|
26.2.2009
|
PL
|
Dziennik Urzędowy Unii Europejskiej
|
L 54/1
|
ROZPORZĄDZENIE KOMISJI (WE) nr 152/2009
z dnia 27 stycznia 2009 r.
ustanawiające metody pobierania próbek i dokonywania analiz do celów urzędowej kontroli pasz
(Tekst mający znaczenie dla EOG)
KOMISJA WSPÓLNOT EUROPEJSKICH,
uwzględniając Traktat ustanawiający Wspólnotę Europejską,
uwzględniając rozporządzenie (WE) nr 882/2004 Parlamentu Europejskiego i Rady z dnia 29 kwietnia 2004 r. w sprawie kontroli urzędowych przeprowadzanych w celu sprawdzenia zgodności z prawem paszowym i żywnościowym oraz regułami dotyczącymi zdrowia zwierząt i dobrostanu zwierząt (1), w szczególności jego art. 11 ust. 4 lit. a), b) i c),
a także mając na uwadze, co następuje:
|
(1)
|
Poniższe akty prawne przyjęto w celu wdrożenia dyrektywy 70/373/EWG i pozostają one w mocy zgodnie z art. 61 ust. 2 rozporządzenia (WE) nr 882/2004:
|
—
|
pierwsza dyrektywa Komisji 71/250/EWG z dnia 15 czerwca 1971 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (2),
|
|
—
|
druga dyrektywa Komisji 71/393/EWG z dnia 18 listopada 1971 r. ustanawiająca wspólnotowe metody analiz do celów urzędowej kontroli pasz (3),
|
|
—
|
trzecia dyrektywa Komisji 72/199/EWG z dnia 27 kwietnia 1972 r. ustalająca wspólnotowe metody analiz do celów urzędowej kontroli pasz (4),
|
|
—
|
czwarta dyrektywa Komisji 73/46/EWG z dnia 5 grudnia 1972 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (5),
|
|
—
|
pierwsza dyrektywa Komisji 76/371/EWG z dnia 1 marca 1976 r. ustanawiająca wspólnotowe metody pobierania próbek i dokonywania analizy do celów urzędowej kontroli pasz (6),
|
|
—
|
siódma dyrektywa Komisji 76/372/EWG z dnia 1 marca 1976 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (7),
|
|
—
|
ósma dyrektywa Komisji 78/633/EWG z dnia 15 czerwca 1978 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (8),
|
|
—
|
dziewiąta dyrektywa Komisji 81/715/EWG z dnia 31 lipca 1981 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (9),
|
|
—
|
dziesiąta dyrektywa Komisji 84/425/EWG z dnia 25 lipca 1984 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (10),
|
|
—
|
dyrektywa Komisji 86/174/EWG z dnia 9 kwietnia 1986 r. określająca metodę obliczania wartości energetycznej mieszanek paszowych dla drobiu (11),
|
|
—
|
jedenasta dyrektywa Komisji 93/70/EWG z dnia 28 lipca 1993 r. ustanawiająca wspólnotowe metody analiz do celów urzędowej kontroli pasz (12),
|
|
—
|
dwunasta dyrektywa Komisji 93/117/WE z dnia 17 grudnia 1993 r. ustanawiająca wspólnotowe metody analizy do celów urzędowej kontroli pasz (13),
|
|
—
|
dyrektywa Komisji 98/64/WE z dnia 3 września 1998 r. ustanawiająca wspólnotowe metody analiz do oznaczenia aminokwasów, surowych olejów i tłuszczów oraz olaquindoksu w paszach i zmieniająca dyrektywę 71/393/EWG (14),
|
|
—
|
dyrektywa Komisji 1999/27/WE z dnia 20 kwietnia 1999 r. ustanawiająca wspólnotowe metody analiz dla oznaczania amprolium, diklazurilu i karbadoksu w paszach oraz zmieniająca dyrektywy 71/250/EWG, 73/46/EWG i uchylająca dyrektywę 74/203/EWG (15),
|
|
—
|
dyrektywa Komisji 1999/76/WE z dnia 23 lipca 1999 r. ustanawiająca wspólnotową metodę analizy dla oznaczania lasalocidu soli sodowej w paszach (16),
|
|
—
|
dyrektywa Komisji 2000/45/WE z dnia 6 lipca 2000 r. ustanawiająca wspólnotowe metody analizy do celów oznaczania witaminy A, witaminy E i tryptofanu w paszach (17),
|
|
—
|
dyrektywa Komisji 2002/70/WE z dnia 26 lipca 2002 r. ustanawiająca wymagania dotyczące określania poziomów dioksyn i dioksynopochodnych polichlorowanych bifenyli (PCB) w paszach (18),
|
|
—
|
dyrektywa Komisji 2003/126/WE z dnia 23 grudnia 2003 r. w sprawie analitycznej metody określania składników pochodzenia zwierzęcego do celów urzędowej kontroli pasz (19).
|
|
|
(2)
|
Ponieważ dyrektywę 70/373/EWG zastąpiono rozporządzeniem (WE) nr 882/2004, należy zastąpić akty wykonawcze do tej dyrektywy jednym rozporządzeniem. Jednocześnie metody powinny zostać dostosowane w świetle rozwoju wiedzy naukowej i technicznej. Metody, które straciły ważność w odniesieniu do ich celu, powinny zostać usunięte. Przewiduje się, że przepisy dotyczące pobierania próbek zostaną w odpowiednim czasie zaktualizowane w celu uwzględnienia nowych osiągnięć w zakresie produkcji, przechowywania, transportu i sprzedaży pasz, jednakże do tego czasu należy utrzymać istniejące przepisy dotyczące pobierania próbek.
|
|
(3)
|
Należy zatem uchylić dyrektywy 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/WE, 98/64/WE, 1999/27/WE, 1999/76/WE, 2000/45/WE, 2002/70/WE i 2003/126/WE.
|
|
(4)
|
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Stałego Komitetu ds. Łańcucha Żywnościowego i Zdrowia Zwierząt,
|
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Pobieranie próbek do celów urzędowej kontroli pasz, w zakresie oznaczania składników, dodatków oraz niepożądanych substancji, z wyjątkiem pozostałości pestycydów i mikroorganizmów, przeprowadzane jest zgodnie z metodami opisanymi w załączniku I.
Artykuł 2
Przygotowanie próbek do analizy i wyrażanie wyników odbywa się zgodnie z metodami opisanymi w załączniku II.
Artykuł 3
Badania do celów urzędowej kontroli pasz przeprowadzane są z wykorzystaniem metod określonych w załącznikach III (Metody analizy składu materiałów i mieszanek paszowych), IV (Metody analizy do celów kontroli poziomu dopuszczonych dodatków w paszach), V (Metody analizy do celów kontroli zawartości niepożądanych substancji w paszach) oraz VI (Metody analizy dotyczące oznaczania składników pochodzenia zwierzęcego do celów urzędowej kontroli pasz).
Artykuł 4
Wartość energetyczna mieszanek paszowych dla drobiu obliczana jest zgodnie z załącznikiem VII.
Artykuł 5
Określone w załączniku VIII metody analizy mające na celu kontrolę nielegalnej obecności w paszach dodatków, które już nie są dopuszczone, stosowane są do celów potwierdzania.
Artykuł 6
Dyrektywy 71/250/EWG, 71/393/EWG, 72/199/EWG, 73/46/EWG, 76/371/EWG, 76/372/EWG, 78/633/EWG, 81/715/EWG, 84/425/EWG, 86/174/EWG, 93/70/EWG, 93/117/WE, 98/64/WE, 1999/27/WE, 1999/76/WE, 2000/45/WE, 2002/70/WE i 2003/126/WE tracą moc.
Odesłania do uchylonych dyrektyw rozumiane są jako odesłania do niniejszego rozporządzenia i interpretuje zgodnie z tabelami korelacji w załączniku IX.
Artykuł 7
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia 26 sierpnia 2009 r.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 27 sierpnia 2009 r.
W imieniu Komisji
Androulla VASSILIOU
Członek Komisji
(1) Dz.U. L 165 z 30.4.2004, s. 1.
(2) Dz.U. L 155 z 12.7.1971, s. 13.
(3) Dz.U. L 279 z 20.12.1971, s. 7.
(4) Dz.U. L 123 z 29.5.1972, s. 6.
(5) Dz.U. L 83 z 30.3.1973, s. 21.
(6) Dz.U. L 102 z 15.4.1976, s. 1.
(7) Dz.U. L 102 z 15.4.1976, s. 8.
(8) Dz.U. L 206 z 29.7.1978, s. 43.
(9) Dz.U. L 257 z 10.9.1981, s. 38.
(10) Dz.U. L 238 z 6.9.1984, s. 34.
(11) Dz.U. L 130 z 16.5.1986, s. 53.
(12) Dz.U. L 234 z 17.9.1993, s. 17.
(13) Dz.U. L 329 z 30.12.1993, s. 54.
(14) Dz.U. L 257 z 19.9.1998, s. 14.
(15) Dz.U. L 118 z 6.5.1999, s. 36.
(16) Dz.U. L 207 z 6.8.1999, s. 13.
(17) Dz.U. L 174 z 13.7.2000, s. 32.
(18) Dz.U. L 209 z 6.8.2002, s. 15.
(19) Dz.U. L 339 z 24.12.2003, s. 78.
ZAŁĄCZNIK I
METODY POBIERANIA PRÓBEK
1. CEL I ZAKRES STOSOWANIA METODY
Próbki przeznaczone do urzędowej kontroli pasz pobierane są zgodnie z metodami opisanymi poniżej. Próbki otrzymane w ten sposób uważa się za reprezentatywne dla kontrolowanych partii.
2. PERSONEL POBIERAJĄCY PRÓBKI
Próbki pobierane są przez osoby upoważnione do tego celu przez państwa członkowskie.
3. DEFINICJE
Kontrolowana partia: ilość produktu stanowiąca partię, posiadająca cechy wystarczające do uznania jej za jednolitą.
Próbka pierwotna: ilość produktu pobrana z jednego miejsca kontrolowanej partii.
Próbka zbiorcza: sumaryczna ilość pierwotnych próbek pobranych z tej samej kontrolowanej partii.
Próbka zredukowana: reprezentatywna część zbiorczej próbki otrzymana z niej w procesie redukcyjnym.
Próbka końcowa: część zredukowanej próbki lub zhomogenizowanej próbki zbiorczej.
4. APARATURA I SPRZĘT
|
4.1.
|
Sprzęt do pobierania próbek musi być wykonany z materiałów, które nie zanieczyszczą kontrolowanego produktu. Taki sprzęt może być urzędowo zatwierdzony przez państwa członkowskie.
|
4.2. Sprzęt zalecany do pobierania próbek pasz stałych
4.2.1. Ręczne pobieranie próbek
|
4.2.1.1.
|
Szufelka o płaskim dnie i prostopadłych ściankach bocznych.
|
|
4.2.1.2.
|
Sonda z otwieranymi okienkami lub przegrodami, której wymiary muszą być dostosowane do charakterystyki kontrolowanej partii (głębokość pojemnika, wymiary worka itp.) i do wielkości cząstek paszy.
|
4.2.2. Mechaniczne pobieranie próbek
Zatwierdzone urządzenie mechaniczne może być użyte do pobierania próbek paszy w ruchu.
4.2.3. Rozdzielacz
Urządzenie przeznaczone do dzielenia próbek w przybliżeniu na równe części może być stosowane do pobierania próbek pierwotnych oraz w celu przygotowania próbek zredukowanych i końcowych.
5. WYMAGANIA ILOŚCIOWE
|
5.A.
|
W odniesieniu do kontroli zawartości substancji lub produktów jednolicie rozmieszczonych w paszy
|
|
5.A.1.
|
Kontrolowana partia
Rozmiar kontrolowanej partii musi umożliwiać pobranie próbek każdej części składowej.
|
|
5.A.2.
|
Próbki pierwotne
|
|
5.A.2.1.
|
Pasze sypkie:
|
Minimalna liczba próbek pierwotnych:
|
|
5.A.2.1.1.
|
waga kontrolowanych partii nie większa niż 2,5 tony
|
siedem
|
|
5.A.2.1.2.
|
waga kontrolowanych partii większa niż 2,5 tony
|
√ dwudziestokrotność liczby ton metrycznych stanowiących wagę kontrolowanej partii (1), maksymalnie do 40 próbek pierwotnych
|
|
5.A.2.2.
|
Pasze opakowane:
|
Minimalna liczba opakowań, z których pobiera się próbki (2):
|
|
5.A.2.2.1.
|
Opakowania o wadze większej niż 1 kg:
|
|
5.A.2.2.1.1.
|
kontrolowana partia zawierająca od 1 do 4 opakowań
|
wszystkie opakowania
|
|
5.A.2.2.1.2.
|
kontrolowana partia zawierająca od 5 do 16 opakowań
|
cztery
|
|
5.A.2.2.1.3
|
kontrolowana partia zawierająca ponad 16 opakowań
|
√ liczba opakowań stanowiących kontrolowaną partię (1), maksymalnie do 20 opakowań
|
|
5.A.2.2.2.
|
Opakowania o wadze nie większej niż 1 kg
|
cztery
|
|
5.A.2.3.
|
Pasze płynne lub półpłynne:
|
Minimalna liczba pojemników, z których pobiera się próbki (2):
|
|
5.A.2.3.1.
|
Pojemniki o objętości większej niż 1 l:
|
|
5.A.2.3.1.1.
|
kontrolowana partia zawierająca od 1 do 4 pojemników
|
wszystkie pojemniki
|
|
5.A.2.3.1.2.
|
kontrolowana partia zawierająca od 5 do 16 pojemników
|
cztery
|
|
5.A.2.3.1.3.
|
kontrolowana partia zawierająca ponad 16 pojemników
|
√ liczba pojemników stanowiących kontrolowaną partię (1), maksymalnie do dwudziestu pojemników
|
|
5.A.2.3.2.
|
Pojemniki o objętości nie większej niż 1 l
|
cztery
|
|
5.A.2.4.
|
Bloki paszy i lizawki mineralne
|
Minimalna liczba bloków lub lizawek, z których pobiera się próbki (2):
jeden blok lub jedna lizawka na kontrolowaną partię 25 sztuk, maksymalnie do czterech bloków lub lizawek
|
|
5.A.3.
|
Próbka zbiorcza
Wymagana jest jedna próbka zbiorcza na kontrolowaną partię. Całkowita waga próbek pierwotnych tworzących próbkę zbiorczą jest nie mniejsza niż:
|
|
5.A.3.1.
|
Pasze sypkie
|
4 kg
|
|
5.A.3.2.
|
Pasze opakowane:
|
|
|
5.A.3.2.1.
|
opakowania o wadze większej niż 1 kg
|
4 kg
|
|
5.A.3.2.2.
|
opakowania o wadze nie większej niż 1 kg
|
waga zawartości czterech oryginalnych opakowań
|
|
5.A.3.3.
|
Pasze płynne lub półpłynne:
|
|
|
5.A.3.3.1.
|
pojemniki o objętości większej niż 1 l
|
4 l
|
|
5.A.3.3.2.
|
pojemniki o objętości nie większej niż 1 l
|
objętość zawartości czterech oryginalnych opakowań
|
|
5.A.3.4.
|
Bloki paszy i lizawki mineralne:
|
|
5.A.3.4.1.
|
o wadze większej niż 1 kg
|
4 kg
|
|
5.A.3.4.2.
|
o wadze nie większej niż 1 kg
|
waga czterech oryginalnych bloków lub lizawek
|
|
5.A.4.
|
Próbki końcowe
Próbki końcowe otrzymywane są z próbki zbiorczej w wyniku redukcji, jeśli jest to konieczne. Wymagana jest analiza co najmniej jednej próbki końcowej. Ilość próbki końcowej potrzebnej do analizy jest nie mniejsza niż:
|
|
|
Pasze stałe
|
500 g
|
|
|
Pasze płynne lub półpłynne
|
500 ml
|
|
5.B.
|
W odniesieniu do kontroli niepożądanych substancji lub produktów, które mogą być rozmieszczone niejednolicie w paszy, takich jak aflatoksyny, sporysz, rącznik pospolity i krotalaria w materiałach paszowych (3)
|
|
5.B.1.
|
Kontrolowana partia: zob. 5.A.1.
|
|
5.B.2.
|
Próbki pierwotne
|
|
5.B.2.1.
|
Pasze sypkie: zob. 5.A.2.1.
|
|
5.B.2.2.
|
Pasze pakowane:
|
Minimalna liczba opakowań:
|
|
5.B.2.2.1.
|
kontrolowana partia zawierająca od 1 do 4 opakowań
|
wszystkie opakowania
|
|
5.B.2.2.2.
|
kontrolowana partia zawierająca od 5 do 16 opakowań
|
cztery
|
|
5.B.2.2.3.
|
kontrolowana partia zawierająca ponad 16 opakowań
|
√ liczba opakowań stanowiących kontrolowaną partię (1), maksymalnie do czterdziestu opakowań
|
|
5.B.3.
|
Próbki zbiorcze
Liczba próbek zbiorczych różni się w zależności od rozmiaru kontrolowanej partii. Minimalna liczba próbek zbiorczych na kontrolowaną partię jest podana poniżej. Całkowita waga próbek pierwotnych tworzących każdą próbkę zbiorczą jest nie mniejsza niż 4 kg
|
|
5.B.3.1.
|
Pasze sypkie
|
|
|
|
Waga kontrolowanej partii w tonach:
|
Minimalna liczba próbek zbiorczych na kontrolowaną partię:
|
|
|
do 1
|
1
|
|
|
od powyżej 1 do 10
|
2
|
|
|
od powyżej 10 do 40
|
3
|
|
|
powyżej 40
|
4
|
|
5.B.3.2.
|
Pasze opakowane
|
|
|
|
Rozmiar kontrolowanej partii w ilości opakowań:
|
Minimalna liczba próbek zbiorczych na kontrolowaną partię:
|
|
|
1 do 16
|
1
|
|
|
17 do 200
|
2
|
|
|
201 do 800
|
3
|
|
|
powyżej 800
|
4
|
|
5.B.4.
|
Próbki końcowe
Próbki końcowe otrzymywane są z każdej próbki zbiorczej w wyniku redukcji. Wymagana jest analiza co najmniej jednej próbki końcowej na próbkę zbiorczą. Waga próbki końcowej do analizy nie może być mniejsza niż 500 g.
|
6. INSTRUKCJE POBIERANIA, PRZYGOTOWANIA I PAKOWANIA PRÓBEK
6.1. Uwaga ogólna
Próbki należy pobierać i przygotowywać tak szybko, jak to możliwe, przy zachowaniu środków ostrożności gwarantujących, że produkt nie ulegnie zmianie lub zanieczyszczeniu. Stosowany sprzęt, a także powierzchnie i pojemniki przeznaczone na próbki muszą być czyste i suche.
6.2. Próbki pierwotne
6.2.A. W odniesieniu do kontroli substancji lub produktów jednolicie rozmieszczonych w paszy
Próbki pierwotne należy pobrać losowo z całej partii przy zachowaniu w przybliżeniu jednakowej ich wielkości.
6.2.A.1. Pasze sypkie
Kontrolowaną partię należy podzielić na równe w przybliżeniu części. Liczbę części odpowiadającą liczbie próbek pierwotnych wymaganej zgodnie z pkt 5.A.2. należy wybrać losowo i pobrać co najmniej jedną próbkę z każdej części.
W stosownych przypadkach próbki mogą być pobierane, gdy kontrolowana partia paszy jest w ruchu (załadunek lub wyładunek).
6.2.A.2. Pasze opakowane
Po wybraniu wymaganej liczby opakowań w celu pobrania próbek zgodnie z pkt 5.A.2, część zawartości każdego opakowania należy pobrać przy użyciu sondy lub szufelki. W miarę potrzeby, próbki należy pobierać po opróżnieniu każdego z opakowań oddzielnie. Jakiekolwiek zbrylenia należy rozbić (jeśli to konieczne odseparować je, po rozbryleniu, zwrócić do próbki), oddzielnie dla każdej próbki zbiorczej.
6.2.A.3. Jednorodne lub nadające się do homogenizacji pasze płynne lub półpłynne
Po wybraniu wymaganej liczby pojemników w celu pobrania próbek zgodnie z pkt 5.A.2 należy w razie potrzeby zhomogenizować zawartość i pobrać próbkę z każdego pojemnika.
Próbki pierwotne mogą być pobierane w trakcie opróżniania zawartości pojemnika.
6.2.A.4. Nienadające się do homogenizacji pasze płynne lub półpłynne
Po wybraniu wymaganej liczby pojemników w celu pobrania próbek zgodnie z pkt 5.A.2, należy pobrać próbki z różnych poziomów pojemnika.
Próbki mogą być także pobierane w trakcie opróżniania pojemników, przy czym pierwszą frakcję należy odrzucić.
W każdym przypadku całkowita pobrana objętość nie może być mniejsza niż 10 l.
6.2.A.5. Bloki paszy i lizawki mineralne
Po wybraniu wymaganej liczby bloków i lizawek w celu pobrania próbek zgodnie z pkt 5.A.2. należy pobrać część każdego bloku lub lizawki.
6.2.B. W odniesieniu do kontroli niepożądanych substancji lub produktów, które mogą być rozmieszczone niejednolicie w paszy, takich jak aflatoksyny, sporysz, rącznik pospolity i krotalaria w materiałach paszowych
Kontrolowaną partię należy podzielić na pewną liczbę równych w przybliżeniu części, odpowiadającą liczbie próbek zbiorczych, określonej w pkt 5.B.3. Jeżeli liczba ta jest większa niż jeden, całkowita liczba pierwotnych próbek określona w pkt 5.B.2. musi być rozdzielona równo w przybliżeniu na wszystkie części. Następnie należy pobrać próbki o zbliżonej wielkości (4) tak, aby całkowita masa próbek z każdej części była nie mniejsza niż wymagane 4 kg dla każdej próbki zbiorczej. Próbki pierwotne pobierane z różnych części nie mogą być łączone.
6.3. Przygotowanie próbek zbiorczych
6.3.A. W odniesieniu do kontroli substancji lub produktów jednolicie rozmieszczonych w paszy
Próbki pierwotne miesza się w jedną próbkę zbiorczą.
6.3.B. W odniesieniu do kontroli niepożądanych substancji lub produktów, które mogą być rozmieszczone niejednolicie w paszy, takich jak aflatoksyny, sporysz, rącznik pospolity i krotalaria w materiałach paszowych
Pierwotne próbki z każdej części kontrolowanej partii należy zmieszać i podzielić na próbki zbiorcze w liczbie określonej w pkt 5.B.3, przy czym należy zanotować pochodzenie każdej próbki zbiorczej.
6.4. Przygotowanie próbek końcowych
Materiał każdej próbki zbiorczej należy dokładnie wymieszać w celu otrzymania próbki jednolitej (5). Jeśli jest to konieczne, próbkę zbiorczą należy najpierw zredukowana do wielkości nie mniejszej niż 2 kg lub 2 l (próbka zredukowana) przy użyciu mechanicznego rozdzielacza lub metodą kwadratów.
Należy przygotować co najmniej trzy próbki końcowe tej samej wielkości i dostosowane do ilościowych wymagań określonych w pkt 5.A.4 lub 5.B.4. Każdą próbkę należy umieścić w odpowiednim pojemniku. Należy podjąć wszelkie środki ostrożności, aby zapobiec zmianie składu próbki, zanieczyszczeniu lub sfałszowaniu, które mogą nastąpić w czasie transportu lub przechowywania.
6.5. Pakowanie próbek końcowych
Pojemniki opakowań należy opieczętować i oznakować (wszelkie etykiety muszą być opieczętowane) w taki sposób, aby nie można było otworzyć pojemnika bez naruszania pieczęci.
7. PROTOKÓŁ POBIERANIA PRÓBEK
Po każdym pobraniu próbek należy sporządzić protokół umożliwiający jednoznaczną identyfikację każdej kontrolowanej partii.
8. PRZEZNACZENIE PRÓBEK
Z każdej próbki zbiorczej co najmniej jedną próbkę końcową należy niezwłocznie przesłać do laboratorium upoważnionego do prowadzenia badań wraz z informacjami niezbędnymi dla analityka.
(1) Jeżeli uzyskana liczba nie jest liczbą całkowitą, należy ją zaokrąglić do następującej po niej liczby całkowitej.
(2) W przypadku opakowań lub pojemników, których zawartość nie przekracza 1 kg lub 1 l, oraz bloków i lizawek o wadze nie większej niż 1 kg, próbkę pierwotną stanowi zawartość jednego oryginalnego opakowania lub pojemnika bądź jeden blok lub jedna lizawka.
(3) Metody przewidziane w odniesieniu do 5.A stosuje się w kontroli aflatoksyny, sporyszu, rącznika pospolitego i krotalarii w paszach pełnoporcjowych i uzupełniających.
(4) W przypadku pasz opakowanych pobiera się część zawartości opakowań przy użyciu sondy lub szufelki po uprzednim oddzielnym opróżnieniu opakowań, jeśli jest to konieczne.
(5) Jakiekolwiek zbrylenia należy rozbić (jeśli to konieczne odseparować je, po rozbryleniu, zwrócić do próbki) oddzielnie dla każdej próbki zbiorczej.
ZAŁĄCZNIK II
PRZEPISY OGÓLNE DOTYCZĄCE METODYKI ANALIZY PASZ
A. PRZYGOTOWANIE PRÓBEK DO BADAŃ
1. Cel
Poniżej opisany tryb postępowania dotyczy przygotowania do badań próbek końcowych, przesyłanych do laboratoriów kontrolnych po pobraniu zgodnie z przepisami określonymi w załączniku I.
Próbkę do badań należy przygotować tak, aby odważone ilości, zgodnie z metodyką postępowania analitycznego, były jednorodne i reprezentatywne w odniesieniu do próbki końcowej.
2. Zalecane środki ostrożności
Wybór trybu postępowania przy przygotowaniu próbek zależy od stosowanych metod analizy. Z tego względu bardzo ważne jest upewnienie się, czy wybrany tryb postępowania jest właściwy w odniesieniu do stosowanej metody analizy.
Wszystkie konieczne czynności należy wykonywać w taki sposób, aby zapobiec zanieczyszczeniu i zmianie składu próbki do badań.
Rozdrabnianie, mieszanie, przesiewanie należy wykonywać możliwie szybko, przy ograniczonym dostępie powietrza i światła do próbki do badań. Nie należy stosować młynków i rozdrabniaczy, które podczas pracy powodują nagrzewanie próbki do badań.
W przypadku pasz szczególnie wrażliwych na ciepło zaleca się ręczne rozdrabnianie. Ponadto zastosowany sprzęt nie może stanowić źródła zanieczyszczenia pasz mikroelementami.
Jeżeli przygotowanie próbki do badań nie może być przeprowadzone bez znacznych zmian poziomu wilgotności próbki, poziom jej wilgotności oznacza się przed i po przygotowaniu próbki do badań, zgodnie z metodą, o której mowa w załączniku III część A.
3. Sposób postępowania
Podzielić próbkę na odpowiednie mniejsze próbki do celów analizy i celów referencyjnych, stosując stosowne techniki podziału, np. naprzemienne przekopywanie, przerzucanie miejscowe lub rotacyjne. Stożkowanie i dzielenie na ćwiartki nie są zalecane, ponieważ mogą dawać próbki o dużym błędzie podziału. Próbkę referencyjną przechowywać w odpowiednim czystym i suchym pojemniku, wyposażonym w hermetyczny korek i przygotować próbki do analizy o masie co najmniej 100 g w sposób opisany poniżej.
3.1. Pasze, które mogą być zmielone
Jeżeli w metodyce postępowania analitycznego nie ustalono inaczej, po zmieleniu, jeżeli to konieczne, całą próbkę przesiać przez sito o otworach w kształcie kwadratów o bokach 1 mm (zgodnie z zaleceniem ISO R565). Unikać nadmiernego rozdrobnienia.
Przesianą próbkę zamieszać i umieścić w odpowiednim, czystym i suchym pojemniku, wyposażonym w hermetyczny korek. Bezpośrednio przed odważeniem próbki do badań – powtórnie zamieszać.
3.2. Pasze, które mogą być zmielone po wysuszeniu
Jeżeli w metodyce postępowania analitycznego nie ustalono inaczej, wysuszyć próbkę w celu obniżenia poziomu wilgotności do 8–12 %, zgodnie ze wstępną procedurą suszenia opisaną w pkt 4.3 w metodyce oznaczania wilgotności, o której mowa w załączniku III część A. Następnie postępować w sposób określony w pkt 3.1.
3.3. Pasze płynne lub półpłynne
Próbkę umieścić w odpowiednim, czystym i suchym pojemniku, wyposażonym w hermetyczny korek. Bezpośrednio przed odważeniem próbki do badań – dokładnie zamieszać.
3.4. Inne pasze
Próbki, które nie mogą być przygotowane w jeden z wyżej opisanych sposobów, należy przygotować w taki sposób, aby odważone ilości wymagane do metodyki postępowania analitycznego były jednorodne i reprezentatywne dla próbki końcowej.
4. Przechowywanie próbek
Próbki należy przechowywać w temperaturze, która nie spowoduje zmiany ich składu. Próbki przeznaczone do badania witamin lub substancji, które są szczególnie wrażliwe na światło, należy przechowywać w brązowych, szklanych pojemnikach.
B. PRZEPISY DOTYCZĄCE ODCZYNNIKÓW I APARATURY UŻYWANEJ W METODYCE POSTĘPOWANIA ANALITYCZNEGO
|
1.
|
Jeżeli w metodyce postępowania analitycznego nie ustalono inaczej, wszystkie stosowane odczynniki muszą mieć stopień czystości do analizy. W przypadku oznaczania mikroelementów czystość odczynników należy sprawdzić przez wykonanie ślepej próby. Zależnie od uzyskanych wyników, może być konieczne dalsze oczyszczanie odczynników.
|
|
2.
|
Jeżeli w metodyce postępowania analitycznego nie podano konkretnego rozpuszczalnika lub rozcieńczalnika, w przypadku czynności wymagających przygotowania roztworów, rozcieńczania, spłukiwania lub przemywania należy stosować wodę. Podstawowym wymogiem jest stosowanie wody demineralizowanej lub destylowanej. W szczególnych przypadkach, wymienionych w metodyce postępowania analitycznego, wodę należy oczyścić w specjalny sposób.
|
|
3.
|
Ze względu na aparaturę i sprzęt, które zwykle stosuje się w laboratoriach kontrolnych, w metodyce postępowania analitycznego wymienia się tylko tę aparaturę i sprzęt, które wymagają szczególnego zastosowania lub mają specjalny charakter. Wszystkie urządzenia muszą być czyste, zwłaszcza gdy są oznaczane bardzo małe ilości substancji.
|
C. STOSOWANIE METODYKI POSTĘPOWANIA ANALITYCZNEGO I WYRAŻANIE WYNIKÓW
1. Postępowanie ekstrakcyjne
W kilku metodach określono konkretne postępowanie ekstrakcyjne. Z reguły można zastosować inne postępowanie ekstrakcyjne, niż to, o którym mowa w danej metodyce, pod warunkiem, że dowiedziono, iż w porównaniu z postępowaniem określonym w metodyce stosowane postępowanie ma równoważną wydajność ekstrakcji w odniesieniu do analizowanej matrycy.
2. Postępowanie oczyszczające
W kilku metodach określono konkretne postępowanie oczyszczające. Z reguły można zastosować inne postępowanie oczyszczające, niż to, o którym mowa w danej metodyce, pod warunkiem, że dowiedziono, iż w porównaniu z postępowaniem określonym w metodyce stosowane postępowanie daje równoważne wyniki analityczne w odniesieniu do analizowanej matrycy.
3. Podawanie zastosowanych metod analitycznych w sprawozdaniu
Na ogół w odniesieniu do oznaczania zawartości każdej substancji w paszy określona jest jedna metoda analityczna. Jeżeli podanych jest kilka metod, w sprawozdaniu z badań należy wymienić konkretną metodę zastosowaną w laboratorium kontrolnym.
4. Liczba oznaczeń
Wynik podany w sprawozdaniu z badań stanowi średnią wartość co najmniej z dwóch oznaczeń wykonanych z oddzielnych części tej samej próbki i charakteryzuje się zadowalającą powtarzalnością.
Jednakże w przypadku substancji niepożądanych, jeżeli wynik pierwszego oznaczenia jest znacznie (o > 50 %) niższy niż ustalona zawartość maksymalna, nie są konieczne dodatkowe oznaczenia, pod warunkiem, że stosowane jest odpowiednie postępowanie w odniesieniu do jakości.
W przypadku kontroli deklarowanej zawartości substancji lub składnika, jeżeli wynik pierwszego oznaczenia potwierdza deklarowaną zawartość, tj. wyniki analizy mieszczą się w dopuszczalnym zakresie zmienności co do deklarowanej zawartości, nie są konieczne dodatkowe oznaczenia, pod warunkiem, że stosowane jest odpowiednie postępowanie w odniesieniu do jakości.
W niektórych przypadkach dopuszczalny zakres zmienności określony jest w prawodawstwie, np. w dyrektywie Rady 79/373/EWG (1).
5. Podawanie wyników analizy w sprawozdaniu
Wynik analizy należy wyrażać w sposób podany w metodyce postępowania analitycznego, zawierać odpowiednią liczbę cyfr znaczących i, jeżeli to konieczne, musi być korygowany do wilgotności próbki końcowej przed przygotowaniem.
6. Niepewność pomiaru i stopień odzysku w przypadku analizy substancji niepożądanych
W odniesieniu do substancji niepożądanych w rozumieniu dyrektywy 2002/32/WE, w tym dioksyn i dioksynopodobnych polichlorowanych bifenyli (PCB), uznaje się, że produkt przeznaczony do żywienia zwierząt jest niezgodny pod względem ustalonej zawartości maksymalnej, w przypadku gdy wynik analizy wskazuje na przekroczenie zawartości maksymalnej z uwzględnieniem rozszerzonej niepewności pomiaru i poprawki na stopień odzysku. Badane stężenie po uwzględnieniu poprawki na stopień odzysku i odjęciu rozszerzonej niepewności pomiaru od wyniku stanowi podstawę oceny zgodności. Postępowanie to ma zastosowanie jedynie w przypadkach, gdy stosowana metoda analizy pozwala na ustalenie wartości niepewności pomiaru i stopnia odzysku (nie jest to możliwe np. w przypadku analizy mikroskopowej).
Wynik analizy przedstawia się w sposób następujący (o ile stosowana metoda analizy pozwala na ustalenie wartości niepewności pomiaru i stopnia odzysku):
|
a)
|
jako skorygowany na stopień odzysku, przy czym stopień ten należy wskazać. Korekta na odzysk nie jest konieczna, jeśli stopień odzysku wynosi 90–110 %;
|
|
b)
|
jako x +/- U, gdzie x oznacza wynik analizy, a U rozszerzoną niepewność pomiaru, przy zastosowaniu współczynnika rozszerzenia w wysokości 2, który daje wynik w przedziale ufności ok. 95 %.
|
Jeżeli wynik analizy jest jednak znacznie (o > 50 %) niższy niż ustalona zawartość maksymalna oraz pod warunkiem, że stosowane jest odpowiednie postępowanie w odniesieniu do jakości, a analiza wykonywana jest jedynie do celów sprawdzenia zgodności z przepisami prawnymi, wynik analizy można podać bez korekty na odzysk; w takim przypadku można pominąć podawanie stopnia odzysku i niepewności pomiaru.
(1) Dz.U. L 86 z 6.4.1979, s. 30.
ZAŁĄCZNIK III
METODY ANALIZY SKŁADU MATERIAŁÓW I MIESZANEK PASZOWYCH
A. OZNACZANIE WILGOTNOŚCI
1. Cel i zakres stosowania metody
Metoda służy do oznaczania wilgotności pasz. Należy zauważyć, że w przypadku pasz zawierających substancje lotne, np. kwasy organiczne, podczas oznaczania wilgotności oznaczana jest również znaczna ilość substancji lotnych.
Metody nie stosuje się do badania produktów mlecznych występujących jako materiały paszowe, związków mineralnych, mieszanin składających się głównie ze związków mineralnych, tłuszczów i olejów pochodzenia zwierzęcego i roślinnego oraz nasion i owoców roślin oleistych.
2. Sposób przeprowadzenia metody
Próbka jest suszona w warunkach dostosowanych do właściwości pasz. Obniżenie masy jest określane przez ważenie. Jeżeli pasze stałe zawierają duże ilości wilgotności, konieczne jest przeprowadzenie wstępnego suszenia.
3. Aparatura i sprzęt
|
3.1.
|
Rozdrabniacz wykonany z materiałów niepochłaniających wilgotności, łatwy do czyszczenia, pozwalający na szybkie i równomierne rozdrobnienie materiału bez nadmiernego ogrzewania, w sposób ograniczający do minimum kontakt z powietrzem, spełniający wymagania określone w pkt 4.1.1 i pkt 4.1.2 (np. młotek, chłodzony wodą mikrorozdrabniacz, składany stożkowy młynek albo rozdrabniacz wolnoobrotowy lub wyposażony w koło zębate).
|
|
3.2.
|
Waga analityczna ważąca z dokładnością do 1 mg.
|
|
3.3.
|
Suche naczynka z nierdzewnego metalu lub ze szkła z przykrywkami umożliwiającymi hermetyczne zamknięcie, o powierzchni roboczej umożliwiającej równomierne rozprowadzenie badanej próbki w warstwie 0,3 g/cm2.
|
|
3.4.
|
Suszarka elektryczna z termostatem (± 2 oC), dobrze wentylowana i pozwalająca na szybką regulację temperatury (1).
|
|
3.5.
|
Termostatowana suszarka próżniowa zaopatrzona w pompę olejową oraz mechanizm umożliwiający nawiew gorącego suchego powietrza lub wprowadzenie czynnika suszącego (np. tlenku wapnia).
|
|
3.6.
|
Eksykator z płytką z grubego perforowanego metalu lub porcelany, zawierający efektywny czynnik suszący.
|
4. Sposób postępowania
|
Uwaga
|
:
|
Czynności opisane w tej części należy wykonywać natychmiast po otworzeniu opakowania próbki. Analizę należy wykonać co najmniej w 2 powtórzeniach.
|
4.1. Przygotowanie
4.1.1. Pasze inne niż określone w pkt 4.1.2 i pkt 4.1.3
Pobrać co najmniej 50 g próbki. Jeżeli to konieczne, rozdrobnić lub podzielić w taki sposób, aby zapobiec jakimkolwiek zmianom zawartości wilgotności (zob. pkt 6).
4.1.2. Zboża i kasze
Pobrać co najmniej 50 g próbki. Rozdrobnić na cząstki, które przechodzą przez oczka sita o średnicy 0,5 mm co najmniej w 50 % i pozostają w ilości nie wyższej niż 10 % na sicie o oczkach o średnicy 1 mm.
4.1.3. Pasze ciekłe lub w postaci pasty, pasze z przewagą olejów lub tłuszczów
Pobrać 25 g próbki, zważyć z dokładnością do 10 mg, dodać odpowiednią ilość bezwodnego piasku odważonego z dokładnością do 10 mg i zmieszać do uzyskania homogennego produktu.
4.2. Suszenie
4.2.1. Pasze inne niż określone w pkt 4.2.2 i 4.2.3
Zważyć, z dokładnością do 1 mg, naczynko z przykrywką (pkt 3.3). Do naczynka odważyć, z dokładnością do 1 mg, około 5 g próbki i równo rozmieścić. Wstawić naczynko bez przykrywki do suszarki podgrzanej do temperatury 103 oC. Aby zapobiec nadmiernemu spadkowi temperatury, umieścić naczynko w suszarce tak szybko, jak to możliwe. Suszyć przez cztery godziny od czasu, gdy suszarka ponownie osiągnie temperaturę 103 oC. Nałożyć przykrywkę na naczynko, wyjąć je z suszarki, pozostawić do schłodzenia w eksykatorze (pkt 3.6) na 30 do 45 minut i zważyć z dokładnością do 1 mg.
Pasze z przeważającym udziałem oleju lub tłuszczu suszyć ponownie w suszarce przez 30 minut w temperaturze 130 oC. Różnica pomiędzy dwoma ważeniami nie może przekraczać 0,1 % wilgotności.
4.2.2. Zboża, mąki, kasze i mączki
Zważyć, z dokładnością do 0,5 mg, naczynko z przykrywką (pkt 3.3). Do naczynka odważyć, z dokładnością do 1 mg, około 5 g rozdrobnionej próbki i równo rozmieścić. Wstawić naczynko bez przykrywki do suszarki podgrzanej do temperatury 130 oC. Aby zapobiec nadmiernemu spadkowi temperatury, umieścić naczynko w suszarce tak szybko, jak to możliwe. Suszyć przez dwie godziny od czasu, gdy suszarka ponownie osiągnie temperaturę 130 oC. Nałożyć przykrywkę na naczynko, wyjąć je z suszarki, pozostawić do schłodzenia w eksykatorze (pkt 3.6) na 30 do 45 minut i zważyć z dokładnością do 1 mg.
|
4.2.3.
|
Mieszanki paszowe zawierające ponad 4 % sacharozy lub laktozy: materiały paszowe, takie jak chleb świętojański, hydrolizowane produkty zbożowe, słód jęczmienny, susz buraczany, hydrolizaty rybne i cukrowe, mieszanki z ponad 25 % udziałem soli mineralnych zawierających wodę krystalizacyjną.
Zważyć, z dokładnością do 0,5 mg, naczynko z przykrywką (pkt 3.3). Do naczynka odważyć, z dokładnością do 1 mg, około 5 g próbki i równo rozmieścić. Wstawić naczynko bez przykrywki do suszarki próżniowej (pkt 3.5), podgrzanej do temperatury od 80 do 85 oC. Aby zapobiec nadmiernemu spadkowi temperatury, umieścić naczynko w suszarce tak szybko, jak to możliwe.
Podwyższyć ciśnienie do 100 Torr i pozostawić w celu wysuszenia na cztery godziny pod działaniem tego ciśnienia, w strumieniu gorącego, suchego powietrza albo stosując czynnik suszący (ok. 300 g na 20 próbek). W drugim przypadku odłączyć pompę próżniową po uzyskaniu odpowiedniego ciśnienia. Czas suszenia liczyć od chwili, gdy suszarka ponownie osiągnie temperaturę od 80 do 85 oC. Ostrożnie zrównać ciśnienie w suszarce z ciśnieniem atmosferycznym. Otworzyć suszarkę, szybko nałożyć przykrywkę na naczynko, wyjąć je z suszarki, pozostawić do schłodzenia w eksykatorze (pkt 3.6) na 30–45 minut i zważyć z dokładnością do 1 mg. Suszyć ponownie w suszarce próżniowej przez 30 minut o temperaturze od 80 do 85 oC i zważyć powtórnie. Różnica pomiędzy dwoma ważeniami nie może przekraczać 0,1 % wilgotności.
|
4.3. Suszenie wstępne
4.3.1. Pasze inne niż określone w pkt 4.3.2
Pasze stałe o wysokim poziomie wilgotności, trudne do rozdrabniania, należy poddać wstępnemu suszeniu w następujący sposób:
Odważyć 50 g, z dokładnością do 10 mg, nierozdrobnionej próbki (pasze sprasowane lub zbrylone można w razie potrzeby wstępnie rozdzielić) do odpowiedniego naczynia (np. płytka aluminiowa o wymiarach 20 cm × 12 cm z obwódką o wysokości 0,5 cm). Suszyć w suszarce o temperaturze od 60 do 70 oC, aż poziom wilgotności obniży się do 8–12 %. Wyjąć próbkę z suszarki, pozostawić bez przykrycia do schłodzenia w laboratorium przez godzinę i zważyć z dokładnością do 10 mg. Szybko rozdrobnić, postępując w sposób określony w pkt 4.1.1, i suszyć w sposób określony w pkt 4.2.1 lub 4.2.3, w zależności od rodzaju paszy.
4.3.2. Zboża
Ziarno zbóż o wilgotności wyższej niż 17 % należy poddać wstępnemu suszeniu w następujący sposób:
Odważyć, z dokładnością do 10 mg, 50 g nierozdrobnionego ziarna do odpowiedniego naczynia (np. płytka aluminiowa o wymiarach 20 × 12 cm z obwódką o wysokości 0,5 cm). Suszyć w suszarce od 5 do 7 minut w temperaturze 130 oC. Wyjąć próbkę z suszarki, pozostawić bez przykrycia do schłodzenia w laboratorium przez dwie godziny i zważyć z dokładnością do 10 mg. Szybko rozdrobnić, postępując w sposób określony w pkt 4.1.2, i suszyć w sposób określony w pkt 4.2.2.
5. Obliczanie wyników
Wilgotność (X) jako udział procentowy w próbce obliczana jest według następujących wzorów:
5.1. Suszenie bez wstępnego podsuszania

gdzie:
|
m
|
=
|
początkowa masa w g badanej próbki
|
|
m0
|
=
|
masa w g badanej próbki po wysuszeniu
|
5.2. Suszenie z podsuszaniem

gdzie:
|
m
|
=
|
początkowa masa w g badanej próbki
|
|
m1
|
=
|
masa w g badanej próbki po wstępnym podsuszaniu
|
|
m2
|
=
|
masa w g badanej próbki po rozdrobnieniu
|
|
m0
|
=
|
masa w g badanej próbki po wysuszeniu
|
5.3. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,2 % bezwzględnej wartości wilgotności.
6. Objaśnienia
Jeżeli rozdrabnianie próbki jest konieczne i wpływa na poziom wilgotności w produkcie, wyniki analizy składników paszowych należy skorygować o poziom wilgotności w początkowym stanie próbki.
B. OZNACZANIE WILGOTNOŚCI OLEJÓW I TŁUSZCZÓW POCHODZENIA ZWIERZĘCEGO I ROŚLINNEGO
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości wody i substancji lotnych w tłuszczach i olejach pochodzenia zwierzęcego oraz roślinnego.
2. Sposób przeprowadzenia metody
Próbka jest suszona w temperaturze 103 oC do uzyskania stałej masy (ubytek masy między dwoma kolejnymi ważeniami musi być równy lub mniejszy niż 1 mg). Ubytek masy jest określany metodą wagową.
3. Aparatura i sprzęt
|
3.1.
|
Naczynie z płaskim dnem wykonane z materiału nierdzewnego, o średnicy od 8 do 9 cm i wysokości około 3 cm.
|
|
3.2.
|
Termometr rtęciowy ze wzmocnionym zbiornikiem i ekspansyjną rurką w górnym końcu, z podziałką od 80 do co najmniej 110 oC i o długości około 10 cm.
|
|
3.3.
|
Łaźnia piaskowa lub elektryczna płyta grzewcza.
|
|
3.4.
|
Eksykator zawierający efektywny środek suszący.
|
4. Sposób postępowania
Odważyć, z dokładnością do 1 mg, około 20 g homogennej próbki do suchego, zważonego naczynia (3.1), zawierającego termometr (3.2). Ogrzewać na łaźni piaskowej lub płycie grzewczej (3.3) ciągle mieszając termometrem, tak aby uzyskać wzrost temperatury do 90 oC w czasie około 7 minut.
Zmniejszyć ogrzewanie, obserwując częstotliwość odrywania się pęcherzyków powietrza od dna naczynia. Temperatura nie może przekroczyć 105 oC. Kontynuować mieszanie, pocierając dno naczynia do czasu, aż przestaną się tworzyć pęcherzyki.
W celu całkowitego odparowania wilgotności, ogrzewać próbkę kilka razy do temperatury 103 oC ± 2 oC, schładzając do 93 oC pomiędzy kolejnymi ogrzewaniami. Następnie schłodzić próbkę do temperatury pokojowej w eksykatorze (3.4) i zważyć. Powtarzać ogrzewanie, dopóki ubytek masy pomiędzy dwoma kolejnymi ważeniami nie będzie większy niż 2 mg.
|
Uwaga
|
:
|
Wzrost masy próbki po powtórnym ogrzewaniu wskazuje na utlenienie się tłuszczu. W takim przypadku do obliczenia wyniku wziąć ostatnią masę próbki, przed wzrostem jej masy.
|
5. Obliczanie wyników
Wilgotność (X) jako udział procentowy w próbce obliczana jest według następującego wzoru:

gdzie:
|
m
|
=
|
masa w g badanej próbki
|
|
m1
|
=
|
masa w g naczynia z zawartością przed ogrzewaniem
|
|
m2
|
=
|
masa w g naczynia z zawartością po ogrzewaniu
|
Wyniki oznaczania niższe niż 0,05 % należy zapisać, używając określenia „niższe niż 0,05 %”.
Powtarzalność
Różnica wilgotności pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,05 % wartości bezwzględnej.
C. OZNACZANIE ZAWARTOŚCI BIAŁKA SUROWEGO
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości białka surowego w paszach na podstawie zawartości azotu oznaczonego metodą Kjeldahla.
2. Sposób przeprowadzenia metody
Próbka jest mineralizowana kwasem siarkowym w obecności katalizatora. Kwaśny roztwór jest alkalizowany z użyciem roztworu wodorotlenku sodu. Amoniak destyluje się i zbiera w znanej ilości roztworu kwasu siarkowego, którego nadmiar jest miareczkowany mianowanym roztworem wodorotlenku sodu.
Ewentualnie uwolniony amoniak jest destylowany do nadmiaru roztworu kwasu borowego, a następnie miareczkowany roztworem kwasu chlorowodorowego lub siarkowego.
3. Odczynniki i roztwory
|
3.2.
|
Katalizator: tlenek miedzi(II) CuO lub siarczan miedzi(II) CuSO4 5H2O pentahydrat.
|
|
3.4.
|
Kwas siarkowy, ρ20 = 1,84 g/ml.
|
|
3.5.
|
Kwas siarkowy, mianowany roztwór objętościowy o stężeniu c(H2SO4) = 0,25 mol/l.
|
|
3.6.
|
Kwas siarkowy, mianowany roztwór objętościowy o stężeniu c(H2SO4) = 0,10 mol/l.
|
|
3.7.
|
Kwas siarkowy, mianowany roztwór objętościowy o stężeniu c(H2SO4) = 0,05 mol/l.
|
|
3.8.
|
Czerwień metylowa, wskaźnik: rozpuścić 300 mg czerwieni metylowej w 100 ml etanolu, σ = 95–96 % (v/v).
|
|
3.9.
|
Wodorotlenek sodu (może być techniczny), roztwór o stężeniu β = 40 g/100 ml (m/v: 40 %).
|
|
3.10.
|
Wodorotlenek sodu, mianowany roztwór objętościowy o stężeniu c(NaOH) = 0,25 mol/l.
|
|
3.11.
|
Wodorotlenek sodu, mianowany roztwór objętościowy o stężeniu c(NaOH) = 0,10 mol/l.
|
|
3.12.
|
Granulowany pumeks, przemyty kwasem chlorowodorowym i wyprażony.
|
|
3.13.
|
Acetanilid (temperatura topnienia = 114 oC, zawartość N = 10,36 %).
|
|
3.14.
|
Sacharoza, niezawierająca azotu.
|
|
3.15.
|
Kwas borowy (H3BO3).
|
|
3.16.
|
Roztwór czerwieni metylowej, wskaźnik: rozpuścić 100 mg czerwieni metylowej w 100 ml etanolu lub metanolu.
|
|
3.17.
|
Roztwór zieleni bromokrezolowej: rozpuścić 100 mg zieleni bromokrezolowej w 100 ml etanolu lub metanolu.
|
|
3.18.
|
Roztwór kwasu borowego (od 10 g/l do 40 g/l, w zależności od stosowanej aparatury i sprzętu)
Przy stosowaniu kolorymetrycznego punktu końcowego do roztworów kwasu borowego należy dodać wskaźniki czerwieni metylowej i zieleni bromokrezolowej. Jeżeli przygotowuje się 1 l roztworu kwasu borowego, przed dostosowaniem objętości należy dodać 7 ml roztworu czerwieni metylowej (3.16) i 10 ml roztworu zieleni bromokrezolowej (3.17).
Odczyn pH roztworu kwasu borowego może różnić się między seriami w zależności od użytej wody. Często do uzyskania pozytywnej próby ślepej konieczne jest dostosowanie pH przy pomocy małej objętości zasady.
|
Uwaga
|
:
|
Dodanie ok. 3–4 ml NaOH (3.11) do 1 l kwasu borowego o stężeniu 10 g/l zwykle zapewnia dobre dostosowanie. Roztwór należy przechowywać w temperaturze pokojowej oraz chronić przed światłem i źródłami oparów amoniaku.
|
|
|
3.19
|
Kwas chlorowodorowy, mianowany roztwór objętościowy o stężeniu c(HCl) = 0,10 mol/l.
|
Uwaga
|
:
|
Można zastosować inne stężenia roztworów objętościowych (3.5, 3.6, 3.7, 3.10, 3.11 i 3.19), jeżeli zostanie to uwzględnione w obliczeniach. Stężenie należy podawać zawsze z dokładnością do czterech miejsc po przecinku.
|
|
4. Aparatura i sprzęt
Aparatura i sprzęt odpowiedni do przeprowadzenia mineralizacji, destylacji i miareczkowania według metody Kjeldahla.
5. Sposób postępowania
5.1. Mineralizacja
Odważyć, z dokładnością do 0,001 g, 1g próbki i przenieść do kolby aparatu do mineralizacji. Dodać 15 g siarczanu potasu (3.1), odpowiednią ilość katalizatora (3.2) (od 0,3 do 0,4 g tlenku miedzi(II) lub od 0,9 do 1,2 g pentahydratu siarczanu miedzi(II)), 25 ml kwasu siarkowego (3.4) oraz, w razie potrzeby, kilka granulek pumeksu (3.12) i zamieszać.
Początkowo ogrzewać kolbę ostrożnie, w razie konieczności mieszając od czasu do czasu, aż do zwęglenia substancji i zaniku piany; następnie zwiększyć intensywność ogrzewania aż do trwałego wrzenia roztworu. Ogrzewanie jest właściwe, jeżeli wrzący kwas kondensuje się na ściankach kolby. Zapobiegać przegrzewaniu się ścianek i przylepianiu do nich organicznych cząstek.
Od chwili gdy roztwór stanie się klarowny i przyjmie jasnozieloną barwę, kontynuować ogrzewanie przez dwie godziny, a następnie pozostawić do schłodzenia.
5.2. Destylacja
Ostrożnie dodać do kolby ilość wody wystarczającą do całkowitego rozpuszczenia siarczanów. Pozostawić do schłodzenia, a następnie w razie potrzeby dodać kilka granulek cynku (3.3). Postępować zgodnie z pkt 5.2.1 lub 5.2.2.
5.2.1. Destylacja do kwasu siarkowego
Umieścić w odbieralniku aparatu destylacyjnego odmierzone 25 ml kwasu siarkowego (3.5 lub 3.7), zależnie od przewidywanej zawartości azotu. Dodać kilka kropli wskaźnika czerwieni metylowej (3.8).
Podłączyć kolbę mineralizacyjną do chłodnicy aparatu destylacyjnego i zanurzyć koniec chłodnicy w cieczy znajdującej się w kolbie odbieralnika na głębokość co najmniej 1 cm (zob. objaśnienia pkt 8.3). Powoli wlać 100 ml roztworu wodorotlenku sodu (3.9) do kolby mineralizacyjnej, nie dopuszczając do strat amoniaku (zob. objaśnienia pkt 8.1). Ogrzewać kolbę aż do całkowitego oddestylowania amoniaku.
5.2.2. Destylacja do kwasu borowego
Jeżeli miareczkowanie zawartości amoniaku w destylacie wykonywane jest ręcznie, zastosowanie ma poniższe postępowanie. Jeżeli aparat destylacyjny jest w pełni zautomatyzowany i obejmuje miareczkowanie zawartości amoniaku w destylacie, należy stosować się do instrukcji producenta aparatu.
Umieścić kolbę odbieralnika zawierającą od 25 do 30 ml roztworu kwasu borowego (3.18) pod ujściem chłodnicy w taki sposób, aby rurka doprowadzająca znajdowała się pod powierzchnią nadmiaru roztworu kwasu borowego. Ustawić aparat destylacyjny, aby podał 50 ml roztworu wodorotlenku sodu (3.9). Obsługiwać aparat destylacyjny zgodnie z instrukcjami producenta i oddestylować amoniak uwolniony w wyniku dodania roztworu wodorotlenku sodu. Zebrać destylat w roztworze kwasu borowego. Ilość destylatu (czas destylacji z parą wodną) zależy od zawartości azotu w próbce. Postępować zgodnie z instrukcjami producenta.
|
Uwaga
|
:
|
W przypadku półautomatycznego aparatu destylacyjnego dodawanie nadmiaru wodorotlenku sodu i destylacja z parą wodną przeprowadzane są automatycznie.
|
5.3. Miareczkowanie
Postępować zgodnie z pkt 5.3.1 lub 5.3.2.
5.3.1. Kwas siarkowy
Miareczkować nadmiar kwasu siarkowego w kolbie odbieralnika roztworem wodorotlenku sodu (pkt 3.10 lub 3.11), w zależności od stężenia zastosowanego kwasu siarkowego, do uzyskania punktu końcowego.
5.3.2. Kwas borowy
Miareczkować zawartość kolby odbieralnika mianowanym roztworem objętościowym kwasu chlorowodorowego (3.19) lub kwasu siarkowego (3.6) przy pomocy biurety i odczytać ilość zużytego titrantu.
Przy stosowaniu kolorymetrycznego punktu końcowego punkt końcowy osiąga się w momencie pojawienia się pierwszego śladu różowego zabarwienia cieczy. Określić odczyt biurety z dokładnością do 0,05 ml. W wizualizacji punktu końcowego pomocna jest podświetlana płyta mieszadła magnetycznego lub detektor fotometryczny.
Można to przeprowadzić automatycznie za pomocą parowego aparatu destylacyjnego z funkcją automatycznego miareczkowania.
Przy obsłudze konkretnego aparatu destylacyjnego lub aparatu do miareczkowania stosować się do instrukcji producenta.
|
Uwaga
|
:
|
Jeżeli stosowany jest system automatycznego miareczkowania, rozpoczyna się ono bezpośrednio po destylacji i stosowany jest 1 % roztwór kwasu borowego (3.18).
Jeżeli stosowany jest w pełni automatyczny aparat destylacyjny, automatyczne miareczkowanie amoniaku można przeprowadzić również z wykryciem punktu końcowego przy pomocy potencjometrycznego systemu pH.
W tym przypadku stosowany jest automatyczny aparat do miareczkowania z pehametrem. Pehametr musi być odpowiednio skalibrowany w zakresie od pH 4 do pH 7 za pomocą zwykłych laboratoryjnych metod kalibracji pH.
Punkt końcowy pH w miareczkowaniu osiąga się przy wartości pH równej 4,6, co stanowi najwyższy punkt krzywej miareczkowania (punkt przegięcia).
|
5.4. Ślepa próba
W celu potwierdzenia, że zastosowane odczynniki nie zawierają azotu, przeprowadzić ślepą próbę (mineralizację, destylację i miareczkowanie), stosując 1 g sacharozy (3.14) zamiast próbki.
6. Obliczanie wyników
Obliczenia wykonywane są zgodnie z pkt 6.1 lub 6.2.
6.1. Obliczenia do miareczkowania zgodnie z pkt 5.3.1
Zawartość białka surowego jako udział procentowy w masie obliczana jest według następującego wzoru:
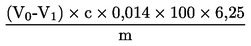
gdzie:
|
V0
|
=
|
objętość w ml NaOH (3.10 lub 3.11) zużytego do ślepej próby
|
|
V1
|
=
|
objętość w ml NaOH (3.10 lub 3.11) zużytego do miareczkowania próbki
|
|
c
|
=
|
stężenie w mol/l wodorotlenku sodu (3.10 lub 3.11)
|
|
m
|
=
|
masa w g próbki
|
6.2 Obliczenia do miareczkowania zgodnie z pkt 5.3.2
6.2.1. Miareczkowanie kwasem chlorowodorowym
Zawartość białka surowego jako udział procentowy w masie obliczana jest według następującego wzoru:
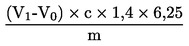
gdzie:
|
m
|
=
|
masa naważki w g
|
|
c
|
=
|
stężenie w mol/l mianowanego roztworu objętościowego kwasu chlorowodorowego (3.19)
|
|
V0
|
=
|
objętość w ml kwasu chlorowodorowego użytego do próby ślepej
|
|
V1
|
=
|
objętość w ml kwasu chlorowodorowego użytego do naważki
|
6.2.2. Miareczkowanie kwasem siarkowym
Zawartość białka surowego jako udział procentowy w masie obliczana jest według następującego wzoru:
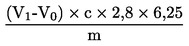
gdzie:
|
m
|
=
|
masa naważki w g
|
|
c
|
=
|
stężenie w mol/l mianowanego roztworu objętościowego kwasu siarkowego (3.6)
|
|
V0
|
=
|
objętość w ml kwasu siarkowego (3.6) użytego do próby ślepej
|
|
V1
|
=
|
objętość w ml kwasu siarkowego (3.6) użytego do naważki
|
7. Sprawdzenie metody
7.1. Powtarzalność
Różnica między wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
0,2 % wartości bezwzględnej, dla zawartości białka surowego niższej niż 20 %,
|
|
—
|
1,0 % najwyższego wyniku, dla zawartości białka surowego od 20 do 40 %,
|
|
—
|
0,4 % wartości bezwzględnej, dla zawartości białka surowego wyższej niż 40 %.
|
7.2. Dokładność
Przeprowadzić analizę (mineralizację, destylację i miareczkowanie), stosując od 1,5 do 2,0 g acetanilidu (3.13) w obecności 1 g sacharozy (3.14); 1 g acetanilidu zużywa 14,80 ml kwasu siarkowego (3.5). Stopień odzysku musi wynosić co najmniej 99 %.
8. Objaśnienia
|
8.1.
|
Sprzęt i aparatura mogą być obsługiwane ręcznie, półautomatycznie lub być całkowicie zautomatyzowane. W przypadku konieczności przeniesienia urządzenia pomiędzy etapami mineralizacji i destylacji, czynność tę należy wykonać bez jakichkolwiek strat. Jeżeli kolba aparatu destylacyjnego nie jest wyposażona we wkraplacz, dodać wodorotlenek sodu natychmiast przed podłączeniem kolby do chłodnicy, wlewając ciecz powoli po ściankach.
|
|
8.2.
|
Jeżeli roztwór przechodzi w postać stałą, ponownie przeprowadzić oznaczanie, stosując większe ilości kwasu siarkowego (3.4) niż podano wyżej.
|
|
8.3.
|
W przypadku produktów o niskiej zawartości azotu objętość kwasu siarkowego (3.7), która ma zostać dodana do kolby odbieralnika, może być w miarę potrzeby zmniejszona do 10 lub 15 ml i uzupełniona wodą do objętości 25 ml.
|
|
8.4.
|
Do celów rutynowego badania można stosować inne metody analizy w odniesieniu do oznaczania białka surowego, ale metoda Kjeldahla opisana w niniejszej części C stanowi metodę referencyjną. Równoważność wyników uzyskanych inną metodą (np. DUMAS) w porównaniu z metodą referencyjną musi zostać wykazana odrębnie dla każdej matrycy. Ze względu na fakt, iż nawet po potwierdzeniu równoważności wyniki uzyskane inną metodą mogą się nieznacznie różnić od wyników uzyskanych metodą referencyjną, konieczne jest podanie w sprawozdaniu z badań metody analitycznej zastosowanej do oznaczenia białka surowego.
|
D. OZNACZANIE MOCZNIKA
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości mocznika w paszach.
2. Sposób przeprowadzenia metody
Próbka ze środkiem klarującym tworzy w wodzie zawiesinę. Zawiesina jest filtrowana. Zawartość mocznika w filtracie jest oznaczana po dodaniu 4-dwumetyloaminobenzaldehydu (4-DMAB) poprzez pomiar gęstości optycznej przy długości fali 420 nm.
3. Odczynniki i roztwory
|
3.1.
|
Roztwór 4-dwumetyloaminobezaldehydu: rozpuścić 1,6 g 4-DMAB w 100 ml 96 % etanolu i dodać 10 ml kwasu chlorowodorowego (ρ201,19 g/ml). Trwałość tego odczynnika wynosi nie więcej niż dwa tygodnie.
|
|
3.2.
|
Roztwór Carreza I: rozpuścić w wodzie 21,9 g octanu cynkowego Zn(CH3COO)2 2H2O i 3 g lodowatego kwasu octowego. Uzupełnić do objętości 100 ml wodą.
|
|
3.3.
|
Roztwór Carreza II: rozpuścić w wodzie 10,6 g żelazocyjanku potasowego K4Fe(CN)6 3H2O. Uzupełnić do objętości 100 ml wodą.
|
|
3.4.
|
Aktywny węgiel nieabsorbujący mocznika (sprawdzony).
|
|
3.5.
|
Roztwór 0,1 % (w/v) mocznika.
|
4. Aparatura i sprzęt
|
4.1.
|
Mikser (tumbler): ok. 35 do 40 obr./min.
|
|
4.2.
|
Probówki: 160 × 16 mm ze szklanymi, szlifowanymi korkami.
|
5. Sposób postępowania
5.1. Analiza próbki
Odważyć, z dokładnością do 1 mg, 2 g próbki i umieścić razem z 1 g aktywnego węgla (3.4) w kolbie miarowej o pojemności 500 ml. Dodać 400 ml wody i 5 ml roztworu Carreza I (3.2), mieszać przez ok. 30 sekund, a następnie dodać 5 ml roztworu Carreza II (3.3). Mieszać przez 30 minut w mikserze. Uzupełnić do pełnej objętości kolby wodą, wstrząsnąć i przefiltrować.
Do probówek ze szklanymi szlifowanymi korkami przenieść po 5 ml przezroczystego, bezbarwnego filtratu, dodać 5 ml roztworu 4-DMAB (3.1) i zamieszać. Wstawić probówki do łaźni wodnej ustawionej na temperaturę 20 oC (+/- 4 oC). Po 15 minutach zmierzyć gęstość optyczną roztworu próbki spektrofotometrem przy długości fali 420 nm. Pomiar porównać ze ślepą próbą odczynnikową.
5.2. Krzywa kalibracyjna
Do kolb miarowych o pojemności 100 ml nalać 1, 2, 4, 5 i 10 ml roztworu mocznika (3.5) i uzupełnić do pełnej objętości kolb wodą. Odlać 5 ml każdego z roztworów, dodać do każdej kolby po 5 ml roztworu 4-DMAB (3.1), wymieszać i zmierzyć gęstość optyczną w podany wyżej sposób, porównując z roztworem kontrolnym zawierającym 5 ml 4-DMAB i 5 ml wody pozbawionej mocznika. Wykreślić krzywą kalibracyjną.
6. Obliczanie wyników
Na podstawie krzywej kalibracyjnej oznaczyć zawartość mocznika w próbce.
Wynik wyrazić jako udział procentowy w próbce.
7. Objaśnienia
|
7.1.
|
W przypadku gdy zawartość mocznika jest wyższa niż 3 %, zmniejszyć masę próbki do 1 g lub rozcieńczyć pierwotny roztwór tak, aby w 500 ml nie znajdowało się więcej niż 50 mg mocznika.
|
|
7.2.
|
W przypadku niskiej zawartości mocznika zwiększać masę próbki tak, aby filtrat pozostał przezroczysty i bezbarwny.
|
|
7.3.
|
Jeśli próbka zawiera proste związki azotowe takie jak aminokwasy, gęstość optyczną należy mierzyć przy długości fali 435 nm.
|
E. OZNACZANIE LOTNYCH ZWIĄZKÓW AZOTOWYCH
I. METODA MIKRODYFUZYJNA
1. Cel i zakres stosowania metody
Metoda służy do oznaczania w paszach zawartości lotnych związków azotowych, w przeliczeniu na amoniak.
2. Sposób przeprowadzenia metody
Próbka jest ekstrahowana wodą, a roztwór – klarowany i filtrowany. Lotne związki azotowe są oddzielane metodą mikrodyfuzji z użyciem roztworu węglanu potasu, zbierane w roztworze kwasu borowego i miareczkowane kwasem siarkowym.
3. Odczynniki i roztwory
|
3.1.
|
Kwas trójchlorooctowy, roztwór 20 % (w/v).
|
|
3.2.
|
Wskaźnik: rozpuścić 33 mg zieleni bromokrezolowej i 65 mg czerwieni metylowej w 100 ml etanolu o stężeniu od 95 do 96 % (v/v).
|
|
3.3.
|
Kwas borowy, roztwór: w kolbie miarowej o pojemności 1 l rozpuścić 10 g kwasu borowego w 200 ml etanolu o stężeniu od 95 do 96 % (v/v) i 700 ml wody. Dodać 10 ml wskaźnika (3.2). Zamieszać i, jeżeli to konieczne, doprowadzić barwę roztworu do jasnoczerwonej przez dodanie roztworu wodorotlenku sodu. 1 ml tego roztworu będzie pochłaniał maksymalnie 300 μg NH3.
|
|
3.4.
|
Węglan potasu, roztwór nasycony: rozpuścić 100 g węglanu potasu w 100 ml wrzącej wody. Schłodzić i przefiltrować.
|
|
3.5.
|
Kwas siarkowy o stężeniu 0,01 mol/l.
|
4. Aparatura i sprzęt
|
4.1.
|
Mikser (tumbler): ok. 35 do 40 obr./min.
|
|
4.2.
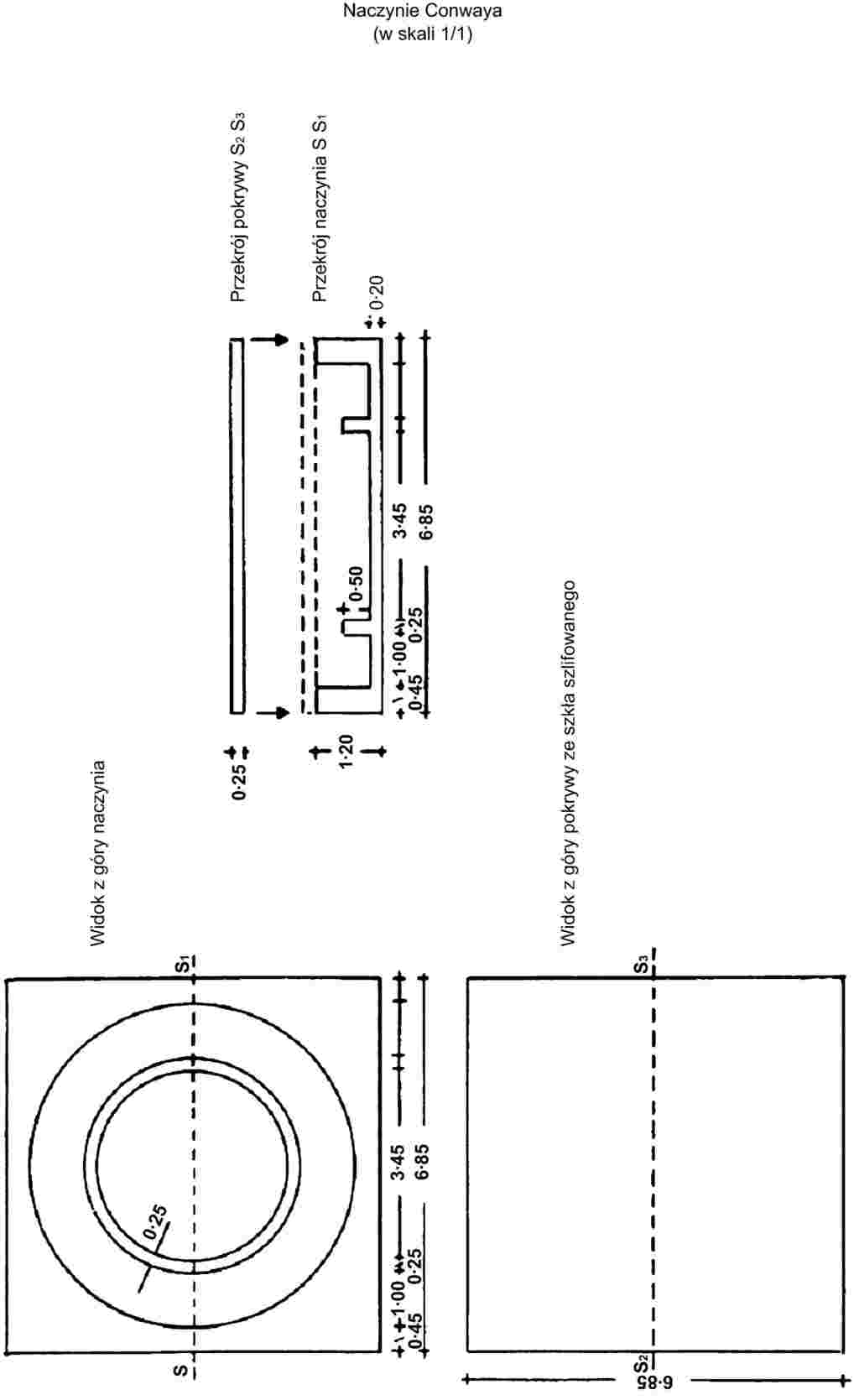
|
Szklane lub plastykowe naczynka Conwaya (zob. rysunek).
|
|
4.3.
|
Mikrobiurety ze skalą 1/100 ml.
|
5. Sposób postępowania
Odważyć, z dokładnością do 1 mg, 10 g próbki do kolby miarowej o pojemności 200 ml i dodać 100 ml wody. Miksować w mikserze przez 30 minut. Dodać 50 ml roztworu kwasu trójchlorooctowego (3.1), uzupełnić do pełnej objętości kolby wodą, energicznie wstrząsnąć i przefiltrować przez karbowany filtr.
Pipetą wprowadzić 1 ml roztworu kwasu borowego (3.3) do środkowej części naczynka Conwaya i 1 ml filtratu próbki na obrzeże naczynka. Częściowo przykryć nasmarowaną przykrywką. Szybko dodać kroplami 1 ml nasyconego roztworu węglanu potasu (3.4) na obrzeże naczynka i zamknąć pokrywką, tak aby naczynko było hermetyczne. Obracać ostrożnie naczynkiem w płaszczyźnie poziomej, aby doprowadzić do zmieszania dwóch odczynników. Inkubować przez co najmniej cztery godziny w temperaturze pokojowej lub godzinę w temperaturze 40 oC.
Mikrobiuretą (4.3) miareczkować lotne związki zaabsorbowane w kwasie borowym, stosując roztwór kwasu siarkowego (3.5).
Ślepą próbę przeprowadzić w ten sam sposób, lecz bez próbki przeznaczonej do analizy.
6. Obliczanie wyników
1 ml H2SO4 o stężeniu 0,01 mol/l odpowiada 0,34 mg amoniaku.
Przedstawić wynik jako udział procentowy w próbce.
Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
10 % wartości względnej, dla zawartości amoniaku poniżej 1,0 %,
|
|
—
|
0,1 % wartości bezwzględnej, dla zawartości amoniaku równej lub wyższej niż 1,0 %.
|
7. Objaśnienia
Jeżeli zawartość amoniaku w próbce przekracza 0,6 %, rozcieńczyć filtrat.
II. METODA DESTYLACYJNA
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości lotnych związków azotowych, w przeliczeniu na amoniak, w mączkach rybnych niezawierających mocznika. Metoda ma zastosowanie dla zawartości amoniaku niższej niż 0,25 %.
2. Sposób przeprowadzenia metody
Próbka jest ekstrahowana wodą, a uzyskany roztwór klarowany i filtrowany. Lotne związki azotowe są oddzielane w punkcie wrzenia poprzez dodanie tlenku magnezu i zbierane w określonej ilości kwasu siarkowego, którego nadmiar jest miareczkowany z użyciem roztworu wodorotlenku sodu.
3. Odczynniki i roztwory
|
3.1.
|
Kwas trójchlorooctowy, roztwór 20 % (w/v).
|
|
3.3.
|
Emulsja zapobiegająca spienieniu (np. silikon).
|
|
3.4.
|
Kwas siarkowy o stężeniu 0,05 mol/l.
|
|
3.5.
|
Wodorotlenek sodu, roztwór o stężeniu 0,1 mol/l.
|
|
3.6.
|
Czerwień metylowa, roztwór 0,3 % w alkoholu etylowym 95 %–96 % (v/v).
|
4. Aparatura i sprzęt
|
4.1.
|
Mikser (tumbler): ok. 35–40 obr./min.
|
|
4.2.
|
Aparat do destylacji typu Kjeldahla.
|
5. Sposób postępowania
Odważyć, z dokładnością do 1 mg, 10 g próbki do kolby miarowej o pojemności 200 ml i dodać 100 ml wody. Miksować w mikserze przez 30 minut. Dodać 50 ml roztworu kwasu trójchlorooctowego (3.1), uzupełnić do pełnej objętości kolby wodą, energicznie wstrząsnąć i filtrować przez karbowany filtr.
Pobrać taką ilość klarownego filtratu, która odpowiada spodziewanej zawartości lotnych związków azotowych, co zwykle odpowiada objętości 100 ml. Rozcieńczyć do 200 ml, dodać 2 g tlenku magnezu (3.2) i kilka kropli emulsji zapobiegającej spienieniu (3.3). Roztwór musi wykazywać alkaliczność wobec papierka lakmusowego, w przeciwnym razie dodać nieco tlenku magnezu (3.2). Postępować zgodnie z pkt 5.2 i 5.3 metody analitycznej dotyczącej oznaczania zawartości białka surowego (część C niniejszego załącznika).
Ślepą próbę przeprowadzić w ten sam sposób, lecz bez próbki przeznaczonej do analizy.
6. Obliczanie wyników
1 ml H2SO4 o stężeniu 0,05 mol/l odpowiada 1,7 mg amoniaku.
Wynik wyrazić jako udział procentowy w próbce.
Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń, wykonanych dla tej samej próbki, nie może przekraczać 10 % wartości względnej amoniaku.
F. OZNACZANIE AMINOKWASÓW (Z WYJĄTKIEM TRYPTOFANU)
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości wolnych (syntetycznych i naturalnych) oraz całkowitych (związanych i wolnych peptydów) aminokwasów w paszach z użyciem analizatora aminokwasów. Metoda ma zastosowanie do następujących aminokwasów: cystyny (cysteiny), metioniny, lizyny, treoniny, alaniny, argininy, kwasu asparaginowego, kwasu glutaminowego, glicyny, histydyny, izoleucyny, leucyny, fenyloalaniny, proliny, seryny, tyrozyny i waliny.
Metoda nie pozwala odróżnić soli aminokwasów, a także form D i L aminokwasów. Nie można jej stosować do oznaczania tryptofanu lub hydroksyanalogów aminokwasów.
2. Sposób przeprowadzenia metody
2.1. Wolne aminokwasy
Wolne aminokwasy są ekstrahowane rozcieńczonym kwasem chlorowodorowym. Wyekstrahowane jednocześnie makrocząsteczki związków azotowych są wytrącane kwasem sulfosalicylowym i usuwane przez filtrowanie. Filtrowany roztwór jest dostosowany do pH 2,20. Aminokwasy są oddzielane w drodze chromatografii jonowymiennej i oznaczane z zastosowaniem reakcji z ninhydryną, poprzez fotometryczną detekcję przy 570 nm.
2.2. Aminokwasy całkowite
Wybór sposobu postępowania zależy od oznaczanych aminokwasów. Cystynę (cysteinę) i metioninę przed hydrolizą poddać utlenianiu, odpowiednio do kwasu cysteinowego i sulfonu metioniny. Tyrozyna musi być oznaczana w hydrolizatach próbek niepoddanych utlenieniu. Pozostałe aminokwasy wymienione w pkt 1 mogą być oznaczane w próbce utlenionej albo niepoddanej utlenianiu.
Utlenianie jest przeprowadzane w temperaturze 0 oC z użyciem mieszaniny kwasu nadmrówkowego i fenolu. Nadmiar odczynnika utleniającego jest rozkładany z użyciem dwusiarczku sodu. Utleniona albo niepoddana utlenianiu próbka jest hydrolizowana kwasem chlorowodorowym (3.20) przez 23 godziny. Hydrolizat jest dostosowany do pH 2,20. Aminokwasy są oddzielane w drodze chromatografii jonowymiennej i oznaczane na drodze reakcji z ninhydryną, przez fotometryczną detekcję przy 570 nm (440 nm w przypadku proliny).
3. Odczynniki i roztwory
Należy stosować wodę podwójnie destylowaną lub podobnej jakości (przewodność < 10 μS).
|
3.1.
|
Nadtlenek wodoru, w (w/w) = 30 %.
|
|
3.2.
|
Kwas mrówkowy, w (w/w) = 98–100 %.
|
|
3.6.
|
Kwas 5-sulfosalicylowy dwuhydrat.
|
|
3.7.
|
Kwas chlorowodorowy, gęstość około 1,18 g/ml.
|
|
3.8.
|
Cytrynian trisodu, dwuhydrat.
|
|
3.9.
|
2,2' tiodwuetanol (tiodwuglikol).
|
|
3.12.
|
Eter naftowy o temperaturze wrzenia 40–60 oC.
|
|
3.13.
|
Norleucyna lub inny związek odpowiedni do stosowania jako wzorzec wewnętrzny.
|
|
3.14.
|
Azot gazowy (< 10 ppm tlenu).
|
|
3.16.1.
|
Substancje wzorcowe określone w pkt 1. Czyste związki, niezawierające wody krystalizacyjnej. Suszyć w warunkach próżni w obecności P2O5 lub H2SO4 przez tydzień przed użyciem.
|
|
3.16.3.
|
Sulfon metioniny.
|
|
3.17.
|
Roztwór wodorotlenku sodu, c = 7,5 mol/l:
Rozpuścić 300 g NaOH (3.5) w wodzie i uzupełnić kolbę do objętości 1 l wodą.
|
|
3.18.
|
Roztwór wodorotlenku sodu, c = 1 mol/l:
Rozpuścić 40 g NaOH (3.5) w wodzie i uzupełnić kolbę do objętości 1 l wodą.
|
|
3.19.
|
Roztwór kwasu mrówkowego i fenolu:
Zmieszać 889 g kwasu mrówkowego (3.2) z 111 g wody i dodać 4,73 g fenolu (3.3).
|
|
3.20.
|
Mieszanina hydrolizująca, c = 6 mol HCl/l, zawierająca 1 g fenolu/l:
Dodać 1 g fenolu (3.3) do 492 ml HCl (3.7) i uzupełnić kolbę do objętości 1 l wodą.
|
|
3.21.
|
Mieszanina ekstrakcyjna, c = 0,1 mol HCl/l, zawierająca 2 % tiodwuglikolu: Pobrać 8,2 ml HCl (3.7), rozpuścić w około 900 ml wody, dodać 20 ml tiodwuglikolu (3.9) i uzupełnić kolbę do objętości 1 l wodą (nie mieszać bezpośrednio odczynników określonych w pkt 3.7 i 3.9.).
|
|
3.22.
|
Kwas 5-sulfosalicylowy, ß = 6 %:
Rozpuścić 60 g kwasu 5-sulfosalicylowego (3.6) w wodzie i uzupełnić kolbę do objętości 1 l wodą.
|
|
3.23.
|
Mieszanina utleniająca (kwas nadmrówkowy – fenol):
Zmieszać w małej zlewce 0,5 ml nadtlenku wodoru (3.1) z 4,5 ml roztworu kwasu mrówkowego i fenolu (3.19). Inkubować w temperaturze od 20 do 30 oC przez godzinę w celu utworzenia kwasu nadmrówkowego, następnie schłodzić w lodowej łaźni wodnej (15 minut) przed dodaniem tej mieszaniny do próbki.
Uwaga: Unikać kontaktu ze skórą i nosić odzież ochronną.
|
|
3.24.
|
Bufor cytrynianowy, c = 0,2 mol Na+/l, pH 2,20:
Rozpuścić 19,61 g cytrynianu trisodu (3.8), 5 ml tiodwuglikolu (3.9), 1 g fenolu (3.3) i 16,50 ml HCl (3.7) w około 800 ml wody. Dostosować pH do 2,20. Uzupełnić do objętości 1 l wodą.
|
|
3.25.
|
Bufor wymywający, przygotowany zgodnie z warunkami stosowanego analizatora (4.9).
|
|
3.26.
|
Odczynnik ninhydrynowy, przygotowany zgodnie z warunkami stosowanego analizatora (4.9).
|
|
3.27.
|
Wzorcowe roztwory aminokwasów. Roztwory te należy przechowywać w temperaturze niższej niż 5 oC.
|
|
3.27.1.
|
Podstawowy wzorcowy roztwór aminokwasów (3.16.1).
Każdy o stężeniu c = 2,5 μmol/ml, w kwasie chlorowodorowym.
Mogą być otrzymane w handlu.
|
|
3.27.2.
|
Podstawowe wzorcowe roztwory kwasu cysteinowego i sulfonu metioniny, c = 1,25 μmol/ml.
Rozpuścić 0,2115 g kwasu cysteinowego (3.16.2) i 0,2265 g sulfonu metioniny (3.16.3) w buforze cytrynianowym (3.24) w kolbie miarowej o pojemności 1 l i uzupełnić do pełnej objętości kolby buforem cytrynianowym. Przechowywać w temperaturze niższej niż 5 oC nie dłużej niż przez 12 miesięcy. Roztworu nie stosuje się, jeżeli podstawowy wzorcowy roztwór (3.27.1) zawiera kwas cysteinowy i sulfon metioniny.
|
|
3.27.3.
|
Podstawowy wzorcowy roztwór wzorca wewnętrznego, proponuje się zastosować norleucynę, c = 20 μmol/ml.
Rozpuścić 0,6560 g norleucyny (3.13) w buforze cytrynianowym (3.24) w kolbie miarowej i uzupełnić kolbę do objętości 250 ml buforem cytrynianowym. Przechowywać w temperaturze niższej niż 5 oC nie dłużej niż przez 6 miesięcy.
|
|
3.27.4.
|
Kalibracyjny roztwór wzorca aminokwasów do użycia z hydrolizatami, c = 5 nmol/50 μl kwasu cysteinowego i sulfonu metioniny i c = 10 nmol/50 μl innych aminokwasów. Rozpuścić 2,2 g chlorku sodu (3.10) w zlewce o pojemności 100 ml z 30 ml buforu cytrynianowego (3.24). Dodać 4,0 ml podstawowego roztworu wzorcowego aminokwasów (3.27.1), 4,0 ml podstawowego roztworu wzorcowego kwasu cysteinowego i sulfonu metioniny (3.27.2) i, jeżeli jest stosowany, 0,5 ml podstawowego wzorcowego roztworu wzorca wewnętrznego (3.27.3). Dostosować pH do 2,20 z użyciem roztworu wodorotlenku sodu (3.18).
Przenieść ilościowo do kolby miarowej o pojemności 50 ml, uzupełnić do pełnej objętości kolby buforem cytrynianowym (3.24) i zmieszać.
Przechowywać w temperaturze niższej niż 5 oC nie dłużej niż przez 3 miesiące.
Zob. również objaśnienia pkt 9.1.
|
|
3.27.5.
|
Kalibracyjny roztwór wzorca aminokwasów do stosowania z hydrolizatami przygotowanymi w sposób określony w pkt 5.3.3.1 i do użycia z ekstraktami (5.2). Roztwór kalibracyjny przygotowuje się w sposób określony w pkt 3.27.4, z pominięciem chlorku sodu.
Przechowywać w temperaturze niższej niż 5 oC nie dłużej niż przez 3 miesiące.
|
4. Aparatura i sprzęt
|
4.1.
|
Kolba okrągłodenna o pojemności 100 lub 250 ml z chłodnicą zwrotną.
|
|
4.2.
|
Butelka ze szkła borokrzemowego o pojemności 100 ml z nakrętką pokrytą gumą/teflonem (np. Duran, Schott) do stosowania w suszarce.
|
|
4.3.
|
Suszarka z silnym nawiewem i możliwością regulacji temperatury z dokładnością większą niż ±2 oC.
|
|
4.4.
|
Pehametr (dokładność do trzech miejsc po przecinku).
|
|
4.5.
|
Filtr membranowy (0,22 μm).
|
|
4.7.
|
Obrotowa wyparka próżniowa.
|
|
4.8.
|
Mechaniczna wytrząsarka lub mieszadło magnetyczne.
|
|
4.9.
|
Analizator aminokwasów lub wyposażenie do HPLC z kolumną jonowymienną, przystawką do ninhydryny, postkolumnową derywatyzacją i detektorem fotometrycznym.
Kolumna jest wypełniona sulfonowaną żywicą polistyrenową mającą zdolność rozdziału aminokwasów od siebie i od innych składników reagujących z ninhydryną. Przepływ buforu i ninhydryny jest przeprowadzany przez pompy o stabilności przepływu ±0,5 % w czasie testowania kalibracyjnych roztworów, jak i analizy próbek.
W niektórych analizatorach aminokwasów mogą być stosowane metody hydrolizy, w których hydrolizat zawiera stężenie sodu c = 0,8 mol/l i całą pozostałość kwasu mrówkowego z etapu utleniania. Inne analizatory nie dają zadowalającego rozdziału niektórych aminokwasów, jeżeli hydrolizat zawiera nadmiar kwasu mrówkowego lub wysokie stężenie jonów sodowych. W tym przypadku objętość kwasu jest zmniejszana przez odparowanie do objętości około 5 ml, które wykonuje się po hydrolizie i przed dostosowaniem pH. Odparowywanie należy prowadzić w warunkach próżni w temperaturze nie wyższej niż 40 oC.
|
5. Sposób postępowania
5.1. Przygotowanie próbki
Rozdrobnić próbkę, tak aby przesiewała się przez oczka sita o średnicy 0,5 mm. Próbki o dużej zawartości wilgotności przed rozdrobnieniem należy wysuszyć albo suchym powietrzem o temperaturze nieprzekraczającej 50 oC, albo przez zamrożenie. Próbki o wysokiej zawartości tłuszczu przed rozdrobnieniem należy poddać ekstrakcji eterem naftowym (3.12).
5.2. Oznaczanie wolnych aminokwasów w paszach i premiksach
Odważyć, z dokładnością do 0,2 mg, odpowiednią ilość (1–5 g) próbki przygotowanej w sposób określony w pkt 5.1 do kolby stożkowej i dodać 100 ml mieszaniny ekstrakcyjnej (3.21). Wstrząsać mieszaninę przez 60 minut, używając mechanicznej wytrząsarki lub mieszadła magnetycznego (4.8). Odstawić do sedymentacji osadu i pobrać pipetą 10 ml supernatantu roztworu do zlewki o objętości 100 ml.
Dodać, mieszając, 5,0 ml roztworu kwasu sulfosalicylowego (3.22) i kontynuować mieszanie z użyciem mieszadła magnetycznego przez 5 minut. Przefiltrować lub odwirować supernatant w celu usunięcia osadu. Umieścić 10,0 ml otrzymanego roztworu w zlewce o objętości 100 ml, dostosować pH do 2,20 z użyciem roztworu wodorotlenku sodu (3.18), przenieść do wolumetrycznej kolby o odpowiedniej pojemności, z użyciem buforu cytrynianowego (3.24) i uzupełnić roztworem buforu cytrynianowego do pełnej objętości tej kolby (3.24).
Jeżeli jest stosowany wzorzec wewnętrzny, dodać 1,00 ml podstawowego wzorcowego roztworu wzorca wewnętrznego (3.27.3) na każde 100 ml końcowego roztworu i uzupełnić roztworem buforu cytrynianowego (3.24) do pełnej objętości kolby.
Przeprowadzić rozdział chromatograficzny w sposób określony w pkt 5.4.
Jeżeli ekstrakty nie będą oznaczane tego samego dnia, należy przechowywać je w temperaturze niższej niż 5 oC.
5.3. Oznaczanie całkowitej zawartości aminokwasów
5.3.1. Utlenianie
Odważyć, z dokładnością do 0,2 mg, od 0,1 do 1 g próbki przygotowanej w sposób określony w pkt 5.1 do:
|
—
|
kolby okrągłodennej o pojemności 100 ml (4.1) do hydrolizy otwartej (5.3.2.3), lub
|
|
—
|
kolby okrągłodennej o pojemności 250 ml (4.1), jeżeli wymagane jest niskie stężenie sodu (5.3.3.1), lub
|
|
—
|
butelki o pojemności 100 ml z nakrętką (4.2), w przypadku hydrolizy zamkniętej (5.3.2.4).
|
Odważona próbka musi zawierać około 10 mg azotu, a poziom wilgotności nie może przekraczać 100 mg.
Umieścić kolbę lub butelkę w lodowej łaźni wodnej i oziębić do temperatury 0 oC, dodać 5 ml mieszaniny utleniającej (3.23) i zamieszać, używając szklanej bagietki z wygiętym końcem. Uszczelnić kolbę/butelkę wraz z bagietką, stosując hermetyczny film, umieścić wraz z lodową łaźnią wodną w lodówce o temperaturze 0 oC i pozostawić na 16 godzin. Po 16 godzinach wyjąć z lodówki i rozłożyć nadmiar odczynnika utleniającego przez dodanie 0,84 g dwusiarczynu sodu (3.4).
Następnie postępować w sposób określony w pkt 5.3.2.1.
5.3.2. Hydroliza
5.3.2.1.
Hydroliza próbek utlenionych
Do utlenionej próbki, przygotowanej w sposób określony w pkt 5.3.1, dodać 25 ml mieszaniny hydrolizującej (3.20), zwracając uwagę, aby zmyć pozostałości próbki przywierające do bocznych ścianek naczynia i bagietki.
W zależności od sposobu przeprowadzonej hydrolizy postępować w sposób określony w pkt 5.3.2.3 lub 5.3.2.4.
5.3.2.2.
Hydroliza próbek nieutlenionych
Odważyć, z dokładnością do 0,2 mg, od 0,1 do 1 g próbki przygotowanej w sposób określony w pkt 5.1 do kolby okrągłodennej o pojemności 100 lub 250 ml (4.1) lub do butelki o pojemności 100 ml z nakrętką (4.2). Odważona próbka musi zawierać około 10 mg azotu. Ostrożnie dodać 25 ml mieszaniny hydrolizującej (3.20) i zmieszać z próbką. Następnie postępować w sposób określony w pkt 5.3.2.3 lub 5.3.2.4.
5.3.2.3.
Hydroliza otwarta
Do kolby z mieszaniną, przygotowaną w sposób określony w pkt 5.3.2.1 lub 5.3.2.2, dodać 3 szklane perełki i gotować, utrzymując stan wrzenia ze zwrotem skroplin przez 23 godziny. Po zakończonej hydrolizie przemyć chłodnicę od dołu 5 ml buforu cytrynianowego (3.24). Odłączyć kolbę i schłodzić w łaźni lodowej.
Następnie postępować w sposób określony w pkt 5.3.3.
5.3.2.4.
Hydroliza zamknięta
Umieścić butelkę zawierającą mieszaninę, przygotowaną w sposób określony w pkt 5.3.2.1 lub 5.3.2.2, w suszarce (4.3) w temperaturze 110 oC. W czasie 1 godziny hydrolizy, aby uniknąć gwałtownego wzrostu ciśnienia w wyniku powstawania substancji gazowych i wybuchu, umieścić nakrętkę na naczyniu. Nie zamykać naczynia korkiem. Po godzinie dokręcić nakrętkę i pozostawić w suszarce (4.3) przez 23 godziny. Po zakończeniu hydrolizy wyjąć butelkę z suszarki, ostrożnie odkręcić nakrętkę i umieścić butelkę w lodowej łaźni wodnej. Pozostawić do schłodzenia.
W zależności od sposobu dostosowania pH (5.3.3) przenieść ilościowo zawartość butelki do zlewki lub kolby okrągłodennej o pojemności 250 ml, stosując bufor cytrynianowy (3.24).
Następnie postępować w sposób określony w pkt 5.3.3.
5.3.3. Dostosowanie pH
W zależności od tolerancji analizatora aminokwasów (4.9) na sód pH dostosowuje się w sposób określony w pkt 5.3.3.1 lub 5.3.3.2.
5.3.3.1.
Dla systemów chromatograficznych (4.9) wymagających niskiego stężenia sodu
Wskazane jest zastosowanie podstawowego wzorcowego roztworu wzorca wewnętrznego (3.27.3), gdy analizatory aminokwasów wymagają niskiego stężenia sodu, w przypadku gdy należy zredukować objętość kwasu.
W tym przypadku dodać 2,00 ml podstawowego roztworu wzorca wewnętrznego (3.27.3) do hydrolizatu przed odparowaniem.
Dodać 2 krople 1-oktanolu (3.15) do hydrolizatu otrzymanego w sposób określony w pkt 5.3.2.3 lub 5.3.2.4.
Stosując obrotową wyparkę próżniową (4.7) zmniejszyć w warunkach próżni i w temperaturze 40 oC objętość do 5–10 ml. Jeżeli objętość roztworu zostanie przypadkowo zredukowana poniżej 5 ml, hydrolizat należy wyrzucić i powtórzyć analizę.
Dostosować pH do 2,20 z użyciem roztworu wodorotlenku sodu (3.18) i postępować w sposób określony w pkt 5.3.4.
5.3.3.2.
Dla pozostałych analizatorów aminokwasów (4.9)
Pobrać hydrolizaty otrzymane w sposób określony w pkt 5.3.2.3 lub 5.3.2.4 i częściowo je zneutralizować poprzez mieszanie, ostrożnie dodając 17 ml roztworu wodorotlenku sodu (3.17), zwracając uwagę, aby temperatura roztworu nie była wyższa niż 40 oC.
Dostosować pH do 2,20 w temperaturze pokojowej, dodając roztwór wodorotlenku sodu (3.17), a pod koniec roztwór wodorotlenku sodu (3.18). Następnie postępować w sposób określony w pkt 5.3.4.
5.3.4. Roztwór próbki do analizy chromatograficznej
Przenieść ilościowo hydrolizaty o dostosowanym pH (5.3.3.1 lub 5.3.3.2) z użyciem buforu cytrynianowego (3.24) do kolby miarowej o pojemności 200 ml i uzupełnić do pełnej objętości kolby buforem cytrynianowym (3.24).
Jeżeli wcześniej nie dodawano wzorca wewnętrznego, dodać 2,00 ml podstawowego wzorcowego roztworu wzorca wewnętrznego (3.27.3) i uzupełnić buforem cytrynianowym (3.24) do pełnej objętości kolby. Dokładnie zmieszać.
Przejść do analizy chromatograficznej (5.4).
Jeżeli roztwory próbek nie będą analizowane tego samego dnia, należy przechowywać je w temperaturze niższej niż 5 oC.
5.4. Chromatografia
Przed analizą chromatograficzną doprowadzić ekstrakt (5.2) lub hydrolizat (5.3.4) do temperatury pokojowej. Wstrząsnąć mieszaniną i przefiltrować jej odpowiednią ilość przez filtr membranowy 0,22 μm (4.5). Otrzymany klarowny roztwór jest poddawany chromatografii jonowymiennej przy wykorzystaniu analizatora aminokwasów (4.9).
Dozowanie wykonuje się ręcznie lub automatycznie. Ważne jest, aby na kolumnę nanieść takie same ilości roztworów z dokładnością ±0,5 % zarówno w przypadku analizy wzorców, jak i badanych próbek, z wyjątkiem gdy jest stosowany wzorzec wewnętrzny, oraz aby stosunek sodu do aminokwasu w roztworze badanej próbki był jak najbardziej zbliżony do tego w roztworze wzorcowym.
Zasadniczo częstość przeprowadzania kalibracji zależy od stabilności odczynnika ninhydrynowego i od systemu analitycznego. Wzorzec lub próbka są wymywane buforem cytrynianowym (3.24), tak aby powierzchnia piku wzorca odpowiadała od 30 do 200 % powierzchni piku badanej próbki aminokwasu.
Analiza chromatograficzna aminokwasów będzie różnić się nieznacznie w zależności od typu stosowanego analizatora i żywicy. Wybrany system musi mieć zdolność oddzielenia od siebie aminokwasów oraz od innych substancji dających z ninhydryną reakcje. W badanym zakresie stosowany system chromatograficzny musi dawać liniową odpowiedź na zmiany ilości aminokwasów dodawanych na kolumnę.
W czasie analizy chromatograficznej opisany poniżej stosunek doliny do wysokości piku odnosi się do przypadku, gdy jest analizowany równomolowy roztwór oznaczanego aminokwasu. Równomolowy roztwór aminokwasu musi zawierać co najmniej 30 % maksymalnej zawartości każdego aminokwasu, który można aktualnie oznaczyć z użyciem danego analizatora aminokwasów (4.9).
Dla oddzielenia treoniny od seryny stosunek doliny do wysokości piku niższego z pokrywających się aminokwasów na chromatografie nie może przekraczać 2:10, w przypadku gdy oznaczane są tylko cystyna (cysteina), metionina, treonina i lizyna, niewystarczające oddzielenie od sprzężonych pików będzie ujemnie wpływało na oznaczenie. Dla wszystkich innych aminokwasów rozdział musi być lepszy niż 1:10.
System musi zapewniać oddzielenie lizyny od „artefaktów lizyny” i od ornityny.
6. Obliczanie wyników
Powierzchnię pików próbki i wzorca mierzy się dla każdego pojedynczego aminokwasu i obliczana jest zawartość aminokwasu (X) w g/kg próbki.
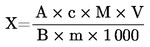
Jeżeli jest stosowany wzorzec wewnętrzny, wyrażenie należy pomnożyć przez: 
gdzie:
|
A
|
=
|
powierzchnia piku hydrolizatu lub ekstraktu
|
|
B
|
=
|
powierzchnia piku wzorcowego roztworu kalibracyjnego
|
|
C
|
=
|
powierzchnia piku wzorca wewnętrznego w hydrolizacie lub ekstrakcie
|
|
D
|
=
|
powierzchnia piku wzorca wewnętrznego w roztworze wzorcowym kalibracyjnym
|
|
M
|
=
|
masa cząsteczkowa oznaczanego aminokwasu
|
|
c
|
=
|
stężenie wzorca w μmol/ml
|
|
m
|
=
|
masa próbki w g przeliczona na masę oryginalnej próbki w przypadku odtłuszczania lub podsuszania
|
|
V
|
=
|
hydrolizat ogółem w ml (5.3.4) lub obliczona ogólna objętość w ml rozcieńczonego ekstraktu (6.1)
|
Cystyna i cysteina są oznaczane jako kwas cysteinowy w hydrolizatach utlenionej próbki, ale są obliczane jak cystyna (C6H12N2O4S2, M 240,30 g/mol), stosując M 120,15 g/mol (= 0,5 × 240,30 g/mol).
Metionina jest oznaczana jako sulfon metioniny w hydrolizatach utlenionej próbki, ale jest obliczana jak metionina, stosując M metioniny: 149,21 g/mol.
Dodana wolna metionina jest oznaczana po ekstrakcji jako metionina, przy czym dla obliczeń stosowana jest taka sama wartość M.
|
6.1.
|
Ogólną objętość rozcieńczonych ekstraktów (F) w przypadku oznaczania zawartości wolnych aminokwasów (5.2) obliczyć według następującego wzoru:

|
7. Ocena metody
Międzynarodowe badania porównawcze metody były przeprowadzone w 1990 r. z użyciem czterech pasz (mieszanki paszowej dla świń, mieszanki paszowej dla brojlerów, koncentratu białkowego i premiksu). Po odrzuceniu wyników odbiegających, średnie wyniki i odchylenia standardowe są przedstawione w tabelach zawartych w niniejszym punkcie:
Średnie wielkości w g/kg
|
Materiał badany
|
Aminokwas
|
|
Treonina
|
Cystyna/cysteina
|
Metionina
|
Lizyna
|
|
Mieszanka paszowa dla świń
|
6,94
n = 15
|
3,01
n = 17
|
3,27
n = 17
|
9,55
n = 13
|
|
Mieszanka paszowa dla brojlerów
|
9,31
n = 16
|
3,92
n = 18
|
5,08
n = 18
|
13,93
n = 16
|
|
Koncentrat białkowy
|
22,32
n = 16
|
5,06
n = 17
|
12,01
n = 17
|
47,74
n = 15
|
|
Premiks
|
58,42
n = 16
|
—
|
90,21
n = 16
|
98,03
n = 16
|
|
n
|
=
|
liczba uczestniczących laboratoriów.
|
|
7.1. Powtarzalność
Powtarzalność wyrażona jako „odchylenie standardowe wewnątrz laboratorium” omawianego powyżej porównawczego badania jest przedstawiona w poniższych tabelach:
Odchylenie standardowe wewnątrz laboratorium (Sr) w g/kg
|
Materiał badany
|
Aminokwas
|
|
Treonina
|
Cystyna/cysteina
|
Metionina
|
Lizyna
|
|
Mieszanka paszowa dla świń
|
0,13
n = 15
|
0,10
n = 17
|
0,11
n = 17
|
0,26
n = 13
|
|
Mieszanka paszowa dla brojlerów
|
0,20
n = 16
|
0,11
n = 18
|
0,16
n = 18
|
0,28
n = 16
|
|
Koncentrat białkowy
|
0,48
n = 16
|
0,13
n = 17
|
0,27
n = 17
|
0,99
n = 15
|
|
Premiks
|
1,30
n = 16
|
—
|
2,19
n = 16
|
2,06
n = 16
|
|
n
|
=
|
liczba uczestniczących laboratoriów.
|
|
Współczynnik zmienności (%) dla odchylenia standardowego wewnątrz laboratorium (Sr)
|
Materiał badany
|
Aminokwas
|
|
Treonina
|
Cystyna/cysteina
|
Metionina
|
Lizyna
|
|
Mieszanka paszowa dla świń
|
1,9
n = 15
|
3,3
n = 17
|
3,4
n = 17
|
2,8
n = 13
|
|
Mieszanka paszowa dla brojlerów
|
2,1
n = 16
|
2,8
n = 18
|
3,1
n = 18
|
2,1
n = 16
|
|
Koncentrat białkowy
|
2,7
n = 16
|
2,6
n = 17
|
2,2
n = 17
|
2,4
n = 15
|
|
Premiks
|
2,2
n = 16
|
—
|
2,4
n = 16
|
2,1
n = 16
|
|
n
|
=
|
liczba uczestniczących laboratoriów.
|
|
7.2. Odtwarzalność
Wyniki odchylenia standardowego między laboratoriami otrzymane w wyniku przeprowadzenia omawianego powyżej porównawczego badania są przedstawione w poniższej tabeli:
Odchylenie standardowe między laboratoriami (SR) w g/kg
|
Materiał badany
|
Aminokwas
|
|
Treonina
|
Cystyna/cysteina
|
Metionina
|
Lizyna
|
|
Mieszanka paszowa dla świń
|
0,28
n = 15
|
0,30
n = 17
|
0,23
n = 17
|
0,30
n = 13
|
|
Mieszanka paszowa dla brojlerów
|
0,48
n = 16
|
0,34
n = 18
|
0,55
n = 18
|
0,75
n = 16
|
|
Koncentrat białkowy
|
0,85
n = 16
|
0,62
n = 17
|
1,57
n = 17
|
1,24
n = 15
|
|
Premiks
|
2,49
n = 16
|
—
|
6,20
n = 16
|
6,62
n = 16
|
|
n
|
=
|
liczba uczestniczących laboratoriów.
|
|
Współczynnik zmienności (%) dla odchylenia standardowego między laboratoriami (SR)
|
Materiał badany
|
Aminokwas
|
|
Treonina
|
Cystyna/cysteina
|
Metionina
|
Lizyna
|
|
Mieszanka paszowa dla świń
|
4,1
n = 15
|
9,9
n = 17
|
7,0
n = 17
|
3,2
n = 13
|
|
Mieszanka paszowa dla brojlerów
|
5,2
n = 16
|
8,8
n = 18
|
10,9
n = 18
|
5,4
n = 16
|
|
Koncentrat białkowy
|
3,8
n = 16
|
12,3
n = 17
|
13,0
n = 17
|
3,0
n = 15
|
|
Premiks
|
4,3
n = 16
|
—
|
6,9
n = 16
|
6,7
n = 16
|
|
n
|
=
|
liczba uczestniczących laboratoriów.
|
|
8. Stosowanie materiałów odwoławczych
Prawidłowe stosowanie metody należy weryfikować przez wykonywanie powtarzalnych oznaczeń certyfikowanych materiałów odwoławczych, jeżeli takie są dostępne. Zalecana jest także kalibracja z zastosowaniem certyfikowanych kalibracyjnych roztworów aminokwasów.
9. Objaśnienia
|
9.1.
|
Z powodu różnic pomiędzy analizatorami aminokwasów końcowe stężenia kalibracyjnych roztworów aminokwasów wzorcowych (zob. 3.27.4 i 3.27.5) oraz hydrolizatu (zob. 5.3.4) należy traktować jako przykładowe.
Zakres liniowej odpowiedzi aparatu należy sprawdzić dla wszystkich aminokwasów.
Roztwór wzorcowy jest rozcieńczany buforem cytrynianowym, tak aby otrzymać powierzchnię piku o średnim zasięgu.
|
|
9.2.
|
Jeżeli do analizy produktów hydrolizy jest wykorzystywany wysoko sprawny chromatograf cieczowy, należy zoptymalizować warunki doświadczalne analizy zgodnie z zaleceniami producenta.
|
|
9.3.
|
Przy stosowaniu tej metody do pasz zawierających więcej niż 1 % chlorków (koncentraty, mieszanki mineralne, mieszanki paszowe uzupełniające) wyniki oznaczania metioniny mogą być zaniżone i należy stosować specjalne postępowanie.
|
G. OZNACZANIE TRYPTOFANU
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości całkowitego i wolnego tryptofanu w paszach. Metoda nie wyróżnia form D i L.
2. Sposób przeprowadzenia metody
W celu oznaczenia całkowitego tryptofanu próbka jest hydrolizowana w środowisku zasadowym z użyciem nasyconego roztworu wodorotlenku baru i ogrzewana do 110 oC przez 20 godzin. Po hydrolizie dodawany jest wzorzec wewnętrzny.
W celu oznaczenia wolnego tryptofanu próbka jest ekstrahowana w warunkach lekko kwaśnych w obecności wzorca wewnętrznego.
Tryptofan i wzorzec wewnętrzny w hydrolizacie lub w ekstrakcie jest oznaczony metodą HPLC z detekcją fluorescencyjną.
3. Odczynniki i roztwory
|
3.1.
|
Należy stosować wodę podwójnie destylowaną lub o porównywalnej jakości (przewodnictwo < 10 μS/cm).
|
|
3.2.
|
Substancja wzorcowa: tryptofan (czystość/zawartość ≥ 99 %) suszony w próżni nad pięciotlenkiem fosforu.
|
|
3.3.
|
Substancja wzorcowa wewnętrzna: α-metylo-tryptofan (czystość/zawartość ≥ 99 %) suszony w próżni nad pięciotlenkiem fosforu.
|
|
3.4.
|
Wodorotlenek baru, oktahydrat (należy uważać, aby nie wystawiać Ba(OH)2 · 8H2O na nadmierne działanie powietrza, aby uniknąć tworzenia się BaCO3, który mógłby utrudniać oznaczenie) (zob. objaśnienia pkt 9.3).
|
|
3.6.
|
Kwas ortofosforowy, w (w/w) = 85 %.
|
|
3.7.
|
Kwas chlorowodorowy, ρ20 = 1,19 g/ml.
|
|
3.9.
|
Eter naftowy o temperaturze wrzenia od 40 do 60 oC.
|
|
3.10.
|
Roztwór wodorotlenku sodu, c = 1 mol/l:
Rozpuścić 40,0 g NaOH (3.5) w wodzie i uzupełnić do objętości 1 l wodą (3.1).
|
|
3.11.
|
Kwas chlorowodorowy, c = 6 mol/l:
Odmierzyć 492 ml HCl (3.7) i uzupełnić do objętości 1 l wodą.
|
|
3.12.
|
Kwas chlorowodorowy, c = 1 mol/l:
Odmierzyć 82 ml HCl (3.7) i uzupełnić do objętości 1 l wodą.
|
|
3.13.
|
Kwas chlorowodorowy, c = 0,1 mol/l:
Odmierzyć 8,2 ml HCl (3.7) i uzupełnić do objętości 1 l wodą.
|
|
3.14.
|
Kwas ortofosforowy, c = 0,5 mol/l:
Odmierzyć 34 ml kwasu ortofosforowego (3.6) i uzupełnić do objętości 1 l wodą (3.1).
|
|
3.15.
|
Stężony roztwór tryptofanu (3.2), c = 2,50 μmol/ml:
W kolbie miarowej o pojemności 500 ml rozpuścić 0,2553 g tryptofanu (3.2) w kwasie chlorowodorowym (3.13) i uzupełnić do pełnej objętości kolby kwasem chlorowodorowym (3.13). Przechowywać w temperaturze - 18 oC nie dłużej niż przez 4 tygodnie.
|
|
3.16.
|
Stężony roztwór wzorca wewnętrznego, c = 2,50 μmol/ml:
W kolbie miarowej o pojemności 500 ml rozpuścić 0,2728 g α-metylo-tryptofanu (3.3) w kwasie chlorowodorowym (3.13) i uzupełnić do pełnej objętości kolby kwasem chlorowodorowym (3.13). Przechowywać w temperaturze - 18 oC nie dłużej niż przez 4 tygodnie.
|
|
3.17.
|
Kalibracyjny roztwór wzorcowy tryptofanu i wzorca wewnętrznego:
Odmierzyć 2,00 ml stężonego roztworu tryptofanu (3.15) i 2,00 ml stężonego roztworu wzorca wewnętrznego (α-metylo-tryptofanu) (3.16). Rozcieńczyć wodą (3.1) i metanolem (3.8) w przybliżeniu do tej samej objętości i tego samego stężenia metanolu (10–30 %) jak w końcowym hydrolizacie.
Roztwór należy przygotować bezpośrednio przed użyciem.
Chronić przed bezpośrednim działaniem światła podczas przygotowywania.
|
|
3.19.
|
1,1,1-trichloro-2-metylo-2-propanol.
|
|
3.20.
|
Etanoloamina w (w/w) > 98 %.
|
|
3.21.
|
Roztwór 1 g 1,1,1-trichloro-2-metylo-2-propanolu (3.19) w 100 ml metanolu (3.8).
|
|
3.22.
|
Faza ruchoma do HPLC: 3,00 g kwasu octowego (3.18) + 900 ml wody (3.1) +50,0 ml roztworu (3.21) 1,1,1-trichloro-2-metylo-2-propanolu (3.19) w metanolu (3.8) (1 g/100 ml). Dostosować pH do 5,00, stosując etanoloaminę (3.20). Uzupełnić do objętości 1 000 ml wodą (3.1).
|
4. Aparatura i sprzęt
|
4.1.
|
Wyposażenie HPLC z detektorem spektrofluorometrycznym.
|
|
4.2.
|
Kolumna do chromatografii cieczowej, 125 mm × 4 mm, C18, wypełnienie 3 μm lub równoważne.
|
|
4.4.
|
Kolba propylenowa o pojemności 125 ml, z szeroką szyjką i nakrętką.
|
|
4.5.
|
Filtr membranowy, 0,45 μm.
|
|
4.6.
|
Autoklaw, 110 (±2) oC, 1,4 (±0,1) bar.
|
|
4.7.
|
Wstrząsarka mechaniczna lub mieszadło magnetyczne.
|
5. Sposób postępowania
5.1. Przygotowanie próbek
Rozdrobnić próbkę tak, aby przesiewała się przez oczka sita o średnicy 0,5 mm. Próbki o dużej zawartości wilgotności przed rozdrobnieniem wysuszyć albo na powietrzu w temperaturze nieprzekraczającej 50 oC, albo przez wymrażanie. Próbki o wysokiej zawartości tłuszczu przed rozdrobnieniem należy poddać ekstrakcji eterem naftowym (3.9).
5.2. Oznaczanie wolnego tryptofanu (ekstrakt)
Odważyć, z dokładnością do 1 mg, odpowiednią ilość (1–5 g) próbki przygotowanej w sposób określony w 5.1 do kolby stożkowej. Dodać 100,0 ml kwasu chlorowodorowego (3.13) i 5,00 ml stężonego roztworu wzorca wewnętrznego (3.16). Wstrząsać lub mieszać przez 60 minut z użyciem wstrząsarki mechanicznej lub mieszadła magnetycznego (4.7). Pozostawić do oddzielenia się osadu i przenieść pipetą 10,0 ml supernatantu roztworu do zlewki. Dodać 5 ml kwasu ortofosforowego (3.14). Dostosować pH do 3 z użyciem roztworu wodorotlenku sodu (3.10). Dodać odpowiednią ilość metanolu (3.8), aby uzyskać stężenie od 10 do 30 % metanolu w końcowej objętości. Przenieść do kolby miarowej o odpowiedniej pojemności i rozcieńczyć wodą do objętości właściwej dla chromatografii (objętość zbliżona do objętości kalibracyjnego roztworu wzorcowego (3.17)).
Przefiltrować kilka ml roztworu przez filtr membranowy 0,45 μm (4.5) przed dozowaniem na kolumnę HPLC. Przeprowadzić chromatografię w sposób określony w pkt 5.4.
Roztwór wzorcowy i ekstrakty chronić przed bezpośrednim działaniem światła słonecznego. Jeżeli jest niemożliwe dokonanie analizy ekstraktów tego samego dnia, ekstrakty można przechowywać w temperaturze 5 oC nie dłużej niż przez 3 dni.
5.3. Oznaczanie całkowitego tryptofanu (hydrolizat)
Odważyć, z dokładnością do 0,2 mg, od 0,1 do 1 g próbki przygotowanej w sposób określony w pkt 5.1 do kolby propylenowej (4.4). Odważona próbka musi zawierać około 10 mg azotu. Dodać 8,4 g oktahydratu wodorotlenku baru (3.4) i 10 ml wody. Mieszać z użyciem mieszadła Vortex (4.8) lub mieszadła magnetycznego (4.7). Pozostawić osłonięty teflonem magnes w mieszaninie. Spłukać ściany naczynia z użyciem 4 ml wody. Nałożyć nakrętkę i zamknąć luźno naczynie. Przenieść kolbę do autoklawu (4.6) z wrzącą wodą i parą i utrzymywać ją w czasie od 30 do 60 minut. Zamknąć autoklaw i autoklawować w temperaturze 110 oC (±2) oC przez 20 godzin.
Przed otwarciem autoklawu obniżyć temperaturę nieco poniżej 100 oC. W celu uniknięcia krystalizacji Ba(OH)2 · 8H2O dodać do ciepłej mieszaniny 30 ml wody o temperaturze pokojowej. Łagodnie wstrząsać lub mieszać. Dodać 2,00 ml stężonego roztworu wzorca wewnętrznego (α-metylo-tryptofanu) (3.16). Studzić naczynia w łaźni wodnej lub lodowej przez 15 minut.
Następnie dodać 5 ml kwasu ortofosforowego (3.14). Utrzymując naczynie w zimnej łaźni, zobojętnić z użyciem HCl (3.11), mieszając, i dostosować pH do 3,0 z HCl (3.12). Dodać ilość metanolu konieczną do uzyskania stężenia od 10 do 30 % metanolu w końcowej objętości. Przenieść do kolby miarowej o odpowiedniej pojemności i rozcieńczyć wodą do objętości, koniecznej dla chromatografii (np. 100 ml). Dodanie metanolu nie może powodować wytrącania.
Przefiltrować kilka ml roztworu przez filtr membranowy 0,45 μm (4.5) przed dozowaniem na kolumnę HPLC. Przeprowadzić chromatografię w sposób określony w 5.4.
Roztwór wzorcowy i produkty hydrolizy chronić przed bezpośrednim działaniem światła słonecznego. Jeżeli jest niemożliwe dokonanie analizy produktów hydrolizy tego samego dnia, można je przechowywać w temperaturze 5 oC nie dłużej niż przez 3 dni.
5.4. Oznaczanie HPLC
Poniższe parametry izokratycznej elucji są przykładowe. Dopuszczalne jest zastosowanie innych parametrów gwarantujących uzyskanie równoważnych wyników (zob. objaśnienia 9.1 i 9.2):
|
Kolumna do chromatografii cieczowej (4.2):
|
125 mm × 4 mm, C18, wypełnienie 3 μm lub równoważne
|
|
Temperatura kolumny:
|
Temperatura pokojowa
|
|
Faza ruchoma (3.22):
|
3,00 g kwasu octowego (3.18) + 900 ml wody (3.1) +50,0 ml roztworu (3.21) 1,1,1-trichloro-2-metylo-2-propanolu (3.19) w metanolu (3.8) (1g/100ml). Dostosować pH do 5,00 z użyciem etanoloaminy (3.20). Uzupełnić do objętości 1 l wodą (3.1)
|
|
Prędkość przepływu:
|
1 ml/min
|
|
Całkowity czas analizy chromatograficznej:
|
około 34 minuty
|
|
Długość fali przy detekcji:
|
wzbudzenie: 280 nm, emisja: 356 nm.
|
|
Dozowana objętość:
|
20 μl
|
6. Obliczanie wyników
Zawartość tryptofanu (X) w g na 100 g próbki obliczana jest według poniższego wzoru:

|
A
|
=
|
powierzchnia piku wzorca wewnętrznego, kalibracyjny roztwór wzorcowy (3.17)
|
|
B
|
=
|
powierzchnia piku tryptofanu, ekstraktu (5.2) lub hydrolizatu (5.3)
|
|
V1
|
=
|
objętość w ml (2 ml) stężonego roztworu tryptofanu (3.15) dodanego do roztworu kalibracyjnego (3.17)
|
|
c
|
=
|
stężenie w μmol/ml (= 2,50) stężonego roztworu tryptofanu (3.15) dodanego do roztworu kalibracyjnego (3.17)
|
|
V2
|
=
|
objętość w ml stężonego roztworu wzorca wewnętrznego (3.16) dodanego przy ekstrakcji (5.2) (= 5,00 ml) lub do hydrolizatu (5.3) (= 2,00 ml)
|
|
C
|
=
|
powierzchnia piku wzorca wewnętrznego, ekstraktu (5.2) lub hydrolizatu (5.3)
|
|
D
|
=
|
powierzchnia piku tryptofanu, kalibracyjnego roztworu wzorcowego (3.17)
|
|
V3
|
=
|
objętość w ml (= 2,00 ml) stężonego roztworu wzorca wewnętrznego (3.16) dodanego do kalibracyjnego roztworu wzorcowego (3.17)
|
|
m
|
=
|
naważka próbki w g (skorygowana względem masy wyjściowej, jeżeli była podsuszana lub odtłuszczana)
|
|
M
|
=
|
ciężar cząsteczkowy tryptofanu (= 204,23 g/mol)
|
7. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 10 % względem najwyższego wyniku.
8. Wyniki badań porównawczych
We Wspólnocie Europejskiej przeprowadzono badania porównawcze (czwarte porównanie wyników różnych laboratoriów), w których trzy próbki były analizowane przez maksymalnie 12 laboratoriów w celu sprawdzenia metody hydrolizy. Na każdej próbce powtórzono analizy pięciokrotnie. Ich wyniki są przedstawione w poniższej tabeli:
|
|
Próbka 1
Pasza dla świń
|
Próbka 2
Pasza dla świń uzupełniona L-tryptofanem
|
Próbka 3
Koncentrat paszowy dla świń
|
|
L
|
12
|
12
|
12
|
|
n
|
50
|
55
|
50
|
|
Średnia [g/kg]
|
2,42
|
3,40
|
4,22
|
|
sr [g/kg]
|
0,05
|
0,05
|
0,08
|
|
r [g/kg]
|
0,14
|
0,14
|
0,22
|
|
CV r [%]
|
1,9
|
1,6
|
1,9
|
|
SR [g/kg]
|
0,15
|
0,20
|
0,09
|
|
R [g/kg]
|
0,42
|
0,56
|
0,25
|
|
CVR [%]
|
6,3
|
6,0
|
2,2
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych zachowanych wyników eliminujących wartości oddalone (według Cochrana, próba wartości oddalonych Dixona)
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
r
|
=
|
powtarzalność wyników
|
|
R
|
=
|
odtwarzalność wyników
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności, w %
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności, w %
|
W innym badaniu porównawczym przeprowadzonym we Wspólnocie Europejskiej (trzecie porównanie wyników różnych laboratoriów) dwie próbki były analizowane przez maksymalnie 13 laboratoriów w celu sprawdzenia metody ekstrakcji wolnego tryptofanu. Na każdej próbce powtórzono analizy pięciokrotnie. Ich wyniki są przedstawione w poniższej tabeli:
|
|
Próbka 4
Mieszanka pszenicy i soi
|
Próbka 5
Mieszanka pszenicy i soi (= próbka 4) z dodatkiem tryptofanu (0,457 g/kg)
|
|
L
|
12
|
12
|
|
n
|
55
|
60
|
|
Średnia [g/kg]
|
0,391
|
0,931
|
|
sr [g/kg]
|
0,005
|
0,012
|
|
r [g/kg]
|
0,014
|
0,034
|
|
CVr [%]
|
1,34
|
1,34
|
|
SR [g/kg]
|
0,018
|
0,048
|
|
R [g/kg]
|
0,050
|
0,134
|
|
CVR [%]
|
4,71
|
5,11
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych zachowanych wyników eliminujących wartości oddalone (według Cochrana, próba wartości oddalonych Dixona)
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
r
|
=
|
powtarzalność wyników
|
|
R
|
=
|
odtwarzalność wyników
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności, w %
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności, w %
|
W kolejnym badaniu porównawczym przeprowadzonym we Wspólnocie Europejskiej cztery próbki były analizowane przez maksymalnie 7 laboratoriów w celu sprawdzenia metody hydrolizy tryptofanu. Na każdej próbce powtórzono analizy pięciokrotnie. Ich wyniki są przedstawione w poniższej tabeli.
|
|
Próbka 1
Mieszanka paszowa dla świń
(CRM 117)
|
Próbka 2
Niskotłuszczowa mączka rybna
(CRM 118)
|
Próbka 3
Mączka sojowa
(CRM 119)
|
Próbka 4
Odtłuszczone mleko w proszku
(CRM 120)
|
|
L
|
7
|
7
|
7
|
7
|
|
n
|
25
|
30
|
30
|
30
|
|
Średnia [g/kg]
|
2,064
|
8,801
|
6,882
|
5,236
|
|
sr [g/kg]
|
0,021
|
0,101
|
0,089
|
0,040
|
|
r [g/kg]
|
0,059
|
0,283
|
0,249
|
0,112
|
|
CVr [%]
|
1,04
|
1,15
|
1,30
|
0,76
|
|
SR [g/kg]
|
0,031
|
0,413
|
0,283
|
0,221
|
|
R [g/kg]
|
0,087
|
1,156
|
0,792
|
0,619
|
|
CVR [%]
|
1,48
|
4,69
|
4,11
|
4,22
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych zachowanych wyników eliminujących wartości oddalone (według Cochrana, próba wartości oddalonych Dixona)
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
r
|
=
|
powtarzalność wyników
|
|
R
|
=
|
odtwarzalność wyników
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności, w %
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności, w %
|
9. Objaśnienia
|
9.1.
|
Lepszy rozdział tryptofanu i α-metylo-tryptofanu może nastąpić przy uwzględnieniu następujących parametrów.
Izokratyczna elucja po gradientowym czyszczeniu kolumny:
|
Kolumna do chromatografii cieczowej:
|
125 mm × 4 mm, C18, wypełnienie 5 μm lub równoważne
|
|
Temperatura kolumny:
|
32 oC
|
|
Faza ruchoma:
|
A: 0,01 mol/l KH2PO4/metanol, 95+5 (V+V)
B: Metanol
|
|
Program gradientowy:
|
0 min
|
100 % A
|
0 % B
|
|
|
15 min
|
100 % A
|
0 % B
|
|
|
17 min
|
60 % A
|
40 % B
|
|
|
19 min
|
60 % A
|
40 % B
|
|
|
21 min
|
100 % A
|
0 % B
|
|
|
33 min
|
100 % A
|
0 % B
|
|
Prędkość przepływu:
|
1,2 ml/min
|
|
Całkowity czas analizy chromatograficznej:
|
około 33 minuty
|
|
|
9.2.
|
Analiza chromatograficzna będzie się zmieniać w zależności od typu HPLC i materiału użytego do wypełnienia kolumny. Wybrany system musi umożliwić uzyskanie oddzielającej linii bazowej pomiędzy tryptofanem i wzorcem wewnętrznym. Ponadto ważne jest, aby produkty degradacji zostały dobrze oddzielone od tryptofanu i wzorca wewnętrznego. Hydrolizaty bez wzorca wewnętrznego należy poddać analizie w celu sprawdzenia linii bazowej stosownie do wzorca wewnętrznego względem zanieczyszczeń. Ważne jest, aby czas analizy był wystarczająco długi dla elucji wszystkich produktów degradacji, w przeciwnym razie ostatnie eluujące piki na chromatogramie mogą zakłócać kolejną analizę.
W zakresie badanych stężeń układ chromatograficzny musi zapewniać liniową odpowiedź. Liniową odpowiedź należy mierzyć przy stałym (normalnym) stężeniu wzorca wewnętrznego i zmieniających się stężeniach tryptofanu. Ważne jest, aby wielkość pików tryptofanu i wzorca wewnętrznego zawierała się w liniowym zakresie układu HPLC z detekcją fluorescencyjną. Jeżeli piki tryptofanu lub wzorca wewnętrznego są zbyt małe lub zbyt duże, analizę należy powtórzyć przy zmienionej wielkości próbki lub jej objętości końcowej.
|
|
9.3.
|
Wodorotlenek baru
Fakt, że wodorotlenek baru z czasem jest coraz trudniejszy do rozpuszczenia, wpływa na nieklarowność roztworu do oznaczania HPLC, która może być przyczyną zaniżania wyników oznaczania tryptofanu.
|
H. OZNACZANIE SUROWEGO OLEJU I TŁUSZCZU
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości surowych olejów i tłuszczów w paszach. Nie stosuje się jej do analizy nasion i owoców roślin oleistych.
Zastosowanie dwóch sposobów postępowania opisanych poniżej zależy od rodzaju i składu paszy oraz celu prowadzenia analizy.
1.1. Postępowanie A – Bezpośrednio ekstrahowane oleje i tłuszcze
Postępowanie ma zastosowanie do materiałów paszowych pochodzenia roślinnego, z wyjątkiem tych, które zostały objęte zakresem postępowania B.
1.2. Postępowanie B – Całkowite surowe oleje i tłuszcze
Postępowanie ma zastosowanie do materiałów paszowych pochodzenia zwierzęcego i wszystkich mieszanek paszowych. Jest stosowane do wszystkich materiałów, z których oleje i tłuszcze nie mogą być w całości wyekstrahowane bez uprzedniej hydrolizy (np. gluten, drożdże, białka ziemniaczane i produkty przetworzone np. poprzez ekstruzję, płatkowanie i ogrzewanie).
1.3. Interpretacja wyników
We wszystkich przypadkach, w których wynik otrzymany przy zastosowaniu postępowania B jest wyższy od wyniku otrzymanego przy zastosowaniu postępowania A, wynik otrzymany przy zastosowaniu postępowania B należy traktować jako rzeczywisty.
2. Sposób przeprowadzenia metody
2.1. Postępowanie A
Próbka jest poddawana ekstrakcji eterem naftowym. Roztwór jest oddestylowany, a pozostałość jest suszona i ważona.
2.2. Postępowanie B
Próbka jest ogrzewana kwasem chlorowodorowym. Mieszanina jest oziębiana i filtrowana. Pozostałość jest przemywana i suszona, a następnie poddawana oznaczaniu przy zastosowaniu postępowania A.
3. Odczynniki i roztwory
|
3.1.
|
Eter naftowy o zakresie temperatur wrzenia od 40 do 60 oC. Wartość bromowa musi być niższa niż 1, a pozostałość po odparowaniu niższa niż 2 mg/100 ml.
|
|
3.2.
|
Siarczan sodu bezwodny.
|
|
3.3.
|
Kwas chlorowodorowy, c = 3 mol/l.
|
|
3.4.
|
Materiał wspomagający filtrację, np. Kieselgur, Hyflo-supercel.
|
4. Aparatura i sprzęt
|
4.1.
|
Aparat do ekstrakcji. Jeżeli aparat jest wyposażony w syfon (jak np. aparat Soxhleta), prędkość skraplania musi wynosić około 10 cykli na godzinę. Jeżeli aparat nie ma syfonu, wówczas prędkość skraplania musi wynosić około 10 ml na minutę.
|
|
4.2.
|
Gilzy do ekstrakcji, niezawierające substancji rozpuszczalnych w eterze naftowym, o porowatości odpowiadającej wymaganiom określonym w pkt 4.1.
|
|
4.3.
|
Suszarka albo suszarka próżniowa, z możliwością ustawienia temperatury suszenia na 75 ± 3 oC, lub suszarka nawiewna z możliwością ustawienia temperatury na 100 ± 3 oC.
|
5. Sposób postępowania
5.1. Postępowanie A (zob. pkt 8.1)
Odważyć, z dokładnością do 1 mg, 5 g próbki, przenieść do gilzy ekstrakcyjnej (4.2) i przykryć beztłuszczowym zwitkiem waty bawełnianej.
Umieścić gilzę w aparacie do ekstrakcji (4.1) i ekstrahować przez sześć godzin eterem naftowym (3.1). Zebrać ekstrakt eterowy do suchej, zważonej kolby zawierającej kawałki pumeksu (2).
Oddestylować rozpuszczalnik. Suszyć pozostałość, umieszczając kolbę w suszarce (4.3) na półtorej godziny. Schłodzić w eksykatorze i zważyć. Suszyć ponownie przez 30 minut w celu zapewnienia stałej masy olejów i tłuszczów (ubytek masy między dwoma kolejnymi ważeniami nie może przekraczać 1 mg).
5.2. Postępowanie B
Odważyć, z dokładnością do 1 mg, 2,5 g próbki (zob. pkt 8.2), umieścić w zlewce o pojemności 400 ml lub w kolbie stożkowej o pojemności 300 ml i dodać 100 ml kwasu chlorowodorowego (3.3) oraz kawałki pumeksu. Przykryć zlewkę szkiełkiem zegarkowym lub dopasować kolbę stożkową z chłodnicą zwrotną. Doprowadzić mieszaninę do spokojnego wrzenia w małym płomieniu palnika lub na płytce grzewczej i utrzymywać wrzenie przez godzinę. Nie dopuszczać do osadzania się produktu na ścianach naczynia.
Schłodzić i dodać materiał wspomagający filtrację (3.4) w ilości zapobiegającej jakimkolwiek stratom oleju i tłuszczu podczas filtracji. Przefiltrować przez zwilżony, beztłuszczowy podwójny filtr papierowy. Przemywać pozostałość zimną wodą do uzyskania obojętnego filtratu. Sprawdzić, czy filtrat nie zawiera śladów olejów lub tłuszczów. Ich obecność wskazuje na konieczność przeprowadzenia przed hydrolizą ekstrakcji eterem naftowym, przy zastosowaniu postępowania A.
Umieścić na szkiełku zegarkowym podwójny filtr papierowy z pozostałością i suszyć przez 1,5 godziny w suszarce nawiewnej (4.3) w temperaturze 100 ± 3 oC.
Umieścić podwójny filtr papierowy zawierający suchą pozostałość w gilzie ekstrakcyjnej (4.2) i przykryć zwitkiem beztłuszczowej waty bawełnianej. Umieścić gilzę w aparacie do ekstrakcji (4.1) i postępować w sposób określony w pkt 5.1, akapity drugi i trzeci.
6. Wyrażenie wyników
Masę pozostałości wyrazić jako procent wagowy próbki.
7. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
0,2 % w wartości bezwzględnej, dla zawartości surowego oleju i tłuszczu niższej niż 5 %,
|
|
—
|
4,0 % względem najwyższego wyniku, dla zawartości oleju i tłuszczu od 5 do 10 %,
|
|
—
|
0,4 % w wartości bezwzględnej, dla zawartości oleju i tłuszczu wyższej niż 10 %.
|
8. Objaśnienia
|
8.1.
|
Dla produktów o wysokiej zawartości olejów i tłuszczów, które są trudne do rozdrobnienia lub osiągnięcia homogenności badanej próbki, postępować w sposób podany poniżej.
Odważyć, z dokładnością do 1 mg, 20 g próbki i zmieszać z co najmniej 10 g bezwodnego siarczanu sodu (3.2). Ekstrahować eterem naftowym (3.1) w sposób określony w pkt 5.1. Otrzymany ekstrakt uzupełnić do objętości 500 ml eterem naftowym (3.1) i zmieszać. Pobrać 50 ml roztworu i przenieść do małej, suchej i zważonej kolby zawierającej kawałki pumeksu. Oddestylować rozpuszczalnik, suszyć i postępować, jak wskazano w pkt 5.1 akapit ostatni.
Usunąć rozpuszczalnik z pozostałości po ekstrakcji w gilzie, rozdrobnić pozostałość na 1 mm cząstki, ponownie umieścić w gilzie (nie dodawać siarczanu sodu) i postępować, jak wskazano w pkt 5.1 akapity drugi i trzeci.
Zawartość oleju i tłuszczu obliczyć w procentach wagowych próbki, według następującego wzoru:
(10 m1 + m2) × 5
gdzie:
|
m1
|
=
|
masa w g pozostałości po pierwszej ekstrakcji, podzielnej części ekstraktu
|
|
m2
|
=
|
masa w g pozostałości po drugiej ekstrakcji
|
|
|
8.2.
|
Dla produktów o niskiej zawartości olejów i tłuszczów próbkę można zwiększyć do 5 g.
|
|
8.3.
|
W przypadku karmy dla zwierząt domowych o wysokiej zawartości wody dla zwierząt domowych może wystąpić potrzeba zmieszania tej karmy przed hydrolizą i ekstrakcją z bezwodnym siarczanem sodu, z zastosowaniem postępowania B.
|
|
8.4.
|
W przypadku postępowania określonego w pkt 5.2 do przemywania pozostałości po filtracji skuteczniejsze może być użycie wody gorącej zamiast zimnej.
|
|
8.5.
|
W przypadku niektórych pasz czas suszenia wynoszący 1,5 godziny może być przedłużony. Należy jednak unikać nadmiernego suszenia, gdyż może to prowadzić do zaniżania wyników. Można również stosować kuchenki mikrofalowe.
|
|
8.6.
|
Jeżeli zawartość surowego oleju lub tłuszczu jest wyższa niż 15 %, zaleca się wstępną ekstrakcję przed hydrolizą, z zastosowaniem postępowania A, i powtórną ekstrakcję, z zastosowaniem postępowania B. Postępowanie to zależy od rodzaju paszy i oleju lub tłuszczu w paszy.
|
I. OZNACZANIE WŁÓKNA SUROWEGO
1. Cel i zakres stosowania metody
Metoda służy do oznaczania w paszach zawartości nietłuszczowych substancji organicznych, które nie rozpuszczają się ani w środowisku kwaśnym, ani zasadowym i które zwyczajowo określa się mianem włókna surowego.
2. Sposób przeprowadzenia metody
Próbkę, w razie konieczności odtłuszczoną, gotuje się w roztworach kwasu siarkowego i wodorotlenku potasu o odpowiednim stężeniu. Pozostałość jest rozdzielana przez filtrację na filtrze ze szklanego spieku, przemywana, suszona, ważona i spopielana w temperaturze od 475 do 500 oC. Ubytek masy po spopielaniu odpowiada zawartości włókna surowego w badanej próbce.
3. Odczynniki i roztwory
|
3.1.
|
Kwas siarkowy, c = 0,13 mol/l.
|
|
3.2.
|
Odczynnik przeciwpieniący (np. n-oktanol).
|
|
3.3.
|
Materiał wspomagający filtrowanie (Celit 545 lub podobny), wyprażony w temperaturze 500 oC przez cztery godziny (8.6).
|
|
3.5.
|
Eter naftowy o temperaturze wrzenia od 40 do 60 oC.
|
|
3.6.
|
Kwas chlorowodorowy, c = 0,5 mol/l.
|
|
3.7.
|
Roztwór wodorotlenku potasu, c = 0,23 mol/l.
|
4. Aparatura i sprzęt
|
4.1.
|
Urządzenie grzewcze do roztwarzania kwasem siarkowym lub roztworem wodorotlenku potasu, wyposażone w podstawkę mocującą tygiel filtracyjny (4.2) i rurkę odprowadzającą z kranem służącą do odprowadzenia cieczy i próżni, ewentualnie ze sprzężonym powietrzem. Codziennie przed użyciem podgrzewa się urządzenie, przemywając je wrzącą wodą przez pięć minut.
|
|
4.2.
|
Szklany tygiel filtracyjny ze spiekiem o porach od 40 do 90 μm. Przed pierwszym użyciem ogrzewać przez kilka minut w temperaturze 500 oC i schłodzić (8.6).
|
|
4.3.
|
Cylinder o pojemności co najmniej 270 ml z chłodnicą zwrotną, przystosowany do gotowania.
|
|
4.4.
|
Suszarka z termostatem.
|
|
4.5.
|
Piec muflowy z termostatem.
|
|
4.6.
|
Urządzenie do ekstrakcji składające się z podstawki mocującej tygiel filtracyjny (4.2) i rurki odprowadzającej z kranem, służącej do ujścia cieczy i próżni.
|
|
4.7.
|
Pierścienie łączące, służące do połączenia urządzenia grzewczego (4.1) z tyglem filtracyjnym (4.2) i cylindrem (4.3) oraz do dołączenia urządzenia ekstrakcyjnego do ekstrakcji na zimno (4.6) i tygla.
|
5. Sposób postępowania
Odważyć, z dokładnością do 1 mg, 1 g przygotowanej próbki do tygla (4.2) (zob. objaśnienia 8.1, 8.2 i 8.3) i dodać 1 g materiału wspomagającego filtrowanie (3.3).
Połączyć urządzenie grzewcze (4.1) z tyglem filtracyjnym (4.2), a następnie dołączyć cylinder (4.3) do tygla. Wlać 150 ml wrzącego kwasu siarkowego (3.1) do cylindra połączonego z tyglem i, w razie potrzeby, dodać kilka kropli odczynnika przeciwpieniącego (3.2).
Doprowadzić ciecz do wrzenia w czasie 5 ± 2 minut i gotować przez dokładnie 30 minut.
Otworzyć kran rurki odprowadzającej (4.1) i w warunkach próżni filtrować kwas siarkowy przez tygiel filtracyjny i przemywać pozostałość trzykrotnie 30 ml porcjami wrzącej wody, upewniając się, że pozostałość przefiltrowano do sucha po każdym przemyciu.
Zamknąć ujście kranu i wlać 150 ml wrzącego roztworu wodorotlenku potasu (3.7) do cylindra połączonego z tyglem i dodać kilka kropli odczynnika przeciwpieniącego (3.2). Doprowadzić ciecz do punktu wrzenia w czasie 5 ± 2 minut i gotować przez 30 minut. Przefiltrować i powtórzyć procedurę przemywania wykorzystaną w odniesieniu do kwasu siarkowego.
Po ostatnim przemyciu i wysuszeniu odłączyć tygiel i jego zawartość i podłączyć go do urządzenia do zimnej ekstrakcji (4.6). W warunkach próżni przemywać pozostałość w tyglu trzema kolejnymi porcjami 25 ml acetonu (3.4), upewniając się, że pozostałość przefiltrowano do sucha po każdym przemyciu.
Wysuszyć tygiel do stałej masy w suszarce o temperaturze 130 oC. Po każdym suszeniu schłodzić w eksykatorze i niezwłocznie zważyć. Umieścić tygiel w piecu muflowym i spopielać do stałej masy (ubytek masy między dwoma kolejnymi ważeniami musi być równy lub mniejszy niż 2 mg) w temperaturze od 475 do 500 oC przez co najmniej 30 minut.
Przed ważeniem, po każdym ogrzewaniu, najpierw schłodzić w piecu, a następnie w eksykatorze.
Przeprowadzić test ślepej próby bez próbki. Ubytek masy po spopielaniu nie może przekraczać 4 mg.
6. Obliczanie wyników
Procentową zawartość włókna surowego w próbce oblicza się według następującego wzoru:

gdzie:
|
m
|
=
|
masa próbki w g
|
|
m0
|
=
|
ubytek masy po spopieleniu w trakcie oznaczania, w g
|
|
m1
|
=
|
ubytek masy po spopieleniu w trakcie ślepej próby, w g
|
7. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
0,6 % wartości bezwzględnej, dla zawartości włókna surowego niższej niż 10 %,
|
|
—
|
6 % względem wyższego wyniku, dla zawartości włókna surowego równej lub wyższej niż 10 %.
|
8. Objaśnienia
|
8.1.
|
Pasze zawierające więcej niż 10 % tłuszczu surowego przed oznaczaniem należy odtłuścić eterem naftowym (3.5). Podłączyć tygiel filtracyjny (4.2) wraz z jego zawartością do urządzenia do zimnej ekstrakcji (4.6), zastosować próżnię i przemyć pozostałość trzykrotnie 30 ml porcjami eteru naftowego, upewniając się, że pozostałość jest sucha. Podłączyć tygiel wraz z jego zawartością do urządzenia grzewczego (4.1) i postępować w sposób określony w pkt 5.
|
|
8.2.
|
Pasze zawierające tłuszcze, które nie mogą być bezpośrednio ekstrahowane eterem naftowym (3.5), odtłuścić w sposób określony w pkt 8.1 i ponownie odtłuścić po gotowaniu z kwasem. Po gotowaniu z kwasem, a następnie przemyciu podłączyć tygiel wraz z jego zawartością do urządzenia do zimnej ekstrakcji (4.6) i przemyć trzykrotnie 30 ml porcjami acetonu, a następnie trzykrotnie 30 ml porcjami eteru naftowego. Przefiltrować do sucha w warunkach próżni i postępować w sposób określony w pkt 5, rozpoczynając postępowanie z wodorotlenkiem potasu.
|
|
8.3.
|
Jeżeli pasze zawierają więcej niż 5 % węglanów, wyrażonych jako węglan wapnia, podłączyć tygiel (4.2) z odważoną próbką do urządzenia grzewczego (4.1). Przemyć próbkę trzykrotnie 30 ml porcjami kwasu chlorowodorowego (3.6). Po dodaniu każdej porcji kwasu pozostawić próbkę na około minutę przed filtrowaniem. Przemyć 30 ml wody i postępować w sposób określony w pkt 5.
|
|
8.4.
|
Jeżeli używana jest aparatura stojąca (kilka tygli podłączonych do jednego urządzenia grzewczego), nie należy wykonywać dwóch pojedynczych oznaczeń tej samej próbki w tej samej partii.
|
|
8.5.
|
Jeżeli po gotowaniu filtrowanie roztworu kwasowego i zasadowego jest trudne, przepuścić sprężone powietrze przez rurkę podłączaną do urządzenia grzewczego i następnie kontynuować filtrowanie.
|
|
8.6.
|
Temperatura spopielania nie może być wyższa niż 500 oC, ze względu na wytrzymałość szklanego tygla. Postępować w taki sposób, aby uniknąć szoku termicznego podczas ogrzewania i schładzania.
|
J. OZNACZANIE CUKRÓW
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości cukrów redukujących i całkowitej zawartości cukrów po inwersji, wyrażonych jako glukoza lub w razie potrzeby jako sacharoza, z zastosowaniem współczynnika 0,95. Metodę stosuje się do mieszanek paszowych. Dla innych pasz stosuje się specjalne metody. W razie potrzeby laktozę należy mierzyć oddzielnie, uwzględniając ją w obliczeniach.
2. Sposób przeprowadzenia metody
Cukry ekstrahuje się w rozcieńczonym etanolu. Otrzymany roztwór klaruje się roztworami Carreza I i Carreza II. Po wyeliminowaniu etanolu zawartość cukrów przed i po inwersji oznacza się metodą Luff-Schoorla.
3. Odczynniki i roztwory
|
3.1.
|
Etanol, roztwór 40 % (v/v), gęstość d = 0,948 g/ml przy temperaturze 20 oC, obojętny wobec fenoloftaleiny.
|
|
3.2.
|
Roztwór Carreza I: rozpuścić w wodzie 21,9 g octanu cynku, Zn (CH3COO)2 2H2O i 3 g lodowatego kwasu octowego. Uzupełnić do objętości 100 ml wodą.
|
|
3.3.
|
Roztwór Carreza II: rozpuścić w wodzie 10,6 g heksacyjanożelazianu(II) potasu K4Fe (CN)6 3H2O. Uzupełnić do objętości 100 ml wodą.
|
|
3.4.
|
Roztwór 0,1 % (w/v) oranżu metylowego.
|
|
3.5.
|
Kwas chlorowodorowy, 4 mol/l.
|
|
3.6.
|
Kwas chlorowodorowy, 0,1 mol/l.
|
|
3.7.
|
Roztwór wodorotlenku sodu, 0,1 mol/l.
|
|
3.8.
|
Odczynnik Luff-Schoorla:
Ostrożnie mieszając, dodać roztwór kwasu cytrynowego (3.8.2) do roztworu węglanu sodu (3.8.3). Następnie dodać roztwór siarczanu miedzi (3.8.1) i uzupełnić do objętości 1 l wodą. Zostawić na noc do osadzenia i przefiltrować.
Sprawdzić normalność otrzymanego odczynnika (Cu 0,05 mol/l; Na2 CO3 1 mol/l). Zob. pkt 5.4, ostatni akapit. pH roztworu musi wynosić około 9,4.
|
|
3.8.1.
|
Roztwór siarczanu miedzi: rozpuścić 25 g siarczanu miedzi Cu SO4 5H2O, wolnego od żelaza, w 100 ml wody.
|
|
3.8.2.
|
Roztwór kwasu cytrynowego: rozpuścić 50 g kwasu cytrynowego C6H8O7·H2O w 50 ml wody.
|
|
3.8.3.
|
Roztwór węglanu sodu: rozpuścić 143,8 g bezwodnego węglanu sodu w około 300 ml ciepłej wody. Pozostawić do schłodzenia.
|
|
3.9.
|
Roztwór tiosiarczanu(VI) sodu, 0,1 mol/l.
|
|
3.10.
|
Roztwór skrobi: do 1 l wrzącej wody dodać 5 g skrobi rozpuszczonej w 30 ml wody. Gotować trzy minuty, pozostawić do schłodzenia, a jeżeli to konieczne, dodać 10 mg jodku rtęci jako środka konserwującego.
|
|
3.11.
|
Kwas siarkowy, 3 mol/l.
|
|
3.12.
|
30 % (w/v) roztwór jodku potasu.
|
|
3.13.
|
Granulki pumeksu gotowane w kwasie chlorowodorowym, przemyte wodą i wysuszone.
|
|
3.14.
|
3-metylobutan-l-ol.
|
4. Aparatura i sprzęt
Mikser (tumbler): ok. 35–40 obr./min.
5. Sposób postępowania
5.1. Ekstrakcja próbki
Odważyć, z dokładnością do 1 mg, 2,5 g próbki i umieścić w kolbie miarowej o pojemności 250 ml. Dodać 200 ml etanolu (3.1) i mieszać w mikserze przez godzinę. Dodać 5 ml roztworu Carreza I (3.2) i mieszać przez ok. 30 sekund. Dodać 5 ml roztworu Carreza II (3.3), i ponownie mieszać przez minutę. Uzupełnić do pełnej objętości kolby etanolem (3.1), homogenizować i przefiltrować. Pobrać 200 ml filtratu i odparować do około połowy objętości w celu usunięcia większości etanolu. Przenieść ilościowo pozostałość po odparowaniu do kolby miarowej o pojemności 200 ml z użyciem ciepłej wody, schłodzić, uzupełnić do pełnej objętości kolby wodą, homogenizować i, jeżeli to konieczne, przefiltrować. Roztwór ten będzie użyty do oznaczenia zawartości cukrów redukujących i, po inwersji, całkowitej zawartości cukrów.
5.2. Oznaczanie cukrów redukujących
Używając pipety, pobrać nie więcej niż 25 ml roztworu zawierającego mniej niż 60 mg cukrów redukujących wyrażonych jako glukoza. Jeżeli to konieczne, uzupełnić do objętości 25 ml wodą destylowaną i oznaczyć zawartość cukrów redukujących metodą Luff-Schoorla. Wynik wyrazić jako procentową zawartość glukozy w próbce.
5.3. Oznaczanie całkowitej zawartości cukru po inwersji
Używając pipety, pobrać 50 ml roztworu i przenieść go do kolby miarowej o pojemności 100 ml. Dodać kilka kropli roztworu oranżu metylowego (3.4), i następnie, ostrożnie mieszając, dodać kwas chlorowodorowy (3.5), aż do zmiany barwy cieczy na czerwoną. Dodać 15 ml kwasu chlorowodorowego (3.6) i zanurzyć kolbę we wrzącej łaźni wodnej na 30 minut. Szybko schłodzić do temperatury około 20 oC i dodać 15 ml roztworu wodorotlenku sodu (3.7). Uzupełnić kolbę do objętości 100 ml wodą i homogenizować. Pobrać nie więcej niż 25 ml roztworu zawierającego mniej niż 60 mg cukrów redukujących wyrażonych jako glukoza. Jeżeli to konieczne, uzupełnić do objętości 25 ml wodą destylowaną i oznaczyć zawartość cukrów redukujących metodą Luff-Schoorla. Wynik wyrazić jako procentową zawartość glukozy lub, w stosownych przypadkach, sacharozy, mnożąc przez współczynnik 0,95.
5.4. Miareczkowanie metodą Luff-Schoorla
Używając pipety, pobrać 25 ml odczynnika Luff-Schoorla (3.8), przenieść go do kolby Erlenmeyera o pojemności 300 ml i dodać dokładnie 25 ml klarownego roztworu cukru. Dodać dwie granulki pumeksu (3.13), ogrzewać nad średnim płomieniem, ręcznie mieszając, i doprowadzić ciecz do wrzenia w czasie około dwóch minut. Natychmiast postawić kolbę Erlenmeyera na siatce azbestowej z otworem o średnicy około 6 cm, pod którym pali się płomień. Płomień należy ustawić tak, aby ogrzewane było tylko dno kolby. Dopasować chłodnicę zwrotną do kolby Erlenmeyera. Gotować dokładnie dziesięć minut. Schłodzić natychmiast zimną wodą i po około pięciu minutach miareczkować w następujący sposób:
Dodać 10 ml roztworu jodku potasu (3.12) i od razu po tej czynności (zachowując ostrożność ze względu na ryzyko powstania piany) dodać 25 ml kwasu siarkowego (3.11). Miareczkować roztworem tiosiarczanu(VI) sodu (3.9) do pojawienia się mętnej żółtej barwy, dodać wskaźnik skrobiowy (3.10) i dokończyć miareczkowanie.
Przeprowadzić takie samo miareczkowanie na mieszaninie 25 ml odczynnika Luff-Schoorla (3.8) i 25 ml wody, po dodaniu 10 ml roztworu jodku potasu (3.12) i 25 ml kwasu siarkowego (3.11), bez gotowania.
6. Obliczanie wyników
Wykorzystując tabelę, określić ilość glukozy w mg, która odpowiada różnicy między wynikami dwóch miareczkowań, wyrażonych w mg tiosiarczanu(VI) sodu 0,1 mol/l. Wynik wyrazić jako udział procentowy w próbce.
7. Postępowanie specjalne
|
7.1.
|
W przypadku pasz bogatych w melasę i innych pasz, które nie są szczególnie homogenizowane, odważyć 20 g próbki i umieścić z 500 ml wody w kolbie miarowej o pojemności 1 l. Mieszać przez godzinę w mikserze. Sklarować odczynnikami Carreza I (3.2) i II (3.3) w sposób określony w pkt 5.1, z użyciem czterokrotnie większych ilości każdego odczynnika. Uzupełnić kolbę do objętości 80 % etanolem (v/v).
Homogenizować i przefiltrować. Usunąć etanol w sposób określony w pkt 5.1. Jeżeli nie ma dekstrynowanej skrobi, uzupełnić do pełnej objętości kolby wodą destylowaną.
|
|
7.2.
|
W przypadku melasy i materiałów paszowych, bogatych w cukier i prawie wolnych od skrobi (np. chleb świętojański, suszone krajane buraki ćwikłowe itp.), odważyć 5 g próbki, umieścić w kolbie miarowej o pojemności 250 ml, dodać 200 ml wody destylowanej, mieszać w mikserze przez godzinę lub, jeśli to konieczne, dłużej. Roztwór klarować odczynnikami Carreza I (3.2) i II (3.3) w sposób określony w pkt 5.1. Uzupełnić do pełnej objętości kolby zimną wodą, homogenizować i przefiltrować. Następnie postępować w sposób określony w pkt 5.3, w celu określenia zawartości cukrów.
|
8. Objaśnienia
|
8.1.
|
Aby zapobiec powstawaniu piany, wskazane jest dodanie (niezależnie od objętości) około 1 ml 3-metylobutan-1-olu (3.14) przed gotowaniem z odczynnikiem Luff-Schoorla.
|
|
8.2.
|
Różnica pomiędzy zawartością całkowitą cukrów po inwersji, wyrażonych jako glukoza, a zawartością cukrów redukujących, wyrażonych jako glukoza, pomnożona przez 0,95 daje procentową zawartość sacharozy.
|
|
8.3.
|
Aby oznaczyć zawartość cukrów redukujących, z wyłączeniem laktozy, można zastosować dwie następujące metody:
|
|
8.3.1.
|
Dla szacunkowych obliczeń pomnożyć zawartość laktozy oznaczoną inną metodą przez 0,675 i uzyskany wynik odjąć od zawartości cukrów redukujących.
|
|
8.3.2.
|
Aby dokładnie obliczyć zawartość cukrów redukujących, z wyłączeniem laktozy, ta sama próbka musi być użyta do dwóch końcowych oznaczeń. Jedną analizę przeprowadza się na części roztworu otrzymanego w sposób określony w pkt 5.1, drugą na części roztworu otrzymanego podczas oznaczania laktozy metodą służącą do jej oznaczania (po fermentacji innych rodzajów cukrów i sklarowaniu).
W obu przypadkach ilość cukru jest oznaczana metodą Luff-Schoorla i przeliczana na mg glukozy Jedna wartość jest odejmowana od drugiej, a różnica jest wyrażana jako udział procentowy w próbce.
Przykład
Dwie pobrane objętości odpowiadają próbce o masie próbki 250 mg dla każdego oznaczenia.
W pierwszym przypadku 17 ml roztworu tiosiarczanu(VI) sodu o stężeniu 0,1 mol/l odpowiada 44,2 mg zużytej glukozy, w drugim przypadku 11 ml odpowiada 27,6 mg glukozy.
Różnica wynosi 16,6 mg glukozy.
Zawartość cukrów redukujących (z wyłączeniem laktozy), w przeliczeniu na glukozę, wynosi zatem:

|
Tabela wartości dla 25 ml odczynnika Luff-Schoorla
ml Na2 S2 O3, 0,1 mol/l, podgrzewanie przez 2 minuty, gotowanie przez 10 minut
|
Na2 S2 O3
0,1 mol/l
|
Glukoza, fruktoza, cukry inwertowane
C6 H12 O6
|
Laktoza
C12 H22 O11
|
Maltoza
C12 H22 O11
|
Na2 S2 O3
0,1 mol/l
|
|
ml
|
mg
|
różnica
|
mg
|
różnica
|
mg
|
różnica
|
ml
|
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
2,4
4,8
7,2
9,7
12,2
14,7
17,2
19,8
22,4
25,0
27,6
30,3
33,0
35,7
38,5
41,3
44,2
47,1
50,0
53,0
56,0
59,1
62,2
|
2,4
2,4
2,5
2,5
2,5
2,5
2,6
2,6
2,6
2,6
2,7
2,7
2,7
2,8
2,8
2,9
2,9
2,9
3,0
3,0
3,1
3,1
|
3,6
7,3
11,0
14,7
18,4
22,1
25,8
29,5
33,2
37,0
40,8
44,6
48,4
52,2
56,0
59,9
63,8
67,7
71,7
75,7
79,8
83,9
88,0
|
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,8
3,8
3,8
3,8
3,8
3,8
3,9
3,9
3,9
4,0
4,0
4,1
4,1
4,1
|
3,9
7,8
11,7
15,6
19,6
23,5
27,5
31,5
35,5
39,5
43,5
47,5
51,6
55,7
59,8
63,9
68,0
72,2
76,5
80,9
85,4
90,0
94,6
|
3,9
3,9
3,9
4,0
3,9
4,0
4,0
4,0
4,0
4,0
4,0
4,1
4,1
4,1
4,1
4,1
4,2
4,3
4,4
4,5
4,6
4,6
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
K. OZNACZANIE LAKTOZY
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości laktozy w paszach zawierających ponad 0,5 % laktozy.
2. Sposób przeprowadzenia metody
Cukry są rozpuszczane w wodzie. Roztwór jest poddawany fermentacji w obecności drożdży Saccharomyces cerevisiae, które nie rozkładają laktozy. Po sklarowaniu i przefiltrowaniu zawartość laktozy w filtracie jest oznaczana metodą Luff-Schoorla.
3. Odczynniki i roztwory
|
3.1.
|
Suspensja drożdży Saccharomyces cerevisiae: 25 g świeżych drożdży umieścić w 100 ml wody. Zawiesinę przechowywać w lodówce nie dłużej niż przez 7 dni od jej sporządzenia.
|
|
3.2.
|
Roztwór Carreza I: rozpuścić w wodzie 21,9 g octanu cynku, Zn (CH3 COO)2 2H2O i 3 g lodowatego kwasu octowego. Uzupełnić kolbę do objętości 100 ml wodą.
|
|
3.3.
|
Roztwór Carreza II: rozpuścić w wodzie 10,6 g heksacyjanożelazianu(II) potasu K4Fe (CN)6 3H2O. Uzupełnić kolbę do objętości 100 ml wodą.
|
|
3.4.
|
Odczynnik Luff-Schoorla:
Ostrożnie mieszając, dodać roztwór kwasu cytrynowego (3.4.2) do roztworu węglanu sodu (3.4.3). Następnie dodać roztwór siarczanu miedzi (3.4.1) i uzupełnić kolbę do objętości 1 l wodą. Zostawić na noc do osadzenia i przefiltrować. Sprawdzić normalność otrzymanego odczynnika (Cu 0,05 mol/l; Na2 CO3 1 mol/l). Wartość pH roztworu musi wynosić około 9,4.
|
|
3.4.1.
|
Roztwór siarczanu miedzi: rozpuścić 25 g siarczanu miedzi Cu SO4 5H2O wolnego od żelaza w 100 ml wody.
|
|
3.4.2.
|
Roztwór kwasu cytrynowego: rozpuścić 50 g kwasu cytrynowego C6H8O7 · H2O w 50 ml wody.
|
|
3.4.3.
|
Roztwór węglanu sodu: rozpuścić 143,8 g bezwodnego węglanu sodu w około 300 ml ciepłej wody. Pozostawić do schłodzenia.
|
|
3.5.
|
Granulki pumeksu gotowane w kwasie chlorowodorowym, przemyte wodą i wysuszone.
|
|
3.6.
|
30 % roztwór jodku potasu (w/v).
|
|
3.7.
|
Kwas siarkowy, 3 mol/l.
|
|
3.8.
|
Roztwór tiosiarczanu sodu, 0,1 mol/l.
|
|
3.9.
|
Roztwór skrobi: do 1 l wrzącej wody dodać 5 g skrobi rozpuszczonej w 30 ml wody. Gotować trzy minuty, pozostawić do schłodzenia, a jeżeli to konieczne, dodać 10 mg jodku rtęci jako środka konserwującego.
|
4. Aparatura i sprzęt
Łaźnia wodna z termostatem umożliwiającym utrzymanie temperatury od 38 do 40 oC.
5. Sposób postępowania
Odważyć, z dokładnością do 1 mg, 1 g próbki i umieścić w kolbie miarowej o pojemności 100 ml. Dodać od 25 do 30 ml wody. Umieścić kolbę we wrzącej łaźni wodnej na 30 minut, a następnie schłodzić do temperatury około 35 oC. Dodać 5 ml zawiesiny drożdży (3.1) i homogenizować. Pozostawić kolbę na dwie godziny w łaźni wodnej o temperaturze od 38 do 40 oC. Schłodzić do temperatury około 20 oC.
Dodać 2,5 ml roztworu Carreza I (3.2) i mieszać przez 30 sekund, następnie dodać 2,5 ml roztworu Carreza II (3.3) i ponownie mieszać przez 30 sekund. Uzupełnić kolbę do objętości 100 ml wodą, zmieszać i przefiltrować. Używając pipety, pobrać nie więcej niż 25 ml filtratu, który zawiera od 40 do 80 mg laktozy, i umieścić go w kolbie Erlenmeyera o pojemności 300 ml. W miarę potrzeby uzupełnić do objętości 25 ml wodą.
W ten sam sposób przeprowadzić test ślepej próby z 5 ml suspensji drożdżowej (3.1). Oznaczyć zawartość laktozy według Luff-Schoorla, w następujący sposób: dodać 25 ml odczynnika Luff-Schoorla (3.4) i dwie granulki pumeksu (3.5). Ogrzewać nad średnim płomieniem, ręcznie mieszając, i doprowadzić ciecz do wrzenia w czasie około 2 minut. Natychmiast postawić kolbę Erlenmeyera na siatce azbestowej z otworem o średnicy około 6 cm, pod którym pali się płomień. Płomień należy ustawić tak, aby ogrzewane było tylko dno kolby Erlenmeyera. Dopasować chłodnicę zwrotną do kolby Erlenmeyera. Gotować dokładnie 10 minut. Schłodzić natychmiast zimną wodą i po około 5 minutach miareczkować w następujący sposób:
Dodać 10 ml roztworu jodku potasu (3.6) i niezwłocznie (zachowując ostrożność ze względu na ryzyko powstania piany) dodać 25 ml kwasu siarkowego (3.7). Miareczkować roztworem tiosiarczanu sodowego (3.8) do pojawienia się mętnej żółtej barwy, dodać wskaźnik skrobiowy (3.9) i dokończyć miareczkowanie.
Przeprowadzić takie samo miareczkowanie na mieszaninie 25 ml odczynnika Luff-Schoorla (3.4) i 25 ml wody, po dodaniu 10 ml roztworu jodku potasu (3.6) i 25 ml kwasu siarkowego (3.7), bez gotowania.
6. Obliczanie wyników
Wykorzystując załączoną tabelę, określić ilość laktozy w mg, która odpowiada różnicy między wynikami dwóch miareczkowań, wyrażonych w ml roztworu tiosiarczanu sodu 0,1 mol/l.
Wynik wyrazić jako udział procentowy bezwodnej laktozy w próbce.
7. Objaśnienia
W przypadku produktów zawierających ponad 40 % fermentujących cukrów dodać więcej niż 5 ml zawiesiny drożdży (3.1).
Tabela wartości dla 25 ml odczynnika Luff-Schoorla
ml Na2 S2 O3, 0,1 mol/l, podgrzewanie przez 2 minuty, gotowanie przez 10 minut
|
Na2 S2 O3
0,1 mol/l
|
Glukoza, fruktoza, cukry inwertowane
C6 H12 O6
|
Laktoza
C12 H22 O11
|
Maltoza
C12 H22 O11
|
Na2 S2 O3
0,1 mol/l
|
|
ml
|
mg
|
różnica
|
mg
|
różnica
|
mg
|
różnica
|
ml
|
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
2,4
4,8
7,2
9,7
12,2
14,7
17,2
19,8
22,4
25,0
27,6
30,3
33,0
35,7
38,5
41,3
44,2
47,1
50,0
53,0
56,0
59,1
62,2
|
2,4
2,4
2,5
2,5
2,5
2,5
2,6
2,6
2,6
2,6
2,7
2,7
2,7
2,8
2,8
2,9
2,9
2,9
3,0
3,0
3,1
3,1
|
3,6
7,3
11,0
14,7
18,4
22,1
25,8
29,5
33,2
37,0
40,8
44,6
48,4
52,2
56,0
59,9
63,8
67,7
71,7
75,7
79,8
83,9
88,0
|
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,8
3,8
3,8
3,8
3,8
3,8
3,9
3,9
3,9
4,0
4,0
4,1
4,1
4,1
|
3,9
7,8
11,7
15,6
19,6
23,5
27,5
31,5
35,5
39,5
43,5
47,5
51,6
55,7
59,8
63,9
68,0
72,2
76,5
80,9
85,4
90,0
94,6
|
3,9
3,9
3,9
4,0
3,9
4,0
4,0
4,0
4,0
4,0
4,0
4,1
4,1
4,1
4,1
4,1
4,2
4,3
4,4
4,5
4,6
4,6
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
L. OZNACZANIE SKROBI
METODA POLARYMETRYCZNA
1. Cel i zakres stosowania metody
Metoda służy do polarymetrycznego oznaczania zawartości skrobi i wysokocząsteczkowych produktów jej rozkładu w paszach, w celu sprawdzenia zgodności z zadeklarowaną wartością energetyczną (przepisy zawarte w załączniku VII) oraz dyrektywą Rady 96/25/WE (3).
2. Sposób przeprowadzenia metody
Metoda składa się z dwóch oznaczeń. W pierwszym oznaczeniu próbka jest traktowana rozcieńczonym kwasem chlorowodorowym. Po sklarowaniu i przefiltrowaniu mierzy się polarymetrycznie skręcalność optyczną roztworu.
W drugim oznaczeniu próbka jest ekstrahowana 40 % etanolem. Po zakwaszeniu filtratu kwasem chlorowodorowym, sklarowaniu i przefiltrowaniu mierzy się skręcalność optyczną, tak jak w pierwszym oznaczeniu.
Różnica pomiędzy dwoma pomiarami, pomnożona przez znany współczynnik, daje zawartość skrobi w próbce.
3. Odczynniki i roztwory
|
3.1.
|
Kwas chlorowodorowy, roztwór 25 % (w/w), gęstość: 1,126 g/ml.
|
|
3.2.
|
Kwas chlorowodorowy, roztwór 1,13 % (w/v)
Stężenie należy sprawdzić poprzez miareczkowanie przy użyciu roztworu wodorotlenku sodu 0,1 mol/l w obecności czerwieni metylowej 0,1 % (w/V) w 94 % (v/v) etanolu. Do neutralizacji 10 ml potrzeba 30,94 ml NaOH 0,1 mol/l.
|
|
3.3.
|
Roztwór Carreza I: rozpuścić w wodzie 21,9 g octanu cynku, Zn(CH3COO)2 2H2O i 3 g lodowatego kwasu octowego. Uzupełnić do objętości 100 ml wodą.
|
|
3.4.
|
Roztwór Carreza II: rozpuścić w wodzie 10,6 g heksacyjanożelazianu(II) potasu K4 (Fe(CN)6 3H2O. Uzupełnić do objętości 100 ml wodą.
|
|
3.5.
|
Etanol, roztwór 40 % (v/v), gęstość: 0,948 g/ml w temperaturze 20 oC.
|
4. Aparatura i sprzęt
|
4.1.
|
Kolba Erlenmeyera o pojemności 250 ml ze szlifem szklanym i chłodnicą zwrotną.
|
|
4.2.
|
Polarymetr lub sacharymetr.
|
5. Sposób postępowania
5.1. Przygotowanie próbki
Rozdrobnić próbkę tak, aby przesiewała się przez oczka sita o średnicy 0,5 mm.
5.2. Oznaczanie całkowitej skręcalności optycznej (P lub S) (zob. objaśnienia 7.1)
Odważyć, z dokładnością do 1 mg, 2,5 g rozdrobnionej próbki i umieścić w kolbie miarowej o pojemności 100 ml. Dodać 25 ml kwasu chlorowodorowego (3.2), wstrząsnąć do równomiernego rozprowadzenia próbki i dodać następną porcję 25 ml kwasu chlorowodorowego (3.2). Kolbę zanurzyć we wrzącej łaźni wodnej. Aby zapobiec aglomeracji, przez pierwsze 3 minuty nieprzerwanie i energicznie wstrząsać kolbą. Ilość wody w łaźni wodnej musi być wystarczająca, aby pozostała ona w stanie wrzenia, kiedy kolba zostanie do niej włożona. Nie należy wyjmować kolby z wody w trakcie wstrząsania. Dokładnie po 15 minutach wyjąć kolbę z łaźni wodnej, dodać 30 ml zimnej wody i natychmiast schłodzić do temperatury 20 oC.
Dodać 5 ml roztworu Carreza I (3.3) i wstrząsać przez ok. 30 sekund. Następnie dodać 5 ml roztworu Carreza II (3.4) i ponownie wstrząsać przez ok. 30 sekund. Uzupełnić do pełnej objętości kolby wodą, zmieszać i przefiltrować. W przypadku gdy filtrat nie jest idealnie klarowny (co zdarza się rzadko), powtórzyć oznaczanie z użyciem większej ilości roztworów Carreza I i II, np. 10 ml.
Zmierzyć skręcalność optyczną roztworu w 200 mm rurce przy zastosowaniu polarymetru lub sacharymetru.
5.3. Oznaczanie skręcalności optycznej (P' lub S') produktów rozpuszczonych w 40 % etanolu
Odważyć, z dokładnością do 1 mg, 5 g próbki, umieścić w kolbie miarowej o pojemności 100 ml i dodać około 80 ml etanolu (3.5) (zob. objaśnienia 7.2). Pozostawić kolbę na godzinę w temperaturze pokojowej, sześciokrotnie energicznie wstrząsając w tym czasie kolbą tak, aby próbka została dobrze zmieszana z etanolem. Uzupełnić do pełnej objętości kolby etanolem (3.5), zmieszać i przefiltrować.
Pobrać pipetą 50 ml filtratu, co jest równe 2,5 g próbki, do kolby Erlenmeyera o pojemności 250 ml, dodać 2,1 ml kwasu chlorowodorowego (3.1) i energicznie wstrząsnąć. Przymocować chłodnicę zwrotną do kolby Erlenmeyera i kolbę tę zanurzyć we wrzącej łaźni wodnej. Po dokładnie 15 minutach wyjąć kolbę Erlenmeyera z łaźni, przenieść zawartość do kolby miarowej o pojemności 100 ml, spłukując kolbę Erlenmeyera niewielką ilością zimnej wody, i schłodzić do temperatury 20 oC.
Sklarować z użyciem roztworów Carreza I (3.3) i Carreza II (3.4), uzupełnić do pełnej objętości kolby wodą, zmieszać, przefiltrować i zmierzyć skręcalność optyczną, w sposób określony w drugim i trzecim akapicie pkt 5.2.
6. Obliczanie wyników
Zawartość skrobi (w %) oblicza się według następujących wzorów:
6.1. Pomiary z użyciem polarymetru

|
P
|
=
|
całkowita skręcalność optyczna w stopniach kątowych
|
|
P'
|
=
|
skręcalność optyczna w stopniach kątowych substancji rozpuszczonych w 40 % (V/V) etanolu
|
|

|
=
|
określona skręcalność optyczna czystej skrobi. Umowne wartości liczbowe przyjęte dla tego współczynnika są następujące:
|
+ 185,9o:
|
skrobia ryżowa
|
|
+ 185,7o:
|
skrobia ziemniaczana
|
|
+ 184,6o:
|
skrobia kukurydziana
|
|
+ 182,7o:
|
skrobia pszenna
|
|
+ 181,5o:
|
skrobia jęczmienna
|
|
+ 181,3o:
|
skrobia owsiana
|
|
+ 184,0o:
|
inne typy skrobi i jej mieszaniny w mieszankach paszowych
|
|
6.2. Pomiary z użyciem sacharymetru
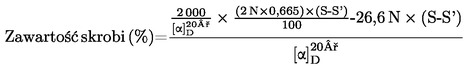
|
S
|
=
|
całkowita skręcalność optyczna w stopniach sacharymetru
|
|
S'
|
=
|
skręcalność optyczna w stopniach sacharymetru substancji rozpuszczonych w 40 % (v/v) etanolu
|
|
N
|
=
|
masa w g sacharozy w 100 ml wody ulegająca skręcalności optycznej 100 stopni sacharymetrycznych, podczas pomiaru z użyciem rurki o długości 200 mm
16,29 g dla sacharymetrów francuskich
26,00 g dla sacharymetrów niemieckich
20,00 g dla sacharymetrów połączonych
|
|
P
|
=
|
całkowita skręcalność optyczna w stopniach kątowych
|
|
P'
|
=
|
skręcalność optyczna w stopniach kątowych substancji rozpuszczonych w 40 % (V/V) etanolu
|
|
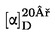
|
=
|
określona skręcalność optyczna czystej skrobi (zob. pkt 6.1)
|
6.3. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,4 w wartości bezwzględnej dla zawartości skrobi niższej niż 40 % i 1 % względem zawartości skrobi równej lub wyższej niż 40 %.
7. Objaśnienia
|
7.1.
|
Jeżeli próbka zawiera więcej niż 6 % węglanów, w przeliczeniu na węglan wapnia, należy rozłożyć je przy użyciu odpowiedniej ilości rozcieńczonego kwasu siarkowego przed określeniem całkowitej skręcalności optycznej.
|
|
7.2.
|
W przypadku produktów o wysokiej zawartości laktozy, takich jak serwatka w proszku lub mleko odtłuszczone w proszku, postępować w następujący sposób: po dodaniu 80 ml etanolu (3.5) przymocować chłodnicę zwrotną do kolby i kolbę tę zanurzyć w łaźni wodnej o temperaturze 50 oC na 30 minut. Pozostawić do schłodzenia i kontynuować analizę w sposób określony w pkt 5.3.
|
|
7.3.
|
Następujące materiały paszowe – w przypadku obecności w znacznych ilościach w paszy – powodują interferencje podczas określania zawartości skrobi metodą polarymetryczną i mogą wpływać na otrzymywanie nieprawidłowych wyników:
|
—
|
produkty z buraków cukrowych, takie jak wysłodki buraczane, melasa buraczana, wysłodki buraczane melasowane, wywar buraczany melasowany,
|
|
—
|
miazga (pulpa, wysłodki) cytrusowa,
|
|
—
|
siemię lniane, ekspelery z siemienia lnianego, siemię lniane ekstrahowane,
|
|
—
|
nasiona rzepaku, ekspelery z nasion rzepaku, ekstrahowane nasiona rzepaku, łuski nasion rzepaku,
|
|
—
|
nasiona słonecznika, ekstrahowane nasiona słonecznika, nasiona słonecznika częściowo łuskane, ekstrahowane,
|
|
—
|
ekspelery kopry, kopra ekstrahowana,
|
|
—
|
miazga (pulpa) ziemniaczana,
|
|
—
|
produkty bogate w inulinę, a w szczególności chipsy i mączka z karczochów jerozolimskich,
|
|
M. OZNACZANIE POPIOŁU SUROWEGO
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości popiołu surowego w paszach.
2. Sposób przeprowadzenia metody
Próbka jest spopielana w temperaturze 550 oC, a pozostałość jest ważona.
3. Odczynniki i roztwory
20 % roztwór azotanu amonu (w/v).
4. Aparatura i sprzęt
|
4.2.
|
Elektryczny piec muflowy z termostatem.
|
|
4.3.
|
Kwarcowe, porcelanowe lub platynowe tygle do spalań, prostokątne (ok. 60 × 40 × 25 mm) lub okrągłe (średnica: 60–75 mm, wysokość: 20–40 mm).
|
5. Sposób postępowania
Odważyć, z dokładnością do 1 mg, około 5 g próbki (2,5 w przypadku substancji pęczniejących), i umieścić w tyglu do spalań, który został podgrzany do temperatury 550 oC, schłodzony i wytarowany. Tygiel postawić na płycie grzewczej i ogrzewać stopniowo aż do zwęglenia substancji. Spopielić zgodnie z pkt 5.1 lub 5.2.
|
5.1.
|
Wstawić tygiel do skalibrowanego pieca muflowego o temperaturze ustawionej na 550 oC. Utrzymywać w tej temperaturze aż do uzyskania popiołu o barwie białej, jasnoszarej lub jasnoczerwonej, świadczącej o niewystępowaniu cząsteczek węglowych. Umieścić tygiel w eksykatorze, pozostawić do schłodzenia i niezwłocznie zważyć.
|
|
5.2.
|
Wstawić tygiel do skalibrowanego pieca muflowego o temperaturze ustawionej na 550 oC. Spopielać przez 3 godziny. Umieścić tygiel w eksykatorze, pozostawić do schłodzenia i niezwłocznie zważyć. Ponownie spopielać przez 30 minut w celu zapewnienia stałej masy popiołu (ubytek masy między dwoma kolejnymi ważeniami musi być równy lub mniejszy niż 1 mg).
|
6. Obliczanie wyników
Obliczyć masę pozostałości, odejmując tarę.
Wynik wyrazić jako udział procentowy w próbce.
7. Objaśnienia
|
7.1.
|
Substancje, które trudno się spopielają, należy wstępnie spopielać co najmniej przez 3 godziny, schłodzić, a następnie dodać do nich – ostrożnie, w taki sposób, aby nie spowodować strat popiołu i tworzenia się zbryleń – kilka kropli 20 % roztworu azotanu amonu. Po wysuszeniu kontynuować zwęglanie w piecu. Powtarzać czynność w razie potrzeby aż do całkowitego spopielenia.
|
|
7.2.
|
W przypadku gdy badane produkty trudno poddają się postępowaniu określonemu w pkt 7.1, postępuje się w następujący sposób: po spopielaniu przez 3 godziny popiół umieścić w ciepłej wodzie i przefiltrować przez mały, bezpopiołowy filtr. Spopielić filtr z zawartością w tyglu. Filtrat umieścić w schłodzonym tyglu, odparować do sucha, spopielić i zważyć.
|
|
7.3.
|
W przypadku olejów i tłuszczów należy dokładnie odważyć próbkę o masie 25 g i umieścić ją w tyglu o odpowiedniej wielkości. Zwęglić, przenosząc płomień na substancję skrawkiem bezpopiołowej bibuły filtracyjnej. Po spaleniu pozostałość zwilżyć jak najmniejszą ilością wody. Wysuszyć i spopielać w sposób określony w pkt 5.
|
N. OZNACZANIE POPIOŁU NIEROZPUSZCZALNEGO W KWASIE CHLOROWODOROWYM
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości substancji mineralnych nierozpuszczalnych w kwasie chlorowodorowym w paszach. W zależności od rodzaju próbki stosuje się dwie metody.
|
1.1.
|
Metoda A: mająca zastosowanie do materiałów paszowych i do większości mieszanek paszowych;
|
|
1.2.
|
Metoda B: mająca zastosowanie do mieszanek paszowych mineralnych, mieszanin oraz do mieszanek paszowych zawierających substancje nierozpuszczalne w kwasie chlorowodorowym w ilości wyższej niż 1 %, co sprawdza się, stosując wcześniej metodę A.
|
2. Sposób przeprowadzenia metody
|
2.1.
|
Metoda A: próbka jest spopielana, popiół gotowany w kwasie chlorowodorowym, a nierozpuszczalna pozostałość jest filtrowana i ważona.
|
|
2.2.
|
Metoda B: próbka jest traktowana kwasem chlorowodorowym. Roztwór się filtruje, pozostałość spopiela i z otrzymanym popiołem postępuje się w sposób opisany w metodzie A.
|
3. Odczynniki i roztwory
|
3.1.
|
Kwas chlorowodorowy, 3 mol/l.
|
|
3.2.
|
20 % roztwór kwasu trichlorooctowego (w/v).
|
|
3.3.
|
1 % roztwór kwasu trichlorooctowego (w/v).
|
4. Aparatura i sprzęt
|
4.2.
|
Elektryczny piec muflowy z termostatem.
|
|
4.3.
|
Kwarcowe, porcelanowe lub platynowe tygle do spalań, prostokątne (ok. 60 × 40 × 25 mm) lub okrągłe (średnica: 60–75 mm, wysokość: 20–40 mm).
|
5. Sposób postępowania
5.1. Metoda A
Spopielić próbkę metodą opisaną w odniesieniu do oznaczania popiołu surowego. Popiół uzyskany w wyniku zastosowanej analizy może być także wykorzystany do badania.
Popiół przenieść do zlewki o pojemności od 250 do 400 ml z użyciem 75 ml kwasu chlorowodorowego (3.1). Powoli doprowadzić do wrzenia i gotować ostrożnie przez 15 minut. Przefiltrować gorący roztwór przez bibułę bez popiołu i przemywać pozostałość gorącą wodą, aż reakcja z kwasem przestanie być widoczna. Filtr z osadem wysuszyć i spopielić w uprzednio wytarowanym tyglu w temperaturze nie niższej niż 550 oC i nie wyższej niż 700 oC. Ostudzić w eksykatorze i zważyć.
5.2. Metoda B
Odważyć, z dokładnością do 1 mg, 5 g próbki i umieścić w zlewce o pojemności od 250 do 400 ml. Dodać kolejno 25 ml wody i 25 ml kwasu chlorowodorowego (3.1), wymieszać i odczekać, aż roztwór przestanie się burzyć. Dodać kolejne 50 ml kwasu chlorowodorowego (3.1). Odczekać do ulotnienia się gazu, następnie umieścić zlewkę we wrzącej łaźni wodnej i trzymać ją tam przez co najmniej 30 minut, w celu całkowitej hydrolizy skrobi. Gorący roztwór przefiltrować przez bibułę bez popiołu i przemyć 50 ml gorącej wody (zob. objaśnienia pkt 7). Filtr z osadem umieścić w tyglu do spalań, wysuszyć i spopielać w temperaturze nie niższej niż 550 oC i nie wyższej niż 700 oC. Popiół umieścić w zlewce o pojemności od 250 do 400 ml z użyciem 75 ml kwasu chlorowodorowego (3.1). Postępować w sposób określony w drugim akapicie pkt 5.1.
6. Obliczanie wyników
Masę pozostałości obliczyć, odejmując tarę. Wynik wyrazić jako udział procentowy w próbce.
7. Objaśnienia
Jeżeli filtracja ma trudny przebieg, zaleca się powtórne przeprowadzenie analizy, zastępując 50 ml kwasu chlorowodorowego (3.1) 50 ml 20 % kwasu trichlorooctowego (3.2) i przemywając filtr ciepłym 1 % roztworem kwasu trichlorooctowego (3.3).
O. OZNACZANIE WĘGLANÓW
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości węglanów, umownie wyrażanych jako węglan wapnia, w większości pasz.
Jednakże w niektórych przypadkach (np. węglan żelaza) należy stosować szczególną metodę.
2. Sposób przeprowadzenia metody
Węglany rozkłada się kwasem chlorowodorowym. Powstały dwutlenek węgla zbiera się do probówki miarowej, a jego objętość jest porównywana z objętością uwolnioną ze znanej ilości węglanu wapnia w tych samych warunkach.
3. Odczynniki i roztwory
|
3.1.
|
Kwas chlorowodorowy, gęstość 1,10 g/ml.
|
|
3.3.
|
Kwas siarkowy o stężeniu ok. 0,05 mol/l, zabarwiony czerwienią metylową.
|
4. Aparatura i sprzęt
Zestaw Scheiblera-Dietriecha (zob. rysunek) lub podobny.
5. Sposób postępowania
W zależności od zawartości węglanów w próbce, zważyć próbkę o masie:
|
—
|
0,5 g w przypadku produktów zawierających od 50 do 100 % węglanów, wyrażanych jako węglan wapnia,
|
|
—
|
1 g w przypadku produktów zawierających od 40 do 50 % węglanów, wyrażanych jako węglan wapnia,
|
|
—
|
2 do 3 g w przypadku innych produktów.
|
Próbkę umieścić w specjalnej kolbie aparatu (4) wyposażonej w małą probówkę wykonaną z nietłukącego się szkła zawierającą 10 ml kwasu chlorowodorowego (pkt 3.1) i podłączyć kolbę do aparatury. Kran trójdrożny (5) ustawić tak, aby rurka (1) była podłączona z wyjściem. Używając ruchomej rurki (2) wypełnionej barwnym kwasem siarkowym (pkt 3.3) i połączonej z wykalibrowaną rurką (1), doprowadzić poziom cieczy do znaku zero, który wyznaczony jest na podziałce. Przekręcić kran (5) tak, aby połączyć rurki (1) i (3), i sprawdzić, czy poziom cieczy doprowadzony został do znaku 0.
Przechylając kolbę (4), powoli wlewać kwas chlorowodorowy (3.1) do próbki. Wyrównać ciśnienie, obniżając rurkę (2). Potrząsać kolbą (4), dopóki nie ustanie wydzielanie dwutlenku węgla.
Przywrócić ciśnienie, doprowadzając ciecz w rurkach (1) i (2) do tego samego poziomu. Po kilku minutach, kiedy ustali się objętość gazu, wykonać odczyt.
Przeprowadzić test kontrolny w tych samych warunkach z 0,5 g węglanu wapnia (3.2).
6. Obliczanie wyników
Wyrażoną jako węglan wapnia zawartość węglanów oblicza się według następującego wzoru:
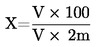
gdzie:
|
X
|
=
|
zawartość w % (w/w) węglanów w próbce wyrażona jako węglan wapnia
|
|
V
|
=
|
ilość CO2 w ml uwolniona z próbki
|
|
V1
|
=
|
ilość CO2 w ml uwolniona z 0,5 g CaCO3
|
|
m
|
=
|
masa w g próbki.
|
7. Objaśnienia
|
7.1.
|
Jeżeli masa próbki jest większa niż 2 g, do kolby (4) wlać najpierw 15 ml wody destylowanej i zmieszać przed rozpoczęciem badania. Do testu kontrolnego zastosować taką samą objętość wody.
|
|
7.2.
|
Jeżeli objętość zastosowanej aparatury różni się od objętości aparatu Scheiblera-Dietricha, należy dopasować porcje badanej próbki i substancji kontrolnej i odpowiednio obliczyć wyniki.

|
P. OZNACZANIE CAŁKOWITEJ ZAWARTOŚCI FOSFORU
METODA FOTOMETRYCZNA
1. Cel i zakres stosowania metody
Metoda służy do oznaczania całkowitej zawartości fosforu w paszach. Metodę należy stosować do analizy pasz o niskiej zawartości fosforu. W niektórych przypadkach (produkty bogate w fosfor) dopuszcza się stosowanie metody wagowej.
2. Sposób przeprowadzenia metody
Próbka jest mineralizowana poprzez suche spalanie (w przypadku pasz organicznych) lub przez rozpuszczenie kwasem (w przypadku związków mineralnych i pasz ciekłych), a następnie wprowadzona do kwaśnego roztworu. Roztwór jest traktowany odczynnikiem molibdenowanadowym. Gęstość optyczna utworzonego żółtego roztworu jest mierzona w spektrofotometrze przy 430 nm.
3. Odczynniki i roztwory
|
3.2.
|
Kwas chlorowodorowy, ρ20 = 1,10 g/ml (ok. 6 mol/l).
|
|
3.3.
|
Kwas azotowy, ρ20 = 1,045 g/ml.
|
|
3.4.
|
Kwas azotowy, ρ20 od 1,38 do 1,42 g/ml.
|
|
3.5.
|
Kwas siarkowy, ρ201,84 g/ml.
|
|
3.6.
|
Odczynnik molibdenowanadowy: zmieszać 200 ml roztworu heptamolibdenianu amonu (3.6.1), 200 ml roztworu wanadanu amonu (3.6.2) i 134 ml kwasu azotowego (3.4) w kolbie miarowej o pojemności 1 l. Uzupełnić do pełnej objętości kolby wodą.
|
|
3.6.1.
|
Roztwór heptamolibdenianu (VI) amonu: rozpuścić w gorącej wodzie 100 g heptamolibdenianu (VI) amonu (NH4) 6Mo7O24·4H2O. Dodać 10 ml amoniaku (o gęstości 0,91 g/ml) i uzupełnić do objętości 1 l wodą.
|
|
3.6.2.
|
Roztwór wanadanu (V) amonu: 2,35 g wanadanu (V) amonu NH4VO3 rozpuścić w 400 ml gorącej wody. Stale mieszając, dodawać powoli 20 ml rozcieńczonego kwasu azotowego (7 ml HNO3 (3.4) + 13 ml H2O) i uzupełnić do objętości 1 l wodą.
|
|
3.7.
|
Roztwór wzorcowy fosforu o stężeniu 1 mg/ml: rozpuścić 4,387 g dwuwodorofosforanu potasu KH2PO4 w wodzie. Uzupełnić do objętości 1 l wodą.
|
4. Aparatura i sprzęt
|
4.1.
|
Kwarcowe, porcelanowe lub platynowe tygle do spopielania.
|
|
4.2.
|
Elektryczny piec muflowy z termostatem nastawionym na temperaturę 550 oC.
|
|
4.3.
|
Kolba Kjeldahla o pojemności 250 ml.
|
|
4.4.
|
Kolby i pipety miarowe.
|
|
4.6.
|
Probówki o średnicy 16 mm i pojemności od 25 do 30 ml z korkami stopniowanymi do średnicy 14,5 mm.
|
5. Sposób postępowania
5.1. Przygotowanie roztworu
W zależności od rodzaju próbki przygotować roztwór w sposób określony w pkt 5.1.1 lub 5.1.2.
5.1.1. Zwykły sposób postępowania
Odważyć, z dokładnością do 1 mg, co najmniej 1 g próbki. Umieścić próbkę w kolbie Kjeldahla, dodać 20 ml kwasu siarkowego (3.5), wstrząsnąć do całkowitego nasycenia kwasem i nie dopuścić do przyklejania cząstek do ścianek kolby, podgrzać i utrzymywać w stanie wrzenia przez 10 minut. Nieznacznie schłodzić, dodać 2 ml kwasu azotowego (3.4), ostrożnie podgrzać, ponownie nieznacznie schłodzić i dodać nieco więcej kwasu azotowego (3.4), a następnie ponownie doprowadzić do wrzenia. Powtarzać tę czynność aż do uzyskania bezbarwnego roztworu. Schłodzić, dodać trochę wody, zdekantować ciecz do kolby miarowej o pojemności 500 ml, spłukać kolbę Kjeldahla gorącą wodą. Schłodzić, uzupełnić do pełnej objętości kolby wodą, homogenizować i przefiltrować.
5.1.2. Próbki zawierające substancje organiczne, wolne od dwuwodorofosforanu magnezu i wapnia
Odważyć, z dokładnością do 1 mg, około 2,5 g próbki i umieścić w tyglu do spopielania. Zmieszać dokładnie próbkę z 1 g węglanu wapnia (3.1). Spopielać w piecu w temperaturze 550 oC do uzyskania popiołu o białej lub szarej barwie (niewielka ilość węgla drzewnego nie ma znaczenia). Przenieść popiół do zlewki o pojemności 250 ml. Dodać 20 ml wody i kwasu chlorowodorowego (3.2) aż do zaprzestania pienienia się. Następnie dodać kolejne 10 ml kwasu chlorowodorowego (3.2). Umieścić zlewkę na łaźni piaskowej i odparować do sucha do powstania nierozpuszczalnej krzemionki. Pozostałość rozpuścić w 10 ml kwasu azotowego (3.3) i gotować na łaźni piaskowej lub płycie przez 5 minut, nie doprowadzając do zupełnego odparowania cieczy. Przenieść ciecz do kolby miarowej o pojemności 500 ml, spłukując kilkakrotnie zlewkę gorącą wodą. Pozostawić do schłodzenia, uzupełnić do pełnej objętości kolby wodą, homogenizować i przefiltrować.
5.2. Wywołanie zabarwienia i gęstości optycznej
Rozcieńczyć podzielną część filtratu otrzymanego w sposób określony w pkt 5.1.1 lub 5.1.2 do uzyskania stężenia fosforu nie większego niż 40 μg/ml. Umieścić 10 ml tego roztworu w probówce (4.6) i dodać 10 ml odczynnika molibdenowanadowego (3.6). Homogenizować i pozostawić co najmniej na 10 minut w temperaturze 20 oC. Zmierzyć gęstość optyczną w spektrofotometrze przy 430 nm w stosunku do roztworu otrzymanego przez dodanie 10 ml odczynnika molibdenowanadowego (3.6) do 10 ml wody.
5.3. Krzywa kalibracyjna
Z roztworu wzorcowego fosforu (3.7) przygotować roztwory zawierające odpowiednio 5, 10, 20, 30 i 40 μg fosforu w 1 ml. Pobrać 10 ml każdego z tych roztworów i dodać do nich po 10 ml odczynnika molibdenowanadowego (3.6). Homogenizować i pozostawić co najmniej na 10 minut w temperaturze 20 oC. Zmierzyć gęstość optyczną w sposób określony w pkt 5.2. Wykreślić krzywą kalibracyjną, odkładając wartości gęstości optycznej w stosunku do odpowiadających jej ilości fosforu. W zakresie stężeń od 0 do 40 μg/ml krzywa jest liniowa.
6. Obliczanie wyników
Określić zawartość fosforu w badanej próbce z zastosowaniem krzywej kalibracyjnej.
Wynik wyrazić jako udział procentowy w próbce.
Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
3 % względem wyższego wyniku dla zawartości fosforu niższej niż 5 %,
|
|
—
|
0,15 % wartości bezwzględnej, dla zawartości fosforu równej lub wyższej niż 5 %.
|
Q. OZNACZANIE CHLORU Z CHLORKÓW
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości chloru w chlorkach rozpuszczalnych w wodzie, umownie wyrażanego jako chlorku sodu w paszach. Metodę tę stosuje się do wszystkich pasz.
2. Sposób przeprowadzenia metody
Chlorki są rozpuszczane w wodzie. Jeżeli badany produkt zawiera substancje organiczne, roztwór jest klarowany. Po nieznacznym zakwaszeniu kwasem azotowym chlorki strąca się w postaci chlorku srebra przy użyciu roztworu azotanu srebra. Nadmiar azotanu srebra jest miareczkowany roztworem tiocyjanianu amonu według metody Volharda.
3. Odczynniki i roztwory
|
3.1.
|
Roztwór tiocyjanianu amonu 0,1 mol/l.
|
|
3.2.
|
Roztwór azotanu srebra 0,1 mol/l.
|
|
3.3.
|
Nasycony roztwór siarczanu (VI) amonu i żelaza (NH4)Fe(SO4)2.
|
|
3.4.
|
Kwas azotowy, gęstość: 1,38 g/ml.
|
|
3.7.
|
Roztwór Carreza I: rozpuścić w wodzie 21,9 g octanu cynku, Zn (CH3COO)2·2H2O i 3 g lodowatego kwasu octowego. Uzupełnić do objętości 100 ml wodą.
|
|
3.8.
|
Roztwór Carreza II: rozpuścić w wodzie 10,6 g heksacyjanożelazianu (II) potasu K4Fe(CN)6·3H2O. Uzupełnić do objętości 100 ml wodą.
|
|
3.9.
|
Węgiel aktywny, wolny od chlorków i nieabsorbujący chlorków.
|
4. Aparatura i sprzęt
Mikser (tumbler): ok. 35–40 obr./min.
5. Sposób postępowania
5.1. Przygotowanie roztworu
Zależnie od rodzaju próbki, przygotować roztwór w sposób określony w pkt 5.1.1, 5.1.2 lub 5.1.3.
W tym samym czasie przeprowadzić ślepą próbę, pomijając analizowaną próbkę.
5.1.1. Próbki niezawierające substancji organicznej
Odważyć, z dokładnością do 1 mg, nie więcej niż 10 g próbki zawierającej nie więcej niż 3 g chloru w postaci chlorków. Odważkę umieścić z 400 ml wody o temperaturze około 20 oC w kolbie miarowej o pojemności 500 ml. Mieszać przez 30 minut w mikserze, uzupełnić do pełnej objętości kolby, homogenizować i przefiltrować.
5.1.2. Próbki zawierające substancje organiczne, z wyłączeniem produktów opisanych w pkt 5.1.3
Odważyć, z dokładnością do 1 mg, około 5 g próbki i wraz z 1 g węgla aktywnego umieścić w kolbie miarowej o pojemności 500 ml. Dodać 400 ml wody o temperaturze około 20 oC i 5 ml roztworu Carreza I (3.7), mieszać przez 30 sekund i dodać 5 ml roztworu Carreza II (3.8). Mieszać przez 30 minut w mikserze, uzupełnić do pełnej objętości kolby, homogenizować i przefiltrować.
5.1.3. Pasze po obróbce cieplnej, makuchy lniane, mączka lniana, produkty bogate w mączkę lnianą i inne produkty bogate w śluz lub substancje koloidalne (np. dekstryna skrobi)
Przygotować roztwór w sposób określony w pkt 5.1.2, ale nie filtrować. Zdekantować (w razie potrzeby odwirować), pobrać 100 ml płynnego supernatantu i przenieść go do kolby miarowej o pojemności 200 ml. Zmieszać z acetonem (3.6) i uzupełnić do pełnej objętości kolby tym samym rozpuszczalnikiem, homogenizować i przefiltrować.
5.2. Miareczkowanie
W zależności od przewidywanej zawartości chlorku przenieść pipetą do kolby Erlenmeyera od 25 do 100 ml filtratu, otrzymanego w sposób określony w pkt 5.1.1, 5.1.2 lub 5.1.3. Podzielna część nie może zawierać więcej niż 150 mg chloru (Cl). Rozcieńczyć w razie potrzeby wodą do objętości nie mniejszej niż 50 ml, dodać 5 ml kwasu azotowego (3.4), 20 ml nasyconego roztworu siarczanu amonu i żelaza (3.3) i 2 krople roztworu tiocyjanianu amonu (3.1) przeniesionego z zastosowaniem biurety wypełnionej do znaku 0. Stosując biuretę, przenieść roztwór azotanu srebra (3.2), tak aby nadmiar wyniósł 5 ml. Dodać 5 ml eteru dwuetylowego (3.5) i mocno wstrząsać do skoagulowania się osadu. Nadmiar azotanu srebra miareczkować roztworem tiocyjanianu amonu (3.1), aż do uzyskania czerwonobrązowego odcienia utrzymującego się przez minutę.
6. Obliczanie wyników
Ilość chlorku (X), wyrażonego jako udział procentowy chlorku sodu, oblicza się według następującego wzoru:
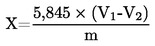
gdzie:
|
V1
|
=
|
objętość w ml dodanego roztworu azotanu srebra o stężeniu 0,1 mol/l
|
|
V2
|
=
|
objętość w ml użytego do miareczkowania roztworu tiocyjanku amonu o stężeniu 0,1 mol/l
|
|
m
|
=
|
masa próbki
|
Jeżeli ślepa próba wykaże, że roztwór azotanu srebra o stężeniu 0,1 mol/l został zużyty, odjąć jego objętość od objętości (V1 - V2).
7. Objaśnienia
|
7.1.
|
Można także zastosować miareczkowanie potencjometryczne.
|
|
7.2.
|
W przypadku produktów bogatych w oleje i tłuszcze najpierw odtłuścić je eterem dwuetylowym lub eterem naftowym.
|
|
7.3.
|
W przypadku mączki rybnej miareczkowanie można prowadzić metodą Mohra.
|
(1) Do suszenia roślin zbożowych, mąki, kaszy i mączki piec musi mieć pojemność cieplną taką, żeby po wstępnym nastawieniu na temperaturę 131 oC powrócił do tej temperatury w czasie krótszym niż 45 minut po umieszczeniu w nim maksymalnej liczby próbek do jednoczesnego wysuszenia. Wentylacja musi być taka, aby wyniki suszenia przez dwie godziny maksymalnej liczby próbek pszenicy zwyczajnej nie różniły się od wyników suszenia przez cztery godziny o więcej niż o 0,15 %.
(2) Jeśli olej lub tłuszcz musi zostać poddany późniejszym próbom badania jakości, kawałki pumeksu należy zastąpić paciorkami szklanymi.
(3) Dz.U. L 125 z 23.5.1996, s. 35.
ZAŁĄCZNIK IV
METODY ANALIZY DO CELÓW KONTROLI POZIOMU DOPUSZCZONYCH DODATKÓW W PASZACH
A. OZNACZANIE WITAMINY A
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości witaminy A (retinolu) w paszach i premiksach. Metoda pozwala na oznaczenie witaminy A obejmującej wszystkie formy alkoholowe trans-retinolu i wszystkie jej izomery cis. Zawartość witaminy A jest wyrażana w jednostkach międzynarodowych (IU) na kg. Jedna IU odpowiada aktywności 0,300 μg wszystkich form alkoholowych trans-witaminy A lub 0,344 μg wszystkich form octanowych trans-witaminy A lub 0,550 μg wszystkich form palmitynianowych trans-witaminy A.
Granica oznaczalności metody wynosi 2 000 IU witaminy A/kg.
2. Sposób przeprowadzenia metody
Próbka jest hydrolizowana w etanolowym roztworze wodorotlenku potasu, a witamina A ekstrahowana eterem naftowym. Rozpuszczalnik jest usuwany przez odparowanie, a pozostałość rozpuszczana w metanolu i, w razie konieczności, rozcieńczana do wymaganego stężenia. Zawartość witaminy A jest oznaczana metodą wysokosprawnej chromatografii cieczowej z fazą odwróconą (RP-HPLC) z wykorzystaniem detektora UV lub detektora fluorescencyjnego. Parametry chromatograficzne zostały tak dobrane, że nie zachodzi rozdział pomiędzy wszystkimi formami alkoholowymi trans-witaminy A i jej cis izomerami.
3. Odczynniki i roztwory
|
3.2.
|
Eter naftowy, zakres wrzenia od 40 do 60 oC
|
|
3.4.
|
Roztwór wodorotlenku potasu, c = 50 g/100 ml
|
|
3.5.
|
Roztwór askorbinianu sodu, c = 10 g/100 ml (zob. objaśnienia pkt 7.7)
|
|
3.6.
|
Siarczek sodu, Na2S· x H2O (x = 7 – 9)
|
|
3.6.1.
|
Roztwór siarczku sodu, c = 0,5 mol/l w glicerolu, ß = 120 g/l (dla x = 9) (zob. objaśnienia pkt 7.8)
|
|
3.7.
|
Roztwór fenoloftaleiny, c = 2 g/100 ml w etanolu (pkt 3.1)
|
|
3.9.
|
Faza ruchoma do HPLC: mieszanina metanolu (3.3) i wody, np. 980 + 20 (v + v). Dokładny współczynnik zostanie określony przez charakterystykę stosowanej kolumny.
|
|
3.10.
|
Azot wolny od tlenu
|
|
3.11.
|
Wszystkie formy octanowe trans-witaminy A, o specjalnej czystości i certyfikowanej aktywności, np. 2,80 × 106 IU/g
|
|
3.11.1.
|
Roztwór podstawowy wszystkich form octanowych trans-witaminy A: odważyć, z dokładnością do 0,1 mg, 50 mg octanu witaminy A (3.11) do kolby miarowej o pojemności 100 ml. Rozpuścić w 2-propanolu (3.8) i uzupełnić tym samym rozpuszczalnikiem do pełnej objętości kolby. Nominalne stężenie tego roztworu wynosi 1 400 IU witaminy A w 1 ml. Dokładną zawartość oznaczyć w sposób określony w pkt 5.6.3.1.
|
|
3.12.
|
Wszystkie formy palmitynianowe-trans-witaminy A, o specjalnej czystości i certyfikowanej aktywności, np. 1,80 × 106 IU/g
|
|
3.12.1.
|
Roztwór podstawowy wszystkich form palmitynianowych trans-witaminy A: odważyć, z dokładnością do 0,1 mg, 80 mg palmitynianu witaminy A (3.12) do kolby miarowej o pojemności 100 ml. Rozpuścić w 2-propanolu (3.8) i uzupełnić do pełnej objętości tej kolby tym samym rozpuszczalnikiem. Nominalne stężenie tego roztworu wynosi 1 400 IU witaminy A w 1 ml. Dokładną zawartość oznaczyć w sposób określony w pkt 5.6.3.2.
|
|
3.13.
|
2,6-dwu-tetr-butylo-4-metylofenol (BHT) (zob. objaśnienia pkt 7.5)
|
4. Aparatura i sprzęt
|
4.1.
|
Rotacyjna wyparka próżniowa
|
|
4.2.
|
Szkło laboratoryjne oranżowe
|
|
4.2.1.
|
Kolby płaskodenne lub stożkowe o pojemności 500 ml, ze szlifem
|
|
4.2.2.
|
Kolby miarowe o wąskich szyjkach o pojemnościach 10, 25, 100 i 500 ml, ze szklanymi korkami
|
|
4.2.3.
|
Rozdzielacze stożkowe o pojemności 1 000 ml, ze szklanymi korkami
|
|
4.2.4.
|
Kolby gruszkowe o pojemności 250 ml, ze szlifem
|
|
4.3.
|
Chłodnica zwrotna kulkowa o długości płaszcza 300 mm, z połączeniami szlifowanymi, z nasadką do podłączenia gazu
|
|
4.4.
|
Karbowany filtr bibułowy do rozdzielania faz o średnicy 185 mm (np. Schleicher & Schuell 597 HY 1/2)
|
|
4.5.
|
Wyposażenie HPLC z systemem dozowania
|
|
4.5.1.
|
Kolumna do chromatografii cieczowej, 250 × 4 mm, C18, wypełnienie 5 lub 10 μm lub równoważne (kryterium poprawności: pojedynczy pik dla wszystkich izomerów retinolu w warunkach HPLC)
|
|
4.5.2.
|
Detektor UV lub fluorescencyjny, z możliwością zmiany długości fali
|
|
4.6.
|
Spektrofotometr z kwarcowymi kuwetami o długości drogi optycznej 10 mm
|
|
4.7.
|
Łaźnia wodna z mieszadłem magnetycznym
|
|
4.8.
|
Aparat do ekstrakcji (zob. rys. 1) zawierający:
|
|
4.8.1.
|
Szklany cylinder o pojemności 1 l, ze szlifem i korkiem
|
|
4.8.2.
|
Nasadkę szklaną ze szlifem z bocznikiem i ruchomą nastawną rurką przesuwającą się przez środek nasadki. Nastawna rurka musi mieć zakończenie w kształcie litery U i przeciwległy otwór skierowany ku górze, tak aby górna warstwa cieczy w cylindrze mogła być przeniesiona do rozdzielacza stożkowego.
|
5. Sposób postępowania
|
Uwaga:
|
Witamina A jest wrażliwa na światło (UV) i utlenianie. Wszystkie czynności należy przeprowadzać bez dostępu światła (z zastosowaniem szkła oranżowego lub szkła zabezpieczonego folią aluminiową) i bez dostępu tlenu (płukanie strumieniem azotu). Podczas ekstrakcji powietrze znad cieczy należy zastąpić azotem (zapobiegać zbyt wysokiemu ciśnieniu, zwalniając od czasu do czasu korek).
|
5.1. Przygotowanie próbki
Rozdrobnić próbkę tak, aby przesiewała się przez oczka sita o średnicy 1 mm, uważając, aby podczas rozdrabniania nie wydzielało się ciepło. Rozdrabnianie musi być przeprowadzone tuż przed ważeniem i zmydlaniem, gdyż w przeciwnym razie mogą wystąpić straty witaminy A.
5.2. Zmydlanie
W zależności od zawartości witaminy A zważyć, z dokładnością do 1 mg, od 2 do 25 g próbki do kolby płaskodennej lub stożkowej o pojemności 500 ml (4.2.1). Dodać kolejno, mieszając, 130 ml etanolu (3.1), około 100 mg BHT (3.13), 2 ml roztworu askorbinianu sodu (3.5) i 2 ml roztworu siarczku sodu (3.6). Założyć chłodnicę (4.3) na kolbę i umieścić kolbę w łaźni wodnej z mieszadłem magnetycznym (4.7). Ogrzać do wrzenia i skraplać przez 5 minut. Następnie dodać 25 ml roztworu wodorotlenku potasu (3.4) przez chłodnicę (4.3) i skraplać przez 25 minut, mieszając w trakcie powolnego przepływu azotu. Następnie spłukać chłodnicę około 20 ml wody i schłodzić zawartość kolby do temperatury pokojowej.
5.3. Ekstrakcja
Przenieść, dekantując, zmydlony roztwór, ilościowo spłukując 250 ml wody, do rozdzielacza o pojemności 1 000 ml (4.2.3) lub do aparatu do ekstrakcji (4.8). Spłukać kolbę po zmydlaniu kolejno 25 ml etanolu (3.1) i 100 ml eteru naftowego (3.2) i przenieść popłuczyny do rozdzielacza lub do zestawu do ekstrakcji. Proporcja wody i etanolu w łączonych roztworach musi wynosić około 2:1. Wstrząsać energicznie przez 2 minuty i pozostawić w celu wytrącenia osadu na 2 minuty.
5.3.1. Ekstrakcja z użyciem rozdzielacza stożkowego (4.2.3)
Po rozdzieleniu warstw (zob. objaśnienia pkt 7.3) przenieść warstwę eteru naftowego do innego rozdzielacza (4.2.3). Powtórzyć ekstrakcję dwukrotnie, używając 100 ml eteru naftowego (3.2) i dwukrotnie używając 50 ml eteru naftowego (3.2).
Przemyć dwukrotnie połączone ekstrakty w rozdzielaczu stożkowym 100 ml porcjami wody, ostrożnie mieszając, aby zapobiec powstawaniu zawiesiny, a następnie, wielokrotnie wstrząsając, powtórzyć przemywanie kolejnymi 100 ml porcjami wody, aż woda pozostanie bezbarwna po dodaniu roztworu fenoloftaleiny (3.7) (wystarczy zwykle czterokrotne przemycie). W celu usunięcia suspensji wodnej filtrować przemyty ekstrakt przez suchy karbowany filtr do rozdzielania faz (4.4) do kolby miarowej o pojemności 500 ml (4.2.2). Spłukać rozdzielacz i filtr przy użyciu 50 ml eteru naftowego (3.2), uzupełnić eterem naftowym (3.2) do pełnej objętości kolby i dobrze zmieszać.
5.3.2. Ekstrakcja z użyciem aparatu do ekstrakcji (4.8)
Gdy warstwy zostaną rozdzielone (zob. objaśnienia pkt 7.3) zastąpić korek szklanego cylindra (4.8.1) nasadką szklaną ze szlifem (4.8.2) i ustawić niższy koniec rurki w kształcie litery U tak, aby znajdował się tuż ponad granicą rozdziału faz. Przez aplikację ciśnienia azotu dopływającego przez nasadkę przenieść górną warstwę eteru naftowego do rozdzielacza o pojemności 1 000 ml (4.2.3). Do szklanego cylindra dodać 100 ml eteru naftowego (3.2), zamknąć i dobrze wstrząsnąć. Pozostawić do rozdzielenia się faz i przenieść górną warstwę do rozdzielacza w sposób opisany powyżej. Powtórzyć postępowanie ekstrakcyjne, stosując kolejną porcję 100 ml eteru naftowego (3.2), a następnie stosując dwukrotnie porcje 50 ml eteru naftowego (3.2) i dodać warstwy eteru naftowego do rozdzielacza.
Przemyć połączone ekstrakty eteru naftowego w sposób określony w pkt 5.3.1 i dalej postępować w sposób tam określony.
5.4. Przygotowanie roztworu próbki do HPLC
Pobrać pipetą podzielną część roztworu eteru naftowego (z pkt 5.3.1 lub 5.3.2) do kolby gruszkowej o pojemności 250 ml (4.2.4). Odparować rozpuszczalnik prawie do sucha na rotacyjnej wyparce próżniowej (4.1) przy obniżonym ciśnieniu na łaźni wodnej w temperaturze nie wyższej niż 40 oC. Przywrócić ciśnienie atmosferyczne przez wpuszczenie azotu (3.10) i zdjąć kolbę z rotacyjnej wyparki próżniowej. Usunąć pozostały rozpuszczalnik strumieniem azotu (3.10) i szybko rozpuścić pozostałość w znanej objętości metanolu (10–100 ml) (3.3) (stężenie witaminy A musi wynosić od 5 IU/ml do 30 IU/ml).
5.5. Oznaczanie HPLC
Witamina A jest rozdzielana na kolumnie z fazą odwróconą C18 (4.5.1), a stężenie jest mierzone z użyciem detektora UV (325 nm) lub detektora fluorescencyjnego (wzbudzanie: 325 nm; emisja 475 nm) (4.5.2).
Zadozować podzielną część roztworu metanolowego (np. 20 μl), otrzymanego w sposób określony w pkt 5.4 i wymywać fazą ruchomą (3.9). Obliczyć średnią wysokość (powierzchnię) piku kilku kolejnych dozowań tego samego roztworu próbki i średnie wysokości (powierzchnie) pików kilku dozowań kalibracyjnych roztworów (5.6.2).
Warunki HPLC
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
|
Kolumna do chromatografii cieczowej (4.5.1):
|
250 mm × 4 mm, C18, wypełnienie 5 lub 10 μm lub równoważne
|
|
Faza ruchoma (3.9):
|
Mieszanina metanolu (3.3) i wody, np. 980 + 20 (v + v)
|
|
Prędkość przepływu:
|
1–2 ml/min
|
|
Detektor (4.5.2):
|
Detektor UV (325 nm) lub detektor fluorescencyjny
(wzbudzenie: 325 nm/emisja: 475 nm)
|
5.6. Kalibracja
5.6.1. Przygotowanie roboczych roztworów wzorcowych
Pobrać pipetą 20 ml podstawowego roztworu octanowej witaminy A (3.11.1) lub 20 ml podstawowego roztworu palmitynianowej witaminy A (3.12.1) do kolby płaskodennej lub stożkowej o pojemności 500 ml (4.2.1) i hydrolizować w sposób określony w pkt 5.2, lecz bez dodawania BHT. Następnie ekstrahować eterem naftowym (3.2) w sposób określony w pkt 5.3 i uzupełnić do objętości 500 ml eterem naftowym (3.2). Odparować prawie do sucha 100 ml tego ekstraktu na rotacyjnej wyparce próżniowej (zob. pkt 5.4), usunąć pozostały rozpuszczalnik strumieniem azotu (3.10) i ponownie rozpuścić pozostałość w 10,0 ml metanolu (3.3). Nominalne stężenie tego roztworu wynosi 560 IU witaminy A na 1 ml. Oznaczyć dokładną zawartość ekstraktu w sposób określony w pkt 5.6.3.3. Roboczy roztwór wzorcowy przygotować bezpośrednio przed użyciem.
Przenieść pipetą 2,0 ml tego roboczego roztworu wzorcowego do kolby miarowej o pojemności 20 ml, uzupełnić metanolem (3.3) do pełnej objętości kolby i zmieszać. Nominalne stężenie tego rozcieńczonego roboczego roztworu wzorcowego wynosi 56 IU witaminy A na 1 ml.
5.6.2. Przygotowanie kalibracyjnych roztworów i krzywej kalibracyjnej
Przenieść 1,0, 2,0, 5,0 i 10,0 ml rozcieńczonego roboczego roztworu wzorcowego do partii kolb miarowych o pojemności 20 ml, uzupełnić do pełnej objętości kolb metanolem (3.3) i zmieszać. Nominalne stężenia tych roztworów wynoszą 2,8, 5,6, 14,0 i 28,0 IU witaminy A na 1 ml.
Zadozować kilka razy 20 μl każdego z roztworów kalibracyjnych i określić średnie wysokości (powierzchnie) piku. Wykorzystując średnie wysokości (powierzchnie) piku, wykreślić krzywą kalibracyjną, uwzględniając wyniki kontroli UV (5.6.3.3).
5.6.3. Standaryzacja UV wzorcowych roztworów
5.6.3.1.
Roztwór podstawowy octanowej witaminy A
Pobrać pipetą 2,0 ml roztworu podstawowego octanowej witaminy A (3.11.1) do kolby miarowej o pojemności 50 ml (4.2.2) i uzupełnić do pełnej objętości kolby 2-propanolem (3.8). Nominalne stężenie tego roztworu wynosi 56 IU witaminy A na 1 ml. Pobrać pipetą 3,0 ml tego rozcieńczonego roztworu octanowej witaminy A do kolby miarowej o pojemności 25 ml i uzupełnić do pełnej objętości kolby 2-propanolem (3.8). Nominalne stężenie tego roztworu wynosi 6,72 IU witaminy A na ml. Zmierzyć spektrum UV tego roztworu względem 2-propanolu (3.8) w spektrofotometrze (4.6) pomiędzy 300 nm a 400 nm. Maksimum wygaśnięcia musi znajdować się pomiędzy 325 nm a 327 nm.
Obliczenie zawartości witaminy A:
IU witaminy A/ml = E326 × 19,0
( dla octanowej witaminy A = 1 530 przy 326 nm w 2-propanolu)
dla octanowej witaminy A = 1 530 przy 326 nm w 2-propanolu)
5.6.3.2.
Roztwór podstawowy palmitynianowej witaminy A
Pobrać pipetą 2,0 ml roztworu podstawowego palmitynianowej witaminy A (3.12.1) do kolby miarowej o pojemności 50 ml (4.2.2) i uzupełnić do pełnej objętości kolby 2-propanolem (3.8). Nominalne stężenie tego roztworu wynosi 56 IU witaminy A na 1 ml. Pobrać pipetą 3,0 ml tego rozcieńczonego roztworu palmitynianowej witaminy A do kolby miarowej o pojemności 25 ml i uzupełnić do pełnej objętości kolby 2-propanolem (3.8). Nominalne stężenie tego roztworu wynosi 6,72 IU witaminy A na 1 ml. Zmierzyć spektrum UV tego roztworu względem 2-propanolu (3.8) w spektrofotometrze (4.6) pomiędzy 300 nm a 400 nm. Maksimum wygaśnięcia musi znajdować się pomiędzy 325 nm a 327 nm.
Obliczenie zawartości witaminy A:
IU witaminy A/ml = E326 × 19,0
(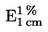 palmitynianowej witaminy A = 957 przy 326 nm w 2- propanolu)
palmitynianowej witaminy A = 957 przy 326 nm w 2- propanolu)
5.6.3.3.
Roboczy roztwór wzorcowy witaminy A
Pobrać pipetą 3,0 ml nierozcieńczonego roboczego roztworu wzorcowego witaminy A, przygotowanego w sposób określony w pkt 5.6.1, do kolby miarowej o pojemności 50 ml (4.2.2) i uzupełnić 2-propanolem (3.8) do pełnej objętości kolby. Pobrać pipetą 5,0 ml tego roztworu do kolby miarowej o pojemności 25 ml i uzupełnić do pełnej objętości kolby 2-propanolem (3.8). Nominalne stężenie tego roztworu wynosi 6,72 IU witaminy A na 1 ml. Zmierzyć spektrum UV tego roztworu względem 2-propanolu (3.8) w spektrofotometrze (4.6) pomiędzy 300 nm a 400 nm. Maksimum wygaśnięcia musi znajdować się pomiędzy 325 nm a 327 nm.
Obliczenie zawartości witaminy A:
IU witaminy A/ml = E325 × 18,3
( dla alkoholowej formy witaminy A = 1 821 przy 325 nm w 2-propanolu)
dla alkoholowej formy witaminy A = 1 821 przy 325 nm w 2-propanolu)
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików witaminy A roztworu próbki określić stężenie roztworu próbki w IU/ml, odnosząc się do krzywej kalibracyjnej (5.6.2).
Zawartość witaminy A w IU/kg próbki oblicza się według następującego wzoru:
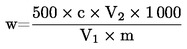 [UI/kg]
[UI/kg]
gdzie:
|
c
|
=
|
stężenie w IU/ml witaminy A w roztworze próbki (5.4)
|
|
V1
|
=
|
objętość w ml roztworu próbki (5.4)
|
|
V2
|
=
|
objętość w ml pobranej podzielnej części z roztworu próbki, o którym mowa w pkt 5.4
|
|
m
|
=
|
masa naważki w g
|
7. Objaśnienia
|
7.1.
|
W przypadku próbek o niskim stężeniu witaminy A jest wskazane połączenie ekstraktów eteru naftowego dwóch zmydlanych naważek (o wadze 25 g każda) w jeden roztwór próbki przeznaczony do oznaczania HPLC.
|
|
7.2.
|
Próbka pobrana do analizy nie może zawierać więcej niż 2 g tłuszczu.
|
|
7.3.
|
Jeżeli nie dochodzi do rozdzielenia faz, dodać około 10 ml etanolu (3.1), aby rozbić emulsję.
|
|
7.4.
|
W przypadku oleju z wątroby dorsza i innych czystych tłuszczów czas zmydlania należy przedłużyć do od 45 do 60 minut.
|
|
7.5.
|
Zamiast BHT może być użyty hydrochinon.
|
|
7.6.
|
Przy zastosowaniu zwykłej kolumny możliwe jest oddzielenie izomerów retinolu. W tym wypadku do obliczeń należy jednak zsumować wysokości (powierzchnie) pików wszystkich izomerów trans i cis.
|
|
7.7.
|
Zamiast roztworu askorbinianu sodu można zastosować około 150 mg kwasu askorbinowego.
|
|
7.8.
|
Zamiast roztworu siarczku sodu można zastosować około 50 mg EDTA.
|
|
7.9.
|
W przypadku analizy witaminy A w preparatach mlekozastępczych należy zwrócić szczególną uwagę na:
|
—
|
zmydlanie (5.2): ze względu na zawartość tłuszczu w próbce konieczne może być zwiększenie ilości roztworu wodorotlenku potasu (3.4),
|
|
—
|
ekstrakcję (5.3): ze względu na obecność emulsji konieczne może być dostosowanie proporcji wody i etanolu do 2:1.
|
Aby sprawdzić, czy zastosowana metoda analizy daje wiarygodne wyniki w odniesieniu do tej konkretnej matrycy (preparat mlekozastępczy), na dodatkowej naważce należy przeprowadzić test odzysku. Jeżeli stopień odzysku jest niższy niż 80 %, wyniki analizy należy skorygować o odzysk.
|
8. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 15 % względem wyższego wyniku.
9. Wyniki badań międzylaboratoryjnych
(1)
|
|
Premiks
|
Pasza z premiksem
|
Mieszanka paszowa mineralna
|
Pasza białkowa
|
Pasza dla prosiąt
|
|
L
|
13
|
12
|
13
|
12
|
13
|
|
n
|
48
|
45
|
47
|
46
|
49
|
|
średnia [IU/kg]
|
17,02 × 106
|
1,21 × 106
|
537 100
|
151 800
|
18 070
|
|
sr [IU/kg]
|
0,51 × 106
|
0,039 × 106
|
22 080
|
12 280
|
682
|
|
r [IU/kg]
|
1,43 × 106
|
0,109 × 106
|
61 824
|
34 384
|
1 910
|
|
CVr [%]
|
3,0
|
3,5
|
4,1
|
8,1
|
3,8
|
|
sR [IU/kg]
|
1,36 × 106
|
0,069 × 106
|
46 300
|
23 060
|
3 614
|
|
R [IU/kg]
|
3,81 × 106
|
0,193 × 106
|
129 640
|
64 568
|
10 119
|
|
CVR [%]
|
8,0
|
6,2
|
8,6
|
15
|
20
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych wartości
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
sR
|
=
|
odchylenie standardowe odtwarzalności
|
|
r
|
=
|
powtarzalność wyników
|
|
R
|
=
|
odtwarzalność wyników
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|

B. OZNACZANIE WITAMINY E
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości witaminy E w paszach i premiksach. Zawartość witaminy E jest wyrażana w mg octanu DL-α-tokoferolu na kg. 1 mg octanu DL-α-tokoferolu odpowiada 0,91 mg DL-α-tokoferolu (witamina E).
Granica oznaczalności metody wynosi 2 mg witaminy E/kg. Granica oznaczalności jest osiągalna jedynie przy pomocy detektora fluorescencyjnego. W przypadku detektora UV granica oznaczalności wynosi 10 mg/kg.
2. Sposób przeprowadzenia metody
Próbka jest hydrolizowana etanolowym roztworem wodorotlenku potasu, a witamina E jest ekstrahowana eterem naftowym. Rozpuszczalnik jest usuwany przez odparowanie, a pozostałość rozpuszczana w metanolu i, w razie konieczności, rozcieńczana do wymaganego stężenia. Zawartość witaminy E jest oznaczana metodą wysokosprawnej chromatografii cieczowej z fazą odwróconą (RP-HPLC) przy wykorzystaniu detektora UV lub detektora fluorescencyjnego.
3. Odczynniki i roztwory
|
3.2.
|
Eter naftowy, zakres wrzenia od 40 do 60 oC
|
|
3.4.
|
Roztwór wodorotlenku potasu, c = 50 g/100 ml
|
|
3.5.
|
Roztwór askorbinianu sodu, c = 10 g/100 ml (zob. objaśnienia pkt 7.7)
|
|
3.6.
|
Siarczek sodu, Na2S· x H2O (x = 7 – 9)
|
|
3.6.1.
|
Roztwór siarczku sodu, c = 0,5 mol/l w glicerolu, β = 120 g/l (dla x = 9) (zob. objaśnienia pkt 7.8)
|
|
3.7.
|
Roztwór fenoloftaleiny, c = 2 g/100 ml w etanolu (3.1)
|
|
3.8.
|
Faza ruchoma do HPLC: mieszanina metanolu (3.3) i wody, np. 980 + 20 (v + v). Dokładny współczynnik jest określony przez charakterystykę stosowanej kolumny.
|
|
3.10.
|
Octan DL-α-tokoferolu o specjalnej czystości i certyfikowanej aktywności
|
|
3.10.1.
|
Roztwór podstawowy octanu DL-α-tokoferolu: odważyć, z dokładnością do 0,1 mg, 100 mg octanu DL-α-tokoferolu (3.10) do kolby miarowej o pojemności 100 ml. Rozpuścić w etanolu (3.1) i uzupełnić do pełnej objętości kolby tym samym rozpuszczalnikiem. 1 ml tego roztworu zawiera 1 mg octanu DL-α-tokoferolu. (kontrola UV – zob. pkt 5.6.1.3; stabilizacja – zob. objaśnienia pkt 7.4).
|
|
3.11.
|
DL-α-tokoferol o specjalnej czystości i certyfikowanej aktywności
|
|
3.11.1.
|
Roztwór podstawowy DL-α-tokoferolu: odważyć, z dokładnością do 0,1 mg, 100 mg DL-α-tokoferolu (3.11) do kolby miarowej o pojemności 100 ml. Rozpuścić w etanolu (3.1) i uzupełnić do pełnej objętości kolby tym samym rozpuszczalnikiem. 1 ml tego roztworu zawiera 1 mg DL-α-tokoferolu. (kontrola UV – zob. pkt 5.6.2.3; stabilizacja – zob. objaśnienia pkt 7.4).
|
|
3.12.
|
2,6-dwu-tetr-butylo-4-metylofenol (BHT) (zob. objaśnienia pkt 7.5)
|
4. Aparatura i sprzęt
|
4.1.
|
Rotacyjna wyparka warstwowa
|
|
4.2.
|
Szkło laboratoryjne oranżowe
|
|
4.2.1.
|
Kolby płaskodenne lub stożkowe o pojemności 500 ml, ze szlifem
|
|
4.2.2.
|
Kolby miarowe o wąskich szyjkach o pojemnościach 10, 25, 100 i 500 ml, ze szklanymi korkami
|
|
4.2.3.
|
Rozdzielacze stożkowe o pojemności 1 000 ml, ze szklanymi korkami
|
|
4.2.4.
|
Kolby gruszkowe o pojemności 250 ml, ze szlifem
|
|
4.3.
|
Chłodnica zwrotna kulkowa o długości płaszcza 300 mm, z połączeniami szlifowanymi, z nasadką do podłączenia gazu
|
|
4.4.
|
Karbowany filtr bibułowy do rozdzielania faz o średnicy 185 mm (np. Schleicher & Schuell 597 HY 1/2)
|
|
4.5.
|
Wyposażenie HPLC z systemem dozowania
|
|
4.5.1.
|
Kolumna do chromatografii cieczowej, 250 mm × 4 mm, C18, wypełnienie 5 lub 10 μm lub równoważne
|
|
4.5.2.
|
Detektor UV lub fluorescencyjny, z możliwością zmiany długości fali
|
|
4.6.
|
Spektrofotometr z kwarcowymi kuwetami o długości drogi optycznej 10 mm
|
|
4.7.
|
Łaźnia wodna z mieszadłem magnetycznym
|
|
4.8.
|
Aparat do ekstrakcji (zob. rys. 1) zawierający:
|
|
4.8.1.
|
Cylinder szklany o pojemności 1 l, ze szlifem i korkiem
|
|
4.8.2.
|
Nasadkę szklaną ze szlifem z bocznikiem i ruchomą nastawną rurką przesuwającą się przez środek nasadki. Nastawna rurka musi mieć zakończenie w kształcie litery U i przeciwległy otwór skierowany ku górze, tak aby górna warstwa cieczy w cylindrze mogła być przeniesiona do rozdzielacza stożkowego.
|
5. Sposób postępowania
|
Uwaga:
|
Witamina E jest wrażliwa na światło (UV) i utlenianie. Wszystkie czynności należy przeprowadzać bez dostępu światła (z zastosowaniem szkła oranżowego lub szkła zabezpieczonego folią aluminiową) i bez dostępu tlenu (płukanie strumieniem azotu). Podczas ekstrakcji powietrze znad cieczy należy zastąpić azotem (zapobiegać zbyt wysokiemu ciśnieniu, zwalniając od czasu do czasu korek).
|
5.1. Przygotowanie próbki
Rozdrobnić próbkę tak, aby przesiewała się przez oczka sita o średnicy 1 mm, uważając, aby podczas rozdrabniania nie wydzielało się ciepło. Rozdrabnianie musi być przeprowadzone tuż przed ważeniem i zmydlaniem, gdyż w przeciwnym razie mogą wystąpić straty witaminy E.
5.2. Zmydlanie
W zależności od zawartości witaminy E odważyć, z dokładnością do 0,01 g, od 2 do 25 g próbki do kolby płaskodennej lub stożkowej o pojemności 500 ml (4.2.1). Dodać kolejno, mieszając, 130 ml etanolu (3.1), około 100 mg BHT (3.12), 2 ml roztworu askorbinianu sodu (3.5) i 2 ml roztworu siarczku sodu (3.6). Założyć chłodnicę (4.3) na kolbę i umieścić kolbę na łaźni wodnej z mieszadłem magnetycznym (4.7). Ogrzać do wrzenia i skraplać przez 5 minut. Następnie dodać 25 ml roztworu wodorotlenku potasu (3.4) przez chłodnicę (4.3) i skraplać przez 25 minut, mieszając pod powolnym przepływem azotu. Następnie spłukać chłodnicę około 20 ml wody i schłodzić zawartość kolby do temperatury pokojowej.
5.3. Ekstrakcja
Przenieść, dekantując, zmydlony roztwór, ilościowo spłukując 250 ml wody, do rozdzielacza o pojemności 1 000 ml (4.2.3) lub do aparatu do ekstrakcji (4.8). Spłukać kolbę po zmydlaniu kolejno 25 ml etanolu (3.1) i 100 ml eteru naftowego (3.2) i przenieść popłuczyny do rozdzielacza lub do aparatu do ekstrakcji. Proporcja wody i etanolu w łączonych roztworach musi wynosić około 2:1. Wstrząsać energicznie przez 2 minuty i pozostawić na 2 minuty.
5.3.1. Ekstrakcja przy użyciu rozdzielacza stożkowego (4.2.3)
Po rozdzieleniu warstw (zob. objaśnienia pkt 7.3) przenieść warstwę eteru naftowego do innego rozdzielacza (4.2.3). Powtórzyć ekstrakcję dwukrotnie, używając 100 ml eteru naftowego (3.2) i używając dwukrotnie 50 ml eteru naftowego (3.2).
Przemyć dwukrotnie połączone ekstrakty w rozdzielaczu stożkowym 100 ml porcjami wody, ostrożnie mieszając, aby zapobiec powstawaniu zawiesiny, a następnie, wielokrotnie wstrząsając, powtórzyć przemywanie kolejnymi 100 ml porcjami wody, aż woda pozostanie bezbarwna po dodaniu roztworu fenoloftaleiny (3.7) (wystarczy zwykle czterokrotne przemycie). Aby usunąć suspensję wodną, filtrować przemyty ekstrakt przez suchy karbowany filtr do rozdzielania faz (4.4) do kolby miarowej o pojemności 500 ml (4.2.2). Spłukać rozdzielacz i filtr przy użyciu 50 ml eteru naftowego (3.2), uzupełnić do pełnej objętości kolby eterem naftowym (3.2) i dobrze zmieszać.
5.3.2. Ekstrakcja przy użyciu aparatu do ekstrakcji (4.8)
Gdy warstwy zostaną rozdzielone (zob. objaśnienia pkt 7.3), zastąpić korek szklanego cylindra (4.8.1) nasadką szklaną ze szlifem (4.8.2) i ustawić niższy koniec rurki w kształcie litery U tak, aby znajdował się tuż ponad granicą rozdziału faz. Przez aplikację ciśnienia azotu dopływającego przez nasadkę przenieść górną warstwę eteru naftowego do rozdzielacza o pojemności 1 000 ml (4.2.3). Do szklanego cylindra dodać 100 ml eteru naftowego (3.2), zamknąć i dobrze wstrząsnąć. Pozostawić do rozdzielenia się faz i przenieść górną warstwę do rozdzielacza w sposób opisany powyżej. Powtórzyć postępowanie ekstrakcyjne, stosując kolejną porcję 100 ml eteru naftowego (3.2), a następnie stosując dwukrotnie porcję 50 ml eteru naftowego (3.2), i dodać warstwy eteru naftowego do rozdzielacza.
Przemyć połączone ekstrakty eteru naftowego, w sposób określony w pkt 5.3.1 i postępować w sposób tam określony.
5.4. Przygotowanie roztworu próbki do HPLC
Pobrać pipetą podzielną część roztworu eteru naftowego (z pkt 5.3.1 lub 5.3.2) do kolby gruszkowej o pojemności 250 ml (4.2.4). Odparować rozpuszczalnik prawie do sucha na rotacyjnej wyparce próżniowej (4.1) przy obniżonym ciśnieniu na łaźni wodnej w temperaturze nie wyższej niż 40 oC. Przywrócić ciśnienie atmosferyczne przez wpuszczenie azotu (3.9) i zdjąć kolbę z rotacyjnej wyparki próżniowej. Usunąć pozostały rozpuszczalnik strumieniem azotu (3.9) i szybko rozpuścić pozostałość w znanej objętości metanolu (10–100 ml) (3.3) (stężenie DL-α-tokoferolu musi wynosić od 5 μg/ml do 30 μg/ml).
5.5. Oznaczanie HPLC
Witamina E jest rozdzielana na kolumnie z fazą odwróconą C18 (4.5.1), a stężenie jest mierzone przy użyciu detektora fluorescencyjnego (wzbudzanie: 295 nm; emisja: 330 nm) lub detektora UV (292 nm) (4.5.2).
Zadozować podzielną część roztworu metanolowego (np. 20 μl), otrzymanego w sposób określony w pkt 5.4, i wymywać fazą ruchomą (3.8). Obliczyć średnie wysokości (powierzchnie) pików kilku kolejnych dozowań tego samego roztworu próbki i średnie wysokości (powierzchnie) pików kilku dozowań kalibracyjnych roztworów (5.6.2).
Warunki HPLC
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
|
Kolumna do chromatografii cieczowej (4.5.1):
|
wypełnienie 5 lub 10 μm lub 250 mm × 4 mm, C18, lub równoważne
|
|
Faza ruchoma (3.8):
|
Mieszanina metanolu (3.3) i wody np. 980 + 20 (v + v).
|
|
Prędkość przepływu:
|
1–2 ml/min
|
|
Detektor (4.5.2):
|
detektor fluorescencyjny
(wzbudzenie: 295 nm/emisja: 330 nm) lub detektor UV (292 nm)
|
5.6. Kalibracja (octan DL-α-tokoferolu lub DL-α-tokoferol)
5.6.1. Wzorzec octanu DL-α-tokoferolu
5.6.1.1.
Przygotowanie roboczego roztworu wzorcowego
Pobrać pipetą 25 ml podstawowego roztworu octanu DL-α-tokoferolu (3.10.1) do kolby płaskodennej lub stożkowej o pojemności 500 ml (4.2.1) i hydrolizować w sposób określony w pkt 5.2. Następnie ekstrahować eterem naftowym (3.2) w sposób określony w pkt 5.3 i uzupełnić do pełnej objętości kolby 500 ml eterem naftowym. Odparować prawie do sucha 25 ml tego ekstraktu na rotacyjnej wyparce próżniowej (zob. pkt 5.4), usunąć pozostały rozpuszczalnik strumieniem azotu (3.9) i ponownie rozpuścić pozostałość w 25,0 ml metanolu (3.3). Nominalne stężenie tego roztworu wynosi 45,5 μg DL-α-tokoferolu na 1 ml, co odpowiada 50 μg octanu DL-α-tokoferolu na 1 ml. Roztwór wzorcowy roboczy należy przygotować bezpośrednio przed użyciem.
5.6.1.2.
Przygotowanie kalibracyjnych roztworów i krzywej kalibracyjnej
Przenieść 1,0, 2,0, 4,0 i 10,0 ml roboczego roztworu wzorcowego do kolb miarowych o pojemności 20 ml, uzupełnić do pełnej objętości kolby metanolem (3.3) i zmieszać. Nominalne stężenia tych roztworów wynoszą 2,5, 5,0, 10,0 i 25,0 μg/ml octanu DL-α-tokoferolu, co odpowiada 2,28, 4,55, 9,10 i 22,8 μg/ml DL-α-tokoferolu.
Zadozować kilka razy 20 μl każdego roztworu kalibracyjnego i określić średnie wysokości (powierzchnie) pików. Wykorzystując średnie wysokości (powierzchnie) pików, wykreślić krzywą kalibracyjną.
5.6.1.3.
Standaryzacja UV roztworu podstawowego octanu DL-α-tokoferolu (3.10.1)
Rozcieńczyć 5,0 ml roztworu podstawowego octanu DL-α-tokoferolu (3.10.1) w 25,0 ml etanolu i zmierzyć spektrum UV tego roztworu względem etanolu (3.1) w spektrofotometrze (4.6) pomiędzy 250 nm i 320 nm.
Maksimum absorpcji musi wystąpić przy 284 nm:
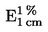 = 43,6 przy 284 nm w etanolu
= 43,6 przy 284 nm w etanolu
Przy tym rozcieńczeniu wartość wygaśnięcia musi wynosić od 0,84 do 0,88.
5.6.2. Wzorzec DL-α-tokoferolu
5.6.2.1.
Przygotowanie roboczego roztworu wzorcowego
Pobrać pipetą 2 ml podstawowego roztworu wzorcowego DL-α-tokoferolu (3.11.1) do kolby miarowej o pojemności 50 ml, rozpuścić w metanolu (3.3) i uzupełnić do pełnej objętości kolby metanolem. Nominalne stężenie tego roztworu wynosi 40 μg DL-α-tokoferolu na 1 ml, co odpowiada 44,0 μg octanu DL-α-tokoferolu na 1 ml. Wzorcowy roztwór roboczy przygotowuje się bezpośrednio przed użyciem.
5.6.2.2.
Przygotowanie kalibracyjnych roztworów i krzywej kalibracyjnej
Przenieść 1,0, 2,0, 4,0 i 10,0 ml roboczego roztworu wzorcowego do kolb miarowych o pojemności 20 ml, uzupełnić do pełnej objętości kolb metanolem (3.3) i zmieszać. Nominalne stężenia tych roztworów wynoszą 2,0, 4,0, 8,0 i 20,0 μg/ml DL-α-tokoferolu, czyli 2,20, 4,40, 8,79 i 22,0 μg/ml octanu DL-α-tokoferolu.
Zadozować kilka razy 20 μl każdego roztworu kalibracyjnego i określić średnie wysokości (powierzchnie) pików. Wykorzystując średnie wysokości (powierzchnie) pików, wykreślić krzywą kalibracyjną.
5.6.2.3.
Standaryzacja UV roztworu podstawowego DL-α-tokoferolu (3.11.1)
Rozcieńczyć 2,0 ml roztworu podstawowego DL-α-tokoferolu (3.11.1) w 25,0 ml etanolu i zmierzyć spektrum UV tego roztworu względem etanolu (3.1) w spektrofotometrze (4.6) pomiędzy 250 nm i 320 nm. Maksimum absorpcji musi wystąpić przy 292 nm:
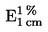 = 75,8 przy 292 nm w etanolu
= 75,8 przy 292 nm w etanolu
Przy tym rozcieńczeniu wartość wygaśnięcia musi wynosić 0,6.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików witaminy E roztworu próbki określić stężenie roztworu próbki w μg/ml (obliczonego jako octan DL-α-tokoferolu) z krzywej wzorcowej (5.6.1.2 lub 5.6.2.2).
Zawartość witaminy E w mg/kg próbki oblicza się według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
c
|
=
|
stężenie w μg/ml witaminy E (jako octan DL-α-tokoferolu) w roztworze próbki (5.4)
|
|
V1
|
=
|
objętość w ml roztworu próbki (5.4)
|
|
V2
|
=
|
objętość w ml podzielnej części pobranej z roztworu próbki, o którym mowa w pkt 5.4
|
|
m
|
=
|
masa naważki w g
|
7. Objaśnienia
|
7.1.
|
W przypadku próbek o niskim stężeniu witaminy E wskazane jest połączenie ekstraktów eteru naftowego z 2 zmydlonych naważek (o wadze 25 g każda) w jeden roztwór próbki przeznaczony do oznaczania HPLC.
|
|
7.2.
|
Próbka pobrana do analizy nie może zawierać więcej niż 2 g tłuszczu.
|
|
7.3.
|
Jeżeli nie dochodzi do rozdzielenia faz, dodać około 10 ml etanolu (3.1), aby usunąć emulsję.
|
|
7.4.
|
Po pomiarze spektrofotometrycznym roztworu octanu DL-α-tokoferolu lub DL-α-tokoferolu, wykonanym odpowiednio w sposób określony w pkt 5.6.1.3 lub 5.6.2.3, dodać 10 mg BHT (3.12) do roztworu (3.10.1 lub 3.10.2) i roztwór przechowywać w lodówce (nie dłużej niż przez 4 tygodnie).
|
|
7.5.
|
Zamiast BHT może być użyty hydrochinon.
|
|
7.6.
|
Przy zastosowaniu zwykłej kolumny możliwe jest oddzielenie α-, β-, γ- i δ-tokoferolu.
|
|
7.7.
|
Zamiast roztworu askorbinianu sodu można zastosować około 150 mg kwasu askorbinowego.
|
|
7.8.
|
Zamiast roztworu siarczku sodu można zastosować około 50 mg EDTA.
|
|
7.9.
|
Octanowa witamina E ulega bardzo szybkiej hydrolizie w warunkach zasadowych i dlatego jest ona wrażliwa na utlenianie, szczególnie w obecności takich pierwiastków śladowych jak żelazo lub miedź. W przypadku oznaczania witaminy E w premiksach na poziomach wyższych niż 5 000 mg/kg, konsekwencją może być degradacja witaminy E. Dlatego też do celów potwierdzenia zaleca się metodę HPLC obejmującą enzymatyczny rozkład witaminy E bez zmydlania zasadą.
|
8. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 15 % względem wyższego wyniku.
9. Wyniki badań międzylaboratoryjnych
(2)
|
|
Premiks
|
Pasza z premiksem
|
Mieszanka paszowa mineralna
|
Pasza białkowa
|
Pasza dla prosiąt
|
|
L
|
12
|
12
|
12
|
12
|
12
|
|
n
|
48
|
48
|
48
|
48
|
48
|
|
Średnia [mg/kg]
|
17 380
|
1 187
|
926
|
315
|
61,3
|
|
sr [mg/kg]
|
384
|
45,3
|
25,2
|
13,0
|
2,3
|
|
r [mg/kg]
|
1 075
|
126,8
|
70,6
|
36,4
|
6,4
|
|
CVr [%]
|
2,2
|
3,8
|
2,7
|
4,1
|
3,8
|
|
sR mg/kg]
|
830
|
65,0
|
55,5
|
18,9
|
7,8
|
|
R [mg/kg]
|
2 324
|
182,0
|
155,4
|
52,9
|
21,8
|
|
CVR [%]
|
4,8
|
5,5
|
6,0
|
6,0
|
12,7
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych wartości
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
sR
|
=
|
odchylenie standardowe odtwarzalności
|
|
r
|
=
|
powtarzalność wyników
|
|
R
|
=
|
odtwarzalność wyników
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|

C. OZNACZANIE PIERWIASTKÓW ŚLADOWYCH: ŻELAZA, MIEDZI, MANGANU I CYNKU
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości pierwiastków śladowych żelaza, miedzi, manganu i cynku w paszach. Dolne granice oznaczalności metody wynoszą dla:
|
—
|
manganu (Mn): 20 mg/kg,
|
2. Sposób przeprowadzenia metody
Próbkę wprowadza się do roztworu kwasu chlorowodorowego po zniszczeniu ewentualnej substancji organicznej. Pierwiastki śladowe: żelazo, miedź, mangan i cynk są oznaczane, po odpowiednim rozcieńczeniu, z użyciem spektrometrii absorpcji atomowej.
3. Odczynniki i roztwory
Uwagi wstępne
Do przygotowania odczynników i roztworów używanych w postępowaniu analitycznym stosuje się wodę wolną od oznaczanych kationów, otrzymaną w drodze podwójnej destylacji w destylarce ze szkła borokrzemowego lub kwarcowego lub w wyniku podwójnego traktowania na żywicy jonowymiennej.
Stosować odczynniki o czystości co najmniej analitycznej. Nieobecność oznaczanych pierwiastków w stosowanych odczynnikach i roztworach należy sprawdzać poprzez ślepą próbę. Odczynnik, jeżeli to konieczne, poddaje się oczyszczeniu przed zastosowaniem.
Zamiast przygotowania wzorcowych roztworów opisanych poniżej dopuszcza się stosowanie innych dostępnych wzorcowych roztworów, pod warunkiem że mają one gwarancję i zostały sprawdzone przed użyciem.
|
3.1.
|
Kwas chlorowodorowy (d: 1,19 g/ml).
|
|
3.2.
|
Kwas chlorowodorowy (6 mol/l).
|
|
3.3.
|
Kwas chlorowodorowy (0,5 mol/l).
|
|
3.4.
|
Kwas fluorowodorowy od 38 do 40 % (v/v), o zawartości żelaza (Fe) niższej niż 1 mg/l i pozostałości po odparowaniu niższej niż 10 mg (jako siarczanu)/l.
|
|
3.5.
|
Kwas siarkowy (d: 1,84 g/ml).
|
|
3.6.
|
Nadtlenek wodoru (około 100 objętości tlenu (30 % wagowych)).
|
|
3.7.
|
Roztwór wzorcowy żelaza (1 000 μg Fe/ml) przygotowany w następujący sposób lub równoważny roztwór dostępny w handlu: rozpuścić 1 g drutu żelaznego w 200 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2), dodać 16 ml nadtlenku wodoru (3.6) i uzupełnić do objętości 1 l wodą.
|
|
3.7.1.
|
Roboczy roztwór wzorcowy żelaza (100 μg Fe/ml) przygotowany przez rozcieńczenie roztworu wzorcowego żelaza (3.7) wodą w stosunku 1:9.
|
|
3.8.
|
Roztwór wzorcowy miedzi (1 000 μg Cu/ml) przygotowany w następujący sposób lub równoważny roztwór dostępny w handlu:
|
—
|
rozpuścić 1 g sproszkowanej miedzi w 25 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2), dodać 5 ml nadtlenku wodoru (3.6) i uzupełnić do objętości 1 l wodą.
|
|
|
3.8.1.
|
Roboczy roztwór wzorcowy miedzi (10 μg Cu/ml) przygotowany przez rozcieńczenie roztworu wzorcowego miedzi (3.8) wodą w stosunku 1:9, a następnie przez rozcieńczenie jednej części uzyskanego roztworu wodą stosunku 1:9.
|
|
3.9.
|
Roztwór wzorcowy manganu (1 000 μg Mn/ml) przygotowany w następujący sposób lub równoważny roztwór dostępny w handlu:
|
—
|
rozpuścić 1 g sproszkowanego manganu w 25 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2) i uzupełnić do objętości 1 l wodą.
|
|
|
3.9.1.
|
Roboczy roztwór wzorcowy manganu (10 μg Mn/ml) przygotowany przez rozcieńczenie roztworu wzorcowego manganu (3.9) wodą w stosunku 1:9, a następnie przez rozcieńczenie jednej części uzyskanego roztworu wodą w stosunku 1:9.
|
|
3.10.
|
Roztwór wzorcowy cynku (1 000 μg Zn/ml) przygotowany w następujący sposób lub równoważny roztwór dostępny w handlu:
|
—
|
rozpuścić 1 g cynku w postaci paska lub listka w 25 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2) i uzupełnić do objętości 1 l wodą.
|
|
|
3.10.1.
|
Roboczy roztwór wzorcowy cynku (10 μg Zn/ml) przygotowany przez rozcieńczenie roztworu wzorcowego cynku (3.10) wodą w stosunku 1:9, a następnie przez rozcieńczenie jednej części uzyskanego roztworu wodą w stosunku 1:9.
|
|
3.11.
|
Roztwór chlorku lantanu: rozpuścić 12 g tlenku lantanu w 150 ml wody, dodać 100 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2) i uzupełnić do objętości 1 l wodą.
|
4. Aparatura i sprzęt
|
4.1.
|
Piec muflowy z regulacją temperatury i rejestratorem.
|
|
4.2.
|
Naczynie szklane z odpornego borokrzemowego szkła; zalecane jest stosowanie sprzętu wyłącznie do oznaczania mikroelementów.
|
|
4.3.
|
Spektrofotometr absorpcji atomowej o czułości i precyzji w zakresie prezentowanej metody.
|
5. Sposób postępowania
(3)
5.1. Próbki zawierające substancję organiczną
5.1.1. Spopielanie i przygotowanie roztworu do analizy
(4)
|
5.1.1.1.
|
Odważyć, z dokładnością do 0,2 mg, od 5 do 10 g próbki i umieścić w kwarcowym lub platynowym tyglu (zob. uwaga b)), wysuszyć w suszarce o temperaturze 105 oC i wstawić tygiel do zimnego pieca muflowego (4.1). Zamknąć piec, (zob. uwaga c)) i stopniowo podwyższać temperaturę do 450 do 475 oC w czasie około 90 minut. Utrzymywać tę temperaturę od 4 do 16 godzin, np. przez noc, aby usunąć związki węglowe, następnie otworzyć piec i pozostawić do schłodzenia (zob. uwaga d)).
Zwilżyć spopielałe pozostałości wodą i przenieść do zlewki o pojemności 250 ml. Przemyć tygiel z użyciem około 5 ml kwasu chlorowodorowego (3.1) i dodawać kwas powoli i ostrożnie do zlewki (może zajść gwałtowna reakcja w wyniku powstania CO2). Dodawać kroplami kwas chlorowodorowy (3.1) mieszając do zaniku burzenia się mieszaniny. Odparować do sucha, od czasu do czasu mieszając szklaną bagietką.
Następnie dodać do pozostałości 15 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2), a następnie około 120 ml wody. Zamieszać szklaną bagietką, którą należy pozostać w zlewce, i przykryć zlewkę szkiełkiem zegarkowym. Doprowadzić ostrożnie do wrzenia i pozostawić w tym stanie aż do całkowitego rozpuszczenia rozpuszczalnych cząstek popiołu. Przefiltrować przez bezpopiołowy filtr i zebrać filtrat w kolbie miarowej o pojemności 250 ml. Spłukać zlewkę i filtr z użyciem 5 ml gorącego kwasu chlorowodorowego o stężeniu 6 mol/l (3.2) i dwukrotnie wrzącą wodą. Uzupełnić kolbę miarową do pełnej objętości wodą tak, aby uzyskać stężenie HCl około 0,5 mol/l.
|
|
5.1.1.2.
|
Jeżeli pozostałość na filtrze jest czarna (węgiel), wstawić z powrotem do pieca i ponownie spopielać w temperaturze od 450 do 475 oC. Spopielanie, które wymaga kilku godzin (od trzech do pięciu), uważa się za zakończone, gdy popiół ma barwę białą lub białawą. Rozpuścić pozostałość w około 2 ml kwasu chlorowodorowego (3.1), odparować do sucha i dodać 5 ml kwasu chlorowodorowego o stężeniu 6 mol/l (3.2). Podgrzać, przefiltrować roztwór do kolby miarowej i uzupełnić do pełnej objętości kolby wodą tak, aby uzyskać stężenie HCl około 0,5 mol/l.
Uwagi:
|
a)
|
Przy oznaczaniu pierwiastków śladowych należy postępować tak, aby nie dopuścić do zanieczyszczenia, zwłaszcza cynkiem, miedzią i żelazem. Dlatego sprzęt stosowany podczas przygotowania próbki musi być wolny od zanieczyszczeń tymi metalami.
W celu zmniejszenia ogólnego ryzyka zanieczyszczenia wykonywać oznaczenie w środowisku wolnym od kurzu, dokładnie czyszcząc wyposażenie i myjąc naczynia szklane. Zwłaszcza przy oznaczaniu cynku występuje ryzyko zanieczyszczeń, np. z naczynia szklanego, odczynników, kurzu.
|
|
b)
|
Masę próbki, która ma być spopielona, szacuje się na podstawie przybliżonej zawartości pierwiastka śladowego w paszy, w stosunku do czułości stosowanego spektrofotometru. W przypadku pasz ubogich w pierwiastki śladowe może zaistnieć konieczność rozpoczęcia oznaczania od odważenia od 10 do 20 g próbki i sporządzenia roztworu o końcowej objętości 100 ml.
|
|
c)
|
Spopielanie należy przeprowadzać w zamkniętym piecu bez dostępu powietrza lub tlenu.
|
|
d)
|
Temperatura według wskazań pirometru nie może być wyższa niż 475 oC.
|
|
5.1.2. Spektrofotometryczne oznaczanie
5.1.2.1.
Przygotowanie roztworów do kalibracji
Dla każdego z oznaczanych pierwiastków śladowych przygotować, z roboczych roztworów wzorcowych określonych w pkt 3.7.1, 3.8.1, 3.9.1 i 3.10.1, roztwory do kalibracji o stężeniu kwasu chlorowodorowego około 0,5 mol/l i – w przypadku żelaza, manganu i cynku – chlorek lantanu o stężeniu 0,1 % La (w/v).
Wybrane stężenia pierwiastków śladowych muszą mieścić się w zakresie czułości stosowanego spektrofotometru. W poniższych tabelach podano przykładowy skład typowych zakresów stężeń kalibracyjnych roztworów; w zależności od typu i czułości stosowanego spektrofotometru może zachodzić konieczność wyboru innych stężeń.
Żelazo
|
μg Fe/ml
|
0
|
0,5
|
1
|
2
|
3
|
4
|
5
|
|
ml roboczego roztworu wzorcowego (3.7.1) (1 ml= 100 μg Fe)
|
0
|
0,5
|
1
|
2
|
3
|
4
|
5
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml roztworu chlorku lantanu (3.11) i uzupełnić do objętości 100 ml wodą.
|
Miedź
|
μg Cu/ml
|
0
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
1,0
|
|
ml roboczego roztworu wzorcowego (3.8.1) (1 ml= 10 μg Cu)
|
0
|
1
|
2
|
4
|
6
|
8
|
10
|
|
ml HCl (3.2)
|
8
|
8
|
8
|
8
|
8
|
8
|
8
|
Mangan
|
μg Mn/ml
|
0
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
1,0
|
|
ml roboczego roztworu wzorcowego (3.9.1) (1 ml= 10 μg Mn)
|
0
|
1
|
2
|
4
|
6
|
8
|
10
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml roztworu chlorku lantanu (3.11) i uzupełnić do objętości 100 ml wodą.
|
Cynk
|
μg Zn/ml
|
0
|
0,05
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
|
ml roboczego roztworu wzorcowego (3.10.1) (1 ml= 10 μg Zn)
|
0
|
0,5
|
1
|
2
|
4
|
6
|
8
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml roztworu chlorku lantanu (3.11) i uzupełnić do objętości 100 ml wodą.
|
5.1.2.2.
Przygotowanie roztworu do analizy
W przypadku oznaczania miedzi może być wykorzystany roztwór przygotowany w sposób określony w pkt 5.1.1. W przypadku konieczności dostosowania jego stężenia do zakresu stężeń kalibracyjnych roztworów, pobrać pipetą podzielną część roztworu do kolby miarowej o pojemności 100 ml i uzupełnić do pełnej objętości kolby kwasem chlorowodorowym o stężeniu 0,5 mol/l (3.3).
Przy oznaczaniu żelaza, manganu i cynku pobrać pipetą podzielną część roztworu przygotowanego w sposób określony w pkt 5.1.1 do kolby miarowej o pojemności 100 ml, dodać 10 ml roztworu chlorku lantanu (3.11) i uzupełnić do pełnej objętości kolby kwasem chlorowodorowym o stężeniu 0,5 mol/l (3.3) (zob. objaśnienia pkt 8).
5.1.2.3.
Ślepa próba
Ślepą próbę przeprowadzić zgodnie z kolejnością wykonywania czynności metody, pomijając te czynności, w których materiał próbki został pominięty. Nie stosować roztworu kalibracyjnego „0” jako ślepej próby.
5.1.2.4.
Pomiar absorpcji atomowej
Zmierzyć absorpcję atomową kalibracyjnych roztworów i badanego roztworu z użyciem utleniającego płomienia powietrzno-acetylenowego przy następujących długościach fal:
Każdy pomiar przeprowadzić czterokrotnie.
5.2. Pasze mineralne
Jeżeli próbka paszy nie zawiera substancji organicznych, wcześniejsze spopielanie nie jest konieczne. Postępować w sposób określony w pkt 5.1.1.1, poczynając od akapitu drugiego. Można pominąć etap odparowania z kwasem fluorowodorowym.
6. Obliczanie wyników
Przy zastosowaniu krzywej kalibracyjnej obliczyć zawartość oznaczanego pierwiastka śladowego w roztworze do analizy i wyrazić wynik w mg pierwiastka śladowego na kg próbki (ppm).
7. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
5 mg/kg wartości bezwzględnej, dla zawartości pierwiastka śladowego nie wyższej niż 50 mg/kg,
|
|
—
|
10 % wyższego wyniku, dla zawartości pierwiastka śladowego od 50 do 100 mg/kg,
|
|
—
|
10 mg/kg wartości bezwzględnej, dla zawartości pierwiastka śladowego od 100 do 200 mg/kg,
|
|
—
|
5 % wyższego wyniku, dla zawartości pierwiastka śladowego wyższej niż 200 mg/kg.
|
8. Objaśnienia
Obecność znacznych ilości fosforanów może interferować przy oznaczaniu żelaza, manganu i cynku. Takie interferencje należy skorygować przez dodanie chlorku lantanu (3.11). Jeżeli jednak w próbce stosunek wagowy Ca + Mg/P jest > 2, to dodanie roztworu chlorku lantanu (3.11) do analizowanego roztworu i kalibracyjnych roztworów może być pominięte.
D. OZNACZANIE HALOFUGINONU
DL-trans-7-bromo-6-chloro-3-[3-(3-hydroksy-2-piperydylo)acetonylo]-chinazoliny-4-(3H)-1 bromowodorek
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości halofuginonu w paszach. Dolna granica oznaczalności wynosi 1 mg/kg.
2. Sposób przeprowadzenia metody
Po potraktowaniu gorącą wodą halofuginon ekstrahuje się w postaci wolnej zasady w octanie etylowym, a następnie rozdziela jako chlorowodorek w wodnym roztworze kwasu. Ekstrakt jest oczyszczany z wykorzystaniem chromatografii jonowymiennej. Zawartość halofuginonu określa się metodą wysokosprawnej chromatografii cieczowej w układzie faz odwróconych (HPLC) przy użyciu detektora UV.
3. Odczynniki i roztwory
|
3.1.
|
Acetonitryl do HPLC.
|
|
3.2.
|
Żywica Amberlit XAD-2.
|
|
3.5.
|
Lodowaty kwas octowy.
|
|
3.6.
|
Substancja wzorcowa halofuginonu (DL-trans-7-bromo-6-chloro-3-[3-(3-hydroksy-2-piperydylo)acetonylo]-chinazoliny-4-(3H)-1 bromowodorek, E 764).
|
|
3.6.1.
|
Roztwór wzorcowy podstawowy halofuginonu, 100 μg/ml
Odważyć, z dokładnością do 0,1 mg, 50 mg halofuginonu (3.6) do kolby miarowej o pojemności 500 ml, rozpuścić w roztworze buforu octanu amonu (3.18), uzupełnić do pełnej objętości kolby roztworem buforu i zmieszać. Roztwór zachowuje stabilność do 3 tygodni, jeżeli jest przechowywany bez dostępu światła w temperaturze 5 oC.
|
|
3.6.2.
|
Roztwory kalibracyjne
Do partii kolb miarowych o pojemności 100 ml przenieść kolejno 1,0, 2,0, 3,0, 4,0 i 6,0 ml roztworu wzorcowego podstawowego halofuginonu (3.6.1). Uzupełnić do pełnej objętości kolb fazą ruchomą (3.21) i zmieszać. Roztwory te odpowiadają odpowiednio 1,0, 2,0, 3,0, 4,0 i 6,0 μg/ml halofuginonu. Roztwory te przygotować bezpośrednio przed użyciem.
|
|
3.7.
|
Kwas chlorowodorowy (ρ 20 = około 1,16 g/ml).
|
|
3.13.
|
EDTA (etylenodwuaminoczterooctowy kwas, sól dwusodowa).
|
|
3.15.
|
Roztwór węglanu sodu, c = 10 g/100 ml.
|
|
3.16.
|
Roztwór chlorku sodu nasyconego węglanem sodu, c = 5g/100 ml.
Rozpuścić 50 g węglanu sodu (3.11) w wodzie, rozcieńczyć do objętości 1 l i dodać chlorek sodu (3.12), aż do otrzymania roztworu nasyconego.
|
|
3.17.
|
Kwas chlorowodorowy, stężenie około 0,1 mol/l.
Rozcieńczyć 10 ml HCI (3.7) wodą do objętości 1 l.
|
|
3.18.
|
Roztwór buforu octanu amonu, około 0,25 mol/l.
Rozpuścić 19,3 g octanu amonu (3.3) i 30 ml kwasu octowego (3.5) w wodzie (3.14), i rozcieńczyć do objętości 1 l.
|
|
3.19.
|
Przygotowanie żywicy Amberlit XAD-2.
Żywicę (3.2) przemywać odpowiednią ilością wody aż do zaniku wszystkich jonów chlorku, co sprawdza się roztworem azotanu srebra (3.20) w odrzucanej fazie wodnej. Następnie przemyć żywicę 50 ml metanolu (3.8), odrzucić metanol i przechowywać żywicę w świeżym metanolu.
|
|
3.20.
|
Roztwór azotanu srebra około 0,1 mol/l.
Rozpuścić 0,17 g azotanu srebra (3.9) w 10 ml wody.
|
|
3.21.
|
Faza ruchoma HPLC.
Zmieszać 500 ml acetonitrylu (3.1) z 300 ml roztworu buforu octanu amonu (3.18) i 1 200 ml wody (3.14). Przy użyciu kwasu octowego (3.5) dostosować pH do 4,3. Przefiltrować przez filtr membranowy 0,22 μm (4.8), a następnie odgazować roztwór (np. przy zastosowaniu łaźni ultradźwiękowej przez 10 minut). Roztwór zachowuje stabilność do miesiąca, jeżeli jest przechowywany w zamkniętym naczyniu, bez dostępu światła.
|
4. Aparatura i sprzęt
|
4.1.
|
Łaźnia ultradźwiękowa
|
|
4.2.
|
Rotacyjna wyparka warstwowa
|
|
4.4.
|
Wyposażenie do HPLC z detektorem UV o zmiennej długości fali miarowej lub z detektorem diodowym
|
|
4.4.1.
|
Kolumna do chromatografii cieczowej o wymiarach 300 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
4.5.
|
Kolumna szklana o wymiarach 300 mm × 10 mm z kranem i filtrem ze spiekanego szkła
|
|
4.6.
|
Filtry z włóknem szklanym o średnicy 150 mm
|
|
4.7.
|
Filtry membranowe, 0,45 μm
|
|
4.8.
|
Filtry membranowe, 0,22 μm
|
5. Sposób postępowania
|
Uwaga:
|
Halofuginon w formie wolnej zasady jest niestabilny w roztworach zasadowych i octanu etylowego. Nie może on pozostawać w octanie etylu dłużej niż przez 30 minut.
|
5.1. Wskazówki ogólne
|
5.1.1.
|
Ślepą próbę paszy należy przeanalizować w celu sprawdzenia, czy nie zawiera ona halofuginonu i innych substancji interferujących.
|
|
5.1.2.
|
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy fortyfikowanej przez dodanie znanej ilości halofuginonu, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Aby fortyfikować na poziomie 3 mg/kg, dodać 300 μl podstawowego roztworu wzorcowego (3.6.1) do 10 g ślepej próby paszy, zmieszać i pozostawić na 10 minut przed przejściem do etapu ekstrakcji (5.2).
|
Uwaga:
|
dla celów niniejszej metody ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności halofuginonu.
|
|
5.2. Ekstrakcja
Odważyć, z dokładnością do 0,1 g, 10 g przygotowanej próbki i umieścić w probówce wirówkowej o pojemności 200 ml. Dodać 0,5 g askorbinianu sodu (3.10), 0,5 g EDTA (3.13), 20 ml wody i zmieszać. Probówkę umieścić na 5 minut w łaźni wodnej o temperaturze 80 oC. Po schłodzeniu do temperatury pokojowej dodać 20 ml roztworu węglanu sodu (3.15) i zmieszać. Niezwłocznie dodać 100 ml octanu etylowego (3.4) i ręcznie, energicznie wstrząsać przez 15 sekund. Następnie umieścić probówkę w łaźni ultradźwiękowej (4.1) na 3 minuty i odkorkować. Wirować przez 2 minuty i zdekantować fazę octanu etylowego przez filtr z włóknem szklanym (4.6) do rozdzielacza o pojemności 500 ml. Powtórzyć ekstrakcję tej próbki drugą porcją 100 ml octanu etylowego. Przemywać połączone ekstrakty przez minutę 50 ml roztworu chlorku sodu nasyconego węglanem sodu (3.16) i odrzucić warstwę wodną.
Warstwę organiczną ekstrahować przez minutę 50 ml kwasu chlorowodorowego (3.17). Dolną kwasową warstwę spuścić do rozdzielacza o pojemności 250 ml. Przez 1,5 minuty powtarzać ekstrakcję warstwy organicznej przy użyciu kolejnej ilości 50 ml kwasu chlorowodorowego i połączyć z ekstraktem uzyskanym z pierwszego procesu. Przemyć połączone ekstrakty kwasu, wirując z 10 ml octanu etylowego (3.4) przez około 10 sekund.
Warstwę wodną przenieść ilościowo do kolby okrągłodennej o pojemności 250 ml i odrzucić warstwę organiczną. Znajdujący się jeszcze w roztworze kwasu octan etylowy odparować na rotacyjnej wyparce warstwowej (4.2). Temperatura wody w łaźni nie może przekraczać 40 oC. Pozostały octan etylu zostanie usunięty w próżni o ciśnieniu około 25 mbar i temperaturze 38 oC, w czasie 5 minut.
5.3. Oczyszczanie
5.3.1. Przygotowanie kolumny z Amberlitem
Dla każdej próbki ekstraktu przygotować kolumnę XAD-2. Przygotowany Amberlit o masie 10 g (3.19) umieścić w szklanej kolumnie (4.5) z metanolem (3.8). Wprowadzić mały zwitek szklanej waty w górną część podłoża żywicy. Wydrenować metanol z kolumny, a następnie przemyć żywicę 100 ml wody, zatrzymując proces, gdy ciecz dosięgnie górnej części powierzchni żywicy. Pozostawić kolumnę do ustabilizowania na 10 minut przed użyciem. Nie dopuścić do osuszenia kolumny.
5.3.2. Oczyszczanie próbki
Uzyskany ekstrakt (5.2) przenieść ilościowo na górną powierzchnię kolumny z Amberlitem (5.3.1) i eluować, odrzucając eluent. Prędkość elucji nie może przekraczać 20 ml/min. Przemyć kolbę okrągłodenną 20 ml kwasu chlorowodorowego (3.17). Użyć tego kwasu do przemycia kolumny z żywicą. Usunąć pozostałości roztworu kwasu strumieniem powietrza. Wylać popłuczyny. Dodać 100 ml metanolu (3.8) na kolumnę i pozostawić od 5 do 10 ml w celu wymycia, zebrać eluent w kolbę okrągłodenną o pojemności 250 ml. Pozostawić pozostały metanol na 10 minut w celu ustabilizowania z żywicą, a następnie kontynuować elucję, której prędkość nie może przekraczać 20 ml/min, zbierając eluent do tej samej kolby okrągłodennej. Odparować metanol na rotacyjnej wyparce (4.2), przy czym temperatura wody w łaźni nie może być wyższa niż 40 oC. Za pomocą fazy ruchomej (3.21) przenieść ilościowo pozostałość do kolby miarowej o pojemności 10 ml. Uzupełnić do pełnej objętości kolb fazą ruchomą i zmieszać. Podzielna część jest filtrowana przez filtr membranowy (4.7). Roztwór zastosować do oznaczania HPLC (5.4).
5.4. Oznaczanie HPLC
5.4.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
Kolumna do chromatografii cieczowej (4.4.1)
Faza ruchoma do HPLC (3.21)
Prędkość przepływu: od 1,5 do 2 ml/min
Długość fali przy detekcji: 243 nm
Dozowana objętość: od 40 do 100 μl
Sprawdzić stabilność systemu chromatograficznego, dozując kilka razy roztwór kalibracyjny (3.6.2) o stężeniu 3 μg/ml, aż do uzyskania stałych wysokości (powierzchni) piku i czasów retencji.
5.4.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z roztworów kalibracyjnych (3.6.2) i określić wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików kalibracyjnych roztworów na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.4.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.3.2) stosując taką samą objętość jak przy kalibracyjnych roztworach, i określić średnią wysokość (powierzchnię) piku dla pików halofuginonu.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików halofuginonu roztworu próbki, określić stężenie roztworu próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.4.2).
Zawartość halofuginonu w mg/kg w próbce oblicza się według następującego wzoru:

gdzie:
|
c
|
=
|
stężenie w μg/ml halofuginonu w roztworze próbki
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzenie wyników
7.1. Sprawdzanie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki i roztworu kalibracyjnego (3.6.2) zawierającego 6,0 μg/ml.
7.1.1. Kochromatografia
Ekstrakt próbki jest fortyfikowany przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.6.2). Ilość dodanego halofuginonu musi być podobna do tej w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i stopnia rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku halofuginonu. Szerokość piku w połowie jego wysokości musi mieścić się w granicach ±10 % pierwotnej szerokości.
7.1.2. Detekcja diodowa
Wyniki sprawdza się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach ±2 nm;
|
|
b)
|
w zakresie pomiędzy 225 a 300 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między dwoma widmami nie jest większa niż 15 % absorbancji wzorcowego analitu;
|
|
c)
|
w zakresie od 225 do 300 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między widmami nie jest wyższa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami 2 równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 0,5 mg/kg dla zawartości halofuginonu do 3 mg/kg.
7.3. Odzysk
W przypadku fortyfikowanej ślepej próby odzysk nie może być mniejszy niż 80 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy ośmiu laboratoriów przeprowadzono analizę (5) trzech próbek.
Wyniki
|
|
Próbka A
(ślepa)
Przy odbiorze
|
Próbka B (mączka)
|
Próbka C (granulki)
|
|
|
|
Przy odbiorze
|
Po 2 miesiącach
|
Przy odbiorze
|
Po 2 miesiącach
|
|
Średnia [mg/kg]
|
N.W.
|
2,80
|
2,42
|
2,89
|
2,45
|
|
SR [mg/kg]
|
—
|
0,45
|
0,43
|
0,40
|
0,42
|
|
CVR [%]
|
—
|
16
|
18
|
14
|
17
|
|
Odzysk [%]
|
|
86
|
74
|
88
|
75
|
|
N.W.
|
=
|
nie wykryto
|
|
SR
|
=
|
odchylenie standardowe powtarzalności
|
|
CVR
|
=
|
współczynnik zmienności powtarzalności (%)
|
|
Odzysk (%)
|
=
|
odzysk (%)
|
E. OZNACZANIE ROBENIDYNY
Chlorowodorek 1,3-bis[(4-chlorobenzylideno)amino]guanidyny
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości robenidyny w paszach. Granica oznaczalności wynosi 5 mg/kg.
2. Sposób przeprowadzenia metody
Próbkę poddaje się ekstrakcji zakwaszonym metanolem. Ekstrakt osusza się, a podzielną część oczyszcza na kolumnie z tlenkiem glinu. Robenidynę z kolumny wymywa się metanolem, zatęża i uzupełnia do odpowiedniej objętości fazą ruchomą. Robenidynę oznacza się metodą chromatografii cieczowej (HPLC) z odwróconymi fazami przy użyciu detektora UV.
3. Odczynniki i roztwory
|
3.2.
|
Zakwaszony metanol
W kolbie miarowej o pojemności 500 ml umieścić 4 ml kwasu chlorowodorowego (ρ 20 = 1,18 g/ml), uzupełnić do pełnej objętości kolby metanolem (3.1) i zmieszać. Roztwór należy przygotować bezpośrednio przed użyciem.
|
|
3.4.
|
Sito molekularne
Typ 3A, granulki 8–12 mesh (granulki z glinokrzemianu krystalicznego 1,6–2,5 mm, średnica porów 0,3 mm).
|
|
3.5.
|
Tlenek glinu 1 stopnia aktywności kwasowej do chromatografii
100 g tlenku glinu umieścić w naczyniu i dodać 2 ml wody. Naczynie zakorkować i wstrząsać przez około 20 minut. Przechowywać w dobrze zamkniętym naczyniu.
|
|
3.6.
|
Roztwór dwuwodorofosforanu potasu (KH2PO4), c = 0,025 mol/l
Rozpuścić 3,40 g dwuwodorofosforanu potasu w wodzie do HPLC w kolbie miarowej o pojemności 1 000 ml, uzupełnić do pełnej objętości kolby i zmieszać.
|
|
3.7.
|
Roztwór wodorofosforanu dwusodu, c = 0,025 mol/l
Rozpuścić 3,55 g bezwodnego lub 4,45 g dwuhydratu, lub 8,95 g dekahydratu wodorofosforanu dwusodu w wodzie do HPLC w kolbie miarowej o pojemności 1 l, uzupełnić do pełnej objętości kolby wodą do HPLC i zmieszać.
|
|
3.8.
|
Faza ruchoma do HPLC
Zmieszać razem:
|
|
650 ml acetonitrylu (3.3),
|
|
|
50 ml roztworu dwuwodorofosforanu potasu (3.6),
|
|
|
50 ml roztworu wodorofosforanu dwusodu (3.7).
|
Filtrować przez filtr 0,22 μm (4.6) i odgazować roztwór, np. poprzez zastosowanie ultradźwięków przez 10 minut.
|
|
3.9.
|
Substancja wzorcowa
Czysta robenidyna: chlorowodorek 1,3-bis[(4-chlorobenzylideno)amino]guanidyny.
|
|
3.9.1.
|
Roztwór wzorcowy podstawowy robenidyny: 300 μg/ml
Odważyć, z dokładnością do 0,1 mg, 30 mg substancji wzorcowej robenidyny (3.9). Rozpuścić w zakwaszonym metanolu (3.2) w kolbie miarowej o pojemności 100 ml, uzupełnić do pełnej objętości kolby tym rozpuszczalnikiem i zmieszać. Kolbę owinąć folią aluminiową i przechowywać w ciemnym miejscu.
|
|
3.9.2.
|
Roztwór wzorcowy pośredni robenidyny: 12 μg/ml
Przenieść 10,0 ml roztworu wzorcowego podstawowego (3.9.1) do kolby miarowej o pojemności 250 ml, uzupełnić do pełnej objętości kolby fazą ruchomą (3.8) i zmieszać. Kolbę owinąć folią aluminiową i przechowywać w ciemnym miejscu.
|
|
3.9.3.
|
Roztwory kalibracyjne
Do kolb miarowych o pojemności 50 ml przenieść 5,0, 10,0, 15,0, 20,0 i 25,0 ml roztworu wzorcowego pośredniego robenidyny (3.9.2). Uzupełnić do pełnej objętości kolb fazą ruchomą (3.8) i zmieszać. Roztwory te odpowiadają odpowiednio 1,2, 2,4, 3,6, 4,8 i 6,0 μg/ml robenidyny. Roztwory te przygotować bezpośrednio przed użyciem.
|
4. Aparatura i sprzęt
|
4.1.
|
Kolumna szklana
Przygotować kolumnę ze szkła oranżowego o średnicy wewnętrznej od 10 do 15 mm i długości 250 mm, wyposażoną w kran i zbiorniczek o pojemności około 150 ml.
|
|
4.2.
|
Wstrząsarka mechaniczna lub mieszadło magnetyczne
|
|
4.3.
|
Rotacyjna wyparka warstwowa
|
|
4.4.
|
Wyposażenie do HPLC z detektorem UV lub diodowym pracującym w zakresie od 250 do 400 mm
|
|
4.4.1.
|
Kolumna do chromatografii cieczowej: 300 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
4.5.
|
Filtr szklany (Whatman GF/A lub podobny)
|
|
4.6.
|
Filtry membranowe, 0,22 μm
|
|
4.7.
|
Filtry membranowe, 0,45 μm
|
5. Sposób postępowania
|
Uwaga:
|
Robenidyna jest wrażliwa na światło. Wszystkie czynności należy przeprowadzać z zastosowaniem szkła oranżowego.
|
5.1. Wskazówki ogólne
|
5.1.1.
|
Ślepą próbę paszy należy przebadać w celu sprawdzenia, czy nie zawiera robenidyny ani innych substancji interferujących.
|
|
5.1.2.
|
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy (5.1.1) fortyfikowanej przez dodanie znanej ilości robenidyny, o wadze zbliżonej do tej, która znajduje się w próbce. Aby fortyfikować na poziomie 60 mg/kg, przenieść 3 ml roztworu wzorcowego podstawowego robenidyny (3.9.1) do kolby stożkowej o pojemności 250 ml. Odparować roztwór w strumieniu azotu do około 0,5 ml. Dodać 15 g ślepej próby paszy, zmieszać i pozostawić na 10 minut przed przejściem do etapu ekstrakcji (5.2).
|
Uwaga:
|
Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności robenidyny.
|
|
5.2. Ekstrakcja
Odważyć, z dokładnością do 0,01 g, 15 g wcześniej przygotowanej próbki. Odważkę umieścić w kolbie stożkowej o pojemności 250 ml i dodać 100,0 ml zakwaszonego metanolu (3.2), zakorkować i wstrząsać przez godzinę na wytrząsarce (4.2). Roztwór filtrować przez filtr szklany (4.5), a filtrat zebrać do kolby stożkowej o pojemności 150 ml. Dodać 7,5 g sita molekularnego (3.4), zamknąć i wstrząsać przez 5 minut. Natychmiast filtrować przez filtr szklany. Roztwór poddać oczyszczaniu, w sposób określony w pkt 5.3.
5.3. Oczyszczanie
5.3.1. Przygotowanie kolumny z tlenkiem glinu
Umieścić w dolnym końcu szklanej kolumny chromatograficznej (4.1) zwitek szklanej waty i wbić ją, używając szklanego pręcika. Odważyć 11,0 g tlenku glinu (3.5) i przenieść do kolumny. Czynność tę należy wykonywać tak, aby ograniczyć do minimum działanie powietrza atmosferycznego. Delikatnie postukać w napełnioną kolumnę od strony dna w celu osadzenia tlenku glinu.
5.3.2. Oczyszczanie próbki
Używając pipety, przenieść do kolumny 5,0 ml ekstraktu próbki otrzymanego w sposób określony w pkt 5.2. Zakończenie pipety przyłożyć blisko do ścianki kolumny i pozwolić, aby roztwór został wchłonięty przez tlenek glinu. Wymyć robenidynę z kolumny przy użyciu 100 ml metanolu (3.1) przy prędkości przepływu od 2 do 3 ml na minutę, a eluent zebrać do kolby okrągłodennej o pojemności 250 ml. Na rotacyjnej wyparce warstwowej (4.3) odparować roztwór metanolu do sucha, przy pomniejszonym ciśnieniu w temperaturze 40 oC. Pozostałość ponownie rozpuścić w od 3 do 4 ml fazy ruchomej (3.8) i przenieść ilościowo do kolby miarowej o pojemności 10 ml. Kolbę przemyć kilkakrotnie od 1 do 2 ml porcjami fazy ruchomej, a popłuczyny wlać do kolby miarowej. Uzupełnić do pełnej objętości kolby tym samym rozpuszczalnikiem i zmieszać. Podzielna część jest filtrowana przez filtr membranowy 0,45 μm (4.7). Roztwór zastosować do oznaczania HPLC (5.4).
5.4. Oznaczanie HPLC
5.4.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników:
Kolumna do chromatografii cieczowej (4.4.1),
Faza ruchoma do HPLC (3.8),
Prędkość przepływu: 1,5 do 2 ml/min.,
Długość fali przy detekcji: 317 nm,
Dozowana objętość: 20 do 50 μl.
Sprawdzić stabilność systemu chromatograficznego, dozując kilka razy roztwór kalibracyjny (3.9.3) zawierający 3,6 μg/ml, aż do uzyskania stałych wysokości (powierzchni) piku i czasów retencji.
5.4.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z roztworów kalibracyjnych (3.9.3) i określić wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików kalibracyjnych roztworów na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.4.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.3.2), stosując taką samą objętość jak przy roztworach kalibracyjnych, i określić średnią wysokość (powierzchnię) piku dla pików robenidyny.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików robenidyny roztworu próbki określić stężenie roztworu próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.4.2).
Zawartość robenidyny (w mg/kg) w próbce oblicza się według następującego wzoru:

gdzie:
|
c
|
=
|
stężenie w μg/ml robenidyny w roztworze próbki
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzenie wyników
7.1. Sprawdzanie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki i roztworu kalibracyjnego (3.9.3) zawierającego 6 μg/ml.
7.1.1. Kochromatografia
Ekstrakt próbki jest fortyfikowany przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.9.3). Ilość dodanej robenidyny musi być porównywalna do ilości robenidyny w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku robenidyny. Szerokość piku w połowie jego maksymalnej wysokości musi mieścić się w granicach ± 10 % pierwotnej szerokości.
7.1.2. Detekcja diodowa
Wyniki sprawdza się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach 2 nm;
|
|
b)
|
w zakresie pomiędzy 250 a 400 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między 2 widmami nie jest większa niż 15 % absorbancji wzorcowego analitu;
|
|
c)
|
w zakresie od 250 do 400 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między 2 widmami nie jest wyższa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 10 % najwyższego wyniku dla zawartości robenidyny powyżej 15 mg/kg.
7.3. Odzysk
W przypadku fortyfikowanej ślepej próby paszy odzysk nie może być mniejszy niż 85 %.
8. Wyniki badań międzylaboratoryjnych
W ramach prowadzonej z inicjatywy Wspólnoty współpracy 12 laboratoriów przeprowadzono analizę 4 próbek paszy dla drobiu i królików, w postaci mączki lub granulatu. Analizę każdej próbki przeprowadzono dwukrotnie. W poniższej tabeli podano wyniki przeprowadzonych badań:
|
|
Pasza dla drobiu
|
Pasza dla królików
|
|
|
Mączka
|
Granulat
|
Mączka
|
Granulat
|
|
Średnia [mg/kg]
|
27,00
|
27,99
|
43,6
|
40,1
|
|
sr [mg/kg]
|
1,46
|
1,26
|
1,44
|
1,66
|
|
CVr [%]
|
5,4
|
4,5
|
3,3
|
4,1
|
|
SR [mg/kg]
|
4,36
|
3,36
|
4,61
|
3,91
|
|
CVR [%]
|
16,1
|
12,0
|
10,6
|
9,7
|
|
Odzysk [%]
|
90,0
|
93,3
|
87,2
|
80,2
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności, %
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności, %
|
F. OZNACZANIE DIKLAZURILU
(+)-4-chlorofenylo[2,6-dwuchloro-4-(2,3,4,5-tetrahydro-3,5-diokso-1,2,4-triazyno-2-yl)fenylo]acetonitryl
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości diklazurilu w paszach i premiksach. Granica wykrywalności wynosi 0,1 mg/kg, granica oznaczalności 0,5 mg/kg.
2. Sposób przeprowadzenia metody
Po dodaniu wzorca wewnętrznego próbka jest ekstrahowana zakwaszonym metanolem. W przypadku pasz podzielna część ekstraktu zostaje oczyszczona na wypełnieniu ekstrakcyjnym fazy stałej C18. Diklazuril jest eluowany z wypełnienia mieszaniną zakwaszonego metanolu i wody. Po odparowaniu pozostałość jest rozpuszczana w DMF/woda. W przypadku premiksów ekstrakt jest odparowywany, a pozostałość rozpuszczana w DMF/woda. Zawartość diklazurilu jest oznaczana w układzie podwójnego gradientu faz odwróconych wysokosprawnej chromatografii cieczowej (HPLC) z zastosowaniem detektora UV.
3. Odczynniki i roztwory
|
3.3.
|
Kwaśny siarczan tetrabutyloamoniowy (TBHS)
|
|
3.6.
|
N, N-dwumetyloformamid (DMF)
|
|
3.7.
|
Kwas chlorowodorowy, ρ20 = 1,19 g/ml
|
|
3.8.
|
Substancja wzorcowa: diklazuril II-24: (+)-4-chlorofenylo[2,6-dwuchloro-4-(2,3,4,5-tetrahydro-3,5-diokso-1,2,4-triazyno-2-yl) fenylo]acetonitryl o gwarantowanej czystości, E 771.
|
|
3.8.1.
|
Roztwór wzorcowy podstawowy diklazurilu, 500 μg/ml
Odważyć, z dokładnością do 0,1 mg, 25 mg substancji wzorcowej (3.8), do kolby miarowej o pojemności 50 ml. Rozpuścić w DMF (3.6), uzupełnić do pełnej objętości kolby tym samym DMF (3.6) i zmieszać. Kolbę owinąć folią aluminiową lub użyć kolby ze szkła oranżowego i przechowywać w lodówce. W temperaturze ≤ 4 oC roztwór zachowuje stabilność do miesiąca.
|
|
3.8.2.
|
Roztwór wzorcowy diklazurilu, 50 μg/ml
Przenieść 5,00 ml roztworu wzorcowego podstawowego (3.8.1) do kolby miarowej o pojemności 50 ml, uzupełnić do pełnej objętości kolby DMF (3.6) i zmieszać. Kolbę owinąć folią aluminiową lub użyć kolby ze szkła oranżowego i przechowywać w lodówce. W temperaturze ≤ 4 oC roztwór zachowuje stabilność do miesiąca.
|
|
3.9.
|
Substancja wzorcowa wewnętrzna: 2,6-dwuchloro-α-(4-chlorofenylo)-4-(4,5dihydro-3,5-diokso-1,2,4-triazyno-2(3H) -yl) α-metylobenzeno-acetonitryl
|
|
3.9.1.
|
Roztwór wewnętrznego wzorca podstawowego, 500 μg/ml
Odważyć, z dokładnością do 0,1 mg, 25 mg substancji wzorcowej wewnętrznej (3.9), do kolby miarowej o pojemności 50 ml. Rozpuścić w DMF (3.6), uzupełnić do pełnej objętości kolby tym samym DMF (3.6) i zmieszać. Kolbę owinąć folią aluminiową lub użyć kolby ze szkła oranżowego i przechowywać w lodówce. W temperaturze ≤ 4 oC roztwór zachowuje stabilność przez miesiąc.
|
|
3.9.2.
|
Roztwór wzorca wewnętrznego, 50 μg/ml
Przenieść 5,00 ml roztworu wewnętrznego wzorca podstawowego (3.9.1) do kolby miarowej o pojemności 50 ml, uzupełnić do pełnej objętości kolby DMF (3.6) i zmieszać. Kolbę owinąć folią aluminiową lub użyć kolby ze szkła oranżowego i przechowywać w lodówce. W temperaturze ≤ 4 oC roztwór zachowuje stabilność przez miesiąc.
|
|
3.9.3.
|
Roztwór wzorca wewnętrznego dla premiksów, p/1 000 mg/ml
(p = nominalna zawartość diklazurilu w premiksie w mg/kg)
Odważyć, z dokładnością do 0,1 mg, p/10 mg substancji wzorca wewnętrznego do kolby miarowej o pojemności 100 ml, rozpuścić w DMF (3.6) w łaźni ultradźwiękowej (4.6), uzupełnić do pełnej objętości kolby tym samym DMF i zmieszać. Kolbę owinąć folią aluminiową lub użyć kolby ze szkła oranżowego i przechowywać w lodówce. W temperaturze ≤ 4 oC roztwór zachowuje stabilność przez miesiąc.
|
|
3.10.
|
Roztwór kalibracyjny, 2 μg/ml.
Odmierzyć pipetą 2,00 ml roztworu wzorcowego diklazurilu (3.8.2) i 2,00 ml roztworu wzorca wewnętrznego (3.9.2) do kolby miarowej o pojemności 50 ml. Dodać 16 ml DMF (3.6), uzupełnić do pełnej objętości kolby wodą i zmieszać. Roztwór sporządzać bezpośrednio przed użyciem.
|
|
3.11.
|
C18, wypełnienie ekstrakcyjne fazy stałej; proponuje się zastosować Bond Elut, rozmiar: 1 cc, masa sorbentu: 100 mg.
|
|
3.12.
|
Rozpuszczalnik ekstrakcyjny: zakwaszony metanol.
Odmierzyć pipetą 5,0 ml kwasu chlorowodorowego (3.7) do 1 000 ml metanolu (3.5) i zmieszać.
|
|
3.13.
|
Faza ruchoma do HPLC.
|
|
3.13.1.
|
Eluent A: octan amonu – roztwór kwaśnego siarczanu tetrabutyloamonu.
Rozpuścić 5 g octanu amonu (3.2) i 3,4 g TBHS (3.3) w 1 000 ml wody (3.1) i zmieszać.
|
|
3.13.2.
|
Eluent B: acetonitryl (3.4).
|
|
3.13.3.
|
Eluent C: metanol (3.5).
|
4. Aparatura i sprzęt
|
4.1.
|
Wstrząsarka mechaniczna
|
|
4.2.
|
Sprzęt do HPLC przy potrójnym gradiencie
|
|
4.2.1.
|
Kolumna do chromatografii cieczowej, Hypersil ODS, wypełnienie 3 μm, 100 mm × 4,6 mm lub równoważne
|
|
4.2.2.
|
Detektor UV z regulacją długości fal lub detektor diodowy
|
|
4.3.
|
Rotacyjna wyparka warstwowa
|
|
4.4.
|
Filtr membranowy, 0,45 μm
|
|
4.5.
|
Rozdzielnik próżni do jednoczesnej próżniowej izolacji
|
|
4.6.
|
Łaźnia ultradźwiękowa
|
5. Sposób postępowania
5.1. Wskazówki ogólne
5.1.1. Ślepa próba paszy
Należy przeprowadzić analizę ślepej próby paszy w celu sprawdzenia, czy nie zawiera ona diklazurilu ani substancji interferujących. Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności diklazurilu i substancji interferujących.
5.1.2. Badanie odzysku
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy, fortyfikowaną przez dodanie znanej ilości diklazurilu, o wadze zbliżonej do tej, która znajduje się w próbce. Aby fortyfikować na poziomie 1 mg/kg, dodać 0,1 ml roztworu wzorcowego podstawowego (3.8.1) do 50 g ślepej próby paszy, zmieszać i pozostawić na 10 minut, kilkakrotnie mieszając, przed przejściem do etapu ekstrakcji (5.2).
Jeżeli nie można przeprowadzić badania ślepej próby paszy podobnej do badanej próbki (zob. pkt 5.1.1), badanie odzysku można przeprowadzić stosując metodę dodawania wzorca. W tym przypadku przeznaczona do analizy próbka jest fortyfikowana przez dodanie znanej ilości diklazurilu o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Próbkę tę poddaje się analizie wraz z niefortyfikowaną próbką, a odzysk może być obliczony przez odjęcie od próbki fortyfikowanej masy próbki niefortyfikowanej.
5.2. Ekstrakcja
5.2.1. Pasze
Odważyć, z dokładnością do 0,01 g, 50 g próbki. Przenieść do kolby stożkowej o pojemności 500 ml, dodać 1 ml roztworu wzorca wewnętrznego (3.9.2), 200 ml rozpuszczalnika ekstrakcyjnego (3.12) i zamknąć kolbę korkiem. Wstrząsać mieszaninę na wstrząsarce (4.1) przez noc. Pozostawić na 10 minut do odstania. Przelać 20 ml podzielnej części supernatantu do odpowiedniego szklanego naczynia i rozcieńczyć 20 ml wody. Przenieść ten roztwór na wypełnienie ekstrakcyjne (3.11) i przepuścić przez to wypełnienie, stosując rozdzielnik próżni (4.5). Przepłukać wypełnienie 25 ml mieszaniny rozpuszczalnika ekstrakcyjnego (3.12) i wody, 65 + 35 (V + V). Odrzucić zebrane frakcje i eluować związki, stosując 25 ml mieszaniny rozpuszczalnika ekstrakcyjnego (3.12) i wody, 80 + 20 (V + V). Odparować tę frakcję do osuszenia przy zastosowaniu rotacyjnej wyparki (4.3) w temperaturze 60 oC. Rozpuścić suchą pozostałość w 1,0 ml DMF (3.6), dodać 1,5 ml wody (3.1) i zmieszać. Przefiltrować przez filtr membranowy (4.4). Przystąpić do oznaczania HPLC (5.3).
5.2.2. Premiksy
Odważyć, z dokładnością do 0,001 g, 1 g próbki. Przenieść do kolby stożkowej o pojemności 500 ml, dodać 1,00 ml roztworu wzorca wewnętrznego (3.9.3), 200 ml rozpuszczalnika ekstrakcyjnego (3.12) i zamknąć kolbę korkiem. Wstrząsać mieszaninę na wstrząsarce (4.1) przez noc. Pozostawić na 10 minut do odstania. Przenieść podzielną część 10 000/p ml (p = nominalna zawartość diklazurilu w premiksie w mg/kg) supernatantu do kolby okrągłodennej o odpowiedniej pojemności. Odparować do osuszenia, pod obniżonym ciśnieniem, w temperaturze 60 oC przy użyciu rotacyjnej wyparki (4.3). Ponownie rozpuścić suchą pozostałość w 10,0 ml DMF (3.6), dodać 15,0 ml wody (3.1) i zmieszać. Przystąpić do oznaczania HPLC (5.3).
5.3. Oznaczanie HPLC
5.3.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
|
Kolumna do chromatografii cieczowej (4.2.1):
|
100 × 4,6 mm, Hypersil ODS, wypełnienie 3 μm lub równoważne
|
|
|
Faza ruchoma:
|
Eluent A (3.13.1):
|
wodny roztwór octanu amonu i kwaśnego siarczanu tetrabutyloamoniowego
|
|
|
Eluent B (3.13.2):
|
acetonitryl
|
|
|
Eluent C (3.13.3):
|
metanol
|
|
Elucja:
|
|
—
|
warunki początkowe: A + B + C = 60 + 20 + 20 (V + V + V)
|
|
—
|
po 10 minutach frakcjonowanie przez 30 minut do: A + B + C = 45 + 20 + 35 (V + V + V).
|
Płukać eluentem B przez 10 minut.
|
|
Prędkość przepływu:
|
1,5–2 ml/min
|
|
|
Dozowana objętość:
|
20 μl
|
|
|
Długość fali przy detekcji:
|
280 nm
|
|
Sprawdzić stabilność systemu chromatograficznego, dozując kilka razy roztwór kalibracyjny (3.10) zawierający 2 μg/ml, aż do uzyskania stałych wysokości (powierzchni) piku i czasów retencji.
5.3.2. Roztwór kalibracyjny
Zadozować kilka razy 20 μl roztworu kalibracyjnego (3.10) i określić średnią wysokość (powierzchnię) piku diklazurilu i wewnętrzne wzorcowe pików.
5.3.3. Roztwór próbki
Zadozować kilka razy 20 μl roztworu próbki (5.2.1 lub 5.2.2) i określić średnią wysokość (powierzchnię) piku diklazurilu i wewnętrzne wzorcowe pików.
6. Obliczanie wyników
6.1. Pasze
Zawartość diklazurilu w próbce (w mg/kg) oblicza się według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
hd,s
|
=
|
wysokość (powierzchnia) piku diklazurilu w roztworze próbki (5.2.1)
|
|
hi,s
|
=
|
wysokość (powierzchnia) piku wzorca wewnętrznego w roztworze próbki (5.2.1)
|
|
hd,c
|
=
|
wysokość (powierzchnia) piku diklazurilu w roztworze kalibracyjnym (3.10)
|
|
hi,c
|
=
|
wysokość (powierzchnia) piku wzorca wewnętrznego w roztworze kalibracyjnym (3.10)
|
|
cd,c
|
=
|
stężenie w μg/ml diklazurilu w roztworze kalibracyjnym (3.10)
|
|
m
|
=
|
masa naważki w g
|
|
V
|
=
|
objętość ekstraktu próbki zgodna z pkt 5.2.1 (tj. 2,5 ml)
|
6.2. Premiksy
Zawartość diklazurilu w próbce (w mg/kg) obliczyć według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
hd,c
|
=
|
wysokość (powierzchnia) piku diklazurilu w roztworze kalibracyjnym (3.10)
|
|
hi,c
|
=
|
wysokość (powierzchnia) piku wzorca wewnętrznego w roztworze kalibracyjnym (3.10)
|
|
hd,s
|
=
|
wysokość (powierzchnia) piku diklazurilu w roztworze próbki (5.2.2)
|
|
hi,s
|
=
|
wysokość (powierzchnia) piku wzorca wewnętrznego w roztworze próbki (5.2.2)
|
|
cd,c
|
=
|
stężenie w μg/ml diklazurilu w roztworze kalibracyjnym (3.10)
|
|
m
|
=
|
masa naważki w g
|
|
V
|
=
|
objętość ekstraktu próbki zgodna z pkt 5.2.2 (tj. 25 ml)
|
|
p
|
=
|
nominalna zawartość w mg/kg diklazurilu w premiksie
|
7. Sprawdzenie wyników
7.1. Sprawdzenie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki (5.2.1 lub 5.2.2) i roztworu kalibracyjnego (3.10).
7.1.1. Kochromatografia
Ekstrakt próbki (5.2.1 lub 5.2.2) fortyfikuje się przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.10) Ilość dodanego diklazurilu musi odpowiadać ilości diklazurilu znajdującej się w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i stopnia rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku diklazurilu i piku wzorca wewnętrznego. Szerokość piku w połowie jego wysokości musi mieścić się w granicach ± 10 % pierwotnej szerokości piku diklazurilu lub piku wzorca wewnętrznego niefortyfikowanego ekstraktu próbki.
7.1.2. Detekcja diodowa
Wyniki sprawdza się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach ±2 nm;
|
|
b)
|
w zakresie od 230 do 320 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między 2 widmami nie jest większa niż 15 % absorbancji wzorcowego agalitu;
|
|
c)
|
w zakresie od 230 do 320 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między 2 widmami nie jest większa niż 15 % absorbancji widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
30 % względem wyższej wartości, dla zawartości diklazurilu od 0,5 mg/kg do 2,5 mg/kg,
|
|
—
|
0,75 mg/kg, dla zawartości diklazurilu od 2,5 do 5 mg/kg,
|
|
—
|
15 % względem wyższej wartości, dla zawartości diklazurilu powyżej 5 mg/kg.
|
7.3. Odzysk
W przypadku fortyfikacji ślepej próby lub próby paszy odzysk musi wynosić co najmniej 80 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy 11 laboratoriów przeprowadzono analizę 5 próbek. Były to próbki z dwóch premiksów; jeden był wymieszany z głównym składnikiem organicznym (O 100), a drugi z głównym składnikiem nieorganicznym (A 100). Zawartość teoretyczna diklazurilu wynosiła 100 mg/kg, trzy mieszanki pasz dla drobiu zostały sporządzone przez 3 różnych producentów (NL) (L1/Z1/K1). Teoretyczna zawartość diklazurilu wynosiła 1 mg/kg. Analizę każdej próbki przeprowadzono raz lub dwukrotnie. (Bardziej szczegółowe informacje dotyczące tej analizy można znaleźć w Journal of AOAC International, tom 77, nr 6, 1994, s. 1359–1361). W poniższej tabeli podano wyniki przeprowadzonych badań.
|
|
Próbka 1
A 100
|
Próbka 2
O 100
|
Próbka 3
L1
|
Próbka 4
Z1
|
Próbka 5
K1
|
|
L
|
11
|
11
|
11
|
11
|
6
|
|
n
|
19
|
18
|
19
|
19
|
12
|
|
Średnia
|
100,8
|
103,5
|
0,89
|
1,15
|
0,89
|
|
Sr (mg/kg)
|
5,88
|
7,64
|
0,15
|
0,02
|
0,03
|
|
CVr (%)
|
5,83
|
7,38
|
17,32
|
1,92
|
3,34
|
|
SR (mg/kg)
|
7,59
|
7,64
|
0,17
|
0,11
|
0,12
|
|
CVR (%)
|
7,53
|
7,38
|
18,61
|
9,67
|
13,65
|
|
Zawartość nominalna (mg/kg)
|
100
|
100
|
1
|
1
|
1
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych wartości
|
|
Sr
|
=
|
odchylenie standardowe powtarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|
9. Objaśnienia
Należy uprzednio wykazać, że reakcja diklazurilu jest liniowa w zakresie mierzonych stężeń.
G. OZNACZANIE SOLI SODOWEJ LASALOCIDU
Sól sodowa polieterowego monokarboksylowego kwasu wytwarzanego przez Streptomyces lasaliensis
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości soli sodowej lasalocidu w paszach i premiksach. Granica wykrywalności wynosi 5 mg/kg, granica oznaczalności 10 mg/kg.
2. Sposób przeprowadzenia metody
Sól sodowa lasalocidu jest ekstrahowana z próbki zakwaszonym metanolem i oznaczana metodą wysokosprawnej chromatografii cieczowej z fazą odwróconą (HPLC) przy wykorzystaniu detektora spektrofluorometrycznego.
3. Odczynniki i roztwory
|
3.1.
|
Dwuwodorofosforan potasu (KH2PO4).
|
|
3.2.
|
Kwas ortofosforowy, w (w/w) = 85 %.
|
|
3.3.
|
Roztwór kwasu ortofosforowego, c = 20 %.
Rozcieńczyć 23,5 ml kwasu ortofosforowego (3.2) wodą do objętości 100 ml.
|
|
3.4.
|
6-metylo-2-heptyloamina(1,5-dwumetyloheksyloamina), w (w/w) = 99 %.
|
|
3.6.
|
Kwas chlorowodorowy, gęstość = 1,19 g/ml.
|
|
3.7.
|
Roztwór buforu fosforanowego, c = 0,01 mol/l.
Rozpuścić 1,36 g KH2PO4 (3.1), w 500 ml wody (3.11), dodać 3,5 ml kwasu ortofosforowego (3.2) i 10,0 ml 6-metylo-2-heptyloaminy (3.4). Dostosować pH do 4,0 przy użyciu roztworu kwasu ortofosforowego (3.3) i rozcieńczyć wodą (3.11) do objętości 1 000 ml.
|
|
3.8.
|
Zakwaszony metanol.
Przenieść 5,0 ml kwasu chlorowodorowego (3.6) do kolby miarowej o pojemności 1 000 ml, uzupełnić do pełnej objętości kolby metanolem (3.5) i zmieszać. Roztwór przygotować bezpośrednio przed użyciem.
|
|
3.9.
|
Faza ruchoma HPLC: mieszanina roztworu buforu fosforanowego i metanolu w stosunku 5 + 95 (V +V).
Zmieszać 5 ml roztworu buforu fosforanowego (3.7) i 95 ml metanolu (3.5).
|
|
3.10.
|
Substancja wzorcowa soli sodowej lasalocidu o gwarantowanej czystości, C34H53O8Na (sól sodowa polieterowego monokarboksylowego kwasu wytwarzanego przez Streptomyces lasaliensis), E 763.
|
|
3.10.1.
|
Podstawowy roztwór wzorcowy soli sodowej lasalocidu, 500 μg/ml
Zważyć, z dokładnością do 0,1 mg, 50 mg soli sodowej lasalocidu (3.10) do kolby miarowej o pojemności 100 ml, rozpuścić w zakwaszonym metanolu (3.8), uzupełnić do pełnej objętości kolby tym samym rozpuszczalnikiem i zmieszać. Roztwór przygotować bezpośrednio przed użyciem.
|
|
3.10.2.
|
Roztwór wzorcowy pośredni soli sodowej lasalocidu, 50 μg/ml
Przenieść pipetą 10,0 ml podstawowego roztworu wzorcowego soli sodowej lasalocidu (3.10.1) do kolby miarowej o pojemności 100 ml, uzupełnić do pełnej objętości kolby zakwaszonym metanolem (3.8) i zmieszać. Roztwór przygotować bezpośrednio przed użyciem.
|
|
3.10.3.
|
Roztwory kalibracyjne
Do kolb miarowych o pojemności 50 ml przenieść 1,0, 2,0, 4,0, 5,0 i 10,0 ml roztworu wzorcowego pośredniego (3.10.2). Uzupełnić do pełnej objętości kolb zakwaszonym metanolem (3.8) i zmieszać. Roztwory te odpowiadają odpowiednio 1,0, 2,0, 4,0, 5,0 i 10,0 μg soli sodowej lasalocidu w 1 ml. Roztwory przygotować bezpośrednio przed użyciem.
|
4. Aparatura i sprzęt
|
4.1.
|
Łaźnia ultradźwiękowa lub wibracyjna łaźnia wodna, z możliwością kontroli temperatury.
|
|
4.2.
|
Filtry membranowe, 0,45 μm.
|
|
4.3.
|
Zestaw HPLC z systemem dozowania, umożliwiający dozowanie 20 μl.
|
|
4.3.1.
|
Kolumna do chromatografii cieczowej: 125 mm × 4 mm, faza odwrócona C18, wypełnienie 5 μm lub równoważne
|
|
4.3.2.
|
Spektrofluorymetr o zmiennej fali, z możliwością zmiany długości fal wzbudzenia i emisji
|
5. Sposób postępowania
5.1. Objaśnienia
5.1.1. Ślepa próba
W celu przeprowadzenia badania odzysku (5.1.2) należy przeprowadzić analizę ślepej próby paszy w celu sprawdzenia, czy badana pasza nie zawiera lasalocidu ani substancji interferujących. Pasza użyta do ślepej próby musi być podobna do próbki paszy badanej i nie może zawierać lasalocidu sodu ani interferujących substancji.
5.1.2. Badanie odzysku
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy fortyfikowaną przez dodanie znanej ilości soli sodowej lasalocidu, o wadze zbliżonej do tej, która znajduje się w próbce. Aby fortyfikować na poziomie 100 mg/kg, dodać 10,0 ml podstawowego roztworu wzorcowego (3.10.1) do kolby stożkowej o pojemności 250 ml i odparować roztwór do objętości około 0,5 ml. Dodać 50 g ślepej próby paszy, zmieszać i pozostawić na 10 minut, a następnie kilkakrotnie zamieszać i przejść do etapu ekstrakcji (5.2).
Jeżeli nie można przeprowadzić badania ślepej próby paszy podobnej do badanej próbki (zob. pkt 5.1.1), badanie odzysku można przeprowadzić, stosując metodę dodawania wzorca. W tym przypadku przeznaczona do analizy próbka jest fortyfikowana przez dodanie znanej ilości soli sodowej lasalocidu, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Próbkę tę poddaje się analizie wraz z niefortyfikowaną próbką, a odzysk może być obliczony przez odjęcie od próbki fortyfikowanej masy próbki niefortyfikowanej.
5.2. Ekstrakcja
5.2.1. Pasze
Zważyć, z dokładnością do 0,01 g, od 5 g do 10 g próbki do wyposażonej w korek kolby stożkowej o pojemności 250 ml. Dodać przy użyciu pipety 100,0 ml zakwaszonego metanolu (3.8). Zamknąć luźno korkiem i zamieszać w celu rozproszenia. Umieścić kolbę w łaźni ultradźwiękowej (4.1) w temperaturze około 40 oC na 20 minut, następnie wyjąć i schłodzić do temperatury pokojowej. Pozostawić kolbę na około godzinę, dopóki suspensja nie opadnie, następnie przefiltrować podzielną część przez filtr membranowy 0,45 μm (4.2) do odpowiedniego naczynia. Przystąpić do oznaczania HPLC (5.3).
5.2.2. Premiksy
Odważyć, z dokładnością do 0,001 g, około 2 g nierozdrobnionego premiksu do kolby miarowej o pojemności 250 ml. Dodać 100,0 ml zakwaszonego metanolu (3.8) i zamieszać w celu rozproszenia. Umieścić kolbę z zawartością w łaźni ultradźwiękowej (4.1) w temperaturze około 40 oC na 20 minut, następnie wyjąć i schłodzić do temperatury pokojowej. Uzupełnić do pełnej objętości kolb zakwaszonym metanolem (3.8) i dokładnie zmieszać. Pozostawić kolbę na około godzinę, dopóki suspensja nie opadnie, następnie przefiltrować podzielną część przez filtr membranowy 0,45 μm (4.2). Rozcieńczyć odpowiednią objętość klarownego filtratu przy użyciu zakwaszonego metanolu (3.8) w celu otrzymania finalnego testowego roztworu zawierającego około 4 μg/ml soli sodowej lasalocidu. Przystąpić do oznaczania HPLC (5.3).
5.3. Oznaczanie HPLC
5.3.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników:
|
Kolumna do chromatografii cieczowej (4.3.1):
|
125 mm × 4 mm, faza odwrócona C18, wypełnienie 5 μm lub równoważne
|
|
Faza ruchoma (3.9):
|
Mieszanina roztworu buforu fosforanowego (3.7) i metanolu (3.5), 5+95 (V+V)
|
|
Prędkość przepływu:
|
1,2 ml/min
|
|
Długość fali przy detekcji:
|
|
|
Wzbudzenie:
|
310 nm
|
|
Emisja:
|
419 nm
|
|
Dozowana objętość:
|
20 μl
|
Sprawdzić stabilność systemu chromatograficznego, dozując kilkakrotnie roztwór kalibracyjny (3.10.3) zawierający 4 μg/ml, aż do uzyskania stałych wysokości (powierzchni) piku i czasów retencji.
5.3.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z kalibracyjnych roztworów (3.10.3) i określić średnie wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) piku na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.3.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.2.1 lub 5.2.2) stosując taką samą objętość jak przy kalibracyjnych roztworach, i określić średnią wysokość (powierzchnię) piku dla pików soli sodowej lasalocidu.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) piku otrzymanej po zadozowaniu roztworu próbki (5.3.3) określić stężenie soli sodowej lasalocidu w μg/ml, odnosząc się do krzywej kalibracyjnej.
6.1. Pasze
Zawartość soli sodowej lasalocidu w mg/kg w próbce oblicza się według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
c
|
=
|
stężenie w μg/ml soli sodowej lasalocidu w roztworze próbki (5.2.1)
|
|
V1
|
=
|
objętość w ml ekstraktu próbki (5.2.1) (tj. 100 ml)
|
|
m
|
=
|
masa naważki w g
|
6.2. Premiksy
Zawartość soli sodowej lasalocidu, w mg/kg, w próbce obliczyć według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
c
|
=
|
stężenie w μg/ml soli sodowej lasalocidu w roztworze próbki (5.2.2)
|
|
V2
|
=
|
objętość w ml ekstraktu próbki zgodny z pkt 5.2.2 (tj. 250 ml)
|
|
f
|
=
|
współczynnik rozcieńczania zgodny z pkt 5.2.2
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzenie wyników
7.1. Sprawdzenie tożsamości
Metody bazujące na detekcji spektrofluorometrycznej są mniej podatne na interferencje niż metody z zastosowaniem detekcji UV. Tożsamość analitu może być potwierdzona przez kochromatografię.
7.1.1. Kochromatografia
Ekstrakt próbki (5.2.1 lub 5.2.2) jest fortyfikowany przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.10.3). Ilość dodanej soli sodowej lasalocidu musi odpowiadać ilości soli sodowej lasalocidu w ekstrakcie próbki. Po uwzględnieniu ilości dodanej soli sodowej lasalocidu i rozcieńczenia ekstraktu zwiększyć się może tylko wysokość piku soli sodowej lasalocidu. Szerokość piku w połowie wysokości musi mieścić się w zakresie ± 10 % oryginalnej szerokości piku niefortyfikowanego ekstraktu.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
15 % względem wyższej wartości, dla zawartości soli sodowej lasalocidu od 30 mg/kg do 100 mg/kg,
|
|
—
|
15 mg/kg dla zawartości soli sodowej lasalocidu od 100 mg/kg do 200 mg/kg,
|
|
—
|
7,5 % względem wyższej wartości, dla zawartości soli sodowej lasalocidu powyżej 200 mg/kg.
|
7.3. Odzysk
W przypadku fortyfikowanej (ślepej) próby paszy lub próby paszy odzysk nie może być niższy niż 80 %. W przypadku fortyfikowanych próbek premiksów odzysk nie może być niższy niż 90 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy 12 laboratoriów (6) przeprowadzono analizę 2 premiksów (próbki 1 i 2) oraz 5 pasz (próbki 3–7). Analizę każdej próbki przeprowadzono dwukrotnie. W poniższej tabeli podane są wyniki przeprowadzonych badań:
|
|
Próbka 1
Premiks dla kurcząt
|
Próbka 2
Premiks dla indyków
|
Próbka 3
Śruty dla indyków
|
Próbka 4
Granulki dla kurcząt
|
Próbka 5
Pasza dla indyków
|
Próbka 6
Pasza dla drobiu A
|
Próbka 7
Pasza dla drobiu B
|
|
L
|
12
|
12
|
12
|
12
|
12
|
12
|
12
|
|
n
|
23
|
23
|
23
|
23
|
23
|
23
|
23
|
|
Średnia [mg/kg]
|
5 050
|
16 200
|
76,5
|
78,4
|
92,9
|
48,3
|
32,6
|
|
sr [mg/kg]
|
107
|
408
|
1,71
|
2,23
|
2,27
|
1,93
|
1,75
|
|
CVr [%]
|
2,12
|
2,52
|
2,24
|
2,84
|
2,44
|
4,00
|
5,37
|
|
sR [mg/kg]
|
286
|
883
|
3,85
|
7,32
|
5,29
|
3,47
|
3,49
|
|
CVR [%]
|
5,66
|
5,45
|
5,03
|
9,34
|
5,69
|
7,18
|
10,70
|
|
Zawartość nominalna [mg/kg]
|
5 000 (7)
|
16 000 (7)
|
80 (7)
|
105 (7)
|
120 (7)
|
50 (8)
|
35 (8)
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba poszczególnych wyników
|
|
sr
|
=
|
standardowe odchylenie powtarzalności
|
|
sR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVr
|
=
|
odchylenie standardowe zmienne powtarzalności, %
|
|
CVR
|
=
|
odchylenie standardowe zmienne odtwarzalności, %
|
(1) Przeprowadzone przez grupę roboczą ds. pasz Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Przeprowadzone przez grupę roboczą ds. pasz Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Inne metody rozkładu mogą być stosowane, pod warunkiem, iż wykazano, że zapewniają podobne wyniki (np. rozkład mikrofalami).
(4) Zielonki świeże lub suszone mogą zawierać duże ilości krzemionki roślinnej, która może zawierać pierwiastki śladowe i którą trzeba usunąć. Dlatego też w przypadku analizy próbek takich pasz należy zastosować następujące, zmodyfikowane postępowanie. Postępować w sposób określony w pkt 5.1.1.1 do etapu filtrowania. Następnie należy dwukrotnie wrzącą wodą przemyć filtr papierowy zawierający nierozpuszczalną pozostałość i umieścić w platynowym lub kwarcowym tyglu. Należy włączyć piec muflowy (4.1), nastawić temperaturę poniżej 550 oC i poczekać, aż całkowicie zniknie wszelki materiał zawierający węgiel. Pozostawić do ostygnięcia, dodać kilka kropli wody, a następnie 10–15 ml kwasu fluorowodorowego (3.4) i odparować do osiągnięcia suchości w temperaturze około 150 oC. Jeśli w pozostałościach pozostanie krzemionka, to należy ponownie rozpuścić tę pozostałość w kilku mililitrach kwasu fluorowodorowego (3.4) i odparować aż do osiągnięcia suchości. Dodać pięć kropli kwasu siarkowego (3.5) i podgrzewać do momentu pokazania się białego dymu. Po dodaniu 5 ml kwasu solnego o stężeniu 6 mol/l (3.2) i około 30 ml wody podgrzać, przefiltrować roztwór do kolby miarowej o pojemności 250 ml i dopełnić wodą do pełnej objętości (stężenie HCl około 0,5 mol/l). Następnie należy wykonać oznaczenie zgodnie z pkt 5.1.2.
(5) „The Analyst” 108 z 1983 r., s. 1 252–1 256
(6) „The Analyst” nr 120 z 1995 r., s. 2175–2180.
(7) Zawartość zadeklarowana przez producenta.
(8) Pasza przygotowana w laboratorium.
ZAŁĄCZNIK V
METODY ANALIZY DO CELÓW KONTROLI NIEPOŻĄDANYCH SUBSTANCJI W PASZACH
A. OZNACZANIE WOLNEGO I CAŁKOWITEGO GOSSYPOLU
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości wolnego gossypolu, całkowitego gossypolu i chemicznie związanych substancji w nasionach bawełny, w makuchach, w mączce z nasion bawełny i w mieszankach paszowych zawierających te materiały paszowe, w których zawartość wolnego gossypolu, całkowitego gossypolu i chemicznie związanych substancji przekracza 20 mg/kg.
2. Sposób przeprowadzenia metody
Gossypol ekstrahuje się w obecności 3-aminopropan-1-olu lub mieszaniny propan-2-olu i heksanu w przypadku oznaczania wolnego gossypolu lub dwumetyloformamidem w przypadku oznaczenia całkowitego gossypolu. Gossypol, reagując z aniliną, tworzy gossypol-dianilinę, której gęstość optyczna mierzona jest przy 440 nm.
3. Odczynniki i roztwory
|
3.1.
|
Mieszanina propan-2-ol-heksanu: Zmieszać 60 części objętościowych propan-2-olu z 40 częściami objętościowymi n-heksanu.
|
|
3.2.
|
Rozpuszczalnik A: Umieścić w kolbie o pojemności 1 l około 500 ml mieszaniny propan-2-ol-heksanu (3.1), 2 ml 3-aminopropan-1-olu, 8 ml lodowatego kwasu octowego i 50 ml wody. Uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1). Odczynnik zachowuje stabilność przez tydzień.
|
|
3.3.
|
Rozpuszczalnik B: odpipetować 2 ml 3-aminopropan-1-olu i 10 ml lodowatego kwasu octowego do kolby miarowej o pojemności 100 ml. Schłodzić do temperatury pokojowej i uzupełnić do pełnej objętości kolby N,N-dwumetyloformamidem. Odczynnik zachowuje stabilność przez tydzień.
|
|
3.4.
|
Anilina: Jeżeli gęstość optyczna ślepej próby przekracza 0,022, przedestylować anilinę nad proszkiem cynku, odrzucając pierwsze i ostatnie 10 % frakcji destylatu. Odczynnik można przechowywać przez kilka miesięcy, jeżeli jest trzymany w lodówce w brązowej butelce zamkniętej korkiem.
|
|
3.5.
|
Roztwór A wzorcowego gossypolu: 27,9 mg octanu gossypolu umieścić w kolbie miarowej o pojemności 250 ml. Rozpuścić i uzupełnić do pełnej objętości kolby rozpuszczalnikiem A (3.2). Przenieść pipetą 50 ml tego roztworu do kolby miarowej o pojemności 250 ml i uzupełnić do pełnej objętości kolby rozpuszczalnikiem A. Stężenie gossypolu w tym roztworze wynosi 0,02 mg/ml. Przed użyciem pozostawić na godzinę w temperaturze pokojowej.
|
|
3.6.
|
Roztwór B wzorcowego gossypolu: 27,9 mg octanu gossypolu umieścić w kolbie miarowej o pojemności 50 ml. Rozpuścić i uzupełnić do pełnej objętości kolby rozpuszczalnikiem B (3.3). Stężenie gossypolu w tym roztworze wynosi 0,5 mg/ml.
Wzorcowe roztwory gossypolu A i B nadają się do użycia przez 24 godziny, jeżeli są chronione przed światłem.
|
4. Aparatura i sprzęt
|
4.1.
|
Mikser lub tumbler: około 35 obr./min.
|
5. Sposób postępowania
5.1. Analiza próbki
Masa próbki pobrana do analizy zależy od przewidywanej zawartości gossypolu w próbce. Zalecana jest praca na małych ilościach próby i stosunkowo dużej ilości filtratu po to, aby otrzymać wystarczającą ilość gossypolu do precyzyjnego oznaczenia fotometrycznego. W przypadku oznaczania wolnego gossypolu w nasionach bawełny, w makuchach i mączce z nasion bawełny masa próbki do analizy nie może przekraczać 1 g. W przypadku mieszanek paszowych może wynosić do 5 g. W większości przypadków wystarczające jest 10 ml podzielnej części filtratu. Musi on zawierać od 50 do 100 μg gossypolu. Jeżeli oznaczamy całkowity gossypol, ilość badanej próbki musi wynosić od 0,5 do 5 g, wówczas 2 ml podzielnej części filtratu będzie zawierać od 40 do 200 μg gossypolu.
Analizę należy przeprowadzać w temperaturze pokojowej, za którą uważa się temperaturę około 20 oC.
5.2. Oznaczanie wolnego gossypolu
Umieścić próbkę w kolbie ze szlifowaną szyjką o pojemności 250 ml. Dno kolby pokryć pokruszonym szkłem. Przy użyciu pipety dodać 50 ml rozpuszczalnika A (3.2), zamknąć kolbę korkiem i mieszać przez godzinę w mikserze. Filtrować przez suchy filtr, a filtrat zebrać do małej kolby ze szlifowaną szyjką. Podczas filtracji przykryć lejek szkiełkiem zegarkowym.
Odmierzyć pipetą identyczne podzielne części filtratu zawierające od 50 do 100 μg gossypolu i umieścić w 2 kolbach miarowych o pojemności 25 ml (A i B). Jeżeli to konieczne, uzupełnić zawartość kolby do 10 ml rozpuszczalnikiem A (3.2). Następnie uzupełnić zawartość kolby (A) do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1). Roztwór ten będzie użyty jako roztwór odniesienia względem pomiaru roztworu próbki.
Przenieść pipetą po 10 ml rozpuszczalnika A (3.2) do 2 następnych kolb miarowych o pojemności 25 ml (C i D). Zawartość kolby (C) uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1). Roztwór ten będzie używany jako roztwór odniesienia względem pomiaru roztworu ślepej próby.
Dodać po 2 ml aniliny (3.4) do każdej z kolb (D) i (B). Ogrzewać na wrzącej łaźni przez 30 minut, do wywołania reakcji barwnej. Schłodzić do temperatury pokojowej, uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1), homogenizować i odstawić na godzinę.
Oznaczyć gęstość optyczną roztworu ślepej próby (D), używając odpowiedniego roztworu (C) jako roztworu odniesienia, oraz gęstość optyczną roztworu próbki (B) przez porównanie z odpowiednim roztworem (A), jako roztworem odniesienia w spektrofotometrze przy 440 nm, stosując 1-centymetrowe szklane kuwety.
Od odpowiadającej gęstości optycznej wartości roztworu próbki odjąć wartość gęstości optycznej roztworu ślepej próby (= skorygowana gęstość optyczna). Z użyciem tej różnicy obliczyć zawartość wolnego gossypolu, w sposób określony w pkt 6.
5.3. Oznaczanie całkowitego gossypolu
Próbkę zawierającą od 1 do 5 mg gossypolu umieścić w kolbie miarowej o pojemności 50 ml i dodać 10 ml rozpuszczalnika B (3.3). W tym samym czasie przygotować ślepą próbę, umieszczając 10 ml rozpuszczalnika B (3.3) w następnej kolbie miarowej o pojemności 50 ml. Ogrzewać obie kolby przez 30 minut na wrzącej łaźni wodnej. Następnie schłodzić do temperatury pokojowej i uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1). Homogenizować i odstawić na 10 do 15 minut. Następnie filtrować, a filtrat zebrać do kolb ze szlifowaną szyjką.
Przenieść pipetą po 2 ml filtratu próbki do każdej z dwóch kolb miarowych o pojemności 25 ml i oraz po 2 ml filtratu do próby ślepej do pozostałych dwóch kolb. Uzupełnić zawartość jednej kolby z każdej partii do objętości 25 ml mieszaniną 1-propan-2-ol-heksanu (3.1). Roztwory te będą używane jako roztwory odniesienia.
Dodać po 2 ml aniliny (3.4) do 2 następnych kolb miarowych. Ogrzewać obie kolby przez 30 minut na wrzącej łaźni wodnej do wywołania reakcji barwnej. Schłodzić do temperatury pokojowej, uzupełnić do objętości 25 ml mieszaniną propan-2-ol-heksanu (3.1), homogenizować i odstawić na godzinę.
Oznaczyć gęstość optyczną, tak jak dla wolnego gossypolu (5.2). Używając tej wartości, obliczyć zawartość całkowitego gossypolu, w sposób określony w pkt 6.
6. Obliczanie wyników
Wyniki mogą być obliczone na podstawie określonej gęstości optycznej (6.1) lub na podstawie odniesienia do krzywej kalibracyjnej (6.2).
6.1. Wyniki obliczane na podstawie określonej gęstości optycznej
W podanych warunkach określone gęstości optyczne są następujące:
|
Wolny gossypol:
|
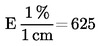
|
|
Całkowity gossypol:
|

|
Zawartość wolnego lub całkowitego gossypolu w próbce oblicza się według następującego wzoru:
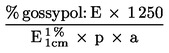
gdzie:
|
E
|
=
|
skorygowana gęstość optyczna określona w pkt 5.2
|
|
p
|
=
|
masa próbki w g
|
|
a
|
=
|
podzielna część filtratu w ml
|
6.2. Wyniki obliczane na podstawie krzywej kalibracyjnej
6.2.1. Wolny gossypol
Przygotować 2 partie po 5 kolb miarowych o pojemności 25 ml. Do każdej partii kolb wprowadzić pipetą odpowiednio 2,0, 4,0, 6,0, 8,0 i 10,0 ml roztworu A wzorcowego gossypolu (3.5). Uzupełnić do objętości 10 ml rozpuszczalnikiem A (3.2). Uzupełnić każdą partię kolbą miarową o pojemności 25 ml zawierającą jedynie 10 ml rozpuszczalnika A (3.2) (próba ślepa).
Uzupełnić do objętości 25 ml kolby w pierwszej partii, włączając kolbę ślepej próby, mieszaniną propan-2-ol-heksanu (3.1) (partia odniesienia).
Dodać po 2 ml aniliny (3.4) do każdej kolby drugiej partii, stanowiącej partię wzorcową, włączając kolbę ślepej próby. Ogrzewać przez 30 minut na wrzącej łaźni wodnej do wywołania reakcji barwnej. Schłodzić do temperatury pokojowej, uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1), homogenizować i odstawić na godzinę.
Oznaczyć w sposób określony w 5.2 gęstość optyczną roztworów w partii wzorcowej przez porównanie z odpowiadającymi im roztworami w partii odniesienia. Wykreślić krzywą kalibracyjną, nanosząc gęstości optyczne względem ilości gossypolu (w μg).
6.2.2. Całkowity gossypol
Przygotować 6 kolb miarowych o pojemności 50 ml. Do pierwszej kolby dodać 10 ml rozpuszczalnika B (3.3), a do pozostałych odpowiednio 2,0, 4,0, 6,0, 8,0 i 10,0 ml roztworu B wzorcowego gossypolu (3.6). Każdą kolbę uzupełnić do objętości 10 ml rozpuszczalnikiem B (3.3). Ogrzewać przez 30 minut na wrzącej łaźni wodnej. Schłodzić do temperatury pokojowej, uzupełnić do pełnej objętości kolby mieszaniną propan-2-olheksanu (3.1) i homogenizować.
Do każdej z 2 partii po 6 kolb miarowych o pojemności 25 ml wprowadzić po 2,0 ml wyżej wspomnianych roztworów. 6 kolb pierwszej partii uzupełnić do objętości 25 ml mieszaniną propan-2-ol-heksanu (3.1) (partia odniesienia).
Do każdej kolby drugiej partii, stanowiącej partię wzorcową, dodać po 2 ml aniliny (3.4). Ogrzewać przez 30 minut na wrzącej łaźni wodnej. Schłodzić do temperatury pokojowej, uzupełnić do pełnej objętości kolby mieszaniną propan-2-ol-heksanu (3.1), homogenizować i odstawić na godzinę (partia standardowa).
Oznaczyć w sposób określony w pkt 5.2 gęstość optyczną roztworów w partii wzorcowej przez porównanie z odpowiadającymi im roztworami w partii odniesienia. Wykreślić krzywą kalibracyjną, nanosząc gęstości optyczne względem ilości gossypolu (w μg).
6.3. Powtarzalność
Różnica pomiędzy wynikami 2 równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
15 % najwyższej wartości względnej, dla zawartości gossypolu do 500 ppm,
|
|
—
|
75 ppm w wartości bezwzględnej, dla zawartości gossypolu od 500 do 750 ppm,
|
|
—
|
10 % najwyższej wartości względnej, dla zawartości gossypolu powyżej 750 ppm.
|
B. OZNACZANIE POZIOMÓW ZAWARTOŚCI DIOKSYN (PCDD/PCDF) I DIOKSYNOPODOBNYCH PCB
I. METODY POBIERANIA PRÓBEK I INTERPRETACJA WYNIKÓW
1. Cel i zakres stosowania metody
Próbki przeznaczone do urzędowej kontroli poziomu zawartości dioksyn (polichlorowanych dibenzo-p-dioksyn (PCDD) i polichlorowanych dibenzofuranów (PCDF)) oraz dioksynopodobnych bifenyli polichlorowanych (PCB) (1) w paszach pobiera się zgodnie z przepisami załącznika I. Należy stosować wymagania ilościowe w stosunku do kontroli substancji lub produktów jednolicie rozmieszczonych w paszy, zgodnie z załącznikiem I pkt 5.A. Uzyskane w ten sposób próbki zbiorcze uważa się za reprezentatywne dla partii lub podpartii, z których pochodzą. Zgodność z maksymalnymi poziomami określonymi w dyrektywie 2002/32/WE Parlamentu Europejskiego i Rady (2) ustala się na podstawie zawartości stwierdzonych w próbkach laboratoryjnych.
2. Zgodność partii lub podpartii z wymaganiami
Badaną partię uznaje się za spełniającą wymagania, jeżeli wynik pojedynczej analizy nie przekracza dopuszczalnej zawartości określonej w dyrektywie 2002/32/WE, z uwzględnieniem niepewności pomiaru.
Badaną partię uznaje się za niespełniającą wymagań w zakresie dopuszczalnych zawartości określonych w dyrektywie 2002/32/WE, jeżeli górny (3) wynik analizy potwierdzony drugą analizą (4) ponad wszelką wątpliwość przekracza dopuszczalną zawartość, z uwzględnieniem przy tym niepewności pomiaru.
Niepewność pomiaru uwzględnia się z zastosowaniem jednego z następujących sposobów:
|
—
|
obliczając niepewność rozszerzoną z użyciem współczynnika rozszerzenia 2, która określa poziom zaufania na poziomie około 95 %. Partia wykazuje niezgodność, jeśli mierzona wartość po odjęciu U jest wyższa niż poziom maksymalny. W przypadku odrębnego oznaczania dioksyn i dioksynopodobnych PCB należy zastosować sumę szacowanej niepewności rozszerzonej dla odrębnych wyników badań dioksyn i dioksynopodobnych PCB w odniesieniu do sumy dioksyn i dioksynopodobnych PCB,
|
|
—
|
ustalając decyzyjną wartość graniczną (CCα) zgodnie z decyzją Komisji 2002/657/WE (5) (pkt 3.1.2.5 załącznika – przypadek substancji z ustaloną dopuszczalną wartością graniczną). Partia wykazuje niezgodność, jeśli mierzona wartość jest równa lub wyższa niż CCα.
|
Niniejsze zasady interpretacji odnoszą się do wyniku analizy uzyskanego z próbki przeznaczonej do urzędowej kontroli. Nie ma to wpływu na prawo państw członkowskich do stosowania przepisów krajowych do celów obrony lub arbitrażu.
II. PRZYGOTOWANIE PRÓBEK I WYMAGANIA DOTYCZĄCE METOD ANALIZ WYKORZYSTYWANYCH W URZĘDOWEJ KONTROLI DIOKSYN (PCDD/PCDF) I DIOKSYNOPODOBNYCH PCB
1. Cel i zakres stosowania metody
Niniejsze wymagania należy stosować w analizie pasz i materiałów paszowych w zakresie oznaczania zawartości dioksyn polichlorowanych dibenzo-p-dioksyn (PCDD) i polichlorowanych dibenzofuranów (PCDF) oraz dioksynopodobnych polichlorowanych bifenyli (PCB).
Monitorowanie obecności dioksyn w paszach można wykonać przy zastosowaniu metody skriningowej w celu wyselekcjonowania tych próbek, które zawierają dioksyny i dioksynopodobne PCB na poziomie o 25 % niższym lub wyższym niż poziom zainteresowania. Stężenia dioksyn w tych próbkach, które zawierają znaczące ich poziomy, należy oznaczyć lub potwierdzić z zastosowaniem metody potwierdzającej.
Metody skriningowe służą do wykrywania obecności dioksyn i dioksynopodobnych PCB na poziomie zainteresowania. Metody te mają zdolność związaną z wysoką wydajnością próbek i są stosowane do oddzielenia wielu próbek potencjalnie pozytywnych. Są tak zaprojektowane, aby ograniczały liczbę wyników fałszywie negatywnych.
Metody potwierdzające są metodami dostarczającymi pełnej lub uzupełniającej informacji, pozwalającej na jednoznaczną identyfikację i oznaczanie ilościowe dioksyn oraz dioksynopodobnych PCB na poziomie zainteresowania.
2. Informacje ogólne
Ze względu na fakt, że próbki środowiskowe i biologiczne (włączając w to próbki pasz i materiałów paszowych) zazwyczaj zawierają złożone mieszaniny różnych kongenerów dioksyn, w celu ułatwienia oceny ryzyka wprowadzono pojęcie współczynników równoważnej toksyczności – TEF (Toxic Equivalency Factors). Współczynniki te zostały opracowane w celu wyrażenia stężeń mieszanin posiadających podstawniki chloru w pozycjach 2,3,7,8- PCDD i PCDF, a także niektórych PCB posiadających podstawnik chloru w pozycjach non-orto i mono-orto, które posiadają podobną do dioksyn aktywność w postaci równoważników toksyczności – TEQ (Toxic Equivalent) o 2,3,7,8-TCDD. Stężenia poszczególnych substancji w próbce są mnożone przez ich TEF, a następnie wartości te są sumowane w celu obliczenia całkowitego stężenia związków dioksynopodobnych, wyrażonego w wartości TEQ.
Wyłącznie do celów niniejszego rozporządzenia akceptowalną granicą oznaczalności danego kongeneru jest stężenie analitu w ekstrakcie próbki, które wywołuje instrumentalną reakcję na impuls dla dwóch różnych jonów, rejestrowaną przy współczynniku S/N (sygnał/szum) w stosunku 3:1 dla sygnału mniej wrażliwego i przy spełnieniu podstawowych wymagań, takich jak czas retencji oraz stosunek izotopów, zgodnie z procedurą oznaczania metodą EPA 1613, korekta B.
3. Wymagania jakościowe dotyczące przygotowania próbek do analizy
Stosuje się ogólne przepisy dotyczące przygotowania próbek do analizy, określone w załączniku II.
Ponadto należy przestrzegać następujących wymagań:
|
—
|
Próbki należy przechowywać i transportować w pojemnikach szklanych, aluminiowych, polipropylenowych lub polietylenowych. Pojemnik na próbki należy oczyścić z pyłków papieru. Szkło laboratoryjne należy wypłukać rozpuszczalnikami, wcześniej zbadanymi na obecność dioksyn.
|
|
—
|
Wykonać analizę ślepej próby, obejmującą przeprowadzenie pełnej procedury analitycznej z wyłączeniem próbki.
|
|
—
|
Masa próbki użytej do ekstrakcji musi być wystarczająca do spełnienia wymagań w zakresie czułości metody.
|
4. Wymagania dotyczące laboratoriów
|
—
|
Laboratorium muszą wykazać możliwości wykonawcze metody na poziomie zainteresowania, np.0,5-, 1- i 2-krotny poziom zainteresowania wraz z akceptowalną wartością współczynnika zmienności obliczonego dla wielokrotnych powtórzeń analizy. Szczegółowe kryteria akceptacji są określone w pkt 5.
|
|
—
|
Granica oznaczalności metody potwierdzającej musi wynosić około 1/5 poziomu zainteresowania, aby gwarantować, że akceptowalne współczynniki zmienności znajdą się w zakresie danego poziomu zainteresowania.
|
|
—
|
W ramach wewnątrzlaboratoryjnej kontroli jakości należy regularnie przeprowadzać ślepe próby i badania z wykorzystaniem próby wzbogaconej lub analizy próbek kontrolnych (zwłaszcza certyfikowanych materiałów odniesienia, jeżeli materiały takie są dostępne).
|
|
—
|
Potwierdzeniem biegłości laboratorium w zakresie danych analiz są pozytywne wyniki uzyskiwane w międzylaboratoryjnych badaniach biegłości. Jednakże pozytywne wyniki uzyskane w badaniach międzylaboratoryjnych, np. w badaniach próbek gleby lub osadów dennych, nie oznaczają jednocześnie podobnej kompetencji w analizie próbek żywności lub pasz, które zawierają niższe poziomy skażeń. Dlatego obowiązkowe jest stałe uczestnictwo w międzylaboratoryjnych badaniach biegłości w zakresie oznaczania dioksyn i dioksynopodobnych PCB w odpowiednich próbkach środków spożywczych lub pasz.
|
|
—
|
Laboratoria muszą być akredytowane przez uznawany organ działający zgodnie z ISO Guide 58, w celu dopilnowania że w przeprowadzaniu analiz stosują one system zapewnienia jakości. Laboratoria muszą posiadać akredytację zgodną z normą ISO/IEC/17025.
|
5. Wymagania dotyczące procedur analitycznych stosowanych przy oznaczaniu dioksyn i dioksynopochodnych PCB
Podstawowe wymagania dotyczące przyjęcia procedur analitycznych:
|
—
|
Wysoka czułość i niska granica wykrywalności. Ilości PCDD i PCDF muszą być wykrywane na poziomie pikogramów TEQ (10-12 g) ze względu na bardzo dużą toksyczność niektórych związków. PCB występują w wyższych stężeniach niż PCDD i PCDF. Dla większości kongenerów PCB za wystarczającą uznaje się czułość rzędu nanogramów (10-9 g). Jednakże w przypadku oznaczania bardziej toksycznych dioksynopodobnych PCB kongenerów (w szczególności non-orto podstawionych kongenerów), musi być uzyskana taka sama czułość, jak dla PCDD i PCDF.
|
|
—
|
Wysoka selektywność (specyficzność). Konieczne jest odróżnienie PCDD, PCDF i dioksynopodobnych PCB od wielu współekstrahujących i ewentualnie interferujących substancji występujących w stężeniach do kilku wielkości wyższych od analizowanych związków. Przy stosowaniu metody chromatografii gazowej ze spektrometrią mas (GC/MS) konieczne jest odróżnienie kongenerów toksycznych (np. siedemnastu 2,3,7,8-podstawionych kongenerów PCDD i PCDF oraz dioksynopodobnych PCB) od pozostałych kongenerów. Testy biologiczne muszą być w stanie oznaczyć wartości TEQ selektywnie jako sumę PCDD, PCDF i dioksynopodobnych PCB.
|
|
—
|
Wysoka dokładność (poprawność i precyzja). Oznaczenie musi dostarczyć wiarygodnego oszacowania rzeczywistego stężenia analitu w próbce. Wysoka dokładność (dokładność pomiaru: stopień zgodności między wynikiem badania a rzeczywistą lub wyznaczoną wartością pomiaru) jest warunkiem uniknięcia odrzucenia wyniku analizy próbki na podstawie słabej wiarygodności oszacowanej wartości TEQ. Dokładność jest wyrażona jako prawdziwość (różnica między średnią wartością mierzoną w odniesieniu do przedmiotu analizy w certyfikowanym materiale, a jego certyfikowaną wartością, wyrażoną jako odsetek tej wartości) i precyzja (RSDR – względne standardowe odchylenie obliczone na podstawie wyników osiągniętych w zakresie odtwarzalności).
|
Metody skriningowe mogą obejmować testy biologiczne oraz metody GC/MS. Metody potwierdzające to metody wysokorozdzielczej chromatografii gazowej sprzężonej z wysokorozdzielczą spektometrią masową (HRGC/HRMS).
Przy oznaczaniu sumarycznej wartości TEQ muszą zostać spełnione następujące kryteria:
|
|
Metody skriningowe (przesiewowe)
|
Metody potwierdzające
|
|
Częstość wyników fałszywie negatywnych
|
< 1 %
|
|
|
Prawdziwość
|
|
od – 20 % do + 20 %
|
|
Precyzja RSDR
|
< 30 %
|
< 15 %
|
6. Szczególne wymagania dla metod GC/MS w metodach skriningowych lub potwierdzających
|
—
|
Wzorce wewnętrzne 2,3,7,8-chloropodstawionych PCDD/F znakowane izotopem węgla 13C oraz wzorce wewnętrzne dioksynopodobnych PCB, znakowane izotopem węgla 13C należy dodawać na początku metody analitycznej, np. przed ekstrakcją, w celu potwierdzenia postępowania analitycznego. Należy dodać co najmniej 1 kongener dla każdej grupy homologicznej, od tetra- do oktachloro PCDD/F oraz co najmniej 1 kongener dla każdej grupy homologicznej dioksynopodobnych PCB (alternatywnie co najmniej 1 kongener na każdy wybrany jon masy spektrometrycznej, rejestrujący funkcję służącą do monitorowania PCDD/F i dioksynopodobnych PCB). Pierwszeństwo zastosowania, szczególnie dla metod potwierdzających, mają wszystkie siedemnaście 2,3,7,8-podstawionych wzorców wewnętrznych PCDD/F znakowanych izotopem węgla 13C oraz wszystkie dwanaście wzorców wewnętrznych dioksynopodobnych PCB znakowanych izotopem węgla 13C.
|
|
—
|
Względne współczynniki odpowiedzi należy również oznaczyć dla tych kongenerów, dla których nie zastosowano analogów znakowanych izotopem węgla 13C poprzez odpowiedni roztwór kalibracyjny.
|
|
—
|
Przed procedurą ekstrakcji w przypadku pasz pochodzenia roślinnego i zwierzęcego o zawartości tłuszczu poniżej 10 % należy dodać wzorce wewnętrzne. W przypadku pasz pochodzenia zwierzęcego o zawartości tłuszczu powyżej 10 % wzorce wewnętrzne mogą być dodane zarówno przed, jak i po ekstrakcji tłuszczu. Należy przeprowadzić sprawdzenie skuteczności ekstrakcji, w zależności od etapu, na którym dodaje się wzorce wewnętrzne, a także od tego, czy wyniki są wyrażane na jednostkę masy produktu czy tłuszczu.
|
|
—
|
Przed analizą GC/MS dodać do próbki 1 lub 2 wzorce odzysku (zastępcze).
|
|
—
|
Konieczne jest sprawdzenie odzysku. Odzysk dla poszczególnych wzorców wewnętrznych w metodach potwierdzających musi zawierać się w granicach od 60 do 120 %. Dopuszczalne są niższe lub wyższe wartości odzysku dla poszczególnych kongenerów, w szczególności dla niektórych hepta- i oktachlorodibenzo-p-dioksyn i dibenzofuranów, jeżeli ich udział w TEQ nie przekracza 10 % całkowitego TEQ (na podstawie PCDD/F oraz dioksynopodobnych PCB). W przypadku metod skriningowych odzysk musi zawierać się w granicach od 30 do 140 %.
|
|
—
|
Rozdzielenie dioksyn od interferujących chlorowanych związków, takich jak dioksynopodobne PCB czy polichlorowane difenyloetery, należy osiągnąć przez zastosowanie odpowiednich technik chromatograficznych; (najlepiej stosując florisil, tlenek glinu lub kolumnę węglową).
|
|
—
|
Rozdzielanie izomerów przy chromatografii gazowej musi być wystarczające (< 25 % nakładania się pików pomiędzy 1,2,3,4,7,8-HxCDF i 1,2,3,6,7,8-HxCDF).
|
|
—
|
Oznaczanie musi być wykonywane zgodnie z metodą EPA 1613, korekta B: tetra- poprzez oktachlorowane dioksyny i furany zastosować rozcieńczenie izotopowe HRGC/HRMS lub inne o równoważnych parametrach wydajności.
|
|
—
|
Różnica między poziomem górnej i dolnej granicy oznaczalności nie może przekraczać 20 % w odniesieniu do pasz o skażeniu dioksynami równym lub wyższym niż dopuszczalny poziom. W przypadku pasz o zawartości dioksyn wyraźnie niższej od dopuszczalnej ich zawartości różnica ta może zawierać się w granicach od 25 do 40 %.
|
7. Skriningowe metody analizy
7.1. Wprowadzenie
Z zastosowaniem metody skriningowej można przeprowadzić różne podejścia analityczne: podejście czysto skriningowe i ilościowe.
Podejście skriningowe
Wartość sygnału uzyskanego w badanej próbce porównuje się z wartością sygnału uzyskanego w próbce referencyjnej zawierającej analit na poziomie zainteresowania. Próbki, dla których wartość sygnału jest niższa niż wartość sygnału próbki referencyjnej, uznaje się za negatywne, natomiast próbki o wyższej wartości sygnału uznaje się za potencjalnie pozytywne. Wymagania:
|
—
|
W każdej partii badanych próbek musi znaleźć się próbka ślepa i referencyjna, które ekstrahuje się i bada w tym samym czasie i w identycznych warunkach. Wartość sygnału uzyskanego w próbce referencyjnej musi być wyraźnie wyższa niż wartość sygnału uzyskanego w próbce ślepej.
|
|
—
|
Należy zbadać dodatkowe próbki referencyjne zawierające 0,5- i 2-krotny poziom zainteresowania, aby wykazać poprawność wykonania badania na poziomie zainteresowania w badaniach kontrolnych.
|
|
—
|
W przypadku badania innych matryc wykazać, że stosowane próbki referencyjne są odpowiednie, szczególnie przez włączenie próbek, dla których z użyciem metody HRGC/HRMS wykazano, że równoważnik toksyczności TEQ jest zbliżony do próbki referencyjnej lub ślepej, wzbogaconej na tym samym poziomie.
|
|
—
|
Ponieważ w testach biologicznych niemożliwe jest zastosowanie wzorców wewnętrznych, szczególnie istotne jest badanie powtarzalności pozwalającej na uzyskanie informacji o odchyleniu standardowym w obrębie jednej partii próbek. Wartość współczynnika zmienności musi być niższa niż 30 %.
|
|
—
|
W przypadku testów biologicznych należy zdefiniować badane związki, możliwe interferencje oraz najwyższą dopuszczalną wielkość sygnału próbki ślepej.
|
Podejście ilościowe
Analiza ilościowa wymaga użycia serii wzorcowych rozcieńczeń, dwu- lub trzystopniowego procesu oczyszczania, pomiarów oraz ślepych prób i odzyskowych kontroli. Wyniki mogą być wyrażone jako równoważnik toksyczności (TEQ), tym samym zakładając, że związki odpowiedzialne za sygnał odpowiadają regule TEQ. Można tego dokonać przez zastosowanie TCDD (lub mieszaniny wzorców dioksyn/furanów/dioksynopodobnych PCB) do sporządzenia krzywej kalibracyjnej umożliwiającej obliczenie poziomu TEQ w ekstrakcie oraz w próbce. Wynik jest następnie korygowany o wartość TEQ uzyskanej w wyniku analizy próbki ślepej (do obliczenia zanieczyszczeń z zastosowanych rozpuszczalników i użytych odczynników) oraz odzysk (obliczony z wartości TEQ oznaczonej w jakościowej próbce kontrolnej w zakresie poziomu zainteresowania). Należy zauważyć, że część obserwowanych strat odzysku może być spowodowana oddziaływaniem matrycy lub różnicami między wartościami współczynników toksyczności (TEF) wykorzystywanymi w testach biologicznych oraz oficjalnymi wartościami TEF przyjętymi przez Światową Organizację Zdrowia (WHO).
7.2. Wymagania dotyczące skriningowych metod analizy
|
—
|
Do badań skriningowych można stosować metody GC/MS oraz testy biologiczne. Należy przestrzegać wymagań dla metod GC/MS, określonych w pkt 6. Wymagania dla biologicznych testów komórkowych są określone w pkt 7.3, a dla zestawów do testów biologicznych w pkt 7.4.
|
|
—
|
Konieczne jest podanie informacji o liczbie wyników fałszywie pozytywnych i wyników fałszywie negatywnych, poniżej i powyżej maksymalnego dopuszczalnego poziomu, uzyskanych w dużych seriach próbek w porównaniu do zawartości TEQ ustalonej z zastosowaniem potwierdzającej metody analizy. Rzeczywista częstość występowania wyników fałszywie negatywnych musi być niższa niż 1 %. Częstość uzyskiwania wyników fałszywie pozytywnych musi być na tyle niska, aby można było wykorzystać zalety metod skriningowych.
|
|
—
|
Wyniki pozytywne należy zawsze potwierdzać z zastosowaniem potwierdzającej metody analizy (HRGC/HRMS). Ponadto próbki z szerokim zakresem TEQ należy potwierdzać HRGC/HRMS (ok. 2–10 % próbek negatywnych). Należy udostępnić informacje na temat korelacji pomiędzy testem biologicznym a wynikami HRGC/HRMS.
|
7.3. Szczegółowe wymagania dotyczące biologicznych testów komórkowych
|
—
|
W czasie wykonywania testów biologicznych przeprowadzenie każdego testu wymaga referencyjnych stężeń TCDD lub mieszaniny dioksyn i furanów (pełna dawka – krzywa reakcji z R2 > 0,95). Do celów badań skriningowych można jednakże zastosować rozszerzoną krzywą niskiego poziomu dla analizy próbek niskopoziomowych.
|
|
—
|
Do oceny sprawności testu biologicznego w stałych odstępach czasu należy stosować referencyjne stężenia TCDD (około trzykrotna granica oznaczalności) nanoszone na karty kontrolne. Alternatywnie można zastosować względną odpowiedź próbki referencyjnej w porównaniu do linii krzywej kalibracyjnej TCDD, ponieważ odpowiedź komórkowa może zależeć od wielu czynników.
|
|
—
|
Dla każdego rodzaju materiału referencyjnego należy prowadzić i sprawdzać wykresy kontroli jakości w celu zapewnienia, że wyniki są zgodne z deklarowanymi wytycznymi.
|
|
—
|
Przy obliczeniach ilościowych należy szczególnie zwracać uwagę, aby zastosowane rozcieńczenie próbki mieściło się w zakresie liniowym krzywej kalibracji. Próbki, których sygnał przekracza liniowy zakres krzywej kalibracji, rozcieńczyć i ponownie poddać oznaczeniu. Dlatego też w czasie oznaczania zaleca się przeprowadzenie co najmniej 3 lub więcej rozcieńczeń.
|
|
—
|
Procentowe odchylenie standardowe nie może być wyższe niż 15 % w trzykrotnym oznaczaniu dla każdego rozcieńczenia próbek i nie może być wyższe niż 30 % dla wyników uzyskanych w trzech niezależnych eksperymentach.
|
|
—
|
Granicę wykrywalności można ustalić jako trzykrotne odchylenie standardowe rozpuszczalnika zerowego lub sygnału tła. Innym podejściem jest wykorzystanie sygnału wyższego od tła (wskaźnik indukcji jest pięciokrotnością rozpuszczalnika zerowego), wyznaczonego na podstawie krzywej kalibracji przygotowanej tego samego dnia. Granicę oznaczalności można ustalić jako 5–6-krotną wartość odchylenia standardowego rozpuszczalnika zerowego lub sygnału tła lub zastosować sygnał powyżej tła (wskaźnik indukcji jest 10-krotnością rozpuszczalnika zerowego), wyznaczony na podstawie krzywej kalibracji przygotowanej tego samego dnia.
|
7.4. Szczegółowe wymagania dotyczące zestawów do testów biologicznych
|
—
|
Zapewnia się odpowiednią czułość i wiarygodność zestawów do testów biologicznych w zakresie stosowania ich do pasz.
|
|
—
|
Należy przestrzegać dostarczonych przez producenta instrukcji przygotowania próbek i sposobu przeprowadzenia ich analizy.
|
|
—
|
Zestawy do testów nie mogą być wykorzystywane po upływie daty ważności.
|
|
—
|
Nie należy stosować materiałów lub składników przeznaczonych do stosowania z innymi zestawami.
|
|
—
|
Zestawy do testów należy przechowywać oraz używać do badań w określonej temperaturze.
|
|
—
|
W przypadku testów immunologicznych granicę wykrywalności określa się jako sumę średniej i trzykrotnego odchylenia standardowego obliczonego na podstawie wyników 10 analiz zerowych podzieloną przez współczynnik nachylenia krzywej wzorcowej wyznaczonej według równania regresji liniowej.
|
|
—
|
W badaniach laboratoryjnych należy stosować wzorce referencyjne w celu kontroli, czy ich odpowiedź mieści się w akceptowanym zakresie.
|
8. Przedstawianie wyników
Jeżeli pozwala na to zastosowana procedura analityczna, wyniki analiz muszą zawierać poziomy poszczególnych kongenerów PCDD/F i PCB oraz być przedstawione zgodnie z dolną, średnią i górną granicą w celu dostarczenia w sprawozdaniu z badań jak największej ilości informacji, umożliwiając tym samym interpretację uzyskanych wyników w zależności od określonych wymagań.
Sprawozdanie musi również zawierać informację o zawartości lipidów w próbce oraz o zastosowanej metodzie ich ekstrakcji.
Na żądanie należy podać informacje na temat odzysku poszczególnych wzorców wewnętrznych, jeżeli wartości odzysku wykraczają poza granice określone w pkt 6, w razie przekroczenia dopuszczalnej zawartości lub w innych przypadkach.
Ponieważ przy ustalaniu zgodności próbki uwzględnia się niepewność pomiaru, parametr ten również należy podawać. Wobec tego wyniki badań należy podawać jako x +/- U, gdzie x oznacza wynik analizy, a U rozszerzoną niepewność pomiaru, przy zastosowaniu współczynnika rozszerzenia w wysokości 2, który daje wynik w przedziale ufności ok. 95 %. W przypadku odrębnego oznaczania dioksyn i dioksynopodobnych PCB należy zastosować sumę szacowanej niepewności rozszerzonej dla odrębnych wyników badań dioksyn i dioksynopodobnych PCB w odniesieniu do sumy dioksyn i dioksynopodobnych PCB.
Jeżeli niepewność pomiaru będzie uwzględniana przez zastosowanie CCα (zob. część B, pkt I.2), parametr ten również należy podać.
(1) Tabela TEF (= współczynników równoważności toksycznej) dla dioksyn, furanów i dioksynopodobnych PCB
|
Kongener
|
Wartość TEF
|
Kongener
|
Wartość TEF
|
|
Dibenzo-p-dioksyny (PCDD)
|
|
Dioksynopodobne PCB:
|
|
|
2,3,7,8-TCDD
|
1
|
|
|
|
1,2,3,7,8-PeCDD
|
1
|
Non-orto PCB
|
|
|
1,2,3,4,7,8-HxCDD
|
0,1
|
PCB 77
|
0,0001
|
|
1,2,3,6,7,8-HxCDD
|
0,1
|
PCB 81
|
0,0001
|
|
1,2,3,7,8,9-HxCDD
|
0,1
|
PCB 126
|
0,1
|
|
1,2,3,4,6,7,8-HpCDD
|
0,01
|
PCB 169
|
0,01
|
|
OCDD
|
0,0001
|
Mono-orto PCB
|
|
|
|
|
PCB 105
|
0,0001
|
|
Dibenzofurany (PCDF)
|
|
PCB 114
|
0,0005
|
|
2,3,7,8-TCDF
|
0,1
|
PCB 118
|
0,0001
|
|
1,2,3,7,8-PeCDF
|
0,05
|
PCB 123
|
0,0001
|
|
2,3,4,7,8-PeCDF
|
0,5
|
PCB 156
|
0,0005
|
|
1,2,3,4,7,8-HxCDF
|
0,1
|
PCB 157
|
0,0005
|
|
1,2,3,6,7,8-HxCDF
|
0,1
|
PCB 167
|
0,00001
|
|
1,2,3,7,8,9-HxCDF
|
0,1
|
PCB 189
|
0,0001
|
|
2,3,4,6,7,8-HxCDF
|
0,1
|
|
|
|
1,2,3,4,6,7,8-HpCDF
|
0,01
|
|
|
|
1,2,3,4,7,8,9-HpCDF
|
0,01
|
|
|
|
OCDF
|
0,0001
|
|
|
|
Użyte skróty oznaczają: T = tetra; Pe = penta; Hx = hexa; Hp = hepta; O = octa; CDD = chlorodibenzo-p-dioksyna; CDF = chlorodibenzofuran; CB = chlorobifenyl.
|
(2) Dz.U. L 140 z 30.5.2002, s. 10.
(3) Koncepcja górnej granicy (upperbound) wymaga podania granicy oznaczalności danego kongeneru do obliczenia TEQ, w przypadku stwierdzenia zawartości tego kongeneru poniżej granicy oznaczalności.
Koncepcja dolnej granicy (lowerbound) wymaga podania zerowej zawartości danego kongeneru do obliczenia TEQ, w przypadku stwierdzenia zawartości tego kongeneru poniżej granicy oznaczalności.
Koncepcja średniej granicy (mediumbound) wymaga podania zawartości równej połowie granicy oznaczalności danego kongeneru do obliczenia TEQ, w przypadku stwierdzenia zawartości tego kongeneru poniżej granicy oznaczalności.
(4) Druga analiza konieczna jest do wykluczenia możliwości wewnętrznego zanieczyszczenia krzyżowego lub przypadkowego wymieszania się próbek. Pierwsza analiza używana jest do weryfikacji zgodności przy uwzględnieniu niepewności pomiaru.
W przypadku analizy wykonywanej w kontekście zanieczyszczenia dioksynami można pominąć drugą analizę, jeżeli próbki wybrane do badania są związane z tym przypadkiem zanieczyszczenia poprzez identyfikowalność.
(5) Dz.U. L 221 z 17.8.2002, s. 8.
ZAŁĄCZNIK VI
METODY ANALIZY DOTYCZĄCE OZNACZANIA SKŁADNIKÓW POCHODZENIA ZWIERZĘCEGO DO CELÓW URZĘDOWEJ KONTROLI PASZ
Warunki dotyczące mikroskopowego wykrywania, identyfikacji lub szacowania występowania składników pochodzenia zwierzęcego w paszach
1. Cel i zakres stosowania metody
Niniejsze warunki stosuje się podczas wykrywania występowania w paszach składników pochodzenia zwierzęcego (zdefiniowanych jako produkty pozyskane podczas przetwarzania tusz lub części tusz ssaków, drobiu i ryb) przeprowadzanego z zastosowaniem badania mikroskopowego w ramach wspólnego programu inspekcji w dziedzinie żywienia zwierząt zgodnie z rozporządzeniem (WE) 882/2004 Parlamentu Europejskiego i Rady (1). O ile we wszystkich badaniach urzędowych stosuje się metody określone w niniejszym załączniku, dodatkowo w celu zwiększenia wykrywalności lub aby określić pochodzenie pewnych rodzajów składników pochodzenia zwierzęcego można przeprowadzić również drugie badanie, stosując metody wariantowe lub alternatywne. W przypadku badania zawartości specyficznych składników pochodzenia zwierzęcego, takich jak badania zawartości plazmy lub kości w łoju (zob. pkt 9), można ponadto zastosować inne postępowanie, o ile badania te są przeprowadzane oprócz analiz przewidzianych we wspólnym programie inspekcji.
2. Czułość
Metodą mogą być wykrywane bardzo małe ilości (< 0,1 %) składników pochodzenia zwierzęcego zależnie od rodzaju tych składników w paszach.
3. Sposób przeprowadzenia metody
Do badań jest wykorzystywana reprezentatywna próbka, pobrana zgodnie z przepisami określonymi w załączniku I i odpowiednio przygotowana. Metoda ma zastosowanie do paszy o niskiej wilgotności. Pasza o wilgotności wyższej niż 14 % musi być przed obróbką wysuszona (zagęszczona). Pewne pasze lub materiały paszowe (np. tłuszcze i oleje) wymagają określonej obróbki (9). Składniki pochodzenia zwierzęcego są identyfikowane na podstawie typowych, mikroskopowo identyfikowalnych cech charakterystycznych (np. włókien mięśniowych i innych cząstek tkanki mięsnej, chrząstki, kości, rogów, włosów, szczeciny, krwi, piór, skorup jaj, ości ryb, łusek). Identyfikacji poddać zarówno frakcję sitową (6.1), jak i zagęszczony osad (6.2).
4. Odczynniki i roztwory
4.1. Odczynniki zanurzeniowe
|
4.1.1.
|
Hydrat chloralu (roztwór wodny, 60 % w/v)
|
|
4.1.2.
|
Ług (NaOH 2,5 %, w/v lub KOH 2,5 %, w/v) do badania frakcji sitowych
|
|
4.1.3.
|
Olej parafinowy lub gliceryna (lepkość: 68–81) do badania osadu pod mikroskopem
|
4.2. Odczynniki chemiczne do płukania
4.3. Odczynnik zagęszczający
|
4.3.1.
|
Czterochloroetylen (gęstość: 1,62)
|
4.4. Odczynniki barwiące
|
4.4.1.
|
Roztwór jodu w jodku potasu (rozpuścić 2 g jodku potasu w 100 ml wody i dodać 1 g jodu, często wstrząsając)
|
|
4.4.2.
|
Czerwień alizarynowa (rozcieńczyć 2,5 ml 1 M kwasu chlorowodorowego w 100 ml wody i dodać 200 mg czerwieni alizarynowej)
|
|
4.4.3.
|
Odczynnik cystynowy (2 g octanu ołowiu, 10 g NaOH/100 ml H2O)
|
|
4.4.4.
|
Roztwór jodu w jodku potasu (rozpuszczony w 70 % etanolu)
|
4.5. Odczynnik bielący
|
4.5.1
|
Handlowy roztwór podchlorynu sodowego będący 9,6 % aktywnym chlorem
|
5. Aparatura i sprzęt
|
5.1.
|
Waga analityczna (dokładność do 0,01 g, a w przypadku badania zagęszczonego osadu – 0,001 g).
|
|
5.2.
|
Sprzęt do rozdrabniania (młynek lub moździerz, szczególnie w przypadku paszy zawierającej > 15 % tłuszczu).
|
|
5.3.
|
Sito o otworach w kształcie kwadratów, o bokach maksimum 0,50 mm.
|
|
5.4.
|
Rozdzielacz lub zlewka osadowa ze stożkowym dnem.
|
|
5.5.
|
Mikroskop stereoskopowy (powiększenie minimum 40 x).
|
|
5.6.
|
Mikroskop optyczny (powiększenie minimum 400 x), światło przechodzące lub spolaryzowane.
|
|
5.7.
|
Standardowe szkło laboratoryjne.
Cały sprzęt szklany należy starannie umyć. Rozdzielacze i szkło wymagają mycia w zmywarce. Sita czyści się z użyciem szczotki o twardym włosiu.
|
6. Sposób postępowania
Granulowane pasze mogą być wstępnie przesiane, jeżeli obie frakcje są analizowane jako oddzielna próbka.
Obróbce poddać co najmniej 50 g próbki, starannie zmielonej, z zastosowaniem sprzętu do rozdrabniania (5.2) jeżeli to konieczne, aby uzyskać odpowiednie rozdrobnienie. Ze zmielonego materiału pobrać dwie reprezentatywne próbki, każda o masie co najmniej 5 g, jedną do badania frakcji sitowej (6.1), a drugą do badania zagęszczonego osadu (6.2). Dodatkowo, aby ułatwić identyfikację, można zastosować barwienie odczynnikami barwiącymi (6.3).
Aby wykazać charakter białek zwierzęcych i pochodzenie cząsteczek, można zastosować pomocniczy system ARIES i udokumentować próbki referencyjne.
6.1. Identyfikacja składników pochodzenia zwierzęcego we frakcjach sitowych
Przesiać przez sito (5.3) co najmniej 5 g próbki w celu otrzymania dwóch frakcji.
Frakcję sitową lub reprezentatywną jej część, zawierającą duże cząstki, rozsypać równomiernie na odpowiednim podłożu i przeglądać systematycznie pod mikroskopem stereoskopowym (5.5) stosując różne powiększenia, poszukując składników pochodzenia zwierzęcego.
Preparaty wykonane z frakcji sitowej drobnej przeglądać systematycznie pod mikroskopem optycznym (5.6), stosując różne powiększenia, poszukując składników pochodzenia zwierzęcego.
6.2. Identyfikacja składników pochodzenia zwierzęcego w zagęszczonym osadzie
Odważyć, z dokładnością do 0,01 g, co najmniej 5 g próbki, przenieść do rozdzielacza lub zlewki osadowej o stożkowym dnie, dodać co najmniej 50 ml czterochloroetylenu (4.3.1). Mieszaninę wielokrotnie wstrząsać lub mieszać.
|
—
|
Jeżeli jest stosowany zamknięty rozdzielacz, osad pozostawić na pewien czas (co najmniej na 3 minuty) przed oddzieleniem osadu. Powtórzyć wytrząsanie i pozostawić osad co najmniej na 3 minuty. Osad musi oddzielić się ponownie.
|
|
—
|
Jeżeli stosuje się zlewkę otwartą, osad należy pozostawić co najmniej na 5 minut, zanim się oddzieli.
|
Cały uzyskany osad wysuszyć i następnie zważyć, z dokładnością do 0,001 g. Ważenie jest konieczne tylko w przypadku ilościowego oznaczania. Jeżeli osad zawiera wiele dużych cząstek, może być przesiany przez sito (5.3) w dwóch frakcjach. Zbadać wysuszony osad na obecność składników kostnych pod mikroskopem stereoskopowym (5.5) i mikroskopem optycznym (5.6).
6.3. Stosowanie odczynników zanurzeniowych i barwiących
Identyfikacja mikroskopowa składników pochodzenia zwierzęcego może być wspomagana przez zastosowanie specjalnych środków zanurzeniowych i odczynników do barwienia
Hydrat chloralu (4.1.1): Ostrożnie ogrzewając, można zobaczyć wyraźniej struktury komórkowe, ponieważ ziarna skrobi żelują i niepożądana zawartość komórek zostaje usunięta.
Ług (4.1.2): Wodorotlenek sodu albo wodorotlenek potasu oczyszcza materiał paszowy, wspomagając wykrycie włókien mięśniowych, włosów i innych keratynowych struktur.
Olej parafinowy i gliceryna (4.1.3): Składniki kostne mogą być dobrze zidentyfikowane, gdyż większość lakun wypełnia się powietrzem i są widoczne jako czarne otwory o rozmiarach od 5 do 15 μm.
Roztwór jodu w jodku potasu (4.4.1): Stosowany do wykrywania zawartości skrobi (kolor niebiesko-fioletowy) i białka (kolor żółto-pomarańczowy). Roztwory można rozcieńczyć, jeżeli to konieczne.
Roztwór czerwieni alizarynowej (4.4.2): Czerwono-różowe zabarwienie kości, ości ryb i łusek. Przed wysuszeniem (6.2) cały osad przenieść do szklanej probówki i przepłukać dwukrotnie około 5 ml alkoholu (4.2.1). Za każdym razem stosować wytrząsarkę typu Vortex, pozostawić rozpuszczalnik przez minutę do osadzenia osadu i następnie go odlać. Przed zastosowaniem tego odczynnika osad wybielić przez dodanie co najmniej 1 ml roztworu podchlorynu sodowego (4.5.1). Pozwolić na przebieg reakcji przez 10 minut. Probówkę napełnić wodą, osad musi osadzać się od 2 do 3 minut, a wodę z zawieszonymi cząsteczkami wylać. Osad przepłukać dwukrotnie około 10 ml wody. Zastosować wytrząsarkę typu Vortex, pozwolić na osadzenie się osadu i za każdym razem wylać wodę. Dodać od 2 do 10 lub więcej kropli w zależności od ilości pozostałego roztworu czerwieni alizarynowej. Mieszaninę wytrząsać i pozwolić na przebieg reakcji przez kilka sekund. Zabarwiony osad przepłukać dwukrotnie około 5 ml alkoholu (4.2.1), a następnie raz acetonem (4.2.2). Za każdym razem stosować wytrząsarkę typu Vortex, doprowadzić do osadzania się rozpuszczalnika przez minutę i wylać go. Osad jest wówczas gotowy do wysuszenia.
Odczynnik cystynowy (4.4.3): Po ostrożnym ogrzewaniu składniki zawierające cystynę, zwłaszcza włosy i pióra, przybierają czarno-brązową barwę.
6.4. Badanie paszy na obecność mączki rybnej
Zbadać pod mikroskopem optycznym co najmniej 1 preparat z frakcji sitowej drobnej i z frakcji osadu (6.1 i 6.2).
Jeżeli na etykiecie jest informacja, że w składzie jest mączka rybna, lub gdy istnieje podejrzenie występowania mączki rybnej lub została ona wykryta w badaniu wstępnym, zbadać dodatkowo co najmniej dwa preparaty z drobnej frakcji sitowej i cały osad.
7. Obliczanie i ocena
Państwa członkowskie zapewniają stosowanie postępowania opisanego w niniejszym ustępie w przypadku przeprowadzania urzędowej analizy mającej na celu ocenę ilościową (a nie tylko obecność) składników pochodzenia zwierzęcego.
Obliczenia mogą być przeprowadzane tylko w przypadku, gdy składniki pochodzenia zwierzęcego zawierają fragmenty kostne.
Fragmenty kostne lądowych gatunków zwierząt stałocieplnych (tj. ssaków i ptaków) można odróżnić od różnych typów ości rybich na podstawie występowania typowych lakun. Proporcję składników pochodzenia zwierzęcego w materiale próbki szacuje się, biorąc pod uwagę:
|
—
|
oszacowaną proporcję (% wagowy) fragmentów kości w zagęszczonym osadzie, oraz
|
|
—
|
proporcję kości (% wagowy) w składnikach pochodzenia zwierzęcego.
|
Szacowania składników pochodzenia zwierzęcego w materiale próbki dokonać z wykorzystaniem co najmniej 3 preparatów, jeżeli to możliwe, zawierających co najmniej 5 pól każdy. W mieszankach paszowych zagęszczony osad zawiera zwykle nie tylko fragmenty kości zwierząt lądowych i fragmenty ości ryb, ale także inne drobiny o specyficznym ciężarze właściwym, m.in. związki mineralne, piasek, fragmenty zdrewniałych roślin.
7.1. Oszacowanie procentowej zawartości fragmentów kości
% zawartość fragmentów kości zwierząt lądowych = (S × c)/W
% zawartość fragmentów ości i łusek ryb = (S × d)/W
(S = masa osadu w mg, c = współczynnik korekcyjny (%) dla szacowanej części kości zwierząt lądowych w osadzie, d = współczynnik korekcyjny (%) dla szacowanej części ości i łusek ryb w osadzie, W = masa w mg materiału próbki służącej do wytworzenia osadu).
7.2. Szacowanie zawartości składników pochodzenia zwierzęcego
Proporcja kości w produktach zwierzęcych może znacznie się zmieniać. Zawartość kości w przypadku mączek kostnych wynosi od 50 do 60 %, a w przypadku mączek mięsnych od 20 do 30 %. W przypadku mączek rybnych zawartości ości i łusek zmieniają się w zależności od kategorii i pochodzenia mączki rybnej i wynoszą zwykle od 10 do 20 %.
Jeżeli rodzaj mączki zwierzęcej obecnej w próbce jest znany, możliwe jest szacunkowe określenie zawartości:
Szacunkowa zawartość składników produktów pochodzących od zwierząt lądowych (%) = (S × c)/(W × f) × 100
Szacunkowa zawartość składników produktów rybnych (%) = (S × d)/(W × f) × 100
(S = masa osadu w mg, c = współczynnik korekcyjny (%) dla szacowanej części kości zwierząt lądowych w osadzie, d = współczynnik korekcyjny (%) dla szacowanej części ości ryb i łusek w osadzie, f = współczynnik korekcyjny (%) dla proporcji kości w składnikach pochodzenia zwierzęcego w badanej próbce, W = masa w mg materiału próbki służącej do wytworzenia osadu).
8. Wyrażanie wyników
Sprawozdanie musi zawierać co najmniej informacje o występowaniu składników pochodzenia zwierzęcego ze zwierząt lądowych i z mączki rybnej. Informacje należy sformułować w podany niżej sposób:
|
8.1.
|
W odniesieniu do występowania składników pochodzących ze zwierząt lądowych:
|
—
|
Nie stwierdzono, z użyciem mikroskopu, w badanej próbce żadnych składników pochodzących ze zwierząt lądowych,
lub
|
|
—
|
Stwierdzono, z użyciem mikroskopu, w badanej próbce składniki pochodzące ze zwierząt lądowych.
|
|
|
8.2.
|
Oraz, w odniesieniu do występowania mączki rybnej:
|
—
|
Nie stwierdzono, z użyciem mikroskopu, w badanej próbce żadnych składników pochodzących z ryb,
lub
|
|
—
|
Stwierdzono, z użyciem mikroskopu, w badanej próbce składniki pochodzące z ryb.
|
W przypadku gdy zostanie stwierdzone występowanie składników pochodzących z ryb lub ze zwierząt lądowych, w sprawozdaniu z badań, jeżeli to konieczne, wskazać oszacowaną ilość znalezionych składników (x %, < 0,1 %, 0,1–0,5 %, 0,5–5 % lub > 5 %), dalszą specyfikację typu zwierzęcia lądowego, jeżeli to jest możliwe, oraz zidentyfikowane składniki zwierzęce (włókna mięśniowe, chrząstkę, kości, rogi, sierść, szczecinę, pióra, krew, skorupy jaj, ości ryb, łuski).
Jeżeli szacuje się ilość składników zwierzęcych, wymienić zastosowany współczynnik korekcyjny f.
Jeżeli są zidentyfikowane składniki kostne zwierząt lądowych, w sprawozdaniu umieścić dodatkową informację o następującej treści:
Nie można wykluczyć, że powyższe składniki pochodzą od ssaków.
Dodatkowa informacja nie jest konieczna w przypadku, gdy fragmenty kostne zwierząt lądowych zostały wyszczególnione jako fragmenty kości drobiu lub ssaków.
|
9. Dodatkowa procedura w przypadku analizy tłuszczu lub oleju
W celu dokonania analizy tłuszczu lub oleju można zastosować następującą procedurę:
|
—
|
Jeżeli tłuszcz występuje w postaci stałej, ogrzewać, np. w kuchence mikrofalowej, aż do uzyskania postaci płynnej.
|
|
—
|
Pipetą przenieść 40 ml tłuszczu z dna próbki do probówki wirówkowej.
|
|
—
|
Wirować przez 10 minut przy 4 000 obr./min.
|
|
—
|
Jeżeli tłuszcz jest zestalony po odwirowaniu, ogrzać go jeszcze raz, aż do uzyskania postaci płynnej. Ponownie wirować przez 5 minut przy 4 000 obr./min.
|
|
—
|
Małą łyżką lub łopatką laboratoryjną przenieść połowę zdekantowanych zanieczyszczeń na płytkę Petriego lub na szkiełko mikroskopowe w celu dokonania mikroskopowej identyfikacji ewentualnej zawartości składników pochodzenia zwierzęcego (włókna mięśniowe, pióra, fragmenty kostne itp.). Jako środek zanurzeniowy stosowany w badaniach mikroskopowych zaleca się olej parafinowy lub glicerynę.
|
|
—
|
Pozostałe zanieczyszczenia poddać sedymentacji w sposób określony w pkt 6.2.
|
(1) Dz.U. L 165 z 30.4.2004, s. 1.
ZAŁĄCZNIK VII
METODA OBLICZANIA WARTOŚCI ENERGETYCZNEJ PASZ DLA DROBIU
1. Metoda obliczania i wyrażania wartości energetycznej
Wartość energetyczna mieszanek paszowych dla drobiu musi być obliczana zgodnie ze wzorem określonym poniżej, na podstawie procentowej zawartości niektórych składników analitycznych paszy. Wartość ta musi być wyrażana w megadżulach (MJ) energii metabolicznej (EM), skorygowanej azotem, na kilogram mieszanki paszowej:
MJ/kg EM = 0,1551 × % białka surowego +0,3431 × % tłuszczu surowego +0,1669 × % skrobi +0,1301 × % całkowitej zawartości cukru (wyrażonego jako sacharoza).
2. Tolerancje mające zastosowanie do podawanych wartości
Jeżeli urzędowa kontrola wykaże rozbieżność (wyższą lub niższą wartość energetyczną paszy) między wynikiem kontroli a podaną wartością energetyczną, dopuszczalna jest tolerancja minimalna wynosząca 0,4 MJ/kg EM.
3. Wyrażanie wyników
Wynik otrzymany po zastosowaniu podanego powyżej wzoru należy podawać z dokładnością do jednego miejsca po przecinku.
4. Pobieranie próbek i metody analizy
Pobieranie próbek mieszanki paszowej oraz określanie zawartości składników analitycznych wskazanych w metodzie obliczania muszą odbywać się odpowiednio zgodnie ze wspólnotowymi metodami pobierania próbek oraz metodami analizy dla urzędowej kontroli pasz.
Należy stosować następujące metody:
|
—
|
do oznaczania zawartości tłuszczu surowego: postępowanie B z metody oznaczania surowych olejów i tłuszczów, określone w załączniku III część H,
|
|
—
|
do oznaczania zawartości skrobi: metoda polarymetryczna, określona w załączniku III część L.
|
ZAŁĄCZNIK VIII
METODY ANALIZY DO CELÓW KONTROLI NIELEGALNEJ OBECNOŚCI W PASZACH DODATKÓW, KTÓRE JUŻ NIE SĄ DOPUSZCZONE
Ważne uwagi:Do wykrywania w paszach dodatków nielegalnych, które już nie są dopuszczone, można stosować bardziej wrażliwe metody analizy, niż określone w niniejszym załączniku.Metody analizy, o których mowa w niniejszym załączniku, należy stosować do celów potwierdzania.
A. OZNACZANIE METYLOBENZOQUATU
7-benzyloksy-6-butylo-3-metoksykarbonylo-4-chinolon
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości metylobenzoquatu w paszach. Granica oznaczalności wynosi 1 mg/kg.
2. Sposób przeprowadzenia metody
Metylobenzoquat ekstrahuje się z próbki metanolowym roztworem kwasu metanosulfonowego. Ekstrakt oczyszcza się dwuchlorometanem metodą chromatografii jonowymiennej, a następnie ponownie dwuchlorometanem. Zawartość metylobenzoquatu jest oznaczana metodą wysokosprawnej chromatografii cieczowej z odwróconą fazą (HPLC) z użyciem detektora UV.
3. Odczynniki i roztwory
|
3.3.
|
Faza ruchoma HPLC.
Mieszanina metanolu, o którym mowa w 3.2, i wody do HPLC, w stosunku 75 + 25 (v+v).
Przefiltrować przez filtr 0,22 μm (4.5) i odgazować roztwór, np. przy zastosowaniu ultradźwięków przez 10 minut.
|
|
3.4.
|
Roztwór kwasu metanosulfonowego, c = 2 %.
Rozcieńczyć 20,0 ml kwasu metanosulfonowego metanolem (3.2) do objętości 1 000 ml.
|
|
3.5.
|
Roztwór kwasu chlorowodorowego, c = 10 %.
Rozcieńczyć 100 ml kwasu chlorowodorowego (ρ20 = 1,18 g/ml) wodą do objętości 1 000 ml.
|
|
3.6.
|
Żywica kationowowymienna Amberlit CG-120 (Na), 100 do 200 oczek.
Żywicę częściowo przygotowuje się przed użyciem, sporządzając zawiesinę ze 100 g żywicy i 500 ml roztworu kwasu chlorowodorowego (3.5), i ogrzewa na płycie grzewczej do wrzenia, ciągle mieszając. Pozostawić do schłodzenia i zdekantować kwasem. Przefiltrować w warunkach próżni na filtrze z bibuły. Żywicę przemyć dwukrotnie 500 ml porcjami wody, a następnie 250 ml metanolu (3.2). Żywicę przemyć ponownie 250 ml metanolu i wysuszyć, przepuszczając powietrze przez warstwę żywicy na filtrze. Wysuszoną żywicę przechowywać w zakorkowanej butelce.
|
|
3.7.
|
Substancja wzorcowa: czysty metylobenzoquat (7-benzyloksy-6-butylo-3-metoksykarbonylo-4-chinolon).
|
|
3.7.1.
|
Roztwór wzorcowy podstawowy metylobenzoquatu, 500 μg/ml
Odważyć, z dokładnością do 0,1 mg, 50 mg substancji wzorcowej (3.7), rozpuścić w roztworze kwasu metanosulfonowego (3.4) w kolbie miarowej o pojemności 100 ml, uzupełnić do pełnej objętości kolby kwasem i zmieszać.
|
|
3.7.2.
|
Rozwór wzorcowy pośredni metylobenzoquatu, 50 μg/ml
Do kolby miarowej o pojemności 50 ml przenieść 5,0 ml podstawowego roztworu wzorcowego metylobenzoquatu (3.7.1) uzupełnić do pełnej objętości kolby metanolem (3.2) i zmieszać.
|
|
3.7.3.
|
Roztwory kalibracyjne
Do kolb miarowych o pojemności 25 ml przenieść kolejno 1,0, 2,0, 3,0, 4,0 i 5,0 ml roztworu wzorcowego pośredniego metylobenzoquatu (3.7.2). Uzupełnić do pełnej objętości kolb fazą ruchomą (3.3) i zmieszać. Roztwory te mają odpowiednio stężenia 2,0, 4,0, 6,0, 8,0 i 10,0 μg/ml metylobenzoquatu. Roztwory te przygotować bezpośrednio przed użyciem.
|
4. Aparatura i sprzęt
|
4.1.
|
Wstrząsarka laboratoryjna
|
|
4.2.
|
Próżniowa wyparka rotacyjna
|
|
4.3.
|
Kolumna szklana (250 mm × 15 mm) z kranem i zbiornikiem o pojemności około 200 ml
|
|
4.4.
|
Sprzęt do HPLC z detektorem UV o zmiennej długości fali miarowej lub z detektorem diodowym
|
|
4.4.1.
|
Kolumna do chromatografii cieczowej: 300 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
4.5.
|
Filtry membranowe, 0,22 μm
|
|
4.6.
|
Filtry membranowe, 0,45 μm
|
5. Sposób postępowania
5.1. Wskazówki ogólne
|
5.1.1.
|
Ślepą próbę paszy należy poddać analizie w celu sprawdzenia, czy nie zawiera ona metylobenzoquatu i substancji interferujących.
|
|
5.1.2.
|
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy fortyfikowaną przez dodanie znanej ilości metylobenzoquatu, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Aby fortyfikować na poziomie 15 mg/kg, dodać 600 μl roztworu wzorcowego podstawowego (3.7.1) do 20 g ślepej próby paszy, zmieszać i odczekać 10 minut do rozpoczęcia ekstrakcji (5.2).
Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności metylobenzoquatu.
|
5.2. Ekstrakcja
Odważyć, z dokładnością do 0,01 g, 20 g przygotowanej próby i umieścić w kolbie stożkowej o pojemności 250 ml. Dodać 100,0 ml roztworu kwasu metanosulfonowego (3.4) i wstrząsać mechanicznie przy zastosowaniu wstrząsarki (4.1) przez 30 minut. Roztwór filtrować przez filtr z bibuły i zachować filtrat do rozdziału w układzie ciecz-ciecz (5.3).
5.3. Rozdział w układzie ciecz-ciecz
Do rozdzielacza o pojemności 500 ml zawierającego 100 ml roztworu kwasu chlorowodorowego (3.5) wprowadzić 25,0 ml otrzymanego wcześniej filtratu (5.2). Dodać 100 ml dwuchlorometanu (3.1) i wstrząsać przez minutę. Pozostawić do rozdzielenia się warstw, a następnie dolną warstwę (dwuchlorometanową) spuścić do kolby okrągłodennej o pojemności 500 ml. Powtórzyć ekstrakcję fazy wodnej, dodając 2 porcje 40 ml dwuchlorometanu, i połączyć je z pierwszym ekstraktem w kolbie okrągłodennej. Odparować ekstrakt dwuchlorometanu do sucha na próżniowej wyparce rotacyjnej (4.2) przy zmniejszonym ciśnieniu w temperaturze 40 oC. Pozostałość rozpuścić w 20 do 25 ml metanolu (3.2), zamknąć kolbę i zachować całość ekstraktu do chromatografii jonowymiennej (5.4).
5.4. Chromatografia jonowymienna
5.4.1. Przygotowanie kolumny kationowowymiennej
Dolny koniec kolumny szklanej (4.3) zatkać zwitkiem waty szklanej. Przygotować zawiesinę 5,0 g żywicy kationitowowymiennej (3.6) i 50 ml roztworu kwasu chlorowodorowego (3.5). Wlać ją do kolumny i pozostawić do ustabilizowania się. Zlać nadmiar kwasu znad powierzchni żywicy i przemywać kolumnę wodą do obojętnego odczynu wycieku wobec lakmusu. Przenieść 50 ml metanolu (3.2) do kolumny i pozwolić, aby przesączył się do powierzchni żywicy.
5.4.2. Chromatografia kolumnowa
Filtrat uzyskany w sposób, o którym mowa w pkt 5.3, przenieść ostrożnie pipetą do kolumny. Kolbę okrągłodenną przemyć 2 porcjami od 5 do 10 ml metanolu (3.2), a następnie roztwór z przemycia nanieść na kolumnę. Poczekać aż ekstrakt dojdzie do powierzchni żywicy, następnie przemyć kolumnę 50 ml metanolu, tak aby prędkość wzbierania ekstraktu nie była wyższa niż 5 ml na minutę. Wyciek odrzucić. Metylobenzoquat wymyć z kolumny przy użyciu 150 ml roztworu kwasu metanosulfonowego (3.4) i zebrać eluent w kolbie stożkowej o pojemności 250 ml.
5.5. Rozdział w układzie ciecz-ciecz
Eluent otrzymany w sposób określony w pkt 5.4.2 przenieść do rozdzielacza o pojemności 1 l. Przemyć kolbę stożkową od 5 do 10 ml metanolu (3.2) i roztwór po przemyciu dołączyć do zawartości rozdzielacza. Dodać 300 ml roztworu kwasu chlorowodorowego (3.5) i 130 ml dwuchlorometanu (3.1). Wstrząsać przez minutę i pozostawić do rozdzielenia faz. Dolną warstwę (dwuchlorometan) przenieść do kolby okrągłodennej o pojemności 500 ml. Powtórzyć ekstrakcję fazy wodnej następnymi 2 porcjami 70 ml dwuchlorometanu i ekstrakty dołączyć do zawartości pierwszej kolby okrągłodennej.
Ekstrakt dwuchlorometanu odparować do sucha na próżniowej wyparce rotacyjnej (4.2) przy zmniejszonym ciśnieniu w temperaturze 40 oC. Pozostałość rozpuścić w kolbie zawierającej około 5 ml metanolu (3.2) i roztwór przenieść ilościowo do kolby miarowej o pojemności 10 ml. Przemyć dwa razy kolbę okrągłodenną następnymi 2 porcjami od 1 do 2 ml metanolu, a następnie przenieść do kolby miarowej. Uzupełnić do pełnej objętości kolby metanolem i zmieszać. Podzielna część jest filtrowana przez filtr membranowy (4.6). Roztwór zachować do oznaczania HPLC (5.6).
5.6. Oznaczanie HPLC
5.6.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników:
|
—
|
kolumna do chromatografii cieczowej (4.4.1),
|
|
—
|
faza ruchoma do HPLC: mieszanina metanolu z wodą (3.3),
|
|
—
|
prędkość przepływu: 1 do 1,5/min,
|
|
—
|
długość fali przy detekcji: 265 nm,
|
|
—
|
dozowana objętość: 20 do 50 μl.
|
Sprawdzić stabilność układu chromatograficznego, dozując kilkakrotnie roztwór kalibracyjny (3.7.3) zawierający 4 μg/ml, aż do ustalenia stałych wysokości (powierzchni) piku i czasów retencji.
5.6.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z kalibracyjnych roztworów (3.7.3) i określić wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików kalibracyjnych roztworów na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.6.3. Badany roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.5) stosując taką samą objętość jak przy roztworach kalibracyjnych, i wyznaczyć średnią wysokość (powierzchnię) piku metylobenzoquatu.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików metylobenzoquatu roztworu próbki określić stężenie roztworu próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.6.2).
Zawartość metylobenzoquatu w (mg/kg) w próbce oblicza się według następującego wzoru:

gdzie:
|
c
|
=
|
stężenie metylobenzoquatu w g/ml w roztworze próbki
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzenie metody
7.1. Sprawdzanie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki i roztworu kalibracyjnego (3.7.3) zawierającego 10 μg/ml metylobenzoquatu.
7.1.1. Kochromatografia
Ekstrakt próbki jest fortyfikowany przez dodanie odpowiedniej ilości pośredniego roztworu wzorcowego (3.7.2). Ilość dodanego metylobenzoquatu musi być podobna do ilości metylobenzoquatu w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i stopnia rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku metylobenzoquatu. Szerokość piku w połowie jego wysokości nie może przekraczać około 10 % pierwotnej szerokości.
7.1.2. Detekcja diodowa
Wyniki sprawdza się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach 2 nm;
|
|
b)
|
w zakresie od 220 do 350 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między dwoma widmami nie jest większa niż 15 % absorbancji wzorcowego analitu;
|
|
c)
|
w zakresie od 220 do 350 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między widmami nie jest większa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać: 10 % względem wyższego wyniku, dla zawartości metylobenzoquatu od 4 do 20 mg/kg.
7.3. Odzysk
W przypadku fortyfikowanej ślepej próby odzysk nie może być mniejszy niż 90 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy 10 laboratoriów przeprowadzono analizę pięciu próbek. Analizę każdej próbki przeprowadzono dwukrotnie.
|
|
Ślepa próba
|
Mączka 1
|
Granulat 1
|
Mączka 2
|
Granulat 2
|
|
Średnia [mg/kg]
|
n.w.
|
4,50
|
4,50
|
8,90
|
8,70
|
|
sr [mg/kg]
|
—
|
0,30
|
0,20
|
0,60
|
0,50
|
|
CVr [%]
|
—
|
6,70
|
4,40
|
6,70
|
5,70
|
|
sR [mg/kg]
|
—
|
0,40
|
0,50
|
0,90
|
1,00
|
|
CVR [%]
|
—
|
8,90
|
11,10
|
10,10
|
11,50
|
|
Odzysk [%]
|
—
|
92,00
|
93,00
|
92,00
|
89,00
|
|
n.w.
|
=
|
nie wykryto
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności w %
|
|
sR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności w %
|
B. OZNACZANIE OLAQUINDOKSU
2-[N-2'-(hydroksyetyl)karbamoil]-3-metylochinoksalino-N1,N4-dioksyd
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości olaquindoksu w paszach. Granica oznaczalności wynosi 5 mg/kg.
2. Sposób przeprowadzenia metody
Próbka jest ekstrahowana mieszaniną wody i metanolu. Zawartość olaquindoksu jest oznaczana metodą wysokosprawnej chromatografii cieczowej (HPLC) z fazą odwróconą przy użyciu detektora UV.
3. Odczynniki i roztwory
|
3.4.
|
Faza ruchoma do HPLC.
Mieszanina wody (3.3) i metanolu (3.2) w stosunku 900 + 100 (V + V).
|
|
3.5.
|
Substancja wzorcowa: czysty olaquindoks 2-[N-2'-(hydroksyetyl)karbamoil]-3-metylochinoksalino-N1,N4-dioksyd, E 851.
|
|
3.5.1.
|
Roztwór wzorcowy podstawowy olaquindoksu, 250 μg/ml
Odważyć, z dokładnością do 0,1 mg, 50 mg olaquindoksu (3.5) do kolby miarowej o pojemności 200 ml i dodać około 190 ml wody. Następnie umieścić kolbę w łaźni ultradźwiękowej (4.1) na 20 minut. Po zastosowaniu ultradźwięków roztwór doprowadzić do temperatury pokojowej, uzupełnić do pełnej objętości kolby wodą i zmieszać. Przykryć kolbę folią aluminiową i przechowywać w lodówce. Roztwór należy przygotowywać raz w miesiącu.
|
|
3.5.2.
|
Roztwór wzorcowy pośredni olaquindoksu, 25 μg/ml
Przenieść 10,0 ml roztworu wzorcowego podstawowego (3.5.1) do kolby miarowej o pojemności 100 ml, uzupełnić do pełnej objętości kolby fazą ruchomą (3.4) i zmieszać. Przykryć kolbę folią aluminiową i przechowywać w lodówce. Roztwór należy przygotowywać codziennie.
|
|
3.5.3.
|
Roztwory kalibracyjne
Do kolb miarowych o pojemności 50 ml przenieść 1,0, 2,0, 5,0, 10,0, 15,0 i 20,0 ml roztworu wzorcowego pośredniego (3.5.2). Uzupełnić do pełnej objętości kolb fazą ruchomą (3.4) i zmieszać. Przykryć kolbę folią aluminiową. Roztwory te odpowiadają odpowiednio 0,5, 1,0, 2,5, 5,0, 7,5, i 10,0 μg olaquindoksu/ml.
Roztwory należy przygotowywać codziennie.
|
4. Aparatura i sprzęt
|
4.1.
|
Łaźnia ultradźwiękowa.
|
|
4.2.
|
Wstrząsarka mechaniczna.
|
|
4.3.
|
Aparat do HPLC z detektorem o zmiennej długości fali lub detektorem diodowym.
|
|
4.3.1.
|
Kolumna do chromatografii cieczowej o wymiarach 250 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
4.4.
|
Filtr membranowy, 0,45 μm.
|
5. Sposób postępowania
|
Uwaga:
|
Olaquindoks jest wrażliwy na światło. Wszystkie czynności należy przeprowadzać przy ograniczonym dostępie światła lub z zastosowaniem szkła oranżowego.
|
5.1. Wskazówki ogólne
|
5.1.1.
|
Ślepą próbę paszy należy poddać analizie w celu potwierdzenia braku olaquindoksu i substancji interferujących.
|
|
5.1.2.
|
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy fortyfikowaną przez dodanie znanej ilości olaquindoksu, o wadze zbliżonej do tej, która znajduje się w próbce. Aby fortyfikować na poziomie 50 mg/kg, przenieść 10,0 ml roztworu wzorcowego podstawowego (3.5.1) do kolby stożkowej o pojemności 250 ml i odparować roztwór do około 0,5 ml. Dodać 50 g ślepej próby paszy, dokładnie zmieszać i pozostawić na 10 minut, kilkakrotnie mieszając przed przejściem do etapu ekstrakcji (5.2).
|
Uwaga:
|
Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności olaquindoksu.
|
|
5.2. Ekstrakcja
Odważyć, z dokładnością do 0,01 g, 50 g próbki. Przenieść do kolby stożkowej o pojemności 1 000 ml, dodać 100 ml metanolu (3.1) i umieścić kolbę na 5 minut w łaźni ultradźwiękowej (4.1). Dodać 410 ml wody i pozostawić w łaźni ultradźwiękowej na 15 minut. Wyjąć kolbę z łaźni i wstrząsać nią przez 30 minut z użyciem wstrząsarki mechanicznej (4.2), następnie przefiltrować przez pofałdowany filtr. Przenieść 10,0 ml filtratu do kolby miarowej o pojemności 20 ml, uzupełnić do pełnej objętości kolby wodą i zmieszać. Przefiltrować podzielną część przez filtr membranowy (4.4) (zob. objaśnienia pkt 9). Przystąpić do oznaczania HPLC (5.3).
5.3. Oznaczanie HPLC
5.3.1. Parametry:
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
|
Kolumna analityczna (4.3.1)
|
|
|
Faza ruchoma (3.4):
|
mieszanina wody (3.3) i metanolu (3.2), 900 + 100 (V + V)
|
|
Prędkość przepływu:
|
1,5–2 ml/min
|
|
Długość fali przy detekcji:
|
380 nm
|
|
Dozowana objętość:
|
20 μl–100 μl
|
Sprawdzić stabilność systemu chromatograficznego, dozując kilkakrotnie roztwór kalibracyjny (3.5.3) zawierający 2,5 μg/ml, aż do uzyskania stałych wysokości piku i czasów retencji.
5.3.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z roztworów kalibracyjnych (3.5.3) i określić średnie wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików roztworów kalibracyjnych na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.3.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.2) stosując taką samą objętość jak przy roztworach kalibracyjnych, i określić średnią wysokość (powierzchnię) piku dla pików olaquindoksu.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików olaquindoksu roztworu próbki określić stężenie roztworu próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.3.2).
Zawartość olaquindoksu w próbce w mg/kg oblicza się według następującego wzoru:

gdzie:
|
c
|
=
|
stężenie olaquindoksu w μg/ml w ekstrakcie próbki (5.2)
|
|
m
|
=
|
masa naważki w g/ml (5.2)
|
7. Sprawdzenie metody
7.1. Sprawdzenie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki (5.2) i roztworu kalibracyjnego (3.5.3) zawierającego 5 μg/ml olaquidoksu.
7.1.1. Kochromatografia
Ekstrakt próbki (5.2) fortyfikuje się przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.5.3). Ilość dodanego olaquindoksu musi odpowiadać ilości olaquindoksu w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku olaquindoksu. Szerokość piku w połowie jego wysokości musi mieścić się w granicach ± 10 % pierwotnej szerokości piku niefortyfikowanego ekstraktu próbki.
7.1.2. Detekcja diodowa
Wyniki ocenia się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach ± 2 nm;
|
|
b)
|
w zakresie pomiędzy 220 a 400 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między dwoma widmami nie jest większa niż 15 % absorbancji wzorcowego agalitu;
|
|
c)
|
w zakresie od 220 do 400 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między widmami nie jest większa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 15 % najwyższego wyniku, dla zawartości olaquindoksu od 10 do 200 mg/kg.
7.3. Odzysk
Odzysk olaquindoksu dodanego do ślepej próby paszy nie może być niższy niż 90 %.
8. Wyniki badań międzylaboratoryjnych
We Wspólnocie Europejskiej przeprowadzono badania porównawcze, w których cztery próbki pasz dla prosiąt, w tym jedna ślepa, były analizowane przez maksymalnie 13 laboratoriów. Wyniki są podane poniżej:
|
|
Próbka 1
|
Próbka 2
|
Próbka 3
|
Próbka 4
|
|
L
|
13
|
10
|
11
|
11
|
|
n
|
40
|
40
|
44
|
44
|
|
Średnia [mg/kg]
|
—
|
14,6
|
48,0
|
95,4
|
|
Sr [mg/kg]
|
—
|
0,82
|
2,05
|
6,36
|
|
SR [mg/kg]
|
—
|
1,62
|
4,28
|
8,42
|
|
CVr [%]
|
—
|
5,6
|
4,3
|
6,7
|
|
CVR [%]
|
—
|
11,1
|
8,9
|
8,8
|
|
Nominalna zawartość
|
|
|
|
|
|
[mg/kg]
|
—
|
15
|
50
|
100
|
|
Odzysk %
|
—
|
97,3
|
96,0
|
95,4
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych wartości
|
|
Sr
|
=
|
odchylenie standardowe powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|
9. Objaśnienia
Metoda nie była sprawdzana dla pasz zawierających więcej niż 100 mg/kg olaquindoksu, zakłada się jednak otrzymanie zadowalających wyników poprzez zmniejszenie odważki analitycznej lub rozcieńczenie ekstraktu (5.2) w celu otrzymania stężenia w zakresie krzywej kalibracyjnej (5.3.2).
C. OZNACZANIE AMPROLIUM
chlorowodorek chlorku 1-[(4-amino-2-propylopirymidyno-5-yl)metyl]-2-metylo-pirydyny
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości amprolium w paszach i premiksach. Granica wykrywalności wynosi 1 mg/kg, granica oznaczalności wynosi 5 mg/kg.
2. Sposób przeprowadzenia metody
Próbka jest ekstrahowana mieszaniną metanolu i wody. Po rozcieńczeniu fazą ruchomą i filtrowaniu przez filtr membranowy zawartość amprolium jest oznaczana metodą kationowo wymienną wysokosprawnej chromatografii cieczowej HPLC przy wykorzystaniu detektora UV.
3. Odczynniki i roztwory
|
3.2.
|
Acetonitryl do HPLC.
|
|
3.4.
|
Roztwór dwuwodorofosforanu sodu, c = 0,1 mol/l.
Rozpuścić 13,80 g monohydratu dwuwodorofosforanu sodu w wodzie (3.3) w kolbie miarowej o pojemności 1 000 ml, uzupełnić do pełnej objętości kolby wodą (3.3) i zmieszać.
|
|
3.5.
|
Roztwór nadchloranu sodu, c = 1,6 mol/l.
Rozpuścić 224,74 g monohydratu nadchloranu sodu w wodzie (3.3) w kolbie miarowej o pojemności 1 000 ml, uzupełnić do pełnej objętości kolby wodą (3.3) i zmieszać.
|
|
3.6.
|
Faza ruchoma do HPLC (zob. objaśnienia pkt 9.1).
Mieszanina, w stosunku 450+450+100 (v+v+v): acetonitrylu (3.2), roztworu dwuwodorofosforanu sodu (3.4), oraz roztworu nadchloranu sodu (3.5). Przed użyciem przefiltrować przez filtr membranowy 0,22 μm (4.3) i odgazować roztwór, np. przy zastosowaniu łaźni ultradźwiękowej (4.4) co najmniej przez 15 minut.
|
|
3.7.
|
Substancja wzorcowa: czyste amprolium, chlorowodorek chlorku 1-[(4-amino-2-propylopirymidyno-5-yl)metyl]-2-metylo-pirydyny, E 750 (zob. pkt 9.2)
|
|
3.7.1.
|
Roztwór wzorcowy podstawowy amprolium, 500 μg/ml
Odważyć, z dokładnością do 0,1 mg, 50 mg amprolium (3.7) do kolby miarowej o pojemności 100 ml, rozpuścić w 80 ml metanolu (3.1) i umieścić kolbę na 10 minut w łaźni ultradźwiękowej (4.4). Następnie doprowadzić roztwór do temperatury pokojowej, uzupełnić do pełnej objętości kolby wodą i zmieszać. Roztwór przechowywany w temperaturze ≤ 4 oC zachowuje stabilność nie dłużej niż miesiąc.
|
|
3.7.2.
|
Pośredni wzorcowy roztwór amprolium, 50 μg/ml
Przenieść pipetą 5,0 ml roztworu wzorcowego podstawowego (3.7.1) do kolby miarowej o pojemności 50 ml, uzupełnić do pełnej objętości kolby roztworem do ekstrakcji (3.8) i zmieszać. Roztwór przechowywany w temperaturze ≤ 4 oC zachowuje stabilność nie dłużej niż miesiąc.
|
|
3.7.3.
|
Roztwory kalibracyjne
Przenieść 0,5, 1,0 i 2,0 ml pośredniego wzorcowego roztworu (3.7.2) do partii kolb miarowych o pojemności 50 ml. Uzupełnić do pełnej objętości kolb fazą ruchomą (3.6) i zmieszać. Roztwory odpowiadają odpowiednio 0,5, 1,0 i 2,0 μg/ml amprolium. Roztwory sporządzić bezpośrednio przed użyciem.
|
|
3.8.
|
Rozpuszczalnik ekstrakcyjny.
Mieszanina metanolu (3.1) i wody, 2 + 1 (v + v)
|
4. Aparatura i sprzęt
|
4.1.
|
Zestaw do HPLC z dozownikiem umożliwiającym dozowanie 100 μl cieczy.
|
|
4.1.1.
|
Kolumna do chromatografii cieczowej o wymiarach 125 mm × 4 mm, z Nucleosil 10 SA, wypełnienie 5 lub 10 μm, lub równoważne
|
|
4.1.2.
|
Detektor UV z regulacją długości fal lub detektor diodowy
|
|
4.2.
|
Filtr membranowy, PTFE, 0,45 μm.
|
|
4.3.
|
Filtr membranowy, 0,22 μm.
|
|
4.4.
|
Łaźnia ultradźwiękowa.
|
|
4.5.
|
Wstrząsarka mechaniczna lub mieszadło magnetyczne.
|
5. Sposób postępowania
5.1. Wskazówki ogólne
5.1.1. Ślepa próba paszy
W celu zbadania odzysku (5.1.2) należy przeprowadzić analizę ślepej próby paszy, aby sprawdzić, czy nie zawiera ona amprolium ani substancji interferujących. Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności amprolium lub substancji interferujących.
5.1.2 Badanie odzysku
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy fortyfikowaną przez dodanie znanej ilości amprolium, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Aby fortyfikować na poziomie 100 mg/kg, dodać 10,0 ml roztworu wzorcowego podstawowego (3.7.1) do kolby stożkowej o pojemności 250 ml i odparować roztwór do objętości około 0,5 ml. Dodać 50 g ślepej próby paszy, zmieszać i pozostawić na 10 minut, mieszając kilkakrotnie przed przejściem do etapu ekstrakcji (5.2).
Jeżeli nie można przeprowadzić badania ślepej próby paszy podobnej do badanej próbki (5.1.1), badanie odzysku można przeprowadzić, stosując metodę dodawania wzorca. W tym przypadku przeznaczona do analizy próbka jest fortyfikowana przez dodanie znanej ilości amprolium, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Próbkę tę poddaje się analizie wraz z niefortyfikowaną próbką, a odzysk może być obliczony przez odjęcie od próbki fortyfikowanej masy próbki niefortyfikowanej.
5.2. Ekstrakcja
5.2.1. Premiksy (zawartość amprolium < 1 %) i pasze
Odważyć, z dokładnością do 0,01 g, od 5 do 40 g próbki, w zależności od zawartości amprolium, do kolby stożkowej o pojemności 500 ml i dodać 200 ml rozpuszczalnika ekstrakcyjnego (3.8). Umieścić kolbę w łaźni ultradźwiękowej (4.4) na 15 minut. Wyjąć kolbę z łaźni i wstrząsać nią przez godzinę na wstrząsarce lub mieszadle magnetycznym (4.5). Rozcieńczyć podzielną część ekstraktu fazą ruchomą (3.6) do zawartości amprolium od 0,5 do 2 μg/ml i zmieszać (zob. objaśnienia pkt 9.3). Przefiltrować od 5 do 10 ml rozcieńczonego roztworu przez filtr membranowy (4.2). Przystąpić do oznaczania HPLC (5.3).
5.2.2. Premiksy (zawartość amprolium ≥ 1 %)
Odważyć, z dokładnością do 0,001 g, od 1 do 4 g premiksu, w zależności od zawartości amprolium, do kolby stożkowej o pojemności 500 ml, dodać 200 ml rozpuszczalnika ekstrakcyjnego (3.8). Umieścić kolbę w łaźni ultradźwiękowej (4.4) na 15 minut. Wyjąć kolbę z łaźni i wstrząsać nią przez godzinę na wstrząsarce lub mieszadle magnetycznym (4.5). Rozcieńczyć podzielną część ekstraktu fazą ruchomą (3.6) do zawartości amprolium od 0,5 do 2 μg/ml i zmieszać. Przefiltrować od 5 do 10 ml rozcieńczonego roztworu przez filtr membranowy (4.2). Przystąpić do oznaczania HPLC (5.3).
5.3. Oznaczanie HPLC
5.3.1. Parametry:
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników.
|
Kolumna do chromatografii
|
|
|
cieczowej (4.1.1):
|
125 mm × 4 mm, wymienny kation Nucleosil 10 SA, wypełnienie 5 lub 10 μm lub równoważne
|
|
Faza ruchoma (3.6):
|
Mieszanina acetonitrylu (3.2),roztworu dwuwodorofosforanu sodu (3.4) i roztworu nadchloranu sodu (3.5), 450+450+100 (v+v+v).
|
|
Prędkość przepływu:
|
0,7–1 ml/min
|
|
Długość fali przy detekcji:
|
264 nm
|
|
Dozowana objętość:
|
100 μl
|
Sprawdzić stabilność systemu chromatograficznego, dozując kilkakrotnie roztwór kalibracyjny (3.7.3) zawierający 1,0 μg/ml, aż do uzyskania stałych wysokości piku i czasów retencji.
5.3.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z roztworów kalibracyjnych (3.7.3) i określić średnie wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików roztworów kalibracyjnych na osi rzędnych i odpowiadające im stężenia w μg/ml na osi odciętych.
5.3.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki (5.2), stosując taką samą objętość jak przy kalibracyjnych roztworach, i określić średnią wysokość (powierzchnię) piku dla pików amprolium.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików amprolium roztworu próbki określić stężenie roztworu próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.3.2).
Zawartość amprolium w mg/kg próbki obliczyć według następującego wzoru:
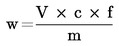 [mg/kg]
[mg/kg]
gdzie:
|
V
|
=
|
objętość rozpuszczalnika ekstrakcyjnego w ml (3.8); zgodnie z pkt 5.2 (np. 200 ml)
|
|
c
|
=
|
stężenie amprolium w μg/ml w ekstrakcie próbki (5.2)
|
|
f
|
=
|
współczynnik rozcieńczania zgodny z pkt 5.2
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzanie metody
7.1. Sprawdzenie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki (5.2) i roztworu kalibracyjnego (3.7.3) zawierającego 2,0 μg/ml.
7.1.1. Kochromatografia
Ekstrakt próbki (5.2) fortyfikuje się przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.7.3). Ilość dodanego amprolium musi odpowiadać ilości amprolium znajdującego się w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku amprolium. Szerokość piku w połowie jego wysokości musi mieścić się w granicach ± 10 % pierwotnej szerokości piku amprolium niefortyfikowanego ekstraktu próbki.
7.1.2. Detekcja diodowa
Wyniki sprawdza się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach ±2 nm;
|
|
b)
|
w zakresie od 210 do 320 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między dwoma widmami nie jest większa niż 15 % absorbancji wzorcowego agalitu;
|
|
c)
|
w zakresie od 210 do 320 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między widmami nie jest większa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać:
|
—
|
15 % względem wyższej wartości, dla zawartości amprolium od 25 mg/kg do 500 mg/kg,
|
|
—
|
75 mg/kg, dla zawartości amprolium od 500 mg/kg do 1 000 mg/kg,
|
|
—
|
7,5 % względem wyższej wartości, dla zawartości amprolium powyżej 1 000 mg/kg.
|
7.3. Odzysk
W przypadku fortyfikacji (ślepej) próby paszy odzysk musi wynosić co najmniej 90 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy kilku laboratoriów przeprowadzono analizę 3 pasz dla drobiu (próbki 1–3), paszy mineralnej (próbka 4) i premiksu (próbka 5). W poniższej tabeli podano wyniki przeprowadzonych badań.
|
|
Próbka 1 (ślepa)
|
Próbka 2
|
Próbka 3
|
Próbka 4
|
Próbka 5
|
|
L
|
14
|
14
|
14
|
14
|
15
|
|
n
|
56
|
56
|
56
|
56
|
60
|
|
Średnia [mg/kg]
|
—
|
45,5
|
188
|
5 129
|
25 140
|
|
sr [mg/kg]
|
—
|
2,26
|
3,57
|
178
|
550
|
|
CVr [%]
|
—
|
4,95
|
1,90
|
3,46
|
2,20
|
|
sR [mg/kg]
|
—
|
2,95
|
11,8
|
266
|
760
|
|
CVR [%]
|
—
|
6,47
|
6,27
|
5,19
|
3,00
|
|
Zawartość nominalna [mg/kg]
|
—
|
50
|
200
|
5 000
|
25 000
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych oznaczeń
|
|
sr
|
=
|
odchylenie standardowe powtarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
sR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|
9. Objaśnienia
|
9.1.
|
Jeżeli próbka zawiera tiaminę, pik tiaminy na chromatogramie ukazuje się na krótko przed pikiem amprolium. W tej metodzie amprolium i tiamina należy rozdzielić. Jeżeli amprolium i tiamina nie zostaną rozdzielone w kolumnie (4.1.1) użytej w tej metodzie, zastąpić do 50 % acetonitrylu fazy ruchomej (3.6) metanolem.
|
|
9.2.
|
Według Farmakopei Brytyjskiej widmo roztworu amprolium: (c = 0,02 mol/l) w kwasie chlorowodorowym (c = 0,1 mol/l) wykazuje maksima przy 246 nm i 262 nm. Absorbancja musi wynieść 0,84 przy 246 nm i 0,80 przy 262 nm.
|
|
9.3.
|
Ekstrakt zawsze rozcieńczyć fazą ruchomą, gdyż w przeciwnym razie czas retencji piku amprolium może się znacznie przesunąć, z uwagi na zmiany siły jonowej.
|
D. OZNACZANIE KARBADOKSU
Metylo 3-(2-chinoksalinylometyleno)pikrynian N
1
,N
4
-dwutlenek
1. Cel i zakres stosowania metody
Metoda służy do oznaczania zawartości karbadoksu w paszach, premiksach i preparatach. Granica wykrywalności wynosi 1 mg/kg. Granica oznaczalności wynosi 5 mg/kg.
2. Sposób przeprowadzenia metody
Próbka jest równoważona wodą i poddawana ekstrakcji przy użyciu mieszaniny metanolu i acetonitrylu. W przypadku pasz podzielną część przefiltrowanego ekstraktu zostaje oczyszczona na kolumnie z tlenkiem glinu. W przypadku premiksów i preparatów podzielną część przefiltrowanego ekstraktu zostaje rozcieńczona do odpowiedniego stężenia wodą, metanolem i acetonitrylem. Zawartość karbadoksu oznacza się metodą wysokosprawnej chromatografii cieczowej w układzie faz odwróconych (HPLC) przy użyciu detektora UV.
3. Odczynniki i roztwory
|
3.2.
|
Acetonitryl do HPLC.
|
|
3.3.
|
Kwas octowy, w = 100 %.
|
|
3.4.
|
Tlenek glinu: obojętny, stopień aktywności I.
|
|
3.5.
|
Metanol-acetonitryl 1 + 1 (v + v).
Zmieszać 500 ml metanolu (3.1) z 500 ml acetonitrylu (3.2).
|
|
3.6.
|
Kwas octowy, σ = 10 %.
Rozcieńczyć 10 ml kwasu octowego (3.3) wodą do objętości 100 ml.
|
|
3.9.
|
Roztwór buforu octanowego, c = 0,01 mol/l, pH = 6,0.
Rozpuścić 0,82 g octanu sodu (3.7) w 700 ml wody (3.8) i dostosować pH do 6 kwasem octowym (3.6). Przenieść do kolby miarowej o pojemności 1000 ml, uzupełnić do pełnej objętości kolby wodą (3.8) i zmieszać.
|
|
3.10.
|
Faza ruchoma do HPLC.
Zmieszać 825 ml roztworu buforu octanowego (3.9) i 175 ml acetonitrylu (3.2).
Przefiltrować przez filtr 0,22 μm (4.5) i odgazować roztwór, np. przy zastosowaniu łaźni ultradźwiękowej przez 10 minut.
|
|
3.11.
|
Substancja wzorcowa.
Czysty karbadoks: Metylo 3-(2-chinoksalinylometyleno)pikrynianN1, N4-dwutlenek, E 850.
|
|
3.11.1.
|
Roztwór wzorcowy podstawowy karbadoksu, 100 μg/ml, (zob. uwaga pkt 5). Sposób postępowania:
Odważyć, z dokładnością do 0,1 mg, 25 mg substancji wzorcowej (3.11) do kolby miarowej o pojemności 250 ml. Rozpuścić w mieszaninie metanolu i acetonitrylu (3.5) na łaźni ultradźwiękowej (4.7). Po zastosowaniu ultradźwięków doprowadzić roztwór do temperatury pokojowej, uzupełnić do pełnej objętości kolby mieszaniną metanolu i acetonitrylu (3.5) i zmieszać. Kolbę owinąć folią aluminiową lub zastosować oranżowe szklane naczynie laboratoryjne i przechowywać w lodówce. Roztwór w temperaturze ≤ 4 oC zachowuje stabilność do miesiąca.
|
|
3.11.2.
|
Roztwory kalibracyjne
Przenieść 2,0, 5,0, 10,0 i 20,0 ml roztworu wzorcowego podstawowego karbadoksu (3.11.1) do partii kolb miarowych o pojemności 100 ml. Dodać 30 ml wody, uzupełnić mieszaniną metanolu i acetonitrylu (3.5) do pełnej objętości kolb i zmieszać. Kolby owinąć folią aluminiową. Roztwory te odpowiadają odpowiednio 2,0, 5,0, 10,0 i 20,0 μg/ml karbadoksu.
Roztwory kalibracyjne sporządza się bezpośrednio przed użyciem.
|
Uwaga:
|
Do oznaczenia karbadoksu w paszach zawierających mniej niż 10 mg/kg sporządza się roztwory kalibracyjne o stężeniu niższym niż 2,0 μg/ml.
|
|
|
3.12.
|
Mieszanina wody i mieszanina metanolu z acetonitrylem (3.5), 300 + 700 (v + v)
Zmieszać 300 ml wody z 700 ml mieszaniny metanolu i acetonitrylu (3.5).
|
4. Aparatura i sprzęt
|
4.1.
|
Wstrząsarka laboratoryjna lub mieszadło magnetyczne.
|
|
4.2.
|
Bibuła filtracyjna z włóknem szklanym (Whatman GF/A lub podobna).
|
|
4.3.
|
Szklana kolumna o długości od 300 do 400 mm, wewnętrznej średnicy około 10 mm, ze spiekiem szklanym oraz zaworem odpływowym.
|
Uwaga:
|
Dopuszcza się użycie szklanej kolumny z kurkiem lub ze ściętym końcem i w takim przypadku w dolnym końcu umieszcza się zwitek waty szklanej, ubity szklanym prętem.
|
|
|
4.4.
|
Sprzęt do HPLC z systemem do dozowania umożliwiającym dozowanie 20 μl.
|
|
4.4.1.
|
Kolumna do chromatografii cieczowej o wymiarach 300 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
4.4.2.
|
Detektor UV z regulacją długości fal lub detektor diodowy pracujący w zakresie do 225 do 400 nm
|
|
4.5.
|
Filtr membranowy, 0,22 μm.
|
|
4.6.
|
Filtr membranowy, 0,45 μm.
|
|
4.7.
|
Łaźnia ultradźwiękowa.
|
5. Sposób postępowania
|
Uwaga:
|
Karbadoks jest wrażliwy na światło. Wszystkie czynności należy przeprowadzać przy ograniczonym dostępie światła lub z zastosowaniem szkła oranżowego lub szkła zabezpieczonego folią aluminiową.
|
5.1. Wskazówki ogólne
5.1.1. Ślepa próba paszy
Do badania odzysku (5.1.2) należy przeprowadzić analizę ślepej próby paszy w celu sprawdzenia, czy nie zawiera ona karbadoksu i substancji interferujących. Ślepa próba paszy musi być podobna do badanej próbki, a jej analiza nie może potwierdzać obecności karbadoksu i substancji interferujących.
5.1.2. Badanie odzysku
Badanie odzysku należy przeprowadzić, analizując ślepą próbę paszy (5.1.1) fortyfikowaną przez dodanie znanej ilości karbadoksu, o wadze zbliżonej do tej, która znajduje się w próbce. Aby fortyfikować na poziomie 50 mg/kg, dodać 5,0 ml roztworu wzorcowego podstawowego karbadoksu (3.11.1) do kolby stożkowej o pojemności 200 ml. Odparować roztwór w strumieniu azotu do objętości około 0,5 ml. Dodać 10 g ślepej próby paszy, zmieszać i pozostawić na 10 minut przed przejściem do etapu ekstrakcji (5.2).
Jeżeli nie można przeprowadzić badania ślepej próby paszy podobnej do badanej próbki (zob. pkt 5.1.1) badanie odzysku można przeprowadzić, stosując metodę dodawania wzorca. W tym przypadku przeznaczona do analizy próbka jest fortyfikowana przez dodanie znanej ilości karbadoksu, o wadze zbliżonej do tej, która znajduje się w analizowanej próbce. Próbkę tę poddaje się analizie wraz z niefortyfikowaną próbką, a odzysk może być obliczony przez odjęcie od próbki fortyfikowanej masy próbki niefortyfikowanej.
5.2. Ekstrakcja
5.2.1. Pasze
Odważyć, z dokładnością do 0,01 g, 10 g próbki i przenieść do kolby stożkowej o pojemności 200 ml. Dodać 15,0 ml wody, zmieszać i równoważyć przez 5 minut. Dodać 35,0 ml mieszaniny metanolu i acetonitrylu (3.5), zamknąć korkiem i wstrząsać przez 30 minut z zastosowaniem wstrząsarki lub mieszadła magnetycznego (4.1). Przefiltrować roztwór przez bibułę filtracyjną z włóknem szklanym (4.2). Roztwór zachowuje się do etapu oczyszczania (5.3).
5.2.2. Premiksy (zawartość karbadoksu od 0,1 do 2 %)
Odważyć, z dokładnością do 0,001 g, 1 g niezmielonej próbki i przenieść do kolby stożkowej o pojemności 200 ml. Dodać 15,0 ml wody, zmieszać i równoważyć przez 5 minut. Dodać 35,0 ml mieszaniny metanolu i acetonitrylu (3.5), zamknąć korkiem i wstrząsać przez 30 minut z użyciem wstrząsarki lub mieszadła magnetycznego (4.1). Przefiltrować ten roztwór przez bibułę filtracyjną z włóknem szklanym (4.2).
Pipetą przenieść podzielną część filtratu do kolby miarowej o pojemności 50 ml. Dodać 15,0 ml wody, uzupełnić do pełnej objętości kolby mieszaniną metanolu z acetonitrylem (3.5) i zmieszać. Stężenie karbadoksu w roztworze końcowym musi wynieść około 10 μg/ml. Podzielną część roztworu przefiltrować przez filtr 0,45 μm (4.6).
Przystąpić do oznaczania HPLC (5.4).
5.2.3. Preparaty (zawartość karbadoksu > 2 %)
Odważyć, z dokładnością do 0,001 g, 0,2 g niezmielonej próbki i przenieść do kolby stożkowej o pojemności 250 ml. Dodać 45,0 ml wody, zmieszać i równoważyć przez 5 minut. Dodać 105,0 ml mieszaniny metanolu z acetonitrylem (3.5), zamknąć korkiem i homogenizować. Poddać próbkę działaniu ultradźwięków przez 15 minut (4.7), a następnie wstrząsać z zastosowaniem wstrząsarki lub mieszadła magnetycznego (4.1) przez 15 minut. Przefiltrować roztwór przez bibułę filtracyjną z włóknem szklanym (4.2).
Rozcieńczyć podzielną część filtratu mieszaniną wody i mieszaniną metanolu z acetonitrylem (3.12), aby uzyskać ostateczne stężenie karbadoksu od 10 do 15 μg/ml (w przypadku preparatów 10 %, współczynnik rozcieńczania wynosi 10). Podzielną część przefiltrować przez filtr (4.6).
Przystąpić do analizy HPLC (5.4).
5.3. Oczyszczanie
5.3.1. Przygotowanie kolumny tlenku glinu
Odważyć 4 g tlenku glinu (3.4) i przenieść do szklanej kolumny (4.3).
5.3.2. Czyszczenie próbki
Umieścić 15 ml odfiltrowanego ekstraktu (5.2.1) w kolumnie z tlenkiem glinu i usunąć pierwsze 2 ml eluentu. Następnie zebrać kolejne 5 ml i przefiltrować podzielną część przez filtr 0,45 μm (4.6).
Przystąpić do oznaczania HPLC (5.4).
5.4. Oznaczanie HPLC
5.4.1. Parametry
Poniższe parametry podano jako przykładowe; inne parametry mogą być zastosowane, o ile gwarantują otrzymanie równoważnych wyników:
|
Kolumna do chromatografii
|
|
|
cieczowej (4.4.1):
|
300 mm × 4 mm, C18, wypełnienie 10 μm lub równoważne
|
|
Faza ruchoma (3.10):
|
Mieszanina roztworu buforu octanowego (3.9) i acetonitrylu (3.2), 825 + 175 (v + v)
|
|
Prędkość przepływu:
|
1,5–2 ml/min
|
|
Długość fali przy detekcji:
|
365 nm
|
|
Dozowana objętość:
|
20 μl
|
Sprawdzić stabilność systemu chromatograficznego, dozując kilka razy roztwór kalibracyjny (3.11.2) zawierający 5,0 μg/ml, aż do uzyskania stałych wysokości (powierzchni) piku i czasów retencji.
5.4.2. Krzywa kalibracyjna
Zadozować kilka razy każdy z roztworów kalibracyjnych (3.11.2) i określić wysokości (powierzchnie) piku dla każdego stężenia. Wykreślić krzywą kalibracyjną, odkładając średnie wysokości (powierzchnie) pików kalibracyjnych roztworów na osi rzędnych oraz odpowiadające im stężenia w μg/ml na osi odciętych.
5.4.3. Roztwór próbki
Zadozować kilka razy ekstrakt próbki otrzymany dla pasz (5.3.2), dla premiksów (5.2.2) oraz dla preparatów (5.2.3), i określić średnią wysokość (powierzchnię) piku dla pików karbadoksu.
6. Obliczanie wyników
Na podstawie średniej wysokości (powierzchni) pików karbadoksu roztworu próbki, określić stężenie karbadoksu w roztworze próbki w μg/ml, odnosząc się do krzywej kalibracyjnej (5.4.2).
6.1. Pasze
Zawartość karbadoksu w próbce, wyrażoną w mg/kg, oblicza się według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
c
|
=
|
stężenie karbadoksu w ekstrakcie próbki w μg/ml (5.3.2)
|
|
V1
|
=
|
objętość ekstraktu w ml (np. 50 ml)
|
|
m
|
=
|
masa naważki w g
|
6.2. Premiksy i preparaty
Zawartość karbadoksu w próbce, wyrażoną w mg/kg, oblicza się według następującego wzoru:
 [mg/kg]
[mg/kg]
gdzie:
|
c
|
=
|
stężenie karbadoksu w ekstrakcie próbki w μg/ml (5.2.2 lub 5.2.3)
|
|
V2
|
=
|
objętość ekstraktu w ml (np. 50 ml w przypadku premiksów lub 150 ml w przypadku preparatów)
|
|
f
|
=
|
współczynnik rozcieńczenia zgodny z pkt 5.2.2 (premiksy) lub z pkt 5.2.3 (preparaty)
|
|
m
|
=
|
masa naważki w g
|
7. Sprawdzenie metody
7.1. Sprawdzenie tożsamości
Tożsamość analitu może być potwierdzona przez kochromatografię lub zastosowanie detektora diodowego, przy użyciu którego porównuje się widma ekstraktu próbki i roztworu kalibracyjnego (3.11.2) zawierającego 10,0 μg/ml karbadoksu.
7.1.1. Kochromatografia
Ekstraky próbki fortyfikuje się przez dodanie odpowiedniej ilości roztworu kalibracyjnego (3.11.2). Ilość dodanego karbadoksu musi odpowiadać ilości karbadoksu w ekstrakcie próbki.
Po uwzględnieniu zarówno dodanej ilości, jak i stopnia rozcieńczenia ekstraktu, zwiększyć się może tylko wysokość piku karbadoksu. Szerokość piku w połowie jego wysokości musi mieścić się w granicach ok. 10 % pierwotnej szerokości.
7.1.2. Detekcja diodowa
Wyniki ocenia się według następujących kryteriów:
|
a)
|
długość fali odpowiadająca maksymalnej absorpcji próbki i widm wzorcowych zapisana w punkcie wierzchołka piku zarejestrowanego na chromatogramie musi być taka sama, po uwzględnieniu marginesu określonego zdolnością rozdzielczą systemu detekcji. W przypadku detekcji diodowej zdolność rozdzielcza mieści się zazwyczaj w granicach ±2 nm;
|
|
b)
|
w zakresie pomiędzy 225 a 400 nm próbka i widma wzorcowe zapisane w punkcie wierzchołka piku na chromatogramie nie mogą się różnić dla tych części widma w zakresie od 10 do 100 % względnej absorbancji. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między dwoma widmami nie jest wyższa niż 15 % absorbancji wzorcowego analitu;
|
|
c)
|
w zakresie od 225 do 400 nm widma wartości rosnących, szczytowych i malejących ekstraktu próbki nie mogą różnić się od siebie dla tych części widma w zakresie od 10 do 100 % absorbancji względnej. Kryterium uważa się za spełnione, jeżeli występują te same maksymalne wartości i w ramach żadnego z obserwowanych punktów rozbieżność między widmami nie jest wyższa niż 15 % widma wartości szczytowej.
|
Jeżeli jedno z tych kryteriów nie jest spełnione, obecność analitu nie jest potwierdzona.
7.2. Powtarzalność
Dla zawartości karbadoksu równej lub wyższej niż 10 mg/kg różnica pomiędzy wynikami dwóch równoległych oznaczeń wykonanych dla tej samej próbki nie może przekraczać 15 % najwyższego wyniku.
7.3. Odzysk
W przypadku fortyfikacji (ślepej) próby paszy odzysk musi wynosić co najmniej 90 %.
8. Wyniki badań międzylaboratoryjnych
W ramach współpracy 8 laboratoriów przeprowadzono analizę 6 próbek pasz, 4 premiksów i 3 preparatów. Analizę każdej próbki przeprowadzono dwukrotnie. (Bardziej szczegółowe informacje dotyczące tych badań można znaleźć w Journal of the AOAC, tom 71, 1988, s. 484–490). Wyniki (z wyłączeniem wartości oddalonych) podano poniżej:
Tabela 1
Wyniki badań porównawczych dla pasz
|
|
Próbka 1
|
Próbka 2
|
Próbka 3
|
Próbka 4
|
Próbka 5
|
Próbka 6
|
|
L
|
8
|
8
|
8
|
8
|
8
|
8
|
|
n
|
15
|
14
|
15
|
15
|
15
|
15
|
|
Średnia (mg/kg)
|
50,0
|
47,6
|
48,2
|
49,7
|
46,9
|
49,7
|
|
Sr (mg/kg)
|
2,90
|
2,69
|
1,38
|
1,55
|
1,52
|
2,12
|
|
CVr (%)
|
5,8
|
5,6
|
2,9
|
3,1
|
3,2
|
4,3
|
|
SR (mg/kg)
|
3,92
|
4,13
|
2,23
|
2,58
|
2,26
|
2,44
|
|
CVR (%)
|
7,8
|
8,7
|
4,6
|
5,2
|
4,8
|
4,9
|
|
Zawartość nominalna (mg/kg)
|
50,0
|
50,0
|
50,0
|
50,0
|
50,0
|
50,0
|
Tabela 2
Wyniki badań porównawczych dla premiksów i preparatów
|
|
Premiksy
|
Preparaty
|
|
A
|
B
|
C
|
D
|
A
|
B
|
C
|
|
L
|
7
|
7
|
7
|
7
|
8
|
8
|
8
|
|
n
|
14
|
14
|
14
|
14
|
16
|
16
|
16
|
|
Średnia (mg/kg)
|
8,89
|
9,29
|
9,21
|
8,76
|
94,6
|
98,1
|
104
|
|
Sr (g/kg)
|
0,37
|
0,28
|
0,28
|
0,44
|
4,1
|
5,1
|
7,7
|
|
CVr (%)
|
4,2
|
3,0
|
3,0
|
5,0
|
4,3
|
5,2
|
7,4
|
|
SR (g/kg)
|
0,37
|
0,28
|
0,40
|
0,55
|
5,4
|
6,4
|
7,7
|
|
CVR (%)
|
4,2
|
3,0
|
4,3
|
6,3
|
5,7
|
6,5
|
7,4
|
|
Zawartość nominalna (g/kg)
|
10,0
|
10,0
|
10,0
|
10,0
|
100
|
100
|
100
|
|
L
|
=
|
liczba laboratoriów
|
|
n
|
=
|
liczba pojedynczych wartości
|
|
Sr
|
=
|
odchylenie standardowe powtarzalności
|
|
CVr
|
=
|
współczynnik zmienności powtarzalności
|
|
SR
|
=
|
odchylenie standardowe odtwarzalności
|
|
CVR
|
=
|
współczynnik zmienności odtwarzalności
|
ZAŁĄCZNIK IX
TABELE KORELACJI, O KTÓRYCH MOWA W ART. 6
1. Dyrektywa 71/250/EWG
|
Dyrektywa 71/250/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1, pierwszy akapit
|
Artykuł 3
|
|
Artykuł 1, drugi akapit
|
Artykuł 2
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik, część 1
|
Załącznik II
|
|
Załącznik, część 2
|
—
|
|
Załącznik, część 3
|
—
|
|
Załącznik, część 4
|
Załącznik III, część O
|
|
Załącznik, część 5
|
Załącznik III, część M
|
|
Załącznik, część 6
|
Załącznik III, część N
|
|
Załącznik, część 7
|
Załącznik III, część Q
|
|
Załącznik, część 9
|
Załącznik III, część K
|
|
Załącznik, część 10
|
—
|
|
Załącznik, część 11
|
—
|
|
Załącznik, część 12
|
Załącznik III, część J
|
|
Załącznik, część 14
|
Załącznik III, część D
|
|
Załącznik, część 16
|
—
|
2. Dyrektywa 71/393/EWG
|
Dyrektywa 71/393/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik, część I
|
Załącznik III, część A
|
|
Załącznik, część II
|
Załącznik III, część E
|
|
Załącznik, część III
|
Załącznik III, część P
|
|
Załącznik, część IV
|
Załącznik III, część H
|
3. Dyrektywa 72/199/EWG
|
Dyrektywa 72/199/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Załącznik I, część 1
|
Załącznik III, część L
|
|
Załącznik I, część 2
|
Załącznik III, część C
|
|
Załącznik I, część 3
|
—
|
|
Załącznik I, część 4
|
—
|
|
Załącznik I, część 5
|
Załącznik V, część A
|
|
Załącznik II
|
—
|
4. Dyrektywa 73/46/EWG
|
Dyrektywa 73/46/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Załącznik I, część 1
|
Załącznik III, część B
|
|
Załącznik I, część 2
|
—
|
|
Załącznik I, część 3
|
Załącznik III, część I
|
5. Dyrektywa 76/371/EWG
|
Dyrektywa 76/371/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 1
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
Załącznik I
|
6. Dyrektywa 76/372/EWG
|
Dyrektywa 76/372/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
—
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
—
|
7. Dyrektywa 78/633/EWG
|
Dyrektywa 78/633/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik, część 1
|
—
|
|
Załącznik, część 2
|
—
|
|
Załącznik, część 3
|
Załącznik IV, część C
|
8. Dyrektywa 81/715/EWG
|
Dyrektywa 81/715/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
—
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
—
|
9. Dyrektywa 84/425/EWG
|
Dyrektywa 84/425/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
—
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
—
|
10. Dyrektywa 86/174/EWG
|
Dyrektywa 86/174/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 4
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
Załącznik VII
|
11. Dyrektywa 93/70/EWG
|
Dyrektywa 93/70/EWG
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik
|
Załącznik IV, część D
|
12. Dyrektywa 93/117/WE
|
Dyrektywa 93/117/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuły 3 i 5
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Załącznik, część 1
|
Załącznik IV, część E
|
|
Załącznik, część 2
|
Załącznik VIII, część A
|
13. Dyrektywa 98/64/WE
|
Dyrektywa 98/64/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuły 3 i 5
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Załącznik, część A
|
Załącznik III, część F
|
|
Załącznik, część C
|
Załącznik VIII, część B
|
14. Dyrektywa 1999/27/WE
|
Dyrektywa 1999/27/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuły 3 i 5
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Artykuł 5
|
—
|
|
Artykuł 6
|
—
|
|
Artykuł 7
|
—
|
|
Załącznik, część A
|
Załącznik VIII, część C
|
|
Załącznik, część B
|
Załącznik IV, część F
|
|
Załącznik, część C
|
Załącznik VIII, część D
|
15. Dyrektywa 1999/76/WE
|
Dyrektywa 1999/76/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Załącznik
|
Załącznik IV, część G
|
16. Dyrektywa 2000/45/WE
|
Dyrektywa 2000/45/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Załącznik, część A
|
Załącznik IV, część A
|
|
Załącznik, część B
|
Załącznik IV, część B
|
|
Załącznik, część C
|
Załącznik III, część G
|
17. Dyrektywa 2002/70/WE
|
Dyrektywa 2002/70/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 1
|
|
Artykuł 2
|
Artykuły 2 i 3
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Artykuł 5
|
—
|
|
Załącznik I
|
Załącznik I i załącznik V, część B (I)
|
|
Załącznik II
|
Załącznik II i załącznik V, część B (II)
|
18. Dyrektywa 2003/126/WE
|
Dyrektywa 2003/126/WE
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 3
|
|
Artykuł 2
|
—
|
|
Artykuł 3
|
—
|
|
Artykuł 4
|
—
|
|
Artykuł 5
|
—
|
|
Artykuł 6
|
—
|
|
Załącznik
|
Załącznik VI
|