

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 52016XC0726(02)
Commission Notice — The ‘Blue Guide’ on the implementation of EU products rules 2016 (Text with EEA relevance)
Zawiadomienie Komisji – Niebieski przewodnik – wdrażanie unijnych przepisów dotyczących produktów 2016 (Tekst mający znaczenie dla EOG)
Zawiadomienie Komisji – Niebieski przewodnik – wdrażanie unijnych przepisów dotyczących produktów 2016 (Tekst mający znaczenie dla EOG)
C/2016/1958
OJ C 272, 26.7.2016, p. 1–149
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
|
26.7.2016 |
PL |
Dziennik Urzędowy Unii Europejskiej |
C 272/1 |
ZAWIADOMIENIE KOMISJI
Niebieski przewodnik – wdrażanie unijnych przepisów dotyczących produktów 2016
(Tekst mający znaczenie dla EOG)
(2016/C 272/01)
SPIS TREŚCI
SŁOWO WSTĘPNE
UWAGA
|
1. |
REGULACJA SWOBODNEGO PRZEPŁYWU TOWARÓW | 5 |
|
1.1. |
Ujęcie historyczne | 5 |
|
1.1.1. |
Stare podejście | 6 |
|
1.1.2. |
Wzajemne uznawanie | 7 |
|
1.1.3. |
„Nowe podejście” i „globalne podejście” | 7 |
|
1.2. |
Nowe ramy prawne | 9 |
|
1.2.1. |
Koncepcja | 9 |
|
1.2.2. |
Charakter prawny aktów nowych ram prawnych i ich związek z innymi przepisami UE | 10 |
|
1.2.3. |
Dopasowanie elementów systemu | 11 |
|
1.3. |
Dyrektywa w sprawie ogólnego bezpieczeństwa produktów | 12 |
|
1.4. |
Prawodawstwo dotyczące odpowiedzialności za produkt | 12 |
|
1.5. |
Zakres przewodnika | 13 |
|
2. |
KIEDY MA ZASTOSOWANIE UNIJNE PRAWODAWSTWO HARMONIZACYJNE? | 15 |
|
2.1. |
Zakres produktów | 15 |
|
2.2. |
Udostępnienie na rynku | 17 |
|
2.3. |
Wprowadzenie do obrotu | 18 |
|
2.4. |
Produkty importowane z państw spoza UE | 20 |
|
2.5. |
Oddawanie do użytku (i instalacja) | 21 |
|
2.6. |
Równoczesne stosowanie unijnych aktów harmonizacyjnych | 22 |
|
2.7. |
Użytkowanie zgodne z przeznaczeniem/niewłaściwe użytkowanie | 23 |
|
2.8. |
Zastosowanie geograficzne (państwa EOG, EFTA, kraje i terytoria zamorskie, Turcja) | 24 |
|
2.8.1. |
Państwa członkowskie oraz kraje i terytoria zamorskie | 24 |
|
2.8.2. |
Państwa EOG i EFTA | 25 |
|
2.8.3. |
Monako, San Marino i Andora | 25 |
|
2.8.4. |
Turcja | 26 |
|
2.9. |
Okresy przejściowe w przypadku nowych lub zmienionych zasad UE | 27 |
|
2.10. |
Ustalenia przejściowe dotyczące deklaracji zgodności UE wynikające z dostosowania do decyzji nr 768/2008/WE | 27 |
|
3. |
UCZESTNICY ŁAŃCUCHA DOSTAW PRODUKTÓW I ICH OBOWIĄZKI | 28 |
|
3.1. |
Producent | 28 |
|
3.2. |
Upoważniony przedstawiciel | 32 |
|
3.3. |
Importer | 33 |
|
3.4. |
Dystrybutor | 34 |
|
3.5. |
Inni pośrednicy: usługodawcy będący pośrednikami na podstawie dyrektywy o handlu elektronicznym | 37 |
|
3.6. |
Użytkownik | 38 |
|
4. |
WYMAGANIA DOTYCZĄCE PRODUKTÓW | 39 |
|
4.1. |
Zasadnicze wymagania dotyczące produktów | 39 |
|
4.1.1. |
Definicja zasadniczych wymagań | 39 |
|
4.1.2. |
Zgodność z zasadniczymi wymaganiami: normy zharmonizowane | 40 |
|
4.1.3. |
Zgodność z zasadniczymi wymaganiami: inne możliwości | 51 |
|
4.2. |
Wymagania w zakresie identyfikowalności | 51 |
|
4.2.1. |
Dlaczego identyfikowalność jest ważna? | 52 |
|
4.2.2. |
Przepisy dotyczące identyfikowalności | 52 |
|
4.3. |
Dokumentacja produktu | 56 |
|
4.4. |
Deklaracja zgodności UE | 57 |
|
4.5. |
Wymogi w zakresie oznakowania | 58 |
|
4.5.1. |
Oznakowanie CE | 58 |
|
4.5.2. |
Inne obowiązkowe oznakowania | 64 |
|
5. |
OCENA ZGODNOŚCI | 65 |
|
5.1. |
Moduły oceny zgodności | 65 |
|
5.1.1. |
Czym jest ocena zgodności? | 65 |
|
5.1.2. |
Modułowa struktura oceny zgodności w unijnym prawodawstwie harmonizacyjnym | 65 |
|
5.1.3. |
Podmioty zaangażowane w ocenę zgodności – umiejscowienie oceny zgodności w łańcuchu dostaw | 66 |
|
5.1.4. |
Moduły i ich warianty | 69 |
|
5.1.5. |
Procedury jedno- i dwumodułowe – procedury oparte na typie (badanie typu UE) | 69 |
|
5.1.6. |
Moduły oparte na zapewnianiu jakości | 70 |
|
5.1.7. |
Przegląd modułów | 70 |
|
5.1.8. |
Przegląd procedur | 73 |
|
5.1.9. |
Uzasadnienie wyboru odpowiednich modułów | 74 |
|
5.2. |
Jednostki oceniające zgodność | 75 |
|
5.2.1. |
Jednostki oceniające zgodność oraz jednostki notyfikowane | 75 |
|
5.2.2. |
Role i obowiązki | 76 |
|
5.2.3. |
Kompetencje jednostek notyfikowanych | 78 |
|
5.2.4. |
Koordynacja między jednostkami notyfikowanymi | 79 |
|
5.2.5. |
Zlecanie prac podwykonawcy przez jednostki notyfikowane | 79 |
|
5.2.6. |
Akredytowane jednostki własne | 81 |
|
5.3. |
Notyfikacja | 81 |
|
5.3.1. |
Organy notyfikujące | 81 |
|
5.3.2. |
Proces notyfikacji | 82 |
|
5.3.3. |
Publikacja przez Komisję – strona internetowa NANDO | 85 |
|
5.3.4. |
Monitorowanie kompetencji jednostek notyfikowanych – zawieszenie – wycofanie – odwołanie | 86 |
|
6. |
AKREDYTACJA | 87 |
|
6.1. |
Dlaczego akredytacja? | 87 |
|
6.2. |
Czym jest akredytacja? | 88 |
|
6.3. |
Zakres akredytacji | 89 |
|
6.4. |
Akredytacja zgodnie z rozporządzeniem (WE) nr 765/2008 | 89 |
|
6.4.1. |
Krajowe jednostki akredytujące | 90 |
|
6.4.2. |
Niekonkurencyjny i niekomercyjny charakter działalności krajowych jednostek akredytujących | 91 |
|
6.5. |
Europejska infrastruktura akredytacyjna | 92 |
|
6.5.1. |
Sektorowe systemy akredytacji | 92 |
|
6.5.2. |
Ocena wzajemna | 92 |
|
6.5.3. |
Domniemanie zgodności dla krajowych jednostek akredytujących | 93 |
|
6.5.4. |
Rola EA we wspieraniu i harmonizacji praktyki akredytacji w Europie | 93 |
|
6.6. |
Akredytacja transgraniczna | 93 |
|
6.7. |
Akredytacja w kontekście międzynarodowym | 95 |
|
6.7.1. |
Współpraca między jednostkami akredytującymi | 95 |
|
6.7.2. |
Wpływ na stosunki handlowe w obszarze oceny zgodności między UE a państwami trzecimi | 96 |
|
7. |
NADZÓR RYNKU | 97 |
|
7.1. |
Dlaczego nadzór rynku jest potrzebny? | 98 |
|
7.2. |
Kontrole prowadzone przez organy nadzoru rynku | 99 |
|
7.3. |
Kontrola produktów z państw trzecich przez organy celne | 101 |
|
7.4. |
Obowiązki państw członkowskich | 103 |
|
7.4.1. |
Infrastruktury krajowe | 103 |
|
7.4.2. |
Krajowe programy nadzoru rynku oraz przeglądy działalności | 104 |
|
7.4.3. |
Publiczne udostępnianie informacji | 105 |
|
7.4.4. |
Procedury nadzoru rynku | 105 |
|
7.4.5. |
Środki naprawcze – zakazy – wycofanie z obrotu – wycofanie od użytkowników | 107 |
|
7.4.6. |
Sankcje | 108 |
|
7.5. |
Współpraca między państwami członkowskimi i Komisją Europejską | 108 |
|
7.5.1. |
Mechanizmy ochronne | 109 |
|
7.5.2. |
Stosowanie mechanizmów ochronnych krok po kroku | 110 |
|
7.5.3. |
Wzajemna pomoc, współpraca administracyjna i wymiana informacji między państwami członkowskimi | 112 |
|
7.5.4. |
System wczesnego ostrzegania o niebezpiecznych produktach nieżywnościowych | 114 |
|
7.5.5. |
ICSMS | 115 |
|
7.5.6. |
Wyroby medyczne: system nadzoru | 117 |
|
8. |
SWOBODNY PRZEPŁYW PRODUKTÓW W UE | 117 |
|
8.1. |
Klauzula dotycząca swobodnego przepływu | 117 |
|
8.2. |
Ograniczenia i restrykcje | 118 |
|
9. |
MIĘDZYNARODOWE ASPEKTY PRZEPISÓW UNIJNYCH DOTYCZĄCYCH PRODUKTÓW | 118 |
|
9.1. |
Układy w sprawie oceny zgodności oraz uznawania (ACAA) | 118 |
|
9.2. |
Umowy o wzajemnym uznawaniu (MRA) | 119 |
|
9.2.1. |
Główne cechy umów | 119 |
|
9.2.2. |
MRA między UE a Szwajcarią | 120 |
|
9.2.3. |
Państwa EFTA należące do EOG: umowy o wzajemnym uznawaniu i układy w sprawie oceny zgodności oraz uznawania | 121 |
ZAŁĄCZNIKI
|
ZAŁĄCZNIK I – |
Unijne akty prawne, o których mowa w niniejszym przewodniku (niewyczerpująca lista) | 122 |
|
ZAŁĄCZNIK II – |
Dodatkowe wytyczne | 127 |
|
ZAŁĄCZNIK III – |
Przydatne adresy internetowe | 129 |
|
ZAŁĄCZNIK IV – |
Procedury oceny zgodności (moduły z decyzji nr 768/2008/WE) | 130 |
|
ZAŁĄCZNIK V – |
Zależność między ISO 9000 a modułami wymagającymi systemu zapewnienia jakości | 140 |
|
ZAŁĄCZNIK VI – |
Stosowanie norm zharmonizowanych do oceny kompetencji jednostek oceniających zgodność | 142 |
|
ZAŁĄCZNIK VII – |
Często zadawane pytania dotyczące oznakowania CE | 147 |
SŁOWO WSTĘPNE
W 2000 r. oddaliśmy w Państwa ręce publikację „Wdrażanie dyrektyw opartych na koncepcji nowego i globalnego podejścia – przewodnik” („Niebieski przewodnik”). Od tamtej pory stała się ona jednym z głównych dokumentów referencyjnych dla osób odpowiedzialnych za wdrażanie przepisów opartych na koncepcji nowego podejścia, ujętych aktualnie w nowych ramach prawnych.
Wiele informacji zawartych w wydaniu „Niebieskiego przewodnika” z 2000 r. jest nadal ważny, ale dokument wymaga aktualizacji uwzględniającej zmiany, które zaszły od tamtego czasu, i zapewniającej możliwie najszersze zrozumienie kwestii związanych z wdrożeniem nowych ram prawnych regulujących wprowadzanie produktów do obrotu. Konieczne jest również uwzględnienie zmian wprowadzonych Traktatem lizbońskim (obowiązującym od dnia 1 grudnia 2009 r.) w zakresie źródeł prawa i terminologii znajdujących zastosowanie w unijnych dokumentach, procedurach itp.
W związku z tym niniejsza nowa wersja przewodnika będzie oparta na poprzednim wydaniu, ale będzie zawierać nowe rozdziały dotyczące np. obowiązków podmiotów gospodarczych czy akredytacji albo całkowicie zmienione rozdziały, jak np. rozdział o normalizacji czy nadzorze rynku. Przewodnik otrzymał również nową nazwę, która odzwierciedla fakt, że nowe ramy prawne mogą być stosowane, przynajmniej w części, we wszystkich typach unijnego prawodawstwa harmonizacyjnego, a nie tylko w tzw. dyrektywach „nowego podejścia”.
UWAGA
Niniejszy przewodnik ma przyczynić się do lepszego zrozumienia przepisów Unii Europejskiej dotyczących produktów oraz bardziej spójnego zastosowania ich w różnych sektorach jednolitego rynku. Jest skierowany do państw członkowskich i innych podmiotów poszukujących informacji o przepisach mających na celu zapewnienie swobodnego przepływu produktów i zapewnienie wysokiego poziomu ochrony na terenie całej Unii (np. dla stowarzyszeń handlowych, organizacji konsumenckich, jednostek normalizacyjnych, producentów, importerów, dystrybutorów, jednostek oceniających zgodność, związków zawodowych).
Niniejszy dokument ma charakter wyłącznie informacyjny – tylko tekst unijnego aktu harmonizacyjnego ma moc wiążącą. Czasami treść niniejszego przewodnika może różnić się od przepisów wybranego unijnego aktu harmonizacyjnego, w szczególności tam, gdzie specyfika przewodnika nie pozwala na pełne omówienie nieznacznie różniących się przepisów takiego aktu. Wiążąca interpretacja przepisów unijnych leży w wyłącznej kompetencji Trybunału Sprawiedliwości Unii Europejskiej. Poglądy przedstawione w tym przewodniku nie mogą przesądzać o stanowisku, jakie Komisja może zająć przed Trybunałem Sprawiedliwości. Ani Komisja Europejska, ani żadna osoba działająca w jej imieniu nie ponosi odpowiedzialności za sposób wykorzystania poniższych informacji.
Niniejszy przewodnik dotyczy państw członkowskich UE, a także Islandii, Liechtensteinu i Norwegii jako sygnatariuszy Porozumienia o Europejskim Obszarze Gospodarczym, a w wybranych przypadkach również Turcji. Odniesienia do Unii lub jednolitego rynku należy zatem rozumieć jako odniesienia do EOG lub rynku EOG.
Ponieważ przewodnik jest odzwierciedleniem stanu w chwili jego powstawania, zawarte w nim wskazówki mogą być modyfikowane w późniejszym terminie (1). Obecnie toczą się przede wszystkim szczegółowe dyskusje dotyczące różnych aspektów unijnych ram prawnych mających zastosowanie do sprzedaży internetowej, a niniejszy przewodnik pozostaje bez uszczerbku dla wszelkich przyszłych interpretacji i wskazówek, które mogą zostać opracowane w odniesieniu do tych kwestii.
1. REGULACJA SWOBODNEGO PRZEPŁYWU TOWARÓW
1.1. UJĘCIE HISTORYCZNE
Pierwsze dyrektywy harmonizacyjne dotyczyły eliminacji barier, a także swobodnego przepływu towarów w ramach jednolitego rynku. Obecnie cele te uzupełniono o kompleksową politykę, która gwarantuje, że na rynku dostępne będą tylko bezpieczne produkty spełniające stosowne wymagania prawne, co daje równe szanse uczciwym podmiotom gospodarczym i jednocześnie promuje skuteczną ochronę unijnych konsumentów i profesjonalnych użytkowników oraz jednolity rynek UE oparty na konkurencji.
W ciągu ostatnich 40 lat integracji europejskiej rozwinęły się polityki i techniki prawodawcze, w szczególności w dziedzinie swobodnego przepływu towarów, co przyczyniło się do obecnego sukcesu jednolitego rynku.
Historycznie prawodawstwo UE dotyczące towarów przeszło cztery główne fazy:
|
— |
podejście tradycyjne lub „stare podejście” ze szczegółowymi dokumentami zawierającymi wszystkie niezbędne wymagania techniczne i administracyjne, |
|
— |
„nowe podejście” opracowane w 1985 r., w którym treść przepisów prawa była ograniczona do „zasadniczych wymagań”, a szczegóły techniczne uregulowano za pomocą zharmonizowanych norm. To z kolei doprowadziło do rozwoju europejskiej polityki normalizacyjnej uzupełniającej to prawodawstwo, |
|
— |
rozwój instrumentów oceny zgodności konieczny przy wdrażaniu różnych unijnych aktów harmonizacyjnych, zarówno w ramach nowego, jak i starego podejścia, |
|
— |
„nowe ramy prawne” (2) przyjęte w lipcu 2008 r., które opierały się na nowym podejściu i uzupełniły ogólne ramy prawne wszystkimi elementami potrzebnymi do skutecznej oceny zgodności, akredytacji i nadzoru rynku, w tym kontroli produktów spoza Unii. |
1.1.1. STARE PODEJŚCIE
Stare podejście odzwierciedlało tradycyjny sposób, w jaki organy krajowe tworzyły prawodawstwo techniczne. Wprowadzanie szczegółowych przepisów często było spowodowane brakiem zaufania do samodyscypliny podmiotów gospodarczych w kwestiach zdrowia i bezpieczeństwa publicznego. W pewnych sektorach (np. metrologii prawnej) doprowadziło to do tego, że organy same wydawały certyfikaty zgodności. Wymagana do 1986 r. jednomyślność w tym obszarze sprawiła, że przyjmowanie przepisów przebiegało bardzo ociężale, a ciągły powrót do dawnej techniki w wielu sektorach jest często uzasadniony polityką publiczną (np. przepisami prawa dotyczącymi żywności) lub tradycjami i/lub porozumieniami międzynarodowymi, które nie mogą być zmienione jednostronnie (np. prawodawstwo dotyczące przemysłu samochodowego lub, ponownie, żywności).
Pierwszą próbę zmiany tej sytuacji podjęto, przyjmując dyrektywę 83/189/EWG (3) z dnia 28 marca 1983 r. ustanawiającą procedurę przekazywania informacji pomiędzy państwami członkowskimi a Komisją w celu uniknięcia tworzenia nowych barier technicznych w swobodnym przepływie towarów, których wyeliminowanie w ramach procesu harmonizacji byłoby długotrwałe.
Zgodnie z tą dyrektywą państwa członkowskie mają obowiązek powiadamiania innych państw członkowskich i Komisji o projektach krajowych przepisów technicznych (a krajowe jednostki normalizacyjne zostały zobowiązane do powiadamiania Komisji, europejskich organizacji normalizacyjnych i innych krajowych jednostek normalizacyjnych o projektach norm krajowych (4)). W okresie zawieszenia nie można przyjmować takich przepisów, a Komisja i inne państwa członkowskie mają czas na ewentualną reakcję. Jeśli w początkowym okresie zawieszenia wynoszącym trzy miesiące Komisja lub inne państwa członkowskie nie zareagują, projekt regulacji technicznych może zostać przyjęty. W przeciwnym przypadku, jeśli zostanie zgłoszony sprzeciw, okres zawieszenia zostanie przedłużony o kolejne trzy miesiące.
Okres zawieszenia wynosi 12 miesięcy w przypadku istnienia projektu unijnego aktu harmonizacyjnego dla danego obszaru. Jednak okres ten nie znajduje zastosowania w sytuacji, gdy państwo członkowskie jest zobowiązane do pilnego wprowadzenia przepisów technicznych w celu ochrony zdrowia i bezpieczeństwa publicznego, zwierząt lub roślin.
1.1.2. WZAJEMNE UZNAWANIE
Obok inicjatyw legislacyjnych podejmowanych w celu uniknięcia tworzenia nowych barier oraz wspierania swobodnego przepływu towarów, zwracano także uwagę na systemowe stosowanie zasady wzajemnego uznawania zawartej w prawie Unii Europejskiej. Krajowe przepisy techniczne podlegają przepisom art. 34–36 Traktatu o funkcjonowaniu Unii Europejskiej (TFUE), które zakazują ograniczeń ilościowych lub środków o skutku równoważnym. Orzecznictwo Europejskiego Trybunału Sprawiedliwości, w szczególności sprawa 120/78 (sprawa „Cassis de Dijon” (5)) zawiera kluczowe elementy związane ze wzajemnym uznawaniem. Skutek tego orzecznictwa jest następujący:
|
— |
Swobodny przepływ w całej Unii powinien być z zasady zagwarantowany w odniesieniu do produktów legalnie wytwarzanych lub wprowadzanych do obrotu w jednym państwie członkowskim, jeżeli spełniają one takie same wymagania ochrony jak wymagania nałożone przez docelowe państwo członkowskie. |
|
— |
Wobec braku unijnego prawodawstwa harmonizacyjnego, państwa członkowskie mają swobodę w zakresie uchwalania przepisów obowiązujących na ich terytorium, o ile są one zgodne z przepisami Traktatu w zakresie swobodnego przepływu towarów (art. 34–36 TFUE). |
|
— |
Bariery w swobodnym przepływie wynikające z różnic w prawodawstwie krajowym można zaakceptować tylko wtedy, gdy środki krajowe:
|
W celu ułatwienia wdrożenia tych zasad w 2008 r. Parlament Europejski i Rada przyjęły w ramach pakietu towarowego rozporządzenie (WE) nr 764/2008 z dnia 9 lipca 2008 r. ustanawiające procedury dotyczące stosowania niektórych krajowych przepisów technicznych do produktów wprowadzonych legalnie do obrotu w innym państwie członkowskim oraz uchylające decyzję nr 3052/95/WE (6).
Jednak chociaż zasada wzajemnego uznawania w dużej mierze przyczynia się do swobodnego przepływu towarów w ramach jednolitego rynku, nie może ona rozwiązać wszystkich problemów i nawet dziś, jak podkreślono komentarzach do sprawozdania Montiego (7), istnieje pole do dalszej harmonizacji.
1.1.3. „NOWE PODEJŚCIE” I „GLOBALNE PODEJŚCIE”
Sprawa Cassis de Dijon jest dobrze znana ze względu na rolę, jaką odegrała w propagowaniu zasady wzajemnego uznawania. Miała jednak także ogromne znaczenie w modyfikowaniu podejścia UE do harmonizacji technicznej pod trzema najważniejszymi względami:
|
— |
uznając, że państwa członkowskie mogą uzasadnić zakaz lub ograniczenie wprowadzenia do obrotu produktów z innych państw członkowskich wyłącznie niezgodnością z „zasadniczymi wymaganiami”, Trybunał dał impuls do przemyślenia treści przyszłego prawodawstwa harmonizacyjnego: skoro niespełnienie wymagań innych niż zasadnicze nie może uzasadniać nakładania ograniczeń na wprowadzanie produktu do obrotu, to takie wymagania nie muszą pojawiać się w aktach harmonizacyjnych UE. To otworzyło drogę do nowego podejścia i późniejszej refleksji nad tym, co stanowi zasadnicze wymaganie i w jaki sposób należy je formułować, aby służyło jako punkt odniesienia przy wykazywaniu zgodności produktu z przepisami prawa, |
|
— |
uznając tę zasadę Trybunał jasno wskazał, że ciężar udowodnienia niezgodności produktu z zasadniczymi wymaganiami spoczywa na organach krajowych, ale również przesądził sprawę odpowiednich, proporcjonalnych środków wykazania zgodności, |
|
— |
zauważając, że państwa członkowskie są zobowiązane do przyjmowania produktów z innych państw członkowskich z wyjątkiem określonych sytuacji, Trybunał ustalił regułę prawną, ale nie stworzył środków budujących zaufanie władz do produktów, które mają zaakceptować, a za które nie mogą ręczyć. Spowodowało to potrzebę rozwinięcia polityki w zakresie oceny zgodności. |
Technika legislacyjna nowego podejścia przyjęta Uchwałą Rady Ministrów dnia 7 maja 1985 r. w sprawie harmonizacji technicznej i norm (8) była logicznym następstwem legislacyjnym sprawy Cassis de Dijon. Za pomocą tej techniki regulacyjnej ustanowiono następujące zasady:
|
— |
harmonizacja legislacyjna powinna ograniczać się do zasadniczych wymagań (najlepiej wymagań technicznych lub użytkowych), które muszą być spełnione przez produkty wprowadzane na rynek UE, jeśli ich wytwórcy chcą korzystać ze swobodnego przepływu towarów w ramach UE, |
|
— |
specyfikacje techniczne produktów spełniających zasadnicze wymagania określone w prawodawstwie powinny być ustalone w normach zharmonizowanych, które mogą być stosowane wraz z przepisami prawa, |
|
— |
w przypadku produktów wytworzonych zgodnie z normami zharmonizowanymi zachodzi domniemanie zgodności z odpowiednimi zasadniczymi wymaganiami stosownego prawodawstwa, a w niektórych przypadkach producent może skorzystać z uproszczonej procedury oceny zgodności (w wielu przypadkach wydania przez producenta deklaracji zgodności, która jest chętniej przyjmowana przez władze dzięki istnieniu przepisów prawa dotyczących odpowiedzialności za produkt (9)), |
|
— |
stosowanie norm zharmonizowanych lub innych pozostaje dobrowolne, a producent zawsze może zastosować inne specyfikacje techniczne, aby spełnić wymagania dla produktów (ale będzie musiał wykazać, że takie specyfikacje spełniają zasadnicze wymagania, najczęściej w ramach procedury z udziałem zewnętrznej jednostki oceniającej zgodność). |
Funkcjonowanie unijnego prawodawstwa harmonizacyjnego nowego podejścia wymaga, aby normy zharmonizowane gwarantowały określony poziom ochrony w odniesieniu do zasadniczych wymagań ustalonych w prawodawstwie. Jest to jedno z głównych zadań, nad którymi pracuje Komisja, realizując politykę skutecznego procesu i solidnej infrastruktury europejskiej normalizacji. Rozporządzenie (UE) nr 1025/2012 w sprawie normalizacji europejskiej (10) daje Komisji możliwość wnioskowania, po konsultacji z państwami członkowskimi, do europejskich organizacji normalizacyjnych o przygotowanie norm zharmonizowanych i ustala procedury oceny norm zharmonizowanych i wnoszenia wobec nich sprzeciwu.
Ponieważ nowe podejście wymaga, aby wspólne zasadnicze wymagania zostały wprowadzone w prawie, można je stosować tylko wtedy, gdy możliwe jest rozróżnienie pomiędzy zasadniczymi wymaganiami a specyfikacjami technicznymi. Co więcej, ponieważ zakres takiego prawodawstwa jest związany z ryzykiem, szeroka gama produktów musi być wystarczająco jednorodna, aby można było zastosować wspólne zasadnicze wymagania. Grupa produktów lub zagrożenia z nimi związane muszą również spełniać wymagania normalizacji.
Zasady nowego podejścia dały podstawę europejskiej normalizacji wspierającej unijne prawodawstwo harmonizacyjne. Rola zharmonizowanych norm oraz obowiązki europejskich organizacji normalizacyjnych są obecnie określone w rozporządzeniu (UE) nr 1025/2012 oraz w stosownym unijnym prawodawstwie harmonizacyjnym.
Zasada polegania na normach w przypadku przepisów technicznych została również przyjęta przez Światową Organizację Handlu (WTO). W swoim Porozumieniu w sprawie barier technicznych w handlu (TBT – ang. Technical Barriers to Trade) WTO propaguje stosowanie norm międzynarodowych (11).
Podczas negocjowania pierwszych unijnych aktów harmonizujących w ramach nowego podejścia natychmiast na jaw wyszedł fakt, że ustalenie zasadniczych wymagań i tworzenie norm zharmonizowanych nie jest wystarczające do zbudowania odpowiedniego zaufania pomiędzy państwami członkowskimi i że należy opracować odpowiednią horyzontalną politykę oceny zgodności i stosowne instrumenty. Tworzono je równocześnie z przyjmowaniem dyrektyw (12).
Dlatego też w latach 1989 i 1990 Rada podjęła Uchwałę w sprawie globalnego podejścia i decyzję 90/683/EWG (zaktualizowaną i zastąpioną decyzją 93/465/EWG) (13) ustalającą ogólne wskazówki i szczegółowe procedury w zakresie oceny zgodności. Zostały one uchylone i zaktualizowane decyzją Parlamentu Europejskiego i Rady nr 768/2008/WE z dnia 9 lipca 2008 r. w sprawie wspólnych ram dotyczących wprowadzania produktów do obrotu (14).
Główną ideą, dla której stworzono te instrumenty polityki, było opracowanie wspólnych narzędzi oceny zgodności we wszystkich obszarach (zarówno uregulowanych, jak i nieuregulowanych).
Polityka dotycząca norm produktowych została początkowo opracowana w celu zagwarantowania, że normy te ustalą specyfikacje techniczne, na podstawie których może być sprawdzana zgodność. Jednak na wniosek Komisji CEN i CENELEC przyjęły serię norm EN 45000 w celu ustalenia kompetencji podmiotów zewnętrznych zaangażowanych w ocenę zgodności. Seria ta stała się od tamtego czasu zharmonizowaną serią norm EN ISO/IEC 17000. Na podstawie dyrektyw nowego podejścia stworzono mechanizm, w ramach którego organy krajowe notyfikowały podmioty zewnętrzne wyznaczone do przeprowadzania oceny zgodności w oparciu o wspomniane normy.
Na podstawie dokumentacji ISO/IEC Rada w swoich decyzjach stworzyła skonsolidowane procedury oceny zgodności i zasady ich wyboru i zastosowania w dyrektywach (moduły). Moduły zostały zorganizowane w taki sposób, aby wybierać je od najprostszego („wewnętrzna kontrola produkcji”) w przypadku prostych produktów lub produktów niekoniecznie stwarzających poważne zagrożenie, do najbardziej kompleksowego (pełne zapewnienie jakości, w tym badanie projektu UE) tam, gdzie są poważniejsze zagrożenia lub bardziej skomplikowane produkty/technologie. Aby sprostać potrzebom nowoczesnych procesów produkcyjnych, w modułach przewidziano zarówno ocenę zgodności produktów z wymogami prawnymi, jak i ocenę systemu zarządzania jakością, pozostawiając prawodawcy wybór, która z nich jest właściwsza w danym sektorze, ponieważ np. zapewnianie indywidualnej certyfikacji każdego produktu masowego może być nieskuteczne. Aby poprawić przejrzystość modułów i ich skuteczność, na wniosek Komisji zharmonizowano na poziomie europejskim serię norm ISO 9001 dotyczącą zapewnienia jakości i włączono ją do modułów. Dlatego też podmioty gospodarcze, które stosują te narzędzia w dobrowolnie przyjętych politykach zarządzania jakością, aby wzmocnić swoją pozycję na rynku, mogą odnieść korzyści ze stosowania tych samych narzędzi w sektorach regulowanych.
Wszystkie te inicjatywy podjęto z myślą o bezpośrednim wzmocnieniu procesu oceny zgodności produktów przed wprowadzeniem ich do obrotu. Oprócz tego, Komisja w bliskiej współpracy z państwami członkowskimi oraz krajowymi jednostkami akredytującymi rozwinęła współpracę europejską na polu akredytacji, aby ustanowić ostatni szczebel kontroli i wzmocnić wiarygodność podmiotów zewnętrznych zaangażowanych w przeprowadzanie ocen zgodności produktów i zapewniania jakości. Była to inicjatywa bardziej polityczna niż legislacyjna, ale pomimo to skutecznie tworząca pierwszą europejską infrastrukturę w tym obszarze oraz dająca europejskim graczom dużą przewagę na arenie międzynarodowej.
Współpraca ta skutkowała przyjęciem na podstawie elementów nowego podejścia około 27 dyrektyw. Liczba ta jest zdecydowanie niższa niż w przypadku tradycyjnych dyrektyw dotyczących produktów przemysłowych (ok. 700), ale szeroki zakres objętych zagrożeń oznacza, że dzięki tej technice legislacyjnej wszystkie sektory przemysłowe czerpią korzyści ze swobodnego przepływu towarów.
1.2. NOWE RAMY PRAWNE
1.2.1. KONCEPCJA
Pod koniec lat 90. Komisja zaczęła zastanawiać się nad skutecznym wdrożeniem nowego podejścia. W 2002 r. rozpoczęto proces szerokich konsultacji, a 7 maja 2003 r. Komisja wystosowała komunikat do Rady i Parlamentu Europejskiego sugerujący możliwą rewizję określonych elementów nowego podejścia. To z kolei skutkowało rezolucją Rady z dnia 10 listopada 2003 r. w sprawie komunikatu Komisji Europejskiej „Poprawa wdrażania dyrektyw nowego podejścia” (15).
Konsensus w sprawie potrzeby aktualizacji i przeglądu elementów był jasny i zdecydowany. Jasne były także najważniejsze elementy wymagające uwagi: ogólna spójność i zgodność, procedura notyfikacji, akredytacja, procedury oceny zgodności (moduły), oznakowanie CE oraz nadzór rynku (w tym rewizja procedur klauzul ochronnych).
Rozporządzenie i decyzja stanowiące część „pakietu towarowego Ayrala” (16) zostały przyjęte przez Parlament Europejski i Radę dnia 9 lipca 2008 r. (17).
Rozporządzenie (WE) nr 765/2008 i decyzja nr 768/2008/WE skupiły w nowych ramach prawnych wszystkie elementy potrzebne do skutecznego funkcjonowania kompleksowych ram prawnych zapewniających bezpieczeństwo produktów i ich zgodność z wymaganiami przyjętymi w celu ochrony różnych interesów publicznych oraz poprawnego funkcjonowania jednolitego rynku.
Za pomocą rozporządzenia (WE) nr 765/2008 ustalono podstawy prawne akredytacji i nadzoru rynku oraz ujednolicono znaczenie oznakowania CE, a tym samym wypełniono istniejącą lukę. Decyzją nr 768/2008/WE zaktualizowano, zharmonizowano i skonsolidowano różne instrumenty techniczne stosowane już w istniejącym unijnym prawodawstwie harmonizacyjnym (nie tylko w dyrektywach nowego podejścia): definicje, kryteria wyznaczania i notyfikacji jednostek oceniających zgodność, zasady procedury notyfikacji, procedury oceny zgodności (moduły) oraz zasady ich stosowania, mechanizmy ochronne, obowiązki podmiotów gospodarczych oraz wymagania dotyczące identyfikowalności.
Nowe ramy prawne uwzględniają istnienie wszystkich podmiotów gospodarczych w łańcuchu dostaw – producentów, upoważnionych przedstawicieli, dystrybutorów i importerów – oraz odpowiadające im role w odniesieniu do produktu. Obecnie importer ma jasno określone obowiązki dotyczące zapewnienia zgodności produktów z wymogami. Jeśli dystrybutor lub importer modyfikuje produkt lub wprowadza go do obrotu pod własną marką, staje się jego producentem i ma w odniesieniu do produktu obowiązki przypisane do tej roli.
Nowe ramy prawne uwzględniają także różne aspekty obowiązków organów krajowych: organów regulacyjnych, organów ds. notyfikacji, organów nadzorujących krajową jednostkę akredytującą, organów nadzoru rynku, organów odpowiedzialnych za kontrolę produktów pochodzących z państw trzecich itp., i podkreślają jednocześnie, że takie obowiązki zależą od podejmowanych działań.
Za pomocą nowych ram prawnych zmieniono w prawodawstwie UE nacisk na kwestie związane z dostępem do rynku. Wcześniej język unijnego prawodawstwa harmonizacyjnego koncentrował się na „wprowadzaniu do obrotu”, co jest przykładem tradycyjnego języka swobodnego przepływu towarów, tj. skupienia się na pierwszym udostępnieniu produktu na rynku UE. Nowe ramy prawne, uznając istnienie jednolitego rynku wewnętrznego, kładą nacisk na udostępnianie produktu, a przez to większą wagę przywiązują do tego, co dzieje się po wprowadzeniu produktu po raz pierwszy do obrotu. Jest to zgodne również z logiką wprowadzenia przepisów regulujących nadzór rynku UE. Wprowadzenie koncepcji udostępniania ułatwia wsteczne prześledzenie drogi produktu niezgodnego z wymogami do samego producenta. Należy zauważyć, że zgodność jest oceniana w odniesieniu do wymogów prawnych obowiązujących w czasie pierwszego udostępnienia.
Najważniejszą zmianą wprowadzoną przez nowe ramy prawne do otoczenia regulacyjnego UE było wprowadzenie kompleksowej polityki nadzoru rynku. Przesunęła ona znacząco środek ciężkości przepisów prawnych UE: początkowo przepisy koncentrowały się na ustalaniu związanych z produktami wymagań, które należało spełnić w chwili wprowadzania produktów do obrotu, później jednak jednakowy nacisk położono na spełnianie takich wymagań przez cały cykl życia produktów.
1.2.2. CHARAKTER PRAWNY AKTÓW NOWYCH RAM PRAWNYCH I ICH ZWIĄZEK Z INNYMI PRZEPISAMI UE
1.2.2.1. Rozporządzenie (WE) nr 765/2008
Rozporządzenie (WE) nr 765/2008 nakłada jasne obowiązki na państwa członkowskie, które nie muszą transponować jego przepisów (chociaż wiele z nich może wprowadzać środki krajowe, aby dostosować krajowe ramy prawne). Przepisy rozporządzenia mają bezpośrednie zastosowanie w państwach członkowskich względem wszystkich stosownych podmiotów gospodarczych (producentów, dystrybutorów, importerów) oraz jednostek oceniających zgodność i jednostek akredytujących. Podmioty gospodarcze mają teraz nie tylko obowiązki, ale również bezpośrednie prawa, których mogą dochodzić w sądach krajowych zarówno w stosunku do organów krajowych, jak i innych podmiotów gospodarczych, jeśli te nie zastosują się do przepisów rozporządzenia.
Wobec istnienia innych przepisów UE, rozporządzenie ma zastosowanie przede wszystkim a) na podstawie faktu, że stosuje się bezpośrednio, tj. organy krajowe i podmioty gospodarcze muszą stosować przepisy rozporządzenia jako takie (większość innych przepisów jest zawarta w dyrektywach) oraz b) na zasadzie lex specialis, tj. jeśli sprawę regulują dwa przepisy, pierwszeństwo ma przepis bardziej szczegółowy.
W przypadku braku bardziej szczegółowego prawodawstwa w kwestiach regulowanych przepisami rozporządzenia (WE) nr 765/2008 akt ten znajduje zastosowanie jednocześnie z istniejącym prawodawstwem oraz jako jego uzupełnienie. Jeśli istniejące prawodawstwo zawiera podobne przepisy jak wspomniane rozporządzenie, należy zbadać każdy przepis, aby ustalić, który jest najbardziej szczegółowy.
Ogólnie rzecz biorąc, stosunkowo niewiele dokumentów legislacyjnych mówi o akredytacji, można więc stwierdzić, że rozporządzenie (WE) nr 765/2008 ma ogólne zastosowanie w tym obszarze. W obszarze nadzoru rynku (w tym kontroli produktów z państw trzecich) sytuacja jest bardziej złożona, ponieważ w unijnym prawodawstwie harmonizacyjnym istnieją różne przepisy związane z kwestiami objętymi rozporządzeniem (np. prawo dotyczące farmaceutyków i sprzętu medycznego, które przewiduje określoną procedurę informacyjną).
1.2.2.2. Decyzja nr 768/2008/WE
Decyzja nr 768/2008/WE to tzw. decyzja sui generis, co oznacza, że nie ma adresatów i dlatego nie stosuje się ani pośrednio, ani bezpośrednio. Stanowi jedynie polityczne zobowiązanie ze strony trzech instytucji UE: Parlamentu Europejskiego, Rady i Komisji.
Aby takie przepisy znalazły zastosowanie w prawie unijnym, musi pojawić się wyraźne odwołanie do nich w przyszłym prawodawstwie lub muszą zostać do niego włączone.
Wspomniane trzy instytucje w rzeczywistości zobowiązały się jak najczęściej stosować zapisy tej decyzji i odwoływać się do nich podczas tworzenia prawodawstwa dotyczącego produktów. Dlatego też odpowiednie przyszłe projekty należy zbadać w świetle omawianej decyzji, a odstępstwa od jej treści należycie uzasadnić.
1.2.3. DOPASOWANIE ELEMENTÓW SYSTEMU
Rozwój technik legislacyjnych UE w tym obszarze był stopniowy, kwestiami zajmowano się po kolei, chociaż czasami odbywało się to równocześnie, natomiast punkt kulminacyjny nastąpił w chwili przyjęcia nowych ram prawnych: zasadniczych lub innych wymagań prawnych, norm produktowych, norm i reguł dotyczących kompetencji jednostek oceniających zgodność, a także akredytacji, norm zarządzania jakością, procedur oceny zgodności, oznakowania CE, polityki akredytacyjnej i ostatnio polityki nadzoru rynku, w tym kontroli produktów z państw trzecich.
Nowe ramy prawne to kompletny system łączący różne elementy niezbędne w prawodawstwie dotyczącym bezpieczeństwa produktów w jednym spójnym, kompleksowym instrumencie prawnym, który może być stosowany na szeroką skalę we wszystkich sektorach przemysłowych i nie tylko (również polityki środowiskowe i zdrowotne odwołują się do wielu tych elementów) za każdym razem, gdy potrzebne są przepisy unijne.
W systemie tym przepisy prawa muszą ustalać docelowe poziomy ochrony związanej z danymi produktami oraz podstawowe charakterystyki bezpieczeństwa; powinny ustalać obowiązki podmiotów gospodarczych i dotyczące ich wymagania; muszą ustalać – tam, gdzie to konieczne – poziom kompetencji zewnętrznych jednostek oceniających zgodność, które dokonują oceny produktów lub systemów zarządzania jakością, oraz mechanizmy kontrolne dla tych jednostek (notyfikacja i akredytacja); muszą ustalać, które procesy oceny zgodności (moduły, które obejmują również deklarację zgodności producenta) należy stosować i wreszcie muszą przewidywać odpowiednie mechanizmy nadzoru rynku (wewnętrzne i zewnętrzne), aby zagwarantować skuteczne i niezakłócone działanie całego instrumentu legislacyjnego.
Wszystkie te elementy są ze sobą powiązane, funkcjonują razem i wzajemnie się uzupełniają, tworząc łańcuch jakości (18) UE. Jakość produktu zależy od jakości produkcji, na którą w wielu przypadkach wpływ ma jakość badania wewnętrznego lub przeprowadzanego przez zewnętrzne jednostki, która zależy od jakości procesów oceny zgodności, zależnej od jakości jednostek, która z kolei zależy od jakości ich kontroli, która zależy od jakości notyfikacji lub akredytacji; cały system zaś zależy od jakości nadzoru rynku i kontroli produktów z państw trzecich.
Wszystkie te jakości powinny być w ten czy inny sposób uregulowane w odpowiednich przepisach UE dotyczących bezpieczeństwa produktów. Jeśli zabraknie któregokolwiek elementu lub okaże się on słaby, siła i skuteczność całego „łańcucha jakości” jest zagrożona.
1.3. DYREKTYWA W SPRAWIE OGÓLNEGO BEZPIECZEŃSTWA PRODUKTÓW
Celem dyrektywy 2001/95/WE (19) w sprawie ogólnego bezpieczeństwa produktów (GPSD) jest zapewnienie wysokiego poziomu bezpieczeństwa produktów w całej UE w przypadku produktów konsumenckich, których nie obejmuje sektorowe prawodawstwo harmonizacyjne UE. GPSD uzupełnia również w pewnych aspektach przepisy prawodawstwa sektorowego. Kluczowy przepis GPSD stanowi, że producenci są zobowiązani do wprowadzania do obrotu tylko produktów, które są bezpieczne (20). GPSD zawiera również przepisy dotyczące nadzoru rynku mające na celu zagwarantowanie wysokiego poziomu ochrony zdrowia i bezpieczeństwa konsumentów.
Na mocy dyrektywy w sprawie ogólnego bezpieczeństwa produktów utworzono system wczesnego ostrzegania o niebezpiecznych produktach nieżywnościowych (RAPEX – wspólnotowy system szybkiej informacji) stosowany w kontaktach między państwami członkowskimi a Komisją. Wspólnotowy system szybkiej informacji gwarantuje szybkie powiadamianie odpowiednich organów o niebezpiecznych produktach. W określonych okolicznościach powiadomienia tego systemu mogą być również wymieniane z państwami spoza Unii. W przypadku produktów stwarzających poważne zagrożenia dla zdrowia i bezpieczeństwa konsumentów w różnych państwach członkowskich GPSD przewiduje możliwość podejmowania przez Komisję tymczasowych decyzji w sprawie ogólnounijnych środków, tzw. „środków nadzwyczajnych”. W określonych okolicznościach Komisja może przyjąć formalne decyzje (ważne przez rok, ale odnawialne na kolejny rok) nakładające na państwa członkowskie obowiązek ograniczenia lub uniemożliwienia wprowadzenia do obrotu produktu stwarzającego poważne zagrożenie dla zdrowia i bezpieczeństwa konsumentów. Wspólnotowy system szybkiej informacji został następnie rozszerzony rozporządzeniem (WE) nr 765/2008 i stosuje się do wszystkich zharmonizowanych produktów przemysłowych, niezależnie od ich użytkownika końcowego (tj. produktów specjalistycznych) oraz do produktów stwarzających zagrożenia dla innych chronionych interesów niż zdrowie i bezpieczeństwo, np. zagrożenia dla środowiska.
1.4. PRAWODAWSTWO DOTYCZĄCE ODPOWIEDZIALNOŚCI ZA PRODUKT
Koncepcja producenta według unijnego prawodawstwa harmonizacyjnego w kształcie nadanym mu przez nowe ramy prawne różni się od koncepcji przedstawionej w dyrektywie w sprawie odpowiedzialności za produkty wadliwe 85/374/EWG (21). W tym drugim przypadku pojęcie „producent” (22) odnosi się do większej liczby osób niż pojęcie „producent” w nowych ramach prawnych.
Postępowanie prawne lub administracyjne może być wszczęte przeciwko dowolnemu podmiotowi w łańcuchu dostaw lub dystrybucji, który może ponosić odpowiedzialność za niezgodny produkt. Może tak się stać w szczególności wtedy, gdy producent ma swoją siedzibę poza Unią. Dyrektywa w sprawie odpowiedzialności za produkty wadliwe dotyczy wszystkich ruchomości (23) i urządzeń elektrycznych oraz materiałów i komponentów produktów gotowych. Usługi jako takie są aktualnie wyłączone z tego zakresu. Po drugie, dyrektywa ma zastosowanie tylko do produktów wadliwych, tj. produktów niezapewniających takiego poziomu bezpieczeństwa, jakiego można się spodziewać. Nie wystarczy, aby produkt nie był zdatny do wykorzystania w spodziewany sposób. Dyrektywa ma zastosowanie tylko wtedy, gdy produkt nie jest bezpieczny. Stworzenie w późniejszym terminie lepszego produktu nie oznacza, że starsze modele są wadliwe.
Odpowiedzialność, obowiązek zapłaty odszkodowania, spoczywa na producencie. Producent to producent produktu gotowego lub części produktu gotowego, producent surowca lub osoba deklarująca się jako producent (np. poprzez umieszczenie znaku towarowego). Importerzy wprowadzający na rynek unijny produkty z państw trzecich są uważani za producentów w rozumieniu dyrektywy w sprawie odpowiedzialności za produkty wadliwe. Jeśli nie można ustalić producenta, odpowiedzialność ponosi każdy dostawca produktu, chyba że w rozsądnym czasie poinformuje osobę poszkodowaną o tożsamości producenta lub osoby, która dostarczyła mu produkt. Jeżeli za tę samą szkodę odpowiedzialnych jest kilka podmiotów, wszystkie one są odpowiedzialne solidarnie za taką szkodę.
Producent jest zobowiązany do wypłacenia odszkodowania za spowodowane wadliwym produktem szkody osobowe (śmierć, uszczerbek na zdrowiu) i majątkowe (dobra do użytku osobistego). Jednak dyrektywa nie obejmuje szkody majątkowej o wartości poniżej 500 EUR (24) za pojedyncze zdarzenie. Prawo krajowe może regulować szkody niematerialne (takie jak ból i cierpienie). Dyrektywa nie dotyczy zniszczenia samego produktu wadliwego i dlatego nie przewiduje obowiązku wypłaty odszkodowania za takie zdarzenie. Pozostaje to bez uszczerbku dla prawa krajowego.
Dyrektywa w sprawie odpowiedzialności za produkty wadliwe daje państwom członkowskim możliwość ustalenia limitu finansowego dla zdarzeń powtarzających się na poziomie minimum 70 mln euro (25). Jednak większość państw członkowskich nie korzysta z tej możliwości.
Producent nie ponosi automatycznie odpowiedzialności za szkodę spowodowaną produktem. Aby otrzymać odszkodowanie, osoba poszkodowana, bez względu na to, czy jest nabywcą lub użytkownikiem wadliwego produktu, czy nie, musi dochodzić swoich praw. Ofiary otrzymają wypłatę tylko wtedy, gdy udowodnią, że produkt był wadliwy i że to właśnie ten produkt spowodował szkodę. Jeśli osoba poszkodowana przyczyniła się do szkody, odpowiedzialność producenta może być ograniczona lub nawet zniesiona. Jednak ofiary nie muszą udowadniać, że producent zaniedbał swoje obowiązki, ponieważ dyrektywa w sprawie odpowiedzialności za produkty wadliwe jest oparta na zasadzie odpowiedzialności za szkodę bez ustalenia winy. Dlatego producent nie zostanie uniewinniony nawet w przypadku, gdy udowodni, że nie zaniedbał swoich obowiązków, gdy do powstałej szkody przyczyni się działanie lub zaniechanie osoby trzeciej, gdy stosował normy lub gdy produkt podlegał kontroli. Producent nie będzie zobowiązany do wypłaty odszkodowania, jeżeli udowodni, że:
|
— |
nie wprowadził produktu do obrotu (np. skradziono produkt), |
|
— |
produkt nie był wadliwy w chwili wprowadzenia go do obrotu (tj. udowodni, że wada powstała później), |
|
— |
produkt nie został wyprodukowany jako przeznaczony do sprzedaży lub dystrybucji w celach ekonomicznych, |
|
— |
wada powstała w wyniku zastosowania się do obowiązkowych regulacji wydanych przez organy publiczne (co wyłącza normy krajowe, europejskie i międzynarodowe) (26), |
|
— |
stan wiedzy naukowej i technicznej w chwili wprowadzenia produktu do obrotu nie pozwalał na wykrycie wady (obrona wykorzystująca ryzyko związane z rozwojem) (27), lub |
|
— |
jeśli jest podwykonawcą, że wada wynikała z projektu produktu gotowego lub wadliwych instrukcji wydanych mu przez producenta produktu gotowego. |
Odpowiedzialność producenta wygasa po upływie dziesięciu lat od wprowadzenia produktu na rynek, chyba że trwa postępowanie prawne. Co więcej, ofiara musi wnieść pozew w ciągu trzech lat od ujawnienia szkody, wady i tożsamości producenta. Producent nie może zrzec się odpowiedzialności.
Dyrektywa w sprawie odpowiedzialności za produkty wadliwe nie nakłada na państwa członkowskie obowiązku uchylenia żadnych przepisów dotyczących odpowiedzialności. Pod tym względem stanowi uzupełnienie krajowych zasad. To od ofiary zależy wybór podstawy, na jakiej wniesie pozew.
1.5. ZAKRES PRZEWODNIKA
Niniejszy przewodnik dotyczy produktów innych niż żywność i produkty rolne – produktów określanych mianem produktów przemysłowych lub produktów wykorzystywanych zarówno przez konsumentów, jak i profesjonalistów. Prawodawstwo związane z produktami regulujące te produkty będzie nazywane w tekście bez rozróżnienia unijnym prawodawstwem harmonizacyjnym, sektorowym prawodawstwem harmonizacyjnym Unii lub unijnymi aktami harmonizacyjnymi.
Nowe ramy prawne stanowią zestaw dokumentów prawnych. W szczególności decyzja nr 768/2008/WE zawiera elementy, które są częściowo lub całkowicie włączone w unijne prawodawstwo harmonizacyjne regulujące różne interesy publiczne. Przewodnik zawiera wskazówki dotyczące wdrożenia przepisów i koncepcji zawartych w nowych ramach prawnych (28). Jeśli istnieją odstępstwa lub przepisy charakterystyczne dla danego produktu, przewodnik odnosi się do wytycznych sektorowych, które istnieją dla prawie całego sektorowego prawodawstwa harmonizacyjnego Unii.
Niniejszy przewodnik ma na celu szczegółowe wyjaśnienie różnych elementów nowych ram prawnych i przyczyni się do lepszego całościowego zrozumienia systemu, co pozwoli na prawidłowe wdrożenie przepisów prawa i skuteczną ochronę interesów publicznych, takich jak ochrona zdrowia i bezpieczeństwa, konsumentów, środowiska naturalnego oraz odpowiedniego funkcjonowania rynku wewnętrznego podmiotów gospodarczych. Co więcej, przewodnik pomaga w realizacji celów komisyjnej polityki lepszych uregulowań prawnych, ponieważ przyczynia się do rozwoju bardziej kompleksowego, spójnego i proporcjonalnego prawodawstwa.
Każdy z poniższych rozdziałów należy interpretować przy uwzględnieniu wyjaśnień przedstawionych powyżej, innymi słowy w odniesieniu do ogólnej sytuacji i w połączeniu z innymi rozdziałami, ponieważ są one ze sobą powiązane i nie należy ich rozpatrywać osobno.
|
Niniejszy przewodnik odnosi się głównie do unijnych przepisów prawa dotyczących:
|
Jednak części niniejszego przewodnika mogą być istotne dla innego unijnego prawodawstwa harmonizacyjnego wychodzącego również poza zakres produktów przemysłowych. Dzieje się tak szczególnie w przypadku różnych definicji zawartych w niniejszym przewodniku oraz rozdziałów poruszających kwestie normalizacji, oceny zgodności, akredytacji i nadzoru rynku. Chociaż stworzenie wyczerpującej listy stosownych przepisów prawa nie jest ani poprawne, ani pożądane, w załączniku 1 przedstawiono dłuższą listę odpowiednich przepisów prawa.
W przewodniku nie podjęto się uwzględnienia:
|
— |
dyrektywy w sprawie ogólnego bezpieczeństwa produktów (29). Szczegółowe wskazówki dotyczące zastosowania GPSD (30) w praktyce przedstawiły służby Komisji, |
|
— |
unijnego prawodawstwa dotyczącego pojazdów silnikowych, wyrobów budowlanych, REACH i substancji chemicznych. |
2. KIEDY MA ZASTOSOWANIE UNIJNE PRAWODAWSTWO HARMONIZACYJNE?
2.1. ZAKRES PRODUKTÓW
|
Unijne prawodawstwo harmonizacyjne ma zastosowanie w przypadku produktów, które mają być wprowadzone do obrotu (31) (lub oddane do użytku (32)). Co więcej, unijne prawodawstwo harmonizacyjne jest stosowane wtedy, gdy produkt jest wprowadzany do obrotu (lub użytku) i udostępniany do momentu, gdy produkt dotrze do użytkownika końcowego (33) (34) (35). Produkt pozostający w łańcuchu dystrybucyjnym nadal podlega obowiązkom określonym w unijnym prawodawstwie harmonizacyjnym, pod warunkiem, że jest to nowy produkt (36). W chwili dotarcia do użytkownika końcowego produkt przestaje być traktowany jako nowy, a unijne prawodawstwo harmonizacyjne nie ma zastosowania (37). Użytkownik nie jest jednym z podmiotów gospodarczych, które ponoszą odpowiedzialność w ramach unijnego prawodawstwa harmonizacyjnego, tj. żadne działanie lub transakcja dokonane przez użytkownika w związku z produktem nie podlega unijnemu prawodawstwu harmonizacyjnemu. Jednak takie działanie lub transakcja może podlegać innym przepisom prawa, w szczególności na szczeblu krajowym.
Produkt musi spełniać wymogi prawne obowiązujące w chwili wprowadzania go do obrotu (lub oddania do użytku).
Unijne prawodawstwo harmonizacyjne znajduje zastosowanie w odniesieniu do wszystkich form dostawy, w tym sprzedaży na odległość i środków handlu elektronicznego. Stąd bez względu na technikę sprzedaży, produkty przeznaczone do udostępnienia na rynku unijnym muszą spełniać wymogi stosownych przepisów prawa.
Produkt przeznaczony do wprowadzenia na rynek unijny, oferowany w katalogu lub poprzez środki handlu elektronicznego musi być zgodny z unijnym prawodawstwem harmonizacyjnym, jeżeli katalog lub strona internetowa są skierowane na rynek unijny i zawierają system składania zamówień i wysyłki (38). Jeżeli produkt nie jest przeznaczony na rynek unijny lub nie spełnia wymagań stosownych przepisów unijnych, należy to jasno wskazać (np. za pomocą ostrzeżenia graficznego).
Unijne prawodawstwo harmonizacyjne znajduje zastosowanie nie tylko w odniesieniu do nowo wyprodukowanych produktów, ale też do produktów używanych, w tym produktów będących wynikiem przygotowania do ponownego użycia zużytego sprzętu elektrycznego lub elektronicznego, importowanych z państwa trzeciego w chwili, gdy po raz pierwszy są wprowadzane na rynek unijny (39) (40). Dotyczy to również produktów używanych importowanych z państw trzecich, które zostały wytworzone przed wejściem w życie stosownych przepisów prawa (41).
Unijne prawodawstwo harmonizacyjne dotyczy produktów gotowych. Jednak koncepcja produktu różni się w poszczególnych elementach unijnego prawodawstwa harmonizacyjnego. Przedmioty objęte przepisami prawa są określane np. jako produkty, urządzenia, aparaty, wyposażenie, sprzęt, przyrządy, materiały, zespoły, komponenty lub elementy bezpieczeństwa, części, osprzęt, akcesoria, systemy lub częściowo ukończone maszyny. Dlatego w rozumieniu danego unijnego aktu harmonizacyjnego komponenty, części zamienne lub podzespoły mogą być uznawane za produkty gotowe, a ich końcowym zastosowaniem może być montaż lub włączenie w produkt gotowy. Obowiązek zweryfikowania, czy dany produkt mieści się w zakresie danego aktu unijnego prawodawstwa harmonizacyjnego, leży po stronie producenta (42) (43).
Połączenie produktów i części, z których każdy(-a) spełnia wymogi odpowiednich przepisów prawa, nie zawsze stanowi produkt gotowy, który jako całość musi być zgodny z unijnym prawodawstwem harmonizacyjnym. Jednak w niektórych przypadkach połączenie różnych produktów i części zaprojektowane lub złożone przez tę samą osobę jest uważane za jeden produkt gotowy, który, jako taki, musi być zgodny z przepisami. W szczególności producent takiego połączenia jest odpowiedzialny za wybór odpowiednich produktów tworzących połączenie, za złożenie części w sposób zgodny z odpowiednimi przepisami prawa i za spełnienie wymogów prawnych związanych z montażem, deklarację zgodności UE i oznakowanie CE. Fakt, że komponenty lub części posiadają znak CE nie oznacza automatycznie, że produkt gotowy jest również zgodny z przepisami. Producenci muszą wybrać komponenty i części w taki sposób, aby produkt gotowy był także zgodny z przepisami. Producent musi w każdym przypadku sprawdzić, czy daną kombinację produktów i części należy uznać za jeden produkt gotowy objęty zakresem stosowania odpowiednich przepisów.
Produkt, w którym po oddaniu do użytku dokonano poważnych zmian lub napraw w celu zmodyfikowania oryginalnego działania, zastosowania lub typu, mających istotny wpływ na jego zgodność z unijnym prawodawstwem harmonizacyjnym, musi zostać uznany za nowy produkt. Oceny należy dokonywać w odniesieniu do poszczególnych przypadków, w szczególności biorąc pod uwagę cel przepisów prawa i typ produktów objętych omawianymi przepisami prawa. Jeżeli przebudowany (44) lub zmodyfikowany produkt zostanie uznany za nowy produkt, w chwili udostępnienia lub oddania do użytku musi on być zgodny z odpowiednimi przepisami prawa. Należy zweryfikować tę zgodność za pomocą odpowiedniej procedury oceny zgodności określonej w omawianych przepisach. W szczególności, jeżeli ocena ryzyka prowadzi do wniosku, że zmienił się charakter zagrożenia lub wzrósł poziom ryzyka, zmodyfikowany produkt należy uznać za nowy produkt, tj. należy na nowo ocenić zgodność zmodyfikowanego produktu ze stosownymi zasadniczymi wymaganiami, a osoba przeprowadzająca modyfikację musi spełnić takie same wymogi, jak oryginalny producent – np. należy przygotować dokumentację produktu, sporządzić deklarację zgodności UE i umieścić oznakowanie CE na produkcie.
Za każdym razem zmodyfikowany produkt sprzedawany pod nazwą lub znakiem towarowym osoby fizycznej lub prawnej innej niż oryginalny producent należy traktować jak nowy produkt podlegający unijnemu prawodawstwu harmonizacyjnemu. Osoby, które dokonują poważnych zmian w produkcie, mają obowiązek zweryfikować, czy taki produkt należy uznać za nowy produkt w odniesieniu do stosownego unijnego prawodawstwa harmonizacyjnego. Jeżeli tak, osoba dokonująca zmian staje się producentem i ma związane z tym obowiązki. Co więcej, w przypadku gdy taki produkt zostanie uznany za nowy, musi zostać poddany pełnej ocenie zgodności zanim zostanie udostępniony na rynku. Jednak dokumentacja produktu musi zostać zaktualizowana w zakresie, w jakim zmiana ma wpływ na wymogi odpowiednich przepisów prawa. Nie jest wymagane powtarzanie testów i tworzenie nowej dokumentacji w odniesieniu do aspektów, na które zmiana nie miała wpływu, pod warunkiem że producent posiada kopie (lub dostęp do kopii) oryginalnych sprawozdań z badań dotyczących niezmienionych aspektów. Osoba fizyczna lub prawna, która dokonuje lub dokonała zmian w produkcie, musi wykazać, że nie wszystkie elementy dokumentacji produktu wymagają aktualizacji.
Produktów, które zostały naprawione lub wymienione (np. w wyniku wykrycia wady) w sposób niezmieniający oryginalnego działania, przeznaczenia lub typu, nie uznaje się za nowe produkty w rozumieniu unijnego prawodawstwa harmonizacyjnego. Dlatego też takie produkty nie muszą być na nowo poddawane ocenie zgodności, bez względu na to, czy oryginalny produkt został wprowadzony do obrotu przed wejściem w życie przepisów czy później. Dzieje się tak nawet wtedy, gdy produkt zostanie wyeksportowany do państw trzecich na czas naprawy. Najczęściej polega ona na wymianie wadliwej lub zużytej części na część zapasową, która jest albo identyczna, albo przynajmniej podobna do części oryginalnej (np. zmiany mogły wyniknąć z postępu technicznego lub zakończenia produkcji starej części), czyli na wymianie kart, komponentów, podzespołów lub nawet całych identycznych zespołów. Jeżeli pierwotny sposób działania produktu zostało zmodyfikowany (w ramach planowanego wykorzystania, zakresu działania i utrzymania pierwotnie przewidzianych na etapie projektu), ponieważ części zamienne wykorzystane do naprawy działają lepiej dzięki postępowi technicznemu, produktu tego nie uznaje się za nowy zgodnie z unijnym prawodawstwem harmonizacyjnym. Dlatego też działania konserwacyjne są zasadniczo wyłączone z zakresu unijnego prawodawstwa harmonizacyjnego. Jednak na poziomie projektowania produktu należy wziąć pod uwagę jego przeznaczenie i konserwację (45).
Aktualizacje lub naprawy oprogramowania można zaliczyć do działań konserwacyjnych, pod warunkiem że w ich wyniku produkt już wprowadzony do obrotu nie zostanie zmieniony w sposób naruszający zgodność ze stosownymi wymogami prawa.
2.2. UDOSTĘPNIENIE NA RYNKU
|
Produkt zostaje udostępniony na rynku, gdy zostanie dostarczony, bezpłatnie lub za opłatą, do dystrybucji, konsumpcji lub użytku na rynku unijnym w ramach działalności handlowej (46). Dostawa obejmuje wszelkie oferty dystrybucji, konsumpcji lub użytku na rynku unijnym, które mogą skutkować rzeczywistą dostawą (np. zaproszenie do kupna, kampanie reklamowe).
Dostarczanie produktu uznaje się za udostępnianie na rynku unijnym tylko wtedy, gdy produkt jest przeznaczony do użytku na rynku unijnym. Dostarczanie produktów, czy to do dalszej dystrybucji, włączenia do produktu gotowego, czy do dalszego przetwarzania lub rafinowania w zamiarem wyeksportowania produktu gotowego poza Unię, nie jest uznawane za udostępnianie. Działalność handlowa jest rozumiana jako dostarczanie towarów w kontekście biznesowym. Można uznać, że organizacje non-profit prowadzą działalność handlową, jeżeli działają w takim kontekście. Ustalenie tego wymaga zbadania każdego konkretnego przypadku przy uwzględnieniu regularności dostaw, charakterystyki produktu, intencji dostawcy itp. Zasadniczo uznaje się, że okazjonalne dostawy dokonywane przez organizacje charytatywne lub hobbystów nie odbywają się w kontekście biznesowym.
„Użytek” odnosi się do stosowania produktu zgodnie z określonym przez producenta przeznaczeniem w warunkach, które można przewidzieć z dużym prawdopodobieństwem. Najczęściej jest to końcowe użytkowanie produktu.
Główna rola, jaką koncepcja udostępniania odgrywa w unijnym prawodawstwie harmonizacyjnym, wiąże się z faktem, że na wszystkie podmioty gospodarcze w łańcuchu dostaw nałożony jest obowiązek identyfikowalności i zagwarantowania, że tylko produkty zgodne z przepisami prawa będą wprowadzone na rynek unijny.
Udostępnianie odnosi się do każdego egzemplarz produktu, a nie typu produktu czy tego, czy został wyprodukowany jako pojedynczy egzemplarz czy w ramach serii.
Udostępnianie produktu zakłada ofertę lub umowę (pisemną lub ustną) pomiędzy dwoma lub kilkoma osobami prawnymi lub fizycznymi w zakresie przeniesienia tytułu własności, posiadania lub innego prawa (47) związanego z danym produktem po zakończeniu etapu produkcji. Przeniesienie niekoniecznie wymaga fizycznego przekazania produktu.
Może odbyć się za opłatą lub bezpłatnie i może opierać się na dowolnym instrumencie prawnym. Stąd też uznaje się, że doszło do przeniesienia własności produktu np. w sytuacji sprzedaży, pożyczki, wynajmu (48), leasingu i prezentu. Przeniesienie własności oznacza, że produkt został przekazany do dyspozycji innej osoby prawnej lub fizycznej.
2.3. WPROWADZENIE DO OBROTU
|
W rozumieniu unijnego prawodawstwa harmonizacyjnego produkt zostaje wprowadzony do obrotu w chwili jego pierwszego udostępnienia na rynku unijnym. Działanie to jest zarezerwowane dla producenta lub importera, tj. producent lub importer to jedyne podmioty gospodarcze, które wprowadzają produkty do obrotu (49). Kiedy producent lub importer po raz pierwszy dostarcza produkt do dystrybutora (50) lub użytkownika, takie działanie jest zawsze nazywane w kontekście prawnym „wprowadzeniem do obrotu”. Wszelkie późniejsze operacje, np. pomiędzy dystrybutorami lub między dystrybutorem a użytkownikiem, są określane jako udostępnianie.
Jeśli chodzi o udostępnianie, koncepcja wprowadzania na rynek odnosi się do każdego egzemplarza produktu, a nie typu produktu czy tego, czy został wyprodukowany jako pojedynczy egzemplarz czy w ramach serii. W konsekwencji nawet jeśli model lub typ produktu został dostarczony przed wejściem w życie nowego unijnego prawodawstwa harmonizacyjnego określającego nowe obowiązkowe wymagania, poszczególne egzemplarze tego samego modelu lub typu, które są wprowadzane na rynek po wprowadzeniu nowych wymagań, muszą być zgodne z takimi wymaganiami.
Wprowadzenie produktu do obrotu wymaga złożenia oferty zawarcia lub umowy (pisemnej lub ustnej) pomiędzy dwoma lub kilkoma osobami prawnymi lub fizycznymi w zakresie przeniesienia tytułu własności, posiadania lub innego prawa własności związanego z danym produktem po zakończeniu etapu produkcji (51). Takie przeniesienie może odbyć się za opłatą lub bezpłatnie. Nie wymaga fizycznego przekazania produktu.
Uznaje się, że nie doszło do wprowadzenia do obrotu, jeżeli produkt jest:
|
— |
produkowany na własny użytek. Jednak część unijnego prawodawstwa harmonizacyjnego obejmuje swym zakresem produkty wytwarzane na własny użytek (52) (53), |
|
— |
nabyty przez konsumenta w państwie trzecim podczas fizycznej obecności w tym państwie (54) i przywieziony przez tego konsumenta do UE do osobistego użytku tej osoby, |
|
— |
przekazywany od producenta w państwie trzecim do upoważnionego przedstawiciela w Unii, którego producent zaangażował w celu zapewnienia zgodności produktu z unijnym prawodawstwem harmonizacyjnym (55), |
|
— |
sprowadzony z państwa trzeciego na obszar celny UE w tranzycie, umieszczony w wolnych obszarach celnych, magazynach, miejscach czasowego składowania towarów lub w ramach innych specjalnych procedur celnych (odprawa czasowa lub uszlachetnianie czynne) (56), |
|
— |
produkowany w państwie członkowskim z zamiarem wyeksportowania go do państwa trzeciego (obejmuje to komponenty dostarczane producentowi do włączenia do produktu gotowego, który będzie eksportowany do państwa trzeciego), |
|
— |
przewieziony w celu badania lub walidacji prefabrykowanych zespołów, w przypadku których uznaje się, że nadal znajdują się w fazie produkcji, |
|
— |
pokazywany lub użytkowany w kontrolowanych warunkach (57) na targach, pokazach lub w celach demonstracyjnych (58), lub |
|
— |
w magazynie producenta (lub upoważnionego przedstawiciela mającego swoją siedzibę w Unii) lub importera, jeśli produkt nie został jeszcze udostępniony, tj. jeżeli nie jest dostarczany do dystrybucji, konsumpcji lub użycia, chyba że odpowiednie unijne prawodawstwo harmonizacyjne przewiduje inaczej. |
Produkty oferowane do sprzedaży przez podmioty internetowe (59) (60) mające siedzibę w UE uważa się za wprowadzone do obrotu w Unii, niezależnie od tego, kto wprowadził je do obrotu (operator internetowy, importer itp.). Produkty oferowane on-line przez sprzedawców mających siedzibę poza UE są uważane za wprowadzane do obrotu w Unii w przypadku gdy sprzedaż ukierunkowana jest na konsumentów i innych użytkowników końcowych z UE. Ocenę, czy strona internetowa prowadzona w UE lub poza UE skierowana jest do unijnych konsumentów, należy przeprowadzić indywidualnie dla każdego przypadku, biorąc pod uwagę wszelkie stosowne czynniki, takie jak obszary geograficzne, do których wysyłka jest możliwa, języki, w jakich przedstawiona jest oferta i w jakich można składać zamówienie, możliwe formy płatności, itd. (61) Jeżeli podmiot internetowy dostarcza produkty w UE, przyjmuje płatności od konsumentów/użytkowników końcowych z UE i używa języków unijnych, można uznać, że jednoznacznie kieruje swoją ofertę do konsumentów i innych użytkowników z Unii Europejskiej. Podmioty internetowe mogą oferować internetową sprzedaż danego rodzaju produktu lub pojedynczych egzemplarzy produktów, które zostały już wytworzone. Jeżeli oferta dotyczy rodzaju produktu, wprowadzenie do obrotu nastąpi dopiero po zakończeniu etapu produkcji.
Ponieważ produkty wystawione na sprzedaż przez podmiot internetowy prawdopodobnie zostaną (lub już zostały) zamówione przez konsumentów lub przedsiębiorstwa w UE, są one dostarczane w kontekście działalności handlowej w drodze handlu elektronicznego. Produkty w sprzedaży internetowej są na ogół oferowane odpłatnie. Niemniej jednak bezpłatne dostarczanie produktów również może stanowić działalność komercyjną (62). Jeśli chodzi o sprzedaż między konsumentami, zasadniczo nie jest ona uznawana za działalność handlową. Niemniej jednak ocenę, czy dany produkt będący przedmiotem takiej sprzedaży jest dostarczany w ramach działalności handlowej, należy przeprowadzać indywidualnie dla każdego przypadku z uwzględnieniem wszelkich stosownych kryteriów, takich jak regularność dostaw, intencje dostawcy itp. (63).
Prawnym skutkiem powyższego jest to, że produkty wystawiane na sprzedaż przez podmioty internetowe w momencie wprowadzania do obrotu muszą być zgodne ze wszystkimi mającymi zastosowanie przepisami unijnymi (64). Taką zgodność z przepisami właściwe organy mogą fizycznie zweryfikować (jeżeli dane produkty są w ich jurysdykcji) najwcześniej na etapie kontroli celnej.
Ponadto produkty oferowane przez podmioty internetowe są na ogół przechowywane w centrach realizacji zamówień znajdujących się w UE w celu zapewnienia ich szybkiej dostawy do konsumentów w UE. W związku z tym produkty przechowywane w takich centrach uważa się za dostarczone w celu dystrybucji, konsumpcji lub użytkowania na rynku unijnym, a zatem wprowadzone do obrotu w UE. Jeżeli podmiot internetowy korzysta z centrum realizacji zamówień, wysyłka produktów do takiego centrum w UE stanowi etap dystrybucji produktów w łańcuchu dostaw (65).
Wprowadzenie do obrotu jest najważniejszym momentem decydującym o zastosowaniu unijnego prawodawstwa harmonizacyjnego (66). W chwili udostępnienia na rynku produkty muszą być zgodne z unijnym prawodawstwem harmonizacyjnym obowiązującym w chwili wprowadzenia ich do obrotu. W związku z powyższym nowe produkty wytworzone w Unii oraz wszystkie produkty importowane z państw trzecich – czy to nowe, czy używane – muszą spełniać stosowne przepisy unijnego prawodawstwa harmonizacyjnego w chwili udostępniania ich na rynku unijnym. Zgodne z przepisami produkty po wprowadzeniu do obrotu mogą być następnie udostępniane w całym łańcuchu dostawy bez dodatkowych uwag, nawet w przypadku zmian w stosownych przepisach prawa lub odpowiednich normach zharmonizowanych, o ile przepisy nie stanowią inaczej.
W ramach nadzoru rynku państwa członkowskie mają obowiązek zagwarantować, że tylko bezpieczne i zgodne z wymogami produkty będą dostępne na rynku (67). Produkty używane, które są dostępne na rynku unijnym, podlegają swobodnemu przepływowi zgodnie z zasadami określonymi w art. 34 i 36 TFUE. Należy odnotować, że produkty używane udostępniane konsumentom w ramach działalności handlowej podlegają przepisom GPSD.
2.4. PRODUKTY IMPORTOWANE Z PAŃSTW SPOZA UE
|
Unijne prawodawstwo harmonizacyjne ma zastosowanie wtedy, gdy produkt jest udostępniany (lub oddawany do użytku (68)) na rynku unijnym po raz pierwszy. Ma ono także zastosowanie do produktów używanych importowanych z państwa trzeciego – w tym produktów będących wynikiem przygotowania do ponownego użycia zużytego sprzętu elektrycznego lub elektronicznego – w chwili, gdy po raz pierwszy są wprowadzane na rynek unijny, ale nie ma zastosowania w przypadku takich produktów obecnych już na rynku. Dotyczy ono również produktów używanych importowanych z państw trzecich, które zostały wyprodukowane przed wejściem w życie stosownego unijnego prawodawstwa harmonizacyjnego.
Podstawową zasadą przepisów dotyczących produktów w Unii Europejskiej jest to, że, bez względu na swoje pochodzenie, produkty muszą być zgodne ze stosownym unijnym prawodawstwem harmonizacyjnym, jeżeli są udostępniane na rynku unijnym. Produkty wytworzone w UE oraz produkty spoza UE są traktowane tak samo.
Produkty pochodzące z spoza UE, zanim będą mogły dotrzeć do użytkowników końcowych w UE, zostaną przedstawione do odprawy celnej w ramach procedury dopuszczenia do swobodnego obrotu. Celem dopuszczenia do swobodnego obrotu jest wypełnienie wszystkich formalności przywozowych, tak aby towary mogły zostać udostępnione na rynku unijnym jak każdy inny produkt wytworzony w UE. Zatem gdy produkty są przedstawiane do odprawy celnej w ramach procedury dopuszczenia do swobodnego obrotu, można zasadniczo uznać, że towary te są wprowadzane do obrotu w UE i dlatego muszą być zgodne z mającymi zastosowanie wymogami unijnego prawodawstwa harmonizacyjnego. Niemniej jednak możliwe jest również, że dopuszczenie do swobodnego obrotu i wprowadzenie do obrotu nie odbywają się równocześnie. Wprowadzeniem do obrotu jest moment, w którym produkt został przekazany do dystrybucji, konsumpcji lub użytkowania do celów zapewnienia zgodności z unijnym prawodawstwem harmonizacyjnym. Wprowadzenie do obrotu może odbyć się przed dopuszczeniem do swobodnego obrotu, na przykład w przypadku sprzedaży przez internet przez podmioty gospodarcze spoza UE, nawet jeżeli fizyczna kontrola zgodności produktów z wymogami może się odbyć najwcześniej w momencie przybycia towarów do odprawy celnej w UE. Wprowadzenie do obrotu może nastąpić również po dopuszczeniu do swobodnego obrotu.
Organy celne i organy nadzoru rynku są zobowiązane i uprawnione do przeprowadzania, na podstawie analizy ryzyka, kontroli produktów przywożonych z państw trzecich i odpowiedniej interwencji przed ich dopuszczeniem do swobodnego obrotu, niezależnie od tego, kiedy zostały one uznane za wprowadzone do obrotu w Unii. Ma to zapobiec dopuszczaniu do swobodnego obrotu, a tym samym udostępnianiu na terytorium UE produktów, które nie spełniają wymogów stosownych przepisów unijnego prawodawstwa harmonizacyjnego (69).
W przypadku produktów importowanych spoza państw UE unijne prawodawstwo harmonizacyjne przewiduje specjalną rolę importera. Importer przejmuje obowiązki, które do pewnego stopnia odzwierciedlają obowiązki producentów z UE (70).
W przypadku produktów importowanych z państw spoza Unii Europejskiej upoważniony przedstawiciel może wypełniać pewne obowiązki w imieniu producenta (71). Jednak jeżeli upoważniony przedstawiciel producenta z państwa trzeciego dostarcza produkt do dystrybutora lub konsumenta w UE, nie występuje wtedy jedynie jako upoważniony przedstawiciel, ale staje się importerem i podlega obowiązkom importerów.
2.5. ODDAWANIE DO UŻYTKU (I INSTALACJA)
|
Oddanie do użytku następuje w chwili pierwszego użycia produktu przez użytkownika w Unii w celu, do którego produkt został przeznaczony (72) (73). Pojęcie to, oprócz pojęcia wprowadzenia do obrotu, jest stosowane np. w odniesieniu do dźwigów, maszyn, urządzeń radiowych, przyrządów pomiarowych, wyrobów medycznych, wyrobów do diagnostyki in vitro lub produktów objętych dyrektywami EMC lub ATEX, skutkiem czego jest rozszerzenie zakresu unijnego prawodawstwa harmonizacyjnego poza moment udostępnienia produktu (74).
W sytuacji gdy produkt jest oddawany przez pracodawcę do użytku jego pracowników, za użytkownika końcowego uznaje się pracodawcę.
Państwa członkowskie nie mogą zakazać, ograniczyć lub utrudniać oddawania do użytku produktów, które spełniają wymogi stosownego unijnego prawodawstwa harmonizacyjnego (75). Jednak zgodnie z Traktatem (w szczególności art. 34 i 36 TFUE) i z zastrzeżeniem przepisów unijnego prawodawstwa harmonizacyjnego mogą utrzymać w mocy i przyjąć dodatkowe przepisy krajowe dotyczące oddawania do użytku, instalacji lub użytkowania produktów przeznaczonych do ochrony pracowników i innych użytkowników albo innych produktów. Takie przepisy krajowe nie mogą wymagać zmian w produkcie wytworzonym zgodnie z przepisami stosownego unijnego prawodawstwa harmonizacyjnego.
Potrzeba wykazania zgodności produktów z wymogami w momencie oddania do użytku i – w stosownych przypadkach – ich poprawnej instalacji, konserwacji i użytkowania zgodnie z założonymi celami powinna ograniczać się do produktów:
|
— |
które nie zostały wprowadzone do obrotu przed ich oddaniem do użytku, lub które mogą być wykorzystane dopiero po przeprowadzeniu montażu, instalacji lub innych czynności, lub |
|
— |
na których zgodność z wymogami wpływ mogą mieć warunki dystrybucji (np. magazynowanie lub transport). |
2.6. RÓWNOCZESNE STOSOWANIE UNIJNYCH AKTÓW HARMONIZACYJNYCH
|
Unijne prawodawstwo harmonizacyjne obejmuje szeroki zakres produktów, zagrożeń i oddziaływań (76), które zarówno pokrywają się, jak i uzupełniają. W konsekwencji w przypadku jednego produktu może zaistnieć potrzeba uwzględnienia kilku dokumentów prawnych, ponieważ udostępnianie lub oddawanie do użytku może nastąpić tylko wtedy, gdy produkt jest zgodny z wszystkimi stosownymi przepisami i gdy zostanie przeprowadzona ocena zgodności według wszystkich stosownych przepisów unijnego prawodawstwa harmonizacyjnego.
Zagrożenia regulowane wymogami różnych unijnych aktów harmonizacyjnych są przeważnie związane z różnymi aspektami produktu, które w wielu przypadkach uzupełniają się nawzajem (np. dyrektywy dotyczące kompatybilności elektromagnetycznej i urządzeń ciśnieniowych obejmują aspekty nieuregulowane dyrektywami dotyczącymi sprzętu niskonapięciowego lub maszyn). Taka sytuacja wymaga równoczesnego zastosowania różnych aktów prawnych. Dlatego też produkt musi być zaprojektowany i wyprodukowany zgodnie ze wszystkimi stosownymi przepisami unijnego prawodawstwa harmonizacyjnego i musi przejść procedurę oceny zgodności zgodnie ze wszystkimi stosownymi przepisami prawa, chyba że przepisy stanowią inaczej.
Pewne unijne akty harmonizacyjne wyłączają ze swojego zakresu produkty objęte innymi aktami (77) lub włączają zasadnicze wymagania określone w innych aktach (78), co pozwala na uniknięcie równoczesnego stosowania pokrywających się wymagań. W innych przypadkach tak się nie dzieje i jeśli wymogi unijnych aktów harmonizacyjnych wzajemnie się uzupełniają, nadal stosuje się ogólne zasady równoczesnego zastosowania.
Ten sam produkt, zagrożenie lub oddziaływanie może być regulowany dwoma lub więcej unijnymi aktami harmonizacyjnymi. W takim przypadku kwestia pokrywania się zakresu aktów prawnych może zostać rozwiązana dzięki zastosowaniu w pierwszej kolejności bardziej szczegółowego unijnego aktu harmonizacyjnego (79). Wymaga to często analizy ryzyka związanego z produktem lub czasem analizy przeznaczenia produktu, w wyniku której można następnie ustalić odpowiednie przepisy prawne. Podczas ustalania niebezpieczeństw związanych z produktem producent może wykorzystać odpowiednie mające zastosowanie normy zharmonizowane związane z danym produktem.
2.7. UŻYTKOWANIE ZGODNE Z PRZEZNACZENIEM/NIEWŁAŚCIWE UŻYTKOWANIE
|
Producenci muszą dopasować poziom ochrony do zalecanego przez siebie sposobu użytkowania produktu w warunkach, które można z dużym prawdopodobieństwem przewidzieć. |
Unijne prawodawstwo harmonizacyjne ma zastosowanie w przypadku, gdy produkty udostępniane lub oddawane do użytku (80) na rynku unijnym są stosowane zgodnie z przeznaczeniem. Użytkowanie zgodne z przeznaczeniem oznacza użytkowanie, do którego produkt jest przeznaczony zgodnie z informacjami przedstawionymi przez osobę wprowadzającą go do obrotu albo zwykłe użytkowanie wynikające z projektu i konstrukcji produktu.
Często takie produkty są gotowe do użytku lub wymagają tylko dostrojenia do docelowego użycia. Produkty są „gotowe do użytku”, jeżeli można ich używać bez dodatkowych części. Produkty są również uważane za gotowe do użytku, jeśli wszystkie części, z których mają być zmontowane, są wprowadzone do obrotu przez tylko jedną osobę lub trzeba je jedynie złożyć lub podłączyć albo są wprowadzone do obrotu bez części, które są często dostarczane osobno i dodawane w celu użytkowania zgodnego z przeznaczeniem (np. kabel zasilający).
Producenci są zobowiązani do dopasowania poziomu ochrony użytkowników produktu do przeznaczenia określonego w informacji o produkcie. Jest to szczególnie ważne wtedy, gdy może dojść do niewłaściwego wykorzystania produktu (81).
Jeśli chodzi o działania w zakresie nadzoru rynku, organy nadzoru rynku są zobowiązane do sprawdzania zgodności produktu:
|
— |
z przeznaczeniem (ustalonym przez producenta) oraz |
|
— |
z przepisami w warunkach użytkowania, które można racjonalnie przewidzieć, tj. gdy taki sposób użytkowania może być wynikiem zgodnego z prawem i łatwo przewidywalnego zachowania ludzkiego. |
Konsekwencją dla producentów jest to, że muszą wziąć pod uwagę warunki użytkowania, które mogą być racjonalnie przewidziane przed wprowadzeniem produktu do obrotu.
Producenci muszą umieć przewidzieć nie tylko to, co uważają za użytkowanie produktu zgodnie z przeznaczeniem, lecz postawić się w sytuacji przeciętnego użytkownika danego produktu i wyobrazić sobie, w jaki sposób prawdopodobnie używaliby tego produktu (82).
Ważne jest również, aby organy nadzoru rynku brały pod uwagę, że nie wszystkich zagrożeń można uniknąć, odpowiednio projektując produkt. Nadzorowanie docelowych użytkownikami i zapewnienie im pomocy należy uznać za część warunków, które można racjonalnie przewidzieć. Na przykład niektóre profesjonalne narzędzia mechaniczne mogą być używane pod nadzorem pracodawcy przez pracowników posiadających przeciętne umiejętności i przeszkolenie. Producent nie ponosi odpowiedzialności, jeżeli takie narzędzia są wynajmowane przez dystrybutora lub zewnętrznego usługodawcę do użytku przez konsumentów nieposiadających odpowiednich umiejętności lub przeszkolenia.
W każdym razie producent nie musi zakładać, że użytkownicy będą korzystać z produktu niezgodnie z określonymi warunkami.
2.8. ZASTOSOWANIE GEOGRAFICZNE (PAŃSTWA EOG, EFTA, KRAJE I TERYTORIA ZAMORSKIE, TURCJA)
|
2.8.1. PAŃSTWA CZŁONKOWSKIE ORAZ KRAJE I TERYTORIA ZAMORSKIE
Celem unijnego prawodawstwa harmonizacyjnego odnoszącego się do towarów, przyjętego zgodnie z art. 114 i 115 TFUE, jest ustanowienie i zapewnienie funkcjonowania rynku wewnętrznego towarów. W konsekwencji unijne prawodawstwo harmonizacyjne nie może zostać oddzielone od przepisów Traktatu dotyczących swobodnego przepływu towarów, a terytorialny zakres zastosowania takiego prawodawstwa powinien być zbieżny z terytorialnym zakresem zastosowania art. 30 oraz 34 do 36 TFUE.
Zgodnie z art. 355 TFUE i w związku z art. 52 Traktatu o Unii Europejskiej, Traktat, a co za tym idzie unijne prawodawstwo harmonizacyjne, ma zastosowanie do państw członkowskich Unii Europejskiej. Zgodnie z art. 355 ust. 1 TFUE dotyczy to także Gwadelupy, Gujany Francuskiej, Martyniki, Réunion, Saint-Martin, Azorów, Madery i Wysp Kanaryjskich. Ponadto Traktat i prawodawstwo harmonizacyjne związane z produktami przyjętymi na podstawie art. 114 i 115 TFUE znajdują zastosowanie w przypadku określonych terytoriów europejskich w zakresie niezbędnym do zrealizowania ustaleń określonych w stosownym traktacie akcesyjnym (83).
Jednak nie dotyczy to ani Wysp Owczych, Grenlandii, Akrotiri i Dhekelii, ani krajów i terytoriów zamorskich mających szczególne stosunki ze Zjednoczonym Królestwem Wielkiej Brytanii i Irlandii Północnej, takich jak Gibraltar. Unijne prawodawstwo harmonizacyjne nie ma zastosowania do krajów i terytoriów zamorskich, w szczególności: Nowej Kaledonii i terytoriów zależnych, Polinezji Francuskiej, Francuskich Terytoriów Południowych i Antarktycznych, Wysp Wallis i Futuna, Saint Pierre i Miquelon, Aruba, Curaçao, Sint Maarten, Holandii Karaibskiej (Bonaire, Saba, Sint Eustatius), Anguilli, Kajmanów, Falklandów, Georgii Południowej i Sandwicha Południowego, Montserrat, Pitcairn, Wyspy Świętej Heleny i terytoriów zależnych, Brytyjskiego Terytorium Antarktycznego, Brytyjskiego Terytorium Oceanu Indyjskiego, Wysp Turks i Caicos, Brytyjskich Wysp Dziewiczych, Bermudów.
2.8.2. PAŃSTWA EOG I EFTA
2.8.2.1. Podstawowe aspekty Porozumienia o Europejskim Obszarze Gospodarczym
Porozumienie o Europejskim Obszarze Gospodarczym obowiązujące od 1 stycznia 1994 r. obejmuje wszystkie przepisy unijnego prawodawstwa harmonizacyjnego, do których odnosi się niniejszy przewodnik. Stąd też unijne prawodawstwo harmonizacyjne omówione w niniejszym przewodniku ma zastosowanie również w przypadku tzw. państw EOG EFTA: Islandii, Liechtensteinu i Norwegii.
Celem Porozumienia EOG jest ustanowienie dynamicznego i jednolitego Europejskiego Obszaru Gospodarczego opartego na wspólnych zasadach i równej konkurencji.
Prawa przyznane państwom członkowskim, ich publicznym podmiotom, przedsiębiorstwom lub osobom prywatnym oraz nałożone na nie zobowiązania wobec siebie nawzajem są, zgodnie z Porozumieniem EOG, w ten sam sposób przyznane lub nałożone na państwa EOG EFTA. Dzięki temu państwa EOG EFTA i ich podmioty gospodarcze mają te same prawa i obowiązki, co ich unijne odpowiedniki. Na przykład dyrektywy nowego podejścia i inne unijne przepisy harmonizacyjne są wdrażane i stosowane w państwach EOG EFTA dokładnie w ten sam sposób, jak w państwach członkowskich – chociaż klauzula ochronna jest modyfikowana. Dlatego też wszystkie wskazówki dotyczące państw członkowskich mają również zastosowanie w przypadku państw EOG EFTA.
Do celów Porozumienia EOG odniesienia do rynku wspólnotowego (lub unijnego) lub wspólnego rynku w aktach UE/EOG należy rozumieć jako odniesienia do terytoriów umawiających się stron. Zgodnie z powyższym produkt nie jest wprowadzany na rynek unijny, ale na rynek EOG (tj. rynki krajowe państw członkowskich oraz Islandii, Liechtensteinu i Norwegii).
Porozumienie EOG jest stale zmieniane decyzjami Wspólnego Komitetu EOG, wynikającymi ze zmian w odpowiednim prawodawstwie unijnym. W celu ustalenia i utrzymania jednolitej interpretacji i zastosowania porozumienia ustanowiono Trybunał EFTA oraz Urząd Nadzoru EFTA.
Porozumienie EOG gwarantuje bliską współpracę pomiędzy Komisją a administracją państw EOG–EFTA. Komisja korzysta z nieformalnego doradztwa ekspertów z tych państw, podobnie jak ekspertów z państw członkowskich. Zacieśniono współpracę z komitetami wspierającymi Komisję w jej pracach. Rada EOG spotyka się co dwa lata, także Wspólny Komitet Parlamentarny EOG i Komitet Konsultacyjny EOG spotykają się regularnie.
2.8.2.2. Procedura klauzuli ochronnej
Urząd Nadzoru EFTA jest odpowiedzialny za badanie powiadomień o zastosowaniu klauzuli ochronnej od państw EOG–EFTA. Urząd konsultuje się ze wszystkimi stronami, których dotyczy dana sprawa i wymienia informacje z Komisją dotyczące postępowania w tej sprawie. Urząd przekazuje swoją decyzję państwom EOG–EFTA i Komisji w celu podjęcia dalszych działań. Jeżeli wybrane państwo EOG–EFTA nie zastosuje się do decyzji, Urząd Nadzoru może wszcząć postępowanie w sprawie naruszenia.
W przypadkach, gdy państwo członkowskie powołuje się na klauzulę ochronną, przewidziane są konsultacje pomiędzy Komisją a Urzędem Nadzoru. Komisja przekazuje swoją decyzję Urzędowi Nadzoru EFTA, który przesyła ją do państw EOG–EFTA w celu podjęcia dalszych działań. Jeżeli wybrane państwo EOG–EFTA nie zastosuje się do decyzji, Urząd Nadzoru może wszcząć postępowanie w sprawie naruszenia.
2.8.3. MONAKO, SAN MARINO I ANDORA
Handel dwustronny pomiędzy UE a Monako, San Marino i Andorą jest łatwiejszy dzięki porozumieniom o unii celnej: Monako jest w unii celnej z Francją i jest częścią obszaru celnego UE; natomiast San Marino i Andora zawarły porozumienia o unii celnej bezpośrednio z UE.
Jednak aby produkty z tych krajów mogły być udostępniane na rynku UE, muszą być zgodne z unijnymi przepisami (84).
2.8.4. TURCJA
Turcja i UE stworzyły unię celną w 1995 r. (decyzja 1/95 Rady Stowarzyszenia UE-Turcja, 96/142/WE). Decyzja w sprawie unii celnej dotyczy handlu produktami przetwórstwa przemysłowego oraz przetworzonymi produktami rolnymi pomiędzy Turcją a UE i obejmuje dostosowanie przepisów obowiązujących w Turcji do wszystkich przepisów dotyczących produktów w UE. Porozumienie ma na celu zapewnienie swobodnego przepływu produktów przetwórstwa przemysłowego oraz przetworzonych produktów rolnych pomiędzy UE i Turcją dzięki zniesieniu kontroli przywozowych na granicy UE–Turcja dla takich produktów.
Art. 5–7 decyzji dotyczą wyeliminowania środków mających ten sam skutek co cła pomiędzy Unią Europejską i Turcją i odzwierciedlają postanowienia art. 34–36 TFUE. Zgodnie z art. 66 decyzji w celu wdrożenia i zastosowania jej art. 5–7 w odniesieniu do produktów objętych decyzją w sprawie unii celnej, artykuły te muszą być interpretowane zgodnie z odpowiednim orzecznictwem Trybunału Sprawiedliwości, zwłaszcza sprawą Cassis de Dijon dotyczącą wzajemnego uznawania.
W konsekwencji w przypadku sektorów, w których Turcja dostosowała swoje przepisy do prawa unijnego, produkt zgodnie z prawem wyprodukowany i/lub wprowadzony do obrotu w Turcji powinien być traktowany tak samo jak produkt zgodnie z prawem wyprodukowany i/lub wprowadzony do obrotu w UE i nie należy stosować wobec niego kontroli przywozowych. To samo dotyczyłoby sektorów niezharmonizowanych, jeśli Turcja dostosowała swoje prawo do art. 34–36 TFUE.
Decyzja nakłada również na Turcję obowiązek przyjęcia przepisów Unii Europejskiej dotyczących produktów i infrastruktury jakości, w szczególności wymagań oznakowania CE, jednostek notyfikowanych, nadzoru rynku, akredytacji, normalizacji, metrologii i wzajemnego uznawania w obszarze niezharmonizowanym.
Inna decyzja (decyzja nr 2/97 Rady Stowarzyszenia WE–Turcja) podpisana w 1997 r. określa listę unijnych instrumentów prawnych, w tym część dorobku w dziedzinie produktów przemysłowych związaną z usuwaniem barier technicznych, a także ustalenia regulujące ich przyjęcie przez Turcję. Załącznik I do tej decyzji gwarantuje, że jeśli Turcja przyjęłaby przepisy prawa wymienione w załączniku II do decyzji, to w UE i Turcji obowiązywałyby takie same zasady i procedury dotyczące produktów mieszczących się w zakresie przepisów prawa wymienionych w załączniku II do decyzji. Jednak wiele instrumentów prawnych określonych w załączniku II zostało stopniowo zastąpionych nowymi dyrektywami i rozporządzeniami unijnymi.
W 2006 r. Rada Stowarzyszenia UE–Turcja przyjęła nową decyzję (1/2006), w której przewidziano wyznaczanie tureckich jednostek notyfikowanych i uznawanie sprawozdań z badań i certyfikatów wystawionych przez takie jednostki w Turcji. Strony podpisały oświadczenia potwierdzające, że tureckie prawodawstwo odpowiada przepisom unijnym w zakresie wielu dyrektyw i rozporządzeń w ramach nowego podejścia.
W obszarze niezharmonizowanym prawa i obowiązki podmiotów gospodarczych dostarczających produkty na rynek UE z Turcji zostały określone w komunikacie wyjaśniającym Komisji w „sprawie ułatwienia dostępu produktów do rynków innych państw członkowskich: praktyczne stosowanie zasady wzajemnego uznawania” (85).
Turecka Agencja Akredytacyjna (TURKAK) jest członkiem organizacji Europejska Współpraca w Dziedzinie Akredytacji (EA) i podpisała wiele umów o wzajemnym uznawaniu z EA. Certyfikaty wydawane przez tureckie jednostki oceniające zgodność akredytowane przez TURKAK powinny być zatem uznawane za równoważne z certyfikatami wydawanymi przez jednostki oceniające zgodność mające siedzibę w UE i akredytowane przez krajowe jednostki akredytujące UE.
W obszarze normalizacji zarówno CEN, jak i CENELEC dnia 1 stycznia 2012 r. przyznały pełne członkostwo Tureckiemu Instytutowi Norm (TSE).
2.9. OKRESY PRZEJŚCIOWE W PRZYPADKU NOWYCH LUB ZMIENIONYCH ZASAD UE
|
W przypadku nowych lub zmienionych przepisów prawa podmioty gospodarcze mogą otrzymać dodatkowy czas na dostosowanie się do nowych zasad – tzw. okres przejściowy – który odpowiada okresowi od chwili wejścia w życie nowego przepisu do momentu, gdy zacznie on obowiązywać. |
Okres przejściowy oznacza, że dotychczasowe przepisy prawa są nadal stosowane, chociaż nowe zasady zostały już przyjęte. Okres przejściowy może zostać wprowadzony przez prawodawcę, jeśli przepisy dotyczące produktów w UE ulegają zmianom lub zastępują przepisy krajowe.
Celem okresu przejściowego jest umożliwienie producentom, organom krajowym i jednostkom notyfikowanym stopniowego dostosowania się do procedur oceny zgodności i zasadniczych lub innych wymagań prawnych ustalonych nowymi lub zmienionymi przepisami prawa i tym samym uniknięcie ryzyka zablokowania produkcji. Co więcej, producenci, importerzy i dystrybutorzy muszą mieć czas, aby skorzystać z praw, które nabyli zgodnie z wcześniejszymi, krajowymi lub unijnymi przepisami, np. sprzedać zapasy produktów wytworzonych zgodnie z poprzednimi zasadami. Wreszcie okres przejściowy daje dodatkowy czas na rewizję i przyjęcie norm zharmonizowanych, nawet jeśli nie jest to warunek konieczny wdrożenia unijnego prawodawstwa harmonizacyjnego
Każdy unijny akt harmonizacyjny przewidujący możliwość zastosowania okresu przejściowego określa termin „zamrożenia” obowiązującego systemu. Jest to zasadniczo dzień, kiedy takie przepisy prawa wchodzą w życie, ale czasami data przyjęcia przepisów.
Po upływie okresu przejściowego produkty wytworzone przed lub w trakcie tego okresu zgodnie z przepisami, które mają zostać uchylone, nie mogą być już wprowadzane do obrotu. W przypadku produktu, który zostanie wprowadzony do obrotu przed końcem okresu przejściowego, powinna istnieć możliwość udostępniania go na rynku lub oddania do użytku (86). Jednak mimo to, pewne unijne przepisy harmonizacyjne mogą zakazać udostępniania takich produktów, jeśli wymagają tego względy bezpieczeństwa lub inne cele określone w prawodawstwie.
Produkty, które nie zostaną wprowadzone do obrotu przed końcem okresu przejściowego, mogą być wprowadzone do obrotu lub oddane do użytku tylko wtedy, gdy w pełni spełniają wymagania nowych przepisów prawa (87).
Zgodnie z ogólną zasadą oznakowanie CE stanowi informację, że produkty, które są regulowane przez jeden lub kilka unijnych aktów harmonizacyjnych określających umieszczanie takiego oznakowania, są zgodne ze wszystkimi mającymi zastosowanie przepisami. Jednak jeśli co najmniej jeden akt prawny pozwala producentowi w okresie przejściowym wybierać ustalenia, do których będzie się stosować, oznakowanie CE wskazuje tylko na zgodność z aktami prawnymi, do których stosuje się producent. W konsekwencji w okresie przejściowym oznakowanie CE niekoniecznie wskazuje, że produkt jest zgodny ze wszystkimi mającym zastosowanie przepisami dotyczącymi jego umieszczania. Informacje dotyczące wszystkich unijnych przepisów harmonizacyjnych stosowanych przez producenta muszą znaleźć się w deklaracji zgodności UE (88).
2.10. USTALENIA PRZEJŚCIOWE DOTYCZĄCE DEKLARACJI ZGODNOŚCI UE WYNIKAJĄCE Z DOSTOSOWANIA DO DECYZJI NR 768/2008/WE
Unijne prawodawstwo harmonizacyjne nie musi przewidywać rozwiązania przejściowego dotyczącego informacji, które należy zawrzeć w deklaracji zgodności UE, w przypadku gdy obowiązujące prawodawstwo zostało zastąpione nowym. Ma to miejsce w przypadku dyrektyw, które zostały zmienione w celu dostosowania do przepisów odniesienia zawartych w decyzji 768/2008/WE (89). Zasadnicze wymagania w większości tych dyrektyw nie zostały zmienione i ma okresu przejściowego w odniesieniu stosowania starych lub nowych dyrektyw. Ponadto w stosownych przypadkach dostosowane dyrektywy precyzują, że certyfikaty wydane na podstawie starej dyrektywy zachowują ważność na podstawie nowej dyrektywy. Od dnia ich wejścia w życie deklaracja zgodności UE musi zawierać odniesienie do nowych dyrektyw, aby produkty wprowadzane do obrotu mogły zostać uznane za zgodne z wymogami.
Unijne prawodawstwo harmonizacyjne w większości przypadków określa tylko obowiązkowy minimalny zakres treści deklaracji zgodności UE, ale zazwyczaj akceptuje się też inne przydatne informacje. Producenci mogą skorzystać z tej elastyczności i zacząć używać nowego wzoru określonego w załącznikach do dostosowanych dyrektyw jeszcze przed ich wejściem w życie. Jeżeli produkty spełniają wymogi zarówno starych, jak i nowych dyrektyw, podmioty gospodarcze mogą odnosić się w deklaracji zgodności UE do obu grup dyrektyw („starych” i dostosowanych), podając odpowiednie okresy stosowania dla każdej z dyrektyw. Przykładowo, w odniesieniu do produktu objętego zakresem dyrektywy 2014/30/UE, deklaracja zgodności UE może zawierać następujące stwierdzenie:
„Opisany powyżej przedmiot niniejszej deklaracji jest zgodny z odnośnymi wymogami unijnego prawodawstwa harmonizacyjnego: dyrektywa 2004/108/WE (do dnia 19 kwietnia 2016 r.) i dyrektywa 2014/30/UE (od dnia 20 kwietnia 2016 r.)”.
3. Uczestnicy łańcucha dostaw produktów i ich obowiązki
Unijne prawodawstwo harmonizacyjne nazywa producenta, upoważnionego przedstawiciela, importera i dystrybutora „podmiotami gospodarczymi” (90).
3.1. PRODUCENT
|
Producent to osoba fizyczna lub prawna, która jest odpowiedzialna za zaprojektowanie lub wytworzenie produktu i wprowadza go do obrotu pod własną nazwą lub znakiem towarowym (91). Definicja ta obejmuje dwa warunki, które muszą być spełnione łącznie: osoba ta musi wytwarzać produkt (lub zlecać jego wytwarzanie) i wprowadzać go do obrotu pod własną nazwą lub znakiem towarowym. Jeśli produkt jest sprzedawany pod nazwą lub znakiem towarowym innej osoby, to producentem jest właśnie ta osoba.
Obowiązki producenta są wiążące również dla każdej osoby fizycznej lub prawnej zajmującej się montażem, pakowaniem, przetwarzaniem lub etykietowaniem gotowych produktów i wprowadzaniem ich do obrotu pod własną nazwą lub znakiem towarowym. Odpowiedzialność producenta spoczywa również na osobie, która zmienia przeznaczenie produktu w taki sposób, że zastosowanie znajdą inne zasadnicze lub inne wymagania prawne, albo istotnie zmieni lub przebuduje produkt (a tym samym stworzy nowy produkt) z myślą o wprowadzeniu go do obrotu lub oddaniu do użytku w przypadkach gdy unijne prawodawstwo harmonizacyjne mające zastosowanie do produktu obejmuje swoim zakresem również oddawanie do użytku (92).
Producent może sam zaprojektować i wytworzyć produkt. Może również zlecić jego zaprojektowanie, wytworzenie, montaż, pakowanie, przetworzenie i etykietowanie z myślą o wprowadzeniu go do obrotu pod własną nazwą lub znakiem towarowym, tym samym przedstawiając siebie jako producenta (93). W przypadku podwykonawstwa producent musi zachować całkowitą kontrolę nad produktem i dopilnować, aby otrzymywał wszelkie informacje niezbędne do spełnienia swoich obowiązków wynikających z istotnych unijnych aktów harmonizacyjnych. Producent, który zleca wykonanie części lub wszystkich czynności w żadnym wypadku nie może zwolnić się ze swoich obowiązków, przenosząc je np. na upoważnionego przedstawiciela, dystrybutora, użytkownika lub podwykonawcę.
Producent ponosi ostateczną odpowiedzialność za zgodność produktu ze stosownymi przepisami unijnego prawodawstwa harmonizacyjnego, bez względu na to, czy wytworzył produkt samodzielnie, czy jest uznawany za producenta, ponieważ wprowadził produkt do obrotu pod własną nazwą lub znakiem towarowym.
Dlatego też jeżeli produkt zostanie przekazany producentowi w celu przeprowadzenia dalszych działań, takich jak montaż, pakowanie, przetwarzanie lub etykietowanie, w chwili wprowadzania produktu do obrotu ponosi on wyłączną i ostateczną odpowiedzialność za zapewnienie zgodności produktu z mającymi zastosowanie przepisami i musi być w stanie taką zgodność zapewnić.
Producent jest odpowiedzialny za zaprojektowanie i wytworzenie produktu zgodnie z zasadniczymi lub innymi wymaganiami prawnymi określonymi w odpowiednich unijnych przepisach harmonizacyjnych oraz za przeprowadzenie oceny zgodności zgodnie z procedurą (procedurami) określoną(-ymi) w unijnym prawodawstwie harmonizacyjnym (94).
Producent musi znać zarówno projekt, jak i konstrukcję produktu, aby móc wziąć odpowiedzialność za produkt zgodny ze wszystkimi stosownymi przepisami unijnego prawodawstwa harmonizacyjnego. Dotyczy to zarówno sytuacji, gdy producent projektuje, wytwarza, pakuje i etykietuje produkt samodzielnie, jak i sytuacji, gdy część lub wszystkie powyższe czynności są wykonywanie przez podwykonawcę. Producent musi dysponować odpowiednimi informacjami, które pozwalają mu wykazać zgodność produktu z wymogami.
W związku z tym podmiot gospodarczy wprowadzający produkt do obrotu pod własną nazwą lub znakiem towarowym automatycznie staje się producentem w rozumieniu przepisów unijnego prawodawstwa harmonizacyjnego. Bierze zatem na siebie całą odpowiedzialność za ocenę zgodności (projektowanie i wytwarzanie) produktu, nawet w przypadku gdy została on faktycznie przeprowadzona przez kogoś innego. Ponadto musi on posiadać wszystkie dokumenty i zaświadczenia niezbędne do wykazania zgodności danego produktu z wymogami, lecz nie muszą być one wydane w jego imieniu.
W dyrektywie 95/16/WE dotyczącej dźwigów instalatora dźwigu definiuje się jako „osobę fizyczną lub prawną, która bierze odpowiedzialność za konstrukcję, wykonanie, zainstalowanie oraz wprowadzenie do obrotu dźwigu i która umieszcza oznakowanie zgodności CE oraz sporządza deklarację zgodności WE”. Dlatego instalator to osoba, która przyjmuje obowiązki, które w kontekście innych unijnych przepisów harmonizacyjnych są zazwyczaj przypisane producentowi.
Unijne prawodawstwo harmonizacyjne nie wymaga, aby producent miał siedzibę w Unii Europejskiej. Dlatego też w momencie wprowadzania produktu do obrotu w Unii producent ma takie same obowiązki, bez względu na to, czy ma swoją siedzibę poza Unią Europejską, czy w dowolnym państwie członkowskim.
Zasadniczo przy wprowadzaniu produktu do obrotu producent musi dołożyć wszelkich starań, aby proces produkcji zapewniał zgodność produktów (95) z wymogami, a w szczególności musi:
|
1) |
przeprowadzić stosowną ocenę zgodności lub zlecić jej przeprowadzenie zgodnie z procedurą(-ami) określoną(-ymi) w odnośnym unijnym prawodawstwie harmonizacyjnym. W zależności od unijnego aktu harmonizacyjnego producent może być zobowiązany do przedłożenia produktu podmiotowi zewnętrznemu (zazwyczaj jednostce notyfikowanej) w celu przeprowadzenia oceny zgodności lub do uzyskania zatwierdzenia systemu jakości przez jednostkę notyfikowaną. W każdym razie producent ponosi pełną odpowiedzialność za zgodność produktu; |
|
2) |
sporządzić wymaganą dokumentację produktu; |
|
3) |
sporządzić deklarację zgodności UE; |
|
4) |
dołączyć do produktu instrukcje i informacje dotyczące bezpieczeństwa (96) (97) zgodnie z wymogami odpowiedniego unijnego prawodawstwa harmonizacyjnego (98) w języku łatwo zrozumiałym dla konsumentów i innych użytkowników, zgodnie z ustaleniami zainteresowanego państwa członkowskiego (99). O ile nie wskazano inaczej w przepisach szczegółowych, instrukcje i informacje dotyczące bezpieczeństwa muszą być podane (100) bez względu na to, czy produkt jest przeznaczony dla konsumentów lub innych użytkowników końcowych. Powinny być to wszystkie informacje niezbędne dla bezpiecznego użytkowania produktu, umożliwiające konsumentowi montaż, instalację, obsługę, przechowywanie, konserwację i usunięcie produktu. Instrukcja montażu lub instalacji powinna zawierać wykaz części i potrzebnych specjalnych umiejętności lub narzędzi. Instrukcja obsługi powinna zawierać informacje dotyczące ograniczenia użytkowania, potrzeby stosowania środków ochrony indywidualnej, konserwacji i czyszczenia lub naprawy. Na producencie spoczywa obowiązek określenia odpowiednich informacji, które powinny być zawarte w instrukcji, oraz informacji na temat bezpieczeństwa dotyczących danego produktu. Producenci muszą umieć przewidzieć nie tylko to, co uważają za użytkowanie produktu zgodnie z przeznaczeniem, lecz postawić się w sytuacji przeciętnego użytkownika danego produktu i wyobrazić sobie, w jaki sposób prawdopodobnie używaliby tego produktu. Ponadto narzędzie zaprojektowane i przeznaczone wyłącznie dla profesjonalistów może być również używane przez inne osoby, co należy uwzględnić w jego projekcie i instrukcjach dołączonych do urządzenia; |
|
5) |
spełnić następujące wymagania identyfikowalności:
|
|
6) |
umieścić znak zgodności (oznakowanie CE, a w stosownych przypadkach inne oznakowania (108)) na produkcie zgodnie z odpowiednimi przepisami; |
|
7) |
dopilnować, aby w przypadku produkcji seryjnej wprowadzono procedury gwarantujące stałą zgodność produktów z wymogami. Należy odpowiednio uwzględnić zmiany w projekcie lub właściwościach produktu i zmiany w normach zharmonizowanych lub specyfikacjach technicznych, w odniesieniu do których zadeklarowano zgodność produktu. Rodzaj działania, które producent powinien podjąć, zależy od charakteru zmian w normach zharmonizowanych lub specyfikacjach technicznych, a w szczególności od tego, czy dane zmiany są istotne, jeśli chodzi o spełnianie zasadniczych lub innych wymagań prawnych, i czy dotyczą danego produktu. Może to wymagać np. aktualizacji deklaracji zgodności UE, zmiany projektu produktu, skontaktowania się z jednostką notyfikowaną (109) itp.; |
|
8) |
w stosownych przypadkach certyfikować produkt i/lub system jakości. |
Zgodnie z określonymi unijnymi aktami harmonizacyjnymi producent może mieć obowiązek przeprowadzenia badań próbek na końcu łańcucha produkcji lub produktów wprowadzonych już do obrotu w celu udzielenia konsumentom lub innym użytkownikom dodatkowej ochrony (110) (111).
Producenci, którzy uważają lub mają powód, by sądzić, że wprowadzony przez nich produkt jest niezgodny z mającymi zastosowanie unijnymi przepisami harmonizacyjnymi, muszą niezwłocznie podjąć konieczne środki naprawcze, aby zapewnić zgodność produktu z wymogami albo wycofać go z obrotu lub od użytkowników. Ponadto, jeśli producenci mają powód, by wierzyć, że produkt stwarza zagrożenie dla zdrowia, bezpieczeństwa, środowiska lub zagraża innemu interesowi publicznemu chronionego stosownymi przepisami (112), muszą oni natychmiast poinformować o tym właściwe organy państwa członkowskiego, w którym udostępnili produkt, podając szczegółowe informacje głównie na temat niezgodności i wszelkich zastosowanych środków naprawczych. Komisja udostępnia narzędzie informatyczne „GPSD Business Application”, aby ułatwić wypełnianie tego obowiązku od strony praktycznej (113).
Na uzasadnione żądanie (114) producent ma obowiązek przedstawić właściwym organom krajowym wszelkie informacje i dokumentację niezbędną do wykazania zgodności produktu w języku, który będzie łatwo zrozumiały dla tego organu. Producenci, na żądanie danego organu, muszą z nim współpracować we wszelkich działaniach mających na celu wyeliminowanie zagrożeń powodowanych przez wprowadzone przez nich produkty. Na żądanie organów nadzoru rynku producenci muszą wskazać podmiot gospodarczy, któremu dostarczyli produkty. Muszą być w stanie udzielić tego rodzaju informacji przez 10 lat od chwili dostarczenia produktu.
Z założenia organ krajowy może dopuścić język, który jest dla niego zrozumiały, a który jest inny od języka(-ów) narodowego. Wybrany język jest przedmiotem negocjacji z organem i może być językiem trzecim, jeśli tylko zostanie przez niego zaakceptowany.
W przypadku uzasadnionego żądania wystarczy, że producent przedstawi część dokumentacji produktu dotyczącą rzekomej niezgodności, pozwalającą stwierdzić, czy producent zajął się tą kwestią. Dlatego wszelkie żądania przetłumaczenia dokumentacji produktu powinny ograniczać się do takich części. W zależności od unijnego prawodawstwa harmonizacyjnego, któremu podlega produkt, w żądaniu może być wskazany termin dostarczenia żądanych dokumentów. Można ustalić krótszy termin, jeśli organ krajowy uzasadni pośpiech bezpośrednim, poważnym zagrożeniem.
Jeżeli unijne prawodawstwo harmonizacyjne obejmuje oddanie do użytku, osoba fizyczna lub prawne, która oddaje produkt do użytku, ma takie same obowiązki, jak producent wprowadzający produkt do obrotu. Musi zapewnić zgodność produktu z unijnym prawodawstwem harmonizacyjnym i dopilnować przeprowadzenia odpowiedniej procedury oceny zgodności (115).
Ponadto osoba, która wprowadza na wspólny rynek produkty używane z państw trzecich lub produkty zaprojektowane lub wyprodukowane z myślą o rynkach innych niż unijny, musi przyjąć rolę producenta.
Jeżeli importer lub dystrybutor modyfikuje produkt w takim stopniu, że może to wpłynąć na jego zgodność z mającymi zastosowanie wymogami lub dostarcza go pod własną nazwą lub swoim znakiem towarowym, jest wtedy traktowany jak producent i musi wywiązać się ze wszystkich obowiązków nałożonych na producenta (116). W związku z powyższym musi zagwarantować, że produkt jest zgodny z odpowiednimi unijnymi przepisami harmonizacyjnymi i że przeprowadzono odpowiednią procedurę oceny zgodności (117).
3.2. UPOWAŻNIONY PRZEDSTAWICIEL
|
Bez względu na to, czy producent posiada siedzibę w UE czy poza nią, może wyznaczyć upoważnionego przedstawiciela w Unii, który będzie działać w jego imieniu podczas wykonywania określonych zadań. |
Bez względu na to, czy producent posiada siedzibę w UE czy poza nią, może wyznaczyć upoważnionego przedstawiciela w Unii, który będzie działać w jego imieniu podczas wykonywania określonych zadań wymaganych stosownym unijnym prawodawstwem harmonizacyjnym (118). Producent mający siedzibę poza Unią Europejską nie jest zobowiązany do posiadania upoważnionego przedstawiciela (119).
W rozumieniu unijnego prawodawstwa harmonizacyjnego, aby upoważniony przedstawiciel mógł występować w imieniu producenta, musi mieć swoją siedzibę w Unii. Nie należy mylić przedstawicieli handlowych producenta (takich jak autoryzowani dystrybutorzy lub agenci) z upoważnionym przedstawicielem w rozumieniu unijnego prawodawstwa harmonizacyjnego.
Oddelegowanie przez producenta zadań upoważnionemu przedstawicielowi musi być wyraźne i ustalone na piśmie, w szczególności musi określać rodzaj i zakres zadań przedstawiciela. Zadania, które można oddelegować upoważnionemu przedstawicielowi zgodnie z unijnym prawodawstwem harmonizacyjnym mają charakter administracyjny. Dlatego też producent nie może oddelegować ani działań służących zagwarantowaniu, że proces produkcji zapewnia zgodność produktów z przepisami, ani przygotowania dokumentacji produktu, chyba że przepisy przewidują inaczej. Ponadto upoważniony przedstawiciel nie może modyfikować produktu z własnej inicjatywy w celu dostosowania go do stosownego unijnego prawodawstwa harmonizacyjnego.
Jeżeli producent wyznaczył upoważnionego przedstawiciela, pełnomocnictwo to powinno przynajmniej umożliwiać temu przedstawicielowi wykonywanie następujących zadań:
|
— |
przechowywanie deklaracji zgodności UE i dokumentacji produktu do dyspozycji krajowego organu nadzoru i współpraca z nim na jego żądanie, |
|
— |
na uzasadnione żądanie właściwego krajowego organu udzielanie mu wszelkich informacji i udostępnianie dokumentacji koniecznej do wykazania zgodności danego produktu z wymogami, |
|
— |
na żądanie właściwych organów krajowych – podejmowanie z nimi współpracy we wszelkich objętych pełnomocnictwem działaniach ukierunkowanych na usunięcie zagrożeń, jakie stwarzają produkty. |
W zależności od danej procedury oceny zgodności i unijnego aktu harmonizacyjnego upoważniony przedstawiciel może również zostać wyznaczony do wykonywania takich zadań, jak:
|
— |
umieszczanie na produkcie oznakowania CE (i gdzie stosowne innego oznakowania) i numeru jednostki notyfikowanej, |
|
— |
sporządzanie i podpisywanie deklaracji zgodności UE, |
Upoważniony przedstawiciel wyznaczony przez producenta może być importerem lub dystrybutorem w rozumieniu unijnego prawodawstwa harmonizacyjnego – w tym przypadku musi również spełniać obowiązki importera lub dystrybutora (120).
3.3. IMPORTER
|
Importer to podmiot gospodarczy mający siedzibę w Unii, który wprowadza do obrotu w Unii produkt z państwa trzeciego. Ma ważne obowiązki, które są jasno zdefiniowane w unijnym prawodawstwie harmonizacyjnym (121) (122). Obowiązki te w dużej mierze opierają się na rodzaju zobowiązań nałożonych na producenta z siedzibą w UE.
Importer musi upewnić się, że producent poprawnie wypełnił swoje obowiązki. Importer nie jest po prostu odsprzedawcą produktu, ale odgrywa ważną rolę w zagwarantowaniu zgodności importowanych produktów.
Importer to osoba fizyczna lub prawna mająca siedzibę w Unii i wprowadzająca do obrotu w Unii produkt z państwa trzeciego. Co do zasady przed wprowadzeniem produktu na rynek importer musi upewnić się:
|
1) |
że producent przeprowadził odpowiednią procedurę oceny zgodności. Jeśli ma obawy co do zgodności produktu z wymogami, musi powstrzymać się od wprowadzania go do obrotu. Jeżeli produkt został już wprowadzony do obrotu, musi podjąć działania naprawcze (123). W obydwu przypadkach może być potrzebny kontakt z producentem w celu wyjaśnienia wątpliwości dotyczących zgodności produktu; |
|
2) |
że producent sporządził dokumentację produktu, umieścił odpowiednie oznakowanie potwierdzające zgodność (np. oznakowanie CE), spełnił swoje obowiązki dotyczące identyfikowalności i w stosownych przypadkach dołączył instrukcje i informacje dotyczące bezpieczeństwa w języku łatwo zrozumiałym dla konsumentów i innych użytkowników, zgodnie z ustaleniami odpowiedniego państwa członkowskiego (124). |
Zobowiązania te mają uświadomić importerom spoczywający na nich obowiązek wprowadzenia do obrotu tylko produktów zgodnych z przepisami (125). Nie oznaczają one potrzeby systematycznego stosowania przez importerów dodatkowych procedur kontrolnych lub zlecania badań podmiotom zewnętrznym ani też nie wykluczają takiej możliwości.
Importer ma również obowiązek:
|
— |
podać następujące trzy elementy: swoją 1) nazwę, 2) zarejestrowaną nazwę handlową lub zarejestrowany znak towarowy i 3) adres kontaktowy na produkcie lub, jeśli nie jest to możliwe ze względu na rozmiar lub właściwości fizyczne produktu lub dlatego, że opakowanie będzie musiało zostać otwarte – na opakowaniu i/lub (126) w dołączonej dokumentacji (127). Należy przy tym zachować czytelność informacji dot. bezpieczeństwa wydrukowanych na produkcie lub w dołączonych dokumentach, |
|
— |
dopilnować, aby w czasie, gdy jest odpowiedzialny za produkt, warunki magazynowania lub transportu nie naruszały jego zgodności z wymogami określonymi w mających zastosowanie przepisach, |
|
— |
przechowywać kopię deklaracji zgodności UE przez 10 lat od chwili wprowadzenia produktu do obrotu (128) lub przez okres wskazany w odpowiednim unijnym akcie harmonizacyjnym, |
|
— |
zagwarantować możliwość udostępnienia dokumentacji produktu na żądanie właściwego organu krajowego (129). Importer ma obowiązek współpracować z tym organem i na uzasadnione żądanie (130) przedstawić mu w języku dla niego zrozumiałym wszelkie informacje i dokumenty konieczne do wykazania zgodności produktu z wymogami. Z założenia organ krajowy może dopuścić język, który jest dla niego zrozumiały, a który jest inny od języka (języków) narodowego. Wybrany język jest przedmiotem negocjacji z organem i może być językiem trzecim, jeśli tylko zostanie przez niego zaakceptowany. |
|
— |
W przypadku uzasadnionego żądania wystarczy, że importer przedstawi część dokumentacji produktu dotyczącą rzekomej niezgodności, pozwalającą stwierdzić, czy producent zajął się tą kwestią. Dlatego wszelkie żądania przetłumaczenia dokumentacji produktu powinny ograniczać się do takich części, |
|
— |
na żądanie organów nadzoru rynku importer jest zobowiązany wskazać podmiot gospodarczy, który był dostawcą i odbiorcą jego produktu. Musi być w stanie udzielić tej informacji przez 10 lat od chwili otrzymania produktu od dostawcy i przez 10 lat od chwili dostarczenia produktu do odbiorcy. |
Zgodnie z pewnymi unijnymi aktami harmonizacyjnymi importer – podobnie jak producent – może mieć obowiązek przeprowadzenia badań próbek produktów wprowadzonych już do obrotu (131).
Podobnie importerzy, którzy uważają lub mają powód, by wierzyć, że wprowadzony przez nich produkt jest niezgodny z mającymi zastosowanie unijnymi przepisami harmonizacyjnymi, muszą niezwłocznie podjąć konieczne środki naprawcze, aby zapewnić zgodność produktu z wymogami albo wycofać go z obrotu lub od użytkowników. Ponadto, jeśli produkt stwarza zagrożenie, importerzy powinni niezwłocznie poinformować o tym właściwe organy krajowe.
Importer nie musi mieć ani upoważnienia od producenta, ani preferencyjnych zasad współpracy z nim, tak jak upoważniony przedstawiciel. Jednak do obowiązków importera należy zapewnienie możliwości kontaktu z producentem (np. w celu udostępnienia dokumentacji produktu na żądanie właściwego organu).
Importer, jeżeli chce, może wykonywać zadania administracyjne w imieniu producenta. W takim przypadku producent musi wyraźnie wskazać, że importer będzie pełnił funkcję upoważnionego przedstawiciela.
3.4. DYSTRYBUTOR
|
Obok producentów i importerów dystrybutorzy są trzecią kategorią podmiotów gospodarczych, na którą nakładane są określone obowiązki. Dystrybutor to osoba fizyczna lub prawna w łańcuchu dostaw, niebędąca producentem ani importerem, która udostępnia produkt na rynku.
Sprzedawcy detaliczni, hurtownicy i inni dystrybutorzy w łańcuchu dostaw nie muszą mieć preferencyjnych zasad współpracy z producentem, tak jak upoważniony przedstawiciel. Dystrybutor nabywa produkt do dalszej dystrybucji od producenta, importera lub innego dystrybutora.
Dystrybutor ma obowiązek działać z należytą starannością (132) w odniesieniu do odpowiednich wymagań (133). Musi np. wiedzieć, które produkty muszą posiadać oznakowanie CE, jakie informacje powinny być dołączone do produktu (np. deklaracja zgodności UE), jakie są wymagania dotyczące języka etykiet, instrukcji dla użytkownika i innych dołączonych dokumentów, a także jakie są ewidentne oznaki niezgodności produktu z przepisami. Dystrybutorzy mają obowiązek przedstawić krajowym organom nadzoru rynku dowód na to, że działali z należytą starannością, i zagwarantować, że producent, jego upoważniony przedstawiciel lub osoba, która dostarczyła mu produkt podjął(-ęła) działania wymagane stosownymi unijnymi przepisami harmonizacyjnymi, w których wymieniono obowiązki dystrybutorów.
Ocena zgodności, sporządzenie i przechowywanie deklaracji zgodności UE oraz dokumentacji produktu nadal pozostają obowiązkiem producenta i/lub w przypadku produktów z państw trzecich – importera. Dystrybutor nie ma obowiązku sprawdzać, czy produkt wprowadzony już do obrotu jest nadal zgodny z aktualnie obowiązującymi wymaganiami prawnymi, jeśli te wymagania uległy zmianie. Obowiązki dystrybutora ograniczają się do przepisów obowiązujących w chwili wprowadzenia produktu do obrotu przez producenta lub importera, chyba że w szczegółowych przepisach przewidziano inaczej.
Dystrybutor jest zobowiązany do wskazania producenta, jego upoważnionego przedstawiciela, importera lub osoby, która dostarczyła mu produkt, aby pomóc organowi nadzoru rynku w uzyskaniu deklaracji zgodności UE i niezbędnych części dokumentacji produktu. Organy nadzoru rynku mogą skierować swoje żądanie dokumentacji produktu bezpośrednio do dystrybutora. Od dystrybutora nie należy jednak oczekiwać, że będzie posiadać stosowne dokumenty.
Przed udostępnieniem produktu na rynku dystrybutor musi zweryfikować następujące wymogi formalne (134):
|
— |
obecność na produkcie wymaganych oznakowań zgodności (np. oznakowania CE), |
|
— |
dołączenie do produktu istotnych informacji (np. deklaracja zgodności UE (135)) oraz instrukcji i informacji dotyczących bezpieczeństwa (136) w języku łatwo zrozumiałym dla konsumentów i innych użytkowników, jeśli wymagają tego mające zastosowanie przepisy, |
|
— |
wskazanie przez producenta i importera następujących trzech elementów: swojej 1) nazwy, 2) zarejestrowanej nazwy handlowej lub znaku towarowego i 3) adresu kontaktowego na produkcie lub, jeśli nie jest to możliwe ze względu na rozmiar lub właściwości fizyczne produktu – na opakowaniu i/lub dołączonej dokumentacji (137), oraz obecności na produkcie informacji dotyczącej typu, numeru partii lub numeru seryjnego lub innych elementów pozwalających na identyfikację produktu. |
Dystrybutorom nie wolno dostarczać produktów, o których wiedzą lub powinni przypuszczać na podstawie posiadanych informacji i wiedzy fachowej, że są niezgodne z przepisami prawa. Dystrybutorzy muszą współpracować z właściwym organem przy działaniach podejmowanych w celu uniknięcia lub zminimalizowania takich zagrożeń, a także informować producenta lub importera i właściwe organy krajowe (138).
Podobne obowiązki mają dystrybutorzy po udostępnieniu produktu. Jeśli mają podstawy, aby sądzić, że produkt jest niezgodny z przepisami, muszą upewnić się, czy producent lub importer podjął środki naprawcze, aby zapewnić zgodność produktu, a także poinformować o takiej sytuacji właściwe organy krajowe. Dystrybutorzy mają obowiązek kontaktować się z importerem lub producentem w celu wyjaśnienia wszelkich wątpliwości dotyczących zgodności produktu z przepisami.
Oprócz kontrolowania zgodności produktu z wymogami formalnymi, dystrybutor ma obowiązek:
|
1) |
podjąć środki naprawcze, jeśli ma podejrzenie, że produkt jest niezgodny z przepisami (139); |
|
2) |
pomagać organom nadzoru rynku w ustaleniu producenta lub importera odpowiedzialnego za produkt; |
|
3) |
na uzasadnione żądanie (140) właściwego organu krajowego współpracować z nim i przedstawić mu wszelkie informacje i dokumentację niezbędne do wykazania zgodności produktu z przepisami (141); |
|
4) |
na żądanie organów nadzoru rynku wskazać podmiot gospodarczy, który był dostawcą i odbiorcą produktu. Muszą być w stanie udzielić tej informacji przez 10 lat od chwili otrzymania produktu od dostawcy i przez 10 lat od chwili dostarczenia produktu do odbiorcy (142). |
Warunki dystrybucji (np. transport lub magazynowanie) mogą mieć wpływ na utrzymanie zgodności produktu z przepisami stosownego unijnego prawodawstwa harmonizacyjnego. Dlatego osoba odpowiedzialna za dystrybucję ma obowiązek podjąć niezbędne kroki w celu ochrony zgodności produktu. Musi zagwarantować, że produkt jest zgodny z zasadniczymi lub innymi wymaganiami prawnymi w chwili jego pierwszego użycia w Unii (143).
W przypadku braku unijnych przepisów harmonizacyjnych warunki dystrybucji mogą być w pewnym stopniu regulowane na poziomie krajowym zgodnie z art. 34 i 36 TFUE. Krajowe przepisy, które gwarantują osobom wykonującym określony zawód wyłączne prawo do dystrybucji określonych produktów, mogą – o ile ograniczają sprzedaż do określonych kanałów – wpłynąć na możliwości wprowadzania do obrotu produktów importowanych. Zgodnie z powyższym takie przepisy mogą stanowić środek mający skutki równoznaczne z ograniczeniem ilościowym importu. Jednak taka sytuacja może być uzasadniona np. ochroną zdrowia publicznego, jeśli środek jest odpowiedni do celu i nie wykracza poza zakres niezbędny do osiągnięcia tego celu (144).
Podmioty świadczące usługi realizacji zamówień
Centra realizacji zamówień (145) stanowią nowy model działalności gospodarczej powstały na potrzeby handlu elektronicznego. Ponadto produkty oferowane przez podmioty internetowe są na ogół przechowywane w centrach realizacji zamówień znajdujących się w UE w celu zapewnienia ich szybkiej dostawy do konsumentów w UE. Centra te są podmiotami świadczącymi usługi na rzecz innych podmiotów gospodarczych. Zajmują się one składowaniem produktów i – po otrzymaniu zlecenia – pakowaniem i przesyłaniem ich klientom. Czasem zajmują się również zwrotami. Istnieje wiele różnych scenariuszy świadczenia usług w zakresie realizacji zamówień. Niektóre centra realizacji zamówień oferują wszystkie wymienione wyżej usługi, natomiast inne świadczą tylko część z nich. Różnią się także pod względem wielkości i skali – od światowych operatorów do mikroprzedsiębiorstw.
Działalność dostawców opisanych powyżej usług realizacji zamówień jest szersza niż w przypadku podmiotów zajmujących się dostarczaniem paczek, które świadczą usługi rozliczenia, sortowania, transportu i doręczania przesyłek. Złożoność tego modelu biznesowego sprawia, że podmioty realizujące zamówienia są niezbędnym elementem łańcucha dostaw i dlatego mogą zostać uznane za uczestniczące w dostarczaniu produktu, a następnie we wprowadzaniu go do obrotu. Jeżeli zatem usługodawcy zajmujący się realizacją zamówień świadczą opisane wyżej usługi wykraczające poza zakres usług dostawców paczek, powinni być traktowani jako dystrybutorzy i powinni wypełniać odpowiednie obowiązki prawne.
Ze względu na zróżnicowanie centrów realizacji zamówień oraz usług, jakie oferują, analiza modelu działalności niektórych podmiotów może wykazać, że są one importerami lub upoważnionymi przedstawicielami.
3.5. INNI POŚREDNICY: USŁUGODAWCY BĘDĄCY POŚREDNIKAMI NA PODSTAWIE DYREKTYWY O HANDLU ELEKTRONICZNYM
Dyrektywa o handlu elektronicznym (146) ustanawia ramy prawne dla handlu elektronicznego w UE. Wprowadza ona zharmonizowane zasady dotyczące takich kwestii, jak wymogi w zakresie przejrzystości i informacji dotyczące usługodawców internetowych, informacji handlowych, umów zawieranych drogą elektroniczną.
W dyrektywie o handlu elektronicznym nie określa się kategorii podmiotów gospodarczych, lecz opisuje różne kategorie działalności. Najważniejszą kategorią działalności, z punktu widzenia bezpieczeństwa produktów i ich zgodności z wymogami, jest działalność w zakresie hostingu (147). Hosting obejmuje takie czynności, jak przechowywanie informacji przekazanych przez usługobiorców, np. sklepy i markety internetowe oraz platformy handlu internetowego.
Usługodawcy będący pośrednikami, prowadzący działalność opisaną powyżej, są zwolnieni z odpowiedzialności odszkodowawczej lub sankcji karnych związanych z treściami dostarczanymi przez strony trzecie korzystające z ich sieci. Jednakże zwolnienie od odpowiedzialności nie ma charakteru bezwzględnego. W przypadku działalności hostingowej, która jest najważniejsza z punktu widzenia bezpieczeństwa i zgodności produktów, zwolnienie to ma zastosowanie wyłącznie w sytuacji, gdy usługodawca będący pośrednikiem 1) nie posiada rzeczywistej wiedzy o nielegalnym charakterze przechowywanych informacji i 2) uzyskawszy taką wiedzę lub świadomość nielegalnych treściach (np. w wyniku „wystarczająco precyzyjnego i właściwie uzasadnionego” powiadomienia (148)) niezwłocznie je usuwa lub blokuje do nich dostęp. Jeżeli nie spełnia powyższych warunków, nie może być objęty zwolnieniem, a zatem może ponosić odpowiedzialność za treści, które przechowuje.
Zgodnie z art. 15 dyrektywy o handlu elektronicznym państwa członkowskie nie mogą nakładać na tych usługodawców ogólnego obowiązku nadzorowania informacji ani ogólnego obowiązku aktywnego poszukiwania faktów i okoliczności wskazujących na bezprawną działalność. Oznacza to, że organy krajowe nie mogą zobowiązać pośredników do aktywnego monitorowania całego ruchu internetowego oraz poszukiwania elementów wskazujących na bezprawną działalność, np. niebezpieczne produkty.
Zakaz żądania ogólnego nadzoru nie stawia jednak organom publicznym ograniczeń pod względem ustalania konkretnych wymogów dotyczących monitorowania, chociaż zakres takich wymogów musi być ukierunkowany. Jeśli chodzi o odrębny, choć niepozbawiony pewnych podobieństw obszar polityki, sąd może nakazać usługodawcom, by dopilnowali, aby niektóre strony internetowe, które zawierają wyłącznie treści naruszające prawa autorskie lub podrobione produkty, były zablokowane dla użytkowników w danym państwie członkowskim.
W praktyce oznacza to, że organy krajowe mogą zwrócić się do dostawców usług hostingowych, którzy – powiadomieni o bezprawnym działaniu – mogą skorzystać ze zwolnienia z odpowiedzialności, o ile usuną lub zablokują daną treść, co oznacza, że produkty niebezpieczne lub niespełniające wymogów nie będą już dostępne dla klientów z UE za pośrednictwem ich usług. Niemniej jednak organy nadzoru rynku powinny opierać się na mających zastosowanie przepisach rozporządzenia (WE) nr 765/2008 oraz odpowiednich przepisach unijnego prawodawstwa harmonizacyjnego, a zatem w pierwszej kolejności zwracać się do odpowiedzialnego podmiotu gospodarczego. Organy nadzoru rynku powinny ocenić najbardziej odpowiednie działania, które należy podjąć, indywidualnie dla każdego przypadku i z uwzględnieniem zasady proporcjonalności oraz biorąc pod uwagę poziom ryzyka, możliwość identyfikacji podmiotu gospodarczego, pilność sprawy oraz ewentualne środki zastosowane już wcześniej wobec danego produktu itd.
Termin „treść” obejmuje ofertę produktu w internecie (np. zdjęcie, opis itp.). Termin „bezprawna działalność” oznacza działania objęte zarówno prawem karnym, jak i prawem administracyjnym. Zwolnienie z odpowiedzialności odnosi się do cywilnej, administracyjnej i karnej odpowiedzialności za wszelkiego rodzaju bezprawne działania podejmowane przez osoby trzecie w internecie, takie jak naruszanie praw autorskich i praw do znaków towarowych, nieuczciwe praktyki handlowe itp. Celem dyrektywy jest odpowiednie wyważenie interesów wszystkich zainteresowanych stron. Podstawę prawną dla powiadamiania dostawców usług hostingowych oraz wymagania od nich usunięcia/dezaktywacji dostępu do nielegalnych treści stanowią przepisy transponujące dyrektywę o handlu elektronicznym do prawa krajowego. Ponadto większość pośredników internetowych opracowała własne mechanizmy dokonywania zgłoszeń.
3.6. UŻYTKOWNIK
|
Unijne prawodawstwo harmonizacyjne nie nakłada obowiązków na użytkowników produktów (149). Dotyczy to nawet sytuacji, gdy na terytorium UE nie ma odpowiedzialnych podmiotów gospodarczych (na przykład w odniesieniu do produktów sprzedawanych przez internet). W konsekwencji termin ten nie ma swojej prawnej definicji. Jednak pewne jest, że pojęcie to obejmuje zarówno użytkowników profesjonalnych, jak i konsumentów. Koncepcja „użytkowania” przez profesjonalistę lub konsumenta jest z natury związana z koncepcją „użytkowania zgodnie z przeznaczeniem” (150).
Wiele produktów regulowanych unijnymi przepisami harmonizacyjnymi jest używanych w pracy. Zgodnie z przepisami opartymi na art. 153 TFUE na pracodawców nakładane są obowiązki, jeśli chodzi o korzystanie przez pracowników ze sprzętu roboczego w miejscu pracy. Pracodawca to osoba fizyczna lub prawna, która pozostaje w stosunku pracy z pracownikiem (tj. osobą zatrudnioną przez pracodawcę) i odpowiada za przedsiębiorstwo lub organizację.
Zgodnie z dyrektywą dotyczącą minimalnych wymagań w dziedzinie bezpieczeństwa i higieny użytkowania sprzętu roboczego przez pracowników w pracy (2009/104/WE) pracodawca ma obowiązek zastosować wszelkie środki niezbędne do zapewnienia, by sprzęt roboczy (np. maszyny lub aparatura) udostępniony pracownikom był odpowiedni do wykonywanej przez nich pracy i może być przez nich używany bez narażania ich bezpieczeństwa i zdrowia. Pracodawca może pozyskać i stosować tylko taki sprzęt roboczy, który jest zgodny z zapisami odpowiednich przepisów prawa w chwili pierwszego użycia lub, jeśli zastosowania nie mają inne przepisy lub są stosowane częściowo, z minimalnymi wymaganiami określonymi w załączniku I do dyrektywy 2009/104/WE. Pracodawca jest również zobowiązany do zagwarantowania, że sprzęt roboczy jest utrzymywany w takim stanie. Pracodawca ma obowiązek przekazać pracownikom informacje i przeszkolić ich w zakresie użytkowania sprzętu roboczego.
Zgodnie z dyrektywą dotyczącą minimalnych wymagań w dziedzinie bezpieczeństwa i higieny użytkowania środków ochrony indywidualnej przez pracowników w miejscu pracy (89/656/EWG) takie wyposażenie musi być zgodne z odpowiednimi przepisami unijnymi dotyczącymi projektu i produkcji w zakresie ich bezpieczeństwa i higieny (tj. unijnym aktem harmonizacyjnym dotyczącym środków ochrony indywidualnej). Wyposażenie musi być odpowiednie do istniejących zagrożeń, warunków w miejscu pracy, uwzględniać wymagania ergonomiczne i stan zdrowia pracownika, być dopasowane do użytkownika i, jeśli jednocześnie stosowane są przynajmniej dwa elementy wyposażenia, muszą być ze sobą kompatybilne. Przed dokonaniem wyboru środków ochrony indywidualnej pracodawca musi ocenić, czy spełniają one te wymagania.
Zgodnie z dyrektywą w sprawie minimalnych wymagań w dziedzinie bezpieczeństwa i ochrony zdrowia przy pracy z urządzeniami wyposażonymi w monitory ekranowe (90/270/EWG) pracodawcy są zobowiązani do oceny stanowisk pracy pod względem bezpieczeństwa i higieny pracy, w szczególności przy uwzględnieniu możliwych zagrożeń dla wzroku, problemów fizycznych i problemów ze stresem. Dyrektywa określa również minimalne wymagania dotyczące monitorów ekranowych i innego sprzętu.
Zgodnie z dyrektywą w sprawie wprowadzenia środków w celu poprawy bezpieczeństwa i zdrowia pracowników w miejscu pracy (89/391/EWG) pracownicy mają ogólny obowiązek zadbania w miarę możliwości o bezpieczeństwo i zdrowie własne i innych osób, na które wpływ ma ich praca. Muszą np. korzystać z maszyn, aparatury i innych środków produkcji, a także środków ochrony indywidualnej w sposób zademonstrowany na szkoleniu i zgodnie z przekazanymi instrukcjami.
Minimalne wymagania określono w dyrektywach 89/391/EWG, 2009/104/WE, 89/656/EWG i 90/270/EWG. Dlatego też państwa członkowskie mają możliwość przyjąć lub utrzymać surowsze przepisy, jeśli tylko są one zgodne z TFUE. Należy jednak przestrzegać unijnych przepisów harmonizacyjnych i dlatego dodatkowe przepisy krajowe nie mogą nakładać obowiązku zmiany produktu objętej zakresem unijnego aktu harmonizacyjnego ani wpływać na warunki udostępniania takich produktów na rynku.
4. WYMAGANIA DOTYCZĄCE PRODUKTÓW
4.1. ZASADNICZE WYMAGANIA DOTYCZĄCE PRODUKTÓW
4.1.1. DEFINICJA ZASADNICZYCH WYMAGAŃ
|
Podstawową cechą większości unijnych przepisów harmonizacyjnych jest ograniczenie harmonizacji prawnej do zasadniczych wymagań, które leżą w interesie publicznym. Wymagania te dotyczą ochrony zdrowia i bezpieczeństwa użytkowników (zazwyczaj konsumentów i pracowników), ale mogą także obejmować podstawowe wymagania (np. ochronę własności, ograniczonych zasobów lub środowiska naturalnego).
Zasadnicze wymagania zostały stworzone w celu osiągnięcia i zapewnienia wysokiego poziomu ochrony. Wynikają one albo z pewnych zagrożeń związanych z produktem (np. odporność fizyczna i mechaniczna, łatwopalność, właściwości chemiczne, elektryczne lub biologiczne, higiena, radioaktywność, dokładność), albo odnoszą się do produktu lub jego charakterystyki (np. przepisy dotyczące materiałów, projektu, budowy, procesu produkcji, instrukcji stworzonych przez producenta), albo określają główny cel ochrony (np. za pomocą przykładowego wykazu). Często łączą te cechy. W konsekwencji do jednego produktu zastosowanie może mieć jednocześnie kilka unijnych aktów harmonizacyjnych. Może zachodzić potrzeba, by zasadnicze wymagania wskazane w różnych unijnych aktach harmonizacyjnych były stosowane jednocześnie w celu uwzględnienia wszystkich odpowiednich aspektów interesu publicznego.
Zasadnicze wymagania muszą być stosowane jako funkcja zagrożenia związanego z danym produktem. W związku z tym producenci muszą przeprowadzać analizy ryzyka, aby najpierw zidentyfikować wszelkie możliwe zagrożenia, które może stwarzać produkt oraz określić zasadnicze wymagania mające zastosowanie do tego produktu. Analizę tę należy udokumentować i włączyć do dokumentacji produktu (151). Ponadto producent musi udokumentować ocenę tego, w jaki sposób radzi sobie z zidentyfikowanym ryzykiem, aby zapewnić zgodność produktu z odpowiednimi zasadniczymi wymaganiami (na przykład stosowanie norm zharmonizowanych). Jeżeli stosowana jest tylko część normy zharmonizowanej lub norma nie obejmuje wszystkich mających zastosowanie zasadniczych wymagań, należy udokumentować sposób spełnienia stosownych zasadniczych wymagań nieobjętych normą (152).
Zasadnicze wymagania określają wyniki, które należy osiągnąć, lub zagrożenia, którymi należy się zająć, ale nie wskazują rozwiązań technicznych, które można przy tym wykorzystać. Dokładne rozwiązania techniczne mogą być wedle uznania producenta przedstawione w normie lub innych specyfikacjach technicznych lub opracowane zgodnie z ogólną wiedzą inżynieryjną lub naukową dostępną w literaturze fachowej. Dzięki tej elastyczności producent może wybierać sposób, w jaki spełni wymagania. Pozwala ona także np. na dostosowanie materiałów i projektu do postępu technologicznego. W związku z powyższym unijne prawodawstwo harmonizacyjne oparte na zasadniczych wymaganiach nie wymaga regularnego dostosowywania do postępu technicznego, ponieważ spełnienie wymagań jest oceniane w odniesieniu do stanu wiedzy technicznej w chwili wprowadzania produktu do obrotu.
Zasadnicze wymagania są określone w odpowiednich rozdziałach lub załącznikach do unijnych harmonizacyjnych aktów prawnych. Chociaż zasadnicze wymagania nie zawierają szczegółowych specyfikacji dotyczących produkcji, stopień dokładności sformułowań różni się w poszczególnych unijnych aktach harmonizacyjnych (153). Treść aktów jest z założenia na tyle precyzyjna, aby nakładały one, po przeniesieniu do prawa krajowego, prawnie wiążące obowiązki, które mogą być egzekwowane, i aby ułatwiały Komisji składanie wniosków o normalizację do europejskich organizacji normalizacyjnych w celu opracowania norm zharmonizowanych. Zasadnicze wymagania formułowane również w taki sposób, aby umożliwić ocenę zgodności z nimi nawet w przypadku braku norm zharmonizowanych lub jeśli producent zdecyduje się ich nie stosować.
4.1.2. ZGODNOŚĆ Z ZASADNICZYMI WYMAGANIAMI: NORMY ZHARMONIZOWANE
|
4.1.2.1. Definicja normy zharmonizowanej
Definicje terminów „norma”, „norma krajowa”, „norma europejska”, „norma zharmonizowana” i „norma międzynarodowa” zawarte są w rozporządzeniu (UE) nr 1025/2012 (154).
|
— |
„Normy” to specyfikacje techniczne o nieobowiązkowym charakterze przyjęte przez uznane jednostki normalizacyjne do wielokrotnego lub ciągłego stosowania. |
|
— |
„Normy europejskie” to „normy” przyjęte przez europejskie organizacje normalizacyjne wymienione w załączniku I do rozporządzenia (UE) nr 1025/2012 (155). |
|
— |
Biorąc pod uwagę powyższe dwie definicje, „normy zharmonizowane” to „normy europejskie” przyjęte na wniosek Komisji na potrzeby stosowania unijnego prawodawstwa harmonizacyjnego. Normy zharmonizowane zachowują swój nieobowiązkowy charakter. |
Definicja „normy zharmonizowanej” w kontekście rozporządzenia (UE) nr 1025/2012 nie ogranicza się do norm zharmonizowanych będących częścią zharmonizowanego prawodawstwa dotyczącego produktów, ponieważ rozporządzenie uwzględnia zastosowanie prawodawstwa harmonizacyjnego w odniesieniu do usług na podobnych zasadach jak w przypadku produktów.
4.1.2.2. Rola norm zharmonizowanych
Normy zharmonizowane są tworzone i publikowane, podobnie jak inne normy europejskie, zgodnie z wewnętrznymi regulaminami europejskich organizacji normalizacyjnych. Według nich wszystkie normy europejskie muszą być przeniesione na poziom krajowy przez krajowe jednostki normalizacyjne. Oznacza to, że w taki sam sposób należy wprowadzić stosowne normy europejskie jako normy krajowe i że krajowe normy pozostające w konflikcie z normami europejskimi muszą w określonym czasie zostać wycofane.
Normy zharmonizowane to normy europejskie, którym rozporządzenie (UE) nr 1025/2012 i sektorowe prawodawstwo harmonizacyjne Unii nadają specjalne znaczenie (156). Normy zharmonizowane zachowują swój nieobowiązkowy charakter (157). Jednak należy zauważyć, że w definicji normy zharmonizowanej nie wspomina się o publikowaniu tytułu tejże normy w Dzienniku Urzędowym Unii Europejskiej. Dopóki tytuł normy zharmonizowanej nie zostanie opublikowany w Dzienniku Urzędowym Unii Europejskiej, norma zharmonizowana lub jej część nie stanowi podstawy domniemania zgodności z wymogami zasadniczymi lub innymi, które ma obejmować. Składając wnioski o normalizację (zlecenia normalizacji), Komisja formalnie zwraca się do europejskich organizacji normalizacyjnych o przedstawienie norm zharmonizowanych. Rolę i sposób opracowania przez Komisję wniosków o normalizację kierowanych do europejskich organizacji normalizacyjnych opisano szczegółowo w „Vademecum nt. normalizacji europejskiej” (158). Wcześniej Komisja przeprowadza konsultacje z państwami członkowskimi (159). Wypracowanie norm opartych na konsensusie w rozumieniu rozporządzenia (UE) nr 1025/2012 (160) wiąże się z szerokimi konsultacjami organów sektorowych na poziomie krajowym. W związku z powyższym wniosek o normalizację stanowi silny przejaw oczekiwań organów publicznych.
Europejskie organizacje normalizacyjne zajmą oficjalne stanowisko w sprawie wniosku o normalizację złożonego przez Komisję, zgodne ze swoimi wewnętrznymi regulaminami. Na podstawie tych regulaminów, a w przypadku norm zharmonizowanych także rozporządzenia (UE) nr 1025/2012, przyjęcie wniosku o normalizację i późniejsza praca normalizacyjna są początkiem okresu zawieszenia działań krajowych jednostek normalizacyjnych.
Opracowywanie i przyjmowanie zharmonizowanych norm opiera się na rozporządzeniu (UE) nr 1025/2012 (161) oraz ogólnych wytycznych dotyczących współpracy między europejskimi organizacjami normalizacyjnymi a Komisją i EFTA z dnia 28 marca 2003 r. (162). Istnieje szereg wymagań, zasad i zobowiązań dotyczących normalizacji, takich jak udział wszystkich zainteresowanych stron (np. producentów, w tym MŚP, stowarzyszeń konsumenckich, zainteresowanych podmiotów z dziedziny ochrony środowiska i związków zawodowych), roli organów publicznych, jakości norm i jednolitej transpozycji norm europejskich w całej Unii przez krajowe organy normalizacyjne.
Europejskie organizacje normalizacyjne mają obowiązek ustalenia – zgodnie ze stosownymi wnioskami o normalizację – i opracowania norm zharmonizowanych w rozumieniu odpowiednich przepisów regulujących rynek wewnętrzny. Są także odpowiedzialne za przedstawienie listy przyjętych norm zharmonizowanych Komisji. Za techniczne zagadnienia norm zharmonizowanych odpowiadają wyłącznie europejskie organizacje normalizacyjne. Gdy organy publiczne dojdą do porozumienia w sprawie wniosku o normalizację, ustalenie rozwiązań technicznych powinno z zasady leżeć po stronie zainteresowanych stron. W określonych obszarach, takich jak środowisko naturalne, zdrowie i bezpieczeństwo na poziomie technicznym procesu normalizacji ważne jest uczestnictwo organów publicznych. Jednak unijne prawodawstwo harmonizacyjne dotyczące produktów nie przewiduje procedury, która nakładałaby na organy publiczne obowiązek systematycznej weryfikacji i zatwierdzania na poziomie unijnym lub krajowym treści norm zharmonizowanych, które zostały przyjęte przez europejskie organizacje normalizacyjne (163). Mimo to dialog pomiędzy jednostkami normalizacyjnymi a organami publicznymi oraz, gdy to stosowne, ich udział w procesie normalizacji powinny być częścią gwarancji, że warunki wniosku o normalizację są prawidłowo rozumiane i że w procesie wzięto pod uwagę publiczne obawy.
Europejskie organizacje normalizacyjne podejmują decyzję w sprawie programu pracy nad normami zharmonizowanymi zgodnie z odpowiednim wnioskiem o normalizację. Mogą również wskazać istniejące normy, które według ich oceny po zbadaniu i ewentualnych poprawkach będą spełniać warunki określone we wniosku o normalizację. Mogą także zmienić istniejące normy w celu spełnienia tych warunków. Na tej samej zasadzie mogą wskazać normy międzynarodowe lub krajowe i przyjąć je jako normy europejskie, a następnie przedstawić je Komisji jako normy zharmonizowane. Czasem może się również zdarzyć, że tylko pewne części lub przepisy normy zharmonizowanej dotyczą zasadniczego wymagania i tylko te części lub przepisy będą przewidywać domniemanie zgodności po opublikowaniu stosownych odniesień w Dzienniku Urzędowym Unii Europejskiej.
Norma zharmonizowana musi odpowiadać zasadniczym lub innym wymaganiom prawnym stosownego obszaru prawodawstwa zgodnie z danym wnioskiem o normalizację. Norma zharmonizowana może zawierać specyfikacje odnoszące się nie tylko do zasadniczych wymagań, ale również obejmować kwestie nieuregulowane. W takim przypadku specyfikacje te należy w jasny sposób odróżnić od przepisów dotyczących zasadniczych wymagań. Norma zharmonizowana niekoniecznie musi dotyczyć wszystkich zasadniczych wymagań, ale zawsze musi być wiadomo, które wymagania ma obejmować (164); w przeciwnym wypadku producent stosujący się do normy zharmonizowanej wskazanej w Dzienniku Urzędowym Unii Europejskiej nie wie, względem których wymagań zachodzi „domniemanie zgodności”, a organy publiczne nie wiedzą, względem których zasadniczych wymagań muszą takie domniemanie przyjąć.
Stosowne zasadnicze lub inne wymagania prawne, które mają być objęte daną normą wskazuje się zazwyczaj w oddzielnym załączniku informacyjnym (165) do normy zharmonizowanej. Jeżeli wymagania zasadnicze są uwzględnione tylko częściowo, należy to wyraźnie wskazać w normie. W niektórych przypadkach także zakres normy zharmonizowanej może jasno wskazywać na odpowiednie wymagania (np. jeśli istnieje wyraźne odniesienie do objętych normą zagrożeń związanych z bezpieczeństwem). Podane w normie zharmonizowanej informacje dotyczące ujęcia zasadniczych lub innych wymagań ustalają tym samym zakres tzw. „domniemania zgodności z wymaganiami prawnymi”.
Należy wyraźnie rozróżnić pomiędzy „zgodnością z normą” a „domniemaniem zgodności (przy stosowaniu normy zharmonizowanej)”. „Zgodność z normą” zazwyczaj odnosi się do sytuacji, gdy norma jest „stosowana w pełni”. Taka sytuacja ma miejsce np. w przypadku dobrowolnej certyfikacji w związku z wdrożeniem danej normy. Do celów „domniemania zgodności” wystarczy stosować tylko przepisy odnoszące się do zasadniczych lub innych wymagań prawnych, które mają zostać ujęte w normie.
Normy zharmonizowane nigdy nie zastępują prawnie wiążących zasadniczych wymagań. Specyfikacja podana w normie zharmonizowanej nie stanowi alternatywy dla odpowiedniego zasadniczego lub innego wymagania prawnego, a jedynie możliwy środek techniczny, który pozwoli je spełnić. W szczególności w prawodawstwie harmonizacyjnym dotyczącym ryzyka oznacza to, że producent zawsze – nawet gdy stosuje normy zharmonizowane – musi ocenić wszystkie rodzaje ryzyka związane ze swoim produktem, aby ustalić stosowne zasadnicze (lub inne) wymagania. Po dokonaniu takiej oceny producent może zdecydować się na zastosowanie specyfikacji określonych w normach zharmonizowanych, aby wdrożyć „środki zmniejszania ryzyka” (166), które są określone w normach zharmonizowanych. W prawodawstwie harmonizacyjnym dotyczącym ryzyka normy zharmonizowane najczęściej przewidują określone środki zmniejszenia lub wyeliminowania ryzyka, przy czym producenci pozostają w pełni odpowiedzialni za ocenę ryzyka podejmowaną w celu zidentyfikowania stosownych zasadniczych wymagań i wyboru odpowiednich norm zharmonizowanych lub innych specyfikacji.
Rola norm zharmonizowanych w zapewnianiu zgodności z odpowiednimi zasadniczymi wymaganiami określonymi przez producenta – ogólny sposób rozumowania, w przypadku gdy producent musi określić mające zastosowanie zasadnicze wymagania:

Jeśli w normach zharmonizowanych nie wskazano jasno uwzględnianych zasadniczych wymagań, to normy takie mogą stać się mniej przydatne dla producentów, ponieważ mniejsza jest pewność prawa co do rzeczywistego „zakresu domniemania zgodności”. Również niejasne lub niepoprawne wskazanie uwzględnianych zasadniczych wymagań może w niektórych przypadkach prowadzić do formalnych sprzeciwów wobec norm zharmonizowanych (zob. pkt 4.1.2.5). Jeżeli dana norma zharmonizowana obejmuje jedynie część zasadniczych wymagań określonych odpowiednio przez producenta lub tylko niektóre ich aspekty, producent musi dodatkowo zastosować inne odpowiednie specyfikacje techniczne lub opracować rozwiązania zgodne z ogólną wiedzą inżynieryjną lub naukową dostępną w literaturze fachowej w celu spełnienia zasadniczych wymagań przedmiotowego ustawodawstwa. Podobnie jeśli producent zdecyduje się nie stosować do wszystkich przepisów danej normy zharmonizowanej, które w innym przypadku stwarzałyby domniemanie zgodności, musi na podstawie własnej oceny ryzyka wskazać w dokumentacji produktu, w jaki sposób osiągnięto zgodność z przepisami prawa lub że produkt nie podlega stosownym zasadniczym wymaganiom.
Czasami normy mogą zawierać błędy lub można je interpretować w różny sposób. Jeśli producent znajdzie taki błąd lub wątpliwość, powinien najpierw skontaktować się z krajową jednostką normalizacyjną w celu uzyskania wyjaśnienia.
4.1.2.3. Proces tworzenia norm zharmonizowanych powodujących domniemanie zgodności
Cała procedura, na podstawie której norma zharmonizowana powoduje domniemanie zgodności, jest opisana na schemacie 1.
Warunkiem przygotowania wniosku o normalizację, który jest wnioskiem o opracowanie norm zharmonizowanych, jest istnienie unijnych przepisów harmonizacyjnych (lub toczący się proces ich przygotowywania) (167) przewidujących wykorzystanie norm zharmonizowanych jako środka do osiągnięcia zgodności z zasadniczymi lub innymi wymaganiami prawnymi, tj. wyrażenie przez prawodawcę politycznej zgody na opracowanie i opublikowanie norm zharmonizowanych w ramach prawnych określonych w rozporządzeniu (UE) nr 1025/2012.
|
1. |
Planowanie przez Komisję wniosku o normalizację: Komisja publikuje swoje plany dotyczące przyszłych wniosków o normalizację w rocznym programie prac Unii w zakresie normalizacji europejskiej zgodnie z art. 8 rozporządzenia (UE) nr 1025/2012. Program ten określa również potrzeby w zakresie normalizacji związane z przyszłym prawodawstwem harmonizacyjnym. |
|
2. |
Przygotowanie wniosku o normalizację: Komisja przygotowuje zgodnie z art. 10 ust. 1 rozporządzenia (UE) nr 1025/2012 projekt wniosku o normalizację, konsultując się, zgodnie z art. 10 ust. 2 i art. 12 tego rozporządzenia, z europejskimi organizacjami normalizacyjnymi, ekspertami z poszczególnych sektorów z państw członkowskich oraz odpowiednimi stronami zainteresowanymi na poziomie europejskim. |
|
3. |
Przyjęcie wniosku o normalizację i powiadomienie o nim: Komisja przyjmuje wniosek o normalizację w formie decyzji wykonawczej Komisji skierowanej do europejskich organizacji normalizacyjnych po uzyskaniu pozytywnej opinii od państw członkowskich zgodnie z procedurą określoną w art. 22 ust. 3 rozporządzenia (UE) nr 1025/2012. Następnie odpowiednie europejskie organizacje normalizacyjne są powiadamiane o wniosku o normalizację. |
|
4. |
Przyjęcie wniosku o normalizację: Odpowiednia europejska organizacja normalizacyjna wskazuje, czy przyjmuje wniosek (168) w terminie określonym w art. 10 ust. 3 rozporządzenia (UE) nr 1025/2012. Krajowe jednostki normalizacyjne są zobowiązane do przestrzegania okresu zawieszenia zgodnie z art. 3 ust. 6 rozporządzenia (UE) nr 1025/2012. Odpowiednia europejska organizacja normalizacyjna może wnioskować o finansowanie unijne (grant na działanie) na podstawie rozdziału V rozporządzenia (UE) nr 1025/2012. Komisja informuje odpowiednią europejską organizację normalizacyjną o przyznaniu grantu w terminie określonym w art. 10 ust. 4 rozporządzenia (UE) nr 1025/2012. |
|
5. |
Przygotowanie i zatwierdzenie programu prac: Odpowiednia europejska organizacja normalizacyjna (lub kilka organizacji) przygotowuje (przygotowują wspólny) program prac zgodny z danym wnioskiem o normalizację i przedstawia (przedstawiają) go Komisji. W stosownych przypadkach Komisja może podać informacje na temat priorytetów w pracach normalizacyjnych. |
|
6. |
Prace nad projektem: Odpowiedzialny komitet techniczny europejskiej organizacji normalizacyjnej (169) opracowuje projekt normy europejskiej. Europejskie organizacje normalizacyjne stosują zasady uznane przez Światową Organizację Handlu (WTO) w zakresie normalizacji (spójność, przejrzystość, otwartość, konsensus, dobrowolność stosowania i skuteczność). Oprócz tego w art. 3–5 rozporządzenia (UE) nr 1025/2012 odniesiono się do bezpośrednio stosowanych wymagań dotyczących udziału zainteresowanych stron i przejrzystości programów pracy oraz projektów norm. Przyjęty wniosek o normalizację jest jednym z dokumentów referencyjnych, do których musi odnosić się komitet techniczny podczas przygotowywania projektu normy. Zgodnie z art. 10 ust. 5 rozporządzenia (UE) nr 1025/2012 odpowiednia europejska organizacja normalizacyjna musi poinformować Komisję o podjętych działaniach (sprawozdawczość) i wdrożyć wraz z Komisją i pod jej przewodnictwem odpowiednie środki (170) w celu oceny zgodności projektów norm z pierwotnym wnioskiem. |
|
7. |
Ankieta powszechna: Europejskie organizacje normalizacyjne wraz z krajowymi jednostkami normalizacyjnymi organizują ankietę powszechną, w której wszystkie zainteresowane strony mogą zgłosić swoje uwagi za pośrednictwem krajowych jednostek normalizacyjnych. W art. 4 ust. 3 rozporządzenia (UE) nr 1025/2012 określono procedurę do stosowania w przypadku, gdy krajowa jednostka normalizacyjna otrzyma uwagi wskazujące na możliwy negatywny wpływ na rynek wewnętrzny. |
|
8. |
Uwzględnianie otrzymanych uwag: Odpowiedzialny komitet techniczny rozpatruje uwagi zebrane w czasie ankiety powszechnej i przygotowuje ostateczny projekt normy europejskiej. |
|
9. |
Formalne głosowanie: Krajowe jednostki normalizacyjne głosują nad ostatecznym projektem w formalnym głosowaniu, w którym krajowe jednostki normalizacyjne dysponują głosami ważonymi. Ostateczny projekt jest przyjmowany, jeżeli zostanie przegłosowany zwykłą większością i jeżeli oddano przynajmniej 71,00 % ważonych głosów za jego przyjęciem (nieobecni nie są uwzględniani). |
|
10. |
Ratyfikacja i publikacja normy europejskiej (EN): Jeśli głosowanie zakończy się pozytywnym wynikiem, odpowiednia europejska organizacja normalizacyjna ratyfikuje i publikuje normę europejską (EN). Ponieważ w tym przypadku norma europejska jest w tym samym duchu co unijne prawodawstwo harmonizacyjne i została przygotowana na podstawie wniosku o normalizację ze strony Komisji, jest ona normą zharmonizowaną w rozumieniu art. 2 ust. 1 lit. c) rozporządzenia (UE) nr 1025/2012 – jednak nie daje jeszcze podstaw do domniemania zgodności. |
|
11. |
Przedstawienie Komisji danych identyfikacyjnych: Stosowna europejska organizacja normalizacyjna automatycznie przekazuje Komisji dane identyfikacyjne przygotowanej normy zharmonizowanej. Informacje te zawierają w szczególności numer identyfikacyjny i tytuł we wszystkich oficjalnych językach UE. |
|
12. |
Weryfikacja spełnienia warunków publikacji w Dzienniku Urzędowym Unii Europejskiej: Zgodnie z art. 10 ust. 5 rozporządzenia (UE) nr 1025/2012 Komisja musi zweryfikować, czy dana norma zharmonizowana jest zgodna z pierwotnym wnioskiem o normalizację. W czasie tej weryfikacji Komisja sprawdza w szczególności, czy dana norma zharmonizowana mieści się w ramach stosownego wniosku o normalizację i czy zasadnicze lub inne wymagania prawne, które miały zostać uwzględnione są jasno określone i ujęte w normie. Podczas weryfikacji Komisja nie musi oceniać normy od strony technicznej, ponieważ do jej zadań nie należy zatwierdzanie zagadnień technicznych czy branie za nie odpowiedzialności. Jednak już na tym etapie Komisja może także ocenić, czy specyfikacje techniczne zawarte w normie zharmonizowanej pozwalają na spełnienie odpowiednich zasadniczych wymagań. Taka ocena może prowadzić do nieopublikowania odniesień do normy w Dzienniku Urzędowym Unii Europejskiej). |
|
13. |
Publikacja odniesień w Dzienniku Urzędowym Unii Europejskiej: Zgodnie z art. 10 ust. 6 rozporządzenia (UE) nr 1025/2012 Komisja publikuje odniesienia do normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej. Publikacja ta powoduje domniemanie zgodności z zasadniczymi lub innymi wymaganiami prawnymi objętymi daną normą zharmonizowaną. Domniemanie zgodności obowiązuje często od dnia publikacji w Dzienniku Urzędowym Unii Europejskiej i, w najczęstszych przypadkach, (zob. też pkt 4.1.2.5), można je uchylić w drodze formalnego sprzeciwu lub publikacji odniesień do zmienionej wersji normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej. |
|
14. |
Transpozycja krajowa: Krajowe jednostki normalizacyjne mają obowiązek transponować stosowną normę europejską (171) jako identyczną normę krajową na podstawie wewnętrznych regulaminów europejskich organizacji normalizacyjnych. Zgodnie z art. 3 ust. 6 rozporządzenia (UE) nr 1025/2012 są również zobowiązane do wycofania norm krajowych, które są sprzeczne z normą zharmonizowaną. |
|
15. |
Formalny sprzeciw: Zgodnie z art. 11 rozporządzenia (UE) nr 1025/2012 (172) państwo członkowskie lub Parlament Europejski może zakwestionować publikację odniesień do normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej. W ramach tej procedury państwo członkowskie lub Parlament Europejski może poprosić Komisję o przygotowanie decyzji Komisji w celu uniemożliwienia lub uchylenia domniemania zgodności. Formalny sprzeciw może zostać złożony natychmiast po tym, jak norma została przyjęta i ratyfikowana (w przypadku CEN i Cenelec) lub przyjęta (w przypadku ETSI) zgodnie z regulaminem danej organizacji. |
Schemat 1
Proces prowadzący do tworzenia norm zharmonizowanych i domniemania zgodności

4.1.2.4. Domniemanie zgodności
Zakłada się, że normy zharmonizowane są zgodne z określonymi w nich zasadniczymi wymaganiami, jeżeli odniesienia do nich zostały opublikowane w Dzienniku Urzędowym Unii Europejskiej. Odniesienia do norm zharmonizowanych są publikowane jako komunikaty Komisji w serii C Dziennika Urzędowego Unii Europejskiej (173).
Normy europejskie, w tym normy zharmonizowane są często w całości lub w części oparte na międzynarodowych normach ISO lub IEC. Jednak czasami domniemanie zgodności zachodzi tylko w przypadku stosowania europejskich wersji tych norm ze względu na wprowadzone zmiany.
Celem publikacji odniesień w Dzienniku Urzędowym jest ustalenie daty, od której zachodzi domniemanie zgodności. Publikacja odniesień do norm zharmonizowanych należy do administracyjnych zadań Komisji, które są wykonywane bez dalszych konsultacji z państwami członkowskimi lub odpowiednimi komitetami sektorowymi. Jest to ostateczny cel normy zharmonizowanej oraz koniec procesu zapoczątkowanego wydaniem stosownego wniosku o normalizację przez Komisję. Zanim Komisja opublikuje informacje dotyczące normy musi jednak ocenić wspólnie z europejskimi organizacjami normalizacyjnymi, zgodnie z art. 10 ust. 5 rozporządzenia (UE) nr 1025/2012, czy spełniono warunki danego(-ych) wniosku(-ów) o normalizację i czy norma zharmonizowana rzeczywiście uwzględnia wskazane (174) w niej zasadnicze lub inne wymagania.
Publikacja odniesień nie odbywa się automatycznie – Komisja musi przeprowadzić określone kontrole i oceny przed opublikowaniem informacji o normie. Komisja może zatem odmówić opublikowania danych identyfikacyjnych lub, w pewnych przypadkach, może wprowadzić określone ograniczenia, które zostaną opublikowane wraz z danymi identyfikacyjnymi.
W sytuacji gdy rozpoczęto już procedurę formalnego sprzeciwu, pojawiają się wątpliwości, czy norma zharmonizowana w całości spełnia wskazane w niej wymagania w rozumieniu art. 11 ust. 1 rozporządzenia (UE) nr 1025/2012. Ze względu na te wątpliwości zgodnie z art. 10 ust. 6 rozporządzenia (UE) nr 1025/2012 Komisja nie może opublikować odniesienia; taka sytuacja wymaga podjęcia przez Komisję decyzji wykonawczej w rozumieniu art. 11 ust. 1.
Również w innych sytuacjach informacje o normie mogą nie być opublikowane. Ocena przeprowadzona zgodnie z art. 10 ust. 5 może wykazać, że nie spełniono warunków danego wniosku o normalizację i że norma zawiera oczywiste błędy. W takich przypadkach warunki wszczęcia procedury sprzeciwu zgodnie z art. 11 (175) rozporządzenia (UE) nr 1025/2012 nie są zazwyczaj spełnione.
Inne powody niepublikowania normy mogą być następujące: norma nie mieści się w ramach odpowiedniego wniosku o normalizację; produkty, których dotyczy norma, nie wchodzą w zakres odpowiedniego unijnego prawodawstwa harmonizacyjnego; norma nie wskazuje, które (zasadnicze) wymagania prawne obejmuje (176); norma nie obejmuje wskazanych w niej (zasadniczych) wymagań prawnych; norma zawiera specyfikacje, które nie są w tym samym duchu, co zasadnicze wymagania, i nie oddziela ich wyraźnie od specyfikacji zgodnych z zasadniczymi wymaganiami; norma dotyczy innych wymagań prawnych niż określone we wniosku o normalizację; norma zawiera odniesienia normatywne do innych specyfikacji, które są nie do przyjęcia ze względu na ich pochodzenie lub brak odpowiedniego opartego na konsensusie procesu ich przyjmowania, lub odniesienia normatywne nie są jeszcze dostępne; inne powody wynikające z niezastosowania wewnętrznych regulaminów europejskich organizacji normalizacyjnych lub nieprzestrzeganie wymogu rozporządzenia (UE) nr 1025/2012 w czasie przygotowywania danej normy zharmonizowanej.
W takich przypadkach Komisja poprzez niepublikowanie informacji o normie zapewnia prawidłowe zastosowanie odnośnego unijnego prawodawstwa harmonizacyjnego oraz spójne i właściwe funkcjonowanie jednolitego rynku. W tej sytuacji Komisja może jedynie prosić odpowiednie europejskie organizacje normalizacyjne o skorygowanie norm, odnosząc się do wymagań określonych w danym wniosku o normalizację i innych uznanych i ustalonych zasad, na podstawie których takie organizacje powinny działać. W niektórych przypadkach Komisja może rozważać opublikowanie odniesień z ograniczeniami, mając jednak na uwadze, że ograniczenia te nie powinny pokrywać się z powodami, dla których należy wszcząć właściwą procedurę sprzeciwu. Uzasadnienie niepublikowania odniesień wynika z samego wniosku o normalizację, jednak Komisja może odmówić publikacji również w celu ochrony funkcjonowania jednolitego rynku.
Odwoływanie się do norm zharmonizowanych zacytowanych w Dzienniku Urzędowym Unii Europejskiej i dających domniemanie zgodności pozostaje dobrowolne (177). Producent może zdecydować, czy będzie się do nich odwoływał. Jednak jeśli producent zdecyduje się nie stosować norm zharmonizowanych, musi wykazać, że jego produkty są zgodne z zasadniczymi wymaganiami za pomocą innych, wybranych przez siebie środków (np. za pomocą dowolnych istniejących specyfikacji technicznych, w tym wszystkich innych dostępnych norm). Jeśli producent stosuje tylko część normy zharmonizowanej lub gdy stosowana przez niego norma zharmonizowana nie obejmuje w całości wszystkich stosownych zasadniczych wymagań, domniemanie zgodności ma tylko taki zakres, w jakim norma zharmonizowana odpowiada zasadniczym wymaganiom. Z tego powodu niezbędne jest, aby każda norma zharmonizowana zawierała jasne i poprawne informacje na temat ujętych w niej (zasadniczych) wymagań prawnych.
Według niektórych unijnych aktów harmonizacyjnych zgodność z normami zharmonizowanymi jest opcją wpływającą na stosowną procedurę oceny zgodności i czasami daje możliwość oceny zgodności bez udziału podmiotów zewnętrznych lub większy wybór procedur (178).
4.1.2.5. Wycofanie, ograniczenie lub uniemożliwienie domniemania zgodności
Rozporządzenie (UE) nr 1025/2012 zawiera przepis, zgodnie z którym można podważać publikację tytułów norm zharmonizowanych w Dzienniku Urzędowym Unii Europejskiej (179). Może do tego dojść przed opublikowaniem odniesienia do normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej lub w przypadku normy zharmonizowanej, o której informacje zostały już tam opublikowane.
W obydwu przypadkach, jeśli państwo członkowskie lub Parlament Europejski (180) uważa, że norma zharmonizowana nie spełnia w całości określonych w niej i zapisanych w stosownym unijnym prawodawstwie harmonizacyjnym wymagań, powinno (powinien) poinformować o tym Komisję. Po konsultacjach z państwami członkowskimi (181) Komisja może podjąć decyzję wykonawczą o:
|
— |
opublikowaniu, niepublikowaniu lub opublikowaniu z ograniczeniami odniesień do danej normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej lub |
|
— |
utrzymaniu lub utrzymaniu z ograniczeniami odniesień do danej normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej albo wycofaniu z niego tych odniesień. |
We wszystkich przypadkach Komisja musi opublikować na swojej stronie internetowej (182) informacje na temat norm zharmonizowanych, w stosunku do których wydano takie decyzje.
W ramach tych obowiązków zgodnie z rozporządzeniem (UE) nr 1025/2012 i stosownym prawodawstwem sektorowym Komisja może również przygotować i zaproponować takie decyzje wykonawcze, aby z własnej inicjatywy zgłosić sprzeciw wobec norm zharmonizowanych. Komisja ma także obowiązek zgłosić sprzeciw względem danej normy zharmonizowanej, jeśli państwo członkowskie na podstawie klauzuli ochronnej (183) zakwestionowało zgodny z nią produkt i jeśli takie postępowanie jest uzasadnione.
Procedura kwestionowania normy zharmonizowanej i jej skutków nie wpływa na nią jako na normę zharmonizowaną lub normę europejską, ponieważ tylko europejskie organizacje normalizacyjne mogą podejmować decyzje o zmianie lub wycofaniu efektów swojej pracy. Procedura ta daje prawodawcy możliwość kontroli domniemania zgodności, tj. skutku prawnego, który wynika z opublikowania odniesienia do normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej. Może ona prowadzić tylko do tego, że publikacja w Dzienniku Urzędowym zostanie wycofana, ograniczona lub do niej nie dojdzie. W pierwszych dwóch przypadkach oznacza to, że dana norma nie będzie już stanowić podstawy domniemania zgodności z zasadniczymi wymaganiami lub domniemanie zgodności z zasadniczymi wymaganiami zostanie ograniczone. W ostatnim przypadku (gdy nie dochodzi do publikacji) taka norma w ogóle nie stanie się normą zharmonizowaną powodującą domniemanie zgodności.
Normę zharmonizowaną można zakwestionować w dowolnym momencie po jej przyjęciu przez CEN, CENELEC lub ETSI jako normę europejską.
Ponadto odniesienie może zostać usunięte z Dziennika Urzędowego UE przez Komisję bez zastosowania procedury formalnego sprzeciwu w pewnych wyjątkowych przypadkach, gdy dana wersja normy zharmonizowanej nie jest już rewidowana i aktualizowana przez europejską organizację normalizacyjną i jeżeli sama ta organizacja nie uznaje jej za normę. Takie przypadki mogą być np. następujące: dana zharmonizowana norma została wycofana przez właściwą europejską organizację normalizacyjną bez zamiaru przyjęcia zrewidowanej normy zharmonizowanej; krajowe normy transponujące zharmonizowaną normę nie są już dostępne lub obowiązujące jako normy krajowe. Pojęcie zasadniczych wymagań opiera się na założeniu, że normy zharmonizowane odzwierciedlają powszechnie uznawany stan wiedzy, a europejska organizacja normalizacyjna regularnie rewiduje normy. Jeżeli jest oczywiste, że norma zharmonizowana nie jest już uznawana za normę przez właściwą europejską organizację normalizacyjną lub gdy norma ta nie jest już rewidowana lub dostępna jako norma krajowa, takiego dokumentu nie można już co do zasady wykorzystywać w celu domniemania zgodności. Celem art. 11 rozporządzenia (UE) nr 1025/2012 jest zapewnienie procedury umożliwiającej kwestionowanie jedynie obowiązujących norm zharmonizowanych, a nie wycofanych norm zharmonizowanych lub projektów norm zharmonizowanych, które nie mogą być postrzegane jako przyjęte normy europejskie w kontekście definicji zawartych w art. 2 rozporządzenia (UE) nr 1025/2012.
Inne szczególne sytuacje, w których Komisja może być zmuszona do usunięcia odniesień z Dziennika Urzędowego bez formalnego sprzeciwu, to przypadki gdy publikacja w Dzienniku Urzędowym została dokonana omyłkowo lub gdy opublikowano odniesienie do dokumentu, który nie może być uważany za normę zharmonizowaną. Ten ostatni przypadek może obejmować sytuacje, gdy norma nie jest objęta wnioskiem o normalizację lub gdy nie obejmuje ona żadnych zasadniczych wymagań lub gdy norma ta nie została prawidłowo przyjęta przez europejską organizację normalizacyjną zgodnie z uznanymi zasadami normalizacyjnymi.
Zgodnie z rozporządzeniem (UE) nr 1025/2012 przed podjęciem formalnych decyzji Komisja jest zobowiązana do poinformowania zainteresowanych stron (184) o wszystkich wniesionych formalnych sprzeciwach w odniesieniu do norm zharmonizowanych.
4.1.2.6. Rewizja norm zharmonizowanych
Normy zharmonizowane przekładają zasadnicze wymagania na szczegółowe specyfikacje techniczne, metody pomiaru wykorzystywane w celu oceny i/lub zadeklarowania zgodności z zasadniczymi wymaganiami, a w niektórych przypadkach wartości liczbowe pozwalające na zgodność z zasadniczymi wymaganiami. Podobnie jak inne dokumenty techniczne, podlegają one zmianom czyli rewizji.
Formalną decyzję o przeprowadzeniu rewizji normy zharmonizowanej zasadniczo podejmują europejskie organizacje normalizacyjne. Rewizja jest przeprowadzana na ich własnej inicjatywy (185), bezpośrednio w odpowiedzi na wniosek o normalizację ze strony Komisji lub pośrednio na podstawie decyzji Komisji po wniesieniu formalnego sprzeciwu. Potrzeba rewizji może wynikać ze zmian w zakresie unijnego aktu harmonizacyjnego (np. rozszerzenie zakresu o inne produkty lub zmiana zasadniczych wymagań), z faktu, że Komisja lub państwo członkowskie kwestionuje treść normy zharmonizowanej, twierdząc, że nie może ona już stanowić podstawy domniemania zgodności z zasadniczymi wymaganiami lub z rozwoju technologicznego.
Rewizja normy zharmonizowanej musi odbywać się na podstawie wniosku o normalizację, dzięki czemu będzie mogło zaistnieć domniemanie zgodności. Warunki oryginalnego wniosku o normalizację dotyczą również rewizji normy zharmonizowanej, chyba że można z nich wywnioskować inaczej. Nie wyklucza to możliwości wydania nowego wniosku o normalizację, w szczególności jeśli rewizja jest związana z brakami w zasadniczych wymaganiach.
Aby zachodziło domniemanie zgodności, norma podlegająca rewizji musi spełniać ogólne warunki unijnego prawodawstwa harmonizacyjnego: norma musi opierać się na wniosku o normalizację, być przedstawiona Komisji przez odpowiednią europejską organizację normalizacyjną, a odniesienia do niej muszą zostać opublikowane przez Komisję w Dzienniku Urzędowym.
Europejska organizacja normalizacyjna, zgodnie ze swoim wewnętrznym regulaminem, określa dla krajowych organów normalizacyjnych ostateczny termin wycofania danej wersji normy krajowej – w tym przypadku normy krajowej stanowiącej transpozycję poprzedniej wersji normy zharmonizowanej. Okres przejściowy, w którym zarówno wycofana, jak i zrewidowana norma zharmonizowana może stanowić podstawę domniemania zgodności, jest ustalany przez Komisję i publikowany w Dzienniku Urzędowym. Jest to zwykle okres pomiędzy datą publikacji odniesienia do nowej wersji normy w Dzienniku Urzędowym a datą wycofania sprzecznych z nią norm krajowych, tj. norm krajowych, które transponują poprzednie wersje norm zharmonizowanych. Komisja ma obowiązek dopilnować, aby takie okresy przejściowe były wystarczające i ustalone spójnie dla wszystkich norm zharmonizowanych. Po okresie przejściowym tylko zrewidowana norma zharmonizowana daje podstawę domniemania zgodności.
Ze względów bezpieczeństwa lub innych Komisja może uznać, że domniemanie zgodności na podstawie zastąpionej wersji normy zharmonizowanej musi zakończyć się jeszcze przed datą wycofania ustaloną przez daną europejską organizację normalizacyjną albo po tej dacie. W takich przypadkach Komisja ustala wcześniejszą lub późniejszą datę, kiedy wycofana norma zharmonizowana przestanie stanowić podstawę domniemania zgodności, i opublikuje tę informację w Dzienniku Urzędowym. Jeśli pozwolą na to okoliczności, przed podjęciem decyzji o skróceniu lub wydłużeniu okresu, w którym obie wersje normy są podstawą domniemania zgodności, Komisja może skonsultować się z państwami członkowskimi.
Jeśli na podstawie propozycji Komisji nie podjęto innej decyzji, usunięcie odniesienia do normy zharmonizowanej z Dziennika Urzędowego po jej rewizji nie unieważnia automatycznie dotychczasowych certyfikatów wydanych przez jednostki notyfikowane; dotyczy to tylko zgodności ustalonej podczas nowych ocen zgodności przeprowadzanych zgodnie z nowymi normami zharmonizowanymi. Produkt wytworzony zgodnie ze starymi certyfikatami może być nadal zgodny z zasadniczymi wymaganiami i może pozostawać w obrocie do końca ważności stosownych certyfikatów wydanych przez jednostki notyfikowane. Producenci powinni jednak oszacować zakres zmian w zastąpionej wersji normy. Rodzaj działania, które powinien podjąć producent zależy od charakteru zmian w normach zharmonizowanych, a w szczególności od tego, czy dane zmiany są istotne, jeśli chodzi o spełnianie zasadniczych wymagań, i czy dotyczą danego produktu. Ponadto jednostka notyfikowana na bieżąco śledzi wszelkie zmiany w powszechnie uznanym stanie wiedzy technicznej wskazujące, że zatwierdzony typ może nie spełniać już mających zastosowanie wymagań, oraz ustala, czy zmiany takie wymagają dalszego badania. Jeśli tak jest, jednostka notyfikowana informuje o tym producenta. Odniesienie do zrewidowanej normy zharmonizowanej, wraz z informacją o zastąpionej wersji normy zharmonizowanej, oraz data, kiedy zastąpiona norma przestanie stanowić podstawę domniemania zgodności, są publikowane razem w Dzienniku Urzędowym. W interesie producenta leży zapoznawanie się z każdym wydaniem wykazu norm zharmonizowanych i sprawdzanie, czy normy zharmonizowane stosowane przez niego w celu zapewnienia zgodności produktu zachowują swoją ważność. Jest to szczególnie ważne w przypadkach, gdy producent sam deklaruje zgodność (w przypadku wewnętrznej kontroli produkcji) i gdy producent chce zapewnić stałe domniemanie zgodności dla produktów, które wprowadza do obrotu.
W związku z wytycznymi (186) uzgodnionymi między Komisją a europejskimi organizacjami normalizacyjnymi oczekuje się, że wszystkie zrewidowane normy zharmonizowane będą zawierać konkretne informacje na temat istotnych modyfikacji w zrewidowanych lub zmienionych normach zharmonizowanych, a informacje te powinny być udostępniane publicznie (nieodpłatnie) przez organizacje normalizacyjne.
Norma zharmonizowana może zawierać odniesienia normatywne do innych norm. Ze względu na wspomniane odniesienia normy te lub ich części stają się nieodzowne przy stosowaniu danej normy zharmonizowanej. Przy odnoszeniu się do innych norm zastosowanie mają wewnętrzne regulaminy europejskich organizacji normalizacyjnych. Ze względu na charakter norm zharmonizowanych nie należy w normalnych okolicznościach używać niedatowanych odniesień do innych norm, jeżeli stosowne przepisy mają być zgodne z zasadniczymi lub innymi wymaganiami prawnymi. Niedatowane odniesienia mogą powodować sytuacje, gdy zmiany w specyfikacjach zawartych w normach zharmonizowanych stanowiących podstawę domniemania zgodności są poza kontrolą i pozostają niejasne – zmiany w odniesieniach normatywnych nie mogą być kontrolowane w rozumieniu art. 10 ust. 6 rozporządzenia (UE) nr 1025/2012, chociaż w rzeczywistości zmiany takie powodują rewizję normy zharmonizowanej (lub jej części).
4.1.3. ZGODNOŚĆ Z ZASADNICZYMI WYMAGANIAMI: INNE MOŻLIWOŚCI
|
Stosowanie norm zharmonizowanych nie jest jedynym sposobem wykazania zgodności produktu – jednak tylko normy zharmonizowane (187), po opublikowaniu odniesień w Dzienniku Urzędowym Unii Europejskiej, mogą stanowić podstawę domniemania zgodności z zasadniczymi wymaganiami ujętymi w takich normach.
Zgodnie z niektórymi unijnymi aktami harmonizacyjnymi normy krajowe mogą stanowić podstawę domniemania zgodności – jako środki przejściowe – jeśli nie istnieje norma zharmonizowana regulująca ten sam obszar (188). Państwa członkowskie mogą przekazywać Komisji treść norm krajowych, które ich zdaniem spełniają zasadnicze wymagania. Po konsultacjach z państwami członkowskimi (189) Komisja powiadamia je, czy krajowe normy powinny powodować domniemanie zgodności. Jeśli opinia jest pozytywna, państwa członkowskie mają obowiązek publikować odniesienia do takich norm. Odniesienie jest również publikowane w Dzienniku Urzędowym Unii Europejskiej. Procedura ta nie była dotychczas stosowana, aby dać pierwszeństwo tworzeniu norm europejskich.
Producent może wybierać, czy będzie stosował takie normy zharmonizowane i czy będzie się do nich odwoływał. Jednak jeśli producent zdecyduje się nie stosować norm zharmonizowanych, musi wykazać, że jego produkty są zgodne z zasadniczymi wymaganiami za pomocą innych, wybranych przez siebie środków, które zapewniają poziom bezpieczeństwa lub ochrony innych interesów wymagany na mocy mających zastosowanie przepisów. Mogą to być specyfikacje techniczne takie jak normy krajowe, niezharmonizowane normy europejskie lub międzynarodowe, tj. normy niepublikowane w Dzienniku Urzędowym Unii Europejskiej lub specyfikacje własne producenta. W tych przypadkach producent nie korzysta z domniemania zgodności, ale musi sam wykazać zgodność produktu z przepisami. Oznacza to, że ma obowiązek szczegółowo wykazać w dokumentacji technicznej danego produktu, w jaki sposób stosowane przez niego specyfikacje zapewniają zgodność z zasadniczymi wymaganiami (190).
Należy podkreślić, że unijne przepisy harmonizacyjne dotyczące produktów na ogół nie nakładają obowiązku stosowania norm zharmonizowanych. Prawnie wiążące są tylko zasadnicze wymagania, a producenci mogą stosować dowolne normy i specyfikacje techniczne – jednak tylko normy zharmonizowane stanowią podstawę domniemania zgodności.
Co więcej, nawet jeśli producent nie stosował norm zharmonizowanych, zmiana w danej normie zharmonizowanej może oznaczać zmianę w aktualnym stanie wiedzy technicznej, co z kolei świadczy o tym, że jego produkt może być niezgodny z przepisami.
4.2. WYMAGANIA W ZAKRESIE IDENTYFIKOWALNOŚCI
|
4.2.1. DLACZEGO IDENTYFIKOWALNOŚĆ JEST WAŻNA?
Identyfikowalność to możliwość prześledzenia historii produktu.
Z perspektywy prawodawcy identyfikowalność jest ważna dlatego, że pozwala na skuteczne wykonanie prawa poprzez nadzór rynku wykorzystujący środki naprawcze, w tym wycofanie z obrotu i wycofanie od użytkowników. Pozwala na prześledzenie drogi niebezpiecznych lub niezgodnych z wymogami produktów w górę łańcucha dystrybucji oraz określa rolę i obowiązki podmiotów gospodarczych w całym łańcuchu dostaw. Identyfikowalność pozwala organom nadzoru rynku w pewnych sytuacjach prześledzić drogę produktów z powrotem do bramy fabryki i od fabryki do użytkownika.
Z perspektywy producenta identyfikowalność jest ważna, ponieważ pozwala na skuteczną kontrolę procesu produkcji i dostawców przed wprowadzeniem produktów do obrotu, a także na kontrolę ich dystrybucji po wprowadzeniu ich na rynek. W przypadku niezgodności producenci mogą zmniejszyć skutki odzyskiwania lub wycofywania produktu od użytkowników, w zależności od tego, jak dokładny mają system identyfikowalności.
4.2.2. PRZEPISY DOTYCZĄCE IDENTYFIKOWALNOŚCI
Unijne prawodawstwo harmonizacyjne narzuca cele, ale nie reguluje środków, którymi te cele należy osiągnąć. Oznacza to, że przewiduje wymagania w zakresie identyfikowalności produktu udostępnianego na rynku, nie określając sposobu, w jaki należy spełnić lub wdrożyć te wymagania. Unijne prawodawstwo harmonizacyjne jest również neutralne pod względem technologii – nie określa technologii, która powinna być wykorzystana, np. drukowanie czy formowanie. Producenci powinni wybrać system identyfikowalności, który uznają za najlepszy dla swoich produktów, metod produkcji i systemu dystrybucji.
Podanie na produkcie nazwy i adresu producenta, a w przypadku produktów importowanych również nazwy importera, stanowi podstawowy wymóg identyfikowalności. W razie potrzeby pozwala to organom nadzoru rynku na szybki kontakt z podmiotem gospodarczym odpowiedzialnym za wprowadzenie do obrotu na rynku unijnym niebezpiecznego produktu lub produktu niezgodnego z przepisami.
Nie ma wyraźnego obowiązku, aby adresy te poprzedzać słowami: „Wyprodukowany przez” („Manufactured by”), „Importowany przez” („Imported by”) lub „Reprezentowany przez” („Represented by”). Informacje te nie mogą jednak wprowadzać w błąd użytkowników końcowych i organów nadzoru rynku co do miejsca wytworzenia produktu oraz adresu każdego podmiotu gospodarczego (191). Jeśli słowa te się nie pojawią, organy nadzoru rynku zdecydują, jaką rolę odgrywają poszczególne podmioty gospodarcze; wówczas to do podmiotu gospodarczego należy udowodnienie, że jego rola jest inna.
Nie ma obowiązku tłumaczenia na wszystkie stosowne języki słów „manufactured by”, „imported by” lub „represented by”. Uważa się, że są to słowa łatwe do zrozumienia we wszystkich urzędowych językach UE.
Rozporządzenie (WE) nr 765/2008 ustanawiające wymagania w zakresie akredytacji i nadzoru rynku odnoszące się do warunków wprowadzania produktów do obrotu oraz decyzja nr 768/2008/WE w sprawie wspólnych ram dotyczących wprowadzania produktów do obrotu ustalają aktualne praktyki dotyczące identyfikowalności, nakładając obowiązek umieszczania odpowiednich etykiet. Przepisy odniesienia decyzji nr 768/2008/WE odzwierciedlone w unijnym prawodawstwie harmonizacyjnym wymagają, aby:
|
1) |
producenci podawali następujące trzy elementy: swoją 1) nazwę, 2) zarejestrowaną nazwę handlową lub zarejestrowany znak towarowy (192) i 3) adres kontaktowy na produkcie lub, jeśli nie jest to możliwe, na opakowaniu i/lub dołączonej dokumentacji. Adres musi wskazywać pojedynczy punkt kontaktowy, w którym można skontaktować się z producentem (193); |
|
2) |
importerzy podawali następujące trzy elementy: swoją 1) nazwę, 2) zarejestrowaną nazwę handlową lub zarejestrowany znak towarowy i 3) adres kontaktowy na produkcie lub, jeśli nie jest to możliwe, na opakowaniu i/lub dołączonej dokumentacji (194); |
|
3) |
producenci gwarantowali, że na ich produktach znajduje się numer typu, partii, serii lub modelu albo inny element pozwalający na identyfikację produktu, lub jeśli rozmiar lub właściwości produktu na to nie pozwalają, że taka informacja znajduje się na opakowaniu lub dołączonej dokumentacji (195); oraz |
|
4) |
podmioty gospodarcze wskazywały każdy podmiot gospodarczy, który dostarczył im produkt i każdy podmiot gospodarczy, którym dostarczyły produkt (196). |
4.2.2.1. Wymóg podania nazwy i adresu przez producentów
Producenci muszą podawać następujące trzy elementy: swoją 1) nazwę, 2) zarejestrowaną nazwę handlową lub zarejestrowany znak towarowy i 3) adres kontaktowy na produkcie lub, jeśli nie jest to możliwe na opakowaniu i/lub dołączonej dokumentacji.
Nazwa i adres muszą z zasady znaleźć się na produkcie. Jednak w wyjątkowych sytuacjach, gdy nie można postąpić zgodnie z tą zasadą, można nie umieścić wspomnianych wyżej informacji na produkcie. Jest to uzasadnione w sytuacji, gdy umieszczenie ich na produkcie jest niemożliwe z racjonalnych przyczyn technicznych lub ekonomicznych, wyłączając jednak kwestie estetyczne. Ocena należy do producenta. Należy jej dokonać z uwzględnieniem wielkości i charakteru produktu (197). Niektóre produkty, np. aparaty słuchowe czy czujniki, są za małe, aby umieszczać na nich takie informacje. W takich przypadkach w pierwszej kolejności informacja powinna być umieszczona na opakowaniu, a w drugiej w dokumentacji dołączonej do produktu, z wyjątkiem przypadków, gdy unijne prawodawstwo harmonizacyjne wymaga, aby informacja znalazła się zarówno na opakowaniu, jak i w dołączonej dokumentacji.
Producent musi spełnić ten obowiązek bez względu na swoją lokalizację (w UE lub poza nią). Przepis ten oznacza, że w przypadku produktów sprzedawanych bez opakowania czy dołączonej dokumentacji nazwę i adres producenta należy umieścić na samym produkcie.
W adresie należy podać jeden punkt do kontaktu z producentem, zwłaszcza dla organów nadzoru rynku. Prawo zobowiązuje producenta do podania na produkcie pojedynczego punktu kontaktowego. Dopuszczalny jest tylko jeden taki punkt kontaktowy w UE. Niekoniecznie musi to być adres, pod którym zarejestrowany jest producent. Może to być np. adres jednego z upoważnionych przedstawicieli lub adres biura obsługi klienta.
Nie ma potrzeby ustalania pojedynczego punktu kontaktowego dla każdego państwa członkowskiego, w którym udostępniany jest dany produkt. Jednak producent może podać również inne adresy (198), pod warunkiem że wiadomo, który z nich jest pojedynczym punktem kontaktowym. Powinien go wskazać na produkcie/w dokumentacji jako „pojedynczy punkt kontaktowy”. Nie ma obowiązku tłumaczenia adresu czy nazwy państwa na język państwa członkowskiego, w którym produkt jest udostępniany na rynku, ale muszą one być podane w języku, którego alfabet umożliwia zidentyfikowanie miejsca pochodzenia i nazwy przedsiębiorstwa.
Adres strony jest dodatkową informacją, ale sam w sobie nie jest wystarczający. Zazwyczaj adres pocztowy składa się z nazwy ulicy i numeru lub skrzynki pocztowej i numeru oraz kodu pocztowego i miejscowości, ale w niektórych państwach może różnić się od tego modelu.
4.2.2.2. Wymóg podania nazwy i adresu przez importerów
Importerzy mają obowiązek podawać następujące trzy elementy: swoją 1) nazwę, 2) zarejestrowaną nazwę handlową lub zarejestrowany znak towarowy i 3) adres kontaktowy na produkcie lub, jeśli nie jest to możliwe na opakowaniu i/lub dołączonej dokumentacji. Przepis ten odnosi się do adresu będącego punktem kontaktowym, zwłaszcza dla organów nadzoru rynku. Nie musi to być koniecznie adres siedziby importera, ale może to być np. adres punktu obsługi klienta.
Zasadniczo nazwę i adres importera należy wskazać na produkcie. Tylko jeśli nie jest to możliwe, nazwa i adres importera mogą zostać wskazane na opakowaniu i/lub w dokumentacji dołączonej do produktu. Taka sytuacja może mieć miejsce, gdy importer musiałby otworzyć opakowanie, aby umieścić swoją nazwę i adres. Dodatkowe informacje podane przez importera nie powinny zakrywać danych umieszczonych przez producenta.
Adres strony internetowej jest dodatkową informacją, ale nie wystarcza jako adres kontaktowy. Zazwyczaj adres pocztowy składa się z nazwy ulicy i numeru lub skrzynki pocztowej i numeru oraz kodu pocztowego i miejscowości, ale w niektórych państwach może różnić się od tego modelu.
Na produkcie zawsze musi znajdować się nazwa i adres producenta. Na produktach importowanych musi także znajdować się nazwa i adres importera. Dlatego zazwyczaj na produkcie umieszcza się jeden lub dwa adresy (199):
|
— |
Jeśli producent ma siedzibę w Unii Europejskiej, na produkcie będzie umieszczony tylko jeden adres (producenta), ponieważ w procesie udostępniania produktu nie bierze udziału żaden importer. |
|
— |
Jeśli producent (deklarujący się jako producent poprzez umieszczenie swojej nazwy i adresu na produkcie) ma siedzibę poza UE, a jego produkty są wprowadzane do obrotu w Unii przez importera, na produktach będą umieszczone dwa adresy: jeden producenta i jeden importera. |
|
— |
Jeśli oryginalny producent ma siedzibę poza UE, a importer wprowadza produkt do obrotu pod własną nazwą lub znakiem towarowym lub dokonuje zmian w produkcie już wprowadzonym do obrotu (w taki sposób, że może to wpłynąć na zgodność produktu ze stosownymi wymaganiami), to taki importer jest uważany za producenta. W tej sytuacji jedyny adres, który będzie widniał na produkcie (albo opakowaniu lub dołączonej dokumentacji) to adres importera, którego uważa się za producenta (200) (201). |
|
— |
Jeżeli producent ma siedzibę w UE (firma z siedzibą w UE deklarująca się jako producent poprzez umieszczenie swojej nazwy i adresu na produkcie), chociaż produkty są wytwarzane poza UE, to firma ta jest uznawana za producenta, który wprowadza produkt na unijny rynek, nawet jeśli właściwego importu dokonuje inna firma. W tej sytuacji nie istnieje importer w rozumieniu definicji importera i wystarczy umieścić tylko adres producenta. |
4.2.2.3. Elementy identyfikacyjne
Na produkcie musi znajdować się numer typu, partii, serii modelu albo inny element pozwalający na jego identyfikację. Dane identyfikacyjne muszą co do zasady znaleźć się na produkcie. Jednak w wyjątkowych sytuacjach, gdy nie można postąpić zgodnie z tą zasadą, można nie umieścić wspomnianych wyżej informacji na produkcie. Jest to uzasadnione w sytuacji, gdy wielkość i/lub charakter produktu sprawiają, że dane identyfikacyjne byłyby nieczytelne lub ich umieszczenie byłoby technicznie niemożliwe (202). W takich przypadkach dane należy umieścić na opakowaniu, jeśli istnieje, i/lub w dołączonej dokumentacji. Informacji identyfikacyjnych nie można pominąć czy przenieść na opakowanie lub do dołączonej dokumentacji z powodów czysto estetycznych czy ekonomicznych. Ocena sytuacji należy do producenta.
Przepis ten oznacza, że jeżeli produkt nie ma opakowania i nie jest do niego dołączana dokumentacja, dane identyfikacyjne muszą znaleźć się na samym produkcie.
Wymóg ten daje producentom wolność wyboru elementu, którego chcą użyć do identyfikacji produktu, o ile zagwarantowana jest jego identyfikowalność. Elementy identyfikacyjne muszą mieć wyraźny odniesienie do odpowiedniej dokumentacji, która wykazuje zgodność z wymogami danego rodzaju produktu, w szczególności z deklaracją zgodności UE. Element identyfikacyjny musi być taki sam, jak element podany w deklaracji zgodności UE. Element identyfikacyjny wybrany przez producenta jest istotny również w przypadku wycofania produktu lub odzyskania go od użytkowników, ponieważ dotyczy ono wszystkich produktów noszących ten sam element identyfikacyjny.
W niektórych przypadkach, np. gdy produkt składa się z kilku części lub elementów, jego właściwości nie pozwalają na umieszczenie elementu identyfikacji. W takich przypadkach dane identyfikacyjne należy umieścić na opakowaniu (lub w dołączonej dokumentacji). Opierając się na swoich wewnętrznych zasadach i chcąc zminimalizować zakres potencjalnego wycofywania produktów od użytkowników dzięki zaawansowanemu systemowi identyfikowalności pojedynczych sztuk (np. kodów partii, dat produkcji), producent, oprócz oznakowania elementem identyfikacyjnym opakowania produktu, może umieścić dodatkowe oznakowania na poszczególnych produktach/częściach/komponentach.
Według niektórych podmiotów gospodarczych sposobem identyfikacji produktów jest zastosowanie numeru produktu (tzw. „jednostki magazynowej”). Numeru produktu można także użyć jako identyfikatora w deklaracji zgodności UE obok innych elementów pozwalających na identyfikację produktu.
Produkt składa się z kilku części/komponentów
Każdy produkt jest zamknięty w jednym opakowaniu, ale często niektóre części/komponenty mogą być/są sprzedawane w innym opakowaniu jako oddzielne części/komponenty lub w ramach innych zestawów części/komponentów. Może się okazać, że wybrane części/komponenty w tych opakowaniach pozwalają na ich oznakowanie, podczas gdy inne są zbyt małe lub mają kształt, który nie pozwala na umieszczenie na nich oznakowania. Z tego powodu dopuszcza się nadanie numeru produktu całemu zestawowi/opakowaniu i użycie tego samego numeru w deklaracji zgodności UE.
Głównym celem umieszczania elementu identyfikacyjnego jest umożliwienie organom nadzoru rynku zidentyfikowania konkretnego produktu i połączenie go z odpowiednią deklaracją zgodności UE. Jeśli w chwili wykonywania czynności nadzoru rynku produkt nadal znajduje się w opakowaniu, łatwo będzie zidentyfikować dany element, a tym samym zagwarantować, że odpowiednia deklaracja zgodności UE odnosi się do danego produktu. Konieczność otwarcia opakowania i znalezienia elementów identyfikacji na poszczególnych częściach, a następnie odniesienie ich do odpowiedniej deklaracji zgodności UE byłoby bardziej skomplikowane.
Produkt stanowi całość złożoną z kilku elementów
Również kiedy produkt obejmuje tylko jedną „całość”, często zdarza się, że został złożony przez producenta z kilku innych części (ale zakłada się, że konsument nie będzie go z powrotem rozkładał na części). Części tworzące daną całość (produkt) są często wykorzystywane w projektach również innych produktów. Często zdarza się, że niektóre części są zbyt małe, aby można było umieścić na nich element identyfikacyjny, podczas gdy inne nie pozwalają na umieszczenie takiego elementu z przyczyn technicznych (nierówna powierzchnia, kulisty kształt, itp.). Również w tym przypadku dopuszcza się umieszczenie numeru produktu na opakowaniu i użycie tego samego numeru w deklaracji zgodności UE.
Produkt stanowi całość, która nie została złożona z kilku elementów
Jest to przypadek, gdy może się wydawać, że najprościej będzie oznaczyć sam produkt, używając do tego elementu identyfikacyjnego, który jest taki sam jak element w deklaracji zgodności UE (np. numer produktu). Jednak ten sam produkt może być sprzedawany w zestawie wraz z innymi produktami/elementami. Ponieważ w chwili produkcji nie wiadomo, które sztuki będą sprzedawane samodzielnie, a które znajdą się w opakowaniu wraz z innymi produktami, łatwiej jest umieścić numer produktu (zgodny z deklaracją zgodności UE) na opakowaniu. Ułatwi to także organom nadzoru rynku połączenie produktu z odpowiednią deklaracją zgodności UE.
4.2.2.4. Identyfikacja podmiotów gospodarczych
Podmioty gospodarcze są zobowiązane do przechowywania informacji o innych podmiotach gospodarczych będących ich dostawcami lub odbiorcami przez 10 lat. Należy pamiętać, że wymóg ten nie dotyczy użytkowników (konsumentów) ponieważ nie są oni uznawani za podmioty gospodarcze.
Unijne prawodawstwo harmonizacyjne nie określa sposobu, w jaki należy spełnić ten wymóg, ale należy zauważyć, że organy nadzoru rynku mogą poprosić o stosowne dokumenty, w tym faktury pozwalające na sprawdzenie pochodzenia produktu. Dlatego też aby spełnić warunki identyfikowalności, warto przechowywać faktury przez dłuższy okres niż przewidują to przepisy księgowe.
4.3. DOKUMENTACJA PRODUKTU
|
Unijne prawodawstwo harmonizacyjne nakłada na producenta obowiązek sporządzenia dokumentacji produktu zawierającej informacje świadczące o zgodności produktu ze stosownymi wymaganiami. Dokumentacja ta może być częścią dokumentacji systemu jakości, jeśli prawodawstwo przewiduje procedurę oceny zgodności opartą na systemie jakości (moduły D, E, H i ich warianty). Dokumentacja produktu musi być dostępna w chwili wprowadzania produktu do obrotu, bez względu na jego pochodzenie czy położenie geograficzne (203).
Dokumentację produktu należy przechowywać przez 10 lat od chwili wprowadzenia produktu na rynek, chyba że stosowne unijne przepisy harmonizacyjne wyraźnie określają inny termin (204). Jest to obowiązek producenta lub upoważnionego przedstawiciela posiadającego swoją siedzibę w Unii. Ponieważ koncepcja „wprowadzania do obrotu” dotyczy każdego indywidualnego produktu, okres przechowywania dokumentów należy obliczać od chwili wprowadzenia do obrotu każdego produktu objętego dokumentacją produktu.
W każdym unijnym dokumencie harmonizacyjnym treść dokumentacji produktu jest określona w zależności od produktów. Z zasady dokumentacja musi zawierać opis produktu i jego przeznaczenia, a także projektu, produkcji i działania produktu. Szczegóły zawarte w dokumentacji zależą od właściwości produktu i od tego, co jest uważane za niezbędne z technicznego punktu widzenia do wykazania zgodności produktu z zasadniczymi wymaganiami stosownego unijnego prawodawstwa harmonizacyjnego lub, jeśli stosowano normy zharmonizowane, zgodności produktu z tymi normami poprzez wskazanie zasadniczych wymagań ujętych w tych normach. Wymagania załącznika II do decyzji nr 768/2008/WE odnoszą się do treści dokumentacji produktu, która jest istotna z punktu widzenia zapewnienia zgodności produktu ze stosownymi unijnymi przepisami harmonizacyjnymi. Ponadto z uwagi na wymóg „odpowiedniej analizy i oceny ryzyka” producent musi w pierwszej kolejności określić wszystkie potencjalne zagrożenia związane z produktem i ustalić, które zasadnicze wymagania mają zastosowanie. Analizę tę należy udokumentować i włączyć do dokumentacji produktu. Ponadto producent musi udokumentować ocenę tego, w jaki sposób radzi sobie z zidentyfikowanym ryzykiem, aby zapewnić zgodność produktu z odpowiednimi zasadniczymi wymaganiami (na przykład stosowanie norm zharmonizowanych). Jeżeli stosowana jest tylko część normy zharmonizowanej lub norma nie obejmuje wszystkich mających zastosowanie zasadniczych wymagań, w dokumentacji produktu należy udokumentować również sposób spełnienia stosownych zasadniczych wymagań nieobjętych normą.
W przypadku gdy zmieniono projekt produktu i poddano go ponownej ocenie zgodności, w dokumentacji produktu należy uwzględnić wszystkie wersje produktu: opisać wprowadzone zmiany, sposób identyfikacji poszczególnych wersji i podać informacje dotyczące poszczególnych ocen zgodności. Ma to na celu uniknięcie sytuacji, gdzie w czasie całego cyklu życia produktu organ nadzoru rynku napotyka poprzednie wersje produktu, do których otrzymana przez niego wersja dokumentacji produktu nie znajduje zastosowania.
Niektóre unijne akty harmonizacyjne wymagają, aby dokumentacja produktu była w języku zaakceptowanym przez jednostkę notyfikowaną (205). W celu właściwego przeprowadzenia procedur oceny zgodności wymagających weryfikacji ze strony podmiotu zewnętrznego dokumentacja zawsze powinna być sporządzona w języku rozumianym przez jednostkę notyfikowaną, nawet jeśli nie jest to wyraźnie wskazane w unijnym prawodawstwie harmonizacyjnym.
4.4. DEKLARACJA ZGODNOŚCI UE
|
Unijne prawodawstwo harmonizacyjne nakłada na przedsiębiorcę obowiązek sporządzenia i podpisania deklaracji zgodności UE przed wprowadzeniem produktu do obrotu (206). Producent lub jego upoważniony przedstawiciel posiadający swoją siedzibę w Unii ma obowiązek sporządzić i podpisać deklarację zgodności UE w ramach procedury oceny zgodności przewidzianej w unijnym prawodawstwie harmonizacyjnym. Deklaracja zgodności UE to dokument, w którym stwierdza się, że produkt spełnia wszystkie odpowiednie wymagania stosownych przepisów prawa.
Sporządzając i podpisując deklarację zgodności UE, producent bierze odpowiedzialność za zgodność produktu z przepisami.
Tak jak w przypadku dokumentacji produktu (207), europejską deklarację UE należy przechowywać przez dziesięć lat od daty wprowadzenia produktu do obrotu, chyba że w przepisach prawa określono inny termin (208). Jest to obowiązek producenta lub upoważnionego przedstawiciela posiadającego swoją siedzibę w Unii. W przypadku produktów importowanych importer ponosi odpowiedzialność za deklarację zgodności (209).
Treść deklaracji zgodności UE odnosi się do wzoru deklaracji przedstawionego w załączniku III do decyzji nr 768/2008/WE lub wzoru deklaracji załączonego bezpośrednio do odpowiednich unijnych przepisów harmonizacyjnych. Norma EN ISO/IEC 17050-1 została sporządzona w celu ustalenia ogólnych kryteriów dotyczących deklaracji zgodności; może być ona również traktowana jako dokument referencyjny, pod warunkiem że jest zgodna ze stosownymi unijnymi przepisami harmonizacyjnymi. Deklaracja może mieć formę dokumentu, etykiety lub równorzędną i musi zawierać wystarczające informacje, aby umożliwić skojarzenie z nią wszystkich objętych nią produktów.
Wzór deklaracji w decyzji nr 768/2008/WE zawiera:
|
1) |
numer identyfikujący produkt. Nie musi on być unikalny dla każdego produktu. Może to być numer produktu, partii, typu lub serii (210). Zależy to od decyzji producenta (211); |
|
2) |
nazwę i adres producenta lub upoważnionego przedstawiciela wydającego deklarację; |
|
3) |
oświadczenie, że deklaracja jest wydana na wyłączną odpowiedzialność producenta; |
|
4) |
dane identyfikacyjne produktu umożliwiające jego identyfikowalność. Są to zasadniczo wszelkie istotne informacje uzupełniające pkt 1, opisujące produkt i umożliwiające jego identyfikowalność. Jeśli to ważne z punktu widzenia identyfikacji produktu, dane identyfikacyjne mogą zawierać element graficzny, ale jeśli nie określono tego jako wymagania w unijnych przepisach harmonizacyjnych, decyzja o umieszczeniu obrazu zależy od producenta; |
|
5) |
wszystkie istotne unijne przepisy harmonizacyjne, które produkt musi spełniać; przywoływane normy lub inne specyfikacje techniczne (takie jak krajowe normy i specyfikacje techniczne) w precyzyjny, pełny i jasno zdefiniowany sposób – oznacza to, że należy wskazać wersję i/lub datę wydania stosownej normy; |
|
6) |
nazwę i numer identyfikacyjny jednostki notyfikowanej, jeśli brała udział w procedurze oceny zgodności (212) (213) oraz odniesienie do odpowiedniego certyfikatu (w stosownych przypadkach); |
|
7) |
wszystkie dodatkowe informacje, które mogą być wymagane (np. stopień, kategorię), jeśli to stosowne; |
|
8) |
datę wydania deklaracji; podpis i stanowisko lub równoważne oznaczenie upoważnionej osoby (214) (215); może to być dowolna data po zakończeniu oceny zgodności. |
Jeśli jeden produkt reguluje kilka przepisów harmonizacyjnych, producent lub upoważniony przedstawiciel musi przedstawić jedną deklarację zgodności w odniesieniu do wszystkich odpowiednich aktów unijnych (216). Aby zmniejszyć obciążenie administracyjne podmiotów gospodarczych i ułatwić dostosowanie deklaracji w przypadku zmiany jednego mającego zastosowanie aktu unijnego, dopuszcza się, aby jedna deklaracja miała formę dossier zawierającego poszczególne odpowiednie deklaracje zgodności (217).
Deklaracja zgodności UE musi być dostępna na żądanie organu nadzoru rynku. Ponadto unijne przepisy harmonizacyjne w sprawie maszyn, urządzeń w atmosferach potencjalnie wybuchowych, urządzeń radiowych i końcowych urządzeń telekomunikacyjnych, przyrządów pomiarowych, rekreacyjnych jednostek pływających, dźwigów, systemów kolei dużych prędkości i kolei konwencjonalnych oraz elementów Europejskiej Sieci Zarządzania Ruchem Lotniczym nakładają obowiązek dołączenia do produktów deklaracji zgodności UE.
Deklarację zgodności UE należy przetłumaczyć na język lub języki wymagane przez państwo członkowskie, w którym produkt jest wprowadzany do obrotu lub udostępniany na rynku (218). Unijne prawodawstwo harmonizacyjne zawsze określa, kto jest zobowiązany do przetłumaczenia deklaracji. Logicznie rzecz biorąc, powinien to być producent lub inny podmiot gospodarczy udostępniający produkt. Deklaracja zgodności UE musi być podpisana przez producenta lub jego upoważnionego przedstawiciela. Jeżeli tłumaczenie deklaracji zgodności UE zostało przygotowane przez inny podmiot gospodarczy i nie jest podpisane przez producenta, wraz z tłumaczeniem należy dostarczyć również kopię deklaracji zgodności UE podpisaną przez producenta.
4.5. WYMOGI W ZAKRESIE OZNAKOWANIA
4.5.1. OZNAKOWANIE CE
4.5.1.1. Definicja i rola oznakowania CE
|
Oznakowanie CE stanowi kluczowy wskaźnik (ale nie dowód) zgodności produktu z przepisami UE i umożliwia swobodny przepływ produktów na rynku EOG i tureckim bez względu na to, czy zostały wyprodukowane w EOG, Turcji czy w innym państwie.
Państwa członkowskie Europejskiego Obszaru Gospodarczego (EOG – państwa członkowskie UE i określone kraje EFTA: Islandia, Norwegia, Liechtenstein) nie mają prawa ograniczać wprowadzania do obrotu produktów oznaczonych znakiem CE, chyba że takie środki są uzasadnione i poparte dowodami niezgodności produktu. Dotyczy to również produktów wytworzonych w państwach trzecich, które są sprzedawane w EOG.
Oznakowanie CE nie oznacza, że produkt został wyprodukowany w Unii Europejskiej. Oznakowanie CE wskazuje na zgodność produktu z wymaganiami przewidzianymi w stosownych unijnych dokumentach harmonizacyjnych. Dlatego należy je rozpatrywać jako istotną informację dla organów państw członkowskich i innych zainteresowanych stron (np. dystrybutorów). Oznakowanie CE nie służy celom komercyjnym, tj. nie jest narzędziem marketingowym.
Oznakowanie CE jest widocznym rezultatem całego procesu obejmującego ocenę zgodności w szerokim ujęciu i oznaką, że producent zadeklarował zgodność produktu z unijnym prawodawstwem harmonizacyjnym.
4.5.1.2. Związek z dotychczasowym prawodawstwem
|
Rozporządzenie (WE) nr 765/2008 zawiera definicję i format oznakowania CE i ogólne zasady regulujące jego przyznawanie. Decyzja nr 768/2008/WE reguluje procedury oceny zgodności, które prowadzą do umieszczenia omawianego oznakowania na produkcie.
Sektorowe dokumenty harmonizacyjne Unii regulujące umieszczanie oznakowania CE są w większości zgodne z zasadami rozporządzenia (WE) nr 765/2008 i decyzji nr 768/2008/WE.
Przyjmuje się zasadniczo (219), że unijne akty prawne mogą wprowadzać oznakowanie CE jako zgodne z prawem oznakowanie zgodności, jeżeli:
|
— |
stosuje się metodę całkowitej harmonizacji, co oznacza, że stosowanie rozbieżnych regulacji krajowych dotyczących tego samego zakresu co odpowiedni unijny akt prawny jest zabronione, |
|
— |
unijny akt harmonizacyjny zawiera procedury oceny zgodności zgodne z decyzją nr 768/2008/WE. |
Jednak istnieje wyjątek od tej zasady.
W dobrze uzasadnionych przypadkach w pełni zharmonizowane przepisy prawa, które są zgodne z decyzją nr 768/2008/WE mogą przewidywać inne oznaczenia niż oznakowanie CE. Np. dyrektywa w sprawie wyposażenia statków nie przewiduje oznakowania CE, ale specjalne oznakowania zgodności – znak koła sterowego. Stosowanie znaku koła sterowego również podlega ogólnym zasadom określonym w rozporządzeniu (WE) nr 765/2008 i decyzji nr 768/2008/WE, a wszelkie odniesienia do oznakowania CE należy rozumieć jako odniesienia do znaku koła sterowego. Podobnie w przypadku ciśnieniowych urządzeń transportowych zamiast oznakowania CE wymagany jest znak zgodności Pi.
4.5.1.3. Kto ma obowiązek umieszczania oznakowania CE, a komu nie wolno tego robić
|
Producent, bez względu na to, czy posiada siedzibę w Unii czy poza nią, jest ostatecznie odpowiedzialny za zgodność produktu z unijnym prawodawstwem harmonizacyjnym i za umieszczanie oznakowania CE. Producent może wyznaczyć upoważnionego przedstawiciela, aby ten umieszczał oznakowanie CE w jego imieniu.
Umieszczając oznakowanie CE, producent na swoją wyłączną odpowiedzialność (i bez względu na to, czy w procesie oceny zgodności brał udział podmiot zewnętrzny, czy nie) deklaruje zgodność produktu ze wszystkimi stosownymi wymaganiami prawnymi.
Jeśli importer, dystrybutor lub inny podmiot wprowadza produkty na rynek pod własną nazwą lub znakiem towarowym albo wprowadza w nich zmiany, przejmuje wtedy obowiązki producenta. Obejmują one obowiązek zapewnienia zgodności produktu z przepisami i umieszczanie oznakowań CE na produkcie. W takim przypadku podmiot ten musi posiadać wystarczające informacje dotyczące projektu i procesu wytwarzania produktu, ponieważ umieszczając oznakowanie CE, przyjmuje odpowiedzialność prawną.
4.5.1.4. Zasady umieszczania oznakowania CE
|
Oznakowanie CE musi mieć poniższą formę. Jeśli oznakowanie CE jest zmniejszane lub powiększane, należy zachować odpowiednie proporcje. |

Oznakowanie CE należy umieścić na produkcie lub tabliczce znamionowej w sposób widoczny, czytelny i nieusuwalny. Jednak jeżeli nie jest to możliwe albo nie można tego zagwarantować ze względu na charakter produktu, oznakowanie CE należy umieścić na opakowaniu i/lub w dołączonej dokumentacji. Z zasady oznakowanie CE nie może być umieszczone na produkcie do momentu zakończenia procedury oceny zgodności, co ma na celu zagwarantowanie, że produkt spełnia wszystkie przepisy stosownych unijnych aktów harmonizacyjnych. Zazwyczaj następuje to na koniec fazy produkcji. Nie stwarza to problemów, jeśli np. oznakowanie CE znajduje się na tabliczce znamionowej, która jest umieszczana na produkcie dopiero po ostatecznej kontroli produktu. Jednak jeśli (np.) oznakowanie CE jest umieszczane techniką tłoczenia lub odlewu, można je umieścić na dowolnym etapie produkcji, jeśli w ramach procesu produkcji weryfikowana jest zgodność produktu.
Wymóg widoczności oznacza, że oznakowanie CE musi być łatwo dostępne dla wszystkich zainteresowanych stron. Może zostać umieszczone np. na tylnej lub spodniej stronie produktu. Wymóg widoczności niekoniecznie oznacza, że oznakowanie CE musi być widoczne przed otwarciem opakowania produktu, ponieważ umieszczenie oznakowania CE również na opakowaniu jest konieczne jedynie w przypadku gdy jest to wyraźnie wymagane w stosownych przepisach unijnych. Aby oznakowanie było czytelne, musi mieć przynajmniej 5 mm wysokości. Jednak zgodnie z niektórymi przepisami harmonizacyjnymi (220) w przypadku małych urządzeń lub komponentów możliwe jest uzyskanie zwolnienia z obowiązku zapewnienia minimalnego wymiaru oznakowania CE.
Oznakowanie CE może przyjmować różne formy (tj. różne kolory, pogrubione/niewypełnione), o ile jest widoczne, czytelne i ma odpowiednie proporcje. Musi także być nieusuwalne, tak aby w normalnych okolicznościach nie można było go usunąć bez pozostawienia widocznych śladów (np. niektóre normy produktowe przewidują test ścierania przy użyciu wody i benzyny lakowej). Mimo to nie oznacza to, że oznakowanie CE musi być integralną częścią produktu.
Jednak w określonych przypadkach umieszczenie oznakowania CE na produkcie jest niemożliwe (np. na określonych rodzajach materiałów wybuchowych) lub niemożliwe w racjonalnych warunkach technicznych lub ekonomicznych. Co więcej, mogą zdarzyć się przypadki, gdy niemożliwe będzie dotrzymanie minimalnych wymiarów lub spełnienie warunku widoczności, czytelności i nieusuwalności oznakowania CE.
W takich przypadkach oznakowanie CE można umieścić na opakowaniu, jeśli istnieje i/lub w dołączonej dokumentacji, jeśli dane unijne przepisy harmonizacyjne przewidują dołączanie dokumentów. Oznakowania CE produktu nie można pominąć czy umieścić tylko na opakowaniu lub w dołączonej dokumentacji z powodów czysto estetycznych czy ekonomicznych.
W rozporządzeniu (WE) nr 765/2008 i decyzji nr 768/2008/WE określono, że oznakowanie CE musi mieć wymiary, format i proporcje określone w załączniku II do rozporządzenia (WE) nr 765/2008, a także być czytelne i widoczne. Rozporządzenie (WE) nr 765/2008 i decyzja nr 768/2008/WE nie zakazują żadnego rodzaju projektu (np. „niewypełnionego”), o ile spełnione są powyższe warunki. Jednak niedozwolone jest stosowanie wyłącznie etykiet elektronicznych.
4.5.1.5. Umieszczanie oznakowania CE wraz z numerem identyfikacyjnym jednostki notyfikowanej
Jeśli zgodnie ze stosownymi unijnymi przepisami harmonizacyjnymi w kontroli produkcji udział bierze jednostka notyfikowana, za oznakowaniem CE musi znaleźć się jej numer identyfikacyjny. Jeśli wymaga tego prawo, producent lub upoważniony przedstawiciel umieszcza na odpowiedzialność jednostki notyfikowanej jej numer.
Jednostka notyfikowana może być zaangażowana w fazę produkcji w zależności od stosowanych procedur oceny zgodności. Numer identyfikacyjny jednostki notyfikowanej należy umieścić za oznakowaniem CE tylko wtedy, gdy bierze ona udział w fazie produkcji. Dlatego nie umieszcza się za oznakowaniem CE numeru jednostki notyfikowanej zaangażowanej w ocenę zgodności w fazie projektu zgodnie z modułem B. Czasami w fazę produkcji zaangażowanych jest kilka jednostek notyfikowanych, co jest możliwe w sytuacji, gdy zastosowanie ma więcej niż jeden unijny dokument harmonizacyjny. W takiej sytuacji za oznakowaniem CE umieszczanych jest kilka numerów identyfikacyjnych.
Dlatego też, jeśli oznakowanie CE pojawia się na produkcie bez numeru identyfikacyjnego, oznacza to, że:
|
— |
w fazie projektu lub produkcji nie uczestniczyła żadna jednostka notyfikowana (moduł A), lub |
|
— |
z wyboru producenta w fazie produkcji uczestniczyła akredytowana jednostka własna (moduły A1 i A2), lub |
|
— |
jednostka notyfikowana uczestniczyła w fazie projektowania (moduł B), ale żadna jednostka notyfikowana nie uczestniczyła w fazie produkcji, |
|
— |
(moduł C następujący po module B), lub |
|
— |
jednostka notyfikowana uczestniczyła w fazie projektowania (moduł B), a w fazie produkcji z wyboru producenta uczestniczyła akredytowana jednostka własna (moduł C1, C2 następujący po module B). |
Jeśli natomiast oznakowanie CE pojawia się na produkcie z numerem identyfikacyjnym (221), oznacza to, że:
|
— |
z wyboru producenta w fazie produkcji uczestniczyła jednostka notyfikowana (moduły A1 i A2), lub |
|
— |
w fazie projektowania (moduł B), a z wyboru producenta także w fazie produkcji uczestniczyła jednostka notyfikowana (nie musi to być ta sama jednostka; należy wskazać numery identyfikacyjny) (moduły C1, C2 następujące po module B), lub |
|
— |
w fazie projektowania (moduł B) i w fazie produkcji uczestniczyła jednostka notyfikowana (nie musi to być ta sama jednostka; należy wskazać numery identyfikacyjne) (moduły C1, C2 następujące po module B), lub |
|
— |
jednostka notyfikowana uczestniczyła w fazie projektu i produkcji (moduły D1, E1, F1, G1 H, H1). |
Oznakowanie CE wraz z numerem identyfikacyjnym jednostki notyfikowanej niekoniecznie musi być umieszczane na produkcie na terytorium Unii. Można je umieszczać w państwie trzecim, np. jeśli produkt jest tam wytwarzany, a jednostka notyfikowana przeprowadziła w tym państwie ocenę zgodności stosownie do odpowiednich unijnych przepisów harmonizacyjnych. Oznakowanie CE i numer identyfikacyjny mogą być umieszczane osobno, o ile będą znajdowały się obok siebie.
4.5.1.6. Które produkty muszą posiadać oznakowanie CE, a które nie mogą go mieć
|
Nie wszystkie produkty muszą być opatrzone znakiem CE (222). Obowiązek umieszczania oznakowania CE obejmuje wszystkie produkty objęte zakresem aktów prawnych regulujących umieszczanie tego znaku i przeznaczone na rynek unijny. Dlatego też oznakowanie CE należy umieścić:
|
— |
na wszystkich nowo wyprodukowanych produktach, które podlegają przepisom prawa regulującym kwestie oznakowania CE, bez względu na to, czy są produkowane w państwach członkowskich, czy w państwach trzecich, |
|
— |
na produktach używanych importowanych z państw trzecich, które podlegają przepisom regulującym kwestię oznakowania CE, |
|
— |
na produktach zmodyfikowanych, które, jako nowe produkty, podlegają przepisom regulującym kwestię oznakowania CE i które zostały zmienione w sposób, który może mieć wpływ na bezpieczeństwo produktu i jego zgodność z przepisami harmonizacyjnymi. |
W niektórych przypadkach do celów określonego unijnego aktu harmonizacyjnego produkt uznawany jest za produkt gotowy i musi zostać oznaczony znakiem CE. Ten sam produkt jest następnie włączany do innego produktu gotowego podlegającego innemu unijnemu aktowi harmonizacyjnemu, który również przewiduje obowiązek umieszczenia oznakowania CE. W konsekwencji na produkcie można znaleźć więcej niż jedno oznakowanie CE (223).
Unijne przepisy harmonizacyjne w ogólny sposób regulujące kwestie oznakowania CE mogą nie przewidywać użycia oznakowań CE na określonych produktach. Ogólna zasada mówi, że produkty są dopuszczone do swobodnego obrotu, jeżeli:
|
a) |
dołączono do nich:
|
|
b) |
dołączono świadectwa zgodności w przypadku komponentów zgodnie z przepisami dyrektywy w sprawie urządzeń i systemów ochronnych przeznaczonych do użytku w atmosferach potencjalnie wybuchowych (ATEX); |
|
c) |
dołączono oświadczenie w przypadku:
|
|
d) |
dołączono certyfikat zgodności w przypadku osprzętu wskazanego w dyrektywie w sprawie urządzeń spalających paliwa gazowe; |
|
e) |
w przypadku przyrządów niepodlegających ocenie zgodności według dyrektywy w sprawie wag nieautomatycznych na produkcie należy wskazać nazwę producenta i maksymalną ładowność; |
|
f) |
w przypadku określonych zbiorników wskazanych w dyrektywie w sprawie prostych zbiorników ciśnieniowych i urządzeń ciśnieniowych produkt jest wytwarzany zgodnie z uznaną praktyką inżynieryjną. |
Dodatkowo dyrektywa w sprawie urządzeń ciśnieniowych daje państwom członkowskim prawo zezwolenia na ich terytorium na wprowadzanie do obrotu i oddawanie do użytku przez użytkowników urządzeń ciśnieniowych lub zespołów nieopatrzonych oznakowaniem CE, ale podlegających ocenie zgodności przeprowadzanej przez inspektorat ds. użytkowników zamiast jednostkę notyfikowaną.
4.5.1.7. Oznakowanie CE i inne oznakowania
|
Oznakowanie CE zastępuje wszystkie obowiązkowe oznakowania zgodności, które mają to samo znaczenie i które istniały przed harmonizacją. Takie krajowe oznakowania zgodności są niekompatybilne z oznakowaniem CE i stanowiłyby naruszenie mających zastosowanie przepisów unijnych. Podczas transpozycji unijnego prawodawstwa harmonizacyjnego państwa członkowskie są zobowiązane do włączenia oznakowania CE do prawodawstwa krajowego i procedur administracyjnych. Nie mogą także wprowadzać do prawa krajowego żadnego innego oznakowania zgodności, które miałoby takie same znaczenie jak oznakowanie CE.
Jednak inne oznakowania mogą być stosowane, o ile przyczyniają się do ochrony interesu publicznego, nie są objęte unijnym prawodawstwem harmonizacyjnym, a ich umieszczanie nie ma wpływu na widoczność, czytelność i znaczenie oznakowań CE. Umieszczanie dodatkowych oznakowań (takich jak chroniony znak towarowy producenta lub inne prywatne/krajowe oznakowania) jest dozwolone w zakresie, w nie można pomylić ich z oznakowaniem CE. Podobieństwo powodujące taką pomyłkę może odnosić się do znaczenia lub formy oznakowania CE.
Z tego powodu inne oznakowania, dodatkowe w stosunku do oznakowania CE, muszą spełniać inne funkcje niż oznakowanie CE. Dlatego powinny one być źródłem informacji na temat zgodności z innymi założeniami niż te, do których odnosi się oznakowanie CE (np. kwestie środowiskowe nieujęte w stosownym unijnym prawodawstwie harmonizacyjnym).
Ponadto kilka unijnych aktów harmonizacyjnych przewiduje dodatkowe oznakowania, które uzupełniają oznakowanie CE, ale nie pokrywają się z nim (zob. pkt 4.5.2).
4.5.1.8. Sankcje
|
Oznakowanie CE to pierwszy znak informujący, że można założyć, że zanim dany produkt został wprowadzony do obrotu, przeprowadzono niezbędne kontrole w celu zagwarantowania jego zgodności ze stosownymi wymaganiami prawa. Organy nadzoru rynku mają prawo przeprowadzić dodatkowe kontrole w celu ochrony interesu publicznego. Decyzje o działaniach podejmowanych przez organy nadzoru rynku muszą być podejmowane w każdym przypadku indywidualnie, zgodnie z zasadą proporcjonalności.
Państwa członkowskie muszą wprowadzić do swojego prawodawstwa krajowego odpowiednie środki zarówno zapobiegające nadużyciu/niewłaściwemu użyciu oznakowania CE, jak również gwarantujące reakcję w sytuacji, gdy dojdzie do takiego nadużycia/niewłaściwego użycia. Środki te muszą być skuteczne, współmierne i odstraszające oraz mogą zostać zaostrzone, w przypadku gdy dany podmiot gospodarczy w przeszłości popełnił podobne naruszenie. Mogą obejmować, w zależności od potrzeb, wycofanie z obrotu takich produktów, wycofanie ich od użytkowników, kary i sankcje karne (takie jak kary pieniężne i kara pozbawienia wolności).
Środki te są nakładane bez uszczerbku dla innych środków podejmowanych w przypadku, gdy organy nadzoru rynku uznają, że produkt stwarza ryzyko lub jest niezgodny z odpowiednimi przepisami prawa. Co więcej, państwa członkowskie muszą zagwarantować, że środki te zostaną wdrożone.
Pod tym względem umieszczenie oznakowania CE na produkcie, który nie mieści się w zakresie żadnych unijnych przepisów harmonizacyjnych przewidujących umieszczanie takich oznakowań, uznaje się za wprowadzanie w błąd, ponieważ konsumenci lub użytkownicy mogą z dużym prawdopodobieństwem np. odnieść wrażenie, że produkt spełnia określone przepisy unijnego prawodawstwa harmonizacyjnego. Dlatego też właściwe organy muszą dysponować instrumentami prawnymi, które pozwalają im na podjęcie działań mających na celu walkę z wprowadzającym w błąd zastosowaniem oznakowania CE. Muszą także podejmować działania przeciwko podmiotom odpowiedzialnym za umieszczenie oznakowania CE na produkcie niezgodnym z przepisami.
Umieszczanie dodatkowych oznakowań, innych niż oznakowanie CE, podlega określonym ograniczeniom (224). Organy nadzoru rynku muszą podjąć niezbędne środki w celu zagwarantowania, że zasady te są przestrzegane, a tam, gdzie to konieczne, podjąć odpowiednie działania.
Państwo członkowskie jest zobowiązane powiadomić Komisję i inne państwa członkowskie, jeśli zdecyduje się ograniczyć swobodny przepływ towarów ze względu na niewłaściwe umieszczenie oznakowania CE lub podejmuje działania przeciwko podmiotom odpowiedzialnym za umieszczenie oznakowania CE na produkcie niezgodnym z przepisami. Następnie od innych państw członkowskich zależy decyzja, czy podejmą podobne działania. Państwa członkowskie powinny poinformować Komisję i inne państwa członkowskie o przypadkach niewłaściwego umieszczenia oznakowania CE na produktach niepodlegających wymogowi oznakowania CE.
4.5.2. INNE OBOWIĄZKOWE OZNAKOWANIA
|
Niektóre unijne akty harmonizacyjne przewidują dodatkowe oznakowania, które uzupełniają oznakowanie CE, ale nie pokrywają się z nim. |
Piktogramy lub inne oznakowania wskazujące np. kategorię zastosowania stanowią, według niektórych unijnych przepisów harmonizacyjnych, uzupełnienie oznakowania CE, ale nie są jego częścią, ani go nie zastępują. Ogólnie rzecz biorąc, oznaczenia te opierają się na tych samych zasadach co oznakowanie CE. Kilka przykładów:
|
— |
etykiety energetyczne UE dla produktów związanych z energią, |
|
— |
specjalne oznakowania dotyczące ochrony przed wybuchem w przypadku urządzeń i systemów ochronnych przeznaczonych do użytku w atmosferach potencjalnie wybuchowych, |
|
— |
identyfikator klasy sprzętu wymagany przy sprzęcie radiowym (klasa 2), |
|
— |
dodatkowe oznakowanie metrologiczne wymagane w przypadku przyrządów pomiarowych i wag nieautomatycznych. |
5. OCENA ZGODNOŚCI
5.1. MODUŁY OCENY ZGODNOŚCI
5.1.1. CZYM JEST OCENA ZGODNOŚCI?
|
Dwa istotne elementy każdego aktu prawnego (czy to starego, czy nowego podejścia) dotyczącego produktów to:
|
— |
wymagania prawne regulujące właściwości danego produktu, oraz |
|
— |
procedury oceny zgodności przeprowadzane przez producenta w celu wykazania przed wprowadzeniem produktu do obrotu, że jest on zgodny ze wspominanymi wymaganiami prawnymi. |
W niniejszym przewodniku poruszono kwestie oceny zgodności określonej w decyzji nr 768/2008/WE (w szczególności dla unijnego prawodawstwa harmonizacyjnego zgodnego z nowym podejściem, a teraz nowymi ramami prawnymi).
Produkt jest poddawany ocenie zgodności zarówno w czasie fazy projektowej, jak i fazy produkcji. Ocena zgodności należy do obowiązków producenta. Jeśli producent zleca projektowanie lub produkcję innemu podmiotowi, nadal pozostaje odpowiedzialny za przeprowadzenie oceny zgodności.
Oceny zgodności nie należy mylić z nadzorem rynku, który obejmuje kontrole przeprowadzane przez krajowe organy nadzoru rynku po wprowadzeniu produktu do obrotu. Jednak obie techniki uzupełniają się nawzajem i są równie potrzebne do ochrony interesu publicznego i płynnego funkcjonowania rynku wewnętrznego.
Zasadniczym celem procedury oceny zgodności jest wykazanie, że produkty wprowadzone do obrotu są zgodne z wymaganiami określonymi w stosownych przepisach.
5.1.2. MODUŁOWA STRUKTURA OCENY ZGODNOŚCI W UNIJNYM PRAWODAWSTWIE HARMONIZACYJNYM
|
Zgodnie z unijnym prawodawstwem harmonizacyjnym procedury oceny zgodności składają się z jednego lub dwóch modułów oceny zgodności. Ponieważ produkty są poddawane ocenie zgodności zarówno w czasie fazy projektowej, jak i fazy produkcji, procedury oceny zgodności dotyczą fazy projektowania i produkcji, podczas gdy moduł może obejmować:
|
— |
jedną z dwóch faz (w tym przypadku procedura oceny zgodności składa się z dwóch modułów), albo |
|
— |
jedną fazę (w tym przypadku procedura oceny zgodności składa się z jednego modułu). |
Decyzja nr 768/2008/WE określa „zestaw horyzontalny” modułów oceny zgodności oraz to, w jaki sposób moduły te składają się na odpowiednie procedury.
Prawodawca wybiera z zestawu modułów oceny zgodności/ procedur (określonych w decyzji nr 768/2008/WE) elementy najbardziej odpowiadające specyficznym potrzebom danego sektora (225). Powinien wybrać najmniej uciążliwy moduł, biorąc pod uwagę typ produktów i związanych z nimi zagrożeń, ich wpływ na ochronę interesu publicznego, infrastrukturę gospodarczą danego sektora, metodę produkcji itp.; tam, gdzie to możliwe, należy zapewnić wybór modułów kontroli, certyfikacji i/lub zapewnienia jakości.
Procedury oceny zgodności są równoważne z prawnego punktu widzenia, ale nie są technicznie identyczne pod względem metod. Ich zastosowanie w prawodawstwie sektorowym ma na celu zagwarantowanie wysokiego poziomu pewności, jeśli chodzi o zgodność produktów ze stosownymi zasadniczymi wymaganiami.
Celem modułów określonych w decyzji nr 768/2008/WE jest umożliwienie zastosowania ograniczonej liczby możliwych procedur.
Mimo to możliwy wybór powinien być wystarczająco szeroki, co pozwoli na zastosowanie środków w przypadku możliwie największego zakresu produktów.
Unijne prawodawstwo harmonizacyjne ustala procedury oceny zgodności, nie pozostawiając producentowi wyboru lub ustalając zakres procedur, z którego producent musi wybierać. Ponieważ procedury oceny zgodności w unijnym prawodawstwie harmonizacyjnym wywodzą się z decyzji nr 768/2008/WE, są jednolite i spójne. Dlatego też ocena zgodności produktu staje się bardziej przejrzysta w przypadku, gdy do danego produktu zastosowanie ma więcej niż jeden akt harmonizacyjny.
5.1.3. PODMIOTY ZAANGAŻOWANE W OCENĘ ZGODNOŚCI – UMIEJSCOWIENIE OCENY ZGODNOŚCI W ŁAŃCUCHU DOSTAW
|
Ocena zgodności należy do obowiązków producenta. Jednak jeśli wymagają tego przepisy, w procedurę oceny zgodności musi zaangażować się podmiot zewnętrzny.
Ogólnie istnieją trzy możliwości:
|
— |
Nie jest zaangażowany żaden podmiot zewnętrzny. Może tak stać się w przypadku, gdy zdaniem prawodawcy, aby zagwarantować zgodność produktu (produktów) z mającymi zastosowanie wymogami prawa, wystarczy, że producent sporządzi deklarację (wraz ze stosownymi badaniami i dokumentacją techniczną). W takim przypadku producent sam przeprowadza wszystkie kontrole i testy, tworzy dokumentację produktu i gwarantuje zgodność procesu produkcji. |
|
— |
Ocenę zgodności przeprowadza się przy udziale akredytowanej jednostki własnej, która stanowi część organizacji producenta. Jednak taka jednostka własna nie może mieć innych zadań niż ocena zgodności i musi być niezależna od jednostek handlowych, projektujących i produkujących (szczegółowe dane znajdują się w art. R21 załącznika I do decyzji nr 768/2008/WE). Poprzez akredytację musi ona wykazać taki sam poziom kompetencji technicznych i bezstronności jak zewnętrzne jednostki oceniające zgodność. Jeśli to stosowne w przypadku określonego sektora, prawodawca może uznać fakt, że producenci prowadzą bardzo dobrze wyposażone laboratoria badawcze lub zakłady. Może się tak zdarzyć w przypadku innowacyjnych, skomplikowanych produktów, dla których wiedza na temat sposobu przeprowadzania badań pozostaje u producenta. |
|
— |
Jednak w innych przypadkach prawodawca może uznać, że konieczna jest interwencja podmiotu zewnętrznego, tj. zewnętrznej jednostki oceniającej zgodność. Taka jednostka musi być bezstronna i w pełni niezależna od organizacji lub produktu, który ocenia (zob. również art. R17 ust. 3 załącznika I do decyzji nr 768/2008/WE), nie może angażować się w żadne działania, które mogą podważyć jej niezależność (zob. również art. R21 ust. 2 lit. c) załącznika I do decyzji nr 768/2008/WE) i nie może mieć interesów jako użytkownik lub innych interesów w związku z ocenianym produktem. |
Obowiązek notyfikowania takich zewnętrznych jednostek oceniających zgodność spoczywa na państwach członkowskich. Muszą one wskazać takie jednostki w ramach swojej jurysdykcji, które ich zdaniem mają techniczne kompetencje do oceny zgodności produktu z odpowiednimi unijnymi przepisami harmonizacyjnymi. Jednostki własne nie mogą być notyfikowane, ale nadal poprzez akredytację muszą wykazać taki sam poziom kompetencji technicznych jak jednostki zewnętrzne. Państwa członkowskie muszą również zagwarantować, że jednostki (własne lub zewnętrzne) utrzymają swoje kompetencje techniczne.
Biorąc powyższe pod uwagę, podmiotami zainteresowanymi procedurą oceny zgodności są:
|
a) |
prawodawca, który:
|
|
b) |
producent, który:
Należy jasno zaznaczyć, że to producent jest zawsze odpowiedzialny za zgodność produktów z odpowiednimi wymaganiami prawa. W związku z tym podmiot gospodarczy wprowadzający produkt do obrotu pod własną nazwą lub znakiem towarowym automatycznie staje się producentem w rozumieniu przepisów unijnego prawodawstwa harmonizacyjnego. Bierze zatem na siebie całą odpowiedzialność za ocenę zgodności (projektowanie i wytwarzanie) produktu, nawet w przypadku gdy została on faktycznie przeprowadzona przez kogoś innego. Ponadto musi on posiadać wszystkie dokumenty i zaświadczenia niezbędne do wykazania zgodności danego produktu z wymogami, lecz nie muszą być one wydane w jego imieniu; |
|
c) |
jednostka oceniająca zgodność (własna lub zewnętrzna), która:
|
Jednostka oceniająca zgodność, która chce przeprowadzić ocenę zgodności w zakresie jednego lub kilku modułów zgodnie z określonymi unijnymi przepisami harmonizacyjnymi, musi zostać oceniona pod kątem wszystkich wymagań dla różnych modułów, w ramach których oferuje swoje usługi (zob. pkt 5.2.3). Jednostka oferująca dokonanie oceny zgodności w ramach danego unijnego aktu harmonizacyjnego musi oferować usługi w zakresie przynajmniej jednego modułu wskazanego w takim akcie. Należy zauważyć, że jednostka nie ma obowiązku oferować usług z zakresu więcej niż jednego modułu, ale odpowiada za cały oferowany moduł.
Dokładne umiejscowienie oceny zgodności w łańcuchu dostaw zostało przedstawione na schemacie 2.
Schemat 2
Ocena zgodności
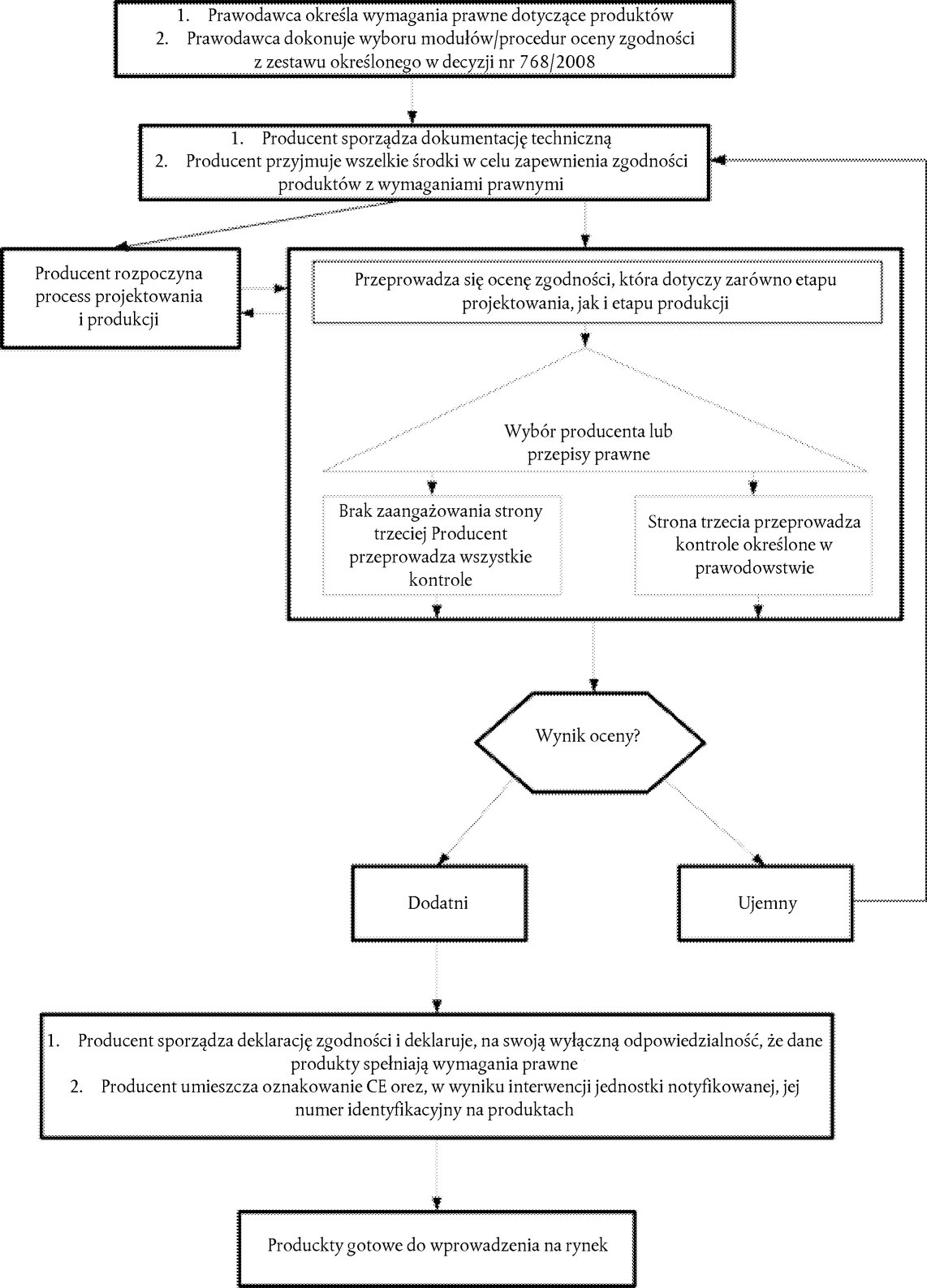
5.1.4. MODUŁY I ICH WARIANTY
|
Istnieje osiem modułów. Niektóre z nich mają swoje warianty. |
Istnieje osiem modułów (nazwanych od A do H). Określono w nich obowiązki producenta (i jego upoważnionego przedstawiciela) oraz stopień zaangażowania akredytowanej jednostki własnej lub jednostki notyfikowanej oceniającej zgodność. Są to elementy procedury oceny zgodności określone w decyzji nr 768/2008/WE – „zestaw horyzontalny”.
Niektóre moduły mają swoje warianty. Warianty modułów (dotyczy to wszystkich wariantów modułów określonych w decyzji nr 768/2008/WE) jest umożliwienie zagwarantowania niezbędnego poziomu ochrony w przypadku produktów stwarzających wyższy poziom zagrożenia bez konieczności stosowania cięższego modułu. Założeniem jest możliwie jak największe ograniczenie ciężaru nakładanego na producenta.
5.1.5. PROCEDURY JEDNO- I DWUMODUŁOWE – PROCEDURY OPARTE NA TYPIE (BADANIE TYPU UE)
|
W niektórych przypadkach procedura oceny zgodności ma dwa etapy:
|
W niektórych przypadkach (np. produkcja masowa w oparciu o typ/próbkę „reprezentatywną dla przewidywanej produkcji”), gdy projekt danego produktu jest skomplikowany, prawodawstwo UE może przewidywać dwa etapy procedury oceny zgodności:
|
— |
najpierw następuje badanie zgodności typu/próbki ze stosownymi wymaganiami prawnymi (tzw. badanie typu UE – moduł B), |
|
— |
a następnie ustalana jest zgodność wytworzonych produktów z zatwierdzonym typem UE. |
W takich przypadkach procedury oceny zgodności składają się z dwóch modułów. Pierwszy jest zawsze moduł B.
Taka metoda nie tylko zmniejsza ciężar i koszty związane z badaniem, ale jest również bardziej wydajna w porównaniu do tradycyjnych badań zgodności produktów przeprowadzanych bezpośrednio w odniesieniu do wymagań prawnych. Gdy ustalony zostanie typ (robi się to tylko raz dla konkretnej próbki), trzeba tylko sprawdzić, czy produkt, który ma zostać wprowadzony do obrotu jest zgodny z zatwierdzonym typem.
Jednostka oceniająca zgodność podejmująca działania w ramach modułu B to niekoniecznie ta sama jednostka, która jest zaangażowana w moduł zestawiony z modułem B.
W przypadkach, w których nie jest przeprowadzane badanie typu UE, procedury oceny zgodności obejmują jeden moduł dwufazowy (projektowanie i produkcja).
Producent przeprowadzający moduł (226) stosowany razem z modułem B nie musi być tą samą osobą, która posiada certyfikat badania typu UE na podstawie modułu B. Jednak taki producent, wprowadzając dany produkt na rynek, ponosi całkowitą odpowiedzialność za ocenę zgodności (projektowanie i produkcję) produktu. W konsekwencji musi posiadać obydwa certyfikaty, przy czym certyfikat badania typu UE nie musi być wystawiony na niego, oraz pełną historię produktu. Musi także posiadać informacje oraz dane administracyjne i techniczne, przeprowadzić badania typu, zarządzać dokumentacją techniczną związaną z badaniami typu i przeprowadzić badania partii. Powyższe argumenty odnoszą się w rzeczywistości do wszystkich modułów i procedur, niezależnie od tego, czy jest to jedno- czy dwuetapowa procedura oceny zgodności. W przypadkach gdy producent korzysta z usług jednego producenta lub kilku innych producentów w celu zaprojektowania i wytworzenia produktu, musi istnieć dowód na to, że producent jest w pełni informowany o wszelkich zmianach w projekcie, produkcji i oceny zgodności produktu.
5.1.6. MODUŁY OPARTE NA ZAPEWNIANIU JAKOŚCI
|
Niektóre moduły i ich warianty opierają się na technikach zapewnienia jakości i powstały na podstawie norm EN ISO 9000 (227), EN ISO 9001 (228). Moduły oparte na technikach zapewnienia jakości (moduły D, E, H i ich warianty) określają elementy, które producent musi wdrożyć w swojej organizacji, aby wykazać, że produkt spełnia zasadnicze wymagania stosownych przepisów.
Oznacza to, że producent ma możliwość zastosowania zatwierdzonego systemu jakości w celu wykazania zgodności z wymaganiami regulacyjnymi. System jakości jest oceniany przez jednostkę notyfikowaną.
System jakości wdrożony na podstawie normy EN ISO 9000, EN ISO 9001 daje podstawę domniemania zgodności z odpowiednimi modułami w odniesieniu do zapisów modułów, które te normy obejmują, pod warunkiem że system jakości uwzględnia specyficzne właściwości objętych nim produktów.
Jednak aby spełnić wymagania modułów, producent może stosować inne modele systemów jakości niż te oparte na normie EN ISO 9001.
Podczas stosowania systemu jakości producent musi zawsze uwzględnić wszystkie przepisy regulacyjne, w szczególności:
|
— |
Cele jakościowe, planowanie jakości i księga jakości muszą w pełni uwzględniać cel dostarczenia produktu zgodnego z zasadniczymi wymaganiami. |
|
— |
Producent ma obowiązek zidentyfikowania i udokumentowania zasadniczych wymagań istotnych z punktu widzenia produktu, a także norm zharmonizowanych lub innych rozwiązań technicznych, które zagwarantują spełnienie tych wymagań. |
|
— |
Zidentyfikowane normy lub inne rozwiązania techniczne muszą być zastosowane jako podstawa projektowania, a także jako potwierdzenie, że spełnione zostaną zasadnicze wymagania. |
|
— |
Środki podjęte w celu kontrolowania produkcji muszą gwarantować, że produkty będą zgodne z ustalonymi zasadniczymi wymaganiami. |
|
— |
Zapisy dotyczące jakości, takie jak sprawozdania z badań i dane z badań, dane dotyczące wzorcowania, sprawozdania dotyczące kwalifikacji odpowiedniego personelu, muszą gwarantować spełnienie odpowiednich zasadniczych wymagań. |
5.1.7. PRZEGLĄD MODUŁÓW
|
Moduły |
Opis |
|
A Wewnętrzna kontrola produkcji |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent we własnym zakresie zapewnia zgodność produktów z wymogami prawnymi (brak badania typu UE). |
|
A1 Wewnętrzna kontrola produkcji oraz badanie produktów pod nadzorem |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. A + badania określonych aspektów produktu przeprowadzone przez akredytowaną jednostkę własną lub jednostkę notyfikowaną wybraną przez producenta. |
|
A2 Wewnętrzna kontrola produkcji oraz nadzorowana kontrola produktów w losowych odstępach czasu |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. A + kontrola produktów w losowych odstępach czasu przeprowadzona przez jednostkę notyfikowaną lub akredytowaną jednostkę własną. |
|
B Badanie typu UE |
Obejmuje fazę projektowania. Zawsze następuje po nim inny moduł, w ramach którego wykazywana jest zgodność produktów z zatwierdzonym typem UE. Jednostka notyfikowana bada projekt techniczny i/lub próbkę danego typu, a także weryfikuje i poświadcza, że spełnia on/ona wymagania określone w instrumencie prawnym, który ma zastosowanie w jego/ jej przypadku poprzez wydanie certyfikatu badania typu UE. Badanie typu UE można przeprowadzić na trzy sposoby: 1) typ produkcji, 2) połączenie typu produkcji i typu projektu oraz 3) typ projektu. |
|
C Zgodność z typem UE w oparciu o wewnętrzną kontrolę produkcji |
Obejmuje produkcję i następuje po module B. Producent musi prowadzić wewnętrzną kontrolę produkcji w celu zapewnienia zgodności produktu z typem UE zatwierdzonym w ramach modułu B. |
|
C1 Zgodność z typem UE na podstawie wewnętrznej kontroli produkcji oraz badania produktów pod nadzorem |
Obejmuje produkcję i następuje po module B. Producent musi prowadzić wewnętrzną kontrolę produkcji w celu zapewnienia zgodności produktu z typem UE zatwierdzonym w ramach modułu B. C + badania określonych aspektów produktu przeprowadzone przez akredytowaną jednostkę własną lub jednostkę notyfikowaną wybraną przez producenta (*). |
|
C2 Zgodność z typem UE na podstawie wewnętrznej kontroli produkcji oraz badania produktów pod nadzorem w losowych odstępach czasu |
Obejmuje produkcję i następuje po module B. Producent musi prowadzić wewnętrzną kontrolę produkcji w celu zapewnienia zgodności produktu z typem UE zatwierdzonym w ramach modułu B. C + kontrola produktów w losowych odstępach czasu w zakresie określonych aspektów produktu przeprowadzona przez jednostkę notyfikowaną lub akredytowaną jednostkę własną. |
|
D Zgodność z typem UE na podstawie systemu zapewnienia jakości procesu produkcji |
Obejmuje produkcję i następuje po module B. Producent stosuje system zapewniania jakości produkcji (faza produkcji i kontrola produktu gotowego) w celu zagwarantowania zgodności z typem UE. Jednostka notyfikowana ocenia system jakości. |
|
D1 Zapewnienie jakości procesu produkcji |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent stosuje system zapewniania jakości produkcji (faza produkcji i kontrola produktu gotowego) w celu zagwarantowania zgodności z wymogami prawnymi (brak typu UE, stosowany jak D bez modułu B). Jednostka notyfikowana ocenia system jakości produkcji (faza produkcji i kontrola produktu gotowego). |
|
E Zgodność z typem UE na podstawie systemu zapewnienia jakości produktu |
Obejmuje produkcję i następuje po module B. Producent stosuje system zapewniania jakości produktu (= jakość produkcji bez fazy produkcji) do kontroli i badań produktu gotowego w celu zagwarantowania zgodności z typem UE. Jednostka notyfikowana ocenia system jakości. Moduł E opiera się na tych samych założeniach co moduł D: oba są oparte na systemie jakości i następują po module B. Różnica między nimi polega na tym, że system jakości zgodny z modułem E ma na celu zapewnić jakość produktu gotowego, natomiast system jakości zgodny z modułem D (i D1) ma na celu zagwarantować jakość całego procesu produkcji (tj. obejmuje zarówno fazę produkcji, jak i badanie produktu gotowego). Dlatego też moduł E jest podobny do D z wyłączeniem przepisów dotyczących procesu produkcji. |
|
E1 Zapewnienie jakości kontroli i badania produktu gotowego |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent stosuje system zapewniania jakości produktu (= jakość produkcji bez fazy produkcji) do kontroli i badań produktu gotowego w celu zagwarantowania zgodności z wymogami prawnymi (bez modułu B (typ UE), stosowany tak jak moduł E bez B). Jednostka notyfikowana ocenia system jakości. Moduł E1 opiera się na tych samych założeniach co moduł D1: oba opierają się na systemie zapewnienia jakości. Różnica między nimi polega na tym, że system jakości zgodny z modułem E1 ma na celu zapewnić jakość produktu gotowego, natomiast system jakości zgodny z modułem D1 ma na celu zagwarantować jakość całego procesu produkcji (tj. obejmuje zarówno fazę produkcji, jak i badanie produktu gotowego). Dlatego też moduł E1 jest podobny do D1 z wyłączeniem przepisów dotyczących procesu produkcji. |
|
F Zgodność z typem UE w oparciu o weryfikację produktu |
Obejmuje produkcję i następuje po module B. Producent zapewnia zgodność wytworzonych produktów z zatwierdzonym typem UE. Jednostka notyfikowana przeprowadza badanie produktów (testowanie każdego produktu lub kontrole statystyczne) w celu zapewnienia zgodności z typem UE. Moduł F jest podobny do modułu C2, ale jednostka notyfikowana przeprowadza bardziej systematyczne kontrole produktów. |
|
F1 Zgodność na podstawie weryfikacji produktu |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent zapewnia zgodność wytworzonych produktów z wymogami prawnymi. Jednostka notyfikowana przeprowadza badania produktów (testowanie każdego produktu lub kontrole statystyczne) w celu zapewnienia zgodności z wymogami prawnymi (brak typu UE, stosowane jak F bez modułu B). Moduł F1 jest podobny do A2, ale jednostka notyfikowana przeprowadza bardziej szczegółowe kontrole produktów. |
|
G Zgodność w oparciu o weryfikację jednostkową |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent zapewnia zgodność wytworzonych produktów z wymogami prawnymi. Jednostka notyfikowana weryfikuje każdy egzemplarz produktu w celu zapewnienia zgodności z wymogami prawnymi (brak typu UE). |
|
H Zgodność oparta na pełnym zapewnieniu jakości |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent stosuje system pełnego zapewnienia jakości w celu zagwarantowania zgodności z wymogami prawnymi (brak typu UE). Jednostka notyfikowana ocenia system jakości. |
|
H1 Zgodność oparta na pełnym zapewnieniu jakości oraz badaniu projektu |
Obejmuje zarówno fazę projektowania, jak i fazę produkcji. Producent stosuje system pełnego zapewnienia jakości w celu zagwarantowania zgodności z wymogami prawnymi (brak typu UE). Jednostka notyfikowana ocenia system jakości i projekt produktu, a także wydaje świadectwo badania projektu UE. W porównaniu do modułu H moduł H1 zakłada dodatkowo, że jednostka notyfikowana przeprowadza bardziej szczegółowe badanie projektu produktu. Nie należy mylić świadectwa badania projektu UE z certyfikatem badania typu UE na podstawie modułu B, który potwierdza zgodność próbki „reprezentatywnej dla przewidywanej produkcji”, dzięki czemu można sprawdzać zgodność produktów względem takiej próbki. W przypadku świadectwa badania projektu UE zgodnie z modułem H1 nie ma takiej próbki. Świadectwo badania projektu UE stanowi potwierdzenie, że zgodność projektu produktu została sprawdzona i poświadczona przez jednostkę notyfikowaną. |
5.1.8. PRZEGLĄD PROCEDUR
Możliwe są następujące procedury:
|
— |
A – Wewnętrzna kontrola produkcji |
|
— |
A1 – Wewnętrzna kontrola produkcji oraz badanie produktów pod nadzorem |
|
— |
A2 – Wewnętrzna kontrola produkcji oraz nadzorowana kontrola produktów w losowych odstępach czasu |
|
— |
B+C – Badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o wewnętrzną kontrolę produkcji (C) |
|
— |
B+C1 – Badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o wewnętrzną kontrolę produkcji oraz badanie produktów pod nadzorem (C1) |
|
— |
B+C2 – Badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o wewnętrzną kontrolę produkcji oraz kontrola produktów w losowych odstępach czasu (C2) |
|
— |
B+D – Badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o system zapewnienia jakości procesu produkcji (D) |
|
— |
D1 – Zapewnienie jakości procesu produkcji |
|
— |
B+E – Badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o system zapewnienia jakości produktu (E) |
|
— |
E1 – Zapewnienie jakości kontroli i badania gotowych produktów |
|
— |
B+F – badanie typu UE (B), a następnie badanie zgodności z typem UE w oparciu o weryfikację produktu (F) |
|
— |
F1 – Zgodność na podstawie weryfikacji produktu |
|
— |
G – Zgodność na podstawie weryfikacji jednostkowej |
|
— |
H – Zgodność oparta na pełnym zapewnieniu jakości |
|
— |
H1 – Zgodność oparta na pełnym zapewnieniu jakości oraz badaniu projektu |

5.1.9. UZASADNIENIE WYBORU ODPOWIEDNICH MODUŁÓW
|
Wybierając moduły do swojego instrumentu prawnego prawodawca powinien stosować się do następujących zasad:
|
— |
Zasadniczo przed wprowadzeniem produktów do obrotu, należy poddać je ocenie zgodnie z zasadami określonymi w modułach dla fazy projektowania i produkcji. |
|
— |
Jeśli to możliwe z punktu widzenia interesu publicznego, producentowi należy zostawić możliwie najszerszy wybór modułów. |
|
— |
Jeśli do zapewnienia zgodności produktów z przepisami prawa wystarczy, aby producent samodzielnie przeprowadził kontrole, prawodawca może wybrać moduł A. Jest to możliwe w przypadku nieskomplikowanych produktów (prosty mechanizm projektowania i produkcji), które nie stwarzają dużego zagrożenia dla interesu publicznego. |
|
— |
W przypadkach produkcji masowej w oparciu o typ/próbkę i gdy projekt danego produktu jest skomplikowany lub istnieje większe ryzyko, że będzie niezgodny z wymogami prawnymi, prawodawstwo UE może przewidywać dwuetapową procedurę oceny zgodności: najpierw badanie zgodności prototypu/ próbki z odpowiednimi przepisami prawa (badanie typu UE – moduł B), a następnie ustalenie zgodności produktów z zatwierdzonym typem UE (moduł C i jego warianty, moduły D, E, F). |
|
— |
Jeśli prawodawca wybrał przeprowadzanie oceny zgodności w odniesieniu do próbki (moduł B), musi sprawdzić, czy producent może samodzielnie przeprowadzić wszystkie kontrole mające na celu zagwarantowanie zgodności z wymogami prawnymi w fazie produkcji. W takim przypadku prawodawca może wybrać moduł C. |
|
— |
W wielu przypadkach prawodawca musi uznać, że dość często producenci prowadzą bardzo dobrze wyposażone laboratoria lub pomieszczenia badawcze. Najczęściej ma to miejsce w przypadku innowacyjnych, skomplikowanych produktów, gdy wiedza na temat sposobu przeprowadzania badań pozostaje u producenta. W takiej sytuacji prawodawca może zastanowić się nad wyborem modułów A1, A2 albo C1, C2 (w przypadku ostatnich dwóch, jeśli wybrał przeprowadzanie oceny zgodności w odniesieniu do próbki – moduł B), które pozwalają na wykorzystanie akredytowanej jednostki własnej. |
|
— |
Jeśli producent nie może samodzielnie wykazać zgodności produktów z zatwierdzonym typem UE, a wymagany jest nadzór procesu produkcji ze strony jednostki notyfikowanej, to prawodawca może nałożyć na producenta obowiązek wdrożenia zatwierdzonego systemu jakości (moduły D, E) lub weryfikacji zgodności wytwarzanych przez niego produktów za pomocą testów/kontroli (moduł F). Jeśli mechanizm produkcji jest stosunkowo „prosty”, prawodawca może uznać, że wystarczy, aby system jakości producenta koncentrował się wyłącznie na kontroli produktu gotowego z pominięciem właściwej fazy produkcji. W takiej sytuacji najbardziej odpowiedni jest moduł E. |
|
— |
Jeśli chodzi o produkty o prostym projekcie, ale skomplikowanym procesie produkcji, prawodawca może rozważyć wybór modułów D1, E1, F1 z zastosowaniem korzyści płynących odpowiednio z modułów D, E i F i bez potrzeby odwoływania się do bardziej formalnego badania próbki (zgodnie z przepisami modułu B, który poprzedza moduł D, E i F). |
|
— |
W przypadku produktów wytwarzanych w małych seriach prawodawca może wybrać moduł G. |
|
— |
W skomplikowanych przypadkach, gdy producent ma obowiązek wdrożyć system pełnego zapewnienia jakości obejmujący zarówno fazę projektowania, jak i fazę produkcji, prawodawca może wybrać moduł H. |
|
— |
Jeśli producent wdrożył system pełnego zapewnienia jakości, ale wymagana jest weryfikacja zgodności projektu i wydanie certyfikatu badania projektu UE przez jednostkę notyfikowaną, prawodawca może wybrać moduł H1. |
5.2. JEDNOSTKI OCENIAJĄCE ZGODNOŚĆ
5.2.1. JEDNOSTKI OCENIAJĄCE ZGODNOŚĆ ORAZ JEDNOSTKI NOTYFIKOWANE
|
Jednostki notyfikowane wykonują zadania związane z procedurami oceny zgodności, o których mowa w obowiązującym prawodawstwie dotyczącym harmonizacji technicznej, gdy wymagana jest strona trzecia. |
Jednostka oceniająca zgodność jest jednostką, która przeprowadza jeden lub więcej elementów oceny zgodności, w tym jedną lub więcej następujących czynności: wzorcowanie, badanie, certyfikacja, inspekcja. Jednostki notyfikowane są jednostkami oceniającymi zgodność, które zostały oficjalnie wyznaczone przez swoje organy krajowe do przeprowadzania procedur oceny zgodności w rozumieniu obowiązującego unijnego prawodawstwa harmonizacyjnego, gdy wymagana jest strona trzecia. W prawodawstwie UE są nazywane „jednostkami notyfikowanymi”.
Jednostki notyfikowane pełnią obowiązki w obszarach interesu publicznego, zatem muszą być odpowiedzialne przed właściwymi organami krajowymi. Aby kwalifikować się na jednostkę notyfikowaną, jednostka musi być podmiotem prawnym posiadającym swoją siedzibę na terytorium państwa członkowskiego, a w związku z tym podlegać jego jurysdykcji. Państwa członkowskie mają wolny wybór w zakresie notyfikowania, bądź nie, jednostki, która spełnia wymagania ustanowione w stosownym unijnym przepisie harmonizacyjnym.
5.2.2. ROLE I OBOWIĄZKI
|
Mimo że jednostka notyfikowana musi posiadać swoją siedzibę na terytorium państwa członkowskiego udzielającego notyfikacji, może ona prowadzić działalność lub zatrudniać pracowników poza państwem członkowskim lub nawet poza UE. Certyfikaty oraz inne poświadczenia oceny zgodności są jednak zawsze wydawane przez jednostkę notyfikowaną oraz w jej imieniu (229). W związku z tym, że jednostka notyfikowana zawsze przeprowadza swoją ocenę w ramach jurysdykcji wyznaczającego państwa członkowskiego, musi ona informować organ udzielający notyfikacji, który musi być zdolny do zapewnienia kontroli całej jednostki jako organ odpowiedzialny za jej działania. Jeżeli kontrola nie jest możliwa, organ notyfikujący powinien wycofać lub ograniczyć zakres notyfikacji w koniecznym zakresie.
Jednostki notyfikowane muszą na bieżąco informować krajowe organy notyfikujące o swojej działalności (m.in. o przeprowadzaniu oceny zgodności, dostępności zasobów, zlecaniu prac podwykonawcy, sytuacjach konfliktu interesów) bezpośrednio lub za pośrednictwem upoważnionej jednostki (np. krajowej jednostki akredytującej). Muszą być również przygotowane na dostarczenie wszelkich informacji dotyczących właściwej realizacji warunków, na podstawie których zostały notyfikowane, zarówno na żądanie ich organów notyfikujących, jak i Komisji.
Jednostki notyfikowane zasadniczo zobowiązane są do informowania organu notyfikującego o wszystkich certyfikatach, których nie wydały, które ograniczyły, zawiesiły lub wycofały z powodu niezgodności wiązanych z bezpieczeństwem oraz, na wniosek, o wydanych świadectwach lub innych prowadzonych działaniach w zakresie oceny zgodności. Ponadto jednostki notyfikowane muszą przekazywać pozostałym notyfikowanym na mocy tego samego unijnego prawodawstwo harmonizacyjnego jednostkom prowadzącym podobną działalność w zakresie oceny zgodności tych samych produktów istotne informacje na temat kwestii dotyczących negatywnych wyników oceny zgodności, a na żądanie, również na temat kwestii dotyczących wyników pozytywnych. Biorąc pod uwagę wymogi dotyczące poufności, których muszą przestrzegać jednostki notyfikowane przy wypełnianiu swych zadań, informacje, które mają być udostępniane innym jednostkom notyfikowanym nie mogą dotyczyć poufnych informacji handlowych o produkcie. Podlegające wymianie informacje na temat negatywnych wyników oceny zgodności powinny zatem dotyczyć głównie odmowy wydania poświadczenia oceny zgodności, zawierającej identyfikację danego produktu i producenta.
Jednostki notyfikowane muszą również dostarczać organowi nadzoru rynku oraz, zgodnie z pewnymi unijnymi przepisami harmonizacyjnymi, również organom nadzoru rynku innych państw członkowskich stosowne informacje do celów prowadzenia nadzoru rynku. Jednostki notyfikowane jako takie nie są odpowiedzialne za dostarczanie deklaracji zgodności UE ani dokumentacji produktu. Jednakże, zgodnie z obowiązującą procedurą oceny zgodności, mogą być zobowiązane do przechowywania dokumentacji produktu jako części swojego zbioru dokumentów technicznych oraz przedstawienia jej na żądanie Komisji lub państw członkowskich (230). Ponadto jednostki notyfikowane muszą przedstawić niezbędne informacje dotyczące produktu lub oceny zgodności na żądanie departamentu Komisji odpowiedzialnego za stosowanie klauzuli ochronnej.
Jednostki notyfikowane są i muszą pozostać stronami trzecimi niezależnymi od swoich klientów oraz innych zainteresowanych stron. Status prawny jednostek starających się o notyfikację, tzn. czy są to jednostki prywatne czy państwowe, jest nieistotny, o ile zagwarantowana jest ich niezależność, bezstronność i rzetelność oraz o ile stanowią niezależne podmioty prawne posiadające stosowne prawa i obowiązki.
Wymóg niezależności dotyczy całej organizacji, łącznie z zarządem, oraz ma też zastosowanie do jednostek należących do stowarzyszeń przedsiębiorców lub zrzeszeń zawodowych.
W celu zapewnienia bezstronności jednostka notyfikowana oraz jej pracownicy nie mogą znajdować się pod żadną presją o charakterze komercyjnym, finansowym lub innym, która mogłaby mieć wpływ na ich osąd. Jednostka musi wdrożyć procedury gwarantujące, ze czynniki zewnętrzne nie będą miały wpływu na jej pracę. Struktura jednostki musi gwarantować jej bezstronność, w szczególności jeżeli jednostka prowadzi inną działalność oprócz tej, którą realizuje jako jednostka notyfikowana.
Ponadto jednostka musi posiadać strategię i procedury pozwalające na rozróżnienie między zadaniami realizowanymi przez nią jako przez jednostkę notyfikowaną a inną działalnością, w którą jest zaangażowana, a rozróżnienie to musi być zrozumiałe dla jej klientów. Z tego względu materiały marketingowe nie mogą stwarzać wrażenia, że ocena lub inna działalność prowadzona przez jednostkę jest związana z zadaniami określonymi w stosownym unijnym prawodawstwie harmonizacyjnym.
Gdy jednostka oceniająca zgodność dostarcza sprawozdanie z badań, leży to w jej kompetencjach jako jednostki oceniającej zgodność; w roli jednostki notyfikowanej może wyłącznie dostarczać certyfikaty badania typu UE – certyfikaty zawierające w szczególności nazwę oraz numer identyfikacyjny jednostki notyfikowanej. W żadnych okolicznościach jednostka notyfikowana nie może wydać sprawozdania z badań opatrzonego jej numerem jednostki notyfikowanej (231) w odniesieniu do testów, które nie są wyszczególnione w przepisach, niezależnie od tego, czy testy te zostały przeprowadzone samodzielnie przez jednostkę, czy przez inny organ. Ponadto jednostka notyfikowana może używać swojego numeru wyłącznie w związku z czynnościami dotyczącymi oceny zgodności wykonywanymi w ramach konkretnego modułu oceny zgodności, który wymaga interwencji jednostki notyfikowanej oraz do których została notyfikowana.
Jednostka notyfikowana musi zobowiązać producenta do zastosowania stosownego środka naprawczego oraz w razie konieczności zawiesić lub wycofać wydany przez siebie certyfikat, jeżeli w trakcie kontroli zgodności po wydaniu certyfikatu uzna, że produkt przestał spełniać wymagania zgodności (232).
W ramach swoich kompetencji jako jednostki notyfikowane nie mogą one oferować ani świadczyć dodatkowych usług, chyba że mają one wartość dodaną dla oceny zgodności produktu. Jednakże jednostki notyfikowane mogą oferować dowolny typ usług oceny zgodności oraz oznakowania w przypadku, gdy produkty są przeznaczone na rynki krajów trzecich spoza Unii Europejskiej, na przykład w kontekście umów o wzajemnym uznawaniu (MRA) (233). Takie czynności muszą być wyraźnie oddzielone od działań jednostki wykonywanych jako jednostka notyfikowana. Jednostki notyfikowane muszą również dopilnować, aby ich działania wykraczające poza zakres przepisów dotyczących harmonizacji technicznej nie miały wpływu na ich kompetencje, obiektywność, bezstronność lub rzetelność ich działalności jako jednostek notyfikowanych. Jednostki notyfikowane nie mogą stosować swojej nazwy ani numeru jednostki notyfikowanej w celu wykonywania tych działań.
Jednostka notyfikowana nie może być producentem, upoważnionym przedstawicielem, dostawcą lub jego konkurentem, nie może (ani nie mogła w przeszłości) oferować lub świadczyć usług doradczych żadnej z tych stron w odniesieniu do projektu, produkcji, praktyk marketingowych lub konserwacji przedmiotowych produktów. Nie wyklucza to jednak możliwości wymiany informacji oraz wskazówek technicznych pomiędzy producentem, jego upoważnionym przedstawicielem, dostawcami i jednostką notyfikowaną.
Aby zagwarantować bezstronność oraz uniknąć konfliktów interesów ważne jest, aby wyraźnie oddzielić ocenę zgodności przeprowadzaną przed jednostki notyfikowane przed wprowadzeniem produktu do obrotu od nadzoru rynku. Ponadto organy nadzoru rynku muszą wykonywać swoje obowiązki w sposób niezależny, bezstronny i wolny od uprzedzeń. Dlatego należy uznać, że organy nadzoru rynkowego nie powinny być wyznaczane jako jednostki notyfikowane, a także należy wprowadzić niezbędne zabezpieczenia w celu zagwarantowania bezstronności i braku konfliktu interesów, w przypadku gdy jednemu podmiotowi powierzono obydwa zadania (234) (235). Jednostki notyfikowane muszą posiadać udokumentowane procedury pozwalające na rozpoznanie, analizę i rozwiązywanie wszelkich przypadków podejrzewanego lub potwierdzonego konfliktu interesów. Jednostka notyfikowana powinna również wymagać od wszystkich pracowników działających w jej imieniu, aby informowali o każdym potencjalnym konflikcie interesów.
Jednostki notyfikowane muszą zatrudniać niezbędny personel, który jest odpowiednio przeszkolony oraz posiada wystarczającą wiedzę techniczną i doświadczenie związane z danymi produktami i procedurą oceny zgodności. Ich wiedza i doświadczenie powinny w szczególności dotyczyć stosownych wymagań prawnych i polityki egzekwowania przepisów, europejskich i międzynarodowych działań normalizacyjnych, odpowiednich technologii, metod produkcji i procedur weryfikacji oraz normalnych warunków użytkowania danego produktu. Jednostka musi być zdolna do zarządzania swoimi wszystkimi zasobami, kontrolowania ich oraz ponoszenia odpowiedzialności za ich działalność oraz musi prowadzić kompleksową ewidencję należytych kwalifikacji wszystkich pracowników, których angażuje w poszczególnych obszarach, niezależnie od tego, czy są to pracownicy zatrudnieni na podstawie umowy o pracę czy delegowani przez jednostki zewnętrzne. Jednostka musi również mieć dostęp do odpowiedniej infrastruktury oraz być zdolna do przeprowadzania testów lub ponownych testów w UE. W przeciwnym przypadku organ notyfikujący nie będzie mógł sprawdzić jej kompetencji.
Jednostki notyfikowane muszą zapewniać poufność wszelkich informacji uzyskanych w trakcie przeprowadzania oceny zgodności. Muszą wdrożyć odpowiednie środki gwarantujące, że żadne wyniki ani inne informacje nie zostaną ujawnione żadnej stronie poza właściwym organem oraz producentem lub jego upoważnionym przedstawicielem.
Jednostki notyfikowane muszą posiadać odpowiednie ubezpieczenie obejmujące ich działalność związaną z prowadzeniem oceny zgodności. Zakres i całkowita wartość finansowa ubezpieczenia od odpowiedzialności musi być adekwatna do poziomu ryzyka związanego z działaniami jednostki notyfikowanej. Jednakże to przede wszystkim producent ponosi ogólną odpowiedzialność za zgodność produktu ze wszystkimi wymaganiami stosownych przepisów, nawet jeżeli niektóre etapy oceny zgodności są przeprowadzane na odpowiedzialność jednostki notyfikowanej.
Jednostki notyfikowane są zobowiązane do uczestniczenia w działaniach koordynacyjnych (236). Muszą również bezpośrednio brać udział lub być reprezentowane w europejskich działaniach normalizacyjnych lub w inny sposób zdobywać wiedzę na temat sytuacji dotyczącej stosownych norm (237).
5.2.3. KOMPETENCJE JEDNOSTEK NOTYFIKOWANYCH
|
Podstawowym zadaniem jednostki notyfikowanej jest świadczenie usług związanych z oceną zgodności na warunkach ustanowionych w stosownym unijnym prawodawstwie harmonizacyjnym. Oznacza to świadczenie producentom usług leżących w interesie publicznym. |
Jednostki notyfikowane są wyznaczane do dokonywania oceny zgodności z zasadniczymi wymaganiami oraz zapewniania konsekwentnego stosowania tych wymagań pod względem technicznym zgodnie ze procedurami przewidzianymi w stosownych unijnych przepisach harmonizacyjnych. Jednostki notyfikowane muszą posiadać odpowiednią infrastrukturę oraz personel techniczny, aby móc wykonywać techniczne i administracyjne zadania związane z oceną zgodności. Muszą również stosować odpowiednie procedury kontroli jakości w związku z takimi usługami. Producenci mogą dowolnie wybierać spośród jednostek notyfikowanych, które zostały wyznaczone do przeprowadzania danej procedury oceny zgodności zgodnie z obowiązującym unijnym prawodawstwem harmonizacyjnym.
Jednostka notyfikowana, która chciałaby oferować usługi zgodnie z kilkoma procedurami oceny zgodności, musi spełniać stosowne wymagania dotyczące poszczególnych zadań, co musi zostać ocenione według wymogów dla poszczególnych procedur. Jednakże, w związku z tym, że zakres wielu przepisów dotyczących harmonizacji technicznej może być stosunkowo szeroki i różnorodny, jednostka notyfikowana nie musi być zakwalifikowana w ramach wszystkich produktów objętych zakresem tego prawodawstwa, lecz może być notyfikowana wyłącznie w zakresie określonej grupy produktów.
Jednostki notyfikowane muszą posiadać odpowiednie struktury i procedury pozwalające zagwarantować, że prowadzenie oceny zgodności oraz wydawanie certyfikatów podlega procesowi weryfikacji. Stosowne procedury muszą w szczególności obejmować obowiązki i zakres odpowiedzialności związane z zawieszaniem oraz wycofywaniem certyfikatów, wnioskami kierowanymi do producenta o podjęcie środków naprawczych oraz składaniem sprawozdań do właściwego organu.
Poza wykonywaniem pewnych zobowiązań w obszarze interesu publicznego jednostki notyfikowane muszą uznać, że świadczą również usługi dla przemysłu. W związku z tym powinny dostarczać producentowi oraz jego upoważnionemu przedstawicielowi stosowne informacje dotyczące przedmiotowych przepisów, przeprowadzać procedurę oceny zgodności bez niepotrzebnego obciążenia dla podmiotów gospodarczych oraz nie proponować dodatkowej certyfikacji lub oznakowania, które nie stanowi wartości dodanej dla oceny zgodności produktu. Ostatnie z wymienionych czynności muszą być wyraźnie oddzielone od działań jednostki wykonywanych jako jednostka notyfikowana. Jednostki notyfikowane nie mogą stosować swojej nazwy ani numeru jednostki notyfikowanej w celu wykonywania tych działań.
Aby uniknąć niepotrzebnych utrudnień dla podmiotów gospodarczych oraz pomóc w zapewnianiu ochrony poufnych danych lub praw własności intelektualnej, dokumentacja produktu przedstawiana jednostkom notyfikowanym musi ograniczać się do informacji, które są wymagane wyłącznie do celu oceny zgodności z przepisami prawa.
5.2.4. KOORDYNACJA MIĘDZY JEDNOSTKAMI NOTYFIKOWANYMI
Ze względu na fakt, że jednostki notyfikowane wykonują zadania wyznaczone im przez organy publiczne, są one zobowiązane do uczestniczenia w działaniach koordynacyjnych organizowanych przez Komisję. Komisja wraz z państwami członkowskimi zapewnia koordynację między jednostkami notyfikowanymi.
Grupa koordynacyjna składająca się z jednostek notyfikowanych jest ustanawiana dla każdego unijnego aktu harmonizacyjnego lub kilku powiązanych aktów, a jej praca ogranicza się do problemów technicznych związanych z oceną zgodności, aby zapewnić jednolite stosowanie technicznych przepisów danego aktu prawnego. W tym celu grupa koordynacyjna powinna być w stanie swobodnie określać swoje zasady pracy oraz swój statut. Każda grupa jednostek notyfikowanych posiada sekretariat do spraw technicznych oraz przewodniczącego.
Zasadniczo grupy jednostek notyfikowanych składają się z przedstawicieli jednostek notyfikowanych. Aby osiągnąć wyższą wydajność swojej pracy, grupy mogą tworzyć podgrupy o ograniczonej liczbie uczestników, których zadaniem jest omawianie konkretnych kwestii natury technicznej. Komisja jest w tych grupach reprezentowana. Eksperci rządowi oraz przedstawiciele organów bezpośrednio odpowiedzialnych za skuteczne wdrażanie unijnego prawodawstwa harmonizacyjnego mogą uczestniczyć w grupach jako obserwatorzy. Europejskie organizacje normalizacyjne (CEN, CENELEC oraz ETSI) są reprezentowane w grupach w przypadku pojawienia się kwestii związanych z normami. Grupy mogą również zapraszać do współpracy stosowne europejskie federacje lub inne zainteresowane strony. Jeżeli grupy jednostek notyfikowanych muszą zajmować się zagadnieniami o charakterze poufnym, uczestnictwo w spotkaniach jest odpowiednio ograniczane.
Jeżeli jednostka odmówi współpracy, notyfikacja może zostać cofnięta. Jednakże jednostki notyfikowane nie są zobowiązane do uczestniczenia w spotkaniach na poziomie europejskim, jeżeli pozostają na bieżąco oraz wdrażają decyzje administracyjne oraz dokumenty wydawane przez ich grupę. Stosowne dokumenty robocze, sprawozdania ze spotkań, zalecenia oraz wytyczne wydawane przez sektorowe oraz międzysektorowe grupy jednostek notyfikowanych lub ich podgrupy powinny być udostępniane wszystkim jednostkom notyfikowanym należącym do tych grup, niezależnie od tego, czy biorą one udział w spotkaniach. Wymianę informacji oraz komunikację można usprawnić, korzystając z platformy takiej jak udostępniana przez Komisję CIRCABC.
Zachęca się również do tworzenia krajowych grup koordynacyjnych, a jeżeli takie istnieją, jednostki notyfikowane z danego państwa członkowskiego mogą być zobowiązane do uczestnictwa w ich działalności.
5.2.5. ZLECANIE PRAC PODWYKONAWCY PRZEZ JEDNOSTKI NOTYFIKOWANE
|
Jednostka notyfikowana może zlecać podwykonawcy wyłącznie zadania, które mieszczą się w zakresie jej kompetencji. Nie są dozwolone sytuacje, w których jednostka notyfikowana zleca część swoich zadań, ponieważ nie posiada wymaganych kompetencji i wiedzy.
Jednostki działające jako podwykonawcy jednostek notyfikowanych same nie muszą być notyfikowane. Niemniej jednostka notyfikowana musi zawiadomić odpowiednie państwo członkowskie o swoim zamiarze zlecenia pracy podwykonawcy. Państwo członkowskie może zadecydować, że jako organ notyfikujący nie może ponosić ogólnej odpowiedzialności za takie ustalenie i cofnąć notyfikację lub ograniczyć jej zakres. Jednostka notyfikowana musi prowadzić ewidencję wszystkich zadań zleconych podwykonawcy i systematycznie ją aktualizować.
Jednostka, której jednostka notyfikowana zleciła pracę na zasadzie podwykonawstwa musi posiadać kompetencje techniczne oraz wykazywać niezależność i obiektywność zgodnie z tymi samymi kryteriami oraz na tych samych warunkach co jednostka notyfikowana. Państwo członkowskie, które notyfikowało jednostkę, która zleca część swoich zadań, musi być zdolne do zapewnienia skutecznego monitorowania kompetencji jednostki zaangażowanej przez jednostkę notyfikowaną. Poszczególni audytorzy zewnętrzni lub specjaliści muszą spełniać warunki podwykonawcy.
Jednostka notyfikowana musi gwarantować, że jej podwykonawcy posiadają wymagane kompetencje oraz że utrzymują te kompetencje, na przykład przeprowadzając regularne oceny oraz będąc na bieżąco ze szczegółami dotyczącymi przeprowadzania ich zadań. Jednostka notyfikowana musi również być zdolna do wykazania zgodności swoich podwykonawców z wymaganiami ustanowionymi w stosownych unijnych przepisach harmonizacyjnych.
Informacje dotyczące podwykonawstwa oraz kompetencji podwykonawców i/lub spółek zależnych muszą być w każdej chwili dostępne, aby organ notyfikujący mógł podjąć każde potrzebne działanie oraz bezzwłocznie powiadomić o nim Komisję i inne państwa członkowskie na ich żądanie. Zgodność z serią norm EN ISO/IEC 17000 stanowi podstawę domniemania zgodności podwykonawcy z większością wymagań, podobnie jak w przypadku samej jednostki notyfikowanej. W przypadku gdy akredytacja nie jest stosowana do oceny kompetencji jednostek notyfikowanych, właściwy organ powinien przeprowadzać kontrole podwykonawcy na miejscu w takim samym zakresie, jaki przewidywałaby akredytacja.
Kolejnym warunkiem zlecania prac podwykonawcy jest to, że procedura oceny zgodności może zostać podzielona na czynności techniczne oraz czynności związane z oceną oraz że metodologia stosowana do przeprowadzenia czynności technicznych jest wystarczająco precyzyjna. Jednostka notyfikowana może zlecać podwykonawcy ściśle ograniczone zadania o charakterze technicznym (takie jak testy i badania), o ile można określić je jako istotne i spójne elementy działań technicznych. Mimo to jednostka zaangażowana przez jednostkę notyfikowaną na zasadzie podwykonawstwa musi wykonać istotne i spójne części tych działań technicznych. Pracownicy jednostki notyfikowanej muszą posiadać odpowiednie kwalifikacje techniczne, aby być w stanie ocenić wyniki testów podwykonawców. Jednostki notyfikowane nie mogą ograniczać swojej działalności wyłącznie do funkcji administracyjnych.
Jednostki notyfikowane mogą na przykład zlecać podwykonawcom przeprowadzanie badań, podczas gdy same będą w dalszym ciągu dokonywać oceny ich wyników oraz, w szczególności, zatwierdzać sprawozdanie z badań, aby sprawdzić, czy zostały spełnione wymagania unijnego prawodawstwa harmonizacyjnego. Podobnie zlecanie prac podwykonawcy jest możliwe w zakresie certyfikacji systemów jakości, pod warunkiem że jednostka notyfikowana przeprowadza ocenę wyników z audytu. Jednostka notyfikowana nie może w żadnym wypadku zlecać podwykonawstwa wszystkich swoich zadań, ponieważ notyfikacja straciłaby wówczas swoje znaczenie.
Zlecona podwykonawcy praca musi być wykonywana zgodnie z wcześniej ustanowioną specyfikacją techniczną zawierającą szczegółową procedurę opartą na obiektywnych kryteriach gwarantujących pełną przejrzystość. W przypadku gdy jednostka zatrudniona przez jednostkę notyfikowaną w charakterze podwykonawcy uczestniczy w ocenie zgodności z normami, należy je stosować, jeżeli służą do ustanowienia procedur. Jeżeli ta jednostka uczestniczy w ocenie zgodności z zasadniczymi wymaganiami, należy stosować procedurę używaną przez jednostkę notyfikowaną lub procedurę, którą jednostka notyfikowana uzna za równoważną.
Jednostka notyfikowana musi w każdym przypadku posiadać wiążącą umowę ze swoimi podwykonawcami gwarantującą, że jej zadania zostaną wykonane (238). Jednostki notyfikowane muszą przechowywać do dyspozycji organu notyfikującego stosowne dokumenty dotyczące oceny kwalifikacji podwykonawcy lub jednostki zależnej oraz wykonywanych przez nich zadań na podstawie stosownego unijnego prawodawstwa harmonizacyjnego (239).
Jednostka notyfikowana zlecająca prace podwykonawcy w dalszym ciągu ponosi odpowiedzialność za wszystkie czynności objęte notyfikacją. Zlecanie prac podwykonawcy nie wiąże się z przekazaniem uprawnień lub obowiązków. Certyfikaty oraz inne poświadczenia oceny zgodności są zawsze wydawane w imieniu jednostki notyfikowanej i na jej odpowiedzialność. Dlatego też jednostka notyfikowana zlecająca podwykonawstwo musi posiadać kompetencje do weryfikowania pracy podwykonawcy we wszystkich jej aspektach oraz musi podejmować ostateczne decyzje.
Warunki zlecania podwykonawstwa prac mają zastosowanie do dowolnego podwykonawcy, niezależnie od tego, czy ma on siedzibę na terytorium Unii Europejskiej. Jednostka notyfikowana pozostaje w pełni odpowiedzialna za pracę wykonywaną w jej imieniu przez podwykonawcę.
Jednostka notyfikowana musi posiadać odpowiednią infrastrukturę oraz personel, aby być w stanie weryfikować wyniki wszelkich badań, inspekcji lub innych zadań wykonywanych przez podwykonawcę. Ponadto jeżeli wybraną ścieżką do notyfikacji jest akredytacja, musi ona obejmować jednostki zależne jednostek notyfikowanych, z których pomocy korzystają. Jednostki akredytujące muszą to uwzględnić, właściwie stosując międzynarodowe wytyczne dotyczące akredytacji transgranicznej albo wyszczególniając je w dokumentach akredytacyjnych. Jeżeli notyfikacja nie jest oparta na akredytacji, wtedy w celu zapewnienia właściwego i spójnego nadzoru nad takimi jednostkami zależnymi oraz podwykonawcami informacje, które należy przedstawić organowi notyfikującemu powinny zostać doprecyzowane w kontekście odpowiednich praktyk akredytacyjnych.
Producent może dostarczyć sprawozdania z badań lub inne elementy dokumentacji produktu. Jednostka notyfikowana może uwzględnić te sprawozdania, pod warunkiem że przyjmuje pełną odpowiedzialność za wyniki. Jednostka notyfikowana może uznać wyniki badań producenta za ocenę zgodności, pod warunkiem że uzasadni, dlaczego uwzględnia te badania.
5.2.6. AKREDYTOWANE JEDNOSTKI WŁASNE (240)
Akredytowana jednostka własna może być wykorzystana do przeprowadzenia czynności w zakresie oceny zgodności w imieniu przedsiębiorstwa, którego jest częścią w ramach realizacji procedur oceny zgodności, modułu A1, A2, C1 lub C2 wyłącznie w przypadkach, w których przewiduje to sektorowe prawodawstwo harmonizacyjne Unii. Jednostka ta musi stanowić niezależną i odrębną część przedsiębiorstwa oraz nie może uczestniczyć w projektowaniu, produkcji, dostawie, instalacji, używaniu lub konserwacji produktów, które ocenia.
Akredytowana jednostka własna musi spełniać szereg wymagań. Musi być akredytowana zgodnie z rozporządzeniem (WE) nr 765/2008 (241). Jednostka oraz jej personel muszą być wyodrębnione w strukturze organizacji oraz posiadać metody składania sprawozdań w ramach przedsiębiorstwa, którego są częścią, zapewniające ich bezstronność, co mogą wykazać przed właściwą krajową jednostką akredytującą. Ani jednostka ani jej personel nie mogą być odpowiedzialne za projekt, produkcję, dostawę, instalację, używanie ani konserwację produktów, które oceniają. Nie mogą też angażować się w żadne czynności, które mogą naruszać niezależność ich osądu lub rzetelność w związku z ich działalnością w zakresie oceny zgodności. Akredytowana jednostka własna może świadczyć usługi wyłącznie na rzecz przedsiębiorstwa, którego jest częścią.
Akredytowana jednostka własna nie podlega notyfikacji państwom członkowskim lub Komisji, natomiast informacja o jej akredytacji musi zostać przekazana przez przedsiębiorstwo, którego jest częścią lub przez krajową jednostkę akredytującą organowi notyfikującemu na jego żądanie.
5.3. NOTYFIKACJA
5.3.1. ORGANY NOTYFIKUJĄCE
|
Organ notyfikujący jest jednostką rządową lub publiczną, której zadaniem jest wyznaczanie i notyfikowanie jednostek oceniających zgodność na podstawie unijnego prawodawstwa harmonizacyjnego. |
Organ notyfikujący jest jednostką rządową lub publiczną, której zadaniem jest wyznaczanie i notyfikowanie jednostek oceniających zgodność na podstawie unijnego prawodawstwa harmonizacyjnego. Przeważnie organem odpowiedzialnym za wdrażanie unijnego aktu harmonizacyjnego, na podstawie którego jednostka jest notyfikowana oraz zarządzanie nim jest administracja krajowa. Każde państwo członkowskie musi wyznaczyć organ notyfikujący, który jest odpowiedzialny za ocenę, notyfikację oraz monitorowanie jednostek oceniających zgodność. Organ notyfikujący ponosi pełną odpowiedzialność za kompetencje jednostek, które notyfikuje.
Każde państwo członkowskie musi ustanowić swoje organy notyfikujące w sposób wykluczający konflikt interesów z jednostkami oceniającymi zgodność. Muszą być zorganizowane oraz funkcjonować tak, aby zapewniać obiektywność i bezstronność ich działań. Każda decyzja dotycząca notyfikacji jednostki oceniającej zgodność musi zostać podjęta przez kompetentne osoby, które osobiście nie przeprowadzały oceny.
Zgodnie z dalszymi wymaganiami dotyczącymi organu notyfikującego nie może on oferować ani świadczyć usług wykonywanych przez jednostki oceniające zgodność, ani usług doradczych na zasadach komercyjnych bądź konkurencyjnych. Musi zapewniać poufność uzyskiwanych informacji oraz musi dysponować wystarczającą liczbą pracowników, aby móc należycie wykonywać swoje zadania.
Państwa członkowskie muszą informować Komisję o swoich procedurach służących do oceny i notyfikacji jednostek oceniających zgodność oraz kontroli jednostek notyfikowanych. Komisja publicznie udostępnia te informacje na swojej stronie internetowej.
5.3.2. PROCES NOTYFIKACJI
|
5.3.2.1. Zasady notyfikacji
Status jednostki notyfikowanej mogą uzyskać jednostki oceniające zgodność ustanowione na terytorium Unii Europejskiej. Państwa członkowskie są odpowiedzialne za notyfikację jednostek notyfikowanych, natomiast wybór i odpowiedzialność za jednostki notyfikowane leży po stronie organów krajowych. Mogą one wybrać jednostki, które notyfikują spośród jednostek ustanowionych na ich terytorium, spełniających wymagania zawarte w prawodawstwie oraz posiadających kompetencje niezbędne do uzyskania notyfikacji. Notyfikacja stanowi akt powiadomienia Komisji oraz innych państw członkowskich przez organ notyfikujący, że taka jednostka została wyznaczona do przeprowadzania oceny zgodności zgodnie z unijnym aktem harmonizacyjnym oraz spełnia wymagania dotyczące jednostek notyfikowanych ustanowione w tym unijnym akcie harmonizacyjnym.
Samo wyznaczenie uznawane jest za akt organu wyznaczającego – który może być tą samą jednostką co organ notyfikujący, natomiast dopiero akt notyfikowania Komisji oraz pozostałych państw członkowskich sprawia, że „jednostka wyznaczona” staje się „jednostką notyfikowaną”.
W związku z tym, że notyfikacja zależy od uznania państw członkowskich, nie są one zobowiązane do notyfikowania wszystkich jednostek wykazujących kompetencje techniczne. Państwa członkowskie nie są też zobowiązane do notyfikowania jednostek w zakresie każdej procedury, która ma być stosowana zgodnie z określonym unijnym aktem harmonizacyjnym.
Państwa członkowskie mogą notyfikować jednostkę w dowolnym momencie po przyjęciu unijnego aktu harmonizacyjnego. Niemniej jednak powinny podjąć wszelkie niezbędne działania w celu udzielenia notyfikacji, zanim unijny akt harmonizacyjny zacznie obowiązywać (242). Przyczyni się to do efektywnego wykorzystania okresu przejściowego przewidzianego w danym unijnym akcie harmonizacyjnym oraz umożliwi działanie jednostkom notyfikowanym i przyznawanie certyfikatów od dnia pierwszego zastosowania unijnego aktu harmonizacyjnego. Jeżeli na podstawie nowych przepisów wymagana jest ponowna notyfikacja jednostek notyfikowanych, gdy tylko państwa członkowskie przetransponują odpowiednie przepisy do prawa krajowego i wyznaczą organ notyfikujący dla danego unijnego aktu harmonizującego, ten organ zgłaszający może dokonać notyfikacji. Jednostka notyfikowana może być zatem notyfikowana na podstawie zarówno starych, jak i nowych przepisów w okresie przejściowym, lecz powiadomienie na mocy starych przepisów wygasa automatycznie z dniem rozpoczęcia stosowania nowych przepisów, chyba że przepisy szczegółowe stanowią inaczej. Należy jednak podkreślić, że w takich przypadkach jednostki notyfikowane, chociaż mogą wykonywać prace przygotowawcze, nie są upoważnione do wydawania certyfikatów przed wejściem w życie unijnego aktu harmonizacyjnego, o ile prawodawstwo sektorowe nie przewiduje inaczej.
5.3.2.2. Ocena jednostek oceniających zgodność
Ocena jednostki oceniającej zgodność dążącej do uzyskania notyfikacji określa, czy posiada ona odpowiednie kompetencje techniczne, czy jest zdolna do przeprowadzania przedmiotowych procedur oceny zgodności oraz czy jest w stanie wykazać wymagany poziom niezależności, bezstronności i rzetelności.
Państwa członkowskie przyjmują ostateczną odpowiedzialność za kompetencje swoich jednostek notyfikowanych względem pozostałych państw członkowskich oraz instytucji UE. Dlatego też muszą zweryfikować kompetencje jednostek chcących uzyskać notyfikację na podstawie kryteriów ustanowionych w unijnym prawodawstwie harmonizacyjnym, które ma zastosowanie, w połączeniu z zasadniczymi wymaganiami oraz przedmiotowymi procedurami oceny zgodności. Zasadniczo kryteria dotyczące kompetencji określone w unijnych aktach harmonizacyjnych obejmują:
|
— |
dostępność personelu oraz sprzętu, |
|
— |
niezależność oraz bezstronność w odniesieniu do podmiotów bezpośrednio lub pośrednio związanych z produktem – (takich jak projektant, producent, upoważniony przedstawiciel producenta, dostawca, monter, instalator, użytkownik), |
|
— |
kompetencje techniczne personelu, które mają związek z przedmiotowymi produktami oraz procedurą oceny zgodności, |
|
— |
zachowanie zasad poufności zawodowej oraz rzetelności, oraz |
|
— |
posiadanie ubezpieczenia od odpowiedzialności cywilnej, o ile nie jest ono zapewnione przez państwo na mocy prawa krajowego. |
Organy notyfikujące lub jednostki akredytujące muszą przeprowadzać okresowe monitorowanie, aby oceniać ciągłość kompetencji jednostek notyfikowanych po uzyskaniu przez nie notyfikacji.
5.3.2.3. Akredytacja na podstawie rozporządzenia (WE) nr 765/2008
Akredytacja przeprowadzana zgodnie z serią norm EN ISO/IEC 17000 przez uznawane na poziomie krajowym jednostki akredytujące należące do Europejskiej Współpracy w Dziedzinie Akredytacji (EA) stanowi techniczną ocenę kompetencji jednostki oceniającej zgodność dążącej do uzyskania notyfikacji. Mimo iż nie jest ona wymaganiem, pozostaje istotnym i preferowanym instrumentem służącym do oceniania kompetencji oraz rzetelności jednostek, które mają zostać notyfikowane. Z tego względu akredytacja powinna być uznawana przez krajowe organy notyfikujące jako najbardziej preferowana podstawa techniczna dla oceny jednostek oceniających zgodność, co pozwoli zmniejszyć różnice w kryteriach stosowanych do notyfikacji.
Akredytacja stanowi wiarygodne potwierdzenie kompetencji, profesjonalnej rzetelności oraz bezstronności jednostek, które mają zostać notyfikowane do Komisji oraz pozostałych państw członkowskich. Wiąże się również z regularną kontrolą oraz nadzorem nad jednostkami akredytowanymi. Zawsze gdy krajowa jednostka akredytująca stwierdza, że jednostka oceniająca zgodność, której przyznała certyfikat akredytacji przestała być kompetentna lub nie wypełnia swoich obowiązków, certyfikat akredytacji może zostać cofnięty. W takim przypadku notyfikacja jednostki powinna zostać wycofana, a jednostka traci upoważnienie do przeprowadzania czynności związanych z oceną zgodności na podstawie przedmiotowego prawodawstwa.
Pierwszeństwo akredytacji przyznawane jest na podstawie procesu wzajemnej oceny, który gwarantuje, że jednostka akredytująca odpowiednio nadzoruje jednostki oceniające zgodność, którym udziela akredytacji. Mogą jednak wystąpić przypadki, gdy krajowa jednostka akredytująca nie przeszła pozytywnie wzajemnej oceny, ale być może oceniła jednostki notyfikowane (243). Jeśli krajowa jednostka akredytująca nie została poddana wzajemnej ocenie w odniesieniu do danej działalności akredytacyjnej, ale w dalszym ciągu ocenia kompetencje jednostki oceniającej zgodność w zakresie tej działalności, wówczas notyfikacji tej jednostki oceniającej zgodność nie należy uznawać za akredytację do celów unijnego prawodawstwa harmonizacyjnego.
Jeśli krajowa jednostka akredytująca pozytywnie przeszła poprzednią wzajemną ocenę w odniesieniu do danej działalności, ale została zawieszona w kolejnej wzajemnej ocenie, nowe notyfikacje jednostek oceniających zgodność ocenianych przez tę krajową jednostkę akredytującą należy także uważać za nieakredytowane. Zasadniczo certyfikaty akredytacji wydane do momentu zawieszenia krajowej jednostki akredytującej w wyniku wzajemnej oceny, powinny nadal być uznawane przez organy krajowe.
Jeżeli powody zawieszenia krajowej jednostki akredytującej skutkują poważnymi wątpliwościami co do kompetencji jednostek notyfikowanych, odpowiedzialny organ notyfikujący musi poinformować Komisję i inne państwa członkowskie o tym, jak zamierza on zapewnić kompetencje jednostek notyfikowanych, oraz o wszelkich podjętych środkach naprawczych, łącznie z odebraniem notyfikacji.
Pomimo że akredytacja stanowi preferowany instrument weryfikacji kompetencji jednostek oceniających zgodność, państwa członkowskie mogą przeprowadzać ocenę samodzielnie. Od wejścia w życie rozporządzenia (WE) nr 765/2008 w dniu 1 stycznia 2010 r. w takich przypadkach należy przedstawić Komisji oraz pozostałym państwom członkowskim dowody, że oceniana jednostka spełnia wszystkie stosowne wymogi regulacyjne. Ponadto jednostka notyfikowana musi podlegać regularnemu nadzorowi podobnemu do praktyki ustanowionej przez organizacje akredytujące.
5.3.2.4. Art. 5 ust. 2 rozporządzenia (WE) nr 765/2008
Zgodnie z art. 5 ust. 2 rozporządzenia (WE) nr 765/2008, jeżeli państwo członkowskie nie opiera swojej notyfikacji na akredytacji, wtedy „dostarcza Komisji i innym państwom członkowskim wszelkich pisemnych dowodów koniecznych do sprawdzenia kompetencji jednostek oceniających zgodność, które wybiera do wdrożenia tego wspólnotowego prawodawstwa harmonizacyjnego” (244).
Aby zapewnić wymagany poziom zaufania do bezstronności oraz kompetencji technicznych jednostek oceniających zgodność oraz wydawanych przez nie certyfikatów, organy krajowe, przeprowadzając ocenę bez akredytacji, powinny przedstawić szczegółowe i pełne informacje opisujące, w jaki sposób kandydat na jednostkę notyfikowaną został oceniony jako należycie wykwalifikowany do przeprowadzania zadań objętych notyfikacją oraz wykazujące, że spełnia on stosowne kryteria dotyczące jednostek notyfikowanych. Te informacje, dołączone do danej notyfikacji, są udostępniane Komisji oraz pozostałym państwom członkowskim za pośrednictwem elektronicznego narzędzia do notyfikacji NANDO.
Procedura oceny powinna opierać się co najmniej na następujących elementach:
|
— |
formalna procedura składania wniosku, |
|
— |
ocena zgodności z obowiązującymi wymaganiami, |
|
— |
sporządzenie sprawozdania z oceny, |
|
— |
przejrzysty proces podejmowania decyzji, |
|
— |
istnienie systematycznego nadzoru oraz powiązanego mechanizmu sankcji przewidującego okresową kontrolę obejmującą, |
|
— |
wizyty w celu weryfikacji ciągłego spełniania wymagań przez jednostkę notyfikowaną, |
|
— |
wykazanie własnych kompetencji technicznych organu krajowego do przeprowadzania oceny jednostek oceniających zgodność do celów notyfikacji w ramach prawodawstwa dotyczącego harmonizacji technicznej. Wykazanie to musi dawać gwarancję równoważną z systemem oceny wzajemnej EA (245), |
|
— |
kandydaci na jednostkę notyfikowaną powinni zostać poinformowani o ogólnych warunkach, swoich prawach i obowiązkach oraz o wymaganiach związanych z oceną przeprowadzaną do celów notyfikacji. |
Sama ocena powinna obejmować:
|
— |
przegląd dokumentów potwierdzający ich kompletność i stosowność w odniesieniu do obowiązujących wymagań pod kątem merytorycznym, |
|
— |
audyt na miejscu sprawdzający aspekty techniczne i proceduralne – takie jak dostępność i stosowność infrastruktury i wyposażenia, kompetencje techniczne pracowników, istnienie odpowiedniego systemu zarządzania – oraz inne aspekty wykazujące, że zgodność z wymaganiami jest należycie wdrożona. Ocena musi obejmować osobistą obserwację czynności technicznych. |
Wybierając proces oceny inny niż formalna akredytacja, organy notyfikujące muszą wskazać powody, dla których akredytacji nie wybrano jako podstawy procesu notyfikacji. Ponadto organy notyfikujące nie mogą powierzyć krajowej jednostce akredytującej przeprowadzenia oceny nieakredytowanych jednostek oceniających zgodność, które dążą do uzyskania notyfikacji bez przeprowadzenia całego procesu akredytacji łącznie z dostarczeniem certyfikatu akredytacji.
Jeżeli nie stosuje się akredytacji, organy notyfikujące muszą przeprowadzać okresowe weryfikacje, podobnie jak robią to krajowe jednostki akredytujące, aby zapewnić ciągłość kompetencji jednostki notyfikowanej.
5.3.2.5. Etapy notyfikacji jednostki notyfikowanej
Aby uzyskać notyfikację, jednostka oceniająca zgodność składa wniosek o notyfikację do organu notyfikującego państwa członkowskiego, w którym jest ustanowiona. Do wniosku powinien być dołączony opis czynności związanych z oceną zgodności, procedur lub modułów oceny zgodności oraz produktu lub produktów wchodzących w zakres kompetencji danej jednostki, jak również certyfikat akredytacji, jeżeli istnieje, wydany przez krajową jednostkę akredytującą, potwierdzający, że jednostka oceniająca zgodność spełnia wymagania przewidziane w stosownych przepisach harmonizacyjnych.
Jeżeli dana jednostka nie może przedstawić certyfikatu akredytacji, musi dostarczyć organowi notyfikującemu wszelkie dokumenty stanowiące dowody niezbędne do weryfikacji, uznania oraz regularnej kontroli jej zgodności z wymaganiami określonymi w stosownym prawodawstwie harmonizacyjnym. Po dokonaniu weryfikacji państwo członkowskie informuje Komisję oraz pozostałe państwa członkowskie o szczegółach dotyczących jednostki.
Notyfikacja jednostki notyfikowanej jest wysyłana przez organ notyfikujący do Komisji oraz pozostałych państw członkowskich za pośrednictwem NANDO (ang. New Approach Notified and Designated Organisations), elektronicznego narzędzia do notyfikacji opracowanego i zarządzanego przez Komisję. Powinna ona zawierać dokładne informacje o jednostce, wykonywanych przez nią czynnościach związanych oceną zgodności, procedurami lub modułami oceny zgodności i przedmiotowym produkcie lub produktach oraz stosowne poświadczenie kompetencji jednostki. Powinna ona zawierać również wyznaczony termin ponownej oceny jednostek notyfikowanych przez krajowe jednostki akredytujące lub, w przypadku notyfikacji nieakredytowanej, termin kolejnej oceny wykonywanej przez organ notyfikujący.
Jeżeli notyfikacja nie jest oparta na certyfikacie akredytacji, organ notyfikujący musi przedstawić Komisji oraz pozostałym państwom członkowskim dokumenty potwierdzające kompetencje jednostki oceniającej zgodność, sposób przeprowadzenia jej oceny oraz wdrożone środki mające na celu zagwarantowanie, że dana jednostka będzie regularnie kontrolowana oraz nieprzerwanie zgodna z wymaganiami.
Notyfikacja wchodzi w życie po tym, jak e-mail z notyfikacją zostanie wysłany z NANDO do Komisji oraz pozostałych państw członkowskich oraz opublikowany na stronie internetowej NANDO. Wtedy przedmiotowa jednostka może wykonywać zadania jednostki notyfikowanej. Zgodnie z przepisami dostosowanymi do postanowień decyzji nr 768/2008/WE notyfikacja jest publikowana po okresie przeznaczonym na wnoszenie zastrzeżeń przez inne państwa członkowskie lub Komisję – wynoszącym dwa tygodnie w przypadku wykorzystania akredytacji i dwa miesiące, jeżeli nie stosowano akredytacji – oraz wyłącznie w wypadku gdy nie zgłoszono żadnych zastrzeżeń.
Komisja oraz pozostałe państwa członkowskie muszą zostać w podobny sposób powiadomione o wszelkich późniejszych zmianach w notyfikacji, które są istotne, takich jak zmiana zakres lub okresu ważności notyfikacji bądź zmiany w informacjach dotyczących samej jednostki.
5.3.3. PUBLIKACJA PRZEZ KOMISJĘ – STRONA INTERNETOWA NANDO
Komisja publicznie udostępnia wykazy jednostek notyfikowanych (oraz innych kategorii jednostek oceniających zgodność, takich jak inspektoraty użytkowników oraz uznane organizacje strony trzeciej) na stronie internetowej NANDO na jej serwerze Europa w celach informacyjnych. Wykazy te są aktualizowane przy okazji publikacji nowych notyfikacji, natomiast strona internetowa jest odświeżana każdego dnia.
Wraz z pierwszą notyfikacją jednostka notyfikowana otrzymuje swój numer identyfikacyjny w systemie NANDO. Numer ten jest automatycznie generowany przez system w chwili walidacji notyfikacji w bazie danych NANDO. Podmiot prawny może posiadać wyłącznie jeden numer identyfikacyjny jednostki notyfikowanej, bez względu na liczbę unijnych przepisów harmonizacyjnych, w odniesieniu do których jest notyfikowany. Przyznanie numeru jest czynnością czysto administracyjną mającą zapewnić sprawne zarządzanie wykazami jednostek notyfikowanych i nie nadaje żadnych praw ani w żaden sposób nie zobowiązuje Komisji. System numerowania w NANDO ma charakter ciągły i numery nie są ponownie wykorzystywane, gdy jednostka notyfikowana zostaje wycofana z wykazu. W przypadku zawieszenia lub wycofania notyfikacji informacje dotyczące notyfikacji pozostają w bazie danych i zostają przeniesione do części strony internetowej o nazwie „Wycofane/wygasłe notyfikacje/jednostki notyfikowane” (246).
Zmiany (rozszerzenie lub ograniczenie) zakresu, modyfikacje okresu ważności notyfikacji lub anulowanie notyfikacji są również podawane do wiadomości państw członkowskich e-mailem i publikowane na stronie internetowej NANDO. Stronę internetową można przeszukiwać według unijnego aktu harmonizacyjnego, kraju, numeru jednostki notyfikowanej lub słów kluczowych.
5.3.4. MONITOROWANIE KOMPETENCJI JEDNOSTEK NOTYFIKOWANYCH – ZAWIESZENIE – WYCOFANIE – ODWOŁANIE
Ważne jest, aby jednostki notyfikowane utrzymywały swoje kompetencje i aby fakt ten był znany pozostałym państwom członkowskim oraz Komisji. Prawodawstwo na poziomie UE wyraźnie nakłada na właściwe organy krajowe obowiązek regularnego monitorowania i oceny kompetencji notyfikowanych przez siebie organów wymienionych w wykazie NANDO. Strona internetowa NANDO powinna być przejrzysta, jeśli chodzi o te bieżące procesy, które stanowią wsparcie systemu powiadomień.
Wszystkie notyfikacje jednostek notyfikowanych, akredytowanych lub nie, figurujących w bazie NANDO, należy aktualizować – w ciągu maksymalnie pięciu lat od daty pierwszej notyfikacji lub ostatniej aktualizacji – podając informacje na temat stałego monitorowania kompetencji jednostki notyfikowanej. Aktualizacje te powinny obejmować stosowne nowe dane dotyczące akredytacji lub, jeżeli notyfikacja jest nieakredytowana, informacje dotyczące wymaganego monitorowania jednostki przez organ notyfikujący – w szczególności sprawozdanie z procesu oceny, tj. przegląd dokumentacji, ocenę na miejscu, opis mechanizmu systematycznego nadzoru, w tym kontroli na miejscu, oraz dowód kompetencji technicznych organu w zakresie prowadzenia oceny. Jeżeli powiadomienie nie zostało zaktualizowane po upływie 5 lat, Komisja uznaje, że istnieją powody, aby zakwestionować ciągłość kompetencji jednostki notyfikowanej (247) i zwraca się do notyfikującego państwa członkowskiego o przekazanie wszelkich informacji dotyczących utrzymania kompetencji przedmiotowej jednostki.
Komisja oraz państwa członkowskie mają obowiązek zareagować, jeżeli pojawią się wątpliwości dotyczące kompetencji jednostki notyfikowanej w chwili notyfikacji lub po jej przyznaniu. Jeżeli Komisja z własnej inicjatywy lub po otrzymaniu skargi stwierdzi, że dana jednostka notyfikowana nie spełnia wymagań lub nie wywiązuje się ze swoich obowiązków, informuje o tym krajowy organ notyfikujący i żąda właściwie udokumentowanych podstaw notyfikowania jednostki oraz utrzymania jej kompetencji. Jeżeli państwo członkowskie nie przedstawi takich informacji, Komisja może zwrócić na to uwagę innych państw członkowskich w celu przedyskutowania tej kwestii lub wszcząć procedurę przeciwko notyfikującemu państwu członkowskiemu zgodnie z art. 258 TFUE.
Gdy organ notyfikujący stwierdzi lub zostanie poinformowany, że jednostka notyfikowana przestała spełniać wymagania przewidziane w stosownych przepisach lub przestała wypełniać swoje obowiązki, organ notyfikujący musi – w zależności od powagi tych zaniedbań – zawiesić lub wycofać notyfikację natychmiast po skontaktowaniu się z przedmiotową jednostką. Musi on również odpowiednio natychmiast poinformować Komisję oraz pozostałe państwa członkowskie. Państwo członkowskie musi opublikować tę informację i powiadomić Komisję oraz pozostałe państwa członkowskie zgodnie z procedurą podobną jak w przypadku notyfikacji. Przedmiotowa jednostka powinna mieć możliwość odwołania się od takiej decyzji. To, czy takie odwołanie opóźnia odebranie notyfikacji czy nie, zależy od prawa krajowego.
Wycofanie notyfikacji ma miejsce, gdy jednostka notyfikowana przestaje spełniać wymagania lub swoje obowiązki. Można go dokonać na wniosek zgłaszającego państwa członkowskiego, jeśli otrzymało ono dowody niespełniania wymogów przez jednostkę notyfikowaną, uzyskane podczas okresowego nadzoru (wykonywanego przez jednostkę akredytującą lub organ notyfikujący), lub jeśli otrzymało skargi dotyczące kompetencji lub zachowania jednostki notyfikowanej. Wycofanie może również być wynikiem działań Komisji, jeżeli ma ona powód, by wątpić, czy jednostka notyfikowana spełnia lub w dalszym ciągu spełnia wymagania notyfikacji. W takich przypadkach Komisja odpowiednio informuje notyfikujące państwo członkowskie oraz nakłada na nie obowiązek podjęcia koniecznych środków naprawczych, w tym w razie konieczności odebrania notyfikacji danej jednostce. Organ notyfikujący musi podjąć odpowiednie środki. Innym powodem wycofania notyfikacji może być wniosek samej jednostki notyfikowanej, na przykład ze względu na planowane zmiany w polityce, organizacji lub własności jednostki. Wycofanie notyfikacji może również stanowić wynik końcowy procedury w sprawie uchybienia zobowiązaniom państwa członkowskiego.
Wycofanie notyfikacji wchodzi w zakres odpowiedzialności notyfikującego państwa członkowskiego. Jedynie organy krajowe są upoważnione do wycofania notyfikacji. Komisja może wycofać jednostkę notyfikowaną z wykazu NANDO wyłącznie wówczas, gdy na zakończenie procedury w sprawie uchybienia zobowiązaniom państwa członkowskiego na podstawie art. 258 TFUE Trybunał uzna, że państwo członkowskie uchybiło zobowiązaniom na podstawie przepisów danego unijnego aktu harmonizacyjnego, w wyniku czego uzna notyfikację za nieważną. We wszystkich takich przypadkach Komisja zapewni, aby wszelkie szczególnie chronione informacje uzyskane w trakcie jej dochodzeń były traktowane z zachowaniem poufności.
Bez uszczerbku dla specyfiki różnych sektorów zawieszenie lub wycofanie notyfikacji nie ma wpływu na ważność certyfikatów wydanych przez jednostkę notyfikowaną do czasu udowodnienia, że certyfikaty te powinny zostać cofnięte. W celu zapewnienia ciągłości w przypadku zawieszenia lub wycofania notyfikacji lub zawieszenia działalności przez jednostkę notyfikowaną notyfikujące państwo członkowskie musi dopilnować, aby inna jednostka notyfikowana przejęła dokumentację tej jednostki lub aby dokumentacja ta pozostawała dostępna do wglądu na żądanie odpowiedzialnych organów notyfikujących oraz organów nadzoru rynku.
6. AKREDYTACJA
Rozporządzenie (WE) nr 765/2008 stanowi ramy prawne dla akredytacji na poziomie krajowym oraz unijnym oraz ustanawia ogólną politykę wraz z zasadami, procedurami i infrastrukturami. Wzmocnienie akredytacji jako środka stanowiącego podstawę kompetencji jednostek oceniających zgodność, a zatem również wiarygodności i akceptowalności certyfikatów oraz innych poświadczeń, niezbędne do zapewnienia swobodnego przepływu towarów, jest przedmiotem zainteresowania Komisji od końca lat 70. W latach 90. istniała tendencja, zgodnie z którą akredytacja miała stać się działalnością komercyjną i konkurencyjną, co zmniejszyłoby jej wiarygodność jako ostatniego szczebla kontroli. Jednak nowe ramy prawne potwierdziły, że akredytacja w UE jest niekomercyjną oraz niekonkurencyjną działalnością publiczną podlegającą zarówno organom krajowym, jak i unijnym.
Dzięki temu wzmocniony system akredytacyjny UE jest zgodny z normami, zasadami oraz praktykami organizacji międzynarodowych w tym zakresie. Celem rozporządzenia (WE) nr 765/2008 jest zapewnienie, aby akredytacja służyła interesowi publicznemu. Europejska Współpraca w dziedzinie Akredytacji (EA), czyli europejska organizacja krajowych jednostek akredytujących, jest uznana w rozporządzeniu, w wytycznych podpisanych z państwami członkowskimi (w tym z krajami EFTA) oraz Komisją w dniu 1 kwietnia 2009 r. oraz korzysta z uprzywilejowanych relacji z Komisją na podstawie podpisanej ramowej umowy o partnerstwie. Zgodnie z tymi ramami współpracy główną rolą EA jest przyczynianie się do harmonizacji europejskich usług akredytacyjnych w celu wsparcia wzajemnego uznawania i akceptacji certyfikatów akredytacji w Unii oraz prowadzenie rygorystycznego systemu oceny wzajemnej, który kontroluje kompetencje krajowych jednostek akredytujących oraz równoważność ich usług.
W zakresie akredytacji rozporządzeniem (WE) nr 765/2008 ustanowiono jednolity system europejski obejmujący zarówno obszar regulowany, w którym akredytacja jest wymagana przez przepisy prawa, jak i sferę nieregulowaną. W drugim przypadku, gdy jednostka dobrowolnie chce zostać akredytowana, może zwrócić się wyłącznie do jednostek akredytujących działających na podstawie rozporządzenia (WE) nr 765/2008, dzięki czemu unika się konkurujących systemów, bez względu na to, na jakich zasadach się opierają.
6.1. DLACZEGO AKREDYTACJA?
|
Akredytacja stanowi ostatni szczebel kontroli publicznej w łańcuchu jakości, na którym opiera się swobodny przepływ towarów w Unii. |
Rozporządzeniem (WE) nr 765/2008 po raz pierwszy wprowadzono ramy prawne dotyczące akredytacji. Akredytacja jednostek oceniających zgodność była wcześniej wykorzystywana zarówno w obszarach regulowanych, jak i nieregulowanych, natomiast nie podlegała ramom prawnym na szczeblu unijnym.
Idea uregulowania akredytacji na poziomie Unii ma dwa aspekty. Z jednej strony całościowe unijne ramy prawne dotyczące akredytacji zapewniają ostatni szczebel kontroli publicznej w unijnym łańcuchu oceny zgodności, stanowiąc istotny element gwarantujący zgodność produktów. Z drugiej strony ulepszają one swobodny przepływ produktów i usług w UE, zwiększając zaufanie do ich bezpieczeństwa oraz zgodności z innymi aspektami ochrony interesów publicznych.
Zanim rozporządzenie weszło w życie, brak wspólnych zasad dotyczących akredytacji w państwach członkowskich oznaczał, że akredytacja była stosowana bardzo różnie, w związku z czym certyfikaty akredytacji niekoniecznie były uznawane przez poszczególne organy krajowe oraz operatorów rynku, a to z kolei prowadziło do wielokrotnych akredytacji i zwiększało koszty ponoszone przez przedsiębiorstwa oraz jednostki oceniające zgodność, nie dając opisanych wyżej korzyści.
Wprowadzenie ram prawnych dla akredytacji zmniejszyło obciążenia administracyjne na jednolitym rynku oraz usprawniło kontrolę publiczną nad akredytacją, dzięki czemu odgrywa ona rolę zasadniczego narzędzia w funkcjonowaniu rynku wewnętrznego.
Ramy akredytacji ustanowione rozporządzeniem mają wyraźnie zastosowanie do sfery zarówno regulowanej, jak i dobrowolnej. Rozróżnienie między tymi dwoma sferami może być bowiem niejasne, gdyż jednostki oceniające zgodność prowadzą działalność, a produkty są wykorzystywane w obu obszarach. Rozróżnienie doprowadziłoby zatem do niepotrzebnego obciążenia dla organów publicznych oraz podmiotów rynkowych, skutkując sprzecznościami między domeną dobrowolną a regulowaną.
6.2. CZYM JEST AKREDYTACJA?
|
Akredytacja stanowi oparte na normach zharmonizowanych poświadczenie krajowej jednostki akredytującej, że jednostka oceniająca zgodność posiada kompetencje techniczne do wykonywania określonych zadań związanych z oceną zgodności. |
Akredytacja stanowi poświadczenie przez krajową jednostkę akredytującą, że jednostka oceniająca zgodność spełnia wymagania ustanowione przez normy zharmonizowane oraz, w stosownych przypadkach, wszelkie dodatkowe wymagania, łącznie z określonymi w odpowiednich systemach sektorowych, dotyczące przeprowadzania określonych zadań związanych z oceną zgodności.
Zewnętrznej ocenie zgodności podlega szeroki zakres produktów. Obejmuje to produkty niepodlegające regulacji oraz produkty regulowane na poziomie krajowym lub unijnym. W przypadku produktów regulowanych na poziomie UE, tj. na obszarze zharmonizowanym, zazwyczaj oznacza to, że wyznaczone na poziomie krajowym jednostki oceniające zgodność – jednostki notyfikowane – badają produkt oraz wydają poświadczenie zgodności, zanim produkt może zostać wprowadzony do obrotu.
Dokładniej rzecz ujmując, aby doszło do akredytacji, musi istnieć podlegająca akredytacji jednostka oceniająca zgodność (niezależnie od jej osobowości prawnej), prowadząca określoną działalność z zakresu oceny zgodności.
Akredytacja jest działaniem opartym na normach, które służy zagwarantowaniu i poświadczeniu, że jednostki oceniające zgodność posiadają kompetencje techniczne niezbędne do wykonywania swoich obowiązków określonych w stosownych regulacjach i normach. Ocenia ona zdolność jednostek oceniających zgodność do wykonywania ich obowiązków w określonych obszarach, ponieważ akredytacja jest zawsze związana z określonym zakresem działalności jednostki oceniającej zgodność. Działając w interesie publicznym, akredytacja ocenia kompetencje techniczne, wiarygodność oraz rzetelność jednostek oceniających zgodność. W tym celu stosuje się proces przejrzystej i bezstronnej oceny według uznanych na poziomie międzynarodowym norm oraz innych wymagań. Rozporządzenie (WE) nr 765/2008 zobowiązuje krajowe jednostki akredytujące do weryfikowania, czy oceny zgodności są przeprowadzane w należyty sposób oraz czy uwzględnia się rozmiar i strukturę przedsiębiorstw, stopień złożoności przedmiotowej technologii produktu oraz specyfikę procesu produkcji.
Akredytacja jest oparta na międzynarodowych normach dotyczących jednostek oceniających zgodność, które zostały zharmonizowane w nowych ramach prawnych i do których odniesienia opublikowano w Dzienniku Urzędowym UE. Stanowi ona poświadczenie przez krajową jednostkę akredytującą, że jednostka oceniająca zgodność spełnia wymagania ustanowione przez normy zharmonizowane oraz, w stosownych przypadkach, wszelkie dodatkowe wymagania, łącznie z wymaganiami określonymi w odpowiednich systemach sektorowych. Zgodnie z rozporządzeniem (WE) nr 765/2008 wyłącznie krajowe jednostki akredytujące są upoważnione do prowadzenia akredytacji jednostek oceniających zgodność.
Poleganie na normach zharmonizowanych opartych na odpowiadających im normach międzynarodowych ma na celu zapewnienie koniecznego poziomu przejrzystości i zaufania do kompetencji jednostek oceniających zgodność oraz zagwarantowanie, że unijny system akredytacyjny ustanowiony przez rozporządzenie (WE) nr 765/2008 jest spójny z międzynarodowym systemem akredytacyjnym – co pozwoli usprawnić handel międzynarodowy.
Biorąc pod uwagę znaczącą rolę w systemie oceny zgodności, jaką to rozporządzenie nadało krajowym jednostkom akredytującym, jednostki akredytujące muszą stosować się ściśle do przepisów rozporządzenia przy ocenie kompetencji jednostek oceniających zgodność. Prawodawca postanowił wyraźnie ograniczyć zakres działań, jakie może prowadzić jednostka akredytująca, sprawując ścisłą kontrolę nad ich kompetencjami poprzez bezpośrednie odniesienie do norm zharmonizowanych. Oznacza to również, że organy krajowe nie mogą wymagać od jednostek akredytujących wykonywania usług oceny niezwiązanych z procedurą pełnej akredytacji lub korzystać z norm oceny zgodności, które nie są zharmonizowane – powinny wręcz temu zapobiegać.
6.3. ZAKRES AKREDYTACJI
|
Akredytacji udziela się lub stara się o nią zawsze w odniesieniu do określonego zakresu, tj. do konkretnych działań związanych z oceną zgodności. |
Akredytacja jest opartą na normach metodą oceniania oraz poświadczania kompetencji jednostek oceniających zgodność. Polityka Unii wykorzystuje akredytację jako instrument do tworzenia warunków wzajemnego zaufania, ponieważ opiera się ona na normach wynikających z konsensusu. Wzajemne zaufanie można osiągnąć wyłącznie dzięki stosowaniu kryteriów, które można obiektywnie zweryfikować, zapewniając w ten sposób przejrzystość oraz porównywalność oceny zgodności. Odpowiednie normy dla jednostek oceniających zgodność (248) zostały opracowane z zamiarem wsparcia wprowadzenia procedur oceny zgodności przewidzianych w unijnym prawodawstwie harmonizacyjnym (249). Normy te z założenia obejmują ogólne wymagania dotyczące kompetencji jednostek przeprowadzających ocenę zgodności ze szczegółowymi wymaganiami, niezależnie czy są one zawarte w regulacjach, normach czy innych specyfikacjach technicznych ani czy takie specyfikacje dotyczą sposobu postępowania czy konkretnego produktu. Koncepcja ta stanowi wsparcie dla roli akredytacji jako narzędzia usprawniającego swobodny przepływ produktów na rynku wewnętrznym i została przejęta przez normy ISO/IEC 17000 na poziomie międzynarodowym.
Jak określono w odpowiednich klauzulach obejmujących ich zakres, normy precyzują kryteria dotyczące jednostek niezależnie od przedmiotowego sektora. Jednakże akredytacji udziela się lub stara się o nią zawsze w odniesieniu do określonego zakresu, tj. do konkretnych działań związanych z oceną zgodności oraz, w stosownych przypadkach, typów przeprowadzanych testów i wykorzystywanych metod (np. „Jednostka X posiada odpowiednie kompetencje do przeprowadzania kontroli/inspekcji jako jednostka typu A w zakresie kategorii urządzeń ciśnieniowych zgodnie z dyrektywą nr 97/23”) oraz akredytacja nigdy nie jest ograniczona jedynie do zgodności z ogólnymi normami 17000. Dlatego też akredytacja na podstawie zgodności z normami 17000 zawsze wiąże się z potrzebą uzupełnienia tych ogólnych kryteriów oraz uszczegółowienia ich za pomocą wszelkich specyfikacji technicznych, które odnoszą się do konkretnego zakresu technicznego, w ramach którego wnioskująca jednostka oceniająca zgodność chce uzyskać akredytację. Zatem akredytacja wiąże się z weryfikacją kompetencji w odniesieniu do najnowszego stanu wiedzy oraz obejmuje ocenę na podstawie norm dotyczących jednostek oceniających zgodność oraz wszystkich stosownych regulacji dotyczących produktów oraz/lub technologii, norm i innych specyfikacji.
6.4. AKREDYTACJA ZGODNIE Z ROZPORZĄDZENIEM (WE) NR 765/2008
|
6.4.1. KRAJOWE JEDNOSTKI AKREDYTUJĄCE
Rozporządzenie przewiduje, że każde państwo członkowskie może wyznaczyć tylko jedną krajową jednostkę akredytującą. Wyłącznie krajowe jednostki akredytujące są upoważnione do prowadzenia akredytacji jednostek oceniających zgodność. Żadne inne organy nie mogą rościć sobie prawa do świadczenia takich usług, czy to według norm zharmonizowanych czy niezharmonizowanych. Przepis ten jest kluczowym elementem funkcjonowania akredytacji w UE oraz ram akredytacyjnych ustanowionych w rozporządzeniu. Państwa członkowskie nie są zobowiązane do ustanowienia własnej krajowej jednostki akredytującej, jeżeli uznają, że nie jest to ekonomicznie uzasadnione lub jeżeli uważają, że oferowanie akredytacji dla wszystkich czynności nie jest przydatne. Oznacza to, że na terytorium państwa członkowskiego dla danej czynności w żadnym czasie nie może funkcjonować więcej niż jedna jednostka akredytująca. Aby zapewnić przejrzystość, państwa członkowskie są zobowiązane do informowania Komisji oraz pozostałych państw członkowskich, z usług której krajowej jednostki akredytującej innego państwa członkowskiego korzystają.
Lista krajowych jednostek akredytujących jest dostępna w internecie (250). Krajowe jednostki akredytujące muszą publicznie udostępniać informacje o czynnościach, dla których przeprowadzają akredytację.
Rozporządzenie nie określa formy prawnej krajowej jednostki akredytującej. Oznacza to, że krajowa jednostka akredytująca może funkcjonować w ramach ministerstwa, stanowić agencję rządową lub być zorganizowana jako podmiot prywatny. Rozporządzenie jednak bardzo wyraźnie określa, że akredytacja musi być prowadzona jako działalność organów publicznych, a w tym celu musi być formalnie uznana przez państwo członkowskie.
Ponadto obowiązki i zadania krajowej jednostki akredytującej muszą być wyraźnie oddzielone od działalności innych organów krajowych. Przepis ten ma na celu zwiększenie niezależności krajowej jednostki akredytującej oraz umocnienie bezstronności i obiektywności jej działań. Jeżeli krajowa jednostka akredytująca jest częścią większej struktury publicznej, np. ministerstwa, pozostałe departamenty nie mogą wywierać wpływu na decyzje dotyczące akredytacji. Proces akredytacji musi pozostać oddzielony od innych funkcji. Należy bezwzględnie unikać konfliktu interesów krajowej jednostki akredytującej. Ma to również zastosowanie do pewnych zadań, które krajowa jednostka akredytująca może na siebie przyjmować. W związku z tym, iż decyzja nr 768/2008/WE przewiduje, że krajowa jednostka akredytująca może prowadzić działalność jako organ notyfikujący (251), przekazanie uprawnień musi być wyraźnie udokumentowane oraz należy zapewnić warunki gwarantujące bezstronność, mianowicie rozdzielenie zadań w ramach jednostki akredytującej.
W przypadku gdy zadania mają być przekazywane krajowej jednostce akredytującej, jej obowiązki na mocy rozporządzenia nadal mają zastosowanie. Oznacza to, że jej zadaniem pozostaje ocena kompetencji technicznych organów oceny zgodności zgodnie z procedurą pełnej akredytacji, a certyfikat akredytacji musi zostać wydany, jeżeli kompetencje techniczne jednostki oceniającej zgodność zostały potwierdzone. Krajowa jednostka akredytująca nie może wykonywać żadnych innych ocen, które nie są zgodne z tymi wymogami lub spełniają mniej rygorystyczne wymogi nie uzasadniające wydania certyfikatu akredytacji.
Innymi słowy, jeżeli obowiązek notyfikacji ma zostać przekazany krajowej jednostce akredytującej, możliwa jest jedynie notyfikacja akredytowanych jednostek oceniających zgodność. Notyfikacja jednostek oceniających zgodność, których kompetencje nie zostały ocenione według kryteriów pełnej akredytacji, nie będzie możliwa w przypadkach, gdy podjęto decyzję o takim przekazaniu obowiązków. Oznacza to również, że krajowa jednostka akredytująca nie miałaby żadnej swobody decydowania przy notyfikacji jednostki – wydanie certyfikatu akredytacji prowadziłoby automatycznie do notyfikacji (252).
Ponadto prowadząc akredytację, krajowa jednostka akredytująca musi spełniać szereg warunków w zakresie reprezentacji zainteresowanych stron, swojego zarządzania wewnętrznego oraz kontroli wewnętrznych. Decyzje dotyczące oceny muszą być podejmowane przez inną osobę niż ta, która przeprowadziła ocenę jednostki oceniającej zgodność. Jednostka akredytująca musi dysponować personelem posiadającym wystarczające kompetencje, aby umożliwić wykonywanie zadań jednostki. Należy wdrożyć procedury gwarantujące, że personel należycie wykonuje pracę oraz jest wystarczająco kompetentny do wykonywania swoich zadań. Ponadto należy podjąć odpowiednie środki, aby zapewnić poufność informacji uzyskiwanych od jednostek oceniających zgodność, zaś jednostka akredytująca jest zobowiązana nie nakładać zbędnych obciążeń na swoich klientów. Jednostki akredytujące muszą również posiadać mechanizm obsługi reklamacji.
Ponadto rozporządzenie przewiduje, że krajowa jednostka akredytująca musi posiadać wystarczające zasoby do wykonywania swoich zadań. Obejmuje to z jednej strony wystarczającą liczbę pracowników, ale również wykonywanie zadań specjalnych, takich jak działalność w ramach europejskiej i międzynarodowej współpracy akredytacyjnej oraz działania konieczne do wspierania polityki publicznej, które nie mają samofinansującego się charakteru. W tym zakresie odpowiednie uczestnictwo w EA, jej komitetach oraz procesie oceny wzajemnej jest najistotniejsze. Państwa członkowskie powinny umożliwić udział swoich krajowych jednostek akredytujących w tego typu działaniach.
W tym kontekście krajowe jednostki akredytujące są również zobowiązane do publikowania swoich zbadanych rocznych sprawozdań finansowych. Założenia tego przepisu wykraczają poza wykazanie należytego zarządzania finansami do celów oceny wzajemnej. Krajowe jednostki akredytujące muszą wyraźnie wykazać, że stosują się do przewodnich zasad niekomercyjności oraz dysponowania wystarczającymi zasobami, aby zapewnić kompetencje do wykonywania wszystkich działań. Mając na uwadze ogólne założenie rozporządzenia polegające na ustanowieniu akredytacji ostatnim szczeblem kontroli w systemie oceny zgodności, w przypadkach, w których jednostka akredytująca jest częścią większej struktury wymóg ten należy rozumieć jako narzędzie do wykazywania zgodności z tymi zasadami, a nie do tworzenia niepotrzebnych obciążeń biurokratycznych dla państw członkowskich. W związku z tym jednostki akredytujące mieszczące się w strukturach ministerstw muszą być w stanie przedstawić przynajmniej swoje ogólne dane budżetowe i finansowe dotyczące całkowitych zasobów oraz wydatków globalnych i operacyjnych, a ponadto wszelkie strategie finansowe, które mają do nich zastosowanie, aby móc wykazać, że posiadają wystarczające zasoby na wykonywanie swoich zadań w odpowiedni sposób przy zachowaniu zasady niekomercyjności.
Państwa członkowskie są zobowiązane do zapewnienia, aby ich krajowe jednostki akredytujące nieprzerwanie spełniały wymagania przewidziane w rozporządzeniu oraz do podjęcia działania naprawczego w przeciwnym przypadku. Z tego względu powinny one przede wszystkim uwzględniać wyniki oceny wzajemnej organizowanej przez europejską infrastrukturę akredytacyjną.
6.4.2. NIEKONKURENCYJNY I NIEKOMERCYJNY CHARAKTER DZIAŁALNOŚCI KRAJOWYCH JEDNOSTEK AKREDYTUJĄCYCH
Podstawą realizacji celu rozporządzenia polegającego na utworzeniu spójnych ram akredytacji, które czynią akredytację ostatnim szczeblem kontroli, jest zasada niekonkurowania oraz niekomercyjnego charakteru działalności.
W związku z tym, że akredytacja ma być działalnością o charakterze samofinansującym, należy ją prowadzić bez osiągania zysków. Oznacza to, że celem krajowych jednostek akredytujących nie jest maksymalizacja ani dystrybucja zysków. Mogą one świadczyć usługi w zamian za opłaty lub uzyskiwać dochody, natomiast każda nadwyżka dochodów powinna być inwestowana w dalszy rozwój ich działań akredytacyjnych, które odpowiadają ogólnym zadaniom jednostek akredytujących. Głównym celem akredytacji pozostaje nie generowanie zysków, lecz wykonywanie zadań w interesie publicznym.
Regularne nadwyżki dochodów mogą być sygnałem, że istnieje możliwość obniżenia opłat za akredytację i zachęcenia mniejszych jednostek oceniających zgodność do ubiegania się o akredytację. Ponieważ w rozporządzeniu przywiązuje się duże znaczenie do niekomercyjnego charakteru akredytacji, w motywie 14 wyjaśniono, że akredytacja nie może przynosić żadnych zysków właścicielom ani członkom organizacji. Jeżeli mimo to pojawiają się jakiekolwiek zyski, można temu zaradzić, obniżając wysokość opłat lub ponownie wykorzystując dochody do dalszego rozwoju akredytacji, tak aby uniknąć sprzeczności z zasadą niekomercyjności ustanowioną w rozporządzeniu. Można racjonalnie oczekiwać, że wszelkie nadwyżki dochodów generowanych przez jednostkę akredytującą mogą być również wykorzystywane do wspierania udziału jednostki akredytującej w działalności akredytacyjnej na poziomie europejskim, międzynarodowym lub w sferze publicznej.
Niezależnie od struktury prawnej danej krajowej jednostki akredytującej nie powinien występować jednak regularny transfer nadwyżki dochodów do właścicieli lub członków krajowej jednostki akredytującej – bez względu na to, czy są oni podmiotami publicznymi czy prywatnymi Wykorzystywanie akredytacji jako jednego ze źródeł dochodów budżetowych państwa wywołałoby poważne wątpliwości co do przestrzegania przepisów rozporządzenia dotyczących nienastawionego na zysk charakteru działalności akredytacyjnej.
Zgodnie się tą samą logiką akredytacja musi być ustanowiona jako działalność wyraźnie oddzielona od wszelkich działań związanych z oceną zgodności. Krajowa jednostka akredytująca nie jest zatem upoważniona do oferowania ani świadczenia żadnych usług oferowanych lub świadczonych przez jednostkę oceniającą zgodność. Nie może również świadczyć usług doradczych, posiadać udziałów w jednostce oceniającej zgodność lub innych powiązań finansowych z tą jednostką ani konkurować z jednostkami oceniającymi zgodność, aby uniknąć jakiegokolwiek konfliktu interesów.
Ponadto, aby chronić zasadę niekomercyjnego charakteru działalności, rozporządzenie przewiduje również, że jednostki akredytujące nie mogą konkurować z innymi jednostkami akredytującymi. W obrębie UE jednostki akredytujące mogą prowadzić działalność wyłącznie na terytorium własnego państwa członkowskiego. Akredytację transgraniczną przewidziano wyłącznie w wyjątkowych przypadkach określonych w art. 7 ust. 1 rozporządzenia (WE) nr 765/2008. Jeżeli te warunki nie zostaną spełnione, jednostki oceniające zgodność muszą dążyć do uzyskania akredytacji od krajowej jednostki akredytującej państwa członkowskiego, w którym prowadzą działalność. Ma to zastosowanie do wszystkich działań związanych z oceną zgodności, które odbywają się w Europie oraz dotyczą produktów lub usług, które mają zostać wprowadzone do obrotu (253).
6.5. EUROPEJSKA INFRASTRUKTURA AKREDYTACYJNA
Rozporządzenie stanowi o uznaniu europejskiej infrastruktury akredytującej. Obecnie jest nią Europejska Współpraca w dziedzinie Akredytacji, regionalna organizacja europejskich krajowych jednostek akredytujących. EA ma kluczowe znaczenie dla wdrażania rozporządzenia, a poprzez system oceny wzajemnej jest jednostką, która sprawuje największy nadzór nad praktycznym funkcjonowaniem akredytacji w Europie. Komisja oraz EA podpisały ramową umowę o partnerstwie, na podstawie której EA wykonuje swoje zadania. Jednym z głównych zadań EA jest prowadzenie oceny wzajemnej krajowych jednostek akredytujących zgodnie z normami oraz praktyką międzynarodową. Organizacja działa również na rzecz dalszego rozwoju, utrzymania oraz wdrażania akredytacji w UE. |
6.5.1. SEKTOROWE SYSTEMY AKREDYTACJI
Na wniosek Komisji zadania EA mogą również obejmować tworzenie nowych sektorowych systemów akredytacji lub uznawanie istniejących. System sektorowy jest systemem opartym na odpowiedniej normie dotyczącej danego produktu, procesu, usługi itp. oraz dodatkowych wymogach specyficznych dla konkretnego sektora i/lub prawodawstwa. Akredytacja może być konieczna do oceny kompetencji jednostek oceniających zgodność w zakresie przeprowadzania ocen takich systemów.
EA może przyczyniać się do rozwoju takich systemów sektorowych oraz ich odpowiednich kryteriów oceny i procedur oceny wzajemnej. EA może również uznawać już istniejące systemy, które określają swoje kryteria oceny i procedury oceny wzajemnej.
W przypadku systemów sektorowych związanych z prawodawstwem UE Komisja musi zagwarantować, że proponowany system spełnia niezbędne wymogi przedmiotowego prawodawstwa w zakresie interesu publicznego określonego w tym konkretnym prawodawstwie.
6.5.2. OCENA WZAJEMNA
Jednym z najważniejszych zadań EA jest organizacja systemu oceny wzajemnej krajowych jednostek akredytujących, który stanowi podstawę europejskiego systemu akredytacji.
Krajowe jednostki akredytujące są poddawane wzajemnym ocenom swoich systemów, procedur oraz struktur co najmniej co cztery lata. Celem systemu oceny wzajemnej jest zapewnienie spójności i równoważności praktyk akredytacyjnych w Europie, aby szeroki zakres organizacji, w tym krajowe organy władzy publicznej (254), wzajemnie uznawał usługi świadczone przez jednostki, które pozytywnie przeszły ocenę wzajemną, a w związku z tym akceptował certyfikaty akredytacji oraz poświadczenia wydane przez jednostki oceniające zgodność przez nie akredytowane. EA prowadzi odpowiedni system szkoleniowy w celu zapewnienia spójności działań związanych z oceną wzajemną oraz spójności wyników w Europie. Pozytywny wynik oceny wzajemnej umożliwia krajowej jednostce akredytującej podpisanie wielostronnego porozumienia EA lub zachowanie statusu sygnatariusza. W ramach wielostronnego porozumienia o współpracy realizowanego przez EA wszyscy sygnatariusze są zobowiązani do wzajemnego uznania równoważności swoich systemów akredytacji oraz jednakowej wiarygodności poświadczeń wydawanych przez akredytowane przez nich jednostki oceniające zgodność.
System oceny wzajemnej funkcjonuje na kilku poziomach. Po pierwsze krajowe jednostki akredytujące muszą spełniać wymagania normy zharmonizowanej EN ISO/IEC 17011 „Ocena zgodności – Wymagania ogólne dla jednostek akredytujących prowadzących akredytację jednostek oceniających zgodność” oraz wymagania rozporządzenia, które nie są zawarte w międzynarodowej normie dotyczącej jednostki akredytującej – są to mianowicie zasady dotyczące jednej krajowej jednostki akredytującej działającej jako organ władzy publicznej, niekomercyjnego charakteru tej działalności oraz niekonkurowania.
Jednostki akredytujące muszą następnie wykazać, że posiadają stosowne kompetencje oraz są zdolne do prowadzenia akredytacji w różnych obszarach oceny zgodności przez nie obsługiwanych. Same te działania są określone przez szereg norm zharmonizowanych (takich jak EN ISO/IEC 17025 dotycząca laboratoriów badawczych i wzorcujących, EN ISO/IEC 17020 dotycząca jednostek kontrolujących lub norma EN ISO/IEC 17065 dotycząca jednostek certyfikujących produkty, usługi i procesy). Ponadto podmioty dokonujące oceny wzajemnej muszą upewnić się, że jednostka akredytująca uwzględnia w swoich ocenach wszelkie inne wymagania istotne dla konkretnych działań związanych z oceną zgodności, które mają być wykonywane przez jednostki, które akredytują. Mogą to być szczegółowe wymogi zawarte w systemach oceny zgodności, w tym systemach krajowych i europejskich.
6.5.3. DOMNIEMANIE ZGODNOŚCI DLA KRAJOWYCH JEDNOSTEK AKREDYTUJĄCYCH
Jeżeli krajowa jednostka akredytująca w wyniku oceny wzajemnej może wykazać, że spełnia wymagania stosownej normy zharmonizowanej (255), zakłada się, że spełnia ona wymagania dla krajowych jednostek akredytujących określone w art. 8 rozporządzenia.
Co ważniejsze – i ma to szczególne znaczenie dla sfery regulacyjnej – jeżeli krajowa jednostka akredytująca pozytywnie przeszła ocenę wzajemną w zakresie konkretnego działania związanego z oceną zgodności, organy krajowe są zobowiązane do akceptowania certyfikatów akredytacji wydanych przez tę jednostkę oraz wszelkich poświadczeń (np. sprawozdań z badań lub kontroli, certyfikatów) wydanych przez jednostki oceniające zgodność akredytowane przez tę jednostkę akredytującą.
6.5.4. ROLA EA WE WSPIERANIU I HARMONIZACJI PRAKTYKI AKREDYTACJI W EUROPIE
Z uwagi na rolę EA jako organizacji zawiadującej oceną wzajemną krajowych jednostek akredytujących istnieje potrzeba opracowania spójnego i równoważnego podejścia do akredytacji, które zagwarantuje wzajemne uznawanie i akceptowanie poświadczeń oceny zgodności. Oznacza to, że EA musi ułatwiać stosowanie wspólnego podejścia do praktyki akredytacji oraz norm zharmonizowanych i wymagań, które mogą być zawarte w dowolnych systemach sektorowych. Dlatego też, przy zaangażowaniu wszystkich zainteresowanych stron oraz organów krajowych, zadaniem EA jest opracowanie przejrzystych wytycznych, których muszą przestrzegać jej członkowie przy prowadzeniu akredytacji.
6.6. AKREDYTACJA TRANSGRANICZNA
|
Możliwość wnioskowania o akredytację przez jednostkę oceniającą zgodność do krajowej jednostki akredytującej w innym państwie członkowskim jest dozwolona wyłącznie w określonych przypadkach. |
Zgodnie z art. 7 ust. 1 rozporządzenia (WE) nr 765/2008 jednostki oceniające zgodność, niezależnie od tego, czy są to jednostki zewnętrzne czy własne, składając wniosek o akredytację, powinny kierować go do krajowej jednostki akredytującej w państwie członkowskim, w którym prowadzą działalność. Ta ogólna zasada dopuszcza wyjątki: możliwość wnioskowania o akredytację przez jednostkę oceniającą zgodność do krajowej jednostki akredytującej w innym państwie członkowskim jest ograniczona do przypadków, gdy:
|
— |
dane państwo członkowskie nie ustanowiło własnej krajowej jednostki akredytującej i nie korzysta z usług krajowej jednostki akredytującej w innym państwie członkowskim (art. 7 ust. 1 lit. a)), |
|
— |
krajowa jednostka akredytująca nie oferuje usługi akredytacji w przedmiotowym zakresie (art. 7 ust. 1 lit. b)), |
|
— |
krajowa jednostka akredytująca nie przeszła dotychczas pozytywnej weryfikacji w trybie oceny wzajemnej w odniesieniu do czynności z zakresu oceny zgodności będącej przedmiotem starań o akredytację, tj. krajowa jednostka akredytująca nie jest sygnatariuszem wielostronnego porozumienia EA w zakresie akredytacji przedmiotowej czynności dotyczącej oceny zgodności (art. 7 ust. 1 lit. c)). |
Art. 7 ust. 1 rozporządzenia jest ściśle związany z zasadą niekonkurowania oraz stanowi jej logiczną konsekwencję.
Przepis dotyczący akredytacji transgranicznej ustanowiony w art. 7 jest postrzegany jako bardzo rygorystyczny oraz nadmiernie uciążliwy dla jednostek oceniających zgodność prowadzących działalność w kilku krajach, posiadających siedzibę w jednym państwie członkowskim oraz przez podmioty/placówki lokalne ustanowione w innych państwach członkowskich działające pod nadzorem siedziby głównej oraz podlegające temu samemu systemowi jakości i kierownictwu, ze względu na fakt, że powoduje on kosztowne powielanie ocen. Istnieje obawa przed ryzykiem niekorzystnej sytuacji konkurencyjnej w porównaniu z organizacjami w państwach trzecich. Przy ścisłej interpretacji prawnej art. 7, transgraniczne jednostki oceniające zgodność, przez wzgląd na ich struktury, nie mogą mieć dostępu do korzyści w postaci jednego certyfikatu akredytacji wystarczającego na całe terytorium UE, mimo że unikanie wielokrotnych akredytacji jest jednym z założeń rozporządzenia.
Powinno unikać się powielania zbędnych ocen i obciążeń dla transgranicznych jednostek oceniających zgodność, zapewniając odpowiednią kontrolę lokalnych podmiotów jednostek oceniających zgodność. Powinna funkcjonować wymiana informacji oraz skuteczna współpraca pomiędzy krajowymi jednostkami akredytującymi w celu prowadzenia, w razie konieczności, oceny, ponownej oceny oraz nadzoru lokalnych placówek transgranicznych jednostek oceniających zgodność. Na podstawie wzajemnego uznawania wszystkich ocen przeprowadzonych przez członków EA powinno się bezwzględnie unikać powielania ocen aspektów organizacyjnych lub wymagań.
W razie konieczności oraz na podstawie uzasadnionego wniosku lokalna krajowa jednostka akredytująca powinna przekazać stosowne informacje dotyczące prowadzenia akredytacji niezgodnie z krajowymi wymaganiami prawnymi innego państwa członkowskiego i/lub wymaganiami określonymi w stosownych krajowych systemach sektorowych organom krajowym danego innego państwa członkowskiego. Organy krajowe państw członkowskich, w których lokalna krajowa jednostka akredytująca prowadzi działalność powinny być o tym stale informowane.
Jednostki oceniające zgodność posiadające lokalne placówki (niezależnie od ich osobowości prawnej), po warunkiem, że placówki te podlegają temu samemu globalnemu systemowi jakości oraz kierownictwu oraz że siedziba główna dysponuje środkami do wywierania istotnego wpływu na ich działania oraz kontrolowania ich, można uznawać za stanowiące tylko jedną organizację w odniesieniu do przeprowadzanej czynności w zakresie oceny zgodności. Taka jednostka oceniająca zgodność jest zatem upoważniona do wnioskowania o akredytację do krajowej jednostki akredytującej właściwej dla siedziby głównej, której zakres może również obejmować czynności wykonywane przez oddział lokalny, łącznie z oddziałami mieszczącymi się w innym państwie członkowskim.
Akredytacja wielu placówek jest jednak dozwolona na mocy rozporządzenia wyłącznie wówczas, gdy akredytowana jednostka oceniająca zgodność zachowa ostateczną odpowiedzialność za działania wykonywane przez placówki lokalne objęte zakresem akredytacji wielu placówek. Certyfikat akredytacji wydany przez krajową jednostkę akredytującą państwa, w którym znajduje się siedziba główna zawiera nazwę jednego podmiotu prawnego – siedziby głównej – i jest to podmiot prawny, który posiada akredytację oraz ponosi odpowiedzialność za akredytowane czynności jednostki oceniającej zgodność, łącznie z wszelkimi czynnościami wykonywanymi przez placówkę lokalną objętą zakresem akredytacji. Jeżeli te placówki lokalne wykonują kluczowe czynności (wymienione w normie EN ISO/IEC 17011), certyfikat akredytacji (w swoich załącznikach) musi wyraźnie wskazać adresy tych placówek.
Placówka lokalna jest upoważniona do bezpośredniego oferowania na rynku lokalnym atestów zgodności na podstawie akredytacji wielu placówek, jednakże wyłącznie w imieniu akredytowanej jednostki oceniającej zgodność. Te akredytowane certyfikaty i sprawozdania są zatem wydawane na podstawie akredytacji siedziby głównej, pod nazwą oraz adresem siedziby głównej i nie posiadają logo oddziału lokalnego. To jednak nie wyklucza zamieszczenia na certyfikacie lub sprawozdaniu z oceny zgodności danych kontaktowych lokalnego oddziału wydającego przedmiotowy certyfikat lub sprawozdanie.
Akredytacja wielu oddziałów jest przeznaczona wyłącznie dla podmiotów stanowiących część tej samej organizacji, której siedziba główna ponosi odpowiedzialność za czynności wykonywane przez oddziały lokalne oraz wydawane przez nie certyfikaty/sprawozdania. Odpowiedzialność ta musi być wykazana na podstawie umownych lub równoważnych stosunków prawnych między siedzibą główną a podmiotem lokalnym oraz przepisów wewnętrznych, które szczegółowo określają te stosunki w zakresie kierownictwa oraz obowiązków.
Rozwiązanie polegające na akredytacji wielu placówek może być stosowane do wszystkich typów podmiotów lokalnych (spółek zależnych, oddziałów, agencji, biur itp.), niezależnie od ich osobowości prawnej oraz jest z zasady ważne dla wszystkich typów jednostek oceniających zgodność, w tym laboratoriów, jednostek kontrolujących oraz certyfikujących, jeżeli tylko prowadzą one wyraźnie określone i stosowne czynności do celów akredytacji.
Rozwiązanie polegające na akredytacji wielu placówek jest wykluczone, jeżeli wyżej wspomniane warunki nie zostaną spełnione, tzn. jeżeli jednostki oceniającej zgodność nie można uznać za jedną organizację w odniesieniu do oceny zgodności, a siedziba główna nie ponosi ostatecznej odpowiedzialności za działania podmiotów lokalnych. W takim przypadku placówki lokalne stanowiące oddzielne podmioty prawne powinny złożyć wniosek do lokalnej krajowej jednostki akredytującej o własną akredytację. Wtedy można uznać, że podmiot lokalny wykonuje usługę oceny zgodności całkowicie niezależnie od siedziby głównej.
W przypadku akredytacji wielu placówek pierwsza ocena oraz kolejne oceny muszą być przeprowadzane przy ścisłej współpracy między daną lokalną krajową jednostką akredytującą a krajową jednostką akredytującą właściwą dla siedziby głównej, podejmującą decyzję o akredytacji, natomiast nadzór musi być prowadzony przez lokalną krajową jednostką akredytującą lub we współpracy z nią. Jednostka oceniająca zgodność prowadząca działalność w wielu krajach musi ściśle współpracować z zaangażowanymi krajowymi jednostkami akredytującymi. Podmioty lokalne nie mogą odmówić udziału lokalnej krajowej jednostki akredytującej w procesie oceny, ponownej oceny lub nadzoru. Zharmonizowane zasady współpracy między krajowymi jednostkami akredytującymi istnieją w formie transgranicznej polityki EA. Akredytacja wielu placówek musi być prowadzona zgodnie z transgraniczną polityką EA, aby zagwarantować udział lokalnej krajowej jednostki akredytującej.
Akredytacja wielu placówek nie zastępuje podwykonawstwa, które pozostaje możliwym rozwiązaniem, w przypadku gdy jednostka oceniająca zgodność chce zlecić część swoich zadań podmiotom prawnym zlokalizowanym i prowadzącym działalność w tym samym państwie członkowskim lub w innych, które z kolei nie należą do tej samej organizacji, tj. nie stanowią części transgranicznej jednostki oceniającej zgodność. W takiej sytuacji podwykonawca nie jest objęty akredytacją jednostki oceniającej zgodność. Akredytowana jednostka oceniająca zgodność może zlecać podwykonawstwo poszczególnych części swoich czynności związanych z oceną zgodności innemu podmiotowi prawnemu zgodnie z obowiązującą normą dotyczącą jednostki oceniającej zgodność, w ramach której jest akredytowana oraz wyłącznie w zakresie dozwolonym w tej normie. Jednostka oceniająca zgodność musi być w stanie wykazać przed krajową jednostką akredytującą, że czynności zlecone podwykonawcy są wykonywane w kompetentny i rzetelny sposób zgodny z obowiązującymi wymaganiami dotyczącymi przedmiotowych czynności. Akredytowane poświadczenie oceny zgodności musi być wydane wyłącznie pod nazwą i na odpowiedzialność akredytowanej jednostki oceniającej zgodność, tj. podmiotu prawnego posiadającego akredytację. Stosunek umowny z klientem ma nadal akredytowana jednostka oceniająca zgodność.
6.7. AKREDYTACJA W KONTEKŚCIE MIĘDZYNARODOWYM
|
Na szczeblu międzynarodowym współpraca między jednostkami akredytującymi odbywa się w ramach Międzynarodowego Forum Akredytacji (IAF) oraz Międzynarodowej Współpracy w dziedzinie Akredytacji Laboratoriów (ILAC). |
6.7.1. WSPÓŁPRACA MIĘDZY JEDNOSTKAMI AKREDYTUJĄCYMI
Akredytacja jako bezstronny środek oceny i formalnego wykazania kompetencji technicznych, bezstronności oraz profesjonalnej rzetelności jednostek oceniających zgodność stanowi skuteczne narzędzie infrastruktury jakości stosowane na całym świecie.
Na szczeblu międzynarodowym współpraca między jednostkami akredytującymi odbywa się w ramach dwóch organizacji: w ramach Międzynarodowego Forum Akredytacji (IAF) między jednostkami akredytującymi udzielającymi akredytacji jednostkom certyfikującym (produkty i systemy zarządzania) oraz w ramach Międzynarodowej Współpracy w dziedzinie Akredytacji Laboratoriów (ILAC) między jednostkami akredytującymi udzielającymi akredytacji laboratoriom i jednostkom kontrolującym. Obie organizacje zapewniają wielostronne porozumienia o wzajemnym uznawaniu między jednostkami akredytującymi będącymi ich członkami. IAF zarządza wielostronną umową o uznawaniu (MLA), natomiast ILAC – umową o wzajemnym uznawaniu (MRA). Te wielostronne uzgodnienia/umowy o wzajemnym uznawaniu dotyczące kompetencji na poziomie technicznym między jednostkami akredytującymi mają na celu umożliwianie wprowadzania produktów i usług posiadających akredytowane poświadczenia zgodności na rynki zagraniczne bez potrzeby ponownego przeprowadzania badań lub certyfikacji w państwie importującym. Takie uzgodnienia/umowy o uznawaniu między jednostkami akredytującymi mają zatem przyczyniać się do zwiększenia akceptowalności wyników oceny zgodności.
Na poziomie regionalnym organizacje współpracy między jednostkami akredytującymi powstały dotychczas (256) w:
|
— |
Europie: European co-operation for Accreditation (EA) (Europejska Współpraca w Dziedzinie Akredytacji), |
|
— |
Ameryce: Inter America Accreditation Cooperation (IAAC), |
|
— |
regionie Azji i Pacyfiku: Asia Pacific Laboratory Accreditation Cooperation (APLAC) oraz Pacific Accreditation Cooperation (PAC), |
|
— |
Afryce: Southern African Development Community Accreditation (SADCA), |
|
— |
Afryce: African Accreditation Cooperation (AFRAC), |
|
— |
regionie Bliskiego Wschodu: Arab Accreditation Cooperation (ARCA). |
Z wyjątkiem SADCA, AFRAC i ARAC, które obecnie opracowują swoje regionalne umowy o wzajemnym uznawaniu, wyżej wymienione organizacje współpracy posiadają uzgodnienia/umowy w ramach swojego regionu, na których opierają się porozumienia ILAC/IAF. Udzielając specjalnego uznania, IAF akceptuje umowy o wzajemnym uznawaniu ustanowione w ramach EA, IAAC oraz PAC: jednostki akredytujące będące członkami IAF oraz sygnatariuszami wielostronnej umowy EA (MLA EA) lub wielostronnej umowy o uznawaniu PAC (PAC MLA) są automatycznie przyjmowane do MLA IAF. ILAC akceptuje umowy o wzajemnym uznawaniu oraz związane z nimi procedury oceny EA, APLAC oraz IAAC. Jednostki akredytujące, które nie są stowarzyszone z żadną uznaną regionalną organizacją współpracy mogą wnioskować o ocenę oraz uznanie bezpośrednio do ILAC oraz/lub IAF.
Wymagania dla jednostek akredytujących przewidziane w rozporządzeniu są zgodne z akceptowanymi na całym świecie wymaganiami określonymi w stosownych normach międzynarodowych, chociaż niektóre z nich można postrzegać jako bardziej rygorystyczne. W szczególności:
|
— |
akredytacja jest prowadzona przez jedną krajową jednostkę akredytującą wyznaczoną przez jej państwo członkowskie (art. 4 ust. 1), |
|
— |
akredytacja jest prowadzona jako działalność władz publicznych (art. 4.5), |
|
— |
krajowe jednostki akredytujące prowadzą działalność bez jakichkolwiek motywacji komercyjnych (art. 8 ust. 1) oraz bez osiągania zysków (art. 4 ust. 7), |
|
— |
krajowe jednostki akredytujące nie konkurują z jednostkami oceniającymi zgodność ani między sobą (art. 6 ust. 1 oraz art. 6 ust. 2), |
|
— |
akredytacja transgraniczna w ramach UE oraz EOG (art. 7). |
6.7.2. WPŁYW NA STOSUNKI HANDLOWE W OBSZARZE OCENY ZGODNOŚCI MIĘDZY UE A PAŃSTWAMI TRZECIMI
Ostateczna akceptacja poświadczeń oceny zgodności podlega decyzji organów władzy publicznej w sferze regulacyjnej oraz użytkowników z sektora przemysłu i konsumentów w sferze ekonomicznej. Dobrowolne wielostronne umowy o wzajemnym uznawaniu między jednostkami akredytującymi zawierane na poziomie wsparcia technicznego przyczyniają się do rozwoju i umacniania międzyrządowych umów handlowych.
Wyżej wspomniane wymagania wpływają na akceptację nieeuropejskich certyfikatów oraz wyników testów akredytowanych przez jednostki akredytujące spoza UE niespełniające wymagań unijnych, lecz będące sygnatariuszami MRA/MLA ILAC/IAF w następujący sposób:
|
— |
Dobrowolna ocena zgodności Decyzja o tym, czy oraz gdzie uzyskać akredytację leży w gestii nieeuropejskiej jednostki oceniającej zgodność prowadzącej działalność na rynku UE. Aby poszerzyć akceptowalność swoich poświadczeń oceny zgodności na rynku unijnym (przez nabywców usług oceny zgodności oraz ostatecznie przez konsumentów), nieeuropejska jednostka oceniająca zgodność, która decyduje się na uzyskanie akredytacji, może zdecydować, czy skorzysta z usługi jednostki akredytującej w państwie trzecim, która niekoniecznie spełnia nowe wymagania unijne, lecz jest sygnatariuszem MRA/MLA ILAC/IAF, czy raczej z usług jednostki akredytującej prowadzącej działalność w Unii. Nieeuropejskie poświadczenia oceny zgodności wydane na podstawie akredytacji przez nieeuropejskie jednostki akredytujące niespełniające wymagań unijnych mogą nadal być wykorzystywane na rynku unijnym, lecz wyłącznie na zasadzie dobrowolności. |
|
— |
Obowiązkowa ocena zgodności Jeżeli ocena zgodności jest wymagana w przepisach, państwa członkowskie UE mogą odmówić akceptacji poświadczeń zgodności wydanych na podstawie akredytacji przez nieeuropejskie jednostki akredytujące niespełniające wymagań unijnych, nawet jeżeli są one sygnatariuszami MRA/MLA ILAC/IAF. Jednakże taka odmowa nie może być oparta wyłącznie na argumencie niespełniania wymagań UE przez jednostkę akredytującą państwa trzeciego. Spełnianie wymagań unijnych przez jednostkę akredytującą państwa trzeciego nie jest warunkiem akceptacji wyników oceny zgodności, natomiast takie niespełnianie wymagań może zwiększyć wątpliwości co do jakości i wartości akredytacji, a zatem co do jakości i wartości akredytowanych certyfikatów lub sprawozdań. |
Jednak w przypadku gdy międzyrządowe umowy o wzajemnym uznawaniu (MRA) między UE a państwem trzecim w odniesieniu do oceny zgodności zostały zawarte, organy krajowe państw członkowskich UE będą akceptowały sprawozdania z badań oraz certyfikaty wydane przez jednostki, które strona trzecia wyznaczyła w ramach MRA do oceniania zgodności w kategoriach produktów lub sektorów objętych MRA. Produkty posiadające takie poświadczenia zgodności mogą być eksportowane oraz wprowadzane do obrotu na rynku drugiej strony umowy bez przechodzenia dodatkowych procedur oceny zgodności. Każda strona importująca zobowiązuje się, zgodnie z warunkami MRA, do uznawania poświadczeń oceny zgodności wydanych przez uzgodnione jednostki oceniające zgodność strony eksportującej, niezależnie od tego, czy akredytacja została wykorzystana do wsparcia procesu wyznaczenia jednostek oceniających zgodność w ramach MRA, czy nie, oraz w przypadku gdy akredytacja jest wykorzystywana przez stronę nieeuropejską, bez względu na spełnianie wymagań UE przez jednostkę akredytującą strony trzeciej.
7. NADZÓR RYNKU
Zgodnie z rozporządzeniem (WE) nr 765/2008 krajowe organy nadzoru rynku są wyraźnie zobowiązane do prowadzenia proaktywnej kontroli produktów udostępnianych na rynku, organizowania swojej działalności i zapewniania koordynacji między sobą na szczeblu krajowym oraz do angażowania się we współpracę na szczeblu UE (257). Podmioty gospodarcze są wyraźnie zobowiązane do współpracy z krajowymi organami nadzoru rynku oraz podejmowania w razie konieczności działań naprawczych. Krajowe organy nadzoru rynku są upoważnione do stosowania sankcji, które mogą obejmować niszczenie produktów.
Rozporządzenie (WE) nr 765/2008 obejmuje przepisy rozporządzenia nr 339/93 w sprawie kontroli produktów pochodzących z państw trzecich. Takie kontrole stanowią obecnie część działań w zakresie nadzoru rynku, zaś rozporządzenie (WE) nr 765/2008 zobowiązuje krajowe organy nadzoru rynku oraz organy celne do współpracy w celu zapewnienia sprawnego funkcjonowania systemu. Kontrole muszą być przeprowadzane w sposób niedyskryminujący, zgodnie z zasadami WTO oraz na podstawie tych samych zasad i warunków, które dotyczą kontroli w ramach nadzoru rynku wewnętrznego.
Komisja Europejska jest zobowiązana do usprawniania wymiany informacji między organami krajowymi (w odniesieniu do ich krajowych programów nadzoru rynku, metodologii oceny ryzyka, itp.), aby zagwarantować, że nadzór rynku jest skutecznie prowadzony w całej Unii oraz by państwa członkowskie mogły łączyć swoje środki.
7.1. DLACZEGO NADZÓR RYNKU JEST POTRZEBNY?
|
Państwa członkowskie muszą stosować odpowiednie środki zapobiegające udostępnianiu na rynku oraz użytkowaniu (258) produktów niezgodnych z wymaganiami. |
Celem nadzoru rynku jest zapewnianie, aby produkty spełniały obowiązujące wymagania, przyczyniając się do wysokiego poziomu ochrony interesów publicznych, takich jak zdrowie i ogólnie rozumiane bezpieczeństwo, bezpieczeństwo i higiena w miejscu pracy, ochrona konsumentów oraz ochrona środowiska naturalnego, a jednocześnie zagwarantowanie, że swobodny przepływ produktów nie jest ograniczony w żadnym większym stopniu niż ten przewidziany w unijnym prawodawstwie harmonizacyjnym lub innej stosownej zasadzie unijnej. Nadzór rynku upoważnia obywateli do równoważnego poziomu ochrony na całym jednolitym rynku, niezależnie od pochodzenia produktu. Ponadto nadzór rynku jest istotny z punktu widzenia interesu podmiotów gospodarczych, ponieważ pomaga w eliminowaniu nieuczciwej konkurencji.
Działania w zakresie nadzoru rynku nie są ukierunkowane wyłącznie na ochronę zdrowia i bezpieczeństwa, lecz są dodatkowo podejmowane w celu egzekwowania przepisów UE, które również mają na celu ochronę innych interesów publicznych – na przykład regulują dokładność pomiarów, kompatybilność elektromagnetyczną, wydajność energetyczną, ochronę konsumenta oraz środowiska naturalnego zgodnie z zasadą „wysokiego poziomu ochrony” przewidzianą w art. 114 ust. 3 TFUE.
Państwa członkowskie muszą zapewnić skuteczny nadzór nad swoim rynkiem. Są zobowiązane do organizowania i prowadzenia kontroli produktów udostępnianych na rynku lub importowanych. Państwa członkowskie muszą podejmować odpowiednie środki, aby zagwarantować, że postanowienia rozporządzenia (WE) nr 765/2008, dyrektywy nr 2001/95/WE oraz innych unijnych przepisów harmonizacyjnych, jak również niezharmonizowanych krajowych przepisów prawa obowiązują i są przestrzegane w Unii Europejskiej oraz, w szczególności, aby zapobiegać udostępnianiu na rynku oraz użytkowaniu produktów niezgodnych z wymaganiami i/lub niebezpiecznych.
Nadzór rynku powinien umożliwiać identyfikację produktów niebezpiecznych lub produktów, które w inny sposób nie spełniają obowiązujących wymagań określonych w unijnym prawodawstwie harmonizacyjnym, wyłączanie ich z rynku oraz karanie nieuczciwych, a nawet prowadzących działalność przestępczą podmiotów gospodarczych. Powinien również pełnić funkcję skutecznego czynnika odstraszającego (259). W tym celu państwa członkowskie muszą:
|
— |
należycie wdrażać postanowienia stosownych przepisów oraz ustanawiać sankcje proporcjonalne do naruszeń, |
|
— |
badać produkty (niezależnie od pochodzenia) wprowadzane na ich rynek, aby zagwarantować, że zostały one poddane wymaganym procedurom, że spełniono wymagania dotyczące oznakowania oraz dokumentacji oraz że produkty te zostały zaprojektowane i wytworzone zgodnie z wymaganiami unijnego prawodawstwa harmonizacyjnego. |
Starania w zakresie nadzoru rynku, aby były skuteczne, powinny być jednakowe we wszystkich państwach członkowskich. Jest to tym bardziej ważne z uwagi fakt, że każdy punkt zewnętrznej granicy Unii stanowi punkt dostępu dla ogromnej ilości produktów z państw trzecich. Jeżeli nadzór rynku będzie „łagodniejszy” w niektórych częściach UE niż w innych, powstaną słabe punkty, które zagrażają interesowi publicznemu oraz stwarzają nieuczciwe warunki handlu. W związku z tym skuteczny nadzór rynku musi odbywać się na całej długości zewnętrznej granicy UE.
Aby zagwarantować niezbędną obiektywność oraz bezstronność, nadzór rynku musi być prowadzony przez organy państw członkowskich. Pewne kontrole (np. testy, inspekcje) mogą być przekazywane innym jednostkom, natomiast ograny władz krajowych muszą ponosić pełną odpowiedzialność za decyzje podejmowane w następstwie tych badań. Kontrole w ramach nadzoru rynku mogą być przeprowadzane na różnych etapach cyklu życiowego produktu po wprowadzeniu go do obrotu, na przykład na etapie dystrybucji, oddawania do użytku lub końcowego użytkowania. Nadzór rynku może być zatem sprawowany w różnych miejscach, np. w przedsiębiorstwach importerów, u dystrybutorów hurtowych lub detalicznych, firm zajmujących się wynajmem, użytkowników, itp.
7.2. KONTROLE PROWADZONE PRZEZ ORGANY NADZORU RYNKU
|
Organy prowadzące nadzór rynku powinny badać zgodność produktu z wymogami prawnymi obowiązującymi w chwili wprowadzania do obrotu lub, w stosownych przypadkach, w chwili oddawania do użytku.
W związku z tym nadzór rynku formalnie nie odbywa się na etapie projektowania oraz wytwarzania produktów, czyli jeszcze zanim producent formalnie przyjmuje na siebie odpowiedzialność za zgodność produktów, zwykle umieszczając na nich oznakowanie CE. Jednak nic nie stoi na przeszkodzie współpracy organów nadzoru rynku z podmiotami gospodarczymi na etapie projektowania i wytwarzania. Taka współpraca może pomóc w podejmowaniu działań zapobiegawczych oraz możliwie najszybszym identyfikowaniu problemów związanych z bezpieczeństwem i zgodnością (260).
Innymi wyjątkami od zasady, że nadzór rynku może mieć miejsce dopiero po przyjęciu formalnej odpowiedzialności za produkty przez producenta, są targi, wystawy i pokazy. Większość unijnych przepisów harmonizacyjnych zezwala na wystawianie produktów bez oznakowania CE na targach, wystawach i pokazach, pod warunkiem że widoczny znak wyraźnie wskazuje, że produkty nie mogą zostać wprowadzone na rynek ani oddane do użytku dopóki nie będą spełniać wymagań zgodności oraz że podczas pokazów, w stosownych przypadkach, są stosowane odpowiednie środki zapewniające ochronę interesów publicznych. Organy nadzoru rynku muszą monitorować przestrzeganie tego obowiązku.
Aby nadzór rynku był skuteczny, jego środki powinny być skoncentrowane w miejscach, gdzie prawdopodobieństwo wystąpienia ryzyka jest większe, przypadki niezgodności częstsze lub gdzie można zidentyfikować określony interes. W tym celu można stosować statystyki oraz procedury oceny ryzyka. Organy nadzoru rynku, aby być zdolne do monitorowania produktów na rynku, muszą dysponować uprawnieniami, kompetencjami oraz zasobami niezbędnymi do:
|
— |
regularnego wizytowania zakładów handlowych, przemysłowych oraz miejsc składowania, |
|
— |
regularnego wizytowania, w stosownych przypadkach, miejsc pracy oraz innych miejsc, w których produkty są użytkowane (261), |
|
— |
organizowania kontroli wyrywkowych oraz kontroli na miejscu, |
|
— |
pobierania próbek produktów oraz poddawania ich badaniom i testom oraz |
|
— |
wymagania udzielenia, na uzasadnione żądanie, wszelkich potrzebnych informacji. |
Pierwszym poziomem kontroli są badania dokumentacji oraz oględziny na przykład w odniesieniu do oznakowania CE i umieszczania go na produktach, dostępności deklaracji zgodności UE, informacji dołączonych do produktu oraz właściwego doboru procedur oceny zgodności. Mogą być jednak konieczne bardziej szczegółowe badania w celu zweryfikowania zgodności produktu, dotyczące na przykład należytego stosowania procedury oceny zgodności, zgodności z obowiązującymi zasadniczymi wymaganiami oraz treści deklaracji zgodności UE.
W praktyce poszczególne działania w zakresie nadzoru rynku mogą skupiać się na konkretnych aspektach wymagań. Poza działaniami w zakresie nadzoru rynku, których wyraźnym założeniem jest weryfikacja produktów udostępnianych na runku, istnieją również inne mechanizmy publiczne, które, mimo iż nie zostały bezpośrednio opracowane w tym celu, mogą w efekcie służyć wykazaniu niezgodności (262). Inspektoraty pracy, które sprawdzają bezpieczeństwo w zakładzie pracy mogą na przykład odkryć, że projekt lub konstrukcja maszyny lub środka ochrony indywidualnej posiadających oznakowanie CE są niezgodne z obowiązującym wymogiem (263).
Informację o zgodności produktu w chwili, gdy był on wprowadzany do obrotu można również uzyskać podczas inspekcji w trakcie użytkowania produktu lub analizując czynniki, które spowodowały wypadek. Reklamacje od konsumentów lub innych użytkowników dotyczące produktu lub od producentów lub dystrybutorów dotyczące nieuczciwej konkurencji mogą również dostarczyć informacji do celów nadzoru rynku.
Monitorowanie produktów udostępnionych na rynku może być podzielone między kilka organów na poziomie krajowym, na przykład pod względem funkcjonalnym lub geograficznym. Jeżeli te same produkty podlegają kontroli więcej niż jednego organu (np. organu celnego oraz sektorowego lub organów lokalnych), niezbędna jest koordynacja między służbami państwa członkowskiego.
Dobrowolne inicjatywy, takie jak certyfikacja produktów lub stosowanie systemu zarządzania jakością nie mogą być traktowane na równi z działaniami w zakresie nadzoru rynku prowadzonymi przez dany organ. Mogą jednak przyczynić się do eliminacji czynników ryzyka i przypadków niezgodności z wymogami. Niemniej organy nadzoru rynku muszą być bezstronne w odniesieniu do wszelkich dobrowolnych oznakowań, etykiet i ustaleń, które mogą być brane pod uwagę w przejrzysty i niedyskryminujący sposób wyłącznie w celu oceny ryzyka i zgodności. Dlatego też produktów nie należy wyłączać z operacji w zakresie nadzoru rynku nawet jeżeli zostały poddane nieobowiązkowej certyfikacji lub innym dobrowolnym inicjatywom.
Unijne prawodawstwo harmonizacyjne przewiduje dwa różne narzędzia, które umożliwiają organom nadzoru rynku uzyskanie informacji o produkcie: deklaracja zgodności UE oraz dokumentacja produktu. Muszą być one udostępnione przez producenta, upoważnionego przedstawiciela ustanowionego na terytorium Unii lub w pewnych okolicznościach przez importera (264).
Inne osoby fizyczne lub prawne, takie jak dystrybutorzy, nie mogą zostać zobowiązane do udostępnienia tych dokumentów (265). Oczekuje się jednak, że pomogą organowi nadzoru rynku w ich uzyskaniu. Ponadto, organ nadzoru rynku może zażądać od jednostki notyfikowanej dostarczenia informacji o przebiegu oceny zgodności przedmiotowego produktu.
Deklaracja zgodności UE musi zostać udostępniona organowi nadzoru rynku bezzwłocznie na uzasadnione żądanie (266). Powinna być dołączona do produktu, gdy jest to wymagane w określonych unijnych przepisach harmonizacyjnych. Może zostać udostępniona do celów nadzoru w każdym państwie członkowskim, na przykład w drodze współpracy administracyjnej.
Dokumentacja produktu musi zostać udostępniona organowi nadzoru rynku na jego uzasadnione żądanie w rozsądnym czasie. Organ nie może systematycznie żądać jej przedstawienia. Zasadniczo przedstawienia dokumentacji można wymagać podczas wyrywkowych kontroli przeprowadzanych do celów nadzoru rynku lub gdy istnieją powody, by sądzić, że produkt nie zapewnia wymaganego poziomu bezpieczeństwa we wszystkich aspektach.
Można jednak żądać przedstawienia bardziej szczegółowych informacji (na przykład certyfikatów i decyzji jednostki notyfikowanej) w przypadkach wątpliwości co do zgodności produktu z obowiązującym unijnym prawodawstwem harmonizacyjnym. Przedstawienia pełnej dokumentacji produktu powinno się wymagać wyłącznie, gdy jest to wyraźnie konieczne, a nie gdy na przykład istnieje tylko potrzeba sprawdzenia pewnego szczegółu.
Takie żądanie należy ocenić zgodnie z zasadą proporcjonalności, a zatem biorąc pod uwagę potrzebę zapewnienia zdrowia i bezpieczeństwa osób lub innych interesów publicznych przewidzianych w stosownym unijnym przepisie harmonizacyjnym oraz chronienia podmiotów gospodarczych przed niepotrzebnym obciążeniem. Ponadto nieprzedstawienie dokumentacji w odpowiedzi na uzasadnione żądanie krajowego organu nadzoru rynku w możliwym do przyjęcia terminie może stanowić wystarczające podstawy do zakwestionowania zgodności produktu z zasadniczymi wymaganiami stosownego unijnego przepisu harmonizacyjnego.
W przypadku uzasadnionego żądania wystarczy, że producent przedstawi część dokumentacji produktu dotyczącą rzekomej niezgodności, pozwalającą stwierdzić, czy producent zajął się tą kwestią. Dlatego żądanie przetłumaczenia dokumentacji produktu powinno ograniczać się do tych części dokumentacji. Jeżeli organ nadzoru rynku uzna, że tłumaczenie jest potrzebne, musi wyraźnie wskazać część dokumentacji, która ma zostać przetłumaczona oraz wyznaczyć rozsądny termin dostarczenia tłumaczenia. Na tłumaczenie nie można nałożyć żadnych dalszych warunków, takich jak wykonywanie go przez tłumacza akredytowanego lub uznanego przez organy władzy publicznej.
Organ krajowy może dopuścić język, który jest dla niego zrozumiały, a który jest inny od języka narodowego lub języków narodowych. Wybrany język może być językiem trzecim, jeżeli tylko zostanie zaakceptowany przez ten organ.
Musi istnieć możliwość udostępnienia dokumentacji produktu w Unii. Nie musi być ona jednak przechowywana na terytorium UE, o ile nie zostało przewidziane inaczej w stosownym unijnym prawodawstwie harmonizacyjnym. Wymóg udostępnienia jej nie oznacza, że osoba, która jest za to odpowiedzialna musi sama przechowywać tę dokumentację (267), o ile jest w stanie przedstawić ją na żądanie organu krajowego. Nazwa i adres osoby przechowującej dokumentację nie muszą być wyraźnie zawarte na produkcie lub jego opakowaniu, o ile nie określono inaczej. Ponadto dokumentacja produktu może być przechowywana oraz przesyłana do organów nadzoru rynku w formie papierowej lub elektronicznej, dzięki czemu może zostać udostępniona w czasie proporcjonalnym do przedmiotowego ryzyka lub stopnia niezgodności. Państwa członkowskie muszą zapewnić, że każdy, kto otrzymuje informacje dotyczące zawartości dokumentacji produktu podczas działań w zakresie nadzoru rynku jest zobowiązany do zachowania poufności zgodnie z zasadami określonymi w krajowych przepisach prawa.
Informacje dotyczące procedur stosowanych przez organy nadzoru rynku, jak również środków naprawczych i sankcji można znaleźć w pkt 7.4.4–7.4.6.
7.3. KONTROLA PRODUKTÓW Z PAŃSTW TRZECICH PRZEZ ORGANY CELNE
Punkty graniczne Unii pełnią istotną rolę w kontekście zatrzymywania niezgodnych z wymaganiami i niebezpiecznych produktów pochodzących z krajów trzecich. Muszą przez nie przejść wszystkie produkty z krajów trzecich, dlatego są one idealnym miejscem do zatrzymywania produktów niezgodnych z wymaganiami i stwarzających zagrożenie zanim zostaną dopuszczone na rynek wewnętrzny i w efekcie staną się przedmiotem swobodnego obrotu na terytorium całej UE. W związku z tym organy celne odgrywają bardzo ważną rolę, wspierając organy nadzoru rynku w prowadzeniu kontroli bezpieczeństwa i zgodności produktów na granicach zewnętrznych.
Najbardziej skutecznym sposobem, aby zapobiec udostępnianiu na rynku unijnym niezgodnych z wymaganiami lub niebezpiecznych produktów importowanych z państw trzecich jest przeprowadzanie odpowiednich badań w ramach procedury kontroli przywozu. Wymaga to zaangażowania organów celnych oraz współpracy między organami celnymi a organami nadzoru rynku.
Organy odpowiedzialne za kontrolę produktów wprowadzanych na rynek Unii Europejskiej, organy celne lub organy nadzoru rynku, w zależności od krajowej struktury organizacyjnej, są właściwymi podmiotami do prowadzenia wstępnych kontroli bezpieczeństwa i zgodności importowanych produktów w pierwszym miejscu ich wprowadzenia na rynek unijny. Istnieją szczegółowe wytyczne dotyczące kontroli przywozu w zakresie bezpieczeństwa i zgodności produktów (268). Aby zapewnić takie kontrole, organy odpowiedzialne za kontrolę produktów na granicach zewnętrznych potrzebują odpowiedniego wsparcia technicznego niezbędnego do prowadzenia kontroli właściwości produktów w stosownym zakresie. Mogą one prowadzić kontrole dokumentacji, kontrole fizyczne oraz badania laboratoryjne. Potrzebują również odpowiednich zasobów osobowych i finansowych.
Rozporządzenie (WE) nr 765/2008 w kwestii kontroli zgodności z unijnym prawodawstwem harmonizacyjnym w przypadku produktów importowanych z krajów trzecich stanowi, że organy celne mają być wyraźnie zaangażowane w działania w zakresie nadzoru rynku, a systemy informacyjne przewidziane w zasadach krajowych i unijnych. Art. 27 ust. 2 rozporządzenia (WE) nr 765/2008 przewiduje obowiązkową współpracę między urzędnikami organów celnych a urzędnikami organów nadzoru rynku. Zobowiązania do współpracy są również zawarte w art. 13 Wspólnotowego kodeksu celnego, który stanowi, że kontrole prowadzone przez organy celne oraz inne organy mają odbywać się w ścisłej współpracy między nimi. Ponadto zasady współpracy między państwami członkowskimi a Komisją ustanowione w art. 24 rozporządzenia są rozszerzone, w stosownych przypadkach, o organy odpowiedzialne za kontrole zewnętrzne (art. 27 ust. 5).
Współpraca na szczeblu krajowym powinna umożliwiać przyjęcie wspólnego podejścia przez organy celne oraz organy nadzoru rynku podczas procesu kontroli. Fakt, że różne ministerstwa i organy mogą być odpowiedzialne za wdrażanie rozporządzenia (WE) nr 765/2008 nie powinien mieć na to wpływu.
Na podstawie rozporządzenia (WE) nr 765/2008 organy celne są zobowiązane do:
|
— |
zawieszenia dopuszczenia produktów na rynek, jeżeli istnieje podejrzenie, że produkty stanowią poważne zagrożenie dla zdrowia, bezpieczeństwa, środowiska naturalnego lub innego interesu publicznego oraz/lub nie spełniają wymagań dotyczących dokumentacji i oznakowania oraz/lub że oznakowanie CE jest fałszywe lub może wprowadzać w błąd (art. 27 ust. 3), |
|
— |
nieudzielenia pozwolenia na dopuszczenie do swobodnego obrotu z przyczyn wymienionych w art. 29, |
|
— |
udzielenia pozwolenia na dopuszczenie do swobodnego obrotu w odniesieniu do każdego produktu, który jest zgodny z odpowiednimi unijnymi przepisami harmonizacyjnymi i/lub nie stanowi zagrożenia dla żadnego interesu publicznego, |
|
— |
jeżeli dopuszczenie do swobodnego obrotu zostało zawieszone, organy celne muszą niezwłocznie powiadomić właściwy krajowy organ nadzoru rynku, który ma trzy dni robocze na przeprowadzenie wstępnego badania produktów i podjęcie decyzji w kwestii:
|
Organy celne muszą przekazywać informacje o swoich decyzjach w sprawie zawieszenia wprowadzenia produktu do obrotu do organów nadzoru rynku, które z kolei muszą być zdolne do podjęcia odpowiedniego działania. Od chwili takiego powiadomienia należy rozpatrywać cztery hipotezy. Przedmiotowe produkty stanowią poważne i bezpośrednie zagrożenie.
|
1. |
Produkty stanowią poważne zagrożenie Jeżeli organ nadzoru rynku stwierdzi, że produkty stanowią poważne zagrożenie, musi zakazać dopuszczenia go do obrotu na rynku UE. Organy nadzoru rynku muszą zwrócić się do organów celnych o umieszczenie na fakturze handlowej towarzyszącej produktowi oraz na każdym innym stosownym towarzyszącym dokumencie następującej informacji: „Produkt niebezpieczny – niedopuszczony do swobodnego obrotu – rozporządzenie (WE) nr 765/2008” (269). Organy państwa członkowskiego mogą również podjąć decyzję o zniszczeniu produktów lub w sprawieniu w inny sposób, że staną się bezużyteczne, jeżeli uznają takie działanie za niezbędne i proporcjonalne. W takich przypadkach organ nadzoru rynku musi skorzystać z systemu szybkiej wymiany informacji – RAPEX (270). Organy nadzoru rynku we wszystkich państwach członkowskich zostają wtedy poinformowane i mogą one z kolei powiadomić krajowe organy celne o produktach importowanych z krajów trzecich wykazujących cechy, które dają podstawy do poważnych wątpliwości, czy produkty te nie stwarzają poważnego zagrożenia. Ta informacja jest szczególnie ważna dla organów celnych, gdy obejmuje zakazanie lub wycofanie z obrotu produktów importowanych z państw trzecich. Informacje otrzymane od organów nadzoru rynku o tym, czy towary są uznane za niebezpieczne lub niezgodne z wymogami, mają zasadnicze znaczenie dla zarządzania ryzykiem celnym i procedur kontroli. Dzięki nim kontrole mogą koncentrować się na przesyłkach stwarzających ryzyko, co pomaga usprawnić legalny handel. Ponadto, jeżeli na rynku wewnętrznym wykryto niezgodne z wymogami lub niebezpieczne produkty, często bardzo trudno jest stwierdzić, w jaki sposób zostały one wprowadzone do UE. Aby ułatwić kontrolę w takich przypadkach, zachęca się do współpracy między organami celnymi i organami nadzoru rynku. |
|
2. |
Przedmiotowe produkty nie spełniają wymogów unijnych przepisów harmonizacyjnych W takim przypadku organy nadzoru rynku muszą podjąć odpowiednie działania, w razie konieczności zakazując wprowadzania do obrotu na podstawie przedmiotowych zasad. W przypadkach gdy wprowadzanie do obrotu jest zakazane, muszą poprosić organy celne o umieszczenie na fakturze handlowej towarzyszącej produktowi oraz na każdym innym stosownym towarzyszącym dokumencie następującej informacji: „Produkt niezgodny z wymogami – niedopuszczony do swobodnego obrotu – rozporządzenie (WE) nr 765/2008” (271). |
|
3. |
Przedmiotowe produkty nie stanowią poważnego zagrożenia i nie można uznać, że są niezgodne z unijnymi przepisami harmonizacyjnymi W takim przypadku produkty muszą zostać dopuszczone do swobodnego obrotu, pod warunkiem że wszystkie inne warunki i wymagania formalne dotyczące dopuszczenia do swobodnego obrotu są spełnione. |
|
4. |
Organy celne nie zostały powiadomione o żadnym działaniu podjętym przez organy nadzoru rynku. Jeżeli w ciągu trzech dni roboczych od zawieszenia dopuszczenia do swobodnego obrotu organ nadzoru rynku nie powiadomi organów celnych o żadnym podjętym przez siebie działaniu, produkt musi zostać dopuszczony do swobodnego obrotu, pod warunkiem że wszystkie inne warunki i wymagania formalne dotyczące takiego dopuszczenia do obrotu są spełnione. |
Cała procedura od zawieszenia do dopuszczenia lub niedopuszczenia do swobodnego obrotu przez organy celne powinna zakończyć się bez zwłoki, aby uniknąć tworzenia barier dla handlu zgodnego z prawem, co nie oznacza jednak, że musi ona zostać koniecznie zakończona w ciągu trzech dni roboczych. Zawieszenie dopuszczenia do obrotu może pozostać w mocy na czas wymagany przez organ nadzoru rynku do przeprowadzenia odpowiednich badań produktu oraz podjęcia ostatecznej decyzji. Organy nadzoru rynku muszą zapewnić, aby swobodny przepływ produktów nie był ograniczony w większym stopniu niż ten przewidziany w unijnym prawodawstwie harmonizacyjnym lub innych stosownych przepisach UE. W tym celu organy nadzoru rynku wykonują swoje czynności związane z produktami pochodzącymi z krajów trzecich, w tym prowadzą interakcję ze stosownymi podmiotami gospodarczymi, z taką samą pilnością oraz z zastosowaniem tych samych metodologii, jak w przypadku produktów pochodzących z Unii.
W takim przypadku organ nadzoru rynku w ciągu trzech dni roboczych powiadamia organy celne, że prace nad podjęciem ostatecznej decyzji w sprawie produktów są w toku. Dopuszczenie do swobodnego obrotu musi pozostać zawieszone do czasu podjęcia ostatecznej decyzji przez organ nadzoru rynku. Takie powiadomienie upoważnia organy celne do wydłużenia wstępnego okresu zawieszenia. Produkty pozostaną pod nadzorem organów celnych, nawet jeżeli zezwolono na przechowywanie ich w innym miejscu zatwierdzonym przez organy celne.
7.4. OBOWIĄZKI PAŃSTW CZŁONKOWSKICH
|
7.4.1. INFRASTRUKTURY KRAJOWE
Prowadzenie nadzoru rynku jest obowiązkiem organów publicznych. Ma to w szczególności na celu zagwarantowanie bezstronności działań w zakresie nadzoru rynku. Każde państwo członkowskie może samodzielnie decydować o infrastrukturze nadzoru rynku. Między innymi nie ma żadnego ograniczenia w zakresie podziału obowiązków między organami na zasadzie funkcjonalnej lub geograficznej, o ile nadzór jest skuteczny i obejmuje całe terytorium kraju. Państwa członkowskie organizują i prowadzą nadzór rynku poprzez ustanowienie organów nadzoru rynku (272). Organy nadzoru rynku są organami państwa członkowskiego odpowiedzialnymi za prowadzenie nadzoru rynku na jego terytorium. Prowadzenie nadzoru rynku przez organy publiczne stanowi fundamentalny element dobrego wdrożenia unijnego prawodawstwa harmonizacyjnego.
Państwa członkowskie muszą zapewnić, aby obywatele wiedzieli o istnieniu, znali obowiązki oraz tożsamość krajowych organów nadzoru rynku, jak również sposób skontaktowania się z nimi. Muszą również zapewnić, aby konsumenci oraz inne zainteresowane strony miały możliwość składania do właściwych organów skarg oraz aby skargi te były odpowiednio rozpatrywane.
Państwa członkowskie muszą powierzyć organom nadzoru rynku uprawnienia, zasoby oraz wiedzę niezbędną do należytego wykonywania ich zadań. Zadania te wiążą się z monitorowaniem produktów udostępnianych na rynku oraz, w przypadku produktów stwarzających zagrożenie lub wykazujących inną formę niezgodności, z podejmowaniem odpowiednich działań w celu usunięcia zagrożenia oraz wyegzekwowania zgodności. W odniesieniu do zasobów personalnych organ musi dysponować lub mieć dostęp do wystarczającej liczby odpowiednio wykwalifikowanych i doświadczonych pracowników cechujących się niezbędną uczciwością zawodową. Organ nadzoru rynku powinien również być niezależny oraz prowadzić swoje zadania w sposób bezstronny i niedyskryminujący. Ponadto organ nadzoru rynku powinien prowadzić nadzór rynku z przestrzeganiem zasady proporcjonalności, na przykład działanie musi być stosowne do stopnia ryzyka wystąpienia niezgodności, a wpływ na swobodny przepływ produktów nie może wykraczać poza konieczny dla osiągnięcia założeń nadzoru rynku.
Organ nadzoru rynku może zlecać wykonanie zadań technicznych (takich jak badania lub inspekcje) innej jednostce, pod warunkiem że zachowuje odpowiedzialność za swoje decyzje oraz o ile nie zachodzi żaden konflikt interesów między działaniami z zakresu oceny zgodności prowadzonymi przez tę inną jednostkę w imieniu podmiotów gospodarczych a oceną zgodności prowadzoną przez organ nadzoru rynku. Zlecając zadania podwykonawcy, organ nadzoru rynku powinien zachować najwyższą staranność, aby zagwarantować, że bezstronność uzyskanych w ten sposób informacji jest niepodważalna. Odpowiedzialność za wszelkie decyzje podjęte na podstawie tak uzyskanych informacji spoczywa na organie nadzoru rynku.
7.4.2. KRAJOWE PROGRAMY NADZORU RYNKU ORAZ PRZEGLĄDY DZIAŁALNOŚCI
Organy krajowe są zobowiązane na mocy art. 18 ust. 5 rozporządzenia (WE) nr 765/2008 do utworzenia, wdrożenia, okresowego aktualizowania oraz przekazywania informacji na temat swoich programów nadzoru rynku (273). Programy mogą mieć charakter ogólny i/lub sektorowy. Powinny zapewniać przestrzeganie ogólnych unijnych ram nadzoru rynku. Państwa członkowskie muszą również przekazywać informacje dotyczące programów innym państwom członkowskim i Komisji oraz publicznie udostępniać je w internecie, z wyłączeniem informacji, które mogą zmniejszyć skuteczność programu, jeżeli zostaną opublikowane. Celem tych programów jest umożliwienie organom innych państw oraz ogólnie obywatelom zrozumienie w jaki sposób, kiedy, gdzie oraz w jakich obszarach prowadzony jest nadzór rynku. Programy krajowe zawierają informacje na temat planowanych ulepszeń w ogólnej organizacji nadzoru rynku na szczeblu krajowym (np. mechanizmów koordynacji między poszczególnymi organami, przydzielonych im zasobów, metod pracy itp.) oraz inicjatyw w określonych obszarach interwencji (np. kategorii produktów, kategorii ryzyka, typów użytkowników itp.) (274). Oba rodzaje informacji są konieczne.
Komisja pomogła państwom członkowskim, proponując wspólne szablony programów. Zaleca się korzystanie z odpowiednich szablonów, aby zapewnić kompletność przedkładanych informacji. Ułatwia to również porównywanie krajowych programów nadzoru rynku w zakresie konkretnych produktów lub przepisów oraz umożliwia organom nadzoru rynku planowanie współpracy transgranicznej we wspólnych obszarach zainteresowania.
Ustanawiając krajowe programy nadzoru rynku, organy nadzoru rynku powinny brać pod uwagę potrzeby organów celnych. Programy powinny uwzględniać równowagę między proaktywnymi i rutynowymi działaniami w ramach kontroli oraz wszystkimi innymi czynnikami, które mogą mieć wpływ na priorytety w dziedzinie egzekwowania przepisów. W tym celu należy zapewnić odpowiednie zasoby na granicy kraju.
Zgodnie z art. 18 ust. 6 rozporządzenia (WE) nr 765/2008 państwa członkowskie co najmniej raz na cztery lata dokonują przeglądu i oceny funkcjonowania działalności w zakresie nadzoru. O wynikach tej oceny powiadamia się pozostałe państwa członkowskie i Komisję, a także udostępnia się je opinii publicznej (275).
7.4.3. PUBLICZNE UDOSTĘPNIANIE INFORMACJI
Mając na uwadze, że nadzór rynku ma na celu zapewnianie wysokiego poziomu ochrony pewnych interesów publicznych, upublicznianie informacji jest jego istotnym elementem. Dlatego też państwa członkowskie powinny zapewnić dostępność informacji dla obywateli i stron zainteresowanych, jak również zapewnić publiczny dostęp do informacji dotyczących zgodności produktów posiadanych przez organy. Zgodnie z zasadą przejrzystości informacje w posiadaniu organów państw członkowskich lub Komisji dotyczące zagrożeń dla zdrowia i bezpieczeństwa lub innych interesów publicznych chronionych na podstawie unijnego prawodawstwa harmonizacyjnego, stwarzanych przez produkty zasadniczo powinny być publicznie udostępniane, bez naruszania ograniczeń wymaganych do celów ochrony patentów i innych poufnych informacji handlowych, jak również ochrony danych osobowych oraz prowadzenia działań w zakresie monitorowania, badania i dochodzenia (276).
Obywatele powinni wiedzieć o istnieniu, znać obowiązki oraz tożsamość krajowych organów nadzoru rynku, jak również sposób skontaktowania się z nimi. Ponadto krajowe programy nadzoru rynku oraz wyniki przeglądów działalności muszą być upubliczniane drogą komunikacji elektronicznej oraz, w stosownych przypadkach, w inny sposób.
Jednym z obowiązkowych działań organów nadzoru rynku jest zobowiązanie do ostrzegania użytkowników na terytorium swojego państwa w odpowiednim terminie o odkrytym przez nie zagrożeniu związanym z danym produktem w celu zmniejszenia ryzyka urazu lub innych szkód, zwłaszcza jeżeli nie uczyni tego odpowiedzialny podmiot gospodarczy.
7.4.4. PROCEDURY NADZORU RYNKU
Nadzór rynku jest prowadzony poprzez wdrażanie szeregu procedur, których celem jest zapewnienie, żeby we wszystkich państwach członkowskich był ustanowiony skuteczny i spójny system nadzoru rynku. Organy nadzoru rynku stosują te procedury, gdy zajmują się produktami stwarzającymi zagrożenie dla zdrowia i bezpieczeństwa osób lub innych aspektów ochrony interesu publicznego zgodnie z art. 16 ust. 2 rozporządzenia (WE) nr 765/2008 oraz art. R31 i R32 załącznika I do decyzji nr 768/2008/WE, a także produktami stwarzającymi poważne zagrożenie wymagające szybkiej interwencji zgodnie z art. 20 i 22 rozporządzenia (WE) nr 765/2008.
Pierwsze zdarzenie sugerujące organom nadzoru rynku, że produkt stwarza zagrożenie dla zdrowia i bezpieczeństwa osób lub innych aspektów interesów publicznych może wywołać potrzebę przeprowadzenia dokładniejszej kontroli produktu. Może to być wypadek, otrzymanie skarg, inicjatywy organów nadzoru rynku z urzędu (w tym kontrola celna produktów wprowadzanych na rynek UE), jak również informacje o produktach stwarzających zagrożenie otrzymane od podmiotów gospodarczych. Jeżeli istnieją wystarczające powody, by uważać, że produkt stwarza zagrożenie, organy nadzoru rynku przeprowadzają ocenę spełniania wymagań stosownych unijnych przepisów harmonizacyjnych. Organy muszą przeprowadzić odpowiednie badania (zarówno kontrole dokumentacji, jak również badania fizyczne/laboratoryjne, w zależności od przypadku) właściwości produktów, należycie uwzględniając sprawozdania z oceny zgodności oraz przedstawione przez podmioty gospodarcze certyfikaty oceny zgodności wydane przez akredytowaną jednostkę oceniającą zgodność.
Organy nadzoru rynku przeprowadzają ocenę ryzyka, aby zweryfikować, czy produkty stwarzają poważne zagrożenie. Zgodnie z art. 20 ust. 2 rozporządzenia odpowiednia ocena ryzyka uwzględnia „charakter zagrożenia i prawdopodobieństwo jego wystąpienia” (277).
Jeżeli produkt stanowi zagrożenie dla zdrowia lub bezpieczeństwa osób lub innych aspektów interesu publicznego, organy nadzoru rynku muszą niezwłocznie zobowiązać właściwe podmioty gospodarcze do:
|
a) |
podjęcia wszelkich działań w celu doprowadzenia produktu do zgodności ze stosownymi wymaganiami przewidzianymi w unijnym prawodawstwie harmonizacyjnym; i/lub |
|
b) |
wycofania produktu z rynku; i/lub |
|
c) |
wycofania produktu od użytkowników; i/lub |
|
d) |
zatrzymania lub ograniczenia dostawy produktu w rozsądnym czasie. |
Jeżeli zagrożenie zostanie uznane za „poważne”, organy nadzoru rynku muszą podjąć szybką interwencję, stosując się do szczegółowych postanowień art. 20 i 22 rozporządzenia.
Podmioty gospodarcze muszą zagwarantować, że działanie naprawcze zostanie podjęte w całej UE. Organy nadzoru rynku muszą również informować stosowną jednostkę notyfikowaną (jeżeli istnieje) o podjętej decyzji. W przypadku poważnego zagrożenia wymagającego niezwłocznej interwencji organ nadzoru rynku może zastosować restrykcyjne środki, nie czekając aż podmiot gospodarczy podejmie działanie naprawcze mające na celu doprowadzenie produktu do zgodności z wymaganiami. Zgodnie z art. 21 rozporządzenia środki podejmowane przez organy nadzoru rynku muszą być proporcjonalne oraz należy o nich niezwłocznie powiadomić podmiot gospodarczy. Organy nadzoru rynku muszą również skonsultować się z podmiotem gospodarczym przed podjęciem tych środków oraz, jeżeli taka konsultacja nie jest możliwa ze względu na pilność środków, podmiot gospodarczy powinien mieć możliwość jak najszybszego przedstawienia swojego stanowiska. Organy nadzoru rynku muszą wycofać lub zmienić podjęte środki, jeżeli podmiot gospodarczy wykaże, że podjął skuteczne działania.
Jeżeli brak zgodności nie jest ograniczony do terytorium krajowego, organy nadzoru rynku muszą powiadomić Komisję oraz pozostałe państwa członkowskie o wynikach oceny zgodności oraz o działaniach, do których został zobowiązany podmiot gospodarczy lub o podjętych środkach. W przypadku poważnego zagrożenia organy nadzoru rynku powiadamiają Komisję za pośrednictwem systemu RAPEX o każdym dobrowolnym lub obowiązkowym działaniu zgodnie z procedurą określoną w art. 22 rozporządzenia i/lub art. 12 dyrektywy w sprawie ogólnego bezpieczeństwa produktów. W przypadku produktów, które nie stwarzają poważnego zagrożenia Komisja, inne państwa członkowskie zostają powiadomione poprzez system wspierający wymianę informacji określony w art. 23 rozporządzenia i/lub art. 11 dyrektywy w sprawie ogólnego bezpieczeństwa produktów. Organy nadzoru rynku muszą zweryfikować, czy odpowiednie środki naprawcze zostały podjęte. W przeciwnym przypadku podejmują one stosowne środki tymczasowe, informując Komisję oraz pozostałe państwa członkowskie zgodnie z procedurami określonymi powyżej.
Aby zwiększyć skuteczność działania w zakresie nadzoru rynku podjętego przez notyfikujące państwo członkowskie, pozostałe państwa członkowskie są wezwane do podjęcia dalszych działań związanych z powiadomieniem polegających na sprawdzeniu, czy ten sam produkt został wprowadzony do obrotu na ich terytoriach oraz podjęciu odpowiednich środków. Powinny one poinformować Komisję oraz pozostałe państwa członkowskie zgodnie z procedurami pierwotnego powiadomienia.
Zgodnie z unijnym prawodawstwem harmonizacyjnym dostosowanym do decyzji nr 768/2008/WE, jeżeli Komisja oraz pozostałe państwa członkowskie nie zgłoszą żadnych zastrzeżeń przez określony czas, restrykcyjne środki zostają uznane za uzasadnione i muszą one zostać niezwłocznie przyjęte przez państwa członkowskie. W przypadku niezgodności z powodu uchybień w normach zharmonizowanych Komisja informuje stosowne jednostki normalizacyjne i przedstawia sprawę przed komitetem ustanowionym na podstawie art. 22 rozporządzenia (UE) nr 1025/2012. W świetle opinii komitetu Komisja może podjąć decyzję o: a) zachowaniu odniesienia do norm zharmonizowanych w Dzienniku Urzędowym Unii Europejskiej; b) zachowaniu odniesienia do norm zharmonizowanych w Dzienniku Urzędowym Unii Europejskiej z ograniczeniami; c) wycofaniu odniesienia do norm zharmonizowanych z Dziennika Urzędowego Unii Europejskiej. Komisja informuje również stosowne europejskie organizacje normalizacyjne oraz, w stosownych przypadkach, zobowiązuje je do zrewidowania przedmiotowych norm zharmonizowanych.
W razie zastrzeżeń zastosowanie ma mechanizm ochronny.
Dodatkowe informacje na temat procedury umożliwiającej państwom członkowskim wymianę informacji na temat środków przyjętych wobec produktów stwarzających zagrożenie oraz, w stosownych przypadkach, do celów ich oceny przez Komisję Europejską są określone w pkt 7.5.1 i 7.5.2.
7.4.5. ŚRODKI NAPRAWCZE – ZAKAZY – WYCOFANIE Z OBROTU – WYCOFANIE OD UŻYTKOWNIKÓW
Zgodnie z unijnym prawodawstwem harmonizacyjnym państwa członkowskie mają obowiązek dopilnować, aby produkty były udostępniane na rynku jedynie wówczas, gdy spełniają stosowne wymogi. Te ostatnie obejmują zasadnicze wymagania oraz wymogi administracyjne i formalne. Jeżeli właściwe organy krajowe stwierdzą, że dany produkt nie jest zgodny z mającymi zastosowanie wymogami unijnego prawodawstwa harmonizacyjnego, muszą podjąć działania prowadzące do dostosowania ich do wymogów lub usunięcia z obrotu.
Działanie naprawcze zależy od rodzaju zagrożenia lub niezgodności, a zatem musi być zgodne z zasadą proporcjonalności. Niezgodność z zasadniczymi wymaganiami należy uznawać za niezgodność zasadniczą, ponieważ może ona wiązać się z potencjalnym lub rzeczywistym zagrożeniem stwarzanym przez produkt dla zdrowia i bezpieczeństwa osób lub innych aspektów interesu publicznego. W przypadku poważnego zagrożenia w art. 20 rozporządzenia (WE) nr 765/2008 określono konieczność zakazu udostępniania produktów na rynku, wycofywania ich z obrotu lub odzyskiwania od użytkowników.
Jeżeli produkt objęty unijnym prawodawstwem harmonizacyjnym nie posiada oznakowania CE, jest to wskazówka, że produkt nie spełnia zasadniczych wymagań lub że procedura oceny zgodności nie została zastosowana i w rezultacie produkt może zagrażać zdrowiu i bezpieczeństwu osób lub szkodzić innym interesom publicznym chronionym tym prawodawstwem. Wyłącznie w sytuacji gdy w wyniku dalszego dochodzenia okaże się, że produkt jest zgodny z zasadniczymi wymaganiami, brak oznakowania CE należy uznać za niezgodność formalną, a zatem produkt nie stanowi zagrożenia.
Jeśli nie istnieją powody, by uważać, że produkt stwarza zagrożenie, w niektórych przypadkach niezgodność z wieloma wymogami administracyjnymi lub formalnymi jest zdefiniowana w unijnym prawodawstwie harmonizacyjnym jako niezgodność pod względem formalnym. Dotyczy to przypadku niewłaściwego umieszczenia oznakowania CE np. pod względem jego projektu, wielkości, widoczności, czytelności lub nieusuwalności, co może zwykle być uznawane za niezgodność formalną. Przykładami typowej niezgodności formalnej mogą być również sytuacje, gdy inne znaki zgodności przewidziane w unijnym prawodawstwie harmonizacyjnym są niewłaściwie umieszczone, gdy deklaracja zgodności UE nie może zostać natychmiast przedstawiona lub nie została dołączona do produktu, choć jest to obowiązkowe, gdy wymóg dołączenia innych informacji przewidziany w sektorowym prawodawstwie harmonizacyjnym UE nie został całkowicie spełniony lub, w stosownych przypadkach, gdy numer identyfikacyjny jednostki notyfikowanej nie został dołączony do oznakowania CE.
Wyegzekwowanie zgodności można osiągnąć poprzez zobowiązanie producenta, jego upoważnionego przedstawiciela lub innych odpowiedzialnych osób (importerów, dystrybutorów) do podjęcia wymaganych działań. Działanie naprawcze może również mieć miejsce, jeżeli konieczne środki zostały podjęte (na przykład produkt został zmodyfikowany lub wycofany z rynku) w wyniku konsultacji przeprowadzonych przez organ nadzoru rynku lub w wyniku formalnych lub nieformalnych ostrzeżeń. W każdym przypadku organ nadzoru rynku musi ustanowić środki towarzyszące, aby zapewnić wyegzekwowanie zgodności. „Wytyczne dla firm dotyczące postępowania w przypadku wycofania produktu oraz innych działań naprawczych” (278) organizacji PROSAFE zostały opracowane, aby wesprzeć podmioty gospodarcze w zapewnianiu, w razie potrzeby, odpowiednich działań naprawczych oraz następczych, gdy produkt został już udostępniony na rynku UE lub pochodzi z krajów trzecich.
Działania mające na celu zakazanie lub ograniczenie wprowadzenia do obrotu mogą początkowo być tymczasowe, aby umożliwić organowi nadzoru rynku uzyskanie wystarczających dowodów na zagrożenie stwarzane przez produkt lub inną zasadniczą niezgodność produktu.
W przypadku jedynie niezgodności formalnej (tj. bez zagrożenia) organ nadzoru rynku powinien najpierw zobowiązać producenta lub jego upoważnionego przedstawiciela do dostosowania produktu, który ma zostać wprowadzony do obrotu lub, w razie konieczności, produktu już wprowadzonego na rynek do wymogów stosownych przepisów oraz do usunięcia skutków naruszenia w rozsądnym terminie. Jeżeli nie można osiągnąć żadnego skutku, organ nadzoru rynku musi ostatecznie podjąć dalsze kroki polegające na ograniczeniu lub zakazaniu wprowadzenia produktu do obrotu oraz w stosownych przypadkach zapewnić, że zostanie on również wycofany z rynku lub od użytkowników.
Każda decyzja podjęta przez krajowe organy nadzoru rynku o ograniczeniu lub zakazaniu wprowadzenia do obrotu lub oddania do użytku, wycofaniu produktu z obrotu lub wycofaniu od użytkowników musi zawierać dokładne powody, na których jest oparta. Zainteresowana strona – w szczególności producent lub jego upoważniony przedstawiciel ustanowiony na terytorium Unii – musi zostać powiadomiona. Muszą również zostać poinformowani o przysługujących im środkach prawnych obowiązujących w przedmiotowym państwie członkowskim oraz o ograniczeniach czasowych, którym te środki podlegają (279).
O ile sprawa nie jest pilna (np. produkt nie stwarza poważnego zagrożenia), producent lub jego upoważniony przedstawiciel ustanowiony na terytorium UE powinien mieć możliwość konsultacji z wyprzedzeniem, zanim właściwy organ podejmie działanie mające na celu ograniczenie swobodnego przepływu produktów. W praktyce należy uznać to za wystarczające, jeżeli producent lub jego upoważniony przedstawiciel otrzyma możliwość zareagowania w swojej sprawie (280). Jednakże nie powinno to opóźniać procedur, jeżeli producent lub jego upoważniony przedstawiciel pozostaje bezczynny.
Decyzja o ograniczeniu swobodnego przepływu produktu posiadającego oznakowanie CE w przypadku niezgodności z zasadniczymi wymaganiami zazwyczaj uruchamia procedurę klauzuli ochronnej. Celem tej procedury jest umożliwienie Komisji prowadzenia przeglądu takich środków, rozważenie, czy są uzasadnione oraz dopilnowanie, aby wszystkie państwa członkowskie stosowały podobne środki w odniesieniu do tych samych produktów. Producent, upoważniony przedstawiciel lub inny podmiot gospodarczy może uznać, że poniósł stratę w wyniku zastosowania nieodpowiedniego środka krajowego, który ograniczył swobodny przepływ produktu. W takim przypadku może mieć on prawo ubiegać się o odszkodowanie w ramach jurysdykcji państwa członkowskiego, które rozpoczęło procedurę oraz odpowiednio w ramach Komisji, pod koniec procedury klauzuli ochronnej, jeżeli środek krajowy zostaje uznany za nieuzasadniony. W związku z tym może nasunąć się pytanie, czy może zostać wniesiona sprawa z tytułu odpowiedzialności za niewłaściwe wdrożenie prawa UE.
7.4.6. SANKCJE
Rozporządzenie (WE) nr 765/2008 zobowiązuje państwa członkowskie do zapewnienia prawidłowego wdrożenia jego postanowień oraz podejmowania odpowiedniego działania w przypadku naruszenia. Zgodnie z rozporządzeniem kary mają być proporcjonalne do powagi wykroczenia oraz mają stanowić skuteczny środek odstraszający przed nadużyciami.
Określanie i wdrażanie mechanizmów do egzekwowania postanowień rozporządzenia na terytorium kraju leży w gestii państw członkowskich. Zgodnie z art. 41 rozporządzenia „przewidziane kary są skuteczne, współmierne i odstraszające oraz mogą zostać zaostrzone w przypadku, gdy dany podmiot gospodarczy w przeszłości naruszył w podobny sposób przepisy niniejszego rozporządzenia”.
Ponadto unijne prawodawstwo harmonizacyjne dostosowane do decyzji nr 768/2008/WE zawiera także przepis zobowiązujący państwa członkowskie do ustanowienia kar za naruszanie przez podmioty gospodarcze przepisów danego aktu prawnego.
Sankcje są nakładane w postaci kar finansowych, których kwoty różnią się między państwami członkowskimi. Mogą również obejmować sankcje karne w odniesieniu do poważnych wykroczeń.
Najpowszechniejszymi instrumentami prawnymi ustanawiającymi sankcje są ustawy w sprawie ogólnego bezpieczeństwa produktów oraz/lub przepisy dotyczące poszczególnych sektorów. Jednakże w niektórych państwach członkowskich sankcje są określone w ustawach w sprawie oznakowania CE, kodeksie celnym lub ustawach w sprawie systemu oceny zgodności.
7.5. WSPÓŁPRACA MIĘDZY PAŃSTWAMI CZŁONKOWSKIMI I KOMISJĄ EUROPEJSKĄ
Współpraca i koordynacja działań między organami krajowymi jest niezbędna, aby uzyskać skuteczny i spójny nadzór jednolitego rynku. Ramy prawne UE przewidują szereg instrumentów służących osiągnięciu tego celu. Mechanizm ochronny zawarty w unijnym prawodawstwie harmonizacyjnym zobowiązuje do wymiany informacji na temat środków ograniczających przyjętych przez organy krajowe, aby w stosownych przypadkach inne organy mogły podjąć działania następcze. Wzajemna pomoc na podstawie rozporządzenia (WE) nr 765/2008 pozwala organom egzekwować informacje żądane od podmiotów gospodarczych mających siedzibę w innym państwie członkowskim. Grupy współpracy administracyjnej (ADCO), baza danych ICSMS oraz wspólnotowy system szybkiej informacji RAPEX stanowią podstawowe narzędzia służące wymianie informacji i optymalizacji podziału pracy między organami.
7.5.1. MECHANIZMY OCHRONNE
|
Procedura klauzuli ochronnej, oparta na art. 114 ust. 10 TFUE oraz zawarta w większości sektorowych przepisów harmonizacyjnych UE, upoważnia państwa członkowskie do podejmowania restrykcyjnych środków w odniesieniu do produktów stwarzających zagrożenie dla zdrowia i bezpieczeństwa lub innych aspektów ochrony interesu publicznego oraz zobowiązuje je do powiadamiania o tych środkach Komisji oraz pozostałych państw członkowskich. Procedura klauzuli ochronnej została opracowana, aby zapewnić środki do informowania wszystkich krajowych organów nadzoru rynku o niebezpiecznych produktach oraz, co za tym idzie, umożliwiać objęcie wszystkich państw członkowskich koniecznymi restrykcjami w celu zapewnienia jednakowego poziomu ochrony w całej Unii. Ponadto umożliwia ona Komisji zajęcie stanowiska w sprawie środków krajowych ograniczających swobodny przepływ produktów w celu zapewnienia sprawnego funkcjonowania rynku wewnętrznego.
Należy zauważyć, że procedura ochronna różni się od procedury wspólnotowego systemu szybkiej informacji RAPEX pod względem celów, kryteriów notyfikacji oraz metod stosowania (281).
Jeżeli po przeprowadzeniu oceny państwo członkowskie stwierdzi, że produkt jest niezgodny lub że produkt jest zgodny, lecz stwarza zagrożenie dla zdrowia lub bezpieczeństwa osób lub innych aspektów ochrony interesów publicznych, musi zobowiązać dany podmiot gospodarczy do podjęcia wszystkich stosownych środków, aby zapewnić, że przedmiotowy produkt po udostępnieniu na rynku nie będzie dłużej stwarzał tego zagrożenia, do wycofania produktu z obrotu lub do wycofania go od użytkowników w wyznaczonym przez siebie rozsądnym terminie, stosownym do charakteru zagrożenia.
Procedura ta będzie stosowana, chyba że zostanie ustalone, iż zagrożenie nie wpływa na całą serię wytwarzanych produktów, jakkolwiek ograniczona byłaby ta seria, lub że ryzyko nie jest związane z produktem, lecz jego nieprawidłowym użyciem – czyli sytuacją gdy nie jest on używany zgodnie z przeznaczeniem lub w przewidywalnych warunkach oraz gdy nie jest prawidłowo zainstalowany i konserwowany. W przypadku odizolowanego błędu ograniczonego do terytorium państwa członkowskiego, które odkryło niezgodność nie ma potrzeby odwoływania się do klauzuli ochronnej, ponieważ nie ma potrzeby podjęcia działania na szczeblu UE. Ponadto zagrożenie musi być spowodowane samym produktem, a nie jego niewłaściwym użyciem.
Zgodność można wyegzekwować, jeżeli organ krajowy zobowiąże producenta lub jego upoważnionego przedstawiciela do zastosowania koniecznych środków lub jeżeli produkt zostanie zmodyfikowany lub dobrowolnie wycofany w obrotu. Procedura klauzuli ochronnej nie ma zastosowania dopóki w tych przypadkach nie zostanie podjęta decyzja o zakazie lub ograniczeniu udostępniania produktu na rynku lub o wycofaniu go z obrotu. W przypadku gdy nie ma obowiązkowego środka, nie ma potrzeby stosowania klauzuli ochronnej (282).
Jednakże jeżeli podmiot gospodarczy nie podejmie stosownych działań naprawczych w czasie wskazanym przez organ nadzoru rynku, organy nadzoru rynku muszą podjąć wszelkie odpowiednie środki tymczasowe mające na celu zakazanie lub ograniczenie udostępnienia produktu na ich rynku krajowym, wycofanie produktu z obrotu lub wycofanie go od użytkowników.
7.5.2. STOSOWANIE MECHANIZMÓW OCHRONNYCH KROK PO KROKU
|
7.5.2.1. Zastosowanie obowiązkowego środka restrykcyjnego
Klauzula ochronna może być zastosowana, gdy właściwy organ krajowy zastosuje obowiązkowy środek w celu ograniczenia lub zakazania udostępnienia produktu na rynku oraz, w stosownych przypadkach, oddania go do użytku, lub gdy wycofał go z obrotu, w sytuacji gdy zainteresowany podmiot gospodarczy sam nie podjął właściwych działań naprawczych. Treść decyzji powinna nawiązywać do wszystkich produktów należących do tego samego typu, tej samej partii lub serii. Musi również posiadać wiążący skutek prawny: powodować nałożenie sankcji, jeżeli nie będzie przestrzegana oraz podlegać procedurze odwoławczej. Decyzje sądowe, które ograniczają swobodny przepływ produktu z oznakowaniem CE w zakresie stosownego unijnego prawodawstwa harmonizacyjnego nie oznaczają zastosowania klauzuli ochronnej. Jednakże jeżeli postępowanie administracyjne rozpoczęte przez organ nadzoru rynku zgodnie z prawem krajowym musi być potwierdzone przez sąd, takie decyzje sądowe nie są wyłączone z procedury klauzuli ochronnej.
Ustalenia, które uzasadniają środek krajowy są dokonywane przez organ nadzoru rynku z jego własnej inicjatywy lub w oparciu o informacje otrzymane od strony trzeciej (np. konsumentów, konkurentów, organizacji konsumenckich, inspektoratów pracy). Ponadto środek krajowy musi być poparty dowodami (na przykład testami lub badaniami), które stanowią wystarczające potwierdzenie błędów w projekcie produktu lub procesie jego wytwarzania, aby wykazać przewidywalne potencjalne lub rzeczywiste zagrożenie lub inną zasadniczą niezgodność, nawet jeżeli produkty są prawidłowo wykonane, zainstalowane, konserwowane oraz wykorzystywane zgodnie z ich przeznaczeniem lub w możliwych do przewidzenia warunkach. Istnieje szara strefa między prawidłowym a nieprawidłowym konserwowaniem i użytkowaniem i można uznać, że do pewnego stopnia produkty powinny być bezpieczne, nawet jeżeli są konserwowane i używane zgodnie z przeznaczeniem, lecz w nieprawidłowy sposób, który jest możliwy do przewidzenia. Oceniając tę kwestię należy uwzględniać dane dołączone przez producenta na etykietach, ulotkach, w instrukcjach użytkownika oraz na materiałach promocyjnych.
Powód do podjęcia restrykcyjnych środków może między innymi wynikać z różnic lub błędów w zastosowaniu zasadniczych wymagań, niewłaściwego zastosowania norm zharmonizowanych lub uchybień w tych normach. Stosując klauzulę ochronną, organ nadzoru rynku może dodać lub sprecyzować inne motywy (na przykład nieprzestrzeganie dobrej praktyki inżynierskiej), pod warunkiem że są one bezpośrednio związane z trzema powyższymi powodami.
Gdy niezgodność z normami zharmonizowanymi stanowiącymi podstawę domniemania zgodności zostanie ustalona, producent lub jego upoważniony przedstawiciel musi zostać zobowiązany do przedstawienia dowodów na zgodność z zasadniczymi wymaganiami. Decyzja właściwego organu o podjęciu działania naprawczego musi być zawsze oparta na ustalonej niezgodności z zasadniczymi wymaganiami.
Środki zastosowane przez organy muszą być proporcjonalne do stopnia zagrożenia oraz niezgodności produktu, a informacja o nich musi zostać przekazana Komisji.
7.5.2.2. Powiadamianie Komisji
Gdy tylko właściwy organ krajowy ograniczy swobodny przepływ lub zakaże swobodnego przepływu produktu w sposób, który wiąże się z zastosowaniem klauzuli ochronnej, państwo członkowskie musi natychmiast powiadomić (283) Komisję, podając powody oraz uzasadnienie tej decyzji.
Informacje muszą zawierać wszelkie dostępne dane, a w szczególności:
|
— |
nazwę i adres producenta, upoważnionego przedstawiciela oraz – w stosownych przypadkach – nazwę i adres importera lub innej osoby odpowiedzialnej za udostępnianie produktu na rynku, |
|
— |
dane niezbędne do identyfikacji przedmiotowego produktu, pochodzenie oraz łańcuch dostaw produktu, |
|
— |
charakter przedmiotowego zagrożenia oraz charakter podjętych środków krajowych, |
|
— |
odniesienie do unijnego prawodawstwa harmonizacyjnego, a w szczególności do zasadniczych wymagań, których dotyczy stwierdzona niezgodność, |
|
— |
szczegółową ocenę oraz dowody uzasadniające zastosowanie środka (na przykład normy zharmonizowane lub inne specyfikacje techniczne wykorzystane przez organ, sprawozdania z badań oraz tożsamość laboratorium przeprowadzającego badania). W szczególności organy nadzoru rynku muszą wskazać, czy niezgodność jest spowodowana:
|
|
— |
argumenty przedstawione przez dany podmiot gospodarczy. |
Jeśli to możliwe, powiadomienie powinno zawierać również:
|
— |
kopię deklaracji zgodności, |
|
— |
nazwę i numer identyfikacyjny jednostki notyfikowanej, która brała udział w procedurze oceny zgodności, jeżeli ma to zastosowanie, |
|
— |
kopię decyzji podjętej przez właściwe organy państwa członkowskiego. |
7.5.2.3. Zarządzanie procedurą ochronną przez Komisję
W przypadku gdy są zgłaszane zastrzeżenia dotyczące środka podjętego przez państwo członkowskie (284) lub gdy Komisja uznaje, że środek krajowy jest sprzeczny z unijnym prawodawstwem harmonizacyjnym, Komisja musi niezwłocznie przystąpić do konsultacji z państwami członkowskimi oraz danym podmiotem lub podmiotami gospodarczymi oraz musi dokonać oceny środka krajowego. Na podstawie wyników tej oceny Komisja decyduje, czy środek krajowy jest uzasadniony, czy też nie.
Komisja kieruje swoją decyzję do wszystkich państw członkowskich i natychmiast przekazuje ją państwom członkowskim oraz stosownemu podmiotowi lub podmiotom gospodarczym.
Jeżeli środek krajowy jest uznany za uzasadniony, państwa członkowskie muszą podjąć środki niezbędne do zapewnienia, że niezgodny z wymogami produkt zostanie wycofany z ich rynku oraz muszą odpowiednio zawiadomić Komisję. Jeżeli natomiast środek krajowy zostaje uznany za nieuzasadniony, przedmiotowe państwo członkowskie musi wycofać środek.
Gdy środek krajowy zostaje uznany za zasadny, a niezgodność produktu jest związana z uchybieniami w normach zharmonizowanych, Komisja powinna zastosować procedurę przewidzianą w art. 11 rozporządzenia (UE) nr 1025/2012 dotyczącą formalnego zastrzeżenia do normy zharmonizowanej.
Państwa członkowskie inne niż państwo członkowskie rozpoczynające procedurę muszą niezwłocznie powiadomić Komisję oraz inne państwa członkowskie o wszelkich przyjętych środkach, wszelkich dodatkowych informacjach w ich posiadaniu dotyczących niezgodności przedmiotowego produktu oraz, w przypadku niezgadzania się ze zgłoszonym środkiem krajowym, o swoich zastrzeżeniach. Państwa członkowskie muszą zapewnić zastosowanie odpowiednich restrykcyjnych środków w odniesieniu do przedmiotowego produktu, takich jak niezwłoczne wycofanie produktu z ich rynku.
Jeżeli przez pewien czas od otrzymania informacji żadne państwo członkowskie ani Komisja nie zgłosi zastrzeżeń w odniesieniu do środka tymczasowego podjętego przez państwo członkowskie, środek ten powinno uznać się za uzasadniony.
Natomiast jeżeli Komisja nie widzi uzasadnienia dla działania krajowego, które wywołało klauzulę ochronną, zobowiązuje dane państwo członkowskie do wycofania tego działania oraz podjęcia natychmiast odpowiednich działań w celu przywrócenia swobodnego przepływu przedmiotowych produktów na jego terytorium.
Niezależnie od tego, czy działanie podjęte przez państwo członkowskie zostanie uznane za uzasadnione, czy nie, w każdym przypadku Komisja na bieżąco informuje państwa członkowskie o postępach i wynikach procedury.
Po podjęciu decyzji przez Komisję państwa członkowskie mają prawo podważyć tę decyzję na podstawie art. 263 TFUE. Podmiot gospodarczy, którego decyzja bezpośrednio dotyczy może również ją podważyć na podstawie art. 263 TFUE.
Jeżeli państwo członkowskie, które rozpoczęło procedurę, nie wycofa środka w przypadku uznania braku jego zasadności, Komisja rozważy wszczęcie postępowania w sprawie uchybienia zobowiązaniom państwa członkowskiego przewidzianego w art. 258 TFUE.
7.5.3. WZAJEMNA POMOC, WSPÓŁPRACA ADMINISTRACYJNA I WYMIANA INFORMACJI MIĘDZY PAŃSTWAMI CZŁONKOWSKIMI
|
Właściwe stosowanie przepisów unijnych jest uzależnione od sprawnej współpracy administracyjnej zapewniającej jednakowe i skuteczne egzekwowanie prawodawstwa unijnego we wszystkich państwach członkowskich. Zobowiązanie do współpracy jest zgodne z art. 20 Traktatu o Unii Europejskiej (TUE), który przewiduje, że państwa członkowskie muszą podjąć wszystkie odpowiednie środki do wypełnienia swoich obowiązków (285) oraz z art. 24 rozporządzenia (WE) nr 765/2008. Mimo że harmonizacja techniczna stworzyła jednolity rynek, w ramach którego produkty przemieszczają się między granicami krajowymi, nadzór rynku jest prowadzony na poziomie krajowym. Dlatego też mechanizmy współpracy administracyjnej między krajowymi organami nadzoru muszą zostać rozwinięte, aby zwiększyć skuteczność nadzoru, zminimalizować skutki różnych praktyk prowadzenia nadzoru oraz ograniczyć zachodzenie na siebie krajowych działań w zakresie nadzoru. Współpraca między organami nadzoru rynku może również rozpowszechniać dobre praktyki oraz techniki nadzoru w państwach Unii, ponieważ umożliwia organom krajowym porównywanie swoich metod z metodami stosowanymi przez inne organy, na przykład w ramach porównań i wspólnych sondaży oraz wizyt studyjnych. Ponadto współpraca może być przydatna do wymiany spostrzeżeń oraz rozwiązywania problemów o charakterze praktycznym.
Współpraca administracyjna wymaga wzajemnego zaufania oraz przejrzystości między krajowymi organami nadzoru. Państwa członkowskie oraz Komisja muszą być informowane o sposobie organizacji egzekwowania unijnego prawodawstwa harmonizacyjnego, a w szczególności rynkowego nadzoru produktów, na jednolitym rynku. Dotyczy to informacji o organach krajowych odpowiedzialnych za nadzór rynku w różnych sektorach produktów oraz o krajowych mechanizmach nadzoru rynku, które wyjaśniają, jak odbywa się monitorowanie produktów udostępnionych na rynku oraz do podejmowania jakich działań naprawczych i innych czynności organy nadzoru są upoważnione.
Przejrzystość jest również niezbędna w odniesieniu do krajowych zasad zachowania poufności. Aby osiągnąć skuteczny nadzór rynku w Unii, ważne jest, aby krajowe organy nadzoru rynku zapewniały sobie nawzajem wsparcie. Organ krajowy powinien udostępnić informacje na żądanie innego organu oraz udzielać innego rodzaju pomocy. Bez wcześniejszego żądania organ krajowy może rozważyć przesłanie innym organom krajowym wszelkich stosownych informacji dotyczących działań, które stanowią lub prawdopodobnie stanowią naruszenia unijnego prawodawstwa harmonizacyjnego i które mogą mieć znaczenie na terytorium innych państw członkowskich. Ponadto organy krajowe powinny spontanicznie przekazywać Komisji wszelkie informacje, które uznają za stosowne w odpowiedzi na uzasadnione żądanie Komisji. Komisja może wtedy przekazać te informacje innym organom krajowym, gdy uzna to za konieczne.
Współpraca oraz wzajemne wsparcie zgodnie z art. 24 ust. 2 rozporządzenia (WE) nr 765/2008 są szczególnie potrzebne, aby zapewnić możliwość podjęcia działań w stosunku do wszystkich podmiotów odpowiedzialnych za udostępnienie na rynku produktu niezgodnego z wymogami. W niektórych przypadkach należy skontaktować się z organem państwa członkowskiego, w którym producent, jego upoważniony przedstawiciel lub inna osoba odpowiedzialna ma siedzibę. Ma to na celu egzekwowanie informacji żądanych od tych podmiotów gospodarczych, między innymi deklaracji zgodności UE, określonych szczegółowych informacji z dokumentacji produktu lub informacji dotyczących łańcucha dostaw, których podmioty gospodarcze nie dostarczyły. Należy również skontaktować się z państwem członkowskim, w którego jurysdykcji prowadzi działalność jednostka notyfikowana (w stosownych przypadkach). Gdy organ krajowy podejmuje działania na podstawie informacji, które otrzymał od innej jednostki krajowej, powinien przekazać temu organowi informację zwrotną o wyniku tych działań.
Ponadto nadzór rynku mógłby być bardziej skuteczny na szczeblu Unii, gdyby krajowe organy nadzoru uzgodniły, w jaki sposób rozdzielać swoje zasoby, aby w każdym sektorze objąć maksymalną ilość różnych typów produktów. W celu uniknięcia powielania badań produktów lub innych dochodzeń w ramach nadzoru rynku organy krajowe powinny wymieniać między sobą streszczenia sprawozdań z badań. Jest to możliwe za pośrednictwem systemu informacyjnego i komunikacyjnego do celów nadzoru rynku (ICSMS) (286). Krajowe organy nadzoru powinny również rozważyć, czy istnieje specjalna potrzeba przeprowadzania analiz technicznych lub badań laboratoryjnych, gdy inny organ nadzoru już takie wykonał, a wyniki są dostępne dla tych organów lub mogą zostać im udostępnione na żądanie (287). Przydatna może być również wymiana wyników okresowych inspekcji urządzeń będących w eksploatacji w zakresie informacji o zgodności produktów w czasie wprowadzania ich do obrotu.
Informacje wymieniane między krajowymi organami nadzoru muszą być objęte poufnością zawodową zgodnie z zasadami przedmiotowego krajowego systemu prawnego oraz ochroną obejmującą podobne informacje na mocy prawa krajowego. Jeżeli państwo członkowskie posiada zasady zezwalające na swobodny dostęp osób do informacji będących w posiadaniu organów nadzoru, fakt ten musi być ujawniony w czasie wnioskowania do innego organu nadzoru o dostarczenie informacji lub podczas wymiany informacji, jeżeli taki wniosek nie ma miejsca. Jeżeli organ przekazujący zasygnalizuje, że informacje zawierają kwestie objęte poufnością zawodową lub handlową, organ otrzymujący powinien zapewnić zachowanie poufności. W przeciwnym przypadku organ przekazujący jest upoważniony do wstrzymania tych informacji. Współpraca oraz wymiana informacji między krajowymi organami nadzoru musi być uzgodniona przez strony zaangażowane oraz powinna uwzględniać potrzeby przedmiotowego sektora. W stosownych przypadkach należy uwzględnić następujące zasady:
|
— |
wyznaczenie krajowego punktu kontaktowego lub korespondenta dla każdego sektora do prowadzenia wewnętrznej koordynacji w miarę potrzeb, |
|
— |
uzgodnienie typów spraw, w których przekazywanie informacji z zakresu nadzoru byłoby przydatne, |
|
— |
opracowanie wspólnego podejścia do kwestii takich jak klasyfikacja ryzyka i zagrożeń oraz ich kodowanie, |
|
— |
określenie szczegółowych informacji, które należy przekazywać w każdym przypadku, łącznie z wnioskiem o dalsze informacje, |
|
— |
akceptacja obowiązku udzielenia odpowiedzi na zapytania w określonym czasie (288), |
|
— |
przekazywanie informacji (wniosków i odpowiedzi) w możliwie najprostszej formie, za pośrednictwem wiadomości e-mail lub systemu teleinformatycznego udostępnianego przez Komisję (ICSMS) lub jednostkę zewnętrzną oraz z wykorzystaniem standardowych formularzy wielojęzycznych, |
|
— |
korzystanie z nowoczesnych technik zapisu danych, aby umożliwić łatwą obsługę zapytań oraz |
|
— |
postępowanie z otrzymanymi informacjami z zachowaniem pełnej poufności. |
Współpraca między administracjami krajowymi odbywa się w ramach grup roboczych ustanowionych na mocy unijnego prawodawstwa harmonizacyjnego. Dyskusje dotyczą głównie kwestii interpretacyjnych, choć grupy robocze zajmują się również kwestiami związanymi z nadzorem rynku oraz współpracą administracyjną. Współpraca administracyjna między organami krajowymi prowadzącymi nadzór rynku odbywa się w następujących sektorach: przyrządów pomiarowych i wag nieautomatycznych (WELMEC), sprzętu niskonapięciowego (LVD ADCO), ekoprojektów (Grupa ADCO), kompatybilności elektromagnetycznej (współpraca administracyjna EMC), maszyn, wyrobów medycznych (Grupa Robocza ds. Nadzoru oraz COEN – Grupa ds. Zgodności i Egzekwowania), PEMSAC (Platforma europejskich organów nadzoru rynku ds. kosmetyków), Toy-ADCO (Grupa współpracy administracyjnej ds. zabawek), końcowych urządzeń telekomunikacyjnych (TCAM), rekreacyjnych jednostek pływających, środków ochrony indywidualnej, maszyn oraz urządzeń ATEX, urządzeń radiowych i końcowych urządzeń telekomunikacyjnych (R&TTE), kolei linowych (CABLE), etykietowania energetycznego (ENERLAB), urządzeń gazowych (GAD), dźwigów (LIFTS), wyposażenia morskiego (MED), hałasu, sektora urządzeń ciśnieniowych (PED/SVPD), pirotechniki (PYROTEC), chemikaliów (REACH), ograniczenia stosowania niektórych niebezpiecznych substancji (ROHS), ciśnieniowych urządzeń transportowych (TPED), oznakowania opon. Istnieją również grupy zajmujące się bardziej horyzontalnymi kwestiami, takie jak PROSAFE (Forum ds. egzekwowania bezpieczeństwa produktów w Europie), Grupa ekspercka ds. wewnętrznego rynku produktów (IMP-MSG) oraz komitet horyzontalny, w ramach którego omawiane są ogólne kwestie związane z wdrażaniem i egzekwowaniem unijnych przepisów harmonizacyjnych, takie jak horyzontalne aspekty nadzoru rynku. Komitety ds. nagłych wypadków ustanowione na mocy GPSD regularnie omawiają kwestie dotyczące współpracy administracyjnej będące przedmiotem ogólnego zainteresowania.
7.5.4. SYSTEM WCZESNEGO OSTRZEGANIA O NIEBEZPIECZNYCH PRODUKTACH NIEŻYWNOŚCIOWYCH
|
System wczesnego ostrzegania w zakresie produktów nieżywnościowych umożliwia 31 uczestniczącym państwom (państwa EOG) i Komisji Europejskiej wymianę informacji na temat produktów stwarzających zagrożenie dla zdrowia i bezpieczeństwa lub innych chronionych interesów oraz o działaniach podjętych przez te państwa w celu zlikwidowania takiego ryzyka. |
Art. 12 GPSD stanowi podstawę prawną ogólnego i horyzontalnego systemy służącego do szybkiej wymiany informacji na temat poważnych zagrożeń związanych z użytkowaniem produktów (RAPEX, wspólnotowy system szybkiej informacji).
Wspólnotowy system szybkiej informacji obejmuje produkty konsumenckie i specjalistyczne (289). Ma zastosowanie do produktów niezharmonizowanych, jak również produktów objętych unijnymi przepisami harmonizacyjnymi (290).
Wspólnotowy system szybkiej informacji działa zgodnie ze szczegółowymi procedurami określonymi w załączniku II do GPSD i w wytycznych dotyczących wspólnotowego systemu szybkiej informacji (291).
Wraz z wejściem w życie rozporządzenia (WE) nr 765/2008 zakres wspólnotowego systemu szybkiej informacji został rozszerzony o zagrożenia inne niż dotyczące zdrowia i bezpieczeństwa (tj. zagrożenia dla środowiska naturalnego, w miejscu pracy oraz zagrożenia dla bezpieczeństwa) oraz o produkty przeznaczone do użytku specjalistycznego (w przeciwieństwie do użytku konsumenckiego). Państwa członkowskie powinny dopilnować, aby produkty stwarzające poważne zagrożenie wymagające szybkiej interwencji, w tym poważne zagrożenie, którego skutki nie są natychmiastowe, zostały wycofane od użytkowników, wycofane z obrotu lub aby ich wprowadzenie na rynek zostało zakazane oraz aby Komisja była niezwłocznie powiadamiana za pośrednictwem wspólnotowego systemu szybkiej informacji na podstawie art. 22 rozporządzenia (WE) nr 765/2008.
W dniu 16 grudnia 2009 r. Komisja przyjęła decyzję 2010/15/UE (292) ustanawiającą nowe wytyczne dotyczące zarządzania wspólnotowym systemem szybkiej informacji. Wytyczne zostały sporządzone przed 1 stycznia 2010 r., dlatego też wyraźnie odnoszą się wyłącznie do notyfikacji na podstawie GPSD. Niemniej stanowią one również główne odniesienie do notyfikacji opartych na rozporządzeniu (WE) nr 765/2008 (zob. art. 22 ust. 4) – dotyczących produktów specjalistycznych oraz zagrożeń innych niż dla zdrowia i bezpieczeństwa.
Procedura wspólnotowego systemu szybkiego informowania jest następująca:
|
— |
gdy produkt (np. zabawka, artykuł pielęgnacyjny dla dzieci lub urządzenie gospodarstwa domowego) zostaje uznany np. za niebezpieczny, właściwy organ krajowy podejmuje odpowiednie działanie mające na celu eliminację zagrożenia. Może wycofać produkt z obrotu, wycofać go od użytkowników lub wydać ostrzeżenia. Podmioty gospodarcze mogą stosować takie środki także dobrowolnie, co również powinno być zgłaszane przez właściwe organy. Krajowy punkt kontaktowy zawiadamia wtedy Komisję Europejską (za pośrednictwem systemu IT GRAS w ramach wspólnotowego systemu szybkiej informacji (293)) o produkcie, zagrożeniu, jakie stwarza oraz o działaniach podjętych przez organ lub podmiot gospodarczy, aby zapobiec zagrożeniu i wypadkom, |
|
— |
Komisja rozpowszechnia informacje, które otrzymuje od krajowych punktów kontaktowych we wszystkich pozostałych państwach członkowskich UE i EOG. Publikuje cotygodniowe przeglądy produktów stwarzających zagrożenie oraz działań podjętych w celu eliminacji ryzyka na komisyjnej stronie internetowej dotyczącej wspólnotowego systemu szybkiej informacji (294), |
|
— |
krajowe punkty kontaktowe w każdym państwie UE i EOG zapewniają, aby odpowiedzialne organy sprawdzały, czy nowo opublikowany produkt jest obecny na rynku. Jeżeli tak, organy podejmują działania mające na celu eliminację zagrożenia, polegające na nakazaniu wycofania produktu z obrotu, wycofaniu produktu od użytkowników lub wydaniu ostrzeżeń. |
Procedury klauzuli ochronnej w ramach unijnego prawodawstwa harmonizacyjnego mają zastosowanie w dodatku do RAPEX. Dlatego też nie jest konieczne, aby uruchamiać wspólnotowy system szybkiej informacji przed zastosowaniem procedury klauzuli ochronnej. Jednakże procedura klauzuli ochronnej musi zostać zastosowana w dodatku do wspólnotowego systemu szybkiej informacji, jeżeli państwo członkowskie podejmie decyzję o trwałym zakazie lub ograniczeniu swobodnego przepływu produktów z oznakowaniem CE na podstawie zagrożenia lub innego poważnego zagrożenia stwarzanego przez produkt.
7.5.5. ICSMS
|
7.5.5.1. Rola
ICSMS stanowi szybki i efektywny środek komunikacji dla organów nadzoru rynku służący do wymiany informacji w krótkim czasie. ICSMS umożliwia szybką i skuteczną wymianę informacji między organami na temat produktów niezgodnych z wymogami (wyników testów, danych umożliwiających identyfikację produktu, zdjęć, informacji o podmiocie gospodarczym, ocen ryzyka, informacji o wypadkach, informacji o środkach podjętych przez organy nadzoru itp.).
Ma na celu nie tylko unikanie przypadków, gdy niebezpieczny produkt wycofany z rynku w jednym państwie pozostaje przez długi czas w sprzedaży w innym państwie, lecz przede wszystkim ma stanowić narzędzie polityki nadzoru rynku, które umożliwia ustanowienie mechanizmu współpracy między organami.
Mając świadomość faktu, że sama rzetelna wymiana informacji jest kluczowa dla nadzoru rynku, trzeba uznać, że system ICSMS wnosi wartość dodaną w tym sensie, że może on stanowić platformę do wdrażania unijnej polityki nadzoru rynku.
W tym kontekście ilekroć organ krajowy chce wymienić informacje na temat produktu podlegającego kontroli z innymi organami w celu podziału zasobów (np. do badań produktów), przeprowadzenia wspólnych działań lub konsultacji z innymi organami, musi wprowadzić stosowne informacje do ICSMS. Należy to zrobić możliwie jak najszybciej, a z pewnością na długo przed decyzją o podjęciu środków w stosunku do produktów, które stwarzają zagrożenie. Na przykład jeżeli organ krajowy nie może określić poziomu zagrożenia stwarzanego przez dany produkt i prowadzi badania, musi skorzystać z ICSMS do komunikacji z właściwymi organami innych państw członkowskich.
ICSMS nie jest ograniczony tylko do produktów niezgodnych z wymogami, ale zawiera również informacje również dotyczące wszystkich produktów kontrolowanych przez organy, nawet jeżeli w wyniku kontroli nie stwierdzono niezgodności. Pomaga to organom unikać powtórnego (lub wielokrotnego) kontrolowania tych samych produktów.
Zatem najważniejszą rolą ICSMS jest wspieranie Unii Europejskiej w realizacji jednego z jej najistotniejszych założeń politycznych, zapewnienia rzetelności i spójności we wdrażaniu i egzekwowaniu prawodawstwa unijnego, aby podmioty gospodarcze i obywatele mogli czerpać korzyści z pierwotnego założenia, jakim jest swobodny dostęp do rynku wewnętrznego.
W szczególności ICSMS pomaga organom nadzoru rynku w:
|
— |
prowadzeniu szybkiej wymiany informacji na temat środków w ramach nadzoru rynku, |
|
— |
skuteczniejszym koordynowaniu ich działań i kontroli, głównie poprzez skupienie uwagi na produktach, które jeszcze nie przeszły kontroli ani testów, |
|
— |
dzieleniu się zasobami, a dzięki temu poświęcaniu więcej czasu na inne produkty, które muszą jeszcze zostać zbadane, |
|
— |
prowadzeniu szeroko zakrojonych interwencji rynkowych w przypadku produktów o wątpliwej jakości z wykorzystaniem najnowszych informacji, a zatem w unikaniu zbędnego powielania kontroli, |
|
— |
opracowywaniu najlepszych praktyk, |
|
— |
zapewnianiu skutecznego i jednakowo rygorystycznego nadzoru rynku we wszystkich państwach członkowskich, a zatem w unikaniu zakłócania konkurencji, |
|
— |
tworzeniu encyklopedii informacji o nadzorze rynku UE. |
7.5.5.2. Struktura
Część wewnętrzna jest przeznaczona dla organów nadzoru rynku, organów celnych oraz UE. Zawiera ona wszystkie dostępne informacje (opisy produktów, wyniki badań, informacje o zastosowanych środkach itd.). Dostęp do tej części mają wyłącznie posiadacze konta w systemie ICSMS.
Część publiczna jest przeznaczona dla konsumentów, użytkowników i producentów. Publicznie dostępne informacje obejmują jedynie dane odnoszące się do produktu i jego niezgodności, natomiast nie obejmują żadnych dokumentów wewnętrznych (tj. wymiany informacji między organem a importerem/producentem).
ICSMS umożliwia wyszukiwanie niezgodnych z wymogami produktów pod kątem szczegółowych kryteriów. Informacje poufne są chronione za pomocą systemu autoryzacji dostępu.
Każdy organ nadzoru rynku może wprowadzać dane dotyczące badanych produktów, które nie figurują jeszcze w bazie danych, jak również dodawać informacje (np. dodatkowe wyniki testów, podjęte środki) do istniejącego już katalogu informacji o produkcie.
Komisja jest odpowiedzialna za prawidłowe funkcjonowanie systemu ICSMS. Korzystanie z ICSMS jest bezpłatne.
7.5.6. WYROBY MEDYCZNE: SYSTEM NADZORU
|
W przypadku wyrobów medycznych zastosowanie ma specjalny system nadzoru. |
Zagrożenie stwarzane przez wyroby medyczne wywołało konieczność opracowania kompleksowego systemu monitorowania służącego do odnotowywania wszystkich poważnych wypadków związanych z użytkowaniem produktów (295). System nadzoru wyrobów medycznych ma zastosowanie do wszystkich incydentów, które mogą lub mogły doprowadzić o śmierci pacjenta lub użytkownika lub do poważnego pogorszenia ich stanu zdrowia, a do których dochodzi:
|
— |
w wyniku wadliwego działania lub pogorszenia się właściwości lub wydajności wyrobu, |
|
— |
na skutek podania niewłaściwych informacji na wyrobie lub w instrukcji obsługi lub |
|
— |
z przyczyn technicznych lub medycznych związanych z właściwościami lub wydajnością wyrobu, skłaniających producenta do systematycznego wycofywania wszystkich wyrobów tego samego typu od użytkowników. |
Producent jest odpowiedzialny za uruchomienie systemu nadzoru, w związku z czym jest zobowiązany do powiadomienia organu nadzoru rynku o zdarzeniach, które spowodowały wykorzystanie systemu. Po takiej notyfikacji producent rozpoczyna dochodzenie, wysyła sprawozdanie organowi nadzoru oraz rozważa we współpracy z tym organem, jakie środki należy podjąć.
Po otrzymaniu notyfikacji od producenta organ nadzoru przeprowadza ocenę sytuacji, w miarę możliwości wraz z producentem. Po dokonaniu oceny organ musi natychmiast zawiadomić Komisję oraz pozostałe państwa członkowskie o incydentach, w związku z którymi zostały lub zostaną podjęte stosowne działania. Komisja może następnie poczynić dowolne kroki mające na celu koordynację, ułatwienie oraz wsparcie działań podjętych przez krajowe organy nadzoru wobec tego samego rodzaju incydentów lub, w razie potrzeby, przedsięwziąć środki na szczeblu Unii (np. przewidujące przeklasyfikowanie urządzenia). Baza danych zawierająca między innymi informacje uzyskane zgodnie z systemem nadzoru zostaje utworzona i udostępniona właściwym organom. System nadzoru różni się od procedury klauzuli ochronnej, ponieważ wymaga notyfikacji nawet wtedy, gdy producent dobrowolnie podejmuje niezbędne środki. Niezależnie od tego, stosując system nadzoru, organ nadzoru jest również zobowiązany do zastosowania restrykcyjnego środka w stosunku do niezgodnego produktu posiadającego oznakowanie CE, jeżeli zachodzą okoliczności uzasadniające zastosowanie klauzuli ochronnej, a następnie do powiadomienia o zastosowanym środku zgodnie z procedurą klauzuli ochronnej. Jednakże nie jest konieczne, aby uruchamiać system nadzoru przed zastosowaniem procedury klauzuli ochronnej.
8. SWOBODNY PRZEPŁYW PRODUKTÓW W UE (296)
8.1. KLAUZULA DOTYCZĄCA SWOBODNEGO PRZEPŁYWU
Założenie polegające na eliminacji barier handlowych między państwami członkowskimi oraz wzmocnieniu swobodnego przepływu produktów jest ustanowione w klauzuli dotyczącej swobodnego przepływu zamieszczonej w unijnych przepisach harmonizacyjnych, która gwarantuje swobodny przepływ produktów zgodnych z przepisami. Klauzule swobodnego przepływu stanowią przepisy zawarte w aktach prawnych UE, które wyraźnie zabraniają państwom członkowskim podejmowania bardziej restrykcyjnych środków w danej sprawie, jeżeli ta sprawa spełnia wymagania przedmiotowego prawa. Dlatego też państwa członkowskie nie mogą utrudniać udostępnienia na rynku produktu, który spełnia wszystkie postanowienia sektorowego prawodawstwa harmonizacyjnego.
Oznakowanie CE symbolizuje zgodność ze wszystkimi obowiązkami nałożonymi na producentów na mocy unijnego prawodawstwa harmonizacyjnego. Państwa członkowskie muszą zakładać, że produkty posiadające oznakowanie CE są zgodne ze wszystkimi wymaganiami stosownego przepisu regulującego jego umieszczenie. W związku z tym państwa członkowskie nie mogą zakazać, ograniczać ani utrudniać udostępniania na rynku na swoim terytorium produktów posiadających oznakowanie CE, chyba że przepisy dotyczące oznakowania CE zostały zastosowane nieprawidłowo.
8.2. OGRANICZENIA I RESTRYKCJE
Unijne prawodawstwo harmonizacyjne ma na celu zapewnienie swobodnego przepływu produktów, które spełniają wymóg wysokiego poziomu ochrony ustanowiony w stosownych przepisach. Dlatego też państwa członkowskie nie mogą zakazać, ograniczyć lub utrudniać wprowadzania takich produktów do obrotu. Jednakże zgodnie z Traktatem (w szczególności art. 34 i 36 TFUE) państwa członkowskie mogą utrzymać w mocy lub przyjąć dodatkowe przepisy krajowe dotyczące użytkowania produktów przeznaczonych do ochrony pracowników i innych użytkowników lub środowiska naturalnego. Takie przepisy krajowe nie mogą wymagać dokonania zmian w produkcie wytworzonym zgodnie z przepisami stosownego prawodawstwa ani wpływać na warunki udostępniania takiego produktu na rynku.
Ograniczenie swobodnego przepływu produktu może zostać nałożone w przypadku niezgodności produktu z zasadniczymi lub innymi wymaganiami prawnymi. Może również zdarzyć się, że produkty spełniające wymagania przepisów zharmonizowanych mimo to stwarzają zagrożenie dla zdrowia lub bezpieczeństwa osób lub innych aspektów ochrony interesu publicznego. W takim przypadku państwa członkowskie muszą zobowiązać stosowny podmiot gospodarczy do podjęcia działań naprawczych. Jest zatem możliwe, aby ograniczyć swobodny przepływ produktu nie tylko w przypadku niezgodności produktu z wymaganiami ustanowionymi przez stosowne przepisy prawa, lecz także w przypadku zgodności, jeżeli zasadnicze lub inne wymagania nie obejmują całkowicie wszystkich zagrożeń związanych z produktem (297).
9. MIĘDZYNARODOWE ASPEKTY PRZEPISÓW UNIJNYCH DOTYCZĄCYCH PRODUKTÓW
W swoich relacjach z państwami trzecimi UE między innymi wspiera międzynarodowy handel uregulowanymi produktami. Warunki otwartego handlu obejmują porównywalność podejścia, spójność przepisów i norm, przejrzystość zasad, odpowiednie poziomy i środki regulacji, bezstronność w certyfikacji, porównywalność środków w zakresie nadzoru rynku oraz praktyk nadzoru oraz odpowiedni poziom infrastruktury technicznej i administracyjnej.
W związku z tym, w zależności od statusu powyższych warunków, wiele różnych środków może zostać zastosowanych w celu ułatwienia wymiany handlowej. Ekspansja jednolitego rynku produktów jest realizowana poprzez kilka międzynarodowych instrumentów prawnych, które umożliwiają osiągnięcie odpowiedniego poziomu współpracy, zbieżności lub harmonizacji prawodawstwa, a zatem umożliwiają swobodny przepływ towarów. Do tych instrumentów należy:
|
— |
pełna integracja państw EFTA należących do EOG na rynku wewnętrznym na mocy porozumienia EOG (298), |
|
— |
dostosowanie systemu legislacyjnego oraz infrastruktury krajów kandydujących z systemem prawodawczym oraz infrastrukturą UE, |
|
— |
podobne dostosowanie przez kraje sąsiadujące poprzez zawarcie bilateralnych układów w sprawie oceny zgodności oraz uznawania produktów przemysłowych (ACAA), |
|
— |
zawarcie bilateralnych (międzyrządowych) umów o wzajemnym uznawaniu (MRA) w odniesieniu do oceny zgodności, certyfikatów i znakowania, które mają na celu zmniejszenie kosztów przeprowadzania testów i certyfikacji na innych rynkach, |
|
— |
oraz opieranie się na porozumieniu WTO w sprawie barier technicznych w handlu (299). |
9.1. UKŁADY W SPRAWIE OCENY ZGODNOŚCI ORAZ UZNAWANIA (ACAA)
|
Układy w sprawie oceny zgodności oraz uznawania są zawierane między Unią a krajami objętymi europejską polityką sąsiedztwa. |
Unia Europejska zawsze zajmowała wiodącą pozycję we wspieraniu współpracy międzynarodowej w zakresie regulacji technicznych, norm, oceny zgodności oraz eliminacji barier technicznych w handlu produktami. W ramach europejskiej polityki sąsiedztwa, Komisja Europejska wyraźnie określiła swój zamiar wzmocnienia współpracy ze wschodnimi oraz południowymi sąsiadami Unii w zakresie handlu, dostępu do rynku oraz struktur regulacyjnych.
Korzystanie z unijnego systemu normalizacji i oceny zgodności przez państwa trzecie ma na celu umożliwienie handlu i dostępu do rynku w obu kierunkach.
Układy w sprawie oceny zgodności oraz uznawania produktów przemysłowych mają być z założenia zawierane między Unią a krajami objętymi europejską polityką sąsiedztwa (państwa regionu Morza Śródziemnego – Algieria, Egipt, Izrael, Jordania, Libia, Maroko, Autonomia Palestyńska, Tunezja oraz państwa wschodnie – Armenia, Azerbejdżan, Białoruś, Gruzja, Mołdawia i Ukraina).
Wzajemne uznawanie równoważności regulacji technicznych, normalizacji oraz oceny zgodności, na którym opierają się te porozumienia funkcjonuje na podstawie dorobku UE, który został transponowany przez dane państwo partnerskie, w taki sam sposób, jak miałby zastosowanie do produktów wprowadzonych na rynek państwa członkowskiego. Umożliwia to wprowadzanie produktów objętych porozumieniami oraz atestowanych jako zgodne z procedurami w Unii Europejskiej do obrotu na rynku danego państwa partnerskiego bez potrzeby przeprowadzania dalszych procedur zatwierdzania i odwrotnie.
ACAA wymaga pełnego dostosowania ram prawnych państwa partnerskiego do prawodawstwa oraz norm UE oraz modernizacji jego infrastruktury wdrażania zgodnie z modelem systemu unijnego w odniesieniu do normalizacji, akredytacji, oceny zgodności, systemu miar i wag oraz nadzoru rynku.
ACAA składa się z umowy ramowej oraz co najmniej jednego załącznika określającego objęte układem produkty oraz środki przyjęte w celu rozszerzenia korzyści handlowych w tym sektorze. Umowa ramowa przewiduje dwa mechanizmy: a) uznawanie równoważności regulacji technicznych, normalizacji oraz oceny zgodności w odniesieniu do produktów przemysłowych podlegających równoważnym przepisom w prawie unijnym oraz prawie krajowym państwa partnerskiego oraz b) wzajemne uznawanie produktów przemysłowych, które spełniają wymagania jednej ze stron dotyczące zgodnego z prawem wprowadzenia do obrotu w przypadku braku europejskich regulacji technicznych dotyczących stosownych produktów. Do umowy można sukcesywnie dodawać kolejne załączniki sektorowe.
Pierwszy ACAA, zawarty z Izraelem i dotyczący produktów farmaceutycznych, wszedł w życie w styczniu 2013 r. W czasie powstawania niniejszego przewodnika inne państwa partnerskie z regionu Morza Śródziemnego kończą prace przygotowawcze do rozpoczęcia negocjacji w niektórych sektorach objętych nowym podejściem (produktów elektrycznych, materiałów budowlanych, zabawek, urządzeń gazowych oraz ciśnieniowych).
9.2. UMOWY O WZAJEMNYM UZNAWANIU (MRA)
|
9.2.1. GŁÓWNE CECHY UMÓW
Jednym z instrumentów służących do promowania międzynarodowego handlu uregulowanymi produktami jest zawieranie umów o wzajemnym uznawaniu (MRA) na mocy art. 207 i 218 TFUE. MRA to umowy zawierane między Unią a państwami trzecimi w celu wzajemnego uznawania oceny zgodności uregulowanych produktów.
MRA przewidują, że każda strona uznaje sprawozdania, certyfikaty oraz oznakowania, które zostały wydane w państwie partnerskim zgodnie z jego prawodawstwem. Umowy te są sporządzane i wydawane przez jednostki, która druga strona wyznaczyła w ramach MRA do przeprowadzania oceny zgodności w zakresie lub zakresach objętych daną MRA. Można to osiągnąć, ponieważ MRA obejmują wszystkie wymagania dotyczące oceny zgodności, które strony muszą spełnić, aby uzyskać pełen dostęp do rynku, a produkty są oceniane w państwie ich produkcji pod kątem wymagań regulacyjnych drugiej strony. Umowy te zwykle nazywa się „tradycyjnymi MRA”.
MRA obejmują całe terytorium stron porozumienia, aby zagwarantować pełen swobodny przepływ produktów certyfikowanych jako zgodne z wymogami, w szczególności w państwach o strukturze federalnej. Zasadniczo MRA są ograniczone do produktów, które zostały wytworzone na terytorium jednej ze stron.
MRA mają zastosowanie do jednej lub więcej kategorii produktów lub sektorów wchodzących w zakres uregulowanego obszaru (objętych obowiązującym unijnym prawodawstwem harmonizacyjnym) oraz, w pewnych przypadkach, objętych niezharmonizowanym prawem krajowym. Z zasady MRA powinny obejmować wszystkie produkty przemysłowe, w stosunku do których przepisy przynajmniej jednej ze stron wymagają przeprowadzenia oceny zgodności przez osobę trzecią.
MRA składają się z umowy ramowej oraz załączników sektorowych. Umowa ramowa ustanawia podstawowe zasady tradycyjnego porozumienia. Załączniki sektorowe określają w szczególności zakres, wymagania regulacyjne, wykaz wyznaczonych jednostek oceniających zgodność, procedury, organy odpowiedzialne za wyznaczanie tych jednostek oraz, w stosownych przypadkach, okresy przejściowe. Do umowy można sukcesywnie dodawać kolejne załączniki sektorowe.
MRA nie są oparte na konieczności wzajemnego uznawania norm lub przepisów technicznych drugiej strony, ani uznawania prawodawstwa obu stron za równoważne. Zakładają one tylko wzajemne uznawanie sprawozdań, certyfikatów oraz oznakowań, które zostały wydane w państwie partnerskim zgodnie z jego prawodawstwem. Jednakże MRA mogą torować drogę do zharmonizowania przez strony systemów normalizacji i certyfikacji. Niemniej w stosunku do obu systemów prawnych zasadniczo uznaje się, że zapewniają porównywalny poziom ochrony zdrowia, bezpieczeństwa, środowiska naturalnego oraz innych interesów publicznych. Ponadto MRA zwiększają przejrzystość systemów regulacyjnych. Po zawarciu MRA musi być ono podtrzymywana, na przykład poprzez prowadzenie wykazów uznanych jednostek certyfikujących oraz norm lub zasad, według których muszą prowadzić certyfikację.
Korzyścią MRA jest eliminacja powielanych kontroli lub certyfikacji. W przypadku gdy produkt przeznaczony na dwa rynki wciąż podlega podwójnej ocenie (jeżeli wymagania techniczne lub normy się różnią), ocena jest tańsza, jeżeli przeprowadza ją ta sama jednostka. Czas na wprowadzenie do obrotu jest krótszy, ponieważ kontakty między producentem a jedną jednostką oceniającą zgodność oraz pojedyncza ocena przyspieszają ten proces. Nawet jeżeli leżące u podstaw przepisy są zharmonizowane, na przykład dlatego, że odnoszą się do normy międzynarodowej, potrzeba uznania certyfikatów pozostaje bez zmian i w takich przypadkach korzyść będzie wyraźna: produkt jest oceniany względem wspólnie przyjętej normy jeden raz, a nie dwukrotnie.
Obecnie w mocy są MRA z Australią, Nową Zelandią, Stanami Zjednoczonymi, Kanadą, Japonią i Szwajcarią.
Powyższe umowy są zawarte w odniesieniu do określonych sektorów, które mogą różnić się w poszczególnych krajach. Więcej informacji o porozumieniach można znaleźć pod adresem: http://ec.europa.eu/growth/single-market/goods/international-aspects/mutual-recognition-agreements/index_en.htm. Jednostki wyznaczone w ramach MRA figurują w dedykowanej części NANDO.
9.2.2. MRA MIĘDZY UE A SZWAJCARIĄ
MRA zawarta ze Szwajcarią, która weszła w życie 1 czerwca 2002 r. (Dz.U. L 114 z 30.4.2002) stanowi kompleksową umowę opartą na równoważności prawodawstwa UE oraz Szwajcarii (300). Obejmuje uznawanie ocen zgodności niezależnie od pochodzenia produktów, z wyjątkiem rozdziału 15 dotyczącego, produktów leczniczych, dobrej praktyki wytwarzania (GMP), inspekcji i poświadczania partii. Taki typ MRA zazwyczaj określa się mianem „rozszerzonego MRA”. Jednak przypadek Szwajcarii pozostaje dość wyjątkowy.
Przepisy umowy oraz harmonizacja szwajcarskich przepisów technicznych z przepisami unijnymi zapewniają stały dostęp produktów UE do rynku szwajcarskiego oraz produktów szwajcarskich do rynku UE/EOG. Jednakże, pomimo zawartej MRA, pomiędzy UE a Szwajcarią nie istnieje unia celna.
Zgodnie z umową Szwajcarski Urząd Akredytacyjny (SAS) jest pełnoprawnym członkiem Europejskiej Współpracy w dziedzinie Akredytacji (EA) oraz sygnatariuszem wszystkich umów z EA o wzajemnym uznawaniu. W obszarze normalizacji Szwajcaria jest pełnoprawnym członkiem CEN, CENELEC i ETSI oraz aktywnie uczestniczy w pracach nad normalizacją europejską.
Ponadto unijna jednostka oceniająca zgodność jest upoważniona do wydawania certyfikatów w UE zgodnie z przepisami UE, które są uznawane za równoważne przepisom szwajcarskim. To samo odnosi się również odwrotnie do szwajcarskich jednostek oceniających zgodność. Dlatego też certyfikaty wydawane przez szwajcarskie jednostki oceniające zgodność akredytowane przez SAS dotyczące produktów objętych MRA są uznawane za równoważne certyfikatom wydawanym przez jednostki oceniające zgodność z UE.
Było to możliwe wyłącznie dlatego, że Szwajcaria posiada infrastrukturę techniczną (m.in. instytucje publiczne lub prywatne zajmujące się normami, akredytacją, oceną zgodności, nadzorem rynku oraz ochroną konsumentów), która jest jednakowo rozwinięta oraz uznawana za równoważną infrastrukturze istniejącej w UE. Po drugie Szwajcaria zdecydowała się zmodyfikować swoje przepisy dotyczące sektorów objętych umową w celu dostosowania ich do przepisów unijnych. Ponadto zobowiązała się do dostosowywania swoich przepisów ilekroć Unia Europejska wprowadzi zmiany do mających zastosowanie ram prawnych UE.
Tak zwane „rozszerzona MRA” ze Szwajcarią obejmuje obecnie dwadzieścia sektorów produktów, takich jak: maszyny, środki ochrony indywidualnej (PPE), bezpieczeństwo zabawek, wyroby medyczne, urządzenia gazowe i kotły grzewcze, urządzenia ciśnieniowe, końcowe urządzenia telekomunikacyjne, urządzenia i systemy ochronne przeznaczone do użytku w przestrzeniach zagrożonych wybuchem (ATEX), bezpieczeństwo elektryczne i kompatybilność elektromagnetyczna (EMC), instalacje i urządzenia budowlane, przyrządy pomiarowe i opakowania jednostkowe, pojazdy silnikowe, ciągniki rolnicze i leśne, dobra praktyka laboratoryjna (GLP), inspekcja GMP (dobra praktyka wytwarzania) i certyfikacja partii, produkty budowlane, dźwigi, produkty biobójcze, urządzenia kolei linowych oraz materiały wybuchowe przeznaczone do użytku cywilnego.
Podobna MRA obejmująca dokładnie ten sam zakres została zawarta między Szwajcarią a państwami EFTA należącymi do EOG (załącznik I do konwencji z Vaduz, która weszła w życie w dniu 1 czerwca 2002 r.) i zapewnia ona jednolity dostęp do rynku na całym rynku wewnętrznym UE, EOG oraz Szwajcarii.
9.2.3. PAŃSTWA EFTA NALEŻĄCE DO EOG: UMOWY O WZAJEMNYM UZNAWANIU I UKŁADY W SPRAWIE OCENY ZGODNOŚCI ORAZ UZNAWANIA
Upoważnienie nadane Komisji przez Radę do negocjowania umów o wzajemnym uznawaniu oraz układów w sprawie oceny zgodności oraz uznawania produktów przemysłowych miało założenie, aby zainteresowane kraje trzecie zawarły z państwami EFTA należącymi do EOG podobne umowy, równoważne tym, które mają zostać zawarte z UE oraz które możliwie będą miały tę samą datę wejścia w życie.
System równoległych umów formalnie zapewnia zainteresowanemu państwu trzeciemu taki sam dostęp do rynku na całym Europejskim Obszarze Gospodarczym w odniesieniu do produktów objętych umowami o wzajemnym uznawaniu lub układami w sprawie oceny zgodności oraz uznawania produktów przemysłowych. Jeżeli chodzi o praktyczne wdrażanie tych umów, będą organizowane łączone posiedzenia Wspólnego Komitetu oraz przedstawicieli zainteresowanego państwa trzeciego.
(1) Dnia 13 lutego 2013 r. Komisja przyjęła projekt nowego samodzielnego rozporządzenia w sprawie nadzoru rynku, zbierającego wszystkie przepisy dotyczące nadzoru rynku z rozporządzenia (WE) nr 765/2008, dyrektywy w sprawie ogólnego bezpieczeństwa produktów (GPSD – ang. General Product Safety Directive), a także prawodawstwa sektorowego. Ostateczna wersja COM(2013) 75 jest dostępna pod adresem http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=COM:2013:0075:FIN:pl:PDF
(2) Rozporządzenie Parlamentu Europejskiego i Rady (WE) nr 765/2008 z dnia 9 lipca 2008 r. ustanawiające wymagania w zakresie akredytacji i nadzoru rynku odnoszące się do warunków wprowadzania produktów do obrotu i uchylające rozporządzenie (EWG) nr 339/93 oraz decyzja Parlamentu Europejskiego i Rady nr 768/2008/WE z dnia 9 lipca 2008 r. w sprawie wspólnych ram dotyczących wprowadzania produktów do obrotu, uchylająca decyzję Rady 93/465/EWG.
(3) Dyrektywa (UE) 2015/1535 Parlamentu Europejskiego i Rady z dnia 9 września 2015 r. ustanawiająca procedurę udzielania informacji w dziedzinie przepisów technicznych oraz zasad dotyczących usług społeczeństwa informacyjnego (Dz.U. L 241 z 17.9.2015, s. 1).
(4) Od 1 stycznia 2013 r. na mocy rozporządzenia (UE) nr 1025/2012 każda krajowa jednostka normalizacyjna jest zobowiązana do publicznego udostępniania programu swojej pracy oraz powiadamiania innych krajowych jednostek normalizacyjnych, europejskiej organizacji normalizacyjnej i Komisji o istnieniu takich programów.
(5) Wyrok Trybunału Sprawiedliwości z dnia 20 lutego 1979 r. — Rewe-Zentral AG przeciwko Bundesmonopolverwaltung für Branntwein. Sprawa 120/78. Rec. 1979 s. 649.
(6) Dz.U. L 218 z 13.8.2008, s. 21.
(7) http://ec.europa.eu/internal_market/strategy/docs/monti_report_final_10_05_2010_pl.pdf
(8) Dz.U. C 136 z 4.6.1985, s. 1.
(9) Więcej informacji na temat przepisów prawa dotyczących odpowiedzialności za produkt znajduje się w rozdziale 1.4.
(10) Dz.U. L 316 z 14.11.2012, s. 12.
(11) Art. 2.4 Porozumienia TBT WTO.
(12) Początkowo przepisy przyjęte w ramach nowego podejścia miały zasadniczo formę dyrektyw.
(13) Decyzja Rady 93/465/EWG z dnia 22 lipca 1993 r. w sprawie modułów do różnych faz procedur oceny zgodności oraz zasad umieszczania i stosowania oznakowania CE, które mają być stosowane w dyrektywach harmonizacji technicznej (Dz.U. L 220 z 30.8.1993, s. 23).
(14) Dz.U. L 218 z 13.8.2008, s. 82.
(15) Dz.U. C 282 z 25.11.2003, s. 3.
(16) Parlament Europejski nadał pakietowi taką nazwę, aby upamiętnić dyrektora w Dyrekcji Generalnej ds. Przedsiębiorstw i Przemysłu Michela Ayrala, który był odpowiedzialny za scalenie pakietu.
(18) Słowo „jakość” oznacza poziom bezpieczeństwa i inne cele polityki publicznej, których dotyczy unijne prawodawstwo harmonizacyjne – nie mylić ze znaczeniem słowa „jakość” w kontekście handlowym, pozwalającym na rozróżnianie pomiędzy różnymi poziomami jakości produktów.
(19) Dz.U. L 11 z 15.1.2002, s. 4.
(20) Szczegółowe wskazówki dotyczące zastosowania GPSD można znaleźć pod adresem: http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm
(21) Dz.U. L 210 z 7.8.1985, s. 29.
(22) Zob. art. 3 dyrektywy 85/374/EWG.
(23) Dla porównania, unijne prawodawstwo harmonizacyjne może dotyczyć „ruchomości”, takich jak sprzęt elektroniczny, środki ochrony osobistej itp. lub „rzeczy nieruchomych” (np. winda, gdy jest zamontowana w budynku).
(24) Równowartość w walucie krajowej jest obliczana według kursu z dnia 25 lipca 1985 r.
(25) Zob. przypis 24.
(26) Stosownie normy zharmonizowane – chociaż zapewniają domniemanie zgodności – nie zwalniają z odpowiedzialności, ale mogą zmniejszać prawdopodobieństwo szkody. Informacje na temat zastosowania norm zharmonizowanych i domniemania zgodności, zob. punkt 4.1.2.
(27) Według Trybunału Sprawiedliwości (sprawa C-300/95) odnosi się to do obiektywnego stanu wiedzy, związanego nie tylko z normami bezpieczeństwa przyjętymi w określonym sektorze, ale do wszystkich wysokich standardów, o których istnieniu producent powinien wiedzieć i do których miał dostęp. Odpowiedzialność za ryzyko związane z rozwojem istnieje tylko w dwóch państwach członkowskich.
(28) Decyzja nr 768/2008/WE i rozporządzenie (WE) nr 765/2008.
(29) W przewodniku można jednak znaleźć odniesienia do GPSD w określonych sytuacjach, np. w przypadku produktów używanych.
(30) http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm
(31) Dyrektywa 2014/90/UE w sprawie wyposażenia morskiego odnosi się do umieszczania wyposażenia na statku pływającym pod banderą państwa członkowskiego UE.
(32) Część unijnego prawodawstwa harmonizacyjnego obejmuje również „oddawanie do użytku” (np. dźwigów) lub „własny użytek” (np. maszyny wykorzystywane przez samego producenta) jako termin równoznaczny „wprowadzaniu do obrotu”.
(33) Wprowadzanie do obrotu, udostępnianie na rynku i oddawanie do użytku – zob. rozdziały 2.2, 2.3 i 2.5.
(34) Dyrektywa 1999/44 w sprawie niektórych aspektów sprzedaży towarów konsumpcyjnych i związanych z tym gwarancji (Dz.U. L 171 z 7.7.1999, s. 12) jest poza zakresem zainteresowań niniejszego przewodnika. Zgodnie z tą dyrektywą sprzedawcy produktów konsumpcyjnych w UE są zobowiązani do zagwarantowania zgodności produktów z umową w okresie dwóch lat od daty ich dostarczenia. Jeżeli produkty nie są zgodne z umową sprzedaży, konsumenci mogą domagać się naprawy lub wymiany produktów, obniżenia ceny lub unieważnienia umowy. Sprzedawca końcowy, który ponosi odpowiedzialność przed konsumentem, może również pociągnąć do odpowiedzialności producenta w ramach swoich relacji biznesowych.
(35) W przypadku dyrektyw w sprawie wyrobów medycznych obowiązki mają zastosowanie tylko do wprowadzania do obrotu i/lub oddawania do użytku, lecz nie do jakiegokolwiek dalszego udostępniania.
(36) Zob. rozdział 3.4 Dystrybutorzy.
(37) Fakt, że produkt musi być oferowany zgodnie z unijnym prawodawstwem harmonizacyjnym obowiązującym w chwili wprowadzenia go do obrotu pozostaje bez uszczerbku dla ochrony poziomu bezpieczeństwa lub innego interesu publicznego.
(38) Oznacza to, że podmiot gospodarczy, który oferuje produkt, musi być w stanie przedstawić dowody na to, że produkt spełnia odpowiednie wymagania, tj. przedstawić dokumentację techniczną na żądanie organu nadzoru rynku W rozdziale 2.3 można znaleźć dodatkowe wskazówki dotyczące wprowadzania do obrotu produktów w internecie.
(39) W unijnym prawodawstwie harmonizacyjnym nie przewidziano zakazu wytwarzania produktów spełniających wymagania państw spoza Unii Europejskiej, jeżeli takie produkty nie będą wprowadzane do obrotu i oddawane do użytku na rynku wewnętrznym. Unijne przepisy harmonizacyjne nie zakazują importu produktów, które nie spełniają wymogów odpowiedniego unijnego prawodawstwa harmonizacyjnego, jeżeli takie produkty nie będą wprowadzone do obrotu lub oddane do użytku na rynku wewnętrznym (ale np. rafinowane/przetwarzane/łączone na rynku wewnętrznym), a eksportowane poza EOG.
(40) W tym kontekście jako Unię należy rozumieć obecne państwa członkowskie, w których swobodny przepływ produktów używanych odbywa się zgodnie z art. 34 i 36 TFUE.
(41) Produkty używane dostarczane do konsumentów podlegają przepisom GPSD i muszą być bezpieczne, chyba że są dostarczane jako antyki lub produkty wymagające naprawy przed użyciem, pod warunkiem że dostawca w jasny sposób poinformował o tym odbiorcę swojego produktu.
(42) W pewnych sytuacjach obowiązki pierwotnego producenta przejmuje inny podmiot, zob. rozdział 3.
(43) Będące poza zakresem właściwych przepisów unijnego prawodawstwa harmonizacyjnego części zamienne lub części, które są dostępne i wprowadzane do obrotu osobno jako produkty przeznaczone dla konsumentów w celu włączenia do innych produktów, takie jak części serwisowe lub komponenty przeznaczone do konserwacji lub napraw, muszą jednak spełniać ogólne wymogi bezpieczeństwa określone w GPSD.
(44) W przepisach dotyczących sprzętu medycznego istnieje pojęcie „całkowicie odtworzony”. „Całkowicie odtworzone” produkty są traktowane podobnie do nowych produktów.
(45) W przypadku produktów wykorzystywanych w miejscu pracy pracodawca musi podjąć wszelkie środki, aby sprzęt potrzebny do pracy był odpowiedni i bezpieczny i żeby naprawione maszyny nie były mniej bezpieczne niż oryginalne. Zob. rozdział 3.5.
(46) Zob. art. 2 rozporządzenia (WE) nr 765/2008 oraz art. R1 załącznika I do decyzji nr 768/2008/WE.
(47) Z wyłączeniem praw własności intelektualnej.
(48) W przypadku udostępnienia produktu w drodze wynajmu, wielokrotne wynajmowanie tego samego produktu nie stanowi nowego wprowadzenia do obrotu. Produkt ten musi być zgodny z obowiązującym unijnym prawodawstwem harmonizacyjnym w momencie pierwszego wynajęcia.
(49) W dyrektywie w sprawie dźwigów 95/16/WE wprowadzone jest również pojęcie „instalatora”, który również wprowadza produkty do obrotu.
(50) Łańcuchem dystrybucji może również być łańcuch handlowy producenta lub uprawnionego przedstawiciela.
(51) Oferta lub umowa zawarta przed zakończeniem etapu produkcji nie może być uznana za wprowadzenie do obrotu (np. oferta dotycząca wytworzenia produktu zgodnie z pewnymi specyfikacjami uzgodnionymi przez strony umowy, jeżeli produkt zostanie wyprodukowany i dostarczony dopiero na późniejszym etapie).
(52) Zob. np. dyrektywy w sprawie maszyn, przyrządów pomiarowych, dyrektywa ATEX, dyrektywa w sprawie materiałów wybuchowych przeznaczonych do użytku cywilnego.
(53) W przypadku gdy unijne prawodawstwo harmonizacyjne reguluje użytek własny, nie odnosi się to do sporadycznej produkcji na własny użytek przez osobę prywatną w kontekście niekomercyjnym.
(54) Wyjątek ten nie obejmuje produktów, które są dostarczane przez podmiot gospodarczy konsumentom w UE, jak to ma miejsce w przypadku produktów nabywanych w internecie i wysłanych do UE.
(55) Upoważniony przedstawiciel – zob. rozdział 3.2.
(56) Zob. rozporządzenie Rady (EWG) nr 2913/92 ustanawiające Wspólnotowy kodeks celny. Zgodnie z tym rozporządzeniem towary spoza Wspólnoty podlegające zawieszającej procedurze celnej lub umieszczone w wolnym obszarze celnym są objęte nadzorem celnym i nie można nimi swobodnie obracać na rynku wewnętrznym. Przed wprowadzeniem ich do wolnego obrotu na rynku wewnętrznym towary te muszą zostać zadeklarowane w celu dopuszczenia do obrotu. To obejmuje zastosowanie środków polityki handlowej, zakończenie innych formalności określonych w odniesieniu do importu dóbr i nałożenie wszelkich należnych ceł.
(57) Prototyp musi być bezpieczny, w pełni kontrolowany i nadzorowany. Warunki kontrolowane oznaczają profesjonalnych operatorów, ograniczenia w dostępie do produktu osób postronnych, zapobieganie nieodpowiedniej interakcji z innymi sąsiadującymi produktami itp.
(58) Jednak w takich okolicznościach widoczne oznakowanie musi jasno wskazywać, że dany produkt nie może zostać wprowadzony do obrotu lub do użytku do chwili, gdy nie spełni wymogów prawa.
(59) Jak już odnotowano we wprowadzeniu, toczą się szczegółowe dyskusje dotyczące różnych aspektów unijnych ram prawnych mających zastosowanie do sprzedaży internetowej, a niniejszy przewodnik pozostaje bez uszczerbku dla wszelkich przyszłych interpretacji i wskazówek, które mogą zostać opracowane w odniesieniu do tych kwestii.
(60) Podmiot internetowy nie jest nową kategorię podmiotu gospodarczego, lecz termin ten stosowany jest w odniesieniu do tradycyjnych podmiotów gospodarczych (producentów, importerów, dystrybutorów) działających tylko/głównie w internecie.
(61) Wyrok Trybunału Sprawiedliwości Unii Europejskiej z dnia 12 lipca 2011 r. w sprawie C-324/09, L'Oréal/eBay, s. 65. Chociaż kontekst prawny jest inny, w tej sytuacji można uwzględnić ten element wyroku.
(62) Na przykład w kontekście ofert wiązanych w dyrektywie 2005/29/WE dotyczącej nieuczciwych praktyk handlowych stosowanych przez przedsiębiorstwa wobec konsumentów na rynku wewnętrznym zdefiniowano „praktyki handlowe stosowane przez przedsiębiorstwa wobec konsumentów” jako każde działanie przedsiębiorcy, jego zaniechanie, sposób postępowania, oświadczenie lub komunikat handlowy, w tym reklamę i marketing, bezpośrednio związane z promocją, sprzedażą lub dostawą produktu do konsumentów.
(63) Można również uwzględnić fakt, że dyrektywa 2005/29/WE dotycząca nieuczciwych praktyk handlowych definiuje „przedsiębiorcę” jako każdą osobę fizyczną lub prawną, która w ramach praktyk handlowych objętych tą dyrektywą działa w celu związanym z jej działalnością handlową, gospodarczą, rzemieślniczą lub wolnym zawodem, oraz każdą osobę działającą w imieniu lub na rzecz przedsiębiorcy. Podobnie w dyrektywie 2011/83/UE w sprawie praw konsumentów zdefiniowano „przedsiębiorcę” jako każdą osobę fizyczną lub każdą osobę prawną, niezależnie od tego, czy jest to podmiot publiczny czy prywatny, która działa – w tym również za pośrednictwem każdej innej osoby działającej w jej imieniu lub na jej rzecz – w celach związanych z jej działalnością handlową, gospodarczą, rzemieślniczą lub wykonywaniem wolnego zawodu, w związku z umowami objętymi zakresem tej dyrektywy.
(64) Jeżeli produkty są sprzedawane w internecie, oznakowanie CE oraz wszelkie ostrzeżenia, informacje i etykiety wymagane zgodnie z obowiązującymi przepisami należy podawać na stronie internetowej; powinny one być wyraźnie widoczne w całości przed dokonaniem zakupu przez konsumenta.
(65) Wyjaśnienie to nie rozwiązuje kwestii odpowiedzialności pośredników i termin „podmiot internetowy” użyty w tym kontekście może nie obejmować takich pośredników.
(66) W chwili wprowadzania produktu do obrotu producent musi spełnić wymóg projektu zgodnego z zasadniczymi wymaganiami stosownego aktu prawnego, wynikającej z nich oceny ryzyka i oceny zgodności, wydania deklaracji zgodności, oznakowania (oznakowanie CE, nazwa, adres producenta itp.) i skompletowania dokumentacji technicznej.
(67) Nadzór rynku – zob. rozdział 7.
(68) Oddawanie do użytku – zob. rozdział 2.5.
(69) Zob. art. 27–29 rozporządzenia (WE) nr 765/2008.
(70) Rola importera, zob. pkt 3.3.
(71) Należy zauważyć, że w dziedzinie wyrobów medycznych z rolą upoważnionego przedstawiciela wiąże się więcej obowiązków. Autoryzowany przedstawiciel jest głównym partnerem kontaktowym dla organu nadzoru rynku w przypadku produktów dla państw trzecich.
(72) Koncepcja „oddania do użytku” nie jest istotna dla wszystkich przepisów unijnego prawodawstwa harmonizacyjnego. Np. w przypadku materiałów wybuchowych nie dochodzi do „oddania do użytku”.
(73) Jeśli chodzi o dźwigi i równoważne produkty, uznaje się, że oddanie do użytku następuje w momencie, gdy możliwe jest ich pierwsze użycie na terenie Unii.
(74) W przypadku wejścia w życie nowego unijnego prawodawstwa harmonizacyjnego produkt zgodny z wymogami, który wprowadzono do obrotu przed końcem okresu przejściowego określonego w przepisach, które mają zostać zastąpione nowymi, powinien móc zostać oddany do użytku, chyba że przepisy szczegółowe stanowią inaczej.
(75) W przypadku dyrektywy w sprawie urządzeń radiowych i końcowych urządzeń telekomunikacyjnych art. 7. tego aktu reguluje ograniczenia w oddawaniu produktów do użytku. Państwa członkowskie mogą ograniczyć oddawanie do użytku urządzeń radiowych z przyczyn związanych ze skutecznym i odpowiednim wykorzystaniem częstotliwości radiowych, unikaniem szkodliwych interferencji lub sprawami dotyczącymi zdrowia publicznego.
(76) Np. zużycie energii.
(77) Na przykład: dyrektywa dotycząca sprzętu niskonapięciowego nie znajduje zastosowania w przypadku elektrycznych urządzeń medycznych – zamiast tego stosuje się przepisy regulujące wyroby medyczne; dyrektywy dotyczącej kompatybilności elektromagnetycznej nie stosuje się w przypadku produktów objętych szczegółowymi przepisami, które harmonizują wymagania ochrony określone w dyrektywie w sprawie kompatybilności elektromagnetycznej; dyrektywa dotycząca dźwigów nie ma zastosowania w przypadku dźwigów połączonych z maszynami i przeznaczonych wyłącznie do zapewnienia dostępu do miejsca pracy – zamiast tego stosuje się dyrektywę w sprawie maszyn; wyposażenie statków, które znajduje się również w zakresie dyrektyw innych niż dyrektywa w sprawie wyposażenia statków, zostało wykluczone z zakresu zastosowana takich dyrektyw.
(78) Np. dyrektywa w sprawie urządzeń radiowych i końcowych urządzeń telekomunikacyjnych (R&TTE) reguluje bezpośrednio kwestie kompatybilności elektromagnetycznej i bezpieczeństwa. Aby uniknąć tworzenia zbędnych przepisów, dyrektywa ta włącza zasadnicze wymagania dyrektywy w sprawie kompatybilności elektromagnetycznej (EMCD) i dyrektywy w sprawie sprzętu niskonapięciowego (LVD) bez dolnej granicy napięcia i dopuszcza, aby producent zastosował niektóre określone w nich procedury oceny zgodności. Dodatkowo, normy zharmonizowane określone w EMCD i LVD mają ten sam status także w przypadku dyrektywy R&TTE. Dyrektywa dotycząca dźwigów zawiera istotne wymogi dyrektywy w sprawie maszyn.
(79) Na przykład: dyrektywa w sprawie maszyn reguluje wszystkie zagrożenia, które wiążą się z maszynami, w tym zagrożenia elektryczne. Jednak jeśli chodzi o zagrożenia elektryczne związane z maszynami, dyrektywa w sprawie maszyn odsyła do celów bezpieczeństwa określonych w dyrektywie w sprawie sprzętu niskonapięciowego.
(80) Udostępnianie – zob. rozdział 2.2; oddawanie do użytku – zob. rozdział 2.5.
(81) Należy zauważyć, że w dyrektywie w sprawie maszyn na producenta nałożono obowiązek uwzględnienia „racjonalnie przewidywalnego niewłaściwego użytkowania”.
(82) Ponadto narzędzie zaprojektowane i przeznaczone wyłącznie dla profesjonalistów może być ostatecznie używane przez inne osoby; dlatego też należy uwzględnić tę ewentualność w projekcie produktu i dołączonych do niego instrukcjach.
(83) W Zjednoczonym Królestwie są to Wyspy Normandzkie i Wyspa Man.
(84) Więcej szczegółów znajduje się w dokumencie roboczym służb Komisji w sprawie przeszkód w dostępie Andory, Monako i San Marino do rynku wewnętrznego UE i współpracy w innych dziedzinach (SWD(2012) 388 final), dostępnym pod adresem: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=SWD:2012:0388:FIN:EN:PDF
(85) Dz.U. L 265 z 4.11.2003, s. 2.
(86) Np. taki produkt może być nadal legalnie sprzedawany po upływie okresu przejściowego, jeśli produkt jest w magazynach dystrybutora, tj. produkt został już wprowadzony do obrotu i nastąpiła zmiana właściciela.
(87) Ponieważ dyrektywa w sprawie urządzeń ciśnieniowych nie określa limitów czasowych na oddanie do użytku, produkty objęte tą dyrektywą mogą być oddane do użytku w dowolnym czasie bez konieczności spełnienia dodatkowych określonych w niej warunków. Wprowadzanie do obrotu i oddawanie do użytku – zob. rozdziały 2.3 i 2.5.
(88) Deklaracja zgodności UE – zob. rozdział 4.4; oznakowanie CE – zob. rozdział 4.5.1.
(89) W lutym 2014 r. przyjęto „pakiet dostosowawczy” obejmujący osiem dyrektyw. Dyrektywy z pakietu dostosowawczego zaczną obowiązywać w dniu 20 kwietnia 2016 r., a ich zasadnicze wymagania nie uległy zmianie. Pakiet ten obejmuje dyrektywę 2014/35/UE (niskie napięcie); dyrektywę 2014/30/UE (kompatybilność elektromagnetyczna); dyrektywę 2014/34/UE (ATEX); dyrektywę 2014/33/UE (dźwigi); dyrektywę 2014/29/UE (proste zbiorniki ciśnieniowe); dyrektywę 2014/32/UE (przyrządy pomiarowe); dyrektywę 2014/31/UE (wagi nieautomatyczne); dyrektywę 2014/28/UE (materiały wybuchowe do użytku cywilnego). Dyrektywa 2013/29/UE (wyroby pirotechniczne) również została dostosowana do decyzji 768/2008/WE i zaczęła obowiązywać z dniem 1 lipca 2015 r.
(90) Zob. art. R1 pkt 7 załącznika I do decyzji nr 768/2008/WE.
(91) Zob. art. R1 pkt 3 załącznika I do decyzji nr 768/2008/WE.
(92) Zob. art. R6 załącznika I do decyzji nr 768/2008/WE.
(93) Tacy producenci są często określani mianem „own brand labellers” (producentów OBL) lub „private labellers” [przyp. tłum.: oba terminy oznaczają podmioty umieszczające swoje etykiety na produktach].
(94) W dyrektywie w sprawie dźwigów 95/16/WE stosuje się pojęcie instalatora jako osoby, która ponosi odpowiedzialność za przygotowanie produktu do działania i oddanie go do użytku. Rola instalatora łączy w sobie elementy produkcji i oddania do użytku i postrzegana jest jako zasadnicza dla dostarczenia produktu końcowego.
(95) Art. R2 ust. 1 załącznika I do decyzji nr 768/2008/WE.
(96) Alternatywą dla pisemnych oświadczeń może być zastosowanie symboli zgodnych z międzynarodowymi normami.
(97) W niektórych szczególnych przypadkach, gdy producent zamierza sprzedać użytkownikowi końcowemu kilka identycznych produktów w pakiecie lub sprzedać je w jednym opakowaniu do jednoczesnego zastosowania (np. sprzęt instalacyjny), do takiej wysyłki wystarczy dołączyć jeden egzemplarz instrukcji. Jednakże w przypadku gdy pakiet taki jest rozkładany na części, a poszczególne identyczne produkty sprzedawane pojedynczo, podmiot gospodarczy rozpakowujący pakiet i udostępniający pojedyncze produkty musi upewnić się, że instrukcje i informacje dotyczące bezpieczeństwa towarzyszą każdemu produktowi.
(98) Nie wszystkie unijne przepisy harmonizacyjne nakładają obowiązek dostarczenia instrukcji i informacji dotyczących bezpieczeństwa, ponieważ nie wszystkie dotyczą spraw związanych z bezpieczeństwem.
(99) Producent, importer lub dystrybutor ma obowiązek dopilnować, aby do produktu były dołączone instrukcje w języku łatwo zrozumiałym dla konsumentów i innych użytkowników końcowych, określonym przez zainteresowane państwo członkowskie. Obowiązek zapewnienia instrukcji w wymaganych językach spoczywa na każdym podmiocie gospodarczym udostępniającym produkt w danym państwie członkowskim.
(100) Chociaż informacje dotyczące bezpieczeństwa muszą być udostępniane w formie papierowej, to, o ile nie określono inaczej w przepisach szczegółowych, nie jest wymagane, aby wszystkie pozostałe instrukcje były również dostarczane na papierze – mogą być również dostępne na w formie elektronicznej lub innym formacie gromadzenia danych. Wersja papierowa powinna jednak być zawsze bezpłatnie dostępna dla konsumentów na żądanie.
(101) Należy to rozumieć jako ostatni egzemplarz modelu produktu wprowadzony do obrotu.
(102) W przypadku dyrektyw w sprawie wyrobów medycznych producent musi wskazać miejsce prowadzenia działalności.
(103) Nie obejmuje to względów estetycznych.
(104) Należy zauważyć, że niektóre unijne przepisy harmonizacyjne wykluczają możliwość wykorzystania opakowania do spełnienia tego wymagania (np. dyrektywa o prostych zbiornikach ciśnieniowych).
(105) Producent może wedle uznania dodać stronę internetową. Adres strony jest dodatkową informacją, ale sam w sobie nie jest wystarczający. Zazwyczaj adres pocztowy składa się z nazwy ulicy i numeru lub skrzynki pocztowej i numeru oraz kodu pocztowego i miejscowości, ale w niektórych państwach może różnić się od tego modelu.
(106) Zob. unijne prawodawstwo harmonizacyjne odnoszące się do sprzętu niskonapięciowego, zabawek, maszyn, wag nieautomatycznych, wyrobów medycznych aktywnego osadzania, urządzeń gazowych, wyrobów medycznych, przestrzeni zagrożonych wybuchem, rekreacyjnych jednostek pływających, dźwigów, urządzeń ciśnieniowych, wyrobów medycznych używanych do diagnostyki in vitro oraz urządzeń radiowych i końcowych urządzeń telekomunikacyjnych. Zgodnie z dyrektywą w sprawie wyrobów medycznych używanych do diagnostyki in vitro producent wprowadzający urządzenia do obrotu w Unii pod własną nazwą jest zobowiązany zarejestrowania siedziby w państwie członkowskim, w którym prowadzi interesy.
(107) Więcej informacji na temat wymogów dotyczących nazwy i adresu znajduje się w pkt 4.2.2.1.
(108) Np. oznakowanie ATEX, identyfikator klasy zgodnie z dyrektywą R&TTE lub uzupełniające oznakowanie metrologiczne w przypadku wag nieautomatycznych i przyrządów pomiarowych.
(109) Obowiązek informacyjny w przypadku certyfikatów badania typu UE – zob. załącznik II do decyzji nr 768/2008/WE, moduł B, pkt 7.
(110) Np. dyrektywa w sprawie prostych zbiorników ciśnieniowych i ATEX.
(111) Takie badania należy przeprowadzać w stosownych przypadkach w odniesieniu do zagrożeń stwarzanych przez produkt w celu ochrony zdrowia i bezpieczeństwa konsumentów (zob. art. R2 ust. 4 załącznika I do decyzji 768/2008/WE).
(112) Dopuszczalny poziom ryzyka dla produktu określają zasadnicze wymagania ustanowione w mającym zastosowanie unijnym prawodawstwie harmonizacyjnym. W związku z tym producenci muszą informować właściwe organy w przypadkach, gdy mają powód, by sądzić, że produkt nie jest zgodny z mającymi zastosowanie zasadniczymi wymaganiami.
(113) https://webgate.ec.europa.eu/gpsd-ba/index.do
(114) Uzasadnione żądanie niekoniecznie oznacza formalną decyzję organu. Zgodne z art. 19 ust. 1 akapit drugi rozporządzenia (WE) nr 765/2008 „organy nadzoru rynku mogą wymagać udostępnienia przez podmioty gospodarcze dokumentów i informacji, które w ich ocenie są niezbędne do celów prowadzenia przez nie działań”. Aby żądanie było uzasadnione, wystarczy, że organ nadzoru rynku wyjaśni kontekst, w jakim wymagane są informacje (np. kontrola dotycząca szczególnych cech produktów, wyrywkowe kontrole itp.).
(115) Nie dotyczy to produktów objętych unijnymi przepisami harmonizacyjnymi dotyczącymi zabawek, sprzętu niskonapięciowego, materiałów wybuchowych przeznaczonych do użytku cywilnego i urządzeń chłodniczych, ponieważ dyrektywy te obejmują tylko udostępnianie produktów na rynku. Nie dotyczy to rekreacyjnych jednostek pływających zbudowanych na własny użytek, pod warunkiem że w ciągu pięciu lat nie zostaną wprowadzone do obrotu, oraz jednostek pływających zaprojektowanych przed rokiem 1950.
(116) Art. R6 załącznika I do decyzji nr 768/2008/WE.
(117) Zgodnie z dyrektywami dotyczącymi maszyn i dźwigów obowiązki związane z procedurą oceny zgodności są nakładane na dowolną osobę wprowadzającą produkt do obrotu, jeśli ani producent, ani upoważniony przedstawiciel, ani instalator dźwigu nie spełnia tych obowiązków.
(118) Należy zwrócić uwagę, że nie wszystkie unijne przepisy harmonizacyjne przewidują istnienie upoważnionego przedstawiciela.
(119) W ramach wyjątku zgodnie z dyrektywami w sprawie wyrobów medycznych i wyrobów medycznych używanych do diagnostyki in vitro, producent musi wskazać osobę mającą siedzibę w Unii, odpowiedzialną za wprowadzanie do obrotu wyrobów medycznych, jeżeli nie zarejestrował miejsca prowadzenia działalności w dowolnym państwie członkowskim i wprowadza wyroby na rynek unijny pod własną nazwą.
(120) Obowiązki importera – zob. rozdział 3.3.
(121) Do celów niniejszego przewodnika produkty importowane to produkty wytwarzane w państwach trzecich i wprowadzane do obrotu w Unii. Produkty wytworzone w jednym państwie członkowskim i wprowadzane do obrotu w innym nie stanowią „importu”, ponieważ działania te odbywają się na rynku unijnym.
(122) Importer niekoniecznie jest osobą, która przewozi produkt, ale może być osobą, w imieniu której przeprowadzane jest to działanie logistyczne.
(123) Zob. rozdział o nadzorze rynku.
(124) Nie wszystkie unijne przepisy harmonizacyjne nakładają obowiązek dostarczenia instrukcji i informacji dotyczących bezpieczeństwa, ponieważ nie wszystkie dotyczą spraw związanych z bezpieczeństwem.
(125) Biorąc pod uwagę te obowiązki, za dobrą praktykę importerów uznaje się zasadniczo: odniesienie się do stosownego prawodawstwa UE w umowie z dostawcą (i określenie obowiązków producentów wynikających z prawa unijnego); zagwarantowanie sobie dostępu do dokumentacji produktu lub dopilnowanie, aby producent podpisał zobowiązanie dostarczenia dokumentacji produktu na żądanie organów nadzoru rynku.
(126) Zależy od mającego zastosowanie unijnego prawodawstwa harmonizacyjnego.
(127) Należy zwrócić uwagę, że sektorowe prawodawstwo harmonizacyjne Unii może zawierać surowsze wymagania.
(128) Należy to rozumieć jako ostatni egzemplarz modelu produktu wprowadzony do obrotu.
(129) Importerzy nie mają obowiązku posiadać kopii dokumentacji produktu, lecz muszą zapewnić jej udostępnienie na żądanie właściwych organów. Nawet jeśli nie ma wyraźnego obowiązku, zaleca się, aby importer żądał od producenta formalnego pisemnego zapewnienia, że dokumenty zostaną udostępnione w przypadku żądania ze strony organu nadzoru.
(130) Uzasadnione żądanie niekoniecznie oznacza formalną decyzję organu. Zgodne z art. 19 ust. 1 akapit drugi rozporządzenia (WE) nr 765/2008 „organy nadzoru rynku mogą wymagać udostępnienia przez podmioty gospodarcze dokumentów i informacji, które w ich ocenie są niezbędne do celów prowadzenia przez nie działań”. Aby żądanie było uzasadnione, wystarczy, że organ nadzoru rynku wyjaśni kontekst, w jakim wymagane są informacje (np. kontrola dotycząca szczególnych cech produktów, wyrywkowe kontrole itp.).
(131) Art. R4 ust. 6 załącznika I do decyzji nr 768/2008/WE.
(132) Należyta staranność odnosi się do starań podjętych racjonalnie i zgodnie z zasadą ostrożności przez jedną stronę, aby uniknąć wyrządzenia krzywdy drugiej stronie, przy uwzględnieniu okoliczności. Odnosi się do poziomu osądu, staranności, rozwagi, determinacji i czynności, których racjonalnie oczekiwałoby się od danej osoby w określonych okolicznościach.
(133) Art. R5 pkt 1 załącznika I do decyzji nr 768/2008/WE.
(134) Art. R5 ust. 2 akapit pierwszy załącznika I do decyzji nr 768/2008/WE.
(135) Jeśli w unijnym prawodawstwie harmonizacyjnym jest jasno zapisany obowiązek, że do danego produktu musi być dołączona deklaracja zgodności UE, dystrybutor musi dopilnować, by taka deklaracja została dołączona.
(136) Nie wszystkie unijne przepisy harmonizacyjne nakładają obowiązek dostarczenia instrukcji i informacji dotyczących bezpieczeństwa, ponieważ nie wszystkie dotyczą spraw związanych z bezpieczeństwem.
(137) Zob. obowiązki producenta w pkt 3.1, oraz obowiązki importera w pkt 3.3.
(138) Art. R5 ust. 2 akapit drugi załącznika I do decyzji nr 768/2008/WE.
(139) Art. R5 ust. 2 akapit drugi i art. R5 ust. 4 w załącznika I do decyzji nr 768/2008/WE.
(140) Uzasadnione żądanie niekoniecznie oznacza formalną decyzję organu. Zgodne z art. 19 ust. 1 akapit drugi rozporządzenia (WE) nr 765/2008 „organy nadzoru rynku mogą wymagać udostępnienia przez podmioty gospodarcze dokumentów i informacji, które w ich ocenie są niezbędne do celów prowadzenia przez nie działań”. Aby żądanie było uzasadnione, wystarczy, że organ nadzoru rynku wyjaśni kontekst, w jakim wymagane są informacje (np. kontrola dotycząca szczególnych cech produktów, wyrywkowe kontrole itp.).
(141) Art. R5 pkt 5 załącznika I do decyzji nr 768/2008/WE.
(142) Art. R7 ust. 2 załącznika I do decyzji nr 768/2008/WE.
(143) Art. R5 ust. 3 załącznika I do decyzji nr 768/2008/WE.
(144) Zob. wyrok Trybunału: sprawa C-271/92
(145) Jak już odnotowano we wprowadzeniu, toczą się szczegółowe dyskusje dotyczące różnych aspektów unijnych ram prawnych mających zastosowanie do sprzedaży internetowej, a niniejszy przewodnik pozostaje bez uszczerbku dla wszelkich przyszłych interpretacji i wskazówek, które mogą zostać opracowane w odniesieniu do tych kwestii.
(146) Dyrektywa 2000/31/WE Parlamentu Europejskiego i Rady z dnia 8 czerwca 2000 r. w sprawie niektórych aspektów prawnych usług społeczeństwa informacyjnego, w szczególności handlu elektronicznego w ramach rynku wewnętrznego (dyrektywa o handlu elektronicznym) (Dz.U. L 178 z 17.7.2000, s. 1).
(147) Inne działania opisane w dyrektywie to: 1) „zwykły przekaz”, taki jak przekazywanie informacji (dostarczonych przez usługobiorcę) lub zapewnienie dostępu do sieci łączności (np. dostawcy usług internetowych) oraz 2) „caching”, czyli usprawnienie przekazywania informacji, np. powielanie bazy danych, która stanowi kopię treści początkowego serwera w celu zapewnienia globalnego zasięgu.
(148) W sprawie C-324/09, L'Oréal przeciwko eBay, Europejski Trybunał Sprawiedliwości orzekł, że istotną kwestią, jeśli chodzi o warunki korzystania ze zwolnienia od odpowiedzialności jest to, czy eBay wiedział o faktach i okolicznościach, z których jasno wynikał nielegalny charakter działalności (zob. pkt 120–123).
(149) Dyrektywa 2013/53/UE dotycząca rekreacyjnych jednostek pływających nakłada jednak obowiązki na prywatnych importerów.
(150) Koncepcja „użytkowania zgodnego z przeznaczeniem” – zob. wyżej rozdział 2.7.
(151) Dokumentacja produktu – zob. pkt 4.3.
(152) Ocena ryzyka powinna być przeprowadzona nawet wtedy, gdy producent stosuje normę zharmonizowaną (jeśli odniesienie do niej podano w Dzienniku Urzędowym Unii Europejskiej i jeśli ma na celu uregulowanie określonych rodzajów ryzyka) w celu spełnienia zasadniczych wymagań; producent musi wówczas również sprawdzić, czy norma zharmonizowana obejmuje wszystkie rodzaje ryzyka stwarzanego przez produkt. Dzieje się tak dlatego, że nie można zakładać, iż norma zharmonizowana obejmuje wszystkie zawarte we wszystkich aktach prawnych wymagania mające zastosowanie do danego produktu (lub wszystkie wymagania aktu, na podstawie którego została opracowana) ani tego, czy dany produkt wiąże się również z innymi zagrożeniami nieuregulowanymi normą zharmonizowaną.
(153) Zgodnie z dyrektywą 2008/57/WE w sprawie interoperacyjności systemu kolei, każdy podsystem ma swoje techniczne specyfikacje interoperacyjności (TSI), które określają zasadnicze wymagania. Zgodnie z rozporządzeniem (WE) nr 552/2004 w sprawie interoperacyjności Europejskiej Sieci Zarządzania Ruchem Lotniczym w razie potrzeby zasadnicze wymagania są przeformułowywane i uzupełniane w przepisach wykonawczych dotyczących interoperacyjności.
(154) Dz.U. L 316 z 14.11.2012, s. 12.
(155) CEN (Europejski Komitet Normalizacyjny); CENELEC (Europejski Komitet Normalizacyjny Elektrotechniki); ETSI (Europejski Instytut Norm Telekomunikacyjnych).
(156) W wyjątkowych sytuacjach dokumenty harmonizacyjne przyjęte przez europejskie organizacje normalizacyjne można również uznać za normy zharmonizowane (np. w przypadku dyrektywy o niskim napięciu). Różnice pomiędzy normami europejskimi (EN) a dokumentami harmonizacyjnymi odnoszą się zasadniczo do stopnia zobowiązań po stronie krajowych jednostek normalizacyjnych. Dokumenty harmonizacyjne należy wdrażać na poziomie krajowym przynajmniej za pomocą publicznego powiadomienia o tytule i numerze dokumentu oraz wycofania sprzecznych norm krajowych. Jednak dopuszcza się utrzymanie w mocy lub opublikowanie krajowej normy dotyczącej przedmiotu dokumentu harmonizacyjnego, pod warunkiem że dotyczy tych samych zagadnień technicznych. Dodatkowo, w określonych okolicznościach, dopuszcza się odejście od dokumentów harmonizacyjnych na szczeblu krajowym, co mogłoby powodować pewne problemy związane z ich zastosowaniem, jeśli byłyby przyjęte jako normy zharmonizowane.
(157) Nieobowiązkowy charakter norm odnosi się do faktu, że zastosowanie norm jako takich, publikowanych przez organizacje normalizacyjne, jest zawsze nieobowiązkowe. Zasada ta jest często stosowana w prawodawstwie w przypadku odniesień do norm.
(158) SWD(2015) 205 final z 27.10.2015, dostępny na stronie http://ec.europa.eu/growth/single-market/european-standards/vademecum/index_en.htm
(159) Po konsultacjach z europejskimi organizacjami normalizacyjnymi, zainteresowanymi stronami i ekspertami z poszczególnych sektorów (z tymi ostatnimi za pośrednictwem komitetu utworzonego na podstawie przepisów, o ile taki istnieje) Komisja konsultuje się z komitetem państw członkowskich ustanowionym rozporządzeniem (UE) nr 1025/2012 (rozporządzenie w sprawie normalizacji) zgodnie z procedurą badania określoną w rozporządzeniu nr 182/2011 (Dz.U. L 55 z 28.2.2011, s. 13).
(160) Zob. art. 10 rozporządzenia (UE) nr 1025/2012.
(161) Aby ustanowić spójne zasady opracowywania i przyjmowania, w tym rewizji, norm zharmonizowanych, w „Vademecum nt. normalizacji europejskiej” (SWD(2015) 205 final z 27.10.2015, część III) zawarto wytyczne dotyczące realizacji wniosków o normalizację przyjętych przez europejskie organizacje normalizacyjne.
(162) Dz.U. C 91 z 16.4.2003, s. 7.
(163) Komisja na podstawie rozporządzenia (UE) nr 1025/2012 ma jednak obowiązek weryfikować i oceniać, czy spełniane są warunki wniosku o normalizację, co ma na celu zapewnienie właściwego funkcjonowania jednolitego rynku (zob. pkt 4.1.2.4).
(164) W rzeczywistości europejskie organizacje normalizacyjne mogą tylko zadeklarować zamiar ujęcia niektórych wymagań, a intencję taką podtrzymuje się (lub odrzuca) poprzez opublikowanie (lub usunięcie) odniesienia w (z) Dzienniku Urzędowym (Dziennika Urzędowego) Unii Europejskiej (zob. pkt 4.1.2.4 i 4.1.2.5).
(165) Europejskie organizacje normalizacyjne zazwyczaj określają ten załącznik jako załącznik ZA, ZB lub ZZ itp.
(166) W tym kontekście warunek ten należy rozumieć zgodnie z ISO/IEC Guide 51 Safety aspects – Guidelines for their inclusion in standards (Przewodnikiem ISO/IEC nr 51 Kwestie bezpieczeństwa – wytyczne dotyczące włączenia kwestii bezpieczeństwa do norm), które stanowią ogólne wytyczne w zakresie opracowywania norm dotyczących kwestii bezpieczeństwa.
(167) Przygotowanie wniosku o normalizację może rozpocząć się jednocześnie z procesem legislacyjnym. Jednak w chwili, gdy europejskie organizacje normalizacyjne otrzymują wniosek o normalizację, wymagania prawne ujęte w normach zharmonizowanych muszą być ustalone.
(168) Nie narusza to prawa organizacji do odrzucenia wniosku o normalizację.
(169) Europejska organizacja normalizacyjna może także współpracować z innymi jednostkami odpowiedzialnymi za prace nad projektami norm.
(170) W art. 10 ust. 5 wskazano, że proces budowy konsensusu zgodny z wewnętrznymi regulaminami europejskich organizacji normalizacyjnych nie jest jako taki wystarczającą gwarancją, aby zakładać, że wymagania określone we wniosku o normalizację są spełnione.
(171) Transpozycja normy jest zależna od regulaminów europejskich organizacji normalizacyjnych. Zazwyczaj dokonuje się jej przed opublikowaniem odniesień do normy zharmonizowanej w Dzienniku Urzędowym Unii Europejskiej. Jednak transpozycja krajowa nie jest warunkiem koniecznym do domniemania zgodności. W praktyce normy zharmonizowane są zazwyczaj dostępne jako normy przeniesione na grunt krajowy, natomiast wykaz norm zharmonizowanych opublikowany w Dzienniku Urzędowym Unii Europejskiej i stosowne unijne prawodawstwo harmonizacyjne zawierają bezpośrednie odniesienia do oryginalnych norm europejskich.
(172) Zgodnie z art. 28 rozporządzenia (UE) nr 1025/2012 przepisy dotyczące formalnego sprzeciwu przewidziane w niektórych przepisach sektorowych zachowują swoją ważność jeszcze przez pewien czas.
(173) Serwis internetowy udostępniający najnowsze wykazy norm zharmonizowanych i innych norm europejskich opublikowanych w Dzienniku Urzędowym Unii Europejskiej jest dostępny pod adresem: http://ec.europa.eu/growth/single-market/european-standards/harmonised-standards/index_en.htm
(174) „Wskazane” oznacza zazwyczaj wymagania wymienione w załączniku informacyjnym do normy zharmonizowanej.
(175) W pewnych przypadkach niektóre przepisy sektorowe mogą nadal przewidywać sprzeciw. W takich przypadkach art. 11 rozporządzenia (UE) nr 1025/2012 nie ma zastosowania – zob. art. 28 akapit drugi rozporządzenia (UE) nr 1025/2012.
(176) Domniemanie zgodności byłoby bez znaczenia, gdyby nieznane były zasadnicze wymagania ujęte w normie.
(177) Dyrektywa 1999/5/WE dotycząca końcowych urządzeń telekomunikacyjnych dopuszcza, aby normy zharmonizowane były przekształcane we wspólne regulacje techniczne, zgodność z którymi jest obligatoryjna. Rozporządzenie (WE) nr 552/2004 w sprawie interoperacyjności Europejskiej Sieci Zarządzania Ruchem Lotniczym nakłada obowiązek stosowania specyfikacji wspólnotowych.
(178) Zob. dyrektywy w sprawie prostych zbiorników ciśnieniowych, zabawek, kompatybilności elektromagnetycznej, urządzeń radiowych i końcowych urządzeń telekomunikacyjnych, maszyn, dźwigów i rekreacyjnych jednostek pływających. Brak norm zharmonizowanych może prowadzić do zastosowania określonej procedury, zob. np. dyrektywa w sprawie urządzeń ciśnieniowych (europejskie uznanie może być przyznawane materiałom nieobjętym normą zharmonizowaną, które mają być stale wykorzystywane przy produkcji urządzeń ciśnieniowych).
(179) Art. 11 rozporządzenia (UE) nr 1025/2012 znajduje stopniowo zastosowanie wraz z usuwaniem zapisów dotyczących sprzeciwu zawartych w prawodawstwie sektorowym. Tymczasem część unijnego prawodawstwa harmonizacyjnego może nadal zawierać specjalne procedury, jak np. dyrektywa w sprawie urządzeń radiowych i końcowych urządzeń telekomunikacyjnych, która w przypadku braków w normach zharmonizowanych daje Komisji możliwość opublikowania w Dzienniku Urzędowym wytycznych dotyczących interpretacji norm zharmonizowanych lub warunków, w jakich możliwa jest zgodność.
(180) Parlament Europejski może przedstawić swoje wątpliwości w przypadkach gdy zastosowanie ma art. 11 rozporządzenia (UE) nr 1025/2012.
(181) Zgodnie z art. 11 ust. 1 i art. 11 ust. 4 i 5 rozporządzenia (WE) nr 1025/2012.
(182) http://ec.europa.eu/growth/single-market/european-standards/notification-system/index_en.htm
(183) Klauzula ochronna – zob. rozdział 7.4.
(184) http://ec.europa.eu/growth/single-market/european-standards/notification-system/index_en.htm
(185) Bez względu na to, czy normy zostały opracowane na podstawie wniosku o normalizację, czy nie, nie rzadziej niż raz na pięć lat europejskie organizacje normalizacyjne dokonują rewizji norm zgodnie ze swoimi regulaminami wewnętrznymi. Ta okresowa ocena może skutkować potwierdzeniem (niepodjęciem żadnych działań), rewizją lub wycofaniem danej normy.
(186) Przewodnik po normalizacji europejskiej (SWD(2015) 205 final z 27.10.2015, część III).
(187) Część unijnych przepisów harmonizacyjnych może jednak oferować inne podstawy domniemania zgodności niż normy zharmonizowane, np. możliwość stosowania europejskiego programu oznakowania ekologicznego przewidzianego w dyrektywie w sprawie ekoprojektu; w sektorze wyrobów medycznych używanych do diagnostyki in vitro zgodność z tzw. „wspólnymi specyfikacjami technicznymi” (CTS – ang. common technical specifications) powoduje domniemanie zgodności ze stosownymi zasadniczymi wymaganiami. Innym przykładem jest odniesienie do dokumentów normatywnych Międzynarodowej Organizacji Metrologii Prawnej w dyrektywie 2004/22/WE w sprawie przyrządów pomiarowych.
(188) Zob. np. dyrektywa dotycząca urządzeń spalających paliwa gazowe.
(189) Komitet państw członkowskich na podstawie rozporządzenia (UE) nr 1025/2012 oraz, jeśli przewidziano, komitet sektorowy.
(190) W przypadku rozporządzenia (WE) nr 552/2004 w sprawie interoperacyjności Europejskiej Sieci Zarządzania Ruchem Lotniczym, jeśli producent decyduje się nie stosować norm zharmonizowanych, deklaracja nosi nazwę deklaracji przydatności do stosowania.
(191) Taka niejasność może zdarzyć się np. gdy nazwa dystrybutora pojawia się na opakowaniu, a nazwa producenta na produkcie, który jest w środku.
(192) Znak towarowy to znak wyróżniający lub oznaka stosowana przez osobę prywatną, organizację biznesową lub inny podmiot gospodarczy, aby klienci rozpoznali, że dane produkty lub usługi pochodzą z jednego źródła, i aby odróżnić swoje produkty lub usługi od tych oferowanych przez inne podmioty. Znak towarowy jest rodzajem własności intelektualnej, zazwyczaj nazwą, słowem, frazą, logo, symbolem, projektem, obrazem lub kombinacją tych elementów.
(193) Art. R2 ust. 6 załącznika I do decyzji nr 768/2008/WE.
(194) Art. R4 ust. 3 załącznika I do decyzji nr 768/2008/WE.
(195) Art. R2 ust. 5 załącznika I do decyzji nr 768/2008/WE.
(196) Art. R7 załącznika I do decyzji nr 768/2008/WE.
(197) Zob. motyw 25 decyzji nr 768/2008/WE.
(198) Np. adres służący jako punkt informacyjny dla konsumentów i innych użytkowników w państwie członkowskim, w którym produkt jest udostępniany.
(199) W sektorze wyrobów medycznych na produktach musi znaleźć się także nazwa i adres upoważnionego przedstawiciela.
(200) Jeśli importer umieszcza tylko swoją nazwę i adres, zostawiając przy tym znak towarowy oryginalnego producenta, nadal uważa się go za importera. Adresy importera i producenta pojawią się na produkcie (ew. na opakowaniu lub w dołączonej dokumentacji).
(201) Dzieje się tak również w przypadku, gdy producent i importer należą do tej samej grupy spółek, a przedsiębiorstwo mające siedzibę w UE importujące produkt do UE przyjmuje pełną odpowiedzialność producenta za produkt.
(202) W przypadku zabawek może się tak zdarzyć, jeśli dana zabawka składa się z kilku części lub stanowi zestaw kilki elementów.
(203) Wprowadzanie do obrotu – zob. rozdział 2.3.
(204) Zgodnie z dyrektywami w sprawie wyrobów medycznych i wyrobów medycznych do diagnostyki in vitro dokumenty te muszą być przechowywane przez 5 lat, a w przypadku wyrobów medycznych aktywnego osadzania – przez 15 lat.
(205) Zob. dyrektywy w sprawie prostych zbiorników ciśnieniowych, maszyn (moduł B), wag nieautomatycznych, wyrobów medycznych aktywnego osadzania, urządzeń gazowych, końcowych urządzeń telekomunikacyjnych, wyrobów medycznych, przestrzeni zagrożonych wybuchem, dźwigów (moduły B, C, D, G, H), urządzeń ciśnieniowych, wyrobów medycznych do diagnostyki in vitro oraz urządzeń radiowych i końcowych urządzeń telekomunikacyjnych.
(206) Należy zwrócić uwagę, że dyrektywa w sprawie maszyn 2006/42/WE przewiduje wprowadzanie do obrotu „maszyn nieukończonych” z towarzyszącą im tzw. deklaracją włączenia, która różni się od deklaracji zgodności UE. Zgodnie z rozporządzeniem (WE) nr 552/2004, do elementów Europejskiej Sieci Zarządzania Ruchem Lotniczym dołączana jest deklaracja zgodności albo deklaracja przydatności do stosowania.
(207) Więcej informacji na temat dokumentacji produktu – zob. pkt 4.3.
(208) Zgodnie z dyrektywami w sprawie wyrobów medycznych i wyrobów medycznych do diagnostyki in vitro deklaracje zgodności muszą być przechowywane przez 5 lat, a w przypadku wyrobów medycznych aktywnego osadzania – przez 15 lat.
(209) Obowiązki producenta, uprawnionego przedstawiciela i importera – zob. rozdział 3.
(210) „Numer” może być również kodem alfanumerycznym.
(211) Dodatkowo, bez względu na to, czy przewidziano to wyraźnie w unijnym prawodawstwie harmonizacyjnym, czy nie, producenci mogą wedle własnego uznania samodzielnie dodać numer identyfikacyjny deklaracji zgodności UE zgodnie z EN ISO/IEC 17050-2.
(212) Nie wszystkie unijne przepisy harmonizacyjne wymagają interwencji jednostki notyfikowanej. Np. nie ma tego wymagania w dyrektywie niskonapięciowej i dyrektywie w sprawie zabawek.
(213) Niektóre unijne przepisy harmonizacyjne mogą wymagać podania nazwy i adresu osoby przechowującej dokumentację produktu, ponieważ zgodnie z nimi dokumenty powinny pozostawać nie tylko w dyspozycji producenta.
(214) Może to być dyrektor zarządzający firmy lub inny przedstawiciel firmy, któremu powierzono ten obowiązek.
(215) Sygnatariusz nie musi mieć siedziby w Unii Europejskiej. Producent posiada siedzibę poza Unią jest uprawniony do przeprowadzenia procedur oceny zgodności na terenie swojego przedsiębiorstwa i podpisania deklaracji zgodności UE, chyba że inaczej przewidziano w stosownym unijnym prawodawstwie harmonizacyjnym.
(216) Art. 5 decyzji nr 768/2008/WE.
(217) Zob. np. motyw 22 dyrektywy 2014/35/UE lub podobny motyw 24 dyrektywy 2014/34/UE.
(218) Art. R10 ust. 2 załącznika I do decyzji nr 768/2008/WE.
(219) Ocena zgodności według prawodawstwa dotyczącego wyrobów budowlanych nie jest zgodna z decyzją nr 768/2008/WE, chociaż przewiduje przyznawanie oznakowania CE. Różnica polega na tym, że oznakowanie CE zgodne z przepisami dotyczącymi wyrobów budowlanych określa poziom wydajności wyrobu, a nie zgodności z przepisami w ścisłym tego słowa znaczeniu, co ma miejsce w przypadku innych aktów prawnych regulujących kwestię oznakowania CE.
(220) Takich jak przepisy dotyczące maszyn, środków ochrony indywidualnej, wyrobów medycznych aktywnego osadzania, wyrobów medycznych, atmosfery potencjalnie wybuchowej, dźwigów – w przypadku komponentów bezpieczeństwa, wyrobów medycznych do diagnostyki in vitro, urządzeń radiowych i końcowych urządzeń telekomunikacyjnych lub wyposażenia statków.
(221) Należy zauważyć, że w przypadku gdy produkt podlega przepisom kilku unijnych aktów harmonizacyjnych, a oznakowanie CE pojawia się wraz z numerem identyfikacyjnym, nie oznacza to, że jednostka notyfikowana uczestniczy w procesie oceny zgodności wymaganym przez każdy z takich aktów. Niektóre unijne przepisy harmonizacyjne mogą nie wymagać interwencji jednostki notyfikowanej.
(222) Rozporządzenie (WE) nr 552/2004 w sprawie interoperacyjności Europejskiej Sieci Zarządzania Ruchem Lotniczym nie przewiduje oznakowania CE.
(223) Typowym przykładem może być komputer.
(224) Zob. pkt 4.5.1.7 i 4.5.2.
(225) Zgodnie z dyrektywą w sprawie ekoprojektu procedury oceny zgodności (określone w środku wykonawczym) są określone w samej dyrektywie, ale w dobrze uzasadnionych przypadkach stosuje się moduły z decyzji nr 768/2008/WE.
(226) Stosownymi modułami są moduły C, C1, C2, D, E i F.
(227) Systemy zarządzania jakością – podstawy i słownictwo.
(228) Systemy zarządzania jakością – wymagania.
(*) Prawodawca może ograniczyć wybór producenta.
(229) Zlecanie prac podwykonawcy przez jednostki notyfikowane, zob rozdział 5.2.5.
(230) Zob. załącznik II do decyzji nr 768/2008/WE, moduł B, pkt 8, akapit trzeci.
(231) Więcej informacji dotyczących numeru jednostki notyfikowanej w NANDO, pkt 5.3.3.
(232) Art. R27 ust. 4 załącznika I do decyzji nr 768/2008/WE.
(233) Umowy o wzajemnym uznawaniu, zob. rozdział 9.2.
(234) Nadzór rynku – zob. rozdział 7.
(235) Jest to częstą praktyką w niektórych sektorach (np. sektorze materiałów wybuchowych i artykułów pirotechnicznych), że organy nadzoru rynku powołują się na badania jednostek notyfikowanych, o ile nie zachodzi konflikt interesów.
(236) Koordynacja między jednostkami notyfikowanymi, zob. pkt 5.2.4.
(237) Artykuł R17 ust. 11 w załączniku I do decyzji nr 768/2008/WE.
(238) Rola i obowiązki jednostek notyfikowanych, zob. pkt 5.2.2.
(239) Art. R20 ust. 4 załącznika I do decyzji nr 768/2008/WE.
(240) Należy zwrócić uwagę, iż tylko nieliczne unijne przepisy harmonizacyjne przewidują akredytowane jednostki własne.
(241) Zob. załącznik VI zawierający szczegółowe informacje o normach zharmonizowanych, wg których organy wewnętrzne muszą być akredytowane, zależnie od danego modułu.
(242) Unijne prawodawstwo harmonizacyjne dostosowane do decyzji nr 768/2008/WE zawiera zmienione przepisy dotyczące jednostek notyfikowanych. Jeśli chodzi o organy notyfikujące w rozumieniu tych przepisów, konieczne jest, by przynajmniej odpowiednie przepisy dotyczące jednostek notyfikowanych (obejmujące w szczególności wymogi i obowiązki tych jednostek) zostały przetransponowane do prawa krajowego.Ponadto procedury notyfikacyjne należy przekazać Komisji i innym państwom członkowskim, a państwa członkowskie muszą wyznaczyć organ notyfikujący dla danych aktów unijnego prawodawstwa harmonizacyjnego.
(243) Art. 7 rozporządzenia określa to jako sytuacje, w której jednostka oceniająca zgodność może ubiegać się o akredytację poza państwem członkowskim, w którym jest ustanowiona.
(244) Podobny przepis znajduje się w większości dyrektyw dostosowanych do decyzji nr 768/2008/WE.
(245) Rola EA, zob. pkt 6.5.2 oraz 6.5.4.
(246) Więcej informacji na temat wycofania oraz odebrania notyfikacji, zob. pkt 5.3.4.
(247) Zgodnie z art. R26 załącznika I decyzji nr 768/2008/WE.
(248) Pierwotnie seria norm EN 45000, która została zrewidowana i zastąpiona serią norm EN ISO/IEC 17000.
(249) Zbiór procedur oceny zgodności, które mają być wykorzystywane w unijnym prawodawstwie harmonizacyjnym został po raz pierwszy określony w decyzji Rady nr 93/465/EWG (tzw. „decyzji dotyczącej modułów”).
(250) Na stronie internetowej NANDO: http://ec.europa.eu/enterprise/newapproach/nando/ oraz na stronie internetowej EA: http://www.european-accreditation.org/
(251) Art. R14 ust. 2 załącznika I do decyzji nr 768/2008/WE.
(252) Większość unijnego prawodawstwa harmonizacyjnego zgodnego z decyzją (WE) nr 768/2008 zawiera przepis stanowiący, że organ notyfikujący może przekazać zadania notyfikacji pod pewnymi warunkami. W takim przypadku może on powierzyć notyfikację akredytowanych jednostek oceniających zgodność krajowej jednostce akredytującej, natomiast organ notyfikujący powinien notyfikować nieakredytowane jednostki oceniające zgodność (gdyby zadecydował o utrzymaniu nieakredytowanych notyfikacji). System taki wymagałby dobrej koordynacji wewnętrznej w państwie członkowskim.
(253) Akredytacja transgraniczna, zob. pkt 6.6.
(254) Art. 11 ust. 2 rozporządzenia (WE) nr 765/2008.
(255) ISO/IEC 17011.
(256) Najnowsze informacje są dostępne pod adresem www.ilac.org i www.iaf.nu, gdzie można znaleźć wykazy obecnych regionalnych członków ILAC oraz IAF.
(257) Dyrektywa w sprawie ogólnego bezpieczeństwa produktów również zawiera wymagania dotyczące nadzoru rynku. Związek między rozporządzeniem (WE) nr 765/2008 a dyrektywą w sprawie ogólnego bezpieczeństwa produktów został szczegółowo opisany w dokumencie roboczym z dnia 3 marca 2010 r. dostępnym pod adresem: http://ec.europa.eu/consumers/safety/prod_legis/docs/20100324_guidance_gspd_reg_en.pdf.
(258) Zgodnie z określonymi unijnymi przepisami harmonizacyjnymi.
(259) Zgodnie z art. 16 rozporządzenia (WE) nr 765/2008 „nadzór rynku gwarantuje, że objęte wspólnotowym prawodawstwem harmonizacyjnym produkty, które przy zastosowaniu zgodnie z ich przeznaczeniem i w przewidywalnych warunkach oraz przy właściwym montażu i konserwacji zagrażają zdrowiu i bezpieczeństwu użytkowników lub które są niezgodne ze stosownymi wymaganiami ustanowionymi we wspólnotowym prawodawstwie harmonizacyjnym, zostaną wycofane z obrotu lub ich udostępnianie na rynku zostanie zakazane lub ograniczone, a opinia publiczna, Komisja i pozostałe państwa członkowskie zostaną należycie o tym poinformowane”. Państwa członkowskie zapewniają możliwość podjęcia skutecznych działań w odniesieniu do każdej kategorii produktów podlegającej unijnemu prawodawstwu harmonizacyjnemu.
(260) W takim przypadku oczekuje się, że organ wprowadzi niezbędne środki (np. stosując zasadę „murów chińskich”) w celu zachowania obiektywizmu i bezstronności podczas kontroli produktów po ich wprowadzeniu do obrotu.
(261) Jest to ważne w przypadku produktów (na przykład maszyn lub urządzeń ciśnieniowych), które bezpośrednio po ich wyprodukowaniu są instalowane oraz uruchamiane u klienta.
(262) Zgodnie z dyrektywą w sprawie systemów kolei dużych prędkości każde państwo członkowskie autoryzuje oddanie podsystemów strukturalnych do użytku na swoim terytorium. Jest to systematyczny mechanizm monitorowania zgodności podsystemów oraz ich składników interoperacyjności.
(263) Zgodnie z dyrektywą w sprawie wprowadzenia środków w celu poprawy bezpieczeństwa i zdrowia pracowników w miejscu pracy (89/391/EWG) państwa członkowskie są zobowiązane do zapewnienia stosownej kontroli i nadzoru.
(264) Zgodnie z decyzją nr 768/2008/WE, moduł B, jednostki notyfikowane są zobowiązane do przedstawienia kopii dokumentacji produktu na żądanie państw członkowskich, Komisji Europejskiej lub innych jednostek notyfikowanych.
(265) Chyba że deklaracja zgodności UE musi towarzyszyć produktowi; w takim przypadku dystrybutor powinien przedstawić taki dokument organom nadzoru rynku.
(266) Uzasadnione żądanie niekoniecznie oznacza formalną decyzję organu. Zgodne z art. 19 ust. 1 akapit drugi rozporządzenia (WE) nr 765/2008 „organy nadzoru rynku mogą wymagać udostępnienia przez podmioty gospodarcze dokumentów i informacji, które w ich ocenie są niezbędne do celów prowadzenia przez nie działań”. Aby żądanie było uzasadnione, wystarczy, że organ nadzoru rynku wyjaśni kontekst, w jakim wymagane są informacje (np. kontrola dotycząca szczególnych cech produktów, wyrywkowe kontrole itp.).
(267) Przykładowo przechowywanie dokumentacji produktu można zlecić upoważnionemu przedstawicielowi.
(268) Wytyczne te są dostępne pod adresem: http://ec.europa.eu/taxation_customs/resources/documents/common/publications/info_docs/customs/product_safety/guidelines_pl.pdf
(269) Jeżeli po odmowie dopuszczenia do obrotu produkty zostały zgłoszone do procedury celnej lub objęte innym przeznaczeniem celnym niż dopuszczenie do swobodnego obrotu oraz pod warunkiem, że organy nadzoru rynku nie zgłaszają sprzeciwu, na dokumentach związanych z tą procedurą lub przeznaczeniem należy umieścić tę samą informację na tych samych warunkach.
(270) System RAPEX, zob. pkt 7.5.2.
(271) Również w tym przypadku, jeżeli po odmowie dopuszczenia do obrotu produkty zostały zgłoszone do procedury celnej lub objęte innym przeznaczeniem celnym niż dopuszczenie do swobodnego obrotu oraz pod warunkiem, że organy nadzoru rynku nie zgłaszają sprzeciwu, na dokumentach związanych z tą procedurą lub przeznaczeniem należy umieścić tę samą informację na tych samych warunkach.
(272) Wykaz organów nadzoru rynku wyznaczonych przez państwa członkowskie można znaleźć na stronie: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(273) Podobny przepis można znaleźć w dyrektywie w sprawie ogólnego bezpieczeństwa produktów.
(274) Krajowe programy nadzoru rynku są publicznie dostępne pod adresem: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(275) Krajowe przeglądy i oceny można znaleźć pod adresem: http://ec.europa.eu/growth/single-market/goods/building-blocks/market-surveillance/organisation/index_en.htm
(276) Zob. dyrektywa w sprawie ogólnego bezpieczeństwa produktów, motywy 24 i 35 oraz art. 16; zob. też art. 19 ust. 5 rozporządzenia (WE) nr 765/2008.
(277) Zob. bardziej szczegółowe definicje „zagrożenia” i „poważnego zagrożenia” w wytycznych dotyczących wspólnotowego systemu szybkiej informacji (RAPEX).
(278) http://ec.europa.eu/consumers/archive/safety/rapex/docs/corrective_action_guide_march2012.pdf
(279) Zob. dyrektywy dotyczące prostych zbiorników ciśnieniowych, zabawek, maszyn, wyposażenia ochrony osobistej, wag nieautomatycznych, wyrobów medycznych aktywnego osadzania, urządzeń gazowych, przestrzeni zagrożonych wybuchem, wyrobów medycznych, rekreacyjnych jednostek pływających, dźwigów, urządzeń chłodniczych, urządzeń ciśnieniowych, wymogów ekoprojektu dla produktów związanych z energią oraz wyrobów medycznych używanych do diagnostyki in vitro.
(280) Jednoznaczny przepis dotyczący konsultacji został zawarty w art. 21 rozporządzenia (WE) nr 765/2008, jak również w dyrektywach dotyczących wyrobów medycznych oraz wyrobów medycznych używanych do diagnostyki in vitro.
(281) Procedury klauzuli ochronnej w ramach unijnego prawodawstwa harmonizacyjnego stosuje się niezależnie od wspólnotowego systemu szybkiej informacji. Dlatego też nie jest konieczne, aby uruchamiać wspólnotowy system szybkiej informacji przed zastosowaniem procedury klauzuli ochronnej. Jednakże procedura klauzuli ochronnej musi zostać zastosowana w dodatku do wspólnotowego systemu szybkiej informacji, jeżeli państwo członkowskie podejmie decyzję o trwałym zakazie lub ograniczeniu swobodnego przepływu zharmonizowanych produktów na podstawie zagrożenia lub innego poważnego zagrożenia stwarzanego przez produkt.
(282) Nawet jeśli nie mogą ustanowić klauzuli ochronnej, organy nadzoru rynku powiadamiają Komisję i inne państwa członkowskie o działaniach podjętych wobec produktów niezgodnych z wymogami, jeżeli niezgodność nie ogranicza się wyłącznie do terytorium kraju (zob. art. R31 ust. 2 załącznika I do decyzji nr 768/2008/WE).
(283) Powiadomienie to należy przesyłać za pośrednictwem ICSMS. Powiązanie między bazą danych ICSMS i narzędziem informatycznym GRS RAPEX pozwoli uniknąć podwójnego rejestrowania informacji przez organy krajowe do celów, odpowiednio, procedury klauzuli ochronnej i szybkiego informowania zgodnie z art. 22 rozporządzenia (WE) nr 765/2008.
(284) Unijne przepisy harmonizacyjne dostosowane do decyzji nr 768/2008/WE przewidują procedurę ochronną, która ma zastosowanie wyłącznie w przypadku nieporozumienia między państwami członkowskimi dotyczącego środków podjętych przez państwo członkowskie. Ma to na celu zagwarantowanie, że, w przypadku gdy na terytorium państwa członkowskiego znajduje się niezgodny produkt, stosuje się odpowiednie środki oraz że podobne podejścia stosuje się w różnych państwach członkowskich. W przeszłości notyfikacja o zagrożeniu oznaczała, że Komisja musiała otworzyć sprawę i opracować opinię, natomiast obecnie to obciążenie zostało zniesione, a sprawa ochronna jest otwierana wyłącznie, gdy państwo członkowskie lub Komisja ma zastrzeżenia do środka zastosowanego przez organ notyfikujący. Jeżeli państwa członkowskie oraz Komisja zgadzają się co do zasadności środka zastosowanego przez państwo członkowskie, dalsze zaangażowanie Komisji nie jest wymagane, z wyjątkiem sytuacji, gdy niezgodność jest związana z niedociągnięciami w normie zharmonizowanej.
(285) Wyraźny obowiązek współpracy administracyjnej jest zawarty w dyrektywach dotyczących urządzeń ciśnieniowych oraz wyrobów medycznych używanych do diagnostyki in vitro: państwa członkowskie są zobowiązane do podejmowania odpowiednich środków, aby zachęcić organy odpowiedzialne za wdrażanie dyrektywy do współpracy i zapewnić, że będą ze sobą współpracować i przekazywać sobie nawzajem (oraz Komisji) informacje, aby wesprzeć funkcjonowanie dyrektywy.
(286) System ICSMS, zob. pkt 7.5.3.
(287) Zob. wyrok Trybunału, sprawy 272/80 oraz 25/88.
(288) Wniosek o udzielenie informacji nie ogranicza prawa organu krajowego do podjęcia wszelkich niezbędnych działań w celu zapewnienia zgodności z unijnym prawodawstwem harmonizacyjnym w ramach jego jurysdykcji.
(289) Na mocy art. 22 rozporządzenia (WE) nr 765/2008 system szybkiej informacji stosuje się do produktów objętych unijnym prawodawstwem harmonizacyjnym.
(290) W obszarze wyrobów oraz urządzeń medycznych istnieje specjalny system wymiany informacji.
(291) Przyjętych jako decyzja Komisji 2010/15/UE z dnia 16 grudnia 2009 r. ustanawiająca wytyczne dotyczące zarządzania wspólnotowym systemem szybkiej informacji „RAPEX” utworzonym na mocy art. 12 oraz procedurą zgłoszeniową ustanowioną na mocy art. 11 dyrektywy 2001/95/WE (dyrektywa w sprawie ogólnego bezpieczeństwa produktów) (Dz.U. L 22 z 26.1.2010, s. 1). Komisja jest w trakcie opracowywania ogólnounijnej metodyki oceny ryzyka, która opiera się na wytycznych RAPEX opracowanych na podstawie GPSD i rozszerza oceny ryzyka na produkty, które mogą zagrozić zdrowiu i bezpieczeństwu użytkowników profesjonalnych lub innym interesom publicznym.
(292) Decyzja 2010/15/UE jest dostępna pod adresem: http://ec.europa.eu/consumers/archive/safety/rapex/docs/rapex_guid_26012010_pl.pdf
(293) General Rapid Alert System (ogólny system szybkiego ostrzegania) dla zgłoszeń RAPEX. GRAS-RAPEX zastąpił RAPEX-REIS (system szybkiej wymiany informacji dla aplikacji systemu szybkiej informacji) oraz rozszerzył zakres stosowania systemu szybkiej informacji na produkty specjalistyczne i czynniki ryzyka inne niż zagrożenia dla zdrowia i bezpieczeństwa.
(294) http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm
(295) Zob. dyrektywy w sprawie wyrobów medycznych aktywnego osadzania, wyrobów medycznych oraz wyrobów medycznych do diagnostyki in vitro.
(296) Niniejszy rozdział dotyczy wyłącznie produktów objętych unijnymi przepisami harmonizacyjnymi. Swobodny przepływ produktów nieobjętych unijnym prawodawstwem harmonizacyjnym jest opisany w „Przewodniku stosowania postanowień traktatowych regulujących swobodny przepływ towarów” dostępnym pod adresem: http://ec.europa.eu/DocsRoom/documents/104
(297) Bardziej szczegółowy opis procedur, które należy zastosować w przypadku produktów stwarzających zagrożenie dla zdrowia i bezpieczeństwa osób lub innych aspektów ochrony interesów publicznych przedstawiono w rozdziale 7.
(298) Porozumienie o Europejskim Obszarze Gospodarczym, zob. pkt 2.8.2.
(299) Kwestie związane z porozumieniem WTO wykraczają poza zakres niniejszego przewodnika.
(300) Pełen tekst MRA między UE a Szwajcarią oraz przepisy szczególne można znaleźć na stronie głównej Komisji: http://ec.europa.eu/growth/single-market/goods/international-aspects/mutual-recognition-agreements/index_en.htm
ZAŁĄCZNIK I
UNIJNE AKTY PRAWNE, O KTÓRYCH MOWA W NINIEJSZYM PRZEWODNIKU (NIEWYCZERPUJĄCA LISTA)
|
Horyzontalny unijny akt harmonizacyjny |
Numer (zmiana) |
Odniesienie w Dzienniku Urzędowym Unii Europejskiej |
|
Rozporządzenie Parlamentu Europejskiego i Rady (WE) nr 765/2008 z dnia 9 lipca 2008 r. ustanawiające wymagania w zakresie akredytacji i nadzoru rynku odnoszące się do warunków wprowadzania produktów do obrotu i uchylające rozporządzenie (EWG) nr 339/93 |
(WE) nr 765/2008 |
|
|
Decyzja Parlamentu Europejskiego i Rady nr 768/2008/WE z dnia 9 lipca 2008 r. w sprawie wspólnych ram dotyczących wprowadzania produktów do obrotu |
768/2008/WE |
|
|
Rozporządzenie Parlamentu Europejskiego i Rady (WE) nr 764/2008 z dnia 9 lipca 2008 r. ustanawiające procedury dotyczące stosowania niektórych krajowych przepisów technicznych do produktów wprowadzonych legalnie do obrotu w innym państwie członkowskim oraz uchylające decyzję nr 3052/95/WE |
(WE) nr 764/2008 |
|
|
Dyrektywa Rady 85/374/EWG z dnia 25 lipca 1985 r. w sprawie zbliżenia przepisów ustawowych, wykonawczych i administracyjnych państw członkowskich dotyczących odpowiedzialności za produkty wadliwe |
85/374/EWG (1999/34/WE) |
|
|
Dyrektywa 2001/95/WE Parlamentu Europejskiego i Rady z dnia 3 grudnia 2001 r. w sprawie ogólnego bezpieczeństwa produktów |
2001/95/WE |
|
|
Rozporządzenie Parlamentu Europejskiego i Rady (UE) nr 1025/2012 z dnia 25 października 2012 r. w sprawie normalizacji europejskiej |
(UE) nr 1025/2012 |
|
Sektorowy unijny akt harmonizacyjny |
Numer (zmiana) |
Odniesienie w Dzienniku Urzędowym Unii Europejskiej |
|
Dyrektywa Rady z dnia 19 lutego 1973 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do sprzętu elektrycznego przewidzianego do stosowania w określonych granicach napięcia |
73/23/EWG 93/68/EWG 2006/95/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/35/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku sprzętu elektrycznego przewidzianego do stosowania w określonych granicach napięcia |
2014/35/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2009/48/WE z dnia 18 czerwca 2009 r. w sprawie bezpieczeństwa zabawek |
2009/48/WE |
|
|
Dyrektywa Rady 89/336/EWG z dnia 3 maja 1989 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do kompatybilności elektromagnetycznej |
89/336/EWG 92/31/EWG 93/68/EWG 2004/108/WE (98/13/WE) |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/30/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do kompatybilności elektromagnetycznej (wersja przekształcona) |
2014/30/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady z dnia 22 czerwca 1998 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do maszyn |
98/37/WE 98/79/WE |
|
|
Dyrektywa Rady z dnia 21 grudnia 1989 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do wyposażenia ochrony osobistej |
89/686/EWG 93/68/EWG 93/95/EWG 96/58/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2009/23/WE z dnia 23 kwietnia 2009 r. w sprawie wag nieautomatycznych |
90/384/EWG 93/68/EWG 2009/23/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/31/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku wag nieautomatycznych (wersja przekształcona) |
2014/31/UE |
|
|
Dyrektywa 2004/22/WE Parlamentu Europejskiego i Rady z dnia 31 marca 2004 r. w sprawie przyrządów pomiarowych |
2004/22/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/32/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku przyrządów pomiarowych (wersja przekształcona) |
2014/32/UE |
|
|
Dyrektywa Rady 93/42/EWG z dnia 14 czerwca 1993 r. dotycząca wyrobów medycznych |
93/42/EWG 98/79/WE 2000/70/WE 2001/104/WE 2007/97/WE |
|
|
Dyrektywa Rady z dnia 20 czerwca 1990 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do wyrobów medycznych aktywnego osadzania |
90/385/EWG 93/42/EWG 93/68/EWG |
|
|
Dyrektywa 98/79/WE Parlamentu Europejskiego i Rady z dnia 27 października 1998 r. w sprawie wyrobów medycznych używanych do diagnozy in vitro |
98/79/WE |
|
|
Dyrektywa Rady 90/396/EWG z dnia 29 czerwca 1990 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do urządzeń spalania paliw gazowych |
90/396/EWG 93/68/EWG 09/142/WE |
|
|
Dyrektywa Rady z dnia 5 kwietnia 1993 r. w sprawie harmonizacji przepisów dotyczących wprowadzania do obrotu i kontroli materiałów wybuchowych przeznaczonych do użytku cywilnego |
93/15/EWG |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/28/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku i kontroli materiałów wybuchowych przeznaczonych do użytku cywilnego (wersja przekształcona) |
2014/28/UE |
|
|
Dyrektywa 2007/23/WE Parlamentu Europejskiego i Rady z dnia 23 maja 2007 r. w sprawie wprowadzania do obrotu wyrobów pirotechnicznych |
2007/23/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2013/29/UE z dnia 12 czerwca 2013 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku wyrobów pirotechnicznych (wersja przekształcona) |
2013/29/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 94/9/WE z dnia 23 marca 1994 r. w sprawie zbliżenia ustawodawstw państw członkowskich dotyczących urządzeń i systemów ochronnych przeznaczonych do użytku w przestrzeniach zagrożonych wybuchem |
94/9/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/34/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do urządzeń i systemów ochronnych przeznaczonych do użytku w atmosferze potencjalnie wybuchowej (wersja przekształcona) |
2014/34/UE |
|
|
Dyrektywa 94/25/WE Parlamentu Europejskiego i Rady z dnia 16 czerwca 1994 r. w sprawie zbliżenia przepisów ustawowych, wykonawczych i administracyjnych Państw Członkowskich odnoszących się do rekreacyjnych jednostek pływających |
94/25/WE 03/44/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2013/53/UE z dnia 20 listopada 2013 r. w sprawie rekreacyjnych jednostek pływających i skuterów wodnych i uchylająca dyrektywę 94/25/WE |
2013/53/UE |
|
|
Dyrektywa 95/16/WE Parlamentu Europejskiego i Rady z dnia 29 czerwca 1995 r. w sprawie zbliżenia ustawodawstw państw członkowskich dotyczących dźwigów |
95/16/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/33/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich dotyczących dźwigów i elementów bezpieczeństwa do dźwigów (wersja przekształcona) |
2014/33/UE |
|
|
Dyrektywa 2000/9/WE Parlamentu Europejskiego i Rady z dnia 20 marca 2000 r. odnosząca się do urządzeń kolei linowych przeznaczonych do przewozu osób |
2000/9/WE |
|
|
Dyrektywa 97/23/WE Parlamentu Europejskiego i Rady z dnia 29 maja 1997 r. w sprawie zbliżenia ustawodawstw państw członkowskich dotyczących urządzeń ciśnieniowych |
97/23/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/68/UE z dnia 15 maja 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku urządzeń ciśnieniowych (wersja przekształcona) |
2014/68/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2009/105/WE z dnia 16 września 2009 r. odnosząca się do prostych zbiorników ciśnieniowych |
2009/105/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/29/UE z dnia 26 lutego 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich odnoszących się do udostępniania na rynku prostych zbiorników ciśnieniowych (wersja przekształcona) |
2014/29/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2010/35/UE z dnia 16 czerwca 2010 r. w sprawie ciśnieniowych urządzeń transportowych |
2010/35/WE |
|
|
Dyrektywa Rady z dnia 20 maja 1975 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do dozowników aerozoli |
75/324/EWG 94/1/WE 2008/47/WE |
|
|
Dyrektywa 99/5/WE Parlamentu Europejskiego i Rady w sprawie urządzeń radiowych i końcowych urządzeń telekomunikacyjnych oraz wzajemnego uznawania ich zgodności |
99/5/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/53/UE z dnia 16 kwietnia 2014 r. w sprawie harmonizacji ustawodawstw państw członkowskich dotyczących udostępniania na rynku urządzeń radiowych i uchylająca dyrektywę 1999/5/WE |
2014/53/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2009/125/WE z dnia 21 października 2009 r. ustanawiająca ogólne zasady ustalania wymogów dotyczących ekoprojektu dla produktów związanych z energią |
2009/125/WE |
|
|
Dyrektywa 97/68/WE Parlamentu Europejskiego i Rady z dnia 16 grudnia 1997 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do środków dotyczących ograniczenia emisji zanieczyszczeń gazowych i pyłowych z silników spalinowych montowanych w maszynach samojezdnych nieporuszających się po drogach |
97/68/WE 2002/88/WE 2004/26/WE 2006/105/WE 2010/26/UE 2011/88/UE 2012/46/UE |
|
|
Dyrektywa 2000/14/WE Parlamentu Europejskiego i Rady z dnia 8 maja 2000 r. w sprawie zbliżenia ustawodawstw państw członkowskich odnoszących się do emisji hałasu do środowiska przez urządzenia używane na zewnątrz pomieszczeń |
2000/14/WE 2005/88/WE 219/2009/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2011/65/UE z dnia 8 czerwca 2011 r. w sprawie ograniczenia stosowania niektórych niebezpiecznych substancji w sprzęcie elektrycznym i elektronicznym (RoHS) |
2011/65/UE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2012/19/UE z dnia 4 lipca 2012 r. w sprawie zużytego sprzętu elektrycznego i elektronicznego (WEEE) |
2012/19/UE |
|
|
Dyrektywa Rady 96/98/WE z dnia 20 grudnia 1996 r. w sprawie wyposażenia statków |
96/98/WE |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2014/90/UE z dnia 23 lipca 2014 r. w sprawie wyposażenia morskiego i uchylająca dyrektywę Rady 96/98/WE |
2014/90/UE |
|
|
Dyrektywa Rady 2008/57/WE z dnia 17 czerwca 2008 r. w sprawie interoperacyjności systemu kolei we Wspólnocie |
2008/57/WE 2009/131/WE 2011/18/UE 2013/9/UE |
|
|
Dyrektywa 94/62/WE Parlamentu Europejskiego i Rady z dnia 20 grudnia 1994 r. w sprawie opakowań i odpadów opakowaniowych |
94/62/WE 2004/12/WE 2005/20/WE |
|
|
Rozporządzenie (WE) nr 552/2004 Parlamentu Europejskiego i Rady z dnia 10 marca 2004 r. w sprawie interoperacyjności Europejskiej Sieci Zarządzania Ruchem Lotniczym |
(WE) nr 552/2004 (WE) nr 1070/2009 |
|
|
Dyrektywa Parlamentu Europejskiego i Rady 2010/30/UE z dnia 19 maja 2010 r. w sprawie wskazania poprzez etykietowanie oraz standardowe informacje o produkcie, zużycia energii oraz innych zasobów przez produkty związane z energią |
2010/30/UE |
|
|
Rozporządzenie Parlamentu Europejskiego i Rady (WE) nr 1222/2009 z dnia 25 listopada 2009 r. w sprawie etykietowania opon pod kątem efektywności paliwowej i innych zasadniczych parametrów |
(WE) nr 1222/2009 |
ZAŁĄCZNIK II
DODATKOWE WYTYCZNE
|
— |
Wytyczne Grupy Ekspertów ds. Bezpieczeństwa Zabawek: http://ec.europa.eu/growth/sectors/toys/safety/guidance/index_en.htm |
|
— |
Przyrządy pomiarowe i wagi nieautomatyczne: http://ec.europa.eu/growth/single-market/goods/building-blocks/legal-metrology/measuring-instruments/guidance-standards/index_en.htm |
|
— |
Chemikalia: http://echa.europa.eu/support/guidance |
|
— |
Dyrektywa niskonapięciowa – Wytyczne dotyczące stosowania i zalecenia: http://ec.europa.eu/growth/sectors/electrical-engineering/lvd-directive/index_en.htm |
|
— |
Kompatybilność elektromagnetyczna (EMC) – Wytyczne: http://ec.europa.eu/growth/sectors/electrical-engineering/emc-directive/index_en.htm |
|
— |
Urządzenia radiowe i końcowe urządzenia telekomunikacyjne (R&TTE) – Wytyczne: http://ec.europa.eu/growth/sectors/electrical-engineering/rtte-directive/index_en.htm |
|
— |
Wyroby medyczne – Dokumenty interpretacyjne: http://ec.europa.eu/growth/sectors/medical-devices/guidance/index_en.htm |
|
— |
Często zadawane pytania dotyczące rozporządzenia o wyrobach budowlanych (CPR): http://ec.europa.eu/growth/sectors/construction/product-regulation/faq/index_en.htm |
|
— |
Przemysł motoryzacyjny – Często zadawane pytania: http://ec.europa.eu/growth/sectors/automotive/index_en.htm |
|
— |
RoHS 2 – Często zadawane pytania: http://ec.europa.eu/environment/waste/rohs_eee/events_rohs3_en.htm |
|
— |
Dyrektywa w sprawie urządzeń ciśnieniowych (PED): wytyczne: http://ec.europa.eu/growth/sectors/pressure-gas/pressure-equipment/guidelines/index_en.htm |
|
— |
Maszyny – Dokumenty zawierające wytyczne: http://ec.europa.eu/growth/sectors/mechanical-engineering/machinery/index_en.htm |
|
— |
Dyrektywa w sprawie kolei linowych – Wytyczne dotyczące stosowania: http://ec.europa.eu/growth/sectors/mechanical-engineering/cableways/index_en.htm |
|
— |
Dyrektywa w sprawie dźwigów – Wytyczne dotyczące stosowania: http://ec.europa.eu/growth/sectors/mechanical-engineering/lifts/index_en.htm |
|
— |
Dyrektywa w sprawie wyposażenia ochrony osobistej – Wytyczne dotyczące stosowania: http://ec.europa.eu/growth/sectors/mechanical-engineering/personal-protective-equipment/index_en.htm |
|
— |
Dyrektywa w sprawie emisji hałasu do środowiska przez urządzenia używane na zewnątrz pomieszczeń – Wytyczne dotyczące stosowania, publikacje, badania: http://ec.europa.eu/growth/sectors/mechanical-engineering/noise-emissions/index_en.htm |
|
— |
Wytyczne dotyczące stosowania dyrektywy 94/9/WE z dnia 23 marca 1994 r. w sprawie zbliżenia ustawodawstw państw członkowskich dotyczących urządzeń i systemów ochronnych przeznaczonych do użytku w przestrzeniach zagrożonych wybuchem (czwarta edycja z września 2012 r.): http://ec.europa.eu/growth/sectors/mechanical-engineering/atex/index_en.htm |
|
— |
Branże związane z ochroną zdrowia – Często zadawane pytania: http://ec.europa.eu/growth/sectors/healthcare/index_en.htm |
|
— |
Przewodnik stosowania dyrektywy w sprawie ogólnego bezpieczeństwa produktów w praktyce: http://ec.europa.eu/consumers/safety/prod_legis/index_en.htm |
|
— |
Wytyczne dotyczące wspólnotowego systemu szybkiej informacji RAPEX: http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm |
|
— |
Normy europejskie – Ogólne ramy: http://ec.europa.eu/growth/single-market/european-standards/policy/framework/index_en.htm |
|
— |
Vademecum nt. normalizacji europejskiej wspierającej prawodawstwo i politykę Unii (SWD(2015) 205 final z 27.10.2015): http://ec.europa.eu/growth/single-market/european-standards/vademecum/index_en.htm |
ZAŁĄCZNIK III
PRZYDATNE ADRESY INTERNETOWE
|
— |
Jednolity rynek towarów http://ec.europa.eu/growth/single-market/goods/index_en.htm |
|
— |
Rynek wewnętrzny produktów http://ec.europa.eu/growth/single-market/goods/index_en.htm |
|
— |
Normy europejskie http://ec.europa.eu/growth/single-market/european-standards/index_en.htm |
|
— |
System wczesnego ostrzegania o niebezpiecznych produktach nieżywnościowych stanowiących poważne zagrożenie http://ec.europa.eu/consumers/consumers_safety/safety_products/rapex/index_en.htm |
ZAŁĄCZNIK IV
PROCEDURY OCENY ZGODNOŚCI (MODUŁY Z DECYZJI NR 768/2008/WE)
|
Moduły |
Producent |
Producent lub upoważniony przedstawiciel |
Jednostka oceniająca zgodność |
||||||||||||||||||||||||||||||||||||||||
|
A (wewnętrzna kontrola produkcji)
|
|
|
Brak zaangażowania jednostki oceniającej zgodność. Producent sam przeprowadza wszystkie kontrole, które wykonałaby jednostka notyfikowana |
||||||||||||||||||||||||||||||||||||||||
|
A1 (wewnętrzna kontrola produkcji oraz badanie produktów pod nadzorem)
|
|
|
Jednostka notyfikowana lub akredytowana jednostka własna (zależnie od wyboru producenta) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
A2 (wewnętrzna kontrola produkcji oraz nadzorowana kontrola produktów w losowych odstępach czasu)
|
|
|
Jednostka notyfikowana lub akredytowana jednostka własna (zależnie od wyboru producenta) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
B (badanie typu WE)
|
|
|
Jednostka notyfikowana
W tym zakresie prawodawca ustala następujące sposoby badania:
|
||||||||||||||||||||||||||||||||||||||||
|
C (zgodność z typem w oparciu o wewnętrzną kontrolę produkcji)
|
|
|
|
||||||||||||||||||||||||||||||||||||||||
|
C1 (zgodność z typem w oparciu o wewnętrzną kontrolę produkcji oraz badanie produktów pod nadzorem)
|
|
|
Jednostka notyfikowana lub akredytowana jednostka własna (zależnie od wyboru producenta) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
C2 (zgodność z typem WE w oparciu o wewnętrzną kontrolę produkcji oraz badanie produktów pod nadzorem w losowych odstępach czasu)
|
|
|
Jednostka notyfikowana lub akredytowana jednostka własna (zależnie od wyboru producenta) (*):
|
||||||||||||||||||||||||||||||||||||||||
|
D (zgodność z typem WE na podstawie systemu zapewnienia jakości procesu produkcji)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
D1 (zapewnienie jakości procesu produkcji)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
E (zgodność z typem WE w oparciu o zapewnienie jakości produktu)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
E1 (zapewnienie jakości kontroli i badania gotowych produktów)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
F (zgodność z typem WE w oparciu o weryfikację produktu)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
F1 (zgodność w oparciu o weryfikację produktu)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
G (zgodność w oparciu o weryfikację jednostkową)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
H (zgodność oparta na pełnym zapewnieniu jakości)
|
|
|
Jednostka notyfikowana
|
||||||||||||||||||||||||||||||||||||||||
|
H1 (zgodność oparta na pełnym zapewnieniu jakości oraz badaniu projektu)
|
|
|
Jednostka notyfikowana
|
(*) Prawodawca może ograniczyć wybór producenta.
ZAŁĄCZNIK V
ZALEŻNOŚĆ MIĘDZY ISO 9000 A MODUŁAMI WYMAGAJĄCYMI SYSTEMU ZAPEWNIENIA JAKOŚCI
|
Wymagania dotyczące jakości, o których mowa w modułach w decyzji 768/2008 |
Moduł D |
Moduł D1 |
Moduł E |
Moduł E1 |
Moduł H |
Moduł H1 |
||||
|
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
EN ISO 9001:2008, §5.1, §5.3, §5.4 (bez odniesienia do §7.1), §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
EN ISO 9001:2008, §5.1, §5.3, §5.4 (bez odniesienia do §7.1), §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
EN ISO 9001:2008, §5.1, §5.3, §5.4, §5.5, §5.6 (bez §5.6.2.b – informacja zwrotna od konsumentów) |
||||
|
nie dotyczy – moduł D nie obejmuje fazy projektu |
nie dotyczy – w ramach modułu D1 kwestie związane z projektem są objęte dokumentacją produktu |
nie dotyczy – moduł E nie obejmuje fazy projektu |
nie dotyczy – w ramach modułu E1 kwestie związane z projektem są objęte dokumentacją produktu |
EN ISO 9001:2008, §7.3.1, §7.3.2, §7.3.3 |
EN ISO 9001:2008, §7.3.1, §7.3.2, §7.3.3 |
||||
|
nie dotyczy – moduł D nie obejmuje fazy projektu |
nie dotyczy – w ramach modułu D1 kwestie związane z projektem są objęte dokumentacją produktu |
nie dotyczy – moduł E nie obejmuje fazy projektu |
nie dotyczy – w ramach modułu E1 kwestie związane z projektem są objęte dokumentacją produktu |
EN ISO 9001:2008, §7.3.4 – §7.3.7 |
EN ISO 9001:2008, §7.3.4 – §7.3.7 |
||||
|
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
nie dotyczy – moduł E nie obejmuje części wytwarzania |
nie dotyczy – moduł E1 nie obejmuje części wytwarzania |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
EN ISO 9001:2008, §7.5.1, §7.5.2, §7.5.3 |
||||
|
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
EN ISO 9001:2008, §7.6, §8.2.2, §8.2.3, §8.2.4 (tylko pierwszy punkt), §8.3, §8.4 (bez §8.4.a – zadowolenie klienta), §8.5 |
||||
|
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
EN ISO 9001:2008, §4 |
ZAŁĄCZNIK VI
STOSOWANIE NORM ZHARMONIZOWANYCH DO OCENY KOMPETENCJI JEDNOSTEK OCENIAJĄCYCH ZGODNOŚĆ
Należy zwrócić uwagę, że niniejszy załącznik przedstawia jedynie orientacyjne wskazówki. Nie ustanawia procedur oceniania kompetencji jednostek oceniających zgodność.
1. WYMAGANIA NAŁOŻONE NA JEDNOSTKI OCENIAJĄCE ZGODNOŚĆ
Jednostka oceniająca zgodność, która chce zostać notyfikowana na mocy unijnego aktu harmonizacyjnego w ramach jednego lub kilku modułów oceny zgodności zawartych w decyzji nr 768/2008/WE musi zostać poddana ocenie w celu zweryfikowania, czy jest przygotowana pod względem technicznym do wykonywania czynności wymaganych przez dany moduł.
Równie ważne jest prowadzenie ciągłego nadzoru kompetencji jednostki notyfikowanej. Nadzór musi być przeprowadzany w regularnych odstępach czasu i odbywać się zgodnie z praktyką ustanowioną przez organizacje akredytujące.
Proces oceny musi określić, czy jednostka oceniająca zgodność dysponuje należycie wykwalifikowaną kadrą posiadającą wiedzę i doświadczenie w zakresie stosownej technologii, odpowiednią infrastrukturą i sprzętem, posiada polityki i procedury zapewniające integralność i bezstronność, wykazuje prawidłowe zrozumienie dyrektywy itp.
Ocena zgodności obejmuje takie zadania jak badania (prowadzone przez laboratoria), inspekcje, certyfikacja itp. Inspekcję oraz certyfikację produktów można uważać za podobne czynności, ich definicje częściowo się pokrywają. Obie wykraczają poza zwyczajne badania, ponieważ obejmują zadania związane ze zdolnością dokonania oceny wyników testów oraz podjęcia decyzji o zgodności. Inspekcja i certyfikacja mają ten sam cel (tj. ocena zgodności produktu), który jest realizowany na trochę różne sposoby.
Zasadniczo inspekcja polega na bezpośrednim określeniu zgodności ze specyfikacją unikalnej lub małej serii produktów. Certyfikacja produktu polega przede wszystkim na określaniu zgodności produktów wytwarzanych w długich seriach.
W praktyce inspekcja może również obejmować profesjonalną ocenę w oparciu o ogólne wymagania, natomiast certyfikacja produktów odbywa się na podstawie norm lub innych specyfikacji technicznych.
Dlatego też różne kryteria mają zastosowanie do jednostek oceniających zgodność w zależności od tego, czy są one laboratoriami, jednostkami kontrolującymi/inspekcyjnymi, czy jednostkami certyfikującymi.
2. ZESTAW PODSTAWOWYCH NORM WYZNACZAJĄCYCH KRYTERIA KOMPETENCJI JEDNOSTEK OCENIAJĄCYCH ZGODNOŚĆ
Ogólne, niezależne od sektora kryteria, które jednostki notyfikowane muszą spełniać, aby uzyskać ocenę pozytywną są przedstawione w normach zharmonizowanych opublikowanych w Dzienniku Urzędowym Unii Europejskiej po wydaniu mandatu M417.
Przepisy sektorowe mogą, w razie potrzeby, ustanawiać dodatkowe szczegółowe kryteria dotyczące wiedzy związanej z sektorem, którą jednostka musi posiadać.
Normy EN ISO/IEC 17025, EN ISO/IEC 17020, EN ISO/IEC 17021 oraz EN ISO/IEC 17065 stanowią podstawowe standardy oceny kompetencji jednostek oceniających zgodność. Norma EN ISO/IEC 17020 oraz EN ISO/IEC 17065 skupia się na kryteriach przeprowadzania oceny zgodności, natomiast EN ISO/IEC 17025 dotyczy bardziej szczegółowo aspektu testowania.
|
— |
Norma EN ISO/IEC 17025 (dotyczy laboratoriów – zastępuje normę EN 45001 oraz Przewodnik ISO 25) przedstawia ogólne wymagania, które laboratorium (pierwszej, drugiej i trzeciej strony, niezależnie od liczby pracowników i zakresu działalności) musi spełniać, jeżeli ma zostać uznane za posiadające odpowiednie kwalifikacje do przeprowadzania testów oraz/lub wzorcowania, w tym do pobierania próbek (norma ISO 45001 nie obejmowała pobierania próbek). |
Czynności te obejmują określenie jednej lub kilku cech charakterystycznych produktu zgodnie z określoną metodą (standardową, niestandardową, opracowaną laboratoryjnie). Zgodność funkcjonowania laboratoriów z wymaganiami regulacyjnymi oraz wymaganiami bezpieczeństwa nie jest objęta tą normą.
Jeżeli laboratorium nie prowadzi jednej lub więcej czynności objętych tą normą międzynarodową, takich jak pobieranie próbek oraz projektowanie/opracowywanie nowych metod, wymagania zawarte w tych klauzulach nie mają zastosowania.
|
— |
Norma EN ISO/IEC 17020 (ma zastosowanie do jednostek kontrolujących/inspekcyjnych – zastępuje normę EN 45004). Ta norma określa ogólne kryteria dotyczące kompetencji bezstronnych jednostek przeprowadzających inspekcję niezależnie od przedmiotowego sektora. |
Kontrola obejmuje ocenę projektu produktu, produktu, usługi, procesu lub zakładu oraz określenie ich zgodności ze szczegółowymi wymaganiami lub, na podstawie profesjonalnej oceny, z wymaganiami ogólnymi. W normie wyszczególniono również kryteria niezależności. Dokument ten nie obejmuje laboratoriów badawczych, jednostek certyfikujących, ani deklaracji zgodności dostawcy.
|
— |
Norma EN ISO/IEC 17065 (ma zastosowanie do jednostek certyfikujących – zastępuje normę EN 45011) określa ogólne wymagania, które musi spełniać osoba trzecia prowadząca system certyfikacji produktów, jeżeli ma zostać uznana za kompetentną oraz wiarygodną. |
Certyfikacja produktu wiąże się z uzyskaniem pewności, że dany produkt spełnia określone wymagania takie jak przepisy, normy lub inne specyfikacje techniczne. System certyfikacji produktu może obejmować m.in. test lub badanie typu, test lub kontrolę każdego produktu lub konkretnego produktu, test lub kontrolę partii, bądź ocenę projektu, które mogą być połączone z nadzorem produkcji lub oceną i nadzorem systemu jakości producenta. Norma ta nie obejmuje laboratoriów badawczych, jednostek kontrolujących, ani deklaracji zgodności. Norma ISO/IEC 17065 określa zasadę stosowania dwóch par oczu, co oznacza, że kontroler i osoba podejmująca decyzję są innymi osobami niż oceniający.
|
— |
Norma ISO/IEC 17021 (zastępująca normę EN 45012) zawiera zasady i wymagania dotyczące kompetencji i bezstronności jednostek przeprowadzających audyt oraz certyfikację systemów zarządzania wszelkiego typu (np. systemów zarządzania jakością lub systemów zarządzania środowiskowego). |
Jednostki działające zgodnie z tą normą nie muszą oferować certyfikacji wszystkich typów systemów zarządzania. Certyfikacja systemu jakości obejmuje ocenę, określenie zgodności z normą systemu jakości w określonym zakresie działalności oraz nadzór systemu jakości producenta.
3. ODPOWIEDNIE NORMY ZWIĄZANE Z KOMPETENCJAMI JEDNOSTEK OCENIAJĄCYCH ZGODNOŚĆ DLA KAŻDEGO MODUŁU
W poniższych częściach opisano, które z przedstawionych powyżej norm są najbardziej odpowiednie do zadań w modułach ustanowionych w decyzji nr 768/2008/WE.
3.1 Moduły A1, A2, C1, C2
W ramach tych modułów jednostka musi posiadać wiedzę techniczną, doświadczenie oraz umiejętność przeprowadzania testów. Nawet jeżeli sprzęt do przeprowadzania testów znajduje się u producenta, wymagania dotyczące stosowności, działania, obsługi (np. programy wzorcowania) oraz spójności pomiarowej muszą być zapewnione, za co odpowiedzialność ponosi jednostka notyfikowana. Ponadto, jeżeli producent nie zastosował odpowiednich norm zharmonizowanych, należy przeprowadzić równoważne testy lub, w razie ich braku, opracować odpowiednie metody. W każdym przypadku jednostka notyfikowana musi zatwierdzić stosowane testy.
W przypadku modułu A2 i C2 jednostka musi dodatkowo być zdolna do wykorzystywania metod statystycznych, planu pobierania próbek, metod losowych oraz charakterystyk operacyjnych stanowiących część kontroli produktu oraz opisanych w stosownych unijnych przepisach harmonizacyjnych.
W tym zakresie oraz w odniesieniu do wszystkich tych modułów, tak, jak norma EN ISO/IEC 17025, EN ISO/IEC 17020 lub EN ISO/IEC 17065 (w zależności od tego, czy dana jednostka jest laboratorium, jednostką kontrolującą/inspekcyjną, czy jednostką certyfikującą produkty) ustanawia kryteria kompetencji i deontologii dotyczące przeprowadzania badania produktu, ich wymagania mogą być uznane za najbardziej odpowiednie do oceny jednostek dążących do uzyskania notyfikacji do przeprowadzania zadań zawartych w module.
Jednakże, jeżeli ocena jest oparta na EN ISO/IEC 17025, w związku z tym, że norma ta ustanawia jedynie kryteria testowania/wzorcowania bez dokonywania oceny wyników testów przez jednostkę notyfikowaną, jednostka ta musi oddzielnie wykazać swoją zdolność oraz procedury do oceniania i decydowania na podstawie wyników tych testów, czy zasadnicze lub inne wymagania prawne zostały spełnione oraz/lub czy zastosowano normy zharmonizowane.
Z drugiej strony, jeżeli stosuje się normę EN ISO/IEC 17020 lub EN ISO/IEC 17065, w związku z tym, że normy te nie obejmują kryteriów testowania/wzorcowania, należy uwzględnić wymagania dotyczące przeprowadzania testów określone w normie EN ISO/IEC 17025. We wszystkich przypadkach jednostka notyfikowana musi być zdolna do przeprowadzenia oceny produktu niezależnie od tego, czy producent zastosował stosowne normy zharmonizowane.
3.2 Moduł B
Jednostka notyfikowana musi określić, czy projekt produktu jest zgodny ze stosownymi wymaganiami prawnymi.
W tym zakresie jedynie normę EN ISO/IEC 17025 należy uznać za nieodpowiednią do celów modułu B, ponieważ norma ta dotyczy kwestii związanych jedynie z przeprowadzaniem testów, natomiast nie obejmuje ważnych funkcji modułu B dotyczących oceny projektu produktu, która ze względu na swoją złożoność (znacznie wykracza poza zwyczajne badanie dokumentacji produktu jak w module D1, E1 i F1) wymaga od jednostki notyfikowanej dodatkowych kompetencji (podobnie jak w module G i H1).
Wymagania zarówno normy EN ISO/IEC 17020, jak i EN ISO/IEC 17065 można uznać za odpowiednie do oceny jednostek dążących do uzyskania notyfikacji do przeprowadzania zadań zawartych w module B, ponieważ normy te ustanawiają kryteria kompetencji oraz deontologii dotyczące przeprowadzania badania produktu oraz oceny zgodności. Jednakże, w związku z tym, że normy te nie obejmują kryteriów testowania/wzorcowania, dla wymaganych testów należy zawsze uwzględnić stosowne wymagania zawarte w normie EN ISO/IEC 17025.
3.3 Moduły D, D1, E, E1, H
Jednostka notyfikowana ocenia i decyduje, czy system jakości producenta gwarantuje, że produkty są zgodne lub zapewniają zgodność z instrumentem legislacyjnym, który ma do nich zastosowanie (w przypadku modułów D1, E1, H) lub z zatwierdzonym typem WE (w przypadku modułów D, E).
W związku z tym wymagania normy EN ISO/IEC 17021 można uznać za odpowiednie do oceny jednostek dążących do uzyskania notyfikacji do przeprowadzania zadań zawartych w tym module. Należy podkreślić, że funkcjonowanie systemu jakości producenta musi zapewniać zgodność gotowych produktów z wymaganiami stosownych unijnych przepisów harmonizacyjnych. Dlatego też jednostka notyfikowana musi dodatkowo posiadać odpowiednią zdolność oceniania zdolności producenta do określania stosownych wymagań dla produktów oraz przeprowadzania niezbędnych kontroli i testów.
EN ISO/IEC 17065 jest również właściwa dla oceny modułów D, D1, E, E1, które oceniają zdolność systemu zarządzania producenta do zapewnienia zgodności produktów z odpowiednimi przepisami i utrzymania zgodności produktów z typem. Te moduły oceny są ściśle zorientowane na procesy produkcji i kontrole związanych z produktem(-ami), zatem wymagania EN ISO/IEC 17065 obejmują aspekty związane z procesem oraz produktem, a także ocenę systemu zarządzania (zgodnie z wymaganiami normy EN ISO/IEC 17065 ocena systemu zarządzania musi być przeprowadzana zgodnie z normą ISO/IEC 17021).
3.4 Moduły F, F1
Jednostka notyfikowana przeprowadza odpowiednie badania i testy w drodze badania i testowania każdego produktu lub badania i testowania produktów na zasadzie statystycznej. W ramach modułu F1 Jednostka notyfikowana musi dodatkowo zbadać dokumentację produktu.
W tym zakresie oraz w odniesieniu do wszystkich tych modułów, ponieważ norma EN ISO/IEC 17025, EN ISO/IEC 17020 lub EN ISO/IEC 17065 (w zależności, czy dana jednostka jest laboratorium, jednostką kontrolującą, czy jednostką certyfikującą produkty) ustanawia kryteria kompetencji i deontologii dotyczące przeprowadzania badania produktu, ich wymagania mogą być uznane za najbardziej odpowiednie do oceny jednostek dążących do uzyskania notyfikacji w ramach tych modułów.
Należy zauważyć, że mimo że norma EN ISO/IEC 17025 nie obejmuje badania projektu produktu oraz mimo że moduł F1 obejmuje również fazę projektu, norma ta jako jedyna pozostaje odpowiednia w odniesieniu do tego modułu, ponieważ badanie projektu w ramach modułu F1 jest stosunkowo proste i jest przeprowadzane wyłącznie w drodze badania dokumentacji produktu, a nie w drodze badania dowolnej próbki lub krytycznych części projektu, co wymagałoby dodatkowych kompetencji od jednostki notyfikowanej, jak jest w przypadku modułu B (lub G – zob. niżej).
Jednakże, jeżeli ocena jest oparta na normie EN ISO/IEC 17025, w związku z tym, że norma ta ustanawia jedynie kryteria testowania/wzorcowania bez dokonywania oceny wyników testów przez jednostkę notyfikowaną, jednostka ta musi oddzielnie wykazać swoją zdolność oraz procedury do oceniania i decydowania na podstawie wyników tych testów, czy zasadnicze wymagania zostały spełnione oraz/lub czy zastosowano normy zharmonizowane.
Z drugiej strony, jeżeli stosuje się normę EN ISO/IEC 17020 lub EN ISO/IEC 17065, w związku z tym, że normy te nie obejmują kryteriów testowania/wzorcowania, należy uwzględnić wymagania dotyczące przeprowadzania testów określone w normie EN ISO/IEC 17025. We wszystkich przypadkach jednostka notyfikowana musi być zdolna do przeprowadzenia oceny produktu niezależnie od tego, czy producent zastosował stosowne normy zharmonizowane.
3.5 Moduł G
Jednostka notyfikowana bada kompletny jednostkowy produkt zarówno w fazie projektu, jak i w fazie produkcji.
W tym zakresie jedynie normę EN ISO/IEC 17025 należy uznać za nieodpowiednią do celów modułu G, ponieważ norma ta dotyczy kwestii związanych jedynie z przeprowadzaniem testów, natomiast nie obejmuje ważnych funkcji modułu G dotyczących oceny projektu produktu, która ze względu na swoją złożoność (znacznie wykracza poza zwyczajne badanie dokumentacji produktu jak w module D1, E1 i F1) wymaga od jednostki notyfikowanej dodatkowych kompetencji (podobnie jak w module B i H1).
Wymagania zarówno normy EN ISO/IEC 17020, jak i EN ISO/IEC 17065 można uznać za odpowiednie do oceny jednostek dążących do uzyskania notyfikacji do przeprowadzania zadań zawartych w module G, ponieważ normy te ustanawiają kryteria kompetencji oraz deontologii dotyczące przeprowadzania badania produktu oraz oceny zgodności. Jednakże, w związku z tym, że normy te nie obejmują kryteriów testowania/wzorcowania, dla wymaganych testów należy zawsze uwzględnić stosowne wymagania zawarte w normie EN ISO/IEC 17025.
3.6 Moduł H1
Jednostka notyfikowana ocenia i decyduje, czy system jakości producenta gwarantuje, że produkty są zgodne z instrumentami legislacyjnymi, które mają do nich zastosowanie. Ponadto jednostka notyfikowana bada specyfikację techniczna projektu producenta, w tym wymagane dowody potwierdzające oraz wyniki testów przeprowadzonych przez producenta.
W związku z tym wymagania normy EN ISO/IEC 17021 można uznać za odpowiednie do oceny jednostek dążących do uzyskania notyfikacji w ramach tego modułu. Należy podkreślić, że funkcjonowanie systemu jakości producenta musi zapewniać zgodność gotowych produktów z wymaganiami stosownych unijnych przepisów harmonizacyjnych. Dlatego też jednostka notyfikowana musi dodatkowo posiadać odpowiednią zdolność oceniania zdolności producenta do określania stosownych wymagań dla produktów oraz przeprowadzania niezbędnych kontroli i testów.
Ponadto, w związku z tym, że jednostka notyfikowana bada również projekt produktu, aby go certyfikować, wydając certyfikat badania projektu WE, wymagania zarówno normy EN ISO/IEC 17020, jak i EN ISO/IEC 17065 można uznać za odpowiednie do oceny jednostek dążących do uzyskania notyfikacji w ramach modułu H1, ponieważ normy te ustanawiają kryteria kompetencji oraz deontologii dotyczące przeprowadzania badania produktu oraz oceny zgodności. Norma ISO/IEC 17065 obejmuje elementy testów w taki sposób, że nakłada wymóg przeprowadzania wszelkich testów zgodnie z ISO/IEC 17025. Jednakże, w związku z tym, że EN ISO/IEC 17020 nie obejmuje kryteriów testowania/wzorcowania, dla wymaganych testów należy zawsze uwzględnić stosowne wymagania zawarte w normie EN ISO/IEC 17025.
W tym kontekście należy zauważyć, że jedynie normę EN ISO/IEC 17025 trzeba uznać za nieodpowiednią do celów modułu H1, ponieważ norma ta dotyczy kwestii związanych jedynie z przeprowadzaniem testów, natomiast nie obejmuje ważnych funkcji modułu H dotyczących oceny projektu produktu, która ze względu na swoją złożoność (znacznie wykracza poza zwyczajne badanie dokumentacji produktu jak w module D1, E1 i F1) wymaga od jednostki notyfikowanej dodatkowych kompetencji (podobnie jak w module G i H1).
4. PODSUMOWANIE
Poniższa tabela przedstawia pożądane podejście do wyboru norm w odniesieniu do poszczególnych modułów.
|
Moduł |
Normy EN, które mają zastosowanie |
|
A1, A2 |
EN ISO/IEC 17025 (+ zdolność decydowania o zgodności) lub EN ISO/IEC 17020 z uwzględnieniem EN ISO/IEC 17025 w przypadku, gdy wymagane są testy lub EN ISO/IEC 17065 z uwzględnieniem EN ISO/IEC 17025 w przypadku, gdy wymagane są testy |
|
B |
EN ISO/IEC 17020 z uwzględnieniem EN ISO/IEC 17025 w przypadku, gdy wymagane są testy lub EN ISO/IEC 17065 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy |
|
C1, C2 |
EN ISO/IEC 17025 (+ zdolność decydowania o zgodności) lub EN ISO/IEC 17020 z uwzględnieniem EN ISO/IEC 17025 w przypadku, gdy wymagane są testy lub EN ISO/IEC 17065 z uwzględnieniem EN ISO/IEC 17025 w przypadku, gdy wymagane są testy |
|
D, D1 |
EN ISO/IEC 17021 (+ wiedza związana z produktem) lub EN ISO/IEC 17065 |
|
E, E1 |
EN ISO/IEC 17021 (+ wiedza związana z produktem) lub EN ISO/IEC 17065 |
|
F, F1 |
EN ISO/IEC 17025 (+ zdolność decydowania o zgodności) lub EN ISO/IEC 17020 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy lub EN ISO/IEC 17065 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy |
|
G |
EN ISO/IEC 17020 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy lub EN ISO/IEC 17065 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy |
|
H |
EN ISO/IEC 17021 (+ wiedza związana z produktem) |
|
H1 |
EN ISO/IEC 17021 (+ wiedza związana z produktem) lub EN ISO/IEC 17065 lub EN ISO/IEC 17020 z uwzględnieniem EN 17025 w przypadku, gdy wymagane są testy |
ZAŁĄCZNIK VII
CZĘSTO ZADAWANE PYTANIA DOTYCZĄCE OZNAKOWANIA CE
Co oznacza oznakowanie CE umieszczone na produkcie?
Umieszczając oznakowanie CE na produkcie, producent oświadcza na swoją wyłączną odpowiedzialność, że produkt jest zgodny z zasadniczymi wymaganiami stosownych unijnych przepisów harmonizacyjnych regulujących umieszczanie tego oznakowania oraz że dopełniono odpowiednich procedur oceny zgodności. Zakłada się, że produkty z oznakowaniem CE są zgodne z obowiązującym prawodawstwem harmonizacyjnym UE, w związku z czym mogą one brać udział w swobodnym przepływie na rynku europejskim.
Czy produkt opatrzony oznakowaniem CE jest zawsze wytwarzany w UE?
Nie. Oznakowanie CE sygnalizuje jedynie, że w procesie wytwarzania produktu spełniono wszystkie zasadnicze wymagania. Oznakowanie CE nie jest znakiem pochodzenia, ponieważ nie wskazuje, że produkt został wytworzony w Unii Europejskiej. W związku z tym produkt opatrzony oznakowaniem CE mógł zostać wytworzony w dowolnym miejscu na świecie.
Czy wszystkie produkty opatrzone oznakowaniem CE są badane i zatwierdzane przez organy państwowe?
Nie. Ściśle rzecz ujmując, producent ponosi wyłączną odpowiedzialność za ocenę zgodności produktów z wymaganiami prawnymi mającymi do nich zastosowanie. Producent umieszcza oznakowanie CE i sporządza unijną deklarację zgodności. Jedynie produkty stwarzające znaczące zagrożenie dla interesu publicznego, takie jak zbiorniki ciśnieniowe, dźwigi oraz niektóre narzędzia mechaniczne wymagają przeprowadzenia oceny zgodności przez osobę trzecią, np. jednostkę notyfikowaną.
Czy będąc producentem mogę samodzielnie umieścić oznakowanie CE na własnych produktach?
Tak, oznakowanie CE jest zawsze umieszczane przez producenta lub jego upoważnionego przedstawiciela po przeprowadzeniu niezbędnej procedury oceny zgodności. Oznacza to, że produkt przed umieszczeniem na nim oznakowania CE i wprowadzeniem go do obrotu musi zostać poddany procedurze oceny zgodności przewidzianej w jednym lub kilku obowiązujących unijnych aktach harmonizacyjnych. Przepisy te stanowią, czy ocena zgodności może zostać przeprowadzona przez producenta samodzielnie, czy wymagana jest interwencja osoby trzeciej (jednostki notyfikowanej).
Gdzie powinno być umieszczone oznakowanie CE?
Oznakowanie powinno być umieszczone na produkcie lub na tabliczce znamionowej produktu. Jeżeli nie jest to możliwe ze względu na charakter produktu, oznakowanie CE umieszcza się na opakowaniu i/lub na dokumentach towarzyszących.
Czym jest deklaracja zgodności producenta?
Deklaracja zgodności UE jest dokumentem, w którym producent lub jego upoważniony przedstawiciel na terytorium Europejskiego Obszaru Gospodarczego (EOG) wykazuje, że produkt spełnia wszelkie niezbędne wymagania unijnych przepisów harmonizacyjnych mających zastosowanie do danego produktu. Deklaracja zawiera również nazwę i adres producenta oraz informacje o produkcie, takie jak marka i numer seryjny. Deklaracja zgodności musi być podpisana przez osobę zatrudnioną przez producenta lub jego upoważnionego przedstawiciela, przy czym należy również wskazać stanowisko zajmowane przez tego pracownika.
Niezależnie od tego, czy jednostka notyfikowana była zaangażowana, producent musi sporządzić i podpisać deklarację zgodności WE.
Czy oznakowanie CE jest obowiązkowe, a jeżeli tak, to dla jakich produktów?
Tak, oznakowanie CE jest obowiązkowe. Jednakże oznakowanie umieszcza się wyłącznie na produktach objętych zakresem jednego lub więcej unijnych aktów harmonizacyjnych regulujących oznakowanie CE, które mają zostać wprowadzone na rynek UE. Przykładem produktów, które podlegają pod unijne akty harmonizacyjne regulujące oznakowanie CE są zabawki, produkty elektryczne, maszyny, środki ochrony indywidualnej oraz dźwigi. Na produktach, które nie są objęte przepisami dotyczącymi oznakowania CE nie umieszcza się tego oznakowania.
Informacje o produktach, które podlegają oznakowaniu CE oraz unijnych przepisach harmonizacyjnych regulujących oznakowanie CE można znaleźć na stronie internetowej:
http://ec.europa.eu/growth/single-market/ce-marking/index_en.htm
Jaka jest różnica między oznakowaniem CE a innymi oznakowaniami oraz czy na produkcie opatrzonym oznakowaniem CE można umieszczać inne oznakowania?
Oznakowanie CE jest jedynym oznakowaniem, które wskazuje na zgodność ze wszystkimi zasadniczymi wymaganiami unijnego prawodawstwa harmonizacyjnego, które przewiduje jego umieszczanie. Produkt może posiadać dodatkowe oznakowania pod warunkiem, że nie mają tego samego znaczenia co oznakowanie CE, nie mogą być pomylone z oznakowaniem CE oraz że nie utrudniają rozpoznawania i widoczności oznakowania CE. W związku z tym inne oznakowania mogą być stosowane wyłącznie jeżeli przyczyniają się do poprawy ochrony konsumenta oraz nie są objęte prawodawstwem harmonizacyjnym Unii Europejskiej.
Kto nadzoruje prawidłowe stosowanie oznakowania CE?
Aby zapewnić bezstronność działań związanych z nadzorem rynku, odpowiedzialność za nadzór nad oznakowaniem CE leży po stronie organów publicznych w państwach członkowskich we współpracy z Komisją Europejską.
Jakie są sankcje za fałszowanie oznakowania CE?
Procedury, środki oraz sankcje, które mają zastosowanie do fałszowania oznakowania CE są ustanowione w krajowym prawie administracyjnym i karnym państwa członkowskiego. Zależnie od powagi przestępstwa na podmioty gospodarcze może zostać nałożona kara grzywny lub, w niektórych okolicznościach, kara pozbawienia wolności. Jeżeli jednak produkt nie stwarza bezpośredniego zagrożenia dla bezpieczeństwa, producent może otrzymać drugą możliwość zagwarantowania, że produkt jest zgodny z obowiązującymi przepisami, zanim zostanie zobowiązany do wycofania produktu z rynku.
Co może oznaczać umieszczenie oznakowania CE dla producenta/importera/dystrybutora?
Podczas gdy producenci ponoszą odpowiedzialność za zapewnienie zgodności produktu oraz umieszczenie oznakowania CE, importerzy i dystrybutorzy również odgrywają istotną rolę w zapewnianiu, aby na rynek trafiały wyłącznie produkty zgodne z przepisami oraz posiadające oznakowanie CE. Nie tylko pomaga to we wzmacnianiu skuteczności unijnych wymagań dotyczących ochrony zdrowia, bezpieczeństwa i środowiska naturalnego, lecz także wspiera uczciwą konkurencję między wszystkimi podmiotami podlegającymi tym samym zasadom.
Gdy produkty są wytwarzane w krajach trzecich, a producent nie posiada przedstawiciela na terytorium EOG, importerzy muszą upewnić się, że produkty wprowadzane przez nich na rynek są zgodne z obowiązującymi wymaganiami oraz nie stanowią zagrożenia dla obywateli UE. Importer musi sprawdzić, czy producent spoza UE podjął niezbędne kroki oraz że stosowna dokumentacja jest dostępna na żądanie.
W związku z tym importerzy muszą posiadać ogólną wiedzę na temat stosownych unijnych aktów harmonizacyjnych oraz są zobowiązani do wspierania organów krajowych w razie problemów. Importerzy powinni posiadać pisemne zapewnienie uzyskane od producenta, że będą mieli dostęp do potrzebnej dokumentacji – takiej jak deklaracja zgodności UE oraz dokumentacja produktu – oraz że będą w stanie przedstawić ją organom krajowym, jeżeli zostaną o to poproszeni. Importerzy powinni również dopilnować, aby kontakt z producentem zawsze był możliwy.
Dalej w łańcuchu dostaw ważną rolę w pilnowaniu, aby w obrocie były wyłącznie produkty spełniające wymagania, odgrywają dystrybutorzy. Muszą oni działać z należytą starannością, aby zapewnić, że ich sposób postępowania z produktem nie wpływa niekorzystnie na jego zgodność. Dystrybutor musi również posiadać podstawową wiedzę dotyczącą wymagań prawnych – łącznie z tym, które produkty muszą posiadać oznakowanie CE oraz towarzyszącą dokumentację – oraz powinien być w stanie rozpoznać produkty, które wyraźnie nie spełniają wymagań.
Dystrybutorzy muszą być w stanie wykazać przed organami krajowymi, że postępują z należytą starannością oraz posiadają potwierdzenie od producenta lub importera, że wymagane działania zostały podjęte. Ponadto dystrybutor musi być w stanie pomóc organom krajowym w uzyskaniu wymaganej dokumentacji.
Jeżeli importer lub dystrybutor wprowadza produkty do obrotu pod własną nazwą, przejmuje obowiązki producenta. W takim przypadku musi posiadać wystarczające informacje dotyczące projektu i procesu wytwarzania produktu, ponieważ umieszczając oznakowanie CE przyjmuje odpowiedzialność prawną.
Gdzie można znaleźć więcej informacji?
Informacje o oznakowaniu CE, produktach podlegających oznakowaniu CE, unijnych przepisach harmonizacyjnych regulujących oznakowanie CE oraz krokach, które należy podjąć można znaleźć na stronie internetowej:
http://ec.europa.eu/growth/single-market/ce-marking/index_en.htm
Podmioty gospodarcze mogą nawiązać kontakt z Enterprise Europe Network za pośrednictwem strony:
http://een.ec.europa.eu/index_pl.htm