|
29.3.2008
|
PL
|
Dziennik Urzędowy Unii Europejskiej
|
L 88/1
|
ROZPORZĄDZENIE KOMISJI (WE) NR 273/2008
z dnia 5 marca 2008 r.
ustanawiające szczegółowe zasady stosowania rozporządzenia Rady (WE) nr 1255/1999 w odniesieniu do metod analizy oraz oceny jakości mleka i przetworów mlecznych
KOMISJA WSPÓLNOT EUROPEJSKICH,
uwzględniając Traktat ustanawiający Wspólnotę Europejską,
uwzględniając rozporządzenie Rady (WE) nr 1255/1999 z dnia 17 maja 1999 r. w sprawie wspólnej organizacji rynku mleka i przetworów mlecznych (1), w szczególności jego art. 10 i 15 oraz art. 26 ust. 3, art. 29 ust. 1 i art. 31 ust. 4,
a także mając na uwadze, co następuje:
|
(1)
|
Rozporządzenie Komisji (WE) nr 213/2001 (2) ustanawia szczegółowe zasady stosowania rozporządzenia Rady (WE) nr 1255/1999 w odniesieniu do metod analizy oraz oceny jakości mleka i przetworów mlecznych. W świetle postępu technicznego w dziedzinie metodyki analitycznej niezbędne jest dokonanie dalszych istotnych zmian. W interesie przejrzystości i efektywności, jak również biorąc pod uwagę liczbę oraz techniczny charakter poprawek, rozporządzenie (WE) nr 213/2001 powinno zostać uchylone i zastąpione nowym rozporządzeniem.
|
|
(2)
|
Skład oraz wymogi jakości w odniesieniu do mleka i przetworów mlecznych, ustanowione w ramach uzgodnień przewidzianych w rozporządzeniu (WE) nr 1255/1999, muszą zostać zweryfikowane w celu zapewnienia ich bezwzględnego przestrzegania.
|
|
(3)
|
Metodami referencyjnymi wykorzystywanymi do takich weryfikacji są często metody publikowane przez organizacje międzynarodowe, takie jak Europejski Komitet Normalizacyjny (CEN), Międzynarodowa Federacja Mleczarska (IDF), Międzynarodowa Organizacja Normalizacyjna (ISO) oraz stowarzyszenie naukowe na rzecz doskonałości analitycznej (AOAC International), i regularnie przez te organizacje aktualizowane. W niektórych przypadkach ustanowiona jest wspólnotowa metoda referencyjna, podczas gdy w innych przypadkach taka metoda referencyjna nie jest wymieniona w zasadach wspólnotowych. W celu zapewnienia jednolitego stosowania metod referencyjnych należy opracować ich wykaz oraz umożliwić Komisji dostosowanie tego wykazu w razie potrzeby.
|
|
(4)
|
Korzystanie z metod rutynowych nie powinno być wykluczone, dlatego też należy określić warunki ich stosowania.
|
|
(5)
|
Należy również ustanowić wspólne procedury w celu zapewnienia jednolitej praktyki przy ocenie wyników analiz, w ocenie sensorycznej danych produktów oraz w ponownym badaniu wyników spornych.
|
|
(6)
|
W odniesieniu do niektórych analiz nie istnieją aktualnie zaakceptowane na szczeblu międzynarodowym, zatwierdzone metody referencyjne, zatem nie są dostępne informacje o różnicach wyników analitycznych między laboratoriami. Z tej przyczyny należy ustanowić metody wspólnotowe, zatwierdzone zgodnie z międzynarodowymi regułami i stosować jako metody referencyjne.
|
|
(7)
|
Rozporządzenie Komisji (WE) nr 1898/2005 (3) ustanawia szczegółowe zasady wprowadzenia w życie rozporządzenia Rady (WE) nr 1255/1999 w odniesieniu do środków w zakresie zbytu śmietanki, masła i koncentratu masła i przewiduje w niektórych okolicznościach znakowanie śmietanki, masła i koncentratu masła w celu zapewnienia poprawnego końcowego wykorzystywania tych produktów. Znaczniki są ważne z punktu widzenia poprawnego działania systemu. Aby zapewnić, że podmioty gospodarcze biorące w nim udział są traktowane jednakowo, należy ustalić wspólne metody oznaczania niektórych z tych znaczników.
|
|
(8)
|
Na mocy art. 9 rozporządzenia (WE) nr 1255/1999 może zostać przyznana pomoc w odniesieniu do prywatnego przechowywania serów z mleka owczego. W odniesieniu do tych samych produktów może zostać przyznana specjalna refundacja na mocy art. 31 wspomnianego rozporządzenia. Sery z mleka owczego, mleka koziego, mleka bawolego oraz z mieszanin mleka owczego, koziego i bawolego mogą być przywożone z niektórych krajów trzecich do Wspólnoty w ramach porozumień preferencyjnych. W świetle powyższych przepisów niezbędne są właściwe kontrole w celu zapewnienia, że powyższe produkty nie zawierają mleka krowiego, dlatego należy określić wspólnotową metodę referencyjną wykrywania mleka krowiego, bez uszczerbku dla stosowania metod rutynowych, pod warunkiem że są one zgodne z pewnymi kryteriami.
|
|
(9)
|
Na mocy rozporządzenia Komisji (EWG) nr 2921/90 z dnia 10 października 1990 r. w sprawie pomocy do produkcji kazeiny i kazeinianów z mleka odtłuszczonego (4), niezbędne jest wykrycie braku bakterii z grupy coli. Uznaną na szczeblu międzynarodowym metodą wykrywania bakterii z grupy coli w mleku i przetworach mlecznych jest norma ISO 4831. Wspólnotowa metoda referencyjna wykrywania bakterii z grupy coli została ustanowiona w oparciu o wyżej wymienioną normę.
|
|
(10)
|
Rozporządzenie Rady (EWG) nr 2658/87 z dnia 23 lipca 1987 r. w sprawie nomenklatury taryfowej i statystycznej oraz w sprawie Wspólnej Taryfy Celnej (5) przewiduje różne stawki celne w odniesieniu do mieszanek paszowych objętych pozycją taryfową 2309, w zależności od ilości zawartych w nich przetworów mlecznych. W celu zapewnienia jednolitego stosowania omawianych zasad należy ustanowić ogólnie obowiązującą metodę analizy zawartości laktozy do obowiązkowego stosowania we wszystkich państwach członkowskich.
|
|
(11)
|
Na mocy rozporządzenia (WE) nr 1255/1999 masło i mleko odtłuszczone w proszku przeznaczone do skupu interwencyjnego lub, w przypadku mleka odtłuszczonego w proszku, na paszę dla zwierząt, muszą spełniać pewne wymogi jakości. Należy ustanowić metody referencyjne w celu sprawdzania, czy powyższe wymogi są spełniane.
|
|
(12)
|
Niektóre metody są wprowadzane po raz pierwszy w niniejszym rozporządzeniu. Należy zapewnić dostatecznie długi okres od wejścia w życie niniejszego rozporządzenia, aby umożliwić laboratoriom prawidłowe wprowadzenie i zastosowanie wspomnianych metod. Ilekroć metoda, do której poczyniono odniesienie w załączniku I, jest zmieniona, a następnie w zmienionej formie opublikowana przez organizację normalizacyjną, laboratoriom powinien zostać przyznany okres sześciu miesięcy na uaktualnienie ich procedur analitycznych w celu dostosowania do nowej normy.
|
|
(13)
|
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Komitetu Zarządzającego ds. Mleka i Przetworów Mlecznych,
|
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
ROZDZIAŁ I
PRZEPISY OGÓLNE
Artykuł 1
Przedmiot i zakres
1. Niniejsze rozporządzenie ustanawia niektóre metody referencyjne w odniesieniu do analizy chemicznej, fizycznej i mikrobiologicznej oraz sensorycznej oceny mleka i przetworów mlecznych, które mają być stosowane w ramach uzgodnień przewidzianych we wspólnej organizacji rynku mleka i przetworów mlecznych, określonej rozporządzeniem (WE) nr 1255/1999, jak również zasady stosowania tych metod.
2. Wykaz metod referencyjnych stosowanych w analizach określonych w ust. 1 jest zapisany w załączniku I do niniejszego rozporządzenia.
3. Komisja dokonuje aktualizacji wykazu zgodnie z procedurą ustanowioną w art. 42 rozporządzenia (WE) nr 1255/1999.
Artykuł 2
Metody rutynowe
Metody rutynowe mogą być wykorzystywane w celu wykonywania analiz wymaganych zgodnie z zasadami wspólnotowymi, pod warunkiem że metody te są odpowiednio kalibrowane i regularnie porównywane z metodą referencyjną. Wyniki są porównywane z uwzględnieniem stałego błędu, powtarzalności i odtwarzalności.
W przypadkach spornych wynik uzyskany przy użyciu metody referencyjnej jest ostateczny.
Państwa członkowskie powiadamiają Komisję o stosowaniu metod rutynowych w analizie określonej w art. 1.
ROZDZIAŁ II
METODY ANALIZY
Artykuł 3
Ocena zgodności partii towaru z dopuszczalną prawnie wartością graniczną
Z wyjątkiem analizy znaczników załącznik II do niniejszego rozporządzenia stosuje się w celu określenia zgodności z wymaganiami prawnymi dotyczącymi składu.
Artykuł 4
Ocena sensoryczna
1. W odniesieniu do mleka i przetworów mlecznych innych niż masło przeznaczone do składowania w magazynach państwowych metodą referencyjną, która ma być stosowana przez państwa członkowskie do oceny sensorycznej, jest norma IDF 99C/1997 lub inne porównywalne metody, które są zgłaszane Komisji.
Procedury opisane w załączniku III są stosowane w celu sprawdzenia sprawności osób oceniających oraz wiarygodności wyników analiz sensorycznych.
2. W odniesieniu do masła przeznaczonego do składowania w magazynach państwowych procedury opisane w załączniku III są stosowane w celu sprawdzenia sprawności osób oceniających oraz wiarygodności wyników analiz sensorycznych.
Procedura określona w załączniku IV ma zastosowanie jako metoda referencyjna dla oceny sensorycznej.
Artykuł 5
Znaczniki
1. Metoda analizy opisana w załączniku V jest stosowana jako metoda referencyjna oznaczania zawartości triglicerydów kwasu enantowego w maśle, oleju mlecznym i śmietance.
2. Metoda analizy opisana w załączniku VI jest stosowana jako metoda referencyjna oznaczania waniliny w koncentracie masła, maśle i śmietance.
3. Metoda analizy opisana w załączniku VII jest stosowana jako metoda referencyjna oznaczania zawartości estru etylowego kwasu beta-apo-8’ karotenowego w maśle i koncentracie masła.
4. Metoda analizy opisana w załączniku VIII jest stosowana jako metoda referencyjna w celu oznaczenia zawartości beta-sitosterolu lub stigmasterolu w maśle i koncentracie masła.
5. Uznaje się, że do koncentratu masła, masła i śmietanki zostały dodane znaczniki zgodnie z odpowiednimi zasadami wspólnotowymi, jeżeli otrzymane wyniki są zgodne ze specyfikacjami określonymi w pkt 10 i 11 załącznika V oraz pkt 8 załączników VI, VII i VIII.
Artykuł 6
Wykrywanie kazeiny mleka krowiego
1. Metoda referencyjna analizy opisana w załączniku IX jest stosowana w celu zapewnienia, że ser wyprodukowany wyłącznie z mleka owczego, mleka koziego lub mleka bawolego, lub z mieszanin mleka owczego, koziego i bawolego, nie zawiera kazeiny mleka krowiego.
Uznaje się, że kazeina mleka krowiego jest obecna w produkcie, jeżeli jej zawartość w analizowanej próbce jest równa zawartości w próbce odniesienia zawierającej 1 % mleka krowiego lub od niej większa, jak określono w załączniku IX.
2. Rutynowe metody wykrywania kazeiny mleka krowiego w serach, określone w ust. 1, mogą być stosowane pod warunkiem, że:
|
a)
|
granica wykrywalności wynosi nie więcej niż 0,5 %; oraz
|
|
b)
|
nie występują fałszywie pozytywne wyniki; oraz
|
|
c)
|
kazeina mleka krowiego jest wykrywalna, z wymaganą czułością, również po upływie długich okresów dojrzewania, które mogą wystąpić w zwykłych warunkach handlowych.
|
Jeżeli którekolwiek ze wspomnianych powyżej wymagań nie jest spełnione, stosuje się metodę referencyjną opisaną w załączniku IX.
Artykuł 7
Wykrywanie bakterii z grupy coli
Bakterie z grupy coli w maśle, odtłuszczonym mleku w proszku, kazeinie i kazeinianach wykrywa się przy użyciu metody referencyjnej opisanej w załączniku X.
Artykuł 8
Oznaczanie zawartości laktozy
Zawartość laktozy w produktach objętych kodem CN 2309 oznacza się przy użyciu metody referencyjnej opisanej w załączniku XI.
Artykuł 9
Wykrywanie serwatki podpuszczkowej
1. Serwatkę podpuszczkową w odtłuszczonym mleku w proszku przeznaczonym do składowania w magazynach państwowych wykrywa się przy użyciu metody referencyjnej opisanej w załączniku XII.
2. Serwatkę podpuszczkową w odtłuszczonym mleku w proszku i mieszankach przeznaczonych na pasze dla zwierząt wykrywa się przy użyciu metody referencyjnej opisanej w załączniku XII. W przypadku wykrycia serwatki podpuszczkowej należy zastosować załącznik XIII.
Artykuł 10
Wykrywanie maślanki
Maślankę w odtłuszczonym mleku w proszku wykrywa się przy użyciu metody referencyjnej opisanej w załączniku XIV.
Artykuł 11
Wykrywanie pozostałości antybiotyków
Pozostałości antybiotyków w odtłuszczonym mleku w proszku wykrywa się przy użyciu metody referencyjnej opisanej w załączniku XV.
Artykuł 12
Ustalanie zawartości odtłuszczonego mleka w proszku
Zawartość odtłuszczonego mleka w proszku w mieszankach paszowych ustala się przy użyciu metody referencyjnej opisanej w załączniku XVI.
Artykuł 13
Wykrywanie skrobi
Skrobię w odtłuszczonym mleku w proszku, zdenaturowanym mleku w proszku oraz w mieszankach paszowych wykrywa się przy użyciu metody referencyjnej opisanej w załączniku XVII.
Artykuł 14
Ustalanie zawartości wilgoci w śmietance w proszku
Zawartość wilgoci w śmietance w proszku ustala się przy użyciu metody referencyjnej opisanej w załączniku XVIII.
Artykuł 15
Ustalanie zawartości wilgoci w kwasowej maślance w proszku
Zawartość wilgoci w kwasowej maślance w proszku przeznaczonej do wykorzystania w paszach ustala się przy użyciu metody referencyjnej opisanej w załączniku XIX.
Artykuł 16
Ustalanie czystości tłuszczu mlekowego
Czystość tłuszczu mlekowego ustala się przy użyciu metody referencyjnej opisanej w załączniku XX.
ROZDZIAŁ III
PRZEPISY OGÓLNE I KOŃCOWE
Artykuł 17
Zapewnianie jakości
Analizy są prowadzone w laboratoriach, w których istnieje system zapewniania jakości analitycznej włącznie z wewnętrznymi procedurami kontroli jakości. Laboratoria nieakredytowane uczestniczą w programach badania biegłości co najmniej raz w roku, a ich wyniki nie odbiegają od wartości zgodnej o więcej niż 2σR (odchylenie standardowe odtwarzalności metody referencyjnej). Szczegółowy opis stosowanych systemów jest dostępny w laboratorium w celach konsultacji.
Laboratoria, które są akredytowane zgodnie z normami określonymi w art. 12 rozporządzenia (WE) nr 882/2004 Parlamentu Europejskiego i Rady z dnia 29 kwietnia 2004 r. w sprawie kontroli urzędowych przeprowadzanych w celu sprawdzenia zgodności z prawem paszowym i żywnościowym oraz regułami dotyczącymi zdrowia zwierząt i dobrostanu zwierząt (6), są zwolnione z obowiązku uczestniczenia w badaniu biegłości.
Artykuł 18
Pobieranie próbek oraz spory dotyczące wyników analiz
1. Pobieranie próbek przeprowadzane jest zgodnie z odpowiednimi przepisami dotyczącym rozpatrywanego produktu. Jeżeli nie przedstawiono żadnych przepisów w odniesieniu do pobierania próbek, stosuje się przepisy zamieszczone w normie ISO 707 | IDF 50, Mleko i przetwory mleczne — Wytyczne do pobierania próbek.
2. Laboratoryjne sprawozdania na temat wyników analiz muszą zawierać informacje wystarczające dla oceny wyników przeprowadzanej zgodnie z załącznikiem II i załącznikiem XXI.
3. W celu wykonania analiz wymaganych zgodnie z zasadami wspólnotowymi należy pobierać dwie próbki.
4. W przypadku gdy wyniki analiz nie zostaną zaakceptowane przez podmiot, stosuje się procedurę określoną w załączniku XXI.
5. Jeżeli w ciągu pięciu dni roboczych od momentu pobrania próbek producent może udowodnić, że procedura pobierania próbek nie została przeprowadzona prawidłowo, pobranie próbek należy w miarę możliwości powtórzyć. Jeżeli pobranie próbek nie może być powtórzone, partię towaru należy dopuścić.
Artykuł 19
Okres przejściowy
Oceny zgodności zgodnie z załącznikiem II do niniejszego rozporządzenia dokonuje się w terminie 12 miesięcy od jego wejścia w życie. Państwa członkowskie niezwłocznie informują Komisję, w razie konieczności, o wszelkich napotkanych w trakcie tego okresu poważnych problemach w związku z procedurą kontroli statystycznej.
Artykuł 20
Uchylenia
Rozporządzenie (WE) nr 213/2001 zostaje uchylone.
Odesłania do uchylonego rozporządzenia traktuje się jako odesłania do niniejszego rozporządzenia i odczytuje się je zgodnie z tabelą korelacji w załączniku XXII.
Artykuł 21
Wejście w życie
Niniejsze rozporządzenie wchodzi w życie trzeciego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia 31 marca 2008 r.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 5 marca 2008 r.
W imieniu Komisji
Mariann FISCHER BOEL
Członek Komisji
(1) Dz.U. L 160 z 26.6.1999, s. 48. Rozporządzenie ostatnio zmienione rozporządzeniem (WE) nr 1152/2007 (Dz.U. L 258 z 4.10.2007, s. 3). Rozporządzenie (WE) nr 1255/1999 zastępuje się rozporządzeniem (WE) nr 1234/2007 (Dz.U. L 299 z 16.11.2007, s. 1) od dnia 1 lipca 2008 r.
(2) Dz.U. L 37 z 7.2.2001, s. 1.
(3) Dz.U. L 308 z 25.11.2005, s. 1. Rozporządzenie ostatnio zmienione rozporządzeniem (WE) nr 1546/2007 (Dz.U. L 337 z 21.12.2007, s. 68).
(4) Dz.U. L 279 z 11.10.1990, s. 22. Rozporządzenie ostatnio zmienione rozporządzeniem (WE) nr 1487/2006 (Dz.U. L 278 z 10.10.2006, s. 8).
(5) Dz.U. L 256 z 7.9.1987, s. 1. Rozporządzenie ostatnio zmienione rozporządzeniem Komisji (WE) nr 1352/2007 (Dz.U. L 303 z 21.11.2007, s. 3).
(6) Dz.U. L 165 z 30.4.2004, s. 1.
ZAŁĄCZNIK i
(Artykuł 1)
WYKAZ METOD REFERENCYJNYCH
Indeks Min. = minimalnie, Maks. = maksymalnie, załącznik = załącznik do przytoczonego rozporządzenia, SNF = sucha masa beztłuszczowa, LN = liczba nadtlenkowa, A = wygląd, F = smak, C = konsystencja, OLD = ogólna liczba drobnoustrojów, Bakt. Term. = liczba bakterii termofilnych, MS = państwo członkowskie, IDF = Międzynarodowa Federacja Mleczarska, ISO = Międzynarodowa Organizacja Normalizacyjna, IUPAC = Międzynarodowa Unia Chemii Czystej i Stosowanej, ADPI = Amerykański Instytut Produktów Mleczarskich, SCM = mleko zagęszczone słodzone, EMC = zagęszczone mleko lub śmietanka.
CZĘŚĆ A
|
Rozporządzenie Komisji
|
Produkt
|
Parametr
|
Wartość graniczna (1)
|
Metoda referencyjna
|
Uwagi
|
|
Rozporządzenie (WE) nr 2771/1999 — Składowanie w magazynach państwowych
|
Masło niesolone
|
Tłuszcz
|
Min. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
Kwasowość tłuszczu
|
1,2 mmol/100g tłuszczu
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
LN (maks.)
|
0,3 meq tlenu/1 000 g tłuszczu
|
ISO 3976:2006|IDF 74:2006
|
Uwaga 1
|
|
|
|
Bakterie z grupy coli
|
Niewykrywalne w 1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Tłuszcz niepochodzący z mleka
|
Niewykrywalny poprzez analizę triglicerydów
|
Załącznik XX
|
|
|
|
|
Znaczniki sterolowe
|
Niewykrywalne, beta-sitosterol ≤ 40 mg/kg
|
Załącznik VIII
|
|
|
|
|
Pozostałe znaczniki
|
|
|
|
|
|
|
|
Niewykrywalna
|
Załącznik VI
|
|
|
|
|
|
—
|
ester etylowy kwasu karotenowego
|
|
≤ 6 mg/kg
|
Załącznik VII
|
|
|
|
|
|
—
|
triglicerydy kwasu enantowego
|
|
Niewykrywalny
|
Załącznik V
|
|
|
|
|
Charakterystyka sensoryczna
|
Co najmniej 4 na 5 punktów dla A, F i C
|
Załącznik iV
|
|
|
|
|
Dyspersja w wodzie
|
Co najmniej 4 punkty
|
ISO 7586:1985|IDF 112A:1989
|
|
|
Rozporządzenie (WE) nr 2771/1999 — Prywatne składowanie
|
Masło niesolone
|
Tłuszcz
|
Min. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
Rozporządzenie (WE) nr 2771/1999 — Prywatne składowanie
|
Masło solone
|
Tłuszcz
|
Min. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF (z wyjątkiem soli)
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
Sól
|
Do 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iI
|
Masło niesolone
|
Tłuszcz
|
Min. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Tłuszcz niepochodzący z mleka
|
|
Załącznik XX
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1 2001|IDF 80–1:2001
|
|
|
|
|
SNF
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
Znaczniki:
|
|
|
|
|
|
|
|
Zob. załącznik VIII
|
Załącznik VIII
|
|
|
|
|
|
Zob. załącznik VI
|
Załącznik VI
|
|
|
|
|
|
—
|
ester etylowy kwasu karotenowego
|
|
Zob. załącznik VII
|
Załącznik VII
|
|
|
|
|
|
—
|
triglicerydy kwasu enantowego
|
|
Zob. załącznik V
|
Załącznik V
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iI
|
Masło solone
|
Tłuszcz
|
Min. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Tłuszcz niepochodzący z mleka
|
|
Załącznik XX
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF (z wyjątkiem soli)
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
Sól
|
Do 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
Znaczniki:
|
|
|
|
|
|
|
|
Zob. załącznik VIII
|
Załącznik VIII
|
|
|
|
|
|
Zob. załącznik VI
|
Załącznik VI
|
|
|
|
|
|
—
|
ester etylowy kwasu karotenowego
|
|
Zob. załącznik VII
|
Załącznik VII
|
|
|
|
|
|
—
|
triglicerydy kwasu enantowego
|
|
Zob. załącznik V
|
ZAŁĄCZNIK V
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iI
|
Koncentrat masła
|
Tłuszcz
|
Min. 99,8 % m/m
|
IDF 24:1964
|
|
|
|
|
Woda i SNF
|
Do 0,2 % m/m
|
ISO 5536:2002|IDF 23:2002 (wilgoć)
IDF 24:1964 (SNF)
|
|
|
|
|
Kwasowość tłuszczu
|
1,2 mmol/100g tłuszczu
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
LN (maks.)
|
0,5 meq tlenu/1 000 g tłuszczu
|
ISO 3976:2006|IDF 74:2006
|
Uwaga 1
|
|
|
|
Tłuszcz niepochodzący z mleka
|
Brak
|
Załącznik XX
|
|
|
Smak
|
Czysty
|
|
Zapach
|
Brak obcych zapachów
|
|
Inne
|
Brak środków zobojętniających, przeciwutleniaczy i środków konserwujących
|
|
|
|
Znaczniki:
|
|
|
|
|
|
Zob. załącznik VIII
|
Załącznik VIII
|
|
|
Zob. załącznik VI
|
Załącznik VI
|
|
—
|
ester etylowy kwasu karotenowego
|
|
Zob. załącznik VII
|
Załącznik VII
|
|
—
|
triglicerydy kwasu enantowego
|
|
Zob. załącznik V
|
Załącznik V
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iI
|
Śmietana
|
Tłuszcz
|
Min. 35 % m/m
|
ISO 2450:1999|IDF 16 C:1987
|
|
|
|
|
Tłuszcz niepochodzący z mleka
|
|
Załącznik XX
|
|
|
|
|
Znaczniki:
|
|
|
|
|
|
Zob. załącznik VIII
|
|
Uwaga 2
|
|
|
|
|
Zob. załącznik VI
|
Załącznik VI
|
|
|
|
|
|
—
|
ester etylowy kwasu karotenowego
|
|
Zob. załącznik VII
|
|
Uwaga 2
|
|
|
|
|
—
|
triglicerydy kwasu enantowego
|
|
Zob. załącznik V
|
Załącznik V
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iII
|
Koncentrat masła
|
Tłuszcz
|
Min. 96 % m/m
|
|
Uwaga 2
|
|
|
|
Tłuszcz niepochodzący z mleka
|
|
Załącznik XX
|
|
|
|
|
SNF
|
Do 2 % m/m
|
|
Uwaga 2
|
|
|
|
Znaczniki:
|
|
|
|
|
—
|
stigmasterol (95 % m/m)
|
|
15 g/100 kg koncentratu masła
|
Załącznik VIII
|
|
|
|
|
—
|
stigmasterol (85 % m/m)
|
|
17 g/100 kg koncentratu masła
|
Załącznik VIII
|
|
|
|
|
|
—
|
triglicerydy kwasu enantowego
|
|
10,34 kg/t koncentratu masła
|
Załącznik V
|
|
|
|
|
|
—
|
ester etylowy kwasu masłowego i stigmasterol
|
|
|
|
—
|
ester etylowy kwasu masłowego
|
|
—
|
stigmasterol: załącznik VIII
|
|
Uwaga 2
|
|
|
|
|
—
|
ester etylowy kwasu masłowego i triglicerydy kwasu enantowego
|
|
|
|
—
|
ester etylowy kwasu masłowego
|
|
—
|
triglicerydy kwasu enantowego: załącznik V
|
|
Uwaga 2
|
|
|
|
lecytyna (E 322)
|
Do 0,5 % m/m
|
|
Uwaga 2
|
|
|
|
NaC1
|
Do 0,75 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
Kwasowość tłuszczu
|
1,2 mmol/100g tłuszczu
|
ISO 1740:2004|IDF 6:2004
|
|
|
|
|
LN (maks.)
|
Do 0,5 meq tlenu/1 000 g tłuszczu
|
ISO 3976:2006|IDF 74:2006
|
Uwaga 1
|
|
|
|
Smak
|
Czysty
|
|
|
|
|
|
Zapach
|
Brak obcych zapachów
|
|
|
|
|
|
Inne
|
Brak środków zobojętniających, przeciwutleniaczy i środków konserwujących
|
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iV
|
Masło niesolone
|
Tłuszcz
|
Min. 82 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
Rozporządzenie (WE) nr 1898/2005, rozdział iV
|
Masło solone
|
Tłuszcz
|
Min. 80 % m/m
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Woda
|
Do 16 % m/m
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
SNF (z wyjątkiem soli)
|
Do 2 % m/m
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
Sól
|
Do 2 % m/m
|
ISO 15648:2004|IDF 179:2004
|
|
|
Artykuł 9 i tytuł iI rozporządzenia (WE) nr 1255/1999
|
Ser wytworzony z mleka owczego i/lub koziego
|
Mleko krowie
|
< 1 % m/m
|
Załącznik iX
|
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik i — Kazeina kwasowa
|
Woda
|
Do 12,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Tłuszcz
|
Do 1,75 % m/m
|
ISO 5543:2004|IDF127:2004
|
|
|
|
|
Kwasowość wolna
|
Do 0,30 ml 0,1 N roztworu NaOH/g
|
ISO 5547:1978|IDF 91:1979
|
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik i — Kazeina podpuszczkowa
|
Woda
|
Do 12,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Tłuszcz
|
Do 1,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Popiół
|
Min. 7,50 % m/m
|
ISO 5545:1978|IDF 90:1979
|
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik i — Kazeiniany
|
Woda
|
Do 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Białka mleka
|
Min. 88,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Tłuszcz i popiół
|
Do 6,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Popiół związany
|
|
ISO 5544:1978|IDF 89:1979
|
|
|
|
|
Popiół
|
|
ISO 5545:1978|IDF 90:1979
|
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik iI — Kazeina kwasowa
|
Woda
|
Do 10,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Tłuszcz
|
Do 1,50 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Kwasowość wolna
|
Do 0,20 ml 0,1 N roztworu NaOH/g
|
ISO 5547:1978|IDF 91:1979
|
|
|
|
|
OLD (maks.)
|
30 000/g
|
ISO 4833:2003
|
Uwaga 3
|
|
|
|
Bakterie z grupy coli
|
Brak w 0,1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Bakt. Term. (maks.)
|
5 000/g
|
ISO 4833:2003
|
Uwaga 3 i 4
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik iI — Kazeina podpuszczkowa
|
Woda
|
Do 8,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Tłuszcz
|
Do 1,00 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Popiół
|
Min. 7,50 % m/m
|
ISO 5545:1978|IDF 90:1979
|
|
|
|
|
OLD (maks.)
|
30 000/g
|
ISO 4833:2003
|
Uwaga 3
|
|
|
|
Bakterie z grupy coli
|
Brak w 0,1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Bakt. Term. (maks.)
|
5 000/g
|
ISO 4833:2003
|
Uwaga 3 i 4
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik iI — Kazeiniany
|
Woda
|
Do 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Białka mleka
|
Min. 88,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Tłuszcz i popiół
|
Do 6,00 % m/m
|
ISO 5543:2004|IDF 127:2004
ISO 5544:1978|IDF 89:1979 lub
ISO 5545:1978|IDF 90:1979
|
|
|
|
|
OLD (maks.)
|
30 000/g
|
ISO 4833:2003
|
Uwaga 3
|
|
|
|
Bakterie z grupy coli
|
Brak w 0,1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Bakt. Term. (maks.)
|
5 000/g
|
ISO 4833:2003
|
Uwaga 3 i 4
|
|
Rozporządzenie (EWG) nr 2921/90
|
Załącznik iII — Kazeiniany
|
Woda
|
Do 6,00 % m/m
|
ISO 5550:2006|IDF 78:2006
|
|
|
|
|
Białka mleka
|
Min. 85,00 % m/m
|
ISO 5549:1978|IDF 92:1979
|
|
|
|
|
Tłuszcz
|
Do 1,50 % m/m
|
ISO 5543:2004|IDF 127:2004
|
|
|
|
|
Laktoza
|
Do 1,00 % m/m
|
ISO 5548:2004|IDF 106:2004
|
|
|
|
|
Popiół
|
Do 6,50 % m/m
|
ISO 5544:1978|IDF 89:1979 lub ISO 5545:1978|IDF 90:1979
|
|
|
|
|
OLD (maks.)
|
30 000/g
|
ISO 4833:2003
|
Uwaga 3
|
|
|
|
Bakterie z grupy coli
|
Brak w 0,1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Bakt. Term. (maks.)
|
5 000/g
|
ISO 4833:2003
|
Uwaga 3 i 4
|
|
Rozporządzenie (WE) nr 2799/1999
|
Mieszanki paszowe i odtłuszczone mleko w proszku (OMP) (do stosowania w paszach)
|
Woda (kwasowa maślanka w proszku)
|
Do 5 % m/m
|
Załącznik XIX
|
|
|
|
|
Białko
|
31,4 % m/m (min.) suchej substancji beztłuszczowej
|
ISO 8968–1|2|3:2001|IDF 20–1|2|3:2001
|
|
|
|
|
Woda (OMP)
|
Do 5 % m/m
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Tłuszcze (OMP)
|
Do 11 % m/m
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
Serwatka podpuszczkowa (OMP)
|
Brak
|
Załącznik XIII
|
Uwaga 6
|
|
|
|
Skrobia (OMP)
|
Brak
|
Załącznik XVII
|
|
|
|
|
Woda (mieszanki)
|
Do 5 % m/m substancji beztłuszczowej
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Tłuszcz (mieszanki)
|
|
Dyrektywa Komisji 84/4/EWG (Dz.U. L 15 z 18.1.1984, s. 29)
|
|
|
|
|
Serwatka podpuszczkowa (mieszaniny)
|
Brak
|
Załącznik XIII
|
|
|
|
|
Zawartość OMP (w produkcie końcowym)
|
Min. 50 % m/m
|
Załącznik XVI
|
|
|
|
|
Tłuszcz (w produkcie końcowym)
|
Min. 2,5 % m/m lub 5 % m/m
|
Dyrektywa Komisji 84/4/EWG (Dz.U. L 15 z 18.1.1984, s. 29)
|
Uwaga 7
|
|
|
|
Skrobia (w produkcie końcowym)
|
Min. 2 % m/m
|
Załącznik XVII
|
Uwaga 8
|
|
|
|
Miedź (w produkcie końcowym)
|
25 ppm
|
Dyrektywa Komisji 78/633/EWG (Dz.U. L 206 z 26.7.1987, s. 43)
|
|
|
Rozporządzenie (WE) nr 214/2001
|
OMP (suszone rozpryskowo)
|
Tłuszcz
|
Do 1,0 % m/m
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
Białko
|
31,4 % (2)m/m (min.) suchej substancji beztłuszczowej
|
ISO 8968–1/2:2001|IDF 20–1/2:2001
|
|
|
|
|
Woda
|
Do 3,5 % m/m
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
Kwasowość
|
Do 19,5 ml 0,1 N NaOH, 10 g suchej masy beztłuszczowej
|
ISO 6091:1980|IDF 86:1981
|
|
|
|
|
Mleczany
|
Do 150 mg/100 g suchej masy beztłuszczowej
|
ISO 8069:2005|IDF 69:2005
|
|
|
|
|
Fosfataza
|
Negatywny
|
ISO 11816–1:2006|IDF 155–1:2006
|
|
|
|
|
Wskaźnik nierozpuszczalności
|
Do 0,5 ml w temp. 24 °C
|
ISO 8156:2005|IDF 129:2005
|
|
|
|
|
Cząstki przypalone
|
Krążek A lub B (15,0 mg)
|
ADPI (1990)
|
|
|
|
|
OLD
|
40 000/g
|
ISO 4833:2003
|
Uwaga 3
|
|
|
|
Bakterie z grupy coli
|
Negatywny/ 0,1 g
|
Załącznik X
|
Uwaga 3
|
|
|
|
Maślanka
|
Negatywny
|
Załącznik XIV
|
|
|
|
|
Serwatka podpuszczkowa
|
Negatywny
|
Załącznik XII
|
|
|
|
|
Serwatka kwasowa
|
Negatywny
|
|
Uwaga 2
|
|
|
|
Antybiotyki
|
|
Załącznik XV
|
|
CZĘŚĆ B
Metody referencyjne wymienione w części B mogą być stosowane do analizowania produktów objętych każdym z rozporządzeń wymienionych w kolumnie 1.
|
Rozporządzenie Komisji
|
Produkt
|
Kod CN
|
Parametr
|
Wartość graniczna
|
Metoda referencyjna
|
Uwagi
|
|
Rozporządzenie (EWG) nr 2658/87
Rozporządzenie (WE) nr 2535/2001
Rozporządzenie (WE) nr 1282/2006
|
Mleko i śmietana niezagęszczone ani niezawierające dodatku cukru lub innego środka słodzącego
|
0401
|
Tłuszcz (≤ 6 % m/m)
|
Obowiązują wartości graniczne określone w opisie do kodu CN dla określonego produktu, dodatkowo wymienionego, gdzie stosowne, w rozporządzeniu Komisji (EWG) nr 3846/87 (Dz.U. L 366 z 24.12.1987, s. 1) część 9 nomenklatury wywozowej lub w rozporządzeniu (WE) nr 2535/2001 (Dz.U. L 341 z 22.12.2001, s. 29)
|
ISO 1211:2001|IDF 1D:1996
|
|
|
|
|
|
Tłuszcz (> 6 % m/m)
|
|
ISO 2450:1999|IDF 16C:1987
|
|
|
|
Mleko i śmietana zagęszczone lub zawierające dodatek cukru albo innego środka słodzącego
|
0402
|
Tłuszcze (w postaci płynnej)
|
|
ISO 1737:1999|IDF 13C:1987
|
|
|
|
|
|
Tłuszcze (w postaci stałej)
|
|
ISO 1736:2000|IDF 9C:1987
|
|
|
|
|
|
Białko
|
|
ISO 8968–1|2|3:2001|IDF 20–1|2|3:2001
|
|
|
|
|
|
Sacharoza (zawartość normalna)
|
|
ISO 2911:2004|IDF 35:2004
|
|
|
|
|
|
Sacharoza (zawartość niska)
|
|
|
Uwaga 2
|
|
|
|
|
Substancje stałe (SCM)
|
|
ISO 6734:1989|IDF 15B:1991
|
|
|
|
|
|
Substancje stałe (EMC)
|
|
ISO 6731:1989|IDF 21B:1987
|
|
|
|
|
|
Woda (mleko w proszku)
|
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
|
Woda (śmietana w proszku)
|
|
Załącznik XVIII
|
|
|
|
Maślanka, sfermentowane lub zakwaszone mleko i śmietana, zagęszczone lub niezagęszczone, zawierające dodatek cukru lub innego środka słodzącego
|
0403
|
Tłuszcz
|
|
ISO 1211:2001|IDF 1D:1996
ISO 1736:2000|IDF 9C:1987
ISO 2450:1999|IDF 16 C:1987
ISO 7208:1999|IDF 22B:1987
ISO 8262–3:2005|IDF 124–3:2005
|
|
|
|
|
|
Białko
|
|
ISO 8968–1|2|3:2001|IDF 20–1|2|3:2001
|
|
|
|
|
|
Sacharoza (zawartość normalna)
|
|
ISO 2911:2004|IDF 35:2004
|
|
|
|
|
|
Sacharoza (zawartość niska)
|
|
|
Uwaga 2
|
|
|
|
|
Woda (kwasowa maślanka w proszku)
|
|
Załącznik XIX
|
|
|
|
|
|
Woda (słodka maślanka w proszku)
|
|
ISO 5537:2004|IDF 26:2004
|
|
|
|
|
|
Substancje stałe (inne produkty)
|
|
Metody zatwierdzone przez właściwe władze
|
|
|
|
Serwatka, zagęszczona lub niezagęszczona albo zawierająca lub niezawierająca dodatku cukru lub innego środka słodzącego; produkty składające się ze składników naturalnego mleka
|
0404
|
Tłuszcz
|
|
ISO 1736:2000|IDF 9C:1987
ISO 2450:1999|IDF 16C:1987
ISO 7208:1999|IDF 22B:1987
|
|
|
|
|
|
Białko
|
|
ISO 8968–1|2|3:2001|IDF 20–1|2|3:2001
|
|
|
|
|
|
Sacharoza (zawartość normalna)
|
|
ISO 2911:2004|IDF 35:2004
|
|
|
|
|
|
Sacharoza (zawartość niska)
|
|
|
Uwaga 2
|
|
|
|
0404 90
|
Białko
|
|
ISO 8968 1/2 2001|IDF 20–1/2:2001
|
|
|
|
|
|
Woda
|
|
IDF 21B:1987
|
|
|
|
|
|
Substancje stałe
|
|
ISO 6734:1989|IDF 15B:1991
|
|
|
|
|
|
(Produkty zagęszczone)
|
|
ISO 6731:1989|IDF 21B:1987
|
|
|
|
Masło i pozostałe tłuszcze otrzymane z mleka; produkty mleczarskie do smarowania
|
0405
|
Tłuszcz (jeżeli ≤ 85 % m/m)
|
|
ISO 17189:2003|IDF 194:2003
|
|
|
|
|
Masło
|
Woda
|
|
ISO 3727–1:2001|IDF 80–1:2001
|
|
|
|
|
|
SNF
|
|
ISO 3727–2:2001|IDF 80–2:2001
|
|
|
|
|
|
NaCl
|
|
ISO 15648:2004|IDF 179:2004
|
|
|
|
|
|
Tłuszcz (jeżeli > 99 % m/m)
|
|
IDF 24:1964
|
|
|
|
Bezwodny tłuszcz mleczny
|
|
Woda (jeżeli tłuszcze < 99 % m/m)
|
|
ISO 5536:2002|IDF 23:2002
|
|
|
|
Ser i twaróg
|
0406
|
Tłuszcz
|
|
ISO 1735:2004|IDF 5:2004
|
|
|
|
|
|
Substancje stałe
|
|
ISO 5534:2004|IDF 4:2004
|
|
|
|
|
|
Substancje stałe (Ricotta)
|
|
ISO 2920:2004|IDF 58:2004
|
|
|
|
|
|
NaCl
|
|
ISO 5943:2006|IDF 88:2006
|
|
|
|
|
|
Laktoza
|
|
ISO 5765–1/2:2002|IDF 79–1/2:2002
|
|
|
Rozporządzenie (EWG) nr 2658/87.
|
Mieszanki paszowe
|
2309
|
Laktoza
|
|
Załącznik XI
|
|
Uwagi do wykazu metod referencyjnych Unii Europejskiej
Uwaga 1: Oddzielenie tłuszczu mlecznego zgodnie z opisem zawartym w iSO 1740:1991 (ochrona przed światłem).
Uwaga 2: Nie określono metody referencyjnej. Metody zatwierdzone przez właściwe władze.
Uwaga 3: Próbkę należy przygotować zgodnie z normą ISO 8261:2001|IDF 122:2001.
Uwaga 4: Inkubacja w ciągu 48 godzin w temp. 55 °C, należy podjąć działania w celu zapobieżenia wysuszeniu podłoża hodowli.
Uwaga 5: % m/m SNF = % m/m substancji stałych — % m/m tłuszczu.
Uwaga 6: Dyrektywa Komisji 84/4/EWG.
Uwaga 7: Rozporządzenie Komisji (WE) nr 2799/1999 (Dz.U. L 340 z 31.12.1999, s. 3-27).
Uwaga 8: Dyrektywa Komisji 78/633/EWG.
(1) Bez uszczerbku dla wymogów zamieszczonych w określonym rozporządzeniu.
(2) Z dniem 1 września 2009 r. minimalna zawartość białka wynosiłaby 34 %.
ZAŁĄCZNIK II
(Artykuł 3)
OCENA ZGODNOŚCI PARTII TOWARU Z PRAWNIE OBOWIĄZUJĄCĄWARTOŚCIĄ GRANICZNĄ
1. ZASADA
W przypadkach gdy w odnośnym prawodawstwie podane są szczegółowe procedury dotyczące pobierania próbek, procedur tych należy przestrzegać. We wszystkich pozostałych przypadkach wykorzystuje się próbkę spośród co najmniej 3 próbek pobranych wyrywkowo z partii towaru przedłożonej do kontroli. Dopuszcza się przygotowanie próbki złożonej. Otrzymany wynik jest porównywany z prawnie obowiązującymi wartościami granicznymi poprzez obliczenie przedziału ufności wynoszącego 95 % jako podwójnego odchylenia standardowego, przy czym odnośne odchylenie standardowe zależy od tego, czy 1) metoda jest zwalidowana w drodze współpracy międzynarodowej z uwzględnieniem wartości σr i σR, lub 2) w przypadku walidacji wewnętrznej, czy obliczono odtwarzalność wewnętrzną. Wspomniany przedział ufności będzie wówczas równy niepewności pomiaru danego wyniku.
2. METODA JEST ZWALIDOWANA W DRODZE WSPÓŁPRACY MIĘDZYNARODOWEJ
W tym przypadku odchylenie standardowe powtarzalności σr oraz odchylenie standardowe odtwarzalności σR zostały określone, a laboratorium jest w stanie wykazać zgodność z cechami wydajności zwalidowanej metody.
Należy obliczyć średnią arytmetyczną  liczby n powtórzonych pomiarów.
liczby n powtórzonych pomiarów.
Należy obliczyć niepewność rozszerzoną (k = 2) dla  ze wzoru
ze wzoru

Jeżeli wynik końcowy x pomiaru jest obliczany przy użyciu wzoru w postaci x = y
1 + y
2, x = y
1 – y2, x = y
1. y2 lub x = y
1 / y
2, w takich przypadkach należy przestrzegać normalnych procedur łączenia odchyleń standardowych.
Partia towaru jest uznana za niezgodną z górną prawnie obowiązującą wartością graniczną UL, jeżeli
 ;
;
w przeciwnym razie jest ona uznana za zgodną z UL.
Partia towaru jest uznana za niezgodną z dolną prawnie obowiązującą wartością graniczną LL, jeżeli

w przeciwnym razie jest ona uznana za zgodną z LL.
3. WALIDACJA WEWNĘTRZNA UWZGLĘDNIAJĄCA OBLICZENIE WEWNĘTRZNEGO ODCHYLENIA STANDARDOWEGO ODTWARZALNOŚCI
W przypadkach gdy stosowane są metody nieokreślone w niniejszym rozporządzeniu, a miary precyzji nie zostały ustanowione, należy przeprowadzić walidację wewnętrzną. We wzorach na obliczenie niepewności rozszerzonej U niezbędne jest zastosowanie wewnętrznego odchylenia standardowego powtarzalności sir oraz wewnętrznego odchylenia standardowego odtwarzalności siR
zamiast, odpowiednio, σr i σ
R
.
Zasady podejmowania decyzji określa pkt 1. Jeżeli jednak partia towaru zostanie uznana za niezgodną z prawnie obowiązującą wartością graniczną, pomiary należy powtórzyć przy użyciu metody określonej w niniejszym rozporządzeniu oraz w oparciu o decyzję podjętą zgodnie z pkt 1.
ZAŁĄCZNIK III
(Artykuł 4)
OCENA OSÓB OCENIAJĄCYCH I WIARYGODNOŚĆ WYNIKÓW ANALIZ SENSORYCZNYCH
Jeżeli wykorzystywane są metody oceny punktowej (norma IDF 99C:1997), stosuje się poniższe procedury.
A. OKREŚLENIE „WSKAŹNIKA POWTARZALNOŚCI”
Co najmniej dziesięć próbek zostanie poddanych analizie jako ślepe duplikaty przez osobę oceniającą w okresie 12 miesięcy. Zazwyczaj będzie się to odbywać w czasie kilku sesji. Wyniki dla poszczególnych cech produktu są oceniane z zastosowaniem poniższego wzoru:

gdzie:
|
wI
|
:
|
wskaźnik powtarzalności,
|
|
xi1
|
:
|
wynik (liczba punktów) pierwszej oceny próbki xi,
|
|
xi2
|
:
|
wynik (liczba punktów) drugiej oceny próbki xi,
|
|
n
|
:
|
liczba próbek,
|
Oceniane próbki powinny odzwierciedlać szeroki zakres jakości. Wartość wI nie powinna przekroczyć 1,5 (w skali pięciopunktowej).
B. OKREŚLENIE „WSKAŹNIKA ODCHYLEŃ”
Wskaźnik ten powinien być wykorzystywany w celu sprawdzenia, czy osoba oceniająca wykorzystuje dla oceny jakości tę samą skalę, co doświadczona grupa osób oceniających. Punkty otrzymane przez osobę oceniającą są porównywane ze średnią punktów otrzymanych przez grupę osób oceniających.
Do oceny wyników ma zastosowanie poniższy wzór:

gdzie:
|
xi1; xi2
|
:
|
zob. część A)
|
|

;
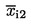
|
:
|
średni wynik grupy osób oceniających, odpowiednio w pierwszej i drugiej ocenie próbki xi,
|
|
n
|
:
|
liczba próbek (co najmniej dziesięć na 12 miesięcy).
|
Oceniane próbki powinny odzwierciedlać szeroki zakres jakości. Wartość DI nie powinna przekroczyć 1,5 (w skali pięciopunktowej).
Państwa członkowskie są zobowiązane do powiadamiania Komisji o wszelkich trudnościach napotkanych podczas stosowania niniejszej procedury.
Jeżeli stwierdzi się, że poszczególne osoby oceniające przekroczyły wartość graniczną wynoszącą 1,5 dla wskaźników odchyleń lub powtarzalności, ekspert (eksperci) ze strony właściwych władz zobowiązany jest (zobowiązani są) do przeprowadzenia przynajmniej jednej wyrywkowej „ponownej kontroli” wydajności na próbkach ocenionych przez te osoby w ciągu ostatnich kilku tygodni, lub do przeprowadzenia przynajmniej jednej „połączonej” kontroli z udziałem tych osób oceniających. Do podjęcia decyzji co do dalszego korzystania z usług tych osób niezbędny jest ścisły monitoring. Ustalenia należy dokumentować i zachować jako dowód działań następczych.
C) PORÓWNANIE WYNIKÓW OTRZYMANYCH W RÓŻNYCH REGIONACH PAŃSTWA CZŁONKOWSKIEGO I W RÓŻNYCH PAŃSTWACH CZŁONKOWSKICH
Gdzie stosowne, badanie należy zorganizować co najmniej raz w roku w celu porównania wyników otrzymanych przez osoby oceniające w różnych regionach. Jeżeli stwierdza się występowanie znacznych różnic, powinny zostać podjęte konieczne kroki w celu zidentyfikowania przyczyn oraz uzyskania porównywalnych wyników.
Państwa członkowskie mogą organizować badania w celu porównania wyników otrzymanych przez ich własne osoby oceniające oraz osoby oceniające z sąsiednich państw członkowskich. Znaczące różnice powinny prowadzić do pogłębionego badania w celu uzyskania porównywalnych wyników.
Państwa członkowskie powinny powiadomić Komisję o wynikach tych porównań.
ZAŁĄCZNIK IV
(Artykuł 4)
SENSORYCZNA OCENA MASŁA
1. ZAKRES
Celem niniejszej procedury w zakresie sensorycznej oceny masła jest ustalenie jednolitej metody stosowanej we wszystkich państwach członkowskich.
W celu uzyskania dalszych szczegółów zob. norma międzynarodowa IDF dla mleka i przetworów mlecznych, IDF 99 — części 1, 2, 3, dotyczące oceny sensorycznej.
2. DEFINICJE
„Ocena sensoryczna” oznacza badanie właściwości produktu za pomocą narządów zmysłu.
„Zespół oceniający” oznacza grupę wybranych osób oceniających, które podczas przeprowadzania oceny nie mają możliwości komunikowania się między sobą oraz nie mają możliwości wywierania wpływu jedna na drugą.
„Osoba oceniająca” jest określona jako osoba wybrana z powodu jej predyspozycji do przeprowadzenia badania sensorycznego. Osoba oceniająca tego rodzaju może mieć ograniczone doświadczenie.
„Biegła osoba oceniająca” jest określona jako osoba o wysokiej wrażliwości sensorycznej oraz doświadczeniu w zakresie metodologii sensorycznej, która jest w stanie dokonać spójnej i wiarygodnej oceny sensorycznej rozmaitych produktów. Osoba oceniająca tego rodzaju musi być obdarzona dobrą długoterminową pamięcią sensoryczną.
„Ocena punktowa” oznacza sensoryczną ocenę przeprowadzaną przez zespół oceniający, przy użyciu skali numerycznej. Należy stosować nomenklaturę dotyczącą wad.
„Stopniowanie” oznacza klasyfikację jakościową, przeprowadzaną w oparciu o ocenę punktową.
„Dokumenty kontroli”: dokumenty używane do rejestrowania indywidualnych punktacji dla każdej cechy oraz końcowej klasy produktu. (Dokument ten może zostać również wykorzystany do rejestrowania składu chemicznego).
3. PRACOWNIA ANALIZY SENSORYCZNEJ
W celu uzyskania dalszych szczegółów zob. ISO 8589 i ISO/DIS 22935–2 | IDF 99–2 ust. 7.
Należy podjąć środki ostrożności, aby osoby oceniające znajdujące się w pracowni analizy sensorycznej nie były poddawane oddziaływaniu czynników zewnętrznych.
Pracownia analizy sensorycznej musi być wolna od obcych zapachów i łatwa do utrzymania w czystości. Ściany muszą być w jasnym kolorze i nie mogą odbijać światła.
Pracownia analizy sensorycznej oraz jej oświetlenie nie mogą wywierać wpływu na właściwości ocenianych produktów.
Pomieszczenie musi być kontrolowane termostatycznie, aby umożliwić utrzymywanie stałej temperatury masła. Masło w czasie stopniowania powinno mieć temp. 12 °C (±2 °C).
4. WYBÓR OSÓB OCENIAJĄCYCH
Osoba oceniająca musi mieć dobrą znajomość produktów z masła oraz posiadać kwalifikacje do przeprowadzenia stopniowania sensorycznego. Jej kwalifikacje powinny być regularnie kontrolowane (co najmniej raz w roku) przez właściwe władze.
4.1. Aby zapoznać się ze szczegółami dotyczącymi ogólnych wymogów i badań przesiewowych, które można stosować przed oficjalnym korzystaniem z usług nowej osoby oceniającej, należy zapoznać się z normą ISO/DIS 22935–1 | IDF 99–1 ust. 4 (rekrutacja) i ust. 5.1
Niezbędne jest, aby szkolenie było ciągłe, a sesje ogólne odbywały się regularnie. W sprawie informacji dotyczących szkolenia zespołu oceniającego, zob. ISO 8586–1.
4.2. Szkolenie wstępne powinno obejmować następujące zagadnienia:
|
—
|
ogólna teoria oraz praktyczne znaczenie oceny sensorycznej,
|
|
—
|
metody, skale i opis wrażeń sensorycznych,
|
|
—
|
wykrywanie i rozpoznawanie cech sensorycznych oraz określona terminologia sensoryczna,
|
|
—
|
podstawowe szkolenie w zakresie produkcji masła,
|
|
—
|
zwalidowane materiały referencyjne i próbki, mające za zadanie pomóc osobie oceniającej zidentyfikowanie określonych smaków i intensywności smaku danego produktu.
|
5. WYMOGI DOTYCZĄCE ZESPOŁU OCENIAJĄCEGO
Liczba osób oceniających w zespole powinna być nieparzysta i wynosić co najmniej trzy osoby. Większość z nich muszą stanowić pracownicy zatrudnieni przez właściwe władze bądź upoważnione osoby niezatrudnione w branży mleczarskiej.
Kierownik zespołu oceniającego jest odpowiedzialny za całą procedurę i może brać udział w pracach zespołu.
Aby zapewnić jak najlepsze wykonanie zadania przez uczestników oceny, przed jej podjęciem należy uwzględnić szereg czynników:
|
—
|
uczestnicy nie mogą cierpieć na żadną chorobę, która mogłaby wpłynąć na ich sprawność. W razie wystąpienia takiego przypadku dana osoba oceniająca powinna zostać zastąpiona w zespole przez inną osobę,
|
|
—
|
uczestnicy muszą punktualnie przystąpić do oceny i upewnić się, że mają wystarczająco dużo czasu na jej wykonanie,
|
|
—
|
uczestnicy nie mogą używać substancji o silnym zapachu, takich jak perfumy, płyny po goleniu, dezodoranty itd., jak również powinni unikać spożywania aromatycznych (np. mocno przyprawionych) pokarmów itd.,
|
|
—
|
uczestnicy nie mogą palić ani jeść, nie mogą również pić niczego poza wodą na pół godziny przed rozpoczęciem oceny.
|
6. PRZEPROWADZENIE OCENY
Wszystkie osoby oceniające powinny uczestniczyć w regularnych pracach zespołu oceniającego w celu utrzymania kwalifikacji. Częstotliwość będzie uzależniona od ilości masła oraz wydajności; tam gdzie jest to możliwe, w miesiącu powinna odbywać się co najmniej jedna sesja zespołu oceniającego.
Wysokie rangą osoby oceniające powinny brać udział w kilku sesjach zespołów oceniających w ciągu roku i, jeżeli istnieje taka możliwość, co najmniej raz na kwartał.
7. POBIERANIE PRÓBEK ORAZ PRZYGOTOWANIE PRÓBKI
Niezbędne jest, aby tożsamość próbek była ukryta podczas przeprowadzania oceny, aby uniknąć wszelkiego potencjalnego wpływu na wyniki badania. Próbki powinny być kodowane.
Należy to zorganizować przed przystąpieniem do oceny. Wymagane jest aby temperatura masła podczas transportu do pracowni analizy sensorycznej była stabilna (6 °C ± 2 °C).
W przypadku gdy ocena sensoryczna przeprowadzana jest w chłodni składowej, próbkę pobiera się za pomocą próbnika do masła. Jeżeli ocena sensoryczna przeprowadzana jest w miejscu innym niż chłodnia składowa, należy pobrać próbkę o masie co najmniej 500 g. W trakcie oceny masło powinno mieć temp. 12 °C (±2 °C) (uwaga: w normie ISO/DIS 22935–2 | IDF 99–2 temperatura wymagana do oceny masła wynosi 14 °C ± 2 °C). Za wszelką cenę należy unikać dużych odchyleń od tych wartości temperatury.
8. OCENA WARTOŚCI DLA KAŻDEJ CECHY
8.1. Przy ocenie sensorycznej należy uwzględnić trzy cechy: wygląd, konsystencję oraz smak.
„Wygląd” wiąże się z następującymi cechami: kolor, widoczna czystość, brak zanieczyszczeń fizycznych, brak porostu pleśni oraz jednolita dyspersja w wodzie. Dyspersję w wodzie bada się zgodnie z normą IDF 112A/1989.
„Konsystencja” wiąże się z następującymi cechami: zwartość, struktura oraz twardość. Smarowność może być monitorowana przy użyciu środków fizycznych, gdyby takie było życzenie któregokolwiek z państw członkowskich w celu zaspokojenia wymagań klientów. Komisja może podjąć w przyszłości decyzję w sprawie ujednolicenia metodologii.
„Zwartość” to termin odnoszący się do spoistości produktu w czasie jego spożywania. Ma ona zazwyczaj związek z twardością i smarownością, powinna też być jednolita w obrębie całej porcji produktu. Cecha ta ma ścisły związek ze strukturą i stanowi zdolność produktu do utrzymania się pod własnym ciężarem. Zwartość wykazywana jest jako opór stawiany podczas krojenia produktu i może być zmierzona mechanicznie, jak również za pośrednictwem ust i palców.
„Smak” jest cechą charakterystyczną odczuwaną w ustach, przede wszystkim za pośrednictwem kubków smakowych języka.
„Aromat” jest cechą charakterystyczną odczuwaną za pośrednictwem nosa i zmysłu węchu.
Znaczne odchylenie od zalecanej temperatury uniemożliwia przeprowadzenie wiarygodnej oceny konsystencji oraz smaku. Temperatura jest parametrem najwyższej wagi.
Jeżeli temperatura jest poza zalecanym zakresem, stopniowanie masła należy przełożyć na inny termin.
8.2. Sensoryczna ocena każdej cechy musi być przeprowadzona oddzielnie. Ocena punktowa musi być dokonana zgodnie z tabelą 1.
8.3. Dla celu osiągnięcia jednolitości w ocenie punktowej może być wskazane, aby osoby oceniające, przed rozpoczęciem przeprowadzania oceny, wspólnie przyznały punkty dla jednej lub większej ilości próbek odniesienia za ich wygląd, konsystencję i smak.
8.4. Ocena punktowa dopuszczalności przedstawia się, jak następuje:
Zob. część 7 — Nomenklatura oraz opis kryteriów odnoszących się do punktów w czasie dokonywania oceny punktowej.
|
|
Ocena maksymalna
|
Ocena wymagana
|
|
Wygląd
|
5
|
4
|
|
Konsystencja
|
5
|
4
|
|
Smak/aromat
|
5
|
4
|
|
—
|
W przypadku gdy nie zostaje uzyskana wymagana liczba punktów, należy koniecznie podać opis wady.
|
|
—
|
Liczba punktów przyznanych przez każdą z osób oceniających dla każdej cechy musi zostać zarejestrowana w dokumencie kontroli.
|
|
—
|
Produkt zostaje dopuszczony bądź odrzucony w oparciu o decyzję większości.
|
|
—
|
Przypadki, w których różnice między indywidualną oceną punktową dla każdej cechy są większe niż wartości mieszczące się między sąsiednimi punktami, nie powinny występować często (nie częściej niż raz na 20 próbek). W przeciwnym przypadku kierownik zespołu oceniającego powinien sprawdzić kompetencje danego zespołu.
|
9. NADZÓR
Kierownik zespołu oceniającego, który musi być pracownikiem właściwych władz i może być członkiem zespołu, jest zobowiązany do ponoszenia ogólnej odpowiedzialności za całą procedurę. Musi on rejestrować indywidualnie przyznane wyniki dla każdej cechy w dokumencie kontrolnym oraz uwierzytelniać dopuszczenie bądź odrzucenie produktu.
10. NOMENKLATURA
Zob. dołączona tabela 2.
11. ODNIESIENIE
FIL-IDF 99C:1997 Sensoryczna ocena produktów mleczarskich w drodze oceny punktowej — Metoda referencyjna
ISO/DIS 22935 | IDF 99 Międzynarodowa norma dla mleka i przetworów mlecznych — Analiza sensoryczna — Części 1–3
ISO 8586–1 Analiza sensoryczna — Ogólne wytyczne wyboru, szkolenia i monitorowania oceniających — Część 1
ISO 8589 Analiza sensoryczna — Ogólne wytyczne dotyczące projektowania pracowni analizy sensorycznej
FIL-IDF 112A:1989 Masło — Oznaczanie wartości dyspersji w wodzie
Tabela 1
Punktowa ocena masła
|
Wygląd
|
Konsystencja
|
Smak i aromat
|
|
Punkty
|
Nr (1)
|
Uwagi
|
Punkty (klasa jakości)
|
Nr (1)
|
Uwagi
|
Punkty (klasa jakości)
|
Nr (1)
|
Uwagi
|
|
5
|
|
Bardzo dobry
idealny rodzaj
najwyższa jakość
(równomiernie suche)
|
5
|
|
Bardzo dobra
idealny rodzaj
najwyższa jakość
(dobrze smarowne)
|
5
|
|
Bardzo dobry
idealny rodzaj
najwyższa jakość
(absolutnie czysty, najlepszy aromat)
|
|
4
|
|
Dobry
(2)
bez wyraźnych wad
|
4
|
17
18
|
Dobra
(2)
twarde
miękkie
|
4
|
|
Dobry
(2)
bez wyraźnych wad
|
|
3
|
1
2
3
4
5
6
7
8
|
Odpowiedni (niewielkie wady)
luźne (niezwiązane), wilgotne
niejednorodne, dwukolorowe
smugowate
nakrapiane, marmurkowe
plamiste
oddzielony olej
przebarwione
słaba, luźna struktura
|
3
|
14
15
16
17
18
|
Odpowiednia (niewielkie wady)
kruche, łamliwe, rozpadające się
ciastowate, ciągnące się, maziste
lepkie
twarde
miękkie
|
3
|
21
22
25
27
33
34
35
|
Odpowiedni (niewielkie wady)
nieczysty
obcy smak
kwaśny
smak gotowania, smak spalenizny
smak paszy
niedelikatny, gorzki
nadmiernie słony
|
|
2
|
1
3
4
5
6
10
11
12
|
Zły (wyraźne wady)
luźne (niezwiązane), wilgotne
smugowate
nakrapiane, marmurkowe
plamiste
oddzielony olej
zanieczyszczenia
zapleśniałe
nierozpuszczona sól
|
2
|
14
15
16
17
18
|
Zła (wyraźne wady)
kruche, łamliwe, rozpadające się
ciastowate, ciągnące się, maziste
klejące się
twarde
miękkie
|
2
|
21
22
23
25
32
33
34
35
36
38
|
Zły (wyraźne wady)
nieczysty
obcy smak
zatęchły
kwaśny
posmak utlenienia, smak metaliczny
smak paszy
niedelikatny, gorzki
nadmiernie słony
zatęchły, zgniły
smak chemikaliów
|
|
1
|
1
3
4
5
6
7
9
10
11
12
|
Bardzo zły (mocne wady)
luźne (niezwiązane), wilgotne
smugowate
nakrapiane, marmurkowe
plamiste
oddzielony olej
przebarwione
granulowate
zanieczyszczenia
zapleśniałe
nierozpuszczona sól
|
1
|
14
15
16
17
18
|
Bardzo zła (mocne wady)
kruche, łamliwe, rozpadające się
ciastowate, ciągnące się, maziste
lepkie
twarde
miękkie
|
1
|
22
24
25
26
28
29
30
31
32
34
35
36
37
38
|
Bardzo zły (mocne wady)
obcy smak
smak serowy, sera twarogowego
kwaśny
drożdżowy
smak pleśni
zjełczały
oleisty, rybi
łojowaty
posmak utlenienia, smak metaliczny
niedelikatny, gorzki
nadmiernie słony
zatęchły, zgniły
słodowy
smak chemikaliów
|
Tabela 2
Wykaz wad masła
|
|
|
1.
|
luźne (niezwiązane), wilgotne
|
|
|
2.
|
niejednorodne, dwukolorowe
|
|
|
|
|
4.
|
nakrapiane, marmurkowe
|
|
|
|
|
|
|
|
|
8.
|
słabe (luźna struktura)
|
|
|
|
|
|
|
|
|
|
|
|
|
14.
|
kruche, łamliwe, rozpadające się
|
|
|
15.
|
ciastowate, ciągnące się, maziste
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24.
|
smak serowy, sera twarogowego
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) Tabela 2.
(2) Wady wymienione pod oceną „dobrą” stanowią jedynie bardzo drobne odchylenia od idealnego rodzaju.
(3) Określenie to powinno być wykorzystywane jak najrzadziej i wyłącznie wówczas, gdy wada nie może być dokładniej opisana.
ZAŁĄCZNIK V
(Artykuł 5)
OZNACZANIE ZAWARTOŚCI TRIGLICERYDÓW KWASU ENANTOWEGO W MAŚLE, BEZWODNYM TŁUSZCZU MLECZNYM I ŚMIETANIE W DRODZE GAZOWO-CHROMATOGRAFICZNEJ ANALIZY TRIGLICERYDÓW
1. ZAKRES
Niniejsza metoda określa metodę oznaczania zawartości triglicerydów kwasu enantowego w bezwodnym tłuszczu mlecznym, maśle i śmietanie.
2. POJĘCIA I DEFINICJA
Zawartość kwasu enantowego: zawartość triglicerydów kwasu enantowego określona w drodze procedury wyszczególnionej w niniejszej metodzie.
Uwaga: Zawartość kwasu enantowego wyrażona jest w kg na tonę produktu w przypadku bezwodnego tłuszczu mlecznego i masła, natomiast w przypadku śmietany wyrażona jest w kg na tonę tłuszczu mlecznego.
3. ZASADA
Tłuszcz mleczny ekstrahuje się z różnych produktów zgodnie z normą ISO 14156 | IDF 172:2001. Oznaczanie ilościowe zawartości triglicerydów kwasu enantowego w wyekstrahowanym tłuszczu przeprowadza się w drodze chromatografii gazowej (GC) na kolumnach kapilarnych. Wynik otrzymany dla danej próbki jest poddawany ocenie poprzez odniesienie do triglicerydów kwasu kapronowego jako wzorca wewnętrznego.
Uwaga: Stwierdzono, że tributyryna również stanowi zadowalający wzorzec wewnętrzny.
4. ODCZYNNIKI
Należy używać wyłącznie odczynników o uznanej klasie analitycznej.
4.1. n-heksan
4.2. Wzorcowe triglicerydy kwasu kapronowego o czystości co najmniej 99 %
4.3. Wzorcowe triglicerydy kwasu enantowego o czystości co najmniej 99 %
4.4. Bezwodny siarczan sodu (Na2SO4).
5. APARATURA
Typowe wyposażenie laboratoryjne, w szczególności:
5.1. Waga analityczna o dokładności do 1 mg
5.2. Kolby miarowe poj. 10 ml i 20 ml
5.3. Probówki wirówkowe poj. 30 ml
5.4. Wyparka obrotowa
5.5. Piec umożliwiający utrzymanie temp. 50 °C ± 5 °C
5.6. Sączki papierowe o średniej porowatości, o średnicy ok. 15 cm
Sprzęt do chromatografii gazowej
5.7.1. Chromatograf gazowy wyposażony w dozownik z podziałem/bez podziału strumienia (ang. split/splitless) lub nakolumnowy (ang. on-column) oraz detektor płomieniowo-jonizacyjny (FID)
Kolumna do GC z fazą stacjonarną, którą z powodzeniem użyto do rozdzielenia triglicerydów (100 % dimetylopolisiloksanu lub 5 % fenylu–95 % metylopolisiloksanu). Wybrać fazę stacjonarną, długość kolumny (4–15 m), średnicę wewnętrzną (0,22–0,50 mm) oraz grubość warstwy (0,12 μm lub grubsza), biorąc pod uwagę doświadczenie w pracy laboratoryjnej oraz zastosowany system wprowadzania. W każdym przypadku wybrana kolumna powinna prowadzić zarówno do uzyskania całkowitego oddzielenia piku rozpuszczalnika od piku triglicerydów kwasu kapronowego, jak i umożliwić rozróżnienie na linii podstawowej pików triglicerydów kwasu kapronowego i enantowego. Przykłady stosownych warunków zamieszczono poniżej.
5.7.2.1. Przykład stosownych warunków z wykorzystaniem dozownika z podziałem strumienia:
|
—
|
ciśnienie w głowicy kolumny: 100 KPa
|
|
—
|
kolumna: kolumna ze stopionej krzemionki o długości 12 m, średnicy wewnętrznej 0,5 mm i grubości warstwy 0,1 μm
|
|
—
|
faza stacjonarna: 100 % dimetylopolisiloksanu lub 5 % fenylu–95 % dimetylopolisiloksanu (np. HT5)
|
|
—
|
temperatura kolumny: temperatura początkowa 130 °C, utrzymana przez 1 minutę, podwyższana stopniowo o 20 °C/min. do 260 °C, a następnie podwyższana stopniowo o 30 °C/min. do 360 °C; utrzymać przez 10 minut w temp. 360 °C
|
|
—
|
temperatura detektora: 370 °C
|
|
—
|
temperatura dozownika: 350 °C
|
|
—
|
ilość wprowadzonej próbki: 1 μl.
|
5.7.2.2. Przykład stosownych warunków z wykorzystaniem dozownika nakolumnowego:
|
—
|
gaz nośny: wodór (system utrzymujący stały przepływ gazu)
|
|
—
|
ciśnienie w głowicy kolumny: 89 kPa
|
|
—
|
kolumna: kolumna ze stopionej krzemionki o długości 4 m, średnicy wewnętrznej 0,32 mm i grubości warstwy 0,25 µm
|
|
—
|
faza stacjonarna: 5 % fenylu, 95 % dimetylopolisiloksanu
|
|
—
|
temperatura kolumny: temperatura początkowa 60 °C, utrzymana przez 2 minuty, podwyższana stopniowo o 35 °C/min. do 340 °C, utrzymana w tej temperaturze przez 5 minut
|
|
—
|
temperatura detektora: 350 °C
|
|
—
|
ilość wprowadzonej próbki: 1 μl
|
5.8. Strzykawka wprowadzająca poj. 5 μl.
6. POBIERANIE PRÓBEK
Istotne jest, aby laboratorium otrzymało próbkę, która jest rzeczywiście reprezentatywna i nie uległa uszkodzeniu ani nie zmieniła właściwości w czasie transportu lub przechowywania.
Pobieranie próbek nie jest częścią metody określonej w niniejszej normie międzynarodowej. Zalecana metoda pobierania próbek przedstawiona jest w IDF: norma 50C:1995 lub ISO 707–1997 — Mleko i przetwory mleczne — Metody pobierania próbek.
7. PROCEDURA
7.1. Przygotowanie próbki do badań i porcji do badań
Postępować zgodnie z normą ISO 14156 | IDF 172:2001
7.1.1. Bezwodny tłuszcz mleczny, masło
7.1.1.1. Roztopić 50 g — 100 g próbki badanej w piecu (ppkt 5.5)
7.1.1.2. Umieścić 0,5 g — 1,0 g bezwodnego siarczanu sodu (ppkt 5.4) w złożonym sączku papierowym
7.1.1.3. Przesączyć tłuszcz przez sączek papierowy z umieszczonym w nim bezwodnym siarczanem sodu, zbierając przesącz w zlewce przetrzymywanej w piecu (ppkt 5.5). W czasie dekantacji roztopionego masła na sączek papierowy należy uważać, aby nie przenieść serwatki
7.1.2. Śmietana
7.1.2.1. Próbkę badaną doprowadzić do temp. 20 °C ± 2 °C.
7.1.2.2. Dokładnie wymieszać lub zmiksować próbkę
7.1.2.3. Rozcieńczyć odpowiednią ilość próbki badanej, tak aby otrzymać 100 ml porcji badanej zawierającej ułamek masowy tłuszczu wynoszący ok. 4 %
7.1.2.4. Postępować jak w przypadku mleka surowego i mleka homogenizowanego (zob. ISO 14156 | IDF 172:2001, § 8.3) w celu wyekstrahowania tłuszczu ze śmietany
7.1.2.5. Odmierzyć wagowo do kolby miarowej poj. 10 ml (ppkt 5.2), z dokładnością do 1 mg, 1 g wyekstrahowanego tłuszczu. Dodać 1 ml roztworu (ppkt 7.2.2). Dopełnić n-heksanem (ppkt 5.1) do objętości 10 ml i homogenizować
7.1.2.6. Przenieść 1 ml roztworu (ppkt 7.1.1.2) do kolby miarowej poj. 10 ml (ppkt 5.2) i uzupełnić n-heksanem do objętości 10 ml (ppkt. 4.1)
7.2. Przygotowanie wzorców kalibracji
7.2.1. Rozpuścić 100 mg triglicerydów kwasu enantowego (ppkt 4.3) w 10 ml n-heksanu (ppkt 4.1)
7.2.2. Rozpuścić 100 mg triglicerydów kwasu kapronowego (ppkt 4.2) w 10 ml n-heksanu (ppkt 4.1)
7.2.3. Przenieść 1 ml roztworu (ppkt 7.2.2) do kolby miarowej poj. 10 ml (ppkt 5.2). Dopełnić n-heksanem (ppkt 4.1) do objętości 10 ml
7.2.4. Przenieść 1 ml roztworu (ppkt 7.2.1) oraz 1 ml roztworu (ppkt 7.2.2) do kolby miarowej poj. 10 ml (ppkt 5.2). Dopełnić n-heksanem (ppkt 4.1) do objętości 10 ml
7.2.5. Przenieść 1 ml roztworu (ppkt 7.2.4) do kolby miarowej poj. 10 ml (ppkt 5.2) i uzupełnić n-heksanem (ppkt 4.1) do objętości 10 ml
7.3. Oznaczenie chromatograficzne
7.3.1. Wprowadzić dwukrotnie 1 µl roztworu wzorcowego (ppkt 7.2.5)
7.3.2. Wprowadzić 1 µl roztworu każdej próbki
Uwaga: W razie przyjęcia systemu z dozownikiem nakolumnowym należy zastosować większe rozcieńczenie zarówno w odniesieniu do roztworu wzorcowego, jak i roztworu próbki.
7.3.3. Powtarzać operację (ppkt 7.3.1) każdorazowo po wprowadzeniu trzech kolejnych porcji roztworu próbek w celu oddzielenia próbek podwójnymi wprowadzeniami roztworu wzorcowego. Wyniki oparte są na średnich arytmetycznych współczynnikach odpowiedzi wyliczonych z chromatogramów wzorcowych
8. OBLICZANIE WYNIKÓW
Dla każdego chromatogramu przeprowadzić całkowanie powierzchni pików związanych z triglicerydami kwasu enantowego i kwasu kapronowego.
Postępować zgodnie z powyższymi instrukcjami w przypadku każdej oddzielonej sekwencji, tzn. dla każdego zbioru trzech oddzielonych próbek wzorzec wprowadzony dwukrotnie bezpośrednio przed nimi to STD1, a wzorzec wprowadzony dwukrotnie bezpośrednio po nich to STD2.
8.1. Kalibracja
8.1.1. Obliczyć współczynnik odpowiedzi dla każdego duplikatu STD1, Rf1(a) i Rf1(b).
Rf1 (a) lub (b) = (Powierzchnia piku triglicerydów kwasu kapronowego / Powierzchnia piku triglicerydów kwasu enantowego) × 100
Obliczyć średni arytmetyczny współczynnik odpowiedzi, Rfl
Rf1 = (Rf1(a) + Rf1(b)) / 2
8.1.2. Podobnie obliczyć średni arytmetyczny współczynnik odpowiedzi STD2, Rf2
8.1.3. Obliczyć średni arytmetyczny współczynnik odpowiedzi, Rf
Rf = (Rf1 + Rf2) / 2
8.2. Próbki badane
Dla chromatogramu każdej próbki, otrzymanego między STD1 i STD2, obliczyć zawartość kwasu enantowego C (kg/t):
C = (Powierzchnia piku triglicerydów kwasu enantowego × Rf × 100) / (Powierzchnia piku triglicerydów kwasu kapronowego × Wt × 1 000)
gdzie:
|
—
|
Wt = masa pobranego tłuszczu (g),
|
|
—
|
100 = objętość, do jakiej rozcieńczono próbkę,
|
|
—
|
1 000 = mnożnik przeliczeniowy (z µg/g na kg/t).
|
W przypadku próbek masła uwzględnić zawartość tłuszczu w maśle i obliczyć skorygowaną wartość stężenia Cmasła (kg/t masła)
Cmasła = Ctłuszczu × F
gdzie F stanowi zawartość tłuszczu w maśle.
9. PRECYZJA
Dane szczegółowe dotyczące międzylaboratoryjnego badania masła zgodnie z normą ISO 5725–1 i ISO 5725–2 w sprawie precyzji metod pomiarowych przedstawiono w ppkt 12.
Wartości w odniesieniu do granicy powtarzalności i odtwarzalności są wyrażone dla poziomu ufności wynoszącego 95 % i nie mogą mieć zastosowania do zakresów stężenia oraz matryc innych niż podane.
9.1. Powtarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi przy użyciu tej samej metody na identycznym materiale badanym w tym samym laboratorium przez ten sam podmiot wykorzystujący to samo wyposażenie w krótkim odstępie czasu, nie będzie większa niż 0,35 kg/t dla więcej niż 5 % przypadków.
9.2. Odtwarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi przy użyciu tej samej metody na identycznym materiale badanym w różnych laboratoriach przez różne podmioty wykorzystujące różne wyposażenie, nie będzie większa niż 0,66 kg/t dla więcej niż 5 % przypadków.
10. GRANICE TOLERANCJI: DOLNE WARTOŚCI GRANICZNE (W PRZYPADKU NIEDOSTATECZNYCH ILOŚCI)
10.1. Z produktu znakowanego należy pobrać trzy próbki w celu sprawdzenia prawidłowości znakowania produktu.
10.2. Masło i koncentrat masła
10.2.1. Stopień włączenia wynosi 11 kg triglicerydów kwasu enantowego o czystości co najmniej 95 % na tonę masła, tzn. 10,45 kg/t
10.2.2. Wyniki dla trzech próbek, otrzymane z analizy produktu są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
9,51 kg/t (95 % minimalnego stopnia włączenia triglicerydów kwasu enantowego o czystości 95 %, pojedyncze oznaczenie),
|
|
—
|
6,89 kg/t (70 % minimalnego stopnia włączenia triglicerydów kwasu enantowego o czystości 95 %, pojedyncze oznaczenie),
|
|
—
|
stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między, odpowiednio, 9,51 kg/t i 6,89 kg/t.
|
10.3. Śmietana
10.3.1. Stopień włączenia wynosi 10 kg triglicerydów kwasu enantowego o czystości co najmniej 95 % na tonę tłuszczu mlecznego, tzn. 9,50 kg/t znakowanego tłuszczu mlecznego.
10.3.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
8,60 kg/t (95 % minimalnego stopnia włączenia triglicerydów kwasu enantowego o czystości 95 %, pojedyncze oznaczenie),
|
|
—
|
6,23 kg/t (70 % minimalnego stopnia włączenia triglicerydów kwasu enantowego o czystości 95 %, pojedyncze oznaczenie),
|
|
—
|
stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między, odpowiednio, 8,60 kg/t i 6,23 kg/t.
|
11. GRANICE TOLERANCJI: GÓRNE WARTOŚCI GRANICZNE (W PRZYPADKU ILOŚCI PRZEKROCZONEJ O PONAD 20 %)
11.1. Z produktu znakowanego należy pobrać trzy próbki w celu sprawdzenia prawidłowości znakowania produktu.
11.2. Masło i koncentrat masła
11.2.1. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
górna wartość graniczna wynosi 12,96 kg/t.
|
11.3. Śmietana
11.3.1. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a średnią z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
górna wartość graniczna wynosi 11,82 kg/t.
|
12. INFORMACJE DODATKOWE: ANALIZA STATYSTYCZNA WYNIKÓW OZNACZANIA TRIENANTANU W TŁUSZCZU MAŚLANYM W DRODZE ANALIZY TRIGLICERYDÓW
Przeprowadzono cztery badania międzylaboratoryjne w celu oznaczenia zawartości trienantanu w znakowanym maśle.
W pierwszej próbie pierścieniowej wzięło udział dziewięć laboratoriów, przy czym nie udostępniono żadnych specyfikacji dotyczących metod analitycznych, które miały być wykorzystane:
W drugiej próbie pierścieniowej wzięło udział dziesięć laboratoriów i zastosowano 4 różne metody:
|
—
|
oznaczenie ilościowe heptanianu metylu przy użyciu n-nonanu lub nonanianu metylu jako wzorca wewnętrznego
|
|
—
|
oznaczenie ilościowe trienantanu przy użyciu trikaprylanu jako wzorca wewnętrznego
|
|
—
|
oznaczenie ilościowe heptanianu metylu przy użyciu próbki kalibracyjnej/mieszaniny kalibracyjnej
|
|
—
|
oznaczenie ilościowe heptanianu metylu przy użyciu mieszaniny kalibracyjnej.
|
Ponadto, jeżeli przeprowadzono analizę estrów metylowych kwasów tłuszczowych (FAME), zastosowano dwie różne procedury metylowania (De Francesco i Christopherson & Glass).
W oparciu o otrzymane wyniki wybrano dwie metody w celu przeprowadzenia trzeciej próby pierścieniowej:
|
—
|
oznaczenie ilościowe heptanianu metylu przy użyciu n-nonanu lub nonanianu metylu jako wzorca wewnętrznego
|
|
—
|
oznaczenie ilościowe trienantanu przy użyciu trikaprylanu jako wzorca wewnętrznego.
|
Wyniki z 7 laboratoriów wykazały, że dzięki metodzie FAME uzyskano wyższą zmienność, i w rezultacie podjęto decyzję o wykorzystaniu jedynie oznaczenia trienantanu jako triglicerydu, zgodnie z procedurą oznaczania trienantanu przy użyciu trikaprylanu jako wzorca wewnętrznego. Ponadto analizę triglicerydów należy przeprowadzić z wykorzystaniem kolumny kapilarnej.
W czwartej próbie pierścieniowej wprowadzono do obiegu cztery próbki (A, B, C, D), a wyniki dostarczyło dziewięć laboratoriów (tabele 1 i 2).
Dwa laboratoria (DE i UE) przeprowadziły analizy próbek przy użyciu metody FAME.
Z racji ograniczonej liczby laboratoriów obliczenia statystyczne przeprowadzono zarówno na pełnym zestawie (ryc. 1 i 2) danych, włącznie z wynikami analizy FAME, jak i na danych otrzymanych w wyniku analizy triglicerydów.
Testy wykrywające wyniki odbiegające:
|
—
|
próbka A. Testy Dixona, Cochrana i Grubbsa na poziomie 1 i 5 % wykazały jeden laboratoryjny wynik odbiegający.
|
|
—
|
próbka B. Test Grubbsa na poziomie 5 % wykazał jeden laboratoryjny wynik odbiegający.
|
|
—
|
próbka C. Testy Dixona i Grubbsa na poziomie 1 i 5 % wykazały jeden laboratoryjny wynik odbiegający.
|
|
—
|
próbka D. Testy Dixona i Grubbsa na poziomie 1 i 5 % wykazały jeden laboratoryjny wynik odbiegający.
|
Wynik odbiegający został wyłączony z obliczeń.
Warto zwrócić uwagę na fakt, że wyniki otrzymane przy użyciu metody FAME nigdy nie były uznawane za wyniki odbiegające w ramach zastosowanych badań.
Parametry precyzji
W tabelach 1 i 2 przedstawiono wyniki ze wszystkich laboratoriów oraz parametry precyzji obliczone w dopuszczalnej liczbie (8) laboratoriów, które nie zostały jednak niestety otrzymane przy zastosowaniu tej samej metody analitycznej.
W tabelach 3 i 4 przedstawiono wyniki otrzymane wyłącznie przy zastosowaniu metody oznaczania TG oraz stosownych parametrów precyzji. Przyjęcie wspomnianych parametrów jest uzależnione od zaakceptowania niskiej liczby laboratoriów (6).
Na ryc. 2 i 3 przedstawiono tendencje Sr i SR, obliczonych dla 4 próbek z 2 zestawów danych opisanych powyżej.
W tabeli 5 przedstawiono wartości Sr i SR wraz ze stosownymi wartościami zbiorczymi i ogólnymi parametrami r i R.
Ponadto obliczono różnicę krytyczną przy poziomie ufności wynoszącym 95 %.
Tabela 1
Wyniki statystyczne metody oznaczania TG + analizy FAME*
|
Próbka A
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
8
|
|
RENNES
|
FR1
|
11,0
|
11,1
|
11,1
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
11,2
|
11,2
|
11,2
|
Wyniki odbiegające
|
DК
|
|
ZPLA
|
DE*
|
11,6
|
11,8
|
11,7
|
Wartość średnia
|
11,3
|
|
ADAS
|
GB
|
11,4
|
11,2
|
11,3
|
Wartość rzeczywista
|
11,0
|
|
CNEVA
|
FR2
|
11,4
|
11,4
|
11,4
|
Odchylenie standardowe powtarzalności (Sr)
|
0,09
|
|
LODI
|
IT
|
11,1
|
11,3
|
11,2
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
0,80
|
|
EELA
|
FI
|
11,3
|
11,2
|
11,3
|
Powtarzalność r (95 %)
|
0,26
|
|
ISPRA
|
UE*
|
11,0
|
11,0
|
11,0
|
Względna powtarzalność r %
|
2,24
|
|
D.V.F.A.
|
DK
|
13,3
|
11,8
|
12,6
|
Odchylenie standardowe odtwarzalności (SR)
|
0,23
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
2,04
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,84
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
5,71
|
|
Próbka B
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
8
|
|
RENNES
|
FR1
|
12,7
|
12,8
|
12,8
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
13,5
|
13,3
|
13,4
|
Wyniki odbiegające
|
DK
|
|
ZPLA
|
DE*
|
14,0
|
13,8
|
13,9
|
Wartość średnia
|
13,4
|
|
ADAS
|
GB
|
13,4
|
13,5
|
13,5
|
Wartość rzeczywista
|
13,5
|
|
CNEVA
|
FR2
|
13,3
|
13,4
|
13,4
|
Odchylenie standardowe powtarzalności (Sr)
|
0,14
|
|
LODI
|
IT
|
13,9
|
13,5
|
13,7
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
1,04
|
|
EELA
|
FI
|
13,4
|
13,2
|
13,3
|
Powtarzalność r (95 %)
|
0,40
|
|
ISPRA
|
UE*
|
13,2
|
13,3
|
13,3
|
Względna powtarzalność r %
|
2,91
|
|
D.V.F.A.
|
DK
|
14,1
|
14,8
|
14,5
|
Odchylenie standardowe odtwarzalności (SR)
|
0,35
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
2,61
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,99
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
7,31
|
Tabela 2
Wyniki statystyczne metody oznaczania TG + analizy FAME*
|
Próbka C
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
8
|
|
RENNES
|
FR1
|
8,9
|
9,2
|
9,1
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
9,2
|
9,3
|
9,3
|
Wyniki odbiegające
|
DK
|
|
ZPLA
|
DE*
|
9,2
|
9,4
|
9,3
|
Wartość średnia
|
9,3
|
|
ADAS
|
GB
|
9,5
|
9,3
|
9,4
|
Wartość rzeczywista
|
9,3
|
|
CNEVA
|
FR2
|
9,4
|
9,4
|
9,4
|
Odchylenie standardowe powtarzalności (Sr)
|
0,14
|
|
LODI
|
IT
|
9,2
|
9,5
|
9,4
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
1,50
|
|
EELA
|
FI
|
9,4
|
9,6
|
9,5
|
Powtarzalność r (95 %)
|
0,40
|
|
ISPRA
|
UE*
|
9,4
|
9,3
|
9,4
|
Względna powtarzalność r %
|
4,20
|
|
D.V.F.A.
|
DK
|
10,7
|
10,9
|
10,8
|
Odchylenie standardowe odtwarzalności (SR)
|
0,17
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
1,82
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,47
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
5,10
|
|
Próbka D
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
8
|
|
RENNES
|
R1
|
1,6
|
1,6
|
1,6
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
2,1
|
2,1
|
2,1
|
Wyniki odbiegające
|
DK
|
|
ZPLA
|
DE*
|
2,3
|
2,3
|
2,3
|
Wartość średnia
|
2,1
|
|
ADAS
|
GB
|
2,1
|
2,2
|
2,2
|
Wartość rzeczywista
|
2,1
|
|
CNEVA
|
FR2
|
2,1
|
2,1
|
2,1
|
Odchylenie standardowe powtarzalności (Sr)
|
0,08
|
|
LODI
|
IT
|
2,2
|
1,9
|
2,1
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
3,81
|
|
EELA
|
FI
|
2,3
|
2,3
|
2,3
|
Powtarzalność r (95 %)
|
0,22
|
|
ISPRA
|
UE*
|
2,3
|
2,3
|
2,3
|
Względna powtarzalność r %
|
10,67
|
|
D.V.F.A.
|
DK
|
3,4
|
2,9
|
3,2
|
Odchylenie standardowe odtwarzalności (SR)
|
0,24
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
11,43
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,67
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
32,00
|
Tabela 3
Wyniki statystyczne metody oznaczania TG + analizy FAME*
|
Próbka A
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
6
|
|
RENNES
|
FR1
|
11,0
|
11,1
|
11,1
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
11,2
|
11,2
|
11,2
|
Wyniki odbiegające
|
DК
|
|
ADAS
|
GB
|
11,4
|
11,2
|
11,3
|
Wartość średnia
|
11,2
|
|
CNEVA
|
FR2
|
11,4
|
11,4
|
11,4
|
Wartość rzeczywista
|
11,0
|
|
LODI
|
IT
|
11,1
|
11,3
|
11,2
|
Odchylenie standardowe powtarzalności (Sr)
|
0,09
|
|
EELA
|
FI
|
11,3
|
11,2
|
11,3
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
0,80
|
|
D.V.F.A.
|
DK
|
13,3
|
11,8
|
12,6
|
Powtarzalność r (95 %)
|
0,25
|
|
|
|
|
|
|
Względna powtarzalność r %
|
2,24
|
|
|
|
|
|
|
Odchylenie standardowe odtwarzalności (SR)
|
0,13
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
1,16
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,36
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
3,25
|
|
Próbka B
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
6
|
|
RENNES
|
FR1
|
12,7
|
12,8
|
12,8
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
13,5
|
13,3
|
13,4
|
Wyniki odbiegające
|
DК
|
|
ADAS
|
GB
|
13,4
|
13,5
|
13,5
|
Wartość średnia
|
13,3
|
|
CNEVA
|
FR2
|
13,3
|
13,4
|
13,4
|
Wartość rzeczywista
|
13,5
|
|
LODI
|
IT
|
13,9
|
13,5
|
13,7
|
Odchylenie standardowe powtarzalności (Sr)
|
0,15
|
|
EELA
|
FI
|
13,4
|
13,2
|
13,3
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
1,13
|
|
D.V.F.A.
|
DK
|
14,1
|
14,8
|
14,5
|
Powtarzalność r (95 %)
|
0,42
|
|
|
|
|
|
|
Względna powtarzalność r %
|
3,16
|
|
|
|
|
|
|
Odchylenie standardowe odtwarzalności (SR)
|
0,33
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
2,48
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,93
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
6,94
|
Tabela 4
Wyniki statystyczne metody oznaczania TG
|
Próbka C
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
6
|
|
RENNES
|
FR1
|
8,9
|
9,2
|
9,1
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
9,2
|
9,3
|
9,3
|
Wyniki odbiegające
|
DК
|
|
ADAS
|
GB
|
9,5
|
9,3
|
9,4
|
Wartość średnia
|
9,3
|
|
CNEVA
|
FR2
|
9,4
|
9,4
|
9,4
|
Wartość rzeczywista
|
9,3
|
|
LODI
|
IT
|
9,2
|
9,5
|
9,4
|
Odchylenie standardowe powtarzalności (Sr)
|
0,15
|
|
EELA
|
FI
|
9,4
|
9,6
|
9,5
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
1,61
|
|
D.V.F.A.
|
DK
|
10,7
|
10,9
|
10,8
|
Powtarzalność r (95 %)
|
0,42
|
|
|
|
|
|
|
Względna powtarzalność r %
|
4,51
|
|
|
|
|
|
|
Odchylenie standardowe odtwarzalności (SR)
|
0,19
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
2,04
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,53
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
5,71
|
|
Próbka D
|
|
R1
|
R2
|
Średnia
|
Liczba laboratoriów pozostałych po odrzuceniu wyników odbiegających
|
6
|
|
RENNES
|
FR1
|
1,6
|
1,6
|
1,6
|
Liczba wyników odbiegających
|
1
|
|
RIKILT
|
NL
|
2,1
|
2,1
|
2,1
|
Wyniki odbiegające
|
DK
|
|
|
|
|
|
|
Wartość średnia
|
2,1
|
|
ADAS
|
GB
|
2,1
|
2,2
|
2,2
|
Wartość rzeczywista
|
2,1
|
|
CNEVA
|
FR2
|
2,1
|
2,1
|
2,1
|
Odchylenie standardowe powtarzalności (Sr)
|
0,09
|
|
LODI
|
IT
|
2,2
|
1,9
|
2,1
|
Względne odchylenie standardowe powtarzalności (RSDr%)
|
4,29
|
|
EELA
|
FI
|
2,3
|
2,3
|
2,3
|
Powtarzalność r (95 %)
|
0,26
|
|
D.V.F.A.
|
DK
|
3,4
|
2,9
|
3,2
|
Względna powtarzalność r %
|
12,01
|
|
|
|
|
|
|
Odchylenie standardowe odtwarzalności (SR)
|
0,25
|
|
|
|
|
|
|
Względne odchylenie standardowe odtwarzalności (RSDR%)
|
11,90
|
|
|
|
|
|
|
Odtwarzalność R (95 %)
|
0,69
|
|
|
|
|
|
|
Względna odtwarzalność R %
|
33,32
|
Tabela 5
Powtarzalność i odtwarzalność (przy zastosowaniu analizy FAME*)
|
|
Liczba laboratoriów
|
Wynik odbiegający
|
Powtarzalność
Sr (95 %)
|
Odtwarzalność
SR (95 %)
|
|
Próbka A
|
8
|
1
|
0,09
|
0.23
|
|
Próbka B
|
8
|
1
|
0,14
|
0,35
|
|
Próbka C
|
8
|
1
|
0,14
|
0,17
|
|
Próbka D
|
8
|
1
|
0,08
|
0,24
|
|
Wartość zbiorcza
|
|
|
0,116
|
0,256
|
|
|
|
|
R
|
R
|
|
Wartość zbiorcza* 2.8
|
|
|
0,324
|
0,716
|
|
CrD95 = 0,40
Minimalna czystość określona dla trienantanu = 95 %
Minimalna wartość graniczna określona dla trienantianu w tłuszczu maślanym = 11 kg/t
Przy uwzględnieniu różnicy krytycznej dla poziomu ufności wynoszącego 95 %, średnia wartość obu wyników nie może być niższa niż:
|
|
w przypadku włączenia trienantanu o czystości 95 % 10,05 kg/t.
|
|
Powtarzalność i odtwarzalność (bez stosowania analizy FAME)
|
|
Liczba laboratoriów
|
Wynik odbiegający
|
Powtarzalność
Sr (95 %)
|
Odtwarzalność
SR (95 %)
|
|
Próbka A
|
6
|
1
|
0,09
|
0,13
|
|
Próbka B
|
6
|
1
|
0,15
|
0,33
|
|
Próbka C
|
6
|
1
|
0,15
|
0,19
|
|
Próbka D
|
6
|
1
|
0,09
|
0,25
|
|
Wartość zbiorcza
|
|
|
0,124
|
0,237
|
|
|
|
|
r
|
R
|
|
Wartość zbiorcza* 2,8
|
|
|
0,347
|
0,663
|
|
CrD95 = 0,36
Minimalna czystość określona dla trienantanu = 95 %
Minimalna wartość graniczna określona dla trienantanu w tłuszczu maślanym = 11 kg/t
Przy uwzględnieniu różnicy krytycznej dla poziomu ufności wynoszącego 95 %, średnia wartość obu wyników nie może być niższa niż:
|
|
w przypadku włączenia trienantanu o czystości 95 % 10,09 kg/t.
|
|
Rysunek 1 (1)
Wyniki doświadczalne: próbka A

Wyniki doświadczalne: próbka B

Wyniki doświadczalne: próbka C

Wyniki doświadczalne: próbka D

Rysunek 2
Odchylenie standardowe powtarzalności i odtwarzalności na różnych poziomach (TG + FAME)

Rysunek 3
Odchylenie standardowe powtarzalności i odtwarzalności na różnych poziomach (TG)
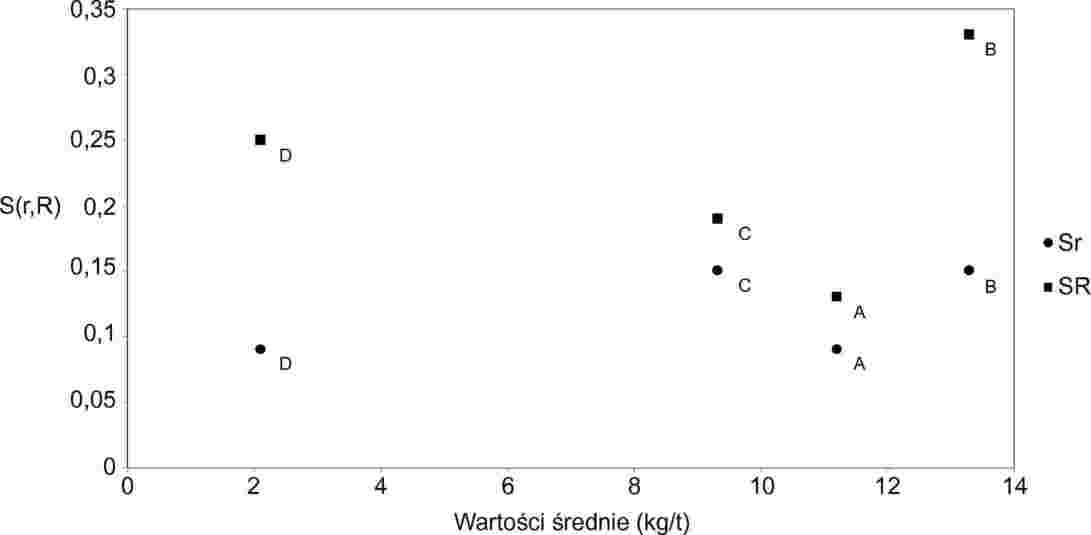
Rysunek 4
Przykład z wykorzystaniem dozownika nakolumnowego
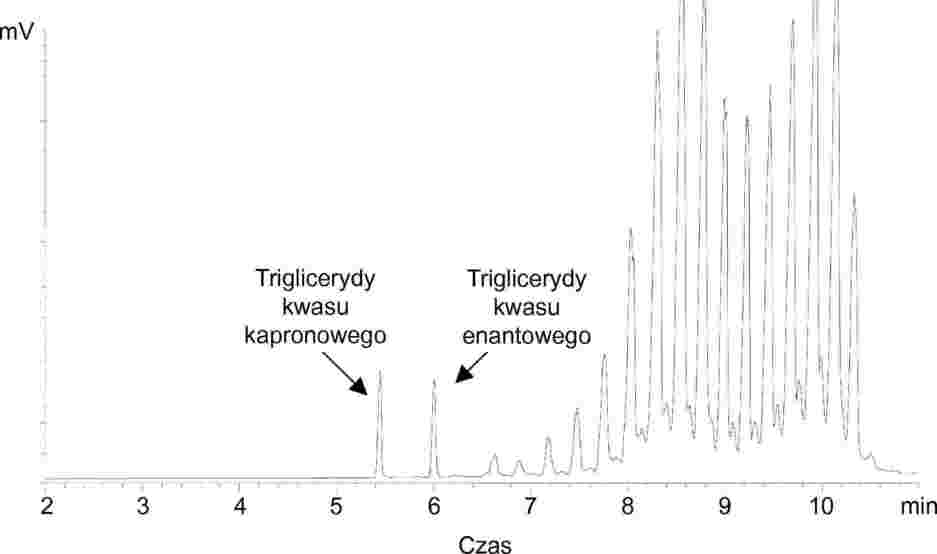
(1) = metoda FAME
ZAŁĄCZNIK VI
(Artykuł 5)
OZNACZANIE ZAWARTOŚCI WANILINY W KONCENTRACIE MASŁA, MAŚLE LUB ŚMIETANIE ZA POMOCĄ WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ HPLC
1. ZAKRES I DZIEDZINA STOSOWANIA
Niniejsza metoda opisuje procedurę ilościowego oznaczania waniliny w koncentracie masła, maśle lub śmietanie.
2. ZASADA
Ekstrakcja znanej ilości próbki za pomocą mieszaniny izopropanolu/etanolu/acetonitrylu (1:1:2). Wytrącenie większości tłuszczu przez schłodzenie do temperatury w granicach między –15 °C a –20 °C, a następnie wirowanie.
Po rozcieńczeniu wodą oznaczenie zawartości waniliny za pomocą wysokosprawnej chromatografii cieczowej chromatografii (HPLC).
3. APARATURA
Typowa aparatura laboratoryjna, w szczególności:
3.1. Zamrażarka, umożliwiająca pracę w zakresie temperatur od –15 °C do –20 °C
3.2. Strzykawki jednorazowe poj. 2 ml
3.3. Mikrosączki membranowe o średnicy porów 0,45 μm, odporne na działanie roztworu zawierającego 5 % roztwór ekstrakcyjny (ppkt 4.4)
3.4. Zestaw do chromatografii cieczowej składający się z pompy (natężenie przepływu 1,0 ml/min.), dozownika (możliwość wprowadzenia 20 μl, automatycznie lub ręcznie), detektora UV (obsługiwanego przy długości fali 306 nm, 0,01 Å pełna skala), rejestratora lub integratora oraz termostatu kolumny działającego w temp. 25 °C
3.5. Kolumna analityczna (250 mm × 4,6 mm śr. wewn.) z wypełnieniem LiChrospher RP 18 (Merck, 5 μm) lub równoważnym
3.6. Kolumna ochronna (ok. 20 mm × 3 mm śr. wewn.) z suchym wypełnieniem LiChrospher RP 18 (5–10 μm) lub równoważnym
3.7. Wirówka o prędkości obrotowej 2 000 obr./min.
4. ODCZYNNIKI
Wszystkie wykorzystywane odczynniki muszą być odczynnikami o uznanej klasie analitycznej.
4.1. Izopropanol
4.2. Etanol 96 % (v/v)
4.3. Acetonitryl
4.4. Roztwór ekstrakcyjny
Wymieszać izopropanol (ppkt 4.1), etanol (ppkt 4.2) i acetonitryl (ppkt 4.3) w stosunku objętościowym 1:1:2.
Wanilina (4-hydroksy-3-metoksybenzaldehyd) ≥ 98 %;
4.5.1. Roztwór podstawowy waniliny (= 500 μg/ml)
Odmierzyć wagowo z dokładnością do 0,1 mg ok. 50 mg (CM mg) waniliny (ppkt 4.5) do kolby miarowej poj. 100 ml, dodać 25 ml roztworu ekstrakcyjnego (ppkt 4.4) i uzupełnić wodą.
4.5.2. Roztwór wzorcowy waniliny (= 10 μg/ml)
Odmierzyć pipetą 5 ml roztworu podstawowego waniliny (ppkt 4.5.1) do kolby miarowej poj. 250 ml i uzupełnić wodą.
4.5.3. Metanol o jakości wymaganej dla HPLC
4.5.4. Kwas octowy lodowaty
4.5.5. Woda o jakości wymaganej dla HPLC
4.5.6. Faza ruchoma HPLC
Zmieszać 300 ml etanolu (ppkt 4.5.3) z ok. 500 ml wody (ppkt 4.5.5) i 20 ml kwasu octowego (ppkt 4.5.4) w kolbie miarowej poj. 1 000 ml, a następnie uzupełnić wodą (ppkt 4.5.5). Przesączyć przez sączek 0,45 μm (ppkt 3.3).
5. PROCEDURA
5.1. Przygotowanie badanej próbki
5.1.1. Masło
Podgrzewać próbkę do momentu, gdy zacznie się topić. Unikać lokalnego przegrzania powyżej 30 °C. Masło w żadnym razie nie może rozdzielić się na dwie fazy. Gdy próbka stanie się dostatecznie plastyczna, homogenizować ją przez wytrząsanie. Przed pobraniem próbki mieszać masło przez 15 s. Odmierzyć wagowo z dokładnością do 1 mg ok. 5 g (SM g) masła do kolby miarowej poj. 100 ml.
5.1.2. Koncentrat masła
Bezpośrednio przed pobraniem próbki umieścić pojemnik z koncentratem masła w piecu o temp. 40–50 °C, dopóki nie ulegnie całkowitemu stopieniu. Przygotować próbkę przez mieszanie ruchem okrężnym lub mieszanie, unikając tworzenia pęcherzyków powietrza na skutek zbyt energicznego mieszania. Odmierzyć wagowo z dokładnością do 1 mg ok. 4 g (SM g) koncentratu masła do kolby miarowej poj. 100 ml.
5.1.3. Śmietana
Podgrzać próbkę w łaźni wodnej lub w inkubatorze w temp. 35–40 °C. Rozprowadzić tłuszcz równomiernie przez mieszanie ruchem okrężnym lub, w razie potrzeby, przez mieszanie. Schłodzić szybko próbkę do temp. 20 °C ± 2 °C. Próbka powinna mieć jednorodny wygląd; w przeciwnym razie powtórzyć procedurę. Odmierzyć wagowo, z dokładnością do 1 g, 10 g (SM g) śmietany do kolby miarowej poj. 100 ml.
5.2. Przygotowanie roztworu badanego
Dodać ok. 75 ml roztworu ekstrakcyjnego (ppkt 4.4) do porcji badanej (ppkt 5.1.1, 5.1.2 lub 5.1.3), energicznie mieszać lub wstrząsać przez 15 minut i uzupełnić roztworem ekstrakcyjnym (ppkt 4.4). Przenieść ok. 10 ml tego wyciągu do probówki z korkiem. Umieścić probówkę w zamrażarce (ppkt 3.1) i pozostawić na ok. 30 minut. Wirować zimny wyciąg przez 5 minut z prędkością ok. 2 000 obr./min., a następnie niezwłocznie zdekantować. Pozostawić zdekantowany roztwór do czasu osiągnięcia temperatury pokojowej. Odmierzyć pipetą 5 ml zdekantowanego roztworu do kolby miarowej poj. 100 ml i uzupełnić wodą. Przesączyć podwielokrotność przez mikrosączek membranowy (ppkt 3.3) za pomocą strzykawki (ppkt 3.2). Przesącz jest gotowy do oznaczania za pomocą HPLC.
5.3. Kalibracja
Odmierzyć pipetą 5 ml roztworu wzorcowego waniliny (ppkt 4.5.2) do kolby miarowej poj. 100 ml. Dodać 5 ml roztworu ekstrakcyjnego (ppkt 4.4) i uzupełnić wodą do kreski. Otrzymany roztwór zawiera 0,5 μg/ml waniliny.
5.4. Oznaczanie za pomocą HPLC
Pozostawić układ chromatograficzny na 30 minut w celu ustabilizowania. Wprowadzić roztwór wzorcowy (ppkt 5.3). Powtarzać tę czynność, dopóki różnica pomiędzy powierzchniami pików lub wysokościami pików w dwóch kolejnych wprowadzeniach jest mniejsza niż 2 %. W opisanych warunkach czas retencji waniliny wynosi ok. 9 minut. Poddać analizie roztwór wzorcowy (ppkt 5.3) w dwóch egzemplarzach przez wprowadzenie 20 μl. Wprowadzić 20 μl roztworów badanych (ppkt 5.2). Określić powierzchnię lub wysokość otrzymanego piku waniliny. Powtórzyć podwójne wprowadzenie roztworu wzorcowego (ppkt 5.3) po 10 kolejnych wprowadzeniach próbek badanych (ppkt 5.2).
6. OBLICZANIE WYNIKÓW
Obliczyć średnią wielkość powierzchni (lub wysokość) (AC) pików waniliny powiązanych z granicznymi podwójnymi wprowadzeniami dla każdej partii roztworów badanych (łącznie cztery powierzchnie lub wysokości).
Obliczyć współczynnik odpowiedzi (R):

gdzie CM jest masą waniliny w mg (ppkt 4.5.1).
Zawartość (w mg/kg) waniliny (C) w próbce badanej określa wzór:
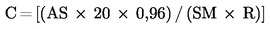
gdzie:
|
AS
|
=
|
powierzchnia lub wysokość piku waniliny w badanej próbce,
|
|
SM
|
=
|
masa badanej próbki w g (ppkt 5.1.1, 5.1.2 lub 5.1.3).
|
Uwaga: W przypadku gdy śmietana jest badana na obecność waniliny, stężenie znacznika musi być wyrażone w mg znacznika/kg tłuszczu mlecznego. Jest to obliczane przez pomnożenie C przez 100/f. Wartość f stanowi procentową zawartość tłuszczu w śmietanie (m/m).
|
20
|
=
|
współczynnik uwzględniający rozcieńczenie próbki wzorcowej i badanej
|
|
0,96
|
=
|
współczynnik korygujący dla zawartości tłuszczu w pierwszym rozcieńczeniu badanej próbki
|
Uwaga: Zamiast powierzchni pików można wykorzystać wysokości pików (zob. ppkt 8.3).
7. DOKŁADNOŚĆ METODY
7.1. Powtarzalność (r)
Różnica między wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez ten sam podmiot z zastosowaniem tej samej aparatury na identycznym materiale badanym nie może przekraczać 16 mg/kg.
7.2. Odtwarzalność (R)
Różnica między wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach z wykorzystaniem różnej aparatury na identycznym materiale badanym nie może przekraczać 27 mg/kg.
8. GRANICE TOLERANCJI
8.1. Z produktu znakowanego należy pobrać trzy próbki w celu sprawdzenia jednorodności.
Znacznik otrzymany z wanilii albo z waniliny syntetycznej
8.2.1. Stopień włączenia 4-hydroksy-3-metoksybenzaldehydu wynosi 250 g na tonę masła lub koncentratu masła. Jeżeli znacznik dodawany jest do śmietany, stopień włączenia wynosi 250 g na tonę tłuszczu mlecznego.
8.2.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
220,8 mg/kg (95 % minimalnego stopnia włączenia),
|
|
—
|
158,3 mg/kg (70 % minimalnego stopnia włączenia).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 220,8 mg/kg i 158,3 mg/kg.
Znacznik otrzymany wyłącznie ze strąków wanilii lub jej integralnych wyciągów:
8.3.1. Stopień włączenia 4-hydroksy-3-metoksybenzaldehydu wynosi 100 g na tonę koncentratu masła lub masła. Jeżeli znacznik dodawany jest do śmietany, stopień włączenia wynosi 100 g na tonę tłuszczu mlecznego.
8.3.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z tych wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
78,3 mg/kg (95 % minimalnego stopnia włączenia),
|
|
—
|
53,3 mg/kg (70 % minimalnego stopnia włączenia).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 78,3 mg/kg i 53,3 mg/kg.
9. UWAGI
9.1. Odzysk dodanej waniliny przy poziomie 250 mg/kg bezwodnego tłuszczu mlecznego waha się w granicach 97,0–103,8. Stwierdzona średnia zawartość wynosiła 99,9 % przy odchyleniu standardowym 2,7 %.
9.2. Roztwór wzorcowy zawiera 5 % roztworu ekstrakcyjnego w celu wyrównania poszerzenia piku spowodowanego obecnością 5 % roztworu ekstrakcyjnego w badanych próbkach. Umożliwia to oznaczenie ilościowe według wysokości pików.
9.3. Analiza oparta jest na liniowej krzywej kalibracji przechodzącej przez początek układu współrzędnych.
9.4. Przy użyciu odpowiednich rozcieńczeń roztworu wzorcowego (ppkt 4.5.2) należy sprawdzać liniowość — po raz pierwszy podczas dokonywania analizy, a następnie w regularnych odstępach czasu i po zmianach lub naprawach sprzętu HPLC. Wanilina może ulec rozkładowi do kwasu wanilinowego, diwaniliny i innych związków pod wpływem działania enzymów obecnych w niepasteryzowanej śmietanie lub jej produktach.
ZAŁĄCZNIK VII
(Artykuł 5)
OZNACZANIE ZAWARTOŚCI ESTRU ETYLOWEGO KWASU BETA-APO-8’-KAROTENOWEGO W KONCENTRACIE MASŁA ORAZ W MAŚLE METODĄ SPEKTROMETRYCZNĄ
1. ZAKRES I DZIEDZINA STOSOWANIA
W metodzie opisano procedurę ilościowego oznaczania zawartości estru etylowego kwasu beta-apo-8’-karotenowego (ester apokarotenowy) w koncentracie masła i w maśle. Ester apokarotenowy stanowi sumę wszystkich substancji obecnych w wyciągu próbek otrzymanych zgodnie z warunkami określonymi w metodzie, które absorbują światło przy 440 nm.
2. ZASADA
Tłuszcz maślany jest rozpuszczany w eterze naftowym i mierzy się absorbancję przy 440 nm. Zawartość estru apokarotenowego jest ustalona poprzez odniesienie do normy zewnętrznej.
3. APARATURA
3.1. Pipety miarowe poj. 0,25 ml, 0,50 ml, 0,75 ml i 1,0 ml.
3.2. Spektrofotometr — odpowiedni do użycia przy 440 nm (oraz przy 447–449 nm) i wyposażony w naczynka o długości drogi optycznej 1 cm.
3.3. Kolby miarowe poj. 20 ml i 100 ml.
3.4. Waga analityczna o czułości 0,1 mg, ważąca z dokładnością do 1 mg, o dokładności odczytu 0,1 mg.
3.5. Piec: 45 °C ± 1 °C.
3.6. Szybkosączące bezpopiołowe sączki papierowe.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej.
Zawiesina estru apokarotenowego (w przybliżeniu 20 %).
4.1.1. Ustalić zawartość zawiesiny w następujący sposób:
Podgrzać zawiesinę w temp. 45 °C–50 °C i homogenizować w zamkniętym oryginalnym pojemniku. Odmierzyć wagowo ok. 400 mg do kolby miarowej (100 ml); rozpuścić w 20 ml chloroformu (ppkt 4.4) i uzupełnić cykloheksanem (ppkt 4.5). Uzupełnić cykloheksanem 5 ml tego roztworu do całkowitej objętości 100 ml (roztwór A). Uzupełnić cykloheksanem 5,0 ml roztworu A do całkowitej objętości 100 ml. Zmierzyć absorbancję przy 447–449 nm (zmierzyć maksimum w stosunku do cykloheksanu, używając jako ślepej próby naczynek o długości drogi optycznej 1 cm).
Zawartość estru apokarotenowego P (%) = (Absmaks × 40 000)/(Mzaw × 2 550) lub przekształcić: (Absmaks/2 550) × (100/5) × (100/5) × (100/Mzaw)
|
Absmaks
|
=
|
absorbancja roztworu pomiarowego, wartość maksymalna
|
|
Mzaw
|
=
|
masa zawiesiny (g)
|
|
2 550
|
=
|
wartość odniesienia Abs (1 %, 1 cm)
|
|
P
|
=
|
czystość (zawartość) zawiesiny (%)
|
Uwaga: Zawiesina estru apokarotenowego jest wrażliwa na oddziaływanie powietrza, ciepła i światła. W zamkniętym oryginalnym pojemniku (w atmosferze azotu) i w chłodnym miejscu może być przechowywana przez okres ok. 12 miesięcy. Po otwarciu zawartość powinna być w krótkim czasie zużyta.
4.1.2. Roztwór wzorcowy estru apokarotenowego, w przybliżeniu 0,2 mg/ml
Odmierzyć wagowo z dokładnością do 1 mg ok. 0,100 g zawiesiny estru apokarotenowego (ppkt 4.1.1) (W), rozpuścić w eterze naftowym (ppkt 4.2), przenieść ilościowo do kolby miarowej poj. 100 ml i uzupełnić eterem naftowym do kreski.
Roztwór ten zawiera (W × P)/10 mg/ml estru apokarotenowego.
Uwaga: Roztwór należy przechowywać w chłodnym, ciemnym miejscu. Niewykorzystany roztwór należy wyrzucić po upływie jednego miesiąca.
4.2. Eter naftowy (40–60 °C)
4.3. Siarczan sodu, bezwodny, granulowany, wcześniej suszony przez dwie godziny w temp. 102 °C
4.4. Chloroform
4.5. Cykloheksan
5. PROCEDURA
5.1. Przygotowanie próbki badanej
5.1.1. Koncentrat masła
Roztopić próbkę w piecu w temp. ok. 45 °C.
5.1.2. Masło
Roztopić próbkę w piecu w temp. ok. 45 °C i przesączyć reprezentatywną porcję przez sączek zawierający ok. 10 g bezwodnego siarczanu sodu (ppkt 4.3) w środowisku osłoniętym przed silnym światłem naturalnym lub sztucznym i utrzymywanym w stałej temp. 45 °C. Zebrać odpowiednią ilość tłuszczu maślanego.
5.2. Oznaczanie
Odmierzyć wagowo z dokładnością do 1 mg ok. 1 g koncentratu masła (lub ekstrahowanego tłuszczu maślanego (ppkt 5.1.2)), (M). Przenieść ilościowo do kolby miarowej poj. 20 ml (V), wykorzystując eter naftowy (ppkt 4.2), uzupełnić do kreski i dokładnie wymieszać.
Przenieść podwielokrotność do 1 cm naczynka i zmierzyć absorbancję przy 440 nm w stosunku do ślepej próby eteru naftowego. Obliczyć stężenie estru apokarotenowego w roztworze poprzez odniesienie do otrzymanej krzywej wzorcowej (C μ/ml).
5.3. Kalibracja
Odmierzyć pipetą 0, 0,25, 0,5, 0,75 i 1,0 ml roztworu wzorcowego estru apokarotenowego (ppkt 4.1.2) do pięciu kolb miarowych poj. 100 ml. Uzupełnić eterem naftowym (ppkt 4.2) do pełnej objętości i wymieszać.
Przybliżone zakresy stężeń roztworów wynoszą 0–2 μg/ml i są dokładnie obliczone przez odniesienie do stężenia roztworu wzorcowego (ppkt 4.1.2) (W × P)/10 mg/ml. Zmierzyć absorbancję przy 440 nm w stosunku do ślepej próby eteru naftowego (ppkt 4.2).
Wykreślić wartości absorbancji na osi y, a stężenia estru apokarotenowego na osi x. Wyliczyć równanie krzywej wzorcowej.
6. OBLICZANIE WYNIKÓW
6.1. Zawartość estru apokarotenowego, wyrażona w mg/kg produktu, jest podawana jako:
Koncentrat masła: (C × V)/M
Masło: 0,82 (C × V)M
gdzie:
|
C
|
=
|
zawartość estru apokarotenowego, μg/ml, odczytana z wykresu kalibracji (ppkt 5.3),
|
|
V
|
=
|
objętość (ml) roztworu badanego (ppkt 5.2),
|
|
M
|
=
|
masa (g) porcji badanej (ppkt 5.2),
|
|
0,82
|
=
|
współczynnik korygujący zawartość tłuszczu maślanego w maśle.
|
7. DOKŁADNOŚĆ METODY
7.1. Powtarzalność
7.1.1. Analiza masła
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez jeden podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 1,4 mg/kg.
7.1.2. Analiza koncentratu masła
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez jeden podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 1,6 mg/kg.
7.2. Odtwarzalność
7.2.1. Analiza masła
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, wykorzystujące różną aparaturę, na identycznym materiale badanym, nie może przekraczać 4,7 mg/kg.
7.3. Analiza koncentratu masła
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, wykorzystujące różną aparaturę, na identycznym materiale badanym, nie może przekraczać 5,3 mg/kg.
7.4. Źródło danych dotyczących precyzji
Dane dotyczące precyzji zostały określone na podstawie doświadczenia przeprowadzonego w 1995 r. z udziałem 11 laboratoriów i przy użyciu 12 znakowanych próbek masła (sześć ślepych duplikatów) masła oraz 12 znakowanych próbek koncentratu masła (sześć ślepych duplikatów).
8. GRANICE TOLERANCJI
8.1. Z produktu znakowanego należy pobrać trzy próbki w celu sprawdzenia prawidłowości znakowania produktu.
8.2. Masło
8.2.1. Stopień włączenia w przypadku masła, przy uwzględnieniu absorbancji tła, wynosi 22 mg/kg.
8.2.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
17,7 mg/kg (95 % minimalnego stopnia włączenia),
|
|
—
|
12,2 mg/kg (70 % minimalnego stopnia włączenia).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 17,7 mg/kg i 12,2 mg/kg.
8.3. Koncentrat masła
8.3.1. Stopień włączenia w przypadku koncentratu masła, przy uwzględnieniu absorbancji tła, wynosi 24 mg/kg.
Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
19,2 mg/kg (95 % minimalnego stopnia włączenia),
|
|
—
|
13,2 mg/kg (70 % minimalnego stopnia włączenia).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 19,2 mg/kg i 13,2 mg/kg.
ZAŁĄCZNIK VIII
(Artykuł 5)
OZNACZANIE SITOSTEROLU I STIGMASTEROLU W MAŚLE LUB KONCENTRACIE MASŁA METODĄ CHROMATOGRAFII GAZOWEJ NA KOLUMNACH KAPILARNYCH
1. ZAKRES I DZIEDZINA STOSOWANIA
W metodzie opisano procedurę ilościowego oznaczania zawartości sitosterolu lub stigmasterolu w maśle i koncentracie masła. Przyjmuje się, że sitosterol stanowi sumę beta-sitosterolu i 22-dihydro-beta-sitosterolu, przy czym zakłada się, że zawartość pozostałych sitosteroli jest nieznaczna.
2. ZASADA
Masło lub koncentrat masła jest zmydlane przy użyciu wodorotlenku potasu w roztworze etanolu, a części nieulegające zmydleniu ekstrahuje się eterem dietylowym.
Sterole są przekształcane w etery trimetylosililowe i poddawane analizie metodą chromatografii gazowej na kolumnach kapilarnych w odniesieniu do wzorca wewnętrznego/betuliny.
3. APARATURA
3.1. Kolba do zmydlania poj. 150 ml połączona z chłodnicą zwrotną (złącza szlifowe)
3.2. Rozdzielacze poj. 500 ml
3.3. Kolby poj. 250 ml
3.4. Lejki wyrównujące ciśnienie, poj. 250 ml lub podobne, do zbierania odpadów eteru dietylowego
3.5. Kolumna szklana, 350 mm × 20 mm, wyposażona w korek ze spiekanego szkła
3.6. Łaźnia wodna lub płaszcz izolujący
3.7. Probówki reakcyjne poj. 2 ml
Chromatograf gazowy umożliwiający wykorzystanie kolumny kapilarnej i wyposażony w układ rozdzielający, w skład którego wchodzą:
3.8.1. komora termostatyczna do kolumn, umożliwiająca utrzymanie żądanej temperatury z dokładnością do ±1 °C;
3.8.2. wyparka z regulowaną temperaturą;
3.8.3. detektor jonizacji płomieniowej oraz konwerter-wzmacniacz;
3.8.4. integrator-rejestrator odpowiedni do wykorzystania z konwerterem-wzmacniaczem (ppkt 3.8.3).
3.9. Kolumna kapilarna ze stopionej krzemionki całkowicie pokryta powłoką BP1 lub równoważną (albo dowolna inna kolumna o co najmniej równej zdolności rozdzielczej), o jednolitej grubości 0,25 μm; kolumna musi umożliwiać rozdzielenie trimetylosililowych pochodnych lanosterolu i sitosterolu. Odpowiednia jest BP1 o długości 12 m i średnicy wewnętrznej 0,2 mm
3.10. Mikrostrzykawka do chromatografii gazowej poj. 1 μl z hartowaną igłą
4. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej. Należy używać wody destylowanej lub wody o równoważnym stopniu czystości.
4.1. Etanol, o czystości co najmniej 95 %
4.2. Wodorotlenek potasu, roztwór 60 %; rozpuścić 600 g wodorotlenku potasu (co najmniej 85 %) w wodzie i uzupełnić wodą do objętości 1 litra
Betulina, o czystości co najmniej 99 %
Roztwory betuliny w eterze dietylowym (ppkt 4.4)
4.3.1.1. Stężenie roztworu betuliny używane do oznaczania sitosterolu powinno wynosić 1,0 mg/ml
4.3.1.2. Stężenie roztworu betuliny używane do oznaczania stigmasterolu powinno wynosić 0,4 mg/ml
4.4. Eter dietylowy, o czystości analitycznej (bez nadtlenków lub pozostałości)
4.5. Siarczan sodu, bezwodny, granulowany, wcześniej suszony przez dwie godziny w temp. 102 °C
4.6. Odczynnik sililujący, np. TRI-SIL (produkowany przez Pierce Chemical Co, nr kat. 49001) lub równoważny. (Ważne: TRI-SIL jest łatwopalny, toksyczny, żrący i prawdopodobnie rakotwórczy. Pracownicy laboratorium muszą być zaznajomieni z parametrami bezpieczeństwa TRI-SIL i stosować właściwe środki ostrożności).
4.7. Lanosterol
Sitosterol o znanej czystości nie mniejszej niż 90 % (P)
Uwaga 1: Czystość materiałów wzorcowych wykorzystywanych do kalibracji musi być ustalona metodą standaryzacji. Należy założyć, że wszystkie sterole występujące w próbce zostają wykazane na chromatogramie, całkowita powierzchnia pików reprezentuje 100 % składników sterolowych, oraz że sterole wywołują taką samą reakcję detektora. Liniowość układu musi być potwierdzona przy zakresach stężeń będących przedmiotem zainteresowania.
4.8.1. Roztwór wzorcowy sitosterolu — przygotować roztwór zawierający, z dokładnością do 0,001 mg/ml, w przybliżeniu 0,5 mg/ml (W1) sitosterolu (ppkt 4.8) w eterze dietylowym (ppkt 4.4)
Stigmasterol o znanej czystości nie mniejszej niż 90 % (P)
4.9.1. Roztwór wzorcowy stigmasterolu — przygotować roztwór zawierający, z dokładnością do 0,001 mg/ml, w przybliżeniu 0,2 mg/ml (W1) stigmasterolu (ppkt 4.9) w eterze dietylowym (ppkt 4.4)
4.10. Mieszanina do badania zdolności rozdzielczej. Przygotować roztwór zawierający 0,05 mg/ml lanosterolu (ppkt 4.7) i 0,5 mg sitosterolu (ppkt 4.8) w eterze dietylowym (ppkt 4.4)
5. METODA
5.1. Przygotowanie roztworów wzorcowych do chromatografii
Wewnętrzny roztwór wzorcowy (ppkt 4.3.1) musi być dodany do odpowiedniego wzorcowego roztworu sterolu jednocześnie z dodaniem go do zmydlonej próbki (zob. ppkt 5.2.2).
5.1.1. Chromatograficzny roztwór wzorcowy sitosterolu: przenieść 1 ml roztworu wzorcowego sitosterolu (ppkt 4.8.1) do każdej z dwóch probówek reakcyjnych (ppkt 3.7) i usunąć eter dietylowy za pomocą strumienia azotu. Dodać 1 ml wewnętrznego roztworu wzorcowego (ppkt 4.3.1.1) i usunąć eter dietylowy za pomocą strumienia azotu
5.1.2. Chromatograficzny wzorcowy roztwór stigmasterolu: przenieść 1 ml roztworu wzorcowego stigmasterolu (ppkt 4.9.1) do każdej z dwóch probówek reakcyjnych (ppkt 3.7) i usunąć eter dietylowy za pomocą strumienia azotu. Dodać 1 ml wewnętrznego roztworu wzorcowego (ppkt 4.3.1.2) i usunąć eter dietylowy za pomocą strumienia azotu
5.2. Przygotowanie substancji niezmydlających się
5.2.1. Roztopić próbkę masła w temperaturze nieprzekraczającej 35 °C, po czym rozrobić, starannie mieszając
Odmierzyć wagowo, z dokładnością do 1 mg, w przybliżeniu 1 g masła (W2) lub koncentratu masła (W2) do kolby poj. 150 ml (ppkt 3.1). Dodać 50 ml etanolu (ppkt 4.1) i 10 ml roztworu wodorotlenku potasu (ppkt 4.2). Zainstalować chłodnicę zwrotną i podgrzewać przez 30 minut w temp. ok. 75 °C. Odłączyć chłodnicę i schłodzić kolbę do temperatury zbliżonej do temperatury otoczenia.
5.2.2. Dodać do kolby poj. 1,0 ml wewnętrznego roztworu wzorcowego (ppkt 4.3.1.1) w przypadku oznaczania sitosterolu, lub wewnętrznego roztworu wzorcowego (ppkt 4.3.1.2) w przypadku oznaczania stigmasterolu. Dokładnie wymieszać. Przenieść ilościowo zawartość kolby do rozdzielacza poj. 500 ml (ppkt 3.2), przemywając kolbę kolejno 50 ml wody i 250 ml eteru dietylowego (ppkt 4.4). Wstrząsać energicznie rozdzielaczem przez dwie minuty i pozostawić do rozdzielenia się faz. Usunąć niższą warstwę wodną i przemyć warstwę eteru, wstrząsając z czterema kolejnymi podwielokrotnościami 100 ml wody.
Uwaga 2: W celu uniknięcia utworzenia się emulsji istotne jest, aby pierwsze dwa przemycia wodą przeprowadzić ostrożnie (10 odwróceń). Podczas trzeciego przemywania można wstrząsać energicznie przez 30 sekund. W przypadku utworzenia się emulsji można ją rozbić, dodając w tym celu 5–10 ml etanolu. W razie dodania etanolu konieczne jest dwukrotne dodatkowe energiczne przemycie wodą.
5.2.3. Przepuścić klarowną, oddzieloną od mydła warstwę eteru przez szklaną kolumnę (ppkt 3.5) zawierającą 30 g bezwodnego siarczanu sodu (ppkt 4.5). Zebrać eter do kolby poj. 250 ml (ppkt 3.3). Dodać jedną granulkę zapobiegającą wrzeniu burzliwemu i odparować prawie do sucha w łaźni wodnej lub płaszczu izolującym, zwracając uwagę na zebranie odpadów rozpuszczalników.
Uwaga 3: Jeżeli wyciągi próbki zostaną całkowicie wysuszone w zbyt wysokiej temperaturze, mogą wystąpić straty sterolu.
5.3. Przygotowanie eterów trimetylosililowych
5.3.1. Przenieść pozostały w kolbie roztwór eteru do probówki reakcyjnej poj. 2 ml (ppkt 3.7) wraz z 2 ml eteru dietylowego, po czym usunąć eter za pomocą strumienia azotu. Przemyć kolbę dwoma dodatkowymi podwielokrotnościami 2 ml eteru dietylowego, przenosząc je do probówki i usuwając za każdym razem eter za pomocą azotu.
5.3.2. Sililować próbkę, dodając 1 ml TRI-SIL (ppkt 4.6). Zamknąć probówkę i wstrząsać energicznie w celu rozpuszczenia. Jeżeli całkowite rozpuszczenie nie nastąpi, podgrzać do temp. 65–70 °C. Odstawić na co najmniej pięć minut przed wprowadzeniem do chromatografu gazowego. Sililować wzorce w taki sam sposób jak próbki. Sililować mieszaninę do badania zdolności rozdzielczej (ppkt 4.10) w taki sam sposób jak próbki.
Uwaga 4: Proces sililowania musi być prowadzony w środowisku bezwodnym. Niepełne sililowanie betuliny uwidacznia się przez pojawienie się drugiego piku usytuowanego w pobliżu piku odpowiadającego betulinie.
Obecność etanolu na etapie sililowania będzie kolidować z procesem sililowania. Może to być wynikiem niedostatecznego przemycia na etapie ekstrakcji. Jeżeli problem utrzymuje się, na etapie ekstrakcji można przeprowadzić piąte przemycie przy energicznym wstrząsaniu przez 30 sekund.
5.4. Analiza metodą chromatografii gazowej
5.4.1. Dobór warunków roboczych
Ustawić chromatograf gazowy zgodnie z instrukcjami producenta.
Wskazówki dla warunków roboczych są następujące:
|
—
|
temperatura kolumny: 265 °C,
|
|
—
|
temperatura dozownika: 265 °C,
|
|
—
|
temperatura detektora: 300 °C,
|
|
—
|
natężenie przepływu strumienia gazu nośnego: 0,6 ml/min.,
|
|
—
|
ciśnienie wodoru: 84 kPa,
|
|
—
|
ciśnienie powietrza: 155 kPa,
|
|
—
|
rozdział próbki od 10:1 do 50:1; stosunek rozdziału musi być dobrany optymalnie zgodnie z instrukcją producenta i liniowością odpowiedzi detektora, a następnie potwierdzony przy zakresie stężenia będącym przedmiotem zainteresowania.
Uwaga 5: Szczególnie ważne jest regularne czyszczenie wkładki w dozowniku.
|
|
—
|
ilość wprowadzonej substancji: 1 μl roztworu TMSE.
|
Przed rozpoczęciem jakiejkolwiek analizy należy poczekać na osiągnięcie przez system stanu równowagi i uzyskanie jego dostatecznie stabilnej reakcji.
Powyższe warunki muszą być zróżnicowane w zależności od parametrów technicznych kolumny i chromatografu gazowego, tak aby otrzymać chromatogramy spełniające poniższe wymagania:
|
—
|
pik sitosterolu musi być odpowiednio oddzielony od piku lanosterolu. Na ryc. 1 przedstawiono typowy chromatogram, jaki powinna dawać sililowana mieszanina do badania zdolności rozdzielczej (ppkt 4.10),
|
|
—
|
względne czasy retencji poniższych steroli powinny wynosić w przybliżeniu:
|
|
—
|
czas retencji betuliny powinien wynosić w przybliżeniu 24 minuty.
|
5.4.2. Procedura analityczna
Wprowadzić 1 μl sililowanego roztworu wzorcowego (stigmasterolu lub sitosterolu) i dostosować parametry kalibracyjne integratora.
Wprowadzić kolejną porcję 1 μl sililowanego roztworu wzorcowego w celu określenia współczynników odpowiedzi w odniesieniu do betuliny.
Wprowadzić 1 μl sililowanego roztworu próbki i zmierzyć powierzchnie pików. Każda seria chromatograficzna musi być oddzielona przez wprowadzenie wzorców.
Wskazówka: poszczególne serie muszą zawierać sześć wprowadzeń próbki.
Uwaga 6: Całkowanie piku stigmasterolu powinno obejmować każdy przypadek ogonowania zgodnie z pkt 1, 2 i 3 na ryc. 2b.
Przy dokonywaniu oceny całkowitego sitosterolu całkowanie piku sitosterolu powinno obejmować powierzchnię piku 22 dihydro-beta-sitosterolu (stigmastanolu), który jest eluowany bezpośrednio po sitosterolu (zob. ryc. 3b).
6. OBLICZANIE WYNIKÓW
6.1. Określić powierzchnię pików sterolu i pików betuliny w obu wzorcach oddzielających poszczególne serie i obliczyć R1:
R1 = (średnia powierzchnia piku sterolu we wzorcu)/(średnia powierzchnia piku betuliny we wzorcu)
Określić powierzchnię piku sterolu (stigmasterolu i sitosterolu) i piku betuliny w próbce i obliczyć R2:
R2 = (powierzchnia piku sterolu w próbce)/(powierzchnia piku betuliny w próbce)
|
W1
|
=
|
zawartość sterolu we wzorcu (mg) zawarta w 1 ml roztworu wzorcowego (ppkt 4.8.1 lub 4.9.1)
|
|
W2
|
=
|
masa próbki (g) (ppkt 5.2.1)
|
|
P
|
=
|
czystość sterolu wzorcowego (ppkt 4.8 lub 4.9)
|
Zawartość sterolu w próbce (mg/kg) = ((R
2)/(R
1)) × ((W
1)/(W
2)) × P × 10.
7. DOKŁADNOŚĆ METODY
7.1. Masło
7.1.1. Powtarzalność
7.1.1.1. Stigmasterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez ten sam podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 19,3 mg/kg.
7.1.1.2. Sitosterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez ten sam podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 23,0 mg/kg.
7.1.2. Odtwarzalność
7.1.2.1. Stigmasterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, przy użyciu różnej aparatury, na identycznym materiale badanym, nie może przekraczać 31,9 mg/kg.
7.1.2.2. Sitosterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, przy użyciu różnej aparatury, na identycznym materiale badanym, nie może przekraczać 8,7 % względem średniej wartości oznaczenia.
7.1.3. Źródło danych dotyczących precyzji
Dane dotyczące precyzji zostały określone na podstawie doświadczenia przeprowadzonego w 1992 r. z udziałem ośmiu laboratoriów i przy użyciu sześciu próbek stigmasterolu (trzy ślepe duplikaty) oraz sześciu próbek sitosterolu (trzy ślepe duplikaty).
7.2. Koncentrat masła
7.2.1. Powtarzalność
7.2.1.1. Stigmasterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez ten sam podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 10,2 mg/kg.
7.2.1.2. Sitosterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych w możliwie najkrótszym odstępie czasu przez ten sam podmiot wykorzystujący tę samą aparaturę na identycznym materiale badanym nie może przekraczać 3,6 mg/kg względem średniej wartości oznaczeń.
7.2.2. Odtwarzalność
7.2.2.1. Stigmasterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, przy użyciu różnej aparatury, na identycznym materiale badanym, nie może przekraczać 25,3 mg/kg.
7.2.2.2. Sitosterol
Różnica pomiędzy wynikami dwóch oznaczeń przeprowadzonych przez podmioty w różnych laboratoriach, przy użyciu różnej aparatury, na identycznym materiale badanym, nie może przekraczać 8,9 % względem średniej wartości oznaczeń.
7.2.3. Źródło danych dotyczących precyzji
Dane dotyczące precyzji zostały określone na podstawie doświadczenia przeprowadzonego w 1991 r. z udziałem dziewięć laboratoriów i przy użyciu sześciu próbek stigmasterolu (trzy ślepe duplikaty) oraz sześciu próbek sitosterolu (trzy ślepe duplikaty).
8. GRANICE TOLERANCJI
8.1. Z produktu znakowanego należy pobrać trzy próbki w celu sprawdzenia prawidłowości znakowania produktu.
8.2. Masło
8.2.1. Stigmasterol
8.2.1.1. Stopień włączenia stigmasterolu wynosi 150 g stigmasterolu o czystości co najmniej 95 % na tonę masła, tzn. 142,5 mg/kg, lub 170 g stigmasterolu o czystości co najmniej 85 % na tonę masła, tzn. 144,5 mg/kg.
8.2.1.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
115,8 mg/kg (95 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 95 %),
|
|
—
|
117,7 mg/kg (95 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 85 %),
|
|
—
|
80,1 mg/kg (70 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 95 %),
|
|
—
|
81,5 mg/kg (70 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 85 %).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją pomiędzy, odpowiednio, 115,8 mg/kg i 80,1 mg/kg lub 117,7 mg/kg i 81,5 mg/kg.
8.2.2. Sitosterol
8.2.2.1. Stopień włączenia sitosterolu wynosi 600 g sitosterolu o czystości co najmniej 90 % na tonę masła, tzn. 540 mg/kg
8.2.2.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
482,6 mg/kg (95 % minimalnego stopnia włączenia w przypadku sitosterolu o czystości 90 %),
|
|
—
|
347,6 mg/kg (70 % minimalnego stopnia włączenia w przypadku sitosterolu o czystości 90 %).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 482,6 mg/kg i 347,6 mg/kg.
8.3. Koncentrat masła
8.3.1. Stigmasterol
8.3.1.1. Stopień włączenia stigmasterolu wynosi 150 g stigmasterolu o czystości co najmniej 95 % na tonę koncentratu masła, tzn. 142,5 mg/kg, lub 170 g stigmasterolu o czystości co najmniej 85 % na tonę koncentratu masła, tzn. 144,5 mg/kg.
8.3.1.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
118,5 mg/kg (95 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 95 %),
|
|
—
|
120,4 mg/kg (95 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 85 %),
|
|
—
|
82,9 mg/kg (70 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 95 %),
|
|
—
|
84,3 mg/kg (70 % minimalnego stopnia włączenia w przypadku stigmasterolu o czystości 85 %).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją pomiędzy, odpowiednio, 118,5 mg/kg i 82,9 mg/kg lub 120,4 mg/kg i 84,3 mg/kg.
8.3.2. Sitosterol
8.3.2.1. Stopień włączenia sitosterolu wynosi 600 g sitosterolu o czystości co najmniej 90 % na tonę koncentratu masła, tzn. 540 mg/kg.
8.3.2.2. Wyniki dla trzech próbek, otrzymane z analizy produktu, są wykorzystywane do sprawdzenia szybkości i równomierności włączenia znacznika, a najniższy z wyników porównuje się z poniższymi wartościami granicznymi:
|
—
|
480,9 mg/kg (95 % minimalnego stopnia włączenia w przypadku sitosterolu o czystości 90 %),
|
|
—
|
345,9 mg/kg (70 % minimalnego stopnia włączenia w przypadku sitosterolu o czystości 90 %).
|
Stężenie znacznika w próbce, która daje najniższy wynik, jest wykorzystywane w powiązaniu z interpolacją między 480,9 mg/kg i 345,9 mg/kg.
Ryc. 1
Chromatogram mieszaniny do badania zdolności rozdzielczej
Najbardziej odpowiednie jest całkowite rozdzielenie, tzn. linia tworząca zarys piku lanosterolu powinna wrócić do linii podstawowej przed przekształceniem się w pik sitosterolu, jednak niepełne rozdzielenie jest dopuszczalne.

|
Ryc. 2a
|
Ryc. 2b
|
|
Wzorzec stigmasterolu
|
Próbka masła skażona stigmasterolem
|
|

|

|
|
Uwaga: Całkowanie piku stigmasterolu powinno uwzględnić każde ogonowanie zgodnie z definicją podaną w pkt 1, 2 i 3.
|
|
Ryc. 3a
|
Ryc. 3b
|
|
Wzorzec sitosterolu
|
Próbka masła skażona beta-sitosterolem
|
|

|

|
|
Uwaga: Beta-sitosterol często zawiera zanieczyszczenie (wykrywane jako stigmasterol), które jest eluowane bezpośrednio po beta-sitosterolu. Przy dokonywaniu oceny całkowitego obecnego beta-sitosterolu powierzchnie obu tych pików należy zsumować.
|
ZAŁĄCZNIK IX
(Artykuł 6)
REFERENCYJNA METODA WYKRYWANIA OBECNOŚCI MLEKA KROWIEGO I KAZEINIANÓW W SERACH Z MLEKA OWCZEGO, MLEKA KOZIEGO, MLEKA BAWOLEGO LUB MIESZANEK MLEKA OWCZEGO, KOZIEGO I BAWOLEGO
1. ZAKRES
Wykrywanie obecności mleka krowiego i kazeinianów w serach wyprodukowanych z mleka owczego, mleka koziego, mleka bawolego lub mieszanek mleka owczego, koziego i bawolego przez wyznaczanie punktu izoelektrycznego gamma-kazeiny po plazminolizie.
2. DZIEDZINA STOSOWANIA
Metoda jest odpowiednia dla czułego i specyficznego wykrywania naturalnego oraz poddanego obróbce cieplnej mleka krowiego i kazeinianów w świeżych i dojrzałych serach wyprodukowanych z mleka owczego, mleka koziego, mleka bawolego lub mieszanin mleka owczego, koziego i bawolego. Metoda nie jest odpowiednia do wykrywania przypadków fałszowania mleka i sera przy użyciu poddanych obróbce cieplnej koncentratów białkowych serwatki pochodzącej od krów.
3. ZASADA METODY
3.1. Oddzielenie kazeiny z sera i wzorców odniesienia
3.2. Rozpuszczenie oddzielonej kazeiny i poddanie jej rozszczepieniu za pomocą plazminy (EC.3.4.21.7)
3.3. Wyznaczenie punktu izoelektrycznego kazeiny poddanej działaniu plazminy w obecności mocznika oraz barwienie białka
3.4. Ocena zabarwionych wzorów gamma3-kazeiny i gamma2-kazeiny (dowód na występowanie mleka krowiego) poprzez porównanie wzoru otrzymanego z próbki z wzorami otrzymanymi w tym samym żelu ze wzorców odniesienia zawierających 0 % i 1 % mleka krowiego.
4. ODCZYNNIKI
Jeżeli nie wskazano inaczej, należy stosować chemikalia klasy analitycznej. Woda musi być redestylowana lub o równoważnym stopniu czystości.
Uwaga: Poniższe dane szczegółowe odnoszą się do przygotowanych w laboratorium żeli poliakryloamidowych zawierających mocznik, o wymiarach 265 × 125 × 0,25 mm. W przypadku gdy stosuje się inne rozmiary i rodzaje żelu, niezbędne może się okazać odpowiednie dostosowanie warunków rozdzielania.
Wyznaczenie punktu izoelektrycznego
4.1. Odczynniki do produkcji żeli poliakryloamidowych zawierających mocznik.
4.1.1. Roztwór podstawowy żelu
Rozpuścić:
4,85 g akryloamidu
0,15 g N, N’-metyleno-bis-akryloamidu (BIS)
48,05 g mocznika
15,00 g gliceryny (87 % w/w)
w wodzie, uzupełnić do 100 ml i przechowywać w butelce z brązowego szkła w chłodziarce.
Uwaga: Można stosować dostępny w handlu wstępnie zestawiony roztwór akryloamid/BIS zamiast podanych mas neurotoksycznych akryloamidów. W przypadku gdy taki roztwór zawiera 30 % w/v akryloamidu i 0,8 % w/v BIS, należy użyć do preparatu objętości 16,2 ml zamiast podanych mas. Okres przechowywania roztworu podstawowego wynosi maksymalnie 10 dni; jeżeli przewodnictwo roztworu jest wyższe niż 5 μS, dejonizować poprzez mieszanie z 2 g Amberlitu MB-3 przez 30 minut, a następnie przesączyć przez sączek membranowy o średnicy porów 0,45 μm.
4.1.2. Roztwór żelu
Przygotować roztwór żelu przez wymieszanie dodatków i amfolitów z podstawowym roztworem żelu (zob. ppkt 4.1.1).
9,0 ml roztworu podstawowego
24 mg beta-alaniny
500 μl amfolitu o pH 3,5–9,5 (1)
250 μl amfolitu o pH 5–7 (1)
250 µl amfolitu o pH 6–8 (1)
Wymieszać roztwór żelu i odgazowywać przez dwie do trzech minut w łaźni ultradźwiękowej lub w próżni.
Uwaga: Przygotować roztwór żelu bezpośrednio przed wlaniem (zob. ppkt 6.2).
4.1.3. Roztwory katalityczne
4.1.3.1. N, N, N’ N’ — tetrametyloetylenodiamina (Temed)
4.1.3.2. nadsiarczan amonu (PER) 40 % w/v:
rozpuścić 800 mg PER w wodzie i uzupełnić do 2 ml.
Uwaga: Zawsze stosować świeżo przygotowany roztwór PER.
4.2. Płyn kontaktowy
Nafta lub parafina ciekła.
4.3. Roztwór anodowy
Rozpuścić 5,77 g kwasu ortofosforowego (85 % w/w) w wodzie i uzupełnić do 100 ml.
4.4. Roztwór katodowy
Rozpuścić 2,00 g wodorotlenku sodu w wodzie i uzupełnić wodą do 100 ml.
Przygotowanie próbki
4.5. Odczynniki do oddzielenia białka
4.5.1. Rozcieńczyć kwas octowy (objętość 25,0 ml kwasu octowego lodowatowego uzupełniona wodą do objętości 100 ml)
4.5.2. Dichlorometan
4.5.3. Aceton
4.6. Bufor do rozpuszczania białka
Rozpuścić:
5,75 g gliceryny (87 % w/w)
24,03 g mocznika
250 mg ditiotreitolu
w wodzie i uzupełnić do 50 ml.
Uwaga: Przechowywać w chłodziarce; maksymalny okres przechowywania wynosi jeden tydzień.
4.7. Odczynniki do rozszczepienia kazeiny za pomocą plazminy
4.7.1. Bufor z węglanem amonu
Miareczkować roztwór wodorowęglanu amonu o stężeniu 0,2 mol/l (1,58 g/100 ml wody) zawierający 0,05 mol/l kwasu etylenodiaminotetraoctowego (EDTA, 1,46 g/100 ml) roztworem węglanu amonu o stężeniu 0,2 mol/l (1,92 g/100 ml wody) zawierającym 0,05 mol/l EDTA do osiągnięcia ph 8.
4.7.2. Plazmina bydlęca (EC. 3.4.21.7) o aktywności co najmniej 5 U/ml
4.7.3. Roztwór kwasu ε-aminokapronowego do inhibicji enzymów
Rozpuścić 2,624 g kwasu ε-aminokapronowego (kwas 6-aminoheksanowy) w 100 ml etanolu o stężeniu 40 % (v/v).
4.8. Wzorce
4.8.1. Certyfikowane wzorce odniesienia mieszanki odtłuszczonego mleka owczego i koziego z dodatkiem podpuszczki, zawierające 0 % i 1 % mleka krowiego, dostępne są w Instytucie Materiałów Referencyjnych i Pomiarów przy Komisji, B-2440 Geel, Belgia
4.8.2. Przygotowanie laboratoryjnych wzorców pośrednich mleka bawolego z dodatkiem podpuszczki, zawierających 0 % i 1 % mleka krowiego
Mleko odtłuszczone przygotowuje się przez odwirowanie surowego mleka bawolego lub krowiego w temp. 37 °C przy 2 500 g przez 20 minut. Po szybkim schłodzeniu probówki wraz z jej zawartością do 6–8 °C, górna warstwa tłuszczu usuwana jest w całości. Aby przygotować wzorzec 1 % należy dodać 5,00 ml odtłuszczonego mleka krowiego do 495 ml odtłuszczonego mleka bawolego w jednolitrowej zlewce, doprowadzić pH do wartości 6,4 przez dodanie rozcieńczonego kwasu mlekowego (10 % w/v). Doprowadzić temperaturę do 35 °C i dodać 100 μl podpuszczki cielęcej (aktywność podpuszczki 1:10 000, ok. 3 000 U/ml), mieszać przez 1 minutę, po czym odstawić zlewkę przykrytą folią aluminiową na jedną godzinę w temp. 35 °C, aby uformował się twaróg. Po uformowaniu twarogu całe mleko z podpuszczką jest suszone sublimacyjnie bez uprzedniej homogenizacji lub odsączania serwatki. Po wysuszeniu sublimacyjnym mleko jest dokładnie rozdrabniane do uzyskania jednorodnego proszku. Aby przygotować wzorzec 0 % należy przeprowadzić taką samą procedurę przy użyciu prawdziwego odtłuszczonego mleka bawolego. Wzorce należy przechowywać w temp. –20 °C.
Uwaga: Przed przygotowaniem wzorców zaleca się zbadanie mleka bawolego pod względem jego czystości poprzez wyznaczenie punktu izoelektrycznego kazeiny poddanej działaniu plazminy.
Odczynniki do barwienia białka
4.9. Utrwalacz
Rozpuścić 150 mg kwasu trichlorooctowego w wodzie i uzupełnić do 1 000 ml.
4.10. Roztwór odbarwiający
Uzupełnić 500 ml metanolu i 200 ml kwasu octowego lodowatego wodą destylowaną do objętości 2 000 ml.
Uwaga: Należy przygotowywać świeży roztwór odbarwiający każdego dnia; można go przygotować poprzez zmieszanie jednakowych objętości roztworów podstawowych 50 % (v/v) metanolu i 20 % (v/v) kwasu octowego lodowatego.
4.11. Roztwory barwiące
4.11.1. Roztwór barwiący (roztwór podstawowy 1)
Rozpuścić 3,0 g barwnika Coomassie Brilliant Blue G-250 (C.I. 42655) w 1 000 ml metanolu o stężeniu 90 % (v/v) za pomocą mieszadła magnetycznego (ok. 45 minut), przesączyć przez dwa złożone sączki o średniej prędkości sączenia.
4.11.2. Roztwór barwiący (roztwór podstawowy 2)
Rozpuścić 5,0 g siarczanu miedzi pięciowodnego w 1 000 ml kwasu octowego o stężeniu 20 % (v/v).
4.11.3. Roztwór barwiący (roztwór roboczy)
Zmieszać razem 125 ml każdego z roztworów podstawowych (ppkt 4.11.1, 4.11.2) bezpośrednio przed barwieniem.
Uwaga: Roztwór barwiący powinien być przygotowany w dniu, w którym ma zostać użyty.
5. WYPOSAŻENIE
5.1. Szklane płytki (265 × 125 × 4 mm); wałek gumowy (szerokość 15 cm); stół poziomujący
5.2. Arkusz nośny żelu (265 × 125 mm)
5.3. Arkusz przykrywający (280 × 125 mm). Przykleić pasek taśmy klejącej (280 × 6 × 0,25 mm) wzdłuż obu dłuższych krawędzi (zob. ryc. 1)
5.4. Komora elektroogniskowania z płytą chłodzącą (na przykład 265 × 125 mm) oraz odpowiednim układem zasilania (≥ 2,5 kV) lub automatyczny system elektroforezy
5.5. Kriostat cyrkulacyjny, regulowany termostatem w temp. 12 ± 0,5 °C
5.6. Wirówka z regulacją do 3 000 g
5.7. Elektrody paskowe (o długości ≥ 265 mm)
5.8. Plastikowe butelki z wkraplaczem do roztworu anodowego i katodowego
5.9. Aplikatory próbek (10 × 5 mm, wiskoza lub sączek papierowy o niskim współczynniku adsorpcji białka)
5.10. Nożyczki, skalpele i szczypce ze stali nierdzewnej
5.11. Naczynia do barwienia i odbarwiania ze stali nierdzewnej lub ze szkła (na przykład tace na narzędzia o wymiarach 280 × 150 mm)
5.12. Nastawny homogenizator obrotowy (średnica wału 10 mm), liczba obr./min. od 8 000 do 20 000
5.13. Mieszadło magnetyczne
5.14. Łaźnia ultradźwiękowa
5.15. Zgrzewarka do folii
5.16. Mikropipety poj. 25 μl
5.17. Koncentrator próżniowy lub suszarka sublimacyjna
5.18. Łaźnia wodna kontrolowana termostatycznie, nastawna do 35 °C i 40 ± 1 °C, z wytrząsarką
5.19. Aparatura densytometryczna umożliwiająca odczyt przy λ = 634 nm
6. PROCEDURA
6.1. Przygotowanie próbki
6.1.1. Oddzielenie kazeiny
Odmierzyć wagowo równowartość 5,0 g suchej masy sera lub wzorców odniesienia do probówki wirówkowej poj. 100 ml, dodać 60 ml destylowanej wody i homogenizować za pomocą homogenizatora obrotowego (8 000–10 000 obr./min.). Doprowadzić pH do wartości 4,6 za pomocą rozcieńczonego kwasu octowego (ppkt 4.5.1) i odwirować (5 minut, 3 000 g). Zdekantować tłuszcz i serwatkę, homogenizować pozostałość przy prędkości 20 000 obr./min. w 40 ml destylowanej wody o pH dostosowanym do wartości 4,5 za pomocą rozcieńczonego kwasu octowego (ppkt 4.5.1), dodać 20 ml dichlorometanu (ppkt 4.5.2), ponownie homogenizować i odwirować (5 minut, 3 000 g). Usunąć warstwę kazeiny znajdującą się pomiędzy fazą wodną i organiczną (zob. ryc. 2) za pomocą szpatułki i zdekantować obydwie fazy. Ponownie homogenizować kazeinę w 40 ml destylowanej wody (zob. wyżej) oraz 20 ml dichlorometanu (ppkt 4.5.2) i odwirować. Powtarzać tę procedurę do czasu, gdy obie fazy ekstrakcyjne staną się bezbarwne (dwa do trzech razy). Homogenizować pozostałości białka wraz z 50 ml acetonu (ppkt 4.5.3) i przesączyć przez złożony sączek papierowy o średniej prędkości sączenia. Przemyć pozostałości znajdujące się na sączku, każdorazowo za pomocą dwóch oddzielnych 25 ml porcji acetonu, i pozostawić do wysuszenia na powietrzu lub suszyć w strumieniu azotu, na koniec dokładnie sproszkować w moździerzu.
Uwaga: Oddzieloną suchą kazeinę należy przechowywać w temp. –20 °C.
6.1.2. Hydrolizowanie beta-kazeiny za pomocą plazminy w celu zwiększenia intensywności uwidocznienia gamma-kazeiny
Zdyspergować 25 mg oddzielonej kazeiny (ppkt 6.1.1) w 0,5 ml buforu z węglanem amonu (ppkt 4.7.1) i homogenizować przez 20 minut, stosując, na przykład, obróbkę ultradźwiękową. Podgrzać do 40 °C i dodać 10 μl plazminy (ppkt 4.7.2), wymieszać i inkubować przez jedną godzinę w temp. 40 °C, nieprzerwanie wstrząsając. Aby doprowadzić do inhibicji enzymu, dodać 20 μl roztworu kwasu ε-aminokapronowego (ppkt 4.7.3), następnie dodać 200 g mocznika w postaci stałej i 2 mg ditiotreitolu.
Uwaga: Aby otrzymać lepszą symetrię w skupionych pasmach kazeiny, zaleca się suszenie sublimacyjne roztworu po dodaniu kwasu ε-aminokapronowego, a następnie rozpuszczenie pozostałości w 0,5 ml buforu do rozpuszczania białka (ppkt 4.6).
6.2. Przygotowanie żeli poliakryloamidowych zawierających mocznik
Dodając kilka kropli wody, rozwałkować arkusz nośny żelu (ppkt 5.2) na szklanej płytce (ppkt 5.1), usuwając resztki wody ręcznikiem papierowym lub bibułą. W ten sam sposób rozwałkować arkusz przykrywający (ppkt 5.3) z przekładkami (0,25 mm) na drugiej płytce szklanej. Położyć płytkę poziomo na stole poziomującym.
Dodać 10 μl Temedu (ppkt 4.1.3.1) do przygotowanego i odpowietrzonego roztworu żelu (ppkt 4.1.2), wymieszać i dodać 10 μl roztworu PER (ppkt 4.1.3.2), starannie wymieszać i natychmiast wylać równomiernie na środek arkusza przykrywającego. Umieścić jedną krawędź płytki pokrytej arkuszem nośnym żelu (stroną pokrytą do dołu) na arkuszu przykrywającym i opuszczać ją powoli, tak aby między arkuszami uformowała się cienka powłoka żelu, rozprowadzona równomiernie i pozbawiona pęcherzyków (ryc. 3). Ostrożnie opuścić płytkę pokrytą arkuszem nośnym żelu, pomagając sobie cienką szpatułką, tak by przylegała do arkusza przykrywającego, i umieścić na wierzchu trzy dodatkowe płytki szklane w charakterze obciążników. Po zakończeniu polimeryzacji (ok. 60 minut) zdjąć spolimeryzowany żel na płytkę pokrytą arkuszem nośnym żelu razem z arkuszem przykrywającym, obstukując lekko płytki szklane. Ostrożnie wyczyścić drugą stronę płytki celem usunięcia pozostałości żelu oraz mocznika. Zgrzać „kanapkę” żelu w tubie foliowej i przechowywać w chłodziarce (maksymalnie przez sześć tygodni).
Uwaga: Arkusz przykrywający z przekładkami może być użyty ponownie. Żel poliakryloamidowy może być pocięty na mniejsze fragmenty, co jest zalecane w przypadku, kiedy jest kilka próbek, lub kiedy używa się automatycznego systemu elektroforezy (dwa żele, rozmiar 4,5 × 5 cm).
6.3. Wyznaczenie punktu izoelektrycznego
Nastawić termostat chłodzący na 12 °C. Przetrzeć drugą stronę arkusza nośnego żelu naftą, po czym nanieść kilka kropel nafty (ppkt 4.2) na środkową część bloku chłodzącego. Następnie rozwałkować na nim „kanapkę” żelu, stroną pokrytą arkuszem nośnym do dołu, nie dopuszczając do powstawania pęcherzyków. Wytrzeć nadmiar nafty i usunąć arkusz przykrywający. Nasączyć elektrody paskowe roztworami elektrodowymi (ppkt 4.3, 4.4), przyciąć do długości żelu i umieścić w określonym położeniu (odległość między elektrodami 9,5 cm).
Warunki wyznaczenia punktu izoelektrycznego:
6.3.1. Rozmiar żelu 265 × 125 × 0,25 mm
|
Etap
|
Czas
(minuty)
|
Napięcie elektryczne
(V)
|
Natężenie elektryczne
(mA)
|
Moc
(W)
|
Woltogodziny
(Vh)
|
|
|
30
|
maksymalnie 2 500
|
maksymalnie 15
|
stała 4
|
ok. 300
|
|
2.
|
Ogniskowanie próbki (2)
|
|
60
|
maksymalnie 2 500
|
maksymalnie 15
|
stała 4
|
ok. 1 000
|
|
|
60
|
maksymalnie 2 500
|
maksymalnie 5
|
maksymalnie 20
|
ok. 3 000
|
|
40
|
maksymalnie 2 500
|
maksymalnie 6
|
maksymalnie 20
|
ok. 3 000
|
|
30
|
maksymalnie 2 500
|
maksymalnie 7
|
maksymalnie 25
|
ok. 3 000
|
Uwaga: Jeżeli grubość lub szerokość żeli zostały zmienione, wartości natężenia elektrycznego i mocy należy odpowiednio dostosować (np. w przypadku stosowania żelu o rozmiarach 265 × 125 × 0,5 mm należy podwoić wartości natężenia elektrycznego i mocy).
6.3.2. Przykładowe programowanie napięcia elektrycznego dla urządzenia do automatycznej elektroforezy (dwa żele 5,0 × 4,5 cm), elektrody bez pasków przyłożone bezpośrednio do żelu.
|
Etap
|
Napięcie elektryczne
|
Natężenie elektryczne
|
Moc
|
Temperatura
|
Woltogodziny
|
|
|
1 000 V
|
10,0 mA
|
3,5 W
|
8 °C
|
85 Vh
|
|
|
250 V
|
5,0 mA
|
2,5 W
|
8 °C
|
30 Vh
|
|
|
1 200 V
|
10,0 mA
|
3,5 W
|
8 °C
|
80 Vh
|
|
|
1 500 V
|
5,0 mA
|
7,0 W
|
8 °C
|
570 Vh
|
Umieścić aplikator próbki w etapie 2 przy 0 Vh.
Usunąć aplikator próbki w etapie 2 przy 30 Vh.
6.4. Barwienie białka
6.4.1. Utrwalanie białka
Usunąć paski elektrod natychmiast po wyłączeniu zasilania i niezwłocznie włożyć żel do naczynia do barwienia/odbarwiania, wypełnionego 200 ml utrwalacza (ppkt 4.9); pozostawić na 15 minut, nieprzerwanie wstrząsając.
6.4.2. Przemywanie i barwienie płytki żelu
Starannie osączyć utrwalacz i dwukrotnie przemywać płytkę żelu przez 30 sekund w 100 ml roztworu odbarwiającego (ppkt 4.10). Wylać roztwór odbarwiający i napełnić naczynie 250 ml roztworu barwiącego (ppkt 4.11.3); pozostawić do zabarwienia na 45 minut, lekko wstrząsając.
6.4.3. Odbarwianie płytki żelu
Wylać roztwór barwiący, przemyć dwukrotnie płytkę żelu, używając za każdym razem 100 ml roztworu odbarwiającego (ppkt 4.10), następnie wstrząsać z 200 ml roztworu odbarwiającego przez 15 minut i powtórzyć etap odbarwiania co najmniej dwa lub trzy razy do czasu uzyskania przezroczystego i bezbarwnego tła. Następnie płukać płytkę żelu wodą destylowaną (dwa razy po 2 minuty) i suszyć na powietrzu (2–3 godziny) lub za pomocą suszarki do włosów (10–15 minut).
Uwaga 1: Czynności utrwalania, przemywania, barwienia i odbarwiania wykonywać w temp. 20 °C. Unikać podwyższonych temperatur.
Uwaga 2: Jeżeli preferowane jest bardziej czułe barwienie srebrem (na przykład zestaw do barwienia srebrem Silver Staining Kit, Protein, Pharmacia Biotech, kod nr 17–1150–01), próbki kazeiny poddanej działaniu plazminy należy rozcieńczyć do uzyskania stężenia 5 mg/ml.
7. OCENA
Ocena dokonywana jest poprzez porównanie wzorów białek nieznanej próbki z wzorcami odniesienia na tym samym żelu. Wykrywanie mleka krowiego w serach wyprodukowanych z mleka owczego, mleka koziego, mleka bawolego lub mieszanek mleka owczego, koziego i bawolego dokonywane jest za pośrednictwem gamma2-kazeiny i gamma3-kazeiny, których punkty izoelektryczne mieszczą się w zakresie od pH 6,5 do pH 7,5 (ryc. 4 a, b, ryc. 5). Granica wykrywalności jest niższa niż 0,5 %.
7.1. Ocena wizualna
W odniesieniu do oceny wizualnej ilości mleka krowiego zaleca się dostosowanie stężeń próbek i wzorców celem uzyskania tego samego poziomu intensywności uwidocznienia owczej, koziej i/lub bawolej gamma2-kazeiny i gamma3-kazeiny (zob. „γ2 E, G, B” i „γ3 E, G, B” na ryc. 4 a, b i na ryc. 5). Po takim dostosowaniu ilość mleka krowiego (mniejsza, równa lub większa od 1 %) w nieznanej próbce może być oceniona bezpośrednio poprzez porównanie intensywności uwidocznienia gamma2-kazeiny i gamma3-kazeiny krowiej (zob. „γ2 C” i „γ3 C” na ryc. 4 a, b i na ryc. 5) z intensywnością uwidocznienia gamma2-kazeiny i gamma3-kazeiny wzorców odniesienia 0 % i 1 % (owczej, koziej) lub laboratoryjnych wzorców pośrednich (bawolej).
7.2. Ocena densytometryczna
Jeżeli istnieje taka możliwość, zastosować densytometrię (ppkt 5.19) celem określenia stosunku powierzchni pików gamma2-kazeiny i gamma3-kazeiny krowiej do gamma2-kazeiny i gamma3-kazeiny owczej, koziej i/lub bawolej (zob. ryc. 5). Porównać tę wartość do stosunku powierzchni pików gamma2-kazeiny i gamma3-kazeiny 1 % wzorca odniesienia (owczej, koziej) lub laboratoryjnego wzorca pośredniego (bawolej), poddanych analizie na tym samym żelu.
Uwaga: Metoda sprawdza się należycie w przypadkach, gdy występuje wyraźny dodatni sygnał zarówno w odniesieniu do krowiej gamma2-kazeiny, jak i krowiej gamma3-kazeiny w 1 % wzorcu odniesienia, ale nie w 0 % wzorcu odniesienia. Jeżeli sygnał nie występuje, należy optymalizować procedurę przestrzegając dokładnie szczegółów metody.
Próbka jest oceniana jako pozytywna, jeżeli krowia gamma2-kazeina oraz krowia gamma3-kazeina, lub stosunek powierzchni odpowiednich pików, są równe lub wyższe od poziomu 1 % wzorca odniesienia.
8. PIŚMIENNICTWO
|
1.
|
Addeo F., Moio L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Bocca A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708–711 (1990).
|
|
2.
|
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Bocca A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83–85 (1995).
|
|
3.
|
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte — and carrier ampholyte/immobilized pH gradient — isoelectric focusing of γ-caseins using plasmin as signal amplifier. w: Electrophoresis-Forum 89 (B. J. Radola, ed.) s. 389–393, Bode-Verlag, München (1989).
|
|
4.
|
Krause Ι., Belitz H.–D., Kaiser K.–P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. -käse durch isoelektrische Fokussierung in harnstoffhaltigen Polyacrylamidgelen. Z. Lebensm. Unters. Forsch. 174, 195–199 (1982).
|
|
5.
|
Radola B.J.: Ultrathin-layer isoelectric focusing in 50–100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43–56 (1980).
|
Rycina 1
Schematyczny rysunek arkusza przykrywającego
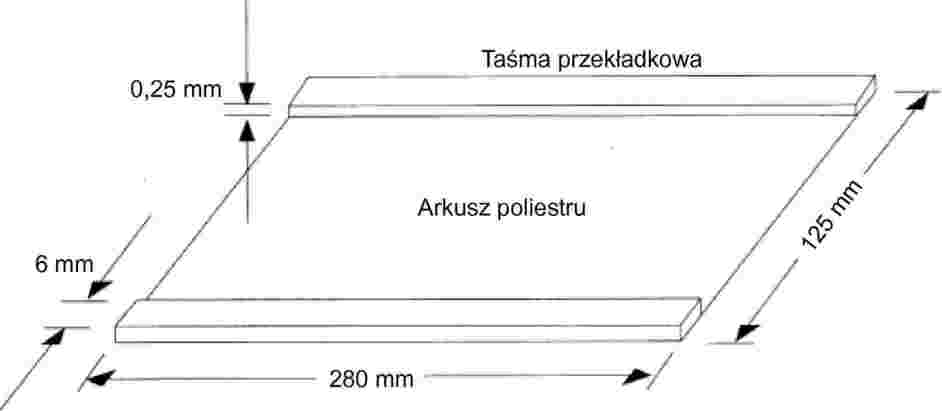
Rycina 2
Warstwa kazeiny utrzymująca się między fazą wodną i fazą organiczną po odwirowaniu
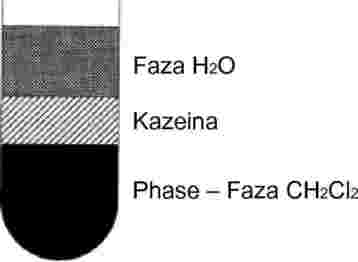
Rycina 3
Technika „przykrywania klapą”, której celem jest otrzymanie ultracienkich żeli poliakryloamidowych

a = taśma przekładkowa (0,25 mm); b = arkusz przykrywający (ppkt 5.3); c, e = płytki szklane (ppkt 5.1); d = roztwór żelu (ppkt 4.1.2); f = arkusz nośny żelu (ppkt 5.2)
Rycina 4a
Wyznaczanie punktu izoelektrycznego kazeiny poddanej działaniu plazminy z sera z mleka owczego i koziego, zawierającego różne ilości mleka krowiego

% CM = udział procentowy mleka krowiego, C = krowie, E = owcze, G = kozie
Przedstawiona jest górna połowa żelu IEF.
Rycina 4b
Wyznaczanie punktu izoelektrycznego kazeiny poddanej działaniu plazminy z sera wyprodukowanego z mieszanek mleka owczego, koziego i bawolego, zawierających różne ilości mleka krowiego

% CM = udział procentowy mleka krowiego; 1 + = próbka zawierająca 1 % mleka krowiego z dodatkiem czystej kazeiny krowiej w środku ścieżki. C = krowie, E = owcze, G = kozie, B = bawole.
Przedstawiona jest całkowita odległość rozdziału w żelu IEF.
Rycina 5
Zestawienie nałożonych densytogramów wzorców (STD) oraz próbek sera wyprodukowanego z mieszanki mleka owczego i koziego po wyznaczeniu punktu izoelektrycznego.

a, b = wzorce zawierające 0 i 1 % mleka krowiego; c–g = próbki sera zawierające 0, 1, 2, 3 i 7 % mleka krowiego. C = krowie, E = owcze, G = kozie.
Górna połowa żelu IEF została zeskanowana przy λ = 634 nm.
(1) Produkty Ampholine® pH 3,5–9,5 (Pharmacia) i Resolyte® pH 5–7 oraz pH 6–8 (BDH, Merck) okazały się szczególnie odpowiednie do uzyskania wymaganego rozdzielenia gamma-kazein.
(2) Zastosowanie próbki: Po ogniskowaniu wstępnym (etap 1) odmierzyć przy użyciu pipety 18 μl próbki i roztworów wzorcowych na aplikatory próbek (10 × 5 mm), rozmieścić je w odległości 1 mm od siebie na żelu i w odległości 5 mm w pozycji poziomej względem anody, po czym lekko docisnąć. Kontynuować ogniskowanie w przedstawionych powyżej warunkach, ostrożnie usuwając aplikatory próbek po 60 minutach ogniskowania próbek.
ZAŁĄCZNIK X
(Artykuł 7)
METODA REFERENCYJNA WYKRYWANIA BAKTERII Z GRUPY COLI W MAŚLE, ODTŁUSZCZONYM MLEKU W PROSZKU, KAZEINIE I KAZEINIANACH
1. PRZYGOTOWANIE PRÓBEK
Norma ISO 8261
2. PROCEDURA
Norma ISO 4831
Próbki odpowiadające 1 g masła lub 0,1 g odtłuszczonego mleka w proszku lub kazeiny/kazeinianów są posiewane na podłożu hodowli.
Materiał z każdej próbki posiewany jest w trzech probówkach.
3. WYNIKI
Jeżeli 3 probówki dają 3 wyniki negatywne, próbka „spełnia wymogi”.
Jeżeli 3 probówki dają 2 lub 3 wyniki pozytywne, próbka „nie spełnia wymogów”.
Jeżeli 3 probówki dają 2 wyniki negatywne, analizę należy powtórzyć dwukrotnie (z użyciem dwóch probówek).
|
—
|
Jeżeli 2 wyniki są negatywne, próbka „spełnia wymogi”.
|
|
—
|
Jeżeli co najmniej 1 wynik jest pozytywny, próbka „nie spełnia wymogów”.
|
ZAŁĄCZNIK XI
(Artykuł 8)
OZNACZANIE LAKTOZY W MIESZANKACH PASZOWYCH
1. ZAKRES I DZIEDZINA STOSOWANIA
Oznaczanie laktozy w mieszankach paszowych.
2. ODNIESIENIE
Zawartość laktozy określona jest jako procent masowy oznaczony w ramach opisanej procedury.
3. DEFINICJA
Zawartość bezwodnej laktozy jest wyrażona w g/100 g.
4. ZASADA
Mieszankę paszową rozpuszcza się w wodzie. Do rozcieńczonej odważonej podwielokrotności dodaje się roztwór „Biggsa” w celu wytrącenia z mieszanki paszowej części składowych tłuszczu i białka. Próbka jest następnie sączona (lub wirowana), a przesącz (lub nadsącz) jest wprowadzany do kolumny kationowymiennej do HPLC wypełnionej jonowymienną żywicą ołowiową przy zastosowaniu wody o jakości wymaganej dla HPLC jako fazy ruchomej. Eluowana laktoza jest wykrywana za pomocą refraktometru różnicowego (1).
5. ODCZYNNIKI
5.1. Uwagi ogólne
Należy korzystać wyłącznie z odczynników o uznanej klasie analitycznej, o ile nie ustalono inaczej, i odgazowanej wody o jakości wymaganej dla HPLC.
5.2. Laktoza
Monohydrat D-laktozy ((C12H22)O11. H2O) może wchłonąć dodatkową wilgoć. Przed użyciem zmierzyć rzeczywistą ilość wody metodą Karla-Fishera lub usunąć dodatkową wilgoć, umieszczając laktozę w piecu w temp. 105 °C na 8 godzin (laktoza nie traci w takim procesie swojej wody krystalicznej).
5.3. Stężony roztwór Biggsa/Szijarto (2)
Rozpuścić 9,10 g dwuwodnego octanu cynku (Zn(CH3COOH)2.2H2O) i 5,46 g monohydratu kwasu fosfowolframowego (H3[P(W3O10)4.xH2O]) w ok. 70 ml wody o jakości wymaganej dla HPLC (ppkt 6.8) w kolbie miarowej poj. 100 ml.
Dodać 5,81 ml kwasu octowego lodowatego (CH3COOH). Uzupełnić do kreski 100 ml wodą o jakości wymaganej dla HPLC (ppkt 6.8) i wymieszać. Roztwór można przechowywać w temperaturze pokojowej przez okres 1 roku.
5.4. Rozcieńczony roztwór Biggsa/Szijarto
Uzupełnić 25 ml stężonego roztworu Biggsa/Szijarto (ppkt 5.3) wodą do objętości 500 ml, w kolbie miarowej. Roztwór można przechowywać w temperaturze pokojowej przez okres 1 miesiąca.
5.5. Przygotowanie wody o jakości wymaganej dla HPLC
Przesączyć ultra czystą wodę (ppkt 6.8), korzystając z zestawu do sączenia próżniowego (ppkt 6.9). Aby usprawnić funkcjonowanie pompy i otrzymać stabilną linię podstawową, należy codziennie odgazowywać fazę ruchomą, wybierając jedną z dostępnych technik, takich jak przepłukiwanie helem, ultrasonikacja, albo system odgazowywania próżniowego lub bezpośredniego (in-line).
Uwaga: W celu przedłużenia żywotności kolumny istotne jest, aby zawartość dwutlenku węgla w eluencie była możliwie najniższa, nie należy również dopuścić do wychwytu zwrotnego.
6. APARATURA
Typowe wyposażenie laboratoryjne, w szczególności:
6.1. Kolumna do HPLC z żywicą jonowymienną
Wypełnienie kolumny: usieciowany kopolimer polistyren-diwinylobenzen (8 %), funkcjonalizowany grupami kationowymiennymi w postaci jonowymiennej żywicy ołowiowej.
Wymiary kolumny: Długość 300 mm, średnica wewnętrzna ok. 8 mm.
Możliwe jest wykorzystanie kolumny o innej średnicy pod warunkiem odpowiedniego dostosowania natężenia przepływu.
6.2. Kolumna ochronna
Kolumna ochronna stanowi połączenie odrębnego wymieniacza kationowego (H+) i wymieniacza anionowego (CO3-), każdy zapakowany w kolumny o wymiarach ok. 30 mm × 4,6 mm (dł. × śr. wewn.) (np. mikrokolumny ochronne w mikrouchwycie ochronnym) i połączone szeregowo lub w postaci złoża mieszanego, składających się z AG 50W–X4, — 400 mesh (H+) oraz AG3-X4A, 200–400 mesh (OH-) w stosunku 35:65 (m/m), ręcznie upakowanych w kolumnie o wymiarach ok. 20 mm × 9 mm (dł. × śr. wewn.).
6.3. Piec do kolumn
Piec umożliwiający utrzymanie stałej temp. 85 °C ± 1 °C.
6.4. Pompa HPLC
Pompa umożliwiająca wytworzenie stałego natężenia przepływu (< 0,5 % wahań) w zakresie 0,2–1,0 m/min.
6.5. Urządzenie wprowadzające do HPLC
Automatyczny dozownik próbek umożliwiający wprowadzenie 25 µl i o powtarzalności < 0,5 %.
Można ewentualnie zastosować ręczne urządzenie wprowadzające (wymagania identyczne jak w stosunku do automatycznego dozownika próbek).
6.6. Detektor HPLC
Wysokoczuły detektor refraktometryczny o poziomie szumów < 5,10–9 jednostek RI.
6.7. Integrator
Oprogramowanie lub wyspecjalizowany integrator do akwizycji i przetwarzania danych oraz obliczania powierzchni pików i wysokości pików, które można przeliczyć na stężenia laktozy.
6.8. Zespół oczyszczania wody
System umożliwiający uzyskanie ultra czystej wody (typu 1) o rezystywności > 14 MΩ.cm.
6.9. Zespół sączenia rozpuszczalnika
Układ umożliwiający przesączenie wody przy zastosowaniu sączka membranowego o średnicy porów 0,45 µm.
Uwaga: Duża liczba zespołów oczyszczania wody (ppkt 6.8) posiada wbudowane zespoły sączące o średnicy porów 0,45 lub 0,2 µm. Dodatkowe sączenie można pominąć, jeżeli woda ta jest wykorzystywana bezpośrednio.
6.10. Waga analityczna
Waga o dokładności odczytu 0,1 mg.
6.11. Łaźnia wodna
Łaźnia wodna umożliwiająca utrzymanie temp. 40 °C (±0,5 °C).
6.12. Wirówka
Umożliwiająca wytworzenie co najmniej 3 000 g w przypadku zastosowania probówek Eppendorfa lub równoważnych albo większych probówek.
6.13. Kolba miarowa poj. 50 ml
Pojemność 50 ml, klasa A.
Uwaga: Można wykorzystać kolby o innej pojemności przy uwzględnieniu czynnika objętości.
6.14. Kolba miarowa poj. 100 ml
Pojemność 100 ml, klasa A.
6.15. Pipeta miarowa
Pipeta miarowa poj. 10 ml
Uwaga: Można ewentualnie wykorzystać pipetę automatyczną poj. 5 ml, dodając dwukrotnie objętość 5 ml odczynnika (ppkt 5.3).
7. POBIERANIE PRÓBEK
Istotne jest, aby laboratorium otrzymało próbkę pobraną zgodnie z normą ISO 707/IDF 50 (3), która jest rzeczywiście reprezentatywna i nie uległa uszkodzeniu w czasie transportu lub przechowywania.
8. PRZYGOTOWANIE ROZTWORU WZORCOWEGO LAKTOZY
8.1. Wzorzec 1
Rozpuścić dokładnie odważoną (odczyt z dokładnością do 0,1 mg) ilość ok. 50 mg monohydratu laktozy (ppkt 5.2) w kolbie miarowej poj. 100 ml (ppkt 6.14) i dopełnić wodą do kreski.
8.2. Wzorzec 2
Rozpuścić dokładnie odważoną (odczyt z dokładnością do 0,1 mg) ilość ok. 100 mg monohydratu laktozy (ppkt 5.2) w kolbie miarowej poj. 100 ml (ppkt 6.14) i dopełnić wodą do kreski.
Uwaga: Roztwory wzorcowe można przechowywać maksymalnie przez okres 1 tygodnia w temp. ok. 5 °C.
9. PRZYGOTOWANIE PRÓBKI DO BADAŃ
9.1. Odtworzenie próbki
Odmierzyć wagowo ok. 5 g. proszku do kolby poj. 50 ml (ppkt 6.13) i zanotować masę z dokładnością do 1 mg (W1, (ppkt 11)). Dodać 50 ml wody i zanotować przyrost masy (W2, (ppkt 11)) z dokładnością do 0,01 g. Umieścić zamkniętą kolbę w łaźni wodnej (ppkt 6.11) na 30 minut i w tym czasie kilkakrotnie ją odwrócić, po czym pozostawić kolbę do ostygnięcia do temperatury pokojowej.
9.2. Obróbka próbki
Pobrać ok. 1 g powyższego roztworu i umieścić w kolbie miarowej poj. 50 ml (ppkt 6.13), zanotować masę z dokładnością do 1 mg (W3 (ppkt 11)), dodać 20 ml wody, po czym dodać 10 ml rozcieńczonego odczynnika Biggsa/Szijarto (ppkt 5.4) i dopełnić wodą do kreski. Ostrożnie odwrócić kolbę 5 razy w ciągu pierwszych 30 minut.
Po upływie 1 godziny pobrać podwielokrotność i wirować (ppkt 6.12) przy 3 000 g przez 10 minut (można zastosować wyższe g przy odpowiednio krótszym czasie wirowania). Wykorzystać podwielokrotność nadsączu do wykonania analizy HPLC.
10. OZNACZANIE HPLC
10.1. Wstępne przygotowanie HPLC
10.1.1. Zainstalowanie kolumny i przedkolumny
Zainstalować przedkolumnę (ppkt 6.2) na zewnątrz pieca do kolumn (ppkt 6.3), a kolumnę (ppkt 6.1) wewnątrz pieca.
Uwaga: W przypadku gdy piec nie zawiera rurek do podgrzania eluentu, niezbędne jest przepuszczenie eluentu przez rurkę ze stali nierdzewnej o długości ok. 15 cm w piecu przed podaniem na kolumnę (jest bezwzględnie konieczne, aby eluent został podgrzany przed podaniem na kolumnę, gdyż w innym wypadku dojdzie do poszerzenia piku).
10.1.2. Detektor i przepływ początkowy
Aby uzyskać stabilną linię podstawową, należy włączyć detektor (ppkt 6.6) co najmniej 24 godziny przez rozpoczęciem analizy. Ustawić temperaturę wewnętrzną detektora na 35 °C. Ustawić natężenie przepływu strumienia gazu nośnego na 0,2 ml/min. (ppkt 6.4) na okres co najmniej 20 minut, natomiast piec do kolumn (ppkt 6.3) jest ustawiony na temperaturę pokojową.
10.1.3. Piec do kolumn i końcowe natężenie przepływu strumienia gazu nośnego
Ustawić temperaturę pieca do kolumn (ppkt 6.3) na 85 °C. Po osiągnięciu zalecanej temperatury zwiększać stopniowo po upływie 30 minut natężenie przepływu strumienia gazu nośnego z 0,2 ml/min. do 0,6 ml/min. (ppkt 6.4). Pozostawić układ do osiągnięcia stanu równowagi przy podanym natężeniu przepływu strumienia gazu nośnego i w temp. 85 °C na 2 godziny lub do czasu uzyskania stabilnej linii podstawowej.
10.1.4. Całkowanie
Starannie dobrać parametry akwizycji i całkowania (ppkt 6.7), takie jak prędkość transmisji danych, czułość, stała czasowa, szerokość piku i wartości progowe.
Czas retencji laktozy wynosi ok. 11 min.
Uwaga: Wiele programów komputerowych do akwizycji danych (ppkt 6.7) umożliwia dokonanie pomiaru liczby półek teoretycznych. Należy mierzyć regularnie liczbę półek teoretycznych dla wzorca 1 (ppkt 8.1) i wymienić kolumnę (ppkt 6.1), gdy liczba półek jest o 25 % niższa niż wartość początkowa dla nowej kolumny.
10.1.5. Sprawdzanie kolumny ochronnej
Sprawdzać regularnie (co najmniej raz w każdej sekwencji) zdolność kolumny ochronnej (ppkt 6.2) do usuwania soli z próbki, wprowadzając 25 µl 0,05 % roztworu chlorku sodu. Ilekroć pojawiają się piki, należy wymienić kolumnę ochronną.
10.2. Przepuszczenie wzorców
Wprowadzić na początku każdej serii analiz 25 µL (ppkt 6.5) wzorca 1 (ppkt 8.1), a następnie taką samo objętość wzorca 2 (ppkt 8.2). Powtarzać operację co 10–20 próbek, zastosować również na koniec sekwencji.
10.3. Przepuszczenie próbek
Wprowadzić 25 µl nadsączu (ppkt 9.2) próbki.
11. OBLICZANIE I PREZENTACJA WYNIKÓW
11.1. Kalibracja
Zazwyczaj do obliczania wyników wykorzystuje się wysokości pików, jeżeli jednak sygnał zawiera zbyt wiele szumów, można wykorzystać powierzchnię pików (piki składników występujących w niskich stężeniach, które są częściowo, ale niedostatecznie oddzielone od piku laktozy, wywierają mniejszy wpływ na pomiar ilościowy w oparciu o wysokość piku).
Oprogramowanie (ppkt 6.7) powinno wyliczyć liniową krzywą kalibracyjną przesuniętą do początku układu współrzędnych. Sprawdzić krzywą pod kątem ewentualnej nieliniowości (ewidentna nieliniowość jest najprawdopodobniej spowodowana błędem przy przygotowywaniu wzorca 1 (ppkt 8.1) lub 2 (ppkt 8.2), nieprawidłowym całkowaniem oraz, co mniej prawdopodobne, wadliwym działaniem dozownika).
Jako dane wejściowe należy wykorzystać obliczone stężenia laktozy w mg/ml we wzorcu 1 (ppkt 8.1) oraz 2 (ppkt 8.2) jako bezwodnej laktozy.
Nachylenie (RF) linii kalibracyjnej jest określone stosunkiem powierzchni do stężenia wyrażonego w mg/ml.
11.2. Próbki
Otrzymany wynik analizy jest wyrażony w g/100 g i obliczony przy użyciu oprogramowania (ppkt 6.7) lub poniższego wzoru:

Gdzie:
|
C
|
:
|
stężenie laktozy wyrażone w g/100 g proszku
|
|
H
|
:
|
wysokość piku laktozy w próbce
|
|
RF
|
:
|
współczynnik odpowiedzi (lub nachylenie) wykresu kalibracji, wyrażony w mV/mg/ml
|
|
W1
|
:
|
masa próbki proszku wyrażona w gramach (ppkt 9.1)
|
|
W2
|
:
|
masa wody dodanej do próbki proszku wyrażona w gramach (ppkt 9.1)
|
|
W3
|
:
|
masa próbki odtworzonego roztworu proszku wyrażona w gramach (ppkt 9.2)
|
|
50
|
:
|
pojemność kolby miarowej zastosowanej w (ppkt 9.2)
|
|
0,1
|
:
|
współczynnik przeliczenia wyniku na g/100 g
|
12. PRECYZJA
Wartości uzyskane w przedstawionym badaniu międzylaboratoryjnym nie mogą mieć zastosowania do zakresów stężeń i matryc innych niż podane. Wartości powtarzalności i odtwarzalności zostaną uzyskane w oparciu o wynik badania międzylaboratoryjnego przeprowadzonego zgodnie z normą ISO 5725 (4).
12.1. Powtarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi przy użyciu tej samej metody na identycznym materiale badanym w tym samym laboratorium przez ten sam podmiot wykorzystujący to samo wyposażenie w krótkim odstępie czasu nie będzie dla więcej niż 5 % przypadków większa niż xxx (wartość zostanie określona w oparciu o wspólne badanie) (5).
12.2. Odtwarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi przy użyciu tej samej metody na identycznym materiale badanym w różnych laboratoriach przez różne podmioty wykorzystujące różne wyposażenie nie będzie dla więcej niż 5 % przypadków większa niż 0,5 g/100 g (wartość zostanie określona w oparciu o wspólne badanie).
13. PIŚMIENNICTWO
(1) J. Koops en C. Olieman, Netherlands Milk and Dairy Journal, 39 (1985) 89–106.
(2) D.A. Biggs en L. Szijarto, Journal of Dairy Science, 46 (1963) 1196.
(3) ISO 707 (IDF 50), Mleko i przetwory mleczne — Wytyczne do pobierania próbek.
(4) ISO 5725–1, Dokładność (poprawność i precyzja) metod pomiarowych i wyników pomiarów. Część 1: Ogólne zasady i definicje.
(5) ISO 5725–2, Dokładność (poprawność i precyzja) metod pomiarowych i wyników pomiarów. Część 2: Podstawowa metoda określania powtarzalności i odtwarzalności standardowej metody pomiarowej.
ZAŁĄCZNIK XII
(Artykuł 9)
WYKRYWANIE OBECNOŚCI SERWATKI PODPUSZCZKOWEJ W ODTłUSZCZONYM MLEKU W PROSZKU PRZEZNACZONYM DO SKłADOWANIA W MAGAZYNACH PAŃSTWOWYCH ZA POMOCĄ OZNACZANIA MAKROPEPTYDÓW KAZEINOWYCH METODĄ WYSOKOSPRAWNEJ CHROMATOGRAFII CIECZOWEJ (HPLC)
1. ZAKRES I DZIEDZINA STOSOWANIA
Omawiana metoda umożliwia wykrywanie obecności serwatki podpuszczkowej w odtłuszczonym mleku w proszku przeznaczonym do składowania w magazynach państwowych za pomocą oznaczania makropeptydów kazeinowych.
2. ODNIESIENIE
Międzynarodowa norma ISO 707 — Mleko i przetwory mleczne — Metody pobierania próbek, zgodnie z wytycznymi zawartymi w załączniku I pkt 2 lit. c), ostatni akapit.
3. DEFINICJA
Zawartość suchej masy serwatki podpuszczkowej jest zdefiniowana jako procent masowy, określony w oparciu o zawartość makropeptydów kazeinowych w ramach opisanej procedury.
4. ZASADA
|
—
|
Odtworzenie odtłuszczonego mleka w proszku, usunięcie tłuszczu i białka za pomocą kwasu trichlorooctowego, a następnie wirowanie lub sączenie.
|
|
—
|
Oznaczenie ilości makropeptydów kazeinowych (CMP) w nadsączu metodą wysokosprawnej chromatografii cieczowej (HPLC).
|
|
—
|
Ocena wyniku otrzymanego dla próbek poprzez odniesienie do próbek wzorcowych składających się z odtłuszczonego mleka w proszku z dodatkiem znanego udziału procentowego ilości serwatki w proszku lub bez takiego dodatku.
|
5. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej. Należy używać wody destylowanej lub wody o równoważnym stopniu czystości.
5.1. Roztwór kwasu trichlorooctowego
Rozpuścić 240 g kwasu trichlorooctowego (CCl3COOH) w wodzie i uzupełnić do 1 000 ml. Roztwór powinien być klarowny i bezbarwny.
5.2. Roztwór eluentu, pH 6,0
Rozpuścić 1,74 g ortofosforanu dipotasu (K2HPO4), 12,37 g ortofosforanu potasu (KH2PO4) oraz 21,41 g siarczanu sodu (Na2SO4) w ok. 700 ml wody. W razie potrzeby doprowadzić pH do wartości 6,0, używając roztworu kwasu ortofosforowego lub wodorotlenku potasu.
Uzupełnić wodą do 1 000 ml i homogenizować.
Uwaga: Skład eluentu można aktualizować w celu zachowania zgodności z certyfikatem norm lub zaleceniami producenta materiału wypełniającego kolumnę.
Przed użyciem przesączyć roztwór eluentu przez sączek membranowy o średnicy porów 0,45 μm.
5.3. Rozpuszczalnik przepłukujący
Zmieszać jedną objętość acetonitrylu (C3CN) z dziewięcioma objętościami wody. Przed użyciem przesączyć mieszaninę przez sączek membranowy o średnicy porów 0,45 µm.
Uwaga: Można zastosować każdy inny rozpuszczalnik przepłukujący o działaniu bakteriobójczym, który nie wpływa niekorzystnie na zdolność rozdzielczą kolumn.
5.4. Próbki wzorcowe
5.4.1. Odtłuszczone mleko w proszku spełniające wymogi niniejszego rozporządzenia (tzn. [0])
5.4.2. To samo odtłuszczone mleko w proszku zafałszowane 5 % (m/m) serwatki typu podpuszczkowego w proszku o składzie standardowym (tzn. [5])
6. APARATURA
6.1. Waga analityczna
6.2. Opcjonalnie wirówka umożliwiająca osiągnięcie siły odśrodkowej 2 200 g, wyposażona w probówki wirówkowe z korkiem lub zatyczką, poj. ok. 50 ml.
6.3. Wstrząsarka mechaniczna
6.4. Mieszadło magnetyczne
6.5. Lejki szklane o średnicy ok. 7 cm
6.6. Sączki papierowe, średnia prędkość sączenia, o średnicy ok. 12,5 cm
6.7. Szklany zestaw do sączenia z sączkiem membranowym o średnicy porów 0,45 μm
6.8. Pipety miarowe pozwalające na dozowanie 10 ml (ISO 648, klasa A lub ISO/R 835) lub system dozujący umożliwiający dostarczenie 10 ml w ciągu dwóch minut
6.9. System dozujący umożliwiający dostarczenie 20 ml wody w temp. ok. 50 °C
6.10. Łaźnia wodna kontrolowana termostatycznie, ustawiona na temp. 25 °C ± 0,5 °C
Zestaw do HPLC, w skład którego wchodzi:
6.11.1. Pompa
6.11.2. Dozownik, ręczny lub automatyczny, poj. 15–30 μl
6.11.3. Dwie kolumny TSK 2 000-SW połączone szeregowo (długość 30 cm, średnica wewnętrzna 0,75 cm) lub kolumny równoważne (np. jedna kolumna TSK 2 000-SWxl, jedna kolumna Agilent Technologies Zorbax GF 250), oraz przedkolumna (3 cm × 0,3 cm) wypełnione I 125 lub materiałem o równoważnej efektywności
6.11.4. Piec do kolumn kontrolowany termostatycznie, ustawiony na temp. 35 °C ± 1 °C
6.11.5. Detektor UV o zmiennej długości fali, umożliwiający pomiary przy 205 nm, o czułości 0,008 A
6.11.6. Integrator umożliwiający całkowanie wykresu między kolejnymi dolinami
Uwaga: Praca z kolumnami utrzymywanymi w temperaturze pokojowej jest możliwa, ale ich zdolność rozdzielcza jest wtedy nieco niższa. W takim wypadku temperatura nie powinna podlegać wahaniom większym niż ± 5 °C w ramach żadnej z analiz.
7. POBIERANIE PRÓBEK
7.1. Próbki należy pobierać zgodnie z procedurą określoną w międzynarodowej normie ISO 707. Państwa członkowskie mogą jednak stosować inną metodę pobierania próbek pod warunkiem, że jest ona zgodna z zasadami powyższej normy.
7.2. Próbkę przechowywać w warunkach wykluczających spadek jakości lub zmianę składu.
8. PROCEDURA
8.1. Przygotowanie próbki do badań
Przenieść mleko w proszku do pojemnika o pojemności w przybliżeniu dwukrotnie większej niż objętość proszku, wyposażonego w hermetyczną pokrywę. Natychmiast zamknąć pojemnik. Wymieszać dobrze mleko w proszku poprzez wielokrotne odwracanie pojemnika do góry dnem.
8.2. Porcja badana
Odmierzyć wagowo 2 000 ± 0,001 g badanej próbki i umieścić w probówce wirówkowej (ppkt 6.2) bądź w odpowiedniej kolbie z zatyczką (50 ml).
8.3. Usuwanie tłuszczu i białek
8.3.1. Do porcji badanej dodać 20,0 ml ciepłej wody (50 °C). Rozpuścić proszek, wstrząsając przez 5 minut przy pomocy wstrząsarki mechanicznej (ppkt 6.3). Umieścić probówkę w łaźni wodnej (ppkt 6.10) i pozostawić do wyrównania się temperatury do 25 °C.
8.3.2. Dodawać stopniowo w ciągu dwóch minut 10,0 ml roztworu kwasu trichlorooctowego (ppkt 5.1) o temp. ok. 25 °C, mieszając jednocześnie energicznie mieszadłem magnetycznym (ppkt 6.4). Umieścić probówkę w łaźni wodnej (ppkt 6.10) i pozostawić tam na 60 minut.
8.3.3. Wirować (ppkt 6.2) przez 10 minut przy 2 200 g lub przesączyć przez sączek papierowy (ppkt 6.6), odrzucając pierwsze 5 ml przesączu.
8.4. Oznaczenie chromatograficzne
8.4.1. Wprowadzić 15–30 μl dokładnie odmierzonego nadsączu lub przesączu (ppkt 8.3.3) do aparatury HPLC (ppkt 6.11), pracując przy natężeniu przepływu wynoszącym 1,0 ml roztworu eluentu (ppkt 5.2) na minutę.
Uwaga 1: Można zastosować inne natężenie przepływu w zależności od średnicy wewnętrznej wykorzystywanej kolumny lub instrukcji producenta kolumny.
Uwaga 2: Utrzymywać roztwór eluentu (ppkt 5.2) w temp. 85 °C przez cały czas trwania analizy chromatograficznej w celu utrzymania go w stanie odgazowanym i zapobieżenia rozwojowi bakterii. Można stosować wszelkie inne środki ostrożności o podobnym działaniu.
Uwaga 3: Podczas każdej przerwy przepłukiwać kolumny wodą. Nie należy nigdy pozostawiać roztworu eluentu (ppkt 5.2) w kolumnach.
Przed każdą przerwą dłuższą niż 24 godziny przepłukać kolumny wodą, a następnie przemywać je roztworem (ppkt 5.3) przez co najmniej trzy godziny przy natężeniu przepływu równym 0,2 ml/min.
8.4.2. Wyniki chromatograficznej analizy badanej próbki [E] otrzymuje się w postaci chromatogramu, na którym poszczególne piki określa się za pomocą ich czasu retencji RT w następujący sposób:
|
Pik II:
|
Drugi pik chromatogramu z RT wynoszącym ok. 12,5 minuty.
|
|
Pik III:
|
Trzeci pik chromatogramu, odpowiadający CMP, z RT wynoszącym 15,5 minuty.
|
Dobór kolumny lub kolumn może mieć znaczący wpływ na czasy retencji poszczególnych pików.
Integrator (ppkt 6.11.6) automatycznie oblicza powierzchnię A każdego piku.
|
AII:
|
powierzchnia piku II
|
|
AIII:
|
powierzchnia piku III
|
Konieczne jest zbadanie wyglądu każdego chromatogramu przed dokonaniem interpretacji ilościowej w celu wykrycia ewentualnych nieprawidłowości, będących skutkiem wadliwego działania aparatury lub kolumn albo też pochodzenia i rodzaju analizowanej próbki.
W razie wątpliwości analizę należy powtórzyć.
8.5. Kalibracja
8.5.1. Do próbek wzorcowych (ppkt 5.4) zastosować dokładnie procedurę opisaną w ppkt 8.2–8.4.2.
Używać świeżo przygotowanych roztworów, ponieważ w środowisku 8 % kwasu trichlorooctowego następuje rozpad CMP. Straty szacuje się na 0,2 % na godzinę w temp. 30 °C.
8.5.2. Przed oznaczeniem chromatograficznym próbek należy przygotować kolumny, wprowadzając wielokrotnie próbkę wzorcową (ppkt 5.4.2) w roztworze (ppkt 8.5.1) do momentu, gdy powierzchnia i czas retencji piku odpowiadającego CMP są stałe.
8.5.3. Oznaczyć współczynniki odpowiedzi R poprzez wprowadzenie takiej samej objętości przesączu (ppkt 8.5.1), jak objętość wykorzystana do próbek.
9. PREZENTACJA WYNIKÓW
9.1. Metoda obliczania i wzory
9.1.1. Obliczanie współczynników odpowiedzi R:
|
Pik II:
|
RII = 100/(AII[0])
|
gdzie:
|
RII
|
=
|
współczynniki odpowiedzi pików II,
|
|
AII [0]
|
=
|
powierzchnie pików II próbki wzorcowej [0] otrzymane w ppkt 8.5.3.
|
|
Pik III:
|
RIII = W/(AIII[5] – AIII[0])
|
gdzie:
|
RIII
|
=
|
współczynniki odpowiedzi piku III,
|
|
AIII [0] i AIII [5]
|
=
|
powierzchnie piku III w próbkach wzorcowych, odpowiednio [0] i [5], otrzymane w ppkt 8.5.3.
|
|
W
|
=
|
ilość serwatki w próbce wzorcowej [5], tzn. 5.
|
9.1.2. Obliczanie względnej powierzchni pików w próbce [E]
SII[E] = RII × AII[E]
SIII[E] = RIII × AIII[E]
SIV[E] = RIV × AIV[E]
gdzie:
|
SII [E], SIII [E], SIV [E]
|
=
|
względne powierzchnie pików, odpowiednio, II, III i IV, w próbce [E],
|
|
AII [E], AIII [E]
|
=
|
powierzchnie pików, odpowiednio, II i III w próbce [E], otrzymane w ppkt 8.4.2,
|
|
RII, RIII
|
=
|
współczynniki odpowiedzi obliczone w ppkt 9.1.1.
|
9.1.3. Obliczanie względnego czasu retencji piku III w próbce [E]: RRTIII[E] = (RTIII[E])/(RTIII[5])
gdzie:
|
RRTIII [E]
|
=
|
względny czas retencji piku III w próbce [E],
|
|
RTIII [E]
|
=
|
względny czas retencji piku III w próbce [E] otrzymany w ppkt 8.4.2,
|
|
RTIII [5]
|
=
|
czas retencji piku III w próbce kontrolnej [5] otrzymany w ppkt 8.5.3.
|
9.1.4. Doświadczenia wykazały, że istnieje liniowa zależność między względnym czasem retencji piku III, tzn. RRTIII [E] i zawartością procentową serwatki w proszku w zakresie do 10 %.
|
—
|
RRTIII [E] jest < 1,000, gdy zawartość serwatki wynosi > 5 %,
|
|
—
|
RRTIII [E] jest ≥ 1,000 gdy zawartość serwatki wynosi ≤ 5 %.
|
Dopuszczalna niepewność w odniesieniu do wartości RRTIII wynosi ±0,002.
Zazwyczaj wartość RRTIII [0] odbiega nieco od 1,034. W zależności od stanu kolumn wartość ta może zbliżać się do 1,000, ale zawsze musi ją przewyższać.
9.2. Obliczanie zawartości procentowej serwatki podpuszczkowej w proszku w próbce:
W = SIII[E] – [1,3 + (SIII[0] – 0,9)]
gdzie:
|
W
|
=
|
zawartość procentowa m/m serwatki podpuszczkowej w próbce [E];
|
|
SIII [E]
|
=
|
względna powierzchnia piku III próbki badanej [E] otrzymana w ppkt 9.1.2;
|
|
1,3
|
=
|
stanowi średnią powierzchnię względną piku III wyrażoną w gramach serwatki podpuszczkowej na 100 g, oznaczoną w niezafałszowanym odtłuszczonym mleku w proszku różnego pochodzenia. Wielkość tę uzyskano doświadczalnie;
|
|
SIII [0]
|
=
|
stanowi względną powierzchnię piku III, która jest równa RIII × AIII [0]. Wartości te uzyskano odpowiednio w ppkt 9.1.1 i pkt. 8.5.3;
|
|
(SIII [0] – 0,9)
|
=
|
stanowi poprawkę, jaką należy wprowadzić w odniesieniu do średniej powierzchni względnej 1,3, gdy SIII [0] nie jest równe 0,9. Doświadczalnie określono, że średnia powierzchnia piku III próbki kontrolnej [0] wynosi 0,9.
|
9.3. Dokładność procedury
9.3.1. Powtarzalność
Różnica między wynikami dwóch oznaczeń wykonanych równocześnie lub w bezpośrednim następstwie przez tego samego analityka przy użyciu tej samej aparatury na identycznym materiale badanym nie przekracza 0,2 % m/m.
9.3.2. Odtwarzalność
Różnica między dwoma pojedynczymi i niezależnymi wynikami, otrzymanymi w dwóch różnych laboratoriach na identycznym materiale badanym, nie przekracza 0,4 % m/m.
9.4. Interpretacja
9.4.1. Należy przyjąć, że serwatka jest nieobecna, jeżeli względna powierzchnia piku III, SIII [E], wyrażona w gramach serwatki podpuszczkowej na 100 g produktu, wynosi ≤ 2,0 + (SIII[0] – 0,9), gdzie
|
2,0
|
=
|
maksymalna wartość dopuszczalna dla względnej powierzchni piku III przy uwzględnieniu względnej powierzchni piku III, tzn. 1,3; niepewność wynika ze zmian składu odtłuszczonego mleka w proszku oraz odtwarzalności metody (ppkt 9.3.2),
|
|
(SIII [0] – 0,9)
|
=
|
poprawka, jaką należy wprowadzić, jeżeli powierzchnia SIII [0] jest różna od 0,9 (zob. ppkt 9.2)
|
9.4.2. Jeżeli względna powierzchnia piku III, SIII [E], jest > 2,0 + (SIII[0] – 0,9), a względna powierzchnia piku II, SII [E] ≤ 160, należy oznaczyć zawartość serwatki podpuszczkowej w sposób wskazany w ppkt 9.2.
Jeżeli względna powierzchnia piku III, SIII [E], jest > 2,0 + (SIII[0] – 0,9), a względna powierzchnia piku II, SII [E] ≤ 160, należy oznaczyć zawartość białka (P %); następnie zbadać wykresy 1 i 2.
9.4.3.1. Dane otrzymane po analizie próbek niezafałszowanego odtłuszczonego mleka w proszku o wysokiej całkowitej zawartości białka zostały zgromadzone na wykresach 1 i 2.
Linia ciągła ilustruje regresję liniową, której współczynniki obliczane są metodą najmniejszych kwadratów.
Linia prosta przerywana wyznacza górną granicę względnej powierzchni piku III z prawdopodobieństwem nieprzekroczenia jej w 90 % przypadków.
Równania w odniesieniu do prostych linii przerywanych na wykresach 1 i 2 są następujące:
|
SIII = 0,376 P % – 10,7
|
(wykres 1),
|
|
SIII = 0,0123 SII [E] + 0,93
|
(wykres 2),
|
gdzie, odpowiednio:
|
SIII
|
=
|
względna powierzchnia piku III wyliczona albo według całkowitej zawartości białka, albo według względnej powierzchni piku SII [E],
|
|
P %
|
=
|
całkowita zawartość białka wyrażona jako zawartość procentowa, masowo,
|
|
SII [E]
|
=
|
względna powierzchnia piku próbki obliczona w ppkt 9.1.2.
|
Równania te odpowiadają liczbie 1,3 wymienionej w ppkt 9.2.
Rozbieżność (T1 i T2) pomiędzy stwierdzoną względną powierzchnią SIII [E] a względną powierzchnią SIII jest podawana w sposób następujący: T1 = SIII[E] – [(0,376 P% – 10,7) + (SIII[0] – 0,9)]T2 = SIII[E] – [(0,0123 SII[E] + 0,93) + (SIII[0] — 0,9)]
|
Jeżeli T1 i/lub T2
|
są równe lub mniejsze od zera, nie można ustalić obecności serwatki podpuszczkowej.
|
|
Jeżeli T1 i T2
|
są większe od zera, serwatka podpuszczkowa jest obecna.
|
Zawartość serwatki podpuszczkowej jest obliczana zgodnie ze wzorem: W = T2 +0,91
gdzie:
0,91 stanowi odległość na osi pionowej pomiędzy ciągłymi i przerywanymi liniami prostymi.
OdtŁuszczone mleko w proszku

OdtŁuszczone mleko w proszku

ZAŁĄCZNIK XIII
(Artykuł 9)
OZNACZANIE SUCHEJ MASY SERWATKI PODPUSZCZKOWEJ W ODTŁUSZCZONYM MLEKU W PROSZKU ORAZ W MIESZANKACH ZGODNIE Z ROZPORZĄDZENIEM (WE) NR 2799/1999
1. CEL: WYKRYCIE DODATKU SUCHEJ MASY SERWATKI PODPUSZCZKOWEJ DO:
|
a)
|
odtłuszczonego mleka w proszku określonego w art. 2 rozporządzenia (WE) nr 2799/1999; oraz
|
|
b)
|
mieszanek określonych w art. 4 rozporządzenia (WE) nr 2799/1999.
|
2. ODNIESIENIA: MIĘDZYNARODOWA NORMA ISO 707
3. DEFINICJA
Zawartość suchej masy serwatki podpuszczkowej zdefiniowana jest jako procent masowy, określony na podstawie zawartości makropeptydów kazeinowych za pomocą opisanej procedury.
4. ZASADA
Zawartość makropeptydów kazeinowych jest oznaczana zgodnie z załącznikiem XII. Próbki dające pozytywne wyniki poddaje się badaniu na makropeptyd kazeinowy A za pomocą procedury wysokosprawnej chromatografii cieczowej (procedura HPLC) z odwróconymi fazami. Próbki można ewentualnie poddawać bezpośredniemu badaniu za pomocą procedury HPLC z odwróconymi fazami. Ocena wyników otrzymywana jest poprzez odniesienie do próbek wzorcowych składających się z odtłuszczonego mleka w proszku zawierającego znany udział procentowy serwatki w proszku i odtłuszczonego mleka w proszku niezawierającego tego udziału. Wyniki wyższe niż 1 % (m/m) wskazują na obecność suchej masy serwatki podpuszczkowej.
5. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej. Należy używać wody destylowanej lub wody o równoważnym stopniu czystości. Acetonitryl powinien mieć jakość spektroskopową lub jakość wymaganą dla HPLC.
Odczynniki konieczne do przeprowadzenia procedury są opisane w załączniku XII do niniejszego rozporządzenia.
Odczynniki do HPLC z odwróconymi fazami.
5.1. Roztwór kwasu trichlorooctowego
Rozpuścić 240 g kwasu trichlorooctowego (CCl3COOH) w wodzie i uzupełnić do 1 000 ml. Roztwór powinien być klarowny i bezbarwny.
5.2. Eluenty A i B
Eluent A: 150 ml acetonitrylu (CH3CN), 20 ml izopropanolu (CH3CHOHCH3) oraz 1,00 ml kwasu trifluorooctowego (TFA, CF3COOH) umieszcza się w kolbie miarowej poj. 1 000 ml. Uzupełnić wodą do 1 000 ml.
Eluent B: 550 ml acetonitrylu, 20 ml izopropanolu oraz 1,00 ml TFA umieszcza się w kolbie miarowej poj. 1 000 ml. Uzupełnić wodą do 1 000 ml. Przed zastosowaniem przesączyć roztwór eluentu przez sączek membranowy o średnicy porów 0,45 µm.
5.3. Konserwacja kolumny
Po przeprowadzeniu analiz kolumna przepłukiwana jest eluentem B (metodą gradientową), a następnie przepłukiwana acetonitrylem (metodą gradientową przez 30 minut). Kolumna przechowywana jest w acetonitrylu.
5.4. Próbki wzorcowe
5.4.1. Odtłuszczone mleko w proszku spełniające wymogi składowania w magazynach państwowych (tzn. [0]).
5.4.2. To samo odtłuszczone mleko w proszku zafałszowane 5 % (m/m) serwatką typu podpuszczkowego w proszku o składzie standardowym (tzn. [5]).
5.4.3. To samo odtłuszczone mleko w proszku zafałszowane 50 % (m/m) serwatką typu podpuszczkowego w proszku o składzie standardowym (tzn. [50]) (1)..
6. APARATURA
Aparatura wymagana do przeprowadzenia opisanej procedury określona jest w załączniku XII do niniejszego rozporządzenia.
6.1. Waga analityczna
6.2. Opcjonalnie wirówka umożliwiająca osiągnięcie siły odśrodkowej 2 200 g, wyposażona w probówki wirówkowe z korkiem lub zatyczką, poj. ok. 50 ml.
6.3. Wstrząsarka mechaniczna
6.4. Mieszadło magnetyczne
6.5. Lejki szklane o średnicy ok. 7 cm
6.6. Sączki papierowe, średnia prędkość sączenia, o średnicy ok. 12,5 cm
6.7. Szklany zestaw do sączenia z sączkiem membranowym o średnicy porów 0,45 µm
6.8. Pipety miarowe pozwalające na dozowanie 10 ml (ISO 648, klasa A lub ISO/R 835) albo system dozujący umożliwiający dostarczenie 10,0 ml w ciągu dwóch minut.
6.9. System dozujący umożliwiający dostarczenie 20 ml wody w temp. ok. 50 °C
6.10. Łaźnia wodna kontrolowana termostatycznie, ustawiona na 25 °C ± 0,5 °C.
Zestaw do HPLC, w skład którego wchodzi:
6.11.1. Układ pompowy do dwuskładnikowej elucji gradientowej
6.11.2. Dozownik, ręczny lub automatyczny, poj. 100 µl
6.11.3. Kolumna Agilent Technologies Zorbax 300 SB-C3 (długość 25 cm, średnica wewnętrzna 0,46 cm) bądź równoważna kolumna do chromatografii z odwróconymi fazami z wypełnieniem na bazie krzemionki o szerokich porach.
6.11.4. Piec do kolumn kontrolowany termostatycznie, ustawiony na temp. 35 °C ± 1 °C
6.11.5. Detektor UV o zmiennej długości fali, umożliwiający pomiary przy 210 nm (w razie konieczności można stosować długość fal do 220 nm), o czułości 0,02 Å.
6.11.6. Integrator umożliwiający całkowanie względem wspólnej linii podstawowej lub wykresu między kolejnymi dolinami
Uwaga: Działanie kolumny w temperaturze pokojowej jest możliwe pod warunkiem, że temperatura pokojowa nie podlega wahaniom większym niż 1 °C, w przeciwnym razie ma miejsce zbyt duże wahanie w czasie retencji CMPA.
7. POBIERANIE PRÓBEK
7.1. Próbki należy pobierać zgodnie z procedurą określoną w międzynarodowej normie ISO 707. Państwa członkowskie mogą jednak stosować inną metodę pobierania próbek pod warunkiem, że jest ona zgodna z zasadami powyższej normy.
7.2. Próbkę przechowywać w warunkach wykluczających spadek jakości lub zmianę składu.
8. PROCEDURA
8.1. Przygotowanie próbki do badań
Przenieść mleko w proszku do pojemnika o pojemności w przybliżeniu dwukrotnie większej niż objętość proszku, wyposażonego w hermetyczną pokrywę. Natychmiast zamknąć pojemnik. Wymieszać dobrze mleko w proszku poprzez wielokrotne odwracanie pojemnika do góry dnem.
8.2. Porcja badana
Odmierzyć wagowo 2,00 ± 0,001 g badanej próbki i umieścić w probówce wirówkowej (ppkt 6.2) bądź w odpowiedniej kolbie z korkiem (50 ml).
Uwaga: W przypadku mieszanek odmierzyć wagowo taką ilość badanej próbki, aby odtłuszczona porcja próbki odpowiadała 2,00 g.
8.3. Usuwanie tłuszczu i białek
8.3.1. Do porcji badanej dodać 20,0 ml ciepłej wody (50 °C). Rozpuścić proszek, wstrząsając przez 5 minut lub przez 30 minut w przypadku maślanki kwasowej, za pomocą wstrząsarki mechanicznej (ppkt 6.3). Umieścić probówkę w łaźni wodnej (ppkt 6.10) i pozostawić do wyrównania się temperatury do 25 °C.
8.3.2. Dodawać stopniowo przez dwie minuty 10,0 ml roztworu kwasu trichlorooctowego (ppkt 5.1) o temp. ok. 25 °C, mieszając jednocześnie energicznie mieszadłem magnetycznym (ppkt 6.4). Umieścić probówkę w łaźni wodnej (ppkt 6.10) i pozostawić na 60 minut.
8.3.3. Wirować (ppkt 6.2) przez 10 minut przy 2 200 g lub przesączyć przez sączek papierowy (ppkt 6.6), odrzucając pierwsze 5 ml przesączu.
8.4. Oznaczenie chromatograficzne
8.4.1. Wykonać analizę HPLC w sposób opisany w załączniku XII. Jeżeli otrzymany wynik jest negatywny, analizowana próbka nie zawiera suchej masy serwatki podpuszczkowej w ilościach wykrywalnych. Jeżeli wynik jest pozytywny, należy zastosować procedurę HPLC z odwróconymi fazami, opisaną poniżej. Można ewentualnie zastosować bezpośrednio metodę HPLC z odwróconymi fazami Obecność maślanki kwasowej w proszku może dać fałszywie pozytywny wynik. Metoda HPLC z odwróconymi fazami wyklucza taką możliwość.
8.4.2. Przed przeprowadzeniem analizy HPLC z odwróconymi fazami należy zoptymalizować warunki gradientu. Czas retencji wynoszący 26 ± 2 minuty dla CMPA jest czasem optymalnym w systemach gradientowych z martwą objętością wynoszącą ok. 6 ml (objętość od punktu, w którym łączą się rozpuszczalniki, do pętli dozownika włącznie). W przypadku systemów gradientowych z niższą martwą objętością (np. 2 ml) należy stosować optymalny czas retencji wynoszący 22 minuty.
Pobrać roztwory próbek wzorcowych (ppkt 5.4) z dodatkiem 50 % serwatki podpuszczkowej oraz bez takiego dodatku.
Wprowadzić 100 μl nadsączu lub przesączu (ppkt 8.3.3) do aparatury HPCL działającej w warunkach gradientu rozpoznawczego podanych w tabeli 1.
Tabela 1
Warunki gradientu rozpoznawczego dla optymalizacji chromatografii
|
Czas
(minuty)
|
Przepływ
(ml/min.)
|
% A
|
% B
|
Krzywa
|
|
Początkowy
|
1,0
|
90
|
10
|
*
|
|
27
|
1,0
|
60
|
40
|
liniowa
|
|
32
|
1,0
|
10
|
90
|
liniowa
|
|
37
|
1,0
|
10
|
90
|
liniowa
|
|
42
|
1,0
|
90
|
10
|
liniowa
|
Porównanie dwóch chromatogramów powinno ujawnić położenie piku CMPΑ.
Wykorzystując podany niżej wzór, można obliczyć początkowy skład rozpuszczalnika, który ma zostać użyty do gradientu normalnego (zob. ppkt 8.4.3) % B = 10 – 2,5 + (13,5 + (RTcmpA – 26) / 6) × 30 / 27 % B = 7,5 + (13,5 + (RTcmpA – 26) / 6) × 1,11
gdzie:
|
RTcmpA
|
:
|
czas retencji CMPΑ w gradiencie rozpoznawczym
|
|
10
|
:
|
początkowy % B gradientu rozpoznawczego
|
|
2,5
|
:
|
% B w punkcie środkowym minus % B w początku gradientu normalnego
|
|
13,5
|
:
|
czas w punkcie środkowym gradientu rozpoznawczego
|
|
26
|
:
|
wymagany czas retencji CMPΑ
|
|
6
|
:
|
stosunek nachylenia gradientu rozpoznawczego i normalnego
|
|
30
|
:
|
% B na początku minus % B w 27 minucie gradientu rozpoznawczego
|
|
27
|
:
|
czas przebiegu gradientu rozpoznawczego
|
8.4.3. Pobranie roztworów badanych próbek:
Wprowadzić 100 µl dokładnie odmierzonego nadsączu lub przesączu (ppkt 8.3.3) do aparatury HPCL, pracując przy natężeniu przepływu roztworu eluentu (ppkt 5.2) wynoszącej 1,0 ml na minutę.
Skład eluentu w chwili rozpoczęcia analizy otrzymuje się z ppkt 8.4.2. Zazwyczaj jest on zbliżony do A: B = 76: 24 (ppkt 5.2). Natychmiast po wprowadzeniu uruchamia się gradient liniowy, który daje zwiększenie wartości procentowej B o 5 % po 27 minutach. Następnie uruchamia się gradient liniowy, który pozwala na uzyskanie składu eluentu w wysokości 90 % B w ciągu pięciu minut. Skład ten utrzymywany jest przez pięć minut, po czym zostaje zmieniony poprzez gradient liniowy w ciągu pięciu minut do składu początkowego. W zależności od wewnętrznej pojemności układu pompującego następne wprowadzenie można wykonać 15 minut po uzyskaniu warunków początkowych.
Uwaga 1: Czas retencji CMPA powinien wynosić 26 ± 2 minuty. Można to osiągnąć poprzez zmianę początkowych i końcowych warunków pierwszego gradientu. Jednakże różnica w % B dla początkowych i końcowych warunków pierwszego gradientu musi pozostać na poziomie 5 % B.
Uwaga 2: Eluenty powinny być odgazowane w sposób wystarczający i pozostawać w takim stanie. Jest to istotne dla właściwego funkcjonowania gradientowego układu pompowego. Odchylenie standardowe dla czasu retencji piku CMPA powinno być mniejsze niż 0,1 minuty (n = 10).
Uwaga 3: Po wprowadzeniu każdej serii pięciu próbek należy wprowadzić próbkę odniesienia (5) i wykorzystać ją do obliczenia nowego współczynnika odpowiedzi R. (ppkt 9.1.1).
8.4.4. Wyniki analizy chromatograficznej badanej próbki (E) otrzymuje się w postaci chromatogramu, na którym pik CMPA określa się na podstawie jego czasu retencji wynoszącego ok. 26 minut.
Integrator (ppkt 6.11.6) automatycznie oblicza wysokość H piku CMPA. W każdym chromatogramie należy sprawdzić położenie linii podstawowej. W przypadku nieprawidłowego położenia linii podstawowej analizę lub całkowanie należy powtórzyć.
Uwaga: Jeżeli pik CMPA jest wystarczająco oddzielony od innych pików, należy zastosować wyznaczenie linii podstawowej na podstawie położenia dolin między pikami, w przeciwnym razie zastosować spuszczenie prostopadłych na wspólną linię podstawową, której punkt wyjścia powinien być położony w pobliżu piku CMPA (co wyklucza t = 0 min.!). W odniesieniu do wzorca i do próbek należy zastosować tę samą metodę całkowania, jak również sprawdzić, w przypadku wspólnej linii podstawowej, jej spójność w odniesieniu do próbek i do wzorca.
Przed dokonaniem interpretacji ilościowej niezbędne jest zbadanie wyglądu każdego chromatogramu w celu wykrycia ewentualnych nieprawidłowości, będących skutkiem wadliwego działania aparatury lub kolumny albo też pochodzenia i rodzaju analizowanej próbki. W razie wątpliwości analizę należy powtórzyć.
8.5. Kalibracja
8.5.1. Do próbek wzorcowych (ppkt 5.4.1–5.4.2) zastosować dokładnie procedurę opisaną od ppkt 8.2 do ppkt 8.4.4. Używać świeżo przygotowanych roztworów, ponieważ w środowisku 8 % kwasu trichlorooctowego w temperaturze pokojowej następuje rozkład CMP. W temp. 4 °C roztwór pozostaje stabilny przez 24 godziny. W przypadku długich serii analiz wskazane jest zastosowanie schłodzonego zasobnika na próbki w dozowniku automatycznym.
Uwaga: Podpunkt 8.4.2. można opuścić w przypadku, gdy % B w warunkach początkowych jest znany z poprzednich analiz.
Chromatogram próbki odniesienia [5] powinien być analogiczny do pokazanego na ryc. 1. Na tym rysunku pik CMPA poprzedzony jest przez dwa niewielkie piki. Istotne jest, aby uzyskać podobny rozdział.
8.5.2. Przed oznaczeniem chromatograficznym próbek wprowadzić 100 μl próbki wzorcowej bez serwatki podpuszczkowej [0] (ppkt 5.4.1).
Chromatogram nie powinien wykazywać piku w czasie retencji piku CMPA.
8.5.3. Określić współczynniki odpowiedzi R poprzez wprowadzenie takiej samej objętości przesączu (ppkt 8.5.1), jak objętość wykorzystana do próbek.
9. PREZENTACJA WYNIKÓW
9.1. Metoda obliczania i wzory
9.1.1. Obliczanie współczynnika odpowiedzi R:
Pik CMPA: R = W/H
Gdzie:
|
R
|
=
|
współczynnik odpowiedzi piku CMPA
|
|
H
|
=
|
wysokość piku CMPA
|
|
W
|
=
|
ilość serwatki w próbce wzorcowej [5].
|
9.2. Obliczanie zawartości procentowej serwatki podpuszczkowej w proszku w próbce
W(E) = R × H(E)
Gdzie:
|
W(E)
|
=
|
zawartość procentowa (m/m) serwatki podpuszczkowej w próbce (E).
|
|
R
|
=
|
współczynnik odpowiedzi piku CMPA (ppkt 9.1.1)
|
|
H(E)
|
=
|
wysokość piku CMPA próbki (E)
|
W przypadku gdy wartość W(E) jest większa niż 1 %, a różnica między czasem retencji próbki badanej a czasem retencji próbki wzorcowej [5] jest mniejsza niż 0,2 minuty, wówczas stwierdzona jest obecność suchej masy serwatki podpuszczkowej.
9.3. Dokładność procedury
9.3.1. Powtarzalność
Różnica między wynikami dwóch oznaczeń wykonanych równocześnie lub w bezpośrednim następstwie przez tego samego analityka przy użyciu tej samej aparatury na identycznym materiale badanym nie może przekraczać 0,2 % m/m.
9.3.2. Odtwarzalność
Nieustalona.
9.3.3. Liniowość
W zakresie 0–16 % serwatki podpuszczkowej należy otrzymać zależność liniową przy współczynniku korelacji > 0,99.
9.4. Interpretacja
Wartość graniczna wynosząca 1 % jest ustalona zgodnie z przepisami ppkt 9.2 i 9.4.1. w załączniku XXI do rozporządzenia (EWG) nr 214/2001, które uwzględnia niepewność związaną z odtwarzalnością.
Tabela 1
Ni — wzorzec 4.6

(1) Serwatka typu podpuszczkowego o składzie standardowym, jak również zafałszowane odtłuszczone mleko w proszku, są dostępne w NIZO, Kernhemseweg 2, skrzynka pocztowa 20 — NL-6710 BA Ede. Można jednak również użyć proszków dających wyniki równoważne z proszkami NIZO.
ZAŁĄCZNIK XIV
(Artykuł 10)
ODTłUSZCZONE MLEKO W PROSZKU: ILOŚCIOWE OZNACZANIE ZAWARTOŚCI FOSFATYDYLOSERYNY I FOSFATYDYLOETANOLAMINY
Metoda: HPLC z odwróconymi fazami.
1. ZAKRES I DZIEDZINA STOSOWANIA
Metoda opisuje procedurę ilościowego oznaczania fosfatydyloseryny (PS) oraz fosfatydyloetanolaminy (PE) w odtłuszczonym mleku w proszku (OMP) i jest odpowiednia do wykrywania suchej masy maślanki w odtłuszczonym mleku w proszku.
2. DEFINICJA
Zawartość PS + PE: ułamek masowy substancji oznaczony z wykorzystaniem procedury określonej w niniejszej metodzie. Wynik wyrażony jest w miligramach dipalmitoilofosfatydyloetanolaminy (PEDP) na 100 g proszku.
3. ZASADA METODY
Ekstrakcja aminofosfolipidów z odtworzonego mleka w proszku przy użyciu metanolu. Oznaczenie PS i PE jako pochodnych dialdehydu o-ftalowego (OPA) metodą HPLC z odwróconymi fazami i detekcją fluorescencyjną. Oznaczenie ilościowe zawartości PS i PE w badanej próbce przez odniesienie do próbki wzorcowej zawierającej znaną ilość PEDP.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej. Woda musi być destylowana lub o co najmniej równoważnym stopniu czystości, jeżeli nie określono inaczej.
4.1. Materiał wzorcowy: PEDP, czystość co najmniej 99 %
Uwaga: Materiał wzorcowy należy przechowywać w temp. – 18 °C.
4.2. Odczynniki do przygotowania próbki wzorcowej i próbki badanej
4.2.1. Metanol o jakości wymaganej dla HPLC
4.2.2. Chloroform o jakości wymaganej dla HPLC
4.2.3. Monochlorowodorek tryptaminy
4.3. Odczynniki do derywatyzacji dialdehydu o-ftalowego
4.3.1. Wodorotlenek sodu, roztwór wodny 12 M
4.3.2. Kwas borny, roztwór wodny 0,4 M, o pH doprowadzonym do 10,0 przy użyciu wodorotlenu sodu (ppkt 4.3.1)
4.3.3. 2-merkaptoetanol
4.3.4. Dialdehyd o-ftalowy (OPA)
4.4. Rozpuszczalniki eluujące do HPLC
4.4.1. Rozpuszczalniki eluujące należy przygotować przy użyciu odczynników o jakości wymaganej dla HPLC.
4.4.2. Woda o jakości wymaganej dla HPLC
4.4.3. Metanol o czystości zbadanej fluorometrycznie
4.4.4. Tetrahydrofuran
4.4.5. Diwodorofosforan sodu
4.4.6. Octan sodu
4.4.7. Kwas octowy
5. APARATURA
5.1. Waga analityczna umożliwiająca ważenie z dokładnością do 1 mg, o dokładności odczytu 0,1 mg
5.2. Zlewki poj. 25 ml i 100 ml
5.3. Pipety umożliwiające dozowanie 1 ml i 10 ml
5.4. Mieszadło magnetyczne
5.5. Pipety miarowe umożliwiające dozowanie 0,2 ml, 0,5 ml i 5 ml
5.6. Kolby miarowe poj. 10 ml, 50 ml i 100 ml
5.7. Strzykawki poj. 20 μl i 100 μl
5.8. Łaźnia ultradźwiękowa
5.9. Wirówka umożliwiająca osiągnięcie 27 000 × g
5.10. Fiolki szklane poj. ok. 5 ml
5.11. Cylinder miarowy poj. 25 ml
5.12. Pehametr o dokładności pomiaru do
Sprzęt do HPLC
5.13.1. Układ pompowy do elucji gradientowej umożliwiający pracę przy natężeniu 1,0 ml/min. i ciśnieniu 200 barów
5.13.2. Automatyczny podajnik do próbek umożliwiający derywatyzację
5.13.3. Ogrzewacz kolumnowy, umożliwiający utrzymanie kolumny w temp. 30 °C ± 1 °C
5.13.4. Detektor fluorescencji, umożliwiający pracę przy długości fali wzbudzenia 330 nm i długości fali emisji 440 nm
5.13.5. Integrator lub oprogramowanie do przetwarzania danych umożliwiające pomiar powierzchni pików
5.13.6. Kolumna Lichrosphere–100 (250 × 4,6 mm) lub równoważna kolumna wypełniona oktadecylosilanem (C 18) o uziarnieniu 5 μm
6. POBIERANIE PRÓBEK
Pobieranie próbek należy przeprowadzić zgodnie z normą ISO 707.
7. PROCEDURA
7.1. Przygotowanie wewnętrznego roztworu wzorcowego
7.1.1. Odmierzyć wagowo 30,0 ± 0,1 mg monochlorowodorku tryptaminy (ppkt 4.2.3) do kolby miarowej poj. 100 ml (ppkt 5.6) i uzupełnić do kreski metanolem (ppkt 4.2.1).
7.1.2. Odmierzyć pipetą 1 ml (ppkt 5.3) tego roztworu do kolby miarowej poj. 10 ml (ppkt 5.6) i uzupełnić do kreski metanolem (ppkt 4.2.1) w celu uzyskania stężenia 0,15 mM tryptaminy.
7.2. Przygotowanie roztworu próbki badanej
7.2.1. Odmierzyć wagowo 1,000 ± 0,001 g próbki OMP do zlewki poj. 25 ml (ppkt 5.2). Odmierzyć pipetą (ppkt 5.3) 10 ml wody destylowanej o temp. 40 °C i mieszać mieszadłem magnetycznym (ppkt 5.4) przez 30 minut do całkowitego rozpuszczenia grudek.
7.2.2. Odmierzyć pipetą (ppkt 5.5) 0,2 ml odtworzonego mleka do kolby miarowej poj. 10 ml (ppkt 5.6), za pomocą strzykawki (ppkt 5.7) dodać 100 μl roztworu 0,15 mM tryptaminy (ppkt 7.1) i uzupełnić do kreski metanolem (ppkt 4.2.1). Wymieszać dokładnie, odwracając kolbę i poddawać sonikacji (ppkt 5.8) przez 15 minut.
7.2.3. Wirować (ppkt 5.9) przy 27 000 × g przez 10 minut i zebrać nadsącz do szklanej fiolki (ppkt 5.10).
Uwaga: Do chwili przeprowadzenia analizy HPLC roztwór próbki badanej należy przechowywać w temp. 4 °C.
7.3. Przygotowanie zewnętrznego roztworu wzorcowego
7.3.1. Odmierzyć wagowo 55,4 mg PEDP (ppkt 4.1) do kolby miarowej poj. 50 ml (ppkt 5.6) i dodać ok. 25 ml chloroformu (ppkt 4.2.2), używając cylindra miarowego (ppkt 5.11). Podgrzać zakorkowaną kolbę do 50 °C i wymieszać dokładnie aż do rozpuszczenia PEDP. Schłodzić kolbę do 20 °C, uzupełnić do kreski metanolem (ppkt 4.2.1) i wymieszać przez odwracanie.
7.3.2. Odmierzyć pipetą 1 ml (ppkt 5.3) tego roztworu do kolby miarowej poj. 100 ml (ppkt 5.6), uzupełnić do kreski metanolem (ppkt 4.2.1). Odmierzyć pipetą 1 ml (ppkt 5.3) tego roztworu do kolby miarowej poj. 10 ml (ppkt 5.6), dodać 100 μl (ppkt 5.7) roztworu 0,15 mM tryptaminy (ppkt 7.1) i uzupełnić do kreski metanolem (ppkt 4.2.1). Wymieszać przez odwracanie.
Uwaga: Do chwili przeprowadzania analizy HPLC roztwór próbki odniesienia należy przechowywać w temp. 4 °C.
7.4. Przygotowanie odczynnika do derywatyzacji
Odmierzyć wagowo 25,0 ± 0,1 mg OPA (ppkt 4.3.4) do kolby miarowej poj. 10 ml (ppkt 5.6), dodać 0,5 ml (ppkt 5.5) metanolu (ppkt 4.2.1) i wymieszać dokładnie w celu rozpuszczenia OPA. Uzupełnić do kreski roztworem kwasu bornego (ppkt 4.3.2) i dodać 20 μl 2-merkaptoetanolu (ppkt 4.3.3) za pomocą strzykawki (ppkt 5.7).
Uwaga: Odczynnik do derywatyzacji należy przechowywać w temp. 4 °C w fiolce z brązowego szkła; w takich warunkach zachowuje on stabilność przez okres jednego tygodnia.
7.5. Oznaczenie za pomocą HPLC
7.5.1. Rozpuszczalniki eluujące (ppkt 4.4)
Rozpuszczalnik A: Roztwór 0,3 mM diwodorofosforanu sodu i roztwór 3 mM octanu sodu (pH doprowadzone do wartości 6,5 za pomocą kwasu octowego):metanol:tetrahydrofuran = 558:440:2 (v/v/v)
Rozpuszczalnik B: metanol
7.5.2. Zalecany gradient elucji
|
Czas
(minuty)
|
Rozpuszczalnik A
(%)
|
Rozpuszczalnik B
(%)
|
Natężenie przepływu
(ml/min.)
|
|
Początkowy
|
40
|
60
|
0
|
|
0,1
|
40
|
60
|
0,1
|
|
5,0
|
40
|
60
|
0,1
|
|
6,0
|
40
|
60
|
1,0
|
|
6,5
|
40
|
60
|
1,0
|
|
9,0
|
36
|
64
|
1,0
|
|
10,0
|
20
|
80
|
1,0
|
|
11,5
|
16
|
84
|
1,0
|
|
12,0
|
16
|
84
|
1,0
|
|
16,0
|
10
|
90
|
1,0
|
|
19,0
|
0
|
100
|
1,0
|
|
20,0
|
0
|
100
|
1,0
|
|
21,0
|
40
|
60
|
1,0
|
|
29,0
|
40
|
60
|
1,0
|
|
30,0
|
40
|
60
|
0
|
Uwaga: W celu uzyskania rozdzielczości przedstawionej na ryc. 1 niezbędna może się okazać drobna modyfikacja gradientu elucji.
Temperatura kolumny: 30 °C.
7.5.3. Objętość wprowadzona: 50 μl odczynnika do derywatyzacji i 50 μl roztworu próbki.
7.5.4. Równoważenie kolumny
Uruchamiając układ każdego dnia, należy przepłukiwać kolumny rozpuszczalnikiem B o stężeniu 100 % przez 15 minut, następnie ustalić rozpuszczalniki A i B w proporcji 40:60 i równoważyć przy 1 ml/min. przez 15 minut. Przeprowadzić ślepą próbę poprzez wprowadzenie metanolu (ppkt 4.2.1).
Uwaga: Przed długotrwałym przechowywaniem przepłukiwać kolumnę mieszanką metanolu i chloroformu w proporcji 80:20 (v/v) przez 30 minut.
7.5.5. Oznaczyć zawartość PS + PE w badanej próbce.
7.5.6. Przeprowadzić sekwencję analiz chromatograficznych, utrzymując niezmieniony czas pomiędzy pojedynczymi analizami w celu uzyskania stałych czasów retencji. Wprowadzać zewnętrzny roztwór wzorcowy (ppkt 7.3) co 5–10 wprowadzeń roztworu badanej próbki w celu obliczenia współczynnika odpowiedzi.
Uwaga: Kolumna musi być czyszczona przez przepłukiwanie 100 % rozpuszczalnikiem B (ppkt 7.5.1), przez co najmniej 30 minut, co 20–25 pojedynczych analiz.
7.6. Metoda całkowania
7.6.1. Pik PEDP
PEDP jest eluowana w postaci pojedynczego piku. Określić powierzchnię piku przez całkowanie wykresu między kolejnymi dolinami.
7.6.2. Pik tryptaminy
Tryptamina jest eluowana w postaci pojedynczego piku (ryc. 1). Określić powierzchnię piku przez całkowanie wykresu między kolejnymi dolinami.
7.6.3. Grupy pików PS i PE
W opisanych warunkach (ryc. 1) PS jest eluowana w postaci dwóch głównych, częściowo nierozdzielonych pików, poprzedzonych mniejszym pikiem. PE jest eluowana w postaci trzech głównych, częściowo nierozdzielonych pików. Określić całkowitą powierzchnię każdego skupiska pików przez ustawienie linii podstawowej zgodnie z ryc. 1.
8. OBLICZANIE I PREZENTACJA WYNIKÓW
Zawartość PS i PE w badanej próbce oblicza się, jak następuje: C = 55,36 × ((A2)/(A1)) × ((T1)/(T2))
gdzie:
|
C
|
=
|
zawartość PS lub PE (mg/100 g proszku) w badanej próbce
|
|
A1
|
=
|
powierzchnia piku PEDP roztworu próbki wzorcowej (ppkt 7.3)
|
|
A2
|
=
|
powierzchnia piku PS lub PE roztworu próbki badanej (ppkt 7.2)
|
|
T1
|
=
|
powierzchnia piku tryptaminy w roztworze próbki wzorcowej (ppkt 7.3)
|
|
T2
|
=
|
powierzchnia piku tryptaminy w roztworze próbki badanej (ppkt 7.2)
|
9. DOKŁADNOŚĆ METODY
Uwaga: Wartości powtarzalności zostały obliczone zgodnie z międzynarodową normą IDF (1). Tymczasowa granica odtwarzalności została obliczona zgodnie z procedurą określoną w załączniku III lit. b) do wspomnianej normy.
9.1. Powtarzalność
Względne odchylenie standardowe powtarzalności, wyrażające zmienność niezależnych wyników analitycznych otrzymanych przez ten sam podmiot, wykorzystujący tę samą aparaturę, w takich samych warunkach, na takiej samej próbce badanej oraz w krótkim odstępie czasu, nie powinno przekraczać 2 % wartości względnej. Jeżeli w tych warunkach otrzymane są dwa oznaczenia, względna różnica między dwoma wynikami nie powinna być większa niż 6 % średniej arytmetycznej wyników.
9.2. Odtwarzalność
Jeżeli dwa oznaczenia otrzymane są przez podmioty w różnych laboratoriach, wykorzystujące różną aparaturę w różnych warunkach, podczas analizy tej samej próbki badanej, względna różnica między dwoma wynikami nie powinna być większa niż 11 % średniej arytmetycznej wyników.
10. PIŚMIENNICTWO
10.1. Resmini P., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., „Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids”. Sci. Tecn. Latt.-Cas., 39,395 (1988).
Ryc. 1
Uzyskany przy użyciu HPLC wzór pochodnych OPA fosfatydyloseryny (PS) i fosfatydyloetanolaminy (PE) w wyciągu metanolowym odtworzonego odtłuszczonego mleka w proszku. Podano tryb całkowania dla pików PS, PE i tryptaminy (wzorzec wewnętrzny).
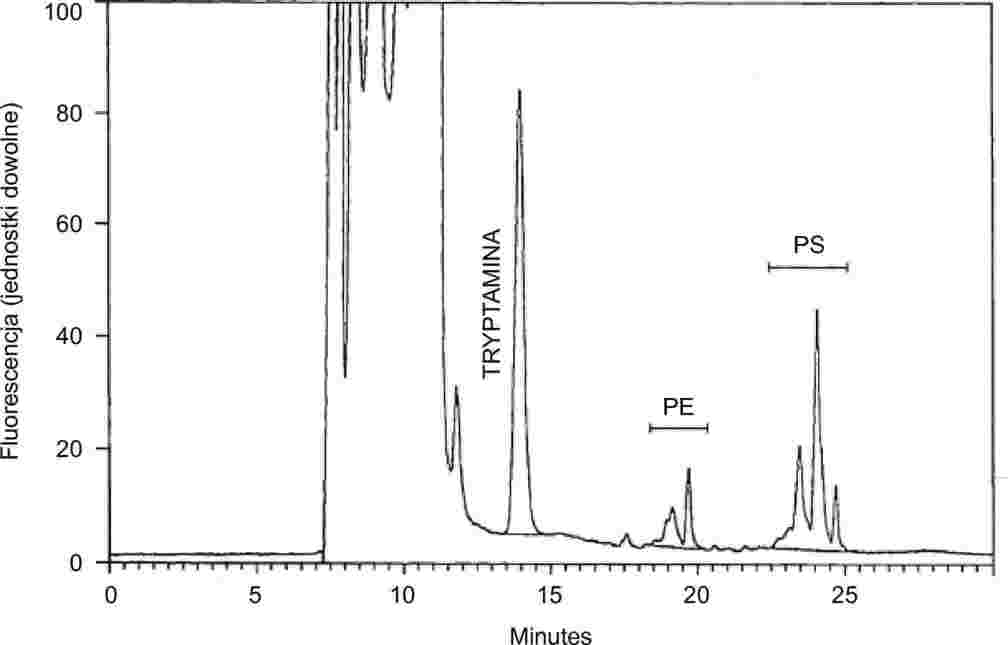
(1) Norma międzynarodowa 135B/1991. Mleko i przetwory mleczne. Charakterystyka precyzji metod analitycznych. Zarys procedury współpracy badawczej.
ZAŁĄCZNIK XV
(Artykuł 11)
WYKRYWANIE POZOSTAŁOŚCI ANTYBIOTYKÓW W ODTŁUSZCZONYM MLEKU W PROSZKU
Stosuje się badanie przesiewowe na obecność antybiotyków, przy użyciu Geobacillus
stearothermophilus var. calidolactis ATCC 10149 (identyczny ze szczepem C953) jako mikroorganizmu badanego, o wrażliwości odpowiedniej do wykrycia w mleku benzylopenicyliny w ilości 4 μg/kg mleka oraz sulfadymidyny w ilości 100 μg/kg mleka. W handlu dostępne są zestawy do badań, które można wykorzystać, o ile mają wymaganą wrażliwość w odniesieniu do benzylopenicyliny i sulfadymidyny.
Do badania używa się odtworzonego odtłuszczonego mleka w proszku (l g proszku +9 ml wody destylowanej). Badanie przeprowadza się w sposób opisany w normie ISO/TS 26844:2006 — IDF Bulletin No 258/1991, sekcja 1, rozdział 2, bądź zgodnie z instrukcją producenta zestawu do badań (1).
Wyniki pozytywne należy interpretować w sposób następujący:
|
1.
|
Obecność beta-laktamów może być potwierdzona w drodze powtórzenia badania i dodania penicylinazy do układu badawczego (2):
Wynik negatywny: Substancją hamującą jest antybiotyk beta-laktamowy.
Wynik pozytywny utrzymuje się: Za pomocą tej procedury nie można zidentyfikować substancji hamującej, postępować zgodnie z pkt. 2.
|
|
2.
|
Obecność sulfonamidów może być potwierdzona w drodze powtórzenia badania i dodania kwasu p-aminobenzoesowy do układu badawczego.
Wynik negatywny: Substancją hamującą jest sulfonamid.
Wynik pozytywny utrzymuje się: Za pomocą tej procedury nie można zidentyfikować substancji hamującej, postępować zgodnie z pkt 3.
|
|
3.
|
Obecność połączenia beta-laktamu i sulfonamidu może być potwierdzona w drodze powtórzenia badania i dodania penicylinazy i kwasu p-aminobenzoesowy do układu badawczego.
Wynik negatywny: Substancjami hamującymi są antybiotyk beta-laktamowy i sulfonamid.
Wynik pozytywny: Za pomocą tej procedury nie można zidentyfikować substancji hamującej.
|
(1) Ważna uwaga: Przy analizie odtłuszczonego mleka w proszku można uzyskać wyniki fałszywie pozytywne. Z tej przyczyny ważne jest sprawdzenie, czy wykorzystywany układ badawczy nie daje wyników fałszywie pozytywnych.
(2) Niektóre beta-laktamy są mniej wrażliwe na beta-laktamazę. W takich przypadkach zaleca się dodatkową wstępną obróbkę próbki (1 ml próbki badanej plus 0,3 ml koncentratu beta-laktamaz Penase Concentrate w temperaturze 37 °C przez 2 godziny).
ZAŁĄCZNIK XVI
(Artykuł 12)
ILOŚCIOWE OZNACZANIE ZAWARTOŚCI ODTŁUSZCZONEGO MLEKA W PROSZKU W MIESZANKACH PASZOWYCH METODĄ ENZYMATYCZNEJ KOAGULACJI PARAKAZEINY
1. CEL
Ilościowe oznaczanie zawartości odtłuszczonego mleka w proszku w mieszankach paszowych metodą enzymatycznej koagulacji parakazeiny.
2. ZAKRES
Metoda ma zastosowanie do mieszanek paszowych zawierających co najmniej 10 % odtłuszczonego mleka w proszku; duże ilości maślanki i/lub niektórych białek niepochodzących z mleka mogą prowadzić do powstania zakłóceń.
3. ZASADA METODY
3.1. Rozpuszczenie kazeiny zawartej w mieszance paszowej poprzez ekstrahowanie za pomocą roztworu cytrynianu sodu.
3.2. Dostosowanie stężenia jonów wapnia do wymaganego poziomu w celu strącenia parakazeiny poprzez dodanie podpuszczki.
3.3. Zawartość azotu w osadzie parakazeiny jest określana metodą Kjeldahla, zgodnie z opisem w normie ISO 8968–2:2001/IDF 20–2:2001; ilość odtłuszczonego mleka w proszku jest obliczana na podstawie minimalnej zawartości kazeiny wynoszącej 27,5 % (zob. ppkt 8.1).
4. ODCZYNNIKI
Wykorzystywane odczynniki muszą być odczynnikami o uznanej klasie analitycznej. Należy używać wody destylowanej lub wody o równoważnym stopniu czystości. Z wyjątkiem podpuszczki (ppkt 4.5), wszystkie odczynniki i roztwory muszą być wolne od substancji azotowych.
4.1. Cytrynian trisodu, dihydrat (roztwór 1 % w/v)
4.2. Chlorek wapnia (roztwór ok. 5 M)
Rozpuścić 75 g CaCl2 · 2 H2O w 100 ml wody destylowanej poprzez wstrząsanie (uwaga na reakcję egzotermiczną). Pozostawić do następnego dnia, a następnie przesączyć roztwór. Przechowywać roztwór w chłodziarce.
4.3. Wodorotlenek sodu 0,1 N
4.4. Kwas chlorowodorowy 0,1 N
4.5. Płynna podpuszczka cielęca (o sile ok. 100 IMCU/ml zgodnie z normą ISO 11815/IDF 157). Przechowywać w chłodziarce w temperaturze od 4 °C do 6 °C.
4.6. Odczynniki do ilościowego oznaczania azotu według metody Kjeldahla, zgodnie z opisem w normie ISO 8968–2:2001/IDF 20–2:2001.
5. APARATURA
Typowa aparatura laboratoryjna, w skład której wchodzi:
5.1. Moździerz lub homogenizator
5.2. Waga analityczna, umożliwiająca ważenie z dokładnością do 1 mg, o dokładności odczytu 0,1 mg
5.3. Wirówka przenośna (500 g lub 2 000–3 000 obr./min.) z probówkami poj. 50 ml oraz 2 000 g
5.4. Mieszadło magnetyczne z dipolami (10–15 mm)
5.5. Zlewki poj. 150–200 ml
5.6. Kolby poj. 250 ml i 500 ml
5.7. Lejki szklane o średnicy 60–80 mm
5.8. Sączki bezpopiołowe o dużej szybkości sączenia, o średnicy 150 mm (Whatman N° 41 lub równoważne)
5.9. Pipety o różnych pojemnościach nominalnych
5.10. Łaźnia wodna kontrolowana termostatycznie w temp. 37 °C ± 1 °C
5.11. Pehametr o dokładności pomiaru do
5.12. Termometry umożliwiające pomiar z dokładnością do 1 °C
6. PROCEDURA
6.1. Przygotowanie próbki
Rozetrzeć w moździerzu lub zhomogenizować w młynku 10–20 g próbki w celu uzyskania jednorodnej mieszanki.
6.2. Rozpuszczenie mleka w proszku i oddzielenie nierozpuszczalnych pozostałości
6.2.1. Odmierzyć wagowo 1,000 ± 0,002 g dobrze zhomogenizowanej mieszanki paszowej (ppkt 6.1) bezpośrednio do probówki wirówkowej poj. 50 ml. Dodać 30 ml roztworu cytrynianu trisodu (ppkt 4.1) podgrzanego uprzednio do temp. 45 °C ± 2 °C. Mieszać za pomocą mieszadła magnetycznego przez co najmniej pięć minut lub energicznie wstrząsając ręcznie.
6.2.2. Wirować przy 500 g (2 000–3 000 obr./min.) przez 10 minut, a następnie zdekantować klarowny fazę wodną nadsączu do zlewki poj. 150–200 ml, uważając, aby nie przelać żadnej ilości luźnego materiału pozostającego na dnie.
6.2.3. Przeprowadzić dwie kolejne ekstrakcje pozostałości, według tej samej procedury, dodając uzyskane wyciągi do pierwszego.
6.2.4. Jeżeli na powierzchni utworzy się warstwa oleju, schłodzić wyciąg w chłodziarce do zestalenia się tłuszczu i usunąć zestaloną warstwę za pomocą szpatułki.
6.3. Koagulacja kazeiny za pomocą enzymów podpuszczki
6.3.1. Nie przerywając mieszania, dodawać po kropli 2 ml chlorku wapnia (ppkt 4.2) do całości wyciągu wodnego (ok. 100 ml). Doprowadzić pH do wartości 6,4–6,5 przy użyciu roztworu NaOH (ppkt 4.3) lub HCl (ppkt 4.4). Umieścić w kontrolowanej termostatycznie łaźni wodnej w temp. 37 °C ± 1 °C na 15–20 minut w celu uzyskania równowagi w zakresie zasolenia. Stanie się to bardziej wyraźne dzięki utworzeniu się lekkiego zmętnienia.
6.3.2. Przenieść płyn do jednej probówki wirówkowej i wirować przy 2 000 g przez 10 minut w celu usunięcia wytrąconego materiału. Przenieść nadsącz, nie spłukując osadu, do następnej probówki wirówkowej.
6.3.3. Doprowadzić temperaturę nadsączu ponownie do 37 °C ± 1 °C. Mieszając wyciąg, dodawać po kropli 0,5 ml płynnej podpuszczki (ppkt 4.5). Koagulacja zachodzi w ciągu dwóch minut.
6.3.4. Umieścić z powrotem próbkę w łaźni wodnej i pozostawić ją w temp. 37 °C ± 1 °C na 15 minut. Wyjąć próbkę z łaźni i rozbić koagulat poprzez mieszanie. Wirować przy 2 000 g przez 10 minut. Przesączyć nadsącz przez odpowiedni sączek papierowy (ppkt 5.8) i zachować sączek. Przemyć osad, mieszając go w probówce wirówkowej z 50 ml wody o temp. ok. 35 °C.
Wirować ponownie przy 2 000 g przez 10 minut. Przesączyć nadsącz przez zachowany poprzednio sączek papierowy.
6.4. Oznaczanie zawartości azotu w kazeinie
6.4.1. Po przemyciu przenieść ilościowo osad na zachowany poprzednio sączek papierowy (ppkt 6.3.4), używając wody destylowanej. Przenieść wysuszony sączek papierowy do kolby Kjeldahla. Oznaczyć zawartość azotu metodą Kjeldahla, zgodnie z opisem w normie ISO 8968–2:2001/IDF 20–2:2001.
7. ŚLEPA PRÓBA
7.1. Ślepą próbę przeprowadza się regularnie przez poddanie mineralizacji metodą Kjeldahla zgodnie z opisem w normie ISO 8968–2:2001/IDF 20–2:2001. Bezpopiołowy sączek papierowy (ppkt 5.8) jest zwilżony mieszaniną składającą się z 90 ml roztworu cytrynianu sodu (ppkt 4.1), 2 ml roztworu chlorku wapnia (ppkt 4.2), 0,5 ml płynnej podpuszczki (ppkt 4.5) i przemyty przy użyciu 3 × 15 ml wody destylowanej.
7.2. Objętość kwasu użytego do przeprowadzenia ślepej próby należy odjąć od objętości kwasu (ppkt 4.4) użytego do miareczkowania próbki.
8. PREZENTACJA WYNIKÓW
8.1. Zawartość procentowa odtłuszczonego mleka w proszku w mieszankach paszowych jest obliczana za pomocą poniższego wzoru:

gdzie:
N stanowi zawartość procentową azotu w parakazeinie,
27,5 stanowi współczynnik służący przeliczeniu oznaczonej kazeiny na zawartość procentową odtłuszczonego mleka w proszku,
2,81 oraz 0,908 są współczynnikami korygującymi otrzymanymi z analizy regresji.
9. DOKŁADNOŚĆ METODY
9.1. Powtarzalność
W co najmniej 95 % przebadanych przypadków analiza tej samej próbki wykonana w dwóch powtórzeniach przez ten sam podmiot w tym samym laboratorium musi dawać różnice wyników równoważne wielkości nie większej niż 2,3 g odtłuszczonego mleka w proszku w 100 g mieszanki paszowej.
9.2. Odtwarzalność
W co najmniej 95 % przebadanych przypadków ta sama próbka analizowana przez dwa laboratoria musi dawać różnice wyników nie większe niż 6,5 g odtłuszczonego mleka w proszku w 100 g mieszanki paszowej.
10. UWAGI
10.1. Dodanie dużych wartości procentowych niektórych białek niepochodzących z mleka, w szczególności białek soi, podczas podgrzewania z odtłuszczonym mlekiem w proszku, może prowadzić do uzyskania zbyt wysokich wyników z powodu współwytrącania z parakazeiną mleka.
10.2. Dodanie maślanki może prowadzić do uzyskania nieco zaniżonych wartości liczbowych z racji, że oznaczona jest wyłącznie część beztłuszczowa. Dodanie niektórych maślanek kwasowych może dawać znacznie niższe wartości liczbowe z powodu niepełnego rozpuszczenia w roztworze cytrynianu.
10.3. Dodatki lecytyny rzędu 0,5 % lub większe mogą również prowadzić do niskich wyników.
10.4. Dodanie mocno podgrzanego odtłuszczonego mleka w proszku może prowadzić do uzyskania zbyt wysokich wartości liczbowych z powodu współwytrącania niektórych białek serwatki z parakazeiną mleka.
ZAŁĄCZNIK XVII
(Artykuł 13)
OZNACZANIE SKROBI W ODTŁUSZCZONYM MLEKU W PROSZKU, ZDENATUROWANYM MLEKU W PROSZKU ORAZ W MIESZANKACH PASZOWYCH
1. ZAKRES
Metoda służy do wykrywania skrobi, która jest wydzielana jako wskaźnik w zdenaturowanym mleku w proszku.
Granica wykrywalności metody wynosi w przybliżeniu 0,05 g skrobi na 100 g próbki.
2. ZASADA
Reakcja oparta jest na reakcji wykorzystywanej w jodometrii:
|
—
|
wiązanie przez koloidy wolnego jodu w roztworze wodnym,
|
|
—
|
absorpcja na micelach skrobi i tworzenie zabarwienia.
|
3. ODCZYNNIKI
3.1. Roztwór jodu:
|
—
|
woda destylowana: 100 ml,
|
|
—
|
rozpuścić 1,0 g jodu i 2,0 g jodku potasu w wodzie w kolbie miarowej poj. 100 ml. Uzupełnić wodą do kreski 100 ml i wymieszać.
|
4. APARATURA
4.1. Waga analityczna
4.2. Wrząca łaźnia wodna
4.3. Probówki, 25 mm × 200 mm
5. PROCEDURA
Odmierzyć wagowo 1 g próbki z dokładnością do 0,1 g i przenieść ją do probówki (ppkt 4.3).
Dodać 20 ml wody destylowanej i wstrząsać w celu dyspersji próbki.
Umieścić we wrzącej łaźni wodnej (ppkt 4.2) i pozostawić na 5 minut.
Wyjąć z łaźni i schłodzić do temperatury pokojowej.
Dodać 0,5 ml roztworu jodu (ppkt 3.1), wstrząsnąć i obserwować pojawiające się zabarwienie.
6. PREZENTACJA WYNIKÓW
Zabarwienie niebieskie wskazuje na obecność naturalnej skrobi w próbce.
W przypadku gdy próbka zawiera skrobię modyfikowaną, zabarwienie może nie być niebieskie.
7. UWAGI
Zabarwienie, intensywność zabarwienia oraz obecność skrobi widziana pod mikroskopem będą się różnić w zależności od pochodzenia skrobi naturalnej (np. kukurydza lub ziemniak) oraz od rodzaju skrobi modyfikowanej obecnej w próbce.
W obecności skrobi zmodyfikowanych wytworzone zabarwienie zmienia się na fioletowe, czerwone lub brązowe, odpowiednio do stopnia modyfikacji struktury krystalicznej skrobi naturalnej.
ZAŁĄCZNIK XVIII
(Artykuł 14)
OZNACZANIE ZAWARTOŚCI WILGOCI W ŚMIETANIE W PROSZKU
1. ZAKRES
Niniejszy załącznik określa metodę oznaczania zawartości wilgoci w śmietanie w proszku.
2. POJĘCIA I DEFINICJE
Do celów niniejszego załącznika stosuje się następujące definicje:
Zawartość wilgoci: Ubytek masy oznaczony w drodze procedury określonej w niniejszej normie międzynarodowej.
Jest ona wyrażona jako procent masy.
3. ZASADA
Suszenie porcji badanej w temp. 102 °C ± 2 °C do uzyskania stałej masy i ważenie w celu określenia ubytku masy.
4. APARATURA
Typowe wyposażenie laboratoryjne, a w szczególności:
4.1. Waga analityczna, umożliwiająca ważenie z dokładnością do 1 mg, o dokładności odczytu 0,1 mg.
4.2. Piec suszarniczy, dobrze wentylowany, umożliwiający termostatyczne utrzymanie temp. 102 °C ± 2 °C w całej przestrzeni roboczej.
4.3. Eksykator, zawierający świeżo wysuszony żel krzemionkowy ze wskaźnikiem wilgotności lub inny skuteczny środek suszący.
4.4. Płaskodenne naczynia o głębokości ok. 25 mm i średnicy ok. 50 mm, wykonane z odpowiedniego materiału (na przykład szkło, stal nierdzewna, nikiel lub aluminium), wraz z dopasowanymi, łatwymi do zdjęcia przykrywkami.
4.5. Butelki, z dopasowanymi korkami, do mieszania próbek laboratoryjnych.
5. POBIERANIE PRÓBEK
Istotne jest, aby laboratorium otrzymało do badań próbkę, która jest rzeczywiście reprezentatywna i nie uległa uszkodzeniu lub zmianie w czasie transportu lub przechowywania.
Sposób pobierania próbek nie jest określony w metodzie wyszczególnionej w niniejszej normie międzynarodowej. Zalecaną metodę pobierania próbek podano w normie ISO 707|IDF 50.
Przechowywać próbkę w sposób uniemożliwiający obniżenie jakości i zmianę składu.
6. PRZYGOTOWANIE PRÓBKI DO BADAŃ
Dokładnie wymieszać próbkę badaną, wielokrotnie potrząsając i odwracając pojemnik (jeżeli zajdzie taka potrzeba, po przeniesieniu wszystkich próbek badanych do hermetycznego pojemnika o dostatecznej pojemności, aby umożliwić przeprowadzenie tej operacji).
W razie nieuzyskania w drodze tej procedury całkowitej homogeniczności, pobrać porcje badane (do dwóch pojedynczych oznaczeń) z przygotowanej próbki badanej, z dwóch możliwie najbardziej oddalonych miejsc.
7. PROCEDURA
7.1. Przygotowanie naczynia
7.1.1. Podgrzewać odkryte naczynie oraz pokrywkę (ppkt 4.4) w piecu (ppkt 4.2), ustawionym na temp. 102 °C ± 2 °C, przez co najmniej 1 godzinę.
7.1.2. Przykryć naczynie przykrywką, przenieść przykryte naczynie do eksykatora (ppkt 4.3), pozostawić do wystygnięcia do temperatury pokojowej i zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg.
7.2. Porcja badana
Przenieść ok. 1–3 g przygotowanej próbki badanej (ppkt 6) do naczynia, przykryć pokrywką i zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg.
7.3. Oznaczenie
7.3.1. Zdjąć z naczynia przykrywkę i umieścić je wraz z przykrywką w piecu (ppkt 4.2) ustawionym na temp. 102 °C ± 2 °C na 2 godziny.
7.3.2. Ponownie przykryć naczynie przykrywką, przenieść przykryte naczynie do eksykatora, pozostawić do wystygnięcia do temperatury pomieszczenia i zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg.
7.3.3. Zdjąć z naczynia przykrywkę i ponownie podgrzewać wraz z przykrywką w piecu przez 1 godzinę. Następnie powtórzyć operację opisaną w ppkt 7.3.2.
7.3.4. Powtarzać procedurę podgrzewania i ważenia do czasu, gdy masa ulegnie zmniejszeniu o 1 mg lub mniej, lub gdy ulegnie zwiększeniu między dwoma następującymi po sobie ważeniami.
Do obliczenia zastosować najniższą zapisaną masę.
8. OBLICZANIE I PREZENTACJA WYNIKÓW
8.1. Obliczanie
Zawartość wilgoci, wyrażona w g/100 g, jest równa:

gdzie:
m0
stanowi wyrażoną w gramach masę naczynia i przykrywki (ppkt 7.1.2),
m1
stanowi wyrażoną w gramach masę naczynia, przykrywki i porcji badanej przed suszeniem (ppkt 7.2),
m2
stanowi wyrażoną w gramach masę naczynia, przykrywki i porcji badanej po suszeniu (ppkt 7.3.4).
Wynik należy przedstawić z dokładnością do dwóch miejsc po przecinku.
9. PRECYZJA
Uwaga: Wartości powtarzalności i odtwarzalności zostały uzyskane z wyników badania międzylaboratoryjnego (zob. Steiger, G., Bulletin of IDF No 285/1993, s. 21–28), przeprowadzonego zgodnie z normą IDF 135B:1991. Mleko i przetwory mleczne — Charakterystyka precyzji metod analitycznych — Zarys procedury współpracy badawczej.
9.1. Powtarzalność
Bezwzględna różnica pomiędzy wynikami dwóch niezależnych pojedynczych badań, uzyskanymi z wykorzystaniem tej samej metody na identycznym materiale badanym w tym samym laboratorium przez ten sam podmiot wykorzystujący to samo wyposażenie w krótkim odstępie czasu nie będzie dla więcej niż 5 % przypadków większa niż 0,20 g wilgoci na 100 g produktu.
9.2. Odtwarzalność
Bezwzględna różnica pomiędzy wynikami dwóch niezależnych pojedynczych badań, uzyskanymi z wykorzystaniem tej samej metody na identycznym materiale badanym w różnych laboratoriach przez różne podmioty wykorzystujące różne wyposażenie nie będzie dla więcej niż 5 % przypadków większa niż 0,40 g wilgoci na 100 g produktu.
10. SPRAWOZDANIE Z BADAŃ
W sprawozdaniu z badań należy określić:
|
—
|
wszelkie informacje niezbędne do pełnej identyfikacji próbki,
|
|
—
|
zastosowaną metodę pobierania próbek, o ile jest znana,
|
|
—
|
metodę badawczą zastosowaną w odniesieniu do niniejszej normy międzynarodowej,
|
|
—
|
wszelkie szczegółowe dane operacyjne niewyszczególnione w niniejszej normie międzynarodowej lub uznane za opcjonalne, wraz z danymi szczegółowymi na temat wszelkich zdarzeń, które mogły wywrzeć wpływ na wynik(i) badania,
|
otrzymany wynik (wyniki) badania oraz w przypadku, gdy sprawdzono powtarzalność, ostateczny przytoczony otrzymany wynik.
ZAŁĄCZNIK XIX
(Artykuł 15)
OZNACZANIE ZAWARTOŚCI WILGOCI W KWASOWEJ MAŚLANCE W PROSZKU
1. ZAKRES
Oznaczyć zawartość wilgoci w kwasowej maślance w proszku przeznaczonej na pasze dla zwierząt.
2. ZASADA
Próbka jest suszona w próżni. Ubytek masy jest oznaczony w drodze ważenia.
3. APARATURA
3.1. Waga analityczna, umożliwiająca ważenie z dokładnością do 1 mg, o dokładności odczytu 0,1 mg
3.2. Naczynia z niekorodującego metalu lub ze szkła, zaopatrzone w pokrywki zapewniające hermetyczne zamknięcie; powierzchnia robocza umożliwiająca rozprowadzenie badanej próbki w ilości ok. 0,3 g/cm2.
3.3. Regulowany, ogrzewany elektrycznie piec próżniowy, wyposażony w pompę olejową oraz w mechanizm służący do wprowadzania gorącego suchego powietrza przez kolumnę zawierającą na przykład tlenek wapnia lub siarczan wapnia (zawierającą wskaźnik wilgoci).
3.4. Eksykator ze skutecznym środkiem suszącym.
3.5. Piec suszarniczy, wentylowany, regulowany termostatycznie, w temp. 102 ± 2 °C.
4. PROCEDURA
Podgrzewać naczynie (ppkt 3.2) oraz pokrywkę w piecu (ppkt 3.5) przez co najmniej jedną godzinę. Przykryć naczynie pokrywką, natychmiast przenieść je do eksykatora (ppkt 3.4) i pozostawić do schłodzenia do temperatury pokojowej, następnie zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg.
Zdjąć pokrywkę z naczynia, przenieść do naczynia ok. 5 g próbki i zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg. Umieścić naczynie oraz pokrywkę w piecu próżniowym (ppkt 3.3) podgrzanym wcześniej do temp. 83 °C. Aby zapobiec nadmiernemu spadkowi temperatury w piecu należy umieścić w nim naczynie możliwie jak najszybciej.
Doprowadzić ciśnienie do 100 Tr (13,3 kPa) i pozostawić do wysuszenia do stałej masy (ok. 4 godzin) w takim ciśnieniu w strumieniu gorącego, suchego powietrza.
Odliczać czas suszenia od momentu, kiedy temperatura pieca powróci do 83 °C. Ostrożnie przywrócić w piecu ciśnienie atmosferyczne. Otworzyć piec, natychmiast przykryć naczynie pokrywką, wyjąć naczynie z pieca, pozostawić do ostygnięcia na 30–45 minut w eksykatorze (ppkt 3.4) i zważyć z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg. Suszyć przez kolejne 30 minut w piecu próżniowym (ppkt 3.3) w temp. 83 Powtarzać procedurę podgrzewania i ważenia do czasu, aż masa naczynia z pokrywką ulegnie zmniejszeniu o 1 mg lub mniej, lub ulegnie zwiększeniu, pomiędzy dwoma następującymi po sobie ważeniami. Do obliczenia wykorzystać najniższą zapisaną masę.
5. OBLICZANIE
% wilgoci = (m1 – m2) / (m1 – m0) × 100 %
Gdzie:
|
m0
|
stanowi masę naczynia oraz pokrywki,
|
|
m1
|
stanowi masę naczynia, pokrywki i porcji badanej przed suszeniem,
|
|
m2
|
stanowi masę naczynia, pokrywki i porcji badanej po suszeniu.
|
Zapisać wynik z dokładnością do 0,1 g / 100 g.
6. PRECYZJA
6.1. Granica powtarzalności
Bezwzględna różnica pomiędzy wynikami dwóch niezależnych pojedynczych badań, uzyskanymi przy wykorzystaniu tej samej metody na identycznym materiale badanym w tym samym laboratorium przez ten sam podmiot wykorzystujący to samo wyposażenie w krótkim odstępie czasu nie będzie dla więcej niż 5 % przypadków większa niż 0,4 g wody/100 g kwasowej maślanki w proszku.
6.2. Granica odtwarzalności
Bezwzględna różnica pomiędzy wynikami dwóch niezależnych pojedynczych badań, uzyskanymi przy wykorzystaniu tej samej metody na identycznym materiale badanym w różnych laboratoriach przez różne podmioty wykorzystujące różne wyposażenie nie będzie dla więcej niż 5 przypadków większa niż 0,6 g wody/100 g kwasowej maślanki w proszku.
6.3. Źródło danych dotyczących precyzji
Dane dotyczące precyzji zostały określone w czasie doświadczeń przeprowadzonych w 1995 r., które obejmowały osiem laboratoriów i 12 próbek (6 ślepych duplikatów).
ZAŁĄCZNIK XX
(Artykuł 16)
METODA REFERENCYJNA OZNACZANIA CZYSTOŚCI TłUSZCZU MLECZNEGO W DRODZE GAZOWO-CHROMATOGRAFICZNEJ ANALIZY TRIGLICERYDÓW — WERSJA POPRAWIONA 2
1. ZAKRES I DZIEDZINA STOSOWANIA
Niniejsza norma ustanawia metodę referencyjną oznaczania czystości tłuszczu mlecznego w drodze gazowo-chromatograficznej analizy triglicerydów. Umożliwia ona wykrycie zarówno tłuszczów roślinnych, jak i zwierzęcych, np. łoju wołowego i smalcu.
Przy użyciu określonych równań triglicerydowych określa się czystość tłuszczu mlecznego. Zasadniczo metoda ma zastosowanie do dużych ilości mleka krowiego lub jego przetworów, niezależnie od warunków żywienia, hodowli lub laktacji. Jedynie wyjątkowo wysoka zawartość w paszy czystych olejów roślinnych, np. oleju rzepakowego, może prowadzić do uzyskania fałszywie pozytywnego wyniku. Przetwory mleczne otrzymane od poszczególnych krów również mogą spowodować uzyskanie fałszywie pozytywnego wyniku.
W szczególności metoda ma zastosowanie do tłuszczu wyekstrahowanego z przetworów mlecznych, rzekomo zawierających czysty tłuszcz mleczny o niezmienionym składzie, takich jak masło, śmietana, mleko oraz mleko w proszku. Technologiczna obróbka tłuszczu mlecznego, np. usuwanie cholesterolu lub frakcjonowanie, mogą spowodować uzyskanie fałszywie pozytywnego wyniku. Powyższe odnosi się również do tłuszczu mlecznego uzyskanego z mleka odtłuszczonego lub maślanki. Omawiana metoda nie zawsze ma zastosowanie do tłuszczu wyekstrahowanego z sera, ponieważ proces dojrzewania może wywierać tak silny wpływ na skład tłuszczu, że uzyskany wynik jest fałszywie pozytywny.
Uwaga 1: Kwas masłowy (C4) występuje wyłącznie w tłuszczu mlecznym i umożliwia przeprowadzenie szacunków ilościowych niskich oraz średnich ilości tłuszczu mlecznego w tłuszczach roślinnych i zwierzęcych. Z powodu dużego zróżnicowania C4 wahającego się przeciętnie w zakresie 3,1–3,8 % (procent masowy), trudno jest jednak przekazać informacje o charakterze jakościowym lub ilościowym, dotyczące obcego tłuszczu, w odniesieniu do ułamków masowych czystego tłuszczu mlecznego wynoszących do 20 %[1].
Uwaga 2: Praktycznie wyników ilościowych nie można uzyskać z zawartości steroli w tłuszczach roślinnych, ponieważ zależy ona od warunków produkcji i przetwarzania. Co więcej, oznaczenie jakościowe tłuszczu obcego z wykorzystaniem steroli jest niejednoznaczne.
2. DEFINICJA
Czystość tłuszczu mlecznego: brak obecności tłuszczów roślinnych i zwierzęcych, określony w drodze procedury wyszczególnionej w niniejszej normie.
Uwaga: Czystość określana jest za pośrednictwem wartości S, które wyliczane są ze składu triglicerydów. Ułamki masowe triglicerydów są wyrażone w postaci wartości procentowej.
3. ZASADA METODY
Tłuszcz wyekstrahowany z mleka lub przetworów mlecznych jest poddawany analizie metodą chromatografii gazowej przy użyciu kolumny z wypełnieniem lub krótkiej kolumny kapilarnej w celu oznaczenia triglicerydów (TG), podzielonych w zależności od całkowitej ilości atomów węgla. Poprzez wprowadzenie ułamka masowego, wyrażonego jako wartość procentowa, cząsteczek tłuszczu różnej wielkości (C24 do C54, wykorzystując jedynie parzyste liczby atomów węgla), do odpowiednich równań TG, obliczane są wartości S. Jeżeli wartości S przekraczają wartości graniczne przyjęte dla czystego tłuszczu mlecznego, oznacza to wykrycie obecności tłuszczu obcego.
Uwaga 1: Przydatność i równoważność zarówno kolumny z wypełnieniem, jak i kolumny kapilarnej, zostały wykazane uprzednio [2–4].
Uwaga 2: Wartość S stanowi sumę ułamków masowych TG pomnożonych odpowiednio przez określone współczynniki.
4. ODCZYNNIKI
Wszystkie odczynniki muszą być odczynnikami o uznanej klasie analitycznej.
4.1. Gaz nośny: azot lub, ewentualnie, hel albo wodór, każdy o czystości co najmniej 99,995 %.
Wzorce tłuszczów, do normalizacji wzorca tłuszczu mlecznego zgodnie z ppkt 7.3.3.
4.2.1. Wzorce triglicerydów, nasycone; odpowiednie produkty są dostępne w handlu.
4.2.2. Wzorzec cholesterolu.
4.3. Metanol (CH3OH), bezwodny.
4.4. n-heksan (CH3(CH2)4CH3).
4.5. n-heptan (CH3(CH2)5CH3).
4.6. Inne gazy: wodór o czystości co najmniej 99,995 %, wolny od zanieczyszczeń organicznych (CnHm < 1 µl/l); syntetyczne powietrze, wolne od zanieczyszczeń organicznych (CnHm < 1 µl/l).
4.7. Bezwodny siarczan sodu (Na2SO4).
5. APARATURA
Typowe wyposażenie laboratoryjne, w szczególności:
5.1. Wysokotemperaturowy chromatograf gazowy
Wysokotemperaturowy chromatograf gazowy powinien być przystosowany do temperatur co najmniej 400 °C i wyposażony w detektor płomieniowo-jonizacyjny (FID). Membrana zastosowana w dozowniku musi być odporna na działanie wysokich temperatur i wykazywać bardzo niski stopień wymywania fazy nieruchomej. W przypadku chromatografii gazowej na kolumnach kapilarnych należy zastosować dozownik nakolumnowy. Przy podłączeniu kolumny należy zawsze używać uszczelek grafitowych, a także (w stosownym przypadku) wkładek do dozownika i/lub detektora.
5.2. Kolumna chromatograficzna
5.2.1. Kolumna z wypełnieniem
Zastosować kolumnę szklaną o średnicy wewnętrznej 2 mm i długości 500 mm, wypełnioną fazą stacjonarną 3 % OV-1 na 125 µm do 150 µm (mesh 100–120) Gas ChromQ (1). Przygotowanie, silanizacja, wypełnianie i kondycjonowanie kolumny z wypełnieniem zostały opisane w załączniku A.
Można ewentualnie zastosować kolumnę kapilarną (ppkt 5.2.2).
5.2.2. Kolumna kapilarna
Zastosować krótką kolumnę kapilarną, np. długości 5 m, z niepolarną fazą stacjonarną odporną na działanie temperatur do 400 °C lub wyższych (2)). Kondycjonować kolumnę przeprowadzając 20 analiz roztworu tłuszczu mlecznego (ppkt 7.2) w ciągu 2–3 dni, stosując ustawienia podane w ppkt 7.3.4.2. W następstwie powyższej operacji współczynniki odpowiedzi (ppkt 7.3.3) są zbliżone do 1 i niższe niż 1,20.
Uwaga: Można zastosować kolumny o różnych wymiarach i zawierające różne niepolarne, odporne na działanie wysokich temperatur fazy, o ile ich sprawność jest zgodna z niniejszą normą. Zob. również: ppkt 7.3.4.2.
5.3. Kolumna Extrelut poj. 1–3 ml, wypełniona żelem krzemionkowym, niezbędna jedynie do ekstrakcji mającej na celu uzyskanie tłuszczu mlecznego zgodnie z ppkt 7.1.3.
5.4. Uszczelki grafitowe, odporne na działanie temperatur co najmniej 400 °C; stosowane przy podłączeniu kolumny chromatografu gazowego, jak również do wkładek dozownika i/lub detektora.
5.5. Łaźnia wodna, umożliwiająca utrzymanie temp. 50 °C ± 2 °C.
5.6. Piec, umożliwiający działanie w temp. 50 °C ± 2 °C i 100 °C ± 2 °C.
5.7. Pipeta mikrolitrowa
5.8. Pipeta miarowa poj. 5 ml.
5.9. Kolba okrągłodenna poj. 50 ml.
5.10. Kolba Erlenmeyera poj. nominalna 250 ml.
5.11. Lejek.
5.12. Sączki papierowe o niewielkiej średnicy porów.
5.13. Wyparka obrotowa.
5.14. Ampułki o pojemności nominalnej 1 ml, wyposażone w aluminiowe zatyczki zaciskowe lub nakrętki pokryte warstwą politetrafluoroetylenu.
5.15. Strzykawka wprowadzająca z tłokiem niedochodzącym do końcówki igły (kolumna do chromatografii gazowej z wypełnieniem).
Uwaga: Opisane powyżej strzykawki umożliwiają lepszą powtarzalność wyników.
5.16. Waga analityczna, umożliwiająca ważenie z dokładnością do 1 mg, o dokładności odczytu 0,1 mg
6. POBIERANIE PRÓBEK
Do laboratorium należy przesłać próbkę reprezentacyjną. W czasie transportu lub przechowywania nie powinna ona ulec uszkodzeniu ani zmianom.
Sposób pobierania próbek nie został ujęty w metodzie określonej w niniejszej normie międzynarodowej. Zalecaną metodę pobierania próbek przedstawiono w normie ISO 707IDF 50 [5].
7. PROCEDURA
7.1. Przygotowanie próbek do badań
Do przygotowania próbek stosuje się jedną z trzech przedstawionych poniżej metod ekstrahowania tłuszczu mlecznego.
7.1.1. Oddzielenie z masła lub bezwodnego tłuszczu mlecznego
Roztopić 50 g–100 g próbki badanej w temp. 50 °C przy użyciu łaźni wodnej (ppkt 5.5) lub pieca (ppkt 5.6). Umieścić 0,5–1,0 g siarczanu sodu (ppkt 4.7) w złożonym sączku papierowym (ppkt 5.12). Podgrzać kolbę Erlenmeyera poj. 250 ml (ppkt 5.10) oraz lejek (ppkt 5.11) z założonym sączkiem papierowym w piecu (ppkt 5.6) w temp. 50 °C. Przesączyć, utrzymując podgrzaną kolbę, lejek i założony sączek w piecu, warstwę tłuszczu roztopionej próbki. Nie dopuścić do przeniesienia serwatki.
Jedynie w przypadkach gdy dostępna jest ograniczona ilość próbki do badań, można wykorzystać mniejszą próbkę, przy czym należy wówczas odpowiednio dostosować procedurę. Posłużenie się mniejszą porcją badaną wiąże się jednak z ryzykiem otrzymania próbki niereprezentatywnej.
Uwaga 1: Masło można uzyskać ze śmietany w drodze gwałtownego mieszania i dokładnego przemycia powstałych grudek masła.
Uwaga 2: Tłuszcz mleczny otrzymany przy zastosowaniu procedury przedstawionej w ppkt 7.1.1 będzie niemal całkowicie wolny od fosfolipidów.
7.1.2. Ekstrakcja według metody grawimetrycznej Röse–Gottlieba
Wyekstrahować frakcję tłuszczową z próbki badanej przy użyciu metody grawimetrycznej opisanej w jednej z norm ISO 1211IDF 001D, ISO 2450IDF 016C lub ISO 7328IDF 116A.
Uwaga: Jeżeli w otrzymanym tłuszczu mlecznym obecne są fosfolipidy, uzyskany zostanie pik cholesterolu, powiększony w przybliżeniu o 0,1 %. Na skład TG, znormalizowany do 100 % włącznie z cholesterolem, wpływ jest tym samym wywierany jedynie w znikomym stopniu.
7.1.3. Ekstrakcja z mleka przy użyciu kolumn z żelem krzemionkowym
Przenieść za pomocą pipety mikrolitrowej (ppkt 5.7) 0,7 ml próbki badanej podgrzanej do temp. 20 °C do kolumny Extrelut poj. 1–3 ml (ppkt 5.3). Pozostawić na ok. 5 minut, aby umożliwić równomierne rozprowadzenie na żelu krzemionkowym.
W celu zdenaturowania kompleksów białkowo-lipidowych dodać, za pomocą pipety miarowej (ppkt 5.8), 1,5 ml metanolu (ppkt 4.3) do kolumny Extrelut. Następnie ekstrahować frakcję tłuszczową z próbki badanej za pomocą 20 ml n-heksanu (ppkt 4.4). Dodawać n-heksan powoli w niewielkich ilościach. Zebrać odprowadzony rozpuszczalnik do okrągłodennej kolby poj. 50 ml (ppkt 5.9), uprzednio wysuszonej do stałej, znanej masy zważonej z dokładnością do 1 mg i zapisanej z dokładnością do 0,1 mg.
Po dokonaniu ekstrakcji całkowicie opróżnić kolumnę. Oddestylować rozpuszczalniki z odcieku na wyparce obrotowej (ppkt 5.13) przy użyciu jej łaźni wodnej ustawionej na temp. 40 °C–50 °C. Po oddestylowaniu rozpuszczalników wysuszyć, a następnie zważyć kolbę okrągłodenną wraz z jej zawartością z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg. Oznaczyć uzysk masy tłuszczu, odejmując masę wysuszonej pustej kolby okrągłodennej od otrzymanej masy.
Uwaga: Ekstrakcja tłuszczu z użyciem metod Gerbera, Weibulla–Berntropa lub Schmida–Bondzynskiego–Ratzlaffa, jak również metoda oddzielania tłuszczu mlecznego przy użyciu detergentów (metoda BDI) nie są odpowiednie dla analizy triglicerydów, ponieważ przy stosowaniu tych metod znaczące ilości glicerydów cząstkowych lub fosfolipidów mogą przeniknąć do fazy tłuszczowej. Tym samym stosowanie niniejszej normy międzynarodowej jest ograniczone w odniesieniu do niektórych produktów, zwłaszcza sera.
7.2. Przygotowanie roztworu próbki
Do celów chromatografii gazowej z wypełnioną kolumną przygotować 5 % (stężenie objętościowe) roztwór tłuszczu (uzyskanego zgodnie z ppkt 7.1) w n-heksanie (ppkt 4.4) lub n-heptanie (ppkt. 4.5). W zależności od wymiarów kolumny zastosować stężenie 1 % (0,53 mm średnicy wewn.) lub mniejsze w przypadku bezpośredniego wprowadzania do kolumny (kolumna kapilarna).
W oparciu o zastosowaną kolumnę oraz masę tłuszczu otrzymanego w ppkt 7.1.3 oznaczyć ilość rozpuszczalnika (ppkt 4.4 lub 4.5), jaki należy dodać do materiału próbki badanej w kolbie na podstawie ważenia z dokładnością do 1 mg, którego wynik został zapisany z dokładnością do 0,1 mg. Rozpuścić całkowicie pozostałość.
Przenieść ok. 1 ml roztworu próbki do ampułki (ppkt 5.14).
7.3. Chromatograficzne oznaczanie triglicerydów
7.3.1. Dryf linii podstawowej
Aby zminimalizować podnoszenie się linii podstawowej, kolumna jest kondycjonowana zgodnie z opisem w ppkt 5.2.2 (kolumna kapilarna) lub w załączniku A.4 (kolumna z wypełnieniem).
Uwaga: Z powodu wysokiej temperatury kolumny analiza TG ma szczególną tendencję do podnoszenia linii podstawowej w zakresie TG o dużej liczbie atomów węgla.
7.3.2. Technika wprowadzania
7.3.2.1. Kolumna z wypełnieniem
Aby uniknąć efektu niewrażliwości należy zastosować technikę „gorącej igły” w celu udoskonalenia ilościowego oznaczania wysokowrzących składników TG. Napełnić igłę powietrzem poprzez nabranie roztworu tłuszczu do strzykawki. Wsunąć igłę do dozownika. Przed wprowadzeniem podgrzewać igłę przez ok. 3 sekundy. Następnie szybko wprowadzić zawartości strzykawki.
7.3.2.2. Kolumna kapilarna
W przypadku wprowadzenia na zimno bezpośrednio do kolumny (ppkt 7.3.4.2), wsunąć igłę strzykawki i natychmiast wprowadzić. Czas przebywania igły w otworze wprowadzającym powinien być odpowiedni, aby uniemożliwić szerokie ogonowanie piku rozpuszczalnika.
Uwaga: Optymalny czas przebywania igły wynosi zazwyczaj ok. 3 sekund.
7.3.3. Kalibracja
7.3.3.1. Uwagi ogólne
Do celów kalibracji próbek badanych należy wykonać dwie lub trzy analizy znormalizowanego tłuszczu mlecznego na początku każdego dnia. Wykorzystać ostatnio wykonaną analizę znormalizowanego tłuszczu mlecznego do określenia współczynników odpowiedzi RFsi
(ułamek masowy/ułamek powierzchni) TG i cholesterolu, i zastosować je w odniesieniu do następnych próbek badanych (zob. ppkt 9.1):
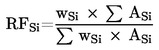 (1)
(1)
gdzie:
|
w
si
|
to ułamek masowy, wyrażony jako wartość procentowa, każdego TG lub cholesterolu w znormalizowanym tłuszczu mlecznym,
|
|
A
si
|
to wartość liczbowa powierzchni piku każdego TG lub cholesterolu w znormalizowanym tłuszczu mlecznym.
|
Zastosować ppkt 7.3.3.2 lub ppkt. 7.3.3.3, aby otrzymać znormalizowany tłuszcz mleczny o znanym składzie TG.
7.3.3.2. Handlowy wzorzec tłuszczu mlecznego
Najlepszym sposobem na określenie współczynnika odpowiedzi każdego składnika próbki badanej jest zastosowanie znormalizowanego tłuszczu mlecznego o potwierdzonym składzie TG.
Uwaga: Odpowiednim wzorcem jest CRM 519 (bezwodny tłuszcz mleczny), dostępny w Instytucie Materiałów Referencyjnych i Pomiarów (IRMM), Geel, Belgia (3)).
7.3.3.3. Laboratoryjny wzorzec tłuszczu mlecznego
Przygotować ok. 1 g mieszaniny wzorców tłuszczów (zob. ppkt 4.2), zawierającej co najmniej TG nasycone, C24, C30, C36, C42, C48 i C54, jak również cholesterol; dodatkowo, co wskazane, C50 i C52), ważąc z dokładnością do 1 mg, zapisując masę z dokładnością do 0,1 mg, tak aby uzyskać względny skład TG zbliżony do składu tłuszczu mlecznego.
Poddać wielokrotnie analizie roztwór mieszaniny wzorców tłuszczów w n-heksanie (ppkt 4.4) lub n-heptanie (ppkt 4.5) zgodnie z ppkt 7.3.4. W tej samej sekwencji poddać wielokrotnie analizie tłuszcz mleczny o przeciętnym składzie.
Określić współczynniki odpowiedzi TG z mieszaniny wzorców tłuszczów. Pośrednie współczynniki odpowiedzi TG nieobecnych w mieszaninie można wyliczyć przy użyciu metody interpolacji matematycznej. Zastosować uzyskane współczynniki odpowiedzi w odniesieniu do tłuszczu mlecznego w celu uzyskania znormalizowanego składu. Otrzymany tym sposobem znormalizowany tłuszcz mleczny może być przechowywany przez kilka lat w atmosferze azotu w temperaturze nie wyższej niż -18 °C.
7.3.4. Warunki chromatograficzne
Uwaga: Zastosowanie kolumn z wypełnieniem lub kolumn kapilarnych prowadzi na ogół do rozdzielczości podobnej do przedstawionej na ryc. 1. Nie obserwuje się zazwyczaj rozszczepienia pików TG o parzystej liczbie atomów węgla i należy go unikać.
7.3.4.1. Kolumna z wypełnieniem
|
a)
|
Program temperaturowy: Ustawić temperaturę początkową pieca na 210 °C. Utrzymać piec w tej temperaturze przez 1 minutę. Następnie podnosić temperaturę stopniowo o 6 °C/min. do 350 °C. Utrzymać piec w tej (końcowej) temperaturze przez 5 minut.
|
|
b)
|
Temperatura detektora i dozownika: Ustawić detektor i dozownik na temp. 370 °C.
|
|
c)
|
Gaz nośny: Zastosować azot przy stałym natężeniu przepływu strumienia ok. 40 ml/min. Dostosować precyzyjne natężenie przepływu strumienia gazu nośnego w taki sposób, aby C54 był eluowany w temp. 341 °C.
|
|
d)
|
Czas trwania analizy: 29,3 min.
|
|
e)
|
Objętość wprowadzona: Wprowadzić 0,5 µl 5 % (stężenie objętościowe) roztworu próbki.
|
Jeżeli nie są wykonywane analizy triglicerydów, należy utrzymywać początkową temperaturę pieca stosownie do lit. a), temperaturę detektora i dozownika stosownie do lit. b), a natężenie przepływu strumienia gazu nośnego stosownie do lit. c), na stałym poziomie, również w nocy oraz podczas weekendów lub świąt. Zapewnia to najwyższą sprawność kolumn.
7.3.4.2. Kolumna kapilarna
|
a)
|
Program temperaturowy: Ustawić temperaturę początkową pieca na 80 °C. Utrzymać piec w tej temperaturze przez 0,5 min. Następnie podnosić temperaturę stopniowo o 50 °C/min. do 190 °C, po czym stopniowo o 6 °C/min. do 350 °C. Utrzymać piec w tej (końcowej) temperaturze przez 5 min.
|
|
b)
|
Temperatura detektora: Ustawić na 370 °C.
|
|
c)
|
Gaz nośny: Zastosować azot przy stałym natężeniu przepływu strumienia ok. 3 ml/min.
|
|
d)
|
Czas trwania analizy: 34,4 min.
|
|
e)
|
Objętość wprowadzona: Wprowadzić 0,5 µl 1 % (stężenie objętościowe) roztworu próbki.
|
Utrzymać powyższe ustawienia w trybie czuwania w celu zapewnienia najwyższej sprawności (zob. ppkt 7.3.4.1).
Ustawienia parametrów analizy zamieszczone w ppkt 7.3.4.2 są odpowiednie do wykorzystania w kolumnie typu „wide bore” (0,53 mm śr. wewn.) zgodnie z wyszczególnieniem podanym w ppkt 5.2.2. Inne warunki można zastosować w przypadku korzystania z kolumny o innych wymiarach lub wypełnione inną fazą.
8. CAŁKOWANIE, OCENA ORAZ KONTROLA SPRAWNOŚCI ANALITYCZNEJ
Ocenę pików chromatogramu należy przeprowadzić za pomocą systemu całkującego umożliwiającego wykreślenie linii podstawowej i ponowne całkowanie. Na ryc. 1 przedstawiono poprawnie zintegrowany chromatogram, podczas gdy na ryc. 2 przedstawiono pojawiający się sporadycznie nieprawidłowy przebieg linii podstawowej, kończący się po C54, który wpływa na zawartość procentową wszystkich TG. Mimo wszystko należy wyłączyć z oceny piki eluowane po C54.
Połączyć TG o nieparzystej liczbie atomów węgla grup acylowych (2n + 1) z poprzedzającymi je TG o liczbie parzystej atomów węgla (2n). Nie należy uwzględniać niskiej zawartości TG o liczbie C56. Pomnożyć wartości procentowe powierzchni pików pozostałych TG włącznie z cholesterolem przez odpowiednie współczynniki odpowiedzi znormalizowanego tłuszczu mlecznego (ostatnia kalibracja) i unormować całkowicie do 100 % zgodnie z ppkt 9.1.
Rycina 1
Przykładowy chromatogram rozdziału triglicerydów tłuszczu mlecznego z poprawnie przeprowadzoną linią podstawową
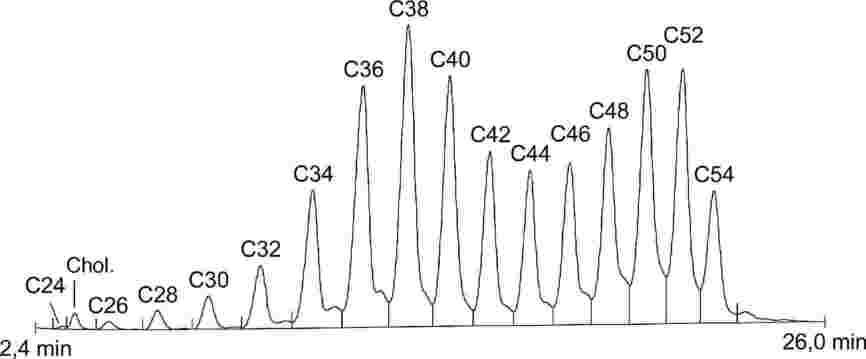
Rycina 2
Przykładowy chromatogram rozdziału triglicerydów tłuszczu mlecznego z niepoprawnie przeprowadzoną linią podstawową

Aby sprawdzić warunki pomiarów, należy je porównać ze współczynnikami zmienności (CV), wyrażonymi jako wartości procentowe, różnych TG podanych w tabeli 1, uzyskanymi w oparciu o 19 sukcesywnych analiz tej samej próbki tłuszczu mlecznego.
W przypadku gdy wartości CV są znacząco wyższe od wartości podanych w tabeli 1, warunki chromatograficzne nie są odpowiednie.
Uwaga: Wartości podane w tabeli 1 nie są obowiązkowe, lecz jedynie orientacyjne do celów kontroli jakości.
Jednakże w przypadku gdy dopuszczalne są wyższe wartości CV, należy mimo wszystko przestrzegać granic powtarzalności i odtwarzalności podanych w pkt 10.
Tabela 1
Współczynniki zmienności zawartości triglicerydów (19 sukcesywnych analiz)
|
Trigliceryd
|
CV %
|
|
C24
|
10,00
|
|
C26
|
2,69
|
|
C28
|
3,03
|
|
C30
|
1,76
|
|
C32
|
1,03
|
|
C34
|
0,79
|
|
C36
|
0,25
|
|
C38
|
0,42
|
|
C40
|
0,20
|
|
C42
|
0,26
|
|
C44
|
0,34
|
|
C46
|
0,37
|
|
C48
|
0,53
|
|
C50
|
0,38
|
|
C52
|
0,54
|
|
C54
|
0,60
|
9. OBLICZANIE I PREZENTACJA WYNIKÓW
9.1. Skład triglicerydów
9.1.1. Obliczanie
Obliczyć ułamek masowy każdego TG (dla i = C24, C26, C28, C30, C32, C34, C36, C38, C40, C42, C44, C46, C48, C50, C52 i C54) oraz cholesterolu, wi
, wyrażony jako wartość procentowa, całkowitego składu triglicerydów w próbce badanej, korzystając z poniższego równania:
 (2)
(2)
gdzie:
|
Ai
|
jest wartością liczbową powierzchni piku każdego TG w próbce badanej,
|
|
RF
si
|
jest współczynnikiem odpowiedzi każdego TG, określonym w wyniku kalibracji (ppkt 7.3.3).
|
9.1.2. Prezentacja wyników badań
Wyniki należy podać z dokładnością do drugiego miejsca po przecinku.
9.2. Wartości S
9.2.1. Obliczanie
9.2.1.1. Obliczyć wartości S, wyrażone jako wartość procentowa, podstawiając obliczoną wartość wi
(ppkt 9.1.1) odpowiednich wartości procentowych TG do równań (3)–(7) Należy zastosować wszystkie równania bez względu na podejrzenia co do rodzaju tłuszczu obcego.
9.2.1.2. Olej sojowy, słonecznikowy, oliwa z oliwek, rzepakowy, lniany, z kiełków pszenicy, z kiełków kukurydzy, z nasion bawełny oraz olej rybny
S = 2,098 3 · w
C30 + 0,728 8 · w
C34 + 0,692 7 · w
C36 + 0,635 3 · w
C38 + 3,745 2 · w
C40 – 1,292 9 · w
C42 + 1,354 4 · w
C44 + 1,701 3 · w
C46 + 2,528 3 · w
C50 (3)
9.2.1.3. Tłuszcz kokosowy i tłuszcz z ziaren palmowych
S = 3,745 3 · w
C32 + 1,113 4 · w
C36 + 1,364 8 · w
C38 + 2,154 4 · w
C42 + 0,427 3 · w
C44 + 0,580 9 · w
C46 + 1,292 6 · w
C48 + 1,030 6 · w
C50 + 0,995 3 · w
C52 + 1,239 6 · w
C54 (4)
9.2.1.4. Olej palmowy i łój wołowy
S = 3,664 4 · w
C28 + 5,229 7 · w
C30 – 12,507 3 · w
C32 + 4,428 5 · w
C34 – 0,201 0 · w
C36 + 1,279 1 · w
C38 + 6,743 3 · w
C40 – 4,271 4 · w
C42 + 6,373 9 · w
C46 (5)
9.2.1.5. Smalec
S = 6,512 5 · w
C26 + 1,205 2 · w
C32 + 1,733 6 · w
C34 + 1,755 7 · w
C36 + 2,232 5 · w
C42 + 2,800 6 · w
C46 + 2,543 2 · w
C52 + 0,989 2 · w
C54 (6)
9.2.1.6. Ogółem.
S = – 2,757 5 · w
C26 + 6,407 7 · w
C28 + 5,543 7 · w
C30 – 15,324 7 · w
C32 + 6,260 0 · w
C34 + 8,010 8 · w
C40 – 5,033 6 · w
C42 + 0,635 6 · w
C44 + 6,017 1 · w
C46 (7)
9.2.2. Prezentacja wyników badań
Wyniki należy podać z dokładnością do drugiego miejsca po przecinku.
9.3. Wykrywanie tłuszczu obcego
Porównać pięć wartości S otrzymanych w ppkt 9.2.1 z odpowiednimi wartościami granicznymi S podanymi w tabeli 2.
Należy przyjąć, że próbka badana jest czystym tłuszczem mlecznym, gdy wszystkie pięć wartości S mieszczą się w zakresie wartości granicznych podanych w tabeli 2. Jeżeli jednak którakolwiek z wartości S nie mieści się w zakresie odpowiednich wartości granicznych, uznaje się, że próbka zawiera tłuszcz obcy.
Mimo, że poszczególne równania (3)–(6) są bardziej wrażliwe na niektóre tłuszcze obce niż równanie ogólne (7) (zob. tabela B.1), wynik pozytywny otrzymany tylko z jednego równania spośród (3)–(6) nie pozwala na wyciągnięcie wniosków co do rodzaju tłuszczu obcego.
W załączniku B opisano procedurę obliczania zawartości tłuszczu roślinnego lub zwierzęcego w zafałszowanym tłuszczu mlecznym. Procedura ta nie jest zwalidowana i podano ją jedynie dla celów informacyjnych.
Tabela 2
Wartości S dla czystych tłuszczów mlecznych
|
Tłuszcz obcy
|
Równanie
|
Wartości S
(4)
|
|
Olej sojowy, słonecznikowy, oliwa z oliwek, rzepakowy, lniany, z kiełków pszenicy, z kiełków kukurydzy, z nasion bawełny, olej rybny
|
(3)
|
98,05 do 101,95
|
|
Tłuszcz kokosowy i tłuszcz z ziaren palmowych
|
(4)
|
99,42 do 100,58
|
|
Olej palmowy i łój wołowy
|
(5)
|
95,90 do 104,10
|
|
Smalec
|
(6)
|
97,96 do 102,04
|
|
Ogółem
|
(7)
|
95,68 do 104,32
|
10. PRECYZJA
10.1. Badanie międzylaboratoryjne
Wartości powtarzalności i odtwarzalności zostały określone na podstawie równań (3)–(7) przy użyciu czystego tłuszczu mlecznego i nie mogą mieć zastosowania do matryc innych niż podane.
10.2. Powtarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi z wykorzystaniem tej samej metody na identycznym materiale badanym w tym samym laboratorium przez ten sam podmiot wykorzystujący to samo wyposażenie w krótkim odstępie czasu nie będzie przekraczać wartości granicznych wymienionych w tabeli 3 w więcej niż 5 % przypadków.
Tabela 3
Granice powtarzalności r dla równań (3)–(7)
|
Tłuszcz obcy
|
Równanie
|
r %
|
|
Olej sojowy, słonecznikowy, oliwa z oliwek, rzepakowy, lniany, z kiełków pszenicy, z kiełków kukurydzy, z nasion bawełny, olej rybny
|
(3)
|
0,67
|
|
Tłuszcz kokosowy i tłuszcz z ziaren palmowych
|
(4)
|
0,12
|
|
Olej palmowy i łój wołowy
|
(5)
|
1,20
|
|
Smalec
|
(6)
|
0,58
|
|
Ogółem
|
(7)
|
1,49
|
10.3. Odtwarzalność
Bezwzględna różnica pomiędzy wynikami dwóch pojedynczych badań, uzyskanymi z wykorzystaniem tej samej metody na identycznym materiale badanym w różnych laboratoriach przez różne podmioty wykorzystujące różne wyposażenie nie będzie przekraczać wartości granicznych wymienionych w tabeli 4 w więcej niż 5 przypadków.
Tabela 4
Granice odtwarzalności R dla równań (3)–(7)
|
Tłuszcz obcy
|
Równanie
|
R %
|
|
Olej sojowy, słonecznikowy, oliwa z oliwek, rzepakowy, lniany, z kiełków pszenicy, z kiełków kukurydzy, z nasion bawełny, olej rybny
|
(3)
|
1,08
|
|
Tłuszcz kokosowy i tłuszcz z ziaren palmowych
|
(4)
|
0,40
|
|
Olej palmowy i łój wołowy
|
(5)
|
1,81
|
|
Smalec
|
(6)
|
0,60
|
|
Ogółem
|
(7)
|
2,07
|
11. NIEPEWNOŚĆ POMIARU
Mając do dyspozycji powtarzalność r i odtwarzalność R, można obliczyć niepewność rozszerzoną dla danej wartości S.
Włączenie niepewności rozszerzonej (w oparciu o analizę w dwóch powtórzeniach) do wartości S zamieszczonych w tabeli 2 prowadzi do poszerzenia zakresu wartości granicznych S podanych w tabeli 5.
Tabela 5
Poszerzony zakres wartości S dla czystych tłuszczów mlecznych włącznie z niepewnością rozszerzoną
|
Tłuszcz obcy
|
Równanie
|
Poszerzony zakres wartości S
|
|
Olej sojowy, słonecznikowy, oliwa z oliwek, rzepakowy, lniany, z kiełków pszenicy, z kiełków kukurydzy, z nasion bawełny, olej rybny
|
(3)
|
97,36 do 102,64
|
|
Tłuszcz kokosowy i tłuszcz z ziaren palmowych
|
(4)
|
99,14 do 100,86
|
|
Olej palmowy i łój wołowy
|
(5)
|
94,77 do 105,23
|
|
Smalec
|
(6)
|
97,65 do 102,35
|
|
Ogółem
|
(7)
|
94,42 do 105,58
|
12. SPRAWOZDANIE Z BADAŃ
W sprawozdaniu z badań należy określić:
|
—
|
wszelkie informacje niezbędne do pełnej identyfikacji próbki,
|
|
—
|
zastosowaną metodę pobierania próbek, o ile jest znana,
|
|
—
|
metodę badawczą zastosowaną w odniesieniu do niniejszej normy międzynarodowej,
|
|
—
|
wszelkie szczegółowe dane operacyjne niewyszczególnione w niniejszej normie międzynarodowej lub uznane za opcjonalne, wraz z danymi szczegółowymi na temat wszelkich zdarzeń, które mogły wywrzeć wpływ na wynik(i) badania,
|
|
—
|
otrzymany wynik (wyniki) badania oraz w przypadku, gdy sprawdzono powtarzalność, ostateczny przytoczony otrzymany wynik.
|
(1) Przykład odpowiedniego produktu dostępnego w handlu. Niniejsza informacja jest przekazana dla wygody użytkowników wspomnianej normy międzynarodowej i nie ma na celu zatwierdzania wspomnianego produktu.
(2) CP-Ultimetal SimDist (5 m × 0,53 mm × 0,17 µm) stanowi przykład odpowiedniego produktu dostępnego w handlu. Niniejsza informacja jest przekazana dla wygody użytkowników wspomnianej normy międzynarodowej i nie ma na celu zatwierdzania wspomnianego produktu.
(3) Przykład odpowiedniego produktu dostępnego w handlu. Niniejsza informacja jest przekazana dla wygody użytkowników wspomnianej normy międzynarodowej i nie ma na celu zatwierdzania wspomnianego produktu.
(4) Obliczono przy poziomie ufności wynoszącym 99 %, w związku z czym dodatek tłuszczu obcego jest wykazany jedynie w przypadku, gdy przekroczone zostały granice wykrywalności odpowiedniego równania (zob. tabela B.1).
ZAŁĄCZNIK A
(normatywny)
PRZYGOTOWANIE KOLUMNY Z WYPEŁNIENIEM
A.1 ODCZYNNIKI I APARATURA
A.1.1 Toluen (C6H5CH3).
A.1.2 Roztwór dichlorodimetylosilanu [Si(CH3)2Cl2].
Rozpuścić 50 ml dichlorodimetylosilanu w 283 ml toluenu (ppkt A.1.1).
A.1.3 Roztwór masła kakaowego, o ułamku masowym masła kakaowego wynoszącym 5 % w n-heksanie (ppkt 4.4) lub n-heptanie (ppkt 4.5).
A.1.4 Faza stacjonarna, 3 % OV-1 na 125 µm do 150 µm (mesh 100–120) Gas ChromQ (1)).
Uwaga: Zalecany rozmiar ziaren przeliczono na mikrometry zgodnie z BS 410 (wszystkie części)[6].
A.1.5 Kolumna szklana o średnicy wewnętrznej 2 mm i długości 500 mm, w kształcie litery U.
Aparatura do wypełniania kolumny.
A.1.6.1 Kolumna zasilająca, z nakrętkami, wraz z oznaczeniem, do jakiej wysokości można ją wypełniać wymaganą ilością fazy stacjonarnej.
A.1.6.2 Drobne sito, o wielkości oczek wynoszącej ok. 100 μm, z nakrętką, odpowiednie do uszczelnienia kolumny szklanej zgodnie z ryc. A.3.
A.1.6.3 Wata szklana silanizowana, zdezaktywowana.
A.1.6.4 Wibrator do jednolitego rozprowadzenia fazy stacjonarnej podczas wypełniania.
A.1.6.5 Urządzenia do silanizacji szklanej powierzchni kolumny.
A.1.6.6 Butelka Woulffa.
A.1.6.7 Wodna pompa ssąca
A.2 SILANIZACJA (DEZAKTYWACJA POWIERZCHNI SZKLANEJ)
Po podłączeniu butelki Woulffa (ppkt A.1.6.6) do wodnej pompy ssącej (ppkt A.1.6.7) zanurzyć rurkę 2 (zob. ryc. A.1) w roztworze dichlorodimetylosilanu (ppkt A.1.2). Wypełnić kolumnę wspomnianym roztworem poprzez zamknięcie zaworu odcinającego. Otworzyć ponownie zawór odcinający, a następnie usunąć obie rurki. Umocować kolumnę na statywie. Napełnić kolumnę całkowicie roztworem dichlorodimetylosilanu (ppkt A.1.2) za pomocą pipety.
Rycina A.1
Zestaw do silanizacji
Legenda
|
6
|
dimetylodichlorosilan i toluen
|
Odstawić kolumnę na 20–30 minut. Następnie zastąpić butelkę Woulffa kolbą filtracyjną. Opróżnić kolumnę, podłączając ją do wodnej pompy ssącej (ppkt A.1.6.7) (zob. ryc. A.2). Przepłukać opróżnioną kolumnę używając kolejno 75 ml toluenu (ppkt A.1.1) i 50 ml metanolu (ppkt 4.3), zanurzając rurkę 2 w rozpuszczalniku. Suszyć przepłukaną kolumnę w piecu (ppkt 5.6) ustawionym na temp. 100 °C przez ok. 30 minut.
Rycina A.2
Zestaw do przepłukiwania

Legenda
A.3 WYPEŁNIANIE
Wypełnić kolumnę przy użyciu zestawu przedstawionego na ryc. A.3. Napełnić kolumnę zasilającą (ppkt A.1.6.1) fazą stacjonarną (ppkt A.1.4) do kreski. Uszczelnić niższy koniec kolumny szklanej, która ma zostać wypełniona, za pomocą korka o długości ok. 1 cm, wykonanego z silanizowanej, sprasowanej waty szklanej (ppkt A.1.6.3). Zamknąć koniec kolumny przy użyciu drobnego sita (ppkt A.1.6.2).
Rycina A.3
Wypełnianie kolumny szklanej

Legenda
|
2
|
kolumna zasilająca, która zostanie wypełniona do kreski OV-1
|
|
3
|
kolumna szklana, która zostanie wypełniona
|
|
4
|
nakrętka z sączkiem, w którą wciska się włókno szklane, stanowiąca barierę dla wprowadzanej fazy stacjonarnej
|
Wypełnić kolumnę pod ciśnieniem (300 kPa i przepływ strumienia azotu) fazą stacjonarną. W celu uzyskania jednorodnego, nieprzerwanego oraz zwartego upakowania, podczas wypełniania przesuwać wibrator w górę oraz w dół wzdłuż powierzchni kolumny szklanej. Po wypełnieniu zakorkować drugi koniec kolumny z wypełnieniem litym korkiem z silanizowanej wełny szklanej (ppkt A.1.3.6). Obciąć wystające końcówki. Wcisnąć korek do kolumny na głębokość kilku milimetrów za pomocą szpatułki.
A.4 KONDYCJONOWANIE
Podczas wykonywania czynności a)–c) nie należy podłączać tylnego końca kolumny do detektora, aby uniknąć zanieczyszczeń. Kondycjonować wypełnioną kolumnę (ppkt A.3) w następujący sposób:
|
a)
|
przepłukiwać kolumnę azotem przez 15 min. przy natężeniu przepływu strumienia ustawionym na 40 ml/min. i temperaturze pieca chromatografu gazowego ustawionej na 50 °C;
|
|
b)
|
podgrzewać kolumnę stopniowo o 1 °C/min. do temp. 355 °C przy natężeniu przepływu strumienia azotu ustawionym na 10 ml/min.;
|
|
c)
|
utrzymywać kolumnę w temp. 355 °C przez 12–15 godzin;
|
|
d)
|
wprowadzić dwukrotnie objętość 1 μl roztworu masła kakaowego (ppkt A.1.3), stosując program temperaturowy dla kolumny wypełnionej, podany w ppkt 7.3.4.1;
Uwaga: Masło kakaowe składa się niemal wyłącznie z wysokowrzących TG o liczbie atomów węgla od C50 do C54, dzięki czemu kondycjonowanie kolumny w odniesieniu do odpowiednich współczynników odpowiedzi staje się zbędne.
|
|
e)
|
wprowadzić dwudziestokrotnie objętość 0,5 μm roztworu tłuszczu mlecznego zgodnie z ppkt 7.2 w czasie 2–3 dni, korzystając z ustawień dla wypełnionej kolumny podanych w ppkt 7.3.4.1.
|
—
|
Do analizy próbek badanych stosować wyłącznie kolumny o współczynnikach odpowiedzi zbliżonych do 1. Wartość współczynników odpowiedzi nie powinna być wyższa niż 1,20.
|
|
(1) Przykład odpowiedniego produktu dostępnego w handlu. Niniejsza informacja jest przekazana dla wygody użytkowników wspomnianej normy międzynarodowej i nie ma na celu zatwierdzania wspomnianego produktu przez ISO lub IDF.
ZAŁĄCZNIK B
(informacyjny)
OZNACZANIE ILOŚCIOWE ZAWARTOŚCI TłUSZCZU OBCEGO
B.1 UWAGI OGÓLNE
W tabeli B.1 przedstawiono granice wykrywalności dla różnych tłuszczów obcych, wyliczone przy poziomie ufności wynoszącym 99 %. W środkowej kolumnie przedstawiono granice wykrywalności najbardziej odpowiednich poszczególnych równań (3)–(6).
Granice wykrywalności równania ogólnego (7), przedstawione w prawej kolumnie, są nieco wyższe. W zasadzie do ilościowego oznaczenia zawartości tłuszczu obcego niezbędne jest jedynie równanie (7).
Mając do dyspozycji wszystkie równania można wykryć również połączenia różnych tłuszczów obcych. Różnice w składzie TG pomiędzy poszczególnymi próbkami jednego rodzaju tłuszczu obcego nie mają znaczącego wpływu na granice wykrywalności.
Przy korzystaniu zarówno z poszczególnych równań, jak i z równania ogólnego, zastosowanie mają granice wykrywalności poszczególnych równań. Wartość S równania ogólnego jest jednak w niektórych przypadkach niezbędna do ilościowego oznaczenia (ppkt B.2).
Tabela B.1
99 % granice wykrywalności tłuszczu obcego dodanego do tłuszczu mlecznego, wyrażone jako wartości procentowe
|
Tłuszcz obcy
|
Poszczególne równania
%
|
Równanie ogólne
%
|
|
Olej sojowy
|
2,1
|
4,4
|
|
Olej słonecznikowy
|
2,3
|
4,8
|
|
Oliwa z oliwek
|
2,4
|
4,7
|
|
Olej kokosowy
|
3,5
|
4,3
|
|
Olej palmowy
|
4,4
|
4,7
|
|
Tłuszcz z ziaren palmowych
|
4,6
|
5,9
|
|
Olej rzepakowy
|
2,0
|
4,4
|
|
Olej lniany
|
2,0
|
4,0
|
|
Olej z kiełków pszenicy
|
2,7
|
6,4
|
|
Olej z kiełków kukurydzy
|
2,2
|
4,5
|
|
Olej z nasion bawełny
|
3,3
|
4,4
|
|
Smalec
|
2,7
|
4,7
|
|
Łój wołowy
|
5,2
|
5,4
|
|
Uwodorniony olej rybny
|
5,4
|
6,1
|
B.2 OBLICZANIE
Oznaczenie ilościowe zawartości tłuszczu obcego należy wykonać jedynie wówczas, gdy przekroczona jest co najmniej jedna z wartości granicznych S (tabela 2 lub tabela 5). Aby uzyskać informacje ilościowe, obliczyć ułamek masowy tłuszczu obcego lub ułamek masowy mieszaniny tłuszczów obcych, w
f, wyrażony jako wartość procentowa, w próbce badanej, przy użyciu poniższego równania:
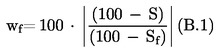
gdzie:
|
S
|
jest wynikiem uzyskanym po podstawieniu danych dotyczących TG w tłuszczu mlecznym, do którego dodano tłuszcz obcy lub mieszaninę tłuszczów obcych, do jednego z równań (3)–(7),
|
|
S
f
|
jest wartością stałą, która zależy od rodzaju dodanego tłuszczu obcego.
|
W przypadku gdy nie jest znany rodzaj tłuszczu obcego dodanego do tłuszczu mlecznego, należy zastosować ogólną wartość S
f wynoszącą 7,46 (tabela B.2). Zawsze należy stosować wartość S otrzymaną z równania (7), nawet jeżeli dotyczące go wartości graniczne S nie są przekroczone, podczas gdy wartości S dotyczące innego równania zostały przekroczone.
Mając do dyspozycji znane tłuszcze obce, należy podstawić ich poszczególne wartości S
f (tabela B.2) do równania (B.1). Aby obliczyć S, należy wybrać odpowiednie równanie tłuszczu obcego spośród równań (3)–(6).
Tabela B.2
Wartości S
f różnych tłuszczów obcych
|
Tłuszcz obcy
|
S
f
|
|
Nieznany tłuszcz
|
7,46
|
|
Olej sojowy
|
8,18
|
|
Olej słonecznikowy
|
9,43
|
|
Oliwa z oliwek
|
12,75
|
|
Olej kokosowy
|
118,13
|
|
Olej palmowy
|
7,55
|
|
Tłuszcz z ziaren palmowych
|
112,32
|
|
Olej rzepakowy
|
3,30
|
|
Olej lniany
|
4,44
|
|
Olej z kiełków pszenicy
|
27,45
|
|
Olej z kiełków kukurydzy
|
9,29
|
|
Olej z nasion bawełny
|
41,18
|
|
Smalec
|
177,55
|
|
Łój wołowy
|
17,56
|
|
Olej rybny
|
64,12
|
B.3 PREZENTACJA WYNIKÓW BADAŃ
Wyniki badań należy podać z dokładnością do drugiego miejsca po przecinku.
Bibliografia
|
(1)
|
Molkentin, J., Precht, D. Representative determination of the butyric acid content in European milk fats. Milchwissenschaft, 52, 1987, s. 82–85.
|
|
(2)
|
Precht, D., Molkentin, J. Quantitative triglyceride analysis using short capillary columns. Chrompack News, 4, 1993, s. 16–17.
|
|
(3)
|
Molkentin, J., Precht, D. Comparison of packed and capillary columns for quantitative gas chromatography of triglycerides in milk fat. Chromatographia, 39, 1994, s. 265–270.
|
|
(4)
|
Molkentin, J., Precht, D. Equivalence of packed and capillary GC columns with respect to suitability for foreign fat detection in butter using the triglyceride formula method. Chromatographia, 52, 2000, s. 791–797.
|
|
(5)
|
ISO 707IDF 50, Mleko i przetwory mleczne — Wytyczne do pobierania próbek.
|
|
(6)
|
BS 410:1988, Test sieves — Technical requirements and testing.
|
|
(7)
|
Precht, D. Control of milk fat purity by gas chromatographic triglyceride analysis. Kieler Milchwirtsch. Forschungsber., 43, 1991, s. 219–242.
|
|
(8)
|
Precht, D. Detection of adulterated milk fat by fatty acid and triglyceride analyses. Fat Sci. Technol., 93, 1991, s. 538–544.
|
|
(9)
|
DIN 10336:1994, Nachweis und Bestimmung von Fremdfetten in Milchfett anhand einer gaschromatographischen Triglyceridanalyse [Wykrywanie i oznaczanie tłuszczów obcych w tłuszczu mlecznym przy użyciu analizy chromatografii gazowej triglicerydów].
|
|
(10)
|
Komisja Wspólnot Europejskich: Consideration of results from the first, second, third, fourth, fifth and sixth EEC collaborative trial: Determination of triglycerides in milk fat; Doc. No VI/2644/91, VI/8.11.91, VI/1919/92, VI 3842/92. VI/5317/92, VI/4604/93.
|
|
(11)
|
Molkentin, J. Detection of foreign fat in milk fat from different continents by triacylglycerol analysis. Eur. J. Lipid Sci. Technol., 109, 2007, s. 505–510.
|
ZAŁĄCZNIK XXI
(Artykuł 18)
PROCEDURA STOSOWANA W PRZYPADKACH SPORNYCH WYNIKÓW ANALIZ (ANALIZA CHEMICZNA)
1. Dalsza analiza jest przeprowadzana w innym laboratorium zatwierdzonym przez właściwe władze przy użyciu odpowiedniej metody na wniosek producenta, pod warunkiem, że zapieczętowane duplikaty próbek produktu są dostępne i były odpowiednio składowane przez właściwe władze. Wniosek jest składany w ciągu 7 dni roboczych od ogłoszenia wyników pierwszej analizy. Analiza jest wykonywana w ciągu 21 dni roboczych od otrzymania wniosku. Właściwe władze prześlą wspomniane próbki do drugiego laboratorium na wniosek i kosztu producenta. Laboratorium musi być uprawnione do przeprowadzania analiz urzędowych i musi posiadać potwierdzone kompetencje do wykonywania wnioskowanych analiz.
2. Niepewności rozszerzone (k = 2) średniej  liczby
liczby  powtórzonych pomiarów w laboratorium 1 oraz średniej
powtórzonych pomiarów w laboratorium 1 oraz średniej  liczby
liczby  powtórzonych pomiarów w laboratorium 2 wynoszą odpowiednio
powtórzonych pomiarów w laboratorium 2 wynoszą odpowiednio
3.  oraz
oraz 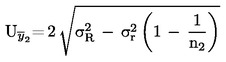 , gdzie
, gdzie 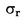 stanowi odchylenie standardowe powtarzalności, a
stanowi odchylenie standardowe powtarzalności, a 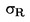 stanowi odchylenie standardowe odtwarzalności stosownej metody. Jeżeli wynik końcowy y pomiaru w laboratorium jest obliczony przy użyciu wzoru w postaci
stanowi odchylenie standardowe odtwarzalności stosownej metody. Jeżeli wynik końcowy y pomiaru w laboratorium jest obliczony przy użyciu wzoru w postaci  ,
,  ,
,  lub
lub  , należy przestrzegać normalnych procedur łączenia odchyleń standardowych w takich przypadkach w celu określenia niepewności.
, należy przestrzegać normalnych procedur łączenia odchyleń standardowych w takich przypadkach w celu określenia niepewności.
4. Aby zbadać, czy wyniki z obu laboratoriów są zgodne z odchyleniem standardowym odtwarzalności 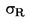 danej metody, oblicza się niepewność rozszerzoną różnicy
danej metody, oblicza się niepewność rozszerzoną różnicy  .
.
5. 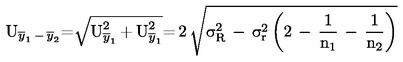 Jeżeli bezwzględna wartość różnicy średnich wartości laboratoryjnych,
Jeżeli bezwzględna wartość różnicy średnich wartości laboratoryjnych,  , nie jest wyższa niż jej niepewność
, nie jest wyższa niż jej niepewność  ,
,
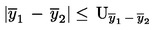 ;
;
wyniki uzyskane w obu laboratoriach są zgodne z odchyleniem standardowym odtwarzalności 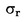 , a średnia arytmetyczna dwóch średnich wartości laboratoryjnych,
, a średnia arytmetyczna dwóch średnich wartości laboratoryjnych,
 ,
,
jest przedstawiany jako wynik końcowy. Niepewność rozszerzona tego wyniku wynosi
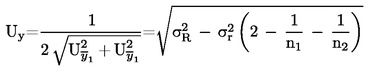 ;
;
Partia towaru jest odrzucona jako niezgodna z górną prawnie obowiązującą wartością graniczną UL, jeżeli
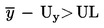 ;
;
w przeciwnym razie partia towaru jest dopuszczona jako zgodna z UL.
Partia towaru jest odrzucona jako niezgodna z dolną prawnie obowiązującą wartością graniczną LL, jeżeli
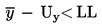 ;
;
w przeciwnym razie partia towaru jest dopuszczona jako zgodna z LL.
Jeżeli bezwzględna wartość różnicy średnich wartości laboratoryjnych, 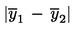 , jest wyższa niż jej niepewność
, jest wyższa niż jej niepewność  ,
,
 ,
,
wyniki uzyskane w obu laboratoriach są niezgodne z odchyleniem standardowym odtwarzalności.
W takim przypadku partia towaru jest odrzucona jako niezgodna, jeżeli druga analiza potwierdza wyniki pierwszej. W przeciwnym razie partia towaru jest dopuszczona jako zgodna.
Wynik końcowy musi zostać przekazany przez właściwe władze do wiadomości producenta możliwie jak najszybciej. Koszty drugiej analizy ponosi producent, o ile partia towaru jest odrzucona.
ZAŁĄCZNIK XXII
TABELA KORELACJI
|
Rozporządzenie (WE) nr 213/2001
|
Niniejsze rozporządzenie
|
|
Artykuł 1
|
Artykuł 1
|
|
Artykuł 2
|
Artykuł 1
|
|
Artykuł 3
|
Artykuł 2
|
|
—
|
Artykuł 3
|
|
Artykuł 4
|
—
|
|
Artykuł 5
|
—
|
|
Artykuł 6
|
Artykuł 4
|
|
Artykuł 7
|
Artykuł 18
|
|
Artykuł 8
|
—
|
|
Artykuł 9
|
Artykuł 5
|
|
Artykuł 10
|
Artykuł 6
|
|
Artykuł 11
|
Artykuł 7
|
|
Artykuł 12
|
Artykuł 8
|
|
Artykuł 13
|
Artykuł 9
|
|
Artykuł 14
|
Artykuł 10
|
|
Artykuł 15
|
Artykuł 11
|
|
Artykuł 16
|
Artykuł 12
|
|
Artykuł 17
|
Artykuł 13
|
|
—
|
Artykuł 14
|
|
Artykuł 18
|
Artykuł 15
|
|
Artykuł 19
|
Artykuł 16
|
|
|
Artykuł 17
|
|
|
Artykuł 19
|
|
Artykuł 20
|
—
|
|
Artykuł 21
|
—
|
|
Artykuł 22
|
Artykuł 20
|
|
Artykuł 23
|
Artykuł 21
|