

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32012R0252
Commission Regulation (EU) No 252/2012 of 21 March 2012 laying down methods of sampling and analysis for the official control of levels of dioxins, dioxin-like PCBs and non-dioxin-like PCBs in certain foodstuffs and repealing Regulation (EC) No 1883/2006 Text with EEA relevance
Rozporządzenie Komisji (UE) nr 252/2012 z dnia 21 marca 2012 r. ustanawiające metody pobierania i analizy próbek do celów urzędowej kontroli poziomów dioksyn, dioksynopodobnych polichlorowanych bifenyli i niedioksynopodobnych polichlorowanych bifenyli w niektórych środkach spożywczych oraz uchylające rozporządzenie (WE) nr 1883/2006 Tekst mający znaczenie dla EOG
Rozporządzenie Komisji (UE) nr 252/2012 z dnia 21 marca 2012 r. ustanawiające metody pobierania i analizy próbek do celów urzędowej kontroli poziomów dioksyn, dioksynopodobnych polichlorowanych bifenyli i niedioksynopodobnych polichlorowanych bifenyli w niektórych środkach spożywczych oraz uchylające rozporządzenie (WE) nr 1883/2006 Tekst mający znaczenie dla EOG
OJ L 84, 23.3.2012, p. 1–22
(BG, ES, CS, DA, DE, ET, EL, EN, FR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
Special edition in Croatian: Chapter 15 Volume 013 P. 282 - 303
 No longer in force, Date of end of validity: 22/06/2014; Uchylony przez 32014R0589
No longer in force, Date of end of validity: 22/06/2014; Uchylony przez 32014R0589
|
23.3.2012 |
PL |
Dziennik Urzędowy Unii Europejskiej |
L 84/1 |
ROZPORZĄDZENIE KOMISJI (UE) NR 252/2012
z dnia 21 marca 2012 r.
ustanawiające metody pobierania i analizy próbek do celów urzędowej kontroli poziomów dioksyn, dioksynopodobnych polichlorowanych bifenyli i niedioksynopodobnych polichlorowanych bifenyli w niektórych środkach spożywczych oraz uchylające rozporządzenie (WE) nr 1883/2006
(Tekst mający znaczenie dla EOG)
KOMISJA EUROPEJSKA,
uwzględniając Traktat o funkcjonowaniu Unii Europejskiej,
uwzględniając rozporządzenie (WE) nr 882/2004 Parlamentu Europejskiego i Rady z dnia 29 kwietnia 2004 r. w sprawie kontroli urzędowych przeprowadzanych w celu sprawdzenia zgodności z prawem paszowym i żywnościowym oraz regułami dotyczącymi zdrowia zwierząt i dobrostanu zwierząt (1), w szczególności jego art. 11 ust. 4,
a także mając na uwadze, co następuje:
|
(1) |
Rozporządzenie Komisji (WE) nr 1881/2006 z dnia 19 grudnia 2006 r. ustalające najwyższe dopuszczalne poziomy niektórych zanieczyszczeń w środkach spożywczych (2) ustanawia najwyższe dopuszczalne poziomy niedioksynopodobnych polichlorowanych bifenyli (PCB), dioksyn i furanów oraz sumy dioksyn, furanów i dioksynopodobnych polichlorowanych bifenyli (PCB) w niektórych środkach spożywczych. |
|
(2) |
W zaleceniu Komisji 2011/516/UE z dnia 23 sierpnia 2011 r. w sprawie ograniczenia obecności dioksyn, furanów i bifenyli polichlorowanych (PCB) w paszy i żywności (3) ustanowiono progi podejmowania działań w celu zachęcenia do proaktywnego podejścia do zmniejszania obecności polichlorowanych dibenzo-para-dioksyn i polichlorowanych dibenzofuranów (PCDD/F) oraz dioksynopodobnych PCB w żywności. Te progi podejmowania działań są narzędziem dla właściwych organów i podmiotów gospodarczych umożliwiającym wyodrębnienie przypadków wymagających zidentyfikowania źródła zanieczyszczeń i podjęcia odpowiednich działań, aby je zredukować lub zlikwidować. |
|
(3) |
W rozporządzeniu Komisji (WE) nr 1883/2006 z dnia 19 grudnia 2006 r. ustanawiającym metody pobierania próbek i metody analizy do celów urzędowej kontroli dioksyn i oznaczania dioksynopodobnych polichlorowanych bifenyli (PCB) w środkach spożywczych (4) ustanowiono przepisy szczegółowe dotyczące procedury pobierania próbek i metod ich analizy, które mają być stosowane do celów kontroli urzędowych. |
|
(4) |
Stosowanie nowych najwyższych poziomów dla niedioksynopodobnych PCB, ustanowionych po udostępnieniu opinii naukowej Europejskiego Urzędu ds. Bezpieczeństwa Żywności (EFSA) w sprawie niedioksynopodobnych PCB w celu harmonizacji na poziomie Unii, a także uaktualnienie kryteriów dla metod przesiewowych wymaga znacznych zmian. W związku z powyższym, w celu zapewnienia jasności, niezbędne jest zastąpienie rozporządzenia (WE) nr 1883/2006 niniejszym rozporządzeniem. |
|
(5) |
Przepisy ustanowione w niniejszym rozporządzeniu odnoszą się wyłącznie do pobierania próbek i analizy dioksyn, dioksynopodobnych PCB i niedioksynopodobnych PCB do celów wykonania rozporządzenia (WE) nr 1881/2006. Nie mają one wpływu na strategię pobierania próbek, zakres i częstotliwość pobierania próbek, określone w załącznikach III i IV do dyrektywy Rady 96/23/WE z dnia 29 kwietnia 1996 r. w sprawie środków monitorowania niektórych substancji i ich pozostałości u żywych zwierząt i w produktach zwierzęcych oraz uchylającej dyrektywy 85/358/EWG i 86/469/EWG oraz decyzje 89/187/EWG i 91/664/EWG (5). Nie mają wpływu na kryteria strategiczne pobierania próbek ustanowione w decyzji Komisji 98/179/WE z dnia 23 lutego 1998 r. ustanawiającej szczegółowe zasady pobierania próbek do celów monitorowania niektórych substancji i ich pozostałości u żywych zwierząt i w produktach pochodzenia zwierzęcego (6). |
|
(6) |
Do identyfikowania próbek o znacznym poziomie PCDD/F oraz dioksynopodobnych PCB można stosować przesiewową metodę analizy z powszechnie akceptowaną walidacją i gwarantującą wysoką przepustowość (najlepiej wybierając próbki przekraczające progi podejmowania działań i zapewniając wybór próbek przekraczających najwyższe poziomy). Następnie należy oznaczyć poziomy PCDD/F i dioksynopodobnych PCB w tych próbkach, stosując potwierdzającą metodę analizy. Należy zatem ustanowić odpowiednie wymogi dotyczące metody przesiewowej, dopilnowując, aby wskaźnik ilości wyników fałszywie ujemnych w odniesieniu do najwyższych dopuszczalnych poziomów nie przekraczał 5 %, a także ustanowić ścisłe wymogi dotyczące potwierdzających metod analizy. Ponadto metody potwierdzające pozwalają na oznaczenie poziomów także przy niskim poziomie tła. Jest to ważne dla śledzenia tendencji w czasie, oceny narażenia oraz ponownej oceny najwyższych poziomów i progów podejmowania działań. |
|
(7) |
Konieczne jest określenie pobierania próbek z bardzo dużych ryb w celu zapewnienia zharmonizowanego podejścia w całej Unii. |
|
(8) |
Poziomy dioksyn, dioksynopodobnych PCB i niedioksynopodobnych PCB w rybach tego samego gatunku, pochodzących z tego samego regionu, mogą się różnić w zależności od wielkości lub wieku ryb. Ponadto poziom dioksyn, dioksynopodobnych PCB i niedioksynopodobnych PCB niekoniecznie jest taki sam we wszystkich częściach ryby. Z tego względu konieczne jest określenie pobierania i przygotowania próbek w celu zapewnienia zharmonizowanego podejścia w całej Unii. |
|
(9) |
Ważne jest, aby wyniki analizy były przekazywane i interpretowane w jednolity sposób w celu zapewnienia zharmonizowanego podejścia w sposobie egzekwowania w całej Unii. |
|
(10) |
Środki przewidziane w niniejszym rozporządzeniu są zgodne z opinią Stałego Komitetu ds. Łańcucha Żywnościowego i Zdrowia Zwierząt i ani Parlament Europejski, ani Rada nie wyraziły wobec nich sprzeciwu, |
PRZYJMUJE NINIEJSZE ROZPORZĄDZENIE:
Artykuł 1
Do celów niniejszego rozporządzenia zastosowanie mają definicje i skróty określone w załączniku I.
Artykuł 2
Pobieranie próbek do celów urzędowej kontroli poziomów dioksyn, furanów, dioksynopodobnych PCB i niedioksynopodobnych PCB w środkach spożywczych wymienionych w sekcji 5 załącznika do rozporządzenia (WE) nr 1881/2006 przeprowadza się zgodnie z metodami określonymi w załączniku II do niniejszego rozporządzenia.
Artykuł 3
Przygotowanie i analiza próbek do celów urzędowej kontroli poziomów dioksyn, furanów i dioksynopodobnych PCB w środkach spożywczych wymienionych w sekcji 5 załącznika do rozporządzenia (WE) nr 1881/2006 przeprowadza się zgodnie z metodami określonymi w załączniku III do niniejszego rozporządzenia.
Artykuł 4
Analizy do celów urzędowej kontroli poziomów niedioksynopodobnych PCB w środkach spożywczych wymienionych w sekcji 5 załącznika do rozporządzenia (WE) nr 1881/2006 przeprowadza się zgodnie z metodami określonymi w załączniku IV do niniejszego rozporządzenia.
Artykuł 5
Rozporządzenie (WE) nr 1883/2006 traci moc.
Odniesienia do uchylonego rozporządzenia są rozumiane jako odniesienia do niniejszego rozporządzenia.
Artykuł 6
Niniejsze rozporządzenie wchodzi w życie dwudziestego dnia po jego opublikowaniu w Dzienniku Urzędowym Unii Europejskiej.
Niniejsze rozporządzenie stosuje się od dnia jego wejścia w życie.
Niniejsze rozporządzenie wiąże w całości i jest bezpośrednio stosowane we wszystkich państwach członkowskich.
Sporządzono w Brukseli dnia 21 marca 2012 r.
W imieniu Komisji
José Manuel BARROSO
Przewodniczący
(1) Dz.U. L 165 z 30.4.2004, s. 1.
(2) Dz.U. L 364 z 20.12.2006, s. 5.
(3) Dz.U. L 218 z 24.8.2011, s. 23.
(4) Dz.U. L 364 z 20.12.2006, s. 32.
(5) Dz.U. L 125 z 23.5.1996, s. 10.
(6) Dz.U. L 65 z 5.3.1998, s. 31.
ZAŁĄCZNIK I
Definicje i skróty
I. DEFINICJE
Do celów niniejszego rozporządzenia zastosowanie mają definicje ustanowione w załączniku I do decyzji Komisji 2002/657/WE z dnia 14 sierpnia 2002 r. wykonującej dyrektywę Rady 96/23/WE dotyczącą wyników metod analitycznych i ich interpretacji (1).
Dodatkowo do celów niniejszego rozporządzenia stosuje się następujące definicje:
|
1.1. |
Próg podejmowania działań oznacza poziom danej substancji, określony w załączniku do zalecenia 2011/516/UE, który prowadzi do rozpoczęcia dochodzenia w celu zidentyfikowania źródła tej substancji w przypadkach, w których wykrywane są podwyższone poziomy tej substancji. |
|
1.2. |
Metody bioanalityczne oznaczają metody oparte na zasadach biologicznych, np. oznaczenia wykorzystujące hodowle komórkowe, oznaczenie wykorzystujące receptor lub oznaczenie immunologiczne. Nie dają one wyników na poziomie kongenerów, a jedynie wskazanie (2) poziomu TEQ wyrażonego w równoważnikach bioanalitycznych (BEQ), aby uwzględnić fakt, że nie wszystkie związki obecne w ekstrakcie próbki, które wywołują odpowiedź w badaniu, spełniają wszystkie wymogi zasady TEQ. |
|
1.3. |
Odzysk uzyskany w oznaczeniu biologicznym oznacza poziom BEQ obliczony z TCDD lub krzywej kalibracyjnej PCB 126, skorygowany o próbę ślepą, a następnie podzielony przez poziom TEQ określony metodą GC/HRMS. Jego celem jest korekta takich czynników, jak strata PCDD/PCDF i związków dioksynopodobnych podczas ekstrakcji i oczyszczania, związki współekstrahowane zwiększające lub zmniejszające odpowiedź (efekt agonistyczny lub antagonistyczny), jakość dopasowania krzywej lub różnice między wartościami TEF a REP. Odzysk uzyskany w oznaczeniu biologicznym jest obliczany z odpowiednich próbek referencyjnych z reprezentatywnymi profilami kongenerów na poziomie zainteresowania. |
|
1.4. |
Metody półilościowe oznaczają metody, które dają przybliżone wskazanie stężenia przypuszczalnie występującego analitu, podczas gdy wynik liczbowy nie spełnia wymogów dla metod ilościowych. |
|
1.5. |
Akceptowalna swoista granica oznaczalności pojedynczego kongeneru oznacza stężenie analitu w ekstrakcie próbki, które wywołuje odpowiedź instrumentu na impuls dla dwóch różnych jonów, monitorowaną przy współczynniku S/N (sygnał/szum) wynoszącym 3:1 dla sygnału mniej wrażliwego i przy spełnieniu kryteriów identyfikacji opisanych na przykład w normie prEN 16215 (Pasze – Oznaczanie dioksyn i dioksynopodobnych PCB metodą GC-HRMS oraz wskaźnikowych PCB metodą GC-HRMS) lub w metodzie EPA 1613, rev. B. |
|
1.6. |
Koncepcja górnej granicy wymaga podania zawartości równej granicy oznaczalności dla każdego kongeneru niemożliwego do oznaczenia. |
|
1.7. |
Koncepcja dolnej granicy wymaga podania ślepej zawartości dla każdego kongeneru niemożliwego do oznaczenia. |
|
1.8. |
Koncepcja średniej granicy wymaga podania zawartości równej połowie granicy oznaczalności dla każdego kongeneru niemożliwego do oznaczenia. |
|
1.9. |
Partia oznacza możliwą do zidentyfikowania ilość żywności dostarczonej w jednym czasie, określoną przez urzędnika jako posiadającą wspólne cechy, takie jak pochodzenie, odmiana, rodzaj opakowania, pakujący, nadawca lub znakowanie. W przypadku ryb i produktów rybołówstwa porównywalna musi być również wielkość ryb. Jeżeli wielkość lub masa ryb w przesyłce nie są porównywalne, przesyłka nadal może być traktowana jako partia, należy jednak zastosować specjalne procedury pobierania próbek. |
|
1.10. |
Podpartia oznacza część wyznaczoną z większej partii w celu zastosowania metody pobierania próbek do tej wyznaczonej części. Każda podpartia musi być fizycznie oddzielona i możliwa do zidentyfikowania. |
|
1.11. |
Próbka pierwotna oznacza ilość materiału pobraną z jednego miejsca w partii lub w podpartii. |
|
1.12. |
Próbka zbiorcza oznacza całość utworzoną przez połączenie wszystkich próbek pierwotnych pobranych z partii lub podpartii. |
|
1.13. |
Próbka laboratoryjna oznacza reprezentatywną część/ilość próbki zbiorczej przeznaczoną dla laboratorium. |
II. STOSOWANE SKRÓTY
|
BEQ |
Równoważnik bioanalityczny |
|
GC |
Chromatografia gazowa |
|
HRMS |
Spektrometria masowa o wysokiej rozdzielczości |
|
LRMS |
Spektrometria masowa o niskiej rozdzielczości |
|
PCB |
Polichlorowane bifenyle |
|
PCDD |
Polichlorowane dibenzo-p-dioksyny |
|
PCDF |
Polichlorowane dibenzofurany |
|
QC |
Kontrola jakości |
|
REP |
Względny potencjał działania |
|
TEF |
Współczynnik równoważny toksyczności |
|
TEQ |
Równoważnik toksyczności |
|
TCDD |
Tetrachlorodibenzodioksyna |
|
U |
Rozszerzona niepewność pomiaru |
(1) Dz.U. L 221 z 17.8.2002, s. 8.
(2) Metody bioanalityczne nie są swoiste dla kongenerów objętych schematem TEF. W ekstrakcie próbki mogą być obecne inne związki strukturalnie podobne do związków aktywujących receptor AhR, co przyczynia się do ogólnej odpowiedzi. Dlatego wyniki uzyskane metodami bioanalitycznymi nie mogą być traktowane jako szacunki, ale raczej jako wskazanie poziomu TEQ w próbce.
ZAŁĄCZNIK II
Metody pobierania próbek do celów kontroli urzędowej poziomów dioksyn (PCDD/PCDF), dioksynopodobnych PCB i niedioksynopodobnych PCB w niektórych środkach spożywczych
I. ZAKRES
Próbki przeznaczone do kontroli urzędowej poziomów dioksyn (PCDD/PCDF), dioksynopodobnych PCB i niedioksynopodobnych PCB, określanych dalej jako „dioksyny i PCB”, w środkach spożywczych pobiera się zgodnie z metodami opisanymi w niniejszym załączniku. Otrzymane w ten sposób próbki zbiorcze uznaje się za próbki reprezentatywne dla partii lub podpartii, z których zostały pobrane. Zgodność z najwyższymi dopuszczalnymi poziomami ustanowionymi w rozporządzeniu Komisji (WE) nr 1881/2006 ustalającym najwyższe dopuszczalne poziomy niektórych zanieczyszczeń w środkach spożywczych określa się na podstawie poziomów oznaczonych w próbkach laboratoryjnych.
II. POSTANOWIENIA OGÓLNE
1. Personel
Próbki pobiera osoba upoważniona, wyznaczona przez państwo członkowskie.
2. Materiał, z którego należy pobrać próbki
Próbki pobierane są odrębnie z każdej partii lub podpartii podlegającej badaniu.
3. Niezbędne środki ostrożności
W trakcie pobierania i przygotowywania próbek należy przestrzegać środków ostrożności, aby uniknąć wszelkich zmian, które mogłyby wpłynąć na zawartość dioksyn i PCB, niekorzystnie wpłynąć na wynik analizy lub uczynić próbkę zbiorczą niereprezentatywną.
4. Próbki pierwotne
W miarę możliwości próbki pierwotne pobiera się z różnych miejsc rozmieszczonych w całej partii lub podpartii. Odstąpienie od tej procedury odnotowywane jest w rejestrze, o którym mowa w pkt II.8 niniejszego załącznika.
5. Przygotowanie próbki zbiorczej
Próbkę zbiorczą tworzy się poprzez połączenie próbek pierwotnych. Masa próbki zbiorczej powinna wynosić co najmniej 1 kg, chyba że nie jest to wykonalne, np. próbka jest pobierana z pojedynczego opakowania lub produkt posiada bardzo wysoką wartość handlową.
6. Kontrpróbki
Kontrpróbki do celów egzekwowania, obrony praw i arbitrażu pobiera się ze zhomogenizowanej próbki zbiorczej, o ile nie jest to sprzeczne z przepisami państw członkowskich w zakresie praw podmiotów prowadzących przedsiębiorstwo spożywcze. Wielkość próbek laboratoryjnych do celów egzekwowania powinna pozwalać na co najmniej powtórne przeprowadzenie analizy.
7. Pakowanie i transport próbek
Każdą próbkę umieszcza się w czystym pojemniku wykonanym z chemicznie obojętnego materiału, zapewniającym odpowiednią ochronę przed zanieczyszczeniem, utratą analitów wskutek adsorpcji na wewnętrznej ściance pojemnika i przed uszkodzeniem w transporcie. Podejmowane są wszelkie niezbędne środki w celu zapobieżenia jakimkolwiek zmianom w składzie próbki, które mogłyby powstać w czasie transportu lub przechowywania.
8. Pieczętowanie i etykietowanie próbek
Każdą próbkę pobraną do celów urzędowych pieczętuje się w miejscu pobrania oraz oznacza zgodnie z przepisami państwa członkowskiego.
Każde pobranie próbki musi zostać odnotowane w celu umożliwienia jednoznacznej identyfikacji każdej partii; należy podać również datę i miejsce pobrania próbki oraz wszelkie dodatkowe informacje, które mogą być przydatne dla analityka.
III. PLAN POBIERANIA PRÓBEK
Stosowana metoda pobierania próbek powinna gwarantować, że próbka zbiorcza jest reprezentatywna dla kontrolowanej partii lub podpartii.
1. Podział partii na podpartie
Duże partie są dzielone na podpartie, pod warunkiem że mogą być one fizycznie wyodrębnione. W przypadku produktów sprzedawanych luzem w dużych ilościach (np. olejów roślinnych) zastosowanie ma tabela 1. W odniesieniu do pozostałych produktów zastosowanie ma tabela 2. Biorąc pod uwagę, że masa partii nie zawsze jest dokładną wielokrotnością masy podpartii, masa podpartii może być większa od wspomnianej masy o nie więcej niż 20 %.
Tabela 1
Podział partii na podpartie w przypadku produktów sprzedawanych luzem
|
Masa partii (w tonach) |
Masa lub liczba podpartii |
|
≥ 1 500 |
500 ton |
|
> 300 oraz < 1 500 |
3 podpartie |
|
≥ 50 oraz ≤ 300 |
100 ton |
|
< 50 |
— |
Tabela 2
Podział partii na podpartie w przypadku pozostałych produktów
|
Masa partii (w tonach) |
Masa lub liczba podpartii |
|
≥ 15 |
15–30 ton |
|
< 15 |
— |
2. Liczba próbek pierwotnych
Masa próbki zbiorczej otrzymanej z połączenia wszystkich próbek pierwotnych powinna wynosić co najmniej 1 kg (zob. pkt II.5 niniejszego załącznika).
Minimalna liczba próbek pierwotnych pobieranych z partii lub podpartii musi być zgodna z liczbą podaną w tabelach 3 i 4.
W przypadku produktów ciekłych luzem partia lub podpartia powinna być jak najdokładniej i w sposób jak najmniej wpływający na jakość produktu wymieszana ręcznie lub mechanicznie, bezpośrednio przed pobraniem próbki. W takim przypadku zakłada się jednorodny rozkład zanieczyszczeń w danej partii lub podpartii. W związku z powyższym do utworzenia próbki zbiorczej wystarczy z partii lub podpartii pobrać trzy próbki pierwotne.
Próbki pierwotne muszą mieć podobną masę. Masa próbki pierwotnej powinna wynosić co najmniej 100 gramów.
Odstąpienie od tej procedury odnotowywane jest w rejestrze, o którym mowa w pkt II.8 niniejszego załącznika. Zgodnie z przepisami decyzji Komisji 97/747/WE z dnia 27 października 1997 r. ustalającej poziomy i częstotliwość pobierania próbek przewidzianych dyrektywą Rady 96/23/WE w sprawie kontroli niektórych substancji i ich pozostałości w niektórych produktach zwierzęcych (1) wielkość próbki zbiorczej jaj kurzych wynosi co najmniej 12 jaj (zarówno dla partii luzem, jak i dla partii składających się z opakowań jednostkowych zastosowanie mają tabele 3 i 4).
Tabela 3
Minimalna liczba próbek pierwotnych pobieranych z partii lub podpartii
|
Masa lub objętość partii lub podpartii (w kg lub litrach) |
Minimalna liczba próbek pierwotnych, które należy pobrać |
|
< 50 |
3 |
|
50–500 |
5 |
|
> 500 |
10 |
W przypadku partii lub podpartii składających się z opakowań jednostkowych lub jednostek, liczbę opakowań lub jednostek, które należy pobrać w celu utworzenia próbki zbiorczej, podano w tabeli 4.
Tabela 4
Liczba opakowań lub jednostek (próbek pierwotnych), które należy pobrać w celu utworzenia próbki zbiorczej, gdy partia lub podpartia składają się z opakowań jednostkowych lub jednostek
|
Liczba opakowań lub jednostek w partii lub podpartii |
Liczba opakowań lub jednostek, które należy pobrać |
|
1–25 |
co najmniej 1 opakowanie lub jednostka |
|
26–100 |
ok. 5 %, co najmniej 2 opakowania lub jednostki |
|
> 100 |
ok. 5 %, maksymalnie 10 opakowań lub jednostek |
3. Przepisy szczegółowe dotyczące pobierania próbek z partii zawierających całe ryby o porównywalnej wielkości lub masie
Uważa się, że ryby mają porównywalną wielkość i masę, jeżeli różnica w wielkości i masie nie przekracza 50 %.
Liczba próbek pierwotnych, które należy pobrać z partii, podana jest w tabeli 3. Masa próbki zbiorczej otrzymanej z połączenia wszystkich próbek pierwotnych powinna wynosić co najmniej 1 kg (zob. pkt II.5).
|
— |
Jeżeli partia, z której należy pobrać próbki, zawiera małe ryby (poszczególne ryby ważą mniej niż ok. 1 kg), jako próbkę pierwotną w celu utworzenia próbki zbiorczej pobiera się całą rybę. Jeżeli otrzymana masa próbki zbiorczej jest większa niż 3 kg, próbki pierwotne mogą składać się z części środkowej ryb tworzących próbkę zbiorczą, ważącej przynajmniej 100 gramów każda. Cała część, do której stosuje się najwyższy dopuszczalny poziom, jest stosowana do homogenizacji próbki. Środkowa część ryby to część, w której znajduje się środek ciężkości. W większości przypadków środek ciężkości ulokowany jest przy płetwie grzbietowej (w przypadku ryb posiadających płetwę grzbietową) lub w połowie odległości między skrzelami a odbytem. |
|
— |
Jeżeli partia, z której należy pobrać próbki, zawiera większe ryby (poszczególne ryby ważące więcej niż ok. 1 kg), próbka pierwotna składa się ze środkowej części ryby. Każda próbka pierwotna powinna ważyć co najmniej 100 gramów. W przypadku ryb średniej wielkości (ok. 1–6 kg) jako próbkę pierwotną pobiera się płat ryby od kręgosłupa do brzucha ze środkowej części ryby. W przypadku bardzo dużych ryb (np. powyżej ok. 6 kg) próbka pierwotna pobierana jest z prawej strony (patrząc od przodu) mięśnia grzbietowo-bocznego w środkowej części ryby. Jeżeli pobranie próbki z takiego miejsca w środkowej części ryby spowodowałoby znaczne szkody ekonomiczne, wystarczające może być pobranie trzech próbek pierwotnych o masie co najmniej 350 gramów każda, niezależnie od wielkości partii, lub alternatywnie pobranie równych części mięśnia w pobliżu ogona oraz mięśnia w pobliżu głowy jednej ryby w celu utworzenia próbki pierwotnej reprezentatywnej dla poziomu dioksyn w całej rybie. |
4. Pobieranie próbek z partii ryb zawierającej całe ryby o różnej wielkości lub masie
|
— |
Zastosowanie mają przepisy pkt III.3 odnoszące się do składu próbki. |
|
— |
Jeżeli przeważa jakaś klasa lub kategoria wielkości lub masy (ok. 80 % partii lub więcej), próbka pobierana jest z ryb, których wielkość lub masa są przeważające. Próbka ta uważana jest za reprezentatywną dla całej partii. |
|
— |
Jeżeli nie przeważa żadna klasa lub kategoria wielkości lub masy, należy dopilnować, aby ryby wybrane do próbki były reprezentatywne dla całej partii. Szczegółowe wytyczne postępowania w takich przypadkach znajdują się w „Wytycznych pobierania próbek z całych ryb o różnej wielkości lub masie” (2). |
5. Pobieranie próbek na etapie sprzedaży detalicznej
Na etapie sprzedaży detalicznej próbki środków spożywczych pobierane są w miarę możliwości zgodnie z zasadami określonymi w pkt III.2 niniejszego załącznika.
Jeżeli to nie jest możliwe, można zastosować inną metodę pobierania próbek na etapie sprzedaży detalicznej, pod warunkiem że zapewnia ona odpowiednią reprezentatywność badanej partii lub podpartii.
IV. ZGODNOŚĆ PARTII LUB PODPARTII ZE SPECYFIKACJĄ
1. W odniesieniu do niedioksynopodobnych PCB
Partia jest akceptowana, jeżeli wynik analizy nie przekracza najwyższego dopuszczalnego poziomu dla niedioksynopodobnych PCB, określonego w rozporządzeniu (WE) nr 1881/2006, przy uwzględnieniu niepewności pomiaru.
Partię uznaje się za niezgodną z wymogami w zakresie najwyższego dopuszczalnego poziomu określonego w rozporządzeniu (WE) nr 1881/2006, jeżeli górna granica wyniku analizy potwierdzonego powtórną analizą (3) ponad wszelką wątpliwość przekracza najwyższy dopuszczalny poziom, przy uwzględnieniu niepewności pomiaru.
Niepewność pomiaru można uwzględnić w jeden z następujących sposobów:
|
— |
obliczając niepewność rozszerzoną z użyciem współczynnika rozszerzenia 2, która określa przedział ufności na poziomie około 95 %. Partia lub podpartia jest niezgodna, jeśli mierzona wartość po odjęciu U jest wyższa niż dopuszczalny poziom, |
|
— |
ustalając decyzyjną wartość graniczną (CCα) zgodnie z przepisami decyzji 2002/657/WE (pkt 3.1.2.5 załącznika I do tej decyzji – przypadki substancji, dla których ustalono dopuszczalny poziom). Partia lub podpartia jest niezgodna, jeżeli wartość mierzona jest równa lub wyższa niż CCα. |
Wymienione wyżej zasady stosuje się do wyniku analitycznego otrzymanego na próbce pobranej do celów kontroli urzędowej. W przypadku analizy do celów obrony praw lub arbitrażu zastosowanie mają przepisy krajowe.
2. W odniesieniu do dioksyn (PCDD/PCDF) i dioksynopodobnych PCB
Partia jest akceptowana, jeżeli wynik pojedynczej analizy
|
— |
przeprowadzonej metodą przesiewową przy wskaźniku ilości wyników fałszywie ujemnych poniżej 5 % wskazuje, że poziom nie przekracza odpowiedniego najwyższego dopuszczalnego poziomu PCDD/F ani sumy PCDD/F i dioksynopodobnych PCB, określonych w rozporządzeniu (WE) nr 1881/2006, |
|
— |
przeprowadzonej metodą potwierdzającą nie przekracza odpowiedniego najwyższego dopuszczalnego poziomu PCDD/F ani sumy PCDD/F i dioksynopodobnych PCB, określonych w rozporządzeniu (WE) nr 1881/2006, przy uwzględnieniu niepewność pomiaru. |
Dla badań przesiewowych należy ustalić wartość odcięcia dla decyzji o zgodności z odpowiednimi poziomami zainteresowania ustanowionymi albo dla PCDD/F, albo dla sumy PCDD/F i dioksynopodobnych PCB.
Partię uznaje się za niezgodną z wymogami w zakresie najwyższego dopuszczalnego poziomu określonego w rozporządzeniu (WE) nr 1881/2006, jeżeli górny wynik analizy uzyskany metodą potwierdzającą i potwierdzony powtórną analizą (3) ponad wszelką wątpliwość przekracza najwyższy dopuszczalny poziom, przy uwzględnieniu niepewności pomiaru.
Niepewność pomiaru można uwzględnić w jeden z następujących sposobów:
|
— |
obliczając niepewność rozszerzoną z użyciem współczynnika rozszerzenia 2, która określa przedział ufności na poziomie około 95 %. Partia lub podpartia jest niezgodna, jeśli mierzona wartość po odjęciu U jest wyższa niż dopuszczalny poziom. W przypadku odrębnego oznaczania PCDD/F i dioksynopodobnych PCB należy zastosować sumę szacowanej niepewności rozszerzonej odrębnych wyników analitycznych dla dioksyn i dioksynopodobnych PCB, w odniesieniu do szacowanej niepewności rozszerzonej sumy dioksyn i dioksynopodobnych PCB, |
|
— |
ustalając decyzyjną wartość graniczną (CCα) zgodnie z przepisami decyzji 2002/657/WE (pkt 3.1.2.5 załącznika I do tej decyzji – przypadki substancji, dla których ustalono dopuszczalny poziom). Partia lub podpartia jest niezgodna, jeżeli wartość mierzona jest równa lub wyższa niż CCα. |
Wymienione wyżej zasady stosuje się do wyniku analitycznego otrzymanego na próbce pobranej do celów kontroli urzędowej. W przypadku analizy do celów obrony praw lub arbitrażu zastosowanie mają przepisy krajowe.
V. PRZEKROCZENIE PROGÓW PODEJMOWANIA DZIAŁAŃ
Progi podejmowania działań są narzędziem wyboru próbek w przypadkach wymagających zidentyfikowania źródła zanieczyszczeń i podjęcia odpowiednich działań, aby je zredukować lub zlikwidować. Metody przesiewowe powinny ustanowić odpowiednie wartości odcięcia dla wyboru tych próbek. Wysiłki niezbędne do zidentyfikowania źródła i zredukowania lub zlikwidowania zanieczyszczenia należy podjąć tylko wówczas, jeżeli przekroczenie progu podejmowania działań jest potwierdzone powtórną analizą przy zastosowaniu metody potwierdzającej i przy uwzględnieniu niepewności pomiaru (4).
(1) Dz.U. L 303 z 6.11.1997, s. 12.
(2) http://ec.europa.eu/food/food/chemicalsafety/contaminants/dioxins_en.htm
(3) Powtórna analiza jest konieczna do wykluczenia możliwości wewnętrznego zanieczyszczenia krzyżowego lub przypadkowego wymieszania się próbek. Pierwsza analiza, przy uwzględnieniu niepewności pomiaru, stosowana jest do weryfikacji zgodności. W przypadku przeprowadzania analizy w ramach incydentu wystąpienia zanieczyszczenia można zrezygnować z przeprowadzania potwierdzenia powtórną analizą, jeżeli próbki wybrane do analizy można powiązać z incydentem wystąpienia zanieczyszczenia na podstawie identyfikowalności.
(4) Identyczne wyjaśnienie i wymagania dla powtórnej analizy w celu kontroli progów podejmowania działań jak w przypisie 3 dla najwyższych dopuszczalnych poziomów.
ZAŁĄCZNIK III
Przygotowanie próbek i wymagania dotyczące metod analizy stosowanych w urzędowej kontroli poziomów dioksyn (PCDD/PCDF) i dioksynopodobnych PCB w niektórych środkach spożywczych
1. ZAKRES ZASTOSOWANIA
Wymogi ustanowione w niniejszym załączniku należy stosować w przypadku środków spożywczych analizowanych do celów urzędowych kontroli poziomów polichlorowanych dibenzo-p-dioksyn z atomami chloru w pozycjach 2,3,7,8 oraz polichlorowanych dibenzofuranów (PCDD/F) i dioksynopodobnych polichlorowanych bifenyli (dioksynopodobnych PCB), a także dla innych celów regulacyjnych.
Monitorowanie obecności PCDD/F i dioksynopodobnych PCB w środkach spożywczych przeprowadza się w dwóch różnych celach:
|
a) |
wybór próbek o poziomach PCDD/F i dioksynopodobnych PCB przekraczających najwyższe dopuszczalne poziomy lub progi podejmowania działań. Podejście to może wymagać zastosowania metody przesiewowej pozwalającej na oszczędną i skuteczną przepustowość próbek i zwiększającej w ten sposób szansę wykrycia nowych incydentów o wysokim stopniu narażenia i ryzyku dla zdrowia konsumentów. Metody przesiewowe mogą obejmować metody bioanalityczne i metody GC/MS. Ich stosowanie powinno mieć na celu unikanie wyników fałszywie ujemnych. Stężenie PCDD/F oraz sum PCDD/F i dioksynopodobnych PCB w próbkach, które zawierają znaczące ich poziomy, należy oznaczyć lub potwierdzić przy zastosowaniu metody potwierdzającej; |
|
b) |
oznaczanie poziomów PCDD/F i dioksynopodobnych PCB w próbkach żywności przy wielu niskich poziomach tła. Jest to ważne dla śledzenia tendencji w czasie, oceny narażenia populacji i stworzenia bazy danych na potrzeby ewentualnej ponownej oceny progów podejmowania działań i najwyższych dopuszczalnych poziomów. Cel ten jest osiągany przy zastosowaniu metody potwierdzającej pozwalającej w sposób jednoznaczny identyfikować i oznaczać ilościowo PCDD/F i dioksynopodobne PCB na poziomie zainteresowania. Metody te można stosować do potwierdzania wyników uzyskanych metodami przesiewowymi oraz do oznaczania niskich poziomów tła w monitorowaniu żywności. Są one ważne także dla ustanowienia profili kongenerów w celu zidentyfikowania źródła ewentualnego zanieczyszczenia. Obecnie metody te wykorzystują chromatografię gazową wysokiej rozdzielczości lub spektrometrię masową wysokiej rozdzielczości (HRGC/HRMS). |
2. KLASYFIKACJA METOD POD WZGLĘDEM STOPNIA OZNACZALNOŚCI (1)
Metody jakościowe dają odpowiedź tak/nie w odniesieniu do obecności interesującego analitu bez wskazania ilościowego stężenia przypuszczalnie występującego analitu. Metody te mogą dostarczać wyników półilościowych, ale wykorzystywane są wyłącznie w celu uzasadniania decyzji tak/nie, jako wskazanie poziomów powyżej lub poniżej pewnych zakresów, np. granicy wykrywalności, granicy oznaczalności lub wartości odcięcia.
Do kontroli najwyższych dopuszczalnych poziomów i progów podejmowania działań dla PCDD/F oraz związków dioksynopodobnych w żywności można zastosować metody przesiewowe, oparte na porównaniu wyniku analitycznego z wartością odcięcia i dające odpowiedź tak/nie dla wskazania ewentualnego przekroczenia poziomów zainteresowania. W tym celu wprowadzono metody bioanalityczne. Zasadniczo można opracować także metody fizykochemiczne, jednak w odniesieniu do najwyższych dopuszczalnych poziomów i progów podejmowania działań opierających się na TEQ oraz w odniesieniu do złożonej analizy wymagającej oznaczenia odpowiednich pojedynczych kongenerów brak jest praktycznych przykładów.
Metody półilościowe dają przybliżone wskazanie stężenia, które może być przydatne jako informacja o zakresie stężenia analitu i pomagać analitykowi przy podejmowaniu decyzji o zakresie kalibracji badania potwierdzającego, które należy następnie wykonać, oraz w celu kontroli jakości. Przykładami takich metod są:
|
— |
metody bioanalityczne, które mogą wykrywać interesujące anality, obejmować krzywą kalibracji, dawać odpowiedź tak/nie dla wskazania ewentualnego przekroczenia poziomu zainteresowania oraz pozwalać na uzyskiwanie wyników w postaci równoważników bioanalitycznych (BEQ), będących wskazaniem wartości TEQ w próbce, |
|
— |
badania fizykochemiczne (np. GC-MS/MS lub GC/LRMS), gdzie wyznaczone parametry precyzji metody nie spełniają kryteriów dla badania ilościowego. |
Metody ilościowe spełniają te same wymogi w odniesieniu do dokładności, zakresu oznaczania i precyzji, co metody potwierdzające. Jeżeli wymagane jest oznaczenie ilościowe, metody te powinny zostać zwalidowane jako metody potwierdzające, zgodnie z opisem w niniejszym dokumencie dla PCDD/F i dioksynopodobnych PCB.
3. INFORMACJE PODSTAWOWE
W celu obliczenia stężeń wyrażonych jako równoważniki toksyczności (TEQ) należy pomnożyć stężenia poszczególnych substancji w danej próbce przez ich odpowiednie współczynniki równoważne toksyczności (TEF), ustanowione przez Światową Organizację Zdrowia i wymienione w dodatku do niniejszego załącznika, a następnie zsumować je, co da łączne stężenie związków dioksynopodobnych wyrażone jako TEQ.
Metody przesiewowe i potwierdzające mogą być stosowane tylko do kontroli niektórych matryc, jeżeli metody te są dostatecznie czułe, aby w sposób wiarygodny wykrywać substancje na poziomie zainteresowania (próg podejmowania działań lub najwyższy dopuszczalny poziom).
4. WYMAGANIA DOTYCZĄCE ZAPEWNIANIA JAKOŚCI
|
— |
Należy podjąć niezbędne środki dla uniknięcia zanieczyszczenia krzyżowego na każdym etapie pobierania i analizy próbek. |
|
— |
Próbki należy przechowywać i transportować w pojemnikach szklanych, aluminiowych, polipropylenowych lub polietylenowych nadających się do tego celu i nie mających wpływu na poziomy PCDD/F i dioksynopodobnych PCB w próbkach. Pojemnik na próbki należy oczyścić z pyłków papieru. |
|
— |
Przechowywanie i przewożenie próbek należy przeprowadzać w sposób zachowujący integralność próbki środka spożywczego. |
|
— |
W razie potrzeby każdą próbkę laboratoryjną należy drobno zemleć i dokładnie wymieszać w sposób gwarantujący pełną homogenizację (np. w sposób umożliwiający przesianie przez sito o oczku 1 mm); jeżeli zawartość wilgoci jest zbyt wysoka, próbki muszą zostać osuszone przed mieleniem. |
|
— |
Duże znaczenie ma kontrola odczynników, aparatury i szkła laboratoryjnego pod kątem ewentualnego wpływu na wyniki oparte na TEQ lub BEQ. |
|
— |
Należy wykonać analizę ślepej próby, obejmującą przeprowadzenie pełnej procedury analitycznej, ale bez próbki. |
|
— |
W przypadku metod bioanalitycznych ważne jest, aby upewnić się, że szkło laboratoryjne i rozpuszczalniki stosowane podczas analizy są wolne od związków interferujących w wykrywaniu związków docelowych w zakresie roboczym. Szkło należy przemyć rozpuszczalnikami lub ogrzać do temperatur, w których z powierzchni usuwane są pozostałości PCDD/F, związków dioksynopodobnych i związków interferujących. |
|
— |
Ilość próbki stosowanej do ekstrakcji musi być wystarczająca do spełnienia wymogów dotyczących wystarczająco niskiego zakresu roboczego, obejmującego stężenia na poziomie zainteresowania. |
|
— |
Konkretne procedury przygotowania próbki stosowane do badanych produktów powinny być zgodne z międzynarodowymi wytycznymi. |
|
— |
W przypadku ryb należy usunąć skórę, ponieważ najwyższy dopuszczalny poziom dotyczy mięsa bez skóry. Konieczne jest jednak dokładne i całkowite zdrapanie całego pozostałego mięsa i tkanki tłuszczowej z wewnętrznej strony skóry i dodanie ich do analizowanej próbki. |
5. WYMAGANIA DOTYCZĄCE LABORATORIÓW
|
— |
Zgodnie z przepisami rozporządzenia (WE) nr 882/2004 laboratoria powinny być akredytowane przez uznany organ działający zgodnie z przewodnikiem ISO 58, aby zagwarantować, że w przeprowadzaniu analiz stosują system zapewniania jakości. Laboratoria muszą posiadać akredytację zgodną z normą EN ISO/IEC 17025. |
|
— |
Profesjonalizm laboratoriów należy udowadniać stałym uczestnictwem w międzylaboratoryjnych badaniach oznaczania PCDD/F i dioksynopodobnych PCB w odpowiednich matrycach żywności i zakresach stężeń. |
|
— |
Laboratoria stosujące przesiewowe metody analizy do rutynowej kontroli próbek powinny nawiązać ścisłą współpracę z laboratoriami stosującymi metody potwierdzające, zarówno w celach kontroli jakości, jak i potwierdzania wyników analitycznych podejrzanych próbek. |
6. PODSTAWOWE WYMAGANIA DOTYCZĄCE PROCEDURY ANALITYCZNEJ DLA DIOKSYN (PCDD/F) I DIOKSYNOPODOBNYCH PCB
6.1. Niski zakres roboczy i granica oznaczalności
|
— |
W przypadku PCDD/F wykrywalne ilości muszą znajdować się na poziomie femtogramów (10–15 g) z uwagi na ekstremalną toksyczność niektórych spośród tych związków. W przypadku większości kongenerów PCB granica oznaczalności rzędu nanogramów (10–9 g) jest już wystarczająca. Jednakże w przypadku oznaczania bardziej toksycznych kongenerów dioksynopodobnych PCB (w szczególności kongenerów non-orto), dolna granica zakresu roboczego musi znajdować się na poziomie pikogramów (10–12 g). |
6.2. Wysoka selektywność (swoistość)
|
— |
Konieczne jest rozróżnienie PCDD/F i dioksynopodobnych PCB od wielu innych współekstrahowanych i ewentualnie interferujących związków występujących w stężeniach do kilku razy wyższych od stężeń interesujących analitów. W przypadku metod GC/MS (chromatografia gazowa/spektometria masowa) konieczne jest rozróżnienie między różnymi kongenerami, takimi jako kongenery toksyczne (np. siedemnaście PCDD/F z atomami chloru w pozycjach 2,3,7,8 oraz dwanaście dioksynopodobnych PCB) i pozostałe kongenery. |
|
— |
Metody bioanalityczne powinny być w stanie wykrywać docelowe związki jako sumę PCDD/F oraz dioksynopodobnych PCB. Oczyszczanie próbki ma na celu usunięcie związków dających wyniki fałszywie ujemne lub związków, które mogą zmniejszać odpowiedź, powodując wyniki fałszywie ujemne. |
6.3. Wysoka dokładność (prawdziwość i precyzja, odzysk uzyskany w oznaczeniu biologicznym)
|
— |
W przypadku metod GC/MS oznaczenie musi dostarczyć wiarygodnych szacunków rzeczywistego stężenia w próbce. Wysoka dokładność (dokładność pomiaru: stopień zgodności między wynikiem pomiaru a rzeczywistą lub domniemaną wartością mierzoną) jest konieczna, aby uniknąć odrzucenia wyniku analizy próbki z powodu słabej wiarygodności oznaczonego poziomu TEQ. Dokładność jest wyrażona jako prawdziwość (różnica między średnią wartością mierzoną w odniesieniu do analitu w certyfikowanym materiale a jego certyfikowaną wartością, wyrażona w procentach) i precyzja (RSDR – względne odchylenie standardowe obliczone na podstawie wyników osiągniętych w warunkach odtwarzalności). |
|
— |
W przypadku metod bioanalitycznych należy oznaczyć odzysk uzyskany w oznaczeniu biologicznym. |
6.4. Walidacja w zakresie poziomu zainteresowania oraz pomiary ogólnej kontroli jakości
|
— |
Laboratoria muszą wykazać zdolność metody w zakresie poziomu zainteresowania, np. 0,5-, 1- i 2-krotność poziomu zainteresowania wraz z akceptowalną wartością współczynnika zmienności obliczonego dla wielokrotnych powtórzeń analizy podczas procedury walidacji lub rutynowej analizy. |
|
— |
W ramach wewnątrzlaboratoryjnej kontroli jakości należy regularnie przeprowadzać ślepe próby i badania z wykorzystaniem próby wzbogaconej lub analizy próbek kontrolnych (zwłaszcza certyfikowanych materiałów referencyjnych, jeżeli materiały takie są dostępne). Wykresy kontroli jakości dla ślepych prób, badań z wykorzystaniem próby wzbogaconej lub analiz próbek kontrolnych należy rejestrować i kontrolować, aby upewnić się, że zdolność jest zgodna z wymaganiami. |
6.5. Granica oznaczalności
|
— |
W przypadku bioanalitycznej metody przesiewowej ustanowienie granicy oznaczalności (LOQ) nie jest niezbędnym wymogiem, ale należy dowieść, że metoda pozwala na rozróżnienie między ślepą próbą a wartością odcięcia. Podając poziom BEQ, należy ustalić poziom zgłaszania, aby uwzględnić próbki wykazujące odpowiedź poniżej tego poziomu. Należy wykazać, że poziom zgłaszania jest różny od procedury ślepej próby co najmniej o współczynnik 3 przy odpowiedzi poniżej zakresu roboczego. W związku z powyższym należy go obliczyć z próbek zawierających związki docelowe w granicach wymaganego poziomu minimalnego, a nie ze współczynnika S/N lub ze ślepej próby. |
|
— |
Granica oznaczalności (LOQ) dla metody potwierdzającej powinna wynosić ok. jednej piątej poziomu zainteresowania. |
6.6. Kryteria analityczne
|
— |
Dla uzyskania wiarygodnych wyników metodą potwierdzającą lub przesiewową należy spełnić poniższe kryteria dla wartości TEQ w odniesieniu do wartości BEQ, niezależnie do tego, czy są oznaczane jako całkowita wartość TEQ (jako suma PCDD/F i dioksynopodobnych PCB), czy oddzielnie dla PCDD/F i dioksynopodobnych PCB.
|
6.7. Szczegółowe wymagania dotyczące metod przesiewowych
|
— |
W metodach przesiewowych można stosować zarówno metody GC/MS, jak i metody bioanalityczne. W przypadku metod GC/MS zastosowanie mają wymagania ustanowione w pkt 7 niniejszego załącznika. W przypadku metod bioanalitycznych opierających się na oznaczeniach komórkowych zastosowanie mają szczegółowe wymogi ustanowione w pkt 8 niniejszego załącznika. |
|
— |
Laboratoria stosujące metody przesiewowe do rutynowej kontroli próbek powinny nawiązać ścisłą współpracę z laboratoriami stosującymi metodę potwierdzającą. |
|
— |
Weryfikacja zdolności metody przesiewowej jest wymagana podczas rutynowej analizy w drodze kontroli jakości analizy i walidacji metody podczas analizy. Należy prowadzić ciągły program kontroli wyników ujemnych. |
|
— |
Należy sprawdzać, czy nie zachodzi ewentualne tłumienie odpowiedzi komórek i cytotoksyczność 20 % ekstraktów próbek należy analizować rutynowo metodą przesiewową z dodanym 2,3,7,8-TCDD i bez, zgodnym z poziomem zainteresowania, aby sprawdzić, czy odpowiedź jest ewentualnie tłumiona przez substancje interferujące obecne w ekstrakcie próbki. Zmierzone stężenie próbki wzbogaconej jest porównywane do sumy stężenia ekstraktu próbki niewzbogaconej i stężenia wzbogacenia. Jeżeli zmierzone stężenie jest o ponad 25 % niższe niż obliczone (zsumowane) stężenie, wskazuje to na ewentualne tłumienie sygnału, a odpowiednia próbka musi zostać przekazana do analizy metodą potwierdzającej GC/HRMS. Wyniki należy monitorować na wykresach kontroli jakości. |
|
— |
Kontrola jakości próbek ujemnych Około 2–10 % próbek ujemnych, zależnie od matrycy próbki i doświadczenia laboratorium, należy potwierdzić metodą GC/HRMS. |
|
— |
Oznaczenie wskaźnika ilości wyników fałszywie ujemnych z danych kontroli jakości Należy oznaczyć wskaźnik ilości wyników fałszywie ujemnych z badań przesiewowych próbek poniżej i powyżej najwyższego dopuszczalnego poziomu lub progu podejmowania działań. Wskaźnik rzeczywistej ilości wyników fałszywie ujemnych nie powinien przekraczać 5 %. Po uzyskaniu co najmniej 20 potwierdzonych wyników na matrycę/grupę matryc z kontroli jakości próbek ujemnych należy z tej bazy danych wyciągnąć wnioski dotyczące wskaźnika ilości wyników fałszywie ujemnych. Do tych 20 wyników w celu oceny wskaźnika ilości wyników fałszywie ujemnych można także włączyć wyniki uzyskane na próbkach analizowanych w badaniach biegłości lub podczas incydentów wystąpienia zanieczyszczenia, obejmujące zakres stężeń do np. 2-krotności najwyższego dopuszczalnego stężenia. Próbki powinny obejmować najczęstsze profile kongenerów, reprezentujące różne źródła. Chociaż badania przesiewowe powinny mieć na celu wykrywanie próbek przekraczających próg podejmowania działań, kryterium określania wskaźnika ilości wyników fałszywie ujemnych jest najwyższy dopuszczalny poziom, przy uwzględnieniu niepewności pomiaru metody potwierdzającej. |
|
— |
Ewentualne dodatnie wyniki z badania przesiewowego powinny zawsze zostać zweryfikowane potwierdzającą metodą analizy (GC/HRMS). Próbki te mogą być też zastosowane do oceny odsetka wyników fałszywie dodatnich. W przypadku metod przesiewowych częstość wyników fałszywie dodatnich to odsetek wyników potwierdzonych jako ujemne metodą potwierdzającą GC/HRMS, które we wcześniejszych badaniach przesiewowych były podejrzewane jako dodatnie. Jednak ocena korzyści metody przesiewowej powinna być oparta na porównaniu próbek fałszywie dodatnich z całkowitą liczbą sprawdzonych próbek. Częstość ta musi być na tyle niska, aby stosowanie narzędzia przesiewowego było korzystne. |
|
— |
Metody bioanalityczne przynajmniej w warunkach walidacji powinny dostarczyć wiarygodnego wskazania poziomu TEQ, obliczonego i wyrażonego jako BEQ. |
|
— |
Także w przypadku metod bioanalitycznych przeprowadzonych w warunkach powtarzalności, wewnątrzlaboratoryjny RSDr byłby zwykle niższy niż odtwarzalność RSDR. |
7. SZCZEGÓŁOWE WYMAGANIA DOTYCZĄCE TECHNIK GC/HRMS W METODACH PRZESIEWOWYCH LUB POTWIERDZAJĄCYCH
7.1. Wymagania ogólne
|
— |
Różnica między poziomem górnej i dolnej granicy nie powinna przekraczać 20 % dla środków spożywczych zanieczyszczonych na poziomie ok. 1 pg WHO-TEQ/g tłuszczu (na podstawie sumy PCDD/F i dioksynopodobnych PCB). W przypadku środków spożywczych o niskiej zawartości tłuszczu stosuje się te same wymagania odnośnie do zanieczyszczeń na poziomie 1 pg WHO-TEQ/g produktu. W przypadku niższych poziomów zanieczyszczeń, np. 0,5 pg WHO-TEQ/g produktu, różnica między górną i dolną granicą może wynosić 25–40 %. |
7.2. Kontrola poziomów odzysku
|
— |
Rozpoczynając analizę, np. przed ekstrakcją, należy dodać wzorce wewnętrzne PCDD/F z atomami chloru w pozycjach 2,3,7,8 i znakowanych izotopem węgla 13C, oraz wzorce wewnętrzne dioksynopodobnych PCB znakowanych izotopem węgla 13C w celu walidacji procedury analitycznej. Należy dodać co najmniej 1 kongener dla każdej grupy homologicznej, od tetra- do oktachlorowanego PCDD/F oraz co najmniej 1 kongener dla każdej grupy homologicznej dioksynopodobnych PCB (alternatywnie, co najmniej 1 kongener na każdy wyselekcjonowany jon widma masowego, rejestrujący funkcję służącą do monitorowania PCDD/F i dioksynopodobnych PCB). W przypadku metod potwierdzających należy zastosować wszystkie siedemnaście wzorców wewnętrznych PCDD/F z atomami chloru w pozycjach 2,3,7,8 i znakowanych izotopem węgla 13C oraz wszystkie dwanaście wzorców wewnętrznych dioksynopodobnych PCB znakowanych izotopem węgla 13C. |
|
— |
Dla tych kongenerów, dla których nie zastosowano analogów znakowanych izotopem węgla 13C, należy również oznaczyć względne współczynniki odpowiedzi przez zastosowanie odpowiednich roztworów kalibracyjnych. |
|
— |
W przypadku środków spożywczych pochodzenia roślinnego i środków spożywczych pochodzenia zwierzęcego zawierających mniej niż 10 % tłuszczu dodawanie wzorców wewnętrznych przed ekstrakcją jest obowiązkowe. W przypadku środków spożywczych pochodzenia zwierzęcego zawierających więcej niż 10 % tłuszczu wzorce wewnętrzne mogą zostać dodane przed albo po ekstrakcji tłuszczu. Zależnie od etapu, na którym wprowadza się wzorce wewnętrzne, a także zależnie od tego, czy wyniki podaje się w odniesieniu do produktu, czy do tłuszczu, należy dokonać właściwej walidacji skuteczności ekstrakcji. |
|
— |
Przed analizą GC/MS należy dodać 1 lub 2 wzorce odzysku. |
|
— |
Konieczna jest kontrola odzysku. W przypadku metody potwierdzającej, odzysk poszczególnych wzorców wewnętrznych musi mieścić się w zakresie od 60 do 120 %. Dopuszczalne są niższe lub wyższe wartości odzysku dla poszczególnych kongenerów, w szczególności dla niektórych hepta- i oktachlorowanych dibenzo-p-dioksyn i dibenzofuranów, jeżeli ich udział w wartości TEQ nie przekracza 10 % całkowitej wartości TEQ (na podstawie PCDD/F oraz dioksynopodobnych PCB). W przypadku metod przesiewowych GC/MS odzysk powinien miesić się w zakresie od 30 do 140 %. |
7.3. Usuwanie substancji interferujących
|
— |
Rozdzielenie PCDD/F od interferujących związków chlorowanych, takich jak niedioksynopodobne PCB czy polichlorowane difenyloetery, należy przeprowadzić przez zastosowanie odpowiednich technik chromatograficznych (najlepiej stosując jako wypełnienie kolumny florisil, tlenek glinu lub węgiel). |
|
— |
Rozdzielanie izomerów metodą chromatografii gazowej musi być wystarczające (< 25% nakładania się pików pomiędzy 1,2,3,4,7,8-HxCDF i 1,2,3,6,7,8-HxCDF). |
7.4. Kalibracja przy zastosowaniu krzywej wzorcowej
|
— |
Zakres krzywej kalibracji powinien obejmować odpowiednie zakresy poziomów zainteresowania. |
8. SZCZEGÓŁOWE WYMAGANIA DOTYCZĄCE METOD BIOANALITYCZNYCH
Metody bioanalityczne oznaczają metody oparte na stosowaniu zasad biologicznych, np. oznaczenie wykorzystujące hodowle komórkowe, oznaczenie wykorzystujące receptor lub oznaczenie immunologiczne. W pkt 8 ustanowiono ogólne wymogi dla metod bioanalitycznych.
Metoda przesiewowa zasadniczo pozwala zaklasyfikować próbkę jako ujemną lub podejrzewaną jako dodatnią. W tym celu obliczony poziom BEQ jest porównywany z wartością odcięcia (zob. 8.3). Próbki poniżej wartości odcięcia są uznawane za ujemne, a próbki równe wartości odcięcia lub ją przekraczające – za podejrzewane jako dodatnie, wymagające analizy metodą potwierdzającą. W praktyce wartość BEQ odpowiadająca 2/3 najwyższego dopuszczalnego poziomu może posłużyć jako najbardziej odpowiednia wartość odcięcia, zapewniająca wskaźnik ilości wyników fałszywie ujemnych poniżej 5 % i dopuszczalną częstość wyników fałszywie dodatnich. Przy odrębnych najwyższych dopuszczalnych poziomach dla PCDD/F i dla sumy PCDD/F i dioksynopodobnych PCB sprawdzanie zgodności próbek bez frakcjonowania wymaga odpowiednich wartości odcięcia metodami biologicznymi dla PCDD/F. Dla sprawdzenia próbek przekraczających progi podejmowania działań jako wartość odcięcia wystarczyłby odpowiedni odsetek właściwego poziomu zainteresowania.
Ponadto w przypadku niektórych metod bioanalitycznych można podać indykatywny poziom wyrażony w BEQ dla próbek w zakresie roboczym i przekraczającym poziom zgłaszania (zob. 8.1.1. i 8.1.6.).
8.1. Ocena czułości badania
8.1.1. Wymagania ogólne
|
— |
Podczas obliczania stężenia z krzywej kalibracji TCDD wartości na dolnej i górnej granicy krzywej wykazywać będą wysoką zmienność (wysoki współczynnik zmienności, CV). Zakres roboczy to obszar, w którym CV jest mniejszy niż 15 %. Dolna granica zakresu roboczego (poziom zgłaszania) musi następnie zostać ustalona znacznie (co najmniej o współczynnik 3) powyżej procedury próby ślepej. Górna granica zakresu roboczego jest zwykle reprezentowana przez wartość EC70 (70 % najwyższego skutecznego stężenia), ale jest niższa, jeżeli CV jest wyższy niż 15 % w tym zakresie. Zakres roboczy powinien być ustalony podczas walidacji. Wartości odcięcia (pkt 8.3) muszą mieścić się w zakresie roboczym. |
|
— |
Roztwory standardowe i ekstrakty próbek należy zbadać co najmniej w duplikacie. Przy stosowaniu duplikatów roztwór standardowy lub ekstrakt kontrolny badany w 4–6 dołkach rozmieszczonych na płytce powinien wywołać odpowiedź lub dać stężenie (możliwe tylko w zakresie roboczym) w oparciu o CV < 15 %. |
8.1.2. Kalibracja
8.1.2.1.
|
— |
Poziomy w próbkach można oszacować przez porównanie czułości badania z krzywą kalibracyjną TCDD (lub PCB 126 albo mieszaniny wzorcowej PCDD/F/dioksynopodobnych PCB) w celu obliczenia poziomu BEQ w ekstrakcie, a następnie w próbce. |
|
— |
Krzywe kalibracji powinny zawierać 8–12 stężeń (co najmniej w duplikacie) z wystarczającą ilością stężeń w dolnych zakresach krzywej (zakres roboczy). Szczególną uwagę należy zwrócić na jakość dopasowania krzywej w roboczym zakresie. Wartość R2 jako taka ma niewielką lub żadną wartość w szacowaniu dopasowania w regresji nieliniowej. Lepsze dopasowanie jest osiągane poprzez minimalizowanie różnicy między obliczanym a obserwowanym poziomem w zakresie roboczym krzywej (np. przez minimalizowanie sumy kwadratów reszt). |
|
— |
Szacowany poziom w ekstrakcie próbki jest następnie korygowany o poziom BEQ obliczany dla ślepej próby matrycy/rozpuszczalnika (aby uwzględnić zanieczyszczenia z zastosowanych rozpuszczalników i chemikaliów) oraz pozorny odzysk (obliczany z poziomu BEQ odpowiednich próbek referencyjnych z reprezentatywnymi profilami kongenerów w granicach poziomu zainteresowania). Dla przeprowadzenia korekty odzysku pozorny odzysk musi zawsze mieścić się w wymaganym zakresie (zob. pkt 8.1.4.). Próbki referencyjne stosowane do korekty odzysku muszą być zgodne z wymogami podanymi w pkt 8.2. |
8.1.2.2.
Alternatywnie można zastosować krzywą kalibracji przygotowaną na co najmniej 4 próbkach referencyjnych (zob. pkt 8.2.: jedna ślepa próba matrycowa plus trzy próbki referencyjne o wartości 0,5-, 1- i 2-krotnego poziomu zainteresowania) w granicach poziomu zainteresowania, eliminując potrzebę korekty o próbę ślepą i odzysk. W takim przypadku czułość badania odpowiadająca 2/3 najwyższego dopuszczalnego poziomu (zob. 8.3.) może zostać obliczona bezpośrednio z tych próbek i zastosowana jako wartość odcięcia. Dla sprawdzenia próbek przekraczających progi podejmowania działań jako wartość odcięcia wystarczyłby odpowiedni procent tych progów podejmowania działań.
8.1.3. Oddzielne oznaczanie PCDD/F i dioksynopodobnych PCB
Ekstrakty można rozdzielać na frakcje zawierające PCDD/F i dioksynopodobne PCB, umożliwiając oddzielne oznaczanie poziomów TEQ dla PCDD/F i dioksynopodobnych PCB (wyrażonych w BEQ). Do oceny wyników dla frakcji zawierającej dioksynopodobne PCB najlepiej zastosować standardową krzywą kalibracyjną PCB 126.
8.1.4. Odzysk uzyskany w oznaczeniu biologicznym
„Odzysk uzyskany w oznaczeniu biologicznym” powinien zostać obliczony na odpowiednich próbkach referencyjnych z reprezentatywnymi profilami kongenerów w granicach poziomu zainteresowania i wyrażony jako odsetek poziomu BEQ w porównaniu do poziomu TEQ. Zależnie od typu badania i zastosowanych TEF (3) różnice między czynnikami TEF i REP dla dioksynopodobnych PCB mogą spowodować niski odzysk dla dioksynopodobnych PCB w porównaniu z PCDD/F. Dlatego, jeżeli przeprowadzane jest osobne oznaczenie PCDD/F i dioksynopodobnych PCB, odzysk uzyskany w oznaczeniu biologicznym powinien wynosić: dla dioksynopodobnych PCB: 25-60 %, dla PCDD/F: 50–130 % (zakresy stosowane są do krzywej kalibracji TCDD). Ponieważ udział dioksynopodobnych PCB w sumie PCDD/F i dioksynopodobnych PCB może różnić się zależnie od różnych matryc i próbek, odzysk uzyskany w oznaczeniu biologicznym dla sumy odzwierciedla te zakresy i powinien wynosić od 30 % do 130 %.
8.1.5. Kontrola poziomów odzysku na potrzeby oczyszczania
|
— |
Strata związków podczas oczyszczania powinna zostać sprawdzona podczas walidacji. Ślepą próbę wzbogaconą mieszaniną różnych kongenerów należy przekazać do oczyszczania (co najmniej n = 3), a odzysk i zmienność sprawdzić metodą GC/HRMS. Odzysk powinien wynosić od 60 do 120 % szczególnie dla kongenerów mających ponad 10 % udział w poziomie TEQ w różnych mieszaninach. |
8.1.6. Poziom zgłaszania
|
— |
Przy zgłaszaniu poziomów BEQ należy określić poziom zgłaszania na odpowiednich próbkach matrycy, obejmujących typowe profile kongenerów, ale nie z krzywej kalibracji wzorców ze względu na niską precyzję w dolnym zakresie krzywej. Należy uwzględnić wpływy z ekstrakcji i oczyszczania. Poziom zgłaszania należy ustalić znacznie (co najmniej o współczynnik 3) powyżej procedury próby ślepej. |
8.2. Stosowanie próbek referencyjnych
|
— |
Próbki referencyjne reprezentują matrycę próbki, profile kongenerów i zakresy stężeń dla PCDD/F i dioksynopodobnych PCB w granicach poziomu zainteresowania (najwyższy dopuszczalny poziom lub próg podejmowania działań). |
|
— |
Ślepą próbę procedury lub najlepiej ślepą próbę matrycową oraz próbkę referencyjną na poziomie zainteresowania należy włączyć do każdej serii badanej. Próbki te muszą zostać ekstrahowane i zbadane w tym samym czasie w identycznych warunkach. Próbka referencyjna musi dawać wyraźnie podwyższoną odpowiedź w porównaniu ze ślepą próbą, gwarantując przydatność badania. Próbki te można zastosować do korekty ślepej próby i korekty odzysku. |
|
— |
Próbki referencyjne wybrane do przeprowadzenia korekty odzysku powinny być reprezentatywne dla próbek badanych, co oznacza, że profile kongenerów nie powinny prowadzić do niedoszacowania poziomów. |
|
— |
Można włączyć dodatkowe próbki referencyjne zawierające np. 0,5- i 2-krotność poziomu zainteresowania, aby wykazać właściwą zdolność badania w zakresie zainteresowania do celów kontroli poziomu zainteresowania. Razem próbki te można zastosować do obliczania poziomów BEQ w próbkach badanych (8.1.2.2). |
8.3. Oznaczenie wartości odcięcia
Należy ustalić relację między wynikami bioanalitycznymi wyrażonymi w BEQ a wynikami GC/HRMS wyrażonymi w TEQ (np. w drodze kalibracji przy zastosowaniu dopasowanej matrycy, z użyciem próbek referencyjnych wzbogaconych o wartość 0-, 0,5-, 1- i 2-krotną najwyższego dopuszczalnego poziomu przy 6 powtórzeniach na każdym poziomie (n = 24)). Z tej relacji można oszacować współczynniki korekcji (ślepa próba i odzysk), jednak należy je sprawdzić w każdej serii badanej przez włączenie ślepej próby procedury/matrycowej i próbek odzysku (8.2).
Wartości odcięcia należy ustalić dla decyzji o zgodności próbki z najwyższymi dopuszczalnymi poziomami lub dla celów kontroli progów podejmowania działań, jeżeli ma to znaczenie, przy odpowiednich poziomach zainteresowania ustanowionych albo dla PCDD/F i dla dioksynopodobnych PCB osobno, albo dla sumy PCDD/F i dioksynopodobnych PCB. Są one reprezentowane przez dolną granicę dystrybucji wyników bioanalitycznych (skorygowaną o próbę ślepą i odzysk), odpowiadającą poziomowi decyzji GC/HRMS w oparciu o 95 % przedział ufności, zakładając wskaźnik ilości wyników fałszywie ujemnych na poziomie < 5 % oraz RSDR < 25 %. Poziom decyzji GC/HRMS to najwyższy dopuszczalny poziom, przy uwzględnieniu niepewności pomiaru.
W praktyce wartość odcięcia (w BEQ) może zostać obliczona w następujący sposób (zob. rysunek 1):
8.3.1. Zastosowanie dolnego pasma 95 % przedziału predykcji przy poziomie decyzji GC/HRMS

gdzie:
|
BEQDL |
wartość BEQ odpowiadająca poziomowi decyzji GC/HRMS, będącemu najwyższym dopuszczalnym poziomem przy uwzględnieniu niepewności pomiaru |
|
sy,x |
odchylenie standardowe reszt |
|
t α,f=m-2 |
czynnik Studenta (α = 5 %, f = stopnie swobody, jednostronne) |
|
m |
całkowita liczba punktów kalibracji (indeks j) |
|
n |
liczba powtórzeń na każdym poziomie |
|
xi |
stężenie próbki GC/HRMS (w TEQ) w punkcie kalibracji i |
|
|
średnia stężeń (w TEQ) wszystkich próbek kalibracji |

 parametr kwadratu sumy, i = indeks punktu kalibracji i
parametr kwadratu sumy, i = indeks punktu kalibracji i
8.3.2. Obliczenie z wyników bioanalitycznych (skorygowanych o próbę ślepą i odzysk) wielokrotnych analiz próbek (n ≥ 6) zanieczyszczonych na poziomie decyzji GC/HRMS, jako dolna granica dystrybucji danych przy odpowiadającej średniej wartości BEQ:
Wartość odcięcia = BEQDL – 1,64 × SDR
gdzie:
|
SDR |
odchylenie standardowe wyników badania biologicznego przy BEQDL, mierzone w warunkach odtwarzalności wewnątrzlaboratoryjnej |
8.3.3. Obliczenie średniej wartości wyników bioanalitycznych (w BEQ, skorygowanych o próbę ślepą i odzysk) z wielokrotnej analizy próbek (n ≥ 6) zanieczyszczonych na 2/3 poziomu zainteresowania. Jest to oparte na obserwacji, że ten poziom będzie mieścił się w granicach odcięcia oznaczonych zgodnie z pkt 8.3.1 lub 8.3.2.
Rysunek 1
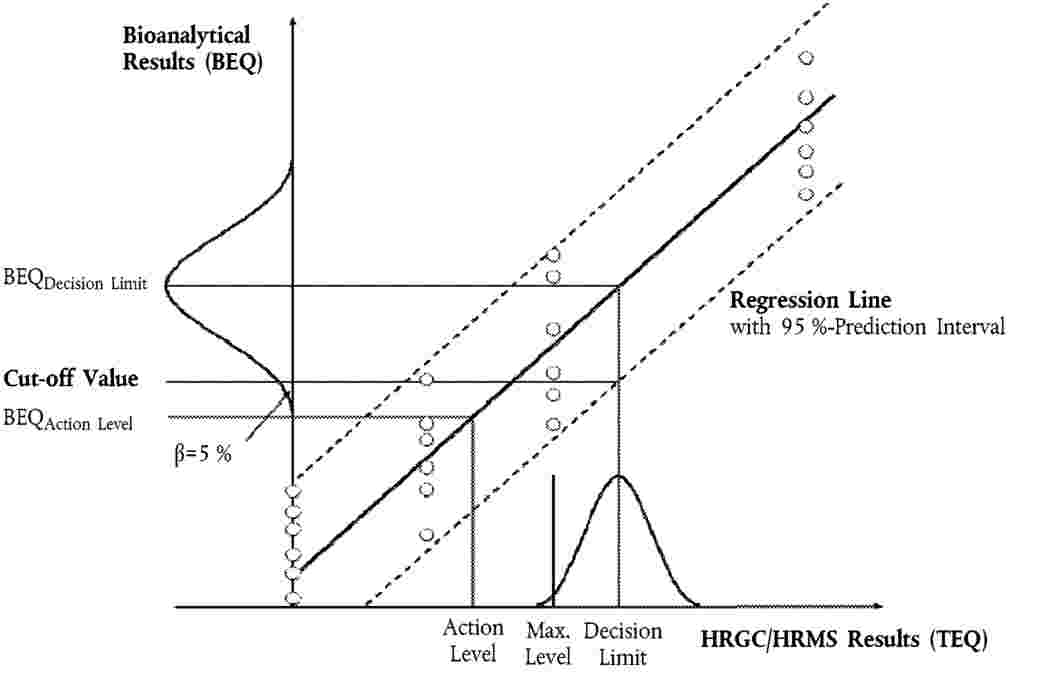
Obliczenie wartości odcięcia w oparciu o 95 % przedział ufności przy założeniu wskaźnika ilości wyników fałszywie ujemnych na poziomie < 5 % oraz RSDR < 25 %: 1. z dolnego pasma 95 % przedziału predykcji na poziomie decyzji HRGC/HRMS, 2. z wielokrotnej analizy próbek (n ≥ 6) zanieczyszczonych na poziomie decyzji HRGC/HRMS, jako dolna granica dystrybucji danych (reprezentowana na rysunku przez krzywą dzwonową) przy odpowiadającej średniej wartości BEQ.
8.3.4. Ograniczenia wartości odcięcia:
Wartości odcięcia oparte na BEQ obliczone z RSDR uzyskanego podczas walidacji przy zastosowaniu ograniczonej liczby próbek o różnych profilach matryc/kongenerów mogą być wyższe niż wartości zainteresowania oparte na TEQ ze względu na lepszą precyzję niż precyzja osiągana rutynowo, kiedy trzeba kontrolować nieznane spektrum możliwych profili kongenerów. W takich przypadkach wartości odcięcia należy obliczać z RSDR = 25 % lub należy wybrać 2/3 poziomu zainteresowania.
8.4. Parametry zdolności metody
|
— |
Ponieważ w metodach bioanalitycznych niemożliwe jest stosowanie wzorców wewnętrznych, należy przeprowadzić badania powtarzalności pozwalające na uzyskanie informacji o odchyleniu standardowym w obrębie jednej serii badanej i między tymi seriami. Powtarzalność powinna być na poziomie poniżej 20 %, odtwarzalność wewnątrzlaboratoryjna – poniżej 25 %. Wyniki te powinny opierać się na obliczonych poziomach w BEQ po korekcie próby ślepej i odzysku. |
|
— |
Jako część procesu walidacji należy wykazać, że badania pozwalają na odróżnienie między próbą ślepą a poziomem wartości odcięcia, umożliwiając identyfikację próbek powyżej odpowiadającej wartości odcięcia (zob. 8.1.2). |
|
— |
Należy zdefiniować docelowe związki, możliwe interferencje oraz najwyższy dopuszczalny poziom sygnału próby ślepej. |
|
— |
Procent odchylenia standardowego w odpowiedzi lub stężeniu obliczonym z odpowiedzi (możliwy tylko w zakresie roboczym) potrójnego oznaczenia ekstraktu próbki nie powinien przekraczać 15 %. |
|
— |
Do oceny zdolności metody bioanalitycznej w stałym okresie należy zastosować nieskorygowane wyniki próbek referencyjnych wyrażone w BEQ (próba ślepa i poziom zainteresowania). |
|
— |
Wykresy kontroli jakości dla próby ślepej procedury i każdego typu próbki referencyjnej należy zarejestrować i sprawdzić, aby upewnić się, że zdolność analityczna jest zgodna z wymogami, w szczególności dla próby ślepej procedury w odniesieniu do wymaganej minimalnej różnicy między dolną granicą zakresu roboczego oraz dla próbek referencyjnych w odniesieniu do odtwarzalności wewnątrzlaboratoryjnej. Próby ślepe procedury muszą być dobrze kontrolowane, aby uniknąć wyników fałszywie ujemnych podczas odejmowania. |
|
— |
Należy zgromadzić wyniki z analiz GC/HRMS podejrzanych próbek oraz 2-10 % próbek ujemnych (co najmniej 20 próbek na matrycę) i zastosować je do oceny zdolności metody przesiewowej oraz związku między BEQ i TEQ. Ta baza danych może zostać zastosowana do ponownej oceny wartości odcięcia stosowanej do rutynowych próbek dla zwalidowanych matryc. |
|
— |
Zdolność metody można także wykazać przez uczestnictwo w badaniach biegłości. Wyniki z próbek analizowanych w badaniach biegłości, obejmujących zakres stężeń do np. 2-krotności najwyższego dopuszczalnego poziomu, mogą zostać włączone do oceny wskaźnika ilości wyników fałszywie ujemnych, jeżeli laboratorium może wykazać zdolność metody. Próbki powinny obejmować najczęstsze profile kongenerów, reprezentujące różne źródła. |
|
— |
Podczas incydentów można ponownie ocenić wartości odcięcia, odzwierciedlające konkretne profile matryc i kongenerów pojedynczego incydentu. |
9. PRZEDSTAWIANIE WYNIKÓW
Metody potwierdzające
|
— |
Jeśli zastosowana procedura analityczna to umożliwia, wyniki analiz powinny zawierać poziomy poszczególnych kongenerów PCDD/F i dioksynopodobnych PCB i być przedstawiane jako ich górne, dolne i średnie granice, aby zawrzeć w sprawozdaniu maksymalną ilość informacji umożliwiających dokonanie interpretacji wyników zgodnie z konkretnymi wymaganiami. |
|
— |
Sprawozdanie powinno także zawierać opis metody zastosowanej do ekstrakcji PCDD/F, dioksynopodobnych PCB i tłuszczów. W sprawozdaniu należy oznaczyć i podać zawartość tłuszczu w próbce dla próbek żywności o najwyższym dopuszczalnym poziomie lub progu podejmowania działań ustalonych w odniesieniu do tłuszczu i oczekiwanym stężeniu tłuszczu w zakresie 0-2 % (zgodnie z obowiązującym prawodawstwem); w przypadku pozostałych próbek oznaczenie zawartości tłuszczu jest opcjonalne. |
|
— |
Należy udostępnić poziomy odzysku poszczególnych wzorców wewnętrznych, jeżeli te poziomy odzysku znajdują się poza zakresem wymienionym w pkt 7.2 lub przekraczają najwyższy dopuszczalny poziom, a w pozostałych przypadkach na żądanie. |
|
— |
Ponieważ przy ustalaniu zgodności próbki uwzględnia się niepewność pomiaru, należy podać również ten parametr. W związku z powyższym wyniki badań należy podawać jako x +/- U, gdzie x oznacza wynik analizy, a U rozszerzoną niepewność pomiaru, przy zastosowaniu współczynnika rozszerzenia w wysokości 2, który daje wynik w przedziale ufności ok. 95 %. W przypadku odrębnego oznaczania PCDD/F i dioksynopodobnych PCB należy zastosować sumę szacowanej niepewności rozszerzonej dla odrębnych wyników analitycznych dla PCDD/F i dioksynopodobnych PCB w odniesieniu do sumy PCDD/F i dioksynopodobnych PCB. |
|
— |
Jeżeli niepewność pomiaru jest uwzględniana przez zastosowanie CCα (zob. załącznik II pkt IV.2), należy podać również ten parametr. |
|
— |
Wyniki są wyrażane w tych samych jednostkach i z (co najmniej) tą samą liczbą cyfr znaczących, co najwyższe dopuszczalne poziomy określone w rozporządzeniu (WE) nr 1881/2006. |
Bioanalityczne metody przesiewowe
|
— |
Wynik badań przesiewowych wyrażany jest jako zgodny lub podejrzewany o niezgodność („podejrzany”). |
|
— |
Ponadto można podać wynik dla PCDD/F lub dla dioksynopodobnych PCB wyrażony w równoważnikach bioanalitycznych (BEQ) (nie TEQ) (zob. załącznik III pkt 2). |
|
— |
Jeżeli podawana jest niepewność pomiaru obliczonego poziomu BEQ, np. jako odchylenie standardowe, musi być oparta co najmniej na trzykrotnej analizie (obejmującej ekstrakcję, oczyszczanie i oznaczenie czułości badania) próbki. |
|
— |
Próbki dające odpowiedź poniżej poziomu zgłaszania należy oznaczyć jako poniżej poziomu zgłaszania. |
|
— |
W sprawozdaniu, dla każdego typu matrycy próbki, należy podać poziom zainteresowania (najwyższy dopuszczalny poziom, próg podejmowania działań), na którym opiera się ocena. |
|
— |
W sprawozdaniu należy podać zastosowany rodzaj badania, podstawowe zasady badania i rodzaj kalibracji. |
|
— |
Sprawozdanie powinno także zawierać opis metody zastosowanej do ekstrakcji PCDD/F, dioksynopodobnych PCB i tłuszczów. W sprawozdaniu należy oznaczyć i podać zawartość tłuszczu w próbce dla próbek żywności o najwyższym dopuszczalnym poziomie lub progu podejmowania działań ustalonych w odniesieniu do tłuszczu i oczekiwanym stężeniu tłuszczu w zakresie 0–2 % (zgodnie z obowiązującym prawodawstwem); w przypadku pozostałych próbek oznaczenie zawartości tłuszczu jest opcjonalne. |
(1) Dostosowane do PCDD/F i związków dioksynopodobnych z „Guidelines for the validation of screening methods for residues of veterinary medicines” (Wytyczne walidacji metod przesiewowych dla pozostałości leków weterynaryjnych), Laboratoria referencyjne UE ds. pozostałości leków weterynaryjnych i zanieczyszczeń w żywności pochodzenia zwierzęcego w Fougeres, Berlinie i Bilthoven, 20/1/2010, http://ec.europa.eu/food/food/chemicalsafety/residues/lab_analysis_en.htm
(2) W odniesieniu do najwyższych dopuszczalnych poziomów.
(3) Aktualne wymogi są oparte na TEF opublikowanych w: M. Van den Berg et al, Toxicol Sci 93 (2), 223–241 (2006).
Dodatek do ZAŁĄCZNIKA III
WHO-TEF dla oceny zagrożenia dla ludzi, na podstawie konkluzji Światowej Organizacji Zdrowia (WHO) – spotkanie ekspertów Międzynarodowego Programu Bezpieczeństwa Chemicznego (IPCS), które odbyło się w Genewie w czerwcu 2005 r. (Martin van den Berg et al., „(Ponowna ocena współczynników równoważnych toksyczności dla ludzi i ssaków w odniesieniu do dioksyn i związków dioksynopodobnych, przeprowadzona w 2005 r. przez Światową Organizację Zdrowia)”, Toxicological Sciences 93(2), 223–241 (2006))
|
Kongener |
Wartość TEF |
|
Dibenzo-p-dioksyny („PCDD”) |
|
|
2,3,7,8-TCDD |
1 |
|
1,2,3,7,8-PeCDD |
1 |
|
1,2,3,4,7,8-HxCDD |
0,1 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
|
OCDD |
0,0003 |
|
Dibenzofurany („PCDF”) |
|
|
2,3,7,8-TCDF |
0,1 |
|
1,2,3,7,8-PeCDF |
0,03 |
|
2,3,4,7,8-PeCDF |
0,3 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
OCDF |
0,0003 |
|
„Dioksynopodobne” PCB non-orto PCB oraz mono-orto PCB |
|
|
Non-orto PCB |
|
|
PCB 77 |
0,0001 |
|
PCB 81 |
0,0003 |
|
PCB 126 |
0,1 |
|
PCB 169 |
0,03 |
|
Mono-orto PCB |
|
|
PCB 105 |
0,00003 |
|
PCB 114 |
0,00003 |
|
PCB 118 |
0,00003 |
|
PCB 123 |
0,00003 |
|
PCB 156 |
0,00003 |
|
PCB 157 |
0,00003 |
|
PCB 167 |
0,00003 |
|
PCB 189 |
0,00003 |
|
Zastosowane skróty: „T” - tetra (cztero); „P” - penta (pięcio); „Hx” - heksa (sześcio); „Hp” - hepta (siedmio); „O” - okta (ośmio); „CDD” - chlorodibenzodioksyna; „CDF” - chlorodibenzofuran; „CB” - chlorobifenyl. |
|
ZAŁĄCZNIK IV
Przygotowanie próbki i wymagania dotyczące metod analizy stosowane w kontroli urzędowej poziomów niedioksynopodobnych PCB (PCB # 28, 52, 101, 138, 153, 180) w niektórych środkach spożywczych
1. Możliwe metody oznaczania
Chromatografia gazowa / detekcja wychwytu elektronów (GC/ECD), GC/LRMS, GC/MS-MS, GC/HRMS lub metody równorzędne.
2. Identyfikacja i potwierdzenie interesujących analitów
|
— |
Względny czas retencji w stosunku do wzorców wewnętrznych lub referencyjnych (akceptowane odchylenia +/- 0,25 %). |
|
— |
ROZDZIELENIE metodą chromatografii gazowej wszystkich sześciu wskaźnikowych PCB (PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 oraz PCB 180) od substancji interferujących, przede wszystkich wymywających PCB, w szczególności jeżeli poziomy próbek mieszczą się w zakresie granic ustanowionych w przepisach i jeżeli należy potwierdzić niezgodność. Uwaga: Kongenery, które są często wymywane, to np. PCB 28/31, PCB 52/69 oraz PCB 138/163/164. W przypadku GC/MS należy rozważyć także ewentualne interferencje z fragmentów wyżej chlorowanych kongenerów. |
|
— |
W przypadku technik GC/MS:
|
|
— |
W przypadku GC/ECD: Potwierdzenie wyników przekraczających tolerancję przy pomocy dwóch kolumn GC z fazami stacjonarnymi o różnej polarności. |
3. Wykazanie zdolności metody
Walidacja w zakresie poziomu zainteresowania (0,5- do 2-krotności poziomu zainteresowania) przy akceptowanym współczynniku zmienności dla powtarzanej analizy (zob. wymagania dotyczące precyzji pośredniej w pkt 8).
4. Granica oznaczania
Wartości próby ślepej nie mogą być wyższe niż 30 % poziomu zanieczyszczenia odpowiadającego najwyższemu dopuszczalnemu poziomowi (2).
5. Kontrola jakości
Regularne analizy próby ślepej, analiza próbek wzbogaconych, kontrola jakości próbek, uczestnictwo w międzylaboratoryjnych badaniach odpowiednich matryc.
6. Kontrola poziomów odzysku
|
— |
Stosowanie właściwych wzorców wewnętrznych o właściwościach fizycznych i chemicznych porównywalnych z właściwościami interesujących analitów. |
|
— |
Dodanie wzorców wewnętrznych:
|
|
— |
Wymagania dla metod z wykorzystaniem wszystkich sześciu wskaźnikowych kongenerów PCB znakowanych izotopowo:
|
|
— |
Wymagania dla metod z wykorzystaniem nie wszystkich sześciu wzorców wewnętrznych znakowanych izotopowo lub innych wzorców wewnętrznych:
|
|
— |
Poziomy odzysku kongenerów nieznakowanych należy sprawdzić przez próbki wzbogacone lub kontrolę jakości próbek o stężeniach w zakresie poziomu zainteresowania. Akceptowalne poziomy odzysku dla tych kongenerów wynoszą między 70 a 120 %. |
7. Wymagania dotyczące laboratoriów
Zgodnie z przepisami rozporządzenia (WE) nr 882/2004 laboratoria powinny być akredytowane przez uznany organ działający zgodnie z przewodnikiem ISO 58, aby zagwarantować, że w przeprowadzaniu analiz stosują system zapewniania jakości. Laboratoria muszą posiadać akredytację zgodną z normą EN ISO/IEC 17025.
8. Parametry zdolności: kryteria dla sumy sześciu wskaźnikowych PCB na poziomie zainteresowania
|
Prawdziwość |
– 30 % do + 30 % |
|
Precyzja pośrednia (RSD%) |
≤ 20 % |
|
Różnica między wyższą a niższą granicą obliczenia |
≤ 20 % |
9. Przedstawianie wyników
|
— |
Jeśli zastosowana procedura analityczna to umożliwia, wyniki analiz powinny zawierać poziomy poszczególnych kongenerów PCB i być przedstawiane jako ich górne, dolne i średnie granice, aby zawrzeć w sprawozdaniu maksymalną ilość informacji umożliwiających dokonanie interpretacji wyników zgodnie z konkretnymi wymaganiami. |
|
— |
Sprawozdanie powinno także zawierać opis metody zastosowanej do ekstrakcji PCB i tłuszczów. W sprawozdaniu należy oznaczyć i podać zawartość tłuszczu w próbce dla próbek żywności o najwyższym dopuszczalnym poziomie ustalonych w odniesieniu do tłuszczu i oczekiwanym stężeniu tłuszczu w zakresie 0-2 % (zgodnie z obowiązującym prawodawstwem); w przypadku pozostałych próbek oznaczenie zawartości tłuszczu jest opcjonalne. |
|
— |
Należy udostępnić poziomy odzysku poszczególnych wzorców wewnętrznych, jeżeli te poziomy odzysku znajdują się poza zakresem wymienionym w pkt 6 lub przekraczają najwyższy dopuszczalny poziom, a w pozostałych przypadkach na żądanie. |
|
— |
Ponieważ przy ustalaniu zgodności próbki uwzględnia się niepewność pomiaru, należy podać również ten parametr. W związku z powyższym wyniki badań należy podawać jako x +/- U, gdzie x oznacza wynik analizy, a U rozszerzoną niepewność pomiaru, przy zastosowaniu współczynnika rozszerzenia w wysokości 2, który daje wynik w przedziale ufności ok. 95 %. |
|
— |
Jeżeli niepewność pomiaru jest uwzględniana przez zastosowanie CCα (zob. załącznik II pkt IV.1), należy podać również ten parametr. |
|
— |
Wyniki są wyrażane w tych samych jednostkach i z (co najmniej) tą samą liczbą cyfr znaczących, co najwyższe dopuszczalne poziomy określone w rozporządzeniu (WE) nr 1881/2006. |
(1) Dostępna wystarczająca liczba fragmentów masy o względnej intensywności > 10 %, dlatego nie zaleca się stosowania jonów pomocniczych o względnej intensywności mniejszej niż 10 % w porównaniu z jonem docelowym.
(2) Zaleca się, aby w próbce znajdował się niższy poziom odczynnika próby ślepej względem poziomu zanieczyszczenia. Laboratorium odpowiada za kontrolowanie zróżnicowania poziomów próby ślepej, w szczególności, jeżeli poziomy próby ślepej są odejmowane.