ISSN 1977-0715
Eiropas Savienības
Oficiālais Vēstnesis
L 426

Izdevums latviešu valodā
Tiesību akti
64. gadagājums
2021. gada 29. novembris
|
ISSN 1977-0715 |
||
|
Eiropas Savienības Oficiālais Vēstnesis |
L 426 |
|
|
|
||
|
Izdevums latviešu valodā |
Tiesību akti |
64. gadagājums |
|
|
|
|
|
(1) Dokuments attiecas uz EEZ. |
|
LV |
Tiesību akti, kuru virsraksti ir gaišajā drukā, attiecas uz kārtējiem jautājumiem lauksaimniecības jomā un parasti ir spēkā tikai ierobežotu laika posmu. Visu citu tiesību aktu virsraksti ir tumšajā drukā, un pirms tiem ir zvaigznīte. |
II Neleģislatīvi akti
REGULAS
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/1 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2076
(2021. gada 26. novembris)
par atļauju L-triptofānu, kas iegūts ar Escherichia coli KCCM 80210, izmantot par barības piedevu visu sugu dzīvniekiem
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1831/2003 (2003. gada 22. septembris) par dzīvnieku ēdināšanā lietotām piedevām (1) un jo īpaši tās 9. panta 2. punktu,
tā kā:
|
(1) |
Regulā (EK) Nr. 1831/2003 noteikts, ka piedevu lietošanai dzīvnieku ēdināšanā jāsaņem atļauja, un paredzēts šādas atļaujas piešķiršanas pamatojums un kārtība. |
|
(2) |
Saskaņā ar Regulas (EK) Nr. 1831/2003 7. pantu tika iesniegts pieteikums piešķirt atļauju L-triptofānam, kas iegūts ar Escherichia coli KCCM 80210. Pieteikumam ir pievienotas minētās regulas 7. panta 3. punktā prasītās ziņas un dokumenti. |
|
(3) |
Pieteikums attiecas uz atļauju lietot L-triptofānu, kas iegūts ar Escherichia coli KCCM 80210, par barības piedevu visu sugu dzīvniekiem, kuru paredzēts klasificēt piedevu kategorijā “uzturfizioloģiskās piedevas”, funkcionālajā grupā “aminoskābes, to sāļi un analogi”. |
|
(4) |
Eiropas Pārtikas nekaitīguma iestāde (“Iestāde”) 2021. gada 27. janvāra atzinumā (2) secināja, ka ar piedāvātajiem lietošanas nosacījumiem L-triptofāns, kas iegūts ar Escherichia coli KCCM 80210, kaitīgi neietekmē nedz neatgremotāju dzīvnieku veselību, nedz patērētāju drošību vai vidi. Lai L-triptofāns būtu drošs atgremotājiem, tas būtu jāpasargā no noārdīšanās spureklī. Iestāde norādīja, ka novērtējamā piedeva tiek uzskatīta par vieglu acu kairinātāju. Piedevas endotoksīna aktivitāte un tās putekļu veidošanās potenciāls norāda uz risku to ieelpojot. Tāpēc Komisija uzskata, ka būtu jāveic pienācīgi aizsargpasākumi, lai nepieļautu nelabvēlīgu ietekmi uz cilvēka, jo īpaši uz piedevas lietotāju, veselību. |
|
(5) |
Iestāde uzskatīja, ka L-triptofāns, kas iegūts ar Escherichia coli KCCM 80210, ir iedarbīgs neaizstājamās aminoskābes triptofāna avots neatgremotājiem dzīvniekiem; un lai papildu L-triptofāns, kas iegūts ar Escherichia coli KCCM 80210, būtu tikpat iedarbīgs atgremotājiem kā neatgremotājiem dzīvniekiem, tas būtu jāaizsargā no noārdīšanās spureklī. Iestāde neuzskata, ka būtu jānosaka īpašas prasības uzraudzībai pēc piedevas laišanas tirgū. Turklāt tā verificēja ar Regulu (EK) Nr. 1831/2003 izveidotās references laboratorijas iesniegto ziņojumu par dzīvnieku barības piedevas analīzes metodi. |
|
(6) |
L-triptofāna, kas iegūts ar Escherichia coli KCCM 80210, novērtējums liecina, ka Regulas (EK) Nr. 1831/2003 5. pantā paredzētie atļaujas piešķiršanas nosacījumi ir izpildīti. Tādēļ šo vielu būtu jāļauj lietot tā, kā norādīts šīs regulas pielikumā. |
|
(7) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Augu, dzīvnieku, pārtikas aprites un dzīvnieku barības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Pielikumā minēto vielu, kas iekļauta barības piedevu kategorijā “uzturfizioloģiskās piedevas” un funkcionālajā grupā “aminoskābes, to sāļi un analogi”, ir atļauts lietot par dzīvnieku barības piedevu, ņemot vērā pielikumā izklāstītos nosacījumus.
2. pants
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 268, 18.10.2003., 29. lpp.
(2) EFSA Journal 2021; 19(3):6425.
PIELIKUMS
|
Piedevas identifikācijas numurs |
Atļaujas turētāja nosaukums |
Piedeva |
Sastāvs, ķīmiskā formula, apraksts, analītiskā metode |
Dzīvnieku suga vai kategorija |
Maksimālais vecums |
Minimālais saturs |
Maksimālais saturs |
Citi noteikumi |
Atļaujas derīguma termiņš |
||||||||
|
mg uz kg kompleksās barības ar mitruma saturu 12 % |
|||||||||||||||||
|
Kategorija: uzturfizioloģiskās piedevas Funkcionālā grupa: aminoskābes, to sāļi un analogi |
|||||||||||||||||
|
3c440i |
- |
L-triptofāns |
Piedevas sastāvs: Pulveris, kurā ir vismaz 98 % L-triptofāna (sausnā) un ar maksimālo mitruma saturu 1 %. Maksimālais 1,1′-etilidēn-bis-L-triptofāna (EBT) saturs: 10 mg/kg. |
Visu sugu dzīvnieki |
- |
- |
- |
|
2031. gada 19. decembris |
||||||||
|
Aktīvās vielas raksturojums: L-triptofāns, kas iegūts fermentācijā ar Escherichia coli KCCM 80210 Ķīmiskā formula: C11H12N2O2 CAS Nr.: 73-22-3 |
|||||||||||||||||
|
Analītiskās metodes: (1) L-triptofāna noteikšanai barības piedevā:
|
|||||||||||||||||
(1) Sīkāka informācija par analītiskajām metodēm ir pieejama references laboratorijas tīmekļa vietnē: https://ec.europa.eu/jrc/en/eurl/feed-additives/evaluation-reports.
(2) Ekspozīcija aprēķināta, balstoties uz endotoksīnu līmeni un piedevas putekļu veidošanas potenciālu, saskaņā ar metodi, ko izmanto EFSA (EFSA Journal 2015;13(2):4015); analītiskā metode: Eiropas Farmakopeja 2.6.14. (bakteriālie endotoksīni).
(3) Komisijas Regula (EK) Nr. 152/2009 ( 2009. gada 27. janvāris), ar ko nosaka paraugu ņemšanas un analīzes metodes barības oficiālajai kontrolei (OV L 54, 26.2.2009., 1. lpp.).
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/5 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2077
(2021. gada 26. novembris)
par atļauju ar Corynebacterium glutamicum CGMCC 7.366 iegūtu L-valīnu lietot par barības piedevu visu sugu dzīvniekiem
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1831/2003 (2003. gada 22. septembris) par dzīvnieku ēdināšanā lietotām piedevām (1) un jo īpaši tās 9. panta 2. punktu,
tā kā:
|
(1) |
Regulā (EK) Nr. 1831/2003 noteikts, ka piedevu lietošanai dzīvnieku ēdināšanā ir vajadzīga atļauja, un paredzēts šādas atļaujas piešķiršanas pamatojums un kārtība. |
|
(2) |
Saskaņā ar Regulas (EK) Nr. 1831/2003 7. pantu ir iesniegts pieteikums uz atļauju izmantot L-valīnu. Pieteikumam ir pievienotas minētās regulas 7. panta 3. punktā prasītās ziņas un dokumenti. |
|
(3) |
Pieteikumā lūgts atļaut ar Corynebacterium glutamicum CGMCC 7.366 iegūtu L-valīnu lietot par barības piedevu visu sugu dzīvniekiem, šo piedevu klasificējot piedevu kategorijā “uzturfizioloģiskās piedevas” un funkcionālajā grupā “aminoskābes, to sāļi un analogi.” |
|
(4) |
Eiropas Pārtikas nekaitīguma iestāde (“Iestāde”) 2021. gada 17. marta atzinumā (2) secināja, ka ar piedāvātajiem lietošanas nosacījumiem ar Corynebacterium glutamicum CGMCC 7.366 iegūts L-valīns, ja atbilstošā daudzumā pievienots uzturam, nelabvēlīgi neietekmē nedz dzīvnieku veselību, nedz patērētāju drošību vai vidi. Iestāde norādīja, ka attiecībā uz L-valīna lietotāju drošību tā nevar izslēgt ne ieelpošanas radītu risku, ne to, ka šī piedeva varētu būt kairinoša ādai vai acīm vai ādas sensibilizators. Tāpēc Komisija uzskata, ka būtu jāveic pienācīgi aizsargpasākumi, lai nepieļautu nelabvēlīgu ietekmi uz cilvēka, jo īpaši uz piedevas lietotāju, veselību. Iestāde turklāt secinājusi, ka minētā piedeva uzskatāma par iedarbīgu neaizstājamās aminoskābes L-valīna avotu dzīvnieku barībā un ka, lai piedeva būtu iedarbīga atgremotājiem, tā būtu jāpasargā no noārdīšanās spureklī. Iestāde neuzskata, ka būtu vajadzīgas īpašas prasības veikt uzraudzību pēc preparāta laišanas tirgū. Tā arī verificējusi ar Regulu (EK) Nr. 1831/2003 izveidotās references laboratorijas iesniegtos ziņojumus par barībā esošās barības piedevas analīzes metodi. |
|
(5) |
Ar Corynebacterium glutamicum CGMCC 7.366 iegūtā L-valīna novērtējums liecina, ka Regulas (EK) Nr. 1831/2003 5. panta nosacījumi atļaujas piešķiršanai ir izpildīti. Tāpēc šo vielu būtu jāatļauj izmantot tā, kā norādīts šīs regulas pielikumā. |
|
(6) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Augu, dzīvnieku, pārtikas aprites un dzīvnieku barības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Pielikumā specificēto vielu, kas pieder pie piedevu kategorijas “uzturfizioloģiskās piedevas” un funkcionālās grupas “aminoskābes, to sāļi un analogi”, ir atļauts lietot par dzīvnieku barības piedevu, ievērojot pielikumā noteiktos nosacījumus.
2. pants
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 268, 18.10.2003., 29. lpp.
(2) EFSA Journal 2021;19(4):6521.
PIELIKUMS
|
Piedevas identifikācijas numurs |
Atļaujas turētāja nosaukums |
Piedeva |
Sastāvs, ķīmiskā formula, apraksts, analītiskā metode |
Dzīvnieku suga vai kategorija |
Maksimālais vecums |
Minimālais saturs |
Maksimālais saturs |
Citi noteikumi |
Atļaujas derīguma termiņš |
||||||||
|
mg uz kg kompleksās barības ar mitruma saturu 12 % |
|||||||||||||||||
|
Kategorija: uzturfizioloģiskās piedevas Funkcionālā grupa: aminoskābes, to sāļi un analogi |
|||||||||||||||||
|
3c371i |
— |
L-valīns |
Piedevas sastāvs Pulveris, kurā ir vismaz 98 % L-valīna (sausnā) un ūdens saturs nepārsniedz 1,5 %. |
Visas sugas |
— |
|
|
|
2031. gada 19. decembris |
||||||||
|
Aktīvās vielas raksturojums Ar Corynebacterium glutamicum CGMCC 7.366 iegūts L-valīns ((2S)-2-amino-3-metilbutānskābe). Ķīmiskā formula: C5H11NO2 CAS numurs: 72-18-4 |
|||||||||||||||||
|
Analītiskā metode (1) L-valīna noteikšanai barības piedevā:
Valīna kvantitatīvai noteikšanai premiksos, barības sastāvdaļās un barības maisījumos:
Valīna kvantitatīvai noteikšanai ūdenī:
|
|||||||||||||||||
(1) Sīkāka informācija par analītiskajām metodēm ir pieejama references laboratorijas tīmekļa vietnē: https://ec.europa.eu/jrc/en/eurl/feed-additives/evaluation-reports.
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/9 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2078
(2021. gada 26. novembris),
ar ko paredz noteikumus par to, kā Eiropas Parlamenta un Padomes Regulu (ES) 2017/745 piemērot Eiropas Medicīnisko ierīču datubāzei (Eudamed)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (ES) 2017/745 (2017. gada 5. aprīlis), kas attiecas uz medicīniskām ierīcēm, ar ko groza Direktīvu 2001/83/EK, Regulu (EK) Nr. 178/2002 un Regulu (EK) Nr. 1223/2009 un atceļ Padomes Direktīvas 90/385/EEK un 93/42/EEK (1), un jo īpaši tās 33. panta 8. punktu,
tā kā:
|
(1) |
Regula (ES) 2017/745 paredz, ka Komisijai ir jānosaka detalizēta kārtība, kas vajadzīga, lai izveidotu un uzturētu Eiropas Medicīnisko ierīču datubāzi (Eudamed). |
|
(2) |
Eiropas Parlamenta un Padomes Regula (ES) 2017/746 (2) paredz, ka Komisijai jāizveido, jāuztur un jāpārvalda Eudamed atbilstoši Regulā (ES) 2017/745 noteiktajiem nosacījumiem un detalizētajai kārtībai. |
|
(3) |
Kā noteikts Regulā (ES) 2017/745 un Regulā (ES) 2017/746, Komisijai, kompetentajām iestādēm, par paziņotajām struktūrām atbildīgajām iestādēm, paziņotajām struktūrām, ražotājiem, pilnvarotajiem pārstāvjiem, importētājiem, fiziskajām vai juridiskajām personām, kas minētas Regulas (ES) 2017/745 22. panta 1. punktā (sistēmas vai procedūras komplektu ražotāji), un klīnisko pētījumu un veiktspējas pētījumu sponsoriem vajadzētu būt piekļuvei Eudamed un būtu tā jāizmanto, lai saskaņā ar minētajām regulām izpildītu savas saistības un veiktu savus uzdevumus. Tāpēc piekļuve Eudamed ir jānodrošina ierobežotas piekļuves vietnē. Turklāt Eudamed būtu jāsniedz sabiedrībai pienācīga informācija par tirgū laistajām ierīcēm, paziņoto struktūru izdotajiem atbilstošajiem sertifikātiem, attiecīgajiem uzņēmējiem un klīniskajiem pētījumiem. Tāpēc arī jānodrošina, ka Eudamed ir piekļūstama publiskā vietnē. Turklāt, lai būtu iespējama datu apmaiņa starp Eudamed un valstu datubāzēm, Eudamed ir jābūt piekļūstamai, izmantojot mašīnas-mašīnas datu apmaiņas pakalpojumus. |
|
(4) |
Attiecībā uz fiziskām un juridiskām personām, kurām ir jāspēj piekļūt Eudamed ierobežotas piekļuves vietnē, ir jāparedz šādas piekļuves piešķiršanas nosacījumi un kārtība. |
|
(5) |
Komisija ir izveidojusi Eiropas Medicīnisko ierīču nomenklatūru (EMDN), kā noteikts Regulā (ES) 2017/745 un Regulā (ES) 2017/746. Tāpēc Eiropas Medicīnisko ierīču nomenklatūra būtu jāpadara pieejama Eudamed bez maksas un tā būtu jāizmanto, lai sniegtu informāciju par medicīniskajām ierīcēm Eudamed. |
|
(6) |
Lai nodrošinātu, ka Eudamed lietotāji saņem datubāzes lietošanai vajadzīgo atbalstu, Komisijai būtu tiem laikus jāsniedz tehniska un administratīva palīdzība saistībā ar Eudamed. |
|
(7) |
Eudamed tehniskas nepieejamības vai darbības traucējumu gadījumā pilnvarotajiem lietotājiem tomēr vajadzētu būt iespējai izpildīt savas saistības. Tāpēc ir jānosaka alternatīvi mehānismi datu apmaiņai šādos gadījumos un jāparedz ārkārtas rīcības noteikumi šādiem mehānismiem. |
|
(8) |
Uz Eudamed attiecas Komisijas Lēmumā (ES, Euratom) 2017/46 (3) paredzētie noteikumi par IT drošību. Lai Eudamed darbotos droši, būtu aizsargāta pret apdraudējumu tās funkciju un datu pieejamībai, integritātei un konfidencialitātei, būtu jānosaka papildu drošības noteikumi. |
|
(9) |
Lai mazinātu risku un nepieļautu Eudamed iespējamu krāpniecīgu izmantošanu, būtu jānosaka īpaši noteikumi pret krāpniecīgu lietotāju darbībām Eudamed. |
|
(10) |
Saskaņā ar Eiropas Parlamenta un Padomes Regulas (ES) 2018/1725 (4) 42. panta 1. punktu ir notikusi apspriešanās ar Eiropas Datu aizsardzības uzraudzītāju, kas 2021. gada 9. jūlijā sniedza atzinumu. |
|
(11) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Medicīnisko ierīču komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Definīcijas
Šajā regulā piemēro šādas definīcijas:
|
1) |
“izpildītājs” ir Komisija, kompetentā iestāde, par paziņotajām struktūrām atbildīgā iestāde, paziņotā struktūra, ražotājs, pilnvarotais pārstāvis, importētājs, sistēmas vai procedūras komplekta ražotājs vai sponsors, kas ir reģistrēts Eudamed atbilstoši šīs regulas 3. pantam, lai izpildītu Regulā (ES) 2017/745 un Regulā (ES) 2017/746 noteiktās saistības; |
|
2) |
“pilnvarots lietotājs” ir fiziska persona, kurai, lai rīkotos izpildītāja vārdā, ir piešķirta piekļuve Eudamed ierobežotas piekļuves vietnē; |
|
3) |
“vietējais izpildītāja administrators” (LAA) ir pilnvarots lietotājs, kuram ir tiesības pārvaldīt konkrētu izpildītāja informāciju un piešķirt piekļuvi Eudamed ierobežotas piekļuves vietnē citām fiziskām personām, kas rīkosies minētā izpildītāja vārdā; |
|
4) |
“vietējais lietotāju administrators” (LUA) ir pilnvarots lietotājs, kuram ir tiesības piešķirt piekļuvi Eudamed ierobežotas piekļuves vietnē citām fiziskām personām, kas rīkosies izpildītāja vārdā; |
|
5) |
“darbības traucējums” ir Eudamed darbības būtiska atteice, tai skaitā atteice neparedzētu apstākļu vai force majeure apstākļu dēļ, kas var negatīvi ietekmēt IT drošību vai kavēt Regulas (ES) 2017/745 33. panta 2. punktā minēto Eudamed elektronisko sistēmu funkcionalitāšu pieejamību. |
2. pants
Piekļuves veidi
1. Pilnvaroti lietotāji Eudamed var piekļūt ierobežotas piekļuves vietnē (“ierobežotas piekļuves vietne”), bet neidentificēti lietotāji – publiski pieejamā vietnē (“publiskā vietne”).
2. Kompetentajām iestādēm, kas minētas Regulas (ES) 2017/745 101. pantā un Regulas (ES) 2017/746 96. pantā (“kompetentās iestādes”), un paziņotajām struktūrām, kas Eudamed reģistrētas atbilstoši šīs regulas 3. pantam, Eudamed ir piekļūstama mašīnas-mašīnas datu apmaiņas pakalpojumiem. Komisija katrai dalībvalstij un paziņotajai struktūrai nodrošina datu apmaiņas piekļuves punktus, kas dod iespēju pēc pieprasījuma izmantot šādus datu apmaiņas pakalpojumus.
Eudamed ar mašīnas-mašīnas datu apmaiņas pakalpojumiem ir piekļūstama izpildītājiem, kas nav kompetentās iestādes un paziņotās struktūras, ja attiecīgā izpildītāja LAA iesniedz šādas piekļuves pieprasījumu, kā noteikts 3. panta 8. punkta pirmajā daļā. Komisija minēto pieprasījumu apstiprina atbilstoši nosacījumiem, kas noteikti 3. panta 8. punkta otrajā daļā.
3. pants
Reģistrācija Eudamed un piekļuve Eudamed ierobežotas piekļuves vietnē
1. Lai saņemtu piekļuvi Eudamed ierobežotas piekļuves vietnē, fiziska persona izveido kontu Komisijas autentifikācijas sistēmas vietnē.
2. Komisija reģistrē kompetentās iestādes un par paziņotajām struktūrām atbildīgās iestādes un piešķir piekļuvi ierobežotas piekļuves vietnei pirmajai fiziskajai personai, kas rīkojas to vārdā. Šajā nolūkā dalībvalstis sniedz Komisijai informāciju par kompetentajām iestādēm, par paziņotajām struktūrām atbildīgajām iestādēm un fiziskajām personām, kas kļūs par minēto iestāžu pirmajiem pilnvarotajiem lietotājiem.
3. Komisija paziņotās struktūras Eudamed reģistrē, pamatojoties uz informāciju, kas ir Komisijas izstrādātajā un pārvaldītajā paziņoto struktūru datubāzē (NANDO).
Lai saņemtu piekļuvi Eudamed ierobežotas piekļuves vietnē, pirmā fiziskā persona, kura rīkojas tāda izpildītāja vārdā, kas ir paziņotā struktūra, piekļuves pieprasījumu iesniedz ierobežotas piekļuves vietnē. Pieprasījumu apstiprina par paziņoto struktūru atbildīgā iestāde.
4. Lai Eudamed reģistrētu citas organizācijas, kas nav minētas 2. un 3. punktā, fiziska persona, kas rīkojas potenciālā izpildītāja vārdā, iesniedz izpildītāja reģistrācijas pieprasījumu ierobežotas piekļuves vietnē. Izpildītāja reģistrācijas pieprasījumā iekļauj parakstītu 10. panta 1. punktā minēto deklarāciju par pienākumiem attiecībā uz informācijas drošību. Izpildītāja reģistrācijas pieprasījumu apstiprina valsts kompetentā iestāde, izņemot, ja pieprasījums attiecas uz klīniskā pētījuma vai veiktspējas pētījuma sponsoru.
Pēc izpildītāja reģistrācijas pieprasījuma apstiprināšanas vai – sponsora gadījumā – pēc izpildītāja reģistrācijas pieprasījuma iesniegšanas, fiziskajai personai, kura iesniedza pirmajā daļā minēto pieprasījumu, automātiski tiek piešķirta piekļuve ierobežotas piekļuves vietnei un tā kļūst par pirmo pilnvaroto lietotāju, ja ir izpildīti 6. punktā paredzētie nosacījumi.
Šā punkta vajadzībām valsts kompetentā iestāde ir potenciālā izpildītāja iedibināšanas vietas iestāde. To ražotāju gadījumā, kas iedibināti ārpus Savienības, valsts kompetentā iestāde ir iestāde, kas ir atbildīga par izpildītāja reģistrācijas pieprasījumā norādīto pilnvaroto pārstāvi. Attiecībā uz sistēmas un procedūras komplektu ražotājiem, kas iedibināti ārpus Savienības, valsts kompetentā iestāde ir tās dalībvalsts iestāde, kurā tiek laists tirgū attiecīgā ražotāja pirmais sistēmas vai procedūras komplekts.
5. Lai fiziska persona saņemtu piekļuvi ierobežotas piekļuves vietnei un varētu rīkoties izpildītāja vārdā, tā iesniedz piekļuves pieprasījumu ierobežotas piekļuves vietnē. Šo piekļuves pieprasījumu apstiprina minētā izpildītāja LAA vai LUA.
6. Lai kļūtu par pilnvarotiem lietotājiem, fiziskās personas piekrīt lietotāja tiesībām un pienākumiem, kas izklāstīti 10. panta 1. punkta a) apakšpunktā minētajā dokumentā, un iepazīstas ar minētā panta c) apakšpunktā minēto paziņojumu par privātumu.
7. Izpildītāja pirmais pilnvarotais lietotājs automātiski kļūst par minētā izpildītāja pirmo LAA.
8. LAA ierobežotas piekļuves vietnē var iesniegt Komisijai pieprasījumu par mašīnas-mašīnas savienojumu, lai varētu veikt datu apmaiņu starp izpildītāja datubāzi un Eudamed.
Komisija pirmajā daļā minēto pieprasījumu apstiprina, ja LAA ir apstiprinājis, ka minētais izpildītājs atbilst 10. panta 1. punktā minētajām datu apmaiņas informācijas drošības prasībām.
4. pants
Nomenklatūra
Iesniedzot Eudamed informāciju par medicīniskajām ierīcēm, pilnvarotie lietotāji izmanto Eiropas Medicīnisko ierīču nomenklatūras (EMDN) atvērtos piekļuves kodus.
Komisija EMDN padara pieejamu Eudamed bez maksas.
5. pants
Tehniskais un administratīvais atbalsts
1. Komisija izveido pieteikumu atbalsta grupu, kas Eudamed lietotājiem sniedz savlaicīgu palīdzību, izmantojot īpaši šim nolūkam izveidotu funkcionālo pastkastīti.
2. Komisija Eudamed lietotājiem padara pieejamu attiecīgo tehnisko dokumentāciju par Eudamed, bieži uzdotos jautājumus par Eudamed un dokumentāciju par mašīnas-mašīnas datu apmaiņas pakalpojumiem.
6. pants
Īpašumtiesības un persondatu apstrāde
1. Komisija ir Eudamed īpašniece, un tai ir visas administratora tiesības.
2. Persondatus Eudamed apstrādā, lai izpildītu Regulā (ES) 2017/745 un Regulā (ES) 2017/746 noteiktās saistības.
3. Apstrādā šādas persondatu kategorijas:
|
a) |
izpildītāju nosaukums un pilnvaroto lietotāju vārds, uzvārds; |
|
b) |
izpildītāju un pilnvaroto lietotāju kontaktinformācija; |
|
c) |
to fizisko un juridisko personu identifikācija, kontaktinformācija un profesionālās kvalifikācijas dati, par kurām ir ziņots Eudamed, lai izpildītu Regulā (ES) 2017/745 un Regulā (ES) 2017/746 noteiktās saistības. |
7. pants
Darbības noteikumi
1. Datu iesniegšanu Eudamed uzskata par pabeigtu dienā un laikā, kad dati ir sekmīgi reģistrēti Eudamed. Iesniegšanas dienu un laiku nosaka pēc attiecīgi Centrāleiropas laika (CET) vai Centrāleiropas vasaras laika (CEST).
2. Eudamed ir piekļūstama jebkurā laikā, izņemot nepieciešamos un iepriekš paziņotos traucējumu periodus, kad tiek veiktas apkopes darbības, tai skaitā atjauninājumi. Komisija šajā sakarā iepriekš attiecīgi ierobežotas piekļuves vietnē vai publiskajā vietnē publicē paziņojumu.
8. pants
Darbības traucējumi
1. Komisija veic visus vajadzīgos pasākumus, lai nepieļautu darbības traucējumus un bez liekas kavēšanās tos identificētu, ja tie radušies.
2. Ja izpildītājam vai pilnvarotam lietotājam ir aizdomas par darbības traucējumiem, tas par tiem nekavējoties informē Komisiju.
3. Ja Komisija ir identificējusi darbības traucējumu, tā veic šādus pasākumus:
|
a) |
nekavējoties šajā sakarā publicē paziņojumu (“paziņojums par darbības traucējumiem”) attiecīgi ierobežotas piekļuves vietnē vai publiskajā vietnē, ja vien darbības traucējums neliedz Komisijai to publicēt, un šādā gadījumā tā, ja vien iespējams, šādu paziņojumu publicē Komisijas specializētajā ar medicīniskajām ierīcēm saistītajā vietnē; |
|
b) |
aptur termiņu piemērošanu to datu iesniegšanai Eudamed, kas minēti Regulā (ES) 2017/745 un Regulā (ES) 2017/746, ja darbības traucējums kavē attiecīgo datu ievadīšanu. |
Ja Komisija aptur termiņu piemērošanu datu iesniegšanai Eudamed, kā noteikts pirmās daļas b) punktā, paziņojumā par darbības traucējumiem norāda paziņojuma publicēšanas laiku un iespējamo apturēšanas ilgumu.
4. Ja darbības traucējums kavē kādu to saistību izpildi, kuras minētas Regulas (ES) 2017/745 80. pantā, 87. panta 1. punktā, 89. panta 5., 7., 8. un 9. punktā, 95. panta 2., 4. un 6. punktā vai 98. panta 2. punktā vai Regulas (ES) 2017/746 76. pantā, 82. panta 1. punktā, 84. panta 5., 7., 8. un 9. punktā, 90. panta 2., 4. un 6. punktā vai 93. panta 2. punktā, papildus tam, ka saskaņā ar šā panta 3. punkta pirmās daļas b) punktu aptur termiņa piemērošanu, piemēro vienu no šādām procedūrām:
|
a) |
ja darbības traucējums ilgst vairāk nekā 12 stundas no brīža, kad publicēts paziņojums par darbības traucējumiem, izpildītājs nekavējoties Komisijai, attiecīgajām valsts kompetentajām iestādēm un paziņotajai struktūrai, kas izdevusi attiecīgi Regulas (ES) 2017/745 56. pantā vai Regulas (ES) 2017/746 51. pantā minēto atbilstības sertifikātu, sniedz vispārīgu informāciju par attiecīgajiem datiem un paziņo, ka datu iesniegšana ir aizkavējusies darbības traucējuma dēļ; |
|
b) |
ja darbības traucējums ilgst vairāk nekā 24 stundas no brīža, kad publicēts paziņojums par darbības traucējumiem, vai ja darbības traucējums ilgst vairāk nekā 24 stundas un attiecīgās valsts kompetentās iestādes pēc šā punkta a) apakšpunktā minētās informācijas saņemšanas to pieprasa, izpildītājs nekavējoties tam norādītājā veidā minētajām iestādēm iesniedz attiecīgos datus. |
5. Ja darbības traucējums kavē kādu to saistību izpildi, kas minētas Regulā (ES) 2017/745 vai Regulā (ES) 2017/746 un kas nav saistības, kuras minētas šā panta 4. punktā, papildus tam, ka saskaņā ar šā panta 3. punkta pirmās daļas b) punktu aptur termiņa piemērošanu, piemēro šādu procedūru:
|
a) |
ja darbības traucējums ilgst vairāk nekā 36 stundas no brīža, kad publicēts paziņojums par darbības traucējumiem, izpildītājs nekavējoties Komisijai, attiecīgajām valsts kompetentajām iestādēm un paziņotajai struktūrai, kas izdevusi attiecīgi Regulas (ES) 2017/745 56. pantā vai Regulas (ES) 2017/746 51. pantā minēto atbilstības sertifikātu, sniedz vispārīgu informāciju par minētajiem datiem un paziņo, ka datu iesniegšana ir aizkavējusies darbības traucējuma dēļ; |
|
b) |
ja darbības traucējums ilgst vairāk nekā piecas dienas no brīža, kad publicēts paziņojums par darbības traucējumiem, izpildītājs par to informē attiecīgās valsts kompetentās iestādes un, ja tās to pieprasa, tam norādītājā veidā minētajām iestādēm iesniedz attiecīgos datus. |
6. Ja Komisija ir konstatējusi, ka darbības traucējums ir novērsts, tā šo informāciju paziņo kompetentajām iestādēm. Turklāt Komisija šajā sakarā attiecīgi ierobežotas piekļuves vietnē un/vai publiskajā vietnē publicē paziņojumu. Abos paziņojumos norāda darbības traucējuma ilgumu un laiku, uz kādu tikusi apturēta 3. punkta b) apakšpunktā minētā termiņa piemērošana.
7. Ja Komisija ir publicējusi 6. punktā minēto paziņojumu, izpildītāji nekavējoties Eudamed ievada datus, kurus tie nevarēja iesniegt Eudamed darbības traucējumu laikā.
9. pants
Testēšanas un apmācības vietnes
1. Komisija dara izpildītājiem pieejamas vietnes testēšanas un apmācības vajadzībām saistībā ar Eudamed lietošanu (“testēšanas un apmācības vietnes”).
Datus, kas ievadīti testēšanas un apmācības vietnēs, uzskata par izdomātiem un nedara pieejamus sabiedrībai.
2. Pirms mašīnas-mašīnas datu apmaiņas pakalpojumu pirmās izmantošanas reizes izpildītājs, izmantojot mašīnas-mašīnas datu apmaiņas pakalpojumu, vismaz vienu reizi sekmīgi iesniedz datus testēšanas un apmācības vietnē.
3. Jebkādas izmaiņas, ko Komisija iecerējusi ieviest Eudamed mašīnas-mašīnas datu apmaiņas pakalpojumos, tā sākotnēji ievieš testēšanas un apmācības vietnēs, un šīs izmaiņas minētajās vietnēs ir pieejamas uz laiku, ko Komisija iepriekš noteikusi sadarbībā ar Medicīnisko ierīču koordinācijas grupu, kura izveidota saskaņā ar Regulas (ES) 2017/745 103. pantu.
Komisija, izmantojot Eudamed, laikus informē attiecīgos izpildītājus par iecerētajām izmaiņām un laiku, uz kādu tās būs pieejamas testēšanas un apmācības vietnēs.
10. pants
IT drošība
1. Komisija ierobežotas piekļuves vietnē dara pieejamus šādus dokumentus:
|
a) |
dokuments par lietotāja tiesībām un pienākumiem; |
|
b) |
deklarācija par pienākumiem attiecībā uz informācijas drošību; |
|
c) |
paziņojums par privātumu; |
|
d) |
informācijas drošības prasības attiecībā uz datu apmaiņu. |
2. Izpildītāji ievēro noteikumus, kas izklāstīti dokumentos, kuri minēti 1. punkta b) apakšpunktā un attiecīgā gadījumā minētā punkta d) apakšpunktā.
3. Ja Komisijai ir aizdomas, ka ir noticis IT drošības incidents vai ka pastāv IT drošības risks vai apdraudējums IT drošībai, kā tie definēti Lēmuma (ES, Euratom) 2017/46 2. panta 15), 22) un 25) punktā un kurus tā uzskata par potenciāli kaitīgiem Eudamed, tās datiem vai to konfidencialitātei (“IT drošības incidents, apdraudējums IT drošībai vai IT drošības risks”), Komisija var apturēt piekļuvi Eudamed.
4. Ja Komisija identificē IT drošības incidentu, apdraudējumu IT drošībai vai IT drošības risku, tā var apturēt visas Eudamed elektronisko sistēmu funkcionalitātes vai daļu no tām.
Ja pirmajā daļā minētā apturēšana kavē datu ievadīšanu Eudamed, mutatis mutandis piemēro 8. panta 3., 4. un 5. punktu.
5. Izpildītājs vai pilnvarots lietotājs, kurš ieguvis informāciju vai kuram ir aizdomas par IT drošības incidentu, apdraudējumu IT drošībai vai IT drošības risku, nekavējoties par to informē Komisiju un attiecīgās dalībvalstis.
11. pants
Krāpniecīgu lietotāju darbība Eudamed
1. Ja kompetentajai iestādei, LAA vai LUA ir aizdomas par krāpniecīgu Eudamed piekļuves pieprasījumu, tās šādu pieprasījumu noraida un nekavējoties par šādu noraidījumu informē Komisiju, sazinoties ar 5. panta 1. punktā minēto pieteikumu atbalsta grupu un norādot, ka radušās aizdomas par krāpniecīgu piekļuves pieprasījumu.
2. Ja Komisijai ir pamatotas aizdomas par pilnvarota lietotāja krāpniecīgu darbību, kas ietekmē Eudamed IT drošību, tā uz laiku aptur attiecīgā pilnvarotā lietotāja piekļuvi Eudamed. Šādā gadījumā Komisija nekavējoties par piekļuves apturēšanu un pamatojumu informē visas dalībvalstis un iesaistītos izpildītājus.
3. Izpildītājs vai pilnvarots lietotājs, kuram ir aizdomas par pilnvarota lietotāja krāpniecīgu darbību, par iespējamo krāpniecīgo darbību nekavējoties informē Komisiju un dalībvalstis, sazinoties ar 5. panta 1. punktā minēto pieteikumu atbalsta grupu.
4. Ja Komisija Eudamed konstatē krāpniecīgu darbību, tā nekavējoties attiecīgajiem pilnvarotajiem lietotājiem atņem piekļuvi Eudamed un veic nepieciešamos pasākumus, tai skaitā attiecīgā gadījumā novērš iespēju turpmāk piekļūt Eudamed no saistītajiem Komisijas autentifikācijas sistēmas vietnē izveidotajiem kontiem. Komisija nekavējoties informē attiecīgās valsts kompetentās iestādes un iesaistītos izpildītājus par pasākumiem, kas veikti saskaņā ar šo punktu.
12. pants
Stāšanās spēkā
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 117, 5.5.2017., 1. lpp.
(2) Eiropas Parlamenta un Padomes Regula (ES) 2017/746 (2017. gada 5. aprīlis) par in vitro diagnostikas medicīniskām ierīcēm un ar ko atceļ Direktīvu 98/79/EK un Komisijas Lēmumu 2010/227/ES (OV L 117, 5.5.2017., 176. lpp.).
(3) Komisijas Lēmums (ES, Euratom) 2017/46 (2017. gada 10. janvāris) par komunikācijas un informācijas sistēmu drošību Eiropas Komisijā (OV L 6, 11.1.2017., 40. lpp.).
(4) Eiropas Parlamenta un Padomes Regula (ES) 2018/1725 (2018. gada 23. oktobris) par fizisku personu aizsardzību attiecībā uz personas datu apstrādi Savienības iestādēs, struktūrās, birojos un aģentūrās un par šādu datu brīvu apriti un ar ko atceļ Regulu (EK) Nr. 45/2001 un Lēmumu Nr. 1247/2002/EK (OV L 295, 21.11.2018., 39. lpp.).
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/16 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2079
(2021. gada 26. novembris),
ar ko atbilstoši Eiropas Parlamenta un Padomes Regulai (ES) 2015/2283 atļauj kā jaunu pārtikas produktu laist tirgū D2 vitamīnu saturošu sēņu pulveri un groza Komisijas Īstenošanas regulu (ES) 2017/2470
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (ES) 2015/2283 (2015. gada 25. novembris) par jauniem pārtikas produktiem un ar ko groza Eiropas Parlamenta un Padomes Regulu (ES) Nr. 1169/2011 un atceļ Eiropas Parlamenta un Padomes Regulu (EK) Nr. 258/97 un Komisijas Regulu (EK) Nr. 1852/2001 (1), un jo īpaši tās 12. pantu,
tā kā:
|
(1) |
Regula (ES) 2015/2283 paredz, ka Savienības tirgū drīkst laist tikai tādus jaunus pārtikas produktus, kas ir atļauti un iekļauti Savienības sarakstā. |
|
(2) |
Saskaņā ar Regulas (ES) 2015/2283 8. pantu tika pieņemta Komisijas Īstenošanas regula (ES) 2017/2470 (2), ar ko izveido atļauto jauno pārtikas produktu Savienības sarakstu. |
|
(3) |
2019. gada 29. jūlijā uzņēmums MBio, Monaghan Mushrooms (“pieteikuma iesniedzējs”) Komisijai saskaņā ar Regulas (ES) 2015/2283 10. panta 1. punktu iesniedza pieteikumu, kurā lūgts atļaut kā jaunu pārtikas produktu Savienības tirgū laist D2 vitamīnu saturošu sēņu pulveri. Pieteikums attiecas uz D2 vitamīnu saturoša sēņu pulvera izmantošanu vairākos plaša patēriņa pārtikas produktos. Pieteikuma iesniedzējs arī lūdza atļaut jauno pārtiku produktu izmantot uztura bagātinātājos, kas definēti Eiropas Parlamenta un Padomes Direktīvā 2002/46/EK (3), izņemot zīdaiņu uztura bagātinātājos, un īpašiem medicīniskiem nolūkiem paredzētā pārtikā, kas definēta Eiropas Parlamenta un Padomes Regulā (ES) Nr. 609/2013 (4), izņemot īpašiem medicīniskiem nolūkiem paredzētu pārtiku zīdaiņiem. Pieteikuma iesniegšanas procesā pieteikuma iesniedzējs piekrita neiekļaut bērnus līdz triju gadu vecumam atļaujas pieprasījumā attiecībā uz jauno pārtikas produktu uztura bagātinātājos. |
|
(4) |
Pieteikuma iesniedzējs iesniedza Komisijai arī pieprasījumu par īpašniekdatu aizsardzību attiecībā uz vairākiem sākotnējiem datiem, kas iesniegti pieteikuma pamatošanai, proti, datiem par ražošanas procesu (5); datiem par produkta sastāvu: daļiņu izmēru (6), fizikāli ķīmiskajām īpašībām (7), D vitamīna analīzi (8), uztura analīzi (9), D2 vitamīna analīzi (10), D vitamīna analīzes validēšanu (11), stabilitātes pētījumiem (12), toksikoloģisko analīzi (13), datiem par tahisterīnu un lumisterīnu (14), ergosterīna attiecības analīzi (15), D vitamīna attiecības analīzi (16), datiem par ergosterīnu (17); svaigu sēņu specifikācijām (18); datiem par alergēniskumu (19). |
|
(5) |
2020. gada 24. janvārī Komisija saskaņā ar Regulas (ES) 2015/2283 10. panta 3. punktu konsultējās ar Eiropas Pārtikas nekaitīguma iestādi (“Iestāde”) un lūdza to sniegt zinātnisku atzinumu, veicot D2 vitamīnu saturoša sēņu pulvera kā jauna pārtikas produkta nekaitīguma novērtējumu. |
|
(6) |
2021. gada 24. februārī Iestāde pieņēma zinātnisko atzinumu par D2 vitamīnu saturoša sēņu pulvera (Agaricus bisporus) kā jauna pārtikas produkta nekaitīgumu saskaņā ar Regulu (ES) 2015/2283 (20). Minētais atzinums atbilst Regulas (ES) 2015/2283 11. panta prasībām. |
|
(7) |
Minētajā atzinumā Iestāde secināja, ka D2 vitamīnu saturošs sēņu pulveris, ja lietots ierosinātajos veidos un daudzumos, ir nekaitīgs. Tāpēc Iestādes atzinums ir pietiekams pamats, lai konstatētu, ka D2 vitamīnu saturošs sēņu pulveris konkrētajos lietošanas apstākļos atbilst Regulas (ES) 2015/2283 12. panta 1. punktam. |
|
(8) |
Lai pienācīgi informētu patērētājus par to, ka zīdaiņiem un bērniem līdz triju gadu vecumam nevajadzētu lietot uztura bagātinātājus, kuru sastāvā ir D2 vitamīnu saturošs sēņu pulveris, būtu jāparedz marķēšanas prasība. |
|
(9) |
Iestāde atzinumā norādīja, ka par pamatu jaunā pārtikas produkta nekaitīguma noteikšanai tika izmantoti dati par ražošanas procesu un sastāvu. Tāpēc Komisija uzskata, ka bez minētajiem datiem secinājumus par D2 vitamīnu saturoša sēņu pulvera nekaitīgumu nebūtu bijis iespējams izdarīt. |
|
(10) |
Komisija lūdza pieteikuma iesniedzēju sīkāk precizēt, kāds ir pamatojums prasībai aizsargāt īpašumtiesības uz minētajiem datiem, un precizēt prasību attiecībā uz ekskluzīvām tiesībām atsaukties uz minētajiem datiem, kā noteikts Regulas (ES) 2015/2283 26. panta 2. punkta b) apakšpunktā. |
|
(11) |
Pieteikuma iesniedzējs paziņoja, ka pieteikuma iesniegšanas brīdī tam saskaņā ar valsts tiesību aktiem bijušas īpašumtiesības un ekskluzīvas tiesības atsaukties uz minētajiem datiem un tāpēc trešās personas nevar likumīgi piekļūt šiem datiem vai tos izmantot, vai atsaukties uz minētajiem datiem. |
|
(12) |
Komisija novērtēja visu pieteikuma iesniedzēja sniegto informāciju un konstatēja, ka pieteikuma iesniedzējs ir pietiekami pamatojis Regulas (ES) 2015/2283 26. panta 2. punktā noteikto prasību izpildi. Tāpēc pieteikuma iesniedzēja dokumentācijā ietvertie dati par ražošanas procesu; produkta sastāvu: daļiņu izmēru, fizikāli ķīmiskajām īpašībām, D vitamīna analīzi, uztura analīzi, D2 vitamīna analīzi, D vitamīna analīzes validēšanu, stabilitātes pētījumiem, toksikoloģisko analīzi, datiem par tahisterīnu un lumisterīnu, ergosterīna attiecības analīzi, D vitamīna attiecības analīzi, datiem par ergosterīnu, uz kuriem Iestāde balstīja savu secinājumu par jaunā pārtikas produkta nekaitīgumu un bez kuriem tā nevarēja novērtēt jauno pārtikas produktu, Iestādei nebūtu jāizmanto neviena cita pieteikuma iesniedzēja labā piecus gadus pēc šīs regulas spēkā stāšanās dienas. Tādēļ atļauja minētajā periodā Savienībā tirgū laist D2 vitamīnu saturošu sēņu pulveri būtu jādod tikai pieteikuma iesniedzējam. |
|
(13) |
Tomēr, lai gan atļauja uz D2 vitamīnu saturoša sēņu pulvera izmantošanu un tiesības atsaukties uz pieteikuma iesniedzēja dokumentācijā ietvertajiem datiem ir vienīgi pieteikuma iesniedzējam, citiem pieteikuma iesniedzējiem neliedz pieteikties uz atļauju laist tirgū to pašu jauno pārtikas produktu ar nosacījumu, ka pieteikuma pamatā ir likumīgi iegūta informācija, kas pamato šādas atļaujas piešķiršanu saskaņā ar Regulu (ES) 2015/2283. |
|
(14) |
Tāpēc Īstenošanas regula (ES) 2017/2470 būtu attiecīgi jāgroza. |
|
(15) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Augu, dzīvnieku, pārtikas aprites un dzīvnieku barības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
1. D2 vitamīnu saturošu sēņu pulveri, kura specifikācijas dotas šīs regulas pielikumā, iekļauj Īstenošanas regulā (ES) 2017/2470 izveidotajā atļauto jauno pārtikas produktu Savienības sarakstā.
2. Piecus gadus no 2021. gada 19. decembra tikai sākotnējam pieteikuma iesniedzējam,
|
|
uzņēmumam MBio, Monaghan Mushrooms, |
|
|
adrese: Tullygony, Tyholland, Co. Monaghan, Īrija, |
|
|
ir atļauts laist Savienības tirgū 1. punktā minēto jauno pārtikas produktu, ja vien vēlāk atļauju attiecībā uz jauno pārtikas produktu nesaņem cits pieteikuma iesniedzējs, neatsaucoties uz datiem, kas aizsargāti saskaņā ar 2. pantu, vai vienojoties ar MBio, Monaghan Mushrooms. |
3. Šā panta 1. punktā minētajā ierakstā, ko iekļauj Savienības sarakstā, ietver pielikumā noteiktos lietošanas nosacījumus un marķēšanas prasības.
2. pants
Pieteikuma dokumentācijā iekļautos datus, uz kuriem pamatojoties Iestāde ir novērtējusi 1. pantā minēto jauno pārtikas produktu un uz kuriem pieteikuma iesniedzējs pieteicis īpašumtiesības, un bez kuriem jaunais pārtikas produkts nebūtu atļauts, bez uzņēmuma MBio, Monaghan Mushrooms piekrišanas neizmanto neviena cita pieteikuma iesniedzēja labā piecus gadus no 2021. gada 19. decembra.
3. pants
Īstenošanas regulas (ES) 2017/2470 pielikumu groza saskaņā ar šīs regulas pielikumu.
4. pants
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 327, 11.12.2015., 1. lpp.
(2) Komisijas Īstenošanas regula (ES) 2017/2470 (2017. gada 20. decembris), ar ko saskaņā ar Eiropas Parlamenta un Padomes Regulu (ES) 2015/2283 izveido jauno pārtikas produktu Savienības sarakstu (OV L 351, 30.12.2017., 72. lpp.).
(3) Eiropas Parlamenta un Padomes Direktīva 2002/46/EK (2002. gada 10. jūnijs) par dalībvalstu tiesību aktu tuvināšanu attiecībā uz uztura bagātinātājiem (OV L 183, 12.7.2002., 51. lpp.).
(4) Eiropas Parlamenta un Padomes Regula (ES) Nr. 609/2013 (2013. gada 12. jūnijs) par zīdaiņiem un maziem bērniem paredzētu pārtiku, īpašiem medicīniskiem nolūkiem paredzētu pārtiku un par pilnīgiem uztura aizstājējiem svara kontrolei, un ar ko atceļ Padomes Direktīvu 92/52/EEK, Komisijas Direktīvas 96/8/EK, 1999/21/EK, 2006/125/EK un 2006/141/EK, Eiropas Parlamenta un Padomes Direktīvu 2009/39/EK un Komisijas Regulas (EK) Nr. 41/2009 un (EK) Nr. 953/2009 (OV L 181, 29.6.2013., 35. lpp.).
(5) 2.3.1 Production Process Confidential_Final.
(6) Annex 1 Particle Size Report.
(7) Annex 3 NIZO Report physico-chemical properties.
(8) Annex 4 COA vitamin D analysis.
(9) Annex 5 COA nutritional analysis.
(10) Annex 7 MBio SOP Vitamin D2 analysis.
(11) Annex 8 MBio Vit D analysis validation report.
(12) Annex 9 Stability Study Report UCC; Annex 14 COA Vit D stability study; Annex 24 Stability study Report CampdenBRI; Annex 25 Stability study report meat free product; Annex 29 COAs Stability Meat free.
(13) Annex 16 COA Toxicological analysis.
(14) Annex 17 Report Tachysterol and lumisterol.
(15) Annex 20 COA Ergosterol ratio analysis.
(16) Annex 21 COA Vitamin D ratio analysis.
(17) Annex 22 MBio Ergosterol.
(18) Annex 13 COA fresh mushrooms analysis.
(19) Annex 12 MBio Allergen Policy.
(20) EFSA Journal 2021;19(4):6516.
PIELIKUMS
Īstenošanas regulas (ES) 2017/2470 pielikumu groza šādi:
|
1) |
pielikuma 1. tabulā (“Atļautie jaunie pārtikas produkti”) iekļauj šādu ierakstu:
|
||||||||||||||||||||||||||||||||||||||||||
|
2) |
pielikuma 2. tabulā (“Specifikācijas”) iekļauj šādu ierakstu:
|
(1) KVV: kolonijas veidojošas vienības.
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/23 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2080
(2021. gada 26. novembris)
par atļauju L-histidīna monohidrohlorīda monohidrātu, kas producēts fermentācijā ar Escherichia coli NITE SD 00268, lietot par barības piedevu visu sugu dzīvniekiem, izņemot zivis
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1831/2003 (2003. gada 22. septembris) par dzīvnieku ēdināšanā lietotām piedevām (1) un jo īpaši tās 9. panta 2. punktu,
tā kā:
|
(1) |
Regulā (EK) Nr. 1831/2003 noteikts, ka piedevu lietošanai dzīvnieku ēdināšanā ir vajadzīga atļauja, un paredzēts šādas atļaujas piešķiršanas pamatojums un kārtība. |
|
(2) |
Saskaņā ar Regulas (EK) Nr. 1831/2003 7. pantu tika iesniegts pieteikums, kurā lūgts L-histidīna monohidrohlorīda monohidrāta, kas producēts fermentācijā ar Escherichia coli NITE SD 00268, lietojumu par barības piedevu zivīm paplašināt, to atļaujot lietot par barības piedevu visu sugu dzīvniekiem. Minētajam pieteikumam ir pievienotas Regulas (EK) Nr. 1831/2003 7. panta 3. punktā prasītās ziņas un dokumenti. |
|
(3) |
Pieteikumā lūgts L-histidīna monohidrohlorīda monohidrāta, kas producēts fermentācijā ar Escherichia coli NITE SD 00268, lietojumu par barības piedevu zivīm paplašināt, to atļaujot lietot par barības piedevu visu sugu dzīvniekiem un šo piedevu klasificējot piedevu kategorijā “uzturfizioloģiskās piedevas”, funkcionālajā grupā “aminoskābes, to sāļi un analogi”, un piedevu kategorijā “organoleptiskās piedevas”, funkcionālajā grupā “aromatizējošas sastāvdaļas”. |
|
(4) |
Eiropas Pārtikas nekaitīguma iestāde (“Iestāde”) 2021. gada 5. maija atzinumā (2) secināja, ka, ievērojot ierosinātos izmantošanas nosacījumus, L-histidīna monohidrohlorīda monohidrāts, kas producēts fermentācijā ar Escherichia coli NITE SD 00268, kaitīgi neietekmē ne dzīvnieku veselību, ne patērētāju drošību, ne vidi. Iestāde arī secināja, ka par aplūkojamo piedevu nav iespējams izdarīt secinājumus par tās potenciālu būt inhalatīvi toksiskai, acis kairinošai vai ādu sensibilizējošai. Tāpēc Komisija uzskata, ka būtu jāīsteno pienācīgi aizsargpasākumi, lai nepieļautu nelabvēlīgu ietekmi uz cilvēka, jo īpaši uz piedevas lietotāju, veselību. Iestāde secināja arī to, ka piedeva ir iedarbīgs neaizstājamās aminoskābes histidīna avots un iedarbīga aromatizējoša sastāvdaļa. |
|
(5) |
Iestāde neuzskata, ka būtu vajadzīgas īpašas prasības veikt uzraudzību pēc preparāta laišanas tirgū. Tā arī ir verificējusi ar Regulu (EK) Nr. 1831/2003 izveidotās references laboratorijas iesniegtos ziņojumus par barībā esošās barības piedevas analīzes metodi. |
|
(6) |
Fermentācijā ar Escherichia coli NITE SD 00268 producētā L-histidīna monohidrohlorīda monohidrāta novērtējums liecina, ka Regulas (EK) Nr. 1831/2003 5. pantā paredzētie atļaujas piešķiršanas nosacījumi ir izpildīti. Tāpēc šo piedevu būtu jāatļauj lietot tā, kā norādīts šīs regulas pielikumā. |
|
(7) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Augu, dzīvnieku, pārtikas aprites un dzīvnieku barības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
1. Pielikumā specificēto vielu L-histidīna monohidrohlorīda monohidrātu, kas producēts fermentācijā ar Escherichia coli NITE SD 00268 un kas pieder pie piedevu kategorijas “uzturfizioloģiskās piedevas” un funkcionālās grupas “aminoskābes, to sāļi un analogi”, ir atļauts lietot par dzīvnieku barības piedevu, ievērojot pielikumā noteiktos nosacījumus.
2. Pielikumā specificēto vielu L-histidīna monohidrohlorīda monohidrātu, kas producēts fermentācijā ar Escherichia coli NITE SD 00268 un kas pieder pie piedevu kategorijas “organoleptiskās piedevas” un funkcionālās grupas “aromatizējošas sastāvdaļas”, ir atļauts lietot par dzīvnieku barības piedevu, ievērojot pielikumā noteiktos nosacījumus.
2. pants
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 268, 18.10.2003., 29. lpp.
(2) EFSA Journal 2021; 19(5):6622.
PIELIKUMS
|
Piedevas identifikācijas numurs |
Atļaujas turētāja nosaukums |
Piedeva |
Sastāvs, ķīmiskā formula, apraksts, analītiskā metode |
Dzīvnieku suga vai kategorija |
Maksimālais vecums |
Minimālais saturs |
Maksimālais saturs |
Citi noteikumi |
Atļaujas derīguma termiņš |
||||||||||
|
mg uz kg kompleksās barības ar mitruma saturu 12 % |
|||||||||||||||||||
|
Kategorija: uzturfizioloģiskās piedevas Funkcionālā grupa: aminoskābes, to sāļi un analogi |
|||||||||||||||||||
|
3c351i |
– |
L-histidīna monohidrohlorīda monohidrāts |
Piedevas sastāvs Pulveris, kurā ir vismaz 98 % L-histidīna monohidrohlorīda monohidrāta un 72 % histidīna un kurā ir maks. 100 ppm histamīna |
Visu sugu dzīvnieki, izņemot zivis |
— |
— |
— |
|
2031. gada 19. decembris |
||||||||||
|
Aktīvās vielas raksturojums L-histidīna monohidrohlorīda monohidrāts, kas producēts fermentācijā ar Escherichia coli NITE SD 00268 Ķīmiskā formula: C3H3N2-CH2-CH(NH2)-COΟΗ·ΗCl·Η2O CAS numurs: 5934-29-2 Einecs numurs: 211-438-9 |
|||||||||||||||||||
|
Analītiskā metode (1) Histidīna kvantitatīvajai noteikšanai barības piedevā:
Histidīna kvantitatīvajai noteikšanai premiksos, barības sastāvdaļās un barības maisījumos:
Histamīna kvantitatīvajai noteikšanai barības piedevā:
|
|||||||||||||||||||
|
Kategorija: organoleptiskās piedevas Funkcionālā grupa: aromatizējošas sastāvdaļas |
|||||||||||||||||||
|
3c351i |
— |
L-histidīna monohidrohlorīda monohidrāts |
Piedevas sastāvs Pulveris, kurā ir vismaz 98 % L-histidīna monohidrohlorīda monohidrāta un 72 % histidīna un kurā ir maks. 100 ppm histamīna |
Visu sugu dzīvnieki |
— |
— |
— |
|
2031. gada 19. decembris |
||||||||||
|
Aktīvās vielas raksturojums L-histidīna monohidrohlorīda monohidrāts, kas producēts fermentācijā ar Escherichia coli NITE SD 00268 Ķīmiskā formula: C3H3N2-CH2-CH(NH2)-COΟΗ·ΗCl·Η2O CAS numurs: 5934-29-2 Einecs numurs: 211-438-9 |
|||||||||||||||||||
|
Analītiskā metode (1) Histidīna kvantitatīvajai noteikšanai barības piedevā:
Histidīna kvantitatīvajai noteikšanai premiksos:
Histidīna kvantitatīvajai noteikšanai barības piedevā:
|
|||||||||||||||||||
(1) Sīkāka informācija par analītiskajām metodēm ir pieejama references laboratorijas vietnē: https://ec.europa.eu/jrc/en/eurl/feed-additives/evaluation-reports.
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/28 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2081
(2021. gada 26. novembris)
par darbīgās vielas indoksakarba apstiprinājuma neatjaunošanu, ievērojot Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1107/2009 par augu aizsardzības līdzekļu laišanu tirgū, un par grozījumiem Komisijas Īstenošanas regulā (ES) Nr. 540/2011
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1107/2009 (2009. gada 21. oktobris) par augu aizsardzības līdzekļu laišanu tirgū, ar ko atceļ Padomes Direktīvas 79/117/EEK un 91/414/EEK (1), un jo īpaši tās 20. panta 1. punkta b) apakšpunktu un 78. panta 2. punktu,
tā kā:
|
(1) |
Ar Komisijas Direktīvu 2006/10/EK (2) indoksakarbs kā darbīgā viela tika iekļauts Padomes Direktīvas 91/414/EEK (3) I pielikumā. |
|
(2) |
Darbīgās vielas, kas iekļautas Direktīvas 91/414/EEK I pielikumā, tiek uzskatītas par apstiprinātām uz Regulas (EK) Nr. 1107/2009 pamata un ir norādītas Komisijas Īstenošanas regulas (ES) Nr. 540/2011 (4) pielikuma A daļas sarakstā. |
|
(3) |
Darbīgās vielas indoksakarba apstiprinājums saskaņā ar Īstenošanas regulas (ES) Nr. 540/2011 pielikuma A daļu zaudē spēku 2022. gada 31. oktobrī. |
|
(4) |
Komisijas Īstenošanas regulas (ES) Nr. 844/2012 (5) 1. pantā norādītajā laikposmā saskaņā ar minēto pantu tika iesniegts pieteikums uz indoksakarba apstiprinājuma atjaunošanu. |
|
(5) |
Pieteikuma iesniedzējs iesniedza Īstenošanas regulas (ES) Nr. 844/2012 6. pantā prasīto papildu dokumentāciju. Ziņotāja dalībvalsts atzina, ka pieteikums ir pilnīgs. |
|
(6) |
Ziņotāja dalībvalsts, apspriedusies ar otru ziņotāju dalībvalsti, sagatavoja atjaunošanas novērtējuma ziņojuma projektu un 2016. gada 28. novembrī to iesniedza Eiropas Pārtikas nekaitīguma iestādei (“Iestāde”) un Komisijai. |
|
(7) |
Iestāde papildu kopsavilkuma dokumentāciju darīja publiski pieejamu. Iestāde atjaunošanas novērtējuma ziņojumu nosūtīja arī pieteikuma iesniedzējam un dalībvalstīm komentāru sniegšanai un sāka ziņojuma sabiedrisko apspriešanu. Saņemtos komentārus Iestāde nosūtīja Komisijai. |
|
(8) |
2017. gada 15. decembrī Iestāde nosūtīja Komisijai secinājumu (6) par to, vai ir gaidāms, ka indoksakarbs atbildīs Regulas (EK) Nr. 1107/2009 4. pantā noteiktajiem apstiprināšanas kritērijiem. Šā secinājuma sadaļa par ekotoksiskumu 2018. gadā tika grozīta, lai precizētu riska novērtējumu attiecībā uz bitēm atbilstoši attiecīgajām Eiropas Komisijas pamatnostādnēm (SANCO/10329/2002-rev.2). 2019. gada 15. maijā Komisija lūdza Iestādei veikt atjauninātu zinātnisko recenzēšanu attiecībā uz indoksakarba radīto risku zīdītājiem un bitēm. 2019. gada 28. oktobrī Iestāde pieņēma paziņojumu par atjaunināto zinātnisko recenzējumu attiecībā uz indoksakarba radīto risku zīdītājiem un bitēm (7), un tas tika atspoguļots Iestādes secinājuma otrajā atjauninājumā par to, vai ir gaidāms, ka indoksakarbs atbildīs Regulas (EK) Nr. 1107/2009 4. pantā noteiktajiem apstiprināšanas kritērijiem. |
|
(9) |
Iestāde konstatēja kritisku problēmu saistībā ar augstu ilgtermiņa risku savvaļas zīdītājiem, it īpaši ilgtermiņa risku maziem augēdājiem zīdītājiem. |
|
(10) |
Papildus tika konstatēts augsts risks patērētājiem un strādniekiem attiecībā uz reprezentatīvu izmantošanu dārza salātos un augsts risks bitēm attiecībā uz reprezentatīvu izmantošanu kukurūzā, cukurkukurūzā un dārza salātos, kas paredzēti sēklu iegūšanai. |
|
(11) |
Turklāt vairākas riska novērtēšanas daļas nebija iespējams pabeigt, jo dokumentācijā nebija pietiekamu datu. Riska novērtēšanu patērētājiem pirmām kārtām nevarēja pabeigt tāpēc, ka trūka datu par metabolismu augsekas kultūrās, par metabolismu mājputnu organismā, par atlieku daudzumu galvenajās un augsekas kultūrās un par ūdens attīrīšanas procesu ietekmi uz dzirdināšanas ūdenī esošo atlieku veidu. Nevarēja arī pabeigt novērtēšanu par pazemes ūdeņu eksponētību augsnes metabolītam IN-U8E24, jo trūka datu par augsnes degradāciju un adsorbciju. Tāpat arī nevarēja pabeigt vairāku metabolītu ekotoksiskuma riska novērtēšanu. |
|
(12) |
2018. gada 14. novembrī pieteikuma iesniedzējs informēja Komisiju par savu lēmumu atsaukt atjaunošanas pieteikumu attiecībā uz reprezentatīvo izmantošanu dārza salātos. |
|
(13) |
Komisija aicināja pieteikuma iesniedzēju iesniegt komentārus par Iestādes sagatavoto secinājumu, pārskatīto secinājumu un paziņojumu. Turklāt saskaņā ar Īstenošanas regulas (ES) Nr. 844/2012 14. panta 1. punkta trešo daļu Komisija aicināja pieteikuma iesniedzēju iesniegt komentārus par atjaunošanas ziņojuma projektu. Pieteikuma iesniedzējs komentārus iesniedza, un tie tika rūpīgi izskatīti. |
|
(14) |
Neraugoties uz pieteikuma iesniedzēja argumentiem, bažas par darbīgo vielu tomēr nebija iespējams kliedēt. |
|
(15) |
Līdz ar to nav konstatēts, ka Regulas (EK) Nr. 1107/2009 4. pantā noteiktie apstiprināšanas kritēriji būtu izpildīti par vismaz viena augu aizsardzības līdzekļa vienu vai vairākiem reprezentatīviem lietojumiem. Tāpēc ir lietderīgi darbīgās vielas indoksakarba apstiprinājumu saskaņā ar minētās regulas 20. panta 1. punkta b) apakšpunktu neatjaunot. |
|
(16) |
Tāpēc Īstenošanas regula (ES) Nr. 540/2011 būtu attiecīgi jāgroza. |
|
(17) |
Dalībvalstīm būtu jādod pietiekami ilgs laiks, lai tās varētu atsaukt indoksakarbu saturošu augu aizsardzības līdzekļu atļaujas. |
|
(18) |
Ja attiecībā uz indoksakarbu saturošiem augu aizsardzības līdzekļiem dalībvalstis nosaka pagarinājuma periodu saskaņā ar Regulas (EK) Nr. 1107/2009 46. pantu, šim periodam vajadzētu būt pēc iespējas īsākam. |
|
(19) |
Ar Komisijas Īstenošanas regulu (ES) 2021/1449 (8) indoksakarba apstiprinājuma termiņš tika pagarināts līdz 2022. gada 31. oktobrim, lai atjaunošanas procedūru varētu pabeigt pirms vielas apstiprinājuma termiņa beigām. Tomēr, ņemot vērā to, ka lēmums par atjaunošanu ir pieņemts pirms šā pagarinātā termiņa beigām, šī regula būtu jāpiemēro pēc iespējas drīzāk. |
|
(20) |
Šī regula neliedz iespēju saskaņā ar Regulas (EK) Nr. 1107/2009 7. pantu iesniegt citus pieteikumus uz indoksakarba apstiprināšanu. |
|
(21) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Augu, dzīvnieku, pārtikas aprites un dzīvnieku barības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Darbīgās vielas apstiprinājuma neatjaunošana
Darbīgās vielas indoksakarba apstiprinājumu neatjauno.
2. pants
Grozījums Īstenošanas regulā (ES) Nr. 540/2011
Īstenošanas regulas (ES) Nr. 540/2011 pielikuma A daļā svītro 119. ierakstu par indoksakarbu.
3. pants
Pārejas pasākumi
Dalībvalstis ne vēlāk kā līdz 2022. gada 19. martam atsauc tādu augu aizsardzības līdzekļu atļaujas, kas kā darbīgo vielu satur indoksakarbu.
4. pants
Pagarinājuma periods
Jebkurš pagarinājuma periods, ko dalībvalstis piešķir saskaņā ar Regulas (EK) Nr. 1107/2009 46. pantu, beidzas ne vēlāk kā 2022. gada 19. septembrī.
5. pants
Stāšanās spēkā
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 309, 24.11.2009., 1. lpp.
(2) Komisijas Direktīva 2006/10/EK (2006. gada 27. janvāris), ar ko groza Padomes Direktīvu 91/414/EEK, lai tajā iekļautu forhlorfenuronu un indoksakarbu kā aktīvās vielas (OV L 25, 28.1.2006., 24. lpp.).
(3) Padomes Direktīva 91/414/EEK (1991. gada 15. jūlijs) par augu aizsardzības līdzekļu laišanu tirgū (OV L 230, 19.8.1991., 1. lpp.).
(4) Komisijas Īstenošanas regula (ES) Nr. 540/2011 (2011. gada 25. maijs), ar ko īsteno Eiropas Parlamenta un Padomes Regulu (EK) Nr. 1107/2009 attiecībā uz darbīgo vielu sarakstu (OV L 153, 11.6.2011., 1. lpp.).
(5) Komisijas Īstenošanas regula (ES) Nr. 844/2012 (2012. gada 18. septembris), ar ko nosaka noteikumus, kas vajadzīgi darbīgo vielu apstiprinājumu atjaunošanas procedūras īstenošanai, kā paredzēts Eiropas Parlamenta un Padomes Regulā (EK) Nr. 1107/2009 par augu aizsardzības līdzekļu laišanu tirgū (OV L 252, 19.9.2012., 26. lpp.).
(6) EFSA Journal 2018;16(1):5140, 36 lpp. doi:10.2903/j.efsa.2018.5140. Pieejams tiešsaistē: www.efsa.europa.eu.
(7) EFSA (Eiropas Pārtikas nekaitīguma iestāde), 2019. gads. Statement on the updated peer review concerning the risk to mammals and bees for the active substance indoxacarb. EFSA Journal 2019;17(10):5866, 10 lpp.
(8) Komisijas Īstenošanas regula (ES) 2021/1449 (2021. gada 3. septembris), ar ko attiecībā uz apstiprinājuma termiņu pagarināšanu darbīgajām vielām 2-fenilfenolam (ieskaitot tādus tā sāļus kā nātrija sāls), 8-hidroksihinolīnam, amidosulfuronam, bifenoksam, hlormekvatam, hlortoluronam, klofentezīnam, klomazonam, cipermetrīnam, daminozīdam, deltametrīnam, dikambam, difenokonazolam, diflufenikānam, dimetahloram, etofēnproksam, fenoksapropam-P, fenpropidīnam, fludioksonilam, flufenacetam, fostiazātam, indoksakarbam, lenacilam, MCPA, MCPB, nikosulfuronam, parafīneļļām, parafīneļļai, penkonazolam, piklorāmam, propakvizafopam, prosulfokarbam, etil-kvizalofopam-P, tefuril-kvizalofopam-P, sēram, tetrakonazolam, trialātam, triflusulfuronam un tritosulfuronam groza Īstenošanas regulu (ES) Nr. 540/2011 (OV L 313, 6.9.2021., 20. lpp.).
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/32 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2082
(2021. gada 26. novembris),
ar ko nosaka kārtību Eiropas Parlamenta un Padomes Regulas (ES) Nr. 376/2014 īstenošanai attiecībā uz Eiropas vienoto riska klasifikācijas shēmu
(Dokuments attiecas uz EEZ)
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (ES) Nr. 376/2014 (2014. gada 3. aprīlis) par ziņošanu, analīzi un turpmākajiem pasākumiem attiecībā uz atgadījumiem civilajā aviācijā un ar ko groza Eiropas Parlamenta un Padomes Regulu (ES) Nr. 996/2010 un atceļ Eiropas Parlamenta un Padomes Direktīvu 2003/42/EK, Komisijas Regulas (EK) Nr. 1321/2007 un (EK) Nr. 1330/2007 (1), un jo īpaši tās 7. panta 7. punktu,
tā kā:
|
(1) |
Saskaņā ar Regulu (ES) Nr. 376/2014 gan dalībvalstīm, gan Eiropas Savienības Aviācijas drošības aģentūrai (“Aģentūra”) ir jāizveido mehānisms, lai neatkarīgi vāktu, novērtētu, apstrādātu, analizētu un glabātu informāciju par aviācijas drošības atgadījumiem. Dalībvalstu kompetentajām iestādēm atgadījumu ziņojumi ir jāsagatavo, pamatojoties uz detalizētu informāciju par atgadījumiem, un tie jāglabā valsts datubāzē. Arī Aģentūrai ir tāds pats pienākums sagatavot atgadījumu ziņojumus, pamatojoties uz detalizētu informāciju par atgadījumiem, un saglabāt tos datubāzē. |
|
(2) |
Saskaņā ar Regulas (ES) Nr. 376/2014 9. panta 1. punktu dalībvalstīm un Aģentūrai jāpiedalās informācijas apmaiņā, visu to attiecīgajās ziņošanas datubāzēs glabāto informāciju, kas attiecas uz drošību, darot pieejamu caur Eiropas Centrālo repozitoriju (ECR). |
|
(3) |
Saskaņā ar Regulu (ES) Nr. 376/2014 atgadījumu ziņojumos jāiekļauj drošības riska klasifikācija, kas jāpārskata dalībvalstu kompetentajām iestādēm vai Aģentūrai, un tie jāpārsūta uz ECR. Lai nodrošinātu, ka visi ECR ietvertie ziņojumi par atgadījumiem tiek klasificēti saskaņotā veidā, dalībvalstu kompetentajām iestādēm un Aģentūrai būtu jānodrošina, ka klasifikācija minētajos ziņojumos tiek noteikta saskaņā ar Eiropas vienoto riska klasifikācijas shēmu (ERCS), kā noteikts Komisijas Deleģētajā regulā (ES) 2020/2034 (2). |
|
(4) |
Tagad ir jānosaka kārtība, kādā Aģentūra un dalībvalstis saskaņoti un konsekventi īsteno ERCS. |
|
(5) |
Ja atgadījumu ziņojumos ir ietverta riska klasifikācija, kas noteikta izmantojot citas metodikas, nevis ERCS, tad dalībvalstu kompetentajām iestādēm vai Aģentūrai attiecīgā atgadījuma risks būtu jāklasificē saskaņā ar ERCS, kā noteikts Komisijas Deleģētajā regulā (ES) 2020/2034. |
|
(6) |
Gadījumos, kad dalībvalstu kompetentās iestādes vai Aģentūra nolemj izmantot pārrēķināšanas procedūru, lai 5. apsvērumā minētās riska klasifikācijas pārrēķinātu ERCS klasifikācijā, un ja šāda metodika ir ARMS-ERC 4x4 vai RAT“ATM – kopā”, dalībvalstu kompetentajām iestādēm vai Aģentūrai būtu jāizmanto šajā regulā paredzētā tiešās pārrēķināšanas procedūra. |
|
(7) |
Ja pielikumā noteiktā tiešā pārrēķināšanas procedūra nav piemērojama, dalībvalstu kompetentajām iestādēm un Aģentūrai būtu jāļauj izmantot citas pārrēķināšanas procedūras, ja vien šādi tiek iegūta ekvivalenta ERCS klasifikācija. |
|
(8) |
Lai nodrošinātu ERCS efektīvu piemērošanu, ir nepieciešama tās pastāvīga uzraudzība un uzlabošana. Ir jānosaka šādas uzraudzības un uzlabošanas detalizēti noteikumi, un Aģentūrai būtu jāpalīdz Komisijai veikt šādu pārskatīšanu un uzraudzību. Šajā nolūkā dalībvalstīm regulāri un noteiktajos termiņos būtu jāziņo Aģentūrai un Komisijai par ERCS izmantošanu un tās novērtējumu. |
|
(9) |
Dalībvalstu kompetentajām iestādēm un Aģentūrai ir jāsagatavojas ERCS piemērošanai, jo īpaši pielāgojot savus iekšējos procesus un, iespējams, piešķirot papildu resursus. Tomēr Regulas (ES) Nr. 376/2014 24. panta 3. punkts nosaka, ka minētās regulas 7. panta 2. punkts, kas pilnvaro dalībvalstis un Aģentūru izmantot ERCS, jāpiemēro pēc tam, kad stājas spēkā deleģētie un īstenošanas akti, ar kuriem precizē un izstrādā ERCS. Komisijas Deleģētā regula (ES) 2020/2034, kurā definēta ERCS, jau stājās spēkā 2020. gada 31. decembrī. Tāpēc nav iespējams atlikt ERCS izmantošanas pienākuma piemērošanu pēc šīs regulas spēkā stāšanās dienas. Turklāt saistībā ar ikgadējo drošības pārskatu, ko Aģentūra publicē saskaņā ar Eiropas Parlamenta un Padomes Regulas (ES) 2018/1139 (3) 72. panta 7. punktu, ir būtiski, lai atgadījumu ziņojumiem, kurus ECR augšupielādē viena gada laikā, novērtēšanas rezultāts tiktu piešķirts saskaņotā veidā. Pienākumu klasificēt atgadījumus saskaņā ar ERCS būtu jāsāk piemērot no šīs regulas spēkā stāšanās dienas. Tāpēc šai regulai būtu jāstājas spēkā 2023. gada 1. janvārī. |
|
(10) |
Šajā regulā paredzētie pasākumi ir saskaņā ar atzinumu, ko sniegusi ar Regulas (ES) 2018/1139 127. pantu izveidotā komiteja, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Priekšmets
Ar šo regulu nosaka kārtību, kādā īstenojama Eiropas vienotās riska klasifikācijas shēma (“ERCS”), kas noteikta Deleģētajā regulā (ES) 2020/2034.
2. pants
Definīcijas
Šajā regulā piemēro Deleģētās regulas (ES) 2020/2034 2. pantā iekļautās definīcijas.
Piemēro arī šādas definīcijas:
|
1) |
“ARMS-ERC metodika” ir metodika, ko operacionālo risku novērtēšanai izstrādājusi nozares darba grupa “Aviosabiedrību riska pārvaldības risinājumi”ARMS; |
|
2) |
“ATM” ir gaisa satiksmes pārvaldība, kā definēts Eiropas Parlamenta un Padomes Regulas (EK) Nr. 549/2004 (4) 2. panta 10. punktā; |
|
3) |
“ATM smaguma rezultāts – gaisā” ir tā RAT metodikas daļa, kas novērtē atgadījumu no lidojuma operāciju viedokļa; |
|
4) |
“ATM smaguma rezultāts – uz zemes” ir tā RAT metodikas daļa, kas novērtē ATM sistēmas veiktspēju (procedūras, aprīkojums un cilvēki); |
|
5) |
“ATM smaguma rezultāts – kopā” ir ATM smaguma rezultāts – uz zemes un ATM smaguma rezultāts – gaisā, kuri apvienoti vienā rezultātā; |
|
6) |
“RAT metodika” ir Eirokontroles izstrādāta riska analīzes rīka metodika, ko izmanto, lai klasificētu ar drošību saistītus atgadījumus ATM jomā; |
|
7) |
“Eirokontrole” ir Eiropas Aeronavigācijas drošības organizācija, kas izveidota saskaņā ar 1960. gada 13. decembra Starptautisko Konvenciju par sadarbību aeronavigācijas drošības jomā (5). |
3. pants
Drošības riska klasifikācijas pārskatīšana, grozīšana un apstiprināšana
1. Dalībvalsts kompetentā iestāde vai Aģentūra pārskata un vajadzības gadījumā groza un apstiprina atgadījuma ziņojumā par attiecīgo atgadījumu ietverto drošības riska klasifikāciju saskaņā ar ERCS, kā noteikts Deleģētajā regulā (ES) 2020/2034.
2. Neskarot 1. punktu, dalībvalsts kompetentā iestāde vai Aģentūra, pārrēķinot drošības riska klasifikāciju, kas noteikta, lietojot ARMS/ERC 4x4 vai RAT“ATM – kopā” metodiku, izmanto pielikumā noteikto tiešās pārrēķināšanas procedūru. Ja drošības riska klasifikācija noteikta ar citu metodiku, dalībvalsts kompetentā iestāde vai Aģentūra drīkst izmantot pielikuma 2. punktā noteikto manuālās pārrēķināšanas procedūru vai, pēc vajadzības, citas pārrēķināšanas procedūras, ja vien šādi tiek iegūta ekvivalenta ERCS klasifikācija.
4. pants
ERCS uzraudzība un uzlabošana
1. Katra dalībvalsts 2026. gada 31. martā un pēc tam reizi piecos gados iesniedz Komisijai un Aģentūrai ziņojumu par ERCS izmantošanu.
2. Aģentūra no dalībvalstīm saņemto informāciju, kā arī citu informāciju, ko Aģentūra var saņemt par ERCS īstenošanu, izskata saskaņā ar šā panta 1. punktu. Veicot izskatīšanu, Aģentūra drīkst ņemt vērā Regulas (ES) Nr. 376/2014 14. panta 2. punktā minētā aviācijas drošības analītiķu tīkla (NoA) un attiecīgu ekspertu grupu zinātību, ja Aģentūra tādas ir izveidojusi.
5. pants
Saderības ar citām riska klasifikācijas shēmām uzraudzība
1. Aģentūra regulāri pārskata pielikumā noteiktās pārrēķināšanas procedūras, lai nodrošinātu, ka tās joprojām ir aktuālas. Pārskatīšanā drīkst ņemt vērā NoA un attiecīgu ekspertu grupu zinātību, ja Aģentūra tādas izveidojusi.
2. Attiecīgā gadījumā dalībvalstis paziņo Komisijai un Aģentūrai manuālo pārrēķināšanas procedūru, kas noteikta pielikuma 2. punktā, un citas pārrēķināšanas procedūras, kas minētas šīs regulas 3. panta 2. punktā.
6. pants
Stāšanās spēkā
Šī regula stājas spēkā 2023. gada 1. janvārī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 122, 24.4.2014., 18. lpp.
(2) Komisijas Deleģētā regula (ES) 2020/2034 (2020. gada 6. oktobris), ar ko Eiropas Parlamenta un Padomes Regulu (ES) Nr. 376/2014 papildina attiecībā uz Eiropas vienoto riska klasifikācijas shēmu (OV L 416, 11.12.2020., 1. lpp.).
(3) Eiropas Parlamenta un Padomes Regula (ES) 2018/1139 (2018. gada 4. jūlijs) par kopīgiem noteikumiem civilās aviācijas jomā un ar ko izveido Eiropas Savienības Aviācijas drošības aģentūru, un ar ko groza Eiropas Parlamenta un Padomes Regulas (EK) Nr. 2111/2005, (EK) Nr. 1008/2008, (ES) Nr. 996/2010, (ES) Nr. 376/2014 un Direktīvas 2014/30/ES un 2014/53/ES un atceļ Eiropas Parlamenta un Padomes Regulas (EK) Nr. 552/2004 un (EK) Nr. 216/2008 un Padomes Regulu (EEK) Nr. 3922/91 (OV L 212, 22.8.2018., 1. lpp.).
(4) Eiropas Parlamenta un Padomes Regula (EK) Nr. 549/2004 (2004. gada 10. marts), ar ko nosaka pamatu Eiropas vienotās gaisa telpas izveidošanai (Pamatregula) (OV L 96, 31.3.2004., 1. lpp.).
(5) Konvencija grozīta ar 1981. gada 12. februāra Protokolu un pārskatīta ar 1997. gada 27. jūnija Protokolu.
PIELIKUMS
Procedūras ar riska analīzes rīku (RAT) un ar Aviosabiedrību riska pārvaldības risinājumu – notikumu riska klasifikāciju (ARMS-ERC) iegūto rezultātu pārrēķināšanai Eiropas riska klasifikācijas shēmas (ERCS) rezultātos
Šajā pielikumā ir noteiktas procedūras RAT un ARMS ERC rezultātu pārrēķināšanai ERCS rezultātā (1), kas definēts Deleģētās regulas (ES) 2020/2034 pielikuma 2. posmā.
Turpmāk noteiktās pārrēķināšanas procedūras nodrošina tiešu vai manuālu pārrēķināšanu, lai iegūtu ERCS klasifikāciju, kas ir ekvivalenta RAT un/vai ARMS – ERC rezultātam saskaņā ar šīs regulas 3. pantu.
1. TIEŠA PĀRRĒĶINĀŠANA
Obligātā pārrēķināšanas procedūra sastāv no šādām divām darbplūsmām:
|
— |
1. darbplūsma – nodrošina tiešu pārrēķināšanu ERCS smaguma rezultāta iegūšanai, |
|
— |
2. darbplūsma – nodrošina tiešu pārrēķināšanu ERCS varbūtības rezultāta iegūšanai. |
1. attēlā ir sniegts pārskats par šīm procedūrām. Process sākas ar “atgadījuma ziņojums iesniegts” lodziņu un beidzas ar “Ekvivalentais ERCS rezultāts” lodziņu. Punktētās līnijas 1. attēlā norāda, ka katra procesa rezultātam ir vajadzīgs tikai viens avots.

1.1. 1. DARBPLŪSMA – ERCS smaguma rezultāts
a. “Atgadījuma kategorijas” un “masas grupas” informācija
|
— |
Ja atgadījuma ziņojumā ir informācija par atgadījuma “atgadījuma kategoriju” un “masas grupu”, to var pārrēķināt “nelaimes gadījuma iespējamā iznākuma smaguma”ERCS rezultātā. Nākamais ir 1. attēla b) posms. |
|
— |
Ja atgadījuma ziņojumā nav informācijas par “atgadījuma kategoriju” vai “masas grupu”, vai abām, tad tieša pārrēķināšana nav iespējama. Ja izmanto šā pielikuma 2. punktā aprakstīto manuālo pārrēķināšanu, nākamais solis ir 1. un 5. attēla D posms. |
b. “Atgadījuma kategorija” un pārrēķināšana uz ERCS galvenajām riska jomām (GRJ)
|
— |
Ja atgadījuma ziņojuma “atgadījuma kategorija” tieši atbilst vienai no ERCS galvenajām riska jomām, kas definētas Deleģētās regulas (ES) 2020/2034 pielikuma 1.2. punktā, tad nākamais ir 1. attēla c) posms. |
|
— |
Ja atgadījuma ziņojumu “atgadījuma kategorijas” atšķiras no ERCS galvenajām riska jomām, tiešu pārrēķināšanu nevar veikt. Ja izmanto šā pielikuma 2. punktā aprakstīto manuālo pārrēķināšanu, nākamais solis ir 1. un 5. attēla D punkts. |
c. ERCS“nelaimes gadījuma iespējamā iznākuma smaguma” rezultāts – tieša pārrēķināšana
|
— |
Ja atgadījuma ziņojumā ir informācija par “atgadījuma kategoriju” un “masas grupu”, tad smaguma rezultātu tieši pārrēķina atbilstīgā ERCS“nelaimes gadījuma iespējamā iznākuma smaguma” rezultātā. Rezultāts ir k), kura pirmā zīme atbilst alfabētiskajai vērtībai, kas iegūta, aprēķinot atgadījuma smagumu (smaguma rezultāts no A līdz X). |
1.2. 2. DARBPLŪSMA – ERCS varbūtības rezultāts
e. Atgadījuma ziņojumam rezultāts noteikts, izmantojot RAT
Ja atgadījuma ziņojumam rezultāts ir noteikts, izmantojot RAT metodiku (2):
|
— |
Atgadījumu ziņojumus, kuriem ir RAT“ATM – kopā” smaguma rezultāta klasifikācija, var kartēt tieši ERCS varbūtības slejās, kā paskaidrots 2. attēla g) posmā. |
|
— |
Atgadījumu ziņojumi, kuros ir tikai RAT“ATM – uz zemes” smaguma rezultāts (3), ir manuāli jāpārrēķina, lai iegūtu ERCS varbūtības rezultātu. Ja izmanto šā pielikuma 2. punktā aprakstīto manuālo pārrēķināšanu, nākamais solis ir 5. attēla L posms. |
|
— |
Ja atgadījuma ziņojumiem piešķirts kods “īpašs ATM atgadījums”, pārrēķināšana no RAT rezultāta uz ERCS rezultātu nav iespējama. |
f. RAT“ATM – kopā” smaguma rezultāts
|
— |
Ja atgadījuma ziņojumā ir “ATM – kopā” smaguma rezultāts, tad nākamais ir 1. attēla g) posms. |
g. ERCS sleja “Nelaimes gadījuma iespējamā iznākuma varbūtība”, kas pārrēķināta no RAT“ATM – kopā” vērtības (attiecas tikai uz A, B, C, E vērtību)
Atgadījumu ziņojumiem ar “ATM – kopā” smaguma rezultāta (A, B, C, E) klasifikāciju piemēro šādu tiešu pārrēķināšanu ERCS varbūtības kategorijās:

h. Atgadījumu ziņojumi, kas klasificēti, izmantojot ARMS-ERC metodiku
|
— |
Atgadījumu ziņojumiem, kuriem rezultāts noteikts saskaņā ar ARMS-ERC, nākamais ir 1. attēla i) posms. |
|
— |
Atgadījumu ziņojumiem, kuriem rezultāts nav noteikts saskaņā ar ARMS-ERC metodiku, nākamais ir 5. attēla M) posms. |
i. Standarta 4x4 ARMS-ERC matrica
Ja atgadījuma ziņojuma rezultāta noteikšanai izmanto 3. attēlā redzamo 4x4 ARMS-ERC matricu, nākamais ir 1. attēla j) posms.
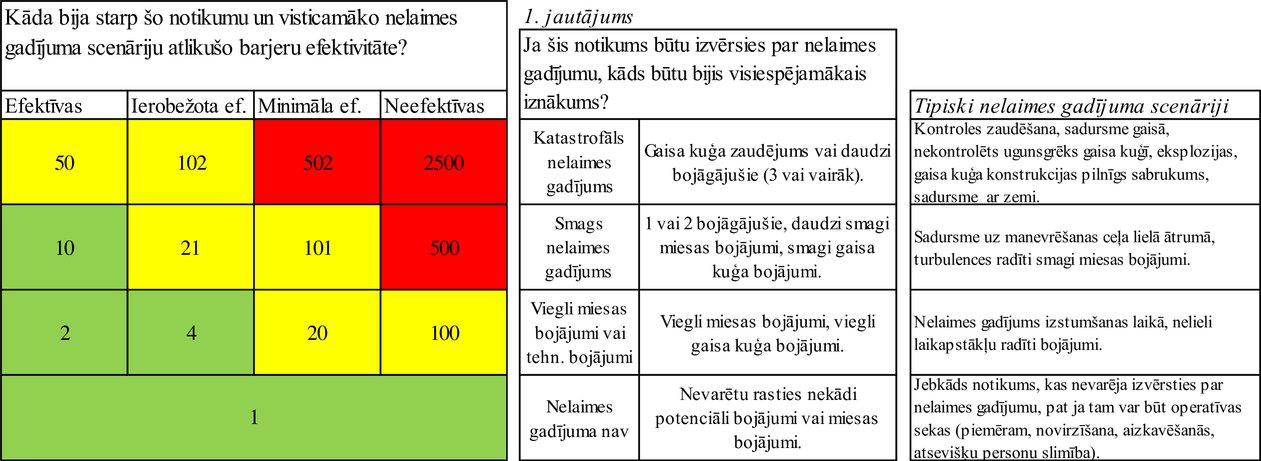
j. ERCS“nelaimes gadījuma iespējamā iznākuma varbūtība” rezultāts – tieša pārrēķināšana
Ja atgadījuma ziņojumā ir ARMS“Barjeru efektivitātes” reitings, tad, lai noteiktu ERCS“Nelaimes gadījuma iespējamā iznākuma varbūtības” rezultātu, izmanto šādu tiešu pārrēķināšanu uz ERCS matricu.

k. Ekvivalentais ERCS rezultāts
ERCS“nelaimes gadījuma iespējamā iznākuma smaguma” un “nelaimes gadījuma iespējamā iznākuma varbūtības” rezultātus kombinē ERCS matricā, lai ģenerētu ekvivalento ERCS rezultātu, kā noteikts Deleģētās regulas (ES) 2020/2034 pielikuma 2. posmā.
2. MANUĀLA PĀRRĒĶINĀŠANA
Šī manuālā pārrēķināšana sastāv no šādām divām darbplūsmām:
|
— |
1. darbplūsma – nodrošina manuālu pārrēķināšanu ERCS smaguma rezultāta iegūšanai, |
|
— |
2. darbplūsma – nodrošina manuālu pārrēķināšanu ERCS varbūtības rezultāta iegūšanai. |

2.1. 1. DARBPLŪSMA
D. ERCS“nelaimes gadījuma iespējamā iznākuma smaguma” rezultāts – manuāla pārrēķināšana
|
— |
Ja atgadījuma ziņojumā nav informācijas par “atgadījuma kategoriju” vai “masas grupu”, vai abām, tad, lai noteiktu “nelaimes gadījuma iespējamo iznākumu” vai galveno riska jomu, piemēro Deleģētās regulas (ES) 2020/2034 pielikumā definēto ERCS metodiku. Galīgais rezultāts ir k), kura pirmā zīme atbilst alfabētiskajai vērtībai, kas iegūta, aprēķinot atgadījuma smagumu (smaguma rezultāts no A līdz X). |
2.2. 2. DARBPLŪSMA
L. ERCS sleja “Nelaimes gadījuma iespējamā iznākuma varbūtība” – manuāla procedūra
|
— |
Atgadījuma ziņojumus bez “ATM – kopā” smaguma nevar tieši pārrēķināt ERCS“Nelaimes gadījuma iespējamā iznākuma varbūtība” rezultātā. |
Tomēr “ATM – uz zemes” smagums var ļaut veikt daļēju pārrēķināšanu, kartējot “ATM – uz zemes” barjeras novērtējumu un izmantojot ERCS barjeras novērtējuma procesu, kas definēts Deleģētās regulas (ES) 2020/2034 pielikuma 2.1.3. punktā.
M. ERCS“nelaimes gadījuma iespējamā iznākuma varbūtība” rezultāts – manuāls process
Ja atgadījumu ziņojumos neizmanto 4x4 ARMS-ERC matricu, lai atgadījumam piešķirtu rezultātu tad, lai ģenerētu ERCS“nelaimes gadījuma iespējamā iznākuma varbūtības” rezultātu, ARMS-ERC barjeras novērtējuma vērtību pārrēķina ERCS barjeras novērtējumā, kā noteikts Deleģētās regulas (ES) 2020/2034 pielikuma 2.1.3. punktā.
k. Ekvivalentais ERCS rezultāts
ERCS“nelaimes gadījuma iespējamā iznākuma smaguma” un “nelaimes gadījuma iespējamā iznākuma varbūtības” rezultātus kombinē ERCS matricā, lai ģenerētu ekvivalento ERCS rezultātu, kā noteikts Deleģētās regulas (ES) 2020/2034 pielikuma 2. posmā.
(1) ERCS rezultāts ir divzīmju vērtība, kur pirmā zīme atbilst alfabētiskajai vērtībai, kas iegūta, aprēķinot atgadījuma smagumu (smaguma rezultāts no A līdz X), un otrā zīme ir skaitliskā vērtība, kas iegūta, aprēķinot atbilstošo atgadījuma rezultātu (varbūtību).
(2) Ar RAT metodiku klasificē ar gaisa satiksmes pārvaldību saistītus atgadījumus. Ar RAT metodiku atgadījumiem nepiešķir rezultātu, jo ar to tikai mēra, cik tuvu ATM atgadījums bijis tam, lai kļūtu par nelaimes gadījumu. RAT metodika ir iedalīta vairākos galvenajos elementos (t. i., “ATM – uz zemes”, “ATM – gaisā”), katrs no kuriem dod ieguldījumu galīgajā RAT“ATM – kopā” smaguma rezultātā. Lai iegūtu “ATM – kopā” smaguma rezultātu, ir jābūt pieejamam gan “ATM – uz zemes”, gan “ATM – gaisā” smaguma rezultātam.
(3) Saskaņā ar RAT metodiku “smagums” RAT norāda, cik slikts faktiskais atgadījums bijis salīdzinājumā ar citiem atgadījumiem. Ar RAT metodiku “smagumu” nosaka, novērtējot aizsardzību/barjeras.
|
29.11.2021 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 426/41 |
KOMISIJAS ĪSTENOŠANAS REGULA (ES) 2021/2083
(2021. gada 26. novembris),
ar kuru aptur tirdzniecības politikas pasākumus, kas attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem noteikti ar Īstenošanas regulām (ES) 2018/886 un (ES) 2020/502
EIROPAS KOMISIJA,
ņemot vērā Līgumu par Eiropas Savienības darbību,
ņemot vērā Eiropas Parlamenta un Padomes Regulu (ES) Nr. 654/2014 (2014. gada 15. maijs) par Savienības tiesību īstenošanu starptautiskās tirdzniecības noteikumu piemērošanai un izpildei un ar kuru groza Padomes Regulu (EK) Nr. 3286/94, ar ko nosaka Kopienas procedūras kopējās tirdzniecības politikas jomā, lai nodrošinātu Kopienas tiesību īstenošanu saskaņā ar starptautiskās tirdzniecības noteikumiem, jo īpaši tiem, kas ieviesti Pasaules Tirdzniecības organizācijas (PTO) aizgādnībā (1), un jo īpaši tās 7. panta 3. punktu,
tā kā:
|
(1) |
Komisija 2018. gada 20. jūnijā pieņēma Īstenošanas regulu (ES) 2018/886 (2) par konkrētiem tirdzniecības politikas pasākumiem attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem, kas paredz vairāku ASV izcelsmes ražojumu importam Savienībā piemērot papildu muitas nodokļus:
|
|
(2) |
Komisija 2020. gada 7. aprīlī pieņēma Īstenošanas regulu (ES) 2020/502 (3), kas paredz konkrētu Amerikas Savienoto Valstu izcelsmes ražojumu importam Savienībā piemērot papildu muitas nodokļus:
|
|
(3) |
Pēc ES un ASV kopīgā paziņojuma, kas publicēts 2021. gada 17. maijā, Komisija 2021. gada 31. maijā pieņēma Īstenošanas regulu (ES) 2021/866 (4) par tirdzniecības politikas pasākumiem attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem, ar kuru līdz 2021. gada 30. novembrim apturēja papildu ad valorem nodokļu piemērošanu ražojumiem, kas uzskaitīti Īstenošanas regulas (ES) 2018/886 II pielikumā. |
|
(4) |
Ja Komisija uzskata, ka tas ir piemēroti, tā Savienības vārdā var grozīt Īstenošanas regulas (ES) 2018/886 (5) un (ES) 2020/502 (6), lai ņemtu vērā jebkādas Amerikas Savienoto Valstu aizsargpasākumu izmaiņas vai grozījumus. |
|
(5) |
Amerikas Savienotās Valstis 2021. gada 31. oktobrī paziņoja par šādiem savu attiecīgo aizsargpasākumu grozījumiem, kas stāsies spēkā 2022. gada 1. janvārī:
|
|
(6) |
Attiecīgi Savienībai būtu jāaptur ar Īstenošanas regulām (ES) 2018/886 un (ES) 2020/502 noteikto papildu ad valorem nodokļu piemērošana uz laikposmu līdz 2023. gada 31. decembrim. Apturēšana būtu jāveic šādi:
|
|
(7) |
Šāda apturēšana ļautu Savienībai un Amerikas Savienotajām Valstīm ievērojami uzlabot pašreizējo sadarbību, tostarp ar mērķi atcelt attiecīgos tarifus. Tomēr jāatzīmē, ka izņēmumi attiecībā uz ASV pasākumiem tiks piemēroti tikai līdz 2023. gada 31. decembrim. Šādi izņēmumi, kas piešķirti Amerikas Savienotajās Valstīs esošiem importētājiem, kuri importē Savienības ražojumus, ievērojami samazina Amerikas Savienoto Valstu aizsargpasākumu negatīvo ietekmi. Tāpēc apturēšanas periods, proti, līdz 2023. gada 31. decembrim, tiek uzskatīts par pietiekamu un saprātīgu, un tajā ir pienācīgi ņemti vērā Amerikas Savienoto Valstu 2021. gada 31. oktobra paziņojumi. |
|
(8) |
Regulas (ES) Nr. 654/2014 4. panta 2. punkta c) apakšpunktā noteikts, ka Savienības rīcībai jābūt praktiski līdzvērtīgai to koncesiju vai citu saistību apjomam, kuras ietekmējuši trešās valsts aizsargpasākumi. |
|
(9) |
Komisijai būtu pastāvīgi jāpārskata apturēšana, ņemot vērā jaunākās norises, piemēram, norises, kas varētu pasliktināt situāciju attiecībā uz Savienības eksportu, uz kuru joprojām attiecas Amerikas Savienoto Valstu aizsargpasākumi, tostarp jebkādi šķēršļi, kas ietekmē Savienības eksportu. Komisija var grozīt šo regulu, lai ņemtu vērā šādas norises un jebkādas izmaiņas Amerikas Savienoto Valstu aizsargpasākumos vai to grozījumus. |
|
(10) |
Apturēšana neskar Savienības nostāju, saskaņā ar kuru Amerikas Savienoto Valstu veiktie aizsargpasākumi joprojām nav saderīgi ar PTO līgumu. |
|
(11) |
Šajā regulā paredzētie pasākumi ir saskaņā ar atzinumu, ko sniegusi Tirdzniecības šķēršļu komiteja, kura izveidota ar Eiropas Parlamenta un Padomes Regulu (ES) 2015/1843 (7), |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
No 2022. gada 1. janvāra līdz 2023. gada 31. decembrim aptur papildu ad valorem nodokļu ar 10 % un 25 % likmi piemērošanu tādu ražojumu importam, kas uzskaitīti Īstenošanas regulas (ES) 2018/886 I pielikumā.
No 2021. gada 1. decembra līdz 2023. gada 31. decembrim aptur papildu ad valorem nodokļu ar 10 %, 25 %, 35 % un 50 % likmi piemērošanu tādu ražojumu importam, kas uzskaitīti Īstenošanas regulas (ES) 2018/886 II pielikumā.
Neskarot turpmāku apturēšanu vai grozīšanu, ieskaitot arī agrāku piemērošanu no jauna, Īstenošanas regulā (ES) 2018/886 paredzētos nodokļus piemēro no 2024. gada 1. janvāra ieskaitot.
2. pants
Īstenošanas regulas (ES) 2020/502 piemērošanu aptur līdz 2023. gada 31. decembrim šādi:
|
a) |
no 2022. gada 1. janvāra aptur papildu ad valorem nodokļus ar 20 % un 7 % likmi Īstenošanas regulas (ES) 2020/502 1. panta 2. punkta a) apakšpunktā minēto ražojumu importam; |
|
b) |
no 2023. gada 8. februāra aptur papildu ad valorem nodokli ar 4,4 % likmi Īstenošanas regulas (ES) 2020/502 1. panta 2. punkta b) apakšpunktā minētā ražojuma importam. |
Neskarot turpmāku apturēšanu vai grozīšanu, ieskaitot arī agrāku piemērošanu no jauna, Īstenošanas regulā (ES) 2020/502 paredzētos nodokļus piemēro no 2024. gada 1. janvāra ieskaitot.
3. pants
Šī regula stājas spēkā 2021. gada 30. novembrī.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2021. gada 26. novembrī
Komisijas vārdā –
priekšsēdētāja
Ursula VON DER LEYEN
(1) OV L 189, 27.6.2014., 50. lpp.; grozījumi izdarīti ar Eiropas Parlamenta un Padomes Regulu (ES) 2015/1843 (2015. gada 6. oktobris) (OV L 272, 16.10.2015., 1. lpp.) un ar Eiropas Parlamenta un Padomes Regulu (ES) 2021/167 (2021. gada 10. februāris) (OV L 49, 12.2.2021., 1. lpp.).
(2) Komisijas Īstenošanas regula (ES) 2018/886 (2018. gada 20. jūnijs) par konkrētiem tirdzniecības politikas pasākumiem attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem un ar ko groza Īstenošanas regulu (ES) 2018/724 (OV L 158, 21.6.2018., 5. lpp.).
(3) Komisijas Īstenošanas regula (ES) 2020/502 (2020. gada 6. aprīlis) par konkrētiem tirdzniecības politikas pasākumiem attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem (OV L 109, 7.4.2020., 10. lpp.).
(4) Komisijas Īstenošanas regula (ES) 2021/866 (2021. gada 28. maijs), ar kuru aptur tirdzniecības politikas pasākumus, kas attiecībā uz konkrētiem Amerikas Savienoto Valstu izcelsmes ražojumiem noteikti ar Īstenošanas regulu (ES) 2018/886 (OV L 190, 31.5.2021., 94. lpp.).
(5) Īstenošanas regulas (ES) 2018/886 7. apsvērums.
(6) Īstenošanas regulas (ES) 2020/502 19. apsvērums.
(7) Eiropas Parlamenta un Padomes Regula (ES) 2015/1843 (2015. gada 6. oktobris), ar ko nosaka Savienības procedūras kopējās tirdzniecības politikas jomā, lai nodrošinātu Savienības tiesību īstenošanu saskaņā ar starptautiskās tirdzniecības noteikumiem, jo īpaši tiem, kas ieviesti Pasaules Tirdzniecības organizācijas aizgādnībā (OV L 272, 16.10.2015., 1. lpp.).