informacje na temat badanej substancji chemicznej są analizowane na każdym poziomie z zastosowaniem procesu uwzględniającego wagę dowodów w celu określenia, czy dostępna jest wystarczająca ilość informacji do podjęcia decyzji o klasyfikacji zagrożenia, przed przejściem do następnego poziomu w obrębie strategii. Jeżeli na podstawie dostępnych informacji można przypisać badanej substancji chemicznej potencjał / siłę działania w zakresie zagrożenia, dodatkowe badania nie są wymagane (18).
ULOQ
: górna granica oznaczalności.
Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ)
: system obejmujący klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz odpowiednie elementy komunikacyjne, takie jak: piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki, mające informować na temat szkodliwego działania tych substancji chemicznych w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) oraz środowiska (1).
Kategoria 1 wg GHS/CLP ONZ
: zob. „Poważne uszkodzenie oczu”.
Kategoria 2 wg GHS/CLP ONZ
: zob. „Działanie drażniące na oczy”.
Brak kategorii wg GHS/CLP ONZ
: Substancje chemiczne, które nie spełniają wymogów w zakresie zaklasyfikowania jako należące do kategorii 1 lub 2 (lub kategorii 2A lub 2B wg GHS) wg GHS/CLP ONZ. Określenie używane zamiennie ze sformułowaniem „Niesklasyfikowane”.
UPLC
: ultrawysokosprawna chromatografia cieczowa.
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
Właściwa metoda badawcza
: metoda badawcza, którą uznano za wystarczająco istotną i wiarygodną dla danego celu i która opiera się na uzasadnionych naukowo podstawach. Metoda badawcza nie może zostać uznana za właściwą w sensie absolutnym, ale tylko w odniesieniu do określonego celu (18).
Zweryfikowana metoda badawcza
: metoda badawcza, w odniesieniu do której zakończono badania walidacyjne w celu określenia jej istotności (w tym dokładności) i wiarygodności w odniesieniu do konkretnego celu. Należy zauważyć, że zweryfikowana metoda badawcza może nie wykazywać dostatecznej efektywności z punktu widzenia dokładności i wiarygodności, aby można było ją uznać za dopuszczalną w odniesieniu do danego celu (18).
Kategoria VRM
: zwalidowana metoda referencyjna.
VRM1
: badanie działania drażniącego na oczy EpiOcular™ jest określone jako zwalidowana metoda referencyjna 1.
VRM2
: badanie działania drażniącego na oczy ludzkiego nabłonka rogówki SkinEthic™ jest określone jako zwalidowana metoda referencyjna 2.
Waga dowodów
: proces analizowania mocnych i słabych stron różnych informacji podczas formułowania wniosku dotyczącego potencjalnego zagrożenia stwarzanego przez badaną substancję oraz uzasadnienia takiego wniosku.
Dodatek 2
GŁÓWNE ELEMENTY BADAWCZE BADAŃ RHCE ZWERYFIKOWANYCH DO IDENTYFIKACJI SUBSTANCJI CHEMICZNYCH NIEWYMAGAJĄCYCH KLASYFIKACJI I OZNAKOWANIA POD WZGLĘDEM DZIAŁANIA DRAŻNIĄCEGO NA OCZY LUB POWODOWANIA POWAŻNYCH USZKODZEŃ OCZU
|
Elementy badawcze
|
badanie działania drażniącego na oczy EpiOcular™
(VRM 1)
|
badanie działania drażniącego na oczy ludzkiego nabłonka rogówki SkinEthic™
(VRM 2)
|
|
Protokoły
|
Ciecze
(pipetowane w temperaturze 37 ±1 °C lub niższej przez 15 min)
|
Ciała stałe
(nie można odmierzać pipetą)
|
Ciecze i substancje lepkie
(pipetowane)
|
Ciała stałe
(nie można odmierzać pipetą)
|
|
Powierzchnia modelu
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Liczba kontrprób tkanki
|
Co najmniej 2
|
Co najmniej 2
|
Co najmniej 2
|
Co najmniej 2
|
|
Wstępna kontrola zakłócenia barw
|
50 μl + 1 ml H2O przez 60 min w temperaturze 37 ±2 °C, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 % (niebarwne badane substancje chemiczne) lub 50 μl + 2 ml izopropanolu mieszanego przez 2–3 godz. w temperaturze pokojowej (barwne badane substancje chemiczne)
|
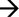 jeżeli gęstość optyczna badanej substancji chemicznej przy 570 ±20 nm po odjęciu gęstości optycznej izopropanolu lub wody wynosi > 0,08 (co odpowiada około 5 % średniej gęstości optycznej kontroli ujemnej), należy przeprowadzić kontrole dostosowane do warunków bytowania.
jeżeli gęstość optyczna badanej substancji chemicznej przy 570 ±20 nm po odjęciu gęstości optycznej izopropanolu lub wody wynosi > 0,08 (co odpowiada około 5 % średniej gęstości optycznej kontroli ujemnej), należy przeprowadzić kontrole dostosowane do warunków bytowania.
|
|
50 mg + 1 ml H2O przez 60 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
(niebarwne badane substancje chemiczne) lub
50 mg + 2 ml izopropanolu mieszanego przez 2–3 godz. w temperaturze pokojowej (barwione i niebarwione badane substancje chemiczne)
|
 jeżeli gęstość optyczna badanej substancji chemicznej przy 570 ±20 nm po odjęciu gęstości optycznej izopropanolu lub wody wynosi > 0,08 (co odpowiada około 5 % średniej gęstości optycznej kontroli ujemnej), należy przeprowadzić kontrole dostosowane do warunków bytowania.
jeżeli gęstość optyczna badanej substancji chemicznej przy 570 ±20 nm po odjęciu gęstości optycznej izopropanolu lub wody wynosi > 0,08 (co odpowiada około 5 % średniej gęstości optycznej kontroli ujemnej), należy przeprowadzić kontrole dostosowane do warunków bytowania.
|
|
10 μl + 90 μl H2O mieszane przez 30 ±2 min w temperaturze pokojowej (tj. 18–28 oC)
|
 jeżeli badana substancja chemiczna uległa zabarwieniu, należy wykonać żywe dostosowane kontrole
jeżeli badana substancja chemiczna uległa zabarwieniu, należy wykonać żywe dostosowane kontrole
|
|
10 mg + 90 μl H2O mieszane przez 30 ±2 min w temperaturze pokojowej
|
 jeżeli badana substancja chemiczna uległa zabarwieniu, należy wykonać żywe dostosowane kontrole
jeżeli badana substancja chemiczna uległa zabarwieniu, należy wykonać żywe dostosowane kontrole
|
|
|
Wstępna kontrola przed bezpośrednią redukcją MTT
|
50 μl + 1 ml roztworu MTT 1 mg/ml przez 180 ±15 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
 jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole dostosowane do uśmiercenia poprzez zamrożenie
jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole dostosowane do uśmiercenia poprzez zamrożenie
(jako kontrolę ujemną stosuje się 50 μl sterylnej wody dejonizowanej w roztworze MTT)
|
|
50 mg + 1 ml roztworu MTT 1 mg/ml przez 180 ±15 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
 jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać dostosowane kontrole uśmiercane przez zamrożenie
jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać dostosowane kontrole uśmiercane przez zamrożenie
(jako kontrolę ujemną stosuje się 50 μl sterylnej wody dejonizowanej w roztworze MTT)
|
|
30 μl + 300 μl roztworu MTT przez 180 ±15 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
 jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole martwych tkanek uśmiercanych wodą
jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole martwych tkanek uśmiercanych wodą
(jako kontrolę ujemną stosuje się 30 μl sterylnej dejonizowanej wody w roztworze MTT)
|
|
30 mg + 300 μl MTT roztwór 1 mg/ml przez 180 ±15 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
 jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole martwych tkanek uśmiercanych wodą
jeżeli roztwór zmienia barwę na niebieską lub fioletową, należy wykonać kontrole martwych tkanek uśmiercanych wodą
(jako kontrolę ujemną stosuje się 30 μl sterylnej dejonizowanej wody w roztworze MTT)
|
|
|
Obróbka wstępna
|
20 μl DPBS wolnego od Ca2+/Mg2+
przez 30 ±2 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %, chronić przed światłem.
|
20 μl DPBS wolnego od Ca2+/Mg2+
przez 30 ±2 min w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %, chronić przed światłem.
|
—
|
—
|
|
Dawkowanie i stosowanie
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2), wykorzystując skalibrowane narzędzie (np. wypoziomowanej łyżki skalibrowanej do pomieszczenia 50 mg chlorku sodu).
|
10 μl DPBS wolnego od Ca2+/Mg2+ + 30 ±2 μl (60 μl/cm2)
W przypadku substancji lepkich, należy zastosować siatkę nylonową
|
30 μl DPBS wolnego od Ca2+/Mg2+ + 30 ±2 mg (60 mg/cm2)
|
|
Czas i temperatura narażania
|
30 min (±2 min)
w podłożu
w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
6 godzin (±0,25 godz.)
w podłożu
w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
30 min (±2 min)
w podłożu
w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
4 godziny (±0,1 godz.)
w podłożu
w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
|
Płukanie w temperaturze pokojowej
|
3 razy w 100 ml DPBS wolnego od Ca2+/Mg2+
|
3 razy w 100 ml DPBS wolnego od Ca2+/Mg2+
|
20 ml DPBS wolnego od Ca2+/Mg2+
|
25 ml DPBS wolnego od Ca2+/Mg2+
|
|
Zanurzanie po zakończeniu narażenia
|
przez 12 min (±2 min) w podłożu w temperaturze pokojowej
|
przez 25 min (±2 min) w podłożu w temperaturze pokojowej
|
przez 30 min (±2 min) w temperaturze 37 oC, atmosferze zawierającej 5 % CO2 i w warunkach względnej wilgotności w podłożu wynoszącej ≥ 95 %
|
przez 30 min (±2 min) w podłożu w temperaturze pokojowej
|
|
Inkubacja po zakończeniu narażenia
|
120 min (±15 min) w podłożu w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
18 godz. (±0,25 godz.) w podłożu w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
brak
|
18 godz. (±0,5 godz.) w podłożu w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
|
Kontrola ujemna
|
50 μl H2O
Badana równolegle
|
50 μl H2O
Badana równolegle
|
30 ±2 μl DPBS wolnego od Ca2+/Mg2+
Badana równolegle
|
30 ±2 μl DPBS wolnego od Ca2+/Mg2+
Badana równolegle
|
|
Kontrola dodatnia
|
50 μl octanu metylu
Badana równolegle
|
50 μl octanu metylu
Badana równolegle
|
30 ±2 μl octanu metylu
Badana równolegle
|
30 ±2 μl octanu metylu
Badana równolegle
|
|
Roztwór MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
Czas i temperatura narażania MTT
|
przez 180 min (±15 min) w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
przez 180 min (±15 min) w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
przez 180 min (±15 min) w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
przez 180 min (±15 min) w temperaturze 37 ±2 oC, atmosferze zawierającej 5 ±1 % CO2 i w warunkach względnej wilgotności wynoszącej ≥ 95 %
|
|
Ekstrahent
|
2 ml izopropanolu
(ekstrakcja z dołu i góry nasadki poprzez przekłucie tkanki)
|
2 ml izopropanolu
(ekstrakcja z dołu nasadki poprzez przekłucie tkanki)
|
1,5 ml izopropanolu
(ekstrakcja z dolnej i górnej części wkładki)
|
1,5 ml izopropanolu
(ekstrakcja z dołu nasadki)
|
|
Czas trwania i temperatura ekstrakcji
|
2–3 godz. ze wstrząsaniem (~120 obr/min) w temperaturze pokojowej lub przez noc w temperaturze 4–10 °C
|
2–3 godz. ze wstrząsaniem (~120 obr/min) w temperaturze pokojowej lub przez noc w temperaturze 4–10 °C
|
4 godz. ze wstrząsaniem (~120 obr/min) w temperaturze pokojowej lub bez wstrząsania co najmniej przez noc w temperaturze 4–10 °C
|
Co najmniej 2 godz. ze wstrząsaniem (~120 obr/min) w temperaturze pokojowej
|
|
Odczyt OD
|
570 nm (550–590 nm)
bez filtra referencyjnego
|
570 nm (550–590 nm)
bez filtra referencyjnego
|
570 nm (540–600 nm)
bez filtra referencyjnego
|
570 nm (540–600 nm)
bez filtra referencyjnego
|
|
Kontrola jakości tkanki
|
Poddawanie działaniu 100 μl Triton X-100 0,3 % (obj./obj.)
12,2 min ≤ ET50 ≤ 37,5 min
|
Poddawanie działaniu 100 μl Triton X-100 0,3 % (obj./obj.)
12,2 min ≤ ET50 ≤ 37,5 min
|
Poddawanie działaniu SDS przez 30 min (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
Poddawanie działaniu SDS przez 30 min (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Kryteria dopuszczalności
|
|
1.
|
Średnia wartość OD kontrprób tkanek poddanych działaniu kontroli ujemnej powinna wynosić > 0,8 i < 2,5
|
|
2.
|
Średnia żywotność kontrprób tkanek poddawanych narażeniu przez 30 min za pomocą kontroli dodatniej, wyrażona jako odsetek kontroli dodatnich, powinna wynosić < 50 %
|
|
3.
|
Różnica w żywotności między dwoma kontrpróbami tkanek powinna wynosić mniej niż 20 %.
|
|
|
1.
|
Średnia wartość OD kontrprób tkanek poddanych działaniu kontroli ujemnej powinna wynosić > 0,8 i < 2,5
|
|
2.
|
Średnia żywotność kontrprób tkanek poddawanych narażeniu przez 6 godzin za pomocą kontroli dodatniej wyrażona jako odsetek kontroli ujemnych powinna wynosić < 50 %
|
|
3.
|
Różnica w żywotności między dwoma kontrpróbami tkanek powinna wynosić mniej niż 20 %.
|
|
|
1.
|
Średnia wartość OD kontrprób tkanek poddanych działaniu kontroli ujemnej powinna wynosić > 1,0 i ≤ 2,5
|
|
2.
|
Średnia żywotność kontrprób tkanek poddawanych narażeniu przez 30 min za pomocą kontroli dodatniej wyrażona jako odsetek kontroli dodatnich powinna wynosić ≤ 30 %
|
|
3.
|
Różnica w żywotności między dwoma kontrpróbami tkanek powinna wynosić mniej niż 20 %.
|
|
|
1.
|
Średnia wartość OD kontrprób tkanek poddanych działaniu kontroli ujemnej powinna wynosić > 1,0 i ≤ 2,5
|
|
2.
|
Średnia żywotność kontrprób tkanek poddawanych narażeniu przez 4 godziny za pomocą kontroli dodatniej wyrażona jako odsetek kontroli ujemnych powinna wynosić ≤ 20 %
|
|
3.
|
Różnica w żywotności między dwoma kontrpróbami tkanek powinna wynosić mniej niż 20 %.
|
|
Dodatek 3
ILUSTRACYJNY SCHEMAT POSTĘPOWANIA, OPIERAJĄC SIĘ NA STANDARDOWEJ PROCEDURZE OPERACYJNEJ ZWALIDOWANEJ METODY WALIDACYJNEJ 1, PRZEDSTAWIA WYTYCZNE ODNOŚNIE DO IDENTYFIKACJI I RADZENIA SOBIE Z BEZPOŚREDNIMI REDUKTORAMI MTT LUB SUBSTANCJAMI CHEMICZNYMI ZAKŁÓCAJĄCYMI BARWĘ

Dodatek 4
ILUSTRACYJNY SCHEMAT POSTĘPOWANIA, OPIERAJĄC SIĘ NA STANDARDOWEJ PROCEDURZE OPERACYJNEJ ZWALIDOWANEJ METODY WALIDACYJNEJ 2, PRZEDSTAWIA WYTYCZNE ODNOŚNIE DO IDENTYFIKACJI I RADZENIA SOBIE Z BEZPOŚREDNIMI REDUKTORAMI MTT LUB SUBSTANCJAMI CHEMICZNYMI ZAKŁÓCAJĄCYMI BARWĘ

Dodatek 5
KLUCZOWE PARAMETRY I KRYTERIA DOPUSZCZALNOŚCI SYSTEMU SPEKTROFOTOMETRYCZNEGO HPLC/UPLC DO POMIARU FORMAZANU MTT POBRANEGO Z WYTWORÓW TKANKOWYCH RHCE
|
Parametr
|
Protokół wyprowadzony z Wytycznej FDA (36) (38)
|
Kryteria dopuszczalności
|
|
Selektywność
|
Analiza izopropanolu, żywej ślepej próby (izopropanolu pobranego z żyjącego wytworu tkankowego RhCE niepoddanego działaniu jakichkolwiek substancji), martwej ślepej próby (izopropanolu pobranego z martwego wytworu tkankowego RhCE niepoddanego działaniu jakichkolwiek substancji) oraz barwika (np. błękitu metylenowego)
|
Obszarinterferencja ≤ 20 % ObszarLLOQ
(87)
|
|
Precyzja
|
Kontrole jakości (tj. formazan MTT w stężeniu 1,6 μg/ml, 16 μg/ml oraz 160 μg/ml) w izopropanolu (n = 5)
|
CV ≤ 15 % lub ≤ 20 % w odniesieniu do LLOQ
|
|
Dokładność
|
Kontrole jakości w izopropanolu (n = 5)
|
% Dev ≤ 15 % lub ≤ 20 % w odniesieniu do LLOQ
|
|
Efekt matrycy
|
Kontrole jakości na żywej ślepej próbie (n = 5)
|
85 % ≤ % efektu macierzystego ≤ 115 %
|
|
Środek przeniesiony
|
Analiza izopropanolu według normy ULOQ (88)
|
Obszarinterferencja ≤ 20 % ObszarLLOQ
|
|
Odtwarzalność (śróddzienna)
|
3 niezależne krzywe wzorcowe (na podstawie 6 kolejnych roztworów 1/3 formazanu MTT w izopropanolu, poczynając od ULOQ, np. 200 μg/ml);
Kontrole jakości w izopropanolu (n = 5)
|
Krzywe kalibracyjne: % Dev ≤ 15 % lub ≤ 20 % w odniesieniu do LLOQ
Kontrole jakości: % Dev ≤ 15 % oraz CV ≤ 15 %
|
|
Odtwarzalność (między kolejnymi dniami)
|
Dzień 1: 1 krzywa wzorcowa oraz kontrole jakości w izopropanolu (n = 3)
Dzień 2: 1 krzywa wzorcowa oraz kontrole jakości w izopropanolu (n = 3)
Dzień 3: 1 krzywa wzorcowa oraz kontrole jakości w izopropanolu (n = 3)
|
|
Krótkookresowa stabilność formazanu MTT w ekstrakcie tkankowym RhCE
|
Kontrole jakości na żywej ślepej próbie (n = 3) analizowane w dniu przygotowania oraz po 24 godzinach składowania w temperaturze pokojowej
|
% Dev ≤ 15 %
|
|
W razie potrzeby – długookresowa stabilność formazanu MTT w ekstrakcie tkankowym RhCE
|
Kontrole jakości w żywej ślepej próbie (n=3) analizowane w dniu przygotowania oraz po kilku dniach przechowywania w temperaturze -20 °C
|
% Dev ≤ 15 %
|
B.70 TESTY IN VITRO LUDZKIEGO REKOMBINOWANEGO RECEPTORA ESTROGENOWEGO (hrER) MAJĄCE NA CELU WYKRYCIE SUBSTANCJI CHEMICZNYCH WYKAZUJĄCYCH POWINOWACTWO WIĄZANIA RECEPTORA ESTROGENOWEGO
WPROWADZENIE OGÓLNE
Wytyczna OECD dotycząca badań w oparciu o wyniki
|
1.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej dotyczącej badań OECD nr 493 (2015). TG 493 jest wytyczną dotyczącą badań w oparciu o wyniki, która opisuje metodykę ludzkich rekombinowanych testów in vitro przeznaczonych do wykrywania substancji wykazujących powinowactwo wiązania receptora estrogenowego (testy wiązania hrER). Zawiera ona dwa testy podobne pod względem mechanistycznym i funkcjonalnym mające na celu określenie substancji wiążących receptora estrogenowego (tj. receptora estrogenowego alfa), które powinny ułatwiać opracowanie nowych – podobnych lub zmienionych – testów zgodnie z zasadami walidacji określonymi w wytycznych OECD pod tytułem Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1). W pełni zwalidowane referencyjne metody badań (dodatek 2 i dodatek 3), które stanowią podstawę tej wytycznej dotyczącej badań w oparciu o wyniki, to:
|
—
|
test in vitro Freybergera–Wilsona dotyczący wiązania receptora estrogenowego i wykorzystujący pełnowymiarowy ludzki rekombinowany receptor estrogenowy alfa (2), oraz
|
|
—
|
test in vitro Instytutu Oceny Chemicznej i Badań (ang. Chemical Evaluation and Research Institute, CERI) dotyczący wiązania receptora estrogenowego i wykorzystujący rekombinowane ludzkie białko domeny wiążącej ligand (2).
|
Dostępność standardów wykonywania badań (3) umożliwia łatwiejszy rozwój i walidację podobnych metod badawczych dotyczących tego samego zagrożenia punktu końcowego, a także pozwalają one na terminową zmianę wytycznej dotyczącej badań w oparciu o wyniki nr 493, aby do zaktualizowanej wytycznej dotyczącej badań w oparciu o wyniki można było dodać nowe podobne testy. Podobne testy badawcze zostaną jednak dodane wyłącznie po przeglądzie przeprowadzonym przez OECD i po uzgodnieniu przez tę organizację, że standardy wykonywania badań zostały spełnione. Testy zawarte w wytycznej dotyczącej badań nr 493 można stosować w sposób nieograniczony w celu spełnienia wymogów państw członkowskich OECD w zakresie wyników badań dotyczących wiązania receptora estrogenowego przy jednoczesnym korzystaniu z wzajemnego uznawania danych OECD.
|
Kontekst i zasady dotyczące testów zawartych w niniejszej metodzie badawczej
|
2.
|
W 1998 r. OECD podjęła działania o wysokim priorytecie, mające na celu zmianę istniejących wytycznych i opracowanie nowych wytycznych dotyczących badań na potrzeby badań klasyfikacyjnych i badań substancji potencjalnie zaburzających funkcjonowanie układu hormonalnego. W 2012 r. zmieniono ramy koncepcyjne OECD dotyczące badania i oceny substancji potencjalnie zaburzających funkcjonowanie układu hormonalnego. Pierwotne i zmienione ramy koncepcyjne uwzględniono jako załączniki do wytycznych o nazwie Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (4). Ramy koncepcyjne składają się z pięciu poziomów, a każdy z nich odpowiada innemu poziomowi złożoności biologicznej. Testy wiązania receptora estrogenowego opisane w niniejszej metodzie badawczej należą do poziomu 2, który obejmuje „testy in vitro dostarczające danych na temat mechanizmów/ścieżek układu hormonalnego”. Niniejsza metoda badawcza dotyczy testów wiązania receptora in vitro stworzonych, aby określić ligandy ludzkiego receptora estrogenowego alfa. α
|
|
3.
|
Wyraźnie wykazano istotność testu wiązania receptora estrogenowego in vitro w kontekście funkcji biologicznych. Testy wiązania receptora estrogenowego zaprojektowano w sposób umożliwiający identyfikację substancji chemicznych, które potencjalnie mogą powodować zaburzenia ścieżek hormonu estrogenu, a także w ciągu ostatnich dwóch dekad były w znacznym stopniu stosowane do charakteryzacji rozkładu tkanek receptora estrogenowego oraz do identyfikacji agonistów/antagonistów. Testy te odzwierciedlają interakcję ligand-receptor, która stanowi początkowy etap estrogenowej ścieżki sygnalizacyjnej i jest niezbędna wszystkim kręgowcom przy pełnieniu funkcji reprodukcyjnej.
|
|
4.
|
Interakcja estrogenów z receptorami estrogenowymi może wpłynąć na transkrypcję genów regulowanych przez estrogen oraz wywoływać skutki pozagenomowe, które mogą doprowadzić do indukcji lub inhibicji procesów komórkowych, w tym tych koniecznych do proliferacji komórek, prawidłowego rozwoju płodu i funkcji reprodukcyjnej (5)(6)(7). Zakłócenie prawidłowego układu estrogenowego może potencjalnie wywołać niekorzystny wpływ na prawidłowy rozwój (ontogenezę), zdrowie reprodukcyjne i integralność układu rozrodczego. Niewłaściwe sygnalizowanie receptora estrogenowego może doprowadzić do skutków takich jak: zwiększone ryzyko hormonozależnego nowotworu, ograniczona płodność oraz zmiany podczas wzrostu i rozwoju płodowego (8).
|
|
5.
|
Testy wiązania in vitro opierają się na bezpośredniej interakcji substancji posiadającej określony obszar wiązania ligand receptora regulującego transkrypcję genu. Główny element testu wiązania ludzkiego receptora estrogenowego alfa mierzy zdolność liganda znakowanego izotopem ([3H]17β-estradiolu) do wiązania się z receptorem estrogenowym w obecności zwiększających się stężeń badanej substancji chemicznej (tj. konkurenta). Badane substancje chemiczne o wysokim powinowactwie z receptorem estrogenowym konkurują ze znakowanym izotopem ligandem w niższym stężeniu w porównaniu z substancjami chemicznymi o niższym powinowactwie z receptorem. Niniejszy test składa się z dwóch głównych elementów: doświadczenie dotyczące wiązania nasycenia mające na celu scharakteryzowanie parametrów interakcji receptor-ligand oraz dokumentację swoistości receptora estrogenowego, po którym następuje doświadczenie dotyczące wiązania konkurencyjnego charakteryzujące konkurencję między badaną substancją chemiczną a ligandem znakowanym izotopem w zakresie wiązania z receptorem estrogenowym.
|
|
6.
|
Badania walidacyjne dotyczące testów wiązania CERI oraz Freybergera–Wilsona wykazały istotność i wiarygodność tych metod w odniesieniu do ich przeznaczenia (2).
|
|
7.
|
Definicje i skróty stosowane w niniejszej metodzie badawczej opisano w dodatku 1.
|
Zakres i ograniczenia związane z testami wiązania receptora
|
8.
|
Testy te proponuje się do celów badań przesiewowych i ustalania substancji priorytetowych, ale mogą one dostarczać również informacji na temat zdarzenia inicjacji molekularnej, które można wykorzystać w ramach podejścia opartego na wadze dowodów. Testy te dotyczą wiązania substancji chemicznej do domeny wiążącej ligand receptora estrogenowego alfa w układzie in vitro. W związku z tym wyników nie należy ekstrapolować bezpośrednio do złożonej sygnalizacji i regulacji nieuszkodzonego układu hormonalnego in vivo.
|
|
9.
|
Wiązanie naturalnego liganda – 17β-estradiolu – stanowi wstęp do serii zdarzeń molekularnych, które aktywują transkrypcję genów docelowych i docelowo kończy się ono fizjologiczną zmianą (9). W związku z tym wiązanie do domeny wiążącej ligand receptora estrogenowego alfa uznaje się za jeden z kluczowych mechanizmów pośredniczących w zaburzeniach funkcjonowania receptora estrogenowego układu hormonalnego, chociaż istnieją także inne mechanizmy, które mogą wywołać zaburzenia funkcjonowania układu hormonalnego, między innymi (i) interakcje z obszarami receptora estrogenowego α innymi niż kieszeń wiązania ligand, (ii) interakcje z innymi receptorami stosownymi do sygnalizacji estrogenów, receptorami estrogenowymi β i białkami G połączonymi z receptorem estrogenowym, inne receptory i układy enzymatyczne będące w zakresie układu hormonalnego, (iii) synteza hormonów, (iv) aktywacja metaboliczna lub dezaktywacja hormonów, (v) dystrybucja hormonów do tkanek docelowych i (vi) usunięcie hormonów z ciała. Żaden z testów wchodzących w skład niniejszej metody badawczej nie dotyczy tych sposobów działania.
|
|
10.
|
Niniejsza metoda badawcza dotyczy zdolności substancji do wiązania się z ludzkim receptorem estrogenowym alfa i nie zachodzi w niej podział na agonistów lub antagonistów receptora estrogenowego alfa. Testy te nie dotyczą ani dalszych zdarzeń niższego stopnia, takich jak transkrypcja genu, ani też zmian psychologicznych. Biorąc pod uwagę, że podczas walidacji wykorzystano jedynie pojedyncze substancja jednoskładnikowe, nie zbadano zastosowania do mieszanin stosowanych w badaniu. Z teoretycznego punktu widzenia testy mogą jednak mieć zastosowanie przy badaniu zarówno substancji wieloskładnikowych, jak i mieszanin. Przed zastosowaniem przedmiotowej metody badawczej z użyciem mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy zastanowić się, czy zastosowanie tej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak, to dlaczego. Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny.
|
|
11.
|
Układy receptorów wolne od komórek nie posiadają wrodzonej zdolności metabolicznej i nie zweryfikowano ich razem z układami enzymów metabolicznych. Możliwe może być jednak włączenie aktywności metabolicznej do projektu badania, aczkolwiek wymagałoby to dołożenia dalszych starań pod względem walidacji.
|
|
12.
|
Substancji chemicznych, które mogą spowodować denaturację białka (tj. białka receptorowe) – takich jak środki powierzchniowo czynne lub substancje chemiczne, których pH bufora testowego może ulec zmianie – nie można badać lub można badać je jedynie w stężeniach, w których takie interakcje nie zachodzą. W innym wypadku zakres stężeń, których badania można przeprowadzić w formie testów dotyczących badanej substancji chemicznej jest ograniczany przez jej rozpuszczalność w buforze testowym.
|
|
13.
|
Do celów informacyjnych w tabeli 1 przedstawiono wyniki badania 24 substancji, które zbadano stosując dwa w pełni zwalidowane testy opisane w niniejszej metodzie badawczej. 17 z tych substancji sklasyfikowano jako substancje wiążące receptor estrogenowy, a 6 substancji innych niż substancje wiążące na podstawie opublikowanych sprawozdań, w tym testów in vitro dotyczących aktywacji transkrypcji lub w teście biologicznego wzrostu macicy (9)(10)(11)(12)(13)(14)(15). Jeżeli chodzi o dane podsumowane w tabeli 1, wyniki obu testów dotyczących klasyfikacji wszystkich substancji do 10-4M były niemal stuprocentowo zgodne, a wszystkie substancje sklasyfikowano prawidłowo jako substancje wiążące lub jako substancje inne niż substancje wiążące receptor estrogenowy. Dodatkowe informacje dotyczące tej grupy substancji, a także dodatkowe substancje zbadane za pomocą testów wiązania receptora estrogenowego podczas badań walidacyjnych zawarto w dodatku 2 (tabele 1, 2 i 3) do standardów wykonywania badań dotyczących testów wiązania hrER (3).
Tabela 1
Klasyfikacja substancji jako „substancji wiążących” lub „substancji innych niż substancji wiążących” receptora estrogenowego w testach wiązania Freybergera–Wilsona oraz CERI dotyczących ludzkiego rekombinowanego receptora estrogenowego w porównaniu z przewidywaną reakcją
|
|
Nazwa substancji
|
NR CAS
|
Przewidywana reakcja
|
Test Freybergera–Wilsona
|
Test CERI
|
Substancja chemiczna MESH Klasa
|
Klasa produktowa
|
|
|
Stężenie Zakres (M)
|
Klasyfikacja
|
Stężenie Zakres (M)
|
Klasyfikacja
|
|
1
|
17β-estradiol
|
50-28-2
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Substancja wiążąca
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
2
|
Noretynodrel
|
68-23-5
|
Substancja wiążąca
|
3×10-9 – 30×10-4
|
Substancja wiążąca
|
3×10-9 – 30×10-4
|
Substancja wiążąca
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
3
|
Noretyndron
|
68-22-4
|
Substancja wiążąca
|
3×10-9 – 30×10-4
|
Substancja wiążąca
|
3×10-9 – 30×10-4
|
Substancja wiążąca
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
4
|
Ftalan di-n-butylu
|
84-74-2
|
Substancja inna niż substancja wiążąca (*5)
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca (*6)
(1)
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca (*6)
(1)
|
Węglowodór (cykliczny), ester
|
Plastyfikator, półprodukt
|
|
5
|
DES
|
56-53-1
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (cykliczny), fenol
|
Produkt leczniczy, środek weterynaryjny
|
|
6
|
17α-etynyloestradiol
|
57-63-6
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
7
|
mezoheksestrol
|
84-16-2
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (cykliczny), fenol
|
Produkt leczniczy, środek weterynaryjny
|
|
8
|
Genisteina
|
446-72-0
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (heterocykliczny), flawonoid
|
Produkt naturalny
|
|
9
|
Equol
|
531-95-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Metabolit fitoestrogenu
|
Produkt naturalny
|
|
10
|
Butyloparaben (n butyl-4-hydroxybenzoate)
|
94-26-8
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Paraben
|
Konserwant
|
|
11
|
Nonylofenol (mieszanina)
|
84852-15-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Alkilofenol
|
Związek pośredni
|
|
12
|
o,p’-DDT
|
789-02-6
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Chloroorganiczny
|
Środek owadobójczy
|
|
13
|
Kortykosteron
|
50-22-6
|
Substancja inna niż substancja wiążąca (*5)
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
Steryd
|
Produkt naturalny
|
|
14
|
Zearalenon
|
17924-92-4
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (heterocykliczny), lakton
|
Produkt naturalny
|
|
15
|
Tamoksyfen
|
10540-29-1
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (cykliczny)
|
Produkt leczniczy, środek weterynaryjny
|
|
16
|
5α-dihydrotestosteron
|
521-18-6
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Steryd, niefenolowy
|
Produkt naturalny
|
|
17
|
Bisfenol A
|
80-05-7
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Fenol
|
Półprodukt
|
|
18
|
4-n-heptylphenol
|
1987-50-4
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Niejednoznaczny (6)
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Alkilofenol
|
Półprodukt
|
|
19
|
Kepon (chlordekon)
|
143-50-0
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Węglowodór (halogenowany)
|
Pestycyd
|
|
20
|
Benz(a)antracen
|
56-55-3
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja inna niż substancja wiążąca (7)
|
1×10-10 – 1×10-3
|
Substancja inna niż substancja wiążąca (7)
|
Węglowodór aromatyczny
|
Półprodukt
|
|
21
|
Enterolakton
|
78473-71-9
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja wiążąca
|
Fitoestrogen
|
Produkt naturalny
|
|
22
|
Progesteron
|
57-83-0
|
Substancja inna niż substancja wiążąca (*5)
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
Steryd
|
Produkt naturalny
|
|
23
|
Oktylotrietoksysilan
|
2943-75-1
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-3
|
Substancja inna niż substancja wiążąca
|
Silan
|
Modyfikator powierzchni
|
|
24
|
Atrazyna
|
1912-24-9
|
Substancja inna niż substancja wiążąca (*5)
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-4
|
Substancja inna niż substancja wiążąca
|
Związek heterocykliczny
|
Herbicyd
|
|
ELEMENTY TESTU WIĄZANIA hrER
Istotne elementy testu
|
14.
|
Niniejsza metoda badawcza ma zastosowanie do testów, w których do receptora mogącego pełnić funkcję markera/znacznika testu oraz posiadającego zdolność przemieszczania się wraz ze wzrostem stężenia badanej substancji chemicznej, stosuje się receptor estrogenowy oraz odpowiednio silny ligand. Testy wiązania obejmują dwa główne elementy: 1) wiązanie nasycenia oraz 2) wiązanie konkurencyjne. Test wiązania nasycenia przeprowadza się, aby potwierdzić swoistość i aktywność preparatów receptora, natomiast doświadczenie dotyczące wiązania konkurencyjnego przeprowadza się, aby ocenić zdolność badanej substancji chemicznej do wiązania się z hrER.
|
Kontrole
|
15.
|
Należy opisać podstawę proponowanych jednocześnie estrogenów odniesienia i kontroli. Jednoczesne kontrole (z rozpuszczalnikiem (nośnikiem), dodatnie (substancje wiążące się z receptorem estrogenowym; o mocnym i słabym powinowactwie wiązania), ujemne (substancje inne niż substancje wiążące)) w stosownych przypadkach służą jako wskazówka, że test działa w warunkach badania, a kontrole te stanowią podstawę porównań między doświadczeniami; kontrole te wchodzą zwykle w skład kryteriów dopuszczalności danego doświadczenia (1). Pełne krzywe stężeń dla estrogenów odniesienia i kontroli (tj. substancji słabo wiążącej i substancji innej niż substancja wiążąca) w każdej serii badawczej należy stosować na jednej płytce. Wszystkie pozostałe płytki powinny zawierać: 1) wysokie (w przybliżeniu pełne przemieszczenie liganda znakowanego izotopem) i średnie (w przybliżeniu IC50) stężenie każdego z E2 oraz substancji słabo wiążącej w trzech powtórzeniach; 2) kontrolę z rozpuszczalnikiem i wiązanie nieswoiste, każde w trzech powtórzeniach.
|
Standardowe procedury kontroli jakości
|
16.
|
Standardowe procedury kontroli jakości należy przeprowadzać tak, jak opisano dla każdego testu w celu zapewnienia czynnych receptorów, właściwych stężeń substancji chemicznych, pułapu tolerancji utrzymującego stabilność przez wiele pasaży oraz zachowania zdolności do uzyskania reakcji wiążących receptor estrogenowy na przestrzeni czasu.
|
Wykazanie biegłości laboratorium
|
17.
|
Przed przeprowadzeniem badania nieznanych substancji chemicznych z wykorzystaniem któregoś z testów przeprowadzanych w ramach niniejszej metody badawczej, każde laboratorium powinno wykazać biegłość w stosowaniu testu, przeprowadzając testy nasycenia pozwalające potwierdzić swoistość i aktywność preparatu z receptorem estrogenowym oraz konkurencyjne testy wiązania zawierające estrogen odniesienia i kontrole (substancję słabo wiążącą i substancję inną niż substancja wiążąca). Laboratorium powinno stworzyć bazę danych historycznych, zawierającą wyniki estrogenu odniesienia i kontroli wygenerowanych w ciągu 3–5 niezależnych, przeprowadzonych w różne dni doświadczeń. Doświadczenia te będą stanowić podstawę w przypadku estrogenu odniesienia i kontroli historycznych w przypadku laboratorium, a w przyszłych seriach badawczych będą stosowane jako częściowa ocena dopuszczalności testu.
|
|
18.
|
Reaktywność układu badawczego zostanie także potwierdzona za pomocą substancji służących do wykazania biegłości, których wykaz znajduje się w tabeli 2. Wykaz substancji służących do wykazania biegłości stanowi podzbiór substancji odniesienia przedstawionych w standardach wykonywania badań dotyczących badań wiązania receptora estrogenowego (3). Substancje te są dostępne na rynku, należą do klas substancji chemicznych łączących się często z aktywnością wiązania receptora estrogenowego, wykazują odpowiedni zakres siły działania oczekiwanej w przypadku substancji wiążących receptora estrogenowego (tj. od wysokiej do słabej) oraz substancji innych niż substancje wiążące (tj. substancji ujemnych). Zbadane stężenia, w przypadku każdej substancji służącej do wykazania biegłości, powinny wchodzić w zakres określony w tabeli 2. W odniesieniu do każdej substancji należy przeprowadzić co najmniej trzy doświadczenia, a ich wyniki powinny być zgodne z przewidywaną aktywnością substancji chemicznej. Każde doświadczenie należy przeprowadzić niezależnie (tj. w świeżych roztworach receptora, badanych substancji chemicznych i odczynnika), stosując trzy kontrpróby na każde stężenie. Biegłość wykazuje się w drodze poprawnej klasyfikacji (dodatnia/ujemna) każdej substancji służącej do wykazania biegłości. Podczas nauki przeprowadzania testów każdy technik powinien wykonać badanie biegłości.
Tabela 2
Wykaz kontroli i substancji służących do wykazania biegłości w odniesieniu do konkurencyjnych testów wiązania hrER
(89)
|
Nie
|
Nazwa substancji
|
NR CAS (90)
|
Przewidywana reakcja (91)
(92)
|
Zakres badanych stężeń (M)
|
Klasa chemiczna MeSH (93)
|
Klasa produktowa (94)
|
|
Kontrole (estrogen odniesienia, substancja słabo wiążąca, substancja inna niż substancja wiążąca)
|
|
1
|
17–εστραδιολ
|
50-28-2
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
2
|
Norethynodrel (lub) Noretyndron
|
68-23-5 (lub) 68-22-4
|
Substancja wiążąca
|
3×10-9 – 30×10-6
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
3
|
Oktylotrietoksysilan
|
2943-75-1
|
Substancja inna niż substancja wiążąca
|
1×10-10 – 1×10-3
|
Silan
|
Modyfikator powierzchni
|
|
Substancje służące do wykazania biegłości
(94)
|
|
4
|
Dietylostylbestrol
|
56-53-1
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Węglowodór (cykliczny), fenol
|
Produkt leczniczy, środek weterynaryjny
|
|
5
|
17α-etynyloestradiol
|
57-63-6
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Steryd
|
Produkt leczniczy, środek weterynaryjny
|
|
6
|
mezoheksestrol
|
84-16-2
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Węglowodór (cykliczny), fenol
|
Produkt leczniczy, środek weterynaryjny
|
|
7
|
Tamoksyfen
|
10540-29-1
|
Substancja wiążąca
|
1×10-11 – 1×10-6
|
Węglowodór (cykliczny)
|
Produkt leczniczy, środek weterynaryjny
|
|
8
|
Genisteina
|
446-72-0
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Związek heterocykliczny, flawonoid
|
Produkt naturalny
|
|
9
|
Bisfenol A
|
80-05-7
|
Substancja wiążąca
|
1×10-10 – 1×10-3
|
Fenol
|
Półprodukt
|
|
10
|
Zearalenon
|
17924-92-4
|
Substancja wiążąca
|
1×10-11 – 1×10-3
|
Związek heterocykliczny, lakton
|
Produkt naturalny
|
|
11
|
Butyloparaben
|
94-26-8
|
Substancja wiążąca
|
1×10-11 – 1×10-3
|
Kwas karboksylowy, fenol
|
Konserwant
|
|
12
|
Atrazyna
|
1912-24-9
|
Substancja inna niż substancja wiążąca
|
1×10-11 – 1×10-6
|
Związek heterocykliczny
|
Herbicyd
|
|
13
|
Ftalan dibutylu (DBP) (95)
|
84-74-2
|
Substancja inna niż substancja wiążąca (96)
|
1×10-10 – 1×10-4
|
Węglowodór (cykliczny), ester
|
Plastyfikator, półprodukt
|
|
14
|
Kortykosteron
|
50-22-6
|
Substancja inna niż substancja wiążąca
|
1×10-11 – 1×10-4
|
Steryd
|
Produkt naturalny
|
|
Badanie rozpuszczalności oraz ustalanie zakresu stężeń badanych substancji chemicznych
|
19.
|
Należy przeprowadzić badanie wstępne, aby ustalić granicę rozpuszczalności każdej badanej substancji chemicznej oraz określić właściwy zakres stężeń, który zostanie zastosowany przy przeprowadzaniu badania. Granicę rozpuszczalności każdej badanej substancji chemicznej należy początkowo oznaczyć w rozpuszczalniku, a następnie potwierdzić w warunkach testowych. Końcowe stężenie badane w teście nie powinno przekraczać 1 mM. Badanie ustalające zakres dawkowania obejmuje kontrolę z rozpuszczalnikiem wraz z ośmioma seriami stężeń wzrastających w postępie logarytmicznym, począwszy od maksymalnego dopuszczalnego stężenia (np. 1 mM lub niższego, na podstawie granicy rozpuszczalności) oraz odnotowaną obecność zmętnienia lub osadu. Stężenia w drugim i trzecim doświadczeniu należy odpowiednio dostosować, aby lepiej scharakteryzować krzywą zależności stężenie-odpowiedź.
|
Kryteria dopuszczalności serii badawczej
|
20.
|
Akceptacji lub odrzucenia serii badawczej dokonuje się na podstawie oceny wyników dotyczących estrogenu i kontroli odniesienia stosowanych w przypadku każdego doświadczenia. Po pierwsze, w przypadku płytki 1 pełne krzywe stężenia dotyczące kontroli odniesienia z każdego doświadczenia powinny odpowiadać miarom efektywności o parametrach dopasowanych do krzywej (np. IC50 i krzywej Hilla) opartych na wynikach zgłoszonych w odniesieniu do właściwych protokołów dotyczących testów CERI i Freybergera-Wilsona (dodatek 2 i 3) oraz danym historycznym dotyczącym kontroli otrzymanym od laboratorium przeprowadzającego badanie. Wszystkie kontrole (estrogen odniesienia, substancję słabo wiążącą oraz substancję inną niż substancja wiążąca) należy odpowiednio zaklasyfikować w przypadku każdego doświadczenia. Po drugie, kontrole na wszystkich kolejnych płytkach należy ocenić pod względem zgodności z płytką 1. Należy zastosować wystarczający zakres stężeń badanej substancji chemicznej, aby wyraźnie określić najwyższy punkt konkurencyjnej krzywej wiązania. Zmienność wśród kontrprób przy każdym stężeniu badanej substancji chemicznej, jak również wśród trzech niezależnych serii badawczych powinna być uzasadniona i poparta naukowo. Zdolność do spójnego przeprowadzania testu należy wykazać poprzez opracowanie i utrzymywanie bazy danych historycznych w odniesieniu do estrogenu odniesienia i kontroli. Odchylenia standardowe (SD) lub współczynniki zmienności (CV) dla środków parametrów dopasowania krzywych estrogenu odniesienia i kontrolnej substancji słabo wiążącej z wielu doświadczeń mogą być stosowane jako miara odtwarzalności wewnątrz laboratorium. Dokonując przeglądu wyników dotyczących płytek kontrolnych z każdej serii badawczej, jak również w przypadku każdej badanej substancji chemicznej, należy przyjąć profesjonalny osąd.
Ponadto powinny być spełnione następujące zasady dotyczące kryteriów dopuszczalności:
|
|
Dane powinny być wystarczające do przeprowadzenia ilościowej oceny wiązania receptora estrogenowego.
|
|
|
Badane stężenia powinny pozostać w zakresie rozpuszczalności badanej substancji chemicznej.
|
|
Analiza danych
|
21.
|
Określona procedura analizy danych dotyczących wiązania nasycenia oraz wiązania konkurencyjnego powinny być zgodne z kluczowymi zasadami charakteryzowania interakcji receptor-ligand. Zazwyczaj dane dotyczące wiązania nasycenia analizuje się, stosując model regresji nieliniowej, który uwzględnia całkowite i nieswoiste wiązanie. Określając Bmax i Kd konieczna może być korekta obniżenia stężenia ligandu (np. Swillens, 1995 r. (19)). Dane uzyskane w wyniku testów wiązania konkurencyjnego zazwyczaj się przekształca (np. odsetek specyficznego wiązania i stężenie badanej substancji chemicznej (logarytm M)). Szacunki logarytmu (IC50) dla każdej badanej substancji chemicznej należy określić wykorzystując właściwe oprogramowanie dopasowane do krzywej nieliniowej, aby pasowały do równania Hilla o czterech parametrach. Po początkowej analizie należy zastosować parametry dopasowane do krzywej oraz dokonać wizualnego przeglądu stopnia, w jakim dane dotyczące wiązania odpowiadają wygenerowanej krzywej konkurencyjnego wiązania. W niektórych przypadkach konieczne może być przeprowadzenie dodatkowej analizy, aby uzyskać najlepsze dopasowanie do krzywej (np. ograniczenie najwyższego lub najniższego punktu krzywej, zastosowanie zasady 10 %, zob. dodatek 4 i 2 pozycja bibliografii (sekcja III.A.2).
|
|
22.
|
Spełnienie kryteriów dopuszczalności (pkt 20) wskazuje, że system testów funkcjonuje prawidłowo, ale nie gwarantuje, że jakiekolwiek konkretne badanie dostarczy dokładnych danych. Powtarzanie się prawidłowych wyników pierwszego badania stanowi najlepszą wskazówkę, że dostarczono dokładnych danych.
|
Ogólne kryteria interpretacji danych
|
23.
|
Obecnie nie istnieje powszechnie uzgodniona metoda interpretacji danych dotyczących wiązania receptora estrogenowego. Zarówno jakościowe (np. substancja wiążąca / substancja inna niż substancja wiążąca) jak i ilościowe (np. logarytm IC50, względne powinowactwo wiązania (RBA) itp.) oceny aktywności przekazywanej przez ludzki receptor estrogenowy alfa powinny jednak być oparte na danych empirycznych i rzetelnej ocenie naukowej.
|
Sprawozdanie z badania
|
24.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
|
Kontrola / wzorzec porównawczy / badana substancja chemiczna
|
—
|
źródło, numer partii, termin przydatności, jeżeli są znane;
|
|
—
|
stabilność badanej substancji chemicznej, jeżeli jest znana;
|
|
—
|
rozpuszczalność i stabilność badanej substancji chemicznej w rozpuszczalniku, jeżeli są znane;
|
|
—
|
pomiar pH, osmolalności i osadu w podłożu, do którego dodano badaną substancję chemiczną, w stosownych przypadkach.
|
|
|
|
Substancja jednoskładnikowa:
|
—
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne;
|
|
—
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa IUPAC lub CAS, numer CAS, kod SMILES lub InChI, wzór strukturalny, czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.
|
|
|
|
Substancja wieloskładnikowa, UVCB i mieszaniny:
|
—
|
opisane w miarę możliwości przez podanie nazwy chemicznej (zob. powyżej), określenie ilości oraz istotnych właściwości fizykochemicznych składników.
|
|
|
|
Rozpuszczalnik/nośnik:
|
—
|
charakterystyka (charakter, dostawca i partia);
|
|
—
|
uzasadnienie wyboru rozpuszczalnika/nośnika;
|
|
—
|
rozpuszczalność i stabilność badanej substancji chemicznej w rozpuszczalniku/nośniku, jeżeli są znane;
|
|
|
|
Receptory:
|
—
|
źródło receptorów (dostawca, nr katalogowy, partia, gatunek receptora, stężenie aktywnego receptora dostarczone przez dostawcę, certyfikat od dostawcy);
|
|
—
|
charakterystyka receptorów (w tym wyniki dotyczące wiązania nasycenia): Kd, Bmax;
|
|
—
|
przechowywanie receptorów;
|
|
—
|
ligand znakowany izotopem promieniotwórczym:
|
|
—
|
dostawca, nr katalogowy, partia, określone działanie.
|
|
|
|
Warunki badania:
|
—
|
granica rozpuszczalności w warunkach testowych;
|
|
—
|
stężenie znacznika (tj. ligandu znakowanego izotopem promieniotwórczym);
|
|
—
|
stężenia badanej substancji chemicznej;
|
|
—
|
odsetek nośnika w teście końcowym;
|
|
—
|
temperatura i czas inkubacji;
|
|
—
|
metoda rozdzielenia związanych/wolnych ligandów;
|
|
—
|
dodatnie i ujemne kontrole / substancje odniesienia;
|
|
—
|
kryteria uznania badania za dodatnie, ujemne lub niejednoznaczne.
|
|
|
|
Kontrola dopuszczalności:
|
—
|
rzeczywiste wartości IC50 i krzywej Hilla w odniesieniu do jednoczesnych kontroli dodatnich / substancji odniesienia.
|
|
|
|
Wyniki:
|
—
|
surowe dane dotyczące związanych i wolnych ligandów;
|
|
—
|
w stosownych przypadkach kontrola potwierdzająca denaturację;
|
|
—
|
jeżeli istnieje, najniższe stężenie efektywne (LEC);
|
|
—
|
wartości względnego powinowactwa wiązania lub IC50, w stosownych przypadkach;
|
|
—
|
w stosownych przypadkach zależność stężenie-odpowiedź;
|
|
—
|
analizy statystyczne, jeśli takie istnieją, wraz z miarą błędu i wiarygodności (np. SEM, SD, CV lub 95 % CI) oraz opisem sposobu uzyskania tych wartości;
|
|
|
|
Omówienie wyników:
|
—
|
zastosowanie zasady 10 %.
|
|
Wniosek
|
BIBLIOGRAFIA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 34. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα) [W:] Publikacje na temat zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 226. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα) [W:] Publikacje na temat zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 222. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(4)
|
OECD (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 150. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, „Climacteric” nr 5, suplement 2, s. 20–6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated?„Endocr. Relat. Cancer” nr 16(4), s. 1073–1089.
|
|
(7)
|
Younes M. i Honma N. (2011). Estrogen Receptor Beta. „Arch. Pathol. Lab. Med”., 135(1): s. 63–66.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement,„Endo Rev” nr 30(4), s. 293–342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (Publikacja NIH 2002 nr 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, Karolina Północna.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, „Toxicol. In Vitro” nr 22(1), s. 225–231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents [W:] Publikacje na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 67. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japonia, s. 1–188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, „Toxicol. Letters” nr 146, s. 111–120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats.„Toxicol. Letters” nr 180, s. 213–221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells.„Toxicol. and Applied Pharmacol.” nr 196, s. 58–67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. [W:] „Chemosphere” nr 34, s. 835–848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis.„Mol Pharmacol” nr 47(6), s. 1197–1203.
|
Dodatek 1
DEFINICJE I SKRÓTY
Zasada 10 %: Możliwość wykluczenia z analiz punktów danych, w przypadku których średnia kontrprób w odniesieniu do odsetka określonego wiązania [3H]17β-estradiolu wynosi 10 % lub więcej niż zaobserwowano w przypadku średniej wartości przy niższym stężeniu (zob. dodatek 4).
Kryteria dopuszczalności: minimalne normy dotyczące przeprowadzania kontroli doświadczalnych i wzorców referencyjnych. Aby doświadczenie zostało uznane za ważne, należy spełnić wszystkie kryteria dopuszczalności.
Dokładność (zgodność): stopień zgodności wyników testu z przyjętymi wartościami odniesienia. Jest to miara efektywności testu i jeden z aspektów jego istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na określenie odsetka prawidłowych wyników testu (1).
CF: ramy koncepcyjne OECD dotyczące testowania i oceny substancji zaburzających funkcjonowanie układu hormonalnego.
Substancja chemiczna: substancja lub mieszanina.
CV: Współczynnik zmienności
E2: 17β-estradiol
ED: zaburzenia endokrynologiczne
hERα: ludzki receptor estrogenowy alfa
ER: receptor estrogenowy
Aktywność estrogenna: zdolność substancji chemicznej do naśladowania 17β-estradiolu w jego zdolności do wiązania się z receptorami estrogenowymi. Wiązanie z ludzkim receptorem estrogenowym alfa można wykryć za pomocą niniejszej metody badawczej.
IC50: połowa maksymalnego stężenia efektywnego badanej hamującej substancji chemicznej.
ICCVAM: Międzyagencyjny Komitet Koordynacyjny ds. Uznawania Metod Alternatywnych.
Odtwarzalność międzylaboratoryjna: miara zakresu, w jakim różne wykwalifikowane laboratoria, przy użyciu tego samego protokołu i testując tę samą badaną substancję, mogą uzyskać jakościowo i ilościowo podobne wyniki. Odtwarzalność międzylaboratoryjna jest ustalana w ramach procesów poprzedzających walidację i obejmujących walidację i wskazuje, w jakim stopniu test można z powodzeniem przekazywać między laboratoriami: nazywana jest również odtwarzalnością międzylaboratoryjną (1).
Odtwarzalność wewnątrzlaboratoryjna: ustalenie stopnia, w jakim wykwalifikowani pracownicy w tym samym laboratorium mogą z powodzeniem odtworzyć wyniki, stosując szczegółowy protokół w różnych momentach. Nazywana również „odtwarzalnością laboratoryjną” (1).
LEC: najniższe stężenie efektywne to najniższe stężenie badanej substancji chemicznej, które powoduje reakcję (tj. najniższe stężenie badanej substancji chemicznej, przy którym krotność indukcji statystycznie różni się od równoległej grupy kontrolnej nośnika).
Badanie me-too: potoczne określenie testu, które jest strukturalnie i funkcjonalnie podobne do zwalidowanej i zaakceptowanej referencyjnej metody badania. Stosowane zamiennie z podobną metodą badań.
PBTG: wytyczne dotyczące badań w oparciu o wyniki
Standardy wykonywania badań (PS): normy oparte na zwalidowanej metodzie badawczej, zapewniające podstawę do oceny porównywalności proponowanego testu, który jest podobny do metody referencyjnej pod względem mechanistycznym i funkcjonalnym. Uwzględnia się: 1) istotne elementy testu; 2) podstawowy wykaz chemicznych substancji odniesienia, wybranych spośród substancji chemicznych wykorzystywanych w celu wykazania akceptowalnej efektywności zwalidowanej metody badania; oraz 3) podobny poziom dokładności i wiarygodności, na podstawie wyników uzyskanych dla zwalidowanej metody badania, który proponowany test powinien osiągnąć przy ocenie z wykorzystaniem podstawowego wykazu chemicznych substancji odniesienia (1).
Substancje służące do wykazania biegłości: podzbiór substancji odniesienia zawartych w standardach wykonywania badań, które mogą być stosowane przez laboratoria w celu wykazania kompetencji technicznych za pomocą znormalizowanego testu. Kryteria kwalifikacji w przypadku tych substancji obejmują zazwyczaj takie czynniki, jak to, że reprezentują one zakres reakcji, są dostępne na rynku i że dostępne są wysokiej jakości dane porównawcze.
Biegłość: wykazana zdolność do prawidłowego wykonania testu przed badaniem nieznanych substancji.
Estrogen odniesienia: 17β-estradiol (E2, CAS 50-28-2).
Referencyjne metody badawcze: testy oparte na PBTG 493.
RBA: względne powinowactwo wiązania. Względne powinowactwo wiązania oblicza się jako odsetek logarytmu (IC50) substancji w stosunku do logarytmu (IC50) 17β-estradiolu
Istotność: opis powiązania testu z oczekiwanym skutkiem oraz czy jest on znaczący i użyteczny z punktu widzenia określonego celu. Jest to zakres, w jakim test pozwala prawidłowo zmierzyć lub przewidzieć biologiczny skutek będący przedmiotem zainteresowania. Istotność obejmuje uwzględnienie dokładności (zgodności) testu (1).
Wiarygodność: miara zakresu, w jakim test może zostać wykonany w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jego przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją w drodze obliczenia odtwarzalności wewnątrz- i międzylaboratoryjnej.
SD: odchylenie standardowe.
Badana substancja chemiczna: dowolna substancja lub mieszanina badana za pomocą niniejszej metody badawczej.
Zweryfikowana metoda badawcza: test, w odniesieniu do którego zakończono badania walidacyjne w celu określenia jej istotności (w tym dokładności) i wiarygodności w odniesieniu do konkretnego celu. Należy zauważyć, że zweryfikowana metoda badawcza może nie wykazywać dostatecznej efektywności z punktu widzenia dokładności i wiarygodności, aby można było ją uznać za dopuszczalną w odniesieniu do danego celu (1).
Walidacja: proces, za pomocą którego ustalana jest wiarygodność i adekwatność danego podejścia, metody, testu, procesu lub oceny w określonym celu (1).
Dodatek 2
TEST IN VITRO NASYCENIA I WIĄZANIA KONKURENCYJNEGO RECEPTORA ESTROGENOWEGO (ERα) FREYBERGERA–WILSONA WYKORZYSTUJĄCY PEŁNOWYMIAROWY REKOMBINOWANY ERα
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
1.
|
Niniejszy test nasycenia i wiązania konkurencyjnego receptora estrogenowego (ERα) in vitro wykorzystuje pełnowymiarowy ludzki receptor ERαα (hrERα), który jest wytwarzany w komórkach owadów zakażonych bakulowirusem i z nich izolowany. Protokół, opracowany przez Freybergera i Wilsona, został poddany międzynarodowemu wielolaboratoryjnemu badaniu walidacyjnemu (2 ), które wykazało jego adekwatność i wiarygodność dla zamierzonego celu testu.
|
|
2.
|
Test ten jest procedurą klasyfikacyjną służącą do identyfikacji substancji, które mogą wiązać się z pełnowymiarowym hrERα. Służy on do określenia zdolności badanej substancji chemicznej do konkurowania z 17β-estradiolem w zakresie wiązania z hrERα. Ilościowe wyniki testu mogą obejmować IC50 (miara stężenia badanej substancji chemicznej potrzebnego do wyparcia połowy [3H]-17β-estradiolu z hrERα) i względne powinowactwa wiązania badanych substancji chemicznych w odniesieniu do hrERα w porównaniu z 17β-estradiolem. Do celów klasyfikacji substancji chemicznych akceptowalne wyniki testów jakościowych mogą obejmować klasyfikację badanych substancji chemicznych jako substancji wiążących hrERα, substancji innych niż substancja wiążąca lub niejednoznacznych w oparciu o kryteria opisane dla krzywych wiązania.
|
|
3.
|
W teście wykorzystuje się ligand promieniotwórczy, który wymaga zezwolenia na stosowanie materiałów promieniotwórczych przez laboratorium. Wszystkie procedury dotyczące radioizotopów i niebezpiecznych substancji chemicznych powinny być zgodne z przepisami i procedurami opisanymi w prawodawstwie krajowym.
|
|
4.
|
Przed rozpoczęciem stosowania tego badania do celów regulacyjnych należy zapoznać się z sekcjami „WPROWADZENIE OGÓLNE” oraz „ELEMENTY TESTU WIĄZANIA hrER”. Definicje i skróty stosowane w niniejszej wytycznej dotyczącej badań opisano w dodatku 1.
|
ZASADY WYKONANIA TESTU (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
5.
|
W teście wiązania hrERα mierzy się zdolność znakowanego izotopem liganda ([3H]17β-estradiolu) do wiązania się z receptorem estrogenowym w obecności zwiększających się stężeń badanej substancji chemicznej (tj. konkurenta). Badane substancje chemiczne o wysokim powinowactwie z receptorem estrogenowym konkurują ze znakowanym izotopem ligandem w niższym stężeniu w porównaniu z substancjami chemicznymi o niższym powinowactwie z receptorem.
|
|
6.
|
Niniejszy test składa się z dwóch głównych elementów: doświadczenie dotyczące wiązania nasycenia mające na celu scharakteryzowanie parametrów interakcji receptor-ligand, po którym następuje doświadczenie dotyczące wiązania konkurencyjnego, charakteryzujący konkurencję pomiędzy badaną substancją chemiczną a znakowanym izotopem ligandem w zakresie wiązania z receptorem estrogenowym.
|
|
7.
|
Celem doświadczenia dotyczącego wiązania nasycenia jest scharakteryzowanie konkretnej partii receptorów pod kątem powinowactwa wiązania i ich liczby w ramach przygotowania do doświadczenia dotyczącego wiązania konkurencyjnego. W doświadczeniu dotyczącym wiązania nasycenia mierzy się, w warunkach równowagi, powinowactwo stałego stężenia receptora estrogenowego w odniesieniu do jego naturalnego ligandu (reprezentowanego przez stałą dysocjacji, Kd) oraz stężenie obszarów czynnego receptora (Bmax).
|
|
8.
|
W doświadczeniu dotyczącym konkurencyjnego wiązania mierzy się powinowactwo substancji do konkurowania z [3H]17β-estradiolem o wiązanie z receptorem estrogenowym. Powinowactwo jest określa się ilościowo za pomocą stężenia badanej substancji chemicznej, która w stanie równowagi hamuje 50 % specyficznego wiązania [3H]17β-estradiolu (nazywanego „stężeniem hamującym 50 %” lub IC50). Można je również ocenić za pomocą względnego powinowactwa wiązania (RBA, w stosunku do IC50 estradiolu mierzonego oddzielnie w tej samej serii badawczej). W doświadczeniu dotyczącym konkurencyjnego wiązania mierzy się wiązanie [3H]17β-estradiolu przy stałym stężeniu w obecności szerokiego zakresu (ośmiu rzędów wielkości) stężeń badanych substancji chemicznych. Dane są następnie dopasowywane, na ile to możliwe, do postaci równania Hilla (Hill 1910), które opisuje przemieszczenie radioligandu przez jednoobszarową konkurencyjną substancję wiążącą. Zakres przemieszczenia estradiolu znakowanego izotopem w stanie równowagi jest wykorzystywany do scharakteryzowania badanej substancji chemicznej jako substancji wiążącej, substancji innej niż substancja wiążąca lub generującej reakcję niejednoznaczną.
|
PROCEDURA
Wykazanie dopuszczalnej efektywności białka hrERα
|
9.
|
Przed rutynowym wykonaniem testów nasycenia i wiązania konkurencyjnego należy wykazać, że efektywność każdej nowej partii hrERα w laboratorium, w którym będzie używana, jest prawidłowa. W celu zademonstrowania efektywności należy zastosować dwuetapowy proces. Etapy te są następujące:
|
—
|
Przeprowadzenie testu wiązania nasycenia [3H]-17β-estradiolu w celu wykazania swoistości i nasycenia hrERα. Analiza regresji nieliniowej tych danych (np. BioSoft; McPherson 1985; Motulsky 1995), a następnie wykres Scatcharda powinien dokumentować powinowactwo wiązania hrERα [3H]-17β-estradiolu (Kd) i liczbę receptorów (Bmax) w odniesieniu do każdej partii hrERα.
|
|
—
|
Wykonanie testu wiązania konkurencyjnego przy użyciu substancji kontrolnych (estrogen odniesienia (17β-estradiol)), substancji słabo wiążącej (np. noretynodrel lub noretyndron) oraz substancji innej niż substancja wiążąca (oktylotrietoksysilan, OTES). Każde laboratorium powinno stworzyć bazę danych historycznych w celu udokumentowania spójności IC50 i innych istotnych wartości dla estrogenów odniesienia i substancji słabo wiążących między doświadczeniami i różnymi partiami hrERα. Parametry krzywych konkurencyjnego wiązania dla substancji kontrolnych powinny mieścić się w granicach 95 % przedziału ufności (zob. tabela 1), które zostały opracowane przy wykorzystaniu danych z laboratoriów uczestniczących w badaniu walidacyjnym w odniesieniu do niniejszego testu (2).
|
|
Tabela 1
Kryteria efektywności opracowane dla estrogenów odniesienia i substancji słabo wiążących, test wiązania FW hrER
|
Substancja
|
Parametr
|
Wartość średnia (8)
|
Odchylenie standardowe (n)
|
Przedziały ufności (9) 95 %
|
|
Dolna wartość graniczna
|
Górna wartość graniczna
|
|
17β-estradiol
|
Najwyższa wartość (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Najniższa wartość (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Krzywa Hilla
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (10)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretynodrel
|
Najwyższa wartość (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Najniższa wartość (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Krzywa Hilla
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
LogIC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretyndronc
|
Najwyższa wartość (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Najniższa wartość (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Krzywa Hilla
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Wykazanie biegłości laboratorium
|
10.
|
Zob. pkt 17 i 18 oraz tabela 2 w rozdziale „ELEMENTY TESTU WIĄZANIA hrER” w niniejszej metodzie badawczej. Każdy test (nasycenia i wiązania konkurencyjnego) powinien składać się z trzech niezależnych serii badawczych (tj. ze świeżymi rozcieńczeniami receptora, substancji chemicznych i odczynników) w różnych dniach, a każda seria badawcza powinna obejmować trzy kontrpróby.
|
Określanie stężenia receptora (hrERα)
|
11.
|
Stężenie czynnego receptora różni się nieznacznie w zależności od partii i warunków składowania. Z tego powodu należy określić stężenie czynnego receptora otrzymanego od dostawcy. Pozwoli to uzyskać odpowiednie stężenie aktywnego receptora w serii badawczej.
|
|
12.
|
W warunkach odpowiadających konkurencyjnym wiązaniom (tj. 1 nM [3H]-estradiolu), nominalne stężenia 0,25, 0,5, 0,75 i 1 nM receptora powinny być inkubowane przy braku (wiązanie całkowite) i obecności (wiązanie nieswoiste) 1 μM nieoznakowanego estradiolu. Określone wiązanie, obliczone jako różnica wiązania całkowitego i nieswoistego, jest wykreślane w funkcji nominalnego stężenia receptora. Stężenie receptora, które daje określone wartości wiązania odpowiadające 20 % dodanego oznaczenia izotopem, jest związane z odpowiadającym mu nominalnym stężeniem receptora, a stężenie receptora powinno być wykorzystywane w doświadczeniach dotyczących nasycenia i konkurencyjnego wiązania. Często końcowe stężenie hrER wynoszące 0,5 nM jest zgodne z tym warunkiem.
|
|
13.
|
Jeżeli kryterium 20 % nie może być wielokrotnie spełnione, należy sprawdzić, czy w zestawie doświadczenia nie ma potencjalnych błędów. Nieosiągnięcie kryterium 20 % może wskazywać, że w partii rekombinowanej jest bardzo mało czynnego receptora, w związku z czym należy rozważyć zastosowanie innej partii receptora.
|
Test nasycenia
|
14.
|
Osiem wzrastających stężeń [3H]17β-estradiolu należy oceniać trzykrotnie, w następujących trzech warunkach (zob. tabela 2):
|
—
|
przy braku nieoznakowanego 17β-estradiolu i w obecności receptora estrogenowego. Jest to wyznaczenie wiązania całkowitego przez pomiar radioaktywności w dołkach, w których znajduje się tylko [3H]17β-estradiol.
|
|
—
|
W obecności 1 000-krotnego przekroczenia stężenia nieoznakowanego 17β-estradiolu nad oznakowanym 17β-estradiolem i obecności receptora estrogenowego. Zamierzeniem tego warunku jest nasycenie aktywnych obszarów wiązania nieoznakowanym 17β-estradiolem, a w wyniku pomiaru radioaktywności w dołkach, określenie wiązania nieswoistego. Każdy pozostały gorący estradiol, który może wiązać się z receptorem uznaje się za wiążący w nieswoistym obszarze, ponieważ zimny estradiol powinien być w tak wysokim stężeniu, aby wiązał się ze wszystkimi dostępnymi swoistymi obszarami na receptorze.
|
|
—
|
Przy braku nieoznakowanego 17β-estradiolu i braku receptora estrogenowego (określenie całkowitej radioaktywności).
|
|
Przygotowanie roztworów [3H]-17β-estradiolu i nieoznakowanego 17β-estradiolu
|
15.
|
Należy przygotować rozcieńczenia [3H]-17β-estradiolu przez dodanie buforu testowego do roztworu podstawowego 12 nM [3H]-17β-estradiolu, aby uzyskać stężenia początkowo w zakresie od 0,12 nM do 12 nM. Dodają 40 μl tych roztworów do odpowiednich dołków testowych płytki 96-dołkowej (w ostatecznej objętości 160 μl), uzyskuje się końcowe stężenia testowe w zakresie od 0,03 do 3,0 nM. Przygotowanie buforu testowego, roztworu podstawowego [3H]-17β-estradiolu oraz rozcieńczeń i określenie stężeń są szczegółowo opisane w protokole Freybergera–Wilsona (2).
|
|
16.
|
Należy przygotować rozcieńczenia etanolowych roztworów 17β-estradiolu przez dodanie buforu testowego, aby uzyskać osiem wzrastających stężeń, początkowo w zakresie od 0,06 μM do 6 μM. Poprzez dodanie 80 μl tych roztworów do odpowiednich dołków testowych płytki 96-dołkowej (w ostatecznej objętości 160 μl), uzyskuje się końcowe stężenia testowe w zakresie od 0,03 μM do 3,0 μM. Ostateczne stężenie nieoznakowanego 17β-estradiolu w poszczególnych dołkach testowych z nieswoistymi wiązaniami powinno być 1 000-krotnie większe od oznakowanego stężenia [3H]-17β- estradiolu. Przygotowanie rozcieńczeń nieoznakowanego 17β-estradiolu jest szczegółowo opisane w protokole Freybergera–Wilsona (2).
|
|
17.
|
Należy stosować nominalne stężenie receptora, które daje specyficzne wiązanie 20±5 % (zob. pkt 12–13). Roztwór hrERα należy przygotować bezpośrednio przed użyciem.
|
|
18.
|
Płytki 96-dołkowe są przygotowywane w sposób przedstawiony w tabeli 2, z 3 kontrpróbami na stężenie. Przykład stężenia płytek i przypisania objętości [3H]-17β-estradiolu, nieoznakowanego 17β-estradiolu, buforu i receptora przedstawiono w dodatku 2.2.
|
Tabela 2
Rozkład na mikropłytce do testu wiązania nasycenia
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3 H] E2 + ER
|
0,06 nM [3 H] E2 + ER
|
0,08 nM [3 H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Wiązanie całkowite (rozpuszczalnik)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER + 0,10 μM E2
|
Wiązanie nieswoiste
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [3H] E2 + ER + 1,0 μM E2
|
3,0 nM [3H] E2 + ER + + 3,0 μM E2
|
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H] E2: [3H]-17β-estradiol
ER: receptor estrogenowy
E2: nieoznakowany 17β-estradiol
|
|
19.
|
Mikropłytki testowe należy inkubować w temperaturze 2–8 °C przez 16–20 godzin i umieszczane na rotatorze podczas okresu inkubacji.
|
Pomiar [3H]-17β-estradiolu związanego z hrERα
|
20.
|
[3H]-17β-estradiol związany z hrERα należy oddzielić od wolnego [3H]-17β-estradiolu poprzez dodanie 80 μl zimnej zawiesiny DCC do każdego dołka, potrząsanie mikropłytkami przez 10 minut i odwirowywanie przez 10 minut z prędkością około 2 500 obr./min. Aby zminimalizować dysocjację związanego [3H]-17β-estradiolu z hrERα podczas tego procesu, jest niezwykle ważne, aby bufory i dołki testowe były utrzymywane w temperaturze 2–8 °C i aby każdy etap był przeprowadzany szybko. Wytrząsarka do mikropłytek jest niezbędna do wydajnej i szybkiej obróbki płytek.
|
|
21.
|
50 μl supernatantu zawierającego [3H]-17β-estradiol związany z hrERα należy wówczas podnieść z najwyższą ostrożnością, aby uniknąć zanieczyszczenia dołków na skutek dotknięcia DCC, i umieścić na drugiej mikropłytce.
|
|
22.
|
200 μl płynu scyntylacyjnego, zdolnego do przekształcenia energii kinetycznej emisji jądrowej w energię świetlną, należy następnie dodać do każdego dołka (A1–B12 i D1 do E12). Dołki G1–H12 (zidentyfikowane jako całkowita liczba rozpadów na minutę) reprezentują serie stężeń wzrastających w postępie geometrycznym [3H]-17β-estradiolu (40 μl), które powinny być dostarczane bezpośrednio do płynu scyntylacyjnego w dołkach płytki pomiarowej, jak wskazano w tabeli 3, tj. dołki te zawierają tylko 200 μl płynu scyntylacyjnego i odpowiednie rozcieńczenie [3H]-17β-estradiolu. Pomiary te pokazują, ile [3H]-17β-estradiolu wyrażonych za pomocą liczby rozpadów na minutę dodano do każdego zestawu dołków w celu uzyskania wiązania całkowitego i wiązania nieswoistego.
|
Tabela 3
Rozkład na mikropłytce do testu wiązania nasycenia, pomiar radioaktywności
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Wiązanie całkowite (rozpuszczalnik)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER + 0,10 μM E2
|
Wiązanie nieswoiste
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [3H] E2 + ER + 1,0 μM E2
|
3,0 nM [3H] E2 + ER + + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (DPM ogółem)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
DPM (*7) ogółem
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [3H]-17β-estradiol
ER: receptor estrogenowy
E2: nieoznakowany 17β-estradiol
DPM: liczba rozpadów na minutę
|
|
23.
|
Pomiar powinien rozpocząć się z opóźnieniem co najmniej 2 godzin, a czas liczenia powinien wynosić 40 minut na dołek. Do określenia liczby rozpadów na minutę na dołek z korekcją tłumienia należy zastosować licznik scyntylacyjny do mikropłytek. Alternatywnie, jeżeli licznik scyntylacyjny dla mikropłytki nie jest dostępny, próbki mogą być mierzone w konwencjonalnym liczniku. W tych warunkach można rozważyć skrócenie czasu liczenia.
|
Test wiązania konkurencyjnego
|
24.
|
W teście wiązania konkurencyjnego mierzy się wiązanie pojedynczego stężenia [3H]17β-estradiolu w obecności zwiększających się stężeń badanej substancji chemicznej. Przy każdym stężeniu w ramach jednej serii badawczej należy zastosować trzy równoległe kontrpróby. Ponadto dla każdej badanej substancji chemicznej należy wykonać trzy niejednoczesne serie badawcze. Test powinien zostać przeprowadzony na jednej płytce 96-dołkowej lub na większej ich liczbie.
|
Kontrole
|
25.
|
Podczas przeprowadzania testu w każdym doświadczeniu należy uwzględnić równoczesny rozpuszczalnik i kontrole (tj. estrogen odniesienia, substancję słabo wiążącą oraz substancję inną niż substancja wiążąca). Pełne krzywe stężeń dla estrogenów odniesienia i kontroli (tj. substancji słabo wiążącej i substancji innej niż substancja wiążąca) w każdej serii badawczej należy stosować na jednej płytce. Wszystkie pozostałe płytki powinny zawierać (i) wysokie (maksymalne przemieszczenie) i średnie (około IC50) stężenie każdego z E2 oraz substancji słabo wiążącej w trzech powtórzeniach; (ii) kontrolę z rozpuszczalnikiem i wiązanie nieswoiste, każde co najmniej w trzech powtórzeniach. Procedury przygotowania buforu testowego, kontroli, [3H]-17β-estradiolu, hrERα i roztworów badanych substancji chemicznych opisano w 2 pozycji bibliografii (załącznik K, zob. Protokół testu Freybergera–Wilsona).
|
Kontrola z rozpuszczalnikiem:
|
26.
|
Kontrola z rozpuszczalnikiem wskazuje, że rozpuszczalnik nie wchodzi w interakcję z układem badawczym, a także umożliwia zmierzenie wiązania całkowitego (TB). Preferowanym rozpuszczalnikiem jest etanol. W przypadku gdy najwyższe stężenie badanej substancji chemicznej nie jest rozpuszczalne w etanolu, alternatywnie można zastosować sulfotlenek dimetylowy. Stężenie etanolu lub sulfotlenku dimetylowego, jeżeli go zastosowano, w dołkach testu końcowego wynosi 1,5 % i nie może przekraczać 2 %.
|
Kontrola z buforem:
|
27.
|
Kontrola z buforem (BC) nie powinna zawierać rozpuszczalnika ani badanej substancji chemicznej, ale wszystkie inne elementy testu. Wyniki kontroli z buforem porównuje się z wynikami kontroli z rozpuszczalnikiem w celu sprawdzenia, czy zastosowany rozpuszczalnik nie ma wpływu na system testu.
|
Substancja silnie wiążąca (estrogen odniesienia)
|
28.
|
17β-estradiol (CAS 50-28-2) jest endogennym ligandem i tworzy wiązanie o wysokim powinowactwie z receptorem estrogenowym, podtyp alfa. Dla każdego testu wiązania konkurencyjnego hrERα należy przygotować krzywą wzorcową z użyciem nieoznakowanego 17β-estradiolu, aby w tym samym laboratorium umożliwić ocenę zmienności podczas wykonywania testu na przestrzeni czasu. W etanolu należy przygotować osiem roztworów nieoznakowanego 17β-estradiolu o stężeniach w dołkach testowych w zakresie od 100 nM do 10 pM (-7[logM] do -11[logM]), w następujących odstępach: (-7[logM], -8[logM], -8,5[logM], -9[logM], -9,5[logM], -10[logM], -11[logM]). Najwyższe stężenie nieoznakowanego 17β-estradiolu (1 μM) służy również jako wskaźnik nieswoistego wiązania. Stężenie to wyróżnia się etykietą „NSB” w tabeli 4, mimo że jest ono również częścią krzywej wzorcowej.
|
Substancja słabo wiążąca
|
29.
|
Należy uwzględnić substancję słabo wiążącą (noretynodrel (CAS 68-23-5) lub noretyndron (CAS 68-22-4)) w celu wykazania czułości każdego doświadczenia i umożliwienia oceny zmienności przy przeprowadzaniu testu z upływem czasu. W etanolu należy przygotować osiem roztworów substancji słabo wiążącej o stężeniach w dołkach testowych w zakresie od 3 nM do 30 μM (-8,5[logM] do -4,5[logM]), w następujących odstępach: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM], -7,5[logM], -8,5[logM].
|
Substancja inna niż substancja wiążąca
|
30.
|
Oktylotrietoksysilan (OTES, CAS 2 943-75-1) powinien być stosowany jako kontrola ujemna (substancja inna niż substancja wiążąca). Daje to pewność, że test w takiej formie umożliwi wykrycie, że badane substancje chemiczne nie wiążą się z hrERα. W etanolu należy przygotować osiem roztworów substancji innej niż substancja wiążąca o stężeniach w dołkach testowych w zakresie 0,1 nM–1 000 μM (-10[logM] do -3[logM]) w skali logarytmicznej. Ftalan dibutylu (DBP) można stosować jako alternatywną kontrolną substancję inną niż substancja wiążąca. Wykazano, że jego maksymalna rozpuszczalność wynosi -4[logM].
|
Stężenie hrERα
|
31.
|
Należy stosować ilość receptora, które daje specyficzne wiązanie 20±5 % 1 nM radioligandu (zob. pkt 12–13 w dodatku 2). Roztwór hrERα należy przygotować bezpośrednio przed użyciem.
|
[3H]-17β-estradiol
|
32.
|
Stężenie [3H]-17β-estradiolu w dołkach testowych powinno wynosić 1,0 nM.
|
Badane substancje chemiczne
|
33.
|
W pierwszej kolejności konieczne jest przeprowadzenie badania rozpuszczalności, aby ustalić granicę rozpuszczalności każdej badanej substancji chemicznej oraz określić właściwy zakres stężeń, który zostanie zastosowany przy realizacji protokołu badania. Granicę rozpuszczalności każdej badanej substancji chemicznej należy początkowo oznaczyć w rozpuszczalniku, a następnie potwierdzić w warunkach testowych. Końcowe stężenie badane w teście nie powinno przekraczać 1 mM. Badanie ustalające zakres dawkowania obejmuje kontrolę z rozpuszczalnikiem wraz z 8 seriami stężeń wzrastających w postępie logarytmicznym, począwszy od maksymalnego dopuszczalnego stężenia (np. 1 mM lub niższego, na podstawie granicy rozpuszczalności) oraz odnotowaną obecność zmętnienia lub osadu (zob. również pkt 35). Badaną substancję chemiczną należy badać przy użyciu 8 krzywych stężeń logarytmicznych, jak określono w poprzednim badaniu zakresu dawkowania. Stężenia w drugim i trzecim doświadczeniu należy odpowiednio dostosować, aby lepiej scharakteryzować krzywą zależności stężenie-odpowiedź.
|
|
34.
|
Rozcieńczenia badanych substancji chemicznych należy przygotować w odpowiednim rozpuszczalniku (zob. pkt 26 dodatku 2). Jeżeli najwyższe stężenie badanej substancji chemicznej nie jest rozpuszczalne ani w etanolu, ani w sulfotlenku dimetylowym, a dodanie większej ilości rozpuszczalnika spowodowałoby, że stężenie rozpuszczalnika w końcowej probówce byłoby wyższe niż dopuszczalna wartość graniczna, najwyższe stężenie można obniżyć do następnego niższego stężenia. W takim przypadku można dodać dodatkowe stężenie na dolnym końcu serii stężeń. Pozostałe stężenia w serii powinny pozostać niezmienione.
|
|
35.
|
Roztwory badanej substancji chemicznej powinny być ściśle monitorowane po dodaniu ich do dołka testowego, ponieważ badana substancja chemiczna może wytrącać się po dodaniu do dołka testowego. Dane dotyczące wszystkich dołków, które zawierają osady, należy wyłączyć z dopasowywania krzywej, a także odnotować powód wyłączenia tych danych.
|
|
36.
|
Jeżeli istnieją wcześniejsze informacje z innych źródeł, które dostarczają logarytm (IC50) badanej substancji chemicznej, właściwe może być geometryczne oddzielenie rozcieńczeń (tj. 0,5 jednostki logarytmicznej wokół oczekiwanego logarytmu (IC50)). Wynik końcowy powinien odzwierciedlać wystarczającą rozpiętość stężeń po obu stronach logarytmu (IC50), włączając „najwyższą wartość” i „najniższa wartość”, tak aby krzywą wiązania można było odpowiednio scharakteryzować.
|
Organizacja płytki testowej
|
37.
|
Oznakowane mikropłytki powinny być przygotowane z uwzględnieniem sześciokrotnej inkubacji z kodami kontroli z rozpuszczalnikiem, najwyższego stężenia estrogenu odniesienia, który służy również jako wskaźnik wiązania nieswoistego (NSB), oraz kontroli z buforem, a także biorąc pod uwagę potrójne inkubacje z kodami dla każdego z ośmiu stężeń kontroli bez wiązań (oktylotrietoksysilan), siedem niższych stężeń estrogenu odniesienia, osiem poziomów dawek stężeń substancji słabo wiążącej oraz osiem stężeń każdej badanej substancji chemicznej (TC). Przykładowy wykres rozkładu płytki dla pełnych krzywych stężeń dla estrogenu odniesienia i kontroli podano w tabeli 4 poniżej. Dodatkowe mikropłytki wykorzystuje się dla badanych substancji chemicznych i powinny one zawierać kontrole płytek, tj. 1) wysokie (maksymalne przemieszczenie) i średnie (około IC50) stężenie każdego z E2 oraz substancji słabo wiążącej w trzech powtórzeniach; 2) kontrolę z rozpuszczalnikiem i wiązanie nieswoiste, każde w sześciu powtórzeniach (tabela 5). Przykład arkusza roboczego rozkładu mikropłytki testowej, wykorzystującego trzy nieznane badane substancje chemiczne, znajduje się w dodatku 2.3. Stężenia wskazane w tabelach 4 i 5 są stężeniami końcowymi testu. Maksymalne stężenie dla E2 powinno wynosić 1×10-7 M, a dla substancji słabo wiążącej należy stosować najwyższe stężenie dla substancji słabo wiążącej na płytce 1. Stężenie IC50 musi być określone przez laboratorium na podstawie jego bazy danych historycznych dotyczących kontroli. Oczekuje się, że wartość ta będzie podobna do wartości zaobserwowanej w badaniach walidacyjnych (zob. tabela 1).
|
Tabela 4
Rozkład na mikropłytce testowej wiązania konkurencyjnego, pełne krzywe stężeń dla estrogenu odniesienia i próby kontrolne (płytka 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (tylko rozpuszczalnik)
|
TB (tylko rozpuszczalnik)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Próba ślepa (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Próba ślepa (dla gorących) (*9)
|
Próba ślepa (dla gorących) (*9)
|
Kontrola z buforem
|
Kontrola z buforem
|
|
W tym przykładzie substancją słabo wiążącą jest noretynodrel (NE)
|
Tabela 5
Rozkład na mikropłytce testowej wiązania konkurencyjnego, pełne krzywe stężeń dla badanych substancji chemicznych i kontrole na płytce
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (tylko rozpuszczalnik)
|
TB (tylko rozpuszczalnik)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
W tym przykładzie substancją słabo wiążącą jest noretynodrel (NE)
|
Zakończenie testu wiązania konkurencyjnego
|
38.
|
Jak pokazano w tabeli 6, do dołków należy dodać 80 μl kontroli z rozpuszczalnikiem, kontroli z buforem, estrogenu odniesienia, substancji słabo wiążącej, substancji innej niż substancja wiążąca i badanych substancji chemicznych przygotowanych w buforze testowym. Następnie do każdego dołka należy dodać 40 μl roztworu 4 nM [3H]-17β-estradiolu. Po łagodnej rotacji przez 10–15 minut w temperaturze od 2–8 °C należy dodać 40 μl roztworu hrERα. Mikropłytki testowe należy inkubować w temperaturze 2–8 °C przez 16–20 godzin i umieścić na rotatorze podczas okresu inkubacji.
|
Tabela 6
Objętość elementów testu w odniesieniu do testu wiązania konkurencyjnego hrER, mikropłytki
|
Objętość (μl)
|
Składnik
|
|
80
|
Nieoznakowany 17β-estradiol, noretynodrel, OTES, badane substancje chemiczne, rozpuszczalnik lub bufor
|
|
40
|
Roztwór 4 nM [3H]-17β-estradiolu
|
|
40
|
Roztwór hrERα, stężenie takie, jak określono
|
|
160
|
Całkowita objętość w każdym dołku testowym
|
|
39.
|
Oznaczenie ilościowe [3H]-17β-estradiolu związanego z hrERα po oddzieleniu [3H]-17β-estradiolu związanego z hrERα od wolnego [3H]-17β-estradiolu poprzez dodanie 80 μl zimnej zawiesiny DCC do każdego dołka należy następnie przeprowadzić jak opisano w pkt 20–23 w odniesieniu do testu wiązania nasycenia.
|
|
40.
|
Dołki H1-6 (oznaczone w tabeli 4 jako próby ślepe (dla gorących)) reprezentują DPM estradiolu oznakowanego [3H] w 40 μl. Podwielokrotność 40 μl należy wprowadzić bezpośrednio do płynu scyntylacyjnego w dołkach H1–H6.
|
Kryteria dopuszczalności
Test wiązania nasycenia
|
41.
|
Krzywa wiązania specyficznego powinna osiągnąć stan równowagi, ponieważ zastosowano zwiększające się stężenia [3H]-17β-estradiolu, wskazujące na nasycenie hrERα ligandem.
|
|
42.
|
Wiązanie specyficzne przy 1 nM [3H]-17β-estradiolu powinno mieścić się w dopuszczalnym zakresie od 15 % do 25 % średniej zmierzonej całkowitej radioaktywności dodanej we wszystkich seriach badawczych. Sporadyczne niewielkie odchylenia poza ten zakres są dopuszczalne, ale jeśli wyniki serii badawczych systematycznie nie mieszczą się w tym zakresie lub wynik danej serii badawczej wychodzi znacznie poza ten zakres, należy dostosować stężenie białka i powtórzyć test nasycenia.
|
|
43.
|
Dane powinny tworzyć liniowy wykres Scatcharda.
|
|
44.
|
Wiązania nieswoiste nie powinny być nadmierne. Wartość dla wiązań nieswoistych powinna zazwyczaj wynosić < 35 % wiązań całkowitych. Stosunek ten może jednak sporadycznie przekraczać tę granicę przy pomiarze bardzo niskich DPM dla najniższego stężenia badanego 17β-estradiolu znakowanego izotopem.
|
Test wiązania konkurencyjnego
|
45.
|
Zwiększające się stężenia nieoznakowanego 17β-estradiolu powinny wyprzeć [3H]-17β-estradiol z receptora w sposób zgodny z jednoobszarowym wiązaniem konkurencyjnym.
|
|
46.
|
Wartość IC50 dla estrogenu odniesienia (tj. 17β-estradiolu) powinna być w przybliżeniu równa stężeniu molowemu [3H]-17β-estradiolu powiększonemu o Kd oznaczony w teście wiązania nasycenia.
|
|
47.
|
Całkowite wiązanie specyficzne powinno być spójne w dopuszczalnym zakresie 20 ± 5 %, gdy średnie zmierzone stężenie całkowitej radioaktywności dodanej do każdego dołka wynosi 1 nM we wszystkich seriach badawczych. Sporadyczne niewielkie odchylenia poza ten zakres są dopuszczalne, ale jeśli wyniki serii badawczych systematycznie nie mieszczą się w tym zakresie lub wynik danej serii badawczej wychodzi znacznie poza ten zakres, należy dostosować stężenie białka.
|
|
48.
|
Rozpuszczalnik nie powinien powodować zmiany czułości ani odtwarzalności testu. Wyniki kontroli z rozpuszczalnikiem (dołki z wiązaniem całkowitym) porównuje się z wynikami kontroli z buforem w celu sprawdzenia, czy zastosowany rozpuszczalnik nie ma wpływu na system testu. Wyniki kontroli wiązania całkowitego i kontroli z buforem powinny być porównywalne, jeżeli rozpuszczalnik nie wpływa na test.
|
|
49.
|
Substancja inna niż substancja wiążąca nie powinna wypierać więcej niż 25 % [3H]-17β-estradiolu z hrERα podczas badania do 10-3 M (OTES) lub 10-4 M (DBP).
|
|
50.
|
Opracowano kryteria efektywności dla estrogenu odniesienia i dwóch substancji słabo wiążących (np. noretynodrel, noretyndron) z wykorzystaniem danych z badania walidacyjnego testu wiązania hrER Freybergera–Wilsona (załącznik N do 2 pozycji bibliografii). 95 % przedziały ufności podano w odniesieniu do średniej (n) +/- SD dla wszystkich serii badawczych kontroli wykonanych w laboratoriach uczestniczących w badaniu walidacyjnym. 95 % przedziały ufności obliczono dla parametrów dopasowania krzywej (tj. najwyższa wartość, najniższa wartość, krzywa Hilla, logarytm IC50) dla estrogenu odniesienia i substancji słabo wiążących oraz dla log10RBA substancji słabo wiążących w stosunku do estrogenu odniesienia i są one dostarczane jako kryteria efektywności dla kontroli dodatnich. W tabeli 1 przedstawiono oczekiwane zakresy parametrów dopasowania krzywej, które mogą być wykorzystane jako kryteria efektywności. W praktyce zakres IC50 może się nieznacznie różnić w zależności od Kd preparatu receptora i stężenia ligandu.
|
|
51.
|
Nie opracowano kryteriów efektywności w odniesieniu do parametrów dopasowania krzywej dla badanych substancji chemicznych ze względu na szeroki wachlarz istniejących potencjalnych badanych substancji chemicznych i zmienność potencjalnych powinowactw i wyników (np. pełna krzywa, krzywa częściowa, brak dopasowania krzywej). Dokonując przeglądu wyników z każdej serii badawczej badanej substancji chemicznej, należy jednak przyjąć profesjonalny osąd. Należy zastosować wystarczający zakres stężeń badanej substancji chemicznej, aby wyraźnie określić najwyższą wartość (np. 90–100 % wiązania) konkurencyjnej krzywej. Zmienność wśród kontrprób przy każdym stężeniu badanej substancji chemicznej, jak również wśród 3 niejednoczesnych serii badawczych powinna być uzasadniona i poparta naukowo. Kontrole z każdej serii badawczej badanej substancji chemicznej powinny zbliżać się do miar efektywności zgłoszonych dla tego testu Freybergera–Wilsona i powinny być spójne z danymi historycznymi dotyczącymi kontroli z każdego odpowiedniego laboratorium.
|
ANALIZA DANYCH
Test wiązania nasycenia
|
52.
|
Dokonuje się pomiaru zarówno wiązania całkowitego, jak i nieswoistego. W przypadku tych wartości wiązanie swoiste wzrastających stężeń [3H]-17β-estradiolu w warunkach równowagi jest obliczane przez odjęcie wiązania nieswoistego od całkowitego. Wykres stężenia wiązań specyficznych w porównaniu ze stężeniem [3H]-17β-estradiolu powinien osiągnąć plateau dla maksymalnego wiązania specyficznego wskazującego na nasycenie hrERα [3H]-17β-estradiolem. Ponadto analiza danych powinna dokumentować wiązanie estradiolu [3H]-17β-estradiolu z pojedynczym obszarem wiązania o wysokim powinowactwie. Wiązanie nieswoiste, całkowite i swoiste powinno być prezentowane na krzywej wiązania nasycenia. W dalszej analizie tych danych należy wykorzystać analizę regresji nieliniowej (np. BioSoft; McPherson 1985; Motulsky 1995) z końcowym prezentowaniem danych w formie wykresu Scatcharda.
|
|
53.
|
W analizie danych należy określić Bmax i Kd wyłącznie na podstawie danych dotyczących wiązania całkowitego, przy założeniu, że wiązanie niespecyficzne jest liniowe, chyba że podano uzasadnienie dla zastosowania innej metody. Ponadto przy określaniu najlepszego dopasowania należy stosować regresję odpornościową, chyba że zostanie podane uzasadnienie. Należy podać metodę wybraną dla regresji odpornościowej. Korektę obniżenia stężenia ligandu (np. przy użyciu metody na podstawie Swillens 1995) należy zawsze stosować przy określaniu Bmax i Kd na podstawie danych dotyczących wiązania nasycenia.
|
Test wiązania konkurencyjnego
|
54.
|
Krzywa wiązania konkurencyjnego jest wykreślona jako krzywa wiązania specyficznego [3H]-17β-estradiolu w stosunku do stężenia (log10 jednostek) konkurenta. Stężenie badanej substancji chemicznej, która hamuje 50 % maksymalnego wiązania specyficznego [3H]-17β-estradiolu, jest wartością IC50.
|
|
55.
|
Szacunki wartości logarytmu (IC50) dla kontroli dodatnich (np. estrogen odniesienia i substancja słabo wiążąca) należy określić, wykorzystując właściwe oprogramowanie dopasowane do krzywej nieliniowej, aby pasowały do równania Hilla o czterech parametrach (np. BioSoft; McPherson 1985; Motulsky 1995). Najwyższą wartość, najniższą wartość, krzywą Hilla i logarytm IC50 należy zasadniczo pozostawić bez ograniczeń przy dopasowywaniu tych krzywych. Regresję odpornościową należy stosować przy określaniu najlepszego dopasowania, chyba że podane zostanie uzasadnienie. Nie należy stosować korekty obniżenia stężenia ligandu. Po przeprowadzeniu analizy wstępnej każda krzywa wiązania powinna zostać poddana przeglądowi w celu zapewnienia odpowiedniego dopasowania do modelu. Względne powinowactwo wiązania (RBA) w odniesieniu do substancji słabo wiążącej należy obliczać jako procent logarytmu (IC50) dla substancji słabo wiążącej względem logarytmu (IC50) dla 17β-estradiolu. Wyniki kontroli dodatnich i kontroli substancji innych niż substancja wiążąca powinny być oceniane przy użyciu pomiarów efektywności testu określonych w pkt 45–50 niniejszego dodatku 2.
|
|
56.
|
Dane dotyczące wszystkich badanych substancji chemicznych należy analizować na zasadzie stopniowego podejścia, aby zapewnić odpowiednią analizę danych i właściwą klasyfikację każdej krzywej wiązania konkurencyjnego. Zaleca się, aby każda seria badawcza w odniesieniu do badanej substancji chemicznej została wstępnie poddana znormalizowanej analizie danych, która jest identyczna z analizą stosowaną w przypadku kontroli estrogenu odniesienia i substancji słabo wiążącej (zob. pkt 55 powyżej). Po zakończeniu należy przeprowadzić przegląd techniczny parametrów dopasowania krzywej, jak również wizualny przegląd stopnia, w jakim dane pasują do wygenerowanej krzywej wiązania konkurencyjnego dla każdej serii badawczej. W trakcie tego przeglądu technicznego obserwacje zależnego od stężenia spadku w procentach związanego specyficznie [3H]-17β-estradiolu, małej zmienności pomiędzy kontrpróbami technicznymi przy każdym stężeniu chemicznym oraz spójności parametrów dopasowania pomiędzy trzema seriami badawczymi stanowią dobrą wskazówkę, że test i analizy danych zostały wykonane prawidłowo.
|
Interpretacja danych
|
57.
|
O ile spełnione są wszystkie kryteria dopuszczalności, badaną substancję chemiczną uważa się za substancję wiążącą w odniesieniu do hrERα, jeżeli krzywa wiązania może być dopasowana, a najniższy punkt na krzywej reakcji w zakresie danych wynosi mniej niż 50 % (rysunek 1).
|
|
58.
|
O ile spełnione są wszystkie kryteria dopuszczalności, badaną substancję chemiczną uważa się za substancję inną niż substancja wiążąca w odniesieniu do hrERα, jeśli:
|
—
|
krzywa wiązania może być dopasowana, a najniższy punkt na dopasowanej krzywej reakcji w zakresie danych wynosi powyżej 75 %, lub
|
|
—
|
krzywa wiązania nie może być dopasowana, a najniższy niewygładzony średni procent wiązania wśród grup stężeń w danych wynosi powyżej 75 %.
|
|
|
59.
|
Badane substancje chemiczne uważa się za niejednoznaczne, jeśli zostanie spełniony żaden z powyższych warunków (np. najniższy punkt na dopasowanej krzywej reakcji mieści się pomiędzy 76 a 51 %).
|
Tabela 7
Kryteria przypisywania klasyfikacji na podstawie krzywej wiązania konkurencyjnego w odniesieniu do badanej substancji chemicznej
|
Klasyfikacja
|
Kryteria
|
|
Substancja wiążącaa
|
Krzywa wiązania może być dopasowana.
Najniższy punkt na krzywej reakcji w zakresie danych wynosi mniej niż 50 %.
|
|
Substancja inna niż substancja wiążącab
|
Jeżeli krzywa wiązania może być dopasowana,
najniższy punkt na dopasowanej krzywej reakcji w zakresie danych wynosi powyżej 75 %.
Jeżeli krzywa wiązania nie może być dopasowana,
najniższy niewygładzony średni odsetek wiązania wśród grup stężeń w danych wynosi powyżej 75 %.
|
|
Wynik niejednoznacznyc
|
Każda sprawdzalna seria badawcza, która nie dotyczy substancji wiążącej, ani substancji innej niż substancja wiążąca
(np. najniższy punkt na dopasowanej krzywej reakcji mieści się w granicach 76–51 %).
|
Rysunek 1
Przykłady klasyfikacji badanej substancji chemicznej z wykorzystaniem krzywej wiązania konkurencyjnego

|
60.
|
Wielokrotne serie badawcze prowadzone w laboratorium w przypadku badanej substancji chemicznej są łączone poprzez przypisanie wartości liczbowych do każdej serii badawczej i uśrednienie wszystkich serii, jak pokazano w tabeli 8. Wyniki połączonych serii badawczych w każdym laboratorium są porównywane z oczekiwaną klasyfikacją każdej badanej substancji chemicznej.
|
Tabela 8
Metoda klasyfikacji badanej substancji chemicznej z wykorzystaniem wielu serii badawczych w laboratorium
|
Przypisanie wartości do każdej serii badawczej:
|
|
|
Klasyfikacja
|
Wartość liczbowa
|
|
Substancja wiążąca
|
2
|
|
Wynik niejednoznaczny
|
1
|
|
Substancja inna niż substancja wiążąca
|
0
|
|
Klasyfikacja średniej wartości liczbowej w poszczególnych seriach badawczych:
|
|
|
Klasyfikacja
|
Wartość liczbowa
|
|
Substancja wiążąca
|
Średnia ≥ 1,5
|
|
Wynik niejednoznaczny
|
0,5 ≤ Średnia < 1,5
|
|
Substancja inna niż substancja wiążąca
|
Średnia < 0,5
|
SPRAWOZDANIE Z BADANIA
|
61.
|
Zob. pkt 24 „ELEMENTÓW TESTU WIĄZANIA hrER” w niniejszej metodzie badawczej.
|
Dodatek 2.1
WYKAZ POJĘĆ
[3H]E2: 17β-estradiol znakowany izotopem z trytem
DCC: Węgiel drzewny pokryty dekstranem
E2
: Nieoznakowany 17β-estradiol (obojętny)
Bufor testowy: 10 mM Tris, 10 mg albuminy surowicy bydlęcej /ml, 2 mM DTT, 10 % glicerol, 0,2 mM leupeptyny, pH 7,5
hrERα: ludzki rekombinowany receptor estrogenowy alfa
Kontrpróba: Jeden z wielu dołków, które mają tę samą zawartość z tym samym stężeniem i są poddawane testowi jednocześnie w ramach jednej serii badawczej. W niniejszym protokole każde stężenie badanej substancji chemicznej jest badane w trzech powtórzeniach; oznacza to, że istnieją trzy kontrpróby, które są testowane jednocześnie przy każdym stężeniu badanej substancji chemicznej.
Seria badawcza: Pełen zestaw poddawanych jednocześnie serii badawczej dołków mikropłytki, który dostarcza wszelkich informacji niezbędnych do scharakteryzowania wiązania badanej substancji chemicznej z hrERα (tj. całkowity [3H]-17β-estradiol dodany do dołka testowego, maksymalne wiązanie [3H]-17β-estradiolu z hrERα, wiązanie niespecyficzne, oraz wiązanie całkowite przy różnych stężeniach badanej substancji chemicznej). W serii badawczej można wykorzystać tylko jeden dołek (tj. kontrpróbę) na każde stężenie, ale ponieważ niniejszy protokół wymaga trzykrotnego przeprowadzania testu, w jednej analizie wykorzystuje się trzy dołki na każde stężenie. Niniejszy protokół wymaga ponadto przeprowadzenia trzech niezależnych (tj. nierównoczesnych) serii badawczych w odniesieniu do każdej substancji chemicznej.
Dodatek 2.2
TYPOWY TEST NASYCENIA [3H]-17Β-ESTRADIOLU Z TRZEMA DOŁKAMI STANOWIĄCYMI KONTRPRÓBĘ
|
Typowy test nasycenia [3H]-17β-estradiolu z trzema dołkami stanowiącymi kontrpróbę
|
|
Pozycja
|
Kontrpróba
|
Kod rodzaju dołka
|
Stężenie początkowe gorącego E2 (nM)
|
Objętość gorącego E2 (μl)
|
Stężenie końcowe gorącego E2 (nM)
|
Stężenie początkowe zimnego E2 (μM)
|
Objętość zimnego E2 (μl)
|
Stężenie końcowe zimnego E2 (μM)
|
Objętość buforu (μl)
|
Objętość receptora (μl)
|
Całkowita objętość w dołkach
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Gorąca
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Gorąca
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Gorąca
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Gorąca
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Gorąca
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Gorąca
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Gorąca
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Gorąca
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Gorąca
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Gorąca
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Gorąca
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Gorąca
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Gorąca
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Gorąca
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Gorąca
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Gorąca
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Gorąca
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Gorąca
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Gorąca
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Gorąca
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Gorąca
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Gorąca
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Gorąca
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Gorąca
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Należy pamiętać, że „gorące” dołki są puste podczas inkubacji. 40 μl dodaje się tylko w celu zliczania scyntylacji.
|
Dodatek 2.3
ROZKŁAD DOŁKÓW W TEŚCIE WIĄZANIA KONKURENCYJNEGO
|
Płytka
|
Pozycja
|
Kontrpróba
|
Rodzaj dołka
|
Kod dołka
|
Kod stężenia
|
Stężenie początkowe konkurenta (M)
|
Roztwór podstawowy hrER (μl)
|
Objętość buforu (μl)
|
Objętość znacznika (gorącego E2) (μl)
|
Objętość z płytki do rozcieńczeń (μl)
|
Ostateczna objętość (μl)
|
Stężenie końcowe konkurenta (M)
|
|
S
|
A1
|
1
|
wiązanie całkowite
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
wiązanie całkowite
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
wiązanie całkowite
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
wiązanie całkowite
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
wiązanie całkowite
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
wiązanie całkowite
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
zimne E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
zimne E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
zimne E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
zimne E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
zimne E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
zimne E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
zimne E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
zimne E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
zimne E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
zimne E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
zimne E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
zimne E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
zimne E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
zimne E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
zimne E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
zimne E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
zimne E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
zimne E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
zimne E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
zimne E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
zimne E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
ślepa próba
|
ślepa próba
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
ślepa próba
|
ślepa próba
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
ślepa próba
|
ślepa próba
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
noretynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
noretynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
noretynodrel
|
NE
|
WP
|
2.00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
noretynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
noretynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
noretynodrel
|
NE
|
WP
|
6.00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
noretynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
noretynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
noretynodrel
|
NE
|
WP
|
6.00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
gorąca
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
kontrola z buforem
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Należy pamiętać, że „gorące” dołki są puste podczas inkubacji. 40 μl dodaje się tylko w celu zliczania scyntylacji.
|
Rozkład dołków w teście wiązania konkurencyjnego
|
|
Płytka
|
Pozycja
|
Kontrpróba
|
Rodzaj dołka
|
Kod dołka
|
Kod stężenia
|
Stężenie początkowe konkurenta (M)
|
Roztwór podstawowy hrER (μL)
|
Objętość buforu (μl)
|
Objętość znacznika (gorącego E2) (μl)
|
Objętość z płytki rozcieńczenia (μL)
|
Ostateczna objętość (μl)
|
Stężenie końcowe konkurenta (M)
|
|
P1
|
A1
|
1
|
wiązanie całkowite
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
wiązanie całkowite
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
wiązanie całkowite
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
wiązanie całkowite
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
wiązanie całkowite
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
wiązanie całkowite
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
zimne E2 (wysokie)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Badana substancja chemiczna nr 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Badana substancja chemiczna nr 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Badana substancja chemiczna nr 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Badana substancja chemiczna nr 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Badana substancja chemiczna nr 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Badana substancja chemiczna nr 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Badana substancja chemiczna nr 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Badana substancja chemiczna nr 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Badana substancja chemiczna nr 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
zimne E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
zimne E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
zimne E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
zimne E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
zimne E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
zimne E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Dodatek 3
TEST IN VITRO WIĄZANIA RECEPTORA ESTROGENOWEGO INSTYTUTU OCENY CHEMICZNEJ I BADAŃ (ANG. CHEMICAL EVALUATION AND RESEARCH INSTITUTE, CERI) WYKORZYSTUJĄCY BIAŁKO DOMENY WIĄŻĄCEJ LIGAND LUDZKIEGO REKOMBINOWANEGO RECEPTORA ESTROGENOWEGO ALFA (2).
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
1.
|
Niniejszy test in vitro nasycenia receptora estrogenowego (ERα) i wiązania konkurencyjnego wykorzystuje domenę wiążącą ligand (LBD) ludzkiego receptora estrogenowego alfa (hrERα). Ten konstrukt białkowy został wyprodukowany przez Instytutu Oceny Chemicznej i Badań (CERI) w Japonii i istnieje jako białko fuzyjne S-transferazy glutationowej (GST) i jest wyrażony w komórkach E. coli. Protokół CERI został poddany międzynarodowemu wielolaboratoryjnemu badaniu walidacyjnemu (2 ), które wykazało jego adekwatność i wiarygodność w odniesieniu do zaplanowanego celu testu.
|
|
2.
|
Test ten jest procedurą klasyfikacyjną służącą do identyfikacji substancji, które mogą wiązać się z hrERα. Służy on do określenia zdolności badanej substancji chemicznej do konkurowania z 17β-estradiolem w zakresie wiązania z hrERα-LBD. Ilościowe wyniki testu mogą obejmować IC50 (miara stężenia badanej substancji chemicznej potrzebnego do wyparcia połowy [3H]-17β-estradiolu z hrERα) i względne powinowactwa wiązania badanych substancji chemicznych w odniesieniu do hrERα w porównaniu z 17β-estradiolem. Do celów klasyfikacji substancji chemicznych akceptowalne wyniki testów jakościowych mogą obejmować klasyfikację badanych substancji chemicznych jako substancji wiążących hrERα, substancji innych niż substancja wiążąca lub niejednoznacznych w oparciu o kryteria opisane dla krzywych wiązania.
|
|
3.
|
W teście wykorzystuje się ligand promieniotwórczy, który wymaga zezwolenia na stosowanie materiałów promieniotwórczych przez laboratorium. Wszystkie procedury dotyczące radioizotopów i niebezpiecznych substancji chemicznych powinny być zgodne z przepisami i procedurami opisanymi w prawodawstwie krajowym.
|
|
4.
|
Przed rozpoczęciem stosowania tego badania do celów regulacyjnych należy zapoznać się z sekcjami „WPROWADZENIE OGÓLNE” oraz „ELEMENTY TESTU WIĄZANIA hrER”. Definicje i skróty stosowane w niniejszej wytycznej dotyczącej badań opisano w dodatku 1.
|
ZASADY WYKONANIA TESTU (ZOB. RÓWNIEŻ WPROWADZENIE OGÓLNE)
|
5.
|
W teście wiązania hrERα mierzy się zdolność znakowanego izotopem liganda ([3H]17β-estradiolu) do wiązania się z receptorem estrogenowym w obecności zwiększających się stężeń badanej substancji chemicznej (tj. konkurenta). Badane substancje chemiczne o wysokim powinowactwie z receptorem estrogenowym konkurują ze znakowanym izotopem ligandem w niższym stężeniu w porównaniu z substancjami chemicznymi o niższym powinowactwie z receptorem.
|
|
6.
|
Niniejszy test składa się z dwóch głównych elementów: doświadczenie dotyczące wiązania nasycenia mające na celu scharakteryzowanie parametrów interakcji receptor-ligand, po którym następuje doświadczenie dotyczące wiązania konkurencyjnego, charakteryzujący konkurencję pomiędzy badaną substancją chemiczną a znakowanym izotopem ligandem w zakresie wiązania z receptorem estrogenowym.
|
|
7.
|
Celem doświadczenia dotyczącego wiązania nasycenia jest scharakteryzowanie konkretnej partii receptorów pod kątem powinowactwa wiązania i ich liczby w ramach przygotowania do doświadczenia dotyczącego wiązania konkurencyjnego. W doświadczeniu dotyczącym wiązania nasycenia mierzy się, w warunkach równowagi, powinowactwo stałego stężenia receptora estrogenowego w odniesieniu do jego naturalnego ligandu (reprezentowanego przez stałą dysocjacji, Kd) oraz stężenie obszarów czynnego receptora (Bmax).
|
|
8.
|
W doświadczeniu dotyczącym konkurencyjnego wiązania mierzy się powinowactwo substancji do konkurowania z [3H]17β-estradiolem o wiązanie z receptorem estrogenowym. Powinowactwo jest określa się ilościowo za pomocą stężenia badanej substancji chemicznej, która w stanie równowagi hamuje 50 % specyficznego wiązania [3H]17β-estradiolu (nazywanego „stężeniem hamującym 50 %” lub IC50). Można je również ocenić za pomocą względnego powinowactwa wiązania (RBA, w stosunku do IC50 estradiolu mierzonego oddzielnie w tej samej serii badawczej). W doświadczeniu dotyczącym konkurencyjnego wiązania mierzy się wiązanie [3H]17β-estradiolu przy stałym stężeniu w obecności szerokiego zakresu (ośmiu rzędów wielkości) stężeń badanych substancji chemicznych. Dane są następnie dopasowywane, na ile to możliwe, do postaci równania Hilla (Hill 1910), które opisuje przemieszczenie radioligandu przez jednoobszarową konkurencyjną substancję wiążącą. Zakres przemieszczenia estradiolu znakowanego izotopem w stanie równowagi jest wykorzystywany do scharakteryzowania badanej substancji chemicznej jako substancji wiążącej, substancji innej niż substancja wiążąca lub generującej reakcję niejednoznaczną.
|
PROCEDURA
Wykazanie dopuszczalnej efektywności białka hrERα
|
9.
|
Przed rutynowym wykonaniem testów nasycenia i wiązania konkurencyjnego należy wykazać, że efektywność każdej nowej partii hrERα w laboratorium, w którym będzie używana, jest prawidłowa. W celu zademonstrowania efektywności należy zastosować dwuetapowy proces. Etapy te są następujące:
|
—
|
Przeprowadzenie testu wiązania nasycenia [3H]-17β-estradiolu w celu wykazania swoistości i nasycenia hrERα. Analiza regresji nieliniowej tych danych (np. BioSoft; McPherson 1985; Motulsky 1995), a następnie wykres Scatcharda powinien dokumentować powinowactwo wiązania hrERα [3H]-17β-estradiolu (Kd) i liczbę receptorów (Bmax) w odniesieniu do konkretnej partii hrERα.
|
|
—
|
Wykonanie testu wiązania konkurencyjnego przy użyciu substancji kontrolnych (estrogen odniesienia (17β-estradiol), substancji słabo wiążącej (np. noretynodrel lub noretyndron) oraz substancji innej niż substancja wiążąca (oktylotrietoksysilan, OTES). Każde laboratorium powinno stworzyć bazę danych historycznych w celu udokumentowania spójności IC50 i wartości istotnych dla estrogenów odniesienia i substancji słabo wiążących między doświadczeniami i różnymi partiami hrERα. Ponadto parametry krzywych konkurencyjnego wiązania substancji kontrolnych powinny mieścić się w przedziale ufności 95 % (zob. tabela 1), który opracowano wykorzystując dane laboratoriów uczestniczących w badaniu walidacyjnym w odniesieniu do celów niniejszego testu (2).
|
Tabela 1
Kryteria efektywności opracowane dla estrogenów odniesienia i substancji słabo wiążących, test wiązania hrER CERI
|
Substancja
|
Parametr
|
Wartość średnia (11)
|
Odchylenie standardowe (n)
|
Przedziały ufności (12) 95 %
|
|
Dolna wartość graniczna
|
Górna wartość graniczna
|
|
17β-estradiol
|
Najwyższa wartość
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Najniższa wartość
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Krzywa Hilla
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretynodrel
|
Najwyższa wartość
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Najniższa wartość
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Krzywa Hilla
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretyndron (13)
|
Najwyższa wartość
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Najniższa wartość
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Krzywa Hilla
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Wykazanie biegłości laboratorium
|
10.
|
Zob. pkt 17 i 18 oraz tabela 2 w rozdziale „ELEMENTY TESTU WIĄZANIA hrER” w niniejszej metodzie badawczej. Każdy test (nasycenia i wiązania konkurencyjnego) powinien składać się z trzech niezależnych serii badawczych (tj. ze świeżymi rozcieńczeniami receptora, substancji chemicznych i odczynników) w różnych dniach, a każda seria badawcza powinna obejmować trzy kontrpróby.
|
Określanie stężenia receptora (hrERα)
|
11.
|
Stężenie czynnego receptora różni się nieznacznie w zależności od partii i warunków składowania. Z tego powodu należy określić stężenie czynnego receptora otrzymanego od dostawcy. Pozwoli to uzyskać odpowiednie stężenie aktywnego receptora w serii badawczej.
|
|
12.
|
W warunkach odpowiadających konkurencyjnym wiązaniom (tj. 0,5 nM [3H]-estradiolu), nominalne stężenia 0,1, 0,2, 0,4 i 0,6 nM receptora powinny być inkubowane przy braku (wiązanie całkowite) i obecności (wiązanie nieswoiste) 1 μM nieoznakowanego estradiolu. Określone wiązanie, obliczone jako różnica wiązania całkowitego i nieswoistego, jest wykreślane w funkcji nominalnego stężenia receptora. Stężenie receptora, które daje określone wartości wiązania odpowiadające 40 % dodanego oznaczenia izotopem, jest związane z odpowiadającym mu stężeniem receptora, a to stężenie receptora należy wykorzystywać w doświadczeniach dotyczących nasycenia i konkurencyjnego wiązania. Często końcowe stężenie hrER wynoszące 0,2 nM jest zgodne z tym warunkiem.
|
|
13.
|
Jeżeli kryterium 40 % nie może być wielokrotnie spełnione, należy sprawdzić, czy w zestawie doświadczenia nie ma potencjalnych błędów. Nieosiągnięcie kryterium 40 % może wskazywać, że w partii rekombinowanej jest bardzo mało aktywnego receptora, w związku z czym należy rozważyć zastosowanie innej partii receptora.
|
Test nasycenia
|
14.
|
Osiem wzrastających stężeń [3H]17β-estradiolu należy oceniać trzykrotnie, w następujących trzech warunkach (zob. tabela 2):
|
a.
|
przy braku nieoznakowanego 17β-estradiolu i w obecności receptora estrogenowego. Jest to wyznaczenie wiązania całkowitego przez pomiar radioaktywności w dołkach, w których znajduje się tylko [3H]17β-estradiol.
|
|
b.
|
W obecności 2000-krotnego przekroczenia stężenia nieoznakowanego 17β-estradiolu nad oznakowanym 17β-estradiolem i w obecności receptora estrogenowego. Zamierzeniem tego warunku jest nasycenie aktywnych obszarów wiązania nieoznakowanym 17β-estradiolem, a w wyniku pomiaru radioaktywności w dołkach, określenie wiązania nieswoistego. Każdy pozostały gorący estradiol, który może wiązać się z receptorem uznaje się za wiążący w nieswoistym obszarze, ponieważ zimny estradiol powinien być w tak wysokim stężeniu, aby wiązał się ze wszystkimi dostępnymi swoistymi obszarami na receptorze.
|
|
c.
|
Przy braku nieoznakowanego 17β-estradiolu i braku receptora estrogenowego (określenie całkowitej radioaktywności).
|
|
Przygotowanie roztworów [3H]-17β-estradiolu, nieoznakowanego 17β-estradiolu i hrERα
|
15.
|
Należy przygotować roztwór o stężeniu 40 nM [3H]-17β-estradiolu z użyciem roztworu podstawowego 1 μM [3H]-17β-estradiolu w sulfotlenku dimetylowym, w temperaturze pokojowej dodając sulfotlenek dimetylowy (aby przygotować roztwór 200 nM) oraz bufor testowy (aby przygotować roztwór 40 nM). Stosując ten roztwór 40 nM, należy w temperaturze pokojowej przygotować serię rozcieńczeń [3H]-17β-estradiolu w zakresie od 0,313 nM do 40 nM i zawierającą roztwór buforowy (co przedstawia pozycja 12 tabeli 2). Dodając 10 μl tych roztworów do odpowiednich dołków płytki 96-dołkowej (w ostatecznej objętości 3 μl), uzyskuje się końcowe stężenia testowe w zakresie od 0,0313 μM do 4,0 μM (zob. tabele 2 i 3). Przygotowanie buforu testowego, obliczenie początkowego roztworu podstawowego [3H]-17β-estradiolu w oparciu na jego właściwej aktywności, preparaty rozcieńczeń i określenie stężeń są szczegółowo opisane w protokole CERI (2).
|
|
16.
|
Należy przygotować rozcieńczenia nieoznakowanych roztworów 17β-estradiolu z użyciem roztworu podstawowego 1 nM 17β-estradiolu dodając bufor testowy, aby uzyskać osiem stężeń wzrastających początkowo od 0,625 μM do 80 μM. Dodając 10 μl tych roztworów do odpowiednich dołków testowych płytki 96-dołkowej przeznaczonej do pomiaru wiązań nieswoistych, uzyskuje się końcowe stężenia testowe w zakresie od 0,0625 μM do 8 μM (zob. tabele 2 i 3). Przygotowanie rozcieńczeń nieoznakowanego 17β-estradiolu jest szczegółowo opisane w protokole CERI (2).
|
|
17.
|
Należy stosować nominalne stężenie receptora, które daje specyficzne wiązanie 40±10 % (zob. pkt 12–13). Roztwór hrERα należy przygotować w lodowatym buforze testowym bezpośrednio przed użyciem, tj. po przygotowaniu wszystkich dołków do wiązania całkowitego, wiązania nieswoistego oraz samodzielnego gorącego ligandu.
|
|
18.
|
Płytki 96-dołkowe są przygotowywane w sposób przedstawiony w tabeli 2, z 3 kontrpróbami na stężenie [3H]-17β-estradiolu. Objętość [3H]-17β-estradiolu, nieoznakowanego 17β-estradiolu, buforu i receptora przedstawiono w tabeli 3.
Tabela 2
Rozkład na mikropłytce do testu wiązania nasycenia
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Do pomiaru TB
|
Do pomiaru NSB
|
Do określenia samodzielnego gorącego ligandu
|
|
Nieoznakowane rozcieńczenia E2 do kolumn płytki 4–6
|
Rozcieńczenia [3H]E2 do kolumn płytki 1–9
|
|
A
|
0,0313 nM [3H] E2+ receptor estrogenowy
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ receptor estrogenowy
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ receptor estrogenowy
|
0,0625 nM [3H] E2+ 0,125 μM E2+ receptor estrogenowy
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ receptor estrogenowy
|
0,125 nM [3H] E2+ 0,25 μM E2+ receptor estrogenowy
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ receptor estrogenowy
|
0,250 nM [3H] E2+ 0,5 μM E2+ receptor estrogenowy
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [ H] E2+ receptor estrogenowy
|
0,50 nM [3H] E2+ 1 μM E2+ receptor estrogenowy
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ receptor estrogenowy
|
1,00 nM [3H] E2+ 2 μM E2+ receptor estrogenowy
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ receptor estrogenowy
|
2,00 nM [3H] E2+ 4 μM E2+ receptor estrogenowy
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ receptor estrogenowy
|
4,00 nM [3H] E2+ 8 μM E2+ receptor estrogenowy
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
wiązanie całkowite;
|
|
NSB
|
:
|
wiązanie nieswoiste;
|
|
[3H] E2
|
:
|
[3H]17β-estradiol
|
|
E2
|
:
|
nieoznakowany 17β-estradiol
|
|
Tabela 3
Objętości odczynnika w odniesieniu do nasycenia mikropłytki
|
Nr pozycji
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Etap przygotowania
|
Dołki TB
|
Dołki NSB
|
Samodzielny gorący ligand
|
|
Objętość elementów w odniesieniu do powyższych dołków reakcyjnych oraz kolejność dodawania
|
Roztwór buforowy
|
60 μl
|
50 μl
|
90 μl
|
|
nieoznakowane E2 z pozycji 11 tabeli 2
|
–
|
10 μl
|
–
|
|
[3H]E2 z pozycji 12 w tabeli 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Całkowita objętość reakcji
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubacja
|
REAKCJA NASTĘPUJĄCA PO 2 GODZINACH INKUBACJI
|
Określenie ilościowe radioaktywności tuż po przygotowaniu. Brak inkubacji
|
|
Poddawanie działaniu substancji za pomocą 0,4 % DCC
|
Tak
|
Tak
|
Nie
|
|
Objętość 0,4 % DCC
|
100 μl
|
100 μl
|
–
|
|
Filtracja
|
Tak
|
Tak
|
Nie
|
|
POMIAR LICZBY ROZPADÓW NA MINUTĘ
|
|
Objętość ilościowa dodawana do koktajlu scyntylacyjnego
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Mikropłytki testowe pozwalające na określenie wiązań całkowitych i wiązań nieswoistych należy przez dwie godziny inkubować w temperaturze pokojowej (22–28 °C).
|
Pomiar [3H]-17β-estradiolu związanego z hrERα
|
20.
|
Po dwugodzinnym okresie inkubacji [3H]-17β-estradiol związany z hrERα należy oddzielić od wolnego [3H]-17β-estradiolu, w tym celu dodając do dołków 100 μl lodowatej zawiesiny DCC 0,4 %. Płytki należy następnie na 10 min umieścić z lodem oraz poprzez przeniesienie ich do filtra mikropłytek należy przeprowadzić filtrację mieszaniny reakcyjnej i zawiesiny DCC; celem tego działania jest usunięcie DCC. Następnie do płynu scyntylacyjnego znajdującego się we fiolkach scyntylacyjnego licznika cieczowego należy dodać 100 μl filtratu, aby za pomocą tego licznika określić liczbę rozpadów na minutę/fiolkę.
|
|
21.
|
W przypadku gdy filtr mikropłytkowy nie jest dostępny, metodą alternatywną może okazać się usunięcie DCC poprzez odwirowanie. 50 μl supernatantu zawierającego [3H]-17β-estradiol związany z hrERα należy wówczas stosować ze szczególną ostrożnością, aby uniknąć zanieczyszczenia dołków przez styczność z DCC, a następnie umieścić na drugiej mikropłytce.
|
|
22.
|
Stan, w jakim znajduje się samodzielny gorący ligand pozwala określić liczbę rozpadów na minutę dodawanego do dołków testowych [3H]-17β-estradiolu. Radioaktywność należy określić ilościowo tuż po przygotowaniu. Dołków tych nie powinno się inkubować, a także nie należy poddawać ich działaniu zawiesiny DCC, natomiast ich zawartość należy wprowadzić bezpośrednio do płynu scyntylacyjnego. Pomiary te pokazują, ile [3H]-17β-estradiolu wyrażonych za pomocą liczby rozpadów na minutę dodano do każdego zestawu dołków w celu uzyskania wiązania całkowitego i wiązania nieswoistego.
|
Test wiązania konkurencyjnego
|
23.
|
W teście wiązania konkurencyjnego mierzy się wiązanie pojedynczego stężenia [3H]17β-estradiolu w obecności zwiększających się stężeń badanej substancji chemicznej. Przy każdym stężeniu w ramach jednej serii badawczej należy zastosować trzy równoległe kontrpróby. Ponadto dla każdej badanej substancji chemicznej należy wykonać trzy niejednoczesne serie badawcze. Test należy przeprowadzić z wykorzystaniem co najmniej jednej płytki 96-dołkowej.
|
Kontrole
|
24.
|
Podczas przeprowadzania testu w każdym doświadczeniu należy uwzględnić równoczesny rozpuszczalnik i kontrole (tj. estrogen odniesienia, substancję słabo wiążącą oraz substancję inną niż substancja wiążąca). Pełne krzywe stężeń dla estrogenów odniesienia i kontroli (tj. substancji słabo wiążącej i substancji innej niż substancja wiążąca) w każdej serii badawczej należy stosować na jednej płytce. Wszystkie pozostałe płytki powinny zawierać (i) wysokie (maksymalne przemieszczenie tj. w przybliżeniu pełne przemieszczenie ligand znakowanych izotopem) i średnie (około IC50) stężenie E2 oraz substancji słabo wiążącej w trzech powtórzeniach; (ii) kontrolę z rozpuszczalnikiem i wiązanie nieswoiste, każde w trzech powtórzeniach. Procedury przygotowania buforu testowego, [3H]-17β-estradiolu, hrERα i roztworów badanych substancji chemicznych szczegółowo opisano w protokole CERI (2).
|
Kontrola z rozpuszczalnikiem:
|
25.
|
Kontrola z rozpuszczalnikiem wskazuje, że rozpuszczalnik nie wchodzi w interakcję z układem badawczym, a także umożliwia zmierzenie wiązania całkowitego (TB). Preferowanym rozpuszczalnikiem jest sulfotlenek dimetylowy. W przypadku gdy najwyższe stężenie badanej substancji chemicznej nie jest rozpuszczalne w sulfotlenku dimetylowym, alternatywnie można zastosować etanol. Stężenie sulfotlenku dimetylowego w dołkach testu końcowego powinno wynosić 2,05 %, a w przypadku braku nierozpuszczalności badanej substancji chemicznej może wzrosnąć do poziomu 2,5 %. Ze względu na interferencję wyższych stężeń rozpuszczalnika z danym testem, nie należy stosować stężeń sulfotlenku dimetylowego wyższych niż 2,5 %. W przypadku substancji chemicznych, które są nierozpuszczalne w sulfotlenku dimetylowym, lecz są rozpuszczalne w etanolu, w teście bez interferencji można zastosować maksymalnie etanol o stężeniu 2 %.
|
Kontrola z buforem:
|
26.
|
Kontrola z buforem (BC) nie powinna zawierać rozpuszczalnika ani badanej substancji chemicznej, ale wszystkie inne elementy testu. Wyniki kontroli z buforem porównuje się z wynikami kontroli z rozpuszczalnikiem w celu sprawdzenia, czy zastosowany rozpuszczalnik nie ma wpływu na system testu.
|
Substancja silnie wiążąca (estrogen odniesienia)
|
27.
|
17β-estradiol (CAS 50-28-2) jest endogennym ligandem i tworzy wiązanie o wysokim powinowactwie z receptorem estrogenowym, podtyp alfa. Dla każdego testu wiązania konkurencyjnego hrERα należy przygotować krzywą wzorcową z użyciem nieoznakowanego 17β-estradiolu, aby w tym samym laboratorium umożliwić ocenę zmienności podczas wykonywania testu na przestrzeni czasu. W sulfotlenku dimetylowym i buforze testowym należy przygotować osiem roztworów nieoznakowanego 17β-estradiolu o stężeniach w dołkach testowych należy stosować na krzywej wzorcowej, w następujących odstępach: 10–6, 10–7, 10–8, 10–8.5, 10–9, 10–9.5, 10–10, 10–11 M. Najwyższe stężenie nieoznakowanego 17β-estradiolu (1 μM) powinno służyć również jako wskaźnik nieswoistego wiązania. Stężenie to wyróżnia się etykietą „NSB” w tabeli 4, mimo że jest ono również częścią krzywej wzorcowej.
|
Substancja słabo wiążąca
|
28.
|
Należy uwzględnić substancję słabo wiążącą (noretynodrel (CAS 68-23-5) lub jego zastępcę – noretyndron (CAS 68-22-4)) w celu wykazania czułości każdego doświadczenia i umożliwienia oceny zmienności przy przeprowadzaniu testu z upływem czasu. W sulfotlenku dimetylowym i buforze testowym należy przygotować osiem roztworów substancji słabo wiążącej o następujących stężeniach w dołkach testowych: 10–4.5, 10–5.5, 10–6, 10–6.5, 10–7, 10–7.5, 10–8 i 10–9 M.
|
Substancja inna niż substancja wiążąca
|
29.
|
Oktylotrietoksysilan (OTES, CAS 2943-75-1) należy stosować jako kontrolę ujemną (substancję inną niż substancja wiążąca). Daje to pewność, że test w takiej formie umożliwi wykrycie, że badane substancji chemiczne nie wiążą się z hrERα. W sulfotlenku dimetylowym i buforze testowym należy przygotować osiem roztworów substancji innej niż substancja wiążąca o następujących stężeniach w dołkach testowych: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Jako alternatywną substancję inną niż substancja wiążąca można zastosować ftalan dibutylu (DBP, nr CAS 84-72-2), można go jednak badać jedynie do 10–4M. Wykazano, że maksymalna rozpuszczalność DBP w teście wynosi 10–4 M.
|
Stężenie hrERα
|
30.
|
Należy stosować taką ilość receptora, która da specyficzne wiązanie 40 ±10 % 1 nM radioligandu (zob. pkt 12–13 dodatku 3). Roztwór hrERα należy przygotować, rozcieńczając funkcjonalne hrERα w lodowatym buforze testowym bezpośrednio przed zastosowaniem.
|
[3H]-17β-estradiol
|
31.
|
Stężenie końcowe [3H]-17β-estradiolu w dołkach testowych powinno wynosić 0,5 nM.
|
Badane substancje chemiczne
|
32.
|
W pierwszej kolejności konieczne jest przeprowadzenie badania rozpuszczalności, aby ustalić granicę rozpuszczalności każdej badanej substancji chemicznej oraz określić właściwy zakres stężeń, który zostanie zastosowany przy realizacji protokołu badania. Granicę rozpuszczalności każdej badanej substancji chemicznej należy początkowo ustalić w rozpuszczalniku, a następnie potwierdzić w warunkach testowych. Stężenia końcowe będące przedmiotem testu nie powinny przekraczać 1 mM. Badanie ustalające zakres dawkowania obejmuje kontrolę z rozpuszczalnikiem wraz z co najmniej ośmioma seriami stężeń wzrastających w postępie logarytmicznym, począwszy od maksymalnego dopuszczalnego stężenia (np. 1 mM lub niższego, na podstawie granicy rozpuszczalności) oraz odnotowaną obecność zmętnienia lub osadu (zob. również pkt 35 dodatku 3). Po ustaleniu zakresu stężeń do celów badań, badaną substancję chemiczną należy badać przy użyciu 8 krzywych stężeń logarytmicznych, ułożonych w odpowiednich odstępach, co określono w poprzednim badaniu dotyczącym zakresu dawkowania. Zbadane stężenia w drugim i trzecim doświadczeniu należy następnie odpowiednio dostosować, aby w razie potrzeby lepiej scharakteryzować krzywą zależności stężenie-odpowiedź.
|
|
33.
|
Rozcieńczenia badanych substancji chemicznych należy przygotować w odpowiednim rozpuszczalniku (zob. pkt 25 dodatku 3). Badana substancja chemiczna w najwyższym stężeniu nie rozpuszcza się ani w sulfotlenku dimetylowym, ani w etanolu, a dodanie większej ilości rozpuszczalnika spowodowałoby, że stężenie rozpuszczalnika w końcowej probówce byłoby wyższe niż dopuszczalna wartość graniczna, najwyższe stężenie można obniżyć do następnego, niższego stężenia. W takim przypadku można dodać dodatkowe stężenie na dolnym końcu serii stężeń. Pozostałe stężenia w serii powinny pozostać niezmienione.
|
|
34.
|
Roztwory badanej substancji chemicznej powinny być ściśle monitorowane po dodaniu ich do dołka testowego, ponieważ badana substancja chemiczna może wytrącać się po dodaniu do dołka testowego. Dane dotyczące wszystkich dołków, które zawierają osady, należy wyłączyć z dopasowywania krzywej, a także odnotować powód wyłączenia tych danych.
|
|
35.
|
Jeżeli istnieją już wcześniejsze informacje pochodzące z innych źródeł, na podstawie których można wyznaczyć logarytm (IC50) badanej substancji chemicznej, właściwe może być geometryczne rozmieszczenie rozcieńczeń bliżej zakładanego log (IC50) (tj. 0,5 jednostki logarytmicznej). Wyniki końcowe powinny dostatecznie wykazywać wystarczającą rozpiętość stężeń po obu stronach logarytmu (IC50), w tym „najwyższa wartość” i „najniższa wartość”, tak aby krzywą wiązania można było odpowiednio scharakteryzować.
|
Organizacja płytki testowej
|
36.
|
Oznakowane mikropłytki powinny być przygotowane z zastosowaniem: sześciokrotnej inkubacji kontroli z rozpuszczalnikiem, najwyższego stężenia estrogenu odniesienia (E2), który służy również za wskaźnik wiązania nieswoistego (NSB), kontroli z buforem, oraz potrójnych inkubacji dla każdego z ośmiu stężeń niewiążących kontroli (oktylotrietoksysilan), siedmiu niższych stężeń estrogenu odniesienia (E2), ośmiu stężeń substancji słabo wiążącej (noretynodrelu lub noretysteronu) oraz ośmiu stężeń każdej badanej substancji chemicznej (TC). W poniższej tabeli 4 przedstawiono przykładowy wykres dotyczący rozkładu na płytce pełnych krzywych stężeń estrogenu odniesienia i kontroli. Dodatkowe mikropłytki wykorzystuje się do badanych substancji chemicznych i powinny one zawierać kontrole płytek, (tj. (i) wysokie (maksymalne przemieszczenie) i średnie stężenie (około IC50) każdego E2 oraz substancji słabo wiążącej w trzech powtórzeniach; (ii) kontrolę z rozpuszczalnikiem (jako wiązanie całkowite) i wiązanie nieswoiste, każde w sześciu powtórzeniach (tabela 5). Przykład arkusza roboczego rozkładu mikropłytki testowej, wykorzystującego trzy nieznane badane substancje chemiczne, znajduje się w dodatku 3.3. Stężenia wskazane w arkuszu roboczym oraz w tabelach 4 i 5 odnoszą się do stężeń końcowych wykorzystywanych w poszczególnych dołkach testowych. Maksymalne stężenie dla E2 powinno wynosić 1×10—7 M, a dla substancji słabo wiążącej należy stosować najwyższe stężenie dla substancji słabo wiążącej na płytce 1. Stężenie IC50 musi być określone przez laboratorium na podstawie jego bazy danych historycznych dotyczących kontroli. Oczekuje się, że wartość ta będzie podobna do wartości zaobserwowanej w badaniach walidacyjnych (zob. tabela 1).
|
Tabela 4
Rozkład na mikropłytce do testu wiązania konkurencyjnego
(97)
(98)
, pełne krzywe stężeń dla estrogenu odniesienia i próby kontrolnej (płytka 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Kontrola z buforem i kontrola dodatnia (E2)
|
Słaba dodatnia (Noretynodrel)
|
Kontrola ujemna (OTES)
|
TB i NSB
|
|
A
|
Próba ślepa (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (kontrola z rozpuszczalnikiem) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Kontrola z buforem
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Próba ślepa (dla gorących) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Tabela 5
Rozkład na mikropłytce testowej wiązania konkurencyjnego, dodatkowe płytki na użytek badanych substancji chemicznych (TC) i próby kontrolne dla płytki
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Badana substancja chemiczna-1 (TC-1)
|
Badana substancja chemiczna-2 (TC-2)
|
Badana substancja chemiczna-3 (TC-3)
|
Kontrole
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10–7M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10–4,5M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (kontrola z rozpuszczalnikiem)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
W tym przykładzie substancją słabo wiążącą jest noretynodrel (NE)
|
Zakończenie testu wiązania konkurencyjnego
|
37.
|
Jak pokazano w tabeli 6, z wyjątkiem dołków dotyczących wiązania całkowitego i prób ślepych (dla gorących), w każdym dołku należy umieścić 50 μl bufora testowego oraz wymieszać z 10 μl kontroli z rozpuszczalnikiem, estrogenu odniesienia (E2), substancji słabo wiążącej, substancji innej niż substancja wiążąca i badanych substancji chemicznych, odpowiednio, 10 μl z 5 nM roztworu [3H]-17β-estriadolu. Następnie do każdej płytki dodaje się 30 μl lodowatego roztworu receptora i miesza się delikatnie. Roztwór hrERα należy dodać na końcu. Mikropłytki testowe należy inkubować w temperaturze pokojowej (22–28 °C) przez dwie godziny.
Tabela 6
Objętość elementów testu w odniesieniu do testu wiązania konkurencyjnego hrER, mikropłytki
|
Etapy przygotowania
|
Inne niż dołki TB
|
Dołki TB
|
Próba ślepa (dla gorących)
|
|
Objętość elementów w odniesieniu do powyższych dołków reakcyjnych oraz kolejność dodawania
|
Bufor testowy w temperaturze pokojowej
|
50 μl
|
60 μl
|
90 μl
|
|
Nieoznakowane E2, substancja słabo wiążąca, substancja inna niż substancja wiążąca, rozpuszczalnik oraz badane substancje chemiczne (*16)
|
10 μl
|
–
|
–
|
|
Stężenie końcowe [3H]-17β-estradiolu względem przyrostu 0,5 nM (tj. 5 nM).
|
10 μl
|
10 μl
|
10 μl
|
|
Ustalone stężenie hrERα (zob. pkt 12–13)
|
30 μl
|
30 μl
|
–
|
|
Całkowita objętość w każdym dołku testowym
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Oznaczenie ilościowe [3H]-17β-estradiolu związanego z hrERα po oddzieleniu [3H]-17β-estradiolu związanego z hrERα od wolnego [3H]-17β-estradiolu poprzez dodanie 100 μl lodowatej zawiesiny DCC do każdego dołka należy następnie przeprowadzić jak opisano w dodatku 3 w pkt 21–23 w odniesieniu do testu wiązania nasycenia.
|
|
39.
|
Dołki G10–12 oraz H10–12 [oznaczone w tabeli 4 jako próby ślepe (dla gorących)] reprezentują DPM estradiolu oznakowanego [3H] w 10 μl. Podwielokrotność 10 μl należy dodać bezpośrednio do płynu scyntylacyjnego.
|
Kryteria dopuszczalności
Test wiązania nasycenia
|
40.
|
Krzywa wiązania specyficznego powinna osiągnąć stan równowagi, ponieważ zastosowano zwiększające się stężenia [3H]-17β-estradiolu, wskazujące na nasycenie hrERα ligandem.
|
|
41.
|
Wiązanie specyficzne przy 0,5 nM [3H]-17β-estradiolu powinno mieścić się w dopuszczalnym zakresie 30–50 % średniej zmierzonej całkowitej radioaktywności dodanej we wszystkich seriach badawczych. Sporadyczne niewielkie odchylenia poza ten zakres są dopuszczalne, ale jeśli wyniki serii badawczych systematycznie nie mieszczą się w tym zakresie lub wynik danej serii badawczej wychodzi znacznie poza ten zakres, należy dostosować stężenie białka i powtórzyć test nasycenia.
|
|
42.
|
Dane powinny tworzyć liniowy wykres Scatcharda.
|
|
43.
|
Wiązania nieswoiste nie powinny być nadmierne. Wartość dla wiązań nieswoistych powinna zazwyczaj wynosić < 35 % wiązań całkowitych. Stosunek ten może jednak sporadycznie przekraczać tę granicę przy pomiarze bardzo niskich DPM dla najniższego stężenia badanego 17β-estradiolu znakowanego izotopem.
|
Test wiązania konkurencyjnego
|
44.
|
Zwiększające się stężenia nieoznakowanego 17β-estradiolu powinny wyprzeć [3H]-17β-estradiol z receptora w sposób zgodny z jednoobszarowym wiązaniem konkurencyjnym.
|
|
45.
|
Wartość IC50 dla estrogenu odniesienia (tj. 17β-estradiolu) powinna być w przybliżeniu równa stężeniu molowemu [3H]-17β-estradiolu powiększonemu o Kd oznaczony w teście wiązania nasycenia.
|
|
46.
|
Całkowite wiązanie specyficzne powinno być spójne w dopuszczalnym zakresie 40 ± 10 %, gdy średnie zmierzone stężenie całkowitej radioaktywności dodanej do każdego dołka wynosi 0,5 nM we wszystkich seriach badawczych. Sporadyczne niewielkie odchylenia poza ten zakres są dopuszczalne, ale jeśli wyniki serii badawczych systematycznie nie mieszczą się w tym zakresie lub wynik danej serii badawczej wychodzi znacznie poza ten zakres, należy dostosować stężenie białka.
|
|
47.
|
Rozpuszczalnik nie powinien powodować zmiany czułości ani odtwarzalności testu. Wyniki kontroli z rozpuszczalnikiem (dołki z wiązaniem całkowitym) porównuje się z wynikami kontroli z buforem w celu sprawdzenia, czy zastosowany rozpuszczalnik nie ma wpływu na system testu. Wyniki kontroli wiązania całkowitego i kontroli z buforem powinny być porównywalne, jeżeli rozpuszczalnik nie wpływa na test.
|
|
48.
|
Substancja inna niż substancja wiążąca nie powinna wypierać więcej niż 25 % [3H]-17β-estradiolu z hrERα podczas badania do 10–3 M (OTES) lub 10–4 M (DBP).
|
|
49.
|
Opracowano kryteria efektywności dla estrogenu odniesienia i dwóch substancji słabo wiążących (np. noretynodrel, noretyndron) z wykorzystaniem danych z badania walidacyjnego dotyczącego testu wiązania hrER CERI (załącznik N do 2 pozycji bibliografii). 95 % przedziały ufności podano w odniesieniu do średniej ± SD (n) wszystkich kontrolnych serii badawczych przeprowadzanych w czterech laboratoriach, które wzięły udział w badaniu walidacyjnym. 95 % przedziały ufności obliczono dla parametrów dopasowania krzywej (tj. najwyższa wartość, najniższa wartość, krzywa Hilla, logarytm IC50) dla estrogenu odniesienia i substancji słabo wiążących oraz dla log10RBA substancji słabo wiążących w stosunku do estrogenu odniesienia. W tabeli 1 przedstawiono oczekiwane zakresy parametrów dopasowania krzywej, które mogą być wykorzystane jako kryteria efektywności. W praktyce zakres IC50 może się nieznacznie różnić w zależności od doświadczalnie wyprowadzonego Kd preparatu receptora i stężenia ligandu wykorzystanego w teście.
|
|
50.
|
Nie opracowano kryteriów efektywności w odniesieniu do parametrów dopasowania krzywej dla badanych substancji chemicznych ze względu na szeroki wachlarz istniejących potencjalnych badanych substancji chemicznych i zmienność potencjalnych powinowactw i wyników (np. pełna krzywa, krzywa częściowa, brak dopasowania krzywej). Dokonując przeglądu wyników z każdej serii badawczej badanej substancji chemicznej, należy jednak przyjąć profesjonalny osąd. Należy zastosować wystarczający zakres stężeń badanej substancji chemicznej, aby wyraźnie określić najwyższą wartość (np. 90–100 % wiązania) konkurencyjnej krzywej. Zmienność wśród kontrprób przy każdym stężeniu badanej substancji chemicznej, jak również wśród 3 niejednoczesnych serii badawczych powinna być uzasadniona i poparta naukowo. Kontrole z każdej serii badawczej badanej substancji chemicznej powinny zbliżać się do miar efektywności zgłoszonych dla tego testu CERI i powinny być spójne z danymi historycznymi dotyczącymi kontroli z każdego odpowiedniego laboratorium.
|
ANALIZA DANYCH
Test wiązania nasycenia
|
51.
|
Dokonuje się pomiaru zarówno wiązania całkowitego, jak i nieswoistego. W przypadku tych wartości wiązanie swoiste wzrastających stężeń [3H]-17β-estradiolu w warunkach równowagi jest obliczane przez odjęcie wiązania nieswoistego od całkowitego. Wykres stężenia wiązań specyficznych w porównaniu ze stężeniem [3H]-17β-estradiolu powinien osiągnąć plateau dla maksymalnego wiązania specyficznego wskazującego na nasycenie hrERα [3H]-17β-estradiolem. Ponadto analiza danych powinna dokumentować wiązanie estradiolu [3H]-17β-estradiolu z pojedynczym obszarem wiązania o wysokim powinowactwie. Wiązanie nieswoiste, całkowite i swoiste powinno być prezentowane na krzywej wiązania nasycenia. W dalszej analizie tych danych należy wykorzystać analizę regresji nieliniowej (np. BioSoft; McPherson 1985; Motulsky 1995) z końcowym prezentowaniem danych w formie wykresu Scatcharda.
|
|
52.
|
W analizie danych należy określić Bmax i Kd wyłącznie na podstawie danych dotyczących wiązania całkowitego, przy założeniu, że wiązanie nieswoiste jest liniowe, chyba że podano uzasadnienie dla zastosowania innej metody. Ponadto przy określaniu najlepszego dopasowania należy stosować regresję odpornościową, chyba że zostanie podane uzasadnienie. Należy podać metodę wybraną dla regresji odpornościowej. Korektę obniżenia stężenia ligandu (np. przy użyciu metody na podstawie Swillens 1995) należy zawsze stosować przy określaniu Bmax i Kd na podstawie danych dotyczących wiązania nasycenia.
|
Test wiązania konkurencyjnego
|
53.
|
Krzywa wiązania konkurencyjnego jest wykreślona jako krzywa wiązania specyficznego [3H]-17β-estradiolu w stosunku do stężenia (log10 jednostek) konkurenta. Stężenie badanej substancji chemicznej, która hamuje 50 % maksymalnego wiązania specyficznego [3H]-17β-estradiolu, jest wartością IC50.
|
|
54.
|
Szacunki wartości logarytmu (IC50) dla kontroli dodatnich (np. estrogen odniesienia i substancja słabo wiążąca) należy określić, wykorzystując właściwe oprogramowanie dopasowane do krzywej nieliniowej, aby pasowały do równania Hilla o czterech parametrach (np. BioSoft; McPherson 1985; Motulsky 1995). Najwyższą wartość, najniższą wartość, krzywą Hilla i logarytm IC50 należy zasadniczo pozostawić bez ograniczeń przy dopasowywaniu tych krzywych. Regresję odpornościową należy stosować przy określaniu najlepszego dopasowania, chyba że podane zostanie uzasadnienie. Nie należy stosować korekty obniżenia stężenia ligandu. Po przeprowadzeniu analizy wstępnej każda krzywa wiązania powinna zostać poddana przeglądowi w celu zapewnienia odpowiedniego dopasowania do modelu. Względne powinowactwo wiązania (RBA) w odniesieniu do substancji słabo wiążącej należy obliczać jako procent logarytmu (IC50) dla substancji słabo wiążącej względem logarytmu (IC50) dla 17β-estradiolu. Wyniki kontroli dodatnich i kontroli substancji innych niż substancja wiążąca powinny być oceniane przy użyciu pomiarów efektywności testu określonych w pkt 44–49 niniejszego dodatku 3.
|
|
55.
|
Dane dotyczące wszystkich badanych substancji chemicznych należy analizować na zasadzie stopniowego podejścia, aby zapewnić odpowiednią analizę danych i właściwą klasyfikację każdej krzywej wiązania konkurencyjnego. Zaleca się, aby każda seria badawcza badanej substancji chemicznej została wstępnie poddana znormalizowanej analizie danych, która jest identyczna z analizą stosowaną w przypadku kontroli estrogenu odniesienia i substancji słabo wiążącej (zob. pkt 54 niniejszego dodatku 3). Po zakończeniu należy przeprowadzić przegląd techniczny parametrów dopasowania krzywej, jak również wizualny przegląd stopnia, w jakim dane pasują do wygenerowanej krzywej wiązania konkurencyjnego dla każdej serii badawczej. W trakcie tego przeglądu technicznego obserwacje zależnego od stężenia spadku w procentach [3H]-17β-estradiolu związanego specyficznie, małej zmienności pomiędzy kontrpróbami technicznymi przy stężeniu każdej badanej substancji chemicznej oraz spójności parametrów dopasowania pomiędzy trzema seriami badawczymi stanowią dobrą wskazówkę, że test i analizy danych zostały przeprowadzone prawidłowo.
|
Interpretacja danych
|
56.
|
O ile spełnione są wszystkie kryteria dopuszczalności, badaną substancję chemiczną uważa się za substancję wiążącą w odniesieniu do hrERα, jeżeli krzywa wiązania może być dopasowana, a najniższy punkt na krzywej reakcji w zakresie danych wynosi mniej niż 50 % (rysunek 1).
|
|
57.
|
O ile spełnione są wszystkie kryteria dopuszczalności, badaną substancję chemiczną uważa się za substancję inną niż substancja wiążąca w odniesieniu do hrERα, jeśli:
|
—
|
krzywa wiązania może być dopasowana, a najniższy punkt na dopasowanej krzywej reakcji w zakresie danych wynosi powyżej 75 %, lub
|
|
—
|
krzywa wiązania nie może być dopasowana, a najniższy niewygładzony średni procent wiązania wśród grup stężeń w danych wynosi powyżej 75 %.
|
|
|
58.
|
Badane substancje chemiczne uważa się za niejednoznaczne, jeśli zostanie spełniony żaden z powyższych warunków (np. najniższy punkt na dopasowanej krzywej reakcji mieści się pomiędzy 76 a 51 %).
|
Tabela 7
Kryteria przypisywania klasyfikacji na podstawie krzywej wiązania konkurencyjnego w odniesieniu do badanej substancji chemicznej
|
Klasyfikacja
|
Kryteria
|
|
Substancja wiążącaa
|
Krzywa wiązania może być dopasowana.
Najniższy punkt na krzywej reakcji w zakresie danych wynosi mniej niż 50 %.
|
|
Substancja inna niż substancja wiążącab
|
Jeżeli krzywa wiązania może być dopasowana,
najniższy punkt na dopasowanej krzywej reakcji w zakresie danych wynosi powyżej 75 %.
Jeżeli krzywa wiązania nie może być dopasowana,
najniższy niewygładzony średni odsetek wiązania wśród grup stężeń w danych wynosi powyżej 75 %.
|
|
Wynik niejednoznacznyc
|
Każda sprawdzalna seria badawcza, która nie dotyczy substancji wiążącej, ani substancji innej niż substancja wiążąca
(np. najniższy punkt na dopasowanej krzywej reakcji mieści się w granicach 76–51 %).
|
Rysunek 1
Przykłady klasyfikacji badanej substancji chemicznej z wykorzystaniem krzywej wiązania konkurencyjnego

|
59.
|
Wielokrotne serie badawcze prowadzone w laboratorium w przypadku badanej substancji chemicznej są łączone poprzez przypisanie wartości liczbowych do każdej serii badawczej i uśrednienie wszystkich serii, jak pokazano w tabeli 8. Wyniki połączonych serii badawczych w każdym laboratorium są porównywane z oczekiwaną klasyfikacją każdej badanej substancji chemicznej.
|
Tabela 8
Metoda klasyfikacji badanej substancji chemicznej z wykorzystaniem wielu serii badawczych w laboratorium
|
Przypisanie wartości do każdej serii badawczej:
|
|
Klasyfikacja
|
Wartość liczbowa
|
|
Substancja wiążąca
|
2
|
|
Wynik niejednoznaczny
|
1
|
|
Substancja inna niż substancja wiążąca
|
0
|
|
Klasyfikacja średniej wartości liczbowej w poszczególnych seriach badawczych:
|
|
Klasyfikacja
|
Wartość liczbowa
|
|
Substancja wiążąca
|
Średnia ≥ 1,5
|
|
Wynik niejednoznaczny
|
0,5 ≤ Średnia < 1,5
|
|
Substancja inna niż substancja wiążąca
|
Średnia < 0,5
|
SPRAWOZDANIE Z BADANIA
|
60.
|
Zob. pkt 24 „ELEMENTÓW TESTU WIĄZANIA hrER” w niniejszej metodzie badawczej.
|
Dodatek 3.1
WYKAZ POJĘĆ
[3H]E2
: 17β-estradiol znakowany izotopem z trytem
DCC
: Węgiel drzewny pokryty dekstranem
E2
: Nieoznakowany 17β-estradiol (obojętny)
Bufor testowy
: 10 mM Tris-HCl, pH 7,4, zawierający 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % gliceryny, 0,2 mM leupeptinu, 1 mM ditiotreitolu i 10 mg/ml albuminy surowicy bydlęcej.
hrERα
: Ludzki rekombinowany receptor estrogenowy alfa (domena wiążąca ligand)
Kontrpróba
: Jeden z wielu dołków, które mają tę samą zawartość z tym samym stężeniem i są poddawane testowi jednocześnie w ramach jednej serii badawczej. W niniejszym protokole każde stężenie badanej substancji chemicznej jest badane w trzech powtórzeniach; oznacza to, że istnieją trzy kontrpróby, które są testowane jednocześnie przy każdym stężeniu badanej substancji chemicznej.
Seria badawcza
: Pełen zestaw poddawanych jednocześnie serii badawczej dołków mikropłytki, który dostarcza wszelkich informacji niezbędnych do scharakteryzowania wiązania badanej substancji chemicznej z hrERα (tj. całkowity [3H]-17β-estradiol dodany do dołka testowego, maksymalne wiązanie [3H]-17β-estradiolu z hrERα, wiązanie niespecyficzne, oraz wiązanie całkowite przy różnych stężeniach badanej substancji chemicznej). W serii badawczej można wykorzystać tylko jeden dołek (tj. kontrpróbę) na każde stężenie, ale ponieważ niniejszy protokół wymaga trzykrotnego przeprowadzania testu, w jednej analizie wykorzystuje się trzy dołki na każde stężenie. Niniejszy protokół wymaga ponadto przeprowadzenia trzech niezależnych (tj. nierównoczesnych) serii badawczych w odniesieniu do każdej substancji chemicznej.
Dodatek 3.2
ROZKŁAD DOŁKÓW W TEŚCIE WIĄZANIA KONKURENCYJNEGO
|
Płytka
|
Pozycja
|
Kontrpróba
|
Rodzaj dołka
|
Kod dołka
|
Kod stężenia
|
Stężenie początkowe konkurenta (M)
|
Roztwór podstawowy hrER (μl)
|
Objętość buforu (μl)
|
Objętość znacznika (gorącego E2) (μl)
|
Objętość z płytki do rozcieńczeń (μl)
|
Ostateczna objętość (μl)
|
Stężenie końcowe konkurenta (M)
|
|
S
|
A1
|
1
|
Ślepa próba
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Ślepa próba
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Ślepa próba
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
zimne E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
zimne E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
zimne E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
zimne E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
zimne E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
zimne E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
zimne E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
zimne E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
zimne E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
zimne E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
zimne E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
zimne E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
zimne E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
zimne E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
zimne E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
zimne E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
zimne E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
zimne E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
zimne E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
zimne E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
zimne E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
wiązanie całkowite
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
wiązanie całkowite
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
wiązanie całkowite
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
wiązanie całkowite
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
wiązanie całkowite
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
wiązanie całkowite
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
zimne E2 (wysokie)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
zimne E2 (wysokie)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
zimne E2 (wysokie)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
zimne E2 (wysokie)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
zimne E2 (wysokie)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
zimne E2 (wysokie)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Kontrola z buforem
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Kontrola z buforem
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Kontrola z buforem
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Kontrola z buforem
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Kontrola z buforem
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Kontrola z buforem
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Próba ślepa (dla gorących)
|
Gorąca
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Próba ślepa (dla gorących)
|
Gorąca
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Próba ślepa (dla gorących)
|
Gorąca
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Próba ślepa (dla gorących)
|
Gorąca
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Próba ślepa (dla gorących)
|
Gorąca
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Próba ślepa (dla gorących)
|
Gorąca
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Nieznany 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Nieznany 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Nieznany 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Nieznany 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Nieznany 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Nieznany 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Nieznany 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Nieznany 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Nieznany 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Nieznany 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Nieznany 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Nieznany 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Nieznany 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Nieznany 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Nieznany 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Nieznany 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Nieznany 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Nieznany 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Nieznany 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Nieznany 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Nieznany 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Nieznany 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Nieznany 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Nieznany 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Nieznany 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Nieznany 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Nieznany 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Nieznany 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Nieznany 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Nieznany 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Nieznany 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Nieznany 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Nieznany 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Nieznany 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Nieznany 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Nieznany 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Nieznany 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Nieznany 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Nieznany 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Nieznany 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Nieznany 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Nieznany 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Nieznany 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Nieznany 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Nieznany 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Nieznany 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Nieznany 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Nieznany 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Nieznany 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Nieznany 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Nieznany 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Nieznany 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Nieznany 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Nieznany 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Nieznany 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Nieznany 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Nieznany 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Nieznany 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Nieznany 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Nieznany 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Nieznany 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Nieznany 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Nieznany 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Nieznany 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Nieznany 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Nieznany 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Nieznany 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Nieznany 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Nieznany 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Nieznany 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Nieznany 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Nieznany 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Kontrola E2 (maks.)
|
S
|
E2max1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
Kontrola E2 (maks.)
|
S
|
E2max2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
Kontrola E2 (maks.)
|
S
|
E2max3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
Kontrola E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Kontrola E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Kontrola E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Kontrola NE (maks.)
|
S
|
Nemax1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
Kontrola NE (maks.)
|
S
|
Nemax2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
Kontrola NE (maks.)
|
S
|
Nemax3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
Kontrola NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Kontrola NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Kontrola NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
zimne E2 (wysokie)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
zimne E2 (wysokie)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
zimne E2 (wysokie)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
zimne E2 (wysokie)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
zimne E2 (wysokie)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
zimne E2 (wysokie)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
wiązanie całkowite
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
wiązanie całkowite
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
wiązanie całkowite
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
wiązanie całkowite
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
wiązanie całkowite
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
wiązanie całkowite
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Dodatek 4
UWAGI DOTYCZĄCE ANALIZY DANYCH UZYSKANYCH Z TESTÓW WIĄZANIA KONKURENCYJNEGO HRER
|
1.
|
W teście wiązania konkurencyjnego hrERα mierzy się wiązanie pojedynczego stężenia [3H]17β-estradiolu w obecności rosnących stężeń badanej substancji chemicznej. Krzywa wiązania konkurencyjnego jest wykreślona jako krzywa wiązania specyficznego [3H]-17β-estradiolu w stosunku do stężenia (log10 jednostek) konkurenta. Stężenie badanej substancji chemicznej, która hamuje 50 % maksymalnego wiązania specyficznego [3H]-17β-estradiolu jest wartością IC50.
|
Analiza danych w odniesieniu do estrogenu referencyjnego i substancji słabo wiążącej (1)
|
2.
|
Dane uzyskane z kontrolnych serii badawczych przekształca się (tj. procent specyficznego wiązania [3H]-17β-estradiolu oraz logarytmu stężenia badanej substancji chemicznej) na potrzeby dalszej analizy. Szacunki wartości logarytmu (IC50) dla kontroli dodatnich (np. estrogen odniesienia i substancja słabo wiążąca) należy określić, wykorzystując właściwe oprogramowanie dopasowane do krzywej nieliniowej, aby pasowały do równania Hilla o czterech parametrach (np. BioSoft; GraphPad Prism) (2). Najwyższą wartość, najniższą wartość, krzywą Hilla i log( IC50) można zasadniczo pozostawić bez ograniczeń przy dopasowywaniu tych krzywych. Regresję odpornościową należy stosować przy określaniu najlepszego dopasowania, chyba że podane zostanie uzasadnienie. Należy podać metodę wybraną dla regresji odpornościowej. Nie było potrzeby zastosowania korekty obniżenia stężenia ligandu w przypadku testów hrER Freybergera–Wilsona lub CERI, ale w stosownych przypadkach można to rozważyć. Po przeprowadzeniu analizy wstępnej każda krzywa wiązania powinna zostać poddana przeglądowi w celu zapewnienia odpowiedniego dopasowania do modelu. Względne powinowactwo wiązania (RBA) w odniesieniu do substancji słabo wiążącej można obliczać jako procent logarytmu (IC50) dla substancji słabo wiążącej względem logarytmu (IC50) dla 17β-estradiolu. Wyniki dotyczące kontroli dodatnich i kontroli substancji innych niż substancja wiążąca należy oceniać przy użyciu pomiarów efektywności testu i kryteriów dopuszczalności opisanych w niniejszej metodzie badawczej (pkt 20), dodatku 2 (test Freybergera–Wilsona, pkt 41–51) oraz w dodatku 3 (test CERI, pkt 41–51). Przykłady trzech prób serii badawczych dotyczących estrogenów odniesienia i substancji słabo wiążących przedstawiono na rysunku 1.
|
Rysunek 1
Przykłady krzywych wiązania konkurencyjnego dla estrogenu odniesienia kontrolnej substancji słabo wiążącej

Analiza danych dotycząca badanych substancji chemicznych
|
3.
|
Dane dotyczące wszystkich badanych substancji chemicznych należy analizować na zasadzie stopniowego podejścia, aby zapewnić odpowiednią analizę danych i właściwą klasyfikację każdej krzywej wiązania konkurencyjnego. Każdą serię badawczą w odniesieniu do badanej substancji chemicznej należy wstępnie poddać znormalizowanej analizie danych, która jest identyczna z analizą stosowaną w przypadku kontroli estrogenu odniesienia i substancji słabo wiążącej. Po zakończeniu należy przeprowadzić przegląd techniczny parametrów dopasowania krzywej, jak również wizualny przegląd stopnia, w jakim dane pasują do wygenerowanej krzywej wiązania konkurencyjnego dla każdej serii badawczej. W trakcie tego przeglądu technicznego obserwacje zależnego od stężenia spadku w procentach związanego specyficznie [3H]-17β-estradiolu, małej zmienności pomiędzy kontrpróbami technicznymi przy każdym stężeniu chemicznym oraz spójności parametrów dopasowania pomiędzy trzema seriami badawczymi stanowią dobrą wskazówkę, że test i analizy danych zostały wykonane prawidłowo. Dokonując przeglądu wyników z każdej serii badawczej w przypadku badanej substancji chemicznej, należy przyjąć profesjonalny osąd, a dane wykorzystane do zaklasyfikowania poszczególnych badanych substancji chemicznych jako wiążące lub niewiążące powinny być uzasadnione i poparte naukowo.
|
|
4.
|
W niektórych przypadkach, dane mogą wymagać większej uwagi w celu właściwego przeanalizowania i zinterpretowania danych dotyczących wiązań hrER. W poprzednich badaniach odnotowano przypadki, w których analiza i interpretacja danych dotyczących konkurencyjnego wiązania receptorów może być utrudniona przez wzrost odsetka wiązania specyficznego podczas badania substancji chemicznych w najwyższym stężeniu (rysunek 2). Ten problem jest dobrze znany i spotykano się z nim podczas stosowania protokołów dotyczących szeregu testów konkurencyjnych wiązań receptorowych (3). W takich sytuacjach przy niższych stężeniach odnotowuje się reakcję zależną od stężenia, ale wraz ze zbliżaniem się stężenia badanej substancji chemicznej do granicy rozpuszczalności, przemieszczenie [3H]17β-estradiolu przestaje się zmniejszać. W takich przypadkach z danych dotyczących wysokich stężeń wynika, że osiągnięto biologiczną granicę testu. Często na przykład wiąże się to zjawisko z nierozpuszczalnością i strąceniem danej substancji chemicznej przy wysokich stężeniach, lub może ono wynikać z przekroczenia zdolności węgla drzewnego pokrytego dekstranem do zbierania niezwiązanego ligandu znakowanego izotopem w trakcie procedury oddzielania przy najwyższych stężeniach substancji chemicznych. Pozostawienie takich punktów danych podczas dopasowywania danych dotyczących wiązań konkurencyjnych do krzywej sigmoidalnej może czasem prowadzić do nieprawidłowej klasyfikacji potencjału wiązania receptora estrogenowego w odniesieniu do badanej substancji chemicznej (rysunek 2). Aby uniknąć takiej sytuacji, protokół testów wiązania hrER Freybergera–Wilsona i CERI obejmuje możliwość wykluczenia z analiz punktów danych, w przypadku których średnia kontrprób w odniesieniu do odsetka określonego wiązania [3H]17β-estradiolu wynosi 10 % lub więcej powyżej średniej wartości zaobserwowanej przy niższym stężeniu (tj. zazwyczaj określa się to jako zasadę 10 %). Zasadę tę można zastosować tylko raz w odniesieniu do danej krzywej i muszą pozostać dane dotyczące co najmniej 6 stężeń, aby można było poprawnie sklasyfikować krzywą.
|
Rysunek 2
Przykłady, krzywe wiązania konkurencyjnego z wykorzystaniem lub bez wykorzystania zasady 10 %
|
5.
|
Należy dokładnie rozważyć zastosowanie zasady 10 % do korekty tych krzywych i stosować ją tylko w przypadkach, w których istnieją wiele wskazuje na substancje wiążącą hrER. Podczas przeprowadzania doświadczeń na użytek badania walidacyjnego testu wiązania hrER Freybergera–Wilsona odnotowano, że zastosowanie zasady 10 % miało czasem niezamierzone i nieprzewidziane konsekwencje. Substancje chemiczne, które nie wchodziły w interakcję z receptorem (tj. rzeczywiste substancje inna niż substancje wiążąca) często odznaczały się zmiennością około 100 % wiązania radioligandu większą niż 10 % w całym zakresie badanych stężeń. Jeżeli najniższą wartość odnotowano przy niskim stężeniu, istnieje możliwość, że dane dotyczące wszystkich wyższych stężeń zostaną usunięte z analizy w wyniku zastosowania zasady 10 %, mimo że stężenia te mogłyby być przydatne do ustalenia, że dana substancja chemiczna to substancja inna niż substancja wiążąca. Na rysunku 3 przedstawiono przykłady, w których nie należy stosować zasady 10 %.
|
Rysunek 3
Przykłady, dane dotyczące wiązania konkurencyjnego, w przypadku których nie należy stosować zasady 10 %


Źródła
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα) [W:] Publikacje na temat zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 226. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju.
|
|
(2)
|
Motulsky H. i Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. San Diego, Kalifornia: GraphPad Software Inc., s. 187–191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
SC Laws, S Yavanhxay, RL Cooper i JC Eldridge (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. „Toxicological. Sci.”nr 94(1), s. 46–56.
|
B.71 TESTY DZIAŁANIA UCZULAJĄCEGO NA SKÓRĘ IN VITRO DOTYCZĄCE KLUCZOWEGO ZDARZENIA AKTYWACJI KOMÓREK DENDRYTYCZNYCH W MECHANIZMIE WYWOŁYWANIA SKUTKÓW SZKODLIWYCH W ZAKRESIE DZIAŁANIA UCZULAJĄCEGO NA SKÓRĘ
WPROWADZENIE OGÓLNE
Metoda badawcza oparta na kluczowym zdarzeniu aktywacji komórek dendrytycznych
|
1.
|
Substancja działająca uczulająco na skórę oznacza substancję, która wywołuje reakcję alergiczną w następstwie kontaktu ze skórą, zgodnie z definicją określoną przez Globalny Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów ONZ (ONZ GHS) (1) oraz w rozporządzeniu Unii Europejskiej (WE) nr 1272/2008 w sprawie klasyfikacji, oznakowania i pakowania substancji i mieszanin (CLP) (99). Panuje ogólna zgoda co do kluczowych zdarzeń biologicznych stanowiących podstawę działania uczulającego na skórę. Istniejącą wiedzę dotyczącą mechanizmów chemicznych i biologicznych powiązanych z działaniem uczulającym na skórę podsumowano w formie mechanizmu wywoływania skutków szkodliwych w ramach programu OECD dotyczącego mechanizmu wywoływania skutków szkodliwych (2), począwszy od molekularnego zdarzenia inicjującego poprzez zdarzenia pośrednie aż do skutku szkodliwego, tj. alergicznego kontaktowego zapalenia skóry. Na przykład molekularnym zdarzeniem inicjującym (tj. pierwszym kluczowym zdarzeniem) jest wiązanie kowalencyjne substancji elektrofilowych z centrami nukleofilowymi w białkach skóry. Drugie kluczowe zdarzenie w tym mechanizmie wywoływania skutków szkodliwych następuje w keratynocytach i obejmuje reakcje zapalne oraz zmiany w ekspresji genów powiązane ze specyficznymi mechanizmami sygnalizacji komórkowej, takimi jak mechanizmy zależne od elementu odpowiedzi antyoksydacyjnej/elektrofilowej (ARE). Trzecim kluczowym zdarzeniem jest aktywacja komórek dendrytycznych, na ogół oceniana na podstawie ekspresji specyficznych markerów powierzchniowych komórki, chemokinów i cytokinów. Czwartym kluczowym zdarzeniem jest aktywacja i proliferacja limfocytów T, która jest oceniana pośrednio w ramach testu lokalnych węzłów chłonnych przeprowadzonego na myszach (LLNA) (3).
|
|
2.
|
Niniejsza metoda badawcza jest równoważna metodzie opisanej w dotyczącej badań wytycznej OECD nr 442E (2017). Opisano w niej testy in vitro, które dotyczą mechanizmów opisanych na podstawie kluczowego zdarzenia aktywacji komórek dendrytycznych w mechanizmie wywoływania skutków szkodliwych w zakresie działania uczulającego na skórę (2). Metoda badawcza uwzględnia badania, które należy stosować do wsparcia rozróżnienia substancji działających uczulająco na skórę i substancji niewykazujących takiego działania zgodnie z GHS ONZ i rozporządzeniem CLP.
|
|
W niniejszej metodzie badawczej opisano następujące badania:
|
—
|
badanie aktywacji ludzkich linii komórkowych (h-CLAT);
|
|
—
|
badanie aktywacji linii komórkowej U937 (U-SENS™)
|
|
—
|
test genu reporterowego Interleukiny 8 (test IL-8 Luc).
|
|
|
|
3.
|
Badania uwzględnione w niniejszej metodzie badawczej i odpowiedniej wytycznej OECD dotyczącej badań mogą różnić się w zależności od procedury wykorzystywanej do generowania danych i odczytów pomiarów, lecz mogą być stosowane bez rozróżnienia na potrzeby spełnienia wymogów poszczególnych państw w zakresie wyników badań nad kluczowym zdarzeniem aktywacji komórek dendrytycznych w mechanizmie wywoływania skutków szkodliwych w zakresie działania uczulającego na skórę, przy jednoczesnym wykorzystaniu wzajemnej akceptacji danych OECD.
|
Kontekst i zasady badań prowadzonych z wykorzystaniem metody badawczej opartej na kluczowym zdarzeniu
|
4.
|
Ocena działania uczulającego na skórę obejmuje na ogół wykorzystanie zwierząt laboratoryjnych. W przypadku klasycznych metod, w których wykorzystuje się świnki morskie, tj. testu maksymalizacji na świnkach morskich (Magnussona-Kligmana) (test GPMT) oraz testu Buehlera (metody badawczej B.6) (4), ocenia się zarówno fazę indukcyjną, jak i fazy wywołania działania uczulającego na skórę. Przeprowadzany na myszach test lokalnych węzłów chłonnych (LLNA) (metoda badawcza B.42) (3) oraz jego dwie nieradioaktywne modyfikacje, LLNA: DA (metoda badawcza B.50) (5) i LLNA: BrdU-ELISA (metoda badawcza B.51) (6), w ramach których ocenia się wyłącznie reakcję indukcyjną, także zyskały akceptację, ponieważ zapewniają one przewagę nad testami prowadzonymi na świnkach morskich pod względem dobrostanu zwierząt i obiektywnego pomiaru fazy indukcyjnej działania uczulającego na skórę.
|
|
5.
|
Niedawno przyjęto, oparte na analizie mechanistycznej, metody badawcze in chemico i in vitro dotyczące pierwszego kluczowego zdarzenia (metoda badawcza B.59; bezpośrednie oznaczanie reaktywności peptydów (7)) i drugiego kluczowego zdarzenia (metoda badawcza B.60; metoda badawcza z wykorzystaniem lucyferazy ARE-Nrf2 (8)) w mechanizmie wywoływania skutków szkodliwych w zakresie działania uczulającego na skórę jako metody ułatwiające ocenę zagrożenia działaniem uczulającym na skórę substancji chemicznych.
|
|
6.
|
Badania opisane w niniejszej metodzie badawczej pozwalają określić ilościowo zmianę ekspresji markera powierzchniowego (markerów powierzchniowych) komórki związanego z procesem aktywacji monocytów i komórek dendrytycznych po narażeniu na działanie czynników uczulających (np. CD54, CD86) albo zmiany ekspresji IL-8 – cytokiny związanej z aktywacją komórek dendrytycznych. Zgłoszono, że substancje działające uczulająco na skórę wywołują ekspresję markerów błony komórkowej, takich jak CD40, CD54, CD80, CD83 i CD86 dodatkowo oprócz indukcji prozapalnych cytokin, takich jak IL-1β i TNF-α, i szeregu chemokinów, takich jak IL-8 (CXCL8) i CCL3 (9)(10)(11)(12), związanych z aktywacją komórek dendrytycznych (2).
|
|
7.
|
Ponieważ aktywacja komórek dendrytycznych stanowi jednak tylko jedno kluczowe zdarzenie inicjujące mechanizm wywoływania skutków szkodliwych w zakresie działania uczulającego na skórę (2)(13), informacje wygenerowane przy użyciu samych tylko markerów badań aktywacji komórek dendrytycznych mogą nie być wystarczające do tego, aby stwierdzić występowanie lub brak występowania potencjalnego działania uczulającego na skórę substancji chemicznych. Dlatego też proponuje się, aby dane wygenerowane przy zastosowaniu testów opisanych w niniejszej metodzie badawczej stosować do wsparcia rozróżnienia substancji działających uczulająco na skórę (tj. należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP) i substancji niewykazujących takiego działania w przypadku ich wykorzystania w ramach zintegrowanych podejść do badań i oceny (IATA), wraz z innymi istotnymi informacjami uzupełniającymi, np. pochodzącymi z testów in vitro, odnoszącymi się do innych kluczowych zdarzeń inicjujących mechanizm wywoływania skutków szkodliwych w zakresie działania uczulającego na skórę, jak również pochodzącymi z metod niebadawczych, w tym danych z podejść przekrojowych z zastosowaniem analogicznych substancji chemicznych (13). Przykłady wykorzystania danych wygenerowanych w tych badaniach w ramach zdefiniowanego podejścia, tj. podejścia ujednoliconego zarówno pod kątem zbioru wykorzystywanych źródeł informacji, jak i procedury stosowanej w odniesieniu do danych w celu uzyskania prognoz, zostały opublikowane (13) i mogą zostać wykorzystane jako użyteczne elementy w ramach IATA.
|
|
8.
|
Badań opisanych w niniejszej metodzie badawczej nie można stosować samodzielnie w celu dalszego klasyfikowania substancji działających uczulająco na skórę do podkategorii 1A i 1B, zgodnie z GHS ONZ / rozporządzeniem CLP przez organy realizujące podział na te dwie kategorie, ani w celu przewidzenia ich siły działania na potrzeby podjęcia decyzji dotyczących oceny bezpieczeństwa. Jednakże w zależności od ram regulacyjnych do zaklasyfikowania substancji chemicznej do kategorii 1 wg GHS ONZ / rozporządzenia CLP można wykorzystać same wyniki dodatnie wygenerowane przy zastosowaniu tych metod.
|
|
9.
|
Termin „badana substancja chemiczna” jest używany w niniejszej metodzie badawczej w odniesieniu do substancji będącej przedmiotem badania (100) i nie jest powiązany z możliwością zastosowania badań do badania substancji jednoskładnikowych, substancji wieloskładnikowych lub mieszanin. Aktualnie dostępne są ograniczone informacje na temat możliwości zastosowania badań do substancji wieloskładnikowych / mieszanin (14)(15). Z technicznego punktu widzenia badania mogą jednak mieć zastosowanie do badania zarówno substancji wieloskładnikowych, jak i mieszanin. Przed zastosowaniem niniejszej metody badawczej z użyciem mieszaniny w celu zgromadzenia danych na potrzeby założonego celu regulacyjnego należy jednak zastanowić się nad tym, czy zastosowanie niniejszej metody może doprowadzić do uzyskania wyników odpowiednich z punktu widzenia tego celu, a jeżeli tak – dlaczego (101). Przeprowadzenie takiej analizy nie jest konieczne, jeżeli ustanowiono wymóg regulacyjny dotyczący badania danej mieszaniny. Ponadto podczas badania substancji wieloskładnikowych lub mieszanin należy rozważyć możliwe zakłócenie obserwowanych reakcji przez składniki cytotoksyczne.
|
BIBLIOGRAFIA
|
(1)
|
Organizacja Narodów Zjednoczonych (ONZ) (2015). Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS). wydanie szóste zmienione, Nowy Jork i Genewa: publikacje Organizacji Narodów Zjednoczonych. ISBN: 978-92-1-117006-1. Dostępne na stronie internetowej: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Seria dotycząca badań i oceny nr 168. Dostępne na stronie internetowej: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(3)
|
Rozdział B.42 niniejszego załącznika: Badanie lokalnych węzłów chłonnych.
|
|
(4)
|
Rozdział B.6 niniejszego załącznika: Sensybilizacja skóry.
|
|
(5)
|
Rozdział B.50 niniejszego załącznika: Działanie uczulające na skórę: badanie lokalnych węzłów chłonnych DA.
|
|
(6)
|
Rozdział B.51 niniejszego załącznika: Działanie uczulające na skórę: badanie lokalnych węzłów chłonnych BrdU-ELISA.
|
|
(7)
|
Rozdział B.59 niniejszego załącznika: Działanie uczulające na skórę in chemico: Bezpośrednie oznaczanie reaktywności peptydów (DPRA).
|
|
(8)
|
Rozdział B.60 niniejszego załącznika: Badanie działania uczulającego na skórę in vitro: metoda badawcza z wykorzystaniem lucyferazy ARE-Nrf2.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL i Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H i Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y i Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Sria dotycząca badań i oceny nr 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, załącznik 1 i załącznik 2. ENV/JM/HA(2016)29. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Dodatek 1
BADANIE DZIAŁANIA UCZULAJĄCEGO NA SKÓRĘ IN VITRO: BADANIE AKTYWACJI LUDZKICH LINII KOMÓRKOWYCH (H-CLAT)
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
1.
|
Badanie h-CLAT pozwala określić ilościowo zmianę ekspresji markerów powierzchniowych komórek związanych z procesem aktywacji monocytów i komórek dendrytycznych (tj. CD86 i CD54) w ludzkiej białaczkowej linii monocytarnej THP-1 po narażeniu na działanie czynników uczulających (1)(2). Pomiary poziomów ekspresji markerów powierzchniowych komórek CD86 i CD54 wykorzystuje się później do wsparcia rozróżnienia substancji działających uczulająco na skórę i substancji niewykazujących takiego działania.
|
|
2.
|
Badanie h-CLAT oceniono w ramach badania walidacyjnego koordynowanego przez laboratorium referencyjne Unii Europejskiej ds. metod alternatywnych wobec testów na zwierzętach (EURL ECVAM) i późniejszej niezależnej wzajemnej oceny przeprowadzonej przez Naukowy Komitet Doradczy EURL ECVAM (ESAC). Uwzględniając wszystkie dostępne dowody i wkład ze strony organów regulacyjnych i zainteresowanych stron, EURL ECVAM (3) zaleciło stosowanie badania h-CLAT w ramach IATA do wsparcia rozróżnienia substancji działających uczulająco i substancji niewykazujących takiego działania na potrzeby klasyfikacji i oznakowania pod względem zagrożeń. Przykłady wykorzystania danych z badania h-CLAT w połączeniu z innymi informacjami są opisane w literaturze (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
Udowodniono, że badanie h-CLAT można przeprowadzać również w laboratoriach posiadających doświadczenie w zakresie technik związanych z kulturą komórkową i analizą przy użyciu cytometrii przepływowej. Poziom odtwarzalności prognoz, którego można oczekiwać od tego badania, ma wartość rzędu 80 % wewnątrz laboratoriów i pomiędzy nimi (3)(12). Wyniki uzyskane w badaniu walidacyjnym (13) i innych opublikowanych badaniach (14) ogólnie wskazują na to, że – w porównaniu z wynikami LLNA – dokładność rozróżniania substancji działających uczulająco na skórę (tj. należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP) i substancji niewykazujących takiego działania wynosi 85 % (N = 142) przy czułości na poziomie 93 % (94/101) oraz swoistości na poziomie 66 % (27/41) (na podstawie ponownej analizy wykonanej przez EURL ECVAM (12) przy uwzględnieniu wszystkich istniejących danych, ale nie uwzględniając ujemnych wyników substancji chemicznych, których log Kow wynosi powyżej 3,5 zgodnie z opisem z pkt 4). Prawdopodobieństwo wystąpienia prognoz fałszywie ujemnych przy zastosowaniu metody h-CLAT jest większe w przypadku substancji chemicznych wykazujących niską lub średnią siłę działania uczulającego na skórę (tj. należących do podkategorii 1B wg GHS ONZ / rozporządzenia CLP) niż w przypadku substancji wykazujących wysoką siłę działania uczulającego na skórę (tj. należących do podkategorii 1A wg GHS ONZ / rozporządzenia CLP) (4)(13)(15). Wszystkie te informacje łącznie wskazują na użyteczność metody h-CLAT w identyfikowaniu zagrożeń związanych z działaniem uczulającym na skórę. Przedstawione tutaj wartości dokładności h-CLAT jako samodzielnego badania są jednak wyłącznie orientacyjne, ponieważ badanie to należy rozpatrywać w połączeniu z innymi źródłami informacji w kontekście IATA i zgodnie z przepisami pkt 7 i 8 wprowadzenia ogólnego. Co więcej, przy ocenie metod niewymagających wykorzystania zwierząt pod kątem działania uczulającego na skórę należy pamiętać, że LLNA, jak również inne badania przeprowadzane na zwierzętach, mogą nie w pełni odzwierciedlać sytuację w odniesieniu do ludzi.
|
|
4.
|
Na podstawie obecnie dostępnych danych wykazano, że metoda h-CLAT ma zastosowanie do badanych substancji chemicznych obejmujących szereg organicznych grup funkcyjnych, mechanizmów reagowania, sił działania uczulającego na skórę (jak określono w badaniach in vivo) oraz właściwości fizykochemicznych (3)(14)(15). Metoda h-CLAT ma zastosowanie do badanych substancji chemicznych, które są rozpuszczalne lub tworzą stabilną dyspersję (tj. koloid lub zawiesinę, w których badana substancja chemiczna nie osadza się ani nie oddziela od rozpuszczalnika/nośnika w różne fazy) w odpowiednim rozpuszczalniku/nośniku (zob. pkt 14). Badane substancje chemiczne o log Kow powyżej 3,5 mają tendencję do generowania wyników fałszywie ujemnych (14). Dlatego nie powinno się uwzględniać wyników ujemnych badanych substancji chemicznych, których log Kow jest wyższy od 3,5. Wyniki dodatnie badanych substancji chemicznych, których log Kow jest wyższy od 3,5, można jednak dalej wykorzystywać do wsparcia identyfikacji badanej substancji chemicznej jako substancji działającej uczulająco na skórę. Ponadto z powodu ograniczonej wydolności metabolicznej zastosowanych linii komórkowych (16) oraz warunków doświadczalnych prohapteny (tj. substancje wymagające aktywacji enzymatycznej np. przez enzymy P450) oraz prehapteny (tj. substancje aktywowane przez utlenianie), w szczególności te o niskim tempie utleniania, również mogą dawać wyniki ujemne w h-CLAT (15). Fluorescencyjne badane substancje chemiczne można ocenić za pomocą badania h-CLAT (17), jednak badane substancje chemiczne o wysokiej fluorescencji emitujące fale tej samej długości co izotiocyjanian fluoresceiny (FITC) lub jodek propidyny będą zakłócać detekcję przy użyciu cytometrii przepływowej, co uniemożliwia ich prawidłową ocenę z wykorzystaniem przeciwciał skoniugowanych z FITC lub jodku propidyny. W takim przypadku można użyć odpowiednio innych przeciwciał znakowanych fluorochromem lub innych markerów cytotoksyczności, o ile można wykazać, że pozwalają one uzyskać wyniki podobne do wyników otrzymywanych w przypadku przeciwciał znakowanych FITC (zob. pkt 24) lub jodku propidyny (zob. pkt 18), np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 1–2. W świetle powyższego wyniki ujemne należy interpretować w kontekście zdefiniowanych ograniczeń i w połączeniu z innymi źródłami informacji w ramach IATA. W przypadkach, w których można przedstawić dowody na brak możliwości zastosowania metody h-CLAT w odniesieniu do innych konkretnych kategorii badanych substancji chemicznych, nie należy jej stosować w odniesieniu do tych konkretnych kategorii.
|
|
5.
|
Jak opisano powyżej, metoda h-CLAT wspiera rozróżnienie substancji działających uczulająco na skórę i substancji niewykazujących takiego działania. Może ona jednak również potencjalnie wpłynąć na ocenę siły działania uczulającego (4)(5)(9), gdy stosuje się ją w ramach podejść zintegrowanych takich jak IATA. Wymagane jest jednak przeprowadzenie dalszych prac, najlepiej na podstawie danych dotyczących ludzi, aby ustalić, w jaki sposób wyniki badania h-CLAT mogą potencjalnie wpłynąć na ocenę siły działania.
|
|
6.
|
Definicje znajdują się w dodatku 1.1.
|
ZASADA BADANIA
|
7.
|
Metoda h-CLAT jest testem in vitro, który pozwala określić ilościowo zmiany ekspresji markera powierzchniowego komórki (tj. CD86 i CD54) w ludzkiej linii komórkowej białaczki monocytarnej, tj. komórkach THP-1, po 24 godzinach narażenia na działanie badanej substancji chemicznej. Te cząsteczki powierzchniowe są typowymi markerami aktywacji monocytarnej THP-1 i mogą imitować aktywację komórek dendrytycznych, która odgrywa kluczową rolę w uczulaniu limfocytów T. Zmiany ekspresji markera powierzchniowego mierzy się za pomocą cytometrii przepływowej po wybarwieniu komórek przeciwciałami znakowanymi fluorochromem. Jednocześnie przeprowadza się również pomiar cytotoksyczności, aby ocenić, czy podwyższona ekspresja markera powierzchniowego zachodzi przy stężeniach niższych niż stężenia cytotoksyczne. Względną intensywność fluorescencji markerów powierzchniowych w porównaniu z kontrolą z rozpuszczalnikiem/nośnikiem wylicza się i wykorzystuje w modelu prognozowania (zob. pkt 26), aby wesprzeć rozróżnienie substancji działających uczulająco i substancji niewykazujących takiego działania.
|
WYKAZANIE BIEGŁOŚCI
|
8.
|
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną z zastosowaniem dziesięciu substancji służących do wykazania biegłości wymienionych w dodatku 1.2. Ponadto użytkownicy badania powinni prowadzić bazę danych historycznych wygenerowanych podczas kontroli reaktywności (zob. pkt 11) oraz kontroli dodatnich i kontroli z rozpuszczalnikiem/nośnikiem (zob. pkt 20–22), a także wykorzystywać te dane do potwierdzenia zachowania odtwarzalności badania w swoim laboratorium w miarę upływu czasu.
|
PROCEDURA
|
9.
|
Podstawę niniejszego badania stanowi protokół nr 158 bazy danych na temat metod alternatywnych wobec testów na zwierzętach (DB-ALM) dotyczący h-CLAT (18), który reprezentuje protokół wykorzystany w badaniu walidacyjnym koordynowanym przez EURL ECVAM. Zaleca się, aby ten protokół był stosowany przy wdrażaniu i stosowaniu metody h-CLAT w laboratorium. Poniżej opisano główne elementy i procedury metody h-CLAT, które składają się z dwóch kroków: testu ustalającego dawkę i pomiaru ekspresji CD86/CD54.
|
Przygotowanie komórek
|
10.
|
Do badania metodą h-CLAT należy wykorzystać ludzką białaczkową linię monocytarną THP-1. Zaleca się pozyskiwanie komórek (TIB-202™) z banku komórek posiadającego niezbędne uprawnienia, takiego jak np. zbiór kultur typu amerykańskiego.
|
|
11.
|
Komórki THP-1 hoduje się w temperaturze 37oC, w obecności 5 % CO2 i w wilgotnym środowisku, w podłożu RPMI-1640 z dodatkiem 10 % bydlęcej surowicy płodowej, 0,05 mM 2-merkaptoetanolu, 100 jednostek/ml penicyliny i 100 μg/ml streptomycyny. Można zrezygnować z użycia penicyliny i streptomycyny w podłożu. W takim przypadku użytkownicy powinni jednak zweryfikować, czy brak antybiotyku w podłożu nie będzie miał wpływu na wyniki, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 1.2. W każdym przypadku, aby zminimalizować ryzyko zanieczyszczenia, należy przestrzegać dobrych praktyk w zakresie kultury komórkowej, niezależnie od tego, czy w podłożu do hodowli kultury komórkowej znajdują się antybiotyki, czy też nie. Komórki THP-1 wysiewa się regularnie co 2–3 dni w gęstości od 0,1 do 0,2 × 106 komórek/ml. Powinny one być utrzymywane w gęstości od 0,1 do 1,0 × 106 komórek/ml. Przed wykorzystaniem komórek do badań należy je zakwalifikować, przeprowadzając kontrolę reaktywności. Kontrolę reaktywności komórek należy przeprowadzić, wykorzystując do tego celu kontrole dodatnie, tj. 2,4-dinitrochlorobenzen (DNCB) (nr CAS 97-00-7, czystość ≥ 99 %) i siarczan niklu (NiSO4) (nr CAS 10101-97-0, czystość ≥ 99 %), oraz kontrolę ujemną, tj. kwas mlekowy (nr CAS 50-21-5, czystość ≥ 85 %), dwa tygodnie po rozmrożeniu. Zarówno 2,4-dinitrochlorobenzen, jak i NiSO4 powinny wywołać reakcję dodatnią markera powierzchniowego komórki CD86, jak również CD54, a kwas mlekowy powinien wywołać reakcję ujemną markera powierzchniowego komórki CD86, jak również CD54. W teście należy wykorzystać wyłącznie te komórki, które pomyślnie przeszły kontrolę reaktywności. Komórki można rozmnażać do dwóch miesięcy po rozmrożeniu. Liczba pasaży nie powinna przekraczać 30. Kontrolę reaktywności należy przeprowadzić zgodnie z procedurami opisanymi w pkt 20–24.
|
|
12.
|
Komórki THP-1 wysiewa się do badań w gęstości 0,1 × 106 komórek/ml albo 0,2 × 106 komórek/ml i dokonuje ich wstępnej hodowli w kolbach z kulturami przez odpowiednio 72 godziny lub 48 godzin. Ważne jest, aby zagęszczenie komórek w kolbie z kulturami tuż po okresie hodowli wstępnej było jak najbardziej spójne w każdym z doświadczeń (stosując jeden z dwóch wyżej wymienionych warunków wstępnej hodowli kultur), ponieważ zagęszczenie komórek w kolbie z kulturami tuż po okresie hodowli wstępnej mogłoby wpłynąć na ekspresję CD86/CD54 wywołaną przez alergeny (19). W dniu badania komórki pobrane z kolby z kulturami zawiesza się ponownie w świeżym podłożu w gęstości 2 × 106 komórek/ml. Następnie komórki umieszcza się w dołkach 24-dołkowej płytki płaskodennej (500 μl, 1 × 106 komórek/dołek) lub w dołkach 96-dołkowej płytki płaskodennej (80 μl, 1,6 × 105 komórek/dołek).
|
Test ustalający dawkę
|
13.
|
Test ustalający dawkę przeprowadza się, aby określić CV75, czyli stężenie badanej substancji chemicznej, dzięki któremu żywotność komórek (CV) wynosi 75 % w porównaniu z kontrolą z rozpuszczalnikiem/nośnikiem. Wartość CV75 wykorzystuje się do określenia stężenia badanych substancji chemicznych dla pomiaru ekspresji CD86/CD54 (zob. pkt 20–24).
|
Przygotowanie badanych substancji chemicznych i substancji kontrolnych
|
14.
|
Badane substancje chemiczne oraz substancje kontrolne przygotowuje się w dniu badania. W metodzie h-CLAT badane substancje chemiczne są rozpuszczane lub tworzą stabilne dyspersje (zob. również pkt 4) w soli fizjologicznej lub podłożu (pierwsze warianty rozpuszczalnika/nośnika) lub w sulfotlenku dimetylu (DMSO, czystość ≥ 99 %) (drugi wariant rozpuszczalnika/nośnika), jeżeli badana substancja chemiczna nie jest rozpuszczalna lub nie tworzy stabilnej dyspersji w dwóch rozpuszczalnikach/nośnikach wymienionych jako pierwsze, do stężeń końcowych wynoszących 100 mg/ml (soli fizjologicznej lub podłoża) lub 500 mg/ml (sulfotlenku dimetylu). Można użyć innych rozpuszczalników/nośników niż te opisane powyżej pod warunkiem przedstawienia wystarczającego uzasadnienia naukowego. Należy uwzględnić stabilność badanej substancji chemicznej w końcowym rozpuszczalniku/nośniku.
|
|
15.
|
Na początku przygotowuje się roztwory podstawowe badanych substancji chemicznych o stężeniach wynoszących 100 mg/ml (w soli fizjologicznej lub podłożu) lub 500 mg/ml (w sulfotlenku dimetylu), a następnie rozcieńcza zgodnie z następującymi krokami:
|
—
|
jeżeli rozpuszczalnikiem/nośnikiem jest sól fizjologiczna lub podłoże: przygotowuje się osiem roztworów podstawowych (osiem stężeń) przez dwukrotne rozcieńczenie w postępie geometrycznym przy użyciu odpowiedniego rozpuszczalnika/nośnika. Następnie otrzymane roztwory podstawowe rozcieńcza się 50-krotnie w podłożu (roztwory robocze). W przypadku gdy najwyższe stężenie końcowe na płytce wynoszące 1 000 μg/ml nie jest toksyczne, najwyższe stężenie należy wyznaczyć ponownie poprzez przeprowadzenie nowego badania cytotoksyczności. Końcowe stężenie na płytce nie powinno przekraczać 5 000 μg/ml dla badanych substancji chemicznych, które zostały rozpuszczone w soli fizjologicznej lub podłożu lub utworzyły w nich stabilną dyspersję;
|
|
—
|
jeżeli rozpuszczalnikiem/nośnikiem jest sulfotlenek dimetylu: przygotowuje się osiem roztworów podstawowych (osiem stężeń) przez dwukrotne rozcieńczenie w postępie geometrycznym przy użyciu odpowiedniego rozpuszczalnika/nośnika. Następnie otrzymane roztwory podstawowe rozcieńcza się 250-krotnie w podłożu (roztwory robocze). Końcowe stężenie na płytce nie powinno przekraczać 1 000 μg/ml, nawet jeżeli stężenie to nie jest toksyczne.
|
Roztwory robocze ostatecznie wykorzystuje się do narażenia na działanie substancji chemicznej poprzez dodanie do zawiesiny komórkowej THP-1 na płytce (zob. również pkt 17) roztworu roboczego w objętości równej objętości tej zawiesiny, aby osiągnąć dodatkowe dwukrotne rozcieńczenie (zazwyczaj końcowy zakres stężeń na płytce wynosi 7,81–1 000 μg/ml).
|
|
16.
|
W ramach kontroli z rozpuszczalnikiem/nośnikiem w metodzie h-CLAT stosuje się podłoże (w przypadku badanych substancji chemicznych rozpuszczonych w podłożu albo soli fizjologicznej lub tworzących w nich stabilną dyspersję (zob. pkt 4)) lub w sulfotlenek dimetylu (w przypadku badanych substancji chemicznych rozpuszczonych w sulfotlenku dimetylu lub tworzących w nim stabilną dyspersję) badane w pojedynczym stężeniu końcowym na płytce wynoszącym 0,2 %. Podlega takiemu samemu rozcieńczeniu jakie opisano dla roztworów roboczych w pkt 15.
|
Podawanie badanych substancji chemicznych i substancji kontrolnych
|
17.
|
Podłoże lub roztwory robocze opisane w pkt 15 i 16 miesza się w stosunku 1:1 (obj.) z zawiesinami komórkowymi przygotowanymi w 24-dołkowej lub 96-dołkowej płytce płaskodennej (zob. pkt 12). Płytki poddane powyższym procesom następnie inkubuje się przez 24 ± 0,5 godziny w temperaturze 37oC w obecności 5 % CO2. Należy dołożyć starań, aby nie dopuścić do odparowania lotnych badanych substancji chemicznych i aby zapobiec zanieczyszczeniu krzyżowemu między dołkami przez badane substancje chemiczne, np. szczelnie zaklejając płytkę przed rozpoczęciem inkubacji z wykorzystaniem badanych substancji chemicznych (20).
|
Barwienie jodkiem propidyny
|
18.
|
Po upływie 24 godzin ±0,5 godziny narażenia komórki przenosi się do probówki i zbiera poprzez odwirowanie. Supernatanty odrzuca się, a pozostałe komórki zawiesza się ponownie w 200 μl (w przypadku płytki 96-dołkowej) lub 600 μl (w przypadku płytki 24-dołkowej) roztworu chlorku sodu buforowanego fosforanami zawierającego 0,1 % albuminy surowicy bydlęcej (roztwór buforowy do wybarwiania). 200 μl zawiesiny komórkowej przenosi się do dołków 96-dołkowej płytki okrągłodennej (w przypadku płytki 96-dołkowej) lub probówki Eppendorfa (w przypadku płytki 24-dołkowej) i dwukrotnie przemywa się w 200 μl (w przypadku płytki 96-dołkowej) lub 600 μl (w przypadku płytki 24-dołkowej) roztworu buforowego do wybarwiania. Później komórki zawiesza się ponownie w roztworze buforowym do wybarwiania (np. w 400 μl) i dodaje roztwór jodku propidyny (np. 20 μl) (przykładowe stężenie końcowe jodku propidyny wynosi 0,625 μg/ml). Można wykorzystać inne markery cytotoksyczności, takie jak 7-aminoaktynomycyna D (7-AAD), błękit trypanu lub innego rodzaju markery, jeżeli można wykazać, że alternatywne barwniki pozwalają uzyskać wyniki podobne do wyników otrzymywanych w przypadku wykorzystania jodku propidyny, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 1.2.
|
Pomiar cytotoksyczności z wykorzystaniem cytometrii przepływowej i oszacowanie wartości CV75
|
19.
|
Wchłanianie jodku propidyny analizuje się za pomocą cytometrii przepływowej z kanałem akwizycji FL-3. Łącznie pozyskuje się 10 000 żywych komórek (nieoznaczonych jodkiem propidyny). Żywotność komórek można obliczyć za pomocą poniższego równania, korzystając z programu do analizy cytometrycznej. Przy niskiej żywotności komórek należy pozyskać do 30 000 komórek, wśród których znajdują się martwe komórki. Ewentualnie dane można pozyskać w ciągu jednej minuty od rozpoczęcia analizy.
Wartość CV75 (zob. pkt 13), tj. stężenie wykazujące przeżywalność komórek THP-1 na poziomie 75 % (cytotoksyczność na poziomie 25 %), oblicza się metodą interpolacji logarytmiczno-liniowej za pomocą następującego równania:

gdzie:
a oznacza minimalną wartość żywotności komórek powyżej 75 %;
c oznacza maksymalną wartość żywotności komórek poniżej 75 %;
b i d oznaczają stężenia, przy których wartość żywotności komórek wynosi odpowiednio a i c.

Do określania CV75 można stosować inne podejścia pod warunkiem wykazania braku wpływu na wyniki (np. poprzez badanie substancji służących do wykazania biegłości).
|
Pomiar ekspresji CD86/CD54
Przygotowanie badanych substancji chemicznych i substancji kontrolnych
|
20.
|
Odpowiedni rozpuszczalnik/nośnik (sól fizjologiczna, podłoże lub sulfotlenek dimetylu; zob. pkt 14) jest używany do rozpuszczania badanych substancji chemicznych lub tworzenia ich stabilnych zawiesin. Badane substancje chemiczne najpierw rozcieńcza się do stężenia odpowiadającego 100-krotności (w przypadku soli fizjologicznej lub podłoża) lub 500-krotności (w przypadku sulfotlenku dimetylu) 1,2 × CV75 określonego w teście ustalającym dawkę (zob. pkt 19). Jeżeli nie można określić wartości CV75 (tj. jeżeli w teście ustalającym dawkę nie można zaobserwować wystarczającej cytotoksyczności), jako stężenia początkowego należy użyć najwyższego rozpuszczalnego stężenia lub stężenia stabilnej zawiesiny badanej substancji chemicznej przygotowanej z użyciem każdego rozpuszczalnika/nośnika. Należy zwrócić uwagę, że końcowe stężenie na płytce nie powinno przekraczać 5 000 μg/ml (w przypadku soli fizjologicznej lub podłoża) lub 1 000 μg/ml (w przypadku sulfotlenku dimetylu). Następnie przy pomocy odpowiedniego rozpuszczalnika/nośnika sporządza się 1,2-krotne rozcieńczenia w postępie geometrycznym, aby uzyskać roztwory podstawowe (osiem stężeń wynoszących od 100 × 1,2 × CV75 do 100 × 0,335 × CV75 (w przypadku soli fizjologicznej lub podłoża) lub od 500 × 1,2 × CV75 do 500 × 0,335 × CV75 (w przypadku sulfotlenku dimetylu)), które zostaną poddane badaniu metodą h-CLAT (przykład schematu dawkowania można znaleźć w protokole DB-ALM nr 158). Następnie roztwory podstawowe rozcieńcza się 50-krotnie (w przypadku soli fizjologicznej lub podłoża) lub 250-krotnie (w przypadku sulfotlenku dimetylu) w podłożu (roztwory robocze). Te roztwory robocze ostatecznie wykorzystuje się do narażenia na działanie substancji chemicznej, stosując dodatkowy końcowy dwukrotny współczynnik rozcieńczenia na płytce. W przypadku gdy wyniki nie spełniają opisanych w pkt 29 i 30 kryteriów dopuszczalności dotyczących żywotności komórek, można powtórzyć test ustalający dawkę, aby wyznaczyć dokładniejszą wartość CV75. Należy zauważyć, że do pomiaru ekspresji CD86/CD54 użyte mogą być jedynie płytki 24-dołkowe.
|
|
21.
|
Kontrolę z rozpuszczalnikiem/nośnikiem przygotowuje się w sposób opisany w pkt 16. 2,4-dinitrochlorobenzen jest stosowany jako kontrola dodatnia w metodzie h-CLAT (zob. pkt 11), w której roztwory podstawowe przygotowuje się w sulfotleneku dimetylu i rozcieńcza w taki sposób, jak opisano w pkt 20 dla roztworów podstawowych. 2,4-dinitrochlorobenzen należy stosować jako kontrolę dodatnią do pomiaru ekspresji CD86/CD54 w końcowym, pojedynczym stężeniu na płytce (zwykle 4,0 μg/ml). Aby uzyskać na płytce stężenie 2,4-dinitrochlorobenzenu wynoszące 4,0 μg/ml, przygotowuje się roztwór podstawowy 2,4-dinitrochlorobenzenu w sulfotleneku dimetylu o stężeniu 2 mg/ml, a następnie rozcieńcza się go 250-krotnie w podłożu do roztworu roboczego o stężeniu 8 μg/ml. Ewentualnie jako stężenie kontroli dodatniej można użyć CV75 2,4-dinitrochlorobenzenu, które jest określane w każdej placówce badawczej. Można stosować inne odpowiednie kontrole dodatnie, jeżeli dostępne są dane historyczne pozwalające na uzyskanie porównywalnych kryteriów dopuszczalności analizy. W przypadku kontroli dodatnich końcowe pojedyncze stężenie na płytce nie powinno przekraczać 5 000 μg/ml (w przypadku soli fizjologicznej lub podłoża) lub 1 000 μg/ml (w przypadku sulfotlenku dimetylu). Kryteria dopuszczalności analizy są takie same jak kryteria opisane w odniesieniu do badanej substancji chemicznej (zob. pkt 29) z wyjątkiem ostatniego kryterium dopuszczalności, ponieważ kontrolę dodatnią bada się przy pojedynczym stężeniu.
|
Podawanie badanych substancji chemicznych i substancji kontrolnych
|
22.
|
W odniesieniu do każdej badanej substancji chemicznej i substancji kontrolnej należy przeprowadzić jedno doświadczenie, aby uzyskać prognozę. Każde doświadczenie składa się co najmniej z dwóch niezależnych analiz pomiaru ekspresji CD86/CD54 (zob. pkt 26–28). Każdą z niezależnych analiz przeprowadza się innego dnia lub tego samego dnia, pod warunkiem że na potrzeby każdej analizy: a) przygotowuje się niezależne, świeże roztwory podstawowe i robocze badanej substancji chemicznej oraz roztwory przeciwciał, a także b) używa się niezależnie zebranych komórek (tj. komórki zbiera się z innych kolb z kulturami); komórki mogą jednak pochodzić z tego samego pasażu. Badana substancja chemiczna i substancje kontrolne przygotowane jako roztwory robocze (500 μl) miesza się z 500 μl zawiesiny komórkowej (1 × 106 komórek) w stosunku 1:1, a komórki inkubuje się przez 24 ± 0,5 godziny, jak opisano w pkt 20 i 21. W każdej analizie wystarczające będzie przeprowadzenie jednej kontrpróby w odniesieniu do każdego stężenia badanej substancji chemicznej i substancji kontrolnej, ponieważ prognozę uzyskuje się z co najmniej dwóch niezależnych analiz.
|
Wybarwianie komórek i analiza
|
23.
|
Po upływie 24 godzin ±0,5 godziny narażenia komórki są przenoszone z płytki 24-dołkowej do probówek, zbierane poprzez odwirowanie, a następnie dwukrotnie przemywane w 1 ml roztworu buforowego do wybarwiania (w razie potrzeby można wykonać dodatkowe kroki związane z przemywaniem). Po przemyciu komórki blokuje się w 600 μl roztworu blokującego (roztworu buforowego do wybarwiania zawierającego 0,01 % (w/v) globuliny (ludzka II, III frakcja Cohna; SIGMA, #G2388-10G lub równoważna)) i inkubuje w temperaturze 4oC przez 15 minut. Po zablokowaniu komórki dzieli się na trzy podwielokrotności po 180 μl i umieszcza się je w dołkach 96-dołkowej płytki okrągłodennej lub w probówce Eppendorfa.
|
|
24.
|
Po odwirowaniu komórki wybarwia się za pomocą 50 μl przeciwciał anty-CD86, anty-CD54 lub mysich przeciwciał IgG1 (izotyp) znakowanych FITC w temperaturze 4oC przez 30 minut. Należy użyć przeciwciał opisanych w protokole nr 158 DB-ALM dotyczącym h-CLAT (18), rozcieńczając w stosunku objętościowym 3:25 (w przypadku CD86 (BD-PharMingen, #555657; klon: Fun-1)) lub w stosunku objętościowym 3:50 (w przypadku CD54 (DAKO, #F7143; klon: 6.5B5) i IgG1 (DAKO, #X0927)) w roztworze buforowym do wybarwiania. Naukowcy opracowujący opisywane badanie wskazali powyższe współczynniki rozcieńczenia przeciwciał jako dające najlepszy stosunek sygnału do szumu. Z doświadczenia naukowców opracowujących badanie wynika, że intensywność fluorescencji przeciwciał jest zwykle stała w różnych partiach. Użytkownicy mogą jednak rozważyć miareczkowanie przeciwciał w warunkach własnego laboratorium, aby określić stężenia, które są najbardziej stosowne dla danego zastosowania. Można wykorzystać inne przeciwciała anty-CD86 lub anty-CD54 znakowane fluorochromem, jeżeli można wykazać, że pozwalają one uzyskać wyniki podobne do wyników otrzymywanych w przypadku wykorzystania przeciwciał skoniugowanych z FITC, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 1.2. Należy zaznaczyć, że zmiana klona lub dostawcy przeciwciał opisanych w protokole nr 158 DB-ALM dotyczącym h-CLAT (18) może wpłynąć na wyniki. Po co najmniej dwukrotnym przemyciu komórek w 150 μl roztworu buforowego do wybarwiania zawiesza się je ponownie w roztworze buforowym do wybarwiania (np. w 400 μl) i dodaje się roztwór jodku propidyny (np. 20 μl, aby uzyskać stężenie końcowe wynoszące 0,625 μg/ml) lub roztwór innego markera cytotoksyczności (zob. pkt 18). Poziomy ekspresji CD86 i CD54 oraz żywotność komórek analizuje się przy wykorzystaniu cytometrii przepływowej.
|
DANE I SPRAWOZDAWCZOŚĆ
Ocena danych
|
25.
|
Ekspresję markerów CD86 i CD54 analizuje się za pomocą cytometrii przepływowej z kanałem pomiarowym FL-1. Na podstawie średniej geometrycznej intensywności fluorescencji (MFI) oblicza względną intensywność fluorescencji (RFI) CD86 i CD54 w odniesieniu do komórek kontroli dodatniej i komórek poddanych działaniu substancji chemicznej, korzystając z poniższego równania:

Żywotność komórek kontroli izotypowej (które barwi się mysimi przeciwciałami IgG1 (izotyp)) również oblicza się zgodnie z równaniem opisanym w pkt 19.
|
Model prognozowania
|
26.
|
Na potrzeby pomiaru ekspresji CD86/CD54 każdą badaną substancję chemiczną poddaje się badaniu w co najmniej dwóch niezależnych analizach, aby uzyskać pojedynczą prognozę (DODATNIĄ lub UJEMNĄ). Prognozę h-CLAT uznaje się za DODATNIĄ, jeżeli co najmniej jeden z przedstawionych poniżej warunków zostanie spełniony w dwóch niezależnych analizach na dwie lub w co najmniej dwóch niezależnych analizach na trzy – w przeciwnym wypadku prognozę h-CLAT uznaje się za UJEMNĄ (rys. 1):
|
—
|
względna intensywność fluorescencji CD86 jest równa lub większa niż 150 % dla każdego badanego stężenia (dla którego żywotność komórek ≥ 50 %);
|
|
—
|
względna intensywność fluorescencji CD54 jest równa lub większa niż 200 % dla każdego badanego stężenia (dla którego żywotność komórek ≥ 50 %).
|
|
|
27.
|
W związku z powyższym jeżeli obie pierwsze analizy dadzą wynik dodatni dla CD86 lub obie dadzą wynik dodatni dla CD54, prognozę h-CLAT uważa się za DODATNIĄ i nie trzeba przeprowadzać trzeciej analizy. Analogicznie – jeżeli pierwsze dwie analizy dadzą wynik ujemny dla obu markerów, prognozę h-CLAT uważa się za UJEMNĄ (przy należytym uwzględnieniu przepisów pkt 30) bez konieczności przeprowadzania trzeciej analizy. Jeżeli jednak wyniki dwóch pierwszych analiz nie są zbieżne w odniesieniu do co najmniej jednego markera (CD54 lub CD86), należy przeprowadzić trzecią analizę, a prognoza końcowa będzie oparta na większości wyników z trzech osobnych analiz (tj. 2 z 3). W tym względzie należy zaznaczyć, że jeżeli przeprowadzono dwie niezależne analizy i jedna jest dodatnia tylko w odniesieniu do CD86 (zwana dalej D1), a druga jest dodatnia tylko w odniesieniu do CD54 (zwana dalej D2), konieczne jest przeprowadzenie trzeciej analizy. Jeżeli trzecia analiza jest ujemna w odniesieniu do obu markerów (zwana dalej U), prognozę h-CLAT uważa się za UJEMNĄ. Jeżeli jednak trzecia analiza jest dodatnia dla jednego z markerów (D1 lub D2) lub dla obu markerów (zwana dalej D12), prognozę h-CLAT uważa się za DODATNIĄ.

Rys. 1: Model prognozowania wykorzystywany w metodzie h-CLAT. Prognozę h-CLAT należy rozpatrywać w ramach IATA i zgodnie z przepisami pkt 7 i 8 wprowadzenia ogólnego.
D1: analiza dodatnia tylko w odniesieniu do CD86; D2: analiza dodatnia tylko w odniesieniu do CD54; D12: analiza dodatnia w odniesieniu do CD86 i CD54; U: analiza niedodatnia ani w odniesieniu do CD86, ani w odniesieniu do CD54.
* W ramkach przedstawiono odpowiednie kombinacje wyników dwóch pierwszych analiz niezależnie od kolejności, w jakiej można je uzyskać.
# W ramkach przedstawiono odpowiednie kombinacje wyników trzech analiz, na podstawie wyników uzyskanych w dwóch pierwszych analizach, przedstawionych w ramce powyżej, nie odzwierciedlając jednak kolejności, w jakiej można je uzyskać.
|
|
28.
|
W odniesieniu do badanych substancji chemicznych, w przypadku których przewiduje się wynik DODATNI względem h-CLAT, opcjonalnie można określić dwie wartości stężenia efektywnego (EC), EC150 dla CD86 i EC200 dla CD54, tj. stężenia, przy którym badane substancje chemiczne indukują względną intensywność fluorescencji wynoszącą 150 lub 200. Te wartości stężenia efektywnego mogą potencjalnie wpłynąć na ocenę siły działania uczulającego (9), gdy stosuje się je w kontekście podejść zintegrowanych takich jak IATA (4)(5)(6)(7)(8). Można je obliczyć, korzystając z poniższych równań:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
gdzie:
Aconc oznacza najniższe stężenie wyrażone w μg/ml przy RFI > 150 (CD86) lub 200 (CD54),
Bconc oznacza najwyższe stężenie wyrażone w μg/ml przy RFI < 150 (CD86) lub 200 (CD54),
ARFI oznacza względną intensywność fluorescencji dla najniższego stężenia przy RFI > 150 (CD86) lub 200 (CD54),
BRFI oznacza względną intensywność fluorescencji dla najwyższego stężenia przy RFI < 150 (CD86) lub 200 (CD54).
Aby uzyskać dokładniejsze wartości EC150 i EC200, konieczne może być przeprowadzenie trzech niezależnych analiz dotyczących pomiaru ekspresji CD86/CD54. Następnie ostateczne wartości EC150 oraz EC200 określa się jako wartość mediany EC wyliczanych na podstawie trzech niezależnych analiz. W przypadku gdy tylko dwie z trzech niezależnych analiz spełniają kryteria uznania za wyniki dodatnie (zob. pkt 26–27), przyjmuje się wyższą z wyliczonych wartości EC150 lub EC200.
|
Kryteria dopuszczalności
|
29.
|
Podczas stosowania metody h-CLAT (22)(27) spełnione muszą zostać następujące kryteria dopuszczalności:
|
—
|
żywotność komórek w podłożu oraz kontrolach z rozpuszczalnikiem/nośnikiem powinna być wyższa niż 90 %;
|
|
—
|
w przypadku kontroli z rozpuszczalnikiem/nośnikiem wartości RFI zarówno w odniesieniu do CD86, jak i CD54 nie powinny przekraczać kryteriów uzyskania wyniku dodatniego (CD86 – RFI ≥ 150 % i CD54 – RFI ≥ 200 %). Wartości RFI kontroli z nośnikiem/rozpuszczalnikiem oblicza się, korzystając ze wzoru przedstawionego w pkt 25 („MFI substancji chemicznej” należy zastąpić „MFI nośnika/rozpuszczalnika”, a „MFI nośnika/rozpuszczalnika” należy zastąpić „MFI kontroli (podłoża)”);
|
|
—
|
stosunek MFI zarówno CD86, jak i CD54 do kontroli izotypowej zarówno w podłożu, jak i w kontroli z rozpuszczalnikiem/nośnikiem powinien wynosić >105 %;
|
|
—
|
w kontroli dodatniej (DNCB) wartości RFI zarówno CD86, jak i CD54 powinny spełniać kryteria uzyskania wyniku dodatniego (CD86 – RFI ≥ 150 % i CD54 – RFI ≥ 200), a żywotność komórek powinna być większa niż 50 %;
|
|
—
|
jeżeli chodzi o badaną substancję chemiczną, żywotność komórek powinna być wyższa niż 50 % w przypadku co najmniej czterech stężeń badanych w każdej analizie.
|
|
|
30.
|
Wyniki ujemne dopuszcza się wyłącznie w przypadku badanych substancji chemicznych charakteryzujących się żywotnością komórek niższą niż 90 % przy najwyższym badanym stężeniu (tj. 1,2 × CV75 zgodnie ze schematem dotyczącym serii rozcieńczeń w postępie geometrycznym opisanym w pkt 20). Jeżeli żywotność komórek przy stężeniu 1,2 × CV75 równa się lub przekracza 90 %, wynik ujemny należy odrzucić. W takim przypadku zaleca się podjęcie próby udoskonalenia dobrania dawki poprzez ponowne określenie wartości CV75. Należy zauważyć, że w przypadku stosowania jako maksymalnego stężenia badanej substancji chemicznej stężenia 5 000 μg/ml w soli fizjologicznej (lub podłożu, lub innych rozpuszczalnikach/nośnikach), stężenia 1 000 μg/ml w sulfotlenku dimetylu lub najwyższego stężenia rozpuszczalnego, wynik ujemny dopuszcza się nawet wówczas, gdy żywotność komórek przekracza 90 %.
|
Sprawozdanie z badania
|
31.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
Badana substancja chemiczna
|
Substancja jednoskładnikowa
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, log Kow, rozpuszczalność w wodzie, rozpuszczalność w sulfotlenku dimetylu, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
Substancja wieloskładnikowa, UVCB i mieszanina
|
opisana w miarę możliwości np. poprzez podanie nazwy chemicznej (zob. powyżej), czystości, przedstawienie informacji dotyczących występowania ilościowego oraz wskazanie istotnych właściwości fizykochemicznych składników (zob. powyżej), w dostępnym zakresie;
|
|
wygląd fizyczny, rozpuszczalność w wodzie, rozpuszczalność w sulfotlenku dimetylu i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
masa cząsteczkowa lub pozorna masa cząsteczkowa w przypadku mieszanin lub polimerów o znanym składzie lub inne informacje istotne dla przebiegu badania;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
|
Kontrole
|
Kontrola dodatnia
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, log Kow, rozpuszczalność w wodzie, rozpuszczalność w sulfotlenku dimetylu, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie i w stosownych przypadkach;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
odniesienie do wyników historycznych kontroli dodatnich wykazujących odpowiednie kryteria dopuszczalności analizy, w stosownych przypadkach.
|
|
|
|
Kontrola ujemna oraz kontrola z rozpuszczalnikiem/nośnikiem
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
wygląd fizyczny, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne w przypadku stosowania innego rozpuszczalnika/nośnika kontrolnego niż te opisane w wytycznej dotyczącej badań, w dostępnym zakresie;
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
Warunki badania
|
nazwa i adres sponsora i placówki przeprowadzającej badanie oraz dane kierownika badań;
|
|
opis zastosowanego badania;
|
|
zastosowana linia komórkowa, warunki jej przechowywania oraz źródło (np. placówka, z której ją otrzymano);
|
|
zastosowana cytometria przepływowa (np. model), w tym informacje na temat zastosowanych ustawień przyrządu, globuliny, przeciwciał i markera cytotoksyczności;
|
|
procedura stosowana w celu wykazania biegłości laboratorium w zakresie przeprowadzania badania poprzez badanie substancji służących do wykazania biegłości lub procedura stosowana do wykazania odtwarzalności badania w czasie, np. dane historyczne dotyczące kontroli lub dane historyczne dotyczące kontroli reaktywności.
|
|
|
Kryteria dopuszczalności badania
|
wartości dotyczące żywotności komórek, MFI oraz RFI uzyskane za pomocą kontroli z rozpuszczalnikiem/nośnikiem w porównaniu z zakresami dopuszczalności;
|
|
wartości dotyczące żywotności komórek oraz RFI uzyskane za pomocą kontroli dodatniej w porównaniu z zakresami dopuszczalności;
|
|
żywotność komórek przy wszystkich badanych stężeniach badanej substancji chemicznej.
|
|
|
Procedura badawcza
|
liczba zastosowanych analiz;
|
|
stosowane stężenia badanej substancji chemicznej, procedura podawania badanej substancji chemicznej i czas narażenia (jeżeli są one inne niż zalecane);
|
|
opis zastosowanych kryteriów oceny i podejmowania decyzji;
|
|
opis wszelkich modyfikacji procedury badawczej.
|
|
|
Wyniki
|
tabelaryczne zestawienie danych, w tym wartości CV75 (w stosownych przypadkach), poszczególnych wartości geometrycznej MFI, RFI i żywotności komórek, wartości EC150/EC200 (w stosownych przypadkach), uzyskanych w odniesieniu do badanej substancji chemicznej oraz kontroli dodatniej w każdej analizie oraz wskazanie oceny badanej substancji chemicznej według modelu prognozowania;
|
|
w stosownych przypadkach opis wszelkich innych istotnych obserwacji.
|
|
|
Omówienie wyników
|
omówienie wyników uzyskanych za pomocą metody h-CLAT;
|
|
uwzględnienie wyników zastosowania badania w kontekście IATA, jeżeli są dostępne inne odpowiednie informacje.
|
|
|
BIBLIOGRAFIA
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EURL-ECVAM UE (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Dostępne na stronie internetowej: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Dostępne na stronie internetowej: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report. Dostępne pod adresem: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). „Protokół 158: human Cell Line Activation Test (h-CLAT)”, s. 23. Dostępne na stronie internetowej: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Seria dotycząca badań i oceny OECD. Francja, Paryż: Organizacja Współpracy Gospodarczej i Rozwoju, 96 s.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Seria dotycząca badań i oceny nr 168. Dostępne na stronie internetowej: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
Organizacja Narodów Zjednoczonych (ONZ) (2013). Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS). Wydanie piąte zmienione, Nowy Jork i Genewa: publikacje Organizacji Narodów Zjednoczonych. ISBN: 978-92-1-117006-1. Dostępne na stronie internetowej: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. Europejskie Centrum ds. Ekotoksykologii i Toksykologii Substancji Chemicznych (sprawozdanie techniczne nr 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Dodatek 1.1
DEFINICJE
Dokładność
: stopień zgodności pomiędzy wynikami badania a przyjętymi wartościami odniesienia. Jest to miara efektywności badania i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na oznaczenie odsetka prawidłowych wyników uzyskiwanych przy przeprowadzeniu badania (21).
Mechanizm wywoływania skutków szkodliwych
: sekwencja zdarzeń, począwszy od struktury chemicznej docelowej substancji chemicznej lub grupy podobnych substancji chemicznych poprzez molekularne zdarzenie inicjujące aż do pożądanego wyniku in vivo (22).
Substancja chemiczna
: substancja lub mieszanina.
CV75
: szacowane stężenie, przy którym żywotność komórek wynosi 75 %.
EC150
: stężenia, przy których wartości RFI w ekspresji CD86 wynoszą 150.
EC200
: stężenia, przy których wartości RFI w ekspresji CD54 wynoszą 200.
Cytometria przepływowa
: technika cytometryczna, w której komórki zawieszone w cieczy przepływają jedna po drugiej przez zogniskowaną wiązkę wzbudzającego światła, które rozprasza się według wzorców charakterystycznych dla komórek i ich składników; komórki często oznacza się znacznikami fluorescencyjnymi, dzięki czemu światło jest najpierw absorbowane, a następnie emitowane w zmienionych częstotliwościach.
Zagrożenie
: nieodłączna właściwość czynnika lub sytuacja, która może potencjalnie doprowadzić do niekorzystnych skutków w przypadku narażenia organizmu, systemu lub (sub)populacji na taki czynnik.
IATA (zintegrowane podejście do badań i oceny)
: usystematyzowane podejście stosowane w odniesieniu do identyfikacji zagrożeń (potencjał), charakterystyki zagrożeń (siła oddziaływania) lub oceny bezpieczeństwa (potencjał / siła oddziaływania oraz narażenie) substancji chemicznej lub grupy substancji chemicznych; w ramach tego podejścia w sposób strategiczny integruje się i waży wszystkie istotne dane, które mogą wpłynąć na podjęcie decyzji regulacyjnej dotyczącej potencjalnego zagrożenia, ryzyka lub konieczności podjęcia dalszych ukierunkowanych, a przez to minimalnych badań.
Kontrola podłoża
: kontrpróba niepoddana działaniu badanej substancji chemicznej zawierająca wszystkie składniki układu badawczego. Próbka ta jest przetwarzana razem z próbkami poddanymi działaniu badanej substancji chemicznej oraz z innymi próbkami kontrolnymi w celu określenia, czy rozpuszczalnik/nośnik wchodzi w reakcję z układem badawczym.
Mieszanina
: mieszanina lub roztwór, które składają się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
Substancja wieloskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥10 % (w/w) i <80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się poprzez zmieszanie przynajmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
Kontrola dodatnia
: kontrpróba zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że wywołuje reakcję dodatnią. Aby zapewnić możliwość oceny zmienności reakcji w kontroli dodatniej w czasie, stopień reakcji dodatniej nie powinien być nadmierny.
Prehapteny
: substancje chemiczne, które stają się czynnikami uczulającymi po transformacji abiotycznej.
Prohapteny
: substancje chemiczne, które wymagają aktywacji enzymatycznej do uwolnienia swojego potencjalnego działania uczulającego na skórę.
Względna intensywność fluorescencji (RFI)
: wartości względne średniej geometrycznej intensywności fluorescencji (MFI) w komórkach poddanych działaniu substancji chemicznej w porównaniu z MFI w komórkach poddanych działaniu rozpuszczalnika/nośnika.
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono znaczące i użyteczne z punktu widzenia określonego celu. Jest to zakres, w jakim badanie pozwala prawidłowo zmierzyć lub przewidzieć biologiczny oczekiwany skutek. Istotność obejmuje uwzględnienie dokładności (zgodności) badania (21).
Wiarygodność
: miary zakresu, w jakim badanie może zostać przeprowadzone w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jego przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją, obliczając odtwarzalność wewnątrz- i międzylaboratoryjną oraz powtarzalność wewnątrzlaboratoryjną (21).
Analiza
: analiza obejmuje jednoczesne badanie jednej badanej substancji chemicznej lub większej liczby takich substancji w kontroli z rozpuszczalnikiem/nośnikiem i kontroli dodatniej.
Czułość
: odsetek wszystkich substancji chemicznych dających wynik dodatni / aktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą danego badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (21).
Roztwór buforowy do wybarwiania
: roztwór chlorku sodu buforowany fosforanami zawierający 0,1 % albuminy surowicy bydlęcej.
Kontrola z rozpuszczalnikiem/nośnikiem
: próbka niepoddana działaniu badanej substancji, zawierająca wszystkie składniki układu badawczego z wyjątkiem badanej substancji chemicznej, ale z uwzględnieniem stosowanego rozpuszczalnika/nośnika. Wykorzystywana jest do określenia wyjściowej reakcji dla próbek poddanych działaniu badanej substancji chemicznej rozpuszczonej lub stabilnie rozproszonej w tym samym rozpuszczalniku/nośniku. W przypadku badania z jednoczesną kontrolą podłoża próbka ta wykazuje również, czy rozpuszczalnik/nośnik wchodzi w reakcję z układem badawczym.
Swoistość
: odsetek wszystkich substancji chemicznych dających wynik ujemny / nieaktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (21).
Substancja
: pierwiastek chemiczny i jego związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania jego trwałości i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem ewentualnych rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody.
Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ)
: system, w ramach którego proponuje się klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz omawia się odpowiednie elementy komunikacyjne, takie jak: piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki substancji i produktów niebezpiecznych, aby przekazać informacje na temat ich szkodliwego działania w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) i środowiska (23).
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
Ważne badanie
: badanie, które uznano za wystarczająco istotne i wiarygodne dla danego celu i które opiera się na uzasadnionych naukowo podstawach. Badanie nie może zostać uznane za ważne w sensie absolutnym, ale tylko w odniesieniu do określonego celu (21).
Dodatek 1.2
SUBSTANCJE SŁUŻĄCE DO WYKAZANIA BIEGŁOŚCI
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo określając prognozę h-CLAT dla 10 substancji wskazanych w tabeli 1 oraz określając wartości CV75, EC150 i EC200, które mieszczą się w odpowiednim zakresie odniesienia, dla co najmniej 8 z 10 substancji służących do wykazania biegłości. Substancje służące do wykazania biegłości wybrano w taki sposób, aby odzwierciedlić zakres reakcji na zagrożenia związane z działaniem uczulającym na skórę. Przy wyborze substancji kierowano się również następującymi kryteriami: dostępnością handlową tych substancji, dostępnością wysokiej jakości danych referencyjnych z badań in vivo oraz dostępnością wysokiej jakości danych in vitro uzyskanych w wyniku zastosowania metody h-CLAT. W odniesieniu do metody h-CLAT można znaleźć również opublikowane dane referencyjne (3)(14).
Tabela 1
Substancje zalecane do wykazania biegłości technicznej w odniesieniu do metody h-CLAT
|
Substancje służące do wykazania biegłości
|
Numer CAS
|
Stan skupienia
|
Prognoza in vivo
(102)
|
CV75 odniesienia CV75 w μg/ml (103)
|
Wynik badania h-CLAT dla CD86 (Zakres odniesienia EC150 w μg/ml) (103)
|
Wynik badania h-CLAT dla CD54 (Zakres odniesienia EC200 w μg/ml) (103)
|
|
2,4-dinitrochlorobenzen
|
97-00-7
|
Stały
|
Czynnik uczulający (wyjątkowo silny)
|
2–12
|
Dodatni (0,5–10)
|
Dodatni (0,5–15)
|
|
4-fenylenodiamina
|
106-50-3
|
Stały
|
Czynnik uczulający (silny)
|
5–95
|
Dodatni (<40)
|
Ujemny (>1,5) (104)
|
|
Siarczan niklu
|
10101-97-0
|
Stały
|
Czynnik uczulający (w stopniu umiarkowanym)
|
30–500
|
Dodatni (<100)
|
Dodatni (10–100)
|
|
2-merkaptobenzotiazol
|
149-30-4
|
Stały
|
Czynnik uczulający (w stopniu umiarkowanym)
|
30–400
|
Ujemny (>10) (104)
|
Dodatni (10–140)
|
|
R(+)-limonen
|
5989-27-5
|
Ciekły
|
Czynnik uczulający (słaby)
|
>20
|
Ujemny (>5) (104)
|
Dodatni (<250)
|
|
Imidazolidynylomocznik
|
39236-46-9
|
Stały
|
Czynnik uczulający (słaby)
|
25–100
|
Dodatni (20–90)
|
Dodatni (20–75)
|
|
Izopropanol
|
67-63-0
|
Ciekły
|
Brak działania uczulającego
|
>5000
|
Ujemny (>5 000 )
|
Ujemny (>5 000 )
|
|
Glicerol
|
56-81-5
|
Ciekły
|
Brak działania uczulającego
|
>5000
|
Ujemny (>5 000 )
|
Ujemny (>5 000 )
|
|
Kwas mlekowy
|
50-21-5
|
Ciekły
|
Brak działania uczulającego
|
1500–5000
|
Ujemny (>5 000 )
|
Ujemny (>5 000 )
|
|
Kwas 4-aminobenzoesowy
|
150-13-0
|
Stały
|
Brak działania uczulającego
|
>1000
|
Ujemny (>1 000 )
|
Ujemny (>1 000 )
|
|
Skróty: Nr CAS = numer w rejestrze Chemical Abstracts Service.
|
Dodatek 2
BADANIE DZIAŁANIA UCZULAJĄCEGO NA SKÓRĘ IN VITRO: BADANIE AKTYWACJI LINII KOMÓRKOWEJ U937 (U-SENS™)
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
1.
|
Badanie U-SENS™ pozwala określić ilościowo zmianę ekspresji markera powierzchniowego komórek związanego z procesem aktywacji monocytów i komórek dendrytycznych (tj. CD86) w ludzkiej linii komórkowej chłoniaka histiocytarnego U937 po narażeniu na działanie czynników uczulających (1). Pomiary poziomów ekspresji markera powierzchniowego komórek CD86 w linii komórkowej U937 wykorzystuje się później do wsparcia rozróżnienia substancji działających uczulająco na skórę i substancji niewykazujących takiego działania.
|
|
2.
|
Badanie U-SENS™ oceniono w ramach badania walidacyjnego (2) koordynowanego przez L’Oreal i późniejszej niezależnej wzajemnej oceny przeprowadzonej przez Naukowy Komitet Doradczy (ESAC) działający przy laboratorium referencyjnym Unii Europejskiej ds. metod alternatywnych wobec testów na zwierzętach (EURL ECVAM) (3). Uwzględniając wszystkie dostępne dowody i wkład ze strony organów regulacyjnych i zainteresowanych stron, EURL ECVAM (4) zaleciło stosowanie badania U-SENS™ w ramach IATA do wsparcia rozróżnienia substancji działających uczulająco i substancji niewykazujących takiego działania na potrzeby klasyfikacji i oznakowania pod względem zagrożeń. W swoich wytycznych dotyczących zgłaszania usystematyzowanych podejść do integracji danych oraz poszczególnych źródeł informacji na temat działania uczulającego na skórę wykorzystywanych w ramach IATA OECD omawia szereg studiów przypadku, opisując różne strategie badań oraz modele prognozowania. Jedno z wielu zdefiniowanych podejść opiera się na teście U-SENS (5). Przykłady wykorzystania danych z badania U-SENS™ w połączeniu z innymi informacjami, w tym z danymi historycznymi oraz istniejącymi ważnymi danymi dotyczącymi ludzi (6), są także zamieszczone w innych pozycjach literatury (4)(5)(7).
|
|
3.
|
Udowodniono, że badanie U-SENS™ można przeprowadzać również w laboratoriach posiadających doświadczenie w zakresie kultury komórkowej i analizy przy użyciu cytometrii przepływowej. Poziom odtwarzalności prognoz, którego można oczekiwać od tego badania, ma wartość rzędu 90 % i 84 % odpowiednio wewnątrz laboratoriów i pomiędzy nimi (8). Wyniki uzyskane w badaniu walidacyjnym (8) i innych opublikowanych badaniach (1) ogólnie wskazują na to, że – w porównaniu z wynikami LLNA – dokładność rozróżniania substancji działających uczulająco na skórę (tj. należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP) i substancji niewykazujących takiego działania wynosi 86 % (N = 166) przy czułości na poziomie 91 % (118/129) oraz swoistości na poziomie 65 % (24/37). W porównaniu z wynikami badań przeprowadzonych na ludziach dokładność rozróżniania substancji działających uczulająco na skórę (tj. należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP) i substancji niewykazujących takiego działania wynosi 77 % (N = 101) przy czułości na poziomie 100 % (58/58) oraz swoistości na poziomie 47 % (20/43). Prawdopodobieństwo wystąpienia prognoz fałszywie ujemnych przy zastosowaniu badania U-SENS™ (w porównaniu do LLNA) jest większe w przypadku substancji chemicznych wykazujących niską lub średnią siłę działania uczulającego na skórę (tj. należących do podkategorii 1B wg GHS ONZ / rozporządzenia CLP) niż w przypadku substancji wykazujących wysoką siłę działania uczulającego na skórę (tj. należących do podkategorii 1A wg GHS ONZ / rozporządzenia CLP) (1)(8)(9). Wszystkie te informacje łącznie wskazują na użyteczność badania U-SENS™ w identyfikowaniu zagrożeń związanych z działaniem uczulającym na skórę. Przedstawione tutaj wartości dokładności U-SENS™ jako samodzielnego badania są jednak wyłącznie orientacyjne, ponieważ badanie to należy rozpatrywać w połączeniu z innymi źródłami informacji w kontekście IATA i zgodnie z przepisami pkt 7 i 8 wprowadzenia ogólnego. Co więcej, przy ocenie metod niewymagających wykorzystania zwierząt pod kątem działania uczulającego na skórę należy pamiętać, że LLNA, jak również inne badania przeprowadzane na zwierzętach, mogą nie w pełni odzwierciedlać sytuację w odniesieniu do ludzi.
|
|
4.
|
Na podstawie obecnie dostępnych danych wykazano, że badanie U-SENS™ ma zastosowanie do badanych substancji chemicznych (w tym do składników kosmetycznych, np. konserwantów, środków powierzchniowo czynnych, substancji czynnych, barwników) obejmujących szereg organicznych grup funkcyjnych, właściwości fizykochemicznych, sił działania uczulającego na skórę (jak określono w badaniach in vivo) oraz spektrum mechanizmów reagowania, o których wiadomo, że mają związek z działaniem uczulającym na skórę (np. akceptor Michaela, powstawanie zasady Schiffa, przenośnik grup acylowych, dwucząsteczkowa substytucja nukleofilowa [SN2] lub aromatyczna substytucja nukleofilowa [SNAr]) (1)(8)(9)(10). Badanie U-SENS™ ma zastosowanie do badanych substancji chemicznych, które są rozpuszczalne lub tworzą stabilną dyspersję (tj. koloid lub zawiesinę, w których badana substancja chemiczna nie osadza się ani nie oddziela od rozpuszczalnika/nośnika w różne fazy) w odpowiednim rozpuszczalniku/nośniku (zob. pkt 13). W badaniu U-SENS™ w sposób prawidłowy przewidziano substancje chemiczne zgłaszane w zbiorze danych jako prehapteny (tj. substancje aktywowane przez utlenianie) lub prohapteny (tj. substancje wymagające aktywacji enzymatycznej, np. przez enzymy P450) (1)(10). Stosowanie substancji powodujących przerwanie błony komórkowej może doprowadzić do uzyskania wyników fałszywie dodatnich z powodu niespecyficznego wzrostu ekspresji CD86 – 3 na 7 wyników fałszywie dodatnich względem klasyfikacji odniesienia in vivo spowodowane było stosowaniem środków powierzchniowo czynnych (1). Do wyników dodatnich uzyskanych przy zastosowaniu środków powierzchniowo czynnych jako takich należy podchodzić z ostrożnością, natomiast wyniki ujemne uzyskane przy zastosowaniu środków powierzchniowo czynnych można dalej wykorzystywać do wsparcia identyfikacji badanej substancji chemicznej jako substancji niewykazującej działania uczulającego na skórę. Fluorescencyjne badane substancje chemiczne można ocenić za pomocą badania U-SENS™ (1), jednak badane substancje chemiczne o wysokiej fluorescencji emitujące fale tej samej długości co izotiocyjanian fluoresceiny (FITC) lub jodek propidyny będą zakłócać detekcję przy użyciu cytometrii przepływowej, co uniemożliwia ich prawidłową ocenę z wykorzystaniem przeciwciał skoniugowanych z FITC (potencjalny wynik fałszywie ujemny) lub jodku propidyny (brak możliwości pomiaru żywotności). W takim przypadku można użyć odpowiednio innych przeciwciał znakowanych fluorochromem lub innych markerów cytotoksyczności, o ile można wykazać, że pozwalają one uzyskać wyniki podobne do wyników otrzymywanych w przypadku przeciwciał znakowanych FITC lub jodku propidyny (zob. pkt 18), np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 2.2. W świetle powyższego wyniki dodatnie uzyskane przy zastosowaniu środków powierzchniowo czynnych oraz wyniki ujemne uzyskane przy zastosowaniu badanych substancji chemicznych o wysokiej fluorescencji należy interpretować w kontekście zdefiniowanych ograniczeń i w połączeniu z innymi źródłami informacji w ramach IATA. W przypadkach, w których można przedstawić dowody na brak możliwości zastosowania badania U-SENS™ w odniesieniu do innych konkretnych kategorii badanych substancji chemicznych, nie należy jej stosować w odniesieniu do tych konkretnych kategorii.
|
|
5.
|
Jak opisano powyżej, badanie U-SENS™ wspiera rozróżnienie substancji działających uczulająco na skórę i substancji niewykazujących takiego działania. Może ono jednak również potencjalnie wpłynąć na ocenę siły działania uczulającego, gdy stosuje się je w ramach podejść zintegrowanych takich jak IATA. Wymagane jest jednak przeprowadzenie dalszych prac, najlepiej na podstawie danych dotyczących ludzi, aby ustalić, w jaki sposób wyniki badania U-SENS™ mogą potencjalnie wpłynąć na ocenę siły działania.
|
|
6.
|
Definicje znajdują się w dodatku 2.1.
|
ZASADA BADANIA
|
7.
|
Badanie U-SENS™ jest testem in vitro, który pozwala określić ilościowo zmiany ekspresji markera powierzchniowego komórki CD86 w ludzkiej linii komórkowej chłoniaka histiocytarnego, tj. komórkach U937, po 45 ± 3 godzinach narażenia na działanie badanej substancji chemicznej. Marker powierzchniowy CD86 jest jednym z typowych markerów aktywacji U937. Wiadomo, że CD86 jest cząsteczką kostymulującą, która może naśladować aktywację monocytów, która odgrywa kluczową rolę w uczulaniu limfocytów T. Zmiany ekspresji markera powierzchniowego CD86 mierzy się za pomocą cytometrii przepływowej, zwykle korzystając z przeciwciał znakowanych izotiocyjanianem fluoresceiny (FITC). Jednocześnie przeprowadza się również pomiar cytotoksyczności (np. z wykorzystaniem jodku propidyny), aby ocenić, czy podwyższona ekspresja markera powierzchniowego komórki CD86 zachodzi przy stężeniach niższych niż stężenia cytotoksyczne. Wskaźnik stymulacji (S.I.) markera powierzchniowego komórki CD86 w porównaniu z kontrolą z rozpuszczalnikiem/nośnikiem wylicza się i wykorzystuje w modelu prognozowania (zob. pkt 19), aby wesprzeć rozróżnienie substancji działających uczulająco i substancji niewykazujących takiego działania.
|
WYKAZANIE BIEGŁOŚCI
|
8.
|
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną z zastosowaniem dziesięciu substancji służących do wykazania biegłości wymienionych w dodatku 2.2 zgodnie z dobrymi praktykami w zakresie metody in vitro (11). Ponadto użytkownicy badania powinni prowadzić bazę danych historycznych wygenerowanych podczas kontroli reaktywności (zob. pkt 11) oraz kontroli dodatnich i kontroli z rozpuszczalnikiem/nośnikiem (zob. pkt 15–16), a także wykorzystywać te dane do potwierdzenia zachowania odtwarzalności badania w swoim laboratorium w miarę upływu czasu.
|
PROCEDURA
|
9.
|
Podstawę niniejszego badania stanowi protokół nr 183 bazy danych na temat metod alternatywnych wobec testów na zwierzętach (DB-ALM) dotyczący U-SENS™ (12). Ze standardowych procedur operacyjnych należy korzystać przy wdrażaniu i wykonywaniu badania U-SENS™ w laboratorium. Badanie U-SENS™ można przeprowadzić, korzystając z zautomatyzowanego systemu, jeżeli można wykazać, że pozwala on uzyskać podobne wyniki, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 2.2. Poniżej opisano główne elementy i procedury badania U-SENS™.
|
Przygotowanie komórek
|
10.
|
Do badania U-SENS™ należy wykorzystać ludzką linię komórkową chłoniaka histiocytarnego – U937 (13). Komórki (klon CRL1593.2) należy pozyskać z banku komórek posiadającego niezbędne uprawnienia, takiego jak np. zbiór kultur typu amerykańskiego.
|
|
11.
|
Komórki U937 hoduje się w temperaturze 37 °C, w obecności 5 % CO2 i w wilgotnym środowisku, w podłożu RPMI-1 640 z dodatkiem 10 % cielęcej surowicy płodowej, 2 mM L-glutaminy, 100 jednostek/ml penicyliny i 100 μg/ml streptomycyny (kompletne podłoże). Pasaż komórek U937 odbywa się regularnie co 2–3 dni w gęstości odpowiednio 1,5 lub 3 × 105 komórek/ml. Zagęszczenie komórek nie powinno przekraczać 2 × 106 komórek/ml, a żywotność komórek mierzona poprzez wykluczenie komórek wybarwionych błękitem trypanu powinna wynosić ≥ 90 % (metody tej nie należy stosować przy pierwszym pasażu następującym po rozmrożeniu). Przed wykorzystaniem komórek do badań każdą partię komórek, płodowej surowicy cielęcej lub przeciwciał należy zakwalifikować do badań, przeprowadzając kontrolę reaktywności. Kontrolę reaktywności komórek należy przeprowadzić, wykorzystując do tego celu kontrolę dodatnią, tj. kwas pikrylosulfonowy (kwas 2,4,6-trinitrobenzenosulfonowy, TNBS) (nr CAS 2 508-19-2, ≥ 99 % czystości), oraz kontrolę ujemną, tj. kwas mlekowy (LA) (nr CAS 50-21-5, ≥ 85 % czystości), co najmniej tydzień po rozmrożeniu. Podczas kontroli reaktywności należy zbadać sześć stężeń końcowych w każdej z 2 kontroli (TNBS: 1; 12,5; 25; 50; 75; 100 μg/ml, oraz LA: 1; 10; 20; 50; 100; 200 μg/ml). Rozpuszczenie TNBS w kompletnym podłożu powinno wywołać reakcję dodatnią CD86, która powinna być powiązana ze stężeniem (np. gdy po uzyskaniu stężenia dodatniego (wskaźnik stymulacji CD86 ≥150) następuje stężenie charakteryzujące się wzrastającą wartością wskaźnika stymulacji CD86), a rozpuszczenie kwasu mlekowego w kompletnym podłożu powinno wywołać reakcję ujemną CD86 (zob. pkt 21). Do analizy należy wykorzystać jedynie partię komórek, która dwukrotnie przeszła kontrolę reaktywności. Komórki można rozmnażać do siedmiu tygodni po rozmrożeniu. Liczba pasaży nie powinna przekraczać 21. Kontrolę reaktywności należy przeprowadzić zgodnie z procedurami opisanymi w pkt 18–22.
|
|
12.
|
Komórki U937 wysiewa się do badań w gęstości 3 x 105 komórek/ml albo 6 × 105 komórek/ml i dokonuje ich wstępnej hodowli w kolbach z kulturami odpowiednio przez 2 dni lub 1 dzień. Można zastosować inne warunki hodowli wstępnej niż warunki opisane powyżej, jeżeli przedstawione zostanie wystarczające uzasadnienie naukowe oraz jeżeli można wykazać, że pozwalają one uzyskać podobne wyniki, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 2.2. W dniu badania komórki pobrane z kolby z kulturami zawiesza się ponownie w świeżym podłożu w gęstości 5 × 105 komórek/ml. Następnie komórki umieszcza się w dołkach 96-dołkowej płytki płaskodennej w objętości po 100 μl (końcowe zagęszczenie komórek – 0,5 × 105 komórek/dołek).
|
Przygotowanie badanych substancji chemicznych i substancji kontrolnych
|
13.
|
Ocenę rozpuszczalności przeprowadza się przed badaniem. W tym celu badane substancje chemiczne są rozpuszczane lub tworzą stabilne dyspersje o stężeniu 50 mg/ml w kompletnym podłożu (pierwszy wariant rozpuszczalnika) lub w sulfotlenku dimetylu (DMSO, czystość ≥99 %) (drugi wariant rozpuszczalnika/nośnika), jeżeli badana substancja chemiczna nie rozpuszcza się w rozpuszczalniku/nośniku kompletnego podłoża. Na potrzeby badania badaną substancję chemiczną rozpuszcza się do uzyskania stężenia końcowego wynoszącego 0,4 mg/ml w kompletnym podłożu, jeżeli substancja chemiczna jest rozpuszczalna w danym rozpuszczalniku/nośniku. Jeżeli substancja chemiczna jest rozpuszczalna jedynie w sulfotlenku dimetylu, rozpuszcza się ją w stężeniu 50 mg/ml. Można użyć innych rozpuszczalników/nośników niż te opisane powyżej pod warunkiem przedstawienia wystarczającego uzasadnienia naukowego. Należy uwzględnić stabilność badanej substancji chemicznej w końcowym rozpuszczalniku/nośniku.
|
|
14.
|
Badane substancje chemiczne oraz substancje kontrolne przygotowuje się w dniu badania. Z uwagi na nieprzeprowadzanie testu ustalającego dawkę na potrzeby pierwszej analizy należy przeprowadzić badania 6 stężeń końcowych (1, 10, 20, 50, 100 i 200 μg/ml) w odpowiednim rozpuszczalniku/nośniku albo w kompletnym podłożu, albo w 0,4 % sulfotlenku dimetylu w podłożu. Na potrzeby kolejnych analiz, przy użyciu odpowiedniego rozpuszczalnika/nośnika, przygotowuje się roztwory badanych substancji chemicznych – co najmniej 4 roztwory robocze (tj. w co najmniej 4 stężeniach), zaczynając od stężenia 0,4 mg/ml w kompletnym podłożu lub stężenia 50 mg/ml w sulfotlenku dimetylu. Roztwory robocze ostatecznie wykorzystuje się w celu poddania działaniu substancji chemicznej poprzez dodanie zawiesiny komórkowej linii U937 (zob. pkt 11 powyżej) w objętości równej objętości roztworu roboczego na płytce, aby osiągnąć dalsze dwukrotne rozcieńczenie (12). Doboru stężeń (co najmniej 4 stężeń), które mają zostać wykorzystane na potrzeby kolejnej analizy, dokonuje się w oparciu o poszczególne wyniki wszystkich poprzednich analiz (8). Użytkowe stężenia końcowe: 1; 2; 3; 4; 5; 7,5; 10; 12,5; 15; 20; 25; 30; 35; 40; 45; 50; 60; 70; 80; 90; 100; 120; 140; 160; 180 i 200 μg/ml. Maksymalne stężenie końcowe wynosi 200 μg/ml. W przypadku zaobserwowania wartości dodatniej CD86 przy stężeniu 1 μg/ml ocenie poddaje się stężenie 0,1 μg/ml, aby ustalić stężenie badanej substancji chemicznej, które nie wywołuje przekroczenia przez CD86 progu uzyskania wyniku dodatniego. Jeżeli zaobserwowano dodatnią zależność stężenie-odpowiedź w odniesieniu do CD86, na potrzeby każdej analizy wylicza się wartość EC150 (stężenie, przy którym substancja chemiczna osiąga próg uzyskania wyniku dodatniego dla CD86 wynoszący 150 %, zob. pkt 19). W przypadku gdy badana substancja chemiczna wywołuje reakcję dodatnią CD86 niepowiązaną ze stężeniem, obliczenie wartości EC150 może nie być istotne, jak opisano w protokole nr 183 DB-ALM dotyczącym U-SENS™ (12). Na potrzeby każdej analizy – gdy tylko jest to możliwe – wylicza się wartość CV70 (stężenie, przy którym substancja chemiczna osiąga próg cytotoksyczności wynoszący 70 %, zob. pkt 19) (12). Aby zbadać wpływ wzrostu ekspresji CD86 na zależność stężenie-odpowiedź, spośród stężeń użytkowych należy wybrać dowolne stężenia, których wartości są równomiernie rozłożone między EC150 (lub najwyższym ujemnym stężeniem niecytotoksycznym CD86) a CV70 (lub najwyższym dopuszczalnym stężeniem, tj. 200 μg/ml). W każdej analizie należy zbadać co najmniej 4 stężenia, przy czym w celu zapewnienia porównywalności co najmniej 2 stężenia powinny być takie same jak w poprzedniej analizie / poprzednich analizach.
|
|
15.
|
W ramach kontroli z rozpuszczalnikiem/nośnikiem w badaniu U-SENS™ stosuje się kompletne podłoże (w przypadku badanych substancji chemicznych rozpuszczonych lub tworzących stabilne dyspersje w kompletnym podłożu) (zob. pkt 4) lub sulfotlenek dimetylu w stężeniu 0,4 % w kompletnym podłożu (w przypadku badanych substancji chemicznych rozpuszczonych lub tworzących stabilne dyspersje w sulfotlenku dimetylu).
|
|
16.
|
W ramach kontroli dodatniej w badaniu U-SENS™ (zob. pkt 11) stosuje się TNBS, przygotowany w kompletnym podłożu. TNBS należy stosować jako kontrolę dodatnią na potrzeby pomiaru ekspresji CD86 w końcowym pojedynczym stężeniu na płytce (50 μg/ml) pozwalającym uzyskać żywotność komórek na poziomie > 70 %. W celu uzyskania na płytce stężenia TNBS wynoszącego 50 μg/ml przygotowuje się 1-molowy (tj. o stężeniu 293 mg/ml) roztwór podstawowy TNBS w kompletnym podłożu, który następnie rozcieńcza się 2 930-krotnie w kompletnym podłożu do uzyskania roztworu roboczego o stężeniu 100 μg/ml. Kwas mlekowy (LA, nr CAS 50-21-5) powinien być stosowany jako kontrola ujemna w stężeniu 200 μg/ml, rozpuszczony w kompletnym podłożu (z roztworu podstawowego o stężeniu 0,4 mg/ml). W każdej płytce z każdej analizy przygotowuje się po trzy kontrpróby: kontroli niepoddanej działaniu substancji chemicznej z kompletnym podłożem, kontroli z rozpuszczalnikiem/nośnikiem oraz kontroli ujemnej i dodatniej (12). Można stosować inne odpowiednie kontrole dodatnie, jeżeli dostępne są dane historyczne pozwalające na uzyskanie porównywalnych kryteriów dopuszczalności analizy. Kryteria dopuszczalności analizy są takie same jak kryteria opisane w odniesieniu do badanej substancji chemicznej (zob. pkt 12).
|
Podawanie badanych substancji chemicznych i substancji kontrolnych
|
17.
|
Kontrolę z rozpuszczalnikiem/nośnikiem lub roztwory robocze opisane w pkt 14–16 miesza się w stosunku 1:1 (obj.) z zawiesinami komórkowymi przygotowanymi w dołkach 96-dołkowej płytki płaskodennej (zob. pkt 12). Następnie płytki poddane działaniu substancji inkubuje się przez 45 ± 3 godziny w temperaturze 37 °C w obecności 5 % CO2. Przed inkubacją płytki uszczelnia się półprzepuszczalną membraną, aby nie dopuścić do odparowania lotnych badanych substancji chemicznych i aby zapobiec zanieczyszczeniu krzyżowemu między komórkami poddanymi działaniu badanych substancji chemicznych (12).
|
Wybarwianie komórek
|
18.
|
Po narażeniu na działanie substancji chemicznej przez 45 godzin ± 3 godziny komórki przenosi się na mikropłytkę z dnem w kształcie litery V i zbiera się przez odwirowanie. Interferencję rozpuszczalności definiuje się jako kryształy lub krople obserwowane pod mikroskopem w po upływie 45 ± 3 godzin po poddaniu działaniu substancji (przed wybarwieniem komórek). Supernatanty odrzuca się, a pozostałe komórki przemywa się raz w 100 μl silnie schłodzonego roztworu chlorku sodu buforowanego fosforanami (PBS) zawierającego 5 % cielęcej surowicy płodowej (roztwór buforowy do wybarwiania). Po odwirowaniu komórki ponownie zawiesza się w 100 μl roztworu buforowego do wybarwiania i wybarwia się za pomocą 5 μl (np. 0,25 μg) przeciwciał anty-CD86 lub mysich przeciwciał IgG1 (izotyp) znakowanych FITC w temperaturze 4 °C przez 30 minut, chronionych przed światłem. Należy wykorzystać przeciwciała opisane w protokole nr 183 DB-ALM dotyczącym U-SENS™ (12) (w odniesieniu do CD86: BD-PharMingen #5556 57 klon: Fun-1 lub Caltag/Invitrogen # MHCD8601 klon: BU63; oraz w odniesieniu do IgG1: BD-PharMingen #5557 48 lub Caltag/Invitrogen # GM4992). Z doświadczenia naukowców opracowujących badanie wynika, że intensywność fluorescencji przeciwciał jest zwykle stała w różnych partiach. Do testu można wykorzystać inne klony lub przeciwciała od innego dostawcy, które pomyślnie przeszły kontrolę reaktywności (zob. pkt 11). Użytkownicy mogą jednak rozważyć miareczkowanie przeciwciał w warunkach własnego laboratorium, aby określić stężenie, które jest najbardziej stosowne dla danego zastosowania. Można wykorzystać inny system detekcji, np. przeciwciała anty-CD86 znakowane fluorochromem, jeżeli można wykazać, że pozwala on uzyskać wyniki podobne do wyników otrzymywanych w przypadku wykorzystania przeciwciał skoniugowanych z FITC, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 2.2. Po dwukrotnym przemyciu w 100 μl roztworu buforowego do wybarwiania i jednokrotnym przemyciu w 100 μl silnie schłodzonego PBS komórki zawiesza się ponownie w silnie schłodzonym PBS (np. w 125 μl w przypadku próbek analizowanych ręcznie probówka po probówce lub w 50 μl przy użyciu płytki do autosamplera) i dodaje się roztwór jodku propidyny (stężenie końcowe wynoszące 3 μg/ml). Można wykorzystać inne markery cytotoksyczności, takie jak 7-aminoaktynomycyna D (7-AAD) lub błękit trypanu, jeżeli można wykazać, że alternatywne barwniki pozwalają uzyskać wyniki podobne do wyników otrzymywanych w przypadku wykorzystania jodku propidyny, np. poprzez badanie substancji służących do wykazania biegłości wymienionych w dodatku 2.2.
|
Analiza przy użyciu cytometrii przepływowej
|
19.
|
Poziom ekspresji CD86 i żywotność komórek analizuje się przy pomocy cytometrii przepływowej. Komórki przedstawia się na wykresie punktowym uwzględniającym parametry wielkości (FSC) i ziarnistości (SSC) w skali logarytmicznej, aby wyraźnie określić populację w pierwszej bramce R1 i wyeliminować pozostałości komórkowe. Na każdy dołek w bramce R1 pozyskuje się docelowo łącznie 10 000 komórek. Komórki z tej samej bramki R1 przedstawia się na wykresie punktowym uwzględniającym parametry FL3 lub FL4 / SSC. Komórki żywotne wyznacza się poprzez umieszczenie drugiej bramki R2, która pozwala wyodrębnić populację komórek niewybarwionych jodkiem propidyny (kanał FL3 lub FL4). Żywotność komórek można obliczyć za pomocą poniższego równania, korzystając z programu do analizy cytometrycznej. Przy niskiej żywotności komórek można pozyskać do 20 000 komórek, wśród których znajdują się martwe komórki. Ewentualnie dane można pozyskać w ciągu jednej minuty od rozpoczęcia analizy.

Następnie mierzy się odsetek tych żywych komórek FL1-dodatnich w bramce R2 (wewnątrz bramki R1). Ekspresję CD86 na powierzchni komórek analizuje się na wykresie punktowym w układzie współrzędnych FL1 / SSC w bramce zawierającej komórki żywotne (R2).
W przypadku dołków zawierających kompletne podłoże / IgG1 znacznik analizy ustawia się blisko głównej populacji, tak aby w kontrolach z kompletnym podłożem przeciwciała IgG1 znajdowały się w strefie docelowej wynoszącej 0,6–0,9 %.
Interferencję barw definiuje się jako przesunięcie wykresu punktowego IgG1 znakowanego FITC (średnia geometryczna wskaźnika symulacji IgG1 FL1 ≥150 %).
Indeks stymulacji (S.I.) CD86 komórek kontrolnych (niepoddanych działaniu substancji lub zawieszonych w sulfotlenku dimetylu o stężeniu 0,4 %) i komórek poddanych działaniu substancji chemicznej oblicza się według następującego równania:.

% komórek IgG1+ w kontroli niepoddanej działaniu substancji chemicznej: oznacza odsetek komórek IgG1 FL1-dodatnich wyznaczonych za pomocą znacznika analizy (dopuszczalny zakres: ≥ 0,6 % i < 1,5 %, zob. pkt 22) w komórkach żywotnych niepoddanych działaniu substancji chemicznej.
% komórek IgG1+/CD86+ kontrolnych / poddanych działaniu substancji chemicznej: oznacza odsetek komórek IgG1/CD86 FL1-dodatnich, mierzony bez przesuwania znacznika analizy, w żywych komórkach kontrolnych / poddanych działaniu substancji chemicznej.
|
DANE I SPRAWOZDAWCZOŚĆ
Ocena danych
|
20.
|
W ramach badania U-SENS™ oblicza się następujące parametry: wartość CV70, tj. stężenie, przy którym przeżywalność komórek linii U937 wynosi 70 % (cytotoksyczność na poziomie 30 %), i wartość EC150, tj. stężenie, przy którym badane substancje chemiczne wywołały indeks stymulacji CD86 (S.I.) wynoszący 150 %.
Do obliczenia wartości CV70 stosuje się interpolację logarytmiczno-liniową według następującego równania:
CV70 = C1 + [(V1 - 70) / (V1 - V2) * (C2 - C1)]
gdzie:
V1 oznacza minimalną wartość żywotności komórek powyżej 70 %;
V2 oznacza maksymalną wartość żywotności komórek powyżej 70 %;
C1 i C2 oznaczają stężenia wskazujące odpowiednio wartość żywotności komórek V1 i V2.

Można stosować inne podejścia do określania wartości CV70 pod warunkiem wykazania braku wpływu na wyniki (np. poprzez badanie substancji służących do wykazania biegłości).
Do obliczenia wartości EC150 stosuje się interpolację logarytmiczno-liniową według następującego równania:
EC150 = C1 + [(150 - S.I.1) / (S.I.2 - S.I.1) * (C2 - C1)]
gdzie:
C1 oznacza najwyższe stężenie wyrażone w μg/ml przy wskaźniku stymulacji CD86 wynoszącym < 150 % (S.I. 1);
C2 oznacza najniższe stężenie wyrażone w μg/ml przy wskaźniku stymulacji CD86 wynoszącym ≥150 % (S.I. 2).
Wartości EC150 i CV70 oblicza się
|
—
|
na potrzeby każdej analizy: poszczególne wartości EC150 i CV70 wykorzystuje się jako narzędzia do badania wpływu wzrostu ekspresji CD86 na zależność stężenie-odpowiedź (zob. pkt 14);
|
|
—
|
na podstawie średniej żywotności określa się ogólną wartość CV70 (12);
|
|
—
|
na podstawie średnich wartości wskaźnika stymulacji CD86 określa się ogólną wartość EC150 dla badanej substancji chemicznej, w odniesieniu do której przewiduje się uzyskanie wyniku DODATNIEGO w badaniu U-SENS™ (zob. pkt 21) (12).
|
|
Model prognozowania
|
21.
|
Na potrzeby pomiaru ekspresji CD86 każdą badaną substancję chemiczną bada się w co najmniej czterech stężeniach i w co najmniej dwóch niezależnych analizach (wykonanych innego dnia) w celu uzyskania pojedynczej prognozy (UJEMNEJ lub DODATNIEJ).
|
—
|
Pojedynczy wynik analizy przeprowadzonej w ramach badania U-SENS™ uznaje się za ujemny (zwany dalej „U”), jeżeli wskaźnik stymulacji CD86 wynosi mniej niż 150 % we wszystkich stężeniach niecytotoksycznych (żywotność komórek ≥70 %) i jeżeli nie zaobserwowano żadnych zakłóceń (cytotoksyczność, rozpuszczalność: zob. pkt 18 lub barwa: zob. pkt 19 niezależnie od wartości stężeń niecytotoksycznych, przy których wykryto zakłócenia). We wszystkich innych przypadkach: gdy zaobserwowano wskaźnik stymulacji CD86 większy lub równy 150 % lub zakłócenia, pojedynczy wynik analizy U-SENS™ uznaje się za dodatni (zwany dalej „D”).
|
|
—
|
Prognozę badania U-SENS™ uważa się za UJEMNĄ, jeżeli wyniki co najmniej dwóch niezależnych analiz są ujemne (U) (rys.1). Jeżeli obie pierwsze analizy dały wyniki ujemne (U), prognozę badania U-SENS™ uznaje się na UJEMNĄ i nie ma potrzeby przeprowadzania trzeciej analizy.
|
|
—
|
Prognozę U-SENS ™ uważa się za DODATNIĄ, jeżeli wyniki co najmniej dwóch niezależnych analiz są dodatnie (D) (rys.1). Jeżeli obie pierwsze analizy dały wyniki dodatnie (D), prognozę badania U-SENS™ uznaje się na DODATNIĄ i nie ma potrzeby przeprowadzania trzeciej analizy.
|
|
—
|
Z uwagi na nieprzeprowadzanie testu ustalającego dawkę wyjątek stanowi sytuacja, w której w pierwszej analizie wskaźnik stymulacji CD86 jest wyższy lub równy 150 % jedynie przy najwyższym stężeniu niecytotoksycznym. Analizę uznaje się wówczas za NIEJEDNOZNACZNĄ (N), a dodatkowe stężenia (pomiędzy najwyższym stężeniem niecytotoksycznym a najniższym stężeniem cytotoksycznym – zob. pkt 20) należy zbadać w ramach dodatkowych analiz. Jeżeli wynik analizy uznano za niejednoznaczny, należy przeprowadzić co najmniej 2 dodatkowe analizy, a czwartą analizę należy przeprowadzić w przypadku, gdy analizy druga i trzecia nie są ze sobą zbieżne (tj. gdy niezależnie uzyskano wyniki U lub D) (rys. 1). Wyniki dalszych analiz zostaną uznane za dodatnie nawet wówczas, gdy tylko jedno stężenie niecytotoksyczne CD86 pozwala uzyskać wynik równy lub wyższy niż 150 %, ponieważ ustalanie stężeń dostosowano do konkretnej badanej substancji chemicznej. O prognozie końcowej przesądzi większość wyników trzech lub czterech osobnych analiz (tj. dwie z trzech lub dwie z czterech) (rys. 1).
|
Rys. 1: Model prognozowania wykorzystywany w badaniu U-SENS™. Prognozę U-SENS™ należy rozpatrywać w ramach IATA oraz zgodnie z przepisami pkt 4 i przepisami pkt 7, 8 i 9 wprowadzenia ogólnego.
U: analiza, w której nie zaobserwowano wyniku dodatniego w odniesieniu do CD86 ani żadnych zakłóceń;
D: analiza, w której zaobserwowano wynik dodatni w odniesieniu do CD86 lub zakłócenia;
N: wynik niejednoznaczny. Pierwsza analiza, której wynik uznano za niejednoznaczny, gdy w odniesieniu do CD86 zaobserwowano wynik dodatni przy najwyższym stężeniu niecytotoksycznym;
#: pojedynczy wynik niejednoznaczny (N) przypisany tylko do pierwszej analizy automatycznie pociąga za sobą konieczność przeprowadzenia trzeciej analizy, aby uzyskać większość wyników dodatnich (D) lub ujemnych (U) w co najmniej dwóch z trzech niezależnych analiz;
$: w polach przedstawiono odpowiednie kombinacje wyników trzech analiz na podstawie wyników uzyskanych w dwóch pierwszych analizach przedstawionych w ramce powyżej;
o: w polach przedstawiono odpowiednie kombinacje wyników czterech analiz na podstawie wyników uzyskanych w trzech pierwszych analizach przedstawionych w ramce powyżej.
|
Kryteria dopuszczalności
|
22.
|
Podczas wykonywania badania U-SENS™ spełnione muszą zostać następujące kryteria dopuszczalności (12):
|
—
|
pod koniec 45 ± 3-godzinnego okresu narażenia na działanie substancji chemicznej średnia żywotność komórek linii U937 niepoddanych działaniu substancji chemicznej w trzech kontrpróbach musiała wynosić >90 % i nie dopuszcza się możliwości zaobserwowania zniesienia ekspresji CD86. Podstawowa ekspresja CD86 w komórkach linii U937 niepoddanych działaniu substancji chemicznej musiała zawierać się w zakresie od ≥ 2 % do ≤25 %;
|
|
—
|
w przypadku stosowania sulfotlenku dimetylu jako rozpuszczalnika zasadność przeprowadzenia kontroli z nośnikiem zawierającej sulfotlenek dimetylu ocenia się, obliczając wartość wskaźnika stymulacji sulfotlenku dimetylu w porównaniu z komórkami niepoddanymi działaniu substancji chemicznej, a średnia żywotność komórek w trzech kontrpróbach musi wynosić >90 %. kontrola z nośnikiem zawierająca sulfotlenek dimetylu jest właściwa, jeżeli średnia wartość wskaźnika stymulacji CD86 w trzech kontrpróbach tej kontroli była mniejsza niż 250 % średniej wartości wskaźnika stymulacji CD86 komórek linii U937 niepoddanych działaniu substancji chemicznej w trzech kontrpróbach;
|
|
—
|
analizy uznaje się za ważne, jeżeli co najmniej dwie z trzech wartości IgG1 komórek linii U937 niepoddanych działaniu substancji chemicznej mieszczą się w przedziale od ≥0,6 % do <1,5 %;
|
|
—
|
równolegle badaną kontrolę ujemną (kwas mlekowy) uważa się za ważną, jeżeli w co najmniej dwóch z trzech kontrprób uzyskano wyniki ujemne (wskaźnik stymulacji CD86 <150 %) i nie odnotowano cytotoksyczności (żywotność komórek ≥70 %);
|
|
—
|
kontrolę dodatnią (TNBS) uznano za ważną, jeżeli w co najmniej dwóch z trzech kontrprób uzyskano wyniki dodatnie (wskaźnik stymulacji CD86 ≥150 %) i nie odnotowano cytotoksyczności (żywotność komórek ≥70 %).
|
|
Sprawozdanie z badania
|
23.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
Badana substancja chemiczna
Substancja jednoskładnikowa
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, rozpuszczalność w kompletnym podłożu, rozpuszczalność w sulfotlenku dimetylu, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
Substancja wieloskładnikowa, UVCB i mieszanina:
|
opisana w miarę możliwości np. poprzez podanie nazwy chemicznej (zob. powyżej), czystości, przedstawienie informacji dotyczących występowania ilościowego oraz wskazanie istotnych właściwości fizykochemicznych składników (zob. powyżej), w dostępnym zakresie;
|
|
wygląd fizyczny, rozpuszczalność w kompletnym podłożu, rozpuszczalność w sulfotlenku dimetylu i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
masa cząsteczkowa lub pozorna masa cząsteczkowa w przypadku mieszanin lub polimerów o znanym składzie lub inne informacje istotne dla przebiegu badania;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
Kontrole
Kontrola dodatnia
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, rozpuszczalność w sulfotlenku dimetylu, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie i w stosownych przypadkach;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
odniesienie do wyników historycznych kontroli dodatnich wykazujących odpowiednie kryteria dopuszczalności analizy, w stosownych przypadkach.
|
Kontrola ujemna oraz kontrola z rozpuszczalnikiem/nośnikiem
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
wygląd fizyczny, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne w przypadku stosowania innego rozpuszczalnika/nośnika kontrolnego niż te opisane w wytycznej dotyczącej badań, w dostępnym zakresie;
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
Warunki badania
|
nazwa i adres sponsora i placówki przeprowadzającej badanie oraz dane kierownika badań;
|
|
opis zastosowanego badania;
|
|
zastosowana linia komórkowa, warunki jej przechowywania oraz źródło (np. placówka, z której ją otrzymano);
|
|
zastosowana cytometria przepływowa (np. model), w tym informacje na temat zastosowanych ustawień przyrządu, przeciwciał i markera cytotoksyczności;
|
|
procedura stosowana w celu wykazania biegłości laboratorium w zakresie przeprowadzania badania poprzez badanie substancji służących do wykazania biegłości lub procedura stosowana do wykazania odtwarzalności badania w czasie, np. dane historyczne dotyczące kontroli lub dane historyczne dotyczące kontroli reaktywności.
|
|
|
Kryteria dopuszczalności badania
|
wartości dotyczące żywotności komórek oraz wskaźnika stymulacji CD86 uzyskane za pomocą kontroli z rozpuszczalnikiem/nośnikiem w porównaniu z zakresami dopuszczalności;
|
|
wartości dotyczące żywotności komórek oraz wskaźnika stymulacji uzyskane za pomocą kontroli dodatniej w porównaniu z zakresami dopuszczalności;
|
|
żywotność komórek przy wszystkich badanych stężeniach badanej substancji chemicznej.
|
|
|
Procedura badawcza
|
liczba zastosowanych analiz;
|
|
stosowane stężenia badanej substancji chemicznej, procedura podawania badanej substancji chemicznej i czas narażenia (jeżeli są one inne niż zalecane);
|
|
czas narażenia na działanie substancji chemicznej;
|
|
opis zastosowanych kryteriów oceny i podejmowania decyzji;
|
|
opis wszelkich modyfikacji procedury badawczej.
|
|
|
Wyniki
|
tabelaryczne zestawienie danych, w tym wartości CV70 (w stosownych przypadkach), wskaźnika stymulacji, wartości żywotności komórek, wartości EC150 (w stosownych przypadkach), uzyskanych w odniesieniu do badanej substancji chemicznej oraz kontroli dodatniej w każdej analizie oraz wskazanie oceny badanej substancji chemicznej według modelu prognozowania;
|
|
w stosownych przypadkach opis wszelkich innych istotnych obserwacji.
|
|
|
Omówienie wyników
|
omówienie wyników uzyskanych za pomocą badania U-SENS™;
|
|
uwzględnienie wyników zastosowania badania w kontekście IATA, jeżeli są dostępne inne odpowiednie informacje.
|
|
Wnioski
|
BIBLIOGRAFIA
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM UE (2017). Sprawozdanie z badania walidacyjnego metody badawczej U-SENS™. Dostępne na stronie internetowej: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Dostępne na stronie internetowej: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Dostępne na stronie internetowej: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373–382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259–270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683–1 696.
|
|
(11)
|
OECD. (2018). Projekt wytycznych: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf
|
|
(12)
|
DB-ALM (2016). (protokół nr 183) Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Dostępne na stronie internetowej: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565–577.
|
|
(14)
|
OECD (2005). Seria dotycząca badań i oceny nr 34. Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(15)
|
Organizacja Narodów Zjednoczonych (ONZ) (2015). Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS). ST/SG/AC.10/30/Rev.6, wydanie szóste zmienione, Nowy Jork i Genewa: publikacje Organizacji Narodów Zjednoczonych. Dostępne na stronie internetowej: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf
|
|
(16)
|
OECD (2012). Seria dotycząca badań i oceny nr 168. The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(17)
|
ECETOC (2003). Sprawozdanie techniczne nr 87: Contact sensitization: Classification according to potency. Europejskie Centrum ds. Ekotoksykologii i Toksykologii Substancji Chemicznych, Bruksela. Dostępne na stronie internetowej: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Dodatek 2.1
DEFINICJE
Dokładność
: stopień zgodności pomiędzy wynikami badania a przyjętymi wartościami odniesienia. Jest to miara efektywności badania i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na oznaczenie odsetka prawidłowych wyników uzyskiwanych przy przeprowadzeniu badania (14).
Mechanizm wywoływania skutków szkodliwych
: sekwencja zdarzeń, począwszy od struktury chemicznej docelowej substancji chemicznej lub grupy podobnych substancji chemicznych poprzez molekularne zdarzenie inicjujące aż do pożądanego wyniku in vivo (15).
Zależność stężenie-odpowiedź w odniesieniu do CD86
: zależność od stężenia (lub zależność stężenie-odpowiedź) występuje wówczas, gdy po uzyskaniu stężenia dodatniego (wskaźnik stymulacji CD86 ≥150) następuje stężenie charakteryzujące się wzrastającą wartością wskaźnika stymulacji CD86.
Substancja chemiczna
: substancja lub mieszanina.
CV70
: szacowane stężenie, przy którym żywotność komórek wynosi 70 %.
Zniesienie
: zniesienie ma miejsce, gdy (i) skorygowana wartość odsetka komórek CD86+ w kontrpróbie stanowiącej próbę kontrolną niepoddawaną działaniu substancji nr 3 jest mniejsza niż 50 % średniej skorygowanej wartości odsetka komórek CD86+ w kontrpróbach stanowiących próbę kontrolną niepoddawaną działaniu substancji nr 1 i 2; oraz (ii) skorygowana wartość odsetka komórek CD86+ w kontrpróbie stanowiącej kontrolę ujemną nr 3 jest mniejsza niż 50 % średniej skorygowanej wartości odsetka komórek CD86+ w kontrpróbach stanowiących kontrolę ujemną nr 1 i 2.
EC150
: szacowane stężenia, przy których wskaźnik stymulacji ekspresji CD86 wynosi 150 %.
Cytometria przepływowa
: technika cytometryczna, w której komórki zawieszone w cieczy przepływają jedna po drugiej przez zogniskowaną wiązkę wzbudzającego światła, które rozprasza się według wzorców charakterystycznych dla komórek i ich składników; komórki często oznacza się znacznikami fluorescencyjnymi, dzięki czemu światło jest najpierw absorbowane, a następnie emitowane w zmienionych częstotliwościach.
Zagrożenie
: nieodłączna właściwość czynnika lub sytuacja, która może potencjalnie doprowadzić do niekorzystnych skutków w przypadku narażenia organizmu, systemu lub (sub)populacji na taki czynnik.
IATA (zintegrowane podejście do badań i oceny)
: usystematyzowane podejście stosowane w odniesieniu do identyfikacji zagrożeń (potencjał), charakterystyki zagrożeń (siła oddziaływania) lub oceny bezpieczeństwa (potencjał / siła oddziaływania oraz narażenie) substancji chemicznej lub grupy substancji chemicznych; w ramach tego podejścia w sposób strategiczny integruje się i waży wszystkie istotne dane, które mogą wpłynąć na podjęcie decyzji regulacyjnej dotyczącej potencjalnego zagrożenia, ryzyka lub konieczności podjęcia dalszych ukierunkowanych, a przez to minimalnych badań.
Mieszanina
: mieszanina lub roztwór, które składają się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
Substancja wieloskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥10 % (w/w) i < 80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się poprzez zmieszanie przynajmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
Kontrola dodatnia
: kontrpróba zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że wywołuje reakcję dodatnią. Aby zapewnić możliwość oceny zmienności reakcji w kontroli dodatniej w czasie, stopień reakcji dodatniej nie powinien być nadmierny.
Prehapteny
: substancje chemiczne, które stają się czynnikami uczulającymi po transformacji abiotycznej, np. poprzez utlenianie.
Prohapteny
: substancje chemiczne, które wymagają aktywacji enzymatycznej do uwolnienia swojego potencjalnego działania uczulającego na skórę.
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono znaczące i użyteczne z punktu widzenia określonego celu. Jest to zakres, w jakim badanie pozwala prawidłowo zmierzyć lub przewidzieć biologiczny oczekiwany skutek. Istotność obejmuje uwzględnienie dokładności (zgodności) badania (14).
Wiarygodność
: miary zakresu, w jakim badanie może zostać przeprowadzone w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jego przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją, obliczając odtwarzalność wewnątrz- i międzylaboratoryjną oraz powtarzalność wewnątrzlaboratoryjną (14).
Analiza
: analiza obejmuje jednoczesne badanie jednej badanej substancji chemicznej lub większej liczby takich substancji w kontroli z rozpuszczalnikiem/nośnikiem i kontroli dodatniej.
Czułość
: odsetek wszystkich substancji chemicznych dających wynik dodatni / aktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą danego badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (14).
S.I.
: wskaźnik stymulacji. Wartości względne średniej geometrycznej intensywności fluorescencji w komórkach poddanych działaniu substancji chemicznej w porównaniu z komórkami poddanymi działaniu rozpuszczalnika.
Kontrola z rozpuszczalnikiem/nośnikiem
: próbka niepoddana działaniu badanej substancji, zawierająca wszystkie składniki układu badawczego z wyjątkiem badanej substancji chemicznej, ale z uwzględnieniem stosowanego rozpuszczalnika/nośnika. Wykorzystywana jest do określenia wyjściowej reakcji dla próbek poddanych działaniu badanej substancji chemicznej rozpuszczonej lub stabilnie rozproszonej w tym samym rozpuszczalniku/nośniku. W przypadku badania z jednoczesną kontrolą podłoża próbka ta wykazuje również, czy rozpuszczalnik/nośnik wchodzi w reakcję z układem badawczym.
Swoistość
: odsetek wszystkich substancji chemicznych dających wynik ujemny / nieaktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (14).
Roztwór buforowy do wybarwiania
: roztwór chlorku sodu buforowany fosforanami zawierający 5 % cielęcej surowicy płodowej.
Substancja
: pierwiastek chemiczny i jego związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania jego trwałości i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem ewentualnych rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszego badania.
Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ)
: system, w ramach którego proponuje się klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz omawia się odpowiednie elementy komunikacyjne, takie jak piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki substancji i produktów niebezpiecznych, aby przekazać informacje na temat ich szkodliwego działania w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) i środowiska (16).
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
Ważne badanie
: badanie, które uznano za wystarczająco istotne i wiarygodne dla danego celu i które opiera się na uzasadnionych naukowo podstawach. Badanie nie może zostać uznane za ważne w sensie absolutnym, ale tylko w odniesieniu do określonego celu (14).
Dodatek 2.2
SUBSTANCJE SŁUŻĄCE DO WYKAZANIA BIEGŁOŚCI
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo określając prognozę U-SENS™ dla 10 substancji wskazanych w tabeli 1 oraz określając wartości CV70 i EC150, które mieszczą się w odpowiednim zakresie odniesienia, dla co najmniej 8 z 10 substancji służących do wykazania biegłości. Substancje służące do wykazania biegłości wybrano w taki sposób, aby odzwierciedlić zakres reakcji na zagrożenia związane z działaniem uczulającym na skórę. Przy wyborze substancji kierowano się również następującymi kryteriami: dostępnością handlową tych substancji, dostępnością wysokiej jakości danych referencyjnych z badań in vivo oraz dostępnością wysokiej jakości danych in vitro uzyskanych w wyniku zastosowania badania U-SENS™. W odniesieniu do badania U-SENS™ można znaleźć również opublikowane dane referencyjne (1)(8).
Tabela 1
Substancje zalecane do wykazania biegłości technicznej w odniesieniu do badania U-SENS™
|
Substancje służące do wykazania biegłości
|
Numer CAS
|
Stan skupienia
|
Prognoza in vivo
(105)
|
U-SENS Rozpuszczalnik/nośnik
|
U-SENS CV70 odniesienia CV70 w μg/ml (106)
|
U-SENS Zakres odniesienia EC150 w μg/ml (106)
|
|
4-fenylenodiamina
|
106-50-3
|
Stały
|
Czynnik uczulający (silny)
|
Kompletne podłoże (107)
|
<30
|
Dodatni (≤10)
|
|
Kwas pikrylosulfonowy
|
2508-19-2
|
Ciekły
|
Czynnik uczulający (silny)
|
Kompletne podłoże
|
>50
|
Dodatni (≤50)
|
|
Maleinian dietylu
|
141-05-9
|
Ciekły
|
Czynnik uczulający (w stopniu umiarkowanym)
|
sulfotlenek dimetylu
|
10-100
|
Dodatni (≤20)
|
|
Rezorcynol
|
108-46-3
|
Stały
|
Czynnik uczulający (w stopniu umiarkowanym)
|
Kompletne podłoże
|
>100
|
Dodatni (≤50)
|
|
Alkohol cynamonowy
|
104-54-1
|
Stały
|
Czynnik uczulający (słaby)
|
sulfotlenek dimetylu
|
>100
|
Dodatni (10–100)
|
|
4-allilamizol
|
140-67-0
|
Ciekły
|
Czynnik uczulający (słaby)
|
sulfotlenek dimetylu
|
>100
|
Dodatni (<200)
|
|
Sacharyna
|
81-07-2
|
Stały
|
Brak działania uczulającego
|
sulfotlenek dimetylu
|
>200
|
Ujemny (>200)
|
|
Glicerol
|
56-81-5
|
Ciekły
|
Brak działania uczulającego
|
Kompletne podłoże
|
>200
|
Ujemny (>200)
|
|
Kwas mlekowy
|
50-21-5
|
Ciekły
|
Brak działania uczulającego
|
Kompletne podłoże
|
>200
|
Ujemny (>200)
|
|
Kwas salicylowy
|
69-72-7
|
Stały
|
Brak działania uczulającego
|
sulfotlenek dimetylu
|
>200
|
Ujemny (>200)
|
|
Skróty: Nr CAS = numer w rejestrze Chemical Abstracts Service.
|
Dodatek 3
BADANIE DZIAŁANIA UCZULAJĄCEGO NA SKÓRĘ IN VITRO: TEST IL-8 LUC
ZAŁOŻENIA WSTĘPNE I OGRANICZENIA
|
1.
|
W przeciwieństwie do analiz, które mają na celu zbadanie ekspresji markerów powierzchniowych komórki, test IL-8-Luc pozwala określić ilościowo zmiany ekspresji IL-8 – cytokiny związanej z aktywacją komórek dendrytycznych. Ekspresję IL-8 w linii komórek reporterowych IL-8 wywodzącej się z THP-1 (THP-G8, ustanowionej z ludzkiej linii komórkowej ostrej białaczki monocytarnej THP-1) mierzy się po narażeniu na działanie czynników uczulających (1). Następnie ekspresję lucyferazy wykorzystuje się do ułatwienia rozróżnienia substancji działających uczulająco na skórę i substancji niewykazujących takiego działania.
|
|
2.
|
Test IL-8 Luc oceniono w ramach badania walidacyjnego (2) przeprowadzonego przez Japońskie Centrum Walidacji Metod Alternatywnych (JaCVAM), Ministerstwo Gospodarki, Handlu i Przemysłu (METI) i Japońskie Towarzystwo ds. Metod Alternatywnych wobec Eksperymentów na Zwierzętach (JSAAE), a następnie poddano niezależnej wzajemnej ocenie (3) pod auspicjami JaCVAM i Ministerstwa Zdrowia, Pracy i Opieki Społecznej (MHLW) przy wsparciu międzynarodowej współpracy w zakresie alternatywnych metod badań (ICATM). Biorąc pod uwagę wszystkie dostępne dowody oraz wkład ze strony organów regulacyjnych i zainteresowanych stron, można stwierdzić, że test IL-8 Luc jest przydatnym narzędziem w ramach IATA, pozwalającym rozróżnić substancje działające uczulająco na skórę i substancje niewykazujące takiego działania na potrzeby klasyfikacji i oznakowania pod względem zagrożeń. Przykłady wykorzystania danych z testu IL-8 Luc w połączeniu z innymi informacjami są opisane w literaturze (4)(5)(6).
|
|
3.
|
Udowodniono, że test IL-8 Luc można przeprowadzać również w laboratoriach posiadających doświadczenie w zakresie kultury komórkowej i pomiaru lucyferazy. Odtwarzalność wyników w badaniu porównawczym wewnątrz- i międzylaboratoryjnym wynosiła odpowiednio 87,7 % i 87,5 % (2). Z danych uzyskanych w badaniu walidacyjnym (2) i innych opublikowanych pracach (1)(6) wynika, że – w porównaniu z LLNA – w teście IL-8 Luc wynik dodatni lub ujemny przypisano 118 spośród 143 substancji chemicznych, a 25 substancjom chemicznym przypisano wynik niejednoznaczny, przy czym dokładność testu IL-8 Luc w rozróżnianiu substancji działających uczulająco na skórę (należących do kategorii 1 wg GHS ONZ / rozporządzenia CLP) i substancji niewykazujących takiego działania (należących do kategorii wg GHS ONZ / rozporządzenia CLP) wynosi 86 % (101/118) przy czułości na poziomie 96 % (92/96) i swoistości na poziomie 41 % (9/22). Wyłączając substancje znajdujące się poza dziedziną zastosowania opisaną poniżej (pkt 5), w teście IL-8 Luc wynik dodatni lub ujemny przypisano 113 spośród 136 substancji chemicznych, a 23 substancjom chemicznym przypisano wynik niejednoznaczny, przy czym dokładność testu IL-8 Luc wynosi 89 % (101/113) przy czułości na poziomie 96 % (92/96) i swoistości na poziomie 53 % (9/17). Wykorzystując dane dotyczące ludzi przytoczone przez Urbisch et al. (7), w teście IL-8 Luc wynik dodatni lub ujemny przypisano 76 spośród 90 substancji chemicznych, a 14 substancjom chemicznym przypisano wynik niejednoznaczny, przy czym dokładność testu wynosi 80 % (61/76), czułość – 93 % (54/58), a swoistość – 39 % (7/18). Wyłączając substancje znajdujące się poza dziedziną zastosowania, w teście IL-8 Luc wynik dodatni lub ujemny przypisano 71 spośród 84 substancji chemicznych, a 13 substancjom chemicznym przypisano wynik niejednoznaczny, przy czym dokładność testu wynosi 86 % (61/71) przy czułości na poziomie 93 % (54/58) i swoistości na poziomie 54 % (7/13). Prawdopodobieństwo wystąpienia prognoz fałszywie ujemnych przy zastosowaniu testu IL-8 Luc jest większe w przypadku substancji chemicznych wykazujących niską/średnią siłę działania uczulającego na skórę (tj. należących do podkategorii 1B wg GHS ONZ / rozporządzenia CLP) niż w przypadku substancji wykazujących wysoką siłę działania uczulającego (tj. należących do podkategorii 1A wg GHS ONZ / rozporządzenia CLP) (6). Łącznie informacje te łącznie wspierają rolę, jaką test IL-8 Luc odgrywa w identyfikacji zagrożeń związanych z działaniem uczulającym na skórę. Dokładność testu IL-8 Luc podana jako dokładność samodzielnego badania ma wyłącznie charakter orientacyjny, ponieważ test należy rozpatrywać w połączeniu z innymi źródłami informacji w kontekście IATA oraz zgodnie z przepisami pkt 7 i 8 wprowadzenia ogólnego. Co więcej, przy ocenie badań niewymagających wykorzystania zwierząt pod kątem działania uczulającego na skórę należy pamiętać, że LLNA, jak również inne badania przeprowadzane na zwierzętach, mogą nie w pełni odzwierciedlać sytuację w odniesieniu do ludzi.
|
|
4.
|
Na podstawie obecnie dostępnych danych wykazano, że test IL-8 Luc ma zastosowanie do badanych substancji chemicznych obejmujących szereg organicznych grup funkcyjnych, mechanizmów reagowania, sił działania uczulającego na skórę (jak określono w badaniach in vivo) oraz właściwości fizykochemicznych (2)(6).
|
|
5.
|
Mimo że w teście IL-8 Luc jako rozpuszczalnik wykorzystuje się X-VIVOTM 15, przy użyciu tego testu poprawnie oceniono substancje chemiczne, których log Kow wynosi > 3,5, oraz te, których rozpuszczalność w wodzie wynosi około 100 μg/ml, zgodnie z obliczeniami przy użyciu EPI SuiteTM, a jego skuteczność w wykrywaniu substancji działających uczulająco słabo rozpuszczalnych w wodzie jest lepsza niż w przypadku testu IL-8 Luc, w którym jako rozpuszczalnik wykorzystuje się sulfotlenek dimetylu (2). Wyniki ujemne uzyskane w odniesieniu do badanych substancji chemicznych, które nie rozpuszczają się w stężeniu 20 mg/ml, mogą jednak dawać wyniki fałszywie ujemne, ponieważ substancje te nie rozpuszczają się w X-VIVOTM 15. Dlatego też nie należy brać pod uwagę wyników ujemnych uzyskanych w odniesieniu do tych substancji chemicznych. Wysoki odsetek wyników fałszywie ujemnych w przypadku bezwodników zaobserwowano w badaniu walidacyjnym. Ponadto z powodu ograniczonej wydolności metabolicznej linii komórkowej (8) i warunków doświadczalnych prohapteny (substancje wymagające aktywacji metabolicznej) i prehapteny (substancje aktywowane przez utlenianie przy użyciu powietrza) mogą dawać w przedmiotowym teście wyniki ujemne. Chociaż wyniki ujemne uzyskane w odniesieniu do substancji, które mogą być pre-/prohaptenami, należy jednak interpretować z pewną dozą ostrożności, w teście IL-8 Luc prawidłowo oceniono jednak 11 spośród 11 prehaptenów, 6/6 prohaptenów i 6/8 pre-/prohaptenów znajdujących się w zestawie danych testu IL-8 Luc (2). Na podstawie najnowszego kompleksowego przeglądu trzech badań niewymagających wykorzystania zwierząt (DPRA, KeratinoSens™ i h-CLAT) przeprowadzonych w celu wykrycia pre- i prohaptenów (9) oraz w oparciu o fakt, że komórki THP-G8 wykorzystywane w teście IL-8 Luc są linią komórkową wywodzącą się z THP-1, którą wykorzystuje się w badaniu h-CLAT, można stwierdzić, że test IL-8 Luc – w połączeniu z innymi badaniami – może również przyczynić się do zwiększenia czułości badań niewymagających wykorzystania zwierząt pod względem wykrywania pre- i prohaptenów. Dotychczas badane środki powierzchniowo czynne dały wyniki (fałszywie) dodatnie niezależnie od ich rodzaju (np. kationowe, anionowe lub niejonowe). Ponadto substancje chemiczne zakłócające działanie lucyferazy mogą zakłócać jej aktywność/pomiar, powodując inhibicję pozorną lub zwiększoną luminescencję (10). Na przykład z dostępnych informacji wynika, że stężenia fitoestrogenu wyższe niż 1 μM zakłócają sygnały luminescencyjne w innych badaniach genów reporterowych opartych na lucyferazie z powodu nadaktywacji lucyferazowego genu reporterowego. W rezultacie należy ostrożnie badać ekspresję lucyferazy uzyskaną przy wysokich stężeniach fitoestrogenów lub związków, co do których przypuszcza się, że wywołują one – podobnie jak fitoestrogeny – aktywację lucyferazowego genu reporterowego (11). W związku z powyższym środki powierzchniowo czynne, bezwodniki oraz substancje chemiczne zakłócające funkcjonowanie działania lucyferazy znajdują się poza dziedziną zastosowania niniejszego testu. W przypadkach, w których można przedstawić dowody na brak możliwości zastosowania testu IL-8 Luc w odniesieniu do innych konkretnych kategorii badanych substancji chemicznych, testu nie należy stosować w odniesieniu do tych konkretnych kategorii.
|
|
6.
|
Jak opisano powyżej, test IL-8 Luc wspiera rozróżnienie substancji działających uczulająco na skórę i substancji niewykazujących takiego działania. Wymagane jest jednak przeprowadzenie dalszych prac, najlepiej na podstawie danych dotyczących ludzi, aby ustalić, w jaki sposób wyniki IL-8 Luc mogą potencjalnie wpłynąć na ocenę siły działania, jeżeli są rozpatrywane w połączeniu z innymi źródłami informacji.
|
|
7.
|
Definicje znajdują się w dodatku 3.1.
|
ZASADA BADANIA
|
8.
|
W teście IL-8 Luc wykorzystuje się ludzką linię komórkową białaczki monocytarnej THP-1, którą otrzymano ze zbioru kultur typu amerykańskiego (Manassas, Wirginia, Stany Zjednoczone). Wydział dermatologii szkoły medycznej Uniwersytetu Tokohu, wykorzystując tę linię komórkową, ustanowił linię komórek reporterowych IL-8 wywodzącą się z THP-1 – THP-G8, która zawiera geny stabilnej lucyferazy pomarańczowej (SLO) i stabilnej lucyferazy czerwonej (SLR), kontrolowane odpowiednio przez promotory IL-8 oraz dehydrogenazy aldehydu 3-fosfoglicerynowego (GAPDH) (1). Pozwala to na dokonanie ilościowego pomiaru indukcji genu lucyferazy przy wykryciu luminescencji z powszechnie stosowanych substratów lucyferazy generujących światło jako wskaźnika aktywności IL-8 i GAPDH w komórkach, które zostały narażone na działanie substancji działających uczulająco.
|
|
9.
|
System testu uwzględniającego dwie barwy zawiera lucyferazę emitującą światło pomarańczowe (SLO; λmaks. = 580 nm) (12) na potrzeby ekspresji genów promotora IL-8, a także lucyferazę emitującą światło czerwone (SLR; λmaks. = 630 nm) (13) na potrzeby ekspresji genów promotora kontroli wewnętrznej – GAPDH. Te dwie lucyferazy emitują różne barwy w reakcji z d-lucyferyną świetlika, a ich luminescencję mierzy się jednocześnie w reakcji jednoetapowej, wykorzystując filtr optyczny do oddzielenia emisji od mieszaniny testowej (14) (dodatek 3.2).
|
|
10.
|
Komórki THP-G8 poddaje się działaniu substancji chemicznej przez 16 godzin, po czym mierzy się aktywność lucyferazy SLO (SLO-LA) odzwierciedlającą aktywność promotora IL-8 oraz aktywność lucyferazy SLR (SLR-LA) odzwierciedlającą aktywność promotora GAPDH. Aby ułatwić zrozumienie skrótów, SLO-LA i SLR-LA wyznaczono odpowiednio jako IL8LA i GAPLA. W tabeli 1 przedstawiono opis pojęć związanych z aktywnością lucyferazy w teście IL-8 Luc. Zmierzone wartości wykorzystuje się do obliczenia: znormalizowanej IL8LA (nIL8LA), która jest stosunkiem IL8LA do GAPLA; indukcji nIL8LA (Ind-IL8LA), która jest stosunkiem średnich arytmetycznych czterokrotnie mierzonych wartości nIL8LA komórek THP-G8 poddanych działaniu substancji chemicznej oraz wartości nIL8LA komórek THP-G8 niepoddanych działaniu substancji chemicznej; oraz inhibicji GAPLA (Inh-GAPLA), która jest stosunkiem średnich arytmetycznych czterokrotnie mierzonych wartości GAPLA komórek THP-G8 poddanych działaniu substancji chemicznej i wartości GAPLA komórek THP-G8 niepoddanych działaniu substancji chemicznej, a także jako wskaźnik cytotoksyczności.
Tabela 1
Opis pojęć związanych z aktywnością lucyferazy w teście IL-8 Luc
|
Skrót
|
Definicja
|
|
GAPLA
|
Aktywność lucyferazy SLR odzwierciedlająca aktywność promotora GAPDH
|
|
IL8LA
|
Aktywność lucyferazy SLO odzwierciedlająca aktywność promotora IL-8
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
nIL8LA komórek THP-G8 poddanych działaniu substancji chemicznych / nIL8LA komórek niepoddanych działaniu substancji chemicznych
|
|
Inh-GAPLA
|
GAPLA komórek THP-G8 poddanych działaniu substancji chemicznych / GAPLA komórek niepoddanych działaniu substancji chemicznych
|
|
CV05
|
Najniższe stężenie substancji chemicznej, w którym Inh-GAPLA przyjmuje wartość < 0,05.
|
|
|
11.
|
Aby usprawnić proces walidacji zmienionych testów in vitro lucyferazy IL-8 podobnych do testu IL-8 Luc i aby umożliwić terminowe modyfikowanie wytycznej OECD nr 442E dotyczącej badań, można skorzystać z dostępnych standardów wykonywania badań (15). Wzajemne uznawanie danych OECD będzie możliwe wyłącznie w przypadku badań zweryfikowanych zgodnie ze standardem wykonywania badań, jeżeli badania te poddano przeglądowi i włączono do wytycznej OECD nr 442E dotyczącej badań (16).
|
WYKAZANIE BIEGŁOŚCI
|
12.
|
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną z zastosowaniem dziesięciu substancji służących do wykazania biegłości wymienionych w dodatku 3.3 zgodnie z dobrymi praktykami w zakresie metody in vitro (17). Ponadto użytkownicy badania powinni prowadzić bazę danych historycznych wygenerowanych podczas kontroli reaktywności (zob. pkt 15) oraz kontroli dodatnich i kontroli z rozpuszczalnikiem/nośnikiem (zob. pkt 21–24), a także wykorzystywać te dane do potwierdzenia zachowania odtwarzalności badania w swoim laboratorium w miarę upływu czasu.
|
PROCEDURA
|
13.
|
Standardowa procedura operacyjna dotycząca testu IL-8 Luc jest dostępna i należy z niej korzystać podczas przeprowadzania badania (18). Laboratoria, które chcą przeprowadzić badanie, mogą otrzymać rekombinowaną linię komórkową THP-G8 w laboratorium GPC Co. Ltd. w Tottori w Japonii, podpisując, w zgodzie z warunkami wzoru OECD, porozumienie o transferze materiału. W poniższych punktach opisano najistotniejsze elementy i procedury związane z testem.
|
Przygotowanie komórek
|
14.
|
Do przeprowadzenia testu IL-8 Luc (zob. pkt 8 i 13) należy wykorzystaćlinię komórkową THP-G8 pochodzącą z laboratorium GPC Co. Ltd. w Tottori w Japonii. Po odebraniu komórki rozmnaża się (2–4 pasaży) i przechowuje zamrożone jako jednorodny zbiór. Komórki pochodzące z tego zbioru można rozmnażać do osiągnięcia maksymalnie 12 pasaży lub przez maksimum 6 tygodni. RPMI-1640 jest podłożem wykorzystywanym do rozmnażania, zawierającym 10 % bydlęcej surowicy płodowej (FBS), roztwór antybiotyczny/przeciwgrzybiczy (100 U/ml penicyliny G, 100 μg/ml streptomycyny i 0,25 μg/ml amfoterycyny B w 0,85 % soli fizjologicznej) (np. GIBCO o numerze kat. 15240-062), 0,15 μg/ml puromycyny (np. nr CAS 58-58-2) oraz 300 μg/ml G418 (np. nr CAS 108321-42-2).
|
|
15.
|
Przed wykorzystaniem komórek do badań należy je zakwalifikować, przeprowadzając kontrolę reaktywności. Kontrolę tę należy przeprowadzić w terminie 1–2 tygodni lub 2–4 pasaży po rozmrożeniu, stosując kontrolę dodatnią, tj. bromek 4-nitrobenzylu (4-NBB) (nr CAS 100-11-8, czystość ≥ 99 %), oraz kontrolę ujemną, tj. kwas mlekowy (LA) (nr CAS 50-21-5, czystość ≥ 85 %). Bromek 4-nitrobenzylu powinien wywołać reakcję dodatnią na Ind-IL8LA (≥1,4), natomiast kwas mlekowy powinien wywołać reakcję ujemną na Ind-IL8LA (<1,4). W teście należy wykorzystać wyłącznie te komórki, które pomyślnie przeszły kontrolę reaktywności. Kontrolę reaktywności należy przeprowadzić zgodnie z procedurami opisanymi w pkt 22–24.
|
|
16.
|
Komórki THP-G8 wysiewa się do badań w gęstości 2–5 × 105 komórek/ml i dokonuje ich wstępnej hodowli w kolbach z kulturami przez 48–96 godzin. W dniu badania komórki pobrane z kolb z kulturami przemywa się w podłożu RPMI-1640 zawierającym 10 % FBS bez antybiotyków, a następnie zawiesza się je ponownie w podłożu RPMI-1640 o zawartości 10 % FBS bez antybiotyków w gęstości 1 × 106 komórek/ml. Następnie komórki umieszcza się w dołkach 96-dołkowej czarnej płytki płaskodennej (np. Costar o numerze kat. 3603) w objętości po 50 μl (5 × 104 komórek/ml).
|
Przygotowanie badanej substancji chemicznej i substancji kontrolnych
|
17.
|
Badaną substancję chemiczną oraz substancje kontrolne przygotowuje się w dniu badania. Na potrzeby testu IL-8 Luc badane substancje chemiczne rozpuszcza się w X-VIVOTM 15, dostępnym na rynku podłożu pozbawionym surowicy (Lonza, 04-418Q), do stężenia końcowego wynoszącego 20 mg/ml. W probówce do mikrowirówki do 20 mg badanej substancji chemicznej (bez względu na rozpuszczalność tej substancji chemicznej) dodaje się X-VIVOTM 15 aż do uzyskania objętości 1 ml, a następnie przez 30 min energicznie wiruje i wstrząsa na wirniku przy maksymalnej prędkości wynoszącej 8 obr./min w temperaturze otoczenia wynoszącej ok. 20 oC. W przypadku gdy stałe substancje chemiczne wciąż są nierozpuszczalne, probówkę dodatkowo poddaje się działaniu ultradźwięków do momentu całkowitego rozpuszczenia substancji chemicznej lub utworzenia jej stabilnej dyspersji. W przypadku badanych substancji chemicznych rozpuszczalnych w X-VIVOTM 15 roztwór rozcieńcza się pięciokrotnie za pomocą X-VIVOTM 15 i wykorzystuje jako roztwór podstawowy X-VIVOTM 15 badanej substancji chemicznej (4 mg/ml). W przypadku badanych substancji chemicznych nierozpuszczalnych w X-VIVOTM 15 mieszaninę ponownie wiruje się przez co najmniej 30 minut, a następnie odwirowuje z prędkością 15000 obr./min. (≈20000 g) przez 5 min; powstały supernatant wykorzystuje się jako roztwór podstawowy X-VIVOTM 15 badanej substancji chemicznej. W przypadku zastosowania innych rozpuszczalników, takich jak sulfotlenek dimetylu, woda lub podłoże, należy przedstawić uzasadnienie naukowe. W dodatku 3.5 szczegółowo opisano procedurę rozpuszczania substancji chemicznych. Roztwory X-VIVOTM 15 opisane w pkt 18–23 miesza się w stosunku 1:1 (obj.) z zawiesinami komórkowymi przygotowanymi w dołkach 96-dołkowej czarnej płytki płaskodennej (zob. pkt 16).
|
|
18.
|
Celem pierwszej analizy testowej jest określenie stężenia cytotoksycznego oraz zbadanie potencjału działania uczulającego na skórę substancji chemicznych. Przy pomocy X-VIVOTM 15 tworzy się serię rozcieńczeń w postępie geometrycznym roztworów podstawowych X-VIVOTM 15 badanych substancji chemicznych, stosując współczynnik rozcieńczenia wynoszący 2 (zob. dodatek 3.5) i wykorzystując do tego celu blok 96-dołkowy (np. Costar o numerze kat. EW-01729-03). Następnie do 50 μl zawiesiny komórkowej znajdującej się w dołkach 96-dołkowej czarnej płytki płaskodennej dodaje się rozcieńczony roztwór w objętości 50 μl/dołek. Dlatego też w przypadku badanych substancji chemicznych rozpuszczalnych w X-VIVOTM 15 stężenia końcowe tych substancji wynoszą 0,002–2 mg/ml (dodatek 3.5). W przypadku badanych substancji chemicznych, które nie rozpuszczają się w X-VIVO TM 15 w stężeniu 20 mg/ml, określa się jedynie współczynniki rozcieńczenia wynoszące 2–210, chociaż rzeczywiste stężenia końcowe badanych substancji chemicznych pozostają niepewne i zależą od stężenia roztworu nasyconego substancji chemicznych w roztworze podstawowym X-VIVO TM 15.
|
|
19.
|
W kolejnych analizach testowych (tj. w drugiej, trzeciej i czwartej kontrpróbie) roztwór podstawowy X-VIVOTM 15 tworzy się w stężeniu czterokrotnie wyższym niż stężenie, przy którym żywotność komórek wynosi 05 (CV05; najniższe stężenie, przy którym Inh-GAPLA przyjmuje wartość < 0,05) w pierwszym doświadczeniu. Jeżeli najwyższe stężenie Inh-GAPLA nie spadnie poniżej 0,05 w pierwszej analizie, roztwór podstawowy X-VIVOTM 15 tworzy się w najwyższym stężeniu użytym w pierwszej analizie. Stężenie CV05 oblicza się, dzieląc stężenie roztworu podstawowego uzyskanego podczas pierwszej analizy przez współczynnik rozcieńczenia CV05 (X) (współczynnik rozcieńczenia CV05 (X); współczynnik rozcieńczenia wymagany do rozcieńczenia roztworu podstawowego do CV05) (zob. dodatek 3.5). W przypadku badanych substancji nierozpuszczalnych w X-VIVO w stężeniu 20 mg/ml wartość CV05 określa się, mnożąc stężenie roztworu podstawowego przez 1/X. Jeżeli chodzi o analizy 2–4, przygotowuje się drugi roztwór podstawowy w postaci 4 × CV05 (dodatek 3.5).
|
|
20.
|
Serie rozcieńczeń w postępie geometrycznym drugich roztworów podstawowych X-VIVOTM 15 tworzy się, stosując współczynnik rozcieńczenia wynoszący 1,5 i wykorzystując do tego celu blok 96-dołkowy. Następnie do 50 μl zawiesiny komórkowej znajdującej się w dołkach 96-dołkowej czarnej płytki płaskodennej dodaje się rozcieńczony roztwór w objętości 50 μl/dołek. Każde stężenie badanej substancji chemicznej należy badać w czterech dołkach. Próbki miesza się wówczas za pomocą wytrząsarki do płytek i inkubuje przez 16 godzin w temperaturze 37 oC i w obecności 5 % CO2, po czym mierzy się aktywność lucyferazy w sposób opisany poniżej.
|
|
21.
|
Kontrola z rozpuszczalnikiem jest mieszaniną X-VIVOTM 15 w objętości 50 μl/dołek i zawiesiny komórkowej w RPMI-1640 zawierającej 10 % bydlęcej surowicy płodowej w objętości 50 μl/dołek.
|
|
22.
|
Zalecaną kontrolą dodatnią jest bromek 4-nitrobenzylu. W 1,5-mililitrowej probówce do mikromiareczkowania przygotowuje się 20 mg bromku 4-nitrobenzylu, a następnie dodaje się X-VIVOTM 15 do uzyskania objętości 1 ml. Probówkę umieszcza się w wirniku, gdzie jej zawartość jest energicznie mieszana i wstrząsana z maksymalną prędkością 8 obr./min przez co najmniej 30 min. Po odwirowywaniu przy 20000 g przez 5 minut supernatant rozcieńcza się czterokrotnie w X-VIVOTM 15, a następnie 500 μl rozcieńczonego supernatantu przenosi się do dołka bloku 96-dołkowego. Rozcieńczony supernatant rozcieńcza się dalej w X-VIVOTM 15, stosując współczynniki rozcieńczenia 2 i 4, po czym 50 μl tego roztworu dodaje się do 50 μl zawiesiny komórkowej THP-G8 znajdującej się w dołkach 96-dołkowej czarnej płytki płaskodennej (dodatek 3.6). Każde stężenie kontroli dodatniej należy badać w czterech dołkach. Płytkę wstrząsa się za pomocą wytrząsarki do płytek i inkubuje w inkubatorze CO2 (37 oC, 5 % CO2) przez 16 godzin, po czym mierzy się aktywność lucyferazy w sposób opisany w pkt 29.
|
|
23.
|
Zaleca się stosowanie kwasu mlekowego jako kontroli ujemnej. W 1,5-mililitrowej probówce do mikromiareczkowania przygotowuje się 20 mg kwasu mlekowego, a następnie dodaje się X-VIVOTM 15 do uzyskania objętości 1 ml (20 mg/ml). Roztwór kwasu mlekowego o stężeniu 20 mg/ml rozcieńcza się pięciokrotnie w X-VIVOTM 15 (4 mg/ml); 500 μl tego roztworu kwasu mlekowego o stężeniu 4 mg/ml przenosi się do dołka bloku 96-dołkowego. Roztwór ten rozcieńcza się dwukrotnie w X-VIVOTM 15, a następnie ponownie rozcieńcza się dwukrotnie, by uzyskać roztwory o stężeniach 2 mg/ml oraz 1 mg/ml. 50 μl tych trzech roztworów oraz kontrolę z nośnikiem (X-VIVOTM 15) dodaje się do 50 μl zawiesiny komórkowej THP-G8 znajdującej się w dołkach 96-dołkowej czarnej płytki płaskodennej. Każde stężenie kontroli ujemnej bada się w czterech dołkach. Płytkę wstrząsa się za pomocą wytrząsarki do płytek i inkubuje w inkubatorze CO2 (37 oC, 5 % CO2) przez 16 godzin, po czym mierzy się aktywność lucyferazy w sposób opisany w pkt 29.
|
|
24.
|
Można stosować inne odpowiednie kontrole dodatnie lub ujemne, jeżeli dostępne są dane historyczne pozwalające na uzyskanie porównywalnych kryteriów dopuszczalności analizy.
|
|
25.
|
Należy dołożyć starań, aby nie dopuścić do odparowania lotnych badanych substancji chemicznych i aby zapobiec zanieczyszczeniu krzyżowemu między dołkami zawierającymi badane substancje chemiczne, np. szczelnie zaklejając płytkę przed rozpoczęciem inkubacji z wykorzystaniem badanych substancji chemicznych.
|
|
26.
|
Uzyskanie prognozy dodatniej lub ujemnej w odniesieniu badanych substancji chemicznych oraz kontroli z rozpuszczalnikiem wymaga przeprowadzenia 2–4 analiz (zob. tabela 2). Każdą analizę przeprowadza się innego dnia, używając świeżego roztworu podstawowego X-VIVOTM 15 badanych substancji chemicznych oraz wykorzystując niezależnie pobrane komórki. Komórki mogą pochodzić z tego samego pasażu.
|
Pomiary aktywności lucyferazy
|
27.
|
Luminescencję mierzy się przy zużyciu 96-dołkowego luminometru mikropłytkowego wyposażonego w filtry optyczne, np. Phelios (ATTO, Tokio, Japonia), Tristan 941 (Berthold, Bad Wildbad, Niemcy) oraz seria ARVO (PerkinElmer, Waltham, Massachusetts, Stany Zjednoczone). Aby zapewnić odtwarzalność, luminometr musi być wykalibrowany na potrzeby każdego badania (19). Na potrzeby tej kalibracji dostępne są rekombinowane lucyferazy emitujące światło pomarańczowe i czerwone.
|
|
28.
|
100μl podgrzanego odczynnika do testu lucyferazy Tripluc® (Tripluc) przenosi się do każdego dołka na płytce, który zawiera zawiesinę komórkową poddaną działaniu substancji chemicznej lub niepoddaną takiemu działaniu. Płytkę wstrząsa się przez 10 minut w temperaturze otoczenia wynoszącej ok. 20 oC. Umieszcza się ją w luminometrze, by zmierzyć aktywność lucyferazy. Bioluminescencję mierzy się każdorazowo przez 3 sekundy zarówno z zastosowaniem filtra optycznego (F1), jak i bez niego (F0). W przypadku zastosowania innych ustawień, np. z uwagi na korzystanie z innego modelu luminometru, należy przedstawić stosowne uzasadnienie.
|
|
29.
|
Parametry każdego stężenia obliczane są na podstawie zmierzonych wartości, np. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, średniego odchylenia standardowego (±SD) IL8LA, średniego ±SD GAPLA, średniego ±SD nIL8LA, średniego ±SD Ind-IL8LA, średniego ±SD Inh-GAPLA oraz 95-procentowego przedziału ufności Ind-IL8LA. Definicje parametrów użytych w niniejszym punkcie znajdują się w dodatkach 3.1 oraz 3.4.
|
|
30.
|
Przed pomiarem rozróżnienie kolorystyczne w testach reporterowych, w których stosuje się kilka barw, uzyskuje się zazwyczaj przy użyciu detektorów (luminometru oraz czytnika płytek) wyposażonych w filtry optyczne, takie jak filtry typu sharp-cut (długoprzepustowe lub krótkoprzepustowe) lub filtry środkowoprzepustowe. Zgodnie z dodatkiem 3.2 współczynniki transmisji filtrów każdego koloru sygnału bioluminescencji należy wykalibrować przed badaniem.
|
DANE I SPRAWOZDAWCZOŚĆ
Ocena danych
|
31.
|
Zgodnie z kryteriami podejmowania decyzji stwierdzającej wynik dodatni/ujemny wymaga się, aby w każdej analizie:
|
—
|
prognozę dotyczącą testu IL-8 Luc uznać za dodatnią, jeżeli Ind-IL8LA badanej substancji chemicznej wynosi ≥1,4 oraz gdy dolna wartość graniczna 95-procentowego przedziału ufności Ind-IL8LA wynosi ≥1,0;
|
|
—
|
prognozę dotyczącą testu IL-8 Luc uznać za ujemną, jeżeli Ind-IL8LA badanej substancji chemicznej wynosi <1,4 lub gdy dolna wartość graniczna 95-procentowego przedziału ufności Ind-IL8LA wynosi <1,0.
|
|
Model prognozowania
|
32.
|
Uznaje się, że badane substancje chemiczne, w odniesieniu do których uzyskano wyniki dodatnie w dwóch analizach spośród analiz pierwszej, drugiej, trzeciej lub czwartej, uzyskują wynik dodatni, natomiast substancje, w odniesieniu do których uzyskano wyniki ujemne w trzech analizach spośród analiz pierwszej, drugiej, trzeciej lub czwartej, uzyskują wynik przypuszczalnie ujemny (tabela 2). Spośród substancji chemicznych, w odniesieniu do których przypuszcza się, że uzyskają wynik ujemny, substancjom chemicznym, które rozpuściły się w X-VIVOTM 15 w stężeniu 20 mg/ml, przyznaje się wynik ujemny, natomiast substancji chemicznych, które nie rozpuściły się w X-VIVOTM 15 w stężeniu 20 mg/ml, nie należy brać pod uwagę (rys. 1).
Tabela 2
Kryteria identyfikacji wyniku dodatniego i przypuszczalnie ujemnego
|
Analiza 1
|
Analiza 2
|
Analiza 3
|
Analiza 4
|
Prognoza końcowa
|
|
Dodatni
|
Dodatni
|
–
|
–
|
Dodatni
|
|
Ujemny
|
Dodatni
|
–
|
Dodatni
|
|
Ujemny
|
Dodatni
|
Dodatni
|
|
Ujemny
|
Przypuszczalnie ujemny
|
|
Ujemny
|
Dodatni
|
Dodatni
|
–
|
Dodatni
|
|
Ujemny
|
Dodatni
|
Dodatni
|
|
Ujemny
|
Przypuszczalnie ujemny
|
|
Ujemny
|
Dodatni
|
Dodatni
|
Dodatni
|
|
Ujemny
|
Przypuszczalnie ujemny
|
|
Ujemny
|
–
|
Przypuszczalnie ujemny
|
|
Rys. 1
Model prognozowania oceny końcowej

Kryteria dopuszczalności
|
33.
|
Podczas wykonywania testu IL-8 Luc spełnione muszą zostać następujące kryteria dopuszczalności:
|
—
|
w każdej analizie wartość Ind-IL8LA powinna wynosić powyżej 5,0 w co najmniej jednym stężeniu kontroli dodatniej, tj. bromku 4-nitrobenzylu;
|
|
—
|
w każdej analizie wartość Ind-IL8LA powinna wynosić poniżej 1,4 w dowolnym stężeniu kontroli ujemnej, tj. kwasie mlekowym;
|
|
—
|
należy odrzucić dane z płytek, w przypadku których wartość GAPLA dołków kontrolnych zawierających komórki i Tripluc, lecz niezawierających substancji chemicznych, jest mniejsza niż pięciokrotność wartości GAPLA dołków zawierających jedynie podłoże do badań (RPMI-1640 zawierające 10 % bydlęcej surowicy płodowej w objętości 50 μl/dołek i X-VIVOTM 15 w objętości 50 μl/dołek);
|
|
—
|
należy odrzucić dane z płytek, w przypadku których wartość Inh-GAPLA wszystkich stężeń badanych lub kontrolnych substancji chemicznych jest mniejsza niż 0,05. W tym przypadku należy powtórzyć pierwsze badanie, tak aby najwyższe stężenie końcowe powtórzonego badania było najniższym stężeniem końcowym poprzedniego badania.
|
|
Sprawozdanie z badania
|
34.
|
Sprawozdanie z badania powinno zawierać następujące informacje:
|
Badane substancje chemiczne
Substancja jednoskładnikowa:
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, rozpuszczalność w wodzie, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
rozpuszczalność w X-VIVOTM 15. w przypadku substancji chemicznych nierozpuszczalnych w X-VIVOTM 15 – czy po odwirowaniu można zaobserwować strącanie lub flotację;
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej, jeśli nie wykorzystano X-VIVOTM 15.
|
Substancja wieloskładnikowa, UVCB i mieszanina:
|
opisana w miarę możliwości np. poprzez podanie nazwy chemicznej (zob. powyżej), czystości, przedstawienie informacji dotyczących występowania ilościowego oraz wskazanie istotnych właściwości fizykochemicznych składników (zob. powyżej), w dostępnym zakresie;
|
|
wygląd fizyczny, rozpuszczalność w wodzie i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie;
|
|
masa cząsteczkowa lub pozorna masa cząsteczkowa w przypadku mieszanin lub polimerów o znanym składzie lub inne informacje istotne dla przebiegu badania;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
rozpuszczalność w X-VIVOTM 15. w przypadku substancji chemicznych nierozpuszczalnych w X-VIVOTM 15 – czy po odwirowaniu można zaobserwować strącanie lub flotację;
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika/nośnika w odniesieniu do każdej badanej substancji chemicznej, jeśli nie wykorzystano X-VIVOTM 15.
|
|
|
Kontrole
Kontrola dodatnia:
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS, kod SMILES lub InChI, wzór strukturalny lub inne identyfikatory;
|
|
wygląd fizyczny, rozpuszczalność w wodzie, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne, w dostępnym zakresie i w stosownych przypadkach;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
postępowanie z daną substancją przed rozpoczęciem badań, w stosownych przypadkach (np. podgrzewanie, mielenie);
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
odniesienie do wyników historycznych kontroli dodatnich wykazujących odpowiednie kryteria dopuszczalności, w stosownych przypadkach.
|
Kontrola ujemna:
|
dane identyfikacyjne substancji chemicznej, takie jak: nazwa(-y) IUPAC lub CAS, numer(-y) CAS lub inne identyfikatory;
|
|
czystość, nazwa chemiczna zanieczyszczeń w stosownych przypadkach i jeśli jest to praktycznie wykonalne itp.;
|
|
wygląd fizyczny, masa cząsteczkowa i dodatkowe istotne właściwości fizykochemiczne w przypadku stosowania innych kontroli ujemnych niż te opisane w wytycznej dotyczącej badań, w dostępnym zakresie;
|
|
warunki przechowywania i stabilność, w dostępnym zakresie;
|
|
uzasadnienie wyboru rozpuszczalnika w odniesieniu do każdej badanej substancji chemicznej.
|
|
|
Warunki badania
|
nazwa i adres sponsora i placówki przeprowadzającej badanie oraz dane kierownika badań;
|
|
opis zastosowanego badania;
|
|
zastosowana linia komórkowa, warunki jej przechowywania oraz źródło (np. placówka, z której ją otrzymano);
|
|
numer partii i pochodzenie bydlęcej surowicy płodowej, nazwa dostawcy, numer partii 96-dołkowej czarnej płytki płaskodennej oraz numer partii odczynnika Tripluc;
|
|
liczba pasaży i gęstość komórek wykorzystanych do badań;
|
|
metoda zliczania komórek zastosowana do posiewania przed badaniem i środki podjęte w celu zapewnienia jednorodnego rozmieszczenia liczby komórek;
|
|
zastosowany luminometr (np. model), w tym ustawienia przyrządu, wykorzystany substrat lucyferazy oraz wykazanie odpowiednich pomiarów luminescencji w oparciu o badanie kontrolne opisane w dodatku 3.2;
|
|
procedura stosowana w celu wykazania biegłości laboratorium w zakresie przeprowadzania badania (np. poprzez badanie substancji służących do wykazania biegłości) lub do wykazania odtwarzalności badania w czasie.
|
|
|
Procedura badawcza
|
liczba zastosowanych kontrprób i wykonanych analiz;
|
|
stosowane stężenia badanej substancji chemicznej, procedura podawania badanej substancji chemicznej i czas narażenia (jeżeli są one inne niż zalecane);
|
|
opis zastosowanych kryteriów oceny i podejmowania decyzji;
|
|
opis zastosowanych kryteriów dopuszczalności badania;
|
|
opis wszelkich modyfikacji procedury badawczej.
|
|
|
Wyniki
|
pomiary wartości IL8LA i GAPLA;
|
|
obliczenia dotyczące nIL8LA, Ind-IL8LA i Inh-GAPLA;
|
|
95-procentowy przedział ufności Ind-IL8LA;
|
|
wykres przedstawiający krzywe dawka-efekt w odniesieniu do indukcji aktywności lucyferazy i żywotności;
|
|
w stosownych przypadkach opis wszelkich innych istotnych obserwacji.
|
|
|
Omówienie wyników
|
omówienie wyników uzyskanych za pomocą testu IL-8 Luc;
|
|
uwzględnienie wyników testu w kontekście IATA, jeżeli są dostępne inne odpowiednie informacje.
|
|
Wniosek
|
BIBLIOGRAFIA
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K i Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359–69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Seria dotycząca badań i oceny nr 267, ENV/JM/MONO(2017)19, Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Seria dotycząca badań i oceny nr 258, ENV/JM/MONO(2017)20. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(4)
|
OECD (2016). Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation. Seria dotycząca badań i oceny nr 256, ENV/JM/MONO(2016)29. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H i Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371–9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y i Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816–30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337–51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N i Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275–84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A i Aschberger K (2016). Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147–155.
|
|
(10)
|
Thorne N, Inglese J i Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646–57.
|
|
(11)
|
OECD (2016). Badanie nr 455: Performance-Based Test Guideline for Stably Transfected TransactivationIn Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, Paryż: OECD Publishing. http://dx.doi.org/10.1787/9789264265295-en
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M i Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286–91.
|
|
(13)
|
Viviani VR, Bechara EJ i Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271–9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M i Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891–4.
|
|
(15)
|
OECD (2017). Przeznaczone do publikacji – Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. Publikacje OECD na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny. Francja, Paryż: OECD.
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Publikacje OECD na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 34. Francja, Paryż: OECD.
|
|
(17)
|
OECD (2018). Projekt wytycznych: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Paryż: Organizacja Współpracy Gospodarczej i Rozwoju. Dostępne na stronie internetowej: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf
|
|
(18)
|
JaCVAM (2016). Protokół z testu IL-8 Luc, dostępny na stronie internetowej: http://www.jacvam.jp/en_effort/effort02.html
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR i Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046–9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. Publikacje OECD na temat środowiska, zdrowia i bezpieczeństwa, seria dotycząca badań i oceny nr 168. Francja, Paryż: OECD.
|
|
(21)
|
Organizacja Narodów Zjednoczonych (2015). Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów (GHS). wydanie szóste zmienione, Nowy Jork i Genewa: publikacje Organizacji Narodów Zjednoczonych. ISBN: 978-92-1-117006-1. Dostępne na stronie internetowej: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
Dodatek 3.1
DEFINICJE
Dokładność
: stopień zgodności pomiędzy wynikami badania a przyjętymi wartościami odniesienia. Jest to miara efektywności badania i jeden z aspektów jej istotności. Pojęcia tego często używa się zamiennie z pojęciem „zgodność” na oznaczenie odsetka prawidłowych wyników uzyskiwanych przy przeprowadzeniu badania (16).
Mechanizm wywoływania skutków szkodliwych
: sekwencja zdarzeń, począwszy od struktury chemicznej docelowej substancji chemicznej lub grupy podobnych substancji chemicznych poprzez molekularne zdarzenie inicjujące aż do pożądanego wyniku in vivo (20).
Substancja chemiczna
: substancja lub mieszanina.
CV05
: żywotność komórek 05, tj. minimalne stężenie, przy którym substancje chemiczne wykazują wartość Inh-GAPLA poniżej 0,05.
FInSLO-LA
: skrót stosowany w sprawozdaniu z walidacji i we wcześniejszych publikacjach dotyczących testu IL-8 Luc w odniesieniu do Ind-IL8LA. Definicja – zob. Ind-IL8LA
GAPLA
: aktywność stabilnej lucyferazy czerwonej (SLR) (λmaks. = 630 nm), regulowana przez promotor GAPDH oraz wykazująca żywotność komórek i liczbę komórek żywotnych.
Zagrożenie
: nieodłączna właściwość czynnika lub sytuacja, która może potencjalnie doprowadzić do niekorzystnych skutków w przypadku narażenia organizmu, systemu lub (sub)populacji na taki czynnik.
IATA (zintegrowane podejście do badań i oceny)
: usystematyzowane podejście stosowane w odniesieniu do identyfikacji zagrożeń (potencjał), charakterystyki zagrożeń (siła oddziaływania) lub oceny bezpieczeństwa (potencjał / siła oddziaływania oraz narażenie) substancji chemicznej lub grupy substancji chemicznych; w ramach tego podejścia w sposób strategiczny integruje się i waży wszystkie istotne dane, które mogą wpłynąć na podjęcie decyzji regulacyjnej dotyczącej potencjalnego zagrożenia, ryzyka lub konieczności podjęcia dalszych ukierunkowanych, a przez to minimalnych badań.
II-SLR-LA
: skrót stosowany w sprawozdaniu z walidacji i we wcześniejszych publikacjach dotyczących testu IL-8 Luc w odniesieniu do Inh-GAPLA. Definicja – zob. Inh-GAPLA.
IL-8 (Interleukina-8)
: cytokina pozyskana z komórek śródbłonka, fibroblastów, keratynocytów, makrofagów i monocytów, która powoduje chemotaksję neutrofili i limfocytów T.
IL8LA
: aktywność stabilnej lucyferazy pomarańczowej (SLO) (λmaks. = 580 nm), regulowana przez promotor IL-8.
Ind-IL8LA
: krotność indukcji nIL8LA. Wartość tę uzyskuje się, dzieląc wartość nIL8LA komórek THP-G8 poddanych działaniu substancji chemicznych przez wartość nil8LA niestymulowanych komórek THP-G8; wartość Ind-IL8LA odzwierciedla indukcję aktywności promotora IL-8 przy pomocy substancji chemicznych.
Inh-GAPLA
: inhibicja GAPLA. Wartość tę uzyskuje się, dzieląc wartość GAPLA komórek THP-G8 poddanych działaniu substancji chemicznych przez wartość GAPLA komórek THP-G8 niepoddanych działaniu substancji chemicznych; Inh-GAPLA odzwierciedla cytotoksyczność substancji chemicznych.
Minimalny próg indukcji
: najniższe stężenie, przy którym substancja chemiczna spełnia kryteria uzyskania wyniku dodatniego.
Mieszanina
: mieszanina lub roztwór, które składają się z co najmniej dwóch substancji.
Substancja jednoskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której jeden główny składnik występuje w stężeniu co najmniej 80 % (w/w).
Substancja wieloskładnikowa
: substancja, zdefiniowana za pomocą jej składu ilościowego, w której więcej niż jeden główny składnik występuje w stężeniu ≥10 % (w/w) i <80 % (w/w). Substancja wieloskładnikowa powstaje w wyniku procesu wytwarzania. Różnica między mieszaniną a substancją wieloskładnikową polega na tym, że mieszaninę uzyskuje się poprzez zmieszanie przynajmniej dwóch substancji, między którymi nie zachodzi reakcja chemiczna. Substancja wieloskładnikowa powstaje w wyniku reakcji chemicznej.
nIL8LA
: aktywność lucyferazy SLO odzwierciedlająca aktywność promotora IL-8 (IL8LA), znormalizowana przez aktywność lucyferazy SLR odzwierciedlającą aktywność promotora GAPDH (GAPLA). Wartość nIL8LA odzwierciedla aktywność promotora IL-8 po uwzględnieniu żywotności komórek lub ich liczby.
nSLO-LA
: skrót stosowany w sprawozdaniu z walidacji i we wcześniejszych publikacjach dotyczących testu IL-8 Luc w odniesieniu do nIL8LA. Definicja – zob. nIL8LA.
Kontrola dodatnia
: kontrpróba zawierająca wszystkie składniki układu badawczego, poddana działaniu substancji chemicznej, o której wiadomo, że wywołuje reakcję dodatnią. Aby zapewnić możliwość oceny zmienności reakcji w kontroli dodatniej w czasie, stopień reakcji dodatniej nie powinien być nadmierny.
Prehapteny
: substancje chemiczne, które stają się czynnikami uczulającymi po transformacji abiotycznej.
Prohapteny
: substancje chemiczne, które wymagają aktywacji enzymatycznej do uwolnienia swojego potencjalnego działania uczulającego na skórę.
Istotność
: opis powiązania badania z oczekiwanym skutkiem oraz czy jest ono znaczące i użyteczne z punktu widzenia określonego celu. Jest to zakres, w jakim badanie pozwala prawidłowo zmierzyć lub przewidzieć biologiczny oczekiwany skutek. istotność obejmuje uwzględnienie dokładności (zgodności) badania (16).
Wiarygodność
: miary zakresu, w jakim badanie może zostać przeprowadzone w sposób odtwarzalny w jednym laboratorium i pomiędzy laboratoriami na przestrzeni czasu w przypadku jego przeprowadzania przy użyciu tego samego protokołu. Ocenia się ją, obliczając odtwarzalność wewnątrz- i międzylaboratoryjną oraz powtarzalność wewnątrzlaboratoryjną (16).
Analiza
: analiza obejmuje jednoczesne badanie jednej badanej substancji chemicznej lub większej liczby takich substancji w kontroli z rozpuszczalnikiem/nośnikiem i kontroli dodatniej.
Czułość
: odsetek wszystkich substancji chemicznych dających wynik dodatni / aktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą danego badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (16).
SLO-LA
: skrót stosowany w sprawozdaniu z walidacji i we wcześniejszych publikacjach dotyczących testu IL-8 Luc w odniesieniu do IL8LA. Definicja – zob. IL8LA.
SLR-LA
: skrót stosowany w sprawozdaniu z walidacji i we wcześniejszych publikacjach dotyczących testu IL-8 Luc w odniesieniu do GAPLA. Definicja – zob. GAPLA.
Kontrola z rozpuszczalnikiem/nośnikiem
: próbka niepoddana działaniu badanej substancji, zawierająca wszystkie składniki układu badawczego z wyjątkiem badanej substancji chemicznej, ale z uwzględnieniem stosowanego rozpuszczalnika/nośnika. Wykorzystywana jest do określenia wyjściowej reakcji dla próbek poddanych działaniu badanej substancji chemicznej rozpuszczonej lub stabilnie rozproszonej w tym samym rozpuszczalniku/nośniku. W przypadku badania z jednoczesną kontrolą podłoża próbka ta wykazuje również, czy rozpuszczalnik/nośnik wchodzi w reakcję z układem badawczym.
Swoistość
: odsetek wszystkich substancji chemicznych dających wynik ujemny / nieaktywnych substancji chemicznych prawidłowo sklasyfikowanych za pomocą badania. Jest to miara dokładności badania, która daje wyniki definitywne i stanowi istotny parametr w ocenie istotności badania (16).
Substancja
: pierwiastki chemiczne i ich związki w stanie naturalnym lub uzyskane w wyniku dowolnego procesu produkcyjnego, w tym wszelkie dodatki konieczne do zachowania trwałości produktu i wszelkie zanieczyszczenia powstałe w wyniku zastosowanego procesu, z wyłączeniem wszelkich rozpuszczalników, które można oddzielić bez wpływu na stabilność substancji i bez zmiany jej składu.
Środek powierzchniowo czynny
: zwany także surfaktantem, jest to substancja, np. detergent, która może zmniejszać napięcie powierzchniowe cieczy, umożliwiając tym samym jej pienienie lub przenikanie w ciała stałe; substancja ta jest także zwana środkiem zwilżającym. (TG437)
Badana substancja chemiczna
: dowolna substancja lub mieszanina badana za pomocą niniejszej metody.
THP-G8
: linia komórek reporterowych IL-8 wykorzystana w teście IL-8 Luc. Ludzką linię komórkową THP-1 typu makrofagowego poddano transfekcji przy użyciu genów lucyferazy SLO i SLR pod kontrolą odpowiednio promotorów IL-8 i GAPDH.
Globalnie Zharmonizowany System Klasyfikacji i Oznakowania Chemikaliów Organizacji Narodów Zjednoczonych (GHS ONZ)
: system, w ramach którego proponuje się klasyfikację substancji chemicznych (substancji i mieszanin) według znormalizowanych rodzajów i poziomów zagrożeń fizycznych, zdrowotnych i środowiskowych oraz omawia się odpowiednie elementy komunikacyjne, takie jak: piktogramy, hasła ostrzegawcze, zwroty wskazujące rodzaj zagrożenia, zwroty wskazujące środki ostrożności i karty charakterystyki substancji i produktów niebezpiecznych, aby przekazać informacje na temat ich szkodliwego działania w celu zapewnienia ochrony ludzi (w tym pracowników, robotników, przewoźników, konsumentów i ratowników) i środowiska (21).
UVCB
: substancje o nieznanym lub zmiennym składzie, złożone produkty reakcji lub materiały biologiczne.
Ważna metoda badawcza
: badanie, które uznano za wystarczająco istotne i wiarygodne dla danego celu i które opiera się na uzasadnionych naukowo podstawach. Badanie nie może zostać uznane za ważne w sensie absolutnym, ale tylko w odniesieniu do określonego celu.
Dodatek 3.2
ZASADA POMIARU AKTYWNOŚCI LUCYFERAZY I OKREŚLANIA WSPÓŁCZYNNIKÓW TRANSMISJI FILTRA OPTYCZNEGO W ODNIESIENIU DO SLO I SLR
Układ do testowania przy użyciu wielu reporterów – Tripluc – można stosować z luminometrem mikropłytkowym posiadającym system detekcji wielu barw, który można wyposażyć w filtr optyczny (np. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Filtrem optycznym stosowanym w pomiarach jest filtr długo- lub krótkoprzepustowy przepuszczający częstotliwości 600–620 nm lub filtr środkowoprzepustowy przepuszczający częstotliwości 600–700 nm.
Pomiar lucyferaz emitujących dwie barwy za pomocą filtra optycznego
W niniejszym przykładzie zastosowano filtr Phelios AB-2350 (ATTO). Przedmiotowy luminometr wyposażony jest w filtr długoprzepustowy przepuszczający częstotliwość 600 nm (R60 HOYA Co., zwany dalej „filtrem długoprzepustowym 600 nm”, filtr 1), do rozdzielania luminescencji SLO (λmaks. = 580 nm) i SLR (λmaks. = 630 nm).
Aby wyznaczyć współczynniki transmisji filtra długoprzepustowego 600 nm, najpierw – korzystając z oczyszczonych enzymów lucyferazy SLO i SLR – należy: (i) zmierzyć intensywność bioluminescencji SLO i SLR bez filtra (F0), (ii) zmierzyć intensywność bioluminescencji SLO i SLR, które przeszły przez filtr długoprzepustowy 600 nm (filtr 1), oraz (iii) obliczyć współczynniki transmisji filtra długoprzepustowego 600 nm w odniesieniu do SLO i SLR wymienionych poniżej.
|
Współczynniki transmisji
|
Skrót
|
Definicja
|
|
SLO
|
Współczynniki transmisji filtra 1
|
=κOR60
|
Współczynnik transmisji filtra dla SLO
|
|
SLR
|
Współczynniki transmisji filtra 1
|
κRR60
|
Współczynnik transmisji filtra dla SLR
|
Jeżeli intensywność SLO i SLR w badanej próbce określono odpowiednio jako O i R, (i) intensywność światła bez filtra (tylko optycznego) F0 i (ii) intensywność światła przechodzącego przez filtr długoprzepustowy 600 nm (filtr 1) F1 opisuje się w sposób określony poniżej:
F0 = O + R
F1 = κOR60 x O + κRR60 x R
Wzory te można wyrazić następująco:

Wówczas za pomocą obliczonych czynników przepuszczalności (κOR60 i κRR60) oraz zmierzonych wartości dla F0 i F1 można obliczyć wartości O i R w następujący sposób:

Materiały i metody wyznaczania czynnika przepuszczalności
|
1)
|
Odczynniki
Pojedyncze oczyszczone enzymy lucyferazy:
|
liofilizowany oczyszczony enzym SLO
|
|
liofilizowany oczyszczony enzym SLR
|
|
(które dla celów walidacji pozyskano z GPC Lab. Co. Ltd. w Tottori w Japonii razem z linią komórkową THP-G8).
|
Odczynnik testowy:
|
odczynnik testowy lucyferazy Tripluc® (np. z TOYOBO Cat#MRA-301).
|
Podłoże: do testu lucyferazy (30 ml, przechowywane w temperaturze 2–8 °C).
|
|
Odczynnik
|
Stężenie
|
Stężenie końcowe w podłożu
|
Wymagana ilość
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
Bydlęca surowica płodowa
|
—
|
10 %
|
3 ml
|
|
2)
|
Przygotowanie roztworu enzymu
Liofilizowany oczyszczony enzym lucyferazy należy rozpuścić w probówce, dodając 200 μl Tris/HCl lub HEPES/HCl o stężeniu 10–100 mM (pH 7,5–8,0) zawierającego 10 % (w/v) glicerolu, podzielić roztwór enzymu na podwielokrotności 10 μl w jednorazowych probówkach o pojemności 1,5 ml i przechowywać je w zamrażarce w temperaturze -80 oC. Zamrożony roztwór enzymu nadaje się do użycia przez okres do 6 miesięcy. Podczas korzystania z roztworu należy dodać 1 ml podłoża do testu lucyferazy (RPMI-1640 zawierającego 10 % bydlęcej surowicy płodowej) do każdej probówki zawierającej roztwory enzymu (rozcieńczony roztwór enzymu), a probówki należy przechowywać na lodzie, by zapobiec dezaktywacji enzymów.
|
|
3)
|
Pomiar bioluminescencji
Rozmrozić odczynnik do testu lucyferazy Tripluc® (Tripluc) i przechowywać w temperaturze pokojowej w łaźni wodnej albo w temperaturze otoczenia. Luminometr należy włączyć 30 minut przed rozpoczęciem pomiaru, aby umożliwić stabilizację fotopowielacza. 100 μl rozcieńczonego roztworu enzymu należy przenieść do dołków 96-dołkowej czarnej płytki (płaskodennej) (próbka referencyjna SLO do #B1, #B2, #B3, próbka referencyjna SLR do #D1, #D2, #D3). Następnie za pomocą pipety należy przenieść 100 μl podgrzanego odczynnika Tripluc do każdego dołka płytki zawierającego rozcieńczony roztwór enzymu. Płytkę należy wytrząsać przez 10 minut w temperaturze pokojowej (około 25 oC) w wytrząsarce do płytek. Jeżeli w dołkach pojawią się pęcherzyki powietrza, należy je usunąć. Płytkę należy umieścić w luminometrze, aby dokonać pomiaru aktywności lucyferazy. Bioluminescencję mierzy się każdorazowo przez 3 sekundy zarówno z zastosowaniem filtra optycznego (F1), jak i bez niego (F0).
Współczynnik transmisji filtra optycznego obliczono w następujący sposób:
współczynnik transmisji (SLO (κOR60)) = (#B1 z F1 + #B2 z F1 + #B3 z F1) / (#B1 z F0 + #B2 z F0 + #B3 z F0)
Współczynnik transmisji (SLR (κRR60))= (#D1 z F1 + #D2 z F1 + #D3 z F1) / (#D1 z F0 + #D2 z F0+ #D3 z F0)
Obliczone współczynniki przepuszczalności wykorzystuje się do wszystkich pomiarów wykonywanych przy pomocy tego samego luminometru.
|
Kontrola jakości sprzętu
Należy stosować procedury opisane w protokole IL-8 Luc (18).
Dodatek 3.3
SUBSTANCJE SŁUŻĄCE DO WYKAZANIA BIEGŁOŚCI
Przed przystąpieniem do rutynowego stosowania badania opisanego w niniejszym dodatku do metody badawczej B.71 laboratoria powinny wykazać swoją biegłość techniczną, prawidłowo określając prognozę testu IL-8 Luc dla 9 substancji wskazanych w tabeli 1 oraz określając wartości, które mieszczą się w odpowiednim zakresie odniesienia, dla co najmniej 8 z 9 substancji służących do wykazania biegłości (wybranych w celu przedstawienia zakresu reakcji na zagrożenia związane z działaniem uczulającym na skórę). Przy wyborze substancji kierowano się również następującymi kryteriami: dostępnością handlową tych substancji, dostępnością wysokiej jakości danych referencyjnych z badań in vivo oraz dostępnością wysokiej jakości danych in vitro uzyskanych w wyniku zastosowania testu IL-8 Luc. W odniesieniu do testu IL-8 Luc można znaleźć również opublikowane dane referencyjne (6)(1).
Tabela 1
Substancje zalecane do wykazania biegłości technicznej w odniesieniu do testu IL-8 Luc
|
Substancje służące do wykazania biegłości
|
Nr CAS
|
Stan skupienia
|
Rozpuszczalność w X-VIVO15 przy stężeniu 20 mg/ml
|
Prognoza in vivo
(108)
|
Prognoza dotycząca IL-8 Luc (109)
|
Zakres odniesienia (μg/ml) (110)
|
|
|
CV05 (111)
|
Minimalny próg indukcji IL-8 Luc (112)
|
|
2,4-dinitrochlorobenzen
|
97-00-7
|
Stały
|
Nierozpuszczalny
|
Czynnik uczulający (wyjątkowo silny)
|
Dodatni
|
2,3–3,9
|
0,5–2,3
|
|
Formaldehyd
|
50-00-0
|
Ciekły
|
Rozpuszczalny
|
Czynnik uczulający (silny)
|
Dodatni
|
9–30
|
4–9
|
|
2-merkaptobenzotiazol
|
149-30-4
|
Stały
|
Nierozpuszczalny
|
Czynnik uczulający (w stopniu umiarkowanym)
|
Dodatni
|
250–290
|
60–250
|
|
Etyleneodiamina
|
107-15-3
|
Ciekły
|
Rozpuszczalny
|
Czynnik uczulający (w stopniu umiarkowanym)
|
Dodatni
|
500–700
|
0,1–0,4
|
|
Dimetakrylan glikolu etylenowego
|
97-90-5
|
Ciekły
|
Nierozpuszczalny
|
Czynnik uczulający (słaby)
|
Dodatni
|
>2 000
|
0,04–0,1
|
|
4-allilamizol (Estragol)
|
140-67-0
|
Ciekły
|
Nierozpuszczalny
|
Czynnik uczulający (słaby)
|
Dodatni
|
>2 000
|
0,01–0,07
|
|
Siarczan streptomycyny
|
3810-74-0
|
Stały
|
Rozpuszczalny
|
Brak działania uczulającego
|
Ujemny
|
>2 000
|
>2 000
|
|
Glicerol
|
56-81-5
|
Ciekły
|
Rozpuszczalny
|
Brak działania uczulającego
|
Ujemny
|
>2 000
|
>2 000
|
|
Izopropanol
|
67-63-0
|
Ciekły
|
Rozpuszczalny
|
Brak działania uczulającego
|
Ujemny
|
>2 000
|
>2 000
|
|
Skróty: Nr CAS = numer w rejestrze Chemical Abstracts Service.
|
Dodatek 3.4
WSKAŹNIKI I KRYTERIA OCENY
nIL8LA (nSLO-LA)
Dokonuje się pomiaru określonego kolejnego powtórzenia j (j = 1–4) określonego kolejnego stężenia i (i = 0–11) odpowiednio dla IL8LA (SLO-LA) oraz GAPLA (SLR-LA). Znormalizowaną IL8LA, zwaną nIL8LA (nSLO-LA), określa się następująco:
nIL8LAij = IL8LAij/GAPLAij
Jest to podstawowa jednostka miary w niniejszym teście.
Ind-IL8LA (FInSLO-LA)
Podstawową miarę w niniejszym teście stanowi wzrost krotności uśrednionego nIL8LA (nSLO-LA) dla powtórzenia określonego kolejnego stężenia i w porównaniu z uśrednionym nIL8LA (nSLO-LA) przy stężeniu wynoszącym 0, Ind-IL8LA. Stosunek ten wyraża się za pomocą następującego wzoru:

Laboratorium wiodące zaproponowało, aby wartość 1,4 odpowiadała dodatniemu wynikowi badanej substancji chemicznej. Wartość tę określono przez zbadanie danych historycznych z laboratorium wiodącego. Następnie zespół ds. zarządzania danymi wykorzystywał tę wartość na wszystkich etapach badania walidacyjnego. Początkowy wynik, Ind-IL8LA, jest stosunkiem 2 średnich arytmetycznych, jak pokazano w równaniu.
95-procentowy przedział ufności (95 % CI)
Na podstawie tego wskaźnika można oszacować 95-procentowy przedział ufności (95 % CI), aby pokazać pokazuje precyzję tego pomiaru początkowego wyniku. Dolna granica 95-procentowego przedziału ufności wynosząca ≥1 wskazuje na to, że wartość nIL8LA przy określonym kolejnym stężeniu i jest znacznie większa niż w kontroli z rozpuszczalnikiem. Istnieje kilka sposobów opracowania 95-procentowego przedziału ufności. W niniejszym badaniu wykorzystano metodę znaną jako twierdzenie Fiellera. 95-procentowy przedział ufności uzyskuje się, korzystając z następującego wzoru:

gdzie:








t0.975(ν) wynosi 97,5 percentyla centralnego rozkładu t przy stopniu swobody ν, gdzie

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA jest stosunkiem uśrednionego GAPLA (SLR-LA) dla powtórzenia określonego kolejnego stężenia i do stężenia w kontroli z rozpuszczalnikiem, a wartość tę wyraża się jako:
 . .
Ponieważ GAPLA jest mianownikiem nIL8LA, bardzo mała wartość powoduje duże zróżnicowanie nIL8LA. Dlatego też wartości Ind-IL8LA przy bardzo małej wartości Inh-GAPLA (poniżej 0,05) można uznać za mało precyzyjne.
Dodatek 3.5
SCHEMAT METOD ROZPUSZCZANIA SUBSTANCJI CHEMICZNYCH W TEŚCIE IL-8 LUC
|
a)
|
W przypadku substancji chemicznych rozpuszczanych w X-VIVOTM 15 w stężeniu 20 mg/ml

|
|
b)
|
W przypadku substancji chemicznych nierozpuszczalnych w X-VIVOTM 15 w stężeniu 20 mg/ml

|
Dodatek 3.6
SCHEMAT METOD ROZPUSZCZANIA BROMKU 4-NITROBENZYLU NA POTRZEBY KONTROLI DODATNIEJ W TEŚCIE IL-8 LUC;

” |