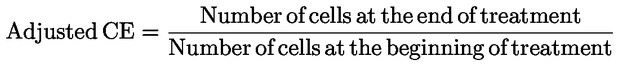|
(8)
|
B daļā pievieno šādas nodaļas:
“B.63. REPRODUKTĪVĀS / ONTOĢENĒTISKĀS TOKSICITĀTES SKRĪNINGA TESTS
IEVADS
|
1.
|
Šī testēšanas metode ir ekvivalenta ESAO testēšanas vadlīnijai (TG) Nr. 421 (2016). ESAO vadlīnijas par ķimikāliju testēšanu periodiski tiek pārskatītas, ņemot vērā zinātnes attīstību. Sākotnējā vadlīnija Nr. 421 par skrīninga testu tika pieņemta 1995. gadā, pamatojoties uz protokolu par “Sākotnējo reproduktīvā toksiskuma skrīninga testu”, kas apspriests divās ekspertu sanāksmēs Londonā 1990. gadā (1) un Tokijā 1992. gadā (2).
|
|
2.
|
Pēc tam, kad 1998. gadā ESAO tika sākta prioritāra darbība ar mērķi pārskatīt esošās testēšanas vadlīnijas un izstrādāt jaunas testēšanas vadlīnijas par endokrīno disruptoru skrīningu un testēšanu (3), šī testēšanas metode tika atjaunināta, iekļaujot tajā endokrīnajiem disruptoriem relevantus beigupunktus. Piemēram, ESAO TG Nr. 407 (Atkārtotas devas 28 dienu orālās toksicitātes pētījums ar grauzējiem, šā pielikuma B.7. nodaļa) 2008. gadā tika papildināta ar parametriem, kas piemēroti testējamo ķimikāliju endokrīnās aktivitātes noteikšanai. TG Nr. 421 tika atjaunināta, lai skrīninga TG, kur ekspozīcijas periodi aptver dažus jutīgos attīstības periodus (prenatālais vai agrīns postnatālais periods), iekļautu dažus endokrīnajiem disruptoriem relevantus beigupunktus.
|
|
3.
|
Izvēlētie endokrīnajiem disruptoriem relevantie papildu beigupunkti, kas ir daļa arī no TG Nr. 443 (Paplašinātais vienas paaudzes reproduktīvās toksicitātes pētījums, šā pielikuma B.56. nodaļa), tika iekļauti TG Nr. 421, pamatojoties uz priekšizpēti, kurā aplūkoti zinātniski un tehniski jautājumi par minēto beigupunktu iekļaušanu, kā arī iespējamie testēšanas plānojuma pielāgojumi, kas vajadzīgi minēto beigupunktu iekļaušanai (4).
|
|
4.
|
Šī testēšanas metode ir izstrādāta, lai iegūtu ierobežotu informāciju par testējamās ķimikālijas ietekmi uz tēviņu un mātīšu reproduktīvo spēju, piemēram, gonadālo funkciju, pārošanās uzvedību, apaugļošanos, miesas augļa attīstību un dzemdībām. Tā nav nedz alternatīva B.31., B.34., B.35. vai B.56. testēšanas metodei, nedz tās aizstāj.
|
SĀKOTNĒJIE APSVĒRUMI
|
5.
|
Šo skrīninga testa metodi var izmantot, lai iegūtu sākotnējo informāciju par iespējamo ietekmi uz reprodukciju un/vai attīstību vai nu ķimikāliju toksikoloģisko īpašību novērtēšanas agrīnā posmā, vai par ķimikālijām, kas rada bažas. To var izmantot arī kā daļu no sākotnējo skrīninga testu kopuma attiecībā uz esošajām ķimikālijām, par kurām pieejams maz toksikoloģiskās informācijas vai tā nav pieejama vispār, vai kā devu diapazona noteikšanas pētījumu plašākiem reprodukcijas/ontoģenēzes pētījumiem; to var izmantot citos relevantos gadījumos. Veicot pētījumu, ir jāievēro galvenie principi un apsvērumi, kas noteikti ESAO norādījumu dokumentā Nr. 19 par klīnisko pazīmju atpazīšanu, novērtēšanu un izmantošanu par humānajiem beigupunktiem eksperimenta dzīvniekiem, kurus izmanto drošības novērtējumā (5).
|
|
6.
|
Šī testēšanas metode nesniedz pilnīgu informāciju par visiem reprodukcijas un attīstības aspektiem. Konkrētāk, tā piedāvā tikai ierobežotus līdzekļus, ar ko detektēt prenatālās ekspozīcijas postnatālās izpausmes vai ietekmi, kas var rasties postnatālās ekspozīcijas laikā. Ņemot vērā dzīvnieku salīdzinoši mazo skaitu devas grupās, beigupunktu selektivitāti, pētījuma īso ilgumu un citus iemeslus, šī metode nesniedz pierādījumus galīgajiem apgalvojumiem par ietekmes neesību. Turklāt, ja nav datu no citiem reproduktīvās/ontoģenētiskās toksicitātes testiem, pozitīvie rezultāti ir noderīgi sākotnējai bīstamības novērtēšanai un palīdz pieņemt lēmumus par papildu testēšanas nepieciešamību un laiku.
|
|
7.
|
Rezultāti, kas iegūti par endokrīnajiem parametriem, ir jāaplūko ESAO endokrīni disruptīvu ķimikāliju testēšanas un novērtēšanas konceptuālā satvara (6) kontekstā. Atjauninātā ESAO TG Nr. 421 ir iekļauta minētā konceptuālā satvara 4. līmenī, kur ietilpst in vivo testi, kas sniedz datus par nelabvēlīgu ietekmi uz endokrīnajiem beigupunktiem. Endokrīnu signālu pašu par sevi tomēr nevar uzskatīt par pietiekamu pierādījumu tam, ka testējamā ķimikālija ir endokrīnais disruptors.
|
|
8.
|
Šajā testēšanas metodē izmanto perorālo testējamās ķimikālijas ievadīšanu. Citu ekspozīcijas ceļu izmantošanas gadījumā var būt vajadzīgas modifikācijas.
|
|
9.
|
Pirms testēšanas metodi attiecībā uz maisījumu izmanto, lai iegūtu datus kādai plānotai regulatīvai vajadzībai, jāapsver, vai un kādēļ tā var sniegt tādam nolūkam piemērotus rezultātus. Šādi apsvērumi nav vajadzīgi, ja pastāv regulatīva prasība maisījumu testēt.
|
|
10.
|
Izmantotās definīcijas sniegtas 1. papildinājumā.
|
TESTA PRINCIPS
|
11.
|
Testējamo ķimikāliju pakāpeniski pieaugošās devās saņem vairākas tēviņu un mātīšu grupas. Tēviņiem testējamās ķimikālijas jādod vismaz četras nedēļas līdz dienai pirms plānotās nonāvēšanas (minēto dienu ieskaitot); minētais laikposms aptver vismaz divas nedēļas pirms pārošanas, pārošanas periodu un aptuveni divas nedēļas pēc pārošanas. Ņemot vērā ierobežoto pirmspārošanas devu došanas periodu tēviņiem, auglība var nebūt precīzs testikulārās toksicitātes rādītājs. Tāpēc būtiska ir sēklinieku detalizēta histopatoloģiska izmeklēšana. Divu nedēļu ilgais pirmspārošanas devu došanas periods apvienojumā ar turpmākiem pārošanās/auglības novērojumiem ar kopējo devu došanas periodu vismaz četras nedēļas, kam seko tēviņu gonādu detalizēta histopatoloģija, tiek uzskatīts par pietiekamu, lai varētu noteikt lielāko daļu ietekmes uz tēviņu auglību un spermatoģenēzi.
|
|
12.
|
Mātītēm devas dod visu pētījuma laiku, kas aptver divas nedēļas pirms pārošanas (aptverot divus pilnus estrālos ciklus), laiku līdz apaugļošanās brīdim, grūsnības laiku un vismaz trīspadsmit dienas pēc dzemdībām, līdz dienai pirms plānotās nonāvēšanas (minēto dienu ieskaitot).
|
|
13.
|
Pētījuma ilgums pēc aklimatizācijas un estrālā cikla izvērtējuma, ko veic pirms devu došanas, ir atkarīgs no mātīšu rādītājiem un ir aptuveni 63 dienas [vismaz 14 dienas pirms pārošanas, (ne vairāk kā) 14 dienas pārošana, 22 dienas gestācija, 13 dienas laktācija].
|
|
14.
|
Devu došanas perioda laikā katru dienu rūpīgi novēro, vai dzīvniekiem neparādās toksiskuma pazīmes. Dzīvniekiem, kas testa periodā nobeigušies vai ir nonāvēti, izdara autopsiju, un izdzīvojušos dzīvniekus testa beigās nonāvē un tiem izdara autopsiju.
|
METODES APRAKSTS
Dzīvnieku sugas izraudzīšanās
|
15.
|
Šo testēšanas metodi paredzēts izmantot ar žurkām. Ja šajā testēšanas metodē noteiktie parametri tiek pētīti citās grauzēju sugās, tas sīki jāpamato. Starptautiskajā validēšanas programmā endokrīno disruptoru detektēšanai saskaņā ar ESAO TG Nr. 407 (šā pielikuma B.7. nodaļa) tika izmantotas tikai žurkas. Nedrīkst izmantot līnijas ar zemu auglību vai līnijas, par kurām ir labi zināms, ka tām ir bieži sastopami attīstības defekti. Jāizmanto veselīgi iepriekš nepāroti dzīvnieki, kuriem eksperimentālas procedūras iepriekš nav veiktas. Jānorāda testa dzīvnieku suga, līnija, dzimums, masa un vecums. Pētījuma sākumā dzīvnieku masas atšķirībām jābūt minimālām un tās nedrīkst pārsniegt 20 % no vidējās masas, kas noteikta katram dzimumam atsevišķi. Ja pētījumu veic kā sākotnēju pētījumu ilgtermiņa pētījumam vai pētījumam, kas aptver visu paaudzi, vēlams abos pētījumos izmantot vienas līnijas un izcelsmes dzīvniekus.
|
Turēšana un barošana
|
16.
|
Visām procedūrām jāatbilst vietējiem laboratorijas dzīvnieku labturības standartiem. Telpā, kurā tur izmēģinājuma dzīvniekus, jābūt 22 ° C (± 3 ° C) temperatūrai. Lai gan relatīvajam mitrumam jābūt vismaz 30 % un, izņemot telpu uzkopšanas laiku, vēlams nepārsniegt 70 %, jāorientējas uz 50–60 %. Dzīvniekus 12 stundas diennaktī tur mākslīgā apgaismojumā, bet 12 stundas – tumsā. Var izmantot parasto laboratorijas barību ar neierobežotu piekļuvi dzirdināmajam ūdenim. Uztura izvēli var ietekmēt vajadzība tam piemaisīt testējamo ķimikāliju, ja ķimikāliju ievada šādā ceļā.
|
|
17.
|
Dzīvniekus ieteicams turēt mazās viendzimuma grupās; ja tas zinātniski pamatoti, dzīvniekus var turēt atsevišķi. Ja dzīvniekus tur grupās, dzīvnieku skaits vienā būrī nedrīkstētu pārsniegt 5. Pārošanas procedūras jāveic attiecīgajam nolūkam piemērotos būros. Grūsnas mātītes jātur atsevišķi, un tām jānodrošina migas ierīkošanai vajadzīgie materiāli. Mātītes laktācijas periodā tiks turētas atsevišķi kopā ar to pēcnācēju.
|
|
18.
|
Barība regulāri jāanalizē, lai noteiktu kontaminantu klātbūtni. Barības paraugs jāglabā līdz ziņojuma pabeigšanai.
|
Dzīvnieku sagatavošana
|
19.
|
Veselīgus jaunus pieaugušus dzīvniekus nejaušināti iedala kontrolgrupās un apstrādes grupas. Būrus izvieto tā, lai būru novietojuma varbūtējā ietekme būtu iespējami maza. Dzīvniekus unikāli identificē un pirms pētījuma sākšanas vismaz piecas dienas tur būros, lai tie aklimatizētos laboratorijas apstākļos.
|
Devu sagatavošana
|
20.
|
Testējamo ķimikāliju ieteicams ievadīt perorāli, ja vien par piemērotākiem netiek uzskatīti citi ievadīšanas ceļi. Ja izvēlēts perorālais ievadīšanas ceļš, testējamo ķimikāliju parasti ievada ar gavāžu; tomēr testējamo ķimikāliju var ievadīt arī ar uzturu vai dzirdināmo ūdeni.
|
|
21.
|
Ja vajadzīgs, testējamo ķimikāliju izšķīdina vai suspendē piemērotā nesējā. Kad vien iespējams, ieteicams lietošanai izvēlēties ūdens šķīdumu/suspensiju vai, ja tas nav iespējams, šķīdumu/emulsiju eļļā (piemēram, kukurūzas eļļā), vai, ja arī tas nav iespējams, šķīdumu citos nesējos. Ja nesējs nav ūdens, ir jāzina šā nesēja toksiskās īpašības. Jānosaka testējamās ķimikālijas stabilitāte un homogenitāte nesējā.
|
PROCEDŪRA
Dzīvnieku skaits un dzimums
|
22.
|
Ieteicams, lai katrā grupā sākumā būtu vismaz 10 tēviņi un 12–13 mātītes. Pirms ekspozīcijas izvērtē mātīšu estrālos ciklus, un dzīvniekus, kam nav novērojami tipiski 4–5 dienu cikli, pētījumā neiekļauj; tāpēc ieteicams izmantot lielāku skaitu mātīšu, lai katrā grupā būtu 10 mātītes. Izņemot izteiktas toksiskās ietekmes gadījumus, sagaidāms, ka katrā grupā būs vismaz 8 grūsnas mātītes, kas parasti ir minimālais pieļaujamais grūsno mātīšu skaits grupā. Mērķis ir nodrošināt pietiekamu daudzumu grūsnu mātīšu un pietiekamu daudzumu pēcnācēju, lai varētu ticami izvērtēt testējamās ķimikālijas spēju ietekmēt auglību, grūsnību, mātišķo uzvedību un zīdīšanu, kā arī F1 pēcnācēju augšanu un attīstību no aizmešanās līdz 13. dienai pēc piedzimšanas.
|
Devas
|
23.
|
Parasti jāizmanto vismaz trīs testa grupas un kontrolgrupa. Devu līmeņus var noteikt, pamatojoties uz informāciju, kas iegūta akūta toksiskuma testos, vai atkārtotas devas pētījumu rezultātiem. Ar kontrolgrupas dzīvniekiem jārīkojas tāpat kā ar testa grupu dzīvniekiem, izņemot attiecībā uz testējamās ķimikālijas ievadīšanu. Ja testējamās ķimikālijas ievadīšanai izmanto nesēju, kontrolgrupa saņem lielāko izmantoto nesēja daudzumu.
|
|
24.
|
Devu līmeņi jāizvēlas, ņemot vērā visus pieejamos toksiskuma un (toksiko)kinētiskos datus. Jāņem vērā arī tas, ka grūsnu un negrūsnu dzīvnieku jutība var atšķirties. Augstākais devas līmenis jāizvēlas tāds, lai tas izraisītu toksisku ietekmi, bet ne nobeigšanos vai smagas ciešanas. Pēc tam jāizvēlas tāda lejupēja devas līmeņu virkne, lai ar to varētu parādīt jebkādas ar devu saistītas atbildreakcijas, kā arī to, ka zemākajā devas līmenī nelabvēlīga ietekme nav novērojama (NOAEL). Devu līmeņu noteikšanai to samazinājuma virzienā optimums bieži vien ir divkārši līdz četrkārši intervāli, un bieži vien ir ieteicamāk pievienot ceturto testa grupu, nevis izmantot ļoti plašu devu intervālu (piemēram, vairāk nekā 10 reizes).
|
|
25.
|
Ja ir novērota vispārēja toksicitāte (piemēram, ķermeņa masas samazināšanās, ietekme uz aknām, sirdi, plaušām vai nierēm utt.) vai citas pārmaiņas, kas var nebūt toksiskas atbildreakcijas (piemēram, samazināts barības patēriņš, aknas palielināšanās), ir jābūt piesardzīgiem, interpretējot novēroto iedarbību uz jutīgiem endokrīnajiem beigupunktiem.
|
Robežtests
|
26.
|
Ja perorālajā pētījumā, kurā izmanto vienu devas līmeni, kas ir vismaz 1 000 mg/kg ķermeņa masas dienā, vai — ja testējamo ķimikāliju ievada ar uzturu vai dzirdināmo ūdeni — līdzvērtīgs procentuālais daudzums uzturā vai dzirdināmajā ūdenī, un kuru veic, ievērojot šajā pētījumā aprakstītās procedūras, novērojama toksiska ietekme nerodas un ja, pamatojoties uz datiem par strukturāli radniecīgām vielām, toksicitāte nav sagaidāma, var uzskatīt, ka pilns pētījums ar vairākiem devu līmeņiem nav vajadzīgs. Robežtestu neizmanto gadījumos, kad cilvēka ekspozīcija norāda uz to, ka ir jāizmanto augstāks perorālās devas līmenis. Ja izmanto citus ievadīšanas veidus, tādus kā ieelpošana vai aplicēšana uz ādas, maksimālā sasniedzamā koncentrācija bieži vien ir atkarīga no testējamo ķimikāliju fizikālķīmiskajām īpašībām.
|
Devu došana
|
27.
|
Dzīvnieki testējamās ķimikālijas devu saņem katru dienu 7 dienas nedēļā. Ja izmanto gavāžu, dzīvniekiem testējamā ķimikālija jāsaņem vienreizējā devā, ko ievada ar kuņģa zondi vai piemērotu intubācijas cauruli. Maksimālais šķidruma tilpums, ko vienā paņēmienā var ievadīt, ir atkarīgs no testa dzīvnieka lieluma. Tilpums nedrīkst pārsniegt 1 ml uz 100 g ķermeņa masas, izņemot tādus ūdens šķīdumus, kur var lietot 2 ml uz 100 g ķermeņa masas. Izņemot kairinošas vai kodīgas testējamās ķimikālijas, kam lielākās koncentrācijās parasti ir saasināta ietekme, testa tilpuma mainība būtu jāsamazina līdz minimumam, koncentrāciju koriģējot tā, lai visos devu līmeņos saglabātos konstants tilpums.
|
|
28.
|
Ja testējamo ķimikāliju ievada ar uzturu vai dzirdināmo ūdeni, ir svarīgi nodrošināt, ka testējamās ķimikālijas daudzums neizjauc normālu uztura vai ūdens līdzsvaru. Ja testējamo ķimikāliju ievada ar uzturu, var izmantot nemainīgu koncentrāciju uzturā (ppm) vai nemainīgu devas līmeni attiecībā pret dzīvnieku ķermeņa masu; izmantotā alternatīva ir jānorāda. Ja testējamo ķimikāliju ievada ar gavāžu, deva jādod katru dienu vienādā laikā un vismaz reizi nedēļā jākoriģē tā, lai uzturētu nemainīgu devas līmeni attiecībā pret dzīvnieka ķermeņa masu.
|
Eksperimenta grafiks
|
29.
|
Devas abu dzimumu dzīvniekiem jāsāk dot vismaz 2 nedēļas pirms pārošanas, pēc tam, kad tie vismaz piecas dienas tikuši aklimatizēti un mātītēm ir novēroti normāli estrālie cikli (divu nedēļu ilgā priekšapstrādes periodā). Pētījums jāplāno tā, lai estrālā cikla izvērtēšana sāktos drīz pēc tam, kad dzīvnieki ir sasnieguši pilnīgu dzimumgatavību. Dažādām žurku līnijām pilnīgas dzimumgatavības sasniegšanas vecums dažādās laboratorijās var nedaudz atšķirties, piemēram, Sprague Dawley līnijas žurkas to sasniedz 10 nedēļu vecumā, Wistar līnijas žurkas — 12 nedēļu vecumā. Mātes ar pēcnācēju jānonāvē 13. pēcdzemdību dienā vai drīz pēc tam. Dzimšanas diena (proti, dienā, kurā dzemdības ir beigušās) ir 0. pēcdzemdību diena. Mātītes, kam nav novērojamas kopulācijas pazīmes, nonāvē 24–26 dienas pēc pēdējās pārošanas perioda dienas. Pārošanas perioda laikā abu dzimumu dzīvniekiem devas turpina dot. Pēc pārošanas perioda tēviņiem devas jāturpina dot vismaz līdz minimālā kopējā devu došanas perioda (28 dienas) beigām. Pēc tam tos vai nu nonāvē, vai arī patur dzīvus un tiem turpina dot devas, lai tos, iespējams, izmantotu otrai pārošanai, ja tāda vajadzīga.
|
|
30.
|
Vecākpaaudzes mātītēm devas turpina dot katru dienu visu grūsnības laiku un vismaz līdz 13. pēcdzemdību dienai vai dienai pirms nonāvēšanas (minētās dienas ieskaitot). Pētījumos, kuros testējamo ķīmisko vielu ievada inhalācijas ceļā vai caur ādu, devas jāturpina dot vismaz līdz gestācijas 19. dienai (ieskaitot) un jāatsāk dot pēc iespējas drīz, bet ne vēlāk kā 4. dienā pēc dzemdībām.
|
|
31.
|
Eksperimenta grafika shēma ir dota 2. papildinājumā.
|
Pārošanas procedūra
|
32.
|
Parasti šajā pētījumā jāizmanto pārošanas shēma 1:1 (viens tēviņš uz vienu mātīti). Izņēmumi pieļaujami tēviņu nāves gadījumā. Mātīte jātur kopā ar to pašu tēviņu, līdz tiek novērotas kopulācijas pazīmes vai ir pagājušas divas nedēļas. Katru rītu mātītēm pārbauda spermas vai embola klātbūtni makstī. Grūsnību sāk skaitīt no dienas, kurā ir apstiprinājies kopulācijas fakts (konstatēts maksts embols vai sperma). Ja pārošana ir neveiksmīga, var apsvērt mātīšu atkārtotu pārošanu ar tās pašas grupas tēviņiem.
|
Metiena lielums
|
33.
|
Ceturtajā dienā pēc piedzimšanas katra metiena lielumu var koriģēt, nejaušināti izņemot liekos mazuļus tā, lai katrā metienā pēc iespējas būtu četri vai pieci katra dzimuma mazuļi atkarībā no izmantotās žurku līnijas parastā metiena lieluma. No diviem liekajiem mazuļiem jāņem asins paraugi, kurus apkopo un izmanto seruma T4 līmeņu noteikšanai. Mazuļu selektīva izņemšana, piemēram, pēc ķermeņa masas vai anoģenitālā attāluma (AGD), nav piemērota. Ja vīrišķā vai sievišķā dzimuma mazuļu skaita dēļ nav iespējams nodrošināt, ka katrā metienā ir četri vai pieci katra dzimuma mazuļi, ir pieļaujama metiena lieluma daļēja koriģēšana (piemēram, seši tēviņi un četras mātītes). Ja metiena lielums ir mazāks par korekcijas robežvērtību (8 vai 10 mazuļi vienā metienā), mazuļus no metiena neizņem. Ja korekcijas robežvērtību pārsniedz tikai viens mazulis, izņem tikai vienu mazuli un no tā ņem asins paraugus, ko izmanto iespējamiem seruma T4 novērtējumiem.
|
|
34.
|
Ja metiena lielums netiek koriģēts, 4. dienā pēc piedzimšanas divus mazuļus no katra metiena nonāvē un no tiem ņem asins paraugus, ko izmanto tiroīdhormona seruma koncentrācijas mērīšanai. Ja iespējams, diviem mazuļiem katrā metienā jābūt sievišķā dzimuma mazuļiem, lai vīrišķā dzimuma mazuļus rezervētu zīdekļu retencijas izvērtējumiem, izņemot gadījumus, kad šo mazuļu izņemšanas rezultātā galējam novērtējumam nepaliek neviena mātīte. Mazuļus no metiena neizņem, ja tā lielums būs mazāks nekā 8 vai 10 mazuļi vienā metienā (atkarībā no izmantotās žurku līnijas parastā metiena lieluma). Ja, pamatojoties uz parasto metiena lielumu, lieks ir tikai viens mazulis, izņem tikai vienu mazuli un no tā ņem asins paraugus, ko izmanto iespējamiem seruma T4 novērtējumiem.
|
NOVĒROJUMI DZĪVES LAIKĀ
Klīniskie novērojumi
|
35.
|
Visā testa periodā vismaz reizi dienā jāizdara klīniskie novērojumi; ja ir novērotas toksiskuma pazīmes, tie jāizdara biežāk. Klīniskie novērojumi katru dienu jāizdara, vēlams, vienā un tajā pašā laikā vai laikos, ņemot vērā gaidāmo ietekmju maksimumperiodu pēc devu došanas. Jāreģistrē acīm redzamas izmaiņas uzvedībā, grūtas vai novēlotas atnešanās pazīmes un visas toksiskuma pazīmes, kā arī mirstība. Šajos datos jāiekļauj toksiskuma pazīmju parādīšanās laiks, pakāpe un ilgums.
|
Ķermeņa masa un barības/ūdens patēriņš
|
36.
|
Tēviņi un mātītes jānosver pirmajā devas došanas dienā, tad vismaz reizi nedēļā un pētījuma beigās. Grūsnības laikā mātītes jānosver 0., 7., 14. un 20. dienā un 24 stundu laikā pēc atnešanās (0. vai 1. pēcdzemdību diena), kā arī vismaz 4. un 13. pēcdzemdību dienā. Novērojumu rezultāti jāreģistrē atsevišķi par katru pieaugušo dzīvnieku.
|
|
37.
|
Pirmspārošanas, grūsnības un laktācijas laikā barības patēriņš jāmēra vismaz reizi nedēļā. Barības patēriņa mērīšana pārošanas laikā ir fakultatīva. Ja testējamo ķimikāliju ievada ar dzirdināmo ūdeni, minētajos laikposmos ir jāmēra arī ūdens patēriņš.
|
Estrālie cikli
|
38.
|
Lai pētījumam atlasītu mātītes ar regulāru cikliskumu, pirms apstrādes sākšanas jānovēro estrālie cikli (sk. 22. punktu). No apstrādes perioda sākuma līdz brīdim, kad ir novērotas pārošanās pazīmes, katru dienu jāveic arī vaginālo uztriepju monitorings. Ja pastāv bažas par akūta stresa ietekmi, kas, uzsākot devu došanu, var mainīt estrālos ciklus, laboratorijas var testa dzīvniekus divas nedēļas pakļaut ekspozīcijai un pēc tam reizi dienā ievākt vaginālās uztriepes, lai pirmspārošanas periodā vismaz divas nedēļas veiktu estrālo ciklu novērošanu, ko turpina veikt arī pārošanas periodā, līdz ir iegūti pārošanās pierādījumi. Iegūstot maksts / dzemdes kakla šūnas, jārūpējas par to, lai nepieļautu gļotādas bojājumus, kas var inducēt viltus grūsnību (7) (8).
|
Pēcnācēju parametri
|
39.
|
Jāreģistrē gestācijas ilgums, ko aprēķina no 0. grūsnības dienas. Pēc atnešanās katru metienu iespējami drīz pārbauda, lai noteiktu mazuļu, nedzīvi dzimušo, dzīvi dzimušo un nīkuļu (mazuļi, kas ir ievērojami mazāki nekā atbilstīgie kontrolmazuļi) skaitu un dzimumu un acīmredzamu anomāliju klātbūtni.
|
|
40.
|
24 stundu laikā pēc atnešanās (0. vai 1. diena pēc piedzimšanas) un vismaz 4. un 13. dienā pēc piedzimšanas jāsaskaita dzīvie mazuļi un jānosaka to dzimums, un jānosver metieni. Papildus 35. punktā aprakstītajiem novērojumiem jāreģistrē pēcnācēju uzvedības anomālijas.
|
|
41.
|
Katra mazuļa AGD jāmēra vienā un tajā pašā dienā pēc piedzimšanas no 0. līdz 4. dienai pēc piedzimšanas. Mazulis ir jānosver tajā pašā dienā, kad tiek mērīts AGD, un AGD ir jāpielāgo mazuļa izmēram, priekšroku dodot kvadrātsaknei no ķermeņa svara (9). Kā ieteikts ESAO GD 151 (10), 12. vai 13. dienā pēc piedzimšanas ir jāsaskaita vīrišķā dzimuma mazuļu zīdekļi/areolas.
|
Klīniskā bioķīmija
|
42.
|
Asins paraugus no noteiktas vietas ņem saskaņā ar šādu grafiku:
|
—
|
4. dienā pēc piedzimšanas — no vismaz diviem mazuļiem katrā metienā, ja mazuļu skaits to ļauj (sk. 33.–34. punktu),
|
|
—
|
pētījuma beigās 13. dienā — no visām mātēm un vismaz diviem mazuļiem katrā metienā, un
|
|
—
|
pētījuma beigās — no visiem pieaugušiem tēviņiem.
|
|
Visus asins paraugus glabā piemērotos apstākļos. Asins paraugos, kas ņemti no mazuļiem 13. dienā un pieaugušiem tēviņiem, nosaka tiroīdhormonu (T4) seruma līmeņus. Ja vajadzīgs, T4 līmeņus nosaka arī asins paraugos, kas ņemti no mātēm un 4. dienas mazuļiem. Attiecīgā gadījumā var fakultatīvi mērīt citus hormonus. Tiroīdhormona analīžu veikšanai mazuļu asinis var apkopot pa metieniem. Tiroīdhormonus (T4 un TSH) vēlams mērīt kopējā formā (kopējais T4 un kopējais TSH).
|
43.
|
Noteikto hormonu mainību un absolūto koncentrāciju var ietekmēt šādi faktori:
|
—
|
nonāvēšanas laiks hormonu koncentrāciju diennakts mainības dēļ,
|
|
—
|
nonāvēšanas metode, kurā dzīvniekiem netiek radīts nevajadzīgs stress, kas varētu ietekmēt hormonu koncentrācijas,
|
|
—
|
hormonu noteikšanas testa komplekti, kas var atšķirties pēc standartlīknēm.
|
|
|
44.
|
Plazmas paraugi, kas paredzēti tieši hormonu noteikšanai, ir jāiegūst salīdzināmā dienas laikā. Skaitliskās vērtības, ko iegūst, analizējot hormonu koncentrācijas, atšķiras atkarībā no tirdzniecībā pieejamajiem testēšanas komplektiem.
|
Patoloģija
Makroskopiskā autopsija
|
45.
|
Tūlīt pēc nonāvēšanas vai nāves, kas iestājusies pētījuma laikā, pieaugušie dzīvnieki makroskopiski jāpārbauda attiecībā uz anomālijām vai patoloģiskām pārmaiņām. Īpaša uzmanība jāpievērš reproduktīvās sistēmas orgāniem. Jāreģistrē implantācijas vietu skaits. Lai noteiktu estrālā cikla fāzi un ļautu to saistīt ar olnīcu histopatoloģiju, vaginālās uztriepes jāizmeklē autopsijas dienas rītā.
|
|
46.
|
Visu pieaugušo tēviņu sēklinieki un sēklinieku piedēkļi, kā arī prostata un sēklas pūslīši ar prostatas priekšējām daivām (kopā) pēc vajadzības jāatbrīvo no pieguļošiem audiem, un to slapjais svars jānosaka iespējami drīz pēc disekcijas, lai izvairītos no izžūšanas. Turklāt fakultatīvos orgānu masas mērījumos var ietvert arī tūpļa cēlājmuskuļa un briedumķermeņa muskuļa kompleksu, Kupera dziedzerus un dzimumlocekļa galviņu tēviņiem un abas olnīcas (slapjais svars) un dzemdi (ar dzemdes kaklu) mātītēm; ietveršanas gadījumā minētie masas mērījumi jāveic iespējami drīz pēc disekcijas.
|
|
47.
|
Nedzīvi mazuļi un mazuļi, kas nonāvēti 13. dienā pēc piedzimšanas vai drīz pēc tam, vismaz rūpīgi ārēji jāpārbauda, lai konstatētu iespējamas acīmredzamas anomālijas. Īpaša uzmanība jāpievērš ārējām reproduktīvajām ģenitālijām, kuras jāpārbauda, meklējot attīstības izmaiņu pazīmes. 13. dienā jāsaglabā vairogdziedzeris, kas izņemts no 1 vīrišķā un 1 sievišķa dzimuma mazuļa katrā metienā.
|
|
48.
|
Jāsaglabā visu pieaugušo dzīvnieku olnīcas, sēklinieki, iekšējie dzimumorgāni (dzemde un dzemdes kakls, sēklinieku piedēkļi, prostata, sēklas pūslīši un prostatas priekšējās daivas), vairogdziedzeris un visi orgāni, kam ir redzami makroskopiski bojājumi. Sēklinieku un sēklinieku piedēkļu parastai pārbaudei nav ieteicams izmantot formalīna fiksāciju. Attiecībā uz šiem audiem pieņemama metode ir izmantot Buēna fiksatoru vai modificētu Davidsona fiksatoru (11). Lai nodrošinātu fiksatora ātru penetrēšanos, bālgano apvalku (tunica albuginea) var abos orgāna polos uzmanīgi un sekli pārdurt ar adatu.
|
Histopatoloģija
|
49.
|
Veic lielākās devas grupas dzīvnieku un kontrolgrupas dzīvnieku olnīcu, sēklinieku un sēklinieku piedēkļu detalizētu histopatoloģisku izmeklēšanu (īpašu uzsvaru liekot uz sēklinieku intersticiālo šūnu struktūras spermatoģenēzi un histopatoloģiju). Vajadzības gadījumā var izmeklēt pārējos saglabātos mazuļu un pieaugušo dzīvnieku orgānus, arī vairogdziedzeri. Vairogdziedzera masu var noteikt pēc fiksācijas. Tas jāatbrīvo no piegulošiem audiem ļoti uzmanīgi un tikai pēc fiksācijas, lai izvairītos no audu bojājumiem. Audu bojājumi var apgrūtināt histopatoloģijas analīzi. Ja lielākās devas grupā ir novērotas pārmaiņas, minētās izmeklēšanas jāveic arī citu devu grupu dzīvniekiem. Norādījumos par histopatoloģiju (11) ir sīki izklāstīta papildu informācija par endokrīno audu disekciju, fiksāciju, secēšanu un histopatoloģiju.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Dati
|
50.
|
Jāsniedz dati par atsevišķiem dzīvniekiem. Turklāt visi dati jāapkopo tabulā, kurā redzams testa sākumā katrā testā grupā esošo dzīvnieku skaits, to dzīvnieku skaits, kas atrasti miruši testa laikā vai nonāvēti humānu apsvērumu dēļ, nāves iestāšanās vai humānās nonāvēšanas laiks, auglīgo dzīvnieku skaits, grūsno mātīšu skaits, to dzīvnieku skaits, kam novērotas toksicitātes pazīmes, novēroto toksicitātes pazīmju apraksts, arī toksiskās darbības sākuma laiks, ilgums un stiprums, histopatoloģisko pārmaiņu veidi un visi relevantie dati par metienu. Tabulas veidā sagatavotā kopsavilkuma ziņojuma paraugs, kas ir ļoti noderīgs ietekmes uz reprodukciju/attīstību izvērtēšanai, ir sniegts 3. papildinājumā.
|
|
51.
|
Tā kā pētījuma tvērums ir ierobežots, statistiskajai analīzei, ko veic, lai izvērtētu rezultātu nozīmīgumu, ir ierobežota vērtība attiecībā uz daudziem beigupunktiem, jo īpaši reproduktīvajiem beigupunktiem. Ja tiek izmantota statistiskā analīze, izvēlētajai metodei jābūt atbilstošai pārbaudāmā mainīgā lieluma sadalījumam, un tā jāizvēlas pirms pētījuma sākšanas. AGD un zīdekļu retencijas statistiskajai analīzei jābalstās uz atsevišķu mazuļu datiem, ņemot vērā ietekmi uz metienu. Vajadzības gadījumā analīzes vienība ir metiens. Mazuļu ķermeņa masas statistiskajai analīzei jābalstās uz atsevišķu mazuļu datiem, ņemot vērā metiena lielumu. Ņemot vērā grupu mazo lielumu, pētījuma interpretēšanā par palīglīdzekli var būt lietderīgi izmantot arī vēsturiskos kontroles datus (piemēram, datus par metiena lielumu), ja tādi ir pieejami.
|
Rezultātu izvērtēšana
|
52.
|
Šā toksicitātes pētījuma konstatējumi jāizvērtē, ņemot vērā novēroto ietekmi, autopsiju un mikroskopiskos konstatējumus. Vērtējums ietver testējamās ķimikālijas devas saistību ar anomāliju — to vidū makroskopisko bojājumu, identificēto mērķorgānu, neauglības, klīnisko anomāliju, ietekmes uz reproduktīvajiem un metiena rādītājiem, ķermeņa masas pārmaiņu, ietekmes uz mirstību un citas toksiskās iedarbības – esību vai neesību, incidenci un smagumu.
|
|
53.
|
Tēviņu apstrādes periods ir īss, tāpēc, novērtējot ietekmi uz tēviņu reproduktīvo funkciju, papildus auglības datiem jāņem vērā sēklinieku un sēklinieku piedēkļu histopatoloģija. Pētījuma interpretēšanā par palīglīdzekli var būt lietderīgi izmantot arī vēsturiskus kontroles datus par reprodukciju/ontoģenēzi (piemēram, datus par metiena lielumu, AGD, zīdekļu retenciju, seruma T4 līmeņiem), ja tādi pieejami.
|
|
54.
|
Kvalitātes kontroles vajadzībām ir ierosināts apkopot vēsturiskus kontroldatus un skaitliskajiem datiem, it sevišķi parametriem, kas saistīti ar endokrīnās sistēmas traucējumu izraisītāju noteikšanu, aprēķināt variācijas koeficientus. Šos datus var izmantot salīdzināšanas nolūkos, kad tiek novērtēti faktiskie pētījumi.
|
Testēšanas pārskats
|
55.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Testējamā ķimikālija:
|
—
|
avots, sērijas numurs, izmantošanas termiņš, ja pieejams,
|
|
—
|
testējamās ķimikālijas stabilitāte, ja tā zināma;
|
|
|
|
vienkomponenta viela:
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības,
|
|
—
|
ķīmiskā identifikācija, piemēram, IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, attiecīgā gadījumā, ja praktiski iespējams, piemaisījumu ķīmiskā identitāte u. tml.;
|
|
|
|
daudzkomponentu viela, UVCB un maisījumi:
|
—
|
iespējami izsmeļoši raksturoti pēc komponentu ķīmiskās identitātes (sk. iepriekš), kvantitatīvās sastopamības un relevantajām fizikālķīmiskajām īpašībām.
|
|
|
|
Nesējs (attiecīgā gadījumā):
|
—
|
nesēja izvēles pamatojums, ja nesējs nav ūdens.
|
|
|
|
Testa dzīvnieki:
|
—
|
izmantotās sugas/līnijas,
|
|
—
|
dzīvnieku skaits, vecums un dzimums,
|
|
—
|
avots, turēšanas apstākļi, uzturs u. tml.,
|
|
—
|
katra dzīvnieka masa testa sākumā,
|
|
—
|
sugas izvēles pamatojums, ja tā nav žurka.
|
|
|
|
Testēšanas nosacījumi:
|
—
|
devu līmeņa izraudzīšanās pamatojums,
|
|
—
|
detalizēta informācija par testējamās ķimikālijas maisījuma/barības preparāta sagatavošanu, sasniegto preparāta koncentrāciju, stabilitāti un homogenitāti,
|
|
—
|
detalizēta informācija par testējamās ķimikālijas devu došanu,
|
|
—
|
attiecīgā gadījumā testējamās ķimikālijas koncentrācijas (ppm) barībā / dzirdināmajā ūdenī pārrēķins faktiskajā devā (mg uz kg ķermeņa masas dienā),
|
|
—
|
detalizēta informācija par barības un ūdens kvalitāti,
|
|
—
|
detalizēts apraksts par nejaušināšanas procedūru, kas izmantota, lai atlasītu mazuļus izņemšanai no metiena, ja šāda izņemšana notikusi.
|
|
|
|
Rezultāti:
|
—
|
ķermeņa masa / ķermeņa masas izmaiņas,
|
|
—
|
barības patēriņš un ūdens patēriņš (ja šādi dati ir pieejami),
|
|
—
|
dati par toksiskajām atbildes reakcijām atkarībā no dzimuma un devas, arī dati par auglību, gestāciju un citām toksicitātes pazīmēm,
|
|
—
|
toksiska vai cita iedarbība uz reprodukciju, pēcnācējiem, postnatālo augšanu utt.,
|
|
—
|
klīnisko novērojumu veids, smagums un ilgums (atgriezenisks vai neatgriezenisks),
|
|
—
|
pieaugušu mātīšu skaits ar normālu vai nenormālu estrālo ciklu un cikla ilgums,
|
|
—
|
dzīvi dzimušo skaits un pēcimplantācijas zudums,
|
|
—
|
mazuļu ķermeņa masas dati,
|
|
—
|
visu mazuļu AGD (un ķermeņa masa AGD mērīšanas dienā),
|
|
—
|
zīdekļu retencija vīrišķā dzimuma mazuļiem,
|
|
—
|
vairogdziedzera hormonu līmenis 13 dienu veciem mazuļiem un pieaugušiem tēviņiem (un mātēm un 4 dienas veciem mazuļiem, ja šādi mērījumi veikti),
|
|
—
|
to mazuļu skaits, kam ir makroskopiskas anomālijas, ārējo ģenitāliju makroskopisks izvērtējums, nīkuļu skaits,
|
|
—
|
nāves iestāšanās laiks testa laikā vai norāde, ka dzīvnieki ir izdzīvojuši līdz pētījuma beigām,
|
|
—
|
implantāciju skaits, metiena lielums un masa datu reģistrēšanas laikā,
|
|
—
|
ķermeņa masa nonāvēšanas laikā un dati par vecākpaaudzes dzīvnieku orgānu masu,
|
|
—
|
autopsijas konstatējumi,
|
|
—
|
histopatoloģiskas izmeklēšanas rezultātu detalizēts apraksts,
|
|
—
|
absorbcijas dati (ja pieejami),
|
|
—
|
rezultātu statistiskā apstrāde (attiecīgā gadījumā).
|
|
Rezultātu iztirzājums.
Secinājumi.
|
Rezultātu interpretēšana
|
56.
|
Pētījums sniegs izvērtējumu par reproduktīvo/ontoģenētisko toksicitāti, kas saistīta atkārtotu devu došanu (sk. 5. un 6. punktu). Tas varētu norādīt uz nepieciešamību veikt turpmākas izmeklēšanas un sniedz norādījumus turpmāko pētījumu plānošanā. Ar reprodukciju un ontoģenēzi saistīto rezultātu interpretēšanā par palīglīdzekli jāizmanto ESAO norādījumu dokuments Nr. 43 (12). ESAO norādījumu dokumentā Nr. 106 par grauzēju endokrīno un reproduktīvo testu histoloģisko izvērtēšanu (11) sniegta informācija par (endokrīno) orgānu un vaginālo uztriepju sagatavošanu un izvērtēšanu; minētā informācija var būt noderīga saistībā ar šo TG.
|
LITERATŪRA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development,.Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
1. papildinājums
DEFINĪCIJAS (SK. ARĪ ESAO GD 150 (6))
Androgenitāte ir ķimikālijas spēja zīdītāja organismā funkcionēt kā dabiskam androgēnu tipa hormonam (piemēram, testosteronam).
Antiandrogenitāte ir ķimikālijas spēja zīdītāja organismā nomākt dabiska androgēnu tipa hormona (piemēram, testosterona) darbību.
Antiestrogenitāte ir ķimikālijas spēja zīdītāja organismā nomākt dabiska estrogēnu tipa hormona (piemēram, estradiola 17ß) darbību.
Antitireoidāla iedarbība ir ķimikālijas spēja zīdītāja organismā nomākt dabiska tiroīdhormona (piemēram, T3) darbību.
Ķimikālija ir viela vai maisījums.
Ontoģenētiskā toksicitāte: reproduktīvās toksicitātes izpausme, kas liecina par pēcnācēju prenatāliem, perinatāliem, postnatāliem, strukturāliem vai funkcionāliem traucējumiem.
Dozējums ir vispārīgs termins, kas aptver devu, tās ievadīšanas biežumu un ilgumu.
Deva ir ievadītās testējamās ķimikālijas daudzums. Devu izsaka ar testējamās ķimikālijas masu uz testa dzīvnieka ķermeņa masas vienību dienā (piemēram, mg uz kg ķermeņa masas dienā) vai kā nemainīgu koncentrāciju barībā.
Izteikta toksicitāte ir vispārīgs termins, kas apzīmē acīmredzamas toksicitātes pazīmes pēc testējamās ķimikālijas ievadīšanas. Šīm pazīmēm jābūt pietiekamām, lai varētu novērtēt bīstamību, un tādām, lai varētu sagaidīt, ka ievadītās devas palielināšana izraisīs smagas toksicitātes pazīmes un, iespējams, mirstību.
Fertilitātes traucējumi ir tēviņu vai mātīšu reproduktīvās funkcijas vai spējas traucējumi.
Maternālā toksicitāte: nelabvēlīgā ietekme uz gravidām mātītēm, kas ir specifiska (tieša ietekme) vai nespecifiska (netieša ietekme).
NOAEL ir saīsinājums, kas apzīmē nenovērojamās nelabvēlīgās ietekmes līmeni. Tas ir augstākais devas līmenis, kurā netiek konstatētas ar vielas saņemšanu saistītas negatīvas parādības.
Estrogenitāte ir ķimikālijas spēja zīdītāja organismā darboties kā dabiskam estrogēnu tipa hormonam (piemēram, estradiolam 17ß).
Reproduktīvā toksicitāte ir kaitīga ietekme uz pēcnācējiem un/vai tēviņu un mātīšu reproduktīvās funkcijas vai spējas traucējumi.
Testējamā ķimikālija ir jebkura viela vai maisījums, ko testē ar šo testēšanas metodi.
Tireoidāla iedarbība ir ķimikālijas spēja zīdītāja organismā darboties kā dabiskam tiroīdhormonam (piemēram, T3).
Validācija ir zinātnisks process, kura mērķis ir raksturot testēšanas metodes darbības prasības un ierobežojumus un apliecināt tās ticamību un piemērotību konkrētam nolūkam.
2. papildinājums
EKSPERIMENTA GRAFIKA SHĒMA, KURĀ NORĀDĪTS MAKSIMĀLAIS PĒTĪJUMA ILGUMS, KURA PAMATĀ IR 14 DIENU ILGS PĀROŠANAS PERIODS

3. papildinājums
TABULAS VEIDĀ SAGATAVOTS KOPSAVILKUMA ZIŅOJUMS PAR IETEKMI UZ REPRODUKCIJU/ONTOĢENĒZI
|
NOVĒROJUMI
|
VĒRTĪBAS
|
|
|
|
Dozējums (vienības)
|
0 (kontrole)
|
…
|
…
|
…
|
…
|
|
Dzīvnieku pāri (N)
|
|
|
|
|
|
|
Estrālais cikls (vismaz vidējais neregulāru ciklu ilgums un biežums)
|
|
|
|
|
|
|
Mātītes, kam novērojamas kopulācijas pazīmes (N)
|
|
|
|
|
|
|
Grūsnas mātītes (N)
|
|
|
|
|
|
|
Apaugļošanās dienas 1–5 (N)
|
|
|
|
|
|
|
Apaugļošanās dienas 6–...(
(21)
) (N)
|
|
|
|
|
|
|
Grūsnība ≤ 21 diena (N)
|
|
|
|
|
|
|
Grūsnība = 22 dienas (N)
|
|
|
|
|
|
|
Grūsnība ≥ 23 dienas (N)
|
|
|
|
|
|
|
Mātes ar dzīviem mazuļiem (N)
|
|
|
|
|
|
|
Mātes ar dzīviem mazuļiem 4. dienā pēc dzemdībām (N)
|
|
|
|
|
|
|
Implanti vienai mātei (vidēji)
|
|
|
|
|
|
|
Dzīvie mazuļi vienai mātei piedzimšanas brīdī (vidēji)
|
|
|
|
|
|
|
Dzīvie mazuļi vienai mātei 4. dienā (vidēji)
|
|
|
|
|
|
|
Dzimumu attiecība (tēv./māt.) piedzimstot (vidēji)
|
|
|
|
|
|
|
Dzimumu attiecība (tēv./māt.) 4. dienā (vidēji)
|
|
|
|
|
|
|
Metiena masa piedzimstot (vidēji)
|
|
|
|
|
|
|
Metiena masa 4. dienā (vidēji)
|
|
|
|
|
|
|
Mazuļu masa piedzimstot (vidēji)
|
|
|
|
|
|
|
Mazuļu masa AGD mērīšanas laikā (tēviņu vidējā masa, mātīšu vidējā masa)
|
|
|
|
|
|
|
Mazuļu AGD tajā pašā postnatālajā dienā, laikā no dzimšanas līdz 4. dienai (tēviņu vidējā masa, mātīšu vidējā masa)
|
|
|
|
|
|
|
Metiena masa 4. dienā (vidēji)
|
|
|
|
|
|
|
Zīdekļu retencija vīrišķā dzimuma mazuļiem 13. dienā (vidēji)
|
|
|
|
|
|
|
Metiena masa 13. dienā (vidēji)
|
|
|
|
|
|
|
|
|
MAZUĻI AR ANOMĀLIJĀM
|
|
Mātes, kam nav neviena šāda mazuļa
|
|
|
|
|
|
|
Mātes ar 1 šādu mazuli
|
|
|
|
|
|
|
Mātes ar ≥ 2 šādiem mazuļiem
|
|
|
|
|
|
|
|
|
PĒCNĀCĒJU ZUDUMI
|
|
|
|
Prenatāli / pēc implantācijām (implantāciju skaits mīnus dzīvi dzimušo skaits)
|
|
Mātītes, kas nav zaudējušas mazuli
|
|
|
|
|
|
|
Mātītes ar vienu zaudētu mazuli
|
|
|
|
|
|
|
Mātītes ar diviem zaudētiem mazuļiem
|
|
|
|
|
|
|
Mātītes ar ≥ 3 zaudētiem mazuļiem
|
|
|
|
|
|
|
|
|
Postnatāli (dzīvi dzimušo skaits mīnus 13. postnatālajā dienā dzīvo mazuļu skaits)
|
|
Mātītes, kas nav zaudējušas mazuli
|
|
|
|
|
|
|
Mātītes ar vienu zaudētu mazuli
|
|
|
|
|
|
|
Mātītes ar diviem zaudētiem mazuļiem
|
|
|
|
|
|
|
Mātītes ar ≥ 3 zaudētiem mazuļiem
|
|
|
|
|
|
B.64. KOMBINĒTS ATKĀRTOTAS DEVAS TOKSISKUMA PĒTĪJUMS AR REPRODUKTĪVĀS/ONTOĢENĒTISKĀS TOKSICITĀTES SKRĪNINGA TESTU
IEVADS
|
1.
|
Šī testēšanas metode ir ekvivalenta ESAO testēšanas vadlīnijai (TG) Nr. 422 (2016). ESAO vadlīnijas par ķimikāliju testēšanu periodiski tiek pārskatītas, ņemot vērā zinātnes attīstību. Sākotnējā vadlīnija Nr. 422 par skrīninga testu tika pieņemta 1996. gadā, pamatojoties uz protokolu par “Kombinēto atkārtotas devas un reproduktīvās/ontoģenētiskās toksicitātes skrīninga testu”, kas apspriests divās ekspertu sanāksmēs Londonā 1990. gadā (1) un Tokijā 1992. gadā (2).
|
|
2.
|
Šī testēšanas metode apvieno reproduktīvās/ontoģenētiskās toksicitātes skrīninga daļu, kuras pamatā ir pieredze, kas gūta dalībvalstīs, izmantojot sākotnējo metodi ar lielā apjomā ražotām esošajām ķimikālijām, un pieredze, kas gūta izpētes testos ar pozitīvās kontroles vielām (3) (4), un atkārtotas devas toksiskuma daļu saskaņā ar ESAO testēšanas vadlīniju Nr. 407 (Atkārtotas devas 28 dienu orālās toksicitātes pētījums ar grauzējiem), kura atbilst šā pielikuma B.7. nodaļai.
|
|
3.
|
Pēc tam, kad 1998. gadā ESAO tika sākta prioritāra darbība ar mērķi pārskatīt esošās testēšanas vadlīnijas un izstrādāt jaunas testēšanas vadlīnijas par endokrīno disruptoru skrīningu un testēšanu (5), šī testēšanas metode tika atjaunināta, iekļaujot tajā endokrīnajiem disruptoriem relevantus beigupunktus. Šajā kontekstā TG Nr. 407 (kas atbilst šā pielikuma B.7. nodaļai) 2008. gadā tika papildināta ar parametriem, kas piemēroti testējamo ķimikāliju endokrīnās aktivitātes noteikšanai. TG Nr. 422 tika atjaunināta, lai skrīninga TG, kur ekspozīcijas periodi aptver dažus jutīgos attīstības periodus (prenatālais vai agrīns postnatālais periods), iekļautu dažus endokrīnajiem disruptoriem relevantus beigupunktus.
|
|
4.
|
Izvēlētie endokrīnajiem disruptoriem relevantie papildu beigupunkti, kas ir daļa arī no TG Nr. 443 (Paplašinātais vienas paaudzes reproduktīvās toksicitātes pētījums, šā pielikuma B.56. nodaļa), tika iekļauti TG Nr. 422, pamatojoties uz priekšizpēti, kurā aplūkoti zinātniski un tehniski jautājumi par minēto beigupunktu iekļaušanu, kā arī iespējamie testēšanas plānojuma pielāgojumi, kas vajadzīgi minēto beigupunktu iekļaušanai (6).
|
|
5.
|
Šī testēšanas metode ir izstrādāta, lai iegūtu ierobežotu informāciju par testējamās ķimikālijas ietekmi uz tēviņu un mātīšu reproduktīvo spēju, piemēram, gonadālo funkciju, pārošanās uzvedību, apaugļošanos, miesas augļa attīstību un dzemdībām. Tā nav nedz alternatīva B.31., B.34., B.35. vai B.56. testēšanas metodei, nedz tās aizstāj.
|
SĀKOTNĒJIE APSVĒRUMI
|
6.
|
Novērtējot un izvērtējot testējamās ķimikālijas toksiskās īpašības, orālo toksicitāti ar atkārtotām devām var noteikt pēc tam, kad akūtās toksicitātes testos ir iegūta sākotnējā informācija par toksicitāti. Šis pētījums sniedz informāciju par iespējamajiem veselības apdraudējumiem, kas varētu rasties no atkārtotas ekspozīcijas relatīvi ierobežotā laikposmā. Metodē ietilpst atkārtotas devas toksiskuma pamatpētījums, ko var izmantot attiecībā uz ķimikālijām, par kurām nav pamatoti veikt 90 dienu pētījumu (piemēram, ja ražošanas apjoms nepārsniedz noteiktas robežvērtības), vai par provizorisku pētījumu ilgtermiņa pētījumam. Veicot pētījumu, ir jāievēro galvenie principi un apsvērumi, kas noteikti ESAO norādījumu dokumentā Nr. 19 par klīnisko pazīmju atpazīšanu, novērtēšanu un izmantošanu par humānajiem beigupunktiem eksperimenta dzīvniekiem, kurus izmanto drošības novērtējumā (7).
|
|
7.
|
Šī metode ietver arī reproduktīvās/ontoģenētiskās toksicitātes skrīninga testu, un tāpēc to var izmantot arī, lai sniegtu sākotnējo informāciju par iespējamo ietekmi uz tēviņu un mātīšu reproduktīvo spēju, piemēram, gonadālo funkciju, pārošanās uzvedību, apaugļošanos, miesas augļa attīstību un dzemdībām, vai nu ķimikāliju toksikoloģisko īpašību novērtēšanas agrīnā posmā, vai par ķimikālijām, kas rada bažas. Šī testēšanas metode nesniedz pilnīgu informāciju par visiem reprodukcijas un attīstības aspektiem. Konkrētāk, tā piedāvā tikai ierobežotus līdzekļus, ar ko detektēt prenatālās ekspozīcijas postnatālās izpausmes vai ietekmi, kas var rasties postnatālās ekspozīcijas laikā. Beigupunktu selektivitātes, pētījuma īsā ilguma un citu iemeslu dēļ šī metode nesniedz pierādījumus galīgajiem apgalvojumiem par reproduktīvās/ontoģenētiskās ietekmes neesību. Turklāt, ja nav datu no citiem reproduktīvās/ontoģenētiskās toksicitātes testiem, pozitīvie rezultāti ir noderīgi sākotnējai bīstamības novērtēšanai un palīdz pieņemt lēmumus par papildu testēšanas nepieciešamību un laiku.
|
|
8.
|
Rezultāti, kas iegūti par endokrīnajiem parametriem, ir jāaplūko ESAO endokrīni disruptīvu ķimikāliju testēšanas un novērtēšanas konceptuālā satvara (8) kontekstā. Atjauninātā ESAO TG Nr. 422 ir iekļauta minētā konceptuālā satvara 4. līmenī, kur ietilpst in vivo testi, kas sniedz datus par nelabvēlīgu ietekmi uz endokrīnajiem beigupunktiem. Endokrīnu signālu pašu par sevi tomēr nevar uzskatīt par pietiekamu pierādījumu tam, ka testējamā ķimikālija ir endokrīnais disruptors.
|
|
9.
|
Šajā testēšanas metodē uzsvars likts arī uz neiroloģisko ietekmi kā īpašu beigupunktu un uzsvērta vajadzība izdarīt rūpīgus dzīvnieku klīniskos novērojumus, lai iegūtu iespējami daudz informācijas. Ar šo metodi jāidentificē ķimikālijas ar neirotoksisku potenciālu, saistībā ar kuru varētu būt pamatoti veikt turpmāku padziļinātu izpēti. Turklāt metode var dot arī pamatnorādes uz imunoloģisko ietekmi.
|
|
10.
|
Ja nav datu no citiem sistēmiskās toksicitātes, reproduktīvās/ontoģenētiskās toksicitātes, neirotoksicitātes un/vai imūntoksicitātes pētījumiem, pozitīvie rezultāti ir noderīgi sākotnējai bīstamības novērtēšanai un palīdz pieņemt lēmumus par papildu testēšanas nepieciešamību un laiku. Tests var būt īpaši noderīgs kā daļa no ESAO skrīninga informācijas datu kopas (SIDS), lai novērtētu esošās ķimikālijas, par kurām ir pieejams maz toksikoloģiskās informācijas vai tā nav pieejama vispār, un tas var kalpot par alternatīvu divu atsevišķu testu, proti, atkārtotas devas toksiskuma testa (ESAO TG Nr. 407, kas atbilst šā pielikuma B.7. nodaļai) un reproduktīvās/ontoģenētiskās toksicitātes testa (ESAO TG Nr. 421, kas atbilst šā pielikuma B.63. nodaļai) veikšanai. To var izmantot arī par devu diapazona noteikšanas pētījumu plašākiem reprodukcijas/ontoģenēzes pētījumiem vai citos relevantos gadījumos.
|
|
11.
|
Parasti tiek pieņemts, ka grūsnu un negrūsnu dzīvnieku jutība atšķiras. Tāpēc šajā kombinētajā testā var būt grūti noteikt devu līmeņus, kas ir pietiekami gan vispārējas sistēmiskas toksicitātes, gan specifiskas reproduktīvās/ontoģenētiskās toksicitātes izvērtēšanai; to vieglāk izdarīt, ja individuālos testus veic atsevišķi. Turklāt testa rezultātus attiecībā uz vispārējo sistēmisko toksicitāti var būt sarežģītāk interpretēt nekā tad, ja veic atsevišķu atkārtotas devas pētījumu, jo īpaši gadījumos, kad seruma un histopatoloģijas parametri pētījumā netiek izvērtēti vienlaicīgi. Šo tehnisko sarežģījumu dēļ šā kombinētā skrīninga testa veikšanai ir vajadzīga liela pieredze toksicitātes testēšanā. No otras puses, papildus tam, ka testā tiek izmantots mazāks skaits dzīvnieku, kombinētais tests var nodrošināt labāku veidu, kā tiešo ietekmi uz reprodukciju/ontoģenēzi nošķir no ietekmes, kas ir sekundāra citiem (sistēmiskas) ietekmes veidiem.
|
|
12.
|
Šajā testā devu došanas periods ir ilgāks nekā parastā 28 dienu ilgā atkārtotas devas pētījumā. Tomēr salīdzinājumā ar situāciju, kad papildus reproduktīvās/ontoģenētiskās toksicitātes skrīninga testam tiek veikts 28 dienu ilgs atkārtotas devas pētījums, šajā testā katrā grupā tiek izmantots mazāks skaits katra dzimuma dzīvnieku
|
|
13.
|
Šajā testēšanas metodē izmanto perorālo testējamās ķimikālijas ievadīšanu. Citu ekspozīcijas ceļu izmantošanas gadījumā var būt vajadzīgas modifikācijas.
|
|
14.
|
Pirms testēšanas metodi attiecībā uz maisījumu izmanto, lai iegūtu datus kādai plānotai regulatīvai vajadzībai, jāapsver, vai un kādēļ tā var sniegt tādam nolūkam piemērotus rezultātus. Šādi apsvērumi nav vajadzīgi, ja pastāv regulatīva prasība maisījumu testēt.
|
|
15.
|
Izmantotās definīcijas sniegtas 1. papildinājumā.
|
TESTA PRINCIPS
|
16.
|
Testējamo ķimikāliju pakāpeniski pieaugošās devās saņem vairākas tēviņu un mātīšu grupas. Tēviņiem testējamās ķimikālijas jādod vismaz četras nedēļas līdz dienai pirms plānotās nonāvēšanas (minēto dienu ieskaitot); minētais laikposms aptver vismaz divas nedēļas pirms pārošanas, pārošanas periodu un aptuveni divas nedēļas pēc pārošanas. Ņemot vērā ierobežoto pirmspārošanas devu došanas periodu tēviņiem, auglība var nebūt īpaši precīzs testikulārās toksicitātes rādītājs. Tāpēc būtiska ir sēklinieku detalizēta histopatoloģiska izmeklēšana. Divu nedēļu ilgais pirmspārošanas devu došanas periods apvienojumā ar turpmākiem pārošanās/auglības novērojumiem ar kopējo devu došanas periodu vismaz četras nedēļas, kam seko tēviņu gonādu detalizēta histopatoloģija, tiek uzskatīts par pietiekamu, lai varētu noteikt lielāko daļu ietekmes uz tēviņu auglību un spermatoģenēzi.
|
|
17.
|
Mātītēm devas dod visu pētījuma laiku, kas aptver divas nedēļas pirms pārošanas (aptverot divus pilnus estrālos ciklus), laiku līdz apaugļošanās brīdim, grūsnības laiku un vismaz trīspadsmit dienas pēc dzemdībām, līdz dienai pirms plānotās nonāvēšanas (minēto dienu ieskaitot).
|
|
18.
|
Pētījuma ilgums pēc aklimatizācijas un estrālā cikla izvērtējuma, ko veic pirms devu došanas, ir atkarīgs no mātīšu rādītājiem un ir aptuveni 63 dienas [vismaz 14 dienas pirms pārošanas, (ne vairāk kā) 14 dienas pārošana, 22 dienas gestācija, 13 dienas laktācija].
|
|
19.
|
Devu došanas perioda laikā katru dienu rūpīgi novēro, vai dzīvniekiem neparādās toksiskuma pazīmes. Dzīvniekiem, kas testa laikā nobeigušies vai ir nonāvēti, izdara autopsiju, un izdzīvojušos dzīvniekus testa beigās nonāvē un tiem izdara autopsiju.
|
METODES APRAKSTS
Dzīvnieku sugas izraudzīšanās
|
20.
|
Šo testēšanas metodi paredzēts izmantot ar žurkām. Ja šajā TG Nr. 422 noteiktie parametri tiek pētīti citās grauzēju sugās, tas sīki jāpamato. Starptautiskajā validēšanas programmā endokrīno disruptoru detektēšanai saskaņā ar TG Nr. 407 izmantoja tikai žurkas. Nedrīkst izmantot līnijas ar zemu auglību vai līnijas, par kurām ir labi zināms, ka tām ir bieži sastopami attīstības defekti. Jāizmanto veselīgi iepriekš nepāroti dzīvnieki, kuriem eksperimentālas procedūras iepriekš nav veiktas. Jānorāda testa dzīvnieku suga, līnija, dzimums, masa un vecums. Pētījuma sākumā dzīvnieku masas atšķirībām jābūt minimālām un tās nedrīkst pārsniegt ± 20 % no vidējās masas, kas noteikta katram dzimumam atsevišķi. Ja pētījumu veic kā sākotnēju pētījumu ilgtermiņa pētījumam vai pētījumam, kas aptver visu paaudzi, vēlams abos pētījumos izmantot vienas līnijas un izcelsmes dzīvniekus.
|
Turēšana un barošana
|
21.
|
Visām procedūrām jāatbilst vietējiem laboratorijas dzīvnieku labturības standartiem. Temperatūrai eksperimentālo dzīvnieku turēšanas telpā jābūt 22 °C (± 3 o). Relatīvajam mitrumam jābūt vismaz 30 % un, vēlams, ne augstākam par 70 %, izņemot telpu uzkopšanas laiku. Dzīvniekus 12 stundas diennaktī tur mākslīgā apgaismojumā, bet 12 stundas – tumsā. Var izmantot parasto laboratorijas barību ar neierobežotu piekļuvi dzirdināmajam ūdenim. Uztura izvēli var ietekmēt vajadzība tam piemaisīt testējamo ķimikāliju, ja ķimikāliju ievada šādā ceļā.
|
|
22.
|
Dzīvniekus ieteicams turēt mazās viendzimuma grupās; ja tas zinātniski pamatoti, dzīvniekus var turēt atsevišķi. Ja dzīvniekus tur grupās, dzīvnieku skaits vienā būrī nedrīkstētu pārsniegt 5. Pārošanas procedūras jāveic attiecīgajam nolūkam piemērotos būros. Grūsnas mātītes jātur atsevišķi, un tām jānodrošina migas ierīkošanai vajadzīgie materiāli. Mātītes laktācijas periodā tiks turētas atsevišķi kopā ar to pēcnācēju.
|
|
23.
|
Barība regulāri jāanalizē, lai noteiktu kontaminantu klātbūtni. Barības paraugs jāglabā līdz ziņojuma pabeigšanai.
|
Dzīvnieku sagatavošana
|
24.
|
Veselīgus jaunus pieaugušus dzīvniekus nejaušināti iedala apstrādes grupās un ievieto būros. Būri jāizvieto tā, lai būru novietojuma varbūtējā ietekme būtu iespējami maza. Dzīvniekus unikāli identificē un pirms pētījuma sākšanas vismaz piecas dienas tur būros, lai tie aklimatizētos laboratorijas apstākļos.
|
Devu sagatavošana
|
25.
|
Testējamo ķimikāliju ieteicams ievadīt perorāli, ja vien par piemērotākiem netiek uzskatīti citi ievadīšanas ceļi. Ja izvēlēts perorālais ievadīšanas ceļš, testējamo ķimikāliju parasti ievada ar gavāžu; tomēr testējamo ķimikāliju var ievadīt arī ar uzturu vai dzirdināmo ūdeni.
|
|
26.
|
Ja vajadzīgs, testējamo ķimikāliju izšķīdina vai suspendē piemērotā nesējā. Kad vien iespējams, ieteicams lietošanai izvēlēties ūdens šķīdumu/suspensiju vai, ja tas nav iespējams, šķīdumu/suspensiju eļļā (piemēram, kukurūzas eļļā), vai, ja arī tas nav iespējams, šķīdumu citos nesējos. Ja nesējs nav ūdens, ir jāzina šā nesēja toksiskās īpašības. Jānosaka testējamās ķimikālijas stabilitāte un homogenitāte nesējā.
|
PROCEDŪRA
Dzīvnieku skaits un dzimums
|
27.
|
Ieteicams, lai katrā grupā sākumā būtu vismaz 10 tēviņi un 12–13 mātītes. Pirms ekspozīcijas izvērtē mātīšu estrālos ciklus, un dzīvniekus, kam nav novērojami tipiski 4–5 dienu cikli, pētījumā neiekļauj; tāpēc ieteicams izmantot lielāku skaitu mātīšu, lai katrā grupā būtu 10 mātītes. Izņemot izteiktas toksiskās ietekmes gadījumus, sagaidāms, ka katrā grupā būs vismaz 8 grūsnas mātītes, kas parasti ir minimālais pieļaujamais grūsno mātīšu skaits grupā. Mērķis ir nodrošināt pietiekamu daudzumu grūsnu mātīšu un pietiekamu daudzumu pēcnācēju, lai varētu ticami izvērtēt testējamās ķimikālijas spēju ietekmēt auglību, grūsnību, mātišķo uzvedību un zīdīšanu, kā arī F1 pēcnācēju augšanu un attīstību no aizmešanās līdz 13. dienai pēc piedzimšanas. Ja ir plānota dzīvnieku nonāvēšana pētījuma gaitā, skaits jāpalielina par dzīvnieku skaitu, ko paredzēts nonāvēt pirms pētījuma pabeigšanas. Jāapsver papildu satelītgrupas (pieci dzīvnieki no katra dzimuma) iekļaušana kontrolgrupā un lielākās devas grupā, lai vismaz 14 dienas pēc apstrādes novērotu sistēmiskas toksiskās ietekmes atgriezeniskumu, noturību vai vēlīnu parādīšanos. Satelītgrupas dzīvniekus nepāro un līdz ar to neizmanto reproduktīvās/ontoģenētiskās toksicitātes novērtēšanai.
|
Devas
|
28.
|
Parasti jāizmanto vismaz trīs testa grupas un kontrolgrupa. Ja piemēroti vispārējās toksicitātes dati nav pieejami, var izdarīt diapazona noteikšanas pētījumu (ar vienas līnijas un izcelsmes dzīvniekiem), kas palīdzētu noteikt izmantojamās devas. Ar kontrolgrupas dzīvniekiem jārīkojas tāpat kā ar testa grupu dzīvniekiem, izņemot attiecībā uz testējamās ķimikālijas ievadīšanu. Ja testējamās ķimikālijas ievadīšanai izmanto nesēju, kontrolgrupa saņem lielāko izmantoto nesēja daudzumu.
|
|
29.
|
Devu līmeņi jāizvēlas, ņemot vērā visus pieejamos toksiskuma un (toksiko)kinētiskos datus. Jāņem vērā arī tas, ka grūsnu un negrūsnu dzīvnieku jutība var atšķirties. Augstākais devas līmenis jāizvēlas tāds, lai tas izraisītu toksisku ietekmi, bet ne nobeigšanos vai acīmredzamas ciešanas. Pēc tam jāizvēlas tāda lejupēja devas līmeņu virkne, lai ar to varētu parādīt jebkādas ar devu saistītas atbildreakcijas, kā arī to, ka zemākajā devas līmenī nelabvēlīga ietekme nav novērojama. Optimums bieži vien ir divkārši līdz četrkārši intervāli, un bieži vien ir ieteicamāk pievienot ceturto testa grupu, nevis izmantot ļoti plašu devu intervālu (piemēram, vairāk nekā 10 reizes).
|
|
30.
|
Ja ir novērota vispārēja toksicitāte (piemēram, ķermeņa masas samazināšanās, ietekme uz aknām, sirdi, plaušām vai nierēm utt.) vai citas pārmaiņas, kas var nebūt toksiskas atbildreakcijas (piemēram, samazināts barības patēriņš, aknas palielināšanās), ir jābūt piesardzīgiem, interpretējot novēroto iedarbību uz jutīgiem endokrīnajiem beigupunktiem.
|
Robežtests
|
31.
|
Ja perorālajā pētījumā, kurā izmanto vienu devas līmeni, kas ir vismaz 1 000 mg/kg ķermeņa masas dienā, vai – ja testējamo ķimikāliju ievada ar uzturu – līdzvērtīgs procentuālais daudzums uzturā vai dzirdināmajā ūdenī (aprēķināts pēc ķermeņa masas), un kuru veic, ievērojot šajā pētījumā aprakstītās procedūras, novērojama toksiska ietekme nerodas un ja, pamatojoties uz datiem par strukturāli radniecīgām vielām, toksicitāte nav sagaidāma, var uzskatīt, ka pilns pētījums ar vairākiem devu līmeņiem nav vajadzīgs. Robežtestu neizmanto gadījumos, kad cilvēka ekspozīcija norāda uz to, ka ir jāizmanto augstāks devas līmenis. Ja izmanto citus ievadīšanas veidus, tādus kā ieelpošana vai aplicēšana uz ādas, maksimālā sasniedzamā ekspozīcija bieži vien ir atkarīga no testējamo ķimikāliju fizikālķīmiskajām īpašībām.
|
Devu došana
|
32.
|
Dzīvnieki testējamās ķimikālijas devu saņem katru dienu 7 dienas nedēļā. Ja izmanto gavāžu, dzīvniekiem testējamā ķimikālija jāsaņem vienreizējā devā, ko ievada ar kuņģa zondi vai piemērotu intubācijas cauruli. Maksimālais šķidruma tilpums, ko vienā paņēmienā var ievadīt, ir atkarīgs no testa dzīvnieka lieluma. Tilpums nedrīkst pārsniegt 1 ml uz 100 g ķermeņa masas, izņemot tādus ūdens šķīdumus, kur var lietot 2 ml uz 100 g ķermeņa masas. Izņemot kairinošas vai kodīgas testējamās ķimikālijas, kam lielākās koncentrācijās parasti ir saasināta ietekme, testa tilpuma mainība būtu jāsamazina līdz minimumam, koncentrāciju koriģējot tā, lai visos devu līmeņos saglabātos konstants tilpums.
|
|
33.
|
Ja testējamās ķimikālijas ievada ar uzturu vai dzirdināmo ūdeni, ir svarīgi nodrošināt, ka testējamās ķimikālijas daudzums neizjauc normālu uztura vai ūdens līdzsvaru. Ja testējamo ķimikāliju ievada ar uzturu, var izmantot nemainīgu koncentrāciju uzturā (ppm) vai nemainīgu devas līmeni attiecībā pret dzīvnieku ķermeņa masu; izmantotā alternatīva ir jānorāda. Ja testējamo ķimikāliju ievada ar gavāžu, deva jādod katru dienu vienādā laikā un vismaz reizi nedēļā jākoriģē tā, lai uzturētu nemainīgu devas līmeni attiecībā pret dzīvnieka ķermeņa masu. Ja kombinēto pētījumu izmanto par provizorisku pētījumu ilgtermiņa vai pilnīgam reproduktīvās toksicitātes pētījumam, abos pētījumos jāizmanto līdzīgs uzturs.
|
Eksperimenta grafiks
|
34.
|
Devas abu dzimumu dzīvniekiem jāsāk dot 2 nedēļas pirms pārošanas, pēc tam, kad tie vismaz piecas dienas tikuši aklimatizēti un mātītēm ir novēroti normāli estrālie cikli (divu nedēļu ilgā priekšapstrādes periodā). Pētījums jāplāno tā, lai estrālā cikla izvērtēšana sāktos drīz pēc tam, kad dzīvnieki ir sasnieguši pilnīgu dzimumgatavību. Dažādām žurku līnijām pilnīgas dzimumgatavības sasniegšanas vecums dažādās laboratorijās var nedaudz atšķirties, piemēram, Sprague Dawley līnijas žurkas to sasniedz 10 nedēļu vecumā, Wistar līnijas žurkas — 12 nedēļu vecumā. Mātes ar pēcnācēju jānonāvē 13. pēcdzemdību dienā vai drīz pēc tam. Lai nodrošinātu, ka mātes nakti pirms asins ņemšanas (ja izvēlēts šis variants) nav barojušās, mātes un to pēcnācējs nav obligāti jānogalina tajā pašā dienā. Dzimšanas diena (proti, dienā, kurā dzemdības ir beigušās) ir 0. pēcdzemdību diena. Mātītes, kam nav novērojamas kopulācijas pazīmes, nonāvē 24–26 dienas pēc pēdējās pārošanas perioda dienas. Pārošanas perioda laikā abu dzimumu dzīvniekiem devas turpina dot. Pēc pārošanas perioda tēviņiem devas jāturpina dot vismaz līdz minimālā kopējā devu došanas perioda (28 dienas) beigām. Pēc tam tos vai nu nonāvē, vai arī patur dzīvus un tiem turpina dot devas, lai tos, iespējams, izmantotu otrai pārošanai, ja tāda vajadzīga.
|
|
35.
|
Vecākpaaudzes mātītēm devas turpina dot katru dienu visu grūsnības laiku un vismaz līdz 13. pēcdzemdību dienai vai dienai pirms nonāvēšanas (minētās dienas ieskaitot). Pētījumos, kuros testējamo ķīmisko vielu ievada inhalācijas ceļā vai caur ādu, devas jāturpina dot vismaz līdz gestācijas 19. dienai (ieskaitot) un jāatsāk dot pēc iespējas drīz, bet ne vēlāk kā 4. dienā pēc dzemdībām.
|
|
36.
|
Satelītgrupas dzīvniekus (ja tie iekļauti), kas paredzēti papildu novērojumiem, nepāro. Pēc pirmās plānotās mātīšu nonāvēšanas tie jātur vēl vismaz 14 dienas bez apstrādes, lai detektētu toksiskās ietekmes vēlīnu parādīšanos vai noturību, vai atgūšanos no tās.
|
|
37.
|
Eksperimenta grafika shēma ir dota 2. papildinājumā.
|
Estrālie cikli
|
38.
|
Lai pētījumam atlasītu mātītes ar regulāru cikliskumu, pirms apstrādes sākšanas jānovēro estrālie cikli (sk. 27. punktu). No apstrādes perioda sākuma līdz brīdim, kad ir novērotas pārošanās pazīmes, katru dienu jāveic arī vaginālo uztriepju monitorings. Ja pastāv bažas par akūta stresa ietekmi, kas, uzsākot devu došanu, var mainīt estrālos ciklus, laboratorijas var testa dzīvniekus divas nedēļas pakļaut ekspozīcijai un pēc tam reizi dienā ievākt vaginālās uztriepes, lai pirmspārošanas periodā vismaz divas nedēļas veiktu estrālo ciklu monitoringu, ko turpina veikt arī pārošanas periodā, līdz ir iegūti pārošanās pierādījumi. Iegūstot maksts / dzemdes kakla šūnas, jārūpējas par to, lai nepieļautu gļotādas bojājumus, kas var inducēt viltus grūsnību (8) (9).
|
Pārošanas procedūra
|
39.
|
Parasti šajā pētījumā jāizmanto pārošanas shēma 1:1 (viens tēviņš uz vienu mātīti). Izņēmumi pieļaujami tēviņu nāves gadījumā. Mātīte jātur kopā ar to pašu tēviņu, līdz tiek novērotas kopulācijas pazīmes vai ir pagājušas divas nedēļas. Katru rītu mātītēm pārbauda spermas vai embola klātbūtni makstī. Grūsnību sāk skaitīt no dienas, kurā ir apstiprinājies kopulācijas fakts (konstatēts maksts embols vai sperma). Ja pārošana ir nesekmīga, var apsvērt mātīšu atkārtotu pārošanu ar tās pašas grupas tēviņiem.
|
Metiena lielums
|
40.
|
Ceturtajā dienā pēc piedzimšanas katra metiena lielumu var koriģēt, nejaušināti izņemot liekos mazuļus tā, lai katrā metienā pēc iespējas būtu četri vai pieci katra dzimuma mazuļi atkarībā no izmantotās žurku līnijas parastā metiena lieluma. No diviem liekajiem mazuļiem jāņem asins paraugi, tie jāapkopo un jāizmanto seruma T4 līmeņu noteikšanai. Mazuļu selektīva izņemšana, piemēram, pēc ķermeņa masas vai anoģenitālā attāluma (AGD), nav piemērota. Ja vīrišķā vai sievišķā dzimuma mazuļu skaita dēļ nav iespējams nodrošināt, ka katrā metienā ir četri vai pieci katra dzimuma mazuļi, ir pieļaujama metiena lieluma daļēja koriģēšana (piemēram, seši tēviņi un četras mātītes). Ja metiena lielums ir mazāks par korekcijas robežvērtību (8 vai 10 mazuļi vienā metienā), mazuļus no metiena neizņem. Ja korekcijas robežvērtību pārsniedz tikai viens mazulis, izņem tikai vienu mazuli un no tā ņem asins paraugus, ko izmanto iespējamiem seruma T4 novērtējumiem.
|
|
41.
|
Ja metiena lielums netiek koriģēts, 4. dienā pēc piedzimšanas divus mazuļus no katra metiena nonāvē un no tiem ņem asins paraugus, ko izmanto tiroīdhormona seruma koncentrācijas mērīšanai. Ja iespējams, diviem mazuļiem katrā metienā jābūt sievišķā dzimuma mazuļiem, lai vīrišķā dzimuma mazuļus rezervētu zīdekļu retencijas izvērtējumiem, izņemot gadījumus, kad šo mazuļu izņemšanas rezultātā galējam novērtējumam nepaliek neviena mātīte. Mazuļus no metiena neizņem, ja tā lielums būs mazāks nekā 8 vai 10 mazuļi vienā metienā (atkarībā no izmantotās žurku līnijas parastā metiena lieluma). Ja, pamatojoties uz parasto metiena lielumu, lieks ir tikai viens mazulis, izņem tikai vienu mazuli un no tā ņem asins paraugus, ko izmanto iespējamiem seruma T4 novērtējumiem.
|
Novērojumi
|
42.
|
Vismaz reizi dienā, vēlams — vienā un tajā pašā laikā vai laikos un ņemot vērā gaidāmo ietekmju maksimumperiodu pēc devu došanas, jāizdara vispārīgi klīniski novērojumi. Jāreģistrē dzīvnieku veselības stāvoklis. Vismaz divreiz dienā visus dzīvniekus novēro attiecībā uz morbiditāti un mirstību.
|
|
43.
|
Vienu reizi pirms pirmās ekspozīcijas (lai varētu izdarīt salīdzinājumus vienam un tam pašam dzīvniekam) un pēc tam vismaz reizi nedēļā jāveic detalizēti klīniskie novērojumi visiem vecākpaaudzes dzīvniekiem. Šie novērojumi jāizdara standartnožogojumā ārpus dzīvnieka turēšanas būra, ieteicams, katru dienu vienā un tajā pašā laikā. Tie rūpīgi jāreģistrē, ieteicams, izmantojot punktu sistēmas, ko skaidri noteikusi testēšanas laboratorija. Jācenšas nodrošināt, lai testēšanas apstākļu mainība būtu minimāla un lai novērojumus izdarītu novērotāji, kas nav informēti par apstrādi. Jāreģistrē šādas pazīmes (neizsmeļošs saraksts): izmaiņas ādā, apmatojumā, acīs, gļotādās, sekrētu un ekskrētu parādīšanās un veģetatīvās nervu sistēmas reakcijas (piemēram, asarošana, piloerekcija, acs zīlītes diametra pārmaiņas, neraksturīga elpošana). Jāreģistrē arī pārmaiņas gaitā, stājā un atbildes reakcijās uz manipulācijām, kā arī kloniskas vai toniskas kustības, stereotipiska uzvedība (piemēram, pārmērīga apmatojuma laizīšana, atkārtota riņķošana), grūtas vai ilgstošas dzemdības vai savāda uzvedība (piemēram, pašsakropļošanās, staigāšana atmuguriski) (10).
|
|
44.
|
Kādā brīdī pētījuma laikā jānovērtē piecu tēviņu un piecu mātīšu, kas nejauši izvēlētas no katras grupas, sensorā reaktivitāte uz dažāda veida kairinājumiem (piemēram, dzirdes, redzes un proprioreceptīvajiem kairinājumiem) (8) (9) (11), satvēriena spēks (12) un kustību aktivitāte (13). Papildinformācija par izmantojamajām procedūrām ir atrodama attiecīgajos atsauces materiālos. Tomēr norādīto procedūru vietā var izmantot arī citas procedūras. Tēviņiem šie funkcionālie novērojumi jāveic, tuvojoties devu došanas perioda beigām, neilgi pirms plānotās nonāvēšanas, bet pirms asins paraugu ņemšanas hematoloģijai vai klīniskās ķīmijas izmeklējumiem (sk. 53.–56. punktu, arī 1. zemsvītras piezīmi). Mātītēm šo funkcionālo testu laikā jābūt fizioloģiski līdzīgā stāvoklī, un tās vēlams testēt vienu reizi pēdējās laktācijas nedēļas laikā (piemēram, 6.–13. laktācijas dienā), neilgi pirms plānotās nonāvēšanas. Māšu un mazuļu atšķiršanas laikiem jābūt pēc iespējas īsākiem.
|
|
45.
|
Funkcionālos novērojumus, kas veicami vienu reizi pētījuma beigās, var neveikt, ja pētījums ir provizorisks pētījums turpmākam subhroniskam (90 dienas) vai ilgtermiņa pētījumam. Tādā gadījumā funkcionālie novērojumi ir jāiekļauj minētajā turpmākajā pētījumā. Tomēr šajā atkārtotas devas pētījumā iegūtie funkcionālo novērojumu dati var atvieglot devu līmeņu izvēli turpmākajam subhroniskajam vai ilgtermiņa pētījumam.
|
|
46.
|
Izņēmuma kārtā funkcionālos novērojumus var neveikt arī tām grupām, kurām toksicitātes pazīmes izpaužas tādā mērā, kas varētu ievērojami traucēt funkcionālā testa veikšanu.
|
|
47.
|
Jāreģistrē gestācijas ilgums, ko aprēķina no 0. grūsnības dienas. Pēc atnešanās katru metienu iespējami drīz pārbauda, lai noteiktu mazuļu, nedzīvi dzimušo, dzīvi dzimušo un nīkuļu (mazuļi, kas ir ievērojami mazāki nekā atbilstīgie kontrolmazuļi) skaitu un dzimumu un acīmredzamu anomāliju klātbūtni.
|
|
48.
|
24 stundu laikā pēc atnešanās (0. vai 1. diena pēc piedzimšanas) un vismaz 4. un 13. dienā pēc piedzimšanas jāsaskaita dzīvie mazuļi un jānosaka to dzimums, un jānosver metieni. Papildus vecākpaaudzes dzīvnieku novērojumiem (sk. 43. un 44. punktu) jāreģistrē pēcnācēju uzvedības anomālijas.
|
|
49.
|
Katra mazuļa AGD jāmēra vienā un tajā pašā dienā pēc piedzimšanas no 0. līdz 4. dienai pēc piedzimšanas. Mazulis ir jānosver tajā pašā dienā, kad tiek mērīts AGD, un AGD ir jāpielāgo mazuļa izmēram, priekšroku dodot kvadrātsaknei no ķermeņa svara (14). Kā ieteikts ESAO GD 151 (15), 12. vai 13. dienā pēc piedzimšanas ir jāsaskaita vīrišķā dzimuma mazuļu zīdekļi/areolas.
|
Ķermeņa masa un barības/ūdens patēriņš
|
50.
|
Tēviņi un mātītes jānosver pirmajā devas došanas dienā, tad vismaz reizi nedēļā un pētījuma beigās. Grūsnības laikā mātītes jānosver 0., 7., 14. un 20. dienā un 24 stundu laikā pēc atnešanās (0. vai 1. pēcdzemdību diena), kā arī vismaz 4. un 13. pēcdzemdību dienā. Novērojumu rezultāti jāreģistrē atsevišķi par katru pieaugušo dzīvnieku.
|
|
51.
|
Pirmspārošanas, grūsnības un laktācijas laikā barības patēriņš jāmēra vismaz reizi nedēļā. Barības patēriņa mērīšana pārošanas laikā ir fakultatīva. Ja testējamo ķimikāliju ievada ar ūdeni, minētajos laikposmos ir jāmēra arī ūdens patēriņš.
|
Hematoloģija
|
52.
|
Pētījuma laikā pieciem tēviņiem un piecām mātītēm, kas nejauši izvēlētas no katras grupas, vienu reizi jāveic šādi hematoloģiski izmeklējumi: hematokrīts, hemoglobīna koncentrācijas, eritrocītu skaits, retikulocīti, leikocītu skaits un leikocitārā formula, trombocītu skaits un asinsreces laika/potenciāla noteikšana. Ja testējamajai ķimikālijai vai tās iespējamajiem metabolītiem ir oksidējošas īpašības vai pastāv aizdomas, ka tiem tādas varētu būt, jānosaka arī methemoglobīna koncentrācija un Heinca ķermenīši.
|
|
53.
|
Asins paraugi jāņem no norādītās vietas. Paraugu ņemšanas laikā mātītēm ir jābūt fizioloģiski līdzīgā stāvoklī. Lai izvairītos no praktiskām grūtībām, kas saistītas ar mainību grūsnības sākumā, asins savākšanu mātītēm var veikt pirms pārošanas perioda beigām kā alternatīvu paraugu ņemšanai tieši pirms dzīvnieku eitanāzijas procedūras vai tās ietvaros. Asins paraugus no tēviņiem vēlams ņemt tieši pirms dzīvnieku eitanāzijas procedūras vai tās ietvaros. Alternatīvi asins savākšanu tēviņiem var veikt arī pārošanas perioda beigās, ja šis brīdis ticis izvēlēts mātītēm.
|
|
54.
|
Asins paraugi jāglabā piemērotos apstākļos.
|
Klīniskā bioķīmija
|
55.
|
Asins paraugiem, kas iegūti no izvēlētiem pieciem tēviņiem un piecām mātītēm katrā grupā, veic klīniskās bioķīmijas analīzes, lai izpētītu būtisku toksisko ietekmi uz audiem un, konkrēti, uz nierēm un aknām. Ir ieteicams dzīvniekus nakti pirms asins paraugu ņemšanas nebarot (22). Plazmā vai serumā jānosaka nātrijs, kālijs, glikoze, kopējais holesterīns, urīnviela, kreatinīns, kopējais olbaltums un albumīns, vismaz divi fermenti, kas norāda uz hepatocelulāro ietekmi, (piemēram, alanīnaminotransferāze, aspartātaminotransferāze un sorbitoldehidrogenāze) un žultsskābes. Noteiktos apstākļos noderīgu informāciju var sniegt papildu fermentu (aknu fermentu vai citu fermentu) un bilirubīna noteikšana.
|
|
56.
|
Asins paraugus no noteiktas vietas ņem saskaņā ar šādu grafiku:
|
—
|
4. dienā pēc piedzimšanas — no vismaz diviem mazuļiem katrā metienā, ja mazuļu skaits to ļauj (sk. 40.-41. punktu),
|
|
—
|
pētījuma beigās 13. dienā — no visām mātēm un vismaz diviem mazuļiem katrā metienā, un
|
|
—
|
pētījuma beigās — no visiem pieaugušiem tēviņiem.
|
Visus asins paraugus glabā piemērotos apstākļos. Asins paraugos, kas ņemti no mazuļiem 13. dienā un pieaugušiem tēviņiem, nosaka tiroīdhormonu (T4) seruma līmeņus. Ja vajadzīgs, T4 līmeņus nosaka arī asins paraugos, kas ņemti no mātēm un 4. dienas mazuļiem. Attiecīgā gadījumā var fakultatīvi mērīt citus hormonus. Tiroīdhormona analīžu veikšanai mazuļu asinis var apkopot pa metieniem. Tiroīdhormonus (T4 un TSH) vēlams mērīt kopējā formā (kopējais T4 un kopējais TSH).
|
|
57.
|
Pēdējā pētījuma nedēļā, izmantojot urīna savākšanu noteiktā laika periodā, pieciem nejauši izvēlētiem tēviņiem katrā grupā pēc izvēles var veikt šādus urīnanalīzes mērījumus: izskats, tilpums, osmolalitāte vai īpatnējais svars, pH, proteīns, glikoze un asinis/ asins šūnas.
|
|
58.
|
Turklāt jāapsver iespēja veikt vispārēja audu bojājuma seruma marķieru izpēti. Ja testējamās ķimikālijas zināmās īpašības var ietekmēt saistītos metaboliskos profilus vai ir aizdomas par šādu ietekmi, jānosaka arī kalcijs, fosfāts, triglicerīdu līmenis tukšā dūšā un glikoze tukšā dūšā, īpaši hormoni, methemoglobīns un holīnesterāze. Tie jānosaka katrā gadījumā atsevišķi.
|
|
59.
|
Noteikto hormonu mainību un absolūto koncentrāciju var ietekmēt šādi faktori:
|
—
|
nonāvēšanas laiks hormonu koncentrāciju diennakts mainības dēļ,
|
|
—
|
nonāvēšanas metode, kurā dzīvniekiem netiek radīts nevajadzīgs stress, kas varētu ietekmēt hormonu koncentrācijas,
|
|
—
|
hormonu noteikšanas testa komplekti, kas var atšķirties pēc standartlīknēm.
|
|
|
60.
|
Plazmas paraugi, kas paredzēti tieši hormonu noteikšanai, ir jāiegūst salīdzināmā dienas laikā. Skaitliskās vērtības, ko iegūst, analizējot hormonu koncentrācijas, atšķiras atkarībā no tirdzniecībā pieejamajiem testēšanas komplektiem.
|
|
61.
|
Ja vēsturiskie bāzlīnijas dati ir nepietiekami, jāapsver iespēja noteikt hematoloģiskos un klīniskās bioķīmijas mainīgos lielumus pirms devu došanas sākšanas vai, vēlams, dzīvniekiem, kas nav ietverti eksperimentālajās grupās. Dati par mātītēm jāiegūst no dzīvniekiem laktācijas periodā.
|
PATOLOĢIJA
Makroskopiskā autopsija
|
62.
|
Visiem pētījumā izmantotajiem pieaugušajiem dzīvniekiem jāveic pilna, detalizēta makroskopiska autopsija, kas ietver ķermeņa ārējās virsmas, visu atveru un galvaskausa, krūšu un vēdera dobumu un to satura rūpīgu izpēti. Īpaša uzmanība jāpievērš reproduktīvās sistēmas orgāniem. Jāreģistrē implantācijas vietu skaits. Autopsijas dienā jāizmeklē vaginālās uztriepes, lai noteiktu estrālā cikla fāzi un ļautu to saistīt ar mātīšu reproduktīvo orgānu histopatoloģiju.
|
|
63.
|
Visu pieaugušo tēviņu sēklinieki un sēklinieku piedēkļi, kā arī prostata un sēklas pūslīši ar prostatas priekšējām daivām (kopā) pēc vajadzības jāatbrīvo no pieguļošiem audiem, un to slapjais svars jānosaka iespējami drīz pēc disekcijas, lai izvairītos no izžūšanas. Turklāt fakultatīvos orgānu masas mērījumos var ietvert arī tūpļa cēlājmuskuļa un briedumķermeņa muskuļa kompleksu, Kupera dziedzerus un dzimumlocekļa galviņu tēviņiem un abas olnīcas (slapjais svars) un dzemdi (ar dzemdes kaklu) mātītēm; ietveršanas gadījumā minētie masas mērījumi jāveic iespējami drīz pēc disekcijas. Jāsaglabā visu pieaugušo dzīvnieku olnīcas, sēklinieki, sēklinieku piedēkļi, iekšējie dzimumorgāni un visi orgāni, kam ir makroskopiski bojājumi.
|
|
64.
|
Vairogdziedzeri, kas ņemti no visiem pieaugušajiem tēviņiem un mātītēm un viena 13 dienas veca vīrišķā dzimuma un sievišķā dzimuma mazuļa katrā metienā, vispiemērotākajā fiksējošajā vidē jāsaglabā paredzētajai turpmākajai histopatoloģiskajai izmeklēšanai. Vairogdziedzera masu var noteikt pēc fiksācijas. Tas jāatbrīvo no piegulošiem audiem ļoti uzmanīgi un tikai pēc fiksācijas, lai izvairītos no audu bojājumiem. Audu bojājumi var apgrūtināt histopatoloģijas analīzi. Asins paraugi jāņem no precīzi norādītas vietas tieši pirms dzīvnieku eitanāzijas procedūras vai tās laikā un jāglabā piemērotos apstākļos (sk. 56. punktu).
|
|
65.
|
Turklāt vismaz piecu pieaugušu tēviņu un mātīšu, kas nejauši izvēlētas no katras grupas (izņemot dzīvniekus, kas ir nāvei tuvā stāvoklī un/vai eitanazēti pirms pētījuma beigām), aknas, nieres, virsnieres, tīms, liesa, smadzenes un sirds pēc vajadzības jāatbrīvo no piegulošiem audiem un to slapjais svars jānosaka iespējami drīz pēc disekcijas, lai izvairītos no izžūšanas. Šādi audi jāglabā audu tipam un paredzētajai turpmākajai histopatoloģiskajai izmeklēšanai vispiemērotākajā fiksējošajā vidē: visi audi ar makroskopiskiem bojājumiem, smadzenes (reprezentatīvas daļas, tostarp galasmadzenes, smadzenītes un tilts), muguras smadzenes, acis, kuņģis, tievā un resnā zarna (arī Peijera plātnīte), aknas, nieres, virsnieres, liesa, sirds, tīms, traheja un plaušas (saglabātas, piepūšot ar fiksatoru un pēc tam iegremdējot), gonādas (sēklinieki un olnīcas), iekšējie dzimumorgāni (dzemde un dzemdes kakls, sēklinieku piedēkļi, prostata un sēklas pūslīši kopā ar prostatas priekšējo daivu), vagīna, urīnpūslis, limfmezgli (saskaņā ar laboratorijas pieredzi papildus proksimālam drenējošam limfmezglam ir jāņem vēl viens limfmezgls (16)), perifērais nervs (sēžas nervs vai apakšstilba nervs), vēlams, tiešā muskuļa tuvumā, skeleta muskulis un kauls ar kaula smadzenēm (izgriezums vai, alternatīvi, svaigs kaula smadzeņu aspirāts). Sēkliniekus ieteicams fiksēt, iegremdējot tos Buēna fiksatorā vai Davidsona fiksatorā (16) (17) (18); šos audus nav ieteicams fiksēt formalīnā. Lai nodrošinātu fiksatora ātru penetrēšanos, bālgano apvalku (tunica albuginea) var abos orgāna polos uzmanīgi un sekli pārdurt ar adatu. Klīnisko un citu konstatējumu rezultātā var rasties vajadzība papildus izmeklēt citus audus. Jāsaglabā arī orgāni, ko, pamatojoties uz zināmajām testējamās ķimikālijas īpašībām, uzskata par iespējamiem mērķorgāniem.
|
|
66.
|
Vērtīgas norādes par endokrīnu iedarbību var gūt no šādiem audiem: gonādas (olnīcas un sēklinieki), iekšējie dzimumorgāni (dzemde un dzemdes kakls, sēklinieku piedēkļi, sēklas pūslīši ar prostatas priekšējām daivām, dorsolaterālā un ventrālā prostata), vagīna, hipofīze, tēviņu krūts dziedzeris un virsnieru dziedzeris. Tēviņu krūts dziedzeru pārmaiņas nav pietiekami dokumentētas, taču šis parametrs var būt ļoti jutīgs pret vielām ar estrogēnisku iedarbību. To orgānu/audu novērošana, kas nav uzskaitīti 65. punktā, nav obligāta.
|
|
67.
|
Nedzīvi mazuļi un mazuļi, kas nonāvēti 13. dienā pēc piedzimšanas vai drīz pēc tam, vismaz rūpīgi ārēji jāpārbauda, lai konstatētu iespējamas acīmredzamas anomālijas. Īpaša uzmanība jāpievērš ārējām reproduktīvajām ģenitālijām, kuras jāpārbauda, meklējot attīstības izmaiņu pazīmes.
|
Histopatoloģija
|
68.
|
Saglabātajiem orgāniem un audiem, kas iegūti no atlasītajiem kontrolgrupas un lielākās devas grupas dzīvniekiem, veic pilnu histopatoloģisko izmeklēšanu (īpašu uzsvaru liekot uz spermatoģenēzes posmiem tēviņu gonādās un sēklinieku intersticiālo šūnu struktūras histopatoloģiju). Vajadzības gadījumā var izmeklēt mazuļu un pārējo pieaugušo dzīvnieku vairogdziedzeri. Ja lielākās devas grupā ir novērotas ar apstrādi saistītas pārmaiņas, minētie izmeklējumi jāattiecina arī uz citu devu grupu dzīvniekiem. Norādījumos par histopatoloģiju (10) ir sīki izklāstīta papildu informācija par endokrīno audu disekciju, fiksāciju, secēšanu un histopatoloģiju.
|
|
69.
|
Jāizmeklē visi makroskopiskie bojājumi. Lai palīdzētu noskaidrot NOAEL, jāizmeklē mērķorgāni citās devu grupās, jo īpaši grupās, kas uzrāda NOAEL.
|
|
70.
|
Ja izmanto satelītgrupu, histopatoloģiskā izmeklēšana jāizdara audiem un orgāniem, uz kuriem apstrādātajās grupās ir konstatēta ietekme.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Dati
|
71.
|
Jāsniedz dati par atsevišķiem dzīvniekiem. Turklāt visi dati jāapkopo tabulā, kurā redzams testa sākumā katrā testā grupā esošo dzīvnieku skaits, to dzīvnieku skaits, kas atrasti miruši testa laikā vai eitanazēti humānu apsvērumu dēļ, nāves iestāšanās vai eitanāzijas laiks, auglīgo dzīvnieku skaits, grūsno mātīšu skaits, to dzīvnieku skaits, kam novērotas toksicitātes pazīmes, novēroto toksicitātes pazīmju apraksts, arī toksiskās darbības sākuma laiks, ilgums un stiprums, histopatoloģisko pārmaiņu veidi un visi relevantie dati par metienu. Tabulas veidā sagatavotā kopsavilkuma ziņojuma paraugs, kas ir ļoti noderīgs ietekmes uz reprodukciju/attīstību izvērtēšanai, ir sniegts 3. papildinājumā.
|
|
72.
|
Ja iespējams, skaitliskie rezultāti jāizvērtē ar piemērotu un vispārpieņemtu statistikas metodi. Salīdzinot ietekmi visā devu diapazonā, jāizvairās no vairāku t-testu izmantošanas. Statistikas metodes jāizvēlas pētījuma plānošanas laikā. AGD un zīdekļu retencijas statistiskajai analīzei jābalstās uz atsevišķu mazuļu datiem, ņemot vērā ietekmi uz metienu. Vajadzības gadījumā analīzes vienība ir metiens. Mazuļu ķermeņa masas statistiskajai analīzei jābalstās uz atsevišķu mazuļu datiem, ņemot vērā metiena lielumu. Tā kā pētījuma tvērums ir ierobežots, statistiskajai analīzei, ko veic, lai izvērtētu rezultātu nozīmīgumu, ir ierobežota vērtība attiecībā uz daudziem beigupunktiem, jo īpaši reproduktīvajiem beigupunktiem. Dažas no visplašāk izmantotajām metodēm, jo īpaši parametriskie testi centrālās tendences mērīšanai, nav piemērotas. Ja tiek izmantota statistiskā analīze, izvēlētajai metodei jābūt atbilstošai pārbaudāmā mainīgā lieluma sadalījumam, un tā jāizvēlas pirms pētījuma sākšanas.
|
Rezultātu izvērtēšana
|
73.
|
Šā toksicitātes pētījuma konstatējumi jāizvērtē, ņemot vērā novēroto ietekmi, autopsiju un mikroskopiskos konstatējumus. Vērtējums ietver testējamās ķimikālijas devas saistību ar anomāliju — to vidū makroskopisko bojājumu, identificēto mērķorgānu, neauglības, klīnisko anomāliju, ietekmes uz reproduktīvajiem un metiena rādītājiem, ķermeņa masas pārmaiņu, ietekmes uz mirstību un citas toksiskās iedarbības – esību vai neesību, incidenci un smagumu.
|
|
74.
|
Tēviņu apstrādes periods ir īss, tāpēc, novērtējot ietekmi uz tēviņu reproduktīvo funkciju, papildus auglības datiem jāņem vērā sēklinieku un sēklinieku piedēkļu histopatoloģija. Pētījuma interpretēšanā par palīglīdzekli var būt lietderīgi izmantot arī vēsturiskus kontroles datus par reprodukciju/ontoģenēzi (piemēram, datus par metiena lielumu, AGD, zīdekļu retenciju, seruma T4 līmeņiem), ja tādi pieejami.
|
|
75.
|
Kvalitātes kontroles vajadzībām ir ierosināts apkopot vēsturiskus kontroldatus un skaitliskajiem datiem, it sevišķi parametriem, kas saistīti ar endokrīnās sistēmas traucējumu izraisītāju noteikšanu, aprēķināt variācijas koeficientus. Šos datus var izmantot salīdzināšanas nolūkos, kad tiek novērtēti faktiskie pētījumi.
|
Testēšanas pārskats
|
76.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Testējamā ķimikālija:
|
—
|
avots, sērijas numurs, izmantošanas termiņš, ja pieejams,
|
|
—
|
testējamās ķimikālijas stabilitāte, ja tā zināma;
|
|
|
|
vienkomponenta viela:
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības,
|
|
—
|
ķīmiskā identifikācija, piemēram, IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, attiecīgā gadījumā, ja praktiski iespējams, piemaisījumu ķīmiskā identitāte u. tml.;
|
|
|
|
daudzkomponentu viela, UVCB un maisījumi:
|
—
|
iespējami izsmeļoši raksturoti pēc komponentu ķīmiskās identitātes (sk. iepriekš), kvantitatīvās sastopamības un relevantajām fizikālķīmiskajām īpašībām
|
. |
|
|
Nesējs (attiecīgā gadījumā):
|
—
|
nesēja izvēles pamatojums, ja nesējs nav ūdens.
|
|
|
|
Testa dzīvnieki:
|
—
|
izmantotās sugas/līnijas,
|
|
—
|
dzīvnieku skaits, vecums un dzimums,
|
|
—
|
avots, turēšanas apstākļi, uzturs u. tml.,
|
|
—
|
katra dzīvnieka masa testa sākumā,
|
|
—
|
sugas izvēles pamatojums, ja tā nav žurka.
|
|
|
|
Testēšanas nosacījumi:
|
—
|
devu līmeņa izraudzīšanās pamatojums,
|
|
—
|
detalizēta informācija par testējamās ķimikālijas maisījuma/barības preparāta sagatavošanu, sasniegto preparāta koncentrāciju, stabilitāti un homogenitāti,
|
|
—
|
detalizēta informācija par testējamās ķimikālijas devu došanu,
|
|
—
|
attiecīgā gadījumā testējamās ķimikālijas koncentrācijas (ppm) barībā / dzirdināmajā ūdenī pārrēķins faktiskajā devā (mg uz kg ķermeņa masas dienā),
|
|
—
|
detalizēta informācija par barības un ūdens kvalitāti,
|
|
—
|
detalizēts apraksts par nejaušināšanas procedūru, kas izmantota, lai atlasītu mazuļus izņemšanai no metiena, ja šāda izņemšana notikusi.
|
|
|
|
Rezultāti:
|
—
|
ķermeņa masa / ķermeņa masas izmaiņas,
|
|
—
|
attiecīgā gadījumā barības patēriņš un ūdens patēriņš,
|
|
—
|
dati par toksiskajām atbildes reakcijām atkarībā no dzimuma un devas, arī dati par auglību, gestāciju un citām toksicitātes pazīmēm,
|
|
—
|
toksiska vai cita iedarbība uz reprodukciju, pēcnācējiem, postnatālo augšanu utt.,
|
|
—
|
klīnisko novērojumu veids, smagums un ilgums (atgriezenisks vai neatgriezenisks),
|
|
—
|
sensoriskās aktivitātes, satvēriena spēka un kustību aktivitātes novērtējums,
|
|
—
|
hematoloģiskie testi ar attiecīgajām bāzes līnijas vērtībām,
|
|
—
|
klīniskās bioķīmijas testi ar attiecīgajām bāzes līnijas vērtībām,
|
|
—
|
pieaugušu mātīšu skaits ar normālu vai nenormālu estrālo ciklu un cikla ilgums,
|
|
—
|
dzīvi dzimušo skaits un pēcimplantācijas zudums,
|
|
—
|
to mazuļu skaits, kam ir makroskopiskas anomālijas, ārējo ģenitāliju makroskopisks izvērtējums, nīkuļu skaits,
|
|
—
|
nāves iestāšanās laiks testa laikā vai norāde, ka dzīvnieki ir izdzīvojuši līdz pētījuma beigām,
|
|
—
|
implantāciju skaits, metiena lielums un masa datu reģistrēšanas laikā,
|
|
—
|
mazuļu ķermeņa masas dati,
|
|
—
|
visu mazuļu AGD (un ķermeņa masa AGD mērīšanas dienā),
|
|
—
|
zīdekļu retencija vīrišķā dzimuma mazuļiem,
|
|
—
|
vairogdziedzera hormonu līmenis 13 dienu veciem mazuļiem un pieaugušiem tēviņiem (un mātēm un 4 dienas veciem mazuļiem, ja šādi mērījumi veikti),
|
|
—
|
ķermeņa masa nonāvēšanas laikā un dati par vecākpaaudzes dzīvnieku orgānu masu,
|
|
—
|
autopsijas konstatējumi,
|
|
—
|
histopatoloģiskas izmeklēšanas rezultātu detalizēts apraksts,
|
|
—
|
absorbcijas dati (ja pieejami),
|
|
—
|
rezultātu statistiskā apstrāde (attiecīgā gadījumā).
|
|
|
Rezultātu interpretēšana
|
77.
|
Pētījums sniegs izvērtējumu par reproduktīvo/ontoģenētisko toksicitāti, kas saistīta atkārtotu devu došanu. Konkrētāk, tā kā uzsvars ir likts uz vispārējās toksicitātes un reproduktīvās/ontoģenētiskās toksicitātes beigupunktiem, pētījuma rezultāti ļaus reproduktīvo/ontoģenētisko ietekmi, kas radusies bez vispārējas toksicitātes, atšķirt no ietekmes, kura izpaužas tikai pie līmeņiem, kas ir toksiski arī vecākpaaudzes dzīvniekiem (sk. 7.–11. punktu). Tas varētu norādīt uz nepieciešamību veikt turpmākas izmeklēšanas un sniegt norādījumus turpmāko pētījumu plānošanā. Ar reprodukciju un ontoģenēzi saistīto rezultātu interpretēšanā par palīglīdzekli jāizmanto ESAO norādījumu dokuments Nr. 43 (19). ESAO norādījumu dokumentā Nr. 106 par grauzēju endokrīno un reproduktīvo testu histoloģisko izvērtēšanu (16) sniegta informācija par (endokrīno) orgānu un vaginālo uztriepju sagatavošanu un izvērtēšanu; minētā informācija var būt noderīga saistībā ar šo testēšanas metodi.
|
LITERATŪRA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). “Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009).Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
1. papildinājums
DEFINĪCIJAS (SK. ARĪ (20) ESAO GD 150)
Androgenitāte ir ķimikālijas spēja zīdītāja organismā funkcionēt kā dabiskam androgēnu tipa hormonam (piemēram, testosteronam).
Antiandrogenitāte ir ķimikālijas spēja zīdītāja organismā nomākt dabiska androgēnu tipa hormona (piemēram, testosterona) darbību.
Antiestrogenitāte ir ķimikālijas spēja zīdītāja organismā nomākt dabiska estrogēnu tipa hormona (piemēram, estradiola 17ß) darbību.
Antitireoidāla iedarbība ir ķimikālijas spēja zīdītāja organismā nomākt dabiska tiroīdhormona (piemēram, T3) darbību.
Ķimikālija ir viela vai maisījums.
Ontoģenētiskā toksicitāte reproduktīvās toksicitātes izpausme, kas liecina par pēcnācēju prenatāliem, perinatāliem, postnatāliem, strukturāliem vai funkcionāliem traucējumiem.
Deva ir ievadītās testējamās ķimikālijas daudzums. Devu izsaka ar testējamās ķimikālijas masu uz testa dzīvnieka ķermeņa masas vienību dienā (piemēram, mg uz kg ķermeņa masas dienā) vai kā nemainīgu koncentrāciju barībā.
Dozējums ir vispārīgs termins, kas aptver devu, tās ievadīšanas biežumu un ilgumu.
Izteikta toksicitāte ir vispārīgs termins, kas apzīmē acīmredzamas toksicitātes pazīmes pēc testējamās ķimikālijas ievadīšanas. Šīm pazīmēm jābūt pietiekamām, lai varētu novērtēt bīstamību, un tādām, lai varētu sagaidīt, ka ievadītās devas palielināšana izraisīs smagas toksicitātes pazīmes un, iespējams, mirstību.
Fertilitātes traucējumi ir tēviņu vai mātīšu reproduktīvās funkcijas vai spējas traucējumi.
Maternālā toksicitāte nelabvēlīgā ietekme uz gravidām mātītēm, kas ir specifiska (tieša ietekme) vai nespecifiska (netieša ietekme) un saistīta ar gravidu stāvokli.
NOAEL ir saīsinājums, kas apzīmē nenovērojamās nelabvēlīgās ietekmes līmeni. Tas ir augstākais devas līmenis, kurā netiek konstatētas ar vielas saņemšanu saistītas negatīvas parādības.
Estrogenitāte ir ķimikālijas spēja zīdītāja organismā darboties kā dabiskam estrogēnu tipa hormonam (piemēram, estradiolam 17ß).
Reproduktīvā toksicitāte ir kaitīga ietekme uz pēcnācējiem un/vai tēviņu un mātīšu reproduktīvās funkcijas vai spējas traucējumi.
Testējamā ķimikālija ir jebkura viela vai maisījums, ko testē, izmantojot šo testēšanas metodi.
Tireoidāla iedarbība ir ķimikālijas spēja zīdītāja organismā darboties kā dabiskam tiroīdhormonam (piemēram, T3).
Validācija ir zinātnisks process, kura mērķis ir raksturot testēšanas metodes darbības prasības un ierobežojumus un apliecināt tās ticamību un piemērotību konkrētam nolūkam.
2. papildinājums
EKSPERIMENTA GRAFIKA SHĒMA, KURĀ NORĀDĪTS MAKSIMĀLAIS PĒTĪJUMA ILGUMS, KURA PAMATĀ IR 14 DIENU ILGS PĀROŠANAS PERIODS


3. papildinājums
TABULAS VEIDĀ SAGATAVOTS KOPSAVILKUMA ZIŅOJUMS PAR IETEKMI UZ REPRODUKCIJU/ONTOĢENĒZI
|
NOVĒROJUMI
|
VĒRTĪBAS
|
|
Dozējums (vienības)
|
0 (kontrole)
|
…
|
…
|
…
|
…
|
|
Dzīvnieku pāri (N)
|
|
|
|
|
|
|
Estrālais cikls (vismaz vidējais neregulāru ciklu ilgums un biežums)
|
|
|
|
|
|
|
Mātītes, kam novērojamas kopulācijas pazīmes (N)
|
|
|
|
|
|
|
Grūsnas mātītes (N)
|
|
|
|
|
|
|
Apaugļošanās dienas 1–5 (N)
|
|
|
|
|
|
|
Apaugļošanās dienas 6–...([1]
(23)
) (N)
|
|
|
|
|
|
|
Grūsnība≤21 diena (N)
|
|
|
|
|
|
|
Grūsnība = 22 dienas (N)
|
|
|
|
|
|
|
Grūsnība ≥ 23 dienas (N)
|
|
|
|
|
|
|
Mātes ar dzīviem mazuļiem (N)
|
|
|
|
|
|
|
Mātes ar dzīviem mazuļiem 4. dienā pēc dzemdībām (N)
|
|
|
|
|
|
|
Implanti vienai mātei (vidēji)
|
|
|
|
|
|
|
Dzīvie mazuļi vienai mātei piedzimšanas brīdī (vidēji)
|
|
|
|
|
|
|
Dzīvie mazuļi vienai mātei 4. dienā (vidēji)
|
|
|
|
|
|
|
Dzimumu attiecība (tēv./māt.) piedzimstot (vidēji)
|
|
|
|
|
|
|
Dzimumu attiecība (tēv./māt.) 4. dienā (vidēji)
|
|
|
|
|
|
|
Metiena masa piedzimstot (vidēji)
|
|
|
|
|
|
|
Metiena masa 4. dienā (vidēji)
|
|
|
|
|
|
|
Mazuļu masa piedzimstot (vidēji)
|
|
|
|
|
|
|
Mazuļu masa AGD mērīšanas laikā (tēviņu vidējā masa, mātīšu vidējā masa)
|
|
|
|
|
|
|
Mazuļu AGD tajā pašā postnatālajā dienā, laikā no dzimšanas līdz 4. dienai (tēviņu vidējā masa, mātīšu vidējā masa)
|
|
|
|
|
|
|
Metiena masa 4. dienā (vidēji)
|
|
|
|
|
|
|
Metiena masa 13. dienā (vidēji)
|
|
|
|
|
|
|
Zīdekļu retencija vīrišķā dzimuma mazuļiem 13. dienā (vidēji)
|
|
|
|
|
|
|
MAZUĻI AR ANOMĀLIJĀM
|
|
Mātes, kam nav neviena šāda mazuļa
|
|
|
|
|
|
|
Mātes ar 1 šādu mazuli
|
|
|
|
|
|
|
Mātes ar ≥ 2 šādiem mazuļiem
|
|
|
|
|
|
|
PĒCNĀCĒJU ZUDUMI
|
|
Prenatāli (implantāciju skaits mīnus dzīvi dzimušo skaits)
|
|
Mātītes, kas nav zaudējušas mazuli
|
|
|
|
|
|
|
Mātītes ar vienu zaudētu mazuli
|
|
|
|
|
|
|
Mātītes ar diviem zaudētiem mazuļiem
|
|
|
|
|
|
|
Mātītes ar ≥ 3 zaudētiem mazuļiem
|
|
|
|
|
|
|
Postnatāli (dzīvi dzimušo skaits mīnus 13. postnatālajā dienā dzīvo mazuļu skaits)
|
|
Mātītes, kas nav zaudējušas mazuli
|
|
|
|
|
|
|
Mātītes ar vienu zaudētu mazuli
|
|
|
|
|
|
|
Mātītes ar diviem zaudētiem mazuļiem
|
|
|
|
|
|
|
Mātītes ar ≥ 3 zaudētiem mazuļiem
|
|
|
|
|
|
B.65.
IN VITRO BARJERMEMBRĀNAS TESTA METODE ATTIECĪBĀ UZ ĀDAS KOROZIJU
IEVADS
|
1.
|
Šī testēšanas metode ir ekvivalenta ESAO testēšanas vadlīnijai (TG) Nr. 435 (2015). Ādas korozija ir nepārejošs ādas bojājums, kas izpaužas kā tāda redzama nekroze visā epidermā, kura ietiecas dermā un radusies pēc tam, kad uz ādas aplicēta testējamā ķimikālija atbilstoši definīcijai, kas dota Apvienoto Nāciju Organizācijas (ANO) Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētajā sistēmā (GHS) (1) un Eiropas Savienības (ES) Regulā Nr. 1272/2008 par vielu un maisījumu klasificēšanu, marķēšanu un iepakošanu (CLP regula) (24). Šī testēšanas metode, kas ir ekvivalenta atjauninātajai ESAO testēšanas vadlīnijai Nr. 435, nosaka in vitro barjermembrānas testa metodi, ko var izmantot kodīgu ķimikāliju identificēšanai. Testēšanas metodē izmanto mākslīgu membrānu, kas izstrādāta tā, lai tā reaģētu uz kodīgām ķimikālijām līdzīgā veidā kā dzīvnieku āda in situ.
|
|
2.
|
Kodīgumu ādai parasti novērtē, aplicējot testējamo ķimikāliju uz dzīvu dzīvnieku ādas un nosakot audu bojājumu pakāpi pēc noteikta laika (2). Papildus šai testēšanas metodei ir pieņemtas vairākas citas in vitro testēšanas metodes, kas ir alternatīvas (3)(4) in vivo standartprocedūrai ar truša ādu (šā pielikuma B.4. nodaļa, ekvivalenta ESAO TG Nr. 404), kuru izmanto kodīgu ķimikāliju identificēšanai (2). ANO GHS secīgu pārbaužu un izvērtēšanas stratēģijā kodīguma ādai novērtēšanai un klasificēšanai un ESAO norādījumu dokumentā par integrētu testēšanas un novērtēšanas pieeju (IATA) attiecībā uz ādas kairinājumu/koroziju ieteikts izmantot validētas un pieņemtas in vitro testēšanas metodes, kas aprakstītas 3. un 4. modulī (1)(5). IATA apraksta vairākus moduļus, kuros tiek grupēti informācijas avoti un analīzes rīki, un, pirmkārt, sniedz norādījumus par to, kā, novērtējot ķimikāliju potenciālu radīt ādas kairinājumu un ādas koroziju, integrēt un izmantot jau esošos testēšanā un citādi iegūtos datus, un, otrkārt, ierosina pieeju, ko izmantot, ja vajadzīga turpmāka testēšana, tostarp gadījumos, kad ir iegūti negatīvi rezultāti (5). Atbilstoši šai modulārajai pieejai pozitīvos rezultātus, kas iegūti, īstenojot in vitro testēšanas metodi, var izmantot ķimikāliju klasificēšanai par kodīgām vielām bez nepieciešamības veikt testus ar dzīvniekiem, tādējādi samazinot un pilnveidojot dzīvnieku izmantošanu un novēršot sāpēs un diskomfortu, ko dzīvniekiem var radīt to izmantošana minētajam mērķim.
|
|
3.
|
In vitro barjermembrānas modeļa, kas komerciāli pieejams ar nosaukumu Corrositex®
, validācijas pētījumi ir pabeigti (6)(7)(8), un tie liecina, ka šā modeļa vispārējā pareizība ādas korozijas paredzēšanā ir 79 % (128/163), jutība — 85 % (76/89) un specifiskums — 70 % (52/74), izmantojot 163 vielu un maisījumu datubāzi (7). Šo validēto referencmetodi (VRM), pamatojoties uz tās atzīto derīgumu, ieteicams izmantot par daļu no secīgu pārbaužu stratēģijas ķimikāliju ādas korozijas briesmu potenciāla novērtēšanai (5)(7). Pirms attiecībā uz ādas koroziju iespējams regulatīvām vajadzībām izmantot kādu in vitro barjermembrānas modeli, saskaņā ar iepriekš noteiktiem veiktspējas standartiem (VS) (10) būtu jānosaka tā ticamība, relevantums (pareizība) un piedāvātā lietojuma ierobežojumi, lai būtu drošība par tā līdzību VRM (9). ESAO datu savstarpējā atzīšana (Mutual acceptance of data) būs garantēta tikai tad, kad jebkāda jauna ierosināta vai atjaunināta VS atbilstoša metode būs izskatīta un iekļauta ekvivalentajā ESAO testēšanas vadlīnijā. Patlaban tikai viena in vitro metode ir iekļauta ESAO testēšanas vadlīnijā Nr. 435 un šajā testēšanas metodē, proti, komerciāli pieejamais Corrositex
® modelis.
|
|
4.
|
Citās testēšanas metodēs kodīguma ādai testēšanai izmanto rekonstruētu cilvēka ādu (ESAO TG Nr. 431) (3) un izolētu žurkas ādu (ESAO TG Nr. 430) (4). Šī testēšanas vadlīnija paredz arī kodīgo ķimikāliju iedalīšanu trīs ANO GHS kodīguma apakškategorijās un trīs ANO transportēšanas iepakojuma grupās attiecībā uz kodīguma risku. Šī testēšanas vadlīnija sākotnēji pieņemta 2006. gadā un 2015. gadā atjaunināta, lai ietvertu atsauci uz IATA norādījumu dokumentu un atjauninātu standartvielu sarakstu.
|
DEFINĪCIJAS
|
5.
|
Izmantotās definīcijas sniegtas papildinājumā.
|
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
6.
|
Šajā testēšanas metodē aprakstītais tests dod iespēju identificēt kodīgas testējamās ķimikālijas un ļauj kodīgas testējamās ķimikālijas iedalīt apakškategorijās saskaņā ar ANO GHS / CLP (1. tabula). Turklāt šādu testēšanas metodi var izmantot, lai konkrētām transportēšanas testu vajadzībām pieņemtu lēmumus par konkrētu ķimikāliju klašu, piemēram, organisku un neorganisku skābju, skābju atvasinājumu (25) un bāžu, kodīgumu vai nekodīgumu (7)(11)(12). Šajā testēšanas metodē aprakstīta vispārēja procedūra, kas ir līdzīga validētajai testēšanas referencmetodei (7). Šī testēšanas metode gan pietiekami neinformē par ādas kairinājumu, taču jānorāda, ka tādai ietekmei uz veselību kā ādas kairinājums in vitro īpaši pievēršas TM B.46., kas ir ekvivalenta ESAO TG Nr. 439 (13). Attiecībā uz to, kā pilnīgi izvērtēt lokālo ietekmi uz ādu pēc vienreizējas dermālas ekspozīcijas, jāizmanto ESAO norādījumu dokuments par integrētajām testēšanas un novērtēšanas pieejām (5).
1. tabula
ANO GHS kategorija “Kodīgs ādai” un tās apakškategorijas (26)
|
Kodīguma kategorija (1. kategorija) (iestādēm, kas neizmanto apakškategorijas)
|
Iespējamās kodīguma apakškategorijas (26) (iestādēm, kas izmanto apakškategorijas, tostarp CLP regulu)
|
Kodīgs ≥ 1 no 3 dzīvniekiem
|
|
Ekspozīcija
|
Novērošana
|
|
Kodīgs
|
Kodīguma apakškategorija 1A
|
≤ 3 minūtes
|
≤ 1 stunda
|
|
Kodīguma apakškategorija 1B
|
> 3 minūtes / ≤ 1 stunda
|
≤ 14 dienas
|
|
Kodīguma apakškategorija 1C
|
> 1 stunda / ≤ 4 stundas
|
≤ 14 dienas
|
|
|
7.
|
Validētās referencmetodes (7) ierobežojums ir tāds, ka daudzas nekodīgas ķimikālijas un dažas kodīgas ķimikālijas, pamatojoties uz sākotnējā saderības testa rezultātiem (sk. 13. punktu), var netikt atzītas par derīgām testēšanai. Ķimikālijas uz ūdens bāzes, kuru pH ir diapazonā no 4,5 līdz 8,5, bieži vien nav derīgas testēšanai; tomēr 85 % no testētajām ķimikālijām, kuru pH ir minētajā diapazonā, nebija kodīgas testos ar dzīvniekiem (7). In vitro barjermembrānas metodi var izmantot, lai testētu cietvielas (ūdenī šķīstošas vai nešķīstošas), šķidrumus (ūdens vai neūdens) un emulsijas. Tomēr ar barjermembrānas metodi nevar testēt tādas testējamās ķimikālijas, kas neizraisa detektējamas izmaiņas saderības testā (piemēram, krāsas maiņu validētās testēšanas referencmetodes ķimikāliju detektēšanas sistēmā (CDS)); tās jātestē, izmantojot citas testēšanas metodes.
|
TESTA PRINCIPS
|
8.
|
Testēšanas sistēmai ir divi komponenti: sintētiska makromolekulāra biobarjera un ķimikāliju detektēšanas sistēma (CDS); ar šo testēšanas metodi, izmantojot CDS, nosaka barjermembrānas bojājumus, kas rodas pēc kodīgas testējamās ķimikālijas aplicēšanas uz sintētiskas makromolekulāras barjermembrānas (7) un ko izraisa, domājams, tie paši korozijas mehānismi, kuri iedarbojas uz dzīvu ādu.
|
|
9.
|
Barjermembrānas penetrāciju (jeb ķimikālijas izlaušanos cauri tai) var mērīt, izmantojot vairākas procedūras vai CDS; to var mērīt arī pēc pH indikatorkrāsvielas krāsas izmaiņām vai kādu citu zem barjeras esoša indikatoršķīduma īpašību izmaiņām.
|
|
10.
|
Jānosaka, vai barjermembrāna ir derīga, t. i., atbilstīga un uzticama, paredzētajam lietojumam. Šajā sakarā ir jāpārliecinās, ka dažādi preparāti nodrošina barjeras īpašības, piemēram, tie spēj saglabāt barjeru pret nekodīgām ķimikālijām, un tie dod iespēju ķimikāliju kodīgās īpašības iedalīt dažādajās ANO GHS kodīguma apakškategorijās (1). Piešķirtā klasifikācija pamatojas uz laiku, kādā ķimikālija caur barjermembrānu nokļūst līdz indikatoršķīdumam.
|
KOMPETENCES PIERĀDĪŠANA
|
11.
|
Pirms šai testēšanas metodei atbilstošo in vitro barjermembrānas metodi sākt izmantot regulāri, laboratorijām jāpierāda tehniskā kompetence, pareizi saklasificējot 2. tabulā ieteiktās 12 standartvielas. Gadījumos, kur kāda sarakstā norādīta viela nav pieejama vai kur tas attaisnojami, var izmantot citu vielu, par kuru pieejami pietiekami in vivo un in vitro references dati (piem., vielu no references ķimikāliju saraksta (10)), ja vien tiek izmantoti tie paši 1. tabulā aprakstītie izraudzīšanās kritēriji.
2. tabula
Standartvielas
(27)
|
Viela (28)
|
CAS Nr.
|
Ķimikāliju klase
|
In Vivo ANO GHS apakškategorija (29)
|
In Vitro ANO GHS apakškategorija (29)
|
|
Bora trifluorīddihidrāts
|
13319-75-0
|
Neorganiskas skābes
|
1A
|
1A
|
|
Slāpekļskābe
|
7697-37-2
|
Neorganiskas skābes
|
1A
|
1A
|
|
Fosfora pentahlorīds
|
10026-13-8
|
Neorganisko skābju prekursori
|
1A
|
1A
|
|
Valerilhlorīds
|
638-29-9
|
Skābju hlorīdi
|
1B
|
1B
|
|
Nātrija hidroksīds
|
1310-73-2
|
Neorganiskas bāzes
|
1B
|
1B
|
|
1-(2-aminoetil) piperazīns
|
140-31-8
|
Alifātiskie amīni
|
1B
|
1B
|
|
Benzolsulfonilhlorīds
|
98-09-9
|
Skābju hlorīdi
|
1C
|
1C
|
|
N, N-dimetilbenzilamīns
|
103-83-3
|
Anilīni
|
1C
|
1C
|
|
Tetraetilēnpentamīns
|
112-57-2
|
Alifātiskie amīni
|
1C
|
1C
|
|
Eigenols
|
97-53-0
|
Fenoli
|
NK
|
NK
|
|
Nonilakrilāts
|
2664-55-3
|
Akrilāti/metakrilāti
|
NK
|
NK
|
|
Nātrija bikarbonāts
|
144-55-8
|
Neorganiski sāļi
|
NK
|
NK
|
|
PROCEDŪRA
|
12.
|
Turpmākajos punktos aprakstīti kodīguma novērtēšanai paredzētās uz pašreizējo VRM, proti, komerciāli pieejamo Corrositex
®, balstītās mākslīgas barjermembrānas testa metodes (7)(15) komponenti un procedūras. Barjermembrānu un saderības šķīdumus vai indikatoršķīdumus, kā arī šķīdumus, ko izmanto ķimikāliju kategorizēšanai, var izstrādāt, sagatavot vai iegūt tirdzniecībā, piemēram, VRM Corrositex
® gadījumā. Validētajai testēšanas referencmetodei ir pieejams parauga testēšanas metodes protokols (7). Testēšana jāveic apkārtējās vides temperatūrā (17–25 °C), un komponentiem jāatbilst šādiem nosacījumiem.
|
Testējamās ķimikālijas saderības tests
|
13.
|
Pirms barjermembrānas testa veikšanas veic saderības testu, lai noteiktu, vai testējamā ķimikālija ir detektējama ar CDS. Ja testējamo ķimikāliju ar CDS nevar detektēt, barjermembrānas testa metode nav piemērota konkrētās testējamās ķimikālijas iespējamā kodīguma izvērtēšanai un jāizmanto cita testēšanas metode. Saderības testā izmantotajai CDS un ekspozīcijas nosacījumiem jāatspoguļo ekspozīcija turpmākajā barjermembrānas testā.
|
Testējamās ķimikālijas laika skalas kategorijas tests
|
14.
|
Ja testēšanas metode to ļauj, testējamai ķimikālijai, kas ir kvalificēta ar saderības testu, jāveic laika skalas kategorijas tests, proti, skrīninga tests, ko izmanto, lai nošķirtu vājas un stipras skābes vai bāzes. Piemēram, validētajā testēšanas referencmetodē laika skalas kategorizēšanas testu izmanto, lai atkarībā no tā, vai ir detektēta būtiska skābju vai sārmu rezerve, noteiktu, kura no divām laika skalām būtu jāizmanto. Kodīguma un ANO GHS kodīguma ādai apakškategorijas noteikšanai būtu jāizmanto divas atšķirīgas izlaušanās laika skalas atkarībā no testējamās ķimikālijas skābju vai sārmu rezerves.
|
BARJERMEMBRĀNAS TESTA METODES KOMPONENTI
Barjermembrāna
|
15.
|
Barjermembrāna sastāv no diviem komponentiem: makromolekulāra ūdens proteīngela un caurlaidīgas pamatmembrānas. Proteīngelam jābūt šķidrumu un cietvielas necaurlaidīgam, taču tas var korodēties un kļūt caurlaidīgs. Gatavā barjermembrāna jāuzglabā iepriekš noteiktos apstākļos, kas novērš gela bojāšanos, piemēram, izžūšanu, mikrobu augšanu, tā nobīdi un plaisāšanu, kas varētu pasliktināt tā funkciju. Jānosaka pieļaujamais glabāšanas termiņš un barjermembrānas preparāti, ko pēc minētā termiņa neizmanto.
|
|
16.
|
Caurlaidīgā pamatmembrāna nodrošina mehānisku balstu proteīngelam sarecēšanas procesā un eksponētību testējamajai ķimikālijai. Pamatmembrānai jānovērš gela noslīdēšana vai nobīde, un tai viegli jālaiž cauri visas testējamās ķimikālijas.
|
|
17.
|
Proteīngels, kas sastāv no proteīna, piemēram, keratīna, kolagēna vai proteīnu maisījumiem, kuri veido gela matrici, kalpo par testējamās ķimikālijas mērķi. Proteīnmateriālu novieto uz pamatmembrānas virsmas, un pirms barjermembrānas novietošanas uz indikatoršķīduma tam ļauj sarecēt. Proteīngelam jābūt viscaur vienādi biezam un blīvam un bez gaisa burbuļiem vai defektiem, kas varētu ietekmēt tā funkcionālo integritāti.
|
Ķimikāliju detektēšanas sistēma (CDS)
|
18.
|
Indikatoršķīdumam, kas ir tas pats šķīdums, ko izmanto saderības testā, būtu jāreaģē uz testējamās ķimikālijas klātbūtni. Jāizmanto pH indikatorkrāsviela vai krāsvielu kombinācija, piemēram, krezolsarkanais un metiloranžais, kas, reaģējot uz testējamās ķimikālijas klātbūtni, maina savu krāsu. Mērīšanas sistēma var būt vizuāla vai elektroniska.
|
|
19.
|
Lai noteiktu detektējamo ķimikāliju klāstu un detektēšanas kvantitatīvos ierobežojumus, jānovērtē, cik relevantas un ticamas ir detektēšanas sistēmas, kas izstrādātas, lai detektētu testējamās ķimikālijas iziešanu caur barjermembrānu.
|
TESTA NORISE
Testēšanas metodes komponentu montāža
|
20.
|
Barjermembrānu ievieto ar indikatoršķīdumu pildītā mēģenē (vai caurulē) tā, lai pamatmembrāna pilnībā saskartos ar indikatoršķīdumu un nerastos gaisa burbuļi. Jāraugās, lai tiktu saglabāta barjeras integritāte.
|
Testējamās ķimikālijas aplicēšana
|
21.
|
Testējamo ķimikāliju pietiekamā daudzumā, piemēram, 500 μl šķidruma vai 500 mg smalki saberztas pulverveida cietvielas veidā (7), rūpīgi uzklāj uz barjermembrānas augšējās virsmas un vienmērīgi izkliedē. Katrai testējamajai ķimikālijai un to atbilstošajām kontrolēm (sk. 23.–25. punktu) sagatavo pietiekamu skaitu replikātu, piemēram, četrus (7). Reģistrē laiku, kad testējamā ķimikālija aplicēta uz barjermembrānas. Lai nodrošinātu, ka tiek precīzi reģistrēti īsi korozijas laiki, testējamās vielas replikātu mēģenēs aplicē dažādos laikos.
|
Barjermembrānas penetrācijas mērījumi
|
22.
|
Katru mēģeni pienācīgi uzrauga, un reģistrē laiku, kad notikušas pirmās izmaiņas indikatoršķīdumā, t. i., barjeras penetrācija, un nosaka laika intervālu no aplicēšanas līdz barjermembrānas penetrācijai.
|
Kontroles
|
23.
|
Testos, kuros testējamo ķimikāliju izmanto kopā ar nesēju vai šķīdinātāju, nesējam vai šķīdinātājam jābūt saderīgam ar barjermembrānas sistēmu, proti, tam nevajadzētu izjaukt barjermembrānas sistēmas integritāti un mainīt testējamās ķimikālijas kodīgumu. Attiecīgā gadījumā, lai noteiktu šķīdinātāja saderību ar barjermembrānas sistēmu, vienlaikus ar testējamo ķimikāliju jātestē šķīdinātāja (vai nesēja) kontrole.
|
|
24.
|
Lai novērtētu, vai testēšanas sistēma darbojas pieņemami, vienlaikus ar testējamo ķimikāliju jātestē arī pozitīva (kodīga) kontrole ar vidēju kodīguma aktivitāti, piemēram, 110 ± 15 mg nātrija hidroksīda (ANO GHS 1.B kodīguma apakškategorija) (7). Lai izvērtētu kodīgas testējamās ķimikālijas relatīvo kodīguma potenciālu, var būt lietderīgi izmantot otru pozitīvo kontroli, kas pieder pie tās pašas ķimikāliju klases kā testējamā ķimikālija. Jāizvēlas vidēji kodīga pozitīvā kontrole vai kontroles (piemēram, ANO GHS 1.B apakškategorija), lai detektētu izmaiņas penetrēšanās laikā, kas var būt nepieņemami ilgāks vai īsāks par noteikto atsauces vērtību un tādējādi norādīt uz to, ka testēšanas sistēma nedarbojas pareizi. Ārkārtīgi kodīgas (ANO GHS 1.A apakškategorija) vai nekodīgas ķimikālijas šim nolūkam nav sevišķi lietderīgas. ANO GHS 1.B apakškategorijas ķimikālija ļautu detektēt pārāk garu vai īsu izlaušanās laiku. Lai noteiktu testēšanas metodes spēju konsekventi atšķirt vāji kodīgas un nekodīgas ķimikālijas, par pozitīvo kontroli var izmantot vāji kodīgu ķimikāliju (ANO GHS 1.C apakškategorija). Neatkarīgi no izmantotās pieejas jānosaka pieņemams pozitīvās kontroles atbildreakciju intervāls, kas balstīts uz izmantotās pozitīvās kontroles vai kontroļu izlaušanās laiku vēsturisko intervālu, piemēram, vidējās standartnovirzes ± 2-3. Lai varētu detektēt novirzes ārpus pieņemamā intervāla, katrā pētījumā jānosaka precīzs pozitīvās kontroles izlaušanās laiks.
|
|
25.
|
Vienlaikus ar testējamo ķimikāliju jātestē arī negatīva (nekodīga) kontrole, piemēram, 10 % citronskābe, 6 % propionskābe (7), kas ir vēl viens kvalitātes kontroles līdzeklis, ar ko pierādīt barjermembrānas funkcionālo integritāti.
|
Pētījuma pieņemamības kritēriji
|
26.
|
Testējamās ķimikālijas kodīguma prognozēšanai izmanto laiku (minūtēs), kas pagājis no testējamās ķimikālijas aplicēšanas uz barjermembrānas līdz barjeras penetrācijai, atbilstīgi katrai ANO GHS kodīguma apakškategorijai noteiktajiem laika parametriem. Lai pētījumu uzskatītu par pieņemamu, paralēlajai pozitīvajai kontrolei jāuzrāda sagaidāmais penetrēšanās atbildreakcijas laiks (piemēram, 8–16 minūtes ilgs izlaušanās laiks, ja par pozitīvo kontroli izmanto nātrija hidroksīdu), paralēlā negatīvā kontrole nedrīkst būt kodīga un paralēlā šķīdinātāja kontrole, ja tāda ir iekļauta, nedrīkst būt kodīga un tā nedrīkst mainīt testējamās ķimikālijas kodīguma potenciālu. Pirms šai testēšanas metodei atbilstošo metodi sākt izmantot regulāri, laboratorijām jāpierāda tehniskā kompetence, izmantojot 2. tabulā ieteiktās 12 vielas. Attiecībā uz jaunām atdarinājuma metodēm, kas izstrādātas saskaņā ar šo testēšanas metodi un ir strukturāli un funkcionāli līdzīgas validētajai referencmetodei (14), pirms jaunās metodes izmantošanas regulatīviem mērķiem ir jāpierāda tās ticamība un pareizība, šim nolūkam izmantojot iepriekš noteiktus veiktspējas standartus (10).
|
Rezultātu interpretēšana un testējamo ķimikāliju kodīguma klasifikācija
|
27.
|
Testējamo ķimikāliju ANO GHS kodīguma apakškategorijās (1) un attiecīgā gadījumā ANO iekopojuma grupā (16) klasificē atbilstoši laikam (minūtēs), kas pagājis no testējamās ķimikālijas aplicēšanas barjermembrānai līdz barjeras penetrācijai. Katrai piedāvātajai testēšanas metodei nosaka laika sliekšņvērtības katrā no trim kodīguma apakškategorijām. Pieņemot galīgos lēmumus par laika sliekšņvērtībām, jāņem vērā nepieciešamība līdz minimumam samazināt korozijas briesmu pārāk zemas klasifikācijas (t. i., šķietami negatīvu rezultātu) skaitu. Pašreizējā testēšanās vadlīnijā jāizmanto metodes Corrositex
® laika sliekšņvērtības, kas norādītās 3. tabulā, jo tā patlaban ir vienīgā testēšanas vadlīnijā ietvertā testēšanas metode (7).
3. tabula
Corrositex® prognozes modelis
|
Vidējais izlaušanās laiks (minūtēs)
|
ANO GHS prognoze (32)
|
|
1. kategorijas testējamās ķimikālijas (30) (nosaka ar metodē paredzēto kategorizēšanas testu)
|
1. kategorijas testējamās ķimikālijas (31) (nosaka ar metodē paredzēto kategorizēšanas testu)
|
|
0–3 min
|
0–3 min
|
Kodīgs fakultatīvā 1.A apakškategorija
|
|
> 3 līdz 60 min
|
> 3 līdz 30 min
|
Kodīgs fakultatīvā 1.B apakškategorija
|
|
> 60 līdz 240 min
|
> 30 līdz 60 min
|
Kodīgs fakultatīvā 1.C apakškategorija
|
|
> 240 min
|
> 60 min
|
Nekodīgs
|
|
DATI UN PĀRSKATA SAGATAVOŠANA
Dati
|
28.
|
Laiks (minūtēs), kas pagājis no testējamās ķimikālijas un pozitīvās kontroles vai kontroļu aplicēšanas līdz barjeras penetrācijai, jānorāda tabulā katram replikātam atsevišķi, kā arī jānorāda katra izmēģinājuma vidējā ± standartnovirze.
|
Testēšanas pārskats
|
29.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Testējamā ķimikālija un kontroles vielas:
|
—
|
vienkomponenta viela: ķīmiskā identifikācija, piemēram, IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, attiecīgā gadījumā un ja praktiski iespējams — piemaisījumu ķīmiskā identitāte u. c.;
|
|
—
|
daudzkomponentu viela, UVCB un maisījums: iespējami izsmeļoši raksturoti pēc komponentu ķīmiskās identitātes (sk. iepriekš), kvantitatīvās sastopamības un relevantajām fizikālķīmiskajām īpašībām;
|
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības;
|
|
—
|
avots, sērijas numurs, ja zināms;
|
|
—
|
testējamās ķimikālijas / kontrolvielas pirmstestēšanas apstrāde (piem., sildīšana, sasmalcināšana), ja tāda veikta;
|
|
—
|
testējamās ķimikālijas stabilitāte, izmantošanas termiņš vai atkārtotas analīzes datums, ja tas zināms;
|
|
|
|
Nesējs:
|
—
|
identifikācija, koncentrācija (attiecīgā gadījumā), izmantotais tilpums;
|
|
—
|
nesēja izraudzīšanās pamatojums.
|
|
|
|
Izmantotais in vitro barjermembrānas modelis un protokols, arī pierādīta pareizība un ticamība
|
|
|
Testēšanas nosacījumi:
|
—
|
izmantoto iekārtu un sagatavošanas procedūru apraksts;
|
|
—
|
izmantotās in vitro barjermembrānas izcelsme un sastāvs;
|
|
—
|
indikatoršķīduma sastāvs un īpašības;
|
|
—
|
testējamās ķimikālijas un kontroles vielas daudzumi;
|
|
—
|
laika skalas kategorijas testa apraksts un pamatojums;
|
|
—
|
izmantoto izvērtēšanas un klasificēšanas kritēriju apraksts;
|
|
—
|
apliecināta kompetence testēšanas metodes veikšanā, kura pirms attiecīgās testēšanas metodes regulāras izmantošanas pierādīta, veicot standartķimikāliju testēšanu.
|
|
|
|
Rezultāti:
|
—
|
tabulā apkopoti individuāli jēldati, kas iegūti no katra replikāta individuāliem testa un kontroles paraugiem;
|
|
—
|
citas novērotās ietekmes apraksts;
|
|
—
|
atvasinātā klasifikācija ar norādi uz izmantoto prognozes modeli un/vai lēmuma pieņemšanas kritēriju.
|
|
Rezultātu iztirzājums
Secinājumi
|
LITERATŪRA
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Šā pielikuma B.4. nodaļa “Akūts ādas kairinājums/korozija”.
|
|
(3)
|
Šā pielikuma B.40a nodaļa “Ādas korozija in vitro: rekonstruētas cilvēka epidermas RHE tests”.
|
|
(4)
|
Šā pielikuma B.40. nodaļa “Ādas korozija in vitro: transkutānā elektriskā pretestība (TEP)”.
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Chapter B.46 of this Annex, In Vitro Skin Irritation: Reconstructed Human Epidermis Test Method. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Available at: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Available at: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Available at: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā testēšanas metodes rezultāti sakrīt ar pieņemtajām atsauces vērtībām. Tas ir testēšanas metodes veiktspējas mērs un viens no relevantuma aspektiem. Šo terminu bieži izmanto tādā pat nozīmē kā terminu “konkordantums”, ar to saprotot testēšanas metodes pareizo iznākumu īpatsvaru (9).
Ķimikālija
: viela vai maisījums.
Ķimikāliju detektēšanas sistēma (CDS)
: vizuāla vai elektroniska mērīšanas sistēma, kurā izmanto indikatoršķīdumu, kas reaģē uz testējamās ķimikālijas klātbūtni, piemēram, ar izmaiņām pH indikatorkrāsvielā vai krāsvielu kombinācijā, kam, reaģējot uz testējamās ķimikālijas klātbūtni, mainīsies krāsa, vai ar cita veida ķīmiskām vai elektroķīmiskām reakcijām.
Konkordantums
: tādas testēšanas metodes veiktspējas mērs, ar kuru iegūst kategoriālus rezultātus, un viens no relevantuma aspektiem. Šo terminu bieži izmanto tādā pat nozīmē kā terminu “pareizība”, ar to saprotot visu to testēto ķimikāliju īpatsvaru, kas pareizi klasificētas kā pozitīvas vai negatīvas. Konkordantums lielā mērā ir atkarīgs no to pozitīvo rezultātu prevalences, kādi iegūti attiecībā uz pētāmajiem testējamo ķimikāliju veidiem (9).
GHS (Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un izmanto tādus attiecīgus komunikācijas elementus kā piktogrammas, signālvārdi, bīstamības apzīmējumi, drošības prasību apzīmējumi un drošības datu lapas, lai sniegtu informāciju par ķimikāliju nelabvēlīgo ietekmi un tādējādi aizsargātu cilvēkus (arī darba devējus un darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (1).
IATA
: integrētā testēšanas un novērtēšanas pieeja.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairākām vielām.
Vienkomponenta viela
: tāda pēc kvantitatīvā sastāva definēta viela, kurā vismaz 80 % (masa/masa) veido viena galvenā sastāvdaļa.
Daudzkomponentu viela
: tāda pēc kvantitatīvā sastāva definēta viela, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu vielas tiek iegūtas ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu viela rodas ķīmiskā reakcijā.
NK
: nekodīgs.
Veiktspējas standarti
: uz validētu testēšanas metodi bāzēti standarti, pēc kuriem izvērtē pēc noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgu testēšanas metožu salīdzināmību. Tajos ietilpst i) nepieciešamie testēšanas metodes komponenti; ii) minimāls tādu references ķimikāliju saraksts, kuras izraudzītas no citām ķimikālijām, ko izmanto, lai pierādītu pieņemamu validētās testēšanas metodes veiktspēju; kā arī iii), uz attiecīgajā validētajā testēšanas metodē iegūto rezultātu pamata — tādi paši pareizības un ticamības līmeņi, kā būtu jāiegūst ar novērtējamo testēšanas metodi, to izvērtējot ar references vielām no minimālā saraksta (9).
Relevantums
: parametrs, kas raksturo testēšanas metodes un interesējošās ietekmes sakarību un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testēšanas metodes pareizības (konkordantuma) aspektu (9).
Ticamība
: parametrs, kas izsaka, kādā pakāpē, izmantojot vienu un to pašu protokolu, kādu testēšanas metodi vienā un tajā pašā laboratorijā vai dažādās laboratorijās ilgākā laika nogrieznī var izmantot reproducējami. To novērtē, aprēķinot iekšlaboratorisko un starplaboratorisko reproducējamību (9).
Jutība
: tas, cik liela daļa visu pozitīvo/aktīvo ķimikāliju ar šo testēšanas metodi tiek pareizi kvalificētas. Tas ir tādas testēšanas metodes pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (9).
Ādas korozija in vivo
: neatgriezenisku ādas bojājumu rašanās; proti, redzama nekroze, kas caur epidermu skar zemādu un rodas, testējamo ķimikāliju aplicējot uz ādas uz laiku līdz četrām stundām. Tipiskas reakcijas uz kodīgumu ir čūlas, asiņošana, asiņainas kreveles un 14 dienu novērošanas perioda beigās — ādas izbalējuma radīta atkrāsošanās, ādas laukumi, kur izkritis apmatojums, un rētas. Neskaidros gadījumos bojājums jānovērtē histopatoloģiski.
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām ķimikālijām ar testēšanas metodi tiek klasificētas pareizi. Tādas testēšanas metodes pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (9).
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai nepieciešamās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu.
Testējamā ķimikālija
: jebkura viela vai maisījums, kuru testē, izmantojot šo testēšanas metodi.
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kompleksi reakcijas produkti vai bioloģiski materiāli.
B.66 STABILI TRANSFICĒTAS TRANSAKTIVĀCIJAS IN VITRO TESTI, AR KO KONSTATĒ ESTROGĒNU RECEPTORU AGONISTUS UN ANTAGONISTUS
VISPĀRĪGS IEVADS
ESAO rezultātbalstītas testēšanas vadlīnijas
|
1.
|
Šī testēšanas metode ir ekvivalenta ESAO Testēšanas vadlīnijā (TG) Nr. 455 (2016) aprakstītajam. TG 455 ir veiktspējā balstītas testēšanas vadlīnija (PBTG), kurā aprakstīta stabili transficētas transaktivācijas in vitro testēšanas metodika, ar ko konstatē estrogēnu receptoru agonistus un antagonistus (ER TA testi). Tajā ietilpst vairākas mehānistiski un funkcionāli līdzīgas testēšanas metodes, ko izmanto, lai identificētu estrogēnu receptora (t.i., ERα un/vai ERβ) agonistus un antagonistus, un tā varētu atvieglot jaunu līdzīgu vai modificētu testēšanas metožu izstrādi atbilstīgi validēšanas principiem, kas izklāstīti ESAO vadlīniju dokumentā par Bīstamības novērtēšanā izmantojamu jaunu vai atjauninātu testēšanas metožu validēšanu un starptautisku akceptēšanu (1). Pilnībā validētās references testēšanas metodes (2. un 3. papildinājums), kas ir šīs PBTG pamatā, ir šādas:
|
—
|
stabili transficētas TA (STTA) tests (2), izmantojot šūnu līniju (h) ERα-HeLa-9903; un
|
|
—
|
VM7Luc ER TA tests (3), izmantojot šūnu līniju VM7Luc4E2 (33), kur galvenokārt ir ekspresēta līnija hERα un daļēji hERβ(4)(5).
|
Līdzīgu testu izstrādē, kas vērsti uz to pašu apdraudējumu/iznākumu, ir pieejami un būtu jāizmanto snieguma standarti (PS) (6) (7). Tas dod iespēju savlaicīgi grozīt PBTG 455, proti, atjaunināto PBTG papildināt ar jauniem līdzīgiem testiem; tomēr līdzīgus testus pievienos tikai pēc izskatīšanas un apstiprināšanas ESAO, ka snieguma standarti ir izpildīti. TG 455 iekļautos testus tam, lai izpildītu ESAO valstu prasības pēc estrogēnu receptora transaktivācijas testa rezultātiem, vienlaikus izmantojot ESAO datu savstarpējo atzīšanu, šos testus var izmantot bez izšķirības.
|
Testēšanas metodē iekļauto testu konteksts un principi
|
2.
|
ESAO 1998. gadā sāka īstenot prioritārus pasākumus ar mērķi pārskatīt esošās un izstrādāt jaunas testēšanas vadlīnijas par potenciāli endokrīnās sistēmas traucējumus izraisošu ķimikāliju skrīningu un testēšanu. ESAO konceptuālais satvars (KS) potenciāli endokrīnās sistēmas traucējumus izraisošu ķimikāliju testēšanai un novērtēšanai 2012. gadā tika pārskatīts. Gan sākotnējais, gan pārskatītais KS ir iekļauts pielikumā ESAO Vadlīniju dokumentam par standartizētām testēšanas metodēm ķimikāliju vērtēšanai attiecībā uz endokrīnajiem disruptoriem (8). KS aptver piecus līmeņus, no kuriem katrs atbilst citam bioloģiskās sarežģītības līmenim. Šajā testēšanas metodē aprakstītie ER transaktivācijas (TA) testi atbilst 2. līmenim, kur ietilpst “in vitro testi, kas sniedz datus par izvēlētiem endokrīniem mehānismiem/ceļiem”. Šī testēšanas metode ir paredzēta in vitro transaktivācijas testiem, ar ko identificē estrogēnu receptoru (ER) agonistus un antagonistus.
|
|
3.
|
Estrogēnu un ER mijiedarbība var ietekmēt estrogēnu regulētos gēnus, un tā rezultātā var tikt inducēti vai inhibēti šūnu procesi, tostarp tie, kas saistīti ar šūnu vairošanos, normālu augļa attīstību un reproduktīvo funkciju (9)(10)(11). Normālu estrogēnisko sistēmu traucējumi var negatīvi ietekmēt normālu attīstību (ontoģenēzi), reproduktīvo veselību un reproduktīvās sistēmas integritāti.
|
|
4.
|
In vitro TA testu pamatā ir vielu tieša vai netieša mijiedarbība ar specifisku receptoru, kas regulē reportiergēna produkta transkripciju. Šādi testi ir plaši izmantoti, lai izvērtētu gēnu ekspresiju, ko regulē specifiski nukleārie receptori, piem., ER (12) (13) (14) (15) (16). Ir ierosināts, ka tos varētu izmantot, lai detektētu estrogēnisku transaktivāciju, ko regulē ER (17) (18) (19). Pastāv vismaz divi lieli nukleāro ER apakštipi: α un β, un tos kodē atšķirīgi gēni. Attiecīgajiem proteīniem ir atšķirīgas bioloģiskās funkcijas, kā arī atšķirīgs sadalījums audos un atšķirīga ligandu afinitāte (20)(21)(22)(23)(24)(25)(26). Nukleārais ERα mediē klasisko estrogēnisko reakciju (27)(28)(29)(30), tāpēc vairums modeļu, ko pašlaik izstrādā ER aktivācijas vai inhibīcijas mērīšanai, balstās uz ERα. Testus izmanto, lai identificētu ķimikālijas, kas aktivē (vai inhibē) ER ar liganda piesaisti, pēc kuras receptora-liganda komplekss piesaistās specifiskiem DNS reakcijas elementiem un transaktivē reportiergēnu, kā rezultātā palielinās marķierproteīna ekspresija šūnās. Testos var izmantot dažādas reportiergēnu reakcijas. Uz luciferāzes balstītās sistēmās luciferāzes enzīms luciferīna substrātu pārveido bioluminiscējošā produktā, ko var kvantitatīvi izmērīt ar luminometru. Citi izplatīti marķieri ir fluorescējošais proteīns un LacZ gēns, kas kodē β-galaktozidāzi — enzīmu, kas bezkrāsaino substrātu X-gal (5- brom-4-hlor-indolil-galaktopiranozīdu) pārveido zilā produktā, ko var kvantificēt ar spektrofotometru. Šos marķierus var ātri un lēti izvērtēt ar komerciāli pieejamiem testa komplektiem.
|
|
5.
|
STTA un VM7Luc TA testu validācijas pētījumi apliecina, ka šie testi ir ticami un relevanti konkrētajam nolūkam (3)(4)(5)(30). Veiktspējas standarti uz luminiscenci balstītiem ER TA testiem, kuros izmanto krūšu šūnas, ir iekļauti ziņojumā ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (VM7Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Šie veiktspējas standarti ir grozīti tā, lai tos varētu izmantot gan STTA, gan VM7Luc TA testiem (2).
|
|
6.
|
Šajā testēšanas metodē izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
TA testu tvērums un ierobežojumi
|
7.
|
Šos testus ierosināts izmantot skrīninga un prioritizēšanas vajadzībām, tomēr tie var sniegt arī mehānistisku informāciju, ko var izmantot pierādījumu izsvēršamas pieejā. Tie attiecas uz TA, ko inducē ķīmiska piesaiste ER in vitro sistēmā. Tāpēc rezultātus nevajadzētu tieši ekstrapolēt uz neskartas endokrīnās sistēmas kompleksajiem signalizēšanas un regulēšanas mehānismiem in vivo.
|
|
8.
|
Uzskata, ka ER mediēta TA ir viens no nozīmīgākajiem endokrīnās disrupcijas (ED) mehānismiem, lai gan ED izraisa arī citi mehānismi, t. sk. i) mijiedarbība ar citiem endokrīnās sistēmas receptoriem un enzīmu sistēmām, ii) hormonu sintēze, iii) hormonu metaboliskā aktivācija/inaktivācija, iv) hormonu sadalījums mērķaudos un v) hormonu izvadīšana no organisma. Neviens pie šīs testēšanas metodes apskatītais tests neattiecas uz šiem darbības modiem.
|
|
9.
|
Šī testēšanas metode attiecas uz ķimikāliju spēju aktivēt (t. i., darboties kā agonistam) un nomākt (t. i., darboties kā antagonistam) ER regulētu transkripciju. Dažas ķimikālijas — atkarībā no izmantotajām šūnām — var iedarboties gan kā agonisti, gan kā antagonisti, un tās sauc par selektīvajiem estrogēnu receptora modulatoriem (SERM). Ja ķimikālijas šajos testos uzrāda negatīvus rezultātus, tad, pirms izdarīt secinājumu, ka tās receptoram nepiesaistās, tās varētu būt lietderīgi novērtēt ER piesaistīšanas testos. Turklāt šie testi sniedz tikai informāciju par vecākmolekulas aktivitāti, ņemot vērā in vitro šūnu sistēmu ierobežotās metabolizēšanas spējas. Tā kā validācijas pētījumā tika izmantotas tikai atsevišķas vielas, nav informācijas par testa izmantojamību ar maisījumiem. Tomēr šī testēšanas metode teorētiski ir piemērojama daudzkomponentu vielu, UVCB un maisījumu testēšanai. Pirms datu iegūšanai normatīvām vajadzībām šo testēšanas metodi izmanto attiecībā uz daudzkomponentu vielu, UVCB vai maisījumu, jāapsver, vai un kādēļ tā var sniegt tādam nolūkam piemērotus rezultātus. Šādi apsvērumi nav vajadzīgi, ja attiecībā uz maisījuma testēšanu pastāv regulatīva prasība.
|
|
10.
|
Informatīvā nolūkā 1. tabulā ir sniegti agonista testa rezultāti 34 vielām, kas testētas ar abām pilnībā validētajām references testēšanas metodēm, kuras aprakstītas šajā testēšanas metodē. No šīm vielām 26 nešaubīgi klasificētas kā ER agonisti, un, kā iziet no publicētajiem pārskatiem, 8 vielas uzrādīja negatīvus rezultātus, t. sk. in vitro ER piesaistes un TA testos un/vai uterotrofiskajā testā (2)(3)(18)(31)(32)(33)(34). 2. tabulā ir sniegti antagonista testa rezultāti 15 vielām, kas testētas ar abām pilnībā validētajām references testēšanas metodēm, kuras aprakstītas šajā testēšanas metodē. No šīm vielām 4 nešaubīgi/potenciāli klasificētas kā ER antagonisti, un, kā iziet no publicētajiem pārskatiem, 10 vielas uzrādīja negatīvus rezultātus, t. sk. in vitro ER piesaistes un TA testos (2)(3)(18)(31). Runājot par 1. un 2. tabulā apkopotajiem datiem, abas references testēšanas metodes ļauj izdarīt 100 % vienādus secinājumus par visu vielu klasifikāciju (izņemot vienu vielu — mifepristonu — antagonista testā), un katra viela ir pareizi klasificēta kā ER agonists/antagonists vai negatīvs rezultāts. Papildu informācija par šo ķimikāliju grupu un par vēl citām ķimikālijām, kas validācijas pētījumu gaitā testētas STTA VM7Luc ER TA testos, ir sniegta ERTA veiktspējas standartu (6)(7), 2. papildinājumā (1., 2. un 3. tabula).
|
1. tabula
Pārskats par STTA un VM7Luc ER TA testu rezultātiem attiecībā uz vielām, kas testētas abos agonista testos un klasificētas kā ES agonisti (POS) vai kā negatīvi rezultāti (NEG)
|
|
Viela
|
CAS Nr.
|
STTA tests (35)
|
VM7Luc ER TA tests (36)
|
Klasifikācijas datu avots (38)
|
|
ER TA aktivitāte
|
PC10 vērtība (M)
|
PC50 vērtība (2) (M)
|
ER TA aktivitāte
|
EC50 vērtība (2), (37) (M)
|
Citi ER TA testi (34)
|
ER piesaistes testi
|
Uterotrofiskie testi
|
|
1
|
17ß-estradiols (1)
|
50-28-2
|
POZ
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POZ
|
5,63 × 10-12
|
POZ (227/227)
|
POZ
|
POZ
|
|
2
|
17α-estradiols (1)
|
57-91-0
|
POZ
|
7,24 × 10-11
|
6,44 × 10-10
|
POZ
|
1,40 × 10-9
|
POZ (11/11)
|
POZ
|
POZ
|
|
3
|
17α-etinilestradiols (1)
|
57-63-6
|
POZ
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POZ
|
7,31 × 10-12
|
POZ (22/22)
|
POZ
|
POZ
|
|
4
|
17β-trenbolons
|
10161-33-8
|
POZ
|
1,78 × 10-8
|
2,73 × 10-7
|
POZ
|
4,20 × 10-8
|
POZ (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosterons (1)
|
434-22-0
|
POZ
|
9,64 × 10-9
|
2,71 × 10-7
|
POZ
|
1,80 × 10-6
|
POZ (4/4)
|
POZ
|
POZ
|
|
6
|
4-kumilfenols (1)
|
599-64-4
|
POZ
|
1,49 × 10-7
|
1,60 × 10-6
|
POZ
|
3,20 × 10-7
|
POZ (5/5)
|
POZ
|
NT
|
|
7
|
4-terc-oktilfenols (1)
|
140-66-9
|
POZ
|
1,85 × 10-9
|
7,37 × 10-8
|
POZ
|
3,19 × 10-8
|
POZ (21/24)
|
POZ
|
POZ
|
|
8
|
Apigenīns (1)
|
520-36-5
|
POZ
|
1,31 × 10-7
|
5,71 × 10-7
|
POZ
|
1,60 × 10-6
|
POZ (26/26)
|
POZ
|
NT
|
|
9
|
Atrazīns (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Bisfenols A (1)
|
80-05-7
|
POZ
|
2,02 × 10-8
|
2,94 × 10-7
|
POZ
|
5,33 × 10-7
|
POZ (65/65)
|
POZ
|
POZ
|
|
11
|
Bisfenols B (1)
|
77-40-7
|
POZ
|
2,36 × 10-8
|
2,11 × 10-7
|
POZ
|
1,95 × 10-7
|
POZ (6/6)
|
POZ
|
POZ
|
|
12
|
Butilbenzilftalāts (1)
|
85-68-7
|
POZ
|
1,14 × 10-6
|
4,11 × 10-6
|
POZ
|
1,98 × 10-6
|
POZ (12/14)
|
POZ
|
NEG
|
|
13
|
Kortikosterons (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Kumestrols (1)
|
479-13-0
|
POZ
|
1,23 × 10-9
|
2,00 × 10-8
|
POZ
|
1,32 × 10-7
|
POZ (30/30)
|
POZ
|
NT
|
|
15
|
Daidzeīns (1)
|
486-66-8
|
POZ
|
1,76 × 10-8
|
1,51 × 10-7
|
POZ
|
7,95 × 10-7
|
POZ (39/39)
|
POZ
|
POZ
|
|
16
|
Dietilstilbestrols (1)
|
56-53-1
|
POZ
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POZ
|
3,34 × 10-11
|
POZ (42/42)
|
POZ
|
NT
|
|
17
|
Di-n-butilftalāts
|
84-74-2
|
POZ
|
4,09 × 10-6
|
|
POZ
|
4,09 × 10-6
|
POZ (6/11)
|
POZ
|
NEG
|
|
18
|
Etilparabēns
|
120-47-8
|
POZ
|
5,00 × 10-6
|
PC50 nav
|
POZ
|
2,48 × 10-5
|
POZ
|
|
NT
|
|
19
|
Estrons (1)
|
53-16-7
|
POZ
|
3,02 × 10-11
|
5,88 × 10-10
|
POZ
|
2,34 × 10-10
|
POZ (26/28)
|
POZ
|
POZ
|
|
20
|
Genisteīns (1)
|
446-72-0
|
POZ
|
2,24 × 10-9
|
2,45 × 10-8
|
POZ
|
2,71 × 10-7
|
POZ (100/102)
|
POZ
|
POZ
|
|
21
|
Haloperidols
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kampferols (1)
|
520-18-3
|
POZ
|
1,36 × 10-7
|
1,21 × 10-6
|
POZ
|
3,99 × 10-6
|
POZ (23/23)
|
POZ
|
NT
|
|
23
|
Kepons (1)
|
143-50-0
|
POZ
|
7,11 × 10-7
|
7,68 × 10-6
|
POZ
|
4,91 × 10-7
|
POZ (14/18)
|
POZ
|
NT
|
|
24
|
Ketokonazols
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linurons (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
mezo-heksestrols (1)
|
84-16-2
|
POZ
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POZ
|
1,65 × 10-11
|
POZ (4/4)
|
POZ
|
NT
|
|
27
|
Metiltestosterons (1)
|
58-18-4
|
POZ
|
1,73 × 10-7
|
4,11 × 10-6
|
POZ
|
2,68 × 10-6
|
POZ (5/6)
|
POZ
|
NT
|
|
28
|
Morīns
|
480-16-0
|
POZ
|
5,43 × 10-7
|
4,16 × 10-6
|
POZ
|
2,37 × 10-6
|
POZ (2/2)
|
POZ
|
NT
|
|
29
|
Noretinodrels (1)
|
68-23-5
|
POZ
|
1,11 × 10-11
|
1,50 × 10-9
|
POZ
|
9,39 × 10-10
|
POZ (5/5)
|
POZ
|
NT
|
|
30
|
p,p’-metoksihlors (1)
|
72-43-5
|
POZ
|
1,23 × 10-6
|
(PC50 nav) (2)
|
POZ
|
1,92 × 10-6
|
POZ (24/27)
|
POZ
|
POZ
|
|
31
|
Fenobarbitāls (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Rezerpīns
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolaktons (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Testosterons
|
58-22-0
|
POZ
|
2,82 × 10-8
|
9,78 × 10-6
|
POZ
|
1,75 × 10-5
|
POZ (5/10)
|
POZ
|
NT
|
|
Saīsinājumi: CAS Nr. = Chemical Abstracts Service reģistra numurs M = molārs; EC50 = testējamās vielas 50 % efektīvā koncentrācija; NEG = negatīvs; POZ = pozitīvs; NT = nav testēts; PC10 (un PC50) = testējamās vielas koncentrācija, pie kuras aktivitāte ir 10 % (vai 50 % PC50 gadījumā) no aktivitātes, ko izraisa pozitīvs rezultāts (E2, 1nM) katrā platē.
|
2. tabula
Salīdzinājums par STTA un VM7Luc ER TA testu rezultātiem attiecībā uz vielām, kas testētas abos antagonista testos un klasificētas kā ES antagonisti (POS) vai kā negatīvi rezultāti (NEG)
|
|
Viela (3)
|
CAS Nr.
|
ER STTA tests (39)
|
VM7Luc ER TA tests (40)
|
ER STTA sagaidāmie rezultāti (42)
|
ICCVAM (43) saskaņotā klasifikācija
|
MeSH (44) ķimikāliju klase
|
Produktu klase (45)
|
|
ER TA aktivitāte
|
IC50 vērtība (4) (M)
|
ER TA aktivitāte
|
IC50 vērtība (4), (41) (M)
|
|
1
|
4-hidroksitamoksifēns
|
68047-06-3
|
POZ
|
3,97 × 10-9
|
POZ
|
2,08 × 10-7
|
mēreni POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
2
|
Dibenz[a,h]antracēns
|
53-70-3
|
POZ
|
Nav IC50:
|
POZ
|
Nav IC50:
|
POZ
|
PP
|
Policiklisks savienojums
|
Laboratoriju ķimikālija, dabisks produkts
|
|
3
|
Mifepristons
|
84371-65-3
|
POZ
|
5,61 × 10-6
|
NEG
|
—
|
vāji POZ
|
NEG
|
Steroīds
|
Farmaceitisks līdzeklis
|
|
4
|
Raloksifēns HCl
|
82640-04-8
|
POZ
|
7,86 × 10-10
|
POZ
|
1,19 × 10-9
|
mēreni POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
5
|
Tamoksifēns
|
10540-29-1
|
POZ
|
4,91 × 10-7
|
POZ
|
8,17 × 10-7
|
POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
6
|
17β-estradiols
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Steroīds
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
7
|
Apigenīns
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterociklisks savienojums
|
Krāsviela, dabisks produkts, farmaceitisks starpprodukts
|
|
8
|
Atrazīns
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Heterociklisks savienojums
|
Herbicīds
|
|
9
|
Di-n-butilftalāts
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Esteris, ftalskābe
|
Kosmētikas līdzekļu sastāvdaļa, rūpnieciska ķimikālija, plastifikators
|
|
10
|
Fenarimols
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
nav testēts;
|
PN
|
Heterociklisks savienojums, pirimidīns
|
Fungicīds
|
|
11
|
Flavons
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
PN
|
PN
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts, farmaceitisks līdzeklis
|
|
12
|
Flutamīds
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
PN
|
Amīds
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
13
|
Genisteīns
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts, farmaceitisks līdzeklis
|
|
14
|
p-n-nonilfenols
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
nav testēts
|
NEG
|
Fenols
|
Ķimikāliju starpprodukts
|
|
15
|
Resveratrols
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
PN
|
NEG
|
Ogļūdeņradis (ciklisks)
|
Dabisks produkts
|
|
Saīsinājumi: CAS Nr. = Chemical Abstracts Service reģistra numurs M = molārs; EC50 = testējamās vielas 50 % maksimālā inhibējošā koncentrācija; NEG = negatīvs; PN = pieņem, ka negatīvs; POZ = pozitīvs; PP = pieņem, ka pozitīvs.
|
ER TA TESTA KOMPONENTI
Nepieciešamie testa komponenti
|
11.
|
Šo testēšanas metodi piemēro testiem, kuros izmanto stabili transficētu vai endogēnisku ERα receptoru un stabili transficētu reportiergēnu konstruktu viena vai vairāku estrogēniskas reakcijas elementu kontrolē; tomēr klātesoši var būt arī citi receptori, piemēram, ERβ. Tie ir nepieciešamie testa komponenti.
|
Kontroles
|
12.
|
Apraksta ierosinātos paralēlos references standartus katram agonista un antagonista testam. Paralēlās kontroles (negatīvs, šķīdinātājs, pozitīvs) vajadzības gadījumā kalpo kā indikators, ka tests funkcionē pie testēšanas nosacījumiem, un ļauj savstarpēji salīdzināt eksperimentus; parasti tie ir daļa no konkrētā eksperimenta pieņemamības kritērijiem (1).
|
Standarta kvalitātes kontroles procedūras
|
13.
|
Katram testam veic aprakstītās standarta kvalitātes kontroles procedūras, lai nodrošinātu, ka šūnu līnija vairāku pasāžu gaitā saglabājas stabila, nesatur mikoplazmu (t.i., nav kontaminēta ar baktērijām) un saglabā spēju laika gaitā nodrošināt gaidītās ER mediētās reakcijas. Jāpārbauda arī, vai šūnu līnijas identitāte ir pareiza un vai nav citu kontaminantu (piem., sēnīšu, raugu un vīrusu).
|
Laboratorijas kvalifikācijas pierādīšana
|
14.
|
Pirms nezināmu ķimikāliju testēšanas ar kādu no šajā testa metodē aprakstītajiem testiem katrai laboratorijai ir jāpierāda sava kompetence testa izmantošanā. Kompetences pierādīšanas labad katra laboratorija testē 14 standartvielas, ko izmanto agonistu testēšanā un kas norādītas 3 tabulā, un 10 standartvielas, ko izmanto antagonistu testēšanā un kas norādītas 4. tabulā. Kompetences pierādīšana arī ļauj pārliecināties par testa sistēmas reaģētspēju. Standartvielu testēšanas saraksts ir daļa no references vielām, kas norādītas ER TA testu veiktspējas standartos (6). Šīs vielas ir komerciāli pieejamas, tās ir reprezentatīvas tādu ķimikāliju klasēm, ko parasti saista ar ER agonistu vai antagonistu aktivitāti, tām ir pienācīgs potences diapazons, kas sagaidāms no ER agonistiem/antagonistiem (t.i., no spēcīga līdz vājam) un tās ietver negatīvas vielas. Standartvielas testē vismaz divas reizes dažādās dienās. Kompetence ir pierādīta, ja katra standartviela tiek pareizi klasificēta (pozitīva/negatīva reakcija). Kompetences testēšanu atkārto katrs laborants, kas apgūst testēšanas metodi. Atkarībā no šūnu tipa dažas standartvielas var izpausties kā SERM, proti, gan kā agonisti, gan kā antagonisti. Tomēr standartvielas 3. un 4. tabulā ir klasificētas pēc to zināmās dominējošās aktivitātes, un tieši pēc tās vadās kompetences izvērtēšanā.
|
|
15.
|
Kompetences pierādīšanas un kvalitātes kontroles vajadzīgām katra laboratorija izveido agonistu un antagonistu vēsturisko datubāzi ar šādiem datiem: references standartvielas (piem., 17β-estradiols un tamoksifēns), pozitīvās un negatīvās kontrolķimikālijas un šķīdinātāja kontrole (piem., DMSO). Sākumā datubāzi ģenerē no vismaz 10 neatkarīgām testa izpildes reizēm ar agonistiem (piem., 17β-estradiolu) un 10 neatkarīgām testa izpildes reizēm ar antagonistiem (piem., tamoksifēnu). Lai datubāzi paplašinātu un tādējādi nodrošinātu laboratorijas konsekventu sniegumu un kompetenci biotestu veikšanā, datubāzi papildina ar vēlāku references standartvielu un šķīdinātāja kontroļu analīžu rezultātiem.
|
3. tabula
Saraksts ar (14) standartvielām, ko izmanto agonistu testā
(46)
|
Nr. (53)
|
Viela
|
CAS Nr.
|
Sagaidāmā reakcija (47)
|
STTA tests
|
VM7Luc ER TA tests
|
MeSH ķimikāliju klase (51)
|
Produktu klase (52)
|
|
PC10 vērtība (M) (48)
|
PC50 vērtība (M) (48)
|
Testa koncentrācijas diapazons (M)
|
VM7Luc EC50 vērtība (M) (49)
|
Augstākā koncentrācija diapazona noteikšanai (M) (50)
|
|
14
|
Dietilstilbestrols
|
56-53-1
|
POZ
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis, veterinārfarmaceitisks līdzeklis
|
|
12
|
17α-estradiols
|
57-91-0
|
POZ
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroīds
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
15
|
mezo-heksestrols
|
84-16-2
|
POZ
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Ogļūdeņradis (ciklisks), fenols
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
11
|
4-terc-oktilfenols
|
140-66-9
|
POZ
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenols
|
Ķimikāliju starpprodukts
|
|
9
|
Genisteīns
|
446-72-0
|
POZ
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts, farmaceitisks līdzeklis
|
|
6
|
Bisfenols A
|
80-05-7
|
POZ
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenols
|
Ķimikāliju starpprodukts
|
|
2
|
Kampferols
|
520-18-3
|
POZ
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts
|
|
3
|
Butilbenzilftalāts
|
85-68-7
|
POZ
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Karbonskābe, esteris, ftalskābe
|
Plastifikators, rūpnieciska ķimikālija
|
|
4
|
p,p’- metoksihlors
|
72-43-5
|
POZ
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Ogļūdeņradis (halogenēts)
|
Pesticīds, veterinārfarmaceitisks līdzeklis
|
|
1
|
Etilparabēns
|
120-47-8
|
POZ
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Karbonskābe, fenols
|
Farmaceitisks līdzeklis
|
|
17
|
Atrazīns
|
1912-24-9
|
NEG
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Heterociklisks savienojums
|
Herbicīds
|
|
20
|
Spironolaktons
|
52-01-7
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Laktons, steroīds
|
Farmaceitisks līdzeklis
|
|
21
|
Ketokonazols
|
65277-42-1
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Heterociklisks savienojums
|
Farmaceitisks līdzeklis
|
|
22
|
Rezerpīns
|
50-55-5
|
NEG
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Heterociklisks savienojums, indols
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
Saīsinājumi: CAS Nr. = Chemical Abstracts Service reģistra numurs EC50 = testējamās vielas 50 % efektīvā koncentrācija; NEG = negatīvs; POZ = pozitīvs; PC10 (un PC50) = testējamās vielas koncentrācija, pie kuras aktivitāte ir 10 % (vai 50 % PC50 gadījumā) no aktivitātes, ko izraisa pozitīvs rezultāts (E2, 1nM) katrā platē.
|
4. tabula
Saraksts ar (10) standartvielām, ko izmanto antagonistu testā
|
|
Viela (5)
|
CAS Nr.
|
ER STTA tests (54)
|
VM7Luc ER TA tests (55)
|
ER STTA (54) sagaidāmie rezultāti
|
ICCVAM (58) saskaņotā klasifikācija
|
MeSH (59) ķimikāliju klase
|
Produktu klase (60)
|
|
ER TA aktivitāte
|
IC50 (M)
|
Testa koncentrācijas diapazons (M)
|
ER TA aktivitāte
|
IC50
(56) (M)
|
Augstākā koncentrācija diapazona noteikšanai (M) (57)
|
|
1
|
4-hidroksitamoksifēns
|
68047-06-3
|
POZ
|
3,97 × 10-9
|
10-12 – 10-7
|
POZ
|
2,08 × 10-7
|
2,58 × 10-4
|
mēreni POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
2
|
Raloksifēns HCl
|
82640-04-8
|
POZ
|
7,86 × 10-10
|
10-12 – 10-7
|
POZ
|
1,19 × 10-9
|
1,96 × 10-4
|
mēreni POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
3
|
Tamoksifēns
|
10540-29-1
|
POZ
|
4,91 × 10-7
|
10-10 – 10-5
|
POZ
|
8,17 × 10-7
|
2,69 × 10-4
|
POZ
|
POZ
|
Ogļūdeņradis (ciklisks)
|
Farmaceitisks līdzeklis
|
|
4
|
17β-estradiols
|
50-28-2
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,67 × 10-3
|
potenciāli negatīvs (*4)
|
PN
|
Steroīds
|
Farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
5
|
Apigenīns
|
520-36-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterociklisks savienojums
|
Krāsviela, dabisks produkts, farmaceitisks starpprodukts
|
|
6
|
Di-n-butilftalāts
|
84-74-2
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Esteris, ftalskābe
|
Kosmētikas līdzekļu sastāvdaļa, rūpnieciska ķimikālija, plastifikators
|
|
7
|
Flavons
|
525-82-6
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,50 × 10-4
|
potenciāli negatīvs (*4)
|
PN
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts, farmaceitisks līdzeklis
|
|
8
|
Genisteīns
|
446-72-0
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
3,70 × 10-4
|
potenciāli negatīvs (*4)
|
NEG
|
Flavonoīds, heterociklisks savienojums
|
Dabisks produkts, farmaceitisks līdzeklis
|
|
9
|
p-n-nonilfenols
|
104-40-5
|
NEG
|
—
|
10-9 – 10-4
|
NEG
|
—
|
4,54 × 10-4
|
nav testēts
|
NEG
|
Fenols
|
Ķimikāliju starpprodukts
|
|
10
|
Resveratrols
|
501-36-0
|
NEG
|
—
|
10-8 – 10-3
|
NEG
|
—
|
4,38 × 10-4
|
potenciāli negatīvs (*4)
|
NEG
|
Ogļūdeņradis (ciklisks)
|
Dabisks produkts
|
|
Saīsinājumi: CAS Nr. = Chemical Abstracts Service reģistra numurs M = molārs; EC50 = testējamās vielas 50 % maksimālā inhibējošā koncentrācija; NEG = negatīvs; PN = pieņem, ka negatīvs; POZ = pozitīvs.
|
Testa pieņemamības kritēriji
|
16.
|
Testa pieņemšana vai noraidīšana ir atkarīga no rezultātiem, kas iegūti ar katrā eksperimentā izmantotajiem references standartiem un kontrolēm. References standartvielu vērtībām PC50 (EC50) vai IC50 būtu jāatbilst pieņemamības kritērijiem, kas paredzēti izvēlētajam testam (par STTA sk. 2. papildinājumu, par VM7Luc ER TA sk. 3. papildinājumu), un visas pozitīvās/negatīvās kontroles būtu pareizi jāklasificē attiecībā uz katru eksperimentu, kas atzīts par pieņemamu. Laboratorija savu spēju pastāvīgi veikt testu pierāda, izveidojot un uzturot datubāzi, kurā apkopoti references standartvielu un kontroļu vēsturiskie rezultāti (sk. 15. punktu). Kā pasākumu, ar ko nodrošina reproducējamību laboratorijas robežās, var izmantot references standartu līknes piemeklēšanas parametru vidējo vērtību standartnovirzes (SN) vai variācijas koeficientus (VK), kas iegūti daudzos eksperimentos. Bez tam attiecībā uz pieņemamības kritērijiem ir jāievēro šādi principi.
|
—
|
Datiem jābūt pietiekamiem, lai varētu kvantitatīvi novērtēt ER aktivāciju (agonistu testā) vai inhibīciju (antagonistu testā) (t.i., efektivitāti un potenci).
|
|
—
|
Lai garantētu adekvātu jutību, marķiera vidējai aktivitātei pie references estrogēna references koncentrācijas jābūt vismaz testēšanas metodes paredzētajai minimālajai aktivitātei attiecībā pret nesēja (šķīdinātāja) kontroles marķiera vidējo vērtību. Testos STTA un VM7Luc ER TA šī minimālā vērtība ir četras reizes lielāka par nesēja kontroles vidējo vērtību katrā platē.
|
|
—
|
Testētajām koncentrācijām jāsaglabājas testa ķimikāliju šķīdības diapazonā un tās nedrīkst būt citotoksiskas.
|
|
Datu analīze
|
17.
|
Lai noteiktu, vai reakcija ir pozitīva vai negatīva, izmanto katram testam definēto datu interpretēšanas procedūru.
|
|
18.
|
Ja ir izpildīti pieņemamības kritēriji (16. punkts), tas nozīmē, ka tests darbojas pareizi, tomēr tas negarantē, ka konkrētajā izpildes reizē tiks iegūti pareizi dati. Labākais pierādījums iegūto datu pareizībai ir pirmās izpildes reizes rezultātu replicēšana. Ja divās izpildes reizēs tiek iegūti reproducējami rezultāti (piem., rezultāti abās izpildes reizēs liecina, ka testa ķimikālija ir pozitīva), nav nepieciešams testu izpildīt vēl trešo reizi.
|
|
19.
|
Ja divās izpildes reizēs netiek netiek iegūti reproducējami rezultāti (piem., testa ķimikālija ir pozitīva vienā izpildes reizē, bet negatīva otrā) vai ka ir nepieciešama lielāka pārliecība par šā testa iznākumu, vajadzīgas vismaz trīs neatkarīgas izpildes reizes. Tādā gadījumā klasifikācijas pamatā ir divi sakritīgie rezultāti no trim.
|
Vispārīgie datu interpretācijas kritēriji
|
20.
|
Pašlaik nav vispārpieņemtas metodes, kā interpretēt ER TA datus. Tomēr gan kvalitatīvajos (piem., pozitīvs/negatīvs), gan kvantitatīvajos (piem., EC50, PC50, IC50) ER mediētās aktivitātes novērtējumos ir jābalstās uz empīriskiem datiem un pamatotiem zinātniskiem spriedumiem. Ja iespējams, pozitīvie rezultāti jāraksturo gan ar ietekmes intensitāti salīdzinājumā ar nesēja (šķīdinātāja) kontroli vai references estrogēnu, gan ar koncentrāciju, pie kuras novērojama ietekme (piem., EC50, PC50, RPCMax, IC50 u.c.).
|
Testēšanas pārskats
|
21.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Tests:
|
—
|
kontrole/ references standarts/ testējamā ķimikālija,
|
|
—
|
avots, sērijas numurs, izmantošanas termiņš, ja pieejams,
|
|
—
|
pašas testējamās ķimikālijas stabilitāte, ja zināma,
|
|
—
|
testējamās ķimikālijas šķīdība un stabilitāte šķīdinātājā, ja tā zināma,
|
|
—
|
(attiecīgā gadījumā) pH, osmolalitātes un izgulsnēšanās mērījums kultūras barotnē, kurā testējamā ķimikālija pievienota.
|
|
|
|
Vienkomponenta viela.
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības,
|
|
—
|
ķīmiskā identifikācija, piemēram, IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, attiecīgā gadījumā, ja praktiski iespējams, piemaisījumu ķīmiskā identitāte u. tml.
|
|
|
|
Daudzkomponentu viela, UVCB un maisījumi:
|
—
|
iespējami izsmeļoši raksturoti pēc komponentu ķīmiskās identitātes (sk. iepriekš), kvantitatīvās sastopamības un relevantajām fizikālķīmiskajām īpašībām.
|
|
|
|
Šķīdinātājs/nesējs:
|
—
|
raksturojums (iedaba, piegādātājs un sērija),
|
|
—
|
šķīdinātāja/nesēja izvēles pamatojums,
|
|
—
|
testējamās ķimikālijas šķīdība un stabilitāte šķīdinātājā/nesējā, ja tā zināma.
|
|
|
|
Šūnas:
|
—
|
šūnu tips un avots:
|
—
|
Vai ER ir endogēniski ekspresēts? Ja nē, tad kuri receptori tikuši transficēti?
|
|
—
|
Izmantotie reportiergēnu konstrukti (t.sk. avotsugas);
|
|
—
|
Atlases metode, ko izmanto stabilas transfekcijas uzturēšanai (attiecīgā gadījumā);
|
|
—
|
Vai transfekcijas metode ļauj iegūt stabilas līnijas?
|
|
|
—
|
šūnu pasāžu skaits (pēc atkausēšanas);
|
|
—
|
šūnu pasāžu skaits (atkausēšanas brīdī);
|
|
—
|
šūnu kultūru audzēšanas metodes.
|
|
|
|
Testēšanas nosacījumi:
|
—
|
apraksts par izmantotajām dzīvotspējas novērtēšanas metodēm;
|
|
—
|
Barotnes sastāvs, CO2 koncentrācija;
|
|
—
|
testējamās ķimikālijas koncentrācija;
|
|
—
|
pievienotā nesēja un testējamās ķimikālijas tilpums;
|
|
—
|
inkubācijas temperatūra un mitrums;
|
|
—
|
šūnu blīvums apstrādes sākumā un gaitā;
|
|
—
|
pozitīvie un negatīvie references standarti;
|
|
—
|
marķierreaģenti (produkta nosaukums, piegādātājs, sērija);
|
|
—
|
kritēriji, pēc kuriem novērtē, vai testa rezultāts ir pozitīvs, negatīvs vai neskaidrs.
|
|
|
|
Pieņemamības pārbaude
|
—
|
indukcija kārtās katrā testa platē un tas, vai tiek izpildīts testā vajadzīgais minimums, pamatojoties uz vēsturiskajiem datiem par kontrolēm;
|
|
—
|
pieņemamības kritēriju faktiskās vērtības, piem., log10EC50, log10PC50, logIC50 un Hilla virziena koeficienta vērtības attiecībā uz testā paralēli iekļautajām pozitīvajām kontrolēm/references standartiem.
|
|
|
|
Rezultāti:
|
—
|
jēldati un normalizēti dati;
|
|
—
|
maksimālais vairākkāršās indukcijas līmenis;
|
|
—
|
dati par citotoksicitāti;
|
|
—
|
zemākā iedarbīgā koncentrācija (LEC), ja tāda ir;
|
|
—
|
attiecīgā gadījumā — RPCMax, PCMax, PC50, IC50 un/vai EC50 vērtības;
|
|
—
|
ja iespējams, koncentrācijas–atbildreakcijas sakarība,
|
|
—
|
statistiskās analīzes (ja tādas ir), kā arī kļūdas un ticamības mērs (piem., SEM, SN, VK vai 95 % CI) un apraksts par to, kā šīs vērtības iegūtas.
|
|
|
LITERATŪRA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERSTARTSYMBOLFONT003FENDSYMBOLFONTSTARTSYMBOLFONT003FENDSYMBOLFONTIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
1. papildinājums
DEFINĪCIJAS UN SAĪSINĀJUMI
Pieņemamības kritēriji
: minimālie standarti, kas attiecas uz eksperimenta kontrolēm un references standartiem. Lai eksperimentu uzskatītu par derīgu, visiem pieņemamības kritērijiem jābūt izpildītiem.
Pareizība (konkordantums)
: pakāpe, kādā testa rezultāti sakrīt ar pieņemtajām atsauces vērtībām. Tas ir testa veiktspējas raksturlielums un viens no tā relevantuma aspektiem. Ar šo terminu, tāpat kā terminu “konkordantums”, bieži vien saprot testa pareizo iznākumu īpatsvaru (1).
Agonists
: viela, kas piesaistē konkrētam receptoram rada reakciju, piemēram, transkripciju.
Antagonists
: tāda veida receptora ligands vai ķimikālija, kas piesaistē receptoram pats(-i) par sevi neizraisa bioloģisku reakciju, bet gan bloķē vai mīkstina agonista mediētas reakcijas.
Antiestrogēniska aktivitāte
: ķimikālijas spēja nomākt 17β-estradiola aktivitāti, ko mediē estrogēnu receptori.
Šūnu morfoloģija
: tādu šūnu forma un izskats, kas monoslānī audzētas audu kultūras plates vienā iedobē. Mirstošām šūnām bieži novērojama anormāla šūnu morfoloģija.
KS
: ESAO konceptuālais satvars (KS) endokrīnās sistēmas traucējumus izraisošu ķimikāliju testēšanai un novērtēšanai.
Apstrāde ar ogli/dekstrānu
: šūnu kultūrā izmantota seruma apstrāde. Apstrāde ar ogli/dekstrānu (bieži vien to sauc arī par “stripingu”) likvidē endogēniskos hormonus un hormonus piesaistošus proteīnus.
Ķimikālija
: viela vai maisījums.
Citotoksicitāte
: kaitīga ietekme uz šūnas struktūru vai funkcionalitāti, kas galu galā var novest pie šūnas nāves; to var atspoguļot šūnu skaita samazinājums iedobē ekspozīcijas perioda beigās vai šūnu funkcijas kapacitātes samazinājums salīdzinājumā ar paralēlo nesēja kontroli.
VK
: variācijas koeficients.
DCC-FBS
: liellopu embriju serums, kas apstrādāts ar dekstrāna pārklātu ogli.
DMEM
: Dulbeko modificētā Īgla barotne.
DMSO
: dimetilsulfoksīds.
E2
: 17β-estradiols.
EC50
: testējamās ķimikālijas 50 % efektīvā koncentrācija.
ED
: endokrīnās sistēmas darbības traucējumi.
hERα
: cilvēka estrogēnu receptors alfa
hERß
: cilvēka estrogēnu receptors beta
EFM
: bezestrogēna barotne. Dulbeko modificētā Īgla barotne (DMEM), kas papildināta ar 4,5 % DCC-FBS, 1,9 % L-glutamīnu un 0,9 % penicilīna un streptomicīna maisījumu.
ER
: estrogēna receptors
ERE
: estrogēna atbildreakcijas elements
Estrogēniska aktivitāte
: ķimikālijas spēja imitēt 17β-estradiolu, proti, piesaistīties estrogēnu receptoriem un aktivēt tos. Ar šo testēšanas metodi var konstatēt hERα mediētu estrogēnisku aktivitāti.
ERTA
: estrogēna receptora transaktivācija
FBS
: liellopu embriju serums
HeLa
: nemirstīga cilvēka dzemdes kakla šūnu līnija
HeLa9903
: HeLa šūnu apakšklons, kurā stabili transficēts hERαα un luciferāzes reportiergēns
IC
50
: testējamās inhibējošās ķimikālijas 50 % iedarbīgā koncentrācija.
ICCVAM
: Alternatīvo metožu validēšanas koordinēšanas starpaģentūru komiteja.
Starplaboratoriju reproducējamība
: lielums, kas raksturo, kādā mērā vairākas kvalificētas laboratorijas spēj iegūt kvalitatīvi un kvantitatīvi līdzīgus rezultātus, ja tās izmanto vienu un to pašu protokolu un vienas un tās pašas testējamās vielas. Starplaboratoriju reproducējamību (saukta arī par interlaboratoriju reproducējamību) nosaka prevalidēšanas un validēšanas procesā, un šis lielums rāda, cik lielā mērā testēšanu vienlīdz sekmīgi iespējams veikt dažādās laboratorijās (1).
Iekšlaboratorijas reproducējamība
: lielums, kas raksturo, kādā mērā vienā laboratorijā strādājoši kvalificēti darbinieki spēj sekmīgi atkārtot iepriekš iegūtus rezultātus, ja to pašu protokolu izmanto citā laikā. Saukts arī par intralaboratoriju reproducējamību (1).
LEC
: zemākā iedarbīgā koncentrācija, t.i., zemākā koncentrācija, kurā testējamā ķīmikālija izraisa reakciju (t.i., testa ķimikālijas zemākā koncentrācija, pie kuras indukcija kārtās ir statistiski atšķirīga no paralēlās nesēja kontroles).
Atdarinājuma tests
: sadzīvisks apzīmējums testam, kas ir noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgs validētai un pieņemtai testēšanas references metodei. Brīvi aizstājama ar līdzīgu testēšanas metodi.
MT
: metalotionīns
MMTV
: peļu piena dziedzeru audzēja vīruss
OHT
: 4-hidroksitamoksifēns
PBTG
: veiktspējā balstītas testēšanas vadlīnijas
(PC) Pozitīvā kontrole:: ļoti aktīva viela — vēlams, 17ß-estradiols —, ko iekļauj visos testos un kas palīdz garantēt testa pienācīgu funkcionēšanu.
PC
10
: testējamās ķimikālijas koncentrācija, pie kuras izmērītā aktivitāte agonista testā ir 10 % no maksimālās aktivitātes, ko inducē PC (testā STTA — E2 pie 1nM) katrā iedobē.
PC
50
: testējamās ķimikālijas koncentrācija, pie kuras izmērītā aktivitāte agonista testā ir 50 % no maksimālās aktivitātes, ko inducē PC (E2 pie testēšanas metodē norādītās references koncentrācijas) katrā iedobē.
PC
Max
: testējamās ķimikālijas koncentrēšana, kas inducē RPCMax
Veiktspējas standarti
: uz validēta testa bāzēti standarti, pēc kuriem izvērtē pēc noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgu testu salīdzināmību. Tie ietver 1) nepieciešamos testa komponentus; 2) minimālo tādu references ķimikāliju sarakstu, kuras izraudzītas no citām ķimikālijām, ko izmanto, lai pierādītu pieņemamu validētās testēšanas metodes veiktspēju; kā arī 3) uz attiecīgajā validētajā testēšanas metodē iegūto rezultātu pamata — salīdzināmi pareizības un ticamības līmeņi, kā būtu jāiegūst ar ierosināto testu, to izvērtējot ar references ķimikālijām no minimālā saraksta (1).
Standartvielas
: veiktspējas standartos iekļauto references vielu apakškopa, ko laboratorijas var izmantot, lai pierādītu savu tehnisko kompetenci ar standartizētu testēšanas metodi. Šādu vielu atlases kritēriji parasti ir, piemēram šādi: viela ir reprezentatīva dažādām reakcijām, ir komerciāli pieejama un un par to pie pieejami ļoti kvalitatīvi references dati.
Kompetence
: vēl pirms nezināmu vielu testēšanas pierādīta spēja testu pienācīgi veikt.
References estrogēns (pozitīvā kontrole, PC)
: 17β-estradiols (E2, CAS 50-28-2).
References standarts
: references viela, ko izmanto, lai pierādītu testa piemērotību. Testos STTA un VM7Luc ER TA references standarts ir 17β-estradiols.
References testēšanas metodes
: testi, uz kuriem balstās PBTG 455.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Tā ir pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testa pareizības (konkordantuma) aspektu (1).
Ticamība
: parametrs, kas izsaka, kādā pakāpē vienā un tajā pašā laboratorijā vai dažādās laboratorijās pēc vienu un tā paša protokola var ilgstoši reproducējami veikt konkrētu testu. To novērtē, aprēķinot reproducējamību vienā laboratorijā un vairākās laboratorijās.
RLU
: relatīvās gaismas vienības
RNS
: ribonukleīnskābe
RPC
Max
: testējamās ķimikālijas inducētās atbildreakcijas maksimālais līmenis, ko izsaka kā procentus no 1nM, E2 inducētās atbildreakcijas tajā pašā platē.
RPMI
: RPMI 1 640 barotne, kas papildināta ar 0,9 % penicilīna un streptomicīna maisījumu un 8,0 % liellopu embriju serumu (FBS).
Izpildes reize
: atsevišķs eksperiments, kurā izvērtē ķīmisko iedarbību uz testa bioloģisko iznākumu. Katra izpildes reize ir pilns eksperiments, ko izdara replikāta iedobēs vienlaikus un ar šūnām no tā paša šūnu krājuma.
Neatkarīga izpildes reize
: atsevišķs, neatkarīgs eksperiments, kurā izvērtē ķīmisko iedarbību uz testa bioloģisko iznākumu; izmanto šūnas no dažādiem krājumiem, svaigi atšķaidītas ķimikālijas un testu izdara vai nu dažādās dienās, vai tajā pašā dienā dažādi laboranti.
SN
: standartnovirze
Jutība
: tas, cik liela daļa no visām pozitīvajām/aktīvajām vielām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (1).
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām vielām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (1).
Stabila transfekcija
: norise, kuras laikā DNS tiek transficēta kultivētajās šūnās tādā veidā, ka tiek stabili integrēta šūnas genomā, kā rezultātā ir iespējama transficēto gēnu stabila ekspresija. Stabili transficētu šūnu klonus atlasa ar stabiliem marķieriem (piem., pretestība G418).
Tests STTA
: stabili transficētas transaktivācijas tests, ERα transkripcionālās aktivācijas tests, izmantojot šūnu līniju HeLa 9 903.
Pētījums
: visi eksperimentālā darba posmi, kas izpildīti, lai specifiskā testā izvērtētu vienu atsevišķu, specifisku vielu. Pētījumā ietilpst visi eksperimenta posmi, tostarp testējamās vielas atšķaidīšana testa barotnē, provizoriskie diapazona noteikšanas testi, visi nepieciešamie visaptverošie testi, datu analīze, kvalitātes nodrošināšana, citotoksicitātes novērtēšana utt. Pētījuma pabeigšana ļauj klasificēt, kā testējamā ķimikālija iespaido testā novērtēto toksiskumu (t.i., aktīva, neaktīva, neviennozīmīga), un raksturot atbildreakciju attiecībā pret pozitīvo references ķimikāliju.
Viela
: REACH regulā (61) viela definēta kā ķīmisks elements un tā dabiski vai ražošanas procesā iegūti savienojumi, tostarp tās stabilizācijai vajadzīgās piedevas, kā arī izmantotajos procesos radušies piejaukumi, kas nav šķīdinātāji, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu. Ļoti līdzīga definīcija tiek izmantota ANO GHS (1).
TA (transaktivācija):: mRNS sintēzes iniciēšana kā atbildreakcija uz specifisku ķīmisko signālu, piemēram, estrogēna piesaisti estrogēna receptoram.
Tests
: šīs testēšanas metodes kontekstā tests ir viena no metodikām, kas atzīta par derīgu, proti, kas izpilda norādītos veiktspējas kritērijus. Testa komponenti ir, piemēram, specifiska šūnu līnija un attiecīgie augšanas apstākļi, specifiska barotne, kurā veic testu, plašu sagatavošanas nosacījumi, testējamo ķimikāliju sagatavošana un atšķaidīšana, kā arī visi citi vajadzīgie kvalitātes kontroles pasākumi un saistītie datu izvērtēšanas posmi.
Testējamā ķimikālija
: jebkura viela vai maisījums, kuru testē, izmantojot šo testēšanas metodi.
Transkripcija
: mRNS sintēze
UVCB
: ķīmiskas vielas, kuru sastāvs nav zināms vai ir mainīgs, kompleksi reakcijas produkti un bioloģiski materiāli.
Validēta testēšanas metode
: tests, kura relevantums (ieskaitot pareizību) un ticamība attiecībā uz lietošanu konkrētā nolūkā ir pārbaudīta validācijas pētījumos. Jāievēro, ka validētas testēšanas metodes veiktspēja pareizības un ticamības ziņā var nebūt pietiekama, lai to izmantotu paredzētajam nolūkam (1).
Validācija
: Process, kurā nosaka, kādā mērā konkrēta pieeja, metode, tests, process vai novērtējums ir ticams un relevants konkrētajam nolūkam (1).
VC (nesēja kontrole)
: šķīdinātāju, kurā izšķīdina testējamās un kontroles ķimikālijas, testē tikai kā nesēju bez izšķīdinātas ķimikālijas.
VM7
: nemirstīga adenokarcinomas šūna, kas endogēniski ekspresē estrogēnu receptorus.
VM7Luc4E2
: Šūnu līnija VM7Luc4E2 atvasināta no VM7 nemirstīgām cilvēka adenokarcinomas šūnām, kas spēj endogēniski ekspresēt abas estrogēnu receptoru formas (ERα and ERβ) un ir stabili transficētas ar plazmīdu pGudLuc7.ERE. Plazmīds satur četras kopijas no sintezēta oligonukleotīda, kas satur estrogēna atbildreakcijas elementu virs MMTV promotera un jāņtārpiņa luciferāzes gēnu.
Vāji pozitīvā kontrole
: vāji aktīva viela, kas izvēlēta no references ķimikāliju saraksta; to iekļauj visos testos un tā palīdz garantēt testa pienācīgu funkcionēšanu.
2. papildinājums
STABILI TRANSFICĒTAS CILVĒKA ESTROGĒNU RECEPTORU Α TRANSAKTIVĀCIJAS TESTS, KURĀ NOSAKA ĶIMIKĀLIJU AGONISTU UN ANTAGONISTU ESTROGĒNISKO AKTIVITĀTI, IZMANTOJOT ŠŪNU LĪNIJU HERΑ-HELA-9903
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI (SK. ARĪ VISPĀRĪGO IEVADU)
|
1.
|
Šajā transaktivācijas (TA) testā šūnu līniju hERα-HeLa-9 903 izmanto, lai noteiktu estrogēnisku agonista aktivitāti, ko mediē cilvēku estrogēnu receptors alfa (hERα). Japānas Ķimikāliju izvērtēšanas un izpētes institūta (CERI) veiktajā stabili transficētas transaktivācijas (STTA) validācijas pētījumā, kurā šūnu līniju ERα-HeLa-9 903 izmantoja, lai noteiktu estrogēnisku agonista un antagonista aktivitāti, ko mediē cilvēku estrogēnu receptors alfa (hERα), pierādījās, ka tests ir ticams un relevants konkrētajam nolūkam (1).
|
|
2.
|
Šis tests ir īpaši paredzēts hERα-mediētas TA noteikšanai, par mērījuma parametru izmantojot hemiluminiscenci. Tomēr pie tādām fitoestrogēnu koncentrācijām, kas pārsniedz 1 μM, ir novēroti receptoru nemediēti luminiscences signāli, ko izraisa luciferāzes reportiergēna pārmērīga aktivācija. Lai gan devas-efekta līkne liecina, ka patiesa ER sistēmas aktivācija notiek pie zemākām koncentrācijām, ir lietderīgi rūpīgi izpētīt tādu luciferāzes ekspresiju stabili transficētās ER TA testu sistēmās (1. papildinājums), ko iegūst pie augstām fitoestrogēnu koncentrācijām vai tādu savienojumu koncentrācijām, par kuriem ir aizdomas, ka tie līdzīgi kā fitoestrogēni izraisa luciferāzes reportiergēna pārmērīgu aktivāciju.
|
|
3.
|
Pirms šā testa izmantošanas regulatīviem mērķiem ir jāizlasa sadaļas “VISPĀRĪGAIS IEVADS” un “ER TA testa KOMPONENTI”. Šajā testēšanas vadlīnijā izmantotās definīcijas un saīsinājumi ir aprakstīti 2.1. papildinājumā.
|
TESTA PRINCIPS (SK. ARĪ VISPĀRĪGO IEVADU)
|
4.
|
Testu izmanto, lai signalizētu, ka estrogēnu receptors ir piesaistījies ligandam. Pēc liganda piesaistes receptora-liganda komplekss translocējas uz kodolu, kur tas piesaistās specifiskiem DNS reakcijas elementiem un transaktivē jāņtārpiņa luciferāzes reportiergēnu, kā rezultātā palielinās luciferāzes fermenta ekspresija šūnās. Luciferīns ir substrāts, kuru luciferāzes enzīms pārveido bioluminiscējošā produktā, ko var kvantitatīvi izmērīt ar luminometru. Luciferāzes aktivitāti var ātri un lēti izvērtēt ar vairākiem komerciāli pieejamiem testa komplektiem.
|
|
5.
|
Testa sistēmā izmanto šūnu līniju hERα-HeLa-9 903, kas iegūta no cilvēka dzemdes kakla audzēja un kam ir divi stabili insertēti konstrukti: i) hERαα ekspersijas konstrukts (kura cilvēka receptors iekodēts pilnībā) un ii) jāņtārpiņa luciferāzes reportiergēna konstrukts, kam ir pieci vitelogenīna estrogēna atbildreakcijas elementa (ERE) tandēmie atkārtojumi, ko regulē peļu metalotionīna (MT) promoters ar TATA elementu. Peļu MT TATA gēna konstrukts līdz šim izrādījies visrezultatīvākais, tāpēc tieši to parasti izmanto. Tāpēc šī šūnu līnija hERα-HeLa-9 903 ļauj izmērīt testējamās ķimikālijas spēju inducēt luciferāzes gēna ekspresijas hERα mediētu transaktivāciju.
|
|
6.
|
ER agonista testā datu interpretācijas pamatā ir tas, vai testējamās ķimikālijas inducētais maksimālais abildreakcijas līmenis ir vienāds ar vai lielāks par agonista atbildreakciju, kas ir vienāda ar 10 % no atbildreakcijas, ko izraisa koncentrācija, kura inducē pozitīvās kontroles (PC) (17β-estradiols (E2)) maksimālo atbildreakciju (1 nM) (t.i.,. the PK10). ER antagonista testā datu interpretācijas pamatā ir tas, vai atbildreakcija parāda vismaz 30 % aktivitātes samazinājumu salīdzinājumā ar atbildreakciju, ko inducē ar kontroles pastiprinājumu (25 pM, E2), neizraisot citotoksicitāti. Datu analīze un interpretācija sīkāk iztirzāta 34.–48. punktā.
|
PROCEDŪRA
Šūnu līnijas
|
7.
|
Testam izmanto stabili transficēto šūnu līniju hERα-HeLa-9 903. Šūnu līniju var iegūt no Japānas Pētniecības bioresursu kolekcijas (JCRB) šūnu bankas (62), parakstot materiālu nodošanas līgumu.
|
|
8.
|
Testēšanā izmanto tikai tādas šūnas, kas nesatur mikoplazmu. Vislabākā un jutīgākā metode, kā noteikt inficēšanos ar mikoplazmu, ir RT-PCR (polimerāzes ķēdes reakcija reāllaikā) (4) (5) (6).
|
Šūnu līnijas stabilitāte
|
9.
|
Lai kontrolētu šūnu līnijas stabilitāti, agonista testā par references standartu izmanto E2, 17α-estradiolu, 17α-metiltestosteronu un kortikosteronu; tāpat vismaz vienu reizi testa veikšanas laikā mēra visu koncentrācijas–atbildreakcijas līkni pie 1. tabulā norādītajām testa koncentrācijām, un iegūtajiem rezultātiem jābūt saskanīgiem ar 1. tabulā norādītajiem rezultātiem.
|
|
10.
|
Antagonista testā vienlaikus paralēli katrai testa izpildes reizei mēra arī divu references standartu — tamoksifēna un flutamīda — pilnas koncentrācijas līknes. Pārliecinās, vai abu ķimikāliju kvalitatīvā klasifikācija (pozitīvs vai negatīvs) ir pareiza.
|
Šūnu kultivēšanas un uzsēšanas apstākļi
|
11.
|
Šūnas uztur fenolsarkano nesaturošā Īgla minimālajā barotnē (EMEM), kas papildināta ar 60 mg/l antibiotiku (kanamicīnu) un 10 % liellopu embriju serumu, kurš apstrādāts ar dekstrāna pārklātu ogli (DCC-FBS), CO2 inkubatorā (5 % CO2) 37±1°C temperatūrā. Kad sasniegta 75 -90 % konfluence, šūnas var sadalīt 10 ml subkultūrās, kas satur 0,4 x 105 – 1 x 105 šūnas/ml, kultivēšanas trauciņos, kuru diametrs ir100 mm. Šūnas suspendē 10 % FBS-EMEM barotnē (kas ir līdzvērtīga EMEM un DCC-FBS barotnei) un tad uzsēj mikroplates iedobēs tā, lai blīvums būtu 1 x 104 šūnas/(100 μl x iedobe). Tad pirms eksponēšanas ķimikālijai šūnas 3 stundas preinkubē 5 % CO2 inkubatorā 37°±1°C temperatūrā. Plastmasas materiālos nedrīkst notikt nekāda estrogēniska aktivitāte.
|
|
12.
|
Lai saglabātu reakcijas integritāti, šūnas audzē vairākās pasāžās no sasaldēta krājuma kondicionētā barotnē, un pasāžu skaits nedrīkst pārsniegt 40. Attiecībā uz šūnu līniju hERα-HeLa-9 903 tas būs mazāk par trim mēnešiem. Tomēr, ja šūnas audzē nepienācīgos kultivēšanas apstākļos, šūnu sniegums var pasliktināties.
|
|
13.
|
DCC-FBS var vai nu sagatavot tā, kā aprakstīts 2.2. papildinājumā, vai iegūt no komerciāliem avotiem.
|
Pieņemamības kritēriji
Pozitīvi un negatīvi references standarti ER agonistu testā
|
14.
|
Pirms pētījuma un tā laikā ir jāverificē testa sistēmas reaģētspēja,, izmantojot stipru estrogēnu (E2), vāju estrogēnu (17α-estradiolu), ļoti vāju agonistu (17α-metiltestosteronu) un negatīvu vielu (kortikosteronu) piemērotā koncentrācijā. 1. tabulā sniegts pieņemamo vērtību diapazons, kas sagatavots validācijas pētījuma (1) gaitā. Šos 4 paralēlos references standartus iekļauj katrā eksperimentā, un rezultātiem jāietilpst dotajās pieņemamajās robežās. Ja tā nav, tad jānoskaidro, kāpēc pieņemamības kritēriji nav izpildīti (iemesli var būt, piem., manipulācijas ar šūnām vai seruma un antibiotiku kvalitāte un koncentrācija), un tests jāatkārto. Ja pieņemamības kritēriji ir izpildīti, tad ļoti svarīga ir šūnu kultivēšanas materiālu konsekventa izmantošana, lai nodrošinātu minimālu EC50, PC50 un PC10 vērtību mainību. Četri paralēlie references standarti, kas jāiekļauj katrā eksperimentā (ko veic identiskos apstākļos, tostarp ar tādiem pašiem materiāliem, šūnu pasāžas līmeni un laborantiem), var nodrošināt testa jutību, jo pieņemamā diapazonā jābūt gan trīs pozitīvo references standartu PC10, gan PC50 un EC50 vērtībām, ja tās var aprēķināt (sk. 1. tabulu).
|
1. tabula.
ER agonista testa četru references standartu pieņemamo vērtību diapazons
|
Nosaukums
|
logPC50
|
logPC10
|
logEC50
|
Hilla virziena koeficients
|
Testa diapazons
|
|
17β-estradiols (E2) CAS Nr.: 50-28-2
|
-11,4~-10,1
|
<-11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-estradiols CAS Nr.: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Kortikosterons CAS Nr.: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-metiltestosterons CAS Nr.: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Pozitīvi un negatīvi references standarti ER antagonistu testā
|
15.
|
Pirms pētījuma un tā laikā ir jāverificē testa sistēmas reaģētspēja, izmantojot pozitīvu vielu (tamoksifēns) un negatīvu vielu (flutamīds) piemērotā koncentrācijā. 2. tabulā sniegts pieņemamo vērtību diapazons, kas sagatavots validācijas pētījuma (1) gaitā. Šos divus paralēlos references standartus iekļauj katrā eksperimentā, un rezultāti, kā norādīts kritērijos, būtu jānovērtē par pareiziem. Ja tā nav, tad jānoskaidro, kāpēc kritēriji nav izpildīti (iemesli var būt, piem., manipulācijas ar šūnām vai seruma un antibiotiku kvalitāte un koncentrācija), un tests jāatkārto. Bez tam jāaprēķina arī pozitīvās vielas (tamoksifēna) IC50 vērtības, un rezultātiem ir jāietilpst dotajās pieņemamības robežās. Ja pieņemamības kritēriji ir izpildīti, tad ļoti svarīga ir šūnu kultivēšanas materiālu konsekventa izmantošana, lai nodrošinātu minimālu IC50 vērtību mainību. Divi paralēlie references standarti, kas jāiekļauj katrā eksperimentā (ko veic identiskos apstākļos, tostarp ar tādiem pašiem materiāliem, šūnu pasāžas līmeni un laborantiem), var nodrošināt testa jutību (sk. 2. tabulu).
|
2. tabula.
ER antagonista testa divu references standartu pieņemamo vērtību diapazons
|
Nosaukums
|
Kritēriji
|
LogIC50
|
Testa diapazons
|
|
Tamoksifēns CAS Nr.: 10540-29-1
|
Pozitīva: jāaprēķina IC50
|
-5,942 ~ -7,596
|
10-10 ~ 10-5 M
|
|
Flutamīds CAS Nr.: 13311-84-7
|
Negatīva: nav jāaprēķina IC30
|
—
|
10-10 ~10-5M
|
Pozitīvās kontroles un nesēja kontroles
|
16.
|
ER agonista testa (1 nM, E2) un ER antagonista testa (10μM TAM) pozitīvās kontrole (PC) katrā platē jātestē vismaz trīskārši. Nesēju, kurā izšķīdina testējamo ķimikāliju, testē kā nesēja kontroli (VC) katrā platē vismaz trīskārši. Papildus šai VC, ja PC izmanto citu nesēju, nevis testējamo ķimikāliju, šī cita nesēja kontrole ir jātestē vismaz trīskārši tajā pašā platē kopā ar PC.
|
ER agonista testa kvalitātes kritēriji
|
17.
|
Pozitīvās kontroles (1 nM, E2) vidējai luciferāzes aktivitātei jābūt vismaz 4 reizes lielākai par vidējo VC katrā platē. Šis kritērijs izvēlēts, pamatojoties uz novērtēšanas parametru uzticamību validācijas pētījumā (vēsturiski 4 līdz 30 reizes).
|
|
18.
|
Kas attiecas uz testa kvalitātes kontroli, indukcijas palielinājums, kas atbilst paralēlās pozitīvās kontroles (1 nM, E2) PC10 vērtībai, par 1+2SN pārsniedz paralēlās nesēja kontroles indukcijas vērtību (=1). PC10 vērtības var izrādīties nozīmīgas prioritizēšanas vajadzībām, proti, lai vienkāršotu vajadzīgo datu analīzi salīdzinājumā ar statistisko analīzi. Statistiskā analīze sniedz informāciju par nozīmību, tomēr tā tā nav kvantitatīvs parametrs attiecībā uz koncentrācijas potenciālu, tāpēc tas prioritizēšanā ir mazāk noderīgs.
|
ER antagonista testa kvalitātes kritēriji
|
19.
|
Pastiprinātās kontroles (25 nM, E2) vidējai luciferāzes aktivitātei jābūt vismaz 4 reizes lielākai par vidējo VC katrā platē. Šis kritērijs izvēlēts, pamatojoties uz novērtēšanas parametru uzticamību validācijas pētījumā.
|
|
20.
|
Kas attiecas uz testa kvalitātes kontroli, 1 nM, E2 relatīvajai transkripcionālai aktivācijai (RTA) jābūt lielākai par 100 %, RTA 1μM 4-hidroksitamoksifēna (OHT) RTA jābūt mazākai par 40,6 % un 100 μM digitonīna (Dig) RTA jābūt mazākai par 0 %.
Laboratorijas kvalifikācijas pierādīšana (sk. 14. punktu un 3. un 4. tabulu sadaļā “ER TA TESTA KOMPONENTI”).
|
Nesējs
|
21.
|
Par paralēlo VC izmanto dimetilsulfoksīdu (DMSO) vai piemērotu šķīdinātāju tādā pašā koncentrācijā, kādu izmanto pozitīvajām un negatīvajām kontrolēm un testējamām ķimikālijām. Testa ķimikālijas izšķīdina tādā šķīdinātājā, kurā attiecīgā testējamā ķimikālija šķīst un kurš ir sajaucams ar šūnu barotni. Piemēroti nesēji ir ūdens, etanols (tīrība 95 -100 %) un DMSO. Ja izmanto DMSO, tā saturs nedrīkst pārsniegt 0,1 % (tilp/.tilp.). Par katru nesēju ir jāpierāda, ka maksimālais izmantotais tilpums nav citotoksisks un neietekmē testa veikšanu.
|
Testējamo ķimikāliju sagatavošana
|
22.
|
Parasti testējamās ķimikālijas izšķīdina DMSO vai citā piemērotā šķīdinātājā; iegūto šķīdumu pēc tam vairākkārt atšķaida ar to pašu šķīdinātāju attiecībā 1:10, un iegūto atšķaidījumu izmanto atšķaidīšanai ar barotni.
|
Šķīdība un citotoksicitāte Apsvērumi par diapazona noteikšanu
|
23.
|
Ir jāveic sākotnējais tests, lai noteiktu katras testējamās ķimikālijas koncentrācijas diapazonu un noteiktu, vai testējamā ķimikālija varētu uzrādīt kādas šķīdības vai citotoksicitātes problēmas. Sākumā ķimikālijas testē līdz maksimālajai koncentrācijai 1 μl/ml, 1 mg/ml vai 1 mM, atkarībā no tā, kura ir viszemākā. Pamatojoties uz sākotnējā testā novēroto citotoksicitāti vai nešķīdību, pirmajā galīgā testa izpildes reizē ķimikāliju testē sērijveida logaritmiskos atšķaidījumos, sākot ar augstāko pieņemamo koncentrāciju (piem., 1 mM, 100μM, 10μM utt.), un vēro, kad šķīdums kļūst duļķains vai rada nogulsnes. Koncentrācijas otrajā un, ja vajadzīgs, trešajā izpildes reizē pēc vajadzības pielāgo nolūkā labāk raksturot koncentrācijas-atbildreakcijas līkni un izvairīties no koncentrācijām, pie kurām ķimikālija izrādījusies nešķīstoša vai pārmērīgu citotoksicitāti izraisoša.
|
|
24.
|
Kas attiecas uz ER agonistiem un antagonistiem, datus interpretējot, ņem vērā to, ka pieaugošs citotoksicitātes līmenis var mainīt vai izslēgt tipisku sigmoidālu atbildreakciju. Jāizmanto tādas citotoksicitātes testēšanas metodes, kas var sniegt informāciju par 80 % šūnu dzīvotspēju, izmantojot piemērotu, laboratorijā apstiprinātu testu.
|
|
25.
|
Ja citotoksicitātes testa rezultāti liecina, ka testējamās ķimikālijas koncentrācija ir šūnu skaitu samazinājusi par 20 % vai vairāk, šī koncentrācija ir uzskatāma par citotoksisku, un visas koncentrācijas, kas ir vienādas ar vai lielākas par šo robežvērtību, no novērtējuma izslēdz.
|
Eksponēšana ķimikālijai un izkārtojums testa platē
|
26.
|
Ķimikāliju atšķaidīšanās (1. un 2. etaps) un šūnu eksponēšanas (3. etaps) procedūra var būt šāda.
|
1. etaps. Katru testa ķimikāliju seriāli atšķaida DMSO vai piemērotā šķīdinātājā un ievieto mikrotitrēšanas plates iedobēs tā, lai sasniegtu tādas galīgās seriālās koncentrācijas, kādas noteiktas sākotnējā diapazona noteikšanas testā (parasti, piemēram, šādās sērijās: 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM, and 10 pM (10-3-10-11 M)) trīskāršai testēšanai.
2. etaps. Ķimikāliju atšķaidīšana: Vispirms 1,5 μl testējamās ķimikālijas atšķaida ar šķīdinātāju, lai iegūtu barotnes tilpumu 500 μl.
3. etaps. Šūnu ķīmiskā ekspozīcija: 50 μl no 2. etapā sagatavotā atšķaidījuma ievieto testa iedobē, kas satur 104 šūnas/100 μl/iedobē.
Ieteicamais galīgais barotnes tilpums katrā iedobē ir 150 μl. Testa paraugu un references standartu sadalījums var būt tāds, kāds redzams 3. un 4. tabulā.
3. tabula
References standartu koncentrāciju sadalījums ER agonista testa platē
|
Rinda
|
17α-metiltestosterons
|
Kortikosterons
|
17α-estradiols
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: nesēja kontrole (0,1 % DMSO); PC: pozitīvā kontrole ((1 nM, E2)
|
|
27.
|
References standartus (E2, 17α-estradiols, 17-μετιλτεστοστερονσμυνυκορτικοστερονσκκτεστττκατρ®®ιζπιλδεσιρειζρρρρρταβυλατττKatrā testa platē iekļauj arī iedobes, kas satur PC, kura apstrādāta ar 1 nM no E2 un spēj izraisīt maksimālu E2 inducēšanu, un iedobes, kas satur VC, kura apstrādāta tikai ar DMSO (vai piemērotu šķīdinātāju (4. tabula). Ja vienā eksperimentā izmanto šūnas no dažādiem avotiem (piem., atšķirīgs pasāžu skaits vai atšķirīgas partijas utt.), tad references standartus testē ar katru šūnu avotu.
|
4. tabula
Testējamo ķimikāliju un plates kontroles ķimikāliju koncentrāciju sadalījums ER agonista testa platē
|
Rinda
|
Testējamā ķimikālija 1
|
Testējamā ķimikālija 2
|
Testējamā ķimikālija 3
|
Testējamā ķimikālija 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
konc. 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
konc. 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
konc. 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
konc. 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
konc. 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
konc. 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
konc. 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: nesēja kontrole (0,1 % DMSO); PC: pozitīvā kontrole ((1 nM, E2)
|
5. tabula
References standartu koncentrāciju sadalījums ER antagonista testa platē
|
28.
|
Lai novērtētu ķimikāliju antagonistisko aktivitāti, iedobēs, kas atrodas A līdz G rindā, pievieno 25pM, E2. References standartus (tamoksifēns un flutamīds) testē katrā izpildes reizē. Katrā testa platē iekļauj arī iedobes ar PC, kas apstrādātas ar 1 nM, E2 un ko var izmantot par šūnu līnijas hERα-HeLa-9 903 kvalitātes kontroli, iedobes ar VC, kas apstrādātas ar DMSO (vai piemērotu šķīdinātāju), iedobes ar 0,1 % DMSO, kas apstrādātas ar DMSO, kurš pievienots E2 pastiprinātajai koncentrācijai, kas atbilst “pastiprinātajai kontrolei”, iedobes ar 1 μM OHT galīgajā koncentrācijā un iedobes ar 100 μM Dig (5. tabula). Pārējās testa platēs izkārtojums ir tāds pats, taču netiek iekļautas references standartu iedobes (6. tabula). Ja vienā eksperimentā izmanto šūnas no dažādiem avotiem (piem., atšķirīgs pasāžu skaits vai atšķirīgas partijas utt.), tad references standartus testē ar katru šūnu avotu.
|
6. tabula
Testējamo ķimikāliju un plates kontroles ķimikāliju koncentrāciju sadalījums ER antagonista testa platē

|
29.
|
Pienācīgi jāpārliecinās, ka nav nekādu malas efektu; ja ir aizdomas, ka malas efekti varētu rasties, izkārtojums platē ir jāmaina, lai tos novērstu, piemēram, malās esošās iedobes var atstāt tukšas.
|
|
30.
|
Pēc ķimikāliju pievienošanas testa plates 20-24 h inkubē 5 % CO2 inkubatorā 37±1oC temperatūrā, lai inducētu reportiergēnu produktu veidošanos.
|
|
31.
|
Īpaša uzmanība jāpievērš sevišķi gaistošiem savienojumiem. Tādos gadījumos tuvumā esošās kontroliedobes var dot pseidopozitīvus rezultātus, un tie būtu jāsalīdzina ar gaidītajām un vēsturiskajām kontrolvērtībām. Nedaudzajos gadījumos, kad gaistamība var izrādīties problemātiska, ieteicams izmantot iedobju aizlīmes, kas ļauj testēšanas laikā efektīvi izolēt atsevišķas iedobes.
|
|
32.
|
Lai nodrošinātu rezultātu neatkarību, galīgos testus ar to pašu ķimikāliju atkārto dažādās dienās.
|
Luciferāzes tests
|
33.
|
Šajā testā var izmantot tirdzniecībā pieejamus luciferāzes testa reaģentus [piem., luciferāzes testa sistēmu Steady-Glo® (Promega, E2510 vai līdzvērtīgs)] vai standarta luciferāzes testa sistēmu (Promega, E1500 vai līdzvērtīgs), ar nosacījumu, ka tiek izpildīti pieņemamības kritēriji. Testa reaģentus izvēlās atkarībā no izmantojamā luminometra jutības. Ja izmanto standarta luciferāzes testa sistēmu, pirms substrāta pievienošanas izmanto šūnu kultūras līzes reaģentu (piem., Promega, E1531 vai līdzvērtīgs). Luciferāzes reaģentu lieto saskaņā ar ražotāja norādījumiem.
|
DATU ANALĪZE
ER agonista tests
|
34.
|
ER agonista testa gadījumā relatīvo transkripcionālo aktivitāti attiecībā pret PC (1 nM, E2) iegūst, analizējot luminiscences signālus tajā pašā platē šādos etapos (ir pieņemamas arī līdzvērtīgas matemātiskās darbības):
|
1. etaps. Aprēķina VC vidējo vērtību.
2. etaps. Datu normalizācijas nolūkā šo VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi.
3. etaps. Aprēķina normalizētās PC vidējo vērtību.
4. etaps. Plates katras iedobes normalizēto vērtību dala ar normalizētās PC vidējo vērtību (PC=100 %).
Attiecībā uz karu iedobi iegūtā vērtība atbilst šīs iedobes transkripcionālajai aktivitātei salīdzinājumā ar PC atbildreakciju.
5. etaps. Aprēķina testējamās ķimikālijas katras koncentrāciju grupas relatīvās transkripcionālās aktivitātes vidējo vērtību. Iegūtais rezultāts liecina par vidējo transkripcionālo aktivitāti (atbildreakcija) un par to, pie kādas koncentrācijas šī atbildreakcija tiek inducēta (sk. nākamo sadaļu).
EC50, PC50 un PC10 inducēšanas noteikšana
|
35.
|
Lai aprēķinātu EC50, ir vajadzīga pilnīga koncentrācijas-atbildreakcijas līkne, tomēr tas ne vienmēr ir iespējami vai lietderīgi, jo var pastāvēt zināmi koncentrācijas diapazona ierobežojumi (piemēram, citotoksicitātes vai šķīdības problēmu dēļ). Tomēr, tā kā EC50 un maksimālais inducēšanas līmenis (kas atbilst Hilla vienādojuma maksimālajai vērtībai) ir informatīvi parametri, tie parametri pēc iespējas būtu jānorāda. EC50 un maksimālā inducēšanas līmeņa aprēķinā jāizmanto piemērota statistiskā programmatūra (piem., Graphpad Prism).Ja datiem par koncentrāciju un atbildreakciju ir piemērojams Hilla logistiskais vienādojums, EC50 aprēķina ar šādu vienādojumu(7):
Y= bāzes punkts+ (virsotnes punkts- bāzes punkts) / (1+10 exp ((log EC50 -X) x Hilla virziena koeficients)) kur:
X ir koncentrācijas logaritms; un
Y ir reakcija; to mēra no sigmoidālās līknes bāzes līdz virsotnei. Hilla logistiskajā vienādojumā bāzes punkts ir nulle.
|
|
36.
|
Par katru testējamo ķimikāliju sniedz šādus datus:
RPCMax , kas ir testa ķimikālijas inducētās atbildreakcijas maksimālais līmenis un ko izsaka procentos no 1 nM, E2 inducētās atbildreakcijas rajā pašā platē, kā arī PCMax (koncentrācija, kas atbilst RPCMax); un
ja ķimikālijas ir pozitīvas — koncentrācijas, kas inducē PC10 un attiecīgā gadījumā PC50.
|
|
37.
|
PCx vērtību var aprēķināt, interpolējot starp 2 punktiem uz X-Y līknes, no kuriem viens atrodas uzreiz virs, bet otrs uzreiz zem PCx vērtības. Ja datu punktiem, kas atrodas uzreiz virs un uzreiz zem PCx vērtības, ir koordinātas attiecīgi (a,b) un (c,d), tad PCx vērtību var aprēķināt ar šādu vienādojumu:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
PC vērtību apraksts sniegts 1. attēlā.
1. attēls
PC vērtību atvasināšanas piemērs. PC (1 nM, E2) ir katrā testa platē

|
ER antagonista tests
|
39.
|
ER antagonista testa gadījumā relatīvo transkripcionālo aktivitāti (RTA) attiecībā pret pastiprināto kontroli (25 pM, E2) iegūst, analizējot luminiscences signālus tajā pašā platē šādos etapos (ir pieņemamas arī līdzvērtīgas matemātiskās darbības):
1. etaps. Aprēķina VC vidējo vērtību.
2. etaps. Datu normalizācijas nolūkā šo VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi. 3. etaps. Aprēķina normalizētās pastiprinātās kontroles vidējo vērtību.
4. etaps. Plates katras iedobes normalizēto vērtību dala ar normalizētās pastiprinātās kontroles vidējo vērtību (pastiprinātā kontrole=100 %).
Attiecībā uz karu iedobi iegūtā vērtība atbilst šīs iedobes transkripcionālajai aktivitātei salīdzinājumā ar pastiprinātās kontroles atbildreakciju.
5. etaps. Aprēķina katras koncentrāciju grupas relatīvās transkripcionālās aktivitātes vidējo vērtību.
|
IC30 and IC50 inducēšanas noteikšana
|
40.
|
Ja ķimikālijas ir pozitīvas — koncentrācijas, kas inducē IC30 un attiecīgā gadījumā IC50.
|
|
41.
|
ICx vērtību var aprēķināt, interpolējot starp 2 punktiem uz X-Y līknes, no kuriem viens atrodas uzreiz virs, bet otrs uzreiz zem ICx vērtības. Ja datu punktiem, kas atrodas uzreiz virs un uzreiz zem ICx vērtības, ir koordinātas attiecīgi (c,d) un (a,b), tad ICx vērtību var aprēķināt ar šādu vienādojumu:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
2. attēls
IC vērtību atvasināšanas piemērs. Pastiprinātā kontrole (25 pM, E2) ir katrā testa platē

RTA: relatīvā transkripcionālā aktivitāte
|
|
42.
|
Rezultātu pamatā ir divas (vai trīs) neatkarīgas izpildes reizes. Ja divās izpildes reizēs tiek iegūti salīdzināmi un līdz ar to reproducējami rezultāti, nav nepieciešams testu izpildīt vēl trešo reizi. Rezultāti ir pieņemami, ja tie:
|
—
|
atbilst pieņemamības kritērijiem (sk. sadaļu “Pieņemamības kritēriji”, 14.-20. punkts),
|
|
Datu interpretācijas kritēriji
7. tabula
Pozitīvu un negatīvu rezultātu interpretācijas kritēriji ER agonistu testā
|
Pozitīvs
|
Ja ir iegūta tāda RPCMax vērtība, kas ir vienāda ar vai lielāka par 10 % no pozitīvās kontroles atbildreakcijas vismaz divās no divām vai divās no trijām izpildes reizēm.
|
|
Negatīvs
|
Ja RPCMax vērtība nesasniedz vismaz 10 % no pozitīvās kontroles atbildreakcijas vismaz divās no divām vai divās no trijām izpildes reizēm.
|
8. tabula
Pozitīvu un negatīvu rezultātu interpretācijas kritēriji ER antagonistu testā
|
Pozitīvs
|
Ja IC30 ir aprēķināta vismaz divās no divām vai divās no trijām izpildes reizēm.
|
|
Negatīvs
|
Ja IC30 nav aprēķināta divās no divām vai divās no trijām izpildes reizēm.
|
|
43.
|
Datu interpretācijas kritēriji ir redzami 7. un 8. tabulā. Pozitīvos rezultātus raksturo gan ietekmes intensitāte, gan koncentrācija, pie kuras novērojama ietekme Šo divējādo mērķi iespējams sasniegt, ja rezultātus izsaka kā koncentrāciju, pie kuras agonista testā sasniedz 50 % (PC50) vai 10 % (PC10) no PC vērtībām un antagonista testā 50 % (IC50) vai 30 % (IC30) no pastiprinātās kontroles vērtībām tiek inhibētas. Testējamo ķimikāliju uzskata par pozitīvu, ka testējamās ķimikālijas inducētā maksimālā atbildreakcija (RPCMax) ir vienāda ar vai lielāka par 10 % no PC atbildreakcijas vismaz divās no divām vai divās no trijām izpildes reizēm, un par negatīvu, ja RPCMax nesasniedz vismaz 10 % no pozitīvās kontroles atbildreakcijas vismaz divās no divām vai divās no trijām izpildes reizēm.
|
|
44.
|
PC10, PC50 un PCMax ER agonista testā un IC30 un IC50 ER antagonista testā var aprēķināt, izmantojot izklājlapu, kas pieejama kopā ar šīm testēšanas vadlīnijām ESAO publiskajā vietnē (63).
|
|
45.
|
PC10 vai PC50 un IC30 vai IC50 vērtību noteikšanai ir pietiekami testu atkārtot divreiz. Tomēr, ja datu bāzlīnija tajā pašā koncentrāciju diapazonā uzrāda pārlieku augstu variācijas koeficientu (VK, %), šos datus nevar uzskatīt par ticamiem un ir jānoskaidro šādu variāciju iemesls. To triplicēto jēldatu (t.i., luminiscences intensitātes datu) variācijas koeficientam, kurus izmanto kā datu punktus PC10 aprēķinā, ir jābūt mazākam par 20 %.
|
|
46.
|
Ja ir izpildīti pieņemamības kritēriji, tas nozīmē, ka testa sistēma darbojas pareizi, tomēr tas negarantē, ka konkrētajā izpildes reizē tiks iegūti pareizi dati. Labākais pierādījums iegūto datu pareizībai ir pirmās izpildes reizes rezultātu duplicēšana.
|
|
47.
|
ER agonista testa gadījumā, kur ir nepieciešama plašāka informācija papildus tai, kas vajadzīga šajās vadlīnijās paredzētajam skrīningam un prioritizēšanai attiecībā uz pozitīvām testējamām ķimikālijām, jo īpaši PC10-PC49 ķimikālijām, kā arī ķimikālijām, par kurām ir aizdomas, ka tās varētu pārlieku stimulēt luciferāzi, apstiprinājumu tam, ka novērotā luciferāzes aktivitāte ir tikai ERα specifiska atbildreakcija, var gūt, izmantojot ERα antagonistu (sk. 2.1. papildinājumu).
|
TESTĒŠANAS PĀRSKATS
|
48.
|
Sk. 20. punktu sadaļā “ER TA TESTA KOMPONENTI”.
|
LITERATŪRA
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1459-1469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4252-4263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
2.1. papildinājums
PSEIDOPOZITĪVI REZULTĀTI: NOVĒRTĒJUMS PAR RECEPTORU NEMEDIĒTIEM LUMINISCENCES SIGNĀLIEM
|
1.
|
ER agonista testā pseidopozitīvus rezultātus var dot vai nu ER nemediēta luciferāzes gēna aktivācija, vai gēna produkta tieša aktivācija, vai nesaistīta fluorescence. Par šādu efektu liecina nepilnīga vai neparasta devas-atbildreakcijas līkne. Ja rodas šādas aizdomas, izvērtē, kāda ietekme uz atbildreakciju ir ER antagonistam (piem., 4-hidroksitamoksifēnam (OHT)) netoksiskās koncentrācijās. Šim nolūkam var nebūt derīgs tīrais antagonists ICI 182780 , jo pienācīga ICI 182780 koncentrācija var samazināt VK vērtību, bet tas savukārt ietekmē datu analīzi.
|
|
1.
|
Lai garantētu šādas pieejas derīgumu, tajā pašā platē jātestē šādi elementi:
|
—
|
nezināmās ķimikālijas agonistiskā aktivitāte ar OHT (10 μM) un bez tā;
|
|
—
|
1 nM, E2 (trīsreiz) kā agonista PC
|
|
—
|
1 nM, E2 + OHT (trīsreiz)
|
|
Datu interpretācijas kritēriji
Piezīme. Visās iedobēs nesējs ir tādā pašā koncentrācijā.
|
—
|
Ja nezināmās ķimikālijas agonistisko aktivitāti NEIETEKMĒ apstrāde ar ER antagonistu, rezultātu uzskata par “negatīvu”.
|
|
—
|
Ja nezināmās ķimikālijas agonistiskā aktivitāti ir pilnībā inhibēta, piemēro interpretācijas kritērijus.
|
|
—
|
Ja agonistiskā aktivitāte pie zemākās koncentrācijas ir vienāda a vai lielāka par PC10 , nezināmās ķimikālijas atbildreakcija ir inhibēta tādā pašā vai lielākā mērā kā PC10 atbildreakcija. Aprēķina starpību starp atbildreakcijām ar ER antagonistu apstrādātās un neapstrādātās iedobēs; šo starpību uzskata par patieso atbildreakciju un izmanto, lai aprēķinātu parametrus, kas nepieciešami ķimikālijas klasificēšanai.
|
Datu analīze
Pārbauda veiktspējas standartu.
Pārbauda VK starp iedobēm, kas apstrādātas vienādos apstākļos.
|
1.
|
Aprēķina VK vidējo vērtību.
|
|
2.
|
Šo VK vidējo vērtību atskaita no katras ar OHT neapstrādātās iedobes vērtības.
|
|
3.
|
Aprēķina OHT vidējo vērtību.
|
|
4.
|
Šo VK vidējo vērtību atskaita no katras ar OHT apstrādātās iedobes vērtības.
|
|
5.
|
Aprēķina PC vidējo vērtību.
|
|
6.
|
Aprēķina visu pārējo iedobju relatīvo transkripcionālo aktivitāti attiecībā pret PC.
|
2.2. papildinājums
AR DCC (AR DEKSTRĀNU PĀRKLĀTA OGLE) APSTRĀDĀTA SERUMA SAGATAVOŠANA
|
1.
|
Seruma apstrādāšana ar DCC (ar dekstrānu pārklāta ogle) plaši izmantots paņēmiens, kā no seruma atdalīt šūnu barotnei pievienotos estrogēniskos savienojumus, lai izslēgtu, ka atbildreakcija ir nobīdījusies, jo serumā ir estrogēnu atliekas. Ar šo paņēmienu var apstrādāt 500 ml liellopu embriju seruma.
|
Sastāvdaļas
|
2.
|
Nepieciešamie materiāli un aprīkojums
|
Materiāli
|
Magnija hlorīda heksahidrāts (MgCl2 6H2O)
|
|
1 M HEPES buferšķīdums (pH 7,4)
|
|
Filtrācijas ceļā iegūts ultratīrs ūdens
|
Aprīkojums
Autoklāvēts stikla trauks (izmēru attiecīgi pielāgo) Parastā laboratorijas centrifūga (temperatūru iespējams iestatīt uz 4 °C)
PROCEDŪRA
|
2.
|
Šī procedūra domāta centrifūgas mēģenēm ar tilpumu 50 ml.
[1. diena] Ar dekstrānu pārklāto ogli suspendē 1 l ultratīrā ūdens, kam pievienots 1,5 mM MgCl2, 0,25 M saharozes, 2,5 g ogles, 0,25 g dekstrāna un 5 mM HEPES; maisa 4 °C visu nakti.
[2. diena] Suspensiju salej 50 ml centrifūgas mēģenēs un 10 minūtes centrifugē ar 10 000 apgr./min. 4 °C temperatūrā. Atdala supernatantu, un pusi no ogles nogulsnēm atliek 4 °C temperatūrā izmantošanai 3. dienā. Atlikušu pusi suspendē FBS, kas ir lēni atkausēts (lai nepieļautu izgulsnēšanos) un pēc tam 30 minūtes 56 °C temperatūrā inaktivēts; pārlej autoklāvētā stikla traukā (piem., Erlenmeijera kolbā). Šo suspensiju atstāj uz nakti un lēni maisa 4 °C temperatūrā.
[3. diena] FBS suspensiju salej centrifūgas mēģenēs un 10 minūtes centrifugē ar 10 000 apgr./min. 4 °C temperatūrā. Savāc FBS un pārlej to 2. dienā sagatavotajās un atliktajās ogles nogulsnēs. Šīs ogles nogulsnes suspendē FBS un uz nakti lēni maisa 4 °C temperatūrā.
[4. diena] Šo suspensiju salej mēģenēs, 10 min. centrifugē pie 10 000 apgr./min. 4 °C temperatūrā; supernatantu sterilizē, filtrējot caur 0,2 μm sterilu filtru. Šo ar DCC apstrādāto FBS glabā -20 °C temperatūrā, un tas ir jāizmanto gada laikā.
|
3. Papildinājums
VM7LUC ESTROGĒNA RECEPTORU TRANSAKTIVĀCIJAS TESTS ESTROGĒNA RECEPTORU AGONISTU UN ANTAGONISTU NOTEIKŠANAI
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI (SK. ARĪ VISPĀRĪGO IEVADU)
|
1.
|
Šajā testā izmanto VM7Luc4E2 šūnu līniju (64). To ir apstiprinājis Alternatīvu toksikoloģisko metožu novērtēšanas nacionālās toksikoloģijas programmas starpaģentūru centrs (National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods, NICEATM) un Alternatīvo metožu validēšanas starpaģentūru koordinācijas komiteja (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (1). VM7Luc šūnu līnijas galvenokārt ekspresē endogēniskos ERα un nelielu daudzumu endogēnisko ERβ (2) (3) (4).
|
|
2.
|
Šis tests ir izmantojams ļoti daudzām vielām, ja vien tās šķīst dimetilsulfoksīdā (DMSO; CAS Nr. 67-68-5), nereaģē ar DMSO vai šūnu kultūru un testējamajā koncentrācijā nav citotoksiskas. Ja DMSO izmantošana nav iespējama, var izmantot citu nesēju, piem., etanolu vai ūdeni (sk. 12. punktu). Pierādītā VM7Luc ER TA (ant)agonistu testa veiktspēja liecina, ka ar šo testu iegūtie dati var sniegt informāciju par ER mediētiem darbības mehānismiem, un tos var izmantot, nosakot, kuras vielas testējamas prioritāri.
|
|
3.
|
Šis tests ir īpaši paredzēts hERα un hERß mediētas TA noteikšanai, par mērījuma parametru izmantojot hemiluminiscenci. Hemiluminiscence tiek plaši izmantota biotestos, jo luminiscencei ir augsta signāla un fona attiecība. Tomēr vielas, kas inhibē luciferāzes fermentu, var radīt maldīgu priekšstatu par jāņtārpiņu luciferāzes aktivitāti šūnu testos, proteīnu stabilizācijas dēļ izraisot šķietamu inhibīciju vai pastiprinātu luminiscenci. Turklāt dažos uz luciferāzi balstītos ER reportiergēnu testos ir novēroti receptoru nemediēti luminiscences signāli luciferāzes reportiergēna pārmērīgas aktivācijas dēļ, ja fitoestrogēnu koncentrācija ir lielāka par 1 μM (9) (11). Lai gan devas–atbildreakcijas līkne liecina, ka ER sistēmas patiesā aktivācija rodas pie mazākām koncentrācijām, stabili transficētu ER TA testu sistēmās rūpīgi jāpārbauda luciferāzes ekspresija, kas iegūta pie fitoestrogēnu vai tādu līdzīgu savienojumu, par kuriem ir aizdomas, ka tie rada fitoestrogēniem līdzīgu pārmērīgu luciferāzes reportiergēna aktivāciju, augstām koncentrācijām.
|
|
4.
|
Pirms šā testa izmantošanas regulatīviem mērķiem ir jāizlasa sadaļa “VISPĀRĪGAIS IEVADS” un “ER TA TESTA KOMPONENTI”. Šajā testēšanas metodē izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
TESTA PRINCIPS (SK. ARĪ VISPĀRĪGO IEVADU)
|
5.
|
Testu izmanto, lai norādītu ER ligandu saistīšanos, kam seko receptoru–ligandu kompleksa translokācija uz kodolu. Kodolā receptoru–ligandu komplekss saistās ar specifiskiem DNS atbildreakcijas elementiem un transaktivē reportiergēnu (luc), kā rezultātā veidojas luciferāze un pēc tam izstarojas gaisma, kuru var kvantitatīvi noteikt ar luminometru. Luciferāzes aktivitāti var ātri un lēti izvērtēt ar vairākiem komerciāli pieejamiem testa komplektiem. VM7Luc ER TA testā izmanto uz ER reaģējošu cilvēka krūts adenokarcinomas šūnu līniju VM7, kas ir stabili transficēta ar jāņtārpiņu luc reportiera konstruktu, ko regulē četri estrogēnu atbildreakcijas elementi, kas ievietoti pirms peles krūts audzēja vīrusa promotera (MMTV), lai noteiktu vielas, kas in vitro uzrāda ER agonistu vai antagonistu aktivitāti. Šim MMTV promoteram vērojama tikai neliela krusteniskā reakcija ar citiem steroīdajiem un nesteroīdajiem hormoniem (8). Datu interpretācijas kritēriji ir detalizēti aprakstīti 41. punktā. Īsumā, pozitīvu atbildreakciju raksturo tāda koncentrācijas–atbildreakcijas līkne, kurā ir vismaz trīs punkti, kur kļūdu joslas (vidējā vērtība ± standartnovirze) nepārklājas, kā arī tādas amplitūdas izmaiņas (normalizētā relatīvā gaismas vienība (RLU)), kas ir vismaz 20 % no references standarta maksimālās vērtības(17β-estradiols [E2; CAS Nr. 50-28-2] agonistu testam, raloksifena HCl [Ral; CAS Nr. 84449-90-1]/E2 antagonistu testam).
|
PROCEDŪRA
Šūnu līnija
|
6.
|
Testam izmanto stabili transficēto šūnu līniju VM7Luc4E2. Pašlaik šūnu līnija iegādājama tikai ar tehniskās licences līgumu no Kalifornijas universitātes Deivisā (University of California, Davis), Kalifornijā, ASV (65), un no Xenobiotic Detection Systems Inc. Daremā, Ziemeļkarolīnā, ASV (66).
|
Šūnu līnijas stabilitāte
|
7.
|
Lai saglabātu šūnu līnijas stabilitāti un integritāti, šūnas audzē vairākās pasāžās no sasaldēta krājuma šūnu uzturēšanas barotnē (sk. 9. punktu). Šūnas nedrīkst kultivēt vairāk nekā 30 pasāžās. VM7Luc4E2 šūnu līnijai 30 pasāžas nozīmē apmēram trīs mēnešus.
|
Šūnu kultivēšanas un uzsēšanas apstākļi
|
8.
|
Jāievēro Norādījumos par labu šūnu kultivēšanas praksi (Guidance on Good Cell Culture Practice) (5) (6) norādītās procedūras, lai nodrošinātu visu materiālu un metožu kvalitāti un garantētu visa veiktā darba integritāti, derīgumu un reproducējamību.
|
|
9.
|
VM7Luc4E2 šūnas tur RPMI 1 640 barotnē, kam pievienots 0,9 % penicilīna–streptomicīna maisījums (Pen-Strep) un 8,0 % liellopu embriju serums (FBS), īpašā audu kultivēšanas inkubatorā 37 oC ± 1 oC temperatūrā, 90 % ± 5 % mitrumā un 5,0 % ± 1 % CO2/gaisā.
|
|
10.
|
Kad sasniegta ~80 % konfluence, VM7Luc4E2 šūnas 48 stundas subkultivē un kondicionē bezestrogēna vidē pirms šūnu uzsēšanas 96 iedobju platēs, lai eksponētu testējamām ķimikālijām un analizētu no estrogēna atkarīgo luciferāzes aktivitātes inducēšanu. Bezestrogēna barotne ( EFM) satur Dulbeko modificēto Īgla barotni (DMEM) bez fenolsarkanā, kas papildināta ar 4,5 % ar ogli/dekstrānu apstrādātu FBS, 1,9 % L-glutamīnu un 0,9 % Pen-Strep. Nevienam plastmasas izstrādājumam nedrīkst būt estrogēniskas aktivitātes [sk. detalizēto protokolu (7)].
|
Pieņemamības kritēriji
|
11.
|
Testa apstiprināšanas vai noraidīšanas pamatā ir novērtējums par rezultātiem, kas katrā 96 iedobju platē iegūti ar references standartiem un kontroli. Katru references standartu testē vairākās koncentrācijās, un ir vairāki katras references standarta un kontroles koncentrācijas paraugi. Rezultātus salīdzina ar kvalitātes kontrolēm (KK) šiem parametriem, kas iegūti no agonistu un antagonistu vēsturiskajām datubāzēm, ko katra laboratorija izveidojusi kompetences pierādīšanas laikā. Vēsturiskajās datubāzēs pastāvīgi tiek atjauninātas references standartu un kontroles vērtības. Ja mainās aprīkojums vai laboratorijas apstākļi, var būt nepieciešams izveidot atjauninātas vēsturiskās datubāzes.
|
Agonistu tests
Diapazona noteikšanas tests
|
•
|
Inducēšana: nosaka inducēšanu platē, , vidējo augstāko E2 references standarta relatīvās gaismas vienības (RLU) vērtību dalot ar vidējo DMSO kontroles RLU vērtību. Parasti iegūst pieckāršu indukciju, taču eksperimentu uzskata par pieņemamu, ja indukcija ir vismaz četrkārša vai lielāka.
|
|
•
|
DMSO kontroles rezultāti: šķīdinātāja kontroles RLU vērtībām jābūt ne vairāk kā 2,5 reizes lielākām par vēsturiskās šķīdinātāja kontroles vidējās RLU vērtības standartnovirzi.
|
|
•
|
Eksperiments, kas neatbilst kādam no pieņemamības kritērijiem, jāatmet un jāatkārto.
|
Visaptverošs tests
Te ietilpst pieņemamības kritēriji no agonistu diapazona noteikšanas testa un tālāk norādītie kritēriji.
|
•
|
References standarta rezultāti. E2 references standarta koncentrācijas–atbildreakcijas līknei jābūt sigmoidālai un ar vismaz trim vērtībām koncentrācijas–atbildreakcijas līknes lineārajā daļā.
|
|
•
|
Pozitīvās kontroles rezultāti. Metoksihlora kontroles RLU vērtībām jābūt lielākām par DMSO vidējo vērtību plus trīskāršu standartnovirzi no DMSO vidējās vērtības.
|
|
•
|
Eksperiments, kas neatbilst pat vienam pieņemamības kritērijam, jāatmet un jāatkārto.
|
Antagonistu tests
Diapazona noteikšanas tests
|
•
|
Samazinājums Samazinājumu platē nosaka, vidējo augstāko Ral/E2 references standarta RLU vērtību dalot ar vidējo DMSO kontroles RLU vērtību. Parasti iegūst pieckāršu samazinājumu, taču eksperimentu uzskata par pieņemamu, ja samazinājums ir vismaz trīskāršs.
|
|
•
|
E2 kontroles rezultāti. E2 kontroles RLU vērtībām jābūt ne vairāk kā 2,5 reizes lielākām par vēsturiskās E2 kontroles vidējās RLU vērtības standartnovirzi.
|
|
•
|
DMSO kontroles rezultāti: DMSO kontroles RLU vērtībām jābūt ne vairāk kā 2,5 reizes lielākām par vēsturiskās šķīdinātāja kontroles vidējās RLU vērtības standartnovirzi.
|
|
•
|
Eksperiments, kas neatbilst pat vienam pieņemamības kritērijam, jāatmet un jāatkārto.
|
Visaptverošs tests
Te ietilpst pieņemamības kritēriji no antagonistu diapazona noteikšanas testa un tālāk norādītie kritēriji.
|
•
|
References standarta rezultāti. Ral/E2 references standarta koncentrācijas–atbildreakcijas līknei jābūt sigmoidālai un ar vismaz trim vērtībām koncentrācijas–atbildreakcijas līknes lineārajā daļā.
|
|
•
|
Pozitīvās kontroles rezultāti. Tamoksifēna/E2 kontroles RLU vērtībām jābūt mazākām par E2 kontroles vidējo vērtību mīnus trīskāršu standartnovirzi no E2 kontroles vidējās vērtības.
|
|
•
|
Eksperiments, kas neatbilst pat vienam pieņemamības kritērijam, jāatmet un jāatkārto.
|
References standarti, pozitīvās un nesēja kontroles
Nesēja kontrole (agonistu un antagonistu testi)
|
12.
|
Nesēju, kurā izšķīdina testējamo ķimikāliju, testē kā nesēja kontroli (VC). VM7Luc ER TA testa validēšanā izmantotais nesējs bija 1 % (tilpumkoncentrācija) dimetilsulfoksīds (DMSO, CAS Nr. 67-68-5) (sk. 24. punktu). Ja izmanto citu nesēju, nevis DMSO, visi references standarti, kontroles un testējamās ķimikālijas vajadzības gadījumā jātestē tajā pašā nesējā.
|
References standarts (agonistu diapazona noteikšana)
|
13.
|
References standarts ir E2 (CAS Nr. 50-28-2). Diapazona noteikšanas testēšanā references standarts nozīmē, ka četras E2 koncentrācijas (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 un 2,87 x 10-12 M) sērijveidā atšķaida, katru koncentrāciju testējot dubultiedobēs.
|
References standarts (agonistu visaptverošs tests)
|
14.
|
Visaptverošā testēšanā E2 sērijveidā atšķaida attiecībā 1:2, līdz iegūst 11 E2 koncentrācijas (diapazonā no 3,67 x 10-10 līdz 3,59 x 10-13 M) dubultiedobēs.
|
References standarts (antagonistu diapazona noteikšana)
|
15.
|
References standarts ir Ral (CAS Nr. 84449-90-1) un E2 (CAS Nr. 50-28-2) maisījums. Diapazona noteikšanas testēšanā Ral/E2 maisījumu sērijveidā atšķaida, līdz iegūst trīs Ral koncentrācijas (3,06 x 10-9; 7,67 x 10-10 un 1,92 x 10-10M) ar E2 konstantā koncentrācijā (9,18 × 10-11 M) dubultiedobēs.
|
References standarts (antagonistu visaptverošs tests)
|
16.
|
Visaptverošā testēšanā Ral (diapazonā no 2,45x 10-8 līdz 9,57 x 10-11 M) sērijveidā atšķaida attiecībā 1:2 ar E2 konstantā koncentrācijā (9,18 × 10-11 M), līdz iegūst deviņas Ral/E2 koncentrācijas dubultiedobēs.
|
Vāji pozitīvā kontrole (agonisti)
|
17.
|
Vāji pozitīvā kontrole ir 9,06 x 10-6 M p,p’-metoksihlors (metoksihlors; CAS Nr. 72-43-5) EFM barotnē.
|
Vāji pozitīvā kontrole (antagonisti)
|
18.
|
Vāji pozitīvā kontrole ir tamoksifēns (CAS Nr. 10540-29-1) 3,36 x 10-6 M ar E2 9,18 × 10-11 M EFM barotnē.
|
E2 kontrole (tikai antagonistu tests)
|
19.
|
E2 kontrole ir E2 9,18 × 10-11 MEFM barotnē, un to izmanto kā bāzlīnijas negatīvo kontroli.
|
Vairākkāršā indukcija (agonisti)
|
20.
|
References standarta (E2) luciferāzes aktivitātes indukciju nosaka, vidējo augstāko E2 references standarta RLU vērtību dalot ar vidējo DMSO kontroles RLU vērtību, un rezultātam jābūt vairāk nekā četras reizes lielākam.
|
Vairākkāršais samazinājums (antagonisti)
|
21.
|
References standarta (Ral/E2) luciferāzes aktivitātes vidējo vērtību nosaka, vidējo augstāko Ral/E2 references standarta RLU vērtību dalot ar vidējo DMSO kontroles RLU vērtību, tai jābūt vairāk nekā trīs reizes lielākai.
Laboratorijas kompetences pierādīšana (sk. 14. punktu un 3. un 4. tabulu šīs testēšanas metodes sadaļā “ER TA TESTA KOMPONENTI”)
|
Nesējs
|
22.
|
Testa ķimikālijas izšķīdina tādā šķīdinātājā, kurā attiecīgā testējamā ķimikālija šķīst un kurš ir sajaucams ar šūnu barotni. Piemēroti nesēji ir ūdens, etanols (tīrība 95 -100 %) un DMSO. Ja izmanto DMSO, tā saturs nedrīkst pārsniegt 1 % (tilp/.tilp.). Par katru nesēju ir jāpierāda, ka maksimālais izmantotais tilpums nav citotoksisks un neietekmē testa veikšanu. References standartus un kontroles izšķīdina 100 % šķīdinātājā un pēc tam EFM barotnē atšķaida līdz atbilstīgām koncentrācijām.
|
Testējamo ķimikāliju sagatavošana
|
23.
|
Testējamās ķimikālijas izšķīdina 100 % DMSO (vai piemērotā šķīdinātājā) un pēc tam EFM barotnē atšķaida līdz atbilstīgām koncentrācijām. Pirms izšķīdināšanas un atšķaidīšanas jānogaida, līdz visas testējamās ķimikālijas sasilst līdz istabas temperatūrai. Katram eksperimentam jāsagatavo svaigi testējamo ķimikāliju šķīdumi. Šķīdumos nedrīkst būt redzamas nogulsnes vai duļķes. References standartu un kontroļu izejšķīdumus var sagatavot lielā apjomā, taču galīgais references standarts, kontroles atšķaidījumi un testējamās ķimikālijas katram eksperimentam jāsagatavo svaigas un jāizmanto 24 stundās pēc sagatavošanas.
|
Šķīdība un citotoksicitāte Apsvērumi par diapazona noteikšanu
|
24.
|
Diapazona noteikšanas pamatā ir septiņi sērijveida atšķaidījumi attiecībā 1:10 divos atkārtojumos. Sākumā testējamās ķimikālijas testē pie maksimālās koncentrācijas 1 mg/ml (~1 mM) agonistu testēšanā un 20 μg/ml (~10 μM) antagonistu testēšanā. Diapazona noteikšanas eksperimentus izmanto, lai noteiktu:
|
|
—
|
testējamās ķimikālijas sākuma koncentrācijas, kas izmantojamas visaptverošā testēšanā;
|
|
—
|
testējamās ķimikālijas atšķaidījumus (1:2 vai 1:5), kas izmantojami visaptverošā testēšanā.
|
|
25.
|
Šūnu dzīvotspējas/citotoksicitātes novērtējums ir ietverts agonistu un antagonistu testu protokolos (7) un iekļauts diapazona noteikšanas un visaptverošajā testēšanā. Citotoksicitātes metode, ko izmantoja šūnu dzīvotspējas novērtēšanai VM7Luc ER TA testa (1) validēšanas laikā, bija mērogota kvalitatīvā vizuālās novērošanas metode, tomēr citotoksicitātes noteikšanai var izmantot kvantitatīvu metodi (skatīt protokolu (7)). Nevar izmantot datus par testējamās ķimikālijas koncentrācijām, kas izraisa dzīvotspējas samazināšanos par vairāk nekā 20 %.
|
Eksponēšana ķimikālijai un izkārtojums testa platē
|
26.
|
Šūnas saskaita un uzsēj audu kultūras 96 iedobju platēs (2 x 105 šūnas vienā iedobē) EFM barotnē, un inkubē 24 stundas, lai šūnas varētu piestiprināties pie plates. EFM atdala un aizstāj ar testējamajām un references ķimikālijām EFM barotnē, un inkubē 19–24 stundas. Īpaša uzmanība jāpievērš ļoti gaistošām vielām, jo tuvējās kontroliedobes var dot pseidopozitīvus rezultātus. Šādos gadījumos ieteicams izmantot iedobju aizlīmes, kas ļauj testēšanas laikā efektīvi izolēt atsevišķas iedobes.
|
Diapazona noteikšanas tests
|
27.
|
Diapazona noteikšanas testēšanā izmanto visas 96 iedobju plates iedobes; testē līdz pat sešām testējamajām ķimikālijām septiņās dažādās koncentrācijās, ko iegūst sērijveida atšķaidīšanā attiecībā 1:10 divos atkārtojumos (sk. 1. un 2. attēlu).
|
|
—
|
Agonistu diapazona noteikšanas testēšanā izmanto četras E2 koncentrācijas divos atkārtojumos kā references standartu un četras DMSO kontroles replikāta iedobes.
|
|
—
|
Antagonistu diapazona noteikšanas testēšanā izmanto trīs Ral/E2 maisījuma koncentrācijas (E2 ir konstants 9,18 × 10-11 M) divos atkārtojumos kā references standartu un trīs replikātu iedobes, kas paredzētas E2 un DMSO kontrolēm.
|
1. attēls.
Agonistu diapazona noteikšanas testa 96 iedobju plates izkārtojums

Saīsinājumi: no E2-1 līdz E2-4 = E2 references standarta koncentrācijas (no augstas līdz zemai); no TC1-1 līdz TC1-7 = 1. testējamās ķimikālijas (TC1) koncentrācijas (no augstas līdz zemai); no TC2-1 līdz TC2-7 = 2. testējamās ķimikālijas (TC2) koncentrācijas (no augstas līdz zemai); no TC3-1 līdz TC3-7 = 3. testējamās ķimikālijas (TC3) koncentrācijas (no augstas līdz zemai); no TC4-1 līdz TC4-7 = 4. testējamās ķimikālijas (TC4) koncentrācijas (no augstas līdz zemai); no TC5-1 līdz TC5-7 = 5. testējamās ķimikālijas (TC5) koncentrācijas (no augstas līdz zemai); no TC6-1 līdz TC6-7 = 6. testējamās ķimikālijas (TC6) koncentrācijas (no augstas līdz zemai); VC = nesēja kontrole (DMSO [1 % tilpumkoncentrācijas EFM]).
2. attēls.
Antagonistu diapazona noteikšanas testa 96 iedobju plates izkārtojums

Saīsinājumi: E2 = E2 kontrole; no Ral-1 līdz Ral-3 = raloksifena/E2 references standarta koncentrācijas (no augstas līdz zemai); no TC1-1 līdz TC1-7 = 1. testējamās ķimikālijas (TC1) koncentrācijas (no augstas līdz zemai); no TC2-1 līdz TC2-7 = 2. testējamās ķimikālijas (TC2) koncentrācijas (no augstas līdz zemai); no TC3-1 līdz TC3-7 = 3. testējamās ķimikālijas (TC3) koncentrācijas (no augstas līdz zemai); no TC4-1 līdz TC4-7 = 4. testējamās ķimikālijas (TC4) koncentrācijas (no augstas līdz zemai); no TC5-1 līdz TC5-7 = 5. testējamās ķimikālijas (TC5) koncentrācijas (no augstas līdz zemai); no TC6-1 līdz TC6-7 = 6. testējamās ķimikālijas (TC6) koncentrācijas (no augstas līdz zemai); VC = nesēja kontrole (DMSO [1 % tilpumkoncentrācijas EFM]).
Piezīme. Visas testējamās ķimikālijas testē E2 klātbūtnē (9,18 × 10-11 M).
|
28.
|
Ieteicamais galīgais barotnes tilpums katrā iedobē ir 200 μl. Izmanto tikai tādas testa plates, kurās šūnām visās iedobēs dzīvotspēja ir 80 % un lielāka.
|
|
29.
|
Sākuma koncentrāciju noteikšana visaptverošai
agonistu
testēšanai ir detalizēti aprakstīta agonistu protokolā (7). Īsumā, tiek izmantoti tālāk aprakstītie kritēriji.
|
|
—
|
Ja testējamās ķimikālijas koncentrācijas līknē neviens punkts nav lielāks par DMSO kontroles rezultātu vidējo vērtību plus trīskāršu standartnovirzi, visaptverošai testēšanai izmanto 11 atšķaidījumu sēriju attiecībā 1:2, sākot ar maksimālo šķīstošo koncentrāciju.
|
|
—
|
Ja testējamās ķimikālijas koncentrācijas līknē ir punkti, kas ir lielāki par DMSO kontroles rezultātu vidējo vērtību plus trīskāršu standartnovirzi, visaptverošā testēšanā izmanto 11 secīgus atšķaidījumus, un sākuma koncentrācijai jābūt par vienu logaritmu augstākai nekā koncentrācijai, kas diapazona noteikšanā dod augstāko koriģēto RLU vērtību. 11 atšķaidījumu sērijas pamatā ir atšķaidījumi attiecībā 1:2 vai 1:5 atbilstīgi tālāk aprakstītajiem kritērijiem.
|
11 punktu atšķaidījumu sērija attiecībā 1:2 jāizmanto, ja šādi iegūtais koncentrāciju diapazons ļauj novērot atbildreakciju pilnu diapazonu, pamatojoties uz koncentrācijas–atbildreakcijas līkni, kas iegūta diapazona noteikšanas testā. Pretējā gadījumā izmanto atšķaidījumu attiecībā 1:5.
|
|
|
—
|
Ja diapazona noteikšanas testā testējamā ķimikālija uzrāda vielai divfāzu koncentrācijas–atbildreakcijas līkni, arī visaptverošajā testēšanā ir jāapskata abas fāzes.
|
|
30.
|
Sākuma koncentrāciju noteikšana visaptverošai antagonistu testēšanai ir detalizēti aprakstīta antagonistu protokolā (7). Īsumā, tiek izmantoti tālāk aprakstītie kritēriji.
|
|
—
|
Ja testējamās ķimikālijas koncentrācijas līknē neviens punkts nav mazāks par E2 kontroles rezultātu vidējo vērtību mīnus trīskāršu standartnovirzi, visaptverošai testēšanai izmanto 11 punktu atšķaidījumu sēriju attiecībā 1:2, sākot ar maksimālo šķīstošo koncentrāciju.
|
|
—
|
Ja testējamās ķimikālijas koncentrācijas līknē ir punkti, kas ir mazāki par E2 kontroles rezultātu vidējo vērtību mīnus trīskāršu standartnovirzi, visaptverošā testēšanā izmanto 11 secīgus atšķaidījumus, un sākuma koncentrācijai jābūt vienai no šādām:
|
—
|
koncentrācija, kas diapazona noteikšanā dod zemāko koriģēto RLU vērtību;
|
|
—
|
maksimālā šķīstošā koncentrācija (sk. 14-2. attēlu antagonistu protokolā (7));
|
|
—
|
zemākā citotoksiskā koncentrācija (attiecīgu piemēru sk. 14-3. attēlā antagonistu protokolā (7)).
|
|
|
—
|
11 atšķaidījumu sērijas pamatā ir atšķaidījumi attiecībā 1:2 vai 1:5 atbilstīgi tālāk aprakstītajiem kritērijiem.
|
11 punktu atšķaidījumu sērija attiecībā 1:2 jāizmanto, ja šādi iegūtais koncentrāciju diapazons ļauj novērot atbildreakciju pilnu diapazonu, pamatojoties uz koncentrācijas–atbildreakcijas līkni, kas iegūta diapazona noteikšanas testā. Pretējā gadījumā jāizmanto atšķaidījums attiecībā 1:5.
|
|
Visaptverošie testi
|
31.
|
Visaptveroša testēšana sastāv no 11 punktu atšķaidījumu sērijas (atšķaidījumu sērija attiecībā 1:2 vai 1:5 atbilstīgi visaptverošas testēšanas kritēriju sākuma koncentrācijai), katru koncentrāciju testējot trīs replikāta iedobēs 96 iedobju platē (sk. 3. un 4. attēlu).
|
|
—
|
Agonistu visaptverošā testēšanā par references standartu izmanto 11 E2 koncentrācijas divos atkārtojumos. Katrā platē ir četras replikāta iedobes DMSO kontrolei un četras replikāta iedobes metoksihlora kontrolei (9,06 x 10-6 M).
|
|
—
|
Antagonistu visaptverošā testēšanā par references standartu izmanto deviņas Ral/E2 koncentrācijas (E2 ir konstants ar 9,18 × 10-11 M) divos atkārtojumos, četras replikāta iedobes domātas E2 9,18 x 10-11 M kontrolei, četras replikāta iedobes domātas DMSO kontrolēm un vēl četras —tamoksifēnam 3,36 x 10-6M.
|
3. attēls.
Agonistu visaptveroša testa 96 iedobju plates izkārtojums

Saīsinājumi: no TC1-1 līdz TC1-11 =1. testējamās ķimikālijas koncentrācijas (no augstas līdz zemai); no TC2-1 līdz TC2-11 =2. testējamās ķimikālijas koncentrācijas (no augstas līdz zemai); no E2-1 līdz E2-11 = E2 references standarta koncentrācijas (no augstas līdz zemai); Meth = p,p’ metoksihlora vāji pozitīvā kontrole; VC = DMSO (1 % tilpumkoncentrācija) EFM nesēja kontrole.
4. attēls.
Antagonistu visaptveroša testa 96 iedobju plates izkārtojums

Saīsinājumi: E2 = E2 kontrole; no Ral-1 līdz Ral-9 = raloksifena/E2 references standarta koncentrācijas (no augstas līdz zemai); Tam = tamoksifēna/E2 vāji pozitīvā kontrole; no TC1-1 līdz TC1-11 = 1. testējamās ķimikālijas (TC1) koncentrācijas (no augstas līdz zemai); no TC2-1 līdz TC2-11 = 2. testējamās ķimikālijas (TC2) koncentrācijas (no augstas līdz zemai); VC = nesēja kontrole (DMSO [1 % tilpumkoncentrācijas EFM]).
Piezīme. Kā norādīts iepriekš, visās iedobēs, kas satur references standartu un testējamo ķimikāliju, E2 ir konstantā koncentrācijā (9,18 x 10-11M).
|
32.
|
Lai nodrošinātu rezultātu neatkarību, visaptverošos testus ar to pašu ķimikāliju atkārto dažādās dienās. Jāveic vismaz divi visaptveroši testi. Ja testu rezultāti ir savstarpējā pretrunā (piem., viens tests ir pozitīvs, otrs negatīvs) vai ja viens tests ir neadekvāts, jāveic trešais papildu tests.
|
Luminiscences mērīšana
|
33.
|
Luminiscenci mēra diapazonā no 300 līdz 650 nm ar inžekcijas luminometru un programmatūru, kas kontrolē inžekcijas tilpumu un mērījumu intervālu (7). Gaismas emisiju no katras iedobes izsaka kā RLU uz katru iedobi.
|
DATU ANALĪZE
EC50/IC50 noteikšana
|
34.
|
EC50 vērtību (puse no testējamās ķimikālijas maksimālās efektīvās koncentrācijas [agonisti]) un IC50 vērtību (puse no testējamās ķimikālijas maksimālās inhibējošās koncentrācijas [antagonisti]) nosaka, pamatojoties uz koncentrācijas–atbildreakcijas datiem. Testējamajām ķimikālijām, kas ir pozitīvas vienā vai vairākās koncentrācijās, testējamās ķimikālijas koncentrāciju, kas izraisa pusi no maksimālās atbildreakcijas (IC50 vai EC50), aprēķina, izmantojot Hilla funkciju vai piemērotu alternatīvu. Hilla funkcija ir četru parametru logistisks matemātisks modelis, kas testējamās ķimikālijas koncentrāciju saista ar atbildreakciju (parasti pēc sigmoidālas līknes), izmantojot tālāk norādīto vienādojumu.
|

kur:
Y= atbildreakcija (t. i., RLU);
X= koncentrācijas logaritms;
Bāze= minimālā atbildreakcija;
Virsotne= maksimālā atbildreakcija;
lg EC50 (vai lg IC50)= X logaritms, kas atbilst atbildreakcijai, kura atrodas pa vidu starp bāzi un virsotni
Hilla virziena koeficients= līknes stāvums.
Ar šo modeli aprēķina rezultātus, kas ir vissaderīgākie ar vērtībām “Virsotne”, “Bāze”, “Hilla virziena koeficients”, IC50 un EC50. EC50 un IC50 vērtību aprēķināšanai jāizmanto atbilstoša statistiskā programmatūra (piem., Graphpad PrismR).
Izlecošo vērtību noteikšana
|
35.
|
Lai izdarītu kvalitatīvus statistiskus secinājumus, būtu lietderīgi veikt arī (bet ne tikai) Q-testu (sk. agonistu un antagonistu protokolus (7)), lai noteiktu “nederīgās” iedobes, kas izslēdzamas no datu analīzes.
|
|
36.
|
Attiecībā uz E2 references standarta replikātiem (paraugā ir divas iedobes) jebkuru replikāta koriģēto RLU vērtību konkrētā E2 koncentrācijā uzskata par izlecošo vērtību, ja vēsturiskajā datubāzē tās vērtība par vairāk nekā 20 % atšķiras no koriģētās RLU vērtības šai koncentrācijai.
|
Luminometra datu apkopošana un koriģēšana diapazona noteikšanas testēšanai
|
37.
|
Neapstrādātie dati no luminometra jāpārsūta uz testam īpaši izveidotu izklājlapas veidni. Jānosaka, vai ir izlecošo vērtību datu punkti, kas jāizslēdz. (Analīzē noteiktos parametrus sk. sadaļā “Testa pieņemamības kritēriji”.) Veic šādus aprēķinus.
|
Agonists:
|
1. etaps.
|
Aprēķina DMSO nesēja kontroles (VC) vidējo vērtību.
|
|
2. etaps.
|
Datu normalizācijas nolūkā šo DMSO VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi.
|
|
3. etaps.
|
Aprēķina references standarta (E2) vidējo vairākkāršo indukciju.
|
|
4. etaps.
|
Aprēķina testējamo ķimikāliju vidējo EC50 vērtību.
|
Antagonists:
|
1. etaps.
|
Aprēķina DMSO VC vidējo vērtību.
|
|
2. etaps.
|
Datu normalizācijas nolūkā šo DMSO VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi.
|
|
3. etaps.
|
Aprēķina references standarta (Ral/E2) vidējo vairākkāršo samazinājumu.
|
|
4. etaps.
|
Aprēķina E2 references standarta vidējo vērtību.
|
|
5. etaps.
|
Aprēķina testējamo ķimikāliju vidējo IC50 vērtību.
|
Luminometra datu apkopošana un koriģēšana visaptverošai testēšanai
|
38.
|
Neapstrādātie dati no luminometra jāpārsūta uz testam īpaši izveidotu izklājlapas veidni. Jānosaka, vai ir izlecošo vērtību datu punkti, kas jāizslēdz. (Analīzē noteiktos parametrus sk. sadaļā “Testa pieņemamības kritēriji”.) Veic tālāk aprakstītos aprēķinus.
|
Agonists:
|
1. etaps.
|
Aprēķina DMSO VC vidējo vērtību.
|
|
2. etaps.
|
Datu normalizācijas nolūkā šo DMSO VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi.
|
|
3. etaps.
|
Aprēķina references standarta (E2) vidējo vairākkāršo indukciju.
|
|
4. etaps.
|
Aprēķina E2 un testējamo ķimikāliju vidējo EC50 vērtību.
|
|
5. etaps.
|
Aprēķina metoksihlora vidējo koriģēto RLU vērtību.
|
Antagonists:
|
1. etaps.
|
Aprēķina DMSO VC vidējo vērtību.
|
|
2. etaps.
|
Datu normalizācijas nolūkā šo DMSO VC vidējo vērtību atskaita no vērtības, kas iegūta attiecībā uz katru iedobi.
|
|
3. etaps.
|
Aprēķina references standarta (Ral/E2) vidējo vairākkāršo indukciju.
|
|
4. etaps.
|
Aprēķina Ral/E2 un testējamo ķimikāliju vidējo IC50 vērtību.
|
|
5. etaps.
|
Aprēķina tamoksifēna vidējo koriģēto RLU vērtību.
|
|
6. etaps.
|
Aprēķina E2 references standarta vidējo vērtību.
|
Datu interpretācijas kritēriji
|
39.
|
VM7Luc ER TA tests ir viens no rīkiem, ko izmanto pierādījumu izsvēršanas pieejā un kas palīdz noteikt, kuras vielas ir prioritāri jātestē ED in vivo testos. Šajā prioritizēšanas procedūrā ietilpst testējamās ķimikālijas klasificēšana par pozitīvu vai negatīvu attiecībā uz ER agonistu vai antagonistu aktivitāti. VM7Luc ER TA testa validēšanas pētījumā izmantotie pozitīvu un negatīvu rezultātu interpretācijas kritēriji ir aprakstīti 1. tabulā.
|
1. tabula.
Pozitīvu un negatīvu rezultātu interpretācijas kritēriji
|
|
|
AGONISTU AKTIVITĀTE
|
|
Pozitīvs
|
|
—
|
Visas testējamās ķimikālijas, kas klasificētas kā pozitīvas attiecībā uz ER agonistu aktivitāti, ir ar šādu koncentrācijas–atbildreakcijas līkni: bāzlīnija, pozitīvs slīpums un plato vai virsotne. Dažos gadījumos var būt definēti tikai divi no šiem raksturlielumiem (bāzlīnija –slīpums vai slīpums –virsotne).
|
|
—
|
Uz pozitīvā slīpuma līnijas ir vismaz trīs punkti, kur kļūdu joslas ar nepārklājas (vidējais ± SN). Bāzlīniju veidojošos punktus izslēdz, savukārt līknes lineārā daļa var ietvert virsotnes punktu vai plato pirmo punktu.
|
|
—
|
Lai ķimikāliju klasificētu par pozitīvu, atbildreakcijas amplitūdai (starpībai starp bāzlīniju un virsotni) — jābūt vismaz 20 % no references standarta E2 maksimālās vērtības (t. i., 2000 RLU vai vairāk, ja references standarta [E2] maksimālo atbildreakcijas vērtību koriģē uz 10 000 RLU).
|
|
—
|
Ja iespējams, EC50 vērtība jāaprēķina katrai pozitīvai testējamai ķimikālijai.
|
|
|
Negatīvs
|
Vidējā koriģētā RLU konkrētai koncentrācijai ir vienāda ar vai mazāka par DMSO kontroles RLU vidējo vērtību plus trīskārša DMSO RLU standartnovirze.
|
|
Neadekvāts
|
Datus, kurus nozīmīgu kvalitatīvu vai kvantitatīvu ierobežojumu dēļ uzskata par nederīgiem aktivitātes esības vai neesības pierādīšanai, uzskata par neadekvātiem, un tos nevar izmantot, lai noteiktu, vai testējamā ķimikālija ir pozitīva vai negatīva. Tāpēc ķimikālijas jātestē atkārtoti.
|
|
|
|
ANTAGONISTU AKTIVITĀTE
|
|
Pozitīvs
|
|
—
|
No testējamās datiem iegūst šādu koncentrācijas–atbildreakcijas līkni: bāzlīnija un negatīvs slīpums.
|
|
—
|
Uz negatīvā slīpuma līnijas ir vismaz trīs punkti, kur kļūdu joslas ar nepārklājas; bāzlīniju veidojošos punktus izslēdz, savukārt līknes lineārā daļa var ietvert plato pirmo punktu.
|
|
—
|
Aktivitātes samazinājumam jābūt vismaz 20 % no references standarta Ral/E2 maksimālās vērtības (t. i., 8 000 RLU vai mazāk, ja references standarta [Ral/E2] maksimālo atbildreakcijas vērtību koriģē uz 10 000 RLU).
|
|
—
|
Testējamās ķimikālijas augstākajai necitotoksiskajai koncentrācijai jābūt mazākai par vai vienādai ar 1 x 10-5 M.
|
|
—
|
Ja iespējams, IC50 vērtība jāaprēķina katrai pozitīvai testējamai ķimikālijai.
|
|
|
Negatīvs
|
Koncentrācijā, kas mazāka par 1,0 x 10-5 M, visi datu punkti ir virs EC80 vērtības (80 % no E2 atbildreakcijas jeb 8 000 RLU).
|
|
Neadekvāts
|
Datus, kurus nozīmīgu kvalitatīvu vai kvantitatīvu ierobežojumu dēļ uzskata par nederīgiem aktivitātes esības vai neesības pierādīšanai, uzskata par neadekvātiem, un tos nevar izmantot, lai noteiktu, vai testējamā ķimikālija ir pozitīva vai negatīva. Tāpēc ķimikālija jātestē atkārtoti.
|
|
40.
|
Pozitīvos rezultātus raksturo gan ietekmes intensitāte, gan koncentrācija, pie kuras novērojama ietekme, ja iespējams. Pozitīvu, negatīvu un neadekvātu rezultātu piemēri sniegti 5. un 6. attēlā.
|
5. attēls.
Agonisti: pozitīvi, negatīvi un neadekvāti rezultāti
Punktētā līnija norāda 20 % no E2 atbildreakcijas, resp. 2000 RLU (koriģētas un normalizēta vērtība).
6. attēls.
Antagonisti: pozitīvi, negatīvi un neadekvāti rezultāti

Punktētā līnija norāda 80 % no Ral/E2 atbildreakcijas, resp. 8 000 RLU (koriģētas un normalizēta vērtība).
Nepārtrauktā līnija atbilst 1,00 x 10-5 M. Lai atbildreakciju uzskatītu par pozitīvu, tai jābūt zem 8 000 RLU līnijas un koncentrācijā, kas mazāka par 1,00 x 10-5 M.
Ar zvaigznīti atzīmētās koncentrācijas mezoheksestrola grafikā norāda, ka dzīvotspējas rādītāji ir “2” vai lielāki.
Mezoheksestrola testa rezultātus uzskata par neadekvātiem, jo vienīgā atbildreakcija, kas ir mazāka par 8 000 RLU, ir koncentrācijā 1,00 x 10-5 M.
|
41.
|
EC50 un IC50 var aprēķināt, izmantojot četru parametru Hilla funkciju (papildinformāciju sk. agonistu protokolā un antagonistu protokolā (7)). Ja ir izpildīti pieņemamības kritēriji, tas nozīmē, ka testa sistēma darbojas pareizi, tomēr tas negarantē, ka konkrētajā izpildes reizē tiks iegūti pareizi dati. Labākais pierādījums iegūto datu pareizībai ir pirmās izpildes reizes rezultātu duplicēšana (sk. 19. punktu sadaļā “ER TA TESTA KOMPONENTI”).
|
TESTĒŠANAS PĀRSKATS
|
42.
|
Sk. 20. punktu sadaļā “ER TA TESTA KOMPONENTI”.
|
LITERATŪRA
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5367-5373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10): 1459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10): 4252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
4. Papildinājums
STABILI TRANSFICĒTU CILVĒKA ESTROGĒNA Α RECEPTORU TRANSAKTIVĀCIJAS TESTS ĶIMIKĀLIJU ESTROGĒNISKAS AGONISTU UN ANTAGONISTU AKTIVITĀTES NOTEIKŠANAI, IZMANTOJOT ERΑ CALUX ŠŪNU LĪNIJU
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI (SK. ARĪ VISPĀRĪGO IEVADU)
|
1.
|
ERα CALUX transaktivācijas testā izmanto cilvēka U2OS šūnu līniju, lai noteiktu estrogēnu agonistu un antagonistu aktivitāti, ko mediē cilvēka estrogēna alfa receptors (hERα). Stabili transficētu ERα CALUX biotesta, ko piedāvā BioDetection Systems BV (Amsterdama, Nīderlande), validēšanas pētījumā pierādīja testa relevantumu un ticamību paredzētajam nolūkam (1). ERα CALUX šūnu līnija ekspresē tikai stabili transficētus cilvēka ERα (2) (3).
|
|
2.
|
Šis tests ir īpaši paredzēts hERα mediētas transaktivācijas noteikšanai, par mērījuma parametru izmantojot bioluminiscenci. Augstās signāla un trokšņa attiecības dēļ bioluminiscenci plaši izmanto biotestos (4).
|
|
3.
|
Novērots, ka fitoestrogēni koncentrācijā, kas ir lielāka par 1 μM, pārmērīgi aktivē luciferāzes reportiergēnu, tā izraisot receptoru nemediētu luminiscenci (5) (6) (7). Tāpēc stabili transficētu ER transaktivācijas testos rūpīgi jāpārbauda fitoestrogēnu vai citu līdzīgu savienojumu, kas var pārmērīgi aktivēt luciferāzes ekspresiju, augstākas koncentrācijas (sk. 2. papildinājumu).
|
|
4.
|
Pirms šā testa izmantošanas regulatīviem mērķiem ir jāizlasa sadaļa “VISPĀRĪGAIS IEVADS” un “ER TA TESTA KOMPONENTI”. Šajā testēšanas metodē izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
TESTA PRINCIPS (SK. ARĪ VISPĀRĪGO IEVADU)
|
5.
|
Biotestu izmanto, lai novērtētu ER ligandu saistīšanos un receptoru–ligandu kompleksa sekojošu translokāciju uz kodolu. Kodolā receptoru–ligandu komplekss saista īpašus DNS atbildreakcijas elementus un transaktivē jāņtārpiņu luciferāzes reportiergēnu, kā rezultātā palielinās luciferāzes fermenta šūnu ekspresija. Pēc luciferāzes substrāta luciferīna pievienošanas luciferīns pārvēršas par bioluminiscējošu produktu. Radīto gaismu var viegli uztvert un kvantitatīvi noteikt ar luminometru.
|
|
6.
|
Testa sistēmā izmanto stabili transficētu ERα CALUX šūnas. ERα CALUX šūnas izveidotas no cilvēka osteoblastiskas osteosarkomas U2OS šūnu līnijas. Cilvēka U2OS šūnas stabili transficēja ar 3xHRE-TATA-Luc un pSG5-neo-hERα, izmantojot kalcija fosfāta līdznogulsnēšanās metodi. U2OS šūnu līnija tika izvēlēta kā labākais variants, ko izmantot kā reportieršūnu līniju, kas reaģē uz estrogēnu (un citiem steroīdajiem hormoniem), un tā pamatā ir novērojums, ka U2OS šūnu līnijā endogēnisko receptoru aktivitāte bija neliela vai tās nebija vispār. Endogēnisko receptoru neesamību novērtēja tikai ar luciferāzes reportierplazmīdām, un pēc receptoru ligandu pievienošanas netika konstatēta nekāda aktivitāte. Turklāt pēc radniecīgu receptoru pagaidu pievienošanas šī šūnu līnija uzturēja spēcīgas hormonu mediētas atbildreakcijas (2) (3) (8).
|
|
7.
|
Ķimikāliju testēšanā ar ERα CALUX šūnu līniju, lai noteiktu estrogēnisku vai antiestrogēnisku aktivitāti, ietilpst priekšskrīnings un visaptveroši testi. Priekšskrīninga laikā visaptverošās testēšanas vajadzībām nosaka testējamo ķimikāliju šķīdību, citotoksicitāti un precīzāku koncentrāciju diapazonu. Visaptverošajos testos testējamo ķimikāliju precīzāko koncentrāciju diapazonu testē ERα CALUX biotestos, un pēc tam testējamās ķimikālijas klasificē pēc agonisma vai antagonisma.
|
|
8.
|
Datu interpretācijas kritēriji ir detalizēti aprakstīti 59. punktā. Īsumā, testējamo ķimikāliju uzskata par pozitīvu attiecībā uz agonismu, ja vismaz divu secīgu testējamās ķimikālijas koncentrāciju atbildreakcija ir vienāda ar vai lielāka par 10 % no references standarta (17β-estradiola) maksimālās atbildreakcijas (PC10). Testējamo ķimikāliju uzskata par pozitīvu attiecībā uz antagonismu, ja vismaz divu secīgu testējamās ķimikālijas koncentrāciju atbildreakcija ir vienāda ar vai mazāka par 80 % no references standarta (tamoksifēna) maksimālās atbildreakcijas (PC80).
|
PROCEDŪRA
Šūnu līnija
|
9.
|
Testam jāizmanto stabili transficēta U2OS ERα CALUX šūnu līnija. Šūnu līniju var iegādāties ar tehniskās licences līgumu no BioDetection Systems BV, Amsterdama, Nīderlande.
|
|
10.
|
Drīkst izmantot tikai šūnu kultūras bez mikoplazmas. Jābūt apliecinājumam, ka izmantojamajās šūnu partijās nav mikoplazmas piesārņojuma, vai arī pirms lietošanas jāveic mikoplazmas tests. Precīzai mikoplazmas infekcijas noteikšanai jāizmanto RT-PCR (polimerāzes ķēdes reakcija reāllaikā) (9).
|
Šūnu līnijas stabilitāte
|
11.
|
Lai saglabātu CALUX šūnu stabilitāti un integritāti, šūnas jāglabā šķidrajā slāpeklī (–800C). Pēc šūnu atkausēšanas jaunas kultūras aizsākšanai šūnas jāsubkultivē vismaz divreiz, tikai pēc tam tās drīkst izmantot ķimikāliju estrogēniskas agonistu un antagonistu aktivitātes novērtēšanai. Šūnas nedrīkst subkultivēt vairāk nekā 30 pasāžās.
|
|
12.
|
Lai kontrolētu šūnu līnijas stabilitāti laika gaitā, agonistu un antagonistu testa sistēmas reaģētspēju verificē, novērtējot references standarta EC50 vai IC50. Tāpat jākontrolē arī pozitīvās kontroles parauga (PC) un negatīvās kontroles parauga (NC) relatīvā indukcija. Rezultātiem jāatbilst agonistu (3.C tabula) vai antagonistu ERα CALUX biotesta (4.C tabula) pieņemamības kritērijiem. References standarti, pozitīvās un negatīvās kontroles ir norādītas 1. tabulā (agonisms) un 2. tabulā (antagonisms).
|
Šūnu kultivēšanas un uzsēšanas apstākļi
|
13.
|
U2OS šūnas jākultivē barotnē (DMEM/F12 (1:1) barotne ar fenolsarkano kā pH indikatoru, kam pievienots liellopu embriju serums (7,5 %), aizvietojamās aminoskābes (1 %), 10 vienības/ml penicilīna, streptomicīna un geneticīna (G-418) kā atlases marķieris). Šūnas jāievieto CO2 inkubatorā (5 % CO2) 370C temperatūrā un 100 % mitrumā. Kad šūnas sasniedz 85–95 % konfluenci, tās jāsubkultivē vai jāsagatavo uzsēšanai 96 iedobju mikrotitrēšanas platēs. Ja šūnas gatavo uzsēšanai, tās atkārtoti jāsuspendē 1 x 105 šūnas/ml bezestrogēna testa barotnē (DMEM/F12 (1:1) barotne bez fenolsarkanā, kam pievienots liellopu embriju serums (5 % tilpumkoncentrācija), kas apstrādāts ar dekstrāna pārklātu ogli, aizvietojamās aminoskābes (1 % tilpumkoncentrācija), 10 vienības/ml penicilīna un streptomicīna) un jāuzsēj 96 iedobju mikrotitrēšanas plates iedobēs (100 μl homogenizētas šūnu suspensijas). Pirms ekspozīcijas šūnas 24 stundas vispirms jāinkubē CO2 inkubatorā (5 % CO2, 37 0C, 100 % mitrums). Plastmasas izstrādājumi nedrīkst saturēt estrogēnu.
|
Pieņemamības kritēriji
|
14.
|
Testējamās(-o) ķimikālijas(-u) agonistisko un antagonistisko aktivitāti testē testu sērijā. Testu sērijā ietilpst ne vairāk kā 6 mikrotitrēšanas plates. Katrā testu sērijā ietilpst references standarta, pozitīvās kontroles parauga, negatīvās kontroles parauga un šķīdinātāja kontroļu vismaz 1 pilna atšķaidījumu sērija. 1. un 2. attēlā parādīta plašu sagatavošana agonistu un antagonistu testu sērijai.
|
|
15.
|
Katrs references standartu, testējamo ķimikāliju, visu šķīdinātāja kontroļu un pozitīvo un negatīvo kontroļu atšķaidījums jāanalizē trīs atkārtojumos. Katras trīs atkārtojumos veiktās analīzes rezultātiem jāatbilst 3.A un 4.A tabulā norādītajām prasībām.
|
|
16.
|
References standarta (17β-estradiols agonisma noteikšanai; tamoksifēns antagonisma noteikšanai) pilnu atšķaidījumu sēriju mēra katras testu sērijas pirmajā platē. Lai atlikušo 5 mikrotitrēšanas plašu analīzes rezultātus varētu salīdzināt ar pirmās mikrotitrēšanas plates rezultātiem, kas satur references standarta pilnu koncentrācijas–atbildreakcijas līkni, visās platēs jābūt 3 kontroles paraugiem: šķīdinātāja kontrolei, testētajam references standartam visaugstākajā koncentrācijā un references standarta aptuvenās EC50 (agonisms) vai IC50 (antagonisms) koncentrācijas. Vidējo kontroles paraugu attiecībai pirmajā platē un pārējās 5 platēs jāatbilst 3.C tabulā (agonisms) vai 4.C tabulā (antagonisms) norādītajām prasībām.
|
|
17.
|
Katrai mikrotitrēšanas platei testu sērijā aprēķina z faktoru (10). Z faktors jāaprēķina pēc atbildreakcijām pie references standarta augstākajām un zemākajām koncentrācijām. Mikrotitrēšanas plati uzskata par derīgu, ja tā atbilst 3.C tabulā (agonisms) vai 4.C tabulā (antagonisms) norādītajām prasībām.
|
|
18.
|
References standarta devas–atbildreakcijas līknei jābūt sigmoidālai. EC50 vai IC50, kas iegūta references standarta atšķaidījumu sērijā, ir jāatbilst 3.C tabulā (agonisms) vai 4.C tabulā (antagonisms) norādītajām prasībām.
|
|
19.
|
Katrā testu sērijā jābūt pozitīvās kontroles un negatīvās kontroles paraugam. Relatīvajai indukcijai, kas aprēķināta, pamatojoties uz pozitīvās kontroles paraugu un negatīvās kontroles paraugu, jāatbilst 3.C tabulā (agonisms) vai 4.C tabulā (antagonisms) norādītajām prasībām.
|
|
20.
|
Visā mērījumu laikā jānosaka, kāds ir references standarta augstākās koncentrācijas indukcijas koeficients, proti, vidējo augstāko 17β-estradiola references standarta relatīvās gaismas vienības (RLU) atbildreakciju dala ar vidējo references šķīdinātāja kontroles RLU atbildreakciju. Šim indukcijas koeficientam jāatbilst 3.C tabulā (agonisms) vai 4.C tabulā (antagonisms) norādītajām minimālajām prasībām par vairākkāršo indukciju.
|
|
21.
|
Tikai tās mikrotitrēšanas plates, kas atbilst visiem iepriekš minētajiem apstiprināšanas kritērijiem, uzskatāmas par derīgām un izmantojamas testējamo ķimikāliju atbildreakcijas novērtēšanai.
|
|
22.
|
Pieņemamības kritērijus piemēro gan priekšskrīningam, gan visaptverošiem testiem.
|
1. tabula.
References standarta, pozitīvās kontroles (PC) un negatīvās kontroles (NC) koncentrācijas agonistu CALUX biotestam
|
|
Viela
|
CAS reģ. nr.
|
Testa diapazons (M)
|
|
References standarts
|
17β-estradiols
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Pozitīvā kontrole (PC)
|
17α-metiltestosterons
|
58-18-4
|
3*10-6
|
|
Negatīvā kontrole (NC)
|
kortikosterons
|
50-22-6
|
1*10-8
|
2. tabula.
References standarta, pozitīvās kontroles (PC) un negatīvās kontroles (NC) koncentrācijas antagonistu CALUX biotestam
|
|
Viela
|
CAS reģ. nr.
|
Testa diapazons (M)
|
|
References standarts
|
tamoksifēns
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Pozitīvā kontrole (PC)
|
4-hidroksitamoksifēns
|
68047-06-3
|
1*10-9
|
|
Negatīvā kontrole (NC)
|
resveratrols
|
501-36-0
|
1,0*10-5
|
3. tabula.
Pieņemamības kritēriji agonistu ERα CALUX biotestam
|
A — atsevišķi paraugi platē
|
Kritērijs
|
|
1
|
Trīs atkārtojumu iedobju maksimālā %SN (NC, PC, katram testējamās ķimikālijas un references standarta atšķaidījumam, izņemot C0)
|
< 15 %
|
|
2
|
Trīs atkārtojumu iedobju maksimālā %SN (references standartam un testējamās ķimikālijas šķīdinātāja kontrolēm (C0, SC))
|
< 30 %
|
|
3
|
LDH maksimālā noplūde kā citotoksicitātes mērījums
|
< 120 %
|
|
B — vienā mikrotitrēšanas platē
|
|
|
4
|
Attiecība starp references standarta šķīdinātāja kontroli (C0; 1. plate) un testējamās ķimikālijas šķīdinātāja kontroli (SC; 2.–x. plate)
|
no 0,5 līdz 2,0
|
|
5
|
Attiecība starp apt. EC50 un references standarta augstāko koncentrāciju 1. platē un apt. EC50 un references standarta augstāko koncentrāciju 2.–x. platē (C4, C8)
|
no 0,70 līdz 1,30
|
|
6
|
Z faktors katrai platei
|
>0,6
|
|
C — analīžu vienā sērijā (visas plates vienā sērijā)
|
|
|
7
|
References standarta sigmoidāla līkne
|
Jā (17ß-estradiols)
|
|
8
|
References standarta (17ß-estradiols) EC50 diapazons
|
4 * 10-12–4 * 10-11 M
|
|
9
|
Augstākās 17ß-estradiola koncentrācijas minimālā vairākkāršā indukcija attiecībā uz references standarta šķīdinātāja kontroli
|
5
|
|
10
|
Relatīvā indukcija (%) PC
|
> 30 %
|
|
11
|
Relatīvā indukcija (%) NC
|
<10 %
|
Apt.: aptuveni; PC: pozitīvā kontrole; NC: negatīvā kontrole; SC: testējamās ķimikālijas šķīdinātāja kontrole; C0: references standarta šķīdinātāja kontrole; SD: standartnovirze LDH: laktātdehidrogenāze.
4. tabula.
Pieņemamības kritēriji antagonistu ERα CALUX biotestam
|
A — atsevišķi paraugi platē
|
Kritērijs
|
|
1
|
Trīs atkārtojumu iedobju maksimālā %SN (NC, PC, katram testējamās ķimikālijas un references standarta atšķaidījumam, šķīdinātāja kontrolei (C0))
|
< 15 %
|
|
2
|
Trīs atkārtojumu iedobju maksimālā %SN (nesēja kontrolei (VC) un references standarta augstākajai koncentrācijai (C8))
|
< 30 %
|
|
3
|
LDH maksimālā noplūde kā citotoksicitātes mērījums
|
< 120 %
|
|
B — vienā mikrotitrēšanas platē
|
|
|
4
|
Attiecība starp references standarta šķīdinātāja kontroli (C0; 1. plate) un testējamās ķimikālijas šķīdinātāja kontroli (SC; 2.–x. plate)
|
no 0,70 līdz 1,30
|
|
5
|
Attiecība starp apt. IC50 references standarta koncentrāciju 1. platē un apt. IC50 references standarta koncentrāciju 2.–x. platē (C4)
|
no 0,70 līdz 1,30
|
|
6
|
Attiecība starp references standarta augstāko koncentrāciju 1. platē un references standarta augstāko koncentrāciju 2.–x. platē (C8)
|
no 0,50 līdz 2,0
|
|
7
|
Z faktors katrai platei
|
>0,6
|
|
C — analīžu vienā sērijā (visas plates vienā sērijā)
|
|
|
8
|
References standarta sigmoidāla līkne
|
Jā (tamoksifēns)
|
|
9
|
References standarta (tamoksifēns) IC50 diapazons
|
1*10-8 - 1*10-7 M
|
|
10
|
References standarta šķīdinātāja kontroles minimālā vairākkāršā indukcija attiecībā uz tamoksifēna augstāko koncentrāciju
|
2,5
|
|
11
|
Relatīvā indukcija (%) PC
|
<70 %
|
|
12
|
Relatīvā indukcija (%) NC
|
>85 %
|
Apt.: aptuveni; PC: pozitīvā kontrole; NC: negatīvā kontrole; VC: nesēja kontrole (šķīdinātāja kontrole bez agonistu references standarta konstantas koncentrācijas); SC: testējamās ķimikālijas šķīdinātāja kontrole; C0: references standarta šķīdinātāja kontrole; SD: standartnovirze LDH: laktātdehidrogenāze.
Šķīdinātāja/nesēja kontrole, references standarti, pozitīvās kontroles, negatīvās kontroles
|
23.
|
Gan priekšskrīningā, gan visaptverošā testā jāizmanto viena un tā pati šķīdinātāja/nesēja kontrole, references standarti, pozitīvās kontroles un negatīvās kontroles. Arī references standartu, pozitīvo kontroļu un negatīvo kontroļu koncentrācijai jābūt tādai pašai.
|
Šķīdinātāja kontrole
|
24.
|
Šķīdinātāju, kurā izšķīdina testējamo ķimikāliju, testē kā šķīdinātāja kontroli. ERα CALUX biotesta validēšanai kā nesēju izmantoja dimetilsulfoksīdu (DMSO, 1 % (tilpumkoncentrācija); CAS Nr. 67-68-5). Ja izmanto citu šķīdinātāju, nevis DMSO, visi references standarti, kontroles un testējamās ķimikālijas jātestē tajā pašā nesējā. Jāievēro, ka šķīdinātāja kontrole antagonistu pētījumiem satur noteiktas koncentrācijas agonistu references standartu 17β-estradiolu (aptuveni EC50 koncentrācija). Lai testētu antagonistu pētījumiem izmantojamo šķīdinātāju, jāsagatavo un jātestē nesēja kontrole.
|
Nesēja kontrole (antagonisms)
|
25.
|
Lai testētu antagonismu, testa barotnei pievieno agonistu references standartu (17β-estradiolu) konstantā koncentrācijā (aptuveni EC50 koncentrācija). Lai noteiktu testējamo ķimikāliju izšķīdināšanai izmantojamā šķīdinātāja antagonismu, jāsagatavo testa barotne, kas nesatur agonistu references standartu (17β-estradiola) konstantā koncentrācijā. Šis kontroles paraugs paredzēts kā nesēja kontrole. Dimetilsulfoksīds (DMSO, 1 % (tilp./tilp.); ERα CALUX biotesta validēšanai kā nesēju izmantoja dimetilsulfoksīdu (DMSO, 1 % (tilpumkoncentrācija); CAS Nr. 67-68-5). Ja izmanto citu šķīdinātāju, nevis DMSO, visi references standarti, kontroles un testējamās ķimikālijas jātestē tajā pašā nesējā.
|
Izmantojamie standarti
|
26.
|
Agonistu references standarts ir 17β-estradiols (1. tabula). References standarts nozīmē 17β-estradiola astoņu koncentrāciju atšķaidījumu sēriju (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Antagonistu references standarts ir tamoksifēns (2. tabula). References standarts nozīmē tamoksifēna astoņu koncentrāciju atšķaidījumu sēriju (3*10-09, 1*10-08, 3*10-08, 1*10-07, 3*10-07, 1*10-06, 3*10-06, 1*10-05 M). Antagonistu references standarta katru koncentrāciju inkubē kopā ar agonistu references standartu 17β-estradiolu konstantā koncentrācijā (3 * 10-12 M).
|
Pozitīvā kontrole
|
28.
|
Pozitīvā kontrole agonistu pētījumiem ir 17α-metiltestosterons (1. tabula).
|
|
29.
|
Pozitīvā kontrole antagonistu pētījumiem ir 4-hidroksitamoksifēns (2. tabula). Antagonistu pozitīvo kontroli inkubē kopā ar agonistu references standartu 17β-estradiolu konstantā koncentrācijā (3 * 10-12 M).
|
Negatīvā kontrole
|
30.
|
Negatīvā kontrole agonistu pētījumiem ir kortikosterons (1. tabula).
|
|
31.
|
Negatīvā kontrole antagonistu pētījumiem ir resveratrols (2. tabula). Antagonistu negatīvo kontroli inkubē kopā ar agonistu references standartu 17β-estradiolu konstantā koncentrācijā (3 * 10-12 M).
|
Laboratorijas kompetences pierādīšana (sk. 14. punktu un 3. un 4. tabulu šīs testēšanas metodes sadaļā “ER TA TESTA KOMPONENTI”)
Nesējs
|
32.
|
Testējamo ķimikāliju izšķīdināšanai izmantojamajam šķīdinātājam pilnībā jāizšķīdina testējamā ķimikālija, un tam jābūt sajaucamam ar šūnu barotni. Piemēroti šķīdinātāji ir DMSO, ūdens un etanols (tīrība no 95 % līdz 100 %). Ja kā šķīdinātāju izmanto DMSO, inkubēšanas laikā DMSO maksimālā koncentrācija nedrīkst pārsniegt 1 % (tilpumkoncentrācija). Pirms lietošanas jāpārbauda, vai šķīdinātājs nav citotoksisks un neietekmē testa veikšanu.
|
References standartu, pozitīvo kontroļu, negatīvo kontroļu un testējamo ķimikāliju sagatavošana
|
33.
|
References standartus, pozitīvās kontroles, negatīvās kontroles un testējamās ķimikālijas izšķīdina 100 % DMSO (vai piemērotā šķīdinātājā). Pēc tam tajā pašā šķīdinātājā sagatavo attiecīgus (sērijveida) atšķaidījumus. Pirms izšķīdināšanas jānogaida, līdz visas vielas sasilst līdz istabas temperatūrai. Svaigi sagatavotos references standartu, pozitīvo kontroļu, negatīvo kontroļu un testējamo ķimikāliju izejšķīdumos nedrīkst būt redzamas nogulsnes vai duļķes. References standartu un kontroļu izejšķīdumus var sagatavot lielā apjomā. Pirms katra eksperimenta jāsagatavo svaigi testējamo ķimikāliju izejšķīdumi. References standartu, pozitīvo kontroļu, negatīvo kontroļu un testējamo ķimikāliju galīgie atšķaidījumi katram eksperimentam jāsagatavo svaigi un jāizmanto 24 stundās pēc sagatavošanas.
|
Šķīdība, citotoksicitāte un diapazona noteikšana
|
34.
|
Priekšskrīningā nosaka testējamo ķimikāliju šķīdību izvēlētajā šķīdinātājā. Sagatavo izejšķīdumu ar maksimālo koncentrāciju 0,1 M. Ja šādā koncentrācijā ir šķīdības problēmas, jāsagatavo mazākas koncentrācijas izejšķīdumi, līdz testējamās ķimikālijas izšķīst pilnībā. Priekšskrīningā testē testējamo ķimikāliju 1:10 sērijveida atšķaidījumus. Agonistu vai antagonistu testēšanā maksimālā testa koncentrācija ir 1 mM. Pēc priekšskrīninga iegūst precīzāku testējamo ķimikāliju koncentrāciju diapazonu, kas jātestē visaptverošajā testā. Visaptverošā testēšanā izmantojamajiem atšķaidījumiem jābūt 1x, 3x, 10x, 30x, 100x, 300x, 1000x un 3 000x.
|
|
35.
|
Agonistu un antagonistu testu protokolos ir ietverta citotoksicitātes testēšana (11). Citotoksicitātes testēšana ir iekļauta gan priekšskrīningā, gan visaptverošajā testā. ERα CALUX biotesta validēšanas laikā citotoksicitātes novērtēšanai izmantotā metode bija laktātdehidrogenāzes (LDH) noplūdes tests kombinācijā ar šūnu kvalitatīvu vizuālu pārbaudi (sk. 4.1. papildinājumu) pēc eksponēšanas testējamām ķimikālijām. Tomēr citotoksicitātes noteikšanai var izmantot citas kvantitatīvas metodes (piem., uz tetrazolu balstītu kolorimetrisko (MTT) testu vai citotoksicitātes CALUX biotestu). Parasti testējamo ķimikāliju koncentrācijas, pie kādām šūnu dzīvotspēja samazinās par vairāk nekā 20 %, uzskatāmas par citotoksiskām, tāpēc tās nav izmantojamas datu novērtēšanai. Attiecībā uz LDH noplūdes testu testējamās ķimikālijas koncentrācija uzskatāma par citotoksisku, ja LDH noplūdes procents ir lielāks par 120 %.
|
Eksponēšana testējamai ķimikālijai un izkārtojums testa platē
|
36.
|
Pēc saplūdušu kultivētu šūnu kolbas tripsinizācijas šūnas atkārtoti suspendē 1 x 105 šūnas/ml bezestrogēna testa barotnē. Simts μl atkārtoti suspendētu šūnu uzsēj 96 iedobju mikrotitrēšanas plates iekšējās iedobēs. Ārējās iedobēs iepilda 200 μl fosfātu fizioloģiskā buferšķīduma (PBS) (sk. 1. un 2. attēlu). Uzsētās šūnas 24 stundas vispirms inkubē CO2 inkubatorā (5 % CO2, 37 °C, 100 % mitrums).
|
|
37.
|
Pēc preinkubācijas plates vizuāli pārbauda, lai noteiktu citotoksicitāti (sk. 4.1. papildinājumu), kontamināciju un konfluenci. Testēšanai izmanto tikai tās plates, kurās nav redzamas citotoksicitātes un kontaminācijas un ir vismaz 85 % konfluences. No iekšējām iedobēm uzmanīgi izvada barotni, ko aizstāj ar 200 μl bezestrogēna testa barotnes, kas satur references standartu, testējamo ķimikāliju, pozitīvo kontroļu, negatīvo kontroļu un šķīdinātāja kontroļu attiecīgo atšķaidījumu sēriju (5. tabula: agonistu pētījumi; 6. tabula: antagonistu pētījumi). Visus references standartus, testējamās ķimikālijas, pozitīvās kontroles, negatīvās kontroles un šķīdinātāja kontroles testē trīs atkārtojumos. 1. attēlā redzams plates izkārtojums agonistu testēšanai. 2. attēlā redzams plates izkārtojums antagonistu testēšanai. Plates izkārtojums priekšskrīningā un visaptverošajā testēšanā ir vienāds. Antagonistu testēšanā visās iekšējās iedobēs, izņemot nesēja kontroles (VC) iedobes, ir arī agonistu references standarts (17β-estradiols) konstantā koncentrācijā (3 * 10-12 M). Jāievēro, ka katrā TC platē jāpievieno references standarti C8 un C4.
|
|
38.
|
Pēc šūnu eksponēšanas visām ķimikālijām 96 iedobju mikrotitrēšanas plates jāinkubē vēl 24 stundas CO2 inkubatorā (5 % CO2, 37 °C, 100 % mitrums).
|
1. attēls.
Izkārtojums 96 iedobju mikrotitrēšanas platēs priekšskrīningā un agonistiskās iedarbības novērtēšanā
C0 = references standartšķīdinātājs.
C(1-8) = references standarta atšķaidījumu sērija (1–8, no zemas līdz augstai koncentrācijai).
PC = pozitīvā kontrole.
NC: = negatīvā kontrole.
TCx-(1–8) = testējamās ķimikālijas atšķaidījumi (1–8, no zemas līdz augstai koncentrācijai), ko izmanto testējamās ķimikālijas priekšskrīningā un agonistiskās iedarbības novērtēšanā.
SC = testējamās ķimikālijas šķīdinātāja kontrole (optimāli tas pats šķīdinātājs, kas ir C0, taču, iespējams, no citas partijas).
Pelēkās šūnas: = ārējās iedobes, kurās iepilda 200 μl PBS.
2. attēls.
Izkārtojums 96 iedobju mikrotitrēšanas platēs priekšskrīningā un antagonistiskās iedarbības novērtēšanā

C0 = references standartšķīdinātājs.
C(1-8) = references standarta atšķaidījumu sērija (1–8, no zemas līdz augstai koncentrācijai).
NC: = negatīvā kontrole.
PC = pozitīvā kontrole.
TCx-(1–8) = testējamās ķimikālijas atšķaidījumi (1–8, no zemas līdz augstai koncentrācijai), ko izmanto testējamās ķimikālijas priekšskrīningā un agonistiskās iedarbības novērtēšanā.
SC = testējamās ķimikālijas šķīdinātāja kontrole (optimāli tas pats šķīdinātājs, kas ir C0, taču, iespējams, no citas partijas).
VC = nesēja kontrole (šķīdinātāja kontrole bez agonistu references standarta (17β-estradiola) konstantā koncentrācijā.
Pelēkās šūnas: = ārējās iedobes, kurās iepilda 200 μl PBS.
Piezīme. Visās iekšējās iedobēs, izņemot nesēja kontroles (VC) iedobes, ir arī agonistu references standarts (17β-estradiols) konstantā koncentrācijā (3,0 * 10-12 M).
Luminiscences mērīšana
|
39.
|
Luminiscences mērīšana ir detalizēti aprakstīta agonistu un antagonistu testu protokolā (10). Barotne no iedobēm jāizvada, bet šūnas pēc 24 stundu inkubēšanas jālizē, lai atvērtu šūnas membrānu un varētu izmērīt luciferāzes aktivitāti.
|
|
40.
|
Luminiscences izmērīšanai šajā procedūrā nepieciešams luminometrs ar 2 inžektoriem. Luciferāzes reakciju sāk, inžektējot substrāta luciferīnu. Reakciju pārtrauc, pievienojot 0,2 M NaOH. Reakciju pārtrauc, lai nepieļautu luminiscences pārnešanu no vienas iedobes citā.
|
|
41.
|
No katras iedobes izstaroto gaismu izsaka kā relatīvās gaismas vienības (RLU) uz katru iedobi.
|
Priekšskrīnings
|
42.
|
Priekšskrīninga analīzes rezultātus izmanto, lai noteiktu visaptverošajā testēšanā testējamo ķimikāliju precīzāku koncentrāciju diapazonu. Priekšskrīninga analīzes rezultātu novērtēšana un visaptverošajā testēšanā testējamo ķimikāliju precīzāka koncentrāciju diapazona noteikšana ir detalizēti aprakstīta agonistu un antagonistu testu protokolā (10). Šeit ir sniegts īss kopsavilkums par testējamo ķimikāliju precīzāka koncentrāciju diapazona noteikšanu agonistu un antagonistu testēšanā. Norādījumus par sērijveida atšķaidījumiem sk. 5. un 6. tabulā.
|
Koncentrāciju izvēle agonistiskās iedarbības novērtēšanai
|
43.
|
Priekšskrīningā testējamās ķimikālijas jātestē, izmantojot 5. tabulā (agonisms) un 6. tabulā (antagonisms) norādītās atšķaidījumu sērijas. Visas koncentrācijas jātestē trīs atkārtojumu iedobēs atbilstoši 1. attēlā (agonisms) vai 2. attēlā (antagonisms) norādītajam plašu izkārtojumam.
|
|
44.
|
Tikai pieņemamības kritērijiem atbilstoši analīzes rezultāti (3. tabula) uzskatāmi par derīgiem un izmantojami testējamo ķimikāliju atbildreakcijas novērtēšanai. Ja viena vai vairākas mikrotitrēšanas plates analīzes sērijā neatbilst pieņemamības kritērijiem, attiecīgās mikrotitrēšanas plates jāanalizē atkārtoti. Ja pirmā plate, kura satur visu references standarta atšķaidījumu sēriju, neatbilst pieņemamības kritērijiem, visa testa sērija (6 plates) jāanalizē atkārtoti.
|
|
45.
|
Testējamo ķimikāliju sākotnējie koncentrāciju diapazoni jākoriģē un priekšskrīnings jāatkārto, ja:
|
—
|
novēro citotoksicitāti. Priekšskrīninga procedūra jāatkārto ar testējamās ķimikālijas mazākām, necitotoksiskām koncentrācijām;
|
|
—
|
testējamās ķimikālijas priekšskrīningā neiegūst pilnu devas–atbildreakcijas līkni, jo testētās koncentrācijas rada maksimālu indukciju. Priekšskrīnings jāatkārto ar mazākām testējamās ķimikālijas koncentrācijām.
|
|
|
46.
|
Ja novēro derīgu, no devas atkarīgu atbildreakciju, jāizvēlas (mazākā) koncentrācija, kādā novēro maksimālo indukciju un nenovēro citotoksicitāti. Testējamās ķimikālijas augstākajai koncentrācijai, kas jātestē visaptverošajā testā, jābūt 3 reizes lielākai par šo izvēlēto koncentrāciju.
|
|
47.
|
Testējamās ķimikālijas visa precīzāku atšķaidījumu sērija jāsagatavo atbilstoši 5. tabulā norādītajiem atšķaidīšanas etapiem, sākot ar minēto augstāko koncentrāciju.
|
|
48.
|
Testējamā ķimikālija, kas neizraisa nekādu agonistisku iedarbību, jātestē visaptverošajos testos, sākot ar augstāko necitotoksisko koncentrāciju, kas konstatēta priekšskrīningā.
|
Koncentrāciju izvēle antagonistiskās iedarbības novērtēšanai
|
49.
|
Tikai pieņemamības kritērijiem atbilstoši analīzes rezultāti (4. tabula) uzskatāmi par derīgiem un izmantojami testējamo ķimikāliju atbildreakcijas novērtēšanai. Ja viena vai vairākas mikrotitrēšanas plates analīzes sērijā neatbilst pieņemamības kritērijiem, attiecīgās mikrotitrēšanas plates jāanalizē atkārtoti. Ja pirmā plate, kura satur visu references standarta atšķaidījumu sēriju, neatbilst pieņemamības kritērijiem, visa testa sērija (6 plates) jāanalizē atkārtoti.
|
|
50.
|
Testējamo ķimikāliju sākotnējie koncentrāciju diapazoni jākoriģē un priekšskrīnings jāatkārto, ja:
|
—
|
novēro citotoksicitāti. Priekšskrīninga procedūra jāatkārto ar testējamās ķimikālijas mazākām, necitotoksiskām koncentrācijām;
|
|
—
|
testējamās ķimikālijas priekšskrīningā neiegūst pilnu devas–atbildreakcijas līkni, jo testētās koncentrācijas rada maksimālu inhibīciju. Priekšskrīnings jāatkārto ar mazākām testējamās ķimikālijas koncentrācijām.
|
|
|
51.
|
Ja novēro derīgu, no devas atkarīgu atbildreakciju, jāizvēlas (mazākā) koncentrācija, kādā novēro maksimālo inhibīciju un nenovēro citotoksicitāti. Testējamās ķimikālijas augstākajai koncentrācijai, kas jātestē visaptverošajā testā, jābūt 3 reizes lielākai par šo izvēlēto koncentrāciju.
|
|
52.
|
Testējamās ķimikālijas visa precīzāku atšķaidījumu sērija jāsagatavo atbilstoši 6. tabulā norādītajiem atšķaidīšanas etapiem, sākot ar minēto augstāko koncentrāciju.
|
|
53.
|
Testējamā ķimikālija, kas neizraisa nekādu antagonistisku iedarbību, jātestē visaptverošajos testos, sākot ar augstāko necitotoksisko koncentrāciju, kas konstatēta priekšskrīningā..
|
Visaptverošie testi
|
54.
|
Kad noteikts precīzākais koncentrāciju diapazons, testējamās ķimikālijas jātestē visaptverošajā testā, izmantojot 5. tabulā (agonisms) un 6. tabulā (antagonisms) norādītās atšķaidījumu sērijas. Visas koncentrācijas jātestē trīs atkārtojumu iedobēs atbilstoši 1. attēlā (agonisms) vai 2. attēlā (antagonisms) norādītajam plašu izkārtojumam.
|
|
55.
|
Tikai pieņemamības kritērijiem atbilstoši analīzes rezultāti (3.un 4. tabula) uzskatāmi par derīgiem un izmantojami testējamo ķimikāliju atbildreakcijas novērtēšanai. Ja viena vai vairākas mikrotitrēšanas plates analīzes sērijā neatbilst pieņemamības kritērijiem, attiecīgās mikrotitrēšanas plates jāanalizē atkārtoti. Ja pirmā plate, kura satur visu references standarta atšķaidījumu sēriju, neatbilst pieņemamības kritērijiem, visa testa sērija (6 plates) jāanalizē atkārtoti.
|
5. tabula.
References standartu, kontroļu un testējamo ķimikāliju koncentrācija un atšķaidījumi, ko izmanto agonistu testēšanā
|
References standarts 17β-estradiols
|
TCx — priekšskrīnings
|
TCx — visaptverošais tests
|
Kontroles
|
|
konc. (M)
|
atšķaidījums
|
atšķaidījums
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC:
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx —testējamā ķimikālija x.
PC —pozitīvā kontrole (17α-metiltestosterons).
NC —negatīvā kontrole (kortikosterons).
C0 —references standarta šķīdinātāja kontrole
SC —testējamās ķimikālijas šķīdinātāja kontrole
|
6. tabula.
References standartu, kontroļu un testējamo ķimikāliju koncentrācija un atšķaidījumi, ko izmanto antagonistu testēšanā
|
References standarts tamoksifēns
|
TCx — priekšskrīnings
|
TCx — visaptverošais tests
|
Kontroles
|
|
konc. (M)
|
atšķaidījums
|
atšķaidījums
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3 000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1 000 x
|
NC:
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Pievienotais agonists
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
konc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiols
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx —testējamā ķimikālija x.
PC —pozitīvā kontrole (4-hidroksitamoksifēns)
NC —negatīvā kontrole (resveratrols).
C0 —references standarta šķīdinātāja kontrole
SC —testējamās ķimikālijas šķīdinātāja kontrole
VC —nesēja kontrole (nesatur agonistu references standartu 17β-estradiolu konstantā koncentrācijā (3 * 10-12 M)
|
Datu apkopošana un datu analīze
|
56.
|
Agonistu testēšana: [pēc priekšskrīninga un visaptverošajiem testiem nosaka testējamās ķimikālijas EC10, EC50, PC10, PC50 un maksimālo indukciju (TCxmax). Antagonistu testēšana: aprēķina IC20, IC50, PC80, PC50 un minimālo indukciju (TCxmin). 3. attēlā (agonisms) un 4. attēlā (antagonisms) šie parametri attēloti grafiski. Nepieciešamos parametrus aprēķina, pamatojoties uz katras testējamās ķimikālijas relatīvo indukciju (attiecībā pret references standarta maksimālo indukciju (= 100 %)). Datu novērtēšanai jāizmanto nelineārā regresija (mainīgs slīpums, 4 parametri) atbilstoši šim vienādojumam:

kur:
X = devas vai koncentrācijas logaritms;
Y = atbildreakcija (relatīvā indukcija (%));
Virsotne = maksimālā indukcija (%)
Bāze = minimālā indukcija (%);
LogEC50 = koncentrācijas, pie kuras novēro 50 % no maksimālās atbildreakcijas, logaritms
Hilla virziena koeficients = Virziena koeficients vai Hilla virziena koeficients.
|
|
57.
|
Neapstrādātie dati no luminometra, kas izteikti kā relatīvās gaismas vienības (RLU), jāpārsūta uz priekšskrīningam un visaptverošajam testam īpaši izveidotu izklājlapas veidni. Neapstrādātajiem datiem jāatbilst 3.A un 3.B tabulā (agonisms) vai 4.A un 4.B tabulā (antagonisms) norādītajiem pieņemamības kritērijiem. Ja neapstrādātie dati atbilst pieņemamības kritērijiem, nepieciešamo parametru noteikšanai veic tālāk aprakstītos aprēķinus.
|
Agonisms
|
—
|
References standarta šķīdinātāja kontroles vidējo RLU atņem no neapstrādātajiem atsauces datiem par references standartiem.
|
|
—
|
Testējamās ķimikālijas šķīdinātāja kontroles vidējo RLU atņem no neapstrādātajiem datiem par testējamo ķimikāliju.
|
|
—
|
Aprēķina references standarta katras koncentrācijas relatīvo indukciju. References standarta augstākās koncentrācijas indukcija ir 100 %.
|
|
—
|
Aprēķina testējamās ķimikālijas katras koncentrācijas relatīvo indukciju salīdzinājumā ar references standarta augstāko koncentrāciju 100 %.
|
|
—
|
Pēc nelineārās regresijas (mainīgs slīpums, 4 parametri) novērtē analīzes rezultātus.
|
|
—
|
Nosaka references standarta EC50 un EC10.
|
|
—
|
Nosaka testējamo ķimikāliju EC50 un EC10.
|
|
—
|
Nosaka testējamās ķimikālijas maksimālo relatīvo indukciju (TCmax).
|
|
—
|
Nosaka testējamo ķimikāliju PC10 un PC50.
|
Testējamām ķimikālijām ne vienmēr var iegūt pilnu devas–atbildreakcijas līkni, piem., citotoksicitātes vai šķīdības problēmu dēļ. Tad nav iespējams noteikt arī EC50, EC10 un PC50. Tādos gadījumos iespējams noteikt tikai PC10 un TCmax.
Antagonisms
|
—
|
References standarta augstākās koncentrācijas vidējo RLU atņem no neapstrādātajiem atsauces datiem par references standartiem.
|
|
—
|
References standarta augstākās koncentrācijas vidējo RLU atņem no neapstrādātajiem atsauces datiem par testējamām ķimikālijām.
|
|
—
|
Aprēķina references standarta katras koncentrācijas relatīvo indukciju. References standarta zemākās koncentrācijas indukcija ir 100 %.
|
|
—
|
Aprēķina testējamās ķimikālijas katras koncentrācijas relatīvo indukciju salīdzinājumā ar references standarta zemāko koncentrāciju 100 %.
|
|
—
|
Pēc nelineārās regresijas (mainīgs slīpums, 4 parametri) novērtē analīzes rezultātus.
|
|
—
|
Nosaka references standarta IC50 un IC20.
|
|
—
|
Nosaka testējamo ķimikāliju IC50 un IC20.
|
|
—
|
Nosaka testējamās ķimikālijas minimālo relatīvo indukciju (TCmin).
|
|
—
|
Nosaka testējamo ķimikāliju PC80 un PC50.
|
3. attēls.
Pārskats par agonistu testā noteiktajiem parametriem

EC10 = vielas koncentrācija, pie kuras novēro 10 % no tās maksimālās atbildreakcijas.
EC50 = vielas koncentrācija, pie kuras novēro 50 % no tās maksimālās atbildreakcijas.
PC10 = testējamās ķimikālijas koncentrācija, pie kuras tās atbildreakcija ir vienāda ar references standarta EC10.
PC50 = testējamās ķimikālijas koncentrācija, pie kuras tās atbildreakcija ir vienāda ar references standarta EC50.
TCxmax = testējamās ķimikālijas maksimālā relatīvā indukcija.
4. attēls.
Pārskats par antagonistu testā noteiktajiem parametriem
IC20 = vielas koncentrācija, pie kuras novēro 80 % no tās maksimālās atbildreakcijas (20 % inhibīcija).
IC50 = vielas koncentrācija, pie kuras novēro 50 % no tās maksimālās atbildreakcijas (50 % inhibīcija).
PC80 = testējamās ķimikālijas koncentrācija, pie kuras tās atbildreakcija ir vienāda ar references standarta IC20.
PC50 = testējamās ķimikālijas koncentrācija, pie kuras tās atbildreakcija ir vienāda ar references standarta IC50.
TCxmin = testējamās ķimikālijas minimālā relatīvā indukcija.
Testējamām ķimikālijām ne vienmēr var iegūt pilnu devas–atbildreakcijas līkni, piem., citotoksicitātes vai šķīdības problēmu dēļ. Tad nav iespējams noteikt arī IC50, IC20 un PC50. Tādos gadījumos iespējams noteikt tikai PC20 un TCmin.
|
58.
|
Rezultātu pamatā ir divas (vai trīs) neatkarīgas izpildes reizes. Ja divās izpildes reizēs tiek iegūti salīdzināmi un līdz ar to reproducējami rezultāti, nav nepieciešams testu izpildīt vēl trešo reizi. Rezultāti ir pieņemami, ja tie:
|
—
|
atbilst pieņemamības kritērijiem (sk. 14.–22. punktu sadaļā “Pieņemamības kritēriji”);
|
|
Datu interpretācijas kritēriji
|
59.
|
Interpretējot datus un lemjot, vai testējamā ķimikālija ir pozitīva vai negatīva, jāizmanto tālāk aprakstītie kritēriji.
|
Katrā visaptverošajā testā testējamo ķimikāliju uzskata par pozitīvu, ja:
|
1
|
TCmax ir vienāda ar vai lielāka par 10 % no references standarta maksimālās atbildreakcijas (REF10).
|
|
2
|
Vismaz 2 secīgas testējamās ķimikālijas koncentrācijas ir vienādas ar vai lielākas par REF10.
|
|
|
Katrā visaptverošajā testā testējamo ķimikāliju uzskata par negatīvu, ja:
|
1
|
TCmax nepārsniedz 10 % no references standarta maksimālās atbildreakcijas (REF10);
|
|
2
|
mazāk nekā 2 testējamās ķimikālijas koncentrācijas ir vienādas ar vai lielākas par REF10.
|
|
|
Katrā visaptverošajā testā testējamo ķimikāliju uzskata par pozitīvu, ja:
|
1
|
TCmin ir vienāda ar vai mazāka par 80 % no references standarta maksimālās atbildreakcijas (REF80 = 20 % inhibīcija).
|
|
2
|
Vismaz 2 secīgas testējamās ķimikālijas koncentrācijas ir vienādas ar vai mazākas par REF80.
|
|
|
Katrā visaptverošajā testā testējamo ķimikāliju uzskata par negatīvu, ja:
|
1
|
TCmin pārsniedz 80 % no references standarta maksimālās atbildreakcijas (REF80 = 20 % inhibīcija).
|
|
2
|
Mazāk nekā 2 testējamās ķimikālijas koncentrācijas ir vienādas ar vai lielākas par REF80.
|
|
|
|
60.
|
Lai raksturotu testējamās ķimikālijas pozitīvās atbildreakcijas potenci, apskata šādus parametrus: ietekmes intensitāte (agonisms: TCmax; antagonisms: TCmin) un koncentrācija, pie kuras novērojama ietekme (agonisms: EC10, EC50, PC10, PC50; antagonisms: IC20, IC50, PC80, PC50).
|
TESTĒŠANAS PĀRSKATS
|
61.
|
Sk. 20. punktu sadaļā “ER TA TESTA KOMPONENTI”.
|
LITERATŪRA
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
4.1.
PAPILDINĀJUMS. ŠŪNU DZĪVOTSPĒJAS VIZUĀLA PĀRBAUDE
B.67. ZĪDĪTĀJU ŠŪNU GĒNU MUTĀCIJU IN VITRO TESTI AR TIMIDĪNKINĀZES GĒNU
IEVADS
|
1.
|
Šī testēšanas metode (TM) ir ekvivalenta ESAO Testēšanas vadlīnijā (TG) Nr. 490 (2016) aprakstītajam. Testēšanas metodes tiek pastāvīgi pārskatītas zinātnes sasniegumu, normatīvo prasību un dzīvnieku labturības apsvērumu gaismā. Peļu limfomas ķīmiskā analīze (MLA) un TK6 tests ar timidīnkināzes (TK) lokusu sākotnēji bija iekļauti B.17. testēšanas metodē. Vēlāk Genotoksiskuma testēšanas starptautiskā darbsemināra (IWGT) MLA ekspertu darbgrupa sagatavoja starptautiski saskaņotus ieteikumus par MLA ķīmiskās analīzes pieņemamības kritērijiem un datu interpretēšanu (1)(2)(3)(4)(5), un minētie ieteikumi ir iestrādāti šajā jaunajā, B.67. testēšanas metodē. Šī testēšanas metode ir paredzēta MLA un – tā kā tajā izmanto TK lokusu – arī TK6 testam. MLA jau iepriekš plaši izmantota normatīvām vajadzībām, bet TK6 lietots daudz retāk. Lai gan abu šūnu līniju beigupunkti ir līdzīgi, tās nav savstarpēji aizstājamas, un normatīvajās programmās var būt pamatoti dota priekšroka vienas vai otras līnijas izmantošanai kādai konkrētai normatīvai vajadzībai. Piemēram, MLA validācijā pierādījās, ka tā ir piemērota, lai detektētu ne tikai gēnu mutāciju, bet arī testējamās ķimikālijas spēju inducēt strukturālus hromosomu bojājumus. Šī testēšanas metode pieder pie ģenētiski toksikoloģiskās testēšanas metožu sērijas. ESAO nesen izstrādājusi dokumentu, kas īsi informē par ģenētiski toksikoloģisko testēšanu un sniedz pārskatu par neseniem ESAO ģenētiskā toksiskuma testēšanas vadlīniju grozījumiem (6).
|
|
2.
|
Šā zīdītāju šūnu gēnu mutāciju in vitro testa nolūks ir detektēt ķimikāliju izraisītas gēnu mutācijas. Ar šajos testos izmantotajām šūnu līnijām mēra turpvērstas reportiergēnu mutācijas endogēniskajā timidīnkināzes gēnā (cilvēka šūnu TK un grauzēju šūnu Tk; šajā testēšanas metodē abi dēvēti par TK). Šo testēšanas metodi paredzēts izmantot ar divām šūnu līnijām, proti, L5178Y TK+/--3.7.2C peļu limfomas šūnu līniju (vispārīgais nosaukums L5178Y) un TK6 cilvēka limfoblastoīdo šūnu līniju (vispārīgais nosaukums TK6). Kaut arī abas šūnu līnijas atšķiras izcelsmes, šūnu augšanas, p53 statusa un citādā ziņā, TK gēnu mutācijas testēšanu abu tipu šūnās ir iespējams veikt līdzīgā veidā, kas aprakstīts šajā testēšanas metodē.
|
|
3.
|
Timidīnkināzes gēna autosomālā un heterozigotiskā iedaba ļauj detektēt dzīvotspējīgas kolonijas, kuru šūnās pēc mutācijas no TK+/-
uz TK-/-
timidīnkināzes ferments vairs neizpaužas. Šo neesību var radīt TK gēnu ietekmējoši ģenētiski notikumi, un to vidū var būt kā gēnu mutācijas (punktveida mutācijas, nolasīšanas fāzes nobīdes mutācijas, sīkas delēcijas u. c.), tā hromosomāli notikumi (plašas delēcijas, strukturālas hromosomu aberācijas un mitotiskā rekombinācija). Pēdējie minētie notikumi izpaužas kā heterozigotitātes zudums, un cilvēka kanceroģenēzē tā ir parasta ģenētiska tumorsupresorgēnu pārmaiņa. Teorētiski ar MLA ir iespējams detektēt pilnīgu TK nesējas hromosomas zudumu dalīšanās vārpstas traucējumu un/vai mitotiskas neizšķiršanās ietekmē. Citoģenētiskā un molekulārā analīze, izmantotas kopā, skaidri parāda, ka daļa MLA TK mutantu ir neizšķiršanās rezultāts. Tomēr pierādījumu svars rāda, ka, piemērojot citotoksicitātes standartkritērijus (aprakstīti šajā testēšanas metodē), ar TK gēnu mutācijas testiem nav iespējams uzticami detektēt aneigēnus, un tāpēc šie testi aneigēnu detektēšanai nav piemēroti (7)(8)(9).
|
|
4.
|
TK gēnu mutācijas testos tiek ģenerēti divām atšķirīgām fenotipiskajām klasēm piederīgi TK mutanti: normāli augošie mutanti, kas aug tikpat ātri kā TK heterozigotiskās šūnas, un lēnaudzīgie mutanti, kuru augšanu raksturo pagarināts dubultošanās laiks. Normāli augošie un lēnaudzīgie mutanti MLA tiek saukti attiecīgi par lielo un mazo koloniju mutantiem, bet TK6 – par agrīno un vēlīno koloniju mutantiem. Lielo un mazo koloniju MLA mutantu molekulārās un citoģenētiskās īpašības ir sīki izpētītas (8)(10)(11)(12)(13). Visnotaļ sīki pētītas arī agrīno un vēlīno TK6 mutantu molekulārās un citoģenētiskās īpašības (14)(15)(16)(17). Abu šūnu tipu lēnaudzīgos mutantus ietekmējuši ģenētiski bojājumi, kuri izpaužas ar augšanu regulējošu varbūtīgu gēnu vai gēniem TK lokusa tuvumā un kuru rezultāts ir ilgāks dubultošanās laiks un vēlīno jeb mazo koloniju veidošanās (18). Lēnaudzīgo mutantu inducēšana tiek saistīta ar ķimikālijām, kas inducē plašas strukturālas izmaiņas hromosomālā līmenī. Šūnas, kuru bojājumi neizpaužas ar augšanu regulējošu varbūtīgu gēnu vai gēniem TK lokusa tuvumā, aug līdzīgā ātrumā kā vecākšūnas un kļūst par normāli augošiem mutantiem. Tādu mutantu inducēšana, kuri aug galvenokārt normāli, tiek saistīta ar ķimikālijām, kas iedarbojas galvenokārt kā punktveida mutagēni. Tāpēc ir svarīgi pieskaitīt gan lēnaudzīgos, gan normāli augošos mutantus, lai tādā veidā apzinātu tos visus un iegūtu priekšstatu par testējamās ķimikālijas inducēto bojājumu tipu vai tipiem (mutagēni / klastogēni) (10)(12)(18)(19).
|
|
5.
|
Šis testēšanas metodes apraksts strukturēts tā, lai dotu vispārīgu informāciju, kas attiecas gan uz MLA, gan uz TK6, un pēc tam sīkākas vadlīnijas par katru atsevišķo testu.
|
|
6.
|
Izmantotās definīcijas sniegtas 1. pielikumā.
|
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
7.
|
In vitro testos metaboliskai aktivācijai parasti jāizmanto eksogēnisks avots. Eksogēniskā metaboliskās aktivācijas sistēma in vivo apstākļus atdarina nepilnīgi.
|
|
8.
|
Būtu jāraugās, lai nerodas tādi apstākļi (proti, varbūtēja mijiedarbība ar testa sistēmu), kas noved pie artefaktuāli pozitīviem rezultātiem, kurus nerada testējamās ķimikālijas un šūnas ģenētiskā materiāla mijiedarbība; pie tādiem apstākļiem pieder arī pH vai osmolalitātes pārmaiņas, mijiedarbība ar barotnes sastāvdaļām (20)(21) vai pārāk augsti citotoksicitātes līmeņi (22)(23)(24). MLA un TK6 testā par pārāk augstiem uzskata līmeņus, kas pārsniedz 28. punktā definētos ieteicamos augšējos citotoksicitātes līmeņus. Turklāt jāatzīmē, ka testējamās vielas, kuras ir timidīna analogi vai iedarbojas kā timidīna analogi, šūnu apstrādes laikā var izraisīt spontānu fona mutantu selektīvu augšanu un tādējādi palielināt mutantu biežumu, tāpēc šo vielu adekvātas izvērtēšanas labad jāizmanto papildu testēšanas metodes (25).
|
|
9.
|
Attiecībā uz nanomateriāliem, kas iegūti ar inženierijas paņēmieniem, šo testēšanas metodi var būt vajadzīgs pielāgot, taču tas šeit nav konkrēti aprakstīts.
|
|
10.
|
Pirms šo testēšanas metodi izmantot maisījuma testēšanai nolūkā iegūt datus kādam normatīvam mērķim, jāapsver, vai un kādā veidā tā varētu dot minētajam mērķim piemērotus rezultātus. Ja pastāv normatīva prasība maisījumu testēt, šāda apsvēršana nav vajadzīga.
|
|
11.
|
Mutantšūnas, kurās pēc mutācijas no TK
+/- uz TK
-/- vairs neizpaužas timidīnkināzes fermentatīvā darbība, ir rezistentas pret pirimidīna analoga trifluortimidīna (TFT) citostatisko iedarbību. TK standartšūnas ir jutīgas pret TFT, kas izraisa šūnu metabolisma inhibēšanu un aptur šūnu turpmāko dalīšanos. Tātad mutantšūnas TFT klātbūtnē spēj proliferēt un veidot redzamas kolonijas, turpretī TK fermentu saturošās šūnas to nespēj.
|
TESTA PRINCIPS
|
12.
|
Šūnas, kas ir suspensijā, uz piemērotu laiku (sk. 33. punktu) gan ar eksogēnisku metaboliskas aktivācijas avotu, gan bez tā (sk. 19. punktu) eksponē testējamajai ķimikālijai un pēc tam subkultivē, lai noteiktu citotoksicitāti un lai pirms mutantu atlasīšanas varētu notikt fenotipiskā ekspresija. Citotoksicitāti nosaka pēc relatīvā kopējā pieauguma (RTG; sk. 25. punktu) MLA gadījumā un pēc relatīvās izdzīvotības (RS; sk. 26. punktu) TK6 gadījumā. Lai inducēto mutāciju fenotipiskā ekspresija varētu izpausties gandrīz optimāli, apstrādātās kultūras barotnē tur pietiekami ilgu laiku, kas ir raksturīgs katram izvēlētajam šūnu tipam (sk. 37. punktu). Kad notikusi fenotipiskā ekspresija, nosaka mutantu biežumu, noteiktu skaitu šūnu izsējot barotnē, kas satur selektīvo aģentu, ar kuru detektē mutantkolonijas, un barotnē bez selektīvā aģenta, pēc kuras nosaka klonēšanās efektivitāti (dzīvotspēju). Pēc piemērota inkubēšanas laika kolonijas saskaita. Mutantu biežumu aprēķina pēc mutantkoloniju skaita, ko koriģē ar klonēšanās efektivitāti mutantu atlasīšanas brīdī.
|
METODES APRAKSTS
Preparāti
Šūnas
|
13.
|
Attiecībā uz MLA: tā kā MLA ir izstrādāta un aprakstīta, izmantojot L5178Y šūnu TK
+/- -3.7.2C apakšlīniju, jāņem šī konkrētā apakšlīnija. L5178Y šūnu līnija tikusi atvasināta no DBA-2 peļu timīnās limfomas, kas inducēta ar metilholantrēnu (26). Clive un līdzstrādnieki L5178Y šūnas (Clive klasificētas kā TK+/+ -3) apstrādāja ar etilmetāna sulfonātu un izolēja TK-/- (klasificēta kā TK-/- -3.7) klonu, par selektīvo aģentu izmantojot bromdeoksiuridīnu. No TK-/- tika izolēts un izmantošanai MLA aprakstīts spontāns TK+/- klons (klasificēts kā TK+/- -3.7.2.) un apakšklons (klasificēts kā TK+/--3.7.2C) (27). Šīs šūnu līnijas kariotips tika publicēts (28)(29)(30)(31). Tā modālais hromosomu skaits ir 40. Ir viena metacentriska hromosoma (t12 13), kas būtu jāpieskaita kā viena hromosoma. Peļu TK lokuss atrodas 11. hromosomas distālajā galā. L5178Y TK
+/- -3.7.2C šūnu līnijai mutācijas ir abās p53 alēlēs, un tā rada p53 mutantproteīnu (32)(33). Visdrīzāk TK+/--3.7.2C šūnu līnijas p53 statuss ir tas, kas nosaka, cik labi tests spēj detektēt plašus bojājumus (17).
|
|
14.
|
Attiecībā uz TK6: TK6 ir cilvēka limfoblastoīdo šūnu līnija. Vecākšūnu līnija ir Epstein–Barr vīrusa transformēta šūnu līnija, WI-L2, sākotnēji iegūta no piecgadīga zēna, kurš sirga ar pārmantotu sferocitozi. Pirmo izolēto klonu, HH4, mutagenizēja ar ICR191, un rezultātā ieguva TK heterozigotisku šūnu līniju, TK6 (34). TK6 šūnas ir gandrīz diploīdas, un reprezentatīvais kariotips ir 47, XY, 13+, t(14; 20), t(3; 21) (35). Cilvēka TK lokuss atrodas 17. hromosomas garajā plecā. TK6 ir p53 ziņā kompetenta šūnu līnija, jo tai abās alēlēs ir savvaļas tipa p53 sekvence un ekspresijā – tikai savvaļas tipa p53 proteīns (53).
|
|
15.
|
Kad testēšanas laboratorija veic izejkultūras veidošanu vai papildināšanu, gan MLA, gan TK6 gadījumā ieteicams nodrošināt, ka nav kontaminācijas ar Mycoplasma, noteikt šūnu kariotipu vai iekrāsot TK lokusu nesošās hromosomas un pārbaudīt populāciju dubultošanās laikus. Jānosaka, kāds ir testēšanas laboratorijā izmantoto šūnu normālais šūnas cikla ilgums, un tam jāsakrīt ar publicētajiem šūnu raksturlielumiem (16)(19)(37). Šī izejkultūra jāglabā -150 °C vai zemākā temperatūrā, un no tās gatavojamas visas šūnu darbkultūras.
|
|
16.
|
Pirms ar krioprezervācijas palīdzību lielā skaitā veidot darbkultūras vai vienkārši pirms kultūras izmantošanas eksperimentā tā var būt jāattīra no iepriekš pastāvošajām mutantšūnām [, izņemot tad, ja šķīdinātāja kontrolē mutantu biežums (MF) jau tolaik ir pieņemamā diapazonā (attiecībā uz MLA sk. 2. tabulu)]. Tas darāms, izmantojot metotreksātu (aminopterīnu), lai šūnas, kurās TK neizpaužas, tiktu negatīvi atlasītas, un pievienojot kultūrai timidīnu, hipoksantīnu un glicīnu (L5178Y) vai 2’-deoksicitidīnu (TK6), lai tādā veidā nodrošinātu TK ziņā kompetento šūnu optimālu augšanu (19)(38)(39), TK6 gadījumā (40). Vispārīgus ieteikumus par labu praksi šūnu kultūru uzturēšanā, kā arī ieteikumus konkrēti par L5178Y un TK6 šūnām skatīt (19)(31)(37)(39)(41). Laboratorijām, kam šūnu izejkultūras vajadzīgas MLA vai TK6 iniciēšanas nolūkiem vai kam vajadzīgas jaunas šūnu izejkultūras, ir pieejams labi aprakstītu šūnu repozitorijs (37).
|
Barotne un kultivēšanas apstākļi
|
17.
|
Abos testos kultūru uzturēšanai jāizmanto attiecīgas barotnes un attiecīgi inkubācijas apstākļi (piem., kultivēšanas trauki, mitrināta atmosfēra ar CO2 koncentrāciju 5 %, inkubācijas temperatūra 37 °C). Šūnu kultūras vienmēr jātur tādos apstākļos, lai būtu nodrošināts, ka augšana notiek eksponenciālā fāzē. Īpaši svarīgi ir izraudzīties tādu barotni un tādus kultivēšanas apstākļus, kuri nodrošina šūnu optimālu augšanu ekspresijas periodā un gan mutantšūnu, gan nemutējušo šūnu klonēšanos. Gan MLA, gan TK6 gadījumā svarīgi ir arī tas, lai kultivēšanas apstākļi nodrošinātu optimālu augšanu kā lielo/agrīno, tā mazo/vēlīno koloniju TK mutantiem. Vairāk par kultivēšanu, arī par vajadzību pienācīgi termiski inaktivēt zirgu serumu, ja mutantu atlasē tiek izmantota RPMI barotne, skatīt (19)(31)(38)(39)(40)(42).
|
Kultūru sagatavošana
|
18.
|
No izejkultūrām tiek pavairotas šūnas, tās iesētas kultūras barotnē tādā blīvumā, lai apstrādes periodā un ekspresijas periodā kultūras suspensijās joprojām augtu eksponenciāli.
|
Metaboliskā aktivācija
|
19.
|
Ja strādā ar L5178Y un TK6 šūnām – to endogēniskā metaboliskā kapacitāte nav pietiekama –, jāizmanto eksogēniskas metaboliskās aktivācijas sistēmas. Visplašāk izmantotā sistēma, kuru iesaka izmantot pēc noklusējuma, ja vien nav pamata izmantot citu, ir ar kofaktoru papildināta postmitohondriālā frakcija (S9) no grauzēju (parasti žurku) aknām, kas apstrādātas ar fermentus inducējošām vielām, piemēram, Aroclor 1254 (43)(44)(45) vai fenobarbitālu kombinācijā ar β-naftoflavonu (46)(47)(48)(49)(50)(51). Pēdējā minētā kombinācija nav pretrunā ar Stokholmas Konvenciju par noturīgajiem organiskajiem piesārņotājiem (52) un izrādījusies tikpat produktīva jauktas funkcijas oksidāžu inducētāja kā Aroclor 1254 (45)(46)(47)(48)(49). S9 frakciju parasti izmanto 1–2 % tilpumkoncentrācijā, bet galīgajā testa barotnē tilpumkoncentrāciju var palielināt līdz 10 %. Eksogēnās metaboliskās aktivācijas sistēmas vai metaboliskā inducētāja veida un koncentrācijas izvēli var ietekmēt testējamo ķimikāliju klase.
|
Testējamās ķimikālijas sagatavošana
|
20.
|
Cietas testējamās ķimikālijas jāsagatavo attiecīgos šķīdinātājos un attiecīgā gadījumā pirms šūnu apstrādes jāatšķaida (sk. 21. punktu). Šķidras testējamās ķimikālijas var testa sistēmai pievienot tieši un/vai pirms testa sistēmas apstrādes atšķaidīt. Gāzveida vai gaistošas testējamās ķimikālijas jātestē pēc attiecīgām standarta protokolu modifikācijām, piemēram, apstrādi veicot hermētiski noslēgtos kultivēšanas traukos (53)(54)(55). Testējamās ķimikālijas preparāti jāpagatavo tieši pirms apstrādes, ja vien dati par stabilitāti neliecina, ka tos var glabāt.
|
TESTĒŠANAS NOSACĪJUMI
Šķīdinātāji
|
21.
|
Šķīdinātājs jāizvēlas tā, lai optimizētu testējamās ķimikālijas šķīdību, šķīdinātājam negatīvi neietekmējot testa norisi, piemēram, nemainot šūnu augšanu, neskarot testējamās ķimikālijas integritāti, nereaģējot ar kultivēšanas traukiem, nepasliktinot metaboliskās aktivācijas sistēmu. Ja vien iespējams, ieteicams vispirms apsvērt, vai neizmantot šķīdinātāju (vai kultūras barotni) uz ūdens bāzes. Plaši atzīti šķīdinātāji ir ūdens vai dimetilsulfoksīds. Galīgajā apstrādes barotnē organisko šķīdinātāju tilpumkoncentrācijai parasti nevajadzētu pārsniegt 1 %, bet uz (sālsūdens vai saldūdens) bāzes pagatavotu šķīdinātāju tilpumkoncentrācijai – 10 %. Ja tiek izmantoti ne tik plaši atzīti šķīdinātāji (piem., etanols vai acetons), to izmantošana jāpamato ar datiem, kas apliecina to saderību ar testējamajām ķimikālijām, izmantoto testa sistēmu un ģenētiskās toksicitātes neesību izmantotajā koncentrācijā. Ja šādu apliecinošu datu nav, svarīgi ir izmantot arī neapstrādātas kontroles (sk. definīciju 1. papildinājumā), lai ar tām pierādītu, ka izraudzītajam šķīdinātājam nav deletējošas vai mutagēniskas ietekmes.
|
CITOTOKSICITĀTES MĒRĪŠANA UN APSTRĀDES KONCENTRĀCIJU IZRAUDZĪŠANĀS
|
22.
|
Nosakot augstāko testējamās ķimikālijas koncentrāciju, neizraugās koncentrācijas, kas spēj izraisīt tādas artefaktuāli pozitīvas atbildreakcijas kā pārmērīga citotoksicitāte (sk. 28. punktu), izgulsnēšanās (sk. 29. punktu) kultūras barotnē vai manāmas pH vai osmolalitātes pārmaiņas (sk. 8. punktu). Lai nepieļautu artefaktuāli pozitīvus rezultātus un uzturētu pienācīgus kultivēšanas apstākļus gadījumā, ja testējamā ķimikālija pievienošanas brīdī manāmi maina barotnes pH, to var koriģēt, galīgajā apstrādes barotnē pievienojot buferšķīdumu.
|
|
23.
|
Koncentrācijas izraugās pēc citotoksicitātes un citiem apsvērumiem (sk. 27.–30. punktu). Kaut arī citotoksicitātes izvērtēšana sākotnējā testā var noderēt, lai sekmīgāk noteiktu galvenajā eksperimentā izmantojamās koncentrācijas, sākotnējais tests nav jāveic obligāti. Pat ja citotoksicitāte izvērtēta jau no sākuma, galvenajā eksperimentā tā joprojām jānosaka katrai kultūrai. Ja eksperimentā nosaka diapazonu, būtu jāaptver plašs koncentrāciju spektrs un eksperiments jābeidz vai nu pirmajā dienā pēc apstrādes, vai (ja izmantotās koncentrācijas izrādās piemērotas) tas jāturpina divu dienu ilgajā ekspresijas laikā līdz mutantu atlasei.
|
|
24.
|
Citotoksicitāte jānosaka katrā individuālā testa kultūrā un kontrolkultūrā: MLA (2) un TK6 (15) atbilstošās metodes ir definētas ar starptautiski saskaņotu praksi.
|
|
25.
|
Abās (agara un mikroiedobju) MLA versijās citotoksicitāte jāizvērtē, izmantojot relatīvo kopējo pieaugumu (RTG), kuru 1975. gadā sākotnēji definējuši Clive un Spektor (2). Tas ietver relatīvo suspensijas pieaugumu (RSG: testa kultūra / šķīdinātāja kontrole) šūnu apstrādes laikā, ekspresijas laiku un relatīvo klonēšanās efektivitāti (RCE: testa kultūra / šķīdinātāja kontrole) mutantu atlasīšanas brīdī (2). Jāatzīmē, ka ar RSG ir ņemts vērā arī apstrādes laikā novērojamais šūnu zudums testa kultūrā (sk. formulas 2. papildinājumā).
|
|
26.
|
Attiecībā uz TK6 citotoksicitāte jāizvērtē pēc relatīvās izdzīvotības (RS), proti, pēc to šūnu klonēšanās efektivitātes (CE), kuras tūlīt pēc apstrādes uzsētas uz platēm, CE koriģējot atkarībā no šūnu zuduma apstrādes laikā salīdzinājumā ar šūnu skaitu negatīvajā kontrolē (kurā izdzīvotību pieņem par 100 %), sk. formulu 2. papildinājumā.
|
|
27.
|
Jāizvērtē vismaz četras testējamās koncentrācijas (nerēķinot šķīdinātāja un pozitīvās kontroles), kas atbilst pieņemamības kritērijiem (attiecīga citotoksicitāte, šūnu skaits u. c.). Lai gan ir ieteicams izmantot duplikatīvas kultūras, ar katru testējamo koncentrāciju var izmantot vai nu replikatīvas, vai atsevišķas apstrādātas kultūras. Ar replikatīvajām kultūrām pie kādas konkrētas koncentrācijas iegūtie rezultāti pārskatā būtu jāatspoguļo atsevišķi, bet datu analīzes vajadzībām tos drīkst apvienot (55). Attiecībā uz testējamajām ķimikālijām, kas uzrāda nelielu vai neuzrāda nekādu citotoksicitāti, parasti noderēs divkāršotas līdz trīskāršotas koncentrācijas intervāli. Ja citotoksicitāte ir novērojama, koncentrācijas jāizraugās tā, lai citotoksitātes diapazons aptvertu gan koncentrācijas, pie kurām citotoksicitāte ir tāda, kāda aprakstīta 28. punktā, gan koncentrācijas, pie kurām citotoksicitāte ir vidēji liela vai maza, vai arī tās nav. Daudzām testējamajām ķimikālijām ir novērojama stāva koncentrācijatkarīgās atbildreakcijas līkne, un, lai aptvertu visu citotoksicitātes diapazonu vai lai detalizēti izpētītu devas un atbildreakcijas sakarību, var būt nepieciešams izmantot koncentrācijas ar tuvākiem intervāliem un vairāk nekā četras koncentrācijas, jo īpaši situācijās, kur eksperiments jāatkārto (sk. 70. punktu). Vairāk nekā 4 koncentrācijas var būt īpaši vajadzīgas, ja jāizmanto atsevišķas kultūras.
|
|
28.
|
Ja maksimālo koncentrāciju izraugās pēc citotoksicitātes, ar augstāko koncentrāciju MLA gadījumā būtu jācenšas panākt 20 līdz 10 % lielu RTG, bet TK6 gadījumā – 20 līdz 10 % lielu RS (67. punkts).
|
|
29.
|
Mazšķīstošu testējamo ķimikāliju gadījumā, kuras pie koncentrācijām, kas zemākas par zemāko nešķīstošās frakcijas koncentrāciju, nav citotoksiskas, pie augstākās analizētās koncentrācijas pēc apstrādes ar testējamo ķimikāliju jārodas ar neapbruņotu aci vai ar invertēto mikroskopu redzamai duļķainībai vai nogulsnēm. Pat ja citotoksicitāte novērojama pie koncentrācijas, kas augstāka par zemāko nešķīstošās frakcijas koncentrāciju, ieteicams testēt tikai vienu koncentrāciju, kas rada acīmredzamu duļķainību vai nogulsnes, jo nogulsnes var artefaktiski ietekmēt rezultātus. Tā kā MLA un TK6 tiek izmantotas suspensijas kultūras, īpaši būtu jāraugās, lai nogulsnes netraucētu testa izdarīšanā. Turklāt var būt lietderīgi pirms eksperimenta noteikt šķīdību kultūras barotnē.
|
|
30.
|
Ja nogulsnes vai ierobežojoša citotoksicitāte netiek novēroti, augstākajai testējamajai koncentrācijai jāatbilst 10 mM, 2 mg/ml vai 2 μl/ml atkarībā no tā, kura ir zemāka (57)(58). Ja testējamās ķimikālijas sastāvs nav noteikts, piem., tā ir viela, kuras sastāvs nav zināms vai ir mainīgs, tā ir komplekss reakcijas produkts vai bioloģisks materiāls [t. i., ķīmiska viela, kuras sastāvs nav zināms vai ir mainīgs (UVCB)], vidiski ekstrakti u. tml., gadījumos, kur netiek novērota pietiekama citotoksicitāte, augstākā koncentrācija var būt jāpalielina (piem., līdz 5 mg/ml), lai būtu panākta augstāka katras sastāvdaļas koncentrācija. Tomēr jāpiebilst, ka attiecībā uz cilvēkiem paredzētajām zālēm šīs prasības var būt citādas (59).
|
Kontroles
|
31.
|
Ikvienā eksperimentā jāiekļauj paralēlas negatīvās kontroles (sk. 21. punktu), proti, tikai šķīdinātāju apstrādes barotnē, ar ko rīkojas tieši tāpat kā ar apstrādātajām kultūrām.
|
|
32.
|
Paralēlās pozitīvās kontroles ir nepieciešamas, lai pierādītu laboratorijas spēju identificēt mutagēnus izmantotā testēšanas protokola apstākļos, (attiecīgā gadījumā) eksogēnās metaboliskās aktivācijas sistēmas efektivitāti un to, ka tiek adekvāti detektēti kā mazo/vēlīno, tā lielo/agrīno koloniju TK mutanti. Pozitīvo kontroļu piemēri sniegti zemāk 1. tabulā. Var izmantot alternatīvas pozitīvās kontrolvielas, ja tas ir pamatoti. Tā kā zīdītāju šūnu ģenētiskās toksicitātes in vitro testi ir pietiekami standartizēti īslaicīgām (3–4 stundu ilgām) apstrādēm, kas tiek veiktas paralēli ar tikpat ilgu metabolisko aktivāciju un bez tās, ir iespēja pozitīvās kontroles izmantot tik vien kā attiecībā uz mutagēnu, kam nepieciešama metaboliskā aktivācija. Šajā gadījumā viena vienīga pozitīva kontroles atbildreakcija apliecinās gan metaboliskās aktivācijas sistēmas darbību, gan testa sistēmas reaģētspēju. Ilglaicīgas (piem., 24 stundas bez S9) apstrādes gadījumā tomēr vajadzīga atsevišķa pozitīvā kontrole, jo apstrādes ilgums atšķirsies no tā, ko izmanto testā ar metabolisko aktivāciju. Lai varētu pierādīt testa sistēmas jutību, katra pozitīvā kontrole būtu jāizmanto ar vienu vai vairākām tādām koncentrācijām, ar kurām gaidāma reproducējama un detektējama palielināšanās salīdzinājumā ar fonu, turklāt atbildreakciju nedrīkstētu kompromitēt citotoksicitāte, kas pārsniedz šai testēšanas metodei specificētās robežas (sk. 28. punktu).
|
1. tabula
References vielas, ko ieteicams izmantot laboratorijas kompetences novērtēšanai un pozitīvo kontroļu izraudzīšanās vajadzībām
|
Kategorija
|
Viela
|
CAS Nr.
|
|
1. Mutagēni, kuriem metaboliskā aktivācija nav vajadzīga
|
|
Metilmetānsulfonāts
|
66-27-3
|
|
Mitomicīns C
|
50-07-7
|
|
4-Nitrohinolīn-N-oksīds
|
56-57-5
|
|
2. Mutagēni, kuriem metaboliskā aktivācija ir vajadzīga
|
|
Benz(a)pirēns
|
50-32-8
|
|
Ciklofosfamīds (monohidrāts)
|
50-18-0 (6055-19-2)
|
|
7,12-Dimetilbenzantracēns
|
57-97-6
|
|
3-Metilholantrēns
|
56-49-5
|
PROCEDŪRA
Apstrāde ar testējamo ķimikāliju
|
33.
|
Proliferējošas šūnas ar testējamo ķimikāliju apstrādā gan metaboliskās aktivācijas sistēmas klātbūtnē, gan bez tās. Ekspozīcijai jāilgst attiecīgu laiku (parasti pietiek ar 3–4 stundām). Tomēr jāpiebilst, ka attiecībā uz cilvēkiem paredzētajām zālēm šīs prasības var būt citādas (59). MLA gadījumā, ja īslaicīga apstrāde dod negatīvus rezultātus un ir informācija, kas liecina, ka var būt vajadzīga ilgāka apstrāde [piem., nukleozīda analogiem, mazšķīstošām ķimikālijām (5)(59)], jāapsver testa izdarīšana, izmantojot ilglaicīgāku apstrādi, piem., 24 stundas bez S9.
|
|
34.
|
Minimālais šūnu skaits, ko izmanto katrai testa kultūrai (gan kontrolkultūrai, gan apstrādātai kultūrai), katrā testa posmā jābalsta uz spontāno mutantu biežumu. Vispārīgs princips ir šūnas apstrādāt un pasāžām ņemt tādā skaitā, lai visās testa fāzēs (apstrādē, fenotipiskajā ekspresijā un mutantu atlasē) katrā eksperimentālajā kultūrā saglabātos vismaz 10, bet ideālā gadījumā 100 spontāno mutantu (56).
|
|
35.
|
MLA gadījumā ieteicamais pieņemamais spontāno mutantu biežums ir 35–140 × 10-6 (agara versijā) un 50–170 × 10-6 (mikroiedobju versijā) (sk. 2. tabulu). Lai katrā testa kultūrā pēc apstrādes paliktu vismaz 10 un ideāli 100 spontāno mutantu, ir jāapstrādā vismaz 6 × 106 šūnu. Šāda skaita apstrāde un pietiekama šūnu skaita uzturēšana ekspresijas un mutantu atlasei vajadzīgās klonēšanas laikā visās eksperimenta fāzēs nodrošina pietiekami daudz (10 vai vairāk) spontāno mutantu, arī kultūrās, kas apstrādātas ar koncentrācijām, kuras rada 90 % citotoksicitāti (izmērīta ar 10 % RGT) (19)(38)(39).
|
|
36.
|
TK6 gadījumā spontāno mutantu biežums parasti ir 2–10 × 10-6. Lai katrā kultūrā pēc apstrādes paliktu vismaz 10 spontāno mutantu, ir jāapstrādā vismaz 20 × 106 šūnu. Šāda šūnu skaita apstrāde nodrošina pietiekami daudz (10 vai vairāk) spontāno mutantu, arī kultūrās, kas apstrādātas ar koncentrācijām, kuras rada 90 % citotoksicitāti (10 % RS). Turklāt pietiekamā skaitā jābūt arī šūnām, ko kultivē ekspresijas periodā un mutantu atlasīšanas nolūkā uzsēj uz platēm (60).
|
Fenotipiskās ekspresijas laiks un citotoksicitātes un mutantu biežuma mērīšana
|
37.
|
Lai jauninducēto mutantu fenotipiskā ekspresija varētu izpausties gandrīz optimāli, pēc apstrādes šūnas vēl kultivē noteiktu laiku, kas ir katrai šūnu līnijai specifisks. MLA gadījumā fenotipiskās ekspresijas periods ilgst 2 dienas. TK6 gadījumā fenotipiskās ekspresijas periods ir 3–4 dienas. Ja veic 24 stundu ilgu apstrādi, ekspresijas periodu sāk skaitīt no apstrādes beigu brīža.
|
|
38.
|
Fenotipiskās ekspresijas perioda laikā šūnas katru dienu tiek saskaitītas. MLA gadījumā šūnu skaitīšanas rezultātu izmanto, lai aprēķinātu dienišķo suspensijas pieaugumu (SG). Kad beidzies 2 dienu ilgais ekspresijas periods, barotnē ar selektīvo aģentu un bez tā šūnas tiek suspendētas, lai noteiktu attiecīgi mutantu skaitu (selekcijas plates) un klonēšanās efektivitāti (dzīvotspējas plates). Ir divas vienādi pieņemamas metodes, ar kurām MLA tiek klonēti atlasīšanai vajadzīgie mutanti: vienā metodē izmanto mīksto agaru, bet otrā – šķidru barotni 96 iedobju mikrotitrēšanas platēs (19)(38)(39). TK6 gadījumā klonēšana notiek, izmantojot šķidras barotnes un 96 iedobju mikrotitrēšanas plates (16).
|
|
39.
|
Vienīgais ieteicamais selektīvais aģents TK mutantiem ir trifluortimidīns (TFT).
|
|
40.
|
MLA gadījumā agara platēm un mikroiedobju platēm skaitīšanu veic pēc 10–12 dienu ilgas inkubācijas. TK6 gadījumā agrīnos mutantus nosaka, kolonijas uz mikroiedobju platēm pieskaitot pēc 10–14 dienām. Lai parādītos lēnaudzīgie (vēlīnie) TK6 mutanti, pēc agrīno mutantu pieskaitīšanas šūnām no jauna jāpievieno barotne un TFT, un tad plates jāinkubē vēl 7–10 dienas (62). Vairāk par lēnaudzīgo un normāli augošo TK mutantu pieskaitīšanu skatīt 42. un 44. punktā.
|
|
41.
|
Attiecīgās aprēķina formulas abiem testiem, arī abām MLA (agara un mikroiedobju) metodēm skatīt 2. papildinājumā. Pēc MLA agara metodes pieskaita kolonijas, atbilstoši klonēšanās efektivitātei koriģē mutantu koloniju skaitu un tad aprēķina MF. MLA un TK6 mikroiedobju versijā klonēšanās efektivitāti gan selekcijas platēm, gan klonēšanās efektivitātes platēm nosaka pēc t. s. Puasona sadalījuma (63). MF aprēķina, vadoties pēc šīm divām klonēšanās efektivitātes vērtībām.
|
Mutantkoloniju raksturošana
|
42.
|
MLA gadījumā, ja testējamā ķimikālija ir pozitīva (sk. 62.–63. punktu), koloniju lielums vai augšana jāraksturo vismaz vienā no testa kultūrām (parasti pie augstākās pieņemamās pozitīvās koncentrācijas) un negatīvajā un pozitīvajā kontrolē. Ja testējamā ķimikālija ir negatīva (sk. 64. punktu), mutantkoloniju raksturošana jāveic negatīvajā un pozitīvajā kontrolē. Pēc MLA mikroiedobju metodes mazo koloniju mutantus definē kā tādus, kuri klāj mazāk nekā 25 % no iedobes platības, mērot pēc diametra, bet lielo koloniju mutantus – kā tādus, kuri klāj vairāk nekā 25 % no iedobes platības, mērot pēc diametra. Pēc agara metodes mutantkolonijas skaita un to lielumu nosaka automātiski, izmantojot koloniju skaitītāju. Koloniju lieluma noteikšanā iespējamās pieejas sīkāk aprakstītas literatūrā (19)(38)(40). Koloniju raksturošana negatīvajā un pozitīvajā kontrolē vajadzīga tādēļ, lai pierādītu, ka pētījums izdarīts adekvāti.
|
|
43.
|
Testējamo ķimikāliju nevar noteikt par negatīvu, ja pozitīvajā kontrolē nav adekvāti detektēti kā lielo, tā mazo koloniju mutanti. Koloniju raksturošanu var izmantot, lai sniegtu vispārīgu informāciju par testējamās ķimikālijas spēju izraisīt punktveida mutācijas un/vai hromosomālus notikumus (4. punkts).
|
|
44.
|
TK6: normāli augošos un lēnaudzīgos mutantus diferencē pēc inkubācijas laika atšķirības (sk. 40. punktu). TK6 gadījumā parasti saskaita gan agrīnos, gan vēlīnos mutantus visās kultūrās, arī negatīvajā un pozitīvajā kontrolē. Koloniju raksturošana negatīvajā un pozitīvajā kontrolē vajadzīga tādēļ, lai pierādītu, ka pētījums izdarīts adekvāti. Testējamo ķimikāliju nevar noteikt par negatīvu, ja pozitīvajā kontrolē nav adekvāti detektēti kā agrīnie, tā vēlīnie mutanti. Koloniju raksturošanu var izmantot, lai sniegtu vispārīgu informāciju par testējamās ķimikālijas spēju izraisīt punktveida mutācijas un/vai hromosomālus notikumus (4. punkts).
|
Laboratorijas kompetence
|
45.
|
Lai pirms testa pastāvīgas izmantošanas demonstrētu pietiekamu vajadzīgo pieredzi, laboratorijai jābūt veikušai eksperimentu sēriju ar pozitīvās kontroles references vielām, kam ir dažādi iedarbības mehānismi (tās izraugās no 1. tabulas saraksta un vismaz vienai no tām aktīvai jābūt ar metabolisku aktivāciju un vienai — bez tās), un ar dažādām negatīvajām kontrolēm (neapstrādātām kultūrām un dažādiem šķīdinātājiem/nesējiem). Šo pozitīvo un negatīvo kontroļu atbildreakcijām jāsaskan ar literatūras datiem. Šī prasība neattiecas uz laboratorijām, kurām ir pieredze, t. i., kuru rīcībā ir vēsturisko datu bāze, kas aprakstīta 47.–50. punktā. MLA gadījumā pozitīvās un negatīvās kontroles vērtībām jābūt saskaņā ar IWGT ieteikumiem (sk. 2. tabulu).
|
|
46.
|
Lai varētu pierādīt, ka laboratorija ir kompetenta detektēt mutagēniskas ķimikālijas, noteikt metaboliskās aktivācijas sistēmas efektivitāti un pierādīt, ka šūnu augšanas apstākļi apstrādes laikā, fenotipiskā ekspresija un mutantu atlase, kā arī skaitīšanas procedūras atbilst vajadzīgajam, pozitīvas kontroles vielu (sk. 1. tabulu) izlase jāpēta gan pie īslaicīgas un ilglaicīgas (ja izmanto) apstrādes bez metaboliskas aktivācijas, gan pie īslaicīgas apstrādes metaboliskas aktivācijas apstākļos. Lai varētu pierādīt testa sistēmas jutību un dinamisko diapazonu, jāizraugās tāds izlases ķimikāliju koncentrāciju diapazons, kurā palielinājumi virs fona līmeņa būs reproducējami un koncentrācijatkarīgi.
|
Vēsturisko kontroļu dati
|
47.
|
Laboratorijai jānosaka:
|
—
|
vēsturisks pozitīvo kontroļu diapazons un sadalījums,
|
|
—
|
vēsturisks negatīvo (neapstrādātu, ar šķīdinātāju) kontroļu diapazons un sadalījums.
|
|
|
48.
|
Pirmoreiz iegūstot datus par vēsturisko negatīvo kontroļu sadalījumu, paralēlajām negatīvajām kontrolēm jāatbilst publicētajiem datiem par negatīvajām kontrolēm. Kontroļu sadalījumam tiekot papildinātam ar jauniem eksperimentu datiem, paralēlajām negatīvajām kontrolēm ideālā gadījumā jābūt minētā sadalījuma 95 % kontrolrobežās (64)(65).
|
|
49.
|
Laboratorijas vēsturiskā negatīvo kontroļu datubāze sākotnēji jāveido ar vismaz 10 eksperimentiem, bet vēlams, lai tajā būtu vismaz 20 eksperimenti, kas veikti salīdzināmos eksperimentēšanas apstākļos. Lai varētu noteikt, cik mainīgi ir laboratorijas pozitīvo un negatīvo kontroļu dati, un parādīt, ka laboratorijā šī metodika “tiek kontrolēta” (66), laboratorijām jāizmanto tādas kvalitātes kontroles metodes kā kontroldiagrammas (piem., C diagrammas vai X joslu diagrammas (65)). Papildu ieteikumi par vēsturisko datu izveidi un izmantošanu atrodami literatūrā (64).
|
|
50.
|
Negatīvo kontroļu datiem vajadzētu būt mutantu biežumam atsevišķās vai, ieteicams, replikatīvajās kultūrās; sk. aprakstu 27. punktā. Paralēlajām negatīvajām kontrolēm ideālā gadījumā jābūt laboratorijas vēsturisko negatīvo kontroļu datubāzes sadalījuma 95 % kontrolrobežās. Tādus negatīvo kontroļu datus, kas ir ārpus 95 % kontrolrobežas, iekļaušanai vēsturiskajā kontroļu sadalījumā var pieņemt tikai tad, ja šie dati nav pārmērīgi anomālas vērtības un ja ir pierādījumi, ka testa sistēma “tiek kontrolēta” (sk. 49. punktu) un nav gadījusies ne tehniska kļūme, ne cilvēka kļūda.
|
|
51.
|
Jebkādas izmaiņas eksperimenta protokolā jāapsver, ņemot vērā attiecīgo datu atbilstību laboratorijas esošajai vēsturisko kontroļu datubāzei. Būtisku neatbilstību gadījumā jāveido jauna vēsturisko kontroļu datubāze.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Rezultātu izklāsts
|
52.
|
Datu izklāstā gan MLA, gan TK6 gadījumā jāiekļauj zemāk aprakstītie dati, kas vajadzīgi, lai aprēķinātu citotoksicitāti (attiecīgi RTG vai RS) un mutantu biežumu apstrādātajās un kontroles kultūrās.
|
|
53.
|
MLA gadījumā jānorāda individuālu kultūru dati par RSG, RTG, klonēšanās efektivitāti mutantu atlasīšanas brīdī un mutantkoloniju skaits (agara versijā) vai tukšo iedobju skaits (mikroiedobju versijā). Mutantu biežums (MF) jāizteic ar mutantšūnu skaitu uz miljonu izdzīvojušo šūnu. Ja atbildreakcija ir pozitīva, vismaz pie vienas no testējamās ķimikālijas koncentrācijām (parasti pie augstākās pozitīvās koncentrācijas) un negatīvajā un pozitīvajā kontrolē jānorāda mazo un lielo koloniju MF (un/vai to īpatsvars kopējā MF). Ja atbildreakcija ir negatīva, mazo un lielo koloniju MF jānorāda negatīvajā kontrolē un pozitīvajā kontrolē.
|
|
54.
|
TK6 gadījumā jānorāda individuālu kultūru dati par RS, klonēšanās efektivitāti mutantu atlasīšanas brīdī un tukšo iedobju skaits attiecībā uz agrīnajiem un vēlīnajiem mutantiem. MF jāizteic ar mutantšūnu skaitu uz izdzīvojušo šūnu skaitu un jānorāda gan kopējais MF, gan agrīno un vēlīno mutantu MF (un/vai to īpatsvars kopējā MF).
|
Pieņemamības kritēriji
|
55.
|
Lai varētu noteikt konkrētas ķimikālijas testēšanas koprezultātus, gan MLA, gan TK6 gadījumā jābūt izpildītiem šādiem kritērijiem:
|
—
|
testēšana notikusi ar diviem eksperimentēšanas nosacījumiem (īslaicīga apstrāde ar metabolisko aktivāciju un bez tās, sk. 33. punktu), ja vien vienā no tiem rezultāti nav bijuši pozitīvi;
|
|
—
|
pietiekams šūnu un koncentrāciju skaits ir analizēšanai derīgs (sk. 27. punktu un 34.–36. punktu);
|
|
—
|
augstākās koncentrācijas izvēles kritēriji atbilst 28.–30. punktā aprakstītajiem.
|
|
Negatīvo un pozitīvo kontroļu pieņemamības kritēriji
|
56.
|
IWGT izveidotā MLA ekspertu darbgrupa ir analizējusi lielu daudzumu MLA datu, un rezultātā ir panākta starptautiska vienprātība par konkrētiem MLA pieņemamības kritērijiem (1)(2)(3)(4)(5). Tāpēc šajā testēšanas metodē sniegti konkrēti ieteikumi par to, kā MLA gadījumā nosakāma negatīvās un pozitīvās kontroles pieņemamība un izvērtējami individuālu vielu rezultāti. TK6 datubāze ir daudz mazāka un nav tikusi izvērtēta darbgrupā.
|
|
57.
|
MLA gadījumā ikviens eksperiments jāizvērtē to tāda viedokļa, vai neapstrādātā/šķīdinātāja kontrole atbilst IWGT izveidotās MLA darbgrupas noteiktajiem pieņemamības kritērijiem (sk. (4) un zemāk 2. tabulu) attiecībā uz: 1) MF (turklāt MLA agara un mikroiedobju versijās IWGT pieņemamie MF ir atšķirīgi); 2) klonēšanās efektivitāti (CE) mutantu atlasīšanas brīdī un 3) suspensijas pieaugumu (SG) šķīdinātāja kontrolē (formulas sk. 2. papildinājumā).
2. tabula
MLA pieņemamības kritēriji
|
Parametrs
|
Mīkstā agara metode
|
Mikroiedobju metode
|
|
Mutantu biežums
|
35–140 × 10-6
|
50–170 × 10-6
|
|
Klonēšanās efektivitāte
|
65–120 %
|
65–120 %
|
|
Suspensijas pieaugums
|
8 līdz 32-kārtīgs (3–4 stundu apstrādē) 32 līdz 180-kārtīgs (24 stundu apstrādē, ja tādu veic)
|
8 līdz 32-kārtīgs (3–4 stundu apstrādē) 32 līdz 180-kārtīgs (24 stundu apstrādē, ja tādu veic)
|
|
|
58.
|
MLA gadījumā ikviens tests jāizvērtē arī no tā viedokļa, vai pozitīvā kontrole (kontroles) atbilst vismaz vienam no šādiem diviem IWGT darbgrupas izstrādātiem pieņemamības kritērijiem:
|
—
|
pozitīvajai kontrolei jāuzrāda absolūts kopējā MF palielinājums, t. i., palielinājums, kas pārsniedz vismaz 300 × 10-6 lielo spontāno fona MF [inducēto MF jeb IMF]. Vismaz 40 % no IMF jābūt mazo koloniju MF;
|
|
—
|
pozitīvajā kontrolē mazo koloniju MF palielinājumam vismaz par 150 × 10-6 jāpārsniedz tas, kas ir paralēlajā neapstrādātajā/šķīdinātāja kontrolē (150 × 10-6 liels mazo koloniju IMF).
|
|
|
59.
|
TK6 gadījumā tests ir pieņemams, ja paralēlā negatīvā kontrole tiek uzskatīta par pieņemamu iekļaušanai laboratorijas vēsturisko negatīvo kontroļu datubāzē saskaņā ar 48.–49. punktu. Turklāt paralēlajām pozitīvajām kontrolēm (sk. 32. punktu) jāinducē atbildreakcijas, kas atbilst tām, kuras ģenerētas vēsturiskajā pozitīvo kontroļu datubāzē iekļautajās kontrolēs, un jāizsaka statistiski ievērojams palielinājums salīdzinājumā ar paralēlo negatīvo kontroli.
|
|
60.
|
Abos testos pozitīvajā kontroles kultūrā novērotās citotoksicitātes augšējai robežvērtībai jābūt tādai pašai kā eksperimentālajās kultūrās. Tas nozīmē, ka RTG/RS jābūt ne mazākam par 10 %. Lai pierādītu, ka pozitīvās kontroles pieņemamības kritēriji ir izpildīti, pietiek ar vienu koncentrāciju (vai pietiek izmantot vienu no pozitīvās kontroles kultūras koncentrācijām, ja tādas ir vairākas). Turklāt pozitīvās kontroles MF jābūt laboratorijas noteiktā pieņemamā diapazona robežās.
|
Rezultātu izvērtēšana un interpretēšana
|
61.
|
IWGT Peļu limfomas ekspertu darbgrupa ir daudz izdarījusi MLA bioloģiskā relevantuma un pozitīvas atbildreakcijas kritēriju jomā (4). Tāpēc šajā testēšanas metodē sniegti konkrēti ieteikumi par to, kā interpretēt ar MLA testēto ķimikāliju rezultātus (sk. 62.–64. punktu). TK6 datubāze ir daudz mazāka un nav tikusi izvērtēta darbgrupā. Tāpēc ieteikumi par TK6 datu interpretēšanu ir vispārīgāki (sk. 65.–66. punktu). Papildus doti daži ieteikumi, kas attiecas uz abiem testiem (sk. 67.–71. punktu).
|
Tikai MLA
|
62.
|
Lai nodrošinātu MF palielinājuma bioloģisko relevantumu, ieteicama šāda pieeja pozitīvas un negatīvas atbildreakcijas noteikšanā. Tajā neizmanto statistisko analīzi kā vairumā pārējo testu, bet gan iepriekš noteiktu inducēto mutantu biežumu (t. i., MF palielinājumu, kas pārsniedz paralēlās kontroles uzrādīto), ko apzīmē ar vispārējo izvērtējuma koeficientu (GEF), kurš noteikts, analizējot līdzdalīgo laboratoriju iegūto negatīvās kontroles MF datu sadalījumu (4). MLA agara versijā GEF ir 90 × 10-6, un MLA mikroiedobju versijā GEF ir 126 × 10-6.
|
|
63.
|
Ja ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par viennozīmīgi pozitīvu, kad pie kāda no vērtētajiem eksperimenta nosacījumiem (sk. 33. punktu) MF palielinājums, kas pārsniedz paralēlās kontroles uzrādīto, ir lielāks par GEF un palielinājums ir koncentrācijatkarīgs (piem., ar dinamikas rindas trenda testu noteikts). Tādā gadījumā šajā testa sistēmā uzskata, ka testējamā ķimikālija spēj inducēt mutācijas.
|
|
64.
|
Ja ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par viennozīmīgi negatīvu, kad ne pie viena no vērtētajiem eksperimenta nosacījumiem (sk. 33. punktu) nav koncentrācijatkarīgas atbildreakcijas vai, ja MF palielinājums ir, tas nepārsniedz GEF. Tādā gadījumā šajā testa sistēmā uzskata, ka testējamā ķimikālija nespēj inducēt mutācijas.
|
Tikai TK6
|
65.
|
Ja ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par viennozīmīgi pozitīvu, kad pie kāda no vērtētajiem eksperimenta nosacījumiem (sk. 33. punktu):
|
—
|
vismaz vienā no testa koncentrācijām salīdzinājumā ar paralēlo negatīvo kontroli novērojams statistiski nozīmīgs palielinājums,
|
|
—
|
šo palielinājumu izvērtējot ar attiecīgu dinamikas rindas trenda testu, konstatējams, ka tas ir koncentrācijatkarīgs,
|
|
—
|
kāds no rezultātiem ir ārpus vēsturisko negatīvo kontroļu datu sadalījuma (piem., ārpus 95 % kontrolrobežām Puasona sadalījumā; sk. 48. punktu).
|
Ja visi šie kritēriji izpildīti, šajā testēšanas sistēmā uzskata, ka testējamā ķimikālija spēj inducēt mutācijas. Ieteikumi par piemērotākajām statistiskajām metodēm atrodami literatūrā (66)(67).
|
|
66.
|
Ja ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par viennozīmīgi negatīvu, kad ne pie viena no vērtētajiem eksperimenta nosacījumiem (sk. 33. punktu):
|
—
|
nevienā testa koncentrācijā nav statistiski nozīmīga palielinājuma salīdzinājumā ar paralēlo negatīvo kontroli,
|
|
—
|
ar attiecīgu dinamikas rindas trenda testu nav konstatējams palielinājuma koncentrācijatkarīgums,
|
|
—
|
visi rezultāti ir vēsturisko negatīvo kontroļu datu sadalījuma robežās (piem., 95 % kontrolrobežās Puasona sadalījumā); sk. 48. punktu).
|
Tādā gadījumā šajā testa sistēmā uzskata, ka testējamā ķimikālija nespēj inducēt mutācijas.
|
Kopīgi MLA un TK6
|
67.
|
Ja maksimālo koncentrāciju izraugās pēc citotoksicitātes, ar augstāko koncentrāciju būtu jācenšas panākt 20 līdz 10 % lielu RTG/RS. Pastāv vienprātība par to, ka jābūt piesardzīgiem, interpretējot vienīgi 20 līdz 10 % RTG/RS robežās esošos pozitīvos rezultātus, un rezultāts nebūtu jāuzskata par pozitīvu, ja MF palielinājums (ja vērtēts) ir tik tikko 10 % RTG/RS līmenī vai mazāks par to (2)(59).
|
|
68.
|
Ja nav kultūras, kas uzrādītu 10–20 % lielu RGT/RS, noteikt, vai testējamā ķimikālija ir mutagēna, zināmos apstākļos var palīdzēt papildu informācija. Šīs situācijas ir šādas: 1) datu punktu rindas no 100 % līdz 20 % RTG/RS robežās nedod mutagenitātes pierādījumus (t. i., nav konstatējama atbildreakcijas atkarība no devas, mutantu biežums nav lielāks kā paralēlajā negatīvajā kontrolē vai vēsturiskajos fona diapazonos u. c.), un vismaz viens datu punkts atrodas 20 līdz 25 % RTG/RS robežās; 2) datu punktu rindas no 100 % līdz 25 % RTG/RS robežās nedod mutagenitātes pierādījumus (t. i., nav konstatējama atbildreakcijas atkarība no devas, mutantu biežums nav lielāks kā paralēlajā negatīvajā kontrolē vai vēsturiskajos fona diapazonos u. c.), un ir arī viens negatīvs datu punkts nedaudz zem 10 % RTG/RS līmeņa. Abās šajās situācijās var secināt, ka testējamā ķimikālija ir negatīva.
|
|
69.
|
Nepārprotami pozitīva vai negatīva atbildreakcija nav obligāti jāverificē.
|
|
70.
|
Ja atbildreakcija nav atbilstoši iepriekš rakstītajam nedz nepārprotami negatīva, nedz nepārprotami pozitīva un/vai lai rezultāta bioloģiskais relevantums būtu labāk nosakāms, šie dati jāizvērtē ekspertam un/vai jāturpina pētīt. Var būt lietderīgi izdarīt atkārtotu eksperimentu ar modificētiem nosacījumiem [piem., koncentrācijas intervāliem, kas palielinātu varbūtību iegūt datu punktus 10–20 % RTG/RS diapazonā, atšķirīgi metaboliskās aktivācijas nosacījumi (proti, S9 koncentrācija vai S9 avots) un apstrādes ilgums].
|
|
71.
|
Retos gadījumos pat pēc sīkākas izpētes datu kopums neļauj izdarīt slēdzienu par pozitīviem vai negatīviem rezultātiem. Tāpēc būtu jāsecina, ka testējamās ķimikālijas atbildreakcija bijusi neskaidra (to vienlīdz varbūtīgi ir interpretēt gan kā pozitīvu, gan kā negatīvu).
|
TESTĒŠANAS PĀRSKATS
|
72.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Testējamā ķimikālija:
|
—
|
avots, sērijas numurs, izmantošanas termiņš, ja pieejams,
|
|
—
|
pašas testējamās ķimikālijas stabilitāte, ja zināma,
|
|
—
|
testējamās ķimikālijas šķīdība un stabilitāte šķīdinātājā, ja zināma,
|
|
—
|
(attiecīgā gadījumā) pH, osmolalitātes un izgulsnēšanās mērījums kultūras barotnē, kurā testējamā ķimikālija pievienota.
|
|
|
Vienkomponenta viela:
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības,
|
|
—
|
ķīmiskā identifikācija, piemēram, IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, attiecīgā gadījumā, ja praktiski iespējams, piemaisījumu ķīmiskā identitāte u. tml.
|
|
|
|
Daudzkomponentu viela, UVCB un maisījumi:
|
—
|
iespējami izsmeļoši raksturoti pēc komponentu ķīmiskās identitātes (sk. iepriekš), kvantitatīvās sastopamības un relevantajām fizikālķīmiskajām īpašībām.
|
|
|
|
|
Šķīdinātājs:
|
—
|
šķīdinātāja izraudzīšanās pamatojums,
|
|
—
|
šķīdinātāja īpatsvars galīgajā kultūras barotnē.
|
|
|
|
Šūnas:
|
|
Laboratorijas izejkultūru gadījumā:
|
—
|
šūnu tips un avots un to vēsture testēšanas laboratorijā,
|
|
—
|
kariotipiskās īpašības un/vai modālais hromosomu skaits,
|
|
—
|
šūnu kultūru uzturēšanas metodes,
|
|
—
|
šūnu dubultošanās laiks.
|
|
|
|
|
Testēšanas nosacījumi:
|
—
|
koncentrāciju izvēles un šūnu kultūru skaita izraudzīšanās pamatojums, piem., dati par citotoksicitāti un šķīdības ierobežojumi,
|
|
—
|
barotnes sastāvs, CO2 koncentrācija, mitruma līmenis,
|
|
—
|
testējamās ķimikālijas koncentrācija, izteikta kā galīgā koncentrācija kultūras barotnē (piem., kultūras barotnes μg vai mg/ml vai mM),
|
|
—
|
kultūras barotnē pievienotā šķīdinātāja un testējamās ķimikālijas koncentrācija (un/vai tilpums),
|
|
—
|
inkubācijas temperatūra,
|
|
—
|
šūnu blīvums apstrādes laikā,
|
|
—
|
metaboliskās aktivācijas sistēmas veids un sastāvs (S9 avots, S9 maisījuma sagatavošanas metode, S9 maisījuma koncentrācija vai tilpums un S9 koncentrācija vai tilpums galīgajā kultūras barotnē, S9 kvalitātes kontroles),
|
|
—
|
attiecībā uz katru no apstrādes nosacījumiem — pozitīvās un negatīvās kontroles vielas, galīgās koncentrācijas,
|
|
—
|
fenotipiskās ekspresijas periods (norādot uzsēto šūnu skaitu, subkultūras un attiecīgā gadījumā barošanas grafiku),
|
|
—
|
selektīvā aģenta identitāte un koncentrācija,
|
|
—
|
MLA gadījumā norāda izmantoto versiju (agars vai mikroiedobes),
|
|
—
|
testu pieņemamības kritēriji,
|
|
—
|
dzīvotspējīgo šūnu un mutantšūnu skaitīšanas metodes,
|
|
—
|
citotoksicitātes mērīšanas metodes,
|
|
—
|
jebkāda citotoksicitātei un izmantotajai metodei relevanta papildinformācija,
|
|
—
|
inkubācijas laiks pēc uzsēšanas,
|
|
—
|
uzskaitīto koloniju lielums un veids (attiecīgā gadījumā norādot kritērijus, pēc kuriem kolonijas definē kā “mazas” vai “lielas”),
|
|
—
|
kritēriji, pēc kuriem novērtē, vai pētījuma rezultāts ir pozitīvs, negatīvs vai neskaidrs,
|
|
—
|
(attiecīgā gadījumā) pH, osmolalitātes un izgulsnēšanās noteikšanai izmantotās metodes.
|
|
|
|
Rezultāti:
|
—
|
par katru kultūru — apstrādāto šūnu un apakškultūrām izmantoto šūnu skaits,
|
|
—
|
toksicitātes parametri (MLA gadījumā RTG, un TK6 gadījumā RS),
|
|
—
|
izgulsnēšanās pazīmes un noteikšanas laiks,
|
|
—
|
selektīvā un neselektīvā barotnē uzsēto šūnu skaits,
|
|
—
|
koloniju skaits neselektīvajā barotnē un rezistento koloniju skaits selektīvajā barotnē, saistītais mutantu biežums,
|
|
—
|
koloniju lielums negatīvajā un pozitīvajā kontrolē un (ja testējamā ķimikālija ir pozitīva) vismaz pie vienas koncentrācijas un saistītais mutantu biežums,
|
|
—
|
ja iespējams, koncentrācijas–atbildreakcijas sakarība,
|
|
—
|
paralēlo negatīvo (šķīdinātājs) un pozitīvo kontroļu dati (koncentrācijas un šķīdinātāji),
|
|
—
|
vēsturisko negatīvo (šķīdinātājs) un pozitīvo kontroļu dati (koncentrācijas un šķīdinātāji), norādot diapazonus, vidējās vērtības un standartnovirzes, vēsturisko kontroļu pamatā esošo testu skaits,
|
|
—
|
statistiskās analīzes (par atsevišķām kultūrām un attiecīgā gadījumā par apkopotiem replikātiem) un p-vērtības, ja tādas ir. MLA gadījumā – arī GEF izvērtējums.
|
|
|
LITERATŪRA
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411–416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Available at: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
1. papildinājums
DEFINĪCIJAS
Aneigēns
: ķimikālija vai process, kas mijiedarbībā ar mitotiskā un mejotiskā šūnu dalīšanās cikla komponentiem izraisa šūnu vai organismu aneiploīdiju.
Aneiploīdija
: novirze no normālā diploīdā (vai haploīdā) hromosomu skaita par vienu vai vairākām atsevišķām hromosomām, bet ne par veselu hromosomu komplektu vai komplektiem (poliploīdija).
Bāzu pāru maiņas mutagēni
: ķimikālijas, kas izraisa DNS bāzu pāru maiņu.
Ķimikālija
: viela vai maisījums.
Klonēšanās efektivitāte
: to zemā blīvumā uzsēto šūnu procents, no kurām var izaugt pieskaitāma kolonija.
Klastogēns
: ķimikālija vai process, kas šūnu vai organismu populācijās izraisa strukturālas hromosomu aberācijas.
Citotoksicitāte
: attiecībā uz šīs testēšanas metodes aptverto ķīmisko analīzi citotoksicitāte ir relatīvā kopējā pieauguma (RTG) samazinājums MLA gadījumā vai relatīvās izdzīvotības (RS) samazinājums TK6 gadījumā.
Tieša mutācija
: tāda gēna mutācija no vecāktipa uz mutantformu, kura maina vai izdzēš iekodētā proteīna fermentatīvo darbību vai tā funkciju.
Nolasīšanas fāzes nobīdes mutagēni
: ķimikālijas, kas DNS molekulā izraisa viena vai vairāku bāzu pāru pievienošanos vai izzušanu.
Genotoksisks
: vispārīgs termins, kas aptver visu veidu bojājumus DNS vai hromosomās, tostarp DNS pārrāvumus, aduktus, pārkārtojumus, mutācijas, hromosomu aberācijas un aneiploīdiju. Ne visas genotoksiskās ietekmes rada mutācijas vai noturīgus hromosomu bojājumus.
Mitotiskā rekombinācija
: mitozes laikā notiekoša homoloģisku hromatīdu rekombinācija, kuras rezultātā var inducēties DNS dubultpavedienu pārrāvumi vai zust heterozigotāte.
Mutagēnisks
: tāds, kas pārmantojami maina vienu vai vairākas DNS bāzu pāru sekvences gēnos vai hromosomu struktūru (hromosomu aberācijas).
Mutantu biežums (MF)
: novēroto mutantšūnu skaita attiecība pret dzīvotspējīgo šūnu skaitu.
Fenotipiskās ekspresijas laiks
: pēcapstrādes laiks, kad ģenētiskā pārmaiņa nostiprinās genomā un jebkādi līdzšinējā gēna produkti izsīkst tiktāl, ka mainās arī fenotipiskā pazīme.
Relatīvā izdzīvotība (RS)
: RS attiecībā uz TK6 izmanto par apstrādes radīta citotoksicitātes mēru. Tā ir tūlīt pēc apstrādes uz platēm uzsētu šūnu relatīvā klonēšanās efektivitāte (CE) salīdzinājumā ar negatīvās kontroles klonēšanās efektivitāti.
Relatīvais suspensijas pieaugums (RSG)
: MLA gadījumā tas ir relatīvais kopējais suspensijas divdienu pieaugums testa kultūrā attiecībā pret kopējo suspensijas divdienu pieaugumu negatīvajā/šķīdinātāja kontrolē (Clive and Spector, 1975). Ar RSG būtu jāizsaka testa kultūras un negatīvās/šķīdinātāja kontroles relatīvā pieauguma salīdzinājums apstrādes periodā.
Relatīvais kopējais pieaugums (RTG)
: RTG attiecībā uz MLA izmanto par apstrādes radītas citotoksicitātes mēru. Tas ir testa kultūru relatīvā pieauguma mērs (salīdzinājumā ar nesēja kontroli) noteiktās testa fāzēs, proti, apstrādē, divu dienu ilgajā ekspresijas laikā un atlasīšanai vajadzīgo mutantu klonēšanā. Katras testa kultūras RSG tiek reizināts ar testa kultūras relatīvo klonēšanas efektivitāti mutantu atlasīšanas laikā un izteikts attiecībā pret klonēšanas efektivitāti negatīvajā/šķīdinātāja kontrolē (Clive and Spector, 1975).
Aknu S9 frakcijas
: aknu homogenāta supernatants pēc centrifugēšanas pie 9 000 g, t. i., jēlu aknu ekstrakts.
S9 maisījums
: aknu S9 frakcijas un metaboliskajai fermentatīvajai darbībai nepieciešamo kofaktoru maisījums.
Suspensijas pieaugums (SG)
: MLA apstrādes un ekspresijas fāzes laikā konstatētā šūnu skaita palielinājuma reižu skaits. Īslaicīgā (3–4 stundu ilgā) apstrādē SG tiek aprēķināts, reizinot palielinājuma reižu skaitu 1. dienā un palielinājuma reižu skaitu 2. dienā. Ja izmanto 24 stundu ilgu apstrādi, SG ir 24 stundu apstrādes laikā notikušā palielinājuma reižu skaits, reizināts ar ekspresijas 1. un 2. dienā notikušā palielinājuma reižu skaitu.
Šķīdinātāja kontrole
: vispārīgs termins, ar ko definē kontroles kultūras, kuras saņem vienīgi testējamās ķimikālijas šķīdināšanai izmantoto šķīdinātāju.
Testējamā ķimikālija
: viela vai maisījums, kuru testē ar šo testēšanas metodi.
Neapstrādātās kontroles
: kultūras, kuras neapstrādā (t. i., ne ar testējamo ķimikāliju, ne ar šķīdinātāju), bet ar kurām izdara tādas pašas manipulācijas kā ar kultūrām, kuras apstrādā ar testējamo ķimikāliju.
2. papildinājums
FORMULAS
Citotoksicitāte
Abām (agara un mikroiedobju) MLA versijām
Citotoksicitāti definē kā relatīvo kopējo pieaugumu (RTG), un tas ietver relatīvo suspensijas pieaugumu (RSG) divu dienu ilgajā ekspresijas periodā un relatīvo klonēšanās efektivitāti (RCE), kuru nosaka mutantu atlases brīdī. Gan RTG, gan RSG un RCE izsaka procentuāli.
RSG aprēķināšana. Suspensijas pieaugums viens (SG1) ir pieauguma temps laikā no 0. dienas līdz 1. dienai (šūnu koncentrācija 1. dienā, dalīta ar šūnu koncentrāciju 0. dienā), un suspensijas pieaugums divi (SG2) ir pieauguma temps laikā no 1. dienas līdz 2. dienai (šūnu koncentrācija 2. dienā, dalīta ar šūnu koncentrāciju 1. dienā). RSG ir apstrādātās kultūras kopējais SG (SG1 x SG2) salīdzinājumā ar neapstrādāto/šķīdinātāja kontroli. Tas ir: RSG = [SG1(test) x SG2(test)] / [SG1(control) x SG2(control)]. SG1 aprēķināms no sākotnējās šūnu koncentrācijas, kas izmantota šūnu apstrādes sākumā. Šādā veidā tiek ņemta vērā šūnu apstrādes laikā testa kultūrās izveidojusies diferenciāla citotoksicitāte, ja tāda ir.
RCE ir testa kultūras relatīvā klonēšanās efektivitāte mutantu atlasīšanas brīdī attiecībā pret klonēšanās efektivitāti negatīvajā/šķīdinātāja kontrolē.
Relatīvais kopējais pieaugums (RTG):
RTG=RSG x RCE
TK6
Relatīvā izdzīvotība (RS)
Citotoksicitāti vērtē pēc relatīvās izdzīvotības, t. i., pēc tā, kāda ir tūlīt pēc apstrādes uzsētu šūnu klonēšanās efektivitāte (CE), kas koriģēta pēc jebkāda šūnu zuduma apstrādes laikā, salīdzinājumā ar klonēšanās efektivitāti negatīvajās kontrolēs (tajās konstatēto izdzīvotību pieņem par 100 %). Ar apstrādes laikā radušos šūnu zudumu saistīto korekciju var aprēķināt šādi:

RS ar testējamo ķimikāliju apstrādātai kultūrai aprēķina šādi:

Mutantu biežums attiecībā uz MLA un uz TK6
Mutantu biežums (MF) ir mutantkoloniju klonēšanās efektivitāte selektīvā barotnē (CEM), kas koriģēta pēc klonēšanās efektivitātes neselektīvā barotnē mutantu atlasīšanas brīdī (CEV), respektīvi, MF=CEM/CEV. Zemāk aprakstīts, kā šīs divas klonēšanās efektivitātes aprēķināmas agara un mikroiedobju metodes gadījumā.
MLA agara versija.
MLA mīkstā agara versijā koloniju skaitu uz mutantu atlasīšanas plates (CM) un koloniju skaitu uz atlasīšanai neparedzētās jeb klonēšanās efektivitātes (dzīvotspējas uzskaites) plates (CV) nosaka, tiešā veidā saskaitot klonus. Ja klonēšanās efektivitātes (CE) vajadzībai uz mutantu atlasīšanas (CEM) platēm un uz atlasīšanai neparedzētām jeb klonēšanas efektivitātes (dzīvotspējas uzskaites) platēm (CEV) uzsēj 600 šūnas un mutantu atlasīšanai izmanto 3 x 106 šūnas,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
MLA un TK6 mikroiedobju versija.
MLA mikroiedobju versijā CM un CV tiek noteikta, reizinot mikroiedobju kopskaitu (TW) un to koloniju varbūtīgo skaitu, kas ir katrā mikroiedobju plates iedobē (P).
CM = PM x TWM
CV = PV x TWV
Pēc Puasona sadalījuma (Furth et al., 1981) nulles terma P iegūst šādi:
P = - ln (EW / TW),
kur EW ir tukšās iedobes un TW ir iedobju kopskaits. Tādējādi
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
Mazo un lielo koloniju mutantu biežumu MLA mikroiedobju versijā aprēķina tieši tāpat, tukšo iedobju skaitu attiecinot uz mazajām un lielajām kolonijām.
TK6 gadījumā mazo un lielo koloniju mutantu biežumu nosaka, pamatojoties uz agrīnajiem un vēlīnajiem mutantiem.
B.68. ĪSA EKSPOZĪCIJAS LAIKA IN VITRO TESTĒŠANAS METODE, AR KURU IDENTIFICĒT i) ĶIMIKĀLIJAS, KAS IZRAISA NOPIETNUS ACU BOJĀJUMUS, UN ii) ĶIMIKĀLIJAS, KURAS NAV JĀKLASIFICĒ KĀ ACU KAIRINĀJUMA VAI NOPIETNU ACU BOJĀJUMU IZRAISĪTĀJAS
IEVADS
|
1.
|
Šī testēšanas metode (TM) ir ekvivalenta ESAO testēšanas vadlīnijā (TG) Nr. 491 (2017) aprakstītajam. Īsa ekspozīcijas laika (STE) testēšanas metode ir in vitro metode, kuru noteiktos apstākļos un ar zināmiem ierobežojumiem var lietot tādu ķimikāliju (vielu un maisījumu) bīstamības klasificēšanai un marķēšanai, kas izraisa nopietnus acu bojājumus vai kuras nav jāklasificē kā nopietnu acu bojājumu vai acu kairinājuma izraisītājas, atbilstoši definīcijai, kas dota Apvienoto Nāciju Organizācijas (ANO) Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētajā sistēmā (GHS) (1) un Eiropas Savienības (ES) Regulā Nr. 1272/2008 par vielu un maisījumu klasificēšanu, marķēšanu un iepakošanu (CLP regula) (67).
|
|
2.
|
Ilgus gadus ķimikāliju potenciālā bīstamība acīm tikusi vērtēta galvenokārt ar truša acu in vivo testu (TM B.5 (8), ekvivalents OECD TG 405). Ir vispārpieņemts, ka pārskatāmā nākotnē neviens atsevišķs alternatīvs in vitro tests nespēs pilnībā aizstāt truša acu in vivo testu, ar kuru var prognozēt pilnu nopietnu acu bojājumu / acu kairinājuma atbildreakciju spektru un vienlaikus aptvert dažādas ķīmisko vielu klases. Tomēr truša acu testu varētu būt iespējams pilnībā aizstāt, (pakāpjveida) testēšanā stratēģiski kombinējot alternatīvas testēšanas metodes (2). Ar lejupēju pieeju tiktu testētas ķimikālijas, par kurām ir informācija, ka tās ir potenciāli ļoti kairinošas acīm vai izraisa nopietnus acu bojājumus. Ar augšupēju pieeju turpretim testētu ķimikālijas, par kurām ir informācija, ka tās acīm nav tik kairinošas, lai būtu jāklasificē. Kaut arī STE testēšanas metodi nevar uzskatīt par pilnīgu truša acu in vivo testa aizstājēju, normatīvām vajadzībām veiktas klasificēšanas un marķēšanas kontekstā tā ir piemērota izmantošanai tādā pakāpjveida testēšanas stratēģijā kā augšupējā/lejupējā pieeja un ļauj bez turpmākas testēšanas identificēt i) ķimikālijas, kas izraisa nopietnus acu bojājumus (ANO GHS / CLP 1. kategorija), un ii) ķimikālijas (izņemot ļoti gaistošas vielas un visas cietās ķimikālijas, kas nav virsmaktīvās vielas), kuras nav jāklasificē kā acu kairinājuma vai nopietnu acu bojājumu izraisītājas (ANO GHS / CLP nekategorizētas ķimikālijas) (1)(2). Tomēr, ja ar STE testēšanas metodi ķimikālija netiek klasificēta kā nopietnu acu bojājumu izraisītāja (ANO GHS / CLP 1. kategorija) vai kā ANO GHS / CLP nekategorizēta ķimikālija (neizraisa nopietnus acu bojājumus vai acu kairinājumu), būs vajadzīga papildu testēšana, lai noteiktu galīgo klasifikāciju. Turklāt, pirms ar augšupēju pieeju STE testēšanas metodi izmantot klasifikācijas shēmā, kas nav ANO GHS / CLP, jākonsultējas ar attiecīgajām regulatīvajām iestādēm. Piemērotākās testēšanas metodes izvēle un šīs testēšanas metodes izmantošana jāskata kontekstā ar ESAO Norādījumu dokumentu par integrētu pieeju nopietnu acu bojājumu un acu kairinājuma testēšanā un novērtēšanā [OECD Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation] (14).
|
|
3.
|
Šajā testēšanas metodē aprakstītas procedūras, ko izmanto, lai testējamās ķimikālijas potenciālo bīstamību acīm izvērtētu pēc tās spējas inducēt citotoksicitāti, ja pielieto īsa ekspozīcijas laika testēšanas metodi. Ķimikāliju citotoksiskā iedarbība uz radzenes epitēlijšūnām ir nozīmīga veida iedarbība (MOA), kas izraisa radzenes epitēlijšūnu bojājumus un acu kairinājumu. Šūnu dzīvotspēju ar STE testēšanas metodi novērtē pēc tā, cik lielā mērā vitālo krāsvielu MTT (3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolijbromīds, saukts arī par tiazolilzilo tetrazolijbromīdu) dzīvās šūnas fermentatīvi pārvērš par zilas krāsas formazāna sāli, kura daudzumu šūnu ekstraktā nosaka kvantitatīvi (3). Iegūto šūnu dzīvotspēju salīdzina ar šķīdinātāja kontroli (relatīvā dzīvotspēja) un izmanto, lai aplēstu testējamās ķimikālijas potenciālo bīstamību acīm. Ja gan ar 5 %, gan ar 0,05 % koncentrāciju iegūtā šūnu dzīvotspēja ir mazāka par vai vienāda ar (≤) 70 %, testējamo ķimikāliju klasificē ANO GHS / CLP 1. kategorijā. Ja turpretim gan ar 5 %, gan ar 0,05 % koncentrāciju iegūtā šūnu dzīvotspēja ir lielāka par (>) 70 %, pieņem, ka testējamo ķimikāliju nav vajadzības kategorizēt ANO GHS / CLP sistēmā.
|
|
4.
|
Terminu “testējamā ķimikālija” šajā testēšanas metodē izmanto, lai apzīmētu to, kas tiek testēts, bez sasaistes ar STE testēšanas metodes pielietojamību vielu un/vai maisījumu testēšanā. Definīcijas dotas papildinājumā.
|
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
5.
|
Šīs testēšanas metodes pamatā ir Kao korporācijas izstrādātais protokols (4), kas izvērtēts divos dažādos validācijas pētījumos: vienu no tiem veikusi Japānas Dzīvniektestēšanas alternatīvu sabiedrība (JSAAE) (5), bet otru – Japānas Alternatīvo metožu validācijas centrs (JaCVAM) (6). Pamatojoties uz šo validācijas pētījumu pārskatiem un citiem testēšanas metodi pamatojošiem dokumentiem, zinātnisko recenzēšanu veica NICEATM/ICCVAM (7).
|
|
6.
|
Kad STE testēšanas metodi izmantoja, lai identificētu ķimikālijas (vielas un maisījumus), kas izraisa nopietnus acu bojājumus (ANO GHS / CLP 1. kategorija) (1), iegūtie dati par 125 ķimikālijām (gan vielām, gan maisījumiem) salīdzinājumā ar truša acu in vivo testu uzrādīja kopējo pareizību 83 % (104/125), pseidopozitīvu īpatsvaru 1 % (1/86) un pseidonegatīvu īpatsvaru 51 % (20/39) (7). Uzrādītais pseidonegatīvu īpatsvars šajā kontekstā nav kritiski svarīgs, jo visas testējamās ķimikālijas, kuru inducētā šūnu dzīvotspēja pie 5 % koncentrācijas ir ≤ 70 % un pie 0,05 % koncentrācijas > 70 %, vēlāk tiks testētas ar citām pienācīgi validētām in vitro testēšanas metodēm vai – kritiskā gadījumā – ar truša acu in vivo testu, izvēli izdarot atkarībā no normatīvajām prasībām un saskaņā ar patlaban ieteiktajām secīgas testēšanas stratēģijas un pierādījumu izsvēršanas pieejām (1) (8). Testētas tika galvenokārt vienkomponenta vielas, bet ir arī ierobežots daudzums datu par maisījumu testēšanu. Daudzkomponentu vielu un maisījumu testēšanā šī testēšanas metode ir tehniski piemērojama. Tomēr pirms tās izmantošanas maisījuma testēšanai, lai iegūtu datus kādam normatīvajam mērķim, jāapsver, vai un kādēļ tā varētu sniegt minētajam mērķim piemērotus rezultātus. Ja pastāv normatīva prasība maisījumu testēt, tāda apsvēršana nav vajadzīga. Kad STE testēšanas metodi izmantoja ANO GHS / CLP 1. kategorijā klasificējamu testējamo ķimikāliju identificēšanai, nekādus citus īpašus trūkumus tā neuzrādīja. Pētnieki var apsvērt šīs testēšanas metodes izmantošanu ar testējamajām ķimikālijām, un tā tiktu pieņemts, ka šūnu dzīvotspēja ≤ 70 % gan pie 5 %, gan pie 0,05 % koncentrācijas uzrāda nopietnus acu bojājumus inducējošu atbildreakciju, un ķimikālija bez turpmākas testēšanas būtu jāklasificē ANO GHS / CLP 1. kategorijā.
|
|
7.
|
Kad STE testēšanas metodi izmantoja, lai identificētu ķimikālijas (vielas un maisījumus), kuras nav jāklasificē kā acu kairinājuma vai nopietnu acu bojājumu izraisītājas (t. i., ANO GHS / CLP nekategorizētas ķimikālijas), iegūtie dati par 130 ķimikālijām (gan vielām, gan maisījumiem) salīdzinājumā ar truša acu in vivo testu uzrādīja kopējo pareizību 85 % (110/130), pseidonegatīvu īpatsvaru 12 % (9/73) un pseidopozitīvu īpatsvaru 19 % (11/57) (7). Ja no datu kopas izslēdz ļoti gaistošas vielas un cietas vietas, kas nav virsmaktīvās vielas, kopējā pareizība pieaug līdz 90 % (92/102), pseidonegatīvu īpatsvars sarūk līdz 2 % (1/54) un pseidopozitīvu īpatsvars saglabājas 19 % (9/48) (7) Sekas ir tādas, ka tad, ja STE tiek izmantota, lai identificētu testējamās ķimikālijas, kuras nav jāklasificē kā acu kairinājuma vai nopietnu acu bojājumu izraisītājas (ANO GHS / CLP nekategorizētas ķimikālijas), metodes potenciālie trūkumi ir liels pseidonegatīvu īpatsvars i) ļoti gaistošām vielām ar tvaika spiedienu virs 6 kPa un ii) cietām ķimikālijām (vielām un maisījumiem), kas nav virsmaktīvās vielas un maisījumi, kuru sastāvā ir vienīgi virsmaktīvās vielas. Šādas ķimikālijas ir izslēgtas no STE testēšanas metodes piemērojamības jomas (7).
|
|
8.
|
Papildus 6. un 7. punktā minētajām ķimikālijām ar STE testēšanas metodi ģenerētajā datu kopā ietilpst arī pašmāju dati par 40 maisījumiem. Šie dati, kad tos salīdzināja ar Dreiza [Draize] in vivo acu testa spēju paredzēt, kuri maisījumi nav jāklasificē ANO GHS / CLP klasifikācijas sistēmā, uzrādīja pareizību 88 % (35/40), pseidopozitīvu īpatsvaru 50 % (5/10) un pseidonegatīvu īpatsvaru 0 % (0/30) (9). Tāpēc STE metodi var izmantot, lai ar augšupēju pieeju identificētu identificētu maisījumus kā ANO GHS / CLP nekategorizētas ķimikālijas. Tāpat kā cieto vielu gadījumā, izņēmums ir cietie maisījumi, ja vien tie pilnībā nesastāv no virsmaktīvajām vielām. Turklāt, lai nepieļautu to, ka ķimikālijas kaitīgums tiek paredzēts nepietiekami, ar sevišķu rūpību jāvērtē maisījumi, kuru sastāvā ir vielas ar tvaika spiedienu virs 6 kPa, un iegūtie rezultāti jāpamato katrā atsevišķā gadījumā.
|
|
9.
|
Tā kā ar STE testēšanas metodi daudzas ANO GHS / CLP 1. kategorijas ķimikālijas tiek nepietiekami stingri novērtētas, ierindojot tās 2., 2A vai 2B kategorijā, savukārt daudzas ANO GHS / CLP nekategorizētas ķimikālijas tiek novērtētas pārlieku stingri, tāpat ierindojot tās 2., 2A vai 2B kategorijā, STE testēšanas metodi nevar izmantot, lai testējamās ķimikālijas identificētu kā tādas, kas ietilpst ANO GHS / CLP 2.kategorijā vai ANO GHS / CLP 2A kategorijā (acu kairinājums) vai 2B kategorijā (viegls acu kairinājums) (7). Tālab var būt nepieciešams veikt turpmāku testēšanu ar citu piemērotu metodi.
|
|
10.
|
STE testēšanas metode ir derīga testējamajām ķimikālijām, kuras izšķīdinātas vai vismaz 5 minūšu ilgumā vienmērīgi suspendētas fizioloģiskajā šķīdumā, 5 % dimetilsulfoksīda (DMSO), kas izšķīdināts fizioloģiskajā šķīdumā, vai minerāleļļā. STE testēšanas metode nav derīga testējamajām ķimikālijām, kuras ir nešķīstošas vai kuras nevar vismaz 5 minūšu ilgumā vienmērīgi suspendēt fizioloģiskajā šķīdumā, 5 % DMSO, kas izšķīdināts fizioloģiskajā šķīdumā, vai minerāleļļā. Minerāleļļu STE testēšanas metodē ir iespējams izmantot tāpēc, ka ekspozīcijas laiks ir īss. Tāpēc STE testēšanas metode ir piemērota, lai paredzētu ūdenī nešķīstošu testējamo ķimikāliju (piem., garķēdes alifātisko spirtu vai ketonu) potenciālo bīstamību acīm, ja vien šīs ķimikālijas ir iespējams iejaukt kādā no trijiem iepriekš nosauktajiem šķīdinātājiem.
|
|
11.
|
Terminu “testējamā ķimikālija” šajā testēšanas metodē izmanto, lai apzīmētu to, kas tiek testēts (68), bez sasaistes ar STE testēšanas metodes pielietojamību vielu un/vai maisījumu testēšanā.
|
TESTA PRINCIPS
|
12.
|
STE testēšanas metode ir uz citotoksicitātes principa pamatota in vitro ķīmiskā analīze, kuru veic uz saplūstoša SIRC (Statens Seruminstitut Rabbit Cornea) šūnu monoslāņa, kas kultivēts uz 96 mikroiebobju polikarbonāta platēm (4). Testējamajai ķimikālijai ļauj iedarboties piecas minūtes, un tad, izmantojot MTT ķīmisko analīzi, citotoksicitāti kvantitatīvi izmēra SIRC šūnu relatīvās dzīvotspējas izteiksmē (4). Pēc konstatētā šūnu dzīvotspējas samazinājuma paredz potenciālo nelabvēlīgo ietekmi, kas var izraisīt acs bojājumus.
|
|
13.
|
Ir zināms, ka 80 % no truša acī iepilinātā šķīduma trīs līdz četru minūšu laikā tiek izvadīti caur konjunktīvas maisu, bet cilvēka gadījumā vienas līdz divu minūšu laikā šādā veidā tiek izvadīti vairāk nekā 80 % iepilinātā šķīduma (10). Ar STE testēšanas metodi ir mēģināts tuvināties šiem ekspozīcijas laikiem, un citotoksicitāte tiek izmantota par beigupunktu, lai novērtētu kaitējumu, kas SIRC šūnām nodarīts piecās minūtēs, kuru laikā uz tām iedarbojusies testējamā ķimikālija.
|
KOMPETENCES PIERĀDĪŠANA
|
14.
|
Pirms šeit aprakstīto STE testēšanas metodi sākt izmantot regulāri, laboratorijām jāpierāda tehniskā kompetence, pareizi saklasificējot 1. tabulā ieteiktās 11 vielas. Šīs vielas izraudzītas tā, lai būtu pārstāvēts pilns nopietnu acu bojājumu un acu kairinājuma atbildreakciju spektrs, kas noteikts ar truša acu in vivo testiem (TG-405) un atbilst ANO GHS / CLP klasifikācijas sistēmai (1). Citi atlases kritēriji bija vielu komerciālā pieejamība, kvalitatīvu in vivo references datu pieejamība un kvalitatīvu ar STE testēšanas metodi iegūtu in vitro datu pieejamība (3). Gadījumos, kur kāda sarakstā norādīta viela nav pieejama vai kur tas attaisnojami, var izmantot citu vielu, par kuru pieejami pietiekami in vivo un in vitro references dati, ja vien tiek izmantoti šeit aprakstītie kritēriji.
1. tabula
Standartvielu saraksts
|
Viela
|
CAS Nr.
|
Ķimikāliju klase (69)
|
Agregāt-stāvoklis
|
In vivo ANO GHS / CLP Kat. (70)
|
Šķīdinātājs STE testā
|
STE ANO GHS / CLP Kat.
|
|
Benzalkonija hlorīds (10 %, ūdens šķīdums)
|
8001-54-5
|
Onijsavieno-jums
|
Šķidrs
|
1. kategorija
|
Fizioloģiskais šķīdums
|
1.kategorija
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Ēteris
|
Šķidrs
|
1. kategorija
|
Fizioloģiskais šķīdums
|
1.kategorija
|
|
Acid Red 92
|
18472-87-2
|
Heterociklisks savienojums; bromīn-savienojums; hlorīnsavienojums
|
Ciets
|
1. kategorija
|
Fizioloģiskais šķīdums
|
1.kategorija
|
|
Nātrija hidroksīds
|
1310-73-2
|
Sārms; neorganiska ķīmiska viela
|
Ciets
|
1.kategorija (71)
|
Fizioloģiskais šķīdums
|
1.kategorija
|
|
Butirolaktons
|
96-48-0
|
Laktons; heterociklisks savienojums
|
Šķidrs
|
2A kategorija (CLP 2.kategorija)
|
Fizioloģiskais šķīdums
|
Nav iespējams prognozēt
|
|
1-oktanols
|
111-87-5
|
Spirts
|
Šķidrs
|
2A/B kategorija (72) (CLP 2.kategorija)
|
Minerāleļļa
|
Nav iespējams prognozēt
|
|
Ciklopentanols
|
96-41-3
|
Spirts; ogļūdeņradis, ciklisks
|
Šķidrs
|
2A/B kategorija (73) (CLP 2.kategorija)
|
Fizioloģiskais šķīdums
|
Nav iespējams prognozēt
|
|
2-etoksietilacetāts
|
111-15-9
|
Spirts; ēteris
|
Šķidrs
|
Nekategorizēts
|
Fizioloģiskais šķīdums
|
Nekategorizēts
|
|
Dodekāns
|
112-40-3
|
Ogļūdeņradis, aciklisks
|
Šķidrs
|
Nekategorizēts
|
Minerāleļļa
|
Nekategorizēts
|
|
Metilizobutilketons
|
108-10-1
|
Ketons
|
Šķidrs
|
Nekategorizēts
|
Minerāleļļa
|
Nekategorizēts
|
|
1,1-dimetilguanidīnsulfāts
|
598-65-2
|
Amidīns; sēra savienojums
|
Ciets
|
Nekategorizēts
|
Fizioloģiskais šķīdums
|
Nekategorizēts
|
|
Saīsinājumi: CAS Nr. — Chemical Abstracts Service reģistra numurs.
|
|
PROCEDŪRA
Šūnu monoslāņa sagatavošana
|
15.
|
STE testēšanas metodē jāizmanto truša radzenes (SIRC) šūnu līnija. SIRC šūnas ieteicams sagādāt no kvalificētas šūnu bankas, piem., Amerikas tipveida kultūru kolekcijas CCL60.
|
|
16.
|
SIRC šūnas kultivē 37 °C pie 5 % CO2 un mitrinātā atmosfērā kultivēšanas kolbā ar kultūras barotni, kura sastāv no Īgla minimālās barotnes (MEM), kas papildināta ar 10 % liellopu embriju seruma jeb govju fetālā seruma (FBS), 2 mM L-glutamīna, 50–100 vienībām/ml penicilīna un 50–100 μg/ml streptomicīna. Kultivēšanas kolbā kofluējušās šūnas jāatdala, izmantojot tripsīna-etilēndiamīntetraetiķskābes šķīdumu. Atdalīšanai var lietot šūnu skrāpi. Pirms šūnas izmantot rutīnas testiem, kultivēšanas kolbā tās jāpavairo (piem., 2–3 pārsējumos). Pēc atkausēšanas šūnas nebūtu jāpavairo vairāk kā 25 pārsējumos.
|
|
17.
|
Pēc tam izmantošanai STE testā gatavās šūnas sagatavo attiecīgā šūnu slāņa biezumā un uzsēj uz 96 iedobju mikrotitrēšanas plates. Ieteicamais šūnu uzsēšanas blīvums ir 6,0 × 103 šūnas uz vienu iedobi, ja šūnas lieto četras dienas pēc uzsēšanas, vai 3,0 × 103 šūnas uz vienu iedobi, ja šūnas lieto piecas dienas pēc uzsēšanas, pie kultivēšanas tilpuma 200 μl. STE testā izmantojamās šūnas, kas kultūras barotnē uzsētas atbilstošā biezumā, līdz testēšanas brīdim, t. i., četrās vai piecās dienās pēc uzsēšanas, sasniegs vairāk nekā 80 % kofluenci.
|
Testējamās ķimikālijas un kontroles vielu aplicēšana
|
18.
|
Vēlamākais šķīdinātājs, kurā izšķīdināt vai suspendēt testējamo ķimikāliju, ir fizioloģiskais šķīdums. Ja testējamā ķimikālija ir mazšķīstoša vai vismaz uz piecām minūtēm nevar tikt vienmērīgi izšķīdināta vai suspendēta fizioloģiskajā šķīdumā, otrā labākā izvēle ir 5 % DMSO (CAS Nr. 67-68-5), kas izšķīdināts fizioloģiskajā šķīdumā. Ja testējamā ķimikālija vismaz uz piecām minūtēm nevar tikt vienmērīgi izšķīdināta vai suspendēta ne fizioloģiskajā šķīdumā, ne 5 % DMSO, kas izšķīdināts fizioloģiskajā šķīdumā, nākamā izvēle ir minerāleļļa (CAS Nr. 8042-47-5).
|
|
19.
|
Izvēlētajā šķīdinātājā testējamo ķimikāliju vienmērīgi izšķīdina vai suspendē 5 % (masa/masa) koncentrācijā un pēc tam, izmantojot sērijveida 10-kārtēju atšķaidījumu, atšķaida līdz 0,5 % un 0,05 % koncentrācijai. Katra testējamā ķimikālija jātestē gan 5 %, gan 0,05 % koncentrācijā. Uz 96 iedobju plates kultivētās šūnas katrā iedobē piecas minūtes istabas temperatūrā eksponē 200 mikrolitriem (μl) testējamās ķimikālijas šķīduma (vai suspensijas), kuras koncentrācija ir 5 % vai 0,05 %. Testējamās ķimikālijas (vienkomponenta vielas, daudzkomponentu vielas vai maisījumus) uzskata par tīrām vielām un neatkarīgi no to patiesās tīrības atšķaida vai suspendē saskaņā ar aprakstīto metodi.
|
|
20.
|
Katrā atkārtojumā uz katras plates par barotnes kontroli izmanto 16. punktā aprakstīto kultūras barotni. Turklāt katrā atkārtojumā uz katras plates šūnas jāeksponē arī šķīdinātāja kontroles paraugiem. Ir pierādīts, ka 18. punktā aprakstītajiem šķīdinātājiem nav kaitīgas ietekmes uz SIRC šūnu dzīvotspēju.
|
|
21.
|
STE testēšanas metodē par pozitīvo kontroli katrā atkārtojumā uz katras plates jāizmanto 0,01 % nātrija laurilsulfāts (SLS), kas izšķīdināts fizioloģiskajā šķīdumā. Lai varētu aprēķināt šūnu dzīvotspēju pozitīvajā kontrolē, katrā atkārtojumā uz katras plates jāiekļauj arī fizioloģiskā šķīduma kontrole.
|
|
22.
|
Lai noteiktu, kā kompensēt optisko blīvumu, būs vajadzīgs tukšais paraugs, un to pielieto iedobēm, kurās ir tikai fosfātu fizioloģiskais buferšķīdums, kas nesatur kalciju un magniju (PBS-) vai šūnas.
|
|
23.
|
Ikviens paraugs (testējamā ķimikālija 5 % un 0,05 % koncentrācijā, barotnes kontrole, šķīdinātāja kontrole un pozitīvā kontrole) jātestē trīs reizes katrā atkārtojumā, uz piecām minūtēm istabas temperatūrā šūnas eksponējot 200 mikrolitriem attiecīgās testējamās vai kontroles ķimikālijas.
|
|
24.
|
Etalonvielas ir lietderīgi izmantot, lai novērtētu kādai konkrētai ķimikāliju vai produktu klasei piederīgas nezināmas ķimikālijas spēju izraisīt acu kairinājumu vai arī lai novērtētu kāda acu kairinātāja relatīvo spēju izsaukt noteiktas intensitātes atbildreakciju uz kairinājumu.
|
Šūnu dzīvotspējas mērīšana
|
25.
|
Pēc eksponēšanas šūnas divreiz mazgā ar 200 μl PBS un tad pievieno 200 μl MTT šķīduma (0,5 mg MTT uz mililitru kultūras barotnes). Pēc divu stundu ilga reakcijas laika inkubatorā (37°C, 5 % CO2) dekantē MTT šķīdumu, tad MTT formazānu ekstrahē, uz 60 minūtēm istabas temperatūrā tumsā pievienojot 200 mikrolitrus (μl) 0,04 N sālsskābes ar izopropanolu, un pie 570 nm ar mikroplašu lasītāju izmēra MTT formazāna šķīduma absorbciju. Testējamo ķimikāliju interference ar MTT ķīmisko analīzi (ar krāsvielām vai tiešiem MTT reducētājiem) rodas tikai tad, ja pēcekspozīcijas skalošanas laikā no testa sistēmas paliek neizvadīts ievērojams daudzums testējamās ķimikālijas. Tas ir aktuāli gadījumā ar trīsdimensionālajiem rekonstruētas cilvēka radzenes vai rekonstruētas cilvēka epidermas audiem, bet neattiecas uz divdimensionālajām šūnu kultūrām, kuras izmanto STE testēšanas metodē.
|
Rezultātu interpretēšana un prognostiskais modelis
|
26.
|
Par katru testējamo ķimikāliju iegūtās optiskā blīvuma (OB) vērtības izmanto, lai aprēķinātu šūnu dzīvotspēju salīdzinājumā ar šķīdinātāja kontroli, kurai to pieņem par 100 %. Šūnu relatīvo dzīvotspēju izsaka ar procentuālo īpatsvaru, testējamās ķimikālijas OB dalot ar šķīdinātāja kontroles OB (iepriekš no abām vērtībām atņem tukšā parauga OB).

Līdzīgā veidā šūnu relatīvo dzīvotspēju procentuāli izsaka katrai šķīdinātāja kontrolei, tās OB dalot ar barotnes kontroles OB (iepriekš no abām vērtībām atņem tukšā parauga OB).
|
|
27.
|
Jāizdara trīs neatkarīgi atkārtojumi, katrs no tiem ar trijām replikāta iedobēm (t. i., n=9). Ar katras testējamās ķimikālijas un šķīdinātāja kontroles trijām iedobēm iegūto vidējo aritmētisko katrā neatkarīgajā atkārtojumā izmanto, lai aprēķinātu šūnu relatīvās dzīvotspējas vidējo aritmētisko vērtību. Šūnu dzīvotspējas galīgo vidējo aritmētisko vērtību aprēķina no visiem trijiem neatkarīgajiem atkārtojumiem.
|
|
28.
|
Tālāk norādītas šūnu dzīvotspējas robežvērtības tādu testējamo ķimikāliju identificēšanai, kuras inducē nopietnus acu bojājumus (ANO GHS / CLP 1. kategorija), un arī tādu testējamo ķimikāliju identificēšanai, kuras nav jāklasificē kā acīm kairinošas vai nopietnus acu bojājumus izraisošas (ANO GHS / CLP nekategorizētās ķimikālijas).
|
2. tabula
STE testēšanas metodes prognostiskais modelis
|
Šūnu dzīvotspēja
|
ANO GHS / CLP klasifikācija
|
Piemērojamība
|
|
Pie 5 %
|
Pie 0,05 %
|
|
> 70 %
|
> 70 %
|
Nekategorizēta
|
Vielas un maisījumi, izņemot: i) ļoti gaistošas vielas, kuru tvaika spiediens pārsniedz 6 kPa (74), un ii) cietas ķimikālijas (vielas un maisījumus), kas nav virsmaktīvās vielas vai maisījumi, kuri pilnībā sastāv no virsmaktīvām vielām
|
|
≤ 70%
|
> 70 %
|
Nav iespējams prognozēt
|
Nepiemēro
|
|
≤ 70%
|
≤ 70%
|
1. kategorija
|
Vielas un maisījumi (75)
|
Pieņemamības kritēriji
|
29.
|
Testēšanas rezultātus uzskata par pieņemamiem, ja ir izpildīti visi zemāk nosauktie kritēriji.
|
a)
|
(Kultūras barotnei eksponētās) barotnes kontroles optiskajam blīvumam, no kura atņemts tukšā parauga optiskais blīvums, jābūt 0,3 vai lielākam.
|
|
b)
|
Šķīdinātāja kontroles dzīvotspējai attiecībā pret barotnes kontroli jābūt 80 % vai lielākai. Ja katrā atkārtojumā izmanto vairāku šķīdinātāju kontroles: lai ar šiem šķīdinātajiem testētās ķimikālijas būtu kvalificējamas, ikvienā no minētajām kontrolēm šūnu dzīvotspējai jābūt lielākai par 80 %.
|
|
c)
|
Ar pozitīvo kontroli (0,01 % SLS) iegūtajai šūnu dzīvotspējai jābūt vēsturiskās vidējās vērtības divu standartnoviržu robežās. Pozitīvās kontroles augšējās un apakšējās pieņemamības robežvērtības būtu jāatjaunina regulāri, t. i., reizi trijos mēnešos vai ikreiz, kad tests ar pieņemamu rezultātu izdarīts laboratorijā, kur testēšana notiek neregulāri (t. i., retāk nekā reizi mēnesī). Ja laboratorijas veikto eksperimentu nav pietiekami daudz, lai iegūtu statistiski robustu pozitīvo kontroļu sadalījumu, ir pieņemami izmantot metodes izstrādātāja noteiktās augšējās un apakšējās pieņemamības robežvērtības, t. i., vērtības intervālā no 21,1 % līdz 62,3 % atkarībā no izstrādātāja laboratorijas vēsturiskajiem datiem, un pašiem savu sadalījumu izveidot pirmo rutīnas testu laikā.
|
|
d)
|
Testējamās ķimikālijas 5 % un 0,05 % koncentrācijā trijos neatkarīgos atkārtojumos iegūtas galīgās šūnu dzīvotspējas standartnovirzei jābūt mazākai nekā 15 %.
Ja viens vai vairāki no šiem kritērijiem nav izpildīti, rezultātus atmet un izdara trīs citus neatkarīgus atkārtojumus.
|
|
DATI UN PĀRSKATA SAGATAVOŠANA
Dati
|
30.
|
Pārskatā jāiekļauj dati par katru individuālo mikroiebobi (piem., šūnu dzīvotspējas vērtības) visos atkārtojumos, kā arī jānorāda kopējā vidējā vērtība, standartnovirze un klasifikācija.
|
Testēšanas pārskats
|
31.
|
Testēšanas pārskatā norāda šādu informāciju.
|
|
Testējamā ķimikālija un kontroles vielas
|
—
|
Vienkomponenta viela: ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
—
|
Daudzkomponentu viela, UVCB un maisījums: iespējami izsmeļošs raksturojums ar, piem., sastāvdaļu ķīmisko identitāti (sk. iepriekš), tīrību, kvantitatīvo saturu un relevantajām fizikālķīmiskajām īpašībām
|
|
—
|
Agregātstāvoklis, gaistamība, pH, LogP, molekulmasa, ķīmiskā klase un citas pētījuma veikšanai relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
—
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
—
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
—
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
|
|
Testēšanas metodes nosacījumi un procedūras
|
—
|
Sponsora, testējošās iestādes, pētījuma vadītāja vārds/nosaukums un adrese
|
|
—
|
Izmantotās testēšanas metodes apraksts
|
|
—
|
Izmantotā šūnu līnija, tās avots, pasāžas numurs un testēšanā izmantoto šūnu konfluence
|
|
—
|
Sīkas ziņas par izmantoto testēšanas procedūru
|
|
—
|
Izmantoto atkārtojumu un replikātu skaits
|
|
—
|
Izmantotās testējamās ķimikālijas koncentrācijas (ja atšķiras no rekomendētajām)
|
|
—
|
Katras testējamās ķimikālijas šķīdinātāja izraudzīšanās pamatojums
|
|
—
|
Ar testējamo ķimikāliju saistītās ekspozīcijas ilgums (ja atšķiras no rekomendētā)
|
|
—
|
Testēšanas procedūras modifikāciju apraksts, ja tādas bijušas
|
|
—
|
Izmantoto izvērtēšanas un lēmumpieņemšanas kritēriju apraksts
|
|
—
|
Atsauce uz vēsturisko pozitīvo kontroles vidējo vērtību un standartnovirzi (SD)
|
|
—
|
Apliecināta laboratorijas kompetence izmantot šo testēšanas metodi (piem., testējot standartvielas) vai apliecināta testēšanas metodes ilgstoša reproducējamība
|
|
|
|
Rezultāti
|
—
|
Par katru testējamo ķimikāliju un kontroles vielu un par katru testēto koncentrāciju tabulā jānorāda individuālās OD vērtības katrā replikāta mikroiedobē, OD vidējās aritmētiskās vērtības katrā neatkarīgā atkārtojumā, procentuālā šūnu dzīvotspēja katrā neatkarīgā atkārtojumā un procentuālās šūnu dzīvotspējas galīgā vidējā aritmētiskā vērtība un SD, kas aptver trīs atkārtojumus
|
|
—
|
Barotnes, šķīdinātāja un pozitīvās kontroles rezultāti pēc piemērotiem pētījuma pieņemamības kritērijiem
|
|
—
|
Jebkādas citas novērotās ietekmes apraksts
|
|
—
|
Kopējā atvasinātā klasifikācija ar norādi uz izmantoto prognostisko modeli un/vai lēmumpieņemšanas kritēriju
|
|
|
LITERATŪRA
|
(1)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1855-1869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Available at: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Šā pielikuma B.5. nodaļa “Akūts acu kairinājums/korozija”.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1648-1653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā ar testēšanas metodi iegūto mērījumu rezultāti sakrīt ar pieņemtajām atsauces vērtībām. Tas ir testēšanas metodes veiktspējas raksturlielums un viens no relevantuma aspektiem. Šo terminu bieži izmanto tādā pat nozīmē kā terminu “konkordantums”, ar to saprotot testēšanas metodes pareizo iznākumu īpatsvaru (13).
Etalonķimikālija
: ķimikālija, ko salīdzināšanā ar testējamo ķimikāliju izmanto par standartu. Etalonķimikālijai jāpiemīt šādām īpašībām: i) konsekvents un uzticams avots vai avoti; ii) strukturāla un funkcionāla līdzība ar testējamās vielas klases vielām; iii) labi zināmas fizikālās un ķīmiskās īpašības; iv) apliecinoši dati par zināmajām ietekmēm un v) vielas zināmajai potencei jāatbilst vēlamās atbildreakcijas diapazonam.
Augšupēja pieeja
: pakāpeniska pieeja, ko izmanto ķimikālijai, par kuru ir aizdomas, ka tā nav jāklasificē kā acu kairinājuma vai nopietnu acu bojājumu izraisītāja, un kas sākas ar to ķimikāliju nošķiršanu, kuras nav jāklasificē (negatīvs iznākums), no citām ķimikālijām (pozitīvs iznākums).
Ķimikālija
: viela vai maisījums.
Acu kairinājums
: izmaiņas acī, kas rodas pēc testējamās ķimikālijas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas ir pilnībā atgriezeniskas. Nozīmē to pašu, ko “atgriezeniska ietekme uz acīm” un ANO GHS / CLP 2. kategorija.
Pseidonegatīvu īpatsvars
: pozitīvā iznākuma ķimikāliju proporcija, kuras ar testēšanas metodi aplami noteiktas par negatīvām. Tas ir viens no testēšanas metodes veiktspējas raksturlielumiem.
Pseidopozitīvu īpatsvars
: negatīvā iznākuma ķimikāliju proporcija, kuras ar testēšanas metodi aplami noteiktas par pozitīvām. Tas ir viens no testēšanas metodes veiktspējas raksturlielumiem.
Bīstamība
: tāda aģenta vai situācijas iedabiska īpašība, kam piemīt potenciāls atstāt nelabvēlīgu ietekmi uz šim aģentam eksponētu organismu, sistēmu vai (sub)populāciju.
Barotnes kontrole
: neapstrādāts replikāts, kurā ir visi testa sistēmas komponenti. Ar šo paraugu izdara tādas pašas manipulācijas kā ar paraugiem, kas apstrādāti ar testējamo ķimikāliju, un citiem kontrolparaugiem, lai tādējādi noteiktu, vai šķīdinātājs un testa sistēma mijiedarbojas.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairāk vielām.
Vienkomponenta viela
: viela, definēta ar tās kvantitatīvo sastāvu, kurā viena galvenā sastāvdaļa veido vismaz 80 % (masa/masa).
MTT
: 3-(4,5-Dimetiltiazol-2-il)-2,5-difeniltetrazolija bromīds; tiazolilzilais tetrazolija bromīds.
Daudzkomponentu viela
: viela, definēta ar tās kvantitatīvo sastāvu, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu viela tiek iegūta ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu viela rodas ķīmiskā reakcijā.
OB
: optiskais blīvums.
Pozitīvā kontrole
: replikāts, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka tā inducē pozitīvu atbildreakciju. Lai būtu iespēja novērtēt pozitīvās kontroles atbildreakcijas mainību laikā, pozitīvās atbildreakcijas intensitāte nedrīkstētu būt pārlieku liela.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Tā ir pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testēšanas metodes pareizības (konkordantuma) aspektu (10).
Ticamība
: pakāpe, kādā vienā un tajā pašā laboratorijā vai dažādās laboratorijās pēc viena un tā paša protokola var ilgāku laiku reproducējami izmantot konkrētu testēšanas metodi. To novērtē, aprēķinot metodes intralaboratorisko un interlaboratorisko reproducējamību un intralaboratorisko atkārtojamību (13).
Jutība
: tas, cik liela proporcija no visām pozitīvajām/aktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir pareizības mērs testēšanas metodei, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (10).
Nopietni acu bojājumi
: acs audu bojājumi vai nopietna fiziska redzes pasliktināšanās, kas rodas pēc testējamās ķimikālijas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas nav pilnībā atgriezeniska. Nozīmē to pašu, ko “neatgriezeniska ietekme uz acīm” un ANO GHS / CLP 1. kategorija.
Šķīdinātāja/nesēja kontrole
: neapstrādāts paraugs, kurā ir visi testa sistēmas komponenti, to vidū šķīdinātājs vai nesējs, ko izmanto ar testējamo ķimikāliju apstrādātajos paraugos un citos kontrolparaugos, un kuru izmanto, lai noteiktu bāzlīnijas atbildreakciju paraugiem, kas apstrādāti ar attiecīgajā šķīdinātājā vai nesējā izšķīdinātu testējamo ķimikāliju. Ja testā izmanto arī paralēlu barotnes kontroli, šis paraugs parāda arī to, vai šķīdinātājs vai nesējs mijiedarbojas ar testa sistēmu.
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tādas testēšanas metodes pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (13).
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai nepieciešamās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu.
Virsmaktīvā viela
: viela, piem., detergents, kas spēj samazināt šķidruma virsmas spraigumu un tādējādi ļauj tam putot vai penetrēt cietvielas; to dēvē arī par mitrinātāju.
Testējamā ķimikālija
: jebkura viela vai maisījums, ko testē ar šo testēšanas metodi.
Secīgas testēšanas stratēģija
: pakāpeniskas testēšanas stratēģija, kurā noteiktā secībā pārskata visu esošo informāciju par testējamo ķimikāliju, katrā pakāpē izmantojot pierādījumu izsvēršanu, lai pirms pārejas uz nākamo pakāpi noteiktu, vai ir pieejams pietiekami daudz informācijas, lai pieņemtu lēmumu par bīstamības klasifikāciju. Ja testējamās ķimikālijas kairinātājpotenciālu var noteikt, pamatojoties uz esošo informāciju, papildu testēšana nav jāveic. Ja, pamatojoties uz esošo informāciju, testējamās ķimikālijas kairinātājpotenciālu noteikt nevar, ievēro pakāpenisku secīgu procedūru testēšanai ar dzīvniekiem, un to dara tik ilgi, līdz var viennozīmīgi pieņemt lēmumu par klasifikāciju.
Lejupēja pieeja
: pakāpeniska pieeja, ko izmanto testējamai ķimikālijai, par kuru ir aizdomas, ka tā izraisa nopietnus acu bojājumus, un kas sākas ar to ķimikāliju nošķiršanu, kuras inducē nopietnus acu bojājumus (pozitīvs rezultāts), no citām ķimikālijām (negatīvs rezultāts).
Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (ANO GHS)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un kura paredz atbilstošus saziņas elementus, piem., piktogrammas, signālvārdus, bīstamības apzīmējumus, drošības prasību apzīmējumus un drošības datu lapas, ar mērķi sniegt informāciju par ķimikāliju nelabvēlīgo ietekmi, lai aizsargātu cilvēkus (viņu vidū darba devējus, darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (1).
ANO GHS / CLP 1. kategorija
: sk. “Nopietnu acu bojājumu” definīciju.
ANO GHS / CLP 2. kategorija
: sk. “Acu kairinājuma” definīciju.
ANO GHS / CLP nekategorizētas ķimikālijas
: Ķimikālijas, kas nav klasificētas ANO GHS / CLP 1. vai 2. kategorijā (vai ANO GHS 2A vai 2B kategorijā).
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kompleksi reakcijas produkti vai bioloģiski materiāli.
B.69 REKONSTRUĒTA, CILVĒKA RADZENEI LĪDZĪGA EPITĒLIJA (RhCE) TESTĒŠANAS METODE, KURĀ IDENTIFICĒ ĶIMIKĀLIJAS, KURAS NAV JĀKLASIFICĒ UN JĀMARĶĒ KĀ TĀDAS, KAS IZRAISA ACU KAIRINĀJUMU VAI NOPIETNUS ACU BOJĀJUMUS
IEVADS
|
1.
|
Šī testēšanas metode (TM) ir ekvivalenta ESAO testēšanas vadlīnijā (TG) Nr. 492 (2017) aprakstītajam. Nopietni acu bojājumi ir acs audu bojājumi vai nopietna fiziska redzes pasliktināšanās, kas rodas pēc testējamās ķimikālijas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas nav pilnībā atgriezeniska, saskaņā ar Apvienoto Nāciju Organizācijas Ķīmisko vielu klasificēšanas un marķēšanas globāli harmonizēto sistēmu (ANO GHS) (1) un Eiropas Savienības (ES) Regulu (EK) Nr. 1272/2008 par vielu un maisījumu klasificēšanu, marķēšanu un iepakošanu (CLP) (76). Saskaņā ar ANO GHS un CLP acu kairinājums ir izmaiņas acī, kas rodas pēc testējamās ķimikālijas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas ir pilnībā atgriezeniskas. Testējamās ķimikālijas, kas izraisa nopietnus acu bojājumus, klasificē ANO GHS un CLP 1. kategorijā, bet testējamās ķimikālijas, kas izraisa acu kairinājumu, klasificē ANO GHS un CLP 2. kategorijā. Testējamās ķimikālijas, kuras nav klasificētas par tādām, kas izraisa acu kairinājumu vai nopietnus acu bojājumus, saskaņā ar definīciju ir ķīmiskās vielas, kuras neatbilst priekšnosacījumiem klasificēšanai ANO GHS un CLP 1. vai 2. kategorijā (2A vai 2B), t. i., tās tiek uzskatītas par ANO GHS un CLP nekategorizētām ķimikālijām.
|
|
2.
|
Nopietnu acu bojājumu / acu kairinājuma novērtēšanai parasti izmantoti laboratorijas dzīvnieki (TM B.5 (2)). Vispiemērotākās testēšanas metodes izvēle un šīs testēšanas metodes izmantošana jāskata ESAO Vadlīniju par integrētu pieeju nopietnu acu bojājumu un acu kairinājuma testēšanai un novērtēšanai (39) kontekstā.
|
|
3.
|
Šajā testēšanas metodē aprakstīta in vitro procedūra, pēc kuras var noteikt ķimikālijas (vielas un maisījumus), kuras nav jāklasificē un jāmarķē kā tādas, kas saskaņā ar ANO GHS un CLP izraisa acu kairinājumu vai nopietnus acu bojājumus. Tajā izmanto rekonstruētu cilvēka radzenei līdzīgu epitēliju (RhCE), kura īpašības ļoti līdzinās cilvēka radzenes epitēlija histoloģiskajām, morfoloģiskajām, bioķīmiskajām un fizioloģiskajām īpašībām. Vēl četras in vitro testēšanas metodes ir apstiprinātas, uzskatāmas par zinātniski derīgām un pieņemtas kā TM B.47 (3), B.48 (4), B.61 (5) un B.68 (6), lai noteiktu cilvēka veselības beigupunktam atbilstošos nopietnos acu bojājumus / acu kairinājumu.
|
|
4.
|
Šajā testēšanas metodē iekļauti divi apstiprināti testi, kuros izmanto komerciāli pieejamus RhCE modeļus. Validācijas pētījumi acu kairinājuma / nopietnu acu bojājumu novērtēšanai veikti (7)(8)(9)(10)(11)(12)(13) ar EpiOcular™ acu kairinājuma testu (EIT) un SkinEthic™ cilvēka radzenes epitēlija (HCE) acu kairinājuma testu (EIT). Katrā no šiem testiem par testa sistēmu izmanto komerciāli pieejamas RhCE audu konstrukcijas, tālāk tekstā tās sauktas par validētām references metodēm: attiecīgi VRM1 un VRM2. No šiem validācijas pētījumiem un to neatkarīgajiem salīdzinošajiem izvērtējumiem (9)(12) secināja, ka ar EpiOcular™ EIT un SkinEthic™ HCE EIT iespējams pareizi identificēt ķimikālijas (vielas un maisījumus), kuras nav jāklasificē un jāmarķē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus saskaņā ar ANO GHS, un testus ieteica kā zinātniski derīgus šim nolūkam (13).
|
|
5.
|
Patlaban ir vispārpieņemts, ka tuvākajā nākotnē neviena atsevišķa in vitro testēšanas metode nevarēs pilnībā aizstāt Dreiza [Draize] in vivo acu testu (2)(14) un prognozēt visas nopietnu acu bojājumu / acu kairinājuma atbildreakcijas dažādām ķimikāliju klasēm. Tomēr Dreiza acu testu varētu būt iespējams pilnībā aizstāt, (pakāpjveida) testēšanas stratēģijā stratēģiski kombinējot vairākas alternatīvas testēšanas metodes, piem., augšupēju/lejupēju pieeju (15). Augšupējā pieeja (15) ir izstrādāta gadījumiem, kad, pamatojoties uz esošo informāciju, paredzams, ka ķimikālija neizraisīs tādu acu kairinājumu, ka tā būtu jāklasificē, savukārt lejupējā pieeja (15) ir izstrādāta gadījumiem, kad, pamatojoties uz esošo informāciju, ķimikālija izraisīs nopietnus acu bojājumus. EpiOcular™ EIT un SkinEthic™ HCE EIT ieteicams izmantot tādā testēšanas stratēģijā kā Scott et al. ierosinātā augšupējā/lejupējā pieeja, piemēram, par sākotnējo posmu augšupējā pieejā vai kā vienu no pēdējiem posmiem lejupējā pieejā, tādu ķimikāliju identificēšanai, kuras saskaņā ar ANO GHS / CLP (nekategorizētas ķīmiskās vielas) bez papildu testēšanas nav jāklasificē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus (15). Taču EpiOcular™ EIT un SkinEthic™ HCE EIT nav paredzēti ANO GHS / CLP 1. kategorijas (nopietni acu bojājumi) un ANO GHS / CLP 2. kategorijas (acu kairinājums) atšķiršanai. Šī diferenciācija būs jānosaka citā testēšanas stratēģijas pakāpē (15). Tāpēc testējamā ķimikālija, kura ar EpiOcular™ EIT vai SkinEthic™ HCE EIT identificēta par tādu, kas jāklasificē kā acu kairinājumu / nopietnus acu bojājumus izraisoša, jātestē papildus (in vitro un/vai in vivo), lai nonāktu pie galīgā secinājuma (ANO GHS / CLP nekategorizēta, 2. kategorijas vai 1. kategorijas ķimikālija), izmantojot, piem., TM B.47, B.48, B.61 vai B.68.
|
|
6.
|
Šajā testēšanas metodē aprakstīta procedūra, ko izmanto, lai izvērtētu testējamās ķimikālijas potenciālo bīstamību acīm, pamatojoties uz tās spēju izraisīt citotoksicitāti RhCE audu konstrukcijā, ko nosaka ar MTT ķīmisko analīzi (16) (sk. 21. punktu). RhCE audu dzīvotspēju pēc eksponēšanas testējamai ķimikālijai nosaka salīdzinājumā ar audiem, kas apstrādāti ar negatīvās kontroles vielu (% dzīvotspējas), pēc tam to izmanto, lai prognozētu testējamās ķimikālijas potenciālo bīstamību acīm.
|
|
7.
|
Lai saskaņā ar ESAO Vadlīniju Nr. 34 (18) principiem būtu vieglāk validēt jaunus vai grozītus in vitro RhCE testus, kas līdzinās EpiOcular™ EIT un SkinEthic™ HCE EIT, un lai ESAO TG 492 būtu iespējams savlaicīgi grozīt ar mērķi tajā iekļaut šos testus, ir pieejami veiktspējas standarti (17). Attiecībā uz testiem, kas validēti saskaņā ar veiktspējas standartiem, datu savstarpējā atzīšana (MAD) saskaņā ar ESAO nolīgumu būs garantēta tikai tad, ja ESAO šos testus būs izskatījusi un iekļāvusi attiecīgajā testēšanas vadlīnijā.
|
DEFINĪCIJAS
|
8.
|
Definīcijas ir sniegtas 1. papildinājumā.
|
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
9.
|
Šīs testēšanas metodes pamatā ir komerciālas trīsdimensiju RhCE audu konstrukcijas, ko ražo, izmantojot primāros cilvēka epidermas keratinocītus (t. i., EpiOcular™ OCL-200) vai imortalizētas cilvēka radzenes epitēlija šūnas (t. i., SkinEthic™ HCE/S). EpiOcular™ OCL-200 un SkinEthic™ HCE/S RhCE audu konstrukcijas ir līdzīgas in vivo radzenes epitēlija trīsdimensiju struktūrai, un tās ražo no pētāmās sugas šūnām (19)(20). Turklāt ar testiem tieši nosaka citotoksicitāti, ko izraisa ķimikālijas iekļūšana cauri radzenei un šūnu un audu bojājumu rašanās, pēc tam citotoksiskā atbildreakcija nosaka nopietnu acu bojājumu / acu kairinājuma vispārējo iznākumu in vivo. Šūnu bojājumus var izraisīt vairāki iedarbības veidi (sk. 20. punktu), taču citotoksicitātei ir svarīga vai pat primāra mehānistiska nozīme ķimikālijas izraisītas nopietnu acu bojājumu / acu kairinājuma vispārējās atbildreakcijas noteikšanā, kas neatkarīgi no audu bojājumu pamatā esošiem fizikāli ķīmiskajiem procesiem in vivo izpaužas galvenokārt kā radzenes apduļķošanās, irīts, konjunktīvas apsārtums un/vai konjunktīvas hemoze.
|
|
10.
|
Šīs testēšanas metodes pamatā esošajā validēšanas pētījumā testētas ļoti daudzas ķimikālijas, aptverot ļoti dažādus ķīmisko vielu veidus, ķīmisko vielu klases, molekulmasas, LogP, ķīmiskās struktūras utt. EpiOcular™ EIT validēšanas datubāzē kopumā bija ietvertas 113 ķimikālijas, saskaņā ar ESAO QSAR instrumentu kopuma analīzi aptverot 95 dažādas organiskās funkcionālās grupas (8). Vairākums šo ķimikāliju bija vienkomponenta vielas, taču pētījumā bija iekļautas arī vairākas daudzkomponentu vielas (tostarp 3 homopolimēri, 5 kopolimēri un 10 kvazipolimēri). 113 testēto ķimikāliju sadalījums pēc agregātstāvokļa un ANO GHS / CLP kategorijām bija šāds: 131. kategorijas šķidrumi, 151. kategorijas cietvielas, 6 2A kategorijas šķidrumi, 102A kategorijas cietvielas, 7 2B kategorijas šķidrumi, 7 2B kategorijas cietvielas, 27 nekategorizēti šķidrumi un 28 nekategorizētas cietvielas (8). SkinEthic™ HCE EIT validēšanas datubāzē kopumā bija ietvertas 200 ķimikālijas, aptverot 165 dažādas organiskās funkcionālās grupas (8)(10)(11). Vairākums šo ķimikāliju bija vienkomponenta vielas, taču pētījumā bija iekļautas arī vairākas daudzkomponentu vielas (tostarp 10 polimēri). 200 testēto ķimikāliju sadalījums pēc agregātstāvokļa un ANO GHS / CLP kategorijām bija šāds: 271. kategorijas šķidrumi, 241. kategorijas cietvielas, 192A kategorijas šķidrumi, 102A kategorijas cietvielas, 9 2B kategorijas šķidrumi, 8 2B kategorijas cietvielas, 50 nekategorizēti šķidrumi un 53 nekategorizētas cietvielas (10)(11).
|
|
11.
|
Šī testēšanas metode attiecas uz vielām un maisījumiem, kā arī uz cietām vielām, šķidrumiem, puscietām vielām un vaskiem. Šķidrumi var būt ūdens vai neūdens šķīdumi; cietas vielas var būt gan ūdenī šķīstošas, gan nešķīstošas. Ja iespējams, cietas vielas pirms aplicēšanas jāsamaļ smalkā pulverī; nekāda cita priekšapstrāde paraugam nav vajadzīga. Validēšanas pētījumā nav novērtētas gāzes un aerosoli. Kaut arī domājams, ka vispār ir iespējams tos testēt ar RhCE tehnoloģiju, ar pašreizējo testēšanas metodi gāzes un aerosolus testēt nevar.
|
|
12.
|
Testējamās ķimikālijas, kas absorbē gaismu tādā pašā diapazonā kā MTT formazāns (dabiski vai pēc apstrādes), un testējamās ķimikālijas, kas spēj tiešā veidā reducēt vitālo krāsvielu MTT (par MTT formazānu), var traucēt audu dzīvotspējas mērījumiem, un korekcijai jāizmanto pielāgotas kontroles. Iespējami nepieciešamo pielāgoto kontroļu veids atšķirsies atkarībā no testējamās ķimikālijas radītās interferences veida un MTT formazāna kvantitatīvai noteikšanai izmantotās procedūras (sk. 36.–42. punktu).
|
|
13.
|
Pirmsvalidēšanas (21)(22) un pilnās validēšanas (8)(10)(11) pētījumos iegūtie rezultāti pierādījuši, ka EpiOcular™ EIT un SkinEthic™ HCE EIT ir izmantojams laboratorijās, kurās iepriekš nav veikta ķīmiskā analīze, un tie ir reproducējami tajā pašā un citās laboratorijās. Pamatojoties uz šiem pētījumiem, prognožu konkordantuma reproducējamības līmenis, ko var sagaidīt no EpiOcular™ EIT no iegūtajiem datiem par 113 ķimikālijām, ir aptuveni 95 % tajā pašā laboratorijā un 93 % citās laboratorijās. Prognožu konkordantuma reproducējamības līmenis, ko var sagaidīt no SkinEthic™ HCE EIT iegūtajiem datiem par 120 ķimikālijām, ir aptuveni 92 % tajā pašā laboratorijā un 95 % citās laboratorijās.
|
|
14.
|
EpiOcular™ EIT var izmantot tādu ķimikāliju identificēšanai, kuras saskaņā ar ANO GHS un CLP klasifikācijas sistēmu nav jāklasificē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus. Ņemot vērā validēšanas pētījumā (8) iegūtos datus, EpiOcular™ EIT vispārējā pareizība ir 80 % (no datiem par 112 ķimikālijām), jutība ir 96 % (no datiem par 57 ķimikālijām), pseidonegatīvu īpatsvars ir 4 % (no datiem par 57 ķimikālijām), specifiskums ir 63 % (no datiem par 55 ķimikālijām) un pseidopozitīvu īpatsvars ir 37 % (no datiem par 55 ķimikālijām), salīdzinot ar truša acu in vivo references testa datiem (TM B.5) (2)(14), kas klasificēti saskaņā ar ANO GHS un CLP klasifikācijas sistēmu. Pētījumā, kurā ar EpiOcular™ EIT testēja 97 šķidrus agroķīmijas preparātus, pierādīja, ka šā veida maisījumu testēšanas metodes veiktspēja ir līdzīga validēšanas pētījuma veiktspējai (23). 97 preparātu sadalījums bija šāds: 211. kategorijas, 192A kategorijas, 142B kategorijas un 43 nekategorizēti, kas klasificēti saskaņā ar ANO GHS klasifikācijas sistēmu atbilstoši truša acu in vivo references testa datiem (TM B.5) (2)(14). Ieguva vispārējo pareizību 82 % (no datiem par 97 preparātiem), jutību 91 % (no datiem par 54 preparātiem), pseidonegatīvu īpatsvaru 9 % (no datiem par 54 preparātiem), specifiskumu 72 % (no datiem par 43 preparātiem) un pseidopozitīvu īpatsvaru 28 % (no datiem par 43 preparātiem) (23).
|
|
15.
|
SkinEthic™ HCE EIT var izmantot tādu ķimikāliju identificēšanai, kuras saskaņā ar ANO GHS un CLP klasifikācijas sistēmu nav jāklasificē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus. Ņemot vērā validēšanas pētījumā (10)(11) iegūtos datus, SkinEthic™ HCE EIT vispārējā pareizība ir 84 % (no datiem par 200 ķimikālijām), jutība ir 95 % (no datiem par 97 ķimikālijām), pseidonegatīvu īpatsvars ir 5 % (no datiem par 97 ķimikālijām), specifiskums ir 72 % (no datiem par 103 ķimikālijām) un pseidopozitīvu īpatsvars ir 28 % (no datiem par 103 ķimikālijām), salīdzinot ar truša acu in vivo references testa datiem (TM B.5) (2)(14), kas klasificēti saskaņā ar ANO GHS un CLP klasifikācijas sistēmu.
|
|
16.
|
Ar abiem RhCE testiem vielām vai maisījumiem iegūtais pseidonegatīvu īpatsvars atbilst 12 % kopējās varbūtības, ka atkārtotā testēšanā ar in vivo Dreiza acu testu ķimikālijas tiek identificētas kā ANO GHS un CLP 2. kategorijas vai ANO GHS un CLP nekategorizētas ķimikālijas; tā iemesls ir metodei raksturīgā mainība viena testa ietvaros (24). Ar abām RhCE testēšanas metodēm vielām vai maisījumiem iegūtajam pseidopozitīvu īpatsvaram šīs testēšanas metodes kontekstā nav būtiskas nozīmes, jo, izmantojot pierādījumu izsvēršanu pakāpjveida testēšanas stratēģijā, visas testējamās ķimikālijas, kuras rada audu dzīvotspēju, kas atbilst noteiktajām robežvērtībām vai ir mazāka par tām (sk. 44. punktu), atkarībā no regulatīvajām prasībām būs jātestē papildus ar citām in vitro testēšanas metodēm vai, ja citas iespējas nebūtu, ar trušiem. Šīs testēšanas metodes var izmantot visu veidu ķimikālijām, negatīvu rezultātu pieņemot par norādi, ka ķimikālija nav jāklasificē kā tāda, kas izraisa acu kairinājumu vai nopietnus acu bojājumus (ANO GHS un CLP nekategorizēta). Pirms EpiOcular™ EIT un SkinEthic™ HCE EIT izmantošanas klasificēšanas shēmās, kas nav ANO GHS / CLP, jākonsultējas ar attiecīgajām regulatīvajām iestādēm.
|
|
17.
|
Šīs testēšanas metodes ierobežojums ir tāds, ka ar to nevar atšķirt acu kairinājumu / atgriezenisku ietekmi uz acīm (2. kategorija) no nopietniem acu bojājumiem / neatgriezeniskas ietekmes uz acīm (1. kategorija), kā noteikts ANO GHS un CLP, nedz arī acu kairinātājus (fakultatīvā 2A kategorija) no viegliem acu kairinātājiem (fakultatīvā 2B kategorija), kā noteikts ANO GHS (1). Šiem nolūkiem nepieciešama papildu testēšana ar citām in vitro testēšanas metodēm.
|
|
18.
|
Terminu “testējamā ķimikālija” šajā testēšanas metodē izmanto, lai apzīmētu to, kas tiek testēts (77), bez sasaistes ar RhCE testēšanas metodes pielietojamību vielu un/vai maisījumu testēšanā.
|
TESTA PRINCIPS
|
19.
|
Testējamo ķimikāliju vietēji aplicē uz vismaz divām trīsdimensiju RhCE audu konstrukcijām, un audu dzīvotspēju mēra pēc ekspozīcijas un pēcekspozīcijas inkubēšanas laika. RhCE audus rekonstruē no primāriem cilvēka epidermas keratinocītiem vai imortalizētām cilvēka radzenes epitēlija šūnām, kas kultivētas vairākas dienas, tā lai veidotos stratificēts, labi diferencēts plakans epitēlijs, kas ir morfoloģiski līdzīgs epitēlijam cilvēka radzenē. EpiOcular™ RhCE audu konstrukciju veido vismaz 3 dzīvotspējīgi šūnu slāņi un nekeratinizēta virsma, un tai piemīt radzenei līdzīga struktūra, kas ir analoga struktūrai in vivo. SkinEthic™ HCE RhCE audu konstrukciju līdzīgi normālam cilvēka radzenes epitēlijam veido vismaz 4 dzīvotspējīgi šūnu slāņi, to vidū cilindriskās bazālās šūnas, pārejas spārnveida šūnas un virspusējās plakanās šūnas (20)(26).
|
|
20.
|
Ķimikālijas izraisīti nopietni acu bojājumi / acu kairinājums, kas in vivo izpaužas galvenokārt kā radzenes apduļķošanās, irīts, konjunktīvas apsārtums un/vai konjunktīvas hemoze, ir rezultāts virknei notikumu, kas sākas ar ķimikālijas iekļūšanu cauri radzenei un/vai konjunktīvai un šūnu bojājumu rašanos. Šūnu bojājumus var izraisīt vairāki iedarbības veidi: šūnas membrānas līze (piem., virsmaktīvo vielu, organisku šķīdinātāju izraisīta); makromolekulu (īpaši proteīnu) koagulācija (piem., virsmaktīvo vielu, organisku šķīdinātāju, sārmu un skābju izraisīta); lipīdu pārziepošanās (piem., sārmu izraisīta); alkilēšana vai cita kovalenta mijiedarbība ar makromolekulām (piem., balināšanas līdzekļu, peroksīdu un alkilētāju izraisīta) (15)(27)(28). Tomēr pierādīts, ka citotoksicitātei ir svarīga vai pat primāra mehānistiska nozīme ķimikālijas izraisītas nopietnu acu bojājumu / acu kairinājuma vispārējās atbildreakcijas noteikšanā neatkarīgi no audu bojājumu pamatā esošiem fizikāli ķīmiskajiem procesiem (29)(30). Turklāt ķimikālijas potenciālu izraisīt nopietnus acu bojājumus / acu kairinājumu galvenokārt nosaka sākotnējā bojājuma apjoms (31), kas korelē ar šūnu mirstības apjomu (29) un sekojošo atbildreakciju un iespējamo iznākumu apjomu (32). Tādējādi pavisam viegli kairinātāji parasti ietekmē tikai virspusējo radzenes epitēliju, viegli un vidēji smagi kairinātāji bojā galvenokārt epitēliju un virspusējo stromu, bet smagi kairinātāji bojā epitēliju, dziļo stromu un reizēm radzenes endotēliju (30)(33). RhCE audu konstrukcijas dzīvotspējas mērīšana, ko pēc vietējas eksponēšanas testējamai ķimikālijai veic, lai noteiktu ķimikālijas, kuras nav jāklasificē kā tādas, kas izraisa nopietnus acu bojājumus / acu kairinājumu (ANO GHS un CLP nekategorizētas ķimikālijas), pamatojas uz pieņēmumu, ka visas ķimikālijas, kas izraisa nopietnus acu bojājumus vai acu kairinājumu, radzenes epitēlijā un/vai konjunktīvā izraisīs citotoksicitāti.
|
|
21.
|
RhCE audu dzīvotspēju ierasts mērīt pēc audu dzīvotspējīgo šūnu ierosinātas vitālās krāsvielas MTT [3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolija bromīds; tiazolilzilais tetrazolija bromīds; CAS Nr. 298-93-1] fermentatīvās pārvēršanās par zilas krāsas MTT formazāna sāli, kura daudzumu kvantitatīvi nosaka audu ekstraktā (16). Par ķimikālijām, kuras nav jāklasificē un jāmarķē saskaņā ar ANO GHS / CLP (nekategorizētas ķimikālijas), nosaka tādas ķimikālijas, kas nesamazina audu dzīvotspēju zem noteiktas robežvērtības (t. i., audu dzīvotspēja > 60 % EpiOcular™ EIT un SkinEthic™ HCE EITL testā (78) vai > 50 % SkinEthic™ HCE EITS testā (79)) (sk. 44. punktu).
|
KOMPETENCES PIERĀDĪŠANA
|
22.
|
Pirms RhCE testu regulāras izmantošanas normatīviem mērķiem laboratorijām jāpierāda sava tehniskā kompetence, pareizi prognozējot 1. tabulā norādītās piecpadsmit standartķimikālijas. Šīs ķimikālijas atlasītas no VRM validēšanas pētījumos izmantotajām ķimikālijām (8)(10)(11). Atlasē iespējamajā apjomā iekļautas ķimikālijas: i) ar atšķirīgu agregātstāvokli; ii) kas aptver visas nopietnu acu bojājumu / acu kairinājuma atbildreakcijas in vivo atbilstoši augstas kvalitātes rezultātiem, kas iegūti truša acu in vivo references testā (TM B.5) (2)(14) un ANO GHS klasifikācijas sistēmā (t. i., 1., 2A, 2B kategorijas vai nekategorizētas ķimikālijas) (1) un CLP klasifikācijas sistēmā (t. i., 1., 2. kategorijas vai nekategorizētas ķimikālijas); iii) kas aptver klasifikācijas atšķirīgos faktorus in vivo (24)(25); iv) kas atbilst validēšanas pētījumā izmantotajām ķimikāliju klasēm (8)(10)(11); v) kas plaši aptver organiskās funkcionālās grupas (8)(10)(11); vi) kuru ķīmiskā struktūra ir precīzi noteikta (8)(10)(11); vii) kurām ir krāsa un/vai kas ir tiešas MTT reducētājas; viii) ar kurām to validēšanas laikā ar RhCE testēšanas metodēm ieguva reproducējamus rezultātus; ix) kuras to validēšanas pētījumu laikā tika pareizi prognozētas ar RhCE testēšanas metodēm; x) kas aptver visas atbildreakcijas in vitro atbilstoši augstas kvalitātes RhCE testēšanas metožu datiem (dzīvotspēja no 0 līdz 100 %); xi) kas ir komerciāli pieejamas; xii) kuru lietošana nav saistīta ar pārlieku augstām iegādes un/vai atkritumu apsaimniekošanas izmaksām. Gadījumos, kad kāda no norādītajām ķimikālijām nav pieejama vai nav izmantojama citu pamatotu iemeslu dēļ, var izmantot citu ķimikāliju, kas atbilst iepriekš aprakstītajiem kritērijiem, piem., ķimikāliju, kas izmantota VRM validēšanai. Šādas atkāpes jāpamato.
1. tabula
Standartķimikāliju saraksts
|
Ķīmiskais nosaukums
|
CAS Nr.
|
Organiskā funkcionālā grupa (80)
|
Agregātstāvoklis
|
VRM1 dzīvotspēja (%) (81)
|
VRM2 dzīvotspēja (%) (82)
|
VRM prognoze
|
MTT reducētājs
|
Krāsas interf.
|
|
In vivo 1. kategorija (83)
|
|
|
Metiltioglikolāts
|
2365-48-2
|
Karbonskābes esteris; tiospirts
|
Šķidrs
|
10,9±6,4
|
5,5±7,4
|
Nav iespējams prognozēt
|
Jā (spēcīgs)
|
Nē
|
|
Hidroksietilakrilāts
|
818-61-1
|
Akrilāts; spirts
|
Šķidrs
|
7,5±4,7 (84)
|
1,6±1,0
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
2,5-Dimetil-2,5-heksāndiols
|
110-03-2
|
Spirts
|
Ciets
|
2,3±0,2
|
0,2±0,1
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
Nātrija oksalāts
|
62-76-0
|
Oksokarbonskābe
|
Ciets
|
29,0±1,2
|
5,3±4,1
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
In vivo 2A kategorija (83)
|
|
|
2,4,11,13-Tetraazatetradekāndiimidamīds, N,N″-bis(4-hlorfenil)-3,12-diimino-, di-D-glikonāts (20 %, ūdens) (85)
|
18472-51-0
|
Aromātiskais heterocikliskais halogenīds; arilhalogenīds; dihidroksilgrupa; guanidīns
|
Šķidrs
|
4,0±1,1
|
1,3±0,6
|
Nav iespējams prognozēt
|
Nē
|
Jā (vājš)
|
|
Nātrija benzoāts
|
532-32-1
|
Arils; karbonskābe
|
Ciets
|
3,5±2,6
|
0,6±0,1
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
In vivo 2B kategorija (83)
|
|
|
Dietiltoluamīds
|
134-62-3
|
Benzamīds
|
Šķidrs
|
15,6±6,3
|
2,8±0,9
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
2,2-Dimetil-3-metilēnbiciklo [2.2.1] heptāns
|
79-92-5
|
Alkāns, sazarots ar trešējo oglekli; alkēns; bicikloheptāns; karbocikli ar tiltiņu saturošu gredzenu; cikloalkāns
|
Ciets
|
4,7±1,5
|
15,8±1,1
|
Nav iespējams prognozēt
|
Nē
|
Nē
|
|
In vivo nekategorizētas
(83)
|
|
|
1-Etil-3-metilimidazolija etilsulfāts
|
342573-75-5
|
Alkoksils; amonija sāls; arils; imidazols; sulfāts
|
Šķidrs
|
79,9±6,4
|
79,4±6,2
|
Nekategor.
|
Nē
|
Nē
|
|
Dikaprililēteris
|
629-82-3
|
Alkoksils; ēteris
|
Šķidrs
|
97,8±4,3
|
95,2±3,0
|
Nekategor.
|
Nē
|
Nē
|
|
Piperonila butoksīds
|
51-03-6
|
Alkoksils; benzdioksols; benzils; ēteris
|
Šķidrs
|
104,2±4,2
|
96,5±3,5
|
Nekategor.
|
Nē
|
Nē
|
|
Polietilēnglikola (PEG-40) hidrogenēta rīcineļļa
|
61788-85-0
|
Acilals; spirts; alils; ēteris
|
Viskozs
|
77,6±5,4
|
89,1±2,9
|
Nekategor.
|
Nē
|
Nē
|
|
1-(4-Hlorfenil)-3-(3,4-dihlorfenil)urīnviela
|
101-20-2
|
Aromātiskais heterocikliskais halogenīds; arilhalogenīds; urīnvielas atvasinājumi
|
Ciets
|
106,7±5,3
|
101,9±6,6
|
Nekategor.
|
Nē
|
Nē
|
|
2,2′-Metilēn-bis-(6-(2H-benzotriazol-2-il)-4-(1,1,3,3-tetrametilbutil)-fenols)
|
103597-45-1
|
Alkāns, sazarots ar četraizvietoto oglekli; saplūdusi karbocikliskā aromātviela; saplūduši piesātinātie heterocikli; prekursoru hinoīdu savienojumi; terc-butils
|
Ciets
|
102,7±13,4
|
97,7±5,6
|
Nekategor.
|
Nē
|
Nē
|
|
Kālija tetrafluorborāts
|
14075-53-7
|
Neorganisks sāls
|
Ciets
|
88,6±3,3
|
92,9±5,1
|
Nekategor.
|
Nē
|
Nē
|
|
Saīsinājumi:
CAS Nr. = Chemical Abstracts Service reģistra numurs; AMO GHS = Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (1); VRM1 = validēta references metode, EpiOcular™ EIT; VRM2 = validēta references metode, SkinEthic™ HCE EIT; Krāsas interf. = krāsas interference ar MTT formazāna standartabsorbcijas (optiskā blīvuma jeb OB) mērījumu.
|
|
|
23.
|
Ieteicams, lai kompetences testēšanas ietvaros lietotāji pēc audu saņemšanas verificētu šo audu barjerfunkcijas, kā precizējis RhCE audu konstrukcijas ražotājs (sk. 25., 27. un 30. punktu). Īpaši svarīgi tas ir tad, ja audi sūtīti no tālienes / sūtīšana aizņēmusi ilgu laiku. Kad tests ir sekmīgi ieviests un apgūts un tā izmantošanas prasme pierādīta, šādu verifikāciju vairs nav nepieciešams veikt katru reizi. Tomēr, ja testu izmanto parastajā praksē, barjerfunkciju novērtēšanu ieteicams turpināt regulāri.
|
PROCEDŪRA
|
24.
|
Testi, uz kuriem pašlaik attiecas šī testēšanas metode, ir zinātniski derīgie EpiOcular™ EIT un SkinEthic™ HCE EIT (9)(12)(13), ko dēvē par validēto references metodi (attiecīgi VRM1 un VRM2). RhCE testēšanas metodēm ir pieejamas standartprocedūras, kas jāizmanto, ieviešot un izmantojot testēšanas metodes laboratorijā (34)(35). Nākamajos punktos un 2. papildinājumā aprakstītas RhCE testu galvenās sastāvdaļas un procedūras.
|
RHCE TESTĒŠANAS METODES KOMPONENTI
Vispārīgi nosacījumi
|
25.
|
Lai rekonstruētu radzenei līdzīga epitēlija trīsdimensiju audus, jāizmanto attiecīgas cilvēka šūnas, kam jāsastāv no pieaugoši stratificētām, bet ne pārragotām šūnām. RhCE audu konstrukciju gatavo insertos ar porainu sintētisku membrānu, caur kuru šūnām var piekļūt barības vielas. Rekonstruētajā radzenei līdzīgajā epitēlijā jābūt vairākiem dzīvotspējīgu, nekeratinizētu epitēlija šūnu slāņiem. RhCE audu konstrukcijas epitēlija virsmai jābūt tiešā saskarē ar gaisu, lai būtu iespējama tieša vietēja eksponēšana testējamajām ķimikālijām līdzīgi tam, kā in vivo tiktu eksponēts radzenes epitēlijs. RhCE audu konstrukcijai jāveido pietiekami spēcīga funkcionāla barjera, lai aizturētu citotoksisko etalonvielu, piem., Triton X-100 vai nātrija dodecilsulfāta (SDS), ātru penetrāciju. Barjerfunkcija būtu uzskatāmi jāpierāda, un šo funkciju iespējams novērtēt, vai nu nosakot, kāds ekspozīcijas laiks vajadzīgs, lai pēc tam, kad konkrētā noteiktā koncentrācijā aplicēta etalonviela (piem., 100 μl 0,3 % (tilp./tilp.) Triton X-100), par 50 % samazinātos audu dzīvotspēja (ET50), vai nosakot, pie kādas koncentrācijas etalonviela pēc noteikta ekspozīcijas laika (piem., 30 minūšu ilgas apstrādes ar 50 μl SDS) par 50 % samazina audu dzīvotspēju (IC50) (sk. 30. punktu). RhCE audu konstrukcijas aizturošajām īpašībām jānovērš testējamās ķimikālijas nokļūšana pāri dzīvotspējīgo audu malai, jo tā dēļ var tikt nepareizi modelēta faktiskā iedarbība uz radzeni. RhCE audu konstrukcijas izveidē izmantotās cilvēka šūnas nedrīkst būt kontaminētas ar baktērijām, vīrusiem, mikoplazmu vai sēnītēm. Piegādātājam jāpārbauda audu konstrukcijas sterilitāte, lai pārliecinātos, ka nav kontaminācijas ar sēnītēm un baktērijām.
|
Funkcionāli nosacījumi
Dzīvotspēja
|
26.
|
Audu dzīvotspējas kvantitatīvai noteikšanai izmanto MTT ķīmisko analīzi (16). RhCE audu konstrukcijas dzīvotspējīgās šūnas vitālo krāsvielu MTT reducē par zilas krāsas MTT formazāna nogulsnēm, ko pēc tam no audiem ekstrahē ar izopropanolu (vai līdzīgu šķīdinātāju). Ekstrahēto MTT formazānu var kvantitatīvi noteikt ar standarta absorbcijas (optiskā blīvuma jeb OB) mērījumu vai HPLC/UPLC spektrofotometrijas procedūru (36). Tīra ekstrakcijas šķīdinātāja OB jābūt pietiekami zemam, t. i., OB < 0,1. RhCE audu konstrukcijas lietotājiem jānodrošina, ka katra izmantotā RhCE audu konstrukcijas partija atbilst negatīvajai kontrolei definētajiem kritērijiem. Negatīvās kontroles OB vērtību pieņemamības diapazoni attiecīgajām VRM ir norādīti 2. tabulā. HPLC/UPLC spektrofotometrijas lietotājam par negatīvās kontroles apstiprināšanas kritēriju jāizmanto 2. tabulā norādītie negatīvās kontroles OB diapazoni. Testēšanas pārskatā jābūt dokumentētam, ka ar negatīvās kontroles vielu apstrādātie audi kultūrā ir stabili (nodrošina līdzīgus audu dzīvotspējas mērījumus) visas testa ekspozīcijas laikā. Audu ražotājam audu partijas izlaides kvalitātes kontroles ietvaros jāveic līdzīga procedūra, taču šajā gadījumā var būt spēkā no 2. tabulā norādītajiem atšķirīgi apstiprināšanas kritēriji. RhCE audu konstrukcijas izstrādātājam/piegādātājam jānosaka negatīvās kontroles OB vērtību pieņemamības diapazons (augšējā un apakšējā robežvērtība) (QC testēšanas metodes apstākļos).
|
2. tabula
Testa lietotājiem paredzētie negatīvās kontroles optiskā blīvuma vērtību pieņemamības diapazoni
|
Tests
|
Apakšējā pieņemamības robežvērtība
|
Augšējā pieņemamības robežvērtība
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (gan šķidru, gan cietu vielu protokoliem)
|
> 0,8 (86)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (gan šķidru, gan cietu vielu protokoliem)
|
> 1,0
|
≤ 2,5
|
Barjerfunkcija
|
27.
|
RhCE audu konstrukcijai jābūt pietiekami biezai un noturīgai, lai aizturētu citotoksisku etalonvielu ātru penetrāciju, ko nosaka pēc ET50 (Triton X-100) vai pēc IC50 (SDS) (3. tabula). RhCE audu konstrukcijas izstrādātājam/pārdevējam, kad tas piegādā audus gala lietotājam, jāpierāda katras izmantojamās RhCE audu konstrukcijas partijas barjerfunkcija (sk. 30. punktu).
|
Morfoloģija
|
28.
|
RhCE audu konstrukcijas histoloģiskā izmeklējumā jāpierāda līdzība cilvēka radzenes epitēlija struktūrai (jābūt vismaz 3 slāņiem dzīvotspējīgu epitēlija šūnu un nekeratinizētai virsmai). Attiecīgajām VRM izstrādātājs/piegādātājs ir izveidojis atbilstošu morfoloģiju, tāpēc testēšanas metodes lietotājam tā nav atkārtoti jāpierāda katrai izmantojamajai audu partijai.
|
Reproducējamība
|
29.
|
Testēšanas metodes pozitīvās un negatīvās kontroles rezultātiem uzskatāmi jāparāda to reproducējamība laika gaitā.
|
Kvalitātes kontrole (QC)
|
30.
|
RhCE audu konstrukciju drīkst izmantot tikai tad, ja izstrādātājs/piegādātājs pierāda, ka ikviena izmantojamās RhCE audu konstrukcijas partija atbilst definētajiem produkcijas izlaides kritērijiem, no kuriem svarīgākie ir dzīvotspēja (26. punkts) un barjerfunkcija (27. punkts). RhCE audu konstrukcijas izstrādātājam/piegādātājam jānosaka ar ET50 vai IC50 mērītas (sk. 25. un 26. punktu) barjerfunkcijas pieņemamības diapazons (augšējā un apakšējā robežvērtība). ET50 un IC50 pieņemamības diapazons, ko RhCE audu konstrukciju izstrādātājs/piegādātājs izmanto kā QC partijas izlaides kritēriju (izmanto VRM), ir norādīts 3. tabulā. RhCE audu konstrukcijas izstrādātājam/piegādātājam dati, kas pierāda atbilstību visiem produkcijas izlaides kritērijiem, jādara pieejami testēšanas metodes lietotājiem, lai tie šo informāciju varētu iekļaut testēšanas pārskatā. Tikai ar visiem šiem produkcijas izlaides kritērijiem atbilstošiem audiem iegūti rezultāti. ir pieņemami tādu ķimikāliju ticamai prognozēšanai, kuras saskaņā ar ANO GHS / CLP nav jāklasificē un jāmarķē kā acu kairinājuma vai nopietnu acu bojājumu izraisītājas.
|
3. tabula
Partiju izlaides QC kritērijs
|
Tests
|
Apakšējā pieņemamības robežvērtība
|
Augšējā pieņemamības robežvērtība
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3 % (tilpums/tilpums) Triton X-100)
|
ET
50 = 12,2 min
|
ET
50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 minūšu ilga apstrāde ar 50 μl SDS)
|
IC
50 = 1 mg/ml
|
IC
50 = 3,2 mg/ml
|
Testējamās ķimikālijas un kontroles vielu aplicēšana
|
31.
|
Katrai testējamai ķimikālijai un katrai kontroles vielai katrā izpildes reizē vajadzīgi vismaz divi audu replikāti. Izmanto divus atšķirīgus apstrādes protokolus: vienu šķidrām testējamajām ķimikālijām un vienu cietām testējamajām ķimikālijām (34)(35). Atbilstoši abām metodēm un protokoliem pirms testējamo ķimikāliju aplicēšanas audu konstrukcijas virsma jāsamitrina ar kalciju un magniju nesaturošu Dulbeko fosfātu fizioloģisko buferšķīdumu (Ca2+/Mg2+ nesaturošs DPBS), lai atdarinātu cilvēka acs mitruma apstākļus. Audu apstrādi sāk, tos eksponējot testējamai ķimikālijai (ķimikālijām) un kontroles vielām. Atbilstoši abiem apstrādes protokoliem abās VRM testējamā ķimikālija vai kontroles viela uz epitēlija virsmas vienmērīgā slānī jāaplicē pietiekamā daudzumā, taču izvairoties no pārmērīgas devas (sk 32. un 33. punktu) (2. papildinājums).
|
|
32.
|
Testējamās ķimikālijas, kas 37 °C vai zemākā temperatūrā ir pilināmas ar pipeti (ja nepieciešams, izmantojot pozitīvo maināmo uzgaļu virzuļpipeti), saskaņā ar VRM uzskata par šķidrumiem, pārējos gadījumos tās uzskatāmas par cietām vielām (sk 33. punktu). Saskaņā ar VRM šķidras testējamās ķimikālijas vienmērīgi uzklāj audu virsmai (t. i., aplicē vismaz 60 μl/cm2) (sk. 2. papildinājumu, (33)(34)). Lai nodrošinātu audu pareizu devu, ciktāl iespējams, jāizvairās no kapilārās iedarbības (virsmas spraiguma efekta), kas var rasties insertam (uz audu virsmas) aplicētā nelielā tilpuma dēļ. Ar šķidrām testējamajām ķimikālijām apstrādātos audus 30 minūtes inkubē standarta kultivēšanas apstākļos (37 ± 2 °C, 5 ± 1 % CO2, ≥ 95 % RM). Ekspozīcijas laika beigās šķidrā testējamā ķimikālija un kontroles vielas no audu virsmas rūpīgi jānoņem, istabas temperatūrā intensīvi skalojot ar Ca2+/Mg2+ nesaturošu DPBS. Nākamā darbība pēc skalošanas ir pēcekspozīcijas iegremdēšana svaigā istabas temperatūras barotnē (lai atbrīvotos no audos absorbējušās testējamās ķimikālijas) uz iepriekš noteiktu laiku, kas atšķiras atkarībā no izmantotās VRM. Tikai saskaņā ar VMR1 pēcekspozīcijas inkubēšanai svaigā barotnē standarta kultivēšanas apstākļos jānotiek pirms MTT ķīmiskās analīzes (sk. 2. papildinājumu, (34)(35)).
|
|
33.
|
Testējamās ķimikālijas, kas līdz 37 °C temperatūrā nav pilināmas ar pipeti, saskaņā ar VRM uzskata par cietām vielām. Aplicējamajam testējamās ķimikālijas daudzumam jābūt pietiekamam, lai pilnībā nosegtu audu virsmu, t. i., jāaplicē vismaz 60 mg/cm2 (2. papildinājums). Ja iespējams, cietas vielas jātestē smalka pulvera veidā. Ar cietām testējamajām ķimikālijām apstrādātos audus iepriekš noteiktu laiku (atkarībā no izmantotās VRM) inkubē standarta kultivēšanas apstākļos (sk. 2. papildinājumu, (34)(35)). Ekspozīcijas laika beigās cietā testējamā ķimikālija un kontroles vielas no audu virsmas rūpīgi jānoņem, istabas temperatūrā intensīvi skalojot ar Ca2+/Mg2+ nesaturošu DPBS. Nākamā darbība pēc skalošanas un pirms MTT ķīmiskās analīzes ir pēcekspozīcijas iegremdēšana svaigā istabas temperatūras barotnē (lai atbrīvotos no audos absorbējušās testējamās ķimikālijas) uz iepriekš noteiktu laiku, kas atšķiras atkarībā no izmantotās VRM, un pēcekspozīcijas inkubēšana svaigā barotnē standarta kultivēšanas apstākļos (sk. 2. papildinājumu, (34)(35)).
|
|
34.
|
Katrā izpildes reizē jāiekļauj paralēlas negatīvās un pozitīvās kontroles, lai pierādītu, ka audu dzīvotspēja (kas noteikta ar negatīvo kontroli) un jutība (kas noteikta ar pozitīvo kontroli) atbilst pēc vēsturiskajiem datiem noteiktajiem pieņemamības diapazoniem. Paralēlā negatīvā kontrole arī nodrošina pamatdatus (100 % audu dzīvotspēja), lai aprēķinātu ar testējamo ķimikāliju apstrādāto audu relatīvo dzīvotspēju procentos (%Viabilitytest
). Ar VRM lietojamā ieteicamā pozitīvās kontroles viela ir neatšķaidīts metilacetāts (CAS Nr. 79-20-9, komerciāli pieejams, piem., no Sigma-Aldrich, kat. Nr. 45997; šķidrums). Ar VRM1 un VRM2 lietojamās ieteicamās negatīvās kontroles vielas ir attiecīgi ultratīrs H2O un Ca2+/Mg2+ nesaturošs DPBS. Šīs ir VRM validēšanas pētījumos izmantotās kontroles vielas, un par tām ir visvairāk vēsturisko datu. Piemērotu alternatīvu pozitīvās vai negatīvās kontroles vielu izmantošana zinātniski un pienācīgi jāpamato. Negatīvās un pozitīvās kontroles jātestē pēc tiem pašiem protokoliem, ko izmanto izpildes reizē iekļautajām testējamajām ķimikālijām (t. i., šķidrumiem un/vai cietām vielām). Nākamā darbība pēc aplicēšanas un pirms MTT ķīmiskās analīzes attiecīgā gadījumā ir apstrādājamā parauga ekspozīcija, skalošana, pēcekspozīcijas iegremdēšana un pēcekspozīcijas inkubēšana, kā paredzēts kontrolēm, kuras izpilda paralēli šķidrām testējamajām ķimikālijām (sk. 32. punktu), vai kontrolēm, kuras izpilda paralēli cietām testējamajām ķimikālijām (sk. 33. punktu) (sk. 35. punktu) (34)(35). Ar vienu negatīvās un pozitīvās kontroles komplektu pietiek visām vienāda agregātstāvokļa (šķidrumiem vai cietām vielām) testējamajām ķimikālijām vienā un tajā pašā izpildes reizē.
|
Audu dzīvotspējas mērījumi
|
35.
|
Audu dzīvotspējas mērīšanai ar šo testēšanas metodi izmantojamā standartizētā kvantitatīvā metode (16) ir MTT ķīmiskā analīze. Tā ir audu trīsdimensiju konstrukcijai piemērota metode. MTT ķīmisko analīzi veic tūlīt pēc pēcekspozīcijas inkubēšanas perioda. Saskaņā ar VRM RhCE audu konstrukcijas paraugu uz 180 ± 15 minūtēm ievieto 0,3 ml MTT šķīduma koncentrācijā 1 mg/ml standarta kultivēšanas apstākļos. RhCE audu konstrukcijas dzīvotspējīgās šūnas vitālo krāsvielu MTT reducē par zilas krāsas MTT formazāna nogulsnēm. Pēc tam izgulsnēto zilās krāsas MTT formazāna produktu no audiem ekstrahē, piemērotā tilpumā izmantojot izopropanolu (vai līdzīgu šķīdinātāju) (34)(35). Ar šķidrām testējamajām ķimikālijām testētie audi jāekstrahē gan no audu augšpuses, gan apakšpuses, bet ar cietām testējamajām ķimikālijām un krāsainiem šķidrumiem testētos audus pietiek ekstrahēt no audu apakšpuses (lai mazinātu izopropanola ekstrakcijas šķīduma iespējamo kontaminēšanu ar testējamo ķimikāliju, kas varētu būt palikusi uz audiem). Arī ar šķidrām testējamajām ķīmiskajām vielām, kas nav viegli noskalojamas, testētos audus pietiek ekstrahēt tikai no audu apakšpuses. Paralēli testētās negatīvās un pozitīvās kontroles vielas jāapstrādā līdzīgi testējamajai ķimikālijai. Ekstrahēto MTT formazānu var kvantitatīvi noteikt ar standarta absorbcijas (OB) mērījumu pie viļņa garuma 570 nm, izmantojot frekvenču joslas filtru, kas nepārsniedz ± 30 nm, vai ar HPLC/UPLC spektrofotometrijas procedūru (sk. 42. punktu) (11)(36).
|
|
36.
|
Testējamās ķimikālijas optiskās īpašības vai ķīmiskās reakcijas ar MTT var traucēt MTT formazāna noteikšanu, un tāpēc dzīvotspēja var būt noteikta nepareizi. Testējamās ķimikālijas var ietekmēt MTT formazāna noteikšanu, MTT tieši reducējot par zilas krāsas MTT formazānu un/vai krāsas interferences dēļ, ja testējamā ķimikālija dabiski vai apstrādes procedūru rezultātā absorbējas tādā pašā OB diapazonā kā MTT formazāns (t. i., apmēram 570 nm). Pirms testēšanas jāveic priekšpārbaudes, lai varētu identificēt potenciālus tiešos MTT reducētājus un/vai krāsu interferējošas ķimikālijas, un šādu testējamo ķimikāliju radītās iespējamās interferences noteikšanai un korekcijām jāizmanto papildu kontroles (sk. 37.–41. punktu). Īpaši svarīgi tas ir tad, ja kādu konkrētu testējamo ķimikāliju pilnībā nenoskalo no RhCE audu konstrukcijas vai ja tā iespiežas radzenei līdzīgajā epitēlijā un tāpēc MTT ķīmiskās analīzes laikā atrodas RhCE audu konstrukcijās. Testējamām ķimikālijām, kas absorbē gaismu tādā pašā diapazonā kā MTT formazāns (dabiski vai pēc apstrādes) un kurām pārāk lielas interferences, t. i., lielas absorbcijas pie viļņa garuma 570 ± 30 nm, dēļ nav izmantojams MTT formazāna standarta absorbcijas (OB) mērījums, MTT formazāna noteikšanai var izmantot HPLC/UPLC spektrofotometrijas procedūru (sk. 41. un 42. punktu) (11)(36). VRM standartprocedūrās ir pieejams plašs apraksts par to, kā noteikt un koriģēt tiešo MTT redukciju un krāsvielu interferenci (34)(35). III un IV papildinājumā sniegtas arī ilustratīvas diagrammas ar norādījumiem par to, kā identificēt tiešos MTT reducētājus un/vai krāsu interferējošas ķimikālijas un kā ar tām rīkoties attiecīgi VRM1 un VRM2 gadījumā.
|
|
37.
|
Lai noteiktu iespējamo interferenci, ko rada testējamās ķimikālijas, kas absorbē gaismu tādā pašā diapazonā kā MTT formazāns (dabiski vai pēc apstrādes), un lemtu par papildu kontroļu nepieciešamību, testējamo ķimikāliju pievieno ūdenim un/vai izopropanolam un attiecīgo laiku inkubē istabas temperatūrā (sk. 2. papildinājumu, (34)(35)). Ja ar VRM1 testējamā ķimikālija ūdenī un/vai izopropanolā absorbē pietiekamu gaismu diapazonā 570 ± 20 nm (sk. 3. papildinājumu) vai ja ar VRM2, sajaucot testējamo ķimikāliju ar ūdeni, iegūst krāsainu šķīdumu (sk. 4. papildinājumu), pieņem, ka testējamā ķimikālija interferē ar MTT formazāna standarta absorbcijas (OB) mērīšanu, un tad jāizpilda papildu kontroles ar krāsvielu vai arī jāizmanto HPLC/UPLC spektrofotometrijas procedūra; minētās kontroles šādā gadījumā nav vajadzīgas (sk. 41. un 42. punktu un III un IV papildinājumu) (34)(35). Mērot standartabsorbciju (OB), katru interferējošo testējamo ķimikāliju aplicē uz vismaz diviem dzīvotspējīgu audu replikātiem, ar kuriem veic visu testēšanas procedūru, taču, lai iegūtu NSC
living kontroli (nespecifiska krāsa dzīvos audos), MTT inkubācijas posmā tos inkubē barotnē, nevis MTT šķīdumā (34)(35). Ņemot vērā dzīvajiem audiem raksturīgo bioloģisko mainību, NSC
living kontrole jāveic paralēli iekrāsotās ķimikālijas testēšanai un vairākkārtējas testēšanas gadījumā katram veiktajam testam (katrā izpildes reizē) jāveic neatkarīga NSC
living kontrole. Faktisko audu dzīvotspēju tad aprēķina, no audu dzīvotspējas procentiem, kas iegūti ar interferējošajai testējamajai ķimikālijai eksponētiem un MTT šķīdumā inkubētiem dzīviem audiem (%Viabilitytest
), atņemot nespecifiskas krāsas procentus, kas iegūti ar interferējošajai testējamajai ķimikālijai eksponētiem dzīviem audiem, kuri paralēli koriģējamajam testam inkubēti barotnē bez MTT (%NSCliving
). Tātad faktiskā audu dzīvotspēja = [%Viabilitytest
] - [%NSCliving
].
|
|
38.
|
Lai identificētu tiešos MTT reducētājus, katra testējamā ķimikālija jāpievieno svaigi pagatavotam MTT šķīdumam. MTT šķīdumam pievieno attiecīgu daudzumu testējamās ķimikālijas un maisījumu apmēram 3 stundas inkubē standarta kultivēšanas apstākļos (skatīt III un IV papildinājumu) (34)(35). Ja MTT maisījums, kas satur testējamo ķimikāliju (nešķīstošas testējamās ķimikālijas gadījumā, tās suspensiju), kļūst zils/violets, pieņem, ka testējamā ķimikālija tieši reducē MTT, un neatkarīgi no standarta absorbcijas (OB) mērījuma vai HPLC/UPLC spektrofotometrijas procedūras izmantošanas dzīvotnespējīgu RhCE audu konstrukcijām jāveic papildu funkcionāla pārbaude. Šajā papildu funkcionālajā pārbaudē izmanto nonāvētus audus, kuriem piemīt vairs tikai atlikusī metaboliskā aktivitāte, taču kuri testējamo ķimikāliju absorbē un saglabā tāpat kā dzīvotspējīgie audi. VRM1 nonāvētos audus sagatavo, tos eksponējot zemai temperatūrai (“nonāvēti aukstumapstrādāti”). VRM2 nedzīvos audus sagatavo, tos ilgstoši (piem., vismaz 24 stundas ± 1 stundu) inkubējot ūdenī un pēc tam glabājot zemā temperatūrā (“nonāvēti ūdensapstrādāti”). Katru MTT reducējošo testējamo ķimikāliju aplicē uz vismaz diviem nonāvētu audu replikātiem, ar kuriem veic visu testēšanas procedūru, lai iegūtu nespecifisku MTT redukcijas (NSMTT) kontroli (34)(35). Ar vienu NSMTT kontroli pietiek vienai testējamai ķimikālijai neatkarīgi no veikto neatkarīgo testu / izpildes reižu skaita. Audu faktisko dzīvotspēju tad aprēķina, no procentuālās audu dzīvotspējas, kas iegūta ar MTT reducētājam eksponētiem dzīviem audiem (%Viabilitytest
), atņemot procentuālo nespecifisko MTT redukciju, kas iegūta ar tam pašam MTT reducētājam eksponētiem nonāvētiem audiem un aprēķināta attiecībā pret negatīvo kontroli, kura izpildīta paralēli koriģējamam testam (%NSMTT). Tātad faktiskā audu dzīvotspēja = [%Viabilitytest
] - [%NSMTT].
|
|
39.
|
Testējamām ķimikālijām, kas identificētas kā krāsu interferējošas (sk. 37. punktu) un tiešu MTT redukciju izraisošas (sk. 38. punktu), standarta absorbcijas (OB) mērījumu veikšanai papildus iepriekšējos punktos aprakstītajām NSMTT un NSC
living kontrolēm būs nepieciešams arī trešais kontroļu komplekts. Parasti tā ir tumšas krāsas testējamajām ķimikālijām, kas gaismu absorbē 570 ± 30 nm diapazonā (piemēram, zilām, violetām, melnām), jo to sākotnējā krāsa traucē novērtēt to spēju tieši reducēt MTT, kā aprakstīts 38. punktā. Tāpēc pēc noklusējama jālieto NSMTT kontroles kopā ar NSCliving
kontrolēm. Testējamās ķimikālijas, kuras tiek pārbaudītas ar abām – NSMTT un NSCliving
– kontrolēm, var absorbēt un saglabāt gan dzīvie, gan nonāvētie audi. Tāpēc šajā gadījumā NSMTT kontrole var koriģēt ne tikai testējamās ķimikālijas izraisīto iespējamo tiešo MTT redukciju, bet arī krāsas interferenci, kas rodas no tā, ka testējamo ķimikāliju absorbē un saglabā nonāvētie audi. Šādi kļūst iespējama dubulta krāsas interferences koriģēšana, jo NSC
living kontrole jau koriģē krāsas interferenci, kas rodas no tā, ka testējamo ķimikāliju absorbē un saglabā dzīvie audi. Lai novērstu iespējamo dubulto krāsas interferences koriģēšanu, jāveic trešā kontrole attiecībā uz nespecifisku krāsu nedzīvos audos (NSC
killed) (sk. III un IV papildinājumu) (34)(35). Pārbaudē ar šo papildu kontroli testējamo ķimikāliju izmanto vismaz diviem nonāvētu audu replikātiem, ar kuriem veic visu testēšanas procedūru, taču MTT inkubācijas posmā tos inkubē barotnē, nevis MTT šķīdumā. Ar vienu NSCkilled
kontroli pietiek vienai testējamai ķimikālijai neatkarīgi no veikto neatkarīgo testu / izpildes reižu skaita, taču tā jāizdara paralēli NSMTT kontrolei un ar to pašu audu partiju. Audu faktisko dzīvotspēju tad aprēķina, no procentuālās audu dzīvotspējas, kas iegūta ar testējamajai ķimikālijai eksponētiem dzīviem audiem (%Viabilitytest
), atņemot %NSMTT un atņemot %NSCliving
, un pieskaitot procentuālo nespecifisko krāsu, kas iegūta ar interferējošai testējamajai ķimikālijai eksponētiem un barotnē bez MTT inkubētiem nonāvētajiem audiem un aprēķināta attiecībā pret negatīvo kontroli, kura izpildīta paralēli koriģējamam testam (%NSCkilled
). Tātad faktiskā audu dzīvotspēja = [%Viabilitytest
] - [%NSMTT] - [%NSCliving
] + [%NSCkilled
].
|
|
40.
|
Svarīgi ievērot, ka (tad, kad veic standarta absorbcijas mērījumus) nespecifiskā MTT redukcija un nespecifiskā krāsas interference var palielināt audu ekstrakta OB virs spektrofotometra linearitātes diapazona un ka (tad, kad veic HPLC/UPLC spektrofotometrijas mērījumus) nespecifiskā MTT redukcija var palielināt arī audu ekstrakta MTT formazāna smailes laukumu virs spektrofotometra linearitātes diapazona. Pamatojoties uz to, ir svarīgi, lai pirms normatīviem mērķiem veiktas ķimikāliju testēšanas sākuma katra laboratorija noteiktu sava spektrofotometra OB/smailes laukuma linearitātes diapazonu, piem., ar MTT formazānu (CAS Nr. 57360-69-7), kas komerciāli pieejams, piem., no Sigma-Aldrich (kat. Nr. M2003).
|
|
41.
|
Standarta absorbcijas (OB) noteikšana ar spektrofotometru ir piemērota tiešu MTT reducētāju un krāsu interferējošu testējamo ķimikāliju novērtēšanai, ja novērotā interference MTT formazāna noteikšanā nav pārāk spēcīga (t. i., ar testējamo ķimikāliju iegūtie audu ekstraktu OB, nekoriģējot tiešu MTT redukciju un/vai krāsas interferenci, atbilst spektrofotometra lineārajam diapazonam). Tomēr par testējamajām ķimikālijām iegūtie rezultāti, kuriem negatīvās kontroles %NSMTT un/vai %NSCliving
ir ≥ 60 % (VRM1 un VRM2 šķidrumu protokolam) vai 50 % (VRM2 cietu vielu protokolam), būtu jāuztver piesardzīgi, jo tā ir robežvērtība, ko VRM izmanto, lai nošķirtu klasificējamas ķimikālijas no neklasificējamām ķimikālijām (sk. 44. punktu). Standarta absorbciju (OB) tomēr nevar noteikt, ja interference MTT formazāna noteikšanā ir pārāk liela (t. i., izraisa to, ka nekoriģētie testējamo audu ekstraktu OB ir ārpus spektrofotometra lineārā diapazona). Ja krāsainas testējamās ķimikālijas vai testējamās ķimikālijas, kam krāsa izveidojas saskarē ar ūdeni vai izopropanolu, pārāk stipri interferē ar MTT formazāna standarta absorbcijas (OB) mērīšanu, tās tomēr var novērtēt ar HPLC/UPLC spektrofotometriju (sk. III un IV papildinājumu). Tas ir tāpēc, ka ar HPLC/UPLC sistēmu MTT formazānu no ķimikālijas var atdalīt pirms kvantitatīvās noteikšanas (36). Tāpēc neatkarīgi no testējamās ķimikālijas, ja tiek izmantota HPLC/UPLC spektrofotometrija, NSCliving
vai NSCkilled
kontroles nav jāveic. Ja ir aizdomas, ka testējamā ķimikālija tieši reducē MTT, NSMTT kontroles tomēr jāizmanto (atbilstoši 38. punktā aprakstītajai procedūrai). NSMTT kontroles jāizmanto arī testējamām ķimikālijām, kurām ir krāsa (jau sākotnēji vai parādās, kad viela ir ūdenī), kas traucē novērtēt to spēju tieši reducēt MTT, kā aprakstīts 38. punktā. MTT formazāna mērīšanai izmantojot HPLC/UPLC spektrofotometriju, kā MTT formazāna smailes laukuma, kas iegūts ar testējamajai ķimikālijai eksponētiem dzīviem audiem, procentuālo attiecību pret MTT formazāna smailes laukumu, kas iegūts ar paralēlo negatīvo kontroli, aprēķina audu procentuālo dzīvotspēju. Testējamām ķimikālijām, kas spēj tieši reducēt MTT, audu faktisko dzīvotspēju aprēķina, no %Viabilitytest
atņemot %NSMTT, kā aprakstīts 38. punkta pēdējā teikumā. Visbeidzot jāņem vērā, ka tādus tiešus MTT reducētājus vai tiešus MTT reducētājus, kas turklāt ir krāsu interferējoši, kuri pēc apstrādes saglabājas audos un reducē MTT tik spēcīgi, ka testējamo audu ekstraktu OB (noteikti ar standarta OB mērījumu) vai smaiļu laukumi (noteikti ar UPLC/HPLC spektrofotometriju) ir ārpus spektrofotometra lineārā diapazona, nav iespējams novērtēt ar RhCE testēšanas metodēm, lai gan tas sagaidāms tikai ļoti retās situācijās.
|
|
42.
|
HPLC/UPLC spektrofotometriju MTT formazāna mērīšanai var izmantot ar visa veida testējamajām ķimikālijām (ar krāsu, bez krāsas, MTT reducētāji un MTT nereducētāji) (11) (36). HPLC/UPLC spektrofotometrijas sistēmas ir dažādas, tāpēc katram lietotājam nav praktiski iespējams noteikt pilnīgi vienādus sistēmas darbības apstākļus. Tāpēc, pirms HPLC/UPLC spektrofotometrijas sistēmu izmantot, lai kvantificētu no audu ekstraktiem iegūtu MTT formazānu, būtu jāpierāda minētās sistēmas kvalifikācijas atbilstība pieņemamības kritērijiem, kas noteikti kvalifikācijas standartparametru kopumam, kuru pamatā ir parametri, kas aprakstīti ASV Pārtikas un zāļu pārvaldes izstrādātājās nozares vadlīnijās par bioanalītisko metožu validēšanu (36)(38). Šie galvenie parametri un to pieņemamības kritēriji ir norādīti 5. papildinājumā. Kad 5. papildinājumā noteiktie pieņemamības kritēriji izpildīti, HPLC/UPLC spektrofotometrijas sistēmu uzskata par kvalificētu un gatavu tam, lai noteiktu MTT formazāna daudzumu, ievērojot šīs testēšanas metodes aprakstā sniegtos eksperimentēšanas nosacījumus.
|
Pieņemamības kritēriji
|
43.
|
Katrā testēšanas reizē, kad izmanto RhCE audu partijas, kas atbilst kvalitātes kontroles prasībām (sk. 30. punktu), ar negatīvās kontroles vielu apstrādāto audu OB jābūt tādam, kas atspoguļo to audu kvalitāti, kuriem ievērotas prasības sūtīšanas, saņemšanas posmiem un visiem protokola procesiem, un to rezultāti nedrīkst būt ārpus 2. tabulā aprakstītajām vēsturiski noteiktajām robežām (sk. 26. punktu). Tāpat ar pozitīvās kontroles vielu, t. i., metilacetātu, apstrādāto audu vidējai dzīvotspējai jābūt < 50 % attiecībā pret negatīvo kontroli VRM1 šķidrumu vai cietu vielu protokoliem un ≤ 30 % (šķidrumu protokolam) vai ≤ 20 % (cietu vielu protokolam) attiecībā pret negatīvo kontroli VRM2, tādējādi atspoguļojot audu spēju reaģēt uz kairinošu testējamo ķimikāliju šīs testēšanas metodes apstākļos (34)(35). Testējamo ķimikāliju un kontroles vielu audu replikātu mainībai jāatbilst pieņemtajām robežām (t. i., dzīvotspējas atšķirībai starp diviem audu replikātiem jābūt mazākai par 20 % vai arī standartnovirze (SD) starp trim audu replikātiem nedrīkst pārsniegt 18 %). Ja izpildes reizē iekļautā negatīvā kontrole vai pozitīvā kontrole neatbilst pieņemtajiem diapazoniem, izpildes reizi uzskata par “nekvalificētu”, un tā jāatkārto. Ja testējamās ķimikālijas audu replikātu mainība neatbilst pieņemtajam diapazonam, tests jāuzskata par “nekvalificētu”, un testējamā ķimikālija jātestē atkārtoti.
|
Rezultātu interpretēšana un prognostiskais modelis
|
44.
|
Katras testējamās ķimikālijas audu replikātu ekstraktiem iegūtās OB vērtības / smailes laukumi jāizmanto vidējās audu dzīvotspējas procentos (vidējā vērtība starp audu replikātiem) aprēķināšanai salīdzinājumā ar negatīvo kontroli, kas noteikta kā 100 %. Audu procentuālās dzīvotspējas robežvērtības, pēc kurām nosaka, kuras testējamās ķimikālijas nav jāklasificē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus (ANO GHS un CLP nekategorizētas ķimikālijas), norādītas 4. tabulā. Tādējādi rezultāti jāinterpretē, kā aprakstīts tālāk tekstā.
|
—
|
Testējamo ķimikāliju nosaka par tādu, kas nav jāklasificē un jāmarķē saskaņā ar ANO GHS / CLP (nekategorizētas ķimikālijas), ja audu vidējā procentuālā dzīvotspēja pēc ekspozīcijas un pēcekspozīcijas inkubēšanas perioda ir lielāka par (>) audu procentuālajai dzīvotspējai noteikto robežvērtību, kas norādīta 4. tabulā. Šādā gadījumā nav nepieciešama papildu testēšana ar citām testēšanas metodēm.
|
|
—
|
Ja audu vidējā procentuālā dzīvotspēja pēc ekspozīcijas un pēcekspozīcijas inkubēšanas perioda ir mazāka par vai vienāda ar (≤) audu procentuālajai dzīvotspējai noteikto robežvērtību, prognozēšana nav iespējama (norādīts 4. tabulā). Šādā gadījumā būs nepieciešama papildu testēšana ar citām testēšanas metodēm, jo RhCE testēšanas metodes uzrāda zināmu skaitu pseidopozitīvu rezultātu (sk. 14.–15. punktu), un nav iespējams izšķirties starp ANO GHS un CLP 1. un 2. kategoriju (sk. 17. punktu).
|
4. tabula
Prognostiskie modeļi sakaņā ar ANO GHS / CLP klasifikāciju
|
VRM
|
Nekategorizēta
|
Nav iespējams prognozēt
|
|
VRM1 — EpiOcular™ EIT (abiem protokoliem)
|
Audu vidējā dzīvotspēja > 60 %
|
Audu vidējā dzīvotspēja ≤ 60 %
|
|
VRM2 — SkinEthic™ HCE EIT (šķidrumu protokolam)
|
Audu vidējā dzīvotspēja > 60 %
|
Audu vidējā dzīvotspēja ≤ 60 %
|
|
VRM2 — SkinEthic™ HCE EIT (cietvielu protokolam)
|
Audu vidējā dzīvotspēja > 50 %
|
Audu vidējā dzīvotspēja ≤ 50 %
|
|
|
45.
|
Vienam testam ar vismaz diviem audu replikātiem jābūt pietiekamam gadījumos, kad testējamās ķimikālijas uzrādītais rezultāts ir nepārprotams. Tomēr, kad rezultāti ir neskaidri, piem., nesakritīgi mērījumu rezultāti atkārtojumos un/vai vidējā audu dzīvotspēja procentos ir 60 ± 5 % (VRM1, kā arī VRM2 šķidrumu protokolam) vai 50 ± 5 % (VRM2 cietvielu protokolam), jāapsver vajadzība pēc otrā vai pat pēc trešā testa, ja nesakrīt pirmo divu testu rezultāti.
|
|
46.
|
Īpašu veidu maisījumiem, ja tas ir piemēroti un pamatoti, var apsvērt atšķirīgas audu dzīvotspējas procentos robežvērtības, pēc kā klasificējamas ķimikālijas nošķir no neklasificējamām ķimikālijām, lai palielinātu testēšanas metodes vispārējo veiktspēju šādu veidu maisījumiem (sk. 14. punktu). Etalonķimikālijas var noderēt, lai novērtētu nezināmu testējamo ķimikāliju vai produktu klases spēju izraisīt nopietnus acu bojājumus / acu kairinājumu vai novērtētu klasificētas ķimikālijas relatīvo spēju izraisīt acu toksicitāti konkrētā pozitīvu atbildreakciju diapazonā.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Dati
|
47.
|
Datus par atsevišķiem audu replikātiem katrā izpildes reizē, arī par atkārtotiem testiem, ja tos veic, (piem., OB vērtības / MTT formazāna smailes laukumus un aprēķinātās audu procentuālās dzīvotspējas datus par testējamo ķimikāliju un kontrolēm, kā arī galīgo RhCE testēšanas metodes prognozi) tabulas veidā apkopo par katru testējamo ķimikāliju. Pārskatā jāiekļauj arī vidējā audu dzīvotspēja procentos un dzīvotspējas atšķirība starp diviem audu replikātiem (ja n = 2 audu replikāti) vai SN (ja n ≥ 3 audu replikāti) par katru atsevišķu testējamo ķimikāliju un kontroli. Pārskatā jāiekļauj visas katrai testējamai ķimikālijai novērotās interferences MTT formazāna mērīšanā, kuras izpaužas kā tieša MTT redukcija un/vai krāsas interference.
|
Testēšanas pārskats
|
48.
|
Testēšanas pārskatā norāda šādu informāciju.
Testējamā ķimikālija
|
|
Vienkomponenta viela
|
—
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati.
|
|
—
|
Agregātstāvoklis, gaistamība, pH, LogP, molekulmasa, ķīmisko vielu klase un papildu attiecīgās fizikāli ķīmiskās īpašības saistībā ar pētījuma veikšanu, ciktāl šī informācija ir pieejama.
|
|
—
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā un ja šī informācija ir pieejama) utt.
|
|
—
|
Apstrāde pirms testēšanas, ja tāda veikta (piem., sildīšana, smalcināšana).
|
|
—
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama.
|
|
|
|
Daudzkomponentu viela, UVCB un maisījumi
|
—
|
Iespējami izsmeļošs raksturojums ar, piem., sastāvdaļu ķīmisko identitāti (sk. iepriekš), tīrību, kvantitatīvo saturu un relevantajām fizikālķīmiskajām īpašībām.
|
|
—
|
Agregātstāvoklis un papildu attiecīgās fizikāli ķīmiskās īpašības saistībā ar pētījuma veikšanu, ciktāl šī informācija ir pieejama.
|
|
—
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
—
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana).
|
|
—
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama.
|
|
Pozitīvās un negatīvās kontroles vielas
|
—
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati.
|
|
—
|
Agregātstāvoklis, gaistamība, molekulmasa, ķīmisko vielu klase un papildu attiecīgās fizikāli ķīmiskās īpašības saistībā ar pētījuma veikšanu, ciktāl šī informācija ir pieejama.
|
|
—
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
—
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana).
|
|
—
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama.
|
|
—
|
Pamatojums citas tādas negatīvās kontroles izmantošanai, kas nav ultratīrs H2O vai Ca2+/Mg2+ nesaturošs DPBS, ja tāda izmantota.
|
|
—
|
Pamatojums citas tādas pozitīvās kontroles izmantošanai, kas nav neatšķaidīts metilacetāts, ja tāda izmantota.
|
|
—
|
Norāde uz vēsturiskajiem pozitīvo un negatīvo kontroļu rezultātiem, kas apliecina atbilstību testa izpildes pieņemamības kritērijiem.
|
Informācija par sponsoru un testējošo iestādi
|
—
|
Sponsora, testējošās iestādes, pētījuma vadītāja vārds/nosaukums un adrese.
|
|
—
|
RhCE audu konstrukcija un izmantotais protokols (attiecīgā gadījumā ar izvēles pamatojumu)
|
Testēšanas metodes apstākļi
|
—
|
Izmantotā RhCE audu konstrukcija, arī partijas numurs.
|
|
—
|
MTT formazāna noteikšanai izmantotais viļņa garums un frekvenču josla (attiecīgā gadījumā), kā arī mērierīces (piem., spektrofotometra) linearitātes diapazons.
|
|
—
|
MTT formazāna noteikšanai izmantotās metodes apraksts.
|
|
—
|
Izmantotās HPLC/UPLC spektrofotometrijas sistēmas apraksts (attiecīgā gadījumā).
|
|
—
|
Pilnīga papildinformācija par konkrēto izmantoto RhCE audu konstrukciju, arī tās veiktspējas raksturlielumi. No tiem jānorāda vismaz:
|
i)
|
dzīvotspējas kvalitātes kontrole (piegādātājs);
|
|
ii)
|
dzīvotspēja testēšanas metodes apstākļos (lietotājs);
|
|
iii)
|
barjerfunkcijas kvalitātes kontrole;
|
|
iv)
|
morfoloģija, ja ir pieejama informācija;
|
|
v)
|
reproducējamība un prognozētspēja;
|
|
vi)
|
RhCE audu konstrukcijas citas kvalitātes kontroles (QC), ja ir pieejama informācija.
|
|
|
—
|
Norāde uz RhCE audu konstrukcijas vēsturiskajiem datiem. No tiem jānorāda vismaz: QC datu pieņemamība saskaņā ar vēsturiskajiem datiem par partiju kontroles rezultātiem.
|
|
—
|
Paziņojums, ka pirms regulāras izmantošanas testējošā iestāde ir pierādījusi kompetenci testēšanas metodes izmantošanā, testējot standartķimikālijas.
|
Izpildes reizes un testa pieņemamības kritēriji
|
—
|
Pozitīvās un negatīvās kontroles vidējās vērtības un pieņemamības diapazoni, kas balstīti uz vēsturiskajiem datiem.
|
|
—
|
Pieņemamā mainība pozitīvām un negatīvām kontrolēm izmantojamiem audu replikātiem.
|
|
—
|
Pieņemamā mainība testējamai ķimikālijai izmantojamiem audu replikātiem.
|
Testa procedūra
|
—
|
Informācija par izmantoto testēšanas procedūru.
|
|
—
|
Izmantotās testējamās ķimikālijas un kontroles vielu devas.
|
|
—
|
Ekspozīcijas, pēcekspozīcijas iegremdēšanas un pēcekspozīcijas inkubēšanas periodu ilgums un temperatūra (attiecīgā gadījumā).
|
|
—
|
Testa procedūras modifikāciju apraksts, ja tādas bijušas.
|
|
—
|
Kontroles, kas izmantotas tiešajiem MTT reducētājiem un/vai testējamajām ķimikālijām, kurām ir krāsa (attiecīgā gadījumā).
|
|
—
|
Katrai testējamai ķimikālijai un kontrolēm (pozitīvā kontrole, negatīvā kontrole, NSMTT, NSC
living un NSC
killed, ja tādas izmantotas) izmantoto audu replikātu skaits.
|
Rezultāti
|
—
|
Datu apkopojums tabulā par atsevišķām testējamajām ķimikālijām un kontroles vielām par katru izpildes reizi (arī par atkārtotiem eksperimentiem, ja tādi veikti) un par katru replikāta mērījumu, arī OB vērtība vai MTT formazāna smailes laukums, audu dzīvotspēja procentos, vidējā audu dzīvotspēja procentos, atšķirība starp audu replikātiem vai SN un galīgā prognoze.
|
|
—
|
Attiecīgā gadījumā tiešiem MTT reducētājiem un/vai krāsainām testējamajām ķimikālijām izmantoto kontroļu rezultāti, arī OB vērtība vai MTT formazāna smailes laukums, %NSMTT, %NSCliving
, %NSCkilled
, atšķirība starp audu replikātiem vai SN, galīgā pareizā audu dzīvotspēja procentos un galīgā prognoze.
|
|
—
|
Rezultāti, kas iegūti ar testējamo ķimikāliju (ķimikālijām) un kontrolķimikālijām, salīdzinājumā ar noteiktajiem izpildes reizes un testa pieņemamības kritērijiem.
|
|
—
|
Citas novērotās ietekmes, piem., krāsainas testējamās ķimikālijas izraisītas audu iekrāsošanās, apraksts.
|
Rezultātu iztirzājums
Secinājumi
|
LITERATŪRA
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Available at: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Šā pielikuma B.5. nodaļa “Akūts acu kairinājums/korozija”.
|
|
(3)
|
Šā pielikuma B.47. nodaļa “Liellopu radzenes apduļķošanās un caurlaidības testēšanas metode, kurā identificē i) nopietnu acu bojājumu inducētājas ķimikālijas un ii) ķimikālijas, kuras nav jāklasificē kā tādas, kas izraisa acu kairinājumu vai nopietnus acu bojājumus”.
|
|
(4)
|
Šā pielikuma B.48. nodaļa “Izolētas vistas acs testēšanas metode, kurā identificē i) nopietnu acu bojājumu inducētājas ķimikālijas un ii) ķimikālijas, kuras nav jāklasificē”.
|
|
(5)
|
Šā pielikuma B.61. nodaļa “Fluoresceīna caurplūdes testēšanas metode acu korozijas vai nopietna kairinājuma izraisītāju identificēšanai”.
|
|
(6)
|
Šā pielikuma B.68. nodaļa “Īsa ekspozīcijas laika in vitro testēšanas metode, ar kuru identificēt i) ķimikālijas, kas izraisa nopietnus acu bojājumus, un ii) ķimikālijas, kuras nav jāklasificē kā acu kairinājuma vai nopietnu acu bojājumu izraisītājas”.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28125 EN; doi:10.2787/41680. Available at: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28173 EN; doi: 10.2787/043697. Available at: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28175 EN; doi: 10.2787/390390. Available at: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in Preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1476-1488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2610–2625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41–54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Available at: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Available at: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Available at: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
1. papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā ar testēšanas metodi iegūto mērījumu rezultāti sakrīt ar pieņemtajām atsauces vērtībām. Tas ir testēšanas metodes veiktspējas mērs un viens no relevantuma aspektiem. Šo terminu bieži izmanto ar nozīmi “konkordantums”, ar to domājot testēšanas metodes pareizo rezultātu īpatsvaru (18).
Etalonķimikālija
: ķimikālija, ko izmanto par standartu salīdzināšanā ar testējamo ķimikāliju. Etalonķimikālijai jāpiemīt šādām īpašībām: i) konsekvents un uzticams avots vai avoti tās identificēšanas un raksturošanas vajadzībām; ii) strukturāla, funkcionāla un/vai ķīmiska un produktklases līdzība ar testējamo ķimikāliju; iii) labi zināmas fizikālķīmiskās īpašības; iv) papildu dati par zināmajiem iedarbības veidiem; v) vielas zināmais iedarbības stiprums atbilst vēlamās atbildreakcijas diapazonam.
Augšupēja pieeja
: pakāpeniska pieeja, ko izmanto testējamai ķimikālijai, par kuru ir aizdomas, ka tā nav jāklasificē vai jāmarķē kā acu kairinājuma vai nopietnu acu bojājumu izraisītāja, un kas sākas ar to ķimikāliju nošķiršanu, kuras nav jāklasificē vai jāmarķē (negatīvs iznākums), no citām ķimikālijām (pozitīvs iznākums).
Ķimikālija
: viela vai maisījums.
Konkordantums
: sk. “Pareizības” definīciju.
Radzene
: acābola priekšējā caurspīdīgā daļa, kas pārklāj varavīksneni un zīlīti un ielaiž acī gaismu.
VK
: variācijas koeficients.
Nov.
: novirze.
EIT
: acu kairinājuma tests.
EURL-ECVAM
: Eiropas Savienības References laboratorija dzīvniektestēšanas alternatīvu jomā.
Acu kairinājums
: izmaiņas acī, kas rodas pēc testējamās ķimikālijas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas ir pilnīgi atgriezeniskas. Nozīmē to pašu, ko “atgriezeniska ietekme uz acīm” un ANO GHS / CLP 2. kategorija.
ET50
: ekspozīcijas laiks, kas pēc noteiktas koncentrācijas etalonķimikālijas aplicēšanas nepieciešams audu dzīvotspējas samazināšanai par 50 %.
Pseidonegatīvu īpatsvars
: visu to vielu daļa, kurām ir attiecīgā veida iedarbība, bet kuras ar konkrēto testēšanas metodi tiek kļūdaini identificētas par vielām bez attiecīgā veida iedarbības. Tas ir viens no testēšanas metodes veiktspējas raksturlielumiem.
Pseidopozitīvu īpatsvars
: visu to vielu daļa, kurām nav attiecīgā veida iedarbības, bet kuras ar konkrēto testēšanas metodi tiek kļūdaini identificētas par vielām ar attiecīgā veida iedarbību. Tas ir viens no testēšanas metodes veiktspējas raksturlielumiem.
Bīstamība
: tāda aģenta vai situācijas iedabiska īpašība, kam piemīt potenciāls atstāt nelabvēlīgu ietekmi uz šim aģentam eksponētu organismu, sistēmu vai (sub)populāciju.
HCE
: SkinEthic™ cilvēka radzenes epitēlijs.
HPLC
: augstefektīva šķidrumhromatogrāfija.
IC50
: koncentrācija, kādā etalonķimikālija pēc noteikta ekspozīcijas laika (piem., 30 minūšu apstrādes ar SDS) audu dzīvotspēju samazina par 50 %.
Pārmērīga deva
: uz RhCE audu konstrukcijas aplicētās testējamās ķimikālijas daudzums, kas ir lielāks par epitēlija virsmas pilnīgai un vienmērīgai pārklāšanai nepieciešamo daudzumu.
Neatgriezeniska ietekme uz acīm
: sk. “Nopietnu acu bojājumu” definīciju.
LLOQ
: kvantitatīvās noteikšanas apakšējā robeža.
LogP
: oktanola-ūdens sadalījuma koeficienta logaritms.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairāk vielām.
Vienkomponenta viela
: viela, definēta ar tās kvantitatīvo sastāvu, kurā viena galvenā sastāvdaļa veido vismaz 80 % (masa/masa).
Daudzkomponentu viela
: viela, definēta ar tās kvantitatīvo sastāvu, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu viela tiek iegūta ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu viela rodas ķīmiskā reakcijā.
MTT
: 3-(4,5-Dimetiltiazol-2-il)-2,5-difeniltetrazolija bromīds; tiazolilzilais tetrazolija bromīds.
Negatīvā kontrole
: paraugs, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka konkrētajā testa sistēmā tā neinducē pozitīvu atbildreakciju. Ar šo paraugu izdara tādas pašas manipulācijas kā ar paraugiem, kas apstrādāti ar testējamo ķimikāliju, un citiem kontrolparaugiem, un to izmanto, lai noteiktu 100 % audu dzīvotspēju.
Neklasificētas ķimikālijas
: ķimikālijas, kuras nav klasificētas kā acu kairinājuma (ANO GHS / CLP 2. kategorija, ANO GHS 2A vai 2B kategorija) vai nopietnu acu bojājumu (ANO GHS / CLP 1. kategorija) izraisītājas. Nozīmē to pašu, ko “ANO GHS / CLP nekategorizētas ķimikālijas”.
NSCkilled
: nespecifiska krāsa nedzīvos audos.
NSCliving
: nespecifiska krāsa dzīvos audos.
NSMTT
: nespecifiskā MTT redukcija.
OB
: optiskais blīvums.
Veiktspējas standarti
: uz validētu testēšanas metodi (tādu, kura tiek uzskatīta par zinātniski validētu) bāzēti standarti, pēc kuriem izvērtē pēc noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgu testēšanas metožu salīdzināmību. Šeit iekļauti: (i) nepieciešamie testēšanas metodes komponenti; (ii) minimāls tādu references ķimikāliju saraksts, kuras izraudzītas no citām ķimikālijām, ko izmanto, lai pierādītu pieņemamu validētās testēšanas metodes veiktspēju; iii) uz attiecīgajā validētajā testēšanas metodē iegūto rezultātu pamata — salīdzināmi pareizības un ticamības līmeņi, kā būtu jāiegūst ar ierosināto testēšanas metodi, to izvērtējot ar references ķimikālijām no minimālā saraksta (18).
Pozitīvā kontrole
: paraugs, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka konkrētajā testa sistēmā tā inducē pozitīvu atbildreakciju. Ar šo paraugu izdara tādas pašas manipulācijas kā ar paraugiem, kas apstrādāti ar testējamo ķimikāliju, un citiem kontrolparaugiem. Lai būtu iespēja novērtēt pozitīvās kontroles atbildreakcijas mainību laikā, pozitīvās atbildreakcijas intensitāte nedrīkstētu būt pārlieku liela.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Tā ir pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testēšanas metodes pareizības (konkordantuma) aspektu (18).
Ticamība
: pakāpe, kādā vienā un tajā pašā laboratorijā vai dažādās laboratorijās pēc viena un tā paša protokola var ilgāku laiku reproducējami izmantot konkrētu testēšanas metodi. To novērtē, aprēķinot metodes intralaboratorisko un interlaboratorisko reproducējamību un intralaboratorisko atkārtojamību (18).
Aizstājējtests
: ikdienā lietota testa aizstāšanai paredzēts tests, kuru pieņemts izmantot bīstamības identificēšanai un/vai riska novērtēšanai, ar pierādītu spēju ekvivalentā vai labākā veidā, salīdzinot ar pieņemto testu, nodrošināt (pēc vajadzības) cilvēka un dzīvnieku veselības vai vides aizsardzību visās iespējamās testēšanas situācijās un ar visām iespējamām ķimikālijām (18).
Reproducējamība
: tādu rezultātu savstarpējā atbilstība, kas iegūti, ar vienu un to pašu testa protokolu testējot vienu to pašu ķimikāliju (sk. “Ticamības” definīciju) (18).
Atgriezeniska ietekme uz acīm
: sk. “Acu kairinājuma” definīciju.
RhCE
: rekonstruēts cilvēka radzenei līdzīgs epitēlijs.
Izpildes reize
: reize, kurā vienu vai vairākas ķimikālijas testē paralēli negatīvai kontrolei un pozitīvai kontrolei.
SD
: standartnovirze.
Jutība
: tā visu pozitīvo/aktīvo testējamo ķimikāliju daļa, kas ar testu tiek pareizi klasificētas. Tas ir pareizības mērs testēšanas metodei, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (18).
Nopietni acu bojājumi
: acs audu bojājumi vai nopietna fiziska redzes pasliktināšanās, kas rodas pēc testējamās vielas aplicēšanas uz acs priekšējās virsmas un kas 21 dienas laikā pēc aplicēšanas nav pilnībā atgriezeniska. Nozīmē to pašu, ko “neatgriezeniska ietekme uz acīm” un ANO GHS / CLP 1. kategorija.
Darbības standartprocedūras (SOP)
: oficiālas, rakstiskas procedūras, kas detalizēti apraksta, kā veicamas konkrētas rutīnas un konkrētam testam specifiskas laboratorijas darbības. Tās ir nepieciešamas saskaņā ar labu laboratorijas praksi (GLP).
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām testējamajām ķimikālijām ar testu tiek klasificētas pareizi. Tādas testēšanas metodes pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testēšanas metodes relevantuma izvērtēšanā (18).
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai nepieciešamās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu.
Tests
: atsevišķas testējamās ķimikālijas paralēla testēšana ar vismaz diviem audu replikātiem, kā noteikts attiecīgajā SOP.
Audu dzīvotspēja
: parametrs, ar ko šūnu populācijas kopējo aktivitāti rekonstruētos audos mēra kā to spēju reducēt vitālo krāsvielu MTT, kas atkarībā no nosakāmā beigupunkta un izmantotās testēšanas shēmas korelē ar dzīvo šūnu kopskaitu un/vai dzīvotspēju.
Lejupēja pieeja
: pakāpeniska pieeja, ko izmanto ķimikālijai, par kuru ir aizdomas, ka tā izraisa nopietnus acu bojājumus, un kas sākas ar to ķimikāliju nošķiršanu, kuras inducē nopietnus acu bojājumus (pozitīvs rezultāts), no citām ķimikālijām (negatīvs rezultāts).
Testējamā ķimikālija
: viela vai maisījums, kuru testē ar šo testēšanas metodi.
Secīgas testēšanas stratēģija
: pakāpeniskas testēšanas stratēģija, kurā testēšanas metodes pielieto noteiktā secībā. Izmantojot pierādījumu izsvēršanu, visu esošo informāciju par testējamo ķimikāliju pārskata, lai pirms pārejas uz nākamo stratēģijas pakāpi noteiktu, vai pieejamās informācijas apjoms ir pietiekams, lai pieņemtu lēmumu par bīstamības klasifikāciju. Ja testējamās ķimikālijas bīstamības potenciālu/potenci var noteikt, pamatojoties uz konkrētajā pakāpē pieejamo informāciju, papildu testēšana nav jāveic (18).
ULOQ
: kvantitatīvās noteikšanas augšējā robeža.
Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (ANO GHS)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un izmanto tādus attiecīgus komunikācijas elementus kā piktogrammas, signālvārdi, bīstamības apzīmējumi, drošības prasību apzīmējumi un drošības datu lapas, lai sniegtu informāciju par ķimikāliju nelabvēlīgo ietekmi un tādējādi aizsargātu cilvēkus (arī darba devējus un darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (1).
ANO GHS un CLP 1. kategorija
: sk. “Nopietnu acu bojājumu” definīciju.
ANO GHS un CLP 2. kategorija
: sk. “Acu kairinājuma” definīciju.
ANO GHS un CLP nekategorizētas ķimikālijas
: Ķimikālijas, kas neatbilst prasībām klasificēšanai ANO GHS / CLP 1. vai 2. kategorijā (vai ANO GHS 2A vai 2B kategorijā). Nozīmē to pašu, ko “neklasificētas ķimikālijas”.
UPLC
: īpaši augstefektīva šķidrumhromatogrāfija.
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kompleksi reakcijas produkti vai bioloģiski materiāli.
Derīga testēšanas metode
: testēšanas metode, kas tiek uzskatīta par pietiekami relevantu un ticamu konkrētam nolūkam un kas ir balstīta uz zinātniskiem principiem. Testēšanas metode nekad nav derīga absolūtā izteiksmē; tā vienmēr ir derīga tikai kādam konkrētam nolūkam (18).
Validēta testēšanas metode
: testēšanas metode, kuras relevantums (ieskaitot pareizību) un ticamība attiecībā uz lietošanu konkrētā nolūkā ir pārbaudīta validācijas pētījumos. Jāievēro, ka validētas testēšanas metodes veiktspēja pareizības un ticamības ziņā var nebūt pietiekama, lai to izmantotu paredzētajam nolūkam (18).
VRM
: validēta references metode.
VRM1
: EpiOcular™ EIT dēvē par 1. validēto references metodi.
VRM2
: SkinEthic™ HCE EIT dēvē par 2. validēto references metodi.
Pierādījumu izsvēršana
: process, kurā izsver dažādu informācijas elementu stiprās un vājās puses, cenšoties izdarīt un pamatot secinājumu par testējamās vielas bīstamības potenciālu.
2. papildinājums
ĶIMIKĀLIJU, KURAS NAV JĀKLASIFICĒ UN JĀMARĶĒ KĀ TĀDAS, KAS IZRAISA ACU KAIRINĀJUMU VAI NOPIETNUS ACU BOJĀJUMUS, IDENTIFICĒŠANAI VALIDĒTO RHCE TESTU GALVENĀS SASTĀVDAĻAS
|
Testa sastāvdaļas
|
EpiOcular™ EIT
(VRM1)
|
SkinEthic™ HCE EIT
(VRM2)
|
|
Protokoli
|
Šķidrumi
(pilināmi ar pipeti 37 ± 1 °C vai zemākā temperatūrā 15 min)
|
Cietas vielas
(nav pilināmas ar pipeti)
|
Šķidrumi un viskozas vielas
(pilināmi ar pipeti)
|
Cietas vielas
(nav pilināmas ar pipeti)
|
|
Modeļa virsma
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Audu replikātu skaits
|
Vismaz 2
|
Vismaz 2
|
Vismaz 2
|
Vismaz 2
|
|
Priekšpārbaude attiecībā uz krāsas interferenci
|
50 μl + 1 ml H2O 60 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM (bezkrāsainas testējamās ķimikālijas), vai 50 μl + 2 ml izopropanola, maisa 2–3 h RT (krāsainas testējamās ķimikālijas)
|
 ja testējamās ķimikālijas OB pie 570±20 nm pēc izopropanola vai ūdens OB atņemšanas ir > 0,08 (tas atbilst apmēram 5 % no negatīvās kontroles vidējā OB), jāpielieto dzīvas pielāgotas kontroles
ja testējamās ķimikālijas OB pie 570±20 nm pēc izopropanola vai ūdens OB atņemšanas ir > 0,08 (tas atbilst apmēram 5 % no negatīvās kontroles vidējā OB), jāpielieto dzīvas pielāgotas kontroles
|
|
50 mg + 1 ml H2O 60 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
(bezkrāsainas testējamās ķimikālijas) un/vai
50 mg + 2 ml izopropanola, maisa 2–3 h pie RT (krāsainas un bezkrāsainas testējamās ķimikālijas)
|
 ja testējamās ķimikālijas OB pie 570±20 nm pēc izopropanola vai ūdens OB atņemšanas ir > 0,08 (tas atbilst apmēram 5 % no negatīvās kontroles vidējā OB), jāpielieto dzīvas pielāgotas kontroles
ja testējamās ķimikālijas OB pie 570±20 nm pēc izopropanola vai ūdens OB atņemšanas ir > 0,08 (tas atbilst apmēram 5 % no negatīvās kontroles vidējā OB), jāpielieto dzīvas pielāgotas kontroles
|
|
10 mg + 90 μl H2O maisa 30 ± 2 min pie istabas temperatūras (RT, 18-28oC)
|
 ja testējamā ķimikālija ir krāsaina, jāpielieto pielāgotas kontroles ar dzīviem audiem
ja testējamā ķimikālija ir krāsaina, jāpielieto pielāgotas kontroles ar dzīviem audiem
|
|
10 mg + 90 μl H2O maisa 30 ± 2 min pie RT
|
 ja testējamā ķimikālija ir krāsaina, jāpielieto pielāgotas kontroles ar dzīviem audiem
ja testējamā ķimikālija ir krāsaina, jāpielieto pielāgotas kontroles ar dzīviem audiem
|
|
|
Priekšpārbaude attiecībā uz tiešo MTT reducēšanos
|
50 μl + 1 ml MTT 1 mg/ml šķīduma 180 ± 15 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
 ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem aukstumapstrādātiem audiem
ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem aukstumapstrādātiem audiem
(par negatīvo kontroli izmanto 50 μl sterila dejonizēta ūdens MTT šķīdumā)
|
|
50 mg + 1 ml MTT 1 mg/ml šķīduma 180 ± 15 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
 ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem aukstumapstrādātiem audiem
ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem aukstumapstrādātiem audiem
(par negatīvo kontroli izmanto 50 μl sterila dejonizēta ūdens MTT šķīdumā)
|
|
30 μl + 300 μl MTT 1 mg/ml šķīduma 180 ± 15 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
 ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem ūdensapstrādātiem audiem
ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem ūdensapstrādātiem audiem
(par negatīvo kontroli izmanto 30 μl sterila dejonizēta ūdens MTT šķīdumā)
|
|
30 mg + 300 μl MTT 1 mg/ml šķīduma 180 ± 15 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
 ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem ūdensapstrādātiem audiem
ja šķīdums iekrāsojas zils vai violets, jāizdara pielāgotas kontroles ar nonāvētiem ūdensapstrādātiem audiem
(par negatīvo kontroli izmanto 30 μl sterila dejonizēta ūdens MTT šķīdumā)
|
|
|
Priekšapstrāde
|
20 μl Ca2+/Mg2+ nesaturošs DPBS
30 ± 2 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM, sargājot no gaismas
|
20 μl Ca2+/Mg2+ nesaturošs DPBS
30 ± 2 min 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM, sargājot no gaismas
|
—
|
—
|
|
Apstrādes devas un aplicēšana
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) ar kalibrētu instrumentu (piem., pilna karote bez kaudzes, kas kalibrēta 50 mg nātrija hlorīda ietveršanai).
|
10 μl Ca2+/Mg2+ nesaturošs DPBS + 30 ± 2 μl (60 μl/cm2)
viskozām vielām izmanto neilona sietu
|
30 μl Ca2+/Mg2+ nesaturošs DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Ekspozīcijas ilgums un temperatūra
|
30 min (± 2 min)
kultūras barotnē:
37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
6 stundas (± 0,25 h)
kultūras barotnē:
37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
30 min (± 2 min)
kultūras barotnē:
37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
4 stundas (± 0,1 h)
kultūras barotnē:
37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
|
Skalošana istabas temperatūrā
|
3 reizes 100 ml Ca2+/Mg2+ nesaturošā DPBS
|
3 reizes 100 ml Ca2+/Mg2+ nesaturošā DPBS
|
20 ml Ca2+/Mg2+ nesaturoša DPBS
|
25 ml Ca2+/Mg2+ nesaturoša DPBS
|
|
Pēcekspozīcijas iegremdēšana
|
12 min (± 2 min) pie RT, kultūras barotnē
|
25 min (± 2 min) pie RT, kultūras barotnē
|
30 min (± 2 min) 37 oC temperatūrā, 5 % CO2, 95 % RM, kultūras barotnē
|
30 min (± 2 min) pie RT, kultūras barotnē
|
|
Pēcekspozīcijas inkubēšana
|
120 min (± 15 min) kultūras barotnē 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
18 h (± 0,25 h) kultūras barotnē 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
nav
|
18 h (± 0,5 h) kultūras barotnē 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
|
Negatīvā kontrole
|
50 μl H2O
Testē paralēli
|
50 μl H2O
Testē paralēli
|
30 ± 2 μl Ca2+/Mg2+ nesaturoša DPBS
Testē paralēli
|
30 ± 2 μl Ca2+/Mg2+ nesaturoša DPBS
Testē paralēli
|
|
Pozitīvā kontrole
|
50 μl metilacetāta
Testē paralēli
|
50 μl metilacetāta
Testē paralēli
|
30 ± 2 μl metilacetāta
Testē paralēli
|
30 ± 2 μl metilacetāta
Testē paralēli
|
|
MTT šķīdums
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT inkubācijas ilgums un temperatūra
|
180 min (± 15 min) 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
180 min (± 15 min) 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
180 min (± 15 min) 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
180 min (± 15 min) 37 ± 2 oC temperatūrā, 5 ± 1 % CO2, ≥ 95 % RM
|
|
Ekstrakcijas šķīdinātājs
|
2 ml izopropanola
(ekstrakcija no inserta augšas un apakšas, caurdurot audus)
|
2 ml izopropanola
(ekstrakcija no inserta apakšas, caurdurot audus)
|
1,5 ml izopropanola
(ekstrakcija no inserta augšas un apakšas)
|
1,5 ml izopropanola
(ekstrakcija no inserta apakšas)
|
|
Ekstrakcijas ilgums un temperatūra
|
2–3 h ar kratīšanu (~120 apgr./min) pie RT vai pa nakti 4–10 °C temperatūrā
|
2–3 h ar kratīšanu (~120 apgr./min) pie RT vai pa nakti 4–10 °C temperatūrā
|
4 h ar kratīšanu (~120 apgr./min) pie RT vai vismaz pa nakti bez kratīšanas 4–10 °C temperatūrā
|
Vismaz 2 h ar kratīšanu (~120 apgr./min) pie RT
|
|
OB rādījums
|
570 nm (550–590 nm)
bez references filtra
|
570 nm (550–590 nm)
bez references filtra
|
570 nm (540–600 nm)
bez references filtra
|
570 nm (540–600 nm)
bez references filtra
|
|
Audu kvalitātes kontrole
|
Apstrāde ar 100 μl 0,3 % (tilpums/tipums) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
Apstrāde ar 100 μl 0,3 % (tilpums/tipums) Triton X-100
12,2 min ≤ ET50 ≤ 37,5 min
|
30 min apstrāde ar SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 min apstrāde ar SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Pieņemamības kritēriji
|
|
1.
|
Ar negatīvo kontroli apstrādāto audu replikātu vidējam OB jābūt > 0,8 un < 2,5.
|
|
2.
|
Ar pozitīvo kontroli 30 min eksponēto audu replikātu vidējai dzīvotspējai, kas izteikta kā % no negatīvās kontroles, jābūt < 50 %.
|
|
3.
|
Divu audu replikātu dzīvotspējas atšķirībai jābūt mazākai par 20 %.
|
|
|
1.
|
Ar negatīvo kontroli apstrādāto audu replikātu vidējam OB jābūt > 0,8 un < 2,5.
|
|
2.
|
Ar pozitīvo kontroli 6 stundas eksponēto audu replikātu vidējai dzīvotspējai, kas izteikta kā % no negatīvās kontroles, jābūt < 50 %.
|
|
3.
|
Divu audu replikātu dzīvotspējas atšķirībai jābūt mazākai par 20 %.
|
|
|
1.
|
Ar negatīvo kontroli apstrādāto audu replikātu vidējam OB jābūt > 1,0 un ≤ 2,5.
|
|
2.
|
Ar pozitīvo kontroli 30 min eksponēto audu replikātu vidējai dzīvotspējai, kas izteikta kā % no negatīvās kontroles, jābūt ≤ 30 %.
|
|
3.
|
Divu audu replikātu dzīvotspējas atšķirībai jābūt mazākai par 20 %.
|
|
|
1.
|
Ar negatīvo kontroli apstrādāto audu replikātu vidējam OB jābūt > 1,0 un ≤ 2,5.
|
|
2.
|
Ar pozitīvo kontroli 4 stundas eksponēto audu replikātu vidējai dzīvotspējai, kas izteikta kā % no negatīvās kontroles, jābūt ≤ 20 %.
|
|
3.
|
Divu audu replikātu dzīvotspējas atšķirībai jābūt mazākai par 20 %.
|
|
3. papildinājums
ILUSTRATĪVA DIAGRAMMA AR NORĀDĪJUMIEM PAR TO, KĀ IDENTIFICĒT TIEŠOS MTT REDUCĒTĀJUS UN/VAI KRĀSU INTERFERĒJOŠAS ĶIMIKĀLIJAS UN KĀ AR TĀM RĪKOTIES ATBILSTOŠI VRM1 STANDARTPROCEDŪRAI

4. papildinājums
ILUSTRATĪVA DIAGRAMMA AR NORĀDĪJUMIEM PAR TO, KĀ IDENTIFICĒT TIEŠOS MTT REDUCĒTĀJUS UN/VAI KRĀSU INTERFERĒJOŠAS ĶIMIKĀLIJAS UN KĀ AR TĀM RĪKOTIES ATBILSTOŠI VRM2 STANDARTPROCEDŪRAI

5. papildinājums
GALVENIE PARAMETRI UN PIEŅEMAMĪBAS KRITĒRIJI NO RhCE AUDU KONSTRUKCIJĀM EKSTRAHĒTA MTT FORMAZĀNA MĒRĪŠANAI PAREDZĒTAS HPLC/UPLC SPEKTROFOTOMETRIJAS SISTĒMAS KVALIFICĒŠANAI
|
Parametrs
|
Saskaņā ar FDA norādījumiem izstrādāts protokols (36)(38)
|
Pieņemamības kritēriji
|
|
Selektivitāte
|
Izopropanola, dzīva tukšā parauga (izopropanola ekstrakts no neapstrādātu dzīvu RhCE audu konstrukcijām), nedzīva tukšā parauga (izopropanola ekstrakts no neapstrādātu nonāvēRhCE audu konstrukcijām) un krāsas (piem., metilēnzilā) analīze
|
Laukumsinterference ≤ 20 % no laukuma
LLOQ
(87)
|
|
Precizitāte
|
Kvalitātes kontroles (t. i., MTT formazāns koncentrācijā 1,6 μg/ml, 16 μg/ml un 160 μg/ml) izopropanolā (n=5)
|
CV ≤ 15 % vai attiecībā uz LLOQ ≤ 20 %
|
|
Pareizība
|
Kvalitātes kontroles izopropanolā (n=5)
|
% nov. ≤ 15 % vai attiecībā uz LLOQ ≤ 20 %
|
|
Matrices efekts
|
Kvalitātes kontroles dzīvā tukšajā paraugā (n=5)
|
85 % ≤ matrices efekts % ≤ 115 %
|
|
Pārnesums
|
Izopropanola analīze pēc ULOQ
(88) standarta
|
Laukumsinterference ≤ 20 % no laukuma
LLOQ
|
|
Reproducējamība (tajā pašā dienā)
|
3 neatkarīgas kalibrēšanas līknes (balstītas uz 6 secīgiem MTT formazāna 1/3 atšķaidījumiem izopropanolā, sākot no ULOQ, t. i., 200 μg/ml);
Kvalitātes kontroles izopropanolā (n=5)
|
Kalibrēšanas līknes: % nov. ≤ 15 % vai attiecībā uz LLOQ ≤ 20 %
Kvalitātes kontroles: % nov. ≤ 15 % un CV ≤ 15 %
|
|
Reproducējamība (no vienas dienas otrā)
|
1. diena: 1 kalibrēšanas līkne un kvalitātes kontroles izopropanolā (n=3)
2. diena: 1 kalibrēšanas līkne un kvalitātes kontroles izopropanolā (n=3)
3. diena: 1 kalibrēšanas līkne un kvalitātes kontroles izopropanolā (n=3)
|
|
MTT formazāna īslaicīgā stabilitāte RhCE audu ekstraktā
|
Kvalitātes kontroles dzīvā tukšajā paraugā (n=3), kas analizēts sagatavošanas dienā un pēc 24 stundu ilgas glabāšanas istabas temperatūrā
|
% nov. ≤ 15 %
|
|
MTT formazāna ilglaicīgā stabilitāte RhCE audu ekstraktā (ja vajadzīgs)
|
Kvalitātes kontroles dzīvā tukšajā paraugā (n=3), kas analizēts sagatavošanas dienā un pēc vairāku dienu ilgas glabāšanas -20°C temperatūrā
|
% nov. ≤ 15 %
|
B.70. CILVĒKA REKOMBINANTIE ESTROGĒNRECEPTORU (hrER) IN VITRO TESTI TĀDU ĶIMIKĀLIJU NOTEIKŠANAI, KAM PIEMĪT ER AFINITĀTE
VISPĀRĪGS IEVADS
Veiktspējā balstītas testēšanas ESAO vadlīnija
|
1.
|
Šī testēšanas metode ir ekvivalenta ESAO testēšanas vadlīnijai (TG) Nr. 493 (2015). Minētā TG Nr. 493 ir veiktspējā balstītas testēšanas vadlīnija (PBTG), kas apraksta cilvēka rekombinantās in vitro testēšanas metodiku tādu vielu noteikšanai, kam piemīt estrogēnreceptoru afinitāte (hrER saistīšanās testi). Tajā ietilpst divas mehānistiski un funkcionāli līdzīgas testēšanas metodes, ko izmanto, lai identificētu vielas, kas saistās ar estrogēnreceptoru (t. i., ERα), un tā varētu atvieglot jaunu līdzīgu vai modificētu testēšanas metožu izstrādi atbilstīgi validēšanas principiem, kas izklāstīti ESAO vadlīniju dokumentā Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1). Šīs PBTG pamatā ir divas pilnībā validētas references testēšanas metodes (sk. 2. un 3. papildinājumu):
|
—
|
Freibergera–Vilsona (Freyberger–Wilson; turpmāk FW) estrogēnreceptoru (ER) saistīšanās in vitro tests, kurā izmanto pilnu cilvēka rekombinanto ERα (2), un
|
|
—
|
Ķīmiskās izvērtēšanas un pētniecības institūta (Chemical Evaluation and Research Institute and Research Institute; turpmāk “CERI”) estrogēnreceptoru saistīšanās in vitro tests, kurā izmanto cilvēka rekombinanto liganda saistīšanās domēna proteīnu (2).
|
Ir izstrādāti veiktspējas standarti (VS) (3), kas sekmē līdzīgu testēšanas metožu izstrādi un validēšanu attiecībā uz to pašu bīstamības beigupunktu un dod iespēju savlaicīgi grozīt PBTG Nr. 493, atjaunināto PBTG ļaujot papildināt ar jauniem līdzīgiem testiem. Tomēr līdzīgus testus pievieno tikai pēc to izskatīšanas un ESAO apstiprinājuma, ka ir izpildīti veiktspējas standarti. Izmantojot ESAO datu savstarpējo atzīšanu, TG Nr. 493 iekļautos testus bez izšķirības var izmantot, lai nodrošinātu ESAO dalībvalstu prasības pēc testēšanas rezultātiem attiecībā uz estrogēnreceptora saistīšanos.
|
Testēšanas metodē iekļauto testu konteksts un principi
|
2.
|
ESAO 1998. gadā sāka īstenot prioritārus pasākumus ar mērķi pārskatīt esošās un izstrādāt jaunas testēšanas vadlīnijas potenciālo endokrīno disruptoru skrīningam un testēšanai. 2012. gadā tika pārskatīts ESAO potenciālo endokrīno disruptoru testēšanas un novērtēšanas konceptuālais satvars (KS). Gan sākotnējais, gan pārskatītais KS ir iekļauts pielikumā Vadlīniju dokumentam par standartizētām testēšanas metodēm ķimikāliju vērtēšanai attiecībā uz potenciālu endokrīni disruptīvu iedarbību (4). KS aptver piecus līmeņus, no kuriem katrs atbilst citam bioloģiskās sarežģītības līmenim. Šajā testēšanas metodē aprakstītie ER saistīšanās testi atbilst 2. līmenim, kas ietver “in vitro testus, kas sniedz datus par izvēlētiem endokrīniem mehānismiem/ceļiem”. Testēšanas metode paredzēta in vitro receptoru saistīšanās testiem, ar ko identificē cilvēka estrogēnreceptora alfa (ERα) ligandus.
|
|
3.
|
Ir skaidri pierādīts in vitro ER saistīšanās testa relevantums attiecībā uz bioloģiskajām funkcijām. ER saistīšanās testi izstrādāti, lai identificētu ķimikālijas, kam piemīt disruptīvs potenciāls attiecībā uz estrogēnu tipa hormonu ceļu, un pēdējo divdesmit gadu laikā tie plaši izmantoti, lai raksturotu ER audu izkliedi, kā arī identificētu ER agonistus/antagonistus. Šie testi atspoguļo ligandu–receptoru mijiedarbību, kas attiecībā uz reproduktīvo funkciju visiem mugurkaulniekiem ir estrogēnu signālceļa sākumpunkts.
|
|
4.
|
Estrogēnu–ER mijiedarbība var ietekmēt estrogēnu regulēto gēnu transkripciju un inducēt negenomisku ietekmi, un tā rezultātā var tikt inducēti vai inhibēti šūnu procesi, tostarp tādi, kas saistīti ar šūnu vairošanos, normālu augļa attīstību un reproduktīvo funkciju (5)(6)(7). Normālu estrogēnisko sistēmu traucējumi var negatīvi ietekmēt normālu attīstību (ontoģenēzi), reproduktīvo veselību un reproduktīvās sistēmas integritāti. Neatbilstoši ER signāli var radīt, piemēram, palielinātu hormonatkarīgu audzēju risku, pasliktinātu auglību un mainītu augļa augšanas un attīstības gaitu (8).
|
|
5.
|
In vitro saistīšanās testu pamatā ir vielu tieša vai netieša mijiedarbība ar specifisku receptoru liganda saistīšanās domēnu, kas regulē gēnu transkripciju. Cilvēka rekombinantā estrogēnreceptora alfa (hrERα) saistīšanās testa galvenais komponents mēra radioaktīvi iezīmēta liganda ([3H]17β-estradiola) spēju saistīties ar ER arvien pieaugošu testējamās ķimikālijas (t. i., konkurenta) koncentrāciju klātbūtnē. Testējamās ķimikālijas, kam piemīt augsta ER afinitāte, konkurē ar radioaktīvi iezīmēto ligandu pie zemākas koncentrācijas nekā ķimikālijas ar zemāku receptora afinitāti. Šis tests sastāv no diviem svarīgākajiem komponentiem: piesātinājuma–saistīšanās eksperimenta, kas raksturo receptora–liganda mijiedarbības parametrus un dokumentē ER specifiskumu, un konkurējošās saistīšanās eksperimenta, kas attiecībā uz saistīšanos ar ER raksturo konkurenci starp testējamo ķimikāliju un radioaktīvi marķēto ligandu.
|
|
6.
|
CERI validējošie pētījumi un FW saistīšanās testi ir pierādījuši, ka minētie komponenti ir relevanti un ticami paredzētajam nolūkam (2).
|
|
7.
|
Šajā testēšanas metodē izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
Receptora saistīšanās testu tvērums un ierobežojumi
|
8.
|
Šie testi paredzēti skrīninga un prioritizēšanas vajadzībām, tomēr tie var sniegt arī informāciju par molekulāro iniciācijas notikumu (MIE), ko var izmantot pierādījumu svara pieejā. Šie testi in vitro sistēmā aplūko ķīmisko saistīšanos ar ERα liganda saistīšanās domēnu. Tāpēc rezultātus nevajadzētu tieši ekstrapolēt uz neskartas endokrīnās sistēmas kompleksajiem signalizēšanas un regulēšanas mehānismiem in vivo.
|
|
9.
|
Dabīgā liganda (17β-estradiola) saistīšanās ir sākumpunkts molekulāru notikumu sērijā, kas aktivizē mērķa gēnu transkripciju un galu galā kulminē ar fizioloģiskām pārmaiņām (9). Tādējādi saistīšanās ar ERα liganda saistīšanās domēnu tiek uzskatīta par vienu no svarīgākajiem ER mediētiem endokrīnās disrupcijas (ED) mehānismiem, lai gan pastāv arī citi ED izraisoši mehānismi, t. sk.: i) mijiedarbība ar citiem ERα domēniem, ne tikai ar liganda saistīšanās kabatu; ii) mijiedarbība ar citiem estrogēnu signalizēšanā būtiskiem receptoriem, ar ERβ un estrogēnreceptoru, kas sasaistīts ar G proteīnu, ar citiem endokrīnās sistēmas receptoriem un fermentu sistēmām; iii) hormonu sintēze; iv) metaboliska hormonu aktivācija un/vai inaktivācija; v) hormonu izkliede mērķaudos un vi) hormonu izvadīšana no organisma. Neviens šajā testēšanas metodē iekļautais tests neattiecas uz uzskaitītajiem iedarbības veidiem.
|
|
10.
|
Šī testēšanas metode aplūko vielu spēju saistīties ar cilvēka ERα, un tā neizšķir ERα agonistus un antagonistus. Šajos testos netiek aplūkoti tādi tālāki lejupēji notikumi kā gēnu transkripcija vai fizioloģiskas pārmaiņas. Tā kā validācijā tika izmantotas tikai vienkomponenta vielas, nav informācijas par testa izmantojamību ar maisījumiem. Tomēr teorētiski šie testi ir izmantojami daudzkomponentu vielu un maisījumu testēšanai. Pirms testēšanas metodi attiecībā uz maisījumu izmanto, lai iegūtu datus kādai plānotai regulatīvai vajadzībai, jāapsver, vai un kādēļ tā var sniegt tādam nolūkam piemērotus rezultātus. Šādi apsvērumi nav vajadzīgi, ja pastāv regulatīva prasība maisījumu testēt.
|
|
11.
|
Šūnas nesaturošām receptoru sistēmām nepiemīt iekšēja metaboliskā spēja, un tās nav validētas kombinācijā ar metaboliskām fermentu sistēmām. Pētījumu plānā gan varētu iekļaut metabolisku aktivitāti, bet tam būtu vajadzīga papildu validācija.
|
|
12.
|
Nedrīkst testēt tādas ķimikālijas, kas varētu atņemt proteīna, proti, receptorproteīna, dabiskās īpašības (piem., virsmaktīvie aģenti) vai mainīt testa buferšķīduma pH, vai arī tās drīkst testēt tikai koncentrācijās, kas šādu mijiedarbību nepieļauj. Visos pārējos gadījumos testā izmantojamās testējamās ķimikālijas koncentrācijas diapazons ir atkarīgs no tās šķīdības testa buferšķīdumā.
|
|
13.
|
Informatīvā nolūkā 1. tabulā ir sniegti testa rezultāti par 24 vielām, kas testētas ar abiem pilnībā validētajiem references testiem, kas aprakstīti šajā testēšanas metodē. No publicētajiem testēšanas pārskatiem t. sk. no in vitro testiem uz ER transkripcionālo aktivāciju un/vai uterotrofiskā testa izriet, ka 17 no šīm vielām klasificētas kā tādas, kas saistās ar ER, bet 6 vielas ir nesaistās ar ER (9) (10) (11) (12) (13) (14) (15). Runājot par 1. tabulā apkopotajiem datiem, abu testu rezultāti gandrīz 100 % sakrita attiecībā uz visu vielu klasifikāciju līdz pat 10-4M, un visas vielas bija pareizi klasificētas kā saistīgas vai nesaistīgas ar ER. Papildu informācija par šo vielu grupu un par citām vielām, kas validācijas pētījumu gaitā testētas ER saistīšanās testos, ir sniegta hrER saistīšanās testa veiktspējas standartos (3), 2. papildinājumā (1., 2. un 3. tabulā).
1. tabula
Vielu klasifikācija pēc to saistīšanās vai nesaistīšanās ar ER, testējot tās ar FW un CERI hrER saistīšanās testiem, un salīdzinājums ar sagaidāmo rezultātu
|
|
Vielas nosaukums
|
CAS reģ. Nr.
|
Sagaidāmais rezultāts
|
FW tests
|
CERI tests
|
Klase pēc MeSH klasifikācijas
|
Produkta klase
|
|
|
Koncentrācijas diapazons (M)
|
Klasifikācija
|
Koncentrācijas diapazons (M)
|
Klasifikācija
|
|
1
|
17β-estradiols
|
50-28-2
|
saisteklis
|
1×10-11 – 1×10-6
|
saisteklis
|
1×10-11 – 1×10-6
|
saisteklis
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
2
|
Noretinodrels
|
68-23-5
|
saisteklis
|
3×10-9 – 30×10-4
|
saisteklis
|
3×10-9 – 30×10-4
|
saisteklis
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
3
|
Noretindrons
|
68-22-4
|
saisteklis
|
3×10-9 – 30×10-4
|
saisteklis
|
3×10-9 – 30×10-4
|
saisteklis
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
4
|
Di-n-butilftalāts
|
84-74-2
|
nesaisteklis (*5)
|
1×10-10 – 1×10-4
|
nesaisteklis (*6)
(1)
|
1×10-10 – 1×10-4
|
nesaisteklis (*6)
(1)
|
ogļūdeņradis (ciklisks), esteris
|
plastifikators, ķimikāliju starpprodukts
|
|
5
|
DES
|
56-53-1
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (ciklisks), fenols
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
6
|
17α-etinilestradiols
|
57-63-6
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
7
|
Mezo-heksestrols
|
84-16-2
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (ciklisks), fenols
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
8
|
Genisteīns
|
446-72-0
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (heterociklisks), flavonoīds
|
dabisks produkts
|
|
9
|
Ekvols
|
531-95-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
fitoestrogēna metabolīts
|
dabisks produkts
|
|
10
|
Butilparabēns (n butil-4-hidroksibenzoāts)
|
94-26-8
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
parabēns
|
konservants
|
|
11
|
Nonilfenols (maisījums)
|
84852-15-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
alkilfenols
|
starpproduktu savienojums
|
|
12
|
o,p’-DDT
|
789-02-6
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
hlororganiska viela
|
insekticīds
|
|
13
|
Kortikosterons
|
50-22-6
|
nesaisteklis (*5)
|
1×10-10 – 1×10-4
|
nesaisteklis
|
1×10-10 – 1×10-4
|
nesaisteklis
|
steroīds
|
dabisks produkts
|
|
14
|
Zearalenons
|
17924-92-4
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (heterociklisks), laktons
|
dabisks produkts
|
|
15
|
Tamoksifēns
|
10540-29-1
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (ciklisks)
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
16
|
5α-dihidrotestosterons
|
521-18-6
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
nefenolisks steroīds
|
dabisks produkts
|
|
17
|
Bisfenols A
|
80-05-7
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
fenols
|
ķimikāliju starpprodukts
|
|
18
|
4-n-heptilfenols
|
1987-50-4
|
saisteklis
|
1×10-10 – 1×10-3
|
neskaidra (6)
|
1×10-10 – 1×10-3
|
saisteklis
|
alkilfenols
|
starpprodukts
|
|
19
|
Kepons (hlordekons)
|
143-50-0
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
ogļūdeņradis (halogenēts)
|
pesticīds
|
|
20
|
Benz(a)antracēns
|
56-55-3
|
nesaisteklis
|
1×10-10 – 1×10-3
|
nesaisteklis (7)
|
1×10-10 – 1×10-3
|
nesaisteklis (7)
|
aromātiskais ogļūdeņradis
|
starpprodukts
|
|
21
|
Enterolaktons
|
78473-71-9
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
1×10-10 – 1×10-3
|
saisteklis
|
fitoestrogēns
|
dabisks produkts
|
|
22
|
Progesterons
|
57-83-0
|
nesaisteklis (*5)
|
1×10-10 – 1×10-4
|
nesaisteklis
|
1×10-10 – 1×10-4
|
nesaisteklis
|
steroīds
|
dabisks produkts
|
|
23
|
Oktiltrietoksisilāns
|
2943-75-1
|
nesaisteklis
|
1×10-10 – 1×10-3
|
nesaisteklis
|
1×10-10 – 1×10-3
|
nesaisteklis
|
silāns
|
virsmas modifikators
|
|
24
|
Atrazīns
|
1912-24-9
|
nesaisteklis (*5)
|
1×10-10 – 1×10-4
|
nesaisteklis
|
1×10-10 – 1×10-4
|
nesaisteklis
|
heterociklisks savienojums
|
herbicīds
|
|
HrER SAISTĪŠANĀS TESTA KOMPONENTI
Nepieciešamie testa komponenti
|
14.
|
Šī testēšanas metode attiecas uz testiem, kuru galvenais elements ir estrogēnreceptors (ER) un atbilstoša stipruma ligands, ko testā var izmantot kā marķieri / iezīmēto elementu un kas, palielinoties testējamās ķimikālijas koncentrācijai, var tikt pārvietots. Saistīšanās testu divi galvenie komponenti ir šādi: 1) piesātinājuma–saistīšanās tests un 2) konkurējošā saistīšanās. Piesātinājuma–saistīšanās testu izmanto, lai apstiprinātu receptoru preparātu specifiskumu un aktivitāti, savukārt konkurējošās saistīšanās eksperimenta mērķis ir izvērtēt testējamās ķimikālijas spēju saistīties ar hrER.
|
Kontroles
|
15.
|
Apraksta ierosinātos paralēlos references estrogēnus un kontroles. Paralēlās kontroles — šķīdinātājs (nesējs), pozitīvā kontrole (saistīgs ar ER; stipra un vāja afinitāte), negatīvā kontrole (nesaistīgs) — vajadzības gadījumā kalpo kā indikators, ka tests pie konkrētajiem testēšanas nosacījumiem funkcionē un ļauj savstarpēji salīdzināt eksperimentus; parasti paralēlās kontroles ir daļa no konkrētā eksperimenta pieņemamības kritērijiem (1). Katrā testa izpildes reizē vienā platē ir jābūt atsauces estrogēna un kontroļu (t.i. vāja saistekļa un nesaistekļa) pilnai koncentrāciju līknei. Visās pārējās platēs jāiekļauj: 1) katra E2 un vāja saistekļa augstas koncentrācijas (aptuveni tāda, pie kuras radioaktīvi marķētais ligands tiek pilnībā pārvietots) un vidējas koncentrācijas (aptuveni IC50) triplikāti; 2) šķīdinātāja kontroles un nespecifiskās saistīšanās triplikāti.
|
Standarta kvalitātes kontroles procedūras
|
16.
|
Katram testam veic aprakstītās standarta kvalitātes kontroles procedūras, lai nodrošinātu, ka aktīvie receptori, pareizas ķīmiskās koncentrācijas un tolerances intervāli saglabājas stabili vairākkārtējās replikācijās un nezaudē spēju laika gaitā nodrošināt gaidītās ER saistīšanās reakcijas.
|
Laboratorijas kvalifikācijas pierādīšana
|
17.
|
Visām laboratorijām pirms nezināmu ķimikāliju testēšanas ar jebkuru no šīs testēšanas metodes testiem jāpierāda sava kvalifikācija konkrētā testa izmantošanā, veicot piesātinājuma testus, lai apstiprinātu ER preparāta specifiskumu un aktivitāti, un konkurējošās saistīšanās testus ar references estrogēniem un kontrolēm (vājiem saistekļiem un nesaistekļiem). Laboratorijai jāizveido vēsturiska datubāze, kurā apkopti trīs līdz piecu neatkarīgu, dažādās dienās veiktu eksperimentu rezultāti, kas gūti ar references estrogēnu un kontrolēm. Šie eksperimenti būs pamats laboratorijas references estrogēnam un vēsturiskajām kontrolēm, un turpmākās testēšanas reizēs tos izmantos kā testu pieņemamības novērtējuma daļu.
|
|
18.
|
Jāapstiprina arī testa sistēmas reaģētspēja, izdarot 2. tabulā iekļauto standartvielu testēšanu. Standartvielu saraksts ir ER saistīšanās testu veiktspējas standartos norādīto references vielu apakškopa (3). Šīs vielas ir komerciāli pieejamas un ir attiecībā uz tādu ķimikāliju klasēm, ko parasti asociē ar ER afinitāti, reprezentatīvas, kā arī tām ir pienācīgs ER afinitātes diapazons (no spēcīgas līdz vājai), kāds būtu sagaidāms ER saistekļiem un nesaistekļiem (t. i., attiecībā uz negatīvajiem rezultātiem). Attiecībā uz katru standartvielu testētajām koncentrācijām būtu jāaptver 2. tabulā norādītais diapazons. Ar katru vielu jāveic vismaz trīs eksperimenti, un rezultātiem jāatbilst sagaidāmajai ķīmiskajai aktivitātei. Katrs eksperimentu veic neatkarīgi (t. i., ar jauniem receptora atšķaidījumiem, ķimikālijām un reaģentu), katrai koncentrācijai izmantojot trīs replikātus. Kompetence ir pierādīta, ja katra standartviela tiek pareizi klasificēta (pozitīva/negatīva reakcija). Kompetences testēšanu atkārto katrs laborants, kas apgūst testēšanas metodi.
2. tabula
Hr ER konkurējošās saistīšanās testu kontroļu un standartvielu saraksts (89)
|
Nr.
|
Vielas nosaukums
|
CAS reģ. Nr. (90)
|
Sagaidāmais rezultāts (91)
(92)
|
Testa koncentrācijas diapazons (M)
|
MeSH ķimikāliju klase (93)
|
Produkta klase (94)
|
|
Kontroles (references estrogēns, vājš saisteklis, nesaisteklis)
|
|
1
|
17β-estradiols
|
50-28-2
|
saisteklis
|
1×10-11 – 1×10-6
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
2
|
Noretinodrels (vai) Noretindrons
|
68-23-5 (vai) 68-22-4
|
saisteklis
|
3×10-9 – 30×10-6
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
3
|
Oktiltrietoksisilāns
|
2943-75-1
|
nesaisteklis
|
1×10-10 – 1×10-3
|
silāns
|
virsmas modifikators
|
|
Standartvielas (94)
|
|
4
|
Dietilstilbestrols
|
56-53-1
|
saisteklis
|
1×10-11 – 1×10-6
|
ogļūdeņradis (ciklisks), fenols
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
5
|
17α-etinilestradiols
|
57-63-6
|
saisteklis
|
1×10-11 – 1×10-6
|
steroīds
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
6
|
Mezo-heksestrols
|
84-16-2
|
saisteklis
|
1×10-11 – 1×10-6
|
ogļūdeņradis (ciklisks), fenols
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
7
|
Tamoksifēns
|
10540-29-1
|
saisteklis
|
1×10-11 – 1×10-6
|
ogļūdeņradis (ciklisks)
|
farmaceitisks un veterinārfarmaceitisks līdzeklis
|
|
8
|
Genisteīns
|
446-72-0
|
saisteklis
|
1×10-10 – 1×10-3
|
heterociklisks savienojums, flavonoīds
|
dabisks produkts
|
|
9
|
Bisfenols A
|
80-05-7
|
saisteklis
|
1×10-10 – 1×10-3
|
fenols
|
ķimikāliju starpprodukts
|
|
10
|
Zearalonons
|
17924-92-4
|
saisteklis
|
1×10-11 – 1×10-3
|
heterociklisks savienojums, laktons
|
dabisks produkts
|
|
11
|
Butilparabēns
|
94-26-8
|
saisteklis
|
1×10-11 – 1×10-3
|
Karbonskābe, fenols
|
konservants
|
|
12
|
Atrazīns
|
1912-24-9
|
nesaisteklis
|
1×10-11 – 1×10-6
|
heterociklisks savienojums
|
herbicīds
|
|
13
|
Di-n-butilftalāts (DBP) (95)
|
84-74-2
|
nesaisteklis (96)
|
1×10-10 – 1×10-4
|
ogļūdeņradis (ciklisks), esteris
|
plastifikators, ķimikāliju starpprodukts
|
|
14
|
Kortikosterons
|
50-22-6
|
nesaisteklis
|
1×10-11 – 1×10-4
|
steroīds
|
dabisks produkts
|
|
Testējamo ķimikāliju šķīdības un koncentrācijas diapazona noteikšana
|
19.
|
Ir jāveic sagatavošanās tests, lai noteiktu katras testējamās ķimikālijas šķīdības robežu un identificētu piemērotu koncentrācijas diapazonu, kas tiks izmantots testā. Sākotnēji katras testējamās ķimikālijas šķīdības robežu nosaka šķīdinātājā un pēc tam apstiprina testēšanas apstākļos. Galīgā testā izmantojamā koncentrācija nepārsniedz 1 mM. Diapazona noteikšanas testēšanā ietilpst šķīdinātāja kontrole; paralēli tai testē astoņus sērijveida logaritmiskus atšķaidījumus, sākot ar augstāko pieņemamo koncentrāciju (piem., 1 mM vai zemāku, balstoties uz šķīdības robežu), un vēro, kad šķīdums kļūst duļķains vai rada nogulsnes. Trešā un ceturtā eksperimenta koncentrācijas būtu jāpielāgo pēc vajadzības, lai labāk raksturotu koncentrācijas–atbildreakcijas līkni.
|
Pieņemamības kritēriji katrai testēšanas reizei
|
20.
|
Konkrēta eksperimenta pieņemšana vai noraidīšana notiek, izvērtējot rezultātus, kas iegūti ar references estrogēnu un kontroli. Pirmkārt, — attiecībā uz 1. plati — pilnām koncentrāciju līknēm, kas iegūtas ar katra eksperimenta references kontroli, jāatbilst līknei atbilstošajiem veiktspējas mērījumu parametriem (piem., IC50 un Hilla virziena koeficientam), balstoties uz rezultātiem, kas paziņoti attiecīgajos CERI un FW testu protokolos (2. un 3. papildinājums), un vēsturiskajiem kontroles datiem, ko apkopojusi testa veicēja laboratorija. Katrā eksperimentā jābūt pareizi klasificētām visām kontrolēm (references estrogēni, vāji saistekļi un nesaistekļi). Otrkārt, visās turpmākajās platēs ir jāvērtē kontroles konsekventums attiecībā pret 1. plati. Testā jāizmanto pietiekams testējamās ķimikālijas koncentrāciju diapazons, lai skaidri definētu konkurējošās saistīšanās līknes augšējo punktu. Rezultātu mainībai starp katras testējamās ķimikālijas koncentrācijas replikātiem, kā arī starp trim neatkarīgajām testēšanas reizēm jābūt saprātīgi pieļaujamās robežās un zinātniski pamatojamai. Laboratorija savu spēju konsekventi veikt testu pierāda, izveidojot un uzturot vēsturisku datubāzi, kurā apkopoti references estrogēnu un kontroļu rezultāti. Lai nodrošinātu reproducējamību laboratorijas ietvaros, var izmantot references estrogēna un vāja saistekļa kontroles līknēm atbilstošo parametru vidējo vērtību standartnovirzes (SN) vai variācijas koeficientus (VK), kas iegūti daudzos eksperimentos. Katrā testēšanas reizē iegūtos plates kontroļu rezultātus, kā arī ar katru testējamo ķimikāliju iegūtos rezultātus ir jāizvērtē speciālistam.
Bez tam attiecībā uz pieņemamības kritērijiem ir jāievēro šādi principi:
|
—
|
datiem jābūt pietiekamiem, lai varētu kvantitatīvi novērtēt ER saistīšanos;
|
|
—
|
testētajām koncentrācijām jāsaglabājas testējamās ķimikālijas šķīdības diapazonā.
|
|
Datu analīze
|
21.
|
Definētajai datu analīzes procedūrai attiecībā uz piesātinājuma un konkurējošās saistīšanās datiem jāatbilst pamatprincipiem, ko izmanto receptora–liganda mijiedarbības raksturošanai. Parasti piesātinājuma–saistīšanās datus analizē, izmantojot nelineāru regresijas modeli, kas atspoguļo gan kopējo, gan nespecifisko saistīšanos. Iespējams, ka, nosakot Bmax un Kd, vajadzēs izmantot liganda noplicināšanās korekciju (piem., Swillens, 1995 (19)). Datus no konkurējošās saistīšanās testiem parasti transformē (piem., procentuālā specifiskā saistīšanās un testējamās ķimikālijas koncentrācija (log M)). Katras testējamās ķimikālijas aplēstais log (IC50) ir jānosaka, izmantojot piemērotu nelineāras līknes piemeklēšanas programmatūru, lai panāktu atbilstību Hilla vienādojumam ar četriem parametriem. Pēc sākotnējās analīzes jāpārbauda, vai līkne atbilst parametriem, un vizuāli jānosaka, cik precīzi saistīšanās dati atbilst ģenerētajai konkurējošās saistīšanās līknei. Dažos gadījumos, lai panāktu precīzāko atbilstību līknei, var būt jāveic papildu analīze (piem., ierobežojot līknes augšējo un/vai apakšējo punktu, izmantojot 10 % kritēriju, sk. 4. papildinājumu un 2. atsauci (III.A.2. iedaļa).
|
|
22.
|
Ja ir izpildīti pieņemamības kritēriji (20. punkts), tas nozīmē, ka testa sistēma darbojas pareizi, tomēr tas negarantē, ka konkrētajā testā tiks iegūti pareizi dati. Labākais pierādījums iegūto datu pareizībai ir pareizu pirmā izpildītā testa rezultātu replicēšana.
|
Vispārīgie datu interpretācijas kritēriji
|
23.
|
Pašlaik nav vispārpieņemtas metodes, kā interpretēt ER saistīšanās datus. Tomēr visi hrER-mediētas aktivitātes kvalitatīvie (piem., saisteklis/nesaisteklis) un/vai kvantitatīvie (piem., log IC50, relatīvā afinitāte (relative binding affinity — RBA), utt.) novērtējumi jābalsta uz empīriskiem datiem un pamatotiem zinātniskiem spriedumiem.
|
Testēšanas pārskats
|
24.
|
Testēšanas pārskatā norāda šādas ziņas.
|
|
Kontroles/references/testējamā ķimikālija:
|
—
|
avots, sērijas numurs, izmantošanas termiņš, ja pieejams;
|
|
—
|
pašas testējamās ķimikālijas stabilitāte, ja zināma;
|
|
—
|
testējamās ķimikālijas šķīdība un stabilitāte šķīdinātājā, ja zināma;
|
|
—
|
(attiecīgā gadījumā) pH, osmolalitātes un nogulsnēšanās mērījums kultūras barotnē, kurā testējamā ķimikālija pievienota.
|
|
|
|
Vienkomponenta viela:
|
—
|
izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības;
|
|
—
|
ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums, CAS numurs, SMILES vai InChI kods, struktūrformula, tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
|
|
Daudzkomponentu viela, UVCB un maisījumi:
|
—
|
iespējami izsmeļošs raksturojums, norādot sastāvdaļu ķīmisko identitāti (sk. iepriekš), kvantitatīvo saturu un relevantās fizikālķīmiskās īpašības.
|
|
|
|
Šķīdinātājs/nesējs:
|
—
|
raksturojums (veids, piegādātājs un sērija);
|
|
—
|
šķīdinātāja/nesēja izvēles pamatojums;
|
|
—
|
testējamās ķimikālijas šķīdība un stabilitāte šķīdinātājā/nesējā, ja zināma.
|
|
|
|
Receptori:
|
—
|
receptoru avots (piegādātājs, kataloga Nr., sērija, receptora suga, piegādātāja nodrošinātā aktīvo receptoru koncentrācija, piegādātāja sertifikāts);
|
|
—
|
receptoru raksturojums (ieskaitot piesātinājuma–saistīšanās rezultātus): Kd, Bmax;
|
|
—
|
radioaktīvi marķētais ligands:
|
|
—
|
piegādātājs, kataloga Nr., sērija, specifiskā aktivitāte.
|
|
|
|
Testēšanas apstākļi:
|
—
|
šķīdības ierobežojums konkrētā testa apstākļos;
|
|
—
|
saistīšanās buferšķīduma sastāvs;
|
|
—
|
receptora koncentrācija;
|
|
—
|
iezīmētā elementa (t. i., radioaktīvi marķētā liganda) koncentrācija;
|
|
—
|
testējamās ķimikālijas koncentrācijas;
|
|
—
|
nesēja saturs (procentuāli) galīgajā testā;
|
|
—
|
inkubācijas temperatūra un ilgums;
|
|
—
|
metode ar kuru nosaka, vai saistīts/brīvs;
|
|
—
|
pozitīvās un negatīvās kontroles/references vielas;
|
|
—
|
kritēriji, pēc kuriem novērtē, vai testa rezultāts ir pozitīvs, negatīvs vai neskaidrs.
|
|
|
|
Pieņemamības pārbaude
|
—
|
faktiskās IC50 un Hilla virziena koeficienta vērtības paralēlajām pozitīvajām kontroles/references vielām.
|
|
|
|
Rezultāti:
|
—
|
jēldati un saistītie/brīvie dati;
|
|
—
|
denaturēta apstiprinoša pārbaude (attiecīgā gadījumā);
|
|
—
|
zemākā iedarbīgā koncentrācija (LEC), ja tāda ir;
|
|
—
|
RBA un/vai IC50 vērtība (attiecīgā gadījumā);
|
|
—
|
ja iespējams, koncentrācijas–atbildreakcijas sakarība;
|
|
—
|
statistiskās analīzes (attiecīgā gadījumā), kā arī kļūdas un ticamības mērs (piem., vidējās vērtības standartnovirze (SEM), standartnovirze (SN), variācijas koeficients (VK) vai 95 % ticamības intervāls (CI)) un apraksts, kā šīs vērtības iegūtas.
|
|
|
|
Rezultātu iztirzājums:
|
—
|
10 % kritērija piemērošana.
|
|
Secinājums
|
LITERATŪRA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
1. papildinājums
DEFINĪCIJAS UN SAĪSINĀJUMI
10 % kritērijs
: izvēle no analīzes izslēgt tādus datu punktus, kuros ar replikātiem iegūtā [3H]17β-estradiola specifiskās saistīšanās procentuālā vidējā vērtība par 10 % vai vairāk pārsniedz pie zemākas koncentrācijas iegūto vidējo vērtību (sk. 4. papildinājumu).
Pieņemamības kritēriji
: minimālie standarti, kas attiecas uz eksperimenta kontrolēm un references standartiem. Lai eksperimentu uzskatītu par derīgu, visiem pieņemamības kritērijiem jābūt izpildītiem.
Pareizība (konkordantums)
: pakāpe, kādā testa rezultāti sakrīt ar pieņemtajām atsauces vērtībām. Tas ir testa veiktspējas raksturlielums un viens no tā relevantuma aspektiem. Ar šo terminu, tāpat kā terminu “konkordantums”, bieži vien saprot testa pareizo iznākumu īpatsvaru (1).
KS
: ESAO konceptuālais satvars (KS) endokrīno disruptoru testēšanai un izvērtēšanai.
Ķimikālija
: viela vai maisījums.
VK
: variācijas koeficients.
E2
: 17β-estradiols.
ED
: endokrīnā disrupcija.
hER
α
: cilvēka estrogēnreceptors alfa.
ER
: estrogēnreceptors.
Estrogēniskā aktivitāte
: ķimikālijas spēja imitēt 17β-estradiolu, proti, saistīties ar estrogēnreceptoriem. Ar šo testēšanas metodi var konstatēt saistīšanos ar hERα.
IC
50
: testējamās inhibējošās ķimikālijas 50 % efektīvā koncentrācija.
ICCVAM
: Alternatīvo metožu validēšanas koordinēšanas starpaģentūru komiteja.
Starplaboratoriju reproducējamība
: lielums, kas raksturo, kādā mērā vairākas kvalificētas laboratorijas spēj iegūt kvalitatīvi un kvantitatīvi līdzīgus rezultātus, ja tās izmanto vienu un to pašu protokolu un vienas un tās pašas testējamās vielas. Starplaboratoriju reproducējamību (saukta arī par “interlaboratoriju reproducējamību”) nosaka prevalidēšanas un validēšanas procesā, un šis lielums rāda, cik lielā mērā testēšanu vienlīdz sekmīgi iespējams veikt dažādās laboratorijās (1).
Iekšlaboratorijas reproducējamība
: lielums, kas raksturo, kādā mērā vienā laboratorijā strādājoši kvalificēti darbinieki spēj sekmīgi atkārtot iepriekš iegūtus rezultātus, ja to pašu protokolu izmanto citā laikā. Saukts arī par “intralaboratoriju reproducējamību” (1).
LEC
: zemākā iedarbīgā koncentrācija ir zemākā koncentrācija, kurā testējamā ķīmikālija izraisa reakciju (t. i., testējamās ķimikālijas zemākā koncentrācija, pie kuras indukcijas reizinājums ir statistiski atšķirīgs no paralēlās nesēja kontroles).
Atdarinājuma tests
: sadzīvisks apzīmējums testam, kas ir noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgs validētai un pieņemtai testēšanas references metodei. Brīvi aizstājams ar līdzīgu testēšanas metodi.
PBTG
: veiktspējā balstītas testēšanas vadlīnijas.
Veiktspējas standarti
: uz validētu testēšanas metodi bāzēti standarti, pēc kuriem izvērtē noteikšanas mehānisma vai funkcionalitātes ziņā līdzīgu testu salīdzināmību. Tie ietver: 1) nepieciešamos testa komponentus; 2) minimālo tādu references ķimikāliju sarakstu, kuras izraudzītas no citām ķimikālijām, ko izmanto, lai pierādītu pieņemamu validētās testēšanas metodes veiktspēju; kā arī 3) uz attiecīgajā validētajā testēšanas metodē iegūto rezultātu pamata — salīdzināmus pareizības un ticamības līmeņus, kādi būtu jāiegūst ar ierosināto testu, to izvērtējot ar references ķimikālijām no minimālā saraksta (1).
Standartvielas
: veiktspējas standartos iekļauto references vielu apakškopa, ko laboratorijas var izmantot, lai pierādītu savu tehnisko kompetenci ar standartizētu testu. Šādu vielu atlases kritēriji parasti ir, piemēram šādi: viela ir reprezentatīva dažādām reakcijām, ir komerciāli pieejama un par to pieejami ļoti kvalitatīvi references dati.
Kompetence
: vēl pirms nezināmu vielu testēšanas pierādīta spēja pienācīgi veikt testu.
References estrogēns
: 17ß-estradiols (E2, CAS 50-28-2).
References testēšanas metodes
: testi, uz kuriem balstās PBTG Nr. 493.
RBA
: relatīvā afinitāte. Vielas RBA aprēķina kā procentuālu vērtību no vielas log (IC50) attiecībā pret 17β-estradiola log (IC50).
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Tā ir pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testa pareizības (konkordantuma) aspektu (1).
Ticamība
: parametrs, kas izsaka, kādā pakāpē vienā un tajā pašā laboratorijā vai dažādās laboratorijās pēc vienu un tā paša protokola var ilgstoši reproducējami veikt konkrētu testu. To novērtē, aprēķinot reproducējamību vienā laboratorijā un vairākās laboratorijās.
SN
: standartnovirze.
Testējamā ķimikālija
: viela vai maisījums, kuru testē ar šo testēšanas metodi.
Validēta testēšanas metode
: tests, kura relevantums (ieskaitot pareizību) un ticamība attiecībā uz lietošanu konkrētā nolūkā ir pārbaudīta validācijas pētījumos. Jāievēro, ka validētas testēšanas metodes veiktspēja pareizības un ticamības ziņā var nebūt pietiekama, lai to izmantotu paredzētajam nolūkam (1).
Validācija
: Process, kurā nosaka, kādā mērā konkrēta pieeja, metode, tests, process vai novērtējums ir ticams un relevants konkrētajam nolūkam (1).
2. papildinājums
FREIBERGERA–VILSONA IN VITRO TESTI, KAS BALSTĀS UZ PIESĀTINĀJUMU UN KONKURĒJOŠO SAISTĪŠANOS AR ESTROGĒNRECEPTORU (ERΑ) UN KUROS IZMANTO PILNU REKOMBINANTO ERΑ
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI (SK. ARĪ VISPĀRĪGO IEVADU)
|
1.
|
Šis in vitro piesātinājuma un konkurējošās saistīšanās ar estrogēnreceptoriem (ERα) tests izmanto pilnu cilvēka receptoru — ERαα(hrERα) —, iegūtu un izolētu no insektu šūnām, kuras inficētas ar bakulovīrusu. Freibergera un Vilsona izstrādātajam protokolam veikts starptautisks daudzlaboratoriju validēšanas pētījums (2), kas pierādīja testa relevanci un ticamību paredzētajam nolūkam.
|
|
2.
|
Šis tests ir skrīninga procedūra, ar ko identificē vielas, kas spēj saistīties ar pilnu hrERα. To izmanto, lai noteiktu testējamās ķimikālijas spēju konkurēt ar 17β-estradiolu attiecībā uz saistīšanos ar hrERα. Kvantitatīvie testa rezultāti var ietvert IC50 (mērs, kas rāda testējamās ķimikālijas koncentrāciju, kas vajadzīga, lai no hrERα pārvietotu pusi no [3H]-17β-estradiola) un testējamo ķimikāliju relatīvo afinitāti attiecībā uz hrERα salīdzinājumā ar 17β-estradiolu. Ķīmiskā skrīninga vajadzībām pieņemami kvalitatīvie testa rezultāti var ietvert testējamo ķimikāliju klasificēšanu saistīgās vai nesaistīgās ar hrERα vai tādās, kuru saistīgums ir neskaidrs, uz to kritēriju pamata, kas aprakstīti attiecībā uz saistīšanās līknēm.
|
|
3.
|
Testā izmanto radioaktīvi marķētu ligandu, tāpēc laboratorijai būs vajadzīga radioaktīvo materiālu licence. Visas procedūras ar radioaktīvajiem izotopiem un bīstamām ķimikālijām jāveic, ievērojot nacionālajos tiesību aktos paredzētos noteikumus un procedūras.
|
|
4.
|
Pirms šā testa izmantošanas regulatīviem mērķiem jāizlasa sadaļas “VISPĀRĪGAIS IEVADS” un “
HrER SAISTĪŠANĀS TESTA KOMPONENTI”. Šajā testēšanas vadlīnijā izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
TESTA PRINCIPI (SK. ARĪ VISPĀRĪGO IEVADU)
|
5.
|
HrERα saistīšanās tests mēra radioaktīvi iezīmēta liganda ([3H]17β-estradiola) spēju saistīties ar ER arvien pieaugošu testējamās ķimikālijas (t. i., konkurenta) koncentrāciju klātbūtnē. Testējamās ķimikālijas, kam piemīt augsta ER afinitāte, konkurē ar radioaktīvi iezīmēto ligandu pie zemākas koncentrācijas nekā ķimikālijas ar zemāku receptora afinitāti.
|
|
6.
|
Šis tests sastāv no diviem svarīgākajiem komponentiem: piesātinājuma–saistīšanās eksperimenta, kas raksturo receptora–liganda mijiedarbības parametrus, un konkurējošās saistīšanās eksperimenta, kas attiecībā uz saistīšanos ar ER raksturo konkurenci starp testējamo ķimikāliju un radioaktīvi marķēto ligandu.
|
|
7.
|
Piesātinājuma–saistīšanās eksperimenta mērķis ir raksturot konkrētas receptoru partijas afinitāti un konkurējošās saistīšanās eksperimentā — to skaitu preparātā. Piesātinājuma–saistīšanās eksperiments līdzsvara apstākļos mēra fiksētas estrogēnreceptora koncentrācijas afinitāti tā dabīgajam ligandam (to izsaka disociācijas konstante, Kd), un aktīvo receptoru domēnu koncentrāciju (Bmax).
|
|
8.
|
Konkurējošās saistīšanās eksperiments mēra tādas vielas afinitāti, kura attiecībā uz saistīšanos ar ER konkurē ar [3H]17β-estradiolu. Afinitāti kvantitatīvi izsaka kā testējamās ķimikālijas koncentrāciju, kas līdzsvara apstākļos inhibē 50 % [3H]17β-estradiola specifiskās saistīšanās (apzīmē kā “50 % inhibējošo koncentrāciju” vai IC50). To var izvērtēt arī, izmantojot relatīvo afinitāti (RBA, relatīvā salīdzinājumā ar estradiola IC50, ko atsevišķi mēra tajā pašā izpildes reizē). Konkurējošās saistīšanās eksperiments mēra fiksētas koncentrācijas [3H]17β-estradiola spēju saistīties ar ER plaša testējamās ķimikālijas koncentrāciju diapazona (astoņas intensitātes pakāpes) klātbūtnē. Pēc tam, ja iespējams, datus pārveido Hilla vienādojuma (A. Hills, 1910) formā, kas apraksta radioaktīvi marķētā liganda pārvietošanu, ko izraisa viena domēna konkurējošais saisteklis. Radioaktīvi marķētā estradiola pārvietošanas pakāpe līdzsvara apstākļos tiek izmantota, lai testējamo ķimikāliju raksturotu kā saistīgu, nesaistīgu vai tādu, kam ir neskaidra reakcija.
|
PROCEDŪRA
Pieņemamas hrERα proteīna veiktspējas pierādīšana
|
9.
|
Pirms piesātinājuma un konkurējošās saistīšanās testu ieviešanas laboratorijas ikdienas praksē attiecībā uz katru jaunu hrERα partiju ir jāpierāda to veiktspējas pareizība laboratorijā, kurā to izmantos. Veiktspēju pierāda ar divu etapu procedūru. Proti,
|
—
|
veic [3H]-17β-estradiola saistīšanās–piesātinājuma testu, lai pierādītu specifiskumu attiecībā uz hrERα un piesātinājumu. Ar iegūto datu nelineāras regresijas analīzi (piem., BioSoft; McPherson, 1985; Motulsky, 1995) un no tiem izveidotu Skačārda (Scatchard) diagrammu dokumentē hrERα spēju saistīt [3H]-17β-estradiolu (Kd) un katras partijas receptoru skaitu (Bmax);
|
|
—
|
veic konkurējošās saistīšanas testu, izmantojot kontroles vielas (references estrogēnu (17β-estradiolu)), vāju saistekli (piem., noretinodrelu vai noretindronu) un nesaistekli (oktiltrietoksisilānu (OTES)). Katra laboratorija izveido un uztur vēsturisku datubāzi, kurā dokumentē IC50 konsekventumu un citas būtiskas references estrogēna un vāja saistekļa vērtības, kas iegūtas dažādos eksperimentos un ar atšķirīgām hrERα partijām. Ar kontroles vielām iegūto konkurējošās saistīšanās līkņu parametriem jābūt 95 % ticamības intervāla robežās (sk. 1. tabulu); tas izstrādāts, izmantojot to laboratoriju datus, kuras piedalījās šā testa validēšanas pētījumā (2).
|
|
1. tabula.
References estrogēna un vāja saistekļa veiktspējas kritēriji, kas izstrādāti FW hrER saistīšanās testam.
|
Viela
|
Parametrs
|
Vidējā vērtība (8)
|
Standartnovirze (n)
|
95 % ticamības intervāli (9)
|
|
apakšējā robeža
|
augšējā robeža
|
|
17β-estradiols
|
augstākais (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
zemākais (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hilla virziena koeficients
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (10)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrels
|
augstākais (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
zemākais (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hilla virziena koeficients
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindronsc
(10)
|
augstākais (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
zemākais (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hilla virziena koeficients
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50(M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Laboratorijas kvalifikācijas pierādīšana
|
10.
|
Sk. šīs testēšanas metodes sadaļas “
HrER SAISTĪŠANĀS TESTA KOMPONENTI” 17. un 18. punktu un 2. tabulu. Katrā (piesātinājuma un konkurējošās saistīšanās) testā īsteno trīs neatkarīgas testa izpildes reizes (t. i., ar jauniem receptora atšķaidījumiem, ķimikālijām un reaģentiem), tās veic dažādās dienās un katrā izpildes reizē izmanto trīs replikātus.
|
Receptora (hrERα) koncentrācijas noteikšana
|
11.
|
Aktīvā receptora koncentrācija katrā atsevišķā partijā un atkarībā no uzglabāšanas apstākļiem var būt nedaudz atšķirīga. Tāpēc ir jānosaka no piegādātāja saņemtā aktīvā receptora koncentrācija. Tas ļaus panākt atbilstošu aktīvā receptora koncentrāciju testa veikšanas laikā.
|
|
12.
|
Apstākļos, kas atbilst konkurējošās saistīšanās apstākļiem (proti, ar 1 nM [3H]-estradiola), nominālas receptora koncentrācijas — 0,25, 0,5, 0,75 un 1 nM — inkubē bez estradiola (kopējā saistīšanās) un ar 1 μM nemarķēta estradiola (nespecifiskā saistīšanās). Grafiski attēlo sakarību starp specifisko saistīšanos (starpību starp kopējo saistīšanos un nespecifisko saistīšanos) un receptora nominālo koncentrāciju. Receptora koncentrācija, pie kuras iegūst tādas specifiskās saistīšanās vērtības, kas atbilst 40 % pievienotā radioaktīvā marķējuma, ļauj noteikt atbilstošo nominālo receptora koncentrāciju, kuru attiecīgi izmanto piesātinājuma un konkurējošās saistīšanās eksperimentiem. Samērā bieži galīgā hrER koncentrācija, kas atbilst šim nosacījumam, ir 0,5 nM.
|
|
13.
|
Ja pēc atkārtotiem mēģinājumiem neizdodas sasniegt 20 % kritēriju, eksperimenta uzstādījumus pārbauda attiecībā uz iespējamām kļūdām. Nespēja sasniegt 20 % kritēriju var norādīt, ka rekombinantajā partijā ir ļoti maz aktīvā receptora; šādos gadījumos jāapsver iespēja izmantot citu receptora partiju.
|
Piesātinājuma tests
|
14.
|
[3H]17β-estradiola astoņu augoša stipruma koncentrāciju triplikātus vērtē, pie šādiem trim turpmāk uzskaitītajiem nosacījumiem (sk. 2. tabulu):
|
—
|
testu veic bez nemarķēta 17β-estradiola un ER klātbūtnē. Šādi, mērot radioaktivitāti iedobēs, kurās ir tikai [3H]17β-estradiols, nosaka kopējo saistīšanos;
|
|
—
|
testu veic ar nemarķēta 17β-estradiola koncentrāciju, kas tūkstoškārt pārsniedz marķētā 17β-estradiola koncentrāciju, un ER klātbūtnē. Šāda nosacījuma mērķis ir piesātināt aktīvos saistīšanās domēnus ar nemarķētu 17β-estradiolu un, mērot radioaktivitāti iedobēs, noteikt nespecifisko saistīšanos. Visu pārējo karsto estradiolu, kas spēj saistīties ar receptoru, uzskata par nespecifiskā domēnā saistīgu, jo aukstajam estradiolam jābūt tik lielā koncentrācijā, ka tas saistās ar visiem uz receptora pieejamajiem specifiskajiem domēniem;
|
|
—
|
testu veic bez nemarķēta 17β-estradiola un bez ER (kopējās radioaktivitātes noteikšana).
|
|
[3H]-17β-estradiola un nemarķēta 17β-estradiola šķīdumu sagatavošana
|
15.
|
Buferšķīdumam pievienojot 12 nM [3H]-17β-estradiola izejšķīduma, sagatavo [3H]-17β-estradiola atšķaidījumus, kuru sākotnējās koncentrācijas ir 0,12nM–12 nM. Galīgā testa koncentrācijas diapazonā no 0,03 līdz 3,0 nM iegūst, 40 μl šādi sagatavoto šķīdumu iepilinot attiecīgajās 96 iedobju mikrotitrēšanas plates iedobēs (beigu tilpums 160 μl). Testa buferšķīduma, [3H]-17β-estradiola izejšķīduma un atšķaidījumu sagatavošana, kā arī koncentrāciju noteikšana ir sīki izskaidrota FW testa protokolā (2).
|
|
16.
|
Atšķaidījumus no etanolu saturoša 17β-estradiola šķīduma sagatavo, pievienojot tam testa buferšķīdumu, lai iegūtu astoņas augoša stipruma koncentrācijas, kas sākotnēji ir diapazonā no 0,06 μM līdz 6 μM. Galīgā testa koncentrācijas diapazonā no 0,03 μM līdz 3 μM iegūst, 80 μl šādi sagatavoto šķīdumu iepilinot attiecīgajās 96 iedobju mikrotitrēšanas plates iedobēs (beigu tilpums 160 μl). Nemarķētā 17β-estradiola galīgajai koncentrācijai atsevišķās nespecifiskās saistīšanās testa iedobēs jābūt tūkstoškārt lielākai par marķētā [3H]-17β- estradiola koncentrāciju. Nemarķēta 17β-estradiola atšķaidījumu sagatavošana ir sīki izskaidrota FW testa protokolā (2).
|
|
17.
|
Izmanto receptora nominālo koncentrāciju, pie kuras specifiskā saistīšanās ir 20±5 % (sk. 12.-13. punktu). HrERα šķīdumu jāsagatavo tieši pirms lietošanas.
|
|
18.
|
96 iedobju mikrotitrēšanas plates sagatavo saskaņā ar 2. tabulas norādījumiem, katrai koncentrācijai sagatavojot trīs replikātus. 2.2. papildinājumā sniegts piemērs [3H]-17β-estradiola, nemarķēta 17β-estradiola, buferšķīduma un receptora koncentrācijām un tilpumiem testa platē.
|
2. tabula.
Piesātinājuma–saistīšanās testa mikrotitrēšanas plates izkārtojums
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Kopējā saistīšanās (šķīdinātājs)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Nespecifiskā saistīšanās
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2:: [3H]-17β-estradiols
ER:: estrogēnreceptors
E2:: nemarķēts 17β-estradiols
|
|
19.
|
Testa mikrotitrēšanas plates 16 līdz 20 stundas inkubē 2° līdz 8°C temperatūrā un inkubācijas periodā novieto rotatora.
|
Ar hrERα saistītā [3H]-17β-estradiola mērīšana
|
20.
|
[3H]-17β-estradiolu, kas piesaistījies hrERα, no brīvā [3H]-17β-estradiola atdala, katrā iedobē pievienojot 80 μl aukstas DCC suspensijas, tad 10 minūtes kratot mikrotitrēšanas plates un 10 minūtes centrifugējot ar aptuveni 2 500 apgr./min. Lai procedūras gaitā nepieļautu [3H]-17β-estradiola atdalīšanos no hrERα, kam tas piesaistījies, ir ļoti svarīgi buferšķīdumus un testa iedobes turēt 2 līdz 8°C temperatūrā un katru testa soli veikt ātri. Lai plates varētu apstrādāt ātri un efektīvi, ir vajadzīgs mikrotitrēšanas plašu kratītājs.
|
|
21.
|
Ņem 50 μl supernatanta, kas satur ar hrERα saistīto [3H]-17β-estradiolu, un novieto uz otras mikrotitrēšanas plates; to dara ar īpašu piesardzību, lai nepieļautu iedobju kontamināciju, saskaroties DCC.
|
|
22.
|
Pēc tam katrai iedobei (A1–B12 un D1–E12) pievieno 200 μl scintilācijas šķidruma, kas kodolemisiju kinētisko enerģiju spēj pārveidot gaismas enerģijā. Katrā no G1–H12 (kopējam dpm atbilstošajām) iedobēm iepilina 40 μl [3H]-17β-estradiola sērijveida atšķaidījuma; tam jānokļūst tieši scintilācijas šķīdumā mērījumu plates iedobēs atbilstoši 3. tabulas norādījumiem, proti, šajās iedobēs jābūt tikai 200 μl scintilācijas šķidruma un atbilstošam [3H]-17β-estradiola atšķaidījumam. Ar šiem mērījumiem pierāda, cik daudz [3H]-17β-estradiola (izteikta dpm) pievienots katrā iedobju kopumā, kas demonstrē kopējo saistīšanos un nespecifisko saistīšanos.
|
3. tabula.
Piesātinājuma–saistīšanās testa mikrotitrēšanas plates izkārtojums, radioaktivitātes mērījums
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Kopējā saistīšanās (šķīdinātājs)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E22
|
0,10 nM [3H] E2 + ER
+ 0,10 μM E2
|
Nespecifiskā saistīšanās
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E2 (kopējais dpm)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
(kopējais dpm
(*7))
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2:: [3H]-17β-estradiols
ER:: estrogēnreceptors
E2:: nemarķēts 17β-estradiols
dpm:: sabrukumu skaits minūtē
|
|
23.
|
Mērījumu sāk ne mazāk kāpēc divu stundu ilgas nogaidīšanas, un skaitīšanas laiks katrai iedobei ir 40 minūtes. Nosakot dpm uz iedobi un piemērojot slāpēšanas korekciju, izmanto mikrotitrēšanas plates scintilāciju skaitītāju. Ja mikrotitrēšanas plates scintilāciju skaitītājs nav pieejams, paraugu skaitīšanai var izmantot parasto skaitītāju. Šādā gadījumā var apsvērt skaitīšanas laika samazināšanu.
|
Konkurējošās saistīšanās tests
|
24.
|
Konkurējošās saistīšanās tests mēra vienas konkrētas [3H]-17β- estradiola koncentrācijas saistīšanos arvien pieaugošu testējamās ķimikālijas koncentrāciju klātbūtnē. Katrā testa veikšanas reizē izmanto trīs paralēlus katras koncentrācijas replikātus. Papildus tam attiecībā uz katru testējamo ķimikāliju testu izpilda trīs reizes, un tas nenotiek paralēli, bet dažādos laikos. Testu veic vienā vai vairākās 96 iedobju mikrotitrēšanas platēs.
|
Kontroles
|
25.
|
Veicot testu, katrā eksperimentā jāiekļauj paralēlais šķīdinātājs un kontroles (t. i., references estrogēns, vājš saisteklis un nesaisteklis). Katrā testa izpildes reizē vienā platē ir jābūt atsauces estrogēna un kontroļu (t.i. vāja saistekļa un nesaistekļa) pilnai koncentrāciju līknei. Visās pārējās platēs ir jābūt: i) katra E2 un vāja saistekļa triplikātiem augstā koncentrācijā (maksimāla pārvietošana) un vidējā koncentrācijā (aptuveni IC50); ii) šķīdinātāja kontroles un nespecifiskās saistīšanās triplikātiem. Testa buferšķīduma, kontroļu, [3H]-17β-estradiola, hrERα un testējamo ķimikāliju šķīdumu sagatavošanas procedūras ir aprakstītas 2. atsaucē (sk. FW testa protokolu K pielikumā).
|
Šķīdinātāja kontrole
|
26.
|
Šķīdinātāja kontrole rāda, vai nenotiek šķīdinātāja mijiedarbība ar testa sistēmu, kā arī mēra kopējo saistīšanos (apzīmē ar “TB” — total binding). Ieteicamais šķīdinātājs ir etanols. Ja testējamās ķimikālijas visaugstākā koncentrācija etanolā nešķīst, kā alternatīvu var izmantot dimetilsulfoksīdu (“DMSO”). Galīgajā testā etanola vai attiecīgā gadījumā DMSO koncentrācijai iedobēs jābūt 1,5 % un ne vairāk kā 2 %.
|
Buferšķīduma kontrole
|
27.
|
Buferšķīduma kontrole (“BC”) nedrīkst saturēt ne šķīdinātāju, ne testējamo ķimikāliju, bet tā satur visus citus testa komponentus. Buferšķīduma kontroles rezultātus salīdzina ar šķīdinātāja kontroli, lai verificētu, ka izmantotais šķīdinātājs neietekmē testa sistēmu.
|
Stiprs saisteklis (references estrogēns)
|
28.
|
17β-estradiols (CAS 50-28-2) ir endogēnisks ligands ar augstu afinitāti (alfa apakštipa) ER. Katram hrERα konkurējošās saistīšanās testam sagatavo standarta līkni ar nemarķētu 17β-estradiolu, lai varētu novērtēt, kāda ir mainība laika gaitā, veicot testu tajā pašā laboratorijā. Sagatavo astoņus nemarķēta 17β-estradiola šķīdumus etanolā tā, lai testa iedobēs šķīdumu koncentrācija būtu no 100 nM–10 pM (-7[logM] līdz -11[logM]), ar šādiem intervāliem: (-7[logM], -8[logM], -8.5[logM], -9[logM], - 9,5[logM], -10[logM], -11[logM]). Nemarķēta 17β-estradiola (1 μM) augstākā koncentrācija kalpo arī kā nespecifiskās saistīšanās (NSB) indikators. 4. tabulā šo koncentrāciju apzīmē ar kodu “NSB”, lai gan tā ir arī standarta līknes daļa.
|
Vājš saisteklis
|
29.
|
Lai demonstrētu katra eksperimenta jutīgumu un ļautu novērtēt, kāda ir rezultātu mainība, veicot testu laika gaitā, tajā iekļauj vāju saistekli (noretinodrelu (CAS 68-23-5) vai noretindronu (CAS 68-22-4). Sagatavo astoņus vāja saistekļa šķīdumus etanolā tā, lai testa iedobēs šķīdumu koncentrācija būtu 3 nM–30 pM (no -8,5[logM] līdz -4,5[logM]), ar šādiem intervāliem: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM],-7,5[logM], -8,5[logM].
|
Nesaisteklis
|
30.
|
Kā negatīvo kontroli (nesaistekli) izmanto oktiltrietoksisilānu (OTES, CAS2943-75-1). Tas apstiprinās, ka tests spēs noteikt ķimikālijas, kas nesaistās ar hrERα. Sagatavo astoņus vāja saistekļa šķīdumus etanolā tā, lai testa iedobēs šķīdumu koncentrācija būtu 0,1 nM–1 000 μM (no -10[logM] līdz -3[logM]), ar logaritmisku palielinājumu. Alternatīvi par nesaistekļa kontroli var izmantot di-n-butilftalātu (DBP). Ir pierādīts, ka tā maksimālā šķīdība testā ir -4[logM].
|
HrERα koncentrācija
|
31.
|
Izmanto receptora daudzumu, pie kura specifiskā saistīšanās ir 20±5 % no 1 nM radioaktīvi marķētā liganda (sk. 2. papildinājuma 12.-13. punktu). HrERα šķīdumu jāsagatavo tieši pirms lietošanas.
|
[3H]-17β-estradiols
|
32.
|
[3H]-17β-estradiola koncentrācija testa iedobēs ir 1,0 nM.
|
Testējamās ķimikālijas
|
33.
|
Vispirms ir jāveic šķīdības tests, lai noteiktu katras testējamās ķimikālijas šķīdības robežu un identificētu piemērotu koncentrācijas diapazonu, kas tiks izmantots testa protokolā. Sākotnēji katras testējamās ķimikālijas šķīdības robežu nosaka šķīdinātājā un pēc tam apstiprina testēšanas apstākļos. Galīgā testā izmantojamā koncentrācija nepārsniedz 1 mM. Diapazona noteikšanas testēšanā ietilpst šķīdinātāja kontrole; paralēli tai testē astoņus sērijveida logaritmiskus atšķaidījumus, sākot ar augstāko pieņemamo koncentrāciju (piem., 1 mM vai zemāku, balstoties uz šķīdības robežu), un vēro, kad šķīdums kļūst duļķains vai rada nogulsnes (sk. arī 35. punktu). Testējamo ķimikāliju testē, izmantojot līkni ar astoņām log koncentrācijām un diapazona noteikšanas testā identificētajiem intervāliem. Trešā un ceturtā eksperimenta koncentrācijas būtu jāpielāgo pēc vajadzības, lai labāk raksturotu koncentrācijas–atbildreakcijas līkni.
|
|
34.
|
Testējamās ķimikālijas atšķaidījumus sagatavo atbilstošā šķīdinātājā (sk. 2. papildinājuma 26. punktu). Ja testējamās ķimikālijas augstākā koncentrācija nešķīst ne etanolā, ne DMSO un lielāka šķīdinātāja daudzuma pievienošana galīgajā testa stobriņā novestu pie pieņemamās robežas pārsniegšanas, augstāko koncentrāciju var samazināt par vienu intervālu uz leju. Šādā gadījumā sērijveida koncentrācijas var papildināt ar vienu papildkoncentrāciju diapazona apakšā. Visas pārējas sērijveida koncentrācijas paliek nemainīgas.
|
|
35.
|
Pievienojot testējamās ķimikālijas šķīdumus testa iedobēs, tos rūpīgi vēro, jo pievienošanas laikā var rasties nogulsnes. Datus par tām iedobēm, kurās konstatē nogulsnes, neņem vērā, pielāgojot datus līknei, un testa pārskatā norāda izslēgšanas iemeslu.
|
|
36.
|
Ja no citiem avotiem jau ir pieejama informācija par testējamās ķimikālijas log (IC50), atšķaidījumus var būt lietderīgi ģeometriski izkārtot (t. i., pa 0,5 log vienībām ap paredzamo log (IC50). Galīgajam rezultātam būtu jāatspoguļo pietiekama koncentrāciju izkliede abās log (IC50) pusēs, arī līknes augšējā un apakšējā punktā, un koncentrācijām jābūt pietiekami raksturojošām attiecībā uz saistīšanās līkni.
|
Testa plates izkārtojums
|
37.
|
Sagatavo marķētas mikrotitrēšanas plates, kurās ir pa sešiem šādiem kodētiem inkubētiem replikātiem: šķīdinātāja kontrole un references estrogēns visaugstākajā koncentrācijā, kas kalpo arī par nespecifiskās saistīšanās (NSB) rādītāju; un pa trim šādiem kodētiem inkubētiem replikātiem: astoņas nesaistīgās kontroles (oktiltrietoksisilāna) koncentrācijas, septiņas references estrogēna zemākās koncentrācijas, astoņas vāja saistekļa devas līmeņa koncentrācijas un astoņas koncentrācijas no katras testējamās ķimikālijas (TC). Mikrotitrēšanas plates izkārtojuma diagrammas paraugs pilnām references estrogēna un kontroļu koncentrāciju līknēm ir sniegts 4. tabulā. Atsevišķas mikrotitrēšanas plates izmanto testējamām ķimikālijām, un tajās jāiekļauj testa plates kontroles, t. i., 1) E2 un vāja saistekļa triplikāti augstā koncentrācijā (maksimāla pārvietošana) un vidējā koncentrācijā (aptuveni IC50); 2) šķīdinātāja kontrole un nespecifiskā saistīšanās, no katra pa sešiem replikātiem (5. tabula). Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojuma darblapas paraugs ar trijām nezināmām testējamajām ķimikālijām ir dots 2.3. papildinājumā. 4. un 5. tabulā norādītās koncentrācijas ir galīgās testa koncentrācijas. Maksimālā E2 koncentrācija ir 1×10-7 M, un attiecībā uz vāju saistekli jāizmanto augstākā koncentrācija, kas 1. mikrotirēšanas platē izmantota vājam saisteklim. IC50 koncentrāciju nosaka laboratorija, balstoties uz savu vēsturisko kontroles datubāzi. Tiek sagaidīts, ka šī vērtība būs līdzīga validācijas pētījumos konstatētajai vērtībai (sk. 1. tabulu).
|
4. tabula.
Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojums, pilnas atsauces estrogēnu un kontroļu koncentrāciju līknes (1. plate).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (tikai šķīdinātājs)
|
TB (tikai šķīdinātājs)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Tukša iedobe (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
tukša iedobe (karsta) (*9)
|
tukša iedobe (karsta) (*9)
|
Buferšķīduma kontrole
|
Buferšķīduma kontrole
|
|
Šajā piemērā vājais saisteklis ir noretinodrels (apzīmēts “NE”).
|
5. tabula.
Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojums, pilnas testējamo ķimikāliju un plates kontroļu koncentrāciju līknes.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (tikai šķīdinātājs)
|
TB (tikai šķīdinātājs)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
Šajā piemērā vājais saisteklis ir noretinodrels (apzīmēts “NE”).
|
Konkurējošās saistīšanās testa izpilde
|
38.
|
Kā parādīts 6. tabulā, katrā testēšanas iedobē pievieno 80 μl šķīdinātāja kontroles, testa buferšķīduma kontroles, references estrogēna, vāja saistekļa, nesaistekļa un testa buferšķīdumā sagatavotas testējamās ķimikālijas. Pēc tam katrā iedobē pievieno 40 μl 4 nM [3H]-17β-estradiola šķīduma. 10 līdz 15 minūtes viegli rotē 2–8°C temperatūrā, pēc tam pievieno 40 μl hrERα šķīduma. Testa mikrotitrēšanas plates 16 līdz 20 stundas inkubē 2° līdz 8°C temperatūrā un inkubācijas periodā novieto rotatora.
|
6. tabula.
HrER konkurējošās saistīšanās testa komponentu tilpums mikrotitrēšanas platēs
|
Tilpums (μl)
|
Komponents
|
|
80
|
Nemarķēts 17β-estradiols, noretinodrels, OTES, testējamās ķimikālijas, šķīdinātājs vai buferšķīdums
|
|
40
|
4 nM [3H]-17β-estradiola šķīduma
|
|
40
|
hrERα šķīdums ar noteiktu koncentrāciju
|
|
160
|
Kopējais tilpums katrā testa plates iedobē
|
|
39.
|
Pēc tam (saskaņā ar piesātinājuma–saistīšanās testa apraksta 20.–23. punktu) kvantificē ar hrERα saistīto [3H]-17β-estradiolu, taču pirms tam, katrā iedobē pievienojot 80 μl aukstas DCC suspensijas, no brīvā [3H]-17β-estradiola atdala ar hrERα saistīto [3H]-17β-estradiolu/
|
|
40.
|
Iedobes H1–6 (sk. aili “Tukša iedobe (karsta)” 4. tabulā) raksturo marķēta [3H]-estradiola dpm 40 μl. 40 μl alikvotu pārnes tieši scintilācijas šķīdumā iedobēs H1–H6.
|
Pieņemamības kritēriji
Piesātinājuma–saistīšanās tests
|
41.
|
Ja tiek izmantotas pieaugošas [3H]-17β-estradiola koncentrācijas, specifiskās saistīšanās līknei ir jāsasniedz atslābuma periods, kas norāda uz hrERα piesātinājumu ar ligandu.
|
|
42.
|
Pieņemamības diapazons attiecībā uz specifisko saistīšanos pie 1 nM [3H]-17β-estradiola koncentrācijas ir 15–25 % no kopējās pievienotās radioaktivitātes vidējās vērtības, kas mērīta visās izpildes reizēs. Ir pieļaujamas atsevišķas nelielas novirzes no minētā diapazona, tomēr, ja izpildes reizēs pastāvīgi iegūst rezultātus ārpus minētā diapazona vai ja konkrētā izpildes reizē novirze ir ļoti būtiska, ir jāpielāgo proteīnu koncentrācija un piesātinājuma tests jāatkārto.
|
|
43.
|
Datiem jābūt attēlojamiem lineāras Skačārda diagrammas veidā.
|
|
44.
|
Nespecifiskā saistīšanās nedrīkst būt pārmērīga. Nespecifiskās saistīšanās vērtība parasti ir <35 % no kopējās saistīšanās. Tomēr minētā procentuālā attiecība dažkārt šo limitu var pārsniegt, ja testā tiek mērīts ļoti zems dpm pie zemākās radioaktīvi marķētā 17β-estradiola koncentrācijas.
|
Konkurējošās saistīšanās tests
|
45.
|
Pie pieaugošām nemarķēta 17β-estradiola koncentrācijām [3H]-17β-estradiolu pakāpeniski pārvieto no receptora tādā veidā, kas līdzinās viena domēna konkurējošajai saistīšanai.
|
|
46.
|
References estrogēna (t. i., 17β-estradiola) IC50 vērtībai jābūt aptuveni vienādai ar [3H]-17β-estradiola molāro koncentrāciju, kam pieskaitīta piesātinājuma–saistīšanās testā noteiktā diasociācijas konstante (Kd).
|
|
47.
|
Visās testa izpildes reizēs kopējās specifiskās saistīšanās vērtībai pastāvīgi jābūt pieņemamības diapazonā (20 ± 5 %), pie nosacījuma, ka kopējās pievienotās radioaktivitātes vidējā mērītā koncentrācija katrā iedobē ir 1 nM. Ir pieļaujamas atsevišķas nelielas novirzes no minētā diapazona, tomēr, ja izpildes reizēs pastāvīgi iegūst rezultātus ārpus minētā diapazona vai ja konkrētā izpildes reizē novirze ir ļoti būtiska, ir jāpielāgo proteīnu koncentrācija.
|
|
48.
|
Šķīdinātājs nedrīkst mainīt testa jutīgumu vai reproducējamību. Šķīdinātāja kontroles rezultātus (TB iedobes) salīdzina ar buferšķīduma kontroli, lai verificētu, ka izmantotais šķīdinātājs neietekmē testa sistēmu. Ja šķīdinātājs neietekmē testa gaitu, TB un buferšķīduma kontrolēm jābūt salīdzināmām.
|
|
49.
|
Testējot ar koncentrācijām, kas nepārsniedz 10-3 M (OTES) vai 10-4 M (DBP), nesaisteklim nevajadzētu no hrERα pārvietot vairāk nekā 25 % [3H]-17β-estradiola.
|
|
50.
|
Izmantojot FW hrER saistīšanās testa validēšanas pētījuma datus (2. atsauces N pielikums), tika izstrādāti references estrogēna un divu vāju saistekļu (piem., noretinodrels, noretindrons) veiktspējas kritēriji. Ir sniegti 95 % ticamības intervāli vidējām vērtībām (n) +/- SN; tās tika iegūtas no visiem kontroles testiem, ko izpildīja laboratorijas, kuras piedalījās validēšanas pētījumā. 95 % ticamības intervālus aprēķināja references estrogēna un vāju saistekļu līknes pielāgošanas parametriem (t. i., augšējam un apakšējam punktam, Hilla virziena koeficientam un log IC50) un vāju saistekļu log10
RBA attiecībā pret references estrogēnu; tos izmanto par pozitīvās kontroles veiktspējas kritērijiem. 1. tabulā sniegti līknes pielāgošanas parametru paredzamie diapazoni, kurus var izmantot kā veiktspējas kritērijus. Praksē IC50 diapazons var būt nedaudz atšķirīgs atkarībā no receptora preparāta Kd un liganda koncentrācijas.
|
|
51.
|
Testējamo ķimikāliju līknes pielāgošanas parametriem veiktspējas kritēriji izstrādāti netika, jo pastāv pārāk plašs esošu potenciāli testējamu ķimikāliju spektrs un potenciālās afinitātes un rezultātu mainīgums (piem., pilna līkne, daļēja līkne, neatbilstība līknei). Tomēr katrā testēšanas reizē un ar katru testējamo ķimikāliju iegūtos rezultātus būtu jāizvērtē speciālistam. Testā jāizmanto pietiekams testējamās ķimikālijas koncentrāciju diapazons, lai skaidri definētu konkurējošās saistīšanās līknes augšējo punktu (t. i., 90–100 % saistīšanās). Rezultātu mainībai starp katras testējamās ķimikālijas koncentrācijas replikātiem, kā arī starp trim neatkarīgajām izpildes reizēm jābūt saprātīgi pieļaujamās robežās un zinātniski pamatojamai. Izpildot kontroles testus attiecībā uz katru testējamo ķimikāliju, mērījumiem būtu jātuvojas veiktspējas parametriem, kas paziņoti attiecībā uz šo FW testu, un tiem jābūt saskanīgiem ar attiecīgās laboratorijas vēsturiskajiem kontroldatiem.
|
DATU ANALĪZE
Piesātinājuma–saistīšanās tests
|
52.
|
Tiek mērīta gan kopējā, gan nespecifiskā saistīšanās. No šim vērtībām, nespecifisko saistīšanos atņemot no kopējās saistīšanās, tiek aprēķināta pieaugošu [3H]-17β-estradiola koncentrāciju specifiskā saistīšanās līdzsvara apstākļos. Grafikā, kurā specifisko saistīšanos attēlo attiecībā pret [3H]-17β-estradiola koncentrāciju, attiecībā uz maksimālo specifisko saistīšanos ir jāsasniedz atslābuma periods, kas norāda uz hrERα piesātinājumu ar [3H]-17β-estradiolu. Turklāt datu analīzē būtu jākonstatē [3H]-17β- estradiola saistīšanās ar vienu augstas afinitātes saistīšanās domēnu. Piesātinājuma–saistīšanās līknē attēlo nespecifisko, kopējo un specifisko saistīšanos. Šo datu turpmākai analīzei izmanto nelineārās regresijas analīzi (piem., BioSoft; McPherson, 1985; Motulsky, 1995), un galīgos datus attēlo ar Skačārda diagrammu.
|
|
53.
|
Analizējot datus, Bmax un Kd nosaka no kopējās saistīšanās datiem vien, izmantojot pieņēmumu, ka nespecifiskā saistīšanās ir lineāra, ja vien nav pamatota iemesla izmantot citu metodi. Turklāt, meklējot labāko atbilstību līknei, izmanto robustu regresiju, ja vien nav pamatota iemesla rīkoties citādi. Pārskatā norāda izvēlēto robustas regresijas metodi. Nosakot Bmax un Kd no piesātinājuma–saistīšanās datiem, vienmēr jāizmanto liganda noplicināšanās korekcija (piem., Swillens (1995) metode).
|
Konkurējošās saistīšanās tests
|
54.
|
Konkurējošās saistīšanās līkni grafiski attēlo kā specifisko [3H]-17β-estradiola saistīšanos attiecībā pret konkurenta koncentrāciju (log10 vienībās). Testējamās ķimikālijas koncentrācija, kas inhibē 50 % [3H]17β-estradiola maksimālās specifiskās saistīšanās, ir IC50 vērtība.
|
|
55.
|
Pozitīvo kontroļu (t. i., references estrogēna un vāja saistekļa) log (IC50) vērtību aplēses nosaka, izmantojot atbilstošu nelineāru līknes piemeklēšanas programmatūru, lai panāktu atbilstību Hilla vienādojumam ar četriem parametriem (piem., BioSoft; McPherson, 1985; Motulsky, 1995). Veicot pielāgošanu līknei, augšējo un apakšējo punktu, virziena koeficientu un log (IC50) parasti neierobežo. Meklējot labāko atbilstību līknei, izmanto robustu regresiju, ja vien nav pamatota iemesla rīkoties citādi. Liganda noplicināšanās korekciju neizmanto. Pēc sākotnējās analīzes katru saistīšanās līkni pārskata, lai nodrošinātu pienācīgu atbilstību paraugam. Vāja saistekļa relatīvo afinitāti (RBA) aprēķina kā procentuālu vērtību no vāja saistekļa log (IC50) attiecībā pret 17β-estradiola log (IC50). Ar pozitīvajām kontrolēm un nesaistekļu kontrolēm iegūtos rezultātus izvērtē, sekojot testa veikšanas norādēm 2. papildinājuma 45.–50. punktā.
|
|
56.
|
Datus par visam testējamajām ķimikālijām būtu jāanalizē, izmantojot pakāpenisku pieeju, kas nodrošina datu pienācīgu analīzi un to, ka katra konkurējošās saistīšanās līkne tiek klasificēta pareizi. Pirms katras testa veikšanas reizes testējamās ķimikālijas datus sākotnēji analizē standartizētā veidā, identiski tam, kā tiek analizēti references estrogēns un vāja saistekļa kontroles (sk. 55. punktu iepriekš tekstā). Pēc tam katrā testa veikšanas reizē tehniski pārskata, vai līkne atbilst parametriem, un vizuāli nosaka, cik precīzi dati atbilst ģenerētajai konkurējošās saistīšanās līknei. Ja šīs tehniskās pārbaudes laikā novēro, ka [3H]-17β-estradiola specifiskā saistīšanās atkarībā no koncentrācijas procentuāli samazinās, rezultātu mainība tehnisko replikātu starpā pie katras ķīmiskās koncentrācijas ir neliela un līknes atbilstība parametriem trīs testa veikšanas reizēs ir konsekventa, tas ir pietiekami uzticams rādītājs, ka tests un datu analīze ir veikti pareizi.
|
Datu interpretācija
|
57.
|
Pie nosacījuma, ka ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par saistīgu ar hrERα, ja saistīšanās līkne ir pielāgojama un zemākais atbildreakcijas līknes punkts datu diapazonā ir mazāks par 50 % (1. attēls).
|
|
58.
|
Pie nosacījuma, ka ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par nesaistīgu ar hrERα, ja:
|
—
|
saistīšanās līkne ir pielāgojama un pielāgotās atbildreakcijas līknes zemākais punkts datu diapazonā ir lielāks par 75 % vai
|
|
—
|
saistīšanās līkni pielāgot nevar un visu koncentrāciju grupu vidējā zemākā nenoapaļotā procentuālā saistīšanās pēc datiem pārsniedz 75 %.
|
|
|
59.
|
Testējamās ķimikālijas klasifikāciju uzskata par neskaidru, ja nav izpildīts neviens no iepriekšminētajiem nosacījumiem (t. i., zemākais pielāgotās atbildreakcijas līknes punkts ir robežās no 76 – 51 %).
|
7. tabula.
Kritēriji, pēc kuriem klasificē testējamās ķimikālijas, izmantojot konkurējošās saistīšanās līkni.
|
Klasifikācija
|
Kritēriji
|
|
Saisteklisa
|
Saistīšanās līkni var pielāgot.
Zemākais atbildreakcijas līknes punkts datu diapazonā ir mazāks par 50 %.
|
|
Nesaisteklisb
|
Ja saistīšanās līkni var pielāgot,
pielāgotās atbildreakcijas līknes zemākais punkts datu diapazonā ir virs 75 %.
Ja saistīšanās līkni nevar pielāgot,
visu koncentrāciju grupu vidējā zemākā nenoapaļotā procentuālā saistīšanās pēc datiem pārsniedz 75 %.
|
|
Neskaidrac
|
Jebkura testa izpildes reize, kas neuzrāda testējamās ķimikālijas saistīgumu vai nesaistīgumu
(piem., pielāgotās atbildreakcijas līknes zemākais punkts ir robežās no 76–51 %).
|
1. attēls.
Piemēri, kā klasificē testējamās ķimikālijas, izmantojot konkurējošās saistīšanās līkni.
|
60.
|
Laboratorijā attiecībā uz katru testējamo ķimikāliju testu veic vairākas izpildes reizes, un katrai izpildes reizei atkarībā no rezultāta piešķir ciparisku vērtību, no kuras pēc tam aprēķina vidējo vērtību atbilstoši 8. tabulā sniegtajam paraugam. Vidējos rezultātus, kas apkopoti konkrētajā laboratorijā, salīdzina ar gaidīto katras testējamās ķimikālijas klasifikāciju.
|
8. tabula.
Metode, pēc kuras klasificē testējamo ķimikāliju, izmantojot vairākas testēšanas reizes vienā laboratorijā.
|
Katrai izpildes reizei piešķir ciparisku vērtību:
|
|
|
Klasifikācija
|
Cipariska vērtība
|
|
Saisteklis
|
2
|
|
Neskaidrs
|
1
|
|
Nesaisteklis
|
0
|
|
Izpildes reizēs iegūto ciparisko vērtību vidējās vērtības klasificēšana:
|
|
|
Klasifikācija
|
Cipariska vērtība
|
|
Saisteklis
|
Vidēji ≥ 1,5
|
|
Neskaidrs
|
0,5 ≤ Vidējā vērtība < 1,5
|
|
Nesaisteklis
|
Vidēji < 0,5
|
TESTĒŠANAS PĀRSKATS
|
61.
|
Sk. šīs testēšanas metodes sadaļas “
HrER SAISTĪŠANĀS TESTA KOMPONENTI” 24. punktu.
|
2.1. papildinājums
TERMINU SARAKSTS
[3H]E2
: 17β-estradiols, radioaktīvi marķēts ar tritiju.
DCC
: ar dekstrānu pārklāta ogle.
E2
: nemarķēts 17β-estradiols (inerts).
Testa buferšķīdums:: 10 mM tris, 10 mg liellopu seruma albumīna/ml, 2 mM DTT, 10 % glicerīna, 0,2 mM leupeptīna (pH 7,5).
hrERα
: cilvēka rekombinantais estrogēnreceptors alfa.
Replikāts
: viena no vairākām vienāda satura un vienādas koncentrācijas iedobēm, kuras paralēli testē vienā testēšanas reizē. Pēc šī protokola katru testējamās ķimikālijas koncentrāciju testē ar trim replikātiem (triplikātiem); proti, pie katras testējamās ķimikālijas koncentrācijas ir trīs replikāti, kurus testē vienlaicīgi.
Izpildes reize
: viss paralēli testēto miktotitrēšanas plates testa iedobju kopums, kas nodrošina visu informāciju, kura vajadzīga, lai raksturotu testējamās ķimikālijas saistīšanos ar hrERα, (t. i., kopējais testa iedobei pievienotais [3H]-17β-estradiols, maksimālā [3H]-17β-estradiola saistīšanās ar hrERα, nespecifiskā saistīšanās un kopējā saistīšanās pie dažādām testējamās ķimikālijas koncentrācijām). Izpildes reize var nozīmēt pat tikai vienas testēšanas iedobes (t. i., replikāta) testēšanu pie katras koncentrācijas, tomēr šajā konkrētajā gadījumā protokols pieprasa testēt ar triplikātiem, un viena izpildes reize nozīmē trīs testēšanas iedobes pie katras koncentrācijas. Papildus minētajam šis protokols prasa ar katru ķimikāliju veikt trīs neatkarīgas (proti, neveicot paralēli) izpildes reizes.
2.2. papildinājums
TIPISKS [3H]-17Β-ESTRADIOLA PIESĀTINĀJUMA TESTS AR TRIJĀM REPLIKĀTA IEDOBĒM
|
Tipisks [3H]-17β-estradiola piesātinājuma tests ar trijām replikāta iedobēm
|
|
Pozīcija
|
Replikāts
|
Iedobes tipa kods
|
Karsta E2 sākotnēja koncentrācija (nM)
|
Karsta E2 tilpums (μl)
|
Karsta E2 beigu koncentrācija (nM)
|
Auksta E2 sākotnēja koncentrācija (μM)
|
Auksta E2 tilpums (μl)
|
Auksta E2 beigu koncentrācija (μM)
|
Buferšķīduma tilpums (μl)
|
Receptora tilpums (μl)
|
Kopējais tilpums iedobēs
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Karsta
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Karsta
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Karsta
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Karsta
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Karsta
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Karsta
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Karsta
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Karsta
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Karsta
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Karsta
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Karsta
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Karsta
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Karsta
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Karsta
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Karsta
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Karsta
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Karsta
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Karsta
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Karsta
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Karsta
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Karsta
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Karsta
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Karsta
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Karsta
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
Piezīme: “karstās” iedobes inkubācijas laikā ir tukšas. 40 μl pievieno tikai scintilāciju skaitīšanai.
|
2.3. papildinājums
KONKURĒJOŠĀS SAISTĪŠANĀS TESTA IEDOBJU IZKĀRTOJUMS
|
Plate
|
Pozīcija
|
Replikāts
|
Iedobes tips
|
Iedobes kods
|
Koncentrācijas kods
|
Konkurenta sākotnējā koncentrācija (M)
|
hrER izejšķīdums (μl)
|
Buferšķīduma tilpums (μl)
|
Iezīmētā elementa (karsta E2) tilpums (μL)
|
Tilpums atšķaidījuma platē (μL)
|
Beigu tilpums (μl)
|
Konkurenta beigu koncentrācija (M)
|
|
S
|
A1
|
1
|
kopējā saistīšanās
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
kopējā saistīšanās
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
kopējā saistīšanās
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
kopējā saistīšanās
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
kopējā saistīšanās
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
kopējā saistīšanās
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
auksts E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
auksts E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
auksts E2
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
auksts E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
auksts E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
auksts E2
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
auksts E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
auksts E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
auksts E2
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
auksts E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
auksts E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
auksts E2
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
auksts E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
auksts E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
auksts E2
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
auksts E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
auksts E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
auksts E2
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
auksts E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
auksts E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
auksts E2
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
tukša
|
tukša
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
tukša
|
tukša
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
tukša
|
tukša
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
noretinodrels
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
noretinodrels
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
noretinodrels
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
noretinodrels
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
noretinodrels
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
noretinodrels
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
noretinodrels
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
noretinodrels
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
noretinodrels
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
noretinodrels
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
noretinodrels
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
noretinodrels
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
noretinodrels
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
noretinodrels
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
noretinodrels
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
noretinodrels
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
noretinodrels
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
noretinodrels
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
noretinodrels
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
noretinodrels
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
noretinodrels
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
noretinodrels
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
noretinodrels
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
noretinodrels
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
karsta
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
buferšķīduma kontrole
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
Piezīme: “karstās” iedobes inkubācijas laikā ir tukšas. 40 μl pievieno tikai scintilāciju skaitīšanai.
|
Konkurējošās saistīšanās testa iedobju izkārtojums
|
|
Plate
|
Pozīcija
|
Replikāts
|
Iedobes tips
|
Iedobes kods
|
Koncentrācijas kods
|
Konkurenta sākotnējā koncentrācija (M)
|
hrER izejšķīdums (μL)
|
Buferšķīduma tilpums (μL)
|
Iezīmētā elementa (karsta E2) tilpums (μL)
|
Tilpums atšķaidījuma platē (μL)
|
Beigu tilpums (μl)
|
Konkurenta beigu koncentrācija (M)
|
|
P1
|
A1
|
1
|
kopējā saistīšanās
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
kopējā saistīšanās
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
kopējā saistīšanās
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
kopējā saistīšanās
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
kopējā saistīšanās
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
kopējā saistīšanās
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
testējamā ķimikālija 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B2
|
2
|
testējamā ķimikālija 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B3
|
3
|
testējamā ķimikālija 1
|
TC1
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
B4
|
1
|
testējamā ķimikālija 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B5
|
2
|
testējamā ķimikālija 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B6
|
3
|
testējamā ķimikālija 1
|
TC1
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
B7
|
1
|
testējamā ķimikālija 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
testējamā ķimikālija 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
testējamā ķimikālija 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
testējamā ķimikālija 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
testējamā ķimikālija 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
testējamā ķimikālija 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
testējamā ķimikālija 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
testējamā ķimikālija 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
testējamā ķimikālija 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
testējamā ķimikālija 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
testējamā ķimikālija 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
testējamā ķimikālija 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
testējamā ķimikālija 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
testējamā ķimikālija 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
testējamā ķimikālija 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
testējamā ķimikālija 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
testējamā ķimikālija 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
testējamā ķimikālija 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
testējamā ķimikālija 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D2
|
2
|
testējamā ķimikālija 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D3
|
3
|
testējamā ķimikālija 2
|
TC2
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
D4
|
1
|
testējamā ķimikālija 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D5
|
2
|
testējamā ķimikālija 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D6
|
3
|
testējamā ķimikālija 2
|
TC2
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
D7
|
1
|
testējamā ķimikālija 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
testējamā ķimikālija 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
testējamā ķimikālija 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
testējamā ķimikālija 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
testējamā ķimikālija 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
testējamā ķimikālija 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
testējamā ķimikālija 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
testējamā ķimikālija 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
testējamā ķimikālija 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
testējamā ķimikālija 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
testējamā ķimikālija 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
testējamā ķimikālija 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
testējamā ķimikālija 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
testējamā ķimikālija 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
testējamā ķimikālija 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
testējamā ķimikālija 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
testējamā ķimikālija 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
testējamā ķimikālija 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
testējamā ķimikālija 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F2
|
2
|
testējamā ķimikālija 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F3
|
3
|
testējamā ķimikālija 3
|
TC3
|
1
|
2,00E-03
|
40
|
0
|
40
|
80
|
160
|
1,0E-03
|
|
P1
|
F4
|
1
|
testējamā ķimikālija 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F5
|
2
|
testējamā ķimikālija 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F6
|
3
|
testējamā ķimikālija 3
|
TC3
|
2
|
2,00E-04
|
40
|
0
|
40
|
80
|
160
|
1,0E-04
|
|
P1
|
F7
|
1
|
testējamā ķimikālija 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
testējamā ķimikālija 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
testējamā ķimikālija 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
testējamā ķimikālija 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
testējamā ķimikālija 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
testējamā ķimikālija 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
testējamā ķimikālija 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
testējamā ķimikālija 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
testējamā ķimikālija 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
testējamā ķimikālija 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
testējamā ķimikālija 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
testējamā ķimikālija 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
testējamā ķimikālija 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
testējamā ķimikālija 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
testējamā ķimikālija 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
testējamā ķimikālija 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
testējamā ķimikālija 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
testējamā ķimikālija 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
noretinodrels
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
noretinodrels
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
noretinodrels
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
noretinodrels
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
noretinodrels
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
noretinodrels
|
NE
|
|
1,00E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
auksts E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
auksts E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
auksts E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
auksts E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
auksts E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
auksts E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
3. papildinājums
ĶĪMISKĀS IZVĒRTĒŠANAS UN PĒTNIECĪBAS INSTITŪTA (CERI) ESTROGĒNRECEPTORU SAISTĪŠANĀS IN VITRO TESTS, KURĀ IZMANTO CILVĒKA REKOMBINANTO ERα LIGANDA SAISTĪŠANĀS DOMĒNA PROTEĪNU
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI (SK. ARĪ VISPĀRĪGO IEVADU)
|
1.
|
Šis estrogēnreceptora (ERα) piesātinājuma un konkurējošās saistīšanās in vitro tests izmanto cilvēka ERα liganda saistīšanās domēnu (LBD). Šo proteīnu modeli izstrādājis Ķīmiskās izvērtēšanas un pētniecības institūts (CERI) Japānā, un tas eksistē kā glutationa-S-transferāzes (GST) jauktais proteīns, kuru ekpresē E. coli. CERI protokolam veikts starptautisks daudzlaboratoriju validēšanas pētījums (2), kas pierādīja testa relevanci un ticamību paredzētajam nolūkam.
|
|
2.
|
Šis tests ir skrīninga procedūra, ar ko identificē vielas, kas spēj saistīties ar hrERα. To izmanto, lai noteiktu testējamās ķimikālijas spēju konkurēt ar 17β-estradiolu attiecībā uz saistīšanos ar hrERα-LBD. Kvantitatīvie testa rezultāti var ietvert IC50 (mērs, kas rāda testējamās ķimikālijas koncentrāciju, kas vajadzīga, lai no hrERα pārvietotu pusi no [3H]-17β-estradiola) un testējamo ķimikāliju relatīvo afinitāti attiecībā uz hrERα salīdzinājumā ar 17β-estradiolu. Ķīmiskā skrīninga vajadzībām pieņemami kvalitatīvie testa rezultāti var ietvert testējamo ķimikāliju klasificēšanu saistīgās vai nesaistīgās ar hrERα vai tādās, kuru saistīgums ir neskaidrs, uz to kritēriju pamata, kas aprakstīti attiecībā uz saistīšanās līknēm.
|
|
3.
|
Testā izmanto radioaktīvi marķētu ligandu, tāpēc laboratorijai būs vajadzīga radioaktīvo materiālu licence. Visas procedūras ar radioaktīvajiem izotopiem un bīstamām ķimikālijām jāveic, ievērojot nacionālajos tiesību aktos paredzētos noteikumus un procedūras.
|
|
4.
|
Pirms šā testa izmantošanas regulatīviem mērķiem jāizlasa sadaļas “VISPĀRĪGAIS IEVADS” un “
HrER SAISTĪŠANĀS TESTA KOMPONENTI”. Šajā testēšanas vadlīnijā izmantotās definīcijas un saīsinājumi ir aprakstīti 1. papildinājumā.
|
TESTA PRINCIPI (SK. ARĪ VISPĀRĪGO IEVADU)
|
5.
|
HrERα saistīšanās tests mēra radioaktīvi iezīmēta liganda ([3H]17β-estradiola) spēju saistīties ar ER arvien pieaugošu testējamās ķimikālijas (t.i. konkurenta) koncentrāciju klātbūtnē. Testējamās ķimikālijas, kam piemīt augsta ER afinitāte, konkurē ar radioaktīvi iezīmēto ligandu pie zemākas koncentrācijas nekā ķimikālijas ar zemāku receptora afinitāti.
|
|
6.
|
Šis tests sastāv no diviem svarīgākajiem komponentiem: piesātinājuma–saistīšanās eksperimenta, kas raksturo receptora–liganda mijiedarbības parametrus, un konkurējošās saistīšanās eksperimenta, kas attiecībā uz saistīšanos ar ER raksturo konkurenci starp testējamo ķimikāliju un radioaktīvi marķēto ligandu.
|
|
7.
|
Piesātinājuma–saistīšanās eksperimenta mērķis ir raksturot konkrētas receptoru partijas afinitāti un konkurējošās saistīšanās eksperimentā — to skaitu preparātā. Piesātinājuma–saistīšanās eksperiments līdzsvara apstākļos mēra fiksētas estrogēnreceptora koncentrācijas afinitāti tā dabīgajam ligandam (to izsaka disociācijas konstante, Kd) un aktīvo receptoru domēnu koncentrāciju (Bmax).
|
|
8.
|
Konkurējošās saistīšanās eksperiments mēra tādas vielas afinitāti, kura attiecībā uz saistīšanos ar ER konkurē ar [3H]17β-estradiolu. Afinitāti kvantitatīvi izsaka kā testējamās ķimikālijas koncentrāciju, kas līdzsvara apstākļos inhibē 50 % [3H]17β-estradiola specifiskās saistīšanās (apzīmē kā “50 % inhibējošo koncentrāciju” vai IC50). To var izvērtēt arī, izmantojot relatīvo afinitāti (RBA, relatīvā salīdzinājumā ar estradiola IC50, ko atsevišķi mēra tajā pašā izpildes reizē). Konkurējošās saistīšanās eksperiments mēra fiksētas koncentrācijas [3H]17β-estradiola spēju saistīties ar ER plaša testējamās ķimikālijas koncentrāciju diapazona (astoņas intensitātes pakāpes) klātbūtnē. Pēc tam, ja iespējams, datus pārveido Hilla vienādojuma (A. Hills, 1910) formā, kas apraksta radioaktīvi marķētā liganda pārvietošanu, ko izraisa viena domēna konkurējošais saisteklis. Radioaktīvi marķētā estradiola pārvietošanas pakāpe līdzsvara apstākļos tiek izmantota, lai testējamo ķimikāliju raksturotu kā saistīgu, nesaistīgu vai tādu, kam ir neskaidra reakcija.
|
PROCEDŪRA
Pieņemamas hrERα proteīna veiktspējas pierādīšana
|
9.
|
Pirms piesātinājuma un konkurējošās saistīšanās testu ieviešanas laboratorijas ikdienas praksē attiecībā uz katru jaunu hrERα partiju ir jāpierāda to veiktspējas pareizība laboratorijā, kurā to izmantos. Veiktspēju pierāda ar divu etapu procedūru. Proti,
|
—
|
veic [3H]-17β-estradiola saistīšanās piesātinājuma testu, lai pierādītu hrERα specifiskumu un piesātinājumu. Ar iegūto datu nelineāras regresijas analīzi (piem., BioSoft; McPherson, 1985; Motulsky, 1995) un no tiem izveidotu Skačārda diagrammu dokumentē hrERα spēju saistīt [3H]-17β-estradiolu (Kd) un katras partijas receptoru skaitu (Bmax);
|
|
—
|
veic konkurējošās saistīšanas testu, izmantojot kontroles vielas (references estrogēnu (17β-estradiolu), vāji saistīgu vielu (piem., noretinodrelu vai noretindronu) un nesaistīgu vielu (oktiltrietoksisilānu, apzīmē ar “OTES”). Katra laboratorija izveido un uztur vēsturisku datubāzi, kurā dokumentē IC50 konsekventumu un citas būtiskas references estrogēna un vāja saistekļa vērtības, kas iegūtas dažādos eksperimentos un ar atšķirīgām hrERα partijām. Turklāt ar kontroles vielām iegūto konkurējošās saistīšanās līkņu parametriem jābūt 95 % ticamības intervāla robežās (sk. 1. tabulu); tas izstrādāts, izmantojot to laboratoriju datus, kuras piedalījās šā testa validēšanas pētījumā (2).
|
1. tabula
References estrogēna un vāja saistekļa veiktspējas kritēriji, kas izstrādāti CERI hrER saistīšanās testam.
|
Viela
|
Parametrs
|
Vidējā vērtība (11)
|
Standartnovirze (n)
|
95 % ticamības intervāli (12)
|
|
apakšējā robeža
|
augšējā robeža
|
|
17β-estradiols
|
Augšējais
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Apakšējais
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hilla virziena koeficients
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretinodrels
|
Augšējais
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Apakšējais
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hilla virziena koeficients
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindrons (13)
|
Augšējais
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Apakšējais
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hilla virziena koeficients
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Laboratorijas kvalifikācijas pierādīšana
|
10.
|
Sk. šīs testēšanas metodes sadaļas “
HrER SAISTĪŠANĀS TESTA KOMPONENTI” 17. un 18. punktu un 2. tabulu. Katrā (piesātinājuma un konkurējošās saistīšanās) testā īsteno trīs neatkarīgas testa izpildes reizes (t. i., ar jauniem receptora atšķaidījumiem, ķimikālijām un reaģentiem), tās veic dažādās dienās un katrā izpildes reizē izmanto trīs replikātus.
|
Receptora (hrERα) koncentrācijas noteikšana
|
11.
|
Aktīvā receptora koncentrācija katrā atsevišķā partijā un atkarībā no uzglabāšanas apstākļiem var būt nedaudz atšķirīga. Tāpēc ir jānosaka no piegādātāja saņemtā aktīvā receptora koncentrācija. Tas ļaus panākt atbilstošu aktīvā receptora koncentrāciju testa veikšanas laikā.
|
|
12.
|
Apstākļos, kas atbilst konkurējošās saistīšanās apstākļiem (proti, ar 0,5 nM [3H]-estradiola), nominālas receptora koncentrācijas — 0,1, 0,2, 0,4 un 0,6 nM — inkubē bez estradiola (kopējā saistīšanās) un ar 1 μM nemarķēta estradiola (nespecifiskā saistīšanās). Grafiski attēlo sakarību starp specifisko saistīšanos (starpību starp kopējo saistīšanos un nespecifisko saistīšanos) un receptora nominālo koncentrāciju. Receptora koncentrācija, pie kuras iegūst tādas specifiskās saistīšanās vērtības, kas atbilst 40 % pievienotā radioaktīvā marķējuma, ļauj noteikt atbilstošo receptora koncentrāciju, kuru attiecīgi izmanto piesātinājuma un konkurējošās saistīšanās eksperimentiem. Samērā bieži galīgā hrER koncentrācija, kas atbilst šim nosacījumam, ir 0,2 nM.
|
|
13.
|
Ja pēc atkārtotiem mēģinājumiem neizdodas sasniegt 40 % kritēriju, eksperimenta uzstādījumus pārbauda attiecībā uz iespējamām kļūdām. Nespēja sasniegt 40 % kritēriju var norādīt, ka rekombinantajā partijā ir ļoti maz aktīvā receptora; šādos gadījumos jāapsver iespēja izmantot citu receptora partiju.
|
Piesātinājuma tests
|
14.
|
[3H]17β-estradiola astoņu augoša stipruma koncentrāciju triplikātus vērtē, pie šādiem trim turpmāk uzskaitītajiem nosacījumiem (sk. 2. tabulu):
|
a.
|
testu veic bez nemarķēta 17β-estradiola un ER klātbūtnē. Šādi, mērot radioaktivitāti iedobēs, kurās ir tikai [3H]17β-estradiols, nosaka kopējo saistīšanos;
|
|
b.
|
testu veic ar nemarķēta 17β-estradiola koncentrāciju, kas 2000 reižu pārsniedz marķētā 17β-estradiola koncentrāciju, un ER klātbūtnē. Šāda nosacījuma mērķis ir piesātināt aktīvos saistīšanās domēnus ar nemarķētu 17β-estradiolu un, mērot radioaktivitāti iedobēs, noteikt nespecifisko saistīšanos. Visu pārējo karsto estradiolu, kas spēj saistīties ar receptoru, uzskata par nespecifiskā domēnā saistīgu, jo aukstajam estradiolam jābūt tik lielā koncentrācijā, ka tas saistās ar visiem uz receptora pieejamajiem specifiskajiem domēniem;
|
|
c.
|
testu veic bez nemarķēta 17β-estradiola un bez ER (kopējās radioaktivitātes noteikšana).
|
[3H]-17β-estradiola un nemarķēta 17β-estradiola šķīdumu un hrERα sagatavošana
|
15.
|
Lai sagatavotu 40 nM [3H]-17β-estradiola šķīduma, ņem 1 μM izejšķīduma — [3H]-17β-estradiola šķīdums dimetilsulfoksīdā (DMSO) —, kam pievieno DMSO (iegūstot 200 nM) un pēc tam testa buferšķīdumu istabas temperatūrā (iegūstot 40 nM). No šiem 40 nM šķīduma, izmantojot testa buferšķīdumu istabas temperatūrā, sagatavo virkni [3H]-17β-estradiola atšķaidījumu koncentrācijās no 0,313 nM līdz 40 nM (sk. paraugu 2. tabulas 12. ailē). Galīgā testa koncentrācijas diapazonā no 0,0313 līdz 4,0 nM iegūst, 10 μl šādi sagatavoto šķīdumu iepilinot attiecīgajās 96 iedobju mikrotitrēšanas plates iedobēs (sk. 2. un 3. tabulu). Testa buferšķīduma sagatavošana un [3H]-17β-estradiola oriģinālā izejšķīduma aprēķināšanas metode, balstoties uz specifisko aktivitāti, kā arī atšķaidījumu sagatavošana un koncentrāciju noteikšana ir sīki izskaidrota CERI testa protokolā (2).
|
|
16.
|
Atšķaidījumus no nemarķēta 17β-estradiola šķīduma sagatavo, 1 nM 17β-estradiola izejšķīduma pievienojot testa buferšķīdumu, lai iegūtu astoņas augoša stipruma koncentrācijas, kas sākotnēji ir diapazonā no 0,625 μM līdz 80 μM. Galīgā testa koncentrācijas diapazonā 0,0625–8 μM iegūst, 10 μl šādi sagatavoto šķīdumu iepilinot attiecīgajās 96 iedobju mikrotitrēšanas plates iedobēs, kurās paredzēts mērīt nespecifisko saistīšanos (sk. 2. un 3. tabulu). Nemarķēta 17β-estradiola atšķaidījumu sagatavošana ir sīki izskaidrota CERI testa protokolā (2).
|
|
17.
|
Izmanto receptora koncentrāciju, pie kuras specifiskā saistīšanās ir 40±10 % (sk. 12.–13. punktu). HrERα šķīdumu sagatavo ar ledusaukstu testa buferšķīdumu un tieši pirms izmantošanas, t. i., pēc tam, kad ir jau sagatavotas visas pārējās iedobes, kas paredzētas kopējās saistīšanās un nespecifiskās saistīšanās noteikšanai un tikai karstajam ligandam paredzētās iedobes.
|
|
18.
|
96 iedobju mikrotitrēšanas plates sagatavo saskaņā ar 2. tabulas norādījumiem, katrai [3H]-17β-estradiola koncentrācijai sagatavojot trīs replikātus. 3. tabulā sniegti testa protokolā paredzētie [3H]-17β-estradiola, nemarķēta 17β-estradiola, buferšķīduma un receptora tilpumi.
2. tabula
Piesātinājuma–saistīšanās testa mikrotitrēšanas plates izkārtojums
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)*
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
TB mērīšanai
|
NSB mērīšanai
|
Tikai karstā liganda noteikšanai
|
|
Nemarķēta E2 atšķaidījumi plates 4.–6. kolonnai
|
[3H]E2 atšķaidījumi plates 1.–9. kolonnai
|
|
A
|
0,0313 nM [ 3H] E2 + ER
|
0,0313 nM [3H] E2 + 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [ H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
kopējā saistīšanās
|
|
NSB
|
:
|
nespecifiskā saistīšanās
|
|
[3H] E2
|
:
|
[3H]17β-estradiols
|
|
E2
|
:
|
nemarķēts 17β-estradiols
|
|
3. tabula
Reaģenta tilpumi piesātinājuma testa mikrotitrēšanas platē
|
Ailes numurs
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Sagatavošanas soļi
|
TB iedobes
|
NSB iedobes
|
Tikai karstais ligands
|
|
Komponentu tilpums minētajām reakcijas iedobēm un pievienošanas kārtība
|
Buferšķīdums
|
60 μl
|
50 μl
|
90 μl
|
|
nemarķēts E2 (2. tabulas 11. aile)
|
–
|
10 μl
|
—
|
|
[3H]E2 (2. tabulas 12. josla)
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
—
|
|
Kopējais reaģenta tilpums
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubācija
|
REAKCIJA PĒC 2 STUNDU INKUBĀCIJAS
|
Radioaktivitātes kvantificēšana uzreiz pēc sagatavošanas. Neinkubē
|
|
Apstrāde ar 0,4 % DCC
|
Jā
|
Jā
|
Nē
|
|
0,4 % DCC tilpums
|
100 μl
|
100 μl
|
—
|
|
Filtrēšana
|
Jā
|
Jā
|
Nē
|
|
DPM MĒRĪŠANA
|
|
Scintilācijas kokteilim pievienotais kvantificēšanas tilpums
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Kopējas saistīšanās un nespecifiskās saistīšanās noteikšanai paredzētās mikrotitrēšanas testa plates divas stundas inkubē istabas temperatūrā (22–28°C).
|
|
Ar hrERα saistītā [3H]-17β-estradiola mērīšana
|
20.
|
Pēc divu stundu inkubācijas perioda ar hrERα saistītais [3H]-17β-estradiols ir jāatdala no [3H]-17β-estradiola, iedobēm pievienojot 100 μl ledusaukstas 0,4 % DCC suspensijas. Pēc tam testa plates uz 10 minūtēm novieto uz ledus, un reakcijas maisījumu un DCC suspensiju filtrē, pārnesot uz mikrotitrēšanas plates filtra, kā rezultātā tiek atdalīts DCC. Pēc tam LSC mēģenēs esošajam scintilācijas šķīdumam pievieno 100 μl filtrāta, lai ar šķidruma scintilāciju skaitīšanas metodi noteiktu sabrukumu skaitu minūtē (dpm) uz mēģeni.
|
|
21.
|
Ja mikrotitrēšanas plates filtrs nav pieejams, alternatīvs veids DCC atdalīšanai ir centrifugēšana. Ņem 50 μl supernatanta, kas satur ar hrERα saistīto [3H]-17β-estradiolu, un izmanto to scintilāciju skaitīšanai; to dara ar īpašu piesardzību, lai nepieļautu iedobju kontamināciju, saskaroties DCC.
|
|
22.
|
Iedobes ar karsto ligandu vienu pašu izmanto, lai noteiktu testa iedobēm pievienotā [3H]-17β-estradiola sabrukumu skaitu minūtē (dpm). Radioaktivitāti kvantificē uzreiz pēc sagatavošanas. Šīs iedobes neinkubē un neapstrādā ar DCC suspensiju, bet to saturu pārnes tieši scintilācijas šķīdumā. Ar šiem mērījumi pierāda, cik daudz [3H]-17β-estradiola (izteikta dpm) pievienots katrā iedobju kopumā, kas demonstrē kopējo saistīšanos un nespecifisko saistīšanos.
|
Konkurējošās saistīšanās tests
|
23.
|
Konkurējošās saistīšanās tests mēra vienas konkrētas [3H]-17β- estradiola koncentrācijas saistīšanos arvien pieaugošu testējamās ķimikālijas koncentrāciju klātbūtnē. Katrā testa veikšanas reizē izmanto trīs paralēlus katras koncentrācijas replikātus. Papildus tam attiecībā uz katru testējamo ķimikāliju testu izpilda trīs reizes, un tas nenotiek paralēli, bet dažādos laikos. Testu veic vienā vai vairākās 96 iedobju mikrotitrēšanas platēs.
|
Kontroles
|
24.
|
Veicot testu, katrā eksperimentā jāiekļauj paralēlais šķīdinātājs un kontroles (t. i., references estrogēns, vājš saisteklis un nesaisteklis). Katrā testa izpildes reizē vienā platē ir jābūt atsauces estrogēna un kontroļu (t.i. vāja saistekļa un nesaistekļa) pilnai koncentrāciju līknei. Visās pārējās platēs ir jābūt: i) E2 un vāja saistekļa triplikātiem augstā koncentrācijā (maksimāla pārvietošana, t. i., gandrīz pilnīga radioaktīvi marķētā liganda pārvietošana) un vidējā koncentrācijā (aptuveni IC50); ii) šķīdinātāja kontroļu un nespecifiskās saistīšanās triplikātiem. Testa buferšķīduma, kontroļu, [3H]-17β-estradiola, hrERα un testējamo ķimikāliju šķīdumu sagatavošanas procedūras ir sīki aprakstītas CERI testa protokolā (2).
|
Šķīdinātāja kontrole
|
25.
|
Šķīdinātāja kontrole rāda, vai nenotiek šķīdinātāja mijiedarbība ar testa sistēmu, kā arī mēra kopējo saistīšanos (apzīmē ar “TB” — total binding). Ieteicamais šķīdinātājs ir DMSO. Ja testējamās ķimikālijas visaugstākā koncentrācija dimetilsulfoksīdā nešķīst, kā alternatīvu var izmantot etanolu. Galīgajā testā DMSO koncentrācija iedobēs ir 2,05 %, un to var palielināt līdz 2,5 % gadījumos, kad testējamā ķimikālija slikti šķīst. Nevajadzētu izmantot DMSO koncentrācijās, kas pārsniedz 2,5 %, jo augstākas koncentrācijas šķīdinātājs var izraisīt interferenci uz testu. Testējamajām ķimikālijām, kas nešķīst DMSO, bet ir šķīstošas etanolā, bez interferences uz testu var izmantot ne vairāk kā 2 % etanola šķīdumu.
|
Buferšķīduma kontrole
|
26.
|
Buferšķīduma kontrole (“BC”) nedrīkst saturēt ne šķīdinātāju, ne testējamo ķimikāliju, bet tā satur visus citus testa komponentus. Buferšķīduma kontroles rezultātus salīdzina ar šķīdinātāja kontroli, lai verificētu, ka izmantotais šķīdinātājs neietekmē testa sistēmu.
|
Stiprs saisteklis (references estrogēns)
|
27.
|
17β-estradiols (CAS 50-28-2) ir endogēnisks ligands ar augstu afinitāti (alfa apakštipa) ER. Katram hrERα konkurējošās saistīšanās testam sagatavo standarta līkni ar nemarķētu 17β-estradiolu, lai varētu novērtēt, kāda ir mainība laika gaitā, veicot testu tajā pašā laboratorijā. Sagatavo astoņus nemarķēta 17β-estradiola šķīdumus dimetilsulfoksīdā (DMSO) tā, lai testa iedobēs, kuras izmantos standarta līknei, šķīdumu galīgā koncentrācija būtu ar šādiem intervāliem: 10–6, 10–7, 10–8, 10–8,5, 10–9, 10–9,5, 10–10, 10–11 M. Nemarķēta 17β-estradiola (1 μM) augstākā koncentrācija kalpo kā nespecifiskās saistīšanās indikators. 4. tabulā šo koncentrāciju apzīmē ar kodu “NSB”, lai gan tā ir arī standarta līknes daļa.
|
Vājš saisteklis
|
28.
|
Lai demonstrētu katra eksperimenta jutīgumu un ļautu novērtēt, kāda ir rezultātu mainība, veicot testu laika gaitā, tajā iekļauj vāju saistekli (noretinodrelu (CAS 68-23-5) vai — alternatīvi — noretindronu (CAS 68-22-4). Sagatavo astoņus vāja saistekļa šķīdumus dimetilsulfoksīdā (DMSO) un testa buferšķīdumā ar šādām galīgajām koncentrācijām testa iedobēs: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 un 10–9 M.
|
Nesaisteklis
|
29.
|
Kā negatīvo kontroli (nesaistekli) izmanto oktiltrietoksisilānu (OTES, CAS 2943-75-1). Tas apstiprinās, ka tests spēs noteikt ķimikālijas, kas nesaistās ar hrERα. Sagatavo astoņus nesaistekļa šķīdumus dimetilsulfoksīdā (DMSO) un testa buferšķīdumā ar šādām galīgajām koncentrācijām testa iedobēs: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Alternatīvi par nesaistekļa kontroli var izmantot di-n-butilftalātu (DBP, CAS 84-72-2), bet to testē tikai līdz koncentrācijai 10–4M. Ir pierādīts, ka DBP maksimālā šķīdība testā ir 10–4M.
|
HrERα koncentrācija
|
30.
|
Izmanto receptora daudzumu, pie kura specifiskā saistīšanās ir 40±10 % (sk. 3. papildinājuma 12.-13. punktu). HrERα šķīdumu sagatavo, tieši pirms izmantošanas funkcionālo hrERα atšķaidot ar ledusaukstu testa buferšķīdumu.
|
[3H]17β-estradiols
|
31.
|
[3H]-17β-estradiola galīgā koncentrācija testa iedobēs ir 0,5 nM.
|
Testējamās ķimikālijas
|
32.
|
Vispirms ir jāveic šķīdības tests, lai noteiktu katras testējamās ķimikālijas šķīdības robežu un identificētu piemērotu koncentrācijas diapazonu, kas tiks izmantots testa protokolā. Sākotnēji katras testējamās ķimikālijas šķīdības robežu nosaka šķīdinātājā un pēc tam apstiprina testēšanas apstākļos. Galīgā testā izmantojamā koncentrācija nepārsniedz 1 mM. Diapazona noteikšanas testēšanā ietilpst šķīdinātāja kontrole; paralēli tai testē vismaz astoņus sērijveida logaritmiskus atšķaidījumus, sākot ar augstāko pieņemamo koncentrāciju (piem., 1 mM vai zemāku, balstoties uz šķīdības robežu), un vēro, kad šķīdums kļūst duļķains vai rada nogulsnes (sk. arī 3.papildinājuma 35. punktu). Kad testa koncentrāciju diapazons ir noteikts, testējamo ķimikāliju testē, izmantojot astoņas diapazona noteikšanas testā identificētās log koncentrācijas ar attiecīgajiem intervāliem. Otrajā un trešajā eksperimentā izmantotās koncentrācijas pielāgo pēc vajadzības, lai labāk raksturotu koncentrācijas–atbildreakcijas līkni.
|
|
33.
|
Testējamās ķimikālijas atšķaidījumus sagatavo atbilstošā šķīdinātājā (sk. 3.papildinājuma 25. punktu). Ja testējamās ķimikālijas augstākā koncentrācija nešķīst ne DMSO, ne etanolā un lielāka šķīdinātāja daudzuma pievienošana galīgajā testa stobriņā novestu pie pieņemamās robežas pārsniegšanas, augstāko koncentrāciju var samazināt par vienu intervālu uz leju. Šādā gadījumā sērijveida koncentrācijas var papildināt ar vienu papildkoncentrāciju amplitūdas apakšā. Visas pārējas sērijveida koncentrācijas paliek nemainīgas.
|
|
34.
|
Pievienojot testējamās ķimikālijas šķīdumus testa iedobēs, tos rūpīgi vēro, jo pievienošanas laikā var rasties nogulsnes. Datus par tām iedobēm, kurās konstatē nogulsnes, neņem vērā, pielāgojot datus līknei, un testa pārskatā norāda izslēgšanas iemeslu.
|
|
35.
|
Ja no citiem avotiem jau ir pieejama informācija par testējamās ķimikālijas log(IC50), atšķaidījumus var būt lietderīgi ģeometriski izkārtot ciešāk ap paredzamo log(IC50) (t. i., pa 0,5 log vienībām). Galīgajiem rezultātiem būtu jāatspoguļo pietiekama koncentrāciju izkliede abās log (IC50) pusēs, arī līknes augšējā un apakšējā punktā, un koncentrācijām jābūt pietiekami raksturojošām attiecībā uz saistīšanās līkni.
|
Testa plates izkārtojums
|
36.
|
Sagatavo marķētas mikrotitrēšanas plates, kurās ir pa sešiem šādiem inkubētiem replikātiem: šķīdinātāja kontrole, references estrogēns (E2) visaugstākajā koncentrācijā, kas kalpo arī par nespecifiskās saistīšanās (NSB) rādītāju, buferšķīduma kontrole, trīs inkubēti replikāti katram no šiem: astoņas nesaistīgās kontroles (oktiltrietoksisilāna) koncentrācijas, septiņas zemākās references estrogēna (E2) koncentrācijas, astoņas vāja saistekļa (noretinodrela vai noretindrona) koncentrācijas un astoņas koncentrācijas no katras testējamās ķimikālijas (TC). Mikrotitrēšanas plates izkārtojuma diagrammas paraugs pilnām references estrogēna un kontroļu koncentrāciju līknēm ir sniegts 4. tabulā. Atsevišķas mikrotitrēšanas plates izmanto testējamai ķimikālijai, un ir vajadzīgas testa plates kontroles, t. i., i) E2 un vāja saistekļa triplikāti augstā koncentrācijā (maksimāla pārvietošana) un vidējā koncentrācijā (aptuveni IC50); ii) šķīdinātāja kontrole (kopējā saistīšanās) un nespecifiskā saistīšanās, no katra pa sešiem replikātiem (5. tabula). Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojuma paraugs ar trijām nezināmām testējamajām ķimikālijām ir dots 3.3. papildinājumā. Darblapā, kā arī 4. un 5. tabulā norādītās koncentrācijas attiecas uz galīgajām koncentrācijām, ko izmanto katrā testēšanas iedobē. Maksimālā E2 koncentrācija ir 1×10–7 M, un attiecībā uz vāju saistekli jāizmanto augstākā koncentrācija, kas 1. mikrotirēšanas platē izmantota vājam saisteklim. IC50 koncentrāciju nosaka laboratorija, balstoties uz savu vēsturisko kontroles datubāzi. Tiek sagaidīts, ka šī vērtība būs līdzīga validācijas pētījumos konstatētajai vērtībai (sk. 1. tabulu).
|
4. tabula
Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojums (97)
(98), pilnas atsauces estrogēnu un kontroļu koncentrāciju līknes (1. plate).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Buferšķīduma kontrole un pozitīvā kontrole (E2)
|
Vāji pozitīva kontrole (noretinodrels)
|
Negatīvā kontrole (OTES)
|
TB un NSB
|
|
A
|
Tukša iedobe (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (šķīdinātāja kontrole) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Buferšķīduma kontrole
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Tukša iedobe (karsta) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
5. tabula
Konkurējošās saistīšanās testa mikrotitrēšanas plates izkārtojums, papildu plates testējamajām ķimikālijām (TC) un plates kontrolēm.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Testējamā ķimikālija Nr. 1 (TC-1)
|
Testējamā ķimikālija Nr. 2 (TC-2)
|
Testējamā ķimikālija Nr. 3 (TC-3)
|
Kontroles
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10
-7
M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10
-4,5
M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10-6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (šķīdinātāja kontrole)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
Šajā piemērā vājais saisteklis ir noretinodrels (apzīmēts “NE”).
|
Konkurējošās saistīšanās testa izpilde
|
37.
|
Katrā testēšanas plates iedobē (izņemot iedobes, kas attiecas uz kopējo saistīšanos, un tukšās, karstās iedobes, kā parādīts 6. tabulā) iepilina 50 μl testa buferšķīduma, tad samaisa ar 10 μl attiecīgi šķīdinātāja kontroles, references estrogēna (E2), vāja saistekļa, nesaistekļa un testējamās ķimikālijas un 10 μl 5 nM [3H]-17β-estradiola šķīduma. Pēc tam katrā platē pievieno 30 μl ledusauksta receptora šķīduma, un uzmanīgi samaisa. Kā pēdējo reaģentu pievieno hrERα šķīdumu. Testa mikrotitrēšanas plates divas stundas inkubē istabas temperatūrā (22–28°C).
6. tabula
HrER konkurējošās saistīšanās testa komponentu tilpums mikrotitrēšanas platēs
|
Sagatavošanas soļi
|
Visas iedobes, izņemot TB
|
TB iedobes
|
Tukšās iedobes (karstas)
|
|
Komponentu tilpums minētajām reakcijas iedobēm un pievienošanas kārtība
|
Testa buferšķīdums istabas temperatūrā
|
50 μl
|
60 μl
|
90 μl
|
|
Nemarķēts E2, vājš saisteklis, nesaisteklis, šķīdinātājs un testējamās ķimikālijas (*16)
|
10 μl
|
—
|
—
|
|
[3H]-17β-estradiols pietiekamā daudzumā, lai panāktu galīgo koncentrāciju 0,5 nM (t. i., 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Noteiktā hrERα koncentrācija (sk. 12.–13. punktu)
|
30 μl
|
30 μl
|
—
|
|
Kopējais tilpums katrā testa plates iedobē
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Saskaņā ar aprakstu 3. papildinājuma 21.–23. punktā (attiecas uz piesātinājuma–saistīšanās testu) kvantificē ar hrERα saistīto [3H]-17β-estradiolu, taču pirms tam, katrā iedobē pievienojot 100 μl ledusaukstas DCC suspensijas, no brīvā [3H]-17β-estradiola jābūt atdalītam ar hrERα saistītajam [3H]-17β-estradiolam.
|
|
39.
|
Iedobes G10–12 un H10–12 (sk. 4. tabulas aili “Tukša iedobe (karsta)”) raksturo marķēta [3H]-estradiola dpm 10 μl. 10 μl alikvotu pārnes tieši scintilācijas šķīdumā.
|
Pieņemamības kritēriji
Piesātinājuma–saistīšanās tests
|
40.
|
Ja tiek izmantotas pieaugošas [3H]-17β-estradiola koncentrācijas, specifiskās saistīšanās līknei ir jāsasniedz atslābuma periods, kas norāda uz hrERα piesātinājumu ar ligandu.
|
|
41.
|
Pieņemamības diapazons attiecībā uz specifisko saistīšanos pie 0,5 nM [3H]-17β-estradiola koncentrācijas ir 30–50 % no kopējās pievienotās radioaktivitātes vidējās vērtības, kas mērīta visās izpildes reizēs. Ir pieļaujamas atsevišķas nelielas novirzes no minētā diapazona, tomēr, ja izpildes reizēs pastāvīgi iegūst rezultātus ārpus minētā diapazona vai ja konkrētā izpildes reizē novirze ir ļoti būtiska, ir jāpielāgo proteīnu koncentrācija un piesātinājuma tests jāatkārto.
|
|
42.
|
Datiem jābūt attēlojamiem lineāras Skačārda diagrammas veidā.
|
|
43.
|
Nespecifiskā saistīšanās nedrīkst būt pārmērīga. Nespecifiskās saistīšanās vērtība parasti ir <35 % no kopējās saistīšanās. Tomēr minētā procentuālā attiecība dažkārt šo limitu var pārsniegt, ja testā tiek mērīts ļoti zems dpm pie zemākās radioaktīvi marķētā 17β-estradiola koncentrācijas.
|
Konkurējošās saistīšanās tests
|
44.
|
Pie pieaugošām nemarķēta 17β-estradiola koncentrācijām [3H]-17β-estradiolu pakāpeniski pārvieto no receptora tādā veidā, kas līdzinās viena domēna konkurējošajai saistīšanai.
|
|
45.
|
References estrogēna (t. i., 17β-estradiola) IC50 vērtībai jābūt aptuveni vienādai ar [3H]-17β-estradiola molāro koncentrāciju, kam pieskaitīta piesātinājuma–saistīšanās testā noteiktā diasociācijas konstante (Kd).
|
|
46.
|
Visās testa izpildes reizēs kopējās specifiskās saistīšanās vērtībai pastāvīgi jābūt pieņemamības diapazonā (40 ± 10 %), pie nosacījuma, ka kopējās pievienotās radioaktivitātes vidējā mērītā koncentrācija katrā iedobē ir 0,5 nM. Ir pieļaujamas atsevišķas nelielas novirzes no minētā diapazona, tomēr, ja izpildes reizēs pastāvīgi iegūst rezultātus ārpus minētā diapazona vai ja konkrētā izpildes reizē novirze ir ļoti būtiska, ir jāpielāgo proteīnu koncentrācija.
|
|
47.
|
Šķīdinātājs nedrīkst mainīt testa jutīgumu vai reproducējamību. Šķīdinātāja kontroles rezultātus (TB iedobes) salīdzina ar buferšķīduma kontroli, lai verificētu, ka izmantotais šķīdinātājs neietekmē testa sistēmu. Ja šķīdinātājs neietekmē testa gaitu, TB un buferšķīduma kontrolēm jābūt salīdzināmām.
|
|
48.
|
Testējot ar koncentrācijām, kas nepārsniedz 10–3 M (OTES) vai 10–4 M (DBP), nesaisteklim nevajadzētu no hrERα pārvietot vairāk nekā 25 % [3H]-17β-estradiola.
|
|
49.
|
Izmantojot CERI hrER saistīšanās testa validēšanas pētījuma datus (2. atsauces N pielikums), tika izstrādāti veiktspējas kritēriji attiecībā uz references estrogēnu un diviem vājiem saistekļiem (piem., noretinodrels, noretindrons). Ir sniegti 95 % ticamības intervāli vidējām vērtībām ± SN (n), kas tika iegūtas no visiem kontroles testiem, ko izpildīja četras laboratorijas, kuras piedalījās validēšanas pētījumā. 95 % ticamības intervālus aprēķināja references estrogēna un vāju saistekļu līknes pielāgošanas parametriem (t. i., augšējam un apakšējam punktam, Hilla virziena koeficientam un log IC50) un vāju saistekļu log10
RBA attiecībā pret references estrogēnu. 1. tabulā sniegti līknes pielāgošanas parametru paredzamie diapazoni, kurus var izmantot kā veiktspējas kritērijus. Praksē IC50 diapazons var būt nedaudz atšķirīgs atkarībā no eksperimentāli iegūtā receptora preparāta Kd un testā izmantotās liganda koncentrācijas.
|
|
50.
|
Testējamo ķimikāliju līknes pielāgošanas parametriem veiktspējas kritēriji izstrādāti netika, jo pastāv pārāk plašs esošu potenciāli testējamu ķimikāliju spektrs un potenciālās afinitātes un rezultātu mainīgums (piem., pilna līkne, daļēja līkne, neatbilstība līknei). Tomēr katrā testēšanas reizē un ar katru testējamo ķimikāliju iegūtos rezultātus būtu jāizvērtē speciālistam. Testā jāizmanto pietiekams testējamās ķimikālijas koncentrāciju diapazons, lai skaidri definētu konkurējošās saistīšanās līknes augšējo punktu (t. i., 90–100 % saistīšanās). Rezultātu mainībai starp katras testējamās ķimikālijas koncentrācijas replikātiem, kā arī starp trim neatkarīgajām izpildes reizēm jābūt saprātīgi pieļaujamās robežās un zinātniski pamatojamai. Izpildot kontroles testus attiecībā uz katru testējamo ķimikāliju, mērījumiem būtu jātuvojas veiktspējas parametriem, kas paziņoti attiecībā uz šo CERI testu, un tiem jābūt saskanīgiem ar attiecīgās laboratorijas vēsturiskajiem kontroldatiem.
|
DATU ANALĪZE
Piesātinājuma–saistīšanās tests
|
51.
|
Tiek mērīta gan kopējā, gan nespecifiskā saistīšanās. No šim vērtībām, nespecifisko saistīšanos atņemot no kopējās saistīšanās, tiek aprēķināta pieaugošu [3H]-17β-estradiola koncentrāciju specifiskā saistīšanās līdzsvara apstākļos. Grafikā, kurā specifisko saistīšanos attēlo attiecībā pret [3H]-17β-estradiola koncentrāciju, attiecībā uz maksimālo specifisko saistīšanos ir jāsasniedz atslābuma periods, kas norāda uz hrERα piesātinājumu ar [3H]-17β-estradiolu. Turklāt datu analīzē būtu jākonstatē [3H]-17β- estradiola saistīšanās ar vienu augstas afinitātes saistīšanās domēnu. Piesātinājuma–saistīšanās līknē attēlo nespecifisko, kopējo un specifisko saistīšanos. Šo datu turpmākai analīzei izmanto nelineārās regresijas analīzi (piem., BioSoft; McPherson, 1985; Motulsky, 1995), un galīgos datus attēlo ar Skačārda diagrammu.
|
|
52.
|
Analizējot datus, Bmax un Kd nosaka no kopējās saistīšanās datiem vien, izmantojot pieņēmumu, ka nespecifiskā saistīšanās ir lineāra, ja vien nav pamatota iemesla izmantot citu metodi. Turklāt, meklējot labāko atbilstību līknei, izmanto robustu regresiju, ja vien nav pamatota iemesla rīkoties citādi. Pārskatā norāda izvēlēto robustas regresijas metodi. Nosakot Bmax un Kd no piesātinājuma–saistīšanās datiem, vienmēr jāizmanto liganda noplicināšanās korekcija (piem., Swillens (1995) metode).
|
Konkurējošās saistīšanās tests
|
53.
|
Konkurējošās saistīšanās līkni grafiski attēlo kā specifisko [3H]-17β-estradiola saistīšanos attiecībā pret konkurenta koncentrāciju (log10 vienībās). Testējamās ķimikālijas koncentrācija, kas inhibē 50 % [3H]17β-estradiola maksimālās specifiskās saistīšanās, ir IC50 vērtība.
|
|
54.
|
Pozitīvo kontroļu (t. i., references estrogēna un vāja saistekļa) log (IC50) vērtību aplēses nosaka, izmantojot atbilstošu nelineāru līknes piemeklēšanas programmatūru, lai panāktu atbilstību Hilla vienādojumam ar četriem parametriem (piem., BioSoft; McPherson, 1985; Motulsky, 1995). Veicot pielāgošanu līknei, augšējo un apakšējo punktu, virziena koeficientu un log (IC50) parasti neierobežo. Meklējot labāko atbilstību līknei, izmanto robustu regresiju, ja vien nav pamatota iemesla rīkoties citādi. Liganda noplicināšanās korekciju neizmanto. Pēc sākotnējās analīzes katru saistīšanās līkni pārskata, lai nodrošinātu pienācīgu atbilstību paraugam. Vāja saistekļa relatīvo afinitāti (RBA) aprēķina kā procentuālu vērtību no vāja saistekļa log (IC50) attiecībā pret 17β-estradiola log (IC50). Ar pozitīvajām kontrolēm un nesaistekļu kontrolēm iegūtos rezultātus izvērtē, sekojot testa veikšanas norādēm 23. papildinājuma 44.–49. punktā.
|
|
55.
|
Datus par visam testējamajām ķimikālijām būtu jāanalizē, izmantojot pakāpenisku pieeju, kas nodrošina datu pienācīgu analīzi un to, ka katra konkurējošās saistīšanās līkne tiek klasificēta pareizi. Pirms katras testa izpildes reizes testējamās ķimikālijas ieteicams sākotnēji analizēt standartizētā veidā, identiski tam, kā tiek analizēti references estrogēns un vāja saistekļa kontroles (sk. 3. papildinājuma 54.punktu). Pēc tam katrā testa veikšanas reizē tehniski pārskata, vai līkne atbilst parametriem, un vizuāli nosaka, cik precīzi dati atbilst ģenerētajai konkurējošās saistīšanās līknei. Ja šīs tehniskās pārbaudes laikā novēro, ka [3H]-17β-estradiola specifiskā saistīšanās atkarībā no koncentrācijas procentuāli samazinās, rezultātu mainība tehnisko replikātu starpā pie katras testējamās ķīmiskās koncentrācijas ir neliela un līknes atbilstība parametriem trīs testa veikšanas reizēs ir konsekventa, tas ir pietiekami uzticams rādītājs, ka tests un datu analīze ir veikti pareizi.
|
Datu interpretācija
|
56.
|
Pie nosacījuma, ka ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par saistīgu ar hrERα, ja saistīšanās līkne ir pielāgojama un zemākais atbildreakcijas līknes punkts datu diapazonā ir mazāks par 50 % (1. attēls).
|
|
57.
|
Pie nosacījuma, ka ir izpildīti visi pieņemamības kritēriji, testējamo ķimikāliju uzskata par nesaistīgu ar hrERα, ja:
|
—
|
saistīšanās līkne ir pielāgojama un pielāgotās atbildreakcijas līknes zemākais punkts datu diapazonā ir lielāks par 75 % vai
|
|
—
|
saistīšanās līkni pielāgot nevar un visu koncentrāciju grupu vidējā zemākā nenoapaļotā procentuālā saistīšanās pēc datiem pārsniedz 75 %.
|
|
|
58.
|
Testējamās ķimikālijas klasifikāciju uzskata par neskaidru, ja nav izpildīts neviens no iepriekšminētajiem nosacījumiem (t. i., zemākais pielāgotās atbildreakcijas līknes punkts ir robežās no 76 – 51 %).
|
7. tabula
Kritēriji, pēc kuriem klasificē testējamās ķimikālijas, izmantojot konkurējošās saistīšanās līkni.
|
Klasifikācija
|
Kritēriji
|
|
Saisteklisa
|
Saistīšanās līkni var pielāgot.
Zemākais atbildreakcijas līknes punkts datu diapazonā ir mazāks par 50 %.
|
|
Nesaisteklisb
|
Ja saistīšanās līkni var pielāgot,
pielāgotās atbildreakcijas līknes zemākais punkts datu diapazonā ir virs 75 %.
Ja saistīšanās līkni nevar pielāgot,
visu koncentrāciju grupu vidējā zemākā nenoapaļotā procentuālā saistīšanās pēc datiem pārsniedz 75 %.
|
|
Neskaidrac
|
Jebkura testa izpildes reize, kas neuzrāda testējamās ķimikālijas saistīgumu vai nesaistīgumu
(piem., pielāgotās atbildreakcijas līknes zemākais punkts ir robežās no 76–51 %).
|
1. attēls
Piemēri, kā klasificē testējamās ķimikālijas, izmantojot konkurējošās saistīšanās līkni.

|
59.
|
Laboratorijā attiecībā uz katru testējamo ķimikāliju testu veic vairākas izpildes reizes, un katrai izpildes reizei atkarībā no rezultāta piešķir ciparisku vērtību, no kuras pēc tam aprēķina vidējo vērtību atbilstoši 8. tabulā sniegtajam paraugam. Vidējos rezultātus, kas apkopoti konkrētajā laboratorijā, salīdzina ar gaidīto katras testējamās ķimikālijas klasifikāciju.
|
8. tabula
Metode, pēc kuras klasificē testējamo ķimikāliju, izmantojot vairākas testēšanas reizes vienā laboratorijā.
|
Katrai izpildes reizei piešķir ciparisku vērtību:
|
|
Klasifikācija
|
Cipariska vērtība
|
|
Saisteklis
|
2
|
|
Neskaidrs
|
1
|
|
Nesaisteklis
|
0
|
|
Izpildes reizēs iegūto ciparisko vērtību vidējās vērtības klasificēšana:
|
|
Klasifikācija
|
Cipariska vērtība
|
|
Saisteklis
|
Vidēji ≥ 1,5
|
|
Neskaidrs
|
0,5 ≤ Vidējā vērtība < 1,5
|
|
Nesaisteklis
|
Vidēji < 0,5
|
TESTĒŠANAS PĀRSKATS
|
60.
|
Sk. šīs testēšanas metodes sadaļas “HrER SAISTĪŠANĀS TESTA KOMPONENTI” 24. punktu.
|
3.1. papildinājums
TERMINU SARAKSTS
[3H]E2
: 17β-estradiols, kas radioaktīvi marķēts ar tritiju.
DCC
: ar dekstrānu pārklāta ogle.
E2
: nemarķēts 17β-estradiols (inerts).
Testa buferšķīdums
: 10 mM tris-HCl (pH 7,4), kas satur 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glicerīna, 0,2 mM leupeptīna, 1 mM ditiotreitola un 10 mg/ml liellopu seruma albumīna.
hrERα
: cilvēka rekombinantais estrogēnreceptors alfa (liganda saistīšanās domēns).
Replikāts
: viena no vairākām vienāda satura un vienādas koncentrācijas iedobēm, kuras paralēli testē vienā testēšanas reizē. Pēc šī protokola katru testējamās ķimikālijas koncentrāciju testē ar trim replikātiem (triplikātiem); proti, pie katras testējamās ķimikālijas koncentrācijas ir trīs replikāti, kurus testē vienlaicīgi.
Izpildes reize
: viss paralēli testēto miktotitrēšanas plates testa iedobju kopums, kas nodrošina visu informāciju, kura vajadzīga, lai raksturotu testējamās ķimikālijas saistīšanos ar hrERα, (t. i., kopējais testa iedobei pievienotais [3H]-17β-estradiols, maksimālā [3H]-17β-estradiola saistīšanās ar hrERα, nespecifiskā saistīšanās un kopējā saistīšanās pie dažādām testējamās ķimikālijas koncentrācijām). Izpildes reize var nozīmēt pat tikai vienas testēšanas iedobes (t. i., replikāta) testēšanu pie katras koncentrācijas, tomēr šajā konkrētajā gadījumā protokols pieprasa testēt ar triplikātiem, un viena izpildes reize nozīmē trīs testēšanas iedobes pie katras koncentrācijas. Papildus minētajam šis protokols prasa veikt trīs neatkarīgas (proti, neveicot paralēli) izpildes reizes ar katru ķimikāliju.
3.2. papildinājums
KONKURĒJOŠĀS SAISTĪŠANĀS TESTA IEDOBJU IZKĀRTOJUMS
|
Plate
|
Pozīcija
|
Replikāts
|
Iedobes tips
|
Iedobes kods
|
Koncentrācijas kods
|
Konkurenta sākotnējā koncentrācija (M)
|
hrER izejšķīdums (μl)
|
Buferšķīduma tilpums (μl)
|
Iezīmētā elementa (karsta E2) tilpums (μL)
|
Tilpums atšķaidījuma platē (μL)
|
Beigu tilpums (μl)
|
Konkurenta beigu koncentrācija (M)
|
|
S
|
A1
|
1
|
tukša
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
tukša
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
tukša
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
auksts E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
auksts E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
auksts E2
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
auksts E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
auksts E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
auksts E2
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
auksts E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
auksts E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
auksts E2
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
auksts E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
auksts E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
auksts E2
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
auksts E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
auksts E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
auksts E2
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
auksts E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
auksts E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
auksts E2
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
auksts E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
auksts E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
auksts E2
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
noretinodrels
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
noretinodrels
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
noretinodrels
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
noretinodrels
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
noretinodrels
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
noretinodrels
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
noretinodrels
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
noretinodrels
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
noretinodrels
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
noretinodrels
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
noretinodrels
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
noretinodrels
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
noretinodrels
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
noretinodrels
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
noretinodrels
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
noretinodrels
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
noretinodrels
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
noretinodrels
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
noretinodrels
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
noretinodrels
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
noretinodrels
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
noretinodrels
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
noretinodrels
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
noretinodrels
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
kopējā saistīšanās
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
kopējā saistīšanās
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
kopējā saistīšanās
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
kopējā saistīšanās
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
-
|
|
S
|
B11
|
5
|
kopējā saistīšanās
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
kopējā saistīšanās
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
buferšķīduma kontrole
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
buferšķīduma kontrole
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
buferšķīduma kontrole
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
buferšķīduma kontrole
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
buferšķīduma kontrole
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
buferšķīduma kontrole
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
tukša iedobe (karsta)
|
karsta
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
-
|
|
S
|
G11 (*17)
|
2
|
tukša iedobe (karsta)
|
karsta
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
tukša iedobe (karsta)
|
karsta
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
tukša iedobe (karsta)
|
karsta
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
tukša iedobe (karsta)
|
karsta
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
tukša iedobe (karsta)
|
karsta
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
nezināms 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
nezināms 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
nezināms 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
nezināms 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
nezināms 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
nezināms 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
nezināms 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
nezināms 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
nezināms 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
nezināms 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
nezināms 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
nezināms 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
nezināms 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
nezināms 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
nezināms 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
nezināms 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
nezināms 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
nezināms 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
nezināms 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
nezināms 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
nezināms 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
nezināms 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
nezināms 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
nezināms 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
nezināms 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
nezināms 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
nezināms 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
nezināms 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
nezināms 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
nezināms 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
nezināms 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
nezināms 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
nezināms 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
nezināms 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
nezināms 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
nezināms 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
nezināms 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
nezināms 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
nezināms 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
nezināms 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
nezināms 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
nezināms 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
nezināms 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
nezināms 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
nezināms 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
nezināms 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
nezināms 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
nezināms 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
nezināms 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
nezināms 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
nezināms 3
|
U3
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
nezināms 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
nezināms 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
nezināms 3
|
U3
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
nezināms 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
nezināms 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
nezināms 3
|
U3
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
nezināms 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
nezināms 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
nezināms 3
|
U3
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
nezināms 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
nezināms 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
nezināms 3
|
U3
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
nezināms 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
nezināms 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
nezināms 3
|
U3
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
nezināms 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G8
|
2
|
nezināms 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G9
|
3
|
nezināms 3
|
U3
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H7
|
1
|
nezināms 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H8
|
2
|
nezināms 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H9
|
3
|
nezināms 3
|
U3
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A10
|
1
|
E2 kontrole (max)
|
S
|
E2max1
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A11
|
2
|
E2 kontrole (max)
|
S
|
E2max2
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
A12
|
3
|
E2 kontrole (max)
|
S
|
E2max3
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,00E-07
|
|
P1
|
B10
|
1
|
E2 kontrole (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
E2 kontrole (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
E2 kontrole (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
NE kontrole (max)
|
S
|
Nemax1
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C11
|
2
|
NE kontrole (max)
|
S
|
Nemax2
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
C12
|
3
|
NE kontrole (max)
|
S
|
Nemax3
|
1,00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00E-4,5
|
|
P1
|
D10
|
1
|
NE kontrole (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
NE kontrole (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
NE kontrole (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
auksts E2 (augsta koncentrācija)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
kopējā saistīšanās
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
kopējā saistīšanās
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
kopējā saistīšanās
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
kopējā saistīšanās
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
kopējā saistīšanās
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
kopējā saistīšanās
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
-
|
4. papildinājums
APSVĒRUMI, KAS JĀŅEM VĒRĀ, ANALIZĒJOT hrER KONKURĒJOŠĀS SAISTĪŠANĀS TESTA DATUS
|
1.
|
HrERα konkurējošās saistīšanās tests mēra vienas konkrētas [3H]-17β- estradiola koncentrācijas saistīšanos arvien pieaugošu testējamās ķimikālijas koncentrāciju klātbūtnē. Konkurējošās saistīšanās līkni grafiski attēlo kā specifisko [3H]-17β-estradiola saistīšanos attiecībā pret konkurenta koncentrāciju (log10 vienībās). Testējamās ķimikālijas koncentrācija, kas inhibē 50 % [3H]17β-estradiola maksimālās specifiskās saistīšanās, ir IC50 vērtība.
|
References estrogēna un vāja saistekļa ģenerēto datu analīze (1)
|
2.
|
Kontroles testēšanas reizēs iegūtos datus transformē turpmākai analizēšanai (t. i., procentuālā [3H]-17β-estradiola specifiskā saistīšanās un kontroles ķimikālijas log koncentrācija). Pozitīvo kontroļu (t. i., references estrogēna un vāja saistekļa) log (IC50) vērtību aplēses nosaka, izmantojot atbilstošu nelineāru līknes piemeklēšanas programmatūru, lai panāktu atbilstību Hilla vienādojumam ar četriem parametriem, piem., BioSoft; GraphPad Prism) (2). Veicot pielāgošanu līknei, augšējo un apakšējo punktu, virziena koeficientu un log (IC50) ierobežot parasti nav vajadzīgs. Meklējot labāko atbilstību līknei, izmanto robustu regresiju, ja vien nav pamatota iemesla rīkoties citādi. Pārskatā norāda izvēlēto robustas regresijas metodi. FW vai CERI hrER testiem liganda noplicināšanās korekcija nav vajadzīga, bet attiecīgā gadījumā var apsvērt tās lietošanu. Pēc sākotnējās analīzes katru saistīšanās līkni pārskata, lai nodrošinātu pienācīgu atbilstību paraugam. Vāja saistekļa relatīvo afinitāti (RBA) var aprēķināt kā procentuālu vērtību no vāja saistekļa log (IC50) attiecībā pret 17β-estradiola log (IC50). Ar pozitīvajām kontrolēm un nesaistīgām kontrolēm iegūtos rezultātus izvērtē, izmantojot testa veiktspējas un pieņemamības kritērijus, kas aprakstīti šajā testa metodē (20. punkts), 2.papildinājumā (FW testa 41.–51. punkts) un 3. papildinājumā (CERI tests, 41.–51. punkts). 1. attēlā sniegti references estrogēna un vāja saistekļa piemēri, testu veicot trīs reizes.
|
1. attēls.
Konkurējošās saistīšanās līknes piemēri ar references estrogēnu un vāja saistekļa kontroli.

Testējamo ķimikāliju ģenerēto datu analīze
|
3.
|
Datus par visam testējamajām ķimikālijām būtu jāanalizē, izmantojot pakāpenisku pieeju, kas nodrošina datu pienācīgu analīzi un to, ka katra konkurējošās saistīšanās līkne tiek klasificēta pareizi. Pirms katras testa veikšanas reizes testējamās ķimikālijas datus sākotnēji analizē standartizētā veidā, identiski tam, kā tiek analizēti references estrogēns un vāja saistekļa kontroles. Pēc tam katrā testa veikšanas reizē tehniski pārskata, vai līkne atbilst parametriem, un vizuāli nosaka, cik precīzi dati atbilst ģenerētajai konkurējošās saistīšanās līknei. Ja šīs tehniskās pārbaudes laikā novēro, ka [3H]-17β-estradiola specifiskā saistīšanās atkarībā no koncentrācijas procentuāli samazinās, rezultātu mainība tehnisko replikātu starpā pie katras ķīmiskās koncentrācijas ir neliela un līknes atbilstība parametriem trīs testa veikšanas reizēs ir konsekventa, tas ir pietiekami uzticams rādītājs, ka tests un datu analīze ir veikti pareizi. Katrā testēšanas reizē attiecībā uz testējamo ķimikāliju iegūtos rezultātus ir jāizvērtē speciālistam, un datiem, pēc kuriem katru testējamo ķimikāliju klasificē kā saistīgu vai nesaistīgu, jābūt zinātniski pamatojamiem.
|
|
4.
|
Dažkārt var būt situācijas, kad hrER saistīšanās datu pienācīgas analizēšanas un interpretēšanas vajadzībām datiem ir jāveic zināmas korekcijas. Agrākos pētījumos ir novēroti gadījumi, kad receptora konkurējošās saistīšanās datu analīzi un interpretāciju apgrūtina procentuālās specifiskās saistīšanās pieaugums, ja ķimikālijas testē augstākā koncentrācijā (2. attēls). Šī problēma ir plaši zināma dažādu receptora konkurējošās saistīšanās testu protokolu lietotājiem (3). Šādos gadījumos no koncentrācijas atkarīgā atbildreakcija tiek novērota pie zemākām koncentrācijām, bet, kad testējamās ķimikālijas koncentrācija tuvojas šķīdības robežai, [3H]17β-estradiola pārvietošana vairs nesamazinās. Šādos gadījumos pie augstākajām koncentrācijām iegūtie dati norāda uz to, ka ir sasniegta testa bioloģiskā robeža. Piemēram, šis fenomens daudzkārt tiek saistīts ar ķīmisko nešķīdību un izgulsnēšanos pie augstākām koncentrācijām vai arī var atspoguļot to, ka pie visaugstākajām ķīmiskajām koncentrācijām ir pārsniegta ar dekstrānu pārklātas ogles spēja atdalīšanās procedūras laikā uztvert nesaistītu radioaktīvi marķētu ligandu. Ja, veicot konkurējošās saistīšanās datu pielāgošanu divvirzienu līknei, šādus datu punktus saglabā, tas dažkārt var novest pie nepareizas ER saistīšanās potenciāla klasificēšanas attiecībā uz testējamo ķimikāliju (2. attēls). Lai no tā izvairītos, FW un CERI hrER saistīšanās testu protokoli ietver izvēli no analīzes izslēgt tādus datu punktus, kuros ar replikātiem iegūtā [3H]17β-estradiola specifiskās saistīšanās procentuālā vidējā vērtība par 10 % vai vairāk pārsniedz ar kādu zemāku koncentrāciju novēroto vidējo vērtību (to dēvē par 10 % kritēriju). Šo kritēriju attiecībā uz konkrēto līkni var izmantot tikai vienreiz, un, lai līkni varētu pareizi klasificēt, jāpaliek vēl datiem vismaz par sešām koncentrācijām.
|
2. attēls.
Piemēri: konkurējošās saistīšanās līknes, izmantojot 10 % kritēriju un bez tā.

|
5.
|
To, vai ir atbilstoši izmantot 10 % kritēriju, lai labotu šīs līknes, ir rūpīgi jāizsver, un to izmanto vien gadījumos, kad dati stipri liecina par hrER saistekli. Kad FW hrER saistīšanās testa validācijas pētījuma laikā veica eksperimentus, tika novērots, ka dažkārt 10 % kritērija piemērošana izraisa neplānotas un neparedzamas sekas. Attiecībā uz ķimikālijām, kas nemijiedarbojās ar receptoru r (t. i., faktiskie nesaistekļi), pie aptuveni 100 % radioaktīvi marķēto ligandu saistīšanās bieži bija vērojama mainība, kas visā testēto koncentrāciju diapazonā pārsniedz 10 %. Ja viszemākā vērtība tika konstatēta pie zemas koncentrācijas, datus par visām augstākajām koncentrācijām potenciāli varētu izņemt no analizējamiem datiem, izmantojot 10 % kritēriju, lai gan šīs koncentrācijas var izrādīties noderīgas, nosakot, vai attiecīgā ķimikālija ir nesaisteklis. 3. attēlā sniegtie piemēri ilustrē gadījumus, kad 10 % kritērija izmantošana nav piemērota.
|
3. attēls.
Piemēri: konkurējošās saistīšanās dati gadījumos, kad nepiemēro 10 % kritēriju.
ATSAUCES
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71. ĀDAS IN VITRO SENSIBILIZĀCIJAS TESTI, KUROS APSKATĪTS ĀDAS SENSIBILIZĀCIJAS NELABVĒLĪGĀ IZNĀKUMA CEĻA (AOP) ATSLĒGNOTIKUMS “DENDRĪTISKO ŠŪNU AKTIVĀCIJA”
VISPĀRĪGS IEVADS
Testēšanas metode, kuras pamatā ir atslēgnotikums “dendrītisko šūnu aktivācija”
|
1.
|
Ādas sensibilizators ir viela, kas, nonākot saskarē ar ādu, izraisa alerģisku atbildreakciju, atbilstoši definīcijai, kas dota Apvienoto Nāciju Organizācijas (ANO) Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētajā sistēmā (GHS) (1) un Eiropas Savienības (ES) Regulā Nr. 1272/2008 par vielu un maisījumu klasificēšanu, marķēšanu un iepakošanu (CLP regula) (99). Par to, kādi bioloģiskie atslēgnotikumi ir ādas sensibilizācijas pamatā, valda vispārēja vienprātība. Pašreizējās zināšanas par ķīmiskajiem un bioloģiskajiem mehānismiem, kas saistīti ar ādas sensibilizāciju, ir apkopotas, ESAO [OECD] AOP programmas ietvaros izveidojot nelabvēlīga iznākuma ceļu (Adverse Outcome Pathway, AOP) (2) no molekulārā iniciācijas notikuma un starpposma notikumiem līdz nelabvēlīgajai ietekmei, proti, alerģiskam kontaktdermatītam. Šajā gadījumā molekulārais iniciācijas notikums (proti, pirmais atslēgnotikums) ir elektrofilu vielu kovalenta saistīšanās ar nukleofilajiem centriem ādas proteīnos. Otrais atslēgnotikums šajā AOP notiek keratinocītos un ietver iekaisuma atbildreakcijas, kā arī izmaiņas gēnu ekspresijā, kas asociētas ar konkrētiem šūnu signālceļiem, piem., no antioksidanta/elektrofila atbildreakcijas elementa (ARE) atkarīgiem ceļiem. Trešais atslēgnotikums ir dendrītisko šūnu (DŠ) aktivācija, ko parasti novērtē pēc ekspresētajiem specifiskajiem šūnu virsmas marķieriem, hemokīniem un citokīniem. Ceturtais atslēgnotikums ir T šūnu aktivācija un savairošanās, ko netieši novērtē apakšdzimtas Murinae grauzēju limfmezglu lokālās stimulācijas testā (LLNA) (3).
|
|
2.
|
Šī testēšanas metode (TM) ir ekvivalenta ESAO testēšanas vadlīnijai (TG) Nr. 442E (2017). Tā apraksta in vitro testus, kuros apskatīti mehānismi, kas aprakstīti pie ādas sensibilizācijas AOP dendrītisko šūnu aktivācijas atslēgnotikuma (2). Šī TM ietver testus, kas palīdz ādas sensibilizatorus nošķirt no ādas nesensibilizatoriem saskaņā ar ANO GHS un CLP regulu.
|
|
Šajā TM aprakstīti šādi testi:
|
—
|
cilvēka šūnu līnijas aktivācijas tests (h-CLAT);
|
|
—
|
šūnu līnijas U937 aktivācijas tests (U-SENS™);
|
|
—
|
interleikīna-8 reportiergēna tests (tests IL-8 Luc).
|
|
|
|
3.
|
Šajā testēšanas metodē iekļautie testi no attiecīgās ESAO TG testiem var atšķirties ar datu ieguves procedūru un mērītajiem rādījumiem, tomēr tam, lai izpildītu valstu prasības pēc ādas sensibilizācijas AOP dendrītisko šūnu aktivācijas atslēgnotikuma testa rezultātiem, vienlaikus izmantojot ESAO datu savstarpējo atzīšanu, šos testus var izmantot bez izšķirības.
|
Atslēgnotikumā balstītajā testēšanas metodē iekļauto testu konteksts un principi
|
4.
|
Līdz šim ādas sensibilizācijas novērtēšanai parasti izmantoti laboratorijas dzīvnieki. Ar klasiskām metodēm, kurās izmanto jūrascūciņas, — Magnusona–Kligmana [Magnusson–Kligman] maksimalizācijas testu ar jūrascūciņām (Guinea Pig Maximisation Test, GPMT) un Bīlera [Buehler] testu (TM B.6) (4) — tiek pētīta gan ādas sensibilizācijas inducēšanas fāze, gan tās izsaukšanas fāze. Piekrišanu ir guvuši arī apakšdzimtas Murinae grauzēju testi, LLNA (TM B.42) (3) un tā divas bezradiācijas modifikācijas, LLNA: DA (TM B.50) (5) un LLNA: BrdU-ELISA (TM B.51) (6), kuros visos tiek novērtēta vienīgi indukcijas atbildreakcija, jo šie testi dzīvnieku labklājības un objektīva ādas sensibilizācijas inducēšanas fāzes mērījuma ziņā ir pārāki par testiem ar jūrascūciņām.
|
|
5.
|
Pēdējā laikā ķimikāliju ādas sensibilizācijas briesmu potenciāla izvērtēšanas vajadzībām pieņemtas mehānistiskas in chemico un in vitro testēšanas metodes, kurās apskatīts ādas sensibilizācijas AOP pirmais atslēgnotikums (TM B.59; tiešais peptīdreaģētspējas tests — Direct Peptide Reactivity Assay (7)) un otrais atslēgnotikums (TM B.60; ARE-Nrf2 luciferāzes testēšanas metode (8)).
|
|
6.
|
Šajā testēšanas metodē aprakstītie testi kvantificē vai nu izmaiņas tādu šūnu virsmas marķieru ekspresijā, kas asociēti ar monocītu un DŠ aktivāciju pēc ekspozīcijas sensibilizatoriem (piem., CD54, CD86), vai izmaiņas IL-8 (ar DŠ aktivāciju asociēta citokīna) ekspresijā. Ir ziņas, ka ādas sensibilizatori inducē šūnu membrānu marķieru (piem., CD40, CD54, CD80, CD83 un CD86) ekspresiju, līdztekus inducējot arī proinflamatoros citokīnus (piem., IL-1β un TNF-α) un vairākus hemokīnus, tostarp IL-8 (CXCL8) un CCL3 (9) (10) (11) (12), kas asociēti ar DŠ aktivāciju (2).
|
|
7.
|
Tomēr, tā kā DŠ aktivācija ir tikai viens no ādas sensibilizācijas AOP (2) (13) atslēgnotikumiem, informācija, kas iegūta testos, kuros tiek mērīti tikai DŠ aktivācijas marķieri vien, var nebūt pietiekama, lai varētu izdarīt secinājumus par ķimikāliju ādas sensibilizācijas potenciāla esību vai neesību. Tāpēc tiek ierosināts šajā testēšanas metodē aprakstīto testu datus izmantot par palīglīdzekli ādas sensibilizatoru (piem., UN GHS / CLP 1. kategorijas sensibilizatoru) nošķiršanā no nesensibilizatoriem integrētā testēšanas un novērtēšanas pieejā (IATA) kopā ar citu relevantu papildinformāciju, piem., informāciju, kas iegūta in vitro testos, kuros apskatīti citi ādas sensibilizācijas AOP atslēgnotikumi, kā arī ar metodēm, kuras neietver testēšanu un kurās pēc analoģijas principa (“aplūkot līdzīgu”) tiek izmantota informācija par ķīmiskajiem analogiem (13). Ir publicēti (13) piemēri, kā šajos testos iegūtos datus var izmantot definētās pieejās, t. i., pieejās, kurās standartizēta gan izmantoto informācijas avotu kopa, gan procedūra, kādā no datiem iegūst prognozes, un tos var izmantot kā lietderīgus IATA elementus.
|
|
8.
|
Šajā testēšanas metodē aprakstītos testus vienus pašus nevar izmantot ne ādas sensibilizatoru klasificēšanai ANO GHS / CLP apakškategorijās 1.A un 1.B (iestādēm, kuras izmanto šīs divas neobligātās apakškategorijas), ne potences prognozēšanai drošuma novērtējuma lēmumu vajadzībām. Tomēr atkarībā no tiesiskā regulējuma pozitīvus rezultātus, kas iegūti ar šīm metodēm, var izmantot ķimikālijas klasificēšanai ANO GHS / CLP 1. kategorijā arī vienus pašus.
|
|
9.
|
Terminu “testējamā ķimikālija” šajā testēšanas metodē izmanto, lai apzīmētu to, kas tiek testēts (100), un tas nav saistīts ar testu izmantojamību vienkomponenta vielu, daudzkomponentu vielu un/vai maisījumu testēšanā. Informācija par testu izmantojamību daudzkomponentu vielu / maisījumu testēšanai pašlaik ir ierobežota (14) (15). Tomēr šie testi ir tehniski izmantojami daudzkomponentu vielu un maisījumu testēšanai. Taču, pirms šo testēšanas metodi izmantot maisījuma testēšanai nolūkā iegūt datus kādam normatīvam mērķim, jāapsver, vai un kādēļ tā varētu sniegt minētajam mērķim piemērotus rezultātus (101). Ja pastāv normatīva prasība maisījumu testēt, tāda apsvēršana nav vajadzīga. Turklāt, testējot daudzkomponentu vielas vai maisījumus, jāapsver iespējamā ietekme, ko uz novērotajām atbildreakcijām varētu atstāt citotoksiskie komponenti.
|
LITERATŪRA
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Available at: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Available at: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Šā pielikuma B.42. nodaļa. Limfmezglu lokālās stimulācijas tests.
|
|
(4)
|
Šā pielikuma B.6. nodaļa. Ādas sensibilizācija.
|
|
(5)
|
Šā pielikuma B.50. nodaļa. Ādas sensibilizācija: limfmezglu lokālās stimulācijas tests: DA.
|
|
(6)
|
Šā pielikuma B.51. nodaļa. Ādas sensibilizācija: limfmezglu lokālās stimulācijas tests: BrdU-ELISA.
|
|
(7)
|
Šā pielikuma B.59. nodaļa. In chemico ādas sensibilizācija: tiešais peptīdreaģētspējas tests (DPRA).
|
|
(8)
|
Šā pielikuma B.60. nodaļa. Ādas in vitro sensibilizācija: ARE-Nrf2 luciferāzes testēšanas metode.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Available at: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
1. papildinājums
ĀDAS IN VITRO SENSIBILIZĀCIJA: CILVĒKA ŠŪNU LĪNIJAS AKTIVĀCIJAS TESTS (H-CLAT)
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
1.
|
h-CLAT kvantificē izmaiņas tādu šūnu virsmas marķieru ekspresijā, kas asociēti ar monocītu un dendrītisko šūnu (DŠ) aktivāciju (konkrētāk, CD86 un CD54), cilvēka monocītiskās leikēmijas šūnu līnijā THP-1 pēc ekspozīcijas sensibilizatoriem (1) (2). Izmērītos CD86 un CD54 šūnu virsmas marķieru ekspresijas līmeņus pēc tam izmanto par palīglīdzekli ādas sensibilizatoru nošķiršanā no nesensibilizatoriem.
|
|
2.
|
h-CLAT ir izvērtēts validācijas pētījumā, ko koordinēja Eiropas Savienības References laboratorija dzīvniektestēšanas alternatīvu jomā (EURL ECVAM), un pēc tam to neatkarīgi profesionālizvērtējusi EURL ECVAM Zinātniskā konsultatīvā komiteja (ESAC). Ņemot vērā visus pieejamos pierādījumus un regulatoru un ieinteresēto personu sniegtās ziņas, EURL ECVAM ir ieteikusi (3) h-CLAT izmantot IATA ietvaros par palīglīdzekli, kas palīdz ādas sensibilizatorus nošķirt no nesensibilizatoriem apdraudējumu klasificēšanas un marķēšanas vajadzībām. Piemēri, kā h-CLAT dati izmantoti kombinācijā ar citu informāciju, ir pieejami literatūrā (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
Ir pierādīts, ka h-CLAT ir pārnesams uz laboratorijām, kam ir pieredze ar paņēmieniem, kuros izmanto šūnu kultūras, un ar plūsmcitometrisko analīzi. Prognožu reproducējamības līmenis, kas sagaidāms no šā testa, ir aptuveni 80 % gan tajā pašā laboratorijā, gan citās laboratorijās (3) (12). Validācijas pētījuma rezultāti (13) un citi publicētie pētījumi (14) kopumā liecina, ka salīdzinājumā ar LLNA rezultātiem šā testa pareizība ādas sensibilizatoru (t. i., ANO GHS / CLP 1. kat.) nošķiršanā no nesensibilizatoriem ir 85 % (N=142) ar jutību 93 % (94/101) un specifiskumu 66 % (27/41) (balstoties uz EURL ECVAM veikto atkārtoto analīzi (12), kurā ņemti vērā visi līdzšinējie dati un neņemot vērā negatīvos rezultātus ar ķimikālijām, kuru Log Kow ir lielāks par 3,5, kā aprakstīts 4. punktā). Testā h-CLAT pseidonegatīvas prognozes, visticamāk, skars ķimikālijas, kurām novērojama zema līdz vidēja ādas sensibilizācijas potence (t. i., ANO GHS / CLP 1.B apakškategorija), nevis ķimikālijas, kurām novērojama augsta ādas sensibilizācijas potence (t. i., ANO GHS / CLP 1.A apakškategorija) (4) (13) (15). Visa šī informācija kopā norāda uz to, ka metode h-CLAT var palīdzēt identificēt apdraudējumus, kas izraisa ādas sensibilizāciju. Tomēr pareizības vērtības, kas šeit sniegtas attiecībā uz h-CLAT kā atsevišķu testu, ir tikai orientējošas, jo šis tests jāskata kombinācijā ar citiem informācijas avotiem IATA kontekstā un saskaņā ar vispārīgā ievada 7. un 8. punkta noteikumiem. Turklāt, izvērtējot ādas sensibilizācijas novērtēšanas metodes, kurās neizmanto dzīvniekus, jāņem vērā, ka LLNA tests, tāpat kā citi testi ar dzīvniekiem, var nepilnīgi atspoguļot situāciju cilvēka gadījumā.
|
|
4.
|
Balstoties uz pašlaik pieejamajiem datiem, ir pierādīts, ka metodi h-CLAT var izmantot testējamām ķimikālijām, kuru sakarā aktuālas ir dažādas organiskās funkcionālās grupas, reakcijas mehānismi, ādas sensibilizācijas potence (kas noteikta in vivo pētījumos) un fizikālķīmiskās īpašības (3) (14) (15). Metode h-CLAT ir izmantojama testējamajām ķimikālijām, kuras šķīst vai stabili disperģējas (t. i., koloīds vai suspensija, kurā testējamā ķimikālija nenosēžas vai no šķīdinātāja/nesēja neatdalās atsevišķās fāzēs) piemērotā šķīdinātājā/nesējā (sk. 14. punktu). Testējamajām ķimikālijām, kuru Log Kow pārsniedz 3,5, ir tendence dot pseidonegatīvus rezultātus (14). Tāpēc nevajadzētu ņemt vērā negatīvus rezultātus testos ar testējamajām ķimikālijām, kuru Log Kow pārsniedz 3,5. Tomēr pozitīvus rezultātus, kas iegūti testos ar testējamajām ķimikālijām, kuru Log Kow pārsniedz 3,5, tik un tā var izmantot par palīglīdzekli testējamās ķimikālijas atzīšanai par ādas sensibilizatoru. Turklāt izmantotās šūnu līnijas ierobežotās metaboliskās spējas (16) un eksperimenta apstākļu dēļ negatīvus rezultātus testā h-CLAT var dot arī prohaptēni (t. i., vielas, kurām nepieciešama fermentatīva aktivācija, piem., ar P450 fermentiem) un prehaptēni (t. i., vielas, kuras aktivizējas oksidējoties), jo īpaši tad, ja oksidācija norit lēni (15). Ar h-CLAT var vērtēt fluorescējošas testējamās ķimikālijas (17), tomēr ļoti fluorescējošas testējamās ķimikālijas, kuru starojuma viļņa garums ir tāds pats kā fluoresceīna izotiocianātam (FITC) vai propīdija jodīdam (PI), traucē detektēšanu ar plūsmas citometriju, tāpēc ar FITC konjugētām antivielām vai PI tās nevar izvērtēt pareizi. Tādā gadījumā var izmantot ar citiem fluorohromiem marķētas antivielas vai citus citotoksicitātes marķierus, ja vien, testējot 1.2. papildinājumā norādītās standartvielas, var pierādīt, ka tie dod līdzīgus rezultātus kā ar FITC marķētās antivielas (sk. 24. punktu) vai PI (sk. 18. punktu). Ņemot vērā iepriekš minēto, negatīvie rezultāti jāinterpretē noteikto ierobežojumu kontekstā un kopā ar citiem informācijas avotiem IATA satvarā. Gadījumos, kad ir liecības, ka metode h-CLAT nav izmantojama vēl citu konkrētu kategoriju testējamajām ķimikālijām, to attiecīgo kategoriju ķimikālijām neizmanto.
|
|
5.
|
Kā norādīts iepriekš, metode h-CLAT palīdz ādas sensibilizatorus nošķirt no ādas nesensibilizatoriem. Izmantojot integrētu pieeju, piem., IATA, tā var palīdzēt arī novērtēt sensibilizācijas potenci (4) (5) (9). Tomēr, lai noteiktu, kā h-CLAT rezultāti varētu palīdzēt novērtēt potenci, jāiegulda papildu darbs, kura pamatā vēlams būt datiem par cilvēku.
|
|
6.
|
Definīcijas ir sniegtas 1.1. papildinājumā.
|
TESTA PRINCIPS
|
7.
|
Metode h-CLAT ir in vitro tests, kas kvantificē izmaiņas šūnu virsmas marķieru (piem., CD86 un CD54) ekspresijā cilvēka monocītiskās leikēmijas šūnu līnijā THP-124 stundas pēc ekspozīcijas testējamajai ķimikālijai. Šīs virsmas molekulas ir tipiski THP-1 monocītu aktivācijas marķieri un var imitēt DŠ aktivāciju, kam ir izšķirīga funkcija T šūnu praimingā (aktivācijā). Izmaiņas virsmas marķieru ekspresijā mēra ar plūsmas citometriju pēc šūnu iekrāsošanas ar antivielām, kas marķētas ar fluorohromiem. Paralēli izdara arī citotoksiskuma mērījumu, lai noteiktu, vai virsmas marķieru ekspresijas aktivācija ir novērojama arī pie subcitotoksiskām koncentrācijām. Lai būtu vieglāk sensibilizatorus nošķirt no nesensibilizatoriem, aprēķina un prognozes modelī (sk. 26. punktu) izmanto virsmas marķieru relatīvo fluorescences intensitāti salīdzinājumā ar šķīdinātāja/nesēja kontroles fluorescences intensitāti.
|
KOMPETENCES PIERĀDĪŠANA
|
8.
|
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām jāpierāda sava tehniskā kompetence, izmantojot 1.2. papildinājumā uzskaitītās 10 standartvielas. Testa lietotājiem turklāt jāuztur vēsturiska datubāze ar datiem, kas iegūti reaģētspējas pārbaudēs (sk. 11. punktu) un ar pozitīvajām un šķīdinātāja/nesēja kontrolēm (sk. 20.–22. punktu), un ar šo datu palīdzību jāpārliecinās, ka testa reproducējamība konkrētajā laboratorijā laika gaitā saglabājas tāda pati.
|
PROCEDŪRA
|
9.
|
Šis tests ir balstīts uz h-CLAT dzīvniekeksperimentiem alternatīvu metožu datubāzes pakalpojuma (DB-ALM) protokolu Nr. 158 (18), kas ir EURL ECVAM koordinētajā validācijas pētījumā izmantotais protokols. Metodi h-CLAT ieviešot un izmantojot laboratorijā, ieteicams lietot šo protokolu. Tālāk sniegts galveno h-CLAT metodes komponentu un procedūru apraksts; metodei h-CLAT ir divas daļas: devas noteikšanas tests un CD86/CD54 ekspresijas mērījums.
|
Šūnu sagatavošana
|
10.
|
Metodei h-CLAT izmanto cilvēka monocītiskās leikēmijas šūnu līniju THP-1. Šūnas (TIB-202™) ir ieteicams sagādāt no kvalificētas šūnu bankas, piem., Amerikas tipveida kultūru kolekcijas.
|
|
11.
|
THP-1 šūnas kultivē 37oC pie 5 % CO2, mitrinātā atmosfērā, RPMI-1640 barotnē, kas papildināta ar 10 % liellopu embriju seruma jeb govju fetālā seruma (FBS), 0,05 mM 2-merkaptoetanola, 100 vien./ml penicilīna un 100 μg/ml streptomicīna. No penicilīna un streptomicīna izmantošanas kultūras barotnē ir iespējams izvairīties. Tomēr tādā gadījumā lietotājiem jāpārliecinās, ka antibiotiku neesība kultūras barotnē neietekmē rezultātus, piem., veicot testu ar 1.2. papildinājumā uzskaitītajām standartvielām. Jebkurā gadījumā, lai minimalizētu kontaminācijas risku, neatkarīgi no tā, vai šūnu kultūras barotnē ir antibiotikas vai to nav, jāievēro laba šūnu kultivēšanas prakse. THP-1 šūnas parasti uzsēj reizi 2–3 dienās blīvumā 0,1 līdz 0,2 × 106 šūnas/ml. To blīvums jāuztur diapazonā no 0,1 līdz 1,0 × 106 šūnas/ml. Pirms šūnu izmantošanas testēšanai tās jākvalificē, pārbaudot to reaģētspēju. Šūnu reaģētspēja jāpārbauda, izmantojot pozitīvās kontroles, 2,4-dinitrohlorbenzolu (DNHB) (CAS Nr. 97-00-7, ≥ 99 % tīrība) un niķeļa sulfātu (NiSO4) (CAS Nr. 10101-97-0, ≥ 99 % tīrība), un negatīvo kontroli, pienskābi (PS) (CAS Nr. 50-21-5, ≥ 85 % tīrība), divas nedēļas pēc atkausēšanas. Gan DNHB, gan NiSO4 būtu jāizraisa pozitīva atbildreakcija attiecībā uz abiem šūnu virsmas marķieriem — CD86 un CD54 —, un PS būtu attiecībā uz tiem abiem jāizraisa negatīva atbildreakcija. Testam izmanto tikai šūnas, kas reaģētspējas pārbaudi izturējušas. Šūnas var pavairot līdz diviem mēnešiem pēc atkausēšanas. Pasāžu skaitam nevajadzētu pārsniegt 30. Reaģētspējas pārbaude veicama saskaņā ar 20.–24. punktā aprakstītajām procedūrām.
|
|
12.
|
Testēšanas vajadzībām THP-1 šūnas uzsēj vai nu blīvumā 0,1 × 106 šūnas/ml, vai 0,2 × 106 šūnas/ml un kultivēšanas kolbās priekškultivē attiecīgi 72 vai 48 stundas. Ir svarīgi, lai šūnu blīvums kultivēšanas kolbā tieši pēc priekškultivēšanas perioda visos eksperimentos būtu iespējami līdzīgs (izmantojot vienu vai abus iepriekš minētos priekškultivēšanas nosacījumus), jo šūnu blīvums kultivēšanas kolbā tieši pēc priekškultivēšanas var ietekmēt alergēnu inducēto CD86/CD54 ekspresiju (19). No kultivēšanas kolbas ievāktās šūnas testēšanas dienā resuspendē svaigā kultūras barotnē blīvumā 2 × 106 šūnas/ml. Tad šūnas sadala vai nu 24 plakandibena iedobju platē (500 μl, 1 × 106 šūnas uz iedobi), vai 96 plakandibena iedobju platē (80 μl, 1,6 × 105 šūnas uz iedobi).
|
Devas noteikšanas tests
|
13.
|
Lai noteiktu CV75, kas ir testējamās ķimikālijas koncentrācija, pie kuras šūnu dzīvotspēja salīdzinājumā ar to dzīvotspēju šķīdinātāja/nesēja kontrolē ir 75 %, veic devas noteikšanas testu. CV75 izmanto tam, lai noteiktu testējamo ķimikāliju koncentrāciju CD86/CD54 ekspresijas mērījumam (sk. 20.–24. punktu).
|
Testējamo ķimikāliju un kontroles vielu sagatavošana
|
14.
|
Testējamās ķimikālijas un kontroles vielas sagatavo testēšanas dienā. Izmantojot metodi h-CLAT, testējamās ķimikālijas izšķīdina vai stabili disperģē (sk. arī 4. punktu) fizioloģiskajā šķīdumā vai barotnē, kas ir pirmais šķīdinātāja/nesēja variants, vai dimetilsulfoksīdā (DMSO, ≥ 99 % tīrība), kas ir otrais šķīdinātāja/nesēja variants, ja testējamā ķimikālija pirmajos divos šķīdinātājos/nesējos nešķīst vai neveido stabilu dispersiju, līdz galīgajai koncentrācijai 100 mg/ml (fizioloģiskajā šķīdumā vai barotnē) vai 500 mg/ml (DMSO). Ja tam ir sniegts pietiekams zinātniskais pamatojums, var izmantot citus šķīdinātājus/nesējus. Jāņem vērā testējamās ķimikālijas stabilitāte galīgajā šķīdinātājā/nesējā.
|
|
15.
|
Testējamo ķimikāliju izejšķīdumus, kas ir 100 mg/ml (fizioloģiskajā šķīdumā vai barotnē) vai 500 mg/ml (DMSO), atšķaida šādi:
|
—
|
ja šķīdinātājs/nesējs ir fizioloģiskais šķīdums vai barotne: sagatavo astoņus izejšķīdumus (astoņās koncentrācijās), ar attiecīgo šķīdinātāju/nesēju izdarot sērijveida atšķaidījumus ar atšķaidījuma koeficientu 2. Šos izejšķīdumus tad vēl atšķaida kultūras barotnē, izmantojot atšķaidījuma koeficientu 50 (tā iegūstot darba šķīdumus). Ja plates augstākā galīgā koncentrācija 1 000 μg/ml nav toksiska, maksimālā koncentrācija ar jaunu citotoksiskuma testu jānosaka vēlreiz. Ja testējamā ķimikālija fizioloģiskajā šķīdumā vai barotnē izšķīdusi vai stabili disperģēta, galīgajai koncentrācijai platē nevajadzētu pārsniegt 5 000 μg/ml;
|
|
—
|
ja šķīdinātājs/nesējs ir DMSO: sagatavo astoņus izejšķīdumus (astoņās koncentrācijās), ar attiecīgo šķīdinātāju/nesēju izdarot sērijveida atšķaidījumus ar atšķaidījuma koeficientu 2. Šos izejšķīdumus tad vēl atšķaida kultūras barotnē, izmantojot atšķaidījuma koeficientu 250 (tā iegūstot darba šķīdumus). Galīgā koncentrācija platē nedrīkst pārsniegt 1 000 μg/ml, pat ja šī koncentrācija nav toksiska.
|
Visbeidzot darba šķīdumus izmanto ekspozīcijai, THP-1 šūnu suspensijai platē pievienojot tādu pašu tilpumdaudzumu darba šķīduma (sk. 17. punktu), tā iegūstot vēl vienu atšķaidījumu ar atšķaidījuma koeficientu 2 (galīgais koncentrāciju diapazons platē parasti ir 7,81–1 000 μg/ml).
|
|
16.
|
h-CLAT par šķīdinātāja/nesēja kontroli izmanto kultūras barotni (testējamajām ķimikālijām, kas izšķīdinātas vai stabili disperģētas (sk. 4. punktu) barotnē vai fizioloģiskajā šķīdumā) vai DMSO (testējamajām ķimikālijām, kas izšķīdinātas vai stabili disperģētas DMSO), ko platē testē vienā galīgajā koncentrācijā — 0,2 %. To atšķaida tāpat, kā saskaņā ar 15. punktu atšķaida darba šķīdumus.
|
Testējamo ķimikāliju un kontroles vielu aplicēšana
|
17.
|
15. un 16. punktā aprakstīto kultūras barotni vai darba šķīdumus attiecībā 1:1 (tilp./tilp.) samaisa ar 24 plakandibena iedobju vai 96 plakandibena iedobju platē sagatavotajām šūnu suspensijām (sk. 12. punktu). Tad apstrādātās plates 24±0,5 stundas inkubē 37oC temperatūrā pie 5 % CO2. Jāizvairās no gaistošu testējamo ķimikāliju iztvaikošanas un testējamo ķimikāliju krusteniskās starpiedobju kontaminācijas, piem., pirms inkubēšanas ar testējamajām ķimikālijām plati noslēdzot (20).
|
Iekrāsošana ar propīdija jodīdu (PI)
|
18.
|
Pēc 24±0,5 ekspozīcijas stundām šūnas pārliek paraugu stobriņos un ievāc centrifugējot. Supernatantus nolej un atlikušās šūnas resuspendē 200 μl (96 iedobju plates gadījumā) vai 600 μl (24 iedobju plates gadījumā) fosfātu fizioloģiskā buferšķīduma, kas satur 0,1 % liellopu seruma albumīna (iekrāsošanas buferšķīdums). 200 μl šūnu suspensijas pārnes uz 96 apaļdibena iedobju plati (ja paraugi ir no 96 iedobju plates) vai mikrostobriņu (ja paraugi ir no 24 iedobju plates) un divreiz noskalo ar 200 μl (96 iedobju plates gadījumā) vai 600 μl (24 iedobju plates gadījumā) iekrāsošanas buferšķīduma. Visbeidzot šūnas resuspendē iekrāsošanas buferšķīdumā (piem., 400 μl), kam pievieno PI šķīdumu (piem., 20 μl; galīgā PI koncentrācija var būt, piem., 0,625 μg/ml). Citus citotoksicitātes marķierus, piem., 7-aminoaktinomicīnu D (7-AAD), tripānzilo v. tml., var izmantot tad, ja iespējams pierādīt, ka alternatīvie iekrāsotāji dod līdzīgus rezultātus kā PI, piem., veicot testus ar 1.2. papildinājumā uzskaitītajām standartvielām.
|
Citotoksiskuma mērīšana ar plūsmas citometriju un CV75 vērtības aplēšana
|
19.
|
PI uzņemšanu analizē ar plūsmas citometriju, izmantojot ieguves kanālu FL-3. Kopā iegūst (izmēra) 10 000 dzīvu (PI-negatīvu) šūnu. Šūnu dzīvotspēju var aprēķināt ar citometra analīzes programmu, izmantojot nākamo vienādojumu. Ja šūnu dzīvotspēja ir maza, iegūst (izmēra) līdz 30 000 šūnu, ieskaitot nedzīvas šūnas. Alternatīva ir datus iegūt vienas minūtes laikā pēc analīzes sākšanas.

CV75 vērtību (sk. 13. punktu), t. i., koncentrāciju, pie kuras izdzīvo 75 % THP-1 šūnu (25 % citotokscitāte), aprēķina ar log-lineāro interpolāciju saskaņā ar šādu vienādojumu:

kur:
a ir minimālā vērtība šūnu dzīvotspējai virs 75 %;
c ir maksimālā vērtība šūnu dzīvotspējai zem 75 %;
b un d ir koncentrācijas, pie kurām šūnu dzīvotspēja ir attiecīgi a un c.

CV75 var noteikt arī citādi, ja vien pierāda (piem., testējot standartvielas), ka tas neietekmē rezultātus.
|
CD86/CD54 ekspresijas mērījums
Testējamo ķimikāliju un kontroles vielu sagatavošana
|
20.
|
Testējamās ķimikālijas izšķīdina vai stabili disperģē piemērotā šķīdinātājā/nesējā (fizioloģiskajā šķīdumā, barotnē vai DMSO; sk. 14. punktu). Vispirms testējamās ķimikālijas atšķaida līdz koncentrācijai, kas atbilst simtkārtējai (fizioloģiskā šķīduma vai barotnes gadījumā) vai piecsimtkārtējai (DMSO gadījumā) devas noteikšanas testā noteiktajai CV75 × 1,2 (sk. 19. punktu). Ja CV75 noteikt nav bijis iespējams (t. i., devas noteikšanas testā nav novērota pietiekama citotoksicitāte), par sākuma koncentrāciju izmanto augstāko koncentrāciju, kurā testējamā ķimikālija katrā šķīdinātājā/nesējā vēl šķīst vai stabili disperģējas. Jāņem vērā, ka galīgā koncentrācija platē nedrīkst pārsniegt 5 000 μg/ml (fizioloģiskā šķīduma vai barotnes gadījumā) vai 1 000 μg/ml (DMSO gadījumā). Tad sērijveida atšķaidījumos ar attiecīgo šķīdinātāju/nesēju (atšķaidījuma koeficients 1,2) iegūst izejšķīdumus (astoņās koncentrācijās diapazonā no 100 × 1,2 × CV75 līdz 100 × 0,335 × CV75 (fizioloģiskā šķīduma vai barotnes gadījumā) vai no 500 × 1,2 × CV75 līdz 500 × 0,335 × CV75 (DMSO gadījumā)), ko testēt ar metodi h-CLAT (dozēšanas shēmas piemēru sk. DB-ALM protokolā Nr. 158). Šos izejšķīdumus tad vēl atšķaida kultūras barotnē, izmantojot atšķaidījuma koeficientu 50 (fizioloģiskā šķīduma vai barotnes gadījumā) vai 250 (DMSO gadījumā), un tā iegūst darba šķīdumus. Iegūtos darba šķīdumus izmanto ekspozīcijai, platē vēl veicot pēdējo atšķaidījumu ar koeficientu 2. Ja rezultāti neatbilst 29. un 30. punktā aprakstītajiem pieņemamības kritērijiem, devas noteikšanas testu var atkārtot, lai CV75 noteiktu precīzāk. Jāņem vērā, ka CD86/CD54 ekspresijas mērījumam var izmantot tikai 24 iedobju plates.
|
|
21.
|
Šķīdinātāja/nesēja kontroli sagatavo tā, kā aprakstīts 16. punktā. Metodē h-CLAT par pozitīvo kontroli izmanto DNHB (sk. 11. punktu), kura izejšķīdumus sagatavo DMSO un atšķaida tā, kā izejšķīdumu sagatavošana aprakstīta 20. punktā. Par pozitīvo kontroli CD86/CD54 ekspresijas mērījumam platē izmanto DNHB vienā galīgajā koncentrācijā (parasti 4,0 μg/ml). Lai platē DNHB būtu koncentrācijā 4,0 μg/ml, sagatavo 2 mg/ml DNHB izejšķīdumu DMSO un tad kultūras barotnē ar koeficientu 250 atšķaida līdz 8 μg/ml darba šķīdumam. Par pozitīvās kontroles koncentrāciju var izmantot arī DNHB CV75, ko nosaka katrā testējošajā iestādē. Ja ir pieejami vēsturiskie dati, no kuriem var iegūt salīdzināmus izpildes reižu pieņemamības kritērijus, var izmantot citas piemērotas pozitīvās kontroles. Pozitīvajām kontrolēm galīgā koncentrācija platē nedrīkst pārsniegt 5 000 μg/ml (fizioloģiskā šķīduma vai barotnes gadījumā) vai 1 000 μg/ml (DMSO gadījumā). Izpildes reižu pieņemamības kritēriji ir tādi paši kā testējamajai ķimikālijai (sk. 29. punktu), izņemot pēdējo pieņemamības kritēriju, jo pozitīvo kontroli testē tikai vienā koncentrācijā.
|
Testējamo ķimikāliju un kontroles vielu aplicēšana
|
22.
|
Lai iegūtu prognozi, katrai testējamajai ķimikālijai un kontroles vielai ir nepieciešams viens eksperiments. Katrs eksperiments sastāv no vismaz divām neatkarīgām CD86/CD54 ekspresijas mērījuma izpildes reizēm (sk. 26.–28. punktu). Katru neatkarīgo izpildes reizi izdara citā dienā vai tajā pašā dienā, ja attiecībā uz katru izpildes reizi izpildīti šādi nosacījumi: a) tiek neatkarīgi sagatavoti svaigi testējamās ķimikālijas un antivielu šķīdumu izejšķīdumi un darba šķīdumi un b) tiek izmantotas neatkarīgi ievāktas šūnas (t. i., šūnas ir ievāktas no dažādām kultivēšanas kolbām); tomēr šūnas var būt no vienas un tās pašas pasāžas. Testējamās ķimikālijas un kontroles vielas, kas sagatavotas kā darba šķīdumi (500 μl), samaisa ar 500 μl suspendētu šūnu (1 x 106 šūnas) attiecībā 1:1 un šūnas 24±0,5 stundas inkubē, kā aprakstīts 20. un 21. punktā. Katrā izpildes reizē pietiek ar vienu katras testējamās ķimikālijas un kontroles vielas koncentrācijas replikātu, jo prognozi iegūst vismaz divās neatkarīgās izpildes reizēs.
|
Šūnu iekrāsošana un analīze
|
23.
|
Pēc 24±0,5 ekspozīcijas stundām šūnas no 24 iedobju plates pārvieto uz paraugu stobriņiem, ievāc centrifugējot un tad divreiz noskalo ar 1 ml iekrāsošanas buferšķīduma (ja nepieciešams, skalošanu var vēl atkārtot). Pēc skalošanas šūnas nobloķē ar 600 μl bloķēšanas šķīduma (iekrāsošanas buferšķīdums, kas satur 0,01 % (masa/tilp.) globulīna (Kona frakcija II, III, cilvēka; SIGMA, #G2388-10G vai līdzvērtīga)) un inkubē 15 min 4oC. Pēc bloķēšanas šūnas sadala trīs 180 μl alikvotās 96 apaļdibena iedobju platē vai mikrostobriņā.
|
|
24.
|
Pēc centrifugēšanas šūnas 30 min iekrāso 4oC, izmantojot 50 μl ar FITC marķētu anti-CD86, anti-CD54 vai peļu IgG1 (izotips) antivielu. h-CLAT DB-ALM protokolā Nr. 158 (18) aprakstītās antivielas izmanto, tās attiecībā 3:25 (tilp./tilp.) (CD86 (BD-PharMingen, #555657; Clone: Fun-1)) vai 3:50 (tilp./tilp.) (CD54 (DAKO, #F7143; Clone: 6.5B5) un IgG1 (DAKO, #X0927)) atšķaidot ar iekrāsošanas buferšķīdumu. Pēc testa izstrādātāju atzinuma šie antivielu atšķaidījuma koeficienti nodrošina vislabāko signāla un trokšņa attiecību. Testa izstrādātāju pieredzē dažādu partiju antivielu fluorescences intensitāte parasti ir līdzīga. Tomēr, lai noteiktu vispiemērotākās koncentrācijas, lietotāji var apsvērt iespēju antivielas titrēt konkrētās laboratorijas apstākļos. Ar citiem fluorohromiem marķētas anti-CD86 un/vai anti-CD-54 antivielas var izmantot, ja, piem., testējot 1.2. papildinājumā norādītās standartvielas, var pierādīt, ka tās dod līdzīgus rezultātus kā ar FITC konjugētās antivielas. Jāpiezīmē, ka, mainot h-CLAT DB-ALM protokolā Nr. 158 (18) aprakstīto antivielu klonu vai piegādātāju, rezultāti var mainīties. Pēc vismaz divkārtējas noskalošanas ar 150 μl iekrāsošanas buferšķīduma šūnas resuspendē iekrāsošanas buferšķīdumā (piem., 400 μl) un tam pievieno PI šķīdumu (piem., 20 μl tā, lai galīgā koncentrācija būtu 0,625 μl/ml) vai cita citotoksicitātes marķiera šķīdumu (sk. 18. punktu). CD86 un CD54 ekspresijas līmeņus un šūnu dzīvotspēju analizē ar plūsmas citometriju.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Datu izvērtēšana
|
25.
|
CD86 un CD54 ekspresiju analizē ar plūsmas citometriju, izmantojot ieguves kanālu FL-1. Balstoties uz ģeometrisko vidējo fluorescences intensitāti (VFI), CD86 un CD54 relatīvo fluorescences intensitāti (RFI) pozitīvās kontroles (ctrl) šūnām un ar ķimikāliju apstrādātajām šūnām aprēķina ar šādu vienādojumu:

Ar 19. punktā norādīto vienādojumu aprēķina arī izotipa kontroles (ctrl) šūnu (šūnas, kas iekrāsotas ar peļu IgG1 (izotips) antivielām) dzīvotspēju.
|
Prognozes modelis
|
26.
|
Lai iegūtu vienu prognozi (POZITĪVU vai NEGATĪVU), CD86/CD54 ekspresijas mērījumam katru testējamo ķimikāliju testē vismaz divās neatkarīgās izpildes reizēs. Uzskata, ka h-CLAT prognoze ir POZITĪVA, ja vismaz 2 no 2 vai vismaz 2 no 3 neatkarīgām izpildes reizēm ir izpildīti šādi nosacījumi; pretējā gadījumā h-CLAT prognozi uzskata par NEGATĪVU (1. attēls):
|
—
|
CD86 RFI jebkurā testētajā koncentrācijā ir vismaz 150 % (ar šūnu dzīvotspēju ≥ 50 %);
|
|
—
|
CD54 RFI jebkurā testētajā koncentrācijā ir vismaz 200 % (ar šūnu dzīvotspēju ≥ 50 %).
|
|
|
27.
|
Tātad, ja pirmās divas izpildes reizes abas dod pozitīvu rezultātu attiecībā uz CD86 un/vai abas dod pozitīvu rezultātu attiecībā uz CD54, uzskata, ka h-CLAT prognoze ir POZITĪVA un trešā izpildes reize nav vajadzīga. Līdzīgā kārtā, ja pirmās divas izpildes reizes attiecībā uz abiem marķieriem ir negatīvas, uzskata, ka h-CLAT prognoze ir NEGATĪVA (pienācīgi ņemot vērā 30. punktā noteikto) un trešā izpildes reize nav vajadzīga. Savukārt, ja pirmās divas izpildes reizes attiecībā uz vismaz vienu no marķieriem (CD54 vai CD86) nesakrīt, ir vajadzīga trešā izpildes reize, un tad galīgo prognozi nosaka pēc tā, kuru rezultātu pārsvars novērojams trīs atsevišķajās izpildes reizēs (proti, 2 no 3). Šajā aspektā jāņem vērā, ka, ja ir divas neatkarīgas izpildes reizes un viena ir pozitīva tikai attiecībā uz CD86 (turpmāk “P1”) un otra ir pozitīva tikai attiecībā uz CD54 (turpmāk “P2”), ir vajadzīga trešā izpildes reize. Ja trešā izpildes reize ir negatīva attiecībā uz abiem marķieriem (turpmāk “N”), uzskata, ka h-CLAT prognoze ir NEGATĪVA. Savukārt, ja trešā izpildes reize attiecībā uz vienu no marķieriem (P1 vai P2) vai tiem abiem (turpmāk “P12”) ir pozitīva, uzskata, ka h-CLAT prognoze ir POZITĪVA.
1. attēls. Metodē h-CLAT izmantotais prognozes modelis. h-CLAT prognoze jāskata IATA kontekstā un saskaņā ar vispārīgā ievada 7. un 8. punkta noteikumiem.
P1: izpildes reize, kas ir pozitīva tikai attiecībā uz CD86; P2; izpildes reize, kas ir pozitīva tikai attiecībā uz CD54; P12: izpildes reize, kas ir pozitīva gan attiecībā uz CD86, gan CD54; N: izpildes reize, kas nav pozitīva ne attiecībā uz CD86, ne CD54.
* Lodziņos redzamas relevantās pirmo divu izpildes reižu rezultātu kombinācijas neatkarīgi no tā, kādā kārtībā šie rezultāti iegūti.
# Lodziņos redzamas relevantās trīs izpildes reižu rezultātu kombinācijas, kuru pamatā ir iepriekšējā lodziņā norādītie pirmo divu izpildes reižu rezultāti; ir vienalga, kādā kārtībā šie rezultāti iegūti.
|
|
28.
|
Testējamajām ķimikālijām, attiecībā uz kurām ar h-CLAT iegūtā prognoze ir POZITĪVA, var izvēlēties noteikt divas efektīvās koncentrācijas vērtības, EC150 marķierim CD86 un EC200 marķierim CD54, t. i., koncentrācijas, kurās testējamo ķimikāliju inducētā RFI ir 150 vai 200. Izmantotas integrētā pieejā, piem., IATA (4) (5) (6) (7) (8), šīs efektīvās koncentrācijas vērtības var palīdzēt novērtēt sensibilizācijas potenci (9). Tās var aprēķināt ar šādiem vienādojumiem:
EC150 (CD86) = Bkonc.
+ [(150 - BRFI
)/ARFI - BRFI
) × (Akonc. - Bkonc.
)]
EC200 (CD86) = Bkonc.
+ [(200 - BRFI
)/ARFI - BRFI
) × (Akonc. - Bkonc.
)]
kur
Akonc. ir zemākā koncentrācija, izteikta μg/ml, kurā RFI > 150 (CD86) vai 200 (CD54);
Bkonc. ir augstākā koncentrācija, izteikta μg/ml, kurā RFI < 150 (CD86) vai 200 (CD54);
ARFI ir RFI zemākajā koncentrācijā, kurā RFI > 150 (CD86) vai 200 (CD54);
BRFI ir RFI augstākajā koncentrācijā, kurā RFI < 150 (CD86) vai 200 (CD54).
Lai EC150 un EC200 vērtības noteiktu precīzāk, var būt vajadzīgas trīs neatkarīgas CD86/CD54 ekspresijas mērījuma izpildes reizes. Galīgās EC150 un EC200 nosaka kā trīs neatkarīgo izpildes reižu efektīvo koncentrāciju medianālo vērtību. Ja pozitivitātes kritērijiem (sk. 26.–27. punktu) atbilst tikai divas no trim neatkarīgajām izpildes reizēm, no divām aprēķinātajām vērtībām pieņem augstāko EC150 vai EC200.
|
Pieņemamības kritēriji
|
29.
|
Izmantojot metodi h-CLAT, jābūt izpildītiem šādiem pieņemamības kritērijiem (22) (27).
|
—
|
Šūnu dzīvotspējai barotnē un šķīdinātāja/nesēja kontrolē jābūt lielākai nekā 90 %.
|
|
—
|
Šķīdinātāja/nesēja kontrolē ne CD86, ne CD54 RFI vērtības nedrīkst pārsniegt pozitivitātes kritērijus (CD86 RFI ≥ 150 % un CD54 RFI ≥ 200 %). Šķīdinātāja/nesēja kontroles RFI vērtības aprēķina ar 25. punktā norādīto formulu (“ķimikālijas VFI” aizstājot ar “šķīdinātāja/nesēja VFI” un “šķīdinātāja/nesēja VFI” aizstājot ar “(barotnes) kontroles VFI”).
|
|
—
|
Gan barotnes, gan šķīdinātāja/nesēja kontroles CD86 un CD54 (abu divu) VFI attiecībai pret izotipa kontroles VFI jābūt > 105 %.
|
|
—
|
Pozitīvās kontroles (DNHB) CD86 un CD54 VFI vērtībām jāatbilst pozitivitātes kritērijiem (CD86 RFI ≥ 150 un CD54 RFI ≥ 200), un šūnu dzīvotspējai jāpārsniedz 50 %.
|
|
—
|
Testējamās ķimikālijas gadījumā šūnu dzīvotspējai katrā izpildes reizē jāpārsniedz 50 % vismaz četrās testētajās koncentrācijās.
|
|
|
30.
|
Negatīvi rezultāti ir pieņemami tikai attiecībā uz testējamajām ķimikālijām, kuru gadījumā šūnu dzīvotspēja augstākajā testētajā koncentrācijā (piem., 1,2 × CV75 saskaņā ar 20. punktā aprakstīto sērijveida atšķaidīšanas shēmu) nesasniedz 90 %. Ja šūnu dzīvotspēja koncentrācijā 1,2 × CV75 ir vienāda ar vai lielāka par 90 %, negatīvo rezultātu atmet. Tādā gadījumā ir ieteicams mēģināt devu izvēli vēl precizēt, CV75 noteikšanu atkārtojot. Jāņem vērā, ka gadījumos, kad testējamās ķimikālijas maksimālā testējamā koncentrācija ir 5 000 μg/ml fizioloģiskajā šķīdumā (vai barotnē, vai citos šķīdinātājos/nesējos), 1 000 μg/ml DMSO vai augstākā koncentrācija, kurā tā vēl šķīst, negatīvs rezultāts ir pieņemams pat tad, ja šūnu dzīvotspēja pārsniedz 90 %.
|
Testēšanas pārskats
|
31.
|
Testēšanas pārskatā norāda šādas ziņas.
|
Testējamā ķimikālija
|
Vienkomponenta vielas
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, Log Kow, šķīdība ūdenī, šķīdība DMSO, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
Daudzkomponentu vielas, UVCB un maisījumi
|
Iespējami izsmeļošs raksturojums ar, piem., sastāvdaļu ķīmisko identitāti (sk. iepriekš), tīrību, kvantitatīvo saturu un relevantajām fizikālķīmiskajām īpašībām
|
|
Izskats, šķīdība ūdenī, šķīdība DMSO un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Molekulmasa vai šķietamā molekulmasa, ja tas ir maisījums/polimērs ar zināmu sastāvu, vai cita pētījuma norisei relevanta informācija
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
|
Kontroles
|
Pozitīvā kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, Log Kow, šķīdība ūdenī, šķīdība DMSO, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama (attiecīgā gadījumā)
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Norāde uz vēsturiskajiem pozitīvo kontroļu rezultātiem, kas apliecina atbilstību testa izpildes pieņemamības kritērijiem (attiecīgā gadījumā)
|
|
|
|
Negatīvā un šķīdinātāja/nesēja kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā un ja šī informācija ir pieejama) utt.
|
|
Ja tiek izmantoti citi (testēšanas vadlīnijā neminēti) šķīdinātāji/nesēji: izskats, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
Testa apstākļi
|
Sponsora, testējošās iestādes, pētījuma vadītāja vārds/nosaukums un adrese
|
|
Izmantotā šūnu līnija, tās glabāšanas apstākļi un avots (piem., iestāde, no kuras tā saņemta)
|
|
Izmantotais plūsmas citometrs (piem., modelis), arī instrumentu iestatījumi, izmantotais globulīns, antivielas un citotoksicitātes marķieris
|
|
Procedūra, ar kādu, testējot standartvielas, pierādīta laboratorijas kompetence šā testa izpildē, un procedūra, ar kādu pierādīta šā testa rezultātu reproducējamība laika griezumā, piem., vēsturiskie kontroļu dati un/vai vēsturiskie reaģētspējas pārbaužu dati
|
|
|
Testa pieņemamības kritēriji
|
Ar šķīdinātāja/nesēja kontroli iegūtās šūnu dzīvotspējas, VFI un RFI vērtības salīdzinājumā ar pieņemamības diapazoniem
|
|
Ar pozitīvo kontroli iegūtās šūnu dzīvotspējas un RFI vērtības salīdzinājumā ar pieņemamības diapazoniem
|
|
Šūnu dzīvotspēja visās testētajās testētās ķimikālijas koncentrācijās
|
|
|
Testa procedūra
|
Izmantotais izpildes reižu skaits
|
|
Izmantotās testējamās ķimikālijas koncentrācijas, aplicēšana un ekspozīcijas laiks (ja atšķiras no rekomendētā)
|
|
Izmantoto izvērtēšanas un lēmuma pieņemšanas kritēriju apraksts
|
|
Testa procedūras modifikāciju apraksts, ja tādas bijušas
|
|
|
Rezultāti
|
Tabulā apkopoti dati, tostarp CV75 (attiecīgā gadījumā), individuālās ģeometriskās VFI, RFI, šūnu dzīvotspējas vērtības, EC150/EC200 (attiecīgā gadījumā), kas katrā izpildes reizē iegūtas attiecībā uz testējamo ķimikāliju un pozitīvo kontroli, un norāde uz testējamās ķimikālijas novērtējumu saskaņā ar prognozes modeli
|
|
Jebkādu citu relevantu novērojumu apraksts (attiecīgā gadījumā)
|
|
|
Rezultātu iztirzājums
|
Ar metodi h-CLAT iegūto rezultātu iztirzājums
|
|
Testa rezultātu apskats IATA kontekstā, ja pieejama cita relevanta informācija
|
|
|
LITERATŪRA
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767–773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report Accessible at: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Accessible at: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, France, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Available at: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
1.1. papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā testa rezultāti sakrīt ar pieņemtajām references vērtībām. Tas ir testa veiktspējas raksturlielums un viens no tā relevantuma aspektiem. Ar šo terminu, tāpat kā terminu “konkordantums”, bieži vien saprot testa pareizo iznākumu īpatsvaru (21).
AOP (nelabvēlīga iznākuma ceļš)
: no mērķķimikālijas vai līdzīgu ķimikāliju grupas ķīmiskās struktūras izrietoša notikumu sekvence no molekulārā iniciācijas notikuma līdz interesējošajam in vivo iznākumam (22).
Ķimikālija
: viela vai maisījums.
CV75
: aplēstā koncentrācija, pie kuras šūnu dzīvotspēja ir 75 %.
EC150
: koncentrācijas, pie kurām RFI vērtības CD86 ekspresijā ir 150.
EC200
: koncentrācijas, pie kurām RFI vērtības CD54 ekspresijā ir 200.
Plūsmas citometrija
: citometrijas paņēmiens, kurā fluīdā suspendētas šūnas pa vienai laiž caur fokusētu ierosinošu gaismu, kura izkliedējas šūnām un to sastāvdaļām specifiskos rakstos; šūnas nereti tiek marķētas ar fluorescējošiem marķieriem tā, ka tās gaismu vispirms absorbē un tad emitē mainītā frekvencē.
Bīstamība
: tāda aģenta vai situācijas iedabiska īpašība, kam piemīt potenciāls atstāt nelabvēlīgu ietekmi uz šim aģentam eksponētu organismu, sistēmu vai (apakš)populāciju.
IATA (integrētā testēšanas un novērtēšanas pieeja)
: strukturēta pieeja, ko izmanto ķimikālijas vai ķimikāliju grupas bīstamības identificēšanai (potenciāls), bīstamības raksturošanai (potence) un/vai drošuma novērtēšanai (potenciāls/potence un ekspozīcija) un kas stratēģiskā veidā integrē un izsver visus relevantos datus, lai varētu pieņemt normatīvu lēmumu par potenciālo bīstamību un/vai risku, un/vai nepieciešamību veikt turpmāku selektīvu un līdz ar to minimālu testēšanu.
Barotnes kontrole
: neapstrādāts replikāts, kurā ir visi testa sistēmas komponenti. Ar šo paraugu rīkojas tāpat kā ar paraugiem, kas apstrādāti ar testējamo ķimikāliju, un citiem kontrolparaugiem, lai noteiktu, vai šķīdinātājs/nesējs mijiedarbojas ar testa sistēmu.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairāk vielām.
Vienkomponenta viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā viena galvenā sastāvdaļa veido vismaz 80 % (masa/masa).
Daudzkomponentu viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu vielas tiek iegūtas ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu vielas rodas ķīmiskās reakcijās.
Pozitīvā kontrole
: replikāts, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka tā inducē pozitīvu atbildreakciju. Lai būtu iespējams novērtēt pozitīvās kontroles atbildreakcijas mainību laikā, pozitīvās atbildreakcijas intensitāte nedrīkstētu būt pārlieku liela.
Prehaptēni
: ķimikālijas, kas par sensibilizatoriem kļūst abiotiskas transformācijas procesā.
Prohaptēni
: ķimikālijas, kuru ādas sensibilizācijas potenciālu atraisa fermentatīva aktivācija.
Relatīvā fluorescences intensitāte (RFI)
: ģeometriskās vidējās fluorescences intensitātes (VFI) relatīvās vērtības ar ķimikāliju apstrādātās šūnās salīdzinājumā ar VFI ar šķīdinātāju/nesēju apstrādātās šūnās.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testa pareizības (konkordantuma) aspektu (21).
Ticamība
: parametrs, kas izsaka, kādā pakāpē, izmantojot vienu un to pašu protokolu, testu vienā un tajā pašā laboratorijā vai dažādās laboratorijās ilgākā laika nogrieznī var veikt reproducējami. To novērtē, aprēķinot metodes intralaboratorisko un interlaboratorisko reproducējamību un intralaboratorisko atkārtojamību (21).
Izpildes reize
: reize, kurā vienu vai vairākas ķimikālijas testē paralēli ar šķīdinātāja/nesēja kontroli un pozitīvo kontroli.
Jutība
: tas, cik liela daļa no visām pozitīvajām/aktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (21).
Iekrāsošanas buferšķīdums
: fosfātu fizioloģiskais buferšķīdums, kas satur 0,1 % liellopu seruma albumīna.
Šķīdinātāja/nesēja kontrole
: neapstrādāts paraugs, kurā ir visi testa sistēmas komponenti, izņemot testējamo ķimikāliju, bet ieskaitot izmantoto šķīdinātāju/nesēju. To izmanto, lai noteiktu bāzlīnijas atbildreakciju paraugiem, kas apstrādāti ar attiecīgajā šķīdinātājā/nesējā izšķīdinātu vai stabili disperģētu testējamo ķimikāliju. Ja testā izmanto arī paralēlu barotnes kontroli, šis paraugs parāda arī to, vai šķīdinātājs/nesējs mijiedarbojas ar testa sistēmu.
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (21).
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai vajadzīgās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu.
Testējamā ķimikālija
: jebkura viela vai maisījums, ko testē ar šo metodi.
Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (ANO GHS)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un kura paredz atbilstošus saziņas elementus, piem., piktogrammas, signālvārdus, bīstamības apzīmējumus, drošības prasību apzīmējumus un drošības datu lapas, ar ko tiek sniegta informācija par ķimikāliju nelabvēlīgo ietekmi, tā aizsargājot cilvēkus (tostarp darba devējus, darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (23).
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kuras ir kompleksi reakcijas produkti vai bioloģiski materiāli.
Derīgs tests
: tests, kas tiek uzskatīts par pietiekami relevantu un ticamu konkrētam nolūkam un ir balstīts uz zinātniski pareiziem principiem. Tests nekad nav derīgs absolūtā izteiksmē; tas vienmēr ir derīgs tikai kādam konkrētam nolūkam (21).
1.2. papildinājums
STANDARTVIELAS
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām jāapliecina sava tehniskā kompetence, pareizi iegūstot paredzamo h-CLAT prognozi attiecībā uz 1. tabulā ieteiktajām 10 vielām un attiecībā uz vismaz 8 no 10 standartvielām iegūstot CV75, EC150 un EC200 vērtības, kuras ietilpst attiecīgajā references diapazonā. Standartvielas ir atlasītas tā, lai tās reprezentētu visu ādas sensibilizācijas apdraudējumu izraisīto atbildreakciju diapazonu. Citi atlases kritēriji bija vielu komerciālā pieejamība un kvalitatīvu ar metodi h-CLAT iegūtu in vivo references datu, kā arī kvalitatīvu ar metodi h-CLAT iegūtu in vitro datu pieejamība. Par metodi h-CLAT ir pieejami arī publicēti references dati (3) (14).
1. tabula.
Vielas, kuras ieteicams izmantot, lai pierādītu tehnisko kompetenci izmantot metodi h-CLAT
|
Standartvielas
|
CAS Nr.
|
Agregātstāvoklis
|
In vivo prognoze (102)
|
CV75 references diapazons μg/ml (103)
|
h-CLAT rezultāts attiecībā uz CD86 (EC150 references diapazons μg/ml) (103)
|
h-CLAT rezultāts attiecībā uz CD54 (EC200 references diapazons μg/ml) (103)
|
|
2,4-dinitrohlorbenzols
|
97-00-7
|
ciets
|
sensibilizators (ļoti spēcīgs)
|
2–12
|
pozitīvs (0,5–10)
|
pozitīvs (0,5–15)
|
|
4-fenilēndiamīns
|
106-50-3
|
ciets
|
sensibilizators (spēcīgs)
|
5–95
|
pozitīvs (< 40)
|
negatīvs (> 1,5) (104)
|
|
niķeļa sulfāts
|
10101-97-0
|
ciets
|
sensibilizators (vidējs)
|
30–500
|
pozitīvs (< 100)
|
pozitīvs (10–100)
|
|
2-merkaptobenztiazols
|
149-30-4
|
ciets
|
sensibilizators (vidējs)
|
30–400
|
negatīvs (> 10) (104)
|
pozitīvs (10–140)
|
|
R(+)-limonēns
|
5989-27-5
|
šķidrs
|
sensibilizators (vājš)
|
> 20
|
negatīvs (> 5) (104)
|
pozitīvs (< 250)
|
|
urīnvielas imidazolidinils
|
39236-46-9
|
ciets
|
sensibilizators (vājš)
|
25–100
|
pozitīvs (20–90)
|
pozitīvs (20–75)
|
|
izopropanols
|
67-63-0
|
šķidrs
|
nav sensibilizators
|
> 5 000
|
negatīvs (> 5 000 )
|
negatīvs (> 5 000 )
|
|
glicerīns
|
56-81-5
|
šķidrs
|
nav sensibilizators
|
> 5 000
|
negatīvs (> 5 000 )
|
negatīvs (> 5 000 )
|
|
pienskābe
|
50-21-5
|
šķidrs
|
nav sensibilizators
|
1 500 —5 000
|
negatīvs (> 5 000 )
|
negatīvs (> 5 000 )
|
|
4-aminobenzoskābe
|
150-13-0
|
ciets
|
nav sensibilizators
|
> 1 000
|
negatīvs (> 1 000 )
|
negatīvs (> 1 000 )
|
|
Saīsinājumi: CAS Nr. — Chemical Abstracts Service reģistra numurs
|
2. papildinājums
ĀDAS IN VITRO SENSIBILIZĀCIJA: ŠŪNU LĪNIJAS U937 AKTIVĀCIJAS TESTS (U-SENS™)
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
1.
|
Tests U-SENS™ kvantificē izmaiņas tāda šūnu virsmas marķiera ekspresijā, kas asociēts ar monocītu un dendrītisko šūnu (DŠ) aktivāciju (konkrētāk, CD86), cilvēka histiocītiskās limfomas šūnu līnijā U937 pēc ekspozīcijas sensibilizatoriem (1). Pēc tam izmērītos CD86 šūnu virsmas marķieru ekspresijas līmeņus šūnu līnijā U937 izmanto par palīglīdzekli ādas sensibilizatoru nošķiršanā no nesensibilizatoriem.
|
|
2.
|
Tests U-SENS™ ir izvērtēts validācijas pētījumā (2), ko koordinēja uzņēmums L’Oreal, un pēc tam to neatkarīgi profesionālizvērtējusi Eiropas Savienības References laboratorijas dzīvniektestēšanas alternatīvu jomā (EURL ECVAM) Zinātniskā konsultatīvā komiteja (ESAC) (3). Ņemot vērā visus pieejamos pierādījumus un regulatoru un ieinteresēto personu sniegtās ziņas, EURL ECVAM ir ieteikusi (4) U-SENS™ izmantot IATA ietvaros par palīglīdzekli, kas palīdz ādas sensibilizatorus nošķirt no nesensibilizatoriem apdraudējumu klasificēšanas un marķēšanas vajadzībām. Savā norādījumu dokumentā par ziņošanu par strukturētām pieejām datu integrācijai un individuāliem informācijas avotiem, ko izmanto ādas sensibilizācijas IATA ietvaros, ESAO aplūko vairākas gadījumu analīzes, kurās aprakstītas dažādas testēšanas stratēģijas un prognožu modeļi. Vienai no dažādajām definētajām pieejām pamatā ir U-SENS tests (5). Piemēri, kā U-SENS™ dati izmantoti kombinācijā ar citu informāciju, arī vēsturiskajiem datiem un esošiem derīgiem datiem par cilvēku (6), ir pieejami arī citur literatūrā (4) (5) (7).
|
|
3.
|
Ir pierādīts, ka tests U-SENS™ ir pārnesams uz laboratorijām, kam ir pieredze ar paņēmieniem, kuros izmanto šūnu kultūras, un ar plūsmcitometrisko analīzi. Prognožu reproducējamības līmenis, kas sagaidāms no šā testa, ir aptuveni 90 % tajā pašā laboratorijā un 84 % citās laboratorijās (8). Validācijas pētījuma rezultāti (8) un citi publicētie pētījumi (1) kopumā liecina, ka salīdzinājumā ar LLNA rezultātiem šā testa pareizība ādas sensibilizatoru (t. i., ANO GHS / CLP 1. kat.) nošķiršanā no ādas nesensibilizatoriem ir 86 % (N=166) ar jutību 91 % (118/129) un specifiskumu 65 % (24/37). Salīdzinājumā ar rezultātiem, kas iegūti testos ar cilvēkiem, testa pareizība ādas sensibilizatoru (t. i., ANO GHS / CLP 1. kat.) nošķiršanā no ādas nesensibilizatoriem ir 77 % (N=101) ar jutību 100 % (58/58) un specifiskumu 47 % (20/43). Salīdzinājumā ar LLNA testā U-SENS™ pseidonegatīvas prognozes, visticamāk, skars ķimikālijas, kurām novērojama zema līdz vidēja ādas sensibilizācijas potence (t. i., ANO GHS / CLP 1.B apakškategorija), nevis ķimikālijas, kurām novērojama augsta ādas sensibilizācijas potence (t. i., ANO GHS / CLP 1.A apakškategorija) (1) (8) (9). Visa šī informācija kopā norāda uz to, ka tests U-SENS™ var palīdzēt identificēt apdraudējumus, kas var izraisīt ādas sensibilizāciju. Tomēr pareizības vērtības, kas šeit sniegtas attiecībā uz U-SENS™ kā atsevišķu testu, ir tikai orientējošas, jo šis tests jāskata kombinācijā ar citiem informācijas avotiem IATA kontekstā un saskaņā ar vispārīgā ievada 7. un 8. punkta noteikumiem. Turklāt, izvērtējot ādas sensibilizācijas novērtēšanas metodes, kurās neizmanto dzīvniekus, jāņem vērā, ka LLNA tests, tāpat kā citi testi ar dzīvniekiem, var nepilnīgi atspoguļot situāciju cilvēka gadījumā.
|
|
4.
|
Balstoties uz pašlaik pieejamajiem datiem, ir pierādīts, ka testu U-SENS™ var izmantot testējamām ķimikālijām (arī kosmētikas līdzekļu sastāvdaļām, piem., konservantiem, virsmaktīvajiem aģentiem, aktīvajiem aģentiem, krāsām), kuru sakarā aktuālas ir dažādas organiskās funkcionālās grupas, fizikālķīmiskās īpašības, ādas sensibilizācijas potence (kas noteikta in vivo pētījumos) un plašs tādu reakcijas mehānismu spektrs, par kuriem zināms, ka tie asociēti ar ādas sensibilizāciju (t. i., Maikla akceptora mehānisms, Šifa bāzu veidošanās, acilgrupas pārneses aģenta (acilpārneses aģenta) mehānisms, bimolekulārā nukleofilā aizvietošana [SN2] vai aromātiskā nukleofilā aizvietošana[SNAr]) (1) (8) (9) (10). Tests U-SENS™ ir izmantojams testējamajām ķimikālijām, kuras šķīst vai stabili disperģējas (t. i., koloīds vai suspensija, kurā testējamā ķimikālija nenosēžas vai neatdalās no šķīdinātāja/nesēja dažādās fāzēs) piemērotā šķīdinātājā/nesējā (sk. 13. punktu). Ar U-SENS™ izdevies iegūt pareizas prognozes attiecībā uz ķimikālijām, kas ziņotajā datu kopā norādītas kā prehaptēni (t. i., ar oksidāciju aktivētas vielas) vai prohaptēni (t. i., vielas, kam vajadzīga fermentatīva aktivācija, piem., ar P450 fermentiem) (1) (10). Sakarā ar nespecifisku CD86 ekspresijas palielinājumu pseidopozitīvus rezultātus var uzrādīt vielas, kas izraisa membrānu bojājumus, jo 3 no 7 pseidopozitīvajiem rezultātiem (salīdzinājumā ar in vivo references klasifikāciju) deva virsmaktīvi aģenti (1). Tātad pozitīvi rezultāti, kas iegūti, testējot virsmaktīvus aģentus, jāvērtē piesardzīgi, savukārt attiecīgus negatīvus rezultātus tomēr var izmantot par palīglīdzekli testējamās ķimikālijas atzīšanā par nesensibilizatoru. Ar U-SENS™ var vērtēt fluorescējošas testējamās ķimikālijas (1), tomēr ļoti fluorescējošas testējamās ķimikālijas, kuru starojuma viļņa garums ir tāds pats kā fluoresceīna izotiocianātam (FITC) vai propīdija jodīdam (PI), traucē detektēšanu ar plūsmas citometriju, tāpēc ar FITC konjugētām antivielām (potenciāli pseidonegatīvi rezultāti) vai PI (nav izmērāma dzīvotspēja) tās nevar izvērtēt pareizi. Tādā gadījumā var izmantot ar citiem fluorohromiem marķētas antivielas vai citus citotoksicitātes marķierus, ja vien, testējot 2.2. papildinājumā norādītās standartvielas, var pierādīt, ka tie dod līdzīgus rezultātus kā ar FITC marķētās antivielas vai PI (sk. 18. punktu). Ņemot vērā iepriekš minēto, pozitīvi rezultāti, kas iegūti ar virsmaktīviem aģentiem, un negatīvi rezultāti, kas iegūti ar ļoti fluorescējošām testējamajām ķimikālijām, jāinterpretē noteikto ierobežojumu kontekstā un kopā ar citiem informācijas avotiem IATA satvarā. Gadījumos, kad ir liecības, ka tests U-SENS™ nav izmantojams vēl citu konkrētu kategoriju testējamajām ķimikālijām, to attiecīgo kategoriju ķimikālijām neizmanto.
|
|
5.
|
Kā norādīts iepriekš, tests U-SENS™ palīdz ādas sensibilizatorus nošķirt no ādas nesensibilizatoriem. Integrētā pieejā, piem., IATA, tas var palīdzēt novērtēt arī sensibilizācijas potenci. Tomēr, lai noteiktu, kā U-SENS™ rezultāti varētu palīdzēt novērtēt potenci, jāiegulda papildu darbs, kura pamatā vēlams būt datiem par cilvēku.
|
|
6.
|
Definīcijas ir sniegtas 2.1. papildinājumā.
|
Testa Princips
|
7.
|
Tests U-SENS™ ir in vitro tests, kas kvantificē izmaiņas šūnu virsmas marķiera CD86 ekspresijā cilvēka histiocītiskās limfomas šūnu līnijā U937 45±3 stundas pēc ekspozīcijas testējamajai ķimikālijai. Virsmas marķieris CD86 ir tipisks U937 aktivācijas marķieris. Ir zināms, ka CD86 ir līdzstimulējoša molekula, kura var atdarināt monocītisko aktivāciju, kam ir svarīga nozīme T šūnu praimingā. Izmaiņas CD86 šūnu virsmas marķiera ekspresijā mēra ar plūsmas citometriju pēc šūnu iekrāsošanas, parasti ar antivielām, kas marķētas ar fluoresceīna izotiocianātu (FITC). Paralēli izdara arī citotoksiskuma mērījumu (piem., ar PI), lai noteiktu, vai CD86 šūnu virsmas marķiera ekspresijas aktivācija ir novērojama arī pie subcitotoksiskām koncentrācijām. Lai palīdzētu nošķirt sensibilizatorus no nesensibilizatoriem, aprēķina un prognozes modelī (sk. 19. punktu) izmanto CD86 šūnu virsmas marķiera stimulācijas indeksu (SI) salīdzinājumā ar šķīdinātāja/nesēja kontroles stimulācijas indeksu.
|
Kompetences Pierādīšana
|
8.
|
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām, ievērojot labu in vitro metožu praksi (11), jāpierāda sava tehniskā kompetence, izmantojot 2.2. papildinājumā uzskaitītās 10 standartvielas. Testa lietotājiem turklāt jāuztur vēsturiska datubāze ar datiem, kas iegūti reaģētspējas pārbaudēs (sk. 11. punktu) un ar pozitīvajām un šķīdinātāja/nesēja kontrolēm (sk. 15.–16. punktu), un ar šo datu palīdzību jāpārliecinās, ka laika gaitā testa reproducējamība konkrētajā laboratorijā saglabājas tāda pati.
|
PROCEDŪRA
|
9.
|
Šis tests ir balstīts uz U-SENS™ dzīvniekeksperimentiem alternatīvu metožu datubāzes pakalpojuma (DB-ALM) protokolu Nr. 183 (12). Testu U-SENS™ ieviešot un izmantojot laboratorijā, jāizmanto darba standartprocedūras (SOP). Automatizētu U-SENS™ izpildes sistēmu var izmantot tad, ja, piem., testējot 2.2. papildinājumā norādītās standartvielas, var pierādīt, ka tā dod līdzīgus rezultātus. Tālāk sniegts galveno testa U-SENS™ komponentu un procedūru apraksts.
|
Šūnu sagatavošana
|
10.
|
Testam U-SENS™ izmanto cilvēka histiocītiskās limfomas šūnu līniju U937 (13). Šūnas (klons CRL1593.2) ir ieteicams sagādāt no kvalificētas šūnu bankas, piem., Amerikas tipveida kultūru kolekcijas.
|
|
11.
|
U937 šūnas kultivē 37 °C pie 5 % CO2, mitrinātā atmosfērā, RPMI-1 640 barotnē, kas papildināta ar 10 % liellopu embriju seruma jeb govju fetālā seruma (FCS), 2 mM L-glutamīna, 100 vienībām/ml penicilīna un 100 μg/ml streptomicīna (pilnīga barotne). U937 šūnas parasti pārsēj reizi 2–3 dienās blīvumā 1,5 vai 3 × 105 šūnas/ml. Šūnu blīvumam nevajadzētu pārsniegt 2 × 106 šūnas/ml, un šūnu dzīvotspējai, kas izmērīta ar tripānzilo uzņēmušo šūnu izslēgšanu, vajadzētu būt ≥ 90 % (neizmantot pirmajai pasāžai pēc atkausēšanas). Pirms izmantošanas testēšanai katra šūnu, FCS vai antivielu partija jākvalificē, pārbaudot tās reaģētspēju. Šūnu reaģētspēja jāpārbauda, izmantojot pozitīvo kontroli, pikrilsulfonskābi (2,4,6-trinitrobenzolsulfonskābe: TNBS) (CAS Nr.: 2508-19-2, ≥ 99 % tīrība), un negatīvo kontroli, pienskābi (PS) (CAS Nr. 50-21-5, ≥ 85 % tīrība), vismaz nedēļu pēc atkausēšanas. Pārbaudot šūnu reaģētspēju, katrai no abām kontrolēm jātestē sešas galīgās koncentrācijas (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml un PS: 1, 10, 20, 50, 100, 200 μg/ml). Pilnīgā barotnē izšķīdinātai TNBS jāizsauc pozitīva koncentrācijatkarīga CD86 atbildreakcija (kurā, piem., nākamā lielākā koncentrācija aiz koncentrācijas, kas izsauc pozitīvu atbildreakciju, CD86 SI ≥ 150, dod lielāku CD86 SI), bet pilnīgā barotnē izšķīdinātai PS jāizsauc negatīva CD86 atbildreakcija (sk. 21. punktu). Testam izmanto tikai tās šūnu partijas, kas divas reizes izturējušas reaģētspējas pārbaudi. Šūnas var pavairot līdz septiņām nedēļām pēc atkausēšanas. Pasāžu skaitam nevajadzētu pārsniegt 21. Reaģētspējas pārbaude veicama saskaņā ar 18.–22. punktā aprakstītajām procedūrām.
|
|
12.
|
Testēšanas vajadzībām U937 šūnas uzsēj vai nu blīvumā 3 × 105 šūnas/ml, vai 6 × 105 šūnas/ml un kultivēšanas kolbās priekškultivē attiecīgi 2 vai 1 dienu. Ja tam ir sniegts pietiekams zinātniskais pamatojums un var pierādīt (piem., testējot 2.2. papildinājuma standartvielas), ka tas dod līdzīgus rezultātus, nupat aprakstīto priekškultivēšanas apstākļu vietā var izmantot citus. No kultivēšanas kolbas ievāktās šūnas testēšanas dienā resuspendē svaigā kultūras barotnē blīvumā 5 × 105 šūnas/ml. Tad šūnas sadala 96 plakandibena iedobju platē, izmantojot 100 μl (galīgais šūnu blīvums: 0,5 × 105 šūnas uz iedobi).
|
Testējamo ķimikāliju un kontroles vielu sagatavošana
|
13.
|
Pirms testēšanas novērtē šķīdību. Šajā nolūkā testējamās ķimikālijas koncentrācijā 50 mg/ml izšķīdina vai stabili disperģē pilnīgā barotnē, kas ir pirmais šķīdinātāja variants, vai, ja testējamā ķimikālijā pilnīgās barotnes šķīdinātājā/nesējā nešķīst, dimetilsulfoksīdā (DMSO, ≥ 99 % tīrība), kas ir otrais šķīdinātāja/nesēja variants. Ja ķimikālija šķīdinātājā/nesējā šķīst, tad, lai veiktu testēšanu, testējamo ķimikāliju pilnīgā barotnē izšķīdina līdz galīgajai koncentrācijai 0,4 mg/ml. Ja ķimikālija šķīst tikai DMSO, to izšķīdina līdz koncentrācijai 50 mg/ml. Ja tam ir sniegts pietiekams zinātniskais pamatojums, var izmantot citus šķīdinātājus/nesējus. Jāņem vērā testējamās ķimikālijas stabilitāte galīgajā šķīdinātājā/nesējā.
|
|
14.
|
Testējamās ķimikālijas un kontroles vielas sagatavo testēšanas dienā. Tā kā netiek veikts devas noteikšanas tests, pirmajā izpildes reizē jātestē ķimikālijas šķīdums attiecīgajā šķīdinātājā/nesējā (vai nu pilnīgā barotnē, vai 0,4 % DMSO šķīduma barotnē) 6 galīgajās koncentrācijās (1, 10, 20, 50, 100 un 200 μg/ml). Nākamajās izpildes reizēs ar attiecīgo šķīdinātāju/nesēju sagatavo vismaz 4 testējamās ķimikālijas darba šķīdumus (t. i., vismaz 4 koncentrācijas) — sākot ar 0,4 mg/ml pilnīgā barotnē vai 50 mg/ml DMSO. Beigās darba šķīdumus izmanto apstrādei, platē esošajam darba šķīdumam pievienojot tādu pašu tilpumdaudzumu U937 šūnu suspensijas (sk. 11. punktu) un tādējādi panākot vēl vienu atšķaidījumu ar atšķaidījuma koeficientu 2 (12). Koncentrācijas (vismaz 4 koncentrācijas) nākamajām izpildes reizēm izvēlas atkarībā no katras iepriekšējās izpildes reizes rezultātiem (8). Izmantojamās galīgās koncentrācijas ir 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 un 200 μg/ml. Maksimālā galīgā koncentrācija ir 200 μg/ml. Ja pie 1 μg/ml konstatē pozitīvu CD86 vērtību, tad izvērtē 0,1 μg/ml, lai noskaidrotu, kādā koncentrācijā testējamā ķimikālija neinducē CD86 atbildreakciju virs pozitīvās atbildreakcijas sliekšņa. Katrā izpildes reizē, kurā tiek novērota pozitīva CD86 atbildreakcija uz attiecīgo koncentrāciju, aprēķina EC150 (koncentrāciju, pie kuras ķimikālija sasniedz pozitīvās CD86 atbildreakcijas slieksni 150 %; sk. 19. punktu). Ja testējamā ķimikālija inducē pozitīvu CD86 atbildreakciju neatkarīgi no koncentrācijas, aprēķināt EC150 var nebūt relevanti, kā aprakstīts U-SENS™ DB-ALM protokolā Nr. 183 (12). Katrā izpildes reizē, ja iespējams, aprēķina CV70 (koncentrāciju, pie kuras ķimikālija sasniedz citotoksiskuma slieksni 70 %, sk. 19. punktu) (12). Lai noskaidrotu, kā koncentrācija ietekmē CD86 atbildreakcijas palielinājumu, visas koncentrācijas no izmantojamo koncentrāciju vidus jāizvēlas regulāros atstatumos diapazonā no EC150 (vai lielākā necitotoksiskā koncentrācija, pie kuras CD86 atbildreakcija ir negatīva) un CV70 (vai lielākā atļautā koncentrācija, t. i., 200 μg/ml). Vienā izpildes reizē jātestē vismaz 4 koncentrācijas, no kurām vismaz 2 jābūt tādām pašām kā iepriekšējā reizē vai reizēs, lai būtu iespējams rezultātus salīdzināt.
|
|
15.
|
Par šķīdinātāja/nesēja kontroli testā U-SENS™ izmanto pilnīgu barotni (pilnīgā barotnē izšķīdinātām vai stabili disperģētām testējamajām ķimikālijām) (sk. 4. punktu) vai 0,4 % DMSO šķīdumu pilnīgā barotnē (DMSO izšķīdinātām vai stabili disperģētām testējamajām ķimikālijām).
|
|
16.
|
Par pozitīvo kontroli testā U-SENS™ izmanto TNBS (sk. 11. punktu), kas sagatavota pilnīgā barotnē. TNBS izmantojama par pozitīvo kontroli CD86 ekspresijas mērījumam tādā vienā galīgajā koncentrācijā platē (50 μg/ml), pie kuras šūnu dzīvotspēja ir > 70 %. Lai platē TNBS būtu koncentrācijā 50 μg/ml, sagatavo 1 M (t. i., 293 mg/ml) TNBS izejšķīdumu pilnīgā barotnē un tad, izmantojot pilnīgu barotni ar koeficientu 2 930 atšķaida līdz 100 μg/ml darba šķīdumam. Par negatīvo kontroli izmanto pienskābi (PS, CAS Nr. 50-21-5), kas koncentrācijā 200 μg/ml izšķīdināta pilnīgā barotnē (no 0,4 mg/ml izejšķīduma). Katrā izpildes reizē katrā platē sagatavo trīs replikātus ar neapstrādātu pilnīgas barotnes kontroli, šķīdinātāja/nesēja kontroli, negatīvo un pozitīvo kontroli (12). Ja ir pieejami vēsturiskie dati, no kuriem var iegūt salīdzināmus izpildes reižu pieņemamības kritērijus, var izmantot citas piemērotas pozitīvās kontroles. Izpildes reižu pieņemamības kritēriji ir tādi paši kā testējamajai ķimikālijai (sk. 12. punktu).
|
Testējamo ķimikāliju un kontroles vielu aplicēšana
|
17.
|
14.–16. punktā aprakstīto šķīdinātāja/nesēja kontroli vai darba šķīdumus attiecībā 1:1 (tilp./tilp.) samaisa ar 96 plakandibena iedobju platē sagatavotajām šūnu suspensijām (sk. 12. punktu). Tad apstrādātās plates 45±3 stundas inkubē 37 °C temperatūrā pie 5 % CO2. Lai izvairītos no gaistošu testējamo ķimikāliju iztvaikošanas un ar testējamajām ķimikālijām apstrādāto šūnu starpiedobju kontaminācijas, plates pirms inkubēšanas noslēdz ar puscaurlaidīgu membrānu (12).
|
Šūnu iekrāsošana
|
18.
|
Pēc 45±3 ekspozīcijas stundām šūnas pārliek mikrotitrēšanas platē ar V veida iedobēm un ievāc centrifugējot. Uzskata, ka novērojama ar šķīdību saistīta interference, ja 45 ± 3 stundas pēc apstrādes (pirms šūnu iekrāsošanas) zem mikroskopa novērojami kristāli vai pilieni. Supernatantus nolej un atlikušās šūnas vienreiz noskalo ar 100 μl ledusauksta fosfātu buferšķīduma (FBS), kas satur 5 % liellopu embriju seruma (iekrāsošanas buferšķīdums). Pēc centrifugēšanas šūnas resuspendē, izmantojot 100 μl iekrāsošanas buferšķīduma, un 4 °C 30 min iekrāso ar 5 μl (piem., 0,25 μl) ar FITC marķētu anti-CD86 antivielu vai peļu IgG1 (izotips) antivielu, sargājot no gaismas. Izmanto U-SENS™ DB-ALM protokolā Nr. 183 (12) aprakstītās antivielas (CD86: BD-PharMingen #555657, klons: Fun-1, vai Caltag/Invitrogen #MHCD8601, klons: BU63; IgG1: BD-PharMingen #555748 vai Caltag/Invitrogen #GM4992). Testa izstrādātāju pieredzē dažādu partiju antivielu fluorescences intensitāte parasti ir līdzīga. Ja tie izturējuši reaģētspējas pārbaudi, testā var izmantot citus klonus vai antivielu piegādātājus (sk. 11. punktu). Tomēr, lai noteiktu vispiemērotāko koncentrāciju, lietotāji var apsvērt iespēju antivielas titrēt konkrētās laboratorijas apstākļos. Citas detektēšanas sistēmas, piem., ar fluorohromiem marķētas anti-CD86 antivielas, var izmantot, ja, piem., testējot 2.2. papildinājumā norādītās standartvielas, var pierādīt, ka tās dod līdzīgus rezultātus kā ar FITC konjugētās antivielas. Šūnas divreiz noskalo ar 100 μl iekrāsošanas buferšķīduma un vienreiz ar 100 μl ledusauksta FBS un tad resuspendē ledusaukstā FBS (piem., 125 μl paraugiem, ko analizē manuāli pa stobriņam, vai 50 μl autosamplera plates gadījumā), kam pievieno PI šķīdumu (galīgā koncentrācija: 3 μl/ml). Citus citotoksicitātes marķierus, piem., 7-aminoaktinomicīnu D (7-AAD) vai tripānzilo, var izmantot tad, ja iespējams pierādīt, ka alternatīvie iekrāsotāji dod līdzīgus rezultātus kā PI, piem., veicot testus ar 2.2. papildinājumā uzskaitītajām standartvielām.
|
Analīze ar plūsmas citometriju jeb plūsmcitometriskā analīze
|
19.
|
CD86 ekspresijas līmeni un šūnu dzīvotspēju analizē ar plūsmas citometriju. Šūnas pēc lieluma (FSC) un granularitātes (SSC) attēlo logaritmiskā punktdiagrammā, lai skaidri noteiktu pirmā vārtojuma [gate] R1 populāciju un izslēgtu šūnu atliekas. Mērķis ir katrai iedobei vārtojumā R1 iegūt 10 000 šūnu. Šūnas no tā paša vārtojuma R1 attēlo FL3 vai FL4/SSC punktdiagrammā. Dzīvotspējīgas šūnas nošķir ar vēl vieniem vārtiem — R2 —, ar ko atlasa to šūnu populāciju, kuru reakcija uz propīdija jodīdu ir negatīva (kanāls FL3 vai FL4). Šūnu dzīvotspēju var aprēķināt ar citometra analīzes programmu, izmantojot nākamo vienādojumu. Ja šūnu dzīvotspēja ir maza, iegūst līdz 20 000 šūnu, ieskaitot nedzīvas šūnas. Alternatīva ir datus iegūt vienas minūtes laikā pēc analīzes sākšanas.

Pēc tam izmēra, kāda procentuālā daļa šo pēc R2 vārtoto dzīvotspējīgo šūnu (vārtojuma R1 ietvaros) ir FL1-pozitīvas šūnas. CD86 ekspresiju uz šūnu virsmas analizē FL1/SSC punktdiagrammā, kas vārtota pēc dzīvotspējīgām šūnām (vārtojums R2).
Iedobēm ar pilnīgu barotni / IgG1 analīzes marķieri nosaka tuvu pamatpopulācijai, lai pilnīgās barotnes kontrolēs IgG1 būtu mērķzonā 0,6 līdz 0,9 %.
Krāsas interference ir ar FITC marķētā IgG1 nobīde punktdiagrammā (IgG1 FL1 ģeometriski vidējais SI ≥ 150 %).
Kontroļu šūnu (neapstrādāto šūnu vai 0,4 % DMSO šķīduma šūnu) un ar ķimikāliju apstrādāto šūnu CD86 stimulācijas indeksu (SI) aprēķina ar šādu vienādojumu:.
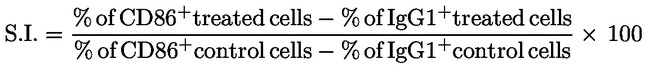
% neapstrādāto kontroles šūnu IgG1+: FL1-pozitīvo IgG1 šūnu procentuālā daļa, kas atlasīta ar analīzes marķieri (pieņemamais diapazons: ≥ 0,6 % un < 1,5 %, sk. 22. punktu), no dzīvotspējīgajām neapstrādātajām šūnām;
% kontroles/apstrādāto šūnu IgG1+/CD86+: FL1-pozitīvo IgG1/CD86 šūnu procentuālā daļa, kas izmērīta, nepārvietojot analīzes marķieri, no dzīvotspējīgajām kontroles/apstrādātajām šūnām.
|
Dati Un Pārskata Sagatavošana
Datu izvērtēšana
|
20.
|
Testā U-SENS™ aprēķina šādus parametrus: CV70 vērtība, t. i., koncentrācija, pie kuras U937 šūnu dzīvotspēja ir 70 % (tātad citotoksicitāte ir 30 %), un EC150 vērtība, t. i., koncentrācija, pie kuras testējamās ķimikālijas inducē CD86 stimulācijas indeksu (SI) 150 % apmērā.
CV70 aprēķina ar log-lineāro interpolāciju saskaņā ar šādu vienādojumu:
CV70 = C1 + [(V1 – 70) / (V1 – V2) * (C2 – C1)]
kur:
V1 ir minimālā vērtība šūnu dzīvotspējai virs 70 %;
V2 ir maksimālā vērtība šūnu dzīvotspējai zem 70 %;
C1 un C2 ir koncentrācijas, pie kurām šūnu dzīvotspēja ir attiecīgi V1 un V2.

CV70 var noteikt arī citādi, ja vien pierāda (piem., testējot standartvielas), ka tas neietekmē rezultātus.
EC150 aprēķina ar log-lineāro interpolāciju saskaņā ar šādu vienādojumu:
EC150 = C1 + [(150 – SI1) / (SI2 – SI1) * (C2 – C1)]
kur:
C1 ir augstākā koncentrācija μg/ml, pie kuras CD86 SI < 150 % (SI1);
C2 ir zemākā koncentrācija μg/ml, pie kuras CD86 SI ≥ 150 % (SI2).

EC150 un CV70 vērtības aprēķina šādi:
|
—
|
katrā izpildes reizē EC150 un CV70 izmanto, lai noskaidrotu, kā koncentrācija ietekmē CD86 atbildreakcijas palielinājumu (sk. 14. punktu);
|
|
—
|
balstoties uz vidējo dzīvotspēju, nosaka kopējo CV70 (12);
|
|
—
|
ja ir prognozēts, ka testā U-SENS™ testējamās ķimikālijas rezultāts būs POZITĪVS, tad, balstoties uz CD86 vērtību vidējo SI, nosaka ķimikālijas kopējo EC150 (sk. 21. punktu) (12).
|
|
Prognozes modelis
|
21.
|
Lai iegūtu vienu prognozi (POZITĪVU vai NEGATĪVU), CD86 ekspresijas mērījumam katru testējamo ķimikāliju testē vismaz četrās koncentrācijās un vismaz divās neatkarīgās izpildes reizēs (dažādās dienās).
|
—
|
Uzskata, ka U-SENS™ izpildes reizes individuālais iznākums ir negatīvs (“N”), ja CD86 SI pie visām necitotoksiskajām koncentrācijām (šūnu dzīvotspēja ≥ 70 %) ir mazāks par 150 % un nav novērojama nekāda interference (citotoksicitāte, šķīdība (sk. 18. punktu) vai krāsa (sk. 19. punktu), lai kādas būtu necitotoksiskās koncentrācijas, pie kurām novērota interference). Visos citos gadījumos: ja CD86 SI ir lielāks par vai vienāds ar 150 % un/vai novērota interference, uzskata, ka U-SENS™ izpildes reizes individuālais iznākums ir pozitīvs (“P”).
|
|
—
|
Uzskata, ka U-SENS™ prognoze ir NEGATĪVA, ja negatīvas (N) ir vismaz divas neatkarīgas izpildes reizes (1. attēls). Ja pirmās divas izpildes reizes abas ir negatīvas (N), uzskata, ka U-SENS™ prognoze ir NEGATĪVA un ka trešā izpildes reize nav vajadzīga.
|
|
—
|
Uzskata, ka U-SENS™ prognoze ir POZITĪVA, ja pozitīvas (P) ir vismaz divas neatkarīgas izpildes reizes (1. attēls). Ja pirmās divas izpildes reizes abas ir pozitīvas (P), uzskata, ka U-SENS™ prognoze ir POZITĪVA un ka trešā izpildes reize nav vajadzīga.
|
|
—
|
Tā kā netiek veikts devas noteikšanas tests, izņēmums ir gadījums, kad pirmajā izpildes reizē CD86 SI tikai pie augstākās necitotoksiskās koncentrācijas ir lielāks par vai vienāds ar 150 %. Tad izpildes reizi uzskata par NEVIENNOZĪMĪGU (NV) un jaunās izpildes reizēs jātestē papildu koncentrācijas (diapazonā starp augstāko necitotoksisko koncentrāciju un zemāko citotoksisko koncentrāciju; sk. 20. punktu). Ja izpildes reize ir NV, vajadzīgas vismaz 2 papildu izpildes reizes, un, ja 2. un 3. izpildes reize nedod vienādu rezultātu (neatkarīgi N un/vai P), vajadzīga vēl ceturtā izpildes reize (1. attēls). Tā kā koncentrācijas ir pielāgotas konkrētajai testējamajai ķimikālijai, papildu izpildes reizes uzskata par pozitīvām pat tad, ja CD86 sasniedz vai pārsniedz 150 % tikai vienas necitotoksiskas koncentrācijas gadījumā. Galīgā prognoze būs atkarīga no trīs vai četru atsevišķo izpildes reižu rezultātu pārsvara (t. i., 2 no 3 vai 2 no 4) (1. attēls).
|

1. attēls. Testā U-SENS™ izmantojamais prognozes modelis. U-SENS™ prognoze jāskata IATA kontekstā un saskaņā ar 4. punkta un vispārīgā ievada 7., 8. un 9. punkta noteikumiem.
N: izpildes reize, kurā nav novērots ne pozitīvs CD86 rezultāts, ne interference.
P: izpildes reize, kurā novērots pozitīvs CD86 rezultāts un/vai interference.
NV: neviennozīmīgs. Pirmā izpildes reize ir neviennozīmīga vienīgi tad, ja CD86 rezultāts ir pozitīvs tikai augstākās necitotoksiskās koncentrācijas gadījumā.
#: neviennozīmīgs (NV) individuālais iznākums, kas konstatēts tikai pirmajā reizē, automātiski nozīmē, ka, lai panāktu pārsvarā pozitīvu (P) vai pārsvarā negatīvu (N) iznākumu vismaz 2 no 3 neatkarīgām izpildes reizēm, vajadzīga trešā izpildes reize.
$: lodziņos redzamas relevantās trīs izpildes reižu rezultātu kombinācijas, kuru pamatā ir iepriekšējā lodziņā norādītie pirmo divu izpildes reižu rezultāti.
°: lodziņos redzamas relevantās četru izpildes reižu rezultātu kombinācijas, kuru pamatā ir iepriekšējā lodziņā norādītie pirmo trīs izpildes reižu rezultāti.
|
Pieņemamības kritēriji
|
22.
|
Izmantojot testu U-SENS™, jābūt izpildītiem šādiem pieņemamības kritērijiem (12).
|
—
|
45±3 h ilgā ekspozīcijas perioda beigās neapstrādāto U937 šūnu triplikāta vidējai dzīvotspējai jābūt > 90 % un nedrīkst būt novērots CD86 ekspresijas dreifs. Neapstrādāto U937 šūnu CD86 bāzlīnijas ekspresijai jābūt diapazonā no ≥ 2 % līdz ≤ 25 %.
|
|
—
|
Ja par šķīdinātāju izmanto DMSO, novērtē DMSO nesēja kontroles derīgumu, aprēķinot DMSO SI salīdzinājumā ar neapstrādāto šūnu SI, un triplikāta šūnu vidējai dzīvotspējai jābūt > 90 %. DMSO nesēja kontrole ir derīga, ja tās triplikāta CD86 SI vidējā vērtība ir mazāka nekā 250 % neapstrādāto U937 šūnu triplikāta CD86 SI vidējās vērtības.
|
|
—
|
Izpildes reizes uzskata par derīgām, ja vismaz divas no trim neapstrādāto U937 šūnu IgG1 vērtībām ir diapazonā no ≥ 0,6 % līdz < 1,5 %.
|
|
—
|
Paralēli testēto negatīvo kontroli (pienskābe) uzskata par derīgu, ja vismaz divi no trim replikātiem dod negatīvu rezultātu (CD86 SI < 150 %) un nav citotoksiski (šūnu dzīvotspēja ≥ 70 %).
|
|
—
|
Pozitīvo kontroli (TNBS) uzskata par derīgu, ja vismaz divi no trim replikātiem dod pozitīvu rezultātu (CD86 SI ≥ 150 %) un nav citotoksiski (šūnu dzīvotspēja ≥ 70 %).
|
|
Testēšanas pārskats
|
23.
|
Testēšanas pārskatā norāda šādas ziņas.
|
Testējamā ķimikālija
Vienkomponenta vielas
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, šķīdība pilnīgā barotnē, šķīdība DMSO, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
Daudzkomponentu vielas, UVCB un maisījumi
|
Iespējami izsmeļošs raksturojums ar, piem., sastāvdaļu ķīmisko identitāti (sk. iepriekš), tīrību, kvantitatīvo saturu un relevantajām fizikālķīmiskajām īpašībām
|
|
Izskats, šķīdība pilnīgā barotnē, šķīdība DMSO un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Molekulmasa vai šķietamā molekulmasa, ja tas ir maisījums/polimērs ar zināmu sastāvu, vai cita pētījuma norisei relevanta informācija
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
Kontroles
Pozitīvā kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, šķīdība DMSO, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama (attiecīgā gadījumā)
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Norāde uz vēsturiskajiem pozitīvo kontroļu rezultātiem, kas apliecina atbilstību testa izpildes pieņemamības kritērijiem (attiecīgā gadījumā)
|
Negatīvā un šķīdinātāja/nesēja kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Ja tiek izmantoti citi (testēšanas vadlīnijā neminēti) šķīdinātāji/nesēji: izskats, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
Testa apstākļi
|
Sponsora, testējošās iestādes, pētījuma vadītāja vārds/nosaukums un adrese
|
|
Izmantotā šūnu līnija, tās glabāšanas apstākļi un avots (piem., iestāde, no kuras tā saņemta)
|
|
Izmantotais plūsmas citometrs (piem., modelis), arī instrumentu iestatījumi, izmantotās antivielas un citotoksicitātes marķieris
|
|
Procedūra, ar kādu, testējot standartvielas, pierādīta laboratorijas kompetence šā testa izpildē, un procedūra, ar kādu pierādīta šā testa rezultātu reproducējamība laika griezumā, piem., vēsturiskie kontroļu dati un/vai vēsturiskie reaģētspējas pārbaužu dati
|
|
|
Testa pieņemamības kritēriji
|
Ar šķīdinātāja/nesēja kontroli iegūtās šūnu dzīvotspējas un CD86 SI vērtības salīdzinājumā ar pieņemamības diapazoniem
|
|
Ar pozitīvo kontroli iegūtās šūnu dzīvotspējas un SI vērtības salīdzinājumā ar pieņemamības diapazoniem
|
|
Šūnu dzīvotspēja visās testētajās testētās ķimikālijas koncentrācijās
|
|
|
Testa procedūra
|
Izmantotais izpildes reižu skaits
|
|
Izmantotās testējamās ķimikālijas koncentrācijas, aplicēšana un ekspozīcijas laiks (ja atšķiras no rekomendētā)
|
|
Izmantoto izvērtēšanas un lēmuma pieņemšanas kritēriju apraksts
|
|
Testa procedūras modifikāciju apraksts, ja tādas bijušas
|
|
|
Rezultāti
|
Tabulā apkopoti dati, tostarp CV70 (attiecīgā gadījumā), šūnu dzīvotspējas vērtības, EC150 vērtības (attiecīgā gadījumā), kas katrā izpildes reizē iegūtas attiecībā uz testējamo ķimikāliju un pozitīvo kontroli, un norāde uz testējamās ķimikālijas novērtējumu saskaņā ar prognozes modeli
|
|
Jebkādu citu relevantu novērojumu apraksts (attiecīgā gadījumā)
|
|
|
Rezultātu iztirzājums
|
Ar testu U-SENS™ iegūto rezultātu iztirzājums
|
|
Testa rezultātu apskats IATA kontekstā, ja pieejama cita relevanta informācija
|
|
Secinājumi
|
LITERATŪRA
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Accessible at: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2787/815737. Available at: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2760/588955. Available at: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Available at: [ http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Accessible at: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. Available at: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Available at: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
2.1. papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā testa rezultāti sakrīt ar pieņemtajām references vērtībām. Tas ir testa veiktspējas raksturlielums un viens no tā relevantuma aspektiem. Ar šo terminu, tāpat kā terminu “konkordantums”, bieži vien saprot testa pareizo iznākumu īpatsvaru (14).
AOP (nelabvēlīga iznākuma ceļš)
: no mērķķimikālijas vai līdzīgu ķimikāliju grupas ķīmiskās struktūras izrietoša notikumu sekvence no molekulārā iniciācijas notikuma līdz interesējošajam in vivo iznākumam (15).
Koncentrācijatkarīga CD86 atbildreakcija
: ja koncentrācijai, pie kuras rezultāts ir pozitīvs (CD86 SI ≥ 150) seko koncentrācija, pie kuras CD86 SI ir vēl lielāks, ir konstatējama koncentrācijatkarība (jeb koncentrācijatkarīga atbildreakcija).
Ķimikālija
: viela vai maisījums.
CV70
: aplēstā koncentrācija, pie kuras šūnu dzīvotspēja ir 70 %.
Dreifs
: gadījums, kad i) 3. neapstrādātā kontroles replikāta koriģētā % CD86+ vērtība ir mazāka par 50 % no 1. un 2. neapstrādātā kontroles replikāta koriģēto % CD86+ vērtību vidējās vērtības un ii) 3. negatīvās kontroles replikāta koriģētā % CD86+ vērtība ir mazāka par 50 % no 1. un 2. negatīvās kontroles replikāta koriģēto % CD86+ vērtību vidējās vērtības.
EC150
: aplēstās koncentrācijas, pie kurām CD86 ekspresijas SI ir 150 %.
Plūsmas citometrija
: citometrijas paņēmiens, kurā fluīdā suspendētas šūnas pa vienai laiž caur fokusētu ierosinošu gaismu, kura izkliedējas šūnām un to sastāvdaļām specifiskos rakstos; šūnas nereti tiek marķētas ar fluorescējošiem marķieriem tā, ka tās gaismu vispirms absorbē un tad emitē mainītā frekvencē.
Bīstamība
: tāda aģenta vai situācijas iedabiska īpašība, kam piemīt potenciāls atstāt nelabvēlīgu ietekmi uz šim aģentam eksponētu organismu, sistēmu vai (apakš)populāciju.
IATA (integrētā testēšanas un novērtēšanas pieeja)
: strukturēta pieeja, ko izmanto ķimikālijas vai ķimikāliju grupas bīstamības identificēšanai (potenciāls), bīstamības raksturošanai (potence) un/vai drošuma novērtēšanai (potenciāls/potence un ekspozīcija) un kas stratēģiskā veidā integrē un izsver visus relevantos datus, lai varētu pieņemt normatīvu lēmumu par potenciālo bīstamību un/vai risku, un/vai nepieciešamību veikt turpmāku selektīvu un līdz ar to minimālu testēšanu.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairāk vielām.
Vienkomponenta viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā viena galvenā sastāvdaļa veido vismaz 80 % (masa/masa).
Daudzkomponentu viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu vielas tiek iegūtas ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu vielas rodas ķīmiskās reakcijās.
Pozitīvā kontrole
: replikāts, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka tā inducē pozitīvu atbildreakciju. Lai būtu iespējams novērtēt pozitīvās kontroles atbildreakcijas mainību laikā, pozitīvās atbildreakcijas intensitāte nedrīkstētu būt pārlieku liela.
Prehaptēni
: ķimikālijas, kas par sensibilizatoriem kļūst abiotiskas transformācijas procesā, piem., oksidācijas procesā.
Prohaptēni
: ķimikālijas, kuru ādas sensibilizācijas potenciālu atraisa fermentatīva aktivācija.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testa pareizības (konkordantuma) aspektu (14).
Ticamība
: parametrs, kas izsaka, kādā pakāpē, izmantojot vienu un to pašu protokolu, testu vienā un tajā pašā laboratorijā vai dažādās laboratorijās ilgākā laika nogrieznī var veikt reproducējami. To novērtē, aprēķinot metodes intralaboratorisko un interlaboratorisko reproducējamību un intralaboratorisko atkārtojamību (14).
Izpildes reize
: reize, kurā vienu vai vairākas ķimikālijas testē paralēli ar šķīdinātāja/nesēja kontroli un pozitīvo kontroli.
Jutība
: tas, cik liela daļa no visām pozitīvajām/aktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (14).
SI
: stimulācijas indekss. ģeometriskās vidējās fluorescences intensitātes relatīvās vērtības ar ķimikāliju apstrādātās šūnās salīdzinājumā ar attiecīgajām vērtībām ar šķīdinātāju apstrādātās šūnās.
Šķīdinātāja/nesēja kontrole
: neapstrādāts paraugs, kurā ir visi testa sistēmas komponenti, izņemot testējamo ķimikāliju, bet ieskaitot izmantoto šķīdinātāju/nesēju. To izmanto, lai noteiktu bāzlīnijas atbildreakciju paraugiem, kas apstrādāti ar attiecīgajā šķīdinātājā/nesējā izšķīdinātu vai stabili disperģētu testējamo ķimikāliju. Ja testā izmanto arī paralēlu barotnes kontroli, šis paraugs parāda arī to, vai šķīdinātājs/nesējs mijiedarbojas ar testa sistēmu.
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (14).
Iekrāsošanas buferšķīdums
: fosfātu fizioloģiskais buferšķīdums, kas satur 5 % liellopu embriju seruma.
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai vajadzīgās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un nemainot tās sastāvu.
Testējamā ķimikālija
: jebkura viela vai maisījums, ko testē ar šo testu.
Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (ANO GHS)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un kura paredz atbilstošus saziņas elementus, piem., piktogrammas, signālvārdus, bīstamības apzīmējumus, drošības prasību apzīmējumus un drošības datu lapas, ar ko tiek sniegta informācija par ķimikāliju nelabvēlīgo ietekmi, tā aizsargājot cilvēkus (tostarp darba devējus, darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (16).
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kuras ir kompleksi reakcijas produkti vai bioloģiski materiāli.
Derīgs tests
: tests, kas tiek uzskatīts par pietiekami relevantu un ticamu konkrētam nolūkam un ir balstīts uz zinātniski pareiziem principiem. Tests nekad nav derīgs absolūtā izteiksmē; tas vienmēr ir derīgs tikai kādam konkrētam nolūkam (14).
2.2. papildinājums
STANDARTVIELAS
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām jāapliecina sava tehniskā kompetence, pareizi iegūstot paredzamo U-SENS™ prognozi attiecībā uz 1. tabulā ieteiktajām 10 vielām un attiecībā uz vismaz 8 no 10 standartvielām iegūstot CV70 un EC150 vērtības, kuras ietilpst attiecīgajā references diapazonā. Standartvielas ir atlasītas tā, lai tās reprezentētu visu ādas sensibilizācijas apdraudējumu izraisīto atbildreakciju diapazonu. Citi atlases kritēriji bija vielu komerciālā pieejamība un kvalitatīvu ar testu U-SENS™ iegūtu in vivo references datu, kā arī kvalitatīvu ar testu U-SENS™ iegūtu in vitro datu pieejamība. Par testu U-SENS™ ir pieejami arī publicēti references dati (1) (8).
1. tabula. 1
Vielas, kuras ieteicams izmantot, lai pierādītu tehnisko kompetenci izmantot testu U-SENS™
|
Standartvielas
|
CAS Nr.
|
Agregātstāvoklis
|
In vivo prognoze (105)
|
U-SENS™ Šķīdinātājs/nesējs
|
U-SENS™ CV70 references diapazons μg/ml (106)
|
U-SENS™ EC150 references diapazons μg/ml (106)
|
|
4-fenilēndiamīns
|
106-50-3
|
ciets
|
sensibilizators (spēcīgs)
|
pilnīga barotne (107)
|
< 30
|
pozitīvs (≤ 10)
|
|
pikrilsulfonskābe
|
2508-19-2
|
šķidrs
|
sensibilizators (spēcīgs)
|
pilnīga barotne
|
> 50
|
pozitīvs (≤ 50)
|
|
dietilmaleāts
|
141-05-9
|
šķidrs
|
sensibilizators (vidējs)
|
DMSO
|
10–100
|
pozitīvs (≤ 20)
|
|
rezorcīns
|
108-46-3
|
ciets
|
sensibilizators (vidējs)
|
pilnīga barotne
|
> 100
|
pozitīvs (≤ 50)
|
|
kanēļspirts
|
104-54-1
|
ciets
|
sensibilizators (vājš)
|
DMSO
|
> 100
|
pozitīvs (10–100)
|
|
4-alilanizols
|
140-67-0
|
šķidrs
|
sensibilizators (vājš)
|
DMSO
|
> 100
|
pozitīvs (< 200)
|
|
saharīns
|
81-07-2
|
ciets
|
nav sensibilizators
|
DMSO
|
> 200
|
negatīvs (> 200)
|
|
glicerīns
|
56-81-5
|
šķidrs
|
nav sensibilizators
|
pilnīga barotne
|
> 200
|
negatīvs (> 200)
|
|
pienskābe
|
50-21-5
|
šķidrs
|
nav sensibilizators
|
pilnīga barotne
|
> 200
|
negatīvs (> 200)
|
|
salicilskābe
|
69-72-7
|
ciets
|
nav sensibilizators
|
DMSO
|
> 200
|
negatīvs (> 200)
|
|
Saīsinājumi: CAS Nr. — Chemical Abstracts Service reģistra numurs
|
3. papildinājums
ĀDAS IN VITRO SENSIBILIZĀCIJA: TESTS IL-8 LUC
SĀKOTNĒJIE APSVĒRUMI UN IEROBEŽOJUMI
|
1.
|
Atšķirībā no testiem, ar ko analizē šūnu virsmas marķieru ekspresiju, tests IL-8 Luc kvantificē izmaiņas IL-8 — ar dendrītisko šūnu (DŠ) aktivāciju asociēta citokīna — ekspresijā. Izmantojot no THP-1 iegūto IL-8 reportieršūnu līniju (THP-G8, kas izveidota no cilvēka akūtās monocītiskās leikēmijas šūnu līnijas THP-1), mēra IL-8 ekspresiju pēc ekspozīcijas sensibilizatoriem (1). Tad luciferāzes ekspresiju izmanto par palīglīdzekli ādas sensibilizatoru nošķiršanā no nesensibilizatoriem.
|
|
2.
|
Tests IL-8 Luc ir izvērtēts validācijas pētījumā (2), ko veica Japānas Alternatīvo metožu validācijas centrs (JaCVAM), Ekonomikas, tirdzniecības un rūpniecības ministrija (METI) un Japānas Dzīvniektestēšanas alternatīvu biedrība (JSAAF), un pēc tam ar Starptautiskās sadarbības alternatīvu testēšanas metožu jomā (ICATM) atbalstu neatkarīgi profesionālizvērtēts (3) JaCVAM un Veselības, darba un labklājības ministrijas (MHLW) paspārnē. Ņemot vērā visus pieejamos pierādījumus un regulatoru un ieinteresēto personu sniegtās ziņas, testu IL-8 Luc ir lietderīgi izmantot IATA ietvaros par palīglīdzekli, kas palīdz ādas sensibilizatorus nošķirt no nesensibilizatoriem apdraudējumu klasificēšanas un marķēšanas vajadzībām. Piemēri, kā testa IL-8 Luc dati izmantoti kombinācijā ar citu informāciju, ir pieejami literatūrā (4) (5) (6).
|
|
3.
|
Ir pierādīts, ka tests IL-8 Luc ir pārnesams uz laboratorijām, kam ir pieredze ar šūnu kultūrām un luciferāzes mērījumiem. Reproducējamības līmenis ir attiecīgi 87,7 % tajā pašā laboratorijā un 87,5 % citās laboratorijās (2). Validācijas pētījuma dati (2) un citi publicētie darbi (1) (6) kopumā liecina, ka salīdzinājumā ar LLNA testā IL-8 Luc attiecībā uz 118 no 143 ķimikālijām slēdziens bija pozitīvs vai negatīvs un attiecībā uz 25 ķimikālijām — neviennozīmīgs, un testa IL-8 Luc pareizība ādas sensibilizatoru (ANO GHS / CLP 1. kat.) nošķiršanā no ādas nesensibilizatoriem (ANO GHS / CLP nekategorizēti) ir 86 % (101/118) ar jutību 96 % (92/96) un specifiskumu 41 % (9/22). Izslēdzot vielas, kas ir ārpus tālāk (5. punktā) aprakstītā izmantojamības lauka, testā IL-8 Luc attiecībā uz 113 no 136 ķimikālijām slēdziens bija pozitīvs vai negatīvs un attiecībā uz 23 ķimikālijām — neviennozīmīgs, un testa IL-8 Luc pareizība bija 89 % (101/113) ar jutību 96 % (92/96) un specifiskumu 53 % (9/17). Izmantojot Urbisch et al. (7) norādītos datus par cilvēku, testā IL-8 Luc attiecībā uz 76 no 90 ķimikālijām slēdziens bija pozitīvs vai negatīvs un attiecībā uz 14 ķimikālijām — neviennozīmīgs, un pareizība bija 80 % (61/76) ar jutību 93 % (54/58) un specifiskumu 39 % (7/18). Izslēdzot vielas, kas ir ārpus izmantojamības lauka, testā IL-8 Luc attiecībā uz 71 no 84 ķimikālijām slēdziens bija pozitīvs vai negatīvs un attiecībā uz 13 ķimikālijām — neviennozīmīgs, un testa pareizība bija 86 % (61/71) ar jutību 93 % (54/58) un specifiskumu 54 % (7/13). Testā IL-8 Luc pseidonegatīvas prognozes, visticamāk, skars ķimikālijas, kurām novērojama zema/vidēja ādas sensibilizācijas potence (ANO GHS / CLP 1.B apakškategorija), nevis ķimikālijas, kurām novērojama augsta ādas sensibilizācijas potence (ANO GHS / CLP 1.A apakškategorija) (6). Visa šī informācija kopā norāda uz to, ka tests IL-8 Luc var palīdzēt identificēt apdraudējumus, kas izraisa ādas sensibilizāciju. Tomēr pareizības vērtības, kas šeit sniegtas attiecībā uz IL-8 Luc kā atsevišķu testu, ir tikai orientējošas, jo šis tests jāskata kombinācijā ar citiem informācijas avotiem IATA kontekstā un saskaņā ar vispārīgā ievada 7. un 8. punkta noteikumiem. Turklāt, izvērtējot ādas sensibilizācijas testus, kuros neizmanto dzīvniekus, jāņem vērā, ka LLNA tests, tāpat kā citi testi ar dzīvniekiem, var nepilnīgi atspoguļot situāciju cilvēka gadījumā.
|
|
4.
|
Balstoties uz pašlaik pieejamajiem datiem, ir pierādīts, ka testu IL-8 Luc var izmantot testējamām ķimikālijām, kuru sakarā aktuālas ir dažādas organiskās funkcionālās grupas, reakcijas mehānismi, ādas sensibilizācijas potence (kas noteikta in vivo pētījumos) un fizikālķīmiskās īpašības (2) (6).
|
|
5.
|
Lai gan testā IL-8 Luc par šķīdinātāju izmanto X-VIVOTM 15, tas pareizi izvērtēja ķimikālijas, kuru Log Kow >3.5, un ķimikālijas, kuru šķīdība ūdenī, aprēķināta ar EPI SuiteTM
, ir apmēram 100 μg/ml; sensibilizatorus, kas ūdenī šķīst vāji, tas detektē labāk nekā tests IL-8 Luc, kurā par šķīdinātāju izmanto dimetilsulfoksīdu (DMSO) (2). Tomēr attiecībā uz testējamām ķimikālijām, kas koncentrācijā 20 mg/ml neizšķīst, rezultāti sakarā ar to vājo šķīdību X-VIVOTM 15 var būt pseidonegatīvi. Tāpēc šādu ķimikāliju gadījumā negatīvus rezultātus ņemt vērā nedrīkst. Validācijas pētījumā tika konstatēts, ka daudz pseidonegatīvu rezultātu iegūst, testējot anhidrīdus. Turklāt šūnu līnijas ierobežotās metaboliskās spējas (8) un eksperimenta apstākļu dēļ negatīvus rezultātus testā var dot arī prohaptēni (vielas, kurām nepieciešama metaboliskā aktivācija) un prehaptēni (vielas, kuras aktivizējas oksidējoties). Tomēr, lai gan domājamiem prehaptēniem/prohaptēniem (ķimikālijām, par kurām ir aizdomas, ka tās varētu būt prehaptēni/prohaptēni) negatīvi rezultāti jāinterpretē piesardzīgi, testa IL-8 Luc datu kopā pareizi ir 11 no 11 slēdzieniem prehaptēnu gadījumā, 6 no 6 slēdzieniem prohaptēnu gadījumā un 6 no 8 slēdzieniem prehaptēnu/prohaptēnu gadījumā (2). Balstoties uz neseno visaptverošo apskatu par trīs bezdzīvnieku testiem (DPRA, KeratinoSens™ un h-CLAT), ko izmanto prehaptēnu un prohaptēnu noteikšanai (9), un arī apstākli, ka testā IL-8 Luc izmantotās THP-G8 šūnas pieder pie h-CLAT izmantotās šūnu līnijas THP-1, testa IL-8 Luc iekļaušana citu testu kombinācijā var uzlabot bezdzīvnieku testu jutību prehaptēnu un prohaptēnu noteikšanā. Līdz šim testētie virsmaktīvie aģenti neatkarīgi no tipa (piem., katjoniski, anjoniski, nejoniski) devuši (pseido)pozitīvus rezultātus. Visbeidzot, ķimikālijas, kas ietekmē luciferāzi, var radīt maldīgu priekšstatu par tās aktivitāti vai ietekmēt mērījumus, izraisot vai nu šķietamu inhibēšanos, vai pastiprinātu luminiscenci (10). Piem., ir ziņots, ka fitoestrogēna koncentrācijas, kas lielākas par 1 μM, citos uz luciferāzi balstītos reportiergēnu testos radījušas luminiscences signālu traucējumus luciferāzes reportiergēna pārmērīgas aktivācijas dēļ. Tāpēc luciferāzes ekspresija, kas iegūta pie augstām fitoestrogēnu koncentrācijām vai augstām tādu savienojumu koncentrācijām, kuri domājami izraisa fitoestrogēnisku luciferāzes reportiergēna aktivāciju, ir rūpīgi jāizmeklē (11). Ņemot vērā visus šos apsvērumus, virsmaktīvie aģenti, anhidrīdi un ķimikālijas, kas ietekmē luciferāzi, ir ārpus šā testa izmantojamības lauka. Gadījumos, kad ir liecības, ka tests IL-8 Luc nav izmantojams vēl citu konkrētu kategoriju testējamajām ķimikālijām, to attiecīgo kategoriju ķimikālijām neizmanto.
|
|
6.
|
Kā norādīts iepriekš, tests IL-8 Luc palīdz ādas sensibilizatorus nošķirt no ādas nesensibilizatoriem. Lai noskaidrotu, vai IL-8 Luc rezultāti kombinācijā ar citiem informācijas avotiem var palīdzēt novērtēt potenci, ir vajadzīga papildu informācija, vēlams, balstīta uz datiem par cilvēku.
|
|
7.
|
Definīcijas ir sniegtas 3.1. papildinājumā.
|
TESTA PRINCIPS
|
8.
|
Tests IL-8 Luc izmanto cilvēka monocītiskās leikēmijas šūnu līniju THP-1, kas iegūta no Amerikas tipveida kultūru kolekcijas (Manasasa, VA, ASV). Tohoku Universitātes Medicīnas augstskolas Dermatoloģijas departaments, izmantojot šo šūnu līniju, izveidoja no THP-1 iegūtu IL-8 reportieršūnu līniju THP-G8, kurā ir luciferāzes gēni Stable Luciferase Orange (SLO) un Stable Luciferase Red (SLR), ko kontrolē attiecīgi IL-8 un gliceraldehīda 3-fosfāta dehidrogenāzes (GAPDH) promoteri (1). Tas ļauj kvantitatīvi izmērīt luciferāzes gēna indukciju, detektējot luminiscenci no labi zināmiem gaismu izstarojošiem luciferāzes substrātiem, kas kalpo par IL-8 un GAPDH aktivitātes rādītāju šūnās pēc to ekspozīcijas sensibilizējošām ķimikālijām.
|
|
9.
|
Divkrāsu testa sistēmu veido viena oranžo izstarojoša luciferāze (SLO; λmaks. = 580 nm) (12) IL-8 promotera gēnu ekspresijai un sarkano izstarojoša luciferāze (SLR; λmaks. = 630 nm) (13) iekšējās kontroles promotera GAPDH gēnu ekspresijai. Reaģējot ar jāņtārpiņu d-luciferīnu, šīs divas luciferāzes izstaro dažādas krāsas, un to luminiscenci vienlaicīgi mēra viensoļa reakcijā, testa maisījuma izstaroto gaismu sadalot ar optisko filtru (14) (3.2. papildinājums).
|
|
10.
|
THP-G8 šūnas 16 stundas apstrādā ar testējamo ķimikāliju; pēc tam mēra SLO luciferāzes aktivitāti (SLO-LA), kas atspoguļo IL-8 promotera aktivitāti, un SLR luciferāzes aktivitāti (SLR-LA), kas atspoguļo GAPDH promotera aktivitāti. Lai saīsinājumus būtu vieglāk uztvert, SLO-LA un SLR-LA sauc attiecīgi par IL8LA un GAPLA. Apzīmējumi, kas saistīti ar luciferāzes aktivitāti testā IL-8 Luc, definēti 1. tabulā. Izmantojot mērījumu vērtības, aprēķina normalizēto IL8LA (nIL8LA), kas IL8LA un GAPLA attiecība, nIL8LA (Ind-IL8LA) indukciju, kas ir ar testējamo ķimikāliju apstrādātu THP-G8 šūnu nIL8LA četrkārt mērītu vērtību un neapstrādātu THP-G8 šūnu nIL8LA vērtību aritmētisko vidējo attiecība, un GAPLA (Inh-GAPLA) inhibēšanos, kas ir ar testējamo ķimikāliju apstrādātu THP-G8 šūnu četrkārt mērītu GAPLA vērtību un neapstrādātu THP-G8 šūnu GAPLA vērtību aritmētisko vidējo attiecība un ko izmanto par citotoksicitātes indikatoru.
1. tabula. 1
Ar luciferāzes aktivitāti testā IL-8 Luc saistīto apzīmējumu definīcijas
|
Abreviatūra
|
Definīcija
|
|
GAPLA
|
SLR luciferāzes aktivitāte, kas atspoguļo GAPDH promotera aktivitāti
|
|
IL8LA
|
SLO luciferāzes aktivitāte, kas atspoguļo IL-8 promotera aktivitāti
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
Ar ķimikālijām apstrādāto THP-G8 šūnu nIL8LA / neapstrādāto šūnu nIL8LA
|
|
Inh-GAPLA
|
Ar ķimikālijām apstrādāto THP-G8 šūnu GAPLA / neapstrādāto šūnu GAPLA
|
|
CV05
|
Zemākā ķimikālijas koncentrācija, pie kuras Inh-GAPLA ir < 0,05
|
|
|
11.
|
Lai būtu vieglāk validēt modificētus in vitro IL-8 luciferāzes testus, kas līdzinās testam IL-8 Luc, un lai ESAO testēšanas vadlīniju Nr. 442E būtu iespējams savlaicīgi attiecīgi grozīt ar mērķi šos testus tajā iekļaut, ir pieejami veiktspējas standarti (VS) (15) Attiecībā uz testiem, kas validēti saskaņā ar VS, ESAO datu savstarpējā atzīšana (Mutual Acceptance of Data, MAD) būs garantēta tikai tad, ja ESAO šos testus būs izskatījusi un iekļāvusi attiecīgajā testēšanas vadlīnijā Nr. 442E (16).
|
KOMPETENCES PIERĀDĪŠANA
|
12.
|
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām, ievērojot labu in vitro metožu praksi (17), jāpierāda sava tehniskā kompetence, izmantojot 3.3. papildinājumā uzskaitītās 10 standartvielas. Testa lietotājiem turklāt jāuztur vēsturiska datubāze ar datiem, kas iegūti reaģētspējas pārbaudēs (sk. 15. punktu) un ar pozitīvajām un šķīdinātāja/nesēja kontrolēm (sk. 21.–24. punktu), un ar šo datu palīdzību jāpārliecinās, ka laika gaitā testa reproducējamība konkrētajā laboratorijā saglabājas tāda pati.
|
PROCEDŪRA
|
13.
|
Testam IL-8 Luc ir pieejama standartprocedūra, kas jāizmanto, veicot šo testu (18). Laboratorijas, kas vēlas šo testu veikt, rekombinanto šūnu līniju THP-G8 var iegūt no GPC Lab. Co. Ltd. (Totori, Japāna), iepriekš parakstot ESAO parauga nosacījumiem atbilstošu materiālu nodošanas līgumu. Galveno testa komponentu un procedūru apraksts sniegts nākamajos punktos.
|
Šūnu sagatavošana
|
14.
|
Testam IL-8 Luc izmanto šūnu līniju THP-G8 no GPC Lab. Co. Ltd. (Totori, Japāna) (sk. 8. un 13. punktu). Saņemtās šūnas pavairo (2–4 pasāžas) un uzglabā sasaldētas kā viendabīgu krājumu. Šā krājuma šūnas var pavairot līdz maksimāli 12 pasāžām vai maksimāli 6 nedēļas. Pavairošanai izmanto kultūras barotni RPMI-1640, kurā ir 10 % liellopu embriju seruma (foetal bovine serum, FBS), antibiotisks/antimikotisks šķīdums (100 U/ml penicilīna G, 100 μg/ml streptomicīna un 0,25 μg/ml amfotericīna B 0,85 % fizioloģiskajā šķīdumā) (piem., GIBCO Cat#15240-062), 0,15 μg/ml puromicīna (piem., CAS Nr. 58-58-2) un 300 μg/ml G418 (piem., CAS Nr. 108321-42-2).
|
|
15.
|
Pirms šūnu izmantošanas testēšanai tās jākvalificē, pārbaudot to reaģētspēju. Šī pārbaude jāveic 1–2 nedēļas vai 2–4 pasāžas pēc atkausēšanas, izmantojot pozitīvo kontroli, 4-nitrobenzilbromīdu (4-NBB) (CAS Nr. 100-11-8, ≥ 99 % tīrība), un negatīvo kontroli, pienskābi (PS) (CAS Nr. 50-21-5, ≥ 85 % tīrība). 4-NBB jāizsauc pozitīva Ind-IL8LA (≥ 1,4) atbildreakcija, savukārt PS — negatīva (< 1,4). Testam izmanto tikai šūnas, kas izturējušas reaģētspējas pārbaudi. Pārbaude veicama saskaņā ar 22.–24. punktā aprakstītajām procedūrām.
|
|
16.
|
Testēšanas vajadzībām THP-G8 šūnas uzsēj blīvumā 2 līdz 5 × 105 šūnas/ml un kultivēšanas kolbās priekškultivē attiecīgi 48 vai 96 stundas. Testēšanas dienā no kultivēšanas kolbas ievāktās šūnas noskalo ar RPMI-1640, kas satur 10 % FBS bez antibiotikām, un resuspendē RPMI-1640, kas satur 10 % FBS bez antibiotikām, blīvumā 1 × 106 šūnas/ml. Tad šūnas sadala melnā 96 plakandibena iedobju platē (piem., Costar Cat#3603), izmantojot 50 μl (5 × 104 šūnas uz iedobi).
|
Testējamās ķimikālijas un kontroles vielu sagatavošana
|
17.
|
Testējamo ķimikāliju un kontroles vielas sagatavo testēšanas dienā. Testā IL-8 Luc testējamās ķimikālijas izšķīdina X-VIVOTM 15, komerciāli pieejamā bezseruma barotnē (Lonza, 04-418Q), līdz galīgajai koncentrācijai 20 mg/ml. X-VIVOTM 15 pievieno 20 mg testējamās ķimikālijas (neatkarīgi no ķimikālijas šķīdības) mikrocentrifūgas stobriņā, papildina līdz 1 ml tilpumam un tad 30 min intensīvi vorteksē un krata ar rotoru, kura maksimālais ātrums ir 8 apgr./min, apkārtējās vides temperatūrā (aptuveni 20 oC). Ja cietas ķimikālijas joprojām nešķīst, stobriņu turklāt sonicē (apstrādā ar skaņu), līdz ķimikālija pilnīgi izšķīdusi vai stabili disperģēta. Ja testējamā ķimikālija X-VIVOTM 15 šķīst, šķīdumu ar X-VIVOTM 15 atšķaida, izmantojot atšķaidījuma koeficientu 5, un izmanto par testējamās ķimikālijas izejšķīdumu X-VIVOTM 15 (4 mg/ml). Ja testējamā ķimikālija X-VIVOTM 15 nešķīst, maisījumu atkal vismaz 30 min rotē un tad 5 min centrifugē ar 15000 apgr./min (≈20000g); iegūto supernatantu izmanto par testējamās ķimikālijas izejšķīdumu X-VIVOTM 15. Citu šķīdinātāju (piem., DMSO, ūdens vai kultūras barotnes) izmantošana zinātniski jāpamato. Ķimikāliju šķīdināšanas procedūra ir detalizēti aprakstīta 3.5. papildinājumā. 18.–23. punktā aprakstītos X-VIVOTM 15 šķīdumus attiecībā 1:1 (tilp./tilp.) samaisa ar melnajā 96 plakandibena iedobju platē sagatavotajām šūnu suspensijām (sk. 16. punktu).
|
|
18.
|
Pirmās testa izpildes reizes mērķis ir noteikt citotoksisko koncentrāciju un novērtēt ķimikāliju ādas sensibilizācijas potenciālu. Izmantojot X-VIVOTM 15, 96 iedobju testa blokā (piem., Costar Cat#EW-01729-03) izdara testējamās ķimikālijas un X-VIVOTM 15 izejšķīdumu sērijveida atšķaidījumus ar atšķaidījuma koeficientu divi (sk. 3.5. papildinājumu). Pēc tam melnā 96 plakandibena iedobju platē 50 μl šūnu suspensijas pievieno 50 μl atšķaidītā šķīduma uz iedobi. Tātad testējamajām ķimikālijām, kas šķīst X-VIVOTM 15, testējamo ķimikāliju galīgās koncentrācijas ir diapazonā no 0,002 līdz 2 mg/ml (3.5. papildinājums). Testējamajām ķimikālijām, kas X-VIVOTM 15 koncentrācijā 20 mg/ml nešķīst, nosaka tikai atšķaidījuma koeficientus diapazonā no 2 līdz 210, lai gan testējamo ķimikāliju faktiskās galīgās koncentrācijas nav droši zināmas un ir atkarīgas no tā, cik piesātināts ir izejšķīdums — testējamās ķimikālijas šķīdums X-VIVOTM 15.
|
|
19.
|
Nākamajās testa izpildes reizēs (t. i., otrais, trešais un ceturtais replikāts) X-VIVOTM 15 izejšķīdumu sagatavo koncentrācijā, kas četrkārtīgi pārsniedz šūnu dzīvotspējas koncentrāciju 05 (CV05 — zemākā koncentrācija, pie kuras Inh-GAPLA ir < 0,05) pirmajā eksperimentā. Ja pirmajā izpildes reizē Inh-GAPLA pie augstākās koncentrācijas nesamazinās zem 0,05, X-VIVOTM 15 izejšķīdumu sagatavo augstākajā pirmās izpildes reizes koncentrācijā. CV05 koncentrāciju aprēķina, pirmās izpildes reizes izejšķīduma koncentrāciju dalot ar CV05 atšķaidījuma koeficientu (X) (atšķaidījuma koeficients CV05 (X) — atšķaidījuma koeficients, kas vajadzīgs izejšķīduma atšķaidīšanai līdz CV05) (3.5. papildinājums). Ja testējamā viela X-VIVO koncentrācijā 20 mg/ml nešķīst, CV05 nosaka pēc izejšķīduma koncentrācijas × 1/X. 2. līdz 4. izpildes reizei sagatavo otru izejšķīdumu — 4 × CV05 (3.5. papildinājums).
|
|
20.
|
Otro X-VIVOTM 15 izejšķīdumu sērijveidā atšķaida ar atšķaidījuma koeficientu 1,5, izmantojot 96 iedobju testa bloku. Pēc tam melnā 96 plakandibena iedobju platē 50 μl šūnu suspensijas pievieno 50 μl atšķaidītā šķīduma uz iedobi. Katra testējamā ķimikālija katrā koncentrācijā jātestē 4 iedobēs. Pēc tam paraugus samaisa ar plates kratītāju un uz 16 h inkubē 37 oC un pie 5 % CO2; tad, kā aprakstīts tālāk, izmēra luciferāzes aktivitāti.
|
|
21.
|
Šķīdinātāja kontrole ir šāds maisījums: 50 μl X-VIVOTM 15 un 50 μl šūnu suspensijas RPMI-1640, kas satur 10 % FBS, uz iedobi.
|
|
22.
|
Par pozitīvo kontroli ieteicams izmantot 4-NBB. 1,5 ml mikrofūgas stobriņā sagatavo 20 mg 4-NBB, kam pievieno X-VIVOTM 15 līdz 1 ml. Stobriņu intensīvi vorteksē un krata ar rotoru, kura maksimālais ātrums ir 8 apgr./min, vismaz 30 min. Pēc 5 min ilgas centrifugēšanas (20000 g) supernatantu atšķaida ar X-VIVOTM 15, izmantojot atšķaidījuma koeficientu 4, un 500 μl atšķaidītā supernatanta pārvieto uz iedobi 96 iedobju testa blokā. Atšķaidīto supernatantu vēl atšķaida ar X-VIVOTM 15, izmantojot atšķaidījuma koeficientus 2 un 4, un 50 μl šķīduma pievieno 50 μl THP-G8 šūnu suspensijai melnas 96 plakandibena iedobju plates iedobēs (3.6. papildinājums). Pozitīvā kontrole katrā koncentrācijā jātestē 4 iedobēs. Plati sakrata ar plates kratītāju un uz 16 h inkubē CO2 inkubatorā (37 oC, 5 % CO2); pēc tam izmēra luciferāzes aktivitāti, kā aprakstīts 29. punktā.
|
|
23.
|
Par negatīvo kontroli ieteicams izmantot pienskābi (PS). 1,5 ml mikrofūgas stobriņā sagatavo 20 mg PS, kam pievieno X-VIVOTM 15 līdz 1 ml (20 mg/ml). 20 mg/ml PS šķīdumu atšķaida ar X-VIVOTM 15, izmantojot atšķaidījuma koeficientu 5 (4 mg/ml); 500 μl šā 4 mg/ml PS šķīduma pārvieto uz 96 iedobju testa bloka iedobi. Šo šķīdumu atšķaida ar X-VIVOTM 15, izmantojot atšķaidījuma koeficientu 2, un tad vēlreiz atšķaida ar atšķaidījuma koeficientu 2, iegūstot šķīdumus ar koncentrāciju 2 mg/ml un 1 mg/ml. 50 μl šo 3 šķīdumu un šķīdinātāja kontroles (X-VIVOTM 15) pievieno 50 μl THP-G8 šūnu suspensijas melnas 96 plakandibena iedobju plates iedobēs. Negatīvo kontroli katrā koncentrācijā testē 4 iedobēs. Plati sakrata ar plates kratītāju un uz 16 h inkubē CO2 inkubatorā (37 oC, 5 % CO2); pēc tam izmēra luciferāzes aktivitāti, kā aprakstīts 29. punktā.
|
|
24.
|
Ja ir pieejami vēsturiskie dati, no kuriem var iegūt salīdzināmus izpildes reižu pieņemamības kritērijus, var izmantot citas piemērotas pozitīvās vai negatīvās kontroles.
|
|
25.
|
Jāizvairās no gaistošu testējamo ķimikāliju iztvaikošanas un testējamo ķimikāliju starpiedobju kontaminācijas, piem., pirms inkubēšanas ar testējamajām ķimikālijām plati noslēdzot.
|
|
26.
|
Lai iegūtu pozitīvu vai negatīvu prognozi, ar testējamajām ķimikālijām un šķīdinātāja kontroli vajadzīgas 2 līdz 4 izpildes reizes (sk. 2. tabulu). Katru izpildes reizi veic citā dienā ar svaigu X-VIVOTM 15 izejšķīdumu un neatkarīgi ievāktām šūnām. Šūnas var būt no vienas un tās pašas pasāžas.
|
Luciferāzes aktivitātes mērījumi
|
27.
|
Luminiscenci mēra, izmantojot 96 iedobju mikroplates luminometru ar optiskajiem filtriem, piem., Phelios (ATTO, Tokija, Japāna), Tristan 941 (Berthold, Bādvildbāde, Vācija) vai ARVO (PerkinElmer, Voltema, MA, ASV). Lai nodrošinātu reproducējamību, luminometrs katram testam jākalibrē (19). Šai kalibrēšanai pieejamas rekombinantās luciferāzes, kas izstaro oranžu un sarkanu gaismu.
|
|
28.
|
Uz katru plates iedobi, kurā ir ar ķimikāliju apstrādāta vai neapstrādāta šūnu suspensija, pārnes 100 μl iepriekš sasildīta testa reaģenta Tripluc® Luciferase (Tripluc). Plati 10 minūtes krata apkārtējās vides temperatūrā — aptuveni 20 oC. Luciferāzes aktivitātes mērīšanai plati ieliek luminometrā. Bioluminiscenci mēra gan bez optiskā filtra (F0), gan ar to (F1), katrreiz 3 s. Citu iestatījumu izmantošanas gadījumā (piem., atkarībā no izmantotā luminometra modeļa) jāsniedz pamatojums.
|
|
29.
|
Parametrus katrai koncentrācijai aprēķina, par pamatu ņemot izmērītās vērtības (piem., IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, IL8LA vidējā vērtība ± standartnovirze, GAPLA vidējā vērtība ± standartnovirze, nIL8LA vidējā vērtība ± standartnovirze, Ind-IL8LA vidējā vērtība ± standartnovirze, Inh-GAPLA vidējā vērtība ± standartnovirze un Ind-IL8LA 95 % ticamības intervāls). Šajā punktā izmantoto parametru definīcijas sk. attiecīgi 3.1 un 3.4. papildinājumā.
|
|
30.
|
Daudzkrāsu reportieru testos pirms mērīšanas parasti panāk krāsu izšķiršanu ar detektoriem (luminometru un plašu lasītāju), kas aprīkoti ar optiskajiem filtriem, piem., sliekšņfiltriem [sharp-cut filters] (garviļņu vai īsviļņu caurlaides filtriem) vai frekvenču joslas filtriem. Pirms testēšanas saskaņā ar 3.2. papildinājumu jākalibrē filtru pārvades koeficienti katrai bioluminiscences signālkrāsai.
|
DATI UN PĀRSKATA SAGATAVOŠANA
Datu izvērtēšana
|
31.
|
Lai iegūtu pozitīvu/negatīvu prognozi, katrai izpildes reizei jāatbilst šādiem kritērijiem:
|
—
|
uzskata, ka testa IL-8 Luc prognoze ir pozitīva, ja testējamās ķimikālijas Ind-IL8LA ≥ 1,4 un Ind-IL8LA 95 % ticamības intervāla apakšējā robeža ≥ 1,0;
|
|
—
|
uzskata, ka testa IL-8 Luc prognoze ir negatīva, ja testējamās ķimikālijas Ind-IL8LA < 1,4 un/vai Ind-IL8LA 95 % ticamības intervāla apakšējā robeža < 1,0.
|
|
Prognozes modelis
|
32.
|
Testējamās ķimikālijas, attiecībā uz kurām 1., 2., 3. un 4. reizē gūti divi pozitīvi rezultāti, uzskata par pozitīvām, savukārt tās, attiecībā uz kurām 1., 2., 3. un 4. reizē gūti trīs negatīvi rezultāti, uzskata par potenciāli negatīvām (2. tabula). No potenciāli negatīvām ķimikālijām ķimikālijas, kas X-VIVOTM 15 izšķīdušas koncentrācijā 20 mg/ml, uzskata par negatīvām, savukārt par ķimikālijām, kas X-VIVOTM 15 koncentrācijā 20 mg/ml nav izšķīdušas, spriedumu neizdara (1. attēls).
2. tabula.
Pozitīvu un potenciāli negatīvu ķimikāliju identificēšanas kritēriji
|
1. izpildes reize
|
2. izpildes reize
|
3. izpildes reize
|
4. izpildes reize
|
Galīgā prognoze
|
|
Pozitīva
|
Pozitīva
|
—
|
—
|
Pozitīva
|
|
Negatīva
|
Pozitīva
|
—
|
Pozitīva
|
|
Negatīva
|
Pozitīva
|
Pozitīva
|
|
Negatīva
|
Potenciāli negatīva
|
|
Negatīva
|
Pozitīva
|
Pozitīva
|
—
|
Pozitīva
|
|
Negatīva
|
Pozitīva
|
Pozitīva
|
|
Negatīva
|
Potenciāli negatīva
|
|
Negatīva
|
Pozitīva
|
Pozitīva
|
Pozitīva
|
|
Negatīva
|
Potenciāli negatīva
|
|
Negatīva
|
—
|
Potenciāli negatīva
|
|
1. attēls.
Galīgā slēdziena prognozes modelis

Pieņemamības kritēriji
|
33.
|
Izmantojot testu IL-8 Luc, jābūt izpildītiem šādiem pieņemamības kritērijiem:
|
—
|
Ind-IL8LA vērtībai katrā izpildes reizē jāpārsniedz 5,0 vismaz vienā pozitīvās kontroles, 4-NBB, koncentrācijā;
|
|
—
|
Ind-IL8LA vērtībai katrā izpildes reizē jābūt mazākai par 1,4 vismaz vienā negatīvās kontroles, pienskābes, koncentrācijā;
|
|
—
|
datus no platēm, kuru kontroles iedobēs ar šūnām un Tripluc, bet bez ķimikālijām GAPLA vērtība mazāk nekā pieckārtīgi pārsniedz šo vērtību iedobēs, kurās ir tikai testa barotne (50 μl RPMI-1640, kas satur 10 % FBS, un 50 μl X-VIVOTM 15 uz iedobi), atmet;
|
|
—
|
datus no platēm, kurās Inh-GAPLA vērtība pie visām testējamo vai kontroles ķimikāliju koncentrācijām ir mazāka nekā 0,05, atmet; tādā gadījumā pirmo testu atkārto tā, lai atkārtotā testa augstākā galīgā koncentrācija būtu iepriekšējā testa zemākā galīgā koncentrācija.
|
|
Testēšanas pārskats
|
34.
|
Testēšanas pārskatā norāda šādas ziņas.
|
Testējamās ķimikālijas
Vienkomponenta viela:
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, šķīdība ūdenī, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Šķīdība X-VIVOTM 15. Ja ķimikālija X-VIVOTM 15 nešķīst, norāda, vai pēc centrifugēšanas novērojama izgulsnēšanās vai flotācija
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Ja neizmanto X-VIVOTM 15: katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
Daudzkomponentu vielas, UVCB un maisījumi
|
Iespējami izsmeļošs raksturojums ar, piem., sastāvdaļu ķīmisko identitāti (sk. iepriekš), tīrību, kvantitatīvo saturu un relevantajām fizikālķīmiskajām īpašībām
|
|
Izskats, šķīdība ūdenī un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Molekulmasa vai šķietamā molekulmasa, ja tas ir maisījums/polimērs ar zināmu sastāvu, vai cita pētījuma norisei relevanta informācija
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Šķīdība X-VIVOTM 15. Ja ķimikālija X-VIVOTM 15 nešķīst, norāda, vai pēc centrifugēšanas novērojama izgulsnēšanās vai flotācija
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Ja neizmanto X-VIVOTM 15: katras testējamās ķimikālijas šķīdinātāja/nesēja izvēles pamatojums
|
|
|
Kontroles
Pozitīvā kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i), SMILES vai InChI kods, struktūrformula un/vai citi identifikācijas dati
|
|
Izskats, šķīdība ūdenī, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama (attiecīgā gadījumā)
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Pirmstestēšanas apstrāde, ja tāda veikta (piem., sildīšana, smalcināšana)
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Norāde uz vēsturiskajiem pozitīvo kontroļu rezultātiem, kas apliecina atbilstību piemērotiem pieņemamības kritērijiem (attiecīgā gadījumā)
|
Negatīvā kontrole
|
Ķīmiskā identifikācija, piem., IUPAC vai CAS nosaukums(-i), CAS numurs(-i) un/vai citi identifikācijas dati
|
|
Tīrība, piemaisījumu ķīmiskā identitāte (attiecīgā gadījumā, ja šī informācija ir pieejama) utt.
|
|
Ja tiek izmantotas citas (testēšanas vadlīnijā neminētas) negatīvās kontroles: izskats, molekulmasa un citas relevantas fizikālķīmiskās īpašības, ciktāl šī informācija ir pieejama
|
|
Glabāšanas apstākļi un stabilitāte, ciktāl šī informācija ir pieejama
|
|
Katras testējamās ķimikālijas šķīdinātāja izvēles pamatojums
|
|
|
Testa apstākļi
|
Sponsora, testējošās iestādes, pētījuma vadītāja vārds/nosaukums un adrese
|
|
Izmantotā šūnu līnija, tās glabāšanas apstākļi un avots (piem., iestāde, no kuras tā saņemta)
|
|
FBS partijas numurs un izcelsme, melnās 96 plakandibena iedobju plates partijas numurs un reaģenta Tripluc partijas numurs
|
|
Testēšanā izmantoto šūnu pasāžu skaits un blīvums
|
|
Šūnu skaitīšanas metode, kas izmantota uzsēšanai pirms testēšanas, un pasākumi, kas veikti, lai nodrošinātu šūnu viendabīgu sadalījumu skaita ziņā
|
|
Izmantotais luminometrs (piem., modelis), arī instrumentu iestatījumi, izmantotais luciferāzes substrāts un pienācīgu luminiscences mērījumu pierādīšana ar 3.2. papildinājumā aprakstīto kontroltestu
|
|
Procedūra, ar kuru pierādīta laboratorijas kompetence izmantot šo testēšanas metodi (piem., testējot standartvielas) vai apliecināta testēšanas metodes ilgstoša reproducējamība
|
|
|
Testa procedūra
|
Replikātu un izpildes reižu skaits
|
|
Testējamās ķimikālijas koncentrācijas, aplicēšanas procedūras un ekspozīcijas laiks (ja atšķiras no rekomendētajiem)
|
|
Izmantoto izvērtēšanas un lēmuma pieņemšanas kritēriju apraksts
|
|
Izmantoto pētījuma pieņemamības kritēriju apraksts
|
|
Testa procedūras modifikāciju apraksts, ja tādas bijušas
|
|
|
Rezultāti
|
nIL8LA, Ind-IL8LA un Inh-GAPLA aprēķini
|
|
Ind-IL8LA 95 % ticamības intervāls
|
|
Grafiks, kurā attēlotas devas–atbildreakcijas līknes attiecībā uz luciferāzes aktivitātes inducēšanos un dzīvotspēju
|
|
Jebkādu citu relevantu novērojumu apraksts (attiecīgā gadījumā)
|
|
|
Rezultātu iztirzājums
|
Ar testu IL-8 Luc iegūto rezultātu iztirzājums
|
|
Testa rezultātu apskats IATA kontekstā, ja pieejama cita relevanta informācija
|
|
Secinājums
|
LITERATŪRA
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). To be published - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, France
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Paris, France.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Available at: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, Available at. http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Paris, France.
|
|
(21)
|
United Nations (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
3.1. papildinājums
DEFINĪCIJAS
Pareizība
: pakāpe, kādā testa rezultāti sakrīt ar pieņemtajām references vērtībām. Tas ir testa veiktspējas raksturlielums un viens no tā relevantuma aspektiem. Ar šo terminu, tāpat kā terminu “konkordantums”, bieži vien saprot testa pareizo iznākumu īpatsvaru (16).
AOP (nelabvēlīga iznākuma ceļš):
: no mērķķimikālijas vai līdzīgu ķimikāliju grupas ķīmiskās struktūras izrietoša notikumu sekvence no molekulārā iniciācijas notikuma līdz interesējošajam in vivo iznākumam (20).
Ķimikālija
: viela vai maisījums.
CV05
: šūnu dzīvotspēja 05, t. i., minimālā koncentrācija, pie kuras ķimikāliju Inh-GAPLA vērtība ir mazāka par 0,05.
FInSLO-LA
: validācijas ziņojumā un iepriekšējās publikācijās par testu IL-8 Luc lietota abreviatūra, ar ko apzīmē Ind-IL8LA. Definīciju sk. ierakstā par Ind-IL8LA.
GAPLA
: luciferāzes aktivitāte, kas raksturīga Stable Luciferase Red (SLR) (λmaks. = 630 nm), ko regulē GAPDH promoters un kas uzrāda šūnu dzīvotspēju un dzīvotspējīgo šūnu skaitu.
Bīstamība
: tāda aģenta vai situācijas iedabiska īpašība, kam piemīt potenciāls atstāt nelabvēlīgu ietekmi uz šim aģentam eksponētu organismu, sistēmu vai (apakš)populāciju.
IATA (integrētā testēšanas un novērtēšanas pieeja):
: strukturēta pieeja, ko izmanto ķimikālijas vai ķimikāliju grupas bīstamības identificēšanai (potenciāls), bīstamības raksturošanai (potence) un/vai drošuma novērtēšanai (potenciāls/potence un ekspozīcija) un kas stratēģiskā veidā integrē un izsver visus relevantos datus, lai varētu pieņemt normatīvu lēmumu par potenciālo bīstamību un/vai risku, un/vai nepieciešamību veikt turpmāku selektīvu un līdz ar to minimālu testēšanu.
II-SLR-LA
: validācijas ziņojumā un iepriekšējās publikācijās par testu IL-8 Luc lietota abreviatūra, ar ko apzīmē Inh-GAPLA. Definīciju sk. ierakstā par Inh-GAPLA.
IL-8 (interleikīns-8)
: no endotēlijšūnām, fibroblastiem, keratinocītiem, makrofāgiem un monocītiem iegūts citokīns, kas izraisa neitrofilu un T šūnu limfocītu hemotaksi.
IL8LA
: luciferāzes aktivitāte, kas raksturīga Stable Luciferase Orange (SLO) (λmaks. = 580 nm), ko regulē IL-8 promoters.
Ind-IL8LA
: inducēšanās pakāpe. To iegūst, ar ķimikālijām apstrādāto THP-G8 šūnu nIL8LA dalot ar nestimulēto THP-G8 šūnu nIL8LA, un šī vērtība izsaka, kādā mērā ķimikālijas inducē IL-8 promotera aktivitāti.
Inh-GAPLA GAPLA
: inhibēšanās. To iegūst, ar ķimikālijām apstrādāto THP-G8 šūnu GAPLA dalot ar neapstrādāto THP-G8 šūnu GAPLA, un šī vērtība izsaka ķimikāliju citotoksiskumu.
Minimālais inducēšanas slieksnis (MIS)
: zemākā koncentrācija, pie kuras ķimikālija atbilst pozitīvuma kritērijiem.
Maisījums
: maisījums vai šķīdums, kas sastāv no divām vai vairāk vielām.
Vienkomponenta viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā viena galvenā sastāvdaļa veido vismaz 80 % (masa/masa).
Daudzkomponentu viela
: tāda viela, definēta ar tās kvantitatīvo sastāvu, kurā vairāk nekā viena no galvenajām sastāvdaļām ir koncentrācijā ≥ 10 % (masa/masa) un < 80 % (masa/masa). Daudzkomponentu vielas tiek iegūtas ražošanas procesā. Atšķirība starp maisījumu un daudzkomponentu vielu ir tāda, ka maisījumu iegūst, divas vai vairākas vielas sajaucot bez ķīmiskas reakcijas. Daudzkomponentu vielas rodas ķīmiskās reakcijās.
nIL8LA SLO
: luciferāzes aktivitāte, kas atspoguļo IL-8 promotera aktivitāti (IL8LA), normalizēta ar SLR luciferāzes aktivitāti, kas atspoguļo GAPDH promotera aktivitāti (GAPLA). Tā izsaka IL-8 promotera aktivitāti pēc tam, kad ņemta vērā šūnu dzīvotspēja vai šūnu skaits.
nSLO-LA
: validācijas ziņojumā un iepriekšējās publikācijās par testu IL-8 Luc lietota abreviatūra, ar ko apzīmē nIL8LA. Definīciju sk. ierakstā par nIL8LA.
Pozitīvā kontrole
: replikāts, kurā ir visi testa sistēmas komponenti un kurš apstrādāts ar vielu, par ko ir zināms, ka tā inducē pozitīvu atbildreakciju. Lai būtu iespējams novērtēt pozitīvās kontroles atbildreakcijas mainību laikā, pozitīvās atbildreakcijas intensitāte nedrīkstētu būt pārlieku liela.
Prehaptēni
: ķimikālijas, kas par sensibilizatoriem kļūst abiotiskas transformācijas procesā.
Prohaptēni
: ķimikālijas, kuru ādas sensibilizācijas potenciālu atraisa fermentatīva aktivācija.
Relevantums
: parametrs, kas raksturo testa attiecināmību uz interesējošo ietekmi un to, vai testu lietot konkrētam nolūkam ir jēgpilni un noderīgi. Pakāpe, kādā ar testu var pareizi izmērīt vai prognozēt interesējošo bioloģisko ietekmi. Relevantums ietver arī testa pareizības (konkordantuma) aspektu (16).
Ticamība
: parametrs, kas izsaka, kādā pakāpē, izmantojot vienu un to pašu protokolu, testu vienā un tajā pašā laboratorijā vai dažādās laboratorijās ilgākā laika nogrieznī var veikt reproducējami. To novērtē, aprēķinot metodes intralaboratorisko un interlaboratorisko reproducējamību un intralaboratorisko atkārtojamību (16).
Izpildes reize
: reize, kurā vienu vai vairākas ķimikālijas testē paralēli ar šķīdinātāja/nesēja kontroli un pozitīvo kontroli.
Jutība
: tas, cik liela daļa no visām pozitīvajām/aktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (16).
SLO-LA
: validācijas ziņojumā un iepriekšējās publikācijās par testu IL-8 Luc lietota abreviatūra, ar ko apzīmē IL8LA. Definīciju sk. ierakstā par IL8LA.
SLR-LA
: validācijas ziņojumā un iepriekšējās publikācijās par testu IL-8 Luc lietota abreviatūra, ar ko apzīmē GAPLA. Definīciju sk. ierakstā par GAPLA.
Šķīdinātāja/nesēja kontrole
: neapstrādāts paraugs, kurā ir visi testa sistēmas komponenti, izņemot testējamo ķimikāliju, bet ieskaitot izmantoto šķīdinātāju/nesēju. To izmanto, lai noteiktu bāzlīnijas atbildreakciju paraugiem, kas apstrādāti ar attiecīgajā šķīdinātājā/nesējā izšķīdinātu vai stabili disperģētu testējamo ķimikāliju. Ja testā izmanto arī paralēlu barotnes kontroli, šis paraugs parāda arī to, vai šķīdinātājs/nesējs mijiedarbojas ar testa sistēmu.
Specifiskums
: tas, cik liela daļa no visām negatīvajām/neaktīvajām ķimikālijām ar testu tiek klasificētas pareizi. Tas ir tāda testa pareizības mērs, ar kuru iegūst kategoriālus rezultātus, un svarīgs faktors testa relevantuma izvērtēšanā (16).
Viela
: ķīmisks elements un tā dabiski radušies vai ražošanas procesā iegūti savienojumi, ieskaitot to stabilizācijai vajadzīgās piedevas, kā arī izmantotajos procesos radušos piejaukumus, bet izņemot šķīdinātājus, kurus var atdalīt, neietekmējot vielas stabilitāti un neizmainot tās sastāvu.
Virsmaktīvs aģents
: viela, piem., detergents, kas var samazināt šķidruma virsmas spraigumu un tādējādi ļauj tam putot vai penetrēt cietvielas; to dēvē arī par mitrinātāju. (TG437)
Testējamā ķimikālija
: jebkura viela vai maisījums, ko testē ar šo metodi.
THP-G8 testā IL-8 Luc
: izmantota IL-8 reportieršūnu līnija. Cilvēka makrofāgiskā šūnu līnija THP-1, kura transficēta ar luciferāzes SLO un SLR gēniem attiecīgi IL-8 un GAPDH promoteru kontrolē.
Apvienoto Nāciju Organizācijas Ķimikāliju klasificēšanas un marķēšanas globāli harmonizētā sistēma (ANO GHS)
: sistēma, kurā ķimikālijas (vielas un maisījumus) klasificē pēc standartizētiem fizisko apdraudējumu, veselības apdraudējumu un vidisko apdraudējumu tipiem un līmeņiem un kura paredz atbilstošus saziņas elementus, piem., piktogrammas, signālvārdus, bīstamības apzīmējumus, drošības prasību apzīmējumus un drošības datu lapas, ar ko tiek sniegta informācija par ķimikāliju nelabvēlīgo ietekmi, tā aizsargājot cilvēkus (tostarp darba devējus, darba ņēmējus, transporta darbiniekus, patērētājus un avārijas dienestu darbiniekus) un vidi (21).
UVCB
: vielas, kuru sastāvs nav zināms vai ir mainīgs, kuras ir kompleksi reakcijas produkti vai bioloģiski materiāli.
Derīga testēšanas metode
: tests, kas tiek uzskatīts par pietiekami relevantu un ticamu konkrētam nolūkam un ir balstīts uz zinātniski pareiziem principiem. Tests nekad nav derīgs absolūtā izteiksmē; tas vienmēr ir derīgs tikai kādam konkrētam nolūkam.
3.2. papildinājums
LUCIFERĀZES AKTIVITĀTES MĒRĪŠANAS PRINCIPS UN OPTISKO FILTRU PĀRVADES KOEFICIENTU NOTEIKŠANA SLO UN SLR
Daudzreportieru testa sistēmu Tripluc var izmantot ar mikroplates tipa luminometru, kam ir daudzu krāsu detektēšanas sistēma un ko var aprīkot ar optisko filtru (piem., Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). Par optisko filtru mērīšanā izmanto 600–620 nm garviļņu vai īsviļņu caurlaides filtru vai 600–700 nm frekvenču joslas filtru.
Divu krāsu luciferāzu mērīšana ar optisko filtru
Šis ir piemērs, kurā izmantots Phelios AB-2350 (ATTO). Šis luminometrs ir aprīkots ar 600 nm garviļņu caurlaides filtru (R60 HOYA Co., 600 nm GCF, 1. filtrs) SLO (λmaks. = 580 nm) un SLR (λmaks. = 630 nm) luminiscences nošķiršanai.
Lai noteiktu 600 nm GCF pārvades koeficientus, vispirms, izmantojot attīrītus luciferāzes SLO un SLR fermentus, i) izmēra SLO un SLR bioluminiscences intensitāti bez filtra (F0), ii) izmēra SLO un SLR bioluminiscences intensitāti caur 600 nm GCF (1. filtrs) un iii) aprēķina tālāk norādītos 600 nm GCF pārvades koeficientus attiecībā uz SLO un SLR.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Ja testa parauga SLO un SLR intensitāte ir attiecīgi O un R, tad i) gaismas intensitāte bez filtra (pilns optiskais spektrs) (F0) un ii) caur 600 nm GCP (1. filtru) pārvadītas gaismas intensitāte (F1) ir tāda, kā redzams šajās formulās.
F0=O+R
F1=κOR60 x O + κRR60 x R
Šīs formulas var izteikt arī šādi:
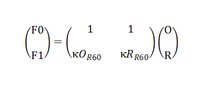
Tad, izmantojot aprēķinātos pārvades koeficientus (κOR60 un κRR60) un izmērīto F0 un F1, O un R vērtību var aprēķināt šādi:

Pārvades koeficientu noteikšanas materiāli un metodes
|
1)
|
Reaģenti
Atsevišķi attīrīti luciferāzes fermenti:
|
liofilizēts attīrīts SLO ferments
|
|
liofilizēts attīrīts SLR ferments
|
|
(kas validācijas darba vajadzībām iegūti no GPC Lab. Co. Ltd., Totori, Japāna, kopā ar šūnu līniju THP-G8)
|
Testa reaģents:
|
testa reaģents Tripluc® Luciferase (piem., no TOYOBO Cat#MRA-301)
|
Barotne: luciferāzes testam (30 ml, turēta 2–8 °C)
|
|
Reaģents
|
Koncentrācija
|
Galīgā koncentrācija barotnē
|
Vajadzīgais daudzums
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
2)
|
Fermenta šķīduma sagatavošana
Liofilizētu attīrītu luciferāzes fermentu izšķīdina stobriņā, tam pievienojot 200 μl 10 ~ 100 mM Tris/HCl vai Hepes/HCl (pH 7,5 ~ 8,0), kas papildināts ar 10 % (masa/tilp.) glicerīna, fermenta šķīdumu sadala 10 μl alikvotās vienreizlietojamos 1,5 ml stobriņos un -80 °C tur saldētavā. Sasaldēto fermenta šķīdumu pēc tam var izmantot 6 mēnešus. Kad šķīdumu izmanto, katram fermenta šķīduma (atšķaidīta fermenta šķīduma) stobriņam pievieno 1 ml luciferāzes testa barotnes (RPMI-1640 ar 10 % FBS) un to tur uz ledus, lai nenotiktu dezaktivācija.
|
|
3)
|
Bioluminiscences mērīšana
Testa reaģentu Tripluc® Luciferase (Tripluc) atkausē un tur istabas temperatūrā vai nu ūdens vannā, vai apkārtējās vides temperatūrā. Luminometru ieslēdz 30 min pirms mērījumu sākšanas, lai fotoelektronpavairotājlampa nostabilizētos. 100 μl atšķaidītā fermenta šķīduma pārnes uz melnu 96 iedobju plati (plakandibena) (SLO referencparaugu uz #B1, #B2, #B3, SLR referencparaugu uz #D1, #D2, #D3). Tad 100 μl iepriekš sasildīta Tripluc ar automātisko pipeti pārnes uz katru plates iedobi, kurā ir atšķaidītais fermenta šķīdums. Ar plates kratītāju plati 10 min krata istabas temperatūrā (apm. 25 °C). Iedobēs šķīdumā likvidē burbuļus, ja tādi parādās. Lai izmērītu luciferāzes aktivitāti, plati ieliek luminometrā. Bioluminiscenci mēra gan bez optiskā filtra (F0), gan ar to (F1), katrreiz 3 s.
Optiskā filtra pārvades koeficientu aprēķina šādi:
pārvades koeficients (SLO (κOR60)) = (#B1 ar F1+ #B2 ar F1+ #B3 ar F1) / (#B1 ar F0+ #B2 ar F0+ #B3 ar F0)
pārvades koeficients (SLR (κRR60)) = (#D1 ar F1+ #D2 ar F1+ #D3 ar F1) / (#D1 ar F0+ #D2 ar F0+ #D3 ar F0)
Aprēķinātos pārvades koeficientus izmanto visiem mērījumiem, ko veic ar to pašu luminometru.
|
Aprīkojuma kvalitātes kontrole
Jāizmanto IL-8 Luc protokolā aprakstītās procedūras (18).
3.3. papildinājums
STANDARTVIELAS
Pirms šajā testēšanas metodes B.71 papildinājumā aprakstītā testa regulāras izmantošanas laboratorijām jāpierāda sava tehniskā kompetence, iegūstot paredzamo testa IL-8 Luc prognozi attiecībā uz 1. tabulā ieteiktajām 9 vielām un attiecībā uz vismaz 8 no 9 standartvielām (kas izraudzītas tā, lai būtu reprezentatīvas ādas sensibilizācijas atbildreakciju diapazonam) iegūstot vērtības, kuras ietilpst attiecīgajā references diapazonā. Citi atlases kritēriji bija vielu komerciālā pieejamība un kvalitatīvu ar testu IL-8 Luc iegūtu in vivo references datu, kā arī kvalitatīvu ar testu IL-8 Luc iegūtu in vitro datu pieejamība. Par testu IL-8 Luc ir pieejami arī publicēti references dati (6) (1).
1. tabula.
Vielas, kuras ieteicams izmantot, lai pierādītu tehnisko kompetenci izmantot testu IL-8 Luc
|
Standartvielas
|
CAS Nr.
|
Agregātstāvoklis
|
Šķīdība X-VIVOTM 15 koncentrācijā 20 mg/ml
|
In vivo prognoze (108)
|
IL-8 Luc prognoze (109)
|
References diapazons (μg/ml) (110)
|
|
|
CV05
(111)
|
IL-8 Luc MIS (112)
|
|
2,4-dinitrohlorbenzols
|
97-00-7
|
ciets
|
nešķīst
|
sensibilizators (ļoti spēcīgs)
|
pozitīva
|
2,3–3,9
|
0,5–2,3
|
|
formaldehīds
|
50-00-0
|
šķidrs
|
šķīst
|
sensibilizators (spēcīgs)
|
pozitīva
|
9–30
|
4–9
|
|
2-merkaptobenztiazols
|
149-30-4
|
ciets
|
nešķīst
|
sensibilizators (vidējs)
|
pozitīva
|
250–290
|
60–250
|
|
etilēndiamīns
|
107-15-3
|
šķidrs
|
šķīst
|
sensibilizators (vidējs)
|
pozitīva
|
500–700
|
0,1–0,4
|
|
etilēnglikola dimetakrilāts
|
97-90-5
|
šķidrs
|
nešķīst
|
sensibilizators (vājš)
|
pozitīva
|
> 2000
|
0,04–0,1
|
|
4-alilanizols (estragols)
|
140-67-0
|
šķidrs
|
nešķīst
|
sensibilizators (vājš)
|
pozitīva
|
> 2000
|
0,01–0,07
|
|
streptomicīna sulfāts
|
3810-74-0
|
ciets
|
šķīst
|
nav sensibilizators
|
negatīva
|
> 2000
|
> 2000
|
|
glicerīns
|
56-81-5
|
šķidrs
|
šķīst
|
nav sensibilizators
|
negatīva
|
> 2000
|
> 2000
|
|
izopropanols
|
67-63-0
|
šķidrs
|
šķīst
|
nav sensibilizators
|
negatīva
|
> 2000
|
> 2000
|
|
Saīsinājumi: CAS Nr. — Chemical Abstracts Service reģistra numurs
|
3.4. papildinājums
INDEKSI UN SLĒDZIENU KRITĒRIJI
nIL8LA (nSLO-LA)
Attiecīgi IL8LA (SLO-LA) un GAPLA (SLR-LA) mēra i-tās koncentrācijas (i = 0–11) j-to atkārtojumu (j = 1–4). Normalizētā IL8LA, ko dēvē par nIL8LA (nSLO-LA), ir definēta šādi:
nIL8LAij = IL8LAij/GAPLAij
Tā ir šā testa pamatmērvienība.
Ind-IL8LA (FInSLO-LA)
Atkārtojuma vidējotās nIL8LA (nSLO-LA) palielinājuma pakāpe pie i-tās koncentrācijas salīdzinājumā ar 0-to koncentrāciju, Ind-IL8LA, ir šā testa pamatmērs. Šo attiecību izsaka ar šādu formulu:

Vadošā laboratorija ierosinājusi uzskatīt, ka pozitīvam testējamās ķimikālijas rezultātam atbilst vērtība 1,4. Šīs vērtības pamatā ir vadošās laboratorijas vēsturisko datu izpēte. Datu pārvaldības grupa šo vērtību izmantojusi visos validācijas pētījuma posmos. Primārais iznākums, Ind-IL8LA, ir divu aritmētisko vidējo attiecība, kā redzams vienādojumā.
95 % ticamības intervāls (95 % TI)
Balstoties uz šo attiecību, var aplēst 95 % ticamības intervālu (95 % TI), kas parāda, cik precīzs ir šis primārā iznākuma mērs. 95 % TI apakšējā robeža ≥ 1 liecina, ka nIL8LA pie i-tās koncentrācijas ir ievērojami lielāka nekā šķīdinātāja kontroles gadījumā. 95 % TI var iegūt vairākos veidos. Šajā pētījumā šādam nolūkam izmantota Fīlera [Fieller] teorēma. Pēc šīs teorēmas 95 % ticamības intervālu iegūst saskaņā ar šādu formulu:

kur: .
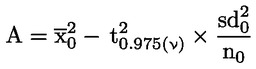
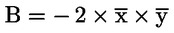



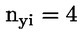


ir centrālā t sadalījuma 97,5. procentile ar brīvības pakāpes ν, kur

Inh-GAPLA (II-SLR-LA)
Inh-GAPLA ir i-tās koncentrācijas atkārtojuma vidējotās GAPLA (SLR-LA) attiecība pret attiecīgo šķīdinātāja kontroles vērtību, un to izsaka
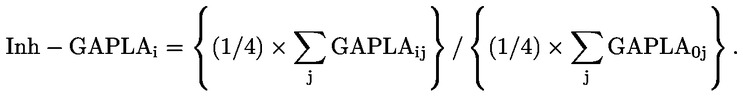
Tā kā GAPLA ir nIL8LA saucējs, ļoti maza vērtība izraisa lielas nIL8LA variācijas. Tāpēc var uzskatīt, ka, ja Inh-GAPLA vērtība ir ļoti maza (mazāka par 0,05), Ind-IL8LA vērtības ir neprecīzas.
3.5. papildinājums
METODES, KĀ IZŠĶĪDINĀT ĶIMIKĀLIJAS TESTA IL-8 LUC VAJADZĪBĀM: SHĒMA
|
a)
|
Ķimikālijas, kas X-VIVOTM 15 šķīst koncentrācijā 20 mg/ml
|
|
b)
|
Ķimikālijas, kas X-VIVOTM 15 koncentrācijā 20 mg/ml nešķīst

|
3.6. papildinājums
METODE, KĀ IZŠĶĪDINĀT 4-NBB IZMANTOŠANAI PAR POZITĪVO KONTROLI TESTA IL-8 LUC VAJADZĪBĀM: SHĒMA

” |


 In force
In force