|
8)
|
In deel B worden de volgende hoofdstukken toegevoegd:
„B.63 REPRODUCTIE-/ONTWIKKELINGSTOXICITEIT, SCREENINGTEST
INLEIDING
|
1.
|
Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 421 (2016) van de OESO. OESO-richtlijnen voor het testen van chemische stoffen worden periodiek in het licht van de wetenschappelijke vorderingen getoetst. De oorspronkelijke screeningtestrichtlijn 421 werd vastgesteld in 1995 op basis van een protocol voor een voorbereidende screeningtest voor reproductietoxiciteit (Preliminary Reproduction Toxicity Screening Test) die was besproken op twee bijeenkomsten van deskundigen, in 1990 in Londen (1) en in 1992 in Tokio (2).
|
|
2.
|
Deze testmethode is bijgewerkt met eindpunten die relevant zijn voor hormoonontregelaars, naar aanleiding van de prioritaire actie die de OESO in 1998 is gestart om bestaande testrichtlijnen te herzien en nieuwe testrichtlijnen te ontwikkelen voor het screenen en testen op potentiële hormoonontregelaars (3). Zo werd OESO TG 407 (toxiciteitsonderzoek (oraal) op knaagdieren bij herhaalde toediening (28 dagen), hoofdstuk B.7 van deze bijlage) in 2008 uitgebreid door er geschikte parameters in op te nemen om de endocriene werking van teststoffen vast te kunnen stellen. Met de bijwerking van TG 421 werd beoogd om in screeningtestrichtlijnen een aantal voor hormoonontregelaars relevante eindpunten op te nemen, waarbij de blootstellingsperioden bepaalde gevoelige perioden in de ontwikkeling (prenatale en vroeg-postnatale perioden) beslaan.
|
|
3.
|
De aanvullende, voor hormoonontregelaars relevante eindpunten die werden geselecteerd en die ook deel uitmaken van TG 443 (uitgebreid onderzoek naar reproductietoxiciteit over één generatie, hoofdstuk B.56 van deze bijlage), werden in TG 421 opgenomen op basis van een haalbaarheidsstudie waarin wetenschappelijke en technische vragen met betrekking tot de opname ervan werden onderzocht, alsook aanpassingen van de testopzet die eventueel voor de opname ervan noodzakelijk zouden zijn (4).
|
|
4.
|
Deze testmethode is ontworpen voor het verkrijgen van beperkte informatie over de effecten van een teststof op het mannelijke en vrouwelijke voortplantingsvermogen, zoals de gonadale functie, paringsgedrag, conceptie, ontwikkeling van de vrucht en geboorte. Deze testmethode is geen alternatief noch een vervanging voor de bestaande testmethoden B.31, B.34, B.35 of B.56.
|
INLEIDENDE OVERWEGINGEN
|
5.
|
Deze screeningtestmethode kan worden gebruikt om oriënterende gegevens te verkrijgen over mogelijke effecten op de voortplanting en/of ontwikkeling, hetzij in een vroeg stadium van de beoordeling van de toxicologische eigenschappen van chemische stoffen, hetzij voor chemische stoffen waarbij dergelijke effecten worden vermoed. De methode kan ook worden gebruikt als deel van een reeks initiële screeningtests voor bestaande chemische stoffen waarvoor weinig of geen toxicologische gegevens beschikbaar zijn, als bereikbepalingsonderzoek voor uitvoeriger onderzoeken naar reproductie-/ontwikkelingstoxiciteit, of wanneer het gebruik ervan anderszins relevant wordt geacht. Bij het uitvoeren van het onderzoek moeten de richtsnoeren en overwegingen worden gevolgd die worden beschreven in de OESO-leidraad nr. 19 over het herkennen, beoordelen en gebruiken van klinische verschijnselen als humane eindpunten voor proefdiergebruik bij veiligheidsbeoordelingen (5).
|
|
6.
|
Deze testmethode verschaft geen volledige informatie over alle aspecten van reproductie en ontwikkeling. Met name voor het detecteren van postnatale gevolgen van prenatale blootstelling of effecten die tijdens postnatale blootstelling zijn veroorzaakt, is de informatie die deze methode oplevert slechts beperkt. Als gevolg van (onder andere) het betrekkelijk kleine aantal dieren in de dosisgroepen, de selectiviteit van de eindpunten en de korte onderzoeksduur, kan met deze methode geen bewijs worden verkregen om definitief te concluderen dat een stof geen effecten veroorzaakt. Als er verder geen gegevens zijn uit andere tests op reproductie-/ontwikkelingstoxiciteit, zijn positieve resultaten evenwel nuttig voor een eerste gevarenbeoordeling en kunnen ze als basis dienen voor beslissingen over de noodzaak en timing van aanvullende tests.
|
|
7.
|
De resultaten die worden verkregen met de parameters die met het endocriene systeem te maken hebben, moeten worden gezien in samenhang met het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen (6). In dit conceptueel kader is de uitgebreide TG 421 van de OESO ondergebracht in niveau 4 als eenin-vivobepaling waarmee gegevens kunnen worden verkregen over schadelijke effecten op eindpunten die relevant zijn voor het endocrien systeem. Een endocrien signaal hoeft op zichzelf echter niet als voldoende bewijs te worden beschouwd dat de teststof een hormoonontregelaar is.
|
|
8.
|
Bij deze testmethode wordt aangenomen dat de teststof oraal wordt toegediend. Als andere blootstellingsroutes worden gebruikt, moet de methode mogelijk worden aangepast.
|
|
9.
|
Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is.
|
|
10.
|
De gebruikte definities zijn opgenomen in aanhangsel 1.
|
PRINCIPE VAN DE TEST
|
11.
|
De teststof wordt in geleidelijk oplopende doseringen aan verschillende groepen mannetjes en vrouwtjes toegediend. De mannetjes moeten ten minste vier weken lang de teststof toegediend krijgen, tot en met de dag voordat ze worden gedood (inclusief twee weken voorafgaand aan het paren, tijdens de paringsperiode en ongeveer twee weken na het paren). Gezien de beperkte doseringsperiode vóór paring, is de vruchtbaarheid mogelijk geen bijzonder gevoelige indicator voor testiculaire toxiciteit. Een uitvoerig histologisch onderzoek van de testes is daarom noodzakelijk. De combinatie van een twee weken durende doseringsperiode voor het paren, gevolgd door waarnemingen op het gebied van paring/vruchtbaarheid, met een totale doseringsperiode van ten minste vier weken gevolgd door een uitvoerige histopathologie van de mannelijke geslachtsklieren, wordt voldoende geacht om het merendeel van de effecten op de mannelijke vruchtbaarheid en de spermatogenese te kunnen detecteren.
|
|
12.
|
De vrouwtjes moeten het hele onderzoek lang de teststof toegediend krijgen. Dit betekent twee weken voorafgaand aan het paren (zodat het onderzoek ten minste twee volledige oestruscycli omvat), de variabele tijd tot conceptie, de duur van de dracht en ten minste dertien dagen na het werpen, tot en met de dag voordat ze worden gedood.
|
|
13.
|
De duur van het onderzoek, na acclimatisering en beoordeling van de oestruscyclus voorafgaand aan de behandeling, is afhankelijk van het gedrag van het vrouwtje en beloopt ongeveer 63 dagen (ten minste 14 dagen vóór het paren, (tot) 14 dagen paring, 22 dagen dracht, en ten slotte een zoogperiode van 13 dagen).
|
|
14.
|
Gedurende de periode dat de stof wordt toegediend, worden de dieren dagelijks zorgvuldig geobserveerd om tekenen van toxiciteit te ontdekken. Bij dieren die tijdens de testperiode sterven of worden gedood, wordt obductie verricht. Dieren die aan het eind van de test nog in leven zijn, worden gedood en ook hierop wordt obductie verricht.
|
BESCHRIJVING VAN DE METHODE
Keuze van de diersoort
|
15.
|
Deze testmethode is ontworpen om te worden toegepast met ratten. Indien de in deze testmethode gespecificeerde parameters worden onderzocht bij een andere knaagdiersoort moet dit uitvoerig worden gemotiveerd. In het internationale valideringsprogramma voor de detectie van hormoonontregelaars in OESO TG 407 (die overeenkomt met hoofdstuk B.7 van deze bijlage) zijn alleen ratten gebruikt. Stammen met een lage vruchtbaarheid of waarvan bekend is dat er vaak ontwikkelingsstoornissen optreden, mogen niet worden gebruikt. Er moet gebruikgemaakt worden van gezonde dieren die nog niet eerder gepaard hebben en nog niet eerder aan experimenten zijn onderworpen. Van de proefdieren moeten soort, stam, geslacht, gewicht en leeftijd bekend zijn. Per geslacht mag bij aanvang van het onderzoek het gewicht van de dieren die worden gebruikt, niet meer dan 20 % van het gemiddelde gewicht afwijken. Wanneer het onderzoek wordt uitgevoerd als voorstudie voor een onderzoek van langere duur of een onderzoek over een volledige generatie, verdient het de voorkeur dat de dieren in beide onderzoeken tot dezelfde stam behoren en dezelfde oorsprong hebben.
|
Huisvesting en voeding
|
16.
|
Alle procedures moeten in overeenstemming zijn met de plaatselijke normen voor de verzorging van proefdieren. De temperatuur in de proefdierruimte moet 22 °C (± 3 °) zijn. De relatieve luchtvochtigheid moet ten minste 30 % zijn en (behalve tijdens het schoonmaken van de ruimte) bij voorkeur niet hoger dan 70 % zijn, maar er moet worden gestreefd naar 50-60 %. Verlichting gebeurt met kunstlicht met een fotoperiode van 12 uur licht en 12 uur donker. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. De keuze van het voer kan worden beïnvloed door de noodzaak om voor een afdoende mengbaarheid met de teststof te zorgen, wanneer de stof in het voer wordt toegediend.
|
|
17.
|
De dieren moeten in kleine groepen van hetzelfde geslacht worden gehuisvest; de dieren kunnen individueel worden gehuisvest als dat wetenschappelijk gerechtvaardigd is. Indien de dieren in groepen in kooien worden ondergebracht, mogen per kooi niet meer dan vijf dieren worden gehuisvest. Het paren moet plaatsvinden in kooien die voor dat doel geschikt zijn. Drachtige vrouwtjes moeten in aparte kooien worden ondergebracht en nestmateriaal krijgen. Zogende vrouwtjes moeten samen met hun jongen in aparte kooien worden ondergebracht.
|
|
18.
|
Het voeder moet regelmatig op verontreinigingen worden onderzocht. Een monster van het voedsel moet worden bewaard totdat het verslag is afgerond.
|
Voorbereiding van de dieren
|
19.
|
Gezonde jonge volwassen dieren worden aselect ingedeeld in de controle- en behandelgroepen. De kooien worden zodanig geplaatst dat mogelijke effecten door de plaatsing van de kooi tot een minimum worden beperkt. De dieren krijgen een unieke identificatie en blijven, voordat de test begint, minimaal vijf dagen in hun kooi om in het laboratorium te acclimatiseren.
|
Voorbereiding van de doses
|
20.
|
Aanbevolen wordt om de teststof oraal toe te dienen, tenzij andere toedieningswegen geschikter worden geacht. Wanneer voor de orale weg wordt gekozen, wordt de teststof gewoonlijk via een sonde toegediend; als alternatieve mogelijkheid kunnen teststoffen echter ook via het voer of het drinkwater worden toegediend.
|
|
21.
|
Waar nodig wordt de teststof in een geschikt vehiculum opgelost of gesuspendeerd. Aanbevolen wordt eerst na te gaan of het mogelijk is een waterige oplossing/suspensie te gebruiken, als dit niet kan een oplossing/emulsie in olie (bv. maïsolie) te overwegen en daarna eventueel andere vehicula. Wanneer een ander vehiculum dan water wordt gebruikt, moeten de toxische kenmerken daarvan bekend zijn. Voorts moeten de stabiliteit en homogeniteit van de teststof in het vehiculum worden bepaald.
|
PROCEDURE
Aantal en geslacht van de dieren
|
22.
|
Aanbevolen wordt dat elke groep aan het begin van de test ten minste 10 mannetjes en 12-13 vrouwtjes telt. Bij de vrouwtjes wordt vóór blootstelling de oestruscyclus beoordeeld. Dieren die geen normale 4-5-daagse cyclus hebben, worden niet het onderzoek opgenomen. Daarom wordt aanbevolen met extra vrouwtjes te beginnen om uiteindelijk op 10 vrouwtjes per groep uit te komen. Behalve in geval van duidelijke toxische effecten wordt verwacht dat dit ten minste 8 drachtige vrouwtjes per groep zal opleveren, hetgeen normaal gesproken het minimaal aanvaardbare aantal drachtige vrouwtjes per groep is. Het doel is voldoende drachten en nakomelingen tot stand te brengen om een betekenisvolle evaluatie mogelijk te maken van de mogelijke invloed van de stof op de vruchtbaarheid, de dracht en het gedrag van moeder en jongen, en op de groei en ontwikkeling van de F1-nakomelingen, vanaf de conceptie tot dag 13 na het werpen.
|
Dosering
|
23.
|
In het algemeen zijn drie testgroepen en een controlegroep vereist. De dosisniveaus kunnen worden gebaseerd op informatie uit acute-toxiciteitstests of op de resultaten van onderzoeken bij herhaalde toediening. Afgezien van de toediening van de teststof worden de dieren in de controlegroep op identieke wijze behandeld als de dieren in de testgroepen. Indien bij de toediening van de teststof van een vehiculum gebruik wordt gemaakt, moet dit aan de controlegroep in het hoogste gebruikte volume toegediend worden.
|
|
24.
|
De dosisniveaus moeten worden gekozen in het licht van de bestaande gegevens over toxiciteit en (toxico)kinetica. Er moet ook rekening mee gehouden worden dat er verschillen kunnen zijn in gevoeligheid tussen drachtige en niet-drachtige dieren. Het hoogste dosisniveau moet zo worden gekozen dat toxische effecten optreden, maar geen sterfte of ernstig lijden. Daarna moet een dalende reeks dosisniveaus worden gekozen teneinde een eventuele doseringsgerelateerde respons en het laagste dosisniveau waarbij geen schadelijk effect werd vastgesteld (No-Observed Adverse Effect Level, NOAEL) aan te tonen. Intervallen van een factor twee of vier zijn vaak optimaal om de dalende dosisniveaus vast te stellen en het is vaak beter een vierde testgroep toe te voegen dan zeer grote intervallen (bv. meer dan een factor 10) tussen de doseringen te gebruiken.
|
|
25.
|
Bij algemene toxiciteit (bv. verminderd lichaamsgewicht, effecten op lever, hart, longen of nieren enz.) of andere veranderingen die mogelijk geen toxische reacties zijn (bv. verminderde voedselinname, leververgroting), moeten de waargenomen effecten op hormoongevoelige eindpunten met de nodige omzichtigheid worden geïnterpreteerd.
|
Limiettest
|
26.
|
Als volgens de hier beschreven procedures een onderzoek met orale toediening wordt uitgevoerd met één dosisniveau van ten minste 1 000 mg/kg lichaamsgewicht/dag of, in het geval van toediening via het voer of het drinkwater, het equivalente percentage in het voer of het drinkwater, en er geen waarneembare toxische effecten optreden, en deze ook niet kunnen worden verwacht op grond van gegevens betreffende stoffen met verwante structuur, is het niet noodzakelijk een volledige test met meerdere dosisniveaus te doen. De limiettest is bruikbaar, behalve wanneer vanwege de verwachte blootstelling van de mens een hogere orale dosis nodig wordt geacht. Voor andere toedieningsvormen, zoals inhalatie of toediening op de huid, zal de maximaal haalbare concentratie vaak worden bepaald door de fysisch-chemische eigenschappen van de teststoffen.
|
Toediening van de doses
|
27.
|
De dieren krijgen de teststof 7 dagen per week dagelijks toegediend. Indien de teststof via een maagsonde aan het dier wordt toegediend, moet dit in één enkele dosis gebeuren met gebruikmaking van een maagbuisje of een geschikte intubatiecanule. Het maximale volume dat in één keer kan worden toegediend, is afhankelijk van de grootte van het dier. Het volume mag niet groter zijn dan 1 ml/100 g lichaamsgewicht, behalve wanneer het om een waterige oplossing gaat: in dat geval mag 2 ml/100 g lichaamsgewicht worden gebruikt. Afgezien van irriterende of bijtende teststoffen die normaliter bij hogere concentraties ergere effecten vertonen, moet de variabiliteit in het voor de proef gebruikte volume zo klein mogelijk worden gehouden door de concentratie zodanig aan te passen dat bij alle dosisniveaus een constant volume wordt gewaarborgd.
|
|
28.
|
Het is belangrijk dat er bij een teststof die via het voer of het drinkwater wordt toegediend, voor wordt gezorgd dat de hoeveelheden teststof de normale voer- of waterbalans niet verstoren. Wanneer de teststof in het voer wordt toegediend, kan een constante concentratie in het voer (in ppm) of een constante dosis in verhouding tot het lichaamsgewicht van de dieren worden gebruikt; daarbij moet worden aangegeven welk alternatief is gebruikt. Wanneer de teststof met een sonde wordt toegediend, moeten de doses dagelijks op vaste tijdstippen worden gegeven. De doses moeten ten minste wekelijks worden aangepast om een constant dosisniveau als functie van het lichaamsgewicht van het dier te verkrijgen.
|
Proefopzet
|
29.
|
Voor beide geslachten moet de toediening ten minste 2 weken voor het paren beginnen, nadat de dieren ten minste vijf dagen lang zijn geacclimatiseerd en de vrouwtjes gescreend zijn op normale oestruscycli (in een voorbehandelingsperiode van 2 weken). Het onderzoek moet zodanig worden ingepland dat de beoordeling van de oestruscyclus begint kort nadat de dieren volledig geslachtsrijp zijn. Dit kan enigszins variëren voor verschillende rattenstammen in verschillende laboratoria, bv. voor Sprague Dawley-ratten op een leeftijd van 10 weken, voor Wistar-ratten op een leeftijd van ongeveer 12 weken. Moederdieren met jongen moeten op dag 13 na het werpen worden gedood of kort daarna. De dag van de geboorte (d.w.z. wanneer het werpen is voltooid) wordt gedefinieerd als dag 0 na het werpen. Vrouwtjes die geen tekenen van copulatie vertonen, worden 24-26 dagen na de laatste dag van de paringsperiode gedood. Voor beide geslachten wordt de toediening gedurende de paringsperiode voortgezet. Daarna moeten de mannetjes de teststof ten minste nog toegediend krijgen tot de minimale totale doseringsperiode van 28 dagen is afgerond. Vervolgens worden ze gedood of, anders, in leven gehouden en verder behandeld voor een eventuele tweede paring.
|
|
30.
|
De dagelijkse toediening aan de moederdieren moet doorgaan gedurende de dracht en ten minste tot en met dag 13 na het werpen of de dag voordat ze gedood worden. Voor onderzoeken waarbij de toediening van de teststof door middel van inhalatie of dermaal plaatsvindt, moet deze ten minste worden voortgezet tot en met dag 19 van de dracht en zo spoedig mogelijk en niet later dan postnatale dag (PND) 4 worden hervat.
|
|
31.
|
In aanhangsel 2 wordt de proefopzet in een diagram weergegeven.
|
De paringsprocedure
|
32.
|
Normaal gesproken moet men voor dit onderzoek één mannetje met één vrouwtje (1:1) laten paren. Hierop zijn uitzonderingen mogelijk in geval van incidentele sterfte van mannetjes. Het vrouwtje moet bij hetzelfde mannetje worden geplaatst totdat aanwijzingen voor copulatie zijn waargenomen of er twee weken zijn verstreken. Elke ochtend worden de vrouwtjes onderzocht op de aanwezigheid van sperma of vaginale plug. Dag 0 van de dracht wordt gedefinieerd als de dag waarop aanwijzingen voor paring worden bevestigd (observatie van een vaginale plug en/of sperma). Wanneer het paren geen succes heeft, kan worden overwogen de vrouwtjes opnieuw te laten dekken door mannetjes van dezelfde groep waarvan de vruchtbaarheid is aangetoond.
|
Nestgrootte
|
33.
|
Op PND 4 kan de grootte van de nesten worden aangepast door eliminatie van extra jongen door willekeurige selectie, zodat elk nest uiteindelijk zoveel mogelijk bestaat uit vier of vijf jongen per geslacht, afhankelijk van de normale nestgrootte van de gebruikte rattenstam. Bij twee van de overtollige jongen moeten bloedmonsters worden afgenomen, gepoold en gebruikt ter bepaling van de serumspiegels van T4. Selectieve eliminatie van jongen, bijvoorbeeld op grond van lichaamsgewicht of anogenitale afstand (AGD), is niet gepast. Als het door het aantal mannelijke of vrouwelijke jongen niet mogelijk is om op vier of vijf per geslacht per nest uit te komen, is gedeeltelijke aanpassing (bv. zes mannetjes en vier vrouwtjes) aanvaardbaar. Er worden geen jongen geëlimineerd als de nestgrootte daardoor onder de streefgrootte (8 of 10 jongen/nest) zou komen. Als er maar één jong beschikbaar is boven de streefgrootte, wordt er slechts één jong geëlimineerd en gebruikt voor het nemen van een bloedmonster ter bepaling van serum-T4.
|
|
34.
|
Als de nestgrootte niet wordt aangepast, worden er op PND 4 twee jongen per nest gedood en worden er bloedmonsters genomen voor het meten van de serumspiegels van schildklierhormonen. De twee jongen per nest moeten zo mogelijk vrouwelijke jongen zijn om de mannelijke jongen te bewaren voor de beoordeling van de tepelretentie, tenzij er na verwijdering van deze jongen geen enkel vrouwtje meer overblijft voor de beoordeling aan het einde van het onderzoek. Er worden geen jongen geëlimineerd als de nestgrootte daardoor onder de 8 of 10 jongen/nest (afhankelijk van de normale nestgrootte van de gebruikte rattenstam) zou komen. Als er maar één jong beschikbaar is boven de normale nestgrootte, wordt er slechts één jong geëlimineerd en gebruikt voor het nemen van een bloedmonster ter bepaling van serum-T4.
|
Waarnemingen bij levende dieren
Klinische waarnemingen
|
35.
|
Gedurende de hele testperiode moeten er minstens eenmaal per dag, en vaker als er tekenen van toxiciteit worden waargenomen, algemene klinische waarnemingen worden verricht. Deze moet bij voorkeur elke dag op dezelfde tijd(en) plaatsvinden, rekening houdend met de piekperiode voor de verwachte effecten na de toediening. Met de teststof samenhangende veranderingen in het gedrag, tekenen van een moeilijke of langdurige worp en alle tekenen van toxiciteit, inclusief sterfte, moeten worden vastgelegd, met inbegrip van de aanvang, de ernst en de duur van de toxiciteitsverschijnselen.
|
Lichaamsgewicht en consumptie van voer en water
|
36.
|
De mannetjes en vrouwtjes moeten worden gewogen op de eerste dag dat zij de teststof krijgen toegediend, vervolgens ten minste eenmaal per week en aan het einde van het onderzoek. De vrouwtjes moeten worden gewogen op de dagen 0, 7, 14 en 20 tijdens de dracht, binnen 24 uur na de geboorte (dag 0 of 1 na het werpen) en ten minste op de dagen 4 en 13 na het werpen. Deze waarnemingen worden voor elk volwassen dier apart gerapporteerd.
|
|
37.
|
Vóór het paren, tijdens de dracht en tijdens het zogen, moet ten minste wekelijks de voerconsumptie worden gemeten. Tijdens de paringsperiode is deze meting facultatief. Wanneer de teststof via het drinkwater wordt toegediend, moet ook de waterconsumptie gedurende deze perioden worden gemeten.
|
Oestruscycli
|
38.
|
Voordat de behandeling begint, moeten de oestruscycli worden gecontroleerd om te selecteren op vrouwelijke proefdieren met regelmatige cycli (zie punt 22). Ook moeten er van het begin van de behandelingsperiode totdat paring is gebleken, dagelijks vaginale uitstrijkjes worden onderzocht. Indien vermoed wordt dat effecten van acute stress bij het begin van de behandeling de oestruscyclus zouden kunnen verstoren, kunnen de laboratoria de proefdieren gedurende 2 weken blootstellen en vervolgens ten minste 2 weken lang dagelijks vaginale uitstrijkjes nemen ter controle van de oestruscycli gedurende de periode vóór paring tot in de paringsperiode, totdat paring gebleken is. Wanneer monsters van vaginale/cervicale cellen worden genomen, moet erop worden gelet verstoring van het slijmvlies, die tot schijndracht kan leiden, te voorkomen (7) (8).
|
Parameters met betrekking tot de nakomelingen
|
39.
|
De drachttijd moet worden geregistreerd en wordt berekend vanaf dag 0 van de dracht. Elke worp moet zo spoedig mogelijk na de geboorte worden onderzocht, teneinde het aantal en geslacht van de jongen vast te stellen, alsmede het aantal doodgeborenen, levendgeborenen, ondermaatse jongen (jongen die aanmerkelijk kleiner zijn dan de corresponderende controlejongen) en de aanwezigheid van macroscopisch zichtbare afwijkingen.
|
|
40.
|
Levende jongen moeten worden geteld en gesekst en de nesten moeten binnen 24 uur na de geboorte (dag 0 of 1 na het werpen) en ten minste op de dagen 4 en 13 na het werpen worden gewogen. Naast de in punt 35 beschreven waarnemingen moet elk afwijkend gedrag van de nakomelingen worden geregistreerd.
|
|
41.
|
De AGD van elk jong moet worden gemeten op dezelfde postnatale dag tussen postnatale dag 0 en het einde van postnatale dag 4. Het lichaamsgewicht van elk jong moet worden bepaald op de dag dat de AGD wordt gemeten en de AGD moet genormaliseerd worden tot een maat voor jonggrootte, bij voorkeur de derdemachtswortel van het lichaamsgewicht (9). Het aantal tepels/tepelhoven bij mannelijke jongen moet worden geteld op postnatale dag 12 of 13, zoals aanbevolen in OESO-leidraad 151 (10).
|
Klinische biochemie
|
42.
|
Er worden bloedmonsters genomen van een gedefinieerde plaats op basis van het volgende schema:
|
—
|
van ten minste twee jongen per nest op PND 4 als het aantal jongen dit toelaat (zie de punten 33-34);
|
|
—
|
van alle moederdieren en ten minste twee jongen per nest op het tijdstip van doden op dag 13, en
|
|
—
|
van alle volwassen mannetjes op het tijdstip van doden.
|
|
Alle bloedmonsters worden onder passende omstandigheden bewaard. In de bloedmonsters van de jongen op dag 13 en de volwassen mannetjes worden de serumspiegels van schildklierhormonen (T4) bepaald. Een verdere bepaling van T4 in bloedmonsters van moederdieren en jongen op dag 4 wordt verricht indien relevant. Eventueel kunnen ook andere hormonen worden gemeten indien relevant. Voor de schildklierhormoonanalyse kan bloed van jongen per nest gepoold worden. De schildklierhormonen (T4 en TSH) worden bij voorkeur als „totaal” gemeten.
|
43.
|
De volgende factoren kunnen van invloed zijn op de variabiliteit en de absolute concentraties van de hormoonbepalingen:
|
—
|
tijdstip waarop de dieren worden gedood, vanwege de schommelingen in de hormoonconcentraties gedurende de dag;
|
|
—
|
wijze waarop de dieren worden gedood: onnodige stress bij de dieren, waardoor de hormoonconcentraties kunnen worden beïnvloed, moet worden voorkomen;
|
|
—
|
testkits voor hormoonbepalingen, die verschillende standaardkrommen kunnen hebben.
|
|
|
44.
|
Plasmamonsters die specifiek voor hormoonbepaling zijn bedoeld, moeten telkens op hetzelfde moment van de dag worden genomen. De verschillende in de handel verkrijgbare „assay kits” kunnen verschillende numerieke waarden opleveren bij de analyse van hormoonconcentraties.
|
Pathologie
Macroscopische obductie
|
45.
|
Op het tijdstip waarop ze worden gedood of tijdens het onderzoek sterven, worden de dieren macroscopisch onderzocht op afwijkingen of pathologische veranderingen. Bijzondere aandacht moet worden geschonken aan de organen van het voortplantingssysteem. Het aantal implantatieplaatsen moet worden geregistreerd. In de ochtend van de dag van obductie moeten er vaginale uitstrijkjes onderzocht worden om de fase van de oestruscyclus te bepalen en deze in verband te kunnen brengen met de histopathologie van de eierstokken.
|
|
46.
|
De testes en epididymides, evenals de prostaat en de zaadblaasjes met coagulans afscheidende klieren als geheel van alle volwassen mannetjes moeten zo nodig worden ontdaan van aanhechtend weefsel en zo spoedig mogelijk na de sectie nat worden gewogen om te voorkomen dat ze uitdrogen. Optionele organen die eventueel ook gewogen kunnen worden, zijn onder meer het m levator ani plus bulbocavernosuscomplex, de Cowperse klieren en de glans penis bij mannetjes en de gepaarde ovaria (nat gewicht) en uterus (inclusief cervix) bij vrouwtjes; indien deze gewichten worden meegenomen, moeten ze zo snel mogelijk na de sectie worden opgenomen.
|
|
47.
|
Dode jongen en jongen die op dag 13 na het werpen, of kort daarna, zijn gedood, moeten in elk geval uitwendig zorgvuldig worden onderzocht op macroscopisch zichtbare afwijkingen. Daarbij wordt met name gelet op de uitwendige voortplantingsorganen, die op tekenen van een afwijkende ontwikkeling worden onderzocht. Op dag 13 moet van 1 mannelijk en 1 vrouwelijk jong per nest de schildklier worden geconserveerd.
|
|
48.
|
Van alle volwassen dieren moeten de ovaria, testes, secundaire geslachtsorganen (uterus en cervix, epididymides, prostaat, zaadblaasjes met coagulans uitscheidende klieren), schildklier en alle organen die macroscopische letsels vertonen, worden geconserveerd. Voor routineonderzoek van testes en epididymides wordt fixatie in formaline niet aanbevolen. Voor deze weefsels is het gebruik van Bouin’s fixatief of gemodificeerde Davidson-oplossing een aanvaardbare methode (11). De tunica albuginea kan voorzichtig en ondiep met een naald worden ingeprikt op de beide polen van het orgaan, zodat het fixatief snel kan binnendringen.
|
Histopathologie
|
49.
|
De ovaria, testes en epididymides van de dieren in de hoogste dosisgroep en de controlegroep moeten aan een uitvoerig histologisch onderzoek worden onderworpen (met speciale aandacht voor de stadia van de spermatogenese en de histopathologie van de interstitiële testiculaire celstructuur). De overige geconserveerde organen, waaronder de schildklier van jongen en van volwassen dieren, kunnen worden onderzocht indien nodig. Het gewicht van de schildklier kan na de fixatie worden bepaald. Aanhechtend weefsel moet zeer voorzichtig en pas na fixatie worden verwijderd, om beschadiging van de weefsels te voorkomen. Weefselbeschadigingen kunnen de histopathologische analyse negatief beïnvloeden. Wanneer er veranderingen worden waargenomen in de groep met de hoogste dosering, moeten ook de dieren in de andere doseringsgroepen worden onderzocht. In de richtsnoeren voor histopathologisch onderzoek (11) zijn nadere gegevens opgenomen over de sectie, de fixatie, het snijden van coupes en de histopathologie van endocriene weefsels.
|
GEGEVENS EN RAPPORTAGE
Gegevens
|
50.
|
Er worden voor elk dier apart gegevens verstrekt. Daarnaast moeten alle gegevens worden samengevat in tabelvorm, waarbij voor elke testgroep worden vermeld: het aantal dieren aan het begin van de test, het aantal dieren dat tijdens de test dood is aangetroffen of met het oog op een humane behandeling is gedood, het tijdstip van sterfte of humane doding, het aantal vruchtbare dieren, het aantal drachtige vrouwtjes, het aantal dieren met toxiciteitsverschijnselen, een beschrijving van de waargenomen toxiciteitsverschijnselen met vermelding van de aanvang, de duur en de ernst van de toxische effecten, de aard van de histopathologische veranderingen en alle relevante gegevens over de nesten. In aanhangsel 3 is een opzet van een overzichtstabel opgenomen die heel praktisch is gebleken voor de beoordeling van effecten op de voortplanting en de ontwikkeling.
|
|
51.
|
Vanwege de beperkte omvang van het onderzoek, zijn statistische analyses in de vorm van toetsen voor „significantie” voor veel eindpunten, vooral die in verband met de voortplanting, van beperkte waarde. Als er statistische analyses worden gebruikt, moeten de gekozen methoden geschikt zijn voor de verdeling van de onderzochte variabele en moeten ze vóór de start van het onderzoek worden gekozen. De statistische analyse van de AGD en de tepelretentie moet worden gebaseerd op de gegevens voor afzonderlijke jongen, waarbij rekening wordt gehouden met nesteffecten. Waar passend is het nest de analyse-eenheid. De statistische analyse van lichaamsgewicht van de jongen moet worden gebaseerd op de gegevens voor afzonderlijke jongen, waarbij rekening wordt gehouden met nestgrootte. Vanwege de geringe groepsgrootte kan het ook nuttig zijn om gebruik te maken van historische controlegegevens (bv. voor nestgrootte), indien beschikbaar, ter ondersteuning van de interpretatie van het onderzoek.
|
Evaluatie van de resultaten
|
52.
|
De resultaten van dit toxiciteitsonderzoek worden beoordeeld aan de hand van de waargenomen effecten en de resultaten van de obductie en het microscopisch onderzoek. De beoordeling omvat het verband tussen de dosis van de teststof en de aanwezigheid of afwezigheid, de frequentie en de ernst van afwijkingen zoals macroscopisch letsel, gespecificeerde doelorganen, onvruchtbaarheid, klinische afwijkingen, aantasting van de reproductie- en nestresultaten, veranderingen in het lichaamsgewicht, effecten op de mortaliteit en andere toxische effecten.
|
|
53.
|
Vanwege de korte behandelperiode van de mannetjes moeten bij de beoordeling van effecten op de voortplanting bij mannetjes niet alleen de vruchtbaarheidsgegevens, maar ook de histopathologie van de testes en de epididymides in aanmerking worden genomen. Ook het gebruik van historische controlegegevens over voortplanting/ontwikkeling (bv. voor nestgrootte, AGD, tepelretentie, T4-serumspiegels), indien beschikbaar, kan nuttig zijn ter ondersteuning van de interpretatie van het onderzoek.
|
|
54.
|
Voor de kwaliteitscontrole wordt voorgesteld historische controlegegevens te verzamelen en variatiecoëfficiënten te berekenen voor numerieke gegevens, met name voor de parameters die verband houden met het opsporen van hormoonontregelaars. Deze gegevens kunnen voor vergelijkingsdoeleinden worden gebruikt wanneer daadwerkelijk uitgevoerd onderzoek wordt geëvalueerd.
|
Testverslag
|
55.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Teststof:
|
—
|
bron, partijnummer, uiterste gebruiksdatum, indien beschikbaar;
|
|
—
|
stabiliteit van de teststof, indien bekend.
|
|
|
|
Stof die uit één component bestaat:
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
chemische identificatiegegevens, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code, structuurformule, zuiverheid, chemische identiteit van onzuiverheden, indien van toepassing en praktisch haalbaar enz.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB’s en mengsels:
|
—
|
voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen.
|
|
|
|
Vehiculum (indien van toepassing):
|
—
|
motivering voor de keuze van het vehiculum, indien geen water wordt gebruikt.
|
|
|
|
Proefdieren:
|
—
|
aantal, leeftijd en geslacht van de dieren;
|
|
—
|
herkomst, leefomstandigheden, voeding enz.;
|
|
—
|
gewicht van elk dier aan het begin van de test;
|
|
—
|
motivering voor het gebruik van een ander soort dan ratten.
|
|
|
|
Testomstandigheden:
|
—
|
achtergrond voor de keuze van de dosisniveaus;
|
|
—
|
gedetailleerde gegevens over de formulering van de teststof, de bereiding van het voer en de bereikte concentratie, stabiliteit en homogeniteit van het preparaat;
|
|
—
|
gedetailleerde gegevens over de toediening van de teststof;
|
|
—
|
omrekening van de concentratie van de teststof in het voer/drinkwater (in ppm) naar de feitelijke dosis (in mg/kg lichaamsgewicht/dag), indien van toepassing;
|
|
—
|
gedetailleerde gegevens over de kwaliteit van het voer en het water;
|
|
—
|
gedetailleerde beschrijving van de randomisatieprocedure voor de selectie van te ruimen jongen, indien van toepassing.
|
|
|
|
Resultaten:
|
—
|
lichaamsgewicht en veranderingen in het lichaamsgewicht;
|
|
—
|
indien beschikbaar, voedselverbruik en waterverbruik;
|
|
—
|
gegevens over de toxische reactie naar geslacht en dosis, met inbegrip van vruchtbaarheid, dracht en andere gegevens over toxiciteit;
|
|
—
|
toxische of andere effecten op de reproductie, de jongen, de postnatale groei enz.;
|
|
—
|
aard, ernst en duur van (al dan niet reversibele) klinische waarnemingen;
|
|
—
|
aantal volwassen vrouwtjes met een normale of abnormale oestruscyclus en cyclusduur van deze vrouwtjes;
|
|
—
|
aantal levend geboren jongen en verliezen na implantatie;
|
|
—
|
gegevens over het lichaamsgewicht van jongen;
|
|
—
|
AGD van alle jongen (en lichaamsgewicht op de dag van de AGD-meting);
|
|
—
|
tepelretentie bij mannelijke jongen;
|
|
—
|
schildklierhormoonspiegels bij dag 13-jongen en volwassen mannetjes (en moederdieren en dag 4-jongen indien gemeten);
|
|
—
|
aantal jongen met macroscopisch zichtbare afwijkingen, macroscopische beoordeling van de uitwendige geslachtsorganen, aantal ondermaatse jongen;
|
|
—
|
tijdstip van sterfte tijdens het onderzoek en of de dieren overleefden tot het einde van het onderzoek;
|
|
—
|
aantal implantaties, nestgrootte en nestgewichten op het moment van registratie;
|
|
—
|
lichaamsgewicht bij het doden van de dieren en gegevens over het gewicht van organen voor de ouderdieren;
|
|
—
|
een gedetailleerde beschrijving van histopathologische bevindingen;
|
|
—
|
resorptiegegevens (indien beschikbaar);
|
|
—
|
een statistische behandeling van de resultaten, indien van toepassing.
|
|
Bespreking van de resultaten.
Conclusies.
|
Interpretatie van resultaten
|
56.
|
Op basis van dit onderzoek kan de reproductie-/ontwikkelingstoxiciteit worden beoordeeld bij de toediening van herhaalde doses (zie de punten 5 en 6). Het onderzoek zou een indicatie kunnen geven of verder onderzoek nodig is en biedt oriëntatiepunten voor de opzet van vervolgonderzoeken. OESO-leidraad nr. 43 moet worden geraadpleegd voor hulp bij de interpretatie van resultaten op het gebied van reproductie- en ontwikkelingstoxiciteit (12). In OESO-leidraad nr. 106 over de histologische beoordeling van endocriene en voortplantingstests bij knaagdieren (11) is informatie te vinden over het prepareren en beoordelen van (endocriene) organen en vaginale uitstrijkjes, die van nut kan zijn voor deze TG.
|
LITERATUUR
|
(1)
|
OESO (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
OESO (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(3)
|
OESO (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(4)
|
OESO (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(5)
|
OESO (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(6)
|
OESO (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OESO (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(11)
|
OESO (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No 106), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(12)
|
OESO (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
Aanhangsel 1
DEFINITIES (ZIE OOK OESO-LEIDRAAD 150 (6))
Androgene werking: het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk androgeen hormoon (bv. testosteron) te werken.
Antiandrogene werking: het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk androgeen hormoon (bv. testosteron) te onderdrukken.
Antioestrogene werking: het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk oestrogeen hormoon (bv. 17β-oestradiol) te onderdrukken.
Antithyreoïde werking: het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk schildklierhormoon (bv. T3) te onderdrukken.
Chemische stof: een stof of mengsel.
Dosering: een algemene term die de dosis, de frequentie en de duur van de toediening omvat.
Dosis: de toegediende hoeveelheid teststof. De dosis wordt uitgedrukt als gewicht van de teststof per eenheid lichaamsgewicht van het proefdier per dag (bv. mg/kg lichaamsgewicht/dag), of als constante voedingsconcentratie.
Manifeste toxiciteit: een algemene term die duidelijke toxiciteitsverschijnselen beschrijft die optreden na de toediening van de teststof. Deze verschijnselen moeten een beoordeling van de gevaren mogelijk maken en van een zodanige aard zijn dat verwacht mag worden dat bij een hogere toegediende dosis ernstige toxische verschijnselen en waarschijnlijk sterfte optreden.
Maternale toxiciteit: schadelijke effecten bij drachtige vrouwtjes, die hetzij specifiek optreden (direct effect), hetzij niet-specifiek (indirect effect).
NOAEL: afkorting van „no-observed-adverse-effect level”, het niveau waarbij geen schadelijk effect werd vastgesteld. Dit is het hoogste dosisniveau waarbij nog geen aan de behandeling gebonden nadelige effecten worden vastgesteld.
Oestrogene werking: het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk oestrogeen hormoon (bv. 17β-oestradiol) te werken.
Ontwikkelingstoxiciteit: de wijze waarop reproductietoxiciteit zich manifesteert, in de vorm van pre-, peri- en postnatale, structurele of functionele afwijkingen bij nakomelingen.
Reproductietoxiciteit: schadelijke effecten op de nakomelingen en/of verstoring van de voortplantingsfuncties of het voortplantingsvermogen bij mannetjes en vrouwtjes.
Teststof: elke volgens deze testmethode getest(e) stof of mengsel.
Thyreoïde werking: het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk schildklierhormoon (bv. T3) te werken.
Validering: een wetenschappelijke methode die erop gericht is om de operationele vereisten en beperkingen van een testmethode te beschrijven en de betrouwbaarheid en relevantie ervan voor een bepaald doel aan te tonen.
Verstoring van de vruchtbaarheid: afwijkingen in de voortplantingsfuncties of het voortplantingsvermogen bij mannetjes of vrouwtjes.
Aanhangsel 2
DIAGRAM VAN DE PROEFOPZET MET DE MAXIMALE ONDERZOEKSDUUR OP BASIS VAN EEN VOLLEDIGE 14-DAAGSE PARINGSPERIODE

Aanhangsel 3
OVERZICHTSTABEL VAN DE EFFECTEN OP REPRODUCTIE/ONTWIKKELING
|
WAARNEMINGEN
|
WAARDEN
|
|
|
|
Dosering (eenheden)
|
0 (controle)
|
…
|
…
|
…
|
…
|
|
Paren bij begin (N)
|
|
|
|
|
|
|
Oestruscyclus (ten minste gemiddelde duur en frequentie van onregelmatige cycli)
|
|
|
|
|
|
|
Vrouwtjes met aanwijzingen voor copulatie (N)
|
|
|
|
|
|
|
Vrouwtjes die drachtig worden (N)
|
|
|
|
|
|
|
Dagen conceptie 1 - 5 (N)
|
|
|
|
|
|
|
Dagen conceptie 6 -... (21) (N)
|
|
|
|
|
|
|
Dracht ≤ 21 dagen (N)
|
|
|
|
|
|
|
Dracht = 22 dagen (N)
|
|
|
|
|
|
|
Dracht ≥ 23 dagen (N)
|
|
|
|
|
|
|
Moederdieren met levend geboren jongen (N)
|
|
|
|
|
|
|
Moederdieren met levende jongen op postnatale dag 4 (N)
|
|
|
|
|
|
|
Implantaten/moederdier (gemiddeld)
|
|
|
|
|
|
|
Levende jongen/moederdier bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Levende jongen/moederdier op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Geslachtsverhouding (m/v) bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Geslachtsverhouding (m/v) op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van het nest bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Gewicht van het nest op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen op het moment van AGD-meting (gemiddeld bij mannetjes, gemiddeld bij vrouwtjes)
|
|
|
|
|
|
|
AGD jongen op dezelfde postnatale dag, geboorte - dag 4 (gemiddeld bij mannetjes, gemiddeld bij vrouwtjes)
|
|
|
|
|
|
|
Gewicht van de jongen op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Tepelretentie bij mannelijke jongen op dag 13 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen op dag 13 (gemiddeld)
|
|
|
|
|
|
|
|
|
AFWIJKENDE JONGEN
|
|
Moederdieren met 0
|
|
|
|
|
|
|
Moederdieren met 1
|
|
|
|
|
|
|
Moederdieren met ≥ 2
|
|
|
|
|
|
|
|
|
VERLIES VAN NAKOMELINGEN
|
|
|
|
Vóór geboorte/na implantatie (implantaties minus levend geborenen)
|
|
Vrouwtjes met 0
|
|
|
|
|
|
|
Vrouwtjes met 1
|
|
|
|
|
|
|
Vrouwtjes met 2
|
|
|
|
|
|
|
Vrouwtjes met ≥ 3
|
|
|
|
|
|
|
|
|
Na geboorte (levend geboren en minus levende jongen op PND 13)
|
|
Vrouwtjes met 0
|
|
|
|
|
|
|
Vrouwtjes met 1
|
|
|
|
|
|
|
Vrouwtjes met 2
|
|
|
|
|
|
|
Vrouwtjes met ≥ 3
|
|
|
|
|
|
B.64 TOXICITEITSONDERZOEK BIJ HERHAALDE TOEDIENING IN COMBINATIE MET DE SCREENINGTEST VOOR REPRODUCTIE/ONTWIKKELINGSTOXICITEIT
INLEIDING
|
1.
|
Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 422 (2016) van de OESO. OESO-richtlijnen voor het testen van chemische stoffen worden periodiek in het licht van de wetenschappelijke vorderingen getoetst. De oorspronkelijke screeningtestrichtlijn 422 werd vastgesteld in 1996 op basis van een protocol voor een gecombineerde screeningtest voor toxiciteit bij herhaalde toediening en reproductie/ontwikkelingstoxiciteit (Combined Repeat Dose and Reproductive/Developmental Screening Test) die was besproken op twee bijeenkomsten van deskundigen, in 1990 in Londen (1) en in 1992 in Tokyo (2).
|
|
2.
|
Deze testmethode combineert een screeninggedeelte voor reproductie-/ontwikkelingstoxiciteit, gebaseerd op de ervaring die in de aangesloten landen is opgedaan bij het gebruik van de oorspronkelijke methode op bestaande chemische stoffen met een hoog productievolume en in verkennende tests met positieve controlestoffen (3) (4), en een gedeelte voor toxiciteit bij herhaalde toediening, in overeenstemming met OESO-testrichtlijn 407 (toxiciteitsonderzoek (oraal) op knaagdieren bij herhaalde toediening (28 dagen), overeenkomend met hoofdstuk B.7 van deze bijlage).
|
|
3.
|
Deze testmethode is bijgewerkt met eindpunten die relevant zijn voor hormoonontregelaars, naar aanleiding van de prioritaire actie die de OESO in 1998 is gestart om bestaande testrichtlijnen te herzien en nieuwe testrichtlijnen te ontwikkelen voor het screenen en testen op potentiële hormoonontregelaars (5). In dit verband werd TG 407 (overeenkomend met hoofdstuk B.7 van deze bijlage) in 2008 uitgebreid met parameters die geschikt zijn om de endocriene activiteit van de teststoffen te detecteren. Met de bijwerking van TG 422 werd beoogd om in screeningtestrichtlijnen een aantal voor hormoonontregelaars relevante eindpunten op te nemen, waarbij de blootstellingsperioden bepaalde gevoelige perioden in de ontwikkeling (prenatale en vroeg-postnatale perioden) beslaan.
|
|
4.
|
De aanvullende, voor hormoonontregelaars relevante eindpunten die werden geselecteerd en die ook deel uitmaken van TG 443 (uitgebreid onderzoek naar reproductietoxiciteit over één generatie, overeenkomend met hoofdstuk B.56 van deze bijlage), werden in TG 422 opgenomen op basis van een haalbaarheidsstudie waarin wetenschappelijke en technische vragen met betrekking tot de opname ervan werden onderzocht, alsook aanpassingen van de testopzet die eventueel voor de opname ervan noodzakelijk zouden zijn (6).
|
|
5.
|
Deze testmethode is ontworpen voor het verkrijgen van beperkte informatie over de effecten van een teststof op het mannelijke en vrouwelijke voortplantingsvermogen, zoals de gonadale functie, paringsgedrag, conceptie, ontwikkeling van de vrucht en geboorte. Deze testmethode is geen alternatief noch een vervanging voor de bestaande testmethoden B.31, B.34, B.35 of B.56.
|
INLEIDENDE OVERWEGINGEN
|
6.
|
Bij de bepaling en evaluatie van de toxische eigenschappen van een teststof moet aan de hand van acute-toxiciteitstests eerst informatie over de toxiciteit worden verkregen. Daarna kan de orale toxiciteit door middel van herhaalde toediening worden bepaald. Dit onderzoek geeft informatie over mogelijke gevaren voor de gezondheid die zich kunnen voordoen bij herhaalde blootstelling gedurende een betrekkelijk beperkte tijd. De methode omvat het basale toxiciteitsonderzoek bij herhaalde toediening dat kan worden gebruikt voor stoffen waarvoor een onderzoek van 90 dagen niet gerechtvaardigd is (bv. als het productievolume binnen bepaalde grenzen blijft), of als voorstudie voor een langetermijnonderzoek. Bij het uitvoeren van het onderzoek moeten de richtsnoeren en overwegingen worden gevolgd die worden beschreven in de OESO-leidraad nr. 19 over het herkennen, beoordelen en gebruiken van klinische verschijnselen als humane eindpunten voor proefdiergebruik bij veiligheidsbeoordelingen (7).
|
|
7.
|
De methode omvat bovendien een screeningtest voor reproductie-/ontwikkelingstoxiciteit en kan daarom ook worden gebruikt om oriënterende gegevens te verkrijgen over mogelijke effecten op het mannelijke en vrouwelijke voortplantingsvermogen, zoals de gonadale functie, paringsgedrag, conceptie, ontwikkeling van de vrucht en geboorte, hetzij in een vroeg stadium van de beoordeling van de toxicologische eigenschappen van teststoffen hetzij voor teststoffen waarbij dergelijke effecten worden vermoed. Deze testmethode verschaft geen volledige informatie over alle aspecten van reproductie en ontwikkeling. Met name voor het detecteren van postnatale gevolgen van prenatale blootstelling of effecten die tijdens postnatale blootstelling zijn veroorzaakt, is de informatie die deze methode oplevert slechts beperkt. Als gevolg van (onder andere) de selectiviteit van de eindpunten en de korte onderzoeksduur kan met deze methode geen bewijs worden verkregen om definitief te concluderen dat een stof geen effecten veroorzaakt op de voortplanting en ontwikkeling. Als er verder geen gegevens zijn uit andere tests op reproductie-/ontwikkelingstoxiciteit, zijn positieve resultaten evenwel nuttig voor een eerste gevarenbeoordeling en kunnen ze als basis dienen voor beslissingen over de noodzaak en timing van aanvullende tests.
|
|
8.
|
De resultaten die worden verkregen met de parameters die met het endocriene systeem te maken hebben, moeten worden gezien in samenhang met het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen (8). In dit conceptueel kader is de uitgebreide TG 422 van de OESO ondergebracht in niveau 4 als eenin-vivobepaling waarmee gegevens kunnen worden verkregen over schadelijke effecten op eindpunten die relevant zijn voor het endocrien systeem. Een endocrien signaal hoeft op zichzelf echter niet als voldoende bewijs te worden beschouwd dat de teststof een hormoonontregelaar is.
|
|
9.
|
Bij deze testmethode ligt het accent ook op neurologische effecten als specifiek eindpunt. Benadrukt moet worden dat het noodzakelijk is de dieren zorgvuldig klinisch te observeren om zoveel mogelijk informatie te verzamelen. Ze moet het mogelijk maken potentieel neurotoxische chemicaliën te identificeren, die in dit opzicht nader verdienen te worden onderzocht. Bovendien kan de methode een ruwe indicatie geven van immunologische effecten.
|
|
10.
|
Als er geen gegevens zijn uit andere onderzoeken naar systemische toxiciteit, reproductie-/ontwikkelingstoxiciteit, neurotoxiciteit en/of immutoxiciteit, zijn positieve resultaten bovendien nuttig voor een eerste gevarenbeoordeling en kunnen ze als basis dienen voor beslissingen over de noodzaak en timing van aanvullende tests. De test kan vooral nuttig zijn als onderdeel van de Screening Information Data Set (SIDS) van de OESO voor de beoordeling van bestaande chemische stoffen waarvoor weinig of geen toxicologische gegevens beschikbaar zijn, en kan als alternatief dienen voor het uitvoeren van twee aparte tests voor respectievelijk toxiciteit bij herhaalde toediening (OSEO-TG 407, die overeenkomt met hoofdstuk B.7 van deze bijlage) en reproductie-/ontwikkelingstoxiciteit (OSEO-TG 421, die overeenkomt met hoofdstuk B.63 van deze bijlage). De test kan ook worden gebruikt als bereikbepalingsonderzoeken voor uitvoeriger onderzoeken naar reproductie-/ontwikkelingstoxiciteit, of wanneer het gebruik ervan anderszins relevant wordt geacht.
|
|
11.
|
In het algemeen wordt aangenomen dat er verschillen in gevoeligheid zijn tussen drachtige en niet-drachtige dieren. In deze gecombineerde test kan het dan ook moeilijker zijn om dosisniveaus te bepalen die geschikt zijn voor zowel de beoordeling van algemene systemische toxiciteit als specifieke reproductie-/ontwikkelingstoxiciteit, ten opzichte van de situatie waarin de afzonderlijke tests apart worden uitgevoerd. Bovendien kunnen de testresultaten voor algemene systemische toxiciteit moeilijker te interpreteren zijn dan wanneer een apart onderzoek bij herhaalde toediening wordt uitgevoerd, vooral wanneer de serum- en histopathologieparameters in het onderzoek niet tegelijkertijd worden beoordeeld. Vanwege de technische complexiteit vergt de uitvoering van deze gecombineerde screeningtest ruime ervaring met testen op toxiciteit. Anderzijds kan de gecombineerde test, afgezien van het geringere aantal dieren dat nodig is, een beter middel bieden om rechtstreekse effecten op voorplanting/ontwikkeling te onderscheiden van effecten die voortvloeien uit andere (systemische) effecten.
|
|
12.
|
In deze test is de doseringsperiode langer dan in een klassiek 28-daags onderzoek bij herhaalde toediening. Er zijn echter minder dieren van elk geslacht per groep nodig dan wanneer een klassiek 28-daags onderzoek bij herhaalde toediening wordt uitgevoerd naast een screeningtest voor reproductie/ontwikkelingstoxiciteit.
|
|
13.
|
Bij deze testmethode wordt aangenomen dat de teststof oraal wordt toegediend. Als andere blootstellingsroutes worden gebruikt, moet de methode mogelijk worden aangepast.
|
|
14.
|
Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is.
|
|
15.
|
De gebruikte definities zijn opgenomen in aanhangsel 1.
|
PRINCIPE VAN DE TEST
|
16.
|
De teststof wordt in geleidelijk oplopende doseringen aan verschillende groepen mannetjes en vrouwtjes toegediend. De mannetjes moeten ten minste vier weken lang de teststof toegediend krijgen, tot en met de dag voordat ze worden gedood (inclusief twee weken voorafgaand aan het paren, tijdens de paringsperiode en ongeveer twee weken na het paren). Gezien de beperkte doseringsperiode voor het paren, is de vruchtbaarheid mogelijk geen bijzonder gevoelige indicator voor testiculaire toxiciteit. Een uitvoerig histologisch onderzoek van de testes is daarom noodzakelijk. De combinatie van een twee weken durende doseringsperiode voor het paren, gevolgd door waarnemingen op het gebied van paring/vruchtbaarheid, met een totale doseringsperiode van ten minste vier weken gevolgd door een uitvoerige histopathologie van de mannelijke geslachtsklieren, wordt voldoende geacht om het merendeel van de effecten op de mannelijke vruchtbaarheid en de spermatogenese te kunnen detecteren.
|
|
17.
|
De vrouwtjes moeten het hele onderzoek lang de teststof toegediend krijgen. Dit betekent twee weken voorafgaand aan het paren (zodat het onderzoek ten minste twee volledige oestruscycli omvat), de variabele tijd tot conceptie, de duur van de dracht en ten minste dertien dagen na het werpen, tot en met de dag voordat ze worden gedood.
|
|
18.
|
De duur van het onderzoek, na acclimatisering en beoordeling van de oestruscyclus voorafgaand aan de behandeling, is afhankelijk van het gedrag van het vrouwtje en beloopt ongeveer 63 dagen (ten minste 14 dagen vóór het paren, (tot) 14 dagen paring, 22 dagen dracht, en ten slotte een zoogperiode van 13 dagen).
|
|
19.
|
Gedurende de periode dat de stof wordt toegediend, worden de dieren dagelijks zorgvuldig geobserveerd om tekenen van toxiciteit te ontdekken. Bij dieren die tijdens het onderzoek sterven of worden gedood, wordt obductie verricht. Dieren die aan het eind van het onderzoek nog in leven zijn, worden gedood en ook hierop wordt obductie verricht.
|
BESCHRIJVING VAN DE METHODE
Keuze van de diersoort
|
20.
|
Deze testmethode is ontworpen om te worden toegepast met ratten. Indien de in TG 422 gespecificeerde parameters worden onderzocht bij een andere knaagdiersoort moet dit uitvoerig worden gemotiveerd. In het internationale valideringsprogramma voor de detectie van hormoonontregelaars voor TG 407 zijn alleen ratten gebruikt. Stammen met een lage vruchtbaarheid of waarvan bekend is dat er vaak ontwikkelingsstoornissen optreden, mogen niet worden gebruikt. Er moet gebruikgemaakt worden van gezonde dieren die nog niet eerder gepaard hebben en nog niet eerder aan experimenten zijn onderworpen. Van de proefdieren moeten soort, stam, geslacht, gewicht en leeftijd bekend zijn. Per geslacht mag bij aanvang van het onderzoek het gewicht van de dieren die worden gebruikt, niet meer dan ± 20 % van het gemiddelde gewicht afwijken. Wanneer het onderzoek wordt uitgevoerd als voorstudie voor een onderzoek van langere duur of een onderzoek over een volledige generatie, verdient het de voorkeur dat de dieren in beide onderzoeken tot dezelfde stam behoren en dezelfde oorsprong hebben.
|
Huisvesting en voeding
|
21.
|
Alle procedures moeten in overeenstemming zijn met de plaatselijke normen voor de verzorging van proefdieren. De temperatuur in de proefdierruimte moet 22 °C (± 3 °C) zijn. De relatieve luchtvochtigheid moet ten minste 30 % bedragen en mag, behalve tijdens het schoonmaken van de ruimte, bij voorkeur niet hoger zijn dan 70 %. Verlichting gebeurt met kunstlicht met een fotoperiode van 12 uur licht en 12 uur donker. Als voeding mag het gewone laboratoriumvoer worden gebruikt met een onbeperkte hoeveelheid drinkwater. De keuze van het voer kan worden beïnvloed door de noodzaak om voor een afdoende mengbaarheid met de teststof te zorgen, wanneer de stof in het voer wordt toegediend.
|
|
22.
|
De dieren moeten in kleine groepen van hetzelfde geslacht worden gehuisvest; de dieren kunnen individueel worden gehuisvest als dat wetenschappelijk gerechtvaardigd is. Indien de dieren in groepen in kooien worden ondergebracht, mogen per kooi niet meer dan vijf dieren worden gehuisvest. Het paren moet plaatsvinden in kooien die voor dat doel geschikt zijn. Drachtige vrouwtjes moeten in aparte kooien worden ondergebracht en nestmateriaal krijgen. Zogende vrouwtjes moeten samen met hun jongen in aparte kooien worden ondergebracht.
|
|
23.
|
Het voeder moet regelmatig op verontreinigingen worden onderzocht. Een monster van het voedsel moet worden bewaard totdat het verslag is afgerond.
|
Voorbereiding van de dieren
|
24.
|
Gezonde jonge volwassen dieren worden gerandomiseerd en in de behandelgroepen en kooien ingedeeld. De kooien worden zodanig geplaatst dat mogelijke effecten door de plaatsing van de kooi tot een minimum worden beperkt. De dieren krijgen een unieke identificatie en blijven, voordat de test begint, minimaal vijf dagen in hun kooi om in het laboratorium te acclimatiseren.
|
Voorbereiding van de doses
|
25.
|
Aanbevolen wordt om de teststof oraal toe te dienen, tenzij andere toedieningswegen geschikter worden geacht. Wanneer voor de orale weg wordt gekozen, wordt de teststof gewoonlijk via een sonde toegediend; als alternatieve mogelijkheid kunnen teststoffen echter ook via het voer of het drinkwater worden toegediend.
|
|
26.
|
Waar nodig wordt de teststof in een geschikt vehiculum opgelost of gesuspendeerd. Aanbevolen wordt eerst te bezien of het mogelijk is een waterige oplossing/suspensie te gebruiken, als dit niet kan een oplossing/suspensie in olie (bv. maïsolie) te overwegen en daarna eventueel andere vehicula. Van niet-waterige vehicula moeten de toxische kenmerken bekend zijn. Voorts moeten de stabiliteit en homogeniteit van de teststof in het vehiculum worden bepaald.
|
PROCEDURE
Aantal en geslacht van de dieren
|
27.
|
Aanbevolen wordt dat elke groep aan het begin van de test ten minste 10 mannetjes en 12-13 vrouwtjes telt. Bij de vrouwtjes wordt vóór blootstelling de oestruscyclus beoordeeld. Dieren die geen normale 4-5-daagse cyclus hebben, worden niet het onderzoek opgenomen. Daarom wordt aanbevolen met extra vrouwtjes te beginnen om uiteindelijk op 10 vrouwtjes per groep uit te komen. Behalve in geval van duidelijke toxische effecten wordt verwacht dat dit ten minste 8 drachtige vrouwtjes per groep zal opleveren, hetgeen normaal gesproken het minimaal aanvaardbare aantal drachtige vrouwtjes per groep is. Het doel is voldoende drachten en nakomelingen tot stand te brengen om een betekenisvolle evaluatie mogelijk te maken van de mogelijke invloed van de stof op de vruchtbaarheid, de dracht en het gedrag van moeder en jongen, en op de groei en ontwikkeling van de F1-nakomelingen, vanaf de conceptie tot dag 13 na het werpen. Indien het de bedoeling is tussentijds dieren te doden, moet het aantal dieren worden verhoogd met het aantal dat tussentijds zal worden gedood. Er moet worden overwogen een aanvullende satellietgroep van vijf dieren per geslacht op te nemen in de controlegroep en de groep met het hoogste dosisniveau, voor waarneming van reversibiliteit, persistentie of vertraagd tot uiting komen van systemische toxische effecten gedurende ten minste 14 dagen na de behandeling. De dieren in de satellietgroepen zullen niet paren en worden dan ook niet gebruikt voor de beoordeling van reproductie-/ontwikkelingstoxiciteit.
|
Dosering
|
28.
|
In het algemeen zijn drie testgroepen en een controlegroep vereist. Als er geen bruikbare algemene toxiciteitsgegevens beschikbaar zijn, kan een bereikbepalingonderzoek worden uitgevoerd (bij dieren van dezelfde stam en dezelfde herkomst) om de orde van grootte van de te gebruiken doses te helpen bepalen. Afgezien van de toediening van de teststof worden de dieren in de controlegroep op identieke wijze behandeld als de dieren in de testgroepen. Indien bij de toediening van de teststof van een vehiculum gebruik wordt gemaakt, moet dit aan de controlegroep in het hoogste gebruikte volume toegediend worden.
|
|
29.
|
De dosisniveaus moeten worden gekozen in het licht van de bestaande gegevens over toxiciteit en (toxico)kinetica. Er moet ook rekening mee gehouden worden dat er verschillen kunnen zijn in gevoeligheid tussen drachtige en niet-drachtige dieren. Het hoogste dosisniveau moet zo worden gekozen dat toxische effecten optreden, maar geen sterfte of duidelijk lijden. Daarna moet een dalende reeks dosisniveaus gekozen worden om het verband tussen dosering en respons aan te tonen en bij het laagste niveau te komen tot een dosis zonder schadelijke effecten. Intervallen van een factor twee tot vier zijn vaak optimaal en het is vaak beter een vierde testgroep toe te voegen dan zeer grote intervallen (bv. meer dan een factor 10) tussen de doseringen te gebruiken.
|
|
30.
|
Bij algemene toxiciteit (bv. verminderd lichaamsgewicht, effecten op lever, hart, longen of nieren enz.) of andere veranderingen die mogelijk geen toxische reacties zijn (bv. verminderde voedselinname, leververgroting), moeten de waargenomen effecten op hormoongevoelige eindpunten met de nodige omzichtigheid worden geïnterpreteerd.
|
Limiettest
|
31.
|
Als volgens de hier beschreven procedures een onderzoek met orale toediening wordt uitgevoerd met één dosisniveau van ten minste 1 000 mg/kg lichaamsgewicht/dag of, in het geval van toediening via het voer, het equivalente percentage in het voer, of het drinkwater (berekend overeenkomstig het lichaamsgewicht), en er geen waarneembare toxische effecten optreden, en deze ook niet kunnen worden verwacht op grond van gegevens betreffende stoffen met verwante structuur, is het niet noodzakelijk een volledig onderzoek met meerdere dosisniveaus te doen. De limiettest is van toepassing tenzij blootstelling bij mensen een hogere dosering noodzakelijk maakt. Voor andere toedieningsvormen, zoals inhalatie of toediening op de huid, zal de maximaal haalbare blootstelling vaak worden bepaald door de fysisch-chemische eigenschappen van de teststoffen.
|
Toediening van de doses
|
32.
|
De dieren krijgen de teststof 7 dagen per week dagelijks toegediend. Indien de teststof via een maagsonde aan het dier wordt toegediend, moet dit in één enkele dosis gebeuren met gebruikmaking van een maagbuisje of een geschikte intubatiecanule. Het maximale volume dat in één keer kan worden toegediend, is afhankelijk van de grootte van het dier. Het volume mag niet groter zijn dan 1 ml/100 g lichaamsgewicht, behalve wanneer het om een waterige oplossing gaat: in dat geval mag 2 ml/100 g lichaamsgewicht worden gebruikt. Afgezien van irriterende of bijtende teststoffen die normaliter bij hogere concentraties ergere effecten vertonen, moet de variabiliteit in het voor de proef gebruikte volume zo klein mogelijk worden gehouden door de concentratie zodanig aan te passen dat bij alle dosisniveaus een constant volume wordt gewaarborgd.
|
|
33.
|
Het is belangrijk dat er bij teststoffen die via het voer of het drinkwater worden toegediend, voor wordt gezorgd dat de hoeveelheden teststof de normale voer- of waterbalans niet verstoren. Wanneer de teststof in het voer wordt toegediend, kan een constante concentratie in het voer (in ppm) of een constante dosis in verhouding tot het lichaamsgewicht van de dieren worden gebruikt; daarbij moet worden aangegeven welk alternatief is gebruikt. Wanneer de teststof met een sonde wordt toegediend, moeten de doses dagelijks op vaste tijdstippen worden gegeven. De doses moeten ten minste wekelijks worden aangepast om een constant dosisniveau als functie van het lichaamsgewicht van het dier te verkrijgen. Indien een gecombineerd onderzoek wordt gebruikt als voorstudie van een langdurig chronisch toxiciteitsonderzoek of een volledig reproductietoxiciteitsonderzoek, moet in beide onderzoeken dezelfde voeding worden gebruikt.
|
Proefopzet
|
34.
|
Voor beide geslachten moet de toediening 2 weken voor het paren beginnen, nadat de dieren ten minste vijf dagen lang zijn geacclimatiseerd en de vrouwtjes gescreend zijn op normale oestruscycli (in een voorbehandelingsperiode van 2 weken). Het onderzoek moet zodanig worden ingepland dat de beoordeling van de oestruscyclus begint kort nadat de dieren volledig geslachtsrijp zijn. Dit kan enigszins variëren voor verschillende rattenstammen in verschillende laboratoria, bv. voor Sprague Dawley-ratten op een leeftijd van 10 weken, voor Wistar-ratten op een leeftijd van ongeveer 12 weken. Moederdieren met jongen moeten op dag 13 na het werpen worden gedood of kort daarna. Om de moederdieren voorafgaand aan de bloedafname een dag te laten vasten (als daaraan de voorkeur wordt gegeven) hoeven de vrouwtjes en hun jongen niet op dezelfde dag gedood te worden. De dag van de geboorte (d.w.z. wanneer het werpen is voltooid) wordt gedefinieerd als dag 0 na het werpen. Vrouwtjes die geen tekenen van copulatie vertonen, worden 24-26 dagen na de laatste dag van de paringsperiode gedood. Voor beide geslachten wordt de toediening gedurende de paringsperiode voortgezet. Daarna moeten de mannetjes de teststof ten minste nog toegediend krijgen tot de minimale totale doseringsperiode van 28 dagen is afgerond. Vervolgens worden ze gedood of, anders, in leven gehouden en verder behandeld voor een eventuele tweede paring.
|
|
35.
|
De dagelijkse toediening aan de moederdieren moet doorgaan gedurende de dracht en ten minste tot en met dag 13 na het werpen of de dag voordat ze gedood worden. Voor onderzoeken waarbij de toediening van de teststof door middel van inhalatie of dermaal plaatsvindt, moet deze ten minste worden voortgezet tot en met dag 19 van de dracht en zo spoedig mogelijk en niet later dan postnatale dag (PND) 4 worden hervat.
|
|
36.
|
Dieren in eventuele satellietgroepen die bestemd zijn voor vervolgwaarnemingen, paren niet. Ze moeten nog 14 dagen nadat de eerste moederdieren gedood worden, zonder behandeling geobserveerd worden om de persistentie van de toxische effecten c.q. het herstel alsmede het eventuele optreden van vertraagde toxiciteit te kunnen waarnemen.
|
|
37.
|
In aanhangsel 2 wordt de proefopzet in een diagram weergegeven.
|
Oestruscycli
|
38.
|
Voordat de behandeling begint, moeten de oestruscycli worden gecontroleerd om te selecteren op vrouwelijke proefdieren met regelmatige cycli (zie punt 27). Ook moeten er van het begin van de behandelingsperiode totdat paring is gebleken, dagelijks vaginale uitstrijkjes worden onderzocht. Indien vermoed wordt dat effecten van acute stress bij het begin van de behandeling de oestruscyclus zouden kunnen verstoren, kunnen de laboratoria de proefdieren gedurende 2 weken blootstellen en vervolgens ten minste 2 weken lang dagelijks vaginale uitstrijkjes nemen ter controle van de oestruscycli gedurende de periode vóór paring tot in de paringsperiode, totdat paring gebleken is. Wanneer monsters van vaginale/cervicale cellen worden genomen, moet erop worden gelet verstoring van het slijmvlies, die tot schijndracht kan leiden, te voorkomen (8) (9).
|
De paringsprocedure
|
39.
|
Normaal gesproken moet men voor dit onderzoek één mannetje met één vrouwtje (1:1) laten paren. Hierop zijn uitzonderingen mogelijk in geval van incidentele sterfte van mannetjes. Het vrouwtje moet bij hetzelfde mannetje worden geplaatst totdat aanwijzingen voor copulatie zijn waargenomen of er twee weken zijn verstreken. Elke ochtend worden de vrouwtjes onderzocht op de aanwezigheid van sperma of vaginale plug. Dag 0 van de dracht wordt gedefinieerd als de dag waarop aanwijzingen voor paring worden bevestigd (observatie van een vaginale plug en/of sperma). Wanneer het paren geen succes heeft gehad, kan worden overwogen de vrouwtjes opnieuw te laten dekken door mannetjes van dezelfde groep waarvan de vruchtbaarheid is aangetoond.
|
Nestgrootte
|
40.
|
Op PND 4 kan de grootte van de nesten worden aangepast door eliminatie van extra jongen door willekeurige selectie, zodat elk nest uiteindelijk zoveel mogelijk bestaat uit vier of vijf jongen per geslacht, afhankelijk van de normale nestgrootte van de gebruikte rattenstam. Bij twee van de overtollige jongen moeten bloedmonsters worden afgenomen, gepoold en gebruikt ter bepaling van de serumspiegels van T4. Selectieve eliminatie van jongen, bijvoorbeeld op grond van lichaamsgewicht of anogenitale afstand (AGD), is niet gepast. Als het door het aantal mannelijke of vrouwelijke jongen niet mogelijk is om op vier of vijf per geslacht per nest uit te komen, is gedeeltelijke aanpassing (bv. zes mannetjes en vier vrouwtjes) aanvaardbaar. Er worden geen jongen geëlimineerd als de nestgrootte daardoor onder de streefgrootte (8 of 10 jongen/nest) zou komen. Als er maar één jong beschikbaar is boven de streefgrootte, wordt er slechts één jong geëlimineerd en gebruikt voor het nemen van een bloedmonster ter bepaling van serum-T4.
|
|
41.
|
Als de nestgrootte niet wordt aangepast, worden er op PND 4 twee jongen per nest gedood en worden er bloedmonsters genomen voor het meten van de serumspiegels van schildklierhormonen. De twee jongen per nest moeten zo mogelijk vrouwelijke jongen zijn om de mannelijke jongen te bewaren voor de beoordeling van de tepelretentie, tenzij er na verwijdering van deze jongen geen enkel vrouwtje meer overblijft voor de beoordeling bij beëindiging van het onderzoek. Er worden geen jongen geëlimineerd als de nestgrootte daardoor onder de 8 of 10 jongen/nest (afhankelijk van de normale nestgrootte van de gebruikte rattenstam) zou komen. Als er maar één jong beschikbaar is boven de normale nestgrootte, wordt er slechts één jong geëlimineerd en gebruikt voor het nemen van een bloedmonster ter bepaling van serum-T4.
|
Waarnemingen
|
42.
|
Algemene klinische waarnemingen moeten ten minste eenmaal per dag plaatsvinden, bij voorkeur steeds op hetzelfde tijdstip en met inachtneming van de piekperiode van de te verwachten effecten na toediening. De gezondheidstoestand van de dieren moet worden geregistreerd. De dieren moeten ten minste tweemaal per dag worden geobserveerd op ziekteverschijnselen en sterfte.
|
|
43.
|
Alle ouderdieren worden eenmaal vóór de eerste blootstelling nauwkeurig klinisch onderzocht (om intra-individuele vergelijking mogelijk te maken) en vervolgens ten minste eenmaal per week. Dit onderzoek moet buiten de kooi op een vaste onderzoekplaats en bij voorkeur steeds op dezelfde dag uitgevoerd worden. De gegevens moeten zorgvuldig worden opgetekend, bij voorkeur met behulp van scoringssystemen die door het testlaboratorium expliciet zijn gedefinieerd. Er moet zo veel mogelijk worden gezorgd dat variaties in de testomstandigheden minimaal zijn en dat de waarnemers niet weten welke behandeling een gegeven dier heeft ondergaan. Tekenen waarop moet worden gelet zijn onder meer (maar niet alleen): veranderingen in huid, vacht, ogen, slijmvliezen, optreden van af- en uitscheiding, en onwillekeurige reacties zoals traanvorming, rechtopstaan van het haar, verandering van de pupilgrootte of een ongewoon ademhalingspatroon. Ook verandering in gang, houding en reactiviteit op vastpakken, alsmede de aanwezigheid van klonische of tonische bewegingen, stereotiep gedrag (bv. overmatige lichaamsverzorging, herhaaldelijk ronddraaien), een moeilijke of langdurige worp of bizar gedrag (bv. zelfverminking, achteruitlopen) moeten worden opgetekend (10).
|
|
44.
|
In de loop van het onderzoek moet op enig moment bij vijf mannetjes en vijf vrouwtjes, die aselect worden gekozen uit elke groep, de sensorische reactiviteit op stimuli van verschillende aard (bv. auditief, visueel of proprioceptief) (8) (9) (11) worden bepaald en moet een bepaling van de grijpkracht (12) en de motorische activiteit (13) plaatsvinden. Meer informatie over de procedures die kunnen worden gevolgd, is te vinden in de desbetreffende literatuur. Er kunnen evenwel ook andere dan de genoemde procedures worden toegepast. Bij mannetjes moeten deze functionele waarnemingen tegen het einde van hun doseringsperiode worden gedaan, kort voordat ze gedood zullen worden, maar ook voordat er bloed wordt afgenomen voor hematologische en klinisch-chemische bepalingen (zie de punten 53-56, waaronder voetnoot 1). Vrouwtjes moeten tijdens deze functionele tests in een vergelijkbare fysiologische toestand verkeren en moeten bij voorkeur eenmaal worden getest in de laatste week van de zoogperiode (bv. zoogdag 6-13), kort voordat ze gedood worden. Probeer moederdieren en jongen steeds zo kort mogelijk van elkaar te scheiden.
|
|
45.
|
Als het onderzoek wordt verricht als voorstudie voor een daaropvolgend subchronisch onderzoek van 90 dagen of langetermijnonderzoek, kunnen de eenmalige functionele waarnemingen tegen het einde van het onderzoek achterwege worden gelaten. In dat geval moeten de functionele waarnemingen in het bedoelde vervolgonderzoek plaatsvinden. Daar staat echter tegenover dat de beschikbaarheid van gegevens betreffende functionele waarnemingen uit het onderzoek met herhaalde toediening, de keuze van de dosisniveaus voor het daaropvolgende subchronische of langetermijnonderzoek ten goede kan komen.
|
|
46.
|
Bij wijze van uitzondering kunnen functionele waarnemingen ook achterwege worden gelaten bij groepen die anders tekenen van toxiciteit vertonen in een mate die aanzienlijk zou interfereren met de uitvoering van de functionele test.
|
|
47.
|
De drachttijd moet worden geregistreerd en wordt berekend vanaf dag 0 van de dracht. Elke worp moet zo spoedig mogelijk na de geboorte te worden onderzocht, teneinde het aantal en geslacht van de jongen vast te stellen, alsmede het aantal doodgeborenen, levendgeborenen, ondermaatse jongen (jongen die aanmerkelijk kleiner zijn dan de corresponderende controlejongen) en de aanwezigheid van macroscopisch zichtbare afwijkingen.
|
|
48.
|
Levende jongen moeten worden geteld en gesekst en de nesten moeten binnen 24 uur na de geboorte (dag 0 of 1 na het werpen) en ten minste op dag 4 en dag 13 na het werpen worden gewogen. Naast de in punt 43 en 44 beschreven waarnemingen voor ouderdieren moet elk afwijkend gedrag van de nakomelingen worden geregistreerd.
|
|
49.
|
De AGD van elk jong moet worden gemeten op dezelfde postnatale dag tussen postnatale dag 0 en het einde van postnatale dag 4. Het lichaamsgewicht van elk jong moet worden bepaald op de dag dat de AGD wordt gemeten en de AGD moet genormaliseerd worden tot een maat voor jonggrootte, bij voorkeur de derdemachtswortel van het lichaamsgewicht (14). Het aantal tepels/tepelhoven bij mannelijke jongen moet worden geteld op postnatale dag 12 of 13, zoals aanbevolen in OESO-leidraad 151 (15).
|
Lichaamsgewicht en consumptie van voer en water
|
50.
|
De mannetjes en vrouwtjes moeten worden gewogen op de eerste dag dat zij de teststof krijgen toegediend, vervolgens ten minste eenmaal per week en aan het einde van het onderzoek. De vrouwtjes moeten worden gewogen op de dagen 0, 7, 14 en 20 tijdens de dracht, binnen 24 uur na de geboorte (dag 0 of 1 na het werpen) en ten minste op dag 4 en dag 13 na het werpen. Deze waarnemingen worden voor elk volwassen dier apart gerapporteerd.
|
|
51.
|
Vóór het paren, tijdens de dracht en tijdens het zogen, moet ten minste wekelijks de voerconsumptie worden gemeten. Tijdens de paringsperiode is deze meting facultatief. Wanneer de teststof via het drinkwater wordt toegediend, moet ook de waterconsumptie gedurende deze perioden worden gemeten.
|
Hematologie
|
52.
|
Tijdens het onderzoek moeten eenmaal bij vijf mannetjes en vijf vrouwtjes, die aselect worden gekozen uit elke groep, de volgende hematologische onderzoeken worden verricht: hematocriet, hemoglobinegehalten, aantal rode bloedcellen, reticulocyten, totaal aantal en gedifferentieerde aantallen witte bloedcellen, aantal bloedplaatjes en bloedstollingstijd/vermogen. Indien de teststof of de vermeende metabolieten ervan oxiderende eigenschappen hebben, of hiervan een vermoeden bestaat, moeten andere bepalingen worden uitgevoerd, zoals van de methemoglobineconcentratie en de Heinz-lichaampjes.
|
|
53.
|
Bij het nemen van de bloedmonsters moet worden genoteerd uit welk lichaamsdeel en hoe de monsters zijn genomen. Tijdens de bloedafname moeten de vrouwtjes in een vergelijkbare fysiologische toestand verkeren. Om praktische problemen in verband met de variabiliteit in het begin van de dracht te vermijden, kan de bloedafname bij vrouwtjes aan het einde van de periode voor het paren worden uitgevoerd als alternatief voor de monstername net voor of tijdens het euthanaseren van de dieren. De bloedmonsters van de mannetjes moeten bij voorkeur net voor of tijdens het euthanaseren van de dieren worden genomen. Als alternatieve mogelijkheid kan de bloedafname bij mannetjes ook aan het einde van de periode voor het paren worden uitgevoerd, wanneer dat tijdstip ook voor de vrouwtjes wordt gekozen.
|
|
54.
|
De bloedmonsters moeten onder passende omstandigheden worden bewaard.
|
Klinische biochemie
|
55.
|
Klinisch-biochemische bepalingen die bedoeld zijn om wezenlijke toxische effecten in weefsels en met name effecten op de nieren en lever te onderzoeken, moeten worden verricht op bloedmonsters die zijn genomen bij de geselecteerde vijf mannetjes en vijf vrouwtjes uit elke groep. Het is aan te bevelen in de nacht voordat de bloedmonsters worden genomen de dieren voedsel te onthouden (22). Onderzoek van plasma of serum moet omvatten: natrium, kalium, glucose, totaal cholesterol, ureum, creatinine, totaal proteïne en albumine, ten minste twee enzymen die een indicatie geven van hepatocellulaire effecten (zoals alanineaminotransferase, aspartaataminotransferase en sorbitoldehydrogenase), en galzuren. Bepaling van andere enzymen (van hepatische of andere oorsprong) en bilirubine kan onder bepaalde omstandigheden nuttige informatie geven.
|
|
56.
|
Er worden bloedmonsters genomen van een gedefinieerde plaats op basis van het volgende schema:
|
—
|
van ten minste twee jongen per nest op PND 4 als het aantal jongen dit toelaat (zie de punten 40-41);
|
|
—
|
van alle moederdieren en ten minste twee jongen per nest op het tijdstip van doden op dag 13, en
|
|
—
|
van alle volwassen mannetjes op het tijdstip van doden.
|
Alle bloedmonsters worden onder passende omstandigheden bewaard. In de bloedmonsters van de jongen op dag 13 en de volwassen mannetjes worden de serumspiegels van schildklierhormonen (T4) bepaald. Een verdere bepaling van T4 in bloedmonsters van moederdieren en jongen op dag 4 wordt verricht indien relevant. Eventueel kunnen ook andere hormonen worden gemeten indien relevant. Voor de schildklierhormoonanalyse kan bloed van jongen per nest gepoold worden. De schildklierhormonen (T4 en TSH) worden bij voorkeur als „totaal” gemeten.
|
|
57.
|
Als optie kunnen tijdens de laatste week van het onderzoek de volgende urineonderzoeken worden verricht bij vijf aselect gekozen mannetjes uit elke groep, waarbij op regelmatige tijdstippen urinemonsters moeten worden genomen: uitzien, volume, osmolaliteit of relatieve dichtheid, pH, eiwit, glucose en bloed/bloedcellen.
|
|
58.
|
Bovendien moet worden overwogen serummarkers van algemene weefselbeschadiging te onderzoeken. Andere onderzoeken die moeten worden uitgevoerd als van de teststof bekend is, of vermoed wordt, dat zij de desbetreffende metabolische profielen beïnvloedt, zijn: calcium, fosfaat, triglyceriden en glucose bij nuchtere toestand, specifieke hormonen, methemoglobine en cholesterinase. Deze moeten per geval geïdentificeerd worden.
|
|
59.
|
De volgende factoren kunnen van invloed zijn op de variabiliteit en de absolute concentraties van de hormoonbepalingen:
|
—
|
tijdstip waarop de dieren worden gedood, vanwege de schommelingen in de hormoonconcentraties gedurende de dag;
|
|
—
|
wijze waarop de dieren worden gedood: onnodige stress bij de dieren, waardoor de hormoonconcentraties kunnen worden beïnvloed, moet worden voorkomen;
|
|
—
|
testkits voor hormoonbepalingen, die verschillende standaardkrommen kunnen hebben.
|
|
|
60.
|
Plasmamonsters die specifiek voor hormoonbepaling zijn bedoeld, moeten telkens op hetzelfde moment van de dag worden genomen. De verschillende in de handel verkrijgbare „assay kits” kunnen verschillende numerieke waarden opleveren bij de analyse van hormoonconcentraties.
|
|
61.
|
In het geval van ontoereikende basisgegevens uit het verleden moet worden overwogen de hematologische en klinisch-biochemische variabelen te bepalen, bij voorkeur bij een groep dieren die niet in de testgroepen zijn opgenomen, of anders voordat met de toediening wordt begonnen. Voor vrouwtjes moeten de gegevens verkregen zijn bij zogende dieren.
|
PATHOLOGIE
Macroscopische obductie
|
62.
|
Bij alle volwassen dieren in het onderzoek moet een volledige macroscopische obductie worden uitgevoerd, waaronder een zorgvuldig onderzoek van het huidoppervlak, alle lichaamsopeningen alsmede de schedelholte, de borstholte en de buikholte, en de inhoud daarvan. Bijzondere aandacht moet worden geschonken aan de organen van het voortplantingssysteem. Het aantal implantatieplaatsen moet worden geregistreerd. Op de dag van obductie moeten er vaginale uitstrijkjes onderzocht worden om de fase van de oestruscyclus te bepalen en deze in verband te kunnen brengen met de histopathologie van de vrouwelijke geslachtsorganen.
|
|
63.
|
De testes en epididymides, evenals de prostaat en de zaadblaasjes met coagulans afscheidende klieren als geheel van alle volwassen mannetjes moeten zo nodig worden ontdaan van aanhechtend weefsel en zo spoedig mogelijk na de sectie nat worden gewogen om te voorkomen dat ze uitdrogen. Optionele organen die eventueel ook gewogen kunnen worden, zijn onder meer het m levator ani plus bulbocavernosuscomplex, de Cowperse klieren en de glans penis bij mannetjes en de gepaarde ovaria (nat gewicht) en uterus (inclusief cervix) bij vrouwtjes; indien deze gewichten worden meegenomen, moeten ze zo snel mogelijk na de sectie worden opgenomen. Van alle volwassen dieren moeten de ovaria, testes, epididymides, secundaire geslachtsorganen en alle organen die macroscopische letsels vertonen, worden geconserveerd.
|
|
64.
|
Van alle volwassen mannetjes en vrouwtjes en één mannelijk en één vrouwelijk dag 13-jong uit elk nest moeten de schildklieren in een fixeermiddel worden bewaard dat geschikt is voor het beoogde latere histopathologische onderzoek. Het gewicht van de schildklier kan na de fixatie worden bepaald. Aanhechtend weefsel moet zeer voorzichtig en pas na fixatie worden verwijderd, om beschadiging van de weefsels te voorkomen. Weefselbeschadigingen kunnen de histopathologische analyse negatief beïnvloeden. De bloedmonsters moeten worden genomen net voor of tijdens het euthanaseren van de dieren; er moet worden genoteerd uit welk lichaamsdeel. De monsters moeten onder de juiste omstandigheden worden bewaard (zie punt 56).
|
|
65.
|
Bovendien moeten van ten minste vijf volwassen mannetjes en vrouwtjes die aselect worden gekozen uit elke groep (afgezien van dieren die stervend zijn aangetroffen en/of zijn gedood vóór beëindiging van het onderzoek) de lever, nieren, bijnieren, thymus, milt, hersenen en het hart zo nodig worden ontdaan van aanhechtend weefsel en zo spoedig mogelijk na de sectie nat worden gewogen om te voorkomen dat ze uitdrogen. De volgende weefsels moeten in een fixeermiddel worden bewaard dat zowel voor het type weefsel als voor het beoogde latere histopathologische onderzoek geschikt is: elk macroscopisch waarneembaar letsel, hersenen (representatieve delen waaronder cerebrum, cerebellum en pons), ruggenmerg, oog, maag, dunne en dikke darm (inclusief Peyerse platen), lever, nieren, bijnieren, milt, hart, thymus, luchtpijp en longen (conserveren door opblazen met een fixatief en dan onderdompelen), geslachtsklieren (testes en ovaria), secundaire geslachtsorganen (uterus en cervix, epididymides, prostaat, zaadblaasjes plus coagulans uitscheidende klieren), vagina, urineblaas, lymfeklieren (naast de meest proximale afvoerende lymfeklier moet ook een andere lymfeklier worden uitgenomen, afhankelijk van de ervaringen van het laboratorium (16)), perifere zenuwen (nervus ischiadicus of nervus tibialis), bij voorkeur dicht bij de spier, skeletspier en bot, met beenmerg (een coupe, of in plaats daarvan een direct op een objectglaasje aangebracht stukje beenmergaspiraat). Aanbevolen wordt de testes te fixeren door middel van onderdompeling in Bouin-oplossing of gemodificeerde Davidson-oplossing (16) (17) (18); fixatie in formaline wordt voor deze weefsels niet aanbevolen. De tunica albuginea kan voorzichtig en ondiep met een naald worden ingeprikt op de beide polen van het orgaan, zodat het fixatief snel kan binnendringen. Uit de klinische en andere bevindingen kan de noodzaak blijken verder weefsel te onderzoeken. Ook organen waarvan op basis van de bekende eigenschappen van de teststof verondersteld wordt dat ze kunnen worden aangetast, moeten worden bewaard.
|
|
66.
|
De volgende weefsels kunnen nuttige aanwijzingen geven voor met het endocrien systeem samenhangende effecten: geslachtsklieren (ovaria en testes), secundaire geslachtsorganen (uterus inclusief cervix, epididymides, zaadblaasjes met coagulans afscheidende klieren, dorsolaterale en ventrale prostaat), vagina, hypofyse, mannelijke borstklier en bijnier. Er bestaat onvoldoende documentatie over veranderingen in de mannelijke borstklieren, maar deze parameter zou kan erg gevoelig kunnen zijn voor stoffen met oestrogene werking. De waarneming van niet in de lijst van punt 65 opgenomen organen/weefsels is facultatief.
|
|
67.
|
Dode jongen en jongen die op dag 13 na het werpen, of kort daarna, zijn gedood, moeten in elk geval uitwendig zorgvuldig worden onderzocht op macroscopisch zichtbare afwijkingen. Daarbij wordt met name gelet op de uitwendige voortplantingsorganen, die op tekenen van een afwijkende ontwikkeling worden onderzocht.
|
Histopathologie
|
68.
|
Bij alle dieren in de groep behandeld met het hoogste dosisniveau en bij de dieren in de controlegroep moet een volledig histopathologisch onderzoek worden verricht op de bewaarde organen en weefsels (met speciale aandacht voor de stadia van de spermatogenese en de histopathologie van de interstitiële testiculaire celstructuur). Van jongen en van de overgebleven volwassen dieren kan de schildklier worden onderzocht indien nodig. Wanneer er behandelingsgerelateerde veranderingen worden waargenomen in de hogedosisgroep, moeten ook de dieren in de andere doseringsgroepen worden onderzocht. In de richtsnoeren voor histopathologisch onderzoek (10) zijn nadere gegevens opgenomen over de sectie, de fixatie, het snijden van coupes en de histopathologie van endocriene weefsels.
|
|
69.
|
Alle macroscopisch waarneembare beschadigingen moeten onderzocht worden. Om bij te dragen tot de bepaling van de NOAEL moeten de doelorganen in andere dosisgroepen worden onderzocht, vooral in groepen waarvoor een NOAEL wordt geclaimd.
|
|
70.
|
Bij het histopathologisch onderzoek van de dieren in de satellietgroepen moet speciaal worden gelet op die organen en weefsels waarin effecten blijken te zijn opgetreden bij de andere behandelde groepen.
|
GEGEVENS EN RAPPORTAGE
Gegevens
|
71.
|
Er worden voor elk dier apart gegevens verstrekt. Daarnaast moeten alle gegevens worden samengevat in tabelvorm, waarbij voor elke testgroep worden vermeld: het aantal dieren aan het begin van de test, het aantal dieren dat tijdens de test dood is aangetroffen of met het oog op een humane behandeling is gedood, het tijdstip van sterfte of doding, het aantal vruchtbare dieren, het aantal drachtige vrouwtjes, het aantal dieren met toxiciteitsverschijnselen, een beschrijving van de waargenomen toxiciteitsverschijnselen met vermelding van de aanvang, de duur en de ernst van de toxische effecten, de aard van de histopathologische veranderingen en alle relevante gegevens over de nesten. In aanhangsel 3 is een opzet van een overzichtstabel opgenomen die heel praktisch is gebleken voor de beoordeling van effecten op de voortplanting en de ontwikkeling.
|
|
72.
|
Waar mogelijk moeten getalsmatige resultaten met behulp van een geschikte en algemeen erkende statistische methode worden geëvalueerd. Het gebruik van meerdere t-tests moet worden voorkomen door vergelijking van het effect over een dosisbereik. De statistische methoden moeten gedurende de opzet van het onderzoek worden gekozen. De statistische analyse van de AGD en de tepelretentie moet worden gebaseerd op de gegevens voor afzonderlijke jongen, waarbij rekening wordt gehouden met nesteffecten. Waar passend is het nest de analyse-eenheid. De statistische analyse van lichaamsgewicht van de jongen moet worden gebaseerd op de gegevens voor afzonderlijke jongen, waarbij rekening wordt gehouden met nestgrootte. Vanwege de beperkte omvang van het onderzoek, zijn statistische analyses in de vorm van toetsen voor „significantie” voor veel eindpunten, vooral die in verband met de voortplanting, van beperkte waarde. Sommige van de meest gebruikte methoden, met name parametrische toetsen voor het meten van de centrale tendens, zijn niet geschikt. Als er statistische analyses worden gebruikt, moeten de gekozen methoden geschikt zijn voor de verdeling van de onderzochte variabele en moeten ze vóór de start van het onderzoek worden gekozen.
|
Evaluatie van de resultaten
|
73.
|
De resultaten van dit toxiciteitsonderzoek worden beoordeeld aan de hand van de waargenomen effecten en de resultaten van de obductie en het microscopisch onderzoek. De beoordeling omvat het verband tussen de dosis van de teststof en de aanwezigheid of afwezigheid, de frequentie en de ernst van afwijkingen zoals macroscopisch letsel, gespecificeerde doelorganen, onvruchtbaarheid, klinische afwijkingen, aantasting van de reproductie- en nestresultaten, veranderingen in het lichaamsgewicht, effecten op de mortaliteit en andere toxische effecten.
|
|
74.
|
Vanwege de korte behandelperiode van de mannetjes, moeten bij de beoordeling van effecten op de voortplanting bij mannetjes niet alleen de vruchtbaarheidsgegevens, maar ook de histopathologie van de testes en de epididymides in aanmerking worden genomen. Ook het gebruik van historische controlegegevens over voortplanting/ontwikkeling (bv. voor nestgrootte, AGD, tepelretentie, T4-serumspiegels), indien beschikbaar, kan nuttig zijn ter ondersteuning van de interpretatie van het onderzoek.
|
|
75.
|
Voor de kwaliteitscontrole wordt voorgesteld historische controlegegevens te verzamelen en variatiecoëfficiënten te berekenen voor numerieke gegevens, met name voor de parameters die verband houden met het opsporen van hormoonontregelaars. Deze gegevens kunnen voor vergelijkingsdoeleinden worden gebruikt wanneer daadwerkelijk uitgevoerd onderzoek wordt geëvalueerd.
|
Testverslag
|
76.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Teststof:
|
—
|
bron, partijnummer, uiterste gebruiksdatum, indien beschikbaar;
|
|
—
|
stabiliteit van de teststof, indien bekend.
|
|
|
|
Stof die uit één component bestaat:
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
chemische identificatiegegevens, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code, structuurformule, zuiverheid, chemische identiteit van onzuiverheden, indien van toepassing en praktisch haalbaar enz.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB’s en mengsels:
|
—
|
voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen
|
. |
|
|
Vehiculum (indien van toepassing):
|
—
|
motivering voor de keuze van het vehiculum, indien geen water wordt gebruikt.
|
|
|
|
Proefdieren:
|
—
|
aantal, leeftijd en geslacht van de dieren;
|
|
—
|
herkomst, leefomstandigheden, voeding enz.;
|
|
—
|
gewicht van elk dier aan het begin van de test;
|
|
—
|
motivering voor het gebruik van een ander soort dan ratten.
|
|
|
|
Testomstandigheden:
|
—
|
achtergrond voor de keuze van de dosisniveaus;
|
|
—
|
gedetailleerde gegevens over de formulering van de teststof, de bereiding van het voer en de bereikte concentratie, stabiliteit en homogeniteit van het preparaat;
|
|
—
|
gedetailleerde gegevens over de toediening van de teststof;
|
|
—
|
omrekening van de concentratie van de teststof in het voer/drinkwater (in ppm) naar de feitelijke dosis (in mg/kg lichaamsgewicht/dag), indien van toepassing;
|
|
—
|
gedetailleerde gegevens over de kwaliteit van het voer en het water;
|
|
—
|
gedetailleerde beschrijving van de randomisatieprocedure voor de selectie van te ruimen jongen, indien van toepassing.
|
|
|
|
Resultaten:
|
—
|
lichaamsgewicht en veranderingen in het lichaamsgewicht;
|
|
—
|
waar van toepassing, voedselverbruik en waterverbruik;
|
|
—
|
gegevens over de toxische reactie naar geslacht en dosis, met inbegrip van vruchtbaarheid, dracht en andere gegevens over
|
|
—
|
duur van de dracht; toxische of andere effecten op de reproductie, de jongen, de postnatale groei enz.;
|
|
—
|
aard, ernst en duur van (al dan niet reversibele) klinische waarnemingen;
|
|
—
|
bepalingen van de sensorische activiteit, grijpkracht en motorische activiteit;
|
|
—
|
uitgevoerde hematologische onderzoeken en de relevante referentiewaarden;
|
|
—
|
uitgevoerde klinisch-biochemische onderzoeken en de relevante referentiewaarden;
|
|
—
|
aantal volwassen vrouwtjes met een normale of abnormale oestruscyclus en cyclusduur van deze vrouwtjes;
|
|
—
|
aantal levend geboren jongen en verliezen na implantatie;
|
|
—
|
aantal jongen met macroscopisch zichtbare afwijkingen; macroscopische beoordeling van de uitwendige geslachtsorganen, aantal ondermaatse jongen;
|
|
—
|
tijdstip van sterfte tijdens het onderzoek en of de dieren overleefden tot het einde van het onderzoek;
|
|
—
|
aantal implantaties, nestgrootte en nestgewichten op het moment van registratie;
|
|
—
|
gegevens over het lichaamsgewicht van jongen;
|
|
—
|
AGD van alle jongen (en lichaamsgewicht op de dag van de AGD-meting);
|
|
—
|
tepelretentie bij mannelijke jongen;
|
|
—
|
schildklierhormoonspiegels bij dag 13-jongen en volwassen mannetjes (en moederdieren en dag 4-jongen indien gemeten);
|
|
—
|
lichaamsgewicht bij het doden van de dieren en gegevens over het gewicht van organen voor de ouderdieren;
|
|
—
|
een gedetailleerde beschrijving van histopathologische bevindingen;
|
|
—
|
resorptiegegevens (indien beschikbaar);
|
|
—
|
een statistische behandeling van de resultaten, indien van toepassing.
|
|
|
|
Bespreking van de resultaten.
|
|
Interpretatie van resultaten
|
77.
|
Op basis van dit onderzoek kan de reproductie-/ontwikkelingstoxiciteit worden beoordeeld bij de toediening van herhaalde doses. Aangezien de nadruk wordt gelegd op zowel het eindpunt algemene toxiciteit als het eindpunt reproductie-/ontwikkelingstoxiciteit, kan met de resultaten van het onderzoek in het bijzonder onderscheid worden gemaakt tussen reproductie-/ontwikkelingseffecten die zich voordoen bij afwezigheid van algemene toxiciteit, en reproductie-/ontwikkelingseffecten die alleen tot uitdrukking komen bij niveaus die ook toxisch zijn voor ouderdieren (zie de punten 7-11). Het onderzoek zou een indicatie kunnen geven of verder onderzoek nodig is en zou oriëntatiepunten kunnen bieden voor de opzet van vervolgonderzoeken. OESO-leidraad nr. 43 moet worden geraadpleegd voor hulp bij de interpretatie van resultaten op het gebied van reproductie- en voortplantingstoxiciteit (19). In OESO-leidraad nr. 106 over de histologische beoordeling van endocriene en voortplantingstests bij knaagdieren (16) is informatie te vinden over het prepareren en beoordelen van (endocriene) organen en vaginale uitstrijkjes, die van nut kan zijn voor deze testmethode.
|
LITERATUUR
|
(1)
|
OESO (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
OESO (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OESO (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Op aanvraag verkrijgbaar bij de Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(6)
|
OESO (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(7)
|
OESO (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl.Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OESO (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environmental Health and Safety Publications, Series on Testing and Assessment (No 151), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(16)
|
OESO (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No 106), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OESO (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(20)
|
OESO (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
Aanhangsel 1
DEFINITIES (ZIE OOK (20) OESO-LEIDRAAD 150)
Androgene werking het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk androgeen hormoon (bv. testosteron) te werken.
Antiandrogene werking het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk androgeen hormoon (bv. testosteron) te onderdrukken.
Antioestrogene werking het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk oestrogeen hormoon (bv. 17β-oestradiol) te onderdrukken.
Antithyreoïde werking het vermogen van een chemische stof om in een zoogdierorganisme de werking van een natuurlijk schildklierhormoon (bv. T3) te onderdrukken.
Chemische stof een stof of mengsel.
Dosering een algemene term die de dosis, de frequentie en de duur van de toediening omvat.
Dosis de toegediende hoeveelheid teststof. De dosis wordt uitgedrukt als gewicht van de teststof per eenheid lichaamsgewicht van het proefdier per dag (bv. mg/kg lichaamsgewicht/dag), of als constante voedingsconcentratie.
Manifeste toxiciteit een algemene term die duidelijke toxiciteitsverschijnselen beschrijft die optreden na de toediening van de teststof. Deze verschijnselen moeten een beoordeling van de gevaren mogelijk maken en van een zodanige aard zijn dat verwacht mag worden dat bij een hogere toegediende dosis ernstige toxische verschijnselen en waarschijnlijk sterfte optreden.
Maternale toxiciteit schadelijke effecten bij drachtige vrouwtjes, die hetzij specifiek optreden (direct effect), hetzij niet-specifiek (indirect effect), en die verband houden met de dracht.
NOAEL afkorting van„no-observed-adverse-effect level”, het niveau waarbij geen schadelijk effect werd vastgesteld. Dit is het hoogste dosisniveau waarbij nog geen aan de behandeling gebonden nadelige effecten worden vastgesteld.
Oestrogene werking het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk oestrogeen hormoon (bv. 17β-oestradiol) te werken.
Ontwikkelingstoxiciteit de wijze waarop reproductietoxiciteit zich manifesteert, in de vorm van pre-, peri- en postnatale, structurele of functionele afwijkingen bij nakomelingen.
Reproductietoxiciteit schadelijke effecten op de nakomelingen en/of verstoring van de voortplantingsfuncties of het voortplantingsvermogen bij mannetjes en vrouwtjes.
Teststof: elke volgens deze testmethode getest(e) stof of mengsel.
Thyreoïde werking het vermogen van een chemische stof om in een zoogdierorganisme als een natuurlijk schildklierhormoon (bv. T3) te werken.
Validering een wetenschappelijke methode die erop gericht is om de operationele vereisten en beperkingen van een testmethode te beschrijven en de betrouwbaarheid en relevantie ervan voor een bepaald doel aan te tonen.
Verstoring van de vruchtbaarheid afwijkingen in de voortplantingsfuncties of het voortplantingsvermogen bij mannetjes of vrouwtjes.
Aanhangsel 2
DIAGRAM VAN DE PROEFOPZET MET DE MAXIMALE ONDERZOEKSDUUR OP BASIS VAN EEN VOLLEDIGE 14-DAAGSE PARINGSPERIODE


Aanhangsel 3
OVERZICHTSTABEL VAN DE EFFECTEN OP REPRODUCTIE/ONTWIKKELING
|
WAARNEMINGEN
|
WAARDEN
|
|
Dosering (eenheden)…….
|
0 (controle)
|
…
|
…
|
…
|
…
|
|
Paren bij begin (N)
|
|
|
|
|
|
|
Oestruscyclus (ten minste gemiddelde duur en frequentie van onregelmatige cycli)
|
|
|
|
|
|
|
Vrouwtjes met aanwijzingen voor copulatie (N)
|
|
|
|
|
|
|
Vrouwtjes die drachtig worden (N)
|
|
|
|
|
|
|
Dagen conceptie 1 - 5 (N)
|
|
|
|
|
|
|
Dagen conceptie 6 -... (23) (N)
|
|
|
|
|
|
|
Dracht ≤ 21 dagen (N)
|
|
|
|
|
|
|
Dracht = 22 dagen (N)
|
|
|
|
|
|
|
Dracht ≥ 23 dagen (N)
|
|
|
|
|
|
|
Moederdieren met levend geboren jongen (N)
|
|
|
|
|
|
|
Moederdieren met levende jongen op postnatale dag 4 (N)
|
|
|
|
|
|
|
Implantaten/moederdier (gemiddeld)
|
|
|
|
|
|
|
Levende jongen/moederdier bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Levende jongen/moederdier op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Geslachtsverhouding (m/v) bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Geslachtsverhouding (m/v) op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van het nest bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Gewicht van het nest op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen bij geboorte (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen op het moment van AGD-meting (gemiddeld bij mannetjes, gemiddeld bij vrouwtjes)
|
|
|
|
|
|
|
AGD jongen op dezelfde postnatale dag, geboorte - dag 4 (gemiddeld bij mannetjes, gemiddeld bij vrouwtjes)
|
|
|
|
|
|
|
Gewicht van de jongen op dag 4 (gemiddeld)
|
|
|
|
|
|
|
Gewicht van de jongen op dag 13 (gemiddeld)
|
|
|
|
|
|
|
Tepelretentie bij mannelijke jongen op dag 13 (gemiddeld)
|
|
|
|
|
|
|
AFWIJKENDE JONGEN
|
|
Moederdieren met 0
|
|
|
|
|
|
|
Moederdieren met 1
|
|
|
|
|
|
|
Moederdieren met ≥ 2
|
|
|
|
|
|
|
VERLIES VAN NAKOMELINGEN
|
|
Vóór geboorte (implantaties minus levend geborenen)
|
|
Vrouwtjes met 0
|
|
|
|
|
|
|
Vrouwtjes met 1
|
|
|
|
|
|
|
Vrouwtjes met 2
|
|
|
|
|
|
|
Vrouwtjes met ≥ 3
|
|
|
|
|
|
|
Na geboorte (levend geboren en minus levende jongen op PND 13)
|
|
Vrouwtjes met 0
|
|
|
|
|
|
|
Vrouwtjes met 1
|
|
|
|
|
|
|
Vrouwtjes met 2
|
|
|
|
|
|
|
Vrouwtjes met ≥ 3
|
|
|
|
|
|
B.65.
IN-VITRO HUIDCORROSIE: TESTMETHODE OP BASIS VAN DE MEMBRAANBARRIÈRE
INLEIDING
|
1.
|
Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 435 (2015) van de OESO. Onder huidcorrosie wordt verstaan: het ontstaan van irreversibele schade aan de huid in de vorm van zichtbare necrose door de epidermis heen in de dermis, na het aanbrengen van een teststof (zoals gedefinieerd door het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (Globally Harmonized System of Classification and Labelling of Chemicals, GHS) van de Verenigde Naties (VN) (1) en Verordening (EU) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels (CLP-verordening) (24)). Deze testmethode, die gelijkwaardig is aan de bijgewerkte testrichtlijn 435 van de OESO, bevat een in-vitrotestmethode op basis van de membraanbarrière die kan worden gebruikt om corrosieve chemische stoffen te identificeren. De testmethode maakt gebruik van een kunstmatige membraan die is ontwikkeld om op een vergelijkbare manier op corrosieve chemische stoffen te reageren als dierenhuid in situ.
|
|
2.
|
De traditionele manier om huidcorrosiviteit te beoordelen is door de teststof op de huid van levende dieren aan te brengen en na een bepaalde tijdsperiode de mate van weefselschade te beoordelen (2). Behalve deze testmethode is er een aantal anderein-vitrotestmethoden vastgesteld als alternatief (3) (4) voor de standaardin-vivoprocedure met konijnenhuid (hoofdstuk B.4 van deze bijlage, gelijkwaardig aan TG 404 van de OESO) die wordt gebruik om corrosieve chemische stoffen te identificeren (2). In de gefaseerde test- en evaluatiestrategie van het VN-GHS voor de beoordeling en classificatie van huidcorrosiviteit en de OESO-leidraad voor een geïntegreerde test- en beoordelingsbenadering (Integrated Approach on Testing and Assessment, IATA) voor huidcorrosie en -irritatie wordt aanbevolen om de gevalideerde en erkende in-vitrotestmethoden uit de modules 3 en 4 te gebruiken (1) (5). De IATA omvat verschillende modules met gedeelde informatiebronnen en analyse-instrumenten en i) biedt richtsnoeren voor de integratie en het gebruik van bestaande gegevens die zijn verzameld met behulp van test- en niet-testmethoden om het potentieel voor huidirritatie en -corrosie van chemische stoffen te beoordelen en ii) draagt een benadering aan voor wanneer verder testen nodig is, onder meer wanneer er negatieve resultaten worden gevonden (5). Volgens deze modulaire aanpak kan een chemische stof op grond van positieve resultaten van in-vitrotestmethoden als corrosief worden ingedeeld, zonder dat er dierproeven nodig zijn, waardoor het gebruik van dieren voor dergelijke proeven wordt verminderd en verfijnd, en pijn en leed waarmee het gebruik van dieren voor dit doel gepaard zou kunnen gaan, worden vermeden.
|
|
3.
|
Er zijn valideringsstudies afgerond voor het in-vitromembraanbarrièremodel dat in de handel verkrijgbaar is als Corrositex®(6) (7) (8). Daarin scoorde het een algemene nauwkeurigheid in het voorspellen van huidcorrosiviteit van 79 % (128/163), een gevoeligheid van 85 % (76/89), en een specificiteit van 70 % (52/74) voor een databank van 163 stoffen en mengsels (7). Op basis van deze erkende validiteit wordt deze gevalideerde referentiemethode (VRM) aanbevolen voor gebruik als onderdeel van een gefaseerde teststrategie voor de beoordeling van het mogelijke huidcorrosiegevaar van chemische stoffen (5) (7). Voordat een in-vitromembraanbarrièremodel voor huidcorrosie kan worden gebruikt met het oog op regelgeving, moeten de betrouwbaarheid, relevantie (nauwkeurigheid) en beperkingen ervan voor het voorgestelde gebruik bepaald worden om te waarborgen dat deze vergelijkbaar zijn met die van de VRM (9), overeenkomstig de vooraf vastgestelde prestatienormen (10). De wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst is pas gewaarborgd wanneer een voorgestelde nieuwe of bijgewerkte methode volgens de prestatienormen is geëvalueerd en zijn opgenomen in de gelijkwaardige OESO-testrichtlijn.Op dit moment bestrijken OESO-testrichtlijn 435 en deze testmethode slechts een enkele in-vitromethode: het model Corrositex®, dat in de handel verkrijgbaar is.
|
|
4.
|
Andere testmethoden voor het testen op huidcorrosiviteit zijn gebaseerd op het gebruik van gereconstrueerde menselijke huid (OESO-TG 431) (3) en geïsoleerde rattenhuid (OESO TG 430) (4). Deze testrichtlijn voorziet ook in de nadere indeling van corrosieve chemische stoffen in de drie VN-GHS-subcategorieën voor corrosiviteit en de drie VN-verpakkingsgroepen voor vervoer betreffende gevaar van corrosiviteit. Deze testrichtlijn werd oorspronkelijk vastgesteld in 2006 en bijgewerkt in 2015 met verwijzingen naar de IATA-leidraad en met een bijgewerkte lijst van stoffen voor bekwaamheidstoetsing.
|
DEFINITIES
|
5.
|
Gebruikte definities worden gegeven in het aanhangsel.
|
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
6.
|
Met de test die in deze testmethode wordt beschreven, kunnen corrosieve teststoffen worden geïdentificeerd en ingedeeld in subcategorieën volgens VN-GHS/CLP (tabel 1). Bovendien kan een dergelijke testmethode worden gebruikt voor het nemen van beslissingen over de corrosieve en niet-corrosieve aard van specifieke klassen van chemische stoffen, bv. organische en anorganische zuren, afgeleide zuren. (25) en basen ten behoeve van bepaalde vervoerstests (7) (11) (12). In deze testmethode wordt een algemene procedure beschreven die vergelijkbaar is met de gevalideerde referentietestmethode (7). Hoewel deze testmethode geen toereikende informatie over huidirritatie oplevert, moet worden opgemerkt dat TM B.46 (gelijkwaardig aan OESO-TG 439) specifiek betrekking heeft op het gezondheidseffect huidirritatie in vitro (13). Voor een volledige beoordeling van lokale huideffecten na één blootstelling op de huid moet de OESO-leidraad voor een geïntegreerde test- en beoordelingsbenadering (Integrated Approach on Testing and Assessment, IATA) worden geraadpleegd (5).
Tabel 1
De VN-GHS-categorie en -subcategorieën voor huidcorrosie (26)
|
Categorie voor corrosiviteit (categorie 1) (voor autoriteiten die geen subcategorieën gebruiken)
|
Eventuele subcategorieën voor corrosiviteit (26) (voor autoriteiten die subcategorieën gebruiken, waaronder de CLP-verordening)
|
Corrosief bij ≥ 1 van de 3 dieren
|
|
Blootstellingstijd
|
Waarneming
|
|
Corrosief
|
Subcategorie voor corrosiviteit 1A
|
≤ 3 minuten
|
≤ 1 uur
|
|
Subcategorie voor corrosiviteit 1B
|
> 3 minuten / ≤ 1 uur
|
≤ 14 dagen
|
|
Subcategorie voor corrosiviteit 1C
|
> 1 uur / ≤ 4 uur
|
≤ 14 dagen
|
|
|
7.
|
De gevalideerde referentiemethode (7) heeft als beperking dat de test op basis van de resultaten van de voorafgaande compatibiliteitstest (zie punt 13) voor veel niet-corrosieve chemische stoffen en sommige corrosieve chemische stoffen mogelijk niet geschikt is. Voor veel waterige chemische stoffen met een pH in het gebied tussen 4,5 en 8,5 is de test niet geschikt; er zij evenwel opgemerkt dat 85 % van de geteste chemische stoffen in dit pH-bereik in dierproeven niet-corrosief bleken (7). De in-vitromembraanbarrièremethode kan worden gebruikt voor het testen van vaste stoffen (al dan niet oplosbaar in water), vloeistoffen (waterig of niet-waterig) en emulsies. Teststoffen die in de compatibiliteitstest geen merkbare verandering teweegbrachten (d.w.z. kleurverandering in het chemische detectiesysteem (CDS) van de gevalideerde testmethode), kunnen niet met de membraanbarrièremethode worden getest en moeten worden getest met andere testmethoden.
|
PRINCIPE VAN DE TEST
|
8.
|
Het testsysteem bestaat uit twee componenten: een synthetische macromoleculaire biobarrière en een chemisch detectiesysteem (CDS); via het CDS detecteert deze testmethode schade aan de membraanbarrière veroorzaakt door corrosieve teststoffen, nadat de teststof op het oppervlak van de synthetische macromoleculaire membraanbarrière is aangebracht (7), schade die waarschijnlijk wordt toegebracht volgens hetzelfde/dezelfde corrosiemechanisme(n) als bij levende huid.
|
|
9.
|
Er zijn meerdere procedures of CDS’en om de doordringing (of doorbraak) van de membraanbarrière te meten, zoals een verandering in de kleur van een pH-indicator of in een andere eigenschap van de indicatoroplossing onder de barrière.
|
|
10.
|
Er moet worden vastgesteld dat de membraanbarrière geschikt is, d.w.z. relevant en betrouwbaar, voor het beoogde doel. Daartoe moet men nagaan dat verschillende preparaten consistent zijn wat betreft hun barrière-eigenschappen, bv. dat ze een barrière in stand kunnen houden tegen niet-corrosieve chemische stoffen en dat de corrosieve eigenschappen van chemische stoffen ermee kunnen worden ingedeeld in de verschillende VN-GHS-subcategorieën voor corrosiviteit (1). De toegekende indeling is gebaseerd op de tijd die een chemische stof erover doet om door de membraanbarrière door te dringen tot de indicatoroplossing.
|
AANTONING VAN BEKWAAMHEID
|
11.
|
Voordat de in-vitromembraanbarrièremethode routinematig wordt gebruikt op een manier die aan deze testmethode voldoet, moeten laboratoria hun technische bekwaamheid aantonen door een correcte indeling van de twaalf in tabel 2 aanbevolen stoffen voor bekwaamheidstoetsing. Wanneer een stof uit de lijst niet beschikbaar is of indien zulks gerechtvaardigd is, kan een andere stof waarvoor voldoende in-vivo- en in-vitroreferentiegegevens beschikbaar zijn, worden gebruikt (bv. uit de lijst met referentiestoffen (10)), mits dezelfde selectiecriteria als in tabel 1 worden toegepast.
Tabel 2
Stoffen voor bekwaamheidstoetsing
(27)
|
Stof (28)
|
CAS RN
|
Chemische klasse
|
In-vivo-VN-GHS-subcategorie (29)
|
In-vitro-VN-GHS-subcategorie (29)
|
|
Boortrifluoridedihydraat
|
13319-75-0
|
Anorganische zuren
|
1A
|
1A
|
|
Salpeterzuur
|
7697-37-2
|
Anorganische zuren
|
1A
|
1A
|
|
Fosforpentachloride
|
10026-13-8
|
Precursoren van anorganische zuren
|
1A
|
1A
|
|
Valerylchloride
|
638-29-9
|
Zuurchloriden
|
1B
|
1B
|
|
Natriumhydroxide
|
1310-73-2
|
Anorganische basen
|
1B
|
1B
|
|
1-(2-Aminoethyl)piperazine
|
140-31-8
|
Alifatische aminen
|
1B
|
1B
|
|
Benzeensulfonylchloride
|
98-09-9
|
Zuurchloriden
|
1C
|
1C
|
|
N,N-Dimethylbenzylamine
|
103-83-3
|
Anilinen
|
1C
|
1C
|
|
Tetraëthyleenpentamine
|
112-57-2
|
Alifatische aminen
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenolen
|
NC
|
NC
|
|
Nonylacrylaat
|
2664-55-3
|
Acrylaten/methacrylaten
|
NC
|
NC
|
|
Natriumbicarbonaat
|
144-55-8
|
Anorganische zouten
|
NC
|
NC
|
|
PROCEDURE
|
12.
|
In de volgende punten worden de componenten en procedures beschreven van een testmethode op basis van een kunstmatige membraanbarrière voor het beoordelen van corrosiviteit (7) (15) op basis van de huidige VRM, d.w.z. de in de handel verkrijgbare Corrositex®. De membraanbarrière en de compatibiliteits-/indicator- en categoriseringsoplossingen kunnen worden opgebouwd, bereid of in de handel worden verkregen zoals in het geval van de VRMCorrositex®. Er is voor de gevalideerde referentietestmethode een voorbeeld van een testmethodeprotocol beschikbaar (7). De test moet worden uitgevoerd bij kamertemperatuur (17-25 °C) en de componenten moeten aan de volgende voorwaarden voldoen.
|
Compatibiliteitstest voor de teststof
|
13.
|
Voordat de membraanbarrièretest wordt uitgevoerd moet een compatibiliteitstest worden verricht om te bepalen of de teststof door het CDS gedetecteerd kan worden. Als het CDS de teststof niet detecteert, is de testmethode op basis van de membraanbarrière niet geschikt voor het beoordelen van de mogelijke corrosiviteit van die teststof en moet er een andere testmethode worden gebruikt. Het CdS en de blootstellingsomstandigheden die voor de compatibiliteitstest worden gebruikt, moeten overeenkomen met de blootstelling in de daarop volgende membraanbarrièretest.
|
Tijdschaalcategorietest voor de teststof
|
14.
|
Indien van toepassing voor de testmethode moet een teststof die geschikt is bevonden in de compatibiliteitstest, aan een tijdschaalcategorietest worden onderworpen, d.w.z. een screeningtest om onderscheid te maken tussen zwakke en sterke zuren of basen. Zo wordt in de gevalideerde referentietestmethode een tijdschaalcategorietest gebruikt om te bepalen welke van de twee tijdschalen moet worden gebruikt al naargelang een significante zuur- of alkalireserve wordt gedetecteerd. Er moeten twee verschillende doorbraaktijdschalen worden gebruikt voor het vaststellen van corrosiviteit en de VN-GHS-subcategorie voor huidcorrosiviteit, afhankelijk van de zuur- of alkalireserve van de teststof.
|
COMPONENTEN VAN DE TESTMETHODE OP BASIS VAN DE MEMBRAANBARRIÈRE
Membraanbarrière
|
15.
|
De membraanbarrière bestaat uit twee componenten: een proteïneachtige, macromoleculaire, waterige gel en een permeabele dragermembraan. De proteïneachtige gel moet ondoordringbaar zijn voor vloeistoffen en vaste stoffen, maar kan gecorrodeerd en permeabel gemaakt worden. De volledig opgebouwde membraanbarrière moet worden bewaard onder van tevoren vastgestelde omstandigheden waarvan is uitgewezen dat de gel niet achteruitgaat, bv. door uitdrogen, groei van micro-organismen, verschuiven of barsten, wat ten koste zou gaan van de prestaties. De uiterste bewaartermijn moet worden vastgesteld en de membraanbarrièrepreparaten mogen na het verstrijken van die termijn niet meer worden gebruikt.
|
|
16.
|
De permeabele dragermembraan levert mechanische ondersteuning aan de proteïneachtige gel gedurende het geleerproces en de blootstelling aan de teststof. De dragermembraan moet doorzakken en verschuiven van de gel voorkomen en moet goed doorlatend zijn voor alle teststoffen.
|
|
17.
|
De proteïneachtige gel, die bestaat uit proteïnen, bv. keratine, collageen of proteïnemengsels, die een gelmatrix vormen, dient als doelwit voor de teststof. Het proteïneachtige materiaal wordt op het oppervlak van de dragermembraan aangebracht en als het gegeleerd is, wordt de membraanbarrière over de indicatoroplossing geplaatst. De proteïneachtige gel moet een egale dikte en dichtheid hebben, zonder luchtbellen of gebreken die de functionele integriteit ervan zouden kunnen aantasten.
|
Chemisch detectiesysteem (CDS)
|
18.
|
De indicatoroplossing, dezelfde oplossing als voor de compatibiliteitstest, moet reageren op de aanwezigheid van de teststof. Er moet een pH-indicator of een combinatie van indicatoren worden gebruikt,bv. cresolrood en methyloranje, die een kleurverandering laten zien in aanwezigheid van de teststof. Het meetsysteem kan zowel visueel of elektronisch zijn.
|
|
19.
|
De detectiesystemen die worden ontwikkeld om te detecteren dat de teststof de barrièremembraan is gepasseerd, moeten worden beoordeeld op relevantie en betrouwbaarheid om de verzameling chemische stoffen die kan worden gedetecteerd, en de kwantitatieve detectiegrenzen aan te tonen.
|
TESTPRESTATIE
Opbouw van de componenten van de testmethode
|
20.
|
De membraanbarrière wordt zodanig in een fles (of buis) met indicatoroplossing geplaatst dat de dragermembraan volledig in contact staat met de indicatoroplossing en er geen luchtbellen aanwezig zijn. Er moet zorgvuldig op worden toegezien dat de barrière volledig intact blijft.
|
Het aanbrengen van de teststof
|
21.
|
Er wordt een passende hoeveelheid van de teststof, bv. 500 μl van een vloeistof of 500 mg van een fijn verpulverde vaste stof (7), in een egale laag over het oppervlak van de membraanbarrière aangebracht. Voor elke teststof worden een passend aantal duplo’s, bv. vier (7), en de bijbehorende controles opgezet (zie de punten 23 tot en met 25). De tijd van het aanbrengen van de teststof op de membraanbarrière wordt geregistreerd. Om te waarborgen dat korte corrosietijden nauwkeurig worden geregistreerd, worden de aanbrengtijden van de teststof in de duploflessen gespreid.
|
Meting van de doordringing door de membraanbarrière
|
22.
|
Elke fles wordt zorgvuldig in de gaten gehouden, de tijd van de eerste verandering in de indicatoroplossing, d.w.z. doordringing van de barrière, wordt geregistreerd en de tijd die is verstreken tussen het aanbrengen van de teststof en het doordringen door de membraanbarrière wordt bepaald.
|
Controles
|
23.
|
In tests waarbij gebruikgemaakt wordt van een vehiculum of oplosmiddel met de teststof, moet het vehiculum of oplosmiddel compatibel zijn met het membraanbarrièresysteem, d.w.z. het membraanbarrièresysteem intact laten, en mag het de corrosiviteit van de teststof niet veranderen. Indien van toepassing moet er tegelijkertijd met de teststof een oplosmiddelcontrole (of vehiculumcontrole) worden getest om de compatibiliteit van het oplosmiddel/vehiculum met het membraanbarrièresysteem aan te tonen.
|
|
24.
|
Om te beoordelen of het testsysteem op aanvaardbare wijze presteert moet er tegelijkertijd met de teststof ook een positieve (corrosieve) controle met matig corrosieve activiteit, bv. 110 ± 15 mg natriumhydroxide (VN-GHS-subcategorie voor corrosiviteit 1B) (7) worden getest. Een tweede positieve controle van dezelfde chemische klasse als de teststof kan nuttig zijn voor het beoordelen van de relatieve corrosiviteit van een corrosieve teststof. De gekozen positieve controle(s) moet(en) matig corrosief zijn (bv. VN-GHS-subcategorie voor corrosiviteit 1B) om veranderingen in de doordringingstijd te kunnen detecteren die mogelijk onaanvaardbaar veel langer of korter zijn dan de vastgestelde referentiewaarde, waaruit blijkt dat het testsysteem niet naar behoren werkt. Voor dit doel zijn extreem corrosieve (VN-GHS-subcategorie voor corrosiviteit 1A) of niet-corrosieve chemische stoffen van beperkt nut. Met een corrosieve VN-GHS-subcategorie 1B-stof kan een te snelle of te langzame doorbraaktijd wel gedetecteerd worden. Er kan een zwak corrosieve stof (VN-GHS-subcategorie voor corrosiviteit 1C) als positieve controle worden gebruikt om te meten in hoeverre de testmethode zwak corrosieve stoffen consistent kan onderscheiden van niet-corrosieve stoffen. Ongeacht de gevolgde aanpak moet er een aanvaardbaar responsbereik voor de positieve controle(s) worden vastgesteld op basis van het historische bereik van doorbraaktijden voor de gebruikte positieve controle(s), zoals het gemiddelde ±2-3 standaarddeviaties. In elk onderzoek moet de precieze doorbraaktijd voor de positieve controle worden bepaald, zodat afwijkingen buiten het aanvaardbare bereik gedetecteerd kunnen worden.
|
|
25.
|
Als verdere kwaliteitscontrolemaatregel om de functionele integriteit van de membraanbarrière aan te tonen moet er tegelijkertijd met de teststof ook een negatieve (niet-corrosieve) controle, bv. 10 % citroenzuur, 6 % propionzuur (7), worden getest.
|
Aanvaardbaarheidscriteria voor het onderzoek
|
26.
|
Volgens de vastgestelde tijdsparameters voor elk van de VN-GHS-subcategorieën voor corrosiviteit, moet de tijd (in minuten) die is verstreken tussen het aanbrengen van de teststof op de membraanbarrière en het doordringen door de barrière worden gebruikt om de corrosiviteit van de teststof te voorspellen. Een onderzoek wordt alleen als aanvaardbaar beschouwd als de gelijktijdige positieve controle de verwachte responstijd voor doordringing (bv. een doorbraaktijd van 8-16 minuten voor natriumhydroxide indien gebruikt als positieve controle) oplevert, de gelijktijdige negatieve controle niet corrosief is en de gelijktijdige oplosmiddelcontrole, indien in het onderzoek opgenomen, niet corrosief is en evenmin de corrosiviteit van de teststof beïnvloedt. Voordat zij een methode routinematig gebruiken op een manier die aan deze testmethode voldoet, moeten laboratoria hun technische bekwaamheid aantonen aan de hand van de twaalf stoffen die in tabel 2 worden aanbevolen. Voordat nieuwe soortgelijke „me too”-methoden die worden ontwikkeld op basis van deze testmethode en die structureel en functioneel vergelijkbaar zijn met de gevalideerde referentiemethode (14) worden gebruikt met het oog op regelgeving, moeten de vooraf vastgestelde prestatienormen worden gebruikt om de betrouwbaarheid en nauwkeurigheid van de nieuwe methode aan te tonen (10).
|
Interpretatie van resultaten en indeling van de teststoffen
|
27.
|
De tijd (in minuten) die is verstreken tussen het aanbrengen van de teststof op de membraanbarrière en het doordringen door de barrière wordt gebruikt om de teststof in te delen in een van de VN-GHS-subcategorieën voor corrosie (1) en, indien van toepassing, een VN-verpakkingsgroep (16). Voor elke voorgestelde testmethode worden de drempeltijden vastgesteld, die als scheiding fungeren tussen elk van de drie subcategorieën voor corrosie. Bij de uiteindelijke beslissing over de drempeltijden moet de noodzaak om onderclassificatie van het corrosiegevaar (d.w.z. vals-negatieve uitkomsten) te voorkomen, in aanmerking worden genomen. In deze testrichtlijn moeten de drempeltijden voor Corrositex®zoals beschreven in tabel 3 worden gebruikt, omdat dit vooralsnog de enige testmethode is die onder deze testrichtlijn valt (7).
Tabel 3
Corrositex®-voorspellingsmodel
|
Gemiddelde doorbraaktijd (minuten)
|
VN-GHS-prognose (32)
|
|
Categorie 1-teststoffen (30) (bepaald door de categorietest van de methode)
|
Categorie 2-teststoffen (31) (bepaald door de categorietest van de methode)
|
|
0-3 min.
|
0-3 min.
|
Corrosief optionele subcategorie 1A
|
|
> 3 tot 60 min.
|
> 3 tot 30 min.
|
Corrosief optionele subcategorie 1B
|
|
> 60 tot 240 min.
|
> 30 tot 60 min.
|
Corrosief optionele subcategorie 1C
|
|
> 240 min.
|
> 60 min.
|
Niet-corrosief
|
|
GEGEVENS EN RAPPORTAGE
Gegevens
|
28.
|
Voor de teststof en de positieve controle(s) moet de tijd (in minuten) die verstrijkt tussen het aanbrengen en het doordringen door de barrière, in tabelvorm worden gerapporteerd, zowel de afzonderlijke gegevens per duplo als het gemiddelde ± de standaarddeviatie voor elke bepaling.
|
Testverslag
|
29.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Teststof en controlestoffen:
|
—
|
stof die uit één component bestaat: chemische identificatiegegevens, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code, structuurformule, zuiverheid, chemische identiteit van onzuiverheden, indien van toepassing en praktisch haalbaar enz.;
|
|
—
|
stof die uit meerdere componenten bestaat, UVCB en mengsel: voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen;
|
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
bron, partijnummer indien beschikbaar;
|
|
—
|
behandeling van de teststof/controlestof voorafgaand aan het testen, indien van toepassing (bv. opwarming, malen);
|
|
—
|
stabiliteit van de teststof, uiterste gebruiksdatum, of datum voor heranalyse, indien bekend;
|
|
|
|
Vehiculum:
|
—
|
identificatiegegevens, de concentratie (indien van toepassing) en het gebruikte volume;
|
|
—
|
een motivering voor de keuze van het vehiculum.
|
|
|
|
In-vitromodel op basis van de membraanbarrière en toegepast protocol, waaronder de aangetoonde nauwkeurigheid en betrouwbaarheid
|
|
|
Testomstandigheden:
|
—
|
beschrijving van de gebruikte apparatuur en voorbereidingsprocedures;
|
|
—
|
herkomst en samenstelling van de gebruikte in-vitromembraanbarrière;
|
|
—
|
samenstelling en eigenschappen van de indicatoroplossing;
|
|
—
|
hoeveelheden van teststof en controlestoffen;
|
|
—
|
een beschrijving en rechtvaardiging van de tijdschaalcategorietest;
|
|
—
|
een beschrijving van de toegepaste beoordelings- en indelingscriteria;
|
|
—
|
aantoning van bekwaamheid in de toepassing van de testmethode voorafgaand aan routinematig gebruik door middel van het testen van de chemische stoffen voor bekwaamheidstoetsing.
|
|
|
|
Resultaten:
|
—
|
een tabel met afzonderlijke ruwe gegevens van afzonderlijke test- en controlemonsters voor elke duplo;
|
|
—
|
beschrijvingen van andere waargenomen effecten;
|
|
—
|
de afgeleide indeling met vermelding van het gebruikte voorspellingsmodel/de gehanteerde beslissingscriteria.
|
|
Bespreking van de resultaten
Conclusies
|
LITERATUUR
|
(1)
|
Verenigde Naties (VN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, VN New York en Genève, 2013. Beschikbaar op: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Hoofdstuk B.4 van deze bijlage, Acute huidirritatie/corrosie.
|
|
(3)
|
Hoofdstuk B.40 bis van deze bijlage, In-vitrohuidcorrosie: testmethode met gereconstrueerde humane epidermis (RhE).
|
|
(4)
|
Hoofdstuk B.40 van deze bijlage, In-vitrohuidcorrosie: transcutane elektrische weerstand (TEW).
|
|
(5)
|
OESO (2015). Guidance document on an Integrated Approach on Testing and Assessment (IATA) for Skin Corrosion and Irritation. Environmental Health and Safety Publications, Series on Testing and Assessment (No 203). Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OESO (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Series on Testing and Assessment, no. Series on Testing and Assessment (No 34).
|
|
(10)
|
OESO (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Transcutaneous Electrical Resistance (TER) Test Method for Skin Corrosion in Relation to TG 435. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
CEVMA (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15e vergadering van het raadgevend wetenschappelijk comité van het Europees Centrum voor de validatie van alternatieve methoden (ESAC), Ispra, Italië. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Hoofdstuk B.46 van deze bijlage, In-vitrohuidirritatie: testmethode met gereconstrueerde humane epidermis. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Beschikbaar op: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Beschikbaar op: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Verenigde Naties (VN) (2013). UN Recommendations on the Transport of Dangerous Goods (aanbevelingen van de VN inzake het vervoer van gevaarlijke goederen), modelreglementen, 18e herziene editie (Deel, Hoofdstuk 2.8), Verenigde Naties, 2013. Beschikbaar op: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Aanhangsel
DEFINITIES
Betrouwbaarheid
: indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid (9).
Chemisch detectiesysteem (CDS)
: een visueel of elektronisch meetsysteem met een indicatoroplossing die reageert op de aanwezigheid van een teststof,bv. door een verandering in een pH-indicator of een combinatie van indicatoren, die een kleurverandering laten zien in aanwezigheid van de teststof, of door andere typen chemische of elektrochemische reacties.
Chemische stof
: een stof of een mengsel.
Concordantie
: dit is een maat voor de prestaties van de testmethode voor testmethoden die een categoriale uitkomst geven en het is één aspect van relevantie. Deze term en de term „nauwkeurigheid” worden soms door elkaar gebruikt; de term wordt gedefinieerd als het percentage van alle geteste chemische stoffen die correct als positief of negatief worden geclassificeerd. De concordantie is sterk afhankelijk van de prevalentie van positieven in de typen teststoffen die worden onderzocht (9).
Gevoeligheid
: het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (9).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen)
: een systeem voor de indeling van chemische stoffen (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (1).
Huidcorrosiein vivo
: het ontstaan van een irreversibele beschadiging van de huid, namelijk zichtbare necrose door deepidermis heen in de dermis, na het aanbrengen van een teststof gedurende ten hoogste vier uur. Corrosie-reacties worden gekenmerkt door zweren, bloedingen, bloedkorsten en, tegen het eind van de observatieperiode van 14 dagen, ontkleuring door bleking van de huid, gebieden met volledige haaruitval en littekens. Om twijfelachtig letsel te evalueren moet histopathologie worden overwogen.
IATA
: Integrated Approach on Testing and Assessment (geïntegreerde test- en beoordelingsbenadering).
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
Nauwkeurigheid
: de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Dit is een maat voor de prestaties van de testmethode en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt (9).
NC
: niet-corrosief.
Prestatienormen
: normen, gebaseerd op een gevalideerde testmethode, die de basis vormen voor de beoordeling van de vergelijkbaarheid van een voorgestelde, in mechanistisch en functioneel opzicht gelijksoortige testmethode. Hieronder begrepen zijn: i) essentiële componenten van de testmethode; ii) een minimumlijst van referentiestoffen die zijn gekozen uit de stoffen die zijn gebruikt om de aanvaardbare prestaties van de gevalideerde testmethode aan te tonen, en iii) de soortgelijke betrouwbaarheids- en nauwkeurigheidsniveaus die door de voorgestelde testmethode moeten worden gehaald wanneer ze worden beoordeeld aan de hand van de minimumlijst van referentiestoffen; deze waarden zijn gebaseerd op de waarden die voor de gevalideerde testmethode zijn verkregen (9).
Relevantie
: beschrijving van het verband van de testmethode met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de testmethode het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (9).
Specificiteit
: het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (9).
Stof
: een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Teststof
: alle volgens deze testmethode geteste stoffen of mengsels.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
B.66
IN-VITRO BEPALINGEN OP BASIS VAN STABIELE TRANSFECTIE EN TRANSACTIVATIE VOOR HET OPSPOREN VAN OESTROGEENRECEPTORAGONISTEN EN -ANTAGONISTEN
ALGEMENE INLEIDING
Op prestaties gebaseerde testrichtlijn van de OESO
|
1.
|
Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 455 (2016) van de OESO. TG 455 is een op prestaties gebaseerde testrichtlijn (performance-based test guideline, PBTG), waarin de methodologie wordt beschreven van in-vitrobepalingen op basis van stabiele transfectie en transactivatie voor het opsporen van oestrogeenreceptoragonisten en -antagonisten (estrogen receptor transactivation assays, ER TA-bepalingen). De PBTG omvat verschillende, in mechanistisch en functioneel opzicht gelijksoortige testmethoden voor het identificeren van agonisten en antagonisten van de oestrogeenreceptor (ERα, en/of ERβ) en moet de ontwikkeling bevorderen van nieuwe gelijksoortige of gewijzigde testmethoden die overeenstemmen met de valideringsbeginselen beschreven in de OESO-leidraad voor de validering en internationale aanvaarding van nieuwe of bijgewerkte testmethoden voor gevarenbeoordeling (Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment) (1) De volledig gevalideerde referentietestmethoden (aanhangsels 2 en 3) die de basis vormen voor deze PBTG zijn:
|
—
|
de bepaling op basis van stabiele transfectie en transactivatie (STTA-bepaling) (2) die gebruikmaakt van de cellijn (h) ERα-HeLa-9903, en
|
|
—
|
de VM7Luc-ER TA-bepaling (3) waarbij gebruikgemaakt wordt van de cellijn VM7Luc4E2 (33), een cellijn die voornamelijk hERα tot expressie brengt met enige bijdrage van hERβ (4) (5).
|
Voor de ontwikkeling en validering van gelijksoortige bepalingen voor hetzelfde gevareneindpunt zijn prestatienormen (6) (7) beschikbaar en moeten deze worden gebruikt. Ze zorgen dat PBTG 455 snel gewijzigd kan worden, zodat nieuwe gelijksoortige bepalingen aan een bijgewerkte PBTG kunnen worden toegevoegd. Gelijksoortige bepalingen zullen evenwel pas worden toegevoegd nadat de OESO ze heeft beoordeeld en onderschrijft dat aan de prestatienormen wordt voldaan. De in TG 455 opgenomen bepalingen kunnen zonder onderscheid worden gebruikt om te voldoen aan de eisen die aan OESO-landen worden gesteld wat betreft de testresultaten voor transactivatie van de oestrogeenreceptor, en in aanmerking te komen voor wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst.
|
Achtergrond en beginselen van de bepalingen die in deze testmethode worden beschreven
|
2.
|
In 1998 is de OESO met hoge prioriteit een actie gestart om bestaande testrichtlijnen te herzien en nieuwe testrichtlijnen te ontwikkelen voor het screenen en testen op potentiële hormoonontregelaars. Het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen werd in 2012 herzien. Het oorspronkelijke en het herziene conceptueel kader zijn als bijlagen opgenomen bij de OESO-leidraad voor gestandaardiseerde testrichtlijnen voor de beoordeling van chemische stoffen op hormoonontregeling (Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption) (8). Het conceptueel kader omvat vijf niveaus, die elk overeenkomen met een ander niveau van biologische complexiteit. De in deze testmethode beschreven ER TA-bepalingen zijn ingedeeld op niveau 2: „in-vitrobepalingen die gegevens moeten opleveren over (een) geselecteerde endocriene mechanisme(n)/weg(en)”. Dit is een testmethode voor in-vitrobepalingen op basis van transactivatie (TA), die zijn ontworpen om agonisten en antagonisten van de oestrogeenreceptor (ER) te identificeren.
|
|
3.
|
De interactie van oestrogenen met ER’s kan de transcriptie van oestrogeengestuurde genen beïnvloeden, wat kan leiden tot de inductie of remming van processen in de cel, waaronder de processen die nodig zijn voor celproliferatie, de normale ontwikkeling van de foetus en de voortplantingsfunctie (9) (10) (11). Verstoring van de normale oestrogene systemen zou kunnen leiden tot schadelijke effecten op de normale ontwikkeling (ontogenese), de reproductieve gezondheid en de integriteit van het voortplantingsstelsel.
|
|
4.
|
In-vitro-TA-bepalingen zijn gebaseerd op een directe of indirecte interactie van de stoffen met een specifieke receptor die de transcriptie van een reportergenproduct reguleert. Dergelijke bepalingen zijn veel gebruikt voor de beoordeling van de genexpressie die wordt gereguleerd door specifieke kernreceptoren zoals ER’s (12) (13) (14) (15) (16). Ze zijn voorgesteld voor het opsporen van door de ER gereguleerde oestrogene transactivatie (17) (18) (19). Er zijn ten minste twee belangrijke subtypen kern-ER’s, α en β, die door afzonderlijke genen gecodeerd worden. De bijbehorende eiwitten hebben elk hun eigen biologische functies en verschillende weefseldistributies en bindingsaffiniteiten voor liganden (20) (21) (22) (23) (24) (25) (26). De kern-ERα medieert de klassieke oestrogeenrespons (27) (28) (29) (30) en daarom zijn de meeste modellen die op dit moment worden ontwikkeld voor het meten van ER-activatie of -remming, specifiek voor ERα. De bepalingen worden gebruikt voor het identificeren van chemische stoffen die de ER activeren (of remmen) na binding van een ligand. Het receptor-ligand-complex bindt vervolgens aan specifieke DNA-responselementen en transactiveert een reportergen, wat leidt tot een verhoogde cellulaire expressie van een markereiwit. Er kunnen in deze bepalingen verschillende reporterresponsen worden gebruikt. In op luciferase gebaseerde systemen zet het enzym luciferase het substraat luciferine om in een bioluminescent product dat kwantitatief bepaald kan worden met een luminometer. Andere voorbeelden van veel gebruikte reporters zijn fluorescent proteïne en het gen LacZ, dat codeert voor β-galactosidase, een enzym dat het kleurloze substraat X-gal (5- broom-4-chloor-indolyl-galactopyranoside) kan omzetten in een blauw product dat kwantitatief bepaald kan worden met een spectrofotometer. Deze reporters kunnen snel en goedkoop worden bepaald met behulp van in de handel verkrijgbare testkits.
|
|
5.
|
De relevantie en de betrouwbaarheid van de STTA-bepaling en de VM7Luc TA-bepaling voor het beoogde doel zijn aangetoond in de valideringsstudies voor deze bepalingen (3) (4) (5) (30). Prestatienormen voor op luminescentie gebaseerde ER TA-bepalingen met borstcellijnen zijn opgenomen in het evaluatieverslag „ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc-ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals” (3). Deze prestatienormen zijn aangepast om te kunnen worden toegepast voor zowel de STTA-bepaling als de bepaling met VM7Luc (2).
|
|
6.
|
De in deze testmethode gebruikte definities en afkortingen staan in aanhangsel 1.
|
Toepassingsgebied en beperkingen voor de TA-bepalingen
|
7.
|
Deze bepalingen worden aangereikt ten behoeve van screening en prioritering, maar kunnen ook mechanistische informatie verschaffen die kan worden gebruikt in een benadering met meervoudige bewijsvoering („weight of evidence”-benadering). De tests berusten op de transactivatie die wordt geïnduceerd door chemische binding aan de ER’s in een in-vitrosysteem. De resultaten mogen dan ook niet rechtstreeks geëxtrapoleerd worden naar de complexe signalering en regulering van het intacte endocriene systeem in vivo.
|
|
8.
|
Door de ER’s gemedieerde transactivatie wordt beschouwd als een van de voornaamste mechanismen voor hormoonontregeling, hoewel er ook andere mechanismen zijn waardoor hormoonontregeling kan optreden, waaronder i) interacties met andere receptoren en enzymsystemen in het endocriene systeem, ii) hormoonsynthese, iii) metabole activering en/of inactivering van hormonen, iv) verdeling van hormonen naar doelweefsels, en v) klaring van hormonen uit het lichaam. In geen van de bepalingen die onder deze testmethode vallen, worden deze werkingsmechanismen in aanmerking genomen.
|
|
9.
|
Deze testmethode richt zich op het vermogen van chemische stoffen om van de ER afhankelijke transcriptie te activeren (d.w.z. als een agonist op te treden) en ook te onderdrukken (d.w.z. als een antagonist op te treden). Sommige chemische stoffen kunnen, afhankelijk van het gebruikte celtype, zowel agonistische als antagonistische activiteit vertonen en staan bekend als selectieve oestrogeenreceptormodulatoren (SERM’s). Overwogen kan worden om chemische stoffen met een negatieve respons in deze bepalingen aan een ER-bindingsbepaling te onderwerpen, alvorens te concluderen dat de chemische stof niet aan de receptor bindt. Bovendien geven de bepalingen slechts een waarschijnlijke indicatie over de activiteit van de uitgangsstof, rekening houdend met het beperkte metabole vermogen van de in-vitrocelsystemen. Aangezien bij de validering alleen zuivere stoffen zijn gebruikt, zijn er geen gegevens over de toepasbaarheid voor testmengsels. In theorie is de testmethode echter wel toepasbaar voor het testen van stoffen die uit meerdere componenten bestaan, UVCB’s en van mengsels. Voordat de testmethode wordt gebruikt op een stof die uit meerdere componenten bestaat, UVCB of mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is.
|
|
10.
|
Ter informatie worden in tabel 1 de resultaten van agonismetests weergegeven voor de 34 stoffen die zijn getest in de beide volledig gevalideerde referentietestmethoden die in deze testmethode worden beschreven. Op basis van de gepubliceerde verslagen, waaronder in-vitrobepalingen voor ER-binding en TA en/of de uterotrofe test, zijn 26 van deze stoffen ingedeeld als definitieve ER-agonisten en 8 als negatief (2) (3) (18) (31) (32) (33) (34). In tabel 2 worden de resultaten van antagonismetests weergegeven voor de 15 stoffen die zijn getest in de beide volledig gevalideerde referentietestmethoden die in deze testmethode worden beschreven. Op basis van de gepubliceerde verslagen, waaronder in-vitrobepalingen voor ER-binding en TA, zijn 4 van deze stoffen ingedeeld als definitieve/waarschijnlijke ER-antagonisten en 10 als negatief (2) (3) (18) (31). Wat betreft de gegevens die zijn weergegeven in tabel 1 en tabel 2 was er 100 % overeenstemming tussen de beide referentietestmethoden over de indeling van alle stoffen, behalve voor één stof (Mifepriston) voor de antagonismebepaling, en elke stof werd correct ingedeeld als ER-(ant)agonist of negatief. Nadere informatie over deze groep chemische stoffen en over de aanvullende chemische stoffen die tijdens de valideringsstudies voor de STTA-bepaling en de VM7Luc-ER TA-bepaling getest zijn, is te vinden in de prestatienormen voor de ER TA (6) (7), aanhangsel 2 (tabellen 1, 2 en 3).
|
Tabel 1
Overzicht van de resultaten van de STTA-bepaling en de VM7Luc-ER TA-bepaling voor stoffen die aan beide agonismebepalingen zijn onderworpen en zijn ingedeeld als ER-agonisten (POS) of negatief (NEG)
|
|
Stof
|
CAS RN
|
STTA-bepaling (35)
|
VM7Luc-ER TA-bepaling (36)
|
Bron van gegevens voor indeling (38)
|
|
ER TA -activiteit
|
PC10-waarde (M)
|
PC50-waarde (2) (M)
|
ER TA-activiteit
|
EC50-waarde (2), (37) (M)
|
Andere ER TA-tests (34)
|
ER Bindmiddel
|
Uterotroof
|
|
1
|
17β-Oestradiol (1)
|
50-28-2
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
5,63 × 10-12
|
POS (227/227)
|
POS
|
POS
|
|
2
|
17α-Oestradiol (1)
|
57-91-0
|
POS
|
7,24 × 10-11
|
6,44 × 10-10
|
POS
|
1,40 × 10-9
|
POS (11/11)
|
POS
|
POS
|
|
3
|
17α-Ethinyloestradiol (1)
|
57-63-6
|
POS
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POS
|
7,31 × 10-12
|
POS (22/22)
|
POS
|
POS
|
|
4
|
17β-Trenbolon
|
10161-33-8
|
POS
|
1,78 × 10-8
|
2,73 × 10-7
|
POS
|
4,20 × 10-8
|
POS (2/2)
|
NT
|
NT
|
|
5
|
19-Nortestosteron (1)
|
434-22-0
|
POS
|
9,64 × 10-9
|
2,71 × 10-7
|
POS
|
1,80 × 10-6
|
POS (4/4)
|
POS
|
POS
|
|
6
|
4-Cumylfenol (1)
|
599-64-4
|
POS
|
1,49 × 10-7
|
1,60 × 10-6
|
POS
|
3,20 × 10-7
|
POS (5/5)
|
POS
|
NT
|
|
7
|
4-tert-Octylfenol (1)
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
POS
|
3,19 × 10-8
|
POS (21/24)
|
POS
|
POS
|
|
8
|
Apigenine (1)
|
520-36-5
|
POS
|
1,31 × 10-7
|
5,71 × 10-7
|
POS
|
1,60 × 10-6
|
POS (26/26)
|
POS
|
NT
|
|
9
|
Atrazine (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenol A (1)
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
POS
|
5,33 × 10-7
|
POS (65/65)
|
POS
|
POS
|
|
11
|
Bisfenol B (1)
|
77-40-7
|
POS
|
2,36 × 10-8
|
2,11 × 10-7
|
POS
|
1,95 × 10-7
|
POS (6/6)
|
POS
|
POS
|
|
12
|
Butylbenzylftalaat (1)
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
POS
|
1,98 × 10-6
|
POS (12/14)
|
POS
|
NEG
|
|
13
|
Corticosteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Coumestrol (1)
|
479-13-0
|
POS
|
1,23 × 10-9
|
2,00 × 10-8
|
POS
|
1,32 × 10-7
|
POS (30/30)
|
POS
|
NT
|
|
15
|
Daidzeïne (1)
|
486-66-8
|
POS
|
1,76 × 10-8
|
1,51 × 10-7
|
POS
|
7,95 × 10-7
|
POS (39/39)
|
POS
|
POS
|
|
16
|
Diëthylstilboestrol (1)
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POS
|
3,34 × 10-11
|
POS (42/42)
|
POS
|
NT
|
|
17
|
Di-n-butylftalaat
|
84-74-2
|
POS
|
4,09 × 10-6
|
|
POS
|
4,09 × 10-6
|
POS (6/11)
|
POS
|
NEG
|
|
18
|
Ethylparabeen
|
120-47-8
|
POS
|
5,00 × 10-6
|
(geen PC50)
|
POS
|
2,48 × 10-5
|
POS
|
|
NT
|
|
19
|
Oestron (1)
|
53-16-7
|
POS
|
3,02 × 10-11
|
5,88 × 10-10
|
POS
|
2,34 × 10-10
|
POS (26/28)
|
POS
|
POS
|
|
20
|
Genisteïne (1)
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
POS
|
2,71 × 10-7
|
POS (100/102)
|
POS
|
POS
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
POS
|
3,99 × 10-6
|
POS (23/23)
|
POS
|
NT
|
|
23
|
Kepone (1)
|
143-50-0
|
POS
|
7,11 × 10-7
|
7,68 × 10-6
|
POS
|
4,91 × 10-7
|
POS (14/18)
|
POS
|
NT
|
|
24
|
Ketoconazool
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POS
|
1,65 × 10-11
|
POS (4/4)
|
POS
|
NT
|
|
27
|
Methyltestosteron (1)
|
58-18-4
|
POS
|
1,73 × 10-7
|
4,11 × 10-6
|
POS
|
2,68 × 10-6
|
POS (5/6)
|
POS
|
NT
|
|
28
|
Morine
|
480-16-0
|
POS
|
5,43 × 10-7
|
4,16 × 10-6
|
POS
|
2,37 × 10-6
|
POS (2/2)
|
POS
|
NT
|
|
29
|
Noretynodrel (1)
|
68-23-5
|
POS
|
1,11 × 10-11
|
1,50 × 10-9
|
POS
|
9,39 × 10-10
|
POS (5/5)
|
POS
|
NT
|
|
30
|
p,p’-Methoxychloor (1)
|
72-43-5
|
POS
|
1,23 × 10-6
|
(geen PC50) (2)
|
POS
|
1,92 × 10-6
|
POS (24/27)
|
POS
|
POS
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reserpine
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolacton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosteron
|
58-22-0
|
POS
|
2,82 × 10-8
|
9,78 × 10-6
|
POS
|
1,75 × 10-5
|
POS (5/10)
|
POS
|
NT
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstract Service; M = molair; EC50 = effectieve-concentratiemediaan van de teststof; NEG = negatief; POS = positief; NT = niet getest; PC10 (en PC50) = de concentratie van een teststof waarbij de respons gelijk is aan 10 % (of 50 % voor de PC50) van de respons die wordt opgewekt door de positieve controle (E2, 1 nm) op elke plaat.
|
Tabel 2
Vergelijking van de resultaten van de STTA-bepaling en de VM7Luc-ER TA-bepaling voor stoffen die aan beide antagonismebepalingen zijn onderworpen en zijn ingedeeld als ER-antagonisten (POS) of negatief (NEG)
|
|
Stof (3)
|
CAS RN
|
ER STTA-bepaling (40)
|
VM7Luc-ER TA-bepaling (41)
|
Verwachte effecten van de ER STTA (43)
|
Consensuele indeling door het ICCVAM (44)
|
Chemische klasse volgens de MeSH (45)
|
Productcategorie (46)
|
|
ER TA -activiteit
|
IC50-waarde (39) (M)
|
ER TA -activiteit
|
IC50-waarde (39), (42) (M)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
POS
|
2,08 × 10-7
|
matig POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
2
|
Dibenzo[a,h]antraceen
|
53-70-3
|
POS
|
Geen IC50
|
POS
|
Geen IC50
|
POS
|
PP
|
Polycyclische verbinding
|
Laboratoriumstof, natuurlijk product
|
|
3
|
Mifepriston
|
84371-65-3
|
POS
|
5,61 × 10-6
|
NEG
|
—
|
mild POS
|
NEG
|
Steroïde
|
Geneesmiddel
|
|
4
|
Raloxifeen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
POS
|
1,19 × 10-9
|
matig POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
5
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
POS
|
8,17 × 10-7
|
POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
6
|
17β-oestradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
7
|
Apigenine
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterocyclische verbinding
|
Kleurstof, natuurlijk product, farmaceutisch tussenproduct
|
|
8
|
Atrazine
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterocyclische verbinding
|
Herbicide
|
|
9
|
Di-n-butylftalaat
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Ester, ftaalzuur
|
Cosmetisch bestanddeel, industriële chemische stof, weekmaker
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
niet getest
|
PN
|
Heterocyclische verbinding, pyrimidine
|
Fungicide
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product, geneesmiddel
|
|
12
|
Flutamide
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amide
|
Geneesmiddel, diergeneesmiddel
|
|
13
|
Genisteïne
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product, geneesmiddel
|
|
14
|
p-n-nonylfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
niet getest
|
NEG
|
Fenol
|
Chemisch tussenproduct
|
|
15
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Koolwaterstof (cyclisch)
|
Natuurlijk product
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstract Service; M = molair; IC50 = concentratie van de teststof die 50 % remming veroorzaakt NEG = negatief; PN = beschouwd als negatief; POS = positief; PP = beschouwd als positief.
|
COMPONENTEN VAN DE ER TA-BEPALING
Essentiële componenten van de bepaling
|
11.
|
Deze testmethode is van toepassing op bepalingen waarbij gebruikgemaakt wordt van een stabiel getransfecteerde of endogene ERα receptor en een stabiel getransfecteerd reportergenconstruct gestuurd door een of meer oestrogeenresponselementen. Er kunnen evenwel ook andere receptoren aanwezig zijn, zoals ERβ. Dit zijn essentiële componenten van de bepaling.
|
Controles
|
12.
|
Voor elke agonisme- en antagonismebepaling moet de keuze van de gelijktijdige referentiestandaarden worden onderbouwd. Gelijktijdige controles (negatieve controle, oplosmiddelcontrole en positieve controle), voor zover van toepassing, dienen als indicatie dat de bepaling werkt onder de testomstandigheden, en zorgen dat de verschillende experimenten met elkaar vergeleken kunnen worden. Ze maken doorgaans deel uit van de aanvaardbaarheidscriteria voor een bepaald experiment (1).
|
Standaard kwaliteitscontroleprocedures
|
13.
|
De standaard kwaliteitscontroleprocedures moeten worden uitgevoerd zoals beschreven voor elke bepaling, om er zeker van te zijn dat de cellijn gedurende meerdere passages stabiel blijft, vrij blijft van mycoplasma (d.w.z. vrij van bacteriële besmetting) en in de loop van de tijd haar vermogen behoudt om de verwachte ER-gemedieerde responsen te geven. De cellijnen moeten bovendien worden gecontroleerd op de juiste identiteit en op andere verontreinigingen (bv. schimmels, gisten en virussen).
|
Aantoning van de bekwaamheid van laboratoria
|
14.
|
Alvorens onbekende chemische stoffen te testen met een van de bepalingen die onder deze testmethode vallen, moet elk laboratorium zijn bekwaamheid in het gebruik van de bepaling aantonen. Om zijn bekwaamheid aan te tonen moet elk laboratorium voor de agonismebepaling de 14 stoffen voor bekwaamheidstoetsing testen die zijn opgesomd in tabel 3, en voor de antagonismebepaling de 10 stoffen voor bekwaamheidstoetsing die zijn opgesomd in tabel 4. Deze bekwaamheidstoetsing dient eveneens ter bevestiging van het responsvermogen van het testsysteem. De lijst van stoffen voor bekwaamheidstoetsing is een subset van de referentiestoffen die worden aangegeven in de prestatienormen voor de ER TA-bepalingen (6). Deze stoffen zijn in de handel verkrijgbaar, vertegenwoordigen chemische klassen waarbij doorgaans sprake is van ER-agonistische of antagonistische activiteit, vertonen een geschikt potentiebereik voor de verwachte potentie van de ER-(ant)agonisten (d.w.z. sterk tot zwak) en voorzien ook in negatieve responsen. De stoffen voor bekwaamheidstoetsing moeten ten minste tweemaal, op verschillende dagen getest worden. De bekwaamheid wordt aangetoond door een correcte indeling (positief/negatief) van elke stof. De bekwaamheidstoetsing moet worden herhaald door elke onderzoeker die de bepalingen leert. Afhankelijk van het celtype kunnen sommige van deze stoffen zich als SERM’s gedragen en zowel agonistische als antagonistische activiteit vertonen. In de tabellen 3 en 4 worden de stoffen voor bekwaamheidstoetsing echter ingedeeld op basis van de activiteit waarvan bekend is dat deze het sterkst is, en dat is de activiteit die moet worden gebruikt om de bekwaamheid te beoordelen.
|
|
15.
|
Als bewijs van de prestaties en ten behoeve van de kwaliteitscontrole moet elk laboratorium historische databanken van agonisten en antagonisten bijhouden met gegevens over de referentiestandaarden (bv. 17β-oestradiol en tamoxifen), de positieve en negatieve controlestoffen en de oplosmiddelcontrole (bv. DMSO). Aanvankelijk moet de databank gegenereerd worden op basis van ten minste 10 onafhankelijke testruns voor agonisten (bv. 17β-oestradiol) en 10 voor antagonisten (bv. tamoxifen). De resultaten van latere analyses van deze referentiestandaarden en oplosmiddelcontroles moeten worden toegevoegd om de databank uit te breiden en de consistentie en prestaties van de bioassay door het laboratorium in de loop van de tijd te waarborgen.
|
Tabel 3
Lijst van (14) stoffen voor bekwaamheidstoetsing voor agonismebepaling
(54)
|
Nr. (53)
|
Stof
|
CAS RN
|
Verwachte respons (47)
|
STTA-bepaling
|
VM7Luc-ER TA-bepaling
|
Chemische klasse volgens de MeSH (51)
|
Productcategorie (52)
|
|
PC10-waarde (M) (48)
|
PC50-waarde (M) (48)
|
Testconcentratiebereik (M)
|
EC50-waarde VM7Luc (M) (49)
|
Hoogste conc. voor bereikbepaling (M) (50)
|
|
14
|
Diëthylstilboestrol
|
56-53-1
|
POS
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Koolwaterstof (cyclisch)
|
Geneesmiddel, diergeneesmiddel
|
|
12
|
17α-oestradiol
|
57-91-0
|
POS
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POS
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Koolwaterstof (cyclisch), fenol
|
Geneesmiddel, diergeneesmiddel
|
|
11
|
4-tert-Octylfenol
|
140-66-9
|
POS
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenol
|
Chemisch tussenproduct
|
|
9
|
Genisteïne
|
446-72-0
|
POS
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product, geneesmiddel
|
|
6
|
Bisfenol A
|
80-05-7
|
POS
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenol
|
Chemisch tussenproduct
|
|
2
|
Kaempferol
|
520-18-3
|
POS
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product
|
|
3
|
Butylbenzylftalaat
|
85-68-7
|
POS
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Carbonzuur, ester, ftaalzuur
|
Weekmaker, industriële chemische stof
|
|
4
|
p,p’-Methoxychloor
|
72-43-5
|
POS
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Koolwaterstof (gehalogeneerd)
|
Pesticide, diergeneesmiddel
|
|
1
|
Ethylparabeen
|
120-47-8
|
POS
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Carbonzuur, fenol
|
Geneesmiddel, conserveringsmiddel
|
|
17
|
Atrazine
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Heterocyclische verbinding
|
Herbicide
|
|
20
|
Spironolacton
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lacton, steroïde
|
Geneesmiddel
|
|
21
|
Ketoconazool
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Heterocyclische verbinding
|
Geneesmiddel
|
|
22
|
Reserpine
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Heterocyclische verbinding, indool
|
Geneesmiddel, diergeneesmiddel
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstract Service; EC50 = effectieve-concentratiemediaan van de teststof; NEG = negatief; POS = positief; PC10 (en PC50) = de concentratie van een teststof waarbij de respons gelijk is aan 10 % (of 50 % voor de PC50) van de respons die wordt opgewekt door de positieve controle (E2, 1 nm) op elke plaat.
|
Tabel 4
Lijst van (10) stoffen voor bekwaamheidstoetsing voor antagonismebepaling
|
|
Stof (4)
|
CAS RN
|
ER STTA-bepaling (55)
|
VM7Luc-ER TA-bepaling (56)
|
Verwachte effecten ER STTA (55)
|
Consensuele indeling door het ICCVAM (59)
|
Chemische klasse volgens de MeSH (60)
|
Productcategorie (61)
|
|
ER TA-activiteit
|
IC50 (M)
|
Testconcentratiebereik (M)
|
ER TA-activiteit
|
IC50
(57) (M)
|
Hoogste conc. voor bereikbepaling (M) (58)
|
|
1
|
4-Hydroxytamoxifen
|
68047-06-3
|
POS
|
3,97 × 10-9
|
10-12 – 10-7
|
POS
|
2,08 × 10-7
|
2,58 × 10-4
|
matig POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
2
|
Raloxifeen HCl
|
82640-04-8
|
POS
|
7,86 × 10-10
|
10-12 – 10-7
|
POS
|
1,19 × 10-9
|
1,96 × 10-4
|
matig POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
3
|
Tamoxifen
|
10540-29-1
|
POS
|
4,91 × 10-7
|
10-10 – 10-5
|
POS
|
8,17 × 10-7
|
2,69 × 10-4
|
POS
|
POS
|
Koolwaterstof (cyclisch)
|
Geneesmiddel
|
|
4
|
17β-Oestradiol
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
naar verwachting NEG (*4)
|
PN
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
5
|
Apigenine
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterocyclische verbinding
|
Kleurstof, natuurlijk product, farmaceutisch tussenproduct
|
|
6
|
Di-n-butylftalaat
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Ester, ftaalzuur
|
Cosmetisch bestanddeel, industriële chemische stof, weekmaker
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
naar verwachting NEG (*4)
|
PN
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product, geneesmiddel
|
|
8
|
Genisteïne
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
naar verwachting NEG (*4)
|
NEG
|
Flavonoïde, heterocyclische verbinding
|
Natuurlijk product, geneesmiddel
|
|
9
|
p-n-Nonylfenol
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
niet getest
|
NEG
|
Fenol
|
Chemisch tussenproduct
|
|
10
|
Resveratrol
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
naar verwachting NEG (*4)
|
NEG
|
Koolwaterstof (cyclisch)
|
Natuurlijk product
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstract Service; M = molair; IC50 = concentratie van de teststof die 50 % remming veroorzaakt NEG = negatief; PN = beschouwd als negatief; POS = positief;
|
Aanvaardbaarheidscriteria voor de testrun
|
16.
|
Of een testrun aanvaard of verworpen wordt, hangt af van de beoordeling van de resultaten voor de referentiestandaarden en de controles die voor elk experiment worden gebruikt. De waarden voor de PC50 (EC50) of IC50 voor de referentiestandaarden moeten voldoen aan de aanvaardbaarheidscriteria voor de gekozen bepaling (voor de STTA-bepaling zie aanhangsel 2, voor de VM7Luc-ER TA-bepaling zie aanhangsel 3) en alle positieve/negatieve controles moeten voor elk aanvaard experiment correct zijn ingedeeld. Het laboratorium moet aantonen de bepaling consistent te kunnen uitvoeren door een historische databank van de referentiestandaarden en controles op te zetten en bij te houden (zie punt 15). Als maatstaf voor de intralaboratoriumreproduceerbaarheid kunnen de standaarddeviaties (SD) of variatiecoëfficiënten (CV) van de gemiddelden van de curve-fittingsparameters voor de referentiestandaarden van meerdere experimenten worden gebruikt. Bovendien moet aan de volgende beginselen in verband met de aanvaardbaarheidscriteria worden voldaan:
|
—
|
er moeten voldoende gegevens zijn voor een kwantitatieve beoordeling van de ER-activering (voor een agonismebepaling) of ER-remming (voor een antagonismebepaling) (d.w.z. effectiviteit en potentie);
|
|
—
|
om zeker te zijn van voldoende gevoeligheid moet de gemiddelde reporteractiviteit voor de referentieconcentratie van het referentieoestrogeen hoger zijn dan of gelijk aan de minimale activiteit zoals gespecificeerd in de testmethodes in verhouding tot de activiteit van de vehiculumcontrole (oplosmiddelcontrole). Voor de STTA-bepaling en de VM7Luc-ER TA-bepaling betekent dit viermaal zo hoog als de gemiddelde activiteit van de vehiculumcontrole voor elke plaat;
|
|
—
|
de geteste controles moeten binnen het oplosbaarheidsbereik van de teststoffen blijven en mogen geen cytotoxiciteit vertonen.
|
|
Analyse van de gegevens
|
17.
|
Om te bepalen of een respons positief of negatief is, moet de voor elke bepaling vastgestelde procedure voor gegevensinterpretatie worden gebruikt.
|
|
18.
|
Als aan de aanvaardbaarheidscriteria (punt 16) wordt voldaan, betekent dit weliswaar dat de bepaling naar behoren functioneert, maar wil dit nog niet zeggen dat een bepaalde testrun kloppende gegevens produceert. Het reproduceren van de resultaten van de eerste testrun is de beste indicatie dat de geproduceerde gegevens kloppen. Als twee testruns reproduceerbare gegevens opleveren (bv. de resultaten van beide testruns wijzen uit dat een teststof positief is), is het niet nodig om een derde testrun uit te voeren.
|
|
19.
|
Als twee testruns geen reproduceerbare resultaten opleveren (bv. een teststof is positief in de ene en negatief in de andere testrun), of als er meer zekerheid nodig is over de uitkomst van deze bepaling, moeten ten minste drie onafhankelijke testruns worden uitgevoerd. In dat geval wordt de indeling gebaseerd op de twee van de drie resultaten die overeenkomen.
|
Algemene criteria voor gegevensinterpretatie
|
20.
|
Er is op vooralsnog geen algemeen erkende methode voor het interpreteren van ER TA-gegevens, maar kwalitatieve (bv. positieve/negatieve) en/of kwantitatieve (bv. EC50, PC50, IC50) beoordelingen van ER-gemedieerde activiteit moeten wel stoelen op empirische gegevens en wetenschappelijk verantwoorde afwegingen. Waar mogelijk moeten positieve resultaten worden gespecificeerd met zowel de grootte van het effect in vergelijking met de vehiculumcontrole (oplosmiddelcontrole) of het referentie-oestrogeen, als de concentratie waarbij het effect optreedt (bv. een EC50, PC50, RPCMax, IC50 enz.).
|
Testverslag
|
21.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Bepaling:
|
—
|
controle/referentiestandaard/teststof;
|
|
—
|
Bron, partijnummer, uiterste gebruiksdatum, indien beschikbaar;
|
|
—
|
stabiliteit van de teststof, indien bekend;
|
|
—
|
oplosbaarheid en stabiliteit van de teststof in het oplosmiddel, indien bekend;
|
|
—
|
meting van pH, osmolaliteit en neerslag in het kweekmedium waaraan de teststof werd toegevoegd, indien van toepassing.
|
|
|
|
Stof die uit één component bestaat:
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
chemische identificatiegegevens, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code, structuurformule, zuiverheid, chemische identiteit van onzuiverheden, indien van toepassing en praktisch haalbaar enz.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB’s en mengsels:
|
—
|
voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen.
|
|
|
|
Oplosmiddel/vehiculum:
|
—
|
karakterisering (aard, leverancier en partij);
|
|
—
|
motivering voor de keuze van het oplosmiddel/vehiculum;
|
|
—
|
oplosbaarheid en stabiliteit van de teststof in het oplosmiddel/vehiculum, indien bekend.
|
|
|
|
Cellen:
|
—
|
type en bron van de cellen:
|
—
|
is er endogene expressie van de ER? Zo niet, welke reporter(s) werd(en) er getransfecteerd?
|
|
—
|
het/de gebruikte reporterconstruct(en) (waaronder brondiersoort(en));
|
|
—
|
selectiemethode voor het handhaven van stabiele transfectie (indien van toepassing);
|
|
—
|
is de transfectiemethode relevant voor stabiele cellijnen?
|
|
|
—
|
aantal passages (na ontdooiing);
|
|
—
|
aantal passages van cellen bij ontdooiing;
|
|
—
|
methoden om de celculturen in leven te houden.
|
|
|
|
Testomstandigheden:
|
—
|
Beperkte oplosbaarheid;
|
|
—
|
Beschrijving van de methoden die zijn toegepast voor de beoordeling van de levensvatbaarheid;
|
|
—
|
mediumsamenstelling, CO2-concentratie;
|
|
—
|
concentraties van de teststof;
|
|
—
|
toegevoegd volume vehiculum en teststof;
|
|
—
|
incubatietemperatuur en vochtigheidsgraad;
|
|
—
|
duur van de behandeling;
|
|
—
|
celdichtheid bij aanvang van en tijdens de behandeling;
|
|
—
|
positieve en negatieve referentiestandaarden;
|
|
—
|
reporterreagentia (productnaam, leverancier en partij);
|
|
—
|
criteria om te bepalen of het resultaat van een testrun positief, negatief of onduidelijk is.
|
|
|
|
Aanvaardbaarheidscontrole:
|
—
|
x-voudige inducties voor elke testplaat en of deze aan het minimum voldoen dat op basis van de historische controles voor de bepaling vereist is;
|
|
—
|
daadwerkelijke waarden voor de aanvaardbaarheidscriteria, bv. log10EC50, log10PC50, log10IC50 en Hill-coëfficiënten, voor de gelijktijdige positieve controles/referentiestandaarden.
|
|
|
|
Resultaten:
|
—
|
ruwe en genormaliseerde gegevens;
|
|
—
|
het maximale x-voudige-inductieniveau;
|
|
—
|
cytotoxiciteitsgegevens;
|
|
—
|
in voorkomend geval, de laagste effectieve concentratie (LEC);
|
|
—
|
RPCMax, PCMax, PC50, IC50en/of EC50, voor zover van toepassing;
|
|
—
|
indien mogelijk het verband tussen concentratie en respons;
|
|
—
|
statistische analyses, indien uitgevoerd, samen met een maat voor de fout en de betrouwbaarheid (bv. SEM, SD, CV of 95 %-BI) en een beschrijving hoe deze waarden zijn verkregen.
|
|
|
|
Bespreking van de resultaten
|
|
LITERATUUR
|
(1)
|
OESO (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
OESO (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OESO (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(7)
|
OESO (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(8)
|
OESO (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor β - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor β (hERβ) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-β: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Aanhangsel 1
DEFINITIES EN AFKORTINGEN
Aanvaardbaarheidscriteria
: minimumnormen voor de prestaties van experimentele controles en referentiestandaarden. Wil een experiment geldig zijn, dan moet aan alle aanvaardbaarheidscriteria worden voldaan.
Agonist
: een stof die bij binding aan een specifieke receptor een respons, bv. transcriptie, opwekt.
Antagonist
: een type receptorligand of chemische stof die zelf geen biologische respons teweegbrengt bij binding aan een receptor, maar die agonist-gemedieerde responsen blokkeert of dempt.
Anti-oestrogene activiteit
: het vermogen van een chemische stof om de werking van 17β-oestradiol te onderdrukken, gemedieerd door oestrogeenreceptoren.
Behandeling met met dextraan beklede houtskool
: behandeling van serum voor celkweek. Door te behandelen met met dextraan beklede kool (vaak „strippen” genoemd) worden endogene hormonen en hormoon-bindende eiwitten verwijderd.
Bekwaamheid
: het bewezen vermogen om een bepaling naar behoren uit te voeren alvorens over te gaan tot het testen van onbekende stoffen.
Bepaling
: in het kader van deze testmethode is een bepaling een van de methoden die als valide zijn aanvaard omdat ze aan de beschreven prestatiecriteria voldoen. Onderdelen van een bepaling zijn bijvoorbeeld onder meer: de specifieke cellijn met bijbehorende groeiomstandigheden, specifieke media waarin de test wordt uitgevoerd, opzet van de kweekplaten, ordening en verdunningen van de teststoffen, en eventuele andere vereiste kwaliteitscontrolemaatregelen en bijbehorende stappen ter evaluatie van de gegevens.
Betrouwbaarheid
: indicatie van de mate waarin een bepaling, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt bepaald door berekening van intra- en interlaboratoriumreproduceerbaarheid.
Celmorfologie
: de vorm en het uiterlijk van cellen die in een monolaag in één putje van een weefselkweekplaat worden gekweekt. Stervende cellen vertonen vaak een afwijkende celmorfologie.
Chemische stof
: een stof of een mengsel.
Conceptueel kader
: het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen.
Cytotoxiciteit
: schadelijke effecten op de celstructuur of celfunctie die uiteindelijk tot celdood kunnen leiden en die kunnen blijken uit een afname van het aantal cellen dat aan het einde van de blootstellingsperiode in het putje aanwezig is, of een afname in sterkte van een maat voor de celfunctie in vergelijking met de gelijktijdige vehiculumcontrole.
CV
: variatiecoëfficiënt
DCC-FBS
: met met dextraan beklede kool behandeld foetaal runderserum.
DMEM
: Dulbecco’s Modification of Eagle’s Medium
DMSO
: dimethylsulfoxide
E2
: 17β-oestradiol
EC50
: de effectieve-concentratiemediaan van een teststof.
EFM
: oestrogeen-vrij medium. DMEM aangevuld met 4,5 % DCC-FBS, 1,9 % L-glutamine en 0,9 % Pen-Strep.
ER
: oestrogeenreceptor.
ERE
: oestrogeenresponselement.
ER TA
: transactivatie van de oestrogeenreceptor.
FBS
: foetaal runderserum.
Gevalideerde testmethode
: een bepaling waarvoor valideringsstudies zijn gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er moet worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel (1).
Gevoeligheid
: het percentage van alle positieve/actieve stoffen dat door middel van de bepaling correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een bepaling die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een bepaling (1).
HeLa
: een onsterfelijke humane baarmoederhalskankercellijn.
HeLa9903
: een subkloon van HeLa waarbij hERααen een luciferasereportergen stabiel getransfecteerd zijn.
hERα
: humane oestrogeenreceptor-alfa
hERβ
: humane oestrogeenreceptor-beta
Hormoonontregeling
: de verstoring van de synthese, de opslag en het metabolisme van hormonen
IC50
: de effectieve-concentratiemediaan van een remmende teststof.
ICCVAM
: het Amerikaanse Interagency Coordinating Committee on the Validation of Alternative Methods.
Interlaboratoriumreproduceerbaarheid
: een maatstaf voor de mate waarin verschillende gekwalificeerde laboratoria die gebruikmaken van hetzelfde protocol en die dezelfde stoffen testen, in kwalitatief en kwantitatief opzicht vergelijkbare resultaten kunnen produceren. De interlaboratoriumreproduceerbaarheid wordt bepaald tijdens de prevaliderings- en valideringsprocessen en geeft aan in hoeverre een bepaling met succes tussen laboratoria kan worden overgedragen; dit wordt ook wel reproduceerbaarheid tussen laboratoria genoemd (1).
Intralaboratoriumreproduceerbaarheid
: een bepaling van de mate waarin gekwalificeerde mensen binnen hetzelfde laboratorium met succes resultaten kunnen repliceren met gebruikmaking van een specifiek protocol op verschillende tijdstippen. Dit wordt ook „reproduceerbaarheid binnen laboratoria” genoemd (1).
LEC
: de laagste effectieve concentratie is de laagste concentratie van de teststof die een respons opwekt (d.w.z.de laagste concentratie van de teststof waarbij de x-voudige inductie statistisch verschilt van de gelijktijdige vehiculumcontrole).
„Me too”-test
: een informele uitdrukking voor een bepaling die in structureel en functioneel opzicht vergelijkbaar is met een gevalideerde en geaccepteerde testmethode. Dit is een alternatieve benaming voor gelijksoortige testmethode.
MMTV
: muizen-borstkankervirus.
MT
: metallothioneïne.
Nauwkeurigheid (concordantie)
: de mate van overeenstemming tussen resultaten verkregen met de bepaling en erkende referentiewaarden. Het is een maat voor de prestaties van de bepaling en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een bepaling wordt verstaan, worden vaak door elkaar gebruikt (1).
Oestrogene activiteit
: het vermogen van een chemische stof om 17β-oestradiol te imiteren door te kunnen binden aan oestrogeenreceptoren en deze te kunnen activeren. Met deze testmethode kan hERα-gemedieerde oestrogene activiteit worden gedetecteerd.
OHT
: 4-hydroxytamoxifen.
Onafhankelijke testruns
: aparte, onafhankelijke experimenten waarin het chemische effect op het biologische resultaat van de bepaling wordt beoordeeld met behulp van cellen uit verschillende pools, met vers verdunde chemische stoffen en uitgevoerd op verschillende dagen door verschillende medewerkers.
Onderzoek
: al het experimentele werk dat wordt uitgevoerd voor het beoordelen van één specifieke stof met behulp van een specifieke bepaling. Een onderzoek omvat alle stappen waaronder tests voor de oplosbaarheid van de teststof in het testmedium, voorbereidende bereikbepalingstestruns, alle nodige volledige testruns, gegevensanalyses, kwaliteitsborging, cytotoxiciteitsbeoordelingen enz. Als het onderzoek is afgerond kan het effect van de teststof op het toxiciteitsdoelwit dat met de gebruikte bepaling wordt beoordeeld, worden ingedeeld (d.w.z. actief, niet-actief of geen uitsluitsel) en kan een schatting worden gemaakt van de potentie ten opzichte van de positieve referentiestof.
PBTG
: op prestaties gebaseerde testrichtlijn.
PC10
: de concentratie van een teststof waarbij de gemeten activiteit in een agonismebepaling 10 % is van de maximale activiteit die wordt opgewekt door de positieve controle (E2 bij 1 nM voor de STTA-bepaling) op elke plaat.
PC50
: de concentratie van een teststof waarbij de gemeten activiteit in een agonismebepaling 50 % is van de maximale activiteit die wordt opgewekt door de positieve controle (E2 bij de in de testmethode gespecificeerde referentieconcentratie) op elke plaat.
PCMax
: de concentratie waarbij een teststof de RPCMax opwekt.
Positieve controle
: een sterk actieve stof, bij voorkeur 17β-oestradiol die in alle bepalingen wordt opgenomen om na te gaan of de bepaling naar behoren functioneert.
Prestatienormen
: normen, gebaseerd op een gevalideerde bepaling die de basis vormen voor de beoordeling van de vergelijkbaarheid van een voorgestelde, in mechanistisch en functioneel opzicht gelijksoortige bepaling. Dit zijn 1) essentiële componenten van de bepaling; 2) een minimumlijst van referentiestoffen die zijn gekozen uit de stoffen die zijn gebruikt om de aanvaardbare prestaties van de gevalideerde testmethode aan te tonen, en 3) de vergelijkbare nauwkeurigheids- en betrouwbaarheidsniveaus die door de voorgestelde bepaling moeten worden gehaald wanneer ze worden beoordeeld aan de hand van de minimumlijst van referentiestoffen; deze waarden zijn gebaseerd op de waarden die voor de gevalideerde testmethode zijn verkregen (1).
Referentie-oestrogeen (Positieve controle)
: 17β-oestradiol (E2, CAS 50-28-2).chk
Referentiestandaard
: een referentiestof die wordt gebruikt om de geschiktheid van een bepaling aan te tonen. De referentiestof voor de STTA-bepaling en de VM7Luc-ER TA-bepaling is 17β-oestradiol.
Referentietestmethoden
: de bepalingen waarop PBTG 455 gebaseerd is.
Relevantie
: beschrijving van het verband van een bepaling met het te onderzoeken effect en of de bepaling betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de bepaling het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een bepaling (1).
RLU
: relatieve lichteenheden.
RNA
: ribonucleïnezuur.
RPCMax
: maximale respons die door een teststof wordt opgewekt, uitgedrukt als percentage van de respons die wordt opgewekt door 1 nM E2 op dezelfde plaat.
RPMI
: RPMI 1640-medium, aangevuld met 0,9 % Pen-Strep en 8,0 % FBS.
SD
: standaarddeviatie.
Specificiteit
: het percentage van alle negatieve/niet-actieve stoffen dat door middel van de test correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een bepaling die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een bepaling (1).
Stabiele transfectie
: wanneer DNA zodanig in gekweekte cellen getransfecteerd wordt dat het stabiel geïntegreerd wordt in het celgenoom, met als resultaat de stabiele expressie van de getransfecteerde genen. Klonen van stabiel getransfecteerde cellen worden geselecteerd aan de hand van stabiele markers (bv. resistentie tegen G418).
Stof
: in het kader van REACH (62) wordt een stof gedefinieerd als een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de vervaardiging ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan. In het kader van het VN-GHS wordt een sterk vergelijkbare definitie gehanteerd (1).
Stoffen voor bekwaamheidstoetsing
: een subset van de referentiestoffen die in de prestatienormen zijn opgenomen. Laboratoria kunnen deze stoffen gebruiken om hun technische bekwaamheid met de gestandaardiseerde testmethode aan te tonen. Doorgaans zijn de selectiecriteria voor deze stoffen onder meer dat ze het hele spectrum van responsen bestrijken, dat ze in de handel verkrijgbaar zijn en dat er kwalitatief hoogwaardige referentiegegevens beschikbaar zijn.
STTA-bepaling
: Stably Transfected Transactivation Assay, bepaling op basis van de transcriptionele activatie van ERα waarbij gebruikgemaakt wordt van de cellijn HeLa 9903.
TA (Transactivatie)
: de initiëring van mRNA-synthese als respons op een specifiek chemisch signaal, zoals de binding van een oestrogeen aan de ER.
Testrun
: een afzonderlijk experiment waarbij het effect van een chemische stof op het biologische resultaat van de bepaling wordt beoordeeld. Elke testrun is een compleet experiment dat wordt uitgevoerd op cellen die vanuit één pool in duploputjes zijn uitgeplaat.
Teststof
: alle volgens deze testmethode geteste stoffen of mengsels.
Transcriptie
: mRNA synthese.
UVCB
: chemische stoffen waarvan de samenstelling niet bekend of variabel is, complexe reactieproducten of biologische materialen.
Validering
: het proces om de betrouwbaarheid en de relevantie van een bepaald(e) benadering, methode, bepaling, proces of beoordeling voor een bepaald doel vast te stellen (1).
VC (vehiculumcontrole)
: het oplosmiddel dat wordt gebruikt voor het oplossen van de test- en controlestoffen wordt afzonderlijk als vehiculum zonder opgeloste chemische stof getest.
VM7
: een geïmmortaliseerde adenocarcinoomcel die endogeen ER tot expressie brengt.
VM7Luc4E2
: de cellijn VM7Luc4E2 is afgeleid van de geïmmortaliseerde, uit mensen verkregen adenocarcinoomcellen VM7 die endogeen de beide vormen van de oestrogeenreceptor (ERα en ERβ) tot expressie brengen, en die stabiel getransfecteerd zijn met het plasmide pGudLuc7.ERE. Dit plasmide bevat vier kopieën van een synthetisch oligonucleotide dat het oestrogeenresponselement stroomopwaarts van de promotor van het muizen-borstkankervirus (MMTV) en het vuurvlieg-luciferasegen bevat.
Zwakke positieve controle
: een zwak actieve stof, geselecteerd vanuit de lijst met referentiestoffen, die in alle bepalingen wordt opgenomen om na te gaan of de bepaling naar behoren functioneert.
Aanhangsel 2
TRANSACTIVATIEBEPALING OP BASIS VAN STABIEL GETRANSFECTEERDE HUMANE OESTROGEENRECEPTOR-Α VOOR HET OPSPOREN VAN OESTROGEENAGONISTISCHE EN -ANTAGONISTISCHE ACTIVITEIT VAN CHEMISCHE STOFFEN, WAARBIJ GEBRUIKGEMAAKT WORDT VAN DE CELLIJN HERΑ-HELA-9903
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN (ZIE OOK ALGEMENE INLEIDING)
|
1.
|
Voor deze transactivatiebepaling (TA-bepaling) wordt gebruikgemaakt van de cellijn hERα-HeLa-9 903 voor het opsporen van oestrogeenagonistische activiteit gemedieerd door de humane oestrogeenreceptor-alfa (hERα). In de valideringsstudie voor de bepaling op basis van stabiele transfectie en transactivatie (STTA) door het Japanse Chemicals Evaluation and Research Institute (CERI), met gebruikmaking van de cellijn hERα-HeLa-9 903 voor het opsporen van oestrogeenagonistische en -antagonistische activiteit gemedieerd door de humane oestrogeenreceptor-alfa (hERα), werden de relevantie en betrouwbaarheid van de bepaling voor het beoogde doel aangetoond (1).
|
|
2.
|
Deze bepaling is speciaal ontworpen voor het opsporen van hERα-gemedieerde TA door meting van chemiluminescentie als eindpunt. Er zijn echter niet-receptor-gemedieerde luminescentiesignalen gemeld bij fyto-oestrogeenconcentraties hoger dan 1 μM als gevolg van overactiviteit van het luciferasereportergen (2) (3). Hoewel de dosis-responscurve aangeeft dat er bij lagere concentraties daadwerkelijk activatie van het ER-systeem optreedt, moet de luciferase-expressie bij hoge concentraties fyto-oestrogenen of soortgelijke verbindingen waarvan vermoed wordt dat zij fyto-oestrogeenachtige overactivatie van het luciferasereportergen kunnen veroorzaken, in ER STTA-bepalingssystemen zorgvuldig worden bestudeerd (aanhangsel 1).
|
|
3.
|
Alvorens deze bepaling te gebruiken met het oog op regelgeving, moeten de punten onder „ALGEMENE INLEIDING” en „COMPONENTEN VAN DE ER TA-BEPALING” worden geraadpleegd. De in deze TG gebruikte definities en afkortingen staan in aanhangsel 2.1.
|
PRINCIPE VAN DE BEPALING (ZIE OOK DE ALGEMENE INLEIDING)
|
4.
|
De bepaling wordt gebruikt om de binding van een ligand aan de ER te signaleren. Na de ligandbinding verplaatst het receptor-ligandcomplex zich naar de nucleus, waar het aan specifieke DNA-responselementen bindt en een vuurvlieg-luciferasereportergen transactiveert, wat leidt tot een verhoogde cellulaire expressie van het luciferase-enzym. Luciferine is een substraat dat door het enzym luciferase wordt omgezet in een bioluminescentieproduct dat kwantitatief gemeten kan worden met een luminometer. De luciferase-activiteit kan snel en goedkoop worden bepaald met behulp van een aantal in de handel verkrijgbare testkits.
|
|
5.
|
Voor het testsysteem wordt gebruikgemaakt van de cellijn hERα-HeLa-9 903, die is afgeleid van een humane baarmoederhalstumor met twee stabiel geïnsereerde constructen: i) het hERαα-expressieconstruct (dat codeert voor de volledige humane receptor), en ii) een vuurvlieg-luciferasereporterconstruct met vijf tandemherhalingen van een oestrogeenresponselement (ERE) van vitellogenine, gestuurd door een promotor-TATA-element van muizen-metallothioneïne (MT). Er wordt doorgaans gebruikgemaakt van het muizen-MT-TATA-genconstruct omdat is aangetoond dat dit de beste prestaties levert. Zo kan deze hERα-HeLa-9 903-cellijn het vermogen van een teststof meten om hERα-gemedieerde transactivatie van de expressie van het luciferasegen te induceren.
|
|
6.
|
In het geval van een ER-agonismebepaling wordt de interpretatie van de gegevens gebaseerd op het criterium of het maximale responsniveau dat door een teststof wordt geïnduceerd, al dan niet gelijk is aan of hoger is dan een agonistische respons die overeenkomt met 10 % van de respons die wordt geïnduceerd door een concentratie die bij de positieve controle een maximale respons induceert, te weten 1 nM 17β-oestradiol (E2) (d.w.z.de PC10). In het geval van een ER-antagonismebepaling wordt de interpretatie van de gegevens gebaseerd op het criterium of de respons al dan niet een afname van de activiteit met ten minste 30 % laat zien ten opzichte van de respons die wordt geïnduceerd door de spike-in-controle (25 pM van E2), zonder cytotoxiciteit. De analyse en de interpretatie van de gegevens worden uitvoerig besproken in de punten 34 tot en met 48.
|
PROCEDURE
Cellijnen
|
7.
|
Voor de bepaling moet de stabiel getransfecteerde cellijn hERα-HeLa-9 903 worden gebruikt. Deze cellijn kan worden verkregen bij de celbank Japanese Collection of Research Bioresources (JCRB) (63) na ondertekening van een overeenkomst inzake overdracht van materiaal (MTA).
|
|
8.
|
Voor het testen mogen alleen cellen worden gebruikt die zijn gekarakteriseerd als vrij van mycoplasma. Voor een gevoelige detectie van mycoplasma-infectie moet bij voorkeur de methode PT-PCR (Real Time Polymerase Chain Reaction) worden gebruikt (4) (5) (6).
|
Stabiliteit van de cellijn
|
9.
|
Om de stabiliteit van de cellijn te controleren moeten E2, 17α-oestradiol, 17α-methyltestosteron en corticosteron worden gebruikt als referentiestandaarden voor de agonismebepaling. Bovendien moet telkens als de bepaling wordt uitgevoerd ten minste eenmaal een volledige concentratie-responscurve worden gemeten in het in tabel 1 aangegeven testconcentratiebereik, waarbij de resultaten in overeenstemming moeten zijn met de resultaten in tabel 1.
|
|
10.
|
In geval van een antagonismebepaling moeten er tegelijkertijd met elke testrun twee volledige concentratiecurven worden gemeten voor twee referentiestandaarden: tamoxifen en flutamide. Voor deze twee stoffen moet worden toegezien op een correcte kwalitatieve indeling als positief of negatief.
|
Omstandigheden voor het kweken en uitplaten van de cellen
|
11.
|
De cellen moeten worden bewaard in Eagle’s Minimum Essential Medium (EMEM) zonder fenolrood, aangevuld met 60 mg/l van het antibioticum kanamycine en 10 % met met dextraan beklede kool behandeld foetaal runderserum (DCC-FBS) in een CO2-incubator (5 % CO2) bij 37 ± 1 °C. Zodra 75-90 % confluentie wordt bereikt, kunnen de cellen worden overgeënt in 10 ml 0,4 × 105– 1 × 105 cellen/ml in petrischaaltjes van 100 mm. De cellen moeten worden gesuspendeerd met 10 % FBS-EMEM (wat gelijk is aan EMEM met DCC-FBS) en vervolgens worden uitgeplaat in de putjes van een microtiterplaat in een dichtheid van 1 × 104cellen/(100 μl × putje). Vervolgens moeten de cellen gedurende 3 uur worden gepre-incubeerd in een 5 % CO2-incubator bij 37 ± 1 °C alvorens te worden blootgesteld aan de teststof. Het kunststof laboratoriumgerei moeten vrij zijn van oestrogene activiteit.
|
|
12.
|
Om de integriteit van de respons te behouden moeten de cellen in meerdere passages uit de bevroren voorraad worden gekweekt in het geconditioneerde medium en mogen ze niet meer dan 40 passages worden gekweekt. Voor de cellijn hERα-HeLa-9 903 betekent dit minder dan drie maanden. De prestaties van de cellen kunnen echter achteruitgaan wanneer ze onder ongeschikte kweekomstandigheden worden gekweekt.
|
|
13.
|
Het DCC-FBS kan worden bereid zoals beschreven in aanhangsel 2.2 of in de handel worden verkregen.
|
Aanvaardbaarheidscriteria
Positieve en negatieve referentiestandaarden voor ER-agonismebepaling
|
14.
|
Voorafgaand aan en gedurende het onderzoek moet het responsvermogen van het testsysteem worden gecontroleerd met behulp van geschikte concentraties van een sterk oestrogeen: E2, een zwak oestrogeen (17α-oestradiol), een zeer zwakke agonist (17α-methyltestosteron) en een negatieve stof (corticosteron). De aanvaardbare bereiken op basis van de valideringsstudie (1) staan in tabel 1. Deze vier gelijktijdige referentiestandaarden moeten in elk experiment worden opgenomen en de resultaten moeten binnen de gegeven grenzen van het aanvaardbare bereik vallen. Is dit niet het geval, dan moet de oorzaak van het niet voldoen aan de aanvaardbaarheidscriteria worden bepaald (bv. hantering van de cellen en kwaliteit en concentratie van serum en antibiotica) en moet de bepaling worden herhaald. Wanneer aan de aanvaardbaarheidscriteria is voldaan, is een consequent gebruik van de materialen voor celkweek van essentieel belang voor een minimale variabiliteit in de EC50, PC50 en PC10. De vier gelijktijdige referentiestandaarden, die in elk experiment moeten worden opgenomen (uitgevoerd onder dezelfde omstandigheden, waaronder materialen, aantal passages van de cellen en uitvoerenden), waarborgen de gevoeligheid van de bepaling, omdat de PC10’s van de drie positieve referentiestandaarden binnen het aanvaardbare bereik moeten vallen, evenals de PC50’s en EC50’s, wanneer deze berekend kunnen worden (zie tabel 1).
|
Tabel 1
Aanvaardbare bereiken voor de vier referentiestandaarden voor de ER-agonismebepaling
|
Naam
|
logPC50
|
logPC10
|
logEC50
|
Hill-coëfficiënt
|
Testbereik
|
|
17β-Oestradiol (E2) CAS-nr.: 50-28-2
|
-11,4~-10,1
|
< -11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8 M
|
|
17α-Oestradiol CAS-nr.: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6 M
|
|
Corticosteron CAS-nr.: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4 M
|
|
17α-Methyltestosteron CAS-nr.: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5 M
|
Positieve en negatieve referentiestandaarden voor ER-antagonismebepaling
|
15.
|
Voorafgaand aan en gedurende het onderzoek moet het responsvermogen van het testsysteem worden gecontroleerd met behulp van geschikte concentraties van een positieve stof (tamoxifen) en een negatieve stof (flutamide). De aanvaardbare bereiken op basis van de valideringsstudie (1) staan in tabel 2. Deze twee gelijktijdige referentiestandaarden moeten in elk experiment worden opgenomen en de resultaten moeten als correct worden beoordeeld zoals aangegeven in de criteria. Is dit niet het geval, dan moet de oorzaak van het niet voldoen aan de criteria worden bepaald (bv. hantering van de cellen en kwaliteit en concentratie van serum en antibiotica) en moet de bepaling worden herhaald. Bovendien moeten de IC50-waarden voor een positieve stof (tamoxifen) worden berekend en de resultaten moeten binnen de opgegeven aanvaardbare grenswaarden liggen. Wanneer aan de aanvaardbaarheidscriteria is voldaan, is een consequent gebruik van de materialen voor celkweek van essentieel belang voor een minimale variabiliteit in de IC50. De twee gelijktijdige referentiestandaarden, die moeten worden opgenomen in elk experiment (dat worden uitgevoerd onder dezelfde omstandigheden, waaronder materialen, aantal passages van de cellen en uitvoerenden), waarborgen de gevoeligheid van de bepaling (zie tabel 2).
|
Tabel 2
Criteria en aanvaardbare bereiken voor de twee referentiestandaarden voor de ER-antagonismebepaling
|
Naam
|
Criterium
|
LogIC50
|
Testbereik
|
|
Tamoxifen CAS-nr.: 10 540 -29-1
|
positief: IC50 moet worden berekend
|
-5,942~-7,596
|
10-10~10-5 M
|
|
Flutamide CAS-nr.: 13 311 -84-7
|
negatief: IC30 hoeft niet te worden berekend
|
-
|
10-10~10-5 M
|
Positieve en vehiculumcontroles
|
16.
|
De positieve controle voor de ER-agonismebepaling (1 nM E2) en de ER-antagonismebepaling (10 μM TAM) moet voor elke plaat ten minste in drievoud worden getest. Het te gebruiken vehiculum voor het oplossen van de teststof moet voor elke plaat ten minste in drievoud worden getest als vehiculumcontrole (VC). Als voor de positieve controle een ander vehiculum wordt gebruikt dan voor de teststof, moet er samen met de positieve controle op dezelfde plaat nog een VC ten minste in drievoud worden getest.
|
Kwaliteitscriteria voor de ER-agonismebepaling
|
17.
|
De gemiddelde luciferase-activiteit van de positieve controle (1 nM E2) moet ten minste het 4-voudige zijn van het gemiddelde voor de VC op elke plaat. Dit criterium is vastgesteld op basis van de betrouwbaarheid van de eindpuntwaarden uit de valideringsstudie (historisch het 4- tot 30-voudige).
|
|
18.
|
Voor de kwaliteitscontrole van de bepaling moet de x-voudige inductie die overeenkomt met de PC10-waarde van de gelijktijdige positieve controle (1 nM E2), groter zijn dan 1+2SD van de x-voudige inductiewaarde (=1) van de gelijktijdige VC. Ten behoeve van de prioritering kan de PC10-waarde nuttig zijn ter vereenvoudiging van de gegevensanalyse die vereist is in vergelijking met een statistische analyse. Hoewel een statistische analyse informatie geeft over de significantie, is een dergelijke analyse geen kwantitatieve parameter voor wat betreft het concentratie-afhankelijke potentieel en daarom minder nuttig voor prioritering.
|
Kwaliteitscriteria voor de ER-antagonismebepaling
|
19.
|
De gemiddelde luciferase-activiteit van de spike-in-controle (25 pM E2) moet ten minste het 4-voudige zijn van het gemiddelde voor de VC op elke plaat. Dit criterium is vastgesteld op basis van de betrouwbaarheid van de eindpuntwaarden uit de valideringsstudie.
|
|
20.
|
Voor de kwaliteitscontrole van de bepaling moet de relatieve transcriptionele activatie (RTA) van 1 nM E2 groter zijn dan 100 %, moet de RTA van 1 μM 4-hydroxytamoxifen (OHT) minder zijn dan 40,6 % en moet de RTA van 100 μM digitonine (Dig) minder zijn dan 0 %.
Aantoning van de bekwaamheid van laboratoria(zie punt 14 en de tabellen 3 en 4 onder „COMPONENTEN VAN DE ER TA-BEPALING” van deze testmethode).
|
Vehiculum
|
21.
|
Als gelijktijdige VC wordt dimethylsulfoxide (DMSO) gebruikt, of een geschikt oplosmiddel, in dezelfde concentratie als voor de verschillende positieve en negatieve controles en voor de teststoffen. De teststoffen moeten worden opgelost in een oplosmiddel dat die teststof oplosbaar maakt en mengbaar is met het celmedium. Geschikte vehicula zijn water, ethanol (95 % tot 100 % zuiverheid) en DMSO. Als DMSO wordt gebruikt, mag het gehalte niet hoger zijn dan 0,1 % (v/v). Voor elk vehiculum moet worden aangetoond dat het maximale gebruikte volume niet cytotoxisch is en de prestaties van de bepaling niet verstoort.
|
Voorbereiding van de teststoffen
|
22.
|
De teststoffen moeten in het algemeen worden opgelost in DMSO, of een ander geschikt oplosmiddel, en serieel worden verdund met hetzelfde oplosmiddel in een gemeenschappelijke verhouding van 1:10 om oplossingen voor verdunning met medium te bereiden.
|
Oplosbaarheid en cytotoxiciteit: overwegingen voor bereikbepaling
|
23.
|
Er moet een inleidende test worden uitgevoerd om het passende concentratiebereik van de te testen chemische stof te bepalen en na te gaan of er oplosbaarheids- of cytotoxiciteitsproblemen zijn met de teststof. In eerste instantie worden de chemische stoffen getest tot een maximale concentratie van 1 μl/ml, 1 mg/ml, of 1 mM, welke van deze het laagste is. Op basis van de mate van cytotoxiciteit die of het gebrek aan oplosbaarheid dat in de inleidende test is waargenomen, moet de chemische stof in de eerste definitieve testrun worden getest bij log-seriële verdunningen die beginnen bij de maximale aanvaardbare concentratie (bv.1 mM, 100 μM, 10 μM enz.), waarbij de aanwezigheid van troebelheid of neerslag of cytotoxiciteit moet worden opgemerkt. De concentraties in de tweede en zo nodig derde testrun moeten voor zover nodig worden aangepast om de concentratie-responscurve beter te kunnen karakteriseren en concentraties waarvan is gebleken dat ze onoplosbaar zijn of overmatige cytotoxiciteit induceren, te vermijden.
|
|
24.
|
Voor ER-agonisten en -antagonisten kan het optreden van toenemende cytotoxiciteit de kenmerkende sigmoïdale respons aanzienlijk verstoren of tenietdoen. Bij het interpreteren van de gegevens moet hier dan ook rekening mee worden gehouden. Er moeten methoden voor het testen op cytotoxiciteit worden toegepast die informatie kunnen opleveren over het punt waarop de cellevensvatbaarheid 80 % is, met behulp van een passende bepaling op basis van laboratoriumervaring.
|
|
25.
|
Wanneer de resultaten van de cytotoxiciteitstest uitwijzen dat de concentratie van de teststof het aantal cellen met 20 % of meer heeft doen afnemen, moet deze concentratie als cytotoxisch worden beschouwd en mogen de concentraties die gelijk zijn aan of hoger zijn dan de cytotoxische concentratie niet in de beoordeling worden meegenomen.
|
Blootstelling aan de teststof en indeling van de testplaat
|
26.
|
De procedure voor de verdunning van de teststoffen (stappen 1 en 2) en de blootstelling van de cellen (stap 3) kan als volgt worden uitgevoerd:
Stap 1: Elke teststof moet serieel worden verdund in DMSO of een geschikt oplosmiddel en worden toegevoegd aan de putjes van een microtiterplaat om uit te komen op de uiteindelijke concentratiereeksen zoals bepaald in de voorbereidende bereikbepalingstest (doorgaans een reeks van bijvoorbeeld 1 mM, 100 μM, 10 μM, 1 μM, 100 nM, 10 nM, 1 nM, 100 pM, en 10 pM (10-3-10-11 M)) voor testen in drievoud.
Stap 2: Verdunning van de teststof: verdun eerst 1,5 μl van de teststof in het oplosmiddel tot een volume van 500 μl medium.
Stap 3: Blootstelling van de cellen aan de teststof: voeg 50 μl van de in medium verdunde teststof (bereid in stap 2) toe aan een putje van de testplaat met 104 cellen/100 μl/putje.
Aanbevolen wordt om uiteindelijk voor elk putje op een volume medium van 150 μl uit te komen. De testmonsters en referentiestandaarden kunnen worden gerangschikt zoals weergegeven in tabel 3 en tabel 4.
Tabel 3
Voorbeeldverdeling van de referentiestandaarden over de testplaat voor de ER-agonismebepaling
|
Rij
|
17α-methyltestosteron
|
Corticosteron
|
17α-oestradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc. 6 (100 nM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc. 7 (10 nM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: vehiculumcontrole (0,1 % DMSO); PC: positieve controle (1 nM E2)
|
|
|
27.
|
De referentiestandaarden (E2, 17α-oestradiol, 17α-methyltestosteron en corticosteron) moeten in elke testrun worden meegenomen (tabel 3)eeDe positievecontroleputjes behandeld met 1 nM E2 die een maximale inductie van E2 kunnen opwekken, en de VC-putjes behandeld met alleen DMSO (of geschikt oplosmiddel) moeten ook in elke testplaat worden opgenomen (tabel 4). Als er cellen van verschillende bronnen (bv.verschillend aantal passages, verschillende partij enz.) voor hetzelfde experiment worden gebruikt, moeten de referentiestandaarden voor elke celbron afzonderlijk worden getest.
Tabel 4
Voorbeeldverdeling van de teststoffen en plaatcontrolestoffen over de testplaat voor de ER-agonismebepaling
|
Rij
|
Teststof 1
|
Teststof 2
|
Teststof 3
|
Teststof 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc. 6 (100 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc. 7 (10 nM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: vehiculumcontrole (0,1 % DMSO); PC: positieve controle (1 nM E2)
|
Tabel 5
Voorbeeldverdeling van de referentiestandaarden over de testplaat voor de ER-antagonismebepaling

|
|
28.
|
Om de antagonistische activiteit van chemische stoffen te bepalen wordt aan de testputjes in de rijen A tot en met G 25 pM E2 toegevoegd om de piek te produceren. De referentiestandaarden (tamoxifen en flutamide) moeten in elke testrun worden meegenomen. In elk testplaat moeten bovendien positievecontroleputjes behandeld met 1 nM E2, die kunnen dienen als kwaliteitscontrole voor de cellijn hERα-HeLa-9 903, VC-putjes behandeld met DMSO (of geschikt oplosmiddel), met 0,1 % DMSO behandelde putjes, die zijn behandeld met DMSO toegevoegd aan de concentratie E2 die de piek produceert, en die zodoende overeenkomen met de „Spike-in-controle” (controle die de piek produceert), putjes behandeld met de eindconcentratie van 1 μM OHT en putjes behandeld met 100 μM Dig worden opgenomen (tabel 5). Bij volgende testplaten moet dezelfde indeling worden aangehouden zonder de referentiestandaardputjes (tabel 6). Als er cellen van verschillende bronnen (bv.verschillend aantal passages, verschillende partij enz.) voor hetzelfde experiment worden gebruikt, moeten de referentiestandaarden voor elke celbron afzonderlijk worden getest.
Tabel 6
Voorbeeldverdeling van de teststoffen en plaatcontrolestoffen over de testplaat voor de ER-antagonismebepaling

|
|
29.
|
Voor zover van toepassing moet worden bevestigd dat er geen randeffecten optreden. Als er een vermoeden bestaat van randeffecten moet de plaatindeling worden aangepast om dergelijke effecten te vermijden. Er kan bijvoorbeeld voor worden gekozen om de putjes aan de randen niet te gebruiken.
|
|
30.
|
Na toevoeging van de chemische stoffen moeten de testplaten gedurende 20-24 uur worden geïncubeerd in een 5 % CO2-incubator bij 37 ± 1 oC om de synthese van de reportergenproducten te induceren.
|
|
31.
|
Indien verbindingen sterk vluchtig zijn is bijzondere aandacht geboden. In zulke gevallen kunnen naburige controleputjes namelijk vals-positieve uitkomsten genereren. Deze moeten dan ook worden bekeken in het licht van de verwachte en historische controlewaarden. In de zeldzame gevallen waarin vluchtigheid een probleem oplevert, kan het gebruik van „plaatsealers” de afzonderlijke putjes effectief helpen isoleren tijdens het testen. Dit wordt voor dergelijke gevallen dan ook aanbevolen.
|
|
32.
|
Met het oog op de onafhankelijkheid van de resultaten moeten de herhalingen van de definitieve tests voor dezelfde chemische stof op een andere dag worden uitgevoerd.
|
Luciferasebepaling
|
33.
|
Voor deze bepaling kan een in de handel verkrijgbaar luciferase-bepalingsreagens [bv.Steady-Glo® Luciferase Assay System (Promega, E2510, of gelijkwaardig)] of een standaard luciferasebepalingssysteem (bv. Promega, E1500, of gelijkwaardig) worden gebruikt, zolang maar aan de aanvaardbaarheidscriteria wordt voldaan. De reagentia voor de bepaling moeten worden geselecteerd op basis van de gevoeligheid van de te gebruiken luminometer. Bij gebruik van een standaard luciferasebepalingssysteem moet een celkweek-lysisreagens (bv. Promega, E1531, of gelijkwaardig) worden gebruikt, voordat het substraat wordt toegevoegd. Het luciferasereagens moet worden toegepast volgens de instructies van de fabrikant.
|
ANALYSE VAN DE GEGEVENS
ER-agonismebepaling
|
34.
|
Bij een ER-agonismebepaling kan de relatieve transcriptionele activiteit ten opzichte van de positieve controle (1 nM E2) worden verkregen door de luminescentiesignalen van een en dezelfde plaat te analyseren aan de hand van de volgende stappen (andere, gelijkwaardige mathematische bewerkingen zijn ook aanvaardbaar):
Stap 1. Bereken de gemiddelde waarde voor de VC.
Stap 2. Trek de gemiddelde waarde voor de VC af van de waarde voor elk putje om de gegevens te normaliseren.
Stap 3. Bereken de gemiddelde waarde voor de genormaliseerde positieve controle.
Stap 4. Deel de genormaliseerde waarde voor elk putje in de plaat door de gemiddelde waarde voor de genormaliseerde positieve controle (positieve controle = 100 %).
Het eindresultaat voor elk putje is de relatieve transcriptionele activiteit voor dat putje in vergelijking met de positievecontrolerespons.
Stap 5. Bereken voor elke concentratiegroep van de teststof de gemiddelde waarde van de relatieve transcriptionele activiteit. De uitkomsten leveren tweeërlei informatie op: de gemiddelde transcriptionele activiteit (respons) en de concentratie waarbij de respons optreedt (zie volgende sectie).
|
Overwegingen bij de EC50, PC50 en PC10
|
35.
|
Voor de berekening van de EC50 is de volledige concentratie-responscurve nodig, maar vanwege beperkingen van het testconcentratiebereik (bijvoorbeeld als gevolg van cytotoxiciteit of oplosbaarheidsproblemen) is het niet altijd mogelijk of praktisch haalbaar om deze op te nemen. Aangezien de EC50 en het maximale-inductieniveau (dat overeenkomt met de maximale waarde van de Hill-vergelijking) slechts ter informatie dienen, hoeven deze parameters echter alleen te worden vermeld indien mogelijk. Voor de berekening van de EC50 en het maximale-inductieniveau moet geschikte statistische software worden gebruikt (bv.Graphpad Prism). Als de logistische vergelijking van Hill niet toepasbaar is op de concentratie-responsgegevens, moet de EC50aan de hand van de volgende vergelijking worden berekend (7):
Y=basis + (top - basis) / (1 + 10 exp ((log EC50- X) × Hill-coëfficiënt)), waarbij:
X = de logaritme van de concentratie, en
Y = de respons; Y begint aan de basis en loopt in een sigmoïde curve naar de top. De basis wordt in de logistische vergelijking van Hill op nul gesteld.
|
|
36.
|
Voor elke teststof moeten de volgende gegevens worden verkregen:
de RPCMax (het maximale responsniveau dat door een teststof wordt opgewekt, uitgedrukt als percentage van de respons die wordt opgewekt door 1 nM E2 op dezelfde plaat) en de PCMax (de concentratie die overeenkomt met de RPCMax), en
voor positieve chemische stoffen, de concentraties die de PC10en, indien van toepassing, de PC50 opwekken.
|
|
37.
|
De PCx-waarde kan worden berekend door te interpoleren tussen 2 punten op de X-Y-curve, het ene direct boven en het andere direct onder een PCx-waarde. Als deze punten direct boven en onder de PCx-waarde respectievelijk de coördinaten (a,b) en (c,d) hebben, kan de PCx worden berekend aan de hand van de volgende vergelijking:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
Beschrijvingen van positievecontrolewaarden worden hieronder weergegeven in figuur 1.
Figuur 1
Voorbeeld van de afleiding van positievecontrolewaarden. De positieve controle (1 nM E2) wordt opgenomen in elke testplaat

|
ER-antagonismebepaling
|
39.
|
Bij een ER-antagonismebepaling kan de relatieve transcriptionele activiteit (RTA) ten opzichte van de spike-in-controle (25 pM E2) worden verkregen door de luminescentiesignalen van een en dezelfde plaat te analyseren aan de hand van de volgende stappen (andere, gelijkwaardige mathematische bewerkingen zijn ook aanvaardbaar):
Stap 1. Bereken de gemiddelde waarde voor de VC.
Stap 2. Trek de gemiddelde waarde voor de VC af van de waarde voor elk putje om de gegevens te normaliseren. Stap 3. Bereken de gemiddelde waarde voor de genormaliseerde spike-in-controle.
Stap 4. Deel de genormaliseerde waard voor elk putje in de plaat door de gemiddelde waarde voor de genormaliseerde spike-in-controle (spike-in-controle = 100 %).
Het eindresultaat voor elk putje is de relatieve transcriptionele activiteit voor dat putje in vergelijking met de spike-in-controlerespons.
Stap 5. Bereken voor elke behandeling de gemiddelde waarde van de relatieve transcriptionele activiteit.
|
Overwegingen bij de IC30 en IC50
|
40.
|
Voor positieve chemische stoffen moeten de concentraties worden gegeven die de IC30 en, indien van toepassing, de IC50 opwekken.
|
|
41.
|
De ICx-waarde kan worden berekend door te interpoleren tussen 2 punten op de X-Y-curve, het ene direct boven en het andere direct onder een ICx-waarde. Als deze punten direct boven en onder de ICx-waarde respectievelijk de coördinaten (c,d) en (a,b) hebben, kan de ICx worden berekend aan de hand van de volgende vergelijking:
lin ICx = a - (b - (100 - x)) (a - c)/(b - d)
Figuur 2
Voorbeeld van de afleiding van IC-waarden. De spike-in-controle (25 pM E2) wordt opgenomen in elke testplaat

RTA: relatieve transcriptionele activiteit
|
|
42.
|
De resultaten moeten op twee (of drie) onafhankelijke testruns worden gebaseerd. Als twee testruns vergelijkbare en dus reproduceerbare resultaten opleveren, is het niet nodig een derde testrun uit te voeren. Om aanvaardbaar te zijn moeten de resultaten:
|
—
|
aan de aanvaardbaarheidscriteria voldoen (zie Aanvaardbaarheidscriteria, punten 14-20),
|
|
Criteria voor gegevensinterpretatie
Tabel 7
Positieve en negatieve beslissingscriteria bij ER-agonismebepaling
|
Positief
|
Als in twee van de twee of ten minste twee van de drie testruns de verkregen RPCMax gelijk is aan of sterker dan 10 % van de respons van de positieve controle.
|
|
Negatief
|
Als in twee van de twee of twee van de drie testruns de verkregen RPCMax lager blijft dan 10 % van de respons van de positieve controle.
|
Tabel 8
Positieve en negatieve beslissingscriteria bij ER-antagonismebepaling
|
Positief
|
Als in twee van de twee of ten minste twee van de drie testruns de IC30 kan worden berekend.
|
|
Negatief
|
Als in twee van de twee of twee van de drie testruns de IC30 niet kan worden berekend.
|
|
43.
|
De criteria voor gegevensinterpretatie staan in de tabellen 7 en 8. Positieve resultaten worden gekarakteriseerd door zowel de grootte van het effect als de concentratie waarbij het effect optreedt. Door de resultaten uit te drukken in de vorm van een concentratie waarbij 50 % (PC50) of 10 % (PC10) van de positievecontrolewaarden bereikt worden voor de agonismebepaling, en 50 % (IC50) of 30 % (IC30) van de spike-in-controlewaarde wordt geremd voor de antagonismebepaling, worden deze beide doelen bereikt. Een teststof wordt echter als positief aangemerkt als in twee van de twee of ten minste twee van de drie testruns de maximale door de teststof opgewekte respons (RPCMax) gelijk is aan of sterker dan 10 % van de respons van de positieve controle, terwijl een teststof als negatief wordt beschouwd als de RPCMax in twee van de twee of twee van de drie testruns lager blijft dan 10 % van de respons van de positieve controle.
|
|
44.
|
De PC10, PC50en PCMaxin een ER-agonismebepaling en de IC30 en IC50 in een ER-antagonismebepaling kunnen worden berekend met behulp van een spreadsheet die samen met de testrichtlijn te vinden is op de publiekswebsite van de OESO (64).
|
|
45.
|
Over het algemeen is het voldoende om de PC10of PC50en de IC30 or IC50ten minste tweemaal te bepalen. Mocht de resulterende basislijn voor gegevens in hetzelfde concentratiebereik echter een variabiliteit laten zien met een onaanvaardbaar hoge variatiecoëfficiënt (CV; %), dan kunnen de gegevens mogelijk niet als betrouwbaar worden beschouwd en moet de bron van de hoge variabiliteit worden opgespoord. De ruwe gegevens in drievoud (d.w.z.de gegevens voor de intensiteit van de luminescentie) van de gegevenspunten die worden gebruikt voor de berekening van de PC10, moeten een CV hebben van minder dan 20 %.
|
|
46.
|
Als aan de aanvaardbaarheidscriteria wordt voldaan, betekent dit weliswaar dat het bepalingssysteem naar behoren functioneert, maar wil dit nog niet zeggen dat een bepaalde testrun kloppende gegevens produceert. Duplicatie van de resultaten van de eerste testrun is de beste garantie dat de geproduceerde gegevens kloppen.
|
|
47.
|
Wanneer er in het geval van de ER-agonismebepaling meer informatie nodig is behalve de screenings- en prioriteringsdoelstellingen van deze TG voor positieve teststoffen, in het bijzonder voor PC10-PC49-stoffen, alsmede stoffen waarbij overstimulatie van luciferase wordt vermoed, kan met behulp van een ERα-antagonist worden bevestigd dat de waargenomen luciferase-activiteit uitsluitend een ERα-specifieke respons is (zie aanhangsel 2.1).
|
TESTVERSLAG
|
48.
|
Zie punt 20 van „COMPONENTEN VAN DE ER TA-BEPALING”.
|
LITERATUUR
|
(1)
|
OESO (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett.,78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med.Sci.,57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Aanhangsel 2.1
VALS-POSITIEVE RESULTATEN: BEOORDELING VAN NIET-RECEPTOR-GEMEDIEERDE LUMINESCENTIESIGNALEN
|
1.
|
In de ER-agonismebepaling kunnen vals-positieve resultaten worden gegenereerd door niet-ER-gemedieerde activatie van het luciferasegen, rechtstreekse activatie van het genproduct of fluorescentie door onbepaalde oorzaak. Dergelijke effecten uiten zich in een onvolledige of ongebruikelijke dosis-responscurve. Als dergelijke effecten vermoed worden, moeten de effecten van een ER-antagonist (bv.4- hydroxytamoxifen (OHT) in een niet-toxische concentratie) op de respons worden onderzocht. De pure antagonist ICI 182780 is mogelijk niet geschikt voor dit doel, omdat een voldoende hoge concentratie van ICI 182780 mogelijk de waarde voor de VC vermindert en daarmee de gegevensanalyse beïnvloedt.
|
|
2.
|
Om de validiteit van deze benadering te waarborgen moeten in dezelfde plaat de volgende elementen worden getest:
|
—
|
agonistische activiteit van de onbekende chemische stof met/zonder 10 μM OHT;
|
|
—
|
1 nM E2 (in drievoud) als agonistische positieve controle;
|
|
—
|
1 nM E2 + OHT (in drievoud).
|
|
Criteria voor gegevensinterpretatie
Opmerking: Alle putjes moeten met dezelfde concentratie van het vehiculum worden behandeld.
|
—
|
Als de agonistische activiteit van de onbekende chemische stof NIET beïnvloed wordt door de behandeling met de ER-antagonist, wordt deze als „Negatief” beschouwd.
|
|
—
|
Als de agonistische activiteit van de onbekende chemische stof volledig geremd wordt, moeten de beslissingscriteria worden toegepast.
|
|
—
|
Als de agonistische activiteit van de laagste concentratie gelijk is aan of sterker is dan de PC10-respons, wordt de onbekende chemische stof in gelijke of sterkere mate geremd ten opzichte van de PC10-respons. Het verschil in de responsen tussen de niet-behandelde en de behandelde putjes met de ER-antagonist wordt berekend en dit verschil wordt beschouwd als de werkelijke respons en moet worden gebruikt voor de berekening van de parameters die nodig zijn om te beslissen over de indeling van de chemische stof.
|
Gegevensanalyse
Controleer de prestatienorm.
Controleer de CV tussen de putjes die onder gelijke omstandigheden behandeld zijn.
|
1.
|
Bereken het gemiddelde voor de VC
|
|
2.
|
Trek het gemiddelde van de VC af van de waarde van elk putje dat niet met OHT is behandeld
|
|
3.
|
Bereken het gemiddelde voor OHT
|
|
4.
|
Trek het gemiddelde van de VC af van de waarde van elk putje dat met OHT is behandeld
|
|
5.
|
Bereken het gemiddelde voor de positieve controle
|
|
6.
|
Bereken de relatieve transcriptionele activiteit van alle andere putjes ten opzichte van de positieve controle.
|
Aanhangsel 2.2
BEREIDING VAN MET MET DEXTRAAN BEKLEDE KOOL (DCC) BEHANDELD SERUM
|
1.
|
Behandeling van serum met met dextraan beklede kool (DCC) is een algemene methode om oestrogene verbindingen te verwijderen uit het serum dat aan het celmedium wordt toegevoegd, om elke invloed op de respons door residuele oestrogenen in het serum uit te sluiten. Met deze procedure kan 500 ml foetaal runderserum (FBS) worden behandeld.
|
Onderdelen
|
2.
|
De volgende materialen en uitrusting zijn nodig:
|
Materiaal
|
Magnesiumchloridehexahydraat (MgCl2· 6H2O)
|
|
1 M HEPES-bufferoplossing (pH 7,4)
|
|
Ultrazuiver water geproduceerd met een filtersysteem
|
Uitrusting
Geautoclaveerde glazen kolf (maat aan te passen naar behoefte) gewone laboratoriumcentrifuge (waarbij de temperatuur kan worden ingesteld op 4 °C)
Procedure
|
3.
|
De volgende procedure is aangepast voor gebruik van centrifugebuizen van 50 ml:
[Dag 1] Bereid een DCC-suspensie met 1 l ultrazuiver water dat 1,5 mM MgCl2, 0,25 M sucrose, 2,5 g kool, 0,25 g dextraan en 5 mM HEPES bevat, en roer gedurende een nacht bij 4 °C.
[Dag 2] Verdeel de suspensie over centrifugebuizen van 50 ml en centrifugeer gedurende 10 minuten bij 10000 rpm bij 4 °C. Verwijder het supernatant en bewaar de helft van het koolsediment bij 4 °C voor gebruik op Dag 3. Suspendeer de andere helft van de kool met FBS dat voorzichtig ontdooid is om precipitatie te vermijden en gedurende 30 minuten bij 56 °C door warmte geïnactiveerd is, en breng het over in een geautoclaveerde glazen kolf zoals een erlenmeyer. Roer deze suspensie voorzichtig gedurende een nacht bij 4 °C.
[Dag 3] Verdeel de suspensie met FBS over centrifugebuizen van 50 ml om gedurende 10 minuten te centrifugeren bij 10000 rpm bij 4 °C. Verzamel het FBS en breng het over bij het op Dag 2 bereide en bewaarde verse koolsediment. Suspendeer het koolsediment en roer de suspensie voorzichtig gedurende een nacht bij 4 °C in een geautoclaveerde glazen kolf.
[Dag 4] Verdeel de suspensie om gedurende 10 minuten te centrifugeren bij 10000 rpm bij 4 °C en steriliseer het supernatant door het te filteren door een 0,2 μm steriel filter. Dit met DCC behandelde FBS moet worden bewaard bij -20 °C en blijft tot een jaar te gebruiken.
|
Aanhangsel 3
TRANSACTIVATIEBEPALING OP BASIS VAN DE OESTROGEENRECEPTOR VAN VM7LUC VOOR HET IDENTIFICEREN VAN AGONISTEN EN ANTAGONISTEN VAN DE OESTROGEENRECEPTOR
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN (ZIE OOK ALGEMENE INLEIDING)
|
1.
|
Voor deze bepaling wordt de cellijn VM7Luc4E2 gebruikt (65). De bepaling is gevalideerd door twee Amerikaanse instanties: het National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) en het Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) (1). De VM7Luc-cellijnen brengen voornamelijk endogene ERα tot expressie en een geringe hoeveelheid endogene ERβ (2) (3) (4).
|
|
2.
|
Deze bepaling is geschikt voor een breed spectrum aan stoffen, mits ze kunnen worden opgelost in dimethylsulfoxide (DMSO; CAS RN 67-68-5), niet met DMSO of het celkweekmedium reageren en in de geteste concentraties niet cytotoxisch zijn. Als het niet mogelijk is om DMSO te gebruiken, kan een ander vehiculum zoals ethanol of water worden gebruikt (zie punt 12). Uit de bewezen prestaties van de VM7Luc-ER TA-(ant)agonismebepaling kan worden opgemaakt dat gegevens die met deze bepaling worden gegenereerd informatie zouden kunnen verschaffen over ER-gemedieerde werkingsmechanismen en gebruikt zouden kunnen worden voor de prioritering van stoffen voor nadere testen.
|
|
3.
|
Deze bepaling is speciaal ontworpen voor het opsporen van hERα- en hERβ-gemedieerde TA door meting van chemiluminescentie als eindpunt. Omdat luminescentie een hoge signaal-achtergrondverhouding heeft, wordt chemiluminescentie veel gebruikt in bioassays. De activiteit van vuurvlieg-luciferase in bepalingen op celbasis kan echter worden verstoord door stoffen die het luciferase-enzym remmen, zodat ze als gevolg van de stabilisatie van het luciferine een schijnbare remming of verhoging van de luminescentie veroorzaken. Bovendien zijn er in sommige ER-reportergenbepalingen op basis van luciferase niet-receptor-gemedieerde luminescentiesignalen gemeld bij fyto-oestrogeenconcentraties hoger dan 1 μM als gevolg van overactiviteit van het luciferasereportergen (9) (11). Hoewel de dosis-responscurve aangeeft dat er bij lagere concentraties daadwerkelijk activatie van het ER-systeem optreedt, moet de luciferase-expressie bij hoge concentraties fyto-oestrogenen of soortgelijke verbindingen waarvan vermoed wordt dat zij fyto-oestrogeenachtige overactivatie van het luciferasereportergen kunnen veroorzaken, in ER STTA-bepalingssystemen zorgvuldig worden bestudeerd.
|
|
4.
|
Alvorens deze bepaling te gebruiken met het oog op regelgeving, moeten de “ALGEMENE INLEIDING” en “COMPONENTEN VAN DE ER TA-BEPALING” worden geraadpleegd. De in deze testmethode gebruikte definities en afkortingen staan in aanhangsel 1.
|
PRINCIPE VAN DE BEPALING (ZIE OOK DE ALGEMENE INLEIDING)
|
5.
|
De bepaling wordt gebruikt voor het detecteren van ER-ligandbinding, gevolgd door de translocatie van het receptor-ligandcomplex naar de nucleus. In de nucleus bindt het receptor-ligandcomplex aan specifieke DNA-responselementen en transactiveert het het reportergen (luc), wat leidt tot de aanmaak van luciferase en vervolgens de emissie van licht, dat kan worden gekwantificeerd met behulp van een luminometer. De luciferase-activiteit kan snel en goedkoop worden bepaald met behulp van een aantal in de handel verkrijgbare kits. In de VM7Luc-ER TA wordt gebruikgemaakt van een ER-responsieve, uit de menselijke borst verkregen adenocarcinoomcellijn, VM7, die stabiel getransfecteerd is met een vuurvlieg-luc reporterconstruct, gestuurd door vier oestrogeenresponselementen die stroomopwaarts van de promotor van het muizen-borstkankervirus (MMTV) zijn geplaatst, voor het detecteren van stoffen met ER-agonistische of -antagonistische activiteit in vitro. Deze MMTV-promotor vertoont slechts geringe kruisreactiviteit met andere steroïde en niet-steroïde hormonen (8). De criteria voor gegevensinterpretatie worden nader beschreven in punt 41. Samengevat wordt een positieve respons gekenmerkt door een concentratie-responscurve die ten minste drie punten bevat met niet-overlappende foutbalken (gemiddelde ± standaarddeviatie), en een verandering in de amplitude (genormaliseerde relatieve lichteenheden [RLU]) van ten minste 20 % van de maximale waarde voor de referentiestandaard (17β-oestradiol [E2; CAS RN 50-28-2] voor de agonismebepaling, raloxifeen HCl [Ral; CAS RN 84 449-90-1]/E2 voor de antagonismebepaling).
|
PROCEDURE
Cellijn
|
6.
|
Voor de bepaling moet de stabiel getransfecteerde cellijn VM7Luc4E2 worden gebruikt. Deze cellijn is op dit moment alleen verkrijgbaar met een technische licentieovereenkomst bij de Universiteit van Californië, Davis, Californië, VS (66) en bij Xenobiotic Detection Systems Inc., Durham, North Carolina, VS (67).
|
Stabiliteit van de cellijn
|
7.
|
Om de stabiliteit en de integriteit van de cellijn te behouden moeten de cellen in meerdere passages uit de bevroren voorraad worden gekweekt in medium voor het in stand houden van cellen (zie punt 9). De cellen mogen niet meer dan 30 passages worden gekweekt. Voor de cellijn VM7Luc4E2 komen 30 passages overeen met ongeveer drie maanden.
|
Omstandigheden voor het kweken en uitplaten van de cellen
|
8.
|
Om de kwaliteit van alle materialen en methoden te waarborgen en zo de integriteit, validiteit en reproduceerbaarheid van het verrichte werk te garanderen moeten de procedures beschreven in de leidraad voor goede celkweekpraktijken (Guidance on Good Cell Culture Practice) (5) (6) worden gevolgd.
|
|
9.
|
De VM7Luc4E2-cellen worden bewaard in RPMI 1 640-medium aangevuld met 0,9 % Pen-Strep en 8,0 % foetaal runderserum (FBS) in een speciaal daarvoor bestemde weefselkweekincubator bij 37 °C ± 1 °C, 90 % ± 5 % vochtigheid en 5,0 % ± 1 % CO2/lucht.
|
|
10.
|
Zodra ~80 % confluentie wordt bereikt, worden de VM7Luc4E2-cellen overgeënt en gedurende 48 uur geconditioneerd in een oestrogeenvrij milieu, alvorens te worden uitgeplaat in microtiterplaten met 96 putjes voor de blootstelling aan de teststoffen en de analyse van de oestrogeen-afhankelijke inductie van luciferase-activiteit. Oestrogeen-vrij medium (EFM) bevat Dulbecco’s Modification of Eagle’s Medium (DMEM) zonder fenolrood, aangevuld met 4,5 % DCC-FBS, 1,9 % L-glutamine en 0,9 % Pen-Strep. Alle kunststof laboratoriumgerei moet vrij zijn van oestrogene activiteit [zie gedetailleerd protocol (7)].
|
Aanvaardbaarheidscriteria
|
11.
|
Of een test aanvaard of verworpen wordt, hangt af van de beoordeling van de resultaten voor de referentiestandaarden en de controles van elk op een plaat met 96 putjes uitgevoerd experiment. Elke referentiestandaard wordt in verschillende concentraties getest en voor elke referentie- en controleconcentratie wordt de bepaling meerdere keren herhaald. De resultaten worden vergeleken met kwaliteitscontroles (QC) voor deze parameters die zijn afgeleid uit de databanken met historische agonisme- en antagonismegegevens die door elk laboratorium zijn gegenereerd bij de bekwaamheidstoetsing. De databanken met historische gegevens worden doorlopend bijgewerkt met nieuwe resultaten voor referentiestandaarden en controles. Bij veranderingen in uitrusting of laboratoriumomstandigheden kan het nodig zijn om bijgewerkte databanken met historische gegevens te genereren.
|
Agonismetest
Bereikbepalingstest
|
•
|
Inductie: het geïnduceerde effect op de plaat wordt gemeten door de gemiddelde maximale RLU-waarde met de referentiestandaard E2 te delen door de gemiddelde RLU-waarde voor de DMSO-controle. Doorgaans wordt een vijfvoudige inductie waargenomen, maar een experiment is aanvaardbaar als ten minste een viervoudige inductie wordt verkregen.
|
|
•
|
Resultaten voor de DMSO-controle: de RLU-waarden voor de oplosmiddelcontrole mogen maximaal 2,5 maal de standaarddeviatie van de gemiddelde historische RLU-waarde voor de oplosmiddelcontrole afwijken.
|
|
•
|
Een experiment dat niet voldoet aan een van beide aanvaardbaarheidscriteria, moet worden verworpen en herhaald.
|
Integrale test
Voor deze test gelden aanvaardbaarheidscriteria van de bereikbepalingstest voor de agonismebepaling, alsmede de volgende criteria:
|
•
|
Resultaten voor de referentiestandaard: de concentratie-responscurve voor de referentiestandaard E2 moet een sigmoïdale vorm hebben en er moeten ten minste drie waarden binnen het lineaire gedeelte van de concentratie-responscurve vallen.
|
|
•
|
Resultaten voor de positieve controle: de RLU-waarden voor de methoxychloorcontrole moeten hoger zijn dan de gemiddelde waarde voor DMSO plus driemaal de standaarddeviatie van de gemiddelde waarde voor DMSO.
|
|
•
|
Een experiment dat niet voldoet aan een van de aanvaardbaarheidscriteria, moet worden verworpen en herhaald.
|
Antagonismetest
Bereikbepalingstest
|
•
|
Remming: het remmende effect op de plaat wordt gemeten door de gemiddelde maximale RLU-waarde met de referentiestandaard E2 te delen door de gemiddelde RLU-waarde voor de DMSO-controle. Doorgaans wordt een vijfvoudige remming waargenomen, maar een experiment is aanvaardbaar als ten minste een drievoudige remming wordt verkregen.
|
|
•
|
Resultaten voor de E2-controle: de RLU-waarden voor de E2-controle mogen maximaal 2,5 maal de standaarddeviatie van de gemiddelde historische RLU-waarde voor de E2-controle afwijken.
|
|
•
|
Resultaten voor de DMSO-controle: de RLU-waarden voor de DMSO-controle mogen maximaal 2,5 maal de standaarddeviatie van de gemiddelde historische RLU-waarde voor de oplosmiddelcontrole afwijken.
|
|
•
|
Een experiment dat niet voldoet aan een van de aanvaardbaarheidscriteria, wordt verworpen en herhaald.
|
Integrale test
Voor deze test gelden aanvaardbaarheidscriteria van de bereikbepalingstest voor de antagonismebepaling, alsmede de volgende criteria:
|
•
|
Resultaten voor de referentiestandaard: de concentratie-responscurve voor de referentiestandaard Ral/E2 moet een sigmoïdale vorm hebben en er moeten ten minste drie waarden binnen het lineaire gedeelte van de concentratie-responscurve vallen.
|
|
•
|
Resultaten voor de positieve controle: de RLU-waarden voor de tamoxifen/E2-controle moeten lager zijn dan de gemiddelde waarde voor de E2-controle minus driemaal de standaarddeviatie van de gemiddelde waarde voor de E2-controle.
|
|
•
|
Een experiment dat niet voldoet aan een van de aanvaardbaarheidscriteria, wordt verworpen en herhaald.
|
Referentiestandaarden, positieve en vehiculumcontroles
Vehiculumcontroles (agonisme- en antagonismebepalingen)
|
12.
|
Het te gebruiken vehiculum voor het oplossen van de teststoffen moet worden getest als vehiculumcontrole. Bij de validering van de VM7Luc-ER TA-bepaling werd 1 % (v/v) dimethylsulfoxide (DMSO, CAS RN 67-68-5) als vehiculum gebruikt (zie punt 24). Als een ander vehiculum dan DMSO wordt gebruikt, moeten alle referentiestandaarden, controles en teststoffen voor zover mogelijk in hetzelfde vehiculum worden getest.
|
Referentiestandaard (bereikbepaling agonisme)
|
13.
|
De referentiestandaard is E2 (CAS RN 50-28-2). Voor de bereikbepalingstest wordt een verdunningsreeks van de referentiestandaard gemaakt bestaande uit vier concentraties E2 (1,84 × 10-10, 4,59 × 10-11, 1,15 ×10-11 en 2,87 × 10-12 M), waarbij elke concentratie in twee putjes wordt getest.
|
Referentiestandaard (integrale agonismetest)
|
14.
|
Voor de integrale test wordt een 1:2 verdunningsreeks van E2 gemaakt bestaande uit 11 concentraties E2 (lopend van 3,67 × 10-10 tot 3,59 × 10-13 M), die elk in twee putjes worden getest.
|
Referentiestandaard (bereikbepaling antagonisme)
|
15.
|
De referentiestandaard is een combinatie van Ral (CAS RN 84 449-90-1) en E2 (CAS RN 50-28-2). Voor de bereikbepalingstest wordt een verdunningsreeks van Ral/E2 gemaakt bestaande uit drie concentraties Ral (3,06 x 10-9, 7,67 x 10-10, en 1,92 x 10-10 M) plus een vaste concentratie E2 (9,18 × 10-11 M), waarbij elke verdunning in twee putjes wordt getest.
|
Referentiestandaard (integrale antagonismetest)
|
16.
|
Voor de integrale test wordt een 1:2 verdunningsreeks van Ral gemaakt (lopend van 2,45 x 10-8 tot 9,57 x 10-11 M) plus een vaste concentratie E2 (9,18 × 10-11 M) om zo tot negen concentraties Ral/E2 te komen, die elk in twee putjes wordt getest.
|
Zwakke positieve controle (agonisme)
|
17.
|
De zwakke positieve controle is 9,06 x 10-6 M p,p’-methoxychloor (methoxychloor; CAS RN 72-43-5) in EFM.
|
Zwakke positieve controle (antagonisme)
|
18.
|
De zwakke positieve controle bestaat uit tamoxifen (CAS RN 10 540-29-1) 3,36 x 10-6 M met 9,18 × 10-11 M E2 in EFM.
|
E2-controle (alleen voor antagonismebepaling)
|
19.
|
De E2-controle is 9,18 × 10-11 M E2 in EFM en wordt gebruikt als negatieve basislijncontrole.
|
X-voudige inductie (agonisme)
|
20.
|
De inductie van luciferaseactiviteit door de referentiestandaard (E2) wordt gemeten door de gemiddelde maximale RLU-waarde voor de referentiestandaard E2 te delen door de gemiddelde RLU-waarde voor de DMSO-controle. Het resultaat moet groter zijn dan viervoudig.
|
X-voudige remming (antagonisme)
|
21.
|
De gemiddelde luciferaseactiviteit voor de referentiestandaard (Ral/E2) wordt gemeten door de gemiddelde maximale RLU-waarde voor de referentiestandaard Ral/E2 te delen door de gemiddelde RLU-waarde voor de DMSO-controle. Het resultaat moet groter zijn dan drievoudig.
Aantoning van de bekwaamheid van laboratoria (zie punt 14 en de tabellen 3 en 4 onder “COMPONENTEN VAN DE ER TA-BEPALING” van deze testmethode).
|
Vehiculum
|
22.
|
De teststoffen moeten worden opgelost in een oplosmiddel dat de teststof oplosbaar maakt en mengbaar is met het celmedium. Geschikte vehicula zijn water, ethanol (95 % tot 100 % zuiverheid) en DMSO. Als DMSO wordt gebruikt, mag het gehalte niet hoger zijn dan 1 % (v/v). Voor elk vehiculum moet worden aangetoond dat het maximale gebruikte volume niet cytotoxisch is en de prestaties van de bepaling niet verstoort. De referentiestandaarden en controles worden eerst opgelost in 100 % oplosmiddel en vervolgens verdund tot de gewenste concentraties in EFM.
|
Voorbereiding van de teststoffen
|
23.
|
De teststoffen worden eerst opgelost in 100 % DMSO (of geschikt oplosmiddel) en vervolgens verdund tot de gewenste concentraties in EFM. Alvorens te worden opgelost en verdund moeten alle teststoffen de gelegenheid krijgen om te equilibreren op kamertemperatuur. De teststofoplossingen moeten voor elk experiment vers bereid worden. Er mag geen neerslag of troebelheid in zichtbaar zijn. Stamoplossingen van referentiestandaarden en controles mogen wel in bulk worden bereid, maar de uiteindelijke referentiestandaard, controleverdunningen en teststoffen moeten voor elk experiment vers worden bereid en binnen 24 uur na bereiding worden gebruikt.
|
Oplosbaarheid en cytotoxiciteit: overwegingen voor bereikbepaling
|
24.
|
De bereikbepalingstests bestaat uit een 1:10 verdunningsreeks van zeven concentraties in duplo. Eerst worden de teststoffen getest tot de maximumconcentratie van 1 mg/ml (~1 mM) voor agonisme en 20 μg/ml (~10 μM) voor antagonisme. De bereikbepalingsexperimenten worden gebruikt voor het bepalen van:
|
|
—
|
de beginconcentraties van de teststof die in de integrale test gebruikt zullen worden;
|
|
—
|
de verdunningen (1:2 of 1:5) van de teststof die in de integrale test gebruikt zullen worden.
|
|
25.
|
In de protocollen voor de agonismebepaling en de antagonismebepaling (7) is een beoordeling opgenomen van de cellevensvatbaarheid en de cytotoxiciteit. Deze maakt zowel deel uit van de bereikbepalingstest als van de integrale test. De cytotoxiciteitstestmethode die werd gebruikt om de cellevensvatbaarheid te beoordelen bij de validering van de VM7Luc-ER TA-bepaling (1), was een kwalitatieve methode op basis van visuele waarneming en een beoordelingsschaal; er kan echter ook een kwantitatieve methode worden gebruikt voor het bepalen van de cytotoxiciteit (zie protocol (7)). Gegevens van teststofconcentraties die meer dan 20 % vermindering van de levensvatbaarheid veroorzaken, zijn niet bruikbaar.
|
Blootstelling aan de teststof en indeling van de testplaat
|
26.
|
De cellen worden geteld en uitgeplaat in weefselkweekplaten met 96 putjes (2 × 105 cellen per putje) in EFM en vervolgens 24 uur geïncubeerd zodat de cellen zich aan de plaat kunnen hechten. Het EFM wordt verwijderd en vervangen door test- en referentiestoffen in EFM, waarna de plaat 19-24 uur wordt geïncubeerd. Bij sterk vluchtige stoffen is bijzondere aandacht geboden omdat naburige controleputjes vals-positieve resultaten kunnen genereren. In zulke gevallen kunnen plaatsealers helpen om de afzonderlijke putjes effectief te isoleren tijdens het testen. Deze worden dan ook aanbevolen.
|
Bereikbepalingstests
|
27.
|
In de bereikbepalingstests worden alle putjes van een plaat met 96 putjes gebruikt om tot zes teststoffen te testen in een 1:10 verdunningsreeks van zeven concentraties in duplo (zie figuren 1 en 2).
|
|
—
|
In de bereikbepalingstest voor de agonismebepaling worden vier concentraties E2 in duplo als referentiestandaard gebruikt en vier duploputjes voor de DMSO-controle.
|
|
—
|
In de bereikbepalingstest voor de antagonismebepaling worden drie concentraties Ral/E2 met 9,18 × 10-11 M E2 in duplo als referentiestandaard gebruikt en drie duploputjes voor de E2- en DMSO-controles.
|
Figuur 1
Indeling van de plaat met 96 putjes voor de bereikbepalingstest voor de agonismebepaling
Afkortingen: E2-1 tot E2-4 = concentraties van de referentiestandaard E2 (van hoog tot laag); TC1-1 tot TC1-7 = concentraties (van hoog tot laag) van teststof 1 (TC1); TC2-1 tot TC2-7 = concentraties (van hoog tot laag) van teststof 2 (TC2); TC3-1 tot TC3-7 = concentraties (van hoog tot laag) van teststof 3 (TC3); TC4-1 tot TC4-7 = concentraties (van hoog tot laag) van teststof 4 (TC4); TC5-1 tot TC5-7 = concentraties (van hoog tot laag) van teststof 5 (TC5); TC6-1 tot TC6-7 = concentraties (van hoog tot laag) van teststof 6 (TC6); VC = vehiculumcontrole (DMSO [1 % v/v/ EFM]).
Figuur 2
Indeling van de plaat met 96 putjes voor de bereikbepalingstest voor de antagonismebepaling

Afkortingen: E2 = E2-controle; Ral-1 tot Ral-3 = concentraties van de referentiestandaard raloxifeen/E2 (van hoog tot laag); TC1-1 tot TC1-7 = concentraties (van hoog tot laag) van teststof 1 (TC1); TC2-1 tot TC2-7 = concentraties (van hoog tot laag) van teststof 2 (TC2); TC3-1 tot TC3-7 = concentraties (van hoog tot laag) van teststof 3 (TC3); TC4-1 tot TC4-7 = concentraties (van hoog tot laag) van teststof 4 (TC4); TC5-1 tot TC5-7 = concentraties (van hoog tot laag) van teststof 5 (TC5); TC6-1 tot TC6-7 = concentraties (van hoog tot laag) van teststof 6 (TC6); VC = vehiculumcontrole (DMSO [1 % v/v/ EFM]).
Opmerking: Alle teststoffen worden getest met E2 in een concentratie van 9,18 × 10-11 M.
|
28.
|
Aanbevolen wordt om uiteindelijk voor elk putje op een volume medium van 200 μl uit te komen. Gebruik alleen testplaten waarin de cellen in alle putjes een levensvatbaarheid van 80 % of meer laten zien.
|
|
29.
|
De bepaling van de beginconcentraties voor de integrale
agonismebepaling
wordt uitvoerig beschreven in het protocol voor deze bepaling (7). Samengevat worden de volgende criteria gebruikt:
|
|
—
|
als er op de concentratiecurve van de teststof geen punten boven het gemiddelde plus driemaal de standaarddeviatie van de DMSO-controle liggen, wordt de integrale test uitgevoerd met een 1:2 verdunningsreeks van 11 concentraties beginnend bij de maximaal oplosbare concentratie;
|
|
—
|
als er op de concentratiecurve van de teststof wel punten boven het gemiddelde plus driemaal de standaarddeviatie van de DMSO-controle liggen, dan moet de beginconcentratie voor de verdunningsreeks van 11 concentraties in de integrale test één log hoger zijn dan de concentratie met de hoogste gecorrigeerde RLU-waarde in de bereikbepalingstest. Voor de 11 concentraties wordt hetzij een 1:2 hetzij een 1:5 verdunningsreeks gekozen, afhankelijk van de volgende criteria:
|
er moet een 1:2 verdunningsreeks van 11 concentraties worden gebruikt als het aldus verkregen concentratiebereik het volledige responsbereik op basis van de in de bereikbepalingstest gegenereerde concentratie-responscurve beslaat. Anders moet een 1:5 verdunningsreeks worden gebruikt;
|
|
|
—
|
als een teststof in de bereikbepalingstest een tweefasige concentratie-responscurve laat zien, moeten beide fasen ook in de integrale test bestudeerd worden.
|
|
30.
|
De bepaling van de beginconcentraties voor de integrale
antagonismebepaling
wordt uitvoerig beschreven in het protocol voor deze bepaling (7). Samengevat worden de volgende criteria gebruikt:
|
|
—
|
als er op de concentratiecurve van de teststof geen punten onder het gemiddelde minus driemaal de standaarddeviatie van de E2-controle liggen, wordt de integrale test uitgevoerd met een 1:2 verdunningsreeks van 11 concentraties beginnend bij de maximaal oplosbare concentratie;
|
|
—
|
als er op de concentratiecurve van de teststof wel punten onder het gemiddelde minus driemaal de standaarddeviatie van de E2-controle liggen, dan moet voor de verdunningsreeks van 11 concentraties in de integrale test een van de volgende beginconcentraties worden gebruikt:
|
—
|
de concentratie met de laagste gecorrigeerde RLU-waarde in de bereikbepalingstest;
|
|
—
|
de maximaal oplosbare concentratie (zie antagonismeprotocol (7), figuur 14-2);
|
|
—
|
de laagste cytotoxische concentratie (zie antagonismeprotocol (7), figuur 14-3 voor een gerelateerd voorbeeld);
|
|
|
—
|
voor de 11 concentraties wordt hetzij een 1:2 hetzij een 1:5 verdunningsreeks gekozen, afhankelijk van de volgende criteria:
|
er moet een 1:2 verdunningsreeks van 11 concentraties worden gebruikt als het aldus verkregen concentratiebereik het volledige responsbereik op basis van de in de bereikbepalingstest gegenereerde concentratie-responscurve beslaat. Anders moet een 1:5 verdunningsreeks worden gebruikt.
|
|
Integrale tests
|
31.
|
De integrale test bestaat uit verdunningsreeksen van 11 concentraties (hetzij 1:2, hetzij 1:5, naargelang van de beginconcentratie op basis van de criteria voor de integrale test), die elk worden getest in drie putjes van de plaat met 96 putjes (zie figuren 3 en 4).
|
|
—
|
Voor de integrale agonismebepaling worden 11 concentraties van E2 in duplo als referentiestandaard gebruikt. In elke plaat worden vier duploputjes voor de DMSO-controle en vier duploputjes voor de methoxychloorcontrole (9,06 × 10-6 M) opgenomen.
|
|
—
|
Voor de integrale antagonismebepaling worden negen concentraties Ral/E2 met 9,18 × 10-11 M E2 in duplo als referentiestandaard gebruikt, met vier duploputjes voor de 9,18 x 10-11 M E2-controle, vier duploputjes voor de DMSO-controles, en vier duploputjes voor 3,36 × 10-6M tamoxifen.
|
Figuur 3
Indeling van de plaat met 96 putjes voor de integrale agonismebepaling

Afkortingen: TC1-1 tot TC1-11 = concentraties (van hoog tot laag) van teststof 1; TC2-1 tot TC2-11 = concentraties (van hoog tot laag) van teststof 2; E2-1 tot E2-11 = concentraties van de referentiestandaard E2 (van hoog tot laag); Meth = zwak positieve controle met p,p’-methoxychloor; VC = vehiculumcontrole (DMSO [1 % v/v/ EFM])
Figuur 4
Indeling van de plaat met 96 putjes voor de integrale antagonismebepaling

Afkortingen: E2 = E2-controle; Ral-1 tot Ral-9 = concentraties van de referentiestandaard raloxifeen/E2 (van hoog tot laag); Tam = zak-positieve controle met tamoxifen/E2; TC1-1 tot TC1-11 = concentraties (van hoog tot laag) van teststof 1 (TC1); TC2-1 tot TC2-11 = concentraties (van hoog tot laag) van teststof 2 (TC2); VC = vehiculumcontrole (DMSO [1 % v/v/ EFM]).
Opmerking: zoals aangegeven bevatten alle referentie- en testputjes een vaste concentratie E2 (9,18 × 10-11M)
|
32.
|
Met het oog op de onafhankelijkheid van de resultaten moeten de herhalingen van de integrale tests voor dezelfde chemische stof op een andere dag worden uitgevoerd. Er moeten ten minste twee integrale tests worden uitgevoerd. Als de testresultaten niet overeenstemmen (d.w.z. de ene test is positief, de andere negatief) of als een van de tests niet aanvaardbaar is, moet een extra derde test worden uitgevoerd.
|
Luminescentiemeting
|
33.
|
De luminescentie wordt gemeten in een bereik van 300 tot 650 nm met behulp van een luminometer met injector en met software die het injectievolume en het meetinterval aanstuurt (7). De lichtemissie uit elk putje wordt uitgedrukt in RLU per putje.
|
ANALYSE VAN DE GEGEVENS
Bepaling van de EC50 en de IC50
|
34.
|
De EC50-waarde (effectieve-concentratiemediaan van een teststof [agonisme]) en de IC50-waarde (de concentratie van een teststof die 50 % remming veroorzaakt [antagonisme]) worden bepaald op basis van de concentratie-responsgegevens. Voor teststoffen die bij een of meer concentraties positief zijn, wordt de concentratie van de teststof die 50 % respons veroorzaakt, (IC50 of EC50) berekend aan de hand van een Hill-functieanalyse of een geschikte alternatieve methode. De Hill-functie is een logistisch wiskundig model met vier parameters die het verband uitdrukt tussen de concentratie van de teststof en de respons (doorgaans in de vorm van een sigmoïdale curve) volgens de volgende vergelijking:
|

Waarbij:
Y= respons (d.w.z. RLU’s);
X= de logaritme van de concentratie;
Basis= minimale respons;
Top= maximale respons;
lg EC50 (of lg IC50)= de logaritme van X als de respons halverwege tussen de Top en de Basis;
Hill-coëfficiënt= helling van de curve.
Het model berekent de beste fit voor de parameters Top, Basis, Hill-coëfficiënt, IC50 en EC50. Voor de berekening van de EC50 en de IC50 moet geschikte statistische software worden gebruikt (bv. Graphpad PrismR).
Bepaling van uitschieters
|
35.
|
Een goede statistische beoordeling zou kunnen worden bevorderd door (onder andere) de Q-test (zie agonisme- en antagonismeprotocollen (7) voor de bepaling van “onbruikbare” putjes die van de gegevensanalyse worden uitgesloten).
|
|
36.
|
Voor de duplo’s van de referentiestandaard E2 (steekproefgrootte twee) wordt elke gecorrigeerde ELU-waarde voor een duplo bij een bepaalde E2-concentratie als uitschieter beschouwd als die meer dan 20 % afwijkt van de gecorrigeerde RLU-waarde voor die concentratie in de databank met historische gegevens.
|
Verzameling en correctie van luminometergegevens voor bereikbepaling
|
37.
|
De ruwe gegevens van de luminometer moeten in een voor de bepaling bestemde spreadsheetsjabloon worden gezet. Vervolgens moet worden vastgesteld of er onder de gegevenspunten uitschieters zijn die verwijderd moeten worden. (Zie aanvaardbaarheidscriteria voor de parameters die in de analyses worden verkregen.) De volgende berekeningen moeten worden uitgevoerd:
|
Agonismebepaling
|
Stap 1
|
Bereken de gemiddelde waarde voor de DMSO-vehiculumcontrole (VC).
|
|
Stap 2
|
Trek de gemiddelde waarde voor de DMSO-VC af van de waarde voor elk putje om de gegevens te normaliseren.
|
|
Stap 3
|
Bereken de gemiddelde x-voudige inductie voor de referentiestandaard (E2).
|
|
Stap 4
|
Bereken de gemiddelde EC50-waarde voor de teststoffen.
|
Antagonismebepaling
|
Stap 1
|
Bereken de gemiddelde waarde voor de DMSO-VC.
|
|
Stap 2
|
Trek de gemiddelde waarde voor de DMSO-VC af van de waarde voor elk putje om de gegevens te normaliseren.
|
|
Stap 3
|
Bereken de gemiddelde x-voudige remming voor de referentiestandaard (Ral/E2).
|
|
Stap 4
|
Bereken de gemiddelde waarde voor de referentiestandaard E2.
|
|
Stap 5
|
Bereken de gemiddelde IC50-waarde voor de teststoffen.
|
Verzameling en correctie van luminometergegevens voor de integrale test
|
38.
|
De ruwe gegevens van de luminometer moeten in een voor de bepaling bestemde spreadsheetsjabloon worden gezet. Vervolgens moet worden vastgesteld of er onder de gegevenspunten uitschieters zijn die verwijderd moeten worden. (Zie aanvaardbaarheidscriteria voor de parameters die in de analyses worden verkregen.) De volgende berekeningen worden uitgevoerd:
|
Agonismebepaling
|
Stap 1
|
Bereken de gemiddelde waarde voor de DMSO-VC.
|
|
Stap 2
|
Trek de gemiddelde waarde voor de DMSO-VC af van de waarde voor elk putje om de gegevens te normaliseren.
|
|
Stap 3
|
Bereken de gemiddelde x-voudige inductie voor de referentiestandaard (E2).
|
|
Stap 4
|
Bereken de gemiddelde EC50-waarde voor E2 en de teststoffen.
|
|
Stap 5
|
Bereken de gemiddelde gecorrigeerde RLU-waarde voor methoxychloor.
|
Antagonismebepaling
|
Stap 1
|
Bereken de gemiddelde waarde voor de DMSO-VC.
|
|
Stap 2
|
Trek de gemiddelde waarde voor de DMSO-VC af van de waarde voor elk putje om de gegevens te normaliseren.
|
|
Stap 3
|
Bereken de gemiddelde x-voudige inductie voor de referentiestandaard (Ral/E2).
|
|
Stap 4
|
Bereken de gemiddelde IC50-waarde voor Ral/E2 en de teststoffen.
|
|
Stap 5
|
Bereken de gemiddelde gecorrigeerde RLU-waarde voor tamoxifen.
|
|
Stap 6
|
Bereken de gemiddelde waarde voor de referentiestandaard E2.
|
Criteria voor gegevensinterpretatie
|
39.
|
De VM7Luc-ER TA-bepaling is bedoeld als onderdeel van een benadering met meervoudige bewijsvoering (“weight of evidence”-benadering) als hulpmiddel bij de prioritering van stoffen voor het testen op hormoonverstorende effecten in vivo. Als onderdeel van deze prioriteringsprocedure wordt de teststof ingedeeld als positief of negatief voor ER-agonistische of ER-antagonistische activiteit. De beslissingscriteria voor positieve of negatieve resultaten die zijn gebruikt in de valideringsstudie voor de VM7Luc-ER TA-bepaling, worden beschreven in tabel 1.
|
Tabel 1
Beslissingscriteria voor positieve en negatieve resultaten
|
|
|
AGONISTISCHE ACTIVITEIT
|
|
Positief
|
|
—
|
Alle teststoffen die als positief voor ER-agonistische activiteit worden ingedeeld, moeten een concentratie-responscurve hebben die bestaat uit een basislijn, gevolgd door een positieve helling en eindigend in een plateau of top. In sommige gevallen kunnen slechts twee van deze kenmerken (baseline-helling of helling-top) worden gedefinieerd.
|
|
—
|
De lijn die de positieve helling definieert, moet ten minste drie punten bevatten waarvan de foutbalken (gemiddelde ± SD) niet overlappen. De punten die de baseline vormen, worden hiervan uitgesloten, maar de top of het eerste punt van het plateau mag wel in het lineaire gedeelte van de curve worden meegenomen.
|
|
—
|
Een positieve indeling vereist een responsamplitude (het verschil tussen basislijn en top) van ten minste 20 % van de maximale waarde voor de referentiestandaard E2 (d.w.z. 2 000 RLU’s of meer wanneer de maximale responswaarde van de referentiestandaard [E2] wordt gecorrigeerd naar 10 000 RLU’s).
|
|
—
|
Zo mogelijk moet voor elke positieve teststof een EC50 worden berekend.
|
|
|
Negatief
|
De gemiddelde gecorrigeerde RLU voor een gegeven concentratie ligt op of onder de gemiddelde RLU-waarde voor de DMSO-controle plus driemaal de standaarddeviatie daarvan.
|
|
Ontoereikend
|
Gegevens die vanwege belangrijke kwalitatieve of kwantitatieve beperkingen niet als geldig kunnen worden geïnterpreteerd voor het vertonen van de aan- of afwezigheid van activiteit, worden beschouwd als ontoereikend en kunnen niet worden gebruikt om te bepalen of de teststof positief of negatief is. De betreffende chemische stoffen moeten opnieuw getest worden.
|
|
|
|
ANTAGONISTISCHE ACTIVITEIT
|
|
Positief
|
|
—
|
De gegevens over de teststof leveren een concentratie-responscurve op die bestaat uit een basislijn, gevolgd door een negatieve helling.
|
|
—
|
De lijn die de negatieve helling definieert, moet ten minste drie punten bevatten waarvan de foutbalken niet overlappen; de punten die de baseline vormen, worden hiervan uitgesloten, maar het eerste punt van het plateau mag wel in het lineaire gedeelte van de curve worden meegenomen.
|
|
—
|
Er moet sprake zijn van een afname in activiteit van minimaal 20 % ten opzichte van de maximale waarde voor de referentiestandaard Ral/E2 (d.w.z. 8 000 RLU’s of minder wanneer de maximale responswaarde van de referentiestandaard [Ral/E2] wordt gecorrigeerd naar 10 000 RLU’s).
|
|
—
|
De hoogste niet-toxische concentraties van de teststof moeten lager zijn dan of gelijk aan 1 × 10-5 M.
|
|
—
|
Zo mogelijk moet voor elke positieve teststof een IC50 worden berekend.
|
|
|
Negatief
|
Alle gegevenspunten liggen boven de EC80-waarde (80 % van de respons voor E2 of 8 000 RLU’s), op concentraties van minder dan 1,0 x 10-5 M.
|
|
Ontoereikend
|
Gegevens die vanwege belangrijke kwalitatieve of kwantitatieve beperkingen niet als geldig kunnen worden geïnterpreteerd voor het vertonen van de aan- of afwezigheid van activiteit, worden beschouwd als ontoereikend en kunnen niet worden gebruikt om te bepalen of de teststof positief of negatief is. De betreffende chemische stof moet opnieuw getest worden.
|
|
40.
|
Positieve resultaten worden, voor zover mogelijk, gekarakteriseerd door zowel de grootte van het effect als de concentratie waarbij het effect optreedt. In figuur 5 en figuur 6 staan voorbeelden van positieve, negatieve en ontoereikende gegevens.
|
Figuur 5
Voorbeelden agonisme: positieve, negatieve en ontoereikende gegevens

De stippellijn geeft 20 % van de respons voor E2 aan, d.w.z. 2 000 gecorrigeerde en genormaliseerde RLU’s.
Figuur 6
Voorbeelden antagonisme: positieve, negatieve en ontoereikende gegevens

De stippellijn geeft 80 % van de respons voor Ral/E2 aan, d.w.z. 8 000 gecorrigeerde en genormaliseerde RLU’s.
De verticale lijn geeft 1,00 x 10-5 M aan. Om als positief te worden beschouwd moet een respons bij concentraties van minder dan 1,00 x 10-5 M onder de lijn voor 8 000 RLU’s liggen.
In de grafiek voor meso-hexestrol staan de concentraties met een asterisk voor levensvatbaarheidsscores van “2” of hoger.
De testresultaten voor meso-hexestrol worden als ontoereikende gegevens beschouwd omdat de enige respons die onder de 8 000 RLU’s ligt, optreedt bij 1,00 x 10-5 M.
|
41.
|
De EC50 en de IC50 kunnen worden berekend met behulp van een Hill-functie met vier parameters (zie voor meer bijzonderheden het agonismeprotocol en het antagonismeprotocol (7)). Als aan de aanvaardbaarheidscriteria wordt voldaan, betekent dit weliswaar dat het systeem naar behoren functioneert, maar wil dit nog niet zeggen dat een bepaalde testrun kloppende gegevens produceert. Duplicatie van de resultaten van de eerste testrun is de beste garantie dat de geproduceerde gegevens kloppen (zie punt 19 van “COMPONENTEN VAN DE ER TA-BEPALING”.
|
TESTVERSLAG
|
42.
|
Zie punt 20 van “COMPONENTEN VAN DE ER TA-BEPALING”.
|
LITERATUUR
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Aanhangsel 4
TRANSACTIVATIEBEPALING OP BASIS VAN STABIEL GETRANSFECTEERDE HUMANE OESTROGEENRECEPTOR-Α VOOR HET OPSPOREN VAN OESTROGEENAGONISTISCHE EN -ANTAGONISTISCHE ACTIVITEIT VAN CHEMISCHE STOFFEN, WAARBIJ GEBRUIKGEMAAKT WORDT VAN DE CELLIJN ERΑ CALUX
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN (ZIE OOK ALGEMENE INLEIDING)
|
1.
|
Voor de ERα CALUX-transactivatiebepaling wordt gebruikgemaakt van de humane cellijn U2OS voor het opsporen van oestrogeenagonistische en -antagonistische activiteit gemedieerd door de humane oestrogeenreceptor-alfa (hERα). In de valideringsstudie voor de stabiel getransfecteerde ERα CALUX-bioassay door BioDetection Systems BV (Amsterdam, Nederland) werden de relevantie en betrouwbaarheid van de bepaling voor het beoogde doel aangetoond (1). De cellijn ERα CALUX brengt enkel de stabiel getransfecteerde humane ERα tot expressie (2) (3).
|
|
2.
|
Deze bepaling is speciaal ontworpen voor het opsporen van hERα-gemedieerde transactivatie door meting van bioluminescentie als eindpunt. In bioassays wordt veel gebruikgemaakt van bioluminescentie vanwege de hoge signaal-ruisverhouding (4).
|
|
3.
|
Van fyto-oestrogeenconcentraties hoger dan 1 μM is echter overactivatie van het luciferasereportergen gerapporteerd, wat leidt tot niet-receptor-gemedieerde luminescentie (5) (6) (7). Bij transactivatiebepalingen op basis van stabiel getransfecteerde ER moeten hogere concentraties van fyto-oestrogenen of andere, vergelijkbare verbindingen die de expressie van luciferase kunnen overactiveren, dan ook zorgvuldig worden bestudeerd (zie aanhangsel 2).
|
|
4.
|
Alvorens deze bepaling te gebruiken met het oog op regelgeving, moeten de „ALGEMENE INLEIDING” en „COMPONENTEN VAN DE ER TA-BEPALING” worden geraadpleegd. De in deze testmethode gebruikte definities en afkortingen staan in aanhangsel 1.
|
PRINCIPE VAN DE BEPALING (ZIE OOK DE ALGEMENE INLEIDING)
|
5.
|
De bioassay wordt gebruikt voor het beoordelen van de ER-ligandbinding en de daarop volgende translocatie van het receptor-ligandcomplex naar de nucleus. In de nucleus bindt het receptor-ligandcomplex aan specifieke DNA-responselementen en transactiveert het een vuurvlieg-luciferasereportergen, wat leidt tot een verhoogde cellulaire expressie van het luciferase-enzym. Na toevoeging van luciferine, het substraat van luciferase, wordt dit luciferine omgezet in een bioluminescent product. Het vrijkomende licht kan gemakkelijk worden gedetecteerd en gekwantificeerd met behulp van een luminometer.
|
|
6.
|
In dit testsysteem worden stabiel getransfecteerde ERα CALUX-cellen gebruikt. ERα CALUX-cellen zijn afgeleid van de humane osteoblastische osteosarcoomcellijn U2OS. Humane U2OS-cellen werden stabiel getransfecteerd met 3xHRE-TATA-Luc en pSG5-neo-hERα volgens de calciumfosfaat-coprecipitatiemethode. De cellijn U2OS werd als beste keuze geselecteerd om als op oestrogenen (en andere steroïde hormonen) reagerende reportercellijn te dienen op basis van de waarneming dat de cellijn U2OS nauwelijks tot geen endogene receptoractiviteit vertoonde. Het ontbreken van endogene receptoren werd vastgesteld met alleen luciferasereporterplasmiden: na toevoeging van receptorliganden werd geen enkele activiteit waargenomen. Bovendien ondersteunde deze cellijn sterke hormoon-gemedieerde responsen bij tijdelijke introductie van verwante receptoren(2) (3) (8).
|
|
7.
|
Voor het testen van chemische stoffen op oestrogene of anti-oestrogene activiteit met behulp van de cellijn ERα CALUX wordt eerst een screeningstestrun uitgevoerd, gevolgd door de integrale testruns. In de screeningstestrun worden de oplosbaarheid, de cytotoxiciteit en een beperkt concentratiebereik van de teststoffen bepaald ten behoeve van de integrale testruns. In de integrale tests worden de beperkte concentratiebereiken van de teststoffen getest in de ERα CALUX-bioassays, waarna de teststoffen worden ingedeeld op agonisme of antagonisme.
|
|
8.
|
De criteria voor gegevensinterpretatie worden nader beschreven in punt 59. Samenvattend wordt een teststof als positief beoordeeld voor agonisme wanneer ten minste twee opeenvolgende concentraties van de teststof een respons opleveren die gelijk is aan of sterker dan 10 % van de maximale respons van de referentiestandaard 17β-oestradiol (PC10). Een teststof wordt als positief beoordeeld voor antagonisme wanneer ten minste twee opeenvolgende concentraties van de teststof een respons opleveren die gelijk is aan of zwakker dan 80 % van de maximale respons van de referentiestandaard tamoxifen (PC80).
|
PROCEDURE
Cellijnen
|
9.
|
Voor de bepaling moet de stabiel getransfecteerde cellijn U2OS-ERα CALUX worden gebruikt. Deze cellijn is verkrijgbaar met een technische licentieovereenkomst bij Detection Systems BV, Amsterdam, Nederland.
|
|
10.
|
Er mogen alleen mycoplasmavrije celculturen worden gebruikt. De gebruikte celpartijen moeten hetzij gecertificeerd negatief zijn voor verontreiniging met mycoplasma of er moet vóór gebruik een mycoplasmatest worden uitgevoerd. Voor een gevoelige detectie van mycoplasma-infectie moet de methode PT-PCR (Real Time Polymerase Chain Reaction) worden gebruikt (9).
|
Stabiliteit van de cellijn
|
11.
|
Om de stabiliteit en de integriteit van de CALUX-cellen te behouden, moeten de cellen worden bewaard in vloeibare stikstof (–80 °C). Nadat ze zijn ontdooid om een nieuwe kweek te beginnen, moeten de cellen ten minste tweemaal worden overgeënt alvorens te worden gebruikt ter beoordeling van de oestrogeenagonistische en -antagonistische activiteit van chemische stoffen. De cellen mogen niet meer dan 30 passages worden overgeënt.
|
|
12.
|
Om de stabiliteit van de cellijn in de loop van de tijd te controleren moeten de responsiviteit van de agonistische en antagonistische testsystemen worden geverifieerd door de EC50 of IC50 van de referentiestandaard te beoordelen. Bovendien moet de relatieve inductie van de positieve controle (PC) en de negatieve controle (NC) worden gemonitord. De resultaten moeten overeenkomen met de aanvaardbaarheidscriteria voor de agonistische (tabel 3C) of antagonistische ERα CALUX-bioassay (tabel 4C). De referentiestandaarden, positieve controles en negatieve controles worden gegeven in tabel 1 voor agonisme en tabel 2 voor antagonisme.
|
Omstandigheden voor het kweken en uitplaten van de cellen
|
13.
|
De U2OS-cellen moeten worden gekweekt in groeimedium (1:1 DMEM/F12-medium met fenolrood als pH-indicator, aangevuld met 7,5 % foetaal runderserum, 1 % niet-essentiële aminozuren, 10 eenheden/ml penicilline, streptomycine en geneticine (G-418) als selectiemarker. De cellen worden in een CO2-incubator (5 % CO2) geplaatst bij 37 °C en 100 % vochtigheid. Wanneer de cellen 85-95 % confluentie bereiken, moeten ze hetzij worden overgeënt, hetzij worden voorbereid om in mictotiterplaten met 96 putjes te worden geënt. In het tweede geval worden de cellen geresuspendeerd in een concentratie van 1 × 105 cellen/ml in oestrogeenvrij medium (1:1 DMEM/F12-medium zonder fenolrood, met met dextraan beklede kool behandeld foetaal runderserum (5 % v/v), niet-essentiële aminozuren (1 % v/v), 10 eenheden/ml penicilline en streptomycine) en uitgeplaat in de putjes van microtiterplaten met 96 putjes (100 μl gehomogeniseerde celsuspensie). Alvorens te worden blootgesteld worden de cellen 24 uur gepre-incubeerd in een CO2-incubator (5 % CO2, 37 °C, 100 % vochtigheid). Het kunststof laboratoriumgerei moet oestrogeenvrij zijn.
|
Aanvaardbaarheidscriteria
|
14.
|
De agonistische activiteit en antagonistische activiteit van de teststof(fen) worden getest in testreeksen. Een testreeks bestaat uit maximaal 6 microtiterplaten. Elke testreeks omvat ten minste 1 volledige verdunningsreeks van een referentiestandaard, een positieve controle, een negatieve controle en oplosmiddelcontroles. In figuur 1 en figuur 2 wordt de plaatindeling weergegeven voor agonistische en antagonistische testreeksen.
|
|
15.
|
Elke verdunning van de referentiestandaarden, teststoffen, alle oplosmiddelcontroles en de positieve en negatieve controles moeten in drievoud worden geanalyseerd. Elke drievoudige analyse moet aan de eisen in tabel 3A en tabel 4A voldoen.
|
|
16.
|
In elke testreeks wordt in de eerste plaat een volledige verdunningsreeks van de referentiestandaard (17β-oestradiol voor agonisme; tamoxifen voor antagonisme) gemeten. Om te zorgen dat de analyseresultaten van de overige 5 microtiterplaten kunnen worden vergeleken met de eerste microtiterplaat, die de volledige concentratie-responscurve van de referentiestandaard bevat, moeten alle platen 3 controles bevatten: oplosmiddelcontrole, de hoogste geteste concentratie van de referentiestandaard en de concentratie van de referentiestandaard die de EC50 (agonisme) of de IC50 (antagonisme) benadert. De verhouding tussen de controlegemiddelden in de eerste plaat en de overige 5 platen moet aan de eisen in tabel 3C (agonisme) of tabel 4C (antagonisme) voldoen.
|
|
17.
|
Voor elk van de microtiterplaten in een testreeks wordt de z-factor berekend (10). De z-factor moet worden berekend aan de hand van de responsen bij de hoogste en de laatste concentratie van de referentiestandaard. Een microtiterplaat wordt als valide beschouwd als hij aan de eisen in tabel 3C (agonisme) of tabel 4C (antagonisme) voldoet.
|
|
18.
|
De referentiestandaard moet een sigmoïdale dosis-responscurve laten zien. De EC50 of IC50 die wordt afgeleid uit de respons van de verdunningsreeks van de referentiestandaard, moet aan de eisen in tabel 3C (agonisme) of tabel 4C (antagonisme) voldoen.
|
|
19.
|
In elke testreeks moet een positieve en een negatieve controle worden opgenomen. De berekende relatieve inductie van zowel de positieve als de negatieve controle moet aan de eisen in tabel 3C (agonisme) of tabel 4C (antagonisme) voldoen.
|
|
20.
|
Tijdens alle metingen moet de inductiefactor van de hoogste concentratie van de referentiestandaard worden gemeten door de gemiddelde hoogste respons voor de referentiestandaard 17β-oestradiol in relatieve lichteenheden (RLU) te delen door de gemiddelde RLU-respons voor de referentie-oplosmiddelcontrole. Deze inductiefactor moet voldoen aan de minimumeisen voor de x-voudige inductie in tabel 3C (agonisme) of tabel 4C (antagonisme).
|
|
21.
|
Alleen microtiterplaten die aan alle bovengenoemde aanvaardbaarheidscriteria voldoen, worden als valide beschouwd en mogen worden gebruikt om de respons van de teststoffen te beoordelen.
|
|
22.
|
De aanvaardbaarheidscriteria gelden voor zowel de screeningstest als de integrale testruns.
|
Tabel 1
Concentraties van de referentiestandaard, PC en NC voor de agonistische CALUX-bioassay
|
|
Stof
|
CAS RN
|
Testbereik (M)
|
|
Referentiestandaard
|
17β-oestradiol
|
50-28-2
|
1 × 10-13 - 1 × 10-10
|
|
Positieve controle (PC)
|
17α-methyltestosteron
|
58-18-4
|
3 × 10-6
|
|
Negatieve controle (NC)
|
corticosteron
|
50-22-6
|
1 × 10-8
|
Tabel 2
Concentraties van de referentiestandaard, positieve controle (PC) en negatieve controle (NC) voor de antagonistische CALUX-bioassay
|
|
Stof
|
CAS RN
|
Testbereik (M)
|
|
Referentiestandaard
|
tamoxifen
|
10540-29-1
|
3 × 10-9 - 1 × 10-5
|
|
Positieve controle (PC)
|
4-hydroxytamoxifen
|
68047-06-3
|
1 × 10-9
|
|
Negatieve controle (NC)
|
resveratrol
|
501-36-0
|
1 × 10-5
|
Tabel 3
Aanvaardbaarheidscriteria voor de agonistische ERα CALUX bioassay
|
A – afzonderlijke monsters in een plaat
|
Criterium
|
|
1
|
Maximale %SD voor putjes in drievoud (voor NC, PC, elke verdunning van de teststof en de referentiestandaard, behalve C0)
|
< 15 %
|
|
2
|
Maximale %SD voor putjes in drievoud (voor referentiestandaard en controles met het oplosmiddel voor de teststof (C0, SC))
|
< 30 %
|
|
3
|
Maximale LDH-lekkage, als maat voor de cytotoxiciteit.
|
< 120 %
|
|
B – binnen een enkele microtiterplaat
|
|
|
4
|
Verhouding tussen de controle met het oplosmiddel van de referentiestandaard (C0; plaat 1) en de controle met het oplosmiddel van de teststof (SC; platen 2 tot x)
|
0,5 tot 2,0
|
|
5
|
Verhouding tussen de EC50 bij benadering en de hoogste concentraties van de referentiestandaard in plaat 1 enerzijds en de EC50 bij benadering en de hoogste concentraties van de referentiestandaard in plaat 2 tot x (C4, C8) anderzijds
|
0,70 tot 1,30
|
|
6
|
Z-factor voor elke plaat
|
> 0,6
|
|
C – binnen een afzonderlijke reeks analyses (alle platen in één reeks)
|
|
|
7
|
Sigmoïdale curve van de referentiestandaard
|
Ja (17β-oestradiol)
|
|
8
|
EC50-bereik referentiestandaard 17β-oestradiol
|
4 × 10-12 - 4 × 10-11 M
|
|
9
|
Minimale x-voudige inductie van de hoogste 17β-oestradiolconcentratie ten opzichte van de controle met het oplosmiddel van de referentiestandaard.
|
5
|
|
10
|
Relatieve inductie (%) PC
|
> 30 %
|
|
11
|
Relatieve inductie (%) NC
|
< 10 %
|
PC: positieve controle; NC: negatieve controle; SC: controle met het oplosmiddel van de teststof; C0: controle met het oplosmiddel van de referentiestandaard; SD: standaarddeviatie; LDH: lactaatdehydrogenase
Tabel 4
Aanvaardbaarheidscriteria voor de antagonistische ERα CALUX bioassay
|
A – afzonderlijke monsters in een plaat
|
Criterium
|
|
1
|
Maximale %SD voor putjes in drievoud (voor NC, PC, elke verdunning van de teststof, de referentiestandaard, en de oplosmiddelcontrole (C0))
|
< 15 %
|
|
2
|
Maximale %SD van de putjes in drievoud (voor VC en hoogste concentratie van de referentiestandaard (C8))
|
< 30 %
|
|
3
|
Maximale LDH-lekkage, als maat voor de cytotoxiciteit.
|
< 120 %
|
|
B – binnen een enkele microtiterplaat
|
|
|
4
|
Verhouding tussen de controle met het oplosmiddel van de referentiestandaard (C0; plaat 1) en de controle met het oplosmiddel van de teststof (SC; platen 2 tot x)
|
0,70 tot 1,30
|
|
5
|
Verhouding tussen de IC50 bij benadering van de referentiestandaard in plaat 1 en de IC50 bij benadering van de referentiestandaard in plaat 2 tot x (C4)
|
0,70 tot 1,30
|
|
6
|
Verhouding van de hoogste concentraties van de referentiestandaard in plaat 1 en de hoogste concentraties van de referentiestandaard in plaat 2 tot x (C8)
|
0,50 tot 2,0
|
|
7
|
Z-factor voor elke plaat
|
> 0,6
|
|
C – binnen een afzonderlijke reeks analyses (alle platen in één reeks)
|
|
|
8
|
Sigmoïdale curve van de referentiestandaard
|
Ja (tamoxifen)
|
|
9
|
IC50-bereik referentiestandaard (tamoxifen)
|
1 × 10-8 - 1 × 10-7 M
|
|
10
|
Minimale x-voudige inductie van de controle met het oplosmiddel van de referentiestandaard ten opzichte van de hoogste concentratie tamoxifen.
|
2,5
|
|
11
|
Relatieve inductie (%) PC
|
< 70 %
|
|
12
|
Relatieve inductie (%) NC
|
> 85 %
|
PC: positieve controle; NC: negatieve controle; VC: vehiculumcontrole (oplosmiddelcontrole zonder vaste concentratie van de agonistische referentiestandaard); SC: controle met het oplosmiddel van de teststof; C0: controle met het oplosmiddel van de referentiestandaard; SD: standaarddeviatie; LDH: lactaatdehydrogenase
Oplosmiddel-/vehiculumcontrole, referentiestandaarden, positieve controles, negatieve controles
|
23.
|
Voor de screeningstestrun en de integrale testruns moeten dezelfde oplosmiddel-/ vehiculumcontrole, referentiestandaarden, positieve controles en negatieve controles worden gebruikt. Bovendien moet de concentratie van de referentiestandaarden, positieve controles en negatieve controles gelijk zijn.
|
Oplosmiddelcontrole
|
24.
|
Het oplosmiddel dat wordt gebruikt voor het oplossen van de teststoffen moet worden getest als oplosmiddelcontrole. Tijdens de validatie van de ERα CALUX-bioassay werd dimethylsulfoxide (DMSO, 1 % (v/v); CAS RN 67-68-5) als vehiculum gebruikt. Als een ander oplosmiddel dan DMSO wordt gebruikt, moeten alle referentiestandaarden, controles en teststoffen in hetzelfde vehiculum worden getest. Er zij op gewezen dat de oplosmiddelcontrole voor antagonisme-onderzoeken een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (bij benadering de EC50-concentratie) bevat. Om het oplosmiddel voor antagonisme-onderzoeken te testen moet een vehiculumcontrole worden opgezet en getest.
|
Vehiculumcontrole (antagonisme)
|
25.
|
Voor antagonismebepalingen wordt aan het testmedium een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (bij benadering de EC50-concentratie) toegevoegd. Om het oplosmiddel dat wordt gebruikt voor het oplossen van de teststoffen in een antagonismebepaling te testen, moet een testmedium zonder vaste concentratie van de agonistische referentiestandaard 17β-oestradiol worden bereid. Deze controle wordt aangeduid als de vehiculumcontrole. Tijdens de validatie van de ERα CALUX-bioassay werd dimethylsulfoxide (DMSO, 1 % (v/v); CAS RN 67-68-5) als vehiculum gebruikt. Als een ander oplosmiddel dan DMSO wordt gebruikt, moeten alle referentiestandaarden, controles en teststoffen in hetzelfde vehiculum worden getest.
|
Referentienormen
|
26.
|
De referentiestandaard voor de agonismebepaling is 17β-oestradiol (tabel 1). De referentiestandaarden bestaan uit een verdunningsreeks van acht concentraties 17β-oestradiol (1 × 10-13, 3 × 10-13, 1 × 10-12, 3 × 10-12, 6 × 10-12, 1 × 10-11, 3 × 10-11, 1 × 10-10 M).
|
|
27.
|
De referentiestandaard voor de antagonismebepaling is tamoxifen (tabel 2). De referentiestandaarden bestaan uit een verdunningsreeks van acht concentraties tamoxifen (3 × 10-9, 1 × 10-8, 3 × 10-8, 1 × 10-7, 3 × 10-7, 1 × 10-6, 3 × 10-6, 1 × 10-5 M). Elke concentratie van de antagonistische referentiestandaard moet geco-incubeerd worden met een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3 × 10-12 M).
|
Positieve controle
|
28.
|
De positieve controle voor agonisme-onderzoeken is 17α-methyltestosteron (tabel 1).
|
|
29.
|
De positieve controle voor antagonisme-onderzoeken is 4-hydroxytamoxifen (tabel 2). De antagonistische positieve controle moet geco-incubeerd worden met een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3 × 10-12 M).
|
Negatieve controle
|
30.
|
De negatieve controle voor agonisme-onderzoeken is corticosteron (tabel 1).
|
|
31.
|
De negatieve controle voor antagonisme-onderzoeken is resveratrol (tabel 2). De antagonistische negatieve controle moet geco-incubeerd worden met een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3 × 10-12 M).
|
Aantoning van de bekwaamheid van laboratoria (zie punt 14 en de tabellen 3 en 4 onder „COMPONENTEN VAN DE ER TA-BEPALING” van deze testmethode)
Vehiculum
|
32.
|
Het oplosmiddel dat wordt gebruikt om de teststof op te lossen, moet de teststof volledig oplosbaar maken en moet mengbaar zijn met het celmedium. Geschikte oplosmiddelen zijn DMSO, water en ethanol (95 % tot 100 % zuiverheid). Bij gebruik van DMSO als oplosmiddel mag de maximale DMSO-concentratie tijdens de incubatie niet hoger zijn dan 1 % (v/v). Het oplosmiddel moet vóór gebruik getest worden op afwezigheid van cytotoxiciteit en interferentie met de prestaties van de bepaling.
|
Bereiding van de referentiestandaarden, positieve controles, negatieve controles en teststoffen
|
33.
|
Referentiestandaarden, positieve controles, negatieve controles en teststoffen worden opgelost in 100 % DMSO (of een geschikt oplosmiddel). Vervolgens moeten in hetzelfde oplosmiddel passende (seriële) verdunningen worden bereid. Alvorens te worden opgelost moeten alle stoffen de gelegenheid krijgen om te equilibreren op kamertemperatuur. In de vers bereide stamoplossingen van de referentiestandaarden, positieve controles, negatieve controles en teststoffen mag geen neerslag of troebelheid zichtbaar zijn. Stamoplossingen van referentiestandaarden en controles mogen in bulk worden bereid. Stamoplossingen van teststoffen moeten voor elk experiment vers worden bereid. De eindverdunningen van de referentiestandaarden, positieve controles, negatieve controles en teststoffen moeten voor elk experiment vers worden bereid en binnen 24 uur na bereiding worden gebruikt.
|
Oplosbaarheid, cytotoxiciteit en bereikbepaling
|
34.
|
De oplosbaarheid van de teststoffen in het gekozen oplosmiddel wordt bepaald tijdens de screeningstestrun. Er wordt een stamoplossing bereid met een maximale concentratie van 0,1 M. Als deze concentratie oplosbaarheidsproblemen vertoont, moeten stamoplossingen met lagere concentraties worden bereid totdat de teststoffen volledig oplosbaar zijn. Tijdens de screeningstestrun worden 1:10-verdunningsreeksen van de teststof getest. De maximale concentratie voor de agonisme- en antagonismebepalingen is 1 mM. Uit de screening wordt een passend, beperkt concentratiebereik voor de teststoffen afgeleid dat tijdens de integrale testruns moet worden getest. Voor de integrale testruns moeten de volgende verdunningen worden gebruikt: 1 x, 3 x, 10 x, 30 x, 100 x, 300 x, 1000 x en 3000 x.
|
|
35.
|
Het testen op cytotoxiciteit is opgenomen in het protocol voor de agonisme- en de antagonismebepaling (11). Deze tests op cytotoxiciteit zijn zowel in de screeningstestrun als in de integrale testruns opgenomen. Bij de validering van de ERα CALUX-bioassay werd de cytotoxiciteit beoordeeld aan de hand van de lactaatdehydrogenase-lekkagetest (LDH-lekkagetest) in combinatie met kwalitatieve visuele inspectie van de cellen (zie aanhangsel 4.1) na blootstelling aan de teststoffen. Er mogen evenwel ook andere kwantitatieve methoden worden gebruikt ter bepaling van de cytotoxiciteit (bv. colorimetrische test op basis van tetrazol (MTT) of CALUX-cytotoxiciteitsbioassay). In het algemeen worden teststofconcentraties die de cellevensvatbaarheid met meer dan 20 % doen afnemen, als cytotoxisch beschouwd, zodat ze niet kunnen worden gebruikt bij de evaluatie van de gegevens. Wat betreft de LDH-lekkagetest wordt de concentratie van de teststof als cytotoxisch beschouwd wanneer het percentage LDH-lekkage hoger is dan 120 %.
|
Blootstelling aan de teststof en indeling van de testplaat
|
36.
|
Nadat een confluente kweekfles met gekweekte cellen is behandeld met trypsine, worden de cellen in een concentratie van 1 × 105 cellen/ml geresuspendeerd in oestrogeenvrij testmedium. 100 μl geresuspendeerde cellen wordt uitgeplaat in de binnenste putjes van een microtiterplaat met 96 putjes. De buitenste putjes worden gevuld met 200 μl fosfaatgebufferde zoutoplossing (PBS) (zie de figuren 1 en 2). De uitgeplate cellen worden 24 uur gepre-incubeerd in een CO2-incubator (5 % CO2, 37 °C, 100 % vochtigheid).
|
|
37.
|
Na de pre-incubatie worden de platen geïnspecteerd op zichtbare cytotoxiciteit (zie aanhangsel 4.1), verontreiniging en confluentie. Voor de bepaling worden alleen platen zonder zichtbare cytotoxiciteit of verontreiniging en met minimaal 85 % confluentie gebruikt. Uit de binnenste putjes wordt het medium zorgvuldig verwijderd en vervangen door 200 μl oestrogeenvrij testmedium met een passende verdunningsreeks van referentiestandaarden, teststoffen, positieve controles, negatieve controles en oplosmiddelcontroles (tabel 5: agonisme-onderzoeken; tabel 6: antagonisme-onderzoeken). Alle referentiestandaarden, teststoffen, positieve controles, negatieve controles en oplosmiddelcontroles worden in drievoud getest. In figuur 1 wordt de indeling van de plaat voor de agonismebepaling weergegeven. In figuur 2 wordt de indeling van de plaat voor de antagonismebepaling weergegeven. Voor screeningstests en integrale tests wordt dezelfde plaatindeling gebruikt. Voor antagonismebepalingen bevatten alle binnenste putjes behalve die voor de vehiculumcontrole (VC) ook een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3 × 10-12 M). Er zij op gewezen dat aan elke plaat die de teststof bevat, de referentiestandaarden C8 en C4 moeten worden toegevoegd.
|
|
38.
|
Nadat de cellen aan alle chemische stoffen zijn blootgesteld, worden de 96-putjesplaten nog eens 24 uur in een CO2-incubator (5 % CO2, 37 °C, 100 % vochtigheid) geïncubeerd.
|
Figuur 1
Indeling van de microtiterplaten met 96 putjes voor de screening en de beoordeling van agonistische effecten.
C0 = oplosmiddel van de referentiestandaard.
C(1-8) = verdunningsreeks (1-8, lage tot hoge concentraties) van de referentiestandaard.
PC = positieve controle.
NC = negatieve controle.
TCx-(1-8) = verdunningen (1-8, lage tot hoge concentraties) van de teststof voor de screeningstestrun en de beoordeling van de agonistische effecten van de teststof x.
SC = controle met het oplosmiddel van de teststof (bij voorkeur hetzelfde oplosmiddel als in C0, maar eventueel van een andere batch).
Grijze cellen: = Buitenste putjes, gevuld met 200 μl PBS.
Figuur 2
Indeling van de microtiterplaten met 96 putjes voor de screening en de beoordeling van antagonistische effecten.
C0 = oplosmiddel van de referentiestandaard.
C(1-8) = verdunningsreeks (1-8, lage tot hoge concentraties) van de referentiestandaard.
NC = negatieve controle.
PC = positieve controle.
TCx-(1-8) = verdunningen (1-8, lage tot hoge concentraties) van de teststof voor de screeningstestrun en de beoordeling van de agonistische effecten van de teststof x.
SC = controle met het oplosmiddel van de teststof (bij voorkeur hetzelfde oplosmiddel als in C0, maar eventueel van een andere batch).
VC = vehiculumcontrole (oplosmiddelcontrole zonder vaste concentratie van de agonistische referentiestandaard 17β-oestradiol).
Grijze cellen: = Buitenste putjes, gevuld met 200 μl PBS.
Opmerking: alle binnenste putjes, behalve die voor de vehiculumcontrole (VC), bevatten ook een vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3,0 × 10-12 M).
Meting van de luminescentie
|
39.
|
Het meten van de luminescentie wordt uitvoerig beschreven in het protocol voor de agonisme- en antagonismebepaling (10). Het medium moet uit de putjes worden verwijderd en de cellen moet worden gelyseerd na 24 uur incubatie om de celmembraan te openen en meting van de luciferaseactiviteit mogelijk te maken.
|
|
40.
|
Voor de procedure voor het meten van de luminescentie is een luminometer nodig met 2 injectoren. De luciferasereactie wordt gestart door het substraat luciferine te injecteren. De reactie wordt gestopt door 0,2 M NaOH toe te voegen. De reactie wordt gestopt om te voorkomen dat de luminescentie van het ene putje op het andere overgaat.
|
|
41.
|
De lichtemissie uit elk putje wordt uitgedrukt als relatieve lichteenheden (RLU) per putje.
|
Screeningstestrun
|
42.
|
De analyseresultaten van de screening worden gebruikt om een beperkt concentratiebereik van de teststoffen vast te stellen voor de integrale tests. Hoe de analyseresultaten van de screening worden beoordeeld en hoe het beperkte concentratiebereik voor de integrale tests wordt bepaald, wordt uitvoerig beschreven in het protocol voor de agonisme- en antagonismebepaling (10). Hier wordt een beknopte samenvatting gegeven van de procedures voor het bepalen van het concentratiebereik van de teststoffen voor de agonisme- en antagonismebepaling. Zie de tabellen 5 en 6 voor richtsnoeren voor het vaststellen van de verdunningsreeksen.
|
Selectie van de concentraties voor de beoordeling van agonistische effecten
|
43.
|
In de screeningstestrun moeten de teststoffen worden getest in een verdunningsreeksen zoals aangegeven in tabel 5 (agonisme) en tabel 6 (antagonisme). Alle concentraties worden in drievoudige putjes getest volgens de plaatindeling die wordt weergegeven in figuur 1 (agonisme) of 2 (antagonisme).
|
|
44.
|
Alleen analyseresultaten die aan de aanvaardbaarheidscriteria (tabel 3) voldoen, worden als valide beschouwd en mogen worden gebruikt om de respons van de teststoffen te beoordelen. Als een of meer microtiterplaten in een analyseserie niet aan de aanvaardbaarheidscriteria voldoen, moeten de betreffende microtiterplaten opnieuw worden geanalyseerd. Indien de eerste plaat met de volledige verdunningsreeks van de referentiestandaard niet aan de aanvaardbaarheidscriteria voldoet, moet de volledige testreeks (6 platen) opnieuw worden geanalyseerd.
|
|
45.
|
De initiële concentratiebereiken van de teststoffen moeten worden aangepast en de screeningstest moet worden herhaald indien:
|
—
|
er cytotoxiciteit wordt waargenomen. De screeningsprocedure moet worden herhaald met lagere, niet-cytotoxische concentraties van de teststof;
|
|
—
|
de screening van de teststof geen volledige dosis-responscurve oplevert, omdat de geteste concentraties maximale inductie vertonen. De screeningstestrun moet worden herhaald met lagere concentraties van de teststof.
|
|
|
46.
|
Wanneer er een valide verband tussen dosis en respons wordt waargenomen, moet de (laagste) concentratie worden geselecteerd waarbij maximale inductie wordt waargenomen en die geen cytotoxiciteit vertoont. De hoogste concentratie van de teststof die in de integrale testruns getest wordt, moet driemaal zo hoog zijn als deze geselecteerde concentratie.
|
|
47.
|
Beginnend met de hoogste concentratie zoals hierboven vastgesteld wordt er een volledige, beperkte verdunningsreeks van de teststof bereid met verdunningsstappen zoals aangegeven in tabel 5.
|
|
48.
|
Een teststof die geen enkel agonistisch effect vertoont, moet in de integrale testruns worden getest vanaf de hoogste, niet-cytotoxische concentratie die in de screening werd vastgesteld.
|
Selectie van de concentraties voor de beoordeling van antagonistische effecten
|
49.
|
Alleen analyseresultaten die aan de aanvaardbaarheidscriteria (tabel 4) voldoen, worden als valide beschouwd en mogen worden gebruikt om de respons van de teststoffen te beoordelen. Als een of meer microtiterplaten in een analyseserie niet aan de aanvaardbaarheidscriteria voldoen, moeten de betreffende microtiterplaten opnieuw worden geanalyseerd. Indien de eerste plaat met de volledige verdunningsreeks van de referentiestandaard niet aan de aanvaardbaarheidscriteria voldoet, moet de volledige testreeks (6 platen) opnieuw worden geanalyseerd.
|
|
50.
|
De initiële concentratiebereiken van de teststoffen moeten worden aangepast en de screeningstest moet worden herhaald indien:
|
—
|
er cytotoxiciteit wordt waargenomen. De screeningsprocedure moet worden herhaald met lagere, niet-cytotoxische concentraties van de teststof;
|
|
—
|
de screening van de teststof geen volledige dosis-responscurve oplevert, omdat de geteste concentraties maximale remming vertonen. De screening moet worden herhaald met lagere concentraties van de teststof.
|
|
|
51.
|
Wanneer er een valide verband tussen dosis en respons wordt gevonden, moet de (laagste) concentratie worden geselecteerd waarbij maximale remming wordt waargenomen en die geen cytotoxiciteit vertoont. De hoogste concentratie van de teststof die in de integrale testruns getest wordt, moet driemaal zo hoog zijn als deze geselecteerde concentratie.
|
|
52.
|
Beginnend met de hoogste concentratie zoals hierboven vastgesteld wordt er een volledige, beperkte verdunningsreeks van de teststof bereid met verdunningsstappen zoals aangegeven in tabel 6.
|
|
53.
|
Teststoffen die geen enkel antagonistisch effect vertonen, moeten in de integrale testruns worden getest vanaf de hoogste, niet-cytotoxische concentratie die in de screening werd vastgesteld.
|
Integrale testruns
|
54.
|
Wanneer de beperkte concentratiebereiken zijn geselecteerd, moeten de teststoffen integraal worden getest in een verdunningsreeksen zoals aangegeven in tabel 5 (agonisme) en tabel 6 (antagonisme). Alle concentraties worden in drievoudige putjes getest volgens de plaatindeling die wordt weergegeven in figuur 1 (agonisme) of 2 (antagonisme).
|
|
55.
|
Alleen analyseresultaten die aan de aanvaardbaarheidscriteria (tabellen 3 en 4) voldoen, worden als valide beschouwd en mogen worden gebruikt om de respons van de teststoffen te beoordelen. Als een of meer microtiterplaten in een analyseserie niet aan de aanvaardbaarheidscriteria voldoen, moeten de betreffende microtiterplaten opnieuw worden geanalyseerd. Indien de eerste plaat met de volledige verdunningsreeks van de referentiestandaard niet aan de aanvaardbaarheidscriteria voldoet, moet de volledige testreeks (6 platen) opnieuw worden geanalyseerd.
|
Tabel 5
Concentraties en verdunningen van de referentiestandaarden, controles en teststoffen voor agonismebepalingen
|
Referentie 17β-oestradiol
|
TCx - verdunning screeningstest
|
TCx - verdunning integrale test
|
Controles
|
|
conc. (M)
|
|
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
3 × 10-6
|
|
C1
|
1 × 10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1 × 10-8
|
|
C2
|
3 × 10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1 × 10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3 × 10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6 × 10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1 × 10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3 × 10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1 × 10-10
|
|
|
|
|
|
|
|
TCx - teststof x
PC - positieve controle (17α-methyltestosteron)
NC - negatieve controle (corticosteron)
C0 - controle met het oplosmiddel van de referentiestandaard
SC - controle met het oplosmiddel van de teststof
|
Tabel 6
Concentraties en verdunningen van de referentiestandaarden, controles en teststoffen voor antagonismebepalingen
|
Referentie tamoxifen
|
TCx - verdunning screeningstest
|
TCx - verdunning integrale test
|
Controles
|
|
conc. (M)
|
|
|
conc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
1 × 10-9
|
|
C1
|
3 × 10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC
|
1 × 10-5
|
|
C2
|
1 × 10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3 × 10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1 × 10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3 × 10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Toegevoegde agonist
|
|
C6
|
1 × 10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
conc. (M)
|
|
C7
|
3 × 10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-oestradiol
|
3,0 × 10-12
|
|
C8
|
1 × 10-5
|
|
|
|
|
|
|
|
TCx -teststof x
PC - positieve controle (4-hydroxytamoxifen)
NC - negatieve controle (resveratrol)
C0 - controle met het oplosmiddel van de referentiestandaard
SC - controle met het oplosmiddel van de teststof
VC - vehiculumcontrole (zonder vaste concentratie van de agonistische referentiestandaard 17β-oestradiol (3,0 × 10-12 M))
|
Verzameling en analyse van de gegevens
|
56.
|
Na de screeningstestruns en de integrale testruns moeten voor een agonismebepaling de EC10, EC50, PC10, PC50 en de maximale inductie (TCxmax) van een teststof worden vastgesteld. Voor een antagonismebepaling moeten de IC20, IC50, PC80, PC50 en minimale inductie (TCxmin) worden berekend. Figuur 3 (agonisme) en figuur 4 (antagonisme) bevatten een grafische weergave van deze parameters. De benodigde parameters worden berekend op basis van de relatieve inductie van elke teststof (ten opzichte van de maximale inductie van de referentiestandaard (=100 %)). De gegevens moeten worden beoordeeld aan de hand van een niet-lineaire regressie (variabele helling, 4 parameters) volgens onderstaande vergelijking:

Waarbij:
X = logaritme van de dosis of concentratie
Y = respons (relatieve inductie (%))
Top = maximale inductie (%)
Basis = minimale inductie (%)
LogEC50 = logaritme van de concentratie waarbij 50 % van de maximale respons wordt waargenomen
Hill-coëfficiënt = hellingfactor of Hill-curve
|
|
57.
|
De ruwe gegevens van de luminometer, uitgedrukt in relatieve lichteenheden (RLU), moeten worden overgezet in de spreadsheet voor gegevensanalyse die is opgezet voor de screeningstestrun en de integrale testruns. De ruwe gegevens moeten voldoen aan de aanvaardbaarheidscriteria in de tabellen 3A en 3B (agonisme) of 4A en 4B (antagonisme). Wanneer de ruwe gegevens aan de aanvaardbaarheidscriteria voldoen, worden de volgende berekeningen uitgevoerd om de benodigde parameters te bepalen:
|
Agonisme
|
—
|
Trek de gemiddelde RLU van de controle met het oplosmiddel van de referentiestandaard af van elk van de ruwe analysegegevens van de referentiestandaarden.
|
|
—
|
Trek de gemiddelde RLU van de controle met het oplosmiddel van de teststof af van elk van de ruwe analysegegevens van de teststoffen.
|
|
—
|
Bereken de relatieve inductie voor elke concentratie van de referentiestandaard. Stel de inductie van de hoogste concentratie van de referentiestandaard op 100 %.
|
|
—
|
Bereken de relatieve inductie van elke concentratie van de teststof, waarbij 100 % overeenkomt met de hoogste concentratie van de referentiestandaard.
|
|
—
|
Beoordeel de analyseresultaten aan de hand van niet-lineaire regressie (variabele helling, 4 parameters).
|
|
—
|
Bepaal de EC50 en de EC10 van de referentiestandaard.
|
|
—
|
Bepaal de EC50 en de EC10 van de teststoffen.
|
|
—
|
Bepaal de maximale relatieve inductie van de teststof (TCmax).
|
|
—
|
Bepaal de PC10 en de PC50 van de teststoffen.
|
Als gevolg van bv. cytotoxiciteit of oplosbaarheidsproblemen is het niet altijd mogelijk om een volledige dosis-responscurve voor teststoffen te verkrijgen. De EC50, EC10 en PC50 kunnen dan niet worden bepaald. In dat geval kunnen alleen de PC10 en de TCmax worden bepaald.
Antagonisme
|
—
|
Trek de gemiddelde RLU van de hoogste concentratie van de referentiestandaard af van elk van de ruwe analysegegevens van de referentiestandaarden.
|
|
—
|
Trek de gemiddelde RLU van de hoogste concentratie van de referentiestandaard af van elk van de ruwe analysegegevens van de teststoffen.
|
|
—
|
Bereken de relatieve inductie voor elke concentratie van de referentiestandaard. Stel de inductie van de laagste concentratie van de referentiestandaard op 100 %.
|
|
—
|
Bereken de relatieve inductie van elke concentratie van de teststof, waarbij 100 % overeenkomt met de laagste concentratie van de referentiestandaard.
|
|
—
|
Beoordeel de analyseresultaten aan de hand van niet-lineaire regressie (variabele helling, 4 parameters).
|
|
—
|
Bepaal de IC50 en de IC20 van de referentiestandaard.
|
|
—
|
Bepaal de IC50 en de IC20 van de teststoffen.
|
|
—
|
Bepaal de minimale relatieve inductie van de teststof (TCmin).
|
|
—
|
Bepaal de PC80 en de PC50 van de teststoffen.
|
Figuur 3
Overzicht van de parameters die worden bepaald in de agonismebepaling
EC10 = concentratie van een stof waarbij 10 % van de maximale respons van de stof wordt waargenomen.
EC50 = concentratie van een stof waarbij 50 % van de maximale respons van de stof wordt waargenomen.
PC10 = concentratie van de teststof waarbij de respons gelijk is aan de EC10 van de referentiestandaard.
PC50 = concentratie van de teststof waarbij de respons gelijk is aan de EC50 van de referentiestandaard.
TCxmax = maximale relatieve inductie van de teststof.
Figuur 4
Overzicht van de parameters die worden bepaald in de antagonismebepaling

IC20 = concentratie van een stof waarbij 80 % van de maximale respons van de stof wordt waargenomen (20 % remming).
IC50 = concentratie van een stof waarbij 50 % van de maximale respons van de stof wordt waargenomen (50 % remming).
PC80 = concentratie van de teststof waarbij de respons gelijk is aan de IC20 van de referentiestandaard.
PC50 = concentratie van de teststof waarbij de respons gelijk is aan de IC50 van de referentiestandaard.
TCxmin = minimale relatieve inductie van de teststof.
Als gevolg van bv. cytotoxiciteit of oplosbaarheidsproblemen is het niet altijd mogelijk om een volledige dosis-responscurve voor teststoffen te verkrijgen. De IC50, IC20 en PC50 kunnen dan niet worden bepaald. In dat geval kunnen alleen de PC20 en de TCmin worden bepaald.
|
58.
|
De resultaten moeten op twee (of drie) onafhankelijke testruns worden gebaseerd. Als twee testruns vergelijkbare en dus reproduceerbare resultaten opleveren, is het niet nodig een derde testrun uit te voeren. Om aanvaardbaar te zijn moeten de resultaten:
|
—
|
aan de aanvaardbaarheidscriteria voldoen (zie Aanvaardbaarheidscriteria, punten 14-22);
|
|
Criteria voor gegevensinterpretatie
|
59.
|
Voor de interpretatie van de gegevens en de beslissing of een teststof als positief of negatief moet worden beschouwd, worden de volgende criteria gehanteerd:
|
Voor elke integrale testrun wordt een teststof als positief beschouwd indien:
|
1
|
de TCmax gelijk is aan of hoger is dan 10 % van de maximale respons van de referentiestandaard (REF10);
|
|
2
|
ten minste 2 opeenvolgende concentraties van de teststof gelijk zijn aan of hoger zijn dan de REF10.
|
|
|
Voor elke integrale testrun wordt een teststof als negatief beschouwd indien:
|
1
|
de TCmax niet hoger is dan 10 % van de maximale respons van de referentiestandaard (REF10);
|
|
2
|
minder dan 2 concentraties van de teststof gelijk zijn aan of hoger zijn dan de REF10.
|
|
|
Voor elke integrale testrun wordt een teststof als positief beschouwd indien:
|
1
|
de TCmin gelijk is aan of lager is dan 80 % van de maximale respons van de referentiestandaard (REF80 = 20 % remming);
|
|
2
|
ten minste 2 opeenvolgende concentraties van de teststof gelijk zijn aan of lager zijn dan de REF80.
|
|
|
Voor elke integrale testrun wordt een teststof als negatief beschouwd indien:
|
1
|
de TCmin hoger is dan 80 % van de maximale respons van de referentiestandaard (REF80 = 20 % remming);
|
|
2
|
minder dan 2 concentraties van de teststof gelijk zijn aan of lager zijn dan de REF80.
|
|
|
|
60.
|
Ter karakterisering van de potentie van de positieve respons van een teststof moeten de sterkte van het effect (agonisme: TCmax; antagonisme: TCmin) en de concentratie waarbij het effect zich voordoet (agonisme: EC10, EC50, PC10, PC50; antagonisme: IC20, IC50, PC80, PC50) in het verslag worden opgenomen.
|
TESTVERSLAG
|
61.
|
Zie punt 20 van „COMPONENTEN VAN DE ER TA-BEPALING”.
|
LITERATUUR
|
(1)
|
OESO (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, Nederland.
|
Aanhangsel 4.1:
VISUELE INSPECTIE OP CELLEVENSVATBAARHEID
B.67. IN-VITROGENMUTATIETESTS MET ZOOGDIERCELLEN AAN DE HAND VAN HET GEN VOOR THYMIDINEKINASE
INLEIDING
|
1.
|
Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 490 (2016) van de OESO. Testmethoden worden periodiek herzien in het licht van de wetenschappelijke vooruitgang, regelgevingsbehoeften en dierenwelzijn. De muizenlymfoombepaling (mouse lymphoma assay, MLA) en de TK6-test met de locus van thymidinekinase (TK) maakten oorspronkelijk deel uit van testmethode B.17. Sindsdien heeft de MLA-deskundigenwerkgroep van de International Workshop on Genotoxicity Testing (IWGT) internationaal geharmoniseerde aanbevelingen opgesteld voor aanvaardbaarheidscriteria en gegevensinterpretatie voor de MLA (1) (2) (3) (4) (5). Deze aanbevelingen worden verwerkt in deze nieuwe testmethode B.67. Deze testmethode is opgesteld voor de MLA, alsmede voor de TK6-test, omdat die ook gebruikmaakt van de TK-locus. Terwijl de MLA algemeen is gebruikt met het oog op regelgeving, is de TK6 vooralsnog veel minder toegepast. Er zij op gewezen dat de beide cellijnen ondanks de vergelijkbare eindpunten niet inwisselbaar zijn en dat regelgevingsprogramma’s voor een bepaald regelgevingsdoel een gegronde voorkeur kunnen geven aan de ene boven de andere. Zo is bij de validering van de MLA aangetoond dat deze bepaling niet alleen geschikt is voor het detecteren van genmutaties, maar ook van het vermogen van een teststof om structurele chromosoombeschadigingen teweeg te brengen. Deze testmethode maakt deel uit van een reeks methoden om de genotoxiciteit te testen. De OESO heeft een document opgesteld met beknopte informatie over genetisch-toxicologische tests en een overzicht van de recente wijzigingen van de OESO-testrichtlijnen voor genetische toxiciteit (6).
|
|
2.
|
De in-vitrogenmutatietests met zoogdiercellen zijn bedoeld voor de detectie van genmutaties die door chemische stoffen zijn geïnduceerd. In de cellijnen die in deze tests worden gebruikt, worden voorwaartse mutaties gemeten in reportergenen, met name het endogene gen voor thymidinekinase (TK in menselijke cellen en Tk in knaagdiercellen, in deze testmethode samen aangeduid als TK). Deze testmethode is bedoeld voor gebruik met twee cellijnen: de muizenlymfoomcellijn L5178Y TK+/--3.7.2C (algemeen aangeduid als L5178Y) en de humane lymfoblastoïdcellijn TK6 (algemeen aangeduid als TK6). Hoewel de beide cellijnen verschillen in herkomst, celgroei, p53-status enz. kunnen de TK-genmutatietests met beide celtypen op vergelijkbare wijze worden uitgevoerd, zoals beschreven in deze testmethode.
|
|
3.
|
Omdat het thymidinekinasegen een autosomaal, heterozygoot gen is, is het mogelijk om levensvatbare kolonies te detecteren waarvan de cellen deficiënt zijn in het enzym thymidinekinase na mutatie van TK+/-
naar TK-/-
. Deze deficiëntie kan het gevolg zijn van genetische gebeurtenissen die effect hebben op het TK-gen, waaronder genmutaties (puntmutaties, leesraammutaties, kleine deleties enz.) en chromosomale gebeurtenissen (grote deleties, chromosoomherschikkingen en mitotische recombinatie). Laatstgenoemde gebeurtenissen komen tot uiting als verlies van heterozygositeit, wat een vaak voorkomende genetische verandering is van tumorsuppressorgenen in de menselijke tumorigenese. Verlies van het volledige chromosoom waarop zich het TK-gen bevindt, als gevolg van verstoring van de spoelvorming en/of mitotische non-disjunctie, kan in theorie worden gedetecteerd in de MLA. Een combinatie van cytogene en moleculaire analyse laat inderdaad duidelijk zien dat bepaalde TK-mutanten in de MLA uit non-disjunctie voortkomen. De bewijskracht wijst evenwel uit datTK-genmutatietests niet betrouwbaar zijn voor het detecteren van aneugenen wanneer de standaard cytotoxiciteitscriteria (zoals beschreven in deze testmethode) worden toegepast, en dat deze tests daarom niet geschikt zijn voor het detecteren van aneugenen(7) (8) (9).
|
|
4.
|
In TK-genmutatietests worden twee onderscheiden fenotypen van TK-mutanten gegenereerd; normaal groeiende mutanten, die even snel groeien als heterozygote TK-cellen, en langzaam groeiende mutanten die groeien met langere verdubbelingstijden. Normaal groeiende en langzaam groeiende mutanten zijn herkenbaar als respectievelijk grote en kleine mutantkolonies in de MLA en vroeg verschijnende en laat verschijnende mutantkolonies in de TK6. Er is een gedetailleerde analyse gemaakt van de moleculaire en cytogenetische aard van zowel grote als kleine MLA-mutantkolonies (8) (10) (11) (12) (13). De moleculaire en cytogenetische aard van de vroeg verschijnende en laat verschijnende TK6-mutanten zijn eveneens uitvoerig onderzocht (14) (15) (16) (17). Langzaam groeiende mutanten voor beide celtypen hebben genetische beschadigingen opgelopen aan een of meer verondersteld groei-regulerende genen dichtbij de TK-locus, wat leidt tot langere verdubbelingstijden en de vorming van laat verschijnende of kleine kolonies (18). Er is een verband gelegd tussen langzaam groeiende mutanten en chemische stoffen die grootschalige structurele veranderingen op chromosoomniveau induceren. Cellen waarvan de beschadigingen geen invloed hebben op het/de verondersteld groei-regulerende gen(en) dichtbij de TK-locus, groeien in vergelijkbaar tempo als de oudercellen en worden normaal groeiende mutanten. De inductie van voornamelijk normaal groeiende mutanten wordt in verband gebracht met chemische stoffen die voornamelijk als puntmutagenen optreden. Het is dan ook essentieel om zowel langzaam groeiende als normaal groeiende mutanten te tellen, teneinde alle mutanten in aanmerking te nemen en enig inzicht te krijgen in het/de type(n) door de teststof veroorzaakte schade (mutagenen vs. clastogenen) (10) (12) (18) (19).
|
|
5.
|
De testmethode is zodanig opgezet dat deze algemene informatie verschaft die zowel voor de MLA als voor de TK6 geldt, evenals gespecialiseerde richtsnoeren voor de afzonderlijke tests.
|
|
6.
|
Gebruikte definities worden gegeven in aanhangsel 1.
|
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
7.
|
Bij in vitro uitgevoerde tests moet er meestal een exogene metabole activeringsbron worden gebruikt. Het exogene metabole activeringssysteem kan de omstandigheden in vivo niet volledig nabootsen.
|
|
8.
|
Er moet goed op worden gelet dat omstandigheden die zouden kunnen leiden tot artefactuele positieve resultaten (d.w.z. mogelijke interactie met het testsysteem), die niet het gevolg zijn van interactie tussen de teststof en het genetische materiaal van de cel, worden vermeden; zulke omstandigheden zijn onder andere veranderingen in de pH of de osmolaliteit, interactie met de bestanddelen van het medium (20) (21) of een te sterke cytotoxiciteit (22) (23) (24). Als de cytotoxiciteit de maximaal aanbevolen cytotoxiciteitsniveaus zoals omschreven in punt 28 overschrijdt, wordt deze als te sterk beschouwd voor de MLA en de TK6. Bovendien zij erop gewezen dat teststoffen die thymidine-analogen zijn of zich als thymidine-analogen gedragen, de mutantfrequentie kunnen verhogen door selectieve groei van de spontane achtergrondmutanten tijdens de behandeling van de cellen en dat het voor een adequate beoordeling van dergelijke stoffen nodig kan zijn om aanvullende testmethoden toe te passen (25).
|
|
9.
|
Voor gefabriceerde nanomaterialen kunnen specifieke aanpassingen van deze testmethode noodzakelijk zijn, maar deze worden in deze testmethode niet beschreven.
|
|
10.
|
Voordat de testmethode wordt gebruikt voor het testen van een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is.
|
|
11.
|
Mutantcellen met een deficiëntie aan thymidinekinase-enzymactiviteit als gevolg van de mutatie van TK
+/- naar TK
-/- zijn resistent voor de cytostatische effecten van de pyrimidine-analoog trifluorthymidine (TFT). Cellen zonder deficiëntie aan TK zijn gevoelig voor TFT, waardoor het celmetabolisme wordt geremd en er geen celdeling meer plaatsvindt. Dit betekent dat mutantcellen zich in aanwezigheid van TFT wel kunnen voortplanten en zichtbare kolonies kunnen vormen, en cellen die het TK-enzym bevatten, niet.
|
PRINCIPE VAN DE TEST
|
12.
|
Cellen in suspensie worden zowel met als zonder een exogene metabole activeringsbron (zie punt 19) gedurende een geschikte periode (zie punt 33) aan de teststof blootgesteld en vervolgens overgeënt om de cytotoxiciteit te bepalen en fenotypische expressie mogelijk te maken alvorens de mutanten te selecteren. Voor de MLA wordt de cytotoxiciteit bepaald aan de hand van de relatieve totale groei (RTG – zie punt 25), voor de TK6 aan de hand van de relatieve overleving (relative survival, RS – zie punt 26). De behandelde culturen blijven een voldoende lange periode, kenmerkend voor elk celtype (zie punt 37), in groeimedium om een vrijwel optimale fenotypische expressie van de geïnduceerde mutaties mogelijk te maken. Na fenotypische expressie wordt de mutantfrequentie bepaald door bekende aantallen cellen te enten in medium met de selecterende stof voor de detectie van mutantkolonies en in medium zonder de selecterende stof voor de bepaling van de kloneringsefficiëntie (levensvatbaarheid). Na een geschikte incubatietijd worden de kolonies geteld. De mutantfrequentie wordt bepaald aan de hand van het aantal mutantkolonies, gecorrigeerd voor de kloneringsefficiëntie op het moment van selecteren van de mutanten.
|
BESCHRIJVING VAN DE METHODE
Voorbereidingen
Cellen
|
13.
|
Voor de MLA: Voor de MLA moet de TK
+/--3.7.2C sublijn van L5178Y-cellen worden gebruikt omdat deze specifieke sublijn ook is gebruikt bij de ontwikkeling en karakterisering van de MLA. De cellijn L5178Y is afgeleid van een door methylcholantreen geïnduceerd thymuslymfoom van een DBA-2-muis (26). Clive et al. behandelden L5178Y-cellen (door Clive TK+/+ -3 genoemd) met ethylmethaansulfonaat en isoleerden een TK-/--kloon (TK-/- -3.7 genoemd) met broomdeoxyuridine als selecterende stof. Van de TK-/--kloon werden een spontane TK+/--kloon (TK+/- -3.7.2. genoemd) en een subkloon (TK+/- -3.7.2C genoemd) geïsoleerd en gekarakteriseerd voor gebruik in de MLA (27). Het karyotype voor de cellijn is gepubliceerd (28) (29) (30) (31). Het modale aantal chromosomen is 40. Er is één metacentrisch chromosoom (t12 13) dat als één chromosoom moet worden geteld. Bij de muis is de locus van TK gelegen op het distale uiteinde van chromosoom 11. De cellijn L5178Y TK
+/- -3.7.2C heeft mutaties in beide p53-allelen en produceert mutant-p53 eiwit (32) (33). Het is waarschijnlijk aan de p53-status van de cellijn TK+/- -3.7.2C te danken dat de test grootschalige schade kan detecteren (17).
|
|
14.
|
Voor de TK6: TK6 is een humane lymfoblastoïdcellijn. De oudercellijn is een door het Epstein-Barr-virus getransformeerde cellijn, WI-L2, die oorspronkelijk afkomstig is van een mannelijke 5-jarige met hereditaire sferocytose. Door de eerste geïsoleerde kloon, HH4, te mutageniseren met ICR191 werd een TK-heterozygote cellijn, TK6, gegenereerd (34). TK6-cellen zijn bijna allemaal diploïde en het representatieve karyotype is 47, XY, 13+, t(14; 20), t(3; 21) (35). Bij de mens is de locus van TK gelegen op de lange arm van chromosoom 17. TK6 is een p53-competente cellijn, omdat het een wildtype p53-sequentie heeft in beide allelen en alleen wildtype p53-eiwit tot expressie brengt (36).
|
|
15.
|
Voor zowel de MLA als de TK6 wordt het testlaboratorium geadviseerd, wanneer het voor het eerst een mastervoorraad aanlegt of deze aanvult, zich ervan te vergewissen dat er geen sprake is van verontreiniging met mycoplasma, de cellen te karyotyperen of de chromosomen met de TK-locus te kleuren, en de populatieverdubbelingstijden te controleren. De normale tijd voor de celcyclus voor de in het testlaboratorium gebruikte cellen moet worden vastgesteld en moet overeenkomen met gepubliceerde celkenmerken (16) (19) (37). Deze mastervoorraad moet worden bewaard bij maximaal –150 °C en moet worden gebruikt voor de bereiding van alle werk-celvoorraden.
|
|
16.
|
Hetzij voorafgaand aan de bereiding van een groot aantal gecryopreserveerde werkvoorraden, hetzij vlak voor gebruik in een experiment is het wellicht nodig reeds aanwezige mutantcellen uit de cultuur te verwijderen (tenzij de mutantfrequentie (MF) van de oplosmiddelcontrole al binnen het aanvaardbare bereik ligt – zie tabel 2 voor de MLA). Hiertoe wordt methotrexaat (aminopterine) gebruikt om TK-deficiënte cellen uit te selecteren en worden thymidine, hypoxanthine en glycine (L5178Y) of 2’-deoxycytidine (TK6) aan de cultuur toegevoegd om een optimale groei van de TK-competente cellen te waarborgen (19) (38) (39), en (40) voor TK6). Algemene adviezen voor goede praktijken om celculturen in leven te houden en speciale adviezen voor L5178Y- en TK6-cellen zijn te vinden in (19) (31) (37) (39) (41). Voor laboratoria die voorraden mastercellen nodig hebben om de MLA of de TK6 op te zetten of om nieuwe voorraden mastercellen te verkrijgen, is een celbank met goed gekarakteriseerde cellen beschikbaar (37).
|
Media en kweekomstandigheden
|
17.
|
Voor beide tests moet er worden gezorgd voor geschikte kweekmedia en incubatieomstandigheden voor de culturen (bv. kweekvaten, vochtige atmosfeer van 5 % CO2, incubatietemperatuur van 37 °C). De celculturen moeten altijd onder zodanige omstandigheden worden gehouden dat ze in log-fase groeien. Het is vooral belangrijk dat de medium- en kweekomstandigheden zodanig worden gekozen dat de celgroei tijdens de expressieperiode en de klonering van zowel de mutantcellen als de gewone cellen optimaal zijn. Voor de MLA en de TK6 is het ook van belang dat de kweekomstandigheden zorgen voor een optimale groei van TK-mutanten die grote kolonies vormen/vroeg verschijnen, als TK-mutanten die kleine kolonies vormen/laat verschijnen. Meer bijzonderheden over de kweek, waaronder de noodzaak om paardenserum goed te verwarmen om het te inactiveren indien bij de selectie van mutanten RPMI-medium wordt gebruikt, is te vinden in (19) (31) (38) (39) (40) (42).
|
Prepareren van de culturen
|
18.
|
Cellen uit stamculturen worden met een zodanige dichtheid in een kweekmedium geënt dat de suspensieculturen tijdens de behandelingsperiode en de expressieperiode exponentieel zullen blijven groeien.
|
Metabole activering
|
19.
|
Bij het gebruik van L5178Y- en TK6-cellen moeten exogene metabole activeringssystemen worden gehanteerd omdat ze onvoldoende endogene metabole capaciteit hebben. Het meest gebruikte systeem dat als standaard wordt aanbevolen, tenzij anders wordt gemotiveerd, is een post-mitochondriale fractie aangevuld met een cofactor (S9), verkregen uit de lever van knaagdieren (doorgaans ratten) die zijn behandeld met enzym-inducerende stoffen als Aroclor 1254 (43) (44) (45) of een combinatie van fenobarbiton en β-naftoflavon (46) (47) (48) (49) (50) (51). De laatste combinatie druist niet in tegen het Verdrag van Stockholm inzake persistente organische verontreinigende stoffen (52). Bovendien is aangetoond dat zij even effectief is als Aroclor 1254 voor de inducering van oxidasen met gemengde functie (45) (46) (47) (48) (49). De S9-fractie wordt meestal gebruikt bij een concentratie van 1-2 %, maar mag worden verhoogd tot 10 % (v/v) in het uiteindelijke testmedium. De keuze van het type en de concentratie van het exogene metabole activeringssysteem dat of de metabole inductor die wordt gebruikt, kan worden beïnvloed door de klasse van de teststoffen.
|
Teststofbereiding
|
20.
|
Vaste teststoffen moeten in geschikte oplosmiddelen worden bereid en eventueel vóór de behandeling van de cellen worden verdund (zie punt 21). Vloeibare teststoffen kunnen direct aan het testsysteem worden toegevoegd en/of worden verdund vóór de behandeling van het testsysteem. Bij het testen van gassen of vluchtige teststoffen moeten geschikte aanpassingen van de standaardprotocollen worden gevolgd, zoals de behandeling in gesloten kweekvaten (53) (54) (55). Preparaten van de teststof moeten worden bereid vlak vóór de behandeling, tenzij met stabiliteitsgegevens is aangetoond dat bewaren van het preparaat aanvaardbaar is.
|
TESTOMSTANDIGHEDEN
Oplosmiddelen
|
21.
|
Het oplosmiddel wordt zo gekozen dat het de oplosbaarheid van de te testen chemische stof vergroot zonder een negatieve invloed te hebben op het verloop van de test, bv. door veranderingen in de celgroei, aantasting van de integriteit van de teststof, reacties met kweekvaten, belemmering van het metabole activeringssysteem. Het wordt aanbevolen waar mogelijk eerst het gebruik van een waterig oplosmiddel (of kweekmedium) te overwegen. Gangbare oplosmiddelen zijn water of dimethylsulfoxide. Organische oplosmiddelen mogen in de regel de 1 % (v/v) en waterige oplosmiddelen (fysiologisch zout of water) de 10 % (v/v) in het uiteindelijke behandelingsmedium niet overschrijden. Als er andere dan de gangbare oplosmiddelen (bv. ethanol of aceton) worden gebruikt, moeten gegevens worden verstrekt waaruit blijkt dat ze verenigbaar zijn met de teststoffen en het testsysteem en dat zij geen genetische toxiciteit vertonen in de gebruikte concentratie. Als die gegevens ontbreken, is het belangrijk ook onbehandelde controles toe te voegen (zie aanhangsel 1, Definities) om aan te tonen dat het gekozen oplosmiddel geen schadelijke of mutagene effecten induceert.
|
METING VAN CYTOTOXICITEIT EN KEUZE VAN BEHANDELINGSCONCENTRATIES
|
22.
|
Bij de bepaling van de hoogste te testen concentratie van de teststof moeten concentraties worden vermeden die kunnen leiden tot artefactuele positieve reacties, zoals reacties die bovenmatige cytotoxiciteit (zie punt 28) voortbrengen, neerslag (zie punt 29) in het kweekmedium of uitgesproken wijzigingen in pH of osmolaliteit (zie punt 8). Indien de teststof op het ogenblik van toevoeging een uitgesproken wijziging in de pH van het medium veroorzaakt, kan de pH worden aangepast door het uiteindelijke behandelingsmedium te bufferen zodat artefactuele positieve reacties worden vermeden en passende kweekomstandigheden in stand worden gehouden.
|
|
23.
|
De concentraties worden gekozen op basis van de cytotoxiciteit en andere overwegingen (zie punten 27-30). Hoewel de evaluatie van cytotoxiciteit in een eerste test nuttig kan zijn om de in het hoofdexperiment te gebruiken concentraties beter te kunnen vaststellen, is een eerste test niet vereist. Ook als er wel een voorevaluatie van cytotoxiciteit wordt uitgevoerd, moet in het hoofdexperiment nog steeds voor elke cultuur de cytotoxiciteit worden gemeten. Als er een bereikbepalingstest wordt uitgevoerd, moet deze een breed concentratiebereik bestrijken en kan deze hetzij worden beëindigd op dag 1 na de behandeling, of worden voortgezet tot en met de expressie op dag 2 en de selectie van de mutanten (als blijkt dat de gebruikte concentraties passend zijn).
|
|
24.
|
Voor elke afzonderlijke testcultuur en controlecultuur moet de cytotoxiciteit worden vastgesteld: de te gebruiken methoden voor de MLA (2) en de TK6 (15) worden bepaald door de internationaal erkende praktijk.
|
|
25.
|
Voor beide versies (agar en microtiterplaat) van de MLA: moet de cytotoxiciteit worden beoordeeld aan de hand van de relatieve totale groei (RTG), zoals oorspronkelijk gedefinieerd door Clive en Spector in 1975 (2). Deze maat voor de cytotoxiciteit omvat de relatieve suspensiegroei (RSG: testcultuur t.o.v. oplosmiddelcontrole) tijdens de behandeling van de cellen, de expressietijd en de relatieve kloneringsefficiëntie (RCE: testcultuur t.o.v. oplosmiddelcontrole) op het moment dat de mutanten worden geselecteerd (2). Er zij op gewezen dat in de RSG elk verlies van cellen in de testcultuur tijdens de behandeling in aanmerking wordt genomen (zie aanhangsel 2 voor de formules).
|
|
26.
|
Voor de TK6: de cytotoxiciteit moet worden beoordeeld aan de hand van de relatieve overleving (RS), d.w.z. de kloneringsefficiëntie van onmiddellijk na de behandeling uitgeplate cellen, gecorrigeerd voor elk verlies van cellen gedurende de behandeling, op basis van het aantal cellen, vergeleken met de negatieve controle (waaraan een overleving van 100 % wordt toegekend) (zie aanhangsel 2 voor de formule).
|
|
27.
|
Er moeten ten minste vier testconcentraties (exclusief het oplosmiddel en positieve controles) die voldoen aan de aanvaardbaarheidscriteria (passende mate van cytotoxiciteit, aantal cellen enz.), worden geëvalueerd. Hoewel het gebruik van duploculturen raadzaam is, kan bij elke geteste concentratie één behandelde cultuur worden gebruikt, maar ook duploculturen. De resultaten verkregen voor duploculturen bij een gegeven concentratie moeten afzonderlijk worden gerapporteerd, maar kunnen voor de gegevensanalyse worden gepoold (55). Voor teststoffen die weinig of geen cytotoxiciteit vertonen, zijn intervallen tussen de testconcentraties van ongeveer een factor 2 tot 3 doorgaans passend. Wanneer er cytotoxiciteit optreedt, moeten de concentraties een cytotoxiciteitsinterval bestrijken van een concentratie waarbij cytotoxiciteit optreedt zoals beschreven in punt 28, tot en met concentraties waarbij er matige en geen of vrijwel geen cytotoxiciteit is. Veel teststoffen vertonen een steile concentratie-responscurve, en om het hele cytotoxiciteitsbereik te bestrijken of in detail de concentratierespons te bestuderen zullen mogelijk dichter bij elkaar liggende concentraties en meer dan vier concentraties moeten worden gebruikt, in het bijzonder in situaties waarin een herhalingsexperiment vereist is (zie punt 70). Vooral bij gebruik van één cultuur kan het van belang zijn om meer dan vier concentraties te gebruiken.
|
|
28.
|
Als de maximale concentratie is gebaseerd op cytotoxiciteit, moet voor de hoogste concentratie worden gestreefd naar een RTG tussen 20 en 10 % voor de MLA en een RS tussen 20 en 10 % voor de TK6 (punt 67).
|
|
29.
|
Voor slecht oplosbare teststoffen die bij concentraties onder de oplosbaarheidsgrens niet cytotoxisch zijn, moet de hoogste geanalyseerde concentratie troebelheid of een met het oog of met behulp van een omgekeerde microscoop aan het eind van de behandeling met de teststof zichtbaar neerslag opleveren. Zelfs indien cytotoxiciteit optreedt boven de oplosbaarheidsgrens, is het raadzaam de test uit te voeren bij slechts één concentratie die troebelheid of een zichtbaar neerslag oplevert, omdat het neerslag kan leiden tot artefactuele effecten. Omdat voor de MLA en de TK6 suspensieculturen worden gebruikt, moet er zorgvuldig op worden toegezien dat het neerslag de uitvoering van de test niet verstoort. Ook kan het nuttig zijn om voorafgaand aan het experiment de oplosbaarheid in het kweekmedium te bepalen.
|
|
30.
|
Indien er geen neerslag of beperkende cytotoxiciteit wordt waargenomen, moet de hoogste testconcentratie overeenkomen met de laagste van de volgende waarden: 10 mM, 2 mg/ml of 2 μl/ml (57) (58). Wanneer de samenstelling van de teststof niet vastgesteld is, bv. in geval van een stof van onbekende of variabele samenstelling, complexe reactieproducten of biologisch materiaal (d.w.z. Chemical Substances of Unknown or Variable Composition of UVCB’s), milieu-extracten enz., moet de hoogste concentratie bij het ontbreken van voldoende cytotoxiciteit mogelijk hoger zijn (bv. 5 mg/ml) om de concentratie van elk van de componenten te verhogen. Er zij echter opgemerkt dat deze eisen voor geneesmiddelen voor menselijk gebruik anders kunnen zijn (59).
|
Controles
|
31.
|
Voor elk experimentele omstandigheid dienen gelijktijdige negatieve controles (zie punt 21) in het experiment te worden opgenomen, waaraan uitsluitend het oplosmiddel wordt toegediend en die verder op dezelfde manier worden behandeld als de culturen met teststof.
|
|
32.
|
Gelijktijdige positieve controles zijn nodig om aan te tonen dat het laboratorium in staat is om mutagenen te identificeren onder de omstandigheden van het gebruikte testprotocol, dat het exogene metabole activeringssysteem doeltreffend is (indien van toepassing) en dat zowel TK-mutanten die kleine kolonies vormen/laat verschijnen, als TK-mutanten die grote kolonies vormen/vroeg verschijnen adequaat gedetecteerd worden. Voorbeelden van voor positieve controle gebruikte chemische stoffen worden gegeven in tabel 1 hieronder. Er kunnen ook alternatieve positieve controlestoffen worden gebruikt, indien dat wordt gemotiveerd. Omdat in-vitrogenotoxiciteitstests op zoogdiercellen voldoende gestandaardiseerd zijn voor korte behandelingen (3-4 uur) die gelijktijdig met en zonder metabole activering met dezelfde behandelingsduur worden toegepast, kan het gebruik van positieve controles worden beperkt tot een mutageen dat metabole activering nodig heeft. In dat geval zal deze ene positievecontrolerespons zowel de activiteit van het metabole activeringssysteem als de gevoeligheid van het testsysteem aantonen. Indien toegepast, moet een langlopende behandeling (d.w.z. 24 uur zonder S9) echter haar eigen positieve controle hebben, omdat de behandelingsduur zal afwijken van de test waarbij metabole activering wordt toegepast. Elke positieve controle moet worden gebruikt bij een of meer concentraties waarvoor een reproduceerbare en detecteerbare stijging ten opzichte van het achtergrondniveau kan worden verwacht, om de gevoeligheid van het testsysteem aan te tonen, en de respons mag niet in het gedrang komen door een mate van cytotoxiciteit die de in deze TM gespecificeerde grenzen overschrijdt (zie punt 28).
|
Tabel 1
Aanbevolen referentiestoffen voor het beoordelen van de bekwaamheid van laboratoria en voor het selecteren van positieve controles
|
Categorie
|
Stof
|
CAS RN
|
|
1. Mutagenen werkzaam zonder metabolische activering
|
|
Methylmethaansulfonaat
|
66-27-3
|
|
Mitomycine C
|
50-07-7
|
|
4-Nitrochinoline-N-oxide
|
56-57-5
|
|
2. Mutagenen waarvoor metabolische activering vereist is
|
|
Benzo[a]pyreen
|
50-32-8
|
|
Cyclofosfamide (monohydraat)
|
50-18-0 (6055-19-2)
|
|
7,12-Dimethylbenzantraceen
|
57-97-6
|
|
3-Methylcholantreen
|
56-49-5
|
PROCEDURE
Behandeling met teststof
|
33.
|
Delende cellen worden zowel met als zonder metabool activeringssysteem met de teststof behandeld. De blootstelling moet gedurende een geschikte periode worden uitgevoerd (meestal is 3 tot 4 uur voldoende). Er zij echter opgemerkt dat deze eisen voor geneesmiddelen voor menselijk gebruik anders kunnen zijn (59). Voor de MLA moet in gevallen waarin een korte behandeling negatieve resultaten oplevert en er informatie is waaruit kan worden opgemaakt dat mogelijk een langere behandeling nodig is [bv. nucleosideanalogen, slecht oplosbare chemische stoffen (5) (59)], worden overwogen om de test uit te voeren met een langere behandeling, d.w.z. 24 uur zonder S9.
|
|
34.
|
Het minimale aantal cellen dat voor elke testcultuur (controle en behandeld) in elke fase van de test wordt gebruikt, moet worden gekozen op basis van de spontane mutantfrequentie. Een algemene vuistregel is om in elke testcultuur voldoende cellen te behandelen en over te enten om in alle fasen van de test (behandeling, fenotypische expressie en selectie van de mutanten) ten minste 10, liefst 100 spontane mutanten over te houden (56).
|
|
35.
|
Voor de MLA ligt de aanbevolen aanvaardbare spontane mutantfrequentie tussen de 35-140 × 10-6 (agarversie) en 50-170 × 10-6 (microtiterplaat-versie) (zie tabel 2). Om voor elke testcultuur ten minste 10 en idealiter 100 spontane mutanten te hebben die de behandeling overleven, moeten er ten minste 6 × 106 cellen worden behandeld. Door dit aantal cellen te behandelen en voldoende cellen over te houden tijdens de expressie en de klonering voor het selecteren van mutanten, zijn er in alle fasen van het experiment voldoende spontane mutanten (10 of meer), ook voor de culturen die worden behandeld met concentraties die 90 % cytotoxiciteit vertonen (zoals gemeten door een RTF van 10 %) (19) (38) (39).
|
|
36.
|
Voor de TK6 ligt de spontane mutantfrequentie meestal tussen de 2 en 10 × 10-6. Om voor elke cultuur ten minste 10 spontane mutanten te hebben die de behandeling overleven, moeten er ten minste 20 × 106 cellen worden behandeld. Door dit aantal cellen te behandelen zijn er steeds voldoende spontane mutanten (10 of meer), ook voor de culturen die worden behandeld met concentraties die tijdens de behandeling 90 % cytotoxiciteit veroorzaken (10 % RS). Bovendien moeten er tijdens de expressieperiode voldoende cellen worden gekweekt en uitgeplaat voor het selecteren van de mutanten (60).
|
Fenotypische expressietijd en meting van de cytotoxiciteit en de mutantfrequentie
|
37.
|
Aan het einde van de behandelingsperiode worden de cellen gedurende een vooraf vastgestelde periode gekweekt om een vrijwel optimale fenotypische expressie van de geïnduceerde mutanten mogelijk te maken. Deze periode is specifiek voor elke cellijn. Voor de MLA is de fenotypische expressieperiode 2 dagen. Voor de TK6 is de fenotypische expressieperiode 3-4 dagen. Als een 24-uursbehandeling wordt toegepast, begint de expressieperiode na het einde van de behandeling.
|
|
38.
|
Gedurende de fenotypische expressieperiode worden de cellen elke dag geteld. Voor de MLA worden de dagelijkse celaantallen gebruikt om de dagelijkse suspensiegroei (SG) te berekenen. Na de expressieperiode van 2 dagen worden de cellen gesuspendeerd in medium met en zonder selecterende stof voor om respectievelijk het aantal mutanten (selectieplaten) en de kloneringsefficiëntie (levensvatbaarheidsplaten) te bepalen. Voor de MLA zijn er twee even aanvaardbare methoden voor de klonering voor het selecteren van de mutanten; de ene met zachte agar en de andere met vloeibaar medium in 96-putjesplaten (19) (38) (39). In de TK6 wordt de klonering uitgevoerd met vloeibare media en 96-putjesplaten (16).
|
|
39.
|
Trifluorthymidine (TFT) is de enig aanbevolen selecterende stof voor TK-mutanten (61).
|
|
40.
|
Voor de MLA worden agarplaten en microtiterplaten na 10-12 dagen incubatie geteld. Voor de TK6 worden kolonies in microtiterplaten na 10-14 dagen gescoord op de vroeg verschijnende mutanten. Om de langzaam groeiende (laat verschijnende) TK6-mutanten in aanmerking te nemen is het nodig de cellen na telling van de vroeg verschijnende mutanten nogmaals met groeimedium en TFT te voeden en de platen vervolgens nog 7-10 dagen te incuberen (62). Zie de punten 42 en 44 voor een bespreking van het tellen van de langzaam en snel groeiende TK-mutanten.
|
|
41.
|
De benodigde berekeningen voor de beide tests, inclusief de twee methoden (agar en microtiterplaat) voor de MLA, worden gegeven in aanhangsel 2. Als de MLA wordt uitgevoerd volgens de agarmethode, worden de kolonies geteld en wordt het aantal mutantkolonies gecorrigeerd met de kloneringsefficiëntie om een MF te berekenen. In de microtiterplaatversie van de MLA en in de TK6 wordt de kloneringsefficiëntie voor zowel de selectieplaten als de kloneringsefficiëntieplaten bepaald aan de hand van de Poisson-verdeling (63). Uit deze beide kloneringsefficiënties wordt de MF berekend.
|
Karakterisering van de mutantkolonies
|
42.
|
Als in de MLA de teststof positief is (zie de punten 62-63), moeten bij ten minste een van de testculturen (normaal gesproken de hoogste aanvaardbare positieve concentratie) en bij de negatieve en positieve controles de kolonies worden gekarakteriseerd door de grootte of de groei van de kolonies te inventariseren. Als de teststof negatief is (zie punt 64), moeten de mutantkolonies worden gekarakteriseerd bij de negatieve en positieve controles. Als de MLA wordt uitgevoerd met microtiterplaten, worden mutanten die kleine kolonies vormen gedefinieerd als mutanten waarvan de kolonies minder dan 25 % van de diameter van het putje beslaan en mutanten die grote kolonies vormen als mutanten waarvan de kolonies meer dan 25 % van de diameter van het putje beslaan. Bij de agarmethode wordt voor het tellen van de mutantkolonies en het inventariseren van de koloniegrootte een automatische kolonieteller gebruikt. Methoden voor het inventariseren van de koloniegrootte worden uitvoerig beschreven in de literatuur (19) (38) (40). Karakterisering van de kolonies van de negatieve en positieve controles is nodig om aan te tonen dat de tests naar behoren worden uitgevoerd.
|
|
43.
|
De teststof kan niet als negatief worden aangemerkt als zowel de mutanten die grote kolonies vormen als de mutanten die kleine kolonies vormen onvoldoende worden gedetecteerd in de positieve controle. De karakterisering van de kolonies kan worden gebruikt om algemene informatie te verkrijgen over het vermogen van de teststof om puntmutaties en/of chromosomale gebeurtenissen te veroorzaken (punt 4).
|
|
44.
|
TK6: normaal groeiende en langzaam groeiende mutanten worden van elkaar onderscheiden door een verschil in incubatietijd (zie punt 40). Voor de TK6 worden normaal gesproken voor alle culturen, inclusief de negatieve en positieve controles, zowel de vroeg als de laat verschijnende mutanten gescoord. Karakterisering van de kolonies van de negatieve en positieve controles is nodig om aan te tonen dat de tests naar behoren worden uitgevoerd. De teststof kan niet als negatief worden aangemerkt als zowel de vroeg verschijnende als de laat verschijnende mutanten onvoldoende worden gedetecteerd in de positieve controle. De karakterisering van de kolonies kan worden gebruikt om algemene informatie te verkrijgen over het vermogen van de teststof om puntmutaties en/of chromosomale gebeurtenissen te veroorzaken (punt 4).
|
Bekwaamheid van het laboratorium
|
45.
|
Om aan te tonen over voldoende ervaring met de test te beschikken vooraleer wordt overgegaan tot routinematig gebruik ervan, moet het laboratorium een reeks experimenten hebben verricht met positieve controlestoffen die via verschillende mechanismen werken (ten minste één die actief is met en één die actief is zonder metabole activering, gekozen uit de stoffen die zijn opgesomd in tabel 1) en met verschillende negatieve controles (waaronder onbehandelde culturen en verschillende oplosmiddelen/vehicula). Deze positieve en negatieve controleresponsen moeten in overeenstemming zijn met de literatuur. Deze vereiste geldt niet voor laboratoria die ervaring hebben, d.w.z. die beschikken over referentiegegevens uit het verleden zoals omschreven in de punten 47-50. Voor de MLA moeten de waarden die worden verkregen met de positieve en negatieve controles, consistent zijn met de aanbevelingen van de IWGT (zie tabel 2).
|
|
46.
|
Om te bewijzen dat het laboratorium bekwaam is om mutagene chemische stoffen op te sporen, de doeltreffendheid van het metabole-activeringssysteem te bepalen en de geschiktheid van de celgroeiomstandigheden tijdens behandeling, fenotypische expressie en selectie van de mutanten en de geschiktheid van de scoringsprocedures aan te tonen, moet er een selectie van positieve controlestoffen (zie tabel 1) worden onderzocht zowel met korte en lange behandeling (indien lange behandelingen worden toegepast) zonder metabole activering, als met korte behandeling met metabole activering. Er moet een bereik van concentraties van de geselecteerde stoffen worden gekozen dat reproduceerbare en concentratiegerelateerde stijgingen tot boven het achtergrondniveau oplevert, om de gevoeligheid en het dynamisch bereik van het testsysteem aan te tonen.
|
Controlegegevens uit het verleden
|
47.
|
Het laboratorium stelt vast:
|
—
|
een historisch bereik en historische spreiding voor de positieve controles;
|
|
—
|
een historisch bereik en historische spreiding voor de negatieve controles (met onbehandelde controlestoffen of oplosmiddelen).
|
|
|
48.
|
Wanneer voor het eerst gegevens worden vergaard voor een historische spreiding voor negatieve controles, dienen gelijktijdige negatieve controles te stroken met gepubliceerde gegevens over negatieve controles. Naarmate meer gegevens uit experimenten aan de controlespreiding worden toegevoegd, dienen gelijktijdige negatieve controles idealiter binnen de controlegrens van 95 % voor die spreiding te vallen (64) (65).
|
|
49.
|
De databank met gegevens over negatieve controles uit het verleden van het laboratorium moet aanvankelijk worden samengesteld op basis van minimaal 10 experimenten, maar bestaat bij voorkeur uit ten minste 20 experimenten die onder vergelijkbare experimentele omstandigheden werden uitgevoerd. Laboratoria dienen methoden voor kwaliteitscontrole, zoals regelkaarten (bv. C-kaart of X-kaart (65)), te gebruiken om na te gaan hoe variabel hun gegevens over positieve en negatieve controles zijn en om aan te tonen dat zij de methodologie „onder controle” hebben (66). Nadere bijzonderheden en aanbevelingen wat betreft de opbouw en het gebruik van een databank met historische gegevens zijn te vinden in de literatuur (64).
|
|
50.
|
Negatieve controlegegevens moeten bestaan uit de mutantfrequenties van enkelvoudige of bij voorkeur duploculturen zoals beschreven in punt 27. Gelijktijdige negatieve controles liggen idealiter binnen de 95 %-controlegrenzen van de verdeling van de verzameling historische negatieve controlegegevens van het laboratorium. Wanneer gegevens van negatieve controles buiten de 95 %-controlegrens vallen, kan opneming ervan in de historische controleverdeling toch aanvaardbaar zijn zolang deze gegevens geen extreme uitschieters zijn, er bewijs is dat het testsysteem „onder controle” is (zie punt 49) en er geen bewijs is van technisch of menselijk falen.
|
|
51.
|
Eventuele aanpassingen van het testprotocol moeten worden bekeken vanuit het oogpunt van de samenhang van de gegevens met de bestaande historische controledatabanken van het laboratorium. Eventuele grote inconsistenties moeten leiden tot de oprichting van een nieuwe databank met historische controlegegevens.
|
GEGEVENS EN RAPPORTAGE
Presentatie van de resultaten
|
52.
|
Voor zowel de MLA als de TK6 moeten in de presentatie van de gegevens voor zowel de behandelde culturen als de controleculturen alle gegevens worden opgenomen die nodig zijn om de cytotoxiciteit (respectievelijk RTG of RS) en de mutantfrequenties te berekenen, zoals hieronder beschreven.
|
|
53.
|
Voor de MLA moeten voor elke cultuur apart gegevens worden verstrekt over de RSG, de RTG, de kloneringsefficiëntie op het moment van selecteren van de mutanten en het aantal mutantkolonies (voor de agarversie) of het aantal lege putjes (voor de microtiterplaatversie). De MF moet worden uitgedrukt als het aantal mutantcellen per miljoen overlevende cellen. Als de respons positief is, moeten voor ten minste één concentratie van de teststof (normaal gesproken de hoogste positieve concentratie) en de negatieve en positieve controles MF’s voor kleine en grote kolonies (en/of percentage van de totale MF) worden verstrekt. Als de respons negatief is, moeten voor de negatieve controle en de positieve controle de MF’s voor kleine en grote kolonies worden verstrekt.
|
|
54.
|
Voor de TK6 moeten voor elke cultuur apart gegevens worden verstrekt over de RS, de kloneringsefficiëntie op het moment van selecteren van de mutanten en het aantal lege putjes voor vroeg verschijnende en laat verschijnende mutanten. De MF’s moeten worden uitgedrukt als de verhouding tussen het aantal mutantcellen en het aantal overlevende cellen, waarbij zowel de totale MF als de MF (en/of het percentage van de totale MF) van de vroeg verschijnende en laat verschijnende mutanten moeten worden verstrekt.
|
Aanvaardbaarheidscriteria
|
55.
|
Voor zowel de MLA als de TK6 moet aan de volgende criteria worden voldaan, voordat de algemene resultaten voor een bepaalde teststof worden vastgesteld:
|
—
|
de test is onder twee experimentele omstandigheden uitgevoerd (korte behandeling met en zonder metabole activering – zie punt 33), tenzij één al positieve resultaten heeft opgeleverd;
|
|
—
|
er moet een voldoende aantal cellen en concentraties geanalyseerd kunnen worden (zie punten 27 en 34-36);
|
|
—
|
de criteria voor de selectie van de hoogste concentratie zijn consistent met de criteria beschreven in de punten 28-30.
|
|
Aanvaardbaarheidscriteria voor de negatieve en positieve controles
|
56.
|
De MLA-deskundigenwerkgroep van de IWGT heeft een grote hoeveelheid MLA-gegevens geanalyseerd, wat geresulteerd heeft in een internationale consensus voor specifieke aanvaardbaarheidscriteria voor de MLA (1) (2) (3) (4) (5). In deze testmethode worden dan ook specifieke aanbevelingen gedaan voor het bepalen van de aanvaardbaarheid van negatieve en positieve controles en voor het beoordelen van de resultaten voor een bepaalde stof in de MLA. De databank voor de TK6 is veel kleiner en is niet door een werkgroep geëvalueerd.
|
|
57.
|
Voor de MLA moet elk experiment worden geëvalueerd om te bepalen of de onbehandelde controle/oplosmiddelcontrole aan de aanvaardbaarheidscriteria van de MLA-deskundigenwerkgroep van de IWGT voldoet ((4) en tabel 2 hieronder) voor de: 1) MF (er zij op gewezen dat de IWGT voor de aanvaardbaarheid van de MF verschil maakt tussen de agarversie en de microtiterplaatversie van de MLA), 2) de kloneringsefficiëntie (CE) op het moment van selecteren van de mutanten en 3) de suspensiegroei (SG) voor de oplosmiddelcontrole (zie aanhangsel 2 voor de formules).
Tabel 2
Aanvaardbaarheidscriteria voor de MLA
|
Parameter
|
Methode met zachte agar
|
Methode met microtiterplaten
|
|
Mutantfrequentie
|
35 – 140 × 10-6
|
50 – 170 × 10-6
|
|
Kloneringsefficiëntie
|
65 – 120 %
|
65 – 120 %
|
|
Suspensiegroei
|
8- tot 32-voudig (behandeling van 3-4 uur) 32- tot 180-voudig (behandeling van 24 uur, indien uitgevoerd)
|
8- tot 32-voudig (behandeling van 3-4 uur) 32- tot 180-voudig (behandeling van 24 uur, indien uitgevoerd)
|
|
|
58.
|
Voor de MLA moet elke test ook worden geëvalueerd om te bepalen of voor de positieve controle(s) aan ten minste één van de volgende twee door de MLA-deskundigenwerkgroep van de IWGT ontwikkelde aanvaardbaarheidscriteria wordt voldaan:
|
—
|
de positieve controle moet een absolute toename in de totale MF vertonen, d.w.z. een toename die hoger is dan de spontane achtergrond-MF (een geïnduceerde MF, of IMF) van ten minste 300 × 10-6. De MF van de kleine kolonies moet goed zijn voor ten minste 40 % van de IMF;
|
|
—
|
de positieve controle vertoont een toename in de MF voor kleine kolonies van ten minste 150 × 10-6 boven de MF in de gelijktijdige onbehandelde controle/oplosmiddelcontrole (een IMF voor kleine kolonies van 150 × 10-6).
|
|
|
59.
|
Voor de TK6 is de test aanvaardbaar als toevoeging van de gelijktijdige negatieve controle aan de databank met historische negatieve controlegegevens van het laboratorium zoals beschreven in de punten 48-49 aanvaardbaar wordt geacht. Bovendien moeten de gelijktijdige positieve controles (zie punt 32) reacties induceren die verenigbaar zijn met de reacties in de databank met historische positieve controlegegevens en moeten een statistisch significante toename opleveren vergeleken met de gelijktijdige negatieve controle.
|
|
60.
|
Voor beide tests moet de bovengrens voor de cytotoxiciteit die wordt waargenomen in de positievecontrolecultuur, gelijk zijn aan die in de experimentele culturen. Met andere woorden: de RTG/RS mag niet lager zijn dan 10 %. Om aan te tonen dat aan de aanvaardbaarheidscriteria voor de positieve controle wordt voldaan, is het voldoende om een enkele concentratie te gebruiken (of een van de concentraties van de positievecontroleculturen als er meer dan een concentratie wordt gebruikt). Bovendien moet de MF van de positieve controle binnen het aanvaardbare bereik liggen dat voor het laboratorium is vastgesteld.
|
Beoordeling en interpretatie van resultaten
|
61.
|
Voor de MLA heeft de MLA-werkgroep van de IWGT veel werk verricht op het vlak van de biologische relevantie en de criteria voor een positieve respons (4). Daarom zijn in deze testmethode specifieke aanbevelingen opgenomen voor het interpreteren van de teststofresultaten van de MLA (zie de punten 62-64). De databank voor de TK6 is veel kleiner en is niet door een werkgroep geëvalueerd. De aanbevelingen voor het interpreteren van de gegevens voor de TK6 worden dan ook in veel algemenere termen geformuleerd (zie de paragrafen 65-66). Bovendien worden er aanvullende aanbevelingen gedaan die voor beide tests gelden (zie de punten 67-71).
|
MLA
|
62.
|
Er wordt een benadering aanbevolen voor het definiëren van positieve en negatieve responsen om er zeker van te zijn dat de toegenomen MF biologisch relevant is. In plaats van de statistische analyse die doorgaans voor andere tests wordt gebruikt, berust deze benadering op het gebruik van een vooraf bepaalde geïnduceerde mutantfrequentie (d.w.z. toename in de MF boven de gelijktijdige controle), die de Global Evaluation Factor (GEF) wordt genoemd en die is gebaseerd op een analyse van de distributie van de MF-gegevens voor de negatieve controle van deelnemende laboratoria (4). Voor de agarversie van de MLA is de GEF 90 × 10-6, voor de microtiterplaatversie 126 × 10-6.
|
|
63.
|
Mits aan alle aanvaardbaarheidscriteria wordt voldaan, wordt een teststof als duidelijk positief beschouwd als in om het even welke van de onderzochte experimentele omstandigheden (zie punt 33) de toename van de MF boven de gelijktijdige achtergrond groter is dan de GEF en er een verband is tussen de toename en de concentratie (bv. aan de hand van een trendtoets). De teststof wordt dan in staat geacht mutatie te induceren in dit testsysteem.
|
|
64.
|
Mits aan alle aanvaardbaarheidscriteria wordt voldaan, wordt een teststof als duidelijk negatief beschouwd als er in alle onderzochte experimentele omstandigheden (zie punt 33) geen enkele concentratiegerelateerde respons is of, wanneer er wel een toename is in de MF, als deze niet groter is dan de GEF. De teststof wordt dan niet in staat geacht mutaties te induceren in dit testsysteem.
|
TK6
|
65.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk positief beschouwd als in een of meer van de onderzochte experimentele omstandigheden (zie punt 33):
|
—
|
ten minste één van de testconcentraties een statistisch significante toename vertoont vergeleken met de gelijktijdige negatieve controle;
|
|
—
|
de toename concentratiegerelateerd is bij evaluatie met een geschikte trendtoets (zie punt 33);
|
|
—
|
een of meer van de resultaten buiten de verdeling van de historische negatieve controlegegevens vallen (bv. op Poisson-verdeling gebaseerde 95 %-controlegrens; zie punt 48).
|
Wanneer aan al deze criteria is voldaan, wordt de teststof in staat geacht mutaties te induceren dit testsysteem. Aanbevelingen voor de meest geschikte statistische methoden zijn te vinden in de literatuur (66) (67).
|
|
66.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt de teststof als duidelijk negatief beschouwd als in alle onderzochte experimentele omstandigheden (zie punt 33):
|
—
|
geen van de testconcentraties een statistisch significante toename vertoont vergeleken met de gelijktijdige negatieve controle;
|
|
—
|
er geen concentratiegerelateerde toename is bij evaluatie met een geschikte trendtoets;
|
|
—
|
alle resultaten binnen de verdeling van de historische negatieve controlegegevens vallen (bv. op Poisson-verdeling gebaseerde 95 %-controlegrens; zie punt 48).
|
De teststof wordt dan niet in staat geacht mutaties te induceren in dit testsysteem.
|
Voor zowel MLA als TK6:
|
67.
|
Als de maximale concentratie is gebaseerd op cytotoxiciteit, moet voor de hoogste concentratie worden gestreefd naar een RTG/RS tussen de 20 en 10 %. Er is consensus over dat er bij de interpretatie van positieve resultaten voor moet worden gezorgd dat deze niet alleen worden gevonden tussen 20 en 10 % RTG/RS en dat een resultaat niet als positief mag worden beschouwd als de toename in de MF alleen is opgetreden bij 10 % RTG/RS of lager (indien beoordeeld) (2) (59).
|
|
68.
|
In sommige omstandigheden kunnen aanvullende gegevens nuttig zijn om vast te stellen dat een teststof niet mutageen is wanneer geen enkele cultuur een RTG-waarde vertoont van 10-20 % RTG/RS. Het betreft de volgende situaties: 1) Er is geen bewijs voor mutageniteit (bv. geen dosis-respons, geen MF’s die hoger zijn dan de MF’s in de gelijktijdige negatieve controle of de historische achtergrondbereiken enz.) in een reeks van gegevenspunten tussen de 100 % en 20 % RTG/RS en er is ten minste één gegevenspunt tussen de 20 en 25 % RTG/RS. 2) Er is geen bewijs voor mutageniteit (bv. geen dosis-respons, geen MF’s die hoger zijn dan de MF’s in de gelijktijdige negatieve controle of de historische achtergrondbereiken enz.) in een reeks van gegevenspunten tussen de 100 % en 25 % RTG/RS en er is ook een negatief gegevenspunt vlak onder de 10 % RTG/RS. In elk van beide situaties kan worden geconcludeerd dat de teststof negatief is.
|
|
69.
|
Een duidelijk positieve of negatieve reactie behoeft niet te worden bevestigd.
|
|
70.
|
In gevallen waarin de reactie noch duidelijk negatief noch duidelijk positief is zoals hierboven beschreven, en/of om de biologische relevantie van een resultaat te helpen vaststellen, moeten de gegevens door een deskundige en/of nader onderzoek worden beoordeeld. Het uitvoeren van een herhalingsexperiment onder mogelijk aangepaste experimentele omstandigheden [bv. concentratie-intervallen om de kans te vergroten dat gegevenspunten worden gevonden die binnen het bereik van 10-20 % RTG/RS liggen, andere omstandigheden bij metabole activering (d.w.z. S9-concentratie of S9-herkomst) en duur van de behandeling] kan nuttig zijn.
|
|
71.
|
In zeldzame gevallen zal op basis van de gegevens, zelfs na nader onderzoek, niet kunnen worden geconcludeerd dat de resultaten positief of negatief zijn. De reactie op de teststof moet dan als moeilijk te interpreteren worden beschouwd (geïnterpreteerd als even waarschijnlijk positief als negatief).
|
TESTVERSLAG
|
72.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Teststof:
|
—
|
bron, partijnummer, uiterste gebruiksdatum, indien beschikbaar;
|
|
—
|
stabiliteit van de teststof, indien bekend;
|
|
—
|
oplosbaarheid en stabiliteit van de teststof in het oplosmiddel, indien bekend;
|
|
—
|
meting van pH, osmolaliteit en neerslag in het kweekmedium waaraan de teststof werd toegevoegd, indien van toepassing.
|
|
|
Stof die uit één component bestaat:
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
chemische identificatiegegevens, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code, structuurformule, zuiverheid, chemische identiteit van onzuiverheden, indien van toepassing en praktisch haalbaar enz.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB’s en mengsels:
|
—
|
voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen.
|
|
|
|
|
Oplosmiddel:
|
—
|
motivering van de keuze van het oplosmiddel;
|
|
—
|
percentage oplosmiddel in het uiteindelijke kweekmedium.
|
|
|
|
Cellen:
|
|
Voor de voorraadculturen van het laboratorium:
|
—
|
type en bron van de cellen en geschiedenis van het testlaboratorium;
|
|
—
|
kenmerken van het karyotype en/of modaal aantal chromosomen;
|
|
—
|
methoden om de celculturen in leven te houden;
|
|
—
|
afwezigheid van mycoplasma;
|
|
|
|
|
Testomstandigheden:
|
—
|
achtergrond voor de keuze van de concentraties en het aantal celculturen, bijvoorbeeld gegevens over de cytotoxiciteit en beperkte oplosbaarheid;
|
|
—
|
samenstelling van het medium, CO2-concentratie, vochtigheidsgraad;
|
|
—
|
concentratie van de teststof uitgedrukt als uiteindelijke concentratie in het kweekmedium (bv. μg of mg/ml of mM van kweekmedium);
|
|
—
|
toegevoegde concentratie (en/of volume) van het oplosmiddel en de teststof in het kweekmedium;
|
|
—
|
duur van de behandeling;
|
|
—
|
celdichtheid bij de behandeling;
|
|
—
|
aard en samenstelling van het metabool activeringssysteem (bron van S9, methode voor de bereiding van de S9-mix, de concentratie of het volume van de S9-mix en S9 in het uiteindelijke kweekmedium, kwaliteitscontroles van S9);
|
|
—
|
positieve en negatieve controlestoffen, uiteindelijke concentraties voor alle behandelingsomstandigheden;
|
|
—
|
duur van de expressieperiode (met vermelding van het aantal geënte cellen en schema’s voor enting en overenting, indien van toepassing;
|
|
—
|
identiteit en concentratie van de selecterende stof;
|
|
—
|
voor de MLA moet worden aangegeven welke versie is gebruikt (agar of microtiterplaat);
|
|
—
|
criteria voor de aanvaardbaarheid van de tests;
|
|
—
|
methoden die zijn gebruikt om het aantal levensvatbare en mutantcellen te bepalen;
|
|
—
|
methoden die zijn gebruikt om de cytotoxiciteit te meten;
|
|
—
|
eventuele aanvullende informatie die betrekking heeft op de cytotoxiciteit en gebruikte methode;
|
|
—
|
duur van de incubatietijden na uitplating;
|
|
—
|
definitie van kolonies van een bepaalde omvang en aard (bv. criteria voor „kleine” en „grote” kolonies);
|
|
—
|
criteria om te bepalen of het resultaat positief, negatief of onduidelijk is;
|
|
—
|
gebruikte methoden voor het bepalen van de pH, osmolaliteit, indien uitgevoerd, en neerslag, indien van toepassing.
|
|
|
|
Resultaten:
|
—
|
aantal behandelde cellen en aantal overgeënte cellen voor elke cultuur;
|
|
—
|
toxiciteitsparameters (RTG voor MLA en RS voor TK6);
|
|
—
|
tekenen van neerslag en tijdstip van de bepaling;
|
|
—
|
aantal uitgeplate cellen in selectief en niet-selectief medium;
|
|
—
|
aantal kolonies in niet-selectief medium en aantal resistente kolonies in selectief medium en bijbehorende mutantfrequenties;
|
|
—
|
inventarisatie van de grootte van de kolonies voor de negatieve en positieve controles en, als de teststof positief is, ten minste een concentratie, en bijbehorende mutantfrequenties;
|
|
—
|
indien mogelijk het verband tussen concentratie en respons;
|
|
—
|
gegevens over tegelijkertijd uitgevoerde negatieve (oplosmiddel) en positieve controles (concentraties en oplosmiddelen);
|
|
—
|
gegevens over in het verleden uitgevoerde negatieve (oplosmiddel) en positieve controles (concentraties en oplosmiddelen) met vermelding van spreiding, gemiddelde en standaardafwijking. aantal tests waarop de historische controles zijn gebaseerd;
|
|
—
|
statistische analyses (voor afzonderlijke culturen en gepoolde duplo’s indien van toepassing), en eventuele p-waarden, en voor de MLA, de GEF-evaluatie.
|
|
|
|
Bespreking van de resultaten
|
|
LITERATUUR
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson,B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OESO (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, nr. 234, OESO, Parijs.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O’Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M.(2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res.,373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in voorbereiding).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N.,Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Beschikbaar op: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OESO (2014). Statistical Analysis Supporting the Revision of the Genotoxicity Test Guidelines. Environment, Health and Safety Publications, Series on Testing and Assessment (No 199), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Aanhangsel 1
DEFINITIES
Aneugeen
: elke chemische stof die of elk proces dat leidt tot aneuploïdie in cellen of organismen door de wisselwerking met componenten van de mitose en meiose in de celdelingscyclus.
Aneuploïdie
: elke afwijking van het normale diploïde (of haploïde) aantal chromosomen met één chromosoom of meer dan één chromosoom, maar niet met (een) volledige reeks(en) chromosomen (polyploïdie).
Chemische stof
: een stof of een mengsel.
Clastogeen
: elke chemische stof die of elk proces dat structurele chromosoomafwijkingen veroorzaakt in populaties van cellen of organismen.
Cytotoxiciteit
: voor de bepalingen bestreken in deze testmethode is cytotoxiciteit gelijkgesteld aan een vermindering van de relatieve totale groei (RTG) voor de MLA of de relatieve overleving (RS) voor de TK6.
Fenotypische expressietijd
: de tijd na behandeling waarin de genetische verandering gefixeerd wordt in het genoom en ongewijzigde genproducten verdwijnen, zodat het fenotypische kenmerk verandert.
Genotoxisch
: een algemene term die alle typen DNA- of chromosoombeschadiging omvat, waaronder DNA-breuken, adductherschikkingen, mutaties, chromosoomafwijkingen en aneuploïdie. Niet alle soorten genotoxische effecten leiden tot mutaties of stabiele chromosoombeschadiging.
Kloneringsefficiëntie
: Percentage van in lage dichtheid uitgeplate cellen die in staat zijn om uit te groeien tot een kolonie die geteld kan worden.
Mitotische recombinatie
: recombinatie tussen homologe chromatiden tijdens de mitose, die mogelijk leidt tot de inductie van breuken in het dubbelstrengig DNA of verlies van heterozygotie.
Mutageen
: produceert een erfelijke wijziging van DNA-basenpaarsequentie(s) in genen of van de structuur van chromosomen (chromosoomafwijkingen).
Mutagenen met basenpaarvervanging
: chemische stoffen die vervanging van een of meer basenparen in het DNA veroorzaken.
Mutagenen met leesraamverschuiving
: chemische stoffen die invoeging of deletie van een of meer basenparen in het DNA-molecuul veroorzaken.
Mutantfrequentie (MF)
: de verhouding tussen het aantal waargenomen mutantcellen en het aantal levensvatbare cellen.
Onbehandelde controles
: culturen die geen behandeling ondergaan (d.w.z. noch teststof noch oplosmiddel), maar op dezelfde wijze worden verwerkt als de culturen die de teststof ontvangen.
Oplosmiddelcontrole
: algemene term om de controleculturen aan te duiden die uitsluitend het oplosmiddel ontvangen dat wordt gebruikt om de teststof op te lossen.
Relatieve overleving (RS)
: de RS wordt gebruikt als maat voor behandelingsgerelateerde cytotoxiciteit in de TK6. De RS is de relatieve kloneringsefficiëntie (CE) van onmiddellijk na de celbehandeling uitgeplate cellen, gecorrigeerd voor eventueel verlies van cellen gedurende de behandeling, vergeleken met de kloneringsefficiëntie van de negatieve controle.
Relatieve suspensiegroei (RSG)
: voor de MLA: de relatieve, totale suspensiegroei over twee dagen van de testcultuur, vergeleken met de totale suspensiegroei over twee dagen van de negatieve/oplosmiddelcontrole (Clive en Spector, 1975). De RSG moet de relatieve groei omvatten van de testcultuur vergeleken met de negatieve/oplosmiddelcontrole tijdens de behandelingsperiode.
Relatieve totale groei (RTG)
: de RTG wordt gebruikt als maat voor behandelingsgerelateerde cytotoxiciteit in de MLA. De RTG is een maat voor de relatieve groei (ten opzichte van de vehiculumcontrole) van de testculturen tijdens de behandelingsfase, de tweedaagse expressiefase en de fase van de klonering voor het selecteren van de mutanten. De RSG van elke testcultuur wordt vermenigvuldigd met de relatieve kloneringsefficiëntie van de testcultuur op het moment van selecteren van de mutanten en uitgedrukt ten opzichte van de kloneringsefficiëntie van de negatieve/oplosmiddelcontrole (Clive en Spector, 1975).
S9-leverfractie
: supernatant van leverhomogenaat na centrifugering bij 9 000 g, d.w.z. ruw leverextract.
S9-mix
: mix van de S9-leverfractie en cofactoren die nodig zijn voor de metabole enzymactiviteit.
Suspensiegroei (SG)
: de x-voudige toename van het aantal cellen in de loop van de behandelingsfase en de expressiefase van de MLA. Voor de korte (3 of 4 uur) behandeling wordt de SG berekend door de x-voudige toename op dag 1 te vermenigvuldigen met de x-voudige toename op dag 2. Als een 24-urige behandeling wordt toegepast, is de SG de x-voudige toename gedurende de 24-urige behandeling, vermenigvuldigd met de x-voudige toenamen op expressiedagen 1 en 2.
Teststof
: alle volgens deze testmethode geteste stoffen of mengsels.
Voorwaartse mutatie
: een genmutatie van het oudertype naar de mutantvorm die leidt tot wijziging of verlies van de enzymactiviteit of de functie van het gecodeerde eiwit.
Aanhangsel 2
FORMULES
Cytotoxiciteit
Voor beide versies (agar en microtiterplaat) van de MLA
De cytotoxiciteit wordt gedefinieerd als de relatieve totale groei (RTG), die de relatieve suspensiegroei (RSG) omvat tijdens de 2-daagse expressieperiode en de relatieve kloneringsefficiëntie (RCE) verkregen op het moment van selecteren van de mutanten. De RTG, RSG en RCE worden alle drie uitgedrukt als percentage.
Berekening van de RSG: de suspensiegroei één (SG1) is de groeisnelheid tussen dag 0 en dag 1 (celconcentratie op dag 1 / celconcentratie op dag 0) en suspensiegroei twee (SG2) is de groeisnelheid tussen dag 1 en dag 2 (celconcentratie op dag 2 / celconcentratie op dag 1). De RSG is de totale SG (SG1 × SG2) voor de behandelde cultuur vergeleken met de onbehandelde/oplosmiddelcontrole.Dus: RSG = [SG1(test) × SG2(test)]/ [SG1(controle) × SG2(controle)] De SG1 moet worden berekend vanaf de oorspronkelijke celconcentratie die is gebruikt aan het begin van de behandeling van de cellen. Aldus wordt rekening gehouden met eventuele verschillen in cytotoxiciteit die optreden in de testcultu(u)r(en) tijdens de behandeling van de cellen.
De RCE is de relatieve kloneringsefficiëntie van de testcultuur vergeleken met de relatieve kloneringsefficiëntie van de onbehandelde/oplosmiddelcontrole, verkregen op het moment van selecteren van de mutanten.
Relatieve totale groei (RTG):
RTG=RSG × RCE
TK6
Relatieve overleving (RS):
De cytotoxiciteit wordt beoordeeld aan de hand van de relatieve overleving, d.w.z. de kloneringsefficiëntie (CE) van onmiddellijk na de behandeling uitgeplate cellen, gecorrigeerd voor eventueel verlies van cellen gedurende de behandeling, vergeleken met de kloneringsefficiëntie in de negatieve controles (waaraan een overleving van 100 % wordt toegekend). De correctie voor verlies van cellen tijdens de behandeling kan worden berekend als:
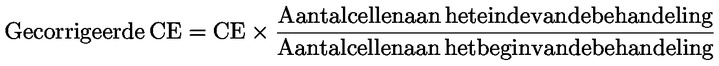
De RS voor een met een teststof behandelde cultuur wordt berekend als:

Mutantfrequentie voor zowel MLA als TK6:
De mutantfrequentie (MF) is de kloneringsefficiëntie van mutantkolonies in selectief medium (CEM) gedeeld door de kloneringsefficiëntie in niet-selectief medium op het moment van selecteren van de mutanten (CEV). Dat wil zeggen: MF = CEM / CEV. De berekening van deze twee kloneringsefficiënties wordt hieronder beschreven voor zowel de kloneringsmethode in agar als die in microtiterplaten.
MLA in Agar: in de versie van de MLA in zachte agar, worden het aantal kolonies op de plaat voor het selecteren van de mutanten (CM) en het aantal kolonies op de ongeselecteerde plaat/de plaat voor het bepalen van de kloneringsefficiëntie (telling van levensvatbare cellen) (CV) verkregen door rechtstreekse telling van de klonen. Wanneer er 600 cellen worden uitgeplaat voor het bepalen van de kloneringsefficiëntie (CE) voor de platen voor het selecteren van mutanten (CEM) en de ongeselecteerde platen/de platen voor het bepalen van de kloneringsefficiëntie (telling van levensvatbare cellen) (CEV) en er voor het selecteren van de mutanten 3 × 106 cellen worden gebruikt,
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
MLA en TK6 in microtiterplaten:
in de microtiterplaatversie van de MLA, worden CM en CV bepaald door het waarschijnlijke aantal kolonies per putje (P) te vermenigvuldigen met het totale aantal putjes (TW) op de microtiterplaten.
CM = PM × TWM
CV = PV × TWV
Vanaf de nulterm van de Poissonverdeling (Furth et al., 1981) wordt de P gegeven door
P = –ln (EW / TW)
Waarbij EW staat voor lege putjes en TW voor het totale aantal putjes. Daaruit volgt:
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
Voor de microtiterplaatversie van de MLA worden de mutantfrequenties voor kleine en grote kolonies op gelijke wijze berekend aan de hand van het relevante aantal lege putjes voor kleine en grote kolonies.
Voor de TK6 worden de mutantfrequenties voor kleine en grote kolonies gebaseerd op de vroeg verschijnende en laat verschijnende mutanten.
B.68 IN-VITROTESTMETHODE MET KORTE BLOOTSTELLING VOOR DE IDENTIFICATIE VAN i) CHEMISCHE STOFFEN DIE ERNSTIG OOGLETSEL VEROORZAKEN, EN ii) CHEMISCHE STOFFEN WAARVOOR CLASSIFICATIE VOOR OOGIRRITATIE OF ERNSTIG OOGLETSEL NIET NODIG IS
INLEIDING
|
1.
|
Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 491 (2017) van de OESO. De testmethode met korte blootstelling (Short Time Exposure, STE) is een in-vitromethode die onder bepaalde voorwaarden en binnen specifieke beperkingen kan worden gebruikt voor de gevarenindeling en etikettering van chemische stoffen (stoffen en mengsels) die ernstig oogletsel veroorzaken, en chemische stoffen waarvoor classificatie voor ernstig oogletsel of oogirritatie niet nodig is, zoals gedefinieerd door het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (Globally Harmonized System of Classification and Labelling of Chemicals, GHS) van de Verenigde Naties (VN) (1) en Verordening (EU) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels (CLP-verordening) (68).
|
|
2.
|
Jarenlang is voor het beoordelen van het mogelijke gevaar van chemische stoffen voor de ogen voornamelijk een in-vivo-oogtest bij konijnen (TM B.5 (8), gelijkwaardig aan TG 405 van de OESO) gebruikt. Naar algemene verwachting zal er in de nabije toekomst geen in-vitro-alternatief komen dat op zichzelf de in-vivo-oogtest bij konijnen volledig zal kunnen vervangen om voorspellingen te doen voor het volledige responsbereik voor ernstig oogletsel of oogirritatie voor verschillende klassen chemische stoffen. Strategische combinaties van alternatieve testmethoden die worden gebruikt in een (trapsgewijze) teststrategie, kunnen de oogtest bij konijnen mogelijk echter wel volledig vervangen (2). De top-downbenadering is bedoeld voor het testen van chemische stoffen waarvan op basis van bestaande informatie verwacht kan worden dat ze een hoog irritatiepotentieel hebben of ernstig oogletsel veroorzaken. Anderzijds is de bottom-upbenadering bedoeld voor het testen van chemische stoffen waarvan op basis van bestaande informatie verwacht kan worden dat ze onvoldoende oogirritatie zullen veroorzaken om classificatie noodzakelijk te maken. Hoewel de STE-testmethode niet geacht wordt de in-vivo-oogtest bij konijnen volledig te kunnen vervangen, is zij wel bruikbaar als onderdeel van een trapsgewijze teststrategie voor classificatie en etikettering volgens de regelgeving, zoals de top-down-/bottom-upbenadering, voor het identificeren zonder verdere tests van i) chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS/CLP-categorie 1), en ii) chemische stoffen (uitgezonderd sterk vluchtige stoffen en alle vaste chemische stoffen, behalve oppervlakteactieve stoffen) waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (VN-GHS/CLP Geen categorie) (1) (2). Voor een chemische stof waarvoor met de STE-testmethode niet wordt voorspeld dat deze ernstig oogletsel veroorzaakt (VN-GHS/CLP-categorie 1), en die evenmin als VN-GHS/CLP Geen categorie (veroorzaakt geen ernstig oogletsel of oogirritatie) wordt aangemerkt, moeten echter aanvullende tests worden uitgevoerd om tot een definitieve classificatie te komen. Bovendien moeten, voordat de STE-testmethode wordt gebruikt in een bottom-upbenadering volgens andere classificatiesystemen dan het VN-GHS/CLP-systeem, de toepasselijke regelgevingsinstanties worden geraadpleegd. Bij de keuze van de meest geschikte testmethode en het gebruik van deze testmethode moet de OESO-leidraad voor een geïntegreerde test- en beoordelingsbenadering (Integrated Approach on Testing and Assessment, IATA) voor ernstig oogletsel en oogirritatie worden gevolgd (14).
|
|
3.
|
Deze testmethode is bedoeld om de procedures te beschrijven om het mogelijke gevaar van een teststof voor de ogen te beoordelen op basis van het vermogen van deze chemische stof om cytotoxiciteit te veroorzaken in de STE-testmethode. Het cytotoxische effect van chemische stoffen op de hoornvliesepitheelcellen is een belangrijk werkingsmechanisme (mode of action, MOA) dat tot schade aan het corneale epitheel en oogirritatie leidt. De levensvatbaarheid van cellen in de STE-testmethode wordt bepaald door kwantitatieve meting, na extractie uit de cellen, van blauw formazanzout dat door de levende cellen wordt geproduceerd door enzymatische omzetting van de vitale kleurstof MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazoliumbromide), die ook bekend is onder de naam thiazolylblauw tetrazoliumbromide (3). De verkregen cellevensvatbaarheid wordt vergeleken met de oplosmiddelcontrole (relatieve levensvatbaarheid) en gebruikt om het mogelijke gevaar van de teststof voor de ogen te bepalen. Een teststof wordt ingedeeld als VN-GHS/CLP-categorie 1, wanneer zowel de concentratie van 5 % als die van 0,05 % tot een cellevensvatbaarheid leiden die gelijk is aan of lager dan (≤) 70 %. Anderzijds wordt voorspeld dat een teststof moet worden ingedeeld als VN-GHS/CLP Geen categorie, wanneer zowel de concentratie van 5 % als die van 0,05 % tot een cellevensvatbaarheid van meer dan (gt;) 70 % leiden.
|
|
4.
|
De term „teststof” wordt in deze teststof gebruikt om te verwijzen naar datgene dat wordt getest en houdt geen verband met de toepasselijkheid van de STE-testmethode op het testen van stoffen en/of mengsels. Definities worden gegeven in het aanhangsel.
|
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
5.
|
Deze testmethode is gebaseerd op een protocol dat is ontwikkeld door Kao Corporation (4). Dit protocol is aan twee verschillende valideringsstudies onderworpen: een studie door de valideringscommissie van de Japanse Vereniging voor alternatieven voor dierproeven (JSAAE) (5) en een studie door het Japans Centrum voor de validering van alternatieve methoden (JaCVAM) (6). Op basis van de verslagen van de valideringsstudies en de Background Review Documents voor de testmethode heeft NICEATM/ICCVAM een onafhankelijke beoordeling door vakgenoten uitgevoerd (7).
|
|
6.
|
Bij gebruik van de STE-testmethode om chemische stoffen (stoffen en mengsels) te identificeren die ernstig oogletsel veroorzaken (VN-GHS/CLP-categorie 1 (1)), is uit de gegevens die werden verkregen voor 125 chemische stoffen (zowel stoffen als mengsels), een algemene nauwkeurigheid gebleken van 83 % (104/125), een percentage vals-positieve uitslagen van 1 % (1/86) en een percentage vals-negatieve uitslagen van 51 % (20/39) in vergelijking met de in-vivo-oogtest bij konijnen (7). Het verkregen percentage vals-negatieve uitslagen is niet kritiek in deze context, omdat alle teststoffen die een cellevensvatbaarheid van ≤ 70 % bij een concentratie van 5 % en > 70 % bij een concentratie van 0,05 % veroorzaken, vervolgens worden getest met andere afdoend gevalideerde in-vitrotestmethoden, of als laatste optie met de in-vivo-oogtest bij konijnen, afhankelijk van de wettelijke voorschriften en volgens de huidige aanbevolen sequentiële teststrategie en op bewijskracht gebaseerde aanpakken (1) (8). Er werden voornamelijk stoffen die bestaan uit één component getest, hoewel er ook een beperkte hoeveelheid gegevens beschikbaar is over het testen van mengsels. De testmethode is vanuit technisch oogpunt evenwel toepasbaar voor het testen van stoffen die uit meerdere componenten bestaan en van mengsels. Voordat deze testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet echter worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. De STE-testmethode vertoont geen andere specifieke tekortkomingen wanneer zij wordt gebruikt om teststoffen te identificeren als VN-GHS/CLP-categorie 1. Onderzoekers zouden kunnen overwegen deze testmethode voor teststoffen te gebruiken, waarbij een cellevensvatbaarheid ≤ 70 % bij zowel een concentratie van 5 % en een concentratie van 0,05 % moet worden geaccepteerd als indicatief voor een respons die ernstig oogletsel veroorzaakt waardoor de teststof zonder verdere tests moet worden ingedeeld in VN-GHS/CLP-categorie 1.
|
|
7.
|
Bij gebruik van de STE-testmethode om chemische stoffen (stoffen en mengsels) te identificeren waarvoor classificatie voor oogirritatie en ernstig oogletsel niet nodig is (d.w.z. VN-GHS/CLP Geen categorie), is uit de gegevens die werden verkregen voor 130 chemische stoffen (zowel stoffen als mengsels), een algemene nauwkeurigheid gebleken van 85 % (110/130), een percentage vals-negatieve uitslagen van 12 % (9/73) en een percentage vals-positieve uitslagen van 19 % (11/57), vergeleken met de in-vivo-oogtest bij konijnen (7). Als sterk vluchtige en vaste stoffen, behalve oppervlakteactieve stoffen, worden uitgesloten van de gegevensset, verbeteren de algemene nauwkeurigheid tot 90 % (92/102), het percentage vals-negatieve uitslagen tot 2 % (1/54) en het percentage vals-positieve uitslagen tot 19 % (9/48) (7). De mogelijke tekortkomingen van de STE-testmethode, wanneer zij wordt gebruikt om teststoffen te identificeren waarvoor classificatie voor oogirritatie en ernstig oogletsel niet nodig is (VN-GHS/CLP Geen categorie), zijn daarom een hoog percentage vals-negatieve uitslagen voor i) sterk vluchtige stoffen met een dampdruk van meer dan 6 kPa en ii) vaste chemische stoffen (stoffen en mengsels) behalve oppervlakteactieve stoffen en mengsels die enkel uit oppervlakteactieve stoffen bestaan. Dergelijke chemische stoffen worden van het toepassingsgebied van de STE-testmethode uitgesloten (7).
|
|
8.
|
Behalve de chemische stoffen die worden genoemd in de punten 6 en 7, bevat de gegevensset die met de STE is gegenereerd, ook gegevens uit eigen laboratorium voor 40 mengsels. Deze hadden, vergeleken met de in-vivo-Draize-oogtest, een algemene nauwkeurigheid van 88 % (35/40), een percentage vals-positieve uitslagen van 50 % (5/10) en een percentage vals-negatieve uitslagen van 0 % (0/30) voor voorspellingen voor mengsels waarvoor classificatie volgens de VN-GHS/CLP-classificatiesystemen niet nodig is (9). De STE-testmethode is daarom wel toepasbaar om mengsels te identificeren als VN-GHS/CLP Geen categorie in een bottom-upbenadering met uitzondering van vaste mengsels, tenzij deze enkel uit oppervlakteactieve stoffen bestaan, als uitbreiding van de beperking ervan voor vaste stoffen. Bovendien moeten mengsels die stoffen bevatten met een dampdruk van meer dan 6 kPa, zorgvuldig worden beoordeeld om onderschatting te vermijden. Het gebruik van de testmethode voor deze mengsels moet per geval gerechtvaardigd worden.
|
|
9.
|
De STE-testmethode mag niet worden gebruikt om teststoffen te identificeren als VN-GHS/CLP-categorie 2 of VN-GHS-categorie 2A (irriterend voor de ogen) of 2B (licht irriterend voor de ogen), vanwege het aanzienlijke aantal chemische stoffen van VN-GHS/CLP-categorie 1 waarvan het mogelijke gevaar voor de ogen werd onderschat als categorie 2, 2A of 2B en chemische stoffen van VN-GHS/CLP Geen categorie waarvan het mogelijke gevaar voor de ogen werd overschat als categorie 2, 2A of 2B (7). Voor dit doel kan het nodig zijn verdere tests met een andere geschikte methode uit te voeren.
|
|
10.
|
De STE-testmethode is geschikt voor teststoffen die opgelost zijn of gedurende ten minste 5 minuten gelijkmatig gesuspendeerd blijven in fysiologische zoutoplossing, 5 % dimethylsulfoxide (DMSO) in zoutoplossing, of minerale olie. De STE-testmethode is niet geschikt voor teststoffen die onoplosbaar zijn of niet gedurende ten minste 5 minuten gelijkmatig gesuspendeerd blijven in fysiologische zoutoplossing, 5 % DMSO in zoutoplossing, of minerale olie. Bij de STE-testmethode kan minerale olie worden gebruikt vanwege de korte duur van de blootstelling. Daarom is de STE-testmethode geschikt om het mogelijke gevaar voor de ogen te voorspellen van teststoffen die onoplosbaar zijn in water (bv. vette alcoholen of ketonen), mits deze mengbaar zijn in ten minste een van de drie hierboven voorgestelde oplosmiddelen (4).
|
|
11.
|
De term „teststof” wordt in deze teststof gebruikt om te verwijzen naar datgene dat wordt getest (69), en houdt geen verband met de toepasselijkheid van de STE-testmethode op het testen van stoffen en/of mengsels.
|
PRINCIPE VAN DE TEST
|
12.
|
De STE-testmethode is een op cytotoxiciteit gebaseerde in-vitrobepaling die wordt uitgevoerd op een confluente monolaag van cellen van SIRC-cellen (Statens Seruminstitut Rabbit Cornea) die worden gekweekt in een microtiterplaat van polycarbonaat met 96 putjes (4). Na vijf minuten blootstelling aan een teststof wordt de cytotoxiciteit kwantitatief gemeten als de relatieve cellevensvatbaarheid van SIRC-cellen met behulp van de MTT-test (4). De afgenomen cellevensvatbaarheid wordt gebruikt voor het voorspellen van mogelijke schadelijke effecten die tot oogschade kunnen leiden.
|
|
13.
|
Er is beschreven dat wanneer een oplossing in het oog van een konijn wordt gedruppeld, 80 % ervan binnen drie à vier minuten via de conjunctivaalzak wordt uitgescheiden, terwijl van een oplossing die in een mensenoog wordt gedruppeld, meer dan 80 % binnen een à twee minuten wordt uitgescheiden (10). Met de STE-testmethode wordt gepoogd deze blootstellingstijden te benaderen en wordt de cytotoxiciteit als eindpunt gebruikt voor de beoordeling van de mate van beschadiging van SIRC-cellen na vijf minuten blootstelling aan de teststof.
|
AANTONING VAN BEKWAAMHEID
|
14.
|
Voordat de STE-testmethode die in deze testmethode wordt beschreven, routinematig wordt gebruikt, moeten laboratoria hun technische bekwaamheid aantonen door een correcte indeling van de elf in tabel 1 aanbevolen stoffen. Deze stoffen zijn zodanig geselecteerd dat ze het volledige responsbereik voor ernstig oogletsel of oogirritatie vertegenwoordigen op basis van de resultaten van de in-vivo-oogtest bij konijnen (TG 405) en het VN-GHS/CLP-classificatiesysteem (1). Andere selectiecriteria waren onder meer dat de stoffen in de handel verkrijgbaar zijn, dat er in-vivoreferentiegegevens van zeer goede kwaliteit beschikbaar zijn en dat er met de STE-testmethode gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn (3). Wanneer een stof uit de lijst niet beschikbaar is of indien zulks gerechtvaardigd is, mag een andere stof waarvoor voldoende in-vivo- en in-vitroreferentiegegevens beschikbaar zijn, worden gebruikt, mits dezelfde criteria als hier beschreven worden toegepast.
Tabel 1
Lijst van stoffen voor bekwaamheidstoetsing
|
Stof
|
CAS RN
|
Chemische klasse (70)
|
Fysische toestand
|
VN-GHS/CLP-cat. in vivo (71)
|
Oplosmiddel in STE-test
|
VN-GHS/CLP-cat. STE
|
|
Benzalkoniumchloride (10 %, waterig)
|
8001-54-5
|
Onium-verbinding
|
Vloeistof
|
Categorie 1
|
Zoutoplossing
|
Categorie 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Ether
|
Vloeistof
|
Categorie 1
|
Zoutoplossing
|
Categorie 1
|
|
Acid Red 92
|
18472-87-2
|
Heterocyclische verbinding; broomverbinding; chloorverbinding
|
Vaste stof
|
Categorie 1
|
Zoutoplossing
|
Categorie 1
|
|
Natriumhydroxide
|
1310-73-2
|
Alkali; Anorganische stof
|
Vaste stof
|
Categorie 1 (72)
|
Zoutoplossing
|
Categorie 1
|
|
Butyrolacton
|
96-48-0
|
Lacton; heterocyclische verbinding
|
Vloeistof
|
Categorie 2A (Categorie 2 in CLP)
|
Zoutoplossing
|
Er kan geen voorspelling worden gedaan
|
|
Octaan-1-ol
|
111-87-5
|
Alcohol
|
Vloeistof
|
Categorie 2A/B (73) (Categorie 2 in CLP)
|
Minerale olie
|
Er kan geen voorspelling worden gedaan
|
|
Cyclopentanol
|
96-41-3
|
Alcohol; Koolwaterstof, cyclisch
|
Vloeistof
|
Categorie 2A/B (74) (Categorie 2 in CLP)
|
Zoutoplossing
|
Er kan geen voorspelling worden gedaan
|
|
2-Ethoxyethylacetaat
|
111-15-9
|
Alcohol; Ether
|
Vloeistof
|
Geen categorie
|
Zoutoplossing
|
Geen categorie
|
|
Dodecaan
|
112-40-3
|
Koolwaterstof, acyclisch
|
Vloeistof
|
Geen categorie
|
Minerale olie
|
Geen categorie
|
|
Methylisobutylketon
|
108-10-1
|
Keton
|
Vloeistof
|
Geen categorie
|
Minerale olie
|
Geen categorie
|
|
1,1-Dimethylguanidinesulfaat
|
598-65-2
|
Amidine; Zwavelverbinding
|
Vaste stof
|
Geen categorie
|
Zoutoplossing
|
Geen categorie
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstracts Service
|
|
PROCEDURE
Bereiding van de monolaag van cellen
|
15.
|
De STE-testmethode moet worden uitgevoerd met de SIRC-cellijn van konijnenhoornvliescellen. Aanbevolen wordt om SIRC-cellen te betrekken van een goed gekwalificeerde celbank, zoals de American Type Culture Collection CCL60.
|
|
16.
|
SIRC-cellen worden gekweekt bij 37 °C in een vochtige atmosfeer met 5 % CO2, in een kweekfles met een kweekmedium dat bestaat uit Eagle’s Minimum Essential Medium (MEM), aangevuld met 10 % foetaal runderserum (FBS), 2 mM L-glutamine, 50-100 eenheden/ml penicilline en 50-100 μg/ml streptomycine. Cellen die confluent zijn geworden in de kweekfles, moeten worden gescheiden met een trypsine-ethyleendiaminetetraazijnzuur-oplossing, al dan niet met behulp van een celschraper. De cellen worden vermeerderd (bv. 2 tot 3 passages) in een kweekfles alvorens te worden gebruikt voor routinetests en mogen na ontdooiing niet meer dan 25 passages ondergaan.
|
|
17.
|
Cellen die gereed zijn voor gebruik in de STE-test, worden vervolgens bereid met een passende dichtheid en geënt in microtiterplaten met 96 putjes. De aanbevolen celdichtheid bij het enten is 6,0 × 103 cellen per putje, wanneer de cellen vier dagen na het enten worden gebruikt, of 3,0 × 103 cellen per putje, wanneer de cellen vijf dagen na het enten worden gebruikt, in een kweekvolume van 200 μl. Cellen die worden gebruikt voor de STE-test en die met de passende dichtheid worden geënt in een kweekmedium, bereiken op het moment van testen, d.w.z. vier of vijf dagen na het enten, een confluentie van meer dan 80 %.
|
Het aanbrengen van de teststoffen en de controlestoffen
|
18.
|
Het eerstekeus-oplosmiddel voor het oplossen of suspenderen van teststoffen is fysiologische zoutoplossing. Als de teststof een lage oplosbaarheid heeft of niet opgelost of gedurende ten minste vijf minuten gelijkmatig gesuspendeerd kan worden in zoutoplossing, wordt als tweedekeus-oplosmiddel 5 % DMSO (CAS RN 67-68-5) in zoutoplossing gebruikt. Voor teststoffen die niet opgelost of gedurende ten minste vijf minuten gelijkmatig gesuspendeerd kunnen worden in zoutoplossing of 5 % DMSO in zoutoplossing, wordt minerale olie (CAS RN 8042-47-5) gebruikt als derdekeus-oplosmiddel.
|
|
19.
|
Teststoffen worden opgelost of gelijkmatig gesuspendeerd in het geselecteerde oplosmiddel in een concentratie van 5 % (w/w) en verder serieel verdund met een factor 10 tot 0,5 % en 0,05 %. Elke teststof wordt getest in zowel een concentratie van 5 % als een concentratie van 0,05 %. De cellen die zijn gekweekt in een microtiterplaat met 96 putjes, worden gedurende vijf minuten bij kamertemperatuur blootgesteld aan 200 μl/putje van de teststofoplossing (of teststofsuspensie) in een concentratie van hetzij 5 %, hetzij 0,05 %. De teststoffen (stoffen die uit één component bestaan, of stoffen die uit meerdere componenten bestaan, of mengsels) worden als zuivere stoffen beschouwd en opgelost of gesuspendeerd volgens deze methode, ongeacht de zuiverheidsgraad.
|
|
20.
|
Het in punt 16 beschreven kweekmedium wordt in elke plaat van elke herhaalde bepaling als mediumcontrole gebruikt. Bovendien worden de cellen eveneens in elke plaat van elke herhaalde bepaling aan oplosmiddelcontrolemonsters blootgesteld. Van de in punt 18 vermelde oplosmiddelen is bevestigd dat ze geen negatieve invloed hebben op de levensvatbaarheid van SIRC-cellen.
|
|
21.
|
In de STE-testmethode wordt in elke plaat van elke herhaalde bepaling 0,01 % natriumlaurylsulfaat (SLS) in zoutoplossing als positieve controle gebruikt. Om de cellevensvatbaarheid van de positieve controle te berekenen, moet elke plaat van elke herhaalde bepaling ook een oplosmiddelcontrole van zoutoplossing bevatten.
|
|
22.
|
Om de compensatie voor de optische dichtheid te bepalen is ook een blanco nodig. Deze moet worden uitgevoerd met putjes die alleen fosfaatgebufferde zoutoplossing bevatten, maar geen calcium en magnesium (PBS-) of cellen.
|
|
23.
|
Elk monster (teststof in 5 % en 0,05 %, mediumcontrole, oplosmiddelcontrole en positieve controle) moet in elke herhaalde bepaling in drievoud worden getest door de cellen gedurende vijf minuten bij kamertemperatuur bloot te stellen aan 200 μl van de betreffende test- of controlestof.
|
|
24.
|
IJkstoffen zijn nuttig voor de beoordeling van potentiële oogirritatie bij onbekende chemische stoffen uit een specifieke chemische klasse of productklasse of voor de beoordeling van potentiële relatieve irritatie van een voor het oog irriterende stof binnen een reeks van irritatieresponsen.
|
Meting van de cellevensvatbaarheid
|
25.
|
Na de blootstelling worden de cellen tweemaal gewassen met 200 μl PBS en wordt er 200 μl MTT-oplossing (0,5 mg MTT/ml kweekmedium) toegevoegd. Na een reactietijd van twee uur in een incubator (37 °C, 5 % CO2) wordt de MTT-oplossing gedecanteerd, wordt het MTT-formazan geëxtraheerd met 200 μl 0,04 N zoutzuur-isopropanol gedurende 60 minuten in een donkere kamer bij kamertemperatuur en wordt met een plaatlezer de extinctie van de MTT-formazanoplossing bij 570 nm gemeten. Interferentie van teststoffen met de MTT-test (door kleurstoffen of rechtstreeks MTT reducerende stoffen) treedt alleen op als er na het wassen (tussen blootstelling en meting) een significante hoeveelheid teststof in het testsysteem achterblijft, hetgeen wel het geval is met 3D-gereconstrueerd menselijk hoornvlies of gereconstrueerde humane epidermisweefsels, maar niet relevant is voor de 2D-celculturen die voor de STE-testmethode worden gebruikt.
|
Interpretatie van de resultaten en voorspellingsmodel
|
26.
|
De waarden voor de optische dichtheid (OD) die voor elke teststof worden verkregen, worden vervolgens gebruikt voor het berekenen van de cellevensvatbaarheid in vergelijking met de oplosmiddelcontrole, die op 100 % wordt gesteld. De relatieve cellevensvatbaarheid wordt uitgedrukt als een percentage en verkregen door de OD van de teststof te delen door de OD van de oplosmiddelcontrole, nadat van beide waarden de OD van de blanco is afgetrokken.

Evenzo wordt de relatieve cellevensvatbaarheid van elke oplosmiddelcontrole uitgedrukt als een percentage en verkregen door de OD van elke oplosmiddelcontrole te delen door de OD van de mediumcontrole, nadat van beide waarden de OD van de blanco is afgetrokken.
|
|
27.
|
Er moeten drie onafhankelijke herhaalde bepalingen worden gedaan, elk met drie duploputjes (d.w.z. n=9). Aan de hand van het meetkundig gemiddelde van de drie putjes voor elke teststof en elke oplosmiddelcontrole in elke onafhankelijke herhaling wordt het meetkundige gemiddelde van de relatieve cellevensvatbaarheid berekend. Het uiteindelijke meetkundige gemiddelde van de cellevensvatbaarheid wordt berekend op basis van de drie onafhankelijke herhalingen.
|
|
28.
|
De drempelwaarden voor de cellevensvatbaarheid om teststoffen te identificeren als chemische stoffen die ernstig oogletsel veroorzaken (VN-GHS/CLP-categorie 1), en teststoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (VN-GHS/CLP Geen categorie), worden hieronder gegeven.
|
Tabel 2
Voorspellingsmodel van de STE-testmethode
|
Cellevensvatbaarheid:
|
VN-GHS/CLP-classificatie
|
Toepassing
|
|
Bij 5 %
|
Bij 0,05 %
|
|
> 70 %
|
> 70 %
|
Geen categorie
|
Stoffen en mengsels, met uitzondering van: i) sterk vluchtige stoffen met een dampdruk van meer dan 6 kPa (75) en ii) vaste chemische stoffen (stoffen en mengsels) behalve oppervlakteactieve stoffen en mengsels die enkel uit oppervlakteactieve stoffen bestaan
|
|
≤ 70 %
|
> 70 %
|
Er kan geen voorspelling worden gedaan
|
Niet van toepassing
|
|
≤ 70 %
|
≤ 70 %
|
Categorie 1
|
Stoffen en mengsels (76)
|
Aanvaardbaarheidscriteria
|
29.
|
De testresultaten worden aanvaardbaar geacht wanneer aan alle onderstaande criteria wordt voldaan:
|
a)
|
de OD van de mediumcontrole (blootgesteld aan kweekmedium) moet, na aftrek van de blanco-OD, 0,3 of hoger zijn;
|
|
b)
|
de levensvatbaarheid van de oplosmiddelcontrole moet 80 % of hoger zijn in ten opzichte van de mediumcontrole. Als er in elke herhaling meerdere oplosmiddelcontroles worden gebruikt, worden de resultaten voor de teststoffen die met die oplosmiddelen zijn getest, aanvaardbaar geacht als elk van deze controles een cellevensvatbaarheid van meer dan 80 % laat zien;
|
|
c)
|
de cellevensvatbaarheid die wordt verkregen met de positieve controle (0,01 % SLS), moet binnen twee standaarddeviaties van het historische gemiddelde liggen. De bovenste en onderste aanvaardbaarheidsgrenzen voor de positieve controle moeten regelmatig worden bijgewerkt, d.w.z. eens in de drie maanden of elke keer dat een aanvaardbare test wordt uitgevoerd in laboratoria waar de tests niet regelmatig (d.w.z. minder dan eens per maand) worden uitgevoerd. Wanneer een laboratorium een onvoldoende aantal experimenten voltooit om een statistisch robuuste verdeling van positieve controles vast te stellen, is het aanvaardbaar om de bovenste en onderste aanvaardbaarheidsgrenzen te gebruiken die door de ontwikkelaar van de methode zijn vastgesteld, d.w.z. 21,1 % en 62,3 % volgens zijn historische laboratoriumgegevens, terwijl er tijdens de eerste routinetests een verdeling wordt opgebouwd voor het eigen laboratorium;
|
|
d)
|
de standaarddeviatie van de uiteindelijke cellevensvatbaarheid die wordt afgeleid uit de drie onafhankelijke herhalingen, moet kleiner zijn dan 15 % voor zowel de concentratie van 5 % als die van 0,05 % van de teststof.
Als aan een of meer van deze criteria niet wordt voldaan, moeten de resultaten worden verworpen en moeten drie nieuwe onafhankelijke herhalingen worden uitgevoerd.
|
|
GEGEVENS EN RAPPORTAGE
Gegevens
|
30.
|
In het verslag moeten de gegevens voor elk afzonderlijk putje (bv. de waarden voor de cellevensvatbaarheid) van elke herhaling, evenals het algemene gemiddelde, de SD en de indeling worden opgenomen.
|
Testverslag
|
31.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Teststof en controlestoffen
|
—
|
stof die uit één component bestaat: chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-registratienummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
—
|
stof die uit meerdere componenten bestaat, UVCB en mengsel: karakterisering, voor zover mogelijk aan de hand van bv. chemische identiteit (zie hierboven), zuiverheid, kwantitatief voorkomen en relevante fysisch-chemische eigenschappen (zie hierboven) van de bestanddelen, voor zover beschikbaar;
|
|
—
|
fysische toestand, vluchtigheid, pH, logP, molecuulgewicht, chemische klassen en aanvullende fysisch-chemische eigenschappen die relevant zijn voor het uitvoeren van het onderzoek, voor zover beschikbaar;
|
|
—
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
—
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
—
|
bewaaromstandigheden en stabiliteit, voor zover beschikbaar.
|
|
|
|
Voorwaarden en procedures voor de testmethode
|
—
|
naam en adres van de opdrachtgever, testinstallatie en de onderzoeksleider;
|
|
—
|
een beschrijving van de gebruikte testmethode;
|
|
—
|
de gebruikte cellijn, de herkomst ervan, het aantal passages en de confluentie van de voor het testen gebruikte cellen;
|
|
—
|
gedetailleerde informatie over de gebruikte testprocedure;
|
|
—
|
het aantal gebruikte herhalingen en duplo’s;
|
|
—
|
de gehanteerde concentraties van de teststof (indien verschillend van de aanbevolen concentraties);
|
|
—
|
motivering van de keuze van het oplosmiddel voor elke teststof;
|
|
—
|
duur van de blootstelling aan de teststof (indien verschillend van de aanbevolen tijd);
|
|
—
|
een beschrijving van eventuele wijzigingen in de testprocedure;
|
|
—
|
een beschrijving van de gebruikte beoordelings- en beslissingscriteria;
|
|
—
|
een verwijzing naar het historische gemiddelde en de bijbehorende standaarddeviatie (SD) voor de positieve controle:
|
|
—
|
aantoning dat het laboratorium bekwaam is om de testmethode toe te passen (bv. door de stoffen voor bekwaamheidstoetsing te testen) of aantoning dat de prestaties van de testmethode in de loop van de tijd kunnen worden gereproduceerd.
|
|
|
|
Resultaten
|
—
|
voor elke teststof en controlestof, en elke geteste concentratie, moeten de afzonderlijke OD-waarden per duploputje, het rekenkundig gemiddelde van de OD-waarden voor elke onafhankelijke herhaling, de procentuele cellevensvatbaarheid voor elke onafhankelijke herhaling en het uiteindelijke rekenkundige gemiddelde van de procentuele cellevensvatbaarheid en de bijbehorende SD over drie herhalingen in een tabel worden weergegeven;
|
|
—
|
resultaten voor medium-, oplosmiddel- en positieve controle waaruit blijkt dat het onderzoek aan de aanvaardbaarheidscriteria voldoet;
|
|
—
|
een beschrijving van andere waargenomen effecten;
|
|
—
|
de uiteindelijk uit de resultaten afgeleide indeling met vermelding van het gebruikte voorspellingsmodel/de gehanteerde beslissingscriteria.
|
|
|
|
Bespreking van de resultaten
|
|
LITERATUUR
|
(1)
|
Verenigde Naties (VN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Beschikbaar op: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Beschikbaar op: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Hoofdstuk B.5 van deze bijlage, Acute oogirritatie/-corrosie.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442-449.
|
|
(13)
|
OESO (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental Health and Safety Publications, Series on Testing and Assessment (No 34), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(14)
|
OESO (2017). Guidance Document on Integrated Approaches to Testing and Assessment (IATA) for Serious Eye Damage and Eye Irritation. Environmental Health and Safety Publications, Series on Testing and Assessment (No 263), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
Aanhangsel
DEFINITIES
Betrouwbaarheid
: indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (13).
Bottom-upbenadering
: een trapsgewijze benadering die wordt gebruikt voor een teststof waarvan wordt vermoed dat classificatie voor oogirritatie of ernstig oogletsel niet nodig is, die begint met het onderscheiden van chemische stoffen waarvoor classificatie niet nodig is (negatief resultaat) van andere chemische stoffen (positief resultaat).
Chemische stof
: een stof of een mengsel.
Ernstig oogletsel
: weefselbeschadiging in het oog of een ernstige fysieke gezichtsvermindering na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen niet volledig omkeerbaar is. Verwisselbaar met „Omkeerbare effecten op ogen” en met „VN-GHS/CLP-categorie 1”.
Gevaar
: inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld.
Gevoeligheid
: het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (10).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN))
: een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar worden gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (1).
IJkstof
: een stof die wordt gebruikt als norm voor vergelijking met een teststof. Een ijkstof moet de volgende eigenschappen hebben: i) (een) constante en betrouwbare bron(nen); ii) een structurele en functionele gelijkenis met de klasse stoffen die getest wordt; iii) hun fysische/chemische kenmerken moeten bekend zijn; iv) ondersteunende gegevens over bekende effecten en v) een bekende werkzaamheid binnen het bereik van de gewenste respons.
Mediumcontrole
: een onbehandeld monster dat alle componenten bevat van een testsysteem. Dit monster wordt samen met de met de teststof behandelde monsters en andere controlemonsters verwerkt om te bepalen of het oplosmiddel interageert met het testsysteem.
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
MTT
: 3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazoliumbromide; thiazolylblauw tetrazoliumbromide.
Nauwkeurigheid
: de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Dit is een maat voor de prestaties van de testmethode en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt (13).
OD
: optische dichtheid.
Oogirritatie
: veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig omkeerbaar zijn. Verwisselbaar met „Omkeerbare effecten op ogen” en met „VN-GHS/CLP-categorie 2”.
Oplosmiddel-/vehiculumcontrole
: een onbehandeld monster dat alle componenten bevat van een testsysteem, met inbegrip van het oplosmiddel of vehiculum, dat tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt wordt om de referentierespons voor de monsters die behandeld werden met de in hetzelfde oplosmiddel of vehiculum opgeloste teststof vast te stellen. Als dit monster getest wordt met een tegelijkertijd gemeten mediumcontrole, toont het ook aan of het oplosmiddel of vehiculum met het testsysteem interageert.
Oppervlakteactieve stof
: ook „agens dat inwerkt op de oppervlakte” genoemd. Dit is een chemische stof, zoals een detergens, die de oppervlaktespanning van een vloeistof kan verlagen en het zo mogelijk maakt dat de vloeistof gaat schuimen of in vaste stoffen doordringt; ook bekend als een bevochtigingsmiddel.
Positieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positievecontrolerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn.
Percentage vals-negatieve uitslagen
: het percentage van alle positieve chemische stoffen dat door een testmethode onterecht als negatief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode.
Percentage vals-positieve uitslagen
: het percentage van alle negatieve chemische stoffen dat door een testmethode onterecht als positief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode.
Relevantie
: beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (10).
Specificiteit
: het percentage van alle negatieve/inactieve chemische stoffen dat door de test correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (13).
Stof
: een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Teststof
: alle volgens deze testmethode geteste stoffen of mengsels.
Top-downbenadering
: een trapsgewijze benadering die gebruikt wordt voor een teststof waarvan wordt vermoed dat deze ernstig oogletsel veroorzaakt, die begint met het onderscheiden van chemische stoffen die ernstig oogletsel veroorzaken (positief resultaat) van andere chemische stoffen (negatief resultaat).
Trapsgewijze teststrategie
: een trapsgewijze teststrategie waarbij alle bestaande informatie over een teststof in een bepaalde volgorde wordt herbezien aan de hand van een proces op basis van bewijskracht op elke trap om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap. Indien het irritatiepotentieel van een teststof kan worden bepaald op basis van de bestaande informatie, dan zijn bijkomende tests niet vereist. Indien het irritatiepotentieel van een teststof niet kan worden bepaald op basis van de bestaande informatie, dan wordt een trapsgewijze sequentiële testprocedure op dieren uitgevoerd tot een ondubbelzinnige indeling mogelijk is.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
VN-GHS/CLP-categorie 1
: zie „Ernstig oogletsel”.
VN-GHS/CLP-categorie 2
: zie „Oogirritatie”.
VN-GHS/CLP Geen categorie
: chemische stoffen die niet worden ingedeeld in VN-GHS/CLP-categorie 1 of 2 (2A of 2B).
B.69 TESTMETHODE MET GERECONSTRUEERD MODEL VAN MENSELIJK HOORNVLIESEPITHEEL VOOR DE IDENTIFICATIE VAN CHEMISCHE STOFFEN WAARVOOR CLASSIFICATIE EN ETIKETTERING VOOR OOGIRRITATIE OF ERNSTIG OOGLETSEL NIET NODIG IS
INLEIDING
|
1.
|
Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 492 (2017) van de OESO. Onder ernstig oogletsel wordt verstaan weefselbeschadiging in het oog of een ernstige fysieke gezichtsvermindering na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen niet volledig omkeerbaar is, zoals gedefinieerd door het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (Globally Harmonized System of Classification and Labelling of Chemicals, GHS) van de Verenigde Naties (VN) (1) en Verordening (EU) nr. 1272/2008 betreffende de indeling, etikettering en verpakking van stoffen en mengsels (CLP-verordening) (77). Eveneens volgens het VN-GHS en de CLP wordt onder oogirritatie verstaan veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig omkeerbaar zijn. Teststoffen die ernstig oogletsel veroorzaken worden ingedeeld in VN-GHS/CLP-categorie 1, terwijl teststoffen die oogirritatie veroorzaken, worden ingedeeld in VN-GHS/CLP-categorie 2. Teststoffen die niet zijn geclassificeerd voor oogirritatie of ernstig oogletsel, worden gedefinieerd als chemische stoffen die niet voldoen aan de voorwaarden voor indeling in VN-GHS/CLP-categorie 1 of 2 (2A of 2B), d.w.z. er wordt naar verwezen als VN-GHS/CLP Geen categorie.
|
|
2.
|
De bepaling van ernstig oogletsel/oogirritatie ging meestal gepaard met het gebruik van proefdieren (TM B.5 (2)). Bij de keuze van de meest geschikte testmethode en het gebruik van deze testmethode moet de OESO-leidraad voor een geïntegreerde test- en beoordelingsbenadering (Integrated Approach on Testing and Assessment, IATA) voor ernstig oogletsel en oogirritatie worden gevolgd (39).
|
|
3.
|
In deze testmethode wordt een in-vitroprocedure beschreven voor het identificeren van chemische stoffen (stoffen en mengsels) waarvoor classificatie en etikettering voor oogirritatie of ernstig oogletsel volgens het VN-GHS en de CLP niet nodig is. Bij deze procedure wordt gebruikgemaakt van een gereconstrueerd model van menselijk hoornvliesepitheel (reconstructed human cornea-like epithelium, RhCE), dat nauwe overeenkomsten heeft met de histologische, morfologische, biochemische en fysiologische eigenschappen van menselijk hoornvliesepitheel. Er zijn voor ernstig oogletsel/oogirritatie als eindpunt voor de menselijke gezondheid nog vier andere in-vitrotestmethoden gevalideerd, wetenschappelijk geldig bevonden en goedgekeurd als TM B.47 (3), B.48 (4), B.61 (5) en B.68 (6)
|
|
4.
|
In deze testmethode zijn twee gevalideerde tests met in de handel verkrijgbare RhCE-modellen opgenomen. Er zijn voor de beoordeling van oogirritatie/ernstig oogletsel valideringsstudies verricht (7) (8) (9) (10) (11) (12) (13) met de EpiOcular™-oogirritatietest (Eye Irritation Test, EIT) en de SkinEthic™ Human Corneal Epithelium (HCE) EIT. Bij elk van deze tests worden in de handel verkrijgbare RhCE-weefselpreparaten als testsysteem gebruikt. Ze worden hierna aangeduid als de Validated Reference Methods – respectievelijk VRM1 en VRM2. Op basis van de valideringsstudies en de onafhankelijke beoordeling daarvan door vakgenoten (9) (12) werd geconcludeerd dat de EpiOcular™ EIT en de SkinEthic™ HCE EIT chemische stoffen (zowel stoffen als mengsels) waarvoor classificatie en etikettering voor oogirritatie of ernstig oogletsel volgens het VN-GHS niet nodig is, correct kunnen identificeren, en werden de tests aanbevolen als wetenschappelijk geldig voor dat doel (13).
|
|
5.
|
Op dit moment wordt algemeen aangenomen dat er in de nabije toekomst geen in-vitrotestmethode zal komen die op zichzelf de in-vivo-Draize-oogtest (2) (14) volledig zal kunnen vervangen om voorspellingen te doen voor het volledige responsbereik voor ernstig oogletsel of oogirritatie voor verschillende klassen chemische stoffen. Strategische combinaties van meerdere alternatieve testmethoden binnen (trapsgewijze) teststrategieën zoals de bottom-up-/top-downbenadering kunnen de Draize-oogtest mogelijk echter wel volledig vervangen (15). De bottom-upbenadering (15) is bedoeld om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof onvoldoende oogirritatie zal veroorzaken om classificatie noodzakelijk te maken, terwijl de top-downbenadering (15) bedoeld is om te worden gebruikt wanneer op basis van bestaande informatie verwacht wordt dat een chemische stof ernstig oogletsel veroorzaakt. De EpiOcular™ EIT en de SkinEthic™ HCE EIT worden aanbevolen voor het zonder verdere tests identificeren van chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is, volgens VN-GHS/CLP Geen categorie, in het kader van een teststrategie zoals de door Scott et al. bv. voorgestelde bottom-up-/top-downbenadering, bijvoorbeeld als eerste stap in een bottom-upbenadering of als een van de laatste stappen in een top-downbenadering (15). De EpiOcular™ EIT en de SkinEthic™ HCE EIT zijn echter niet bedoeld om onderscheid te maken tussen VN-GHS/CLP-categorie 1 (ernstig oogletsel) en VN-GHS/CLP-categorie 2 (oogirritatie). Dit onderscheid zal door een andere trap van een teststrategie moeten worden gemaakt (15). Wanneer een teststof die met de EpiOcular™ EIT of de SkinEthic™ HCE EIT wordt geïdentificeerd als stof die moet worden geclassificeerd voor oogirritatie/ernstig oogletsel, moeten er dus nog aanvullende tests (in vitro en/of in vivo) worden uitgevoerd om tot een definitieve conclusie te komen VN-GHS/CLP Geen categorie, -categorie 2 of -categorie 1), met behulp van bv. TM B.47, B.48, B.61 of B.68.
|
|
6.
|
Deze testmethode is bedoeld om de procedure te beschrijven die wordt gebruikt om het mogelijke gevaar van een teststof voor de ogen te beoordelen op basis van het vermogen van deze chemische stof om cytotoxiciteit te veroorzaken in een RhCE-weefselpreparaat, zoals gemeten met de MTT-test (16) (zie punt 21). De levensvatbaarheid van het RhCE-weefsel na blootstelling aan een teststof wordt bepaald in vergelijking met weefsels die met de negatievecontrolestof zijn behandeld (% levensvatbaarheid), en wordt vervolgens gebruikt om het mogelijke gevaar van de teststof voor de ogen te voorspellen.
|
|
7.
|
Er zijn prestatienormen beschikbaar (17) om de validering van nieuwe of gewijzigde, op RhCE gebaseerde in-vitrotests die vergelijkbaar zijn met de EpiOcular™ EIT en de SkinEthic™ HCE EIT, te vergemakkelijken volgens de beginselen van OESO-leidraad nr. 34 (18) en tijdige wijziging van OESO-TG 492 met het oog op de opname van dergelijke testmethoden mogelijk te maken. De wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst is enkel gewaarborgd voor tests die zijn gevalideerd volgens de prestatienormen, voor zover die tests door de OESO zijn geëvalueerd en zijn opgenomen in de overeenkomstige testrichtlijn.
|
DEFINITIES
|
8.
|
Definities worden gegeven in aanhangsel 1.
|
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
9.
|
Deze testmethode is gebaseerd op in de handel verkrijgbare, driedimensionale RhCE-weefselpreparaten die worden vervaardigd op basis van hetzij primaire menselijke epidermiskeratinocyten (d.w.z. EpiOcular™ OCL-200) of geïmmortaliseerde menselijke hoornvliesepitheelcellen (d.w.z. SkinEthic™ HCE/S). De EpiOcular™ OCL-200) en de SkinEthic™ HCE/S RhCE-weefselpreparaten zijn vergelijkbaar met de driedimensionale in-vivostructuur van hoornvliesepitheel en worden vervaardigd op basis van cellen van de doelsoort (19) (20). Bovendien meten de tests rechtstreeks de cytotoxiciteit als gevolg van de penetratie van de chemische stof door het hoornvlies en de veroorzaking van cel- en weefselschade; deze cytotoxische reactie is bepalend voor de algemene uitkomst voor ernstige oogletsel/oogirritatie in vivo. Er zijn verscheidene werkingsmechanismen waardoor celbeschadiging kan optreden (zie punt 20), maar cytotoxiciteit speelt een belangrijke, zo niet de voornaamste, mechanistische rol die bepalend is voor de algemene respons van een chemische stof als het gaat om ernstig oogletsel/oogirritatie. Deze manifesteert zich in vivo voornamelijk als vertroebeling van het hoornvlies, iritis, conjunctivale roodheid en/of conjunctivale chemosis, ongeacht de fysisch-chemische processen die de weefselschade veroorzaken.
|
|
10.
|
In de valideringsstudie die aan deze testmethode ten grondslag ligt, is een breed spectrum aan chemische stoffen getest, die vele verschillende stoftypen, chemische klassen, molecuulgewichten, LogP’s, chemische structuren enz. vertegenwoordigen. De valideringsdatabank van de EpiOcular™ EIT bevatte in totaal 113 chemische stoffen, die volgens een analyse met de QSAR-toolbox van de OESO 95 verschillende organische functionele groepen bestreken (8). De meeste van deze chemische stoffen waren stoffen die stoffen die uit één component bestaan, maar er werden ook stoffen die uit meerdere componenten bestaan, (waaronder 3 homopolymeren, 5 copolymeren en 10 quasipolymeren) in de studie opgenomen. Wat fysische toestand en VN-GHS/CLP-categorieën betreft, waren de 113 geteste chemische stoffen als volgt verdeeld: 13 categorie 1-vloeistoffen, 15 categorie 1-vaste stoffen, 6 categorie 2A-vloeistoffen, 10 categorie 2A-vaste stoffen, 7 categorie 2B-vloeistoffen, 7 categorie 2B-vaste stoffen, 27 Geen categorie-vloeistoffen en 28 Geen categorie-vaste stoffen (8). De valideringsdatabank van de SkinEthic™ HCE EIT bevatte in totaal 200 chemische stoffen, die 165 verschillende organische functionele groepen bestreken (8) (10) (11). De meeste van deze chemische stoffen waren stoffen die stoffen die uit één component bestaan, maar er werden ook stoffen die uit meerdere componenten bestaan, (waaronder 10 polymeren) in de studie opgenomen. Wat fysische toestand en VN-GHS/CLP-categorieën betreft, waren de 200 geteste chemische stoffen als volgt verdeeld: 27 categorie 1-vloeistoffen, 24 categorie 1-vaste stoffen, 19 categorie 2A-vloeistoffen, 10 categorie 2A-vaste stoffen, 9 categorie 2B-vloeistoffen, 8 categorie 2B-vaste stoffen, 50 Geen categorie-vloeistoffen en 53 Geen categorie-vaste stoffen (10) (11).
|
|
11.
|
Deze testmethode is van toepassing op stoffen en mengsels, en op vaste stoffen, vloeibare stoffen, halfvaste stoffen en wassen. Vloeistoffen mogen waterig of niet-waterig zijn; vaste stoffen mogen al dan niet in water oplosbaar zijn. Waar mogelijk moeten vaste stoffen tot een fijn poeder worden vermalen voordat ze worden aangebracht; verdere voorbehandeling van het monster is niet nodig. Gassen en aerosolen zijn niet gevalideerd in een valideringsstudie. Hoewel het denkbaar is dat deze met behulp van RhCE-technologie kunnen worden getest, staat de huidige testmethode het testen van gassen en aerosolen niet toe.
|
|
12.
|
Teststoffen die (van nature of na behandeling) licht absorberen in hetzelfde bereik als MTT-formazan, en teststoffen die de vitale kleurstof MTT rechtstreeks kunnen reduceren (tot MTT-formazan), kunnen de metingen van de levensvatbaarheid van het weefsel verstoren, zodat het bij deze stoffen nodig is om aangepaste controles te gebruiken met het oog op correcties. Wat voor aangepaste controles er eventueel nodig zijn hangt af van het soort verstoring door de teststof en de procedure die wordt gebruikt voor het kwantificeren van MTT-formazan (zie punten 36-42).
|
|
13.
|
De resultaten van de prevalideringsstudies (21) (22) en de volledige valideringsstudies (8) (10) (11) hebben uitgewezen dat EpiOcular™ EIT en SkinEthic™ HCE EIT beide overdraagbaar zijn naar laboratoria die kunnen worden beschouwd als zonder enige ervaring met het uitvoeren van de bepalingen, en ook reproduceerbaar zijn binnen één laboratorium en tussen laboratoria. Uit deze studies komt naar voren dat voor EpiOcular™ EIT de mate van reproduceerbaarheid wat betreft de concordantie van voorspellingen die kan worden verwacht op basis van de gegevens over 113 chemische stoffen, in de orde van grootte van 95 % ligt binnen laboratoria en 93 % tussen laboratoria. Voor SkinEthic™ HCE EIT ligt de mate van reproduceerbaarheid wat betreft de concordantie van voorspellingen die kan worden verwacht op basis van de gegevens over 120 chemische stoffen, in de orde van grootte van 92 % binnen laboratoria en 95 % tussen laboratoria.
|
|
14.
|
De EpiOcular™ EIT kan ook worden gebruikt om chemische stoffen te identificeren die niet hoeven te worden ingedeeld voor oogirritatie of ernstig oogletsel volgens het VN-GHS/CLP-classificatiesysteem. In het licht van de gegevens die zijn verkregen in de valideringsstudie (8), heeft de EpiOcular™ EIT een algemene nauwkeurigheid van 80 % (op basis van 112 chemische stoffen), een gevoeligheid van 96 % (op basis van 57 chemische stoffen), een percentage vals-negatieve uitslagen van 4 % (op basis van 57 chemische stoffen), een specificiteit van 63 % (op basis van 55 chemische stoffen) en een percentage vals-positieve uitslagen van 37 % (op basis van 55 chemische stoffen), in vergelijking met de referentiegegevens verkregen met de in-vivo-oogtest bij konijnen (TM B.5) (2) (14) en ingedeeld volgens het VN-GHS/CLP-classificatiesysteem. In een studie waarin 97 vloeibare agrochemische formuleringen werden getest met de EpiOcular™ EIT, bleek het prestatieniveau van de testmethode voor dit type mengsels vergelijkbaar ten opzichte van de valideringsstudie (23). De 97 formuleringen waren als volgt verdeeld: 21 categorie 1, 19 categorie 2A, 14 categorie 2B en 43 Geen categorie, ingedeeld volgens het VN-GHS/CLP-classificatiesysteem op basis van referentiegegevens verkregen met de in-vivo-oogtest bij konijnen (TM B.5) (2) (14). De algemene nauwkeurigheid was 82 % (op basis van 97 formuleringen), de gevoeligheid 91 % (op basis van 54 formuleringen), het percentage vals-negatieve uitslagen 9 % (op basis van 54 formuleringen), de specificiteit 72 % (op basis van 43 formuleringen) en het percentage vals-positieve uitslagen 28 % (op basis van 43 formuleringen) (23).
|
|
15.
|
De SkinEthic™ HCE EIT kan ook worden gebruikt om chemische stoffen te identificeren die niet hoeven te worden ingedeeld voor oogirritatie of ernstig oogletsel volgens het VN-GHS/CLP-classificatiesysteem. In het licht van de gegevens die zijn verkregen in de valideringsstudie (10) (11), heeft de SkinEthic™ HCE EIT een algemene nauwkeurigheid van 84 % (op basis van 200 chemische stoffen), een gevoeligheid van 95 % (op basis van 97 chemische stoffen), een percentage vals-negatieve uitslagen van 5 % (op basis van 97 chemische stoffen), een specificiteit van 72 % (op basis van 103 chemische stoffen) en een percentage vals-positieve uitslagen van 28 % (op basis van 103 chemische stoffen), in vergelijking met de referentiegegevens verkregen met de in-vivo-oogtest bij konijnen (TM B.5) (2) (14) en ingedeeld volgens het VN-GHS/CLP-classificatiesysteem.
|
|
16.
|
De percentages vals-negatieve uitslagen die met beide RhCE-tests werden verkregen voor stoffen of mengsels, vallen binnen de algemene waarschijnlijkheid van 12 % dat stoffen in herhaalde tests met de in-vivo-Draize-oogtest als VN-GHS/CLP-categorie 2 of als VN-GHS/CLP Geen categorie worden geïdentificeerd; dit is het gevolg van de inherente intertestvariabiliteit van de methode (24). De percentages vals-positieve uitslagen die met beide RhCE-tests werden verkregen voor stoffen of mengsels, zijn niet kritiek in deze context van deze testmethode, omdat voor alle teststoffen die een weefsellevensvatbaarheid opleveren die gelijk is aan of lager dan de vastgestelde grenswaarden (zie punt 44), aanvullende tests zullen moeten worden uitgevoerd met andere in-vitrotestmethoden of als laatste optie met konijnen, afhankelijk van de wettelijke voorschriften, volgens een sequentiële teststrategie en op bewijskracht gebaseerde aanpakken. Deze testmethoden kunnen worden gebruikt voor alle soorten chemische stoffen, waarbij een negatief resultaat moet worden aangenomen om een chemische stof niet in te delen voor oogirritatie en ernstig oogletsel (VN-GHS/CLP Geen categorie). Voordat de EpiOcular™ EIT en de SkinEthic™ HCE EIT worden gebruikt in een bottom-upbenadering volgens andere classificatiesystemen dan het VN-GHS/CLP-systeem, moeten de toepasselijke regelgevingsinstanties worden geraadpleegd.
|
|
17.
|
Aan deze testmethode kleeft de beperking dat ze geen onderscheid kan maken tussen oogirritatie/omkeerbare effecten op ogen (categorie 2) en ernstig oogletsel/onomkeerbare effecten op ogen (categorie 1) zoals gedefinieerd door het VN-GHS en de CLP, noch tussen stoffen die irriterend zijn voor het oog (optionele categorie 2A), en stoffen die mild irriterend zijn voor het oog (optionele categorie 2B), zoals gedefinieerd door het VN-GHS (1). Hiervoor zijn verdere tests met andere in-vitrotestmethoden nodig.
|
|
18.
|
De term „teststof” wordt in deze teststof gebruikt om te verwijzen naar datgene dat wordt getest (78), en houdt geen verband met de toepasselijkheid van de RhCE-testmethode op het testen van stoffen en/of mengsels.
|
PRINCIPE VAN DE TEST
|
19.
|
De teststof wordt plaatselijk aangebracht op ten minste twee driedimensionale RhE-weefselpreparaten en na de blootstelling en een incubatieperiode na de behandeling wordt de weefsellevensvatbaarheid gemeten. De RhCE-weefsels worden gereconstrueerd uit primaire menselijke epidermiskeratinocyten of geïmmortaliseerde menselijke hoornvliesepitheelcellen, die meerdere dagen zijn gekweekt tot gelaagd, hooggedifferentieerd plaveiselcelepitheel dat morfologisch vergelijkbaar is met het epitheel in het menselijk hoornvlies. Het EpiOcular™ RhCE-weefselpreparaat bestaat uit ten minste 3 levensvatbare cellagen en een niet-gekeratiniseerd oppervlak, met een hoornvliesachtige structuur analoog aan die in vivo wordt aangetroffen. Het SkinEthic™ HCE RhCE-weefselpreparaat bestaat uit ten minste 4 levensvatbare cellagen, bestaande uit cilindrische basaalcellen, tussenliggende wingcellen en plaveiselcellen aan de oppervlakte, vergelijkbaar met de opbouw van normaal menselijk hoornvliesepitheel (20) (26).
|
|
20.
|
Door chemische stoffen geïnduceerd(e) ernstig oogletsel/oogirritatie dat/die zich voornamelijk in vivo manifesteert als vertroebeling van het hoornvlies, irititis, conjunctivale roodheid en/of conjunctivale chemosis, is het gevolg van een cascade van gebeurtenissen, beginnend met de penetratie van de chemische stoffen door het hoornvlies en/of de conjunctiva en de veroorzaking van schade aan de cellen. Er zijn verscheidene werkingsmechanismen waardoor celbeschadiging kan optreden, waaronder: lysis van de celmembraan (bv. door oppervlakteactieve stoffen, organische oplosmiddelen); coagulatie van macromoleculen (vooral eiwitten) (bv. door oppervlakteactieve stoffen, organische oplosmiddelen, alkaliën en zuren); verzeping van lipiden (bv. door alkaliën); en alkylering of andere covalente interacties met macromoleculen (bv. door bleekmiddelen, peroxiden en alkylatoren (15) (27) (28). Er is evenwel aangetoond dat cytotoxiciteit een belangrijke, zo niet de voornaamste, mechanistische rol speelt, die bepalend is voor de algemene respons van een chemische stof als het gaat om ernstig oogletsel/oogirritatie, ongeacht de fysisch-chemische processen die de weefselschade veroorzaken (29) (30). Bovendien wordt het potentieel van een chemische stof voor ernstig oogletsel/oogirritatie hoofdzakelijk bepaald door de omvang van het aanvankelijke letsel (31), dat gecorreleerd is met de mate van celdood (29) en met de omvang van de erop volgende reacties en de uiteindelijke gevolgen (32). Licht irriterende stoffen hebben dus alleen een uitwerking op het oppervlak van het hoornvliesepitheel, mild en matig irriterende stoffen veroorzaken voornamelijk schade aan het epitheel en het oppervlakkige stroma, en hevig irriterende stoffen brengen schade toe aan het epitheel, het diepe stroma en soms het hoornvliesendotheel (30) (33). De levensvatbaarheid van het RhCE-weefselpreparaat na plaatselijke blootstelling aan een teststof voor het identificeren van chemische stoffen waarvoor classificatie voor ernstig oogletsel/oogirritatie niet nodig is (VN-GHS/CLP Geen categorie), wordt gemeten op basis van de aanname dat alle chemische stoffen die ernstig oogletsel of oogirritatie veroorzaken cytotoxiciteit zullen veroorzaken in het hoornvliesepitheel en/of de conjunctiva.
|
|
21.
|
De RhCE-weefsellevensvatbaarheid wordt gemeten aan de hand van de klassieke methode van enzymatische omzetting van de vitale kleurstof MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazoliumbromide; thiazolylblauw tetrazoliumbromide; CAS-nummer 298-93-1] door de levensvatbare cellen van het weefsel in een blauw MTT-formazanzout dat kwantitatief wordt gemeten na extractie uit weefsels (16). Chemische stoffen waarvoor classificatie en etikettering volgens VN-GHS/CLP niet nodig is (Geen categorie), worden geïdentificeerd als stoffen die de weefsellevensvatbaarheid niet tot onder een bepaalde drempelwaarde verlagen (d.w.z. weefsellevensvatbaarheid > 60 % in EpiOcular™ EIT en SkinEthic™ HCE EITL (79), of > 50 % in SkinEthic™ HCE EITS (80)) (zie punt 44).
|
AANTONING VAN BEKWAAMHEID
|
22.
|
Voordat de RhCE-tests routinematig worden gebruikt met het oog op regelgeving, moeten laboratoria hun technische bekwaamheid aantonen door een correcte voorspelling te doen voor de vijftien in tabel 1 aanbevolen chemische stoffen voor bekwaamheidstoetsing. Het betreft een selectie uit de chemische stoffen die werden gebruikt in de valideringsstudies van de VRM’s (8) (10) (11). Voor zover mogelijk omvat de selectie chemische stoffen die: i) verschillende fysische toestanden bestrijken; ii) het volledige responsbereik voor ernstig oogletsel of oogirritatie bestrijken op basis van resultaten van zeer goede kwaliteit die zijn verkregen in de referentie-in-vivo-oogtest bij konijnen (TM B.5) (2) (14), het VN-GHS-classificatiesysteem (d.w.z. categorieën 1, 2A, 2B en Geen categorie) (1) en het CLP-classificatiesysteem (d.w.z. categorieën 1, 2 en Geen categorie); iii) de uiteenlopende in-vivoclassificatiecriteria bestrijken (24) (25); iv) representatief zijn voor de chemische klassen die in de valideringsstudie zijn gebruikt (8) (10) (11); v) een breed en representatief spectrum van organische functionele groepen bestrijken (8) (10) (11); vi) een goed omschreven chemische structuur hebben (8) (10) (11); vii) gekleurd zijn en/of rechtstreeks MTT reduceren; viii) tijdens de validering van de RhCE-testmethoden reproduceerbare resultaten opleverden; ix) tijdens de validering van de RhCE-testmethoden correct werden ingedeeld door deze tests; x) het volledige in-vitroresponsbereik bestrijken op basis van referentiegegevens voor de RhCE-testmethoden van zeer goede kwaliteit (0 tot 100 % levensvatbaarheid); xi) in de handel verkrijgbaar zijn, en waaraan xii) geen buitensporige aanschaf- en/of verwijderingskosten verbonden zijn. In situaties waarin een chemische stof in de lijst niet beschikbaar is of om andere gerechtvaardigde redenen niet kan worden gebruikt, mag een andere chemische stof worden gebruikt, mits deze aan de bovenstaande criteria voldoet, bv. een van de andere chemische stoffen die bij de validering van de VRM zijn gebruikt. Dergelijke afwijkingen moeten echter wel worden verantwoord.
Tabel 1
Lijst van chemische stoffen voor bekwaamheidstoetsing
|
Chemische benaming
|
CAS RN
|
Organische functionele groepen (81)
|
Fysische toestand
|
VRM1-levensvatbaarheid (%) (82)
|
VRM2-levensvatbaarheid (%) (83)
|
VRM-voorspelling
|
MTT reducerende stof
|
Kleurinterf.
|
|
Categorie 1 in vivo (84)
|
|
|
Methylthioglycolaat
|
2365-48-2
|
Carbonzuurester; Thioalcohol
|
Vloeistof
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Er kan geen voorspelling worden gedaan
|
J (sterk)
|
N
|
|
Hydroxyethylacrylaat
|
818-61-1
|
Acrylaat; Alcohol
|
Vloeistof
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
2,5-Dimethyl-2,5-hexaandiol
|
110-03-2
|
Alcohol
|
Vaste stof
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
Natriumoxalaat
|
62-76-0
|
Oxocarbonzuur
|
Vaste stof
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
Categorie 2A in vivo (84)
|
|
|
2,4,11,13-Tetraäzatetradecaan-diimidamide, N,N″-bis(4-chloorfenyl)-3,12-diimino-, di-D-gluconaat (20 %, waterig) (86)
|
18472-51-0
|
Aromatische heterocyclische halide; Arylhalogenide; Dihydroxylgroep; Guanidine
|
Vloeistof
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Er kan geen voorspelling worden gedaan
|
N
|
J (zwak)
|
|
Natriumbenzoaat
|
532-32-1
|
Aryl; Carbonzuur
|
Vaste stof
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
Categorie 2B in vivo
(84)
|
|
|
Diëthyltoluamide
|
134-62-3
|
Benzamide
|
Vloeistof
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
2,2-Dimethyl-3-methyleenbicyclo[2.2.1]heptaan
|
79-92-5
|
Alkaan, vertakt met tertiaire koolstof; Alkeen; Bicycloheptaan; Gebrugde koolstofringen; Cycloalkaan
|
Vaste stof
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Er kan geen voorspelling worden gedaan
|
N
|
N
|
|
Geen categorie in vivo
(84)
|
|
|
1-Ethyl-3-methylimidazoliumethylsulfaat
|
342573-75-5
|
Alkoxy; Ammoniumzout; Aryl; Imidazool; Sulfaat
|
Vloeistof
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Geen categorie
|
N
|
N
|
|
Dicaprylylether
|
629-82-3
|
Alkoxy; Ether
|
Vloeistof
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Geen categorie
|
N
|
N
|
|
Piperonylbutoxide
|
51-03-6
|
Alkoxy; Benzodioxool; Benzyl; Ether
|
Vloeistof
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Geen categorie
|
N
|
N
|
|
Polyethyleenglycol (PEG-40) gehydrogeneerde ricinusolie
|
61788-85-0
|
Acylal; Alcohol; Allyl; Ether
|
Viskeus
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Geen categorie
|
N
|
N
|
|
1-(4-Chloorfenyl)-3-(3,4-dichloorfenyl)ureum
|
101-20-2
|
Aromatische heterocyclische halide; Arylhalogenide; Ureumderivaat
|
Vaste stof
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Geen categorie
|
N
|
N
|
|
2,2′-Methyleenbis(6-(2H-benzotriazool-2-yl)-4-(1,1,3,3-tetramethylbutyl)fenol)
|
103597-45-1
|
Alkaan, vertakt met quaternaire koolstof; Gefuseerde carbocyclische aromaat; Gefuseerde verzadigde heterocyclische verbinding; Chinoïdeprecursor; tert-Butylester
|
Vaste stof
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Geen categorie
|
N
|
N
|
|
Kaliumtetrafluorboraat
|
14075-53-7
|
Anorganisch zout
|
Vaste stof
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Geen categorie
|
N
|
N
|
|
Afkortingen:
CAS RN = registratienummer van de Chemical Abstract Service; VN-GHS = mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (1); VRM1 = gevalideerde referentiemethode 1, EpiOcular™ EIT; VRM2 = gevalideerde referentiemethode 2, SkinEthic™ HCE EIT; Kleurinterf. = kleurinterferentie met de standaard extinctiemeting (optische dichtheid (OD)) van MTT-formazan.
|
|
|
23.
|
Als onderdeel van deze bekwaamheidstoetsing wordt aanbevolen dat de gebruikers na ontvangst de door de fabrikant van het RhCE-weefselpreparaat gespecificeerde barrière-eigenschappen van de weefsels verifiëren (zie de punten 25, 27 en 30). Dit is in het bijzonder van belang indien weefsels over lange afstanden/gedurende lange perioden zijn vervoerd. Zodra een test met succes is opgezet en de bekwaamheid in het gebruik ervan is verworven en aangetoond, behoeft een dergelijke verificatie niet routinematig te worden uitgevoerd. Echter, indien een test routinematig wordt gebruikt, wordt aanbevolen om de barrière-eigenschappen op regelmatige basis te blijven toetsen.
|
PROCEDURE
|
24.
|
De tests die op dit moment onder deze testmethode vallen, zijn de wetenschappelijk geldige EpiOcular™ EIT en SkinEthic™ HCE EIT (9) (12) (13), aangeduid als de Validated Reference Methods (respectievelijk VRM1 en VRM2). De standaardwerkwijzen voor de RhCE-tests zijn beschikbaar en dienen te worden gevolgd wanneer de tests in een laboratorium wordt toegepast en gebruikt (34) (35). In de volgende punten en aanhangsel 2 worden de belangrijkste onderdelen en procedures van de RhCE-tests beschreven.
|
COMPONENTEN VAN DE RHCE-TESTMETHODE
Algemene voorwaarden
|
25.
|
Voor het reconstrueren van het driedimensionale model van hoornvliesepitheelcellen moeten relevante, uit mensen verkregen cellen worden gebruikt. Het model moet worden opgebouwd uit lagen van naar het oppervlak geleidelijk plattere cellen, maar zonder verhoornde cellen. Het RhCE-weefselpreparaat wordt bereid op inzetstukken met een poreus synthetisch membraan dat voedingsstoffen doorlaat naar de cellen. Er moeten in het gereconstrueerde hoornvliesepitheelmodel meerdere lagen van levensvatbare, niet gekeratiniseerde epitheelcellen aanwezig zijn. Het oppervlak van het RhCE-weefselpreparaat moet in rechtstreeks contact staan met de lucht, zodat het rechtstreeks kan worden blootgesteld aan de teststoffen op een manier die vergelijkbaar is met blootstelling van hoornvliesepitheel in vivo. Het RhCE-weefselpreparaat moet een functionele barrière vormen die voldoende robuust is om bestand te zijn tegen snelle penetratie van cytotoxische ijkstoffen, bv. Triton X-100 of natriumdodecylsulfaat (SDS). De barrièrefunctie moet worden aangetoond en kan worden beoordeeld door de blootstellingstijd te bepalen die nodig is om de weefsellevensvatbaarheid met 50 % te doen dalen (ET50) bij aanbrenging van de ijkstof in een bepaalde, vaste concentratie (bv. 100 μl 0,3 % (v/v) Triton X-100), of door de concentratie te bepalen waarin een ijkstof de weefsellevensvatbaarheid met 50 % doet dalen (IC50) na een vaste blootstellingstijd (bv. 30 minuten behandeling met 50 μl SDS) (zie punt 30). De omsluitingseigenschappen van het RhCE-weefselpreparaat moeten voorkomen dat teststof om de rand van het levensvatbare weefsel heen het levende weefsel kan bereiken. Dat zou een slecht model voor hoornvliesblootstelling kunnen opleveren. De bij mensen verkregen cellen die worden gebruikt om het RhCE-weefselpreparaat op te bouwen, mogen niet verontreinigd zijn met bacteriën, virussen, mycoplasma of schimmels. De leverancier moet controleren of het weefselpreparaat steriel is en niet verontreinigd is met schimmels of bacteriën.
|
Functionele voorwaarden
Levensvatbaarheid
|
26.
|
De levensvatbaarheid van het weefsel wordt gekwantificeerd door middel van de MTT-test (16). De levensvatbare cellen van het RhCE-weefselpreparaat reduceren de vitale kleurstof MTT tot een blauw MTT-formazanprecipitaat, dat vervolgens met isopropanol (of een vergelijkbaar oplosmiddel) uit het weefsel wordt geëxtraheerd. Het geëxtraheerde MTT-formazan kan worden gekwantificeerd met behulp van hetzij een standaard extinctiemeting (meting van de optische dichtheid (OD)) of een HPLC/UPLC-spectrofotometrieprocedure (36). De OD van het extractieoplosmiddel alleen moet voldoende klein zijn, d.w.z. OD < 0,1. Gebruikers van het RhCE-weefselpreparaat moeten waarborgen dat iedere gebruikte batch van het RhCE-weefselpreparaat voldoet aan gedefinieerde criteria voor de negatieve controle. In tabel 2 worden de aanvaardbaarheidsbereiken voor de OD-waarden in negatieve controle voor de VRM’s gegeven. Bij gebruik van HPLC/UPLC-spectrofotometrie moeten de OD-bereiken in negatieve controle in tabel 2 als aanvaardbaarheidscriterium voor de negatieve controles worden gebruikt. In het testverslag moet worden gedocumenteerd dat de weefsels die zijn behandeld met de negatievecontrolestof, gedurende de volledige blootstellingsperiode van de test in cultuur stabiel zijn (een vergelijkbare gemeten weefsellevensvatbaarheid opleveren). De weefselproducent moet als onderdeel van de QC-weefselbatchvrijgave een vergelijkbare procedure uitvoeren, maar daarbij gelden mogelijk andere aanvaardbaarheidscriteria dan de criteria in tabel 2. De ontwikkelaar/leverancier van een RhCE-weefselpreparaat moet een aanvaardbaarheidsbereik (boven- en ondergrens) vaststellen voor de OD-waarden in negatieve controle (onder de omstandigheden van de QC-testmethode).
|
Tabel 2
Aanvaardbaarheidsbereiken voor OD-waarden in negatieve controle (voor gebruikers van de test)
|
Test
|
Ondergrens aanvaardbaarheid
|
Bovengrens aanvaardbaarheid
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (voor protocollen voor vloeistoffen en voor vaste stoffen)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (voor protocollen voor vloeistoffen en voor vaste stoffen)
|
> 1,0
|
≤ 2,5
|
Barrièrefunctie
|
27.
|
Het RhCE-weefselpreparaat moet dik en robuust genoeg zijn om bestand te zijn tegen snelle penetratie van cytotoxische ijkstoffen, zoals beoordeeld aan de hand van bv. de ET50 (Triton X-100) of de IC50 (SDS). (tabel 3). Voor elke batch van het gebruikte RhCE-weefselpreparaat moet de ontwikkelaar/leverancier van het RhCE-weefselpreparaat bij levering van de weefsels aan de eindgebruiker de barrièrefunctie aantonen (zie punt 30).
|
Morfologie
|
28.
|
Uit histologisch onderzoek van het RhCE-weefselpreparaat moet blijken dat het een vergelijkbare structuur heeft als menselijk hoornvliesepitheel (met ten minste 3 lagen van levensvatbare epitheelcellen en een niet-gekeratiniseerd oppervlak). Voor de VRM’s is de door de ontwikkelaar/leverancier vastgesteld dat de weefselpreparaten een geschikte morfologie hebben, en hoeft de gebruiker van de testmethode dit dus niet zelf nogmaals voor elke gebruikte weefselbatch aan te tonen.
|
Reproduceerbaarheid
|
29.
|
De resultaten van de positieve en negatieve controles van de testmethode moeten reproduceerbaar zijn in de tijd.
|
Kwaliteitscontrole (QC)
|
30.
|
Het RhCE-weefselpreparaat mag alleen worden gebruikt als de ontwikkelaar/leverancier aantoont dat iedere batch van het gebruikte RhCE-weefselpreparaat voldoet aan gedefinieerde productievrijgavecriteria, waarvan die voor levensvatbaarheid (punt 26) en barrièrefunctie (zie punt 27) de meest relevante zijn. De ontwikkelaar/leverancier van een RhCE-weefselpreparaat moet een aanvaardbaarheidsbereik (boven- en ondergrens) vaststellen voor de barrièrefuncties zoals gemeten aan de hand van de ET50 of de IC50 (zie de punten 25 en 26). Het aanvaardbaarheidsbereik voor de ET50 en de IC50 dat door de ontwikkelaar/leverancier van de (in de VRM’s gebruikte) RhCE-weefselpreparaten als QC-criterium voor batchvrijgave wordt gebruikt, staat in tabel 3. De ontwikkelaar/leverancier van een RhCE-weefselpreparaat moet de gebruikers van de testmethode gegevens verstrekken waaruit blijkt dat aan alle productievrijgavecriteria wordt voldaan, zodat de gebruikers deze informatie in het testverslag kunnen opnemen. Alleen resultaten die zijn verkregen met weefsels die aan alle productievrijgavecriteria voldoen, zijn aanvaardbaar voor een betrouwbare voorspelling voor chemische stoffen waarvoor classificatie en etikettering voor oogirritatie of ernstig oogletsel volgens VN-GHS/CLP niet nodig is.
|
Tabel 3
QC-criterium voor batchvrijgave
|
Test
|
Ondergrens aanvaardbaarheid
|
Bovengrens aanvaardbaarheid
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3 % (v/v) Triton X-100)
|
IC50 = 12,2 min
|
IC50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 minuten behandeling met 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Het aanbrengen van de teststof en de controlestoffen
|
31.
|
Er moeten ten minste twee duploweefsels worden gebruikt voor iedere teststof en iedere controlestof in ieder experiment. Er worden twee verschillende behandelingsprotocollen gebruikt, een voor vloeibare teststoffen en een voor vaste teststoffen (34) (35). Voor beide methoden en protocollen moet het oppervlak van het weefselpreparaat worden bevochtigd met calcium- en magnesiumvrije fosfaatgebufferde zoutoplossing van Dubecco (Ca2+/Mg2+-vrije DPBS) alvorens teststoffen worden aangebracht, om de natte omstandigheden van het menselijk oog na te bootsen. De behandeling van de weefsels wordt begonnen door ze aan de teststof(fen) en controlestoffen bloot te stellen. Voor beide behandelingsprotocollen in beide VRM’s moet er zo veel teststof of controlestof worden aangebracht dat het epitheeloppervlak gelijkmatig bedekt is. Er mag echter geen oneindige dosis worden gebruikt (zie de punten 32 en 33) (aanhangsel 2).
|
|
32.
|
Teststoffen die kunnen worden gepipetteerd bij temperaturen van 37 °C of lager (zo nodig met een plunjerpipet), worden in de VRM’s als vloeistoffen behandeld. Zo niet, dan worden ze als vaste stoffen behandeld (zie punt 33). In de VRM’s wordt een vloeibare teststof gelijkmatig over het weefseloppervlak verspreid (d.w.z. er wordt ten minste 60 μl/cm2 aangebracht) (zie aanhangsel 2, (33) (34)). Om de juiste dosering op het weefsel te garanderen, moeten capillaire effecten (oppervlaktespanningseffecten) die kunnen optreden als gevolg van de geringe volumes die op het inzetstuk (op het weefseloppervlak) worden aangebracht, zo veel mogelijk worden vermeden. Met vloeibare teststoffen behandelde weefsels worden 30 min geïncubeerd onder standaard kweekomstandigheden (37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH). Aan het einde van de blootstellingsperiode moeten de vloeibare teststof en de controlestoffen zorgvuldig van het weefseloppervlak worden verwijderd door grondig te spoelen met Ca2+/Mg2+-vrije DPBS bij kamertemperatuur. Deze spoelstap wordt gevolgd door onderdompeling in vers medium bij kamertemperatuur (om alle in het weefsel geabsorbeerde teststof te verwijderen) gedurende een vooraf bepaalde tijd die varieert afhankelijk van de gebruikte VRM. Alleen bij VRM1 wordt een na-incubatie in vers medium onder standaard kweekomstandigheden toegepast, voordat de MTT-test wordt uitgevoerd (zie aanhangsel 2 (34) (35)).
|
|
33.
|
Teststoffen die niet kunnen worden gepipetteerd bij temperaturen van 37 °C of lager, worden in de VRM’s als vaste stoffen behandeld. De hoeveelheid teststof die wordt aangebracht, moet voldoende zijn om het gehele weefseloppervlak te bedekken, d.w.z. er wordt ten minste 60 mg/cm2 aangebracht (aanhangsel 2). Indien mogelijk moeten vaste stoffen in de vorm van een fijn poeder worden getest. Met vaste teststoffen behandelde weefsels worden gedurende een vooraf bepaalde tijd (afhankelijk van de gebruikte VRM) geïncubeerd onder standaard kweekomstandigheden (zie aanhangsel 2 (34) (35)). Aan het einde van de blootstellingsperiode moeten de vaste teststof en de controlestoffen zorgvuldig van het weefseloppervlak worden verwijderd door grondig te spoelen met Ca2+/Mg2+-vrije DPBS bij kamertemperatuur. Deze spoelstap wordt gevolgd door onderdompeling in vers medium bij kamertemperatuur (om alle in het weefsel geabsorbeerde teststof te verwijderen) gedurende een vooraf bepaalde tijd die varieert afhankelijk van de gebruikte VRM, en een na-incubatie in vers medium onder standaard kweekomstandigheden toegepast, voordat de MTT-test wordt uitgevoerd (zie aanhangsel 2 (34) (35)).
|
|
34.
|
In elke testrun moeten gelijktijdige negatieve en positieve controles worden opgenomen om aan te tonen dat de levensvatbaarheid (bepaald met de negatieve controle) en de gevoeligheid (bepaald met de positieve controle) van de weefsels binnen de op basis van historische gegevens vastgestelde aanvaardbaarheidsgrenzen liggen. De gelijktijdige negatieve controle zorgt ook voor de baseline (100 % levensvatbaarheid) om het relatieve percentage levensvatbaarheid van de met de teststof behandelde weefsels te berekenen (%Levensvatbaarheidtest). De aanbevolen positievecontrolestof voor gebruik met de VRM’s is zuiver methylacetaat (CAS RN 79-20-9, in de handel verkrijgbaar van bv. Sigma-Aldrich, cat. nr. 45997; vloeistof). De aanbevolen negatievecontrolestoffen voor gebruik met VRM1 en VRM2 zijn respectievelijk ultrazuiver H2O en Ca2+/Mg2+-vrije DPBS. Dit zijn de stoffen die als controlestoffen werden gebruikt in de valideringsstudies van de VRM’s en waarvoor de meeste historische gegevens beschikbaar zijn. Het gebruik van geschikte alternatieve positieve- en negatievecontrolestoffen moet afdoende wetenschappelijk gerechtvaardigd worden. De negatieve en positieve controles moeten volgens hetzelfde protocol/dezelfde protocollen worden getest als de teststoffen die in de testrun zijn opgenomen (d.w.z. voor vloeistoffen en/of voor vaste stoffen). Na het aanbrengen volgen de blootstelling aan de behandeling, spoelen, onderdompeling na de behandeling en na-incubatie, voor zover van toepassing, zoals beschreven voor controles die gelijktijdig met vloeibare teststoffen worden gerund (zie punt 32) of voor controles die gelijktijdig met vaste stoffen worden gerund (zie punt 33), alvorens de MTT-test wordt uitgevoerd (zie punt 35) (34) (35). Een enkele set van negatieve en positieve controles is voldoende voor alle teststoffen in dezelfde testrun die dezelfde fysische toestand hebben (vloeistoffen of vaste stoffen).
|
Metingen van de weefsellevensvatbaarheid
|
35.
|
De MTT-omzettingstest is een gestandaardiseerde kwantitatieve methode (16), en de aangewezen methode voor het meten van de weefsellevensvatbaarheid binnen deze testmethode. De test werkt goed met geconstrueerd driedimensionaal weefsel. De MTT-test wordt onmiddellijk na de na-incubatieperiode uitgevoerd. In de VRM’s wordt het RhCE-weefselpreparaat gedurende 180 ± 15 min onder standaard kweekomstandigheden in 0,3 ml 1 mg/ml MTT-oplossing geplaatst. De levensvatbare cellen van het RhCE-weefselpreparaat reduceren de vitale kleurstof MTT tot een blauw MTT-formazanprecipitaat. Het neergeslagen blauwe MTT-formazanproduct wordt vervolgens met een passend volume isopropanol (of een vergelijkbaar oplosmiddel) uit het weefsel geëxtraheerd (34) (35). Bij weefsels die worden getest met vloeibare teststoffen, moet de extractie zowel aan de bovenkant als aan de onderkant van de weefsels plaatsvinden, terwijl bij weefsels die worden getest met vaste teststoffen en gekleurde vloeistoffen, de extractie enkel aan de onderkant van het weefsel moet plaatsvinden (om de kans op verontreiniging van de isopropanol-extractieoplossing met mogelijk op het weefsel achtergebleven teststof zoveel mogelijk te verkleinen). Bij weefsels die worden getest met moeilijk afwasbare teststoffen, kan ook worden gekozen voor extractie enkel aan de onderkant. De gelijktijdig geteste negatieve en positieve controlestoffen moeten op dezelfde manier worden behandeld als de geteste chemische stof. Het geëxtraheerde MTT-formazan kan worden gekwantificeerd met behulp van een meting van de standaard extinctie (OD) bij 570 nm, bij een bandbreedte van maximaal ± 30 nm, of een HPLC/UPLC-spectrofotometrieprocedure (zie punt 42) (11) (36).
|
|
36.
|
De optische eigenschappen van de teststof of de chemische werking ervan op MTT kunnen de meting van MTT-formazan verstoren, met een foutieve bepaling van de weefsellevensvatbaarheid tot gevolg. Soms verstoren teststoffen de meting van MTT-formazan, door rechtstreekse reductie van MTT tot blauw MTT-formazan en/of door kleurinterferentie indien de teststof van nature of als gevolg van de behandelingsprocedures in hetzelfde OD-bereik absorbeert als MTT-formazan (d.w.z. rond 570 nm). Voorafgaand aan de MTT-test moeten teststoffen getoetst worden om mogelijke rechtstreeks MTT reducerende stoffen en/of stoffen die kleurinterferentie veroorzaken, te identificeren. Om mogelijke storing door deze teststoffen te detecteren en ervoor te corrigeren moeten er aanvullende controles worden uitgevoerd (zie de punten 37-41). Dit is vooral van belang als een bepaalde teststof zich niet volledig van het RhCE-weefselpreparaat laat wassen, of als de stof in het hoornvliesepitheelmodel penetreert, en de teststof daardoor in de RhCE-weefselpreparaten aanwezig is wanneer de MTT-test wordt uitgevoerd. Voor teststoffen die (van nature of na behandeling) licht absorberen in hetzelfde bereik als MTT-formazan en niet compatibel zijn met de meting van de standaard extinctie (OD) van MTT-formazan vanwege te sterke interferentie, d.w.z. sterkte extinctie bij 570 ± 30 nm, kan wellicht een HPLC/UPLC-spectrofotometrieprocedure voor het meten van MTT-formazan worden toegepast (zie de punten 41 en 42) (11) (36). Een gedetailleerde beschrijving van hoe rechtstreekse MTT-reductie kan worden gedetecteerd en hoe ervoor kan worden gecorrigeerd, is te vinden in de SOP’s voor de VRM’s (34) (35). Bovendien zijn in de aanhangsels III en IV illustratieve stroomschema’s opgenomen voor respectievelijk VRM1 en VRM2, die als leidraad kunnen dienen voor het identificeren en behandelen van rechtstreeks MTT reducerende stoffen en/of stoffen die kleurinterferentie veroorzaken.
|
|
37.
|
Om na te gaan of teststoffen die (van nature of na behandeling) licht absorberen in hetzelfde bereik als MTT-formazan, de test zouden kunnen verstoren en te beslissen of er aanvullende controles nodig zijn, wordt de teststof toegevoegd aan water en/of isopropanol en voldoende tijd geïncubeerd bij kamertemperatuur (zie aanhangsel 2 (34) (35)). Als de teststof in water en/of isopropanol voldoende licht absorbeert in het bereik van 570 ± 20 nm voor VRM1 (zie aanhangsel 3) of als bij menging van de teststof met water een gekleurde oplossing wordt verkregen voor VRM2 (zie aanhangsel 4), wordt aangenomen dat de teststof de meting van de standaard extinctie (OD) van MTT-formazan verstoort en dat er hetzij extra kleurstofcontroles moeten worden uitgevoerd hetzij, als alternatief, een HPLC/UPLC-spectrofotometrieprocedure moet worden gebruikt, in welk geval deze controles niet nodig zijn (zie de punten 41 en 42) en de aanhangsels III en IV) (34) (35). Bij het uitvoeren van de meting van de standaard extinctie (OD) moet elke verstorende teststof op ten minste twee levensvatbare duploweefsels aangebracht, die de hele testprocedure doorlopen maar tijdens de MTT-incubatiestap geïncubeerd worden met medium in plaats van MTT-oplossing om zo tot een niet-specifieke kleurcontrole (NSClevend-controle) te komen (34) (35). Vanwege de inherente biologische variabiliteit van levende weefsels moet de NSClevend-controle gelijktijdig worden uitgevoerd met het testen van de gekleurde teststof. In geval van meerdere testen moet er voor elke test die wordt uitgevoerd (in elke testrun), een onafhankelijke NSClevend-controle worden verricht. De werkelijke weefsellevensvatbaarheid wordt berekend als: de procentuele levensvatbaarheid verkregen met levende weefsels die zijn blootgesteld aan de verstorende teststof en geïncubeerd met MTT-oplossing (%Levensvatbaarheidtest), minus het percentage niet-specifieke kleur verkregen met levende weefsels die zijn blootgesteld aan de verstorende teststof en geïncubeerd in medium zonder MTT, gelijktijdig uitgevoerd met de te corrigeren test (%NSClevend), d.w.z. werkelijke weefsellevensvatbaarheid = [%Levensvatbaarheidtest] – [%NSClevend]
|
|
38.
|
Om rechtstreeks MTT reducerende stoffen te herkennen moet elke teststof worden toegevoegd aan vers bereide MTT-oplossing. Er wordt een passende hoeveelheid teststof toegevoegd aan een MTT-oplossing en dit mengsel wordt ongeveer 3 uur geïncubeerd onder standaard kweekomstandigheden (zie de aanhangsels III en IV) (34) (35). Als het MTT-mengsel met de teststof (of de suspensie voor onoplosbare teststoffen) blauw/paars wordt, wordt de teststof als een rechtstreeks MTT reducerende stof beschouwd en moet een verdere functionele toetsing op niet-levensvatbare RhCE-weefselpreparaten worden uitgevoerd, ongeacht of de meting van de standaard extinctie (OD) of een HPLC/UPLC-spectrofotometrieprocedure wordt gebruikt. Bij deze aanvullende functionele toetsing worden gedode weefsels gebruikt die slechts een residuele metabole activiteit vertonen, maar de teststof wel absorberen en vasthouden op vergelijkbare wijze als levensvatbare weefsels. Gedode VRM1-weefsels worden bereid door blootstelling aan lage temperatuur („door bevriezing gedood”). Gedode VRM2-weefsels worden bereid door langdurige incubatie (bv. ten minste 24 ± 1 uur) in water, gevolgd door bewaring bij lage temperatuur („in water gedood”). Om een controle voor niet-specifieke MTT-reductie (NSMTT) te genereren wordt elke MTT reducerende teststof op ten minste twee gedode duploweefsels aangebracht, die de gehele testprocedure ondergaan (34) (35). Per teststof is één NSMTT-controle voldoende, ongeacht het aantal onafhankelijk uitgevoerde tests/testruns. De werkelijke weefsellevensvatbaarheid wordt berekend als: de procentuele levensvatbaarheid van het weefsel verkregen met levende weefsels die zijn blootgesteld aan de MTT reducerende stof (%Levensvatbaarheidtest), minus het percentage niet-specifieke MTT-reductie verkregen met de gedode weefsels die aan de zelfde MTT reducerende stof zijn blootgesteld, ten opzichte van de negatieve controle die gelijktijdig met de te corrigeren test is uitgevoerd (%NSMTT), d.w.z. werkelijke weefsellevensvatbaarheid = [%Levensvatbaarheidtest] – [%NSMTT].
|
|
39.
|
Voor teststoffen waarvan wordt vastgesteld dat ze zowel kleurinterferentie (zie punt 37) als rechtstreekse reductie van MTT veroorzaken (zie punt 38), is naast de in de vorige punten beschreven NSMTT- en NSClevend-controles nog een derde reeks controles nodig, wanneer de meting van de standaard extinctie (OD) wordt uitgevoerd. Dit geldt doorgaans voor donkergekleurde teststoffen die licht absorberen in het bereik van 570 ± 30 nm (bv. blauwe, paarse of zwarte stoffen), omdat hun intrinsieke kleur de beoordeling van hun vermogen om rechtstreeks MTT te reduceren zoals beschreven in punt 38 belemmert. Dit maakt een standaard gebruik van NSMTT-controles samen met NSClevend-controles noodzakelijk. Teststoffen waarvoor zowel NSMTT-controles als NSClevend-controles worden uitgevoerd, worden mogelijk zowel door levende als gedode weefsels geabsorbeerd en vastgehouden. Daarom corrigeert de NSMTT-controle in dit geval mogelijk niet alleen voor potentiële rechtstreekse MTT-reductie door de teststof, maar ook voor kleurinterferentie die voortkomt uit de absorptie en retentie van de teststof door de gedode weefsels. Dit zou kunnen leiden tot een dubbele correctie voor kleurinterferentie, aangezien de NSClevend-controle al voor kleurinterferentie als gevolg van de absorptie en retentie van de teststof door levende weefsels corrigeert. Om een mogelijke dubbele correctie voor kleurinterferentie te vermijden moet een derde controle voor niet-specifieke kleur in gedode weefsels (NSCgedood) worden uitgevoerd (zie de aanhangsels III en IV) (34) (35). In deze extra controle wordt de teststof aangebracht op ten minste twee duplo’s van gedood weefsel, die de gehele testprocedure doorlopen maar in de MTT-incubatiestap geïncubeerd worden met medium in plaats van met MTT-oplossing. Per teststof is één NSCgedood-controle voldoende, ongeacht het aantal onafhankelijke tests/testruns dat wordt uitgevoerd, maar deze moet gelijktijdig worden uitgevoerd met de NSMTT-controle en met dezelfde weefselbatch. De werkelijke weefsellevensvatbaarheid wordt berekend als: de procentuele levensvatbaarheid verkregen met levende weefsels die zijn blootgesteld aan de teststof (%Levensvatbaarheidtest) minus %NSMTT minus %NSClevend
plus het percentage niet-specifieke kleur verkregen met gedode weefsels die zijn blootgesteld aan de verstorende teststof en geïncubeerd met medium zonder MTT, ten opzichte van de negatieve controle die gelijktijdig met de te corrigeren test is uitgevoerd (%NSCgedood), d.w.z. werkelijke weefsellevensvatbaarheid = [%Levensvatbaarheidtest] – [%NSMTT] – [%NSClevend] + [%NSCgedood].
|
|
40.
|
Het is van belang op te merken dat niet-specifieke MTT-reductie en niet-specifieke kleurinterferenties de OD van het weefselextract kunnen verhogen tot buiten het lineariteitsbereik van de spectrofotometer (in het geval van standaard extinctiemetingen) en dat niet-specifieke MTT-reductie ook de piekoppervlakte voor MTT-formazan van het weefselextract kan vergroten tot buiten het lineariteitsbereik van de spectrofotometer (in het geval van HPLC/UPLC-spectrofotometriemetingen). Daarom is het van belang dat elk laboratorium, alvorens te beginnen met het testen van teststoffen met het oog op regelgeving, eerst het lineariteitsbereik van de spectrofotometer voor de OD/piekoppervlakte bepaalt met MTT-formazan (CAS RN 57360-69-7), in de handel verkrijgbaar van bv. Sigma-Adrich (cat. nr. M2003).
|
|
41.
|
Standaard extinctiemeting (OD-meting) met een spectrofotometer is een geschikte methode voor de beoordeling van rechtstreeks MTT reducerende stoffen en teststoffen die kleurinterferentie vertonen, als de waargenomen verstoring van de meting van MTT-formazan niet te sterk is (d.w.z. als de OD’s van de weefselextracten die worden verkregen met de teststof zonder correctie voor rechtstreekse MTT-reductie en/of kleurinterferentie, binnen het lineariteitsbereik van de spectrofotometer vallen). Niettemin moeten de resultaten voor teststoffen die een %NSMTT en/of een %NSClevend van ≥ 60 % (VRM1 en VRM2, protocol voor vloeistoffen) of 50 % (VRM2, protocol voor vaste stoffen) van de negatieve controle opleveren, met omzichtigheid worden benaderd, aangezien dit de vastgestelde drempelwaarden zijn die in de VRM’s als scheiding tussen ingedeelde en niet-ingedeelde stoffen fungeren (zie punt 44). De standaard extinctie (OD) kan echter niet worden gemeten wanneer de verstoring van MTT-formazan te sterk is (d.w.z. de verstoring leidt ertoe dat de ongecorrigeerde OD’s van de testweefselextracten buiten het lineariteitsbereik van de spectrofotometer vallen). Gekleurde teststoffen of teststoffen die gekleurd worden wanneer ze in contact komen met water of isopropanol, die de meting van de standaard extinctie (OD) van MTT-formazan te sterk verstoren, kunnen nog altijd worden beoordeeld met behulp van HPLC/UPLC-spectrofotometrie (zie aanhangsels III en IV). In het HPLC/UPLC-systeem kan het MTT-formazan namelijk van de chemische stof worden gescheiden, voordat het gekwantificeerd wordt (36). Daarom zijn er bij gebruik van HPLC/UPLC-spectrofotometrie geen NSClevend- of NSCgedood-controles nodig, ongeacht de geteste chemische stof. Als echter wordt vermoed dat de teststof rechtstreeks MTT reduceert, moeten er wel NSMTT-controles worden toegepast (volgens de in punt 38 beschreven procedure). NSMTT-controles moeten ook worden toegepast als van teststoffen wordt vermoed dat ze (van zichzelf of in water) een kleur hebben die de beoordeling van het vermogen om rechtstreeks MTT te reduceren (zoals beschreven in punt 38) belemmert. Bij gebruik van HPLC/UPLC-spectrofotometrie voor het meten van MTT-formazan wordt de procentuele levensvatbaarheid van het weefsel berekend als percentage van de piekoppervlakte voor MTT-formazan verkregen met levende weefsels die aan de teststof zijn blootgesteld, ten opzichte van de piekoppervlakte voor MTT-formazan verkregen met de gelijktijdige negatieve controle. Voor teststoffen die rechtstreeks MTT kunnen reduceren, wordt de weefsellevensvatbaarheid berekend als: %Levensvatbaarheidtest
minus %NSMTT, zoals beschreven in de laatste zin van punt 38. Tot slot zij erop gewezen dat rechtstreeks MTT reducerende stoffen of rechtstreeks MTT reducerende stoffen die ook kleurinterferentie vertonen, als ze na de behandeling in de weefsels achterblijven en MTT zo sterk reduceren dat dit OD’s (met standaard OD-meting) of piekoppervlakten (met HPLC/UPLC-spectrofotometrie) oplevert voor de geteste weefselextracten die buiten het lineariteitsbereik van de spectrofotometer vallen, niet met RhCE-testmethoden beoordeeld kunnen worden, hoewel dergelijke gevallen naar verwachting slechts zeer zelden voorkomen.
|
|
42.
|
HPLC/UPLC-spectrofotometrie kan voor alle typen teststoffen (al dan niet gekleurd, al dan niet MTT reducerend) worden gebruikt voor het meten van MTT-formazan (11) (36). Vanwege de diversiteit van HPLC/UPLC-spectrofotometriesystemen is het niet mogelijk om voor elke gebruiker een precies gelijke systeemopzet te realiseren. Daarom moet de deugdelijkheid van het HPLC/UPLC-spectrofotometriesysteem worden aangetoond alvorens het wordt gebruikt voor het kwantificeren van MTT-formazan in weefselextracten, door aan de aanvaardbaarheidscriteria te voldoen voor een reeks standaard deugdelijkheidsparameters op basis van de parameters beschreven in de voor de industrie bestemde leidraad van de Amerikaanse Food and Drug Administration voor de validering van bioanalytische methoden (36) (38). Deze kritische parameters en de bijbehorende aanvaardbaarheidscriteria worden weergegeven in aanhangsel 5. Als aan de in aanhangsel 5 gestelde aanvaardbaarheidscriteria is voldaan, wordt het HPLC/UPLC-spectrofotometriesysteem als deugdelijk beschouwd en kan het worden gebruikt voor het meten van MTT-formazan onder de experimentele omstandigheden die worden beschreven in deze testmethode.
|
Aanvaardbaarheidscriteria
|
43.
|
Voor elke testrun met RhCE-batches die de kwaliteitscontrole hebben doorstaan (zie punt 30), moeten de weefsels die met de negatievecontrolestof zijn behandeld, een OD vertonen die overeenkomt met de kwaliteit van de weefsels na vervoer, ontvangst en alle stappen van het protocol, en die niet buiten de in tabel 2 beschreven historisch bepaalde grenzen mag liggen (zie punt 26). Evenzo moet voor weefsels die met de positievecontrolestof, d.w.z. met methylacetaat, zijn behandeld, de gemiddelde weefsellevensvatbaarheid < 50 % zijn ten opzichte van de negatieve controle in VRM1 met het protocol voor vloeistoffen of dat voor vaste stoffen, en ≤ 30 % (protocol voor vloeistoffen) of ≤ 20 % (protocol voor vaste stoffen) ten opzichte van de negatieve controle in VRM2, overeenkomend met het vermogen van de weefsels om op een irriterende teststof te reageren onder de omstandigheden van de testmethode (34) (35). De variabiliteit tussen duploweefsels voor teststoffen en controlestoffen moet binnen de aanvaardbare grenzen liggen (d.w.z. het verschil in levensvatbaarheid tussen twee duploweefsels moet kleiner zijn dan 20 % of de standaarddeviatie (SD) tussen drie duploweefsels mag niet groter zijn dan 18 %). Als de negatieve controle of de positieve controle in een testrun buiten het aanvaarde bereik valt, wordt de testrun als „ondeugdelijk”beschouwd en moet deze worden overgedaan. Als de variabiliteit tussen de duploweefsels voor een teststof buiten het aanvaardbare bereik ligt, moet de test als „ondeugdelijk”worden beschouwd en moet de teststof hertest worden.
|
Interpretatie van de resultaten en voorspellingsmodel
|
44.
|
De OD-waarden/piekoppervlakten die zijn verkregen met de duploweefselextracten voor elke teststof, moeten worden gebruikt voor het berekenen van de gemiddelde procentuele weefsellevensvatbaarheid (gemiddelde over duploweefsels) genormaliseerd ten opzichte van de negatieve controle, die op 100 % wordt gesteld. De drempelwaarde van de procentuele weefsellevensvatbaarheid voor het identificeren van teststoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is (VN-GHS/CLP Geen categorie), wordt gegeven in tabel 4. De resultaten moeten derhalve als volgt worden geïnterpreteerd:
|
—
|
De teststof wordt geïdentificeerd als chemische stof waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is volgens VN-GHS/CLP Geen categorie, als de gemiddelde procentuele weefsellevensvatbaarheid na blootstelling en na-incubatie hoger is dan (gt;) de vastgestelde drempelwaarde van de procentuele weefsellevensvatbaarheid zoals weergegeven in tabel 4. In dat geval is het niet nodig om met andere testmethoden verder te testen.
|
|
—
|
Als de gemiddelde procentuele weefsellevensvatbaarheid na blootstelling en na-incubatie lager is dan of gelijk aan (≤) de vastgestelde drempelwaarde van de procentuele weefsellevensvatbaarheid zoals weergegeven in tabel 4, kan er geen voorspelling worden gedaan. In dat geval zijn er verdere tests met andere testmethoden nodig, omdat de RhCE-testmethoden een zeker aantal vals-positieve resultaten laten zien (zie de punten 14-15) en geen onderscheid kunnen maken tussen de VN-GHS/CLP-categorieën 1 en 2 (zie punt 17).
|
Tabel 4
Voorspellingsmodellen volgens de VN-GHS en CLP-classificatie
|
VRM
|
Geen categorie
|
Er kan geen voorspelling worden gedaan
|
|
VRM1 - EpiOcular™ EIT (voor beide protocollen)
|
Gemiddelde weefsellevensvatbaarheid > 60 %
|
Gemiddelde weefsellevensvatbaarheid ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (voor het protocol voor vloeistoffen)
|
Gemiddelde weefsellevensvatbaarheid > 60 %
|
Gemiddelde weefsellevensvatbaarheid ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (voor het protocol voor vaste stoffen)
|
Gemiddelde weefsellevensvatbaarheid > 50 %
|
Gemiddelde weefsellevensvatbaarheid ≤ 50 %
|
|
|
45.
|
Eén test bestaande uit ten minste twee duploweefsels zou voldoende moeten zijn voor een teststof, indien het resultaat ondubbelzinnig is. Echter, bij grensgevallen zoals niet-concordante metingen van duplo’s en/of een gemiddelde procentuele weefsellevensvatbaarheid van 60 ± 5 % (VRM1 en VRM2, protocol voor vloeistoffen) of 50 ± 5 % (VRM2, protocol voor vaste stoffen), moet een tweede test worden overwogen, evenals een derde testrun in geval van niet-concordante resultaten tussen de eerste twee tests.
|
|
46.
|
Indien van toepassing en gerechtvaardigd kan ter verbetering van de algemene prestaties van de testmethode voor bepaalde typen mengsels worden overwogen om verschillende drempelwaarden van de procentuele weefsellevensvatbaarheid te gebruiken als scheiding tussen geclassificeerde en niet-geclassificeerde teststoffen (zie punt 14). Voor het beoordelen van het potentieel voor ernstig oogletsel/oogirritatie van onbekende teststoffen of productklassen, of van het relatieve toxisch potentieel voor de ogen van een geclassificeerde chemische stof binnen een bepaald spectrum van positieve responsen kan het nuttig zijn om ijkstoffen te gebruiken.
|
GEGEVENS EN RAPPORTAGE
Gegevens
|
47.
|
Voor elke teststof moeten de gegevens van de afzonderlijke duploweefsels in een testrun (bv. OD-waarden/MTT-formazan-piekoppervlakten en berekende gegevens voor de procentuele weefsellevensvatbaarheid voor de teststoffen en controles, en de uiteindelijke voorspelling van de EhCR-testmethode) in tabelvorm in het verslag worden opgenomen, met inbegrip van gegevens uit herhaalde tests, indien van toepassing. Bovendien moeten voor elke afzonderlijke teststof en controle de gemiddelde procentuele weefsellevensvatbaarheid en het verschil in levensvatbaarheid tussen twee duploweefsels (indien n=2 duploweefsels of de SD (indien n ≥ 3 duploweefsels) in het verslag worden opgenomen. Ook eventuele storingen van de MTT-formazanmeting door een teststof door rechtstreekse reductie van MTT en/of kleurinterferentie moeten voor elke geteste chemische stof in het verslag worden opgenomen.
|
Testverslag
|
48.
|
In het testverslag moet de volgende informatie worden opgenomen:
Teststof
|
|
Stof die uit één component bestaat
|
—
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-registratienummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
—
|
fysische toestand, vluchtigheid, pH, LogP, molecuulgewicht, chemische klassen en aanvullende fysisch-chemische eigenschappen die relevant zijn voor het uitvoeren van het onderzoek, voor zover beschikbaar;
|
|
—
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
—
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
—
|
bewaaromstandigheden en stabiliteit, voor zover beschikbaar.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB en mengsel
|
—
|
karakterisering, voor zover mogelijk aan de hand van bv. chemische identiteit (zie hierboven), zuiverheid, kwantitatief voorkomen en relevante fysisch-chemische eigenschappen (zie hierboven) van de bestanddelen, voor zover beschikbaar;
|
|
—
|
fysische toestand en aanvullende fysisch-chemische eigenschappen die relevant zijn voor het uitvoeren van het onderzoek, voor zover beschikbaar;
|
|
—
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
—
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
—
|
bewaaromstandigheden en stabiliteit, voor zover beschikbaar.
|
|
Positieve- en negatievecontrolestoffen
|
—
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-registratienummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
—
|
fysische toestand, vluchtigheid, molecuulgewicht, chemische klassen en aanvullende fysisch-chemische eigenschappen die relevant zijn voor het uitvoeren van het onderzoek, voor zover beschikbaar;
|
|
—
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
—
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
—
|
bewaaromstandigheden en stabiliteit, voor zover beschikbaar;
|
|
—
|
rechtvaardiging voor het gebruik van een andere negatieve controle dan ultrazuiver H2O of Ca2+/Mg2+-vrije DPBS, indien van toepassing;
|
|
—
|
rechtvaardiging voor het gebruik van een andere positieve controle dan zuiver methylacetaat, indien van toepassing;
|
|
—
|
verwijzing naar historische positieve- en negatievecontroleresultaten waaruit de geschiktheid van de aanvaardbaarheidscriteria voor de testrun blijkt.
|
Informatie met betrekking tot de opdrachtgever en de testinstallatie
|
—
|
naam en adres van de opdrachtgever, testinstallatie en de onderzoeksleider.
|
|
—
|
RhCE-weefselpreparaat en gebruikt protocol (met motivering voor keuzes, indien van toepassing)
|
Omstandigheden van de testmethode
|
—
|
het gebruikte RhCE-weefselpreparaat, met inbegrip van het batchnummer;
|
|
—
|
golflengte en bandbreedte (indien van toepassing) die voor het kwantificeren van MTT-formazan zijn gebruikt, en lineariteitsbereik van de meetapparatuur (bv. spectrofotometer);
|
|
—
|
een beschrijving van de methode die voor het kwantificeren van MTT-formazan is gebruikt;
|
|
—
|
een beschrijving van het gebruikte HPLC/UPLC-spectrofotometriesysteem, indien van toepassing;
|
|
—
|
volledige ondersteunende informatie over het specifieke gebruikte RhCE-weefselpreparaat, met inbegrip van de deugdelijkheid daarvan. Hieronder moet ten minste informatie zijn over:
|
i)
|
levensvatbaarheid bij kwaliteitscontrole (QC) (leverancier);
|
|
ii)
|
levensvatbaarheid onder de omstandigheden van de testmethode (gebruiker);
|
|
iii)
|
barrièrefunctie bij QC;
|
|
iv)
|
morfologie, voor zover gegevens beschikbaar zijn;
|
|
v)
|
reproduceerbaarheid en voorspellend vermogen;
|
|
vi)
|
andere QC-gegevens over het RhCE-weefselpreparaat, indien beschikbaar;
|
|
|
—
|
verwijzingen naar historische gegevens over het RhCE-weefselpreparaat. Hieronder moet ten minste informatie zijn over: de aanvaardbaarheid van de QC-gegevens met verwijzing naar historische batchgegevens;
|
|
—
|
verklaring dat voorafgaand aan het routinematig gebruik van de testmethode de bekwaamheid van de testfaciliteit in de toepassing ervan is aangetoond door middel van het testen van de chemische stoffen voor bekwaamheidstoetsing.
|
Aanvaardbaarheidscriteria voor de test en testruns
|
—
|
gemiddelden van de positieve en negatieve controles en aanvaardbaarheidsbereiken op basis van historische gegevens;
|
|
—
|
aanvaardbare variabiliteit tussen de duploweefsels voor de positieve en negatieve controles;
|
|
—
|
aanvaardbare variabiliteit tussen de duploweefsels voor de teststof.
|
Testprocedure
|
—
|
gedetailleerde informatie over de gebruikte testprocedure;
|
|
—
|
de gebruikte doses van de teststof en de controlestoffen;
|
|
—
|
duur en temperatuur van de blootstellings-, onderdompelings- (na de blootstelling) en na-incubatieperioden (voor zover van toepassing);
|
|
—
|
een beschrijving van eventuele wijzigingen van de testprocedure;
|
|
—
|
indicatie van de controles die zijn gebruikt voor rechtstreeks MTT-reducerende stoffen en/of gekleurde teststoffen, indien van toepassing;
|
|
—
|
het aantal gebruikte duploweefsels per teststof en controles (positieve controle, negatieve controle en NSMTT, NSClevend en NSCgedood, indien van toepassing).
|
Resultaten
|
—
|
een tabel met gegevens voor afzonderlijke teststoffen en controlestoffen, voor elke testrun (met inbegrip van herhalingen, voor zover van toepassing) en elke duplometing met inbegrip van OD-waarde of MTT-formazan-piekoppervlakte, procentuele levensvatbaarheid van de weefsels, gemiddelde procentuele levensvatbaarheid van de weefsels, verschil tussen duploweefsels of SD en de uiteindelijke voorspelling;
|
|
—
|
indien van toepassing: resultaten van de gebruikte controles voor rechtstreeks MTT reducerende stoffen en/of gekleurde teststoffen, met inbegrip van OD-waarde of MTT-formazan-piekoppervlakte, %NSMTT, %NSClevend, %NSCgedood, verschil tussen duploweefsels of SD, de uiteindelijke gecorrigeerde procentuele weefsellevensvatbaarheid, en de uiteindelijke voorspelling;
|
|
—
|
resultaten verkregen met de teststof(fen) en controlestoffen ten opzichte van de vastgestelde aanvaardbaarheidscriteria voor de test en testruns;
|
|
—
|
een beschrijving van andere waargenomen effecten, bv. verkleuring van de weefsels door een gekleurde teststof.
|
Bespreking van de resultaten
Conclusie
|
LITERATUUR
|
(1)
|
VN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York en Genève: Verenigde Naties. Beschikbaar op: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Hoofdstuk B.5 van deze bijlage, Acute oogirritatie/-corrosie.
|
|
(3)
|
Hoofdstuk B.47 van deze bijlage, Bovine Corneal Opacity and Permeability- (BCOP-)testmethode voor de identificatie van i) chemische stoffen die ernstig oogletsel veroorzaken en ii) chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is.
|
|
(4)
|
Hoofdstuk B.48 van deze bijlage, Isolated Chicken Eye-testmethode voor de identificatie van i) chemische stoffen die ernstig oogletsel veroorzaken en ii) chemische stoffen waarvoor classificatie niet nodig is.
|
|
(5)
|
Hoofdstuk B.61 van deze bijlage, Fluoresceïnelekkage-testmethode voor de identificatie van voor het oog corrosieve en hevig irriterende stoffen.
|
|
(6)
|
Hoofdstuk B.68 van deze bijlage, in-vitrotestmethode met korte blootstelling voor de identificatie van i) chemische stoffen die ernstig oogletsel veroorzaken en ii) chemische stoffen waarvoor classificatie voor oogirritatie of ernstig oogletsel niet nodig is.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
EC EURL CEVMA (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Beschikbaar op: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
Raadgevend wetenschappelijk comité van het EURL CEVMA (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 EN; doi: 10.2787/043697. Beschikbaar op: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
Raadgevend wetenschappelijk comité van het EURL CEVMA (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. Beschikbaar op: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL CEVMA (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in voorbereiding).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OESO (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(18)
|
OESO (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610-2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan.Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Beschikbaar op: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Versie 1. (20 juli 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Beschikbaar op: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. Mei 2001. Beschikbaar op: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OESO (2017). Guidance Document on Integrated Approaches to Testing and Assessment (IATA) for Serious Eye Damage and Eye Irritation. Series on Testing and Assessment No 263. ENV Publications, Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
Aanhangsel 1
DEFINITIES
Afw.
: afwijking.
Betrouwbaarheid
: indicatie van de mate waarin een testmethode, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (18).
Bewijskracht(proces)
: het proces waarbij de sterke en zwakke punten van verschillende gegevens worden onderzocht bij het komen tot en ondersteunen van een conclusie met betrekking tot het gevarenpotentieel van een teststof.
Bottom-upbenadering
: een trapsgewijze benadering die gebruikt wordt voor een teststof waarvan wordt vermoed dat classificatie en etikettering voor oogirritatie of ernstig oogletsel niet nodig is, die begint met het onderscheiden van chemische stoffen waarvoor classificatie en etikettering niet nodig is (negatief resultaat) van andere chemische stoffen (positief resultaat).
Chemische stof
: een stof of een mengsel.
Concordantie
: zie „Nauwkeurigheid”.
CV
: variatiecoëfficiënt.
EIT
: oogirritatietest (Eye Irritation Test).
Ernstig oogletsel
: het ontstaan van veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig omkeerbaar zijn. Verwisselbaar met „Onomkeerbare effecten op ogen” en met „VN-GHS/CLP-categorie 1”.
ET50
: de blootstellingstijd die nodig is om de weefsellevensvatbaarheid met 50 % te doen dalen bij aanbrenging van een ijkstof in een bepaalde, vaste concentratie.
EURL-CEVMA
: referentielaboratorium van de Europese Unie voor alternatieve methoden ter vervanging van dierproeven.
Gevaar
: inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld.
Geldige testmethode
: een testmethode die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen. Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel (18).
Gevalideerde testmethode
: een testmethode waarvoor valideringsstudies zijn gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er moet worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel (18).
Gevoeligheid
: het percentage van alle positieve/actieve teststoffen dat door de test correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (18).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN))
: een systeem voor de indeling van chemische stoffen (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (1).
HCE
: SkinEthic™ Human Corneal Epithelium (menselijk hoornvliesepitheel).
Hoornvlies
: het doorschijnende gedeelte van het voorste deel van de oogbal dat de iris en pupil bedekt en licht naar binnen toelaat.
HPLC
: hogeprestatievloeistofchromatografie.
IC50
: de concentratie te bepalen waarin een ijkstof de weefsellevensvatbaarheid het weefsel met 50 % doet dalen na een vaste blootstellingstijd (bv. 30 minuten behandeling met SDS).
IJkstof
: een stof die wordt gebruikt als norm voor vergelijking met een teststof. Een ijkstof moet de volgende eigenschappen hebben: i) (een) constante en betrouwbare bron(nen) voor de identificatie en karakterisering ervan; ii) in structureel, functioneel en/of chemisch opzicht of qua productklasse soortgelijk aan de geteste chemische stof(fen); iii) bekende fysisch-chemische eigenschappen; iv) ondersteunende gegevens met betrekking tot bekende effecten, en v) bekende potentie binnen het bereik van de gewenste respons.
LLOQ
: onderste bepaalbaarheidsgrens (Lower Limit of Quantification).
LogP
: logaritme van de octanol-water-verdelingscoëfficiënt.
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
MTT
: 3-(4,5-dimethylthiazol-2-yl)-2,5-difenyltetrazoliumbromide; thiazolylblauw tetrazoliumbromide.
Nauwkeurigheid
: de mate van overeenstemming tussen resultaten verkregen met de testmethode en erkende referentiewaarden. Het is een maat voor de prestaties van de testmethode en één aspect van „relevantie”. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een testmethode wordt verstaan, worden vaak door elkaar gebruikt (18).
Negatieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze in het testsysteem geen positieve respons opwekt. Dit monster wordt samen met de met de teststof behandelde monsters en andere controlemonsters verwerkt en wordt gebruikt voor het vaststellen van 100 % weefsellevensvatbaarheid.
Niet ingedeeld
: stoffen die niet zijn geclassificeerd voor oogirritatie (VN-GHS/CLP-categorie 2, VN-GHS-categorie 2A of 2B) of ernstig oogletsel (VN-GHS/CLP-categorie 1). Verwisselbaar met „VN-GHS/CLP Geen categorie”.
NSCgedood
: niet-specifieke kleur in gedode weefsels.
NSClevend
: niet-specifieke kleur in levende weefsels.
NSMTT
: niet-specifieke MTT-reductie.
OD
: optische dichtheid.
Omkeerbare effecten op ogen
: zie „Oogirritatie”.
Oneindige dosis
: hoeveelheid op het RhCE-weefselpreparaat aangebrachte teststof die groter is dan de hoeveelheid die nodig is om het epitheeloppervlak volledig en gelijkmatig te bedekken.
Onomkeerbare effecten op ogen
: zie „Ernstig oogletsel”.
Oogirritatie
: veranderingen in het oog na het aanbrengen van een teststof op het oppervlak aan de voorzijde van het oog, die binnen 21 dagen na het aanbrengen volledig omkeerbaar zijn. Verwisselbaar met „Omkeerbare effecten op ogen” en met „VN-GHS/CLP-categorie 2”.
Percentage vals-negatieve uitslagen
: het percentage van alle positieve stoffen dat door de testmethode onterecht als negatief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode.
Percentage vals-positieve uitslagen
: het percentage van alle negatieve stoffen dat door een testmethode onterecht als positief wordt geïdentificeerd. Het is één indicator van de prestaties van de testmethode.
Positieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze in het testsysteem een positieve respons opwekt. Dit monster wordt samen met de met de teststof behandelde monsters en andere controlemonsters verwerkt. Om te verzekeren dat variabiliteit in de positievecontrolerespons doorheen de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn.
Prestatienormen
: normen, gebaseerd op een gevalideerde, wetenschappelijk geldig bevonden testmethode, die de basis vormen voor de beoordeling van de vergelijkbaarheid van een voorgestelde, in mechanistisch en functioneel opzicht gelijksoortige testmethode. Hieronder begrepen zijn: i) essentiële componenten van de testmethode; ii) een minimumlijst van referentiestoffen die zijn gekozen uit de stoffen die zijn gebruikt om de aanvaardbare prestaties van de gevalideerde testmethode aan te tonen, en iii) de vergelijkbare nauwkeurigheids- en betrouwbaarheidsniveaus die door de voorgestelde testmethode moeten worden gehaald wanneer ze worden beoordeeld aan de hand van de minimumlijst van referentiestoffen; deze waarden zijn gebaseerd op de waarden die voor de gevalideerde testmethode zijn verkregen (18).
Relevantie
: beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een testmethode (18).
Reproduceerbaarheid
: de overeenkomst tussen resultaten verkregen uit herhaalde tests op dezelfde chemische stof met behulp van hetzelfde testprotocol (zie „Betrouwbaarheid”) (18).
RhCE
: gereconstrueerd model van menselijk hoornvliesepitheel (Reconstructed human Cornea-like Epithelium).
SD
: standaarddeviatie.
Specificiteit
: het percentage van alle negatieve/inactieve teststoffen dat door de test correct wordt ingedeeld. Dit is een maat voor de nauwkeurigheid van een testmethode die categoriale resultaten oplevert en is een belangrijke factor voor de beoordeling van de relevantie van een testmethode (18).
Standard Operating Procedures (SOP)
: formele schriftelijke procedures waarin uitvoerig wordt beschreven hoe specifieke routinematige en testspecifieke laboratoriumhandelingen moeten worden uitgevoerd. Ze zijn verplicht voor GLP.
Stof
: een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Test
: één teststof die gelijktijdig wordt getest op ten minste twee duploweefsels zoals gedefinieerd in de overeenkomstige SOP.
Testrun
: een testrun bestaat uit een of meer teststoffen die gelijktijdig worden getest met een negatieve en een positieve controle.
Teststof
: alle volgens deze testmethode geteste stoffen of mengsels.
Top-downbenadering
: een trapsgewijze benadering die wordt gebruikt voor een chemische stof waarvan wordt vermoed dat deze ernstig oogletsel veroorzaakt, die begint met het onderscheiden van chemische stoffen die ernstig oogletsel veroorzaken (positief resultaat) van andere chemische stoffen (negatief resultaat).
Trapsgewijze teststrategie
: een teststrategie waarbij opeenvolgende testmethoden worden gebruikt. Alle bestaande informatie over een teststof op elke trap wordt herbezien aan de hand van een proces op basis van bewijskracht om te bepalen of er voldoende informatie beschikbaar is voor een beslissing tot gevarenclassificatie, vooraleer wordt overgegaan tot de volgende trap in de strategie. Als op een bepaalde trap op basis van de bestaande informatie het mogelijke gevaar/de potentie van een teststof kan worden bepaald, zijn er geen verdere tests nodig (18).
ULOQ
: bovenste bepaalbaarheidsgrens (Upper Limit of Quantification).
UPLC
: ultrahogeprestatievloeistofchromatografie.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
Vervangende test
: een test, ontworpen ter vervanging van een voor de identificatie van gevaren en/of risicobeoordeling geaccepteerde en routinematig gebruikte test, waarvan is vastgesteld dat hij, in vergelijking met de geaccepteerde test, de gezondheid van mens of dier of het milieu, naargelang het geval, gelijke of betere bescherming biedt voor alle mogelijke testsituaties en chemische stoffen (18).
VN-GHS/CLP-categorie 1
: zie „Ernstig oogletsel”.
VN-GHS/CLP-categorie 2
: zie „Oogirritatie”.
VN-GHS/CLP Geen categorie
: chemische stoffen die niet voldoen aan de criteria voor classificatie in VN-GHS-categorie 1 of 2 (2A of 2B). Verwisselbaar met „Niet ingedeeld”.
VRM
: gevalideerde referentiemethode.
VRM1
: de EpiOcular™ EIT wordt aangeduid als gevalideerde referentiemethode 1.
VRM2
: de SkinEthic™ HCE EIT wordt aangeduid als gevalideerde referentiemethode 2.
Weefsellevensvatbaarheid
: een parameter waarmee de totale activiteit van een celpopulatie in een gereconstrueerd weefsel wordt gemeten als het vermogen om de vitale kleurstof MTT te reduceren, die, afhankelijk van het gemeten eindpunt en de opzet van de test, is gecorreleerd met het totale aantal en/of de vitaliteit van de levende cellen.
Aanhangsel 2
HOOFDCOMPONENTEN VAN DE RHCE-TESTS DIE ZIJN GEVALIDEERD VOOR DE IDENTIFICATIE VAN CHEMISCHE STOFFEN WAARVOOR CLASSIFICATIE EN ETIKETTERING VOOR OOGIRRITATIE OF ERNSTIG OOGLETSEL NIET NODIG IS
|
Testcomponenten
|
EpiOcular™ EIT
(VRM1)
|
SkinEthic™ HCE EIT
(VRM2)
|
|
Protocollen
|
Vloeistoffen
(gedurende 15 minuten pipetteerbaar bij temperaturen van 37 ± 1 °C of lager)
|
Vaste stoffen
(niet pipetteerbaar)
|
Vloeistoffen en viskeuze stoffen
(pipetteerbaar)
|
Vaste stoffen
(niet pipetteerbaar)
|
|
Oppervlak van het model
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Aantal duploweefsels
|
Ten minste 2
|
Ten minste 2
|
Ten minste 2
|
Ten minste 2
|
|
Voorafgaande toetsing op kleurinterferentie
|
50 μl + 1 ml H2O gedurende 60 min bij 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH (niet-gekleurde teststoffen), of 50 μl + 2 ml isopropanol gemengd gedurende 2-3 uur bij RT (gekleurde teststoffen)
|
 als de OD van de teststof bij 570 ± 20 nm na aftrek van de OD voor isopropanol of water > 0,08 is (wat overeenkomt met ongeveer 5 % van de gemiddelde OD van de negatieve controle), moeten er aangepaste controles met levende weefsels worden uitgevoerd.
als de OD van de teststof bij 570 ± 20 nm na aftrek van de OD voor isopropanol of water > 0,08 is (wat overeenkomt met ongeveer 5 % van de gemiddelde OD van de negatieve controle), moeten er aangepaste controles met levende weefsels worden uitgevoerd.
|
|
50 mg + 1 ml H2O gedurende 60 min bij 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % RH (niet-gekleurde teststoffen)
en/of
50 mg + 2 ml isopropanol gemengd gedurende 2-3 uur bij RT (gekleurde en niet-gekleurde teststoffen)
|
 als de OD van de teststof bij 570 ± 20 nm na aftrek van de OD voor isopropanol of water > 0,08 is (wat overeenkomt met ongeveer 5 % van de gemiddelde OD van de negatieve controle), moeten er aangepaste controles met levende weefsels worden uitgevoerd.
als de OD van de teststof bij 570 ± 20 nm na aftrek van de OD voor isopropanol of water > 0,08 is (wat overeenkomt met ongeveer 5 % van de gemiddelde OD van de negatieve controle), moeten er aangepaste controles met levende weefsels worden uitgevoerd.
|
|
10 μl + 90 μl H2O gemengd gedurende 30 ± 2 min bij RT (18-28 oC)
|
 als de teststof gekleurd is, moeten er aangepaste controles met levende weefsels worden uitgevoerd
als de teststof gekleurd is, moeten er aangepaste controles met levende weefsels worden uitgevoerd
|
|
10 mg + 90 μl H2O gemengd gedurende 30 ± 2 min bij RT
|
 als de teststof gekleurd is, moeten er aangepaste controles met levende weefsels worden uitgevoerd
als de teststof gekleurd is, moeten er aangepaste controles met levende weefsels worden uitgevoerd
|
|
|
Voorafgaande toetsing op rechtstreekse MTT-reductie
|
50 μl + 1 ml van een 1 mg/ml MTT-oplossing gedurende 180 ± 15 min bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
 als de oplossing blauw/paars wordt, moeten er aangepaste controles met door bevriezing gedode weefsels worden uitgevoerd
als de oplossing blauw/paars wordt, moeten er aangepaste controles met door bevriezing gedode weefsels worden uitgevoerd
(als negatieve controle wordt 50 μl steriel gedeïoniseerd water in MTT-oplossing gebruikt)
|
|
50 mg + 1 ml van een 1 mg/ml MTT-oplossing gedurende 180 ± 15 min bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
 als de oplossing blauw/paars wordt, moeten er aangepaste controles met door bevriezing gedode weefsels worden uitgevoerd
als de oplossing blauw/paars wordt, moeten er aangepaste controles met door bevriezing gedode weefsels worden uitgevoerd
(als negatieve controle wordt 50 μl steriel gedeïoniseerd water in MTT-oplossing gebruikt)
|
|
30 μl + 300 μl van een 1 mg/ml MTT-oplossing gedurende 180 ± 15 min bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
 als de oplossing blauw/paars wordt, moeten er aangepaste controles met in water gedode weefsels worden uitgevoerd
als de oplossing blauw/paars wordt, moeten er aangepaste controles met in water gedode weefsels worden uitgevoerd
(als negatieve controle wordt 30 μl steriel gedeïoniseerd water in MTT-oplossing gebruikt)
|
|
30 mg + 300 μl van een 1 mg/ml MTT-oplossing gedurende 180 ± 15 min bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
 als de oplossing blauw/paars wordt, moeten er aangepaste controles met in water gedode weefsels worden uitgevoerd
als de oplossing blauw/paars wordt, moeten er aangepaste controles met in water gedode weefsels worden uitgevoerd
(als negatieve controle wordt 30 μl steriel gedeïoniseerd water in MTT-oplossing gebruikt)
|
|
|
Voorbehandeling
|
20 μl Ca2+/Mg2+-vrije DPBS
gedurende 30 ± 2 min bij 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, beschermd tegen het licht.
|
20 μl Ca2+/Mg2+-vrije DPBS
gedurende 30 ± 2 min bij 37 ± 2oC, 5 ± 1 % CO2, ≥ 95 % RH, beschermd tegen het licht.
|
—
|
—
|
|
Doses van de behandeling en wijze van aanbrengen
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) met behulp van een gekalibreerd instrument (bv. een afgestreken maatlepel die is gekalibreerd op een inhoud van 50 mg natriumchloride).
|
10 μl Ca2+/Mg2+-vrije DPBS + 30 ± 2 μl (60 μl/cm2)
Gebruik voor viskeuze stoffen nylongaas
|
30 μl Ca2+/Mg2+-vrije DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Blootstellingstijd en -temperature
|
30 min (± 2 min)
in kweekmedium
bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
6 uur (± 0,25 uur)
in kweekmedium
bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
30 min (± 2 min)
in kweekmedium
bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
4 uur (± 0,1 uur)
in kweekmedium
bij 37 ± 2°C, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Spoelen bij kamertemperatuur
|
3 maal in 100 ml Ca2+/Mg2+-vrije DPBS
|
3 maal in 100 ml Ca2+/Mg2+-vrije DPBS
|
20 ml Ca2+/Mg2+-vrije DPBS
|
25 ml Ca2+/Mg2+-vrije DPBS
|
|
Onderdompeling na blootstelling
|
12 min (± 2 min) bij RT in kweekmedium
|
25 min (± 2 min) bij RT in kweekmedium
|
30 min (± 2 min) bij 37 °C, 5 % CO2, 95 % RH in kweekmedium
|
30 min (± 2 min) bij RT in kweekmedium
|
|
Na-incubatie
|
120 min ( 15 min) in kweekmedium bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
18 uur (± 0,25 uur) in kweekmedium bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
geen
|
18 uur (± 0,5 uur) in kweekmedium bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Negatieve controle
|
50 μl H2O
Gelijktijdig getest
|
50 μl H2O
Gelijktijdig getest
|
30 ± 2 μl Ca2+/Mg2+-vrije DPBS
Gelijktijdig getest
|
30 ± 2 μl Ca2+/Mg2+-vrije DPBS
Gelijktijdig getest
|
|
Positieve controle
|
50 μl methylacetaat
Gelijktijdig getest
|
50 μl methylacetaat
Gelijktijdig getest
|
30 ± 2μl methylacetaat
Gelijktijdig getest
|
30 ± 2μl methylacetaat
Gelijktijdig getest
|
|
MTT-oplossing
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT-incubatieduur en -temperature
|
180 min (± 15 min) bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
180 min (± 15 min) bij 37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RH
|
|
Extractievloeistof
|
2 ml isopropanol
(extractie van boven- en onderkant van inzetstuk door het weefsel door te prikken)
|
2 ml isopropanol
(extractie van onderkant van inzetstuk door het weefsel door te prikken)
|
1,5 ml isopropanol
(extractie van boven- en onderkant van inzetstuk)
|
1,5 ml isopropanol
(extractie van onderkant van inzetstuk)
|
|
Extractieduur en -temperatuur
|
2-3 uur onder schudden (~ 120 rpm) bij RT of gedurende een nacht bij 4-10 °C
|
2-3 uur onder schudden (~ 120 rpm) bij RT of gedurende een nacht bij 4-10 °C
|
4 uur onder schudden (~ 120 rpm) bij RT of ten minste gedurende een nacht zonder schudden bij 4-10 °C
|
Ten minste 2 uur onder schudden (~ 120 rpm) bij RT
|
|
OD-meting
|
570 nm (550 - 590 nm)
zonder referentiefilter
|
570 nm (550 - 590 nm)
zonder referentiefilter
|
570 nm (540 - 600 nm)
zonder referentiefilter
|
570 nm (540 - 600 nm)
zonder referentiefilter
|
|
Weefselkwaliteitscontrole
|
Behandeling met 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
Behandeling met 100 μl 0,3 % (v/v) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
30 min behandeling met SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 min behandeling met SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Aanvaardbaarheidscriteria
|
|
1.
|
De gemiddelde OD van de duploweefsels behandeld met de negatieve controle moet > 0,8 en < 2,5 zijn
|
|
2.
|
De gemiddelde levensvatbaarheid van de duploweefsels die gedurende 30 min zijn blootgesteld aan de positieve controle, uitgedrukt als % van de negatieve controle, moet < 50 % zijn
|
|
3.
|
Het verschil in levensvatbaarheid tussen twee duploweefsels moet kleiner zijn dan 20 %.
|
|
|
1.
|
De gemiddelde OD van de duploweefsels behandeld met de negatieve controle moet > 0,8 en < 2,5 zijn
|
|
2.
|
De gemiddelde levensvatbaarheid van de duploweefsels die gedurende 6 uur zijn blootgesteld aan de positieve controle, uitgedrukt als % van de negatieve controle, moet < 50 % zijn
|
|
3.
|
Het verschil in levensvatbaarheid tussen twee duploweefsels moet kleiner zijn dan 20 %.
|
|
|
1.
|
De gemiddelde OD van de duploweefsels behandeld met de negatieve controle moet > 1,0 en ≤ 2,5 zijn
|
|
2.
|
De gemiddelde levensvatbaarheid van de duploweefsels die gedurende 30 min zijn blootgesteld aan de positieve controle, uitgedrukt als % van de negatieve controle, moet ≤ 30 % zijn
|
|
3.
|
Het verschil in levensvatbaarheid tussen twee duploweefsels moet kleiner zijn dan 20 %.
|
|
|
1.
|
De gemiddelde OD van de duploweefsels behandeld met de negatieve controle moet > 1,0 en ≤ 2,5 zijn
|
|
2.
|
De gemiddelde levensvatbaarheid van de duploweefsels die gedurende 4 uur zijn blootgesteld aan de positieve controle, uitgedrukt als % van de negatieve controle, moet ≤ 20 % zijn
|
|
3.
|
Het verschil in levensvatbaarheid tussen twee duploweefsels moet kleiner zijn dan 20 %.
|
|
Aanhangsel 3
ILLUSTRATIEF STROOMSCHEMA DAT ALS LEIDRAAD KAN DIENEN VOOR HET IDENTIFICEREN EN BEHANDELEN VAN RECHTSTREEKS MTT REDUCERENDE STOFFEN EN/OF STOFFEN DIE KLEURINTERFERENTIE VEROORZAKEN, OP BASIS VAN DE SOP VOOR VRM1

Aanhangsel 4
ILLUSTRATIEF STROOMSCHEMA DAT ALS LEIDRAAD KAN DIENEN VOOR HET IDENTIFICEREN EN BEHANDELEN VAN RECHTSTREEKS MTT REDUCERENDE STOFFEN EN/OF STOFFEN DIE KLEURINTERFERENTIE VEROORZAKEN, OP BASIS VAN DE SOP VOOR VRM2

Aanhangsel 5
KRITISCHE PARAMETERS EN AANVAARDBAARHEIDSCRITERIA VOOR KWALIFICATIES VAN EEN HPLC/UPLC-SPECTROFOTOMETRIESYSTEEM VOOR METING VAN UIT RHCE-WEEFSELPREPARATEN GEËXTRAHEERD MTT-FORMAZAN
|
Parameter
|
Protocol ontleend aan FDA-leidraad (36) (38)
|
Aanvaardbaarheidscriteria
|
|
Selectiviteit
|
Analyse van isopropanol, levende blanco (isopropanolextract van onbehandelde levende RhCE-weefselpreparaten), dode blanco (isopropanolextract van onbehandelde gedode RhCE-weefselpreparaten) en een kleurstof (bv. methyleenblauw)
|
Oppervlakteinterferentie ≤ 20 % van OppervlakteLLOQ
(88)
|
|
Precisie
|
Kwaliteitscontroles (d.w.z. MTT-formazan bij 1,6 μg/ml, 16 μg/ml en 160 μg/ml ) in isopropanol (n=5)
|
CV ≤ 15 % of ≤ 20 % voor de LLOQ
|
|
Nauwkeurigheid
|
Kwaliteitscontroles in isopropanol (n=5)
|
%Afw. ≤ 15 % of ≤ 20 % voor de LLOQ
|
|
Matrixeffect
|
Kwaliteitscontroles in levende blanco (n=5)
|
85 % ≤ %Matrixeffect ≤ 115 %
|
|
Carry-over
|
Analyse van isopropanol na een ULOQ (89)-standaard
|
Oppervlakteinterferentie ≤ 20 % van OppervlakteLLOQ
|
|
Reproduceerbaarheid (binnen één dag)
|
3 onafhankelijke kalibratiecurven (op basis van 6 achtereenvolgende 1/3 verdunningen van MTT-formazan in isopropanol beginnend bij de ULOQ, d.w.z. 200 μg/ml);
Kwaliteitscontroles in isopropanol (n=5)
|
Kalibratiecurven: %Afw. ≤ 15 % of ≤ 20 % voor de LLOQ
Kwaliteitscontroles: %Afw. ≤ 15 % en CV ≤ 15 %
|
|
Reproduceerbaarheid (van dag tot dag)
|
Dag 1: 1 kalibratiecurve en kwaliteitscontroles in isopropanol (n=3)
Dag 2: 1 kalibratiecurve en kwaliteitscontroles in isopropanol (n=3)
Dag 3: 1 kalibratiecurve en kwaliteitscontroles in isopropanol (n=3)
|
|
Kortetermijnstabiliteit van MTT-formazan in RhCE-weefselextract
|
Kwaliteitscontroles op levende blanco (n=3) geanalyseerd op de dag van bereiding en na 24 uur bewaring bij kamertemperatuur
|
%Afw. ≤ 15 %
|
|
Langetermijnstabiliteit van MTT-formazan in RhCE-weefselextract, indien nodig
|
Kwaliteitscontroles op levende blanco (n=3) geanalyseerd op de dag van bereiding en na een aantal dagen bewaring bij –20 °C
|
%Afw. ≤ 15 %
|
B.70
IN-VITRO BEPALINGEN MET EEN MENSELIJKE RECOMBINANTE OESTROGEENRECEPTOR (hrER) VOOR HET OPSPOREN VAN CHEMISCHE STOFFEN MET ER-BINDINGSAFFINITEIT
ALGEMENE INLEIDING
Op prestaties gebaseerde testrichtlijn van de OESO
|
1.
|
Deze testmethode is gelijkwaardig aan testrichtlijn (TG) 493 (2015) van de OESO. TG 493 is een op prestaties gebaseerde testrichtlijn (performance-based test guideline, PBTG), waarin de methodologie wordt beschreven voor menselijke recombinante in-vitrobepalingen (hrER) voor het opsporen van stoffen met oestrogeenreceptorbindingsaffiniteit (hrER-bindingsbepalingen). De PBTG omvat twee, in mechanistisch en functioneel opzicht gelijksoortige bepalingen voor het identificeren van oestrogeenreceptor-α-binders (ERα-binders) en moet de ontwikkeling bevorderen van nieuwe gelijksoortige of gewijzigde testmethoden die overeenstemmen met de valideringsbeginselen beschreven in de OESO-leidraad voor de validering en internationale aanvaarding van nieuwe of bijgewerkte bepalingen voor gevarenbeoordeling (Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment) (1) De volledig gevalideerde referentietestmethoden (aanhangsels 2 en 3) die de basis vormen voor deze op prestaties gebaseerde testrichtlijn zijn:
|
—
|
de in-vitrobepaling van de binding aan de oestrogeenreceptor (ER) met een volledige menselijke recombinante ERα van Freyberger en Wilson (FW-bepaling) (2), en
|
|
—
|
de in-vitrobepaling van de binding aan de oestrogeenreceptor met een menselijk recombinant ligandbindingsdomein-eiwit van het Japanse Chemicals Evaluation and Research Institute (CERI-bepaling) (2).
|
Er zijn prestatienormen (3) beschikbaar om de ontwikkeling en validering van soortgelijke testmethoden voor het zelfde gevareneindpunt te vergemakkelijken en tijdige wijziging van PBTG 493 mogelijk te maken, zodat nieuwe, gelijksoortige bepalingen aan een bijgewerkte PBTG kunnen worden toegevoegd. Gelijksoortige testbepalingen zullen evenwel pas worden toegevoegd nadat de OESO ze heeft beoordeeld en onderschrijft dat aan de prestatienormen wordt voldaan. De in TG 493 opgenomen bepalingen kunnen zonder onderscheid worden gebruikt om te voldoen aan de eisen die aan OESO-landen worden gesteld wat betreft de testresultaten voor oestrogeenreceptorbinding, en in aanmerking te komen voor wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst.
|
Achtergrond en beginselen van de bepalingen die in deze testmethode worden beschreven
|
2.
|
In 1998 is de OESO met hoge prioriteit een actie gestart om bestaande testrichtlijnen te herzien en nieuwe testrichtlijnen te ontwikkelen voor het screenen en testen op potentiële hormoonontregelaars. Het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen werd in 2012 herzien. Het oorspronkelijke en het herziene conceptueel kader zijn als bijlagen opgenomen bij de leidraad voor gestandaardiseerde testrichtlijnen voor de beoordeling van chemische stoffen op hormoonontregeling (Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption) (4). Het conceptueel kader omvat vijf niveaus, die elk overeenkomen met een ander niveau van biologische complexiteit. De in deze testmethode beschreven ER-bindingsbepalingen zijn ingedeeld op niveau 2: „in-vitrobepalingen die gegevens moeten opleveren over (een) geselecteerde endocriene mechanisme(n)/weg(en)”. Deze testmethode betreft in-vitroreceptorbindingsbepalingen die zijn ontworpen voor het identificeren van liganden voor de menselijke oestrogeenreceptor-alfa (ERα).
|
|
3.
|
De relevantie van de in-vitro-ER-bindingsbepaling voor biologische functies is genoegzaam aangetoond. ER-bindingsbepalingen zijn ontworpen voor het identificeren van chemische stoffen die in staat zijn het signaalpad van oestrogene hormonen te verstoren. Ze worden al twee decennia breed toegepast voor het karakteriseren van de weefseldistributie van ER’s en het identificeren van ER-agonisten/antagonisten. Deze bepalingen berusten op de interactie tussen ligand en receptor, die de eerste stap vormt van het signaalpad van oestrogenen en essentieel is voor de voortplantingsfunctie van alle gewervelden.
|
|
4.
|
De interactie van oestrogenen met ER’s kan de transcriptie van oestrogeengestuurde genen beïnvloeden en niet-genomische effecten veroorzaken, wat kan leiden tot de inductie of remming van processen in de cel, waaronder de processen die nodig zijn voor celproliferatie, de normale ontwikkeling van de foetus en de voortplantingsfunctie (5) (6) (7). Verstoring van de normale oestrogene systemen zou kunnen leiden tot schadelijke effecten op de normale ontwikkeling (ontogenese), de reproductieve gezondheid en de integriteit van het voortplantingsstelsel. Een verstoorde ER-signalering kan leiden tot effecten als een verhoogd risico van hormoonafhankelijke kanker, een verstoorde vruchtbaarheid en afwijkingen in de groei en ontwikkeling van de foetus (8).
|
|
5.
|
In-vitro-bindingsbepalingen zijn gebaseerd op een directe interactie van een stof met de ligandbindingsplaats van een specifieke receptor die de gentranscriptie reguleert. De voornaamste component van de bindingsbepaling met een menselijke recombinante oestrogeenreceptor-alfa (hrERα) meet het vermogen van een radioactief gelabelde ligand ([3H]17β-oestradiol) om aan de ER te binden in aanwezigheid van oplopende concentraties van een teststof (een „competitor”). Teststoffen met een hoge affiniteit voor de ER concurreren in een lagere concentratie met de radioactief gelabelde ligand dan chemische stoffen met een lagere affiniteit voor de receptor. Deze bepaling heeft twee hoofdcomponenten: een bepaling van de verzadigingsbinding om de parameters van de receptor-ligandinteractie te bepalen en de ER-specificiteit te documenteren, gevolgd door een bepaling van de competitieve binding die de concurrentie tussen een teststof en een radioactief gelabelde ligand om binding aan de ER karakteriseert.
|
|
6.
|
De relevantie en de betrouwbaarheid van de CERI-bindingsbepaling en de FW-bindingsbepaling voor het beoogde doel zijn aangetoond in de valideringsstudies voor deze bepalingen (2).
|
|
7.
|
De in deze testmethode gebruikte definities en afkortingen staan in aanhangsel 1.
|
Toepassingsgebied en beperkingen voor de receptorbindingsbepalingen
|
8.
|
Deze bepalingen worden aangereikt ten behoeve van screening en prioritering, maar kunnen ook informatie verschaffen voor een moleculaire inleidende gebeurtenis die kan worden gebruikt in een benadering met meervoudige bewijsvoering („weight of evidence”-benadering). De bepalingen berusten op de chemische binding aan het ligandbindingsdomein van de ERα in een in-vitrosysteem. De resultaten mogen dan ook niet rechtstreeks geëxtrapoleerd worden naar de complexe signalering en regulering van het intacte endocriene systeem in vivo.
|
|
9.
|
De binding van de natuurlijke ligand, 17β-oestradiol, is de eerste stap in een reeks moleculaire gebeurtenissen die de transcriptie van doelgenen activeert en uiteindelijk uitmondt in een fysiologische verandering (9). Binding aan het ligandbindingsdomein van de ERα wordt daarom beschouwd als een van de voornaamste mechanismen voor ER-gemedieerde hormoonontregeling, hoewel er ook andere mechanismen zijn waardoor hormoonontregeling kan optreden, waaronder i) interacties met andere delen van de ERα dan de ligandbindingsplaats, ii) interacties met andere receptoren die bij het signaalpad van oestrogenen betrokken zijn, de ERβ en de G-proteïnegekoppelde ER, andere receptoren en enzymsystemen in het endocriene systeem, iii) hormoonsynthese, iv) metabole activering en/of inactivering van hormonen, v) verdeling van hormonen naar doelweefsels, en vi) klaring van hormonen uit het lichaam. In geen van de bepalingen die onder deze testmethode vallen, worden deze werkingsmechanismen in aanmerking genomen.
|
|
10.
|
Deze testmethode richt zich op het vermogen van stoffen om aan de menselijke ERα te binden en maakt geen onderscheid tussen ERα-agonisten en ERα-antagonisten. Gebeurtenissen die op deze binding volgen, zoals gentranscriptie of fysiologische veranderingen, worden door deze bepalingen evenmin in aanmerking genomen. Aangezien bij de validering alleen zuivere stoffen zijn gebruikt die uit één bestanddeel bestaan, zijn er geen gegevens over de toepasbaarheid voor testmengsels. De bepalingen zijn in theorie evenwel toepasbaar voor het testen van stoffen die uit meerdere componenten bestaan en van mengsels.Voordat de testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel. Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is.
|
|
11.
|
De celvrije receptorsystemen hebben geen eigen metaboliseringscapaciteit en zijn niet gevalideerd in combinatie met metabole enzymactiviteit. Niettemin zou het mogelijk kunnen zijn om metabole activiteit in de opzet van een studie te integreren, maar dit zou wel nadere validering vereisen.
|
|
12.
|
Chemische stoffen die het eiwit (d.w.z. receptoreiwit) kunnen denatureren, zoals oppervlakteactieve stoffen of chemische stoffen die de pH van de testbuffer veranderen, kunnen mogelijk niet worden getest of alleen bij concentraties waarbij dergelijke interacties niet optreden. In andere gevallen wordt het concentratiebereik waarin een teststof in deze bepalingen kan worden getest, beperkt door oplosbaarheid ervan in de testbuffer.
|
|
13.
|
Ter informatie worden in tabel 1 de testresultaten weergegeven voor de 24 stoffen die zijn getest in beide volledig gevalideerde bepalingen die in deze testmethode worden beschreven. Op basis van de gepubliceerde verslagen, waaronder in-vitrobepalingen voor de transcriptionele activatie van de ER en/of de uterotrofe test, zijn 17 van deze stoffen ingedeeld als ER-binders en 6 als niet-binders (9) (10) (11) (12) (13) (14) (15). Wat betreft de gegevens die zijn weergegeven in tabel 1 was er bijna 100 % overeenstemming tussen de beide bepalingen over de indeling van alle stoffen tot een concentratie van 10-4 M, en elke stof werd correct ingedeeld als ER-binder of niet-ER-binder. Nadere informatie over deze groep stoffen en over de aanvullende stoffen die tijdens de valideringsstudies voor de ER-bindingsbepalingen getest zijn, is te vinden in de prestatienormen voor de hrER-bindingsbepaling (3), aanhangsel 2 (tabellen 1, 2 en 3).
Tabel 1
Indeling van stoffen als ER-binders of niet-ER-binders op basis van de FW- en CERI-hrER-bindingsbepalingen, vergeleken met de verwachte respons
|
|
Naam van de stof
|
CAS RN
|
Verwachte respons
|
FW-bepaling
|
CERI-bepaling
|
Chemische klasse volgens de MeSH
|
Productcategorie
|
|
|
Concentratie- bereik (M)
|
Indeling
|
Concentratie- bereik (M)
|
Indeling
|
|
1
|
17β-Oestradiol
|
50-28-2
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Binder
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
2
|
Noretynodrel
|
68-23-5
|
Binder
|
3 × 10-9 – 30 × 10-4
|
Binder
|
3 × 10-9 – 30 × 10-4
|
Binder
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
3
|
Norethisteron
|
68-22-4
|
Binder
|
3 × 10-9 – 30 × 10-4
|
Binder
|
3 × 10-9 – 30 × 10-4
|
Binder
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
4
|
Di-n-butylftalaat
|
84-74-2
|
Niet-binder (*5)
|
1 × 10-10 – 1 × 10-4
|
Niet-binder (*6)
(1)
|
1 × 10-10 – 1 × 10-4
|
Niet-binder (*6)
(1)
|
Koolwaterstof (cyclisch), ester
|
Weekmaker, chemisch tussenproduct
|
|
5
|
DES
|
56-53-1
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (cyclisch), fenol
|
Geneesmiddel, diergeneesmiddel
|
|
6
|
17α-Ethinylestradiol
|
57-63-6
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
7
|
meso-Hexestrol
|
84-16-2
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (cyclisch), fenol
|
Geneesmiddel, diergeneesmiddel
|
|
8
|
Genisteïne
|
446-72-0
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (cyclisch), flavonoïde
|
Natuurlijk product
|
|
9
|
Equol
|
531-95-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Fyto-oestrogeenmetaboliet
|
Natuurlijk product
|
|
10
|
Butylparabeen (n-butyl-4-hydroxybenzoaat)
|
94-26-8
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Parabeen
|
Conserveermiddel
|
|
11
|
Nonylfenol (mengsel)
|
84852-15-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Alkylfenol
|
Tussenproduct
|
|
12
|
o,p’-DDT
|
789-02-6
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Organische chloorverbinding
|
Insecticide
|
|
13
|
Corticosteron
|
50-22-6
|
Niet-binder (*5)
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
Steroïde
|
Natuurlijk product
|
|
14
|
Zearalenon
|
17924-92-4
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (cyclisch), lacton
|
Natuurlijk product
|
|
15
|
Tamoxifen
|
10540-29-1
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (cyclisch)
|
Geneesmiddel, diergeneesmiddel
|
|
16
|
5α-dihydrotestosteron
|
521-18-6
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Steroïde, niet-fenolverbinding
|
Natuurlijk product
|
|
17
|
Bisfenol A
|
80-05-7
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Fenol
|
Chemisch tussenproduct
|
|
18
|
4-n-heptylfenol
|
1987-50-4
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Onduidelijk (5)
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Alkylfenol
|
Tussenproduct
|
|
19
|
Kepone (Chloordecon)
|
143-50-0
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Koolwaterstof (gehalogeneerd)
|
Pesticide
|
|
20
|
Benzo[a]antraceen
|
56-55-3
|
Niet-binder
|
1 × 10-10 – 1 × 10-3
|
Niet-binder (6)
|
1 × 10-10 – 1 × 10-3
|
Niet-binder (6)
|
Aromatische koolwaterstof
|
Tussenproduct
|
|
21
|
Enterolacton
|
78473-71-9
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Binder
|
Fyto-oestrogeen
|
Natuurlijk product
|
|
22
|
Progesteron
|
57-83-0
|
Niet-binder (*5)
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
Steroïde
|
Natuurlijk product
|
|
23
|
Octyltriëthoxysilaan
|
2943-75-1
|
Niet-binder
|
1 × 10-10 – 1 × 10-3
|
Niet-binder
|
1 × 10-10 – 1 × 10-3
|
Niet-binder
|
Silaan
|
Oppervlakmodificator
|
|
24
|
Atrazine
|
1912-24-9
|
Niet-binder (*5)
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
1 × 10-10 – 1 × 10-4
|
Niet-binder
|
Heterocyclische verbinding
|
Herbicide
|
|
COMPONENTEN VAN DE hrER-BINDINGSBEPALING
Essentiële componenten van de bepaling
|
14.
|
Deze testmethode is van toepassing op bepalingen waarbij gebruikgemaakt wordt van een ER-receptor en een ligand die voldoende sterk aan de receptor bindt. Deze ligand kan als marker/tracer voor de bepaling worden gebruikt en kan worden verdreven met oplopende concentraties van de teststof. De twee hoofdcomponenten van bindingsbepalingen zijn: 1) verzadigingsbinding en 2) competitieve binding. De bepaling van de verzadigingsbinding wordt gebruikt ter bevestiging van de specificiteit en de activiteit van de receptorpreparaten en aan de hand van de competitievebindingsbepaling wordt het vermogen van de teststof om aan de hrER te binden beoordeeld.
|
Controles
|
15.
|
De keuze van het referentie-oestrogeen en de controles, die gelijktijdig worden getest, moet worden onderbouwd. Gelijktijdige controles (oplosmiddel (vehiculum), positief (ER-binder; sterke en zwakke affiniteit) en negatief (niet-binder)), voor zover van toepassing, dienen als indicatie dat de bepaling werkt onder de testomstandigheden, en zorgen dat de verschillende experimenten met elkaar vergeleken kunnen worden. Ze maken doorgaans deel uit van de aanvaardbaarheidscriteria voor een bepaald experiment (1). Tijdens elke testrun moeten volledige concentratiecurven voor het referentie-oestrogeen en de controles (d.w.z. zwakke binder en niet-binder) in één plaat worden gebruikt. Alle andere platen moeten het volgende bevatten: i) een hoge concentratie (nagenoeg volledige verdrijving van de radioactief gelabelde ligand) en een middelhoge concentratie (bij benadering de IC50) van zowel E2 als de zwakke binder in drievoud; ii) oplosmiddelcontrole en niet-specifieke binding, elk in drievoud.
|
Standaard kwaliteitscontroleprocedures
|
16.
|
Voor elke bepaling moeten de standaard kwaliteitscontroleprocedures worden uitgevoerd zoals beschreven, om er zeker van te zijn dat de receptoren actief zijn, dat de chemische concentraties kloppen en dat de tolerantiegrenzen gedurende meerdere herhaalde bepalingen stabiel blijven, en het vermogen behouden om doorheen de tijd de verwachte ER-bindingsresponsen te geven.
|
Aantoning van de bekwaamheid van laboratoria
|
17.
|
Alvorens onbekende chemische stoffen te testen met een van de bepalingen die onder deze testmethode vallen, moet elk laboratorium zijn bekwaamheid in het gebruik van de bepaling aantonen door verzadigingsbepalingen uit te voeren, ter bevestiging van de specificiteit en de activiteit van het ER-preparaat, en competitievebindingsbepalingen met het referentie-oestrogeen en de controles (zwakke binder en niet-binder). Het laboratorium moet een historische databank aanleggen met resultaten voor het referentie-oestrogeen en de controles, die zijn gegenereerd met 3-5 onafhankelijke, op verschillende dagen uitgevoerde experimenten. Deze experimenten zullen de basis vormen voor het referentie-oestrogeen en de historische controle voor het laboratorium en zullen mede worden gebruikt om de aanvaardbaarheid van de bepaling te beoordelen voor toekomstige testruns.
|
|
18.
|
Door het testen van de in tabel 2 genoemde stoffen voor bekwaamheidstoetsing wordt ook de responsiviteit van het testsysteem bevestigd. De lijst van stoffen voor bekwaamheidstoetsing is een subset van de referentiestoffen die worden aangegeven in de prestatienormen voor de ER-bindingsbepalingen (3). Deze stoffen zijn in de handel verkrijgbaar, vertegenwoordigen chemische klassen waarbij doorgaans sprake is van ER-bindende activiteit, vertonen een geschikt potentiebereik voor de verwachte potentie van ER-binders (d.w.z. sterke tot zwakke binders) en niet-binders (d.w.z. negatieven). Voor elke stof voor bekwaamheidstoetsing moeten de geteste concentraties het bereik in tabel 2 bestrijken. Er moeten voor elke stof ten minste drie experimenten worden uitgevoerd en de resultaten moeten overeenstemmen met de verwachte chemische activiteit. Elk experiment moet onafhankelijk (d.w.z. met verse verdunningen van receptor, chemische stoffen en reagens) worden verricht, met elke concentratie in drievoud. De bekwaamheid wordt aangetoond door een correcte indeling (positief/negatief) van elke stof. De bekwaamheidstoetsing moet worden uitgevoerd door elke onderzoeker die de bepalingen leert.
Tabel 2
Lijst van controles en stoffen voor bekwaamheidstoetsing voor de competitieve hrER-bindingsbepalingen
(90)
|
Nr.
|
Naam van de stof
|
CAS RN (91)
|
Verwachte respons (92)
(93)
|
Testconcentratiebereik (M)
|
Chemische klasse volgens de MeSH (94)
|
Productcategorie (95)
|
|
Controles (referentie-oestrogeen, zwakke binder, niet-binder)
|
|
1
|
17β-Oestradiol
|
50-28-2
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
2
|
Noretynodrel (of) Norethisteron
|
68-23-5 (of) 68-22-4
|
Binder
|
3 × 10-9 – 30 × 10-6
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
3
|
Octyltriëthoxysilaan
|
2943-75-1
|
Niet-binder
|
1 × 10-10 – 1 × 10-3
|
Silaan
|
Oppervlakmodificator
|
|
Stoffen voor bekwaamheidstoetsing (95)
|
|
4
|
Diëthylstilboestrol
|
56-53-1
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Koolwaterstof (cyclisch), fenol
|
Geneesmiddel, diergeneesmiddel
|
|
5
|
17α-Ethinylestradiol
|
57-63-6
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Steroïde
|
Geneesmiddel, diergeneesmiddel
|
|
6
|
meso-Hexestrol
|
84-16-2
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Koolwaterstof (cyclisch), fenol
|
Geneesmiddel, diergeneesmiddel
|
|
7
|
Tamoxifen
|
10540-29-1
|
Binder
|
1 × 10-11 – 1 × 10-6
|
Koolwaterstof (cyclisch)
|
Geneesmiddel, diergeneesmiddel
|
|
8
|
Genisteïne
|
446-72-0
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Heterocyclische verbinding, flavonoïde,
|
Natuurlijk product
|
|
9
|
Bisfenol A
|
80-05-7
|
Binder
|
1 × 10-10 – 1 × 10-3
|
Fenol
|
Chemisch tussenproduct
|
|
10
|
Zearalenon
|
17924-92-4
|
Binder
|
1 × 10-11 – 1 × 10-3
|
Heterocyclische verbinding, lacton
|
Natuurlijk product
|
|
11
|
Butylparabeen
|
94-26-8
|
Binder
|
1 × 10-11 – 1 × 10-3
|
Carbonzuur, fenol
|
Conserveermiddel
|
|
12
|
Atrazine
|
1912-24-9
|
Niet-binder
|
1 × 10-11 – 1 × 10-6
|
Heterocyclische verbinding
|
Herbicide
|
|
13
|
Di-n-butylftalaat (DBP) (96)
|
84-74-2
|
Niet-binder (97)
|
1 × 10-10 – 1 × 10-4
|
Koolwaterstof (cyclisch), ester
|
Weekmaker, Chemisch tussenproduct
|
|
14
|
Corticosteron
|
50-22-6
|
Niet-binder
|
1 × 10-11 – 1 × 10-4
|
Steroïde
|
Natuurlijk product
|
|
Bepaling van de oplosbaarheid en het concentratiebereik van teststoffen
|
19.
|
Er moet voor elke stof een inleidende test worden verricht om de oplosbaarheidsgrens vast te stellen en om het passende concentratiebereik te bepalen dat bij de uitvoering van de test moet worden aangehouden. De oplosbaarheidsgrens van elke teststof wordt eerst bepaald in het oplosmiddel en nader bevestigd onder de omstandigheden van de bepaling. De uiteindelijk in de bepaling te testen concentratie mag niet hoger zijn dan 1 mM. De bereikbepalingstest bestaat uit een oplosmiddelcontrole samen met een logaritmische reeks van acht verdunningen, beginnend bij de maximaal aanvaardbare concentratie (bv. 1 mM of lager, afhankelijk van de oplosbaarheidsgrens), waarbij de aanwezigheid van troebelheid of neerslag moet worden opgemerkt. In het tweede en het derde experiment moeten de concentraties voor zover nodig worden aangepast om de concentratie-responscurve beter te kunnen karakteriseren.
|
Aanvaardbaarheidscriteria voor de testrun
|
20.
|
Of een testrun aanvaard of verworpen wordt, hangt af van de beoordeling van de resultaten voor het referentie-oestrogeen en de controle die voor elk experiment worden gebruikt. Allereerst moeten voor plaat 1 de volledige concentratiecurven voor de referentiecontroles die in elk experiment worden verkregen, voldoen aan de prestatiemaatstaven op basis van de curvefittingsparameters (bv. IC50 en Hill-coëfficiënt) die voortvloeien uit de gerapporteerde resultaten voor de protocollen van de CERI-bepaling en de FW-bepaling (aanhangsels 2 en 3), en de historische controlegegevens van het laboratorium dat de test verricht. Voor elk experiment moeten alle controles (referentie-oestrogeen, zwakke binder en niet-binder) correct zijn ingedeeld. Ten tweede moeten de controles op alle andere platen worden beoordeeld op overeenkomst met plaat 1. Er moet een voldoende breed concentratiebereik van de teststof worden gebruikt om de top van de competitievebindingscurve duidelijk te kunnen bepalen. De variabiliteit tussen de duplo’s van elke concentratie van de teststof en tussen de drie onafhankelijke testruns moet binnen redelijke grenzen blijven en wetenschappelijk verdedigbaar zijn. Het laboratorium moet aantonen de bepaling consistent te kunnen uitvoeren door een historische databank van het referentie-oestrogeen en de controles op te zetten en bij te houden. Als maatstaf voor de intralaboratoriumreproduceerbaarheid kunnen de standaarddeviaties (SD) of variatiecoëfficienten (CV) van de gemiddelden van de curvefittingsparameters voor het referentie-oestrogeen en de zwakkebindercontrole van meerdere experimenten worden gebruikt. De resultaten van de plaatcontroles van elke testrun en voor elke teststof moeten worden geëvalueerd op basis van professioneel oordeel.
Bovendien moet aan de volgende beginselen in verband met de aanvaardbaarheidscriteria worden voldaan:
|
—
|
de gegevens moeten volstaan voor een kwantitatieve beoordeling van de ER-binding;
|
|
—
|
de geteste controles moeten binnen het oplosbaarheidsbereik van de teststof blijven.
|
|
Analyse van de gegevens
|
21.
|
Voor de analyse van de gegevens voor verzadigingsbinding en competitieve binding moet een procedure worden vastgesteld die voldoet aan de grondbeginselen voor de karakterisering van receptor-ligand-interacties. De gegevens voor verzadigingsbinding worden doorgaans geanalyseerd aan de hand van een niet-lineaireregressiemodel dat zowel de totale als de niet-specifieke binding in aanmerking neemt. Bij de bepaling van Bmax- en Kd-gegevens kan het nodig zijn om een correctie voor liganddepletie toe te passen (bv. Swillens, 1995 (19)). De gegevens voor competitieve binding worden doorgaans getransformeerd (bv. percentage specifieke binding en concentratie van de teststof (log M)). De log(IC50) van elke teststof moet worden geschat met behulp van software voor niet-lineaire fitting van de curve die geschikt is voor het fitten van een Hill-vergelijking met vier parameters. Na een eerste analyse worden de curvefittingsparameters bepaald en wordt visueel geverifieerd hoe goed de bindingsgegevens overeenkomen met de gegenereerde competitievebindingscurve. In sommige gevallen kan voor een optimaal gefitte curve een aanvullende analyse nodig zijn (bv. door de top en/of de basis van de curve te begrenzen, de regel van 10 % toe te passen, zie aanhangsel 4 en referentie 2 (sectie III.A.2).
|
|
22.
|
Als aan de aanvaardbaarheidscriteria (punt 20) wordt voldaan, betekent dit weliswaar dat het systeem van de bepaling naar behoren functioneert, maar wil dit nog niet zeggen dat een bepaalde test kloppende gegevens produceert. Het reproduceren van de correcte resultaten van de eerste test is de beste indicatie dat de geproduceerde gegevens kloppen.
|
Algemene criteria voor gegevensinterpretatie
|
23.
|
Er is op vooralsnog geen algemeen erkende methode voor het interpreteren van ER-bindingsgegevens, maar kwalitatieve (bv. binder/niet-binder) en/of kwantitatieve (bv. logIC50, relatieve bindingsaffiniteit (RBA) enz.) beoordelingen van ER-gemedieerde activiteit moeten wel stoelen op empirische gegevens en wetenschappelijk verantwoorde afwegingen.
|
Testverslag
|
24.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
|
Controle-/referentie-/teststof:
|
—
|
bron, partijnummer, uiterste gebruiksdatum, indien beschikbaar;
|
|
—
|
stabiliteit van de teststof, indien bekend;
|
|
—
|
oplosbaarheid en stabiliteit van de teststof in het oplosmiddel, indien bekend;
|
|
—
|
meting van pH, osmolaliteit en neerslag in het kweekmedium waaraan de teststof werd toegevoegd, indien van toepassing.
|
|
|
|
Stof die uit één component bestaat:
|
—
|
fysisch voorkomen, oplosbaarheid in water en aanvullende relevante fysisch-chemische eigenschappen;
|
|
—
|
chemische identificatie, zoals IUPAC- of CAS-naam, CAS-nummer, SMILES- of InChI-code; structuurformule, zuiverheid, chemische identiteit of onzuiverheden, voor zover van toepassing en praktisch haalbaar enz.
|
|
|
|
Stof die uit meerdere componenten bestaat, UVCB’s en mengsels:
|
—
|
voor zover mogelijk, karakterisering aan de hand van chemische identiteit (zie hierboven), kwantitatief voorkomen en relevante fysisch-chemische eigenschappen van de bestanddelen.
|
|
|
|
Oplosmiddel/vehiculum:
|
—
|
karakterisering (aard, leverancier en partij);
|
|
—
|
motivering voor de keuze van het oplosmiddel/vehiculum;
|
|
—
|
oplosbaarheid en stabiliteit van de teststof in het oplosmiddel/vehiculum, indien bekend.
|
|
|
|
Receptoren:
|
—
|
bron van de receptoren (leverancier, catalogusnummer, partij, type receptor, door de leverancier opgegeven concentratie actieve receptoren, certificering van de leverancier);
|
|
—
|
karakterisering van de receptoren (o.a. resultaten verzadigingsbinding): Kd, Bmax,
|
|
—
|
wijze van bewaren van de receptoren.
|
|
—
|
Radioactief gelabelde ligand:
|
|
—
|
leverancier, catalogusnummer, partij, specifieke activiteit.
|
|
|
|
Testomstandigheden:
|
—
|
bepekte oplosbaarheid onder testomstandigheden;
|
|
—
|
samenstelling van de bindingsbuffer;
|
|
—
|
concentratie van de receptor;
|
|
—
|
concentratie van de tracer (d.w.z. de radioactief gelabelde ligand);
|
|
—
|
concentraties van de teststof;
|
|
—
|
percentage vehiculum in de definitieve bepaling;
|
|
—
|
incubatietemperatuur en -tijd;
|
|
—
|
methode voor het scheiden van gebonden en vrije liganden;
|
|
—
|
positieve en negatieve controles/referentiestoffen;
|
|
—
|
criteria om te bepalen of het resultaat positief, negatief of onduidelijk is.
|
|
|
|
Aanvaardbaarheidscontrole:
|
—
|
werkelijke waarden van de IC50 en de Hill-coëfficiënt voor de gelijktijdige positieve controles/referentiestoffen.
|
|
|
|
Resultaten:
|
—
|
ruwe gegevens en gegevens over gebonden/vrije liganden;
|
|
—
|
denatureringscontrole, indien uitgevoerd;
|
|
—
|
in voorkomend geval, de laagste effectieve concentratie (LEC);
|
|
—
|
RBA- en/of IC50-waarden, indien van toepassing;
|
|
—
|
indien mogelijk het verband tussen concentratie en respons;
|
|
—
|
statistische analyses, indien uitgevoerd, samen met een maat voor de fout en de betrouwbaarheid (bv. standaardfout van het gemiddelde, standaarddeviatie, variatiecoëfficiënt of 95 %-betrouwbaarheidsinterval) en beschrijving van de wijze waarop deze waarden zijn verkregen.
|
|
|
|
Bespreking van de resultaten:
|
—
|
toepassing van de regel van 10 %.
|
|
Conclusie
|
LITERATUUR
|
(1)
|
OESO (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
OESO (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(3)
|
OESO (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(4)
|
OESO (2012). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In VivoUterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OESO (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and AppliedPharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Aanhangsel 1
DEFINITIES EN AFKORTINGEN
Aanvaardbaarheidscriteria: minimumnormen voor de prestaties van experimentele controles en referentiestandaarden. Wil een experiment geldig zijn, dan moet aan alle aanvaardbaarheidscriteria worden voldaan.
Bekwaamheid: het bewezen vermogen om een bepaling naar behoren uit te voeren alvorens over te gaan tot het testen van onbekende stoffen.
Betrouwbaarheid: indicatie van de mate waarin een bepaling, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt bepaald door berekening van intra- en interlaboratoriumreproduceerbaarheid.
Chemische stof: een stof of een mengsel.
Conceptueel kader: het conceptueel kader van de OESO voor het testen en beoordelen van hormoonontregelende stoffen.
CV: variatiecoëfficiënt.
E2: 17β-oestradiol.
ER: oestrogeenreceptor.
hERα: humane oestrogeenreceptor-alfa.
Gevalideerde testmethode: een bepaling waarvoor valideringsstudies zijn gedaan om de relevantie (inclusief nauwkeurigheid) en betrouwbaarheid voor een bepaald doel te bepalen. Er moet worden opgemerkt dat een gevalideerde testmethode niet per definitie voldoende nauwkeurig en betrouwbaar is om aanvaardbaar te worden bevonden voor het voorgestelde doel (1).
Hormoonontregeling: de verstoring van de synthese, de opslag en het metabolisme van hormonen.
IC50
: de effectieve-concentratiemediaan van een remmende teststof.
ICCVAM: het Amerikaanse Interagency Coordinating Committee on the Validation of Alternative Methods.
Interlaboratoriumreproduceerbaarheid: een maatstaf voor de mate waarin verschillende gekwalificeerde laboratoria die gebruikmaken van hetzelfde protocol en die dezelfde stoffen testen, in kwalitatief en kwantitatief opzicht vergelijkbare resultaten kunnen produceren. De interlaboratoriumreproduceerbaarheid wordt bepaald tijdens de prevaliderings- en valideringsprocessen en geeft aan in hoeverre een bepaling met succes tussen laboratoria kan worden overgedragen; dit wordt ook wel reproduceerbaarheid tussen laboratoria genoemd (1).
Intralaboratoriumreproduceerbaarheid: een bepaling van de mate waarin gekwalificeerde mensen binnen hetzelfde laboratorium met succes resultaten kunnen repliceren met gebruikmaking van een specifiek protocol op verschillende tijdstippen. Dit wordt ook „reproduceerbaarheid binnen laboratoria” genoemd (1).
LEC: de laagste effectieve concentratie is de laagste concentratie van de teststof die een respons opwekt (d.w.z. de laagste concentratie van de teststof waarbij de x-voudige inductie statistisch verschilt van de gelijktijdige vehiculumcontrole).
„Me too”-test: een informele uitdrukking voor een bepaling die in structureel en functioneel opzicht vergelijkbaar is met een gevalideerde en geaccepteerde testmethode. Dit is een alternatieve benaming voor gelijksoortige testmethode.
Nauwkeurigheid (concordantie): de mate van overeenstemming tussen resultaten verkregen met de bepaling en erkende referentiewaarden. Het is een maat voor de prestaties van de bepaling en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een bepaling wordt verstaan, worden vaak door elkaar gebruikt (1).
Oestrogene activiteit: het vermogen van een chemische stof om 17β-oestradiol na te bootsen voor wat betreft de mogelijkheid om aan oestrogeenreceptoren te binden. Met deze testmethode kan de binding aan de hERα worden gedetecteerd.
PBTG: op prestaties gebaseerde testrichtlijn.
Prestatienormen: normen, gebaseerd op een gevalideerde testmethode, die de basis vormen voor de beoordeling van de vergelijkbaarheid van een voorgestelde, in mechanistisch en functioneel opzicht gelijksoortige bepaling. Dit zijn 1) essentiële componenten van de bepaling; 2) een minimumlijst van referentiestoffen die zijn gekozen uit de stoffen die zijn gebruikt om de aanvaardbare prestaties van de gevalideerde testmethode aan te tonen, en 3) de vergelijkbare nauwkeurigheids- en betrouwbaarheidsniveaus die door de voorgestelde bepaling moeten worden gehaald wanneer ze worden beoordeeld aan de hand van de minimumlijst van referentiestoffen; deze waarden zijn gebaseerd op de waarden die voor de gevalideerde testmethode zijn verkregen (1).
RBA: relatieve bindingsaffiniteit. De RBA van een stof wordt berekend als een percentage van de log(IC50) voor de stof ten opzichte van de log(IC50) voor 17β-oestradiol.
Referentie-oestrogeen: 17ß-oestradiol (E2, CAS 50-28-2).
Referentietestmethoden: de bepalingen waarop PBTG 493 gebaseerd is.
Regel van 10 %: optie om datapunten van de analyse uit te sluiten waarbij het gemiddelde percentage specifiek gebonden [3H]17β-oestradiol van de duplo’s 10 % of meer boven het gemiddelde van de waargenomen binding bij een lagere concentratie ligt (zie aanhangsel 4).
Relevantie: beschrijving van het verband van een bepaling met het te onderzoeken effect en of de bepaling betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de bepaling het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een bepaling (1).
SD: standaarddeviatie.
Stoffen voor bekwaamheidstoetsing: een subset van de referentiestoffen die in de prestatienormen zijn opgenomen. Laboratoria kunnen deze stoffen gebruiken om hun technische bekwaamheid met de gestandaardiseerde bepaling aan te tonen. Doorgaans zijn de selectiecriteria voor deze stoffen onder meer dat ze het hele spectrum van responsen bestrijken, dat ze in de handel verkrijgbaar zijn en dat er kwalitatief hoogwaardige referentiegegevens beschikbaar zijn.
Teststof: alle volgens deze testmethode geteste stoffen of mengsels.
Validering: het proces om de betrouwbaarheid en de relevantie van een bepaald(e) benadering, methode, bepaling, proces of beoordeling voor een bepaald doel vast te stellen (1).
Aanhangsel 2
DE IN-VITROBEPALING VAN DE VERZADIGINGSBINDING EN COMPETITIEVE BINDING AAN DE OESTROGEENRECEPTOR (ERΑ) MET EEN VOLLEDIGE RECOMBINANTE ERΑ VAN FREYBERGER EN WILSON
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN (ZIE OOK ALGEMENE INLEIDING)
|
1.
|
Bij deze in-vitrobepaling van de verzadigingsbinding en competitieve binding aan de oestrogeenreceptor (ERα) wordt gebruikgemaakt van een volledige menselijke ERα (hrERα) die wordt geproduceerd in en geïsoleerd uit met baculovirus geïnfecteerde insectencellen. Het protocol, dat werd ontwikkeld door Freyberger en Wilson, is aan een internationale valideringsstudie in meerdere laboratoria (2) onderworpen, die de relevantie en de betrouwbaarheid van het protocol voor het beoogde doel van de bepaling heeft aangetoond.
|
|
2.
|
Deze bepaling is een screeningsprocedure voor het identificeren van stoffen die aan de volledige hrERα kunnen binden, en wordt gebruikt voor het bepalen van het vermogen van een teststof om met 17β-oestradiol te concurreren om binding aan de hrERα. De kwantitatieve resultaten van de bepaling kunnen onder meer de IC50 (een maat voor de concentratie waarbij een teststof de helft van het [3H]-17β-oestradiol van de hrERα verdrijft) en de relatieve bindingsaffiniteiten van de teststoffen voor de hrERα ten opzichte van 17β-oestradiol omvatten. Ten behoeve van de screening van een chemische stof kunnen aanvaardbare kwalitatieve resultaten van de bepaling onder meer bestaan in de indeling van teststoffen als hrERα-binders, niet-binders, of onduidelijk op basis van criteria die zijn vastgesteld voor de bindingscurven.
|
|
3.
|
Omdat voor de bepaling een radioactief gelabelde ligand wordt gebruikt, heeft het laboratorium een vergunning voor het werken met radioactieve stoffen nodig. Alle procedures met radio-isotopen en gevaarlijke chemische stoffen moeten voldoen aan de regelgeving en procedures zoals beschreven door nationale wetgeving.
|
|
4.
|
Alvorens deze bepaling te gebruiken met het oog op regelgeving, moeten de ALGEMENE INLEIDING en COMPONENTEN VAN DE hrER-BINDINGSBEPALING worden geraadpleegd. De in deze TG gebruikte definities en afkortingen staan in aanhangsel 1.
|
BEGINSELEN VAN DE BEPALING (ZIE OOK DE ALGEMENE INLEIDING)
|
5.
|
De bindingsbepaling met hrERα meet het vermogen van een radioactief gelabelde ligand ([3H]17β-oestradiol) om aan de ER te binden in aanwezigheid van oplopende concentraties van een teststof (een competitor). Teststoffen met een hoge affiniteit voor de ER concurreren in een lagere concentratie met de radioactief gelabelde ligand dan chemische stoffen met een lagere affiniteit voor de receptor.
|
|
6.
|
Deze bepaling heeft twee hoofdcomponenten: een bepaling van de verzadigingsbinding om de parameters van de receptor-ligandinteractie te bepalen, gevolgd door een bepaling van de competitieve binding die de concurrentie tussen een teststof en een radioactief gelabelde ligand om binding aan de ER karakteriseert.
|
|
7.
|
De bepaling van de verzadigingsbinding is bedoeld om de bindingsaffinititeit en het aantal receptoren in een bepaalde partij te bepalen, ter voorbereiding van de bepaling van de competitieve binding. Bij de bepaling van de verzadigingsbinding worden in een evenwichtstoestand de affiniteit van een vaste concentratie oestrogeenreceptoren voor hun natuurlijke ligand (uitgedrukt als de dissociatieconstante Kd) en de concentratie actieve receptorplaatsen (Bmax) gemeten.
|
|
8.
|
Bij de bepaling van de competitieve binding wordt de affiniteit van een stof om met [3H]17β-oestradiol te concurreren om binding aan de ER gemeten. De affiniteit wordt gekwantificeerd door de concentratie waarbij de teststof, in een evenwichtstoestand, 50 % remming van de specifieke binding van [3H]17β-oestradiol veroorzaakt (de zogenaamde inhibitory concentration 50 % of IC50). Ze kan ook worden beoordeeld met behulp van de relatieve bindingsaffiniteit (RBA) ten opzichte van de IC50 van oestradiol, onafhankelijk gemeten in dezelfde testrun. Bij de bepaling van de competitieve binding wordt de binding van [3H]17β-oestradiol in een vaste concentratie gemeten in aanwezigheid van een breed spectrum (acht orden van grootte) aan teststofconcentraties. De gegevens worden vervolgens, voor zover mogelijk, gefit tot een vorm van de Hill-vergelijking (Hill, 1910) die de verdringing van de radioactief gelabelde ligand door een competitieve binder voor één bindingsplaats beschrijft. De mate van verdringing van radioactief gelabeld oestradiol in evenwicht wordt gebruikt om de teststof te karakteriseren als een binder, een niet-binder of een stof met een onduidelijke respons.
|
PROCEDURE
Aantoning van aanvaardbare prestaties van het eiwit hrERα
|
9.
|
Voorafgaand aan de routinematige uitvoering van de bepalingen van de verzadigingsbinding en de competitieve binding moet voor elke nieuwe partij hrERα worden aangetoond dat deze correct presteert in het laboratorium waar de partij zal worden gebruikt. Het proces om de prestaties aan te tonen bestaat uit de volgende twee stappen:
|
—
|
een bepaling van de verzadigingsbinding met [3H]-17β-oestradiol om de specificiteit en de verzadiging voor hrERα aan te tonen. Met behulp van een niet-lineaire regressieanalyse van deze gegevens (bv. BioSoft; McPherson, 1985; Motulsky, 1995) en de daaruit afgeleide Scatchard-plot worden de bindingsaffiniteit voor hrERα van [3H]-17β-oestradiol (Kd) en het aantal receptoren (Bmax) voor elke partij hrERα gedocumenteerd;
|
|
—
|
een bepaling van de competitieve binding met de controlestoffen (het referentie-oestrogeen (17β-oestradiol)), een zwakke binder (bv. noretynodrel of norethisteron), en een niet-binder (octyltriëthoxysilaan, OTES). Elk laboratorium moet een historische databank aanleggen om de consistentie van de IC50 en andere relevante waarden voor het referentie-oestrogeen en de zwakke binder tussen de experimenten en de verschillende partijen hrERα te documenteren. De parameters van de competitievebindingscurven voor de controlestoffen moeten binnen de grenzen van het 95 %-betrouwbaarheidsinterval (zie tabel 1) blijven, die werden vastgesteld op basis van gegevens van de laboratoria die aan de valideringsstudie voor deze bepaling hebben deelgenomen (2).
|
|
Tabel 1
Prestatiecriteria voor het referentie-oestrogeen en de zwakke binder in de FW-hrER-bindingsbepaling
|
Stof
|
Parameter
|
Gemiddelde (7)
|
Standaarddeviatie (n)
|
95 %-Betrouwbaarheidsintervallen (8)
|
|
Ondergrens
|
Bovengrens
|
|
17β-Oestradiol
|
Top (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Basis (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hill-coëfficiënt
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretynodrel
|
Top (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Basis (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hill-coëfficiënt
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
LogIC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Norethisteronc
|
Top (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Basis (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hill-coëfficiënt
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50 (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Aantoning van de bekwaamheid van laboratoria
|
10.
|
Zie de punten 17 en 18 en tabel 2 in COMPONENTEN VAN DE hrER-BINDINGSBEPALING van deze testmethode. Elke bepaling (verzadigingsbinding en competitieve binding) moet bestaan uit drie onafhankelijke testruns (d.w.z. met verse verdunningen van receptor, chemische stoffen en reagentia) op verschillende dagen, en elke testrun moet drie duplo’s omvatten.
|
Bepaling van de concentratie van de receptor (hrERα)
|
11.
|
De concentratie actieve receptoren varieert enigszins, afhankelijk van de partij en de bewaaromstandigheden. Daarom moet de concentratie actieve receptoren in de door de leverancier geleverde partij worden bepaald. Daarmee wordt de precieze concentratie actieve receptoren op het moment van de testrun verkregen.
|
|
12.
|
Onder de omstandigheden van de bepaling van de competitieve binding (d.w.z. 1 nM [3H]-oestradiol) worden nominale concentraties van 0,25, 0,5, 0,75 en 1 nM receptor geïncubeerd in afwezigheid (totale binding) en aanwezigheid (niet-specifieke binding) van 1 μM ongelabeld oestradiol. De specifieke binding, berekend als het verschil tussen de totale en de niet-specifieke binding, wordt uitgezet tegen de nominale receptorconcentratie. Uit de receptorconcentratie waarbij de waarden voor de specifieke binding overeenkomen met 20 % van de toegevoegde radioactief gelabelde ligand kan de nominale receptorconcentratie worden afgeleid en deze moet worden gebruikt voor de bepalingen van de verzadigingsbinding en de competitieve binding. Vaak voldoet een hrER-eindconcentratie van 0,5 nM aan deze voorwaarde.
|
|
13.
|
Als herhaaldelijk niet aan het 20 %-criterium kan worden voldaan, moet de proefopzet worden gecontroleerd op eventuele fouten. Dat het 20 %-criterium niet wordt gehaald, kan erop wijzen dat er zeer weinig actieve receptor in de recombinante partij aanwezig is. In dat geval moet worden overwogen een andere receptorpartij te gebruiken.
|
Verzadigingsbepaling
|
14.
|
Er moeten acht oplopende concentraties [3H]17β-oestradiol in drievoud worden beoordeeld onder de volgende drie omstandigheden (zie tabel 2):
|
—
|
in afwezigheid van ongelabeld 17β-oestradiol en in aanwezigheid van ER. Dit is de bepaling van de totale binding door meting van de radioactiviteit in de putjes die alleen [3H]17β-oestradiol bevatten;
|
|
—
|
in aanwezigheid van een concentratie ongelabeld 17β-oestradiol die 1 000 maal hoger is dan die van gelabeld 17β-oestradiol en in aanwezigheid van ER. De bedoeling hiervan is om de actieve bindingssites te verzadigen met ongelabeld 17β-oestradiol en vervolgens door de radioactiviteit in de putjes te meten, de niet-specifieke binding te bepalen. Van al het resterende gelabelde (’heet’) oestradiol dat aan de receptor kan binden, wordt aangenomen dat het aan een niet-specifieke bindingsplaats bindt, omdat het ongelabelde oestradiol (’koud’) in een dermate hoge concentratie aanwezig is dat het alle beschikbare specifieke bindingsplaaten op de receptor inneemt;
|
|
—
|
in afwezigheid van ongelabeld 17β-oestradiol en in afwezigheid van ER (bepaling van de totale radioactiviteit).
|
|
Bereiding van de oplossingen van [3H]-17β-oestradiol en ongelabeld 17β-oestradiol
|
15.
|
De verdunningen van [3H]-17β-oestradiol moeten worden bereid door toevoeging van testbuffer aan een 12 nM stamoplossing van [3H]-17β-oestradiol om zo concentraties te verkrijgen die in eerste instantie oplopen van 0,12 nM tot 12 nM. De uiteindelijke testconcentraties, oplopend van 0,03 tot 3,0 nM, worden verkregen door 40 μl van deze oplossingen toe te voegen aan de opeenvolgende testputjes van een microtiterplaat met 96 putjes (in een eindvolume van 160 μl). De bereiding van de testbuffer en van de stamoplossing en de verdunningen van [3H]-17β-oestradiol en de bepaling van de concentraties worden uitvoerig beschreven in het protocol van de FW-bepaling, evenals de bepaling van de concentraties (2).
|
|
16.
|
De verdunningen van 17β-oestradiol in ethanol moeten worden bereid door toevoeging van testbuffer om zo acht concentraties te verkrijgen die in eerste instantie oplopen van 0,06 μM tot 6 μM. De uiteindelijke testconcentraties, oplopend van 0,03 tot 3,0 μM, worden verkregen door 80 μl van deze oplossingen toe te voegen aan de opeenvolgende testputjes van een microtiterplaat met 96 putjes (in een eindvolume van 160 μl). De eindconcentraties ongelabeld 17β-oestradiol in elk testputje voor de bepaling van de niet-specifieke binding moeten 1 000 maal zo hoog zijn als de concentratie [3H]-17β-oestradiol. De bereiding van de verdunningen van ongelabeld 17β-oestradiol wordt uitvoerig beschreven in het protocol van de FW-bepaling (2).
|
|
17.
|
Voor de bepaling wordt de nominale receptorconcentratie gebruikt, waarmee een specifieke binding van 20 ± 5 % wordt verkregen (zie de punten 12-13). De hrERα-oplossing moet vlak voor gebruik worden bereid.
|
|
18.
|
De 96-putjesplaten worden ingedeeld zoals weergegeven in tabel 2, met elke concentratie in drievoud. In aanhangsel 2.2 wordt een voorbeeld gegeven van de concentraties en volumes [3H]-17β-oestradiol, ongelabeld 17β-oestradiol, buffer en receptor per testputje.
|
Tabel 2
Microtiterplaatindeling voor de bepaling van de verzadigingsbinding
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [
3
H]-E2+ ER
|
0,06 nM [
3
H]-E2+ ER
|
0,08 nM [
3
H]-E2+ ER
|
0,10 nM [3H]-E2+ ER
|
Totale binding (oplosmiddel)
|
|
B
|
0,30 nM [
3
H]-E2+ ER
|
0,60 nM [
3
H]-E2+ ER
|
1,0 nM [
3
H]-E2+ ER
|
3,0 nM [
3
H]-E2+ ER
|
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3 H]-E2+ ER + 0,03 μM E2
|
0,06 nM [3 H]-E2+ ER + 0,06 μM E2
|
0,08 nM [3 H]-E2+ ER + 0,08 μM E2
|
0,10 nM [3H]-E2+ ER + 0,10 μM E2
|
Niet-specifieke binding
|
|
E
|
0,30 nM [3 H]-E2+ ER + 0,30 μM E2
|
0,60 nM [3 H]-E2+ ER + 0,60 μM E2
|
1,0 nM [
3
H]-E2+ ER + 1,0 μM E2
|
3,0 nM [
3
H]-E2+ ER + 3,0 μM E2
|
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
|
[3H]-E2: [3H]-17β-oestradiol
ER: oestrogeenreceptor
E2: ongelabeld 17β-oestradiol
|
|
19.
|
De testplaten moeten 16 tot 20 uur worden geïncubeerd bij 2 tot 8 °C en moeten gedurende de incubatieperiode op een schudder worden geplaatst.
|
Meting van aan de hrERα gebonden [3H]-17β-oestradiol
|
20.
|
Het aan de hrERα gebonden [3H]-17β-oestradiol wordt gescheiden van het vrije [3H]-17β-oestradiol door aan elk putje 80 μl koude DCC-suspensie toe te voegen, de microtiterplaten 10 minuten te schudden en vervolgens 10 minuten te centrifugeren bij ongeveer 2 500 rpm. Om te voorkomen dat tijdens dit proces het gebonden [3H]-17β-oestradiol loskomt van de hrERα is het van cruciaal belang dat de buffers en de testputjes voortdurend op een temperatuur van 2 tot 8 °C worden gehouden en dat elke stap snel wordt uitgevoerd. Om de platen efficiënt en snel te kunnen verwerken is een schudder voor microtiterplaten nodig.
|
|
21.
|
Vervolgens moet er uiterst voorzichtig, om verontreiniging van de putjes door aanraking van de DCC te vermijden, 50 μl van het supernatans met aan de hrERα gebonden [3H]-17β-oestradiol worden overgebracht naar een tweede microtiterplaat.
|
|
22.
|
Aan elk putje wordt dan 200 μl van een scintillatievloeistof toegevoegd die de kinetische energie van de kernemissies in lichtenergie kan omzetten (A1-B12 en D1 tot E12). De putjes G1-H12 (aangeduid als totaal in dpm) staan een reeks verdunningen van [3H]-17β-oestradiol (40 μl) die direct moeten worden toegevoegd aan de scintillatievloeistof in de putjes van de meetplaat zoals aangegeven in tabel 3. Deze putjes bevatten dus alleen maar 200 μl scintillatievloeistof en de passende verdunning van [3H]-17β-oestradiol. Deze metingen tonen aan hoeveel [3H]-17β-oestradiol er, uitgedrukt in dpm, werd toegevoegd aan elke reeks putjes voor de totale binding en de niet-specifieke binding.
|
Tabel 3
Microtiterplaatindeling voor de bepaling van de verzadigingsbinding, meting van de radioactiviteit
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [
3
H]-E2+ ER
|
0,06 nM [
3
H]-E2+ ER
|
0,08 nM [
3
H]-E2+ ER
|
0,10 nM [3H]-E2+ ER
|
Totale binding (oplosmiddel)
|
|
B
|
0,30 nM [
3
H]-E2+ ER
|
0,60 nM [
3
H]-E2+ ER
|
1,0 nM [
3
H]-E2+ ER
|
3,0 nM [
3
H]-E2+ ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3 H]-E2+ ER + 0,03 μM E2
|
0,06 nM [3 H]-E2+ ER + 0,06 μM E2
|
0,08 nM [3 H]-E2+ ER + 0,08 μM E2
|
0,10 nM [3H]-E2+ ER + 0,10 μM E2
|
Niet-specifieke binding
|
|
E
|
0,30 nM [3 H]-E2+ ER + 0,30 μM E2
|
0,60 nM [3 H]-E2+ ER + 0,60 μM E2
|
1,0 nM [
3
H]-E2+ ER + 1,0 μM E2
|
3,0 nM [
3
H]-E2+ ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H]-E2 (totaal in dpm)
|
0,06 nM [3H]-E2
|
0,08 nM [3H]-E2
|
0,10 nM [3H]-E2
|
Totaal in dpm (*7)
|
|
H
|
0,30 nM [
3
H]-E2
|
0,60 nM [
3
H]-E2
|
1,0 nM [
3
H]-E2
|
3,0 nM [
3
H]-E2
|
|
[3H]-E2: [3H]-17β-oestradiol
ER: oestrogeenreceptor
E2: ongelabeld 17β-oestradiol
dpm: desintegraties per minuut
|
|
23.
|
Er moet ten minste 2 uur worden gewacht, alvorens met meten wordt begonnen, en de teltijd moet 40 minuten per putje bedragen. Het aantal desintegraties per minuut per putje (dpm/putje) wordt bepaald met behulp van een scintillatieteller voor microtiterplaten met dovingscorrectie. Als er geen scintillatieteller voor microtiterplaten beschikbaar is, kunnen de monsters ook met een conventionele teller worden gemeten. In dat geval moet worden overwogen de meettijd te verkorten.
|
Bepaling van de competitieve binding
|
24.
|
Bij de bepaling van de competitieve binding wordt de binding van één concentratie [3H]-17β-oestradiol in aanwezigheid van oplopende concentraties van de teststof gemeten. Binnen een testrun moeten er voor elke concentratie drie gelijktijdige duplobepalingen worden toegepast. Bovendien moeten er voor elke geteste chemische stof drie niet-gelijktijdige testruns worden uitgevoerd. Voor de bepaling moeten een of meer microtiterplaten met 96 putjes worden gebruikt.
|
Controles
|
25.
|
Bij de uitvoering van de bepaling moeten in elk experiment gelijktijdig het oplosmiddel en controles (d.w.z. referentie-oestrogeen, zwakke binder en niet-binder) worden opgenomen. Tijdens elke testrun moeten volledige concentratiecurven voor het referentie-oestrogeen en de controles (d.w.z. zwakke binder en niet-binder) in één plaat worden gebruikt. Alle andere platen moeten i) een hoge (maximale verdrijving) en een middelhoge (bij benadering de IC50) concentratie van zowel E2 als de zwakke binder in drievoud bevatten, en ii) oplosmiddelcontrole en niet-specifieke binding, elk ten minste in drievoud. De procedures voor de bereiding van de testbuffer, controles en [3H]-17β-oestradiol-, hrERα- en teststofoplossingen worden beschreven in referentie 2 (bijlage K, zie protocol FW-bepaling).
|
Oplosmiddelcontrole:
|
26.
|
De oplosmiddelcontrole bevestigt dat er geen interactie optreedt tussen het oplosmiddel en het testsysteem en meet bovendien de totale binding (TB). Als oplosmiddel wordt bij voorkeur ethanol gebruikt. Als de hoogste concentratie teststof niet in ethanol oplosbaar is, mag ook DMSO worden gebruikt. De concentratie ethanol of DMSO, indien gebruikt, is in de putjes aan het eind van de bepaling 1,5 % en mag niet hoger zijn dan 2 %.
|
Buffercontrole:
|
27.
|
De buffercontrole (BC) mag geen oplosmiddel of teststof bevatten, maar wel alle andere componenten van de bepaling. De resultaten van de buffercontrole worden vergeleken met de oplosmiddelcontrole om te verifiëren dat het oplosmiddel het testsysteem niet beïnvloedt.
|
Sterke binder (referentie-oestrogeen)
|
28.
|
17β-oestradiol (CAS 50-28-2) is de endogene ligand en bindt met hoge affiniteit aan de oestrogeenreceptor, alfa-subtype. Voor elke bepaling van de competitieve binding aan de hrERα moet een standaardcurve worden gemaakt met behulp van ongelabeld 17β-oestradiol, om de variabiliteit bij gebruik van de bepaling in de loop van de tijd in hetzelfde laboratorium te kunnen beoordelen. Er moeten acht oplossingen van ongelabeld 17β-oestradiol in ethanol worden bereid, waarbij de concentraties in de testputjes afnemen van 100 nM tot 10 pM (-7[logM] tot -11[logM]), met de volgende verdeling: (-7[logM], -8[logM], -8,5[logM], -9[logM], -9,5[logM], -10[logM], -11[logM]). De hoogste concentratie ongelabeld 17β-oestradiol (1 μM) dient ook als indicator voor niet-specifieke binding. Deze concentratie wordt in tabel 4 aangeduid met het label NSB , maar is ook onderdeel van de standaardcurve.
|
Zwakke binder
|
29.
|
In de test moet een zwakke binder (noretynodrel (CAS RN 68-23-5) of norethisteron (CAS RN 68-22-4)) worden opgenomen om van elk experiment de gevoeligheid aan te tonen en om de variabiliteit te kunnen beoordelen bij gebruik van de bepaling in de loop van de tijd. Er moeten acht oplossingen van de zwakke binder in ethanol worden bereid, waarbij de concentraties in de testputjes oplopen van 3 nM tot 30 μM (-8,5[logM] tot -4,5[logM]), met de volgende verdeling: (-4,5[logM], -5[logM], -5,5[logM], -6[logM], -6.5[logM], -7[logM], -7,5[logM], -8,5[logM]).
|
Niet-binder
|
30.
|
Als negatieve controle (niet-binder) moet octyltriëthoxysilaan (OTES, CAS RN 2943-75-1) worden gebruikt. Hiermee wordt geverifieerd of de bepaling, zoals ze wordt uitgevoerd, teststoffen wel detecteert als ze niet aan de hrERα binden. Er moeten acht oplossingen van de niet-binder in ethanol worden bereid, waarbij de concentraties in de testputjes in logaritmische stappen oplopen van 0,1 nM tot 1 000 μM (-10[logM] tot -3[logM]). Als alternatief kan ook di-n-butylftalaat (DBP) als niet-binder worden gebruikt. De maximale oplosbaarheid daarvan is vastgesteld op -4[logM].
|
hrERα-concentratie
|
31.
|
Voor de bepaling wordt de hoeveelheid receptoren gebruikt waarmee een specifieke binding van 20 ± 5 % aan 1 nM radioligand wordt verkregen (zie de punten 12-13 van aanhangsel 2). De hrERα-oplossing moet vlak voor gebruik worden bereid.
|
[3H]-17β-oestradiol
|
32.
|
De concentratie [3H]-17β-oestradiol in de testputjes moet 1,0 nM bedragen.
|
Teststoffen
|
33.
|
Vooraf moet voor elke stof een oplosbaarheidstest worden verricht om de oplosbaarheidsgrens vast te stellen en om het passende concentratiebereik te bepalen dat bij de uitvoering van het testprotocol moet worden aangehouden. De oplosbaarheidsgrens van elke teststof wordt eerst bepaald in het oplosmiddel en nader bevestigd onder de omstandigheden van de bepaling. De uiteindelijk in de bepaling te testen concentratie mag niet hoger zijn dan 1 mM. De bereikbepalingstest bestaat uit een oplosmiddelcontrole samen met een logaritmische reeks van acht verdunningen, beginnend bij de maximaal aanvaardbare concentratie (bv. 1 mM of lager, afhankelijk van de oplosbaarheidsgrens), waarbij de aanwezigheid van troebelheid of neerslag moet worden opgemerkt (zie ook punt 35). De teststof moet worden getest met de acht curven van de logaritmische reeks van concentraties uit de voorafgaande bereikbepalingstest. In het tweede en het derde experiment moeten de concentraties voor zover nodig worden aangepast om de concentratie-responscurve beter te kunnen karakteriseren.
|
|
34.
|
De verdunningen van de teststof moeten worden bereid in het geschikte oplosmiddel (zie punt 26 van aanhangsel 2). Als de hoogste concentratie van de teststof niet oplosbaar is in ethanol of DMSO en de toevoeging van meer oplosmiddel ertoe zou leiden dat de oplosmiddelconcentratie in het putje aan het eind van de bepaling boven de maximaal aanvaardbare concentratie komt, mag de op een na hoogste concentratie als hoogste concentratie worden genomen. In dat geval kan aan de onderkant van de concentratiereeks een aanvullende concentratie worden toegevoegd. De andere concentraties in de reeks moeten onveranderd blijven.
|
|
35.
|
Als de teststofoplossingen aan de testputjes worden toegevoegd, moet goed worden opgelet of er geen neerslag ontstaat. Bij het fitten van de curven moeten de gegevens voor alle putjes met neerslag worden uitgesloten en moet de reden voor deze uitsluiting worden vermeld.
|
|
36.
|
Als er gegevens uit andere bronnen voorhanden zijn waaraan een log(IC50) van een teststof kan worden ontleend, kan het zinvol zijn om de verdunningen geometrisch rond die waarde te spreiden, bv. met 0,5 logeenheden rond de verwachte log(IC50). De uiteindelijke resultaten moeten een concentratiebereik aan weerszijden van log(IC50) omvatten, inclusief de top en de basis , dat breed genoeg is om de bindingscurve naar behoren te kunnen karakteriseren.
|
Testplaatindeling
|
37.
|
Er moeten gemarkeerde microtiterplaten worden voorbereid voor zes van codes voorziene incubatieduplo’s voor de oplosmiddelcontrole, de hoogste concentratie van het referentie-oestrogeen, die ook als indicator voor niet-specifieke binding (NSB) dient, en de buffercontrole en voor drie van codes voorziene incubatieduplo’s voor alle acht concentraties van de niet-bindende controle (octyltriëthoxysilaan), de zeven lagere concentraties van het referentie-oestrogeen, de acht concentraties/dosisniveaus van de zwakke binder en de acht concentraties van elke teststof (test chemical, TC) In tabel 4 wordt een voorbeeld gegeven van een plaatindelingsdiagram voor de volledige concentratiecurven van het referentie-oestrogeen en de controles. Voor de teststoffen worden aanvullende microtiterplaten gebruikt. Deze moeten ook de volgende plaatcontroles bevatten: 1) een hoge (maximale verdrijving) en een middelhoge (bij benadering de IC50) concentratie van zowel E2 als de zwakke binder in drievoud; 2) oplosmiddelcontrole en niet-specifieke binding, elk in zesvoud (tabel 5). In aanhangsel 2.3 is een voorbeeld opgenomen van een werkblad met de microtiterplaatindeling voor een competitieve bepaling met drie onbekende teststoffen. De in de tabellen 4 en 5 aangegeven concentraties zijn de eindconcentraties van de bepaling. De maximale concentratie E2 moet 1 × 10-7 M bedragen en voor de zwakke binder moet de hoogste concentratie van de zwakke binder in plaat 1 worden gebruikt. De IC50-concentratie moet door het laboratorium worden bepaald op basis van zijn historische controledatabank. Naar verwachting zal deze waarde ongeveer gelijk zijn aan de waarde die in de valideringsstudies werd waargenomen (zie tabel 1).
|
Tabel 4
Microtiterplaatindeling voor de bepaling van de competitieve binding, volledige concentatiecurven voor het referentie-oestrogeen en de controles (plaat 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (alleen oplosmiddel)
|
TB (alleen oplosmiddel)
|
NSB
|
NSB
|
|
B
|
E2 (1 × 10-7)
|
E2 (1 × 10-8)
|
E2 (1 × 10-8,5)
|
E2 (1 × 10-9)
|
|
C
|
E2 (1 × 10-9,5)
|
E2 (1 × 10-10)
|
E2 (1 × 10-11)
|
Blanco (*8)
|
|
D
|
NE (1 × 10-4,5)
|
NE (1 × 10-5)
|
NE (1 × 10-5,5)
|
NE (1 × 10-6)
|
|
E
|
NE (1 × 10-6,5)
|
NE (1 × 10-7)
|
NE (1 × 10-7,5)
|
NE (1 × 10-8,5)
|
|
F
|
OTES (1 × 10-3)
|
OTES (1 × 10-4)
|
OTES (1 × 10-5)
|
OTES (1 × 10-6)
|
|
G
|
OTES (1 × 10-7)
|
OTES (1 × 10-8)
|
OTES (1 × 10-9)
|
OTES (1 × 10-10)
|
|
H
|
Blanco (voor ’heet’) (*9)
|
Blanco (voor ’heet’) (*9)
|
Buffercontrole
|
Buffercontrole
|
|
In dit voorbeeld is de zwakke binder noretynodrel (NE)
|
Tabel 5
Microtiterplaatindeling voor de bepaling van de competitieve binding, volledige concentatiecurven voor de teststoffen en plaatcontroles
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (alleen oplosmiddel)
|
TB (alleen oplosmiddel)
|
NSB
|
NSB
|
|
B
|
TC1 (1 × 10-3)
|
TC1 (1 × 10-4)
|
TC1 (1 × 10-5)
|
TC1 (1 × 10-6)
|
|
C
|
TC1 (1 × 10-7)
|
TC1 (1 × 10-8)
|
TC1 (1 × 10-9)
|
TC1 (1 × 10-10)
|
|
D
|
TC2 (1 × 10-3)
|
TC2 (1 × 10-4)
|
TC2 (1 × 10-5)
|
TC2 (1 × 10-6)
|
|
E
|
TC2 (1 × 10-7)
|
TC2 (1 × 10-8)
|
TC2 (1 × 10-9)
|
TC2 (1 × 10-10)
|
|
F
|
TC3 (1 × 10-3)
|
TC3 (1 × 10-4)
|
TC3 (1 × 10-5)
|
TC3 (1 × 10-6)
|
|
G
|
TC3 (1 × 10-7)
|
TC3 (1 × 10-8)
|
TC3 (1 × 10-9)
|
TC3 (1 × 10-10)
|
|
H
|
NE (IC50)
|
NE (1 × 10-4,5)
|
E2 (IC50)
|
E2 (1 × 10-7)
|
|
In dit voorbeeld is de zwakke binder noretynodrel (NE)
|
Afronding van de bepaling van de competitieve binding
|
38.
|
Zoals in tabel 6 weergegeven moet er 80 μl van de oplosmiddelcontrole, de buffercontrole, het referentie-oestrogeen, de zwakke binder, de niet-binder en de in testbuffer bereide teststoffen aan de putjes worden toegevoegd. Vervolgens wordt er aan elk putje 40 μl van een 4 nM [3H]-17β-oestradioloplossing toegevoegd. Na 10 à 15 minuten voorzichtig roteren bij een temperatuur van 2 tot 8 °C wordt er 40 μl hrERα-oplossing toegevoegd. De testplaten moeten 16 tot 20 uur worden geïncubeerd bij 2 tot 8 °C en moeten gedurende de incubatieperiode op een schudder worden geplaatst.
|
Tabel 6
Het volume van de testcomponenten voor de bepaling van de competitieve binding aan de hrER, microtiterplaten
|
Volume (μl)
|
Bestanddeel
|
|
80
|
Ongelabeld 17β-oestradiol, noretynodrel, OTES, teststoffen, oplosmiddel of buffer
|
|
40
|
4 nM [3H]-17β-oestradioloplossing
|
|
40
|
hrERα-oplossing, in de vooraf bepaalde concentratie
|
|
160
|
Totaal volume in elk testputje
|
|
39.
|
Nadat het aan de hrERα gebonden [3H]-17β-oestradiol door de toevoeging aan elk putje van 80 μl koude DCC-suspensie is gescheiden van het vrije [3H]-17β-oestradiol, moet het aan de hrERα gebonden [3H]-17β-oestradiol worden gekwantificeerd zoals beschreven in de punten 20-23 voor de bepaling van de verzadigingsbepaling.
|
|
40.
|
De putjes H1-6 (in tabel 4 aangeduid als blanco’s (voor ’heet’)) vertegenwoordigen de desintegraties per minuut (dpm) van het [3H]-oestradiol in 40 μl. Het aliquot van 40 μl moet in de putjes H1 - H6 direct aan de scintillatievloeistof worden toegevoegd.
|
Aanvaardbaarheidscriteria
Bepaling van de verzadigingsbinding
|
41.
|
Naarmate de gebruikte concentratie [3H]-17β-oestradiol oploopt, moet de curve voor de specifieke binding een plateau bereiken, waaruit blijkt dat de hrERα verzadigd raakt met ligand.
|
|
42.
|
De specifieke binding bij 1 nM [3H]-17β-oestradiol moet binnen het aanvaardbare bereik van 15 % tot 25 % van het gemiddelde van de in totaal gemeten radioactiviteit over alle testruns blijven. Incidentele, lichte afwijkingen buiten dit bereik zijn aanvaardbaar, maar als testruns structureel buiten dit bereik uitkomen of als voor een bepaalde testrun de specifieke binding ver erbuiten ligt, moet de eiwitconcentratie worden aangepast en moet de bepaling van de verzadigingsbinding worden herhaald.
|
|
43.
|
De gegevens moeten een lineaire Scatchard-plot opleveren.
|
|
44.
|
Er mag niet te veel niet-specifieke binding optreden. In het algemeen moet de waarde voor de niet-specifieke binding < 35 % van de totale binding zijn. Incidenteel mag de verhouding deze grens evenwel overschrijden bij het meten van zeer lage aantallen dpm voor de laagst geteste concentratie radioactief gelabeld 17β-oestradiol.
|
Bepaling van de competitieve binding
|
45.
|
Naarmate de concentratie ongelabeld 17β-oestradiol oploopt, moet het [3H]-17β-oestradiol van de receptor verdrijven op een manier die past bij competitieve binding op één bindingsplaats.
|
|
46.
|
De IC50-waarde voor het referentie-oestrogeen (d.w.z. 17β-oestradiol) moet ongeveer gelijk zijn aan de molaire concentratie [3H]-17β-oestradiol plus de Kd zoals bepaald in de bepaling van de verzadigingsbinding.
|
|
47.
|
De totale specifieke binding moet steeds binnen het aanvaardbare bereik van 20 ± 5 % blijven, wanneer het gemiddelde van de gemeten concentratie van de totale radioactiviteit die aan elk putje is toegevoegd, voor alle testruns 1 nM is. Incidentele, lichte afwijkingen buiten dit bereik zijn aanvaardbaar, maar als testruns structureel buiten dit bereik uitkomen of als voor een bepaalde testrun de specifieke binding ver erbuiten ligt, moet de eiwitconcentratie worden aangepast.
|
|
48.
|
Het oplosmiddel mag geen invloed hebben op de gevoeligheid en reproduceerbaarheid van de bepaling. De resultaten van de oplosmiddelcontrole (TB-putjes) worden vergeleken met de buffercontrole om te verifiëren dat het oplosmiddel het testsysteem niet beïnvloedt. Als het oplosmiddel geen invloed heeft op de bepaling, moeten de resultaten van de oplosmiddel- en de buffercontrole vergelijkbaar zijn.
|
|
49.
|
De niet-binder mag niet meer dan 25 % van het [3H]-17β-oestradiol van de hrERα verdrijven, wanneer wordt getest tot 10-3 M (OTES) of 10-4 M (DBP).
|
|
50.
|
De prestatiecriteria voor het referentie-oestrogeen en de twee zwakke binders (bv. noretynodrel of norethisteron) zijn tot stand gekomen op basis van gegevens uit de valideringsstudie van de FW-hrER-bindingsbepaling (bijlage N van referentie 2). De 95 %-betrouwbaarheidsintervallen komen voort uit de gemiddelden (n) ± SD van alle controletestruns in alle laboratoria die aan de valideringsstudie hebben deelgenomen. Er zijn 95 %-betrouwbaarheidsintervallen berekend voor de curvefittingsparameters (d.w.z. top, basis, Hill-coëfficiënt, log(IC50)) van het referentie-oestrogeen en de zwakke binders en voor de log10RBA van de zwakke binders ten opzichte van het referentie-oestrogeen. Deze worden gegeven als prestatiecriteria voor de positieve controles. In tabel 1 zijn de verwachte bereiken voor de curvefittingsparameters weergegeven, die als prestatiecriteria kunnen worden gebruikt. In de praktijk kan het bereik van de IC50 licht variëren afhankelijk van de Kd van het receptorpreparaat en de ligandconcentratie.
|
|
51.
|
Voor de curvefittingsparameters voor de teststoffen zijn geen prestatiecriteria opgesteld vanwege het brede spectrum aan bestaande potentiële teststoffen en de varia in potentiële affiniteiten en uitslagen (bv. volledig gefitte, gedeeltelijk gefitte of geen gefitte curve). De resultaten van elke testrun voor een teststof moeten evenwel worden geëvalueerd op basis van professioneel oordeel. Er moet een voldoende breed bereik van concentraties van de teststof worden gebruikt om de top van de competitieve curve (bv. 90-100 % binding) duidelijk te kunnen bepalen. De variabiliteit tussen de duplo’s van elke teststofconcentratie en tussen de drie gelijktijdige testruns moet binnen redelijke grenzen blijven en wetenschappelijk verdedigbaar zijn. De controles van elke testrun voor een teststof moeten de voor deze FW-bepaling gerapporteerde prestatiemetingen benaderen en consistent zijn met de historische controlegegevens van elk betreffende laboratorium.
|
ANALYSE VAN DE GEGEVENS
Bepaling van de verzadigingsbinding
|
52.
|
Zowel de totale als de niet-specifieke binding wordt gemeten. Op basis van deze waarden wordt de specifieke binding van oplopende concentraties [3H]-17β-oestradiol in evenwichtstoestand berekend door de niet-specifieke binding van de totale binding af te trekken. Wanneer de specifieke binding wordt uitgezet tegen de concentratie [3H]-17β-oestradiol moet de grafiek een plateau bereiken voor de maximale specifieke binding, die overeenkomt met de verzadiging van de hrERα met het [3H]-17β-oestradiol. Bovendien moet een analyse van de gegevens blijk geven van de binding van het [3H]-17β-oestradiol aan één bindingsplaats met een hoge affiniteit. De curve voor de verzadigingsbinding moet de niet-specifieke, totale en specifieke binding weergeven. Voor de verdere analyse van deze gegevens moet een niet-lineaire regressieanalyse worden gebruikt (bv. BioSoft; McPherson, 1985; Motulsky, 1995), waarna de gegevens worden gepresenteerd in de vorm van een Scatchard-plot.
|
|
53.
|
Bij de analyse van de gegevens moeten Bmax en Kd worden bepaald op basis van alleen de gegevens voor de totale binding, waarbij wordt aangenomen dat de niet-specifieke binding lineair is, tenzij het gebruik van een andere methode wordt onderbouwd. Bovendien moet voor het bepalen van de beste fit robuuste regressie worden toegepast, tenzij een rechtvaardiging wordt gegeven voor een afwijkende keuze. De robuuste-regressiemethode moet worden vermeld. Bij het bepalen van Bmax en Kd uit gegevens voor de verzadigingsbinding moet altijd worden gecorrigeerd voor liganddepletie (bv. met behulp van de methode van Swillens, 1995).
|
Bepaling van de competitieve binding
|
54.
|
De competitievebindingscurve wordt uitgezet als de specifieke binding van [3H]-17β-oestradiol tegen de concentratie (log10-eenheden) van de competitor. De concentratie waarbij de teststof de maximale specifieke binding van [3H]-17β-oestradiol voor 50 % remt, is de IC50-waarde.
|
|
55.
|
De log(IC50)-waarden voor de positieve controles (bv. het referentie-oestrogeen en de zwakke binder) moeten worden geschat met behulp van software voor niet-lineaire fitting van de curve die geschikt is voor het fitten van een Hill-vergelijking met vier parameters (bv. BioSoft; McPherson, 1985; Motulsky, 1995). Bij het fitten van de curven moeten de top, basis, Hill-coëfficiënt, en log(IC50) over het algemeen onbegrensd blijven. Voor het bepalen van de beste fit moet robuuste regressie worden toegepast, tenzij een rechtvaardiging wordt gegeven voor een afwijkende keuze. Er moet niet worden gecorrigeerd voor liganddepletie. Na de eerste analyse moet voor elke bindingscurve worden geverifieerd op een goede fit ten opzichte van het model. De relatieve bindingsaffiniteit (RBA) voor de zwakke binder moet worden berekend als een percentage van de log(IC50) voor de zwakke binder ten opzichte van de log(IC50) voor 17β-oestradiol. De resultaten van de positieve controles en de controle met de niet-binder moeten worden beoordeeld aan de hand van de maatstaven voor de prestaties van de bepaling in de punten 45-50 van aanhangsel 2.
|
|
56.
|
De gegevens voor alle teststoffen moeten worden geanalyseerd volgens een trapsgewijze benadering, zodat de gegevens correct worden beoordeeld en elke competitievebindingscurve correct wordt ingedeeld. Aanbevolen wordt om elke testrun voor een teststof eerst te onderwerpen aan een gestandaardiseerde gegevensanalyse die identiek is aan de gegevensanalyse die werd gebruikt voor het referentie-oestrogeen en de zwakkebindercontroles (zie punt 55 hierboven). Zodra deze is afgerond, moet een technische beoordeling van de curvefittingsparameters worden verricht, alsmede een visuele inspectie van de mate waarin de gegevens overeenkomen met de gegenereerde competitievebindingscurve voor elke testrun. Bij deze technische beoordeling zijn een waargenomen concentratie-afhankelijke afname van het percentage specifiek gebonden [3H]-17β-oestradiol, een geringe variabiliteit tussen de technische duplo’s van elke teststofconcentratie en de consistentie in de fittingsparameters tussen de drie testruns een goede indicatie dat de bepaling en de gegevensanalyses correct zijn uitgevoerd.
|
Gegevensinterpretatie
|
57.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als binder voor de hrERα beschouwd als er een bindingscurve kan worden gefit en het laagste punt op de responscurve binnen het bereik van de gegevens overeenkomt met minder dan 50 % binding (figuur 1).
|
|
58.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als niet-binder voor de hrERα beschouwd als:
|
—
|
er een bindingscurve gefit kan worden en het laagste punt op de gefitte responscurve binnen het bereik van de gegevens overeenkomt met meer dan 75 % binding, of
|
|
—
|
er geen bindingscurve kan worden gefit en het laagste, niet-afgevlakte gemiddelde bindingspercentage van alle concentratiegroepen binnen de gegevens overeenkomt met meer dan 75 % binding.
|
|
|
59.
|
Als aan geen van de bovenstaande voorwaarden wordt voldaan (bv. als het laagste punt op de gefitte responscurve overeenkomt met 76 tot 51 % binding) wordt de teststof als ’onduidelijk’ beschouwd.
|
Tabel 7
Criteria voor de indeling van een teststof op basis van de competitievebindingscurve
|
Indeling
|
Criterium
|
|
Bindera
|
Er kan een curve worden gefit.
Het laagste punt op de responscurve binnen het bereik van de gegevens komt overeen met minder dan 50 % binding.
|
|
Niet-binderb
|
Als er een curve kan worden gefit,
komt het laagste punt op de gefitte responscurve binnen het bereik van de gegevens overeen met meer dan 75 % binding.
Als er geen curve kan worden gefit,
komt het laagste, niet-afgevlakte gemiddelde bindingspercentage van alle concentratiegroepen binnen de gegevens overeen met meer dan 75 % binding.
|
|
Onduidelijkc
|
Een beoordeelbare testrun die noch als binder, noch als niet-binder kan worden ingedeeld
(bv. het laagste punt op de gefitte responscurve komt overeen met 76 tot 51 % binding).
|
Figuur 1
Voorbeelden van de indeling van een teststof met behulp van een competitievebindingscurve

|
60.
|
De verschillende testruns die binnen een laboratorium voor een teststof zijn uitgevoerd, worden gecombineerd door aan elke testrun numerieke waarden toe te kennen en het gemiddelde van deze waarden te nemen, zoals weergegeven in tabel 8. De resultaten van de gecombineerde testruns binnen elk laboratorium worden vergeleken met de verwachte indeling voor elke teststof.
|
Tabel 8
Methode voor de indeling van een teststof op basis van meerdere testruns binnen hetzelfde laboratorium
|
Toekenning van een waarde aan elke testrun:
|
|
|
Indeling
|
Numerieke waarde
|
|
Binder
|
2
|
|
Onduidelijk
|
1
|
|
Niet-binder
|
0
|
|
Indeling op basis van het gemiddelde van de numerieke waarden van de testruns:
|
|
|
Indeling
|
Numerieke waarde
|
|
Binder
|
Gemiddelde ≥ 1,5
|
|
Onduidelijk
|
0,5 ≤ Gemiddelde < 1,5
|
|
Niet-binder
|
Gemiddelde < 0,5
|
TESTVERSLAG
|
61.
|
Zie punt 24 van COMPONENTEN VAN DE hrER-BINDINGSBEPALING van deze testmethode.
|
Aanhangsel 2.1
TERMENLIJST
[3H]E2: met radioactief tritium gelabeld 17β-oestradiol.
DCC: met dextraan beklede kool.
Duplo: een van de meerdere putjes in één testrun die dezelfde inhoud in dezelfde concentraties bevatten en die gelijktijdig worden getest. In dit protocol wordt elke concentratie van de teststof in drievoud getest; dat wil zeggen bij elke concentratie van de teststof worden er drie duplo’s gelijktijdig getest.
E2: ongelabeld 17β-oestradiol (inert).
hrERα: menselijke recombinante oestrogeenreceptor alfa.
Testbuffer: 10 mM tris, 10 mg runderserumalbumine/ml, 2 mM DTT, 10 % glycerol, 0,2 mM leupeptine, pH 7,5.
Testrun: een volledige set van gelijktijdig gerunde testputjes van een microtiterplaat die alle benodigde gegevens oplevert voor het bepalen van de binding van een teststof aan de hrERα, namelijk de totale hoeveelheid aan het testputje toegevoegd [3H]-17β-oestradiol, de maximale binding van [3H]-17β-oestradiol aan de hrERα, de niet-specifieke binding en de totale binding bij verschillende teststofconcentraties). Een testrun zou uit slechts één testputje per concentratie kunnen bestaan, maar aangezien dit protocol testen in drievoud voorschrijft, bestaat een testrun uit drie testputjes per concentratie. Bovendien schrijft dit protocol drie onafhankelijke (d.w.z. niet-gelijktijdige) testruns per chemische stof voor.
Aanhangsel 2.2
VOORBEELD VAN EEN BEPALING VAN DE VERZADIGING MET [3H]-17Β-OESTRADIOL IN DRIEVOUD
|
Voorbeeld van een bepaling van de verzadiging met [3H]-17β-oestradiol in drievoud
|
|
Positie
|
Duplo
|
Code voor type putje
|
Beginconcentratie ’heet’ E2 (nM)
|
Volume ’heet’ E2 (μl)
|
Eindconcentratie ’heet’ E2 (nM)
|
Beginconcentratie ’koud’ E2 (μM)
|
Volume ’koud’ E2 (μl)
|
Eindconcentratie ’koud’ E2 (μM)
|
Volume buffer (μl)
|
Volume receptor (μl)
|
Totaal volume in putjes
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Heet
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Heet
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Heet
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Heet
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Heet
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Heet
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Heet
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Heet
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Heet
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Heet
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Heet
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Heet
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Heet
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Heet
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Heet
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Heet
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Heet
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Heet
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Heet
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Heet
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Heet
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Heet
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Heet
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Heet
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Er zij op gewezen dat de ’hot’ (heet) putjes tijdens de incubatie leeg zijn. De 40 μl wordt alleen toegevoegd voor de scintillatietelling.
|
Aanhangsel 2.3:
PLAATINDELING VOOR DE BEPALING VAN DE COMPETITIEVE BINDING
|
Plaat
|
Positie
|
Duplo
|
Type putje
|
Code putje
|
Code concentratie
|
Beginconcentratie competitor (M)
|
hrER-stamoplossing (μl)
|
Volume buffer (μl)
|
Volume tracer („hete” E2) (μl)
|
Volume verdunningsplaat (μl)
|
Eindvolume (μl)
|
Eindconcentratie competitor (M)
|
|
S
|
A1
|
1
|
totale binding
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
totale binding
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
totale binding
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
totale binding
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
totale binding
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
totale binding
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
„koud” E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
„koud” E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
„koud” E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
„koud” E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
„koud” E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
„koud” E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
„koud” E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
„koud” E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
„koud” E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
„koud” E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
„koud” E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
„koud” E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
„koud” E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
„koud” E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
„koud” E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
„koud” E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
„koud” E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
„koud” E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
„koud” E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
„koud” E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
„koud” E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
blanco
|
blanco
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
blanco
|
blanco
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
blanco
|
blanco
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
noretynodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
noretynodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
noretynodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
noretynodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
noretynodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
noretynodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
noretynodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
noretynodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
noretynodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
noretynodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
noretynodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
noretynodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
noretynodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
noretynodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
heet
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
buffercontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Er zij op gewezen dat de „hot” (heet) putjes tijdens de incubatie leeg zijn. De 40 μl wordt alleen toegevoegd voor de scintillatietelling.
|
Plaatindeling voor de bepaling van de competitieve binding
|
|
Plaat
|
Positie
|
Duplo
|
Type putje
|
Code putje
|
Code concentratie
|
Beginconcentratie competitor (M)
|
hrER-stamoplossing (μl)
|
Volume buffer (μl)
|
Volume tracer („hete” E2) (μl)
|
Volume verdunningsplaat (μl)
|
Eindvolume (μl)
|
Eindconcentratie competitor (M)
|
|
P1
|
A1
|
1
|
totale binding
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
totale binding
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
totale binding
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
totale binding
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
totale binding
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
totale binding
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
„koud” E2 (hoog)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Teststof 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
Teststof 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
Teststof 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
Teststof 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
Teststof 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
Teststof 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
Teststof 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Teststof 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Teststof 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Teststof 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Teststof 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Teststof 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Teststof 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Teststof 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Teststof 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Teststof 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Teststof 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Teststof 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Teststof 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Teststof 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Teststof 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Teststof 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Teststof 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Teststof 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Teststof 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
Teststof 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
Teststof 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
Teststof 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
Teststof 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
Teststof 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
Teststof 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Teststof 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Teststof 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Teststof 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Teststof 2
|
TC2
|
4
|
2.00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Teststof 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Teststof 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Teststof 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Teststof 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Teststof 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Teststof 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Teststof 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Teststof 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Teststof 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Teststof 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Teststof 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Teststof 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Teststof 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Teststof 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
Teststof 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
Teststof 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
Teststof 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
Teststof 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
Teststof 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
Teststof 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Teststof 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Teststof 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Teststof 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Teststof 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Teststof 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Teststof 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Teststof 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Teststof 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Teststof 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Teststof 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Teststof 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Teststof 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Teststof 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Teststof 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Teststof 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Teststof 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Teststof 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
noretynodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
noretynodrel
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
„koud” E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
„koud” E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
„koud” E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
„koud” E2 S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
„koud” E2 S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
„koud” E2 S
|
|
|
1,00E-7
|
40
|
0
|
40
|
80
|
160
|
|
Aanhangsel 3
DE IN-VITROBEPALING VAN DE BINDING AAN DE OESTROGEENRECEPTOR MET EEN MENSELIJK RECOMBINANT ERΑ-LIGANDBINDINGSDOMEIN-EIWIT VAN HET JAPANSE CHEMICALS EVALUATION AND RESEARCH INSTITUTE (CERI-BEPALING)
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN (ZIE OOK ALGEMENE INLEIDING)
|
1.
|
Bij deze in-vitrobepaling van de verzadigingsbinding en competitieve binding aan de oestrogeenreceptor (ERα) wordt gebruikgemaakt van een ligandbindingsdomein (LBD) van de menselijke ERα (hrERα). Dit eiwitpreparaat is geproduceerd door het Japanse Chemicals Evaluation and Research Institute (CERI), Japan, en bestaat in de vorm van een fusie-eiwit met glutathion-S-transferase (GST), tot expressie gebracht in E. coli. Het CERI-protocol is aan een internationale valideringsstudie in meerdere laboratoria (2) onderworpen, die de relevantie en de betrouwbaarheid van het protocol voor het beoogde doel van de bepaling heeft aangetoond.
|
|
2.
|
Deze bepaling is een screeningsprocedure voor het identificeren van stoffen die aan de hrERα kunnen binden, en wordt gebruikt voor het bepalen van het vermogen van een teststof om met 17β-oestradiol te concurreren om binding aan de hrERα-LBD. De kwantitatieve resultaten van de bepaling kunnen onder meer de IC50 (een maat voor de concentratie waarbij een teststof de helft van het [3H]-17β-oestradiol van de hrERα verdrijft) en de relatieve bindingsaffiniteiten van de teststoffen voor de hrERα ten opzichte van 17β-oestradiol omvatten. Ten behoeve van de screening van een chemische stof kunnen aanvaardbare kwalitatieve resultaten van de bepaling onder meer bestaan in de indeling van teststoffen als hrERα-binders, niet-binders, of onduidelijk op basis van criteria die zijn vastgesteld voor de bindingscurven.
|
|
3.
|
Omdat voor de bepaling een radioactief gelabelde ligand wordt gebruikt, heeft het laboratorium een vergunning voor het werken met radioactieve stoffen nodig. Alle procedures met radio-isotopen en gevaarlijke chemische stoffen moeten voldoen aan de regelgeving en procedures zoals beschreven door nationale wetgeving.
|
|
4.
|
Alvorens deze bepaling te gebruiken met het oog op regelgeving, moeten de „ALGEMENE INLEIDING” en „COMPONENTEN VAN DE hrER-BINDINGSBEPALING” worden geraadpleegd. De in deze TG gebruikte definities en afkortingen staan in aanhangsel 1.
|
BEGINSELEN VAN DE BEPALING (ZIE OOK DE ALGEMENE INLEIDING)
|
5.
|
De bindingsbepaling met hrERα meet het vermogen van een radioactief gelabelde ligand ([3H]17β-oestradiol) om aan de ER te binden in aanwezigheid van oplopende concentraties van een teststof (een „competitor”). Teststoffen met een hoge affiniteit voor de ER concurreren in een lagere concentratie met de radioactief gelabelde ligand dan chemische stoffen met een lagere affiniteit voor de receptor.
|
|
6.
|
Deze bepaling heeft twee hoofdcomponenten: een bepaling van de verzadigingsbinding om de parameters van de receptor-ligandinteractie te bepalen, gevolgd door een bepaling van de competitieve binding die de concurrentie tussen een teststof en een radioactief gelabelde ligand om binding aan de ER karakteriseert.
|
|
7.
|
De bepaling van de verzadigingsbinding is bedoeld om de bindingsaffinititeit en het aantal receptoren in een bepaalde partij te bepalen, ter voorbereiding van de bepaling van de competitieve binding. Bij de bepaling van de verzadigingsbinding worden in een evenwichtstoestand de affiniteit van een vaste concentratie oestrogeenreceptoren voor hun natuurlijke ligand (uitgedrukt als de dissociatieconstante Kd) en de concentratie actieve receptorplaatsen (Bmax) gemeten.
|
|
8.
|
Bij de bepaling van de competitieve binding wordt de affiniteit van een stof om met [3H]17β-oestradiol te concurreren om binding aan de ER gemeten. De affiniteit wordt gekwantificeerd door de concentratie waarbij de teststof, in een evenwichtstoestand, 50 % remming van de specifieke binding van [3H]17β-oestradiol veroorzaakt (de zogenaamde „inhibitory concentration 50 %” of IC50). Ze kan ook worden beoordeeld met behulp van de relatieve bindingsaffiniteit (RBA) ten opzichte van de IC50 van oestradiol, onafhankelijk gemeten in dezelfde testrun. Bij de bepaling van de competitieve binding wordt de binding van [3H]17β-oestradiol in een vaste concentratie gemeten in aanwezigheid van een breed spectrum (acht orden van grootte) aan teststofconcentraties. De gegevens worden vervolgens, voor zover mogelijk, gefit tot een vorm van de Hill-vergelijking (Hill, 1910) die de verdringing van de radioactief gelabelde ligand door een competitieve binder voor één bindingsplaats beschrijft. De mate van verdringing van radioactief gelabeld oestradiol in evenwicht wordt gebruikt om de teststof te karakteriseren als een binder, een niet-binder of een stof met een onduidelijke respons.
|
PROCEDURE
Aantoning van aanvaardbare prestaties van het eiwit hrERα
|
9.
|
Voorafgaand aan de routinematige uitvoering van de bepalingen van de verzadigingsbinding en de competitieve binding moet voor elke nieuwe partij hrERα worden aangetoond dat deze correct presteert in het laboratorium waar de partij zal worden gebruikt. Het proces om de prestaties aan te tonen bestaat uit de volgende twee stappen:
|
—
|
een bepaling van de verzadigingsbinding met [3H]-17β-oestradiol om de specificiteit en de verzadiging voor hrERα aan te tonen. Met behulp van een niet-lineaire regressieanalyse van deze gegevens (bv. BioSoft; McPherson, 1985; Motulsky, 1995) en de daaruit afgeleide Scatchard-plot worden de bindingsaffiniteit voor hrERα van [3H]-17β-oestradiol (Kd) en het aantal receptoren (Bmax) voor een bepaalde partij hrERα gedocumenteerd;
|
|
—
|
een bepaling van de competitieve binding met de controlestoffen (het referentie-oestrogeen (17β-oestradiol), een zwakke binder (bv. noretynodrel of norethisteron), en een niet-binder (octyltriëthoxysilaan, OTES). Elk laboratorium moet een historische databank aanleggen om de consistentie van de IC50 en de relevante waarden voor het referentie-oestrogeen en de zwakke binder tussen de experimenten en de verschillende partijen hrERα te documenteren. Bovendien moeten de parameters van de competitievebindingscurven voor de controlestoffen binnen de grenzen van het 95 %-betrouwbaarheidsinterval (zie tabel 1) blijven, die werden vastgesteld op basis van gegevens van de laboratoria die aan de valideringsstudie voor deze bepaling hebben deelgenomen (2).
|
Tabel 1
Prestatiecriteria voor het referentie-oestrogeen en de zwakke binder in de CERI-hrER-bindingsbepaling
|
Stof
|
Parameter
|
Gemiddelde (10)
|
Standaarddeviatie (n)
|
95 %-Betrouwbaarheidsintervallen (11)
|
|
Ondergrens
|
Bovengrens
|
|
17β-Oestradiol
|
Top
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Basis
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hill-coëfficiënt
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretynodrel
|
Top
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Basis
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hill–coëfficiënt
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Norethisteron (12)
|
Top
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Basis
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hill–coëfficiënt
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Aantoning van de bekwaamheid van laboratoria
|
10.
|
Zie de punten 17 en 18 en tabel 2 in „COMPONENTEN VAN DE hrER-BINDINGSBEPALING”van deze testmethode. Elke bepaling (verzadigingsbinding en competitieve binding) moet bestaan uit drie onafhankelijke testruns (d.w.z. met verse verdunningen van receptor, chemische stoffen en reagentia) op verschillende dagen, en elke testrun moet drie duplo’s omvatten.
|
Bepaling van de concentratie van de receptor (hrERα)
|
11.
|
De concentratie actieve receptoren varieert enigszins, afhankelijk van de partij en de bewaaromstandigheden. Daarom moet de concentratie actieve receptoren in de door de leverancier geleverde partij worden bepaald. Daarmee wordt de precieze concentratie actieve receptoren op het moment van de testrun verkregen.
|
|
12.
|
Onder de omstandigheden van de bepaling van de competitieve binding (d.w.z. 0,5 nM [3H]-oestradiol) worden nominale concentraties van 0,1, 0,2, 0,4 en 0,6 nM receptor geïncubeerd in afwezigheid (totale binding) en aanwezigheid (niet-specifieke binding) van 1 μM ongelabeld oestradiol. De specifieke binding, berekend als het verschil tussen de totale en de niet-specifieke binding, wordt uitgezet tegen de nominale receptorconcentratie. Uit de receptorconcentratie waarbij de waarden voor de specifieke binding overeenkomen met 40 % van de toegevoegde radioactief gelabelde ligand kan de receptorconcentratie worden afgeleid en deze moet worden gebruikt voor de bepalingen van de verzadigingsbinding en de competitieve binding. Vaak voldoet een hrER-eindconcentratie van 0,2 nM aan deze voorwaarde.
|
|
13.
|
Als herhaaldelijk niet aan het 40 %-criterium kan worden voldaan, moet de proefopzet worden gecontroleerd op eventuele fouten. Dat het 40 %-criterium niet wordt gehaald, kan erop wijzen dat er zeer weinig actieve receptor in de recombinante partij aanwezig is. In dat geval moet worden overwogen een andere receptorpartij te gebruiken.
|
Verzadigingsbepaling
|
14.
|
Er moeten acht oplopende concentraties [3H]17β-oestradiol in drievoud worden beoordeeld onder de volgende drie omstandigheden (zie tabel 2):
|
a.
|
in afwezigheid van ongelabeld 17β-oestradiol en in aanwezigheid van ER. Dit is de bepaling van de totale binding door meting van de radioactiviteit in de putjes die alleen [3H]17β-oestradiol bevatten;
|
|
b.
|
in aanwezigheid van een concentratie ongelabeld 17β-oestradiol die 2000 maal hoger is dan die van gelabeld 17β-oestradiol en in aanwezigheid van ER. De bedoeling hiervan is om de actieve bindingssites te verzadigen met ongelabeld 17β-oestradiol en vervolgens door de radioactiviteit in de putjes te meten, de niet-specifieke binding te bepalen. Van al het resterende gelabelde („heet”) oestradiol dat aan de receptor kan binden, wordt aangenomen dat het aan een niet-specifieke bindingsplaats bindt, omdat het ongelabelde oestradiol („koud”) in een dermate hoge concentratie aanwezig is dat het alle beschikbare specifieke bindingsplaaten op de receptor inneemt;
|
|
c.
|
in afwezigheid van ongelabeld 17β-oestradiol en in afwezigheid van ER (bepaling van de totale radioactiviteit).
|
Bereiding van de oplossingen van [3H]-17β-oestradiol, ongelabeld 17β-oestradiol en hrERα
|
15.
|
Op basis van een stamoplossing van 1 μM [3H]-17β-oestradiol in DMSO moet een oplossing van 40 nM [3H]-17β-oestradiol worden bereid door toevoeging van DMSO (tot een concentratie van 200 nM) en vervolgens testbuffer bij kamertemperatuur (tot een concentratie van 40 nM). Deze oplossing van 40 nM wordt gebruikt om de [3H]-17β-oestradiol-verdunningsreeks van 0,313 nM tot 40 nM te bereiden door toevoeging van testbuffer bij kamertemperatuur (zoals weergegeven in kolom 12 van tabel 2). De uiteindelijke testconcentraties, oplopend van 0,0313 tot 4,0 nM, worden verkregen door 10 μl van deze oplossingen toe te voegen aan de opeenvolgende testputjes van een microtiterplaat met 96 putjes (zie tabellen 2 en 3). De bereiding van de testbuffer, de berekening van de initiële stamoplossing van [3H]-17β-oestradiol op basis van de specifieke activiteit, de bereiding van de verdunningen en de bepaling van de concentraties worden uitvoerig beschreven in het protocol van de CERI-bepaling (2).
|
|
16.
|
De verdunningen van ongelabeld 17β-oestradiol moeten worden bereid door toevoeging van testbuffer aan een stamoplossing van 1 nM 17β-oestradiol om zo acht concentraties te verkrijgen die in eerste instantie oplopen van 0,625 μM tot 80 μM. De uiteindelijke testconcentraties, oplopend van 0,0625 tot 8 μM, worden verkregen door 10 μl van deze oplossingen toe te voegen aan de opeenvolgende testputjes van een microtiterplaat met 96 putjes voor de meting van de niet-specifieke binding (zie tabellen 2 en 3). De bereiding van de verdunningen van ongelabeld 17β-oestradiol wordt uitvoerig beschreven in het protocol van de CERI-bepaling (2).
|
|
17.
|
Voor de bepaling wordt de receptorconcentratie gebruikt, waarmee een specifieke binding van 40 ± 10 % wordt verkregen (zie de punten 12-13). De hrERα-oplossing moet vlak voor gebruik worden bereid, d.w.z. nadat de putjes voor totale binding, niet-specifieke binding en „hete” ligand afzonderlijk al zijn bereid. Hierbij moet ijskoude testbuffer worden gebruikt.
|
|
18.
|
De 96-putjesplaten worden ingedeeld zoals weergegeven in tabel 2, met elke concentratie van [3H]-17β-oestradiol in drievoud. De volumes [3H]-17β-oestradiol, ongelabeld 17β-oestradiol, buffer en receptor zijn weergegeven in tabel 3.
Tabel 2
Microtiterplaatindeling voor de bepaling van de verzadigingsbinding
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Voor meting van de TB
|
Voor meting van de NSB
|
Voor meting van de „hete” ligand afzonderlijk
|
|
Verdunningen van ongelabeld E2 voor kolommen 4-6 van de plaat
|
Verdunningen van [3H]-E2 voor kolommen 1-9 van de plaat
|
|
A
|
0,0313 nM [3H]-E2+ ER
|
0,0313 nM [3H]-E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H]-E2+ ER
|
0,0625 nM [3H]-E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H]-E2+ ER
|
0,125 nM [3H]-E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H]-E2+ ER
|
0,250 nM [3H]-E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H]-E2+ ER
|
0,50 nM [3H]-E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H]-E2+ ER
|
1,00 nM [3H]-E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H]-E2+ ER
|
2,00 nM [3H]-E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H]-E2+ ER
|
4,00 nM [3H]-E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
totale binding
|
|
NSB
|
:
|
niet-specifieke binding
|
|
[3H]-E2
|
:
|
[3H]-17β-oestradiol
|
|
E2
|
:
|
ongelabeld 17β-oestradiol
|
|
Tabel 3
Reagensvolumes voor de microtiterplaat voor de verzadigingsbepaling
|
Kolomnummer
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Voorbereidende stappen
|
TB-putjes
|
NSB-putjes
|
„Hete” ligand afzonderlijk
|
|
Volume componenten voor betreffende reactieputjes en volgorde toevoeging
|
Buffer
|
60 μl
|
50 μl
|
90 μl
|
|
ongelabelde E2 uit tabel 2, kolom 11
|
-
|
10 μl
|
-
|
|
[3H]-E2 uit tabel 2, kolom 12
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
-
|
|
Totaal reactievolume
|
100 μl
|
100 μl
|
100 μl
|
|
Incubatie
|
NA 2 UUR REACTIE TIJDENS INCUBATIE
|
Kwantificering van de radioactiviteit vlak na bereiding. Geen incubatie
|
|
Behandeling met 0,4 % DCC
|
Ja
|
Ja
|
Nr.
|
|
Volume 0,4 % DCC
|
100 μl
|
100 μl
|
-
|
|
Filtratie
|
Ja
|
Ja
|
Nr.
|
|
METING DPM
|
|
Kwantificering aan het scintillatiemengsel toegevoegd volume
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
De microtiterplaten voor de bepaling van de totale binding en de niet-specifieke binding moeten twee uur worden geïncubeerd bij kamertemperatuur (22 °C to 28 °C).
|
|
Meting van aan de hrERα gebonden [3H]-17β-oestradiol
|
20.
|
Na twee uur incuberen moet het aan de hrERα gebonden [3H]-17β-oestradiol worden gescheiden van het vrije [3H]-17β-oestradiol door 100 μl van een ijskoude 0,4 % DCC-suspensie aan de putjes toe te voegen. Vervolgens moeten de platen 10 minuten op ijs worden geplaatst, waarna het reactiemengsel en de DCC-suspensie moeten worden gefilterd met behulp van een filter voor microtiterplaten om de DCC te verwijderen. 100 μl van het filtraat moet worden toegevoegd aan scintillatievloeistof in scintillatieflesjes om het aantal desintegraties per minuut (dpm) per flesje te meten door middel van vloeistofscintillatietelling.
|
|
21.
|
Als er geen filter voor microtiterplaten beschikbaar is, kan de DCC door middel van centrifugatie worden verwijderd. Vervolgens moet er uiterst voorzichtig, om verontreiniging van de putjes door aanraking van de DCC te vermijden, 50 μl van het supernatans met aan de hrERα gebonden [3H]-17β-oestradiol worden genomen en voor de scintillatietelling worden gebruikt.
|
|
22.
|
De meting van de „hete” ligand afzonderlijk wordt gebruikt om het aantal desintegraties per minuut (dpm) van het aan de testputjes toegevoegde [3H]-17β-oestradiol te bepalen. De radioactiviteit moet direct na de bereiding worden gemeten. Deze putjes moeten worden niet geïncubeerd of met DCC-suspensie behandeld; de inhoud ervan moet rechtstreeks in de scintillatievloeistof worden overgebracht. Deze metingen tonen aan hoeveel [3H]-17β-oestradiol er, uitgedrukt in dpm, werd toegevoegd aan elke reeks putjes voor de totale binding en de niet-specifieke binding.
|
Bepaling van de competitieve binding
|
23.
|
Bij de bepaling van de competitieve binding wordt de binding van één concentratie [3H]-17β-oestradiol in aanwezigheid van oplopende concentraties van de teststof gemeten. Binnen een testrun moeten er voor elke concentratie drie gelijktijdige duplobepalingen worden toegepast. Bovendien moeten er voor elke geteste chemische stof drie niet-gelijktijdige testruns worden uitgevoerd. Voor de bepaling moeten een of meer microtiterplaten met 96 putjes worden gebruikt.
|
Controles
|
24.
|
Bij de uitvoering van de bepaling moeten in elk experiment gelijktijdig het oplosmiddel en controles (d.w.z. referentie-oestrogeen, zwakke binder en niet-binder) worden opgenomen. Tijdens elke testrun moeten volledige concentratiecurven voor het referentie-oestrogeen en de controles (d.w.z. zwakke binder en niet-binder) in één plaat worden gebruikt. Alle andere platen moeten i) een hoge (maximale verdrijving, d.w.z. nagenoeg volledige verdrijving van de radioactief gelabelde ligand) en een middelhoge (bij benadering de IC50) concentratie van E2 en de zwakke binder in drievoud bevatten, en ii) oplosmiddelcontrole en niet-specifieke binding, elk in drievoud. De procedures voor de bereiding van de testbuffer en de [3H]-17β-oestradiol-, hrERα- en teststofoplossingen worden uitvoerig beschreven in het protocol van de CERI-bepaling (2).
|
Oplosmiddelcontrole:
|
25.
|
De oplosmiddelcontrole bevestigt dat er geen interactie optreedt tussen het oplosmiddel en het testsysteem en meet bovendien de totale binding (TB). Als oplosmiddel wordt bij voorkeur DMSO gebruikt. Als de hoogste concentratie teststof niet in DMSO oplosbaar is, mag ook ethanol worden gebruikt. De uiteindelijke DMSO-concentratie in de testputjes moet 2,05 % zijn en mag, als de teststof onvoldoende oplost, worden verhoogd tot maximaal 2,5 %. DMSO-concentraties van meer dan 2,5 % mogen niet worden gebruikt, omdat hogere oplosmiddelconcentraties de bepaling verstoren. Voor teststoffen die niet oplosbaar zijn in DMSO, maar wel in ethanol, kan maximaal 2 % ethanol worden gebruikt zonder dat dit de bepaling verstoort.
|
Buffercontrole:
|
26.
|
De buffercontrole (BC) mag geen oplosmiddel of teststof bevatten, maar wel alle andere componenten van de bepaling. De resultaten van de buffercontrole worden vergeleken met de oplosmiddelcontrole om te verifiëren dat het oplosmiddel het testsysteem niet beïnvloedt.
|
Sterke binder (referentie-oestrogeen)
|
27.
|
17β-oestradiol (CAS 50-28-2) is de endogene ligand en bindt met hoge affiniteit aan de oestrogeenreceptor, alfa-subtype. Voor elke bepaling van de competitieve binding aan de hrERα moet een standaardcurve worden gemaakt met behulp van ongelabeld 17β-oestradiol, om de variabiliteit bij gebruik van de bepaling in de loop van de tijd in hetzelfde laboratorium te kunnen beoordelen. Er moeten acht concentraties ongelabeld 17β-oestradiol in DMSO en testbuffer worden bereid, waarbij de eindconcentraties in de testputjes voor de standaardcurve als volgt zijn gespreid: 10-6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. De hoogste concentratie ongelabeld 17β-oestradiol (1 μM) dient als indicator voor niet-specifieke binding. Deze concentratie wordt in tabel 4 aangeduid met het label „NSB”, maar is ook onderdeel van de standaardcurve.
|
Zwakke binder
|
28.
|
In de test moet een zwakke binder (noretynodrel (CAS RN 68-23-5), of als alternatief norethisteron (CAS RN 68-22-4)) worden opgenomen om van elk experiment de gevoeligheid aan te tonen en om de variabiliteit te kunnen beoordelen bij gebruik van de bepaling in de loop van de tijd. Er moeten acht concentraties zwakke binder in DMSO en testbuffer worden bereid, waarbij de eindconcentraties in de testputjes als volgt zijn gespreid: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 en 10–9 M.
|
Niet-binder
|
29.
|
Als negatieve controle (niet-binder) moet octyltriëthoxysilaan (OTES, CAS RN 2943-75-1) worden gebruikt. Hiermee wordt geverifieerd of de bepaling, zoals ze wordt uitgevoerd, teststoffen die niet aan de hrERα binden, wel detecteert. Er moeten acht concentraties niet-binder in DMSO en testbuffer worden bereid, waarbij de eindconcentraties in de testputjes als volgt zijn gespreid: 10–3, 10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Als alternatief kan ook di-n-butylftalaat (DBP, CAS 84-72-2) als niet-binder worden gebruikt, maar slechts tot 10-4 M. De maximale oplosbaarheid van DBP in de bepaling is vastgesteld op 10–4 M.
|
hrERα-concentratie
|
30.
|
Voor de bepaling wordt de hoeveelheid receptoren gebruikt waarmee een specifieke binding van 40 ± 10 % wordt verkregen (zie de punten 12-13 van aanhangsel 3). De hrERα-oplossing moet vlak voor gebruik worden bereid door verdunning van de functionele hrERα in ijskoude testbuffer.
|
[3H]-17β-oestradiol
|
31.
|
De eindconcentratie [3H]-17β-oestradiol in de testputjes moet 0,5 nM bedragen.
|
Teststoffen
|
32.
|
Vooraf moet voor elke stof een oplosbaarheidstest worden verricht om de oplosbaarheidsgrens vast te stellen en om het passende concentratiebereik te bepalen dat bij de uitvoering van het testprotocol moet worden aangehouden. De oplosbaarheidsgrens van elke teststof wordt eerst bepaald in het oplosmiddel en nader bevestigd onder de omstandigheden van de bepaling. De uiteindelijk in de bepaling te testen concentratie mag niet hoger zijn dan 1 mM. De bereikbepalingstest bestaat uit een oplosmiddelcontrole samen met een logaritmische reeks van acht verdunningen, beginnend bij de maximaal aanvaardbare concentratie (bv. 1 mM of lager, afhankelijk van de oplosbaarheidsgrens), waarbij de aanwezigheid van troebelheid of neerslag moet worden opgemerkt (zie ook punt 35 van aanhangsel 3). Wanneer het concentratiebereik voor de bepaling is bepaald, moet de teststof worden getest in een logaritmische reeks van 8 concentraties met passende intervallen zoals vastgesteld in de voorafgaande bereikbepalingstest. De in het tweede en het derde experiment geteste concentraties moeten voor zover nodig verder worden aangepast om de concentratie-responscurve beter te kunnen karakteriseren.
|
|
33.
|
De verdunningen van de teststof moeten worden bereid in het geschikte oplosmiddel (zie punt 25 van aanhangsel 3). Als de hoogste concentratie van de teststof niet oplosbaar is in DMSO of ethanol en de toevoeging van meer oplosmiddel ertoe zou leiden dat de oplosmiddelconcentratie in het putje aan het eind van de bepaling boven de maximaal aanvaardbare concentratie komt, mag de op een na hoogste concentratie als hoogste concentratie worden genomen. In dat geval kan aan de onderkant van de concentratiereeks een aanvullende concentratie worden toegevoegd. De andere concentraties in de reeks moeten onveranderd blijven.
|
|
34.
|
Als de teststofoplossingen aan de testputjes worden toegevoegd, moet goed worden opgelet of er geen neerslag ontstaat. Bij het fitten van de curven moeten de gegevens voor alle putjes met neerslag worden uitgesloten en moet de reden voor deze uitsluiting worden vermeld.
|
|
35.
|
Als er gegevens uit andere bronnen voorhanden zijn waaraan een log(IC50) van een teststof kan worden ontleend, kan het zinvol zijn om de verdunningen geometrisch dichter rond de verwachte log(IC50) te spreiden,(bv. met 0,5 logeenheden). De uiteindelijke resultaten moeten een voldoende breed concentratiebereik aan weerszijden van log(IC50) bestrijken, inclusief de „top” en de „basis”, dat breed genoeg is om de bindingscurve naar behoren te kunnen karakteriseren.
|
Testplaatindeling
|
36.
|
Er moeten gemarkeerde microtiterplaten worden voorbereid met incubaties in zesvoud voor de oplosmiddelcontrole, de hoogste concentratie van het referentie-oestrogeen (E2), die ook als indicator voor niet-specifieke binding (NSB) dient, de buffercontrole, en drie incubatieduplo’s voor elk van de acht concentraties van de niet-bindende controle (octyltriëthoxysilaan), de zeven lagere concentraties van het referentie-oestrogeen (E2), de acht concentraties van de zwakke binder (noretynodrel of norethisteron) en de acht concentraties van elke teststof (TC). In tabel 4 wordt een voorbeeld gegeven van een plaatindelingsdiagram voor de volledige concentratiecurven van het referentie-oestrogeen en de controles. Voor de teststof worden aanvullende microtiterplaten gebruikt. Deze moeten ook de volgende plaatcontroles bevatten: i) een hoge (maximale verdrijving) en een middelhoge (bij benadering de IC50) concentratie van E2 en de zwakke binder in drievoud; ii) oplosmiddelcontrole (als totale binding) en niet-specifieke binding, elk in zesvoud (tabel 5). In aanhangsel 3.3 is een voorbeeld opgenomen van een werkblad met de microtiterplaatindeling voor een competitieve bepaling met drie onbekende teststoffen. De in het werkblad en in de tabellen 4 en 5 aangegeven concentraties verwijzen naar de eindconcentraties in elk testputje. De maximale concentratie E2 moet 1 × 10–7 M bedragen en voor de zwakke binder moet de hoogste concentratie van de zwakke binder in plaat 1 worden gebruikt. De IC50-concentratie moet door het laboratorium worden bepaald op basis van zijn historische controledatabank. Verwacht wordt dat deze waarde ongeveer gelijk zal zijn aan de waarde die in de valideringsstudies werd waargenomen (zie tabel 1).
|
Tabel 4
Microtiterplaatindeling voor de bepaling van de competitieve binding (98)
(99), volledige concentatiecurven voor het referentie-oestrogeen en de controles (plaat 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Buffercontrole en positieve controle (E2)
|
Zwakke positieve controle (noretynodrel)
|
Negatieve controle (OTES)
|
TB en NSB
|
|
A
|
Blanco (*14)
|
1 × 10–9 M
|
1 × 10–10 M
|
TB (oplosmiddelcontrole) (2,05 % DMSO)
|
|
B
|
E2 (1 × 10–11 M)
|
1 × 10–8 M
|
1 × 10–9 M
|
|
C
|
E2 (1 × 10–10 M)
|
1 × 10–7,5 M
|
1 × 10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1 × 10–9,5 M)
|
1 × 10–7 M
|
1 × 10–7 M
|
|
E
|
E2 (1 × 10–9 M)
|
1 × 10–6,5 M
|
1 × 10–6 M
|
Buffercontrole
|
|
F
|
E2 (1 × 10–8,5 M)
|
1 × 10–6 M
|
1 × 10–5 M
|
|
G
|
E2 (1 × 10–8 M)
|
1 × 10–5,5 M
|
1 × 10–4 M
|
Blanco (voor „heet”) (*15)
|
|
H
|
E2 (1 × 10–7 M)
|
1 × 10–4,5 M
|
1 × 10–3 M
|
Tabel 5
Microtiterplaatindeling voor de bepaling van de competitieve binding, aanvullende platen voor teststoffen (TC) en plaatcontroles
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Teststof 1 (TC-1)
|
Teststof 2 (TC-2)
|
Teststof 3 (TC-3)
|
Controles
|
|
A
|
TC-1 (1 × 10–10 M)
|
TC-2 (1 × 10–10 M)
|
TC-3 (1 × 10–10 M)
|
E2 (1 × 10–7 M)
|
|
B
|
TC-1 (1 × 10–9 M)
|
TC-2 (1 × 10–9 M)
|
TC-3 (1 × 10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1 × 10–8 M)
|
TC-2 (1 × 10–8 M)
|
TC-3 (1 × 10–8 M)
|
NE (1 × 10–4,5 M)
|
|
D
|
TC-1 (1 × 10–7 M)
|
TC-2 (1 × 10–7 M)
|
TC-3 (1 × 10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1 × 10–6 M)
|
TC-2 (1 × 10–6 M)
|
TC-3 (1 × 10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1 × 10–5 M)
|
TC-2 (1 × 10–5 M)
|
TC-3 (1 × 10–5 M)
|
|
G
|
TC-1 (1 × 10–4 M)
|
TC-2 (1 × 10–4 M)
|
TC-3 (1 × 10–4 M)
|
TB (oplosmiddelcontrole)
|
|
H
|
TC-1 (1 × 10–3 M)
|
TC-2 (1 × 10–3 M)
|
TC-3 (1 × 10–3 M)
|
|
In dit voorbeeld is de zwakke binder noretynodrel (NE)
|
Afronding van de bepaling van de competitieve binding
|
37.
|
Met uitzondering van de putjes voor totale binding en de blanco’s (voor „heet”), zoals weergegeven in tabel 6, moet aan elk putje 50 μl testbuffer worden toegevoegd, die eerst gemengd wordt met respectievelijk 10 μl oplosmiddelcontrole, referentie-oestrogeen (E2), zwakke binder, niet-binder, en teststoffen, en dan met 10 μl van een 5 nM [3H]-17β-oestradioloplossing. Vervolgens wordt aan elk putje 30 μl ijskoude receptoroplossing toegevoegd, waarna voorzichtig wordt gemengd. De hrERα-oplossing moet als laatste reagens worden toegevoegd. De microtitertestplaten moeten 2 uur worden geïncubeerd bij kamertemperatuur (22° tot 28 °C).
Tabel 6
Het volume van de testcomponenten voor de bepaling van de competitieve binding aan de hrER, microtiterplaten
|
Stappen van de bereiding
|
Alle putjes behalve TB
|
TB-putjes
|
Blanco (voor „heet”)
|
|
Volume componenten voor betreffende reactieputjes en volgorde toevoeging
|
Testbuffer bij kamertemperatuur
|
50 μl
|
60 μl
|
90 μl
|
|
Ongelabeld E2, zwakke binder, niet-binder, oplosmiddel en teststoffen (*16)
|
10 μl
|
-
|
-
|
|
[3H]-17β-oestradiol om op een eindconcentratie van 0,5 nM te komen (d.w.z. 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Vooraf bepaalde hrERα-concentration (zie punten 12-13)
|
30 μl
|
30 μl
|
-
|
|
Totaal volume in elk testputje
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Nadat het aan de hrERα gebonden [3H]-17β-oestradiol door de toevoeging aan elk putje van 100 μl ijskoude DCC-suspensie is gescheiden van het vrije [3H]-17β-oestradiol, moet het aan de hrERα gebonden [3H]-17β-oestradiol worden gekwantificeerd zoals beschreven in de punten 21-23 van aanhangsel 3 voor de bepaling van de verzadigingsbepaling.
|
|
39.
|
De putjes G10-12 en H10-12 (in tabel 4 aangeduid als blanco’s (voor „heet”)) vertegenwoordigen de desintegraties per minuut (dpm) van het [3H]-oestradiol in 10 μl. Het aliquot van 10 μl moet direct aan de scintillatievloeistof worden toegevoegd.
|
Aanvaardbaarheidscriteria
Bepaling van de verzadigingsbinding
|
40.
|
Naarmate de gebruikte concentratie [3H]-17β-oestradiol oploopt, moet de curve voor de specifieke binding een plateau bereiken, waaruit blijkt dat de hrERα verzadigd raakt met ligand.
|
|
41.
|
De specifieke binding bij 0,5 nM [3H]-17β-oestradiol moet binnen het aanvaardbare bereik van 30 % tot 50 % van het gemiddelde van de in totaal gemeten radioactiviteit over alle testruns blijven. Incidentele, lichte afwijkingen buiten dit bereik zijn aanvaardbaar, maar als testruns structureel buiten dit bereik uitkomen of als voor een bepaalde testrun de specifieke binding ver erbuiten ligt, moet de eiwitconcentratie worden aangepast en moet de bepaling van de verzadigingsbinding worden herhaald.
|
|
42.
|
De gegevens moeten een lineaire Scatchard-plot opleveren.
|
|
43.
|
Er mag niet te veel niet-specifieke binding optreden. In het algemeen moet de waarde voor de niet-specifieke binding < 35 % van de totale binding zijn. Incidenteel mag de verhouding deze grens evenwel overschrijden bij het meten van zeer lage aantallen dpm voor de laagst geteste concentratie radioactief gelabeld 17β-oestradiol.
|
Bepaling van de competitieve binding
|
44.
|
Naarmate de concentratie ongelabeld 17β-oestradiol oploopt, moet het [3H]-17β-oestradiol van de receptor verdrijven op een manier die past bij competitieve binding op één bindingsplaats.
|
|
45.
|
De IC50-waarde voor het referentie-oestrogeen (d.w.z. 17β-oestradiol) moet ongeveer gelijk zijn aan de molaire concentratie [3H]-17β-oestradiol plus de Kd zoals bepaald in de bepaling van de verzadigingsbinding.
|
|
46.
|
De totale specifieke binding moet steeds binnen het aanvaardbare bereik van 40 ± 10 % blijven, wanneer het gemiddelde van de gemeten concentratie van de totale radioactiviteit die aan elk putje is toegevoegd, voor alle testruns 0,5 nM is. Incidentele, lichte afwijkingen buiten dit bereik zijn aanvaardbaar, maar als testruns structureel buiten dit bereik uitkomen of als voor een bepaalde testrun de specifieke binding ver erbuiten ligt, moet de eiwitconcentratie worden aangepast.
|
|
47.
|
Het oplosmiddel mag geen invloed hebben op de gevoeligheid en reproduceerbaarheid van de bepaling. De resultaten van de oplosmiddelcontrole (TB-putjes) worden vergeleken met de buffercontrole om te verifiëren dat het oplosmiddel het testsysteem niet beïnvloedt. Als het oplosmiddel geen invloed heeft op de bepaling, moeten de resultaten van de oplosmiddel- en de buffercontrole vergelijkbaar zijn.
|
|
48.
|
De niet-binder mag niet meer dan 25 % van het [3H]-17β-oestradiol van de hrERα verdrijven, wanneer wordt getest tot 10–3 M (OTES) of 10–4 M (DBP).
|
|
49.
|
De prestatiecriteria voor het referentie-oestrogeen en de twee zwakke binders (bv. noretynodrel of norethisteron) zijn tot stand gekomen op basis van gegevens uit de valideringsstudie voor de CERI-hrER-bindingsbepaling (bijlage N van referentie 2). De 95 %-betrouwbaarheidsintervallen komen voort uit de gemiddelden ± SD (n) van alle controleruns in de vier laboratoria die aan de valideringsstudie hebben deelgenomen. Er zijn 95 %-betrouwbaarheidsintervallen berekend voor de curvefittingsparameters (d.w.z. top, basis, Hill-coëfficiënt en log(IC50)) van het referentie-oestrogeen en de zwakke binders en voor de log10RBA van de zwakke binders ten opzichte van het referentie-oestrogeen. In tabel 1 zijn de verwachte bereiken voor de curvefittingsparameters weergegeven, die als prestatiecriteria kunnen worden gebruikt. In de praktijk kan het bereik van de IC50 licht variëren afhankelijk van de experimenteel afgeleide Kd van het receptorpreparaat en de voor de bepaling gebruikte ligandconcentratie.
|
|
50.
|
Voor de curvefittingsparameters voor de teststoffen zijn geen prestatiecriteria opgesteld vanwege het brede spectrum aan bestaande potentiële teststoffen en de varia in potentiële affiniteiten en uitslagen (bv. volledig gefitte, gedeeltelijk gefitte of geen gefitte curve). De resultaten van elke testrun voor een teststof moeten evenwel worden geëvalueerd op basis van professioneel oordeel. Er moet een voldoende breed bereik van concentraties van de teststof worden gebruikt om de top van de competitieve curve (bv. 90-100 % binding) duidelijk te kunnen bepalen. De variabiliteit tussen de duplo’s van elke teststofconcentratie en tussen de drie gelijktijdige testruns moet binnen redelijke grenzen blijven en wetenschappelijk verdedigbaar zijn. De controles van elke testrun voor een teststof moeten de voor deze CERI-bepaling gerapporteerde prestatiemetingen benaderen en consistent zijn met de historische controlegegevens van elk betreffende laboratorium.
|
ANALYSE VAN DE GEGEVENS
Bepaling van de verzadigingsbinding
|
51.
|
Zowel de totale als de niet-specifieke binding wordt gemeten. Op basis van deze waarden wordt de specifieke binding van oplopende concentraties [3H]-17β-oestradiol in evenwichtstoestand berekend door de niet-specifieke binding van de totale binding af te trekken. Wanneer de specifieke binding wordt uitgezet tegen de concentratie [3H]-17β-oestradiol moet de grafiek een plateau bereiken voor de maximale specifieke binding, die overeenkomt met de verzadiging van de hrERα met het [3H]-17β-oestradiol. Bovendien moet een analyse van de gegevens blijk geven van de binding van het [3H]-17β-oestradiol aan één bindingsplaats met een hoge affiniteit. De curve voor de verzadigingsbinding moet de niet-specifieke, totale en specifieke binding weergeven. Voor de verdere analyse van deze gegevens moet een niet-lineaire regressieanalyse worden gebruikt (bv. BioSoft; McPherson, 1985; Motulsky, 1995), waarna de gegevens worden gepresenteerd in de vorm van een Scatchard-plot.
|
|
52.
|
Bij de analyse van de gegevens moeten Bmax en Kd worden bepaald op basis van alleen de gegevens voor de totale binding, waarbij wordt aangenomen dat de niet-specifieke binding lineair is, tenzij het gebruik van een andere methode wordt onderbouwd. Bovendien moet voor het bepalen van de beste fit robuuste regressie worden toegepast, tenzij een rechtvaardiging wordt gegeven voor een afwijkende keuze. De robuuste-regressiemethode moet worden vermeld. Bij het bepalen van Bmax en Kd uit gegevens voor de verzadigingsbinding moet altijd worden gecorrigeerd voor liganddepletie (bv. met behulp van de methode van Swillens, 1995).
|
Bepaling van de competitieve binding
|
53.
|
De competitievebindingscurve wordt uitgezet als de specifieke binding van [3H]-17β-oestradiol tegen de concentratie (log10-eenheden) van de competitor. De concentratie waarbij de teststof de maximale specifieke binding van [3H]-17β-oestradiol voor 50 % remt, is de IC50-waarde.
|
|
54.
|
De log(IC50)-waarden voor de positieve controles (bv. het referentie-oestrogeen en de zwakke binder) moeten worden geschat met behulp van software voor niet-lineaire fitting van de curve die geschikt is voor het fitten van een Hill-vergelijking met vier parameters (bv. BioSoft; McPherson, 1985; Motulsky, 1995). Bij het fitten van de curven moeten de top, basis, Hill-coëfficiënt, en log(IC50) over het algemeen onbegrensd blijven. Voor het bepalen van de beste fit moet robuuste regressie worden toegepast, tenzij een rechtvaardiging wordt gegeven voor een afwijkende keuze. Er moet niet worden gecorrigeerd voor liganddepletie. Na de eerste analyse moet voor elke bindingscurve worden geverifieerd op een goede fit ten opzichte van het model. De relatieve bindingsaffiniteit (RBA) voor de zwakke binder moet worden berekend als een percentage van de log(IC50) voor de zwakke binder ten opzichte van de log(IC50) voor 17β-oestradiol. De resultaten van de positieve controles en de controle met de niet-binder moeten worden beoordeeld aan de hand van de maatstaven voor de prestaties van de bepaling in de punten 44-49 van aanhangsel 3.
|
|
55.
|
De gegevens voor alle teststoffen moeten worden geanalyseerd volgens een trapsgewijze benadering, zodat de gegevens correct worden beoordeeld en elke competitievebindingscurve correct wordt ingedeeld. Aanbevolen wordt om elke testrun voor een teststof eerst te onderwerpen aan een gestandaardiseerde gegevensanalyse die identiek is aan de gegevensanalyse die werd gebruikt voor het referentie-oestrogeen en de zwakkebindercontroles (zie punt 54 van aanhangsel 3). Zodra deze is afgerond, moet een technische beoordeling van de curvefittingsparameters worden verricht, alsmede een visuele inspectie van de mate waarin de gegevens overeenkomen met de gegenereerde competitievebindingscurve voor elke testrun. Bij deze technische beoordeling zijn een waargenomen concentratie-afhankelijke afname van het percentage specifiek gebonden [3H]-17β-oestradiol, een geringe variabiliteit tussen de technische duplo’s van elke teststofconcentratie en de consistentie in de fittingsparameters tussen de drie testruns een goede indicatie dat de bepaling en de gegevensanalyses correct zijn uitgevoerd.
|
Gegevensinterpretatie
|
56.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als binder voor de hrERα beschouwd als er een bindingscurve kan worden gefit en het laagste punt op de responscurve binnen het bereik van de gegevens overeenkomt met minder dan 50 % binding (figuur 1).
|
|
57.
|
Indien aan alle aanvaardbaarheidscriteria is voldaan, wordt een teststof als niet-binder voor de hrERα beschouwd als:
|
—
|
er een bindingscurve gefit kan worden en het laagste punt op de gefitte responscurve binnen het bereik van de gegevens overeenkomt met meer dan 75 % binding, of
|
|
—
|
er geen bindingscurve kan worden gefit en het laagste, niet-afgevlakte gemiddelde bindingspercentage van alle concentratiegroepen binnen de gegevens overeenkomt met meer dan 75 % binding.
|
|
|
58.
|
Als aan geen van de bovenstaande voorwaarden wordt voldaan (bv. als het laagste punt op de gefitte responscurve overeenkomt met 76 tot 51 % binding) wordt de teststof als ’onduidelijk’ beschouwd.
|
Tabel 7
Criteria voor de indeling van een teststof op basis van de competitievebindingscurve
|
Indeling
|
Criterium
|
|
Bindera
|
Er kan een curve worden gefit.
Het laagste punt op de responscurve binnen het bereik van de gegevens komt overeen met minder dan 50 % binding.
|
|
Niet-binderb
|
Als er een curve kan worden gefit,
komt het laagste punt op de gefitte responscurve binnen het bereik van de gegevens overeen met meer dan 75 % binding.
Als er geen curve kan worden gefit,
komt het laagste, niet-afgevlakte gemiddelde bindingspercentage van alle concentratiegroepen binnen de gegevens overeen met meer dan 75 % binding.
|
|
Onduidelijkc
|
Een beoordeelbare testrun die noch als binder, noch als niet-binder kan worden ingedeeld
(bv. het laagste punt op de gefitte responscurve komt overeen met 76 tot 51 % binding).
|
Figuur 1
Voorbeelden van de indeling van een teststof met behulp van een competitievebindingscurve.
|
59.
|
De verschillende testruns die binnen een laboratorium voor een teststof zijn uitgevoerd, worden gecombineerd door aan elke testrun numerieke waarden toe te kennen en het gemiddelde van deze waarden te nemen, zoals weergegeven in tabel 8. De resultaten van de gecombineerde testruns binnen elk laboratorium worden vergeleken met de verwachte indeling voor elke teststof.
|
Tabel 8
Methode voor de indeling van een teststof op basis van meerdere testruns binnen hetzelfde laboratorium
|
Toekenning van een waarde aan elke testrun:
|
|
Indeling
|
Numerieke waarde
|
|
Binder
|
2
|
|
Onduidelijk
|
1
|
|
Niet-binder
|
0
|
|
Indeling op basis van het gemiddelde van de numerieke waarden van de testruns:
|
|
Indeling
|
Numerieke waarde
|
|
Binder
|
Gemiddelde ≥ 1,5
|
|
Onduidelijk
|
0,5 ≤ Gemiddelde < 1,5
|
|
Niet-binder
|
Gemiddelde < 0,5
|
TESTVERSLAG
|
60.
|
Zie punt 24 van „COMPONENTEN VAN DE hrER-BINDINGSBEPALING” van deze testmethode.
|
Aanhangsel 3.1
TERMENLIJST
[3H]E2
: met radioactief tritium gelabeld 17β-oestradiol.
DCC
: met dextraan beklede kool.
Duplo
: een van de meerdere putjes in één testrun die dezelfde inhoud in dezelfde concentraties bevatten en die gelijktijdig worden getest. In dit protocol wordt elke concentratie van de teststof in drievoud getest; dat wil zeggen bij elke concentratie van de teststof worden er drie duplo’s gelijktijdig getest.
E2
: ongelabeld 17β-oestradiol (inert).
hrERα
: menselijke recombinante oestrogeenreceptor alfa (ligandbindingsdomein).
Testbuffer
: 10 mM tris-HCl, pH 7,4, met 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glycerol, 0,2 mM leupeptine, 1 mM dithiotreïtol en 10 mg/ml runderserumalbumine.
Testrun
: een volledige set van gelijktijdig gerunde testputjes van een microtiterplaat die alle benodigde gegevens oplevert voor het bepalen van de binding van een teststof aan de hrERα, namelijk de totale hoeveelheid aan het testputje toegevoegd [3H]-17β-oestradiol, de maximale binding van [3H]-17β-oestradiol aan de hrERα, de niet-specifieke binding en de totale binding bij verschillende teststofconcentraties). Een testrun zou uit slechts één testputje per concentratie kunnen bestaan, maar aangezien dit protocol testen in drievoud voorschrijft, bestaat een testrun uit drie testputjes per concentratie. Bovendien schrijft dit protocol drie onafhankelijke (d.w.z. niet-gelijktijdige) testruns per chemische stof voor.
Aanhangsel 3.2
PLAATINDELING VOOR DE BEPALING VAN DE COMPETITIEVE BINDING
|
Plaat
|
Positie
|
Duplo
|
Type putje
|
Code putje
|
Code concentratie
|
Beginconcentratie competitor (M)
|
hrER-stamoplossing (μl)
|
Volume buffer (μl)
|
Volume tracer („hete” E2) (μl)
|
Volume verdunningsplaat (μl)
|
Eindvolume (μl)
|
Eindconcentratie competitor (M)
|
|
S
|
A1
|
1
|
Blanco
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Blanco
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Blanco
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
„koud” E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
„koud” E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
„koud” E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
„koud” E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
„koud” E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
„koud” E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
„koud” E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
„koud” E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
„koud” E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
„koud” E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
„koud” E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
„koud” E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
„koud” E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
„koud” E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
„koud” E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
„koud” E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
„koud” E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
„koud” E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
„koud” E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
„koud” E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
„koud” E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
noretynodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
noretynodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
noretynodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
noretynodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
noretynodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
noretynodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
noretynodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
noretynodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
totale binding
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
totale binding
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
totale binding
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
totale binding
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
totale binding
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
totale binding
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
„koud” E2 (hoog)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
„koud” E2 (hoog)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
„koud” E2 (hoog)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
„koud” E2 (hoog)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
„koud” E2 (hoog)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
„koud” E2 (hoog)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Buffercontrole
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Buffercontrole
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Buffercontrole
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Buffercontrole
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Buffercontrole
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Buffercontrole
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Blanco (voor „heet”)
|
Heet
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Blanco (voor „heet”)
|
Heet
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Blanco (voor „heet”)
|
Heet
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Blanco (voor „heet”)
|
Heet
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Blanco (voor „heet”)
|
Heet
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Blanco (voor „heet”)
|
Heet
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Onbekend 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Onbekend 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Onbekend 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Onbekend 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Onbekend 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Onbekend 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Onbekend 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Onbekend 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Onbekend 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Onbekend 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Onbekend 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Onbekend 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Onbekend 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Onbekend 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Onbekend 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Onbekend 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Onbekend 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Onbekend 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Onbekend 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Onbekend 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Onbekend 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Onbekend 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Onbekend 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Onbekend 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Onbekend 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Onbekend 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Onbekend 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Onbekend 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Onbekend 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Onbekend 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Onbekend 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Onbekend 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Onbekend 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Onbekend 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Onbekend 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Onbekend 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Onbekend 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Onbekend 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Onbekend 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Onbekend 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Onbekend 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Onbekend 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Onbekend 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Onbekend 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Onbekend 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Onbekend 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Onbekend 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Onbekend 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Onbekend 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Onbekend 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Onbekend 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Onbekend 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Onbekend 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Onbekend 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Onbekend 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Onbekend 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Onbekend 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Onbekend 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Onbekend 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Onbekend 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Onbekend 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Onbekend 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Onbekend 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Onbekend 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Onbekend 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Onbekend 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Onbekend 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
Onbekend 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
Onbekend 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
Onbekend 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
Onbekend 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
Onbekend 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
Controle E2 (max)
|
S
|
E2max1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
Controle E2 (max)
|
S
|
E2max2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
Controle E2 (max)
|
S
|
E2max3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
Controle E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Controle E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Controle E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Controle NE (max)
|
S
|
Nemax1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
Controle NE (max)
|
S
|
Nemax2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
Controle NE (max)
|
S
|
Nemax3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
Controle NE (IC50)
|
S
|
NEIC501
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Controle NE (IC50)
|
S
|
NEIC502
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Controle NE (IC50)
|
S
|
NEIC503
|
NEIC50 × 10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
„koud” E2 (hoog)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
„koud” E2 (hoog)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
„koud” E2 (hoog)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
„koud” E2 (hoog)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
„koud” E2 (hoog)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
„koud” E2 (hoog)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
totale binding
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
totale binding
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
totale binding
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
totale binding
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
totale binding
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
totale binding
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Aanhangsel 4
OVERWEGINGEN VOOR DE ANALYSE VAN DE GEGEVENS VAN DE BEPALING VAN DE COMPETITIEVE BINDING AAN DE HRER
|
1.
|
Bij de bepaling van de competitieve binding aan de hrERα wordt de binding van één concentratie [3H]-17β-oestradiol in aanwezigheid van oplopende concentraties van de teststof gemeten. De competitievebindingscurve wordt uitgezet als de specifieke binding van [3H]-17β-oestradiol tegen de concentratie (log10-eenheden) van de competitor. De concentratie waarbij de teststof de maximale specifieke binding van [3H]-17β-oestradiol voor 50 % remt, is de IC50.
|
Gegevensanalyse voor het referentie-oestrogeen en de zwakke binder (1)
|
2.
|
Voor verdere analyse worden de gegevens van de controletestruns getransformeerd (d.w.z. procentuele specifieke binding aan [3H]-17β-oestradiol en de log-concentratie van de controlestof). De log(IC50)-waarden voor de positieve controles (bv. het referentie-oestrogeen en de zwakke binder) moeten worden geschat met behulp van software voor niet-lineaire fitting van de curve die geschikt is voor het fitten van een Hill-vergelijking met vier parameters (bv. BioSoft; GraphPad Prism) (2). Bij het fitten van de curven kunnen de top, basis, Hill-coëfficiënt, en log(IC50) doorgaans onbegrensd blijven. Voor het bepalen van de beste fit moet robuuste regressie worden toegepast, tenzij een rechtvaardiging wordt gegeven voor een afwijkende keuze. De robuuste-regressiemethode moet worden vermeld. Correctie voor liganddepletie was voor geen van beide hrER-bepalingen (FW of CERI) nodig, maar kan eventueel worden overwogen. Na de eerste analyse moet voor elke bindingscurve worden gecontroleerd op een goede fit ten opzichte van het model. De relatieve bindingsaffiniteit (RBA) voor de zwakke binder kan worden berekend als een percentage van de log(IC50) voor de zwakke binder ten opzichte van de log(IC50) voor 17β-oestradiol. De resultaten van de positieve controles en de controle met de niet-binder moeten worden beoordeeld aan de hand van de maatstaven voor de prestaties van de bepaling en de aanvaardbaarheidscriteria zoals beschreven in deze testmethode (punt 20), aanhangsel 2 (FW-bepaling, punten 41-51) en aanhangsel 3 (CERI-bepaling, punten 41-51). In figuur 1 staan voorbeelden van 3 testruns voor het referentie-oestrogeen en de zwakke binder.
|
Figuur 1
Voorbeelden van de competitievebindingscurven voor het referentie-oestrogeen en de zwakkebindercontrole

Analyse van de gegevens voor de teststoffen
|
3.
|
De gegevens voor alle teststoffen moeten worden geanalyseerd volgens een trapsgewijze benadering, zodat de gegevens correct worden beoordeeld en elke competitievebindingscurve correct wordt ingedeeld. Elke testrun voor een teststof moet eerst aan een gestandaardiseerde gegevensanalyse worden onderworpen, die identiek is aan de gegevensanalyse die werd gebruikt voor het referentie-oestrogeen en de zwakkebindercontroles. Zodra deze is afgerond, moet een technische beoordeling van de curvefittingsparameters worden verricht, alsmede een visuele inspectie van de mate waarin de gegevens overeenkomen met de gegenereerde competitievebindingscurve voor elke testrun. Bij deze technische beoordeling zijn een waargenomen concentratie-afhankelijke afname van het percentage specifiek gebonden [3H]-17β-oestradiol, een geringe variabiliteit tussen de technische duplo’s van elke teststofconcentratie en de consistentie in de fittingsparameters tussen de drie testruns een goede indicatie dat de bepaling en de gegevensanalyses correct zijn uitgevoerd. De resultaten van elke testrun voor een teststof moeten worden geëvalueerd op basis van professioneel oordeel en de gegevens op basis waarvan elke teststof wordt ingedeeld als binder of niet-binder moeten wetenschappelijk verdedigbaar zijn.
|
|
4.
|
Het kan zo nu en dan gebeuren dat de hrER-bindingsgegevens aanvullende aandacht behoeven om op een passende manier geanalyseerd en geïnterpreteerd te worden. In eerdere onderzoeken zijn gevallen voorgekomen waarin de analyse en interpretatie van de gegevens voor competitieve binding aan de receptor een complex beeld lieten zien door een toename van het percentage specifieke binding wanneer teststoffen bij de hoogste concentraties werden getest (figuur 2). Dit is een bekend verschijnsel dat bij het gebruik van protocollen voor een aantal bepalingen van competitieve receptorbinding is opgetreden (3). In deze gevallen wordt bij lagere concentraties een concentratie-afhankelijke respons waargenomen, maar neemt de verdrijving van [3H]17β-oestradiol niet langer af naarmate de concentratie van de teststof de oplosbaarheidsgrens nadert. In deze gevallen geven de gegevens voor de hogere concentraties aan dat de bepaling haar biologische grens heeft bereikt. Dit verschijnsel is bijvoorbeeld dikwijls gepaard gegaan met chemische onoplosbaarheid en neerslag bij hoge concentraties. Het kan ook betekenen dat bij de hoogste chemische concentraties de capaciteit van de met dextraan beklede kool voor het vasthouden van de ongebonden radioactief gelabelde ligand tijdens de scheidingsprocedure wordt overschreden. Door dergelijke gegevenspunten mee te nemen bij het fitten van competitievebindingsgegevens tot een sigmoïde curve kan het gebeuren dat het ER-bindingspotentieel van een teststof verkeerd wordt ingedeeld (figuur 2). Om dit te vermijden is in zowel het FW-protocol als het CERI-protocol voor de bepaling van hrER-binding een optie opgenomen om gegevenspunten waarbij het gemiddelde percentage specifiek gebonden [3H]-17β-oestradiol van de duplo’s 10 % of meer boven het bij een lagere concentratie gemiddelde ligt, van de analyse uit te sluiten (dit wordt doorgaans de regel van 10 % genoemd). Deze regel mag maar eenmaal per curve worden toegepast en er moeten gegevens voor ten minste 6 concentraties overblijven om de curve correct te kunnen indelen.
|
Figuur 2
Voorbeelden van competitievebindingscurven met en zonder toepassing van de regel van 10 %
|
5.
|
Per geval moet zorgvuldig worden overwogen of voor het corrigeren van de curve de regel van 10 % kan worden toegepast. Toepassing van de regel moet worden gereserveerd voor gevallen waarin er sterke aanwijzingen zijn dat het een hrER-binder betreft. Bij de uitvoering van experimenten voor de valideringsstudie van de FW-hrER-bindingsbepaling bleek de regel van 10 % soms een onbedoeld en onverwacht gevolg te hebben. Chemische stoffen waarbij geen interactie met de receptor optrad (d.w.z. werkelijke niet-binders), vertoonden rond de 100 % binding van de radioactieve ligand vaak een variabiliteit van meer dan 10 % over het hele bereik van geteste concentraties. Als de laagste waarde bij een lage concentratie optrad, kwamen de gegevens voor alle hogere concentraties in aanmerking om aan de hand van de regel van 10 % van de analyse te worden uitgesloten, ook al konden die concentraties dienen om vast te stellen dat de betreffende chemische stof een niet-binder is. In figuur 3 staan voorbeelden waarbij de regel van 10 % niet kan worden toegepast.
|
Figuur 3
Voorbeelden van competitievebindingsgegevens waarbij de regel van 10 % niet kan worden toegepast


Referenties
|
(1)
|
OESO (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71
IN-VITRO HUIDSENSIBILISATIE: TESTS OP BASIS VAN DE BELANGRIJKE GEBEURTENIS VAN ACTIVERING VAN DENDRITISCHE CELLEN OP DE ADVERSE OUTCOME PATHWAY (AOP) VOOR HUIDSENSIBILISATIE
ALGEMENE INLEIDING
Testmethode op basis van de belangrijke gebeurtenis van activering van dendritische cellen
|
1.
|
Een huidsensibiliserende stof is een stof die een allergische reactie uitlokt na contact met de huid, zoals gedefinieerd in het mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (GHS) (1) en Verordening (EU) nr. 1272/2008 van de Europese Unie betreffende de indeling, etikettering en verpakking van stoffen en mengsel (CLP-verordening) (100). Er bestaat algemene overeenstemming over de belangrijkste biologische gebeurtenissen die aan de basis liggen van huidsensibilisatie. De huidige kennis van de chemische en biologische mechanismen die verband houden met huidsensibilisatie is samengevat in een Adverse Outcome Pathway (AOP) in het kader van het AOP-programma van de OESO (2), van de moleculaire inleidende gebeurtenis over de tussenliggende gebeurtenissen tot het schadelijke effect, te weten allergische contactdermatitis. In dit geval is de moleculaire inleidende gebeurtenis (d.w.z. de eerste belangrijke gebeurtenis) de covalente binding van elektrofiele stoffen aan nucleofiele centra in eiwitten in de huid. De tweede belangrijke gebeurtenis in deze AOP vindt plaats in de keratinocyten en omvat zowel ontstekingsreacties als veranderingen in genexpressie die samenhangen met specifieke signaalpaden in de cellen, zoals de paden die afhankelijk zijn van het antioxidant-/elektrofiel responselement (ARE). De derde belangrijke gebeurtenis is de activering van dendritische cellen. Deze gebeurtenis wordt gewoonlijk beoordeeld aan de hand van de expressie van specifieke markers op het celoppervlak. De vierde belangrijke gebeurtenis is de activering en proliferatie van T-cellen, die indirect wordt beoordeeld in de lokale lymfkliertest (LLKT) met muizen (3).
|
|
2.
|
Deze testmethode (TM) is gelijkwaardig aan testrichtlijn (TG) 442E (2017) van de OESO. Hierin worden in-vitrotests beschreven voor mechanismen die worden beschreven onder de belangrijke gebeurtenis van activering van dendritische cellen op de Adverse Outcome Pathway (AOP) voor huidsensibilisatie (2). De TM behelst tests die kunnen worden gebruikt om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven, in overeenstemming met het VN-GHS en de CLP-verordening.
|
|
De in deze TM beschreven tests zijn:
|
—
|
Humanecellijnactiveringstest (h-CLAT)
|
|
—
|
U937-cellijnactiveringstest (U-SENS™)
|
|
—
|
Interleukine-8-reportergenbepaling (IL-8 Luc bepaling)
|
|
|
|
3.
|
De tests die in deze testmethode en de overeenkomstige OESO-testrichtlijn worden beschreven, kunnen verschillen wat betreft de gebruikte procedure voor het genereren van de gegevens en de meetresultaten, maar kunnen zonder onderscheid worden gebruikt om tegelijkertijd te voldoen aan de eisen die aan landen worden gesteld voor de testresultaten voor de belangrijke gebeurtenis van activering van dendritische cellen op de AOP voor huidsensibilisatie, en in aanmerking te komen voor de wederzijdse aanvaarding van gegevens van de OESO.
|
Achtergrond en beginselen van de tests die in deze testmethode op basis van een belangrijke gebeurtenis worden beschreven
|
4.
|
De bepaling van de huidsensibilisatie ging meestal gepaard met het gebruik van proefdieren. Bij de klassieke methoden waarbij cavia’s worden gebruikt, de maximalisatietest met cavia’s (GMPT) van Magnusson-Kligman en de Buehler-test (TM B.6) (4), worden zowel de inductie- als de uitlokkingsfase van huidsensibilisatie beoordeeld. Ook de tests met muizen, de LLKT (TM B.42) (3) en de twee niet-radioactieve aanpassingen ervan, LLKT: AA (TM B.50) (5) en LLKT: BrdU-ELISA (TM B.51) (6), waarbij telkens uitsluitend de inductierespons wordt beoordeeld, zijn gangbaar geworden, aangezien deze beter zijn voor het dierenwelzijn dan tests met cavia’s en het mogelijk maken om de inductiefase van huidsensibilisatie objectief te meten.
|
|
5.
|
Onlangs zijn ook mechanistisch gebaseerde in-chemico- en in-vitrotestmethoden om de eerste belangrijke gebeurtenis (TM B.59; Direct Peptide Reactivity Assay (7)) en de tweede belangrijke gebeurtenis (TM B.60; ARE-Nrf2-luciferasetestmethode (8)) van de AOP voor huidsensibilisatie te bestuderen, aangenomen voor de beoordeling van het gevaar van huidsensibilisatie van chemische stoffen.
|
|
6.
|
De in deze testmethode beschreven tests kwantificeren hetzij de verandering in de expressie van een of meer markers op het celoppervlak die gepaard gaat met de activering van monocyten en dendritische cellen na blootstelling aan sensibiliserende stoffen (bv. CD54, CD86), hetzij de veranderingen in de expressie van IL-8, een cytokine dat verband houdt met de activering van dendritische cellen. Van huidsensibiliserende stoffen is gerapporteerd dat ze de expressie van markers op het celoppervlak zoals CD40, CD54, CD80, CD83 en CD86 induceren, naast de inductie van pro-inflammatoire cytokinen, zoals IL-1β en TNF-α, en verscheidene chemokinen waaronder IL-8 (CXCL8) en CCL3 (9) (10) (11) (12) die verband houden met de activering van dendritische cellen (2).
|
|
7.
|
Desondanks volstaat informatie die werd verzameld met behulp van tests die de activering van dendritische cellen meten, als dusdanig niet altijd om te besluiten dat een chemische stof al dan niet huidsensibilisatie kan veroorzaken, aangezien activering van dendritische cellen slechts een van de belangrijke gebeurtenissen is op de AOP die aanleiding geeft tot huidsensibilisatie (2) (13). De gegevens die de in deze testmethode beschreven tests opleveren, kunnen daarom worden gebruikt ter ondersteuning van het onderscheid tussen huidsensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) en niet-sensibiliserende stoffen in het kader van geïntegreerde test- en beoordelingsbenaderingen (Integrated Approaches to Testing and Assessment of IATA), mits ze worden gecombineerd met andere aanvullende informatie die bijvoorbeeld is afgeleid uit in-vitrotests waarin andere belangrijke gebeurtenissen binnen de AOP van huidsensibilisatie worden bestudeerd, of uit niet-testmethoden zoals een „read-across” van chemische analogen (13). Voorbeelden van het gebruik van gegevens die zijn gegenereerd met behulp van deze tests binnen zogenaamde Defined Approaches – d.w.z. benaderingen die zijn gestandaardiseerd zowel wat betreft de set van gebruikte informatiebronnen als wat betreft de procedure die op de gegevens is toegepast om er voorspellingen uit af te leiden – zijn gepubliceerd (13) en kunnen worden aangewend als bruikbare elementen in IATA’s.
|
|
8.
|
De in deze testmethode beschreven tests kunnen niet afzonderlijk worden gebruikt om huidsensibiliserende stoffen in te delen in de subcategorieën 1A en 1B zoals gedefinieerd door VN-GHS/CLP, voor autoriteiten die deze twee optionele sub categorieën toepassen, of om de potentie te voorspellen met het oog op besluiten inzake veiligheidsbeoordelingen. Naargelang van het regelgevingskader kunnen positieve resultaten op basis van deze methoden echter wel als dusdanig worden gebruikt om een chemische stof in te delen in VN-GHS/CLP-categorie 1.
|
|
9.
|
De term „teststof” wordt in deze testmethode gebruikt om te verwijzen naar datgene dat wordt getest (101), en houdt geen verband met de toepasselijkheid van de tests op het testen van stoffen die uit één component bestaan, stoffen die uit meerdere componenten bestaan en/of mengsels. Er is momenteel slechts beperkte informatie beschikbaar over de toepasselijkheid van de tests op stoffen die uit meerdere componenten bestaan/mengsels (14) (15). De tests zijn vanuit technisch oogpunt evenwel toepasbaar voor het testen van stoffen die uit meerdere componenten bestaan en van mengsels. Voordat deze testmethode wordt gebruikt op een mengsel om gegevens te genereren voor een beoogd regelgevingsdoel, moet echter worden nagegaan of, en zo ja, waarom deze methode betrouwbare resultaten oplevert voor dat doel (102). Zulke overwegingen zijn niet nodig wanneer het testen van het mengsel wettelijk vereist is. Bovendien moet bij het testen op stoffen die uit meerdere componenten bestaan of op mengsels rekening worden gehouden met mogelijke interferentie van cytotoxische componenten met de waargenomen responsen.
|
LITERATUUR
|
(1)
|
Verenigde Naties (VN) (2015). Mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (GHS). Zesde herziene editie. New York & Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Beschikbaar op: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OESO (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Beschikbaar op: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Hoofdstuk B.42 van deze bijlage: huidsensibilisatie: lokale lymfkliertest.
|
|
(4)
|
Hoofdstuk B.6 van deze bijlage: sensibilisatie van de huid.
|
|
(5)
|
Hoofdstuk B.50 van deze bijlage: huidsensibilisatie: lokale lymfkliertest: AA.
|
|
(6)
|
Hoofdstuk B.51 van deze bijlage: huidsensibilisatie: lokale lymfkliertest: BrdU-ELISA.
|
|
(7)
|
Hoofdstuk B.59 van deze bijlage: in-chemicohuidsensibilisatie: Direct Peptide Reactivity Assay (DPRA-test).
|
|
(8)
|
Hoofdstuk B.60 van deze bijlage: in-vitrohuidsensibilisatie: ARE-Nrf2-luciferasetestmethode.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OESO (2016). Series on Testing & Assessment No 256: Guidance Document on the Reporting of Defined Approaches and Individual Information Sources to be Used within Integrated Approaches to Testing and Assessment (IATA) for Skin Sensitisation, bijlage 1 en bijlage 2. ENV/JM/HA(2016)29. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Aanhangsel 1
IN-VITROHUIDSENSIBILISATIE: HUMANECELLIJNACTIVERINGSTEST (H-CLAT)
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
1.
|
De h-CLAT kwantificeert veranderingen in de expressie van markers op het celoppervlak die gepaard gaan met de activering van monocyten en dendritische cellen (d.w.z. CD86 en CD54), in de humane monocytischeleukemiecellijn THP-1, na blootstelling aan sensibiliserende stoffen (1) (2). De gemeten expressieniveaus van de oppervlaktemarkers CD86 en CD54 worden vervolgens gebruikt om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven.
|
|
2.
|
De h-CLAT is geëvalueerd in een valideringsstudie die werd gecoördineerd door het referentielaboratorium voor alternatieven voor dierproeven van de Europese Unie (EURL CEVMA) en in een latere onafhankelijke beoordeling door vakgenoten van het raadgevend wetenschappelijk comité van het CEVMA (ESAC). Op grond van al het beschikbare bewijsmateriaal en de inbreng van regelgevers en belanghebbenden werd de h-CLAT door het EURL CEVMA (3) aanbevolen voor gebruik in het kader van een IATA om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven met het oog op de gevarenindeling en etikettering. Voorbeelden van het gebruik van h-CLAT-gegevens in combinatie met andere informatie worden beschreven in de literatuur (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
De h-CLAT bleek overdraagbaar te zijn naar laboratoria met ervaring in het kweken van cellen en flowcytometrieanalyse. De verwachte reproduceerbaarheid van voorspellingen op basis van deze test binnen laboratoria en tussen verschillende laboratoria bedraagt 80 % (3) (12). De resultaten van de valideringsstudie (13) en andere gepubliceerde onderzoeken (14) duiden er over het geheel genomen op dat, vergeleken met LLKT-resultaten, de nauwkeurigheid in het onderscheiden van huidsensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) en niet-sensibiliserende stoffen 85 % bedraagt (N=142), met een gevoeligheid van 93 % (94/101) en een specificiteit van 66 % (27/41) (op basis van een heranalyse door het EURL CEVMA (12), waarbij rekening werd gehouden met alle bestaande gegevens en waarbij negatieve uitslagen voor chemische stoffen met een log Kow van meer dan 3,5 buiten beschouwing werden gelaten, zoals beschreven in punt 4). Bij de h-CLAT is de kans dat valsnegatieve voorspellingen betrekking hebben op chemische stoffen met een lage tot gemiddelde potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1B) hoger dan dat ze betrekking hebben op chemische stoffen met een hoge potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1A) (4) (13) (15). Gecombineerd geeft deze informatie aan dat de h-CLAT-methode nuttig kan zijn om het gevaar van huidsensibilisatie te bepalen. De waarden die hier worden vermeld voor de nauwkeurigheid van de h-CLAT als losstaande test, vormen echter slechts een indicatie, aangezien de test moet worden bekeken in combinatie met andere informatiebronnen in de context van een IATA en in overeenstemming met hetgeen hierboven bepaald is in de punten 7 en 8 in de algemene inleiding. Bovendien moet bij de beoordeling van methoden zonder dierproeven voor huidsensibilisatie rekening worden gehouden met het feit dat de LLKT en andere dierproeven de situatie van mensen mogelijk niet volledig weerspiegelen.
|
|
4.
|
Op basis van de op dit moment beschikbare gegevens is gebleken dat de h-CLAT kan worden toegepast op teststoffen die verschillende organische functionele groepen, reactiemechanismen, potenties voor huidsensibilisatie (zoals bepaald bij in-vivo-onderzoek) en fysisch-chemische eigenschappen beslaan (3) (14) (15). De h-CLAT-methode is van toepassing op teststoffen die oplosbaar zijn of een stabiele dispersie vormen (d.w.z. een colloïde of suspensie waarin de teststof niet bezinkt of zich van het oplosmiddel/vehiculum afscheidt in verschillende fasen) in een geschikt oplosmiddel/vehiculum (zie punt 14). Teststoffen met een log Kow van meer dan 3,5 leveren vaker valsnegatieve uitslagen op (14). Om die reden moeten negatieve uitslagen bij teststoffen met een log Kow van meer dan 3,5 buiten beschouwing worden gelaten. Positieve uitslagen bij teststoffen met een log Kow van meer dan 3,5 kunnen echter wel worden gebruikt om de identificatie van de teststof als huidsensibiliserende stof te staven. Bovendien kunnen ook prohaptenen (d.w.z. stoffen die enzymactivering nodig hebben, bijvoorbeeld via P450-enzymen) en prehaptenen (d.w.z. stoffen die worden geactiveerd door oxidatie), in het bijzonder met een trage oxidatiesnelheid, negatieve resultaten geven in de h-CLAT (15) vanwege de beperkte metabole capaciteit van de gebruikte cellijn (16) en vanwege de omstandigheden bij het experiment. Fluorescerende teststoffen kunnen worden beoordeeld met de h-CLAT (17). Sterk fluorescerende teststoffen die op dezelfde golflengte uitzenden als fluoresceïne-isothiocyanaat (FITC) of als propidiumjodide (PI) zullen echter interfereren met de flowcytometrische detectie en kunnen dus niet correct worden beoordeeld wanneer gebruikgemaakt wordt van met FITC geconjugeerde antilichamen of PI. In dat geval kunnen respectievelijk andere met fluorochroom gelabelde antilichamen of andere cytotoxiciteitsmarkers worden gebruikt, zolang kan worden aangetoond dat ze vergelijkbare resultaten opleveren als de met FITC gelabelde antilichamen (zie punt 24) of PI (zie punt 18), bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 1.2 te testen. In het licht van het bovenstaande moeten negatieve resultaten worden geïnterpreteerd in de context van de vermelde beperkingen en samen met andere informatiebronnen in het kader van een IATA. In gevallen waarin wordt aangetoond dat de h-CLAT-methode niet toepasselijk is op andere specifieke categorieën teststoffen, dient de methode niet te worden gebruikt voor die specifieke categorieën.
|
|
5.
|
Zoals hierboven beschreven, helpt de h-CLAT-methode om huidsensibiliserende stoffen te onderscheiden van niet-sensibiliserende stoffen. Deze testmethode kan echter ook bijdragen aan de beoordeling van de sensibiliserende potentie (4) (5) (9) wanneer zij wordt gebruikt in het kader van een geïntegreerde aanpak zoals een IATA. Er moet niettemin nog meer onderzoek worden verricht, bij voorkeur op basis van gegevens die zijn verzameld bij mensen, om te bepalen hoe de resultaten van de h-CLAT kunnen worden benut bij de beoordeling van de potentie.
|
|
6.
|
Definities worden gegeven in aanhangsel 1.1.
|
PRINCIPE VAN DE TEST
|
7.
|
De h-CLAT-methode is een in-vitrobepaling die veranderingen in de expressie van markers op het celoppervlak (d.w.z. CD86 en CD54) op een humane monocytischeleukemiecellijn, THP-1, kwantificeert nadat de cellen 24 uur aan de teststof zijn blootgesteld. Deze oppervlaktemoleculen zijn kenmerkende markers van de activering van monocytische THP-1-cellen. Ze kunnen de activering van dendritische cellen, die een cruciale rol speelt bij de priming van T-cellen, nabootsen. De veranderingen in de expressie van oppervlaktemarkers worden gemeten met behulp van flowcytometrie, nadat de cellen zijn gekleurd met fluorochroom-gelabelde antilichamen. Tegelijkertijd wordt een cytotoxiciteitsmeting uitgevoerd om te beoordelen of bij subcytotoxische concentraties opregulatie van de expressie van oppervlaktemarkers plaatsvindt. De relatieve fluorescentie-intensiteit van oppervlaktemarkers vergeleken met de oplosmiddel-/vehiculumcontrole wordt berekend en gebruikt in het voorspellingsmodel (zie punt 26) om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven.
|
AANTONING VAN BEKWAAMHEID
|
8.
|
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen met de 10 stoffen voor bekwaamheidstoetsing die in aanhangsel 1.2 worden genoemd. Gebruikers van de test moeten bovendien een historische gegevensbank bijhouden van de gegevens die het resultaat zijn van de reactiviteitscontroles (zie punt 11), de positieve controles en oplosmiddel-/vehiculumcontroles (zie punten 20-22). Zij moeten deze gegevens gebruiken om te bevestigen dat de reproduceerbaarheid van de test in hun laboratorium in de loop van de tijd gehandhaafd blijft.
|
PROCEDURE
|
9.
|
Deze testmethode is gebaseerd op protocol nr. 158 betreffende h-CLAT van de DataBase service on ALternative Methods to animal experimentation (DB-ALM) (18), het protocol dat werd gebruikt voor de door het EURL CEVMA gecoördineerde valideringsstudie. Wanneer de h-CLAT-methode in het laboratorium wordt ingevoerd en toegepast, wordt aanbevolen om dit protocol te volgen. Hieronder worden de belangrijkste onderdelen en procedures beschreven van de h-CLAT-methode, die uit twee stappen bestaat: dosisbepalingstest en meting van de CD86/CD54-expressie.
|
Celpreparaten
|
10.
|
Voor het toepassen van de h-CLAT-methode moet de humane monocytischeleukemiecellijn, THP-1, worden gebruikt. Aanbevolen wordt cellen (TIB-202™) te betrekken van een goed gekwalificeerde celbank, zoals de American Type Culture Collection.
|
|
11.
|
THP-1-cellen worden gekweekt, bij 37oC in een vochtige atmosfeer met 5 % CO2, in RPMI-1640-medium, aangevuld met 10 % foetaal runderserum (FBS), 0,05 mM 2-mercapto-ethanol, 100 eenheden/ml penicilline en 100 μg/ml streptomycine. Penicilline en streptomycine kunnen in het kweekmedium achterwege worden gelaten. In dat geval moeten gebruikers evenwel verifiëren dat de afwezigheid van antibiotica in het kweekmedium niet van invloed is op de resultaten, bijvoorbeeld door de stoffen voor bekwaamheidstoetsing die in aanhangsel 1.2 worden genoemd, te testen. Om het risico van verontreiniging tot een minimum te beperken, moeten hoe dan ook goede celkweekpraktijken worden gevolgd, ongeacht of het celkweekmedium wel of niet antibiotica bevat. THP-1-cellen worden stelselmatig iedere 2-3 dagen geënt met een dichtheid van 0,1 tot 0,2 × 106 cellen/ml. Ze moeten op een dichtheid van 0,1 tot 1,0 × 106 cellen/ml worden gehouden. Voordat de cellen voor tests worden gebruikt, moet hun geschiktheid worden vastgesteld aan de hand van een reactiviteitscontrole. De reactiviteitscontrole van de cellen moet worden uitgevoerd met behulp van de positieve controles 2,4-dinitrochloorbenzeen (DNCB) (CAS-nr. 97-00-7, zuiverheid ≥ 99 %) en nikkelsulfaat (NiSO4) (CAS-nr. 10101-97-0, zuiverheid ≥ 99 %), en de negatieve controle melkzuur (CAS-nr. 50-21-5, zuiverheid ≥ 85 %), twee weken na het ontdooien. Zowel DNCB als NiSO4 moeten een positieve respons opleveren van beide oppervlaktemarkers CD86 en CD54, en melkzuur moet een negatieve respons opleveren van beide oppervlaktemarkers CD86 en CD54. Voor de test mogen uitsluitend de cellen worden gebruikt die de reactiviteitscontrole met goed gevolg hebben doorstaan. Cellen kunnen tot maximaal twee maanden na het ontdooien worden vermeerderd. Het aantal passages mag niet hoger zijn dan 30. De reactiviteitscontrole moet worden uitgevoerd in overeenstemming met de procedures die in de punten 20-24 worden beschreven.
|
|
12.
|
Voor het testen worden THP-1-cellen geënt met een dichtheid van ofwel 0,1 × 106 cellen/ml ofwel 0,2 × 106 cellen/ml, en gedurende respectievelijk 72 uur of 48 uur voorgekweekt in kweekflessen. Het is belangrijk dat in elk experiment de celdichtheid in de kweekfles direct na het voorkweken zo consistent mogelijk is (door een van de twee hierboven beschreven voorkweekomstandigheden te gebruiken). De celdichtheid in de kweekfles direct na het voorkweken zou namelijk van invloed kunnen zijn op de CD86/CD54-expressie die door allergenen wordt geïnduceerd (19). Op de dag dat de test wordt uitgevoerd, worden cellen die uit de kweekfles worden geoogst, geresuspendeerd met vers kweekmedium bij een dichtheid van 2 × 106 cellen/ml. Vervolgens worden de cellen verdeeld over een 24-putjesplaat met vlakke bodem (500 μl, 1 × 106 cellen/putje) of een 96-putjesplaat met vlakke bodem (80 μl, 1,6 × 105 cellen/putje).
|
Dosisbepalingstest
|
13.
|
Er wordt een dosisbepalingstest uitgevoerd om de CV75 vast te stellen, d.w.z. de concentratie van de teststof waarbij de cellevensvatbaarheid (cell viability, CV) 75 % is, vergeleken met de oplosmiddel-/vehiculumcontrole. De CV75-waarde wordt gebruikt om de concentratie van teststoffen te bepalen voor de meting van de CD86/CD54-expressie (zie punten 20-24).
|
Bereiding van test- en controlestoffen
|
14.
|
De test- en controlestoffen worden bereid op de dag van de tests.Voor de h-CLAT-methode worden teststoffen opgelost of stabiel gedispergeerd (zie ook punt 4). De eerste optie voor een oplosmiddel/vehiculum is een zoutoplossing of medium. Als de teststof niet oplosbaar is in deze twee oplosmiddelen/vehicula is dimethylsulfoxide (DMSO, zuiverheid ≥ 99 %) een tweede optie. De teststoffen worden opgelost of stabiel gedispergeerd tot uiteindelijke concentraties van 100 mg/ml (in zoutoplossing of medium) of 500 mg/ml (in DMSO). Andere oplosmiddelen/vehicula dan die welke hierboven worden beschreven, mogen worden gebruikt als daarvoor een voldoende wetenschappelijke onderbouwing wordt gegeven.De stabiliteit van de teststof in het uiteindelijke oplosmiddel/vehiculum moet in aanmerking worden genomen.
|
|
15.
|
Uitgaande van de 100 mg/ml (in zoutoplossing of medium) of 500 mg/ml (in DMSO) stamoplossingen van de teststoffen moeten de volgende verdunningsstappen worden gevolgd:
|
—
|
Voor zoutoplossing of medium als oplosmiddel/vehiculum: Er worden acht stamoplossingen (acht concentraties) bereid, door middel van seriële verdunningen met een factor twee met behulp van het bijbehorende oplosmiddel/vehiculum. Deze stamoplossingen worden vervolgens 50 keer verdund in kweekmedium (werkoplossingen). Als de hoogste uiteindelijke concentratie in de plaat van 1 000 μg/ml niet-toxisch is, moet de maximumconcentratie opnieuw worden bepaald door een nieuwe cytotoxiciteitstest uit te voeren. De uiteindelijke concentratie in de plaat mag voor teststoffen die opgelost of stabiel gedispergeerd zijn in zoutoplossing of medium, niet hoger zijn dan 5 000 μg/ml.
|
|
—
|
Voor DMSO als oplosmiddel/vehiculum: Er worden acht stamoplossingen (acht concentraties) bereid, door middel van seriële verdunningen met een factor twee met behulp van het bijbehorende oplosmiddel/vehiculum. Deze stamoplossingen worden vervolgens 250 keer verdund in kweekmedium (werkoplossingen). De uiteindelijke concentratie in de plaat mag niet hoger zijn dan 1 000 μg/ml, zelfs als deze concentratie niet-toxisch is.
|
De werkoplossingen worden tot slot gebruikt voor blootstelling, door een gelijke hoeveelheid werkoplossing toe te voegen aan de hoeveelheid THP-1-celsuspensie in de plaat (zie ook punt 17) om op die manier opnieuw een tweevoudige verdunning te verkrijgen (het uiteindelijke concentratiebereik in de plaat is doorgaans 7,81–1 000 μg/ml).
|
|
16.
|
De oplosmiddel-/vehiculumcontrole die in de h-CLAT-methode wordt gebruikt, is kweekmedium (voor teststoffen die hetzij met medium, hetzij met een zoutoplossing opgelost of stabiel gedispergeerd zijn (zie punt 4)) of DMSO (voor teststoffen die opgelost of stabiel gedispergeerd zijn in DMSO). Deze controle wordt getest in één enkele uiteindelijke concentratie in de plaat van 0,2 %. De verdunning die in punt 15 voor de werkoplossingen wordt beschreven, wordt ook op de controle toegepast.
|
Het aanbrengen van de test- en controlestoffen
|
17.
|
Het kweekmedium of de werkoplossingen die in de punten 15 en 16 worden beschreven, worden in een verhouding van 1:1 (v/v) gemengd met de celsuspensies die zijn geprepareerd in de 24- of 96-putjesplaat met vlakke bodem (zie punt 12). De behandelde platen worden vervolgens gedurende 24 ± 0,5 uur geïncubeerd bij 37oC met 5 % CO2. Voorkomen moet worden dat vluchtige teststoffen verdampen en dat er kruisbesmetting met teststoffen tussen putjes optreedt, bijvoorbeeld door de plaat voorafgaand aan de incubatie met de teststoffen af te sluiten (20).
|
Propidiumjodidekleuring (PI-kleuring)
|
18.
|
Na 24 ± 0,5 uur blootstelling worden de cellen overgebracht naar monsterbuisjes en verzameld door centrifugatie. Het supernatans wordt verwijderd en de resterende cellen worden geresuspendeerd met 200 μl (96-putjesplaat) of 600 μl (24-putjesplaat) van een fosfaatgebufferde zoutoplossing met 0,1 % runderserumalbumine (kleuringsbuffer). 200 μl van de celsuspensie wordt overgebracht naar een 96-putjesplaat met ronde bodem (als er een 96-putjesplaat is gebruikt) of microbuisje (als er een 24-putjesplaat is gebruikt) en tweemaal gewassen met 200 μl (96-putjesplaat) of 600 μl (24-putjesplaat) kleuringsbuffer. Tot slot worden de cellen geresuspendeerd in kleuringsbuffer (bv. 400 μl) en er wordt PI-oplossing (bv. 20 μl) toegevoegd (de uiteindelijke concentratie PI is bijvoorbeeld 0,625 μg/ml). Andere cytotoxiciteitsmarkers, zoals 7-aminoactinomycine D (7-AAD) of trypaanblauw, mogen worden gebruikt als de alternatieve kleurstoffen aantoonbaar vergelijkbare resultaten opleveren als PI, bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 1.2 te testen.
|
Cytotoxiciteitsmeting met behulp van flowcytometrie en bepaling van de CV75-waarde
|
19.
|
De PI-opname wordt geanalyseerd met behulp van flowcytometrie waarbij het acquisitiekanaal FL-3 wordt gebruikt. In totaal worden er 10 000 levende cellen (PI-negatief) verzameld. De cellevensvatbaarheid kan worden berekend door in het cytometeranalyseprogramma de onderstaande vergelijking te gebruiken. Wanneer de cellevensvatbaarheid laag is, moeten er maximaal 30 000 cellen, met inbegrip van dode cellen, worden verzameld. Een andere mogelijkheid is dat gedurende één minuut na de start van de analyse gegevens worden verzameld.

De CV75-waarde (zie punt 13), dat wil zeggen een concentratie waarbij 75 % van de THP-1-cellen overleeft (25 % cytotoxiciteit), wordt berekend door middel van loglineaire interpolatie aan de hand van de volgende vergelijking:

waarbij:
a de minimumwaarde is van cellevensvatbaarheid hoger dan 75 %
c de maximumwaarde is van cellevensvatbaarheid lager dan 75 %
b en d de concentraties zijn die de waarde van respectievelijk cellevensvatbaarheid a en c vertonen
Er mogen andere methoden worden gebruikt om de CV75 af te leiden, zolang wordt aangetoond dat dit niet van invloed is op de resultaten (bv. door de stoffen voor bekwaamheidstoetsing te testen).
|
Meting van de CD86/CD54-expressie
Bereiding van de test- en controlestoffen
|
20.
|
Er wordt een oplosmiddel/vehiculum gebruikt (zoutoplossing, medium of DMSO; zie punt 14) dat geschikt is om de teststoffen op te lossen of stabiel te dispergeren. De teststoffen worden eerst verdund tot de concentratie die overeenkomt met 100 maal (voor zoutoplossing of medium) of 500 maal (voor DMSO) de 1,2 × CV75 die is vastgesteld in de dosisbepalingstest (zie punt 19). Als de CV75 niet kan worden bepaald (dat wil zeggen, als er geen voldoende cytotoxiciteit wordt waargenomen in de dosisbepalingstest), moet de hoogste opgeloste of stabiel gedispergeerde concentratie van de teststoffen die met elk oplosmiddel/vehiculum zijn geprepareerd, worden gebruikt als startconcentratie. NB: de uiteindelijke concentratie in de plaat mag niet hoger zijn dan 5 000 μg/ml (in het geval van een zoutoplossing of medium) of 1 000 μg/ml (in het geval van DMSO). Vervolgens worden met het overeenstemmende oplosmiddel/vehiculum 1,2-voudige seriële verdunningen gemaakt om de stamoplossingen te verkrijgen (acht concentraties variërend van 100 × 1,2 × CV75 tot 100 × 0,335 × CV75 (zoutoplossing of medium) of van 500 × 1,2 × CV75 tot 500 × 0,335 × CV75 (DMSO)) die zullen worden getest aan de hand van de h-CLAT-methode (zie DB-ALM-protocol nr. 158 voor een voorbeeld van een doseringsschema). De stamoplossingen worden vervolgens verder verdund – 50 maal (voor zoutoplossing of medium) of 250 maal (voor DMSO) – in het kweekmedium (werkoplossingen). Deze werkoplossingen worden ten slotte gebruikt voor blootstelling met nog eens een laatste tweevoudige verdunningsfactor in de plaat. Als de resultaten niet voldoen aan de aanvaardbaarheidscriteria die in de punten 29 en 30 met betrekking tot de cellevensvatbaarheid worden beschreven, kan de dosisbepalingstest worden herhaald om een nauwkeuriger CV75 vast te stellen. NB: voor de meting van de CD86/CD54-expressie mogen uitsluitend 24-putjesplaten worden gebruikt.
|
|
21.
|
De oplosmiddel-/vehiculumcontrole wordt bereid zoals beschreven in punt 16. De positieve controle die in de h-CLAT-methode wordt gebruikt, is DNCB (zie punt 11). De stamoplossingen daarvoor worden bereid in DMSO en verdund zoals beschreven voor de stamoplossingen in punt 20. DNCB moet worden gebruikt als de positieve controle voor de meting van de CD86/CD54-expressie met één enkele eindconcentratie in de plaat (doorgaans 4,0 μg/ml). Voor een concentratie van 4,0 μg/ml DNCB in de plaat wordt 2 mg/ml stamoplossing van DNCB in DMSO bereid, die weer 250 maal wordt verdund met kweekmedium tot een werkoplossing van 8 μg/ml. Een andere optie is de CV75 van DNCB, die in elke testinstelling wordt bepaald, te gebruiken als positievecontroleconcentratie. Er kunnen andere geschikte positieve controles worden gebruikt als er historische gegevens beschikbaar zijn om vergelijkbare aanvaardbaarheidscriteria voor de testrun experimenten af te leiden. Voor positieve controles mag de uiteindelijke, enkele concentratie in de plaat niet hoger zijn dan 5 000 μg/ml (in het geval van een zoutoplossing of medium) of 1 000 μg/ml (in het geval van DMSO). De aanvaardbaarheidscriteria voor de testrun zijn dezelfde als beschreven voor de teststof (zie punt 29), behalve het laatste aanvaardbaarheidscriterium, aangezien de positieve controle wordt getest bij één enkele concentratie.
|
Het aanbrengen van de test- en controlestoffen
|
22.
|
Voor elke teststof en controlestof is één experiment nodig om een voorspelling te kunnen doen. Elk experiment bestaat uit ten minste twee onafhankelijke testruns voor het meten van de CD86/CD54-expressie (zie punten 26-28). Elke onafhankelijke testrun wordt uitgevoerd op een andere dag of op dezelfde dag mits voor elke testrun: a) afzonderlijke, verse stamoplossingen en werkoplossingen van de teststof en antilichaamoplossingen worden bereid en b) afzonderlijk geoogste cellen worden gebruikt (d.w.z. de cellen worden verzameld uit verschillende kweekflessen); de cellen kunnen echter wel uit dezelfde passage komen. Teststoffen en controlestoffen die zijn bereid als werkoplossingen (500 μl) worden met 500 μl gesuspendeerde cellen (1 × 106 cellen) gemengd in een verhouding van 1:1 en de cellen worden gedurende 24 ± 0,5 uur geïncubeerd zoals beschreven in de punten 20 en 21. In elke testrun volstaat één enkele duplo voor elke concentratie van de teststof en controlestof, omdat een voorspelling wordt verkregen op basis van ten minste twee onafhankelijke testruns.
|
Celkleuring en -analyse
|
23.
|
Na een blootstelling van 24 ± 0,5 uur worden de cellen van de 24-putjesplaat overgebracht naar monsterbuisjes, verzameld door middel van centrifugatie en vervolgens tweemaal gewassen met 1 ml kleuringsbuffer (zo nodig kunnen de cellen vaker worden gewassen). Na het wassen worden de cellen geblokkeerd met 600 μl blokkeeroplossing (kleuringsbuffer die 0,01 % (w/v) globuline bevat (Cohn-fractie II, III, humaan; SIGMA, nr. G2388-10G of equivalent)) en gedurende 15 minuten geïncubeerd bij 4oC. Na het blokkeren worden de cellen in drie aliquots van 180 μl verdeeld over een 96-putjesplaat met ronde bodem of microbuisjes.
|
|
24.
|
Na het centrifugeren worden de cellen gedurende 30 minuten bij 4oC gekleurd met 50 μl met FITC gelabelde anti-CD86-, anti-CD54- of murien IgG1 (isotype)-antilichamen. De antilichamen, die worden beschreven in h-CLAT DB-ALM-protocol nr. 158 (18), moeten worden verdund met kleuringsbuffer in een verhouding van 3:25 v/v (voor CD86 (BD-PharMingen, nr. 5556 57; kloon: Fun-1)) of 3:50 v/v (voor CD54 (DAKO, nr. F7143; kloon: 6.5B5) en IgG1 (DAKO, nr. X0927)). Deze antilichaamverdunningsfactoren zijn door de ontwikkelaars van de test aangemerkt als de factoren die de beste signaal-ruisverhouding geven. De ervaring van de testontwikkelaars leert dat de fluorescentie-intensiteit van de antilichamen gewoonlijk consistent is tussen verschillende partijen. Gebruikers kunnen echter overwegen de antilichamen in de omstandigheden van hun eigen laboratorium te titreren om de beste gebruiksconcentraties vast te stellen. Andere met fluorochroom gelabelde anti-CD86- en/of anti-CD54-antilichamen kunnen worden gebruikt als zij aantoonbaar – bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 1.2 te testen – vergelijkbare resultaten opleveren als met FITC geconjugeerde antilichamen. NB: wijziging van de klonen of leveranciers van de antilichamen die in h-CLAT DB-ALM-protocol nr. 158 (18) worden vermeld, kan de resultaten beïnvloeden. Nadat de cellen tweemaal of vaker zijn gewassen met 150 μl kleuringsbuffer, worden ze geresuspendeerd in kleuringsbuffer (bv. 400 μl), en wordt de PI-oplossing (bv. 20 μl om een uiteindelijke concentratie te verkrijgen van 0,625 μg/ml) of een oplossing van een andere cytotoxiciteitsmarker (zie punt 18) toegevoegd. De expressieniveaus van CD86 en CD54 en de cellevensvatbaarheid worden geanalyseerd met behulp van flowcytometrie.
|
GEGEVENS EN RAPPORTAGE
Evaluatie van de gegevens
|
25.
|
De expressie van CD86 en CD54 wordt geanalyseerd met behulp van flowcytometrie, waarbij het acquisitiekanaal FL-1 wordt gebruikt. Op grond van de geometrisch gemiddelde fluorescentie-intensiteit (MFI) wordt de relatieve fluorescentie-intensiteit (RFI) van CD86 en CD54 voor positievecontrolecellen (ctrl) en met de chemische stof behandelde cellen berekend aan de hand van de volgende vergelijking:

De cellevensvatbaarheid van de isotypecontrolecellen (ctrl), die gekleurd zijn met muriene IgG1 (isotype)-antilichamen, wordt eveneens berekend aan de hand van de vergelijking in punt 19.
|
Voorspellingsmodel
|
26.
|
Voor de meting van de CD86/CD54-expressie wordt elke teststof in ten minste twee onafhankelijke testruns getest om één enkele voorspelling af te leiden (POSITIEF of NEGATIEF). Een h-CLAT-voorspelling wordt POSITIEF geacht als aan ten minste één van de volgende voorwaarden is voldaan in twee van de twee of in ten minste twee van de drie onafhankelijke testruns. Is dat niet het geval, dan wordt de h-CLAT-voorspelling NEGATIEF geacht (figuur 1):
|
—
|
De RFI van CD86 is gelijk aan of groter dan 150 % bij elke willekeurige geteste concentratie (met een cellevensvatbaarheid ≥ 50 %);
|
|
—
|
De RFI van CD54 is gelijk aan of groter dan 200 % bij elke willekeurige geteste concentratie (met een cellevensvatbaarheid ≥ 50 %).
|
|
|
27.
|
Op basis van het bovenstaande wordt de h-CLAT-voorspelling POSITIEF geacht als de eerste twee testruns beide positief zijn voor CD86 en/of beide positief zijn voor CD54. In dat geval hoeft geen derde testrun te worden uitgevoerd. Als de eerste twee testruns voor beide markers negatief zijn, wordt de h-CLAT-voorspelling NEGATIEF geacht (waarbij rekening wordt gehouden met het bepaalde in punt 30), zonder dat een derde testrun hoeft te worden uitgevoerd. Als de eerste twee testruns echter voor ten minste één van beide markers (CD54 of CD86) niet overeenstemmen, is een derde testrun nodig en wordt de uiteindelijke voorspelling gebaseerd op de meerderheidsuitslag van de drie afzonderlijke testruns (d.w.z. twee van de drie). In dit verband moet worden opgemerkt dat als twee onafhankelijke testruns worden uitgevoerd en de ene is alleen positief voor CD86 (hierna P1 genoemd) en de andere is alleen positief voor CD54 (hierna P2 genoemd), een derde testrun vereist is. Als deze derde testrun negatief is voor beide markers (hierna N genoemd) wordt de h-CLAT-voorspelling NEGATIEF geacht. Anderzijds wordt de h-CLAT-voorspelling POSITIEF geacht als de derde testrun positief is voor een van beide markers (P1 of P2) of voor beide markers (hierna P12 genoemd).

Figuur 1: Voorspellingsmodel dat wordt gebruikt in de h-CLAT-methode. Een h-CLAT-voorspelling dient te worden bekeken in.het kader van een IATA en in overeenstemming met hetgeen is bepaald in de punten 7 en 8 in de algemene inleiding.
P1: testrun alleen positief voor CD86; P2; testrun alleen positief voor CD54; P12: testrun positief voor zowel CD86 als CD54; N: testrun noch positief voor CD86, noch voor CD54.
* De vakken tonen de relevante combinaties van resultaten van de eerste twee testruns, onafhankelijk van de volgorde waarin ze worden verkregen.
# De vakken tonen de relevante combinaties van resultaten van de drie testruns op basis van de resultaten in de eerste twee testruns die in het bovenstaande vak worden getoond, maar geven niet de volgorde weer waarin ze zijn verkregen.
|
|
28.
|
Voor teststoffen waarvan de h-CLAT voorspelt dat ze POSITIEF zijn, kan eventueel de waarde van twee Effectieve Concentraties (EC’s) worden bepaald – de EC150 voor CD86 en de EC200 voor CD54 – d.w.z. de concentratie waarbij de teststoffen een RFI van 150 of 200 opleverden. Deze EC-waarden zouden mogelijk ook kunnen bijdragen aan de beoordeling van de sensibiliserende potentie (9), wanneer zij worden gebruikt in het kader van een geïntegreerde aanpak zoals een IATA (4) (5) (6) (7) (8). Ze kunnen worden berekend aan de hand van de volgende vergelijkingen:
EC 150 (for CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (for CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
waarbij:
Aconc de laagste concentratie is in μg/ml met een RFI > 150 (CD86) of 200 (CD54)
Bconc de hoogste concentratie is in μg/ml met een RFI < 150 (CD86) of 200 (CD54)
ARFI de RFI is bij de laagste concentratie met een RFI > 150 (CD86) of 200 (CD54)
BRFI de RFI is bij de hoogste concentratie met een RFI < 150 (CD86) of 200 (CD54)
Om de waarden van de EC150 en EC200 nauwkeuriger af te leiden, kan het nodig zijn drie onafhankelijke testruns uit te voeren voor de meting van de CD86/CD54-expressie. De uiteindelijke EC150- en EC200-waarden worden vervolgens bepaald als de mediane waarde van de EC’s die zijn berekend uit de drie onafhankelijke testruns. Wanneer slechts twee van de drie onafhankelijke testruns aan de criteria voor positiviteit voldoen (zie punten 26-27), wordt de hoogste EC150 of EC200 van de twee berekende waarden genomen.
|
Aanvaardbaarheidscriteria
|
29.
|
Bij gebruik van de h-CLAT-methode moet aan de volgende aanvaardbaarheidscriteria worden voldaan (22) (27).
|
—
|
De cellevensvatbaarheid van controles met medium en oplosmiddel/vehiculum moet groter zijn dan 90 %.
|
|
—
|
In de oplosmiddel-/vehiculumcontrole mogen de RFI-waarden van zowel CD86 als CD54 niet hoger zijn dan de positieve criteria (CD86 RFI ≥ 150 % en CD54 RFI ≥ 200 %). De RFI-waarden van de oplosmiddel-/vehiculumcontrole worden berekend aan de hand van de formule in punt 25 (MFI van chemische stof moet worden vervangen door MFI van oplosmiddel/vehiculum en MFI van oplosmiddel/vehiculum moet worden vervangen door MFI van (medium)controle).
|
|
—
|
Voor zowel medium- als oplosmiddel-/vehiculumcontroles moet de verhouding van de MFI tot de isotypecontrole voor zowel CD86 als CD54 groter zijn dan 105 %.
|
|
—
|
In de positieve controle (DNCB) moeten de RFI-waarden van zowel CD86 als CD54 voldoen aan de positieve criteria (CD86 RFI ≥ 150 en CD54 RFI ≥ 200) en moet de cellevensvatbaarheid groter zijn dan 50 %.
|
|
—
|
De cellevensvatbaarheid van de teststof moet in ten minste vier geteste concentraties in elke testrun groter zijn dan 50 %.
|
|
|
30.
|
Negatieve resultaten zijn uitsluitend aanvaardbaar voor teststoffen die bij de hoogste geteste concentratie (d.w.z. 1,2 × CV75 volgens het seriële verdunningsschema dat in punt 20 wordt beschreven) een cellevensvatbaarheid laten zien van minder dan 90 %. Als de cellevensvatbaarheid bij 1,2 × CV75 90 % of hoger is, moet het negatieve resultaat worden weggelaten. In dat geval wordt aanbevolen te proberen de keuze van de dosis te verfijnen door de CV75-bepaling te herhalen. Opgemerkt moet worden dat een negatief resultaat aanvaardbaar is, zelfs als de cellevensvatbaarheid hoger is dan 90 %, wanneer 5 000 μg/ml in een zoutoplossing (of medium of andere oplosmiddelen/vehicula), 1 000 μg/ml in DMSO of de hoogste oplosbare concentratie wordt gebruikt als de maximale testconcentratie van een teststof.
|
Testverslag
|
31.
|
In het testverslag moet de volgende informatie worden opgenomen.
|
Teststof
|
Stof die uit één component bestaat
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, log Kow, oplosbaarheid in water, oplosbaarheid in DMSO, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
|
|
Stof die uit meerdere componenten bestaat, UVCB en mengsel
|
karakterisering, voor zover mogelijk aan de hand van bv. chemische identiteit (zie hierboven), zuiverheid, kwantitatief voorkomen en relevante fysisch-chemische eigenschappen (zie hierboven) van de bestanddelen, voor zover beschikbaar;
|
|
fysisch voorkomen, oplosbaarheid in water, oplosbaarheid in DMSO en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
molecuulgewicht of schijnbaar molecuulgewicht in het geval van mengsels/polymeren waarvan de samenstelling bekend is, of andere informatie die relevant is voor het verloop van de studie;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
|
|
|
Controles
|
Positieve controle
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, log Kow, oplosbaarheid in water, oplosbaarheid in DMSO, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn en voor zover van toepassing;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
verwijzing naar historische positievecontroleresultaten waaruit de geschiktheid van de aanvaardbaarheidscriteria voor de testrun blijkt, indien van toepassing.
|
|
|
|
Negatieve controle en oplosmiddel-/vehiculumcontrole
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
fysisch voorkomen, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, in het geval er andere controle-oplosmiddelen/-vehicula worden gebruikt dan diegene die worden vermeld in de testrichtlijn, en voor zover beschikbaar;
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
|
|
Testomstandigheden
|
naam en adres van de opdrachtgever, testinstallatie en de onderzoeksleider;
|
|
beschrijving van de gebruikte methode;
|
|
gebruikte cellijn, omstandigheden waarin die werd bewaard en oorsprong (bv. de instelling waarvan de cellijn werd verkregen);
|
|
gebruikte flowcytometrie (bv. model), met inbegrip van de gebruikte instrumentinstellingen, globuline, antilichamen en cytotoxiciteitsmarker;
|
|
de procedure die gebruikt is om aan te tonen dat het laboratorium bekwaam is om de test uit te voeren, door de stoffen voor bekwaamheidstoetsing te testen, en de procedure die gebruikt is om aan te tonen dat de resultaten van de test in de loop van de tijd kunnen worden gereproduceerd, bv. controlegegevens uit het verleden en/of gegevens van reactiviteitscontroles uit het verleden.
|
|
|
Aanvaardbaarheidscriteria voor de test
|
cellevensvatbaarheid, MFI- en RFI-waarden die met de oplosmiddel-/vehiculumcontrole zijn verkregen in vergelijking met het aanvaardbaarheidsbereik;
|
|
cellevensvatbaarheid en RFI-waarden die met de positieve controle zijn verkregen in vergelijking met het aanvaardbaarheidsbereik;
|
|
cellevensvatbaarheid van alle geteste concentraties van de teststof.
|
|
|
Werkwijze
|
aantal gebruikte testruns;
|
|
de gehanteerde concentraties van de teststof, toedienings- en blootstellingstijd (indien verschillend van de aanbevolen tijd);
|
|
een beschrijving van de gebruikte beoordelings- en beslissingscriteria;
|
|
een beschrijving van eventuele modificaties van de testprocedure.
|
|
|
Resultaten
|
tabel van de gegevens, waaronder CV75 (indien van toepassing), afzonderlijke geometrische MFI, RFI, cellevensvatbaarheidswaarden, EC150-/EC200-waarden (indien van toepassing) die zijn verkregen voor de teststof en voor de positieve controle in elke testrun, en een indicatie van de score van de teststof volgens het voorspellingsmodel;
|
|
een beschrijving van eventuele andere relevante waarnemingen.
|
|
|
Bespreking van de resultaten
|
bespreking van de resultaten verkregen met de h-CLAT-methode;
|
|
overweging van de testresultaten in de context van een IATA, als er andere relevante informatie beschikbaar is.
|
|
|
LITERATUUR
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL CEVMA (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Beschikbaar op: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL CEVMA (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Beschikbaar op: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL CEVMA (2012). Human Cell Line Activation Test (h-CLAT) Validation Study Report. Beschikbaar op: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Beschikbaar op: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OESO (2005). Guidance Document No 34 on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Series on Testing and Assessment. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs, Frankrijk, 2005, 96 pp.
|
|
(22)
|
OESO (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Beschikbaar op: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
Verenigde Naties (VN) (2013). Mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (GHS). Vijfde herziene editie. New York & Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Beschikbaar op: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Aanhangsel 1.1
DEFINITIES
AOP (Adverse Outcome Pathway)
: een reeks gebeurtenissen van de chemische structuur van een doelstof of een groep soortgelijke chemische stoffen, van de moleculaire inleidende gebeurtenis tot een te onderzoeken in-vivoresultaat (22).
Betrouwbaarheid
: indicatie van de mate waarin een test, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (21).
Chemische stof
: een stof of een mengsel.
CV75
: de geschatte concentratie waarbij de cellevensvatbaarheid 75 % is.
EC150
: de concentraties waarbij sprake is van de RFI-waarde 150 voor CD86-expressie.
EC200
: de concentraties waarbij sprake is van de RFI-waarde 200 voor CD54-expressie.
Flowcytometrie
: een cytometrische techniek waarbij in een vloeistof gesuspendeerde cellen één voor één door een brandpunt van exciterend licht stromen, dat wordt verstrooid in patronen die kenmerkend zijn voor de cellen en hun bestanddelen; de cellen worden vaak gelabeld met fluorescente markers, zodat het licht eerst wordt geabsorbeerd en vervolgens met gewijzigde frequenties wordt uitgezonden.
Geldige test
: een test die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen. Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel (21).
Gevaar
: inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld.
Gevoeligheid
: het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert, en is een belangrijke overweging bij de beoordeling van de relevantie van een test (21).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN))
: een systeem voor de indeling van chemische stoffen (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (23).
IATA (Integrated Approach on Testing and Assessment, geïntegreerde test- en beoordelingsbenadering)
: een gestructureerde aanpak die wordt gebruikt voor de identificatie van gevaren (potentieel), het kenmerken van gevaren (potentie) en/of het beoordelen van de veiligheid (potentieel/potentie en blootstelling) van een chemische stof of een groep chemische stoffen, waarbinnen alle relevante gegevens strategisch worden geïntegreerd en worden afgewogen als inbreng voor regelgevingsbesluiten inzake potentiële gevaren en/of risico’s en/of de noodzaak van verdere gerichte en bijgevolg minimale tests.
Kleuringsbuffer
: Een fosfaatgebufferde zoutoplossing met 0,1 % runderserumalbumine.
Mediumcontrole
: een onbehandeld monster dat alle componenten bevat van een testsysteem. Dit monster wordt tezamen met de met de teststof behandelde monsters en andere controlemonsters verwerkt om te bepalen of het oplosmiddel interageert met het testsysteem.
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
Nauwkeurigheid
: de mate van overeenstemming tussen testresultaten en erkende referentiewaarden. Het is een maat voor de prestaties van de test en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een test wordt verstaan, worden vaak door elkaar gebruikt (21).
Oplosmiddel-/vehiculumcontrole
: een onbehandeld monster dat alle componenten bevat van een testsysteem met uitzondering van de teststof, maar met inbegrip van het gebruikte oplosmiddel/vehiculum. Dit monster wordt gebruikt om de referentierespons vast te stellen voor de monsters die worden behandeld met de in hetzelfde oplosmiddel/vehiculum opgeloste of stabiel gedispergeerde teststof. Als dit monster getest wordt met een tegelijkertijd gemeten mediumcontrole, toont het ook aan of het oplosmiddel/vehiculum met het testsysteem interageert.
Positieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positievecontrolerespons in de loop van de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn.
Prehaptenen
: chemische stoffen die in sensibiliserende stoffen veranderen na abiotische transformatie.
Prohaptenen
: chemische stoffen die enzymatische activering nodig hebben om hun potentieel voor huidsensibilisatie te realiseren.
Relatieve fluorescentie-intensiteit (RFI)
: Relatieve waarden van de geometrisch gemiddelde fluorescentie-intensiteit (MFI) in de met de chemische stof behandelde cellen vergeleken met de MFI in de met oplosmiddel/vehiculum behandelde cellen.
Relevantie
: beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een test (21).
Specificiteit
: het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert en is een belangrijke overweging bij de beoordeling van de relevantie van een test (21).
Stof
: een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Testrun
: een testrun bestaat uit het gelijktijdig testen van een of meer chemische stoffen met een oplosmiddel-/vehiculumcontrole en met een positieve controle.
Teststof
: alle volgens deze methode geteste stoffen of mengsels.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
Aanhangsel 1.2
STOFFEN VOOR BEKWAAMHEIDSTOETSING
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen door op de juiste wijze de verwachte h-CLAT-voorspelling te verkrijgen voor de 10 stoffen die in tabel 1 worden aanbevolen, en door de CV75-, EC150- en EC200-waarden te verkrijgen die binnen de respectieve referentiebereiken vallen voor ten minste 8 van de 10 stoffen voor bekwaamheidstoetsing. De stoffen voor bekwaamheidstoetsing zijn zodanig geselecteerd dat ze alle mogelijke responsen voor huidsensibilisatiegevaar weerspiegelen. Andere selectiecriteria waren dat stoffen in de handel beschikbaar zijn, en dat er in-vivoreferentiegegevens en met de h-CLAT-methode gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn. Voor de h-CLAT-methode zijn tevens gepubliceerde referentiegegevens beschikbaar (3) (14).
Tabel 1
Aanbevolen stoffen voor het aantonen van technische bekwaamheid met de h-CLAT-methode
|
Stoffen voor bekwaamheidstoetsing
|
CAS RN
|
Fysische toestand
|
In-vivovoorspelling (103)
|
CV75 Referentie-bereik in μg/ml (104)
|
h-CLAT-resultaten voor CD86 (EC150-referentie-bereik in g/ml) (104)
|
h-CLAT-resultaten voor CD54 (EC200-referentie-bereik in g/ml) (104)
|
|
2,4-Dinitrochloorbenzeen
|
97-00-7
|
Vaste stof
|
Sensibiliserend (extreem)
|
2-12
|
Positief (0,5-10)
|
Positief (0,5-15)
|
|
4-Fenyleendiamine
|
106-50-3
|
Vaste stof
|
Sensibiliserend (sterk)
|
5-95
|
Positief (< 40)
|
Negatief (> 1,5) (105)
|
|
Nikkelsulfaat
|
10101-97-0
|
Vaste stof
|
Sensibiliserend (matig)
|
30-500
|
Positief (< 100)
|
Positief (10-100)
|
|
2-Mercaptobenzothiazole
|
149-30-4
|
Vaste stof
|
Sensibiliserend (matig)
|
30-400
|
Negatief (> 10) (105)
|
Positief (10-140)
|
|
R(+)-Limoneen
|
5989-27-5
|
Vloeistof
|
Sensibiliserend (zwak)
|
> 20
|
Negatief (> 5) (105)
|
Positief (< 250)
|
|
Imidazolidinylureum
|
39236-46-9
|
Vaste stof
|
Sensibiliserend (zwak)
|
25-100
|
Positief (20-90)
|
Positief (20-75)
|
|
Propaan-2-ol
|
67-63-0
|
Vloeistof
|
Niet-sensibiliserend
|
> 5 000
|
Negatief (> 5 000 )
|
Negatief (> 5 000 )
|
|
Glycerol
|
56-81-5
|
Vloeistof
|
Niet-sensibiliserend
|
> 5 000
|
Negatief (> 5 000 )
|
Negatief (> 5 000 )
|
|
Melkzuur
|
50-21-5
|
Vloeistof
|
Niet-sensibiliserend
|
1500-5 000
|
Negatief (> 5 000 )
|
Negatief (> 5 000 )
|
|
4-Aminobenzoëzuur
|
150-13-0
|
Vaste stof
|
Niet-sensibiliserend
|
> 1 000
|
Negatief (> 1 000 )
|
Negatief (> 1 000 )
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstracts Service
|
Aanhangsel 2
IN-VITROHUIDSENSIBILISATIE: U937-CELLIJNACTIVERINGSTEST (U-SENS™)
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
1.
|
De U-SENS™-test kwantificeert de verandering in de expressie van een marker op het celoppervlak die gepaard gaat met de activering van monocyten en dendritische cellen (d.w.z. CD86), in de humane histiocytairlymfoomcellijn U937, na blootstelling aan sensibiliserende stoffen (1). De gemeten expressieniveaus van celoppervlaktemarker CD86 in de cellijn U937 wordt vervolgens gebruikt om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven.
|
|
2.
|
De U-SENS™-test is geëvalueerd in een valideringsstudie (2) die werd gecoördineerd door L’Oréal en in een latere onafhankelijke beoordeling door vakgenoten van het Raadgevend wetenschappelijk comité (ESAC) van het referentielaboratorium voor alternatieven voor dierproeven van de Europese Unie (EURL CEVMA) (3). Op grond van al het beschikbare bewijsmateriaal en de inbreng van regelgevers en belanghebbenden werd de U-SENS™ door het EURL CEVMA (4) aanbevolen voor gebruik in het kader van een IATA om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven met het oog op de gevarenindeling en etikettering. In zijn leidraad inzake de rapportage van gestructureerde benaderingen van gegevensintegratie en afzonderlijke informatiebronnen die in het kader van een IATA voor huidsensibilisatie worden gebruikt, bespreekt de OESO momenteel een aantal casestudy’s waarin verschillende teststrategieën en voorspellingsmodellen worden beschreven. Een van de verschillende vastgestelde benaderingen is gebaseerd op de U-SENS™-bepaling (5). Ook elders in de literatuur (4) (5) (7) worden voorbeelden genoemd van het gebruik van U-SENS™-gegevens in combinatie met andere informatie, met inbegrip van gegevens uit het verleden en bestaande, geldige gegevens die bij mensen zijn verzameld (6).
|
|
3.
|
De U-SENS™-test bleek overdraagbaar te zijn naar laboratoria met ervaring in het kweken van cellen en flowcytometrieanalyse. De verwachte reproduceerbaarheid van voorspellingen op basis van deze test binnen laboratoria en tussen verschillende laboratoria bedraagt respectievelijk 90 % en 84 % (8). De resultaten van het valideringsstudie (8) en andere gepubliceerde studies (1) geven over het algemeen aan dat, in vergelijking met de LLKT-resultaten, de nauwkeurigheid in het onderscheiden van sensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) van niet-sensibiliserende stoffen 86 % (N=166) bedraagt, met een gevoeligheid van 91 % (118/129) en een specificiteit van 65 % (24/37). In vergelijking met bij mensen verkregen resultaten, bedraagt de nauwkeurigheid in het onderscheiden van sensibiliserende stoffen (d.w.z. VN-GHS/CLP-categorie 1) van niet-sensibiliserende stoffen 77 % (N=101), met een gevoeligheid van 100 % (58/58) en een specificiteit van 47 % (20/43). Bij de U-SENS™ is, in vergelijking met de LLKT, de kans dat valsnegatieve voorspellingen betrekking hebben op chemische stoffen met een lage tot gemiddelde potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1B) hoger dan dat ze betrekking hebben op chemische stoffen met een hoge potentie voor huidsensibilisatie (d.w.z. VN-GHS/CLP-subcategorie 1A) (1) (8) (9). Gecombineerd geeft deze informatie aan dat de U-SENS™-test nuttig kan zijn om het gevaar van huidsensibilisatie te bepalen. De waarden die hier worden vermeld voor de nauwkeurigheid van de U-SENS™ als losstaande test vormen echter slechts een indicatie, aangezien de test moet worden bekeken in combinatie met andere informatiebronnen in de context van een IATA en in overeenstemming met hetgeen hierboven bepaald is in de punten 7 en 8 in de algemene inleiding. Bovendien moet bij de beoordeling van methoden zonder dierproeven voor huidsensibilisatie rekening worden gehouden met het feit dat de LLKT en andere dierproeven de situatie van mensen mogelijk niet volledig weerspiegelen.
|
|
4.
|
Op basis van de op dit moment beschikbare gegevens is gebleken dat de U-SENS™-test kan worden toegepast op teststoffen (waaronder cosmetica-ingrediënten, zoals conserveermiddelen, oppervlakteactieve stoffen, andere actieve stoffen, kleurstoffen) die verschillende organische functionele groepen, fysisch-chemische eigenschappen en potenties voor huidsensibilisatie (zoals bepaald in in-vivo-onderzoeken) beslaan, evenals het spectrum van reactiemechanismen waarvan bekend is dat ze samenhangen met huidsensibilisatie (d.w.z. Michael-acceptor, vorming van Schiff-basen, acyloverdracht, nucleofiele bimoleculaire substitutie [SN2] of nucleofiele aromatische substitutie [SNAr]) (1) (8) (9) (10). De U-SENS™-test is van toepassing op teststoffen die oplosbaar zijn of een stabiele dispersie vormen (d.w.z. een colloïde of suspensie waarin de teststof niet bezinkt of zich van het oplosmiddel/vehiculum afscheidt in verschillende fasen) in een geschikt oplosmiddel/vehiculum (zie punt 13). De U-SENS™ voorspelde correct welke chemische stoffen in de gegevensreeks prehaptenen waren (d.w.z. stoffen die door oxidatie worden geactiveerd) en welke chemische stoffen prohaptenen waren (d.w.z. stoffen die enzymactivering nodig hebben, bijvoorbeeld via P450-enzymen) (1) (10). Membraanverstorende stoffen kunnen leiden tot valspositieve resultaten als gevolg van een niet-specifieke stijging van de CD86-expressie: 3 van de 7 valspositieven met betrekking tot de in-vivoreferentieclassificatie waren oppervlakteactieve stoffen (1). Positieve resultaten met oppervlakteactieve stoffen moeten dan ook met terughoudendheid in aanmerking worden genomen, terwijl negatieve resultaten met oppervlakteactieve stoffen nog steeds kunnen worden gebruikt om de identificatie van de teststof als niet-sensibiliserende stof te staven. Fluorescerende teststoffen kunnen worden beoordeeld met de U-SENS™ (1). Sterk fluorescerende teststoffen die op dezelfde golflengte uitzenden als fluoresceïne-isothiocyanaat (FITC) of als propidiumjodide (PI), zullen echter interfereren met de flowcytometrische detectie en kunnen dus niet correct worden beoordeeld wanneer gebruikgemaakt wordt van met FITC geconjugeerde antilichamen (potentieel valsnegatief) of PI (levensvatbaarheid niet meetbaar). In dat geval kunnen respectievelijk andere met fluorochroom gelabelde antilichamen of andere cytotoxiciteitsmarkers worden gebruikt, zolang kan worden aangetoond dat ze vergelijkbare resultaten opleveren als de met FITC gelabelde antilichamen of PI (zie punt 18), bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 2.2 te testen. In het licht van het bovenstaande moeten positieve resultaten met oppervlakteactieve stoffen en negatieve resultaten met sterk fluorescerende teststoffen worden geïnterpreteerd in de context van de vermelde beperkingen en samen met andere informatiebronnen in het kader van een IATA. In gevallen waarin kan worden aangetoond dat de U-SENS™-test niet toepasselijk is op andere specifieke categorieën teststoffen, dient de test niet te worden gebruikt voor die specifieke categorieën.
|
|
5.
|
Zoals hierboven beschreven, helpt de U-SENS™-test om huidsensibiliserende stoffen te onderscheiden van niet-sensibiliserende stoffen. Deze testmethode kan echter ook bijdragen aan de beoordeling van de sensibiliserende potentie wanneer zij wordt gebruikt in het kader van een geïntegreerde aanpak zoals een IATA. Er moet niettemin nog meer onderzoek worden verricht, bij voorkeur op basis van gegevens die zijn verzameld bij mensen, om te bepalen hoe de resultaten van de U-SENS™ kunnen worden benut bij de beoordeling van de potentie.
|
|
6.
|
Definities worden gegeven in aanhangsel 2.1.
|
PRINCIPE VAN DE TEST
|
7.
|
De U-SENS™-bepaling is een in-vitrobepaling die veranderingen in de expressie van de celoppervlaktemarker CD86 op een humane histiocytairlymfoomcellijn, U937, kwantificeert nadat de cellen 45 ± 3 uur aan de teststof zijn blootgesteld. De CD86-oppervlaktemarker is één kenmerkende marker van U937-activering. CD86 staat bekend als een co-stimulerend molecuul dat monocytische activatie, die een cruciale rol speelt bij de priming van T-cellen, kan nabootsen. De veranderingen in de expressie van de CD86-celoppervlaktemarker worden gemeten met behulp van flowcytometrie, nadat de cellen zijn gekleurd, doorgaans met antilichamen die zijn gelabeld met fluoresceïne-isothiocyanaat (FITC). Tegelijkertijd wordt een cytotoxiciteitsmeting uitgevoerd (bv. met gebruikmaking van PI) om te beoordelen of bij subcytotoxische concentraties opregulatie van de expressie van de CD86-oppervlaktemarker plaatsvindt. De stimuleringsindex (SI) van de CD86-oppervlaktemarker vergeleken met de oplosmiddel-/vehiculumcontrole wordt berekend en gebruikt in het voorspellingsmodel (zie punt 19) om het onderscheid tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen te staven.
|
AANTONING VAN BEKWAAMHEID
|
8.
|
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen met de 10 stoffen voor bekwaamheidstoetsing die in aanhangsel 2.2 worden genoemd, met inachtneming van de OESO-leidraad „Good in vitro Method Practices” (11). Gebruikers van de test moeten bovendien een historische gegevensbank bijhouden van de gegevens die het resultaat zijn van de reactiviteitscontroles (zie punt 11), de positieve controles en oplosmiddel-/vehiculumcontroles (zie punten 15-16). Zij moeten deze gegevens gebruiken om te bevestigen dat de reproduceerbaarheid van de test in hun laboratorium in de loop van de tijd gehandhaafd blijft.
|
PROCEDURE
|
9.
|
Deze test is gebaseerd op protocol nr. 183 betreffende U-SENS™ van de DataBase service on ALternative Methods to animal experimentation (DB-ALM) (12). Wanneer de testmethode in een laboratorium wordt toegepast en gebruikt, moeten de standaardwerkwijzen voor de U-SENS™-test worden gevolgd. Er kan een geautomatiseerd systeem worden gebruikt om de U-SENS™-test uit te voeren als dit aantoonbaar – bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 2.2 te testen – vergelijkbare resultaten oplevert. Hieronder worden de belangrijkste onderdelen en procedures van de U-SENS™-test beschreven.
|
Celpreparaten
|
10.
|
Voor het toepassen van de U-SENS™-test moet de humane histiocytairlymfoomcellijn, U937 (13), worden gebruikt. Aanbevolen wordt cellen (kloon CRL1593.2) te betrekken van een goed gekwalificeerde celbank zoals de American Type Culture Collection.
|
|
11.
|
U937-cellen worden gekweekt, bij 37 °C in een vochtige atmosfeer met 5 % CO2, in RPMI-1 640-medium, aangevuld met 10 % foetaal runderserum (FBS), 2 mM L-glutamine, 100 eenheden/ml penicilline en 100 μg/ml streptomycine (volledig medium). U937-cellen worden stelselmatig iedere 2-3 dagen gepasseerd met een dichtheid van respectievelijk 1,5 of 3 × 105 cellen/ml. De celdichtheid mag niet hoger zijn dan 2 × 106 cellen/ml en de cellevensvatbaarheid zoals gemeten aan de hand van trypaanblauwuitsluiting moet ≥ 90 % zijn (niet toe te passen tijdens de eerste passage na het ontdooien). Voordat de cellen voor tests worden gebruikt, moet de geschiktheid van elke partij cellen, FBS of antilichamen worden vastgesteld aan de hand van een reactiviteitscontrole. De reactiviteitscontrole van de cellen moet worden uitgevoerd met behulp van de positieve controle picrylsulfonzuur (2,4,6-trinitrobenzeensulfonzuur: TNBS) (CASRN 2 508-19-2, zuiverheid ≥ 99 %) en de negatieve controle melkzuur (CASRN 50-21-5, zuiverheid ≥ 85 %), ten minste één week na het ontdooien. Voor de reactiviteitscontrole moeten voor elk van de twee controles zes uiteindelijke concentraties worden getest (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml en melkzuur: 1, 10, 20, 50, 100, 200 μg/ml). TNBS opgelost in volledig medium moet een positieve en concentratiegerelateerde respons van CD86 opleveren (bijvoorbeeld wanneer een positieve concentratie, CD86-SI ≥ 150, wordt gevolgd door een concentratie met een hogere CD86-SI) en melkzuur opgelost in volledig medium moet een negatieve respons van CD86 opleveren (zie punt 21). Voor de test mag uitsluitend de partij cellen worden gebruikt die de reactiviteitscontrole tweemaal met goed gevolg heeft doorstaan. Cellen kunnen tot maximaal zeven weken na het ontdooien worden vermeerderd. Het aantal passages mag niet hoger zijn dan 21. De reactiviteitscontrole moet worden uitgevoerd in overeenstemming met de procedures die in de punten 18-22 worden beschreven.
|
|
12.
|
Voor het testen worden U937-cellen geënt met een dichtheid van ofwel 3 × 105 cellen/ml ofwel 6 × 105 cellen/ml, en gedurende respectievelijk 2 dagen of 1 dag voorgekweekt in kweekflessen. Het voorkweken kan plaatsvinden onder andere omstandigheden als daarvoor voldoende wetenschappelijke onderbouwing wordt gegeven en deze omstandigheden aantoonbaar – bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 2.2 te testen – vergelijkbare resultaten opleveren. Op de dag dat de test wordt uitgevoerd, worden cellen die uit de kweekfles worden geoogst, geresuspendeerd met vers kweekmedium bij een dichtheid van 5 × 105 cellen/ml. Vervolgens worden de cellen verdeeld over een 96-putjesplaat met vlakke bodem met 100 μl (uiteindelijke celdichtheid 0,5 × 105 cellen/putje).
|
Bereiding van test- en controlestoffen
|
13.
|
Voorafgaand aan de test wordt de oplosbaarheid beoordeeld. Daartoe worden teststoffen bij een concentratie van 50 mg/ml opgelost of stabiel gedispergeerd in volledig medium als eerste oplosmiddel-optie of dimethylsulfoxide (DMSO, zuiverheid ≥ 99 %) als tweede oplosmiddel/vehiculum-optie, indien de teststof niet oplosbaar is in volledig medium als oplosmiddel/vehiculum. Voor het testen wordt de teststof opgelost tot een uiteindelijke concentratie van 0,4 mg/ml in volledig medium als de chemische stof in dit oplosmiddel/vehiculum oplosbaar is. Als de chemische stof alleen in DMSO oplosbaar is, wordt de stof opgelost bij een concentratie van 50 mg/ml. Andere oplosmiddelen/vehicula dan die welke hierboven worden beschreven, mogen worden gebruikt als daarvoor een voldoende wetenschappelijke onderbouwing wordt gegeven. De stabiliteit van de teststof in het uiteindelijke oplosmiddel/vehiculum moet in aanmerking worden genomen.
|
|
14.
|
De test- en controlestoffen worden bereid op de dag van de tests. Omdat er geen dosisbepalingstest wordt uitgevoerd, moeten voor de eerste testrun zes uiteindelijke concentraties worden getest (1, 10, 20, 50, 100 en 200 μg/ml) in het bijbehorende oplosmiddel/vehiculum, ofwel in volledig medium, ofwel in 0,4 % DMSO in medium. Voor de daaropvolgende testruns, beginnend vanaf de 0,4 mg/ml in volledig medium of 50 mg/ml in DMSO, worden oplossingen van de teststoffen (ten minste vier werkoplossingen, d.w.z. ten minste vier concentraties) bereid met behulp van het bijbehorende oplosmiddel/vehiculum. De werkoplossingen worden uiteindelijk gebruikt voor behandeling door een gelijke hoeveelheid U937-celsuspensie (zie punt 11) toe te voegen aan de hoeveelheid werkoplossing in de plaat om zo een verdere tweevoudige verdunning te verkrijgen (12). De concentraties (ten minste vier) voor elke volgende testrun worden gekozen op basis van de afzonderlijke resultaten van alle voorafgaande testruns (8). De bruikbare uiteindelijke concentraties zijn: 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 en 200 μg/ml. De maximale uiteindelijke concentratie is 200 μg/ml. Wanneer bij 1 μg/ml een positieve waarde voor CD86 wordt waargenomen, wordt 0,1 μg/ml beoordeeld om de concentratie van de teststof te vinden die niet leidt tot een CD86-waarde boven de positieve drempelwaarde. Voor elke testrun wordt de EC150 (concentratie waarbij een chemische stof de positieve drempelwaarde van 150 % voor CD86 bereikt, zie punt 19) berekend als een positieve concentratierespons voor CD86 wordt waargenomen. Wanneer de teststof een positieve CD86-respons oplevert die geen verband houdt met de concentratie, is het mogelijk niet zinvol de EC150 te berekenen, zoals beschreven in U-SENS™ DB-ALM protocol nr. 183 (12). Voor elke testrun wordt, waar mogelijk, de CV70 (concentratie waarbij een chemische stof de drempelwaarde van 70 % voor cytotoxiciteit bereikt, zie punt 19) berekend (12). Om het concentratie-responseffect van CD86-toename te onderzoeken, moeten concentraties van de bruikbare concentraties die worden gekozen gelijkmatig verdeeld zijn tussen de EC150 (of de hoogste CD86-negatieve niet-cytotoxische concentratie) en de CV70 (of de hoogste toegestane concentratie, d.w.z. 200 μg/ml). Per testrun moeten ten minste vier concentraties worden getest, waarvan ten minste twee concentraties hetzelfde zijn als in de voorgaande testrun(s), met het oog op vergelijking.
|
|
15.
|
De oplosmiddel-/vehiculumcontrole die in de U-SENS™-test wordt gebruikt, is volledig medium (voor teststoffen die opgelost of stabiel gedispergeerd worden in volledig medium) (zie punt 4) of 0,4 % DMSO in volledig medium (voor teststoffen die opgelost of stabiel gedispergeerd worden in DMSO).
|
|
16.
|
De positieve controle die in de U-SENS™-test wordt gebruikt, is TNBS (zie punt 11), bereid in volledig medium. TNBS moet worden gebruikt als positieve controle voor de meting van de CD68-expressie met één enkele, uiteindelijke concentratie in de plaat (50 μg/ml) die een cellevensvatbaarheid > 70 % oplevert. Om een concentratie van 50 μg/ml TNBS in de plaat te verkrijgen, wordt 1 M (d.w.z. 293 mg/ml) stamoplossing van TNBS in volledig medium bereid, die weer 2 930 maal met volledig medium wordt verdund tot een werkoplossing van 100 μg/ml. Als negatieve controle moet melkzuur (CAS 50-21-5) worden gebruikt bij 200 μg/ml opgelost in volledig medium (van een stamvoorraad van 0,4 mg/ml). In elke plaat van elke testrun worden drie duplo’s van onbehandelde controle met volledig medium, oplosmiddel-/vehiculumcontrole, en negatieve en positieve controles bereid (12). Er kunnen andere geschikte positieve controles worden gebruikt als er historische gegevens beschikbaar zijn om vergelijkbare aanvaardbaarheidscriteria voor de testrun experimenten af te leiden. De aanvaardbaarheidscriteria voor de testrun zijn gelijk aan de aanvaardbaarheidscriteria voor de teststof (zie punt 12).
|
Het aanbrengen van de test- en controlestoffen
|
17.
|
De oplosmiddel-/vehiculumcontrole of de werkoplossingen die in de punten 14-16 worden beschreven, worden in een verhouding van 1:1 (v/v) gemengd met de celsuspensies die zijn geprepareerd in de 96-putjesplaat met vlakke bodem (zie punt 12). De behandelde platen worden vervolgens gedurende 45 ± 3 uur geïncubeerd bij 37 °C met 5 % CO2. Voorafgaand aan de incubatie worden de platen geseald met semipermeabel membraan om verdamping van vluchtige teststoffen en kruisbesmetting tussen behandelde cellen en teststoffen te voorkomen (12).
|
Celkleuring
|
18.
|
Na 45 ± 3 uur blootstelling worden de cellen overgebracht naar een V-vormige microtiterplaat en verzameld door centrifugatie. Oplosbaarheidsinterferentie wordt gedefinieerd als kristallen of druppels die 45 ± 3 uur na de behandeling (voorafgaand aan de celkleuring) onder de microscoop worden waargenomen. Het supernatans wordt afgevoerd en de resterende cellen worden eenmaal gewassen met 100 μl ijskoude fosfaatgebufferde zoutoplossing (PBS) die 5 % foetaal runderserum (kleuringsbuffer) bevat. Na het centrifugeren worden de cellen geresuspendeerd met 100 μl kleuringsbuffer en gedurende 30 minuten bij 4 °C gekleurd met 5 μl (bv. 0,25 μg) FITC-gelabelde anti-CD86- of muriene IgG1 (isotype)-antilichamen, beschermd tegen het licht. De in U-SENS™ DB-ALM protocol nr. 183 (12) beschreven antilichamen moeten worden gebruikt (voor CD86: BD-PharMingen nr. 5556 57, kloon: Fun-1, of Caltag/Invitrogen nr. MHCD8601, kloon: BU63; en voor IgG1: BD-PharMingen nr. 5557 48 of Caltag/Invitrogen nr. GM4992). De ervaring van de testontwikkelaars leert dat de fluorescentie-intensiteit van de antilichamen gewoonlijk consistent is tussen verschillende partijen. Er kan voor de bepaling ook gebruikgemaakt worden van andere klonen of leveranciers van antilichamen die de reactiviteitscontrole met goed gevolg hebben doorstaan (zie punt 11). Gebruikers kunnen echter overwegen de antilichamen in de omstandigheden van hun eigen laboratorium te titreren om de beste gebruiksconcentratie vast te stellen. Andere detectiesystemen, zoals met fluorochroom gelabelde anti-CD86-antilichamen, mogen worden gebruikt als zij aantoonbaar – bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 2.2 te testen – vergelijkbare resultaten opleveren als met FITC geconjugeerde antilichamen. De cellen worden eerst tweemaal gewassen met 100 μl kleuringsbuffer en eenmaal met 100 μl ijskoude PBS. Vervolgens worden ze geresuspendeerd in ijskoude PBS (bv. 125 μl voor monsters die handmatig buisje voor buisje worden geanalyseerd, of 50 μl bij gebruik van een autosamplerplaat) en wordt PI-oplossing toegevoegd (uiteindelijke concentratie 3 μg/ml). Andere cytotoxiciteitsmarkers, zoals 7-aminoactinomycine D (7-AAD) of trypaanblauw, mogen worden gebruikt als de alternatieve kleurstoffen aantoonbaar vergelijkbare resultaten opleveren als PI, bijvoorbeeld door de stoffen voor bekwaamheidstoetsing in aanhangsel 2.2 te testen.
|
Flowcytometrieanalyse
|
19.
|
Het expressieniveau van CD86 en de cellevensvatbaarheid worden geanalyseerd met behulp van flowcytometrie. De grootte (FSC) en de granulariteit (SSC) van de cellen wordt weergegeven in een spreidingsdiagram op een logaritmische schaal, teneinde de populatie in een eerste gate, R1, duidelijk vast te stellen en het debris te elimineren. Voor elke putje wordt een doeltotaal van 10 000 cellen in de R1-gate verzameld. Cellen uit dezelfde R1-gate worden weergegeven in een FL3- of FL4/SSC-spreidingsdiagram. Levensvatbare cellen worden afgebeeld door een tweede gate, R2, te plaatsen, die de populatie propidiumjodide-negatieve cellen selecteert (FL3- of FL4-kanaal). De cellevensvatbaarheid kan worden berekend door in het cytometeranalyseprogramma de onderstaande vergelijking te gebruiken. Wanneer de cellevensvatbaarheid laag is, kunnen er maximaal 20 000 cellen, met inbegrip van dode cellen, worden verzameld. Een andere mogelijkheid is dat gedurende één minuut na de start van de analyse gegevens worden verzameld.

Van deze levensvatbare cellen die gegatet zijn op R2 (binnen R1) wordt vervolgens het percentage FL1-positieve cellen gemeten. De expressie van CD86 op het celoppervlak wordt geanalyseerd in een FL1-/SSC-spreidingsdiagram dat gericht is op levensvatbare cellen (R2).
Voor de putjes met volledig medium/IgG1 wordt de analysemarker dicht bij de hoofdpopulatie gezet, zodat de controles met volledig medium IgG1 bevatten binnen het doelgebied van 0,6 to 0,9 %.
Kleurinterferentie wordt gedefinieerd als een verschuiving van het spreidingsdiagram van de FITC-gelabelde-IgG1 (geometrisch gemiddelde SI van IgG1 FL1 ≥ 150 %).
De stimuleringsindex (SI) van CD86 voor controlecellen (onbehandeld of in 0,4 % DMSO) en met de chemische stof behandelde cellen wordt berekend aan de hand van de volgende vergelijking:.

% van onbehandelde IgG1+-controlecellen: aangeduid als percentage FL1-positieve IgG1-cellen die binnen de levensvatbare onbehandelde cellen zijn gedefinieerd met de analysemarker (aanvaard bereik van ≥ 0,6 % en < 1,5 %, zie punt 22).
% van IgG1+/CD86+-controlecellen/behandelde cellen: aangeduid als percentage FL1-positieve IgG1/CD86-cellen, gemeten zonder de analysemarker binnen de levensvatbare controlecellen/behandelde cellen te verplaatsen.
|
GEGEVENS EN RAPPORTAGE
Evaluatie van de gegevens
|
20.
|
In de U-SENS™-test worden de volgende parameters berekend: de CV70-waarde, d.w.z. een concentratie waarbij 70 % van de U937-cellen overleeft (30 % cytotoxiciteit) en de EC150-waarde, d.w.z. de concentratie waarbij de teststoffen een CD86-SI van 150 % tot resultaat hadden.
De CV70 wordt berekend door middel van loglineaire interpolatie aan de hand van de volgende vergelijking:
CV70 = C1 + [(V1 – 70) / (V1 – V2) * (C2 – C1)]
waarbij:
V1 de minimumwaarde is van cellevensvatbaarheid hoger dan 70 %
V2 de maximumwaarde is van cellevensvatbaarheid lager dan 70 %
C1 en C2 de concentraties zijn die de waarde van respectievelijk cellevensvatbaarheid V1 en V2 vertonen.

Er mogen andere methoden worden gebruikt om de CV70 af te leiden, zolang wordt aangetoond dat dit niet van invloed is op de resultaten (bv. door de stoffen voor bekwaamheidstoetsing te testen).
De EC150 wordt berekend door middel van loglineaire interpolatie aan de hand van de volgende vergelijking:
EC150 = C1 + [(150 – SI1) / (SI2 - SI1) * (C2 - C1)]
waarbij:
C1 de hoogste concentratie in μg/ml is met een CD86-SI < 150 % (SI1)
C2 de laagste concentratie in μg/ml is met een CD86-SI ≥ 150 % (SI2).

De EC150- en CV70-waarden worden berekend
|
—
|
voor elke testrun: de afzonderlijke EC150- en CV70-waarden worden gebruikt als instrument om het concentratie-responseffect van CD86-toename te onderzoeken (zie punt 14),
|
|
—
|
op grond van de gemiddelde levensvatbaarheden wordt de totale CV70 bepaald (12),
|
|
—
|
op grond van de gemiddelde SI van CD86-waarden wordt de totale EC150 bepaald voor de teststof waarvan de U-SENS™ voorspelde dat deze POSITIEF zou zijn (zie punt 21) (12).
|
|
Voorspellingsmodel
|
21.
|
Voor de meting van de CD86-expressie wordt elke teststof in ten minste vier concentraties en in ten minste twee onafhankelijke testruns (uitgevoerd op verschillende dagen) getest om één enkele voorspelling af te leiden (NEGATIEF of POSITIEF).
|
—
|
De afzonderlijke uitslag van een U-SENS™-testrun wordt negatief geacht (hierna N genoemd) als de SI van CD86 lager is dan 150 % bij alle niet-toxische concentraties (cellevensvatbaarheid ≥ 70 %) en als geen interferentie wordt waargenomen (cytotoxiciteit, oplosbaarheid: zie punt 18 of kleur: zie punt 19, ongeacht de niet-cytotoxische concentraties waarbij de interferentie wordt gedetecteerd). In alle andere gevallen: SI van CD86 hoger dan of gelijk aan 150 % en/of interferenties waargenomen, wordt de afzonderlijke uitslag van een U-SENS™-testrun positief geacht (hierna P genoemd).
|
|
—
|
Een U-SENS™-voorspelling wordt NEGATIEF geacht als ten minste twee onafhankelijke testruns negatief (N) zijn (figuur 1). Als de eerste twee testruns beide negatief (N) zijn, wordt de U-SENS™-voorspelling NEGATIEF geacht en is een derde testrun niet nodig.
|
|
—
|
Een U-SENS™-voorspelling wordt POSITIEF geacht als ten minste twee onafhankelijke testruns positief (P) zijn (figuur 1). Als de eerste twee testruns positief (P) zijn, wordt de U-SENS™-voorspelling POSITIEF geacht en is een derde testrun niet nodig.
|
|
—
|
Een uitzondering hierop, omdat er geen dosisbepalingstest wordt uitgevoerd, is wanneer de SI van CD86 in de eerste testrun uitsluitend bij de hoogste niet-cytotoxische concentratie hoger dan of gelijk aan 150 % is. In dat geval wordt de testrun TWIJFELACHTIG (T) geacht en moeten in extra testruns aanvullende concentraties worden getest (tussen de hoogste niet-toxische concentratie en de laagste toxische concentratie – zie punt 20). Als een testrun als T wordt aangemerkt, moeten ten minste twee aanvullende testruns worden uitgevoerd, en een vierde testrun als de testruns 2 en 3 niet overeenstemmen (N en/of P zelfstandig) (figuur 1). Vervolgtestruns worden positief geacht zelfs als slechts één niet-cytotoxische concentratie een CD86 gelijk aan of hoger dan 150 % oplevert, omdat de vastgestelde concentratie voor de specifieke teststof is aangepast. De uiteindelijke voorspelling is gebaseerd op het resultaat van de meerderheid van de drie of vier afzonderlijke testruns (d.w.z. twee van de drie of twee van de vier) (figuur 1).
|

Figuur 1: voorspellingsmodel dat wordt gebruikt in de U-SENS™-test. Een U-SENS™-voorspelling moet worden bekeken in het kader van een IATA en in overeenstemming met hetgeen bepaald is in punt 4 en in de punten 7, 8 en 9 van de algemene inleiding.
N: testrun waarin geen CD86-positief of interferentie wordt waargenomen;
P: terstrun waarin CD86-positief of interferentie wordt waargenomen;
T: twijfelachtig. Eerste testrun met uitslag „Twijfelachtig” uitsluitend wanneer CD86 positief is bij de hoogste niet-toxische concentratie;
#: uitsluitend in de eerste testrun maakt een afzonderlijke uitslag „Twijfelachtig” (T) automatisch een derde testrun noodzakelijk om een meerderheid van Positieve (P) of Negatieve (N) uitslagen in ten minste twee van de drie onafhankelijke testruns te bereiken.
$: de vakken tonen de relevante combinaties van resultaten van de drie testruns op basis van de resultaten in de eerste twee testruns die in het bovenstaande vak worden getoond.
°: de vakken tonen de relevante combinaties van resultaten van de vier testruns op basis van de resultaten in de eerste drie testruns die in het bovenstaande vak worden getoond.
|
Aanvaardbaarheidscriteria
|
22.
|
Bij gebruik van de U-SENS™-test moet aan de volgende aanvaardbaarheidscriteria worden voldaan (12).
|
—
|
Aan het eind van de blootstelling van 45 ± 3 uur moet de gemiddelde levensvatbaarheid van onbehandelde U937-cellen in drievoud > 90 % zijn en mag er geen verschuiving in CD86-expressie worden waargenomen. De basale CD86-expressie van onbehandelde U937-cellen moet binnen een bereik van ≥ 2 % en ≤ 25 % liggen.
|
|
—
|
Wanneer als oplosmiddel DMSO wordt gebruikt, wordt de validiteit van de controle met DMSO-vehiculum beoordeeld door de DMSO-SI in vergelijking met onbehandelde cellen te berekenen, en de gemiddelde cellevensvatbaarheid in drievoud moet > 90 % zijn. De controle met DMSO-vehiculum is geldig als de gemiddelde waarde van de CD86-SI in drievoud ervan kleiner is dan 250 % van het gemiddelde van de CD86-SI in drievoud van onbehandelde U937-cellen.
|
|
—
|
De testruns worden geldig geacht als ten minste twee van de drie IgG1-waarden van onbehandelde U937-cellen binnen een bereik van ≥ 0,6 % en < 1,5 % liggen.
|
|
—
|
De tegelijkertijd geteste negatieve controle (melkzuur) wordt geldig geacht als ten minste twee van de drie duplo’s negatief (CD86-SI < 150 %) en niet-cytotoxisch (cellevensvatbaarheid ≥ 70 %) zijn.
|
|
—
|
De positieve controle (TNBS) wordt geldig geacht als ten minste twee van de drie duplo’s positief (CD86-SI ≥ 150 %) en niet-cytotoxisch (cellevensvatbaarheid ≥ 70 %) zijn.
|
|
Testverslag
|
23.
|
In het testverslag moet de volgende informatie worden opgenomen.
|
Teststof
Stof die uit één component bestaat
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, oplosbaarheid in volledig medium, oplosbaarheid in DMSO, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
Stof die uit meerdere componenten bestaat, UVCB en mengsel:
|
karakterisering, voor zover mogelijk aan de hand van bv. chemische identiteit (zie hierboven), zuiverheid, kwantitatief voorkomen en relevante fysisch-chemische eigenschappen (zie hierboven) van de bestanddelen, voor zover beschikbaar;
|
|
fysisch voorkomen, oplosbaarheid in volledig medium, oplosbaarheid in DMSO en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
molecuulgewicht of schijnbaar molecuulgewicht in het geval van mengsels/polymeren waarvan de samenstelling bekend is, of andere informatie die relevant is voor het verloop van de studie;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
|
|
Controles
Positieve controle
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, oplosbaarheid in DMSO, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn en voor zover van toepassing;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
verwijzing naar historische positievecontroleresultaten waaruit de geschiktheid van de aanvaardbaarheidscriteria voor de testrun blijkt, indien van toepassing.
|
Negatieve controle en oplosmiddel-/vehiculumcontrole
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
fysisch voorkomen, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, in het geval er andere controle-oplosmiddelen/-vehicula worden gebruikt dan diegene die worden vermeld in de testrichtlijn, en voor zover beschikbaar;
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof.
|
|
|
Testomstandigheden
|
naam en adres van de opdrachtgever, testinstallatie en de onderzoeksleider;
|
|
een beschrijving van de gebruikte test;
|
|
gebruikte cellijn, omstandigheden waarin die werd bewaard en oorsprong (bv. de instelling waarvan de cellijn werd verkregen);
|
|
gebruikte flowcytometrie (bv. model), met inbegrip van de gebruikte instrumentinstellingen, antilichamen en cytotoxiciteitsmarker;
|
|
de procedure die gebruikt is om aan te tonen dat het laboratorium bekwaam is om de test uit te voeren, door de stoffen voor bekwaamheidstoetsing te testen, en de procedure die gebruikt is om aan te tonen dat de resultaten van de test in de loop van de tijd kunnen worden gereproduceerd, bv. controlegegevens uit het verleden en/of gegevens van reactiviteitscontroles uit het verleden.
|
|
|
Aanvaardbaarheidscriteria voor de test
|
Cellevensvatbaarheid en CD86-SI-waarden die met de oplosmiddel-/vehiculumcontrole zijn verkregen in vergelijking met het aanvaardbaarheidsbereik;
|
|
cellevensvatbaarheid en SI-waarden die met de positieve controle zijn verkregen in vergelijking met het aanvaardbaarheidsbereik;
|
|
cellevensvatbaarheid van alle geteste concentraties van de teststof.
|
|
|
Werkwijze
|
aantal gebruikte testruns;
|
|
de gehanteerde concentraties van de teststof, toedienings- en blootstellingstijd (indien verschillend van de aanbevolen tijd);
|
|
duur van de blootstelling;
|
|
een beschrijving van de gebruikte beoordelings- en beslissingscriteria;
|
|
een beschrijving van eventuele modificaties van de testprocedure.
|
|
|
Resultaten
|
tabel van de gegevens, waaronder CV70 (indien van toepassing), SI, cellevensvatbaarheidswaarden, EC150-waarden (indien van toepassing) die zijn verkregen voor de teststof en voor de positieve controle in elke testrun, en een indicatie van de score van de teststof volgens het voorspellingsmodel;
|
|
een beschrijving van eventuele andere relevante waarnemingen.
|
|
|
Bespreking van de resultaten
|
bespreking van de resultaten verkregen met de U-SENS™-test;
|
|
overweging van de testresultaten in de context van een IATA, als er andere relevante informatie beschikbaar is.
|
|
Conclusies
|
LITERATUUR
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL CEVMA (2017). The U-SENS™ test method Validation Study Report. Beschikbaar op: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL CEVMA (2016). ESAC Opinion No 2016-03 on the L’Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Beschikbaar op: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL CEVMA (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Beschikbaar op: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OESO (2016). Guidance Document on the Reporting of Defined Approaches and Individual Information Sources to be Used within Integrated Approaches to Testing and Assessment (IATA) for Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OESO (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Beschikbaar op: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OESO (2005). Series on Testing and Assessment No 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
Verenigde Naties (VN) (2015). Mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (GHS). ST/SG/AC.10/30/Rev.6, Zesde herziene editie, New York en Genève: United Nations Publications. Beschikbaar op: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OESO (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Beschikbaar op: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Aanhangsel 2.1
DEFINITIES
AOP (Adverse Outcome Pathway)
: een reeks gebeurtenissen van de chemische structuur van een doelstof of een groep soortgelijke chemische stoffen, van de moleculaire inleidende gebeurtenis tot een te onderzoeken in-vivoresultaat (15).
Betrouwbaarheid
: indicatie van de mate waarin een test, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (14).
CD86-concentratierespons
: Er is sprake van concentratieafhankelijkheid (of concentratierespons) wanneer een positieve concentratie CD86-SI ≥ 150) gevolgd wordt door een concentratie met een hogere CD86-SI.
Chemische stof
: een stof of een mengsel.
CV70
: de geschatte concentratie waarbij de cellevensvatbaarheid 70 % is.
EC150
: de geschatte concentraties waarbij de SI van CD86-expressie 150 % is.
Flowcytometrie
: een cytometrische techniek waarbij in een vloeistof gesuspendeerde cellen één voor één door een brandpunt van exciterend licht stromen, dat wordt verstrooid in patronen die kenmerkend zijn voor de cellen en hun bestanddelen; de cellen worden vaak gelabeld met fluorescente markers, zodat het licht eerst wordt geabsorbeerd en vervolgens met gewijzigde frequenties wordt uitgezonden.
Geldige test
: een test die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen. Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel (14).
Gevaar
: inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld.
Gevoeligheid
: het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert en is een belangrijke overweging bij de beoordeling van de relevantie van een test (14).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN))
: een systeem voor de indeling van chemische producten (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar worden gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (16).
IATA (Integrated Approach on Testing and Assessment, geïntegreerde test- en beoordelingsbenadering)
: een gestructureerde aanpak die wordt gebruikt voor de identificatie van gevaren (potentieel), het kenmerken van gevaren (potentie) en/of het beoordelen van de veiligheid (potentieel/potentie en blootstelling) van een chemische stof of een groep chemische stoffen, waarbinnen alle relevante gegevens strategisch worden geïntegreerd en worden afgewogen als inbreng voor regelgevingsbesluiten inzake potentiële gevaren en/of risico’s en/of de noodzaak van verdere gerichte en bijgevolg minimale tests.
Kleuringsbuffer
: een fosfaatgebufferde zoutoplossing met 5 % foetaal kalverserum.
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
Nauwkeurigheid
: de mate van overeenstemming tussen testresultaten en erkende referentiewaarden. Het is een maat voor de prestaties van de test en één aspect van relevantie. Deze term en de term „concordantie”, waaronder het percentage correcte uitkomsten van een test wordt verstaan, worden vaak door elkaar gebruikt (14).
Oplosmiddel-/vehiculumcontrole
: een onbehandeld monster dat alle componenten bevat van een testsysteem met uitzondering van de teststof, maar met inbegrip van het gebruikte oplosmiddel/vehiculum. Dit monster wordt gebruikt om de referentierespons vast te stellen voor de monsters die worden behandeld met de in hetzelfde oplosmiddel/vehiculum opgeloste of stabiel gedispergeerde teststof. Als dit monster getest wordt met een tegelijkertijd gemeten mediumcontrole, toont het ook aan of het oplosmiddel/vehiculum met het testsysteem interageert.
Positieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positievecontrolerespons in de loop van de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn.
Prehaptenen
: chemische stoffen die in sensibiliserende stoffen veranderen na abiotische transformatie, bv. door oxidatie.
Prohaptenen
: chemische stoffen die enzymatische activering nodig hebben om hun potentieel voor huidsensibilisatie te realiseren.
Relevantie
: beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een test (14).
SI
: stimuleringsindex. Relatieve waarden van de geometrisch gemiddelde fluorescentie-intensiteit in de met de chemische stof behandelde cellen vergeleken met de met oplosmiddel behandelde cellen.
Specificiteit
: het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert en is een belangrijke overweging bij de beoordeling van de relevantie van een test (14).
Stof
: een chemisch element en de verbindingen ervan, zoals zij voorkomen in natuurlijke toestand of bij de productie ontstaan, met inbegrip van alle additieven die nodig zijn voor het behoud van de stabiliteit ervan en alle onzuiverheden ten gevolge van het toegepaste procedé, doch met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder aantasting van de stabiliteit van de stof of wijziging van de samenstelling ervan.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één hoofdcomponent aanwezig is in een concentratie van ≥ 10 % (w/w) en < 80 % (w/w). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Testrun
: een testrun bestaat uit het gelijktijdig testen van een of meer chemische stoffen met een oplosmiddel-/vehiculumcontrole en met een positieve controle.
Teststof
: alle volgens deze test geteste stoffen of mengsels.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
Verschuiving
: een verschuiving wordt als volgt gedefinieerd i) de gecorrigeerde %CD86+-waarde van de onbehandelde controleduplo 3 is minder dan 50 % van het gemiddelde van de gecorrigeerde %CD86+-waarde van de onbehandelde controleduplo’s 1 en 2, en ii) de gecorrigeerde %CD86+-waarde van de negatievecontroleduplo 3 is minder dan 50 % van het gemiddelde van de gecorrigeerde %CD86+-waarde van de negatievecontroleduplo’s 1 en 2.
Aanhangsel 2.2
STOFFEN VOOR BEKWAAMHEIDSTOETSING
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen door op de juiste wijze de verwachte U-SENS™-voorspelling te verkrijgen voor de 10 stoffen die in tabel 1 worden aanbevolen, en door de CV70- en EC150-waarden te verkrijgen die binnen de respectieve referentiebereiken vallen voor ten minste 8 van de 10 stoffen voor bekwaamheidstoetsing. De stoffen voor bekwaamheidstoetsing zijn zodanig geselecteerd dat ze alle mogelijke responsen voor huidsensibilisatiegevaar weerspiegelen. Andere selectiecriteria waren dat stoffen in de handel beschikbaar zijn, en dat er in-vivoreferentiegegevens en met de U-SENS™-test gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn. Voor de U-SENS™-test zijn tevens gepubliceerde referentiegegevens beschikbaar (1) (8).
Tabel 1
Aanbevolen stoffen voor het aantonen van technische bekwaamheid met de U-SENS™-test
|
Stoffen voor bekwaamheidstoetsing
|
CAS RN
|
Fysische toestand
|
In-vivovoorspelling (106)
|
U-SENS Oplosmiddel/Vehiculum
|
U-SENS CV70 Referentie-bereik in μg/ml (107)
|
U-SENS EC150 Referentie-bereik in μg/ml (107)
|
|
4-Fenyleendiamine
|
106-50-3
|
Vaste stof
|
Sensibiliserend (sterk)
|
Volledig medium (108)
|
< 30
|
Positief (≤ 10)
|
|
Picrylsulfonzuur
|
2508-19-2
|
Vloeistof
|
Sensibiliserend (sterk)
|
Volledig medium
|
> 50
|
Positief (≤ 50)
|
|
Diëthylmaleaat
|
141-05-9
|
Vloeistof
|
Sensibiliserend (matig)
|
DMSO
|
10-100
|
Positief (≤ 20)
|
|
Resorcinol
|
108-46-3
|
Vaste stof
|
Sensibiliserend (matig)
|
Volledig medium
|
> 100
|
Positief (≤ 50)
|
|
Cinnamylalcohol
|
104-54-1
|
Vaste stof
|
Sensibiliserend (zwak)
|
DMSO
|
> 100
|
Positief (10-100)
|
|
4-Allylanisol
|
140-67-0
|
Vloeistof
|
Sensibiliserend (zwak)
|
DMSO
|
> 100
|
Positief (< 200)
|
|
Sacharine
|
81-07-2
|
Vaste stof
|
Niet-sensibiliserend
|
DMSO
|
> 200
|
Negatief (> 200)
|
|
Glycerol
|
56-81-5
|
Vloeistof
|
Niet-sensibiliserend
|
Volledig medium
|
> 200
|
Negatief (> 200)
|
|
Melkzuur
|
50-21-5
|
Vloeistof
|
Niet-sensibiliserend
|
Volledig medium
|
> 200
|
Negatief (> 200)
|
|
Salicylzuur
|
69-72-7
|
Vaste stof
|
Niet-sensibiliserend
|
DMSO
|
> 200
|
Negatief (> 200)
|
|
Afkortingen: CAS RN = registratienummer van de Chemical Abstracts Service
|
Aanhangsel 3
IN-VITROHUIDSENSIBILISATIE: IL-8 LUC-BEPALING
INLEIDENDE OVERWEGINGEN EN BEPERKINGEN
|
1.
|
In tegenstelling tot bepalingen die de expressie van celoppervlaktemarkers analyseren, kwantificeert de IL-8 Luc-bepaling veranderingen in de expressie van IL-8, een cytokine die in verband wordt gebracht met de activatie van dendritische cellen. In de van THP-1 afgeleide IL-8-reportercellijn (THP-G8, afkomstig van de humane acute-monocytischeleukemiecellijn THP-1) wordt IL-8-expressie gemeten na blootstelling aan sensibiliserende stoffen (1). De expressie van luciferase wordt vervolgens gebruikt om onderscheid te maken tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen.
|
|
2.
|
De IL-8 Luc-bepaling is in een valideringsstudie (2) beoordeeld door het Japans Centrum voor de validering van alternatieve methoden (JaCVAM), het Japanse ministerie van Economische Zaken, Handel en Industrie en de Japanse Vereniging voor alternatieven voor dierproeven, en vervolgens onderworpen aan een onafhankelijke beoordeling door vakgenoten (3) onder auspiciën van JaCVAM en het ministerie voor Volksgezondheid, Werkgelegenheid en Welzijn met steun van de International Cooperation on Alternative Test Methods (ICATM). Op grond van al het beschikbare bewijsmateriaal en de inbreng van regelgevers en belanghebbenden wordt de IL-8 Luc-bepaling nuttig geacht als onderdeel van een IATA om onderscheid te maken tussen huidsensibiliserende stoffen en niet-sensibiliserende stoffen met het oog op de gevarenindeling en etikettering. Voorbeelden van het gebruik van IL-8 Luc-gegevens in combinatie met andere informatie zijn beschreven in de literatuur (4) (5) (6).
|
|
3.
|
De IL-8 Luc-bepaling bleek overdraagbaar te zijn naar laboratoria met ervaring in het kweken van cellen en luciferasemeting. De reproduceerbaarheid binnen laboratoria en tussen verschillende laboratoria was respectievelijk 87,7 % en 87,5 % (2). Uit gegevens uit de valideringsstudie (2) en andere publicaties (1) (6) blijkt dat met de IL-8 Luc-bepaling, in vergelijking met de LLKT, 118 van de 143 chemische stoffen als positief of negatief werden beoordeeld en 25 chemische stoffen als twijfelachtig, en dat de nauwkeurigheid van de IL-8 Luc-bepaling in het onderscheiden van huidsensibiliserende stoffen (VN-GHS/CLP cat. 1) van niet-sensibiliserende stoffen (VN-GHS/CLP geen categorie) 86 % is (101/118) met een gevoeligheid van 96 % (92/96) en een specificiteit van 41 % (9/22). Als de stoffen die buiten het hierna beschreven toepassingsgebied vallen (punt 5), buiten beschouwing worden gelaten, werden met de IL-8 Luc-bepaling 113 van de 136 chemische stoffen als positief of negatief beoordeeld en 23 chemische stoffen als twijfelachtig, en is de nauwkeurigheid van de IL-8 Luc-bepaling 89 % (101/113) met een gevoeligheid van 96 % (92/96) en een specificiteit van 53 % (9/17). Uitgaande van bij mensen verzamelde gegevens die worden aangehaald in Urbisch et al. (7), werden met de IL-8 Luc-bepaling 76 van de 90 chemische stoffen als positief of negatief beoordeeld en 14 chemische stoffen als twijfelachtig, en is de nauwkeurigheid 80 % (61/76), de gevoeligheid 93 % (54/58) en de specificiteit 39 % (7/18). Als de stoffen die buiten het toepassingsgebied vallen, buiten beschouwing worden gelaten, werden met de IL-8 Luc-bepaling 71 van de 84 chemische stoffen als positief of negatief beoordeeld en 13 chemische stoffen als twijfelachtig, en is de nauwkeurigheid 86 % (61/71), de gevoeligheid 93 % (54/58) en de specificiteit 54 % (7/13). Bij de IL-8 Luc-bepaling is de kans dat valsnegatieve voorspellingen betrekking hebben op chemische stoffen met een lage tot gemiddelde potentie voor huidsensibilisatie (VN-GHS/CLP-subcategorie 1B) hoger dan dat ze betrekking hebben op chemische stoffen met een hoge potentie voor huidsensibilisatie (VN-GHS/CLP-subcategorie 1A) (6). Bij elkaar genomen rechtvaardigt de informatie dat voor de IL-8 Luc-bepaling een rol is weggelegd bij het bepalen van het gevaar van huidsensibilisatie. De nauwkeurigheid die hier wordt vermeld voor de IL-8 Luc-bepaling als losstaande test vormt slechts een indicatie, aangezien de test moet worden bekeken in combinatie met andere informatiebronnen in de context van een IATA en in overeenstemming met hetgeen hierboven bepaald is in de punten 7 en 8 in de algemene inleiding. Bovendien moet bij de beoordeling van tests zonder dierproeven voor huidsensibilisatie in gedachten worden gehouden dat de LLKT en andere dierproeven de situatie van mensen mogelijk niet volledig weerspiegelen.
|
|
4.
|
Op basis van de thans beschikbare gegevens is gebleken dat de IL-8 Luc-bepaling van toepassing is op teststoffen die verschillende organische functionele groepen, reactiemechanismen, potenties voor huidsensibilisatie (zoals bepaald bij in-vivo-onderzoek) en fysisch-chemische eigenschappen beslaan (2) (6).
|
|
5.
|
Bij de IL-8 Luc-bepaling wordt X-VIVO™ 15 als oplosmiddel gebruikt. Met de bepaling werden chemische stoffen met een log Kow > 3,5 en chemische stoffen met een oplosbaarheid in water van ongeveer 100 μg/ml, zoals berekend door EPI Suite™, echter correct beoordeeld. Daarnaast detecteert de bepaling sensibiliserende stoffen die slecht oplosbaar zijn in water, beter dan de IL-8 Luc-bepaling waarbij dimethylsulfoxide (DMSO) als oplosmiddel wordt gebruikt (2). Teststoffen die niet worden opgelost bij 20 mg/ml, kunnen echter valsnegatieve uitslagen opleveren, omdat ze niet oplossen in X-VIVO™ 15. Voor deze chemische stoffen moeten negatieve resultaten dus buiten beschouwing worden gelaten. In de valideringsstudie werd een hoog percentage valsnegatieven waargenomen voor anhydriden. Bovendien zouden prohaptenen (stoffen die metabole activering nodig hebben) en prehaptenen (stoffen die worden geactiveerd door oxidatie aan de lucht) negatieve resultaten kunnen opleveren vanwege de beperkte metabole capaciteit van de cellijn (8) en als gevolg van de testomstandigheden. Hoewel negatieve uitslagen voor vermoedelijke prehaptenen/prohaptenen dus met de nodige voorzichtigheid geïnterpreteerd moeten worden, werden niettemin 11 van de 11 prehaptenen, 6/6 prohaptenen en 6/8 prehaptenen/prohaptenen in de gegevensreeks van de IL-8 Luc-bepaling correct beoordeeld (2). Op basis van het recente uitgebreide overzicht van drie tests zonder dierproeven (de DPRA, de KeratinoSens™ en de h-CLAT) voor het detecteren van pre- en prohaptenen (9), en op basis van het feit dat de THP-G8-cellen die in de IL-8 Luc-bepaling worden gebruikt, een cellijn vormen die is afgeleid van de cellijn THP-1 die in de h-CLAT wordt gebruikt, kan de IL-8 Luc-bepaling ook bijdragen aan een grotere gevoeligheid van tests zonder dierproeven bij het detecteren van pre- en prohaptenen in combinatie met andere tests. Tot dusver leverden oppervlakteactieve stoffen (vals)positieve resultaten op, ongeacht tot welk type ze behoorden (bv. kationisch, anionisch of niet-ionisch). Tot slot kunnen teststoffen die interfereren met luciferase de luciferaseactiviteit/-meting verstoren, doordat zij een schijnbare remming of verhoging van de luminescentie veroorzaken (10). Zo werd bij andere reportergenbepalingen op basis van luciferase gemeld dat fyto-oestrogeenconcentraties boven 1 μM interfereerden met luminescentiesignalen vanwege overactiviteit van het luciferasereportergen. De luciferase-expressie bij hoge concentraties fyto-oestrogenen of soortgelijke verbindingen waarvan vermoed wordt dat zij fyto-oestrogeenachtige activatie van het luciferasereportergen kunnen veroorzaken, moet daarom zorgvuldig worden bestudeerd (11). Op grond van het bovenstaande vallen oppervlakteactieve stoffen, anhydriden en chemische stoffen die interfereren met luciferase buiten het toepassingsgebied van deze bepaling. In gevallen waarin kan worden aangetoond dat de IL-8 Luc-bepaling niet toepasselijk is op andere specifieke categorieën teststoffen, mag de testmethode niet worden gebruikt voor die specifieke categorieën.
|
|
6.
|
Zoals hierboven beschreven, helpt de IL-8 Luc-bepaling om huidsensibiliserende stoffen te onderscheiden van niet-sensibiliserende stoffen. Nader onderzoek, bij voorkeur op basis van bij mensen verzamelde gegevens, is nodig om te bepalen of IL-8 Luc-resultaten, in combinatie met andere informatiebronnen, kunnen bijdragen aan potentiebeoordeling.
|
|
7.
|
Definities worden gegeven in aanhangsel 3.1.
|
PRINCIPE VAN DE TEST
|
8.
|
De IL-8 Luc-bepaling maakt gebruik van een humane monocytischeleukemiecellijn THP-1, afkomstig van de American Type Culture Collection (Manassas, VA, VS). Deze cellijn werd door de faculteit Dermatologie van de Tohoku University School of Medicine in Japan gebruikt om een van THP-1 afgeleide IL-8-reportercellijn, THP-G8, te maken. THP-G8 omvat de luciferasegenen Stable Luciferase Orange (SLO) en Stable Luciferase Red (SLR), die worden gecontroleerd door de promotors van respectievelijk IL-8 en glyceraldehyde-3-fosfaatdehydrogenase (GAPDH) (1). Daardoor wordt een kwantitatieve meting via luminescentiedetectie van de inductie van het luciferasegen met behulp van gangbare lichtproducerende luciferasesubstraten mogelijk, als indicator van de activiteit van IL-8 en GAPDH in cellen die zijn blootgesteld aan sensibiliserende stoffen.
|
|
9.
|
Het op twee kleuren gebaseerde testsysteem omvat een oranje licht uitzendende luciferase (SLO; λmax = 580 nm) (12) voor de genexpressie van de IL-8-promotor, alsmede een rood licht uitzendende luciferase (SLR; λmax = 630 nm) (13) voor de genexpressie van de internecontrolepromotor, GAPDH. De twee luciferasen zenden, wanneer ze reageren met het in vuurvliegen voorkomende d-luciferine, verschillende kleuren licht uit en hun luminescentie wordt gelijktijdig gemeten in een enkelvoudige reactie, door met behulp van een optisch filter de emissie van het testmengsel te scheiden (14) (aanhangsel 3.2).
|
|
10.
|
THP-G8-cellen worden gedurende 16 uur met de teststof behandeld. Vervolgens worden de SLO-luciferase-activiteit (SLO-LA) die de activiteit van de IL-8-promotor weerspiegelt, en de SLR-luciferase-activiteit (SLR-LA), die de activiteit van de GAPDH-promotor weerspiegelt, gemeten. Met het oog op de begrijpelijkheid worden SLO-LA en SLR-LA aangeduid als respectievelijk IL8LA en GAPLA. In tabel 1 worden de termen beschreven die betrekking hebben op luciferase-activiteit in de IL-8 Luc-bepaling. De gemeten waarden worden gebruikt als een indicator voor cytotoxiciteit en om het volgende te berekenen: de genormaliseerde IL8LA (nIL8LA), d.w.z. de verhouding van IL8LA tot GAPLA; de inductie van nIL8LA (Ind-IL8LA), d.w.z. de verhouding van de rekenkundige gemiddelden van viervoudig gemeten waarden van de nIL8LA van THP-G8-cellen die zijn behandeld met een teststof, en de waarden van de nIL8LA van onbehandelde THP-G8-cellen, en de remming van GAPLA (Inh-GAPLA), d.w.z. de verhouding van de rekenkundige gemiddelden van viervoudig gemeten waarden van de GAPLA van THP-G8-cellen die zijn behandeld met een teststof, en de waarden van de GAPLA van onbehandelde THP-G8-cellen. Inh-GAPLA wordt gebruikt als een indicator voor cytotoxiciteit.
Tabel 1
Beschrijving van termen die betrekking hebben op de luciferase-activiteit in de IL-8 Luc-bepaling
|
Afkortingen
|
Definitie
|
|
GAPLA
|
SLR-luciferase-activiteit, die de activiteit van de GAPDH-promotor weerspiegelt
|
|
IL8LA
|
SLO-luciferase-activiteit, die de activiteit van de IL-8-promotor weerspiegelt
|
|
nIL8LA
|
IL8LA / GAPLA
|
|
Ind-IL8LA
|
nIL8LA van met chemische stoffen behandelde THP-G8-cellen / nIL8LA van onbehandelde cellen
|
|
Inh-GAPLA
|
GAPLA van met chemische stoffen behandelde THP-G8-cellen / GAPLA van onbehandelde cellen
|
|
CV05
|
De laagste concentratie van de chemische stof waarbij de Inh-GAPLA < 0,05 wordt.
|
|
|
11.
|
Er zijn prestatienormen (15) beschikbaar om de validering van gewijzigde in-vitro-IL-8-luciferasetests die vergelijkbaar zijn met de IL-8 Luc-bepaling, te vergemakkelijken en tijdige wijziging van OESO-testrichtlijn 442E met het oog op de opname van dergelijke tests mogelijk te maken. De wederzijdse aanvaarding van gegevens in overeenstemming met de OESO-overeenkomst is enkel gewaarborgd voor tests die zijn gevalideerd volgens de prestatienormen, voor zover die tests door de OESO zijn geëvalueerd en opgenomen in testrichtlijn 442E (16).
|
AANTONING VAN BEKWAAMHEID
|
12.
|
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen met de 10 stoffen voor bekwaamheidstoetsing die in aanhangsel 3.3 worden genoemd, met inachtneming van de OESO-leidraad „Good in vitro Method Practices” (17). Gebruikers van de test moeten bovendien een historische gegevensbank bijhouden van de gegevens die het resultaat zijn van de reactiviteitscontroles (zie punt 15), de positieve controles en oplosmiddel-/vehiculumcontroles (zie punten 21-24). Zij moeten deze gegevens gebruiken om te bevestigen dat de reproduceerbaarheid van de test in hun laboratorium in de loop van de tijd gehandhaafd blijft.
|
PROCEDURE
|
13.
|
De standaardwerkwijze voor de IL-8 Luc-bepaling is beschikbaar en moet worden gevolgd wanneer de test wordt uitgevoerd (18). Laboratoria die de test willen uitvoeren, kunnen de recombinante THP-G8-cellijn verkrijgen bij GPC Lab. Co. Ltd., Tottori, Japan, na ondertekening van een overeenkomst inzake overdracht van materiaal (MTA) in overeenstemming met de voorwaarden van het OESO-model. In de volgende punten worden de belangrijkste onderdelen en procedures van de bepaling beschreven.
|
Celpreparaten
|
14.
|
Voor het uitvoeren van de IL-8 Luc-test moet de THP-G8-cellijn van GPC Lab.Co. Ltd., Tottori, Japan, worden gebruikt (zie punten 8 en 13). Bij ontvangst worden de cellen vermeerderd (2-4 passages) en in bevroren toestand bewaard als homogene stam. Cellen van deze stam kunnen maximaal 12 passages of maximaal 6 weken worden vermeerderd. Het medium dat voor het vermeerderen wordt gebruikt, is het RPMI-1640-kweekmedium met 10 % foetaal runderserum (FBS), antibiotica/antimycotica-oplossing (100 U/ml penicilline G, 100 μg/ml streptomycine en 0,25 μg/ml amfotericine B in 0,85 % zoutoplossing) (bv. GIBCO cat. nr. 15240-062), 0,15 μg/ml puromycine (bv. CAS:58-58-2) en 300 μg/ml G418 (bv. CAS:108321-42-2).
|
|
15.
|
Voordat de cellen voor tests worden gebruikt, moet hun geschiktheid worden vastgesteld aan de hand van een reactiviteitscontrole. Deze controle moet 1-2 weken of 2-4 passages na het ontdooien worden uitgevoerd met behulp van de positieve controle 4-nitrobenzylbromide (4-NBB) (CAS:100-11-8, zuiverheid ≥ 99 %) en de negatieve controle melkzuur (CAS:50-21-5, zuiverheid ≥ 85 %). 4-NBB moet een positieve respons op Ind-IL8LA opleveren (≥ 1,4), terwijl melkzuur een negatieve respons op Ind-IL8LA (< 1,4) moet opleveren. Voor de bepaling worden uitsluitend de cellen gebruikt die de reactiviteitscontrole met goed gevolg doorstaan. De controle moet worden uitgevoerd in overeenstemming met de procedures die in de punten 22-24 worden beschreven.
|
|
16.
|
Voor het testen worden THP-G8-cellen geënt met een dichtheid van 2 tot 5 × 105 cellen/ml en gedurende 48 tot 96 uur voorgekweekt in kweekflessen. Op de dag van de test worden de cellen die uit de kweekfles worden geoogst, gewassen met RPMI-1640 dat 10 % FBS en geen antibiotica bevat, en vervolgens met een dichtheid van 1 × 106 cellen/ml geresuspendeerd met RPMI-1640 dat 10 % FBS en geen antibiotica bevat. Vervolgens worden de cellen verdeeld over een zwarte 96-putjesplaat met vlakke bodem (bv. Costar cat. nr. 3603) met 50 μl (5 × 104 cellen/putje).
|
Bereiding van de teststof en de controlestoffen
|
17.
|
De teststof en controlestoffen worden bereid op de dag van de tests. Voor de IL-8 Luc-bepaling worden teststoffen opgelost in X-VIVO™ 15, een in de handel verkrijgbaar, serumvrij medium (Lonza, 04-418Q), tot een uiteindelijke concentratie van 20 mg/ml. X-VIVO™ 15 wordt in a microcentrifugebuisje toegevoegd aan 20 mg van de teststof (ongeacht de oplosbaarheid van de chemische stof) tot een volume van 1 ml is bereikt. Vervolgens wordt het buisje krachtig gevortext en geschud op een rotor met een maximumsnelheid van 8 rpm gedurende 30 minuten bij een omgevingstemperatuur van ongeveer 20 oC. Als vaste chemische stoffen nog steeds niet zijn opgelost, wordt het buisje bovendien ultrasoon behandeld tot de chemische stof volledig is opgelost of stabiel is gedispergeerd. Voor teststoffen die oplosbaar zijn in X-VIVO™ 15 wordt de oplossing met een factor 5 verdund met X-VIVO™ 15 en gebruikt als een X-VIVO™ 15-stamoplossing van de teststof (4 mg/ml). Voor teststoffen die niet oplosbaar zijn in X-VIVO™ 15, wordt het mengsel gedurende ten minste 30 minuten opnieuw geroteerd en vervolgens gedurende 5 minuten gecentrifugeerd met 15000 rpm (≈20000 g); het resulterende supernatans wordt gebruikt als een X-VIVO™ 15-stamoplossing van de teststof. Voor het gebruik van andere oplosmiddelen, zoals DMSO, water of het kweekmedium, moet een wetenschappelijke onderbouwing worden gegeven. De procedure voor het oplossen van chemische stoffen wordt in aanhangsel 3.5 in detail weergegeven. De X-VIVO™ 15-oplossingen die in de punten 18-23 worden beschreven, worden in een verhouding van 1:1 (v/v) gemengd met de celsuspensies die zijn geprepareerd in een zwarte 96-putjesplaat met vlakke bodem (zie punt 16).
|
|
18.
|
De eerste test die wordt uitgevoerd, heeft tot doel de cytotoxische concentratie te bepalen en het huidsensibiliserend potentieel van chemische stoffen te onderzoeken. Met behulp van X-VIVO™ 15 worden seriële verdunningen van de X-VIVO™ 15-stamoplossingen van de teststoffen gemaakt met een verdunningsfactor 2 (zie aanhangsel 3.5), waarbij gebruikgemaakt wordt van een testblok met 96 putjes (bv. Costar cat. nr. EW-01729-03). Vervolgens wordt 50 μl/putje verdunde oplossing toegevoegd aan 50 μl van de celsuspensie in een zwarte 96-putjesplaat met vlakke bodem. Voor teststoffen die in X-VIVO™ 15 oplosbaar zijn, liggen de uiteindelijke concentraties van de teststoffen bijgevolg tussen 0,002 en 2 mg/ml (aanhangsel 3.5). Voor teststoffen die bij 20 mg/ml niet in X-VIVO™ 15 oplosbaar zijn, worden alleen verdunningsfactoren tussen 2 en 210 bepaald, al blijven de daadwerkelijke uiteindelijke concentraties van de teststoffen onzeker en zijn deze afhankelijk van de verzadigde concentratie van de teststoffen in de X-VIVO™ 15-stamoplossing.
|
|
19.
|
In daaropvolgende testruns (d.w.z. de tweede, derde en vierde duplo’s) wordt de X-VIVO™ 15-stamoplossing gemaakt in een concentratie die viermaal hoger is dan de CV05-concentratie (CV05: cellevensvatbaarheid 05; de laagste concentratie waarbij de Inh-GAPLA < 0,05 wordt) in het eerste experiment. Als de Inh-GAPLA in de eerste testrun bij de hoogste concentratie niet onder de 0,05 komt, wordt de X-VIVO™ 15-stamoplossing gemaakt op basis van de hoogste concentratie in de eerste testrun. De CV05-concentratie wordt berekend door de concentratie van de stamoplossing in de eerste testrun te delen door de verdunningsfactor voor CV05 (X) (verdunningsfactor CV05 (X); de verdunningsfactor die vereist is om de stamoplossing te verdunnen tot CV05) (zie aanhangsel 3.5). Voor teststoffen die bij 20 mg/ml niet in X-VIVO™ oplosbaar zijn, wordt de CV05 bepaald door de concentratie van de stamoplossing × 1/X. Voor de testruns 2 tot en met 4 wordt een tweede stamoplossing bereid als 4 × CV05 (aanhangsel 3.5).
|
|
20.
|
Seriële verdunningen van de tweede X-VIVO™ 15-stamoplossingen worden gemaakt met een verdunningsfactor van 1,5 in een testblok met 96 putjes. Vervolgens wordt 50 μl/putje verdunde oplossing toegevoegd aan 50 μl van de celsuspensie in de putjes van een zwarte 96-putjesplaat met vlakke bodem. Elke concentratie van elke teststof moet in 4 putjes worden getest. De monsters worden vervolgens gemengd op een plaatschudder en gedurende 16 uur geïncubeerd bij 37 oC en 5 % CO2, waarna de luciferase-activiteit wordt gemeten zoals verderop beschreven.
|
|
21.
|
De oplosmiddelcontrole is het mengsel van 50 μl/putje X-VIVO™ 15 en 50 μl/putje celsuspensie in 10 % FBS bevattend RPMI-1640.
|
|
22.
|
De aanbevolen positieve controle is 4-NBB. 20 mg 4-NBB wordt bereid in een microcentrifugeerbuisje van 1,5 ml en aangevuld tot 1 ml met X-VIVO™ 15. Het buisje wordt krachtig gevortext en geschud op een rotor met een maximumsnelheid van 8 rpm gedurende ten minste 30 minuten. Na 5 minuten centrifugeren bij 20000 g wordt het supernatans met een factor 4 verdund met X-VIVO™ 15, waarna 500 μl van het verdunde supernatans wordt overgebracht naar een testblok met 96 putjes. Het verdunde supernatans wordt met X-VIVO™ 15 verder verdund met een factor 2 en 4, en 50 μl van de oplossing wordt toegevoegd aan 50 μl THP-G8-celsuspensie in de putjes van een zwarte 96-putjesplaat met vlakke bodem (aanhangsel 3.6). Elke concentratie van de positieve controle moet in 4 putjes worden getest. De plaat wordt geschud op een plaatschudder en gedurende 16 uur geïncubeerd in een CO2-incubator (37 oC, 5 % CO2), waarna de luciferase-activiteit wordt gemeten zoals beschreven in punt 29.
|
|
23.
|
De aanbevolen negatieve controle is melkzuur. 20 mg melkzuur wordt bereid in een microcentrifugebuisje van 1,5 ml en aangevuld tot 1 ml met X-VIVO™ 15 (20 mg/ml). 20 mg/ml melkzuuroplossing wordt met een factor 5 verdund met X-VIVO™ 15 (4 mg/ml); 500 μl van deze 4 mg/ml melkzuuroplossing wordt overgebracht naar een putje van een testblok met 96 putjes. Deze oplossing wordt met een factor 2 verdund met X-VIVO™ 15 en vervolgens opnieuw met een factor 2 verdund tot oplossingen van 2 mg/ml en 1 mg/ml. 50 μl van deze 3 oplossingen en vehiculumcontrole (X-VIVO™ 15) worden toegevoegd aan 50 μl THP-G8-celsuspensie in de putjes van een zwarte 96-putjesplaat met vlakke bodem. Elke concentratie van de negatieve controle wordt in 4 putjes getest. De plaat wordt geschud op een plaatschudder en gedurende 16 uur geïncubeerd in een CO2-incubator (37 oC, 5 % CO2), waarna de luciferase-activiteit wordt gemeten zoals beschreven in punt 29.
|
|
24.
|
Als er historische gegevens beschikbaar zijn om vergelijkbare aanvaardbaarheidscriteria voor de testrun experimenten af te leiden, kunnen er andere geschikte positieve of negatieve controles worden gebruikt.
|
|
25.
|
Er moet worden voorkomen – bijvoorbeeld door de platen voorafgaand aan de incubatie met de teststoffen af te dekken met een folie – dat vluchtige teststoffen verdampen en dat er kruisbesmetting met teststoffen tussen putjes optreedt.
|
|
26.
|
Er zijn 2 tot 4 testruns nodig voordat een positieve of negatieve voorspelling kan worden afgeleid met betrekking tot de teststoffen en de oplosmiddelcontrole (zie tabel 2). Elke testrun wordt op een andere dag verricht met een verse X-VIVO™ 15-stamoplossing van teststoffen en onafhankelijk geoogste cellen. De cellen kunnen wel uit dezelfde passage komen.
|
Metingen van de luciferaseactiviteit
|
27.
|
De luminescentie wordt gemeten met behulp van een 96 putjes-microtiterplaat-luminometer die is uitgerust met optische filters, bv. Phelios (ATTO, Tokio, Japan), Tristan 941 (Berthold, Bad Wildbad, Duitsland) en de ARVO-serie (PerkinElmer, Waltham, MA, VS). Om de reproduceerbaarheid te waarborgen, moet de luminometer voor elke test worden gekalibreerd (19). Voor deze kalibratie zijn recombinante oranje en rood licht uitzendende luciferasen beschikbaar.
|
|
28.
|
Naar elk putje van de plaat met daarin de celsuspensie die al dan niet met een chemische stof is behandeld, wordt 100 μl voorverwarmde Tripluc® Luciferase Assay Reagent overgebracht. De plaat wordt gedurende 10 minuten geschud bij een omgevingstemperatuur van ongeveer 20 oC. De plaat wordt in de luminometer geplaatst om de luciferase-activiteit te meten. De bioluminescentie wordt gedurende 3 seconden zonder (F0) en 3 seconden met (F1) het optisch filter gemeten. Het gebruik van alternatieve instellingen, bv. afhankelijk van het model van de gebruikte luminometer, moet worden gemotiveerd.
|
|
29.
|
De parameters voor elke concentratie worden berekend aan de hand van de gemeten waarden, bv. IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, het gemiddelde ± SD van IL8LA, het gemiddelde ± SD van GAPLA, het gemiddelde ± SD van nIL8LA, het gemiddelde ± SD van Ind-IL8LA, het gemiddelde ± SD van Inh-GAPLA en het 95 %-betrouwbaarheidsinterval van Ind-IL8LA. Definities van de in dit punt gebruikte parameters zijn te vinden in respectievelijk aanhangsel 3.1 en aanhangsel 3.4.
|
|
30.
|
Voorafgaand aan de meting worden kleuren in meerkleurige reporterbepalingen onderscheiden met behulp van detectoren (luminometer en plaatlezer) die zijn uitgerust met optische filters, zoals sharp-cut filters (kortegolf- of langegolffilters) of bandfilters. De transmissiecoëfficiënten van de filters voor elke bioluminescentiesignaalkleur moeten voorafgaand aan het testen worden gekalibreerd, zoals beschreven in aanhangsel 3.2.
|
GEGEVENS EN RAPPORTAGE
Evaluatie van de gegevens
|
31.
|
Bij de beslissing of een resultaat positief of negatief is, gelden voor elke testrun de volgende criteria:
|
—
|
een IL-8 Luc-testvoorspelling wordt positief geacht als een teststof een Ind-IL8LA ≥ 1,4 heeft en de ondergrens van het 95 %-betrouwbaarheidsinterval van Ind-IL8LA ≥ 1,0;
|
|
—
|
een IL-8 Luc-testvoorspelling wordt negatief geacht als een teststof een Ind-IL8LA < 1,4 heeft en de ondergrens van het 95 %-betrouwbaarheidsinterval van Ind-IL8LA < 1,0.
|
|
Voorspellingsmodel
|
32.
|
Teststoffen die twee positieve resultaten opleveren in de 1e, 2e, 3e of 4e testrun, worden aangemerkt als positief, terwijl teststoffen die drie negatieve resultaten opleveren in de 1e, 2e, 3e of 4e testrun, worden aangemerkt als verondersteld negatief (tabel 2). Van de verondersteld negatieve chemische stoffen worden chemische stoffen die bij 20 mg/ml X-VIVO™ 15 oplossen negatief geacht, terwijl chemische stoffen die niet bij 20 mg/ml X-VIVO™ 15 oplossen buiten beschouwing moeten worden gelaten (figuur 1).
Tabel 2
Criteria voor het aanmerken als positief en verondersteld negatief
|
1e testrun
|
2e testrun
|
3e testrun
|
4e testrun
|
Uiteindelijke voorspelling
|
|
Positief
|
Positief
|
—
|
—
|
Positief
|
|
Negatief
|
Positief
|
—
|
Positief
|
|
Negatief
|
Positief
|
Positief
|
|
Negatief
|
Verondersteld negatief
|
|
Negatief
|
Positief
|
Positief
|
—
|
Positief
|
|
Negatief
|
Positief
|
Positief
|
|
Negatief
|
Verondersteld negatief
|
|
Negatief
|
Positief
|
Positief
|
Positief
|
|
Negatief
|
Verondersteld negatief
|
|
Negatief
|
—
|
Verondersteld negatief
|
|
Figuur 1
Voorspellingsmodel voor uiteindelijk oordeel

Aanvaardbaarheidscriteria
|
33.
|
Bij gebruik van de IL-8 Luc-bepaling moet aan de volgende aanvaardbaarheidscriteria worden voldaan:
|
—
|
de Ind-IL8LA moet in elke testrun meer dan 5,0 zijn in ten minste één concentratie van de positieve controle, 4-NBB.
|
|
—
|
de Ind-IL8LA moet in elke testrun minder dan 1,4 zijn in ten minste één concentratie van de negatieve controle, melkzuur.
|
|
—
|
Gegevens van platen waarvoor de GAPLA van controleputjes met cellen en Tripluc, maar zonder chemische stoffen minder dan 5 maal zo hoog is als de GAPLA van putjes die uitsluitend testmedium bevatten (50 μl/putje 10 % FBS bevattend RPMI-1640 en 50 μl/putje X-VIVO™ 15), moeten worden afgekeurd.
|
|
—
|
Gegevens van platen waarvoor de Inh-GAPLA van alle concentraties van de test- of controlestoffen lager is dan 0,05, moeten worden afgekeurd. In dat geval moet de eerste test worden herhaald, zodat de hoogste uiteindelijke concentratie van de herhaalde test de laagste uiteindelijke concentratie van de voorgaande test is.
|
|
Testverslag
|
34.
|
In het testverslag moet de volgende informatie worden opgenomen:
|
Teststoffen
Stof die uit één component bestaat:
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, oplosbaarheid in water, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
oplosbaarheid in X-VIVO™ 15. Voor chemische stoffen die niet oplosbaar zijn in X-VIVO™ 15, ongeacht of na het centrifugeren precipitatie of flotatie wordt waargenomen;
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof, als geen X-VIVO™ 15 is gebruikt.
|
Stof die uit meerdere componenten bestaat, UVCB en mengsel:
|
karakterisering, voor zover mogelijk aan de hand van bv. chemische identiteit (zie hierboven), zuiverheid, kwantitatief voorkomen en relevante fysisch-chemische eigenschappen (zie hierboven) van de bestanddelen, voor zover beschikbaar;
|
|
fysisch voorkomen, oplosbaarheid in water en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn;
|
|
molecuulgewicht of schijnbaar molecuulgewicht in het geval van mengsels/polymeren waarvan de samenstelling bekend is, of andere informatie die relevant is voor het verloop van de studie;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
oplosbaarheid in X-VIVO™ 15. Voor chemische stoffen die niet oplosbaar zijn in X-VIVO™ 15, ongeacht of na het centrifugeren precipitatie of flotatie wordt waargenomen;
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar.
|
|
motivering van de keuze van het oplosmiddel/vehiculum voor elke teststof, als geen X-VIVO™ 15 is gebruikt.
|
|
|
Controles
Positieve controle:
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s), SMILES- of InChI-code(s), structuurformules en/of andere identificatiemiddelen;
|
|
fysisch voorkomen, oplosbaarheid in water, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, voor zover die beschikbaar zijn en voor zover van toepassing;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
behandeling voorafgaand aan de test, indien van toepassing (bv. verwarmen, fijnmalen);
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
verwijzing naar historische positievecontroleresultaten waaruit de geschiktheid van de aanvaardbaarheidscriteria blijkt, indien van toepassing.
|
Negatieve controle:
|
chemische identificatie, zoals IUPAC- of CAS-na(a)m(en), CAS-nummer(s) en/of andere identificatiemiddelen;
|
|
zuiverheid, chemische naam van onzuiverheden waar passend en praktisch haalbaar enz.;
|
|
fysisch voorkomen, molecuulgewicht en andere relevante fysisch-chemische eigenschappen, in het geval er andere negatieve controles worden gebruikt dan diegene die zijn vermeld in deze testmethode, en voor zover beschikbaar;
|
|
opslagomstandigheden en stabiliteit, voor zover beschikbaar;
|
|
motivering van de keuze van het oplosmiddel voor elke teststof.
|
|
|
Testomstandigheden
|
naam en adres van de opdrachtgever, testinstallatie en de onderzoeksleider;
|
|
een beschrijving van de gebruikte test;
|
|
gebruikte cellijn, omstandigheden waarin die werd bewaard en oorsprong (bv. de instelling waarvan de cellijn werd verkregen);
|
|
partijnummer en oorsprong van FBS, naam leverancier, partijnummer van de zwarte 96-putjesplaat met vlakke bodem, en partijnummer van het Tripluc-reagens;
|
|
passagenummer en voor de test gebruikte celdichtheid;
|
|
gebruikte methode voor het tellen van de cellen met het oog op de enting voorafgaand aan de tests en getroffen maatregelen voor een homogene verdeling van het aantal cellen;
|
|
gebruikte luminometer (bv. model), met inbegrip van de instrumentinstellingen, het gebruikte luciferasesubstraat en bewijs van passende luminescentiemetingen op basis van de in aanhangsel 3.2 beschreven controletest;
|
|
gebruikte procedure om aan te tonen dat het laboratorium bekwaam is om de test uit te voeren (bv. door de stoffen voor bekwaamheidstoetsing te testen) of om aan te tonen dat de resultaten van de test in de loop van de tijd kunnen worden gereproduceerd.
|
|
|
Werkwijze
|
het aantal duplo’s en testruns;
|
|
de concentraties van de teststof, gebruikte procedure voor het opbrengen en blootstellingstijd (indien verschillend van wat is aanbevolen);
|
|
een beschrijving van de gebruikte beoordelings- en beslissingscriteria;
|
|
een beschrijving van de gebruikte criteria voor de aanvaardbaarheid van tests;
|
|
een beschrijving van eventuele modificaties van de testprocedure.
|
|
|
Resultaten
|
IL8LA- en GAPLA-metingen;
|
|
berekeningen voor nIL8LA, Ind-IL8LA en Inh-GAPLA;
|
|
het 95 %-betrouwbaarheidsinterval van Ind-IL8LA;
|
|
een grafiek met de dosis-responscurves voor inductie van luciferaseactiviteit en levensvatbaarheid;
|
|
een beschrijving van eventuele andere relevante waarnemingen.
|
|
|
Bespreking van de resultaten
|
bespreking van de resultaten verkregen met de IL-8 Luc-bepaling;
|
|
overweging van de resultaten van de bepaling in de context van een IATA, als er andere relevante informatie beschikbaar is.
|
|
Conclusie
|
LITERATUUR
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OESO (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OESO (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OESO (2016). Guidance Document on the Reporting of Defined Approaches and Individual Information Sources to be Used within Integrated Approaches to Testing and Assessment (IATA) for Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OESO (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Parijs. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OESO (2017). Nog niet gepubliceerd - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OESO, Parijs, Frankrijk
|
|
(16)
|
OESO (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 34. OESO, Parijs, Frankrijk.
|
|
(17)
|
OESO (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisatie voor Economische Samenwerking en Ontwikkeling, Parijs. Beschikbaar op: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Available at. http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OESO (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OESO, Parijs, Frankrijk.
|
|
(21)
|
Verenigde Naties (2015). Mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen (GHS). Zesde herziene editie. New York & Genève: United Nations Publications. ISBN: 978-92-1-117006-1. Beschikbaar op: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Aanhangsel 3.1
DEFINITIES
AOP (Adverse Outcome Pathway)
: een reeks gebeurtenissen van de chemische structuur van een doelstof of een groep soortgelijke chemische stoffen, van de moleculaire inleidende gebeurtenis tot een te onderzoeken in-vivoresultaat (20).
Betrouwbaarheid
: indicatie van de mate waarin een test, uitgevoerd volgens een vast protocol, op reproduceerbare wijze binnen één laboratorium en tussen laboratoria verspreid over de tijd kan worden uitgevoerd. De betrouwbaarheid wordt beoordeeld door berekening van intra- en interlaboratoriumreproduceerbaarheid en intralaboratoriumherhaalbaarheid (16).
Chemische stof
: een stof of een mengsel.
CV05
: cellevensvatbaarheid 05, d.w.z. de minimale concentratie waarbij chemische stoffen een Inh-GAPLA van minder dan 0,05 vertonen.
FInSLO-LA
: in het valideringsrapport en eerdere publicaties over de IL-8 Luc-bepaling gebruikte afkorting waarmee Ind-IL8LA bedoeld wordt. Zie Ind-IL8LA voor de definitie.
GAPLA
: Luciferase-activiteit van Stable Luciferase Red (SLR) (λmax = 630 nm), gereguleerd door GAPDH-promotor, toont cellevensvatbaarheid en het aantal levensvatbare cellen aan. een test die voldoende relevant en betrouwbaar wordt geacht voor een specifiek doel en gebaseerd is op wetenschappelijk verantwoorde beginselen.
Geldige testmethode
: Een testmethode is nooit algemeen geldig, maar enkel met betrekking tot een specifiek doel.
Gevaar
: inherente eigenschap van een agens of situatie die nadelige effecten kan veroorzaken als een organisme, systeem of (sub)populatie aan dit agens wordt blootgesteld.
Gevoeligheid
: het percentage van alle positieve/actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert en is een belangrijke overweging bij de beoordeling van de relevantie van een test (16).
GHS (mondiaal geharmoniseerd classificatie- en etiketteringssysteem voor chemische stoffen van de Verenigde Naties (VN))
: een systeem voor de indeling van chemische stoffen (stoffen en mengsels) op basis van gestandaardiseerde soorten en niveaus van gevaren van fysische aard en gevaren voor de gezondheid en het milieu, en dat de desbetreffende voorlichtingselementen bepaalt, zoals pictogrammen, signaalwoorden, gevarenaanduidingen, voorzorgsmaatregelen en veiligheidsinformatiebladen, waarmee informatie over de schadelijke effecten van de stoffen kenbaar wordt gemaakt teneinde mensen (waaronder werkgevers, werknemers, transporteurs, consumenten en spoedhulpverleners) en het milieu te beschermen (21).
IATA (Integrated Approach on Testing and Assessment, geïntegreerde test- en beoordelingsbenadering)
: een gestructureerde aanpak die wordt gebruikt voor de identificatie van gevaren (potentieel), het kenmerken van gevaren (potentie) en/of het beoordelen van de veiligheid (potentieel/potentie en blootstelling) van een chemische stof of een groep chemische stoffen, waarbinnen alle relevante gegevens strategisch worden geïntegreerd en worden afgewogen als inbreng voor regelgevingsbesluiten inzake potentiële gevaren en/of risico’s en/of de noodzaak van verdere gerichte en bijgevolg minimale tests.
IL-8 (interleukine-8)
: een cytokine die wordt geproduceerd door endotheelcellen, fibroblasten, keratinocyten, macrofagen en monocyten, en die chemotaxis van neutrofielen en T-cellymfocyten veroorzaakt.
IL8LA
: luciferase-activiteit van Stable Luciferase Orange (SLO) (λmax = 580 nm), gereguleerd door IL-8-promotor.
II-SLR-LA
: in het valideringsrapport en eerdere publicaties over de IL-8 Luc-bepaling gebruikte afkorting waarmee Inh- GAPLA bedoeld wordt. Zie Inh-GAPLA voor de definitie.
Ind-IL8LA
: x-voudige inductie van nIL8LA. De Ind-IL8LA wordt verkregen door de nIL8LA van THP-G8-cellen die met chemische stoffen zijn behandeld, te delen door de nIL8LA van niet-gestimuleerde THP-G8-cellen, en staat voor de inductie van IL-8-promotoractiviteit door chemische stoffen.
Inh-GAPLA
: remming van GAPLA. De Inh-GAPLA wordt verkregen door de GAPLA van THP-G8-cellen die met chemische stoffen zijn behandeld, te delen door de GAPLA van niet-behandelde THP-G8-cellen, en staat voor de cytotoxiciteit van chemische stoffen.
Mengsel
: een mengsel of oplossing bestaande uit twee of meer stoffen.
Minimale drempelwaarde voor inductie (MIT, minimum induction threshold)
: de laagste concentratie waarbij een chemische stof voldoet aan de positieve criteria.
Nauwkeurigheid
: de mate van overeenstemming tussen testresultaten en erkende referentiewaarden. Het is een maat voor de prestaties van de test en één aspect van relevantie. Deze term en de term concordantie, waaronder het percentage correcte uitkomsten van een test wordt verstaan, worden vaak door elkaar gebruikt (16).
nIL8LA
: de SLO-luciferase-activiteit die de activiteit van IL-8-promotor (IL8LA) weerspiegelt, genormaliseerd door de SLR-luciferase-activiteit die de activiteit van GAPDH-promotor (GAPLA) weerspiegelt. nIL8LA staat voor de activiteit van IL-8-promotor met inachtneming van de cellevensvatbaarheid en het aantal cellen.
nSLO-LA
: in het valideringsrapport en eerdere publicaties over de IL-8 Luc-bepaling gebruikte afkorting waarmee nIL8LA bedoeld wordt. Zie nIL8LA voor de definitie.
Positieve controle
: een monster met alle bestanddelen van een testsysteem dat met een stof behandeld werd waarvan bekend is dat deze een positieve respons opwekt. Om te verzekeren dat variabiliteit in de positievecontrolerespons in de loop van de tijd kan worden beoordeeld, mag de grootte van de positieve respons niet buitensporig zijn.
Prehaptenen
: chemische stoffen die in sensibiliserende stoffen veranderen na abiotische transformatie.
Prohaptenen
: chemische stoffen die enzymatische activering nodig hebben om hun potentieel voor huidsensibilisatie te realiseren.
Relevantie
: beschrijving van het verband van de test met het te onderzoeken effect en of de test betekenis heeft en nuttig is voor een bepaald doel. Het is de mate waarin de test het te onderzoeken biologische effect correct meet of voorspelt. Relevantie omvat een beoordeling van de nauwkeurigheid (concordantie) van een test (16).
Oplosmiddel-/vehiculumcontrole
: en onbehandeld monster dat alle componenten bevat van een testsysteem met uitzondering van de teststof, maar met inbegrip van het gebruikte oplosmiddel/vehiculum. Dit monster wordt gebruikt om de referentierespons vast te stellen voor de monsters die worden behandeld met de in hetzelfde oplosmiddel/vehiculum opgeloste of stabiel gedispergeerde teststof. Als dit monster getest wordt met een tegelijkertijd gemeten mediumcontrole, toont het ook aan of het oplosmiddel/vehiculum met het testsysteem interageert.
Oppervlakteactieve stof
: ook agens dat inwerkt op de oppervlakte genoemd. Dit is een stof, zoals een detergens, die de oppervlaktespanning van een vloeistof kan verlagen en het zo mogelijk maakt dat de vloeistof gaat schuimen of in vaste stoffen doordringt; ook bekend als een bevochtigingsmiddel. (TG437)
SLO-LA
: in het valideringsrapport en eerdere publicaties over de IL-8 Luc-bepaling gebruikte afkorting waarmee IL8LA bedoeld wordt. Zie IL8LA voor de definitie.
SLR-LA
: in het valideringsrapport en eerdere publicaties over de IL-8 Luc-bepaling gebruikte afkorting waarmee GAPLA bedoeld wordt. Zie GAPLA voor de definitie.
Specificiteit
: het percentage van alle negatieve/niet-actieve chemische stoffen dat door middel van de testmethode correct wordt ingedeeld. Het is een maat voor de nauwkeurigheid van een test die categorale resultaten oplevert en is een belangrijke overweging bij de beoordeling van de relevantie van een test (16).
Stof
: een chemisch element en de verbindingen ervan, zoals zij in natuurlijke toestand voorkomen of bij productie of vervaardiging ontstaan, met inbegrip van alle additieven die nodig zijn om de stabiliteit van dat product te behouden en alle onzuiverheden ten gevolge van het toegepaste procedé, maar met uitzondering van elk oplosmiddel dat kan worden afgescheiden zonder de stabiliteit van de stof aan te tasten of de samenstelling ervan te wijzigen.
Stof die uit één component bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin een hoofdcomponent aanwezig is in een concentratie van ten minste 80 % (w/w).
Stof die uit meerdere componenten bestaat
: een stof die wordt gekenmerkt door haar kwantitatieve samenstelling en waarin meer dan één van haar hoofdcomponenten aanwezig is in een concentratie van ≥ 10 % (g/g) en < 80 % (g/g). Een stof die uit meerdere componenten bestaat, is het resultaat van een fabricageproces. Het verschil tussen mengsels en stoffen die uit meerdere componenten bestaan, is dat mengsels worden verkregen door twee of meer stoffen zonder chemische reactie met elkaar te vermengen. Een stof die uit meerdere componenten bestaat, is het resultaat van een chemische reactie.
Teststof
: alle volgens deze methode geteste stoffen of mengsels.
Testrun
: een testrun bestaat uit het gelijktijdig testen van een of meer chemische stoffen met een oplosmiddel-/vehiculumcontrole en met een positieve controle.
THP-G8
: een IL-8-reportercellijn die in de IL-8 Luc-bepaling wordt gebruikt. De humane macrofaagachtige cellijn THP-1 werd getransfecteerd met de SLO- en SLR-luciferasegenen onder controle van respectievelijk de IL-8- en GAPDH-promotors.
UVCB
: stoffen met een onbekende of variabele samenstelling, complexe reactieproducten en biologische materialen.
Aanhangsel 3.2
PRINCIPE VAN HET METEN VAN LUCIFERASE-ACTIVITEIT EN VAN HET BEPALEN VAN DE TRANSMISSIECOËFFICIËNTEN VAN HET FILTER VOOR SLO EN SLR
MultiReporter Assay System Tripluc kan worden gebruikt in combinatie met een microplaat-luminometer met een detectiesysteem voor meerdere kleuren, dat kan worden uitgerust met een optisch filter (bv. Phelios AB-2350 (ATTO), ARVO (PerkinElmer) of Tristar LB941 (Berthold)). Het optisch filter dat bij de meting wordt gebruikt, is een 600-620 nm langegolf- of kortegolffilter of een 600-700 nm bandfilter.
Meting met een optisch filter van luciferasen die twee kleuren uitzenden.
In dit voorbeeld wordt gebruikgemaakt van Phelios AB-2350 (ATTO). Deze luminometer is uitgerust met een 600 nm langegolffilter (long pass- filter, hierna 600 nm LP genoemd) (R60 HOYA Co., filter 1) voor het splitsen van de luminescentie van SLO (λmax = 580 nm) en SLR (λmax = 630 nm).
Om de transmissiecoëfficiënten van 600 nm LP te bepalen, moet met behulp van gezuiverde SLO- en SLR-luciferasen het volgende worden gedaan: i) meet de intensiteit van de SLO- en SLR-bioluminescentie zonder filter (F0), ii) meet de intensiteit van de SLO- en SLR-bioluminescentie die 600 nm LP (filter 1) is gepasseerd, en iii) bereken de onderstaande transmissiecoëfficiënten van 600 nm LP voor SLO en SLR.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Wanneer de intensiteit van SLO en SLR in het testvoorbeeld worden gedefinieerd als respectievelijk O en R, worden i) de intensiteit van licht zonder filter (volledig optisch) F0 en ii) de intensiteit van licht dat wordt doorgelaten door 600 nm LP (filter 1) F1 als volgt beschreven:
F0 = O + R
F1 = κOR60 × O + κRR60 × R
Deze formules kunnen als volgt worden geherformuleerd:
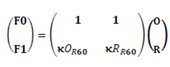
Vervolgens kan de O- en R-waarde, met behulp van de berekende transmissiefactoren (κOR60 en κRR60) en de gemeten F0 en F1, als volgt worden berekend:

Materialen en methoden voor het bepalen van de transmissiefactor
|
1)
|
Reagentia
Enkelvoudige, gezuiverde luciferase-enzymen:
|
Gevriesdroogd, gezuiverd SLO-enzym
|
|
Gevriesdroogd, gezuiverd SLR-enzym
|
|
(die met het oog op de valideringswerkzaamheden zijn gekocht bij GPC Lab. Co. Ltd., Tottori, Japan, met THP-G8-cellijn)
|
Testreagens:
|
Tripluc® Luciferase Assay Reagent (bv. van TOYOBO Cat. nr. MRA-301)
|
Medium: voor luciferasebepaling (30 ml, bewaard bij 2-8 °C)
|
|
Reagens
|
Conc.
|
Uiteindelijke conc. in medium
|
Vereiste hoeveelheid
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
2)
|
Bereiding van de enzymoplossing
Los gevriesdroogd, gezuiverd luciferase-enzym op in een buisje door 200 μl toe te voegen van 10 ~ 100 mM Tris/HCl of Hepes/HCl (pH 7,5 ~ 8,0) aangevuld met 10 % (w/v) glycerol, verdeel de enzymoplossing in aliquots van 10 μl in wegwerpbuisjes van 1,5 ml en sla ze op in een vriezer bij –80 °C. De bevroren enzymoplossing kan maximaal 6 maanden worden gebruikt. Voeg bij gebruik 1 ml medium voor luciferasebepalingen (RPMI-1640 met 10 % FBS) toe aan elk buisje met de enzymoplossing (verdunde enzymoplossing) en plaats ze op ijs om deactivering te voorkomen.
|
|
3)
|
Bioluminescentiemeting
Ontdooi de Tripluc® Luciferase Assay Reagent en bewaar bij kamertemperatuur ofwel in een waterbad ofwel in een ruimte waarin de lucht op kamertemperatuur is. Laat de luminometer gedurende 30 minuten opstarten alvorens te beginnen met de meting, zodat de fotomultiplicator kan stabiliseren. Breng 100 μl van de verdunde enzymoplossing over naar een zwarte plaat met 96 putjes met vlakke bodem (het SLO-referentiemonster naar nr. B1, nr. B2 en nr. B3, en het SLR-referentiemonster naar nr. D1, nr. D2 en nr. D3). Breng vervolgens met behulp van een Pipetman® 100 μl voorverwarmde Tripluc over in elk van de putjes met de verdunde enzymoplossing. Schud de plaat gedurende 10 minuten bij kamertemperatuur (ongeveer 25 oC) in een plaatschudder. Verwijder eventuele luchtbellen uit de oplossingen in de putjes. Zet de plaat in de luminometer om de luciferase-activiteit te meten. De bioluminescentie wordt gedurende 3 seconden zonder (F0) en 3 seconden met (F1) het optisch filter gemeten.
De transmissiecoëfficiënt van het optisch filter wordt als volgt berekend:
Transmissiecoëfficiënt (SLO (κOR60)) = (nr. B1 van F1 + nr. B2 van F1 + nr. B3 van F1) / (nr. B1 van F0 + nr. B2 van F0 + nr. B3 van F0)
Transmissiecoëfficiënt (SLR (κRR60)) = (nr. D1 van F1 + nr. D2 van F1 + nr. D3 van F1) / (nr. D1 van F0 + nr. D2 van F0 + nr. D3 van F0)
De berekende transmissiefactoren worden gebruikt voor alle met dezelfde luminometer uitgevoerde metingen.
|
Kwaliteitscontrole van de apparatuur
De procedures die worden beschreven in het IL-8 Luc-protocol moeten worden gevolgd (18).
Aanhangsel 3.3
STOFFEN VOOR BEKWAAMHEIDSTOETSING
Vooraleer over te gaan tot routinegebruik van de onder testmethode B.71 vallende test die in dit aanhangsel wordt beschreven, moeten laboratoria hun technische bekwaamheid aantonen door de verwachte IL-8 Luc-voorspelling te verkrijgen voor de 9 stoffen die in tabel 1 worden aanbevolen, en door de waarden te verkrijgen die binnen de respectieve referentiebereiken vallen voor ten minste 8 van de 9 stoffen voor bekwaamheidstoetsing (zodanig geselecteerd dat ze alle mogelijke responsen voor huidsensibilisatiegevaar weerspiegelen). Andere selectiecriteria waren dat stoffen in de handel beschikbaar zijn, en dat er in-vivoreferentiegegevens en met de IL-8 Luc-bepaling gegenereerde in-vitrogegevens van zeer goede kwaliteit beschikbaar zijn. Voor de IL-8 Luc-bepaling zijn tevens gepubliceerde referentiegegevens beschikbaar (6) (1).
Tabel 1
Aanbevolen stoffen voor het aantonen van technische bekwaamheid met de IL-8 Luc-bepaling
|
Stoffen voor bekwaamheidstoetsing
|
CAS-nr.
|
Toestand
|
Oplosbaarheid in X-VIVO15 bij 20 mg/ml
|
In-vivovoorspelling (109)
|
IL-8 Luc-voorspelling (110)
|
Referentiebereik (μg/ml) (111)
|
|
|
CV05 (112)
|
IL-8 Luc MIT (113)
|
|
2,4-Dinitrochloorbenzeen
|
97-00-7
|
Vaste stof
|
Onoplosbaar
|
Sensibiliserend (extreem)
|
Positief
|
2,3-3,9
|
0,5-2,3
|
|
Formaldehyde
|
50-00-0
|
Vloeistof
|
Oplosbaar
|
Sensibiliserend (sterk)
|
Positief
|
9-30
|
4-9
|
|
2-Mercaptobenzothiazol
|
149-30-4
|
Vaste stof
|
Onoplosbaar
|
Sensibiliserend (matig)
|
Positief
|
250-290
|
60-250
|
|
Ethyleendiamine
|
107-15-3
|
Vloeistof
|
Oplosbaar
|
Sensibiliserend (matig)
|
Positief
|
500-700
|
0,1-0,4
|
|
Ethyleenglycoldimethacrylaat
|
97-90-5
|
Vloeistof
|
Onoplosbaar
|
Sensibiliserend (zwak)
|
Positief
|
> 2000
|
0,04-0,1
|
|
4-Allylanisol (estragol)
|
140-67-0
|
Vloeistof
|
Onoplosbaar
|
Sensibiliserend (zwak)
|
Positief
|
> 2000
|
0,01-0,07
|
|
Streptomycinesulfaat
|
3810-74-0
|
Vaste stof
|
Oplosbaar
|
Niet-sensibiliserend
|
Negatief
|
> 2000
|
> 2000
|
|
Glycerol
|
56-81-5
|
Vloeistof
|
Oplosbaar
|
Niet-sensibiliserend
|
Negatief
|
> 2000
|
> 2000
|
|
Propaan-2-ol
|
67-63-0
|
Vloeistof
|
Oplosbaar
|
Niet-sensibiliserend
|
Negatief
|
> 2000
|
> 2000
|
|
Afkortingen: CAS-nummer = registratienummer van de Chemical Abstracts Service.
|
Aanhangsel 3.4
INDEXEN EN BEOORDELINGSCRITERIA
nIL8LA (nSLO-LA)
De j-e herhaling (j = 1-4) van de i-e concentratie (i = 0-11) wordt gemeten voor respectievelijk IL8LA (SLO-LA) en GAPLA (SLR-LA). De genormaliseerde IL8LA wordt weergegeven als nIL8LA (nSLO-LA) en wordt als volgt gedefinieerd:
nIL8LAij = IL8LAij/GAPLAij
Dit is de basismeeteenheid in deze bepaling.
Ind-IL8LA (FInSLO-LA)
De primaire meting van deze bepaling is de verhouding tussen de factor waarmee de gemiddelde nIL8LA (nSLO-LA) voor de herhaling bij de i-e concentratie toeneemt, en die bij de concentratie 0, Ind-IL8LA. Deze verhouding wordt weergegeven door de volgende formule:

Het eerstverantwoordelijke laboratorium heeft voorgesteld dat een waarde van 1,4 overeenkomt met een positief resultaat voor de geteste chemische stof. Die waarde is gebaseerd op het onderzoek van de historische gegevens van het eerstverantwoordelijke laboratorium. Vervolgens heeft het team Gegevensbeheer in alle stadia van de valideringsstudie gebruikgemaakt van die waarde. De primaire uitkomst, Ind-IL8LA, is de verhouding van twee rekenkundige gemiddelden zoals in de vergelijking is weergegeven.
95 % betrouwbaarheidsinterval (95 %-BI)
Het 95 % betrouwbaarheidsinterval (95 %-BI) op basis van de verhouding kan worden geschat om te laten zien hoe nauwkeurig de uitkomst van deze primaire meting is. De ondergrens van het 95 %-BI ≥ 1 geeft aan dat de nIL8LA met de i-e concentratie significant groter is dan die voor controle met oplosmiddel. Er zijn diverse manieren om het 95 %-BI te construeren. Wij hebben daarvoor in dit onderzoek gebruik gemaakt van de methode die bekendstaat als de stelling van Fieller. Deze stelling over het 95 %-BI wordt verkregen met de volgende formule:

waarbij:

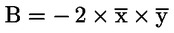

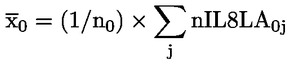




t0.975(ν) het 97,5 percentiel is van de centrale t-verdeling met ν als vrijheidsgraad, met

Inh-GAPLA (II-SLR-LA)
De Inh-GAPLAis een verhouding van de gemiddelde GAPLA (SLR-LA) voor de herhaling van de i-e concentratie vergeleken met die van de controlegroep met oplosmiddel, en wordt geschreven als

Aangezien de GAPLA de noemer van de nIL8LA is, geeft een zeer kleine waarde een grote verandering van de nIL8LA. Daarom zouden Ind-IL8LA-waarden met een zeer kleine waarde van Inh-GAPLA (minder dan 0,05) kunnen worden gezien als onnauwkeurig.
Aanhangsel 3.5
HET SCHEMA VAN DE METHODEN OM CHEMISCHE STOFFEN OP TE LOSSEN VOOR DE IL-8 LUC-BEPALING.
|
a)
|
Voor chemische stoffen die bij 20 mg/ml oplosbaar zijn in X-VIVOTM 15:

|
|
b)
|
Voor chemische stoffen die bij 20 mg/ml onoplosbaar zijn in X-VIVOTM 15:

|
Aanhangsel 3.6
HET SCHEMA VAN DE METHODE OM 4-NBB OP TE LOSSEN VOOR DE POSITIEVE CONTROLE VAN DE IL-8 LUC-BEPALING.

” |























