

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32021R0808
Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to be used for sampling and repealing Decisions 2002/657/EC and 98/179/EC (Text with EEA relevance)
Uitvoeringsverordening (EU) 2021/808 van de Commissie van 22 maart 2021 betreffende de prestaties van analysemethoden voor residuen van farmacologisch werkzame stoffen die bij voedselproducerende dieren worden gebruikt, betreffende de interpretatie van de resultaten en betreffende de toe te passen methoden voor bemonstering, en tot intrekking van de Beschikkingen 2002/657/EG en 98/179/EG (Voor de EER relevante tekst)
Uitvoeringsverordening (EU) 2021/808 van de Commissie van 22 maart 2021 betreffende de prestaties van analysemethoden voor residuen van farmacologisch werkzame stoffen die bij voedselproducerende dieren worden gebruikt, betreffende de interpretatie van de resultaten en betreffende de toe te passen methoden voor bemonstering, en tot intrekking van de Beschikkingen 2002/657/EG en 98/179/EG (Voor de EER relevante tekst)
C/2021/1772
OJ L 180, 21.5.2021, p. 84–109
(BG, ES, CS, DA, DE, ET, EL, EN, FR, GA, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
 In force: This act has been changed. Current consolidated version: 10/06/2021
In force: This act has been changed. Current consolidated version: 10/06/2021
|
21.5.2021 |
NL |
Publicatieblad van de Europese Unie |
L 180/84 |
UITVOERINGSVERORDENING (EU) 2021/808 VAN DE COMMISSIE
van 22 maart 2021
betreffende de prestaties van analysemethoden voor residuen van farmacologisch werkzame stoffen die bij voedselproducerende dieren worden gebruikt, betreffende de interpretatie van de resultaten en betreffende de toe te passen methoden voor bemonstering, en tot intrekking van de Beschikkingen 2002/657/EG en 98/179/EG
(Voor de EER relevante tekst)
DE EUROPESE COMMISSIE,
Gezien het Verdrag betreffende de werking van de Europese Unie,
Gezien Verordening (EU) 2017/625 van het Europees Parlement en de Raad van 15 maart 2017 betreffende officiële controles en andere officiële activiteiten die worden uitgevoerd om de toepassing van de levensmiddelen- en diervoederwetgeving en van de voorschriften inzake diergezondheid, dierenwelzijn, plantgezondheid en gewasbeschermingsmiddelen te waarborgen, tot wijziging van de Verordeningen (EG) nr. 999/2001, (EG) nr. 396/2005, (EG) nr. 1069/2009, (EG) nr. 1107/2009, (EU) nr. 1151/2012, (EU) nr. 652/2014, (EU) 2016/429 en (EU) 2016/2031 van het Europees Parlement en de Raad, de Verordeningen (EG) nr. 1/2005 en (EG) nr. 1099/2009 van de Raad en de Richtlijnen 98/58/EG, 1999/74/EG, 2007/43/EG, 2008/119/EG en 2008/120/EG van de Raad, en tot intrekking van de Verordeningen (EG) nr. 854/2004 en (EG) nr. 882/2004 van het Europees Parlement en de Raad, de Richtlijnen 89/608/EEG, 89/662/EEG, 90/425/EEG, 91/496/EEG, 96/23/EG, 96/93/EG en 97/78/EG van de Raad en Besluit 92/438/EEG van de Raad (verordening officiële controles) (1), en met name artikel 34, lid 6,
Overwegende hetgeen volgt:
|
(1) |
In Verordening (EU) 2017/625 zijn voorschriften vastgelegd voor officiële controles en andere officiële activiteiten die door de bevoegde autoriteiten van de lidstaten worden uitgevoerd om na te gaan of de wetgeving van de Unie op het gebied van onder andere voedselveiligheid wordt nageleefd in alle stadia van de productie, verwerking en distributie. Die verordening voorziet in specifieke regels voor officiële controles in verband met stoffen waarvan het gebruik tot gevolg kan hebben dat residuen in levensmiddelen en diervoeders terechtkomen, en bevat algemene voorschriften voor de methoden die moeten worden gebruikt voor bemonstering, laboratoriumanalyses en tests tijdens officiële controles en andere officiële activiteiten. |
|
(2) |
Beschikking 2002/657/EG van de Commissie (2) bevat voorschriften voor de prestaties van analysemethoden en de interpretatie van de resultaten van analyses van bepaalde stoffen en residuen daarvan in levende dieren en dierlijke producten, en Beschikking 98/179/EG van de Commissie (3) bevat uitvoeringsbepalingen met betrekking tot de officiële bemonstering in het kader van de opsporing van bepaalde stoffen en residuen daarvan in levende dieren en dierlijke producten. Beide beschikkingen zijn vastgesteld op basis van Richtlijn 96/23/EG van de Raad (4), die is ingetrokken bij Verordening (EU) 2017/625. In het licht van nieuwe wetenschappelijke ontwikkelingen moeten die regels worden geactualiseerd en geïntegreerd in het bij Verordening (EU) 2017/625 vastgestelde kader voor officiële controles. |
|
(3) |
Overeenkomstig artikel 1, tweede alinea, van Beschikking 2002/657/EG is die beschikking niet van toepassing op stoffen waarvoor in andere wetgeving van de Unie specifiekere voorschriften zijn vastgesteld. De desbetreffende stoffen zijn mycotoxinen in levensmiddelen, dioxinen en dioxineachtige polychloorbifenylen (pcb’s) in levensmiddelen en lood, cadmium, kwik en benzo[a]pyreen in levensmiddelen. Mycotoxinen in levensmiddelen moeten voldoen aan de voorschriften van Verordening (EG) nr. 401/2006 van de Commissie (5) tot vaststelling van bemonsterings- en analysemethoden voor de officiële controle op het mycotoxinegehalte in levensmiddelen. Verordening (EU) 2017/644 van de Commissie (6) tot vaststelling van bemonsterings- en analysemethoden voor de controle op het gehalte aan dioxinen en dioxineachtige en niet-dioxineachtige pcb’s in bepaalde levensmiddelen is van toepassing in het geval van dioxinen en dioxineachtige pcb’s. Verordening (EG) nr. 333/2007 van de Commissie (7) bevat bepalingen inzake bemonstering en analyse voor de officiële controles op lood, cadmium, kwik en benzo[a]pyreen in levensmiddelen. |
|
(4) |
Omwille van de duidelijkheid en rechtszekerheid is het passend de bepalingen die van toepassing zijn op de bemonstering en analyse met betrekking tot farmacologisch werkzame stoffen samen te voegen tot één rechtshandeling, zoals voor mycotoxinen, dioxinen, dioxineachtige pcb’s, lood, cadmium, kwik en benzo[a]pyreen in levensmiddelen reeds is gebeurd. |
|
(5) |
De Beschikkingen 98/179/EG en 2002/657/EG moeten daarom worden ingetrokken en door deze verordening worden vervangen. |
|
(6) |
Overeenkomstig Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad (8) mogen coccidiostatica en histomonostatica als toevoegingsmiddelen voor diervoeding worden gebruikt. Daarom is Verordening (EG) nr. 152/2009 van de Commissie (9) tot vaststelling van bemonsterings- en analysemethoden voor de officiële controle van diervoeders van toepassing op de analyse van het gehalte van die stoffen in diervoeders. Deze verordening moet echter van toepassing zijn wanneer diervoeders worden geanalyseerd in het kader van vervolgmaatregelen tijdens het onderzoek naar de bron van niet-conforme monsters in gevallen van vermoedelijke of vastgestelde niet-naleving van de Unieregels die van toepassing zijn op het gebruik van in diergeneesmiddelen of als toevoegingsmiddel voor diervoeding toegelaten stoffen of residuen ervan of van de Unieregels die van toepassing zijn op het gebruik van verboden of niet-toegelaten farmacologisch werkzame stoffen of residuen ervan. |
|
(7) |
Om te zorgen voor continuïteit bij de uitvoering van officiële controles en andere officiële activiteiten met betrekking tot residuen van farmacologisch werkzame stoffen en om te voorkomen dat alle methoden tegelijkertijd opnieuw moeten worden gevalideerd, mogen methoden die vóór de datum van inwerkingtreding van deze verordening zijn gevalideerd, gedurende een beperkte periode in gebruik blijven, met inachtneming van de voorschriften van de punten 2 en 3 van bijlage I bij Beschikking 2002/657/EG. Het is daarom passend de lidstaten voldoende tijd te geven om de voorschriften van deze verordening op alle analysemethoden toe te passen. |
|
(8) |
De in deze verordening vervatte maatregelen zijn in overeenstemming met het advies van het Permanent Comité voor planten, dieren, levensmiddelen en diervoeders, |
HEEFT DE VOLGENDE VERORDENING VASTGESTELD:
Artikel 1
Onderwerp en toepassingsgebied
In deze verordening worden regels vastgesteld betreffende de analysemethoden die worden gebruikt voor bemonstering en voor laboratoriumanalyses in verband met residuen van farmacologisch werkzame stoffen in levende voedselproducerende dieren, hun lichaamsdelen en -vloeistoffen, uitwerpselen en weefsels, en van hen afkomstige producten van dierlijke oorsprong en dierlijke bijproducten, alsmede in diervoeders en water. Ook bevat zij regels voor de interpretatie van de analyseresultaten van deze laboratoriumanalyses.
Deze verordening is van toepassing op officiële controles die tot doel hebben na te gaan of de voorschriften inzake de aanwezigheid van residuen van farmacologisch werkzame stoffen worden nageleefd.
Artikel 2
Definities
Voor de toepassing van deze verordening gelden de definities van artikel 2 van Gedelegeerde Verordening (EU) 2019/2090 van de Commissie (10), Verordening (EU) 2019/1871 van de Commissie (11), artikel 2 van Verordening (EG) nr. 470/2009 van het Europees Parlement en de Raad (12) en Verordening (EEG) nr. 315/93 van de Raad (13).
Verder wordt verstaan onder:
|
1) |
“absolute terugvinding”: de opbrengst van het eindstadium van een analyseproces voor een analyt gedeeld door de hoeveelheid analyt in het oorspronkelijke monster, uitgedrukt als een percentage; |
|
2) |
“nauwkeurigheid”: de mate van overeenstemming tussen een testresultaat en de aanvaarde werkelijke referentiewaarde, bepaald door raming van de juistheid en precisie (14); |
|
3) |
“α-fout”: de kans dat het onderzochte monster conform is, ook al is een niet-conform meetresultaat verkregen; |
|
4) |
“analyt”: het te analyseren onderdeel van een systeem; |
|
5) |
“toegelaten stof”: farmacologisch werkzame stof waarvan het gebruik bij voedselproducerende dieren overeenkomstig Richtlijn 2001/82/EG van het Europees Parlement en de Raad (15) is toegestaan; |
|
6) |
“β-fout”: de kans dat het onderzochte monster in werkelijkheid niet-conform is, ook al is een conform meetresultaat verkregen; |
|
7) |
“vertekening”: het verschil tussen de geraamde waarde van het testresultaat en een aanvaarde referentiewaarde; |
|
8) |
“ijkstandaard: een traceerbare referentie voor metingen die de hoeveelheid van de betrokken stof representeert, gerelateerd aan een referentiebasis; |
|
9) |
“gecertificeerd referentiemateriaal” (CRM): referentiemateriaal dat vergezeld gaat van door een daartoe gemachtigde instantie afgegeven documentatie en dat, bij gebruikmaking van geldige procedures, een of meer gespecificeerde eigenschapswaarden met de bijbehorende onzekerheden en traceerbaarheden vertoont (16); |
|
10) |
“co-chromatografie”: een techniek waarbij een onbekende stof wordt aangebracht op een chromatografische drager in combinatie met een of meer bekende verbindingen, in de verwachting dat het relatieve gedrag van de onbekende en bekende stoffen zal bijdragen tot de identificatie van de onbekende stof; |
|
11) |
“ringonderzoek”: analyse van hetzelfde monster of dezelfde monsters met behulp van dezelfde methode om de prestatiekenmerken van de methode in verschillende laboratoria te bepalen, waarbij het onderzoek het mogelijk maakt toevallige meetfouten en de laboratoriumvertekening voor de gebruikte methode te berekenen; |
|
12) |
“bevestigingsmethode”: een methode die volledige of aanvullende informatie levert voor de ondubbelzinnige identificatie en zo nodig de kwantificering van de stof op een van de volgende wijzen:
|
|
13) |
“dekkingsfactor (k)”: een getal dat het gewenste betrouwbaarheidsniveau uitdrukt en dat verband houdt met de uitgebreide meetonzekerheid; |
|
14) |
“beslissingsgrens voor bevestiging (CCα)”: de minimale waarde van waaraf met een foutkans van α kan worden besloten dat een monster niet-conform is, waarbij de waarde 1 – α de statistische zekerheid in procent uitdrukt, dat het toelaatbare gehalte is overschreden; |
|
15) |
“detectievermogen voor screening (CCβ)”: de kleinste hoeveelheid van de analyt die met een foutkans van β in een monster kan worden aangetoond of gekwantificeerd:
|
|
16) |
“monster met toegevoegde analyt”: een monster waaraan een bekende hoeveelheid van de aan te tonen of te kwantificeren analyt is toegevoegd; |
|
17) |
“interlaboratoriumonderzoek”: de organisatie, uitvoering en evaluatie van tests met hetzelfde monster (dezelfde monsters) door twee of meer laboratoria overeenkomstig van tevoren vastgestelde voorwaarden met als doel de testvaardigheid te beoordelen, hetzij in de vorm van een ringonderzoek, hetzij in de vorm van een bekwaamheidstest; |
|
18) |
“interne standaard (IS)”: een stof die niet in het monster voorkomt en die fysisch-chemische eigenschappen heeft die zo vergelijkbaar mogelijk zijn met die van de te identificeren of te kwantificeren analyt; |
|
19) |
“betrokken concentratieniveau”: de concentratie van een stof of analyt in een monster die significant is om te bepalen of dat monster in overeenstemming is met de wetgeving wat betreft:
|
|
20) |
“laagst gekalibreerd gehalte” (LGG): de laagste concentratie waarop het meetsysteem is gekalibreerd; |
|
21) |
“matrix”: het materiaal waaruit een monster wordt genomen; |
|
22) |
“matrixeffect”: het verschil in analytische respons tussen een in het oplosmiddel opgeloste standaard en een matrixgematchte standaard, hetzij zonder correctie met behulp van een interne standaard, hetzij met correctie met behulp van een interne standaard; |
|
23) |
“matrixgematchte standaard”: een blancomatrix (d.w.z. vrij van de analyt) waaraan, na de monstervoorbereiding, de analyt in een reeks concentraties wordt toegevoegd; |
|
24) |
“matrixverrijkte standaard”: een blancomatrix (d.w.z. vrij van de analyt) waaraan, vóór de vloeistof-vloeistofextractie en de monstervoorbereiding, de analyt in een reeks concentraties wordt toegevoegd; |
|
25) |
“te meten grootheid”: een bepaalde aan een meting onderworpen grootheid; |
|
26) |
“meetonzekerheid”: een niet-negatieve parameter die verband houdt met het meetresultaat en die de spreiding van de waarden karakteriseert die redelijkerwijs aan de te meten grootheid kan worden toegeschreven, op basis van de gebruikte informatie; |
|
27) |
“prestatiecriteria”: aan een prestatiekenmerk gestelde eisen aan de hand waarvan kan worden vastgesteld of de analysemethode voor het beoogde doel geschikt is en betrouwbare resultaten oplevert; |
|
28) |
“precisie”: de mate van overeenstemming tussen de onder vastgelegde voorwaarden verkregen resultaten van onafhankelijk van elkaar verrichte tests, die wordt uitgedrukt als de standaardafwijking of de variatiecoëfficiënt van de testresultaten; |
|
29) |
“kwalitatieve methode”: een analysemethode waarmee een stof of een groep stoffen wordt aangetoond of geïdentificeerd aan de hand van zijn chemische, biologische of fysische eigenschappen; |
|
30) |
“kwantitatieve methode”: een analysemethode waarmee de hoeveelheid of massafractie van een stof wordt bepaald, zodat die als numerieke waarde in de juiste eenheid kan worden uitgedrukt; |
|
31) |
“terugvinding”: de voor de terugvinding gecorrigeerde hoeveelheid van een analyt gedeeld door de toegevoegde hoeveelheid analyt in het matrixmonster, uitgedrukt als een percentage; |
|
32) |
“correctie voor terugvinding”: het gebruik van interne standaarden, het gebruik van een matrixkalibratiecurve en het gebruik van een terugvindingscorrectiefactor, alsmede een combinatie van deze benaderingen; |
|
33) |
“referentiemateriaal”: een materiaal dat voldoende homogeen en stabiel is ten aanzien van een of meer gespecificeerde eigenschappen en waarvan is vastgesteld dat het geschikt is voor het beoogde gebruik ervan in een meetproces of bij onderzoek van nominale eigenschappen (19); |
|
34) |
“relatief matrixeffect”: het verschil in analytische respons tussen een in het oplosmiddel opgeloste standaard en een matrixgematchte standaard met correctie met behulp van een interne standaard; |
|
35) |
“herhaalbaarheid”: precisie onder omstandigheden waarbij met dezelfde methode, bij identiek testmateriaal, in hetzelfde laboratorium, door dezelfde analist met dezelfde apparatuur binnen korte tijdsintervallen onafhankelijke testresultaten worden verkregen; |
|
36) |
“reproduceerbaarheid”: precisie onder omstandigheden waarbij testresultaten worden verkregen met behulp van dezelfde methode bij identiek analysemateriaal in verschillende laboratoria door verschillende personen met verschillende apparatuur (20); |
|
37) |
“robuustheid”: de gevoeligheid van een analysemethode voor veranderingen in de proefomstandigheden waaronder de methode zoals beschreven of met gespecificeerde kleine wijzigingen kan worden toegepast; |
|
38) |
“screeningsmethode”: een methode die wordt gebruikt voor het screenen van een stof of klasse van stoffen op het betrokken concentratieniveau; |
|
39) |
“doelconcentratie voor screening” (screening target concentration, STC): de concentratie die lager is dan of gelijk is aan het CCβ waarbij een screeningsmeting leidt tot indeling van het monster als potentieel niet-conform (“positief gescreend”) en aanleiding geeft tot een bevestigende test; |
|
40) |
“selectiviteit”: de geschiktheid van een methode om een onderscheid te maken tussen de te bepalen analyt en andere stoffen; |
|
41) |
“intralaboratoriumonderzoek” of “interne validatie”: een analytisch onderzoek waarbij één laboratorium betrokken is, dat één methode gebruikt om hetzelfde of verschillend analysemateriaal onder verschillende omstandigheden met gerechtvaardigd lange tussentijden te onderzoeken; |
|
42) |
“standaardadditie”: een procedure waarbij een deel van het monster als zodanig wordt geanalyseerd en bekende hoeveelheden van de standaardanalyt vóór de analyse aan de andere testporties worden toegevoegd; |
|
43) |
“standaardanalyt”: een analyt waarvan het gehalte en de zuiverheid bekend en gecertificeerd zijn en die als referentie in de analyse wordt gebruikt; |
|
44) |
“stof”: materie met constante samenstelling die wordt gekenmerkt door de samenstellende entiteiten en door bepaalde fysische eigenschappen; |
|
45) |
“analyseportie”: de hoeveelheid materiaal, genomen uit het monster, waarop de waarneming of de test wordt uitgevoerd; |
|
46) |
“juistheid”: de mate van overeenstemming tussen de gemiddelde waarde die is verkregen uit een lange reeks testresultaten en een aanvaarde referentiewaarde; |
|
47) |
“eenheden”: de eenheden zoals beschreven in de ISO-norm 80000 (21) en Richtlijn 80/181/EEG van de Raad (22); |
|
48) |
“validatie”: het aantonen, door middel van onderzoek en verstrekking van daadwerkelijk bewijs, dat aan de bijzondere eisen van een specifieke beoogde toepassing wordt voldaan (23), met behulp van een intralaboratoriumonderzoek of een ringonderzoek; |
|
49) |
“intralaboratoriumreproduceerbaarheid” of “intermediaire precisie/interne reproduceerbaarheid”: meetprecisie onder een reeks intralaboratoriumomstandigheden in een specifiek laboratorium. |
Artikel 3
Analysemethoden
De lidstaten zien erop toe dat de overeenkomstig artikel 34 van Verordening (EU) 2017/625 genomen monsters worden geanalyseerd met behulp van methoden die aan de volgende voorschriften voldoen:
|
1. |
zij worden gedocumenteerd in testinstructies, bij voorkeur overeenkomstig de bijlagen bij de ISO-norm 78-2:1999 — Chemie — Layouts voor normen — Deel 2: Methoden van chemische analyse (24); |
|
2. |
zij voldoen aan de prestatiecriteria en andere voorschriften voor analysemethoden van bijlage I, hoofdstuk 1, bij deze verordening; |
|
3. |
zij zijn gevalideerd overeenkomstig de voorschriften van bijlage I, hoofdstukken 2 en 4, bij deze verordening; |
|
4. |
zij maken handhaving mogelijk van de in Verordening (EU) 2019/1871 vastgestelde actiedrempels, identificatie van de aanwezigheid van verboden en niet-toegelaten stoffen en handhaving van maximumgehalten zoals vastgesteld op basis van Verordening (EEG) nr. 315/93 en Verordening (EG) nr. 124/2009, en van maximumwaarden voor residuen (MRL’s) zoals vastgesteld op basis van de Verordeningen (EG) nr. 1831/2003 en (EG) nr. 470/2009. |
Artikel 4
Kwaliteitscontrole
De lidstaten waarborgen de kwaliteit van de resultaten van de overeenkomstig Verordening (EU) 2017/625 uitgevoerde analyses, met name door toezicht te houden op tests of kalibratieresultaten overeenkomstig de ISO/IEC-norm 17025:2017 — Algemene eisen voor de bekwaamheid van beproevings- en kalibratielaboratoria en overeenkomstig de voorschriften voor kwaliteitscontrole bij routineanalyses, zoals vastgesteld in bijlage I, hoofdstuk 3, bij deze verordening.
Artikel 5
Interpretatie van de resultaten
1. Het resultaat van een analyse wordt als niet-conform beschouwd wanneer het gelijk is aan of hoger is dan de beslissingsgrens voor bevestiging (CCα).
2. Voor toegelaten stoffen waarvoor een MRL of maximumgehalte is vastgesteld, is de beslissingsgrens voor bevestiging (CCα) de minimale concentratie van waaraf met een statistische zekerheid van numerieke waarde 1 – α kan worden besloten dat het toelaatbare gehalte is overschreden.
3. Voor niet-toegelaten of verboden stoffen of voor toegelaten stoffen waarvoor geen MRL of maximumgehalte in een specifieke soort of in een specifiek product is vastgesteld, is de beslissingsgrens voor bevestiging (CCα) de laagste concentratie waarbij met een statistische zekerheid van numerieke waarde 1 – α kan worden besloten dat de analyt in kwestie aanwezig is.
4. Voor niet-toegelaten of verboden farmacologisch werkzame stoffen mag de α-fout maximaal 1 % bedragen. Voor alle overige stoffen mag de α-fout maximaal 5 % bedragen.
Artikel 6
Bemonsteringsmethoden
De lidstaten zien erop toe dat de monsters worden genomen, gehanteerd en geëtiketteerd overeenkomstig de in bijlage II bij deze verordening vastgestelde gedetailleerde bemonsteringsmethoden.
Artikel 7
Intrekking en overgangsmaatregelen
De Beschikkingen 2002/657/EG en 98/179/EG worden ingetrokken met ingang van de datum van inwerkingtreding van deze verordening.
Tot 10 juni 2026 blijven de voorschriften van de punten 2 en 3 van bijlage I bij Beschikking 2002/657/EG echter van toepassing op methoden die vóór de datum van inwerkingtreding van deze verordening zijn gevalideerd.
Artikel 8
Inwerkingtreding
Deze verordening treedt in werking op de twintigste dag na die van de bekendmaking ervan in het Publicatieblad van de Europese Unie.
Deze verordening is verbindend in al haar onderdelen en is rechtstreeks toepasselijk in elke lidstaat.
Gedaan te Brussel, 22 maart 2021.
Voor de Commissie
De voorzitter
Ursula VON DER LEYEN
(1) PB L 95 van 7.4.2017, blz. 1.
(2) Beschikking 2002/657/EG van de Commissie van 14 augustus 2002 ter uitvoering van Richtlijn 96/23/EG van de Raad wat de prestaties van analysemethoden en de interpretatie van resultaten betreft (PB L 221 van 17.8.2002, blz. 8).
(3) Beschikking 98/179/EG van de Commissie van 23 februari 1998 houdende vaststelling van uitvoeringsbepalingen met betrekking tot de officiële bemonstering in het kader van de opsporing van bepaalde stoffen en residuen daarvan in levende dieren en dierlijke producten (PB L 65 van 5.3.1998, blz. 31).
(4) Richtlijn 96/23/EG van de Raad van 29 april 1996 inzake controlemaatregelen ten aanzien van bepaalde stoffen en residuen daarvan in levende dieren en in producten daarvan en tot intrekking van de Richtlijnen 85/358/EEG en 86/469/EEG en de Beschikkingen 89/187/EEG en 91/664/EEG (PB L 125 van 23.5.1996, blz. 10).
(5) Verordening (EG) nr. 401/2006 van de Commissie van 23 februari 2006 tot vaststelling van bemonsteringswijzen en analysemethoden voor de officiële controle op het mycotoxinegehalte in levensmiddelen (PB L 70 van 9.3.2006, blz. 12).
(6) Verordening (EU) 2017/644 van de Commissie van 5 april 2017 tot vaststelling van bemonsterings- en analysemethoden voor de controle op het gehalte aan dioxinen en dioxineachtige en niet-dioxineachtige pcb’s in bepaalde levensmiddelen en tot intrekking van Verordening (EU) nr. 589/2014 (PB L 92 van 6.4.2017, blz. 9).
(7) Verordening (EG) nr. 333/2007 van de Commissie van 28 maart 2007 tot vaststelling van bemonsteringswijzen en analysemethoden voor de controle op de gehalten aan sporenelementen en procescontaminanten in levensmiddelen (PB L 88 van 29.3.2007, blz. 29).
(8) Verordening (EG) nr. 1831/2003 van het Europees Parlement en de Raad van 22 september 2003 betreffende toevoegingsmiddelen voor diervoeding (PB L 268 van 18.10.2003, blz. 29).
(9) Verordening (EG) nr. 152/2009 van de Commissie van 27 januari 2009 tot vaststelling van de bemonsterings- en analysemethoden voor de officiële controle van diervoeders (PB L 54 van 26.2.2009, blz. 1).
(10) Gedelegeerde Verordening (EU) 2019/2090 van de Commissie van 19 juni 2019 tot aanvulling van Verordening (EU) 2017/625 van het Europees Parlement en de Raad met betrekking tot gevallen van vermoedelijke of vastgestelde niet-naleving van de Unieregels die van toepassing zijn op het gebruik van in diergeneesmiddelen of als toevoegingsmiddel voor diervoeding toegelaten stoffen of residuen ervan of van de Unieregels die van toepassing zijn op het gebruik van verboden of niet-toegelaten farmacologisch werkzame stoffen of residuen ervan (PB L 317 van 9.12.2019, blz. 28).
(11) Verordening (EU) 2019/1871 van de Commissie van 7 november 2019 betreffende actiedrempels voor niet-toegelaten farmacologisch werkzame stoffen in levensmiddelen van dierlijke oorsprong en tot intrekking van Beschikking 2005/34/EG (PB L 289 van 8.11.2019, blz. 41).
(12) Verordening (EG) nr. 470/2009 van het Europees Parlement en de Raad van 6 mei 2009 tot vaststelling van communautaire procedures voor het vaststellen van grenswaarden voor residuen van farmacologisch werkzame stoffen in levensmiddelen van dierlijke oorsprong, tot intrekking van Verordening (EEG) nr. 2377/90 van de Raad en tot wijziging van Richtlijn 2001/82/EG van het Europees Parlement en de Raad en van Verordening (EG) nr. 726/2004 van het Europees Parlement en de Raad (PB L 152 van 16.6.2009, blz. 11).
(13) Verordening (EEG) nr. 315/93 van de Raad van 8 februari 1993 tot vaststelling van communautaire procedures inzake verontreinigingen in levensmiddelen (PB L 37 van 13.2.1993, blz. 1).
(14) ISO 3534-1:2006 — Statistiek — Woordenlijst en symbolen — Deel 1: Algemene statistische termen en termen voor waarschijnlijkheidsrekening (hoofdstuk 1).
(15) Richtlijn 2001/82/EG van het Europees Parlement en de Raad van 6 november 2001 tot vaststelling van een communautair wetboek betreffende geneesmiddelen voor diergeneeskundig gebruik (PB L 311 van 28.11.2001, blz. 1).
(16) JCGM 200:2008, International vocabulary of metrology — Basic and general concepts and associated terms (VIM), derde uitgave (2008): https://www.iso.org/sites/JCGM/VIM-JCGM200.htm (hoofdstuk 5 — Measurement standards (Etalons)).
(17) Verordening (EG) nr. 124/2009 van de Commissie van 10 februari 2009 tot vaststelling van maximumgehalten voor coccidiostatica en histomonostatica in levensmiddelen als gevolg van niet te voorkomen versleping van die stoffen naar niet-doeldiervoeders (PB L 40 van 11.2.2009, blz. 7).
(18) Verordening (EU) nr. 37/2010 van de Commissie van 22 december 2009 betreffende farmacologisch werkzame stoffen en de indeling daarvan op basis van maximumwaarden voor residuen in levensmiddelen van dierlijke oorsprong (PB L 15 van 20.1.2010, blz. 1).
(19) Commissie van de Codex Alimentarius, Voedsel- en Landbouworganisatie van de Verenigde Naties/Wereldgezondheidsorganisatie, Guidelines on analytical terminology (CAC/GL 72-2009).
(20) ISO 5725-1:1994 — Nauwkeurigheid (juistheid en precisie) van meetmethoden en -resultaten — Deel 1: Algemene beginselen en definities (hoofdstuk 3).
(21) ISO 80000-1:2009 — Grootheden en eenheden — Deel 1: Algemeen (inleiding).
(22) Richtlijn 80/181/EEG van de Raad van 20 december 1979 inzake de onderlinge aanpassing van de wetgevingen der lidstaten op het gebied van de meeteenheden, en tot intrekking van Richtlijn 71/354/EEG (PB L 39 van 15.2.1980, blz. 40).
(23) ISO/IEC 17025:2017 — Algemene eisen voor de bekwaamheid van beproevings- en kalibratielaboratoria (hoofdstuk 3).
(24) ISO 78-2:1999 — Chemie — Layouts voor normen — Deel 2: Methoden van chemische analyse (bijlagen).
BIJLAGE I
HOOFDSTUK 1
PRESTATIECRITERIA EN ANDERE VOORSCHRIFTEN VOOR ANALYSEMETHODEN
1.1. Voorschriften voor screeningsmethoden
1.1.1. Categorieën geschikte screeningsmethoden
Kwalitatieve, semikwantitatieve of kwantitatieve methoden worden gebruikt als geschikte screeningsmethoden.
1.1.2. Voorschriften voor biologische, biochemische of fysisch-chemische screeningsmethoden
Voor verboden of niet-toegelaten stoffen moet het CCβ zo laag zijn als redelijkerwijs haalbaar en in ieder geval lager dan de actiedrempel voor stoffen waarvoor overeenkomstig Verordening (EU) 2019/1871 actiedrempels zijn vastgesteld.
Voor toegelaten farmacologisch werkzame stoffen moet het CCβ lager zijn dan de MRL of het maximumgehalte.
Voor screeningsdoeleinden mogen alleen analysemethoden worden gebruikt waarvan op gedocumenteerde en traceerbare wijze kan worden aangetoond dat zij gevalideerd zijn en een fout-conformcijfer hebben van ten hoogste 5 % (β-fout). Wordt een vermoedelijk niet-conforme uitslag verkregen, dan moet die uitslag worden bevestigd door middel van een bevestigingsmethode.
Kwantitatieve screeningsmethoden die zowel voor screening als voor bevestiging worden gebruikt, moeten aan dezelfde voorschriften inzake nauwkeurigheid, bereik en precisie voldoen als beschreven in de punten 1.2.2.1 en 1.2.2.2.
1.2. Voorschriften voor bevestigingsmethoden
1.2.1. Algemene voorschriften voor bevestigingsmethoden
Voor verboden of niet-toegelaten stoffen moet de CCα zo laag zijn als redelijkerwijs haalbaar. Voor verboden of niet-toelaten stoffen waarvoor krachtens Verordening (EU) 2019/1871 een actiedrempel is vastgesteld, moet de CCα ten hoogste gelijk zijn aan de actiedrempel.
Voor toegelaten stoffen moet de CCα boven de MRL of het maximumgehalte liggen, maar zo dicht mogelijk in de buurt daarvan liggen.
Voor bevestigingsdoeleinden worden alleen analysemethoden gebruikt waarvan op gedocumenteerde en traceerbare wijze kan worden aangetoond dat zij gevalideerd zijn en een fout-niet-conformcijfer (α-fout) hebben van ten hoogste 1 % voor verboden of niet-toegelaten stoffen of van ten hoogste 5 % voor toegelaten stoffen.
Bevestigingsmethoden moeten informatie verschaffen over de structurele chemische samenstelling van de analyt. Bevestingsmethoden die alleen gebaseerd zijn op chromatografische analyse, zonder gebruikmaking van massaspectrometrische detectie, zijn daarom op zichzelf niet geschikt als bevestigingsmethode voor verboden of niet-toegelaten farmacologisch werkzame stoffen. Indien massaspectrometrie niet geschikt is voor toegelaten stoffen, kunnen andere methoden, zoals HPLC-DAD en -FLD, of een combinatie daarvan, worden gebruikt.
Indien volgens de bevestigingsmethode vereist, wordt een geschikte interne standaard aan het begin van het extractieprocedé aan de analyseportie toegevoegd. Al naargelang van de beschikbaarheid worden hiervoor met een stabiele isotoop gemerkte vormen van de analyt gebruikt, die bijzonder geschikt zijn voor massaspectrometrische detectie, dan wel analoge verbindingen die structureel nauw met de analyt verwant zijn. Indien geen geschikte interne standaard kan worden gebruikt, wordt de identificatie van de analyt bij voorkeur bevestigd met behulp van co-chromatografie (1). In dat geval mag slechts één piek worden verkregen, waarvan de hoogte (of het oppervlak) evenredig aan de hoeveelheid toegevoegde analyt is toegenomen. Indien dit niet haalbaar is, moeten matrixgematchte of matrixverrijkte standaarden worden gebruikt.
1.2.2. Algemene prestatiecriteria voor bevestigingsmethoden
1.2.2.1. Juistheid aan de hand van terugvinding
Bij herhaalde analyse van een gecertificeerd referentiemateriaal moet de afwijking tussen de experimenteel bepaalde, voor terugvinding gecorrigeerde gemiddelde massafractie en de gecertificeerde waarde voldoen aan de in tabel 1 vermelde intervallen voor de minimale juistheid.
Tabel 1
Minimale juistheid van kwantitatieve methoden
|
Massafractie |
Interval |
|
≤ 1 μg/kg |
tussen – 50 % en + 20 % |
|
> 1 μg/kg tot 10 μg/kg |
tussen – 30 % en + 20 % |
|
≥ 10 μg/kg |
tussen – 20 % en + 20 % |
Zijn er geen gecertificeerde referentiematerialen beschikbaar, dan mag de juistheid van de metingen op andere manieren worden beoordeeld, bijvoorbeeld door gebruik te maken van materialen waaraan door interlaboratoriumonderzoeken waarden zijn toegekend, of door toevoegingen van bekende hoeveelheden van de analyt(en) aan een blancomatrix.
1.2.2.2. Precisie
De variatiecoëfficiënt (VC) voor de herhaalde analyse van een referentiemateriaal of materiaal met toegevoegde analyt onder intralaboratoriumreproduceerbaarheidsomstandigheden mag niet groter zijn dan de waarde die volgt uit de vergelijking van Horwitz. Deze vergelijking luidt:
CV = 2(1 – 0,5 log C)
waarbij C de massafractie is, uitgedrukt als een macht (exponent) van 10 (bijvoorbeeld 1 mg/g = 10-3). Voor massafracties kleiner dan 120 μg/kg geeft de vergelijking van Horwitz onaanvaardbaar hoge waarden. Daarom mag de maximaal toegestane variatiecoëfficiënt niet groter zijn dan de waarden in tabel 2.
Tabel 2
Aanvaardbare variatiecoëfficiënt
|
Massafractie |
VC reproduceerbaarheid (%) |
|
≥ 1 000 μg/kg |
16 (aangepast op basis van vergelijking van Horwitz) |
|
> 120-1 000 μg/kg |
22 (aangepast op basis van vergelijking van Horwitz) |
|
10-120 μg/kg |
25 (*) |
|
< 10 μg/kg |
30 (*) |
Voor analyses die onder herhaalbaarheidsomstandigheden worden uitgevoerd, moet de variatiecoëfficiënt onder herhaalbaarheidsomstandigheden ten hoogste gelijk zijn aan twee derde van de in tabel 2 vermelde waarden.
1.2.3. Voorschriften voor chromatografische scheiding
Bij vloeistofchromatografie (LC) of gaschromatografie (GC) bedraagt de minimaal aanvaardbare retentietijd voor de te bepalen analyt(en) tweemaal de retentietijd die overeenkomt met het dode volume van de kolom. De retentietijd van de analyt in het extract moet overeenkomen met die van de ijkstandaard, een matrixgematchte standaard of een matrixverrijkte standaard met een tolerantie van ± 0,1 minuut. Voor snelle chromatografie, waarbij de retentietijd minder dan 2 minuten bedraagt, is een afwijking van minder dan 5 % van de retentietijd aanvaardbaar. Indien een interne standaard wordt gebruikt, moet de verhouding tussen de chromatografische retentietijd van de analyt en die van de interne standaard, dat wil zeggen de relatieve retentietijd van de analyt, overeenkomen met die van de ijkstandaard, de matrixgematchte standaard of de matrixverrijkte standaard met een maximale afwijking van 0,5 % voor gaschromatografie en 1 % voor vloeistofchromatografie voor methoden die zijn gevalideerd vanaf de datum van inwerkingtreding van deze verordening.
1.2.4. Specifieke prestatiecriteria voor massaspectrometrie
1.2.4.1. Massaspectrometrische detectie
Massaspectrometrische detectie wordt uitgevoerd met behulp van een aantal van de volgende opties:
|
1. |
opnemen van volledige massaspectra (full scans, FS); |
|
2. |
alleen geselecteerde massa’s scannen (selected ion monitoring, SIM); |
|
3. |
technieken voor sequentiële massaspectrometrie (MSn), zoals selected reaction monitoring (SRM, waarbij alleen die ionen die volgens een specifieke manier fragmenteren worden gescand); |
|
4. |
een combinatie van technieken voor massaspectrometrie (MS) of sequentiële massaspectrometrie (MSn) met de juiste ionisatiemethoden. |
Zowel lageresolutiemassaspectrometrie (LRMS, bij een resolutie van één massaeenheid) als hogeresolutiemassaspectrometrie (HRMS), waaronder bijvoorbeeld dubbel focusserende sectorinstrumenten, time-of-flight-instrumenten (TOF) en Orbitrap-instrumenten, zijn geschikt.
Ter bevestiging van de identiteit van een analyt bij hogeresolutiemassaspectrometrie (HRMS) moet de massa-afwijking van alle diagnostische ionen minder dan 5 ppm bedragen (of in het geval van m/z < 200, minder dan 1 mDa). Op basis hiervan moet de effectieve resolutie zo worden geselecteerd dat zij geschikt is voor het beoogde doel, en moet de resolutie normaliter groter zijn dan 10 000 voor het hele massabereik bij een dalwaarde van 10 %, of dan 20 000 bij de halfwaardebreedte (FWHM).
Wanneer massaspectrometrische bepaling wordt uitgevoerd door het opnemen van full-scanspectra (zowel LRMS als HRMS), zijn alleen diagnostische ionen met een relatieve intensiteit van meer dan 10 % in het referentiespectrum van de ijkstandaard, matrixgematchte standaard of matrixverrijkte standaarden geschikt. Diagnostische ionen moeten het molecuul-ion (indien aanwezig bij een intensiteit van ≥ 10 % van de hoofdpiek) en karakteristieke fragment- of product-ionen omvatten.
Selectie van precursor-ionen: Wanneer massaspectrometrische bepaling wordt uitgevoerd door fragmentatie na selectie van precursor-ionen, wordt de selectie van precursor-ionen uitgevoerd bij een resolutie van één massaeenheid of beter. Bij het geselecteerde precursor-ion moet het gaan om het molecuul-ion, kenmerkende adducten van het molecuul-ion, karakteristieke product-ionen of een van hun isotoop-ionen. Als de precursorselectie een massaselectievenster van meer dan één Dalton heeft, bv. in het geval van dataonafhankelijke acquisitie (data-independent acquisition), wordt de techniek beschouwd als een full-scanbevestigingsanalyse.
Fragment- en product-ionen: De geselecteerde fragment- of product-ionen moeten diagnostisch zijn voor het gemeten analyt/product. Niet-selectieve transities (bv. het tropylium-kation of verlies van water) moeten zo veel mogelijk worden weggelaten. De abundantie van diagnostische ionen wordt bepaald aan de hand van het oppervlak of de hoogte van de piek van geïntegreerde geëxtraheerd-ionchromatogrammen (extracted-ion chromatograms). Dit geldt ook wanneer voor de identificatie gebruik wordt gemaakt van full-scanmetingen. De signaal-ruisverhouding van alle diagnostische ionen moet ten minste drie op één (3:1) bedragen.
Relatieve intensiteiten; de relatieve intensiteiten van de diagnostische ionen (de ionverhouding) worden uitgedrukt als percentage van de intensiteit van het meest abundante ion of de meest abundante overgang. De ionverhouding moet worden bepaald door spectra te vergelijken of door de signalen van de geëxtraheerd-ionmassasporen te integreren. De ionverhouding van de te bevestigen analyt moet overeenkomen met die van de matrixgematchte standaarden, matrixverrijkte standaarden of standaardoplossingen bij vergelijkbare concentraties, gemeten onder dezelfde omstandigheden, binnen een relatieve afwijking van ± 40 %.
Bij iedere massaspectrometrische analyse moet ten minste één ionverhouding worden bepaald. Dit zijn bij voorkeur ionen die in één scan worden verkregen, maar de ionen kunnen ook afkomstig zijn van verschillende scans in dezelfde injectie (d.w.z. full- en fragmentatiescan).
1.2.4.2. Identificatie
Er wordt een systeem van identificatiepunten gebruikt om geschikte acquisitiemethoden en evaluatiecriteria te selecteren. Voor de bevestiging van de identiteit van stoffen in een matrix waarvoor een MRL is vastgesteld (toegelaten gebruik), zijn ten minste 4 identificatiepunten vereist. Voor niet-toegelaten of verboden stoffen zijn 5 identificatiepunten vereist. Eén punt kan afkomstig zijn van de chromatografische scheiding. Tabel 3 toont het aantal identificatiepunten dat elk van de technieken oplevert. Om het voor bevestiging vereiste aantal identificatiepunten te bereiken, kunnen de met verschillende technieken verkregen identificatiepunten bij elkaar worden opgeteld.
|
1. |
Alle massaspectrometrische analyses moeten worden gecombineerd met een scheidingstechniek met voldoende scheidend vermogen en selectiviteit voor de specifieke toepassing. Geschikte scheidingstechnieken zijn onder meer vloeistof- en gaschromatografie, capillaire elektroforese (CE) en superkritische chromatografie (supercritical fluid chromatography, SFC). In het geval van een analyt met een isobare of isomere verbinding is een aanvaardbare retentietijd (d.w.z. ± 0,5 % bij GC en ± 1 % bij LC en SFC) vereist om de identiteit ervan te bevestigen. |
|
2. |
Er mogen maximaal drie afzonderlijke technieken worden gecombineerd om het minimumaantal identificatiepunten te verkrijgen. |
|
3. |
Verschillende ionisatiemethoden (bv. elektronenionisatie en chemische ionisatie) worden als verschillende technieken beschouwd. Tabel 3 Identificatiepunten per techniek
Tabel 4 Voorbeelden van het aantal identificatiepunten voor specifieke technieken en combinaties van technieken (n = een geheel getal)
|
1.2.5. Specifieke prestatiecriteria voor de bepaling van een analyt met behulp van vloeistofchromatografie met andere detectietechnieken dan massaspectrometrie
Alleen voor toegelaten stoffen kunnen de volgende technieken worden gebruikt als alternatief voor op massaspectrometrie gebaseerde methoden, mits aan de relevante criteria voor deze technieken wordt voldaan:
|
1. |
full-scan diodearray-detectiespectrofotometrie (DAD) indien gebruikt met HPLC; |
|
2. |
spectometrie met fluorescentiedetectie (FLD) indien gebruikt bij HPLC. |
Vloeistofchromatografie met UV-VIS-detectie (vaste golflengte) is als zodanig niet geschikt als bevestigingsmethode.
1.2.5.1. Prestatiecriteria voor full-scan diodearray-spectrofotometrie
Er moet worden voldaan aan de prestatiecriteria voor chromatografische scheiding in hoofdstuk 1.2.3.
De absorptiemaxima in het uv-spectrum van de analyt moeten bij dezelfde golflengten liggen als die van de ijkstandaard in de matrix, binnen een maximale marge die afhankelijk is van de resolutie van het detectiesysteem. Voor diodearray-detectie is de karakteristieke maximale marge ± 2 nm. Het spectrum van de analyt boven 220 nm mag voor de delen van de twee spectra met een relatieve extinctie van ten minste 10 % niet zichtbaar verschillen van het spectrum van de ijkstandaard. Aan dit criterium wordt voldaan wanneer ten eerste dezelfde maxima aanwezig zijn en ten tweede het verschil tussen de twee spectra op geen enkel punt groter is dan 10 % van de extinctie van de ijkstandaard. Wanneer gebruik wordt gemaakt van computerondersteunde library searching en matching moet de vergelijking tussen de spectrumgegevens van de officiële monsters en die van de ijkoplossing boven een bepaalde kritieke matchfactor liggen. Deze factor wordt tijdens de validatie voor elke analyt vastgesteld aan de hand van spectra die aan de hierboven beschreven criteria voldoen. Nagegaan moet worden of er variaties in de spectra optreden als gevolg van de monstermatrix en de detectorprestaties.
1.2.5.2. Prestatiecriteria voor spectrofotometrie met fluorescentiedetectie
Er moet worden voldaan aan de prestatiecriteria voor chromatografische scheiding in hoofdstuk 1.2.3.
De excitatie- en emissiegolflengte worden in combinatie met de chromatografiecondities zodanig gekozen dat de effecten van storende componenten in blancomonsterextracten zo gering mogelijk zijn. De excitatie- en de emissiegolflengte moeten minimaal 50 nanometer uit elkaar liggen.
De afstand tussen de karakteristieke piek van de analyt en de top van de dichtstbijzijnde piek in het chromatogram moet ten minste eenmaal de volle piekbreedte zijn op 10 % van de maximumhoogte van de piek van de analyt.
Dit geldt voor moleculen die zelf fluoresceren en voor moleculen die na omzetting of derivatisering fluorescentie vertonen.
HOOFDSTUK 2
VALIDATIE
2.1. Te bepalen prestatiekenmerken voor analysemethoden
Door middel van de validatie van de methode moet worden aangetoond dat de analysemethode voldoet aan de criteria voor de desbetreffende prestatiekenmerken. Verschillende controledoeleinden vereisen verschillende categorieën methoden. Tabel 5 geeft aan welk prestatiekenmerk moet worden gecontroleerd voor welk type methode; elk van de parameters wordt in dit hoofdstuk nader toegelicht.
Tabel 5
Indeling van analysemethoden naar de te bepalen prestatiekenmerken
|
Methode |
Bevestiging |
Screening |
|||
|
Kwalitatief |
Kwantitatief |
Kwalitatief |
Semikwantitatief |
Kwantitatief |
|
|
Stoffen |
A |
A, B |
A, B |
A, B |
A, B |
|
Identificatie overeenkomstig punt 1.2 |
x |
x |
|
|
|
|
CCα |
x |
x |
|
|
|
|
CCβ |
— |
|
x |
x |
x |
|
Juistheid |
x |
|
|
x |
|
|
Precisie |
x |
|
(x) |
x |
|
|
Relatief matrixeffect/absolute terugvinding (*) |
x |
|
|
x |
|
|
Selectiviteit/specificiteit |
x |
x |
x |
x |
|
|
Stabiliteit (#) |
x |
x |
x |
x |
|
|
Robuustheid |
x |
x |
x |
x |
|
|
xDoor middel van de validatie moet worden aangetoond dat aan de voorschriften voor het prestatiekenmerk is voldaan. (x)Voor semikwantitatieve screeningsmethoden hoeft niet te worden voldaan aan de precisievoorschrijften van punt 1.2.2.2. De precisie moet echter worden bepaald om aan te tonen dat de methode geschikt is om fout-conforme analyseresultaten te vermijden. AVerboden of niet-toegelaten stoffen. BToegelaten stoffen. |
|||||
2.2. Juistheid, herhaalbaarheid en intralaboratoriumreproduceerbaarheid
Dit hoofdstuk bevat voorbeelden en referenties voor validatieprocedures. Er mogen ook andere methoden worden gebruikt om aan te tonen dat de methode voldoet aan de prestatiecriteria, mits zij dezelfde hoeveelheid informatie van dezelfde kwaliteit opleveren.
2.2.1. Conventionele validatie
Om de parameters volgens conventionele methoden te berekenen moeten verscheidene afzonderlijke proeven gedaan worden. Elk prestatiekenmerk moet voor elke grotere verandering worden bepaald (zie punt 2.4). Bij multianalytmethoden kunnen verschillende analyten tegelijkertijd geanalyseerd worden, mits eventueel relevante storingen zijn uitgesloten. Verscheidene prestatiekenmerken kunnen op een soortgelijke wijze bepaald worden. Om werk te besparen wordt het daarom aangeraden om proeven zo veel mogelijk te combineren (bijvoorbeeld herhaalbaarheid en intralaboratoriumreproduceerbaarheid met specificiteit, analyse van blancomonsters ter bepaling van de beslissingsgrens voor bevestiging en testen op specificiteit).
2.2.1.1. Juistheid op basis van een gecertificeerd referentiemateriaal
Het verdient de voorkeur de juistheid van een analysemethode te bepalen met behulp van gecertificeerd referentiemateriaal (CRM). De procedure hiervoor wordt beschreven in de ISO-norm 5725-4:1994 (2).
Hier volgt een voorbeeld:
|
1. |
analyseer zes identieke monsters van het CRM volgens de testinstructies voor de methode; |
|
2. |
bepaal de concentratie van de analyt in elk van deze monsters; |
|
3. |
bereken het gemiddelde, de standaardafwijking en de variatiecoëfficiënt (in %) voor deze zes identieke monsters; |
|
4. |
bereken de juistheid door de bepaalde gemiddelde concentratie te delen door de gecertificeerde waarde (gemeten als concentratie) en vermenigvuldig met 100 om het resultaat als percentage uit te drukken. |
Juistheid (in %) = (gemiddelde voor de terugvinding gecorrigeerde gemeten concentratie) × (100/gecertificeerde waarde)
2.2.1.2. Juistheid op basis van monsters met toegevoegde analyt
Indien er geen gecertificeerd referentiemateriaal beschikbaar is, wordt de juistheid van de methode bepaald aan de hand van experimenten met behulp van een blancomatrix waaraan analyt is toegevoegd, ten minste overeenkomstig het volgende schema:
|
1. |
voor methoden die zijn gevalideerd vanaf de datum van inwerkingtreding van deze verordening, blancomateriaal selecteren en analyt toevoegen in een concentratie van:
|
|
2. |
de analyse wordt voor elke concentratie in zesvoud uitgevoerd; |
|
3. |
analyseer de monsters; |
|
4. |
bereken de in elk monster gemeten concentratie; |
|
5. |
bereken de juistheid voor elk monster met behulp van onderstaande vergelijking en bereken vervolgens de gemiddelde juistheid en de variatiecoëfficiënt voor de zes resultaten bij elk concentratieniveau. |
Juistheid (in %) = (gemiddelde voor de terugvinding gecorrigeerde gemeten concentratie) × (100/toegevoegde concentratie)
Voor methoden voor toegelaten stoffen die vóór de datum van toepassing van deze verordening zijn gevalideerd, volstaat een bepaling van de juistheid van de methode met 6 aliquots waaraan 0,5, 1,0 en 1,5 maal de MRL dan wel het maximumgehalte is toegevoegd.
2.2.1.3. Herhaalbaarheid
|
1. |
Voor methoden die zijn gevalideerd vanaf de datum van inwerkingtreding van deze verordening, wordt een reeks monsters van identieke blancomatrices van dezelfde soort bereid. De analyt wordt eraan toegevoegd tot concentraties worden bereikt die overeenstemmen met:
|
|
2. |
De analyse wordt voor elke concentratie ten minste in zesvoud uitgevoerd. |
|
3. |
Analyseer de monsters. |
|
4. |
Bereken de in elk monster gemeten concentratie. |
|
5. |
Bereken de gemiddelde concentratie, de standaardafwijking en de variatiecoëfficiënt (in %) van de monsters met toegevoegde analyt. |
|
6. |
Herhaal deze stappen nog ten minste tweemaal. |
|
7. |
Bereken de totale gemiddelde concentraties, standaardafwijkingen (door het gemiddelde van de gekwadrateerde standaardafwijkingen van de afzonderlijke gevallen te berekenen en daar vervolgens de vierkantswortel van te nemen) en de variatiecoëfficiënten voor de monsters met toegevoegde analyt.
Voor methoden voor toegelaten stoffen die vóór de datum van inwerkingtreding van deze verordening zijn gevalideerd, volstaat een bepaling van de herhaalbaarheid met matrices waaraan analyt is toegevoegd tot concentraties van 0,5, 1,0 en 1,5 maal de MRL dan wel het maximumgehalte. Als alternatief kan de herhaalbaarheid worden berekend overeenkomstig de ISO-norm 5725-2:2019 (7). |
2.2.1.4. Intralaboratoriumreproduceerbaarheid
|
1. |
Bereid voor validaties die na de datum van inwerkingtreding van deze verordening worden uitgevoerd, een reeks monsters van gespecificeerd testmateriaal (identieke of verschillende matrices), waaraan de analyt(en) is (zijn) toegevoegd tot concentraties worden bereikt die overeenstemmen met:
|
|
2. |
Voer de analyse bij elk concentratieniveau uit met ten minste zes identieke monsters van blancomateriaal. |
|
3. |
Analyseer de monsters. |
|
4. |
Bereken de in elk monster gemeten concentratie. |
|
5. |
Herhaal deze stappen op ten minste twee andere tijdstippen met andere partijen blancomateriaal, andere analisten en onder zo veel mogelijk andere omgevingsomstandigheden, bv. andere partijen reagentia, oplosmiddelen, andere omgevingstemperaturen, andere apparatuur, of een variatie van andere parameters. |
|
6. |
Bepaal de gemiddelde concentratie, de standaardafwijking en de variatiecoëfficiënt (in %) van de monsters met toegevoegde analyt.
Voor methoden voor toegelaten stoffen die vóór de datum van inwerkingtreding van deze verordening zijn gevalideerd, volstaat een bepaling van de intralaboratoriumreproduceerbaarheid met matrices waaraan analyt is toegevoegd tot concentraties van 0,5, 1,0 en 1,5 maal de MRL dan wel het maximumgehalte. Als alternatief kan de berekening voor intralaboratoriumreproduceerbaarheid/intermediaire precisie ook worden uitgevoerd overeenkomstig de ISO-norm 5725-2:2019, de ISO-norm 11843-1:1997 (8), Codex CAC/GL 59-2006 (9). |
2.2.2. Validatie volgens andere modellen
Om de parameters volgens andere modellen te berekenen, moet met een proefopzet worden gewerkt. De proefopzet moet worden uitgewerkt afhankelijk van het aantal verschillende diersoorten en factoren dat wordt onderzocht. De validatieprocedure moet dus beginnen met te kijken naar de monsterpopulaties die in de toekomst in het laboratorium zullen worden onderzocht, om op basis daarvan te bepalen welke de belangrijkste diersoorten zijn en welke factoren van invloed kunnen zijn op de meetresultaten. Aan de hand van de factoriële benadering kan de meetonzekerheid van de testresultaten, verkregen onder uiteenlopende testomstandigheden in een bepaald laboratorium, zoals andere analisten, andere apparatuur, andere partijen reagentia, andere matrices, andere analysetijden en andere analysetemperaturen, worden beoordeeld. Vervolgens moet het concentratiebereik al naargelang van het doel van de bepalingen en, voor toegelaten stoffen, de MRL, of, voor verboden of niet-toegelaten stoffen, het maximumgehalte of de actiedrempel of het LGG, worden gekozen.
De factoriële benadering is gericht op het vaststellen van betrouwbare precisiegegevens en meetgegevens door gelijktijdige gecontroleerde variatie van de gekozen factoren. Dit maakt het mogelijk het gecombineerde effect van factoriële en willekeurige effecten te beoordelen. De proefopzet maakt het ook mogelijk de robuustheid (10) van de analysemethode te onderzoeken en de standaardafwijking van de interne reproduceerbaarheid over de verschillende matrices te bepalen.
Hieronder wordt een voorbeeld gegeven van een alternatieve benadering waarbij gebruik wordt gemaakt van een uitgewerkte orthogonale proefopzet.
Er kunnen tot zeven factoren (geluidsfactoren) worden onderzocht. De studie is zodanig opgezet dat de precisie, juistheid (op basis van monsters met toegevoegde analyt), gevoeligheid, meetonzekerheid en kritische concentraties gelijktijdig kunnen worden bepaald door volgens de proefopzet te werk te gaan.
Tabel 6
Voorbeeld van een uitgewerkte orthogonale proefopzet met 7 factoren (I-VII) die op twee niveaus (A/B) worden gevarieerd in een validatiestudie met acht analysegangen (combinatie van factorniveaus).
|
Factor |
I |
II |
III |
IV |
V |
VI |
VII |
|
Analysegang 01 |
A |
A |
A |
A |
A |
A |
A |
|
Analysegang 02 |
A |
A |
B |
A |
B |
B |
B |
|
Analysegang 03 |
A |
B |
A |
B |
A |
B |
B |
|
Analysegang 04 |
A |
B |
B |
B |
B |
A |
A |
|
Analysegang 05 |
B |
A |
A |
B |
B |
A |
B |
|
Analysegang 06 |
B |
A |
B |
B |
A |
B |
A |
|
Analysegang 07 |
B |
B |
A |
A |
B |
B |
A |
|
Analysegang 08 |
B |
B |
B |
A |
A |
A |
B |
De kenmerken van de methode worden berekend zoals beschreven door Jülicher et al. (11).
2.2.3. Andere validatiemethoden
Er mogen ook andere methoden worden gebruikt om aan te tonen dat de methode voldoet aan de prestatiecriteria voor het desbetreffende prestatiekenmerk, mits zij dezelfde hoeveelheid informatie van dezelfde kwaliteit opleveren. De validatie kan ook plaatsvinden door middel van een interlaboratoriumonderzoek zoals vastgesteld door de Codex Alimentarius, ISO of IUPAC (12) of volgens alternatieve methoden zoals intralaboratoriumonderzoek of interne validatie (13). Indien andere validatieprocedures worden gevolgd, moeten het model en de strategie waarop deze berusten met de verschillende eisen, aannamen en formules in het validatieprotocol worden vastgelegd of moet hier in elk geval naar worden verwezen.
2.3. Selectiviteit/specificiteit
Het vermogen om de analyt te onderscheiden van nauw verwante stoffen wordt zo goed mogelijk bepaald. Storingen met homologen, isomeren, afbraakproducten, endogene stoffen, analogen, metabolieten van het betrokken residu, matrixverbindingen of andere mogelijk storende stoffen moeten worden bepaald en indien nodig moet de methode worden gewijzigd om de vastgestelde storingen te vermijden. Om de specificiteit van de methode te bepalen, wordt de volgende benadering gevolgd:
|
1. |
kies een reeks chemisch verwante verbindingen of andere stoffen die met enige waarschijnlijkheid kunnen worden aangetroffen met de betrokken verbinding en die in de monsters aanwezig kunnen zijn, en verifieer of zij de analyse van de doelanalyt(en) zouden kunnen verstoren; |
|
2. |
analyseer een voldoende aantal representatieve blancomonsters, bv. verschillende partijen of partijen van verschillende diersoorten (n ≥ 20) en ga na of er storingen van signalen, pieken of ionensporen optreden in het gebied waar de doelanalyt verwacht wordt te elueren; |
|
3. |
voeg aan representatieve blancomonsters in een relevante concentratie een of meer stoffen toe die de identificatie en/of kwantificering van de analyt zouden kunnen verstoren en onderzoek of de toegevoegde stof:
|
2.4. Robuustheid
Getest moet worden of de analysemethode goed blijft presteren onder verschillende proefomstandigheden, zoals verschillende bemonsteringsomstandigheden en kleine veranderingen die zich bij routinetests kunnen voordoen. Om de robuustheid van de methode te testen, moet het bij de wijzigingen van de proefomstandigheden slechts om kleine veranderingen gaan. Het belang van deze veranderingen wordt geëvalueerd. Elk prestatiekenmerk wordt bepaald voor alle kleine veranderingen die een significant effect op de prestaties van de bepaling blijken te hebben.
2.5. Stabiliteit
De stabiliteit van de ijkstandaard, matrixgematchte standaard en/of matrixverrijkte standaarden en van analyt- of matrixbestanddelen in het monster tijdens de opslag of analyse moet worden bepaald, aangezien instabiliteit de testresultaten zou kunnen beïnvloeden.
Doorgaans is de stabiliteit van de analyt onder verschillende opslagcondities goed gekarakteriseerd. De proeven voor het toezicht op de opslagcondities van standaarden en monsters, die in het kader van het normale systeem voor de erkenning en kwaliteitscontrole van laboratoria worden verricht, kunnen de vereiste informatie opleveren. Indien stabiliteitsgegevens voor analyten in de matrix beschikbaar zijn (bv. op basis van informatie van de EURL’s, gepubliceerde gegevens enz.) hoeven deze gegevens niet door elk laboratorium te worden bepaald. Een verwijzing naar beschikbare stabiliteitsgegevens van analyten in oplossing en in matrix kan echter alleen worden aanvaard als voor identieke omstandigheden wordt gezorgd.
Indien de vereiste stabiliteitsgegevens niet beschikbaar zijn, moeten de volgende benaderingen worden gevolgd.
2.5.1. Bepaling van de stabiliteit van de analyt in oplossing
|
1. |
Bereid verse stamoplossingen van de analyt(en) en verdun deze zoals is aangegeven in de testinstructies om voldoende aliquots (bijvoorbeeld 40) te verkrijgen van elke geselecteerde concentratie. Er moeten monsters worden bereid van:
|
|
2. |
Meet het analytgehalte in de versbereide oplossing volgens de testinstructies. |
|
3. |
Breng adequate volumes over in geschikte recipiënten, etiketteer deze en bewaar ze volgens de licht- en temperatuuromstandigheden van het schema in tabel 7. Bij de keuze van de opslagtijd wordt rekening gehouden met de toegepaste analysepraktijk, idealiter totdat de eerste afbraakverschijnselen tijdens de identificatie en/of kwantificering waarneembaar zijn. Indien tijdens het stabiliteitsonderzoek geen afbraak wordt waargenomen, moet de opslagduur voor het stabiliteitsonderzoek gelijk zijn aan de duur van de maximale opslagperiode van de oplossing. |
|
4. |
Bereken de concentratie van de analyt(en) in elk aliquot in vergelijking met de concentratie van de analyt in de versbereide oplossing aan de hand van de onderstaande formule:
Resterende analyt (%) = Ci × 100/Cvers Ci = concentratie op tijdstip i Cvers = concentratie van de verse oplossing De gemiddelde waarde van vijf identiek bereide oplossingen die zijn opgeslagen, mag niet meer dan 15 % afwijken van de gemiddelde waarde van vijf identiek bereide verse oplossingen. De gemiddelde waarde van de vijf verse oplossingen wordt gebruikt als basis voor de berekening van het procentuele verschil. Tabel 7 Schema voor bepaling van de stabiliteit van de analyt in oplossing
|
2.5.2. Bepaling van de stabiliteit van de analyt(en) in de matrix
|
1. |
Gebruik waar mogelijk praktijkmonsters. Is er geen praktijkmatrix beschikbaar, dan moet een blancomatrix worden gebruikt waaraan analyt is toegevoegd. |
|
2. |
Wanneer er wel een praktijkmatrix beschikbaar is, bepaal dan de concentratie in de matrix, terwijl de matrix nog vers is. Sla verdere aliquots van het gehomogeniseerde gevormde materiaal op bij – 20 °C of lager indien nodig, en bepaal de concentraties van de analyt zolang het monster in het laboratorium wordt bewaard. |
|
3. |
Indien er geen praktijkmatrix voorhanden is, neem dan wat blancomatrix en homogeniseer deze. Verdeel de matrix in vijf aliquots. Voeg aan elk aliquot de analyt toe, die bij voorkeur in een kleine hoeveelheid waterige oplossing is bereid. Analyseer één aliquot onmiddellijk. Bewaar de resterende aliquots ten minste bij – 20 °C of lager indien nodig en analyseer ze na opslag op korte, middellange en lange termijn, rekening houdend met de toegepaste analysemethoden. |
|
4. |
Registreer de maximaal aanvaardbare opslagtijd en de optimale opslagcondities.
De gemiddelde waarde van vijf identiek bereide oplossingen die zijn opgeslagen, mag niet meer dan de intralaboratoriumreproduceerbaarheid afwijken van de gemiddelde waarde van vijf identiek bereide verse oplossingen. De gemiddelde waarde van de vijf verse oplossingen wordt gebruikt als basis voor de berekening van het procentuele verschil. |
2.6. Beslissingsgrens voor bevestiging (CCα)
De CCα moet worden bepaald voor bevestigingsmethoden. De CCα wordt vastgesteld onder omstandigheden die voldoen aan de voorschriften voor identificatie of identificatie plus kwantificering zoals gedefinieerd onder “Prestatiecriteria en andere voorschriften voor analysemethoden” in hoofdstuk 1.
Voor de controle op de conformiteit van monsters is in de CCα-waarde (beslissingsgrens) al rekening gehouden met de gecombineerde standaardmeetonzekerheid.
|
1. |
Voor niet-toegelaten of verboden farmacologisch werkzame stoffen wordt de CCα als volgt berekend:
Methode 2 voor de berekening van de CCα mag slechts tot 1 januari 2026 worden gebruikt voor methoden die vóór de datum van inwerkingtreding van deze verordening zijn gevalideerd. Voor methoden die na de inwerkingtreding van deze verordening zijn gevalideerd, mag alleen methoden 1 of 3 worden gebruikt. |
|
2. |
Voor toegelaten stoffen wordt de CCα als volgt berekend:
|
2.7. Detectievermogen voor screening (CCβ)
Het CCβ moet worden bepaald voor screeningsmethoden. Het CCβ wordt vastgesteld zoals gedefinieerd onder “Prestatiecriteria en andere voorschriften voor analysemethoden” in hoofdstuk 1 van deze bijlage en overeenkomstig de voorschriften in tabel 5. De volledige voorschriften voor identificatie (zie 1.2.3, 1.2.4, 1.2.5) hoeven echter niet te worden toegepast voor screeningsmethoden.
|
1. |
Voor niet-toegelaten of verboden farmacologisch werkzame stoffen moet een maximale β-fout van 5 % worden gewaarborgd. Het CCβ wordt als volgt berekend:
|
|
2. |
Voor toegelaten stoffen moet een maximale β-fout van 5 % worden gewaarborgd. Het CCβ wordt als volgt berekend:
Voor farmacologisch werkzame stoffen waarvoor de MRL voor de som van verschillende stoffen is vastgesteld, wordt het CCβ van de stof met de hoogste concentratie in het monster gebruikt als het CCβ om de som van de stoffen in het gemeten monster te beoordelen. |
2.8. IJkcurves
Indien voor de kwantificering ijkcurves worden gebruikt:
|
1. |
moeten ten minste vijf, bij voorkeur equidistante, concentraties (inclusief nulniveau) voor het uitzetten van de curve gebruikt worden; |
|
2. |
wordt het werkbereik van de ijkcurve aangegeven; |
|
3. |
worden de wiskundige formule van de curve en de goodness-of-fit van de gegevens (determinatiecoëfficiënt R2) met de curve aangegeven; |
|
4. |
worden aanvaardbaarheidsintervallen voor de parameters van de curve aangegeven. |
Voor ijkcurves op basis van een standaardoplossing, matrixgematchte standaarden of matrixverrijkte standaarden, worden aanvaardbare intervallen aangegeven voor de parameters van de ijkcurve, die van reeks tot reeks kunnen verschillen.
2.9. Absolute terugvinding
De absolute terugvinding van de methode moet worden bepaald als er geen interne standaard of geen matrixverrijkte ijking wordt gebruikt.
Wanneer aan de in tabel 1 vastgestelde voorschriften inzake juistheid is voldaan, mag een vaste correctiefactor worden gebruikt. Indien dit niet het geval is, moet de voor die specifieke batch verkregen terugvindingsfactor worden gebruikt. Als alternatief wordt de standaardadditieprocedure (16) of een interne standaard gebruikt in plaats van een terugvindingscorrectiefactor.
De absolute terugvinding wordt berekend voor ten minste zes representatieve partijen matrix.
Vóór de extractie wordt analyt toegevoegd aan een aliquot van blancomatrix en na de monstervoorbereiding wordt analyt bij een relevant concentratieniveau toegevoegd aan een tweede aliquot van blancomatrix, waarna de concentratie van de analyt wordt bepaald.
De terugvinding wordt als volgt berekend:
Terugv (analyt) = (matrixverrijkte standaard)/(matrixgematchte standaard) × 100
2.10. Relatieve matrixeffecten
Het relatieve matrixeffect wordt in alle gevallen bepaald. Dit kan gebeuren in het kader van de validatie of in afzonderlijke proeven. Het relatieve matrixeffect wordt berekend voor ten minste twintig verschillende blancopartijen (matrix/soort), afhankelijk van het toepassingsgebied van de methode, bv. de verschillende soorten die moeten worden bestreken.
Aan de blancomatrix moet na extractie de analyt worden toegevoegd bij de actiedrempel, de MRL of het maximumgehalte, waarna zij samen met een zuivere oplossing van de analyt moet worden geanalyseerd.
Het relatieve matrixeffect of de matrixfactor (MF) wordt als volgt berekend:

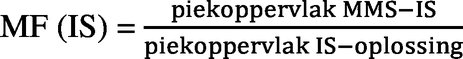
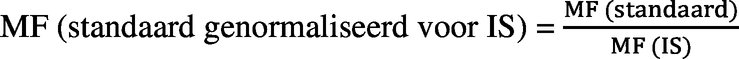
IS: interne standaard
MMS: matrixgematchte standaard
De variatiecoëfficiënt mag niet groter zijn dan 20 % voor de MF (standaard genormaliseerd voor IS).
HOOFDSTUK 3
KWALITEITSCONTROLE TIJDENS ROUTINEANALYSE — DOORLOPENDE VERIFICATIE VAN DE PRESTATIES VAN DE METHODE
Er moet worden voldaan aan de voorschriften voor het waarborgen van de kwaliteit van de analyseresultaten van de ISO/IEC-norm 17025:2017 (17).
Bij routineanalyse wordt bij voorkeur gebruikgemaakt van de analyse van gecertificeerde referentiematerialen (CRM’s) om het bewijs te leveren van de prestaties van de methode. Aangezien er zelden CRM’s beschikbaar zijn die de relevante analyten bevatten bij de vereiste concentratieniveaus, mogen als alternatief ook referentiematerialen worden gebruikt die worden verstrekt en gekenmerkt door de EURL’s of door laboratoria met een accreditatie volgens de ISO/IEC-norm 17043:2010 (18). Een ander toegestaan alternatief is gebruik te maken van intern referentiemateriaal dat regelmatig wordt gecontroleerd.
De doorlopende verificatie van de prestaties van de methode tijdens routineanalyses moet worden uitgevoerd tijdens de screeningsstap en de bevestigingsstap.
|
1. |
Voor de screeningsstap: Voor elke reeks (batch) van uitgevoerde analyses wordt tegelijkertijd een set van de volgende kwaliteitscontrolemonsters geanalyseerd:
|
|
2. |
Voor de bevestigingsstap: Voor elke reeks (batch) van uitgevoerde analyses wordt tegelijkertijd een set van de volgende kwaliteitscontrolemonsters geanalyseerd:
|
Voor de kwaliteitscontrolemonsters wordt de volgende volgorde aanbevolen: controlemonster voor de systeemgeschiktheid van het instrument, conform controlemonster, te bevestigen monster(s), opnieuw conform controlemonster en kwaliteitscontrolemonster met toegevoegde analyt (niet-conforme controlemonsters).
Voor kwantitatieve methoden moet met elke batch officiële monsters vóór of na de bovengenoemde monsters een ijkcurve worden geanalyseerd en gemeten.
Waar haalbaar wordt de juistheid (op basis van monsters met toegevoegde analyt) van alle doelanalyten in de niet-conforme controlemonsters beoordeeld aan de hand van kwaliteitscontrolekaarten overeenkomstig hoofdstuk 7.7 van de ISO/IEC-norm 17025:2017. Indien hiervoor een onevenredig groot aantal juistheidsbepalingen nodig is, mag het aantal analyten worden teruggebracht tot een aantal representatieve analyten.
HOOFDSTUK 4
UITBREIDING VAN HET GEVALIDEERDE TOEPASSINGSGEBIED VAN EEN EERDER GEVALIDEERDE METHODE
Soms is het nodig het toepassingsgebied van een voorheen volledig gevalideerde methode uit te breiden. In deze gevallen moet het toepassingsgebied op efficiënte en analytisch verantwoorde wijze worden uitgebreid. Dit kan worden bereikt door een validatie uit te voeren op een beperkt aantal monsters (bv. het halve aantal monsters) in vergelijking met een volledige validatie.
Om vast te stellen welk type en welk aantal wijzigingen in één enkele beperkte validatieregeling kunnen worden gevalideerd, moet echter altijd een beroep worden gedaan op deskundigheid en eerdere ervaringen; voor een wijziging van de detectietechniek zou bijvoorbeeld in elk geval een volledige validatie nodig zijn.
Om de geldigheid van de methode te blijven waarborgen, moeten de prestaties ervan in het algemeen doorlopend worden gemonitord en vergeleken met de aanvankelijk verkregen validatieparameters. Idealiter is deze doorlopende controle van de prestaties van de methode zodanig opgezet dat de ontbrekende gegevens voor een volledige validatie in de loop der tijd kunnen worden verzameld (bv. met enkele gegevenspunten afkomstig van kwaliteitscontrolemonsters uit elke analysereeks).
4.1. Uitbreidingen van methoden wat betreft de reeks concentraties
Als gevolg van wijzigingen van MRL’s, maximumgehalten en actiedrempels kan het nodig zijn het concentratiebereik waarvoor een methode is gevalideerd, aan te passen. In een dergelijk geval is de toepassing van een beperkte validatieregeling aanvaardbaar.
De ijkcurves voor het gewijzigde bereik moeten volgens de gevalideerde procedure worden opgesteld. Er moeten verschillende batches worden geanalyseerd waaraan analyt is toegevoegd bij verschillende concentratieniveaus (zie 2.2.1, 2.2.2). De juistheid, herhaalbaarheid en intralaboratorium-reproduceerbaarheid/intermediaire precisie moeten binnen een aanvaardbaar bereik liggen ten opzichte van de oorspronkelijk gevalideerde methode. Indien relevant moet een herberekening van het CCβ (screeningsmethoden) en de CCα (bevestigingsmethoden) worden uitgevoerd.
4.2. Uitbreidingen van methoden wat betreft aanvullende stoffen
In het algemeen is uitbreiding van de methode tot aanvullende verbindingen alleen mogelijk voor analyten die qua structuur en kenmerken vergelijkbaar zijn met die welke al in de analysemethode zijn opgenomen. In een dergelijk geval is de toepassing van een beperkte validatieregeling aanvaardbaar. Evenmin mag van de beschrijving van de methode worden afgeweken.
De ijkcurves voor de aanvullende stoffen moeten volgens de gevalideerde procedure worden opgesteld. Er moeten verschillende batches van matrixmaterialen worden geanalyseerd waaraan analyt is toegevoegd bij verschillende concentratieniveaus (zie 2.2.1, 2.2.2). De juistheid, herhaalbaarheid en intralaboratoriumreproduceerbaarheid/intermediaire precisie moeten binnen een bereik liggen dat vergelijkbaar is met dat van de andere analyten van de oorspronkelijk gevalideerde methode en in overeenstemming zijn met de voorschriften van punt 1.2.2. Voor de nieuwe analyten moet een berekening van het CCβ (screeningsmethoden) en de CCα (bevestigingsmethoden) worden gemaakt.
4.3. Uitbreidingen van methoden wat betreft de matrices/soorten
De opname van nieuwe matrices of soorten in een reeds gevalideerde analysemethode wordt altijd per geval afgewogen op basis van de tot dusver opgedane kennis en ervaring met de methode en voorbereidende proeven waarbij potentiële matrixeffecten en storingen worden beoordeeld. In het algemeen is dit alleen mogelijk voor matrices met vergelijkbare eigenschappen en voor niet-kritische analyten (stabiliteit, detecteerbaarheid).
IJkkrommen (standaard of matrix) moeten volgens de gevalideerde procedure worden opgesteld. Er moeten verschillende batches van matrixmateriaal worden geanalyseerd waaraan analyt is toegevoegd bij verschillende concentratieniveaus (zie 2.2.1, 2.2.2). De juistheid, herhaalbaarheid en intralaboratoriumreproduceerbaarheid/intermediaire precisie moeten binnen een bereik liggen dat aanvaardbaar is ten opzichte van de oorspronkelijk gevalideerde methode en in overeenstemming zijn met de voorschriften van punt 1.2.2. Afhankelijk van de validatiemethode kan een herberekening van het CCβ (screeningsmethoden) of de CCα (bevestigingsmethoden) nodig zijn.
Als de resultaten niet binnen een aanvaardbaar bereik liggen ten opzichte van de waarden voor de oorspronkelijke matrix, is een aanvullende volledige validatie nodig om de specifieke prestatieparameters voor de matrix/soort te bepalen.
Wanneer de MRL’s voor een specifieke stof voor bepaalde matrices verschillen, zal het hoogstwaarschijnlijk moeilijk zijn om het toepassingsgebied van de methode aan te passen aan de aanvullende matrix/soort en concentratie, aangezien in dit geval twee wijzigingen moeten worden overwogen. In dergelijke gevallen wordt een volledige validatie aanbevolen.
(1) Co-chromatografie is een procedure waarbij het monsterextract vóór de chromatografiestap(pen) in tweeën gedeeld wordt: een deel wordt als zodanig chromatografisch gescheiden; het andere deel wordt gemengd met de standaardanalyt die moet worden gemeten. Vervolgens wordt dit mengsel ook chromatografisch gescheiden. De hoeveelheid toegevoegde standaardanalyt moet ongeveer gelijk zijn aan de geschatte hoeveelheid analyt in het extract. Co-chromatografie wordt gebruikt om de identificatie van een analyt te verbeteren wanneer chromatografische methoden worden gebruikt, vooral wanneer er geen geschikte interne standaard gebruikt kan worden.
(*) De hier aangegeven VC (%) dient als richtsnoer; hij moet zo laag zijn als redelijkerwijs mogelijk.
(a) Er wordt geen extra identificatiepunt voor de selectie van het precursor-ion verkregen als dit precursor-ion hetzelfde ion is (of een adduct of isotoop) als het HRMS-ion in de full-scanmonitoring.
(#) Indien stabiliteitsgegevens voor analyten in een matrix beschikbaar zijn uit wetenschappelijke literatuur of uit een ander laboratorium, hoeven deze gegevens niet opnieuw door het betrokken laboratorium te worden bepaald. Een verwijzing naar beschikbare stabiliteitsgegevens van analyten in oplossing kan echter alleen worden aanvaard als voor identieke omstandigheden wordt gezorgd.
(*) Relevant voor MS-methoden om door middel van de validatie aan te tonen dat aan de voorschriften voor de prestatiekenmerken is voldaan. Het relatieve matrixeffect van de methode moet worden bepaald wanneer dit effect niet tijdens de validatieprocedure is beoordeeld. De absolute terugvinding van de methode moet worden bepaald als er geen interne standaard of geen matrixverrijkte ijking wordt gebruikt.
(2) ISO 5725-4:2020 Nauwkeurigheid (juistheid en precisie) van meetmethoden en –resultaten — Deel 4: Basismethoden voor het bepalen van de juistheid van een standaard meetmethode (punt 3).
(3) Wanneer voor een niet-toegelaten farmacologisch werkzame stof validatie van een concentratie van 0,5 maal de actiedrempel redelijkerwijs niet haalbaar is, kan de concentratie van 0,5 maal de actiedrempel worden vervangen door de laagste concentratie tussen 0,5 en 1,0 maal de actiedrempel die redelijkerwijs haalbaar is.
(4) Wanneer voor een specifieke farmacologisch werkzame stof validatie van een concentratie van 0,1 maal de MRL redelijkerwijs niet haalbaar is, kan de concentratie van 0,1 maal de MRL worden vervangen door de laagste concentratie tussen 0,1 en 0,5 maal de MRL die redelijkerwijs haalbaar is.
(5) Wanneer voor een niet-toegelaten farmacologisch werkzame stof validatie van een concentratie van 0,5 maal de actiedrempel redelijkerwijs niet haalbaar is, kan de concentratie van 0,5 maal de actiedrempel worden vervangen door de laagste concentratie tussen 0,5 en 1,0 maal de actiedrempel die redelijkerwijs haalbaar is.
(6) Wanneer voor een specifieke farmacologisch werkzame stof validatie van een concentratie van 0,1 maal de MRL redelijkerwijs niet haalbaar is, kan de concentratie van 0,1 maal de MRL worden vervangen door de laagste concentratie tussen 0,1 en 0,5 maal de MRL die redelijkerwijs haalbaar is.
(7) ISO 5725-2:2019 Nauwkeurigheid (juistheid en precisie) van meetmethoden en –resultaten — Deel 2: Basismethode voor de bepaling van herhaalbaarheid en reproduceerbaarheid van een standaard meetmethode (punt 3).
(8) ISO 11843-1:1997 — Detectievermogen — Deel 1: Termen en definities.
(9) Commissie van de Codex Alimentarius, Voedsel- en Landbouworganisatie van de Verenigde Naties, Wereldgezondheidsorganisatie, Guidelines on estimation of uncertainty of results (CAC/GL 59-2006).
(10) De veranderingen van de proefomstandigheden waarnaar daarin wordt verwezen, kunnen betrekking hebben op de monstermaterialen, analyten, opslagcondities en omgevings- en/of monstervoorbereidingsomstandigheden. Voor alle proefomstandigheden die in de praktijk aan fluctuaties onderhevig zouden kunnen zijn (bijvoorbeeld stabiliteit van de reagentia, samenstelling van het monster, pH, temperatuur) worden alle afwijkingen die invloed zouden kunnen hebben op het resultaat van de analyse aangegeven.
(11) Jülicher, B., Gowik, P., and Uhlig, S. (1998), Assessment of detection methods in trace analysis by means of a statistically based in-house validation concept. Analyst, 120, 173.
(12) IUPAC (1995), Protocol for the design, conduct and interpretation of method-performance studies, Pure & Applied Chem, 67, 331.
(13) Gowik, P., Jülicher, B., and Uhlig, S. (1998), Multi-residue method for non-steroidal anti-inflammatory drugs in plasma using high performance liquid chromatography-photodiodearray detection. Method description and comprehensive in-house validation. J. Chromatogr., 716, 221.
(14) ISO 11843-1:1997 — Detectievermogen — Deel 1: Termen en definities.
(15) Uitvoeringsverordening (EU) 2018/470 van de Commissie van 21 maart 2018 betreffende nadere regels voor de maximumwaarde voor residuen die in aanmerking moet worden genomen bij de controle van levensmiddelen die afkomstig zijn van overeenkomstig artikel 11 van Richtlijn 2001/82/EG in de Unie behandelde dieren (PB L 79 van 22.3.2018, blz. 16).
(16) De hoeveelheid toegevoegde standaardanalyt kan bijvoorbeeld ongeveer twee- tot vijfmaal zo groot zijn als de geschatte hoeveelheid analyt in het monster. Deze procedure is bedoeld om het gehalte van een analyt in een monster te bepalen met inachtneming van de terugvinding van de analysemethode.
(17) ISO/IEC 17025:2017 — Algemene eisen voor de bekwaamheid van beproevings- en kalibratielaboratoria (hoofdstuk 7.7).
(18) ISO/IEC 17043:2010 — Conformiteitsbeoordeling — Algemene eisen voor ringtesten.
BIJLAGE II
BEMONSTERINGSPROCEDURES EN OFFICIËLE MONSTERBEHANDELING
1. Omvang van de monsters
De minimale monsterhoeveelheden worden vastgesteld in het nationale residubewakingsprogramma. De minimale monsterhoeveelheden moeten volstaan om de erkende laboratoria in staat te stellen de analyseprocedures uit te voeren die nodig zijn om de screening en de bevestigingsanalyses te voltooien. Specifiek voor pluimvee, aquacultuur, konijnen, gekweekt wild, reptielen en insecten bestaat een monster uit een of meer dieren, afhankelijk van de voorschriften van de analysemethoden. Voor eieren bedraagt de omvang van het te nemen monster ten minste twaalf eieren, of meer, naargelang van de toe te passen analysemethode. Indien verschillende stofcategorieën in één monster met verschillende analysemethoden moeten worden geanalyseerd, wordt de monstergrootte dienovereenkomstig vergroot.
2. Opsplitsing in submonsters
Tenzij het technisch onmogelijk is of niet vereist krachtens de nationale wetgeving, wordt elk monster opgesplitst in ten minste twee equivalente submonsters die elk afzonderlijk kunnen volstaan voor het uitvoeren van de volledige analyseprocedure. De opsplitsing kan plaatsvinden op de plaats van bemonstering of in het laboratorium.
3. Traceerbaarheid
Elk monster wordt zo genomen dat het altijd mogelijk is het terug te traceren naar het bedrijf van oorsprong en de partij dieren of het individuele dier, voor zover van toepassing. Met name kunnen de monsters voor melk naar keuze van de lidstaat op een van de volgende plaatsen worden genomen:
|
1. |
op het bedrijf van de verzameltank; |
|
2. |
in de zuivelfabriek, vóór het lossen van de melk. |
4. Recipiënten voor de monsters
De monsters worden verzameld in daarvoor geschikte recipiënten teneinde te garanderen dat de deugdelijkheid van de monsters niet wordt aangetast en dat de herkomst ervan op elk moment kan worden bepaald. De recipiënten moeten met name zo zijn ontworpen dat vervanging, kruisbesmetting en bederf van de monsters worden voorkomen. De recipiënten worden officieel verzegeld.
5. Bemonsteringsrapport
Na elke bemonstering wordt een rapport opgesteld.
In het bemonsteringsrapport vermeldt de inspecteur ten minste de volgende gegevens:
|
1. |
adres van de bevoegde autoriteit; |
|
2. |
naam van de inspecteur of identificatiecode; |
|
3. |
officieel codenummer van het monster; |
|
4. |
datum van bemonstering; |
|
5. |
naam en adres van de eigenaar van de dieren of de dierlijke producten, of van de persoon die ervoor verantwoordelijk is; |
|
6. |
naam en adres van het bedrijf van herkomst van het dier (bij bemonstering op het bedrijf); |
|
7. |
registratienummer van het bedrijf/nummer van het slachthuis; |
|
8. |
identificatie van het dier of het product; |
|
9. |
diersoort; |
|
10. |
aard van de monstermatrix; |
|
11. |
in voorkomend geval, in de laatste vier weken vóór de bemonstering toegediende geneesmiddelen (bij bemonstering op het bedrijf); |
|
12. |
op te sporen stoffen of groepen stoffen; |
|
13. |
bijzondere opmerkingen. |
Afhankelijk van de bemonsteringsprocedure moeten papieren of elektronische exemplaren van het rapport worden verstrekt. Het bemonsteringsrapport en de kopieën daarvan worden zodanig ingevuld dat hun authenticiteit en rechtsgeldigheid gewaarborgd zijn, waarvoor het nodig kan zijn dat deze documenten door de inspecteur worden ondertekend. In het geval van bemonstering op het bedrijf kan de bedrijfsvoerder of zijn assistent worden verzocht het originele bemonsteringsrapport te ondertekenen.
Het originele exemplaar van het bemonsteringsrapport blijft bij de bevoegde autoriteit, die dient te garanderen dat onbevoegden geen toegang hebben tot dat origineel.
Indien nodig, wordt de bedrijfsvoerder of de eigenaar van het bedrijf ervan in kennis gesteld dat bemonstering zal plaatsvinden.
6. Bemonsteringsrapport voor het laboratorium
Het door de bevoegde autoriteiten opgestelde bemonsteringsrapport voor het laboratorium moet in overeenstemming zijn met de voorschriften van hoofdstuk 7 van de ISO/IEC-norm 17025:2017 (1) en ten minste de volgende informatie bevatten:
|
1. |
adres van de bevoegde autoriteiten of aangewezen instanties; |
|
2. |
naam van de inspecteur of identificatiecode; |
|
3. |
officieel codenummer van het monster; |
|
4. |
datum van bemonstering; |
|
5. |
diersoort; |
|
6. |
aard van de monstermatrix; |
|
7. |
op te sporen stoffen of groepen stoffen; |
|
8. |
bijzondere opmerkingen. |
Het bemonsteringsrapport voor het laboratorium vergezelt het monster wanneer het naar het laboratorium wordt gestuurd.
7. Vervoer en opslag
Residubewakingsprogramma’s moeten voorzien in adequate opslag en vervoer voor elke analyt/matrixcombinatie, teneinde te garanderen dat de analyt stabiel blijft en het monster deugdelijk. Het vervoer moet zo kort mogelijk duren en de temperatuur tijdens het vervoer moet geschikt zijn om de stabiliteit van de analyt te waarborgen.
Bijzondere aandacht moet worden besteed aan de transportdozen, de temperatuur en de termijn voor levering bij het bevoegde laboratorium.
Wanneer niet aan de in het bewakingsprogramma vastgestelde eisen wordt voldaan, stelt het laboratorium de bevoegde autoriteit daarvan onverwijld in kennis.
(1) ISO/IEC 17025:2017 — Algemene eisen voor de bekwaamheid van beproevings- en kalibratielaboratoria (hoofdstuk 7.7).