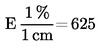ISSN 1725-5112
Eiropas Savienības
Oficiālais Vēstnesis
L 54

Izdevums latviešu valodā
Tiesību akti
52. sējums
2009. gada 26. februāris
|
ISSN 1725-5112 |
||
|
Eiropas Savienības Oficiālais Vēstnesis |
L 54 |
|
|
|
||
|
Izdevums latviešu valodā |
Tiesību akti |
52. sējums |
|
Saturs |
|
I Tiesību akti, kuri pieņemti, piemērojot EK/Euratom līgumus, un kuru publicēšana ir obligāta |
Lappuse |
|
|
|
REGULAS |
|
|
|
* |
Komisijas Regula (EK) Nr. 152/2009 (2009. gada 27. janvāris), ar ko nosaka paraugu ņemšanas un analīzes metodes barības oficiālajai kontrolei ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) Dokuments attiecas uz EEZ |
|
LV |
Tiesību akti, kuru virsraksti ir gaišajā drukā, attiecas uz kārtējiem jautājumiem lauksaimniecības jomā un parasti ir spēkā tikai ierobežotu laika posmu. Visu citu tiesību aktu virsraksti ir tumšajā drukā, un pirms tiem ir zvaigznīte. |
I Tiesību akti, kuri pieņemti, piemērojot EK/Euratom līgumus, un kuru publicēšana ir obligāta
REGULAS
|
26.2.2009 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 54/1 |
KOMISIJAS REGULA (EK) Nr. 152/2009
(2009. gada 27. janvāris),
ar ko nosaka paraugu ņemšanas un analīzes metodes barības oficiālajai kontrolei
(Dokuments attiecas uz EEZ)
EIROPAS KOPIENU KOMISIJA,
ņemot vērā Eiropas Kopienas dibināšanas līgumu,
ņemot vērā Eiropas Parlamenta un Padomes 2004. gada 29. aprīļa Regulu (EK) Nr. 882/2004 par oficiālo kontroli, ko veic, lai nodrošinātu atbilstības pārbaudi saistībā ar dzīvnieku barības un pārtikas aprites tiesību aktiem un dzīvnieku veselības un dzīvnieku labturības noteikumiem (1), un jo īpaši tās 11. panta 4. punkta a), b) un c) apakšpunktu,
tā kā:
|
(1) |
Direktīvas 70/373/EEK īstenošanai tika pieņemti šādi tiesību akti, kas paliek spēkā saskaņā ar Regulas (EK) Nr. 882/2004 61. panta 2. punktu:
|
|
(2) |
Tā kā Direktīvu 70/373/EEK aizstāja ar Regulu (EK) Nr. 882/2004, ir lietderīgi ar vienu regulu aizstāt minētās direktīvas īstenošanas aktus. Vienlaikus jāpielāgo metodes, ņemot vērā jaunākos zinātnes un tehnikas sasniegumus. Metodes, kas vairs neder to paredzētajam nolūkam, jāsvītro. Ir paredzēts pienācīgā laikā atjaunināt paraugu ņemšanas noteikumus, lai ņemtu vērā jaunākos sasniegumus attiecībā uz barības ražošanas, uzglabāšanas, pārvadāšanas un tirgošanas veidu, tomēr ir lietderīgi pagaidām saglabāt spēkā esošos noteikumus par paraugu ņemšanu. |
|
(3) |
Tāpēc jāatceļ Direktīva 71/250/EEK, 71/393/EEK, 72/199/EEK, 73/46/EEK, 76/371/EEK, 76/372/EEK, 78/633/EEK, 81/715/EEK, 84/425/EEK, 86/174/EEK, 93/70/EEK, 93/117/EK, 98/64/EK, 1999/27/EK, 1999/76/EK, 2000/45/EK, 2002/70/EK un 2003/126/EK. |
|
(4) |
Šajā regulā paredzētie pasākumi ir saskaņā ar Pārtikas aprites un dzīvnieku veselības pastāvīgās komitejas atzinumu, |
IR PIEŅĒMUSI ŠO REGULU.
1. pants
Paraugus barības oficiālajai kontrolei, lai noteiktu sastāvdaļas, piedevas un nevēlamas vielas, izņemot pesticīdu un mikroorganismu atliekas, ņem saskaņā ar metodēm, kas izklāstītas I pielikumā.
2. pants
Paraugus analīzei sagatavo un rezultātus izsaka saskaņā ar metodēm, kas izklāstītas II pielikumā.
3. pants
Analīzes barības oficiālajai kontrolei veic, izmantojot metodes, kas izklāstītas III pielikumā (Analīzes metodes, lai kontrolētu barības līdzekļu un barības maisījumu sastāvu), IV pielikumā (Analīzes metodes, lai kontrolētu atļauto piedevu koncentrāciju barībā), V pielikumā (Analīzes metodes, lai kontrolētu nevēlamas vielas barībā) un VI pielikumā (Analīzes metodes, lai noteiktu dzīvnieku izcelsmes sastāvdaļas barības oficiālajai kontrolei).
4. pants
Mājputnu barības maisījumu enerģētisko vērtību aprēķina saskaņā ar VII pielikumu.
5. pants
VIII pielikumā izklāstītās analīzes metodes, lai kontrolētu vairs neatļautu piedevu nelikumīgu atrašanos barībā, izmanto apstiprināšanas nolūkos.
6. pants
Direktīvu 71/250/EEK, 71/393/EEK, 72/199/EEK, 73/46/EEK, 76/371/EEK, 76/372/EEK, 78/633/EEK, 81/715/EEK, 84/425/EEK, 86/174/EEK, 93/70/EEK, 93/117/EK, 98/64/EK, 1999/27/EK, 1999/76/EK, 2000/45/EK, 2002/70/EK un 2003/126/EK atceļ.
Atsauces uz atceltajām direktīvām uzskata par atsaucēm uz šo regulu un lasa saskaņā ar atbilstības tabulām IX pielikumā.
7. pants
Šī regula stājas spēkā divdesmitajā dienā pēc tās publicēšanas Eiropas Savienības Oficiālajā Vēstnesī.
To piemēro no 2009. gada 26. augusta.
Šī regula uzliek saistības kopumā un ir tieši piemērojama visās dalībvalstīs.
Briselē, 2009. gada 27. janvārī
Komisijas vārdā –
Komisijas locekle
Androulla VASSILIOU
(1) OV L 165, 30.4.2004., 1. lpp.
(2) OV L 155, 12.7.1971., 13. lpp.
(3) OV L 279, 20.12.1971., 7. lpp.
(4) OV L 123, 29.5.1972., 6. lpp.
(5) OV L 83, 30.3.1973., 21. lpp.
(6) OV L 102, 15.4.1976., 1. lpp.
(7) OV L 102, 15.4.1976., 8. lpp.
(8) OV L 206, 29.7.1978., 43. lpp.
(9) OV L 257, 10.9.1981., 38. lpp.
(10) OV L 238, 6.9.1984., 34. lpp.
(11) OV L 130, 16.5.1986., 53. lpp.
(12) OV L 234, 17.9.1993., 17. lpp.
(13) OV L 329, 30.12.1993., 54. lpp.
(14) OV L 257, 19.9.1998., 14. lpp.
(15) OV L 118, 6.5.1999., 36. lpp.
(16) OV L 207, 6.8.1999., 13. lpp.
(17) OV L 174, 13.7.2000., 32. lpp.
(18) OV L 209, 6.8.2002., 15. lpp.
(19) OV L 339, 24.12.2003., 78. lpp.
I PIELIKUMS
PARAUGU ŅEMŠANAS METODES
1. MĒRĶIS UN DARBĪBAS JOMA
Paraugus, kas paredzēti barības oficiālajai kontrolei, ņem saskaņā ar tālāk aprakstītajām metodēm. Tādā veidā iegūtus paraugus uzskata par reprezentatīviem tā apjoma sastāvam, no kura tie ņemti.
2. DARBINIEKI, KURI ŅEM PARAUGUS
Paraugus ņem darbinieki, kurus dalībvalstis ir norīkojušas šā darba veikšanai.
3. DEFINĪCIJAS
Apjoms, no kura ņem paraugu, ir vienību veidojoša produkta daļa, kuras īpašības pieņem par viendabīgām.
Elementārparaugs ir daudzums, kas paņemts vienā punktā no apjoma, no kura ņem paraugu.
Kopparaugs ir elementārparaugu kopums no viena un tā paša apjoma, no kura ņem paraugu.
Samazinātais paraugs ir kopparauga reprezentatīva daļa, kas no tā iegūta samazinājuma procesā.
Gala paraugs ir samazinātā parauga daļa vai homogenizētā kopparauga daļa.
4. IERĪCES
|
4.1. |
Paraugu ņemšanas ierīcēm jābūt izgatavotām no materiāliem, kas nevar piesārņot paraugu ņemšanai izmantojamos produktus. Dalībvalstis drīkst oficiāli apstiprināt tādas ierīces. |
4.2. Ierīces, ko iesaka cietās barības paraugu ņemšanai
4.2.1. Manuāla paraugu ņemšana
|
4.2.1.1. |
Plakandibena lāpstiņa ar vertikālām malām. |
|
4.2.1.2. |
Šķēpveida paraugu ņemšanas ierīce ar garu šķēlumu vai nodalījumiem. Šķēpveida paraugu ņemšanas ierīces izmēriem jāatbilst apjoma, no kura ņem paraugu, raksturlielumiem (konteinera dziļumam, maisa izmēriem utt.) un barības daļiņu lielumam. |
4.2.2. Mehāniska paraugu ņemšana
Apstiprinātu mehānisku ierīci var izmantot, lai paņemtu paraugus no barības, kuru tajā brīdī pārvieto.
4.2.3. Sadalītājs
Ierīci, kas paredzēta paraugu sadalīšanai apmēram vienādās daļās, var izmantot elementārparaugu ņemšanai, kā arī samazināto un gala paraugu sagatavošanai.
5. KVANTITATĪVĀS PRASĪBAS
|
5.A. |
Attiecībā uz tādu vielu vai produktu kontroli, kas vienmērīgi sadalīti visā barībā |
|
|
5.A.1. |
Apjoms, no kura ņem paraugu Apjomam, no kura ņem paraugu, jābūt tādam, lai paraugu varētu paņemt no katras tā sastāvdaļas. |
|
|
5.A.2. |
Elementārparaugi |
|
|
5.A.2.1. |
Neiesaiņota barība: |
Elementārparaugu skaita minimums: |
|
5.A.2.1.1. |
apjomi, no kuriem ņem paraugu, nepārsniedz 2,5 metriskās tonnas |
septiņi |
|
5.A.2.1.2. |
apjomi, no kuriem ņem paraugu, pārsniedz 2,5 metriskās tonnas |
√ 20-kārtīgs to metrisko tonnu skaits, no kurām veidots apjoms, no kura ņem paraugu (1), augstākais līdz 40 elementārparaugiem |
|
5.A.2.2. |
Iesaiņota barība: |
Paku, no kurām jāņem paraugs, minimālais skaits (2): |
|
5.A.2.2.1. |
Pakas, kurās ir vairāk par vienu kg: |
|
|
5.A.2.2.1.1. |
apjoms, no kura ņem paraugu, ja paku skaits ir no 1 līdz 4 |
visas pakas |
|
5.A.2.2.1.2. |
apjoms, no kura ņem paraugu, ja paku skaits ir no 5 līdz 16 |
četras |
|
5.A.2.2.1.3. |
apjoms, no kura ņem paraugu, ja paku skaits ir vairāk par 16 |
√ paku skaits, kas veido apjomu, no kura ņem paraugu (1), augstākais līdz 20 pakām |
|
5.A.2.2.2. |
Pakas, kuru masa nepārsniedz 1 kg |
četras |
|
5.A.2.3. |
Šķidrā vai pusšķidrā barība: |
Konteineru, no kuriem jāņem paraugs, minimālais skaits (2): |
|
5.A.2.3.1. |
Konteineri, kuros ir vairāk par vienu litru: |
|
|
5.A.2.3.1.1. |
apjoms, no kura ņem paraugu, ja konteineru skaits ir no 1 līdz 4 |
visi konteineri |
|
5.A.2.3.1.2. |
apjoms, no kura ņem paraugu, ja konteineru skaits ir no 5 līdz 16 |
četras |
|
5.A.2.3.1.3. |
apjoms, no kura ņem paraugu, ja konteineru skaits ir vairāk par 16 |
√ konteineru skaits, kas veido apjomu, no kura ņem paraugu (1), augstākais līdz 20 konteineriem |
|
5.A.2.3.2. |
Konteineri, kuru tilpums nepārsniedz vienu litru |
četras |
|
5.A.2.4. |
Barības bloki un minerālvielu briketes |
Obligātais bloku vai brikešu skaits, no kā jāņem paraugs (2): viens bloks vai brikete no apjoma, no kura ņem paraugu un kas sastāv no 25 vienībām, augstākais līdz četriem blokiem vai briketēm |
|
5.A.3. |
Kopparaugs Prasīts ir viens kopparaugs no apjoma, no kura ņem paraugu. Kopējais elementārparaugu skaits, kas veido kopparaugu, ir ne mazāk kā: |
|
|
5.A.3.1. |
Neiesaiņota barība |
4 kg |
|
5.A.3.2. |
Iesaiņota barība: |
|
|
5.A.3.2.1. |
pakas, kuru masa ir vairāk par 1 kg |
4 kg |
|
5.A.3.2.2. |
pakas, kuru masa nepārsniedz 1 kg |
četru oriģinālpaku satura svars |
|
5.A.3.3. |
Šķidrā vai pusšķidrā barība: |
|
|
5.A.3.3.1. |
konteineri, kuros ir vairāk par vienu litru |
četri litri |
|
5.A.3.3.2. |
konteineri, kuru tilpums nepārsniedz vienu litru |
četru oriģinālkonteineru satura tilpums |
|
5.A.3.4. |
Barības bloki vai minerālvielu briketes: |
|
|
5.A.3.4.1. |
katrs sver vairāk par 1 kg |
4 kg |
|
5.A.3.4.2. |
katrs sver ne vairāk par 1 kg |
četru oriģinālbloku vai brikešu svars |
|
5.A.4. |
Gala paraugi Ja vajadzīgs, no kopparauga pēc samazināšanas iegūst gala paraugus. Jāanalizē vismaz viens gala paraugs. Analīzei paredzētā gala parauga apjoms ir ne mazāk kā: |
|
|
|
Cietā barība |
500 g |
|
|
Šķidrā vai pusšķidrā barība |
500 ml |
|
5.B. |
Attiecībā uz tādu nevēlamu vielu vai produktu kontroli, kuri varētu būt nevienmērīgi izplatīti barībā, piemēram, aflatoksīni, melnie rudzu graudi, rīcinaugs un crotalaria barības līdzekļos (3) |
|
|
5.B.1. |
Apjoms, no kura ņem paraugu: skatīt 5.A.1. punktu |
|
|
5.B.2. |
Elementārparaugi |
|
|
5.B.2.1. |
Neiesaiņota barība: skatīt 5.A.2.1. punktu |
|
|
5.B.2.2. |
Iesaiņota barība: |
Paku, no kurām jāņem paraugs, skaita minimums: |
|
5.B.2.2.1. |
apjoms, no kura ņem paraugu, kurā ietilpst no 1 līdz 4 pakām |
visas pakas |
|
5.B.2.2.2. |
apjoms, no kura ņem paraugu, kurā ietilpst no 5 līdz 16 pakām |
četras |
|
5.B.2.2.3. |
apjoms, no kura ņem paraugu, kurā ietilpst vairāk par 16 pakām |
√ paku skaits, kas veido apjomu, no kura ņem paraugu (1), augstākais līdz 40 pakām |
|
5.B.3. |
Kopparaugi Kopparaugu skaits mainīsies atkarībā no tā, cik liels ir apjoms, no kura ņem paraugu. Kopparaugu minimālais skaits no apjoma, no kura ņem paraugu, ir norādīts tālāk. Kopējais elementārparaugu svars, kas veido katru kopparaugu, ir ne mazāk kā 4 kg. |
|
|
5.B.3.1. |
Neiesaiņota barība |
|
|
|
Apjoma, no kura ņem paraugu, svars metriskajās tonnās: |
Kopparaugu minimālais skaits no viena apjoma, no kura ņem paraugu: |
|
|
līdz 1 |
1 |
|
|
vairāk par 1, līdz 10 |
2 |
|
|
vairāk par 10, līdz 40 |
3 |
|
|
vairāk par 40 |
4 |
|
5.B.3.2. |
Iesaiņota barība |
|
|
|
Apjoms, no kura ņem paraugu, izteikts paku skaitā: |
Kopparaugu minimālais skaits no viena apjoma, no kura ņem paraugu: |
|
|
no 1 līdz 16 |
1 |
|
|
no 17 līdz 200 |
2 |
|
|
no 201 līdz 800 |
3 |
|
|
vairāk par 800 |
4 |
|
5.B.4. |
Gala paraugi No katra kopparauga pēc samazināšanas iegūst gala paraugus. Jāanalizē vismaz viens gala paraugs no kopparauga. Analīzei paredzētā gala parauga svaram jābūt ne mazākam kā 500 g. |
|
6. INSTRUKCIJAS PARAUGU ŅEMŠANAI, SAGATAVOŠANAI UN IESAIŅOŠANAI
6.1. Vispārīgas norādes
Paraugi jāņem un jāsagatavo, cik vien ātri iespējams, ievērojot piesardzību, kas vajadzīga, lai nodrošinātu, ka produkts netiek nedz mainīts, nedz piesārņots. Instrumentiem, tāpat arī virsmām un paraugiem paredzētajiem konteineriem jābūt tīriem un sausiem.
6.2. Elementārparaugi
6.2.A. Attiecībā uz tādu vielu vai produktu kontroli, kas vienmērīgi sadalīti visā barībā
Elementārparaugi jāņem izlases veidā no visa apjoma, no kura ņem paraugu, un tiem jābūt aptuveni vienāda lieluma.
6.2.A.1.
Apjomu, no kura ņem paraugu, iedomāti sadala aptuveni vienādās daļās. Daļu skaits atbilst saskaņā ar 5.A.2. punktu prasītajam elementārparaugu skaitam, paraugus ņem izlases veidā – vismaz vienu paraugu no katras daļas.
Attiecīgā gadījumā paraugu var ņemt laikā, kad apjomu, no kura jāņem paraugs, pārvieto (iekrauj vai izkrauj).
6.2.A.2.
Pēc tam, kad atbilstīgi 5.A.2. punktā minētajām norādēm izraudzīts paraugu ņemšanai vajadzīgais paku skaits, ar šķēpveida ierīci vai lāpstiņu paņem daļu no katras pakas satura. Vajadzības gadījumā paraugus var ņemt laikā, kad katra paka atsevišķi ir iztukšota. Visus gabalus sadala daļās, vajadzības gadījumā atdalot tos un pievienojot atpakaļ paraugam, katrā kopparaugā atsevišķi.
6.2.A.3.
Pēc tam, kad atbilstīgi 5.A.2. punktā minētajām norādēm izraudzīts paraugu ņemšanai vajadzīgais konteineru skaits, to saturu vajadzības gadījumā homogenizē un paņem noteiktu daudzumu no katra konteinera.
Elementārparaugus var ņemt laikā, kad saturu ņem laukā no konteineriem.
6.2.A.4.
Pēc tam, kad atbilstīgi 5.A.2. punktā minētajām norādēm izraudzīts paraugu ņemšanai vajadzīgais konteineru skaits, paraugus paņem no dažādiem līmeņiem.
Paraugus var arī ņemt laikā, kad saturu ņem laukā no konteineriem, bet pirmās frakcijas izmet.
Abos gadījumos kopējam paņemtajam tilpumam jābūt ne mazākam kā 10 litri.
6.2.A.5.
Pēc tam, kad atbilstīgi 5.A.2. punktā minētajām norādēm izvēlēts paraugu ņemšanai vajadzīgais bloku vai brikešu skaits, paraugus ņem no katra bloka vai briketes.
6.2.B. Attiecībā uz tādu nevēlamu vielu vai produktu kontroli, kuri varētu būt nevienmērīgi izplatīti barībā, kā aflatoksīni, melnie rudzu graudi, rīcinaugs un crotalaria barības līdzekļos
Apjomu, no kura ņem paraugu, iedomāti sadala aptuveni vienādās daļās, un daļu skaits atbilst saskaņā ar 5.B.3. punktu prasītajam kopparaugu skaitam. Ja minētais skaits ir vairāk nekā viens, tad kopējo elementārparaugu skaitu, kas paredzēts 5.B.2. punktā, sadala vienmērīgi pa dažādām daļām. Tad ņem aptuveni vienādu izmēru paraugus (4) un tā, lai kopējais no katras daļas ņemtais paraugu daudzums nebūtu mazāks par 4 kg – daudzumu, kas prasīts katram kopparaugam. Elementārparaugus, kas paņemti no dažādām daļām, neapvieno kopā.
6.3. Kopparaugu sagatavošana
6.3.A. Attiecībā uz tādu vielu vai produktu kontroli, kas vienmērīgi sadalīti visā barībā
Elementārparaugus samaisa, lai tie veidotu vienu kopparaugu.
6.3.B. Attiecībā uz tādu nevēlamu vielu vai produktu kontroli, kuri varētu būt nevienmērīgi izplatīti barībā, kā aflatoksīni, melnie rudzu graudi, rīcinaugs un crotalaria barības līdzekļos
Elementārparaugus no katras tā apjoma daļas, no kura ņem paraugu, samaisa un sagatavo 5.B.3. punktā paredzēto kopparaugu skaitu, atzīmējot katra kopparauga izcelsmi.
6.4. Gala paraugu sagatavošana
Materiālu katrā kopparaugā rūpīgi samaisa, līdz iegūst homogenizētu paraugu (5). Vajadzības gadījumā kopparaugu vispirms samazina vismaz līdz diviem kilogramiem vai diviem litriem (samazinātais paraugs), vai nu izmantojot mehānisko vai automātisko sadalītāju, vai ar kvartēšanas metodi.
Tad sagatavo vismaz trīs aptuveni vienāda lieluma gala paraugus, kuri atbilst 5.A.4. vai 5.B.4. punktā norādītajām kvantitatīvajām prasībām. Katru paraugu ieliek atbilstošā konteinerā. Veic visus vajadzīgos piesardzības pasākumus, lai novērstu jebkādas paraugu satura izmaiņas, to piesārņošanu vai atšķaidīšanu, kas varētu rasties pārvadāšanas vai uzglabāšanas laikā.
6.5. Gala paraugu iesaiņošana
Konteinerus vai pakas aizplombē un uz tiem uzliek etiķetes (visa etiķete jāiestrādā plombā) tā, lai konteinerus nevarētu atvērt, nesabojājot plombu.
7. PARAUGU ŅEMŠANAS PIERAKSTI
Par katru parauga ņemšanu ir jāizdara ieraksts, kas ļauj viennozīmīgi identificēt katru apjomu, no kura paņemts paraugs.
8. PARAUGU GALAMĒRĶIS
No katra kopparauga vismaz vienu gala paraugu pēc iespējas drīzāk nosūta pilnvarotajai analīžu laboratorijai, līdzi nosūtot arī laborantam vajadzīgo informāciju.
(1) Ja iegūtais skaitlis ir daļskaitlis, tad to noapaļo uz augšu līdz nākamajam veselajam skaitlim.
(2) Pakām vai konteineriem, kuru saturs nepārsniedz vienu kilogramu vai vienu litru, un blokiem vai briketēm, kas katrs sver ne vairāk par vienu kilogramu, elementārparaugs ir vienas oriģinālpakas vai konteinera saturs, viens bloks vai viena brikete.
(3) Metodes, kas paredzētas 5.A. punktā, jāizmanto, lai kontrolētu aflatoksīnus, melnos rudzu graudus, rīcinaugu un crotalaria kompleksajā barībā un papildbarībā.
(4) Iesaiņotai barībai daļu no to paku satura, no kurām jāņem paraugs, noņem, izmantojot šķēpveida ierīci vai lāpstiņu – vajadzības gadījumā pēc tam, kad pakas ir atsevišķi iztukšotas.
(5) Visus gabalus sadala daļās (vajadzības gadījumā atdalot tos un pievienojot atpakaļ paraugam) katrā kopparaugā atsevišķi.
II PIELIKUMS
VISPĀRĪGI NOTEIKUMI PAR BARĪBAS ANALĪZES METODĒM
A. PARAUGU SAGATAVOŠANA ANALĪZEI
1. Mērķis
Tālāk aprakstītās procedūras attiecas uz gala paraugu sagatavošanu analīzei, kurus nosūta kontroles laboratorijām pēc paraugu paņemšanas, ievērojot I pielikumā paredzētos noteikumus.
Minētie paraugi jāsagatavo tā, lai analīzes metodēs noteiktie iesvara daudzumi būtu homogēni un gala paraugiem reprezentatīvi.
2. Piesardzības pasākumi, kas jāievēro
Paraugu sagatavošanas procedūra ir atkarīga no tā, kādas analīzes metodes tiks izmantotas. Tāpēc ir ārkārtīgi svarīgi nodrošināt, lai ievērotā paraugu sagatavošanas procedūra būtu atbilstoša izmantotajai analīzes metodei.
Visas vajadzīgās darbības jāveic tā, lai pēc iespējas izvairītos no parauga piesārņošanas un novērstu izmaiņas tā sastāvā.
Lai samazinātu gaisa un gaismas iedarbību uz paraugiem, to samalšanu, samaisīšanu un sijāšanu veic iespējami ātri. Neizmanto dzirnavas un dzirnaviņas, kurās paraugs var ievērojami sakarst.
Barību, kas ir īpaši jutīga pret karstumu, ieteicams samalt manuāli. Veic arī visu iespējamo, lai nodrošinātu, ka pašas ierīces nepiesārņo paraugu ar mikroelementiem.
Ja paraugu nevar sagatavot, ievērojami nemainot mitruma saturu tajā, tad mitruma saturu nosaka pirms un pēc parauga sagatavošanas atbilstoši III pielikuma A daļā noteiktajai metodei.
3. Procedūra
Paraugu sadala atbilstošos apakšparaugos analīzei un salīdzināšanai, izmantojot piemērotus sašķelšanas paņēmienus, piemēram, pamīšus iegrābjot ar lāpstiņu, stacionāri vai rotējoši jaucot. Dalīšana konusā un kvartēšana nav ieteicama, jo tad apakšparaugos var būt augsta šķelšanas kļūda. Salīdzināšanas paraugu tur piemērotā tīrā, sausā konteinerā ar hermētisku aizbāzni un analīzei sagatavo vismaz 100 g apakšparaugu, kā norādīts tālāk.
3.1. Barība, ko var samalt dabīgā stāvoklī
Ja vien analīzes metodēs nav noteikts citādi, tad visu paraugu vajadzības gadījumā vispirms samaļ un tad izsijā caur kvadrātacu sietu ar 1 mm acojumu (saskaņā ar ISO R565 ieteikumu). Paraugus nedrīkst samalt pārāk smalki.
Izsijāto paraugu samaisa un savāc piemērotā tīrā, sausā un hermētiski noslēdzamā konteinerā. Paraugu vēlreiz samaisa tieši pirms iesvara ņemšanas analīzēm.
3.2. Barība, ko var samalt pēc kaltēšanas
Ja analīzes metodēs nav noteikts citādi, tad paraugu izkaltē atbilstoši sākotnējās kaltēšanas procedūrai, kas aprakstīta III pielikuma A daļā minētajā mitruma noteikšanas metodē, līdz tā mitruma saturs samazinās un sasniedz 8–12 %. Pēc tam rīkojas, kā noteikts 3.1. punktā.
3.3. Šķidrā vai pusšķidrā barība
Paraugu savāc piemērotā tīrā, sausā un hermētiski noslēdzamā konteinerā. Paraugu vēlreiz rūpīgi samaisa tieši pirms iesvara ņemšanas analīzēm.
3.4. Cita barība
Paraugus, kurus nevar sagatavot pēc kādas no iepriekšminētajām procedūrām, apstrādā, izmantojot jebkuru citu procedūru, ar kuru var nodrošināt, lai analīzēm ņemtais iesvars būtu homogēns un gala paraugiem reprezentatīvs.
4. Paraugu uzglabāšana
Paraugi jāuzglabā temperatūrā, kurā nemainās to sastāvs. Paraugus, kuros paredzēts analizēt vitamīnus vai vielas, kas ir īpaši jutīgas pret gaismu, uzglabā brūna stikla konteineros.
B. NOTEIKUMI ATTIECĪBĀ UZ REAĢENTIEM UN IERĪCĒM, KO IZMANTO ANALĪZES METODĒS
|
1. |
Ja vien analīzes metodēs nav noteikts citādi, tad visiem analītiskajiem reaģentiem jābūt analītiski tīriem (a. t.). Nosakot mikroelementus, reaģentu tīrība jāpārbauda ar tukšo analīzi. Atkarībā no iegūtajiem rezultātiem var būt vajadzīga reaģentu papildu attīrīšana. |
|
2. |
Visās darbībās, kas ietver šķīdumu gatavošanu, atšķaidīšanu, skalošanu vai mazgāšanu un kas analīzes metodēs minētas, nenorādot izmantojamo šķīdinātāju vai atšķaidītāju, jālieto ūdens. Parasti izmanto demineralizētu vai destilētu ūdeni. Atsevišķos gadījumos, kas norādīti analīzes metodēs, ūdens attīrīšanai izmanto īpašus paņēmienus. |
|
3. |
Ņemot vērā kontroles laboratoriju standarta aprīkojumu, analīzes metodēs norādīti tikai specifiski instrumenti un ierīces vai to īpašs izmantošanas veids. Tiem jābūt tīriem, īpaši gadījumos, kad jānosaka ļoti mazi vielu daudzumi. |
C. ANALĪZES METOŽU PIEMĒROŠANA UN REZULTĀTU IZTEIKŠANA
1. Ekstrahēšanas procedūra
Vairākas metodes nosaka specifisku ekstrahēšanas procedūru. Parasti var izmantot citu ekstrahēšanas procedūru, kas nav minēta konkrētajā metodē, ja ir pierādīts, ka izmantotajai ekstrahēšanas procedūrai ir tāda pati ekstrahēšanas efektivitāte kā metodē minētajai procedūrai.
2. Attīrīšanas procedūra
Vairākas metodes nosaka specifisku attīrīšanas procedūru. Parasti var izmantot citu attīrīšanas procedūru, kas nav minēta konkrētajā metodē, ja ir pierādīts, ka izmantotā attīrīšanas metode dod tādus pašus analīžu rezultātus analizētajai matricei kā metodē minētā procedūra.
3. Ziņošana par izmantoto analīzes metodi
Katru barības sastāvā esošo vielu parasti nosaka, izmantojot vienu metodi. Ja ir paredzētas vairākas metodes, tad kontroles laboratorijā izmantotā konkrētā metode jānorāda analīzes ziņojumā.
4. Noteikšanu skaits
Rezultāts, ko norāda analīzes ziņojumā, ir vidējā vērtība, ko iegūst pēc vismaz divām noteikšanām ar pietiekamu atkārtojamību, analizējot atsevišķas parauga daļas.
Tomēr nevēlamu vielu analīzes gadījumā, ja pirmajā noteikšanā rezultāts ir ievērojami (> 50 %) zemāks nekā kontrolējamā specifikācija, nav vajadzīgs veikt papildu noteikšanu, ja tiek piemērotas atbilstošas kvalitātes procedūras.
Ja kontrolē vielas vai sastāvdaļas deklarēto saturu un pirmajā noteikšanā rezultāts apstiprina deklarēto saturu, t. i., analīzes rezultāts atbilst pieņemamajam deklarētā satura variāciju diapazonam, tad nav vajadzīgs veikt papildu noteikšanu, ja piemēro atbilstošas kvalitātes procedūras.
Dažos gadījumos minētais pieņemamais variāciju diapazons ir noteikts tiesību aktos, piemēram, Padomes Direktīvā 79/373/EEK (1).
5. Ziņošana par analīžu rezultātiem
Analīžu rezultātu izsaka veidā, kas noteikts analīzes metodē, līdz attiecīgam būtisko skaitļu skaitam un vajadzības gadījumā koriģē līdz mitruma saturam, kas gala paraugam bija pirms sagatavošanas.
6. Mērījumu neprecizitāte un atgūstamības procents nevēlamu vielu analīzes gadījumā
Attiecībā uz nevēlamām vielām Direktīvas 2002/32/EK nozīmē, tostarp dioksīniem un dioksīniem līdzīgiem polihlorbifeniliem, dzīvnieku barībai paredzētu produktu uzskata par neatbilstīgu šo vielu noteiktajam maksimāli pieļaujamam saturam, ja pēc analīžu rezultātiem secina, ka minēto vielu maksimāli pieļaujamais saturs ir pārsniegts, ņemot vērā mērījumu paplašināto neprecizitāti un atgūstamības korekciju. Lai novērtētu analīzes rezultātā iegūtās vielas koncentrācijas daudzuma atbilstību, to koriģē atbilstoši atgūstamības rādītājam un samazina atbilstoši mērījumu paplašinātās neprecizitātes koeficientam. Šo procedūru izmanto tikai tad, ja saskaņā ar analīzes metodi ir iespējams aplēst mērījumu neprecizitāti un atgūstamības korekciju (piemēram, to nav iespējams izmantot mikroskopiskā analīzē).
Analīžu rezultātus paziņo šādi (ciktāl izmantotā analīzes metode ļauj aplēst mērījumu neprecizitāti un atgūstamības procentu):
|
a) |
ar atgūstamības korekciju, norādot atgūstamības līmeni. Atgūstamības korekcija nav vajadzīga, ja atgūstamības procents ir 90–110 %; |
|
b) |
kā “x +/– U”, kur x ir analīzes rezultāts un U – mērījumu paplašinātās neprecizitātes procents, izmantojot pārklāšanās koeficientu 2, kas nodrošina aptuveni 95 % ticamības pakāpi. |
Tomēr, ja analīzes rezultāts ir ievērojami (> 50 %) zemāks nekā kontrolējamā specifikācija un ja piemēro atbilstošas kvalitātes procedūras un analīzes nolūks ir tikai pārbaudīt atbilstību tiesību normām, tad analīžu rezultātus var paziņot bez atgūstamības korekcijas, un minētajos gadījumos var neziņot par atgūstamības procentu un mērījumu neprecizitāti.
III PIELIKUMS
ANALĪZES METODES, LAI KONTROLĒTU BARĪBAS LĪDZEKĻU UN BARĪBAS MAISĪJUMU SASTĀVU
A. MITRUMA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt mitruma saturu barībā. Ja barība satur gaistošas vielas, piemēram, organiskās skābes, tad jāievēro, ka kopā ar mitruma saturu nosaka arī ievērojamu daudzumu gaistošo vielu.
Tā neietver piena produktu kā barības līdzekļu analīzi, minerālvielu un galvenokārt no minerālvielām sastāvošu maisījumu analīzi, dzīvnieku un augu tauku un eļļu un eļļas augu sēklu un augļu analīzi.
2. Princips
Paraugu žāvē norādītajos apstākļos, kas mainās atkarībā no barības īpašībām. Svara zudumu nosaka sverot. Ja analizē cietu barību, kurai ir augsts mitruma saturs, tad jāveic sākotnējā kaltēšana.
3. Ierīces
|
3.1. |
Drupinātājs no materiāla, kas neabsorbē mitrumu un ir viegli tīrāms, ļauj ātri un vienmērīgi sasmalcināt produktu, būtiski to nesakarsējot, un, cik iespējams, novērš saskari ar apkārtējo gaisu, kā arī atbilst 4.1.1. un 4.1.2. punktā minētajām prasībām (piemēram, āmurdzirnaviņas vai āmurdzirnaviņas ar ūdens dzesēšanu, saliekamās konusveida dzirnaviņas, palēninātu apgriezienu vai zobratu rupja maluma dzirnaviņas). |
|
3.2. |
Analītiskie svari ar precizitāti līdz 1 mg. |
|
3.3. |
Sausi konteineri no nerūsējoša metāla vai stikla ar vākiem, kas nodrošina hermētisku noslēgšanu; darba virsma, kas ļauj izklāt testa paraugu aptuveni 0,3 g/cm2 slānī. |
|
3.4. |
Elektriskā izotermiskā krāsns (± 2 oC), kas tiek pienācīgi ventilēta un nodrošina iespēju ātri regulēt temperatūru (1). |
|
3.5. |
Regulējams elektriskais vakuumžāvēšanas skapis, kas aprīkots ar eļļas sūkni un vai nu ar mehānismu karsta, sausa gaisa ievadīšanai, vai ar vielu, kas uzsūc mitrumu (piemēram, kalcija oksīdu). |
|
3.6. |
Eksikators ar biezu, perforētu metāla vai porcelāna plāksni, kurā ir viela, kas efektīvi uzsūc mitrumu. |
4. Procedūra
|
N.B.! |
Šajā iedaļā aprakstītās darbības jāveic tūlīt pēc paraugu iesaiņojumu atvēršanas. Analīzes jāatkārto vismaz divas reizes. |
4.1. Sagatavošana
4.1.1.
Ņem vismaz 50 g parauga. Vajadzības gadījumā sasmalcina vai sadala tā, lai novērstu mitruma satura izmaiņas (skatīt 6. punktu).
4.1.2.
Ņem vismaz 50 g parauga. Samaļ daļiņās, no kurām vismaz 50 % izsijājas caur sietu ar 0,5 mm acīm, un atlikums sietā ar 1 mm apaļu acojumu nepārsniedz 10 %.
4.1.3.
Ņem aptuveni 25 g parauga, nosver ar precizitāti līdz 10 mg, pievieno atbilstīgu daudzumu bezūdens smilts, kas nosvērta ar precizitāti līdz 10 mg, un maisa, līdz iegūst homogēnu produktu.
4.2. Kaltēšana
4.2.1.
Nosver konteineru (3.3.) ar vāku ar precizitāti līdz 1 mg. Nosvērtajā konteinerā ar precizitāti līdz 1 mg iesver aptuveni 5 g parauga un izlīdzina. Konteineru bez vāka ieliek krāsnī, kas iepriekš sakarsēta līdz 103 oC. Lai novērstu krāsns temperatūras pārmērīgu samazināšanos, konteineru ievieto pēc iespējas ātri. Atstāj kaltēties četras stundas, skaitot no brīža, kad krāsns temperatūra atkal sasniedz 103 oC. Konteineram atkal uzliek vāku, izņem to no krāsns, 30 līdz 45 minūtes atdzesē eksikatorā (3.6.) un nosver ar precizitāti līdz 1 mg.
Barību, kas galvenokārt sastāv no eļļām un taukiem, kaltē krāsnī vēl papildu 30 minūtes 130 oC temperatūrā. Abu svēršanas rezultātu starpība nedrīkst pārsniegt 0,1 % mitruma.
4.2.2.
Nosver konteineru (3.3.) ar vāku ar precizitāti līdz 0,5 mg. Nosvērtajā konteinerā ar precizitāti līdz 1 mg iesver aptuveni 5 g sasmalcinātā parauga un izlīdzina. Konteineru bez vāka ieliek krāsnī, kas iepriekš sakarsēta līdz 130 oC. Lai novērstu krāsns temperatūras pārmērīgu samazināšanos, konteineru ievieto pēc iespējas ātri. Atstāj kaltēties divas stundas, skaitot no brīža, kad krāsns temperatūra atkal sasniedz 130 oC. Konteineram atkal uzliek vāku, izņem to no krāsns, 30 līdz 45 minūtes atdzesē eksikatorā (3.6.) un nosver ar precizitāti līdz 1 mg.
|
4.2.3. |
Barības maisījumi, kas satur vairāk nekā 4 % saharozes vai laktozes: barības līdzekļi, piemēram, ceratoniju augļi, hidrolizēti graudaugu produkti, iesala sēklas, kaltēti biešu graizījumi, zivju un cukura šķīstošie produkti; barības maisījumi, kas satur vairāk nekā 25 % minerālsāļu, tostarp kristalizācijas ūdeni. Nosver konteineru (3.3.) ar vāku ar precizitāti līdz 0,5 mg. Nosvērtajā konteinerā ar precizitāti līdz 1 mg iesver aptuveni 5 g parauga un izlīdzina. Konteineru bez vāka ieliek vakuuma krāsnī (3.5.), kas iepriekš sakarsēta 80 oC līdz 85 oC temperatūrā. Lai novērstu krāsns temperatūras pārmērīgu samazināšanos, konteineru ievieto pēc iespējas ātri. Palielina spiedienu līdz 100 toriem un zem šāda spiediena kaltē četras stundas – vai nu karsta, sausa gaisa plūsmā, vai izmantojot vielu, kas uzsūc mitrumu (aptuveni 300 g uz 20 paraugiem). Pēdējā gadījumā pēc paredzētā spiediena sasniegšanas vakuumsūkni atvieno. Kaltēšanas laiku skaita no brīža, kad krāsns temperatūra atkal sasniedz 80 oC līdz 85 oC. Krāsnī uzmanīgi atjauno atmosfēras spiedienu. Krāsni atver, konteineram tūlīt uzliek vāku, izņem konteineru no krāsns, 30 līdz 45 minūtes atdzesē eksikatorā (3.6.) un nosver ar precizitāti līdz 1 mg. Vēl 30 minūtes kaltē vakuuma krāsnī 80 oC līdz 85 oC temperatūrā un vēlreiz nosver. Abu svēršanas rezultātu starpība nedrīkst pārsniegt 0,1 % mitruma. |
4.3. Sākotnējā kaltēšana
4.3.1.
Cietu barību ar augstu mitruma saturu, ko grūti sasmalcināt, sākotnēji kaltē šādi:
50 g nesasmalcināta parauga (ja vajadzīgs, tad saspiestu vai aglomerētu barību var nedaudz sadalīt) ar precizitāti līdz 10 mg iesver piemērotā konteinerā (piemēram, 20 ×12 cm alumīnija plāksnē ar 0,5 cm apmali). Atstāj kaltēties krāsnī 60 oC līdz 70 oC temperatūrā, līdz mitruma saturs ir samazinājies līdz 8–12 %. Izņem no krāsns, bez vāka atdzesē laboratorijā vienu stundu un nosver ar precizitāti līdz 10 mg. Nekavējoties sasmalcina, kā norādīts 4.1.1. punktā, un kaltē, kā norādīts 4.2.1. vai 4.2.3. punktā, atkarībā no barības īpašībām.
4.3.2.
Graudus, kuru mitruma saturs pārsniedz 17 %, sākotnēji kaltē šādi:
50 g nesamaltu graudu ar precizitāti līdz 10 mg iesver piemērotā konteinerā (piemēram, 20 ×12 cm alumīnija plāksnē ar 0,5 cm apmali). Piecas līdz septiņas minūtes atstāj kaltēties krāsnī 130 oC temperatūrā. Izņem no krāsns, bez vāka atdzesē laboratorijā divas stundas un nosver ar precizitāti līdz 10 mg. Nekavējoties samaļ, kā norādīts 4.1.2. punktā, un izkaltē, kā norādīts 4.2.2. punktā.
5. Rezultātu aprēķināšana
Mitruma saturu (X), izteiktu procentos no parauga, aprēķina, izmantojot šādas formulas:
5.1. Kaltēšana bez sākotnējās kaltēšanas

kur:
|
m |
= |
testa parauga sākotnējais svars gramos, |
|
m0 |
= |
izkaltētā testa parauga svars gramos. |
5.2. Kaltēšana ar sākotnējo kaltēšanu

kur:
|
m |
= |
testa parauga sākotnējais svars gramos, |
|
m1 |
= |
testa parauga svars gramos pēc sākotnējās kaltēšanas, |
|
m2 |
= |
testa parauga svars gramos pēc sasmalcināšanas vai samalšanas, |
|
m0 |
= |
izkaltētā testa parauga svars gramos. |
5.3. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nepārsniedz 0,2 % mitruma absolūtās vērtības.
6. Novērojums
Ja sasmalcināšana ir vajadzīga un ja tās rezultātā mainās produkta mitruma saturs, tad barības sastāvdaļu analīzes rezultāti jākoriģē, pamatojoties uz parauga mitruma saturu tā sākotnējā stāvoklī.
B. MITRUMA NOTEIKŠANA DZĪVNIEKU UN AUGU TAUKOS UN EĻĻĀS
1. Mērķis un darbības joma
Šī metode ļauj noteikt ūdens un gaistošo vielu saturu dzīvnieku un augu taukos un eļļās.
2. Princips
Paraugu žāvē līdz konstantam svaram (svara zudumam starp diviem secīgiem svērumiem jābūt mazākam par vai vienādam ar 1 mg) 103 oC temperatūrā. Svara zudumu nosaka sverot.
3. Ierīces
|
3.1. |
Plakandibena trauks, izgatavots no materiāla, kas izturīgs pret koroziju, apmēram 3 cm augsts un 8–9 cm diametrā. |
|
3.2. |
Apmēram 10 cm garš termometrs ar pastiprinātu bumbiņu un izplešanās cauruli augšgalā, kas graduēts apmēram no 80 oC līdz vismaz 110 oC. |
|
3.3. |
Smilšu vanna vai elektriskā plītiņa. |
|
3.4. |
Eksikators ar vielu, kas efektīvi uzsūc mitrumu. |
|
3.5. |
Analītiskie svari. |
4. Procedūra
Sausā, nosvērtā traukā (3.1.), kurā ir termometrs (3.2.), ar precizitāti līdz tuvākajam mg iesver apmēram 20 g homogenizētā parauga. Pastāvīgi maisot ar termometru, smilšu vannā vai uz elektriskās plītiņas (3.3.) silda tā, lai apmēram septiņās minūtēs temperatūra paaugstinātos līdz 90 oC.
Karstumu samazina, sekojot tam, cik bieži no trauka dibena paceļas burbuļi. Temperatūra nedrīkst pārsniegt 105 oC. Turpina maisīt, skrāpējot gar trauka dibenu, līdz pārstāj veidoties burbuļi.
Lai pilnībā izdalītos viss mitrums, vairākas reizes temperatūru paaugstina līdz 103 oC ± 2 oC, starplaikos starp karsēšanām atdzesējot līdz 93 oC. Pēc tam eksikatorā (3.4.) ļauj atdzist līdz istabas temperatūrai un nosver. Minēto darbību atkārto tikmēr, kamēr svara zudums starp diviem secīgiem svērumiem vairs nepārsniedz 2 mg.
|
N.B.! |
Ja parauga svars pēc atkārtotas karsēšanas palielinās, tas liecina par tauku oksidēšanos, un tādā gadījumā rezultātu aprēķina, ņemot vērā svērumu, kas izdarīts pirms tam, kad svars sāka palielināties. |
5. Rezultātu aprēķināšana
Mitruma saturu (X), izteiktu procentos no parauga, aprēķina, izmantojot šādu formulu:

kur:
|
m |
= |
testa parauga svars gramos, |
|
m1 |
= |
trauka un tā satura kopējais svars gramos pirms karsēšanas, |
|
m2 |
= |
trauka un tā satura kopējais svars gramos pēc karsēšanas. |
Ja rezultāts ir mazāks par 0,05 %, tad tas jāreģistrē kā “mazāks par 0,05 %”.
Atkārtojamība
Mitruma satura starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 0,05 % absolūtās vērtības.
C. JĒLPROTEĪNA SATURA NOTEIKŠANA
1. Mērķis un darbības joma
Ar šo metodi var noteikt jēlproteīna daudzumu barībā, pamatojoties uz slāpekļa saturu, ko nosaka saskaņā ar Kjeldāla metodi.
2. Princips
Paraugu šķeļ ar sērskābi katalizatora klātbūtnē. Skābes šķīdumu neitralizē un noregulē tā reakciju līdz bāziskai ar nātrija hidroksīda šķīdumu. Amonjaku destilē un uztver noteiktā sērskābes daudzumā, kuras pārpalikumu titrē ar nātrija hidroksīda standartšķīdumu.
Alternatīvi – atbrīvoto amonjaku destilē borskābes šķīduma pārpalikumā, pēc tam titrē ar sālsskābes vai sērskābes šķīdumu.
3. Reaģenti
|
3.1. |
Kālija sulfāts. |
|
3.2. |
Katalizators: vara (II) oksīds (CuO) vai vara (II) sulfāta pentahidrāts (CuSO4 5H2O). |
|
3.3. |
Granulētais cinks. |
|
3.4. |
Sērskābe, ρ20 = 1,84 g/ml. |
|
3.5. |
Sērskābe, standarta volumetriskais šķīdums, c(H2SO4) = 0,25 mol/l. |
|
3.6. |
Sērskābe, standarta volumetriskais šķīdums, c(H2SO4) = 0,10 mol/l. |
|
3.7. |
Sērskābe, standarta volumetriskais šķīdums, c(H2SO4) = 0,05 mol/l. |
|
3.8. |
Metilsarkanais indikators; izšķīdina 300 mg metilsarkanā 100 mililitros etanola, σ = 95–96 % (v/v). |
|
3.9. |
Nātrija hidroksīda šķīdums (var izmantot tehnisko) β = 40 g/100 ml (m/v: 40 %). |
|
3.10. |
Nātrija hidroksīds, standarta volumetriskais šķīdums c(NaOH) = 0,25 mol/l. |
|
3.11. |
Nātrija hidroksīds, standarta volumetriskais šķīdums c(NaOH) = 0,10 mol/l. |
|
3.12. |
Sālsskābē mazgāti, izkarsēti pumeka gabaliņi. |
|
3.13. |
Acetanilīds (kušanas temp. = 114 oC, N-saturs = 10,36 %). |
|
3.14. |
Saharoze (nesatur slāpekli). |
|
3.15. |
Borskābe (H3BO3). |
|
3.16. |
Metilsarkanā indikatora šķīdums: izšķīdina 100 mg metilsarkanā 100 mililitros etanola vai metanola. |
|
3.17. |
Bromkrezolzaļā šķīdums: izšķīdina 100 mg bromkrezolzaļā 100 mililitros etanola vai metanola. |
|
3.18. |
Borskābes šķīdums (10 g/l līdz 40 g/l atkarībā no izmantotās ierīces). Ja izmanto kolorimetrisko galarezultāta noteikšanu, tad metilsarkanais un bromkrezolzaļais indikators jāpievieno borskābes šķīdumiem. Ja sagatavo vienu litru borskābes šķīduma, tad pirms tilpuma korekcijas pievieno 7 ml metilsarkanā indikatora šķīduma (3.16.) un 10 ml bromkrezolzaļā šķīduma (3.17.). Atkarībā no izmantotā ūdens borskābes šķīduma pH līmenis dažādās partijās var būt atšķirīgs. Bieži vien, lai iegūtu pozitīvu tukšo analīzi, šķīdums jākoriģē, pievienojot nelielu sārma daudzumu.
|
|
3.19. |
Sālsskābes standarta volumetriskais šķīdums c(HCl) = 0,10 mol/l.
|
4. Ierīces
Ierīces, kas piemērotas šķelšanai, destilēšanai un titrēšanai saskaņā ar Kjeldāla metodi.
5. Procedūra
5.1. Šķelšana
No parauga ar precizitāti līdz 0,001 g ņem 1 g lielu iesvaru, ko pārnes šķelšanas aparāta kolbā. Pievieno 15 g kālija sulfāta (3.1.), atbilstošu daudzumu katalizatora (3.2.) (0,3 līdz 0,4 g vara (II) oksīda vai 0,9 līdz 1,2 g vara (II) sulfāta pentahidrāta), 25 ml sērskābes (3.4.) un, ja vajadzīgs, dažus pumeka gabaliņus (3.12.) un kolbas saturu samaisa.
Kolbu sākumā karsē uzmanīgi, ja vajadzīgs, laiku pa laikam maisot, līdz masa ir karbonizēta un vairs neputo; tad karsēšanu pastiprina, līdz šķidrums sāk pastāvīgi vārīties. Pietiek karsēt, kad vārošā skābe kondensējas uz kolbas sienām. Nedrīkst pieļaut kolbas pārkaršanu un organisko vielu daļiņu pielipšanu pie tās sienām.
Kad šķīdums kļūst dzidrs un gaiši zaļš, to turpina vārīt vēl divas stundas, tad ļauj atdzist.
5.2. Destilēšana
Uzmanīgi pievieno vajadzīgo ūdens daudzumu, lai nodrošinātu, ka sulfāti pilnībā izšķīst. Ļauj atdzist, tad, ja vajadzīgs, pievieno dažas cinka granulas (3.3.). Tad rīkojas, kā noteikts 5.2.1. vai 5.2.2. punktā.
5.2.1.
Atkarībā no sagaidāmā slāpekļa satura destilācijas aparāta uztvērējkolbā precīzi iemēra 25 ml sērskābes (3.5. vai 3.7.). Pievieno dažus pilienus metilsarkanā indikatora (3.8.).
Šķelšanas kolbu pievieno destilācijas aparāta kondensatoram un kondensatora galu iemērc šķidrumā, kas ir uztvērējkolbā, vismaz 1 cm dziļi (skatīt 8.3. novērojumu). Nepieļaujot amonjaka zudumus (skatīt 8.1. novērojumu), šķelšanas kolbā lēnām ielej 100 ml nātrija hidroksīda šķīduma (3.9.). Kolbu karsē, līdz destilējas amonjaks.
5.2.2.
Ja destilāta amonjaka saturu titrē manuāli, tad izmanto tālāk minēto procedūru. Ja destilēšanas vienība ir pilnībā automatizēta un ietver arī destilāta amonjaka satura titrēšanu, tad jāievēro ražotāja norādījumi attiecībā uz destilēšanas vienības darbināšanu.
Savācējkolbu, kurā ir 25 ml līdz 30 ml borskābes šķīduma (3.18.), novieto zem kondensatora izplūdes tā, lai padeves caurule atrastos zem borskābes šķīduma pārākuma virsmas. Destilācijas vienību koriģē, lai tā izdalītu 50 ml nātrija hidroksīda šķīduma (3.9.). Destilācijas vienību darbina saskaņā ar ražotāja norādījumiem un atbrīvoto amonjaku destilē, pievienojot nātrija hidroksīda šķīdumu. Destilātu uztver borskābē, kura uzņem šķīdumu. Destilāta apjoms (destilācijas ar tvaiku laiks) ir atkarīgs no slāpekļa daudzuma paraugā. Ievēro ražotāja norādījumus.
|
Piezīme. |
Pusautomātiskajā destilēšanas vienībā nātrija hidroksīda pārpalikumu pievieno un tvaiku destilē automātiski. |
5.3. Titrēšana
Rīkojas, kā noteikts 5.3.1. vai 5.3.2. punktā.
5.3.1.
Sērskābes pārpalikumu savācējkolbā titrē ar nātrija hidroksīda šķīdumu (3.10. vai 3.11.) atkarībā no izmantotās sērskābes koncentrācijas, līdz tiek sasniegts galapunkts.
5.3.2.
Savācējkolbas saturu titrē ar sālsskābes standarta volumetrisko šķīdumu (3.19.) vai ar sērskābes standarta volumetrisko šķīdumu (3.6.), izmantojot bireti, un nolasa izmantotā titranta daudzumu.
Ja piemēro kolorimetrisko galapunkta noteikšanu, tad galapunkts ir sasniegts, kad parādās pirmās sārtas krāsas zīmes. Provizoriski nosaka biretes rādījumu ar precizitāti līdz 0,05 ml. Izgaismots magnētiskā maisītāja trauks vai fotometrisks detektors var palīdzēt ieraudzīt galapunktu.
To var izdarīt automātiski, izmantojot tvaika destilētāju ar automātisko titrēšanu.
Jāievēro ražotāja norādījumi attiecībā uz konkrētā destilētāja vai destilētāja/titrētāja izmantošanu.
|
Piezīme. |
Ja izmanto automātisko titrēšanas sistēmu, tad titrēšana sākas tūlīt pēc destilācijas sākšanas un kad ir izmantots 1 % borskābes šķīdums (3.18.). Ja izmanto pilnībā automatizētu destilācijas vienību, tad amonjaka automātisko titrēšanu var veikt arī, ja galapunktu nosaka, izmantojot potenciometrisko pH sistēmu. Tādā gadījumā izmanto automātisko titrētāju ar pH mērītāju. pH mērītājs ir attiecīgi kalibrēts pH 4 līdz pH 7 diapazonā, ievērojot parastās laboratorijas pH kalibrēšanas procedūras. Titrēšanas pH galapunktu sasniedz pie pH 4,6, kas ir stāvākais punkts titrēšanas līknē (pārliekuma punkts). |
5.4. Tukšā analīze
Lai apstiprinātu, ka reaģenti nesatur slāpekli, veic tukšo analīzi (šķelšanu, destilēšanu un titrēšanu), parauga vietā izmantojot 1 g saharozes (3.14.).
6. Rezultātu aprēķināšana
Aprēķinus veic atbilstoši 6.1. vai 6.2. punktam.
6.1. Titrēšanas aprēķins atbilstoši 5.3.1. punktam
Jēlproteīna saturu, kas izteikts procentos no svara, aprēķina pēc šādas formulas:

kur:
|
Vo |
= |
NaOH tilpums (ml) (3.10. vai 3.11.), ko izmanto tukšajā analīzē, |
|
V1 |
= |
NaOH tilpums (ml) (3.10. vai 3.11.), ko izmanto parauga titrēšanā, |
|
c |
= |
nātrija hidroksīda (3.10. vai 3.11.) koncentrācija (mol/l), |
|
m |
= |
parauga svars (g). |
6.2. Titrēšanas aprēķins atbilstoši 5.3.2. punktam
6.2.1.
Jēlproteīna saturu, kas izteikts procentos no svara, aprēķina pēc šādas formulas:

kur:
|
m |
= |
analizējamā parauga svars (g), |
|
c |
= |
sālsskābes (3.19.) standarta volumetriskā šķīduma koncentrācija (mol/l), |
|
V0 |
= |
tukšajā analīzē izmantotās sālsskābes tilpums (ml), |
|
V1 |
= |
analizējamam paraugam izmantotās sālsskābes tilpums (ml). |
6.2.2.
Jēlproteīna saturu, kas izteikts procentos no svara, aprēķina pēc šādas formulas:
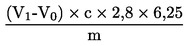
kur:
|
m |
= |
analizējamā parauga svars (g), |
|
c |
= |
sērskābes standarta volumetriskā šķīduma (3.6.) koncentrācija (mol/l), |
|
V0 |
= |
tukšajai analīzei izmantotās sērskābes (3.6.) tilpums (ml), |
|
V1 |
= |
analizējamam paraugam izmantotās sērskābes (3.6.) tilpums (ml). |
7. Metodes verifikācija
7.1. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt:
|
— |
0,2 % absolūtajā vērtībā, ja jēlproteīna saturs ir mazāks par 20 %, |
|
— |
1,0 % no lielākās vērtības, ja jēlproteīna saturs ir no 20 % līdz 40 %, |
|
— |
0,4 % absolūtajā vērtībā, ja jēlproteīna saturs ir lielāks par 40 %. |
7.2. Precizitāte
Analizē (veicot šķelšanu, destilēšanu un titrēšanu) 1,5 līdz 2,0 g acetanilīda (3.13.) 1 g saharozes (3.14.) klātbūtnē; 1 g acetanilīda patērē 14,80 ml sērskābes (3.5.). Atgūstamībai jābūt vismaz 99 %.
8. Novērojumi
|
8.1. |
Ierīce var būt manuāla, pusautomātiska vai automātiska. Ja paraugs pēc šķelšanas jāpārvieto destilēšanai, tad pārnešana jāveic bez zudumiem. Ja destilācijas aparāta kolba nav aprīkota ar pilināmo piltuvi, tad nātrija hidroksīdu, lēnām lejot gar kolbas sienu, pievieno tieši pirms tās savienošanas ar kondensatoru. |
|
8.2. |
Ja šķelšanas produkts sacietē, tad noteikšanu atkārto, lietojot lielāku daudzumu sērskābes (3.4.), nekā norādīts iepriekš. |
|
8.3. |
Analizējot produktus ar zemu slāpekļa saturu, vajadzības gadījumā sērskābes (3.7.) tilpumu uztvērējkolbā var samazināt līdz 10 ml vai 15 ml un papildina līdz 25 ml ar ūdeni. |
|
8.4. |
Regulārai analīzei, lai noteiktu jēlproteīnu, var izmantot alternatīvas analīzes metodes, bet šajā C daļā aprakstītā Kjeldāla metode ir standartmetode. Ar alternatīvo metodi (piemēram, DUMAS) iegūto rezultātu līdzvērtība salīdzinājumā ar standartmetodi jāpierāda attiecībā uz katru matrici atsevišķi. Tā kā ar alternatīvo metodi iegūtie rezultāti pat pēc līdzvērtības verifikācijas var nedaudz atšķirties no rezultātiem, kas iegūti ar standartmetodi, analīžu ziņojumā jāpiemin analīzes metode, kas izmantota jēlproteīna noteikšanai. |
D. URĪNVIELAS NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt urīnvielas daudzumu barībā.
2. Princips
Paraugu kopā ar dzidrināšanas aģentu iejauc ūdenī. Suspensiju filtrē. Urīnvielas saturu filtrātā nosaka pēc 4-dimetilaminobenzaldehīda (4-DMAB) pievienošanas, mērot optisko blīvumu pie 420 nm viļņu garuma.
3. Reaģenti
|
3.1. |
4-dimetilaminobenzaldehīda šķīdums: izšķīdina 1,6 g 4-DAMB 100 mililitros 96 % etanola un pievieno 10 ml sālsskābes (ρ201,19 g/ml). Šo reaģentu var glabāt ne ilgāk par divām nedēļām. |
|
3.2. |
Karesa I šķīdums: ūdenī izšķīdina 21,9 g cinka acetāta, Zn(CH3COO)2 2H2O, un 3 g ledus etiķskābes. Papildina ar ūdeni līdz 100 ml. |
|
3.3. |
Karesa II šķīdums: ūdenī izšķīdina 10,6 g kālija ferocianīda, K4 Fe (CN)6 3H2O. Papildina ar ūdeni līdz 100 ml. |
|
3.4. |
Aktīvā ogle, kas neabsorbē urīnvielu (jāpārbauda). |
|
3.5. |
Urīnviela, 0,1 % šķīdums (w/v). |
4. Ierīces
|
4.1. |
Rotācijas tipa maisītājs: aptuveni 35 līdz 40 apgr./min. |
|
4.2. |
Mēģenes: 160 × 16 mm ar pieslīpēta stikla aizbāžņiem. |
|
4.3. |
Spektrofotometrs. |
5. Procedūra
5.1. Parauga analīze
Nosver 2 g parauga ar precizitāti līdz tuvākajam mg un kopā ar 1 g aktīvās ogles (3.4.) pārnes 500 ml tilpuma mērkolbā. Pievieno 400 ml ūdens un 5 ml Karesa I šķīduma (3.2.), maisa apmēram 30 sekundes un pievieno 5 ml Karesa II šķīduma (3.3.). Trīsdesmit minūtes maisa maisītājā. Papildina ar ūdeni līdz tilpumam, sakrata un filtrē.
Noņem 5 ml caurspīdīgā, bezkrāsainā filtrāta, pārnes mēģenēs ar pieslīpēta stikla aizbāžņiem, pievieno 5 ml 4-DMAB šķīduma (3.1.) un samaisa. Mēģenes ievieto ūdens vannā 20 oC (+/– 4 oC) temperatūrā. Pēc piecpadsmit minūtēm ar spektrofotometru pie 420 nm mēra parauga šķīduma optisko blīvumu. Salīdzina ar reaģentu tukšās analīzes šķīdumu.
5.2. Kalibrēšanas līkne
Ņem pa 1, 2, 4, 5 un 10 ml urīnvielas šķīduma (3.5.), pārnes 100 ml tilpuma mērkolbās un papildina ar ūdeni līdz tilpumam. Ņem 5 ml no katra šķīduma, pievieno pa 5 ml 4-DMAB šķīduma (3.1.), homogenizē un iepriekš aprakstītajā veidā mēra optisko blīvumu pret kontrolšķīdumu, kas sastāv no 5 ml 4-DMAB un 5 ml ūdens, kurš nesatur urīnvielu. Konstruē kalibrēšanas līkni.
6. Rezultātu aprēķināšana
Urīnvielas daudzumu paraugā nosaka, izmantojot kalibrēšanas līkni.
Rezultātu izsaka procentos no parauga masas.
7. Novērojumi
|
7.1. |
Ja urīnvielas saturs pārsniedz 3 %, tad paraugu samazina līdz 1 g vai sākotnēji pagatavoto šķīdumu atšķaida tā, lai 500 mililitros nebūtu vairāk par 50 mg urīnvielas. |
|
7.2. |
Ja urīnvielas saturs ir zems, tad parauga iesvara masu palielina, ievērojot nosacījumu, ka no tā iegūtais filtrāts paliek caurspīdīgs un bezkrāsains. |
|
7.3. |
Ja paraugs satur tādus vienkāršus slāpekļa savienojumus kā aminoskābes, tad optisko blīvumu mēra pie 435 nm. |
E. GAISTOŠO SLĀPEKĻA BĀŽU NOTEIKŠANA
I. AR MIKRODIFŪZIJU
1. Mērķis un darbības joma
Šī metode ļauj barībā noteikt gaistošo slāpekļa bāžu saturu, kas izteikts kā amonjaks.
2. Princips
Paraugu ekstrahē ar ūdeni un šķīdumu dzidrina un filtrē. Gaistošās slāpekļa bāzes atdala mikrodifūzijas ceļā, izmantojot kālija karbonāta šķīdumu, savāc borskābes šķīdumā un titrē ar sērskābi.
3. Reaģenti
|
3.1. |
Trihloretiķskābe, 20 % šķīdums (w/v). |
|
3.2. |
Indikators: izšķīdina 33 mg bromkrezolzaļā un 65 mg metilsarkanā 100 mililitros 95–96 % (v/v) etanola. |
|
3.3. |
Borskābes šķīdums: viena litra mērkolbā 200 mililitros 95–96 % (v/v) etanola un 700 mililitros ūdens izšķīdina 10 g borskābes. Pievieno 10 ml indikatora (3.2.). Samaisa un vajadzības gadījumā koriģē šķīduma krāsu līdz gaiši sarkanai, pievienojot nātrija hidroksīda šķīdumu. 1 ml tāda šķīduma koriģēs līdz 300 μg NH3. |
|
3.4. |
Piesātināts kālija karbonāta šķīdums: 100 mililitros vāroša ūdens izšķīdina 100 g kālija karbonāta. Ļauj atdzist, filtrē. |
|
3.5. |
Sērskābe 0,01 mol/l. |
4. Ierīces
|
4.1. |
Rotācijas tipa maisītājs: aptuveni 35 līdz 40 apgr./min. |
|
4.2. |
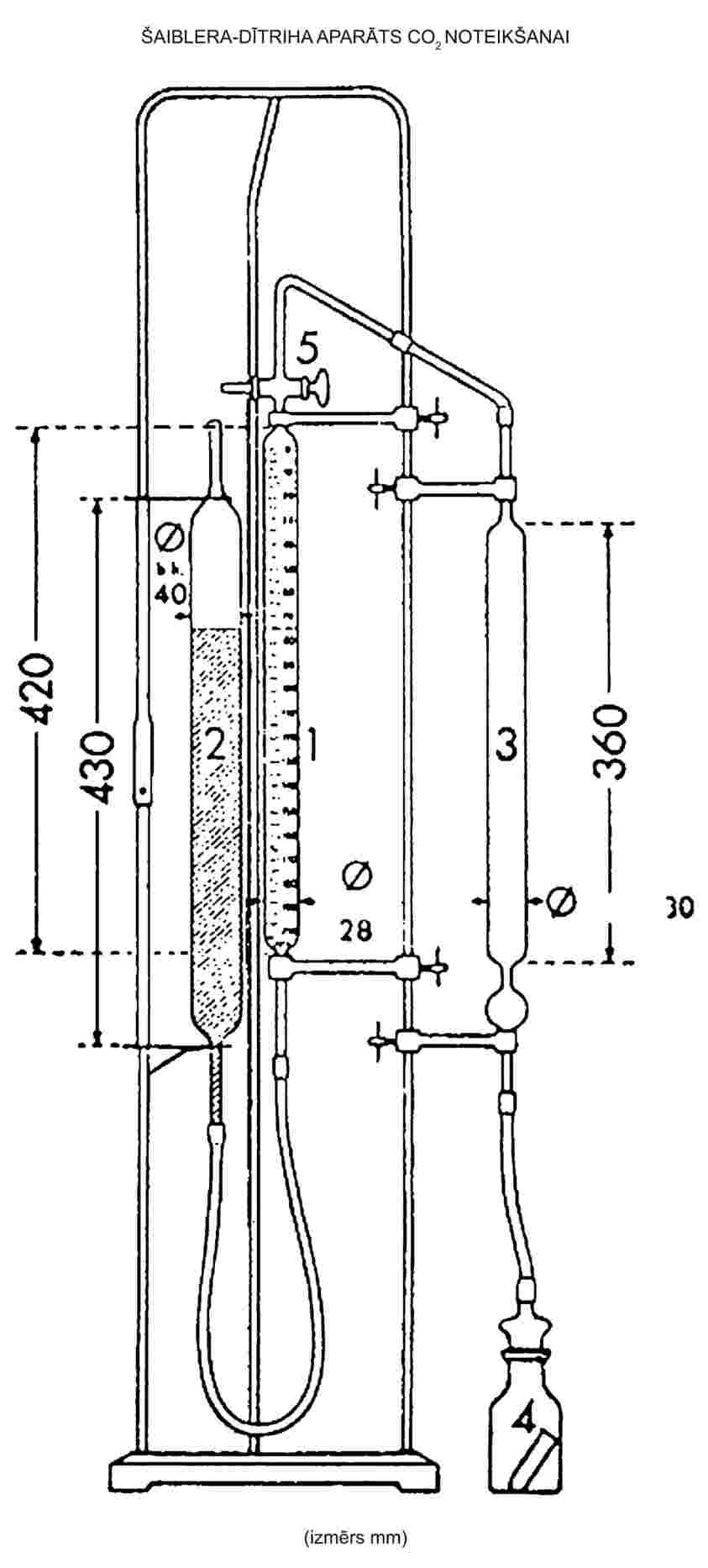
Stikla vai plastmasas Konveja trauks (skatīt diagrammu). |
|
4.3. |
Mikrobiretes, kas kalibrētas 1/100 ml iedaļās. |
5. Procedūra
Nosver 10 g parauga ar precizitāti līdz 1 mg un kopā ar 100 ml ūdens ielej 200 ml mērkolbā. Maisa maisītājā 30 minūtes. Pievieno 50 ml trihloretiķskābes šķīduma (3.1.), papildina līdz vajadzīgajam tilpumam ar ūdeni, spēcīgi sakrata un izfiltrē caur kroku filtru.
Izmantojot pipeti, Konveja trauka centrā iepilina 1 ml borskābes šķīduma (3.3.) un 1 ml parauga filtrāta – trauka ārējā lokā. Daļēji nosedz ar ietaukotu vāku. 1 ml piesātināta kālija karbonāta šķīduma (3.4.) ātri iepilina ārējā lokā un aizver vāku, hermētiski noslēdzot trauku. Trauku uzmanīgi griež, lai tas rotētu horizontālā plaknē un abi reaģenti sajauktos. Iztur vai nu vismaz četras stundas istabas temperatūrā, vai vienu stundu 40 oC.
Ja izmanto mikrobireti (4.3.), tad gaistošās bāzes borskābes šķīdumā titrē ar sērskābi (3.5.).
Veic tukšo analīzi, izmantojot to pašu procedūru, bet bez analizējamā parauga.
6. Rezultātu aprēķināšana
1 ml H2SO40,01 mol/l atbilst 0,34 miligramiem amonjaka.
Rezultātu izsaka procentos no parauga masas.
Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nepārsniedz:
|
— |
10 % relatīvajā vērtībā, ja amonjaka saturs ir mazāks par 1,0 %, |
|
— |
0,1 % absolūtajā vērtībā, ja amonjaka saturs ir 1,0 % vai vairāk. |
7. Novērojums
Ja amonjaka saturs paraugā pārsniedz 0,6 %, tad atšķaida sākotnējo filtrātu.
KONVEJA TRAUKS
Mērogs 1:1

II. DESTILĒJOT
1. Mērķis un darbības joma
Šī metode ļauj zivju miltos, kas praktiski nesatur urīnvielu, noteikt gaistošo slāpekļa bāžu saturu, izteiktu kā amonjaks. To piemēro tikai tad, ja amonjaka saturs ir mazāks par 0,25 %.
2. Princips
Paraugu ekstrahē ar ūdeni un šķīdumu dzidrina un filtrē. Gaistošās slāpekļa bāzes atdala viršanas temperatūrā, pievienojot magnija oksīdu, savāc konkrētā sērskābes daudzumā, kuras pārpalikumu attitrē ar nātrija hidroksīda šķīdumu.
3. Reaģenti
|
3.1. |
Trihloretiķskābe, 20 % šķīdums (w/v). |
|
3.2. |
Magnija oksīds. |
|
3.3. |
Pretputošanas emulsija (piemēram, silikons). |
|
3.4. |
Sērskābe 0,05 mol/l. |
|
3.5. |
Nātrija hidroksīda šķīdums 0,1 mol/l. |
|
3.6. |
Metilsarkanā 0,3 % šķīdums 95–96 % (v/v) etanolā. |
4. Ierīces
|
4.1. |
Rotācijas tipa maisītājs: aptuveni 35 līdz 40 apgr./min. |
|
4.2. |
Kjeldāla tipa destilēšanas ierīce. |
5. Procedūra
Ar precizitāti līdz 1 mg nosver 10 g parauga un kopā ar 100 ml ūdens ievieto 200 ml mērkolbā. Maisa maisītājā 30 minūtes. Pievieno 50 ml trihloretiķskābes šķīduma (3.1.), papildina līdz vajadzīgajam tilpumam ar ūdeni, spēcīgi sakrata un izfiltrē caur kroku filtru.
Ņem tādu tīra filtrāta daudzumu, kas ir piemērots gaistošo slāpekļa bāžu domājamajam saturam (parasti der 100 ml). Atšķaida līdz 200 ml un pievieno 2 g magnija oksīda (3.2.) un dažus pilienus pretputošanas emulsijas (3.3.). Šķīduma reakcijai, pārbaudot ar lakmusa papīru, jābūt sārmainai; pretējā gadījumā pievieno vēl magnija oksīdu (3.2.). Rīkojas, kā noteikts analīzes metodes jēlproteīna satura noteikšanai 5.2. un 5.3. punktā (šā pielikuma C daļa).
Veic tukšo analīzi, izmantojot to pašu procedūru, bet bez analizējamā parauga.
6. Rezultātu aprēķināšana
1 ml H2SO40,05 mol/l atbilst 1,7 miligramiem amonjaka.
Rezultātu izsaka procentos no parauga.
Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nepārsniedz 10 % amonjaka relatīvās vērtības.
F. AMINOSKĀBJU (IZŅEMOT TRIPTOFĀNU) NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt brīvās (sintētiskās un dabīgās) un kopējās (ar peptīdsaitēm saistītās un brīvās) aminoskābes barībā, izmantojot aminoskābju analizētāju. To izmanto, lai noteiktu šādas aminoskābes: cist(e)īns, metionīns, lizīns, treonīns, alanīns, arginīns, asparagīnskābe, glutamīnskābe, glicīns, histidīns, izoleicīns, leicīns, fenilalanīns, prolīns, serīns, tirozīns un valīns.
Pēc šīs metodes neatšķir aminoskābju sāļus un nediferencē D un L formas aminoskābes. To nevar izmantot, lai noteiktu triptofāna un aminoskābju hidroksianalogus.
2. Princips
2.1. Brīvās aminoskābes
Brīvās aminoskābes ekstrahē ar atšķaidītu sālsskābi. Līdzekstrahētās slāpekļa makromolekulas izgulsnē ar sulfosalicilskābi un atdala filtrējot. Filtrēto šķīdumu koriģē līdz pH 2,20. Aminoskābes sadala ar jonu apmaiņas hromatogrāfiju un pēc reakcijas ar ninhidrīnu nosaka fotometriski pie 570 nm.
2.2. Kopējās aminoskābes
Procedūru izvēlas atkarībā no nosakāmajām aminoskābēm. Pirms hidrolīzes cist(e)īns un metionīns jāoksidē attiecīgi līdz cisteīnskābei un metionīna sulfonam. Tirozīns jānosaka neoksidētu paraugu hidrolizātos. Visas pārējās 1. punktā minētās aminoskābes var noteikt oksidētā vai neoksidētā paraugā.
Oksidēšanu veic 0 oC temperatūrā ar peroksiskudrskābes/fenola maisījumu. Oksidēšanas reaģenta pārpalikumu sadala ar nātrija disulfītu. Oksidēto vai neoksidēto paraugu 23 stundas hidrolizē ar sālsskābi (3.20.). Hidrolizātu koriģē līdz pH 2,20. Aminoskābes sadala ar jonu apmaiņas hromatogrāfiju un pēc reakcijas ar ninhidrīnu nosaka fotometriski pie 570 nm (440 nm prolīnam).
3. Reaģenti
Jāizmanto ūdens bidestilāts vai līdzvērtīgas kvalitātes ūdens (vadītspēja < 10 μS).
|
3.1. |
Ūdeņraža peroksīds, w (w/w) = 30 %. |
|
3.2. |
Skudrskābe, w (w/w) = 98–100 %. |
|
3.3. |
Fenols. |
|
3.4. |
Nātrija disulfīts. |
|
3.5. |
Nātrija hidroksīds. |
|
3.6. |
5-sulfosalicilskābes dihidrāts. |
|
3.7. |
Sālsskābe, blīvums apmēram 1,18 g/ml. |
|
3.8. |
Trinātrija citrāta dihidrāts. |
|
3.9. |
2,2'-tiodietanols (tiodiglikols). |
|
3.10. |
Nātrija hlorīds. |
|
3.11. |
Ninhidrīns. |
|
3.12. |
Petrolēteris, viršanas diapazons 40–60 oC. |
|
3.13. |
Norleicīns vai cits savienojums, kas piemērots izmantošanai par iekšējo standartu. |
|
3.14. |
Slāpekļa gāze (< 10 ppm skābekļa). |
|
3.15. |
1-oktanols. |
|
3.16. |
Aminoskābes. |
|
3.16.1. |
Standartvielas, kas minētas 1. punktā. Tīri savienojumi, kuri nesatur kristalizācijas ūdeni. Žāvē vakuumā virs P2O5 vai H2SO4 vienu nedēļu pirms izmantošanas. |
|
3.16.2. |
Cisteīnskābe. |
|
3.16.3. |
Metionīnsulfons. |
|
3.17. |
Nātrija hidroksīda šķīdums, c = 7,5 mol/l. Ūdenī atšķaida 300 g NaOH (3.5.) un papildina līdz vienam litram. |
|
3.18. |
Nātrija hidroksīda šķīdums, c = 1 mol/l. Ūdenī atšķaida 40 g NaOH (3.5.) un papildina līdz vienam litram. |
|
3.19. |
Skudrskābes – fenola šķīdums. Samaisa 889 g skudrskābes (3.2.) ar 111 g ūdens un pievieno 4,73 g fenola (3.3.). |
|
3.20. |
Hidrolīzes maisījums, c = 6 mol HCl/l, kas satur 1 g fenola/l. Pievieno 1 g fenola (3.3.) uz 492 ml HCl (3.7.) un papildina līdz vienam litram ar ūdeni. |
|
3.21. |
Ekstrakcijas maisījums, c = 0,1 mol HCl/l, kas satur 2 % tiodiglikola. 8,2 ml HCl (3.7.) atšķaida apmēram ar 900 ml ūdens, pievieno 20 ml tiodiglikola (3.9.) un papildina līdz vienam litram ar ūdeni (3.7. un 3.9. punktā minētās vielas tieši nejaukt). |
|
3.22. |
5-sulfosalicilskābe, ß = 6 %. Izšķīdina 60 g 5-sulfosalicilskābes (3.6.) ūdenī un papildina līdz vienam litram ar ūdeni. |
|
3.23. |
Oksidēšanas maisījums (peroksiskudrskābe – fenols). Nelielā vārglāzē samaisa 0,5 ml ūdeņraža peroksīda (3.1.) ar 4,5 ml skudrskābes un fenola šķīduma (3.19.). Iztur 20–30 oC temperatūrā vienu stundu, lai izveidojas peroksiskudrskābe, tad atdzesē ledusvannā (15 min), pirms pievieno paraugam. Uzmanību: izvairīties no saskares ar ādu un valkāt aizsargapģērbu! |
|
3.24. |
Citrāta buferšķīdums, c = 0,2 mol Na+/l, pH 2,20 Izšķīdina 19,61 g nātrija citrāta (3.8.), 5 ml tiodiglikola (3.9.), 1 g fenola (3.3.) un 16,50 ml HCl (3.7.) apmēram 800 mililitros ūdens. Koriģē pH līmeni līdz 2,20. Papildina ar ūdeni līdz vienam litram. |
|
3.25. |
Eluācijas buferšķīdumi, sagatavoti atbilstoši izmantotā analizētāja (4.9.) apstākļiem. |
|
3.26. |
Ninhidrīna reaģents, sagatavots atbilstoši izmantotā analizētāja apstākļiem (4.9.). |
|
3.27. |
Aminoskābju standarta šķīdumi. Minētos šķīdumus uzglabā temperatūrā zem 5 oC. |
|
3.27.1. |
Aminoskābju izejas standartšķīdums (3.16.1.). c = 2,5 μmol/ml no katra sālsskābē. Var iegūt komerciāli. |
|
3.27.2. |
Cisteīnskābes un metionīna sulfona izejas standartšķīdums, c = 1,25 μmol/ml. Izšķīdina 0,2115 g cisteīnskābes (3.16.2.) un 0,2265 g metionīna sulfona (3.16.3.) citrāta buferšķīdumā (3.24.) viena litra mērkolbā un papildina līdz zīmei ar citrāta buferšķīdumu. Uzglabā temperatūrā, kas zemāka par 5 oC, ne ilgāk kā 12 mēnešus. Minēto šķīdumu neizmanto, ja izejas standartšķīdums (3.27.1.) satur cisteīnskābi un metionīna sulfonu. |
|
3.27.3. |
Iekšējā standarta izejas standartšķīdums, piemēram, norleicīns, c = 20 μmol/ml. Izšķīdina 0,6560 g norleicīna (3.13.) citrāta buferšķīdumā (3.24.) mērkolbā un papildina līdz 250 ml ar citrāta buferšķīdumu. Uzglabā temperatūrā, kas zemāka par 5 oC, ne ilgāk kā sešus mēnešus. |
|
3.27.4. |
Standarta aminoskābju kalibrēšanas šķīdums izmantošanai ar hidrolizātiem, c = 5 nmol/50 μl cisteīnskābes un metionīnsulfona un c = 10 nmol/50 μl pārējo aminoskābju. Izšķīdina 2,2 g nātrija hlorīda (3.10.) 100 ml tilpuma vārglāzē ar 30 ml citrāta buferšķīduma (3.24.). Pievieno 4,00 ml aminoskābju izejas standartšķīduma (3.27.1.), 4,00 ml cisteīnskābes un metionīnsulfona izejas standartšķīduma (3.27.2.) un 0,50 ml iekšējā standarta izejas standartšķīduma (3.27.3.), ja to izmanto. Koriģē pH līmeni līdz 2,20, izmantojot nātrija hidroksīdu (3,18.). Kvantitatīvi pārnes 50 ml mērkolbā un papildina līdz zīmei ar citrāta buferšķīdumu (3.24.), un samaisa. Uzglabā temperatūrā, kas zemāka par 5 oC, ne ilgāk kā trīs mēnešus. Skatīt arī 9.1. novērojumu. |
|
3.27.5. |
Standarta aminoskābju kalibrēšanas šķīdums izmantošanai ar hidrolizātiem, kas sagatavoti atbilstoši 5.3.3.1. punktam, un izmantošanai ar ekstraktiem (5.2.). Kalibrēšanas šķīdumu sagatavo saskaņā ar 3.27.4. punktu, tikai nepievieno nātrija hlorīdu. Uzglabā temperatūrā, kas zemāka par 5 oC, ne ilgāk kā trīs mēnešus. |
4. Ierīces
|
4.1. |
100 vai 250 ml apaļkolba ar atteces kondensatoru. |
|
4.2. |
100 ml borsilikāta stikla pudele ar skrūvējamu korķi ar gumijas/teflona drīvējumu (piemēram, Duran, Schott) izmantošanai krāsnī. |
|
4.3. |
Krāsns ar mehānisko ventilāciju un temperatūras regulatoru, kura precizitāte ir labāka par ± 2 oC. |
|
4.4. |
pH mērītājs (trīs skaitļi aiz komata). |
|
4.5. |
Membrānfiltrs (0,22 μm). |
|
4.6. |
Centrifūga. |
|
4.7. |
Rotācijas vakuumiztvaicētājs. |
|
4.8. |
Mehāniskais kratītājs vai magnētiskais maisītājs. |
|
4.9. |
Aminoskābju analizētājs vai augstas izšķirtspējas šķidruma hromatogrāfijas iekārta ar jonu apmaiņas kolonnu, ierīce ninhidrīnam, pēckolonnas derivatizācija un fotometrs. Kolonnu piepilda ar sulfonētiem polistirola sveķiem, kas spēj atdalīt aminoskābes citu no citas un no pārējiem ninhidrīnpozitīviem materiāliem. Plūsmu bufera un ninhidrīna līnijā nodrošina sūkņi, kuru plūsmas stabilitāte ir ±0,5 % periodā, kas ietver gan standarta kalibrēšanas norisi, gan parauga analīzi. Ar dažiem aminoskābju analizētājiem var izmantot hidrolīzes procedūras, kurās hidrolizātā nātrija koncentrācija ir c = 0,8 mol/l un hidrolizāts satur visu atlikušo skudrskābi no oksidācijas posma. Pārējās pietiekami labi neatdala dažas aminoskābes, ja hidrolizāts satur skudrskābes pārpalikumu un/vai augstu nātrija jonu koncentrāciju. Tādā gadījumā skābes tilpumu samazina, pēc hidrolīzes un pirms pH līmeņa korekcijas iztvaicējot apmēram līdz 5 ml. Iztvaicēšanu veic vakuumā augstākais 40 oC temperatūrā. |
5. Procedūra
5.1. Parauga sagatavošana
Paraugu samaļ, lai tas izsijātos caur sietu ar 0,5 mm acojumu. Paraugi, kuros ir liels mitruma saturs, pirms malšanas vai nu jāizkaltē temperatūrā, kas nepārsniedz 50 oC, vai jāizžāvē ar izsaldēšanu. Paraugus, kuros ir liels tauku saturs, pirms malšanas ekstrahē ar petrolēteri (3.12.).
5.2. Brīvo aminoskābju noteikšana barībā un premiksos
Attiecīgu apjomu (1–5 g) sagatavotā parauga (5.1.) ar precizitāti līdz 0,2 mg iesver koniskā kolbā un pievieno 100,0 ml ekstrakcijas maisījuma (3.21.). Maisījumu 60 minūtes krata, izmantojot mehānisko kratītāju vai magnētisko maisītāju (4.8.). Ļauj nogulsnēm nogulsnēties un 10,0 ml šķīduma augšējā slāņa ar pipeti iepilina vārglāzē.
Pievieno 5,0 ml sulfosalicilskābes šķīduma (3.22.), vienlaikus maisot, un turpina maisīt ar magnētisko maisītāju piecas minūtes. Šķīduma augšējo slāni filtrē vai centrifugē, lai atdalītu visas nogulsnes. Ievieto 10,0 ml iegūtā šķīduma 100 ml vārglāzē un koriģē pH līmeni līdz 2,20, izmantojot nātrija hidroksīda šķīdumu (3.18.), pārnes uz attiecīga tilpuma mērkolbu, izmantojot citrāta buferšķīdumu (3.24.), un papildina līdz zīmei ar buferšķīdumu (3.24.).
Ja izmanto iekšējo standartu, tad pievieno 1,00 ml iekšējā standarta (3.27.3.) uz katriem 100 ml galīgā šķīduma un papildina līdz zīmei ar buferšķīdumu (3.24.).
Veic nākamo hromatogrāfijas posmu atbilstoši 5.4. punktam.
Ja ekstraktus nepārbauda tajā pašā dienā, tad tie jāuzglabā temperatūrā, kas zemāka par 5 oC.
5.3. Kopējo aminoskābju noteikšana
5.3.1.
No 0,1 līdz 1 g sagatavotā parauga (5.1.) ar precizitāti līdz 0,2 mg iesver:
|
— |
100 ml apaļkolbā (4.1.) vaļējai hidrolīzei (5.3.2.3.) vai |
|
— |
250 ml apaļkolbā (4.1.), ja ir prasīta zema nātrija koncentrācija (5.3.3.1.), vai |
|
— |
100 ml pudelē ar skrūvējamu korķi (4.2.) slēgtai hidrolīzei (5.3.2.4.). |
Nosvērtajā apjomā, no kura ņem paraugu, slāpekļa saturam jābūt apmēram 10 mg un mitruma saturam – ne vairāk par 100 mg.
Kolbu/pudeli ievieto ledusvannā un atdzesē līdz 0 oC, pievieno 5 ml oksidēšanas maisījuma (3.23.) un samaisa, izmantojot stikla lāpstiņu ar liektu galu. Kolbu/pudeli, kurā ir lāpstiņa, cieši noslēdz ar hermētisku plēvi, ievieto ledusvannu ar noslēgto trauku ledusskapī 0 oC temperatūrā un atstāj uz 16 stundām. Pēc 16 stundām izņem no ledusskapja un sadala oksidēšanas reaģenta pārpalikumu, pievienojot 0,84 g nātrija disulfīta (3.4.).
Rīkojas, kā noteikts 5.3.2.1. punktā.
5.3.2.
5.3.2.1.
Oksidētajam paraugam, kas sagatavots atbilstoši 5.3.1. punktam, pievieno 25 ml hidrolīzes maisījuma (3.20.), raugoties, lai tiktu noskalotas visas parauga atliekas, kas pielipušas trauka malām un lāpstiņai.
Atkarībā no izmantotās hidrolīzes procedūras rīkojas, kā noteikts 5.3.2.3. vai 5.3.2.4. punktā.
5.3.2.2.
100 ml vai 250 ml apaļkolbā (4.1.) vai 100 ml pudelē, kas aprīkota ar skrūvējamu vāciņu (4.2.), ar precizitāti līdz 0,2 mg iesver no 0,1 līdz 1 g sagatavotā parauga (5.1.). Slāpekļa saturam iesvērtajā analizējamā paraugā jābūt apmēram 10 mg. Uzmanīgi pievieno 25 ml hidrolīzes maisījuma (3.20.) un samaisa ar paraugu. Rīkojas, kā noteikts 5.3.2.3. vai 5.3.2.4. punktā.
5.3.2.3.
Maisījumam kolbā (kas sagatavots atbilstoši 5.3.2.1. vai 5.3.2.2. punktam) pievieno trīs stikla lodītes un vāra, maisījumam attecē nepārtraukti burbuļojot 23 stundas. Pēc hidrolīzes pabeigšanas kondensatoru noskalo ar 5 ml citrāta buferšķīduma (3.24.). Kolbu atvieno un atdzesē ledusvannā.
Rīkojas, kā noteikts 5.3.3. punktā.
5.3.2.4. S
Pudeli ar maisījumu, kas sagatavots saskaņā ar 5.3.2.1. vai 5.3.2.2. punktu, ievieto krāsnī (4.3.) 110 oC temperatūrā. Pirmajā stundā, lai novērstu spiediena uzkrāšanos (kas notiek gāzveida vielu veidošanās dēļ) un lai novērstu sprādzienu, uz trauka augšas uzliek skrūvējamu vāciņu. Trauku neaizver ar vāciņu. Pēc vienas stundas trauku aizver ar vāciņu un atstāj krāsnī (4.3.) 23 stundas. Pēc hidrolīzes pabeigšanas pudeli izņem no krāsns, uzmanīgi atver pudeles vāciņu un pudeli ieliek ledusvannā. Atstāj atdzist.
Atkarībā no procedūras, kādu izmanto pH līmeņa korekcijai (5.3.3.), kvantitatīvi pārnes pudeles saturu 250 ml vārglāzē vai 250 ml apaļkolbā, izmantojot citrāta buferšķīdumu (3.24.).
Rīkojas, kā noteikts 5.3.3. punktā.
5.3.3.
Lai koriģētu pH līmeni, atkarībā no aminoskābes analizētāja (4.9.) nātrija tolerances rīkojas atbilstoši 5.3.3.1. vai 5.3.3.2. punktam.
5.3.3.1.
Ja izmanto aminoskābju analizētājus, kuriem nepieciešama zema nātrija koncentrācija (kad skābes tilpums jāsamazina), ieteicams izmantot iekšējo izejas standartšķīdumu (3.27.3.).
Tādā gadījumā hidrolizātam pirms iztvaicēšanas pievieno 2,00 ml iekšējā izejas standartšķīduma (3.27.3.).
Pievieno divus pilienus 1-oktanola (3.15.) hidrolizātam, kas iegūts saskaņā ar 5.3.2.3. vai 5.3.2.4. punktu.
Izmantojot rotācijas iztvaicētāju (4.7.), vakuumā un 40 oC grādu temperatūrā samazina tilpumu līdz 5–10 ml. Ja tilpumu nejauši samazina līdz mazāk nekā 5 ml, tad hidrolizāts jāizmet un analīze jāsāk no jauna.
Ar nātrija hidroksīda šķīdumu (3.18.) koriģē pH līmeni līdz 2,20 un rīkojas, kā noteikts 5.3.4. punktā.
5.3.3.2.
Hidrolizātus, kas iegūti saskaņā ar 5.3.2.3. vai 5.3.2.4. punktu, daļēji neitralizē un, uzmanīgi maisot, pievieno 17 ml nātrija hidroksīda šķīduma (3.17.), un nodrošina, lai temperatūra būtu zem 40 oC.
Koriģē pH līmeni līdz 2,20 istabas temperatūrā, izmantojot nātrija hidroksīda šķīdumu (3.17.) un visbeidzot – nātrija hidroksīda šķīdumu (3.18.). Pāriet pie 5.3.4. punkta.
5.3.4.
Kvantitatīvi pārnes pH koriģēto hidrolizātu (5.3.3.1. vai 5.3.3.2.) ar citrāta buferšķīdumu (3.24.) 200 ml tilpuma mērkolbā un papildina līdz zīmei ar buferšķīdumu (3.24.).
Ja iekšējais standarts nav jau izmantots, tad pievieno 2,00 ml iekšējā standarta (3.27.3.) un papildina līdz zīmei ar citrāta buferšķīdumu (3.24.). Rūpīgi samaisa.
Pāriet pie hromatogrāfijas posma (5.4.).
Ja parauga šķīdumus nepārbauda tajā pašā dienā, tad tie jāuzglabā temperatūrā, kas zemāka par 5 oC.
5.4. Hromatogrāfija
Pirms hromatogrāfijas ekstraktu (5.2.) vai hidrolizātu (5.3.4.) sasilda līdz istabas temperatūrai. Maisījumu sakrata un piemērotu daudzumu izfiltrē caur 0,22 μm membrānfiltru (4.5.). Iegūto dzidro šķīdumu pakļauj jonu apmaiņas hromatogrāfijai, izmantojot aminoskābju analizētāju (4.9.).
Injekciju var veikt manuāli vai automātiski. Ir svarīgi, lai kolonnā standartu un paraugu analīzei tiktu pievienots vienāds šķīduma daudzums ±0,5 %, izņemot gadījumus, kad izmanto iekšējo standartu, un lai nātrija/aminoskābes proporcijas standarta un parauga šķīdumos būtu pēc iespējas līdzīgākas.
Parasti kalibrēšanas posmu biežums ir atkarīgs no ninhidrīna reaģenta stabilitātes un analīžu sistēmas. Standartu vai paraugu atšķaida ar citrāta buferšķīdumu (3.24.), lai uzrādītu standarta maksimālo laukumu 30–200 % no parauga aminoskābes maksimālā laukuma.
Aminoskābju hromatogrāfija nedaudz atšķirsies atkarībā no izmantotā analizētāja tipa un sveķiem. Izvēlētajai sistēmai jāspēj atdalīt aminoskābes citu no citas un no ninhidrīnpozitīviem materiāliem. Darbības diapazonā hromatogrāfiskajai sistēmai jāsniedz lineāra atbilde uz pārmaiņām to aminoskābju daudzumos, ko pievieno kolonnā.
Kad analizē (nosakāmo aminoskābju) ekvimolāru šķīdumu, tad hromatogrāfijas posma laikā piemēro tālāk minētās attiecības, kas ir starp maksimālo augstumu un atstarpi starp signāliem. Tādam ekvimolāram šķīdumam jāsatur vismaz 30 % no katras aminoskābes maksimālās slodzes, ko var precīzi izmērīt, izmantojot aminoskābju analizētāja sistēmu (4.9.).
Lai atdalītu treonīnu un serīnu, attiecība starp maksimālo augstumu un atstarpi starp signāliem zemākajā no divām aminoskābēm, kuras pārklājas hromatogrammā, nedrīkst pārsniegt 2:10 (ja nosaka tikai cist(e)īnu, metionīnu, treonīnu un lizīnu, tad nepietiekama atdalīšana no blakusesošajiem maksimālajiem augstumiem negatīvi ietekmēs noteikšanu). Visām pārējām aminoskābēm atdalījumam jābūt precīzākam par 1:10.
Sistēmai jānodrošina, lai lizīns tiktu atdalīts no “lizīna artefaktiem” un ornitīna.
6. Rezultātu aprēķināšana
Parauga un standarta maksimumu laukumu mēra katrai atsevišķai aminoskābei un aprēķina daudzumu (X) gramos, cik aminoskābes ir kilogramā parauga.
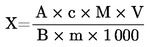
Ja izmanto iekšējo standartu, pareizina ar: 
|
A |
= |
maksimālais laukums, hidrolizāts vai ekstrakts, |
|
B |
= |
maksimālais laukums, kalibrēšanas standartšķīdums, |
|
C |
= |
maksimālais laukums, iekšējais standarts hidrolizātā vai ekstraktā, |
|
D |
= |
maksimālais laukums, iekšējais standarts, kalibrēšanas standartšķīdums, |
|
M |
= |
nosakāmās aminoskābes molmasa, |
|
c |
= |
standarta koncentrācija μmol/ml, |
|
m |
= |
parauga svars (g) (koriģēts līdz sākotnējam svaram, ja kaltēts vai attaukots), |
|
V |
= |
ml kopējā hidrolizāta (5.3.4.) vai ml ekstrakta aprēķinātā kopējā atšķaidījuma tilpuma (6.1.). |
Cistīnu un cisteīnu nosaka kā cisteīnskābi oksidētā parauga hidrolizātos, bet aprēķina kā cistīnu (C6H12N2O4S2, M 240,30 g/mol), izmantojot M 120,15 g/mol (= 0,5 × 240,30 g/mol).
Metionīnu nosaka kā metionīna sulfonu oksidētā parauga hidrolizātos, bet aprēķina kā metionīnu, izmantojot metionīna M 149,21 g/mol.
Pievienoto brīvo metionīnu nosaka pēc tam, kad tas ekstrahēts kā metionīns, aprēķinam izmanto to pašu M.
|
6.1. |
Ekstraktu kopējo atšķaidījuma tilpumu (F), lai noteiktu brīvās aminoskābes (5.2.), aprēķina šādi: 
|
7. Metodes novērtējums
Minēto metodi pārbaudīja starptautiskā līmenī 1990. gadā, salīdzinot rezultātus starp laboratorijām, izmantojot četras dažādas barības (barības maisījumu cūkām, barības maisījumu broileriem, proteīnu koncentrātu un premiksu). Vidējo un standarta noviržu rezultāti, kas iegūti pēc galējo punktu likvidēšanas, ir norādīti tabulās šajā punktā.
Vidējie lielumi g/kg
|
Standartmateriāls |
Aminoskābe |
||||||
|
Treonīns |
Cist(e)īns |
Metionīns |
Lizīns |
||||
|
Barības maisījums cūkām |
6,94 n = 15 |
3,01 n = 17 |
3,27 n = 17 |
9,55 n = 13 |
|||
|
Barības maisījums broileriem |
9,31 n = 16 |
3,92 n = 18 |
5,08 n = 18 |
13,93 n = 16 |
|||
|
Proteīnu koncentrāts |
22,32 n = 16 |
5,06 n = 17 |
12,01 n = 17 |
47,74 n = 15 |
|||
|
Premikss |
58,42 n = 16 |
— |
90,21 n = 16 |
98,03 n = 16 |
|||
|
|||||||
7.1. Atkārtojamība
Iepriekš minētā savstarpējā salīdzinājuma atkārtojamība, kas izteikta kā “laboratorijas standartnovirze”, ir norādīta tālāk tabulā.
Laboratorijas standartnovirze (Sr) g/kg
|
Standartmateriāls |
Aminoskābe |
||||||
|
Treonīns |
Cist(e)īns |
Metionīns |
Lizīns |
||||
|
Barības maisījums cūkām |
0,13 n = 15 |
0,10 n = 17 |
0,11 n = 17 |
0,26 n = 13 |
|||
|
Barības maisījums broileriem |
0,20 n = 16 |
0,11 n = 18 |
0,16 n = 18 |
0,28 n = 16 |
|||
|
Proteīnu koncentrāts |
0,48 n = 16 |
0,13 n = 17 |
0,27 n = 17 |
0,99 n = 15 |
|||
|
Premikss |
1,30 n = 16 |
— |
2,19 n = 16 |
2,06 n = 16 |
|||
|
|||||||
Variācijas koeficients (%) laboratorijas standartnovirzei (Sr)
|
Standartmateriāls |
Aminoskābe |
||||||
|
Treonīns |
Cist(e)īns |
Metionīns |
Lizīns |
||||
|
Barības maisījums cūkām |
1,9 n = 15 |
3,3 n = 17 |
3,4 n = 17 |
2,8 n = 13 |
|||
|
Barības maisījums broileriem |
2,1 n = 16 |
2,8 n = 18 |
3,1 n = 18 |
2,1 n = 16 |
|||
|
Proteīnu koncentrāts |
2,7 n = 16 |
2,6 n = 17 |
2,2 n = 17 |
2,4 n = 15 |
|||
|
Premikss |
2,2 n = 16 |
— |
2,4 n = 16 |
2,1 n = 16 |
|||
|
|||||||
7.2 Atveidojamība
Starplaboratoriju standartnoviržu rezultāti, kas iegūti iepriekš minētajā savstarpējā salīdzinājumā, ir norādīti tālāk tabulā.
Starplaboratoriju standartnovirze (SR) g/kg
|
Standartmateriāls |
Aminoskābe |
||||||
|
Treonīns |
Cist(e)īns |
Metionīns |
Lizīns |
||||
|
Barības maisījums cūkām |
0,28 n = 15 |
0,30 n = 17 |
0,23 n = 17 |
0,30 n = 13 |
|||
|
Barības maisījums broileriem |
0,48 n = 16 |
0,34 n = 18 |
0,55 n = 18 |
0,75 n = 16 |
|||
|
Proteīnu koncentrāts |
0,85 n = 16 |
0,62 n = 17 |
1,57 n = 17 |
1,24 n = 15 |
|||
|
Premikss |
2,49 n = 16 |
— |
6,20 n = 16 |
6,62 n = 16 |
|||
|
|||||||
Variācijas koeficients (%) starplaboratoriju standartnovirzei (SR)
|
Standartmateriāls |
Aminoskābe |
||||||
|
Treonīns |
Cist(e)īns |
Metionīns |
Lizīns |
||||
|
Barības maisījums cūkām |
4,1 n = 15 |
9,9 n = 17 |
7,0 n = 17 |
3,2 n = 13 |
|||
|
Barības maisījums broileriem |
5,2 n = 16 |
8,8 n = 18 |
10,9 n = 18 |
5,4 n = 16 |
|||
|
Proteīnu koncentrāts |
3,8 n = 16 |
12,3 n = 17 |
13,0 n = 17 |
3,0 n = 15 |
|||
|
Premikss |
4,3 n = 16 |
— |
6,9 n = 16 |
6,7 n = 16 |
|||
|
|||||||
8. Standartmateriālu izmantošana
Metodes pareizu piemērošanu verificē, atkārtoti mērot sertificētus standartmateriālus, kad tie ir pieejami. Ir ieteicama kalibrēšana ar sertificētu aminoskābes kalibrēšanas šķīdumu.
9. Novērojumi
|
9.1. |
Tā kā starp aminoskābju analizētājiem ir atšķirības, tad par pamatnostādni ņem standarta aminoskābju (skatīt 3.27.4. un 3.27.5. punktu) un hidrolizāta (skatīt 5.3.4. punktu) kalibrēšanas šķīdumu galīgās koncentrācijas. Aparāta lineārās reakcijas diapazons jāpārbauda attiecībā uz visām aminoskābēm. Standartšķīdumu atšķaida ar citrāta buferšķīdumu, lai uzrādītu maksimālos laukumus diapazona vidū. |
|
9.2. |
Ja izmanto augstas izšķirtspējas šķidruma hromatogrāfijas aprīkojumu, lai analizētu hidrolizātus, tad eksperimenta apstākļi jāoptimizē saskaņā ar ražotāja ieteikumiem. |
|
9.3. |
Saskaņā ar šo metodi analizējot barību, kas satur vairāk par 1 % hlorīdu (barības koncentrātus, minerālbarību, papildbarību), var iegūt pazeminātus rezultātus attiecībā uz metionīnu, un tāpēc tā jāanalizē īpaši. |
G. TRIPTOFĀNA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt kopējo un brīvo triptofānu barībā. Tajā neizšķir D un L formu.
2. Princips
Lai noteiktu kopējo triptofānu, paraugu hidrolizē sārmainos apstākļos ar piesātinātu bārija hidroksīda šķīdumu un 20 stundas karsē līdz 110 oC temperatūrai. Pēc hidrolīzes pievieno iekšējo standartu.
Lai noteiktu brīvo triptofānu, paraugu ekstrahē nedaudz skābā vidē iekšējā standarta klātbūtnē.
Triptofānu un iekšējo standartu hidrolizātā vai ekstraktā nosaka, izmantojot augstas izšķirtspējas šķidruma hromatogrāfiju (HPLC) ar fluorescences noteikšanu.
3. Reaģenti
|
3.1. |
Jāizmanto ūdens bidestilāts vai līdzvērtīgas kvalitātes ūdens (vadītspēja < 10 μS/cm). |
|
3.2. |
Standartviela: triptofāns (tīrība/saturs ≥ 99 %), kas izžāvēts vakuumā virs fosfora pentoksīda. |
|
3.3. |
Iekšējā standartviela: α-metil-triptofāns (tīrība/saturs ≥ 99 %), kas izžāvēts vakuumā virs fosfora pentoksīda. |
|
3.4. |
Bārija hidroksīda oktahidrāts (jārūpējas, lai Ba(OH)2 ·8 H2O pārmērīgi nepakļautu gaisa iedarbībai, lai izvairītos no BaCO3 veidošanās, kas varētu traucēt noteikšanu) (skatīt 9.3. novērojumu). |
|
3.5. |
Nātrija hidroksīds. |
|
3.6. |
Ortofosforskābe, w (w/w) = 85 %. |
|
3.7. |
Sālsskābe, ρ201,19 g/ml. |
|
3.8. |
Metanols, ekvivalents HPLC pakāpei. |
|
3.9. |
Petrolēteris, viršanas diapazons 40-60 oC. |
|
3.10. |
Nātrija hidroksīda šķīdums, c = 1 mol/l. Ūdenī atšķaida 40,0 g NaOH (3.5.) un papildina līdz vienam litram ar ūdeni (3.1.). |
|
3.11. |
Sālsskābe, c = 6 mol/l. 492 ml HCl (3.7.) papildina līdz vienam litram ar ūdeni. |
|
3.12. |
Sālsskābe, c = 1 mol/l. 82 ml HCl (3.7.) papildina līdz vienam litram ar ūdeni. |
|
3.13. |
Sālsskābe, c = 0,1 mol/l. 8,2 ml HCl (3.7.) papildina līdz vienam litram ar ūdeni. |
|
3.14. |
Ortofosforskābe, c = 0,5 mol/l. Paņem 34 ml ortofosforskābes (3.6.) un papildina līdz vienam litram ar ūdeni (3.1.). |
|
3.15. |
Koncentrēts triptofāna šķīdums (3.2.), c = 2,50 μmol/ml. 500 ml tilpuma mērkolbā izšķīdina 0,2553 g triptofāna (3.2.) sālsskābē (3.13.) un papildina līdz zīmei ar sālsskābi (3.13.). Uzglabā –18 oC temperatūrā ne ilgāk kā četras nedēļas. |
|
3.16. |
Koncentrēts iekšējais standartšķīdums, c = 2,50 μmol/ml. 500 ml tilpuma mērkolbā sālsskābē (3.13.) izšķīdina 0,2728 g α-metil-triptofāna (3.3.) un papildina līdz zīmei ar sālsskābi (3.13.). Uzglabā –18 oC temperatūrā ne ilgāk kā četras nedēļas. |
|
3.17. |
Triptofāna kalibrēšanas standartšķīdums un iekšējais standarts. Ņem 2,00 ml koncentrēta triptofāna (3.15.) šķīduma un 2,00 ml koncentrēta iekšējā standarta (α-metil-triptofāna) šķīduma (3.16.). Atšķaida ar ūdeni (3.1.) un metanolu (3.8.) aptuveni līdz tādam pašam tilpumam un aptuveni tādai pašai metanola koncentrācijai (10–30 %) kā gatavajam hidrolizātam. Minētais šķīdums jāsagatavo svaigs pirms izmantošanas. Sagatavošanas laikā jāuzmana, lai nepiekļūst tieša saules gaisma. |
|
3.18. |
Etiķskābe. |
|
3.19. |
1,1,1-trihlor-2-metil-2-propanols. |
|
3.20. |
Etanolamīns w (w/w) > 98 %. |
|
3.21. |
1 g 1,1,1-trihlor-2-metil-2-propanola (3.19.) šķīdums 100 mililitros metanola (3.8.). |
|
3.22. |
Kustīgā fāze priekš HPLC: 3,00 g etiķskābes (3.18.) + 900 ml ūdens (3.1.) +50,0 ml 1,1,1-trihlor-2-metil-2-propanola (3.19.) šķīduma (3.21.) metanolā (3.8.) (1 g/100 ml). Izmantojot etanolamīnu (3.20.), pH līmeni koriģē līdz 5,00. Papildina ar ūdeni līdz 1 000 ml (3.1.). |
4. Ierīces
|
4.1. |
HPLC iekārta ar fluorescences detektoru. |
|
4.2. |
Šķidrumu hromatogrāfa kolonna, 125 × 4 mm, C18, 3 μm iesaiņojums, vai līdzvērtīga. |
|
4.3. |
pH mērītājs. |
|
4.4. |
Polipropilēna kolba, 125 ml tilpums, ar platu kaklu un skrūvējamu korķi. |
|
4.5. |
Membrānfiltrs, 0,45 μm. |
|
4.6. |
Autoklāvs, 110 (± 2) oC, 1,4 (±0,1) bāri. |
|
4.7. |
Mehāniskais kratītājs vai magnētiskais maisītājs. |
|
4.8. |
Virpuļmikseris. |
5. Procedūra
5.1. Paraugu sagatavošana
Paraugu samaļ, lai tas izsijātos caur sietu ar 0,5 mm acojumu. Paraugi, kuros ir liels mitruma saturs, pirms malšanas vai nu jāizkaltē temperatūrā, kas nepārsniedz 50 oC, vai jāizžāvē ar izsaldēšanu. Paraugus, kuros ir liels tauku saturs, pirms malšanas ekstrahē ar petrolēteri (3.9.).
5.2. Brīvā triptofāna noteikšana (ekstrakts)
Attiecīgu apjomu (1–5 g) sagatavotā parauga (5.1.) ar precizitāti līdz 1 mg iesver koniskā kolbā. Pievieno 100,0 ml sālsskābes (3.13.) un 5,00 ml koncentrēta iekšējā standartšķīduma (3.16.). Krata vai maisa 60 minūtes, izmantojot mehānisko kratītāju vai magnētisko maisītāju (4.7.). Ļauj nogulsnēm nogulsnēties un 10,0 ml šķīduma augšējā slāņa ar pipeti iepilina vārglāzē. Pievieno 5 ml ortofosforskābes (3.14.). Koriģē pH līmeni līdz 3, izmantojot nātrija hidroksīdu (3.10.). Pievieno pietiekami daudz metanola (3.8.), lai galīgajā tilpumā metanola koncentrācija būtu no 10 līdz 30 %. Pārnes attiecīga tilpuma kolbā un atšķaida ar ūdeni līdz tilpumam, kas vajadzīgs hromatogrāfijai (aptuveni tikpat, cik ir kalibrēšanas standartšķīdums (3.17.)).
Izfiltrē dažus ml šķīduma caur 0,45 μm membrānfiltru (4.5.), pirms šķīdumu iešpricē HPLC kolonnā. Veic nākamo hromatogrāfijas posmu atbilstoši 5.4. punktam.
Standartšķīdums un ekstrakti jāsargā no tiešas saules gaismas. Ja ekstraktus nav iespējams analizēt tajā pašā dienā, tad tos var uzglabāt 5 oC temperatūrā ne ilgāk kā trīs dienas.
5.3. Kopējā triptofāna (hidrolizāta) noteikšana
No 0,1 līdz 1 g sagatavotā parauga (5.1.) ar precizitāti līdz 0,2 mg iesver polipropilēna kolbā (4.4.). Slāpekļa saturs iesvērtajā analizējamā paraugā ir apmēram 10 mg. Pievieno 8,4 g bārija hidroksīda oktahidrāta (3.4.) un 10 ml ūdens. Samaisa virpuļmikserī (4.8.) vai ar magnētisko maisītāju (4.7.). Atstāj maisījumā ar teflonu pārklātu magnētu. Trauka sienas noskalo ar 4 ml ūdens. Uzliek skrūvējamu korķi un vaļīgi aiztaisa kolbu. Pārvieto uz autoklāvu (4.6.) ar vārošu ūdeni un tvaicē 30–60 minūtes. Aizver autoklāvu un tur karsē 110 (± 2) oC temperatūrā 20 stundas.
Pirms autoklāva atvēršanas temperatūru samazina, lai tā būtu nedaudz zem 100 oC. Lai novērstu Ba(OH)2 ·8 H2O kristalizāciju, siltajam maisījumam pievieno 30 ml istabas temperatūras ūdens. Viegli sakrata vai samaisa. Pievieno 2,00 ml koncentrēta iekšējā standarta (α-metil-triptofāna) šķīduma (3.16.). Atdzesē traukus ūdenī/ledusvannā 15 minūtes.
Tad pievieno 5 ml ortofosforskābes (3.14.). Trauku tur dzesēšanas vannā un neitralizē ar HCl (3.11.), vienlaikus maisot, un pH līmeni koriģē līdz 3,0, izmantojot HCl (3.12.). Pievieno pietiekami daudz metanola, lai galīgajā tilpumā metanola koncentrācija būtu no 10 līdz 30 %. Pārnes atbilstoša tilpuma kolbā un atšķaida ar ūdeni līdz tilpumam, kas noteikts hromatogrāfijas vajadzībām (piemēram, 100 ml). Pievienojot metanolu, neveidojas nogulsnes.
Izfiltrē dažus ml šķīduma caur 0,45 μm membrānfiltru (4.5.), pirms šķīdumu iešpricē HPLC kolonnā. Veic nākamo hromatogrāfijas posmu atbilstoši 5.4. punktam.
Standartšķīdums un hidrolizāti jāsargā no tiešas saules gaismas. Ja hidrolizātus nav iespējams izanalizēt tajā pašā dienā, tad tos var uzglabāt 5 oC temperatūrā ne ilgāk kā trīs dienas.
5.4. HPLC noteikšana
Orientējoši izokrātiskajai eluācijai piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus (skatīt arī 9.1. un 9.2. novērojumu).
|
HPLC kolonna (4.2.): |
125 × 4 mm, C18, 3 μm iesaiņojums, vai līdzvērtīga |
|
Kolonnas temperatūra: |
istabas temperatūra |
|
Kustīgā fāze (3.22.): |
3,00 g etiķskābes (3.18.) + 900 ml ūdens (3.1.) +50,0 ml 1,1,1-trihlor-2-metil-2-propanola (3.19.) šķīdums (3.21.) metanolā (3.8.) 1 g/100 ml). pH līmeni koriģē līdz 5,00, izmantojot etanolamīnu (3.20.). Papildina ar ūdeni līdz 1 000 ml (3.1.) |
|
Plūsmas ātrums: |
1 ml/min |
|
Kopējais norises laiks: |
apmēram 34 min |
|
Noteikšanas viļņa garums: |
ierosa 280 nm, emisija 356 nm |
|
Injekcijas tilpums: |
20 μl |
6. Rezultātu aprēķināšana
Aprēķina triptofāna (X) daudzumu gramos uz 100 g parauga.

|
A |
= |
iekšējā standarta maksimālais laukums, kalibrēšanas standartšķīdums (3.17.), |
|
B |
= |
triptofāna maksimālais laukums, ekstrakts (5.2.) vai hidrolizāts (5.3.), |
|
V1 |
= |
koncentrētā triptofāna šķīduma (3.15.) tilpums mililitros (2 ml), ko pievieno kalibrēšanas šķīdumam (3.17.), |
|
c |
= |
koncentrētā triptofāna šķīduma (3.15.) koncentrācija μmol/ml (= 2,50), ko pievieno kalibrēšanas šķīdumam (3.17.), |
|
V2 |
= |
koncentrētā iekšējā standartšķīduma (3.16.) tilpums mililitros, ko pievieno ekstrahēšanas laikā (5.2.) (= 5,00 ml) vai hidrolizātam (5.3.) (=2,00 ml), |
|
C |
= |
iekšējā standarta maksimālais laukums, ekstrakts (5.2.) vai hidrolizāts (5.3.), |
|
D |
= |
triptofāna maksimālais laukums, kalibrēšanas standartšķīdums (3.17.), |
|
V3 |
= |
koncentrētā iekšējā standartšķīduma (3.16.) tilpums mililitros (= 2,00 ml), ko pievieno kalibrēšanas standartšķīdumam (3.17.), |
|
m |
= |
parauga svars gramos (koriģēts līdz sākotnējam svaram, ja kaltēts un/vai attaukots), |
|
M |
= |
triptofāna molmasa (= 204,23 g/mol). |
7. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 10 % attiecībā pret lielāko rezultātu.
8. Koppētījuma rezultāti
Tika noorganizēts EK koppētījums (4. salīdzinājums starp laboratorijām), kurā līdz 12 laboratorijām analizēja trīs paraugus, lai sertificētu hidrolīzes metodi. Ar katru paraugu tika veiktas atkārtotas (5) analīzes. Rezultāti ir norādīti šajā tabulā.
|
|
1. paraugs Barība cūkām |
2. paraugs Barība cūkām, papildināta ar L-triptofānu |
3. paraugs Barības koncentrāts cūkām |
|
L |
12 |
12 |
12 |
|
n |
50 |
55 |
50 |
|
Vidējais [g/kg] |
2,42 |
3,40 |
4,22 |
|
sr [g/kg] |
0,05 |
0,05 |
0,08 |
|
r [g/kg] |
0,14 |
0,14 |
0,22 |
|
CVr [ %] |
1,9 |
1,6 |
1,9 |
|
SR [g/kg] |
0,15 |
0,20 |
0,09 |
|
R [g/kg] |
0,42 |
0,56 |
0,25 |
|
CVR [ %] |
6,3 |
6,0 |
2,2 |
|
L |
= |
to laboratoriju skaits, kas iesniedza rezultātus, |
|
n |
= |
atsevišķo rezultātu skaits, kas iegūts pēc galējo punktu likvidēšanas (identificē ar Kohrana, Diksona galējo punktu testu), |
|
sr |
= |
atkārtojamības standartnovirze, |
|
SR |
= |
atveidojamības standartnovirze, |
|
r |
= |
atkārtojamība, |
|
R |
= |
atveidojamība, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
CVR |
= |
atveidojamības variācijas koeficients (%). |
Tika noorganizēts cits EK koppētījums (3. salīdzinājums starp laboratorijām), kurā līdz 13 laboratorijām analizēja divus paraugus, lai sertificētu brīvā triptofāna ekstrahēšanas metodi. Ar katru paraugu tika veiktas atkārtotas (5) analīzes. Rezultāti ir norādīti šajā tabulā.
|
|
4. paraugs Kviešu un sojas maisījums |
5. paraugs Kviešu un sojas maisījums (= 4. paraugs), kam pievienots triptofāns (0,457g/kg) |
|
L |
12 |
12 |
|
n |
55 |
60 |
|
Vidējais [g/kg] |
0,391 |
0,931 |
|
sr [g/kg] |
0,005 |
0,012 |
|
r [g/kg] |
0,014 |
0,034 |
|
CVr [ %] |
1,34 |
1,34 |
|
SR [g/kg] |
0,018 |
0,048 |
|
R [g/kg] |
0,050 |
0,134 |
|
CVR [ %] |
4,71 |
5,11 |
|
L |
= |
to laboratoriju skaits, kas iesniedza rezultātus, |
|
n |
= |
atsevišķo rezultātu skaits, kas iegūts pēc galējo punktu likvidēšanas (identificē ar Kohrana, Diksona galējo punktu testu), |
|
sr |
= |
atkārtojamības standartnovirze, |
|
SR |
= |
atveidojamības standartnovirze, |
|
r |
= |
atkārtojamība, |
|
R |
= |
atveidojamība, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
CVR |
= |
atveidojamības variācijas koeficients (%). |
Tika noorganizēts cits EK salīdzinājuma pētījums starp laboratorijām, kurā līdz septiņām laboratorijām analizēja četrus paraugus, lai sertificētu triptofānu hidrolīzei. Rezultāti ir norādīti tālāk. Ar katru paraugu tika veiktas atkārtotas (5) analīzes.
|
|
1. paraugs Barības maisījums cūkām (CRM 117) |
2. paraugs Zivju milti ar zemu tauku saturu (CRM 118) |
3. paraugs Sojas milti (CRM 119) |
4. paraugs Sausais vājpiens (CRM 120) |
|
L |
7 |
7 |
7 |
7 |
|
n |
25 |
30 |
30 |
30 |
|
Vidējais [g/kg] |
2,064 |
8,801 |
6,882 |
5,236 |
|
sr [g/kg] |
0,021 |
0,101 |
0,089 |
0,040 |
|
r [g/kg] |
0,059 |
0,283 |
0,249 |
0,112 |
|
CVr [ %] |
1,04 |
1,15 |
1,30 |
0,76 |
|
SR [g/kg] |
0,031 |
0,413 |
0,283 |
0,221 |
|
R [g/kg] |
0,087 |
1,156 |
0,792 |
0,619 |
|
CVR [ %] |
1,48 |
4,69 |
4,11 |
4,22 |
|
L |
= |
to laboratoriju skaits, kas iesniedza rezultātus, |
|
n |
= |
atsevišķo rezultātu skaits, kas iegūts pēc galējo punktu likvidēšanas (identificē ar Kohrana, Diksona galējo punktu testu), |
|
sr |
= |
atkārtojamības standartnovirze, |
|
SR |
= |
atveidojamības standartnovirze, |
|
r |
= |
atkārtojamība, |
|
R |
= |
atveidojamība, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
CVR |
= |
atveidojamības variācijas koeficients (%). |
9. Novērojumi
|
9.1. |
Ievērojot šādus īpašus hromatogrāfijas apstākļus, var labāk atdalīt triptofānu un α-metil-triptofānu. Izokrātiskā eluācija, pēc kuras notiek gradienta kolonnas tīrīšana:
|
||||||||||||||||||||||||||||||||||||||||||||
|
9.2. |
Hromatogrāfijas rezultāti atšķiras atkarībā no HPLC veida un izmantotā kolonnas iesaiņojuma materiāla. Izvēlētajai sistēmai jānodrošina triptofāna un iekšējā standarta bāzes līnijas atdalīšanās. Turklāt ir ļoti svarīgi sadalīšanās produktus atdalīt no triptofāna un iekšējā standarta. Lai pārbaudītu, vai iekšējais standarts nesatur piemaisījumus, izmanto hidrolizātus bez iekšējā standarta. Norises ilgumam jābūt pietiekamam, lai eluētu visus sadalīšanās produktus, citādi eluēšanas vēlie maksimumi var traucēt turpmāko hromatogrāfijas norisi. Darbības diapazonā hromatogrāfijas sistēma uzrāda lineāru reakciju. Lineāro reakciju mēra nemainīgā (normālā) iekšējā standarta koncentrācijā un mainīgās triptofāna koncentrācijās. Gan triptofāna, gan iekšējā standarta maksimumiem jābūt HPLC/fluorescences sistēmas lineārajā intervālā. Ja triptofāna un/vai iekšējā standarta maksimums(-i) ir pārāk mazs(-i) vai liels(-i), analīzi atkārto ar citu parauga apjomu un/vai citu galīgo tilpumu. |
|
9.3. |
Bārija hidroksīds
Laika gaitā bārija hidroksīda šķīdība samazinās. Līdz ar to HPLC noteiktais šķīdums ir neskaidrs un var uzrādīt zemu triptofāna saturu. |
H. JĒLEĻĻU UN JĒLTAUKU NOTEIKŠANA
1. Mērķis un darbības joma
Ar šo metodi nosaka jēleļļas un jēltaukus barībā. Tā neattiecas uz eļļas augu sēklu un augļu analīzi.
Abu turpmāk aprakstīto procedūru izmantošana ir atkarīga no barības īpašībām un sastāva, kā arī no iemesla, kādēļ tiek veikta analīze.
1.1. A procedūra. Tieši ekstrahējamās jēleļļas un jēltauki
Šo metodi piemēro augu izcelsmes barības līdzekļiem, izņemot tos, kas iekļauti B procedūras darbības jomā.
1.2. B procedūra. Jēleļļas un jēltauki kopā
Šo metodi piemēro dzīvnieku izcelsmes barības līdzekļiem un visiem barību maisījumiem. Tā jāizmanto visiem barības līdzekļiem, no kuriem bez iepriekšējas hidrolīzes nevar pilnībā ekstrahēt eļļas un taukus (piemēram, kviešu lipeklim, raugam, kartupeļu proteīniem un produktiem, kas pārstrādāti ar ekstrūzijas paņēmienu pārslojot vai karsējot).
1.3. Rezultātu interpretēšana
Visos gadījumos, kad, izmantojot B procedūru, iegūst augstāku rezultātu, nekā izmantojot A procedūru, par patieso vērtību uzskata B procedūrā iegūto rezultātu.
2. Princips
2.1. A procedūra
Paraugu ekstrahē ar petrolēteri. Pēc šķīdinātāja atdestilēšanas atlikumu izžāvē un nosver.
2.2. B procedūra
Paraugu apstrādā ar sālsskābi karsējot. Maisījumu atdzesē un filtrē. Atlikumu skalo, žāvē un veic noteikšanu saskaņā ar A procedūru.
3. Reaģenti
|
3.1. |
Petrolēteris, viršanas diapazons 40–60 oC. Broma vērtībai jābūt mazākai par 1, un negaistošajam atlikumam – mazākam par 2 mg/100 ml. |
|
3.2. |
Bezūdens nātrija sulfāts. |
|
3.3. |
Sālsskābe, c = 3 mol/l. |
|
3.4. |
Filtrēšanas palīgmateriāls, piemēram, kīzelgūrs, Hyflo-supercel. |
4. Ierīces
|
4.1. |
Ekstrakcijas aparāts. Ekstraktoros ar sifonu (Soksleta aparātos) atteces ātrums ir tāds, lai tas nodrošinātu apmēram 10 ciklus stundā; ekstraktoros bez sifona atteces ātrums ir apmēram 10 ml minūtē. |
|
4.2. |
Ekstrakcijas kapsulas, kuras nesatur petrolēterī šķīstošas vielas un kuru porozitāte atbilst 4.1. punkta prasībām. |
|
4.3. |
Uz 75 ± 3 oC ieregulēts vakuuma žāvēšanas skapis vai uz 100 ± 3 oC ieregulēts parastais žāvēšanas skapis. |
5. Procedūra
5.1. A procedūra (skatīt 8.1. punktu)
Ar precizitāti līdz 1 mg iesver 5 g parauga, pārnes to uz ekstrakcijas kapsulu (4.2.) un nosedz ar attaukotas kokvilnas vates tamponu.
Kapsulu ieliek ekstraktorā (4.1.) un ekstrahē sešas stundas ar petrolēteri (3.1.). Petrolētera ekstraktu savāc sausā, nosvērtā kolbā, kurā atrodas pumeka gabaliņi (2).
Atdestilē šķīdinātāju. Atlikumu izžāvē, turot kolbu pusotras stundas žāvēšanas skapī (4.3.). Atstāj atdzist eksikatorā un nosver. Atkārtoti žāvē 30 minūtes, lai nodrošinātu, ka eļļu un tauku svars nemainās (divos secīgi veiktos svērumos svara zudumam jābūt mazākam par vai vienādam ar 1 mg).
5.2. B procedūra
Nosver 2,5 g parauga ar precizitāti līdz 1 mg (skatīt 8.2. punktu), ievieto 400 ml vārglāzē vai 300 ml koniskā kolbā un pievieno 100 ml sālsskābes (3.3.) un pumeka gabaliņus. Vārglāzi nosedz ar pulksteņstiklu vai koniskajai kolbai pievieno atteces kondensatoru. Maisījumu uzkarsē līdz viršanas temperatūrai un vienu stundu lēni vāra uz nelielas gāzes degļa liesmas vai elektriskās plītiņas. Neļauj produktam pielipt pie konteinera sienām.
Atdzesē un pievieno tādu filtrēšanas palīgmateriāla (3.4.) daudzumu, kas ir pietiekams, lai novērstu eļļas un tauku zudumus filtrēšanas laikā. Izfiltrē caur samitrinātu, attaukotu, dubultu filtrpapīru. Atlikumu mazgā aukstā ūdenī līdz filtrāta neitrālai reakcijai. Pārbauda, vai filtrātā nav eļļu vai tauku. To klātbūtne liecina, ka paraugs pirms hidrolīzes jāekstrahē ar petrolēteri, izmantojot A procedūru.
Dubulto filtrpapīru ar atlikumu novieto uz pulksteņstikla un žāvē pusotras stundas žāvēšanas skapī (4.3.) 100 ± 3 oC temperatūrā.
Dubulto filtrpapīru ar sauso atlikumu ievieto ekstrakcijas kapsulā (4.2.) un nosedz ar attaukotas kokvilnas vates tamponu. Kapsulu ievieto ekstraktorā (4.1.) un rīkojas, kā norādīts 5.1. punkta otrajā un trešajā daļā.
6. Rezultāta izteikšana
Atlikuma svaru izsaka procentos no parauga.
7. Atkārtojamība
Viena laboranta divās paralēlās noteikšanās no viena parauga iegūtu rezultātu starpība nepārsniedz:
|
— |
0,2 % no absolūtās vērtības, ja jēleļļu un jēltauku saturs ir mazāks par 5 %, |
|
— |
4,0 % no lielākā rezultāta skaitliskās vērtības, ja saturs ir no 5 līdz 10 %, |
|
— |
0,4 % no absolūtās vērtības, ja saturs pārsniedz 10 %. |
8. Novērojumi
|
8.1. |
Attiecībā uz produktiem ar augstu eļļu un tauku saturu, ko ir grūti sasmalcināt vai no kā nevar sagatavot homogēnu samazinātu testa paraugu, rīkojas šādi. Ar precizitāti līdz 1 mg nosver 20 g parauga un samaisa ar 10 g vai vairāk gramiem bezūdens nātrija sulfāta (3.2.). Ekstrahē ar petrolēteri (3.1.), kā norādīts 5.1. punktā. Iegūto ekstraktu papildina līdz 500 ml ar petrolēteri (3.1.) un samaisa. No šķīduma paņem 50 ml un ielej mazā, sausā, nosvērtā kolbā, kurā atrodas pumeka gabaliņi. Atdestilē šķīdinātāju, izžāvē un rīkojas, kā norādīts 5.1. punkta pēdējā daļā. Izdala šķīdinātāju no kapsulā palikušajiem ekstrakcijas atlikumiem, ko sasmalcina līdz 1 mm un pēc tam ievieto atpakaļ ekstrakcijas kapsulā (nepievieno nātrija sulfātu), un rīkojas, kā norādīts 5.1. punkta otrajā un trešajā daļā. Eļļu un tauku saturu aprēķina procentos no parauga, izmantojot šādu formulu: (10m1 + m2) × 5 kur:
|
|
8.2. |
Produktiem ar zemu eļļu un tauku saturu testa paraugu var palielināt līdz 5 g. |
|
8.3. |
Barība lolojumdzīvniekiem ar augstu ūdens saturu pirms hidrolīzes un ekstrakcijas var būt jāsamaisa ar bezūdens nātrija sulfātu, kā noteikts B procedūrā. |
|
8.4. |
5.2. punktā minētajā gadījumā var būt efektīvāk atlikumu pēc filtrēšanas mazgāt ar karstu, nevis aukstu ūdeni. |
|
8.5. |
Dažiem barības veidiem, iespējams, jāpagarina 1½ stundas žāvēšanas laiks. Nedrīkst pārkaltēt, jo tādējādi var iegūt pazeminātus rezultātus. Žāvēšanai var izmantot arī mikroviļņu krāsni. |
|
8.6. |
Ja jēleļļu/jēltauku saturs pārsniedz 15 %, tad iesaka veikt iepriekšēju ekstrakciju pirms hidrolīzes, ievērojot A procedūru, un atkārtotu ekstrakciju, ievērojot B procedūru. Zināmā mērā tas ir atkarīgs no barības īpašībām un barībā esošās eļļas/tauku īpašībām. |
I. JĒLŠĶIEDRAS NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt barībā tādas skābēs un sārmos nešķīstošas organiskās vielas, kas nesatur taukus un ko parasti sauc par jēlšķiedru.
2. Princips
Paraugu, ko attiecīgā gadījumā attauko, secīgi apstrādā ar noteiktas koncentrācijas vārošiem sērskābes un kālija hidroksīda šķīdumiem. Atlikumu ar filtrāciju atdala uz keramiskā stikla filtra, nomazgā, izžāvē, nosver un pārpelno temperatūras diapazonā no 475 līdz 500 oC. Svara zudums pārpelnojot atbilst testa parauga jēlšķiedras saturam.
3. Reaģenti
|
3.1. |
Sērskābe, c = 0,13 mol/l. |
|
3.2. |
Pretputošanas viela (piemēram, n-oktanols). |
|
3.3. |
Filtrēšanas palīgmateriāls (celīts 545 vai līdzvērtīgs), ko četras stundas karsē 500 oC temperatūrā (8.6.). |
|
3.4. |
Acetons. |
|
3.5. |
Petrolēteris, viršanas diapazons no 40 līdz 60 oC. |
|
3.6. |
Sālsskābe, c = 0,5 mol/l. |
|
3.7. |
Kālija hidroksīda šķīdums, c = 0,23 mol/l. |
4. Ierīces
|
4.1. |
Karsēšanas vienība šķelšanai ar sērskābi un kālija hidroksīda šķīdumu, kas aprīkota ar pamatni filtrtīģelim (4.2.) un ar izplūdes cauruli ar krānu šķidrumu izplūdei un vakuumsūkni, var būt ar saspiestu gaisu. Pirms izmantošanas katru dienu vienību iepriekš sakarsē ar vārošu ūdeni piecas minūtes. |
|
4.2. |
Stikla filtrtīģelis ar kausētu keramiskā stikla filtra šķīvi, kura poru lielums ir 40–90 μm. Pirms pirmās izmantošanas dažas minūtes sakarsē līdz 500 oC un atdzesē (8.6.). |
|
4.3. |
Vismaz 270 ml cilindrs ar atteces kondensatoru, kas piemērots vārīšanai. |
|
4.4. |
Žāvēšanas skapis ar termostatu. |
|
4.5. |
Mufeļkrāsns ar termostatu. |
|
4.6. |
Ekstrakcijas vienība ar pamatni filtrtīģelim (4.2.) un izplūdes cauruli ar vakuuma un šķidruma izplūdes krānu. |
|
4.7. |
Savienotājgredzeni, lai samontētu karsēšanas vienību (4.1.), tīģeli (4.2.) un cilindru (4.3.) un savienotu auksto ekstrakcijas vienību (4.6.) un tīģeli. |
5. Procedūra
Ar precizitāti līdz 1 mg iesver 1 g sagatavotā parauga un ievieto to tīģelī (4.2.) (skatīt 8.1., 8.2. un 8.3. novērojumu), un pievieno 1 g filtrēšanas palīgmateriāla (3.3.).
Samontē karsēšanas vienību (4.1.) un filtrtīģeli (4.2.), tad tīģeli savieno ar cilindru (4.3.). Ielej 150 ml vārošas sērskābes (3.1.) samontētajā cilindrā un tīģelī un vajadzības gadījumā pievieno dažus pilienus pretputošanas vielas (3.2.).
5 ± 2 minūtēs šķidrumu uzvāra un ļauj enerģiski vārīties precīzi 30 minūtes.
Atver izplūdes caurules (4.1.) krānu un vakuumā filtrē sērskābi caur filtrtīģeli, un mazgā atlikumu ar trīs secīgām 30 ml vāroša ūdens devām, nodrošinot, lai atlikums uz filtra pēc katras mazgāšanas tiktu izžāvēts sauss.
Aizver izplūdes krānu un ielej 150 ml vāroša kālija hidroksīda šķīduma (3.7.) samontētajā cilindrā un tīģelī, pievienojot dažus pilienus pretputošanas vielas (3.2.). 5 ± 2 minūtēs šķidrumu uzvāra un ļauj enerģiski vārīties precīzi 30 minūtes. Filtrē un atkārto mazgāšanas procedūru, kas izmantota sērskābes posmam.
Pēc pēdējās mazgāšanas un žāvēšanas atvieno tīģeli un tā saturu un atkal savieno to ar auksto ekstrakcijas vienību (4.6.). Atlikumu tīģelī mazgā vakuumā ar trīs secīgām 25 ml acetona (3.4.) devām, nodrošinot, lai atlikums uz filtra pēc katras mazgāšanas tiktu izžāvēts sauss.
Tīģeli žāvē krāsnī 130 oC temperatūrā līdz konstantam svaram. Pēc katras žāvēšanas atdzesē eksikatorā un ātri nosver. Tīģeli ievieto mufeļkrāsnī un vismaz 30 minūtes 475–500 oC temperatūrā pārpelno līdz konstantam svaram (divu secīgu svērumu svara zuduma starpībai jābūt mazākai par vai vienādai ar 2 mg).
Pēc katras karsēšanas un pirms svēršanas vispirms atdzesē krāsnī un tad – eksikatorā.
Veic tukšo analīzi bez parauga. Svara zudums pārpelnošanas rezultātā nedrīkst pārsniegt 4 mg.
6. Rezultātu aprēķināšana
Jēlšķiedras saturu, izteiktu procentos no parauga, aprēķina, izmantojot šādu formulu:

kur:
|
m |
= |
parauga svars gramos, |
|
m0 |
= |
svara zudums gramos pēc pārpelnošanas noteikšanas laikā, |
|
m1 |
= |
svara zudums gramos pēc pārpelnošanas tukšās analīzes laikā. |
7. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt:
|
— |
0,6 % no absolūtās vērtības jēlšķiedru saturam, kas mazāks par 10 %, |
|
— |
6 % no augstākā rezultāta, ja jēlšķiedras saturs ir vienāds ar vai lielāks par 10 %. |
8. Novērojumi
|
8.1. |
Barība, kas satur vairāk par 10 % jēltauku, pirms analīzes jāattauko ar petrolēteri (3.5.). Filtrtīģeli (4.2.) un tā saturu savieno ar auksto ekstrakcijas vienību (4.6.), un atlikumu mazgā vakuumā ar trīs secīgām 30 ml petrolētera devām, nodrošinot, lai atlikums tiktu izžāvēts sauss. Tīģeli un tā saturu savieno ar karsēšanas vienību (4.1.) un rīkojas, kā aprakstīts 5. punktā. |
|
8.2. |
Barība, kas satur taukus, kurus nevar ekstrahēt tieši ar petrolēteri (3.5.), jāattauko, kā norādīts 8.1. punktā, un vēlreiz jāattauko pēc vārīšanas ar skābi. Pēc vārīšanas ar skābi un turpmākas mazgāšanas tīģeli un tā saturu savieno ar auksto ekstrakcijas vienību (4.6.) un trīs reizes mazgā ar 30 ml acetona, un tad trīs reizes mazgā ar 30 ml petrolētera devām. Filtrē vakuumā, līdz paraugs ir sauss, un turpina analīzi, kā aprakstīts 5. punktā, vispirms apstrādājot ar kālija hidroksīdu. |
|
8.3. |
Ja barība satur vairāk par 5 % karbonātu, kas izteikti kā kalcija karbonāts, tad tīģeli (4.2.) ar iesvērto paraugu savieno ar karsēšanas vienību (4.1.). Paraugu trīs reizes mazgā ar 30 ml sālsskābes (3.6.). Pēc katras pievienošanas paraugam ļauj vienu minūti pastāvēt, pirms to filtrē. Mazgā vienu reizi ar 30 ml ūdens, tad rīkojas, kā aprakstīts 5. punktā. |
|
8.4. |
Ja izmanto stendveida aparātu (vairāki tīģeļi savienoti ar vienu un to pašu karsēšanas vienību), tad vienā seansā nedrīkst veikt divas individuālas noteikšanas ar vienu analīzes paraugu. |
|
8.5. |
Ja pēc vārīšanas ir grūti filtrēt skābos un sārmainos šķīdumus, tad caur karsēšanas vienības izplūdes cauruli izpūš saspiestu gaisu un tad turpina filtrēšanu. |
|
8.6. |
Lai pagarinātu stikla filtrtīģeļu kalpošanas laiku, pārpelnošanas temperatūra nepārsniedz 500 oC. Karsēšanas un dzesēšanas ciklos nedrīkst pieļaut pārmērīgas termiskās svārstības. |
J. CUKURU NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt reducējošo cukuru daudzumu un, izdarot inversiju, kopējo cukuru daudzumu, ko izsaka glikozē vai attiecīgā gadījumā saharozē, pārveidojot ar koeficientu 0,95. To piemēro barības maisījumiem. Pārējās barības analīzēm ir paredzētas īpašas metodes. Vajadzības gadījumā laktozi mēra atsevišķi un ņem vērā rezultātu aprēķinos.
2. Princips
Cukurus ekstrahē atšķaidītā etanolā; šķīdumu dzidrina ar Karesa I šķīdumu un Karesa II šķīdumu. Pēc etanola atdalīšanas ar Lufa-Šorla metodi nosaka cukuru daudzumu pirms un pēc inversijas.
3. Reaģenti
|
3.1. |
Etanola šķīdums 40 % (v/v), blīvums 0,948 g/ml 20 oC temperatūrā, neitralizēts līdz fenolftaleīnam. |
|
3.2. |
Karesa I šķīdums: ūdenī izšķīdina 21,9 g cinka acetāta, Zn (CH3COO)2 2H2O, un 3 g ledus etiķskābes. Papildina ar ūdeni līdz 100 ml. |
|
3.3. |
Karesa II šķīdums: ūdenī izšķīdina 10,6 g kālija ferocianīda, K4Fe (CN)6 3H2O. Papildina ar ūdeni līdz 100 ml. |
|
3.4. |
Metiloranžais, 0,1 % šķīdums (w/v). |
|
3.5. |
Sālsskābe 4 mol/l. |
|
3.6. |
Sālsskābe 0,1 mol/l. |
|
3.7. |
Nātrija hidroksīda šķīdums 0,1 mol/l. |
|
3.8. |
Lufa-Šorla reaģents. Uzmanīgi maisot, citronskābes šķīdumu (3.8.2.) ielej nātrija karbonāta šķīdumā (3.8.3.). Pievieno vara sulfāta šķīdumu (3.8.1.) un papildina līdz vienam litram ar ūdeni. Pa nakti atstāj nogulsnēties un filtrē. Pārbauda tādējādi iegūtā reaģenta koncentrāciju (Cu 0,05 mol/litrā; Na2 CO3 1 mol/litrā), skatīt 5.4. punkta pēdējo daļu. Šķīduma pH līmenis ir apmēram 9,4. |
|
3.8.1. |
Vara sulfāta šķīdums: izšķīdina 25 g vara sulfāta, Cu SO4 5H2O, bez dzelzs 100 mililitros ūdens. |
|
3.8.2. |
Citronskābes šķīdums: izšķīdina 50 g citronskābes, C6H8O7 H2O, 50 mililitros ūdens. |
|
3.8.3. |
Nātrija karbonāta šķīdums: izšķīdina 143,8 g bezūdens nātrija karbonāta apmēram 300 mililitros silta ūdens. Atstāj atdzist. |
|
3.9. |
Nātrija tiosulfāta šķīdums 0,1 mol/l. |
|
3.10. |
Cietes šķīdums: 5 g šķīdināmas cietes, kas iemaisīta 30 mililitros ūdens, pievieno vienam litram vāroša ūdens. Vāra trīs minūtes, atstāj atdzist un vajadzības gadījumā pievieno kā konservantu 10 mg dzīvsudraba jodīdu. |
|
3.11. |
Sērskābe 3 mol/l. |
|
3.12. |
Kālija jodīds, 30 % šķīdums (w/v). |
|
3.13. |
Pumeka gabaliņi, vārīti sālsskābē, nomazgāti ūdenī un izžāvēti. |
|
3.14. |
3-metilbutān-1-ols. |
4. Ierīces
Rotācijas tipa maisītājs: aptuveni 35 līdz 40 apgr./min.
5. Procedūra
5.1. Parauga ekstrahēšana
2,5 g parauga ar precizitāti līdz mg iesver 250 ml tilpuma mērkolbā. Pievieno 200 ml etanola (3.1.) un maisa maisītājā vienu stundu. Pievieno 5 ml Karesa I šķīduma (3.2.) un maisa apmēram 30 sekundes. Pievieno 5 ml Karesa II šķīduma (3.3.) un maisa vēl vienu minūti. Papildina ar etanolu (3.1.) līdz vajadzīgajam tilpumam, homogenizē un filtrē. Lai likvidētu lielāko daļu etanola, atdala 200 ml filtrāta un tvaicē, līdz paliek apmēram puse tilpuma. Izmantojot siltu ūdeni, iztvaicēšanas atlikumu kvantitatīvi pārnes 200 ml tilpuma mērkolbā, atdzesē, uzpilda līdz vajadzīgajam tilpumam ar ūdeni, homogenizē un vajadzības gadījumā filtrē. Šo šķīdumu izmantos, lai noteiktu reducējošo cukuru daudzumu un pēc inversijas veikšanas – kopējo cukuru daudzumu.
5.2. Reducējošo cukuru noteikšana
Ar pipeti ņem ne vairāk kā 25 ml šķīduma, kurā, izsakot glikozē, ir ne vairāk kā 60 mg reducējošo cukuru. Vajadzības gadījumā papildina līdz 25 ml ar destilētu ūdeni un nosaka reducējošo cukuru saturu, izmantojot Lufa-Šorla metodi. Rezultātu izsaka kā glikozes procentuālo saturu paraugā.
5.3. Kopējo cukuru noteikšana pēc inversijas
Ar pipeti ņem 50 ml šķīduma un pārnes 100 ml tilpuma mērkolbā. Pievieno dažus pilienus metiloranžā šķīduma (3.4.), tad, nepārtraukti maisot, uzmanīgi pievieno sālsskābi (3.5.), līdz šķidrums kļūst izteikti sarkans. Pievieno 15 ml sālsskābes (3.6.), uz trīsdesmit minūtēm iegremdē kolbu vannā ar ūdeni, kas strauji vārās. Strauji atdzesē apmēram līdz 20 oC temperatūrai un pievieno 15 ml nātrija hidroksīda šķīduma (3.7.). Papildina ar ūdeni līdz 100 ml un homogenizē. Atdala daudzumu, kas nepārsniedz 25 ml un satur mazāk nekā 60 mg reducējošo cukuru, izteiktu kā glikoze. Vajadzības gadījumā papildina līdz 25 ml ar destilētu ūdeni un nosaka reducējošo cukuru saturu, izmantojot Lufa-Šorla metodi. Rezultātu izsaka glikozes vai attiecīgā gadījumā saharozes procentos, reizinot ar koeficientu 0,95.
5.4. Titrēšana, izmantojot Lufa-Šorla metodi
Ar pipeti ņem 25 ml Lufa-Šorla reaģenta (3.8.) un pārnes 300 ml Erlenmeijera kolbā; pievieno precīzi 25 ml dzidrinātā cukura šķīduma. Pievieno divus gabaliņus pumeka (3.13.), ar roku maisot, uz vidēja augstuma vaļējas liesmas šķīdumu apmēram divās minūtēs uzkarsē līdz vārīšanās temperatūrai. Erlenmeijera kolbu nekavējoties novieto uz metāla sieta, kas pārklāts ar azbestu un kam ir apmēram 6 cm liels caurums, zem kura iedegta liesma. Liesmu noregulē tā, lai Erlenmeijera kolba tiktu karsēta tikai no apakšas. Erlenmeijera kolbai pievieno atteces kondensatoru. Vāra precīzi desmit minūtes. Tūlīt atdzesē aukstā ūdenī un apmēram pēc piecām minūtēm titrē šādi.
Pievieno 10 ml kālija jodīda šķīduma (3.12.) un tūlīt pēc tam (uzmanīgi, jo iespējama ievērojama putu veidošanās) pievieno 25 ml sērskābes (3.11.). Titrē ar nātrija tiosulfāta šķīdumu (3.9.), līdz parādās blāvi dzeltens krāsojums, pievieno cietes indikatoru (3.10.) un pabeidz titrēšanu.
Bez vārīšanas tādā pašā veidā titrē 25 ml precīzi mērīta Lufa-Šorla reaģenta (3.8.) un 25 ml ūdens maisījumu, kam pievienoti 10 ml kālija jodīda šķīduma (3.12.) un 25 ml sērskābes (3.11.).
6. Rezultātu aprēķināšana
Izmantojot tabulu, nosaka glikozes daudzumu miligramos, kas atbilst abu titrēšanas vērtību starpībai un ko izsaka mg nātrija tiosulfāta 0,1 mol/l. Rezultātu izsaka procentos no parauga.
7. Īpašas procedūras
|
7.1. |
Ja analizē barību, kurā ir daudz melases, kā arī citu barību, kas nav īpaši homogēna, tad iesver 20 g un ievieto viena litra mērkolbā kopā ar 500 ml ūdens. Vienu stundu maisa maisītājā. Dzidrina, izmantojot Karesa I (3.2.) un Karesa II (3.3.) reaģentu, kā aprakstīts 5.1. punktā, katru reaģentu ņemot četrkārtīgā daudzumā. Uzpilda līdz vajadzīgajam tilpumam ar 80 % etanolu (v/v). Homogenizē un filtrē. Atdala etanolu, kā aprakstīts 5.1. punktā. Ja nav dekstrinētas cietes, tad uzpilda līdz tilpumam ar destilētu ūdeni. |
|
7.2. |
Analizējot melasi un barības līdzekļus, kuri bagāti ar cukuru un kuros gandrīz nemaz nav cietes (ceratonijas, žāvēti biešu graizījumi u. c.), iesver 5 g un ievieto 250 ml tilpuma mērkolbā, pievieno 200 ml destilēta ūdens un maisa maisītājā vienu stundu vai ilgāk, ja vajadzīgs. Dzidrina, izmantojot Karesa I (3.2.) un Karesa II (3.3.) reaģentu, kā aprakstīts 5.1. punktā. Uzpilda līdz tilpumam ar aukstu ūdeni, homogenizē un filtrē. Lai noteiktu kopējo cukuru daudzumu, rīkojas, kā aprakstīts 5.3. punktā. |
8. Novērojumi
|
8.1. |
Lai novērstu putu veidošanos, pirms vārīšanas ar Lufa-Šorla reaģentu ieteicams pievienot (neatkarīgi no tilpuma) apmēram 1 ml 3-metilbutān-1-ola (3.14.). |
|
8.2. |
Starpība starp kopējo cukuru saturu pēc inversijas, kas izteikts kā glikoze, un reducējošo cukuru saturu, kas izteikts kā glikoze, un kas reizināta ar koeficientu 0,95, norāda saharozes satura procentuālo daudzumu. |
|
8.3. |
Lai noteiktu reducējošo cukuru saturu, izņemot laktozi, var izmantot divas metodes. |
|
8.3.1. |
Lai veiktu aptuvenu aprēķinu, laktozes saturu reizina ar 0,675, kas noteikts ar citu analīzes metodi, un iegūto rezultātu atņem no reducējošo cukuru satura. |
|
8.3.2. |
Lai precīzi aprēķinātu reducējošos cukurus, izņemot laktozi, abās galīgās noteikšanās jāizmanto tas pats paraugs. Vienai analīzei ņem daļu šķīduma, kas iegūts saskaņā ar 5.1. punktā doto aprakstu, bet otrai analīzei – daļu šķīduma, ko iegūst laktozes noteikšanā, izmantojot šim nolūkam paredzēto metodi (pēc citu cukuru fermentācijas un dzidrināšanas). Abos gadījumos klātesošā cukura daudzumu nosaka, izmantojot Lufa-Šorla metodi, un aprēķina glikozes miligramos. Vienu vērtību atņem no otras un starpību izsaka procentos no parauga. Piemērs Abi katrā noteikšanā ņemtie šķīdumu tilpumi atbilst 250 mg paraugam. Pirmajā gadījumā patērē 17 ml nātrija tiosulfāta šķīduma 0,1 mol/l, kas atbilst 44,2 mg glikozes; otrā gadījumā – 11 ml, kas atbilst 27,6 mg glikozes. Starpība ir 16,6 mg glikozes. Līdz ar to reducējošo cukuru saturs (izņemot laktozi), kas aprēķināts kā glikoze, ir šāds:
|
25 ml Lufa-Šorla reaģenta vērtību tabula
Na2S2O30,1 mol/l mililitri, karsē divas minūtes, vāra desmit minūtes.
|
Na2S2O3 0,1 mol/l |
Glikoze, fruktoze, invertcukuri C6H12O6 |
Laktoze C12H22O11 |
Maltoze C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
starpība |
mg |
starpība |
mg |
starpība |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
K. LAKTOZES NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt laktozes daudzumu barībā, kas satur vairāk par 0,5 % laktozes.
2. Princips
Cukurus izšķīdina ūdenī. Šķīdumu fermentē ar Saccharomyces cerevisiae raugu, kas laktozi atstāj neskartu. Pēc dzidrināšanas un filtrēšanas laktozes saturu filtrātā nosaka, izmantojot Lufa-Šorla metodi.
3. Reaģenti
|
3.1. |
Saccharomyces cerevisiae suspensija: 25 g svaiga rauga iemaisa 100 mililitros ūdens. Suspensiju var uzglabāt ledusskapī ne ilgāk kā vienu nedēļu. |
|
3.2. |
Karesa I šķīdums: ūdenī izšķīdina 21,9 g cinka acetāta, Zn (CH3 COO)2 2H2O, un 3 g ledus etiķskābes. Papildina ar ūdeni līdz 100 ml. |
|
3.3. |
Karesa II šķīdums: ūdenī izšķīdina 10,6 g kālija ferocianīda, K4Fe (CN)6 3H2O. Papildina ar ūdeni līdz 100 ml. |
|
3.4. |
Lufa-Šorla reaģents. Uzmanīgi maisot, citronskābes šķīdumu (3.4.2.) ielej nātrija karbonāta šķīdumā (3.4.3.). Pievieno vara sulfāta šķīdumu (3.4.1.) un papildina līdz vienam litram ar ūdeni. Pa nakti atstāj nogulsnēties un filtrē. Pārbauda tādējādi iegūtā reaģenta koncentrāciju (Cu 0,05 mol/litrā; Na2 CO3 1 mol/litrā). Šķīduma pH līmenis ir apmēram 9,4. |
|
3.4.1. |
Vara sulfāta šķīdums: izšķīdina 25 g vara sulfāta, Cu SO4 5H2O, bez dzelzs 100 mililitros ūdens. |
|
3.4.2. |
Citronskābes šķīdums: izšķīdina 50 g citronskābes, C6H8O7 H2O, 50 mililitros ūdens. |
|
3.4.3. |
Nātrija karbonāta šķīdums: izšķīdina 143,8 g bezūdens nātrija karbonāta apmēram 300 mililitros silta ūdens. Atstāj atdzist. |
|
3.5. |
Pumeka gabaliņi, vārīti sālsskābē, nomazgāti ūdenī un izžāvēti. |
|
3.6. |
Kālija jodīds, 30 % šķīdums (w/v). |
|
3.7. |
Sērskābe 3 mol/l. |
|
3.8. |
Nātrija tiosulfāta šķīdums 0,1 mol/l. |
|
3.9. |
Cietes šķīdums: 5 g šķīdināmas cietes, kas iemaisīta 30 mililitros ūdens, pievieno vienam litram vāroša ūdens. Vāra trīs minūtes, atstāj atdzist un vajadzības gadījumā pievieno kā konservantu 10 mg dzīvsudraba jodīda. |
4. Ierīces
Ūdens vanna ar termostatu, kas iestatīts uz 38–40 oC.
5. Procedūra
Iesver 1 g parauga ar precizitāti līdz mg un ievieto šo parauga daļu 100 ml tilpuma mērkolbā. Pievieno 25–30 ml ūdens. Uz trīsdesmit minūtēm ievieto kolbu vāroša ūdens vannā, tad atdzesē līdz apmēram 35 oC. Pievieno 5 ml rauga suspensijas (3.1.) un homogenizē. Atstāj kolbu uz divām stundām ūdens vannā 38–40 oC temperatūrā. Atdzesē līdz apmēram 20 oC.
Pievieno 2,5 ml Karesa I šķīduma (3.2.) un maisa trīsdesmit sekundes, tad pievieno 2,5 ml Karesa II šķīduma (3.3.) un maisa vēl trīsdesmit sekundes. Papildina ar ūdeni līdz 100 ml, samaisa un filtrē. Ar pipeti atdala ne vairāk kā 25 ml filtrāta, kas, ja iespējams, satur 40–80 mg laktozes, un pārnes to 300 ml Erlenmeijera kolbā. Vajadzības gadījumā papildina līdz 25 ml ar ūdeni.
Tādā pašā veidā veic tukšo analīzi ar 5 ml rauga suspensijas (3.1.). Šādi nosaka laktozes saturu atbilstoši Lufa-Šorla metodei: pievieno precīzi 25 ml Lufa-Šorla reaģenta (3.4.) un divus pumeka gabaliņus (3.5.). Ar roku maisot, uz vidēja augstuma vaļējas liesmas šķīdumu apmēram divās minūtēs uzkarsē līdz vārīšanās temperatūrai. Erlenmeijera kolbu tūlīt novieto uz metāla sieta, kas pārklāts ar azbestu un kam ir apmēram 6 cm diametra caurums, zem kura iedegta liesma. Liesmu noregulē tā, lai Erlenmeijera kolba tiktu karsēta tikai no apakšas. Erlenmeijera kolbai pievieno atteces kondensatoru. Vāra precīzi desmit minūtes. Tūlīt atdzesē aukstā ūdenī un pēc apmēram piecām minūtēm titrē šādi.
Pievieno 10 ml kālija jodīda šķīduma (3.6.) un tūlīt pēc tam (uzmanīgi, jo iespējama ievērojama putu veidošanās) pievieno 25 ml sērskābes (3.7.). Titrē ar nātrija tiosulfāta šķīdumu (3.8.), līdz parādās blāvi dzeltens krāsojums, pievieno cietes indikatoru (3.9.) un pabeidz titrēšanu.
Bez vārīšanas tādā pašā veidā titrē 25 ml precīzi mērīta Lufa-Šorla reaģenta (3.4.) un 25 ml ūdens maisījumu, kam pievienoti 10 ml kālija jodīda šķīduma (3.6.) un 25 ml sērskābes (3.7.).
6. Rezultātu aprēķināšana
Izmantojot pievienoto tabulu, nosaka laktozes daudzumu miligramos, kas atbilst abu titrēšanu rezultātu starpībai un ko izsaka ml nātrija tiosulfāta 0,1 mol/l.
Rezultātu izsaka bezūdens laktozē kā parauga procentuālo daļu.
7. Novērojums
Produktiem, kuri satur vairāk nekā 40 % fermentējama cukura, izmanto vairāk nekā 5 ml rauga suspensijas (3.1.).
25 ml Lufa-Šorla reaģenta vērtību tabula
Na2S2O30,1 mol/l mililitri, karsē divas minūtes, vāra desmit minūtes.
|
Na2S2O3 0,1 mol/l |
Glikoze, fruktoze, invertcukuri C6H12O6 |
Laktoze C12H22O11 |
Maltoze C12H22O11 |
Na2S2O3 0,1 mol/l |
|||
|
ml |
mg |
starpība |
mg |
starpība |
mg |
starpība |
ml |
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
2,4 4,8 7,2 9,7 12,2 14,7 17,2 19,8 22,4 25,0 27,6 30,3 33,0 35,7 38,5 41,3 44,2 47,1 50,0 53,0 56,0 59,1 62,2 |
2,4 2,4 2,5 2,5 2,5 2,5 2,6 2,6 2,6 2,6 2,7 2,7 2,7 2,8 2,8 2,9 2,9 2,9 3,0 3,0 3,1 3,1 |
3,6 7,3 11,0 14,7 18,4 22,1 25,8 29,5 33,2 37,0 40,8 44,6 48,4 52,2 56,0 59,9 63,8 67,7 71,7 75,7 79,8 83,9 88,0 |
3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,7 3,8 3,8 3,8 3,8 3,8 3,8 3,9 3,9 3,9 4,0 4,0 4,1 4,1 4,1 |
3,9 7,8 11,7 15,6 19,6 23,5 27,5 31,5 35,5 39,5 43,5 47,5 51,6 55,7 59,8 63,9 68,0 72,2 76,5 80,9 85,4 90,0 94,6 |
3,9 3,9 3,9 4,0 3,9 4,0 4,0 4,0 4,0 4,0 4,0 4,1 4,1 4,1 4,1 4,1 4,2 4,3 4,4 4,5 4,6 4,6 |
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 |
L. CIETES NOTEIKŠANA
POLARIMETRISKĀ METODE
1. Mērķis un darbības joma
Šī metode ļauj noteikt cietes un lielmolekulāra svara cietes sadalīšanās produktu daudzumus barībā, lai pārbaudītu atbilstību deklarētajai enerģētiskajai vērtībai (VII pielikuma noteikumi) un Padomes Direktīvai 96/25/EK (3).
2. Princips
Šī metode ietver divas noteikšanas. Pirmajā paraugu apstrādā ar atšķaidītu sālsskābi. Pēc dzidrināšanas un filtrēšanas šķīduma optisko rotāciju mēra, izmantojot polarimetriju.
Otrajā paraugu ekstrahē ar 40 % etanolu. Pēc tam, kad filtrāts ir paskābināts ar sālsskābi, dzidrināts un filtrēts, optisko rotāciju mēra tāpat kā pirmajā noteikšanā.
No abu mērījumu starpības, reizinot ar zināmu koeficientu, iegūst parauga cietes saturu.
3. Reaģenti
|
3.1. |
Sālsskābe, 25 % šķīdums (w/v); blīvums 1,126 g/ml. |
|
3.2. |
Sālsskābe, 1,13 % šķīdums (w/v). Koncentrācija jāpārbauda, titrējot ar 0,1 mol/l nātrija hidroksīda šķīdumu 0,1 % (w/v) metilsarkanā klātbūtnē 94 % (v/v) etanolā. Lai neitralizētu 10 ml, vajag 30,94 ml NaOH 0,1 mol/l. |
|
3.3. |
Karesa I šķīdums: ūdenī izšķīdina 21,9 g cinka acetāta, Zn(CH3COO)2 2H2O, un 3 g ledus etiķskābes. Papildina ar ūdeni līdz 100 ml. |
|
3.4. |
Karesa II šķīdums: ūdenī izšķīdina 10,6 g kālija ferocianīda, K4 Fe(CN)6 3H2O. Papildina ar ūdeni līdz 100 ml. |
|
3.5. |
Etanols, 40 % šķīdums (v/v), blīvums 0,948 g/ml 20 oC temperatūrā. |
4. Ierīces
|
4.1. |
250 ml Erlenmeijera kolba ar standarta pieslīpētu stikla savienojumu un ar atteces kondensatoru. |
|
4.2. |
Polarimetrs vai saharimetrs. |
5. Procedūra
5.1. Parauga sagatavošana
Paraugu sasmalcina, līdz tas ir pietiekami smalks, lai izsijātos caur sietu ar 0,5 mm apaļu acojumu.
5.2. Kopējās optiskās rotācijas (P vai S) noteikšana (skatīt 7.1. novērojumu)
2,5 g sasmalcinātā parauga ar precizitāti līdz mg iesver 100 ml mērkolbā. Pievieno 25 ml sālsskābes (3.2.), krata, lai testa paraugs vienmērīgi sadalītos, un pievieno vēl 25 ml sālsskābes (3.2.). Iegremdē kolbu vāroša ūdens vannā, pirmās trīs minūtes enerģiski un nepārtraukti kratot, lai novērstu aglomerātu veidošanos. Ūdens daudzumam ūdens vannā jābūt pietiekamam, lai vanna turpinātu vārīties, kad tajā ieliek kolbu. Kolbu no vannas nedrīkst izņemt, kad tā tiek kratīta. Precīzi pēc 15 minūtēm kolbu izņem no vannas, pievieno 30 ml auksta ūdens un tūlīt atdzesē līdz 20 oC.
Pievieno 5 ml Karesa I šķīduma (3.3.) un krata apmēram 30 sekundes. Tad pievieno 5 ml Karesa II šķīduma (3.4.) un atkal krata apmēram 30 sekundes. Papildina ar ūdeni līdz tilpumam, samaisa un filtrē. Ja filtrāts nav pilnīgi dzidrs (kas notiek reti), tad atkārto noteikšanu, izmantojot lielāku daudzumu Karesa I un II šķīduma, piemēram, 10 ml.
Ar polarimetru vai saharimetru izmēra šķīduma optisko rotāciju 200 mm mēģenē.
5.3. Optiskās rotācijas (P' vai S') noteikšana vielām, kuras šķīst 40 % etanolā
Ar precizitāti līdz mg iesver 5 g parauga, ievieto 100 ml mērkolbā un pievieno apmēram 80 ml etanola (3.5.) (skatīt 7.2. novērojumu). Atstāj kolbu vienu stundu istabas temperatūrā; šajā laikā sešas reizes kārtīgi sakrata tā, lai testa paraugs labi sajauktos ar etanolu. Papildina līdz tilpumam ar etanolu (3.5.), samaisa un filtrē.
Ar pipeti 50 ml filtrāta (kas atbilst 2,5 g parauga) iepilina 250 ml Erlenmeijera kolbā, pievieno 2,1 ml sālsskābes (3.1.) un kārtīgi sakrata. Erlenmeijera kolbai pievieno atteces kondensatoru un kolbu iegremdē vāroša ūdens vannā. Tieši pēc 15 minūtēm izņem Erlenmeijera kolbu no vannas, saturu pārnes 100 ml mērkolbā, skalojot ar nelielu daudzumu auksta ūdens, un atdzesē līdz 20 oC.
Dzidrina, izmantojot Karesa I (3.3.) un II (3.4.) šķīdumu, papildina līdz tilpumam ar ūdeni, samaisa, filtrē un izmēra optisko rotāciju, kā norādīts 5.2. punkta otrajā un trešajā daļā.
6. Rezultātu aprēķināšana
Cietes saturu (%) aprēķina šādi.
6.1. Mērīšana ar polarimetru

|
P |
= |
kopējā optiskā rotācija leņķa grādos, |
||||||||||||||
|
P' |
= |
40 % (V/V) etanolā šķīstošu vielu optiskā rotācija leņķa grādos, |
||||||||||||||
|
|
= |
tīras cietes specifiskā optiskā rotācija. Vispārīgi pieņemtās ; skaitliskās vērtības šim koeficientam ir šādas:
|
6.2. Mērīšana ar saharimetru

|
S |
= |
kopējā optiskā rotācija saharimetra grādos, |
|
S' |
= |
40 % (v/v) etanolā šķīstošu vielu optiskā rotācija saharimetra grādos, |
|
N |
= |
saharozes svars (g) 100 mililitros ūdens, kas, izmērot ar 200 mm mēģeni, dod 100 saharimetra grādu optisko rotāciju 16,29 g Francijas saharimetriem, 26,00 g Vācijas saharimetriem, 20,00 g jauktajiem saharimetriem.
|
6.3. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 0,4 no absolūtā lieluma, ja cietes saturs ir mazāks par 40 %, un 1 % no relatīvā lieluma, ja cietes saturs ir vienāds ar vai lielāks par 40 %.
7. Novērojumi
|
7.1. |
Ja paraugs satur vairāk nekā 6 % karbonātu, rēķinot kalcija karbonāta izteiksmē, tad tie pirms kopējās optiskās rotācijas noteikšanas ir jāiznīcina, apstrādājot ar precīzi atbilstīgu atšķaidītas sērskābes daudzumu. |
|
7.2. |
Ja produktos ir liels laktozes saturs, piemēram, piena seruma pulverī vai sausajā vājpienā, tad pievieno 80 ml etanola (3.5.) un rīkojas šādi. Kolbai pievieno atteces kondensatoru un kolbu uz 30 minūtēm iegremdē ūdens vannā 50 oC temperatūrā. Atstāj atdzist un turpina analīzi, kā norādīts 5.3. punktā. |
|
7.3. |
Ir zināms, ka šādas barības sastāvdaļas, ja tās barībā ir ievērojamā daudzumā, rada traucējumus, nosakot cietes saturu ar polarimetrisko metodi, un līdz ar to var iegūt nepareizus rezultātus:
|
M. JĒLPELNU NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt jēlpelnu saturu barībā.
2. Princips
Paraugu pārpelno 550 oC temperatūrā; atlikumu nosver.
3. Reaģenti
Amonija nitrāts, 20 % šķīdums (w/v).
4. Ierīces
|
4.1. |
Elektriskā plītiņa. |
|
4.2. |
Elektriskā mufeļkrāsns ar termostatu. |
|
4.3. |
Pārpelnošanai paredzēti kvarca, porcelāna vai platīna tīģeļi, kas ir vai nu taisnstūrveida (apmēram 60 × 40 × 25 mm), vai apaļi (diametrs 60 līdz 75 mm, augstums 20 līdz 40 mm). |
5. Procedūra
Ar precizitāti līdz mg iesver apmēram 5 g parauga (2,5 g produktiem, kam ir tendence uzbriest) un ievieto pārpelnošanai tīģelī, kas iepriekš uzkarsēts līdz 550 oC, atdzesēts un nosvērts. Novieto tīģeli uz elektriskās plītiņas un pakāpeniski karsē, līdz viela pārogļojas. Pārpelno atbilstoši 5.1. vai 5.2. punktam.
|
5.1. |
Tīģeli ievieto kalibrētā mufeļkrāsnī, kas iestatīta uz 550 oC. Iztur šajā temperatūrā, līdz iegūst baltus, gaišpelēkus vai sarkanīgas nokrāsas pelnus, kuros nav acīm redzamu ogles daļiņu. Tīģeli ievieto eksikatorā, atdzesē un tūlīt nosver. |
|
5.2. |
Tīģeli ievieto kalibrētajā mufeļkrāsnī, kas iestatīta uz 550 oC. Pārpelno trīs stundas. Tīģeli ievieto eksikatorā, atstāj atdzist un tūlīt nosver. Atkārtoti pārpelno 30 minūtes, lai nodrošinātu, ka pelnu svars nemainās (divos secīgi veiktos svērumos svara zudumam jābūt mazākam par vai vienādam ar 1 mg). |
6. Rezultātu aprēķināšana
Aprēķina atlikuma masu, atņemot taras masu.
Rezultātu izsaka procentos no parauga.
7. Novērojumi
|
7.1. |
Grūti pārpelnojamām vielām jāveic vismaz trīs stundas ilga sākotnējā pārpelnošana, pēc kuras paraugu atdzesē, tam uzmanīgi pievieno dažus pilienus 20 % amonija nitrāta šķīduma vai ūdens (uzmanīgi, lai pelni neizkaisītos vai nesaķeptu gabalos). Pēc izžāvēšanas krāsnī kalcinēšanu turpina. Vajadzības gadījumā darbību atkārto, līdz pārpelnošana ir pabeigta. |
|
7.2. |
Ar vielām, ko nevar apstrādāt, kā aprakstīts 7.1. punktā, rīkojas šādi: pēc trīs stundu pārpelnošanas pelnus ievieto siltā ūdenī un filtrē caur nelielu bezpelnu filtru. Tajā pašā tīģelī pārpelno filtru kopā ar tā saturu. Filtrātu ievieto atdzesētajā tīģelī, iztvaicē, līdz tas ir sauss, pārpelno un nosver. |
|
7.3. |
Analizējot eļļas un taukus, precīzi iesver 25 g parauga piemērota izmēra tīģelī. Pārogļo, vielu aizdedzinot ar bezpelnu filtrpapīra strēmeli. Pēc sadedzināšanas samitrina ar iespējami mazu ūdens daudzumu. Izžāvē un pārpelno, kā aprakstīts 5. punktā. |
N. SĀLSSKĀBĒ NEŠĶĪSTOŠU PELNU NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt tādu minerālvielu daudzumu barībā, kuras nešķīst sālsskābē. Atkarībā no parauga īpašībām var izmantot divas metodes.
|
1.1. |
A metode: piemērojama organiskiem barības līdzekļiem un lielākajai daļai barības maisījumu. |
|
1.2. |
B metode: piemērojama tādiem minerālu savienojumiem un maisījumiem un barības maisījumiem, kuros sālsskābē nešķīstošo vielu saturs, kā noteikts ar A metodi, pārsniedz 1 %. |
2. Princips
|
2.1. |
A metode: paraugu pārpelno, pelnus uzvāra sālsskābē un nešķīstošo atlikumu filtrē un nosver. |
|
2.2. |
B metode: paraugu apstrādā ar sālsskābi. Šķīdumu filtrē, atlikumu pārpelno un tādējādi iegūtos pelnus apstrādā saskaņā ar A metodi. |
3. Reaģenti
|
3.1. |
Sālsskābe 3 mol/l. |
|
3.2. |
Trihloretiķskābe, 20 % šķīdums (w/v). |
|
3.3. |
Trihloretiķskābe, 1 % šķīdums (w/v). |
4. Ierīces
|
4.1. |
Elektriskā plītiņa. |
|
4.2. |
Elektriskā mufeļkrāsns ar termostatu. |
|
4.3. |
Pārpelnošanai paredzēti kvarca, porcelāna vai platīna tīģeļi, kas ir vai nu taisnstūrveida (apmēram 60 × 40 × 25 mm), vai apaļi (diametrs 60 līdz 75 mm, augstums 20 līdz 40 mm). |
5. Procedūra
5.1. A metode
Paraugu pārpelno, izmantojot metodi, kas aprakstīta jēlpelnu noteikšanai. Var izmantot arī pelnus, kas iegūti no minētās analīzes.
Pelnus ievieto 250–400 ml vārglāzē, izmantojot 75 ml sālsskābes (3.1.). Lēni uzvāra un piecpadsmit minūtes lēni vāra. Silto šķīdumu izfiltrē caur bezpelnu filtrpapīru un atlikumu mazgā ar siltu ūdeni, līdz skābes reakcija vairs nav redzama. Filtru ar atlikumu izžāvē un pārpelno nosvērtajā tīģelī temperatūrā, kas nav zemāka par 550 oC un nav augstāka par 700 oC. Atdzesē eksikatorā un nosver.
5.2. B metode
5 g parauga ar precizitāti līdz mg iesver 250–400 ml vārglāzē. Secīgi pievieno 25 ml ūdens un 25 ml sālsskābes (3.1.), samaisa un gaida, līdz pārstāj izdalīties burbuļi. Pievieno vēl 50 ml sālsskābes (3.1.). Pagaida, līdz gāze pārstāj izdalīties, tad ievieto vārglāzi vāroša ūdens vannā un tur atstāj trīsdesmit minūtes vai ilgāk, ja vajadzīgs, lai rūpīgi hidrolizētu jebkuru tur iespējami klātesošu cieti. Siltu izfiltrē caur bezpelnu filtru un filtru izmazgā 50 ml silta ūdens (skatīt 7. novērojumu). Filtru ar atlikumu ievieto tīģelī pārpelnošanai, izžāvē un pārpelno temperatūrā, kas nav zemāka par 550 oC un nav augstāka par 700 oC. Pelnus ievieto 250–400 ml vārglāzē, izmantojot 75 ml sālsskābes (3.1.); turpina, kā aprakstīts 5.1. punkta otrajā daļā.
6. Rezultātu aprēķināšana
Aprēķina atlikuma masu, atņemot taras masu. Rezultātu izsaka procentos no parauga.
7. Novērojums
Ja atlikums filtrējas slikti, tad analīzi sāk vēlreiz, 50 ml sālsskābes (3.1.) vietā ņemot 50 ml 20 % trihloretiķskābes (3.2.) un filtru mazgājot ar siltu 1 % trihloretiķskābes šķīdumu (3.3.).
O. KARBONĀTU NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj lielākajā daļā barības noteikt karbonātu daudzumu, parasti izteiktu kā kalcija karbonāts.
Tomēr dažos gadījumos (piemēram, ar dzelzs karbonātu) jāizmanto īpaša metode.
2. Princips
Karbonātus sadala ar sālsskābi; izdalījušos oglekļa dioksīdu uztver kalibrētā mēģenē un tā tilpumu salīdzina ar oglekļa dioksīda tilpumu, kas izdalās, tādos pašos apstākļos sadaloties zināmam daudzumam kalcija karbonāta.
3. Reaģenti
|
3.1. |
Sālsskābe, blīvums 1,10 g/ml. |
|
3.2. |
Kalcija karbonāts. |
|
3.3. |
Sērskābe, apmēram 0,05 mol/l, iekrāsota ar metilsarkano. |
4. Ierīces
Šaiblera-Dītriha aparāts (skatīt diagrammu) vai tam līdzvērtīga iekārta.
5. Procedūra
Atkarībā no karbonātu satura paraugā analīzei ņemtā parauga iesvara lielums ir šāds:
|
— |
0,5 g produktiem, kas satur 50 līdz 100 % karbonātu, kas izteikti kā kalcija karbonāts, |
|
— |
1 g produktiem, kas satur 40 līdz 50 % karbonātu, kas izteikti kā kalcija karbonāts, |
|
— |
2 līdz 3 g pārējiem produktiem. |
Parauga iesvaru pārnes aparāta speciālajā kolbā (4), kas aprīkota ar nelielu neplīstoša materiāla mēģeni, kurā ir 10 ml sālsskābes (3.1.), un kolbu pievieno aparātam. Pagriež trīsceļu krānu (5) tā, lai mēģene (1) būtu savienota ar atmosfēru. Izmantojot pārvietojamu mēģeni (2), kas piepildīta ar iekrāsotu sērskābes šķīdumu (3.3.) un pievienota kalibrētajai mēģenei (1), šķidruma līmeni iestata līdz nulles atzīmei. Pagriež krānu (5), savienojot mēģenes (1 un 3), un pārbauda, vai līmenis ir pret nulles atzīmi.
Kolbu (4) pagāžot uz sāna, parauga devai lēni pārlej sālsskābi (3.1.). Nolaižot zemāk mēģeni (2), izlīdzina spiedienu. Kolbu (4) krata, līdz pilnībā pārstāj izdalīties oglekļa dioksīds.
Atjauno spiedienu, izlīdzinot šķidruma līmeni mēģenēs (1 un 2). Pēc dažām minūtēm, kad gāzes tilpums kļuvis nemainīgs, veic nolasījumu.
Tādos pašos apstākļos analizē 0,5 g kalcija karbonāta (3.2.) kontrolparaugu.
6. Rezultātu aprēķināšana
Kalcija karbonātā izteiktu karbonātu saturu aprēķina, izmantojot šādu formulu:

kur:
|
X |
= |
% (w/w) kalcija karbonātā izteiktu karbonātu paraugā, |
|
V |
= |
no parauga devas izdalītais CO2 tilpums ml, |
|
V1 |
= |
no 0,5 g CaCO3 izdalītais CO2 tilpums ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Novērojumi
|
7.1. |
Ja parauga iesvars ir lielāks par 2 g, tad vispirms kolbā (4) iepilda 15 ml destilēta ūdens un pirms testa sākšanas samaisa. Kontrolparauga analīzei ņem tādu pašu ūdens daudzumu. |
|
7.2. |
Ja izmantotajam aparātam ir no Šaiblera-Dītriha aparāta atšķirīgs tilpums, tad attiecīgi jāmaina parauga un kontrolei izmantotās vielas iesvars, kā arī jākoriģē rezultātu aprēķins.
|
P. KOPĒJĀ FOSFORA DAUDZUMA NOTEIKŠANA
FOTOMETRISKĀ METODE
1. Mērķis un darbības joma
Šī metode ļauj noteikt kopējo fosfora saturu barībā. Tā ir īpaši piemērota tādu produktu analīzei, kuros ir mazs fosfora daudzums. Dažos gadījumos (ja produktā ir daudz fosfora) var izmantot gravimetrijas metodi.
2. Princips
Paraugu mineralizē, izmantojot vai nu sauso dedzināšanu (organiskās barības gadījumā), vai skābes šķelšanu (kombinēto minerālu un šķidrās barības gadījumā), un ievieto skābes šķīdumā. Šķīdumu apstrādā ar molibdovanadāta reaģentu. Tādējādi iegūtā dzeltenā šķīduma optisko blīvumu mēra spektrofotometrā pie 430 nm.
3. Reaģenti
|
3.1. |
Kalcija karbonāts. |
|
3.2. |
Sālsskābe, ρ20 = 1,10 g/ml (apmēram 6 mol/l). |
|
3.3. |
Slāpekļskābe, ρ201,045 g/ml. |
|
3.4. |
Slāpekļskābe, ρ20 = 1,38 līdz 1,42 g/ml. |
|
3.5. |
Sērskābe, ρ20 = 1,84 g/ml. |
|
3.6. |
Molibdovanadāta reaģents: 200 ml amonija heptamolibdāta šķīduma (3.6.1.), 200 ml amonija monovanadāta šķīduma (3.6.2.) un 134 ml slāpekļskābes (3.4.) samaisa viena litra mērkolbā. Papildina līdz vajadzīgajam tilpumam ar ūdeni. |
|
3.6.1. |
Amonija heptamolibdāta šķīdums: izšķīdina karstā ūdenī 100 g amonija heptamolibdāta (NH4) 6Mo7O24·4H2O. Pievieno 10 ml amonjaka (blīvums 0,91 g/ml) un papildina līdz vienam litram ar ūdeni. |
|
3.6.2. |
Amonija monovanadāta šķīdums: 2,35 g amonija monovanadāta NH4VO3 izšķīdina 400 mililitros karsta ūdens. Pastāvīgi maisot, lēni pievieno 20 ml atšķaidītas slāpekļskābes (7 ml HNO3 (3.4.) + 13 ml H2O) un papildina līdz vienam litram ar ūdeni. |
|
3.7. |
1 mg fosfora uz ml standartšķīdums: ūdenī izšķīdina 4,387 g kālija dihidrogēnfosfāta KH2PO4. Papildina ar ūdeni līdz vienam litram. |
4. Ierīces
|
4.1. |
Kvarca, porcelāna vai platīna tīģeļi pārpelnošanai. |
|
4.2. |
Elektriskā mufeļkrāsns ar termostatu, kas iestatīts uz 550 oC. |
|
4.3. |
250 ml Kjeldāla kolba. |
|
4.4. |
Mērkolbas un precīzijas pipetes. |
|
4.5. |
Spektrofotometrs. |
|
4.6. |
Apmēram 16 mm diametra mēģenes ar aizbāžņiem un iedaļām līdz 14,5 mm; tilpums 25 līdz 30 ml. |
5. Procedūra
5.1. Šķīduma sagatavošana
Atkarībā no parauga īpašībām sagatavo šķīdumu, kā norādīts 5.1.1. vai 5.1.2. punktā.
5.1.1.
Ar precizitāti līdz 1 mg nosver 1 g vai vairāk parauga. Testa paraugu ievieto Kjeldāla kolbā, pievieno 20 ml sērskābes (3.5.), sakrata, lai vielu pilnībā piesūcinātu ar skābi un neļautu tai pielipt pie kolbas sienām, uzkarsē un uztur viršanas temperatūru 10 minūtes. Nedaudz atdzesē, pievieno 2 ml slāpekļskābes (3.4.), lēnām uzkarsē, nedaudz atdzesē, pievieno vēl nedaudz slāpekļskābes (3.4.) un atjauno viršanas temperatūru. Procedūru atkārto, līdz iegūst bezkrāsainu šķīdumu. Atdzesē, pievieno mazliet ūdens, dekantē šķidrumu 500 ml mērkolbā, izskalojot Kjeldāla kolbu ar karstu ūdeni. Ļauj atdzist, papildina līdz vajadzīgajam tilpumam ar ūdeni, homogenizē un filtrē.
5.1.2.
Apmēram 2,5 g parauga ar precizitāti līdz 1 mg iesver tīģelī pārpelnošanai. Maisa testa paraugu, līdz tas pilnībā sajaucies ar 1 g kalcija karbonāta (3.1.). Pārpelno krāsnī 550 oC temperatūrā, līdz iegūst baltus vai pelēkus pelnus (var būt arī nedaudz ogļu). Pelnus pārnes 250 ml vārglāzē. Pievieno 20 ml ūdens un sālsskābes (3.2.), līdz pārstāj veidoties burbuļi. Pievieno vēl 10 ml sālsskābes (3.2.). Vārglāzi novieto smilšu vannā un tvaicē, līdz tā ir sausa, lai kvarcu padarītu nešķīstošu. Atlikumu atkārtoti šķīdina 10 mililitros slāpekļskābes (3.3.) un piecas minūtes vāra smilšu vannā vai uz elektriskās plītiņas netvaicējot, līdz tas ir sauss. Šķidrumu dekantē 500 ml mērkolbā, vairākas reizes izskalojot vārglāzi ar karstu ūdeni. Ļauj atdzist, papildina līdz vajadzīgajam tilpumam ar ūdeni, homogenizē un filtrē.
5.2. Krāsojuma attīstība un optiskā blīvuma mērījums
Saskaņā ar 5.1.1. vai 5.1.2. punktu iegūtā filtrāta alikvoto daļu atšķaida, lai iegūtu fosfora koncentrāciju, kas nepārsniedz 40 μg/ml. 10 ml šā šķīduma ielej mēģenē (4.6.) un pievieno 10 ml molibdovanadāta reaģenta (3.6.). Homogenizē un atstāj 20 oC temperatūrā vismaz 10 minūtes. Spektrofotometrā pie 430 nm izmēra optisko blīvumu pret šķīdumu, ko iegūst, pievienojot 10 mililitrus molibdovanadāta reaģenta (3.6.) 10 mililitriem ūdens.
5.3. Kalibrēšanas līkne
No standartšķīduma (3.7.) sagatavo šķīdumus, kas satur attiecīgi 5, 10, 20, 30 un 40 μg fosfora uz 1 ml. Ņem 10 ml no katra minētā šķīduma un pievieno 10 ml molibdovanadāta reaģenta (3.6.). Homogenizē un atstāj 20 oC temperatūrā vismaz 10 minūtes. Optisko blīvumu mēra tā, kā norādīts 5.2. punktā. Veido kalibrēšanas līkni, atzīmējot optiskos blīvumus un atbilstīgos fosfora daudzumus. Ja koncentrācija ir no 0 līdz 40 μg/ml, tad līkne būs lineāra.
6. Rezultātu aprēķināšana
Fosfora daudzumu testa paraugā nosaka, izmantojot kalibrēšanas līkni.
Rezultātu izsaka procentos no parauga.
Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nepārsniedz:
|
— |
3 % no lielākā rezultāta, ja fosfora saturs ir mazāks par 5 %, |
|
— |
0,15 % no absolūtās vērtības, ja fosfora saturs ir 5 % vai vairāk. |
Q. HLORĪDU HLORA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt hlora saturu ūdenī šķīstošos hlorīdos, ko parasti izsaka kā nātrija hlorīdu. To piemēro visai barībai.
2. Princips
Hlorīdus izšķīdina ūdenī. Ja produkts satur organisku vielu, tad to dzidrina. Šķīdumu nedaudz paskābina ar slāpekļskābi un hlorīdus ar sudraba nitrāta šķīdumu izgulsnē sudraba hlorīda veidā. Sudraba nitrāta pārpalikumu attitrē ar amonija tiocianāta šķīdumu, izmantojot Volharda metodi.
3. Reaģenti
|
3.1. |
Amonija tiocianāta šķīdums 0,1 mol/l. |
|
3.2. |
Sudraba nitrāta šķīdums 0,1 mol/l. |
|
3.3. |
Piesātināts amonija dzelzs sulfāta šķīdums, (NH4)Fe(SO4)2. |
|
3.4. |
Slāpekļskābe, blīvums 1,38 g/ml. |
|
3.5. |
Dietilēteris. |
|
3.6. |
Acetons. |
|
3.7. |
Karesa I šķīdums: ūdenī izšķīdina 21,9 g cinka acetāta, Zn (CH3COO)2·2H2O, un 3 g ledus etiķskābes. Papildina ar ūdeni līdz 100 ml. |
|
3.8. |
Karesa II šķīdums: ūdenī izšķīdina 10,6 g kālija ferocianīda, K4Fe(CN)6·3H2O. Papildina ar ūdeni līdz 100 ml. |
|
3.9. |
Aktīvā ogle, kurā nav hlorīdu un kura tos neabsorbē. |
4. Ierīces
Rotācijas tipa maisītājs: aptuveni 35 līdz 40 apgr./min.
5. Procedūra
5.1. Šķīduma sagatavošana
Atkarībā no parauga īpašībām sagatavo šķīdumu, kā norādīts 5.1.1., 5.1.2. vai 5.1.3. punktā.
Vienlaikus veic tukšo analīzi, neizmantojot analizējamo paraugu.
5.1.1.
Ar precizitāti līdz mg iesver ne vairāk kā 10 g parauga, kurā ir ne vairāk par 3 g hlora hlorīdu veidā. Kopā ar 400 ml ūdens iepilda 500 ml tilpuma kolbā apmēram 20 oC temperatūrā. Trīsdesmit minūtes maisa maisītājā, papildina līdz vajadzīgajam tilpumam, homogenizē un filtrē.
5.1.2.
Ar precizitāti līdz mg iesver apmēram 5 g parauga, ko kopā ar 1 g aktīvās ogles ievieto 500 ml tilpuma kolbā. Pievieno 400 ml apmēram 20 oC ūdens un 5 ml Karesa I šķīduma (3.7.), maisa 30 sekundes, tad pievieno 5 ml Karesa II šķīduma (3.8.). Trīsdesmit minūtes maisa maisītājā, papildina līdz vajadzīgajam tilpumam, homogenizē un filtrē.
5.1.3.
Sagatavo šķīdumu, kā aprakstīts 5.1.2. punktā, bet nefiltrē. Dekantē (vajadzības gadījumā centrifugē), atdala 100 ml no šķīduma augšējā slāņa un pārnes 200 ml mērkolbā. Samaisa ar acetonu (3.6.) un ar šo šķīdinātāju papildina līdz vajadzīgajam tilpumam, homogenizē un filtrē.
5.2. Titrēšana
Ar pipeti Erlenmeijera kolbā pārnes 25 ml līdz 100 ml filtrāta (atkarībā no sagaidāmā hlorīdu satura), ko iegūst pēc apraksta 5.1.1., 5.1.2. vai 5.1.3. punktā. Alikvotā daļa nedrīkst saturēt vairāk par 150 mg hlora (Cl). Vajadzības gadījumā atšķaida līdz vismaz 50 ml tilpumam ar ūdeni, pievieno 5 ml slāpekļskābes (3.4.), 20 ml piesātināta amonija dzelzs sulfāta šķīduma (3.3.) un divus pilienus amonija tiocianāta šķīduma (3.1.), ko ņem ar bireti, kas uzpildīta līdz nulles atzīmei. Ar bireti pārnes sudraba nitrāta šķīdumu (3.2.) tā, lai tas būtu 5 ml pārākumā. Pievieno 5 ml dietilētera (3.5.) un stipri sakrata, lai koagulētu nogulsnes. Sudraba nitrāta pārākumu titrē ar amonija tiocianāta šķīdumu (3.1.), līdz veidojas sarkanbrūns krāsojums, kas saglabājas vienu minūti.
6. Rezultātu aprēķināšana
Hlora daudzumu (X), izteiktu % no nātrija hlorīda, aprēķina, izmantojot šādu formulu:
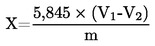
kur:
|
V1 |
= |
pievienotā 0,1 mol/l sudraba nitrāta šķīduma tilpums ml, |
|
V2 |
= |
titrēšanai izmantotais 0,1 mol/l amonija tiocianāta šķīduma tilpums ml, |
|
m |
= |
parauga svars. |
Ja tukšā analīze norāda, ka ir patērēts 0,1 mol/l sudraba nitrāta šķīdums, tad minēto lielumu atskaita no tilpuma (V1 - V2).
7. Novērojumi
|
7.1. |
Titrēšanu var veikt, arī izmantojot potenciometriju. |
|
7.2. |
Analizējot produktus ar ļoti lielu eļļu un tauku saturu, tos vispirms attauko ar dietilēteri vai petrolēteri. |
|
7.3. |
Zivju miltus var titrēt, izmantojot Mora metodi. |
(1) Labības, miltu, putraimu un rupja maluma miltu kaltēšanai krāsns termiskajai jaudai jābūt tādai, lai, kad tā iestatīta uz 131 oC, tā atgrieztos minētajā temperatūrā mazāk nekā 45 minūtēs pēc tam, kad tajā vienlaikus kaltēšanai ir salikts maksimāli iespējamais testa paraugu skaits. Ventilēšanai jābūt tādai, lai tad, kad krāsnī divas un četras stundas ir kaltēts maksimāli iespējamais kviešu paraugu daudzums, abi rezultāti atšķirtos mazāk nekā par 0,15 %.
(2) Ja eļļai vai taukiem jāveic turpmākas kvalitātes kontroles, tad pumeka gabaliņus aizstāj ar stikla lodītēm.
IV PIELIKUMS
ANALĪZES METODES, LAI KONTROLĒTU ATĻAUTO PIEDEVU DAUDZUMU BARĪBĀ
A. A VITAMĪNA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt A vitamīna (retinola) daudzumu barībā un premiksos. Ar A vitamīnu saprot all-trans-retinilspirtu un tā cis-izomērus, ko nosaka ar šo metodi. A vitamīna saturu izsaka starptautiskajās vienībās (SV) uz kg. Viena SV atbilst 0,300 μg all-trans-A vitamīna spirta vai 0,344 μg all-trans-A vitamīna acetāta, vai 0,550 μg all-trans-A vitamīna palmitāta aktivitātei.
Kvantitatīvās noteikšanas robeža ir 2 000 SV A vitamīna/kg.
2. Princips
Paraugu hidrolizē kālija hidroksīda šķīdumā etanolā, un A vitamīnu ekstrahē petrolēterī. Šķīdinātāju atdala iztvaicējot, atlikumu izšķīdina metanolā un vajadzības gadījumā atšķaida līdz nepieciešamajai koncentrācijai. A vitamīna saturu nosaka ar apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfiju (RP-HPLC), izmantojot UV vai fluorescences detektoru. Hromatogrāfijas parametrus izvēlas tā, lai all-trans-A vitamīna spirts neatdalītos no tā cis-izomēriem.
3. Reaģenti
|
3.1. |
Etanols, σ = 96 %. |
|
3.2. |
Petrolēteris, viršanas diapazons 40–60 oC. |
|
3.3. |
Metanols. |
|
3.4. |
Kālija hidroksīda šķīdums, c = 50 g/100 ml. |
|
3.5. |
Nātrija askorbāta šķīdums, c = 10 g/100 ml (skatīt 7.7. novērojumu). |
|
3.6. |
Nātrija sulfīds, Na2S · x H2O (x = 7–9). |
|
3.6.1. |
Nātrija sulfīda šķīdums, c = 0,5 mol/l glicerīnā, ß = 120 g/l, (ja x = 9) (skatīt 7.8. novērojumu). |
|
3.7. |
Fenolftaleīna šķīdums, c = 2 g/100 ml etanolā (3.1.). |
|
3.8. |
2-propanols. |
|
3.9. |
Kustīgā fāze priekš HPLC: metanola (3.3.) un ūdens maisījums, piemēram, 980 + 20 (v + v). Precīzu attiecību nosaka izmantotās kolonnas raksturlielumi. |
|
3.10. |
Slāpeklis, bez skābekļa. |
|
3.11. |
Īpaši tīrs all-trans-A vitamīna acetāts ar sertificētu aktivitāti, piemēram, 2,80 × 106 SV/g. |
|
3.11.1. |
All-trans-A vitamīna acetāta izejas standartšķīdums: ar precizitāti līdz 0,1 mg 100 ml mērkolbā iesver 50 mg A vitamīna acetāta (3.11.). Izšķīdina 2-propanolā (3.8.) un papildina līdz zīmei ar to pašu šķīdinātāju. Šā šķīduma nominālā koncentrācija ir 1 400 SV A vitamīna/ml. Precīzais saturs jānosaka atbilstoši 5.6.3.1. punktam. |
|
3.12. |
Īpaši tīrs all-trans-A vitamīna palmitāts ar sertificētu aktivitāti, piemēram, 1,80 × 106 SV/g. |
|
3.12.1. |
All-trans-A vitamīna palmitāta izejas standartšķīdums: ar precizitāti līdz 0,1 mg 100 ml mērkolbā iesver 80 mg A vitamīna palmitāta (3.12.). Izšķīdina 2-propanolā (3.8.) un papildina līdz zīmei ar to pašu šķīdinātāju. Šā šķīduma nominālā koncentrācija ir 1 400 SV A vitamīna/ml. Precīzais saturs jānosaka atbilstoši 5.6.3.2. punktam. |
|
3.13. |
2,6-Di-tert-butil-4-metilfenols (BHT) (skatīt 7.5 novērojumu). |
4. Ierīces
|
4.1. |
Rotācijas vakuumiztvaicētājs. |
|
4.2. |
Brūnā stikla trauki. |
|
4.2.1. |
500 ml plakandibena vai koniskas kolbas ar pieslīpēta stikla uzmavu. |
|
4.2.2. |
10, 25, 100 un 500 ml šaurkakla mērkolbas ar pieslīpēta stikla aizbāžņiem. |
|
4.2.3. |
1 000 ml koniskas dalāmpiltuves ar pieslīpēta stikla aizbāžņiem. |
|
4.2.4. |
250 ml bumbierveida kolbas ar pieslīpēta stikla uzmavām. |
|
4.3. |
Alīna kondensators, apvalka garums 300 mm, ar pieslīpēta stikla savienojumu un adapteri gāzu padeves caurulei. |
|
4.4. |
185 mm diametra kroku filtrs fāzu atdalīšanai (piemēram, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
HPLC iekārta ar injekcijas sistēmu. |
|
4.5.1. |
Šķidrumu hromatogrāfa kolonna, 250 × 4 mm, C18, 5 vai 10 μm iesaiņojums, vai līdzvērtīga (izpildes kritērijs: viens maksimums visiem retinola izomēriem HPLC apstākļos). |
|
4.5.2. |
UV vai fluorescences detektors ar regulējamu viļņu garumu |
|
4.6. |
Spektrofotometrs ar 10 mm kvarca šūnām. |
|
4.7. |
Ūdens vanna ar magnētisko maisītāju. |
|
4.8. |
Ekstrakcijas aparāts (skatīt 1. attēlu), kurā ietilpst: |
|
4.8.1. |
viena litra tilpuma stikla cilindrs ar pieslīpētu kaklu un aizbāzni; |
|
4.8.2. |
pieslīpēta stikla ieliktnis, kas aprīkots ar sānu nozarojumu un regulējamu cauruli, kura iet caur centru. Regulējamajai caurulei ir U veida apakšējais gals un sprausla pretējā galā, lai virsējo šķidruma slāni cilindrā varētu novadīt uz dalāmpiltuvi. |
5. Procedūra
|
Piezīme. |
A vitamīns ir jutīgs pret (UV) gaismu un oksidāciju. Visas darbības veic, izvairoties no gaismas klātbūtnes (izmantojot brūna stikla traukus vai stikla traukus, kas ir aizsargāti ar alumīnija foliju), kā arī no skābekļa klātbūtnes (noskalo ar slāpekli). Ekstrakcijas laikā gaisu virs šķidruma aizstāj ar slāpekli (periodiski atbrīvojot aizbāzni, lai izvairītos no pārmērīga spiediena). |
5.1. Parauga sagatavošana
Paraugu samaļ tā, lai tas izietu caur sietu ar 1 mm acojumu un lai malšanas laikā nerastos siltums. Paraugs jāsamaļ tieši pirms svēršanas un pārziepjošanas, lai novērstu A vitamīna zudumus.
5.2. Pārziepjošana
Atkarībā no A vitamīna satura svara 500 ml plakandibena vai koniskā kolbā (4.2.1.) ar precizitāti līdz 1 mg iesver 2 g līdz 25 g parauga. Svārstot pakāpeniski pievieno 130 ml etanola (3.1.), apmēram 100 mg BHT (3.13.), 2 ml nātrija askorbāta šķīduma (3.5.) un 2 ml nātrija sulfīda šķīduma (3.6.). Kolbai pievieno kondensatoru (4.3.) un kolbu iegremdē ūdens vannā ar magnētisko maisītāju (4.7.). Uzvāra un ļauj notikt attecei piecas minūtes. Tad caur kondensatoru (4.3.) pievieno 25 ml kālija hidroksīda šķīduma (3.4.) un, maisot lēnā slāpekļa plūsmā, ļauj notikt attecei vēl 25 minūtes. Tad noskalo kondensatoru apmēram ar 20 ml ūdens un atdzesē kolbas saturu līdz istabas temperatūrai.
5.3. Ekstrakcija
Pārziepjoto šķīdumu dekantējot kvantitatīvi pārnes uz 1 000 ml dalāmpiltuvi (4.2.3.) vai ekstrakcijas aparātu (4.8.), noskalojot kopumā ar 250 ml ūdens. Pārziepjošanas kolbu secīgi noskalo ar 25 ml etanola (3.1.) un 100 ml petrolētera (3.2.) un noskalojumus pārnes uz dalāmpiltuvi vai ekstrakcijas iekārtu. Ūdens un etanola attiecība kombinētajos šķīdumos ir apmēram 2:1. Kārtīgi sakrata divas minūtes un ļauj nogulsnēties divas minūtes.
5.3.1.
Kad slāņi ir atdalījušies (skatīt 7.3. novērojumu), petrolētera slāni pārnes uz citu dalāmpiltuvi (4.2.3.). Tādu ekstrakciju divas reizes atkārto ar 100 ml petrolētera (3.2.) un divas reizes – ar 50 ml petrolētera (3.2.).
Kombinētos ekstraktus divas reizes mazgā dalāmpiltuvē ar 100 ml ūdens devām, viegli svārstot, lai neveidotos emulsijas, tad, atkārtoti kratot, ar jaunām 100 ml ūdens devām, līdz ūdens, pievienojot fenolftaleīna šķīdumu (3.7.), kļūst bezkrāsains (parasti pietiek ar četrām mazgāšanas reizēm). Lai atdalītu suspendētu ūdeni, mazgāto ekstraktu caur sausu kroku filtru fāzu atdalīšanai (4.4.) filtrē 500 ml mērkolbā (4.2.2.). Dalāmpiltuvi un filtru noskalo ar 50 ml petrolētera (3.2.), papildina līdz zīmei ar petrolēteri (3.2.) un rūpīgi samaisa.
5.3.2.
Kad slāņi ir atdalījušies (skatīt 7.3. novērojumu), stikla cilindra aizbāzni (4.8.1.) nomaina ar pieslīpēta stikla ieliktni (4.8.2.) un regulējamās caurules U veida apakšējo galu novieto tā, lai tas atrastos tieši virs slāņu saskares līmeņa. Ievadot slāpekli caur sānu nozarojumu, pārnes augšējo petrolētera slāni uz 1 000 ml dalāmpiltuvi (4.2.3.). Stikla cilindrā ievieto 100 ml petrolētera (3.2.), noslēdz ar aizbāzni un kārtīgi sakrata. Ļauj slāņiem atdalīties un pārnes augšējo slāni uz dalāmpiltuvi kā iepriekš. Atkārto ekstrakciju ar papildu 100 ml petrolētera (3.2.), tad divas reizes ar 50 ml petrolētera (3.2.) devām un pārnes petrolētera slāņus uz dalāmpiltuvi.
Mazgā kombinētos petrolētera ekstraktus, kā aprakstīts 5.3.1. punktā, un turpina rīkoties saskaņā ar minēto punktu.
5.4. Parauga šķīduma sagatavošana priekš HPLC
Alikvoto daļu petrolētera šķīduma (no 5.3.1. vai 5.3.2. punkta) ar pipeti pārnes 250 ml bumbierveida kolbā (4.2.4.). Rotācijas iztvaicētājā (4.1.) samazinātā spiedienā ūdens vannas temperatūrā, kas nepārsniedz 40 oC, iztvaicē šķīdinātāju, līdz tas ir gandrīz sauss. Atjauno atmosfēras spiedienu, pievienojot slāpekli (3.10.), un izņem kolbu no rotācijas iztvaicētāja. Atlikušo šķīdinātāju atdala ar slāpekļa (3.10.) plūsmu un tūlīt izšķīdina atlikumu noteiktā tilpumā (10–100 ml) metanola (3.3.) (A vitamīna koncentrācijai jābūt diapazonā no 5 SV/ml līdz 30 SV/ml).
5.5. Noteikšana ar HPLC
A vitamīnu atdala C18 apgrieztās fāzes kolonnā (4.5.1.) un koncentrāciju izmēra, izmantojot UV detektoru (325 nm) vai fluorescences detektoru (ierosa 325 nm, emisija 475 nm) (4.5.2.).
Iešpricē alikvoto daļu (piemēram, 20 μl) šķīduma metilspirtā, kas iegūts 5.4. punktā, un eluē ar kustīgo fāzi (3.9.). Aprēķina vairāku viena parauga šķīduma injekciju vidējo maksimālo augstumu (platību) un vairāku kalibrēšanas šķīdumu (5.6.2.) injekciju vidējos maksimālos augstumus (platības).
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Šķidrumu hromatogrāfa kolonna (4.5.1.): |
250 × 4 mm, C18, 5 vai 10 μm iesaiņojums, vai līdzvērtīga |
|
Kustīgā fāze (3.9.): |
metanola (3.3.) un ūdens maisījums, piemēram, 980 + 20 (v + v) |
|
Plūsmas ātrums: |
1–2 ml/min |
|
Detektors (4.5.2.): |
UV detektors (325 nm) vai fluorescences detektors (ierosa 325 nm/emisija 475 nm) |
5.6. Kalibrēšana
5.6.1.
20 ml A vitamīna acetāta izejas standartšķīduma (3.11.1.) vai 20 ml A vitamīna palmitāta izejas standartšķīduma (3.12.1.) ar pipeti pārnes 500 ml plakandibena vai koniskā kolbā (4.2.1.) un hidrolizē, kā norādīts 5.2. punktā, taču nepievienojot BHT. Tad ekstrahē ar petrolēteri (3.2.) saskaņā ar 5.3. punktu un papildina līdz 500 ml ar petrolēteri (3.2.). Rotācijas iztvaicētājā iztvaicē 100 ml tāda ekstrakta (skatīt 5.4.), līdz tas ir gandrīz sauss, ar slāpekļa plūsmu (3.10.) atdala atlikušo šķīdinātāju un atkārtoti izšķīdina atlikumu 10,0 mililitros metanola (3.3.). Šā šķīduma nominālā koncentrācija ir 560 SV A vitamīna/ml. Precīzais saturs jānosaka atbilstoši 5.6.3.3. punktam. Darba standartšķīdums jāsagatavo svaigs pirms izmantošanas.
2,0 ml tāda darba standartšķīduma ar pipeti pārnes 20 ml mērkolbā, papildina līdz zīmei ar metanolu (3.3.) un samaisa. Šā atšķaidītā darba standartšķīduma nominālā koncentrācija ir 56 SV A vitamīna/ml.
5.6.2.
1,0, 2,0, 5,0 un 10,0 ml atšķaidītā darba standartšķīduma ar pipeti pārnes 20 ml mērkolbās, papildina līdz zīmei ar metanolu (3.3.) un samaisa. Minēto šķīdumu nominālās koncentrācijas ir 2,8, 5,6, 14,0 un 28,0 SV A vitamīna/ml.
20 μl katra kalibrēšanas šķīduma iešpricē vairākas reizes un nosaka vidējos maksimālos augstumus (laukumus). Izmantojot vidējos maksimālos augstumus (laukumus), izveido kalibrēšanas grafiku, kas satur UV kontroles rezultātus (5.6.3.3.).
5.6.3.
5.6.3.1.
2,0 ml A vitamīna acetāta izejas standartšķīduma (3.11.1.) ar pipeti pārnes 50 ml mērkolbā (4.2.2.) un papildina līdz zīmei ar 2-propanolu (3.8.). Šā šķīduma nominālā koncentrācija ir 56 SV A vitamīna/ml. 3,0 ml šā atšķaidītā A vitamīna acetāta šķīduma ar pipeti pārnes 25 ml mērkolbā un papildina līdz zīmei ar 2-propanolu (3.8.). Šā šķīduma nominālā koncentrācija ir 6,72 SV A vitamīna/ml. Šā šķīduma UV spektru spektrofotometrā (4.6.) salīdzina ar 2-propanola (3.8.) spektru intervālā no 300 nm līdz 400 nm. Ekstinkcijas maksimumam jābūt robežās starp 325 nm un 327 nm.
A vitamīna satura aprēķināšana:
A vitamīna SV/ml = E326 × 19,0
( A vitamīna acetātam = 1 530 pie 326 nm 2-propanolā)
A vitamīna acetātam = 1 530 pie 326 nm 2-propanolā)
5.6.3.2.
2,0 ml A vitamīna palmitāta izejas standartšķīduma (3.12.1.) ar pipeti pārnes 50 ml mērkolbā (4.2.2.) un papildina līdz zīmei ar 2-propanolu (3.8.). Šā šķīduma nominālā koncentrācija ir 56 SV A vitamīna/ml. 3,0 ml šā atšķaidītā A vitamīna palmitāta šķīduma ar pipeti pārnes 25 ml mērkolbā un papildina līdz zīmei ar 2-propanolu (3.8.). Šā šķīduma nominālā koncentrācija ir 6,72 SV A vitamīna/ml. Šā šķīduma UV spektru spektrofotometrā (4.6.) salīdzina ar 2-propanola (3.8.) spektru intervālā no 300 nm līdz 400 nm. Ekstinkcijas maksimumam jābūt robežās starp 325 nm un 327 nm.
A vitamīna satura aprēķināšana:
A vitamīna SV/ml = E326 × 19,0
( A vitamīna palmitātam = 957 pie 326 nm 2-propanolā)
A vitamīna palmitātam = 957 pie 326 nm 2-propanolā)
5.6.3.3.
3,0 ml neatšķaidīta A vitamīna darba standartšķīduma, kas sagatavots saskaņā ar 5.6.1. punktu, ar pipeti pārnes 50 ml mērkolbā (4.2.2.) un papildina līdz zīmei ar 2-propanolu (3.8.). 5,0 ml šā šķīduma ar pipeti pārnes 25 ml mērkolbā un papildina līdz zīmei ar 2-propanolu (3.8.). Šā šķīduma nominālā koncentrācija ir 6,72 SV A vitamīna/ml. Šā šķīduma UV spektru spektrofotometrā (4.6.) salīdzina ar 2-propanola (3.8.) spektru intervālā no 300 nm līdz 400 nm. Ekstinkcijas maksimumam jābūt robežās starp 325 nm un 327 nm.
A vitamīna satura aprēķināšana:
A vitamīna SV/ml = E325 × 18,3
( A vitamīna spirtam = 1 821 pie 325 nm 2-propanolā)
A vitamīna spirtam = 1 821 pie 325 nm 2-propanolā)
6. Rezultātu aprēķināšana
Ņemot vērā parauga šķīduma A vitamīna maksimumu vidējo augstumu (laukumu), nosaka parauga šķīduma koncentrāciju SV/ml, izmantojot kalibrēšanas grafiku (5.6.2.).
A vitamīna saturu w (SV/kg) paraugā aprēķina, izmantojot šādu formulu:
 [SV/kg]
[SV/kg]
kur:
|
c |
= |
A vitamīna koncentrācija SV/ml parauga šķīdumā (5.4.), |
|
V1 |
= |
parauga šķīduma (5.4.) tilpums mililitros, |
|
V2 |
= |
5.4. punktā paņemtās alikvotās daļas tilpums ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Novērojumi
|
7.1. |
Paraugiem ar zemu A vitamīna koncentrāciju var būt lietderīgi apvienot divu pārziepjošanas reižu petrolētera ekstraktus (svērtais daudzums 25 g) vienā parauga šķīdumā HPLC noteikšanai. |
|
7.2. |
Analīzei paņemtā parauga svarā nav vairāk par 2 g tauku. |
|
7.3. |
Ja fāzes neatdalās, tad pievieno apmēram 10 ml etanola (3.1.), lai izjauktu emulsiju. |
|
7.4. |
Mencu aknu eļļai un citiem tīriem taukiem pārziepjošanas laiku pagarina līdz 45–60 minūtēm. |
|
7.5. |
BHT vietā var izmantot hidrohinonu. |
|
7.6. |
Izmantojot normālās fāzes kolonnu, var atdalīt retinola izomērus. Tomēr tādā gadījumā, lai veiktu aprēķinus, jāsummē visu cis- un trans-izomēru maksimumu augstumi (laukumi). |
|
7.7. |
Nātrija askorbāta šķīduma vietā var izmantot apmēram 150 mg askorbīnskābes. |
|
7.8. |
Nātrija sulfīda šķīduma vietā var izmantot apmēram 50 mg EDTA. |
|
7.9. |
Analizējot A vitamīnu piena aizstājējbarībā, īpaša uzmanība jāpievērš:
Lai pārbaudītu, vai izmantotā analīzes metode rada ticamus rezultātus par konkrēto matrici (piena aizstājējbarību), papildu analizējamam paraugam veic atgūstamības testu. Ja atgūstamības proporcija ir zemāka par 80 %, tad analīžu rezultāti jākoriģē, ņemot vērā atgūstamību. |
8. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 15 % attiecībā pret lielāko rezultātu.
9. Koppētījuma rezultāti (1)
|
|
Premikss |
Premiksa barība |
Minerālu koncentrāts |
Proteīnbarība |
Sivēnu barība |
|
L |
13 |
12 |
13 |
12 |
13 |
|
n |
48 |
45 |
47 |
46 |
49 |
|
vidējais [SV/kg] |
17,02 × 106 |
1,21 × 106 |
537 100 |
151 800 |
18 070 |
|
sr [SV/kg] |
0,51 × 106 |
0,039 × 106 |
22 080 |
12 280 |
682 |
|
r [SV/kg] |
1,43 × 106 |
0,109 × 106 |
61 824 |
34 384 |
1 910 |
|
CVr [ %] |
3,0 |
3,5 |
4,1 |
8,1 |
3,8 |
|
sR [SV/kg] |
1,36 × 106 |
0,069 × 106 |
46 300 |
23 060 |
3 614 |
|
R [SV/kg] |
3,81 × 106 |
0,193 × 106 |
129 640 |
64 568 |
10 119 |
|
CVR [ %] |
8,0 |
6,2 |
8,6 |
15 |
20 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
sR |
= |
atveidojamības standartnovirze, |
|
r |
= |
atkārtojamība, |
|
R |
= |
atveidojamība, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
CVR |
= |
atveidojamības variācijas koeficients. |

B. E VITAMĪNA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt E vitamīna daudzumu barībā un premiksos. E vitamīna saturu izsaka kā DL-α-tokoferola acetātu mg/kg. 1 mg DL-α-tokoferola acetāta atbilst 0,91 mg DL-α-tokoferola (E vitamīna).
Kvantitatīvās noteikšanas robeža ir 2 mg E vitamīna/kg. Minēto kvantitatīvās noteikšanas robežu var sasniegt tikai ar fluorescences detektoru. Ar UV detektoru kvantitatīvās noteikšanas robeža ir 10 mg/kg.
2. Princips
Paraugu hidrolizē kālija hidroksīda šķīdumā etanolā, un E vitamīnu ekstrahē petrolēterī. Šķīdinātāju atdala iztvaicējot, atlikumu izšķīdina metanolā un vajadzības gadījumā atšķaida līdz nepieciešamajai koncentrācijai. E vitamīna saturu nosaka ar apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfiju (RP-HPLC), izmantojot fluorescences vai UV detektoru.
3. Reaģenti
|
3.1. |
Etanols, σ = 96 %. |
|
3.2. |
Petrolēteris, viršanas diapazons 40–60 oC. |
|
3.3. |
Metanols. |
|
3.4. |
Kālija hidroksīda šķīdums, c = 50 g/100 ml. |
|
3.5. |
Nātrija askorbāta šķīdums, c = 10 g/100 ml (skatīt 7.7. novērojumu). |
|
3.6. |
Nātrija sulfīds, Na2S· x H2O (x = 7–9). |
|
3.6.1. |
Nātrija sulfīda šķīdums, c = 0,5 mol/l glicerīnā, β = 120 g/l, (ja x = 9) (skatīt 7.8. novērojumu). |
|
3.7. |
Fenolftaleīna šķīdums, c = 2 g/100 ml etanolā (3.1.). |
|
3.8. |
Kustīgā fāze priekš HPLC: metanola (3.3.) un ūdens maisījums, piemēram, 980 + 20 (v + v). Precīzu attiecību nosaka izmantotās kolonnas raksturlielumi. |
|
3.9. |
Slāpeklis, bez skābekļa. |
|
3.10. |
Īpaši tīrs DL-α-tokoferola acetāts ar sertificētu aktivitāti. |
|
3.10.1. |
DL-α-tokoferola acetāta izejas standartšķīdums: ar precizitāti līdz 0,1 mg 100 ml mērkolbā iesver 100 mg DL-α-tokoferola acetāta (3.10.). Izšķīdina etanolā (3.1.) un papildina līdz zīmei ar to pašu šķīdinātāju. 1 ml šā šķīduma satur 1 mg DL-α-tokoferola acetāta. (Par UV kontroli skatīt 5.6.1.3. punktu; par stabilizāciju skatīt 7.4. novērojumu.) |
|
3.11. |
Īpaši tīrs DL-α-tokoferols ar sertificētu aktivitāti. |
|
3.11.1. |
DL-α-tokoferola izejas standartšķīdums: ar precizitāti līdz 0,1 mg 100 ml mērkolbā iesver 100 mg DL-α-tokoferola (3.11.). Izšķīdina etanolā (3.1.) un papildina līdz zīmei ar to pašu šķīdinātāju. 1 ml šā šķīduma satur 1 mg DL-α-tokoferola. (Par UV kontroli skatīt 5.6.2.3. punktu; par stabilizāciju skatīt 7.4. novērojumu.) |
|
3.12. |
2,6-Di-tert-butil-4-metilfenols (BHT) (skatīt 7.5 novērojumu). |
4. Ierīces
|
4.1. |
Rotācijas iztvaicētājs. |
|
4.2. |
Brūnā stikla trauki. |
|
4.2.1. |
500 ml plakandibena vai koniskas kolbas ar pieslīpēta stikla uzmavu. |
|
4.2.2. |
10, 25, 100 un 500 ml šaurkakla mērkolbas ar pieslīpēta stikla aizbāžņiem. |
|
4.2.3. |
1 000 ml koniskas dalāmpiltuves ar pieslīpēta stikla aizbāžņiem. |
|
4.2.4. |
250 ml bumbierveida kolbas ar pieslīpēta stikla uzmavām. |
|
4.3. |
Alīna kondensators, apvalka garums 300 mm, ar pieslīpēta stikla savienojumu un adapteri gāzu padeves caurulei. |
|
4.4. |
185 mm diametra kroku filtrs fāzu atdalīšanai (piemēram, Schleicher & Schuell 597 HY 1/2). |
|
4.5. |
HPLC iekārta ar injekcijas sistēmu. |
|
4.5.1. |
Šķidrumu hromatogrāfa kolonna, 250 × 4 mm, C18, 5 vai 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.5.2. |
Fluorescences vai UV detektors ar regulējamu viļņu garumu. |
|
4.6. |
Spektrofotometrs ar 10 mm kvarca šūnām. |
|
4.7. |
Ūdens vanna ar magnētisko maisītāju. |
|
4.8. |
Ekstrakcijas aparāts (skatīt 1. attēlu), kurā ietilpst: |
|
4.8.1. |
viena litra tilpuma stikla cilindrs ar pieslīpētu kaklu un aizbāzni; |
|
4.8.2. |
pieslīpēta stikla ieliktnis, kas aprīkots ar sānu nozarojumu un regulējamu cauruli, kura iet caur centru. Regulējamajai caurulei ir U veida apakšējais gals un sprausla pretējā galā, lai virsējo šķidruma slāni cilindrā varētu novadīt uz dalāmpiltuvi. |
5. Procedūra
|
Piezīme. |
E vitamīns ir jutīgs pret (UV) gaismu un oksidāciju. Visas darbības veic, izvairoties no gaismas klātbūtnes (izmantojot brūna stikla traukus vai stikla traukus, kas ir aizsargāti ar alumīnija foliju), kā arī no skābekļa klātbūtnes (noskalo ar slāpekli). Ekstrakcijas laikā gaisu virs šķidruma aizstāj ar slāpekli (periodiski atbrīvojot aizbāzni, lai izvairītos no pārmērīga spiediena). |
5.1. Parauga sagatavošana
Paraugu samaļ tā, lai tas izietu caur sietu ar 1 mm acojumu un lai malšanas laikā nerastos siltums. Paraugs jāsamaļ tieši pirms svēršanas un pārziepjošanas, lai novērstu E vitamīna zudumus.
5.2. Pārziepjošana
Atkarībā no E vitamīna satura svara 500 ml plakandibena vai koniskā kolbā (4.2.1.) ar precizitāti līdz 0,01 g iesver 2 g līdz 25 g parauga. Svārstot pakāpeniski pievieno 130 ml etanola (3.1.), apmēram 100 mg BHT (3.12.), 2 ml nātrija askorbāta šķīduma (3.5.) un 2 ml nātrija sulfīda šķīduma (3.6.). Kolbai pievieno kondensatoru (4.3.) un kolbu iegremdē ūdens vannā ar magnētisko maisītāju (4.7.). Uzvāra un ļauj notikt attecei piecas minūtes. Tad caur kondensatoru (4.3.) pievieno 25 ml kālija hidroksīda šķīduma (3.4.) un maisot lēnā slāpekļa plūsmā ļauj notikt attecei vēl 25 minūtes. Tad noskalo kondensatoru apmēram ar 20 ml ūdens un atdzesē kolbas saturu līdz istabas temperatūrai.
5.3. Ekstrakcija
Pārziepjoto šķīdumu dekantējot kvantitatīvi pārnes uz 1 000 ml dalāmpiltuvi (4.2.3.) vai ekstrakcijas aparātu (4.8.), noskalojot kopumā ar 250 ml ūdens. Pārziepjošanas kolbu secīgi noskalo ar 25 ml etanola (3.1.) un 100 ml petrolētera (3.2.) un noskalojumus pārnes uz dalāmpiltuvi vai ekstrakcijas iekārtu. Ūdens un etanola attiecība kombinētajos šķīdumos ir apmēram 2:1. Kārtīgi sakrata divas minūtes un ļauj nogulsnēties divas minūtes.
5.3.1.
Kad slāņi ir atdalījušies (skatīt 7.3. novērojumu), petrolētera slāni pārnes uz citu dalāmpiltuvi (4.2.3.). Tādu ekstrakciju divas reizes atkārto ar 100 ml petrolētera (3.2.) un divas reizes – ar 50 ml petrolētera (3.2.).
Kombinētos ekstraktus divas reizes mazgā dalāmpiltuvē ar 100 ml ūdens devām, viegli svārstot, lai neveidotos emulsijas, tad, atkārtoti kratot, ar jaunām 100 ml ūdens devām, līdz ūdens, pievienojot fenolftaleīna šķīdumu (3.7.), kļūst bezkrāsains (parasti pietiek ar četrām mazgāšanas reizēm). Lai atdalītu suspendētu ūdeni, mazgāto ekstraktu caur sausu kroku filtru fāzu atdalīšanai (4.4.) filtrē 500 ml mērkolbā (4.2.2.). Dalāmpiltuvi un filtru noskalo ar 50 ml petrolētera (3.2.), papildina līdz zīmei ar petrolēteri (3.2.) un rūpīgi samaisa.
5.3.2.
Kad slāņi ir atdalījušies (skatīt 7.3. novērojumu), stikla cilindra aizbāzni (4.8.1.) nomaina ar pieslīpēta stikla ieliktni (4.8.2.) un regulējamās caurules U veida apakšējo galu novieto tā, lai tas atrastos tieši virs slāņu saskares līmeņa. Ievadot slāpekli caur sānu nozarojumu, pārnes augšējo petrolētera slāni uz 1 000 ml dalāmpiltuvi (4.2.3.). Stikla cilindrā ievieto 100 ml petrolētera (3.2.), noslēdz ar aizbāzni un kārtīgi sakrata. Ļauj slāņiem atdalīties un pārnes augšējo slāni uz dalāmpiltuvi kā iepriekš. Atkārto ekstrakciju ar papildu 100 ml petrolētera (3.2.), tad divas reizes ar 50 ml petrolētera (3.2.) devām un pārnes petrolētera slāņus uz dalāmpiltuvi.
Mazgā kombinētos petrolētera ekstraktus, kā aprakstīts 5.3.1. punktā, un turpina rīkoties saskaņā ar minēto punktu.
5.4. Parauga šķīduma sagatavošana priekš HPLC
Alikvoto daļu petrolētera šķīduma (no 5.3.1. vai 5.3.2. punkta) ar pipeti pārnes 250 ml bumbierveida kolbā (4.2.4.). Rotācijas iztvaicētājā (4.1.) samazinātā spiedienā ūdens vannas temperatūrā, kas nepārsniedz 40 oC, iztvaicē šķīdinātāju, līdz tas ir gandrīz sauss. Atjauno atmosfēras spiedienu, pievienojot slāpekli (3.9.), un izņem kolbu no rotācijas iztvaicētāja. Atlikušo šķīdinātāju atdala ar slāpekļa plūsmu (3.9.) un tūlīt izšķīdina atlikumu noteiktā tilpumā (10–100 ml) metanola (3.3.) (DL-α-tokoferola koncentrācijai jābūt diapazonā no 5 μg/ml līdz 30 μg/ml).
5.5. Noteikšana ar HPLC
E vitamīnu atdala C18 apgrieztās fāzes kolonnā (4.5.1.) un koncentrāciju izmēra, izmantojot fluorescences detektoru (ierosa 295 nm, emisija 330 nm) vai UV detektoru (292 nm) (4.5.2.).
Iešpricē alikvoto daļu (piemēram, 20 μl) šķīduma metilspirtā, kas iegūts 5.4. punktā, un eluē ar kustīgo fāzi (3.8.). Aprēķina vairāku viena parauga šķīduma injekciju vidējos maksimālos augstumus (platības) un vairāku kalibrēšanas šķīdumu injekciju vidējos maksimālos augstumus (platības) (5.6.2.).
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Šķidrumu hromatogrāfa kolonna (4.5.1.): |
250 × 4 mm, C18, 5 vai 10 μm iesaiņojums, vai līdzvērtīga |
|
Kustīgā fāze (3.8.): |
metanola (3.3.) un ūdens maisījums, piemēram, 980 + 20 (v + v) |
|
Plūsmas ātrums: |
1–2 ml/min |
|
Detektors (4.5.2.): |
fluorescences detektors (ierosa 295 nm/ emisija 330 nm) vai UV detektors (292 nm) |
5.6. Kalibrēšana (DL-α-tokoferola acetāts vai DL-α-tokoferols)
5.6.1.
5.6.1.1.
25 ml DL-α-tokoferola acetāta izejas standartšķīduma (3.10.1.) ar pipeti pārnes 500 ml plakandibena vai koniskā kolbā (4.2.1.) un hidrolizē, kā aprakstīts 5.2. punktā. Tad ekstrahē ar petrolēteri (3.2.) saskaņā ar 5.3. punktu un papildina līdz 500 ml ar petrolēteri (3.2.). Rotācijas iztvaicētājā iztvaicē 25 ml šā ekstrakta (skatīt 5.4. punktu), līdz tas ir gandrīz sauss, ar slāpekļa plūsmu (3.9.) atdala atlikušo šķīdinātāju un atkārtoti izšķīdina atlikumu 25,0 mililitros metanola (3.3.). Šā šķīduma nominālā koncentrācija ir 45,5 μg DL-α-tokoferola/ml, kas atbilst 50 μg DL-α-tokoferola acetāta/ml. Darba standartšķīdums jāsagatavo svaigs pirms izmantošanas.
5.6.1.2.
1,0, 2,0, 4,0 un 10,0 ml darba standartšķīduma pārnes 20 ml mērkolbās, papildina līdz zīmei ar metanolu (3.3.) un samaisa. Minēto šķīdumu nominālās koncentrācijas ir 2,5, 5,0, 10,0 un 25,0 μg/ml DL-α-tokoferola acetāta, t. i., 2,28, 4,55, 9,10 μg/ml un 22,8 μg/ml DL-α-tokoferola.
20 μl katra kalibrēšanas šķīduma iešpricē vairākas reizes un nosaka vidējos maksimālos augstumus (laukumus). Izmantojot vidējos maksimālos augstumus (laukumus), veido kalibrēšanas grafiku.
5.6.1.3.
5,0 ml DL-α-tokoferola acetāta izejas standartšķīduma (3.10.1.) atšķaida ar etanolu līdz 25,0 ml un šā šķīduma UV spektru spektrofotometrā (4.6.) salīdzina ar etanola (3.1.) spektru intervālā no 250 nm līdz 320 nm.
Absorbcijas maksimums ir 284 nm:
 = 43,6 284 nm etanolā
= 43,6 284 nm etanolā
Tādā atšķaidījumā jāiegūst ekstinkcijas vērtība no 0,84 līdz 0,88.
5.6.2.
5.6.2.1.
Ar pipeti pārnes 2 ml DL-α-tokoferola izejas standartšķīduma (3.11.1.) 50 ml mērkolbā, izšķīdina metanolā (3.3.) un papildina līdz zīmei ar metanolu. Šā šķīduma nominālā koncentrācija ir 40 μg DL-α-tokoferola/ml, kas atbilst 44,0 μg DL-α-tokoferola acetāta/ml. Darba standartšķīdums jāsagatavo svaigs pirms izmantošanas.
5.6.2.2.
1,0, 2,0, 4,0 un 10,0 ml darba standartšķīduma pārnes 20 ml mērkolbās, papildina līdz zīmei ar metanolu (3.3.) un samaisa. Minēto šķīdumu nominālās koncentrācijas ir 2,0, 4,0, 8,0 un 20,0 μg/ml DL-α-tokoferola, t. i., 2,20, 4,40, 8,79 μg/ml un 22,0 μg/ml DL-α-tokoferola acetāta.
20 μl katra kalibrēšanas šķīduma iešpricē vairākas reizes un nosaka vidējos maksimālos augstumus (laukumus). Izmantojot vidējos maksimālos augstumus (laukumus), veido kalibrēšanas grafiku.
5.6.2.3.
2,0 ml DL-α-tokoferola izejas standartšķīduma (3.11.1.) atšķaida ar etanolu līdz 25,0 ml un šā šķīduma UV spektru spektrofotometrā (4.6.) salīdzina ar etanola spektru (3.1.) intervālā no 250 nm līdz 320 nm. Absorbcijas maksimums ir 292 nm:
 = 75,8 292 nm etanolā
= 75,8 292 nm etanolā
Tādā atšķaidījumā jāiegūst ekstinkcijas vērtība 0,6.
6. Rezultātu aprēķināšana
Ņemot vērā parauga šķīduma E vitamīna maksimumu vidējo augstumu (laukumu), nosaka parauga šķīduma koncentrāciju μg/ml (aprēķinātu kā α-tokoferola acetātu), izmantojot kalibrēšanas grafiku (5.6.1.2 vai 5.6.2.2.).
E vitamīna saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg]
[mg/kg]
kur:
|
c |
= |
E vitamīna koncentrācija (kā α-tokoferola acetāts) parauga šķīdumā (5.4.) μg/ml, |
|
V1 |
= |
parauga šķīduma (5.4.) tilpums mililitros, |
|
V2 |
= |
5.4. punktā paņemtās alikvotās daļas tilpums ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Novērojumi
|
7.1. |
Paraugiem ar zemu E vitamīna koncentrāciju var būt lietderīgi apvienot divu pārziepjošanas reižu petrolētera ekstraktus (svērtais daudzums 25 g) vienā parauga šķīdumā HPLC noteikšanai. |
|
7.2. |
Analīzei paņemtā parauga svarā nav vairāk par 2 g tauku. |
|
7.3. |
Ja fāzes neatdalās, tad pievieno apmēram 10 ml etanola (3.1.), lai izjauktu emulsiju. |
|
7.4. |
Kad ir veikti DL-α-tokoferola acetāta vai DL-α-tokoferola šķīduma spektrofotometriskie mērījumi (saskaņā ar attiecīgi 5.6.1.3. vai 5.6.2.3. punktu), šķīdumam (3.10.1. vai 3.10.2.) pievieno apmēram 10 mg BHT (3.12.) un tur šķīdumu ledusskapī (ne ilgāk par četrām nedēļām). |
|
7.5. |
BHT vietā var izmantot hidrohinonu. |
|
7.6. |
Izmantojot normālās fāzes kolonnu, var atdalīt α-, β-, γ- un δ-tokoferolu. |
|
7.7. |
Nātrija askorbāta šķīduma vietā var izmantot apmēram 150 mg askorbīnskābes. |
|
7.8. |
Nātrija sulfīda šķīduma vietā var izmantot apmēram 50 mg EDTA. |
|
7.9. |
E vitamīna acetāts sārmainos apstākļos hidrolizējas ļoti ātri, tāpēc ir ļoti jutīgs pret oksidāciju, jo īpaši tādu mikroelementu klātbūtnē kā dzelzs vai varš. Ja E vitamīnu nosaka premiksos, kuros vitamīna daudzums pārsniedz 5 000 mg/kg, tad rezultātā E vitamīns var noārdīties. Tāpēc apstiprinājumam jāiesaka HPLC metode, kurā ietilpst E vitamīna preparāta enzimātiskā šķelšana, neveicot sārmainās pārziepjošanas posmu. |
8. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 15 % no lielākā rezultāta.
9. Koppētījuma rezultāti (2)
|
|
Premikss |
Premiksa barība |
Minerālu koncentrāts |
Proteīnbarība |
Sivēnu barība |
|
L |
12 |
12 |
12 |
12 |
12 |
|
n |
48 |
48 |
48 |
48 |
48 |
|
vidējais [mg/kg] |
17 380 |
1 187 |
926 |
315 |
61,3 |
|
sr [mg/kg] |
384 |
45,3 |
25,2 |
13,0 |
2,3 |
|
r [mg/kg] |
1 075 |
126,8 |
70,6 |
36,4 |
6,4 |
|
CVr [ %] |
2,2 |
3,8 |
2,7 |
4,1 |
3,8 |
|
sR [mg/kg] |
830 |
65,0 |
55,5 |
18,9 |
7,8 |
|
R [mg/kg] |
2 324 |
182,0 |
155,4 |
52,9 |
21,8 |
|
CVR [ %] |
4,8 |
5,5 |
6,0 |
6,0 |
12,7 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
sR |
= |
atveidojamības standartnovirze, |
|
r |
= |
atkārtojamība, |
|
R |
= |
atveidojamība, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
CVR |
= |
atveidojamības variācijas koeficients. |

C. MIKROELEMENTU – DZELZS, VARA, MANGĀNA UN CINKA – NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt barībā mikroelementus – dzelzi, varu, mangānu un cinku. Kvantitatīvās noteikšanas robežas ir šādas:
|
— |
dzelzs (Fe): 20 mg/kg, |
|
— |
varš (Cu): 10 mg/kg, |
|
— |
mangāns (Mn): 20 mg/kg, |
|
— |
cinks (Zn): 20 mg/kg. |
2. Princips
Pēc organisko vielu noārdīšanās, ja tādas ir, paraugu izšķīdina sālsskābē. Dzelzi, varu, mangānu un cinku pēc attiecīgas atšķaidīšanas nosaka atomu absorbcijas spektrometrijā.
3. Reaģenti
Ievadpiezīmes
Reaģentu un analītisko šķīdumu gatavošanā lieto dejonizētu ūdeni, ko iegūst, divreiz destilējot ūdeni borsilikātstikla destilācijas iekārtā vai kvarca destilācijas iekārtā vai divreiz apstrādājot ar jonu apmaiņas sveķiem.
Reaģentiem jābūt vismaz analītiski tīriem. Tas, ka nosakāmā elementa nav, jāpārbauda tukšajā eksperimentā. Vajadzības gadījumā reaģenti vēl jāattīra.
Turpmāk aprakstīto standartšķīdumu vietā var izmantot komerciālus standartšķīdumus, ja tiem ir garantija un tie pirms izmantošanas ir pārbaudīti.
|
3.1. |
Sālsskābe (blīvums 1,19 g/ml). |
|
3.2. |
Sālsskābe (6 mol/l). |
|
3.3. |
Sālsskābe (0,5 mol/l). |
|
3.4. |
Fluorūdeņražskābe (38–40 % (v/v)), kurā dzelzs (Fe) saturs ir mazāks par 1 mg/litrā un atlikums pēc iztvaicēšanas ir mazāks par 10 mg (kā sulfāta)/litrā. |
|
3.5. |
Sērskābe (blīvums 1,84 g/ml). |
|
3.6. |
Ūdeņraža peroksīds (apmēram 100 skābekļa tilpumi (30 % no svara)). |
|
3.7. |
Šādi sagatavots standarta dzelzs šķīdums (1 000 μg Fe/ml) vai līdzvērtīgs tirdzniecībā pieejams šķīdums: izšķīdina 1 g dzelzs stieples 200 mililitros 6 mol/l sālsskābes (3.2.), pievieno 16 ml ūdeņraža peroksīda (3.6.) un papildina līdz vienam litram ar ūdeni. |
|
3.7.1. |
Darba standarta dzelzs šķīdums (100 μg Fe/ml), kas sagatavots, atšķaidot 1 daļu standarta šķīduma (3.7.) ar 9 daļām ūdens. |
|
3.8. |
Šādi sagatavots standarta vara šķīdums (1 000 μg Cu/ml) vai līdzvērtīgs tirdzniecībā pieejams šķīdums:
|
|
3.8.1. |
Darba standarta vara šķīdums (10 μg Cu/ml), kas sagatavots, atšķaidot 1 daļu standarta šķīduma (3.8.) ar 9 daļām ūdens un tad atšķaidot 1 daļu iegūtā šķīduma ar 9 daļām ūdens. |
|
3.9. |
Šādi sagatavots standarta mangāna šķīdums (1 000 μg Mn/ml) vai līdzvērtīgs tirdzniecībā pieejams šķīdums:
|
|
3.9.1. |
Darba standarta mangāna šķīdums (10 μg Mn/ml), kas sagatavots, atšķaidot 1 daļu standarta šķīduma (3.9.) ar 9 daļām ūdens un tad atšķaidot 1 daļu iegūtā šķīduma ar 9 daļām ūdens. |
|
3.10. |
Šādi sagatavots standarta cinka šķīdums (1 000 μg Zn/ml) vai līdzvērtīgs tirdzniecībā pieejams šķīdums:
|
|
3.10.1. |
Darba standarta cinka šķīdums (10 μg Zn/ml), kas sagatavots, atšķaidot 1 daļu standarta šķīduma (3.10.) ar 9 daļām ūdens un tad atšķaidot 1 daļu iegūtā šķīduma ar 9 daļām ūdens. |
|
3.11. |
Lantāna hlorīda šķīdums: izšķīdina 12 g lantāna oksīda 150 mililitros ūdens, pievieno 100 ml 6 mol/l sālsskābes (3.2.) un papildina līdz vienam litram ar ūdeni. |
4. Ierīces
|
4.1. |
Mufeļkrāsns ar regulējamu temperatūru un, vēlams, pašrakstītāju. |
|
4.2. |
Jālieto izturīgi borsilikātstikla trauki, un ieteicams lietot iekārtu, kas paredzēta tikai mikroelementu noteikšanai. |
|
4.3. |
Atomu absorbcijas spektrofotometrs, kas atbilst tām metodes prasībām, kuras attiecas uz jutību un precizitāti vajadzīgajā intervālā. |
5. Procedūra (3)
5.1. Paraugi, kas satur organisku vielu
5.1.1. (4)
|
5.1.1.1. |
Paraugu, kura svars ir 5–10 g un kas nosvērts ar 0,2 mg precizitāti, liek kvarca vai platīna tīģelī (skatīt b) piezīmi), žāvē žāvēšanas skapī 105 oC temperatūrā un liek tīģeli aukstajā mufeļkrāsnī (4.1.). Krāsni aizver (skatīt c) piezīmi) un apmēram 90 minūtēs pakāpeniski paaugstina temperatūru līdz 450–475 oC. Šo temperatūru uztur 4 līdz 16 stundas, piemēram, atstājot pa nakti, lai atdalītu oglekli saturošas vielas, tad atver krāsni un ļauj atdzist (skatīt d) piezīmi). Samitrina pelnus ar ūdeni un pārnes tos 250 ml tilpuma vārglāzē. Tīģeli izmazgā ar apmēram 5 ml sālsskābes (3.1.), ko lēni un uzmanīgi pievieno vārglāzes saturam (CO2 veidošanās var izraisīt spēcīgu reakciju). Sakratot pa pilienam pievieno sālsskābi (3.1.), līdz pārstāj veidoties burbuļi. Iztvaicē, līdz tas ir sauss, pa laikam apmaisot ar stikla stienīti. Tad atlikumam pievieno 15 ml 6 mol/l sālsskābes (3.2.) un pēc tam apmēram 120 ml ūdens. Maisa ar stikla stienīti, ko atstāj vārglāzē, un nosedz vārglāzi ar pulksteņstiklu. Lēni uzvāra un uztur viršanas temperatūrā, līdz vairs neredz, ka pelni izšķīstu. Filtrē uz bezpelnu filtrpapīra un filtrātu savāc 250 ml tilpuma kolbā. Izmazgā vārglāzi un filtru ar 5 ml karstas 6 mol/l sālsskābes (3.2.) un divreiz ar vārošu ūdeni. Uzpilda tilpuma kolbu līdz zīmei ar ūdeni (HCl koncentrācija apmēram 0,5 mol/l). |
|
5.1.1.2. |
Ja atlikums filtrā izskatās melns (ogleklis), tad to liek atpakaļ krāsnī un pārpelno vēlreiz 450–475 oC temperatūrā. Šī pārpelnošana, kurai ir vajadzīgas tikai dažas (apmēram trīs līdz piecas) stundas, ir pabeigta, kad pelni izskatās balti vai gandrīz balti. Izšķīdina atlikumu ar apmēram 2 ml sālsskābes (3.1.), iztvaicē, līdz tas ir sauss, un pievieno 5 ml 6 mol/l sālsskābes (3.2.). Sakarsē, šķīdumu filtrē un iepilda tilpuma kolbā, un papildina līdz zīmei ar ūdeni (HCl koncentrācija apmēram 0,5 mol/l). Piezīmes
|
5.1.2.
5.1.2.1.
Lai noteiktu katru elementu, no darba standartšķīdumiem, kas aprakstīti 3.7.1., 3.8.1., 3.9.1. un 3.10.1. punktā, pagatavo kalibrēšanas šķīdumus, kuru HCl koncentrācija ir 0,5 mol/l, un (dzelzij, mangānam un cinkam) lantāna hlorīda koncentrācija ir līdzvērtīga 0,1 % La (w/v).
Izvēlētajām mikroelementu koncentrācijām jāatrodas izmantotā spektrofotometra jutīguma intervālā. Tālāk tabulās ar piemēriem parādīts tipisku kalibrēšanas šķīdumu sastāvs; tomēr atkarībā no spektrofotometra jutības var būt jāizvēlas citas koncentrācijas.
Dzelzs
|
μg Fe/ml |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml darba standartšķīduma (3.7.1.) (1 ml = 100 μg Fe) |
0 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
ml HCl (3.2.) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantāna hlorīda šķīduma (3.11.) un papildina līdz 100 ml ar ūdeni |
|||||||
Varš
|
μg Cu/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml darba standartšķīduma (3.8.1.) (1 ml = 10 μg Cu) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2.) |
8 |
8 |
8 |
8 |
8 |
8 |
8 |
Mangāns
|
μg Mn/ml |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
ml darba standartšķīduma (3.9.1.) (1 ml = 10 μg Mn) |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
|
ml HCl (3.2.) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantāna hlorīda šķīduma (3.11.) un papildina līdz 100 ml ar ūdeni |
|||||||
Cinks
|
μg Zn/ml |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
ml darba standartšķīduma (3.10.1.) (1 ml = 10 μg Zn) |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
|
ml HCl (3.2.) |
7 |
7 |
7 |
7 |
7 |
7 |
7 |
|
+ 10 ml lantāna hlorīda šķīduma (3.11.) un papildina līdz 100 ml ar ūdeni |
|||||||
5.1.2.2.
Vara noteikšanai parasti var tieši izmantot šķīdumu, kas sagatavots saskaņā ar 5.1.1. punktu. Ja tā koncentrācija jānoregulē atbilstīgi kalibrēšanas šķīdumu intervālam, tad alikvoto daļu ar pipeti var iepilināt 100 ml tilpuma kolbā un papildināt līdz zīmei ar 0,5 mol/l sālsskābi (3.3.).
Dzelzs, mangāna un cinka noteikšanai saskaņā ar 5.1.1. punktu gatavotā šķīduma alikvoto daļu ar pipeti iepilina 100 ml tilpuma kolbā, pievieno 10 ml lantāna hlorīda šķīduma (3.11.) un papildina līdz zīmei ar 0,5 mol/l sālsskābi (3.3.) (skatīt arī 8. punktu “Novērojums”).
5.1.2.3.
Tukšajam eksperimentam jāietver visi iepriekš paredzētie procedūras posmi, izņemot to, ka nav parauga materiāla. Kalibrēšanas šķīdumu “0”nedrīkst lietot kā tukšo šķīdumu.
5.1.2.4.
Izmēra kalibrēšanas šķīdumu un analizējamā šķīduma atomu absorbciju, izmantojot oksidējošu gaisa-acetilēna liesmu šādos viļņu garumos:
|
|
Fe: 248,3 nm, |
|
|
Cu: 324,8 nm, |
|
|
Mn: 279,5 nm, |
|
|
Zn: 213,8 nm, |
Katru mērījumu veic četras reizes.
5.2. Minerālbarība
Ja paraugā nav organisko vielu, tad iepriekšēja pārpelnošana nav vajadzīga. Rīkojas, kā aprakstīts 5.1.1.1. punktā, sākot no otrās daļas. Iztvaicēšanu ar fluorūdeņražskābi var neveikt.
6. Rezultātu aprēķināšana
Izmantojot kalibrēšanas līkni, aprēķina mikroelementu koncentrāciju analizējamajā šķīdumā un rezultātu izsaka mikroelementa miligramos uz parauga kilogramu (ppm).
7. Atkārtojamība
Viena laboranta divās paralēlās noteikšanās no viena parauga iegūtu rezultātu starpība nepārsniedz:
|
— |
5 mg/kg no absolūtā lieluma, ja attiecīgā mikroelementa saturs ir līdz 50 mg/kg, |
|
— |
10 % no lielākā rezultāta, ja attiecīgā mikroelementa saturs ir no 50 līdz 100 mg/kg, |
|
— |
10 mg/kg no absolūtā lieluma, ja attiecīgā mikroelementa saturs ir no 100 līdz 200 mg/kg, |
|
— |
5 % no lielākā rezultāta, ja attiecīgā mikroelementa saturs pārsniedz 200 mg/kg. |
8. Novērojums
Ja barībā ir daudz fosfātu, tas var traucēt noteikt dzelzi, mangānu un cinku. Tādu traucējumu var koriģēt, pievienojot lantāna hlorīda šķīdumu (3.11.). Ja tomēr paraugā svara attiecība Ca + Mg/P ir > 2, tad lantāna hlorīda šķīdumu (3.11.) analizējamajam šķīdumam un kalibrēšanas šķīdumiem var nepievienot.
D. HALOFUGINONA NOTEIKŠANA
DL-trans-7-brom-6-hlor-3-[3-(3-hidroksi-2-piperidil)acetonil]-hinazolīn-4-(3H)-ona hidrogēnbromīds
1. Mērķis un darbības joma
Šī metode ļauj noteikt halofuginona daudzumu barībā. Kvantitatīvās noteikšanas robeža ir 1 mg/kg.
2. Princips
Pēc apstrādes ar karstu ūdeni halofuginonu kā brīvu bāzi ekstrahē etilacetātā un pēc tam atdala kā sālsskābes sāli skābes ūdens šķīdumā. Ekstraktu attīra, izmantojot jonu apmaiņas hromatogrāfiju. Halofuginona saturu nosaka apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Acetonitrils, ekvivalents HPLC tīrības pakāpei. |
|
3.2. |
Amberlīta XAD-2 sveķi. |
|
3.3. |
Amonija acetāts. |
|
3.4. |
Etilacetāts. |
|
3.5. |
Ledus etiķskābe. |
|
3.6. |
Halofuginona standartviela (DL-trans-7-brom-6-hlor-3-[3-(3-hidroksi-2-piperidil)acetonil]-hinazolīn-4-(3H)-ona hidrogēnbromīds, E 764). |
|
3.6.1. |
Ar 0,1 mg precizitāti 500 ml mērkolbā iesver 50 mg halofuginona (3.6.), izšķīdina amonija acetāta buferšķīdumā (3.18.), papildina līdz zīmei ar buferšķīdumu un samaisa. Tumšā vietā 5 oC temperatūrā tādu šķīdumu var glabāt trīs nedēļas. |
|
3.6.2. |
100 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 3,0, 4,0 un 6,0 ml izejas standartšķīduma (3.6.1.) Papildina līdz zīmei ar kustīgo fāzi (3.21.) un samaisa. Minēto šķīdumu koncentrācija ir attiecīgi 1,0, 2,0, 3,0, 4,0 un 6,0 μg/ml halofuginona. Minētie šķīdumi jāsagatavo svaigi pirms izmantošanas. |
|
3.7. |
Sālsskābe (ρ20 apmēram 1,16 g/ml). |
|
3.8. |
Metanols. |
|
3.9. |
Sudraba nitrāts. |
|
3.10. |
Nātrija askorbāts. |
|
3.11. |
Nātrija karbonāts. |
|
3.12. |
Nātrija hlorīds |
|
3.13. |
EDTA (etilēndiamīntetraacetātskābe, dinātrija sāls). |
|
3.14. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
|
3.15. |
Nātrija karbonāta šķīdums, c = 10 g/100 ml. |
|
3.16. |
Ar hloru piesātināts nātrija karbonāta šķīdums, c = 5 g/100 ml. Ūdenī izšķīdina 50 g nātrija karbonāta (3.11.), atšķaida līdz vienam litram un pievieno nātrija hlorīdu (3.12. apmēram), līdz šķīdums ir piesātināts. |
|
3.17. |
Sālsskābe, apmēram 0,1 mol/l. Ar ūdeni atšķaida 10 ml HCI (3.7.) līdz vienam litram. |
|
3.18. |
Amonija acetāta buferšķīdums, apmēram 0,25 mol/l. Ūdenī (3.14.) izšķīdina 19,3 g amonija acetāta (3.3.) un 30 ml etiķskābes (3.5.) un atšķaida līdz vienam litram. |
|
3.19. |
Amberlīta XAD-2 sveķu sagatavošana. Attiecīgu daudzumu amberlīta (3.2.) mazgā ar ūdeni, līdz visi hlorīda joni ir atdalīti, par ko liecina nolietās ūdens fāzes sudraba nitrāta (3.20.) tests. Pēc tam sveķus mazgā ar 50 ml metanola (3.8.), metanolu nolej un sveķus glabā svaigā metanolā. |
|
3.20. |
Sudraba nitrāta šķīdums, apmēram 0,1 mol/l. Izšķīdina 0,17 g sudraba nitrāta (3.9.) 10 mililitros ūdens. |
|
3.21. |
HPLC kustīgā fāze. 500 ml acetonitrila (3.1.) samaisa ar 300 ml amonija acetāta buferšķīduma (3.18.) un 1 200 ml ūdens (3.14.). Izmantojot etiķskābi (3.5.), koriģē pH līmeni līdz 4,3. Šķīdumu filtrē caur 0,22 μm filtru (4.8.) un atgāzo (piemēram, 10 minūtes apstrādājot ar ultraskaņu). Slēgtā konteinerā tumsā tādu šķīdumu var glabāt vienu mēnesi. |
4. Ierīces
|
4.1. |
Ultraskaņas vanna. |
|
4.2. |
Rotācijas iztvaicētājs. |
|
4.3. |
Centrifūga. |
|
4.4. |
HPLC iekārta, kurai ir ultravioletais detektors ar maināmu viļņu garumu vai diodes matricu detektors. |
|
4.4.1. |
Šķidrumu hromatogrāfa kolonna, 300 × 4 mm, C18, 10 μm iesaiņojums, vai līdzvērtīga kolonna. |
|
4.5. |
Stikla kolonna (300 × 10 mm), kas aprīkota ar keramiskā stikla filtru un krānu. |
|
4.6. |
150 mm diametra stikla šķiedras filtri. |
|
4.7. |
Membrānfiltri, 0,45 μm. |
|
4.8. |
Membrānfiltri, 0,22 μm. |
5. Procedūra
|
Piezīme. |
Halofuginons kā brīvā bāze ir nestabils sārmainos un etilacetāta šķīdumos. To neatstāj etilacetātā ilgāk par 30 minūtēm. |
5.1. Vispārīgas norādes
|
5.1.1. |
Lai pārbaudītu, ka barībā nav halofuginona un traucējošu vielu, analizē tukšo paraugu. |
|
5.1.2. |
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz halofuginona, cik ir paraugā. Lai stiprinātu paraugu 3 mg/kg līmenī, 300 μl izejas standartšķīduma (3.6.1.) pievieno 10 gramiem tukšā barības parauga, samaisa un gaida 10 minūtes, tad pāriet pie ekstrakcijas posma (5.2.).
|
5.2. Ekstrakcija
Ar precizitāti līdz 0,1 g iesver 10 g sagatavotā parauga 200 ml centrifūgas mēģenē, pievieno 0,5 g nātrija askorbāta (3.10.), 0,5 g EDTA (3.13.) un 20 ml ūdens un samaisa. Mēģeni uz piecām minūtēm ieliek ūdens vannā (80 oC). Atdzesē līdz istabas temperatūrai, tad pievieno 20 ml nātrija karbonāta šķīduma (3.15.) un samaisa. Tūlīt pievieno 100 ml etilacetāta (3.4.) un ar roku kārtīgi sakrata 15 sekundes. Tad mēģeni uz trīs minūtēm ieliek ultraskaņas vannā (4.1.) un atbrīvo aizbāzni. Centrifugē divas minūtes un caur stikla šķiedras filtru (4.6.) etilacetāta fāzi dekantē 500 ml dalāmpiltuvē. Parauga ekstrakciju atkārto ar otru 100 ml etilacetāta devu. Apvienotos ekstraktus vienu minūti mazgā ar 50 ml nātrija karbonāta šķīduma, kas piesātināts ar nātrija hlorīdu (3.16.), un nolej ūdens slāni.
Organisko slāni ekstrahē vienu minūti ar 50 ml sālsskābes (3.17.). Apakšējo skābes slāni nolaiž 250 ml dalāmpiltuvē. Pusotru minūti ar vēl vienu 50 ml sālsskābes devu atkārtoti ekstrahē organisko slāni un to pievieno pirmajam ekstraktam. Apvienotos skābes ekstraktus mazgā ar 10 ml etilacetāta (3.4.), svārstot apmēram 10 sekundes.
Ūdens slāni kvantitatīvi pārnes 250 ml apaļkolbā un nolaiž organisko fāzi. Izmantojot rotācijas iztvaicētāju (4.2.), iztvaicē visu atlikušo etilacetātu no skābes šķīduma. Ūdens vannā temperatūra nedrīkst pārsniegt 40 oC. Apmēram 25 mbar vakuumā 38 oC viss etilacetāta atlikums atdalīsies apmēram piecās minūtēs.
5.3. Attīrīšana
5.3.1.
Katram parauga ekstraktam sagatavo XAD-2 kolonnu. 10 g sagatavotā amberlīta (3.19.) pārnes uz stikla kolonnu (4.5.) ar metanolu (3.8.). Sveķu slāņa virspusē pieliek nelielu stikla vates tamponu. Notecina metanolu no kolonnas un sveķus mazgā ar 100 ml ūdens, plūsmu apturot, kad šķidrums sasniedz sveķu slāņa virsu. Desmit minūtes pirms izmantošanas ļauj kolonnai nostabilizēties. Kolonna nedrīkst iztecēt sausa.
5.3.2.
Ekstraktu (5.2.) kvantitatīvi pārnes uz sagatavotās amberlīta kolonnas (5.3.1.) augšgalu un eluē, nolejot eluātu. Eluācijas ātrums nedrīkst pārsniegt 20 ml/min. Apaļkolbu izskalo ar 20 ml sālsskābes (3.17.) un ar saskalojumu mazgā sveķu kolonnu. Skābes šķīduma atlikumu izpūš ar gaisa strūklu. Nolej saskalojumus. Kolonnai pievieno 100 ml metanola (3.8.) un 5–10 ml ļauj eluēties, savāc eluātu 250 ml apaļkolbā. Atlikušo metanolu atstāj 10 minūtes, lai nostabilizējas ar sveķiem, un turpina eluāciju ar ātrumu, kas nepārsniedz 20 ml/min, eluātu savāc tajā pašā apaļkolbā. Ar rotācijas iztvaicētāju (4.2.) iztvaicē metanolu, ūdens vannā temperatūra nedrīkst pārsniegt 40 oC. Atlikumu kvantitatīvi pārnes 10 ml mērkolbā, izmantojot kustīgo fāzi (3.21.). Papildina līdz zīmei ar kustīgo fāzi un samaisa. Alikvotu izfiltrē caur membrānfiltru (4.7.). Šo šķīdumu pataupa HPLC noteikšanai (5.4.).
5.4. HPLC noteikšana
5.4.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus:
šķidrumu hromatogrāfa kolonna (4.4.1.),
HPLC kustīgā fāze (3.21.),
plūsmas ātrums 1,5–2 ml/min,
noteikšanas viļņa garums 243 nm,
injekcijas tilpums 40 līdz 100 μl.
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.6.2.) ar koncentrāciju 3,0 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus (vai laukumus) un izdalīšanas laiku.
5.4.2.
Katru kalibrēšanas šķīdumu (3.6.2.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas grafiku, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus vai laukumus un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.4.3.
Iešpricē parauga ekstraktu (5.3.2.) vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumiem, un nosaka halofuginona maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
Nosaka parauga šķīduma koncentrāciju μg/ml, ņemot vērā parauga šķīduma halofuginona maksimumu vidējo augstumu (laukumu), izmantojot kalibrēšanas grafiku (5.4.2.).
Halofuginona saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
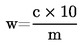
kur:
|
c |
= |
halofuginona koncentrācija parauga šķīdumā μg/ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta spektru ar tā kalibrēšanas šķīduma (3.6.2.) spektru, kura koncentrācija ir 6,0 μg/ml.
7.1.1.
Parauga ekstraktu stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.6.2.). Pievienotā halofuginona daudzumam jābūt tādam pašam kā aplēstajam halofuginona daudzumam, kas atrasts parauga ekstraktā.
Tikai halofuginona maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā maksimālā augstuma jābūt ± 10 % no sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir ± 2 nm; |
|
b) |
no 225 līdz 300 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 225 līdz 300 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
Rezultātu starpība starp divām paralēlām noteikšanām, kas veiktas ar vienu paraugu, nedrīkst pārsniegt 0,5 mg/kg, ja halofuginona saturs ir līdz 3 mg/kg.
7.3. Atgūstamība
Stiprināta tukšā parauga atgūstamība ir vismaz 80 %.
8. Koppētījuma rezultāti
Tika noorganizēts koppētījums (5), kurā astoņas laboratorijas analizēja trīs paraugus.
Rezultāti
|
|
A paraugs (tukšais) saņemot |
B paraugs (rupja maluma milti) |
C paraugs (granulas) |
||
|
|
|
saņemot |
pēc diviem mēnešiem |
saņemot |
pēc diviem mēnešiem |
|
Vidējais [mg/kg] |
NN |
2,80 |
2,42 |
2,89 |
2,45 |
|
SR [mg/kg] |
— |
0,45 |
0,43 |
0,40 |
0,42 |
|
CVR [ %] |
— |
16 |
18 |
14 |
17 |
|
Atg. [ %] |
|
86 |
74 |
88 |
75 |
|
NN |
= |
nav noteikts, |
|
SR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients (%), |
|
Atg. |
= |
atgūstamība (%). |
E. ROBENIDĪNA NOTEIKŠANA
1,3-bis [(4-hlorbenzilidēn)amino]guanidīna – hidrogēnhlorīds
1. Mērķis un darbības joma
Šī metode ļauj noteikt robenidīna daudzumu barībā. Kvantitatīvās noteikšanas robeža ir 5 mg/kg.
2. Princips
Paraugu ekstrahē ar paskābinātu metanolu. Ekstraktu žāvē un alikvoto daļu attīra uz alumīnija oksīda kolonnas. Robenidīnu no kolonnas eluē ar metanolu, kas ir koncentrēts un papildināts līdz attiecīgam tilpumam ar kustīgo fāzi. Robenidīna saturu nosaka apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Metanols. |
|
3.2. |
Paskābināts metanols. 4,0 ml sālsskābes (ρ20 = 1,18 g/ml) pārnes 500 ml mērkolbā, papildina līdz zīmei ar metanolu (3.1.) un samaisa. Minēto šķīdumu sagatavo svaigu pirms izmantošanas. |
|
3.3. |
Acetonitrils, ekvivalents HPLC tīrības pakāpei. |
|
3.4. |
Molekulārais siets. 3.A tips, ar 8 līdz 12 acojuma graudiņiem (1,6 līdz 2,5 mm graudiņi, kristālisks alumosilikāts, poru diametrs 0,3 mm). |
|
3.5. |
I aktivitātes pakāpes skābs alumīnija oksīds kolonnu hromatogrāfijai. 100 ml alumīnija oksīda pārnes piemērotā konteinerā un pievieno 2,0 ml ūdens. Noslēdz ar aizbāzni un krata apmēram 20 minūtes. Glabā cieši noslēgtā konteinerā. |
|
3.6. |
Kālija dihidrogēnfosfāta šķīdums, c = 0,025 mol/l. Izšķīdina 3,40 g kālija dihidrogēnfosfāta HPLC tīrības pakāpes ūdenī 1 000 ml mērkolbā, papildina līdz zīmei un samaisa. |
|
3.7. |
Dinātrija hidrogēnfosfāta šķīdums, c = 0,025 mol/l. Izšķīdina 3,55 g bezūdens (vai 4,45 g dihidrāta, vai 8,95 g dodekahidrāta) dinātrija hidrogēnfosfāta HPLC tīrības pakāpes ūdenī viena litra mērkolbā, papildina līdz zīmei un samaisa. |
|
3.8. |
HPLC kustīgā fāze. Samaisa kopā ar šādiem reaģentiem:
Šķīdumu filtrē caur 0,22 μm filtru (4.6.) un atgāzo (piemēram, 10 minūtes apstrādājot ar ultraskaņu). |
|
3.9. |
Standartviela. Tīrs robenidīns: 1,3-bis [(4-hlorbenzilidēn)amino]guanidīna-hidrogēnhlorīds. |
|
3.9.1. |
Ar precizitāti līdz 0,1 mg iesver 30 mg robenidīna standartvielas (3.9.). Izšķīdina paskābinātā metanolā (3.2.) 100 ml mērkolbā, papildina līdz zīmei ar to pašu šķīdinātāju un samaisa. Ietin kolbu alumīnija folijā un glabā tumšā vietā. |
|
3.9.2. |
10,0 ml izejas standartšķīduma (3.9.1.) pārnes 250 ml mērkolbā, papildina līdz zīmei ar kustīgo fāzi (3.8.) un samaisa. Ietin kolbu alumīnija folijā un glabā tumšā vietā. |
|
3.9.3. |
50 ml mērkolbās pārnes 5,0, 10,0, 15,0, 20,0 un 25,0 ml starpposma standartšķīduma (3.9.2.). Papildina līdz zīmei ar kustīgo fāzi (3.8.) un samaisa. Minētie šķīdumi atbilst attiecīgi 1,2, 2,4, 3,6, 4,8 un 6,0 μg/ml robenidīna. Minētie šķīdumi jāsagatavo svaigi pirms izmantošanas. |
|
3.10. |
Ūdens, ekvivalents HPLC tīrības pakāpei |
4. Ierīces
|
4.1. |
Stikla kolonna. Izgatavota no brūnā stikla, aprīkota ar krānu un rezervuāru, kura tilpums ir apmēram 150 ml, iekšējais diametrs no 10 līdz 15 mm, garums 250 mm. |
|
4.2. |
Mehāniskais kratītājs vai magnētiskais maisītājs. |
|
4.3. |
Rotācijas iztvaicētājs. |
|
4.4. |
HPLC iekārta, kurai ir ultravioletais detektors ar maināmu viļņu garumu vai diodes matricu detektors, kas darbojas 250 līdz 400 mm diapazonā. |
|
4.4.1. |
Šķidrumu hromatogrāfa kolonna: 300 × 4 mm, C18 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.5. |
Stikla šķiedras filtrpapīrs (GF/A Vatmans vai līdzvērtīgs). |
|
4.6. |
Membrānfiltri, 0,22 μm. |
|
4.7. |
Membrānfiltri, 0,45 μm. |
5. Procedūra
|
Piezīme. |
Robenidīns ir gaismjutīgs. Visās darbībās izmanto brūnā stikla traukus. |
5.1. Vispārīgas norādes
|
5.1.1. |
Lai pārbaudītu, ka barībā nav robenidīna un traucējošu vielu, analizē tukšo paraugu. |
|
5.1.2. |
Atgūstamības testu veic, analizējot tukšo barības paraugu (5.1.1.), kas pastiprināts, pievienojot tik daudz robenidīna, cik ir paraugā. Lai pastiprinātu paraugu līdz līmenim 60 mg/kg, 3,0 ml izejas standartšķīduma (3.9.1.) pārnes 250 ml koniskā kolbā. Slāpekļa plūsmā šķīdumu iztvaicē līdz apmēram 0,5 ml. Pirms ekstrakcijas (5.2.) pievieno 15 g tukšā barības parauga, samaisa un atstāj uz 10 minūtēm.
|
5.2. Ekstrakcija
Ar precizitāti līdz 0,01 g iesver apmēram 15 g sagatavotā parauga. Pārnes 250 ml koniskā kolbā un pievieno 100,0 ml paskābināta metanola (3.2.), aiztaisa ar aizbāzni un krata vienu stundu kratītājā (4.2.). Izfiltrē šķīdumu caur stikla šķiedras filtrpapīru (4.5.) un visu filtrātu savāc 150 ml koniskā kolbā. Pievieno 7,5 g molekulārā sieta (3.4.), aiztaisa ar aizbāzni un krata piecas minūtes. Tūlīt izfiltrē caur stikla šķiedras filtrpapīru. Minēto šķīdumu saglabā attīrīšanas posmam (5.3.).
5.3. Attīrīšana
5.3.1.
Nelielu stikla vates tamponu ieliek stikla kolonnas (4.1.) apakšējā galā un saspiež ar stikla spieķīti. Iesver 11,0 g sagatavotā alumīnija oksīda (3.5.) un pārnes kolonnā. Šajā posmā pēc iespējas samazina gaisa iedarbību. Viegli uzsit pa uzpildīto kolonnu tās apakšējā galā, lai alumīnija oksīds sablīvētos.
5.3.2.
Ar pipeti pārnes kolonnā 5,0 ml parauga ekstrakta, kas sagatavots saskaņā ar (5.2.). Pipetes galu pieliek klāt kolonnas sienai un ļauj, lai šķīdums absorbējas uz alumīnija oksīda. Robenidīnu no kolonnas eluē ar 100 ml metanola (3.1.) ar plūsmas ātrumu no 2 līdz 3 ml/minūtē un savāc eluātu 250 ml apaļkolbā. Ar rotācijas iztvaicētāju (4.3.) iztvaicē metanola šķīdumu līdz sausam stāvoklim 40 oC temperatūrā pazeminātā spiedienā. Atlikumu no jauna izšķīdina 3 līdz 4 mililitros kustīgās fāzes (3.8.) un kvantitatīvi pārnes 10 ml mērkolbā. Kolbu izskalo ar vairākām 1 līdz 2 ml kustīgās fāzes devām un saskalojumus pārnes uz mērkolbu. Papildina līdz zīmei ar to pašu šķīdinātāju un samaisa. Alikvotu izfiltrē caur 0,45 μm membrānfiltru (4.7.). Šo šķīdumu pataupa HPLC noteikšanai (5.4.).
5.4. HPLC noteikšana
5.4.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus:
šķidrumu hromatogrāfa kolonna (4.4.1.),
HPLC kustīgā fāze (3.8.),
plūsmas ātrums 1,5 līdz 2 ml/min,
detektora viļņa garums 317 nm,
injekcijas tilpums 20 līdz 50 μl.
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.9.3.) ar koncentrāciju 3,6 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus un izdalīšanas laiku.
5.4.2.
Katru kalibrēšanas šķīdumu (3.9.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas līkni, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus vai laukumus un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.4.3.
Iešpricē parauga ekstraktu (5.3.2.) vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumiem, un nosaka robenidīna maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
Ņemot vērā parauga šķīduma robenidīna maksimumu vidējo augstumu (laukumu), nosaka parauga šķīduma koncentrāciju μg/ml, izmantojot kalibrēšanas grafiku (5.4.2.).
Robenidīna saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:

kur:
|
c |
= |
robenidīna koncentrācija parauga šķīdumā μg/ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta spektru ar tā kalibrēšanas šķīduma (3.9.3.) spektru, kura koncentrācija ir 6 μg/ml.
7.1.1.
Parauga ekstraktu stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.9.3.). Pievienotā robenidīna daudzumam jābūt tādam pašam kā aplēstajam robenidīna daudzumam, kas atrasts parauga ekstraktā.
Tikai robenidīna maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā maksimālā augstuma jābūt apmēram 10 % no sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir apmēram 2 nm; |
|
b) |
no 250 līdz 400 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 250 līdz 400 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nedrīkst pārsniegt 10 % no lielākā rezultāta attiecībā uz robenidīnu, kura saturs ir lielāks par 15 mg/kg.
7.3. Atgūstamība
Stiprināta tukšā parauga atgūstamība ir vismaz 85 %.
8. Koppētījuma rezultāti
EK līmenī tika noorganizēts koppētījums, kurā 12 laboratorijās analizēja četrus saberztas vai granulu formas mājputnu un trušu barības paraugus. Visus paraugus analizēja divas reizes. Rezultāti ir norādīti šajā tabulā.
|
|
Mājputnu barība |
Trušu barība |
||
|
|
Rupja maluma milti |
Granulas |
Rupja maluma milti |
Granulas |
|
Vidējais [mg/kg] |
27,00 |
27,99 |
43,6 |
40,1 |
|
sr [mg/kg] |
1,46 |
1,26 |
1,44 |
1,66 |
|
CVr [ %] |
5,4 |
4,5 |
3,3 |
4,1 |
|
SR [mg/kg] |
4,36 |
3,36 |
4,61 |
3,91 |
|
CVR [ %] |
16,1 |
12,0 |
10,6 |
9,7 |
|
Atgūstamība [ %] |
90,0 |
93,3 |
87,2 |
80,2 |
|
sr |
= |
atkārtojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
SR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients (%). |
F. DIKLAZURILA NOTEIKŠANA
(+)-4-hlorfenil [2,6-dihlor-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazīn-2-il)fenil] acetonitrils
1. Mērķis un darbības joma
Šī metode ļauj noteikt diklazurila daudzumu barībā un premiksos. Noteikšanas robeža ir 0,1 mg/kg, kvantitatīvās noteikšanas robeža ir 0,5 mg/kg.
2. Princips
Pievieno iekšējo standartu, tad paraugu ekstrahē ar paskābinātu metanolu. Barības ekstrakta alikvotu attīra, izmantojot C18 cietās fāzes ekstrakcijas kolonnu. Diklazurilu no kolonnas eluē ar paskābināta metanola un ūdens maisījumu. Pēc iztvaicēšanas atlikumu izšķīdina DMF/ūdenī. Premiksiem ekstraktu iztvaicē un atlikumu izšķīdina DMF/ūdenī. Diklazurila saturu nosaka trīspakāpju gradienta apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
|
3.2. |
Amonija acetāts. |
|
3.3. |
Tetrabutilamonija hidrogēnsulfāts (TBHS). |
|
3.4. |
Acetonitrils, ekvivalents HPLC tīrības pakāpei. |
|
3.5. |
Metanols, ekvivalents HPLC tīrības pakāpei. |
|
3.6. |
N, N-dimetilformamīds (DMFA). |
|
3.7. |
Sālsskābe, ρ20 = 1,19 g/ml. |
|
3.8. |
Standartviela: diklazurils II-24: (+)-4-hlorfenil [2,6-dihlor-4-(2,3,4,5-tetrahidro-3,5-diokso-1,2,4-triazīn-2-il)fenil] acetonitrils ar garantētu tīrību, E 771. |
|
3.8.1. |
Ar precizitāti līdz 0,1 mg 50 ml mērkolbā iesver 25 mg diklazurila standartvielas (3.8.). Izšķīdina DMF (3.6.), papildina līdz zīmei ar DMF (3.6.) un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.8.2. |
5,00 ml izejas standartšķīduma (3.8.1.) pārnes 50 ml mērkolbā, papildina līdz zīmei ar DMF (3.6.) un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.9. |
Iekšējā standartviela: 2,6 dihlor-α-(4-hlorfenil)-4-(4,5 dihidro-3,5-diokso-1,2,4-triazīn-2 (3H) – il) α-metilbenzol-acetonitrils. |
|
3.9.1. |
Ar precizitāti līdz 0,1 mg 50 ml mērkolbā iesver 25 mg iekšējās standartvielas (3.9.). Izšķīdina DMF (3.6.), papildina līdz zīmei ar DMF (3.6.) un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.9.2. |
5,00 ml iekšējā izejas standartšķīduma (3.9.1.) pārnes 50 ml mērkolbā, papildina līdz zīmei ar DMF (3.6.) un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.9.3. |
(p = diklazurila nominālais saturs premiksā mg/kg) Ar precizitāti līdz 0,1 mg iesver p/10 mg iekšējās standartvielas 100 ml mērkolbā, izšķīdina DMF (3.6.) ultraskaņas vannā (4.6.), papildina līdz zīmei ar DMF un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.10. |
Kalibrēšanas šķīdums, 2 μg/ml. Ar pipeti pārnes 2,00 ml diklazurila standartšķīduma (3.8.2.) un 2,00 ml iekšējā standartšķīduma (3.9.2.) 50 ml mērkolbā. Pievieno 16 ml DMF (3.6.), papildina līdz zīmei ar ūdeni un samaisa. Minētais šķīdums jāsagatavo svaigs pirms izmantošanas. |
|
3.11. |
C18 cietās fāzes ekstrakcijas kolonna, piemēram, Bond Elut, kuras izmērs ir 1 cc, sorbenta svars – 100 mg. |
|
3.12. |
Ekstrakcijas šķīdinātājs: paskābināts metanols. Ar pipeti iepilina 5,0 ml sālsskābes (3.7.) 1 000 mililitros metanola (3.5.) un samaisa. |
|
3.13. |
Kustīgā fāze priekš HPLC. |
|
3.13.1. |
A eluents: amonija acetāta-tetrabutilamonija hidrogēnsulfāta šķīdums. Izšķīdina 5 g amonija acetāta (3.2.) un 3,4 g TBHS (3.3.) 1 000 mililitros ūdens (3.1.) un samaisa. |
|
3.13.2. |
B eluents: acetonitrils (3.4.). |
|
3.13.3. |
C eluents: metanols (3.5.). |
4. Ierīces
|
4.1. |
Mehāniskais kratītājs. |
|
4.2. |
Iekārta trīspakāpju gradienta HPLC. |
|
4.2.1. |
Šķidrumu hromatogrāfa kolonna, Hypersil ODS, 3 μm iesaiņojums, 100 × 4,6 mm, vai līdzvērtīga. |
|
4.2.2. |
UV detektors ar maināmu viļņu garumu vai diodes matricu detektors. |
|
4.3. |
Rotācijas iztvaicētājs. |
|
4.4. |
Membrānfiltrs 0,45 μm. |
|
4.5. |
Vakuumlīnija. |
|
4.6. |
Ultraskaņas vanna. |
5. Procedūra
5.1. Vispārīgas norādes
5.1.1.
Lai pārbaudītu, ka barībā nav diklazurila un traucējošu vielu, analizē tukšo paraugu. Tukšais paraugs pēc veida ir līdzīgs analizējamajam paraugam, un, pārbaudot to, neuzrādās diklazurils vai noteikšanu traucējošas vielas.
5.1.2.
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz diklazurila, cik ir paraugā. Lai paraugu pastiprinātu līdz 1 mg/kg, 50 g barības tukšajam paraugam pievieno 0,1 ml izejas standartšķīduma (3.8.1.), rūpīgi samaisa un atstāj uz 10 minūtēm, pirms ekstrakcijas (5.2.) vairākas reizes rūpīgi samaisot.
Ja nav analizējamajam paraugam līdzīga tukšā parauga (skatīt 5.1.1. punktu), tad atgūstamības testu var veikt, izmantojot standarta piedevu metodi. Tādā gadījumā analizējamo paraugu pastiprina ar diklazurila daudzumu, kas ir līdzīgs analizējamajā paraugā jau esošajam daudzumam. Tādu paraugu analizē kopā ar nepastiprināto paraugu un atgūstamību aprēķina pēc starpības.
5.2. Ekstrakcija
5.2.1.
Ar precizitāti līdz 0,01 g iesver apmēram 50 g parauga. Pārnes 500 ml koniskā kolbā, pievieno 1,00 ml iekšējā standarta šķīduma (3.9.2.), 200 ml ekstrakcijas šķīdinātāja (3.12.), kolbu noslēdz ar aizbāzni. Maisījumu krata kratītājā (4.1.) visu nakti. Ļauj nogulsnēties 10 minūtes. Pārnes šķīduma augšējā slāņa 20 ml alikvotu piemērotā stikla traukā un atšķaida ar 20 ml ūdens. Minēto šķīdumu pārnes uz ekstrakcijas kolonnu (3.11.) un izlaiž cauri, izmantojot vakuumu (4.5.). Kolonnu mazgā ar 25 ml ekstrakcijas šķīdinātāja (3.12.) un ūdens maisījumu tilpuma attiecībās 65 + 35 (V + V). Savāktās frakcijas izlej un savienojumus eluē ar 25 ml ekstrakcijas šķīdinātāja (3.12.) un ūdens maisījumu tilpuma attiecībās 80 + 20 (V + V). Šo frakciju iztvaicē sausu ar rotācijas iztvaicētāja (4.3.) palīdzību 60 oC temperatūrā. Atlikumu izšķīdina 1,0 ml DMF (3.6.), pievieno 1,5 ml ūdens (3.1.) un samaisa. Izfiltrē caur membrānfiltru (4.4.). Pāriet pie HPLC noteikšanas (5.3.).
5.2.2.
Ar precizitāti līdz 0,001 g iesver apmēram 1 g parauga. Pārnes 500 ml koniskā kolbā, pievieno 1,00 ml iekšējā standarta šķīduma (3.9.3.), 200 ml ekstrakcijas šķīdinātāja (3.12.) un kolbu noslēdz ar aizbāzni. Maisījumu krata kratītājā (4.1.) visu nakti. Ļauj nogulsnēties 10 minūtes. Šķīduma augšējā slāņa alikvotu daļu 10,000/p ml (p = diklazurila nominālais saturs premiksā mg/kg) pārnes piemērota izmēra apaļkolbā. Iztvaicē to sausu, izmantojot rotācijas iztvaicētāju (4.3.), 60 oC temperatūrā. Atlikumu no jauna izšķīdina 10,0 ml DMF (3.6.), pievieno 15,0 ml ūdens (3.1.) un samaisa. Pāriet pie HPLC noteikšanas (5.3.).
5.3. HPLC noteikšana
5.3.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Šķidrumu hromatogrāfa kolonna (4.2.1.): |
100 × 4,6 mm, Hypersil ODS, 3 μm iesaiņojums, vai līdzvērtīga |
|
||||||
|
Kustīgā fāze: |
A eluents (3.13.1.): |
amonija acetāta un tetrabutil- amonija hidrogēnsulfāta šķīdums ūdenī |
||||||
|
|
B eluents (3.13.2.): |
acetonitrils |
||||||
|
|
C eluents (3.13.3.): |
metanols |
||||||
|
Eluācijas veids: |
10 minūtes skalo ar B. |
|||||||
|
Plūsmas ātrums: |
1,5–2 ml/min |
|
||||||
|
Injekcijas tilpums: |
20 μl |
|
||||||
|
Detektora viļņa garums: |
280 nm |
|
||||||
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.10.) ar koncentrāciju 2 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus un izdalīšanas laiku.
5.3.2.
Vairākas reizes iešpricē pa 20 μl kalibrēšanas šķīduma (3.10.) un nosaka diklazurila un iekšējā standarta maksimumu vidējo maksimālo augstumu (laukumu).
5.3.3.
Vairākas reizes iešpricē pa 20 μl parauga šķīduma (5.2.1. vai 5.2.2.) un nosaka diklazurila un iekšējā standarta maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
6.1. Barības
Diklazurila saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
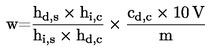 [mg/kg]
[mg/kg]
kur:
|
hd, s |
= |
diklazurila maksimālais augstums (laukums) parauga šķīdumā (5.2.1.), |
|
hi, s |
= |
iekšējā standarta maksimālais augstums (laukums) parauga šķīdumā (5.2.1.), |
|
hd, c |
= |
diklazurila maksimālais augstums (laukums) kalibrēšanas šķīdumā (3.10.), |
|
hi, c |
= |
iekšējā standarta maksimālais augstums (laukums) kalibrēšanas šķīdumā (3.10.), |
|
cd, c |
= |
diklazurila koncentrācija kalibrēšanas šķīdumā, μg/ml (3.10.), |
|
m |
= |
analizējamā parauga svars gramos, |
|
V |
= |
parauga ekstrakta tilpums atbilstoši 5.2.1. punktam (t. i., 2,5 ml). |
6.2. Premiksi
Diklazurila saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
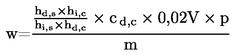 [mg/kg]
[mg/kg]
kur:
|
hd, c |
= |
diklazurila maksimālais augstums (laukums) kalibrēšanas šķīdumā (3.10.), |
|
hi, c |
= |
iekšējā standarta maksimālais augstums (laukums) kalibrēšanas šķīdumā (3.10.), |
|
hd, s |
= |
diklazurila maksimālais augstums (laukums) parauga šķīdumā (5.2.2.), |
|
hi, s |
= |
iekšējā standarta maksimālais augstums (laukums) parauga šķīdumā (5.2.2.), |
|
cd, c |
= |
diklazurila koncentrācija kalibrēšanas šķīdumā, μg/ml (3.10.), |
|
m |
= |
analizējamā parauga svars gramos, |
|
V |
= |
parauga ekstrakta tilpums atbilstoši 5.2.2. punktam (t. i., 25 ml), |
|
P |
= |
diklazurila nominālais saturs mg/kg premiksā. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta spektru (5.2.1. vai 5.2.2.) ar kalibrēšanas šķīduma (3.10.) spektru.
7.1.1.
Parauga ekstraktu (5.2.1. vai 5.2.2.) stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.10.). Pievienotā diklazurila daudzumam jābūt tādam pašam kā aplēstajam diklazurila daudzumam, kas atrasts parauga ekstraktā.
Tikai diklazurila maksimālo augstumu un iekšējā standarta maksimālo augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā augstuma jābūt ± 10 % robežās no nepastiprināta parauga ekstrakta diklazurila maksimuma vai iekšējā standarta maksimuma sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir ± 2 nm; |
|
b) |
no 230 līdz 320 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 230 līdz 320 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt:
|
— |
30 % no lielākās noteiktās vērtības, ja diklazurila saturs ir no 0,5 mg/kg līdz 2,5 mg/kg, |
|
— |
0,75 mg/kg, ja diklazurila saturs ir no 2,5 mg/kg līdz 5 mg/kg, |
|
— |
15 % no lielākās noteiktās vērtības, ja diklazurila saturs pārsniedz 5 mg/kg. |
7.3. Atgūstamība
Stiprināta (tukšā) parauga atgūstamība ir vismaz 80 %.
8. Koppētījuma rezultāti
Tika noorganizēts koppētījums, kurā 11 laboratorijas analizēja piecus paraugus. Divi analizējamie paraugi bija premiksi: viens ar organisku matricu (O 100), otrs ar neorganisku matricu (A 100). Abu paraugu teorētiskais diklazurila saturs ir 100 mg/kg. Trīs mājputniem domāto barības maisījumu paraugus ražoja trīs atšķirīgi ražotāji (NL) (L1/Z1/K1). Abu paraugu teorētiskais diklazurila saturs ir 1 mg/kg. Laboratorijām tika dots norādījums katru paraugu analizēt vienu vai divas reizes. (Sīkāku informāciju par šo laboratoriju koppētījumu skatīt Journal of AOAC International, Volume 77, Nr. 6, 1994. g., 1359.–1361. lpp.). Rezultāti ir norādīti šajā tabulā.
|
|
1. paraugs A 100 |
2. paraugs O 100 |
3. paraugs L1 |
4. paraugs Z1 |
5. paraugs K1 |
|
L |
11 |
11 |
11 |
11 |
6 |
|
n |
19 |
18 |
19 |
19 |
12 |
|
Vidējais |
100,8 |
103,5 |
0,89 |
1,15 |
0,89 |
|
Sr (mg/kg) |
5,88 |
7,64 |
0,15 |
0,02 |
0,03 |
|
CVr (%) |
5,83 |
7,38 |
17,32 |
1,92 |
3,34 |
|
SR (mg/kg) |
7,59 |
7,64 |
0,17 |
0,11 |
0,12 |
|
CVR (%) |
7,53 |
7,38 |
18,61 |
9,67 |
13,65 |
|
Nominālais saturs (mg/kg) |
100 |
100 |
1 |
1 |
1 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
Sr |
= |
atkārtojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
SR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients. |
9. Novērojumi
Vispirms jāparāda, ka diklazurila signāla izmaiņas mērāmo koncentrāciju diapazonā ir lineāras.
G. LAZALOCĪDNĀTRIJA NOTEIKŠANA
Poliētera monokarbonskābes nātrija sāls, kuru ražo Streptomyces lasaliensis
1. Mērķis un darbības joma
Šī metode ļauj noteikt lazalocīdnātrija daudzumu barībā un premiksos. Noteikšanas robeža ir 5 mg/kg, kvantitatīvās noteikšanas robeža ir 10 mg/kg.
2. Princips
Lazalocīdnātriju no parauga ekstrahē ar paskābinātu metanolu un nosaka ar apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfiju (HPLC), izmantojot apektrofluorimetra detektoru.
3. Reaģenti
|
3.1. |
Kālija dihidrogēnfosfāts (KH2PO4). |
|
3.2. |
Ortofosforskābe, w (w/w) = 85 %. |
|
3.3. |
Ortofosforskābes šķīdums, c = 20 %. Atšķaida 23,5 ml ortofosforskābes (3.2.) līdz 100 ml tilpumam ar ūdeni. |
|
3.4. |
6-metil-2-heptilamīns (1,5-dimetilheksilamīns), w (w/w) = 99 %. |
|
3.5. |
Metanols, ekvivalents HPLC tīrības pakāpei. |
|
3.6. |
Sālsskābe, blīvums 1,19 g/ml. |
|
3.7. |
Fosfāta buferšķīdums, c = 0,01 mol/l. Izšķīdina 1,36 g KH2PO4 (3.1.) 500 mililitros ūdens (3.11.), pievieno 3,5 ml ortofosforskābes (3.2.) un 10,0 ml 6-metil-2-heptilamīna (3.4.). Koriģē pH līdz 4,0 ar ortofosforskābes šķīdumu (3.3.) un atšķaida līdz 1 000 ml tilpumam ar ūdeni (3.11.). |
|
3.8. |
Paskābināts metanols. 5,0 ml sālsskābes (3.6.) pārnes 1 000 ml mērkolbā, papildina līdz zīmei ar metanolu (3.5.) un samaisa. Minētais šķīdums jāsagatavo svaigs pirms izmantošanas. |
|
3.9. |
HPLC kustīgā fāze, fosfāta bufera-metanola šķīdums 5 + 95 (V + V). Samaisa 5 ml fosfāta buferšķīduma (3.7.) ar 95 ml metanola (3.5.). |
|
3.10. |
Lazalocīdnātrija standartviela ar garantētu tīrību, C34H53O8Na (poliētera monokarbonskābes nātrija sāls, kuru ražo Streptomyces lasaliensis), E 763. |
|
3.10.1. |
Ar 0,1 mg precizitāti 100 ml mērkolbā iesver 50 mg lazalocīdnātrija (3.10.), izšķīdina paskābinātā metanolā (3.8.), papildina līdz zīmei ar to pašu šķīdinātāju un samaisa. Minētais šķīdums jāsagatavo svaigs pirms izmantošanas. |
|
3.10.2. |
10,0 ml izejas standartšķīduma (3.10.1.) ar pipeti pārnes 100 ml mērkolbā, papildina līdz zīmei ar paskābināto metanolu (3.8.) un samaisa. Minētais šķīdums jāsagatavo svaigs pirms izmantošanas. |
|
3.10.3. |
50 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 4,0, 5,0 un 10,0 ml starpposma standartšķīduma (3.10.2.) Papildina līdz zīmei ar paskābināto metanolu (3.8.) un samaisa. Minētie šķīdumi atbilst attiecīgi šādām lazalocīdnātrija koncentrācijām: 1,0, 2,0, 4,0, 5,0 un 10,0 μg. Minētie šķīdumi jāsagatavo svaigi pirms izmantošanas. |
|
3.11. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
4. Ierīces
|
4.1. |
Ultraskaņas vanna (vai vibrējoša ūdens vanna) ar temperatūras kontroli. |
|
4.2. |
Membrānfiltri, 0,45 μm. |
|
4.3. |
HPLC iekārta ar injekcijas sistēmu, piemērota 20 μl tilpumu injekcijai. |
|
4.3.1. |
Šķidrumu hromatogrāfa kolonna, 125 × 4 mm, apgrieztās fāzes C18, 5 μm iesaiņojums, vai līdzvērtīga. |
|
4.3.2. |
Spektrofluorimetrs ar maināmu ierosas un emisijas viļņu garumu. |
5. Procedūra
5.1. Vispārīgas norādes
5.1.1.
Lai veiktu atpazīšanas testu (5.1.2.), analizē tukšo barības paraugu, lai pārbaudītu, ka barībā nav ne lazalocīdnātrija, ne noteikšanu traucējošu vielu. Tukšais paraugs pēc veida ir līdzīgs analizējamajam paraugam, un, pārbaudot to, neuzrādās lazalocīdnātrijs vai noteikšanu traucējošas vielas.
5.1.2.
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz lazalocīdnātrija, cik ir paraugā. Lai paraugu pastiprinātu līdz līmenim 100 mg/kg, 10,0 ml izejas standartšķīduma (3.10.1.) pārnes 250 ml koniskā kolbā un šķīdumu iztvaicē līdz apmēram 0,5 ml. Pirms ekstrakcijas (5.2.) pievieno 50 g tukšā barības parauga, vairākas reizes kārtīgi samaisa un atstāj uz 10 minūtēm.
Ja nav analizējamajam paraugam līdzīga tukšā parauga (skatīt 5.1.1. punktu), tad atgūstamības testu var veikt, izmantojot standarta piedevu metodi. Tādā gadījumā analizējamo paraugu pastiprina ar lazalocīdnātrija daudzumu, kas ir līdzīgs analizējamajā paraugā jau esošajam daudzumam. Tādu paraugu analizē kopā ar nepastiprināto paraugu, un atgūstamību aprēķina pēc starpības.
5.2. Ekstrakcija
5.2.1.
Ar precizitāti līdz 0,01 mg iesver 5–10 g parauga 250 ml koniskā kolbā ar aizbāzni. Ar pipeti pievieno 100,0 ml paskābināta metanola (3.8.). Vaļīgi aizkorķē korķi un pasvārsta, lai disperģējas. Kolbu uz 20 minūtēm ieliek ultraskaņas vannā (4.1.) apmēram 40 oC temperatūrā, tad izņem un atdzesē līdz istabas temperatūrai. Ļauj nostāties apmēram vienu stundu, kamēr suspendētā viela nogulsnējas, tad alikvotu devu izfiltrē caur 0,45 μm membrānfiltru (4.2.) piemērotā traukā. Pāriet pie HPLC noteikšanas (5.3.).
5.2.2.
Ar precizitāti līdz 0,001 g iesver apmēram 2 g nesamalta premiksa 250 ml mērkolbā. Ar pipeti pievieno 100,0 ml paskābināta metanola (3.8.) un pasvārsta, lai disperģējas. Kolbu ar tās saturu uz 20 minūtēm ieliek ultraskaņas vannā (4.1.) apmēram 40 oC temperatūrā, tad izņem un atdzesē līdz istabas temperatūrai. Atšķaida līdz zīmei ar paskābināto metanolu (3.8.) un rūpīgi samaisa. Ļauj nostāties vienu stundu, kamēr suspendētā viela nogulsnējas, tad alikvotu devu izfiltrē caur 0,45 μm membrānfiltru (4.2.). Atšķaida atbilstošu tilpumu tīrā filtrāta ar paskābinātu metanolu (3.8.), izveidojot galīgā testa šķīdumu, kas satur apmēram 4 μg/ml lazalocīdnātrija. Pāriet pie HPLC noteikšanas (5.3.).
5.3. HPLC noteikšana
5.3.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
HPLC kolonna (4.3.1.): |
125 × 4 mm, apgrieztās fāzes C18, 5 μm iesaiņojums, vailīdzvērtīga |
|
Kustīgā fāze (3.9.): |
fosfāta buferšķīduma (3.7.) un metanola (3.5.) maisījums, 5+95 (V+V) |
|
Plūsmas ātrums: |
1,2 ml/min |
|
Noteikšanas viļņa garums: |
|
|
ierosa |
310 nm |
|
emisija |
419 nm |
|
Injekcijas tilpums: |
20 μl |
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.10.3.) ar koncentrāciju 4,0 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus (vai laukumus) un izdalīšanas laiku.
5.3.2.
Katru kalibrēšanas šķīdumu (3.10.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas līkni, uz ordinātu ass atzīmējot vidējos maksimālos augstumus (laukumus) un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.3.3.
Iešpricē parauga ekstraktu, kas iegūts saskaņā ar 5.2.1. vai 5.2.2. punktu, vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumam, un nosaka lazalocīdnātrija maksimumu vidējos augstumus (laukumus).
6. Rezultātu aprēķināšana
Lazalocīdnātrija šķīduma koncentrāciju (μg/ml) nosaka pēc parauga šķīduma (5.3.3.) vidējā maksimālā augstuma (laukuma), salīdzinot ar kalibrēšanas grafiku.
6.1. Barība
Lazalocīdnātrija saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg]
[mg/kg]
kur:
|
c |
= |
lazalocīdnātrija koncentrācija parauga šķīdumā (5.2.1.) μg/ml, |
|
V1 |
= |
parauga ekstrakta tilpums ml atbilstoši 5.2.1. punktam (t. i., 100 ml), |
|
m |
= |
analizējamā parauga svars gramos. |
6.2. Premiksi
Lazalocīdnātrija saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg]
[mg/kg]
kur:
|
c |
= |
lazalocīdnātrija koncentrācija parauga šķīdumā (5.2.2.) μg/ml, |
|
V2 |
= |
parauga ekstrakta tilpums ml atbilstoši 5.2.2. punktam (t. i., 250 ml), |
|
f |
= |
atšķaidījuma pakāpe atbilstoši 5.2.2. punktam, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Metodes, kuru pamatā ir spektrofluorimetrija, interference ietekmē mazāk nekā tās, kur izmanto UV staru detektoru. Nosakāmās vielas identitāti var apstiprināt ar paralēlu hromatogrāfisko analīzi.
7.1.1.
Parauga ekstraktu (5.2.1. vai 5.2.2.) stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.10.3.). Pievienotā lazalocīdnātrija daudzumam jābūt tādam pašam kā aplēstajam lazalocīdnātrija daudzumam, kas atrasts parauga ekstraktā. Tikai lazalocīdnātrija maksimuma augstumu palielina, ņemot vērā pievienoto lazalocīdnātrija daudzumu un ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā augstuma jābūt ± 10 % robežās no nepastiprinātā parauga ekstrakta sākotnējā maksimuma.
7.2. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt:
|
— |
15 % no lielākās noteiktās vērtības, ja lazalocīdnātrija saturs ir no 30 mg/kg līdz 100 mg/kg, |
|
— |
15 mg/kg, ja lazalocīdnātrija saturs ir no 100 mg/kg līdz 200 mg/kg, |
|
— |
7,5 % no lielākās noteiktās vērtības, ja lazalocīdnātrija saturs pārsniedz 200 mg/kg. |
7.3. Atgūstamība
Stiprināta (tukšā) barības parauga atgūstamība ir vismaz 80 %. Stiprinātu premiksu paraugu atgūstamība ir vismaz 90 %.
8. Koppētījuma rezultāti
Tika noorganizēts koppētījums (6), kurā 12 laboratorijas analizēja divus premiksus (1. un 2. paraugs) un piecus barības paraugus (3. līdz 7. paraugs). Visus paraugus analizēja divas reizes. Rezultāti ir norādīti šajā tabulā.
|
|
1. paraugs Premikss vistām |
2. paraugs Premikss tītariem |
3. paraugs Granulas tītariem |
4. paraugs Drupatas vistām |
5. paraugs Barība tītariem |
6. paraugs A barība mājputniem |
7. paraugs B barība mājputniem |
|
L |
12 |
12 |
12 |
12 |
12 |
12 |
12 |
|
n |
23 |
23 |
23 |
23 |
23 |
23 |
23 |
|
Vidējais [mg/kg] |
5 050 |
16 200 |
76,5 |
78,4 |
92,9 |
48,3 |
32,6 |
|
sr [mg/kg] |
107 |
408 |
1,71 |
2,23 |
2,27 |
1,93 |
1,75 |
|
CVr [ %] |
2,12 |
2,52 |
2,24 |
2,84 |
2,44 |
4,00 |
5,37 |
|
sR [mg/kg] |
286 |
883 |
3,85 |
7,32 |
5,29 |
3,47 |
3,49 |
|
CVR [ %] |
5,66 |
5,45 |
5,03 |
9,34 |
5,69 |
7,18 |
10,70 |
|
Nominālais saturs [mg/kg] |
5 000 (7) |
16 000 (7) |
80 (7) |
105 (7) |
120 (7) |
50 (8) |
35 (8) |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo rezultātu skaits, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
sR |
= |
atveidojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
CVR |
= |
atveidojamības variācijas koeficients (%). |
(1) Veica Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA) Barības darba grupa.
(2) Veica Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA) Barības darba grupa.
(3) Var izmantot citas šķelšanas metodes, ja ir pierādīts, ka tās uzrāda līdzīgus rezultātus (piemēram, šķelšana mikroviļņu spiedienā).
(4) Rupjā lopbarība zaļmasā (svaiga vai kaltēta), kura var saturēt lielus daudzumus dārzeņu kvarca, kas var aizturēt mikroelementus un tāpēc ir jāatdala. Minēto barību paraugiem līdz ar to jāievēro šāda koriģēta procedūra. Veic 5.1.1.1. punktā minēto procedūru līdz filtrēšanai. Divreiz ar vārošu ūdeni nomazgā filtrpapīru, kas satur nešķīstošo atlikumu, un ievieto to kvarca vai platīna tīģelī. Iededzina mufeļkrāsni (4.1.) temperatūrā, kas ir zem 550 oC, līdz viss oglekļa materiāls ir pilnībā izzudis. Ļauj atdzist, pievieno dažus pilienus ūdens un tad 10–15 ml fluorūdeņražskābes (3.4.) un iztvaicē, līdz tas ir sauss, apmēram 150 oC temperatūrā. Ja atlikumā paliek kvarcs, tad to atkārtoti izšķīdina dažos mililitros fluorūdeņražskābes (3.4.) un iztvaicē, līdz tas ir sauss. Pievieno dažus pilienus sērskābes (3.5.) un karsē, līdz vairs nepaceļas balti dūmi. Pievieno 5 ml 6 mol/l sālsskābes (3.2.), tad apmēram 30 ml ūdens, karsē, šķīdumu filtrē un ievieto 250 ml tilpuma kolbā, un papildina līdz zīmei ar ūdeni (HCl koncentrācija apmēram 0,5 mol/l). Pēc tam noteikšanu turpina no 5.1.2. punkta.
(5) Analyst 108, 1983. g., 1252. līdz 1256. lpp.
(6) Analyst, 1995. g., 120., 2175.–2180. lpp.
(7) Ražotāja deklarētais saturs.
(8) Laboratorijā sagatavotā barība.
V PIELIKUMS
ANALĪZES METODES, LAI KONTROLĒTU NEVĒLAMAS VIELAS BARĪBĀ
A. BRĪVĀ UN KOPĒJĀ GOSIPOLA NOTEIKŠANA
1. Mērķis un darbības joma
Šī metode ļauj noteikt brīvā gosipola, kopējā gosipola un ķīmiski radniecīgu vielu daudzumu kokvilnas sēklās, kokvilnas sēklu miltos un kokvilnas sēklu raušos, un barības maisījumos, kas satur šos barības līdzekļus, ja tajos ir vairāk par 20 mg/kg brīvā gosipola, kopējā gosipola un ķīmiski radniecīgu vielu.
2. Princips
Gosipolu ekstrahē 3-amino-1-propanola klātbūtnē ar 2-propanola un heksāna maisījumu brīvā gosipola noteikšanai vai ar dimetilformamīdu kopējā gosipola noteikšanai. Gosipolu ar anilīnu pārvērš gosipoldianilīnā, kura optisko blīvumu mēra pie 440 nm.
3. Reaģenti
|
3.1. |
2-propanola-heksāna maisījums: samaisa 60 tilpuma daļas 2-propanola ar 40 tilpuma daļām n-heksāna. |
|
3.2. |
A šķīdinātājs: viena litra mērkolbā ielej apmēram 500 ml 2-propanola-heksāna maisījuma (3.1.), 2 ml 3-amino-1-propanola, 8 ml ledus etiķskābes un 50 ml ūdens. Papildina līdz vajadzīgajam tilpumam ar 2-propanola-heksāna maisījumu (3.1.). Tādu reaģentu var glabāt nedēļu. |
|
3.3. |
B šķīdinātājs: ar pipeti iepilina 2 ml 3-amino-1-propanola un 10 ml ledus etiķskābes 100 ml mērkolbā. Atdzesē līdz istabas temperatūrai un papildina līdz vajadzīgajam tilpumam ar N, N-dimetilformamīdu. Tādu reaģentu var glabāt nedēļu. |
|
3.4. |
Anilīns: ja optiskais blīvums tukšajā analīzē pārsniedz 0,022, tad anilīnu destilē virs cinka putekļiem, nolejot destilāta pirmo un pēdējo 10 % frakciju. Ledusskapī un ar aizbāzni noslēgtā brūna stikla kolbā tādu reaģentu var glabāt vairākus mēnešus. |
|
3.5. |
Standarta gosipola A šķīdums: ievieto 27,9 mg gosipola acetāta 250 ml mērkolbā. Izšķīdina un papildina līdz vajadzīgajam tilpumam ar A šķīdinātāju (3.2.). Ar pipeti pārnes 50 ml šā šķīduma 250 ml mērkolbā un papildina līdz vajadzīgajam tilpumam ar A šķīdinātāju. Gosipola koncentrācija šajā šķīdumā ir 0,02 mg/ml. Pirms izmantošanas atstāj uz vienu stundu istabas temperatūrā. |
|
3.6. |
Standarta gosipola B šķīdums: ievieto 27,9 mg gosipola acetāta 50 ml mērkolbā. Izšķīdina un papildina līdz vajadzīgajam tilpumam ar B šķīdinātāju (3.3.). Gosipola koncentrācija šajā šķīdumā ir 0,5 mg/ml. Standarta gosipola A un B šķīdumu var uzglabāt 24 stundas, ja tiem nepiekļūst gaisma. |
4. Ierīces
|
4.1. |
Rotācijas tipa maisītājs: apmēram 35 apgr./min. |
|
4.2. |
Spektrofotometrs. |
5. Procedūra
5.1. Testa paraugs
Izmantotā testa parauga daudzums ir atkarīgs no domājamā gosipola satura paraugā. Ieteicams strādāt ar mazu testa paraugu un relatīvi lielu filtrāta alikvoto daļu, lai ar iegūto gosipola daudzumu būtu iespējami precīzi fotometrijas mērījumi. Brīvā gosipola noteikšanai kokvilnas sēklās, kokvilnas sēklu miltos un kokvilnas sēklu raušos testa paraugs nepārsniedz 1 g; barības maisījumam tas var būt līdz 5 g. Vairumā gadījumu piemērota ir 10 ml filtrāta alikvotā daļa; tā satur 50–100 μg gosipola. Kopējā gosipola noteikšanai testa paraugs ir no 0,5 g līdz 5 g tā, lai 2 ml filtrāta alikvotā daļa saturētu 40 līdz 200 μg gosipola.
Analīzi veic istabas temperatūrā – apmēram 20 oC.
5.2. Brīvā gosipola noteikšana
Testa paraugu ievieto 250 ml kolbā ar pieslīpētu kaklu, kuras dibens ir klāts ar stikla drumslām. Ar pipeti pievieno 50 ml šķīdinātāja A (3.2.), aizkorķē kolbu un maisa maisītājā vienu stundu. Izfiltrē caur sausu filtru un savāc filtrātu mazā kolbā ar pieslīpētu kaklu. Filtrēšanas laikā piltuvi apklāj ar pulksteņstiklu.
Ar pipeti iepilina identiskas filtrāta alikvotās daļas, kas satur 50 līdz 100 μg gosipola, katrā no divām 25 ml mērkolbām (A un B). Vajadzības gadījumā papildina tilpumu līdz 10 ml ar A šķīdinātāju (3.2.). Tad papildina kolbas (A) saturu līdz vajadzīgajam tilpumam ar 2-propanola-heksāna maisījumu (3.1.). Tādu šķīdumu izmantos par salīdzināšanas šķīdumu, pret kuru mēra parauga šķīdumu.
Ar pipeti iepilina 10 ml A šķīdinātāja (3.2.) katrā no divām citām 25 ml mērkolbām (C un D). Papildina kolbas (C) saturu līdz vajadzīgajam tilpumam ar 2-propanola-heksāna maisījumu (3.1.). Tādu šķīdumu izmantos kā salīdzināšanas šķīdumu, pret kuru mēra tukšās analīzes šķīdumu.
Katrā no abām kolbām (D) un (B) pievieno 2 ml anilīna (3.4.). Karsē 30 minūtes virs vārošas ūdens vannas, līdz šķīdums iekrāsojas. Atdzesē līdz istabas temperatūrai, papildina līdz vajadzīgajam tilpumam ar 2-propanola-heksāna maisījumu (3.1.), homogenizē un atstāj uz vienu stundu.
Ar spektrofotometru pie 440 nm nosaka optisko blīvumu tukšās analīzes šķīdumam (D), salīdzinot ar salīdzināšanas šķīdumu (C), un optisko blīvumu parauga šķīdumam (B), salīdzinot ar salīdzināšanas šķīdumu (A), izmantojot 1 cm stikla šūnas.
Atņem tukšās analīzes šķīduma optisko blīvumu no parauga šķīduma optiskā blīvuma (= koriģētais optiskais blīvums). No šā lieluma aprēķina brīvā gosipola saturu, kā norādīts 6. punktā.
5.3. Kopējā gosipola noteikšana
Testa paraugu, kas satur 1 mg līdz 5 mg gosipola, ievieto 50 ml mērkolbā un pievieno 10 ml B šķīdinātāja (3.3.). Vienlaikus sagatavo tukšo analīzi, ievietojot 10 ml B šķīdinātāja (3.3.) citā 50 ml mērkolbā. 30 minūtes abas kolbas karsē virs vāroša ūdens vannas. Atdzesē līdz istabas temperatūrai un papildina katras kolbas saturu līdz vajadzīgajam tilpumam ar 2-propanola-heksāna maisījumu (3.1.). Homogenizē un ļauj nogulsnēties 10–15 minūtes, tad izfiltrē un filtrātus savāc kolbās ar pieslīpētu kaklu.
Ar pipeti iepilina katrā no divām 25 ml mērkolbām 2 ml parauga filtrāta un ar pipeti iepilina katrā no divām citām 25 ml kolbām 2 ml tukšās analīzes filtrāta. Tad papildina katra komplekta vienas kolbas saturu līdz 25 ml ar 2-propanola-heksāna maisījumu (3.1.). Minētos šķīdumus izmantos par salīdzināšanas šķīdumiem.
Katrā no abām pārējām kolbām pievieno 2 ml anilīna (3.4.). Karsē 30 minūtes virs vārošas ūdens vannas, līdz šķīdums iekrāsojas. Atdzesē līdz istabas temperatūrai, papildina līdz 25 ml ar 2-propanola-heksāna maisījumu (3.1.), homogenizē un atstāj uz vienu stundu.
Nosaka optisko blīvumu, kā norādīts 5.2. punktā attiecībā uz brīvo gosipolu. No šā lieluma aprēķina kopējā gosipola saturu, kā norādīts 6. punktā.
6. Rezultātu aprēķināšana
Rezultātus var aprēķināt vai nu no specifiskā optiskā blīvuma (6.1.), vai izmantojot kalibrēšanas līkni (6.2.).
6.1. No specifiskā optiskā blīvuma
Specifiskie optiskie blīvumi aprakstītajos apstākļos būs šādi:
|
Brīvais gosipols: |
|
|
Kopējais gosipols: |
|
Brīvā vai kopējā gosipola saturu paraugā aprēķina, izmantojot šādu formulu:

kur:
|
E |
= |
koriģētais optiskais blīvums, kas noteikts, kā norādīts 5.2. punktā, |
|
p |
= |
testa paraugs gramos, |
|
a |
= |
filtrāta alikvotā daļa mililitros. |
6.2. Izmantojot kalibrēšanas līkni
6.2.1.
Sagatavo divus komplektus, katrā pa piecām 25 ml mērkolbām. Katrā kolbu komplektā ar pipeti iepilina standarta gosipola A šķīduma 2,0, 4,0, 6,0, 8,0 un 10,0 ml alikvotus. Papildina tilpumus līdz 10 ml ar A šķīdinātāju (3.2.). Papildina katru komplektu ar 25 ml mērkolbu, kurā ir tikai 10 ml A šķīdinātāja (3.2.) (tukšā analīze).
Papildina pirmā komplekta kolbas (tostarp kolbu tukšajai analīzei) līdz 25 ml tilpumam ar 2-propanola-heksāna maisījumu (3.1.) (salīdzināšanas komplekts).
Katrai otrā komplekta kolbai (tostarp kolbu tukšajai analīzei) pievieno 2 ml anilīna (3.4.). Karsē 30 minūtes virs vārošas ūdens vannas, līdz šķīdums iekrāsojas. Atdzesē līdz istabas temperatūrai, papildina līdz vajadzīgajam tilpumam ar 2-propanola-heksāna šķīdumu (3.1.), homogenizē un atstāj uz vienu stundu (standarta komplekts).
Kā norādīts 5.2. punktā, nosaka standartkomplekta šķīdumu optisko blīvumu, salīdzinot ar attiecīgajiem šķīdumiem salīdzināšanas komplektā. Veido kalibrēšanas līkni, atzīmējot optiskos blīvumus un atbilstīgos gosipola daudzumus (μg).
6.2.2.
Sagatavo sešas 50 ml mērkolbas. Pirmajā kolbā ievieto 10 ml B šķīdinātāja (3.3.) un pārējās – attiecīgi 2,0, 4,0, 6,0, 8,0 un 10,0 ml standarta gosipola B šķīduma (3.6.). Katras kolbas saturu papildina līdz 10 ml ar B šķīdinātāju (3.3.). Karsē 30 minūtes virs vārošas ūdens vannas. Atdzesē līdz istabas temperatūrai, papildina līdz vajadzīgajam tilpumam ar 2-propanola-heksāna šķīdumu (3.1.) un homogenizē.
Ievieto 2,0 ml minēto šķīdumu katrā no sešām abu komplektu 25 ml mērkolbām. Papildina pirmā komplekta kolbu saturu līdz 25 ml ar 2-propanola-heksāna maisījumu (3.1.) (salīdzināšanas komplekts).
Katrai kolbai otrajā komplektā pievieno 2 ml anilīna (3.4.). Karsē 30 minūtes virs vārošas ūdens vannas. Atdzesē līdz istabas temperatūrai, papildina līdz vajadzīgajam tilpumam ar 2-propanola-heksāna šķīdumu (3.1.), homogenizē un atstāj uz vienu stundu (standarta komplekts).
Kā norādīts 5.2. punktā, nosaka standartkomplekta šķīdumu optisko blīvumu, salīdzinot ar attiecīgajiem šķīdumiem salīdzināšanas komplektā. Veido kalibrēšanas līkni, atzīmējot optiskos blīvumus un atbilstīgos gosipola daudzumus (μg).
6.3. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt:
|
— |
15 % relatīvajā vērtībā no lielākā daudzuma, ja gosipola saturs ir mazāk par 500 ppm, |
|
— |
75 ppm absolūtajā vērtībā, ja saturs ir ne mazāk kā 500 ppm un ne vairāk kā 750 ppm, |
|
— |
10 % relatīvajā vērtībā no lielākā daudzuma, ja gosipola saturs ir vairāk par 750 ppm. |
B. DIOKSĪNU (PCDD/PCDF) UN DIOKSĪNIEM LĪDZĪGU PCB DAUDZUMU NOTEIKŠANA
I. PARAUGU ŅEMŠANAS UN ANALĪŽU REZULTĀTU INTERPRETĀCIJAS METODES
1. Mērķis un darbības joma
Paraugus, kas paredzēti dioksīnu (polihlordibenzo-p-dioksīnu (PCDD) un polihlordibenzofurānu (PCDF)) un dioksīniem līdzīgu polihlorbifenilu (PCB) daudzumu oficiālai kontrolei barībā (1), ņem saskaņā ar I pielikuma noteikumiem. Attiecībā uz vielu vai produktu, kas vienmērīgi izplatīti barībā, kontroli jāpiemēro I pielikuma 5.A. punktā paredzētās kvantitatīvās prasības. Tā iegūtos kopparaugus uzskata par raksturīgiem tām partijām vai apakšpartijām, no kurām tie ņemti. Atbilstību maksimāli pieļaujamajai koncentrācijai, kas noteikta Eiropas Parlamenta un Padomes Direktīvā 2002/32/EK (2), nosaka, pamatojoties uz laboratorijas paraugos noteikto daudzumu.
2. Partijas vai apakšpartijas atbilstība specifikācijai
Partiju pieņem, ja vienas analīzes rezultāts nepārsniedz attiecīgo maksimāli pieļaujamo koncentrāciju, kura noteikta Direktīvā 2002/32/EK, ņemot vērā mērījumu neprecizitāti.
Partiju uzskata par neatbilstošu maksimāli pieļaujamajai koncentrācijai, kas noteikta Direktīvā 2002/32/EK, ja augšējā robeža (3) analīžu rezultātiem, kas apstiprināta otrreizējā analīzē (4), acīmredzami pārsniedz maksimāli pieļaujamo koncentrāciju, ņemot vērā mērījumu neprecizitāti.
Mērījumu neprecizitāti var ņemt vērā atbilstoši vienai no šādām pieejām:
|
— |
aprēķinot izvērsto neprecizitāti, izmantojot paplašinājuma koeficientu 2, kas nodrošina aptuveni 95 % ticamības līmeni. Partija nav atbilstoša, ja, atņemot U no izmērītās vērtības, iegūtā vērtība ir virs noteiktās atļautās vērtības. Ja dioksīnus un dioksīniem līdzīgus PCB nosaka atsevišķi, tad atsevišķo dioksīnu un dioksīniem līdzīgu PCB analīžu rezultātu aplēstā izvērstās nenoteiktības summa jāizmanto kā dioksīnu un dioksīniem līdzīgu PCB summa, |
|
— |
nosakot izšķiršanas robežu (CCα) saskaņā ar Komisijas Lēmumu 2002/657/EK (5) (pielikuma 3.1.2.5. punkts – vielām, kurām ir noteikts atļautais līmenis). Partija nav atbilstoša, ja izmērītā vērtība ir vienāda ar vai lielāka par CCα. |
Šos interpretācijas noteikumus piemēro analīžu rezultātiem, kas iegūti parauga oficiālajā kontrolē. Tas neietekmē dalībvalstu tiesības piemērot analīzēm valsts tiesību aktus aizsardzības vai arbitrāžas nolūkos.
II. PARAUGU GATAVOŠANA UN PRASĪBAS ATTIECĪBĀ UZ ANALĪZES METODĒM, KO IZMANTO DIOKSĪNU (PCDD/PCDF) UN DIOKSĪNIEM LĪDZĪGU PCB DAUDZUMU OFICIĀLAI KONTROLEI
1. Mērķis un piemērošanas joma
Minētās prasības piemēro, ja barības līdzekļus un barību analizē, lai kvantitatīvi noteiktu dioksīnus (polihlordibenzo-p-dioksīnus (PCDD) un polihlordibenzofurānus (PCDF)) un dioksīniem līdzīgus polihlorbifenilus (PCB).
Dioksīnu klātbūtnes uzraudzību barībā var veikt atbilstoši stratēģijai, kas paredz izmantot skrīninga metodi, ar kuru atlasa paraugus, kur dioksīnu un dioksīniem līdzīgu PCB koncentrācija ir par 25 % mazāka vai lielāka nekā vajadzīgā koncentrācija. Šajos paraugos, kuros dioksīnu koncentrācija ir ievērojama, tā jānosaka/jāapstiprina, izmantojot apstiprinošu metodi.
Skrīninga metodes ir metodes, ko izmanto dioksīnu un dioksīniem līdzīgu PCB kvalitatīvai noteikšanai ar noteiktu ticamību. Šīs metodes ir jaudīgas, un tās izmanto potenciāli pozitīvu paraugu atlasīšanai. Tās īpaši izveidotas tā, lai nevarētu iegūt šķietami negatīvus rezultātus.
Apstiprinošās metodes ir metodes, ko izmantojot par analizējamo paraugu iegūst pilnīgu informāciju vai papildu informāciju, kas vajadzīga dioksīnu un dioksīniem līdzīgu PCB precīzai kvalitatīvai un kvantitatīvai noteikšanai ar noteiktu ticamību.
2. Pamatinformācija
Tā kā vides un bioloģiskajos paraugos (tostarp barības līdzekļu/barības paraugos) parasti ir dažādu dioksīniem radniecīgu vielu sarežģīti maisījumi, lai atvieglotu riska novērtēšanu, ir radīts toksiskuma ekvivalences faktoru (TEF) jēdziens. Ar TEF izsaka 2,3,7,8-aizvietoto PCDD un PCDF maisījumu koncentrāciju un pēdējā laikā dažu to dioksīniem līdzīgu neorto- un mono-orto- hloraizvietoto PCB koncentrāciju, kuriem ir dioksīniem līdzīga aktivitāte 2,3,7,8-TCDD toksiskuma ekvivalentos (TEQ). Atsevišķo vielu koncentrācijas paraugā reizina ar attiecīgo TEF un pēc tam summē, lai iegūtu kopējo dioksīniem līdzīgo savienojumu koncentrāciju, ko izsaka ar TEQ.
Tikai minētās regulas mērķiem atsevišķas radniecīgas vielas kvantitatīvās noteikšanas pieņemtā specifiskā robeža ir tāda analizējamās vielas koncentrācija parauga ekstraktā, kurai atbilst instrumenta atbilde pie diviem atšķirīgiem joniem, kurus kontrolē pie mazāk jutīgā signāla S/T (signāla/trokšņa) attiecības 3:1, un tādas pamatprasības kā, piemēram, izdalīšanas laiks un izotopu attiecība ir izpildīti atbilstīgi noteikšanas procedūrai, kas aprakstīta EPA metodes 1613 pārskatītajā B variantā.
3. Kvalitātes nodrošināšanas prasības, kas jāievēro, gatavojot paraugus
Piemēro II pielikumā izklāstītos vispārīgos noteikumus par paraugu sagatavošanu analīzei.
Turklāt jāievēro šādas prasības:
|
— |
paraugi jāuzglabā un jātransportē stikla, alumīnija, polipropilēna vai polietilēna konteineros. No parauga konteinera jāaizvāc papīra putekļu pēdas. Stikla traukus skalo ar šķīdinātājiem, kuros iepriekš pārbaudīta dioksīnu klātbūtne, |
|
— |
veic tukšo analīzi, izpildot visu analīzes procedūru bez analizējamā parauga, |
|
— |
ekstrakcijai izmantojamā parauga masai jābūt pietiekami lielai, lai tiktu ievērotas prasības attiecībā uz jutību. |
4. Prasības laboratorijām
|
— |
Laboratorijas pierāda metodes efektivitāti interesējošās koncentrācijas diapazonā, piemēram, 0,5x, 1x un 2x interesējošā koncentrācija, un ka šajā diapazonā ir pieņemams atkārtotu analīžu variāciju koeficients. Sīku informāciju par pieņemšanas kritērijiem skatīt 5. punktā. |
|
— |
Apstiprinājuma metodes kvantitatīvās noteikšanas robeža ir aptuveni viena piektdaļa no nosakāmās koncentrācijas vērtības, lai nodrošinātu, ka vajadzīgā līmeņa diapazonā ir pieņemami variāciju koeficienti. |
|
— |
Kvalitātes iekšējās kontroles nolūkos regulāri izdara tukšo paraugu analīzes un kontrolparaugu analīzes ar standartpiedevu metodi vai kontrolparaugu analīzes (ja iespējams, ieteicams izmantot sertificētu standartmateriālu). |
|
— |
Sekmīga līdzdalība starplaboratoriju pētījumos, kuros novērtē laboratorijas kompetenci, ir labākais paņēmiens, kā apliecināt spēju veikt specifiskas analīzes. Tomēr sekmīga līdzdalība starplaboratoriju pētījumos, kuros analizē, piemēram, augsnes vai notekūdeņu paraugus, ne vienmēr apliecina laboratorijas kompetenci analizēt pārtikas vai barības paraugus, kuros piesārņojuma līmenis ir zemāks. Tāpēc obligāta prasība ir līdzdalība starplaboratoriju pētījumos, kuros kvantitatīvi nosaka dioksīnus un dioksīniem līdzīgus PCB attiecīgajās barības/pārtikas matricās. |
|
— |
Laboratorijas akreditē atzīta organizācija, kas darbojas saskaņā ar ISO Guide 58, lai nodrošinātu to, ka laboratorijās piemēro analīžu kvalitātes nodrošināšanu. Laboratorijas akreditē, ievērojot ISO/IEC/17025 standartu. |
5. Prasības, kas jāievēro, veicot analīzes procedūras dioksīnu un dioksīniem līdzīgu PCB noteikšanai
Analīzes procedūru piemērotības pamatprasības
|
— |
Liela jutība un zema noteikšanas robeža. PCDD un PCDF mazākajam nosakāmajam daudzumam jābūt TEQ pikogramos (10-12 g), jo daži no tādiem savienojumiem ir ārkārtīgi toksiski. Ir zināms, ka PCB koncentrācija ir augstāka par PCDD un PCDF koncentrāciju. Attiecībā uz lielāko daļu PCB radniecīgo vielu pietiekama jutība ir nanogramos (10-9 g). Tomēr, lai kvantitatīvi noteiktu toksiskākas dioksīniem līdzīgo PCB radniecīgās vielas (jo īpaši orto- neizvietotās radniecīgās vielas), vajadzīga tāda pati jutība kā PCDD un PCDF noteikšanai. |
|
— |
Augsta selektivitāte (specifiskums). Jāatšķir PCDD, PCDF un dioksīniem līdzīgi PCB no daudziem citiem vienlaikus ekstrahētiem savienojumiem un varbūtēji traucējošiem savienojumiem, kuru koncentrācija līdz vairākām kārtām pārsniedz nosakāmās vielas koncentrāciju. Ja izmanto gāzu hromatogrāfijas/masspektrometrijas (GC/MS) metodes, tad jādiferencē tādas atšķirīgas radniecīgas vielas kā toksiskās (piemēram, septiņpadsmit 2,3,7,8-aizvietotie PCDD un PCDF un dioksīniem līdzīgie PCB) un citas radniecīgas vielas. Izmantojot biotestus, jāspēj selektīvi noteikt TEQ vērtības, ko izsaka ar PCDD, PCDF un dioksīniem līdzīgo PCB summu. |
|
— |
Liela precizitāte (ticamība un precizitāte). Kvantitatīvajā noteikšanā iegūst pareizu un ticamu patiesās koncentrācijas novērtējumu paraugā. Augsta precizitāte (mērījuma precizitāte: mērāmā lieluma izmērītās vērtības un tā patiesās vai pieņemtās vērtības sakritība) ir vajadzīga, lai noteiktās TEQ mazās ticamības dēļ nevarētu noraidīt parauga analīzes rezultātus. Precizitāti izsaka ar ticamību (starpība starp analizējamās vielas vidējo vērtību, kas izmērīta, analizējot sertificētu standartmateriālu, un tās sertificēto vērtību, kuru izsaka procentos no šīs vērtības) un precizitāti (RSDR relatīvā standartnovirze, ko aprēķina no atkārtojamības apstākļos iegūtiem rezultātiem). |
Par skrīninga metodēm var izmantot gan biotestus, gan GC/MS metodes; par apstiprinošajām metodēm izmanto augstas izšķirtspējas gāzu hromatogrāfijas/augstas izšķirtspējas masspektrometrijas (HRGC/HRMS) metodes.
Kopējās TEQ vērtībai jāatbilst šādiem kritērijiem.
|
|
Skrīninga metodes |
Apstiprinājuma metodes |
|
Šķietami negatīva likme |
< 1 % |
|
|
Ticamība |
|
–20 % līdz + 20 % |
|
Precizitāte RSDR |
< 30 % |
< 15 % |
6. Īpašas prasības, kas jāievēro un kas attiecas uz GC/MS metodēm, kuras izmanto skrīningam vai apstiprināšanai
|
— |
Lai analīzes procedūra būtu derīga, sākot izpildīt analīzi, piemēram, pirms ekstrakcijas, jāpievieno ar 13C iezīmēti 2,3,7,8-hloraizvietoti iekšējie PCDD/F standarti un ar 13C iezīmēti dioksīniem līdzīgu PCB iekšējie standarti. Katrai tetra- līdz oktahloratvasināto PCDD/F homologu grupai jāpievieno vismaz viena radniecīga viela, un vismaz viena radniecīga viela jāpievieno katrai dioksīniem līdzīgu PCB homologu grupai (alternatīvi jāpievieno vismaz viena radniecīga viela uz katru masspektrometrijai izraudzīto reģistrējamo jonu funkciju, ko izmanto PDDD/F un dioksīniem līdzīgu PCB uzraudzībai). Apstiprinošajām metodēm noteikti vislabāk ir izmantot visus 17 ar 13C iezīmētos 2,3,7,8-aizvietoto PCDD/F iekšējos standartus un visus 12 ar 13C iezīmētos dioksīniem līdzīgu PCB iekšējos standartus. |
|
— |
Izmantojot attiecīgus kalibrēšanas šķīdumus, nosaka signāla relatīvo attiecību pret masas vienību arī tām radniecīgajām vielām, kurām nav pievienots ar 13C iezīmēts analogs. |
|
— |
Pirms ekstrakcijas iekšējie standarti obligāti jāpievieno augu un dzīvnieku izcelsmes barībai, kurā ir mazāk nekā 10 % tauku. Analizējot dzīvnieku izcelsmes barību, kas satur vairāk nekā 10 % tauku, iekšējos standartus var pievienot pirms ekstrakcijas vai pēc tauku ekstrakcijas. Atbilstoši izvērtē ekstrakcijas efektivitāti atkarībā no tā, kurā analīzes posmā ievada iekšējos standartus, un no tā, vai rezultātus izsaka uz produkta vai tauku pamata. |
|
— |
Pirms GC/MS analīzes jāpievieno 1 vai 2 atgūstamības (surogāta) standarts(-i). |
|
— |
Nepieciešama atgūstamības kontrole. Attiecībā uz apstiprinājuma metodēm atsevišķo iekšējo standartu atgūstamībai jābūt diapazonā no 60 % līdz 120 %. Lielāka vai mazāka atsevišķu radniecīgo vielu, jo īpaši dažu hepta- un oktahloratvasināto dibenzodioksīnu un dibenzofurānu atgūstamība ir pieņemama ar nosacījumu, ka to daļa TEQ vērtībā nepārsniedz 10 % no kopējās TEQ vērtības (pamatojoties uz PCDD/F un dioksīniem līdzīgu PCB summu). Ja piemēro skrīninga metodes, tad atgūstamībai jābūt diapazonā no 30 % līdz 140 %. |
|
— |
Dioksīnus no tādiem traucējošiem hloratvasinātiem savienojumiem kā PCB, kas nav līdzīgi dioksīniem, un hloratvasinātiem difenilēteriem atdala ar piemērotām hromatogrāfijas metodēm (ieteicams ar florisila, alumīnija oksīda un/vai aktīvās ogles kolonnu). |
|
— |
Izomēru atdalīšana gāzu hromatogrāfijā ir pietiekama (1,2,3,4,7,8-HxCDF signāla attiecība pret 1,2,3,6,7,8-HxCDF signālu < 25 %). |
|
— |
Kvantitatīvo noteikšanu veic atbilstoši metodei EPA Method 1613 revision B: Tetra- līdz oktahlordioksīnus un furānus nosaka pēc izotopu atšķaidīšanas HRGC/HRMS vai citām metodēm, kurām ir līdzvērtīgi efektivitātes kritēriji. |
|
— |
Starpība starp augšējo robežu un apakšējo robežu nedrīkst pārsniegt 20 % attiecībā uz barību, kurā piesārņojums ar dioksīniem ir vienāds ar maksimāli pieļaujamo koncentrāciju vai pārsniedz to. Barībā, kurā piesārņojuma līmenis ir ievērojami zem maksimāli pieļaujamā, starpība drīkst būt diapazonā no 25 % līdz 40 %. |
7. Analīzes skrīninga metodes
7.1. Ievads
Izmantojot skrīninga metodi, var ievērot dažādas analītiskās pieejas: tīra skrīninga pieeja un kvantitatīvā pieeja.
Parauga signālu salīdzina ar standartparauga signālu vajadzīgajā koncentrācijā. Uzskata, ka paraugi, kuru signāls ir zem standartparauga signāla, ir negatīvi, bet paraugi, kuru signāls pārsniedz standartparauga signālu, ir iespējami pozitīvi. Prasības:
|
— |
visās analīžu sērijās jāiekļauj tukšais paraugs un standartparaugs vai paraugi, kurus identiskos apstākļos ekstrahē un analizē vienlaikus. Standartparauga signālam ievērojami jāpārsniedz tukšā parauga signāls, |
|
— |
iekļauj papildu standartparaugus, kuros attiecīgā koncentrācija ir 0,5 un divas reizes lielāka par vajadzīgo koncentrāciju, lai pierādītu analīzes efektivitāti vajadzīgajā diapazonā nolūkā kontrolēt vajadzīgo koncentrāciju, |
|
— |
analizējot citas matricas, jāparāda standartparauga(-u) piemērotība, vēlams, iekļaujot paraugus, kuros ar HRGC/HRMS noteiktā TEQ ir apmēram tāda pati kā standartparaugā vai tukšajā paraugā, kam pievienots standarts, kurā ir šāda koncentrācija, |
|
— |
tā kā biotestos nav iespējams izmantot iekšējos standartus, lai noteiktu vienas analīžu sērijas rezultātu standartnovirzi, ļoti būtiski ir noteikt atkārtojamību. Variāciju koeficients nedrīkst pārsniegt 30 %, |
|
— |
attiecībā uz biotestiem definē noteicamos savienojumus, iespējamie traucējumi un maksimāli pieļaujamā koncentrācija tukšajā paraugā. |
Kvantitatīvai noteikšanai vajadzīga standartatšķaidījumu rinda, divkārša vai trīskārša attīrīšana un mērīšana, kā arī tukšā parauga un atgūstamības kontrole. Rezultātu var izteikt ar TEQ, pieņemot, ka savienojumi, kas dod signālu, atbilst TEQ principam. To var veikt, izmantojot TCDD (vai dioksīnu/furānu/dioksīniem līdzīgu PCB standartmaisījumu), lai iegūtu kalibrēšanas līkni TEQ līmeņa noteikšanai ekstraktā un tātad paraugā. Pēc tam to koriģē atbilstīgi TEQ līmenim, ko aprēķina tukšajam paraugam (ņemot vērā lietoto šķīdinātāju un ķimikāliju piemaisījumus), un atgūstamībai (ko aprēķina pēc TEQ līmeņa kvalitātes kontroles paraugā, kurā ir aptuveni vajadzīgā koncentrācija). Noteikti jānorāda, ka daļa atgūstamības acīmredzamo zudumu var rasties matricas ietekmē un/vai starpības dēļ starp TEF vērtībām biotestos un oficiālajām TEF vērtībām, ko noteikusi PVO.
7.2. Prasības attiecībā uz skrīninga analīzes metodēm
|
— |
Skrīningam var izmantot GC/MS analīzes metodes un biotestus. Attiecībā uz GC/MS metodēm jāievēro 6. punktā noteiktās prasības. Šūnu biotestiem īpašas prasības noteiktas 7.3. punktā un komplektu biotestiem – 7.4 punktā. |
|
— |
Vajadzīga informācija par šķietami pozitīvu un šķietami negatīvu rezultātu skaitu no liela paraugu daudzuma, kurā ir paraugi, kuros nosakāmā koncentrācija ir zem un virs maksimāli pieļaujamās koncentrācijas vai iedarbības koncentrācijas salīdzinājumā ar TEQ saturu, kas kvantitatīvi noteikts ar apstiprinošas analīzes metodi. Faktiskajiem šķietami negatīvajiem rezultātiem jābūt mazāk par 1 %. Lai būtu lietderīgi izmantot skrīningu, šķietami pozitīvo rezultātu skaits ir pietiekami mazs. |
|
— |
Pozitīvie rezultāti vienmēr jāapstiprina, izmantojot apstiprinošas analīzes metodi (HRGC/HRMS). Turklāt plaša TEQ diapazona paraugus apstiprina ar HRGC/HRMS (aptuveni 2 % līdz 10 % negatīvo paraugu). Jāpublisko informācija par biotestu un HRGC/HRMS rezultātu atbilstību. |
7.3. Īpašas prasības attiecībā uz šūnu biotestiem
|
— |
Ja veic biotestus, tad katrā analīzes kārtā jāizmanto TCDD vai dioksīnu/furānu maisījumu standarta koncentrāciju rinda (pilnas devas signāla līkne ar R2 > 0,95). Tomēr, analizējot paraugus ar mazu koncentrāciju, skrīningam var izmantot izvērstu līkni nelielu koncentrāciju apgabalam. |
|
— |
TCDD standartkoncentrāciju (aptuveni trīskārša kvantitatīvās noteikšanas robeža) kvalitātes kontroles lapā izmanto kā biotesta rezultātu konstantā periodā. Alternatīva varētu būt standartparauga relatīvais signāls salīdzinājumā ar TCDD kalibrēšanas līkni, jo šūnu signāls var būt atkarīgs no daudziem faktoriem. |
|
— |
Izveido un pārbauda katra veida standartmateriālu kvalitātes kontroles grafikus, lai pārliecinātos, ka rezultāts atbilst izvirzītajām pamatnostādnēm. |
|
— |
Jo īpaši attiecībā uz kvantitatīvajiem aprēķiniem izmantotā parauga atšķaidījuma inducētajam signālam jābūt signāla līknes lineārajā daļā. Paraugi, kuru signāls ir virs signāla līknes lineārās daļas, jāatšķaida un jāanalizē atkārtoti. Tāpēc ieteicams vienā reizē analizēt vismaz trīs atšķaidījumus vai vairāk. |
|
— |
Katram parauga atšķaidījumam, ja to analizē trijos atkārtojumos, rezultāta procentuālā standartnovirze nepārsniedz 15 %, un, ja to analizē trijās neatkarīgās analīzēs – tad 30 %. |
|
— |
Var paredzēt, ka noteikšanas robeža ir šķīdinātāja tukšā parauga vai fona signāla trīskārša standartnovirze. Cita pieeja ir izmantot signālu, kas pārsniedz fona līmeni (indukcijas koeficients 5x šķīdinātāja tukšā parauga signāls), ko aprēķina pēc dienas kalibrēšanas līknes. Var paredzēt, ka kvantitatīvās noteikšanas robeža ir pieckārša vai seškārša šķīdinātāja tukšā parauga vai fona signāla standartnovirze, vai izmantot signālu, kas pārsniedz fona līmeni (indukcijas koeficients 10x šķīdinātāja tukšā parauga signāls), ko aprēķina pēc dienas kalibrēšanas līknes. |
7.4. Īpašas prasības attiecībā uz biotestiem, kuros izmanto komplektus
|
— |
Jānodrošina, ka komplektu biotesti ir pietiekami jutīgi un uzticami, lai tos varētu piemērot barībai. |
|
— |
Jāievēro izgatavotāja norādījumi par paraugu sagatavošanu un analīzēm. |
|
— |
Testu komplektus neizmanto pēc derīguma termiņa. |
|
— |
Neizmanto materiālus vai komponentus, ko paredzēts izmantot citos komplektos. |
|
— |
Testu komplektus glabā norādītā diapazona glabāšanas temperatūrā un izmanto analīzēm norādītajā darba temperatūrā. |
|
— |
Ir paredzēts, ka imūntestos noteikšanas robeža, pamatojoties uz tukšā parauga desmit atkārtotajām analīzēm, ir vienāda ar vidējās un trīskāršas standartnovirzes summu, kas dalīta ar lineārās regresijas vienādojuma virziena koeficientu. |
|
— |
Laboratorijas analīzēs izmanto etalonstandartus, lai pārliecinātos, ka reakcija uz standartu ir pieņemamās robežās. |
8. Rezultātu ziņošana
Analīžu rezultātos norāda atsevišķu PCDD/F un PCB radniecīgo vielu koncentrāciju – ciktāl to nodrošina izmantotā analīzes procedūra, un, lai analīžu rezultāti būtu iespējami informatīvi un interpretējami saskaņā ar īpašām prasībām, tie jānorāda kā augšējā robeža, apakšējā robeža vai vidējā robeža.
Ziņojumā norāda arī lipīdu saturu paraugā un lipīdu ektrakcijas metodi.
Ja atgūstamība ir ārpus 6. punktā minētā diapazona, gadījumos, kad ir pārsniegta maksimāli pieļaujamā koncentrācija, un citos gadījumos pēc pieprasījuma jānorāda atsevišķu iekšējo standartu atgūstamība.
Tā kā, lemjot par parauga atbilstību, jāņem vērā mērījumu neprecizitāte, sniedz arī šo rādītāju. Līdz ar to analīžu rezultātus paziņo kā “x +/– U”, kur x ir analīzes rezultāts un U – mērījumu paplašinātās neprecizitātes procents, izmantojot pārklāšanās koeficientu 2, kas nodrošina aptuveni 95 % ticamības pakāpi. Ja dioksīnus un dioksīniem līdzīgus PCB nosaka atsevišķi, tad atsevišķo dioksīnu un dioksīniem līdzīgu PCB analīžu rezultātu aprēķinātā izvērstās nenoteiktības summa jāizmanto kā dioksīnu un dioksīniem līdzīgu PCB summa.
Ja, piemērojot CCα (kā aprakstīts šīs B daļas I.2. punktā), tiek ņemta vērā mērījumu neprecizitāte, tad paziņo arī šo rādītāju.
(1) TEF (= toksiskuma ekvivalences faktoru) tabula dioksīniem, furāniem un dioksīniem līdzīgiem PCB.
|
Radniecīga viela |
TEF vērtība |
Radniecīga viela |
TEF vērtība |
|
Dibenzo-p-dioksīni (“PCDD”) |
|
“Dioksīniem līdzīgi” PCB: |
|
|
2,3,7,8-TCDD |
1 |
|
|
|
1,2,3,7,8-PeCDD |
1 |
Neorto PCB |
|
|
1,2,3,4,7,8-HxCDD |
0,1 |
PCB 77 |
0,0001 |
|
1,2,3,6,7,8-HxCDD |
0,1 |
PCB 81 |
0,0001 |
|
1,2,3,7,8,9-HxCDD |
0,1 |
PCB 126 |
0,1 |
|
1,2,3,4,6,7,8-HpCDD |
0,01 |
PCB 169 |
0,01 |
|
OCDD |
0,0001 |
Mono-orto PCB |
|
|
|
|
PCB 105 |
0,0001 |
|
Dibenzofurāni(“PCDF”) |
|
PCB 114 |
0,0005 |
|
2,3,7,8-TCDF |
0,1 |
PCB 118 |
0,0001 |
|
1,2,3,7,8-PeCDF |
0,05 |
PCB 123 |
0,0001 |
|
2,3,4,7,8-PeCDF |
0,5 |
PCB 156 |
0,0005 |
|
1,2,3,4,7,8-HxCDF |
0,1 |
PCB 157 |
0,0005 |
|
1,2,3,6,7,8-HxCDF |
0,1 |
PCB 167 |
0,00001 |
|
1,2,3,7,8,9-HxCDF |
0,1 |
PCB 189 |
0,0001 |
|
2,3,4,6,7,8-HxCDF |
0,1 |
|
|
|
1,2,3,4,6,7,8-HpCDF |
0,01 |
|
|
|
1,2,3,4,7,8,9-HpCDF |
0,01 |
|
|
|
OCDF |
0,0001 |
|
|
|
Izmantotie saīsinājumi: “T” = tetra, “Pe” = penta, “Hx” = heksa, “Hp” = hepta, “O” = okta, “CDD” = hlordibenzo-p-dioksīns, “CDF” = hlordibenzofurāns, “CB” = hlorbifenils. |
|||
(2) OV L 140, 30.5.2002., 10. lpp.
(3) “Augšējās robežas” jēdzienā jāizmanto kvantitatīvās noteikšanas robeža, lai aprēķinātu katras kvantitatīvi nenoteiktās radniecīgās vielas devumu toksiskā ekvivalenta koncentrācijā (TEQ).
“Apakšējās robežas” jēdzienā jāizmanto nulle, lai aprēķinātu katras kvantitatīvi nenoteiktās radniecīgās vielas devumu TEQ.
“Vidējās robežas” jēdzienā jāizmanto puse no kvantitatīvās noteikšanas robežas, lai aprēķinātu katras kvantitatīvi nenoteiktās radniecīgās vielas devumu TEQ.
(4) Otrreizējā analīze ir nepieciešama, lai izslēgtu iekšējas savstarpējas piesārņošanās iespēju vai paraugu nejaušu samainīšanu. Pirmo analīzi, ņemot vērā mērījumu neprecizitāti, izmanto atbilstības pārbaudei.
Ja analīzi veic dioksīna piesārņojuma gadījuma ietvaros, tad apstiprinājumu ar otrreizēju analīzi var neveikt, ja vien analīzei atlasītie paraugi caur izsekojamību ir saistīti ar dioksīna piesārņojuma gadījumu.
VI PIELIKUMS
ANALĪZES METODES, LAI NOTEIKTU DZĪVNIEKU IZCELSMES SASTĀV DAĻAS BARĪBAS OFICIĀLAI KONTROLEI
Nosacījumi dzīvnieku izcelsmes sastāvdaļu mikroskopiskai noteikšanai, identificēšanai vai aplēsēm barībā
1. Mērķis un piemērošanas joma
Minētie nosacījumi jāizmanto, ja ar mikroskopu nosaka dzīvnieku izcelsmes sastāvdaļas (ko definē kā zīdītāju, mājputnu un zivju liemeņu un liemeņu daļu pārstrādes produktus) barībā saskaņā ar koordinētās kontroles programmu dzīvnieku barošanas jomā, ņemot vērā Eiropas Parlamenta un Padomes Regulu (EK) Nr. 882/2004 (1). Ja šā pielikuma metodes izmanto visos oficiālajos testos, tad var veikt arī otru testu, izmantojot atšķirīgas vai alternatīvas metodes, lai uzlabotu dažu dzīvnieku izcelsmes sastāvdaļu veidu noteikšanu vai precizētu sīkāk dzīvnieku izcelsmes sastāvdaļu izcelsmi. Turklāt var izmantot atšķirīgu protokolu, pārbaudot atsevišķas dzīvnieku izcelsmes sastāvdaļas, piemēram, plazmu vai kaulus taukos (skatīt arī 9. punktu), ar noteikumu, ka šīs analīzes veic papildus tām, kas paredzētas koordinētajā kontroles programmā.
2. Jutība
Atkarībā no dzīvnieku izcelsmes sastāvdaļu īpašībām barībā iespējams atklāt ļoti nelielu daudzumu (< 0,1 %).
3. Princips
Identifikācijai izmanto reprezentatīvu paraugu, kas paņemts saskaņā ar I pielikumā paredzētajiem noteikumiem un ir piemēroti sagatavots. Šis protokols ir piemērots barības apstrādei, kurā ir neliels mitruma saturs. Barība, kuras mitruma saturs pārsniedz 14 %, pirms apstrādes jāžāvē (jākondensē). Īpašai barībai vai barības līdzekļiem (piemēram, taukiem, eļļām) vajadzīga speciāla apstrāde (skatīt 9. punktu). Dzīvnieku izcelsmes sastāvdaļas identificē, pamatojoties uz raksturīgiem, ar mikroskopu nosakāmiem elementiem (t. i., muskuļu šķiedras un citas gaļas daļiņas, skrimšļi, kauli, ragi, mati, sari, asinis, spalvas, olu čaumalas, asakas, zvīņas). Identifikācija jāveic gan parauga sijātā frakcijā (6.1.), gan koncentrētās nogulsnēs (6.2.).
4. Reaģenti
4.1. Ieslēdzošs aģents
|
4.1.1. |
Hloralhidrāts (ūdenī, 60 % w/v). |
|
4.1.2. |
Sārms (NaOH 2,5 % w/v vai KOH 2,5 % w/v) sijātām frakcijām. |
|
4.1.3. |
Parafīneļļa vai glicerīns (viskozitāte 68–81) mikroskopiskiem novērojumiem nogulsnēs. |
4.2. Skalošanas aģenti
|
4.2.1. |
Spirts, 96 %. |
|
4.2.2. |
Acetons. |
4.3. Koncentrēšanas aģents
|
4.3.1. |
Tetrahloretilēns (blīvums 1,62). |
4.4. Krāsošanas reaģenti
|
4.4.1. |
Joda/kālija jodīda šķīdums (izšķīdina 2 g kālija jodīda 100 mililitros ūdens un, bieži kratot, pievieno 1 g joda). |
|
4.4.2. |
Alizarīnsarkanais (atšķaida 2,5 ml 1M sālsskābes 100 mililitros ūdens un pievieno šim šķīdumam 200 mg alizarīnsarkanā). |
|
4.4.3. |
Cistīna reaģents (2 g svina acetāta, 10 g NaOH/100 ml H2O). |
|
4.4.4. |
Joda/kālija jodīda šķīdums (izšķīdināts 70 % etanolā). |
4.5. Balināšanas reaģents
|
4.5.1 |
Komerciāls nātrija hipohlorīta šķīdums (9,6 % aktīvā hlora). |
5. Iekārtas un piederumi
|
5.1. |
Analītiskie svari (ar precizitāti līdz 0,01 g, izņemot koncentrētās nogulsnes: 0,001 g). |
|
5.2. |
Materiāls malšanai (smalcinātājs vai piesta, jo īpaši barībai, kas analīzē satur vairāk par 15 % tauku). |
|
5.3. |
Siets ar maksimāli 0,50 mm platām kvadrātveida acīm. |
|
5.4. |
Dalāmpiltuve vai izgulsnēšanas vārglāze ar konisku dibenu. |
|
5.5. |
Stereomikroskops (ar palielinājumu līdz vismaz 40'). |
|
5.6. |
Saliktais mikroskops (ar palielinājumu līdz vismaz 400'), atstarota gaisma vai polarizēta gaisma. |
|
5.7. |
Laboratorijas standarta stikla trauki. Visam aprīkojumam jābūt rūpīgi notīrītam. Dalāmpiltuves un stikla trauki jāmazgā trauku mazgāšanas mašīnā. Sieti jātīra, izmantojot suku ar cietiem sariem. |
6. Procedūra
Granulētu barību var priekšsijāt, ja abas frakcijas analizē kā atsevišķus paraugus.
Apstrādā vismaz 50 g parauga (rūpīgi sasmalcina, izmantojot piemērotu smalcināšanas ierīci (5.2.), ja nepieciešams, lai iegūtu atbilstošu struktūru). No samaltā materiāla paņem divus reprezentatīvus paraugus, vienu sijātai frakcijai (vismaz 5 g) (6.1.) un vienu – koncentrētām nogulsnēm (vismaz 5 g) (6.2.). Identifikācijai papildus var izmantot iekrāsošanu ar krāsošanas reaģentiem (6.3.).
Lai norādītu dzīvnieku proteīnu veidu un daļiņu izcelsmi, var izmantot lēmumatbalsta sistēmu, piemēram, ARIES un var noformēt kontrolparaugus.
6.1. Dzīvnieku izcelsmes sastāvdaļu identifikācija sijātās frakcijās
Vismaz 5 g parauga izsijā caur sietu (5.3.) divās frakcijās.
Sijāto(-ās) frakciju(-as) ar lielām daļiņām (vai frakcijas reprezentatīvu daļu) izmanto plānas kārtiņas veidā uz piemērota balsta un regulāri pārbauda zem stereomikroskopa (5.5.) ar dažādiem palielinājumiem attiecībā uz dzīvnieku izcelsmes sastāvdaļām.
Priekšmetstikliņus ar sijāto(-ām) frakciju(-ām) ar smalkām daļiņām regulāri pārbauda zem saliktā mikroskopa (5.6.) ar dažādiem palielinājumiem attiecībā uz dzīvnieku izcelsmes sastāvdaļām.
6.2. Dzīvnieku izcelsmes sastāvdaļu identifikācija koncentrētās nogulsnēs
Vismaz 5 g parauga (ar precizitāti līdz 0,01 g) pārnes dalāmpiltuvē vai izgulsnēšanas vārglāzē ar konisku dibenu un apstrādā ar vismaz 50 ml tetrahloretilēna (4.3.1.). Maisījumu atkārtoti krata vai samaisa.
|
— |
Ja izmanto slēgtu dalāmpiltuvi, tad nogulsnes atstāj nostāvēties pietiekamu laiku (vismaz trīs minūtes) pirms nogulšņu atdalīšanas. Kratīšanu atkārto un nogulsnes atkal atstāj nostāvēties vismaz trīs minūtes. Nogulsnes atdala vēlreiz. |
|
— |
Ja izmanto vaļēju vārglāzi, tad nogulsnes atstāj nostāvēties vismaz piecas minūtes pirms nogulšņu atdalīšanas. |
Kopējo nogulšņu daudzumu izžāvē un pēc tam nosver (ar precizitāti līdz 0,001 g). Svēršana vajadzīga tikai tad, ja jāveic novērtējums. Ja nogulsnes sastāv no daudz lielām daļiņām, tad tās var izsijāt caur sietu (5.3.) divās frakcijās. Izžāvētās nogulsnēs ar stereomikroskopu (5.5.) un salikto mikroskopu (5.6.) pārbauda, vai nav kaulu sastāvdaļas.
6.3. Ieslēdzošo aģentu un krāsošanas reaģentu izmantošana
Lai mikroskopiski identificētu dzīvnieku izcelsmes sastāvdaļas, var izmantot īpašus ieslēdzošus aģentus un krāsošanas reaģentus.
Hloralhidrāts (4.1.1.). Uzmanīgi karsējot, var skaidrāk saskatīt šūnu struktūras, jo cietes graudi želatinizējas un nevēlamais šūnu saturs tiek atdalīts.
Sārms (4.1.2.). Vai nu nātrija hidroksīds, vai kālija hidroksīds dzidrina barības materiālu, palīdzot noteikt muskuļu šķiedras, matus un citas keratīna struktūras.
Parafīneļļa un glicerīns (4.1.3.). Šajā ieslēdzošajā aģentā var labi identificēt kaulu sastāvdaļas, jo vairākums lakūnu tiek piepildītas ar gaisu un parādās kā apmēram 5–15 μm melni caurumi.
Joda/kālija jodīda šķīdums (4.4.1.). Izmanto cietes (zila – violeta krāsa) un proteīna (dzeltena – oranža krāsa) noteikšanai. Vajadzības gadījumā šķīdumus var atšķaidīt.
Alizarīnsarkanā šķīdums (4.4.2.). Kaulu, asaku un zvīņu krāsojums sarkanā/rozā krāsā. Pirms nogulšņu žāvēšanas (skatīt 6.2. punktu) kopējās nogulsnes pārnes stikla mēģenē un divreiz noskalo apmēram ar 5 ml spirta (4.2.1.) (katru reizi izmanto virpuļmikseri, šķīdinātājam ļauj apmēram vienu minūti nogulsnēties, tad to nolej). Pirms šā krāsošanas reaģenta izmantošanas nogulsnes balina, pievienojot vismaz 1 ml nātrija hipohlorīta šķīdumu (4.5.1.). Reakcijai ļauj turpināties 10 minūtes. Mēģeni piepilda ar ūdeni, nogulsnēm ļauj nogulsnēties 2–3 minūtes un ūdeni un suspendētās daļiņas nolej. Nogulsnes vēl divreiz noskalo apmēram ar 10 ml ūdens (izmanto virpuļmikseri, ļauj nogulsnēties un ūdeni katru reizi nolej). Pievieno divus līdz desmit vai vairāk pilienus (atkarībā no atlikuma daudzuma) alizarīnsarkanā šķīduma. Maisījumu sakrata un reakcijai ļauj turpināties dažas sekundes. Iekrāsotās nogulsnes divreiz noskalo apmēram ar 5 ml spirta (4.2.1.), pēc tam vienreiz ar acetonu (4.2.2.) (katru reizi izmanto virpuļmikseri, šķīdinātājam ļauj nogulsnēties apmēram vienu minūti, tad to nolej). Tad nogulsnes ir gatavas žāvēšanai.
Cistīna reaģents (4.4.3.). Uzmanīgi karsējot, cistīnu saturošās sastāvdaļas (mati, spalvas u.c.) kļūst melni brūnas.
6.4. Tādas barības pārbaude, kas, iespējams, satur zivju miltus
Vismaz vienu priekšmetstikliņu pārbauda no smalki sijātas frakcijas un no nogulšņu smalkās frakcijas zem saliktā mikroskopa (skatīt 6.1. un 6.2. punktu).
Ja marķējums norāda, ka sastāvdaļās ir zivju milti, vai ja uzskata vai atklāj zivju miltus sākotnējā pārbaudē, tad pārbauda vismaz vēl divus smalki sijātās frakcijas priekšmetstikliņus no sākotnējā parauga un no kopējās nogulšņu frakcijas.
7. Aprēķināšana un novērtēšana
Dalībvalstis nodrošina, ka šajā punktā aprakstītās procedūras izmanto, veicot oficiālu analīzi, lai novērtētu dzīvnieku izcelsmes sastāvdaļu daudzumu (ne tikai klātbūtni).
Aprēķinus var veikt vienīgi tad, ja dzīvnieku izcelsmes sastāvdaļās ir kaulu fragmenti.
Sauszemes siltasiņu dzīvnieku sugu (t. i., zīdītāju un putnu) kaulu fragmentus uz mikroskopa priekšmetstikliņa var atšķirt no dažāda veida asakām pēc raksturīgās lakūnas. Dzīvnieku izcelsmes sastāvdaļu proporcionālo daudzumu parauga materiālā novērtē, ņemot vērā:
|
— |
novērtēto kaulu fragmentu proporcionālo daudzumu (svara %) koncentrētās nogulsnēs un |
|
— |
kaulu proporcionālo daudzumu (svara %) dzīvnieku izcelsmes sastāvdaļās. |
Novērtējumā jāņem vērā vismaz trīs (ja iespējams) priekšmetstikliņi un vismaz pieci laukumi katram priekšmetstikliņam. Barības maisījumos koncentrētajās nogulsnēs parasti ir ne vien sauszemes dzīvnieku kaulu un zivju asaku fragmenti, bet arī citas daļiņas, kam piemīt ļoti īpatnējs svars, piemēram, minerāli, smiltis, pārkoksnējušās augu daļas un tamlīdzīgi.
7.1. Kaulu fragmentu novērtētā procentuālā vērtība
% sauszemes dzīvnieku kaulu fragmenti = (S × c)/W
% zivju asaku un zvīņu fragmenti = (S × d)/W
|
(S = |
nogulšņu svars (mg), c = korekcijas koeficients (%) aplēstajai sauszemes dzīvnieku kaulu daļai nogulsnēs, d = korekcijas koeficients (%) aplēstajai zivju asaku un zvīņu daļai nogulsnēs, W = nogulsnēšanās paraugu materiālu svars (mg).) |
7.2. Dzīvnieku izcelsmes sastāvdaļu aplēstā vērtība
Kaulu proporcija dzīvnieku produktos var būt ļoti dažāda. (Kaulu procentuālais daudzums kaulu miltiem ir 50 līdz 60 % un gaļas miltiem ir 20 līdz 30 %; zivju miltiem asaku un zvīņu saturs ir atšķirīgs atkarībā no zivju miltu kategorijas un izcelsmes, parasti tas ir 10 līdz 20 %.)
Ja ir zināms, kāda veida dzīvnieku milti atrodas paraugā, tad ir iespējams aplēst saturu.
Sauszemes dzīvnieku produktu sastāvdaļu aplēstais saturs (%) = (S × c)/(W × f) × 100.
Zivju produktu sastāvdaļu aplēstais saturs (%) = (S × d)/(W × f) × 100.
|
(S = |
nogulšņu svars (mg), c = korekcijas koeficients (%) aplēstajai sauszemes dzīvnieku kaulu daļai nogulsnēs, d = korekcijas koeficients (%) aplēstajai zivju asaku un zvīņu daļai nogulsnēs, f = korekcijas koeficients kaulu daļai pārbaudāmajā paraugā esošajām dzīvnieku izcelsmes sastāvdaļām, W = nogulsnēšanās paraugu materiālu svars (mg) .) |
8. Pārbaudes rezultātu izteikšana
Ziņojumā jābūt vismaz informācijai par to sastāvdaļu klātbūtni, kas radušās no sauszemes dzīvniekiem un zivju miltiem. Dažādos gadījumus paziņo šādi.
|
8.1. |
Attiecībā uz to sastāvdaļu klātbūtni, kas radušās no sauszemes dzīvniekiem:
|
|
8.2. |
Un attiecībā uz zivju miltu klātbūtni:
Ja atrod sastāvdaļas, kas radušās no zivīm vai sauszemes dzīvniekiem, tad pārbaudes rezultātu ziņojumā vajadzības gadījumā var sīkāk norādīt atrasto sastāvdaļu daudzuma novērtējumu (x %, < 0,1 %, 0,1–0,5 %, 0,5–5 % vai > 5 %), sauszemes dzīvnieka veida, ja iespējams, un identificēto sastāvdaļu sīkāku specifikāciju (muskuļu šķiedras, skrimšļi, kauli, ragi, mati, sari, spalvas, asinis, olu čaumalas, asakas, zvīņas). Ja aplēš dzīvnieku izcelsmes sastāvdaļu daudzumu, tad piemin izmantoto korekcijas koeficientu f. Attiecībā uz gadījumiem, kad identificē sauszemes dzīvnieku kaulu sastāvdaļas, ziņojumā iekļauj papildu klauzulu: “Nevar izslēgt iespēju, ka iepriekš minētās sastāvdaļas ir radušās no zīdītājiem.” Šī papildu klauzula nav vajadzīga, ja sauszemes dzīvnieku kaulu fragmenti ir uzskaitīti kā mājputnu vai zīdītāju kaulu fragmenti. |
9. Fakultatīvais protokols tauku vai eļļas analīzei
Šo protokolu var izmantot tauku vai eļļas analīzei.
|
— |
Ja tauki ir cieti, tad tos sasilda, piemēram, mikroviļņu krāsnī, līdz tie ir šķidri. |
|
— |
Izmantojot pipeti, 40 ml tauku no parauga apakšpuses pārnes uz centrifugēšanas mēģeni. |
|
— |
Centrifugē 10 minūtes pie 4 000 apgr./min. |
|
— |
Ja tauki ir cieti pēc centrifugēšanas, tad tos vēlreiz sasilda krāsnī, līdz tie ir šķidri. Vēlreiz centrifugē piecas minūtes pie 4 000 apgr./min. |
|
— |
Izmantojot nelielu karoti vai lāpstiņu, pusi no dekantētā piemaisījuma pārnes uz nelielu Petri trauciņu vai mikroskopa priekšmetstikliņu, lai ar mikroskopu identificētu iespējamo dzīvnieku izcelsmes sastāvdaļu saturu (gaļas šķiedras, spalvas, kaulu fragmenti, …). Kā mikroskopijas ieslēdzošais aģents ieteicama parafīneļļa vai glicerīns. |
|
— |
Pārējos piemaisījumus izmanto nogulsnēšanai, kā aprakstīts 6.2. punktā. |
VII PIELIKUMS
METODE MĀJPUTNU BARĪBAS ENERĢĒTISKĀS VĒRTĪBAS APRĒĶINĀŠANAI
1. Metode enerģētiskās vērtības aprēķināšanai un izteikšanai
Mājputnu barības maisījumu enerģētiskā vērtība jāaprēķina, izmantojot tālāk izklāstīto formulu, pamatojoties uz noteiktu barības analītisko komponentu procentuālajām attiecībām. Šo vērtību izsaka megadžoulos (MJ) no vielmaiņas rezultātā pārveidotās enerģijas (ME), koriģējot to slāpeklim, uz vienu kilogramu barības maisījuma:
MJ/kg ME = 0,1551 × % kopproteīna +0,3431 × % jēltauku +0,1669 × % cietes +0,1301 × % kopējo cukuru (kas izteikti kā saharoze).
2. Deklarētajām vērtībām piemērojamās pielaides
Ja oficiālā pārbaudē atklāj neatbilstību (lielāku vai mazāku barības enerģētisko vērtību) starp pārbaudes rezultātu un deklarēto enerģētisko vērtību, tad minimālā pieļaujamā pielaide ir 0,4 MJ/kg ME.
3. Rezultāta izteikšana
Pēc iepriekš minētās formulas piemērošanas iegūtais rezultāts jānorāda līdz vienam ciparam aiz komata.
4. Paraugu ņemšanas un analīzes metodes
Barības maisījumu paraugu ņemšana un analītisko komponentu satura noteikšana, kas norādīta aprēķināšanas metodē, jāveic attiecīgi atbilstoši Kopienas paraugu ņemšanas metodēm un analīzes metodēm barības oficiālās kontroles veikšanai.
Jāpiemēro:
|
— |
jēltauku satura noteikšanai: III pielikuma H daļā noteiktā B procedūra jēleļļu un jēltauku noteikšanas metodei, |
|
— |
cietes satura noteikšanai: III pielikuma L daļā noteiktā polarimetrijas metode. |
VIII PIELIKUMS
ANALĪZES METODES, LAI KONTROLĒTU VAIRS NEATĻAUTU PIEDEVU NELIKUMĪGU KLĀTBŪTNI BARĪBĀ
Svarīgas piezīmesLai noteiktu vairs neatļautu piedevu nelikumīgu klātbūtni barībā, var izmantot jutīgākas analīzes metodes nekā šajā pielikumā minētās.Šajā pielikumā minētās analīzes metodes var izmantot apstiprināšanas nolūkos.
A. METILBENZOKVĀTA NOTEIKŠANA
7-benziloksi-6-butil-3-metoksikarbonil-4-hinolīns
1. Mērķis un darbības joma
Šī metode ļauj noteikt metilbenzokvāta daudzumu barībā. Kvantitatīvās noteikšanas robeža ir 1 mg/kg.
2. Princips
Metilbenzokvātu ekstrahē no parauga ar metānsulfoskābes šķīdumu metanolā. Ekstraktu attīra ar dihlormetānu, ar jonu apmaiņas hromatogrāfiju un tad atkal ar dihlormetānu. Metilbenzokvāta saturu nosaka apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Dihlormetāns. |
|
3.2. |
Metanols, ekvivalents HPLC tīrības pakāpei. |
|
3.3. |
HPLC kustīgā fāze. Metanola (3.2.) un ūdens (kas ekvivalents HPLC tīrības pakāpei) maisījums, piemēram, 75 +25 (v + v). Šķīdumu filtrē caur 0,22 μm filtru (4.5.) un atgāzo (piemēram, 10 minūtes apstrādājot ar ultraskaņu). |
|
3.4. |
Metānsulfoskābes šķīdums, c = 2 %. 20,0 ml metānsulfoskābes atšķaida līdz 1 000 ml tilpumam ar metanolu (3.2.). |
|
3.5. |
Sālsskābes šķīdums, c = 10 %. 100 ml sālsskābes (ρ201,18 g/ml) atšķaida līdz 1 000 ml tilpumam ar ūdeni. |
|
3.6. |
Katjonu apmaiņas Amberlita sveķi CG-120 (Na), 100 līdz 200 acojuma. Sveķus pirms izmantošanas iepriekš apstrādā. 100 g sveķu apstrādā ar 500 ml sālsskābes šķīduma (3.5.) un, pastāvīgi maisot, karsē uz elektriskās plītiņas līdz viršanai. Ļauj atdzist un dekantē skābi. Vakuumā izfiltrē caur filtrpapīru. Sveķus divas reizes mazgā ar 500 ml ūdens devām un pēc tam ar 250 ml metanola (3.2.). Sveķus skalo vēl ar 250 ml metanola un ar caurplūstošu gaisu žāvē nogulsnes, kas paliek uz filtra. Nožāvētos sveķus glabā ar aizbāzni noslēgtā pudelē. |
|
3.7. |
Standartviela: tīrs metilbenzokvāts (7-benziloksi-6-butil-3-metoksikarbonil-4-hinolīns). |
|
3.7.1. |
Ar precizitāti līdz 0,1 mg iesver 50 mg standartvielas (3.7.), izšķīdina metānsulfoskābes šķīdumā (3.4.) 100 ml mērkolbā, papildina līdz zīmei un samaisa. |
|
3.7.2. |
5,0 ml metilbenzokvāta izejas standartšķīduma (3.7.1.) pārnes 50 ml mērkolbā, papildina līdz zīmei ar metanolu (3.2.) un samaisa. |
|
3.7.3. |
Pārnes 1,0, 2,0, 3,0, 4,0 un 5,0 ml metilbenzokvāta starpposma standartšķīduma (3.7.2.) 25 ml mērkolbu komplektos. Papildina līdz zīmei ar kustīgo fāzi (3.3.) un samaisa. Minēto šķīdumu koncentrācija ir attiecīgi 2,0, 4,0, 6,0, 8,0 un 10,0 μg/ml metilbenzokvāta. Minētie šķīdumi jāsagatavo svaigi pirms izmantošanas. |
4. Ierīces
|
4.1. |
Laboratorijas kratītājs. |
|
4.2. |
Rotācijas iztvaicētājs. |
|
4.3. |
Stikla kolonna (250 × 15 mm), kas aprīkota ar krānu un aptuveni 200 ml tilpuma rezervuāru. |
|
4.4. |
HPLC iekārta, kurai ir ultravioletais detektors ar maināmu viļņu garumu vai diodes matricu detektors. |
|
4.4.1. |
Šķidrumu hromatogrāfa kolonna: 300 × 4 mm, C18, 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.5. |
Membrānfiltri, 0,22 μm. |
|
4.6. |
Membrānfiltri, 0,45 μm. |
5. Procedūra
5.1. Vispārīgas norādes
|
5.1.1. |
Lai pārbaudītu, ka barībā nav metilbenzokvāta un traucējošu vielu, veic tukšo eksperimentu. |
|
5.1.2. |
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz metilbenzokvāta, cik ir paraugā. Lai stiprinātu paraugu 15 mg/kg līmenī, 600 μl izejas standartšķīduma (3.7.1.) pievieno 20 gramiem tukšā barības parauga, samaisa un gaida 10 minūtes, tad pāriet pie ekstrakcijas posma (5.2.). Piezīme. Šīs metodes nolūkos tukšais barības paraugs ir tāda paša veida kā analizējamais paraugs, un analīzē nedrīkst uzrādīties metilbenzokvāts. |
5.2. Ekstrakcija
Ar precizitāti līdz 0,01 g iesver apmēram 20 g sagatavotā parauga un pārnes 250 ml koniskā kolbā. Pievieno 100,0 ml metānsulfoskābes šķīduma (3.4.) un 30 minūtes mehāniski krata (4.1.). Izfiltrē šķīdumu caur filtrpapīru un filtrātu glabā divu šķidro fāžu atdalīšanai (5.3.).
5.3. Divu šķidro fāžu atdalīšana
Uz 500 ml dalāmpiltuvi, kurā ir 100 ml sālsskābes šķīduma (3.5.), pārnes 25,0 ml iegūtā filtrāta (5.2.). Pievieno 100 ml dihlormetāna (3.1.) un krata vienu minūti. Ļauj slāņiem atdalīties un nolaiž apakšējo (dihlormetāna) slāni 500 ml apaļkolbā. Ūdens fāzes ekstrakciju atkārto ar vēl divām 40 ml dihlormetāna devām un apvieno tās ar pirmo ekstraktu apaļkolbā. Ar rotācijas iztvaicētāju (4.2.) pazeminātā spiedienā 40 oC temperatūrā iztvaicē dihlormetāna ekstraktu līdz sausam stāvoklim. Izšķīdina atlikumu 20 līdz 25 mililitros metanola (3.2.), ar aizbāzni noslēdz kolbu un visu ekstraktu glabā jonu apmaiņas hromatogrāfijai (5.4.).
5.4. Jonu apmaiņas hromatogrāfija
5.4.1.
Stikla kolonnas (4.3.) apakšējā galā ieliek stikla vates tamponu. Sagatavo 5,0 g apstrādāto katjonu apmaiņas sveķu (3.6.) un 50 ml sālsskābes (3.5.) uzduļķojuma, ielej stikla kolonnā un ļauj nostāties. Nolaiž lieko skābi tā, lai skābes līmenis ir tikai nedaudz virs sveķu virsmas, un mazgā kolonnu ar ūdeni, līdz izplūde ir neitrāla pret lakmusu. 50 ml metanola (3.2.) pārnes uz kolonnu un ļauj notecēt līdz sveķu virsmai.
5.4.2.
Ar pipeti uzmanīgi pārnes iegūto ekstraktu (5.3.) uz kolonnu. Izskalo apaļkolbu ar divām 5 līdz 10 ml devām metanola (3.2.) un saskalojumus pārnes uz kolonnu. Nodrošinot, ka plūsmas ātrums nepārsniedz 5 ml minūtē, ekstraktu nolaiž līdz sveķu virsmai un kolonnu izmazgā ar 50 ml metanola. Efluentu izlej. Ar 150 ml metānsulfoskābes šķīduma (3.4.) no kolonnas eluē metilbenzokvātu un kolonnas eluātu savāc 250 ml koniskā kolbā.
5.5. Divu šķidro fāžu atdalīšana
Pārnes iegūto eluātu (5.4.2.) viena litra dalāmpiltuvē. Izskalo konisko kolbu ar 5 līdz 10 ml metanola (3.2.) un saskalojumus apvieno ar dalāmpiltuves saturu. Pievieno 300 ml sālsskābes šķīduma (3.5.) un 130 ml dihlormetāna (3.1.). Krata vienu minūti un ļauj fāzēm atdalīties. Nolaiž apakšējo (dihlormetāna) slāni 500 ml apaļkolbā. Ūdens fāzes ekstrakciju atkārto ar vēl divām 70 ml dihlormetāna devām un šos ekstraktus apvieno ar pirmo ekstraktu apaļkolbā.
Ar rotācijas iztvaicētāju (4.2.) pazeminātā spiedienā 40 oC temperatūrā iztvaicē dihlormetāna ekstraktu līdz sausam stāvoklim. Atlikumu izšķīdina kolbā, kur ir apmēram 5 ml metanola (3.2.), un šo šķīdumu kvantitatīvi pārnes uz 10 ml mērkolbu. Izskalo apaļkolbu ar vēl divām 1 līdz 2 ml devām metanola un saskalojumus pārnes uz mērkolbu. Papildina līdz zīmei ar metanolu un samaisa. Alikvotu devu izfiltrē caur membrānfiltru (4.6.). Šo šķīdumu pataupa HPLC noteikšanai (5.6.).
5.6. HPLC noteikšana
5.6.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus:
|
— |
šķidrumu hromatogrāfa kolonna (4.4.1.), |
|
— |
HPLC kustīgā fāze: metanola un ūdens maisījums (3.3.), |
|
— |
plūsmas ātrums: 1 līdz 1,5 ml/min, |
|
— |
noteikšanas viļņa garums: 265 nm, |
|
— |
injekcijas tilpums: 20 līdz 50 μl. |
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.7.3.) ar koncentrāciju 4 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus vai laukumus un izdalīšanas laiku.
5.6.2.
Katru kalibrēšanas šķīdumu (3.7.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas grafiku, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus vai laukumus un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.6.3.
Iešpricē parauga ekstraktu (5.5.) vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumiem, un nosaka metilbenzokvāta maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
Ņemot vērā parauga šķīduma metilbenzokvāta maksimumu vidējo augstumu (laukumu), nosaka parauga šķīduma koncentrāciju μg/ml, izmantojot kalibrēšanas grafiku (5.6.2.).
Metilbenzokvāta saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
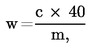
kur:
|
c |
= |
metilbenzokvāta koncentrācija parauga šķīdumā, g/ml, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta spektru ar tā kalibrēšanas šķīduma (3.7.3.) spektru, kura koncentrācija ir 10 μg/ml.
7.1.1.
Parauga ekstraktu stiprina, pievienojot attiecīgu daudzumu starpposma standartšķīduma (3.7.2.). Pievienotā metilbenzokvāta daudzumam jābūt tādam pašam kā aplēstajam metilbenzokvāta daudzumam, kas atrasts parauga ekstraktā.
Tikai metilbenzokvāta maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā maksimālā augstuma jābūt apmēram 10 % no sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir apmēram 2 nm; |
|
b) |
no 220 līdz 350 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 220 līdz 350 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt 10 % no lielākā rezultāta, ja metilbenzokvāta saturs ir starp 4 un 20 mg/kg.
7.3. Atgūstamība
Stiprināta tukšā parauga atgūstamība ir vismaz 90 %.
8. Koppētījuma rezultāti
Desmit laboratorijas analizēja piecus paraugus. Visus paraugus analizēja divas reizes.
|
|
Tukšais |
Milti 1 |
Granulas 1 |
Milti 2 |
Granulas 2 |
|
Vidējais [mg/kg] |
NN |
4,50 |
4,50 |
8,90 |
8,70 |
|
sr [mg/kg] |
— |
0,30 |
0,20 |
0,60 |
0,50 |
|
CVr [ %] |
— |
6,70 |
4,40 |
6,70 |
5,70 |
|
sR [mg/kg] |
— |
0,40 |
0,50 |
0,90 |
1,00 |
|
CVR [ %] |
— |
8,90 |
11,10 |
10,10 |
11,50 |
|
Atgūstamība [ %] |
— |
92,00 |
93,00 |
92,00 |
89,00 |
|
NN |
= |
nav noteikts, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients (%), |
|
sR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients (%), |
B. OLAHINDOKSA NOTEIKŠANA
2-[N-2'-(hidroksietil)karbamoil]-3-metilhinoksalīn-N 1 ,N 4 -dioksīds
1. Mērķis un darbības joma
Šī metode ļauj noteikt olahindoksa daudzumu barībā. Kvantitatīvās noteikšanas robeža ir 5 mg/kg.
2. Princips
Paraugu ekstrahē ar ūdens un metanola maisījumu. Olahindoksa saturu nosaka apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Metanols. |
|
3.2. |
Metanols, ekvivalents HPLC tīrības pakāpei. |
|
3.3. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
|
3.4. |
Kustīgā fāze priekš HPLC. Ūdens (3.3.) un metanola (3.2.) maisījums, 900 + 100 (V + V). |
|
3.5. |
Standartviela: tīrs olahindokss 2-[N-2'-(hidroksietil)karbamoil]-3-metilhinoksalīn-N1,N4-dioksīds, E 851. |
|
3.5.1. |
Ar precizitāti līdz 0,1 mg iesver 50 mg olahindoksa (3.5.) 200 ml mērkolbā un pievieno apmēram 190 ml ūdens. Tad kolbu uz 20 minūtēm ievieto ultraskaņas vannā (4.1.). Pēc apstrādes ar ultraskaņu šķīdumu atdzesē līdz istabas temperatūrai, papildina līdz zīmei ar ūdeni un samaisa. Ietin kolbu alumīnija folijā un glabā ledusskapī. Minētais šķīdums jāsagatavo svaigs katru mēnesi. |
|
3.5.2. |
10,0 ml izejas standartšķīduma (3.5.1.) pārnes 100 ml mērkolbā, papildina līdz zīmei ar kustīgo fāzi (3.4.) un samaisa. Ietin kolbu alumīnija folijā un glabā ledusskapī. Minētais šķīdums jāsagatavo svaigs katru dienu. |
|
3.5.3. |
50 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 5,0, 10,0, 15,0 un 20,0 ml starpposma standartšķīduma (3.5.2.) Papildina līdz zīmei ar kustīgo fāzi (3.4.) un samaisa. Ietin kolbas alumīnija folijā. Minētie šķīdumi atbilst attiecīgi šādām olahindoksa koncentrācijām: 0,5, 1,0, 2,5, 5,0, 7,5 un 10,0 μg/ml. Minētie šķīdumi jāsagatavo svaigi katru dienu. |
4. Ierīces
|
4.1. |
Ultraskaņas vanna. |
|
4.2. |
Mehāniskais kratītājs. |
|
4.3. |
HPLC iekārta, kurai ir ultravioletais detektors ar maināmu viļņu garumu vai diodes matricu detektors. |
|
4.3.1. |
Šķidrumu hromatogrāfa kolonna, 250 × 4 mm, C18, 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.4. |
Membrānfiltri, 0,45 μm. |
5. Procedūra
|
Piezīme. |
Olahindokss ir gaismjutīgs. Visas darbības veic izkliedētā gaismā vai lieto brūnā stikla traukus. |
5.1. Vispārīgas norādes
|
5.1.1. |
Lai pārbaudītu, ka barībā nav olahindoksa un traucējošu vielu, veic tukšo eksperimentu. |
|
5.1.2. |
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz olahindoksa, cik ir paraugā. Lai paraugu pastiprinātu līdz līmenim 50 mg/kg, 10,0 ml izejas standartšķīduma (3.5.1.) pārnes 250 ml koniskā kolbā un šķīdumu iztvaicē līdz apmēram 0,5 ml. Pirms ekstrakcijas (5.2.) pievieno 50 g tukšā barības parauga, vairākas reizes kārtīgi samaisa un atstāj uz 10 minūtēm.
|
5.2. Ekstrakcija
Ar precizitāti līdz 0,01 g iesver apmēram 50 g parauga. Pārnes 1 000 ml koniskā kolbā, pievieno 100 ml metanola (3.1.) un kolbu uz piecām minūtēm ievieto ultraskaņas vannā (4.1.). Pievieno 410 ml ūdens un atstāj ultraskaņas vannā vēl uz 15 minūtēm. Izņem kolbu no ultraskaņas vannas, 30 minūtes krata uz kratītāja (4.2.) un izfiltrē caur kroku filtru. 10,0 ml filtrāta pārnes 20 ml mērkolbā, papildina līdz zīmei ar ūdeni un samaisa. Alikvotu izfiltrē caur membrānfiltru (4.4.) (skatīt 9. novērojumu). Pāriet pie HPLC noteikšanas (5.3.).
5.3. HPLC noteikšana
5.3.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Analītiskā kolonna (4.3.1.) |
|
|
Kustīgā fāze (3.4.): |
ūdens (3.3.) un metanola (3.2.) maisījums, 900 + 100 (V + V) |
|
Plūsmas ātrums: |
1,5–2 ml/min |
|
Noteikšanas viļņa garums: |
380 nm |
|
Injekcijas tilpums: |
20–100 μl |
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.5.3.) ar koncentrāciju 2,5 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus un izdalīšanas laiku.
5.3.2.
Katru kalibrēšanas šķīdumu (3.5.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas grafiku, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus (laukumus) un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.3.3.
Iešpricē parauga ekstraktu (5.2.) vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumiem, un nosaka olahindoksa maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
Ņemot vērā olahindoksa maksimumu vidējo augstumu (laukumu) parauga šķīdumā, nosaka parauga šķīduma koncentrāciju μg/ml, izmantojot kalibrēšanas grafiku (5.3.2.).
Olahindoksa saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
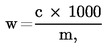
kur:
|
c |
= |
olahindoksa koncentrācija parauga ekstraktā (5.2.), μg/ml, |
|
m |
= |
analizējamā parauga svars g/ml (5.2.). |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta (5.2.) spektru ar tā kalibrēšanas šķīduma (3.5.3.) spektru, kura koncentrācija ir 5,0 μg/ml.
7.1.1.
Parauga ekstraktu (5.2.) stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.5.3.). Pievienotā olahindoksa daudzumam jābūt tādam pašam kā aplēstajam olahindoksa daudzumam, kas atrasts parauga ekstraktā.
Tikai olahindoksa maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā augstuma jābūt ± 10 % robežās no nepastiprināta parauga ekstrakta olahindoksa maksimuma sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir ± 2 nm; |
|
b) |
no 220 līdz 400 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 220 līdz 400 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
Starpība starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas vienam paraugam, nedrīkst pārsniegt 15 % attiecībā pret lielāko rezultātu attiecībā uz olahindoksu, kura saturs ir no 10 līdz 200 mg/kg.
7.3. Atgūstamība
Stiprināta tukšā parauga atgūstamība ir vismaz 90 %.
8. Koppētījuma rezultāti
EK organizētajā koppētījumā līdz 13 laboratorijām analizēja četrus sivēnu barības paraugus, no kuriem viens bija tukšais paraugs. Analīžu rezultāti ir šādi.
|
|
1. paraugs |
2. paraugs |
3. paraugs |
4. paraugs |
|
L |
13 |
10 |
11 |
11 |
|
n |
40 |
40 |
44 |
44 |
|
vidējais [mg/kg] |
— |
14,6 |
48,0 |
95,4 |
|
Sr [mg/kg] |
— |
0,82 |
2,05 |
6,36 |
|
SR [mg/kg] |
— |
1,62 |
4,28 |
8,42 |
|
CVr [ %] |
— |
5,6 |
4,3 |
6,7 |
|
CVR [ %] |
— |
11,1 |
8,9 |
8,8 |
|
Nominālais saturs |
|
|
|
|
|
[mg/kg] |
— |
15 |
50 |
100 |
|
Atgūstamība % |
— |
97,3 |
96,0 |
95,4 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
sR |
= |
atveidojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
CVR |
= |
atveidojamības variācijas koeficients. |
9. Novērojums
Lai gan metode nav validēta tādas barības analīzēm, kas satur vairāk par 100 mg/kg olahindoksa, apmierinošus analīzes rezultātus iespējams iegūt, ņemot mazāku parauga svaru un/vai atšķaidot ekstraktu (5.2.) tā, lai koncentrācija būtu kalibrēšanas grafika ietvaros (5.3.2.).
C. AMPROLIJA NOTEIKŠANA
1-[(4-amino-2-propil-5-pirimidinil)metil]-2-metil-piridīna hlorīda hidrogēnhlorīds
1. Mērķis un darbības joma
Šī metode ļauj noteikt amprolija daudzumu barībā un premiksos. Noteikšanas robeža ir 1 mg/kg, kvantitatīvās noteikšanas robeža ir 5 mg/kg.
2. Princips
Paraugu ekstrahē ar metanola un ūdens maisījumu. Pēc atšķaidīšanas ar kustīgo fāzi un membrānfiltrācijas amprolija saturu nosaka ar katjonu apmaiņas augstas efektivitātes šķidrumu hromatogrāfijas metodi (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Metanols. |
|
3.2. |
Acetonitrils, ekvivalents HPLC tīrības pakāpei. |
|
3.3. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
|
3.4. |
Nātrija dihidrogēnfosfāta šķīdums, c = 0,1 mol/l. Izšķīdina 13,80 g nātrija dihidrogēnfosfāta ūdenī (3.3.) 1 000 ml mērkolbā, papildina līdz zīmei ar ūdeni (3.3.) un samaisa. |
|
3.5. |
Nātrija perhlorāta šķīdums, c = 1,6 mol/l. Izšķīdina 224,74 g nātrija perhlorāta monohidrāta ūdenī (3.3.) 1 000 ml mērkolbā, papildina līdz zīmei ar ūdeni (3.3.) un samaisa. |
|
3.6. |
Kustīgā fāze priekš HPLC (skatīt 9.1. novērojumu). Acetonitrila (3.2.), nātrija dihidrogēnfosfāta šķīduma (3.4.) un nātrija perhlorāta šķīduma (3.5.) maisījums 450 + 450 + 100 (v + v + v). Pirms lietošanas filtrē caur 0,22 μm membrānfiltru (4.3.) un šķīdumu atgāzo (piemēram, ultraskaņas vannā (4.4.) vismaz 15 minūtes). |
|
3.7. |
Standartviela: tīrs amprolijs 1-[(4-amino-2-propil-5-pirimidinil)metil]-2-metil-piridīna hlorīda hidrogēnhlorīds, E 750 (skatīt 9.2. punktu). |
|
3.7.1. |
Ar precizitāti līdz 0,1 mg iesver 50 mg amprolija (3.7.) 100 ml mērkolbā, izšķīdina 80 mililitros metanola (3.1.) un kolbu uz 10 minūtēm ievieto ultraskaņas vannā (4.4.). Pēc apstrādes ar ultraskaņu šķīdumu atdzesē līdz istabas temperatūrai, papildina līdz zīmei ar ūdeni un samaisa. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.7.2. |
5,0 ml izejas standartšķīduma (3.7.1.) ar pipeti iepilina 50 ml mērkolbā, papildina līdz zīmei ar ekstrakcijas šķīdinātāju (3.8.) un samaisa. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.7.3. |
Pārnes 0,5, 1,0 un 2,0 ml starpposma standartšķīduma (3.7.2.) 50 ml mērkolbu komplektos. Papildina līdz zīmei ar kustīgo fāzi (3.6.) un samaisa. Tādi šķīdumi atbilst attiecīgi šādām koncentrācijām: 0,5, 1,0 un 2,0 μg/ml amprolija. Minētie šķīdumi jāsagatavo svaigi pirms izmantošanas. |
|
3.8. |
Ekstrakcijas šķīdinātājs. Metanola (3.1.) un ūdens maisījums 2+1 (v+v). |
4. Ierīces
|
4.1. |
HPLC iekārta ar injekcijas sistēmu, kas piemērota 100 μl tilpumu injekcijai. |
|
4.1.1. |
Šķidrumu hromatogrāfa kolonna, 125 × 4 mm, katjonu apmaiņa Nucleosil 10 SA, 5 vai 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.1.2. |
UV detektors ar maināmu viļņu garumu vai diodes matricu detektors. |
|
4.2. |
Membrānfiltrs, PTFE materiāls, 0,45 μm. |
|
4.3. |
Membrānfiltrs, 0,22 μm. |
|
4.4. |
Ultraskaņas vanna. |
|
4.5. |
Mehāniskais kratītājs vai magnētiskais maisītājs. |
5. Procedūra
5.1. Vispārīgas norādes
5.1.1.
Lai veiktu atgūstamības testu (5.1.2.), analizē tukšo barības paraugu, lai pārbaudītu, ka barībā nav ne amprolija, ne noteikšanu traucējošu vielu. Tukšais paraugs pēc veida ir līdzīgs analizējamajam paraugam, un, pārbaudot to, nav jāuzrādās amprolijam vai noteikšanu traucējošām vielām.
5.1.2
Atgūstamības testu veic, analizējot tukšo barības paraugu, kas pastiprināts, pievienojot tik daudz amprolija, cik ir paraugā. Lai paraugu pastiprinātu līdz līmenim 100 mg/kg, 10,0 ml izejas standartšķīduma (3.7.1.) pārnes 250 ml koniskā kolbā un šķīdumu iztvaicē līdz apmēram 0,5 ml. Pirms ekstrakcijas (5.2.) pievieno 50 g tukšā barības parauga, vairākas reizes kārtīgi samaisa un atstāj uz 10 minūtēm.
Ja nav analizējamajam paraugam līdzīga tukšā parauga (skatīt 5.1.1. punktu), tad atgūstamības testu var veikt, izmantojot standarta piedevu metodi. Tādā gadījumā analizējamo paraugu pastiprina ar amprolija daudzumu, kas ir līdzīgs analizējamajā paraugā jau esošajam daudzumam. Tādu paraugu analizē kopā ar nepastiprināto paraugu un atgūstamību aprēķina pēc starpības.
5.2. Ekstrakcija
5.2.1.
Atkarībā no amprolija satura ar precizitāti līdz 0,01 g iesver 5 līdz 40 g parauga, ko pārnes 500 ml koniskā kolbā, un pievieno 200 ml ekstrakcijas šķīdinātāja (3.8.). Kolbu ievieto ultraskaņas vannā (4.4.) un atstāj uz 15 minūtēm. Kolbu izņem no ultraskaņas vannas un vienu stundu krata kratītājā vai maisa ar magnētisko maisītāju (4.5.). Ekstrakta alikvotu atšķaida ar kustīgo fāzi (3.6.), līdz amprolija koncentrācija sasniedz 0,5–2 μg/ml, un samaisa (skatīt 9.3. novērojumu). Šā atšķaidītā šķīduma 5 līdz 10 ml filtrē ar membrānfiltru (4.2.). Pāriet pie HPLC noteikšanas (5.3.).
5.2.2.
Atkarībā no amprolija satura ar precizitāti līdz 0,001 g iesver 1 līdz 4 g premiksa, ko pārnes 500 ml koniskā kolbā, un pievieno 200 ml ekstrakcijas šķīdinātāja (3.8.). Kolbu ievieto ultraskaņas vannā (4.4.) un atstāj uz 15 minūtēm. Kolbu izņem no ultraskaņas vannas un vienu stundu krata kratītājā vai maisa ar magnētisko maisītāju (4.5.). Ekstrakta alikvotu atšķaida ar kustīgo fāzi (3.6.), līdz amprolija koncentrācija sasniedz 0,5–2 μg/ml, un samaisa. Šā atšķaidītā šķīduma 5 līdz 10 ml filtrē ar membrānfiltru (4.2.). Pāriet pie HPLC noteikšanas (5.3.).
5.3. HPLC noteikšana
5.3.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Šķidrumu hromatogrāfa |
|
|
kolonna (4.1.1.): |
125 × 4 mm, katjonu apmaiņa Nucleosil 10 SA, 5 vai 10 μm iesaiņojums, vai līdzvērtīga |
|
Kustīgā fāze (3.6.): |
acetonitrila (3.2.), nātrija dihidrogēnfosfāta šķīduma (3.4.) un nātrija perhlorāta šķīduma (3.5.) maisījums 450 + 450 + 100 (v + v + v) |
|
Plūsmas ātrums: |
0,7–1 ml/min |
|
Noteikšanas viļņa garums: |
264 nm |
|
Injekcijas tilpums: |
100 μl |
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.7.3.) ar koncentrāciju 1,0 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus un izdalīšanas laiku.
5.3.2.
Katru kalibrēšanas šķīdumu (3.7.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošo vidējos maksimālos augstumus (laukumus). Izveido kalibrēšanas grafiku, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus (laukumus) un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.3.3.
Iešpricē parauga ekstraktu (5.2.) vairākas reizes, izmantojot to pašu tilpumu, kāds bija kalibrēšanas šķīdumiem, un nosaka amprolija maksimumu vidējo augstumu (laukumu).
6. Rezultātu aprēķināšana
Ņemot vērā amprolija maksimumu vidējo augstumu (laukumu) parauga šķīdumā, nosaka parauga šķīduma koncentrāciju μg/ml, izmantojot kalibrēšanas grafiku (5.3.2.).
Amprolija saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg],
[mg/kg],
kur:
|
V |
= |
ekstrakcijas šķīdinātāja (3.8.) tilpums mililitros atbilstoši 5.2. punktam (t. i., 200 ml), |
|
c |
= |
amprolija koncentrācija parauga ekstraktā (5.2.) μg/ml, |
|
f |
= |
atšķaidījuma pakāpe atbilstoši 5.2. punktam, |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta (5.2.) spektru ar tā kalibrēšanas šķīduma (3.7.3.) spektru, kura koncentrācija ir 2,0 μg/ml.
7.1.1.
Parauga ekstraktu (5.2.) stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.7.3.). Pievienotā amprolija daudzumam jābūt tādam pašam kā aplēstajam amprolija daudzumam, kas atrasts parauga ekstraktā.
Tikai amprolija maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā augstuma jābūt ± 10 % robežās no nepastiprināta parauga ekstrakta amprolija maksimuma sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu detektoram minētās robežas parasti ir ± 2 nm; |
|
b) |
no 210 līdz 320 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 210 līdz 320 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
No viena parauga divās paralēlās noteikšanās iegūto rezultātu atšķirība nedrīkst pārsniegt:
|
— |
15 % no lielākās noteiktās vērtības, ja amprolija saturs ir no 25 mg/kg līdz 500 mg/kg, |
|
— |
75 mg/kg, ja amprolija saturs ir no 500 mg/kg līdz 1 000 mg/kg, |
|
— |
7,5 % no lielākās noteiktās vērtības, ja amprolija saturs ir vairāk par 1 000 mg/kg. |
7.3. Atgūstamība
Stiprināta (tukšā) parauga atgūstamība ir vismaz 90 %.
8. Koppētījuma rezultāti
Tika noorganizēts koppētījums, kurā analizēja trīs mājputnu barības paraugus (1. līdz 3. paraugs), vienu minerālbarības paraugu (4. paraugs) un vienu premiksu (5. paraugs). Rezultāti ir norādīti šajā tabulā.
|
|
1. paraugs (tukšā barība) |
2. paraugs |
3. paraugs |
4. paraugs |
5. paraugs |
|
L |
14 |
14 |
14 |
14 |
15 |
|
n |
56 |
56 |
56 |
56 |
60 |
|
vidējais [mg/kg] |
— |
45,5 |
188 |
5 129 |
25 140 |
|
sr [mg/kg] |
— |
2,26 |
3,57 |
178 |
550 |
|
CVr [ %] |
— |
4,95 |
1,90 |
3,46 |
2,20 |
|
sR [mg/kg] |
— |
2,95 |
11,8 |
266 |
760 |
|
CVR [ %] |
— |
6,47 |
6,27 |
5,19 |
3,00 |
|
Nominālais saturs [mg/kg] |
— |
50 |
200 |
5 000 |
25 000 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
sr |
= |
atkārtojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
sR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients. |
9. Novērojumi
|
9.1. |
Ja paraugs satur tiamīnu, tad tiamīna maksimums hromatogrammā parādās neilgi pirms amprolija maksimuma. Izmantojot šo metodi, amprolijs un tiamīns jāatdala. Ja šajā metodē izmantotā kolonna (4.1.1.) amproliju un tiamīnu neatdala, tad līdz 50 % acetonitrila devas kustīgajā fāzē (3.6.) aizstāj ar metanolu. |
|
9.2. |
Saskaņā ar britu farmakopeju amprolija šķīduma (c = 0,02 mol/l) spektrs sālsskābes šķīdumā sasniedz maksimumu pie 246 nm un 262 nm. Absorbcija pie 246 nm ir 0,84, bet pie 262 nm – attiecīgi 0,80. |
|
9.3. |
Parauga ekstrakts noteikti jāatšķaida tikai ar kustīgo fāzi, citādi amprolija maksimuma izdalīšanas laiks var ievērojami mainīties jonu spēka izmaiņu dēļ. |
D. KARBADOKSA NOTEIKŠANA
Metil 3-(2-hinoksalinilmetilēn)karbazāta N 1 ,N 4 -dioksīds
1. Mērķis un darbības joma
Šī metode ļauj noteikt karbadoksa daudzumu barībā, premiksos un preparātos. Noteikšanas robeža ir 1 mg/kg. Kvantitatīvās noteikšanas robeža ir 5 mg/kg.
2. Princips
Paraugu līdzsvaro ar ūdeni un ekstrahē ar metanola un acetonitrila maisījumu. Analizējot barību, filtrēta ekstrakta alikvotu attīra alumīnija oksīda kolonnā. Analizējot premiksus un preparātus, filtrēta ekstrakta alikvotu līdz vajadzīgajai koncentrācijai atšķaida ar ūdeni, metanolu un acetonitrilu. Kabadoksa saturu nosaka apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), izmantojot UV detektoru.
3. Reaģenti
|
3.1. |
Metanols. |
|
3.2. |
Acetonitrils, ekvivalents HPLC tīrības pakāpei. |
|
3.3. |
Etiķskābe, w = 100 %. |
|
3.4. |
Alumīnija oksīds: neitrāls, I aktivitātes pakāpe. |
|
3.5. |
Metanola un acetonitrila maisījums 1 + 1 (v + v). 500 ml metanola (3.1.) samaisa ar 500 ml acetonitrila (3.2.). |
|
3.6. |
Etiķskābe, σ = 10 %. Atšķaida 10 ml etiķskābes (3.3.) līdz 100 ml tilpumam ar ūdeni. |
|
3.7. |
Nātrija acetāts. |
|
3.8. |
Ūdens, ekvivalents HPLC tīrības pakāpei. |
|
3.9. |
Acetāta buferšķīdums, c = 0,01 mol/l, pH = 6,0. Izšķīdina 0,82 g nātrija acetāta (3.7.) 700 mililitros ūdens (3.8.) un koriģē pH līmeni līdz 6,0 ar etiķskābi (3.6.). Pārnes 1 000 ml mērkolbā, papildina līdz zīmei ar ūdeni (3.8.) un samaisa. |
|
3.10. |
Kustīgā fāze priekš HPLC. Samaisa 825 ml acetāta buferšķīduma (3.9.) ar 175 ml acetonitrila (3.2.). Šķīdumu filtrē caur 0,22 μm filtru (4.5.) un atgāzo (piemēram, 10 minūtes apstrādājot ar ultraskaņu). |
|
3.11. |
Standartviela. Tīrs karbadokss: metil 3-(2-hinoksalinilmetilēn)karbazāta N1,N4-dioksīds, E 850. |
|
3.11.1. |
ar precizitāti līdz 0,1 mg 250 ml mērkolbā iesver 25 mg karbadoksa standartvielas (3.11.). Izšķīdina metanola un acetonitrila maisījumā (3.5.), apstrādājot ar ultraskaņu (4.7.). Pēc apstrādes ar ultraskaņu šķīdumu atdzesē līdz istabas temperatūrai, papildina līdz zīmei ar metanola un acetonitrila maisījumu (3.5.) un samaisa. Ietin kolbu alumīnija folijā vai izmanto kolbu no brūnā stikla, uzglabā ledusskapī. Temperatūrā, kas ir ≤ 4 oC, šķīdumu var glabāt vienu mēnesi. |
|
3.11.2. |
Pārnes 2,0, 5,0, 10,0 un 20,0 ml izejas standartšķīduma (3.11.1.) 100 ml mērkolbu komplektos. Pievieno 30 ml ūdens, papildina līdz zīmei ar metanola un acetonitrila maisījumu (3.5.) un samaisa. Ietin kolbas alumīnija folijā. Tādi šķīdumi atbilst attiecīgi 2,0, 5,0, 10,0 un 20,0 μg/ml karbadoksa. Kalibrēšanas šķīdumi jāsagatavo svaigi pirms izmantošanas.
|
|
3.12. |
Ūdens-[metanola-acetonitrila] (3.5.) maisījums, 300 + 700 (v + v). Samaisa 300 ml ūdens ar 700 ml metanola un acetonitrila maisījuma (3.5.). |
4. Ierīces
|
4.1. |
Laboratorijas kratītājs vai magnētiskais maisītājs. |
|
4.2. |
Stikla šķiedras filtrpapīrs (GF/A Vatmans vai līdzvērtīgs). |
|
4.3. |
Stikla kolonna (garums 300 līdz 400 mm, iekšējais diametrs apmēram 10 mm) ar keramikas stikla starpsienu un noteces vārstu.
|
|
4.4. |
HPLC iekārta ar injekcijas sistēmu, kas piemērota 20 μl tilpumu injekcijai. |
|
4.4.1. |
Šķidrumu hromatogrāfa kolonna: 300 × 4 mm, C18, 10 μm iesaiņojums, vai līdzvērtīga. |
|
4.4.2. |
UV detektors, kam ir maināms viļņu garums, vai diodes matricu detektors, kas darbojas 225 līdz 400 mm diapazonā. |
|
4.5. |
Membrānfiltrs, 0,22 μm. |
|
4.6. |
Membrānfiltrs, 0,45 μm. |
|
4.7. |
Ultraskaņas vanna. |
5. Procedūra
|
Piezīme. |
Karbadokss ir gaismjutīgs. Visas darbības veic izkliedētā gaismā vai izmanto brūnā stikla traukus, vai izmanto alumīnija folijā ietītus stikla traukus. |
5.1. Vispārīgas norādes
5.1.1.
Lai veiktu atgūstamības testu (5.1.2.), analizē tukšo barības paraugu, lai pārbaudītu, ka barībā nav ne karbadoksa, ne noteikšanu traucējošu vielu. Tukšais paraugs pēc veida ir līdzīgs analizējamajam paraugam, un, pārbaudot to, nav jāuzrādās karbadoksam vai noteikšanu traucējošām vielām.
5.1.2.
Atgūstamības testu veic, analizējot tukšo barības paraugu (5.1.1.), kas pastiprināts, pievienojot tik daudz karbadoksa, cik ir paraugā. Lai pastiprinātu paraugu līdz līmenim 50 mg/kg, 5,0 ml izejas standartšķīduma (3.11.1.) pārnes 200 ml koniskā kolbā. Slāpekļa plūsmā šķīdumu iztvaicē līdz apmēram 0,5 ml. Pirms ekstrakcijas (5.2.) pievieno 10 g tukšā barības parauga, samaisa un atstāj uz 10 minūtēm.
Ja nav analizējamajam paraugam līdzīga tukšā parauga (skatīt 5.1.1. punktu), tad atgūstamības testu var veikt, izmantojot standarta piedevu metodi. Tādā gadījumā analizējamo paraugu pastiprina ar karbadoksa daudzumu, kas ir līdzīgs analizējamajā paraugā jau esošajam daudzumam. Tādu paraugu analizē kopā ar nepastiprināto paraugu un atgūstamību aprēķina pēc starpības.
5.2. Ekstrakcija
5.2.1.
Ar precizitāti līdz 0,01 g iesver 10 g parauga un pārnes 200 ml koniskā kolbā. Pievieno 15,0 ml ūdens, samaisa un līdzsvaro piecas minūtes. Pievieno 35,0 ml metanola un acetonitrila maisījuma (3.5.), noslēdz ar aizbāzni un 30 minūtes krata kratītājā vai maisa ar magnētisko maisītāju (4.1.). Šķīdumu izfiltrē caur stikla šķiedras filtrpapīru (4.2.). Minēto šķīdumu saglabā attīrīšanas posmam (5.3.).
5.2.2.
Ar precizitāti līdz 0,001 g iesver 1 g nesamalta parauga un pārnes 200 ml koniskā kolbā. Pievieno 15,0 ml ūdens, samaisa un līdzsvaro piecas minūtes. Pievieno 35,0 ml metanola un acetonitrila maisījuma (3.5.), noslēdz ar aizbāzni un 30 minūtes krata kratītājā vai maisa ar magnētisko maisītāju (4.1.). Šķīdumu izfiltrē caur stikla šķiedras filtrpapīru (4.2.).
Ar pipeti iepilina filtrāta alikvotu 50 ml mērkolbā. Pievieno 15,0 ml ūdens, papildina līdz zīmei ar metanola un acetonitrila maisījumu (3.5.) un samaisa. Karbadoksa koncentrācija galīgajā šķīdumā ir apmēram 10 μg/ml. Alikvotu izfiltrē caur 0,45 μm filtru (4.6.).
Pāriet pie HPLC noteikšanas (5.4.).
5.2.3.
Ar precizitāti līdz 0,001 g iesver 0,2 g nesamalta parauga un pārnes 250 ml koniskā kolbā. Pievieno 45,0 ml ūdens, samaisa un līdzsvaro piecas minūtes. Pievieno 105,0 ml metanola un acetonitrila maisījuma (3.5.), noslēdz ar aizbāzni un homogenizē. Paraugu 15 minūtes apstrādā ar ultraskaņu (4.7.), pēc tam 15 minūtes kratot vai maisot (4.1.). Šķīdumu izfiltrē caur stikla šķiedras filtrpapīru (4.2.).
Filtrāta alikvotu atšķaida ar ūdens, metanola un acetonitrila maisījumu (3.12.), līdz karbadoksa galīgā koncentrācija sasniedz 10–15 μg/ml (10 % preparātam atšķaidījuma pakāpe ir 10). Alikvotu izfiltrē caur 0,45 μm filtru (4.6.).
Pāriet pie HPLC noteikšanas (5.4.).
5.3. Attīrīšana
5.3.1.
Iesver 4 g alumīnija oksīda (3.4.) un pārnes to uz stikla kolonnu (4.3.).
5.3.2.
15 ml filtrētā ekstrakta (5.2.1.) pārnes alumīnija oksīda kolonnā un pirmos 2 ml eluāta izlej. Savāc nākamos 5 ml un izfiltrē alikvotu caur 0,45 μm filtru (4.6.).
Pāriet pie HPLC noteikšanas (5.4.).
5.4. HPLC noteikšana
5.4.1.
Orientējoši piedāvā šādus parametrus; var izmantot citus parametrus, ja tie nodrošina līdzvērtīgus rezultātus.
|
Šķidrumu hromatogrāfa |
|
|
kolonna (4.4.1.): |
300 × 4 mm, C18, 10 μm iesaiņojums, vai līdzvērtīga |
|
Kustīgā fāze (3.10.): |
acetāta buferšķīduma (3.9.) un acetonitrila (3.2.) maisījums, 825 + 175 (v+v) |
|
Plūsmas ātrums: |
1,5–2 ml/min |
|
Noteikšanas viļņa garums: |
365 nm |
|
Injekcijas tilpums: |
20 μl |
Pārbauda hromatogrāfiskās sistēmas stabilitāti, vairākas reizes iešpricējot kalibrēšanas šķīdumu (3.11.2.) ar koncentrāciju 5,0 μg/ml, līdz sasniedz nemainīgus maksimālos augstumus (laukumus) un izdalīšanas laiku.
5.4.2.
Katru kalibrēšanas šķīdumu (3.11.3.) iešpricē vairākas reizes un izmēra katrai koncentrācijai atbilstošos maksimālos augstumus (laukumus). Izveido kalibrēšanas līkni, uz ordinātu ass atzīmējot kalibrēšanas šķīdumu vidējos maksimālos augstumus vai laukumus un uz abscisu ass – atbilstīgās koncentrācijas μg/ml.
5.4.3.
Vairākas reizes iešpricē parauga ekstraktu ((5.3.2.) barībai, (5.2.2.) premiksiem un (5.2.3.) preparātiem) un nosaka karbadoksa maksimumu vidējo maksimālo augstumu (laukumu).
6. Rezultātu aprēķināšana
Ņemot vērā karbadoksa maksimumu vidējo augstumu (laukumu) parauga šķīdumā, nosaka karbadoksa koncentrāciju parauga šķīdumā μg/ml, izmantojot kalibrēšanas grafiku (5.4.2.).
6.1. Barība
Karbadoksa saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg],
[mg/kg],
kur:
|
c |
= |
karbadoksa koncentrācija parauga ekstraktā (5.3.2.), μg/ml, |
|
V1 |
= |
ekstrakcijas tilpums ml (t. i., 50), |
|
m |
= |
analizējamā parauga svars gramos. |
6.2. Premiksi un preparāti
Karbadoksa saturu w (mg/kg) paraugā aprēķina, izmantojot šādu formulu:
 [mg/kg],
[mg/kg],
kur:
|
c |
= |
karbadoksa koncentrācija parauga ekstraktā (5.2.2. vai 5.2.3.), μg/ml, |
|
V2 |
= |
ekstrakcijas tilpums ml (t. i., 50 premiksiem; 150 preparātiem), |
|
f |
= |
atšķaidījuma pakāpe atbilstoši 5.2.2. (premiksiem) vai 5.2.3. (preparātiem), |
|
m |
= |
analizējamā parauga svars gramos. |
7. Rezultātu validācija
7.1. Identitāte
Nosakāmās vielas identitāti var apstiprināt paralēlā hromatogrāfiskajā analīzē vai izmantojot diodes matricu detektoru, ar kura palīdzību salīdzina parauga ekstrakta spektru ar tā kalibrēšanas šķīduma (3.11.3.) spektru, kura koncentrācija ir 10,0 μg/ml.
7.1.1.
Parauga ekstraktu stiprina, pievienojot attiecīgu daudzumu kalibrēšanas šķīduma (3.11.2.). Pievienotā karbadoksa daudzumam jābūt tādam pašam kā aplēstajam karbadoksa daudzumam, kas atrasts parauga ekstraktā.
Tikai karbadoksa maksimuma augstumu palielina, ņemot vērā gan pievienoto daudzumu, gan ekstrakta atšķaidījumu. Maksimālajam platumam pusē no tā maksimālā augstuma jābūt apmēram 10 % no sākotnējā platuma.
7.1.2.
Rezultātus novērtē atbilstoši šādiem kritērijiem:
|
a) |
parauga maksimālās absorbcijas viļņa garumam un standarta spektriem, ko reģistrē pīķa smailē hromatogrammā, atklāšanas sistēmas izšķirtspējas noteiktajās robežās jābūt vienādiem. Diodes matricu noteikšanai minētās robežas parasti ir ± 2 nm; |
|
b) |
no 225 līdz 400 nm parauga un standartspektri, ko reģistrē pīķa smailē hromatogrammā, nedrīkst atšķirties tajās spektra daļās, kas ir no 10 līdz 100 % relatīvās absorbcijas diapazonā. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no nosakāmās vielas standarta absorbcijas; |
|
c) |
no 225 līdz 400 nm parauga ekstrakta kāpuma, smailes un krituma spektri cits no cita nedrīkst atšķirties tajās spektra daļās, kurās relatīvā absorbcija ir no 10 līdz 100 %. Atbilstība šim kritērijam ir ievērota, ja maksimumi sakrīt un nevienā novērojumu punktā novirze starp abiem spektriem nepārsniedz 15 % no absorbcijas vērtības maksimuma smailē. |
Ja nav atbilstības kādam no šiem kritērijiem, tad nosakāmās vielas klātbūtne nav apstiprinājusies.
7.2. Atkārtojamība
Ja saturs ir 10 mg/kg un vairāk, tad starpībai starp rezultātiem, kas iegūti divās paralēlās noteikšanās, kuras veiktas ar vienu paraugu, nav jāpārsniedz 15 % no lielākā rezultāta.
7.3. Atgūstamība
Stiprināta (tukšā) parauga atgūstamība ir vismaz 90 %.
8. Koppētījuma rezultāti
Tika organizēts koppētījums, kurā astoņas laboratorijas analizēja sešas barības, četrus premiksus un trīs preparātus. Visus paraugus analizēja divas reizes. (Sīkāku informāciju par šo koppētījumu skatīt Journal of the AOAC, Volume 71, 1988. g., 484.–490. lpp.). Rezultāti (izņemot galējos punktus) ir šādi.
1. tabula
Barības koppētījuma rezultāti
|
|
1. paraugs |
2. paraugs |
3. paraugs |
4. paraugs |
5. paraugs |
6. paraugs |
|
L |
8 |
8 |
8 |
8 |
8 |
8 |
|
n |
15 |
14 |
15 |
15 |
15 |
15 |
|
Vidējais (mg/kg) |
50,0 |
47,6 |
48,2 |
49,7 |
46,9 |
49,7 |
|
Sr (mg/kg) |
2,90 |
2,69 |
1,38 |
1,55 |
1,52 |
2,12 |
|
CVr (%) |
5,8 |
5,6 |
2,9 |
3,1 |
3,2 |
4,3 |
|
SR (mg/kg) |
3,92 |
4,13 |
2,23 |
2,58 |
2,26 |
2,44 |
|
CVR (%) |
7,8 |
8,7 |
4,6 |
5,2 |
4,8 |
4,9 |
|
Nominālais saturs (mg/kg) |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
50,0 |
2. tabula
Premiksu un preparātu koppētījuma rezultāti
|
|
Premiksi |
Preparāti |
|||||
|
A |
B |
C |
D |
A |
B |
C |
|
|
L |
7 |
7 |
7 |
7 |
8 |
8 |
8 |
|
n |
14 |
14 |
14 |
14 |
16 |
16 |
16 |
|
Vidējais (g/kg) |
8,89 |
9,29 |
9,21 |
8,76 |
94,6 |
98,1 |
104 |
|
Sr (g/kg) |
0,37 |
0,28 |
0,28 |
0,44 |
4,1 |
5,1 |
7,7 |
|
CVr (%) |
4,2 |
3,0 |
3,0 |
5,0 |
4,3 |
5,2 |
7,4 |
|
SR (g/kg) |
0,37 |
0,28 |
0,40 |
0,55 |
5,4 |
6,4 |
7,7 |
|
CVR (%) |
4,2 |
3,0 |
4,3 |
6,3 |
5,7 |
6,5 |
7,4 |
|
Nominālais saturs (g/kg) |
10,0 |
10,0 |
10,0 |
10,0 |
100 |
100 |
100 |
|
L |
= |
laboratoriju skaits, |
|
n |
= |
atsevišķo vērtību skaits, |
|
Sr |
= |
atkārtojamības standartnovirze, |
|
CVr |
= |
atkārtojamības variācijas koeficients, |
|
SR |
= |
atveidojamības standartnovirze, |
|
CVR |
= |
atveidojamības variācijas koeficients. |
IX PIELIKUMS
6. PANTĀ MINĒTĀS ATBILSTĪBAS TABULAS
1. Direktīva 71/250/EEK
|
Direktīva 71/250/EEK |
Šī regula |
|
1. panta pirmā daļa |
3. pants |
|
1. panta otrā daļa |
2. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikuma 1. daļa |
II pielikums |
|
Pielikuma 2. daļa |
— |
|
Pielikuma 3. daļa |
— |
|
Pielikuma 4. daļa |
III pielikuma O daļa |
|
Pielikuma 5. daļa |
III pielikuma M daļa |
|
Pielikuma 6. daļa |
III pielikuma N daļa |
|
Pielikuma 7. daļa |
III pielikuma Q daļa |
|
Pielikuma 9. daļa |
III pielikuma K daļa |
|
Pielikuma 10. daļa |
— |
|
Pielikuma 11. daļa |
— |
|
Pielikuma 12. daļa |
III pielikuma J daļa |
|
Pielikuma 14. daļa |
III pielikuma D daļa |
|
Pielikuma 16. daļa |
— |
2. Direktīva 71/393/EEK
|
Direktīva 71/393/EEK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikuma I daļa |
III pielikuma A daļa |
|
Pielikuma II daļa |
III pielikuma E daļa |
|
Pielikuma III daļa |
III pielikuma P daļa |
|
Pielikuma IV daļa |
III pielikuma H daļa |
3. Direktīva 72/199/EEK
|
Direktīva 72/199/EEK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
I pielikuma 1. daļa |
III pielikuma L daļa |
|
I pielikuma 2. daļa |
III pielikuma C daļa |
|
I pielikuma 3. daļa |
— |
|
I pielikuma 4. daļa |
— |
|
I pielikuma 5. daļa |
V pielikuma A daļa |
|
II pielikums |
— |
4. Direktīva 73/46/EEK
|
Direktīva 73/46/EEK |
Šī regula |
|
1. pants |
3. pants |
|
3. pants |
— |
|
4. pants |
— |
|
I pielikuma 1. daļa |
III pielikuma B daļa |
|
I pielikuma 2. daļa |
— |
|
I pielikuma 3. daļa |
III pielikuma I daļa |
5. Direktīva 76/371/EEK
|
Direktīva 76/371/EEK |
Šī regula |
|
1. pants |
1. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
I pielikums |
6. Direktīva 76/372/EEK
|
Direktīva 76/372/EEK |
Šī regula |
|
1. pants |
— |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
— |
7. Direktīva 78/633/EEK
|
Direktīva 78/633/EEK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikuma 1. daļa |
— |
|
Pielikuma 2. daļa |
— |
|
Pielikuma 3. daļa |
IV pielikuma C daļa |
8. Direktīva 81/715/EEK
|
Direktīva 81/715/EEK |
Šī regula |
|
1. pants |
— |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
— |
9. Direktīva 84/425/EEK
|
Direktīva 84/425/EEK |
Šī regula |
|
1. pants |
— |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
— |
10. Direktīva 86/174/EEK
|
Direktīva 86/174/EEK |
Šī regula |
|
1. pants |
4. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
VII pielikums |
11. Direktīva 93/70/EEK
|
Direktīva 93/70/EEK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikums |
IV pielikuma D daļa |
12. Direktīva 93/117/EK
|
Direktīva 93/117/EK |
Šī regula |
|
1. pants |
3. un 5. pants |
|
2. pants |
— |
|
3. pants |
— |
|
Pielikuma 1. daļa |
IV pielikuma E daļa |
|
Pielikuma 2. daļa |
VIII pielikuma A daļa |
13. Direktīva 98/64/EK
|
Direktīva 98/64/EK |
Šī regula |
|
1. pants |
3. un 5. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
Pielikuma A daļa |
III pielikuma F daļa |
|
Pielikuma C daļa |
VIII pielikuma B daļa |
14. Direktīva 1999/27/EK
|
Direktīva 1999/27/EK |
Šī regula |
|
1. pants |
3. un 5. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
5. pants |
— |
|
6. pants |
— |
|
7. pants |
— |
|
Pielikuma A daļa |
VIII pielikuma C daļa |
|
Pielikuma B daļa |
IV pielikuma F daļa |
|
Pielikuma C daļa |
VIII pielikuma D daļa |
15. Direktīva 1999/76/EK
|
Direktīva 1999/76/EK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
Pielikums |
IV pielikuma G daļa |
16. Direktīva 2000/45/EK
|
Direktīva 2000/45/EK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
Pielikuma A daļa |
IV pielikuma A daļa |
|
Pielikuma B daļa |
IV pielikuma B daļa |
|
Pielikuma C daļa |
III pielikuma G daļa |
17. Direktīva 2002/70/EK
|
Direktīva 2002/70/EK |
Šī regula |
|
1. pants |
1. pants |
|
2. pants |
2. un 3. pants |
|
3. pants |
— |
|
4. pants |
— |
|
5. pants |
-– |
|
I pielikums |
I pielikums un V pielikuma B daļas I punkts |
|
II pielikums |
II pielikums un V pielikuma B daļas II punkts |
18. Direktīva 2003/126/EK
|
Direktīva 2003/126/EK |
Šī regula |
|
1. pants |
3. pants |
|
2. pants |
— |
|
3. pants |
— |
|
4. pants |
— |
|
5. pants |
— |
|
6. pants |
— |
|
Pielikums |
VI pielikums |
|
26.2.2009 |
LV |
Eiropas Savienības Oficiālais Vēstnesis |
L 54/s3 |
PIEZĪME LASĪTĀJAMIestādes ir nolēmušas savos tekstos turpmāk nenorādīt jaunākos tiesību aktu grozījumus.Ja vien nav noteikts citādi, par tiesību aktiem, kuri ir norādīti šeit publicētajos tekstos, uzskatāmi tiesību akti to spēkā esošajā redakcijā.


 = tīras cietes specifiskā optiskā rotācija (skatīt 6.1. punktu).
= tīras cietes specifiskā optiskā rotācija (skatīt 6.1. punktu).