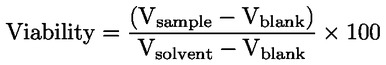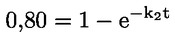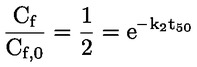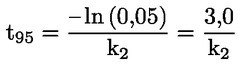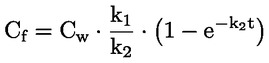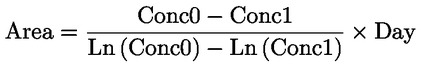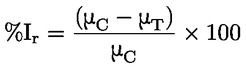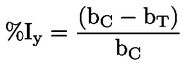ISSN 1977-0707
Gazzetta ufficiale
dell'Unione europea
L 112

Edizione in lingua italiana
Legislazione
60° anno
28 aprile 2017
|
ISSN 1977-0707 |
||
|
Gazzetta ufficiale dell'Unione europea |
L 112 |
|
|
|
||
|
Edizione in lingua italiana |
Legislazione |
60° anno |
|
|
|
|
|
(1) Testo rilevante ai fini del SEE. |
|
IT |
Gli atti i cui titoli sono stampati in caratteri chiari appartengono alla gestione corrente. Essi sono adottati nel quadro della politica agricola ed hanno generalmente una durata di validità limitata. I titoli degli altri atti sono stampati in grassetto e preceduti da un asterisco. |
II Atti non legislativi
REGOLAMENTI
|
28.4.2017 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 112/1 |
REGOLAMENTO (UE) 2017/735 DELLA COMMISSIONE
del 14 febbraio 2017
recante modifica del regolamento (CE) n. 440/2008 che istituisce dei metodi di prova ai sensi del regolamento (CE) n. 1907/2006 del Parlamento europeo e del Consiglio concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH), al fine di adeguarlo al progresso tecnico
(Testo rilevante ai fini del SEE)
LA COMMISSIONE EUROPEA,
visto il trattato sul funzionamento dell'Unione europea,
visto il regolamento (CE) n. 1907/2006 del Parlamento europeo e del Consiglio, del 18 dicembre 2006, concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH), che istituisce un'Agenzia europea per le sostanze chimiche, che modifica la direttiva 1999/45/CE e che abroga il regolamento (CEE) n. 793/93 del Consiglio e il regolamento (CE) n. 1488/94 della Commissione, nonché la direttiva 76/769/CEE del Consiglio e le direttive della Commissione 91/155/CEE, 93/67/CEE, 93/105/CE e 2000/21/CE (1), in particolare l'articolo 13, paragrafo 2,
considerando quanto segue:
|
(1) |
Il regolamento (CE) n. 440/2008 (2) della Commissione istituisce i metodi di prova per determinare le proprietà fisico-chimiche, la tossicità e l'ecotossicità delle sostanze chimiche applicabili ai fini del regolamento (CE) n. 1907/2006. |
|
(2) |
È necessario aggiornare il regolamento (CE) n. 440/2008 per includervi in via prioritaria nuovi e aggiornati metodi di prova adottati di recente dall'Organizzazione per la cooperazione e lo sviluppo economici (OCSE) al fine di tener conto del progresso tecnico e ridurre il numero di animali usati a scopi di sperimentazione, conformemente alla direttiva 2010/63/UE del Parlamento europeo e del Consiglio (3). Le parti interessate sono state consultate in merito alla proposta. |
|
(3) |
L'adeguamento al progresso tecnico contiene venti metodi di prova: un nuovo metodo per la determinazione delle proprietà fisico-chimiche, cinque metodi di prova aggiornati e uno nuovo per la valutazione dell'ecotossicità, due metodi di prova aggiornati per valutare il destino e il comportamento ambientale e quattro metodi di prova nuovi e sette aggiornati per la determinazione degli effetti sulla salute umana. |
|
(4) |
L'OCSE esamina regolarmente le linee guida relative ai metodi di prova, al fine di individuare quelli che sono scientificamente superati. Tale adeguamento al progresso tecnico elimina sei metodi di prova per i quali le linee guida dell'OCSE corrispondenti sono state soppresse. |
|
(5) |
È opportuno pertanto modificare di conseguenza il regolamento (CE) n. 440/2008. |
|
(6) |
Le misure di cui al presente regolamento sono conformi al parere del comitato di cui all'articolo 133 del regolamento (CE) n. 1907/2006, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
L'allegato del regolamento (CE) n. 440/2008 è modificato conformemente all'allegato del presente regolamento.
Articolo 2
Il presente regolamento entra in vigore il ventesimo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 14 febbraio 2017
Per la Commissione
Il presidente
Jean-Claude JUNCKER
(1) GU L 396 del 30.12.2006, pag. 1.
(2) Regolamento (CE) n. 440/2008 della Commissione, del 30 maggio 2008, che istituisce dei metodi di prova ai sensi del regolamento (CE) n. 1907/2006 del Parlamento europeo e del Consiglio concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH) (GU L 142 del 31.5.2008, pag. 1).
(3) Direttiva 2010/63/UE del Parlamento europeo e del Consiglio, del 22 settembre 2010, sulla protezione degli animali utilizzati a fini scientifici (GU L 276 del 20.10.2010, pag. 33).
ALLEGATO
L'allegato del regolamento (CE) n. 440/2008 è così modificato:
|
(1) |
Nella parte A è aggiunto il seguente capitolo: «A.25 COSTANTI DI DISSOCIAZIONE IN ACQUA (METODO DELLA TITOLAZIONE — METODO SPETTROFOTOMETRICO — METODO CONDUTTIMETRICO) INTRODUZIONE Questo metodo di prova è equivalente alla linea guida dell'OCSE n. 112 (1981) Presupposti
Informazioni generali
Condizioni particolari
Documenti di riferimento Il presente metodo di prova si basa sui metodi indicati nei riferimenti citati nella sezione “Bibliografia” e nel documento Preliminary Draft Guidance for Premanufacture Notification dell'EPA, del 18 agosto 1978. INTRODUZIONE, OGGETTO, PORTATA, PERTINENZA, APPLICAZIONE E LIMITI DI PROVA La dissociazione di una sostanza in acqua è importante per valutare il suo impatto sull'ambiente. Essa determina la forma della sostanza che, a sua volta, ne determina il comportamento e il trasporto. Può incidere sull'adsorbimento della sostanza chimica nel suolo e nei sedimenti e sull'assorbimento all'interno di cellule biologiche. Definizioni e unità di misura La dissociazione è la divisione reversibile in due o più specie chimiche che possono essere ionizzate. Il processo è comunemente indicato dall'equilibrio: RX ⇌ R ++ X – e la costante di equilibrio della concentrazione è:
Ad esempio, nel caso specifico in cui R è l'idrogeno (la sostanza è un acido), la costante è:
oppure
Sostanze di riferimento Non è necessario utilizzare le seguenti sostanze di riferimento ogni volta che si esamina una nuova sostanza. Esse sono fornite soprattutto affinché la calibrazione del metodo possa essere effettuata periodicamente e che i risultati possano essere confrontati con i risultati ottenuti con altri metodi.
Sarebbe utile disporre di una sostanza con diversi pK, come indicato nella sezione “Principio del metodo”. Una siffatta sostanza potrebbe essere:
Principio del metodo Il processo chimico descritto dipende generalmente solo in misura ridotta dalla temperatura nell'intervallo delle temperature registrate nell'ambiente. La determinazione della costante di dissociazione implica la misurazione delle concentrazioni delle forme dissociate e non dissociate della sostanza chimica in esame. Conoscendo la stechiometria della reazione di dissociazione indicata nella sezione “Definizioni e unità”, si può determinare la costante corrispondente. Nel caso specifico descritto nel presente metodo di prova, la sostanza si comporta come un acido o una base, ed il metodo più facile consiste nel determinare le relative concentrazioni nelle forme ionizzata e non ionizzata della sostanza e il pH della soluzione. Il rapporto tra queste condizioni è dato dall'equazione per pKa nella sezione “Definizione e unità”. Alcune sostanze presentano più di una costante di dissociazione e possono essere sviluppate formule analoghe. Alcuni dei metodi qui descritti si applicano anche alla dissociazione non-acido/base. Criteri di qualità Riproducibilità La costante di dissociazione va replicata (un minimo di tre determinazioni); i valori devono essere compresi in un intervallo di ± 0,1 unità logaritmiche. DESCRIZIONE DEL METODO Vi sono due differenti approcci per la determinazione del pKa. Uno comporta la titolazione di una quantità nota della sostanza con un acido o una base standard, a seconda dei casi; l'altro consiste nel determinare la concentrazione relativa delle forme ionizzata e non ionizzata e la sua dipendenza dal pH. Preparazioni I metodi basati su questi principi possono essere classificati come metodo della titolazione, metodo spettrofotometrico e metodo conduttimetrico. Soluzioni campione Per il metodo della titolazione e quello conduttimetrico, dissolvere la sostanza chimica in esame in acqua distillata. Per il metodo spettrofotometrico e altri metodi sono utilizzate le soluzioni tampone. La concentrazione della sostanza di prova non deve superare il valore minore tra 0,01 M e la metà della concentrazione di saturazione; per la preparazione delle soluzioni utilizzare la sostanza nella forma più pura disponibile. Se è scarsamente solubile, la sostanza può essere disciolta in una piccola quantità di solvente miscibile con l'acqua prima di essere aggiunta alle concentrazioni sopra indicate. Le soluzioni devono essere sottoposte a controllo per verificare la presenza di emulsioni utilizzando un fascio Tyndall, soprattutto se è stato utilizzato un co-solvente per migliorare la solubilità. Quando si utilizzano soluzioni tampone, la loro concentrazione non deve superare 0,05 M. Condizioni sperimentali Temperatura La temperatura deve essere controllata almeno a ± 1 °C. La determinazione deve essere effettuata, preferibilmente, a 20 °C. Se si ritiene che i risultati varino in modo significativo in funzione della temperatura, ripetere la determinazione ad almeno altre due temperature. In tal caso, gli intervalli di temperatura devono essere di 10°C, con un controllo di ± 0,1 °C circa. Analisi Il metodo sarà determinato dalla natura della sostanza chimica in esame. Esso deve essere sufficientemente sensibile da consentire la determinazione delle diverse specie a ciascuna delle concentrazioni della soluzione campione. Esecuzione della prova Metodo della titolazione La soluzione campione è determinata per titolazione con una soluzione acida o basica standard, secondo il caso. Misurare il pH dopo ogni aggiunta di titolante. Vanno effettuate almeno 10 aggiunte incrementali prima del punto di equivalenza. Se l'equilibrio è raggiunto abbastanza rapidamente, si può utilizzare un potenziometro registratore. Per questo metodo è necessario conoscere con precisione la quantità totale della sostanza e la sua concentrazione. Vanno prese le precauzioni necessarie per escludere la presenza di CO2. I dettagli della procedura, le misure precauzionali da adottare e il metodo di calcolo sono riportati nelle prove standardizzate, ad esempio nei riferimenti (1), (2), (3) e (4) Metodo spettrofotometrico Trovare una lunghezza d'onda in cui le forme ionizzate e non ionizzate della sostanza presentano coefficienti di estinzione significativamente diversi. Lo spettro di assorbimento UV/Vis è ottenuto da soluzioni di concentrazione costante, in condizioni di pH in cui la sostanza è praticamente non ionizzata, completamente ionizzata e a diversi pH intermedi. Ciò può essere ottenuto mediante aggiunta di acido (o di base) concentrato a un volume relativamente significativo di una soluzione della sostanza in un tampone a più componenti, inizialmente a pH elevato (basso) (rif. 5), o aggiungendo volumi uguali della soluzione madre della sostanza stessa, ad esempio in acqua o metanolo, a volumi costanti di varie soluzioni tampone che coprono l'intervallo voluto di pH. A partire dai valori di pH e di assorbanza alla lunghezza d'onda scelta si calcola un numero sufficiente di valori pKa utilizzando i dati ottenuti per almeno 5 pH diversi ai quali il tasso di ionizzazione della sostanza è compreso tra il 10 e il 90 %. Ulteriori dati sperimentali e metodo di calcolo sono riportati nel riferimento (1). Metodo conduttimetrico Utilizzando una cella, la cui costante è nota e ridotta, misurare la conducibilità di circa 0,1 M di soluzione della sostanza in acqua. Sono inoltre misurate le conducibilità di alcune diluizioni, accuratamente preparate, di tale sostanza. La concentrazione è dimezzata ogni volta, e la serie deve comprendere almeno un ordine di grandezza di concentrazione. La conducibilità limite a una diluizione infinita è ottenuta eseguendo una sperimentazione simile con il sale di Na e estrapolando. Si può quindi calcolare il grado di dissociazione a partire dalla conducibilità di ciascuna soluzione utilizzando l'equazione di Onsager; successivamente, applicando la legge di diluizione di Ostwald, la costante di dissociazione può essere calcolata con la seguente formula: K = α2C/(1 – α), dove C è la concentrazione in moli per litro e α è la frazione dissociata. Vanno prese le precauzioni necessarie per escludere la presenza di CO2. Ulteriori dati sperimentali e metodo di calcolo sono indicati nei testi di riferimento e nei riferimenti (1), (6) e (7). DATI E RELAZIONE Calcolo dei risultati Metodo per titolazione Il pKa è calcolato per 10 punti misurati sulla curva di titolazione. Si calcola la media e la deviazione standard di tali valori pKa. Vanno inclusi un grafico del pH in funzione del volume di base o acido standard e una presentazione sotto forma di tabella. Metodi spettrofotometrici L'assorbanza e il pH sono registrati per ciascuno spettro. Partendo dai punti corrispondenti ai dati relativi agli spettri intermedi si calcolano almeno cinque valori pKa, nonché la media e la deviazione standard dei risultati. Metodo conduttimetrico La conducibilità equivalente Λ è calcolata per ciascuna concentrazione dell'acido e per ciascuna concentrazione di una miscela di 1 equivalente di acido più 0,98 equivalente di idrossido di sodio privo di carbonato. L'acido è eccedente per evitare un eccesso di OH– dovuto all'idrolisi. Si rappresenta graficamente 1/Λ in funzione di √C e si può trovare il valore Λo del sale mediante estrapolazione a concentrazione zero. Il valore λo dell'acido può essere calcolato utilizzando i valori forniti dalla letteratura per H+ and Na+. Il pKa può essere ricavato dalle formule α = Λi /Λo e Ka = α2C/(1 – α) per ciascuna concentrazione. Si possono ottenere valori migliori per Ka procedendo a correzione per la mobilità e l'attività. Calcolare le medie e le deviazioni standard dei valori pKa. Relazione sula prova Tutti i dati grezzi e i valori calcolati per il pKa devono essere presentati assieme al metodo di calcolo (preferibilmente sotto forma di tabella, come suggerito nel rif. 1) così come i parametri statistici sopra descritti. Per i metodi per titolazione, indicare informazioni dettagliate sui metodi di standardizzazione dei titolanti. Per i metodi spettrofotometrici, presentare tutti gli spettri. Per il metodo conduttimetrico riportare i dettagli della determinazione della costante di cella. Vanno indicati la tecnica utilizzata, i metodi di analisi e la natura di eventuali tamponi utilizzati. La o le temperature sperimentali devono essere indicate. BIBLIOGRAFIA
|
|
(2) |
Nella parte B, il capitolo B.5 è sostituito dal seguente: «B.5 IRRITAZIONE/CORROSIONE OCULARE ACUTA INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 405 (2012). Le linee guida dell'OCSE per le prove sulle sostanze chimiche sono rivedute periodicamente affinché riflettano le migliori conoscenze scientifiche disponibili. Nelle precedenti revisioni di questa linea guida si è prestata particolare attenzione, mediante la valutazione di tutte le informazioni disponibili sulla sostanza chimica in esame, ai miglioramenti che si possono apportare per evitare prove non necessarie sugli animali da laboratorio e tenere conto in modo più adeguato del benessere degli animali. La linea guida n. 405 (adottata nel 1981 e aggiornata nel 1987, 2002 e 2012) raccomanda, prima di eseguire il saggio in vivo descritto per l'irritazione/corrosione oculare acuta, di effettuare un'analisi basata sul “peso dell'evidenza” (weight-of-the-evidence) (1) dei dati pertinenti esistenti. Qualora i dati disponibili fossero insufficienti, si raccomanda di ottenerli mediante l'applicazione di sperimentazioni sequenziali (2)(3). La strategia sperimentale raccomandata comprende l'esecuzione di prove in vitro validate ed accettate ed è descritta nell'allegato del presente metodo. Ai fini del regolamento (CE) n. 1907/2006 concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH) (2), una strategia sperimentale integrata è inclusa anche nella rispettiva linea guida ECHA (21). La sperimentazione sugli animali dovrebbe essere effettuata solo se ritenuta necessaria, dopo aver preso in considerazione i metodi alternativi disponibili e aver utilizzato quelli ritenuti appropriati. Al momento della stesura del presente metodo di prova aggiornato, esistono ancora casi in cui il ricorso a questo metodo di prova rimane indispensabile o obbligatorio nell'ambito di alcuni quadri regolamentari. L'aggiornamento più recente si è concentrato principalmente sull'uso di analgesici e anestetici senza modificare il concetto di base e la struttura della linea guida. L'ICCVAM (3) e un gruppo internazionale di esperti scientifici indipendenti hanno esaminato l'utilità e i limiti di un ricorso sistematico ad anestetici per uso topico, ad analgesici sistemici e a endpoint umanitari durante la prova di sicurezza in vivo dell'irritazione oculare (12). Da questo esame è emerso che l'impiego di anestetici per uso topico e di analgesici sistemici permette di evitare agli animali la maggior parte, se non la totalità, del dolore e dello stress senza modificare il risultato della prova, e raccomanda che tali sostanze siano utilizzate in modo sistematico. Questo metodo tiene conto delle conclusioni di tale revisione. È pertanto opportuno che gli anestetici per uso topico, gli analgesici sistemici e gli endpoint umanitari siano utilizzati sistematicamente nell'ambito della sperimentazione in vivo dell'irritazione/corrosione oculare acuta. Qualsiasi eccezione deve essere giustificata. Le modifiche di cui al presente metodo contribuiranno in misura significativa a ridurre o evitare il dolore e lo stress degli animali nella maggior parte delle sperimentazioni che richiedono ancora una prova di sicurezza oculare in vivo. Una gestione preventiva ed equilibrata del dolore comprende: i) un pretrattamento di routine con un anestetico a uso topico (ad es. proparacaina o tetracaina) e un analgesico sistemico (ad es. buprenorfina), ii) un programma di trattamento post-esposizione di routine con un analgesico sistemico (ad es. buprenorfina e melossicam), iii) un programma di osservazione, monitoraggio e la registrazione dei segni clinici di dolore o di stress negli animali, e iv) un programma di osservazione e monitoraggio e la registrazione della natura, della gravità e della progressione di tutte le lesioni oculari. Ulteriori dettagli sono forniti nelle procedure aggiornate descritte di seguito. Dopo l'esposizione alla sostanza chimica in esame, non dovrà essere somministrato alcun anestetico o analgesico per uso topico supplementare, in modo da evitare qualsiasi interferenza con la prova. Gli analgesici con proprietà antinfiammatorie (come il melossicam) non saranno oggetto di applicazione topica e le dosi utilizzate sistematicamente non devono interferire con gli effetti oculari. Le definizioni sono riportate nell'Appendice del presente metodo di prova. CONSIDERAZIONI INIZIALI Nell'interesse dell'accuratezza scientifica e del benessere degli animali, non bisogna considerare il ricorso a prove in vivo finché non siano stati valutati, sulla base del peso dell'evidenza, tutti i dati disponibili pertinenti sulla potenziale corrosività/irritazione oculare della sostanza chimica. Tali dati comprendono le evidenze scientifiche derivanti da studi esistenti su soggetti umani e/o animali da laboratorio, le evidenze di corrosività/irritazione di una o più sostanze strutturalmente correlate o di miscele di tali sostanze, dati dimostranti l'elevata acidità o alcalinità della sostanza chimica (4) (5), nonché i risultati di prove in vitro o ex vivo validate ed accettate sulla corrosione e l'irritazione cutanea e oculare (6) (13) (14) (15) (16) (17). Gli studi possono essere stati condotti prima di un'analisi basata sul peso delle evidenze, o in conseguenza di essa. Per alcune sostanze chimiche, un'analisi di questo tipo può indicare la necessità di studi in vivo del potenziale di corrosione/irritazione oculare. In tutti questi casi, prima di considerare il ricorso a prove oculari in vivo, va preferibilmente condotto uno studio sugli effetti della corrosione cutanea in vivo e/o in vitro della sostanza chimica, e valutato in base alla strategia di sperimentazione sequenziale nel metodo di prova B.4 (7) o alla strategia sperimentale integrata descritta nella linea guida ECHA (21). La strategia di sperimentazione sequenziale, che prevede l'esecuzione di prove della corrosione/irritazione oculare in vitro o ex vivo validate, è riportata nell'Allegato al presente metodo di prova e, per le finalità del REACH, nella linea guida ECHA. Si raccomanda di seguire questa strategia prima di passare alla sperimentazione in vivo. Per le nuove sostanze chimiche, si raccomanda di adottare un approccio sperimentale graduale, per sviluppare dati scientificamente validi sulla corrosività/irritazione della sostanza chimica. Se per le sostanze chimiche esistenti i dati sulla corrosione/irritazione oculare e cutanea sono insufficienti, si può usare tale strategia per recuperare i dati mancanti. È necessario giustificare l'uso di una strategia o procedura sperimentale differente, nonché l'eventuale decisione di non usare un approccio sperimentale «per gradi.» PRINCIPIO DELLA PROVA IN VIVO Successivamente al pretrattamento con un analgesico sistemico e della somministrazione di un'adeguata anestesia topica, la sostanza chimica in esame è applicata in un'unica dose su uno degli occhi dell'animale sperimentale; l'occhio non trattato serve da controllo. Il grado di irritazione/corrosione è valutato dando un punteggio alle lesioni di congiuntiva, cornea e iride, a specifici intervalli di tempo. Per fornire una valutazione completa degli effetti sono descritti anche altri effetti sull'occhio ed effetti negativi sistemici. La durata dello studio deve essere sufficiente per valutare la reversibilità o irreversibilità degli effetti. Gli animali che presentano segni di grave stress e/o dolore, in qualsiasi fase della prova, o lesioni compatibili con gli endpoint umanitari descritti nel presente metodo di prova (cfr. paragrafo 26) vanno soppressi con metodi non cruenti e la sostanza chimica va valutata di conseguenza. I criteri da seguire nel decidere la soppressione non cruenta di animali moribondi o che soffrono gravemente formano oggetto di un documento orientativo dell'OCSE (8). PREPARAZIONE DELLA PROVA IN VIVO Selezione delle specie L'animale da laboratorio di elezione è il coniglio albino, del quale vanno utilizzati giovani adulti sani. Giustificare l'eventuale uso di altri ceppi o specie. Preparazione degli animali Entro 24 ore dall'inizio della prova è necessario esaminare entrambi gli occhi di ciascun animale sperimentale provvisoriamente selezionato per la prova. Non vanno utilizzati animali che presentino irritazione oculare, difetti degli occhi o preesistenti lesioni corneali. Condizioni di stabulazione e alimentazione. Gli animali vanno posti in gabbie singole. La temperatura del locale sperimentale deve essere di 20°C (± 3°C) per i conigli. L'umidità relativa dovrebbe raggiungere almeno il 30 % e preferibilmente non superare il 70 % (tranne che nel corso delle pulizie degli ambienti), ma occorre puntare a un valore del 50-60 %. L'illuminazione deve essere artificiale, con una sequenza di 12 ore di luce e 12 d'oscurità. Va evitata un'eccessiva intensità luminosa. Per l'alimentazione, attenersi alle diete convenzionali da laboratorio con una quantità illimitata di acqua potabile. PROCEDURA Utilizzo di anestetici topici e analgesici sistemici Le seguenti procedure sono raccomandate per ridurre o evitare dolore e stress nelle prove di sicurezza sugli occhi. Si può ricorrere a procedure alternative più efficaci o efficienti per ridurre o evitare il dolore e lo stress degli animali.
Applicazione della sostanza chimica in esame Instillare la sostanza chimica in esame nel sacco congiuntivale di un occhio di ciascun animale, dopo aver allontanato delicatamente la palpebra inferiore dal bulbo. Le palpebre vanno poi tenute unite con delicatezza per circa un secondo, per evitare la fuoriuscita del materiale. L'altro occhio, che non viene trattato, serve da controllo. Irrigazione Gli occhi degli animali sperimentali non vanno lavati per almeno 24 ore dopo l'instillazione della sostanza chimica in esame, tranne nel caso di sostanze solide (cfr. paragrafo 18) e nel caso di effetti corrosivi o irritanti immediati. Dopo 24 ore è possibile effettuare un lavaggio, se lo si considera necessario. Non si raccomanda l'uso di un gruppo satellite di animali per studiare l'influenza del lavaggio oculare, a meno che ciò non risulti scientificamente giustificato. Qualora sia necessario un gruppo satellite, vanno usati due conigli. Le condizioni del lavaggio vanno documentate accuratamente: ad esempio, momento del lavaggio, composizione e temperatura della soluzione di lavaggio, durata, volume e velocità di applicazione. Livelli di dose (1) Saggio di liquidi Per testare le sostanze liquide si impiega una dose di 0,1 ml. Non usare spray per instillare la sostanza chimica direttamente nell'occhio; lo spray liquido va spruzzato e raccolto in un contenitore prima di instillarne 0,1 ml nell'occhio. (2) Saggio di solidi Per testare le sostanze solide, paste e sostanze particellari, la quantità impiegata deve avere un volume di 0,1 ml o un peso non superiore a 100 mg. La sostanza chimica in esame va ridotta in polvere fine. Prima della misurazione del volume, il materiale solido va delicatamente compattato, ad esempio picchiettando sul contenitore per la misurazione. Se la sostanza chimica in esame solida non è stata rimossa dall'occhio dell'animale da meccanismi fisiologici, alla prima osservazione che avviene un'ora dopo il trattamento si può sciacquare l'occhio con soluzione salina o acqua distillata. (3) Saggio di aerosol Si raccomanda di raccogliere tutti gli spray e gli aerosol prima dell'instillazione nell'occhio. L'unica eccezione riguarda le sostanze chimiche in contenitori pressurizzati per aerosol, che non possono essere raccolte a causa della vaporizzazione. In questi casi l'occhio va tenuto aperto e la sostanza chimica va somministrata nell'occhio con un unico spruzzo di circa un secondo, da una distanza di 10 cm, direttamente nella parte frontale dell'occhio. La distanza può variare a seconda della pressione dello spray e del suo contenuto. Occorre evitare di danneggiare l'occhio con la pressione dello spray. In alcuni casi può essere necessario valutare il potenziale di danno «meccanico» all'occhio dovuto alla forza dello spray. È possibile ottenere una stima della dose di un aerosol simulando la prova come segue: spruzzare la sostanza chimica attraverso un'apertura delle dimensioni dell'occhio di un coniglio posta esattamente di fronte ad un foglio di carta. L'aumento di peso della carta viene usato quindi per approssimare la quantità spruzzata nell'occhio. Per le sostanze chimiche volatili, la dose può essere stimata pesando un contenitore ricevente prima e dopo la rimozione della sostanza chimica in esame. Prova iniziale (prova di irritazione/corrosione oculare in vivo usando un solo animale) Si raccomanda caldamente di eseguire inizialmente la prova in vivo usando un solo animale (si veda l'Allegato del presente metodo: Strategia di sperimentazione sequenziale per l'irritazione e la corrosione oculare). Le osservazioni devono consentire di determinare la gravità e la reversibilità delle lesioni prima di eseguire una prova di conferma su un secondo animale. Se con la procedura descritta, i risultati di tale prova indicano che la sostanza chimica è corrosiva o gravemente irritante per l'occhio, non eseguire altre prove di irritazione oculare. Prova di conferma (prova di irritazione oculare in vivo con animali supplementari) Se nella prova iniziale non si osservano effetti corrosivi o fortemente irritanti, confermare la reazione irritante o negativa su un massimo di altri due animali. Se nella prova iniziale è stato osservato un effetto irritante si raccomanda di eseguire il saggio di conferma in maniera sequenziale su un solo animale per volta, anziché esporre contemporaneamente i due animali. Se il secondo animale rivela effetti corrosivi o gravemente irritanti, interrompere la prova. Se i risultati delle prove sul secondo animale sono sufficienti per determinare la categoria di pericolo della sostanza chimica, è necessario interrompere la sperimentazione. Periodo di osservazione La durata del periodo di osservazione deve essere sufficiente a valutare completamente l'entità e la reversibilità degli effetti osservati. Occorre tuttavia interrompere l'esperimento in qualsiasi momento se l'animale mostra segni di dolore o stress gravi (8). Per determinare la reversibilità degli effetti, gli animali vanno osservati di norma per 21 giorni successivamente alla somministrazione della sostanza chimica in esame. In caso di reversibilità prima dei 21 giorni, interrompere subito l'esperimento. Osservazioni cliniche e classificazione delle reazioni oculari Gli occhi sono sottoposti ad un esame approfondito per individuare eventuali lesioni oculari un'ora dopo l'applicazione della sostanza chimica in esame, e tale procedura va ripetuta almeno una volta al giorno. Nei primi tre giorni successivi gli animali vanno esaminati più volte al giorno per assicurare che si possa decidere di porre fine alla prova in tempo utile, se necessario. Gli animali sono sottoposti a esami di routine durante tutta la prova alla ricerca di segni clinici di dolore e/o di stress (ad esempio eccessivo scalpitare con le zampe, sfregamento ripetuto dell'occhio, eccessivo sbattere delle palpebre o eccessiva lacrimazione) (9) (10) (11), almeno due volte al giorno con un intervallo minimo di sei, o più frequentemente se necessario. Tali esami sono necessari per i) valutare correttamente i segni di dolore e stress degli animali al fine di dimostrare la necessità di aumentare il dosaggio di analgesici e ii) «valutare» gli animali per riconoscere segni conclamati di endpoint umanitari, in modo da decidere con cognizione di causa la soppressione incruenta degli animali e garantire che tali decisioni siano adottate in tempo utile. Per agevolare l'individuazione e la misurazione delle lesioni oculari e determinare se i criteri stabiliti ai fini della soppressione incruenta sono stati raggiunti, va utilizzata di routine una colorazione con fluoresceina nonché un microscopio con lampada a fessura se ritenuto opportuno (ad esempio per valutare la gravità della lesione in caso di ulcerazione della cornea). Fotografie digitali delle lesioni riscontrate possono essere raccolte come riferimento e per documentare in modo permanente l'estensione della lesione oculare. Gli animali devono essere sottoposti a sperimentazione per il tempo minimo necessario per ottenere informazioni definitive. Gli animali che presentano dolore o stress intensi vanno soppressi al più presto con metodi non cruenti e la sostanza chimica va valutata di conseguenza. Sopprimere con metodi non cruenti gli animali che, dopo l'instillazione, presentano le seguenti lesioni oculari (si veda la tabella 1 per una descrizione dei gradi di lesione): perforazione corneale o ulcerazione corneale di rilievo, compreso stafiloma; sangue nella camera anteriore dell'occhio; opacità corneale di grado 4; assenza di riflesso pupillare alla luce (risposta dell'iride di grado 2) che persista per 72 ore; ulcerazione della membrana congiuntivale; necrosi della congiuntiva o della membrana nittitante; distacco epidermico. Tali lesioni sono infatti generalmente irreversibili. È inoltre opportuno considerare le seguenti lesioni oculari come endpoint umanitari, la cui insorgenza giustifica di porre termine alla prova prima che sia trascorso il periodo di osservazione di 21 giorni previsto. Si ritiene che tali lesioni prefigurino altre reazioni a una sostanza corrosiva o gravemente irritante e altre lesioni che probabilmente non saranno pienamente scomparse entro il periodo di osservazione di 21 giorni: lesioni profonde — come ad esempio un'ulcerazione della cornea oltre gli strati superficiali dello stroma), distruzione del lembo > 50 % (comprovato dallo scoloramento del tessuto congiuntivale) e un'infezione oculare acuta (secrezione purulenta). Una combinazione di: vascolarizzazione della superficie corneale (cioè, panno corneale) associata a una superficie macchiata di fluoresceina che non diminuisce nel tempo, sulla base di esami giornalieri; e/o all'assenza di ricrescita epiteliale 5 giorni dopo l'applicazione della sostanza chimica in esame, può anche costituire un insieme di criteri pertinenti per giustificare la decisione di porre termine alla prova anticipatamente. Tuttavia, ciascuno di questi risultati preso isolatamente non è sufficiente a motivare la conclusione anticipata della sperimentazione. Qualora siano osservati effetti oculari gravi, occorre consultare un veterinario curante o specializzato in animali da laboratorio, o a personale debitamente formato a individuare le lesioni cliniche, per condurre un esame clinico che consenta di stabilire se la combinazione di tali reazioni implica una conclusione anticipata della sperimentazione. Si determinano i gradi della reazione oculare (congiuntiva, cornea e iride) a 1, 24, 48 e 72 ore dopo l'applicazione della sostanza chimica in esame e se ne registrano i risultati (tabella 1). Gli animali che non sviluppano lesioni oculari possono essere soppressi non prima di 3 giorni dopo l'instillazione. Gli animali con lesioni oculari non gravi vanno tenuti sotto osservazione fino alla scomparsa delle lesioni, oppure per 21 giorni, momento in cui lo studio si conclude. Occorre effettuare le osservazioni e le registrazioni con intervalli minimi di 1 ora, 24 ore, 48 ore, 72 ore, 7 giorni, 14 giorni e 21 giorni, con l'obiettivo di determinare lo stato delle lesioni e la loro reversibilità o irreversibilità. Talvolta è necessario eseguire le osservazioni con frequenza maggiore per stabilire se l'animale sperimentale debba essere sottoposto a morte non cruenta per considerazioni etiche o eliminata dalla prova a motivo di risultati negativi. Per ogni esame si deve registrare il livello di lesione oculare (vedi tabella 1). Annotare anche qualsiasi altra lesione dell'occhio (ad es. panno corneale, macchie, alterazioni della camera anteriore) e qualsiasi effetto sistemico negativo. L'esame delle reazioni può essere facilitato usando una lente binoculare, una lampada manuale a fessura, un biomicroscopio o altro dispositivo idoneo. Dopo aver registrato le osservazioni a 24 ore, è possibile esaminare ulteriormente gli occhi con l'ausilio di fluoresceina. La classificazione delle reazioni oculari è necessariamente soggettiva. Per favorirne l'armonizzazione e per assistere i laboratori e le persone che eseguono e interpretano le osservazioni, è necessario istruire adeguatamente il personale responsabile sul sistema di punteggio utilizzato. DATI E RELAZIONE Valutazione dei risultati Valutare i punteggi dell'irritazione oculare insieme alla natura e alla gravità delle lesioni, nonché alla loro reversibilità o irreversibilità. I punteggi individuali non rappresentano uno standard assoluto per le proprietà irritanti di una sostanza chimica, in quanto si valutano anche altri effetti della sostanza chimica in esame. I punteggi individuali vanno invece considerati come valori di riferimento e hanno significato solo se corredati di una descrizione e una valutazione complete di tutte le osservazioni. Relazione sulla prova La relazione sulla prova comprende le informazioni seguenti.
Interpretazione dei risultati L'estrapolazione dei risultati degli studi sull'irritazione oculare negli animali da laboratorio agli esseri umani ha un valore solo limitato. In molti casi, il coniglio albino è più sensibile dell'uomo alle sostanze irritanti o corrosive per l'occhio. È necessario interpretare con attenzione i dati per escludere l'irritazione dovuta a infezione secondaria. BIBLIOGRAFIA
Tabella 1 Classificazione delle lesioni oculari
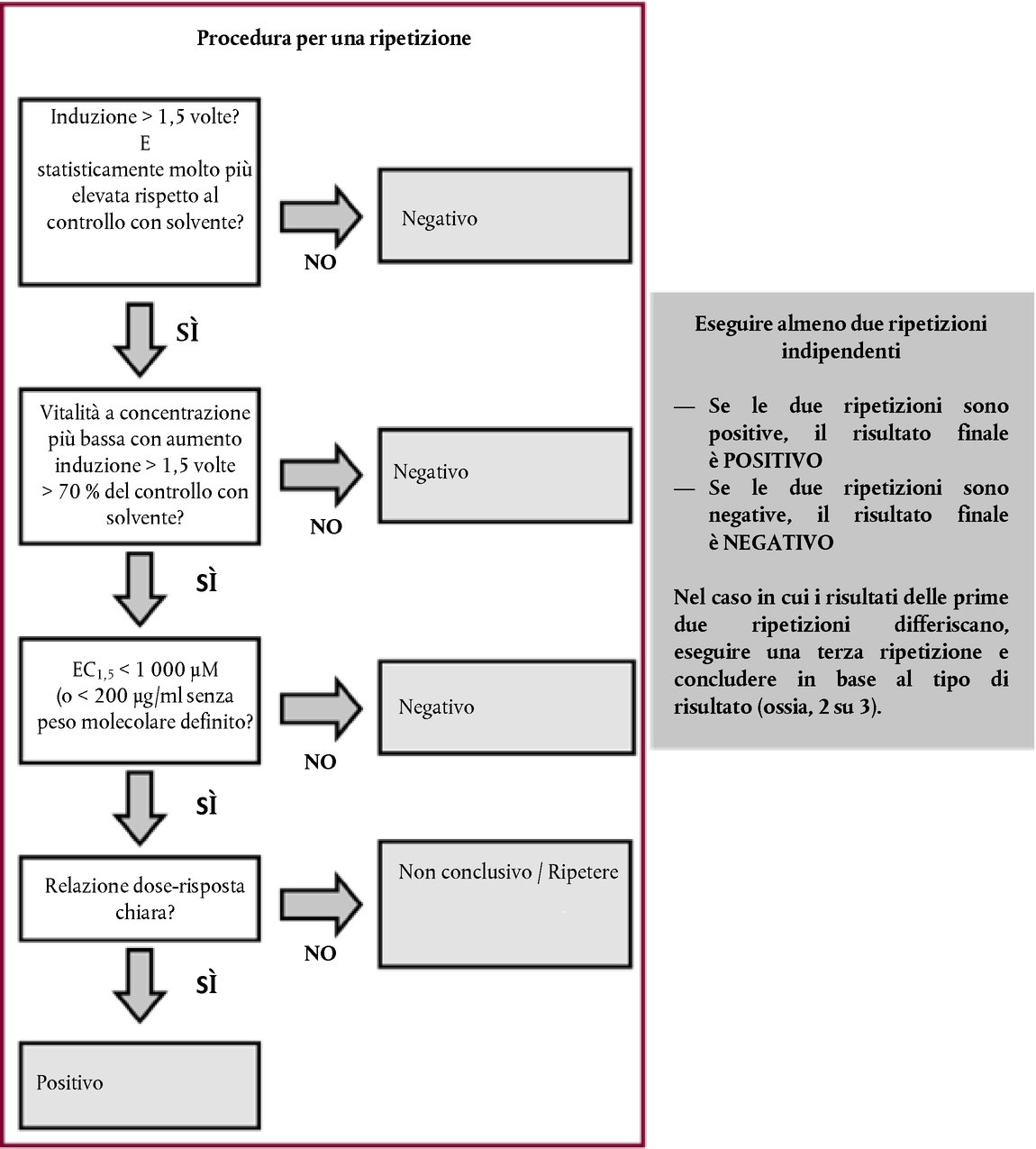
Appendice DEFINIZIONI Rapporto acido/alcalino : per i preparati acidi, si tratta della quantità (g) di idrossido di sodio/100 g di preparazione necessaria per ottenere un determinato pH. Per i preparati alcalini, si tratta della quantità (g) di idrossido di sodio che equivale alla quantità (g) di acido solforico/100 g di preparazione necessaria per ottenere uno specifico pH (Young et al. 1988). Sostanza chimica : una sostanza o una miscela. Non irritanti : sostanze non classificate come irritanti oculari di categoria I, II o III dell'EPA; o irritanti oculari di categoria 1, 2, 2A o 2B nel sistema GHS; o di categoria 1 o 2 dell'UE (17) (18) (19). Corrosivo oculare : a) sostanza chimica che causa lesioni irreversibili dei tessuti oculari; b) sostanza chimica classificata come irritante oculare di categoria 1 nel sistema GHS, categoria I dell'EPA, o categoria 1 dell'UE (17) (18) (19). Irritante oculare : a) sostanza chimica che produce un'alterazione oculare reversibile; b) sostanza classificata come irritante oculare di categoria II o III dell'EPA, di categoria 2, 2A o 2B nel sistema GHS, o di categoria 1 o 2 dell'UE (17) (18) (19). Grave irritante oculare : a) sostanza chimica che causa danni ai tessuti oculari non risolvibili entro 21 giorni dall'applicazione o indebolimento grave della vista; b) sostanze chimiche classificate come irritanti oculari di categoria 1 nel sistema GHS, di categoria I dell'EPA o di categoria 1 dell'UE (17) (18) (19). Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Approccio per gradi : strategia sperimentale realizzata per stadi successivi nella quale sono riesaminate tutte le informazioni disponibili su una sostanza chimica in esame, secondo un ordine preciso, applicando un processo basato sul peso delle evidenze a ciascuno stadio, al fine di stabilire se vi sono informazioni sufficienti per prendere una decisione sulla classificazione del pericolo prima di procedere allo stadio successivo. Se è possibile assegnare un potenziale di irritazione alla sostanza chimica in esame in base alle informazioni disponibili, non è necessario procedere a una prova supplementare. Se non è possibile assegnare un potenziale di irritazione alla sostanza chimica in esame in base alle informazioni disponibili, eseguire una sperimentazione sequenziale su animali fino a che non sarà possibile giungere a una classificazione inequivocabile. Peso dell'evidenza (processo) : i punti forti e deboli di un insieme di informazioni sono utilizzati come base per giungere ad una conclusione che potrebbe non essere evidente a partire dai dati presi individualmente. ALLEGATO DEL METODO DI PROVA B.5 (4) STRATEGIA DI PROVE IN SEQUENZA PER L'IRRITAZIONE E LA CORROSIONE OCULARI Considerazioni generali Nell'interesse dell'accuratezza scientifica e del benessere degli animali, è importante evitare l'uso non necessario di animali e ridurre al minimo le prove atte a provocare reazioni gravi. Valutare tutte le informazioni relative alla potenziale irritazione/corrosività oculare di una sostanza chimica prima di prendere in considerazione le prove in vivo. È possibile che esistano già prove sufficienti per classificare il potenziale di irritazione o corrosione oculare di una sostanza chimica in esame senza bisogno di effettuare prove su animali da laboratorio. L'analisi basata sul peso delle evidenze e l'uso di una strategia di sperimentazione sequenziale ridurranno al minimo la necessità di eseguire prove in vivo, soprattutto se è probabile che la sostanza chimica provochi reazioni gravi. Si raccomanda di svolgere un'analisi basata sul peso delle evidenze per valutare le informazioni esistenti sul potenziale di irritazione e corrosione oculare delle sostanze chimiche e determinare la necessità di altri studi, diversi da quelli in vivo sugli occhi, per meglio caratterizzare tale potenziale. Qualora tali studi fossero necessari, si raccomanda di applicare la strategia di sperimentazione sequenziale per ottenere i dati sperimentali pertinenti. Per le sostanze per le quali non è disponibile una documentazione sperimentale, usare la strategia di sperimentazione sequenziale per ottenere i dati necessari al fine di valutarne la corrosività/irritazione oculari. La strategia sperimentale iniziale descritta nel presente Allegato è stata sviluppata nel corso di un workshop dell'OCSE (1) e successivamente confermata ed ampliata nello Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances (Sistema di classificazione armonizzato integrato dei rischi per la salute umana e gli effetti ambientali delle sostanze chimiche), approvata alla 28a riunione congiunta del comitato sulle sostanze chimiche e dal gruppo di lavoro sulle sostanze chimiche nel novembre 1998 (2) e aggiornata da un gruppo di esperti dell'OCSE nel 2011. Sebbene non sia parte integrante del metodo di prova B.5, questa strategia sperimentale esprime tuttavia l'approccio raccomandato per la determinazione delle proprietà di irritazione/corrosione oculari. Tale approccio rappresenta non solo una “migliore pratica” ma anche un punto di riferimento etico per l'esecuzione di prove in vivo sull'irritazione/corrosione oculari. Il metodo di prova fornisce indicazioni sull'esecuzione della prova in vivo e riassume i fattori da valutare prima di prendere in considerazione il ricorso a tale prova. La strategia di sperimentazione sequenziale fornisce un metodo di analisi basata sul peso delle evidenze per la valutazione dei dati esistenti sulle proprietà di irritazione/corrosione oculare delle sostanze chimiche e un approccio graduale per lo sviluppo di dati pertinenti sulle sostanze chimiche che necessitano ulteriori studi o che non sono mai state oggetto di studio. La strategia prevede dapprima l'esecuzione di prove validate ed accettate in vitro o ex vivo e successivamente di studi in base al metodo di prova B.4 in circostanze specifiche (3) (4). Descrizione della strategia sperimentale «per gradi» Prima di effettuare le prove nell'ambito della strategia di sperimentazione sequenziale (Figura), valutare tutte le informazioni disponibili, per determinare l'effettiva necessità di eseguire prove oculari in vivo. Sebbene sia possibile trarre significative informazioni dalla valutazione di singoli parametri (ad es. pH estremo), è necessario valutare la totalità delle informazioni esistenti. Nel prendere una decisione basata sul peso delle evidenze, valutare tutti i dati pertinenti sugli effetti della sostanza chimica in questione e dei suoi analoghi strutturali; siffatta decisione deve essere giustificata. Si dovrebbe attribuire innanzitutto importanza ai dati esistenti sulla sperimentazione della sostanza chimica su persone e animali, e successivamente ai risultati di prove in vitro o ex vivo. Ove possibile, evitare gli studi in vivo delle sostanze chimiche corrosive. I fattori considerati nella strategia sperimentale sono:
STRATEGIA SPERIMENTALE E VALUTAZIONE DELL'IRRITAZIONE/CORROSIONE OCULARE
BIBLIOGRAFIA
|
|
(3) |
Nella parte B, il capitolo B.10 è sostituito dal seguente: «B.10 Prova in vitro di aberrazione cromosomica nei mammiferi INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 473 (2016) e fa parte di una serie di metodi di prova sulla tossicologia genetica. È stato elaborato un documento OCSE contenente informazioni succinte sulle prove di tossicologia genetica e un compendio delle modifiche recentemente apportate alla rispettiva linea guida (1). La prova in vitro di aberrazione cromosomica è destinata ad identificare le sostanze chimiche che causano aberrazioni cromosomiche strutturali in una coltura di cellule di mammifero (2) (3) (4). Le aberrazioni strutturali possono essere di due tipi: cromosomiche o cromatidiche. Nei test di aberrazione cromosomica in vitro può verificarsi poliploidia (compresa l'endoriduplicazione). Se è vero che gli aneugeni possono provocare poliploidia, quest'ultima di per sé non indica un potenziale aneugenico e può semplicemente rivelare una perturbazione del ciclo cellulare o citotossicità (5). Questa prova non è destinata a misurare l'aneuploidia. Per il rilevamento dell'aneuploidia si raccomanda un test del micronucleo in vitro (6). Nella prova in vitro di aberrazione cromosomica si possono usare colture di linee cellulari stabilizzate o colture cellulari primarie di origine umana o di roditori. Le cellule devono essere scelte in funzione della capacità di crescita in coltura, della stabilità del cariotipo (compreso il numero dei cromosomi) e della frequenza delle aberrazioni cromosomiche spontanee (7). Attualmente i dati disponibili non consentono di elaborare raccomandazioni solide ma rivelano l'importanza di considerare, al momento di valutare i rischi chimici: lo stato della p53, la stabilità genetica (del cariotipo), la capacità di riparazione del DNA e l'origine (da roditori piuttosto che umane) delle cellule scelte per la sperimentazione. Gli utilizzatori del presente metodo di prova sono pertanto incoraggiati a valutare l'influenza di queste ed altre caratteristiche cellulari sul comportamento di una linea cellulare nel rilevare l'induzione di aberrazioni cromosomiche, in funzione dell'evoluzione delle conoscenze in questo campo. Le definizioni utilizzate figurano nell'appendice 1. CONSIDERAZIONI INIZIALI E LIMITI Le prove in vitro richiedono in generale l'uso di una fonte esogena di attivazione metabolica, a meno che le cellule non siano metabolicamente compatibili con le sostanze chimiche in esame. Il sistema esogeno di attivazione metabolica non simula perfettamente le condizioni in vivo. Occorre adoperarsi per evitare condizioni che potrebbero portare a falsi risultati positivi, vale a dire danni cromosomici non causati da un'interazione diretta tra le sostanze chimiche in esame e i cromosomi; tali condizioni comprendono le variazioni di pH o di osmolalità (8) (9) (10), l'interazione con gli ingredienti del mezzo (11) (12) o livelli eccessivi di citotossicità (13) (14) (15) (16). La prova è utilizzata per individuare aberrazioni cromosomiche che possono derivare da eventi clastogenici. L'analisi dell'induzione di aberrazioni cromosomiche deve avvenire utilizzando cellule in metafase. È quindi essenziale che le cellule raggiungano la mitosi sia nelle colture trattate sia in quelle non trattate. Possono essere necessari adattamenti specifici di questo metodo di prova, non descritti in questa sede, per i nanomateriali di sintesi. Prima dell'uso del metodo di prova su una miscela, per la generazione di dati per un determinato scopo normativo, occorre esaminare se, e in caso affermativo perché, può fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie in presenza di un obbligo normativo di prova sulla miscela. PRINCIPIO DELLA PROVA Le colture di cellule umane o di altro mammifero sono esposte alla sostanza chimica in esame con e senza una fonte esogena di attivazione metabolica, a meno di utilizzare cellule con un'adeguata capacità metabolizzante (cfr. il paragrafo 13). Dopo l'inizio dell'esposizione alla sostanza chimica in esame, le colture cellulari sono trattate, a intervalli opportunamente predefiniti, con un inibitore della metafase (per esempio Colcemid o colchicina), raccolte e sottoposte a un processo di colorazione; le cellule in metafase sono esaminate al microscopio per determinare la presenza di aberrazioni di tipo cromatidico e cromosomiche. DESCRIZIONE DEL METODO Preparazioni Cellule Si possono utilizzare varie linee cellulari (ad esempio cellule di ovario di criceto cinese (CHO), di polmone di criceto cinese V79, di polmone di criceto cinese (CHL)/IU, TK 6) o colture cellulari primarie, fra cui linfociti del sangue periferico umano o di altri mammiferi (7). La scelta delle linee cellulari deve essere scientificamente motivata. Nel caso di impiego di cellule primarie, per motivi attinenti al benessere degli animali occorre prendere in considerazione l'uso di cellule di origine umana ove possibile, prelevate in conformità dei principi e delle norme etiche pertinenti. I linfociti del sangue periferico umano devono essere ottenuti da soggetti giovani (circa 18-35 anni di età), non fumatori, non affetti da malattie note e non esposti recentemente ad agenti genotossici (ad esempio sostanze chimiche o radiazioni ionizzanti) a livelli tali da aumentare l'incidenza di base di aberrazioni cromosomiche. In tal modo si garantisce che l'incidenza di base di aberrazioni cromosomiche rimanga lieve e omogenea. L'incidenza di base di aberrazioni cromosomiche aumenta con l'età e questa tendenza è più marcata nelle donne che negli uomini (17) (18). Se si raccolgono cellule da più donatori, il numero dei donatori dev'essere indicato. È necessario dimostrare che le cellule si sono divise fra l'inizio del trattamento con la sostanza chimica in esame e il prelievo. Le colture cellulari vengono mantenute in una fase di crescita esponenziale (linee cellulari) o stimolate a dividersi (colture primarie di linfociti), per esporre le cellule in diversi stadi del ciclo cellulare, essendo potenzialmente ignota la sensibilità dei diversi stadi delle cellule alle sostanze chimiche in esame. In generale, le cellule primarie che per dividersi devono essere stimolate con agenti mitogenici non sono più sincronizzate durante l'esposizione alla sostanza chimica in esame (ad esempio linfociti umani dopo 48 ore dalla stimolazione mitogenica). L'uso di cellule sincronizzate durante il trattamento non è raccomandato, ma può essere ammissibile se giustificato. Mezzo e condizioni di coltura Le colture vanno mantenute in mezzi di coltura e condizioni di incubazione (recipienti di coltura, atmosfera umidificata al 5 % di CO2 se del caso, temperatura di incubazione di 37 °C) adeguati. Occorre controllare periodicamente la stabilità del numero modale dei cromosomi e l'assenza di contaminazione da micoplasma nelle linee cellulari (7) (19); non si devono usare cellule contaminate o che presentano modifiche del numero modale dei cromosomi. Occorre stabilire la durata normale del ciclo cellulare delle linee cellulari o delle colture primarie utilizzate nel laboratorio di prova, che deve essere coerente con le caratteristiche cellulari pubblicate (20). Preparazione delle colture Linee cellulari: le cellule provenienti da colture primarie vengono inoculate in un terreno di coltura ad una densità tale da consentire alle cellule in sospensione o in monostrati di continuare a crescere in maniera esponenziale fino al momento del prelievo (occorre, ad esempio, evitare che le cellule in crescita in monostrati raggiungano la confluenza). Linfociti: sangue intero trattato con un anticoagulante (ad esempio eparina) o linfociti isolati sono posti in un terreno di coltura (ad esempio per 48 ore nel caso di linfociti umani) contenente un mitogeno [ad esempio fitoemoagglutinina (PHA) nel caso di linfociti umani] per ridurre la divisione cellulare prima dell'esposizione alla sostanza chimica in esame. Attivazione metabolica Se si utilizzano cellule prive di un'adeguata capacità di attivazione metabolica endogena si deve ricorrere a sistemi di attivazione metabolica esogeni. Il sistema più comunemente usato, raccomandato in tutti i casi salvo alternativa motivata, è una frazione post-mitocondriale integrata di cofattore (S9), prelevata dal fegato di roditori (solitamente ratti) trattati con induttori enzimatici come Aroclor 1254 (21) (22) (23) o con una combinazione di fenobarbitone e β-naftoflavone (24) (25) (26) (27) (28) (29). Quest'ultima combinazione è conforme alla convenzione di Stoccolma sugli inquinanti organici persistenti (30) e ha dimostrato di essere tanto efficace quanto l'Aroclor 1254 nell'indurre ossidasi a funzione mista (24) (25) (26) (28). Solitamente la frazione S9 viene usata a concentrazioni comprese tra 1 % e 2 % (v/v), ma può essere aumentata al 10 % (v/v) nel terreno di coltura finale. Occorre evitare, durante il trattamento, l'impiego di prodotti che riducono il coefficiente mitotico, soprattutto prodotti di complessazione del calcio (31). La scelta del tipo e della concentrazione del sistema di attivazione metabolica esogeno o dell'induttore metabolico utilizzato può essere influenzata dalla classe delle sostanze chimiche in esame. Preparazione della sostanza chimica in esame Le sostanze chimiche in esame solide devono essere preparate in adeguati solventi e, se necessario, diluite prima del trattamento delle cellule (cfr. il paragrafo 23). Le sostanze chimiche in esame liquide possono essere aggiunte direttamente alla coltura o diluite prima del trattamento della coltura stessa. Le sostanze chimiche in esame gassose o volatili devono essere sottoposte alla prova modificando adeguatamente i protocolli standard (trattamento in recipienti di coltura ermetici) (33) (34). Occorre preparare la sostanza chimica in esame subito prima del trattamento, salvo se i dati sulla stabilità dimostrano che la conservazione è un'alternativa accettabile. Condizioni sperimentali Solventi La scelta del solvente deve favorire l'ottimizzazione della solubilità delle sostanze chimiche in esame, senza nuocere alla conduzione del saggio (ad esempio influenzando la crescita cellulare), compromettere l'integrità della sostanza chimica in esame, reagire con recipienti di coltura o pregiudicare il sistema di attivazione metabolica. Si raccomanda di prendere in considerazione in primo luogo, se possibile, l'uso di un solvente (o mezzo di coltura) acquoso. Solventi di uso consolidato sono, ad esempio, l'acqua o il dimetilsolfossido. In generale è opportuno che i solventi organici non superino l'1 % (v/v) e quelli acquosi (soluzione fisiologica o acqua) il 10 % (v/v) nel terreno di coltura finale. Qualora si utilizzino solventi di uso non consolidato (ad esempio etanolo o acetone), il loro uso dovrebbe essere suffragato da dati che ne comprovino la compatibilità con le sostanze chimiche in esame e con il sistema di prova e l'assenza di tossicità genetica alla concentrazione usata. In assenza di tali dati, è importante includere controlli non trattati (cfr. l'appendice 1) per dimostrare che il solvente scelto non induce effetti nocivi o clastogenici. Misurazione della proliferazione cellulare e della citotossicità e scelta delle concentrazioni di trattamento Nel determinare la concentrazione più elevata della sostanza chimica in esame, occorre evitare le concentrazioni che hanno la capacità di produrre falsi risultati positivi, come quelle che causano eccessiva citotossicità (cfr. il paragrafo 22), precipitazione nel terreno di coltura (cfr. il paragrafo 23) o variazioni marcate del pH o osmolalità (cfr. il paragrafo 5). Se la sostanza chimica in esame provoca una variazione marcata del pH del mezzo al momento dell'aggiunta, il pH può essere adeguato tamponando il terreno di coltura finale in modo da evitare falsi risultati positivi e mantenere adeguate condizioni di coltura. Occorre misurare la proliferazione cellulare per garantire che un numero sufficiente di cellule trattate abbia raggiunto la mitosi durante la prova e che i trattamenti siano condotti ad adeguati livelli di citotossicità (cfr. i paragrafi 18 e 22). La citotossicità deve essere determinata con e senza attivazione metabolica nel test principale, sulla base di un indicatore adeguato della morte e della crescita cellulare. Mentre la valutazione della citotossicità in un saggio iniziale può essere utile per definire meglio le concentrazioni da utilizzare per il test principale, l'effettuazione di un saggio iniziale non è obbligatoria. Se viene eseguito, non deve sostituire la misurazione della citotossicità nel test principale. Il raddoppiamento relativo della popolazione (RPD) o l'aumento relativo delle conte cellulari (RICC) sono metodi adeguati per la valutazione della citotossicità nella prova citogenetica (13) (15) (35) (36) (55) (cfr. l'appendice 2 per le formule). In caso di trattamento a lungo termine e fasi di campionamento dopo l'inizio del trattamento superiori a 1,5 volte la durata normale del ciclo cellulare (ossia oltre 3 cicli cellulari in totale), l'RPD potrebbe sottostimare la citotossicità (37). In tali circostanze l'RICC potrebbe essere una misura migliore, ma una stima utile si potrebbe ottenere anche valutando la citotossicità mediante l'RPD dopo 1,5 cicli cellulari normali. Per i linfociti in colture primarie, il coefficiente mitotico (MI) è una misura degli effetti citotossici/citostatici, ma è anche influenzato dal tempo trascorso fra il trattamento e la misurazione, dal mitogeno usato e dalla possibile interruzione del ciclo cellulare. Tuttavia, il coefficiente mitotico è accettabile perché altre misurazioni della citotossicità potrebbero essere onerose e di difficile esecuzione e potrebbero non risultare adeguate alla popolazione interessata di linfociti in crescita in risposta alla stimolazione con PHA. Mentre RICC e RPD per le linee cellulari e MI per la coltura primaria di linfociti sono i parametri raccomandati di citotossicità, altri indicatori (ad esempio l'integrità delle cellule, l'apoptosi, la necrosi, il ciclo cellulare) potrebbero fornire utili informazioni aggiuntive. Occorre valutare almeno tre concentrazioni di prova (non compreso il solvente e i controlli positivi) che soddisfano i criteri di accettabilità (adeguata citotossicità, numero di cellule, ecc.). A prescindere dal tipo di cellula (linee cellulari o colture primarie di linfociti), ciascuna coltura realizzata singolarmente o in più repliche può essere utilizzata per ciascuna concentrazione di prova. È consigliato l'uso di colture in duplicato, ma colture singole sono accettabili a condizione di analizzare lo stesso numero totale di cellule sia nel caso di coltura singola sia nel caso di colture in duplicato. L'uso di colture singole è particolarmente indicato quando si valutano più di 3 concentrazioni (cfr. il paragrafo 31). I risultati ottenuti con colture replicate indipendenti con una determinata concentrazione possono essere aggregati per l'analisi dei dati (38). Per le sostanze chimiche di prova che dimostrino tossicità assente o debole, sono solitamente indicati intervalli di concentrazione di un fattore da 2 a 3. In caso di citotossicità, le concentrazioni selezionate per la prova devono coprire una gamma comprendente quella che ha prodotto la citotossicità di cui al paragrafo 22 e concentrazioni alle quali la citotossicità è debole o assente. Molte sostanze chimiche in esame presentano curve di concentrazione-risposta accentuate e al fine di ottenere dati a bassa o debole citotossicità o di studiare la relazione dose-risposta nel dettaglio, sarà necessario ricorrere a concentrazioni separate fra loro da intervalli minori e/o a più di tre concentrazioni (colture singole o replicate), in particolare nelle situazioni in cui è necessario ripetere l'esperimento (cfr. il paragrafo 47). Se la concentrazione massima è basata sulla citotossicità, la concentrazione più elevata deve puntare a raggiungere il 55 ± 5 % di citotossicità sulla base dei parametri di citotossicità raccomandati (ossia la riduzione di RICC e RPD per le linee cellulari e la riduzione di MI per le colture primarie di linfociti al 45 ± 5 % nel controllo negativo parallelo). Occorre interpretare con cautela risultati positivi ottenuti nel solo segmento superiore di tale intervallo di citotossicità al 55 ± 5 % (13). Per le sostanze chimiche scarsamente solubili non citotossiche a concentrazioni inferiori alla concentrazione minima insolubile, la più elevata concentrazione analizzata dovrebbe produrre torbidità o la formazione di un precipitato visibile a occhio nudo o con l'aiuto di un microscopio invertito, alla fine del trattamento con la sostanza chimica di prova. Anche se la citotossicità si verifica a concentrazioni superiori a quella minima insolubile, è indicato effettuare la prova a una sola concentrazione che produce torbidità o un precipitato visibile, perché il precipitato può falsare gli effetti. Alla concentrazione che produce un precipitato, occorre adoperarsi per garantire che il precipitato non interferisca nello svolgimento della prova (ad esempio mediante colorazioni o abrasioni). Può essere utile determinare la solubilità nel terreno di coltura prima del test. Se non si osserva nessun precipitato o nessuna citotossicità limitante, la concentrazione massima di prova dovrebbe essere pari al valore più basso fra 10 mM, 2 mg/ml o 2 μl/ml (39) (40) (41). Se la sostanza chimica in esame non ha una composizione definita — quale ad esempio una sostanza di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici (UVCB) (42) o estratti dall'ambiente, ecc. — la concentrazione massima potrebbe dover essere più elevata (ad esempio 5 mg/ml), in mancanza di sufficiente citotossicità, per aumentare la concentrazione di ciascuna componente. Va tuttavia rilevato che tali requisiti possono essere diversi per i prodotti farmaceutici per uso umano (43). Controlli Occorre anche effettuare controlli negativi paralleli (cfr. il paragrafo 15), con il solo solvente sul terreno di coltura, trattato allo stesso modo delle colture di trattamento, per ogni fase di raccolta. Controlli positivi paralleli sono necessari per dimostrare la capacità del laboratorio di individuare clastogeni alle condizioni del protocollo di prova utilizzato e l'efficacia del sistema di attivazione metabolica esogeno, se del caso. Esempi di controlli positivi sono indicati nella tabella 1 in appresso. Si possono utilizzare sostanze chimiche alternative di controllo, se giustificate. Poiché test in vitro su cellule di mammiferi per tossicità genetica sono sufficientemente standardizzati, l'uso dei controlli positivi può limitarsi a un clastogeno che richiede attivazione metabolica. A condizione che si svolga contemporaneamente alla prova senza attivazione con la stessa durata di trattamento, quest'unico risultato di controllo positivo dimostrerà sia l'attività del sistema di attivazione metabolica sia la capacità di risposta del sistema di prova. Tuttavia, il trattamento a lungo termine (senza S9) richiede l'effettuazione di un proprio controllo positivo, in quanto la durata del trattamento differisce da quella della prova con attivazione metabolica. Ciascun controllo positivo deve essere utilizzato a una o più concentrazioni da cui ci si attende un aumento riproducibile e rilevabile rispetto ai valori di fondo, per dimostrare la sensibilità del sistema di prova (ossia effetti chiari che tuttavia non rivelino immediatamente allo sperimentatore l'identità dei vetrini codificati), e la risposta non deve essere compromessa da una citotossicità superiore ai limiti specificati nel metodo di prova. Tabella 1. Sostanze chimiche di riferimento raccomandate per la valutazione della competenza dei laboratori e per la selezione dei controlli positivi.
SVOLGIMENTO DEL METODO Trattamento con la sostanza chimica in esame Le cellule in proliferazione sono trattate con la sostanza chimica in esame in presenza e in assenza di un sistema di attivazione metabolica. Raccolta delle colture Ai fini di una valutazione rigorosa, necessaria per concludere un esito negativo, occorre rispettare tutte e tre le seguenti condizioni sperimentali utilizzando un trattamento di breve durata con e senza attivazione metabolica e un trattamento di lunga durata senza attivazione metabolica (cfr. i paragrafi 43, 44 e 45):
Nel caso in cui una qualsiasi delle suddette condizioni sperimentali porti ad un risultato positivo, può non essere necessario esaminare gli altri regimi di trattamento. Preparazione dei cromosomi Le colture cellulari sono trattate con Colcemid o colchicina, di norma per un periodo variabile da una a tre ore prima della raccolta. Ogni coltura cellulare viene raccolta e trattata separatamente per la preparazione dei cromosomi. La preparazione dei cromosomi comprende il trattamento ipotonico delle cellule, il fissaggio e la colorazione. Nei monostrati possono essere presenti cellule mitotiche (identificabili perché di forma tonda e in fase di distacco dalla superficie) al termine del trattamento di 3-6 ore. Poiché tali cellule mitotiche si staccano facilmente, potrebbero andare perdute al momento della rimozione del terreno di coltura contenente la sostanza chimica in esame. Se si rileva un aumento sostanziale del numero di cellule mitotiche rispetto ai controlli, tale da indicare il probabile arresto mitotico, occorre allora prelevare le cellule mediante centrifugazione e aggiungerle nuovamente alle colture per evitare di perdere cellule in mitosi, e quindi a rischio di aberrazione cromosomica, al momento della raccolta. Analisi Tutti i vetrini, compresi quelli dei controlli positivi e negativi, devono essere codificati indipendentemente prima dell'esame al microscopio per l'aberrazione cromosomica. Poiché le procedure di fissaggio provocano spesso la perdita di cromosomi in una parte delle cellule in metafase, le cellule classificate devono contenere un numero di centromeri pari al numero modale ± 2. Occorre classificare almeno 300 metafasi ben spaziate per ogni concentrazione e per ogni controllo, per rilevare un risultato chiaramente negativo per la sostanza chimica in esame (cfr. il paragrafo 45). Se si usano colture replicate, le 300 cellule devono essere divise equamente tra le repliche. Se si usano colture singole per ogni concentrazione (cfr. il paragrafo 21), occorre classificare almeno 300 metafasi ben spaziate in ogni coltura singola. L'analisi di 300 cellule comporta il vantaggio di aumentare la potenza statistica della prova; inoltre valori pari a zero saranno rari (stimati dell'ordine del 5 %) (44). È possibile ridurre il numero di metafasi classificate se si osserva un numero elevato di cellule con aberrazioni cromosomiche e la sostanza chimica in esame è considerata chiaramente positiva. Le cellule che presentano una o più aberrazioni cromosomiche strutturali con e senza gap devono essere classificate. Le rotture e i gap sono definiti nell'appendice 1 conformemente a (45) (46). Occorre registrare separatamente le aberrazioni cromatidiche e quelle cromosomiche e classificarle in sottotipi (rotture, scambi). Le procedure in uso presso il laboratorio devono assicurare che l'analisi delle aberrazioni cromosomiche sia effettuata da analisti ben preparati e sottoposta valutazione inter pares, se del caso. Sebbene la prova sia destinata a rivelare aberrazioni cromosomiche strutturali, è importante registrare le frequenze di eventuali casi di poliploidia e di endoriduplicazione (cfr. il paragrafo 2). Competenza del laboratorio Al fine di verificare sufficiente esperienza con la prova prima del suo impiego corrente, il laboratorio deve avere eseguito una serie di esperimenti con sostanze chimiche positive di riferimento che agiscono attraverso meccanismi diversi e vari controlli negativi (con diversi solventi/veicoli). Tali risposte dei controlli, positive e negative, devono essere coerenti con la letteratura scientifica. Ciò non si applica ai laboratori che hanno già maturato un'esperienza, vale a dire che dispongono di una banca di dati storici quale definita al paragrafo 37. Occorre analizzare una selezione di sostanze chimiche di controllo a esito positivo (cfr. la tabella 1 al paragrafo 26) con trattamenti brevi e lunghi in assenza di attivazione metabolica e anche con trattamento breve in presenza di attivazione metabolica, al fine di dimostrare la capacità di individuare sostanze chimiche con proprietà clastogeniche e determinare l'efficacia del sistema di attivazione metabolica. Occorre scegliere una serie di concentrazioni delle sostanze chimiche selezionate per fornire aumenti riproducibili e correlati alla concentrazione rispetto ai valori di fondo, dimostrando così la sensibilità e la gamma dinamica del sistema di prova. Dati storici di controllo Il laboratorio deve stabilire:
All'acquisizione dei primi dati di distribuzione dei controlli negativi storici, è necessario che i controlli negativi paralleli siano coerenti con i dati di controllo pubblicati, se esistenti. Con l'aumento dei dati sperimentali aggiunti alla distribuzione dei controlli, i controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % di tale distribuzione (44) (47). La banca dati dei controlli negativi storici del laboratorio deve inizialmente essere costituita con un minimo di 10 esperimenti ma preferibilmente con almeno 20 esperimenti svolti in condizioni sperimentali analoghe. I laboratori devono utilizzare metodi di controllo della qualità, quali diagrammi di controllo (ad esempio carte C o carte X medio (48)), per rilevare la variabilità dei loro dati di controllo positivi e negativi e dimostrare che la metodologia è “sotto controllo” nel laboratorio (44). Ulteriori raccomandazioni su come sviluppare e utilizzare i dati storici (cioè i criteri per l'inclusione e l'esclusione di dati nelle serie storiche e i criteri di accettabilità per un determinato esperimento) sono reperibili nella letteratura scientifica (47). Eventuali modifiche apportate al protocollo sperimentale devono essere considerate in base alla loro coerenza con le esistenti banche dati di controlli storici del laboratorio. L'esistenza di incongruenze significative deve portare alla creazione di una nuova banca dati di controlli storici. I dati sui controlli negativi devono comprendere l'incidenza di cellule con aberrazioni cromosomiche da coltura singola o dalla somma di colture replicate, come descritto nel paragrafo 21. I controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % della distribuzione della banca dati dei controlli negativi storici del laboratorio (44) (47). Qualora i dati dei controlli negativi paralleli non rientrino nei limiti del controllo al 95 %, possono essere accettabili per l'inserimento nella distribuzione dei controlli storici purché non siano valori erratici estremi e sia provato che il sistema di prova è “sotto controllo” (cfr. il paragrafo 37) e che non si sono verificati errori tecnici o umani. DATI E RELAZIONE Presentazione dei risultati Occorre valutare la percentuale di cellule con una o più aberrazioni cromosomiche strutturali. Occorre elencare separatamente le aberrazioni cromosomiche e quelle cromatidiche e classificarle in sottotipi (rotture, scambi) con l'indicazione del numero e della frequenza per le colture sperimentali e di controllo. I gap devono essere registrati e indicati separatamente nella relazione, ma non inclusi nella frequenza totale delle aberrazioni. Le percentuali di poliploidia e/o di cellule endoriduplicate sono segnalate se verificate. Occorre riportare anche le misurazioni di citotossicità condotte in parallelo per tutte le colture trattate di controllo negativo e positivo nei principali test di aberrazione. Occorre fornire dati sulle singole colture. Tutti i dati devono essere riassunti in tabelle. Criteri di accettabilità L'accettazione di una prova è basata sui seguenti criteri:
Analisi e interpretazione dei risultati A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente positiva se, in una qualsiasi delle condizioni sperimentali esaminate (cfr. il paragrafo 28):
Se tutti i criteri sono soddisfatti, la sostanza chimica in esame è ritenuta in grado di indurre aberrazioni cromosomiche in cellule di mammifero coltivate nel sistema di prova. Raccomandazioni dei metodi statistici più appropriati sono reperibili nella letteratura scientifica (49) (50) (51). A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente negativa se, in tutte le condizioni sperimentali esaminate (cfr. il paragrafo 28):
La sostanza chimica in esame è quindi ritenuta non in grado di indurre aberrazioni cromosomiche in cellule di mammifero coltivate nel sistema di prova. Non è necessario verificare una risposta palesemente positiva o negativa. In caso di risposta non chiaramente positiva o negativa come sopra descritto o per contribuire a stabilire la pertinenza biologica di un risultato, i dati devono essere valutati da esperti e/o mediante ulteriori indagini. Potrebbe essere utile analizzare nuove cellule (se appropriato) o ripetere un esperimento modificandone eventualmente le condizioni (ad esempio, intervallazione delle concentrazioni, altre condizioni di attivazione metabolica (ossia concentrazione S9 od origine S9)). In rari casi, anche dopo ulteriori indagini, i dati non consentono di arrivare a una conclusione positiva o negativa e pertanto la risposta della sostanza chimica in esame è considerata ambigua. Un aumento del numero di cellule poliploidi può significare che le sostanze chimiche in esame sono in grado di inibire processi mitotici e di indurre aberrazioni numeriche nei cromosomi (52). Un aumento del numero di cellule con cromosomi endoriduplicati può indicare che le sostanze chimiche in esame sono in grado di inibire la progressione del ciclo cellulare (53) (54) (cfr. il paragrafo 2). Pertanto, l'incidenza delle cellule poliploidi e quella delle cellule con cromosomi endoriduplicati devono essere registrate separatamente. Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti.
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Aneuploidia : qualsiasi deviazione dal normale numero diploide (o aploide) di cromosomi da parte di uno o più cromosomi, ma non dell'intero corredo di cromosomi (poliploidia). Apoptosi : morte cellulare programmata caratterizzata da una serie di fasi che portano alla disintegrazione delle cellule in particelle legate alla membrana, le quali vengono poi eliminate mediante fagocitosi o “shedding” (clivaggio dei ricettori di membrana). Proliferazione cellulare : aumento del numero di cellule dovuto alla divisione mitotica delle cellule. Sostanza chimica : una sostanza o una miscela. Rottura cromatidica : discontinuità di un singolo cromatidio in cui vi è un'evidente disallineamento di uno dei cromatidi. Gap cromatidico : regione non colorata (lesione acromatica) di un singolo cromatidio in cui vi è un disallineamento minimo del cromatidio. Aberrazione di tipo cromatidico : alterazione cromosomica strutturale che si manifesta nella rottura di un singolo cromatidio o nella rottura e ricongiunzione di cromatidi. Aberrazione di tipo cromosomico : alterazione cromosomica strutturale che si manifesta nella rottura, o nella rottura e ricongiunzione, di entrambi i cromatidi in uno stesso punto. Clastogeno : qualsiasi sostanza chimica che causa aberrazioni cromosomiche strutturali in popolazioni di cellule o organismi eucarioti. Concentrazioni : si riferiscono alle concentrazioni finali della sostanza chimica in esame nel mezzo di coltura. Citotossicità : per i saggi di cui al presente metodo di prova con linee cellulari, la citotossicità corrisponde a una riduzione del raddoppiamento relativo della popolazione (RPD) o dell'aumento relativo del numero di cellule (RICC) delle cellule trattate rispetto al controllo negativo (cfr. il paragrafo 17 e l'appendice 2). per i saggi di cui al presente metodo di prova con colture primarie di linfociti, la citotossicità corrisponde a una riduzione del coefficiente mitotico (MI) delle cellule trattate rispetto al controllo negativo (cfr. il paragrafo 18 e l'appendice 2). Endoriduplicazione : processo nel quale, dopo una fase S di replicazione del DNA, il nucleo non inizia la mitosi ma inizia una nuova fase S. Ne risultano cromosomi con 4, 8, 16,… cromatidi. Genotossico : termine generico che comprende tutti i tipi di danno a carico del DNA o dei cromosomi, tra cui rotture, delezioni, modifiche e collegamenti di nucleotidi, riarrangiamenti, mutazioni geniche, aberrazioni cromosomiche e aneuploidia. Non tutti i tipi di effetti genotossici determinano alterazioni cromosomiche o danni permanenti ai cromosomi. Coefficiente mitotico (MI) : numero delle cellule in metafase diviso per il numero totale di cellule della popolazione: costituisce un'indicazione del grado di proliferazione della popolazione cellulare. Mitosi : divisione del nucleo cellulare, solitamente suddivisa in profase, prometafase, metafase, anafase e telofase. Mutageno : un fattore in grado di provocare mutazioni ereditarie delle sequenze di coppie di basi del DNA nei geni o della struttura dei cromosomi (aberrazioni cromosomica). Aberrazione numerica : variazione del numero di cromosomi rispetto al numero diploide caratteristico della specie. Poliploidia : aberrazioni numeriche dei cromosomi che interessa l'intero corredo cromosomico di cellule o organismi, a differenza dall'aneuploidia, che invece interessa un solo cromosoma o più cromosomi, ma non l'intero corredo cromosomico. Stato della p53 : la proteina p53 partecipa alla regolazione del ciclo cellulare, all'apoptosi e alla riparazione del DNA. Le cellule carenti di proteine p53 funzionali, che non sono in grado di arrestare il ciclo cellulare o di eliminare cellule danneggiate tramite apoptosi o altri meccanismi (ad esempio induzione di riparazione del DNA) relativi alle funzioni della p53 in risposta ad alterazioni del DNA, dovrebbero essere teoricamente più soggette a mutazioni geniche o aberrazioni cromosomiche. Aumento relativo delle conte cellulari (RICC) : aumento del numero di cellule nelle colture esposte ad agenti chimici rispetto all'aumento nelle colture non trattate; il rapporto è espresso in percentuale. Raddoppiamento relativo della popolazione (Relative Population Doubling, RPD) : aumento del numero di raddoppiamenti della popolazione nelle colture esposte ad agenti chimici rispetto all'aumento nelle colture non trattate; il rapporto è espresso in percentuale. Frazione S9 del fegato : supernatante di omogenato epatico centrifugato a 9 000 g, cioè estratto di fegato crudo. Miscela S9 : miscela di frazione S9 del fegato con cofattori necessari per l'attività degli enzimi metabolici. Controllo con solvente : termine generico che designa le colture di controllo che ricevono unicamente il solvente utilizzato per disciogliere la sostanza chimica in esame. Aberrazione strutturale : alterazione della struttura cromosomica visibile all'esame microscopico dello stadio di metafase, che si presenta con perdita di segmenti e riordinamenti intercromosomici e intracromosomici. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Controllo non trattato : colture non sottoposte a trattamento (che non ricevono alcuna sostanza chimica in esame né solvente) ma preparate in parallelo e in modo identico alle culture esposte alla sostanza chimica in esame. Appendice 2 FORMULE PER LA VALUTAZIONE DELLA CITOTOSSICITÀ Coefficiente mitotico (MI):
si raccomanda di avvalersi dell'aumento relativo delle conte cellulari (RICC) o del raddoppiamento relativo della popolazione (RPD), poiché entrambi i metodi tengono conto della proporzione di popolazione cellulare che è andata incontro a divisione.
dove: Raddoppiamento della popolazione = [logaritmo (numero di cellule dopo il trattamento ÷ numero di cellule iniziale)] ÷ logaritmo 2 Ad esempio, un RICC o un RPD del 53 % indica una citotossicità/citostasi del 47 % e una citotossicità/citostasi del 55 % misurato con MI significa che il MI reale rappresenta il 45 % del controllo. In ogni caso, occorre misurare il numero di cellule prima del trattamento, che deve essere identico per le colture trattate e per i controlli negativi. L'RCC (ossia il rapporto fra il numero di cellule nelle colture trattate e il numero di cellule nelle colture di controllo) è stato usato come parametro di citotossicità in passato, ma oggi non è più raccomandato perché può sottovalutare la citotossicità. Nelle colture di controllo negativo, il raddoppiamento della popolazione deve essere compatibile con l'esigenza di campionare le cellule dopo il trattamento, trascorso un lasso di tempo pari a circa 1,5 volte la durata del ciclo cellulare normale, e il coefficiente mitotico deve essere sufficientemente elevato per ottenere un numero adeguato di cellule in mitosi e calcolare attendibilmente una riduzione del 50 %. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(4) |
Nella parte B, il capitolo B.11 è sostituito dal seguente: «B.11 Prova di aberrazione cromosomica del midollo osseo nei mammiferi INTRODUZIONE Il presente metodo di prova è equivalente alle linee guida dell'OCSE per le prove sulle sostanze chimiche n. 475 (2016), e fa parte di una serie di metodi di prova di tossicologia genetica. È stato elaborato un documento OCSE contenente informazioni succinte sulle prove di tossicologia genetica e un compendio delle modifiche recentemente apportate alla rispettiva linea guida (1). La prova in vivo di aberrazione cromosomica del midollo osseo nei mammiferi è particolarmente rilevante per valutare la genotossicità. Difatti, benché possano variare a seconda delle specie, i fattori del metabolismo in vivo, gli aspetti farmacocinetici e i processi di riparazione del DNA sono attivi e contribuiscono alle reazioni. La prova in vivo è inoltre utile per studiare più approfonditamente la genotossicità rilevata tramite un sistema in vitro. La prova in vivo di aberrazione cromosomica del midollo osseo nei mammiferi è utilizzato per individuare aberrazioni cromosomiche strutturali indotte dalle sostanze chimiche in esame nelle cellule del midollo osseo di animali, di solito roditori (2) (3) (4) (5). Tali aberrazioni strutturali possono essere di due tipi: cromosomiche o cromatidiche. La maggior parte delle aberrazioni indotte da sostanze chimiche genotossiche è di tipo cromatidico, ma si verificano anche aberrazioni di tipo cromosomico. I danni cromosomici e i fenomeni ad esse connessi sono la causa di molte malattie genetiche umane e vi sono prove concrete del fatto che, quando causano alterazioni degli oncogeni e dei geni soppressori dei tumori, queste lesioni ed i fenomeni ad esse connessi sono implicati nella comparsa delle neoplasie umane e di quelle sperimentalmente indotte negli animali da laboratorio. Nelle prove di aberrazione cromosomica in vivo possono verificarsi casi di poliploidia (inclusa l'endoriduplicazione). Un aumento della poliploidia non è tuttavia, di per sé, un segno di potenziale aneugenico, e può semplicemente indicare una perturbazione del ciclo delle cellule o citotossicità. Questo test non è destinato a misurare l'aneuploidia, per il cui rilevamento sono raccomandati i test in vivo sui micronuclei negli eritrociti di mammifero (capitolo B.12 del presente allegato) o i test del micronucleo in vitro con cellule di mammifero (capitolo B.49 del presente allegato). Le definizioni della terminologia usata figurano nell'Appendice 1. CONSIDERAZIONI INIZIALI Per tale saggio si usano di norma roditori, ma in certi casi si può ricorrere anche ad altre specie se ciò è giustificato sul piano scientifico. Il midollo osseo è il tessuto bersaglio, in quanto è altamente vascolarizzato e contiene una popolazione di cellule a ciclo rapido, che possono essere agevolmente isolate e trattate. L'utilizzazione di specie diverse dai ratti e dai topi deve essere scientificamente giustificata nella relazione. Se si ricorre a specie diverse dai roditori, viene raccomandato di integrare la misurazione delle aberrazioni cromosomiche del midollo osseo in un'altra prova di tossicità pertinente. Se è comprovato che la sostanza o le sostanze chimiche in esame, o i loro metaboliti, non raggiungono il tessuto bersaglio, può non essere opportuno utilizzare questa prova. Prima dell'uso del metodo di prova su una miscela, per la generazione di dati per un determinato scopo normativo, occorre esaminare se, e in caso affermativo perché, può fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie in presenza di un obbligo normativo di prova sulla miscela. PRINCIPIO DELLA PROVA Gli animali sono esposti alla sostanza chimica in esame attraverso un'adeguata via d'esposizione, e al momento opportuno, dopo il trattamento, sono soppressi in modo incruento. Prima della soppressione incruenta, gli animali sono trattati con un inibitore della metafase (ad es. colchicina o colcemid). Le preparazioni cromosomiche approntate dalle cellule di midollo osseo sono poi sottoposte a un processo di colorazione, e le cellule in metafase sono analizzate per individuare aberrazioni cromosomiche. VERIFICA DELLA COMPETENZA DEL LABORATORIO Prove di competenza Per accertare che il laboratorio possieda un'esperienza sufficiente nella conduzione del saggio prima di utilizzarlo nelle prove di routine, il laboratorio deve dimostrare la capacità di riprodurre i risultati attesi dai dati pubblicati (ad es. (6)) riguardanti le frequenze delle aberrazioni cromosomiche, con un minimo di due sostanze per i controlli positivi (incluse le risposte deboli indotte da dosi basse di sostanze per i controlli positivi), come quelle elencate nella tabella 1, e con controlli sui mezzi disperdenti/solventi compatibili (cfr. il paragrafo 22). Le dosi utilizzate nel corso di tali sperimentazioni devono produrre aumenti riproducibili e collegati alle dosi somministrate, e devono dimostrare la sensibilità e l'intervallo dinamico del sistema di analisi sul tessuto in questione (il midollo osseo). Il metodo di conteggio deve essere quello che sarà usato dal laboratorio. Questo requisito non si applica ai laboratori con esperienza, che dispongono cioè di una base di dati storica quale definita ai paragrafi 10-14. Dati di controllo storici Nell'ambito delle prove di competenza il laboratorio deve stabilire:
All'acquisizione dei primi dati di distribuzione dei controlli negativi storici, è necessario che i controlli negativi paralleli siano coerenti con i dati di controllo pubblicati, se esistenti. Con l'aumento dei dati sperimentali aggiunti alla distribuzione dei controlli storici, i controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % di tale distribuzione. La banca dati dei controlli negativi storici del laboratorio deve essere solida sotto il profilo statistico per garantire la capacità del laboratorio di valutare la distribuzione dei suoi dati di controllo negativi. La letteratura suggerisce che possa essere necessario un minimo di 10 esperimenti, ma che sarebbe preferibile contarne almeno 20, svolti in analoghe condizioni sperimentali. I laboratori devono utilizzare metodi di controllo della qualità, quali diagrammi di controllo (ad esempio carte C o carte X medio (7)), per rivelare la variabilità dei loro dati e dimostrare che la metodologia è “sotto controllo” nel laboratorio. Ulteriori raccomandazioni su come sviluppare e utilizzare i dati storici (cioè i criteri per l'inclusione e l'esclusione di dati nelle serie storiche e i criteri di accettabilità per un determinato esperimento) sono reperibili nella letteratura scientifica (8). Se nell'ambito delle prove di competenza (descritte al paragrafo 9) il laboratorio non porta a termine un numero sufficiente di esperimenti per stabilire una distribuzione dei controlli negativi statisticamente solida (cfr. il paragrafo 11), si può accettare che la distribuzione sia definita durante i primi test di routine. Questo approccio deve seguire le raccomandazioni formulate nella letteratura (8), e i risultati dei controlli negativi ottenuti in questi esperimenti devono essere coerenti con i dati di controllo negativi pubblicati. Eventuali modifiche apportate al protocollo sperimentale devono essere considerate in base alle loro ripercussioni sulla coerenza fra i nuovi dati e le esistenti banche dati di controlli storici del laboratorio. Solo le incongruenze significative dovrebbero portare alla creazione di una nuova banca dati di controlli storici, se il parere di un esperto stabilisce che vi è una differenza rispetto alla distribuzione precedente (cfr. il paragrafo 11). Durante la costituzione di questa nuova banca dati, il laboratorio non ha necessariamente bisogno di una banca dati completa di controlli negativi per permettere lo svolgimento di una prova, a condizione che esso possa dimostrare che i valori dei controlli negativi paralleli rimangano coerenti con la precedente banca dati o con i dati corrispondenti pubblicati. I dati sui controlli negativi devono riguardare l'incidenza delle aberrazioni cromosomiche strutturali (senza “gap”) in ogni animale. I controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % della distribuzione della banca dati dei controlli negativi storici del laboratorio. Qualora i dati dei controlli negativi paralleli non rientrino nei limiti del controllo al 95 % possono essere accettabili per l'inserimento nella distribuzione dei controlli storici purché non siano outlier estremi e sia provato che il sistema di prova è “sotto controllo” (cfr. il paragrafo 11) e che non si sono verificati errori tecnici o umani. DESCRIZIONE DEL METODO Preparazioni Selezione delle specie animali Di preferenza sono utilizzati individui adulti, giovani e in buona salute, provenienti da ceppi di animali da laboratorio. Sono comunemente usati i ratti, ma possono essere idonei anche i topi. Si può ricorrere a qualsiasi altra specie idonea di mammiferi, se ciò viene giustificato scientificamente nella relazione. Condizioni di stabulazione e alimentazione degli animali Per i roditori, la temperatura dello stabulario deve essere mantenuta a 22 °C (± 3 °C). Idealmente l'umidità relativa deve essere del 50-60 %. Deve comunque essere come minino del 40 % e non deve preferibilmente superare il 70 %, tranne che nel corso delle pulizie degli ambienti. L'illuminazione deve essere artificiale, con una sequenza di 12 ore di luce e 12 d'oscurità. Per l'alimentazione, attenersi alle diete convenzionali da laboratorio con una quantità illimitata di acqua potabile. La scelta della dieta può essere influenzata dalla necessità di garantire un'adeguata miscela della sostanza in esame, se somministrata per questa via. Se non ci si aspetta alcun comportamento aggressivo, i roditori vanno alloggiati in piccoli gruppi (non più di cinque in ogni gabbia) dello stesso sesso e gruppo di trattamento, preferibilmente in gabbie a fondo pieno con adeguato arricchimento ambientale. Gli animali possono essere alloggiati individualmente solo se ciò è giustificato sotto il profilo scientifico. Preparazione degli animali Sono generalmente utilizzati animali adulti, giovani e sani (i roditori hanno idealmente un'età di 6-10 settimane all'inizio del trattamento, ma sono accettati anche animali di età un pÓ più avanzata), che vengono suddivisi a caso in gruppi di controllo e di trattamento. I singoli animali sono identificati univocamente applicando un metodo incruento e il meno possibile invasivo (cioè inanellamento, etichettatura, applicazione di un microchip o identificazione biometrica, evitando il taglio delle orecchie o la falangectomia) e devono essere acclimatati alle condizioni di laboratorio per almeno 5 giorni. Le gabbie devono essere sistemate in modo da ridurre al minimo eventuali effetti dovuti alla loro collocazione. Deve essere evitata ogni contaminazione incrociata fra il controllo positivo e la sostanza chimica in esame. All'inizio dello studio la variazione di peso degli animali deve essere minima e non superare il ± 20 % del peso medio per ciascun sesso. Preparazione delle dosi Le sostanze solide in esame vanno sciolte o poste in sospensione in appropriati solventi o mezzi disperdenti o mescolate alla dieta o all'acqua da bere prima di essere somministrate agli animali. Le sostanze liquide possono essere somministrate direttamente o diluite prima del trattamento. In caso di esposizione per via inalatoria, le sostanze possono essere somministrate sotto forma di gas, vapore o aerosol solido o liquido, in funzione delle loro proprietà fisico-chimiche. Vanno usati preparati freschi della sostanza, a meno che i dati sulla stabilità dimostrino che la conservazione è accettabile e definiscano le adeguate condizioni di conservazione. Solvente/mezzo disperdente Il solvente/mezzo disperdente non deve produrre effetti tossici ai livelli di dose usati e non deve reagire chimicamente con la sostanza in esame. L'uso di solventi/mezzi disperdenti poco noti è ammesso solo se suffragato da dati che ne provino la compatibilità. Si raccomanda di prendere in primo luogo in considerazione, se possibile, l'uso di un solvente/mezzo disperdente acquoso. Fra gli esempi di solventi/mezzi disperdenti compatibili e comunemente usati si possono menzionare l'acqua, la soluzione fisiologica, la soluzione di metilcellulosa, la soluzione di carbossimetilcellulosa sodica, l'olio di oliva e l'olio di mais. In mancanza di dati di controllo storici o pubblicati che dimostrino che un solvente/mezzo disperdente atipico prescelto non induce aberrazioni strutturali o altri effetti nocivi, va effettuato uno studio iniziale per stabilire l'accettabilità del controllo del solvente/mezzo disperdente. Controlli Controlli positivi Ogni prova deve generalmente includere un gruppo di animali trattati con una sostanza usata per i controlli positivi. Questa condizione può essere eliminata quando il laboratorio di prova ha dimostrato la sua competenza nello svolgimento della prova e ha stabilito l'intervallo dei controlli positivi storici. Quando manca un gruppo di controllo positivo parallelo, in ogni esperimento devono essere inclusi controlli del conteggio (vetrini fissati e non colorati). Questo può avvenire includendo nello studio campioni di riferimento idonei ottenuti e conservati in occasione di un esperimento separato di controllo positivo svolto periodicamente (ad es. ogni 6-18 mesi) nel laboratorio in cui è condotta la prova; ad esempio, durante la verifica della competenze e successivamente, se necessario, su base periodica. Le sostanze chimiche per i controlli positivi devono portare, in modo affidabile, a un aumento individuabile della frequenza delle cellule con aberrazioni cromosomiche strutturali al di là del livello spontaneo. Le dosi dei controlli positivi devono essere scelte in modo che gli effetti siano chiari ma non rivelino immediatamente all'analista l'identità dei campioni codificati. È ammissibile che le sostanze per i controlli positivi siano somministrate per una via diversa da quella della sostanza chimica in esame, usando un programma di trattamento diverso, e che il campionamento avvenga in un unico momento. È anche ammissibile, se appropriato, l'uso per i controlli positivi di sostanze di una classe chimica correlata. Nella tabella 1 figurano esempi di sostanze chimiche per i controlli positivi. Tabella 1. Esempi di sostanze chimiche per i controlli positivi
Controlli negativi In ogni momento del campionamento devono essere inclusi animali del gruppo di controllo negativo, che sono gestiti nello stesso modo dei gruppi di trattamento, ma a cui non viene somministrata la sostanza chimica in esame. Se per la somministrazione della sostanza in esame si usa un solvente/mezzo disperdente, anche il gruppo di controllo deve riceverlo. Tuttavia, se dai dati dei controlli negativi storici risulta una variabilità intraspecifica e una frequenza delle cellule con aberrazioni strutturali coerente ad ogni momento del campionamento per il laboratorio di prova, può essere sufficiente un solo campionamento per il controllo negativo. In tal caso, esso deve avvenire al momento del primo campionamento effettuato nell'ambito dello studio. PROCEDURA Numero e sesso degli animali In generale, la risposta dei micronuclei è simile nei maschi e nelle femmine degli animali (9), ed è da prevedere che ciò valga anche per le aberrazioni cromosomiche strutturali; la maggior parte degli studi potrebbe pertanto venire effettuata sull'uno o sull'altro sesso. L'esistenza di dati che dimostrano differenze rilevanti fra i maschi e le femmine (ad es. differenze riguardanti la tossicità sistemica, il metabolismo, la biodisponibilità, la tossicità del midollo osseo, ecc., incluso ad es. un range-finding test) incoraggerebbero l'uso di entrambi i sessi. Nella fattispecie potrebbe essere appropriato effettuare uno studio su entrambi i sessi, ad es. come parte di uno studio di tossicità a dose ripetuta. Nel caso si effettuino prove su entrambi i sessi potrebbe essere adeguato ricorrere al metodo fattoriale. Nell'appendice 2 figurano precisazioni sulle modalità di analisi dei dati in base a tale metodo. All'inizio dello studio, la dimensione stabilita del gruppo deve consentire di disporre, in ogni gruppo, di almeno 5 animali analizzabili dello stesso sesso, o di ogni sesso se sono usati entrambi. Se l'esposizione umana a sostanze chimiche è specifica per un sesso, ad esempio nel caso di alcuni prodotti farmaceutici, il test deve essere eseguito su animali di tale sesso. A titolo di orientamento sul numero massimo di animali tipicamente necessario, uno studio sul midollo osseo con due momenti di campionamento, con tre gruppi di trattamento e un gruppo di controllo negativo parallelo, più un gruppo di controllo positivo (ogni gruppo composto da cinque animali dello stesso sesso), richiederebbe 45 animali. Livelli di dose Se viene effettuato uno studio preliminare di range-finding poiché non sono già disponibili dati appropriati per orientare la scelta delle dosi, tale studio deve essere svolto nello stesso laboratorio, usando specie, ceppi, sesso e un regime di trattamento identici a quelli che saranno utilizzati nello studio principale (10). Lo studio preliminare serve a individuare la dose massima tollerata (MTD), definita come la dose più alta che sarà tollerata per la durata della prova senza che appaia una tossicità che limita lo studio (inducendo ad esempio un calo del peso corporeo o citotossicità del sistema emopoietico, ma che non provoca morte o segni di dolore, sofferenza o stress che rendano necessaria una soppressione incruenta (11)). La dose massima può essere definita anche come la dose che produce qualche segno di tossicità nel midollo osseo. Le sostanze chimiche che comportano saturazione delle caratteristiche tossicocinetiche o che inducono processi di detossificazione che possono portare a una diminuzione dell'esposizione dopo un trattamento a lungo termine possono essere considerate eccezioni rispetto ai criteri di definizione della dose e vanno valutate caso per caso. Per ottenere informazioni sulla relazione dose-risposta, uno studio completo deve includere un gruppo di controllo negativo e almeno tre livelli di dose, generalmente separati da un fattore 2, ma non più di 4. Se la sostanza chimica in esame non provoca alcuna tossicità nell'ambito di un test di range-finding o secondo quanto emerge dai dati esistenti, la dose massima per una singola somministrazione deve essere di 2 000 mg/kg di peso corporeo. Se la sostanza chimica in esame provoca invece tossicità, la MTD deve corrispondere alla dose più elevata somministrata, e i livelli di dose utilizzati devono essere compresi in un intervallo che va dalla tossicità massima a una tossicità modica o assente. Quando una tossicità del tessuto bersaglio (midollo osseo) è osservata a tutti i livelli di dose di prova, è consigliabile un ulteriore studio con dosi non tossiche. Gli studi volti a descrivere più pienamente le informazioni quantitative sulla relazione dose-risposta possono richiedere gruppi di trattamento supplementari. Per certi tipi di sostanze chimiche in esame (ad es. medicinali per uso umano) per le quali valgono specifici requisiti, i limiti possono variare. Prova limite Se gli esperimenti per determinare gli intervalli di dose o i dati disponibili relativi ai ceppi di animali affini indicano che un trattamento uguale o superiore alla dose limite (sotto descritta) non produce effetti tossici osservabili (comprese nessuna depressione della funzione del midollo osseo o altre manifestazioni di citotossicità del tessuto bersaglio), e se non si prevede genotossicità sulla base degli studi di genotossicità in vitro o dei dati relativi alle sostanze chimiche strutturalmente affini, uno studio completo con tre livelli di dose può non essere considerato necessario, purché sia stato dimostrato che la sostanza o le sostanze chimiche in esame raggiungono il tessuto bersaglio (midollo osseo). In tali casi può essere sufficiente un solo livello di dose, corrispondente alla dose limite. Per un periodo di somministrazione di >14 giorni, la dose limite è 1 000 mg/kg di peso corporeo al giorno. Per periodi di somministrazione di 14 giorni o meno, la dose limite è 2 000 mg/kg/ di peso corporeo al giorno. Somministrazione delle dosi Nella concezione di un saggio va considerata la via d'esposizione umana prevista. Pertanto possono essere scelti, se giustificati, vie di somministrazione quali l'alimentazione, l'acqua potabile, la via sottocutanea locale, la via endovenosa, la via orale (sonda gastrica), inalatoria, intratracheale, o l'impianto. In ogni caso, deve essere scelta la via che garantisce un'adeguata esposizione del o dei tessuti bersaglio. L'iniezione intraperitoneale non è generalmente raccomandata in quanto non si tratta di una via di esposizione umana prevista, e va utilizzata solo con una specifica giustificazione scientifica. Se la sostanza chimica in esame è miscelata nell'alimentazione o nell'acqua occorre fare attenzione, specialmente nei casi di dosaggio singolo, che il lasso di tempo fra l'assunzione del cibo e dell'acqua e il campionamento sia sufficiente per consentire l'individuazione degli effetti (cfr. i paragrafi 33-34). Il volume massimo di liquido somministrabile in un'unica volta con sonda gastrica o con iniezione dipende dalle dimensioni dell'animale da laboratorio. Esso non deve generalmente essere superiore a 1 ml/100 g di peso corporeo, tranne nel caso delle soluzioni acquose che possono essere somministrate in quantità pari a massimo 2 ml/100 g di peso corporeo. L'uso di volumi maggiori deve essere giustificato. Salvo nel caso di sostanze chimiche irritanti o corrosive, i cui effetti di norma tendono a esacerbarsi con l'aumentare della concentrazione, la variabilità del volume somministrato deve essere ridotta al minimo adeguando la concentrazione, in modo da garantire la somministrazione di un volume costante in relazione al peso corporeo per tutti i livelli di dose. Programma di trattamento Le sostanze chimiche in esame sono di norma somministrate in un'unica presa, ma possono essere somministrate anche in prese frazionate (due prese nello stesso giorno, a distanza di qualche ora al massimo) per agevolare la somministrazione di un grosso volume. In queste situazioni, o in caso di somministrazione della sostanza chimica in esame per inalazione, il momento del campionamento deve essere programmato in base al momento di somministrazione dell'ultima dose o della fine dell'esposizione. Vi sono pochi dati disponibili sull'idoneità di un protocollo a dosi ripetute per questa prova. Tuttavia, qualora sia auspicabile integrare la prova in questione con un test di tossicità a dosi ripetute, occorre fare attenzione a evitare la perdita delle cellule mitotiche che presentano danni cromosomici, fenomeno che può verificarsi con le dosi tossiche. Una tale integrazione è accettabile quando la dose massima è superiore o pari alla dose limite (cfr. il paragrafo 29), e la dose limite è somministrata a un gruppo di trattamento per tutta la durata del trattamento. Il test del micronucleo (metodo di prova B.12) va considerato come il test in vivo prescritto per le aberrazioni cromosomiche quando è auspicata un'integrazione con altri studi. I campioni di midollo osseo devono essere prelevati in due momenti diversi dopo i singoli trattamenti. Per i roditori, il primo intervallo di campionamento deve corrispondere al tempo necessario per completare una volta e mezza la durata del ciclo cellulare normale (che è generalmente di 12-18 ore dopo il periodo del trattamento). Poiché il tempo necessario affinché la sostanza o le sostanze chimiche in esame siano assorbite e metabolizzate e producano effetti sulla cinetica del ciclo cellulare può influire sul momento ottimale per l'individuazione delle aberrazioni cromosomiche, si raccomanda di prelevare un altro campione 24 ore dopo. Al momento del primo campionamento, tutti i gruppi di trattamento devono essere trattati e i campioni per l'analisi prelevati, mentre al momento del o dei campionamenti successivi deve essere somministrata solo la dose massima. Se il protocollo di trattamento è più lungo di un giorno in base a una giustificazione scientifica, si deve procedere ad un unico campionamento una volta trascorso un periodo equivalente fino a circa una volta e mezza la durata normale del ciclo cellulare dopo l'ultima somministrazione. Dopo il trattamento e prima della raccolta dei campioni, agli animali viene iniettata per via intraperitoneale un'adeguata dose di un agente inibitore della metafase (ad es. colcemid o colchicina), e i campioni sono prelevati successivamente a un intervallo appropriato. Per i topi questo intervallo è di circa 3-5 ore prima della raccolta, e per i ratti è di 2-5 ore. Le cellule sono prelevate dal midollo osseo, sono ingrandite, fissate e colorate, e analizzate ai fini dell'individuazione di aberrazioni cromosomiche (12). Osservazioni Gli animali da laboratorio devono essere oggetto di osservazioni cliniche generali e i segni clinici devono essere registrati almeno una volta al giorno, preferibilmente alla stessa o alle stesse ore e tenendo conto del periodo di massima intensità degli effetti previsti dopo la somministrazione. Almeno due volte al giorno, durante il periodo di somministrazione, tutti gli animali vengono esaminati al fine di determinare la morbilità e la mortalità. Tutti gli animali devono essere pesati all'inizio dello studio, almeno una volta alla settimana nel corso degli studi a dosi ripetute, e al momento della soppressione incruenta. Negli studi la cui durata è di almeno una settimana, la misurazione del consumo di cibo va eseguita almeno con cadenza settimanale. Se la sostanza in esame viene somministrata con l'acqua, il consumo di acqua va misurato ad ogni cambio dell'acqua e almeno una volta alla settimana. Gli animali che manifestano segni di eccessiva, ma non letale, tossicità vanno soppressi in modo incruento prima della fine del periodo di saggio (11). Esposizione del tessuto bersaglio Ove ciò sia giustificato e non esistano altri dati sull'esposizione (cfr. il paragrafo 44), al o ai momenti idonei va prelevato un campione di sangue per esaminare i livelli delle sostanze chimiche in esame nel plasma, allo scopo di dimostrare l'avvenuta esposizione del midollo osseo. Midollo osseo e preparazioni cromosomiche Immediatamente dopo la soppressione incruenta, dai femori e dalle tibie degli animali vengono prelevate le cellule del midollo osseo, che vengono esposte a una soluzione ipotonica e fissate. Le cellule in metafase vengono poi poste sui vetrini e colorate secondo metodi prestabiliti (cfr. (3) (12)). Analisi Tutti i vetrini, compresi quelli dei controlli positivi e negativi, devono essere codificati indipendentemente prima dell'analisi, e devono essere randomizzati in modo che l'analista non conosca le condizioni del trattamento. Come misura della citotossicità deve essere determinato il coefficiente mitotico in almeno 1 000 cellule per animale, in tutti gli animali trattati (compresi i controlli positivi), e nei controlli negativi non trattati o con somministrazione della sostanza chimica in esame in solvente/mezzo disperdente. Per individuare le aberrazioni cromosomiche strutturali, inclusi ed esclusi i gap, devono essere analizzate almeno 200 metafasi per ogni animale (6). Tuttavia, se la banca dati dei controlli negativi storici indica che la frequenza di fondo media delle aberrazioni cromosomiche strutturali nel laboratorio di prova è <1 %, occorre prendere in considerazione l'opportunità di esaminare cellule supplementari. Le aberrazioni di tipo cromatidico e cromosomico devono essere registrate separatamente e classificate in sottotipi (rotture, scambi). Le procedure in uso nel laboratorio devono garantire che l'analisi delle aberrazioni cromosomiche sia effettuata da analisti qualificati e, se appropriato, che sia oggetto di una revisione inter pares. Poiché le procedure di preparazione dei vetrini causano spesso la rottura di una parte delle cellule in metafase, con perdita di cromosomi, le cellule conteggiate devono quindi contenere un numero di centromeri non inferiore a 2n±2, dove n è il numero aploide di cromosomi per quella specie. DATI E RELAZIONE Trattamento dei risultati I dati relativi a ciascun animale vanno presentati sotto forma di tabella. Per ogni animale devono essere indicati l'indice mitotico, il numero di cellule in metafase conteggiate, il numero di aberrazioni per ogni cellula in metafase e la percentuale di cellule con aberrazioni cromosomiche strutturali. Devono essere elencati i diversi tipi di aberrazioni cromosomiche strutturali, con il relativo numero e la frequenza per i gruppi trattati e i gruppi di controllo. I gap, così come le cellule poliploidi e le cellule con cromosomi endoriduplicati sono registrati separatamente. La frequenza dei gap viene indicata nella relazione, ma non viene generalmente inclusa nell'analisi della frequenza totale delle aberrazioni strutturali. Se non c'è un'evidente differenza di risposta tra i sessi, i dati possono essere combinati nell'analisi statistica. Nella relazione devono anche figurare i dati riguardanti la tossicità animale e i segni clinici. Criteri di accettabilità L'accettabilità delle prove si basa sui seguenti criteri:
Valutazione e interpretazione dei risultati A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è ritenuta chiaramente positiva se:
Se in un particolare momento del campionamento viene esaminata solo la dose massima, la sostanza chimica in esame è ritenuta chiaramente positiva se vi è un aumento statisticamente significativo rispetto al controllo negativo parallelo, e i risultati sono al di fuori della distribuzione dei dati di controllo negativi storici (ad es. limiti del controllo al 95 % in base alla distribuzione di Poisson). Raccomandazioni relative agli appropriati metodi statistici figurano nella letteratura (13). Quando si effettua un'analisi della relazione dose-risposta, devono essere analizzati almeno tre gruppi di trattamento. Nei test statistici l'unità sperimentale deve essere l'animale. L'ottenimento di risultati positivi nella prova di aberrazione cromosomica indica che la sostanza chimica in esame induce aberrazioni cromosomiche strutturali nel midollo osseo della specie esaminata. A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è ritenuta chiaramente negativa se in tutte le condizioni sperimentali esaminate:
Raccomandazioni relative ai metodi statistici più appropriati figurano nella letteratura (13). L'esposizione del midollo osseo alla sostanza chimica in esame può essere dimostrata, ad esempio, da un calo dell'indice mitotico, o dalla misurazione del livello delle sostanze in esame nel plasma o nel sangue In caso di somministrazione per via endovenosa, la prova dell'esposizione non è necessaria. In alternativa, l'esposizione del midollo osseo può essere dimostrata ricorrendo ai dati ADME, ottenuti nell'ambito di uno studio indipendente usando la stessa via di somministrazione e le stesse specie. L'ottenimento di risultati negativi indica che, nelle condizioni di prova, la sostanza chimica in esame non induce aberrazioni cromosomiche strutturali nel midollo osseo delle specie esaminate. Non è necessario verificare una risposta chiaramente positiva o chiaramente negativa. Nei casi in cui la risposta non sia chiaramente negativa né chiaramente positiva, per poter stabilire la rilevanza biologica di un risultato (ad es. un aumento debole o marginale), i dati devono venire sottoposti al giudizio di esperti e/o a indagini più approfondite sugli esperimenti portati a termine. In alcuni casi può essere utile analizzare più cellule o ripetere l'esperienza in condizioni sperimentali modificate. In rari casi, anche dopo ulteriori indagini, i dati non permetteranno di concludere se la sostanza chimica in esame produce risultati positivi o negativi, e lo studio dovrà pertanto essere dichiarato ambiguo. La frequenza delle metafasi poliploidi e delle metafasi con endoriduplicazione, rispetto al numero totale delle metafasi, va registrata separatamente. Un aumento del numero di cellule poliploidi/endoriduplicate può indicare che la sostanza chimica in esame è in grado di inibire il processo mitotico o il ciclo cellulare (cfr. il paragrafo 3). Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti: Sintesi
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Aneuploidia : qualsiasi deviazione dal normale numero diploide (o aploide) di cromosomi da parte di uno o più cromosomi, ma non dai multipli delle intere serie di cromosomi (cfr. poliploidia). Centromero : regione/i di un cromosoma sede di attacco del fuso durante la divisione cellulare, che permette il corretto movimento dei cromosomi figli verso i poli delle cellule figlie. Sostanza chimica : una sostanza o una miscela. Aberrazione di tipo cromatidico : alterazione cromosomica strutturale che si manifesta nella rottura di un singolo cromatidio o nella rottura e ricongiunzione di cromatidi. Aberrazione di tipo cromosomico : alterazione cromosomica strutturale che si manifesta nella rottura, o nella rottura e ricongiunzione, di entrambi i cromatidi in uno stesso punto. Endoriduplicazione : processo nel quale, dopo una fase S di replicazione del DNA, il nucleo non inizia la mitosi ma inizia una nuova fase S. Ne risultano cromosomi con 4, 8, 16,... cromatidi. Gap : regione non colorata (lesione acromatica) di un singolo cromatidio in cui vi è un disallineamento minimo del cromatidio. Indice mitotico : rapporto fra il numero di cellule in mitosi e il numero totale di cellule in una popolazione, che indica lo stato di proliferazione di tale popolazione di cellule. Aberrazione numerica : variazione del numero di cromosomi rispetto al numero diploide caratteristico della specie (aneuploidia). Poliploidia : aberrazioni numeriche dei cromosomi che interessa l'intero corredo cromosomico di cellule o organismi, a differenza dall'aneuploidia, che invece interessa un solo cromosoma o più cromosomi, ma non l'intero corredo. Aberrazione cromosomica strutturale : alterazione della struttura cromosomica visibile all'esame microscopico dello stadio di metafase, che si presenta con delezioni, perdita di segmenti, riordinamenti intercromosomici e intracromosomici. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Appendice 2 METODO FATTORIALE PER INDIVIDUARE LE DIFFERENZE FRA I SESSI NEL SAGGIO IN VIVO SULLE ABERRAZIONI CROMOSOMICHE Metodo fattoriale e analisi Questo modello prevede di esporre 5 maschi e 5 femmine a ciascuna concentrazione sperimentale e comporta pertanto l'utilizzo di un minimo di 40 animali (20 maschi e 20 femmine più i relativi controlli positivi). Il modello qui presentato — una delle forme semplici del modello fattoriale — è equivalente a un'analisi della varianza a due fattori, in cui il sesso e il livello delle concentrazioni costituiscono i fattori principali. I dati possono essere analizzati utilizzando diversi programmi statistici standard, quali SPSS, SAS, STATA, Genstat o il programma R. A partire dall'insieme dei dati si determina la variabilità tra i sessi e le concentrazioni, nonché la variabilità relativa alle interazioni tra sessi e concentrazioni. Ciascuno di questi aspetti è quindi valutato in rapporto alla stima della variabilità tra gli animali ripartiti nei gruppi di animali dello stesso sesso esposti alle stesse concentrazioni. Maggiori informazioni su questa metodologia sono reperibili in diversi manuali standard di statistica (cfr. bibliografia), nonché nelle schede “di aiuto” fornite con i programmi statistici. In seguito viene esaminata l'interazione sesso x concentrazione in una tabella ANOVA (5). In assenza di un termine di interazione significativo la combinazione dei valori inter-sessi o inter-livelli di concentrazione consente di effettuare test statistici validi tra i livelli, basandosi sul termine di variabilità intra-gruppo combinata di 'ANOVA. L'analisi prosegue suddividendo la variabilità stimata tra le concentrazioni in modo da ottenere contrasti che permettano di stabilire contrasti lineari e quadratici delle risposte per l'insieme dei livelli di concentrazione. Quando si riscontra un'interazione significativa sesso x concentrazione, questo termine può essere a sua volta in ripartito in contrasti di interazione lineare x sesso e quadratica x sesso. In questo modo si può verificare se le risposte alle concentrazioni sono parallele per i due sessi o se vi è una differenza riconducibile al sesso. La stima della variabilità intra-gruppo combinata può essere utilizzata per verificare lo scarto tra le medie, confrontandole due a due. I confronti possono essere effettuati tra le medie per i due sessi e tra le medie dei diversi livelli delle concentrazioni (come, ad esempio, confronti tra i livelli dei controlli negativi). Nei casi in cui si registra un'interazione significativa i confronti possono essere effettuati tra le medie di concentrazioni diverse per lo stesso sesso o tra le medie dei due sessi a parità di concentrazione. Riferimenti Numerosi manuali di statistica esaminano la teoria, la concezione, la metodologia, l'analisi e l'interpretazione dei modelli fattoriali, dall'analisi più semplice, a due fattori, alle forme più complesse utilizzate per la metodologia Design of Experiment. Di seguito ne è riportato un elenco non completo. Alcuni manuali forniscono esempi di modelli comparabili e, in alcuni casi, forniscono un codice che permette di effettuare l'analisi utilizzando diversi programmi.
|
|
(5) |
Nella parte B, il capitolo B.12 è sostituito dal seguente: «B.12 Prova sui micronuclei negli eritrociti di mammifero INTRODUZIONE Il presente metodo di prova è equivalente alle linee guida dell'OCSE per le prove sulle sostanze chimiche n. 474 (2016), e fa parte di una serie di metodi di prova di tossicologia genetica. È stato elaborato un documento OCSE contenente informazioni succinte sulle prove di tossicologia genetica e un compendio delle modifiche recentemente apportate alla rispettiva linea guida (1). La prova in vivo sui micronuclei dei mammiferi è particolarmente rilevante per valutare la genotossicità. Difatti, benché possano variare a seconda delle specie, i fattori del metabolismo in vivo, gli aspetti farmacocinetici e i processi di riparazione del DNA sono attivi e contribuiscono alle reazioni. La prova in vivo è inoltre utile per studiare più approfonditamente la genotossicità rilevata tramite un sistema in vitro. La prova in vivo sui micronuclei dei mammiferi è utilizzato per individuare i danni indotti dalla sostanza chimica in esame sui cromosomi o sull'apparato mitotico degli eritoblasti. Esso valuta la formazione di micronuclei in eritrociti provenienti dal midollo osseo o dalle cellule del sangue periferico di animali, di norma roditori. Lo scopo della prova sui micronuclei è individuare le sostanze chimiche che provocano danni citogenetici, che si manifestano nella formazione di micronuclei contenenti frammenti residui di cromosomi o cromosomi interi. Quando da un eritroblasto del midollo osseo si sviluppa un eritrocita immaturo (designato a volte anche come eritrocita policromatico o reticolocito), il nucleo principale viene espulso; i micronuclei formatisi possono rimanere nel citoplasma. In queste cellule la visualizzazione o il rilevamento dei micronuclei sono facilitati poiché manca il nucleo principale. L'aumento della frequenza di eritrociti immaturi contenenti micronuclei negli animali trattati è un'indicazione di aberrazioni cromosomiche strutturali o numeriche indotte. Gli eritrociti contenenti micronuclei di nuova formazione sono individuati e quantificati mediante colorazione, seguita o da un conteggio visivo al microscopio, o da un'analisi automatizzata. Il conteggio di un numero sufficiente di eritrociti immaturi nel sangue periferico o nel midollo osseo degli animali adulti è grandemente facilitato dall'uso di una piattaforma di calcolo automatizzata. Tali piattaforme sono alternative accettabili alla valutazione manuale (2). Studi comparativi hanno mostrato che tali metodi, usando standard di calibrazione adeguati, possono consentire riproducibilità inter- e intra-laboratorio e sensibilità migliore di quella consentita dal conteggio manuale al microscopio (3) (4). I sistemi automatizzati che possono misurare la frequenza degli eritrociti contenenti micronuclei includono (ma non sono limitati a questi): citometri a flusso (5), piattaforme di analisi delle immagini (6) (7), e citometri a scansione laser (8). Benché generalmente non rientri nella prova, i frammenti di cromosomi possono essere distinti dai cromosomi interi in base a una serie di criteri. Questi includono la presenza o l'assenza del cinetocore, o del centromero, entrambi caratteristici dei cromosomi intatti. L'assenza del cinetocore, o del centromero, indica che il micronucleo contiene solo frammenti di cromosomi, mentre la presenza è indicativa di una perdita cromosomica. Le definizioni della terminologia usata figurano nell'Appendice 1. CONSIDERAZIONI INIZIALI In questo saggio il tessuto bersaglio delle lesioni genetiche è il midollo osseo di roditori adulti giovani. In questo tessuto sono difatti prodotti gli eritrociti. La misurazione dei micronuclei negli eritrociti immaturi del sangue periferico può essere effettuata in altre specie di mammiferi per i quali è stata dimostrata una sensibilità sufficiente per l'individuazione di sostanze chimiche che provocano aberrazioni cromosomiche strutturali o numeriche in queste cellule (tramite induzione di micronuclei negli eritrociti immaturi), e a condizione che ciò sia giustificato sul piano scientifico. L'endpoint è la frequenza degli eritrociti immaturi micronucleati. La frequenza degli eritrociti maturi contenenti micronuclei nel sangue periferico può anch'essa essere utilizzata come endpoint nelle specie che non presentano una forte selezione splenica contro le cellule contenenti micronuclei e quando gli animali sono trattati continuativamente per un periodo superiore alla durata di vita di un eritrocita nella specie utilizzata (ad es., nel topo, 4 settimane o più). Se è comprovato che la sostanza o le sostanze chimiche in esame, o i loro metaboliti, non raggiungono il tessuto bersaglio, può non essere opportuno utilizzare questa prova. Prima dell'uso del metodo di prova su una miscela, per la generazione di dati per un determinato scopo normativo, occorre esaminare se, e in caso affermativo perché, può fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie in presenza di un obbligo normativo di prova sulla miscela. PRINCIPIO DELLA PROVA Gli animali sono esposti alla sostanza chimica in esame attraverso un'adeguata via d'esposizione. Se si utilizza il midollo osseo, al momento / ai momenti opportuni, dopo il trattamento, gli animali sono soppressi in modo incruento. Il midollo osseo viene estratto, e si procede alle preparazioni e alle colorazioni (9) (10) (11) (12) (13) (14) (15). Quando si utilizza il sangue periferico, il sangue viene raccolto al momento / ai momenti opportuni dopo il trattamento e si procede alle preparazioni e alle colorazioni (12) (16) (17) (18). In caso di somministrazione acuta del trattamento, è importante effettuare il prelievo del midollo osseo o del sangue in un momento in cui l'induzione di eritrociti immaturi micronucleati dovuta al trattamento possa essere individuata. In caso di prelievo di sangue periferico, perché tali elementi appaiano nella circolazione sanguigna deve essere trascorso un lasso di tempo sufficiente. Le preparazioni sono analizzate ai fini dell'individuazione dei micronuclei, tramite visualizzazione con microscopio, analisi delle immagini, citometri a flusso, oppure citometria a scansione laser. VERIFICA DELLA COMPETENZA DEL LABORATORIO Prove di competenza Per accertare che il laboratorio possieda un'esperienza sufficiente nella conduzione del saggio prima di utilizzarlo nelle prove di routine, il laboratorio deve dimostrare la capacità di riprodurre i risultati attesi dai dati pubblicati (17) (19) (20) (21) (22) riguardanti le frequenze dei micronuclei, con un minimo di due sostanze per i controlli positivi (incluse le risposte deboli indotte da dosi basse di sostanze per i controlli positivi), come quelle elencate nella tabella 1, e con controlli su mezzi disperdenti/solventi compatibili (cfr. il paragrafo 26). Le dosi utilizzate nel corso di tali sperimentazioni devono produrre aumenti riproducibili e collegati alle dosi somministrate, e devono dimostrare la sensibilità e l'intervallo dinamico del sistema di analisi sul tessuto in questione (il midollo osseo o il sangue periferico). Il metodo di conteggio deve essere quello che sarà usato dal laboratorio. Questo requisito non si applica ai laboratori con esperienza, che dispongono cioè di una base di dati storica quale definita ai paragrafi 14-18. Dati di controllo storici Nell'ambito delle prove di competenza il laboratorio deve stabilire:
All'acquisizione dei primi dati di distribuzione dei controlli negativi storici, è necessario che i controlli negativi paralleli siano coerenti con i dati di controllo pubblicati, se esistenti. Con l'aumento dei dati sperimentali aggiunti alla distribuzione dei controlli storici, i controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % di tale distribuzione. La banca dati dei controlli negativi storici del laboratorio deve essere solida sotto il profilo statistico per garantire la capacità del laboratorio di valutare la distribuzione dei suoi dati di controllo negativi. La letteratura suggerisce che possa essere necessario un minimo di 10 esperimenti, ma che sarebbe preferibile contarne almeno 20, svolti in analoghe condizioni sperimentali. I laboratori devono utilizzare metodi di controllo della qualità, quali diagrammi di controllo (ad esempio carte C o carte X medio (23)), per rivelare la variabilità dei loro dati e dimostrare che la metodologia è “sotto controllo” nel laboratorio. Ulteriori raccomandazioni su come sviluppare e utilizzare i dati storici (cioè i criteri per l'inclusione e l'esclusione di dati nelle serie storiche e i criteri di accettabilità per un determinato esperimento) sono reperibili nella letteratura scientifica (24). Se nell'ambito delle prove di competenza (descritte al paragrafo 13) il laboratorio non porta a termine un numero sufficiente di esperimenti per stabilire una distribuzione dei controlli negativi statisticamente solida (cfr. il paragrafo 15), si può accettare che la distribuzione sia definita durante i primi test di routine. Questo approccio deve seguire le raccomandazioni formulate nella letteratura (24), e i risultati dei controlli negativi ottenuti in questi esperimenti devono essere coerenti con i dati di controllo negativi pubblicati. Eventuali modifiche apportate al protocollo sperimentale devono essere considerate in base alle loro ripercussioni sulla coerenza fra i nuovi dati e le esistenti banche dati di controlli storici del laboratorio. Solo le incongruenze significative dovrebbero portare alla creazione di una nuova banca dati di controlli storici, se il parere di un esperto stabilisce che vi è una differenza rispetto alla distribuzione precedente (cfr. il paragrafo 15). Durante la costituzione di questa nuova banca dati, il laboratorio non ha necessariamente bisogno di una banca dati completa di controlli negativi per permettere lo svolgimento di una prova, a condizione che esso possa dimostrare che i valori dei controlli negativi paralleli rimangano coerenti con la precedente banca dati o con i dati corrispondenti pubblicati. I dati sui controlli negativi devono riguardare l'incidenza degli eritrociti immaturi micronucleati in ogni animale. I controlli negativi paralleli dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % della distribuzione della banca dati dei controlli negativi storici del laboratorio. Qualora i dati dei controlli negativi paralleli non rientrino nei limiti del controllo al 95 % possono essere accettabili per l'inserimento nella distribuzione dei controlli storici purché non siano outlier estremi e sia provato che il sistema di prova è “sotto controllo” (cfr. il paragrafo 15) e che non si sono verificati errori tecnici o umani. DESCRIZIONE DEL METODO Preparazione Selezione delle specie animali Di preferenza sono utilizzati individui adulti, giovani e in buona salute, provenienti da ceppi di animali da laboratorio. Si può ricorrere a topi, ratti, o ad altre specie idonee di mammiferi. Quando si utilizza il sangue periferico, occorre accertare che la rimozione splenica delle cellule micronucleate dalla circolazione non comprometta l'individuazione dei micronuclei indotti nelle specie selezionate. Questo è stato chiaramente dimostrato per il sangue periferico del topo e del ratto (2). L'utilizzazione di specie diverse dai ratti e dai topi deve essere scientificamente giustificata nella relazione. Se si ricorre a specie diverse dai roditori, viene raccomandato di integrare la misurazione dei micronuclei indotti dalle aberrazioni cromosomiche del midollo osseo in un'altra prova di tossicità pertinente. Condizioni di stabulazione e alimentazione degli animali Per i roditori, la temperatura dello stabulario deve essere mantenuta a 22 °C (± 3 °C). Idealmente l'umidità relativa deve essere del 50-60 %. Deve comunque essere come minino del 40 % e non deve preferibilmente superare il 70 %, tranne che nel corso delle pulizie degli ambienti. L'illuminazione deve essere artificiale, con una sequenza di 12 ore di luce e 12 d'oscurità. Per l'alimentazione, attenersi alle diete convenzionali da laboratorio con una quantità illimitata di acqua potabile. La scelta della dieta può essere influenzata dalla necessità di garantire un'adeguata miscela della sostanza in esame, se somministrata per questa via. Se non ci si aspetta alcun comportamento aggressivo i roditori vanno alloggiati in piccoli gruppi (non più di cinque in ogni gabbia) dello stesso sesso e gruppo di trattamento, preferibilmente in gabbie a fondo pieno con adeguato arricchimento ambientale. Gli animali possono essere alloggiati individualmente solo se ciò è giustificato sotto il profilo scientifico. Preparazione degli animali Sono generalmente utilizzati animali adulti, giovani e sani (i roditori hanno idealmente un'età di 6-10 settimane all'inizio del trattamento, ma sono accettati anche animali di età un pÓ più avanzata), che vengono suddivisi a caso in gruppi di controllo e di trattamento. I singoli animali sono identificati univocamente applicando un metodo incruento e il meno possibile invasivo (cioè inanellamento, etichettatura, applicazione di un microchip o identificazione biometrica, evitando il taglio delle orecchie o la falangectomia) e devono essere acclimatati alle condizioni di laboratorio per almeno 5 giorni. Le gabbie devono essere sistemate in modo da ridurre al minimo eventuali effetti dovuti alla loro collocazione. Deve essere evitata ogni contaminazione incrociata fra il controllo positivo e la sostanza chimica in esame. All'inizio dello studio la variazione di peso degli animali deve essere minima e non superare il ± 20 % del peso medio per ciascun sesso. Preparazione delle dosi Le sostanze solide in esame vanno sciolte o poste in sospensione in appropriati solventi o mezzi disperdenti o mescolate alla dieta o all'acqua da bere prima di essere somministrate agli animali. Le sostanze liquide possono essere somministrate direttamente o diluite prima del trattamento. In caso di esposizione per via inalatoria, le sostanze possono essere somministrate sotto forma di gas, vapore o aerosol solido o liquido, in funzione delle loro proprietà fisico-chimiche. Vanno usati preparati freschi della sostanza, a meno che i dati sulla stabilità dimostrino che la conservazione è accettabile e definiscano le adeguate condizioni di conservazione. Condizioni sperimentali Solvente/mezzo disperdente Il solvente/mezzo disperdente non deve produrre effetti tossici ai livelli di dose usati e non deve poter reagire chimicamente con la sostanza in esame. L'uso di solventi/mezzi disperdenti poco noti è ammesso solo se suffragato da dati che ne provino la compatibilità. Si raccomanda di prendere in primo luogo in considerazione, se possibile, l'uso di un solvente/mezzo disperdente acquoso. Fra gli esempi di solventi/mezzi disperdenti compatibili e comunemente usati si possono menzionare l'acqua, la soluzione fisiologica, la soluzione di metilcellulosa, la soluzione di carbossimetilcellulosa sodica, l'olio di oliva e l'olio di mais. In mancanza di dati di controllo storici o pubblicati che dimostrino che un solvente/mezzo disperdente atipico prescelto non induce micronuclei o altri effetti nocivi, va effettuato uno studio iniziale per stabilire l'accettabilità del controllo del solvente/mezzo disperdente. Controlli Controlli positivi Ogni prova deve generalmente includere un gruppo di animali trattati con una sostanza usata per i controlli positivi. Questa condizione può essere eliminata quando il laboratorio di prova ha dimostrato la sua competenza nello svolgimento della prova e ha stabilito l'intervallo dei controlli positivi storici. Quando manca un gruppo di controllo positivo parallelo, in ogni esperimento devono essere inclusi controlli del conteggio (vetrini fissati e non colorati o campioni di sospensione cellulare, se appropriato ai fini del metodo di conteggio). Questo può avvenire includendo nello studio campioni di riferimento idonei ottenuti e conservati in occasione di un esperimento separato di controllo positivo svolto periodicamente (ad es. ogni 6-18 mesi); ad esempio, durante la verifica della competenze e successivamente, se necessario, su base periodica. Le sostanze chimiche per i controlli positivi devono portare, in modo affidabile, a un aumento individuabile della frequenza dei micronuclei al di là del livello spontaneo. Quando si ricorre al conteggio manuale al microscopio, le dosi dei controlli positivi devono essere scelte in modo che gli effetti siano chiari ma non rivelino immediatamente all'analista l'identità dei campioni codificati. È ammissibile che le sostanze per i controlli positivi siano somministrate per una via diversa da quella della sostanza chimica in esame, usando un programma di trattamento diverso, e che il campionamento avvenga in un unico momento. È anche ammissibile, se appropriato, l'uso per i controlli positivi di sostanze di una classe chimica correlata. Nella tabella 1 figurano esempi di sostanze chimiche per i controlli positivi. Tabella 1. Esempi di sostanze chimiche per i controlli positivi. Sostanze chimiche e CASRN Metansolfonato di etile [CASRN 62-50-0] Metansolfonato di metile [CASRN 66-27-3] Etilnitrosourea [CASRN 759-73-9] Mitomicina C [CASRN 50-07-7] Ciclofosfamide (monoidrato) [CASRN 50-18-0 (CASRN 6055-19-2)] Trietilenmelammina [CASRN 51-18-3] Colchicina [CASRN 64-86-8] o vinblastina [CASRN 865-21-4] — come aneugeni Controlli negativi In ogni fase del campionamento devono essere inclusi animali del gruppo di controllo negativo, che sono gestiti nello stesso modo dei gruppi di trattamento, ma a cui non viene somministrata la sostanza chimica in esame. Se per la somministrazione della sostanza in esame si usa un solvente / mezzo disperdente, anche il gruppo di controllo deve riceverlo. Tuttavia, se da dati dei controlli negativi storici risulta una variabilità intraspecifica e una frequenza delle cellule con micronuclei coerente ad ogni fase del campionamento per il laboratorio di prova, può essere sufficiente un solo campionamento per il controllo negativo. In tal caso, esso deve avvenire al momento del primo campionamento effettuato nell'ambito dello studio. Se viene usato sangue periferico, un campione prelevato prima del trattamento può essere accettato al posto del controllo negativo parallelo per gli studi di breve durata, qualora i dati ottenuti siano coerenti con la banca dei dati di controllo storici del laboratorio di prova. Per i ratti è stato dimostrato che il campionamento pre-trattamento di piccoli volumi (ad es. < 100 μl/al giorno) ha un impatto minimo sulla frequenza di fondo dei micronuclei (25). PROCEDURA Numero e sesso degli animali In generale, la risposta dei micronuclei è simile nei maschi e nelle femmine degli animali, e, pertanto, la maggior parte degli studi potrebbe pertanto venire effettuata sull'uno o sull'altro sesso (26). L'esistenza di dati che dimostrano differenze rilevanti fra i maschi e le femmine (ad es. differenze riguardanti la tossicità sistemica, il metabolismo, la biodisponibilità, la tossicità del midollo osseo, ecc., incluso ad es. in un range-finding test) incoraggerebbero l'uso di entrambi i sessi. Nella fattispecie potrebbe essere appropriato effettuare uno studio su entrambi i sessi, ad es. come parte di uno studio di tossicità a dose ripetuta. Nel caso si effettuino prove su entrambi i sessi potrebbe essere adeguato ricorrere al metodo fattoriale. Nell'Appendice 2 figurano precisazioni sulle modalità di analisi dei dati in base a tale metodo. All'inizio dello studio, la dimensione stabilita del gruppo deve consentire di disporre, in ogni gruppo, di almeno 5 animali analizzabili dello stesso sesso, o di ogni sesso se sono usati entrambi. Se l'esposizione umana a sostanze chimiche è specifica per un sesso, ad esempio nel caso di alcuni prodotti farmaceutici, il test deve essere eseguito su animali di tale sesso. A titolo di orientamento sul numero massimo di animali tipicamente necessario, uno studio sul midollo osseo effettuato conformemente ai parametri stabiliti al paragrafo 37, con tre gruppi di trattamento e controlli negativi e positivi paralleli (ogni gruppo composto da cinque animali dello stesso sesso), richiederebbe fra i 25 e i 35 animali. Livelli di dose Se viene effettuato uno studio preliminare di range-finding poiché non sono già disponibili dati appropriati per orientare la scelta delle dosi, tale studio deve essere svolto nello stesso laboratorio, usando specie, ceppi, sesso e un regime di trattamento identici a quelli che saranno utilizzati nello studio principale (27). Lo studio preliminare serve a individuare la dose massima tollerata (MTD), definita come la dose più alta che sarà tollerata per la durata della prova senza che appaia una tossicità che limita lo studio (inducendo ad esempio un calo del peso corporeo o citotossicità del sistema emopoietico, ma senza provocare morte o segni di dolore, sofferenza o stress che rendano necessaria una soppressione incruenta (28)). La dose massima può essere definita anche come la dose che produce tossicità nel midollo osseo (ad es. una diminuzione della proporzione di eritrociti immaturi rispetto al numero totale di eritrociti nel midollo osseo o nel sangue periferico di più del 50 %, ma senza che questa proporzione si riduca a meno del 20 % del valore di controllo). Tuttavia, dall'analisi delle cellule con reazione positiva al CD71 nella circolazione sanguigna periferica (con citometria a flusso), emerge che questa frazione di eritrociti immaturi molto giovani risponde alle sostanze tossiche più rapidamente della più ampia schiera di eritrociti immaturi positivi all'RNA. Pertanto, può apparire una tossicità apparente maggiore in caso di sperimentazioni che prevedono un'esposizione acuta seguita dall'esame della frazione di eritrociti immaturi positivi al CD71 rispetto alle metodologie che individuano gli eritrociti immaturi sulla base del contenuto di RNA. Per questa ragione, quando l'esperimento prevede cinque o meno giorni di trattamento, il livello di dose più elevato, e tossico, della sostanza chimica in esame può essere definito come la dose che provoca una riduzione statisticamente significativa della proporzione degli eritrociti immaturi positivi al CD71 rispetto al numero totale di eritrociti, senza che questa proporzione scenda al di sotto del 5 % del valore di controllo (29). Le sostanze chimiche che comportano saturazione delle caratteristiche tossicocinetiche o che inducono processi di detossificazione che possono portare a una diminuzione dell'esposizione dopo una somministrazione a lungo termine possono essere considerate eccezioni rispetto ai criteri di definizione della dose e vanno valutate caso per caso. Per ottenere informazioni sulla relazione dose-risposta, uno studio completo deve includere un gruppo di controllo negativo e almeno tre livelli di dose, generalmente separati da un fattore 2, ma non più di 4. Se la sostanza chimica in esame non provoca alcuna tossicità nell'ambito di un test di range-finding o secondo quanto emerge dai dati esistenti, la dose massima per periodo di somministrazione di 14 giorni o più deve essere di 1 000 mg/kg di peso corporeo al giorno o, per periodi di somministrazione di meno di 14 giorni, di 2 000 mg/kg di peso corporeo al giorno. Se la sostanza chimica in esame provoca invece tossicità, la MTD deve corrispondere alla dose più elevata somministrata, e i livelli di dose utilizzati devono essere compresi in un intervallo che va dalla tossicità massima a una tossicità modica o assente. Quando una tossicità del tessuto bersaglio (midollo osseo) è osservata a tutti i livelli di dose di prova, è consigliabile un ulteriore studio con dosi non tossiche. Gli studi volti a descrivere più pienamente le informazioni quantitative sulla relazione dose-risposta possono richiedere gruppi di trattamento supplementari. Per certi tipi di sostanze chimiche in esame (ad es. medicinali per uso umano) per le quali valgono specifici requisiti, i limiti possono variare. Prova limite Se gli esperimenti per determinare gli intervalli di dose o i dati disponibili relativi ai ceppi di animali affini indicano che un trattamento uguale o superiore alla dose limite (sotto descritta) non produce effetti tossici osservabili (comprese nessuna depressione della funzione del midollo osseo o altre manifestazioni di citotossicità del tessuto bersaglio), e se non si prevede genotossicità sulla base degli studi di genotossicità in vitro o dei dati relativi alle sostanze chimiche strutturalmente affini, uno studio completo con tre livelli di dose può non essere considerato necessario, purché sia stato dimostrato che la sostanza o le sostanze chimiche in esame raggiungono il tessuto bersaglio (midollo osseo). In tali casi può essere sufficiente un solo livello di dose, corrispondente alla dose limite. Quando la somministrazione avviene per 14 giorni o più, la dose limite è 1 000 mg/kg di peso corporeo al giorno. Per periodi di somministrazione di meno di 14 giorni, la dose limite è 2 000 mg/kg/ di peso corporeo al giorno. Somministrazione delle dosi Nella concezione di un saggio va considerata la via d'esposizione umana prevista. Pertanto possono essere scelti, se giustificati, vie di somministrazione quali l'alimentazione, l'acqua potabile, la via sottocutanea locale, la via endovenosa, la via orale (sonda gastrica), inalatoria, intratracheale, o l'impianto. In ogni caso, deve essere scelta la via che garantisce un'adeguata esposizione del o dei tessuti bersaglio. L'iniezione intraperitoneale non è generalmente raccomandata in quanto non si tratta di una via di esposizione umana prevista, e va utilizzata solo con una specifica giustificazione scientifica. Se la sostanza chimica in esame è miscelata nell'alimentazione o nell'acqua occorre fare attenzione, specialmente nei casi di dosaggio singolo, che il lasso di tempo fra l'assunzione del cibo e dell'acqua e il campionamento sia sufficiente per consentire l'individuazione degli effetti (cfr. il paragrafo 37). Il volume massimo di liquido somministrabile in un'unica volta con sonda gastrica o con iniezione dipende dalle dimensioni dell'animale da laboratorio. Esso non deve generalmente essere superiore a 1 ml/100 g di peso corporeo, tranne nel caso delle soluzioni acquose che possono essere somministrate in quantità pari a massimo 2 ml/100 g di peso corporeo. L'uso di volumi maggiori deve essere giustificato. Salvo nel caso di sostanze chimiche irritanti o corrosive, i cui effetti di norma tendono a esacerbarsi con l'aumentare della concentrazione, la variabilità del volume somministrato deve essere ridotta al minimo adeguando la concentrazione, in modo da garantire la somministrazione di un volume costante in relazione al peso corporeo per tutti i livelli di dose. Programma di trattamento Sono di preferenza effettuati 2 o più trattamenti, somministrati a intervalli di 24 ore, specialmente quando la prova viene integrata in altri studi di tossicità. Alternativamente, possono essere somministrati trattamenti singoli, se ciò è giustificato sul piano scientifico (ad es. qualora sia noto che le sostanze chimiche in esame bloccano il ciclo cellulare). Le sostanze chimiche in esame possono anche essere somministrate in prese frazionate, cioè due prese nello stesso giorno a distanza di qualche ora al massimo, per agevolare la somministrazione di un grosso volume. In queste situazioni, o in caso di somministrazione della sostanza chimica in esame per inalazione, il momento del campionamento deve essere programmato in base al momento di somministrazione dell'ultima dose o della fine dell'esposizione. La prova può essere realizzata su topi o su ratti in uno dei tre seguenti modi:
Qualora ciò sia pertinente e scientificamente giustificato, e per facilitare l'integrazione con altri test di tossicità, possono essere utilizzati altri regimi di dosaggio o di campionamento. Osservazioni Gli animali da laboratorio devono essere oggetto di osservazioni cliniche generali e i segni clinici devono essere registrati almeno una volta al giorno, preferibilmente alla stessa o alle stesse ore e tenendo conto del periodo di massima intensità degli effetti previsti dopo la somministrazione. Almeno due volte al giorno, durante il periodo di somministrazione, tutti gli animali vengono esaminati al fine di determinare la morbilità e la mortalità. Tutti gli animali devono essere pesati all'inizio della prova, almeno una volta alla settimana nel corso degli studi a dosi ripetute, e al momento della soppressione incruenta. Nelle prove la cui durata è di almeno una settimana, la misurazione del consumo di cibo va eseguita almeno con cadenza settimanale. Se la sostanza in esame viene somministrata con l'acqua, il consumo di acqua va misurato ad ogni cambio dell'acqua e almeno una volta alla settimana. Gli animali che manifestano segni di eccessiva, ma non letale, tossicità vanno soppressi in modo incruento prima della fine del periodo di saggio (28). In certe circostanze può essere controllata la temperatura corporea degli animali, poiché un'iper- o ipotermia indotta dal trattamento può falsare i risultati (32) (33) (34). Esposizione del tessuto bersaglio Ove ciò sia giustificato e non esistano altri dati sull'esposizione (cfr. il paragrafo 48), al o ai momenti idonei va prelevato un campione di sangue per esaminare i livelli delle sostanze chimiche in esame nel plasma, allo scopo di dimostrare l'avvenuta esposizione del midollo osseo). Preparazione del midollo osseo / del sangue Le cellule del midollo osseo vengono di solito prelevate dal femore o dalla tibia degli animali immediatamente dopo la soppressione incruenta. Generalmente le cellule sono prelevate, preparate e colorate secondo metodi prestabiliti. Conformemente alle norme pertinenti sul benessere degli animali possono essere ottenute piccole quantità di sangue periferico usando un metodo che permetta la sopravvivenza della cavia (come il prelievo dalla vena caudale o da altro vaso sanguigno adeguato), oppure tramite puntura cardiaca o prelievo da un vaso sanguigno principale dopo la soppressione incruenta. Sia per gli eritrociti ottenuti dal midollo osseo che per quelli ottenuti dal sangue periferico, a seconda del metodo di analisi, le cellule possono venire immediatamente sottoposte a un processo di colorazione sopravitale (16) (17) (18), vengono preparati strisci che sono poi colorati per l'analisi al microscopio, o fissati e opportunamente colorati per l'analisi citometrica a flusso. L'uso di un colorante specifico per il DNA [ad esempio, arancio di acridina (35) o Hoechst 33258 più pironina-Y (36)] può eliminare alcuni degli artefatti dovuti all'uso di un colorante non specifico per il DNA. Ciò non esclude l'uso di coloranti convenzionali (per esempio Giemsa per l'analisi microscopica). Si possono usare anche altri sistemi [per esempio colonne di cellulosa per la rimozione delle cellule nucleate (37) (38)], se ne è dimostrata la compatibilità con la preparazione dei campioni in laboratorio. Qualora questi metodi siano applicabili, per individuare la natura dei micronuclei (cromosoma/frammento cromosomico) possono essere usati anticorpi anticinetocore (39), FISH con sonde di DNA pancentromerico (40), o marcatura in situ mediante primer specifici per DNA pancentromerico, unitamente a un'adeguata colorazione del DNA (41), per stabilire se il meccanismo di induzione di micronuclei è dovuto a un'attività clastogenica e/o aneugenica. Possono infine essere usati altri metodi per distinguere i clastogeni dagli aneugeni, purché sia stata dimostrata la loro efficacia. Analisi (manuale e automatizzata) Tutti i vetrini o i campioni per l'analisi, compresi quelli dei controlli positivi e negativi, devono essere codificati indipendentemente prima di ogni tipo di analisi, e devono essere randomizzati in modo che l'analista manuale non conosca le condizioni del trattamento; questa codificazione non è necessaria quando si usano sistemi di conteggio automatizzato che non si basano sull'esame visivo e non possono essere influenzati dall'operatore. Per ogni animale, la percentuale di eritrociti immaturi rispetto agli eritrociti totali (immaturi + maturi) è determinata contando un totale di almeno 500 eritrociti per il midollo osseo e 2 000 eritrociti per il sangue periferico (42). Per determinare l'incidenza degli eritrociti immaturi micronucleati devono essere esaminati almeno 4 000 eritrociti immaturi per animale (43). Se la banca dati dei controlli negativi storici indica che la frequenza di fondo media degli eritrociti immaturi micronucleati nel laboratorio di prova è <0,1 %, occorre prendere in considerazione l'opportunità di esaminare cellule supplementari. Quando si analizzano i campioni, la percentuale di eritrociti immaturi rispetto al numero totale di eritrociti negli animali trattati non deve essere inferiore al 20 % della proporzione verificata nel gruppo di controllo mezzo disperdente/solvente con l'analisi microscopica, e non deve essere inferiore al 5 % circa della proporzione verificata nel gruppo di controllo mezzo disperdente/solvente con l'analisi citometrica e il conteggio degli eritrociti immaturi positivi al CD71+ (cfr. il paragrafo 31) (29). Ad esempio, per un saggio su midollo osseo con esame al microscopio, se la percentuale di controllo degli eritrociti immaturi nel midollo osseo è del 50 %, il limite superiore di tossicità equivarrebbe al 10 % di eritrociti immaturi. Poiché la milza del ratto blocca e distrugge gli eritrociti micronucleati, per mantenere un'alta sensibilità delle analisi per l'esame del sangue periferico del ratto è preferibile limitare l'analisi degli eritrociti immaturi micronucleati alla frazione più giovane. Quando si utilizzano metodi di analisi automatizzata, questi eritrociti più immaturi possono essere individuati in base al loro contenuto elevato di RNA, o all'alto livello di recettori della transferrina (CD71+) espressi sulla loro superficie (31). Tuttavia, il raffronto diretto di vari metodi di colorazione ha mostrato che possono essere ottenuti risultati soddisfacenti con varie metodologie, compresa la colorazione classica con arancio di acridina (3) (4). DATI E RELAZIONE Trattamento dei risultati I dati relativi a ciascun animale vanno presentati sotto forma di tabella. Il numero degli eritrociti immaturi e degli eritrociti immaturi con micronuclei, nonché la percentuale degli eritrociti immaturi sul totale degli eritrociti, devono essere elencati separatamente per ciascun animale. Quando i topi sono trattati per 4 settimane o più, devono essere anche forniti, se raccolti, i dati sul numero e la percentuale degli eritrociti maturi micronucleati. Nella relazione devono anche figurare i dati riguardanti la tossicità animale e i segni clinici. Criteri di accettabilità L'accettabilità delle prove si basa sui seguenti criteri:
Valutazione e interpretazione dei risultati A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è ritenuta chiaramente positiva se:
Se in un particolare momento del campionamento viene esaminata solo la dose massima, la sostanza chimica in esame è ritenuta chiaramente positiva se vi è un aumento statisticamente significativo rispetto al controllo negativo parallelo, e i risultati sono al di fuori della distribuzione dei dati di controllo negativi storici (ad es. limiti del controllo al 95 % in base alla distribuzione di Poisson). Raccomandazioni relative agli appropriati metodi statistici figurano nella letteratura (44) (45) (46) (47). Quando si effettua un'analisi della relazione dose-risposta, devono essere analizzati almeno tre gruppi di trattamento trattati. Nei test statistici l'unità sperimentale deve essere l'animale. L'ottenimento di risultati positivi nel test del micronucleo indica che la sostanza chimica in esame induce la formazione di micronuclei, che sono il risultato di danni cromosomici o danni all'apparato mitotico degli eritroblasti nelle specie in esame. Nel caso in cui sia stata effettuata una prova per individuare i centromeri nei micronuclei, una sostanza chimica in esame che produca micronuclei contenenti centromeri (DNA centromerico o cinetocore, indicativi di un'intera perdita cromosomica) è da considerarsi aneugenica. A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è ritenuta chiaramente negativa se in tutte le condizioni sperimentali esaminate:
Raccomandazioni relative ai metodi statistici più appropriati figurano nella letteratura (44) (45) (46) (47). L'esposizione del midollo osseo alla sostanza chimica in esame può essere dimostrata, ad esempio, da un calo del rapporto fra eritrociti immaturi e maturi, o dalla misurazione del livello delle sostanze in esame nel plasma o nel sangue In caso di somministrazione per via endovenosa, la prova dell'esposizione non è necessaria. In alternativa, l'esposizione del midollo osseo può essere dimostrata ricorrendo ai dati ADME, ottenuti nell'ambito di uno studio indipendente usando la stessa via di somministrazione e le stesse specie. L'ottenimento di risultati negativi indica che, nelle condizioni di prova, la sostanza chimica in esame non induce la formazione di micronuclei negli eritrociti immaturi delle specie utilizzate. Non è necessario verificare una risposta chiaramente positiva o chiaramente negativa. Nei casi in cui la risposta non sia chiaramente negativa né chiaramente positiva, per poter stabilire la rilevanza biologica di un risultato (ad es. un aumento debole o marginale), i dati devono venire sottoposti al giudizio di esperti e/o a indagini più approfondite sugli esperimenti portati a termine. In alcuni casi può essere utile analizzare più cellule o ripetere l'esperienza in condizioni sperimentali modificate. In rari casi, anche dopo ulteriori indagini, i dati non permetteranno di concludere se la sostanza chimica in esame produce risultati positivi o negativi, e lo studio dovrà pertanto essere dichiarato ambiguo. Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti:
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Centromero : regione/i di un cromosoma sede di attacco del fuso durante la divisione cellulare, che permette il corretto movimento dei cromosomi figli verso i poli delle cellule figlie. Sostanza chimica : sostanza o miscela. Eritroblasto : stadio precoce dello sviluppo di un eritrocita, immediatamente precedente la formazione dell'eritrocita immaturo, quando la cellula contiene ancora il nucleo. Cinetocore : struttura proteica che si forma sul centromero delle cellule eucariotiche, che collega il cromosoma ai polimeri microtubolari del fuso mitotico durante la mitosi e la meiosi, e che durante la divisione cellulare serve a separare i cromatidi fratelli. Micronuclei : piccoli nuclei, distinti e soprannumerari rispetto al nucleo principale delle cellule, prodotti durante la telofase della mitosi (meiosi) da frammenti residui di cromosomi o da cromosomi interi. Eritrocita normocromatico o maturo : un eritrocita completamente maturo che ha perso l'RNA residuo che rimane dopo l'enucleazione e/o ha perso altri marcatori cellulari a ciclo vitale breve che generalmente scompaiono a seguito dell'enucleazione, dopo la divisione finale dell'eritroblasto. Eritrocita policromatico o immaturo : eritrocita di nuova formazione, a uno stadio di sviluppo intermedio, che reagisce alle componenti blu e rosse dei coloranti classici del sangue, come la colorazione di Wright-Giemsa, in virtù della presenza di RNA residuo nella cellula di nuova formazione. Queste cellule di nuova formazione sono praticamente quasi identiche ai reticolociti, visualizzabili usando una colorazione vitale che fa precipitare in un “reticolo” l'RNA residuo. Altri metodi, compresa la colorazione monocromatica dell'RNA con colorante fluorescente o l'evidenziazione dei marcatori di superficie a vita breve come il CD71 con anticorpi fluorescenti, sono ora spesso usati per individuare gli eritrociti di nuova formazione. Gli eritrociti policromatici, i reticolociti, e gli eritrociti positivi al marcatore CD71 sono tutti eritrociti immaturi, ciascuno corrispondente a uno stadio di sviluppo leggermente diverso. Reticolocito : eritrocita di nuova formazione. L'RNA cellulare residuo precipita sotto forma di un caratteristico “reticolo”, evidenziabile con un colorante vitale. Nei reticolociti e negli eritrociti policromatici l'età cellulare ha una distribuzione simile. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Appendice 2 METODO FATTORIALE PER INDIVIDUARE LE DIFFERENZE FRA I SESSI NEL SAGGIO IN VIVO DEL MICRONUCLEO Metodo fattoriale e analisi Questo modello prevede di esporre 5 maschi e 5 femmine a ciascuna concentrazione sperimentale e comporta pertanto l'utilizzo di un minimo di 40 animali (20 maschi e 20 femmine più i relativi controlli positivi). Il modello qui presentato — una delle forme semplici del modello fattoriale — è equivalente a un'analisi della varianza a due fattori, in cui il sesso e il livello delle concentrazioni costituiscono i fattori principali. I dati possono essere analizzati utilizzando diversi programmi statistici standard, quali SPSS, SAS, STATA, Genstat o il programma R. A partire dall'insieme dei dati si determina la variabilità tra i sessi e le concentrazioni, nonché la variabilità relativa alle interazioni tra sessi e concentrazioni. Ciascuno di questi aspetti è quindi valutato in rapporto alla stima della variabilità tra gli animali ripartiti nei gruppi di animali dello stesso sesso esposti alle stesse concentrazioni. Maggiori informazioni su questa metodologia sono reperibili in diversi manuali standard di statistica (cfr. bibliografia), nonché nelle schede “di aiuto” fornite con i programmi statistici. In seguito viene esaminata l'interazione sesso x concentrazione in una tabella ANOVA (6). In assenza di un termine di interazione significativo la combinazione dei valori inter-sessi o inter-livelli di concentrazione consente di effettuare test statistici validi tra i livelli, basandosi sul termine di variabilità intra-gruppo combinata di 'ANOVA. L'analisi prosegue suddividendo la variabilità stimata tra le concentrazioni in modo da ottenere contrasti che permettano di stabilire contrasti lineari e quadratici delle risposte per l'insieme dei livelli di concentrazione. Quando si riscontra un'interazione significativa sesso x concentrazione, questo termine può essere a sua volta in ripartito in contrasti di interazione lineare x sesso e quadratica x sesso. In questo modo si può verificare se le risposte alle concentrazioni sono parallele per i due sessi o se vi è una differenza riconducibile al sesso. La stima della variabilità intra-gruppo combinata può essere utilizzata per verificare lo scarto tra le medie, confrontandole due a due. I confronti possono essere effettuati tra le medie per i due sessi e tra le medie dei diversi livelli delle concentrazioni (come, ad esempio, confronti tra i livelli dei controlli negativi). Nei casi in cui si registra un'interazione significativa i confronti possono essere effettuati tra le medie di concentrazioni diverse per lo stesso sesso o tra le medie dei due sessi a parità di concentrazione. Riferimenti Numerosi manuali di statistica esaminano la teoria, la concezione, la metodologia, l'analisi e l'interpretazione dei modelli fattoriali, dall'analisi più semplice, a due fattori, alle forme più complesse utilizzate per la metodologia Design of Experiment. Di seguito ne è riportato un elenco non completo. Alcuni manuali forniscono esempi di modelli comparabili e, in alcuni casi, forniscono un codice che permette di effettuare l'analisi utilizzando diversi programmi.
|
|
6) |
Nella parte B, il capitolo B.15. è soppresso. |
|
7) |
Nella parte B, il capitolo B.16. è soppresso. |
|
8) |
Nella parte B, il capitolo B.18. è soppresso. |
|
9) |
Nella parte B, il capitolo B.19. è soppresso. |
|
10) |
Nella parte B, il capitolo B.20. è soppresso. |
|
11) |
Nella parte B, il capitolo B.24. è soppresso. |
|
12) |
Nella parte B, il capitolo B.47. è sostituito dal seguente: «B.47 Metodo di prova dell'opacità e della permeabilità della cornea nei bovini per l'identificazione di i) sostanze chimiche che inducono gravi lesioni oculari e ii) sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari INTRODUZIONE Questo metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 437 (2013). Il metodo di prova dell'opacità e della permeabilità della cornea nei bovini (Bovine Corneal Opacity and Permeability, BCOP) è un metodo di prova valutato dal Comitato di coordinamento interagenzia per la convalida dei metodi alternativi (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM), in collaborazione con il Centro europeo per la convalida di metodi alternativi (European Centre for the Validation of Alternative Methods, ECVAM) e del corrispondente organismo del Giappone (Japanese Centre for the Validation of Alternative Methods, JaCVAM) nel 2006 e nel 2010 (1) (2). Nella prima valutazione il metodo di prova BCOP è stato valutato per la sua utilità nell'identificare sostanze chimiche (sostanze e miscele) che inducono gravi lesioni oculari (1). Nella seconda valutazione il metodo di prova BCOP è stato valutato per la sua utilità nell'identificare sostanze chimiche (sostanze e miscele) non classificate che inducono irritazione oculare o gravi lesioni oculari (2). La banca dati di validazione del metodo BCOP conteneva complessivamente 113 sostanze e 100 miscele (2) (3). Tali valutazioni e i relativi esami inter pares hanno permesso di concludere che il metodo di prova può identificare correttamente le sostanze chimiche (sostanze e miscele) che inducono gravi lesioni oculari (categoria 1) nonché quelle che non richiedono classificazione per irritazione oculare o gravi lesioni oculari, secondo la definizione del Sistema generale armonizzato di classificazione e di etichettatura dei prodotti chimici delle Nazioni Unite (Globally Harmonized System of Classification and Labelling of Chemicals — UN GHS) (4) e del regolamento (CE) n. 1272/2008 relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele (CLP) (7) e la validità scientifica del metodo di prova è stata riconosciuta per entrambe le finalità. Per gravi lesioni oculari s'intendono lesioni dei tessuti oculari o un grave deterioramento della vista conseguente all'applicazione di una sostanza chimica in esame sulla superficie anteriore dell'occhio, non totalmente reversibili entro 21 giorni dall'applicazione. Le sostanze chimiche in esame che inducono gravi lesioni oculari sono classificate nella categoria UN GHS 1. Le sostanze chimiche non classificate in termini di irritazione oculare o gravi lesioni oculari sono definite come quelle sostanze che non soddisfano quanto prescritto per la classificazione nella categoria UN GHS 1 o 2 (2A o 2B), ovvero sono definite “senza categoria” da tale sistema. Il presente metodo di prova include l'uso raccomandato e le limitazioni del metodo di prova BCOP sulla base delle valutazioni ottenute. Le principali differenze fra la versione originale del 2009 e quella aggiornata nel 2013 della linea guida dell'OCSE riguardano fra l'altro: l'uso del metodo di prova BCOP per identificare le sostanze chimiche che non richiedono classificazione secondo il sistema GHS delle Nazioni Unite (paragrafi 2 e 7); chiarimenti circa l'applicabilità del metodo di prova BCOP per la prova di alcoli, chetoni e sostanze solide (paragrafi 6 e 7) e delle sostanze e delle miscele (paragrafo 8); chiarimenti sulle modalità di prova delle sostanze tensioattive e delle miscele contenenti tensioattivi (paragrafo 28); aggiornamenti e chiarimenti relativi ai controlli positivi (paragrafi 39 e 40); un aggiornamento dei criteri decisionali relativi al metodo di prova BCOP (paragrafo 47); un aggiornamento dei criteri di accettazione dello studio (paragrafo 48); un aggiornamento degli elementi della relazione sulla prova (paragrafo 49); un aggiornamento delle definizioni contenute nell'appendice 1; l'aggiunta dell'appendice 2 relativa alla capacità predittiva del metodo di prova BCOP secondo diversi sistemi di classificazione; un aggiornamento dell'appendice 3 relativa all'elenco delle sostanze chimiche per la verifica della prestazione; e un aggiornamento dell'appendice 4 relativa al supporto corneale per il metodo di prova BCOP (paragrafo 1) e all'opacimetro (paragrafi 2 e 3). Attualmente è generalmente riconosciuto che nel prossimo futuro nessuna singola prova di irritazione oculare in vitro sarà in grado di sostituire il test di Draize in vivo per prevedere tutta la gamma di irritazione per diverse classi chimiche. Tuttavia combinazioni strategiche di diversi metodi alternativi nell'ambito di una strategia di prova (sequenziale) possono sostituire il test oculare di Draize (5). L'approccio top-down (5) è usato quando, in base alle informazioni esistenti, si prevede che una sostanza chimica abbia un elevato potenziale di irritazione, mentre l'approccio bottom-up (5) è usato quando, in base alle informazioni esistenti, si prevede che una sostanza chimica non causi un'irritazione oculare sufficiente da richiedere una classificazione. Il metodo di prova BCOP è un metodo di prova in vitro che può essere usato in alcune circostanze e con limitazioni specifiche per la classificazione dei rischi oculari e l'etichettatura delle sostanze chimiche. Anche se da solo non è considerato un valido sostituto del test in vivo sugli occhi dei conigli, il metodo di prova BCOP è raccomandato come fase iniziale nell'ambito di una strategia sperimentale, quale l'approccio top-down proposto da Scott et al. (5) per identificare le sostanze chimiche che inducono gravi lesioni oculari, ossia le sostanze chimiche da classificare nella categoria UN GHS 1, senza effettuare ulteriori prove (4). Il metodo di prova BCOP è altresì raccomandato per identificare le sostanze chimiche che non richiedono una classificazione per irritazione oculare o per gravi lesioni oculari, secondo la definizione del sistema GHS delle Nazioni Unite (“senza categoria” nel sistema UN GHS) (4) nell'ambito di una strategia di prova quale l'approccio bottom-up (5). Tuttavia, la classificazione definitiva di una sostanza chimica cui non si attribuisce la possibilità di causare gravi lesioni oculari o non classificata per irritazione oculare/gravi lesioni oculari con il metodo di prova BCOP richiederebbe prove supplementari (in vitro e/o in vivo). Scopo di questo metodo di prova è descrivere le procedure impiegate per valutare il potenziale di pericolo per gli occhi di una sostanza chimica in esame sulla base della capacità di tale sostanza di indurre opacità e aumentare la permeabilità di una cornea isolata di bovino. Gli effetti tossici sulla cornea sono misurati attraverso: i) diminuzione della trasmissione della luce (opacità), e ii) aumento del passaggio del colorante a base di fluorosceina sodica (permeabilità). Le valutazioni di opacità e permeabilità della cornea successivamente all'esposizione a una sostanza chimica in esame vengono combinate per ottenere un punteggio di irritazione in vitro (In Vitro Irritancy Score — IVIS), utilizzato per classificare il livello di irritazione provocato dalla sostanza chimica in esame. Le definizioni di questi parametri figurano nell'appendice 1. CONSIDERAZIONI INIZIALI E LIMITI Questo metodo di prova è basato sul protocollo del metodo di prova ICCVAM BCOP (6) (7), in origine elaborato dalle informazioni ottenute dal protocollo dell'Institute for in vitro Sciences (IIVS) e dal protocollo INVITTOX 124 (8). Quest'ultimo rappresenta il protocollo usato nello studio di prevalidazione condotto negli anni 1997-1998 e finanziato dalla Comunità europea. Entrambi i protocolli erano basati sul metodo di prova BCOP inizialmente trattato da Gautheron et al. (9). Il metodo di prova BCOP può essere usato per identificare le sostanze chimiche che inducono gravi lesioni oculari, secondo la definizione UN GHS, ossia le sostanze chimiche da classificare nella categoria UN GHS 1 (4). Se usato a tali fini, il metodo di prova BCOP presenta un'accuratezza complessiva del 79 % (150/191), una percentuale di falsi positivi del 25 % (32/126) e una percentuale di falsi negativi del 14 % (9/65), rispetto ai dati ottenuti con il test in vivo sugli occhi dei conigli classificati secondo il sistema di classificazione UN GHS (3) (cfr. appendice 2, tabella 1). Se le sostanze chimiche in esame nell'ambito di alcune classi chimiche (per es. alcoli, chetoni) o fisiche (per es. solidi) sono escluse dalla banca dati, il metodo di prova BCOP presenta un'accuratezza complessiva dell'85 % (111/131), una percentuale di falsi positivi del 20 % (16/81) e una percentuale di falsi negativi dell'8 % (4/50) secondo il sistema di classificazione UN GHS (3). Le lacune potenziali del metodo di prova BCOP quando è usato per identificare le sostanze chimiche che inducono gravi lesioni oculari (categoria UN GHS 1) sono basate sulle elevate percentuali di falsi positivi per gli alcoli e i chetoni nonché sulle elevate percentuali di falsi negativi per i solidi osservate nella banca dati di validazione (1)(2)(3). Tuttavia, poiché non tutti gli alcoli e i chetoni sono sovrastimati dal metodo di prova BCOP e alcuni sono stimati correttamente nella categoria UN GHS 1, questi due gruppi funzionali organici non sono considerati esterni all'ambito d'applicazione del metodo di prova. Spetta all'utilizzatore di questo metodo di prova decidere se un'eventuale sovrastima di un alcol o di un chetone possa essere accettata o se sia necessario effettuare prove supplementari con un metodo basato sul peso dell'evidenza. Per quanto attiene alle percentuali di falsi negativi per i solidi, è opportuno osservare che i solidi possono generare condizioni di esposizione variabili ed estreme nel test di Draize in vivo, il che può falsare il reale potenziale di irritazione (10). Va altresì osservato che nessuno dei falsi negativi identificati nella banca dati di validazione ICCVAM (2) (3), nell'ambito dell'identificazione delle sostanze chimiche che inducono gravi lesioni oculari (categoria UN GHS 1), ha prodotto un risultato IVIS ≤ 3, ossia il criterio usato per identificare una sostanza chimica in esame come “senza categoria” nel sistema UN GHS. Inoltre, i falsi positivi BCOP in questo contesto non sono fondamentali poiché tutte le sostanze chimiche in esame che producono un risultato 3 ≤ IVIS ≤ 55 sarebbero successivamente sottoposte a prova con altri metodi in vitro adeguatamente validati o, in ultima ratio sui conigli, a seconda dei requisiti regolamentari, mediante una strategia di saggi sequenziali con un metodo basato sul peso dell'evidenza. Considerato che alcuni solidi chimici sono stimati correttamente con il metodo di prova BCOP come appartenenti alla categoria UN GHS 1, nemmeno tale stato fisico è ritenuto esterno all'ambito di applicazione del metodo di prova. Gli sperimentatori potrebbero prendere in considerazione il ricorso a tale metodo per tutti i tipi di sostanze chimiche, laddove un risultato IVIS > 55 dovrebbe essere accettato come indicativo di una risposta che induce gravi lesioni oculari tale da essere classificata nella categoria UN GHS 1, senza ulteriori prove. Come già precisato, i risultati positivi ottenuti con alcoli o chetoni andrebbero tuttavia interpretati con cautela, considerato il rischio di sovrastima. Il metodo di prova BCOP può inoltre essere usato per identificare le sostanze chimiche che non richiedono una classificazione per irritazione oculare o per gravi lesioni oculari nell'ambito del sistema di classificazione UN GHS (4). Se usato a tali fini, il metodo di prova BCOP presenta un'accuratezza complessiva del 69 % (135/196), una percentuale di falsi positivi del 69 % (61/89) e una percentuale di falsi negativi dello 0 % (0/107), rispetto ai dati ottenuti con il test in vivo sugli occhi dei conigli classificati secondo il sistema di classificazione UN GHS (3) (cfr. appendice 2, tabella 2). La percentuale di falsi positivi ottenuta è notevolmente elevata (le sostanze chimiche “senza categoria” nel sistema UN GHS in vivo che producono un risultato IVIS > 3, cfr. paragrafo 47), ma in questo contesto non è fondamentale poiché tutte le sostanze chimiche in esame che producono un risultato 3 ≤ IVIS ≤ 55 sarebbero successivamente sottoposte a prova con altri metodi in vitro adeguatamente validati o, in ultima ratio sui conigli, a seconda dei requisiti regolamentari, mediante una strategia di saggi sequenziali con un metodo basato sul peso dell'evidenza. Il metodo di prova BCOP non mostra lacune specifiche per quanto riguarda le prove di alcoli, chetoni e solidi se il fine è identificare le sostanze chimiche che non richiedono classificazione per l'irritazione oculare o gravi lesioni oculari (“senza categoria” nel sistema UN GHS)(3). Gli sperimentatori potrebbero prendere in considerazione il ricorso a tale metodo di prova per tutti i tipi di sostanze chimiche, laddove un risultato negativo (IVIS ≤ 3) dovrebbe essere accettato come indicativo del fatto che non è necessaria alcuna classificazione (“senza categoria” nel sistema UN GHS). Poiché il metodo di prova BCOP può solo identificare correttamente il 31 % delle sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari, questo metodo di prova non dovrebbe costituire la prima scelta per avviare un approccio bottom-up (5), se sono disponibili altri metodi in vitro validati e accettati aventi un'analoga elevata sensibilità ma una maggiore specificità. La banca dati di validazione del metodo BCOP conteneva complessivamente 113 sostanze e 100 miscele (2) (3). Il metodo di prova BCOP è pertanto ritenuto applicabile per le prove sia delle sostanze che delle miscele. Il metodo di prova BCOP non è raccomandato per identificare le sostanze chimiche in esame da classificarsi come irritanti per gli occhi (categoria UN GHS 2 o 2A) o le sostanze chimiche in esame da classificarsi come moderatamente irritanti per gli occhi (categoria UN GHS 2B) a causa dell'elevato numero di sostanze chimiche appartenenti alla categoria UN GHS 1 sottoclassificate nelle categorie 2, 2A o 2B e di sostanze chimiche “senza categoria” nel sistema UN GHS sovraclassificate nelle categorie 2, 2A o 2B (2) (3). A tal fine possono essere necessarie ulteriori prove con un altro metodo adeguato. Tutte le procedure che prevedono l'impiego di occhi e cornee di bovini devono osservare le regolamentazioni e le procedure in vigore nella struttura che effettua l'analisi relativamente alla gestione dei materiali di derivazione animale che comprendono, tra l'altro, i tessuti e i liquidi tessutali. È consigliabile adottare le comuni norme di cautela osservate nei laboratori (11). Sebbene il metodo di prova BCOP non prenda in considerazione le lesioni della congiuntiva e dell'iride, esso riguarda gli effetti sulla cornea, che sono i principali fattori di classificazione in vivo nel sistema UN GHS. Il metodo di prova BCOP di per sé non consente di valutare la reversibilità delle lesioni corneali. Studi condotti sugli occhi dei conigli suggeriscono di esaminare la profondità iniziale di una lesione corneale per identificare alcuni tipi di effetti irreversibili (12). Tuttavia è necessario un approfondimento scientifico per comprendere in quale modo possano verificarsi effetti irreversibili non collegati all'elevato livello iniziale della lesione. Infine, il metodo di prova BCOP non consente di valutare il potenziale di tossicità sistemica associato all'esposizione attraverso l'occhio. Infine, il metodo di prova BCOP sarà aggiornato con cadenza regolare, mano a mano che si disporrà di ulteriori dati e informazioni. A titolo di esempio, l'istopatologia presenta un'utilità potenziale nei casi in cui è necessaria una caratterizzazione più completa della lesione corneale. Come delineato nel documento orientativo dell'OCSE “OECD Guidance Document n. 160” (13), gli utilizzatori sono invitati a conservare le cornee e a preparare campioni istopatologici che possono servire a elaborare una banca dati e criteri decisionali in grado di migliorare ulteriormente l'accuratezza di questo metodo di prova. Ai laboratori che ricorrono a questo tipo di metodo di prova per la prima volta si consiglia di utilizzare le sostanze chimiche di riferimento indicate nell'appendice 3. Un laboratorio può utilizzare tali sostanze chimiche per dimostrare le proprie competenze tecniche nell'esecuzione del metodo di prova BCOP prima di presentare i dati del metodo di prova BCOP a scopi regolamentari per la classificazione dei rischi. PRINCIPIO DEL METODO Il metodo di prova BCOP è un modello organotipico che mantiene a breve termine in vitro la normale funzionalità fisiologica e biochimica della cornea dei bovini. Questo metodo di prova è utilizzato per la valutazione dei danni prodotti dalla sostanza chimica in esame sulla base di misurazioni quantitative dei cambiamenti di opacità e permeabilità, effettuate rispettivamente con un opacimetro e uno spettrofotometro. Entrambe le misurazioni vengono impiegate per il calcolo del punteggio IVIS, utilizzato per assegnare una categoria di classificazione di pericolo di irritazione in vitro per la previsione del potenziale di irritazione oculare in vivo di una sostanza chimica in esame (cfr. criteri decisionali al paragrafo 48). Il metodo di prova BCOP impiega cornee isolate da occhi di bovini appena macellati. L'opacità corneale si misura con la quantità di luce trasmessa attraverso la cornea. La permeabilità si misura con la quantità di colorante a base di fluoresceina sodica che attraversa l'intero spessore della cornea ed è rilevata nel mezzo della camera posteriore. Le sostanze chimiche in esame vengono applicate alla superficie epiteliale della cornea e mediante aggiunta nella camera anteriore del supporto corneale. L'appendice 4 riporta una descrizione e un diagramma del supporto corneale utilizzato per il metodo di prova BCOP. I supporti corneali possono essere acquistati sul mercato da diversi fornitori o possono essere costruiti. Origine ed età degli occhi dei bovini e selezione delle specie animali I bovini inviati ai macelli vengono di norma abbattuti per il consumo umano o altri usi commerciali. Le cornee utilizzate per il metodo di prova BCOP provengono esclusivamente da animali sani ritenuti idonei a essere immessi nella catena alimentare umana. Poiché i capi di bestiame sono di peso diverso a seconda della razza, dell'età e del sesso, non vi sono raccomandazioni relative al peso dell'animale al momento della macellazione. L'impiego di animali di età diversa può comportare variazioni nelle dimensioni della cornea. Le cornee di diametro orizzontale > 30,5 mm e spessore corneale centrale (central corneal thickness — CCT) ≥ 1 100 μm si ottengono di norma da bovini di età superiore a otto anni, mentre le cornee che presentano un diametro orizzontale < 28,5 mm e un valore di CCT < 900 μm derivano solitamente da capi di bestiame di età inferiore a cinque anni (14). Per questo motivo non si utilizzano di norma occhi di bovini di età superiore a 60 mesi. Gli occhi dei bovini di età inferiore a 12 mesi di norma non sono impiegati, in quanto sono ancora in fase di sviluppo e i valori relativi allo spessore e al diametro della cornea sono notevolmente inferiori rispetto ai corrispondenti parametri rilevati nei bovini adulti. L'impiego di cornee di animali giovani (ovvero di età compresa fra 6 e 12 mesi) è tuttavia ammesso in quanto comporta alcuni vantaggi, quali la maggiore disponibilità, la limitata fascia di età e la riduzione dei rischi legati alla potenziale esposizione del lavoratore all'encefalopatia spongiforme bovina (15). Sarebbe utile valutare ulteriormente gli effetti delle dimensioni o dello spessore corneali sulla capacità di reazione a sostanze chimiche corrosive e irritanti, pertanto si raccomanda agli utilizzatori di indicare la stima dell'età e/o del peso degli animali da cui provengono le cornee utilizzate in uno studio. Prelievo e trasporto degli occhi in laboratorio Gli occhi sono prelevati dai dipendenti del mattatoio. Per minimizzare i danni meccanici e di altro tipo agli occhi, questi dovrebbero essere enucleati non appena possibile dopo la morte e raffreddati immediatamente dopo l'enucleazione e durante il trasporto. Durante il risciacquo della testa dell'animale è necessario che i dipendenti del mattatoio non facciano uso di detergenti al fine di evitare l'esposizione degli occhi a sostanze chimiche potenzialmente irritanti. Gli occhi vanno immersi completamente nella soluzione salina bilanciata di Hank raffreddata (Hank's Balanced Salt Solution, HBBS) in un contenitore di dimensioni adeguate e trasportati in laboratorio in modo da ridurre al minimo il relativo deterioramento e/o la contaminazione batterica. Gli occhi sono prelevati durante il processo di macellazione e potrebbero pertanto essere esposti al sangue o ad altri materiali biologici, compresi batteri e microorganismi di altra natura. Per questo motivo è importante assicurare che il rischio di contaminazione sia ridotto al minimo (per es. tenendo il contenitore degli occhi in ghiaccio durante la raccolta e il trasporto e aggiungendo antibiotici alla soluzione HBSS utilizzata per conservare gli occhi durante il trasporto [per es. penicillina a 100 IU/ml e streptomicina a 100 μg/ml]). È consigliabile ridurre al minimo l'intervallo temporale che intercorre fra il prelievo degli occhi e l'utilizzo delle cornee per il metodo di prova BCOP (idealmente, gli occhi andrebbero raccolti e utilizzati nell'arco della stessa giornata) e accertare che tale intervallo non pregiudichi i risultati del saggio. I risultati in questione si basano sui criteri di selezione degli occhi e sulle reazioni dei controlli positivi e negativi. Si suggerisce di utilizzare per il saggio occhi dello stesso gruppo di prelievo, ottenuti nella medesima giornata. Criteri di selezione egli occhi usati nel metodo di prova BCOP Giunti in laboratorio, gli occhi sono sottoposti a un attento esame volto a evidenziare la presenza di eventuali difetti, quali per es. aumento dell'opacità, graffi e neovascolarizzazione. Possono essere utilizzate solamente cornee provenienti da occhi privi di tali difetti. La qualità di ciascuna cornea è valutata anche durante le fasi successive del saggio. Vanno scartate le cornee che, dopo un periodo iniziale di un'ora per ristabilire l'equilibrio, presentano un valore di opacità superiore a sette unità o equivalente per l'opacimetro e per i supporti corneali (NOTA: l'opacimetro va tarato su valori di opacità standard impiegati per definire le unità di opacità, cfr. appendice 4). Ciascun gruppo di trattamento (sostanza chimica in esame, controlli negativi e positivi paralleli) consiste di un minimo di tre occhi. Tre cornee dovrebbero essere utilizzate per le cornee di controllo negativo nel metodo di prova BCOP. Poiché tutte le cornee sono tagliate dal bulbo intero e montate nelle camere corneali, vi è il rischio che maneggiando tali dispositivi vengano alterati i valori di opacità e permeabilità (compresi i controlli negativi). Inoltre, i valori di opacità e permeabilità delle cornee di controllo negativo sono utilizzati per correggere i valori di opacità e permeabilità corneali relativi alla sostanza chimica in esame e al controllo positivo nei calcoli IVIS. PROCEDURA Preparazione degli occhi Le cornee prive di difetti sono dissezionate lasciando un bordo di 2-3 mm di sclera per permettere di maneggiarle più agevolmente in seguito e facendo attenzione a non danneggiare l'epitelio e l'endotelio corneale. Le cornee così separate sono montate in appositi supporti corneali composti da una sezione anteriore e una posteriore, che si interfacciano rispettivamente con i lati epiteliale ed endoteliale della cornea. Entrambe le camere sono riempite fino all'orlo con mezzo minimo essenziale di Eagle (EMEM) preriscaldato privo di fenolo rosso, riempiendo per prima la camera posteriore e facendo attenzione a evitare la formazione di bolle. L'apparecchio è poi equilibrato a 32 (± 1) °C per almeno un'ora per consentire alle cornee di equilibrarsi con il mezzo e raggiungere per quanto possibile la normale attività metabolica (la temperatura approssimativa della superficie corneale in vivo è di 32 °C). Dopo il periodo di equilibratura si aggiunge il mezzo EMEM fresco preriscaldato privo di fenolo rosso in entrambe le camere e si rilevano i valori base di opacità per ciascuna cornea. Sono scartate le cornee che presentano lesioni macroscopiche dei tessuti (per es. graffi, pigmentazione, neovascolarizzazione) o un'opacità superiore a sette unità di opacità o equivalente per l'opacimetro. Almeno tre cornee sono selezionate come cornee di controllo negativo (o del solvente). Le cornee restanti sono poi allocate al gruppo di trattamento e al gruppo dei controlli positivi. La capacità termica dell'acqua è più elevata di quella dell'aria, di conseguenza l'acqua offre condizioni di temperatura più stabili per l'incubazione. Si suggerisce pertanto di utilizzare un bagno ad acqua per mantenere il supporto corneale e il relativo contenuto a una temperatura pari a 32 (± 1) °C. Si possono comunque utilizzare anche incubatori ad aria, a condizione che si adottino le necessarie cautele per il mantenimento di condizioni di temperatura stabili (per es. preriscaldando i supporti e il mezzo in essi contenuto). Applicazione della sostanza chimica in esame Si seguono due diversi protocolli di trattamento, uno per i liquidi e i tensioattivi (solidi o liquidi) e uno per i solidi non tensioattivi. I liquidi sono analizzati non diluiti. Le sostanze semisolide, le creme e le cere sono di norma analizzate come liquidi. La prova sui tensioattivi non diluiti è eseguita a una concentrazione del 10 % p/v in una soluzione composta da cloruro di sodio allo 0,9 %, acqua distillata o altro solvente che abbia dimostrato di non avere ripercussioni negative sul sistema di prova. L'impiego di diluizioni diverse deve essere opportunamente motivato. Le miscele contenenti tensioattivi possono essere sottoposte a prova non diluite o diluite in opportuna concentrazione secondo il pertinente scenario di esposizione in vivo. Le concentrazioni di prova devono essere opportunamente motivate. Le cornee sono esposte ai liquidi e ai tensioattivi per 10 minuti. La scelta di altri tempi di esposizione va accompagnata da adeguate motivazioni scientifiche. Cfr. appendice 1 per una definizione di tensioattivo e di miscela contenente tensioattivi. I solidi non tensioattivi sono di norma analizzati sotto forma di soluzioni o sospensioni a una concentrazione del 20 % p/v, in una soluzione composta da cloruro di sodio allo 0,9 %, acqua distillata o altro solvente, che abbia dimostrato di non avere ripercussioni negative sul sistema di prova. In determinate circostanze e con opportune giustificazioni scientifiche i solidi possono anche essere analizzati non diluiti e applicati direttamente sulla superficie della cornea utilizzando il metodo della camera aperta (cfr. paragrafo 32). Le cornee sono esposte ai solidi per quattro ore, mentre in caso di applicazione di liquidi e tensioattivi è possibile scegliere tempi di esposizione alternativi a condizione che vengano fornite opportune motivazioni scientifiche. A seconda della natura fisica e delle caratteristiche chimiche della sostanza chimica in esame (per es. solidi, liquidi, liquidi viscosi o liquidi non viscosi) possono essere adottati diversi metodi di trattamento. La criticità sta nel garantire che la sostanza chimica in esame copra adeguatamente la superficie epiteliale e che venga correttamente rimossa durante il risciacquo. Il metodo della camera chiusa è di norma impiegato per sostanze chimiche in esame liquide da non viscose a leggermente viscose, mentre il metodo della camera aperta si utilizza di regola per sostanze chimiche in esame liquide da semiviscose a viscose e per solidi non diluiti. Il metodo della camera chiusa prevede l'introduzione nella camera anteriore di una sostanza chimica in esame in quantità sufficiente (750 μl) a coprire il lato epiteliale della cornea attraverso i fori di dosaggio posti sulla superficie superiore della camera. I fori sono successivamente sigillati con tappi durante l'esposizione. È importante assicurare che ciascuna cornea sia esposta alla sostanza chimica in esame per l'intervallo di tempo necessario. Nel metodo della camera aperta, prima di eseguire il trattamento, occorre rimuovere dalla camera anteriore l'anello di chiusura della finestra e la finestra in vetro. La sostanza di controllo o in esame (750 μl o una quantità della sostanza chimica in esame sufficiente a coprire interamente la cornea) è applicata direttamente alla superficie epiteliale della cornea con una micropipetta. Se è difficile usare la pipetta per una sostanza chimica in esame, questa può essere caricata a pressione in una pipetta a spostamento positivo per agevolare il dosaggio. Il puntale di una pipetta a spostamento positivo è inserito nel puntale di erogazione della siringa al fine di caricare sotto pressione il materiale nel puntale di spostamento. Contemporaneamente, lo stantuffo della siringa è premuto man mano che il pistone della pipetta è tirato verso l'alto. Se si formano bolle d'aria nel puntale della pipetta, occorre rimuovere (espellere) la sostanza chimica e ripetere il processo finché il puntale non sarà riempito senza che si creino bolle d'aria. È possibile all'occorrenza utilizzare una normale siringa (senza ago), poiché tale metodo consente la misurazione di un volume preciso di sostanza chimica in esame e facilita l'applicazione della sostanza alla superficie epiteliale della cornea. Dopo il dosaggio, la finestra è riposizionata sulla camera anteriore per ricreare un sistema chiuso. Incubazione post-esposizione Dopo il periodo di esposizione la sostanza chimica in esame, la sostanza chimica usata per il controllo negativo o la sostanza chimica per il controllo positivo è rimossa dalla camera anteriore e si lava l'epitelio almeno tre volte (o comunque fino a che non restino ulteriori tracce visibili della sostanza in esame) con EMEM (contenente rosso fenolo). Si utilizza per il risciacquo il mezzo contenente rosso fenolo, in quanto è possibile monitorare l'eventuale cambiamento di colore del rosso fenolo per determinare l'efficacia del risciacquo di sostanze chimiche acide o alcaline. Le cornee sono lavate più di tre volte nel caso in cui il rosso fenolo continui a scolorirsi, divenendo giallo o porpora, o se vi siano ancora tracce visibili della sostanza chimica in esame. Non appena la sostanza chimica in esame è stata completamente eliminata, le cornee sono sottoposte a un ultimo risciacquo con EMEM (senza rosso fenolo). Si impiega l'EMEM (senza rosso fenolo) per il risciacquo finale per assicurare che il rosso fenolo sia stato completamente rimosso dalla camera anteriore prima della misurazione dell'opacità. Si riempie la camera anteriore nuovamente con EMEM fresco senza rosso fenolo. Per i liquidi o i tensioattivi, dopo il risciacquo le cornee restano in incubazione per ulteriori due ore a una temperatura di 32 (± 1) °C. Un periodo più lungo di post-esposizione può rivelarsi utile in determinate circostanze e va valutato caso per caso. Le cornee trattate con i solidi sono risciacquate accuratamente al termine delle quattro ore di esposizione, ma non richiedono un ulteriore periodo di incubazione. Al termine del periodo di post-incubazione per i liquidi e i tensioattivi e delle quattro ore di esposizione per i solidi non tensioattivi si registrano i valori di opacità e permeabilità di ciascuna cornea. Inoltre, ogni cornea è sottoposta a esame visivo e si registrano altre osservazioni utili (per es. desquamazione dei tessuti, presenza di residui della sostanza chimica in esame, opacità non uniforme). Queste osservazioni potrebbero essere importanti in quanto possono determinare eventuali variazioni dei dati registrati con l'opacimetro. Sostanze chimiche usate come controlli Ogni prova deve includere controlli positivi o negativi (solvente o mezzo disperdente) paralleli. Nelle prove con sostanze liquide al 100 % il metodo di prova BCOP comprende un controllo negativo parallelo (per es. soluzione di cloruro di sodio allo 0,9 % o acqua distillata) al fine di rilevare eventuali cambiamenti non specifici nel sistema sperimentale e offrire una base di riferimento per i risultati del saggio. La presenza del controllo garantisce inoltre che non si verifichino reazioni irritanti a causa delle condizioni del saggio. Nelle prove con liquidi diluiti, tensioattivi o solidi, il metodo di prova BCOP comprende un gruppo di controllo (solvente o mezzo disperdente) parallelo al fine di rilevare eventuali cambiamenti non specifici nel sistema sperimentale e offrire una base di riferimento per i risultati del saggio. Può essere utilizzato solo un solvente/mezzo disperdente che abbia dimostrato di non influire negativamente sul sistema sperimentale. Una sostanza chimica nota per indurre una risposta positiva è inclusa come controllo positivo parallelo in ogni esperimento per verificare l'integrità del sistema di prova e del suo corretto svolgimento. Tuttavia il grado dell'irritazione non deve essere eccessivo, per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo. L'etanolo al 100 % o la dimetilformammide al 100 % sono esempi di controlli positivi di sostanze chimiche in esame liquide. Un esempio di controllo positivo per le sostanze chimiche in esame solide è l'imidazolo al 20 % p/v in una soluzione composta da cloruro di sodio allo 0,9 %. Le sostanze chimiche di riferimento sono utili per valutare il potenziale di irritazione oculare di sostanze chimiche sconosciute appartenenti a determinate classi di sostanze chimiche o prodotti o per la valutazione del potenziale di irritazione relativo di una sostanza irritante per occhi nell'ambito di una serie specifica di reazioni irritanti. Risultati misurati L'opacità è determinata dalla quantità di luce trasmessa attraverso la cornea. L'opacità corneale è misurata quantitativamente per mezzo di un opacimetro, che consente di ottenere valori di opacità misurati su una scala continua. La permeabilità è determinata dalla quantità di colorante a base di fluoresceina sodica che penetra negli strati cellulari della cornea (ovvero, l'epitelio sulla superficie esterna della cornea attraverso l'endotelio sulla superficie corneale interna). Si aggiunge 1 ml di soluzione di fluoresceina sodica (rispettivamente 4 o 5 mg/ml nella prova con sostanze liquide e tensioattivi o solidi non tensioattivi) alla camera anteriore del supporto corneale. Tale soluzione è a contatto con l'epitelio della cornea, mentre la camera posteriore, che è a contatto con il lato endoteliale della cornea, è riempita con EMEM fresco. Il supporto è poi messo in incubazione in posizione orizzontale per 90 (± 5) min a 32 (± 1) °C. La quantità di fluoresceina sodica che giunge alla camera posteriore è misurata attraverso la spettrofotometria UV/VIS. I dati spettrofotometrici rilevati a 490 nm sono registrati come valori di densità ottica (OD490) o assorbanza e misurati su una scala continua. I valori di permeabilità della fluoresceina sono determinati utilizzando valori OD490 a partire da uno spettrofotometro a luce visibile impostato su una lunghezza di percorso standard di 1 cm. In alternativa è possibile usare un lettore di piastre da microtitolazione a 96 pozzetti, purché: i) sia possibile stabilire l'intervallo lineare del lettore per determinare i valori della fluoresceina OD490e ii), si usi il volume corretto di campioni di fluoresceina nella piastra a 96 pozzetti per risultare in valori OD490 equivalenti alla lunghezza standard del percorso di 1 cm (che potrebbe richiedere un pozzetto completamente riempito, di norma 360 μl). DATI E RELAZIONE Valutazione dei dati Appena saranno stati corretti i valori di opacità e permeabilità media (OD490) con i valori dell'opacità di fondo e i valori di permeabilità OD490dei controlli negativi, i valori medi di opacità e permeabilità OD490 relativi a ciascun gruppo di trattamento saranno utilizzati nella seguente formula empirica per il calcolo del punteggio di irritazione in vitro (IVIS) per ciascun gruppo di trattamento: IVIS = valore medio di opacità + (15 × valore medio di permeabilità OD490) Sina et al. (16) precisano che questa formula è stata derivata durante studi interni e interlaboratorio. I dati generati per una serie di 36 composti nell'ambito di uno studio che ha coinvolto più laboratori sono stati oggetto di un'analisi multivariata per la determinazione dell'equazione più appropriata per la correlazione fra dati in vivo e in vitro. Gli scienziati di due diverse imprese hanno eseguito questa analisi e hanno derivato equazioni quasi identiche. I valori di opacità e permeabilità dovrebbero essere valutati separatamente per determinare se una sostanza chimica in esame ha indotto corrosività o irritazione grave solo per uno dei due risultati (cfr. criteri decisionali). Criteri decisionali I valori limite IVIS per identificare le sostanze chimiche in esame come in grado di indurre gravi lesioni oculari (categoria UN GHS 1) e le sostanze chimiche in esame che non richiedono classificazione per l'irritazione oculare o per gravi lesioni oculari (“senza categoria” nel sistema UN GHS) sono elencati in appresso:
Criteri di accettazione dello studio Una prova è considerata accettabile se il punteggio IVIS risultante dal controllo positivo rientra fra due deviazioni standard dalla media storica, che va aggiornata con cadenza almeno trimestrale o comunque ogni volta si effettui una prova accettabile in laboratori nei quali le prove sono svolte con scarsa frequenza (ovvero, meno di una volta al mese). Le reazioni dei controlli negativi o dei solventi/mezzi disperdenti dovrebbero corrispondere a valori di opacità e permeabilità inferiori ai limiti superiori stabiliti per i valori storici di opacità e permeabilità di cornee di bovini trattati con i relativi controlli negativi o solventi/mezzi disperdenti. Un singolo ciclo di prove costituito da almeno tre cornee dovrebbe essere sufficiente per una sostanza chimica in esame, se la classificazione che ne risulta è univoca. Tuttavia, nei casi in cui sussiste un margine di incertezza nel primo ciclo di prove, è opportuno prendere in considerazione un secondo ciclo di prove (non obbligatorio) nonché un terzo qualora emergessero risultati IVIS discordanti fra i primi due cicli di prova. In questo contesto, un risultato nel primo ciclo di prova è ritenuto incerto se le stime relative alle? tre cornee si sono rivelate divergenti, come:
Relazione sulla prova La relazione sulla prova include le seguenti informazioni, se pertinenti alla conduzione dello studio:
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Accuratezza : grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e rappresenta un aspetto della pertinenza. Il termine è usato spesso in modo intercambiabile con «concordanza», per indicare la proporzione di risultati corretti di un metodo di prova. Sostanza chimica di riferimento : sostanza chimica usata come standard di confronto rispetto a una sostanza chimica in esame. Una sostanza di riferimento dovrebbe presentare le seguenti proprietà: i) fonte o fonti coerenti e affidabili; ii) analogia strutturale e funzionale alla classe delle sostanze in esame; iii) caratteristiche fisiche/chimiche conosciute; iv) dati di supporto relativi agli effetti noti; e v) efficacia nota nell'ambito della reazione auspicata. Approccio bottom-up : approccio graduale applicato nel caso di una sostanza chimica che si ritiene non richieda di essere classificata in termini di irritazione oculare o gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che non richiedono classificazione (esito negativo) rispetto ad altre sostanze chimiche (esito positivo). Sostanza chimica : una sostanza o una miscela. Cornea : parte trasparente frontale del bulbo oculare che copre l'iride e la pupilla e consente il passaggio della luce verso l'interno. Opacità corneale : misurazione del grado di opacità della cornea in seguito all'esposizione a una sostanza chimica in esame. Un aumento dell'opacità corneale è indice di danni alla cornea. L'opacità può essere valutata in modo soggettivo, come nel test di Draize sugli occhi dei conigli, o oggettivo utilizzando uno strumento come un «opacimetro». Permeabilità corneale : misurazione quantitativa dei danni all'epitelio corneale, effettuata attraverso la determinazione della quantità di colorante a base di fluoresceina sodica che attraversa tutti gli strati cellulari della cornea. Irritazione oculare : produzione di alterazioni nell'occhio in seguito all'applicazione di una sostanza chimica in esame sulla superficie anteriore dell'occhio, totalmente reversibili entro 21 giorni dall'applicazione. Sostituibile con “Effetti reversibili sugli occhi” e con la categoria UN GHS 2 (4). Percentuale di falsi negativi : percentuale di tutte le sostanze chimiche positive falsamente identificate come negative da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. Percentuale di falsi positivi : percentuale di tutte le sostanze chimiche negative falsamente identificate come positive da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. Pericolo : proprietà intrinseca di un agente o di una situazione in grado di provocare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti. Punteggio di irritazione in vitro (IVIS) : formula empirica utilizzata nel metodo di prova BCOP con la quale i valori medi di opacità e permeabilità relativi a ciascun gruppo di trattamento sono combinati per ottenere un unico punteggio in vitro per ogni gruppo di trattamento. IVIS = valore di opacità medio + (15 x valore di permeabilità medio) Effetti irreversibili sugli occhi : Cfr. “Gravi lesioni oculari”. Miscela : miscela o soluzione composta da due o più sostanze non reagenti (4). Controllo negativo : una replica non trattata che contiene tutti i componenti di un sistema di prova. Il campione è analizzato con campioni trattati con la sostanza chimica in esame e altri campioni di controllo per determinare se il solvente interagisce con il sistema di prova. Non classificata : sostanza chimica non classificata in termini di irritazione oculare (categoria UN GHS 2, 2A o 2B) o gravi lesioni oculari (categoria UN GHS 1). Termine intercambiabile con “Senza categoria GHS”. Opacimetro : strumento impiegato per la misurazione dell'«opacità corneale» attraverso la determinazione della quantità di luce trasmessa attraverso la cornea. Lo strumento tipico ha due comparti, ciascuno provvisto di una fonte di luce propria e di una fotocellula. Un comparto è utilizzato per la cornea trattata e l'altro per la taratura e l'azzeramento dello strumento. La luce emessa da una lampada alogena è irradiata attraverso un comparto di controllo (rappresentato da una camera vuota senza finestre o liquidi) a una fotocellula e confrontata con la luce trasmessa a una fotocellula attraverso il comparto di prova in cui si trova la camera contenente la cornea. Si confronta la differenza di luce trasmessa dalle fotocellule e un valore numerico di opacità è visualizzato su un display digitale. Controllo positivo : una replica che contiene tutti i componenti di un sistema di prova e che si tratta con una sostanza che notoriamente induce una reazione positiva. Il grado dell'irritazione non deve essere eccessivo per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo. Effetti reversibili sugli occhi : Cfr. “Irritazione oculare”. Affidabilità : misura la possibilità di eseguire un metodo di prova in maniera riproducibile nel tempo all'interno dei laboratori e fra di essi seguendo lo stesso protocollo. Si valuta calcolando la riproducibilità intra- e inter-laboratorio e la ripetibilità intra-laboratorio. Gravi lesioni oculari : produzione di danni ai tessuti oculari o indebolimento grave della vista in seguito all'applicazione di una sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Sostituibile con “Effetti irreversibili sugli occhi” e con “Categoria UN GHS 1” (4). Controllo solvente/disperdente : campione non trattato che contiene tutti i componenti di un sistema di prova, compreso il solvente o il mezzo disperdente usato con la sostanza chimica in esame analizzato con gli altri campioni di controllo al fine di stabilire la reazione di base nei campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente o mezzo disperdente. Nelle prove con controlli negativi paralleli, questo campione dimostra anche se il mezzo disperdente è in grado di interagire con il sistema di prova. Sostanza : elementi chimici e loro composti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a conservare la stabilità del prodotto e le impurità derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza ripercussioni sulla stabilità della sostanza o modifiche della sua composizione (4). Tensioattivo : denominato anche surfattante, è una sostanza, come un detergente, in grado di ridurre la tensione superficiale di un liquido consentendo quindi di formare schiuma o di penetrare solidi; è noto anche come agente umettante. Miscela contenente tensioattivi : nel contesto di questo metodo di prova, si tratta di una miscela contenente uno o più tensioattivi in una concentrazione finale >5 %. Approccio top-down : approccio graduale applicato nel caso di una sostanza chimica sospettata di indurre gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che inducono gravi lesioni oculari (esito positivo) rispetto ad altre sostanze chimiche (esito negativo). Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Strategia di prova in sequenza : strategia di prove graduali in cui sono riesaminate tutte le informazioni disponibili su una sostanza chimica in esame, secondo un ordine ben specificato, seguendo un approccio basato sul peso dell'evidenza disponibile per ciascuna prova, al fine di stabilire se vi sono informazioni sufficienti per una decisione sulla classificazione del pericolo prima di procedere alla fase successiva. Se è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, non è necessario svolgere prove aggiuntive. Se non è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, si svolge una procedura sperimentale graduale in sequenza su animali fino a che non è possibile effettuare una classificazione inequivocabile. Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS) : sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede tecniche di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (4). Categoria UN GHS 1 : cfr. “Gravi lesioni oculari”. Categoria UN GHS 2 : cfr. “Irritazione oculare”. Senza categoria UN GHS : sostanze chimiche che non soddisfano i requisiti di classificazione nelle categorie UN GHS 1 o 2 (2A o 2B). Sostituibile con “Non classificata”. Metodo di prova convalidato : metodo di prova in base al quale sono stati completati studi di validazione per determinare la rilevanza (compresa l'accuratezza) e l'affidabilità per un fine specifico. Va sottolineato che un metodo di prova convalidato potrebbe non avere un rendimento sufficiente in termini di valori di accuratezza e affidabilità ritenuti accettabili per il raggiungimento dell'obiettivo prefissato. Peso dell'evidenza (weight-of-evidence) : il processo che consiste nel tener conto dei punti di forza e di debolezza di informazioni diverse per conseguire e supportare una data conclusione relativa al potenziale di pericolo di una sostanza chimica in esame. Appendice 2 CAPACITÀ PREDITTIVA DEL METODO DI PROVA BCOP Tabella 1 Capacità predittiva del metodo BCOP per identificare le sostanze chimiche che inducono gravi lesioni oculari [UN GHS/ EU CLP Cat 1 vs Not Cat 1 (Cat 2 + No Cat); US EPA Cat I vs Not Cat I (Cat II + Cat III + Cat IV)]
Tabella 2 Capacità predittiva del metodo BCOP per identificare le sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari (“non irritanti”) [UN GHS/ EU CLP No Cat vs Not No Cat (Cat 1 + Cat 2); US EPA Cat IV vs Not Cat IV (Cat I + Cat II + Cat III)]
Appendice 3 SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA NEL METODO DI PROVA BCOP Prima di utilizzare regolarmente questo metodo di prova, i laboratori dovrebbero dimostrare la loro competenza tecnica identificando correttamente la classificazione del pericolo per gli occhi rappresentato dalle 13 sostanze raccomandate nella tabella 1. Tali sostanze chimiche sono state selezionate per rappresentare la gamma di risposte relative al pericolo per gli occhi in base agli esiti del test in vivo sugli occhi dei conigli (TG 405) (17) e del sistema di classificazione UN GHS (ossia le categorie 1, 2A, 2B, o non classificata) (4). Fra gli altri criteri di selezione si annoverano la disponibilità in commercio, la disponibilità di dati di riferimento in vivo di elevata qualità e la disponibilità di dati di riferimento in vitro di elevata qualità ottenuti con il metodo di prova BCOP. I dati di riferimento sono disponibili nei documenti Streamlined Summary Document (3) e ICCVAM Background Review Document for the BCOP test method (2)(18). Tabella 1 Sostanze raccomandate per la verifica della competenza tecnica nel metodo BCOP
Appendice 4 SUPPORTO CORNEALE PER IL METODO DI PROVA BCOP I supporti corneali impiegati per il test BCOP sono fatti di materiale inerte (per es. polipropilene). I supporti constano di due metà (una camera anteriore e una posteriore) e presentano due camere cilindriche interne simili. Ciascuna camera è progettata per contenere un volume di circa 5 ml e sfocia in una finestra di vetro, attraverso la quale sono registrate le misurazioni dell'opacità. Ciascuna camera interna ha un diametro di 1,7 cm ed è profonda 2,2 cm (11). Una guarnizione ad anello posta sulla camera posteriore serve a prevenire fuoriuscite. Le cornee sono posizionate dal lato dell'endotelio in basso sulla guarnizione ad anello delle camere posteriori, mentre le camere anteriori sono poste dal lato dell'epitelio delle cornee. Le camere sono mantenute in posizione da tre viti in acciaio inossidabile poste sui bordi esterni della camera. L'estremità di ciascuna camera presenta una finestra in vetro che può essere rimossa per facilitare l'accesso alla cornea. Una guarnizione ad anello è posizionata anche fra la finestra in vetro e la camera per prevenire fuoriuscite. Due aperture nella parte superiore di ciascuna camera consentono di introdurre ed estrarre il mezzo e le sostanze chimiche di prova. Le aperture sono chiuse con tappi di gomma durante le fasi di trattamento e incubazione. La trasmissione della luce attraverso i supporti corneali può modificarsi mano a mano che gli effetti dell'usura o dell'accumulo di specifici residui di materiale sui fori della camera interna o sulla finestra di vetro possono incidere sulla diffusione luminosa o sulla riflettanza. La conseguenza potrebbe constare in aumenti o diminuzioni della trasmissione luminosa di base (e quindi delle letture dell'opacità di base) attraverso i supporti corneali e può essere evidente se intervengono variazioni importanti delle misurazioni dell'opacità corneale iniziale previste nelle camere individuali (ossia i valori iniziali dell'opacità corneale in singoli specifici supporti corneali possono differire di norma di oltre 2 o 3 unità di opacità dai valori di base previsti). Ciascun laboratorio dovrebbe prendere in considerazione l'istituzione di un programma per valutare le modifiche della trasmissione luminosa attraverso i supporti corneali, secondo la natura dei tipi di sostanze chimiche sottoposte a prova e la frequenza di utilizzo delle camere. Per stabilire i valori di base, i supporti corneali possono essere ispezionati prima dell'uso consueto mediante misurazione dei valori di base dell'opacità (o della trasmissione luminosa) delle camere riempite con il mezzo completo, senza cornee. I supporti corneali sono quindi ispezionati con cadenza regolare per accertare le variazioni della trasmissione luminosa durante i periodi di uso. Ciascun laboratorio può stabilire la frequenza di controllo dei supporti corneali, in base ai tipi di chimica sottoposta a prova, alla frequenza d'uso e alle osservazioni delle variazioni dei valori di base dell'opacità corneale. Se si rilevano variazioni importanti della trasmissione luminosa attraverso i supporti corneali, è opportuno prendere in considerazione idonee procedure di pulizia e/o lucidatura della superficie interna dei supporti corneali o la loro sostituzione. Supporto corneale: diagramma esploso. 
Appendice 5 L'OPACIMETRO L'opacimetro è un dispositivo di misurazione della luce trasmessa. A titolo di esempio, per l'apparecchiatura OP-KIT di Electro Design (Riom, Francia) usata nella validazione del metodo di prova BCOP, la luce emessa da una lampada alogena è irradiata attraverso un comparto di controllo (una camera vuota senza finestre o liquidi) a una fotocellula e confrontata con la luce trasmessa a una fotocellula attraverso il comparto di prova in cui si trova la camera contenente la cornea. Si confronta la differenza di luce trasmessa dalle fotocellule e un valore numerico di opacità è visualizzato su un display digitale. Le unità di opacità sono predefinite. È possibile usare altri tipi di opacimetri con impostazioni diverse (per es. che non richiedono misurazioni parallele dei compartimenti di controllo e di prova) se è dimostrato che forniscono risultati analoghi a quelli dell'apparecchiatura validata. L'opacimetro dovrebbe fornire una risposta lineare a partire da una serie di valori di opacità registrati, che coprono tutti i valori limite utilizzati per le diverse classificazioni descritte dal modello predittivo (ovvero, fino al valore limite che determina la presenza di corrosività/irritazione grave). Per garantire letture lineari e accurate fino a 75-80 unità di opacità occorre tarare l'opacimetro attraverso una serie di calibratori. I calibratori sono disposti nella camera di taratura (una camera corneale progettata per contenere i calibratori) e letti sull'opacimetro. La camera di taratura è concepita per alloggiare i calibratori all'incirca alla stessa distanza fra la luce e la fotocellula, alla quale sarebbero poste le cornee durante le misurazioni di opacità. I valori di riferimento e il valore di taratura iniziale dipendono dal tipo di opacimetro impiegato. La linearità delle misurazioni dell'opacità dovrebbero essere garantite da procedure idonee (specifiche dello strumento). A titolo di esempio, per l'apparecchiatura OP-KIT di Electro Design (Riom, Francia) l'opacimetro è inizialmente tarato a 0 unità di opacità per mezzo della camera di taratura senza calibratore. Tre diversi calibratori sono poi disposti all'interno della camera di taratura uno dopo l'altro e i valori di opacità sono successivamente registrati. I calibratori 1, 2 e 3 dovrebbero corrispondere a valori registrati di opacità pari alla rispettiva serie di valori rispettivamente di 75, 150 e 225 unità di opacità, con una tolleranza di ±5 %. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(13) |
nella parte B, il capitolo B.48 è sostituito dal seguente: «B.48 Metodo di prova sull'occhio isolato dei polli (Isolated Chicken Eye — ICE) per l'identificazione di i) sostanze chimiche che inducono gravi lesioni oculari e ii) sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari INTRODUZIONE Questo metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 438 (2013). Il metodo di prova sull'occhio isolato dei polli (Isolated Chicken Eye — ICE) è un metodo di prova valutato dal Comitato di coordinamento interagenzia per la convalida dei metodi alternativi (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM), insieme con il Centro europeo per la convalida di metodi alternativi (European Centre for the Validation of Alternative Methods, ECVAM) e del corrispondente organismo del Giappone (Japanese Centre for the Validation of Alternative Methods, JaCVAM) nel 2006 e nel 2010 (1) (2) (3). Nella prima valutazione la validità del metodo di prova ICE è stata riconosciuta come test di screening per identificare le sostanze chimiche (e le miscele) che inducono gravi lesioni oculari (categoria 1) secondo la definizione del Sistema generale armonizzato di classificazione e di etichettatura dei prodotti chimici delle Nazioni Unite (Globally Harmonized System of Classification and Labelling of Chemicals — UN GHS) (1) (2) (4) e del regolamento (CE) n. 1272/2008 relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele (CLP) (12). Nella seconda valutazione il metodo di prova ICE è stato valutato per il suo uso come test di screening per identificare sostanze chimiche non classificate che inducono irritazione oculare o gravi lesioni oculari secondo la definizione del sistema UN GHS (3) (4). I risultati dello studio di validazione e il panel di esame inter pares ha ribadito le raccomandazioni originarie di utilizzare il metodo di prova ICE per classificare le sostanze chimiche che inducono gravi lesioni oculari (categoria UN GHS 1) poiché la banca dati disponibile è rimasta invariata dalla validazione originaria dell'ICCVAM. In questa fase non sono state suggerite ulteriori raccomandazioni per un'espansione dell'ambito d'applicazione del metodo di prova ICE al fine di includere altre categorie. È stata condotta una valutazione ulteriore dell'insieme di dati in vitro e in vivo usati nello studio di validazione al fine di valutare l'utilità del metodo di prova ICE per identificare le sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari (5). Questo esercizio ha permesso di concludere che il metodo di prova può inoltre essere usato per identificare le sostanze chimiche che non richiedono una classificazione per l'irritazione oculare o per gravi lesioni oculari secondo il sistema UN GHS (4) (5). Questo metodo di prova include gli usi raccomandati e le limitazioni del metodo di prova ICE sulla base di tali valutazioni. Le principali differenze fra la versione originale del 2009 e la versione aggiornata del 2013 della linea guida dell'OCSE comprende fra l'altro l'uso del metodo di prova ICE per identificare le sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari secondo il sistema di classificazione UN GHS, un aggiornamento degli elementi della relazione, un aggiornamento dell'appendice 1 sulle definizioni nonché un aggiornamento dell'appendice 2 sulle sostanze chimiche di riferimento. Attualmente è generalmente riconosciuto che nel prossimo futuro nessuna singola prova di irritazione oculare in vitro sarà in grado di sostituire il test di Draize in vivo per prevedere tutta la gamma di irritazione per diverse classi chimiche. Tuttavia combinazioni strategiche di diversi metodi alternativi nell'ambito di una strategia di prova (sequenziale) possono sostituire il test oculare di Draize (6). L'approccio top-down (7) è usato quando, in base alle informazioni esistenti, si stima che una sostanza chimica abbia un elevato potenziale di irritazione, mentre l'approccio bottom-up (7) è usato quando, in base alle informazioni esistenti, si stima che una sostanza chimica non causi un'irritazione oculare sufficiente da richiedere una classificazione. Il metodo di prova ICE è un metodo di prova in vitro che può essere usato in alcune circostanze e con limitazioni specifiche, come descritto ai paragrafi da 8 a 10, per la classificazione dei rischi oculari e l'etichettatura delle sostanze chimiche. Anche se da solo non è considerato un valido sostituto del test in vivo sugli occhi dei conigli, il metodo di prova ICE è raccomandato come fase iniziale nell'ambito di una strategia di prova, quale l'approccio top-down proposto da Scott et al. (7) per identificare le sostanze chimiche che inducono gravi lesioni oculari, ossia le sostanze chimiche da classificare nella categoria UN GHS 1, senza effettuare ulteriori prove (4). Il metodo di prova ICE è altresì raccomandato per identificare le sostanze chimiche che non richiedono una classificazione per l'irritazione oculare o per gravi lesioni oculari, secondo la definizione del sistema GHS delle Nazioni Unite (“senza categoria”) (4) e può quindi essere usato come fase iniziale nell'ambito di una strategia di prova quale l'approccio bottom-up (7). Tuttavia, la classificazione definitiva di una sostanza chimica cui non si attribuisce la possibilità di causare gravi lesioni oculari o non classificata per irritazione oculare/gravi lesioni oculari con il metodo di prova BCOP richiederebbe prove supplementari (in vitro e/o in vivo). Si dovrebbero inoltre consultare le autorità di regolamentazione preposte prima di utilizzare il metodo di prova ICE in un approccio bottom-up nell'ambito di sistemi di classificazione diversi dal sistema UN GHS. Scopo di questo metodo di prova è descrivere le procedure impiegate per valutare il potenziale di rischio oculare di una sostanza chimica in esame sulla base della capacità di tale sostanza di causare tossicità in un occhio enucleato di pollo. Gli effetti tossici sulla cornea sono misurati attraverso i) una valutazione qualitativa dell'opacità, ii) una valutazione qualitativa dei danni all'epitelio in base all'applicazione di fluoresceina all'occhio (ritenzione della fluoresceina), iii) una misurazione quantitativa dell'aumento dello spessore (rigonfiamento), e iv) una stima qualitativa di eventuali danni morfologici macroscopici alla superficie. L'opacità, il rigonfiamento e le stime dei danni della cornea in seguito all'esposizione a una sostanza chimica in esame sono valutati individualmente e poi cumulativamente per formulare un sistema di classificazione di irritazione oculare (Eye Irritancy Classification). Le definizioni figurano nell'appendice 1. CONSIDERAZIONI INIZIALI E LIMITI Il presente metodo di prova ICE si basa sul protocollo suggerito nel documento orientativo n. 160 dell'OCSE (8), elaborato in seguito allo studio internazionale di validazione dell'ICCVAM (1) (3) (9) con il contributo dell'ECVAM e del JaCVAM nonché del dipartimento di tossicologia e farmacologia applicata per la qualità della vita (TNO, Paesi Bassi). Il protocollo si basa su informazioni ottenute da protocolli già pubblicati, nonché dal protocollo attualmente impiegato dal TNO (10) (11) (12) (13) (14). Nella validazione del metodo in questione è stato sottoposto a prova un'ampia gamma di sostanze chimiche e la banca dati empirica dello studio di validazione comprende 152 sostanze chimiche, fra cui 72 sostanze e 80 miscele (5). Questo metodo di prova è applicabile ai solidi, ai liquidi, alle emulsioni e ai gel. I liquidi possono essere acquosi o non acquosi; i solidi possono essere solubili o insolubili in acqua. I gas e gli aerosol non sono ancora stati valutati nell'ambito di uno studio di validazione. Il metodo di prova ICE può essere usato per classificare le sostanze chimiche che inducono gravi lesioni oculari, ossia le sostanze chimiche da classificare nella categoria UN GHS 1 (4). Quando è usato a questo fine, le limitazioni identificate per il metodo di prova ICE sono basate su un tasso elevato di falsi positivi per gli alcoli e un elevato tasso di falsi negativi per i solidi e i tensioattivi (1) (3) (9). La percentuale di falsi negativi ottenuta (categoria UN GHS 1) in questo contesto non costituisce un aspetto critico poiché tutte le sostanze chimiche in esame che producono un risultato negativo sono successivamente sottoposte a prova con altri metodi in vitro adeguatamente validati o, in ultima ratio sui conigli, a seconda dei requisiti regolamentari, mediante una strategia di saggi sequenziali con un metodo basato sul peso dell'evidenza. È opportuno osservare che i solidi possono generare condizioni di esposizione variabili ed estreme nel test di Draize in vivo, che potrebbe falsare il reale potenziale di irritazione (15). Gli sperimentatori potrebbero prendere in considerazione il ricorso a tale metodo per tutti i tipi di sostanze chimiche, laddove un risultato positivo dovrebbe essere accettato come indicativo di una risposta che induce gravi lesioni oculari, per es. classificazione nella categoria UN GHS 1, senza ulteriori prove. I risultati positivi ottenuti con gli alcol andrebbero tuttavia interpretati con cautela, considerato il rischio di effettuare previsioni non corrette. Se usato per identificare le sostanze chimiche che inducono gravi lesioni oculari (categoria UN GHS 1), il metodo di prova ICE presenta un'accuratezza dell'86 % (120/140), una percentuale di falsi positivi del 6 % (7/113) e una percentuale di falsi negativi del 48 % (13/27), rispetto ai dati ottenuti con il test in vivo sugli occhi dei conigli classificati secondo il sistema di classificazione UN GHS (4) (5). Il metodo di prova ICE può inoltre essere usato per identificare le sostanze chimiche che non richiedono una classificazione per irritazione oculare o per gravi lesioni oculari nell'ambito del sistema di classificazione UN GHS (4). Si dovrebbero consultare le autorità di regolamentazione preposte prima di utilizzare il metodo di prova ICE in un approccio bottom-up nell'ambito di sistemi di classificazione diversi. Questo metodo di prova può essere usato per tutti i tipi di sostanze chimiche in cui è accettabile un risultato negativo per non classificare una sostanza chimica per irritazione oculare o gravi lesioni oculari. Tuttavia, sulla base di un unico risultato proveniente dalla banca dati di validazione le pitture antivegetative contenenti solventi organici possono essere sottostimate (5). Se usato per identificare le sostanze chimiche che non richiedono classificazione per irritazione oculare e gravi lesioni oculari, il metodo di prova ICE presenta un'accuratezza dell'82 % (125/152), una percentuale di falsi positivi del 33 % (26/79) e una percentuale di falsi negativi dell'1 % (1/73), rispetto ai dati ottenuti con il test in vivo sugli occhi dei conigli classificati secondo il sistema di classificazione UN GHS (4) (5). Se le sostanze chimiche in esame nell'ambito di alcune classi chimiche (per es. pitture antivegetative contenenti solventi organici) sono escluse dalla banca dati, il metodo di prova ICE presenta un'accuratezza complessiva dell'83 % (123/149), una percentuale di falsi positivi del 33 % (26/78) e una percentuale di falsi negativi dello 0 % (0/71) secondo il sistema di classificazione UN GHS (5). Il metodo di prova ICE non è raccomandato per identificare le sostanze chimiche in esame da classificarsi come irritanti per gli occhi (categoria UN GHS 2 o 2A) o le sostanze chimiche in esame da classificarsi come moderatamente irritanti per gli occhi (categoria UN GHS 2B) a causa dell'elevato numero di sostanze chimiche appartenenti alla categoria UN GHS 1 sottoclassificate nelle categorie 2, 2A o 2B e le sostanze chimiche “senza categoria” secondo il sistema UN GHS sovraclassificate nelle categorie 2, 2A o 2B. A tal fine possono essere necessarie ulteriori prove con un altro metodo adeguato. Tutte le procedure che prevedono l'impiego di occhi di polli dovrebbero osservare le regole e le procedure in vigore nella struttura che effettua l'analisi relativamente alla gestione di materiali di derivazione umana o animale che comprendono, tra l'altro, i tessuti e i liquidi tessutali. È consigliabile adottare le comuni norme di cautela osservate nei laboratori (16). Sebbene il metodo di prova ICE non prenda in considerazione le lesioni della congiuntiva e dell'iride valutati secondo il metodo di prova per l'irritazione oculare sui conigli, esso riguarda gli effetti sulla cornea, che sono i principali fattori di classificazione in vivo secondo la classificazione UN GHS. Inoltre, benché il metodo di prova ICE non consente di per sé di valutare la reversibilità delle lesioni corneali, studi condotti sugli occhi dei conigli suggeriscono di esaminare la profondità iniziale di una lesione corneale per identificare alcuni tipi di effetti irreversibili (17). In particolare è necessario un approfondimento scientifico per comprendere in quale modo possano verificarsi effetti irreversibili non collegati all'elevato livello iniziale della lesione. Infine, il metodo di prova ICE non consente di valutare il potenziale di tossicità sistemica associato all'esposizione attraverso l'occhio. Infine, questo metodo di prova sarà aggiornato con cadenza regolare, mano a mano che si disporrà di ulteriori dati e informazioni. A titolo di esempio, l'istopatologia presenta un'utilità potenziale nei casi in cui è necessaria una caratterizzazione più completa della lesione corneale. Gli utilizzatori sono invitati a conservare le cornee e a preparare campioni istopatologici che possono servire a elaborare una banca dati e criteri decisionali in grado di migliorare ulteriormente l'accuratezza di questo metodo di prova. L'OCSE ha elaborato un documento orientativo sull'uso dei metodi di prova in vitro per la tossicità oculare, che comprende procedure dettagliate sulla raccolta di campioni istopatologici e informazioni in merito alla trasmissione di campioni e/o di dati istopatologici (8). Ai laboratori che ricorrono a questo tipo di metodo di prova per la prima volta si consiglia di utilizzare le sostanze chimiche di riferimento per la verifica della competenza tecnica indicate nell'appendice 2. Un laboratorio può utilizzare tali sostanze chimiche per dimostrare le proprie competenze tecniche nell'esecuzione del metodo di prova ICE prima di presentare i dati relativi al metodo di prova ICE a scopi regolamentari per la classificazione dei rischi. PRINCIPIO DELLA PROVA Il metodo di prova ICE è un modello organotipico per il mantenimento di occhi di pollo a breve termine in vitro. Questo metodo di prova consente di valutare i danni prodotti dalla sostanza chimica in esame in base al rigonfiamento corneale, all'opacità e alla ritenzione di fluoresceina. Mentre gli ultimi due parametri richiedono una valutazione qualitativa, l'analisi del rigonfiamento corneale è di tipo quantitativo. Ciascuna misurazione è convertita in un punteggio quantitativo usato per calcolare un indice di irritazione generale oppure è categorizzata qualitativamente al fine di assegnare un pericolo oculare in vitro, alla categoria UN GHS 1 o senza categoria UN GHS. Questi risultati possono essere quindi usati per stimare in vivo il potenziale di gravi lesioni oculari o dell'assenza di classificazione relativamente alla classificazione di pericolo oculare di una sostanza chimica (cfr. criteri decisionali). Con il metodo di prova ICE non è tuttavia possibile attribuire una classificazione delle sostanze non ritenute causare gravi lesioni oculari o non classificate (cfr. paragrafo 11). Origine ed età degli occhi dei polli Storicamente, per questo tipo di saggio sono utilizzati occhi prelevati da polli abbattuti nei mattatoi per il consumo alimentare umano, ovviando in questo modo alla necessità di utilizzare animali da laboratorio. Sono impiegati solamente occhi di animali sani ritenuti idonei a essere immessi nella catena alimentare umana. Sebbene non sia stato condotto alcuno studio con controlli per identificare l'età ottimale del pollo, l'età e il peso degli animali storicamente impiegati per questo metodo di prova corrispondono a quelli dei pollastri tradizionalmente abbattuti nei macelli per pollame (ovvero, pollastri di circa 7 settimane e 1,5 – 2,5 kg di peso). Prelievo e trasporto degli occhi in laboratorio Si suggerisce di rimuovere le teste subito dopo aver stordito i polli, di norma con scossa elettrica, e aver praticato loro un'incisione sul collo per consentire il sanguinamento. È preferibile che l'allevamento da cui provengono i polli sia situato nelle vicinanze del laboratorio, per consentire di trasferire le teste dal macello in tempi sufficientemente rapidi e ridurre così al minimo il deterioramento e/o la contaminazione batterica. L'intervallo temporale fra la raccolta delle teste di pollo e la collocazione degli occhi nella camera di superfusione in seguito all'enucleazione dovrebbe essere minimizzato (di norma, entro due ore) per garantire il rispetto dei criteri di accettazione del saggio. Si suggerisce di utilizzare per il saggio occhi dello stesso gruppo di prelievo, ottenuti nella medesima giornata. Gli occhi sono dissezionati in laboratorio, pertanto il trasporto delle teste intatte dal macello deve avvenire a temperatura ambiente (solitamente 18°-25°) in scatole di plastica umidificate con panni inumiditi di soluzione salina isotonica. Criteri di selezione e numero di occhi usati nel metodo di prova ICE Sono scartati gli occhi che presentano consistenti macchie di fluoresceina (> 0,5) o un punteggio di opacità corneale elevato (> 0,5) dopo l'enucleazione. Ciascun gruppo di trattamento e di controllo positivo parallelo comprende almeno tre occhi. Il gruppo di controllo negativo o del solvente (se si utilizza un solvente diverso dalla soluzione salina) comprende almeno un occhio. Nel caso dei materiali solidi che producono un risultato UN GHS, si raccomanda un secondo ciclo di tre occhi per confermare o smentire il risultato negativo. PROCEDURA Preparazione degli occhi Le palpebre sono asportate con cautela, facendo attenzione a non danneggiare la cornea. L'integrità della cornea è subito valutata con una goccia di fluoresceina sodica al 2 % (p/v) applicata alla superficie della cornea per alcuni secondi e poi risciacquata con soluzione salina isotonica. Gli occhi trattati con la fluoresceina sono poi esaminati al microscopio con lampada a fessura per assicurare che la cornea non presenti danni (per es. ritenzione di fluoresceina e valori di opacità corneale ≤ 0,5). Se non è danneggiato, l'occhio è ulteriormente dissezionato dal cranio facendo attenzione a non danneggiare la cornea. Il bulbo oculare è estratto dall'orbita servendosi di pinze chirurgiche per tenere saldamente ferma la membrana nittitante e i muscoli oculari sono tagliati con forbici chirurgiche curve smusse. È importante non applicare una pressione eccessiva (per es. artefatti da compressione) per evitare di danneggiare la cornea. Nel rimuovere l'occhio dall'orbita, dovrebbe restare attaccata una porzione visibile di nervo ottico. Rimosso dall'orbita, l'occhio è posto su un tappetino assorbente e sono recisi la membrana nittitante e il restante tessuto connettivo. L'occhio enucleato è montato in un morsetto in acciaio inossidabile con la cornea in posizione verticale. Il morsetto è poi trasferito all'interno di una camera dell'apparecchio di superfusione (18). I morsetti dovrebbero essere posizionati nell'apparecchio di superfusione in modo che tutta la cornea sia esposta alla perfusione di soluzione salina isotonica (3-4 gocce al minuto o 0,1-0,15 ml/min). Le camere dell'apparecchio di superfusione devono essere a temperatura controllata di 32 (± 1,5) °C. L'appendice 3 presenta un diagramma esemplificativo di apparecchio di superfusione e dei morsetti oculari, che sono disponibili sul mercato o possono essere costruiti. L'apparecchio può essere modificato a seconda delle esigenze dei singoli laboratori (per es. per alloggiare un numero diverso di occhi). Dopo l'inserimento nell'apparecchio di superfusione, gli occhi sono nuovamente esaminati con un microscopio con lampada a fessura per accertarsi che non siano stati danneggiati durante la procedura di dissezione. È opportuno misurare anche lo spessore corneale, questa volta all'apice della cornea, utilizzando lo strumento di misurazione della profondità del microscopio con lampada a fessura. Occhi con i), un punteggio di ritenzione della fluoresceina > 0,5; ii) opacità corneale > 0,5; o iii) qualsiasi altro segno di lesione, dovrebbero essere sostituiti. Fra gli occhi che non sono stati scartati per nessuno dei criteri suddetti, devono essere scartati gli occhi con una deviazione dello spessore corneale di oltre il 10 % dal valore medio di tutti gli occhi. È opportuno che gli sperimentatori siano consapevoli del fatto che i microscopi con lampade a fessura possono produrre misurazioni dello spessore corneale diverse a seconda delle diverse larghezze su cui è impostata la fessura. La larghezza della fessura dovrebbe essere impostata a 0,095 mm. Dopo essere stati esaminati e selezionati, gli occhi sono incubati per un periodo di tempo compreso fra 45 e 60 minuti circa per l'equilibratura con il sistema prima del dosaggio. Successivamente al periodo di equilibratura è registrato il valore di riferimento per lo spessore e l'opacità della cornea, che funge da base per le altre misurazioni (per es. tempo = 0). Il valore di fluoresceina determinato al momento della dissezione è usato come misurazione di base per questo risultato. Applicazione della sostanza chimica in esame Subito dopo le misurazioni del valore di riferimento (zero) si estrae l'occhio (nel suo supporto) dall'apparecchio di superfusione, lo si dispone in posizione orizzontale e si applica alla cornea la sostanza chimica in esame. Le sostanze chimiche in esame liquide sono di norma analizzate non diluite, ma possono essere diluite se lo si ritiene necessario (per es. se previsto dallo studio). Il solvente più frequentemente utilizzato per le sostanze chimiche in esame diluite è la soluzione salina fisiologica. In determinate condizioni controllate possono tuttavia essere utilizzati anche solventi alternativi diversi dalla soluzione salina fisiologica, ma la loro idoneità deve essere comunque dimostrata. Le sostanze chimiche in esame liquide sono applicate alla cornea in modo che l'intera superficie sia uniformemente coperta con tale sostanza; il volume standard è pari a 0,03 ml. Se possibile, è opportuno triturare le sostanze chimiche in esame solide il più finemente possibile in un mortaio con un pestello o un analogo strumento di macinatura. La polvere è applicata alla cornea in modo che la superficie sia uniformemente coperta con tale sostanza; il quantitativo standard è pari a 0,03 g. La sostanza chimica in esame (liquida o solida) è applicata per 10 secondi e successivamente risciacquata dall'occhio con circa 20 ml di soluzione salina isotonica a temperatura ambiente. L'occhio (nel suo supporto) è in seguito posizionato nuovamente nell'apparecchio di superfusione nella posizione verticale originaria. Se necessario, è possibile risciacquare ulteriormente dopo l'applicazione di 10 secondi e a scadenze temporali successive (per es. se sono presenti residui di sostanza chimica in esame sulla cornea). Di norma il quantitativo di soluzione salina usato in aggiunta per il risciacquo non è rilevante, tuttavia l'osservazione dell'adesione della sostanza chimica alla cornea è importante. Sostanze chimiche di controllo Ogni saggio dovrà comprendere controlli negativi o con solventi/mezzi disperdenti e controlli positivi paralleli. Le prove con i liquidi al 100 % o i solidi prevedono l'impiego di una soluzione salina fisiologica come controllo negativo parallelo nel metodo di prova ICE per rilevare cambiamenti non specifici del sistema di prova e assicurare che le condizioni sperimentali non causino una reazione irritante non desiderata. Le prove con i liquidi diluiti comprendono un gruppo di controllo con solventi/mezzi disperdenti per rilevare cambiamenti non specifici del sistema di prova e assicurare che le condizioni sperimentali non causino una reazione irritante non desiderata. Come indicato al paragrafo 31, può essere utilizzato solo un solvente/mezzo disperdente per il quale si sia dimostrato che non ha ripercussioni negative sul sistema di prova. Ciascuna prova comprende un controllo positivo parallelo (noto irritante per occhi) per verificare la possibilità di indurre una reazione adeguata. Poiché il presente metodo di prova utilizza il saggio ICE per l'identificazione di sostanze corrosive o gravemente irritanti, il controllo positivo ideale dovrebbe essere una sostanza chimica di riferimento che consenta di indurre una reazione grave durante questo metodo di prova. Tuttavia la portata dell'irritazione non dovrebbe essere eccessiva per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo. Dovrebbero essere generati dati in vitro sufficienti per il controllo positivo in modo da poter calcolare una serie statisticamente accettabile di dati per il controllo positivo. Se non sono disponibili dati storici idonei relativi al metodo di prova ICE per un determinato controllo positivo, è opportuno condurre degli studi per ottenere questo tipo di informazioni. L'acido acetico al 10 % o il cloruro di benzalconio al 5 % sono esempi di controlli positivi di sostanze chimiche in esame liquide, mentre esempi di controlli positivi per le sostanze di prova solide comprendono l'idrossido di sodio o l'imidazolo. Le sostanze chimiche di riferimento sono utili per valutare il potenziale di irritazione oculare di sostanze chimiche sconosciute appartenenti a determinate classi di sostanze o prodotti chimici o per valutare il potenziale di irritazione relativo di una sostanza irritante per gli occhi nell'ambito di una serie specifica di reazioni irritanti. Endpoint misurati La valutazione delle cornee trattate avviene prima del trattamento e dopo 30, 75, 120, 180 e 240 minuti (con una tolleranza di ± 5 minuti) dal risciacquo successivo al trattamento. Queste scadenze temporali consentono di svolgere un numero idoneo di misurazioni nel corso del periodo complessivo di quattro ore previsto dal trattamento e lasciano al tempo stesso un periodo sufficiente fra una misurazione e l'altra per le necessarie osservazioni da effettuarsi su tutti gli occhi. Gli endpoint valutati sulla cornea comprendono fenomeni quali opacità e rigonfiamento corneali, ritenzione di fluoresceina ed effetti morfologici (per es. pitting o allentamento dell'epitelio). Tutti gli endpoint sono rilevati a ciascuna delle scadenze temporali sopra menzionate, ad eccezione della ritenzione della fluoresceina che è determinata solo prima del trattamento e 30 minuti dopo l'esposizione alla sostanza chimica in esame. È consigliabile servirsi di fotografie per documentare l'opacità corneale, la ritenzione della fluoresceina, gli effetti morfologici e l'istopatologia, se effettuata. Dopo l'esame finale condotto al termine delle quattro ore, si suggerisce agli sperimentatori di conservare gli occhi in un apposito fissativo (per es. formalina tamponata neutra) per un possibile esame istopatologico (cfr. paragrafo 14 e riferimento 8 per i dettagli). Il rigonfiamento corneale si determina grazie a misurazioni dello spessore della cornea, condotte con un pachimetro ottico montato su un microscopio con lampada a fessura. Il valore relativo è espresso in percentuale ed è calcolato sulla base delle misurazioni dello spessore della cornea secondo la seguente formula:
La percentuale media di rigonfiamento corneale per tutti gli occhi sottoposti al saggio è calcolata a tutte le scadenze temporali di osservazione. In base al punteggio medio più elevato di rigonfiamento corneale, osservato a ciascuna scadenza, è poi assegnato un punteggio complessivo di categoria per ciascuna sostanza chimica in esame (cfr. paragrafo 51). L'opacità corneale è valutata usando l'area della cornea maggiormente opacizzata per stabilire un punteggio secondo la tabella 1. La percentuale media di opacità corneale per tutti gli occhi sottoposti al saggio è calcolata a tutte le scadenze temporali di osservazione. In base al punteggio medio più elevato di opacità corneale, osservato a ciascuna scadenza, è poi assegnato un punteggio complessivo di categoria per ciascuna sostanza chimica in esame (cfr. paragrafo 51). Tabella 1 Punteggi di opacità corneale
Il valore di ritenzione della fluoresceina è valutato solo a 30 minuti, come indicato alla tabella 2. Il valore di ritenzione della fluoresceina medio per tutti gli occhi esaminato è calcolato a 30 minuti dal trattamento e vale come punteggio di categoria assegnato a ciascuna sostanza chimica in esame (cfr. paragrafo 51). Tabella 2 Punteggi di ritenzione della fluoresceina
Gli effetti morfologici comprendono il “pitting” delle cellule epiteliali corneali, l'“allentamento” dell'epitelio, l'“irruvidimento” della superficie corneale e l'“incollamento” della sostanza chimica in esame alla cornea. Questi risultati possono variare in termini di gravità e presentarsi contemporaneamente. La classificazione dei risultati suddetti è soggettiva e dipende dall'interpretazione del ricercatore. DATI E RELAZIONE Valutazione dei dati I risultati delle misurazioni relative a opacità e rigonfiamento corneali e ritenzione di fluoresceina dovrebbero essere valutati separatamente per creare una classe ICE per ciascun endpoint. Le classi ICE relative a ciascun endpoint sono poi associate per generare una classificazione di irritazione per ciascuna sostanza chimica in esame. Criteri decisionali Dopo aver valutato ciascun endpoint, è possibile assegnare le classi ICE sulla base di una serie predefinita. L'interpretazione del rigonfiamento corneale (tabella 3), dell'opacità (tabella 4) e della ritenzione della fluoresceina (tabella 5), utilizzando quattro classi ICE, avviene in base alle seguenti scale di classificazione: è importante osservare che i punteggi di rigonfiamento corneale di cui nella tabella 3 sono applicabili solo se lo spessore è misurato con microscopio con lampada a fessura (per es. del tipo Haag-Streit BP900) con dispositivo di misurazione della profondità n. 1 e larghezza della fessura impostata a 9 Tabella 3 Criteri di classificazione ICE per il rigonfiamento corneale
Tabella 4 Criteri di classificazione ICE per l'opacità
Tabella 5 Criteri di classificazione ICE per la ritenzione media di fluoresceina
La classificazione in vitro di una sostanza chimica è valutata mediante lettura della classificazione UN GHS corrispondente alla combinazione di categorie risultanti per il rigonfiamento corneale, l'opacità e la ritenzione della fluoresceina secondo la tabella 6. Tabella 6 Classificazioni in vitro generali.
Criteri di accettazione dello studio Una prova è ritenuta accettabile se i controlli negativi o con solventi/mezzi disperdenti e i controlli positivi paralleli sono identificati rispettivamente come “senza categoria e di categoria UN GHS 1”. Relazione di prova La relazione di prova dovrebbe includere le seguenti informazioni, se pertinenti alla conduzione dello studio:
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Accuratezza : grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e rappresenta un aspetto della pertinenza. Il termine è usato spesso in modo intercambiabile con «concordanza», per indicare la proporzione di risultati corretti di un metodo di prova. Sostanza chimica di riferimento : sostanza chimica usata come standard di confronto rispetto a una sostanza chimica in esame. Una sostanza di riferimento dovrebbe presentare le seguenti proprietà: i) fonte o fonti coerenti e affidabili; ii) analogia strutturale e funzionale alla classe delle sostanze in esame; iii) caratteristiche fisiche/chimiche note, iv) dati di supporto relativi agli effetti noti; e v) efficacia nota nell'ambito della reazione auspicata. Approccio bottom-up : approccio graduale applicato nel caso di una sostanza chimica che si ritiene non richieda di essere classificata in termini di irritazione oculare o gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che non richiedono classificazione (esito negativo) rispetto ad altre sostanze chimiche (esito positivo). Sostanza chimica : una sostanza o una miscela. Cornea : parte trasparente frontale del bulbo oculare che copre l'iride e la pupilla e consente il passaggio della luce verso l'interno. Opacità corneale : misurazione del grado di opacità della cornea in seguito all'esposizione a una sostanza in esame. Un aumento dell'opacità corneale è indice di danni alla cornea. Rigonfiamento corneale : misurazione oggettiva nel metodo di prova ICE del grado di distensione della cornea in seguito all'esposizione a una sostanza chimica in esame. Si esprime in percentuale ed è calcolato a partire dalle misurazioni di riferimento (pre-dosaggio) dello spessore corneale e dallo spessore registrato a intervalli regolari dopo l'esposizione alla sostanza chimica di prova del metodo di prova ICE. Il grado di rigonfiamento corneale è indice di danni alla cornea. Irritazione oculare : produzione di alterazioni nell'occhio in seguito all'applicazione di una sostanza chimica in esame sulla superficie anteriore dell'occhio, totalmente reversibili entro 21 giorni dall'applicazione. Sostituibile con “Effetti reversibili sugli occhi” e con la categoria UN GHS 2 (4). Percentuale di falsi negativi : percentuale di tutte le sostanze chimiche positive falsamente identificate come negative da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. Percentuale di falsi positivi : percentuale di tutte le sostanze chimiche negative falsamente identificate come positive da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. Ritenzione della fluoresceina : misurazione soggettiva nel metodo di prova ICE consiste nel quantitativo di fluoresceina sodica ritenuto nelle cellule epiteliali della cornea in seguito all'esposizione a una sostanza chimica in esame. Il grado di ritenzione della fluoresceina è indicativo della lesione dell'epitelio corneale. Pericolo : proprietà intrinseca di un agente o di una situazione in grado di provocare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti. Effetti irreversibili sugli occhi : cfr. “Gravi lesioni oculari” e “Categoria UN GHS 1”. Miscela : miscela o soluzione composta da due o più sostanze non reagenti (4). Controllo negativo : una replica non trattata che contiene tutti i componenti di un sistema di prova. Il campione è analizzato con campioni trattati con la sostanza chimica in esame e altri campioni di controllo per determinare se il solvente interagisce con il sistema di prova. Non classificata : sostanza chimica non classificata in termini di irritazione oculare (categoria UN GHS 2) o gravi lesioni oculari (categoria UN GHS 1). Termine intercambiabile con “Senza categoria GHS”. Controllo positivo : una replica che contiene tutti i componenti di un sistema di prova e che si tratta con una sostanza che notoriamente induce una reazione positiva. La gravità della reazione non deve essere eccessiva, per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo. Affidabilità : misura in cui un metodo può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. Si valuta calcolando la riproducibilità intra- e inter-laboratorio e la ripetibilità intra-laboratorio. Effetti irreversibili sugli occhi : cfr. “Irritazione oculare” e “Categoria UN GHS 2”. Gravi lesioni oculari : produzione di danni ai tessuti oculari o indebolimento grave della vista in seguito all'applicazione di una sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Sostituibile con “Effetti irreversibili sugli occhi” e con “Categoria UN GHS 1” (4). Microscopio con lampada a fessura : strumento utilizzato per esaminare direttamente l'occhio sotto l'ingrandimento di un microscopio binoculare e creando un'immagine stereoscopica e diritta. Nel metodo di prova ICE lo strumento è usato per esaminare le strutture anteriori degli occhi dei polli nonché misurare oggettivamente lo spessore della cornea mediante un apposito accessorio. Controllo solvente/disperdente : campione non trattato che contiene tutti i componenti di un sistema di prova, compreso il solvente o il mezzo disperdente usato con i campioni trattati con la sostanza chimica in esame analizzato con gli altri campioni di controllo al fine di stabilire la reazione di base nei campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente o mezzo disperdente. Nelle prove con controlli negativi paralleli, questo campione dimostra anche se il mezzo disperdente è in grado di interagire con il sistema di prova. Sostanza : elementi chimici e loro composti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a conservare la stabilità del prodotto e le impurità derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza ripercussioni sulla stabilità della sostanza o modifiche della sua composizione (4). Tensioattivo : denominato anche surfattante, è una sostanza, come un detergente, in grado di ridurre la tensione superficiale di un liquido consentendo quindi di formare schiuma o di penetrare solidi; è noto anche come agente umettante. Approccio top-down : approccio graduale applicato nel caso di una sostanza chimica sospettata di indurre gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che inducono gravi lesioni oculari (esito positivo) rispetto ad altre sostanze chimiche (esito negativo). Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Strategia di prova in sequenza : strategia di prove graduali in cui sono riesaminate tutte le informazioni disponibili su una sostanza chimica in esame, secondo un ordine ben specificato, seguendo un approccio basato sul peso dell'evidenza disponibile per ciascuna prova, al fine di stabilire se vi sono informazioni sufficienti per una decisione sulla classificazione del pericolo prima di procedere alla fase successiva. Se è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, non è necessario svolgere prove aggiuntive. Se non è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, si svolge una procedura sperimentale graduale in sequenza su animali fino a che non è possibile effettuare una classificazione inequivocabile. Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche (UN GHS) : sistema di classificazione delle sostanze chimiche sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (4). Categoria UN GHS 1 : cfr. “Gravi lesioni oculari” e/o “Effetti irreversibili sugli occhi”. Categoria UN GHS 2 : cfr. “Irritazione oculare” e/o “Effetti reversibili sugli occhi”. Senza categoria UN GHS : sostanze chimiche che non soddisfano i requisiti di classificazione nelle categorie UN GHS 1 o 2 (2A o 2B). Sostituibile con “Non classificata”. Metodo di prova convalidato : metodo di prova in base al quale sono stati completati studi di validazione per determinare la rilevanza (compresa l'accuratezza) e l'affidabilità per un fine specifico. Va sottolineato che un metodo di prova convalidato potrebbe non avere un rendimento sufficiente in termini di valori di accuratezza e affidabilità ritenuti accettabili per il raggiungimento dell'obiettivo prefissato. Peso dell'evidenza (weight-of-evidence) : il processo che consiste nel tener conto dei punti di forza e di debolezza di informazioni diverse per conseguire e supportare una data conclusione relativa al potenziale di pericolo di una sostanza chimica. Appendice 2 SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA NEL METODO DI PROVA ICE Prima di utilizzare regolarmente un metodo di prova che soddisfi i requisiti delle presenti linee guida, i laboratori dovrebbero dimostrare la loro competenza tecnica identificando correttamente la classificazione di corrosività oculare delle 13 sostanze raccomandate nella tabella 1. Tali sostanze chimiche sono state selezionate per rappresentare la gamma di risposte relative ai pericoli per gli occhi in base agli esiti del test in vivo sugli occhi dei conigli (TG 405) (17) e al sistema di classificazione UN GHS (ossia le categorie 1, 2A, 2B, o “senza categoria”) (4) (6). Fra gli altri criteri di selezione si annoverano la disponibilità in commercio delle sostanze chimiche, la disponibilità di dati di riferimento in vivo di elevata qualità e la disponibilità di dati di riferimento in vitro di elevata qualità ottenuti con il metodo di prova ICE. I dati di riferimento sono disponibili nei documenti Streamlined Summary Document (5) e ICCVAM Background Review Document for the ICE test method (9). Tabella 1: ostanze chimiche raccomandate per la verifica della competenza tecnica nel metodo ICE
Appendice 3 DIAGRAMMI RELATIVI ALL'APPARECCHIO ICE DI SUPERFUSIONE E AI MORSETTI OCULARI (cfr Burton et al. (18) per ulteriori descrizioni generiche dell'apparecchio di superfusione e del morsetto oculare) 
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(14) |
Nella parte B, il capitolo B.49 è sostituito dal seguente: «B.49. Prova del micronucleo in vitro con cellule di mammifero INTRODUZIONE Questo metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 487 (2016) e rientra in una serie di metodi di prova intesi a saggiare la tossicità genetica. È stato elaborato un documento OCSE contenente informazioni succinte sulle prove di tossicologia genetica e un compendio delle modifiche recentemente apportate alla rispettiva linea guida (1). Il test del micronucleo in vitro (MNvit) è una prova di genotossicità effettuata per rilevare la presenza di micronuclei (MN) nel citoplasma delle cellule durante l'interfase. I micronuclei possono formarsi a partire da frammenti cromosomici acentrici (ossia privi di centromero) o da cromosomi interi che non sono in grado di migrare verso i poli durante l'anafase (una fase della divisione cellulare). Pertanto, il test MNvit è un metodo in vitro che, grazie alla sua capacità di rilevare agenti sia aneugeni sia clastogeni, costituisce una base completa per studiare il potenziale di danno cromosomico in vitro (2) (3) nelle cellule che hanno iniziato la divisione cellulare durante o dopo l'esposizione alla sostanza chimica in esame (cfr. paragrafo 13 per ulteriori dettagli). I micronuclei rappresentano danni che sono stati trasmessi alle cellule figlie, mentre le aberrazioni cromosomiche conteggiate nelle cellule in metafase potrebbero non essere trasmesse. In entrambi i casi, le modifiche potrebbero essere incompatibili con la sopravvivenza delle cellule Il presente metodo di prova permette il ricorso a protocolli sperimentali con e senza citocalasina B (citoB), un inibitore della polimerizzazione dell'actina. L'aggiunta di citoB prima della mitosi produce cellule binucleate e consente pertanto l'individuazione e l'analisi dei micronuclei solo nelle cellule che hanno completato una mitosi (3) (4). Il presente metodo di prova permette altresì di ricorrere a protocolli senza inibitore della citocinesi, purché si possa dimostrare che la popolazione cellulare analizzata abbia già intrapreso la mitosi. Oltre al test MNvit per individuare le sostanze chimiche che inducono la formazione di micronuclei, anche il ricorso alla marcatura immunochimica dei cinetocori o all'ibridazione con sonde centromeriche o telomeriche [ibridazione fluorescente in situ (FISH)] può fornire informazioni aggiuntive sui meccanismi che inducono danno cromosomico e formazione di micronuclei (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Tali procedure di marcatura e ibridazione possono essere impiegate quando si osserva un incremento della formazione di micronuclei e lo sperimentatore intende stabilire se esso sia la conseguenza di eventi clastogeni e/o aneugeni. Poiché i micronuclei nelle cellule in interfase possono essere valutati in maniera relativamente obiettiva, è sufficiente che il personale del laboratorio determini il numero di cellule binucleate nei trattamenti con citoB nonché l'incidenza delle cellule con micronuclei in tutti i casi. Di conseguenza, i vetrini possono essere analizzati con relativa rapidità e l'analisi può essere automatizzata, il che permetterebbe di analizzare migliaia anziché centinaia di cellule per trattamento, rafforzando la significatività della prova. Infine, poiché i micronuclei possono formarsi anche da cromosomi ritardatari, ciò consentirebbe di individuare agenti che inducono aneuploidia che sono difficili da studiare tramite prove tradizionali sulle aberrazioni cromosomiche (cfr. capitolo B.10 del presente allegato) (18). Tuttavia, il test MNvit come descritto nel presente metodo di prova non permette di distinguere le sostanze chimiche che inducono alterazioni del numero di cromosomi e/o ploidia da quelle che causano clastogenicità senza il ricorso a tecniche speciali come la FISH, menzionata al paragrafo 4. Il test MNvit è una prova affidabile e può essere condotta per vari tipi cellulari, con o senza ricorso a citoB. Sono numerosi i dati a sostegno della validità del test MNvit con diversi tipi di cellule (culture di linee cellulari o culture di cellule primarie) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Tra questi si annoverano, in particolare, gli studi internazionali di convalida coordinati dalla Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23) e le relazioni dell'International Workshop on Genotoxicity Testing (5) (17). Le informazioni disponibili sono anche state riesaminate nell'ambito di uno studio di convalida retrospettivo basato sul peso delle evidenze condotto dal Centro europeo per la convalida di metodi alternativi (ECVAM) della Commissione europea, mentre la validità scientifica del metodo di prova è stata riconosciuta dal comitato scientifico consultivo dell'ECVAM (ESAC) (37) (38) (39). Il test MNvit su cellule di mammifero può impiegare colture di linee cellulari o colture cellulari primarie di origine umana o di roditori. Poiché la frequenza di fondo dei micronuclei influenzerà la sensibilità della prova, si raccomanda di utilizzare tipi cellulari con una frequenza stabile e definita di formazione di micronuclei. Le cellule sono scelte in funzione della loro capacità di crescita correttamente in coltura, della stabilità del cariotipo (compreso il numero dei cromosomi) e della frequenza di aberrazioni cromosomiche spontanee (40). I dati attualmente disponibili non consentono di formulare delle raccomandazioni rigorose ma suggeriscono che è importante tener conto, nel valutare i rischi chimici, dello stato del gene p53, della stabilità genetica (cariotipo), della capacità di riparazione del DNA e dell'origine (roditori o umani) delle cellule utilizzate nella prova. Gli utilizzatori del presente metodo di prova sono pertanto invitati a prendere in considerazione l'influenza di queste e altre caratteristiche cellulari sulle prestazioni di una linea cellulare per individuare l'induzione di micronuclei, poiché le conoscenze evolvono in questo settore. Le definizioni usate sono riportate nell'appendice 1. CONSIDERAZIONI INIZIALI E LIMITI Le prove in vitro richiedono in generale l'uso di una fonte esogena di attivazione metabolica, a meno che le cellule non siano metabolicamente competenti per quanto riguarda le sostanze chimiche in esame. Il sistema esogeno di attivazione metabolica non simula perfettamente le condizioni in vivo. Si deve inoltre prestare attenzione al fine di evitare condizioni che porterebbero a risultati positivi artefatti, che non riflettono la genotossicità delle sostanze chimiche in esame. Tali condizioni possono includere modifiche del pH (41) (42) (43) o dell'osmolalità, un'interazione con il terreno di coltura cellulare (44) (45) o una eccessiva citotossicità (cfr. paragrafo 29). Per esaminare l'induzione di micronuclei è fondamentale che sia avvenuta la mitosi sia nelle colture trattate che in quelle non trattate. Lo stadio più informativo per il conteggio dei micronuclei è quello che si riscontra nelle cellule che hanno completato una mitosi durante o dopo il trattamento con la sostanza chimica in esame. Per i nanomateriali di sintesi, è necessario apportare alcuni adattamenti specifici al presente metodo di prova, che tuttavia non sono descritti nel presente documento. Prima di utilizzarlo su una miscela per generare dati per una determinata finalità regolamentare, occorre valutare se il metodo di prova possa fornire risultati adeguati a tale finalità, e in caso affermativo indicarne i motivi. Tali considerazioni non sono necessarie quando esiste un obbligo regolamentare di testare la miscela. PRINCIPIO DELLA PROVA Le colture di cellule umane o di altro mammifero sono esposte alla sostanza chimica in esame con e senza una fonte esogena di attivazione metabolica, a meno che non si utilizzino cellule con un'adeguata capacità metabolizzante (cfr. paragrafo 19). Durante o dopo l'esposizione alla sostanza chimica in esame, le cellule sono coltivate per un periodo sufficiente a consentire che il danno cromosomico o altro effetto sul ciclo della cellula/divisione cellulare porti alla formazione di micronuclei nelle cellule in interfase. Per l'induzione di aneuploidia, la sostanza chimica in esame dovrebbe solitamente essere presente durante la mitosi. Le cellule in interfase coltivate e colorate sono analizzate per rilevare la presenza di micronuclei. Idealmente, si dovrebbero conteggiare micronuclei soltanto nelle cellule che hanno completato la mitosi durante l'esposizione alla sostanza chimica in esame o nell'eventuale periodo successivo al trattamento. Nelle colture trattate con un inibitore della citocinesi questo risultato si ottiene prendendo in considerazione soltanto le cellule binucleate. In assenza di un inibitore della citocinesi è importante dimostrare che le cellule analizzate hanno molto probabilmente avviato la divisione cellulare, sulla base dell'incremento della popolazione cellulare durante o dopo l'esposizione alla sostanza chimica in esame. Per tutti i protocolli è essenziale dimostrare che la proliferazione cellulare è avvenuta sia nelle colture trattate che nei controlli; inoltre, nelle colture in cui si è riscontrata la presenza di micronuclei dev'essere valutata l'entità della citotossicità o della citostasi indotta dalla sostanza chimica in esame. DESCRIZIONE DEL METODO Cellule Possono essere usati linfociti del sangue periferico primario coltivato di esseri umani o di altri mammiferi (7) (20) (46) (47) e una serie di linee cellulari di roditori quali CHO, V79, CHL/IU e L5178Y e di linee cellulari quali TK6 (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (cfr. paragrafo 6). Altre linee cellulari quali cellule HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2 (52) (53), A549 e le cellule embrionali primarie di criceto siriano (54) sono state utilizzate per la sperimentazione sul micronucleo, ma non sono ancora state pienamente convalidate. Pertanto, l'uso di tali linee cellulari e di altri tipi di cellule deve essere giustificato in base alla loro prestazione accertata nella prova, così come descritta nella sezione relativa ai criteri di accettabilità. È stato riportato che la citocalasina B influenza la crescita delle cellule L5178Y e non è pertanto raccomandata con questa linea cellulare (23). In caso di utilizzo di cellule primarie, per il benessere degli animali, occorrerà considerare, ove possibile, il ricorso a cellule di origine umana prelevate nel rispetto dei principi etici e della regolamentazione in materia. I linfociti del sangue periferico umano devono essere prelevati da individui giovani (circa 18-35 anni di età), non fumatori, che non siano stati esposti di recente ad agenti genotossici (ad es. sostanze chimiche, radiazioni ionizzanti) a livelli che farebbero aumentare l'incidenza di fondo delle cellule micronucleate, al fine di garantire che tale incidenza sia modesta e omogenea. L'incidenza di fondo delle cellule micronucleate aumenta con l'età e questa tendenza è più marcata nelle donne rispetto agli uomini (55). Se si prelevano cellule da più donatori, va indicato il numero dei donatori. Occorre dimostrare che le cellule si sono divise dall'inizio del trattamento con la sostanza chimica in esame fino al campionamento delle cellule. Le colture di cellule vengono mantenute in una fase di crescita esponenziale (linee cellulari) o stimolate a dividersi (colture primarie di linfociti) per esporre le cellule a diversi stadi del ciclo cellulare, dato che la sensibilità delle fasi cellulari alle sostanze chimiche in esame può essere sconosciuta. In genere le cellule primarie la cui divisione deve essere stimolata con agenti mitogeni non sono più sincronizzate durante l'esposizione alla sostanza chimica in esame (ad es. i linfociti umani dopo la stimolazione di 48 ore con agenti mitogeni). L'uso di cellule sincronizzate per il trattamento con la sostanza chimica in esame non è raccomandato, ma può essere accettabile se giustificato. Terreni e condizioni di coltura Per mantenere le colture devono essere garantiti terreni e condizioni di incubazione adeguati (recipienti per coltura, atmosfera umidificata con il 5 % di concentrazione di CO2, se del caso, temperatura di 37°C). Occorre controllare periodicamente la stabilità del numero modale dei cromosomi e l'assenza di contaminazione da micoplasma nelle linee cellulari; non si dovrebbero utilizzare cellule contaminate o che presentano modifiche del numero modale dei cromosomi. La durata normale del ciclo cellulare delle linee o colture primarie utilizzate nel laboratorio di prova deve essere stabilita e deve corrispondere alle caratteristiche cellulari pubblicate. Preparazione delle colture Linee cellulari: le cellule provenienti da colture primarie vengono inoculate in un terreno di coltura ad una densità tale che le cellule in sospensione o in monostrato proseguiranno la loro crescita esponenziale fino al momento della raccolta (occorre ad esempio evitare la confluenza delle cellule che si stanno moltiplicando in monostrato). Linfociti: sangue intero trattato con un anticoagulante (ad esempio, eparina) o linfociti isolati sono posti in un terreno di coltura (ad es. per 48 ore per i linfociti umani) contenente un mitogeno (ad esempio, fitoemoagglutinina, PHA, per i linfociti umani) per indurre la divisione cellulare prima dell'esposizione alla sostanza chimica in esame e alla citocasalina B (detta anche citoB). Attivazione metabolica Se si utilizzano cellule prive di un'adeguata capacità di attivazione metabolica endogena si deve ricorrere a sistemi di attivazione metabolica esogeni. Il sistema più comunemente usato, che è generalmente raccomandato in assenza di un altro sistema giustificato, è una frazione post-mitocondriale integrata di cofattori (S9) ricavata dal fegato di roditori trattati con induttori enzimatici, come Aroclor 1254 (56) (57) o una combinazione di fenobarbitone e β-naftoflavone (58) (59) (60). Quest'ultima combinazione è conforme alla Convenzione di Stoccolma sugli inquinanti organici persistenti (61), e ha dimostrato di essere altrettanto efficace dell'Aroclor 1254 nell'indurre ossidasi a funzione mista (58) (59) (60). La frazione S9 viene di solito usata a concentrazioni comprese tra 1-2 % (v/v) ma può aumentare fino al 10 % (v/v) nel terreno di coltura finale. Durante il trattamento, si dovrà evitare l'impiego di prodotti che riducono l'indice mitotico, in particolare gli agenti complessanti del calcio (62). La scelta del tipo e della concentrazione del sistema di attivazione metabolica esogena o dell'induttore metabolico usato può dipendere dalla classe delle sostanze chimiche in esame. Preparazione della sostanza chimica in esame Le sostanze chimiche solide in esame vanno preparate in adeguati solventi e, se necessario, diluite prima del trattamento delle cellule. Le sostanze chimiche liquide in esame possono essere aggiunte direttamente alla coltura e/o diluite prima del trattamento del sistema di prova. Le sostanze chimiche gassose o volatili vanno testate modificando adeguatamente i protocolli standard (trattamento in contenitori sigillati) (63) (64) (65). Si usino preparati della sostanza chimica approntati immediatamente prima del trattamento, salvo qualora siano disponibili dati sulla sua stabilità che dimostrino che la conservazione è accettabile. Condizioni di prova Solventi Il solvente va scelto in modo da massimizzare la solubilità delle sostanze chimiche in esame, senza determinare effetti negativi sulla conduzione della sperimentazione, in particolare senza modificare la crescita cellulare, compromettere l'integrità della sostanza chimica in esame, reagire con i recipienti di coltura o ostacolare il sistema di attivazione metabolica. Si raccomanda di prendere in considerazione in primo luogo, se possibile, l'uso di un solvente acquoso (o terreno di coltura). Solventi il cui uso è consolidato sono l'acqua e il dimetilsolfossido (DMSO). Di norma, i solventi organici non devono superare l'1 % v/v. Se la citoB è sciolta nel DMSO, il quantitativo totale di solventi organici utilizzato per la sostanza chimica in esame e la citoB non deve superare l'1 % (v/v); in caso contrario, si farà ricorso a controlli non trattati al fine di verificare che la percentuale di solvente organico non abbia effetti dannosi. I solventi acquosi (soluzione salina o acqua) non devono superare 10 % (v/v) nel terreno di coltura finale. L'uso di solventi poco noti (ad esempio, etanolo o acetone) è ammesso purché suffragato da dati che ne provino la compatibilità con la sostanza chimica in esame e l'assenza di tossicità genetica alla concentrazione utilizzata. In mancanza di tali dati, è importante includere nella prova controlli non trattati (cfr. allegato 1) così come controlli con solvente per dimostrare che il solvente scelto non comporta effetti deleteri o cromosomici (aneuploidia o clastogenicità). Uso della citoB come inibitore della citocinesi Una delle considerazioni più importanti riguardo alla prestazione del test MNvit è garantire che le cellule analizzate abbiano completato la micosi durante il trattamento o nell'eventuale periodo di incubazione successiva al trattamento. L'analisi dei micronuclei, pertanto, va limitata alle cellule che hanno subito la mitosi durante o dopo il trattamento. La citoB è l'agente più diffusamente usato per bloccare la citocinesi, poiché inibisce l'aggregazione dell'actina e, di conseguenza, impedisce la separazione delle cellule figlie dopo la mitosi, portando alla formazione di cellule binucleate (6) (66) (67). L'effetto della sostanza chimica in esame sulla proliferazione cellulare può essere misurato simultaneamente, quando viene utilizzata la citoB. La citoB deve essere usata come inibitore della citocinesi quando si impiegano linfociti umani, perché la durata del ciclo cellulare può variare tra donatori e perché non tutti i linfociti rispondono alla simulazione con PHA. La citoB non è obbligatoria per altri tipi di cellule se si può dimostrare che queste cellule hanno subito una divisione come descritto al paragrafo 27. Inoltre, la citoB non è generalmente utilizzata quando si valuta la formazione di micronuclei nei campioni usando metodi di citometria a flusso. Per ciascun tipo cellulare il laboratorio determina la concentrazione adeguata di citoB per ottenere la frequenza ottimale di cellule binucleate nelle colture di controllo con solvente e si deve dimostrare di pervenire a una buona produzione di cellule binucleate ai fini dell'analisi. La concentrazione adeguata di citoB è solitamente compresa tra 3 e 6 μg/ml (19). Misurazione della proliferazione cellulare e della citotossicità e scelta delle concentrazioni di trattamento Nel determinare la concentrazione massima della sostanza chimica in esame si devono evitare concentrazioni che hanno la capacità di produrre false risposte positive, ad esempio le concentrazioni che causano eccessiva citotossicità (cfr. paragrafo 29), precipitazione nel terreno di coltura (cfr. paragrafo 30) e variazioni marcate di pH o osmolalità (cfr. paragrafo 9). Se la sostanza chimica in esame provoca un'alterazione marcata del pH del terreno al momento della sua aggiunta, è possibile adeguare il pH mediante l'azione tampone del terreno di trattamento finale in modo da evitare falsi risultati positivi e da mantenere condizioni di coltura appropriate. La proliferazione cellulare deve essere misurata per assicurarsi che un numero sufficiente di cellule trattate hanno subito la mitosi durante la prova e che i trattamenti sono condotti ad adeguati livelli di citotossicità (cfr. il paragrafo 29). La citotossicità va determinata nell'esperimento principale con e senza attivazione metabolica mediante un indicatore appropriato di morte e crescita cellulari (cfr. paragrafi 26 e 27). Una prova preliminare per valutare la citotossicità può essere utile per definire meglio le concentrazioni da impiegare nella prova principale, ma non è obbligatoria. Se effettuata, essa non dovrebbe sostituire la misurazione della citotossicità effettuata nell'ambito della prova principale. Il trattamento di colture con la citoB e la misurazione delle frequenze relative di cellule mononucleate, binucleate e multinucleate nella coltura rappresentano un metodo accurato per la quantificazione dell'effetto sulla proliferazione cellulare e dell'attività citotossica o citostatica di un trattamento (6), e garantisce che siano analizzate mediante microscopio soltanto le cellule che hanno iniziato la divisione durante o dopo il trattamento. Si raccomanda di misurare l'indice di proliferazione cellulare (CBPI) (6) (27) (68) o l'indice di replicazione (RI) a partire da almeno 500 cellule per coltura (cfr. le formule nell'allegato 2) per stimare l'attività citotossica e citostatica di un trattamento confrontando i valori nelle colture trattate e nei controlli. La valutazione di altri indicatori di citotossicità (ad es. integrità cellulare, apoptosi, necrosi, conteggio in metafase, ciclo cellulare) può fornire informazioni utili, ma non dovrebbe sostituire il CBPI o l'RI. Negli studi condotti senza citoB occorre dimostrare che le cellule nella coltura si sono divise, ossia che una proporzione significativa delle cellule analizzate abbiano avviato il processo di divisione durante o in seguito al trattamento con la sostanza chimica in esame, per evitare che si producano falsi negativi. Si raccomanda la misurazione del raddoppiamento relativo della popolazione (RPD) o dell'aumento relativo della conta cellulare (RICC) per valutare l'attività citotossica e citostatica di un trattamento (17) (68) (69) (70) (71) (cfr. le formule nell'allegato 2). Quando il prelievo dei campioni è distribuito su un lungo periodo (ad es. quando la durata del trattamento equivale a 1,5-2 volte la durata del ciclo cellulare e la raccolta avviene dopo un periodo pari a 1,5-2 cicli cellulari successivamente al trattamento, il che comporta tempi di campionamento totali più lunghi di 3-4 volte la durata del ciclo cellulare, come descritto nei paragrafi 38 e 39), è possibile che il valore RPD sottovaluti la citotossicità (71). In tali circostanze, il RICC potrebbe costituire una migliore misurazione, ma anche la valutazione della citotossicità dopo un periodo equivalente a 1,5-2 volte la durata del ciclo cellulare fornisce una stima valida. La valutazione di altri marcatori della citotossicità o citostasi (ad es. integrità cellulare, apoptosi, necrosi, conteggio in metafase, indice di proliferazione, ciclo cellulare, ponti nucleoplasmatici o vescicole nucleari) può fornire informazioni utili, ma non dovrebbe sostituire l'RPD o il RICC. Si usino almeno tre concentrazioni di prova (senza contare i controlli positivi e i controlli con solvente) che soddisfano i criteri di accettabilità (citotossicità adeguata, numero di cellule, ecc.). Indipendentemente dal tipo di cellule (linee cellulari o colture primarie di linfociti), ciascuna delle colture trattate, uniche o replicate, può essere utilizzata per ciascuna concentrazione di prova. Mentre l'uso di colture duplicate è raccomandato, l'uso di colture uniche è accettato a condizione che il numero totale di cellule analizzate sia lo stesso tanto per le colture uniche quanto per le colture duplicate. L'uso di colture uniche è particolarmente pertinente quando si valutano più di 3 concentrazioni (cfr. paragrafi 44 e 45). I risultati ottenuti per ciascuna delle colture realizzate in più copie indipendenti a una data concentrazione possono essere riuniti per l'analisi dei dati. Per le sostanze chimiche in esame la cui citotossicità è bassa o nulla, sono generalmente adeguati intervalli di concentrazione di un fattore di circa 2 a 3. In caso di citotossicità, le concentrazioni di prova selezionate dovrebbero rientrare in un intervallo di concentrazione a partire dai valori che producono citotossicità come descritto al paragrafo 29 e che include le concentrazioni alle quali si riscontra una citotossità modesta o nulla. Molte sostanze chimiche in esame presentano curve di concentrazione-risposta a forte pendenza e, per ottenere dati nel quadro di una tossicità bassa e media o per valutare il rapporto dose-risposta in dettaglio, si dovranno pertanto utilizzare concentrazioni più ravvicinate e/o più di tre concentrazioni (colture uniche o repliche), in particolare nei casi in cui è necessario ripetere l'esperimento (v. paragrafo 60). Se la concentrazione massima è basata sulla citotossicità, la concentrazione più elevata dovrebbe raggiungere una citotossicità di 55 ± 5 % in base ai parametri di citotossicità raccomandati (cioè riduzione di RICC e RPD per le linee cellulari senza citoB e una riduzione di CBPI o RI quando è usata la citoB a 45 ± 5 % del controllo negativo parallelo) (72). Si devono interpretare con cautela i risultati positivi presenti soltanto nella fascia più alta dell'intervallo di citotossicità 55 ± 5 % (71). Per le sostanze chimiche in esame scarsamente solubili che non sono citotossiche a concentrazioni inferiori alla concentrazione insolubile più bassa, la concentrazione più elevata analizzata dovrebbe presentare una torbidità o un precipitato visibile ad occhio nudo o con un microscopio invertito alla fine del trattamento con la sostanza chimica in esame. Anche se la citotossicità interviene al di sopra della concentrazione insolubile più bassa, è consigliabile saggiare una sola concentrazione che induce torbidità o un precipitato visibile, in quanto dal precipitato potrebbero derivare false risposte. Alla concentrazione che produce un precipitato, occorre assicurarsi che quest'ultimo non interferisca con la conduzione della sperimentazione (ad es. colorazione o conteggio). Può essere utile valutare la solubilità nel terreno di coltura prima della prova. Se non si osserva una citotossicità limitante o si rileva la presenza di un precipitato, la concentrazione massima di prova dovrebbe essere pari a 10 mM, 2 mg/ml or 2 μl/ml, (si scelga il valore più basso) (73) (74) (75). Quando la composizione della sostanza chimica in esame non è definita, ad esempio nel caso di sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici (UVCB) (76), prodotti estratti dall'ambiente, ecc., può essere necessario aumentare la concentrazione massima (5 mg/ml) in assenza di citotossicità sufficiente, per aumentare la concentrazione di ciascun componente. Va tuttavia osservato che le prescrizioni possono essere diverse per i prodotti farmaceutici per uso umano (93). Controlli Al momento di ciascuna raccolta, si effettuino anche controlli negativi paralleli (cfr. paragrafo 21), consistenti in solo solvente nel terreno di coltura; i controlli vanno trattati come le colture di trattamento. I controlli positivi paralleli sono necessari per dimostrare la capacità del laboratorio di individuare clastogeni e aneugeni alle condizioni del protocollo di prova, nonché l'efficacia del sistema di attivazione metabolica esogena, se del caso. Esempi di controlli positivi sono riportati nella tabella 1. Sostanze chimiche alternative possono essere usate per i controlli positivi, se necessario. Attualmente, non sono noti aneugeni che richiedono l'attivazione metabolica per la loro attività genotossica (17). Poiché le prove di genotossicità in vitro su cellule di mammifero sono sufficientemente standardizzate per i trattamenti paralleli di breve durata effettuati con e senza attivazione metabolica per la stessa durata di trattamento, l'uso dei controlli positivi può essere limitato a un clastogeno che richiede attivazione metabolica. In questo caso, una sola risposta clastogena in un controllo positivo dimostrerà sia l'attività del sistema di attivazione metabolica che la capacità di risposta del sistema di prova. Tuttavia, un trattamento di lunga durata (senza S9) dovrebbe disporre di un proprio controllo positivo, poiché la durata del trattamento è diversa da quella della prova con attivazione metabolica. Se un clastogeno è selezionato come controllo positivo unico per il trattamento di breve durata con e senza attivazione metabolica, per il trattamento di lunga durata senza attivazione metabolica occorre scegliere un aneugeno. Nelle cellule metabolicamente competenti che non necessitano di S9 si dovrebbero usare controlli positivi sia per la clastogenicità che per l'aneugenicità. Ciascun controllo positivo va utilizzato con una o più concentrazioni che generalmente danno luogo ad un aumento riproducibile e individuabile rispetto al valore di fondo per dimostrare la sensibilità del sistema sperimentale (vale a dire che gli effetti sono chiari ma che non rivelano immediatamente all'esaminatore l'identità dei vetrini codificati), e la risposta non può essere compromessa da una citotossicità superiore ai limiti stabiliti nel presente metodo di prova. Tabella 1 Sostanze chimiche di riferimento raccomandate per la verifica delle competenze del laboratorio e per la selezione dei controlli positivi.
PROCEDURA Calendario del trattamento Per massimizzare la probabilità di individuare un aneugeno o un clastogeno in azione in una determinata fase del ciclo cellulare è importante che quantitativi sufficienti di cellule che rappresentano tutte le varie fasi del loro ciclo cellulare siano trattati con la sostanza chimica in esame. Tutti i trattamenti devono iniziare e terminare nel momento di crescita esponenziale delle cellule e le cellule devono continuare a crescere fino al momento del campionamento. Il calendario di trattamento per le linee cellulari e per le colture cellulari primarie, pertanto, può differire in parte da quello previsto per i linfociti, i quali necessitano di una stimolazione mitogena per avviare il ciclo cellulare (17). Per i linfociti, l'approccio più efficiente è iniziare il trattamento con la sostanza chimica in esame a distanza di 44-48 ore dopo la stimolazione con PHA, quando le cellule si dividono in modo asincrono (6). I dati pubblicati (19) indicano che la maggior parte degli aneugeni e dei clastogeni sarà rilevata entro un periodo di trattamento breve di 3 — 6 ore, con o senza S9, cui seguirà la rimozione della sostanza chimica in esame e un campionamento al momento equivalente a un periodo di crescita di circa 1,5-2,0 cicli cellulari dall'inizio del trattamento (7). Tuttavia, per una valutazione completa, che sarebbe necessaria per giungere alla conclusione di un risultato negativo, tutte e tre le condizioni sperimentali dovrebbero essere saggiate utilizzando un trattamento di breve durata con e senza attivazione metabolica e un trattamento di lunga durata senza attivazione metabolica (cfr. paragrafi 56, 57 e 58):
Se una di queste condizioni sperimentali porta a una risposta positiva, può non essere necessario esaminare gli altri regimi di trattamento. Se è noto o si sospetta che la sostanza chimica in esame incide sulla durata del ciclo cellulare (ad es., quando si saggiano analoghi nucleosidici), in particolare per quanto riguarda le cellule competenti per il gene p53 (35) (36) (77), i tempi di campionamento e di recupero possono essere prolungati di un periodo equivalente a 1,5-2,0 volte la durata del ciclo cellulare (per un totale di 3,0-4,0 cicli cellulari dopo l'inizio dei trattamenti di breve durata e di lunga durata). Queste alternative permettono di gestire situazioni in cui si temano potenziali interazioni tra la sostanza chimica in esame e la citoB. In caso di ricorso a periodi di campionamento prolungati (ossia un periodo di coltura totale equivalente a 3,0-4,0 cicli cellulari), occorre garantire che le cellule si dividano ancora attivamente. Per i linfociti, ad esempio, la crescita esponenziale può diminuire a partire da 96 ore dopo la stimolazione e le colture cellulari monostrati possono diventare confluenti. I protocolli di trattamento suggeriti sono riassunti nella tabella 2. Si tratta di calendari generici che possono essere modificati (e debitamente giustificati) in base alla stabilità o alla reattività della sostanza chimica in esame ovvero alle particolari caratteristiche di crescita delle cellule utilizzate. Tabella 2 Trattamento delle cellule e fasi di raccolta per i test MNvit
Nelle colture monostrati possono essere presenti cellule mitotiche (identificabili perché di forma tonda e in fase di distacco dalla superficie) al termine del trattamento di 3-6 ore. Poiché tali cellule mitotiche si staccano facilmente, potrebbero andare perdute al momento della rimozione del terreno di coltura che contiene la sostanza chimica in esame. Se si constata un aumento sostanziale del numero di cellule mitotiche rispetto ai controlli (il che indicherebbe il probabile arresto della mitosi), le cellule vanno raccolte mediante centrifugazione e successivamente reintrodotte nelle colture per evitare di perdere le cellule che sono in fase di mitosi e presentano un rischio di aberrazione dei micronuclei/cromosomica, al momento della raccolta. Raccolta delle colture e preparazione dei vetrini Per ogni coltura si procede a una raccolta e a un trattamento separati. Per la preparazione delle cellule può essere necessario un trattamento ipotonico, che tuttavia si può evitare se, con altri mezzi, è possibile ottenere un'adeguata diffusione delle cellule. Possono essere usate tecniche diverse per la preparazione dei vetrini, purché siano garantiti preparati cellulari di elevata qualità ai fini dell'analisi. Le cellule con la membrana cellulare e il citoplasma cellulare intatti vanno conservati per consentire il rilevamento dei micronuclei e (con il metodo di inibizione della citocinesi) l'individuazione affidabile di cellule binucleate. I vetrini possono essere colorati con tecniche diverse, per esempio con il metodo Giemsa o tinture fluorescenti che si legano al DNA. L'uso di coloranti fluorescenti appropriati [ad esempio, arancio di acridina (78) o Hoechst 33258 più pironina-Y (79)] può eliminare alcuni dei falsi risultati dovuti all'uso di un colorante non specifico per il DNA. Per individuare i contenuti (cromosomi interi saranno colorati mentre i frammenti cromosomici acentrici non lo saranno) dei micronuclei si possono utilizzare anche anticorpi anticinetocore, FISH con sonde di DNA pancentromerico o marcatura in situ mediante primer specifici per DNA pancentromerico, unitamente a un'adeguata colorazione del DNA, se interessa acquisire informazioni sui meccanismi di formazione dei micronuclei (16) (17). Possono infine essere usati altri metodi per distinguere i clastogeni dagli aneugeni, purché sia stata dimostrata la loro efficacia e siano validati. Ad esempio, per determinate linee cellulari, la misurazione dei nuclei sub-2N come eventi ipodiploidi utilizzando tecniche quali l'analisi di immagini, la citometria a scansione laser o la citometria a flusso potrebbe anche fornire informazioni utili (80) (81) (82). Le osservazioni morfologiche dei nuclei potrebbero inoltre fornire indicazioni su un'eventuale aneuploidia. Inoltre, potrebbe anche essere utile una prova di aberrazione cromosomica nelle cellule in metafase, di preferenza nello stesso tipo di cellule e secondo un protocollo di sensibilità comparabile, per stabilire se i micronuclei sono dovuti a una rottura cromosomica (sapendo che la perdita di cromosomi non sarebbe rilevata durante la prova di aberrazione cromosomica). Analisi Tutti i vetrini, compresi quelli del solvente e quelli non trattati (se utilizzati) e dei controlli positivi, devono essere codificati indipendentemente prima dell'esame al microscopio delle frequenze micronucleari. È opportuno utilizzare tecniche adeguate per controllare le distorsioni o le derive in caso di utilizzazione di un sistema di analisi automatica come la citometria a flusso, la citometria a scansione laser o l'analisi di immagini. Indipendentemente dalla piattaforma automatizzata utilizzata per valutare i micronuclei, il CBPI, il RI, l'RPD o il RICC dovrebbero essere valutati contestualmente. Nelle colture trattate con citoB, le frequenze dei micronuclei dovrebbero essere analizzate in almeno 2 000 cellule binucleate per concentrazione e controllo (83), ripartite in ugual misura tra le repliche, se sono utilizzate le repliche. Se si utilizza un'unica coltura per ciascuna concentrazione (cfr. paragrafo 28), si valutino almeno 2 000 cellule binucleate per coltura (83). Se per il conteggio è disponibile un numero sostanzialmente inferiore a 1 000 cellule binucleate per coltura (per colture doppie), o a 2 000 cellule (per una coltura singola), per ogni concentrazione, e se non si rileva un aumento significativo dei micronuclei, la prova dev'essere ripetuta con più cellule o con una concentrazione meno tossica, a seconda di quale opzione sia più appropriata. Si deve prestare attenzione a non prendere in considerazione cellule binucleate con forme irregolari o che evidenzino due nuclei di dimensioni marcatamente diverse. Inoltre, le cellule binucleate non vanno confuse con cellule multinucleate a scarsa diffusione. Le cellule contenenti più di due nuclei principali non devono essere considerate ai fini della ricerca di micronuclei, poiché la frequenza di riferimento dei micronuclei può essere superiore in queste cellule (84). Il conteggio delle cellule mononucleate è ammissibile se è dimostrato che la sostanza chimica in esame interferisce con l'attività della citoB. In tal caso, può essere opportuno ripetere la prova senza citoB. Il conteggio delle cellule mononucleate oltre a quello di cellule binucleate potrebbe fornire informazioni pertinenti (85) (86), ma non è obbligatorio. Per quanto riguarda le linee cellulari, se il test è condotto senza l'impiego di citoB, i micronuclei vanno conteggiati in almeno 2 000 cellule per concentrazione di prova (83) e controllo, distribuite equamente tra le repliche, se si usano le repliche. Se si utilizza un'unica coltura per ciascuna concentrazione (cfr. paragrafo 28), si devono valutare almeno 2 000 cellule binucleate per coltura. Se per il conteggio è disponibile un numero notevolmente inferiore a 1 000 cellule per coltura, o a 2 000 cellule se si usa un'unica coltura, per ogni concentrazione, e se non si rileva un aumento significativo dei micronuclei, la prova dev'essere ripetuta con più cellule o con una concentrazione meno tossica, a seconda di quale opzione sia più appropriata. Nei trattamenti con citoB dev'essere determinato un CBPI o un RI per valutare la proliferazione cellulare (cfr. l'appendice 2) a partire da almeno 500 cellule per coltura. Al contrario, nei trattamenti effettuati senza citoB, è fondamentale dimostrare che le cellule nella coltura sono in fase di proliferazione, come si è detto ai paragrafi 24-28. Competenza del laboratorio Al fine di acquisire una sufficiente esperienza con una prova prima di utilizzarla regolarmente, il laboratorio deve aver effettuato una serie di esperienze con sostanze chimiche di riferimento positive che agiscono mediante meccanismi differenti (almeno una con attivazione metabolica e una senza attivazione metabolica, e una secondo un meccanismo aneugeno, con sostanze selezionate dalle sostanze chimiche elencate nella tabella 1) e con vari controlli negativi (comprese le colture non trattate e vari solventi/mezzi disperdenti). Le risposte dei controlli positivi e negativi devono essere coerenti con la letteratura. Questo requisito non si applica ai laboratori con esperienza, ossia che dispongono di una base di dati storici quale definita ai paragrafi da 49 a 52. Una selezione di sostanze chimiche utilizzate come sostanze di controllo positivo (cfr. tabella 1) deve essere verificata nell'ambito di trattamenti di breve e di lungo periodo in assenza di attivazione metabolica, nonché nell'ambito di un trattamento di breve durata in presenza di attivazione metabolica, allo scopo di dimostrare che il laboratorio dispone della competenza necessaria per individuare le sostanze clastogene e aneugene, determinare l'efficacia del sistema di attivazione metabolica e dimostrare l'adeguatezza dei metodi di conteggio (analisi visiva al microscopio, citometria a flusso, citometria a scansione laser o analisi di immagini). Occorrerà definire un intervallo di concentrazione delle sostanze chimiche selezionate che consenta di ottenere aumenti riproducibili e correlati alla concentrazione rispetto ai valori di fondo allo scopo di dimostrare la sensibilità e l'intervallo dinamico del sistema di prova. Dati storici di controllo Il laboratorio deve stabilire:
Al momento della prima acquisizione di dati per stabilire la distribuzione dei controlli negativi storici, i dati dei controlli negativi paralleli devono essere coerenti con i dati pubblicati, se disponibili. Successivamente, man mano che nuovi dati sperimentali ampliano la distribuzione dei controlli, i dati dei controlli negativi paralleli dovrebbero, idealmente, rientrare nei limiti di controllo al 95 % di tale distribuzione (87) (88). La base di dati dei precedenti controlli negativi del laboratorio deve inizialmente essere costituita da almeno 10 esperimenti, ma preferibilmente ne dovrebbe contare almeno 20, realizzati in condizioni di prova comparabili. I laboratori devono applicare metodi di controllo della qualità quali grafici di controllo (ad es. C-chart o X-bar-chart (88)) per stabilire la variabilità dei propri dati di controllo positivi e negativi e dimostrare che il laboratorio “domina” la metodologia (83). Altre raccomandazioni sul modo di costituire e di utilizzare dati storici (criteri di inclusione e di esclusione dei dati nella base e criteri di accettabilità per un esperimento) sono reperibili nella bibliografia (87). Qualsiasi modifica del protocollo sperimentale deve essere valutata in termini di coerenza dei dati con le banche dati storiche esistenti del laboratorio. Qualsiasi incoerenza significativa dovrebbe portare alla creazione di una nuova banca dati di controlli storici. I dati dei controlli negativi dovrebbero consistere nell'incidenza delle cellule con micronuclei da un'unica coltura o dalla somma delle colture replicate, come descritto al paragrafo 28. I controlli negativi paralleli dovrebbero idealmente collocarsi entro limiti di controllo al 95 % della distribuzione dei dati storici dei controlli negativi contenuti nella banca dati del laboratorio (87) (88). Se i dati dei controlli negativi paralleli si situano al di fuori dei limiti di controllo al 95 %, la loro inclusione nella distribuzione dei controlli storici può essere accettata a condizione che tali dati non siano valori erratici estremi e che sia provato che il sistema di prova è «sotto controllo» (cfr. paragrafo 50) e che non vi sono stati problemi tecnici o errori umani. DATI E RELAZIONE Presentazione dei risultati Se si ricorre alla tecnica dell'inibizione della citocinesi, per valutare l'induzione di micronuclei si usano soltanto le frequenze di cellule binucleate con micronuclei (indipendentemente dal numero di micronuclei per cellula). Il conteggio del numero di cellule con uno, duo o più micronuclei può essere registrato separatamente e potrebbe fornire informazioni utili, ma non è obbligatorio. Per tutte le colture di controllo trattate, le colture di controllo positive e negative si determini in parallelo la citotossicità e/o la citostasi (16). Nell'eventualità in cui si ricorra al metodo dell'inibizione della citocinesi, per tutte le colture trattate e per i controlli deve essere calcolato il CBPI o l'RI quale misurazione del ritardo del ciclo cellulare. Nei trattamenti senza citoB si utilizza l'RPD o il RICC (cfr. l'appendice 2). Devono essere forniti dati sulle singole colture. Tutti i dati vanno riportati sinteticamente in una tabella. Criteri di accettabilità L'accettazione delle prove si basa sui seguenti criteri:
Analisi e interpretazione dei risultati A condizione che tutti i criteri di ammissibilità siano soddisfatti, una sostanza chimica in esame si intende chiaramente positiva se, nelle condizioni sperimentali esaminate (cfr. paragrafi 36-39):
La sostanza chimica in esame è in grado di indurre rotture cromosomiche e/o acquisizione o perdita di materiale cromosomico in questo sistema di prova. Raccomandazioni sui metodi statistici più adeguati sono disponibili anche in letteratura (90) (91) (92). A condizione che tutti i criteri di ammissibilità siano soddisfatti, una sostanza chimica in esame si intende chiaramente negativa se, in tutte le condizioni sperimentali esaminate (cfr. paragrafi 36-39):
Si considera allora che sostanza chimica in esame non sia in grado di indurre rotture cromosomiche e/o acquisizione o perdita di materiale cromosomico in questo sistema di prova. Raccomandazioni sui metodi statistici più adeguati sono disponibili anche in letteratura (90) (91) (92). Non è necessario verificare una risposta chiaramente positiva o negativa. Se la risposta non è chiaramente negativa o positiva, come descritto in precedenza, e/o al fine di contribuire a stabilire la rilevanza biologica di un risultato, i dati devono venire sottoposti al giudizio di esperti e/o a indagini più approfondite. Può essere utile esaminare cellule supplementari (se del caso) o ripetere l'esperienza, eventualmente in condizioni sperimentali modificate [ad es. intervallazione delle concentrazioni, altre condizioni di attivazione metabolica (concentrazione o origine di S9)]. In rari casi, anche dopo ulteriori indagini, l'insieme dei dati non consente stabilire se si abbia un risultato positivo o negativo; il risultato dovrà pertanto essere dichiarato equivoco. Le sostanze chimiche che inducono formazione di micronuclei nel test MNvit possono avere questo effetto come conseguenza dell'induzione della rottura cromosomica, della perdita di cromosomi o di entrambi tali eventi. Si può ricorrere a un'ulteriore analisi con anticorpi anticinetocore, sonde centromero-specifiche in situ o altri metodi per stabilire se il meccanismo di induzione di micronuclei è dovuto a un'attività clastogena e/o aneugena. Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti.
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Aneugeno : detto di qualsiasi sostanza chimica o processo che, interagendo con le componenti del ciclo mitotico e meiotico di divisione cellulare, determina aneuploidia nelle cellule o negli organismi. Aneuploidia : qualsiasi deviazione dal normale numero diploide (o aploide) di cromosomi da parte di uno o più cromosomi, ma non dell'intero corredo di cromosomi (poliploidia). Apoptosi : morte cellulare programmata caratterizzata da una serie di fasi che portano alla disintegrazione delle cellule in piccole vescicole chiuse da membrana, le quali vengono poi eliminate mediante fagocitosi o «shedding» (clivaggio dei ricettori di membrana). Proliferazione cellulare : aumento del numero di cellule dovuto alla divisione mitotica delle cellule. Centromero : regione del DNA di un cromosoma in cui i cromatidi appaiati sono mantenuti uniti e nella quale sono localizzati fianco a fianco i due cinetocori. Sostanza chimica : una sostanza o una miscela. Concentrazioni : concentrazioni finali della sostanza chimica in esame nel terreno di coltura. Clastogeno : qualsiasi sostanza chimica o processo in grado di provocare aberrazioni cromosomiche strutturali in popolazioni di cellule o organismi eucarioti. Citocinesi : il processo di divisione cellulare immediatamente successivo alla mitosi e che porta alla formazione di due cellule figlie, ciascuna contenente un unico nucleo. Indice della cinetica di proliferazione cellulare (CBPI) : la proporzione di cellule nella fase della seconda divisione cellulare nella popolazione trattata rispetto al controllo (cfr. l'appendice 2 per la formula). Citostasi : inibizione della crescita cellulare (cfr. l'appendice 2 per la formula). Citotossicità : per le prove contemplate nel presente metodo di prova effettuate in presenza di citocalasina B, la citotossicità corrisponde ad una diminuzione dell'indice della cinetica della proliferazione cellulare (CBPI) o dell'indice di replicazione (RI) delle cellule trattate rispetto al controllo negativo (cfr. paragrafo 26 e appendice 2). Per le prove contemplate nel presente metodo di prova effettuate in assenza di citocalasina B, la citotossicità corrisponde ad una diminuzione del raddoppiamento relativo della popolazione (RPD) o dell'aumento relativo della conta cellulare (RICC) delle cellule trattate rispetto al controllo negativo (cfr. paragrafo 27 e allegato 2). Genotossico : termine generico che comprende tutti i tipi di danno a carico del DNA o dei cromosomi, tra cui rotture, cancellazioni, alterazioni e collegamenti nucleotidi, riarrangiamenti, mutazioni genetiche, aberrazioni cromosomiche e aneuploidia. Non tutti i tipi di effetti genotossici determinano alterazioni cromosomiche o danni permanenti ai cromosomi. Cellule in interfase : cellule non ancora approdate alla fase mitotica. Cinetocore : struttura proteica che si forma nel centromero di un cromosoma e alla quale si agganciano i microtubuli del fuso durante la divisione cellulare, consentendo un movimento ordinato dei cromosomi delle cellule figlie verso i poli di queste ultime. Micronuclei : piccoli nuclei, distinti e soprannumerari rispetto al principale nucleo delle cellule, prodotti durante la telofase della mitosi o della meiosi da frammenti residui di cromosomi o da cromosomi interi. Mitosi : divisione del nucleo cellulare, solitamente suddivisa in profase, prometafase, metafase, anafase e telofase. Indice mitotico : il rapporto tra cellule in metafase e il numero totale di cellule osservate in una popolazione cellulare; un'indicazione del grado di proliferazione cellulare di una popolazione. Mutageno : un fattore in grado di provocare mutazioni ereditarie delle sequenze di coppie di basi del DNA nei geni o della struttura dei cromosomi (aberrazioni cromosomiche). Non disgiunzione : incapacità della coppia di cromatidi di separarsi e di migrare in due cellule figlie distinte in fase di divisione, con la conseguenza che una cellula figlia presenterà un numero anomalo di cromosomi. Stato della p53 : la proteina p53 partecipa alla regolazione del ciclo cellulare, all'apoptosi e alla riparazione del DNA. Le cellule carenti di proteine p53 funzionali, che non sono in grado di arrestare il ciclo cellulare o di eliminare cellule danneggiate tramite apoptosi o altri meccanismi (ad esempio induzione di riparazione del DNA) relativi alle funzioni della p53 in risposta ad alterazioni del DNA, sarebbero in teoria più soggette a mutazioni genetiche o aberrazioni cromosomiche. Poliploidia : aberrazioni numeriche dei cromosomi che interessano l'intero corredo cromosomico di cellule o organismi, a differenza dall'aneuploidia, che invece interessa un solo cromosoma o più cromosomi. Indice di proliferazione (PI) : metodo per la misurazione della citotossicità nel caso in cui non si utilizzi citoB (cfr. l'appendice 2 per la formula). Aumento relativo della conta cellulare (Relative Increase in Cell Count, RICC) : metodo per la misurazione della citotossicità nel caso in cui non si utilizzi citoB (cfr. l'appendice 2 per la formula). Raddoppiamento relativo della popolazione (Relative Population Doubling, RPD) : metodo per la misurazione della citotossicità nel caso in cui non si utilizzi citoB (cfr. l'appendice 2 per la formula). Indice di replicazione (RI) : la proporzione dei cicli di divisione cellulare completati in una coltura trattata rispetto al controllo non trattato, durante il periodo di esposizione e il recupero (cfr. l'appendice 2 per la formula). Frazione S9 di fegato : supernatante dell'omogenato di fegato centrifugato a 9 000 g (estratto di fegato crudo). Miscela di frazione S9 : miscela di frazione S9 di fegato e dei cofattori necessari all'attività degli enzimi metabolici. Controllo con solvente : termine generico che designa le colture di controllo che ricevono unicamente il solvente utilizzato per dissolvere la sostanza chimica in esame. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Controllo non trattato : colture non sottoposte a trattamento (che non ricevono alcuna sostanza chimica in esame né solvente) ma preparate in parallelo e in modo identico alle culture esposte alla sostanza chimica in esame. Appendice 2 FORMULE PER LA VALUTAZIONE DELLA CITOTOSSICITÀ Nei trattamenti con citoB , la valutazione della tossicità deve basarsi sull'indice della cinetica di proliferazione cellulare (CBPI) o sull'indice di replicazione (RI) (17) (69). Il CBPI indica il numero medio di nuclei per singola cellula durante il periodo di esposizione alla citoB e può essere usato per calcolare la proliferazione cellulare. L'RI indica il numero relativo di cicli cellulari per singola cellula durante il periodo di esposizione alla citoB nelle colture trattate rispetto alle colture di controllo e può essere usato per calcolare la percentuale di citostasi: % Citostasi = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)} e:
dove:
Quindi, un CBPI pari a 1 (tutte le cellule sono mononucleate) equivale a una percentuale di citostasi del 100 %. Citostasi = 100 — RI
Quindi, un RI del 53 % significa che, rispetto al numero di cellule che si sono divise per formare cellule binucleate e multinucleate nella coltura di controllo, nella coltura trattata soltanto il 53 % di tale numero si è diviso nella coltura trattata, ossia si ha una percentuale di citostasi pari al 47 %. Nei trattamenti senza citoB , si raccomanda di valutare la citotossicità sulla base dell'aumento relativo delle conte cellulari (RICC) o del raddoppiamento relativo della popolazione (RPD) (69), poiché entrambi i metodi tengono conto della proporzione di popolazione cellulare che è andata incontro a divisione.
dove: Raddoppiamento della popolazione = [log (numero di cellule dopo il trattamento ÷ numero di cellule iniziale)] ÷ log 2 Quindi, un RICC o un RPD del 53 % indica una citotossicità/citostasi del 47 %. Con un indice di proliferazione (PI) è possibile valutare la citotossicità contando il numero di cloni consistenti in 1 cellula (cl1), 2 cellule (cl2), da 3 a 4 cellule (cl4) e da 5 a 8 cellule (cl8)
Il PI è stato usato come parametro di citotossicità valido e affidabile anche per le linee cellulari coltivate in vitro in assenza di citoB (35) (36) (37) (38) e può essere considerato un utile parametro supplementare. In ogni caso, occorre misurare il numero di cellule prima del trattamento, che deve essere identico per le colture trattate e per le colture di controllo negativo. Benché l'RCC (cioè il numero di cellule nelle colture trattate/numero di cellule nelle colture di controllo) fosse utilizzato come parametro di citotossicità in passato, esso non è più raccomandato in quanto può indurre una sottostima della citotossicità. Se si utilizzano sistemi di analisi automatica, come ad esempio la citometria a flusso, la citometria a scansione laser o l'analisi di immagini, il numero di cellule nella formula può essere sostituito dal numero di nuclei. Nelle colture di controllo negativo, il raddoppiamento della popolazione o l'indice di replicazione devono essere compatibili con l'obbligo di campionare le cellule dopo il trattamento al termine di un periodo equivalente a circa 1,5-2 volte la durata del ciclo cellulare normale. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(15) |
Nella parte B sono aggiunti i seguenti capitoli: «B.59 Sensibilizzazione cutanea in chemico: saggio di reattività peptidica diretta (DPRA) INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 442C (2015). Un sensibilizzante cutaneo è una sostanza che determina una risposta allergica a seguito del contatto con la pelle, secondo la definizione del Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche (UN GHS) delle Nazioni Unite (1) e del regolamento (CE) n. 1272/2008 dell'Unione europea relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele (CLP) (18). Il presente metodo di prova descrive una procedura in chemico (saggio di reattività peptidica diretta — DPRA) da utilizzare per distinguere i sensibilizzanti dai non sensibilizzanti, secondo la definizione del sistema UN GHS e della classificazione CLP. Vi è consenso generale circa le principali fasi del processo biologico di sensibilizzazione cutanea. Le conoscenze attuali dei meccanismi chimico-biologici associati alla sensibilizzazione cutanea sono stati sintetizzati nel concetto del meccanismo d'azione degli eventi avversi (Adverse Outcome Pathway — AOP) (2), dall'evento molecolare scatenante fino agli effetti avversi per la salute (dermatite allergica da contatto negli esseri umani o ipersensibilità da contatto nei roditori)n passando attraverso le fasi intermedie. Nel caso dell'AOP relativo alla sensibilizzazione cutanea l'evento molecolare scatenante è il legame covalente tra sostanze chimiche elettrofile e i centri nucleofili nelle proteine della pelle. Generalmente, la valutazione della sensibilizzazione cutanea è effettuata su cavie. I metodi classici che utilizzano le cavie GMPT (Guinea Pig Maximisation Test) di Magnusson e Kligman e il test di Buehler (metodo di prova B.6 (3)), studiano sia le fasi di induzione che quelle di reazione della sensibilizzazione cutanea. Sono utilizzati inoltre un test sui topi, il test sui linfonodi locali (LLNA, metodo di prova B.42 (4)) e le sue due varianti non radioattive, LLNA: DA (TM B.50 (5)) e LLNA: BrdU-ELISA (metodo di prova B.51 (6)), che riguardano esclusivamente la reazione di induzione, garantendo un vantaggio rispetto ai test che utilizzano le cavie per quanto riguarda il benessere degli animali e la possibilità di ottenere una misurazione obiettiva della fase di induzione della sensibilizzazione cutanea. Più di recente sono stati considerati scientificamente validi per la valutazione del rischio di sensibilizzazione cutanea delle sostanze chimiche metodi in chemico e in vitro di tipo meccanicistico. Tuttavia, sarà necessario combinare metodi diversi dalla sperimentazione animale (in silico, in chemico, in vitro) nell'ambito delle metodologie integrate di prova e valutazione (IATA — Integrated Approaches to Testing and Assessment) per poter sostituire integralmente le prove sugli animali attualmente in uso, tenuto conto della limitata copertura meccanicistica dell'AOP di ciascuno dei metodi di prova senza ricorso agli animali (2) (7). Il DPRA viene proposto per lo studio dell'evento molecolare scatenante nell'AOP relativo alla sensibilizzazione cutanea, ovvero la reattività proteica, mediante quantificazione della reattività delle sostanze chimiche in esame a modelli peptidici di sintesi contenenti lisina o cisteina (8). I valori percentuali di deplezione dei peptidi con cisteina e lisina sono quindi utilizzati per classificare le sostanze in una delle quattro classi di reattività al fine di contribuire a distinguere i sensibilizzanti dai non sensibilizzanti cutanei (9). Il DPRA è stato oggetto di uno studio di validazione in uno dei laboratori di riferimento dell'Unione europea per le alternative alla sperimentazione animale (European Union Reference Laboratory for Alternatives to Animal Testing — EURL ECVAM) e di una successiva revisione inter pares sotto la guida del comitato scientifico consultivo dello stesso laboratorio (ESAC) ed è stato considerato scientificamente valido (10) per essere impiegato nell'ambito di una metodologia integrata di tipo IATA, in modo da distinguere tra sensibilizzanti e non sensibilizzanti cutanei a fini di classificazione ed etichettatura del pericolo. Nella letteratura scientifica figurano esempi dell'uso dei dati DPRA in combinazione con altre informazioni (11) (12) (13) (14). Le definizioni sono riportate nell'appendice I. CONSIDERAZIONI PRELIMINARI, APPLICABILITÀ E LIMITI La correlazione tra reattività proteica e potenziale di sensibilizzazione cutanea è ben documentata (15) (16) (17). Tuttavia, poiché il legame alle proteine costituisce solo uno degli eventi fondamentali dell'AOP sensibilizzazione cutanea, pur trattandosi dell'evento molecolare scatenante, le informazioni relative alla reattività alle proteine ottenute con metodi sperimentali e non sperimentali possono non essere di per sé sufficienti a decretare l'assenza di un potenziale di sensibilizzazione cutanea delle sostanze chimiche. È opportuno pertanto considerare i dati generati utilizzando il presente metodo di prova nel contesto di metodologie integrate quali IATA, combinandoli con altre informazioni complementari, ricavate ad esempio da test in vitro che prendano in esame altri eventi fondamentali della sensibilizzazione cutanea AOP come pure da metodi non sperimentali, compreso quello del read-across in relazione a sostanze chimiche analoghe. Il presente metodo di prova può essere utilizzato, in combinazione con altre informazioni complementari, per contribuire a distinguere tra sensibilizzanti cutanei (ad esempio, categoria 1 del GHS dell'ONU/CLP ) e non sensibilizzanti cutanei nell'ambito di un approccio integrato (IATA). Il presente metodo di prova non può essere utilizzato isolatamente, né per dividere i sensibilizzanti cutanei in sottocategorie 1A e 1B, quali definite dal GHS dell'ONU/CLP, né per prevedere la potenza di sensibilizzazione in sede di valutazione della sicurezza. Tuttavia, in funzione del quadro normativo applicabile, un risultato positivo ottenuto applicando il DPRA può essere usato isolatamente per classificare una sostanza chimica nella categoria 1 del GHS dell'ONU/CLP. È stato dimostrato che il metodo di prova DPRA può essere applicato in laboratori con esperienza nel campo dell'analisi mediante cromatografia liquida ad alta prestazione (HPLC). Il livello di riproducibilità delle previsioni che ci può aspettare dal metodo di prova è nell'ordine dell'85 % a livello intra-laboratorio e dell'80 % a livello inter-laboratorio (10). I risultati dello studio di validazione (18) e degli studi pubblicati (19) indicano globalmente che l'accuratezza del DPRA nella distinzione tra sensibilizzanti (ad esempio, categoria 1 del GHS dell'ONU/CLP ) e non sensibilizzanti è pari all'80 % (N=157) per una sensibilità dell'80 % (88/109) e una specificità del 77 % (37/48) rispetto ai risultati ottenuti con il LLNA. È probabile che il metodo DPRA sottostimi le sostanze chimiche con una potenza di sensibilizzazione cutanea da bassa a moderata (ad esempio, sottocategoria 1B del GHS dell'ONU/CLP) rispetto alle sostanze chimiche con una potenza di sensibilizzazione cutanea elevata (ad esempio, sottocategoria 1A del GHS dell'ONU/CLP) (18) (19). Tuttavia, i valori di accuratezza forniti in questa sede per il DPRA in quanto metodo utilizzato isolatamente sono squisitamente indicativi, in quanto il metodo di prova dovrebbe essere considerato in combinazione con altre fonti di informazione nell'ambito di un approccio IATA e in conformità alle disposizioni del precedente punto 9. Inoltre, nel valutare i metodi di studio della sensibilizzazione cutanea che non utilizzano la sperimentazione animale si deve tenere presente che il LLNA, come pure altre prove che utilizzano la sperimentazione animale, possono non riflettere interamente la situazione per la specie di interesse, ovvero gli esseri umani. I dati generali disponibili indicano che il DPRA è applicabile alle sostanze chimiche in esame relative a diversi gruppi funzionali organici, meccanismi di reazione, potenza di sensibilizzazione cutanea (quale determinata dagli studi in vivo) e proprietà fisico-chimiche (8) (9) (10) (19). Queste informazioni, nel loro insieme, indicano l'utilità del DPRA per l'identificazione del rischio di sensibilizzazione cutanea. Il termine “sostanza chimica in esame” utilizzato nel presente metodo di prova designa l'oggetto della prova e non si riferisce all'applicabilità del DPRA alle prove di sostanze e/o miscele. Il presente metodo di prova non è applicabile alle prove di composti di metalli in quanto è noto che essi reagiscono con le proteine con meccanismi differenti dal legame covalente. Una sostanza chimica in esame dovrebbe essere solubile in un solvente adeguato a una concentrazione finale di 100 mM (cfr. il punto 18). Tuttavia, le sostanze chimiche in esame non solubili a tale concentrazione possono essere sottoposte a prova a concentrazioni inferiori. In questo caso un risultato positivo può essere utilizzato per individuare una sostanza chimica in esame come sensibilizzante cutaneo; un risultato negativo, invece, non dovrebbe permettere di trarre conclusioni negative quanto alla mancanza di reattività. Attualmente disponiamo di informazioni limitate quanto all'applicabilità del DPRA alle miscele di composizione conosciuta (18) (19). Si ritiene tuttavia che il DPRA sia tecnicamente applicabile alle prove di sostanze multi-costituenti e alle miscele di composizione conosciuta (cfr. punto 18). Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tale verifica non è necessaria se la miscela viene sottoposta a prova in ottemperanza a un obbligo regolamentare. L'attuale modello predittivo non può essere utilizzato con le miscele complesse la cui composizione è sconosciuta o le sostanze di composizione sconosciuta o variabile, i prodotti di una reazione complessa o i materiali origine biologica (ovvero le sostanze UVCB) a causa del rapporto molare definito della sostanza chimica in esame e del peptide. A tal fine è necessario elaborare un nuovo modello predittivo basato su un metodo gravimetrico. Il metodo di prova non deve essere utilizzato con altre categorie specifiche di sostanze chimiche qualora possa essere dimostrata la sua non applicabilità a tali categorie specifiche. Il presente metodo di prova è del tipo in chemico che non prevede l'intervento di un sistema metabolico. Le sostanze chimiche che necessitano di una bioattivazione enzimatica per dispiegare il loro potenziale di sensibilizzazione cutanea (ad esempio, i pro-apteni) non possono essere individuate applicando il presente metodo di prova. In alcuni casi, le sostanze chimiche che diventano sensibilizzanti in seguito a una trasformazione abiotica (ad esempio, i pre-apteni) sono correttamente individuate dal metodo di prova (18). Alla luce di quanto precede, i risultati negativi ottenuti applicando il metodo di prova dovrebbero essere interpretati nel contesto dei limiti indicati e in combinazione con altre fonti di informazione nell'ambito di un approccio IATA. Le sostanze chimiche in esame che non presentano un legame covalente con il peptide ma che ne favoriscono l'ossidazione (ad esempio, dimerizzazione della cisteina) potrebbero determinare una sopravvalutazione della deplezione peptidica risultante in eventuali falsi positivi e/o nell'assegnazione della sostanza a una classe di reattività più elevata (cfr. punti 29 e 30). Come descritto il precedenza, il DPRA aiuta a distinguere tra sensibilizzanti e non sensibilizzanti cutanei. Tuttavia, esso potrebbe contribuire anche alla valutazione della potenza di sensibilizzazione cutanea (11) se utilizzato nell'ambito di approcci integrati quali IATA. Sono tuttavia necessarie ulteriori ricerche, di preferenza basate su dati relativi all'uomo, per determinare in che modo il DPRA possa contribuire alla valutazione della potenza di sensibilizzazione. PRINCIPIO DELLA PROVA Il DPRA è un metodo di prova in chemico che permette di quantificare la concentrazione residua di peptide contenente cisteina o lisina dopo 24 ore di incubazione con la sostanza chimica in esame a una temperatura di 25(± 2,5) °C. I peptidi di sintesi contengono fenilalanina per facilitare l'individuazione. La concentrazione peptidica relativa è misurata mediante cromatografia liquida ad alta prestazione (HPLC) con eluizione a gradiente e rilevatore UV a 220 nm. I valori percentuali di deplezione dei peptidi con cisteina e lisina sono quindi calcolati e utilizzati in un modello predittivo (cfr. punto 29) che consente di assegnare la sostanza chimica in esame a una delle quattro classi di reattività utilizzate per distinguere la sensibilizzazione dalla non sensibilizzazione cutanea. Prima di utilizzare il presente metodo di prova per prove di routine, i laboratori dovrebbero dimostrare le loro competenze tecniche utilizzando le 10 sostanze di riferimento elencate nell'appendice 2. PROCEDURA Il presente metodo di prova si basa sul protocollo DPRA DB-ALM no 154 (20) utilizzato per lo studio di validazione coordinato dal laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale (EURL ECVAM). Si raccomanda di utilizzare questo protocollo nell'applicazione e nell'impiego del metodo in laboratorio. Di seguito è fornita una descrizione dei principali elementi e procedure del DPRA. Qualora venga utilizzata una configurazione alternativa della HPLC, è necessario dimostrarne l'equivalenza con la configurazione convalidata descritta nel protocollo DB-ALM (ad esempio, sottoponendo a prova le sostanze di riferimento di cui all'appendice 2). Preparazione dei peptidi contenenti cisteina o lisina Soluzioni madre dicisteina (Ac-RFAACAA-COOH) e lisina (Ac-RFAAKAA-COOH) contenenti peptidi di sintesi di purezza superiore all'85 %, ma di preferenza compresa tra il 90 e il 95 %, dovrebbero essere preparate al momento dell'uso e immediatamente prima dell'incubazione con la sostanza chimica in esame. La concentrazione finale del peptide con cisteina deve essere pari a 0,667 mM in tampone fosfato a pH 7,5 mentre la concentrazione finale del peptide con lisina deve essere pari a 0,667 mM in tampone di acetato ammonico a pH 10,2. Le sequenze di analisi HPLC devono essere programmate per fare in modo che l'analisi HPLC non superi le 30 ore. Per l'analisi HPLC utilizzata nello studio di convalida e descritta nel presente metodo di prova possono essere impiegati fino a 26 campioni di analisi (comprendenti la sostanza chimica in esame, il controllo positivo e il numero appropriato di controlli con solvente sulla base del numero di singoli solventi utilizzati nella prova, ciascuno sottoposto a prova in tre repliche) in un'unica sequenza HPLC. Tutte le repliche analizzate nella stessa sequenza devono utilizzare le stesse soluzioni madre di peptide con cisteina e lisina. Si raccomanda di verificare l'adeguata solubilità dei singoli lotti di peptide prima del loro utilizzo. Preparazione della sostanza chimica in esame La solubilità della sostanza chimica in esame in un solvente adeguato deve essere verificata prima della prova applicando il metodo di solubilizzazione descritto nel protocollo DPRA DB-ALM (20). Per solvente adeguato si intende quello che permette di dissolvere completamente la sostanza chimica in esame. Nel DPRA la sostanza chimica in esame è incubata in forte eccesso con i peptidi contenenti cisteina o lisina; pertanto un'ispezione visiva che verifichi la formazione di una soluzione chiara è considerata sufficiente per accertare che la sostanza chimica in esame (e tutti i suoi costituenti qualora si sottoponga a prova una sostanza multi-costituente o una miscela) sia dissolta. Solventi adeguati sono l'acetonitrile, l'acqua, una miscela 1:1 di acqua: acetonitrile, l'isopropanolo, l'acetone o una miscela 1:1 di acetone: acetonitrile. Possono essere utilizzati anche altri solventi, a condizione che non abbiano un impatto sulla stabilità del peptide monitorata con i controlli di riferimento C (ovvero campioni costituiti dal solo peptide dissolto nel solvente appropriato; cfr. appendice 3). Come ultima opzione, qualora la sostanza chimica in esame non sia solubile in nessuno dei citati solventi, si può cercare di solubilizzarla in 300 μL di DMSO diluendo la soluzione risultante in 2 700 μL di acetonitrile; se la sostanza chimica in esame non è solubile nemmeno in questa miscela si può provare a solubilizzare la sostanza chimica in esame in 1 500 μL di DMSO diluendo la soluzione risultante in 1 500 μL di acetonitrile. La sostanza chimica in esame deve essere prepesata in recipienti di vetro e dissolta immediatamente prima della prova in un solvente appropriato per preparare una soluzione a 100mM. Nel caso delle miscele e delle sostanze multi-costituenti di composizione nota, si deve determinare un singolo livello di purezza sommando la proporzione dei costituenti (esclusa l'acqua) e un singolo peso molecolare apparente, considerando il singolo peso molecolare di ciascuno dei costituenti della miscela (esclusa l'acqua) e le relative proporzioni. La purezza e il peso molecolare apparente che ne risultano sono quindi utilizzati per calcolare il peso della sostanza chimica in esame necessario per preparare una soluzione a 100 mM. Nel caso di polimeri per i quali non sia possibile determinare un peso molecolare predominante, per preparare la soluzione a 100 mM si può prendere in considerazione il peso molecolare del monomero (o il peso molecolare apparente dei diversi monomeri che costituiscono il polimero). Tuttavia quando si sottopongono a prova miscele, sostanze multi-costituenti o polimeri di composizione nota, si dovrebbe considerare anche la possibilità di sottoporre a prova la sostanza chimica pura. Nel caso dei liquidi la sostanza chimica pura deve essere sottoposta a prova senza diluirla preventivamente ma incubandola con peptidi contenenti cisteina e lisina aventi un rapporto molare pari a rispettivamente 1:10 e 1:50. Nel caso dei solidi la sostanza chimica in esame deve essere dissolta alla sua concentrazione massima di solubilità nello stesso solvente utilizzato per preparare la soluzione apparente a 100 mM. La sostanza chimica in esame deve essere quindi sottoposta a prova senza diluirla ulteriormente, incubandola con peptidi contenenti cisteina e lisina aventi un rapporto pari a rispettivamente 1:10 e 1:50. Risultati concordanti (reattivi o non reattivi) tra la soluzione apparente a 100 mM e la sostanza chimica pura dovrebbero consentire di formulare conclusioni definitive. Preparazione del controllo positivo, dei controlli di riferimento e dei controlli di co-eluizione Come controllo positivo viene utilizzata l'aldeide cinnamica (CAS 104-55-2; purezza grado alimentare ≥95 %) a una concentrazione di 100 mM in acetonitrile. Altri controlli positivi adeguati, che forniscano di preferenza valori di deplezione medi, possono essere utilizzati in caso di disponibilità di dati storici da cui ricavare criteri di accettabilità comparabili per la sequenza. Nella sequenza di analisi HPLC devono inoltre essere inclusi controlli di riferimento (ovvero campioni contenenti esclusivamente il peptide dissolto nel solvente appropriato) da utilizzare per verificare l'adeguatezza del sistema HPLC prima di procedere all'analisi (controlli di riferimento A), la stabilità nel tempo dei controlli di riferimento (controlli di riferimento B) e accertarsi che il solvente utilizzato per dissolvere la sostanza chimica in esame non abbia un impatto sulla percentuale di deplezione del peptide (controlli di riferimento C) (cfr. appendice 3). Per ciascuna sostanza chimica viene utilizzato un controllo di riferimento appropriato al fine di calcolare la percentuale di deplezione del peptide (cfr. punto 26). Inoltre, per ciascuna sostanza chimica in esame deve essere incluso nella sequenza un controllo di co-eluizione costituito dalla sola sostanza chimica in esame allo scopo di individuare un'eventuale co-eluizione della sostanza chimica in esame con il peptide contenente lisina o cisteina. Incubazione della sostanza chimica in esame con le soluzioni di peptide contenenti cisteina e lisina Le soluzioni di peptide contenenti cisteina e lisina devono essere incubate in dispensatori automatici di vetro con la sostanza chimica in esame con un rapporto pari a rispettivamente 1:10 e 1:50. Se si osserva un precipitato immediatamente dopo l'aggiunta della soluzione con la sostanza chimica in esame alla soluzione con il peptide a causa della ridotta idrosolubilità della sostanza chimica in esame, non è possibile conoscere con certezza il quantitativo di tale sostanza rimasto nella soluzione che potrebbe reagire con il peptide. In questi casi, pertanto, mentre si può utilizzare un risultato positivo, un risultato negativo va interpretato con la dovuta cautela (cfr. anche le disposizioni del punto 11 per le prove delle sostanze chimiche non solubili fino a concentrazioni di 100 mM). La soluzione di reazione deve essere conservata al buio a 25(± 2,5) C per 24 ± 2 ore prima di procedere all'analisi HPLC. Ciascuna sostanza chimica in esame deve essere analizzata in tre repliche per entrambi i peptidi. Prima dell'analisi HPLC i campioni devono essere ispezionati visivamente. Qualora si osservi un precipitato o una separazione di fase, è opportuno centrifugare i campioni a bassa velocità (100-400 × g) per far scendere il precipitato in fondo al recipiente, in quanto un eccesso di precipitato potrebbe ostruire i tubi o le colonne del cromatografo. Qualora si osservi un precipitato o una separazione di fase dopo il periodo di incubazione, la deplezione dei peptidi può essere sottostimata e, in caso di risultato negativo, non è possibile stabilire con sufficiente certezza una mancanza di reattività. Preparazione della curva di taratura standard HPLC Per entrambi i peptidi con cisteina e lisina deve essere generata una curva di taratura standard. Gli standard di peptidi sono preparati in una soluzione del 20 % o 25 % di acetonitrile: tampone utilizzando un tampone di fosfato (pH 7,5) per il peptide con cisteina e un tampone di acetato ammonico (pH 10,2) per il peptide con lisina. Utilizzando standard ottenuti per diluizione in serie della soluzione madre di peptide (0,667 mM), sono preparate 6 soluzioni di taratura comprese tra 0,534 e 0,0167 mM. Nella curva di taratura standard è inserito un bianco costituito dal tampone di diluizione. Una curva di taratura adeguata presenta un coefficiente di r2>0,99. Preparazione e analisi HPLC Prima di effettuare l'analisi deve essere verificata l'adeguatezza del sistema HPLC. La deplezione dei peptidi è misurata con la HPLC associata a un rilevatore UV (un rivelatore a serie di fotodiodi o un rivelatore ad assorbanza UV di lunghezza d'onda pari a 220nm). La colonna appropriata è installata nel sistema HPLC. La configurazione HPLC descritta nel protocollo validato utilizza di preferenza una colonna Zorbax SB-C-18 2,1 mm × 100 mm × 3,5 μm. Con questa colonna HPLC a fase inversa l'intero sistema deve essere equilibrato a 30 °C con il 50 % di fase A (0,1 % (v/v) di acido trifluoroacetico in acqua) e il 50 % di fase B (0,085 % (v/v) di acido trifluoroacetico in acetonitrile) per almeno due ore prima dell'esecuzione dell'analisi. L'analisi HPLC deve essere effettuata utilizzando una portata di 0,35 ml/min e un gradiente lineare compreso tra il 10 % e il 25 % di acetonitrile per 10 minuti seguita da un rapido aumento dell'acetonitrile (fino al 90 %) allo scopo di eliminare gli altri materiali. Devono essere iniettati volumi uguali di ciascuno standard, campione e controllo. Tra le iniezioni la colonna deve essere riequilibrata alle condizioni iniziali per 7 minuti. Qualora venga utilizzata una differente colonna HPLC a fase inversa, può rivelarsi necessario adeguare i parametri di configurazione sopraindicati per assicurare una eluizione e integrazione adeguate dei peptidi con cisteina e lisina, compreso il volume iniettato, che può variare a seconda del sistema utilizzato (di solito compreso tra 3 e 10 μl). Qualora venga utilizzata una configurazione alternativa della HPLC, è essenziale dimostrarne l'equivalenza con la configurazione convalidata sopradescritta (ad esempio, sottoponendo a prova le sostanze di riferimento di cui all'appendice 2). L'assorbanza è misurata a 220 nm. Qualora sia utilizzato un rivelatore a serie di fotodiodi deve essere rilevata anche l'assorbanza a 258nm. Va segnalato che taluni lotti di acetonitrile possono avere un'incidenza negativa sulla stabilità del peptide e che quest'ultima deve essere valutata quando si utilizza un nuovo lotto di acetonitrile. Il rapporto tra l'area di picco a 220 nm e l'area di picco 258 nm può essere utilizzato come indicatore della co-eluizione. Per ciascun campione un rapporto compreso tra il 90 % e il 100 % della media (19) dei rapporti delle superfici ottenuti per i campioni di controllo costituisce un buon indicatore del fatto che non vi è stata co-eluizione. Determinate sostanze chimiche in esame possono favorire l'ossidazione del peptide con cisteina. Il picco del peptide con cisteina dimerizzato può essere verificato visivamente. Qualora si ritenga che si è verificata una dimerizzazione, occorre segnalarlo, in quanto la percentuale della deplezione del peptide può essere sovrastimata e determinare previsioni di falsi positivi e/o l'assegnazione della sostanza a una classe di reattività più elevata (cfr. punti 29 e 30). L'analisi HPLC dei peptidi con cisteina e lisina può essere effettuata nello stesso momento (se sono disponibili due sistemi HPLC) o in giorni diversi. Se l'analisi è effettuata in giorni diversi, è necessario preparare sul momento tutte le soluzioni delle sostanze chimiche in esame prima di ciascuna analisi. L'analisi deve essere programmata in modo da garantire che l'iniezione del primo campione inizi tra 22 e 26 ore dopo la miscelazione della sostanza chimica in esame con la soluzione peptidica. Le sequenze di analisi HPLC devono essere programmate per fare in modo che l'analisi HPLC non superi le 30 ore. Per l'analisi HPLC utilizzata nello studio di validazione e descritta nel presente metodo di prova possono essere impiegati fino a 26 campioni di analisi in un'unica sequenza HPLC (si veda anche il punto 17). L'appendice 3 presenta un esempio di sequenza di analisi HPLC. DATI E RELAZIONE Valutazione dei dati La concentrazione del peptide con cisteina e lisina è determinata con metodo fotometrico a 220 nm in ciascun campione misurando l'area di picco (l'area al di sotto della curva, AUC) nei picchi appropriati e calcolando la concentrazione peptidica mediante la curva lineare di taratura derivata dagli standard. La percentuale di deplezione dei peptidi è determinata in ciascun campione misurando l'area dei picchi dei pertinenti controlli di riferimento C (cfr. appendice 3), applicando la formula riportata di seguito.
Criteri di accettabilità Affinché una sequenza sia considerata valida, devono essere soddisfatti i seguenti criteri:
Se uno di questi criteri non è soddisfatto è necessario ripetere la sequenza. Affinché i risultati per la sostanza chimica in esame siano considerati validi, devono essere soddisfatti i seguenti criteri:
Modello predittivo Il valore del tasso medio di deplezione della cisteina e della lisina è calcolato per ciascuna sostanza chimica in esame. Per il calcolo della media una deplezione negativa è considerata pari a “0”. Se si utilizza il modello predittivo cisteina 1:10/lisina 1:50 di cui alla tabella 1, per l'individuazione dei sensibilizzanti e dei non sensibilizzanti nell'ambito di una approccio IATA si utilizza una soglia del 6,38 % di deplezione media dei peptidi. L'applicazione del modello predittivo per assegnare una sostanza chimica in esame a una classe di reattività (ad esempio, bassa, moderata ed elevata) potrebbe rivelarsi utile per valutare la potenza di sensibilizzazione nell'ambito di un approccio IATE. Tabella 1 Modello predittivo (20) cisteina 1:10/lisina 1:50
Vi possono essere casi in cui la sostanza chimica in esame (la sostanza o uno o più costituenti di una sostanza multi-costituente o una miscela) presenti un assorbanza significativa a 220 nm e abbia lo stesso tempo di ritenzione del peptide (co-eluizione). La co-eluizione può essere risolta con un adeguamento minimo della configurazione HPLC per separare ulteriormente il tempo di eluizione della sostanza chimica in esame e del peptide. Qualora venga utilizzata una configurazione alternativa della HPLC per risolvere il problema della co-eluizione, si deve dimostrarne l'equivalenza con la configurazione convalidata (ad esempio, sottoponendo a prova le sostanze di riferimento di cui all'appendice 2). In caso di co-eluizione, il picco del peptide non può essere integrato e non è possibile calcolarne il tasso di deplezione. Se la co-eluizione della sostanza chimica in esame si verifica con i peptidi contenenti sia cisteina che lisina, l'analisi viene registrata come “non conclusiva”. Nei casi in cui si registra co-eluizione soltanto con il peptide contenente lisina, si può utilizzare il modello predittivo per la cisteina 1:10 di cui alla tabella 2. Tabella 2 Modello predittivo1cisteina 1:10 (22)
Vi possono essere altri casi in cui la sovrapposizione del tempo di ritenzione tra la sostanza chimica in esame e uno dei peptidi è incompleta. In questi casi i valori di deplezione del peptide possono essere stimati e utilizzati nel modello predittivo cisteina1:10/lisina 1:50, per quanto non sia possibile assegnare con accuratezza la sostanza chimica in esame a una classe di reattività. Un'unica analisi HPLC per il peptide contenente cisteina e quello contenente lisina dovrebbe essere sufficiente per la sostanza chimica in esame qualora i risultati siano inequivocabili. Tuttavia, qualora i risultati siano prossimi alla soglia utilizzata per stabilire la positività o negatività (ovvero, risultati borderline), possono rendersi necessarie ulteriori prove. Nei casi in cui la percentuale media di deplezione si collochi nella fascia compresa tra il 3 % e il 10 % per il modello predittivo cisteina 1:10/lisina 1:50 o la percentuale di deplezione della cisteina si collochi nella fascia compresa tra il 9 % e il 17 % per il modello predittivo cisteina1:10, si deve considerare l'eventualità di una seconda analisi o di una terza in caso di risultati discordanti tra le prime due. Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti.
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Accuratezza : grado di concordanza tra i risultati ottenuti con il metodo e i valori di riferimento comunemente accettati. Misura l'efficienza del metodo di prova e uno degli aspetti della sua “pertinenza”. Il termine è spesso utilizzato come sinonimo di “concordanza” a indicare la percentuale di risultati corretti di un metodo di prova (21). AOP ( Adverse Outcome Pathway — meccanismo d'azione degli eventi avversi): sequenza di eventi che, a partire dalla struttura chimica di una sostanza chimica bersaglio o di un gruppo di sostanze chimiche simili, attraverso l'evento molecolare scatenante, produce un effetto avverso in vivo (2). Curva di taratura : relazione tra il valore della risposta sperimentale e la concentrazione analitica (detta anche curva standard) di una sostanza nota. Sostanza chimica : una sostanza o una miscela. Coefficiente di variazione : misura della variabilità calcolata per una serie di dati ottenuti dalle repliche dividendo la deviazione standard per la media. Tale misura può essere moltiplicata per 100 per ottenere una percentuale. Pericolo : proprietà intrinseca di un agente o di una situazione in grado di causare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti. IATA ( Integrated Approach to Testing and Assessment — Approccio integrato di prova e valutazione) : approccio strutturato utilizzato per l'individuazione del rischio (potenziale), la caratterizzazione del pericolo (potenza) e/o per la valutazione di sicurezza (potenziale/potenza e esposizione) di una sostanza o gruppo di sostanze chimiche che integra in modo strategico e ponderato tutti i dati pertinenti per orientare una decisione di tipo regolamentare concernente il pericolo potenziale e/o il rischio e/o la necessità di effettuare altre prove mirate e, pertanto, limitate allo stretto necessario. Evento molecolare scatenante : perturbazione, chimicamente indotta, di un sistema biologico a livello molecolare individuata come l'evento scatenante nell'azione dell'effetto avverso (AOP). Miscela : una miscela o una soluzione composta da due o più sostanze che non interagiscono tra di loro (1). Sostanza mono-costituente : sostanza, definita dalla sua composizione quantitativa, in cui un costituente principale è presente almeno all'80 % (p/p). Sostanza multi-costituente : sostanza, definita dalla sua composizione quantitativa, in cui più di un costituente principale è presente in una concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multi-costituente è il risultato di un processo di fabbricazione. La differenza tra una miscela e una sostanza multi-costituente è data dal fatto che la miscela è ottenuta miscelando due o più sostanze senza che vi sia reazione chimica. Una sostanza multi-costituente è il risultato di una reazione chimica. Controllo positivo : replica che contiene tutti i componenti di un sistema di prova e che è trattata con una sostanza che notoriamente induce una reazione positiva. Per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo, la portata della reazione positiva non dovrebbe essere eccessiva. Controllo di riferimento : un campione non trattato che contiene tutti i componenti di un sistema di prova, compreso il solvente e il mezzo disperdente usati con la sostanza chimica in esame e gli altri campioni di controllo al fine di stabilire la reazione di base nei campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente o mezzo disperdente. Nelle prove con controlli negativi paralleli, questo campione dimostra anche se il solvente o mezzo disperdente è in grado di interagire con il sistema di prova. Pertinenza : descrizione della relazione tra la prova e l'effetto di interesse e indicazione del fatto che la prova sia o meno significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di un metodo di prova (21). Affidabilità : misura in cui un metodo può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna ai laboratori e la ripetibilità fra i laboratori (21). Riproducibilità : concordanza tra i risultati ottenuti saggiando la stessa sostanza chimica in esame in applicazione dello stesso protocollo sperimentale (vedi Affidabilità) (21). Sensibilità : proporzione di tutte le sostanze chimiche in esame positive/attive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo (21). Specificità : proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo di prova (21). Sostanza : elementi chimici e loro componenti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a conservare la stabilità del prodotto e le eventuali impurità derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza ripercussioni sulla stabilità della sostanza o modifiche della sua composizione (1). Adeguatezza del sistema : determinazione dell'efficacia degli strumenti (ad esempio, sensibilità) mediante l'analisi di uno standard di riferimento prima di sottoporre a prova il lotto analitico (22). Sostanza chimica in esame : il termine “sostanza chimica in esame” designa la sostanza oggetto della prova. Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (GHS dell'ONU) : sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (1). UVCB : sostanze la cui composizione non è conosciuta o è variabile, prodotti di una reazione complessa o i materiali origine biologica. Metodo di prova valido : un metodo di prova che si ritiene abbia sufficiente pertinenza e affidabilità per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in termini assoluti ma soltanto in relazione a un obiettivo definito (21). Appendice 2 SOSTANZE DI RIFERIMENTO Sensibilizzazione cutanea in chemico: saggio di reattività peptidica diretta Prima di utilizzare sistematicamente il presente metodo di prova, i laboratori sono tenuti a dimostrare la loro competenza tecnica ottenendo correttamente la predizione attesa con il metodo DPRA per le 10 sostanze di riferimento raccomandate nella tabella 1 e ottenendo valori di deplezione della cisteina e della lisina che si collochino nei rispettivi intervalli di riferimento per 8 delle 10 sostanze di riferimento per ciascun peptide. Le sostanze di riferimento di cui trattasi sono state selezionate per rappresentare la gamma di risposte in caso di rischio di sensibilizzazione cutanea. Altri criteri di selezione erano la disponibilità in commercio delle sostanze, la qualità elevata dei dati di riferimento in vivo disponibili, la qualità elevata dei dati in vitro ottenuti con il DPRA e il loro utilizzo nello studio di convalida coordinato dal laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale (EURL ECVAM) per dimostrare che il metodo di prova è stato attuato con successo dai laboratori partecipanti allo studio. Tabella 1 Sostanze di riferimento raccomandate per la verifica della competenza tecnica con il saggio di reattività peptidica diretta (DPRA).
Appendice 3 ESEMPI DI SEQUENZA DI ANALISI
B.60 Sensibilizzazione cutanea in vitro: metodo di prova della luciferasi ARE-Nrf2 INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 442D (2015). Per sensibilizzante cutaneo s'intende una sostanza che provoca una reazione allergica a contatto con la pelle, secondo la definizione del Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS) (1) e del regolamento (CE) n. 1272/2008 dell'Unione europea relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele (27). Il presente metodo di prova riguarda una procedura in vitro (il saggio della luciferasi ARE-Nrf2) da utilizzare per distinguere tra sostanze sensibilizzanti della pelle e sostanze non sensibilizzanti, secondo la definizione del sistema UN GHS (1) e del regolamento (CE) n. 1272/2008. Vi è consenso generale circa le principali fasi del processo biologico di sensibilizzazione cutanea. Le attuali conoscenze relative ai meccanismi chimici e biologici associati alla sensibilizzazione cutanea sono state riassunte nel concetto del meccanismo d'azione degli eventi avversi (AOP, Adverse Outcome Pathway) (2), dall'evento molecolare scatenante fino agli effetti avversi per la salute (dermatite allergica da contatto negli esseri umani o ipersensibilità da contatto nei roditori) (2) (3), passando attraverso le fasi intermedie. L'evento molecolare scatenante è la costituzione di un legame covalente tra le sostanze elettrofile e i centri nucleofili delle proteine della pelle. Il secondo evento chiave dell'AOP avviene a livello di cheratinociti e comprende risposte infiammatorie e fenomeni di espressione genica, associati a vie di segnalazione intercellulare come le vie dipendenti dall'elemento di risposta antiossidante/elettrofilo (ARE, Antioxidant Response Element). Il terzo evento chiave è l'attivazione di cellule dendritiche, generalmente valutate attraverso l'espressione di specifici marcatori di superficie cellulare, chemochine e citochine. Il quarto evento chiave è la proliferazione dei linfociti T, valutati indirettamente con il saggio LLNA (Local Lymph Node Assay) su topi (4). Tipicamente, la valutazione della sensibilizzazione cutanea è effettuata su cavie. I metodi classici che prevedono l'impiego di cavie — prova GMPT (Guinea Pig Maximisation Test) di Magnusson e Kligman e prova di Buehler (metodo di prova B.6 (5)) — studiano sia la fase di induzione sia la fase di elicitazione della sensibilizzazione cutanea. Anche un saggio su topi, l'LLNA (Local Lymph Node Assay) (metodo di prova B.42 (4)) e le sue due varianti che non utilizzano isotopi radioattivi, LLNA: DA (TM B.50 (6)) e LLNA: BrdU-ELISA (metodo di prova B.51 (7)), che valutano esclusivamente la risposta all'induzione, hanno ottenuto riconoscimento grazie al fatto che rispetto alle prove su cavie presentano il vantaggio di preservare maggiormente il benessere animale e di fornire una misurazione oggettiva della fase di induzione della sensibilizzazione cutanea. Più di recente, i metodi di prova in chemico e in vitro di tipo meccanicistico sono stati considerati scientificamente validi per la valutazione del pericolo di sensibilizzazione cutanea correlato alle sostanze chimiche. Tuttavia saranno necessarie combinazioni di metodi che non utilizzano la sperimentazione sugli animali (in silico, in chemico, in vitro), nel quadro degli approcci integrati in materia di prove e valutazioni (IATA, Integrated Approaches to Testing and Assessment), per poter sostituire completamente le prove sugli animali attualmente in uso, tenuto conto della limitata copertura meccanicistica dell'AOP di ciascuno dei metodi di prova senza ricorso agli animali (2) (3). Il presente metodo di prova (saggio della luciferasi ARE-Nrf2) è proposto per lo studio del secondo evento chiave descritto al paragrafo 2. È stato segnalato che i sensibilizzanti cutanei provocano l'induzione dei geni che sono regolati dall'elemento di risposta antiossidante (ARE) (8) (9). Piccole sostanze elettrofile come i sensibilizzanti cutanei possono agire sulla proteina sensore Keap1 (Kelch-like ECH-associated protein 1), ad esempio attraverso una modifica covalente del suo residuo di cisteina, provocando la sua dissociazione dal fattore di trascrizione Nrf2 (nuclear factor-erythroid 2-related factor 2). Il fattore Nrf2 dissociato può quindi attivare i geni che dipendono dall'ARE, come quelli che rappresentano il codice genetico degli enzimi detossificanti della fase II (8) (10) (11). Attualmente l'unico saggio della luciferasi ARE-Nrf2 in vitro contemplato dal presente metodo di prova è il saggio KeratinoSensTM, per il quale sono stati completati studi di valutazione (9) (12) (13) seguiti da una valutazione inter pares indipendente condotta dal laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale (EURL ECVAM) (14). Il saggio KeratinoSensTM è stato ritenuto scientificamente valido, nel quadro di un approccio integrato di tipo IATA, per poter distinguere tra sostanze sensibilizzanti della pelle e sostanze non sensibilizzanti ai fini della classificazione e dell'etichettatura dei pericoli (14). I laboratori che intendono applicare questo metodo di prova possono ottenere la linea cellulare ricombinante utilizzata nel saggio KeratinoSensTM sottoscrivendo un accordo di licenza con lo sviluppatore del metodo di prova (15). Le definizioni di questi parametri figurano nell'appendice 1. CONSIDERAZIONI INIZIALI, APPLICABILITÀ E LIMITI Poiché l'attivazione della via Keap1-Nrf2-ARE riguarda solo il secondo evento chiave dell'AOP riguardante la sensibilizzazione cutanea, le informazioni ottenute grazie a metodi di prova basati sull'attivazione di questa via probabilmente non bastano da sole a trarre conclusioni sul potenziale di sensibilizzazione cutanea delle sostanze chimiche. Pertanto, i dati generati con l'utilizzo del presente metodo di prova dovrebbero essere considerati nel contesto di un approccio integrato di tipo IATA e combinati con altre informazioni complementari, ad esempio quelle ottenute con i saggi in vitro concernenti altri eventi chiave dell'AOP riguardante la sensibilizzazione cutanea, nonché con metodi che non fanno uso della sperimentazione, compresi i metodi read-across utilizzati con sostanze chimiche analoghe. La letteratura scientifica contiene esempi di utilizzo del metodo di prova della luciferasi ARE-Nrf2 in combinazione con altre informazioni (13) (16) (17) (18) (19). Il presente metodo di prova può essere utilizzato per aiutare a distinguere tra sostanze sensibilizzanti della pelle (categoria 1 del sistema UN GHS/CLP) e non sensibilizzanti nel quadro di un approccio integrato di tipo IATA. Questo metodo di prova non può essere utilizzato da solo, né per la classificazione dei sensibilizzanti cutanei nelle sottocategorie 1A e 1B del sistema UN GHS/CLP né per prevedere la potenza di sensibilizzazione nel quadro delle valutazioni relative alla sicurezza. Tuttavia, in funzione del quadro regolamentare applicabile, un risultato positivo può essere ritenuto sufficiente per classificare una sostanza chimica nella categoria 1 del sistema UN GHS/CLP. Sulla base della serie di dati ottenuta grazie allo studio di validazione e alle sperimentazioni interne effettuate durante la valutazione inter pares indipendente del metodo di prova, si può concludere che il saggio KeratinoSensTM è trasferibile ai laboratori con esperienza di coltura cellulare. Il livello di riproducibilità atteso delle predizioni è dell'ordine dell'85 % a livello intralaboratorio e a livello interlaboratorio (14). L'accuratezza (77 % — 155/201), la sensibilità (78 % — 71/91) e la specificità (76 % — 84/110) delle predizioni ottenute con il saggio KeratinoSensTM per distinguere le sostanze sensibilizzanti della pelle (categoria 1 del sistema UN GHS/CLP) dalle sostanze non sensibilizzanti, rispetto ai risultati dell'LLNA, sono state calcolate prendendo in considerazione tutti i dati trasmessi all'EURL ECVAM per la valutazione, anche inter pares, del metodo di prova (14). Queste cifre sono simili a quelle pubblicate di recente sulla base di sperimentazioni interne relative a circa 145 sostanze (77 % di precisione, 79 % di sensibilità, 72 % di specificità) (13). Il saggio KeratinoSensTM ha una maggiore probabilità di fornire una sottopredizione delle sostanze chimiche aventi una potenza di sensibilizzazione cutanea da debole a moderata (sottocategoria 1B del sistema UN GHS/CLP) rispetto alle sostanze chimiche aventi una potenza di sensibilizzazione cutanea elevata (sottocategoria 1A del sistema UN GHS/CLP) (13) (14). L'insieme di queste informazioni indica l'utilità del saggio KeratinoSensTM come elemento in grado di contribuire all'identificazione del pericolo di sensibilizzazione cutanea. Tuttavia i valori relativi all'accuratezza del saggio KeratinoSensTM come metodo di prova autonomo sono soltanto indicativi, poiché tale metodo dovrebbe essere considerato in combinazione con altre fonti di informazioni, nel quadro di un approccio di tipo IATA, e conformemente alle disposizioni di cui al paragrafo 9. Inoltre, nel valutare i metodi di studio della sensibilizzazione cutanea che non utilizzano la sperimentazione sugli animali, occorre tenere presente che l'LLNA e gli altri metodi che ricorrono alla sperimentazione sugli animali potrebbero non rispecchiare interamente la situazione riscontrata presso la specie di interesse, ossia gli esseri umani. Nel presente metodo di prova, il termine “sostanza chimica in esame” fa riferimento alla sostanza oggetto della sperimentazione e non è correlato all'applicabilità del metodo di prova della luciferasi ARE-Nrf2 all'esecuzione di prove sulle sostanze e/o sulle miscele. I dati attualmente disponibili dimostrano che il saggio KeratinoSensTM è applicabile all'esecuzione di prove su sostanze chimiche che coprono una varietà di gruppi funzionali organici, meccanismi di reazione, valori della potenza di sensibilizzazione cutanea (determinati con gli studi in vivo) e proprietà fisico-chimiche (9) (12) (13) (14). Le prove sono state eseguite soprattutto su sostanze mono-costituente, sebbene sia disponibile anche una quantità limitata di dati relativi alle prove sulle miscele (20). Il metodo di prova è in ogni caso tecnicamente applicabile alle prove sulle sostanze multicostituente e sulle miscele. Tuttavia, prima di ricorrere a questo metodo di prova per testare una miscela per ottenere dati a fini regolamentari, è opportuno chiedersi se, e in caso affermativo perché, i dati ottenuti possono essere ritenuti idonei per i fini regolamentari previsti. Tali considerazioni non sono necessarie se l'esecuzione della prova sulla miscela risponde a un obbligo normativo. Inoltre, quando si testano sostanze multicostituente o miscele, occorre prestare attenzione alle possibili interferenze dei costituenti citotossici con le risposte osservate. Il metodo di prova è applicabile alle sostanze chimiche solubili o che formano una dispersione stabile (colloide o sospensione in cui la sostanza chimica in esame non si deposita né si separa dal solvente formando più fasi) nell'acqua o nel DMSO (ciò vale per tutti i componenti della sostanza chimica in esame se la prova è eseguita su una sostanza multicostituente o su una miscela). Le sostanze chimiche che non soddisfano queste condizioni alla massima concentrazione finale richiesta di 2 000 μM (cfr. il paragrafo 22) possono comunque essere testate a concentrazioni inferiori. In tal caso, i risultati che soddisfano i criteri di positività descritti al paragrafo 39 possono essere comunque utilizzati nel processo di identificazione della sostanza chimica come sensibilizzante cutaneo, mentre un risultato negativo ottenuto con concentrazioni < 1 000 μM dovrebbe essere considerato non conclusivo (cfr. il modello predittivo di cui al paragrafo 39). In generale, le sostanze con un LogP inferiore o uguale a 5 sono state testate con successo, mentre le sostanze estremamente idrofobiche con un LogP superiore a 7 sono al di fuori dei limiti di applicabilità noti del metodo di prova (14). Per le sostanze con un LogP compreso tra 5 e 7 le informazioni disponibili sono limitate. I risultati negativi devono essere interpretati con prudenza perché le sostanze reattive esclusivamente nei confronti dei residui di lisina possono essere identificate come negative dal metodo di prova. Inoltre, a causa della capacità metabolica limitata della linea cellulare utilizzata (21) e a causa delle condizioni sperimentali, anche i pro-apteni (sostanze chimiche che richiedono attivazione enzimatica, ad esempio mediante enzimi P450) e i pre-apteni (sostanze chimiche attivate tramite auto-ossidazione), in particolare quelli a ossidazione lenta, possono dare risultati negativi. D'altra parte le sostanze chimiche in esame che non agiscono come sensibilizzanti, ma che sono comunque fattori di stress chimico, possono dar luogo a falsi positivi (14). Inoltre, se le sostanze chimiche in esame sono altamente citotossiche, la loro valutazione non sempre è affidabile. Infine, le sostanze chimiche che interferiscono con l'enzima luciferasi possono determinare un rischio di confusione con l'attività della luciferasi nei saggi a livello cellulare provocando un'inibizione apparente o una maggiore luminescenza (22). Ad esempio, è stato segnalato che le concentrazioni di fitoestrogeni superiori a 1 μM interferirebbero con i segnali di luminescenza in altri saggi con geni reporter basati sulla luciferasi a causa della sovrattivazione del gene reporter della luciferasi (23). Di conseguenza, è necessario esaminare attentamente l'espressione della luciferasi ottenuta in presenza di concentrazioni elevate di fitoestrogeni o di sostanze chimiche simili che si ritiene provochino una sovrattivazione del gene reporter della luciferasi comparabile a quella causata dai fitoestrogeni (23). Nel caso in cui si possa dimostrare che il metodo di prova non è applicabile ad altre categorie specifiche di sostanze chimiche, è opportuno evitare di utilizzarlo per tali categorie. Oltre ad essere di ausilio nella distinzione tra sostanze sensibilizzanti della pelle e sostanze non sensibilizzanti, il saggio KeratinoSensTM fornisce informazioni sulla relazione concentrazione-risposta che possono contribuire alla valutazione della potenza di sensibilizzazione, nel quadro di un approccio integrato di tipo IATA (19). Tuttavia, è necessario uno studio supplementare, che sia preferibilmente basato su dati affidabili rilevati sulle persone, per stabilire in che modo i risultati del saggio KeratinoSensTM possono contribuire alla valutazione della potenza (24) e alla classificazione dei sensibilizzanti in sottocategorie secondo il sistema UN GHS/CLP. PRINCIPIO DEL METODO Il metodo di prova della luciferasi ARE-Nrf2 utilizza una linea cellulare aderente immortalizzata derivata da cheratinociti umani HaCaT trasferiti in modo stabile con un plasmide selezionabile. La linea cellulare contiene il gene della luciferasi sotto il controllo trascrizionale di un promotore costitutivo fuso con un elemento ARE di un gene di cui è nota la regolazione positiva (up-regulation) della sua espressione sotto l'effetto dei sensibilizzanti cutanei (25) (26). Il segnale della luciferasi riflette l'attivazione da parte dei sensibilizzanti di geni endogeni dipendenti dal fattore Nrf2, e la dipendenza del segnale della luciferasi dal fattore Nrf2 nella linea cellulare ricombinante è stata dimostrata (27). Ciò consente la misurazione quantitativa (mediante rivelazione della luminescenza) dell'induzione del gene della luciferasi, grazie all'uso di substrati di luciferasi che producono una luminescenza soddisfacente, come indicatore dell'attività del fattore di trascrizione Nrf2 nelle cellule in seguito all'esposizione a sostanze elettrofile. Nel saggio KeratinoSens™ le sostanze chimiche in esame sono considerate positive se provocano un'induzione statisticamente significativa dell'attività della luciferasi superiore a una determinata soglia (ossia superiore a 1,5 volte, il che corrisponde a un aumento del 50 %), al di sotto di una concentrazione definita che non incide in misura significativa sulla vitalità cellulare (ossia al di sotto di 1 000 μM e a una concentrazione in cui la vitalità cellulare è superiore al 70 % (9) (12)). A tal fine si stabilisce il fattore massimo (Imax) per il quale è moltiplicata l'induzione dell'attività della luciferasi rispetto al controllo (negativo) con solvente. Inoltre, poiché le cellule sono esposte a una serie di concentrazioni delle sostanze chimiche in esame, la concentrazione necessaria per ottenere un'induzione statisticamente significativa dell'attività della luciferasi superiore alla soglia (ossia il valore EC1.5) dovrebbe essere interpolata a partire dalla curva dose-risposta (cfr. il paragrafo 32 per i calcoli). Infine, dovrebbero essere effettuate in parallelo misurazioni della citotossicità per stabilire se l'induzione dell'attività della luciferasi avviene in presenza di concentrazioni sub-citotossiche. Prima di utilizzare sistematicamente il saggio della luciferasi ARE-Nrf2 conforme al presente metodo di prova, i laboratori dovrebbero dimostrare la loro competenza tecnica utilizzando le sostanze elencate nell'appendice 2. Sono disponibili standard di prestazione (28) che facilitano la validazione di metodi di prova della luciferasi ARE-Nrf2 in vitro nuovi e modificati simili al saggio KeratinoSens™ e permettono di modificare rapidamente il presente metodo di prova per poterveli inserire. L'accettazione reciproca dei dati conformemente all'accordo OCSE sarà garantita solo per i metodi di prova validati secondo gli standard di prestazione, se tali metodi di prova sono stati esaminati e integrati nella corrispondente linea guida dell'OCSE. PROCEDURA Attualmente l'unico metodo coperto dal presente metodo di prova è il saggio scientificamente valido KeratinoSensTM (9) (12) (13) (14). Le procedure operative standard per il saggio KeratinoSensTM sono disponibili e dovrebbero essere applicate quando si utilizza questo metodo di prova in laboratorio (15). I laboratori che intendono applicare questo metodo di prova possono ottenere la linea cellulare ricombinante utilizzata nel saggio KeratinoSensTM sottoscrivendo un accordo di licenza con lo sviluppatore del metodo di prova. Nei seguenti paragrafi viene fornita una descrizione dei componenti e delle procedure principali del metodo di prova della luciferasi ARE-Nrf2. Preparazione delle colture di cheratinociti Bisogna utilizzare una linea cellulare transgenica che comporti un'introduzione stabile del gene reporter della luciferasi sotto il controllo dell'elemento ARE (ad es. la linea cellulare KeratinoSens™). Al ricevimento, le cellule sono moltiplicate (ad es. da 2 a 4 passaggi) al fine di costituire uno stock omogeneo da conservare in stato di congelamento. Le cellule di questo stock originario possono essere moltiplicate fino a un numero massimo di passaggi (ossia 25 nel caso del metodo KeratinoSensTM) e sono utilizzate per prove di routine con il mezzo di mantenimento idoneo (nel caso del metodo KeratinoSensTM, si tratta di DMEM contenente siero e geneticina). Per le prove, le cellule devono avere una confluenza pari all'80-90 % e occorre fare attenzione affinché non venga mai raggiunta la confluenza completa. Il giorno prima della prova, le cellule sono raccolte e ripartite su piastre a 96 pozzetti (10 000 cellule/pozzetto per il metodo KeratinoSensTM). Bisogna fare attenzione ad evitare la sedimentazione delle cellule al momento dell'inoculazione al fine di assicurare la distribuzione omogenea del numero di cellule tra i pozzetti. In caso di distribuzione non omogenea, questa fase potrebbe dar luogo a un'elevata variabilità da un pozzetto all'altro. Per ciascuna ripetizione si utilizzano tre repliche per misurare l'attività della luciferasi e una replica parallela per il saggio della vitalità cellulare. Preparazione della sostanza chimica in esame e delle sostanze di controllo La sostanza chimica in esame e le sostanze di controllo vengono preparate il giorno della prova. Per il saggio KeratinoSensTM, le sostanze chimiche in esame sono disciolte in dimetilsulfossido (DMSO) alla concentrazione finale desiderata (ad esempio 200 mM). Le soluzioni di DMSO possono essere considerate soluzioni autosterilizzanti e quindi non è necessario procedere alla sterilizzazione mediante filtrazione. Le sostanze chimiche non solubili in DMSO sono disciolte in acqua sterile o in un mezzo di coltura e le soluzioni sono sterilizzate, ad esempio mediante filtrazione. Per una sostanza chimica in esame senza un peso molecolare definito (Mw), si prepara una soluzione madre a una concentrazione predefinita (40 mg/ml o 4 % (p/v)) nel saggio KeratinoSensTM. In caso di utilizzo di solventi diversi dal DMSO, dall'acqua o dal mezzo di coltura, è necessario fornire un'adeguata motivazione scientifica. A partire dalle soluzioni madre della sostanza chimica in esame nel DMSO, sono preparate diluizioni in serie con il DMSO per ottenere 12 concentrazioni di riferimento della sostanza chimica da testare (da 0,098 a 200 mM nel saggio KeratinoSensTM). Se la sostanza chimica in esame non è solubile nel DMSO, le diluizioni per ottenere le concentrazioni di riferimento sono ottenute utilizzando acqua sterile o un mezzo di coltura sterile. Indipendentemente dal solvente utilizzato, le concentrazioni di riferimento sono quindi ulteriormente diluite 25 volte in un mezzo di coltura contenente siero e infine utilizzate per il trattamento dopo un'ulteriore diluizione con fattore 4 in modo che le concentrazioni finali della sostanza chimica in esame siano comprese tra 0,98 e 2 000 μM nel saggio KeratinoSensTM. Se giustificato, è possibile utilizzare concentrazioni diverse (ad esempio in presenza di un prodotto citotossico o scarsamente solubile). Il controllo negativo (con solvente) utilizzato nel saggio KeratinoSensTM è il DMSO (n. CAS 67-68-5, purezza ≥ 99 %), per il quale si preparano sei pozzetti per piastra. Esso subisce la stessa diluizione descritta al paragrafo 22 per le concentrazioni di riferimento, in modo tale che la concentrazione finale del controllo negativo (con solvente) sia pari all'1 %, valore che notoriamente non influisce sulla vitalità cellulare e corrisponde alla concentrazione del DMSO nella sostanza chimica in esame e nel controllo positivo. Se la sostanza chimica in esame non è solubile nel DMSO ed è stata diluita in acqua, il livello di DMSO in tutti i pozzetti della soluzione di prova finale deve essere ricondotta all'1 %, come per le altre sostanze chimiche in esame e sostanze di controllo. Il controllo positivo utilizzato nel caso del saggio KeratinoSensTM è l'aldeide cinnamica (n. CAS 14371-10-9, purezza ≥ 98 %), per la quale si prepara una serie di 5 concentrazioni di riferimento che vanno da 0,4 a 6,4 mM nel DMSO (a partire da una soluzione madre di 6,4 mM) che viene diluita seguendo la procedura descritta al paragrafo 22 per le concentrazioni di riferimento, in modo tale che la concentrazione finale del controllo positivo sia compresa tra 4 e 64 μM. Si possono utilizzare altri controlli positivi idonei, preferibilmente controlli che forniscono valori EC1.5 nella gamma dei valori medi, qualora siano disponibili dati storici per ricavare criteri di accettazione risultanti da procedure comparabili. Applicazione della sostanza chimica in esame e delle sostanze di controllo Per ogni sostanza chimica in esame e sostanza di controllo positivo, è necessario un esperimento finalizzato a ricavare una predizione (positiva o negativa) consistente in almeno due ripetizioni indipendenti contenenti ciascuna tre repliche (ossia n = 6). In caso di discordanza di risultati tra le due ripetizioni indipendenti, è necessaria una terza ripetizione contenente tre repliche (ossia n=9). Ogni ripetizione indipendente è eseguita in un giorno diverso con una nuova soluzione madre delle sostanze chimiche in esame e cellule raccolte in modo indipendente. Le cellule tuttavia possono provenire dallo stesso passaggio. Dopo avere effettuato l'inoculazione seguendo la procedura descritta al paragrafo 20, le cellule sono coltivate per 24 ore nelle piastre per microtitolazione a 96 pozzetti. Il mezzo va quindi rimosso e sostituito con un nuovo mezzo di coltura (150 μl di mezzo di coltura contenente siero ma senza geneticina nel caso del metodo KeratinoSensTM) al quale si aggiungono 50 μl della sostanza chimica in esame e delle sostanze di controllo diluite 25 volte. Almeno un pozzetto per piastra deve restare vuoto (senza cellule o trattamenti) al fine di determinare i valori di fondo. Le piastre trattate sono quindi messe in incubazione per 48 ore a una temperatura di 37(± 1) °C e con il 5 % di CO2 nel saggio KeratinoSensTM. È necessario prestare attenzione al fine di evitare l'evaporazione delle sostanze chimiche in esame volatili, come pure la contaminazione incrociata tra i pozzetti, ad esempio coprendo le piastre con un foglio di protezione prima dell'incubazione con le sostanze chimiche in esame. Misurazioni dell'attività della luciferasi I fattori determinanti per una lettura corretta della luminescenza sono tre:
Prima della prova è necessario effettuare un esperimento di controllo secondo la procedura descritta nell'appendice 3 per accertare che queste tre condizioni siano soddisfatte. Dopo 48 ore di esposizione alla sostanza chimica in esame e alle sostanze di controllo nel saggio KeratinoSensTM, le cellule sono lavate con tampone fosfato isotonico, e il tampone di lisi appropriato per la lettura della luminescenza è aggiunto a ciascun pozzetto per 20 minuti a temperatura ambiente. Le piastre contenenti lisato cellulare sono quindi collocate per la lettura nel luminometro, che nel saggio KeratinoSensTM è programmato per: i) aggiungere il substrato di luciferasi a ciascun pozzetto (ossia 50 μl), ii) attendere un secondo e iii) integrare l'attività della luciferasi per 2 secondi. L'eventuale utilizzo di impostazioni alternative, ad esempio a seconda del modello di luminometro utilizzato, deve essere giustificato. Inoltre, è possibile utilizzare anche un substrato luminescente, purché l'esperimento di controllo della qualità soddisfi i criteri descritti nell'appendice 3. Valutazione della citotossicità Per il saggio della vitalità cellulare KeratinoSensTM, dopo le 48 ore di esposizione il mezzo è sostituito con un nuovo mezzo contenente MTT (3-(4,5-dimetiltiazol-2-ile)-2,5-bromuro di difeniltetrazolio, tiazolil blu tetrazolio bromuro; n. CAS 298-93-1) e le cellule sono messe in incubazione per 4 ore a 37°C e con il 5 % di CO2. Il mezzo contenente MTT viene quindi rimosso e le cellule durante la notte sono sottoposte a lisi (ad esempio aggiungendo soluzione SDS al 10 % in ciascun pozzetto). In seguito ad agitazione, l'assorbimento è misurato a 600 nm con un fotometro. DATI E RELAZIONE Valutazione dei dati I seguenti parametri sono calcolati con il saggio KeratinoSensTM:
Equazione 1:
dove
EC1.5 è calcolato mediante interpolazione lineare con l'equazione 2, e il valore risultante di EC1.5 è la media geometrica delle singole ripetizioni. Equazione 2:
dove
La vitalità è calcolata con l'equazione 3: Equazione 3:
dove
IC50 e IC30 sono calcolati mediante interpolazione lineare con l'equazione 4 e i valori risultanti di IC50 e IC30 sono calcolati come media geometrica delle singole ripetizioni. Equazione 4:
dove
Per ciascuna concentrazione che determina un'induzione dell'attività della luciferasi > 1,5 volte, la significatività statistica è calcolata (ad esempio mediante un test t di Student bilaterale) confrontando i valori della luminescenza per le tre repliche di campioni con i valori della luminescenza nei pozzetti contenenti il controllo (negativo) con solvente, al fine di stabilire se l'induzione dell'attività della luciferasi è statisticamente significativa (p < 0,05). La concentrazione più bassa per la quale l'aumento dell'induzione dell'attività della luciferasi è > 1,5 volte è il valore che determina il valore EC1.5. In ciascun caso si verifica se tale valore è inferiore al valore IC30 e dunque se la riduzione della vitalità cellulare è inferiore al 30 % alla concentrazione che determina EC1.5. Si consiglia di verificare i dati visivamente con l'ausilio di grafici. In assenza di una curva dose-risposta chiaramente osservabile, o se la curva dose-risposta ottenuta è bifasica (ossia supera due volte la soglia di 1,5), è opportuno ripetere l'esperimento per verificare se il risultato è specificamente riconducibile alla sostanza chimica in esame o se invece è dovuto a un artefatto sperimentale. Nel caso in cui la risposta bifasica sia riproducibile in un esperimento indipendente, si deve indicare il valore EC1.5 più basso (la concentrazione alla quale la soglia di 1,5 è superata per la prima volta). Nei rari casi in cui si osserva un'induzione statisticamente non significativa superiore a 1,5 volte, seguita da un'induzione statisticamente significativa a una concentrazione più elevata, i risultati di questa ripetizione sono ritenuti validi e positivi solo se l'induzione statisticamente significativa superiore alla soglia di 1,5 è stata ottenuta per una concentrazione non citotossica. Infine, per le sostanze chimiche in esame che producono un aumento dell'induzione pari o superiore a 1,5 volte già alla concentrazione di prova più bassa di 0,98 μM, il valore EC1.5 < 0,98 è fissato sulla base di un esame visivo della curva dose-risposta. Criteri di accettabilità Quando si utilizza il saggio KeratinoSensTM devono essere soddisfatti i criteri di accettabilità elencati di seguito. In primo luogo, l'induzione dell'attività della luciferasi ottenuta con il controllo positivo (aldeide cinnamica) deve essere statisticamente significativa e superiore alla soglia di 1,5 (ad esempio con un test t) ad almeno una delle concentrazioni impiegate (da 4 a 64 μM). In secondo luogo, il valore EC1.5 deve rientrare tra due deviazioni standard dalla media storica dell'infrastruttura utilizzata per la prova (ad es. tra 7 μM e 30 μM, in base ai dati usati per la validazione), da aggiornare regolarmente. Inoltre, l'induzione media delle tre repliche per l'aldeide cinnamica a 64 μM deve essere compresa tra 2 e 8. Se quest'ultimo criterio non è soddisfatto, la curva dose-risposta dell'aldeide cinnamica dovrà essere verificata attentamente, e le prove potranno essere accettate solo in presenza di una curva dose-risposta chiaramente osservabile, con l'induzione dell'attività della luciferasi che aumenta con l'aumentare della concentrazione per il controllo positivo. Infine, il coefficiente medio di variazione della luminescenza per il controllo negativo (con solvente) DMSO deve essere inferiore al 20 % per ciascuna ripetizione con 6 pozzetti testati in triplicato. Se la variabilità è superiore, i risultati non devono essere presi in considerazione. Interpretazione dei risultati e modello predittivo Una predizione KeratinoSensTM è considerata positiva se tutte e 4 le condizioni elencate di seguito sono soddisfatte in 2 ripetizioni su 2 o in 2 ripetizioni su 3; in caso contrario, la predizione KeratinoSensTM è considerata negativa (figura 1):
Se, in una data ripetizione, le prime tre condizioni sono soddisfatte ma non si osserva una curva dose-risposta chiara per l'induzione della luciferasi, il risultato di tale ripetizione deve essere considerato non conclusivo e potrebbero essere necessarie ulteriori prove (figura 1). Inoltre, anche un risultato negativo ottenuto con concentrazioni < 1 000 μM (o < 200 μg/ml per le sostanze chimiche senza un peso molecolare definito) deve essere considerato non conclusivo (cfr. il paragrafo 11). Figura 1 Modello predittivo utilizzato nel saggio KeratinoSensTM. Una predizione KeratinoSensTM deve essere considerata nel quadro di un approccio di tipo IATA e conformemente alle disposizioni dei paragrafi 9 e 11.
In rari casi, le sostanze chimiche in esame che inducono un'attività della luciferasi a livelli molto vicini a quelli delle concentrazioni citotossiche possono essere positive in alcune ripetizioni in presenza di concentrazioni non citotossiche (ossia il valore EC1.5 che determina la concentrazione al di sotto (<) del valore IC30), e in altre ripetizioni unicamente ai livelli citotossici (ossia il valore EC1.5 che determina la concentrazione al di sopra (>) del valore IC30). Tali sostanze chimiche saranno ritestate procedendo a un'analisi dose-risposta più ristretta, con un fattore di diluizione più basso (ad esempio 1,33 o √2 (= 1,41) volte tra i pozzetti), al fine di determinare se l'induzione ha avuto luogo a livelli citotossici oppure no (9). Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti.
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Accuratezza : grado di concordanza tra i risultati ottenuti con il metodo e i valori di riferimento comunemente accettati. È una misura dell'efficienza del metodo di prova e un aspetto della pertinenza. Il termine è usato spesso in modo intercambiabile con «concordanza» per indicare la proporzione di risultati corretti di un metodo di prova (29). AOP (Adverse Outcome Pathway, meccanismo d'azione degli eventi avversi) : sequenza di eventi che, a partire dalla struttura chimica di una sostanza chimica bersaglio o di un gruppo di sostanze chimiche simili, attraverso l'evento molecolare scatenante, produce un effetto avverso in vivo (2). ARE (Antioxidant Response Element) : l'elemento di risposta agli antiossidanti (denominato anche EpRE, elemento di risposta agli elettrofili) è un elemento di risposta presente nella regione promotrice a monte di molti geni citoprotettivi e geni di fase II. Una volta attivato da Nfr2, esso media l'induzione trascrizionale di tali geni. Sostanza chimica : una sostanza o una miscela. Coefficiente di variazione : misura della variabilità calcolata per un gruppo di dati sulle repliche dividendo la deviazione standard per la media. Tale misura può essere moltiplicata per 100 per ottenere una percentuale. EC1.5 : concentrazione, stabilita per interpolazione, alla quale l'induzione della luciferasi è moltiplicata per 1,5. IC30 : concentrazione alla quale la vitalità cellulare è ridotta del 30 %. IC50 : concentrazione alla quale la vitalità cellulare è ridotta del 50 %. Pericolo : proprietà intrinseca di un agente o di una situazione in grado di provocare effetti avversi se un organismo, un sistema o una (sotto)popolazione vi sono esposti. IATA (Integrated Approaches to Testing and Assessment, approcci integrati in materia di prove e valutazioni) : approccio strutturato utilizzato per l'identificazione del pericolo (potenziale), la caratterizzazione del pericolo (potenza) e/o la valutazione della sicurezza (potenziale/potenza ed esposizione) di una sostanza chimica o di un gruppo di sostanze chimiche, che integra in modo strategico e ponderato tutti i dati pertinenti per orientare una decisione di tipo regolamentare concernente il pericolo potenziale e/o il rischio e/o la necessità di effettuare altre prove mirate e, pertanto, limitate allo stretto necessario. Imax : fattore d'induzione massima dell'attività della luciferasi, a qualsiasi concentrazione della sostanza chimica in esame, rispetto al controllo (negativo) con solvente. Keap1 : Keap1 (Kelch-like ECH-associated protein 1) è una proteina sensore in grado di regolare l'attività del fattore Nrf2. In condizioni non indotte la proteina sensore Keap1 attiva il fattore di trascrizione Nrf2 e ne provoca l'ubiquitinazione e la degradazione proteolitica nel proteasoma. In caso di modifica covalente dei residui di cisteina reattiva di Keap1 da parte di molecole di piccole dimensioni, il fattore Nrf2 può dissociarsi da Keap1 (8) (10) (11)). Miscela : miscela o soluzione costituita da due o più sostanze che non reagiscono tra loro (1). Sostanza mono-costituente : sostanza, definita attraverso la sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno l'80 % (p/p). Sostanza multicostituente : sostanza, definita attraverso la sua composizione quantitativa, in cui più costituenti principali sono presenti in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente è che una miscela è ottenuta attraverso la miscelazione di due o più sostanze senza che avvenga una reazione chimica. Una sostanza multicostituente è il risultato di una reazione chimica. Nrf2 (Nuclear factor (erythroid-derived 2)-like 2) : fattore di trascrizione che interviene nel percorso di risposta agli antiossidanti. Quando Nrf2 non è ubiquitinato, esso si sviluppa nel citoplasma e si sposta nel nucleo, dove si combina all'ARE nella regione promotrice a monte di molti geni citoprotettivi, dando inizio alla loro trascrizione (8) (10) (11). Controllo positivo : replica contenente tutti i componenti di un sistema di prova, trattata con una sostanza che notoriamente induce una risposta positiva. Perché sia possibile valutare la variabilità nel tempo della risposta del controllo positivo, l'intensità di tale risposta non dovrebbe essere eccessiva. Pertinenza : descrizione della relazione tra la prova e l'effetto di interesse e indicazione del fatto che la prova sia o meno significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende la valutazione dell'accuratezza (concordanza) di un metodo di prova (29). Affidabilità : misura in cui un metodo di prova può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. L'affidabilità è valutata calcolando la riproducibilità intra- e inter-laboratorio e la ripetibilità intra-laboratorio (29). Riproducibilità : concordanza dei risultati ottenuti dall'esecuzione di prove sulla stessa sostanza chimica in applicazione dello stesso protocollo sperimentale (cfr. Affidabilità) (29). Sensibilità : proporzione di tutte le sostanze chimiche positive/attive correttamente classificate con il metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo (29). Controllo con solvente/disperdente : replica contenente tutti i componenti di un sistema di prova, esclusa la sostanza chimica in esame, ma incluso il solvente utilizzato. È utilizzata per stabilire la risposta di base per i campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente. Specificità : proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate con il metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo (29). Sostanza : elementi chimici e loro composti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a conservare la stabilità del prodotto e le impurità derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza ripercussioni sulla stabilità della sostanza o modifiche della sua composizione (1). Sostanza chimica in esame : il termine “sostanza chimica in esame” designa la sostanza oggetto della prova. Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS) : sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (1). UVCB : sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiale biologico. Metodo di prova valido : metodo di prova la cui pertinenza e affidabilità sono ritenute soddisfacenti per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in assoluto ma solo in relazione a un determinato scopo (29). Appendice 2 SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA Sensibilizzazione cutanea in vitro: metodo di prova della luciferasi ARE-Nrf2 Prima di utilizzare sistematicamente il presente metodo di prova, i laboratori sono tenuti a dimostrare la loro competenza tecnica ottenendo correttamente la predizione attesa con il metodo KeratinoSens™ per le 10 sostanze raccomandate nella tabella 1 e ottenendo i valori EC1.5 e IC50 che rientrano nel rispettivo intervallo di riferimento per almeno 8 delle 10 sostanze. Tali sostanze sono state selezionate per rappresentare la gamma di risposte per quanto riguarda i pericoli di sensibilizzazione cutanea. Altri criteri di selezione sono la disponibilità in commercio, la disponibilità di dati di riferimento in vivo di alta qualità e la disponibilità di dati in vitro di alta qualità ottenuti con il saggio KeratinoSensTM. Tabella 1 Sostanze raccomandate per la verifica della competenza tecnica con il saggio KeratinoSensTM
Appendice 3 CONTROLLO QUALITÀ DELLE MISURAZIONI DELLA LUMINESCENZA Esperimento di base volto a garantire misurazioni ottimali della luminescenza con il saggio KeratinoSensTM I tre parametri elencati di seguito sono essenziali per ottenere risultati affidabili con il luminometro:
Prima di procedere all'esecuzione della prova, si consiglia di accertarsi che le misurazioni della luminescenza siano di qualità adeguata eseguendo una prova su una piastra di controllo predisposta nel modo descritto di seguito (analisi in triplo). Predisposizione della piastra per il primo esperimento preliminare
EGDMA= etilene glicol dimetacrilato (n. CAS: 97-90-5), sostanza chimica che provoca una forte induzione CA= aldeide cinnamica, riferimento positivo (n. CAS: 104-55-2) L'analisi di controllo della qualità deve dimostrare:
B.61 Metodo di prova di diffusione della fluoresceina per l'individuazione di sostanze corrosive e gravemente irritanti per gli occhi INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 460 (2012). Il metodo di prova di diffusione della fluoresceina (FL) è un metodo di prova in vitro che può essere utilizzato, in determinate circostanze e con specifiche limitazioni, per la classificazione delle sostanze chimiche (sostanze e miscele) come sostanze corrosive e gravemente irritanti per gli occhi, secondo la definizione del Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche (GHS) (categoria 1) delle Nazioni Unite (ONU), del regolamento (CE) n. 1272/2008 relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele (regolamento CLP) (31) (categoria 1), e dell'Agenzia per la protezione dell'ambiente degli Stati Uniti (EPA) (categoria I) (1) (2). Ai fini del presente metodo di prova, per sostanze gravemente irritanti per gli occhi si intendono le sostanze chimiche che causano danni ai tessuti oculari non reversibili entro 21 giorni in seguito a somministrazione della sostanza chimica in esame o che causano un grave deterioramento della vista; per sostanze corrosive si intendono le sostanze chimiche che causano danni irreversibili ai tessuti oculari. Tali sostanze chimiche sono classificate come appartenenti alla categoria 1 del sistema GHS dell'ONU e del regolamento CLP e alla categoria I dell'EPA. Il metodo di prova FL, pur non essendo considerato atto a sostituire completamente la prova in vivo sugli occhi del coniglio, è raccomandato quale parte integrante di una strategia di prove in sequenza per la classificazione e l'etichettatura. Pertanto, il metodo FL è raccomandato come prima fase di un approccio top-down (dall'alto verso il basso) per individuare sostanze corrosive / gravemente irritanti, in particolare per alcuni tipi limitati di sostanze chimiche (ossia le sostanze e miscele solubili in acqua) (3) (4). Attualmente è generalmente riconosciuto che nel prossimo futuro nessuna singola prova di irritazione oculare in vitro sarà in grado di sostituire la prova oculare in vivo (metodo di prova B.5 (5)) per prevedere tutta la gamma di irritazione per diverse classi chimiche. Tuttavia, combinazioni strategiche di diversi metodi di prova alternativi nell'ambito di una strategia di prova sequenziale potrebbero essere in grado di sostituire la prova oculare in vivo (4). L'approccio top-down (4) è concepito per essere utilizzato allorché, sulla base delle informazioni disponibili, si prevede che una sostanza chimica abbia un elevato potenziale di irritazione. Sulla base del modello di previsione di cui al paragrafo 35, il metodo di prova FL può rilevare che sostanze chimiche entro un limitato campo di applicabilità siano classificate come corrosive / gravemente irritanti per gli occhi (categoria 1 del sistema GHS dell'ONU; categoria 1 del regolamento CLP; categoria I dell'EPA) senza ulteriori prove. Lo stesso vale per le miscele, anche se non utilizzate nella validazione. Pertanto, il metodo di prova FL può essere usato per determinare il grado di irritazione / corrosività oculare delle sostanze chimiche, applicando la strategia di prova sequenziale di cui al metodo di prova B.5 (5). Tuttavia, una sostanza chimica non ritenuta corrosiva o gravemente irritante con il metodo di prova FL dovrebbe essere sottoposta a prova con uno o più metodi di prova supplementari (in vitro e/o in vivo) capaci di identificare con precisione: i) sostanze chimiche che sono, in vitro, falsi negativi corrosivi / gravi irritanti oculari della prova FL (categoria 1 del sistema GHS dell'ONU; categoria 1 del regolamento CLP; categoria I dell'EPA); ii) sostanze chimiche che non sono classificate per corrosione/irritazione oculare (nessuna categoria del sistema GHS dell'ONU; nessuna categoria del regolamento CLP; categoria IV dell'EPA); e/o iii) sostanze chimiche che hanno caratteristiche irritanti per gli occhi moderate/lievi (categorie 2A e 2B nel sistema GHS dell'ONU; categoria 2 del regolamento CLP; categorie II e III dell'EPA). Scopo di questo metodo di prova è descrivere le procedure impiegate per valutare il potenziale di corrosione o di grave irritazione oculare di una sostanza chimica in esame sulla base della capacità di tale sostanza di indurre lesioni su un monostrato epiteliale confluente impermeabile. L'integrità della permeabilità transepiteliale è una funzione importante dell'epitelio situato nella congiuntiva e nella cornea. Tale permeabilità transepiteliale è controllata da diverse giunzioni strette. Si è osservato che l'aumento della permeabilità dell'epitelio della cornea in vivo è correlato al livello di infiammazione e di lesioni superficiali constatato nell'evoluzione dell'irritazione oculare. Nel metodo di prova FL gli effetti tossici dopo un breve tempo di esposizione alla sostanza chimica in esame sono misurati per l'aumento della permeabilità della fluoresceina sodica attraverso il monostrato epiteliale di cellule renali canine Madin-Darby (MDCK) coltivate su inserti permeabili. La quantità di diffusione della fluoresceina è proporzionale alle lesioni indotte dalle sostanze chimiche alle giunzioni strette, ai desmosomi e alle membrane cellulari e può essere utilizzata per stimare il potenziale di tossicità oculare di una sostanza chimica in esame. L'appendice 1 riporta un diagramma di cellule MDCK coltivate su un inserto di membrana per il metodo di prova FL. Le definizioni figurano nell'appendice 2. CONSIDERAZIONI INIZIALI E LIMITI Il presente metodo di prova si basa sul protocollo INVITTOX n. 71 (6), valutato in uno studio di validazione internazionale dal Centro europeo per la convalida di metodi alternativi (ECVAM), in collaborazione con il Comitato di coordinamento interagenzia per la convalida dei metodi alternativi (ICCVAM) statunitense e con il Centro giapponese per la convalida di metodi alternativi (JACVAM). Il metodo di prova FL non è raccomandato per l'identificazione di sostanze chimiche da classificare come irritanti lievi/moderati o delle sostanze chimiche senza classificazione per l'irritazione oculare (sostanze e miscele) (ossia categoria 2A/2B o senza categoria GHS; categoria 2 o nessuna categoria del regolamento CLP; categoria II/III/IV dell'EPA), come comprovato dallo studio di validazione (3) (7). Il metodo di prova può essere applicato soltanto alle sostanze chimiche solubili in acqua (sostanze e miscele). Generalmente il metodo di prova FL rileva con precisione il potenziale di irritazione oculare grave delle sostanze chimiche idrosolubili e/o il cui effetto tossico non è influenzato dalla diluizione (7). Per essere classificata come solubile in acqua, in condizioni sperimentali, una sostanza chimica deve essere solubile in soluzione salina bilanciata di Hank (HBSS) sterile, contenente calcio (a una concentrazione di 1,0-1,8 mM), priva di rosso fenolo, in una concentrazione ≥ 250 mg/ml (una dose sopra la soglia di 100 mg/ml). Tuttavia, se la sostanza chimica in esame è solubile a una concentrazione inferiore a 100 mg/ml ma provoca un'induzione FL del 20 % già a quella concentrazione (vale a dire FL20 < 100 mg/ml), può essere classificata anche in questo caso come categoria 1 GHS o categoria I dell'EPA. Le limitazioni individuate per questo metodo di prova escludono dal campo di applicabilità acidi e basi forti, fissativi cellulari e sostanze chimiche ad elevata volatilità. Tali sostanze chimiche comportano meccanismi che non sono misurati dal metodo di prova FL, ad esempio coagulazione estesa, saponificazione o reazioni chimiche specifiche. Altre limitazioni identificate per questo metodo si basano sui risultati della capacità predittiva per sostanze chimiche in esame colorate e viscose (7). Entrambi questi tipi di sostanze chimiche sono di difficile rimozione dal monostrato dopo il breve periodo di esposizione e si ritiene che la predittività del metodo di prova potrebbe essere migliorata utilizzando un maggior numero di fasi di lavaggio. Sostanze chimiche solide in sospensione liquida hanno tendenza a precipitare, rendendo difficile determinare la concentrazione finale a cui le cellule sono esposte. Quando si escludono dalla banca dati sostanze appartenenti a queste classi chimiche e fisiche si registra un marcato miglioramento dell'accuratezza del metodo di prova FL nei sistemi di classificazione dell'UE, dell'EPA e GHS (7). In considerazione dell'obiettivo del presente metodo di prova (ossia identificare esclusivamente sostanze corrosive / gravemente irritanti degli occhi), i tassi di falsi negativi (cfr. il paragrafo 13) non sono essenziali perché tali sostanze sono destinate ad essere sottoposte a prove successive adeguatamente validate, in vitro o sui conigli, a seconda delle prescrizioni normative, applicando una strategia di prova sequenziale basata sul peso dell'evidenza disponibile (5) (cfr. anche i paragrafi 3 e 4). Altre limitazioni identificate del metodo di prova FL si basano sui tassi di falsi negativi e falsi positivi. Quando il metodo è utilizzato come prima tappa nell'ambito di un approccio top-down per identificare sostanze e miscele solubili in acqua che sono corrosive / gravemente irritanti per gli occhi (categoria 1 GHS; categoria 1 del regolamento CLP; categoria I dell'EPA), il tasso di falsi positivi del metodo di prova FL varia fra il 7 % (7/103; GHS delle Nazioni Unite e regolamento CLP) e il 9 % (9/99; EPA) e la percentuale di falsi negativi fra il 54 % (15/28; EPA) e il 56 % (27/48; GHS e regolamento CLP) rispetto ai risultati in vivo. I gruppi chimici che presentano falsi positivi e/o falsi negativi con il metodo di prova FL non sono definiti in questa sede. Determinate limitazioni tecniche sono specifiche per la coltura cellulare MDCK. Le giunzioni strette che bloccano il passaggio del colorante fluoresceina sodica attraverso il monostrato sono progressivamente sempre più compromesse con l'aumentare del numero di passaggi delle cellule. La formazione incompleta delle giunzioni strette provoca l'aumento di diffusione della fluoresceina nel controllo non trattato. Pertanto, è importante definire un livello di diffusione massima ammissibile nei controlli non trattati (cfr. il paragrafo 38: diffusione dello 0 %). Come per tutte le prove in vitro esiste un potenziale di trasformazione delle cellule nel corso del tempo ed è pertanto indispensabile indicare l'intervallo del numero di passaggi per le prove. L'attuale campo di applicabilità potrebbe essere maggiore in alcuni casi, ma solo dopo aver analizzato un insieme ampliato di dati di sostanze chimiche in esame sottoposte a prova, acquisito preferibilmente attraverso prove (3). Il presente metodo di prova sarà aggiornato di conseguenza tenendo conto di nuove informazioni e nuovi dati. Per i laboratori che ricorrono a questo tipo di saggio per la prima volta è consigliabile utilizzare le sostanze chimiche di riferimento indicate nell'appendice 3. I laboratori possono utilizzare tali sostanze chimiche per dimostrare le proprie competenze tecniche nell'esecuzione della prova FL prima di presentare i dati della prova FL a scopi normativi per la classificazione dei rischi. PRINCIPIO DELLA PROVA Il metodo di prova FL è un saggio in vitro basato sulla citotossicità e sulla funzione cellulare, eseguito su un monostrato confluente di cellule epiteliali tubolari MDCK BC 997 coltivate su inserti semipermeabili e riproducenti lo stato di non proliferazione dell'epitelio della cornea in vivo. La linea cellulare MDCK è ben consolidata e forma giunzioni strette e desmosomi simili a quelli riscontrati sulla parte apicale degli epiteli della congiuntiva e della cornea. Le giunzioni strette e i desmosomi in vivo impediscono ai soluti e ai corpi estranei di penetrare l'epitelio della cornea. La perdita di impermeabilità transepiteliale dovuta a lesioni subite dalle giunzioni strette e dai desmosomi è uno dei primi eventi che si verificano nell'irritazione oculare indotta da sostanze chimiche. La sostanza chimica in esame è applicata allo strato confluente di cellule coltivate sul lato apicale dell'inserto. Solitamente si usa una breve esposizione di un minuto per rispecchiare la normale velocità di eliminazione nell'esposizione umana. Un vantaggio del breve periodo di esposizione è che le sostanze e le miscele a base di acqua possono essere analizzate non diluite, se sono facilmente rimovibili dopo il periodo di esposizione. Ciò consente un confronto più diretto dei risultati con gli effetti chimici nell'uomo. La sostanza chimica in esame è poi rimossa e il colorante fluoresceina sodica, non tossico e altamente fluorescente, è aggiunto alla parte apicale del monostrato per 30 minuti. La lesione provocata alle giunzioni strette dalla sostanza chimica in esame è determinata dalla quantità di fluoresceina che attraversa lo strato cellulare entro un periodo di tempo definito. La quantità di colorante fluoresceina sodica che attraversa il monostrato e l'inserto di membrana per passare a un volume predeterminato di soluzione presente nel pozzetto (in cui si diffonde il colorante fluoresceina sodica) è determinata misurando la concentrazione di fluoresceina nel pozzetto con l'ausilio di uno spettrofluorimetro. La quantità di diffusione della fluoresceina (FL) è calcolata con riferimento alle letture dell'intensità della fluorescenza (FI) in due controlli: un controllo in bianco e un controllo di diffusione massima. La percentuale di diffusione e dunque l'entità della lesione alle giunzioni strette è espressa, relativamente a tali controlli, per ogni concentrazione della sostanza chimica in esame. Successivamente si calcola l'FL20 (ossia la concentrazione che provoca il 20 % di FL rispetto al valore registrato per il monostrato confluente non trattato e per gli inserti senza cellule). Il valore dell'FL20 (mg/ml) è utilizzato nel modello predittivo per l'identificazione di sostanze corrosive e gravemente irritanti per gli occhi (cfr. il paragrafo 35). La capacità di recupero è un aspetto importante del profilo tossicologico di una sostanza chimica in esame valutata anche mediante la prova di irritazione oculare in vivo. Analisi preliminari hanno indicato che i dati di recupero (fino a 72 ore dopo l'esposizione a sostanze chimiche) potrebbero potenzialmente migliorare la capacità predittiva del protocollo INVITTOX n. 71, ma è necessaria una valutazione più approfondita per la quale occorrerebbero altri dati, preferibilmente acquisiti mediante ulteriori prove (6). Il presente metodo di prova sarà aggiornato di conseguenza tenendo conto di nuove informazioni e nuovi dati. PROCEDURA Preparazione del monostrato cellulare Il monostrato di cellule MDCK CB 997 viene preparato utilizzando cellule subconfluenti coltivate in matracci con DMEM/miscela nutriente F12 (1x concentrato con L-glutammina, 15 mM di HEPES, calcio (a una concentrazione di 1,0-1,8 mM) e 10 % di FCS/FBS inattivati termicamente). È importante che tutti i mezzi/soluzioni utilizzati nel corso della prova FL contengano una concentrazione di calcio tra 1,8 mM (200 mg/l) e 1,0 mM (111 mg/l), per garantire la formazione e l'integrità delle giunzioni strette. Occorre controllare l'intervallo del numero di passaggi cellulari per garantire una formazione regolare e riproducibile delle giunzioni strette. Di preferenza, le cellule dovrebbero situarsi nell'intervallo 3-30 dal decongelamento perché in questo intervallo di passaggio le cellule hanno una funzionalità simile, che contribuisce alla riproducibilità dei risultati della prova. Prima di eseguire il metodo di prova FL, le cellule sono staccate dal matraccio per tripsinizzazione e centrifugate e quindi inoculate in una quantità adeguata negli inserti disposti in piastre a 24 pozzetti (cfr. l'appendice 1). Per l'inoculazione delle cellule si utilizzano inserti di 12 mm di diametro dotati di membrana in esteri di cellulosa misti, di spessore compreso fra 80 e 150 μm e diametro dei pori di 0,45 μm. Nello studio di validazione sono stati utilizzati inserti Millicell-HA da 12 mm. Le caratteristiche del tipo di inserto e di membrana sono importanti perché possono influire sulla crescita cellulare e sui legami chimici. Alcuni tipi di sostanze chimiche possono legarsi alla membrana dell'inserto Millicell-HA, potenzialmente influenzando l'interpretazione dei risultati. In caso di ricorso ad altre membrane, occorre dimostrare l'equivalenza utilizzando le sostanze chimiche di riferimento (cfr. l'appendice 3). La formazione di legami chimici alla membrana dell'inserto è più frequente per i composti cationici, come il cloruro di benzalconio, che subiscono l'attrazione della carica della membrana (7). Il legame chimico alla membrana può aumentare il periodo di esposizione alla sostanza chimica, portando a una sovrastima del potenziale tossico della sostanza stessa, ma può anche ridurre il volume di diffusione della fluoresceina attraverso l'inserto, legando il colorante al composto cationico legato alla membrana dell'inserto e portare così a una sottostima della tossicità. Ciò può essere facilmente controllato esponendo la sola membrana alla concentrazione più alta della sostanza chimica in esame e aggiungendo poi il colorante fluoresceina sodica alla concentrazione normale per il tempo di esposizione standard (senza controllo cellulare). Se si forma il legame con il colorante fluoresceina sodica, la membrana dell'inserto assume colorazione gialla dopo la rimozione per lavaggio della sostanza chimica in esame. È dunque essenziale conoscere le proprietà leganti della sostanza chimica in esame per poterne interpretare gli effetti sulle cellule. L'inoculazione delle cellule sugli inserti deve produrre un monostrato confluente al momento dell'esposizione alla sostanza chimica. Occorre aggiungere 1,6 × 105 cellule a ogni inserto (400 μl di una sospensione cellulare con una densità di 4 × 105 cellule/ml). In queste condizioni, si ottiene generalmente un monostrato confluente dopo 96 ore di coltura. Occorre esaminare visivamente gli inserti prima dell'inoculazione, per assicurare che qualsiasi lesione registrata dal controllo visivo di cui al paragrafo 30 sia dovuta alla manipolazione. Le colture cellulari MDCK vanno conservate in incubatrici in atmosfera umidificata, a 5(± 1) % di CO2 e 37 (± 1) °C. Le cellule non devono essere contaminate da batteri, virus, micoplasmi o funghi. Applicazione delle sostanze chimiche di prova e di controllo Occorre preparare una nuova soluzione madre della sostanza chimica in esame per ciascuna esecuzione dell'esperimento, da utilizzare entro 30 minuti dalla preparazione. Le sostanze chimiche in esame devono essere preparate in HBBS contenente calcio (a una concentrazione di 1,0-1,8 mM), priva di rosso fenolo, per evitare la formazione di legami con le proteine seriche. Prima della prova occorre valutare la solubilità della sostanza chimica a 250 mg/ml in HBSS. Se a questa concentrazione la sostanza chimica forma una sospensione stabile o un'emulsione (ossia rimane uniforme e non si decanta né si separa in più fasi) per 30 minuti, l'HBBS può ancora essere utilizzata come solvente. Tuttavia, se la sostanza chimica risulta insolubile in HBSS a questa concentrazione, occorre prendere in considerazione l'uso di altri metodi di prova diversi dal metodo FL. L'uso di olio minerale come solvente, nei casi in cui la sostanza chimica sia risultata insolubile in HBSS, andrebbe considerato con cautela in quanto i dati disponibili non sono sufficienti per stabilire l'efficacia della prova FL in tali condizioni. Tutte le sostanze chimiche da sottoporre a prova sono preparate in HBBS sterile contenente calcio (a una concentrazione di 1,0-1,8 mM), priva di rosso fenolo, a partire dalla soluzione madre, a cinque concentrazioni fisse diluite in peso/volume: 1, 25, 100,250 mg/ml e una soluzione pura o satura. Durante la prova di una sostanza chimica solida, occorre includere una concentrazione molto elevata di 750 mg/ml. Tale concentrazione della sostanza chimica può dover essere applicata alle cellule utilizzando una pipetta a spostamento positivo. Se si rileva tossicità tra 25 e 100 mg/ml, occorre testare due volte le ulteriori concentrazioni successive: 1, 25, 50, 75, 100 mg/ml. Il valore dell'FL20 deve essere ricavato da tali concentrazioni, a condizione che siano stati soddisfatti i criteri di accettazione. Le sostanze chimiche in esame vengono applicate ai monostrati cellulari confluenti dopo la rimozione del terreno di coltura cellulare e due lavaggi con HBBS sterile, calda (37 °C), contenente calcio (a una concentrazione di 1,0-1,8 mM) e priva di rosso fenolo. In precedenza, i filtri sono stati sottoposti a ispezione visiva per rilevare eventuali danni preesistenti che potrebbero essere erroneamente attribuite a potenziali incompatibilità con le sostanze chimiche in esame. Occorre utilizzare almeno tre repliche per ciascuna concentrazione della sostanza chimica in esame e per i controlli in ciascuna esecuzione della prova. Dopo un minuto di esposizione a temperatura ambiente, la sostanza chimica in esame deve essere rimossa con cura per aspirazione, il monostrato lavato due volte con HBSS sterile, calda (37 °C), contenente calcio (a una concentrazione di 1,0-1,8 mM) e priva di rosso fenolo e la diffusione della fluoresceina misurata immediatamente. Controlli negativi (NC) e positivi (PC) sono allestiti in parallelo in ciascuna esecuzione della prova per dimostrare che l'integrità del monostrato (NC) e la sensibilità delle cellule (PC) rientrano in un definito intervallo storico di accettabilità. La sostanza consigliata per il controllo positivo è il Brij 35 (n. CAS 9002-92-0) a 100 mg/ml. A tale concentrazione dovrebbe corrispondere una diffusione della fluoresceina approssimativa del 30 % (intervallo accettabile del 20-40 % di diffusione della fluoresceina, ossia di danni allo strato cellulare). La sostanza consigliata per il controllo negativo è HBBS contenente calcio (a una concentrazione di 1,0-1,8 mM), priva di rosso fenolo (controllo in bianco non trattato). Occorre includere in ciascuna serie di prove anche un controllo della diffusione massima, per consentire il calcolo dei valori dell'FL20. La diffusione massima si determina utilizzando un inserto di controllo senza cellule. Determinazione della permeabilità alla fluoresceina Immediatamente dopo la rimozione delle sostanze chimiche in esame e delle sostanze di controllo, si aggiungono agli inserti 400 μl di soluzione di fluoresceina sodica (0,01 % (peso/volume) in HBBS contenente calcio [a una concentrazione di 1,0-1,8 mM] e priva di rosso fenolo) (ad esempio Millicell-HA). Le colture sono mantenute per 30 minuti a temperatura ambiente. Al termine dell'incubazione con fluoresceina, gli inserti sono accuratamente rimossi da ciascun pozzetto. Si effettua un controllo visivo su ciascun filtro e si registrano eventuali danni verificatisi durante la manipolazione. La quantità di fluoresceina diffusa attraverso il monostrato e l'inserto è quantificata nella soluzione rimasta nei pozzetti dopo la rimozione degli inserti. Le misurazioni sono effettuate in uno spettrofluorimetro a lunghezze d'onda di eccitazione e di emissione di 485 nm e 530 nm, rispettivamente. Occorre regolare la sensibilità dello spettrofluorimetro in modo da ottenere la massima differenza numerica tra l'FL massima (inserto senza cellule) e l'FL minima (inserto con monostrato confluente trattato con il controllo negativo). A causa delle differenze fra gli spettrofluorimetri utilizzati, si suggerisce una sensibilità tale da provocare un'intensità della fluorescenza > 4 000 al controllo della diffusione massima di fluoresceina. Il valore massimo dell'FL non deve essere superiore a 9 999. La massima intensità di diffusione di fluorescenza deve rientrare nell'intervallo lineare dello spettrofluorimetro utilizzato. Interpretazione dei risultati e modello predittivo L'entità dell'FL è proporzionale alle lesioni indotte dalle sostanze chimiche alle giunzioni strette. La percentuale di FL per ciascuna concentrazione testata della sostanza chimica è calcolata a partire dai valori di FL ottenuti per la sostanza chimica in esame con riferimento ai valori FL del controllo negativo (lettura del monostrato confluente di cellule trattate con il controllo negativo ) e un controllo della diffusione massima (lettura per l'entità dell'FL attraverso un inserto senza cellule). Intensità media della fluorescenza corrispondente alla diffusione massima = x Intensità media della fluorescenza corrispondente alla diffusione dello 0 % = y La media della diffusione del 100 % si ottiene sottraendo la diffusione dello 0 % media dalla diffusione massima media, vale a dire x – y = z La diffusione percentuale per ciascuna dose fissa si ottiene sottraendo la diffusione dello 0 % dall'intensità media della fluorescenza rilevata nelle tre repliche (m) e dividendo tale valore per la diffusione al 100 %, vale a dire %FL = [(m-y) / z] x 100 %, ove:
Si applica la seguente equazione per il calcolo della concentrazione chimica che provoca una FL del 20 %: FLD = [(A-B) / (C-B)] × (MC – MB) + MB ove:
Il valore soglia dell'FL20 che consente di prevedere se le sostanze chimiche sono corrosive / gravemente irritanti per gli occhi è riportato di seguito:
Il metodo di prova FL è raccomandato esclusivamente per l'individuazione di sostanze solubili in acqua corrosive e gravemente irritanti (categoria 1 del sistema GHS dell'ONU, categoria 1 del regolamento CLP e categoria I dell'EPA) (cfr. i paragrafi 1 e 10). Al fine di individuare le sostanze chimiche solubili in acqua (sostanze e miscele) (3) (6) (7) “causanti gravi lesioni oculari” (categoria 1 GHS ONU e categoria 1 del regolamento CLP) o “corrosive o gravemente irritanti per gli occhi” (categoria I dell'EPA USA), la sostanza chimica in esame deve indurre un valore di FL20 ≤ 100 mg/ml. Accettazione dei risultati Il valore medio della diffusione massima di fluoresceina (x) deve essere superiore a 4 000 (cfr. il paragrafo 31), la media della diffusione dello 0 % (y) deve essere pari o inferiore a 300 e la media della diffusione del 100 % (z) deve essere compresa tra 3 700 e 6 000. Una prova si ritiene accettabile se il controllo positivo ha causato lesioni che interessano dal 20 % al 40 % dello strato cellulare (da misurare come % di diffusione della fluoresceina). DATI E RELAZIONE Dati Per ciascuna serie, i dati ottenuti dai pozzetti di ciascuna replica (ad esempio i valori dell'intensità della fluorescenza e le percentuali di FL calcolate per ogni sostanza chimica in esame, compresa la relativa classificazione) sono presentati in forma di tabella. Inoltre, occorre riferire i valori medi ± deviazione standard delle misurazioni corrispondenti a ciascuna replica per ciascuna serie di prove. Relazione sulla prova La relazione sulla prova deve comprendere le informazioni seguenti.
BIBLIOGRAFIA
Appendice 1 DIAGRAMMA DI CELLULE MDCK COLTIVATE SU UN INSERTO DI MEMBRANA PER IL METODO DI PROVA FL Si coltiva uno strato confluente di cellule DMCK sulla membrana semipermeabile di un inserto. Gli inserti sono disposti nei pozzetti di piastre a 24 pozzetti. 
Figura tratta da: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Tesi, Università di Nottingham, Regno Unito. Appendice 2 DEFINIZIONI Accuratezza : grado di concordanza tra i risultati ottenuti con il metodo e i valori di riferimento comunemente accettati. È una misura dell'efficienza del metodo di prova e costituisce un aspetto della “pertinenza”. Questo termine è spesso utilizzato in modo equivalente a “concordanza”, a significare la proporzione di risultati corretti di un metodo di prova. Sostanza chimica : una sostanza o una miscela. Categoria I dell'EPA : sostanze ad effetto corrosivo (distruzione irreversibile del tessuto oculare) o che producono interessamento o irritazione della cornea persistenti per oltre 21 giorni (2). Regolamento CLP : (regolamento (CE) n. 1271/2008 relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele): attua nell'Unione europea (UE) il sistema GHS dell'ONU per la classificazione delle sostanze chimiche (sostanze e miscele). Percentuale di falsi negativi : percentuale di tutte le sostanze chimiche positive falsamente identificate come negative da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. Percentuale di falsi positivi : percentuale di tutte le sostanze chimiche negative falsamente identificate come positive da un metodo di prova. È un indicatore dell'efficienza del metodo di prova. FL20 : può essere calcolata determinando la concentrazione alla quale la sostanza chimica in esame causa la diffusione del 20 % della fluoresceina attraverso lo strato cellulare. Diffusione della fluoresceina : quantità di fluoresceina diffusa attraverso lo strato cellulare, misurata mediante spettrofluorometria. GHS (Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (ONU)) : sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente. Categoria 1 del GHS : produzione di danni ai tessuti oculari o indebolimento grave della vista in seguito all'applicazione di una sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Pericolo : proprietà intrinseca di un agente o di una situazione di causare potenzialmente effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti. Miscela : nel contesto del sistema GHS delle Nazioni Unite, miscela o soluzione composta di due o più sostanze che non interagiscono. Controllo negativo : una replica non trattata che contiene tutti i componenti di un sistema di prova. Il campione è saggiato con campioni trattati con la sostanza chimica in esame e altri campioni di controllo per determinare se il disperdente interagisce con il sistema di prova. Senza classificazione : sostanze chimiche non classificate come irritanti oculari di categoria 1, 2A o 2B del sistema GHS dell'ONU; categoria 1 o 2 del regolamento CLP; o categorie I, II o III dell'EPA. Sostanza corrosiva oculare : a) sostanza chimica che causa danni irreversibili ai tessuti oculari; b) sostanze chimiche classificate come irritanti oculari di categoria 1 del sistema GHS dell'ONU, categoria 1 del regolamento CLP o categoria I dell'EPA. Irritante oculare : a) sostanza chimica che produce cambiamenti reversibili negli occhi in seguito all'applicazione alla superficie anteriore dell'occhio; b) sostanze chimiche classificate come irritanti oculari di categoria 2A o 2B del sistema GHS dell'ONU, categoria 2 del regolamento CLP o categoria II o III dell'EPA di irritanti per gli occhi. Grave irritante oculare : a) sostanza chimica che causa danni ai tessuti oculari in seguito all'applicazione sulla superficie anteriore dell'occhio non risolvibili entro 21 giorni dall'applicazione o indebolimento grave della vista; b) sostanze chimiche classificate come irritanti oculari di categoria 1 del sistema GHS dell'ONU, categoria 1 del regolamento CLP; o categoria I dell'EPA. Controllo positivo : una replica che contiene tutti i componenti di un sistema di prova e che è trattato con una sostanza che notoriamente induce una reazione positiva. L'entità della reazione positiva non dovrebbe essere estrema, per garantire la possibilità di valutare la variabilità della reazione dei controlli positivi nel tempo. Sostanze chimiche per la verifica della competenza : Un sottogruppo dell'elenco di sostanze chimiche di riferimento che può essere utilizzato da un laboratorio per dimostrare la competenza ad eseguire il metodo di prova di riferimento validato che non è mai stato utilizzato dal medesimo laboratorio. Pertinenza : descrizione del rapporto del saggio con l'effetto di interesse e se esso è significativo e utile per uno scopo specifico. È il grado con cui il saggio misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di un metodo di prova (8). Affidabilità : misura in cui un metodo può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna ai laboratori e la ripetibilità fra i laboratori. Prova di sostituzione : una prova progettata per sostituire un saggio usato correntemente e accettato per l'individuazione dei pericoli e/o la valutazione dei rischi, e che è stata determinata per fornire una protezione equivalente o maggiore della salute dell'uomo o degli animali oppure dell'ambiente, se del caso, rispetto alla prova accettata, per tutte le possibili situazioni sperimentali e le sostanze di prova. Sensibilità : proporzione di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo (8). Gravi lesioni oculari : produzione di danni ai tessuti oculari o di indebolimento grave della vista in seguito all'applicazione di una sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Controllo con solvente/disperdente : campione non trattato che contiene tutti i componenti di un sistema di prova, compreso il solvente e il mezzo disperdente (veicolo) usato con la sostanza chimica in esame saggiato con gli altri campioni di controllo al fine di stabilire la reazione di base nei campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente o disperdente. Nelle prove con controlli negativi paralleli, questo campione dimostra anche se il disperdente può interagire con il sistema di prova. Specificità : proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo. Sostanza : nel contesto del sistema GHS dell'ONU, elementi chimici e relativi composti, allo stato naturale o ottenuti mediante qualsiasi procedimento di produzione, compresi gli additivi necessari per preservare la stabilità del prodotto e le impurità derivanti dal procedimento impiegato, ed esclusi i solventi che possono essere separati senza incidere sulla stabilità della sostanza né modificarne la composizione. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Strategia di prova sequenziale : strategia di prove graduali in cui sono riesaminate tutte le informazioni disponibili su una sostanza chimica in esame, secondo un ordine ben specificato, seguendo un approccio basato sul peso dell'evidenza disponibile per ciascuna prova, al fine di stabilire se vi sono informazioni sufficienti per una decisione sulla classificazione del pericolo prima di procedere alla fase successiva. Se è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, non è necessario svolgere prove aggiuntive Se non è possibile assegnare il potenziale di irritazione di una sostanza chimica in esame in base alle informazioni disponibili, è svolta una procedura di prova graduale su animali in sequenza fino a che non sia possibile effettuare una classificazione inequivocabile. Metodo di prova convalidato : metodo di prova in base al quale sono stati completati studi di validazione per determinare la pertinenza (compresa l'accuratezza) e l'affidabilità per un fine specifico. Va sottolineato che un metodo di prova convalidato potrebbe non avere un rendimento sufficiente in termini di valori di accuratezza e affidabilità ritenuti accettabili per il raggiungimento dell'obiettivo prefissato (8). Peso dell'evidenza : il processo che consiste nel tener conto dei punti di forza e di debolezza di informazioni diverse per conseguire e supportare una data conclusione relativa al potenziale di pericolo di una sostanza chimica in esame. Appendice 3 SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA NEL METODO DI PROVA FL Prima di utilizzare regolarmente il presente metodo di prova, i laboratori devono dimostrare di possedere la necessaria competenza tecnica identificando correttamente la classificazione di corrosività oculare delle 8 sostanze chimiche raccomandate nella tabella 1. Tali sostanze chimiche sono state selezionate quali rappresentative di una serie di reazioni locali di irritazione/corrosione degli occhi, sulla base di risultati del saggio sugli occhi del coniglio in vivo (TG 405, TM B.5 (5)) (ovvero, categorie 1, 2A, 2B o senza classificazione per il sistema GHS dell'ONU). Tuttavia, in considerazione dell'utilità validata della prova FL (che consiste nell'identificare esclusivamente corrosivi/gravi irritanti oculari) esistono solo due risultati di prova ai fini di una classificazione (corrosivo/gravemente irritante o non corrosivo/non gravemente irritante) per dimostrare la competenza. Gli altri criteri di selezione comprendevano la disponibilità delle sostanze chimiche sul mercato, la presenza di dati di qualità che fungono da riferimento per gli studi in vivo, nonché di dati di qualità di riferimento concernenti il metodo di prova FL. Per questo motivo, le sostanze chimiche per la verifica della competenza sono tratte dal documento Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing (8), che è stato utilizzato per la validazione del metodo di prova FL. Tabella 1 Sostanze chimiche raccomandate per la verifica della competenza tecnica per il metodo FL
B.62 Test della cometa in vivo in condizioni alcaline su cellule di mammiferi INTRODUZIONE Questo metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 489 (2016). Il test della cometa in vivo in condizioni alcaline su cellule di mammiferi (elettroforesi su gel a singola cellula — single cell gel electrophoresis), nel prosieguo semplicemente il “test della cometa”, è utilizzato per individuare le rotture di filamenti del DNA in cellule o nuclei isolati a partire da tessuti multipli di animali, in genere roditori, esposti a materiali potenzialmente genotossici. Il test della cometa è stato esaminato e diversi gruppi di esperti hanno pubblicato raccomandazioni in materia (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). Il presente metodo di prova è parte integrante di una serie di metodi di prova sulla tossicologia genetica. È stato elaborato un documento OCSE contenente informazioni succinte sulle prove di tossicologia genetica e un compendio delle modifiche recentemente apportate alla rispettiva linea guida (11). Il test della cometa ha l'obiettivo di individuare le sostanze chimiche che provocano lesioni al DNA. In condizioni alcaline (> pH 13), il test della cometa può individuare rotture singole e doppie di filamenti provocate, ad esempio, da interazioni dirette con il DNA, siti alcali-labili o riconducibili a rotture provvisorie del DNA derivanti da una riparazione del DNA per escissione. Le rotture dei filamenti del DNA possono essere riparate, e avere quindi effetti non persistenti, possono essere letali per la cellula o possono essere riparate dando vita a una mutazione permanente funzionale. Possono inoltre provocare lesioni cromosomiche del tipo di quelle associate a molte malattie dell'uomo, quali il cancro. Uno studio formale di validazione del test della cometa in vitro su roditori è stato effettuato nel periodo 2006-2012 con il coordinamento del Centro giapponese per la convalida dei metodi alternativi (JaCVAM) in collegamento con il Centro europeo per la convalida di metodi alternativi (ECVAM), il Comitato di coordinamento inter-agenzia per la convalida dei metodi alternativi (ICCVAM) e il Centro inter-agenzia per la valutazione di metodi tossicologici alternativi del NTP (NICEATM) (12). Il presente metodo di prova indica l'uso e i limiti raccomandati del test della cometa e si basa sul protocollo finale (12) utilizzato nella prova di convalida e su pertinenti dati complementari pubblicati e non pubblicati (dati di proprietà dei laboratori). Le definizioni dei termini fondamentali figurano nell'appendice 1. Va rilevato che per l'esecuzione del presente test possono essere utilizzate molte piattaforme diverse (vetrini per microscopio, gocce di gel, piastre a 96 pozzetti, ecc.). Per motivi di praticità in tutto il presente documento viene utilizzato il termine “vetrini” ma esso è riferito anche a tutti gli altri supporti. CONSIDERAZIONI PRELIMINARI E LIMITI Il test della cometa è un metodo per misurare le rotture di filamenti del DNA nelle cellule eucariote. Singole cellule/nuclei in una sospensione in gel di agarosio collocata sui vetrini sono sottoposte a lisi con un detergente e concentrazioni elevate di sale. La lisi permette di digerire le membrane cellulari e nucleari e consente il rilascio di anse di DNA a spirale, chiamate generalmente nucleotidi, e frammenti di DNA. L'elettroforesi a pH elevato produce strutture che assomigliano a comete e che, utilizzando adeguati coloranti fluorescenti, possono essere esaminate con un microscopio a fluorescenza; i frammenti di DNA migrano dalla testa verso la coda della cometa in base alla loro dimensione e l'intensità della coda della cometa in rapporto all'intensità totale (testa più coda) riflette la portata della rottura del DNA (13) (14) (15). Il test della cometa in vivo in condizioni alcaline è particolarmente indicato per valutare il rischio genotossico, in quanto le risposte al test dipendono dall'ADME in vivo (assorbimento, distribuzione, metabolismo e eliminazione) e anche dai processi di riparazione del DNA. Questi ultimi possono variare a seconda delle specie, dei tessuti e dei tipi di lesioni del DNA. Al fine di rispettare i requisiti in materia di benessere degli animali e, in particolare, la riduzione del loro impiego mediante la sostituzione, la riduzione e il perfezionamento degli esperimenti su animali (in inglese il principio delle tre R — Replacement, Reduction, Refinement), il presente test può essere integrato con altri studi tossicologici (10) (16) (17) oppure l'endpoint può essere combinato con altri endpoint della genotossicità, quali il test micronucleare di eritrocita di mammifero (18) (19) (20). Il test della cometa è generalmente praticato sui roditori, anche se è stato utilizzato con altre specie di mammiferi e non mammiferi. L'uso di specie diverse dai roditori deve essere giustificato caso per caso sul piano etico e scientifico e comunque si raccomanda vivamente di eseguire il test della cometa su specie diverse dai roditori soltanto nell'ambito di un altro studio sulla tossicità e non come test isolato. La selezione della via di esposizione e del o dei tessuti da sottoporre a prova va determinata sulla base di tutte le conoscenze esistenti/disponibili delle sostanze chimiche in esame, ad esempio la via di esposizione umana intesa/attesa, il metabolismo e la distribuzione, i potenziali effetti per il punto di contatto, le allerte strutturali, altri dati sulla tossicità o genotossicità e l'obiettivo dello studio. In questo modo, se del caso, il potenziale genotossico delle sostanze chimiche in esame può essere sottoposto a prova nel o nei tessuti interessati dagli effetti cancerogeni e/o da altri effetti tossici. Il test può essere considerato utile inoltre per esaminare ulteriormente la genotossicità rilevata da un sistema in vitro. È opportuno effettuare un test della cometa in vivo su un tessuto di interesse, se ci si può ragionevolmente attendere che tale tessuto sarà adeguatamente esposto. I test di validazione più completi sull'uso del test della cometa hanno riguardato i tessuti somatici dei ratti maschi e sono stati effettuati in studi interlaboratorio, quali il test del JaCVAM (12) e in Rothfuss et al., 2010 (10). Nello studio internazionale di convalida del JaCVAM sono stati utilizzati lo stomaco e il fegato. Il fegato, perché è l'organo maggiormente attivo nel metabolismo delle sostanze chimiche e perché è sovente bersaglio di cancerogenicità. Lo stomaco perché è di solito il primo punto di contatto delle sostanze chimiche dopo l'esposizione per via orale, per quanto anche altre aree del tratto gastrointestinale, quali il duodeno e il digiuno, vadano considerate tessuti di contatto e siano forse più pertinenti per l'uomo di quanto sia lo stomaco ghiandolare dei roditori. Ci si deve assicurare che tali tessuti non siano esposti a quantitativi eccessivamente elevati della sostanza chimica in esame (21). La tecnica di cui trattasi è in linea di principio applicabile a qualsiasi tessuto dal quale possano essere ricavate sospensioni analizzabili di singole cellule/nuclei. I dati in possesso di molti laboratori dimostrano che il test può essere applicato con successo a molti tessuti differenti e numerose pubblicazioni indicano l'applicabilità della tecnica a organi o tessuti diversi dal fegato e dallo stomaco, ad esempio l'intestino digiuno (22), i reni (23) (24), la pelle (25) (26), o le cellule provenienti dalla vescica (27) (28) e dal lavaggio polmonare o broncoalveolare (pertinente per lo studio delle sostanze chimiche inalate) (29) (30); sono stati realizzati inoltre test su organi multipli (31) (32). Se, da un lato, può essere interessante per studiare gli effetti genotossici nelle cellule germinali, va tuttavia sottolineato, dall'altro, che il presente metodo di prova non è considerato appropriato per misurare le rotture dei filamenti del DNA in cellule germinali mature. Poiché, in materia di lesioni del DNA, la letteratura relativa all'uso del test della cometa per determinare la genotossicità sulle cellule germinali ha evidenziato livelli di fondo elevati e variabili (33), si ritiene necessario modificare il protocollo e migliorare gli studi di standardizzazione e convalida prima di includere nel metodo di prova il test della cometa sulle cellule germinali (ad esempio, quelle dello sperma). Inoltre, il regime di esposizione raccomandato, descritto nel presente metodo di prova, non è ottimale e, per un'applicazione all'analisi delle rotture del filamento di DNA nelle cellule mature dello sperma, sarebbero necessari tempi di esposizione o di campionamento più lunghi. Gli effetti genotossici misurati dal test della cometa nelle cellule dei testicoli a differenti stadi di differenziazione sono illustrati in letteratura (34) (35). Va tuttavia rilevato che le gonadi contengono un misto di cellule somatiche e germinali. Per questo motivo risultati positivi sull'insieme delle gonadi (testicoli) non indicano necessariamente danni alle cellule germinali; essi indicano tuttavia che la o le sostanze in esame e/o i loro metaboliti hanno raggiunto le gonadi. Le condizioni sperimentali standard del test della cometa non permettono di individuare in modo affidabile i legami crociati. In alcune condizioni sperimentali potrebbero essere individuati i legami crociati DNA-DNA e DNA-proteina come pure altre modificazioni di base, quali le basi ossidate (23) (36) (37) (38) (39). Ma ulteriori ricerche sono necessarie per caratterizzare in modo adeguato le necessarie modifiche del protocollo. Pertanto, l'individuazione degli agenti responsabili dei legami crociati non è l'obiettivo principale del test qui descritto. Il test non è appropriato, neanche in versione modificata, per l'individuazione degli aneugeni. Allo stato attuale delle conoscenze, il test della cometa in vivo presenta diversi altri limiti (cfr. appendice 3). Ci si aspetta che in futuro il metodo di prova sarà esaminato e, se necessario, modificato alla luce dell'esperienza acquisita. Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tale verifica non è necessaria se la miscela viene sottoposta a prova in ottemperanza a un obbligo regolamentare. PRINCIPIO DEL METODO Gli animali sono esposti alla sostanza chimica in esame tramite una via adeguata. Ai punti 36-40 viene fornita una descrizione dettagliata di dosaggio e campionamento. Nei momenti scelti per il campionamento i tessuti di interesse sono sezionati e vengono preparate sospensioni di singole cellule/nuclei (se considerato utile, ad esempio per il fegato, può essere realizzata una perfusione in situ) in un gel di agarosio per fissarle sui vetrini. Le cellule/nuclei sono trattate con un tampone di lisi per rimuovere la membrana cellulare e/o nucleare e sono esposte a una base forte (ad esempio, pH ≥13) per consentire lo svolgimento del DNA e il rilascio delle anse e frammenti di DNA “rilassato”. Il DNA nucleare nell'agarosio è quindi sottoposto a elettroforesi. Le molecole normali, non frammentate, di DNA restano nella posizione in cui si trovava il DNA nucleare nell'agarosio, mentre il DNA frammentato e le anse di DNA rilassato si spostano verso l'anodo. Dopo l'elettroforesi il DNA è visualizzato utilizzando un adeguato colorante fluorescente. Le preparazioni sono analizzate utilizzando un microscopio e sistemi di analisi dell'immagine parzialmente o totalmente automatizzati. La portata della migrazione del DNA durante l'elettroforesi e la distanza di tale migrazione danno conto del numero e delle dimensioni dei frammenti di DNA. Il test della cometa presenta diversi endpoint. Per valutare le lesioni al DNA si raccomanda di prendere in considerazione il contenuto di DNA nella coda o l'intensità della coda ( % tail DNA or % tail intensity) (12) (40) (41) (42). Dopo l'analisi di un sufficiente numero di nuclei, i dati sono interpretati utilizzando metodi di analisi appropriati. Va sottolineato che le modifiche apportate a diversi aspetti della metodologia, inclusa la preparazione dei campioni, le condizioni dell'elettroforesi, i parametri dell'analisi visiva (ad esempio, intensità del colorante, intensità luminosa della lampada del microscopio e l'utilizzo di filtri del microscopio e i parametri dinamici della fotocamera), nonché le condizioni ambiente (ad esempio, illuminazione ambiente) sono state oggetto di indagine e potrebbero influenzare la migrazione del DNA (43) (44) (45) (46). VERIFICA DELLA COMPETENZA DEL LABORATORIO Ciascun laboratorio deve provare la propria competenza sperimentale a effettuare il test della cometa, dimostrando la capacità di ottenere sospensioni di singole cellule o nuclei in quantità sufficienti per tutti i tessuti in esame per ciascuna specie utilizzata. La qualità delle preparazioni viene analizzata innanzitutto sulla base della percentuale di DNA nella coda per gli animali trattati con il mezzo disperdente che si collocano in una gamma bassa riproducibile. I dati attuali suggeriscono che la percentuale media di DNA di coda del gruppo (basata sulla media delle mediane — cfr. punto 57 per dettagli su questi termini) nel fegato dei ratti non dovrebbe, di preferenza, superare il 6 %, dato che sarebbe conforme ai valori dello studio di convalida del JaCVAM (12) e di altri dati pubblicati o privati. Al momento non si dispone di dati sufficienti per formulare raccomandazioni relativamente alle gamme ottimali o accettabili per altri tessuti. Ciò non preclude tuttavia l'uso di altri tessuti, se giustificato. La relazione sulla prova deve illustrare in modo appropriato l'esecuzione del test della cometa su tali tessuti in relazione alla letteratura pubblicata o a dati privati. In primo luogo, una percentuale bassa di DNA di coda nei controlli è auspicabile per disporre di un intervallo dinamico sufficiente al fine di individuare un effetto positivo. In secondo luogo, ciascun laboratorio deve essere in grado di riprodurre le risposte attese per i mutageni diretti e promutageni, con differenti modalità di azione, come indicato nella tabella 1 (punto 29). Sostanze positive possono essere selezionate, ad esempio, dallo studio di convalida del JaCVAM (12) o da altri dati pubblicati (cfr. punto 9), se del caso, giustificando tale scelta e dimostrando l'esistenza di risposte positive chiare nei tessuti di interesse. Deve essere inoltre dimostrata la capacità di individuare gli effetti deboli di mutageni conosciuti, quali l'EMS a basso dosaggio, stabilendo, a titolo di esempio, relazioni dose-risposta con adeguati numeri di dosi e intervalli tra le dosi. In un primo tempo le operazioni devono cercare di stabilire competenze nel trattamento dei tessuti di uso più comune, ad esempio il fegato dei roditori, per i quali è possibile operare confronti con i dati esistenti e i risultati attesi (12). Allo stesso tempo possono essere raccolti dati relativi ad altri tessuti, ad esempio stomaco/duodeno/digiuno, sangue ecc. Il laboratorio deve evidenziare la propria competenza per ciascun tessuto di ciascuna specie che prevede di studiare e dimostrare che in tale tessuto può essere ottenuta una risposta positiva accettabile con un mutageno conosciuto (ad esempio, EMS). È necessario raccogliere i dati relativi al mezzo disperdente/controllo negativo per dimostrare la riproducibilità delle risposte negative e accertarsi che gli aspetti tecnici del test siano stati adeguatamente verificati o per suggerire la necessità di ristabilire gli intervalli di controllo storici (cfr. punto 22). Va sottolineato che, se da un lato è possibile raccogliere tessuti multipli in fase di necroscopia e trattarli per il test della cometa, il laboratorio, dall'altro, deve essere in grado di raccogliere differenti tessuti da un singolo animale, assicurandosi così che non vada trascurata nessuna lesione potenziale del DNA e che il test della cometa non sia compromesso. L'intervallo di tempo tra la soppressione dell'animale e la rimozione dei tessuti può essere critico (cfr. punto 44). Nel mettere a punto le competenze nei diversi aspetti del test in questione si deve tenere conto del benessere degli animali; a tal fine è quindi possibile utilizzare tessuti provenienti da animali utilizzati in altri test. Inoltre, può non essere necessario effettuare uno studio completo nelle fasi di definizione di un nuovo metodo di prova in laboratorio e al fine di sviluppare le necessarie abilità si possono utilizzare meno animali e concentrazioni di prova. Dati storici di controllo In sede di verifica delle competenze il laboratorio deve creare una banca di dati storici per stabilire gli intervalli e le distribuzioni dei controlli positivi e negativi per i tessuti e le specie oggetto di indagine. In letteratura (47) figurano raccomandazioni sulle modalità di raccolta e uso dei dati storici (ad esempio, i criteri di inclusione o esclusione di dati nei dati storici e i criteri di accettabilità di un dato esperimento). Differenti tessuti e differenti specie, come pure differenti mezzi disperdenti e vie di somministrazione, possono determinare percentuali di DNA di coda differenti per i controlli negativi. È pertanto importante stabilire intervalli dei controlli negativi per ciascun tessuto e specie. I laboratori devono utilizzare metodi di controllo della qualità, quali carte di controllo (ad esempio, carte C o X medio (48)) per evidenziare la variabilità dei loro dati e dimostrare che la metodologia è “sotto controllo”. Può essere inoltre necessario ottimizzare la selezione delle sostanze appropriate per i controlli positivi, gli intervalli delle dosi e le condizioni sperimentali (ad esempio, le condizioni dell'elettroforesi) per individuare gli effetti deboli (cfr. punto 17). Eventuali modifiche del protocollo sperimentale devono essere valutate in base alla coerenza con le banche dati storiche in relazione ai controlli. Eventuali notevoli incoerenze devono tradursi nella creazione di una nuova banca dati storica sui controlli. DESCRIZIONE DEL METODO Preparazioni Selezione delle specie animali Gli animali utilizzati sono di norma adulti giovani e sani (di età compresa tra 6 e 10 settimane all'inizio del trattamento, benché siano accettabili anche animali di età leggermente superiore) appartenenti a ceppi comunemente usati in laboratorio. La scelta delle specie di roditori deve basarsi i) sulle specie utilizzate in altri studi di tossicità (per poter effettuare la correlazione tra i dati e consentire l'integrazione degli studi), ii) sulle specie che hanno sviluppato tumori in uno studio sulla cancerogenicità (quando si studia il meccanismo della cancerogenesi) oppure iii) sulle specie che presentano il metabolismo più simile a quello dell'uomo, se note. Per il presente test si usano di norma roditori. Possono essere tuttavia utilizzate altre specie, qualora ciò sia eticamente e scientificamente giustificato. Condizioni di stabulazione e alimentazione degli animali Per i roditori la temperatura dello stabulario deve essere idealmente di 22 °C (± 3 °C). L'umidità relativa deve essere idealmente del 50-60 %, non inferiore al 30 % e, preferibilmente, non superiore al 70 %, tranne durante la pulizia dei locali. L'illuminazione deve essere artificiale, con una sequenza di 12 ore di luce e 12 di oscurità. Per quanto concerne l'alimentazione, si possono usare le diete convenzionali da laboratorio con una quantità illimitata d'acqua da bere. La scelta della dieta può essere influenzata dalla necessità di garantire un'adeguata miscela della sostanza in esame, se somministrata con questo metodo. I roditori vanno stabulati in piccoli gruppi (di solito non più di cinque) dello stesso sesso se non si prevede alcun comportamento aggressivo. Gli animali possono essere stabulati individualmente soltanto se ciò è giustificato dal punto di vista scientifico. Laddove possibile, il fondo delle gabbie deve essere compatto in quanto fondi grigliati possono provocare ferite gravi (49). È necessario fornire un adeguato arricchimento ambientale. Preparazione degli animali L'assegnazione degli animali ai gruppi di controllo e di trattamento avviene mediante randomizzazione. Gli animali sono identificati individualmente e acclimatati alle condizioni del laboratorio per almeno cinque giorni prima dell'inizio del trattamento. Per identificare individualmente gli animali deve essere usato il metodo meno invasivo possibile. Tra i metodi appropriati figurano l'apposizione di anelli, di etichette, di microchip e l'identificazione biometrica. L'apposizione di graffette metalliche sulle orecchie o sulle zampe non è scientificamente giustificata in queste prove. Le gabbie devono essere sistemate in modo da ridurre al minimo eventuali effetti dovuti alla loro collocazione. All'inizio dello studio le variazioni di peso tra gli animali devono essere minime e non superare ± 20 %. Preparazione delle dosi Le sostanze solide in esame vanno sciolte o poste in sospensione in appropriati mezzi disperdenti o mescolate alla dieta o all'acqua da bere prima di essere somministrate agli animali. Le sostanze liquide possono essere somministrate direttamente o diluite prima del trattamento. In caso di esposizione per via inalatoria, le sostanze possono essere somministrate sotto forma di gas, vapore o aerosol solido o liquido, in funzione delle loro proprietà fisico-chimiche (50) (51). Le preparazioni della sostanza chimica in esame devono essere predisposte sul momento a meno di non disporre di dati che dimostrino la stabilità delle preparazioni in condizioni di stoccaggio e permettano di definire tali condizioni in modo adeguato. Condizioni di prova Mezzo disperdente Il mezzo disperdente non deve produrre effetti tossici alle dosi usate e non deve reagire chimicamente con le sostanze chimiche in esame. L'uso di mezzi disperdenti poco noti è ammesso solo se suffragato da dati che ne provino la compatibilità per quanto riguarda gli animali utilizzati nella prova, la via si somministrazione e l'endpoint. Se possibile si raccomanda di considerare in primo luogo l'uso di un solvente/mezzo disperdente acquoso. Si deve tenere presente che alcuni mezzi disperdenti (in particolare quelli viscosi) possono provocare infiammazioni e aumentare il livello di fondo delle rotture di filamenti di DNA nel punto di contatto, soprattutto in caso di somministrazioni multiple. Controlli Controlli positivi Attualmente, per ciascuna prova viene di norma utilizzato un gruppo comprendente un minimo di 3 animali analizzabili dello stesso sesso, o dei due sessi se sono utilizzati entrambi (cfr. punto 32), trattato con una sostanza utilizzata come controllo positivo. In futuro i laboratori potranno forse dimostrare che competenze tali da consentire loro di ridurre il numero dei controlli positivi. Qualora siano usati tempi di campionamento multipli (ad esempio, nel caso di un protocollo che prevede un'unica somministrazione), è sufficiente prevedere controlli positivi solo per un campionamento, garantendo al contempo una ripartizione equilibrata (cfr. punto 48). Non è necessario somministrare le sostanze chimiche utilizzate come controllo positivo per la stessa via della sostanza chimica in esame; è importante invece utilizzare la stessa via di somministrazione per misurare gli effetti sul punto di contatto. È necessario dimostrare che le sostanze utilizzate come controllo positivo provocano rotture dei filamenti di DNA in tutti i tessuti di interesse per la sostanza chimica in esame; l'EMS costituisce probabilmente la scelta più logica di controllo positivo in quanto ha prodotto rotture dei filamenti di DNA in tutti i tessuti oggetto di studio. Le dosi delle sostanze chimiche utilizzate come controllo positivo devono essere scelte in modo da produrre effetti moderati che consentano una valutazione dell'efficacia e della sensibilità della prova e possono basarsi sulle curve dose-risposta stabilite dal laboratorio nella fase di dimostrazione delle proprie competenze. La percentuale di DNA di coda nei controlli positivi simultanei deve essere coerente con gli intervalli prestabiliti in laboratorio per ciascun tessuto e tempi di campionamento per la specie considerata (cfr. punto 16). Esempi di sostanze usate per i controlli positivi e di alcuni loro tessuti bersaglio (nei roditori) figurano nella tabella 1. Sostanze diverse da quelle riportate nella tabella 1 possono essere selezionate, se ciò è scientificamente giustificato. Tabella 1 Esempi di sostanze usate come controlli positivi e di alcuni loro tessuti bersaglio Sostanze e n. CAS Metansolfonato di etile (n. CAS 62-50-0), per tutti i tessuti Etilnitrosourea (n. CAS 759-73-9), per il fegato e lo stomaco, il duodeno o il digiuno Metansolfonato di metile (n. CAS 66-27-3), per il fegato, lo stomaco, il duodeno o il digiuno, le cellule provenienti dal lavaggio polmonare o broncoalveolare, reni, vescica, polmoni, testicoli e midollo osseo/sangue N-Metil-N'-nitro-N-nitrosoguanidina (n. CAS: 70-25-7), per lo stomaco, il duodeno o il digiuno 1,2-dimetilidrazina 2HCl (n. CAS 306-37-6), per il fegato e l'intestino N-metil-N-nitrosourea (n. CAS 684-93-5), per il fegato, il midollo osseo, sangue, reni, stomaco, digiuno e cervello Controlli negativi In ciascuna prova e per ciascun momento di campionamento deve essere incluso un gruppo di animali di controllo negativo, cui viene somministrato il solo mezzo disperdente e che sono altrimenti trattati nello stesso modo dei gruppi di trattamento. La percentuale di DNA di coda negli animali del controllo negativo deve rientrare negli intervalli di fondo prestabiliti in laboratorio per ciascun tessuto e momenti di campionamento per la specie considerata (cfr. punto 16). In assenza di dati storici o pubblicati relativi ai controlli, indicanti che il mezzo disperdente scelto, il numero di somministrazioni o la via di somministrazione non inducono effetti deleteri o genotossici, è necessario effettuare studi preliminari prima di avviare lo studio completo al fine di stabilire l'accettabilità del mezzo disperdente da utilizzare per i controlli. PROCEDURA Numero e sesso degli animali Benché vi siano pochi dati su animali di sesso femminile sulla cui base effettuare confronti tra i sessi nel caso del test della cometa, in generale le risposte ai test di genotossicità in vivo sono simili tra gli animali di sesso maschile e femminile e, pertanto, la maggior parte degli studi può essere realizzata con animali dell'uno o dell'altro sesso. I dati che evidenziano differenze significative tra maschi e femmine (ad esempio, differenze sul piano della tossicità sistemica, del metabolismo, della biodisponibilità, ecc., comprendenti, ad esempio, dati provenienti da uno studio per determinare l'intervallo di dosi) incoraggiano a utilizzare entrambi i sessi. In questo caso può essere opportuno realizzare uno studio su entrambi i sessi, ad esempio nell'ambito di uno studio di tossicità a dosi ripetute. In caso di uso di entrambi i sessi può essere opportuno ricorrere a un modello fattoriale. Nell'appendice 2 sono riportate informazioni sulle modalità di analisi dei dati in caso di utilizzo di tale modello. Le dimensioni del gruppo all'inizio dello studio (e in fase di dimostrazione delle competenze) devono essere tali da permettere di disporre di almeno 5 animali analizzabili dello stesso sesso, o di ciascun sesso in caso di utilizzo di entrambi i sessi, per gruppo (e di un numero inferiore nel concomitante gruppo di controllo positivo — cfr punto 29). Se l'esposizione umana a sostanze chimiche è specifica per un sesso, ad esempio nel caso di alcuni prodotti farmaceutici, il test deve essere eseguito su animali di tale sesso. In relazione al numero massimo di animali generalmente richiesto, a titolo informativo uno studio realizzato sulla base dei parametri di cui al punto 33 con tre gruppi di trattamento, e relativi gruppi di controllo positivo e negativo (ognuno composto da cinque animali dello stesso sesso) richiede tra 25 e 35 animali. CALENDARIO DI TRATTAMENTO Gli animali devono ricevere un trattamento quotidiano per un periodo di due giorni o più (ovvero due o più trattamenti a intervalli di circa 24 l'uno dall'altro) e i campioni devono essere raccolti una volta entro 2-6 ore (o a Tmax) dopo l'ultimo trattamento (12). Possono essere accettati anche campioni provenienti da regimi di trattamento prolungato (ad esempio, dosi quotidiane per 28 giorni). È stato dimostrato che il test della cometa e il test micronucleare di eritrocita possono essere combinati con successo (10) (19). Tuttavia, grande attenzione deve essere dedicata agli aspetti logistici che comporta il prelievo di campioni di tessuti per il test della cometa, rispettando al contempo i requisiti relativi al campionamento di tessuti per altri tipi di valutazioni tossicologiche. Il prelievo 24 ore dopo l'ultima dose, tipico di uno studio generale sulla tossicità, non è appropriato nella maggior parte dei casi (cfr punto 40 sui momenti di campionamento). L'utilizzo di altri calendari di trattamento e di campionamento deve essere giustificato (cfr. appendice 3). Ad esempio, è possibile utilizzare un unico trattamento con campionamento multiplo, sapendo tuttavia che per uno studio che preveda una sola somministrazione è necessario un numero maggiore di animali dato il numero di momenti di campionamento necessari; in alcuni casi tale soluzione può essere preferibile, ad esempio quando la sostanza chimica in esame provoca una tossicità eccessiva a seguito di ripetute somministrazioni. Qualunque modalità di esecuzione del test è tuttavia accettabile, a condizione che la sostanza chimica in esame dia una risposta positiva o, in caso di risposta negativa, a condizione che si ottenga una risposta diretta o indiretta dell'esposizione del o dei tessuti bersaglio o della tossicità per tali tessuti o qualora venga raggiunta la dose limite (cfr. punto 36). Per agevolare la somministrazione di un grande volume di sostanze chimiche in esame, quest'ultime possono essere somministrate anche in dosi frazionate, ad esempio in due volte nello stesso giorno, a distanza di 2-3 ore al massimo. In questo caso il campionamento viene fissato in relazione al momento della somministrazione dell'ultima dose (cfr. punto 40). Livelli di dose Qualora venga effettuato uno studio preliminare per determinare l'intervallo di dosi, in quanto non sono disponibili dati adeguati da studi pertinenti che forniscano un orientamento in tal senso, tale studio deve essere effettuato nello stesso laboratorio, utilizzando specie, ceppi, sesso e regime di trattamento identici a quelli da utilizzare nello studio principale sulla base delle metodologie attualmente in uso per gli studi sugli intervalli di concentrazione delle dosi. Lo studio deve essere finalizzato a individuare la dose massima tollerata (DMT), definita come la dose che provoca lievi effetti tossici in relazione alla durata dello studio (ad esempio, segnali clinici chiari, quali reazioni o comportamenti anormali, perdita di peso moderata o citotossicità del tessuto interessato), ma che non provoca la morte dell'animale o segni di dolore e sofferenza tali da renderne necessaria la soppressione. Per una sostanza chimica in esame non tossica, con un periodo di somministrazione pari o superiore a 14 giorni, la dose massima (limite) è di 1 000 mg/kg per peso corporeo/giorno. Per periodi di somministrazione inferiori a 14 giorni, la dose massima (limite) è di 2 000 mg/kg per peso corporeo/giorno. Nel caso di talune sostanze chimiche di prova (ad esempio, prodotti farmaceutici per uso umano), oggetto di regolamentazioni specifiche, questi limiti possono subire variazioni. Le sostanze chimiche che evidenziano saturazione delle caratteristiche tossicocinetiche, o che inducono processi di detossificazione che si traducono in una diminuzione dell'esposizione dopo una somministrazione di lungo termine, possono essere considerate eccezioni rispetto ai criteri di definizione della dose e vanno valutate caso per caso. Nel caso delle versioni acuta e subacuta del test della cometa, oltre alla dose massima (DMT, dose massima possibile, esposizione massima o dose limite), al fine di dimostrare le risposte relative alla dose si deve selezionare, per ciascun momento di campionamento, una serie supplementare di almeno due dosi decrescenti che presentino un intervallo adeguato ( di preferenza inferiore a 10). Tuttavia, i livelli di dose utilizzati devono anche, di preferenza, coprire un intervallo che va dalla dose massima alla dose che produce un effetto tossico limitato o nessun effetto tossico. Quando, per tutti i livelli di dose oggetto di studio, viene rilevata tossicità del tessuto bersaglio, è consigliabile effettuare ulteriori studi con dosi non tossiche (cfr. punti 54-55). Gli studi che intendano approfondire l'andamento della curva dose-risposta possono dover ricorrere a uno o più gruppi di trattamento supplementari. Somministrazione delle dosi Nella concezione di una prova va considerata la via d'esposizione umana prevista. Pertanto, vie di somministrazione quali alimentazione, acqua da bere, inalazione, impianto, vie topiche, sottocutanee, endovenose, via orale (mediante sonda gastrica) o intratracheale possono essere utilizzate nella misura in cui sono giustificate. In ogni caso, si deve optare per la via che garantisce un'adeguata esposizione del o dei tessuti bersaglio. Le iniezioni intraperitoneali non sono in genere raccomandate in quanto non costituiscono una via di esposizione rappresentativa di quella umana e devono essere usate soltanto in presenza di una giustificazione precisa (ad esempio, nel caso di talune sostanze utilizzate per il controllo positivo o di farmaci somministrati per via intraperitoneale). Il volume massimo di liquido somministrabile in una sola volta con sonda gastrica o con iniezione dipende dalle dimensioni dell'animale da laboratorio. Il volume non deve superare 1 ml/100 g di peso corporeo, tranne nel caso delle soluzioni acquose che possono essere somministrate in quantità pari a 2 ml/100 g di peso corporeo. L'uso di volumi maggiori (se consentito dalla legislazione in materia di benessere degli animali) deve essere giustificato. Laddove possibile, i differenti livelli delle dosi devono essere ottenuti adeguando la concentrazione della formulazione somministrata per garantire, a tutti i livelli di dose, un volume costante in relazione al peso corporeo. Momento di campionamento Il momento di campionamento è una variabile fondamentale in quanto dipende dal tempo necessario affinché le sostanze chimiche in esame raggiungano la concentrazione massima nel tessuto bersaglio e provochino la rottura del filamento del DNA, ma prima che tali rotture siano rimosse, riparate o provochino la morte della cellula. La durata di determinate lesioni che provocano la rottura di filamenti del DNA, individuate dal test della cometa, può essere molto breve, almeno per alcune sostanze chimiche testate in vitro (52) (53). Ne consegue che, se si sospetta la presenza di tali lesioni transitorie del DNA, è necessario adottare misure per limitarne la sparizione, assicurandosi che i tessuti siano prelevati precocemente, eventualmente prima dei tempi standard riportati di seguito. I periodi di campionamento ottimali possono dipendere dalla sostanza chimica o dalla via di somministrazione, traducendosi, ad esempio, in una rapida esposizione del tessuto in caso di somministrazione per endovena o inalazione. Di conseguenza, i periodi di campionamento sono determinati in funzione dei dati cinetici, se disponibili (ad esempio, tempo (Tmax) in cui si raggiunge la concentrazione massima (Cmax) nel plasma o tessuto o allo stato stazionario nel caso di somministrazioni multiple). In assenza di dati cinetici un compromesso accettabile per misurare la genotossicità è quello di effettuare il campionamento 2-6 ore dopo l'ultimo trattamento nel caso di due o più trattamenti o dopo 2-6 ore e 16-26 ore nel caso di una singola somministrazione, avendo cura di effettuare la necropsia di tutti gli animali contemporaneamente dopo l'ultima (o la sola) dose. Per selezionare i momenti di campionamento adeguati possono essere utilizzate, se disponibili, anche informazioni sulla presenza di effetti tossici negli organi bersaglio. Osservazioni Osservazioni cliniche generali sulla salute degli animali devono essere effettuate e registrate almeno una volta al giorno, di preferenza ogni giorno alla stessa ora, tenendo conto che, dopo la somministrazione, gli effetti anticipati sono più marcati (54). Almeno due volte al giorno, tutti gli animali vengono esaminati al fine di determinarne la morbilità e la mortalità. Nel caso di studi di più lunga durata tutti gli animali devono essere pesati almeno una volta alla settimana e alla fine della prova. Il consumo di cibo va misurato ad ogni cambio e almeno una volta alla settimana. Se la sostanza chimica in esame è diluita in acqua prima di essere somministrata, il consumo di acqua va misurato ad ogni cambio dell'acqua e almeno una volta alla settimana. Gli animali che manifestano segni di eccessiva, ma non letale, tossicità vanno soppressi prima della fine del test e non sono di norma utilizzati per il test della cometa. Raccolta dei tessuti Poiché è possibile studiare l'induzione delle rotture di filamenti di DNA (comete) in praticamente tutti i tessuti, è necessario definire in modo chiaro le ragioni della selezione dei tessuti, richiamandosi ai motivi alla base dello studio e a eventuali dati relativi all'ADME, alla genotossicità, cancerogenicità e altri dati relativi alla tossicità delle sostanze chimiche in esame. Tra i fattori importanti da tenere in considerazione figurano la via di somministrazione (basata sulle probabili vie di esposizione dell'uomo), la distribuzione e l'assorbimento previsti nei tessuti, il ruolo del metabolismo e i possibili meccanismi di azione delle sostanze chimiche in esame. Tra i tessuti il fegato è quello più frequentemente studiato e per il quale esistono più dati. Pertanto, in assenza di informazioni generali e qualora non sia stato individuato un tessuto specifico di interesse, la scelta del fegato è giustificata in quanto si tratta del sito principale del metabolismo xenobiotico ed è sovente altamente esposto sia alle sostanze madri sia ai metaboliti. In alcuni casi l'esame di un punto di contatto diretto (ad esempio, per le sostanze chimiche somministrate per via orale, lo stomaco ghiandolare o il duodeno/digiuno o, per le sostanze chimiche inalate, i polmoni) può rivelarsi estremamente importante. Tessuti complementari o differenti possono essere selezionati per ragioni attinenti all'esecuzione del test; può essere tuttavia utile esaminare diversi tessuti provenienti dallo stesso animale, purché il laboratorio abbia dimostrato la propria competenza per tali tessuti e la propria capacità di manipolare diversi tessuti allo stesso tempo. Preparazione dei campioni Per le operazioni descritte nei punti che seguono (44-49) è importante che tutte le soluzioni o sospensioni stabili siano utilizzate prima della data di scadenza o, se necessario, siano preparate sul momento. Inoltre, nei punti che seguono, il tempo impiegato per i) rimuovere ciascun tessuto dopo la necropsia, ii) trattare ciascun tessuto per ottenere sospensioni di cellule/nuclei e iii) trattare la sospensione e preparare i vetrini è considerato una variabile critica (cfr. le definizioni nell'appendice 1) e la durata di ciascuna delle operazioni descritte deve essere determinata in fase di definizione del metodo e dimostrazione delle competenze. Gli animali vengono soppressi conformemente alla legislazione in vigore in materia di benessere degli animali e al principio delle tre R nel o nei momenti adeguati dopo l'ultima somministrazione della sostanza chimica in esame. I tessuti selezionati sono prelevati e sezionati; una porzione è prelevata per il test della cometa e contemporaneamente una sezione della stessa parte di tessuto è tagliata e messa in una soluzione di formaldeide o in un'adeguata soluzione fissativa per un'eventuale analisi istopatologica (cfr. punto 55) applicando i metodi standard (12). Il tessuto destinato al test della cometa è messo in un tampone di macerazione, è lavato accuratamente con tale tampone per rimuovere il sangue residuo e conservato in un tampone di macerazione refrigerato fino al trattamento. Si può inoltre realizzare una perfusione in situ, ad esempio, per il fegato e i reni. In letteratura sono pubblicati diversi metodi per l'isolamento delle cellule/nuclei. Tra questi figurano la macerazione di tessuti quali il fegato e i reni, il raschiamento delle mucose nel caso del tratto gastrointestinale, l'omogeneizzazione e la digestione enzimatica. Poiché lo studio di convalida del JaCVAM si è limitato allo studio di cellule isolate, l'uso di queste ultime va privilegiato per poter definire il metodo e fare riferimento ai dati sperimentali del JaCVAM nella fase di dimostrazione delle competenze. Tuttavia è stato dimostrato che l'uso di cellule isolate o di nuclei non produceva differenze significative nei risultati del test della cometa. Anche l'applicazione di metodi differenti per isolare le cellule/nuclei (ad esempio, omogeneizzazione, macerazione, digestione enzimatica e filtrazione con setaccio) ha prodotto risultati comparabili (55). Di conseguenza è possibile utilizzare sia cellule isolate sia nuclei. I laboratori devono valutare e convalidare accuratamente i metodi di isolamento di cellule/nuclei in funzione del tipo di tessuto. Come indicato al punto 40, la durata di determinate lesioni che provocano la rottura di filamenti del DNA, individuate dal test della cometa, può essere molto breve (52) (53). Pertanto, qualunque sia il metodo utilizzato per preparare le sospensioni delle singole cellule/nuclei, è importante che i tessuti siano trattati prima possibile dopo la soppressione degli animali e conservati in condizioni tali da evitare la scomparsa delle lesioni (ad esempio, mantenendo il tessuto a temperature basse). Le sospensioni contenenti le cellule devono essere conservate a temperature molto basse fino a quando possono essere utilizzate, così da poter evidenziare una differenza minima tra i campioni e adeguate risposte dei controlli positivi e negativi. PREPARAZIONE DEI VETRINI La preparazione dei vetrini deve avvenire il prima possibile dopo la preparazione delle cellule /nuclei (idealmente entro un'ora), ma la temperatura e il tempo intercorso tra la soppressione dell'animale e la preparazione dei vetrini devono essere rigorosamente controllati e validati in condizioni di laboratorio. Il volume della sospensione contenente le cellule aggiunta all'agarosio con basso punto di fusione (in genere 0,5-1,0 %) per preparare i vetrini non deve ridurre la percentuale di agarosio con basso punto di fusione a meno di 0,45 %. La densità ottimale delle cellule viene determinata con il sistema di analisi delle immagini utilizzato per il conteggio delle comete. Lisi Anche le condizioni di lisi costituiscono una variabile critica e possono interferire con le rotture di filamenti derivanti dalle modifiche di specifici tipi di DNA (alchilazioni e addotti alla base del DNA). Si raccomanda pertanto di mantenere le condizioni di lisi il più costanti possibile per tutti i vetrini di uno stesso esperimento. Una volta pronti, i vetrini devono essere immersi in una soluzione di lisi refrigerata per almeno un'ora (o tutta la notte) a una temperatura di 2-8 °C in condizioni di luce attenuata, ad esempio luce gialla o ambiente a tenuta di luce, per evitare l'esposizione alla luce bianca che può contenere componenti UV. Dopo il periodo di incubazione i vetrini devono essere lavati per rimuovere il detergente o i sali residui prima della fase alcalina. A tal fine si può utilizzare acqua depurata, un tampone neutralizzante o un tampone fosfato o anche un tampone di elettroforesi. In questo modo è possibile mantenere le condizioni alcaline nella camera di elettroforesi. Svolgimento e elettroforesi I vetrini sono piazzati a caso sulla piattaforma di un'unità di elettroforesi di tipo sottomarino contenente una soluzione di elettroforesi in quantità sufficiente per coprire completamente le superfici dei vetrini (anche la profondità di immersione deve essere costante tra le serie). In altri tipi di unità di elettroforesi utilizzate per il test della cometa, a raffreddamento attivo, circolazione del liquido di raffreddamento e alimentazione in alta tensione, l'intensità della corrente elettrica, a tensione costante, è tanto più elevata quanto più è consistente la copertura della soluzione. È necessario ripartire i vetrini in modo equilibrato nella vaschetta dell'elettroforesi per limitare gli effetti di eventuali tendenze o gli effetti di bordo all'interno della vaschetta o per ridurre al minimo le variazioni tra i lotti; per questi motivi in ciascuna sequenza di elettroforesi si deve utilizzare lo stesso numero di vetrini provenienti dallo stesso animale, nonché campioni di differenti gruppi di trattamento, controlli positivi e negativi. I vetrini devono restare per almeno 20 minuti nella vaschetta per consentire lo svolgimento del DNA e quindi essere sottoposti a elettroforesi in condizioni controllate che permettano di massimizzare la sensibilità e l'intervallo dinamico del test (per ottenere percentuali accettabili di DNA di coda, nei controlli positivi e negativi, e massimizzare così la sensibilità). Il grado di migrazione del DNA è associato in modo lineare alla durata dell'elettroforesi oltre che al potenziale (V/cm). Secondo lo studio del JaCVAM quest'ultimo potrebbe essere pari a 0,7 V/cm per almeno 20 minuti. La durata dell'elettroforesi è considerata una variabile critica e dovrebbe essere impostata in modo da ottimizzare l'intervallo dinamico. Tempi di elettroforesi più lunghi (ad esempio, 30 o 40 minuti per massimizzare la sensibilità) si traducono in genere in risposte positive più nette nel caso di mutageni conosciuti. Tuttavia, tempi di elettroforesi più lunghi possono determinare anche una migrazione eccessiva nei campioni di controllo. Ogni esperimento deve essere realizzato a tensione costante e la variabilità degli altri parametri deve esse contenuta all'interno di un intervallo ristretto e definito; a titolo di esempio, nello studio del JaCVAM, 0,7 V/cm con un'intensità iniziale di 300 mA. La profondità del tampone deve essere in funzione delle condizioni richieste e va mantenuta per tutto l'esperimento. Si deve registrare la corrente all'inizio e alla fine della fase di elettroforesi. Le condizioni ottimali devono pertanto essere determinate nella fase iniziale di dimostrazione delle competenze del laboratorio con ciascun tessuto oggetto di studio. La temperatura della soluzione di elettroforesi in fase di svolgimento e elettroforesi deve essere mantenuta a un livello ridotto, di solito 2-10 °C (10). Si deve registrare la temperatura della soluzione di elettroforesi in fase di svolgimento e all'inizio e alla fine dell'elettroforesi. Una volta conclusa l'elettroforesi i vetrini devono essere immersi/lavati nel tampone neutralizzante per almeno 5 minuti. I gel possono essere colorati e osservati allo stato “fresco” (ad esempio, entro 1-2 giorni) oppure essere disidratati e analizzati in una fase successiva (ad esempio, 1-2 settimane dopo la colorazione) (56). Tuttavia, le condizioni devono essere convalidate durante la dimostrazione di competenza e, per ciascuna delle opzioni, è necessario acquisire, e conservare separatamente, dati storici. Qualora si opti per la seconda possibilità, i vetrini sono disidratati per immersione di almeno cinque minuti in etanolo assoluto, sono lasciati asciugare all'aria aperta e conservati a temperatura ambiente o in un recipiente in frigorifero fino alla lettura. Metodi di misurazione Per la valutazione quantitativa delle comete si utilizzano sistemi di analisi dell'immagine parzialmente o totalmente automatizzati. I vetrini sono colorati utilizzando un adeguato colorante fluorescente, ad esempio SYBR Gold, Green I, ioduro di propidio o bromuro di etidio, e sono misurati con un ingrandimento adeguato (ad esempio, 200x) utilizzando un microscopio a epifluorescenza munito di rilevatori adeguati o di una fotocamera digitale (ad esempio, CCD). Le cellule possono essere classificate in tre categorie, come descritto nell'atlante delle immagini delle comete (57), ovvero cellule misurabili, non misurabili e “fantasma” (cfr. punto 56 per una discussione più approfondita). Per evitare artefatti, soltanto le cellule misurabili (con testa e coda chiaramente definite e nessuna interferenza da parte di cellule vicine) possono essere classificate in funzione della percentuale di DNA di coda. Non è necessario registrare la frequenza delle cellule non misurabili. La frequenza delle cellule fantasma è determinata sulla base di un esame visivo (in quanto l'assenza di una testa chiaramente definita ne rende difficile l'individuazione mediante analisi dell'immagine) di almeno 150 cellule per campione (cfr. punto 56 per ulteriori informazioni) e documentata a parte. Tutti i vetrini dell'analisi, compresi quelli dei controlli positivi e negativi, devono essere codificati individualmente ed esaminati “alla cieca” affinché la persona che effettua l'analisi non sappia a quale trattamento corrispondono. Per ciascun campione (per tessuto e per animale) si devono analizzare almeno 150 cellule (escluse le cellule fantasma — cfr. punto 56). L'analisi di 150 cellule per animale in almeno 5 animali per dose (ma in un numero inferiore nel concomitante controllo positivo — cfr. punto 29) garantisce una potenza statistica adeguata secondo l'analisi di Smith et al., 2008 (5). Se sono utilizzati i vetrini, ciò può corrispondere a 2 o 3 vetrini analizzati per campione quando sono utilizzati cinque animali per gruppo. È necessario osservare diverse aree del vetrino a una densità tale da garantire che non vi sia sovrapposizione delle code. Va evitata la valutazione ai margini del vetrino. Le rotture di filamenti di DNA nel test della cometa possono essere misurate sulla base di parametri indipendenti, quali la percentuale di DNA di coda, la lunghezza della coda e il momento di coda. Le tre misurazioni possono essere effettuate utilizzando un programma adeguato di analisi dell'immagine. Tuttavia, la percentuale del DNA di coda (nota anche come intensità percentuale di coda) è il parametro raccomandato per la valutazione e l'interpretazione dei risultati (12) (40) (41) (42) ed è determinata dall'intensità dei frammenti di DNA nella coda espressa come percentuale dell'intensità totale della cellula (13). Lesioni dei tessuti e citotossicità Risultati positivi nel test della cometa possono non essere dovuti esclusivamente alla genotossicità e la tossicità del tessuto bersaglio può anche tradursi in un aumento della migrazione del DNA (12) (41). Al contrario, una citotossicità bassa o moderata viene spesso riscontrata in presenza di sostanze genotossiche conosciute (12), dimostrando che il test della cometa da solo non consente di distinguere la migrazione del DNA indotta dalla genotossicità rispetto a quella provocata dalla citotossicità. Tuttavia, quando si osserva un aumento nella migrazione del DNA, si raccomanda di effettuare l'esame di uno o più indicatori della citotossicità come ausilio all'interpretazione dei risultati. Un aumento della migrazione del DNA in presenza di chiari segni di citotossicità deve essere interpretato con cautela. Tra i numerosi criteri di valutazione della citotossicità proposti, le alterazioni istopatologiche sono considerate un criterio pertinente per valutare la tossicità dei tessuti. La migrazione del DNA è stata associata anche a infiammazioni, infiltrazioni cellulari, cambiamenti apoptotici o necrotici; tuttavia, come dimostrato dallo studio di convalida del JaCVAM (12), non si dispone di un elenco definitivo dei cambiamenti istopatologici che sono sempre associati a un aumento della migrazione del DNA. Anche i cambiamenti che interessano alcuni parametri biologici (ad esempio, AST, ALT) possono fornire utili informazioni sui danni ai tessuti; possono inoltre essere considerati anche altri indicatori, quali l'attivazione delle caspasi, la rilevazione delle cellule in fase di apoptosi con il metodo TUNEL, la colorazione con annessina V, ecc. Tuttavia i dati pubblicati in relazione all'utilizzo di tali indicatori negli studi in vivo sono pochi e non tutti affidabili. Le cellule fantasma sono cellule la cui immagine al microscopio consiste di una testa di piccole dimensioni, se non inesistente, e di una grande coda diffusa e, per quanto la causa di questo fenomeno sia incerta (cfr. appendice 3), sono considerate cellule fortemente danneggiate. Dato l'aspetto di tali cellule, la misurazione della percentuale di DNA di coda mediante l'analisi dell'immagine è inaffidabile; le cellule fantasma devono pertanto essere analizzate a parte. La presenza di cellule fantasma deve essere rilevata e registrata e un loro eventuale aumento che si ritiene sia dovuto alla sostanza chimica in esame deve essere analizzato e interpretato con attenzione. A tal fine può aiutare la conoscenza del meccanismo di azione potenziale delle sostanze chimiche in esame. DATI E RELAZIONE Trattamento dei risultati Poiché l'unità sperimentale è data dall'animale è opportuno presentare in forma di tabella sia i risultati relativi ai singoli animali sia la sintesi dei risultati. Data la natura gerarchica dei dati, si raccomanda di determinare la percentuale mediana di DNA di coda per ciascun vetrino e di calcolare per ciascun animale la media dei valori della mediana (12). La media di un gruppo è quindi determinata facendo la media delle medie individuali dei gruppi che lo compongono. Tutti questi valori devono essere riportati nella relazione. Metodologie alternative (cfr. punto 53) sono ammesse, purché scientificamente e statisticamente giustificate. L'analisi statistica può essere svolta applicando diverse metodologie (58) (59) (60) (61). Nel selezionare i metodi statistici da utilizzare si deve tenere conto dell'eventuale necessità di trasformare i dati (ad esempio, in logaritmi o radici quadrate) e/o di aggiungere un numero piccolo (ad esempio, 0,001) a tutti i valori (anche a quelli non nulli), per attenuare l'incidenza dei valori nulli, come illustrato nei riferimenti sopracitati. Nell'appendice 2 sono riportate informazioni sulle interazioni trattamento/sesso, quando vengono utilizzati entrambi i sessi e la successiva analisi dei dati a seconda che siano riscontrate o no differenze. Nella relazione devono figurare inoltre dati sulla tossicità e i segni clinici. Criteri di accettabilità L'accettazione dei risultati di una prova si basa sui criteri seguenti:
Analisi e interpretazione dei risultati A condizione che siano stati rispettati tutti i criteri di accettabilità, una sostanza chimica in esame è considerata chiaramente positiva se:
Se sono rispettati tutti i criteri di cui sopra, si ritiene che la sostanza chimica in esame sia in grado di provocare rotture dei filamenti di DNA nei tessuti studiati nel presente sistema di prova. Si veda il punto 62 se sono soddisfatti soltanto uno o due dei criteri summenzionati. A condizione che siano stati rispettati tutti i criteri di accettabilità, una sostanza chimica in esame è considerata chiaramente negativa se:
In questi casi si ritiene che la sostanza chimica in esame non sia in grado di provocare rotture dei filamenti di DNA nei tessuti studiati nel presente sistema di prova. Non è necessario verificare una risposta palesemente positiva o negativa. Qualora la risposta non sia né chiaramente positiva né chiaramente negativa (ovvero non sono rispettati tutti i criteri elencati ai punti 59 e 60), e al fine di stabilire la rilevanza biologica di un risultato, i dati devono essere sottoposti alla valutazione di esperti e/o devono essere effettuati ulteriori accertamenti, se ciò è scientificamente giustificato. Può essere utile, se del caso, analizzare cellule supplementari o ripetere l'esperimento, eventualmente in condizioni sperimentali migliori (ad esempio, intervallo tra le dosi, altre vie di somministrazione, momenti campionamento diversi, altri tessuti). In rari casi, anche dopo ulteriori analisi, quando la serie di dati non consente di valutare i risultati come positivi o negativi, i risultati sono dichiarati ambigui. Per valutare la rilevanza biologica di un risultato positivo o ambiguo, sono necessarie informazioni sulla citotossicità del tessuto bersaglio (cfr. punti 54-55). Se risultati positivi o ambigui sono riscontrati soltanto in presenza di chiare indicazioni di citotossicità, lo studio è dichiarato ambiguo per quanto riguarda la genotossicità, a meno di non possedere sufficienti informazioni che permettano di trarre conclusioni definitive. Qualora lo studio dia un risultato negativo in presenza di segni di tossicità con tutte le dosi sperimentali, può essere consigliabile effettuare un ulteriore studio con dosi non tossiche. Relazione sulla prova La relazione deve comprendere i seguenti dati:
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Elettroforesi di cellule isolate in gel in condizioni alcaline : tecnica sensibile per l'individuazione di lesioni primarie del DNA a livello di singole cellule/nuclei. Sostanza chimica : una sostanza o una miscela. Cometa : la forma, simile a quella di una cometa, che i nucleotidi assumono dopo essere stati esposti a un campo elettroforetico: la testa corrisponde al nucleo e la coda è costituita dal DNA che migra al di fuori del nucleo nel campo elettrico. Parametro critico/variabile critica : la variabile di un protocollo per il quale una piccola modifica può avere un grande impatto sulla conclusione del test. Le variabili critiche possono essere specifiche di un tessuto. Le variabili critiche non devono essere modificate, soprattutto nell'ambito di un test, senza tenere conto dell'incidenza che tali modifiche possono avere sulla risposta al test, come indicato ad esempio dall'ampiezza e dalla variabilità della risposta dei controlli positivi e negativi. La relazione sulla prova deve precisare le modifiche apportate alle variabili critiche nel corso del test o in rapporto al protocollo standard del laboratorio e fornire una giustificazione di ogni modifica. Intensità della coda o percentuale del DNA di coda : corrisponde all'intensità della coda della cometa in rapporto all'intensità totale (testa più coda). Essa riflette la portata (espressa in percentuale) delle lesioni del DNA. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. UVCB : sostanze la cui composizione non è conosciuta o è variabile, prodotti di una reazione complessa o i materiali origine biologica. Appendice 2 MODELLO FATTORIALE PER INDIVIDUARE LE DIFFERENZE TRA I SESSI NEL TEST DELLA COMETA IN VIVO Modello fattoriale e relativa analisi Questo modello prevede di esporre 5 maschi e 5 femmine a ciascuna concentrazione sperimentale e comporta pertanto l'utilizzo di un minimo di 40 animali (20 maschi e 20 femmine più i relativi controlli positivi). Il modello qui presentato — una delle forme semplici del modello fattoriale — è equivalente a un'analisi della varianza a due fattori, in cui il sesso e il livello delle concentrazioni costituiscono i fattori principali. I dati possono essere analizzati utilizzando diversi programmi statistici standard, quali SPSS, SAS, STATA, Genstat o il programma R. A partire dall'insieme dei dati si determina la variabilità tra i sessi e le concentrazioni, nonché la variabilità relativa alle interazioni tra sessi e concentrazioni. Ciascuno di questi aspetti è quindi valutato in rapporto alla stima della variabilità tra gli animali ripartiti nei gruppi di animali dello stesso sesso esposti alle stesse concentrazioni. Maggiori informazioni su questa metodologia sono reperibili in diversi manuali standard di statistica (cfr. bibliografia), nonché nelle schede “di aiuto” fornite con i programmi statistici. In seguito viene esaminata l'interazione sesso x concentrazione in una tabella ANOVA (35). In assenza di un termine di interazione significativo la combinazione dei valori inter-sessi o inter-livelli di concentrazione consente di effettuare test statistici validi tra i livelli, basandosi sul termine di variabilità intra-gruppo combinata di 'ANOVA. L'analisi prosegue suddividendo la variabilità stimata tra le concentrazioni in modo da ottenere contrasti che permettano di stabilire contrasti lineari e quadratici delle risposte per l'insieme dei livelli di concentrazione. Quando si riscontra un'interazione significativa sesso x concentrazione, questo termine può essere a sua volta in ripartito in contrasti di interazione lineare x sesso e quadratica x sesso. In questo modo si può verificare se le risposte alle concentrazioni sono parallele per i due sessi o se vi è una differenza riconducibile al sesso. La stima della variabilità intra-gruppo combinata può essere utilizzata per verificare lo scarto tra le medie, confrontandole due a due. I confronti possono essere effettuati tra le medie per i due sessi e tra le medie dei diversi livelli delle concentrazioni (come, ad esempio, confronti tra i livelli dei controlli negativi). Nei casi in cui si registra un'interazione significativa i confronti possono essere effettuati tra le medie di concentrazioni diverse per lo stesso sesso o tra le medie dei due sessi a parità di concentrazione. Riferimenti Numerosi manuali di statistica esaminano la teoria, la concezione, la metodologia, l'analisi e l'interpretazione dei modelli fattoriali, dall'analisi più semplice, a due fattori, alle forme più complesse utilizzate per la metodologia Design of Experiment. Di seguito ne è riportato un elenco non completo. Alcuni manuali forniscono esempi di modelli comparabili e, in alcuni casi, forniscono un codice che permette di effettuare l'analisi utilizzando diversi programmi.
Appendice 3 LIMITI ATTUALI DEL TEST Allo stato attuale delle conoscenze, il test della cometa in vivo presenta diversi limiti. Ci si aspetta che tali limiti siano ridotti, o che siano definiti in modo più preciso via via che la maggiore esperienza acquisita nell'applicazione del test fornirà risposte alle questioni di sicurezza in un contesto regolamentare.
Riferimenti
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(16) |
Nella parte C, il capitolo C.13 è sostituito dal seguente: «C.13 Bioaccumulo nei pesci: esposizione attraverso l'ambiente acquatico e per via alimentare INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 305 (2012). L'obiettivo principale della presente revisione del metodo di prova è duplice. In primo luogo, essa è intesa a includere una prova di bioaccumulo per via alimentare (36) idonea a determinare il potenziale di bioaccumulo delle sostanze a bassa solubilità in acqua. In secondo luogo, intende elaborare un metodo di prova che utilizzi, ove possibile, un minor numero di pesci nel rispetto del principio del benessere degli animali, e che risulti più efficiente in termini di costi. Negli ultimi anni, dopo l'adozione del metodo di prova C.13 consolidato (1), sono state saggiate numerose sostanze e tanto i laboratori che le autorità di regolamentazione hanno maturato una notevole esperienza. Ciò ha portato alla convinzione che la prova possa essere semplificata in presenza di determinate condizioni (cfr. paragrafo 88) e che sia possibile adottare un approccio graduale. L'esperienza ha anche dimostrato che i fattori biologici quali la crescita e il contenuto lipidico del pesce possono avere un forte impatto sui risultati e che pertanto è necessario tenerne conto. Inoltre, è stato riconosciuto che la sperimentazione su sostanze scarsamente solubili in acqua non è tecnicamente realizzabile. Inoltre, per le sostanze con scarsa idrosolubilità in acqua, l'esposizione attraverso l'ambiente acquatico può essere meno importante rispetto all'esposizione per via alimentare. Per questo motivo è stato sviluppato un metodo di prova in cui i pesci sono esposti alla sostanza da testare attraverso la dieta (cfr. paragrafi 7-14 e 97). La validazione (mediante prova interlaboratorio) del metodo con esposizione per via alimentare è stata eseguita nel 2010 (51). Le principali modifiche apportate al metodo di prova sono le seguenti:
Prima di effettuare le prove di bioaccumulo, devono essere note le seguenti informazioni sulla sostanza in esame:
Indipendente dal sistema di campionamento o metodo di esposizione prescelto, il presente metodo descrive una procedura per caratterizzare il potenziale di bioaccumulo di una sostanza nei pesci. Benché i sistemi di prova a flusso continuo siano ampiamente preferibili, sono ammissibili sistemi semistatici, purché i criteri di validità (cfr. paragrafi 24 e 113) siano soddisfatti. Quando si utilizza il canale dell'esposizione alimentare, il sistema a flusso continuo non è necessario per mantenere concentrazioni acquose della sostanza in esame, ma contribuirà a mantenere un'adeguata concentrazione dell'ossigeno disciolto e ad assicurare acqua pulita e a eliminare le influenze, ad es. prodotti da escrezione. Indipendentemente dal metodo di prova prescelto, il presente metodo di prova fornisce sufficienti dettagli per eseguire la prova e al contempo lasciare la necessaria libertà per adattare il disegno sperimentale alle specifiche condizioni di laboratorio e alla variabilità delle caratteristiche delle sostanze analizzate. La prova relativa all'esposizione in ambiente acquatico è applicata appropriatamente alle sostanze organiche stabili con valori log Kow tra 1,5 e 6,0 (13), ma è applicabile anche a sostanze fortemente idrofobe (con log Kow > 6,0), se può essere dimostrata una concentrazione della sostanza in esame stabile e pienamente disciolta in acqua. Se una siffatta concentrazione non può essere dimostrata, lo studio in acqua non sarebbe adeguato; pertanto, si renderebbe necessario ricorrere all'approccio relativo all'esposizione per via alimentare per saggiare una determinata sostanza nei pesci (anche se l'interpretazione e l'applicazione dei risultati della prova con esposizione alimentare può dipendere dal quadro normativo). Le stime preliminari del fattore di bioconcentrazione (BCF) (indicato talvolta con K B) per le sostanze organiche con valori di log Kow, fino a circa 9,0 si possono ricavare dall'equazione di Bintein et al. (14). La stima preliminare del fattore di bioconcentrazione per tali sostanze fortemente idrofobe può essere più elevata del fattore di bioconcentrazione allo stato stazionario (BCFSS) prevedibilmente ottenuto mediante esperimenti di laboratorio, in particolare quando si utilizza un semplice modello lineare per la stima preliminare. I parametri che caratterizzano il potenziale di bioaccumulo includono la costante cinetica di assorbimento (k 1), costanti del tasso di perdita compresa la costante cinetica di depurazione (k 2), il fattore di bioconcentrazione allo stato stazionario (BCFSS), il fattore di bioconcentrazione cinetico (BCFk) e il fattore di biomagnificazione alimentare (BMF) (38). Le sostanze radiomarcate in esame possono agevolare l'analisi dei campioni di pesce, acqua o cibo e possono essere utilizzate per stabilire se è necessario identificare e quantificare i metaboliti. Se si misurano solo i residui radioattivi totali (per esempio per combustione o solubilizzazione dei tessuti), il BCF o il BMF si basa sulla sostanza madre totale, eventuali metaboliti trattenuti e anche sul carbonio assimilato. I valori BMF o BCF basati sui residui radioattivi totali non sono pertanto confrontabili direttamente con un BCF o BMF ottenuto mediante analisi chimica specifica della sola sostanza madre. Procedure di separazione, quali TLC, HPLC o GC possono essere impiegate negli studi con marcatore radioattivo prima dell'analisi per determinare il BCF o BMF basato sulla sostanza madre (39). Quando si utilizzino tecniche di separazione, vanno eseguite l'identificazione e la quantificazione della sostanza madre e dei relativi metaboliti (cfr. paragrafo 65) se si basano i valori BCF o BMF sulla concentrazione della sostanza madre nei pesci e non sui residui radiomarcati totali (40). È anche possibile combinare uno studio sul metabolismo o studio di distribuzione in vivo con uno studio di bioaccumulo mediante l'analisi e l'identificazione dei residui nei tessuti. La possibilità di metabolismo può essere prevista mediante strumenti adeguati (ad esempio l'OCSE Toolbox (15) e programmi QSAR proprietari). La decisione sull'opportunità di effettuare una prova di esposizione per via alimentare o attraverso l'ambiente acquatico, e con quale dispositivo di prova, dovrebbe essere basata sugli elementi di cui al paragrafo 3 nonché sulla normativa in vigore. Ad esempio, per sostanze che hanno un elevato log Kow, ma continuano a presentare un'elevata solubilità in acqua, per quanto riguarda la sensibilità dei metodi d'analisi disponibili, va considerata in primo luogo un'esposizione attraverso l'ambiente acquatico. Tuttavia è possibile che le informazioni sull'idrosolubilità non siano definitive per questi tipi di sostanze idrofobe, pertanto prima di decidere il metodo da utilizzare (16) va esaminata la possibilità di preparare concentrazioni disciolte nell'ambiente acquatico stabili e misurabili (le emulsioni stabili non sono ammesse) utilizzabili per uno studio sull'esposizione attraverso l'ambiente acquatico. Non è possibile dare istruzioni esatte sulla metodologia da seguire in funzione dei criteri di esclusione della solubilità in acqua e del coefficiente di ripartizione ottanolo/acqua, in quanto altri fattori (tecniche di analisi, degradazione, adsorbimento, ecc.) possono avere notevoli ripercussioni sulla applicabilità del metodo, per i motivi sopra esposti. Tuttavia, a partire da un log Kow superiore a 5 e di una solubilità in acqua inferiore ~ 0.01-0,1 mg/l le prove da esposizione attraverso l'ambiente acquatico diventano sempre più difficili. Devono essere considerati altri fattori che possono influenzare la scelta della prova, tra cui il contenuto del potenziale d'adsorbimento nei recipienti e nei dispositivi di prova, la sua stabilità in soluzione acquosa rispetto alla stabilità nel mangime per pesci (17) (18), ecc. Altri studi in ambiente acquatico possono contenere informazioni sugli aspetti pratici. Maggiori informazioni sulla valutazione di aspetti relativi all'esecuzione di studi di bioaccumulo sono disponibili in letteratura [cfr. ad esempio (19)]. Per le sostanze per le quali la solubilità o il mantenimento della concentrazione acquosa e l'analisi di questa concentrazione non rappresentano alcun vincolo alla realizzazione di un'esposizione in ambiente acquatico, tale metodo deve essere preferito per determinare il potenziale di bioconcentrazione della sostanza. In ogni caso, è necessario verificare che la o le concentrazioni di esposizione in ambiente acquatico da applicare rientrino nell'intervallo della solubilità nel mezzo di prova. Diversi metodi possono essere utilizzati per mantenere stabili le concentrazioni della sostanza in esame disciolta, quali l'uso di soluzioni madre o di sistemi di dosaggio passivo (ad esempio metodo dell'eluizione su colonna), purché si possa dimostrare che le concentrazioni possono essere mantenute stabili e i mezzi di prova rispettano le raccomandazioni di cui al paragrafo 27). Per le sostanze fortemente idrofobe (log Kow > 5 e una solubilità inferiore ~ 0,01-0,1 mg/l), le prove tramite esposizione in ambiente possono diventare sempre più problematiche. La difficoltà può venire dal fatto che non si riesce a mantenere la concentrazione acquosa ad un livello ritenuto sufficientemente costante (ad esempio, a causa di fenomeni di as/desorbimento verso i contenitori di vetro o di un rapido assorbimento dei pesci) o del fatto che le concentrazioni da applicare sono basse e si situano nello stesso ordine di grandezza del limite analitico di quantificazione o sono inferiori allo stesso (41). Per queste sostanze fortemente idrofobe, si raccomanda di effettuare la prova per via alimentare, a condizione che tale prova sia coerente con il quadro normativo pertinente e gli obblighi di valutazione dei rischi. Per i tensioattivi, occorre esaminare la fattibilità della prova di bioconcentrazione in ambiente acquatico, tenendo conto delle proprietà della sostanza; in caso contrario lo studio per via alimentare è probabilmente più adeguato. I tensioattivi sono agenti di superficie, che riducono la tensione interfacciale tra due liquidi. La loro natura anfifilica (vale a dire che contengono sia un gruppo idrofilo sia un gruppo idrofobo) fa sì che si accumulino alle interfacce, quali l'interfaccia acqua-aria, l'interfaccia acqua-cibo e pareti di vetro, il che impedisce di determinare la loro concentrazione acquosa. La sperimentazione per via alimentare può eludere talune difficoltà di esposizione a miscele complesse i cui componenti presentano differenti limiti di solubilità in acqua, in quanto è più probabile ottenere un'esposizione comparabile di tutti i componenti della miscela per via alimentare rispetto ad un'esposizione in ambiente acquatico [cfr. (20)]. Va osservato che il metodo per via alimentare permette di ottenere un fattore di biomagnificazione alimentare (BMF) e non un fattore di bioconcentrazione (BCF) (42). Sono disponibili metodi per stimare un fattore di bioconcentrazione cinetico (BCFk ) a partire dai dati ottenuti nello studio per via alimentare (come discusso nell'appendice 8), ma dovrebbero essere utilizzati con cautela. In generale, tali metodi presuppongono una cinetica di primo ordine e sono applicabili soltanto a determinati gruppi di composti. È improbabile che tali approcci possano essere applicati ai tensioattivi (cfr. paragrafo 12). Un impianto di prova minimo di esposizione in ambiente acquatico con un minor numero di punti di campionamento per ridurre il numero di animali e/o delle risorse (cfr. paragrafi 83 e segg.) dovrebbe essere applicato esclusivamente alle sostanze per le quali vi sia motivo di ritenere che l'assorbimento e la depurazione seguano approssimativamente una cinetica di primo ordine (vale a dire, in generale, le sostanze organiche non ionizzate, cfr. paragrafo 88). C.13 — I Prova di bioconcentrazione nei pesci per esposizione attraverso l'ambiente acquatico PRINCIPIO DEL METODO La prova consta di due fasi: la fase di esposizione (assorbimento) e di post-esposizione (depurazione). Durante la fase di assorbimento, un gruppo di pesci di una stessa specie viene esposto alla sostanza in esame ad una o più concentrazioni prescelte, in funzione delle caratteristiche della sostanza in esame (cfr. paragrafo 49). Essi vengono poi trasferiti in un ambiente esente dalla sostanza in esame per la fase di depurazione. È sempre necessaria una fase di depurazione, salvo che la quantità di sostanza assorbita durante la fase di assorbimento sia insignificante. La concentrazione della sostanza in esame nel o sul pesce (o suo tessuto specificato) viene seguita in entrambe le fasi della prova. Oltre al gruppo di trattamento, un gruppo di controllo di pesci viene mantenuto in condizioni identiche ma senza esposizione alla sostanza in esame, per confrontare possibili effetti dannosi osservati nella prova di bioconcentrazione con un gruppo di controllo analogo e per ottenere la concentrazione di fondo della sostanza in esame (43). Nella prova di esposizione in ambiente acquatico, la fase di assorbimento dura solitamente 28 giorni. Tale periodo può essere prolungato se necessario (cfr. paragrafo 18), o abbreviato qualora sia dimostrato che lo stato stazionario è stato raggiunto anticipatamente (cfr. appendice 1, Definizioni e unità di misura). È possibile prevedere la durata della fase di assorbimento e del tempo necessario al raggiungimento dello stato stazionano attraverso le equazioni di cui all'appendice 5. Inizia quindi la fase di depurazione, in cui i pesci non sono più esposti alla sostanza in esame: i pesci sono trasferiti in un mezzo identico, ma privo della sostanza in esame, contenuto in una vasca pulita. È preferibile, nella misura del possibile, calcolare il fattore di bioconcentrazione in due modi: da un lato il fattore di bioconcentrazione allo stato stazionario (BCFSS; cfr. appendice 1, Definizione), vale a dire il rapporto tra la concentrazione nel pesce (Cf) e la concentrazione nell'acqua (Cw); e, dall'altro, il fattore di bioconcentrazione cinetico (BCFk; cfr. appendice 1, Definizioni e unità di misura), vale a dire il rapporto tra la costante cinetica di assorbimento (k1) e la costante cinetica di depurazione (k2) presupponendo una cinetica di primo ordine (44). Se lo stato stazionario non è raggiunto dopo 28 giorni, occorre calcolare il BCF con il metodo cinetico (cfr. paragrafo 38) oppure si può prorogare la fase di assorbimento. Se il tempo necessario per raggiungere lo stato stazionario è troppo lungo nella pratica (cfr. paragrafi 37 e 38, appendice 5), l'approccio cinetico è preferibile. In alternativa, per le sostanze fortemente idrofobe si esaminerà la possibilità di effettuare la prova per via alimentare, a condizione che quest'ultima rispetti la normativa in vigore (45). La costante cinetica di assorbimento, la costante cinetica di depurazione (perdita) (o le costanti, se sono applicati sistemi più complessi), il fattore di bioconcentrazione (allo stato stazionario e/o cinetico) e, se possibile, gli intervalli di confidenza di ciascuno di questi parametri vengono calcolati sulla base del modello che meglio descrive le concentrazioni misurate di sostanza in esame nel pesce e nell'acqua (cfr. appendice 5). L'aumento della massa dei pesci durante la prova si tradurrà in una diminuzione della concentrazione della sostanza in esame nel pesce (effetto noto come «effetto di diluizione dovuto alla crescita), e pertanto il BCF cinetico sarà sottostimato se non corretto di conseguenza (cfr. paragrafi 72 e 73)». Il BCF è calcolato in base alla concentrazione totale nel pesce (ossia in funzione della massa umida totale dei pesci). Tuttavia, per scopi speciali, se il pesce è sufficientemente grande o può venire diviso in parti commestibili (filetto) e non commestibili (viscere), si possono usare determinati tessuti o organi (per esempio muscolo, fegato). Poiché per molte sostanze organiche esiste una chiara relazione tra il potenziale di bioconcentrazione e l'idrofobia, esiste anche una relazione corrispondente tra il contenuto lipidico dei pesci in esame e il valore osservato di bioconcentrazione di tali sostanze. Pertanto, allo scopo di ridurre questa fonte di variabilità nei risultati sperimentali per le sostanze altamente lipofile (cioè con log Kow > 3), la bioconcentrazione dovrebbe essere espressa in modo standardizzato per un pesce con un contenuto di grassi del 5 % (del peso corporeo totale), oltre alla bioconcentrazione ottenuta direttamente dalla prova. Ciò è necessario per comparare i risultati ottenuti da diverse sostanze e/o specie di prova. La percentuale del 5 % di tenore lipidico viene generalmente utilizzata, poiché ciò rappresenta di fatto il valore medio del contenuto di lipidi del pesce comunemente utilizzato nel presente metodo di prova (21). INFORMAZIONI SULLA SOSTANZA IN ESAME Oltre alle proprietà della sostanza in esame elencate nell'introduzione (paragrafo 3), occorre conoscere anche la tossicità per le specie ittiche usate nella prova, preferibilmente la LC50 asintotica (cioè indipendente dal tempo) e/o la tossicità prevista durante le prove a lungo termine sui pesci [cfr. in particolare gli orientamenti dell'OCSE 210 (22), 212 (23), 215 (24)]. Occorre disporre di un metodo analitico di comprovata accuratezza, precisione e sensibilità, per la quantificazione della sostanza in esame nelle soluzioni di prova e nel materiale biologico, nonché le modalità per la preparazione e la conservazione dei campioni. Dovrebbe essere noto anche il limite analitico di quantificazione della sostanza di prova nell'acqua e nei tessuti del pesce. Se viene utilizzata una sostanza radiomarcata, essa dovrebbe essere della massima purezza (preferibilmente > 98 %) e la percentuale di radioattività associata alle impurità deve essere nota. VALIDITÀ DELLA PROVA Perché una prova sia valida devono essere soddisfatte le seguenti condizioni:
SOSTANZE DI RIFERIMENTO Sarebbe utile disporre di sostanze di riferimento di cui si conosce il potenziale di bioconcentrazione e il metabolismo ridotto per verificare la procedura sperimentale, se del caso (ad esempio se il laboratorio non ha precedenti esperienze con l'esecuzione di tale prova o quando le condizioni sperimentali sono state modificate). DESCRIZIONE DEL METODO Apparecchiatura Occorre evitare l'impiego di materiali — in tutte le parti dell'apparecchiatura sperimentale — che possono essere soggetti a dissoluzione, assorbimento o lisciviatura e avere un effetto dannoso sul pesce. Possono essere utilizzate vasche convenzionali rettangolari o cilindriche di materiale chimicamente inerte e di capacità adeguata al tasso di carico (cfr. paragrafo 43). È opportuno minimizzare l'uso di tubi di materia plastica flessibile; è opportuno scegliere un tubo in teflon®, in acciaio inossidabile o in vetro. L'esperienza ha dimostrato che in presenza di sostanze in esame con elevati coefficienti di adsorbimento, quali i piretroidi sintetici, potrebbe essere necessario utilizzare il vetro silanizzato. In tal caso le apparecchiature non possono essere riutilizzate. È opportuno esporre i sistemi di esame alle concentrazioni adeguate della sostanza in esame per il tempo necessario a dimostrare il mantenimento della stabilità delle concentrazioni di esposizione prima dell'introduzione degli organismi di prova. Acqua Ai fini della prova si usa in genere acqua naturale che dovrebbe essere ottenuta da una fonte non contaminata e di qualità uniforme. Tuttavia, l'acqua ricostituita (acqua demineralizzata in cui specifici nutrienti sono stati aggiunti in quantità note) può essere più adatta per garantire una qualità uniforme nel tempo. La qualità dell'acqua di diluizione, vale a dire l'acqua mescolata con la sostanza in esame prima di essere inserita nel recipiente di prova (cfr. paragrafo 30), deve essere di una qualità che permetta la sopravvivenza delle specie ittiche scelte per la durata del periodo di acclimatazione e del periodo di prova senza che presentino anomalie sul piano del comportamento o dell'apparenza. L'ideale sarebbe dimostrare che la specie in esame è in grado di sopravvivere, crescere e riprodursi nell'acqua di diluizione (per esempio in una coltura di laboratorio o in un saggio di tossicità sull'intero ciclo di vita). L'acqua di diluizione deve essere caratterizzata almeno dal pH, la durezza, i solidi totali, il carbonio organico totale (TOC) (47) e di preferenza anche ammonio, nitriti e alcalinità nonché, per le specie marine, la salinità. I parametri che sono importanti ai fini del benessere dei pesci non sono perfettamente noti, ma l'appendice 2 fornisce concentrazioni massime raccomandate per un certo numero di parametri per le acque dolci e marine usate nella prova. L'acqua di diluizione deve essere di qualità costante per tutta la durata della prova. Il pH deve essere compreso tra 6,0 e 8,5 all'inizio della prova, ma senza variare di oltre ± 0,5 unità di pH nel corso dell'esperimento. Al fine di garantire che l'acqua di diluizione non abbia influenze indesiderate sul risultato sperimentale (per esempio per complessazione della sostanza in esame) o influenze negative sulla performance dello stock di pesce, ad intervalli si dovrebbero prelevare campioni per analisi, almeno all'inizio e alla fine della prova. Occorre determinare i metalli pesanti (ad esempio Cu, Pb, Zn, Hg, Cd, Ni), i principali anioni e cationi (ad es. Ca2+, Mg2+, Na+, K+, Cl–, e SO4 2–), i pesticidi (ad esempio pesticidi organofosforati totali e organoclorurati totali), il carbonio organico totale e i solidi in sospensione, ad esempio, ogni tre mesi, se l'acqua di diluizione è di qualità relativamente costante. Se la qualità dell'acqua si è dimostrata costante per almeno un anno, le titolazioni possono essere effettuate con minore frequenza (ad esempio ogni sei mesi). Il contenuto naturale di particelle in sospensione nonché di carbonio organico totale nell'acqua di diluizione deve essere il più basso possibile per evitare un assorbimento della sostanza in esame su materia organica, il che ridurrebbe la biodisponibilità e porterebbe a sottostimare il BCF. Il valore massimo accettabile è di 5 mg/l per i solidi sospesi (materia secca che non passa attraverso un filtro da 0,45 μm) e di 2 mg/l per il carbonio organico totale (cfr. appendice 2). Se necessario, filtrare l'acqua di diluizione prima dell'uso. È opportuno che il contributo degli escrementi dei pesci sottoposti a prova e dei residui alimentari al contenuto di carbonio organico dell'acqua di prova sia il minore possibile (cfr. paragrafo 46). Soluzioni di prova Preparare una soluzione madre della sostanza in esame alla concentrazione adeguata. La soluzione madre deve essere preparata preferibilmente per semplice miscelazione o agitazione della sostanza in esame nell'acqua di diluizione. Un'alternativa idonea, in alcuni casi, è l'utilizzo di un sistema di dosaggio del desorbimento in fase solida. L'utilizzo di solventi o disperdenti (agenti solubilizzanti) è generalmente sconsigliato (cfr. (25)); il loro uso può essere accettabile per ottenere una soluzione madre alla concentrazione adeguata, ma occorre adoperarsi per ridurre al minimo l'uso di tali materiali e non superare la loro concentrazione micellare critica (se del caso). I solventi che possono essere utilizzati sono acetone, etanolo, metanolo, dimetilformammide e glicole trietilenico; disperdenti utilizzati sono Tween 80, metilcellulosa 0,01 % e HCO-40. La concentrazione di solvente nel mezzo di prova finale deve essere identica in tutti i trattamenti (vale a dire indipendentemente dalla concentrazione della sostanza in esame) e non dovrebbe superare i limiti di tossicità del solvente determinata nelle condizioni sperimentali. La concentrazione massima è di 100 mg/l (o 0,1 ml/l). È improbabile che una concentrazione di solvente di 100 mg/l modifichi significativamente la concentrazione della sostanza in esame disciolta, ottenibile nel mezzo di prova (25). Il contributo del solvente (insieme con la sostanza in esame) al contenuto complessivo di carbonio organico nell'acqua usata per il saggio deve essere noto. Durante l'intera prova, la concentrazione del carbonio organico totale nei recipienti di prova non deve superare la concentrazione di carbonio organico derivata dalla sostanza in esame, solvente o agente solubilizzante, se usato, di più di 10 mg/l (± 20 %) (48). Il contenuto di materia organica può avere un effetto significativo sul volume della sostanza in esame disciolta liberamente durante le prove con metodo a flusso continuo, soprattutto per le sostanze chimiche fortemente lipofile. La microestrazione in fase solida (cfr. paragrafo 60) può fornire importanti informazioni sul rapporto tra composti disciolti liberi e vincolati, considerati come frazione biodisponibile. La concentrazione della sostanza in esame dovrebbe essere inferiore al limite di solubilità della sostanza in esame nel mezzo di prova nonostante l'uso di un solvente o solubilizzante. Occorre utilizzare con cautela i solventi facilmente biodegradabili, poiché potrebbero causare problemi di crescita batterica nelle prove a flusso continuo. Se non è possibile preparare una soluzione madre senza l'impiego di un agente di solubilizzazione, occorre valutare l'opportunità di eseguire una prova di esposizione in ambiente acquatico anziché una prova per via alimentare. Per le prove a flusso continuo occorre un sistema che eroghi e diluisca in continuo una soluzione madre della sostanza in esame (ad esempio pompa dosatrice, diluitore proporzionale, sistema di saturazione) o un sistema di dosaggio del desorbimento in fase solida, per ottenere la concentrazione desiderata nelle vasche sperimentali. Preferibilmente rinnovare il volume almeno cinque volte al giorno in vasca sperimentale. La modalità a flusso continuo va preferita, ma laddove non sia possibile (per esempio se ciò danneggiasse gli organismi sperimentali) si può utilizzare una tecnica semistatica, purché siano rispettati i criteri di validità (cfr. paragrafo 24). Le portate di soluzione madre e acqua di diluizione devono essere controllate 48 ore prima della prova e poi almeno una volta al giorno durante la prova. Nel corso di tale verifica, determinare la portata attraverso ciascuna vasca sperimentale e assicurare che la variazione non superi il 20 % all'interno di ciascuna camera e tra una vasca e l'altra. Selezione della specie Criteri importanti nella scelta delle specie sono la disponibilità, la possibilità di ottenerle di dimensioni adeguate e di mantenerle in modo soddisfacente in laboratorio. Altri criteri per la scelta delle specie ittiche includono un interesse ricreativo o commerciale, l'importanza ecologica nonché una sensibilità comparabile, utilizzi precedenti riusciti, ecc. Le specie sperimentali raccomandate sono indicate nell'appendice 3. Si possono usare anche altre specie, ma in tal caso la procedura di prova potrebbe dover essere adattata per ottenere condizioni sperimentali idonee. In tal caso occorre spiegare i criteri di scelta delle specie e del metodo sperimentale. In generale, l'uso di specie ittiche più piccole ridurrà il tempo di raggiungimento dello stato stazionario; tuttavia un numero maggiore di individui (campioni) sarà necessario per analizzare adeguatamente il contenuto lipidico e la concentrazione della sostanza in esame nel pesce. Inoltre, è possibile che le differenze di ritmo respiratorio e metabolismo tra pesci giovani e meno giovani possano ostacolare il raffronto dei risultati tra le diverse prove e le diverse specie. Si noti che eseguire la prova di tossicità sui pesci nelle prime fasi di vita (giovani) in rapida crescita, può rendere difficile l'interpretazione dei dati. Mantenimento dei pesci (acqua e regime alimentare pertinenti per l'esposizione) La popolazione ittica va acclimatata alle condizioni di laboratorio per almeno due settimane in acqua (cfr. paragrafo 28) alla temperatura di prova ed essere sufficientemente nutrita durante tutto il periodo (cfr. paragrafo 45). L'acqua e il regime alimentare devono essere dello stesso tipo di quelli destinati ad essere usati durante la prova. Dopo un periodo di ambientazione di 48 ore, si registra la mortalità e si applicano i seguenti criteri:
I pesci usati nelle prove devono essere esenti da malattie o anomalie osservabili. È necessario eliminare qualsiasi pesce ammalato. Durante le due settimane precedenti la prova e durante la prova i pesci non devono ricevere trattamenti per malattie. ESECUZIONE DELLA PROVA Prova preliminare Può essere utile condurre un esperimento preliminare allo scopo di ottimizzare le condizioni sperimentali della prova definitiva, per esempio la scelta delle concentrazioni della sostanza in esame e la durata delle fasi di assorbimento e di depurazione o per determinare se è necessario eseguire una prova completa. La prova preliminare deve essere concepita in modo tale da ottenere le informazioni richieste. Si può valutare se una sperimentazione ridotta possa essere sufficiente per ottenere un fattore di bioconcentrazione o se sia necessario uno studio completo sulla prova (cfr. i paragrafi da 83 a 95 sulla sperimentazione ridotta). Condizioni di esposizione Durata della fase di assorbimento La durata prevedibile della fase di assorbimento si può ricavare dall'esperienza pratica (per esempio da uno studio precedente o da un accumulo di studi su sostanze strutturalmente affini) o da certe relazioni empiriche, conoscendo la solubilità in acqua o il coefficiente di ripartizione ottanolo/acqua della sostanza in esame (a condizione che l'assorbimento segua una cinetica di primo ordine, cfr. appendice 5). La fase di assorbimento deve durare 28 giorni, salvo dimostrazione che è stato raggiunto prima lo stato stazionario (cfr. appendice 1, Definizioni e unità di misura). Il raggiungimento dello stato stazionario nel tracciato della sostanza in esame nei pesci (Cf) in funzione del tempo avviene quando la curva diventa parallela all'asse del tempo e tre analisi successive di Cf su campioni prelevati ad intervalli di almeno due giorni danno valori che si collocano entro ± 20 % uno dall'altro, e non vi è alcun aumento significativo di Cf nel periodo di tempo trascorso tra la prima e l'ultima analisi successive. Quando si analizzano campioni raggruppati, sono necessarie almeno quattro analisi successive. Per il controllo di sostanze che vengono assorbite lentamente saranno più opportuni intervalli di sette giorni. Se lo stato stazionario non viene raggiunto entro 28 giorni, il BCF è calcolato utilizzando solo l'approccio cinetico, che non dipende dal raggiungimento dello stato stazionario, oppure può essere prolungata la fase di assorbimento, effettuando ulteriori misure, fino al raggiungimento dello stato stazionario o fino al 60° giorno (a seconda di quale periodo sia più breve). Inoltre, la concentrazione della sostanza in esame nel pesce alla fine della fase di assorbimento deve essere sufficientemente elevata da consentire una stima attendibile della costante k 2 a partire dalla fase di depurazione. Se nessun assorbimento significativo è accertato dopo 28 giorni, la prova può essere interrotta. Durata della fase di depurazione Per le sostanze che seguono una cinetica di primo ordine, un periodo pari a metà della durata della fase di assorbimento è solitamente sufficiente perché si verifichi una riduzione appropriata (per esempio del 95 %) del carico corporeo della sostanza (cfr. appendice 5 per una spiegazione della stima). Se il periodo necessario per raggiungere una perdita del 95 % è eccessivamente lungo, per esempio se supera il doppio della normale durata della fase di assorbimento (cioè oltre 56 giorni), si può utilizzare un periodo più breve (ad esempio, fino a che la concentrazione della sostanza in esame sia inferiore al 10 % della concentrazione allo stato stazionario). Tuttavia, possono essere necessari periodi di depurazione più lunghi per le sostanze che hanno caratteristiche di assorbimento e depurazione più complesse di quelle rappresentate da un modello ittico a compartimento singolo che segue una cinetica di primo ordine. Se si osservano o si prevedono tali fenomeni complessi, si consiglia di ricorrere all'assistenza di un esperto in biostatistica e/o farmacocinetica per garantire una corretta configurazione della prova. Se la fase di depurazione è prorogata, il numero di pesci da prelevare può diventare un ostacolo e le differenze nella crescita dei pesci possono influenzare i risultati. Il periodo dipenderà inoltre dal periodo durante il quale la concentrazione della sostanza in esame nel pesce rimane al di sopra del limite analitico di quantificazione. Numero di pesci di prova Scegliere il numero di pesci per ogni concentrazione di prova in modo da includere almeno quattro pesci per ciascun tempo di campionamento. I pesci sono raggruppati solo se non sia possibile l'analisi di un unico pesce. Se è richiesta una maggiore precisione nell'adattamento della curva (e nei parametri derivati) o se sono necessari gli studi sul metabolismo (ad esempio per distinguere tra i metaboliti e la sostanza madre quando si utilizzano sostanze radiomarcate), è necessario un maggior numero di pesci per punto di campionamento. Il contenuto di lipidi deve essere determinato possibilmente sullo stesso materiale biologico usato per determinare la concentrazione della sostanza in esame. Se ciò non fosse possibile, possono essere necessari ulteriori pesci (cfr. paragrafi 56 e 57). Se si usano pesci adulti (cioè sessualmente maturi), essi non dovrebbero essere in stato di riproduzione o non essersi riprodotti di recente, sia prima che durante la prova. Occorre anche precisare il sesso dei pesci utilizzati. Se si utilizzano pesci di entrambi i sessi, occorre documentare che non vi sono differenze significative tra i sessi in termini di crescita e di contenuto lipidico prima dell'inizio dell'esposizione, in particolare se è previsto che sarà necessaria la messa in comune dei pesci maschi e femmine per ottenere concentrazioni rilevabili della sostanza e/o del contenuto di lipidi. In tutte le prove è necessario scegliere pesci di peso simile, tale che il più piccolo abbia dimensioni non inferiori a due terzi del peso dei più grandi. I pesci dovrebbero essere tutti della stessa classe di età e avere la medesima provenienza. Poiché il peso e l'età di un pesce possono avere un effetto significativo sui valori del BCF (12), tali indicazioni devono essere registrate con precisione. Si raccomanda di pesare un sottocampione dello stock di pesce subito prima dell'inizio della prova per stimare il peso medio (cfr. paragrafo 61). Carico Usare elevati rapporti acqua/pesce per minimizzare la riduzione nella concentrazione della sostanza in esame nell'acqua derivante dall'aggiunta del pesce all'inizio della prova e per evitare riduzioni della concentrazione dell'ossigeno disciolto. È importante che il carico sia appropriato per la specie usata nella prova. In ogni caso si raccomanda normalmente un tasso di carico pesce/acqua di 0,1-1,0 g di pesce (peso umido) per litro d'acqua per giorno. Si possono utilizzare carichi pesce/acqua più elevati se si dimostra che la concentrazione della sostanza in esame non registra una variazione superiore a ± 20 %, e che la concentrazione dell'ossigeno disciolto non scende al di sotto del 60 % della saturazione (cfr. paragrafo 24). Nella scelta di appropriati regimi di carico si deve tener conto dell'habitat normale della specie ittica. Per esempio, le specie bentoniche, a parità di volume d'acqua, possono richiedere un acquario con area di base maggiore rispetto a quello destinato alle specie ittiche pelagiche. Alimentazione Durante i periodi di acclimatazione e di prova, occorre mantenere i pesci ad un regime alimentare appropriato, con un contenuto totale di lipidi e di proteine noto, in quantità sufficiente per mantenerli in buone condizioni di salute e mantenere costante il peso corporeo (è tollerata una certa crescita). Per tutto il periodo di acclimatazione e di prova somministrare ai pesci il cibo in una quantità fissa in funzione della specie utilizzata, alle medesime condizioni sperimentali e di un valore energetico stabile (ad esempio, per la trota iridea approssimativamente dall'1 % al 2 % del peso corporeo al giorno). La razione alimentare è definita in modo da evitare un rapido sviluppo e un forte aumento del tenore di grassi. La quantità di cibo somministrato deve venire ricalcolata ad intervalli appropriati, per esempio una volta alla settimana, per mantenere costanti il peso corporeo e il contenuto di lipidi. Per questo calcolo, si può stimare il peso dei pesci in ciascuna vasca sperimentale in base al peso del pesce campionato più recentemente nella stessa vasca. Non pesare i pesci rimasti nella vasca. Cibo non consumato e feci vanno sifonati giornalmente dalle vasche sperimentali poco dopo la somministrazione del cibo (da 30 minuti a 1 ora). Mantenere vasche più pulite possibile per l'intera prova in modo che la concentrazione di materia organica rimanga per quanto possibile scarsa (cfr. paragrafo 29) perché la presenza di carbonio organico può limitare la biodisponibilità della sostanza in esame (12). Poiché molti mangimi derivano da farina di pesce, occorre assicurarsi che il mangime non influenzi i risultati della prova o induca effetti nocivi, ad esempio se contiene (tracce di) pesticidi, metalli pesanti e /o la stessa sostanza in esame. Illuminazione e temperatura Si raccomanda un fotoperiodo di 12-16 ore e la temperatura dell'acqua (± 2 °C) deve essere adatta alla specie utilizzata (appendice 3). Il tipo e le caratteristiche dell'illuminazione devono essere noti. Fare attenzione ad una possibile fototrasformazione della sostanza in esame alle condizioni di irraggiamento dello studio. Usare un'illuminazione appropriata evitando l'esposizione del pesce a fotoprodotti non naturali. In alcuni casi può essere appropriato utilizzare un filtro per bloccare la radiazione UV al di sotto di 290 nm. Concentrazioni della sostanza in esame Questa prova è stata inizialmente concepita per le sostanze organiche non polari. Per questo tipo di sostanza, l'esposizione del pesce a una concentrazione unica dovrebbe essere sufficiente, poiché non sono previsti effetti di concentrazione, benché il quadro normativo in vigore possa richiedere due concentrazioni. Se si saggiano altri tipi di sostanze, o se vi sono altre indicazioni di dipendenza della concentrazione, la prova deve essere effettuata con due o più concentrazioni. Se viene saggiata una sola concentrazione, occorre giustificare la scelta della concentrazione (cfr. paragrafo 79). Inoltre, la concentrazione deve essere la più bassa possibile o tecnicamente possibile (cioè non vicino al limite di solubilità). In alcuni casi si può prevedere che la bioconcentrazione di una sostanza dipenda dalla concentrazione dell'acqua (ad esempio per i metalli, per i quali l'assorbimento da parte dei pesci può essere almeno in parte regolato). In tal caso può essere necessario analizzare almeno due, e possibilmente più (cfr. paragrafo 49), concentrazioni rilevanti per l'ambiente. Inoltre per le sostanze per le quali le concentrazioni devono, per ragioni pratiche, essere vicine al limite di solubilità, si raccomanda di analizzare almeno due concentrazioni, il che può dare un'idea dell'affidabilità delle concentrazioni di esposizione. Tra le concentrazioni di prova figurano la concentrazione dell'ambiente reale, nonché la concentrazione pertinente per l'argomento specifico della valutazione. La o le concentrazioni della sostanza in esame devono essere inferiori al livello al quale producono effetti cronici o all'1 % della concentrazione LC50 acuta asintotica all'interno di un intervallo di interesse ambientale e superiori di almeno un ordine di grandezza al limite di quantificazione in acqua mediante il metodo analitico utilizzato. Il valore massimo ammissibile di concentrazione di prova può essere determinato anche dividendo la LC50 acuta (96 h) per un appropriato rapporto di concentrazione acuta/cronica (rapporti appropriati per alcuni prodotti chimici si situano intorno a 3, ma alcuni sono oltre 100). Se è utilizzata una seconda concentrazione, essa non deve differire dalla prima di un fattore dieci. Se ciò non è possibile in base al criterio di tossicità (che fissa un limite massimo per la concentrazione di prova) e al limite inferiore di determinazione analitica è opportuno applicare un fattore inferiore a 10 e utilizzare una sostanza radiomarcata, (della purezza più elevata, di preferenza superiore al 98 %). Occorre provvedere a che la concentrazione della sostanza in esame non superi la solubilità nel mezzo di prova. Controlli In aggiunta alle concentrazioni con la sostanza di prova, dovrebbe essere allestita una serie di controllo con l'acqua di diluizione o, se del caso, una serie di controllo contenente il solvente. Frequenza delle misurazioni della qualità dell'acqua Durante la prova, misurare l'ossigeno disciolto, il carbonio organico totale, il pH e la temperatura in tutte le vasche di prova e di controllo. La durezza totale e la salinità (se del caso) vanno misurate almeno nelle vasche di controllo e in una vasca di prova. Se due o più concentrazioni sono sottoposte a prova, misurare tali parametri alla concentrazione massima. Come minimo, l'ossigeno disciolto e, se del caso, la salinità devono essere misurati tre volte — all'inizio, verso la metà e alla fine del periodo di assorbimento — e una volta alla settimana durante il periodo di depurazione. Il TOC deve essere misurato all'inizio del saggio (24 h e 48 h prima dell'inizio della fase di assorbimento), prima dell'aggiunta del pesce e almeno una volta la settimana durante le fasi di assorbimento e depurazione. La temperatura va misurata e registrata giornalmente, il pH all'inizio e al termine di ciascun periodo e la durezza una volta per ogni prova. La temperatura dovrebbe preferibilmente essere controllata in continuo in almeno una vasca. Campionamento e analisi dei pesci e dell'acqua Programma di campionamento del pesce e dell'acqua Per la determinazione della concentrazione della sostanza in esame, l'acqua delle vasche sperimentali deve essere campionata prima dell'aggiunta del pesce e durante le fasi di assorbimento e depurazione. L'acqua deve essere campionata contemporaneamente al campionamento del pesce e prima della somministrazione di cibo. Campionamenti più frequenti possono essere utili per garantire la stabilità delle concentrazioni dopo l'introduzione dei pesci. Occorre determinare le concentrazioni della sostanza in esame durante la fase di assorbimento al fine di verificare il rispetto dei criteri di validità (paragrafo 24). Se l'analisi dell'acqua prelevata all'inizio della fase di depurazione non rileva alcuna sostanza in esame, ciò potrebbe giustificare la sospensione della verifica della presenza di tale sostanza nell'acqua di prova e acqua di controllo per il resto della fase di eliminazione. I pesci devono essere campionati almeno cinque volte durante la fase di assorbimento e almeno quattro volte durante la fase di depurazione per rilevare la sostanza in esame. Poiché in qualche caso risulterà difficile calcolare una stima ragionevolmente precisa del BCF sulla base di questo numero di campioni, in particolare quando la cinetica di depurazione non è una semplice cinetica di primo ordine, è consigliabile prelevare campioni a frequenza più elevata in tutti e due i periodi (cfr. appendice 4). Il contenuto di lipidi e la concentrazione della sostanza in esame sono determinati sullo stesso materiale biologico almeno all'inizio e alla fine della fase di assorbimento e alla fine della fase di depurazione. Qualora ciò non fosse possibile, almeno tre esemplari dovranno essere campionati indipendentemente per determinare il contenuto lipidico in ciascuno dei medesimi tre punti di campionamento. Il numero di pesci per vasca all'inizio dell'esperimento dovrebbe essere adeguato di conseguenza (49). In alternativa, se non sono rilevate quantità significative della sostanza in esame nei pesci di controllo (cioè i pesci dello stock di popolazione), i pesci di controllo della prova possono essere analizzati soltanto per il loro contenuto lipidico e l'analisi della sostanza in esame nel o nei gruppi di prova (nonché la costante cinetica di assorbimento, la costante cinetica di depurazione e valori del BCF) può essere corretta in funzione del contenuto lipidico del gruppo di controllo durante la prova (50). Gli esemplari morti o malati non devono essere esaminati per la sostanza in esame o concentrazione lipidica. L'appendice 4 presenta un esempio di un programma di campionamento accettabile. Si possono facilmente calcolare altri programmi se si usano altri valori di Kow per calcolare il tempo di esposizione necessario per un assorbimento del 95 % (cfr. appendice 5). Il campionamento va proseguito durante la fase di assorbimento fino al raggiungimento dello stato stazionario (cfr. appendice 1, Definizioni e unità di misure) o una volta terminata la fase di assorbimento (dopo 28 o 60 giorni, cfr. paragrafi 37 e 38). Prima dell'inizio della fase di depurazione, i pesci devono essere trasferiti in contenitori puliti. Campionamento e preparazione del campione I campioni d'acqua per l'analisi vengono ottenuti, per esempio, mediante sifonatura attraverso tubature inerti da un punto centrale della vasca sperimentale. Né la filtrazione né la centrifugazione sembrano separare sempre la frazione non-biodisponibile della sostanza in esame da quella biodisponibile. Nel caso si ricorra a una separazione, occorre giustificare tale tecnica o fornirne una validazione nel verbale di prova, tenuto conto delle difficoltà di biodisponibilità (25). In particolare le sostanze fortemente idrofobe (ossia le sostanze il cui log Kow > 5) (12) (26), per le quali può verificarsi un adsorbimento alla matrice del filtro o ai recipienti di centrifugazione, non sono soggette a tali trattamenti. Vanno, invece, adottate le misure atte a mantenere le vasche più pulite possibile (cfr. paragrafo 46) e controllare il contenuto di carbonio organico totale durante le fasi di assorbimento e depurazione (cfr. paragrafo 53). Per evitare i problemi dovuti alla riduzione della biodisponibilità, si possono eseguire, per le sostanze scarsamente solubili e fortemente idrofobe, campionamenti mediante tecniche di microestrazione in fase solida. I pesci campionati devono essere soppressi immediatamente con metodi non cruenti, utilizzando il metodo più appropriato e meno crudele (per i pesci interi, procedendo unicamente a risciacquarli in acqua (cfr. paragrafo 28) e asciugarli tamponando con carta assorbente). Pesare e misurare la lunghezza totale (51). Per ogni singolo pesce, il peso e la lunghezza misurati sono correlati alla concentrazione chimica esaminata (e se del caso al contenuto di grassi), ad esempio assegnando un codice di identificazione univoco per ciascun pesce. È preferibile analizzare il pesce e l'acqua immediatamente dopo il campionamento allo scopo di evitare degradazione o altre perdite e calcolare costanti approssimative della velocità di assorbimento e depurazione nel corso della prova. L'analisi immediata consente inoltre di individuare rapidamente l'inizio di una fase di stato stazionario. In mancanza di un'analisi immediata, tutti i campioni vanno conservati con un metodo appropriato. Prima di iniziare lo studio è opportuno ottenere informazioni sul metodo appropriato di conservazione della particolare sostanza in esame — ad esempio il congelamento, la conservazione a 4 °C, l'estrazione ecc. La durata della conservazione va scelta in modo da garantire che la sostanza non si degradi durante la stessa. Qualità del metodo analitico Poiché tutta la procedura è basata sostanzialmente sull'accuratezza, la precisione e la sensibilità del metodo analitico utilizzato per la sostanza in esame, controllare sperimentalmente che l'accuratezza, la precisione e la riproducibilità dell'analisi della sostanza, nonché il recupero della sostanza in esame dall'acqua e dal pesce, siano soddisfacenti per quel particolare metodo. Tali verifiche si effettuano nel corso delle prove preliminari. Occorre inoltre controllare che la sostanza in esame non sia rilevabile nell'acqua di diluizione usata. Se necessario, correggere i valori di concentrazione della sostanza in esame nell'acqua e nei pesci in funzione dei valori ottenuti nella prova per il recupero della sostanza in esame e la sua concentrazione naturale nei controlli. Manipolare sempre i campioni di pesce e acqua in modo da minimizzare la contaminazione e le perdite (per esempio per assorbimento sul dispositivo di campionamento). Analisi dei campioni di pesce Se nella prova vengono usati materiali radiomarcati, è possibile analizzare il marcatore radioattivo totale (cioè progenitore e metaboliti), oppure i campioni possono venire depurati, così da poter analizzare la sostanza madre separatamente. Se il BCF deve basarsi sulla sostanza madre, i principali metaboliti sono caratterizzati, almeno alla fine della fase di assorbimento (cfr. paragrafo 6). I principali metaboliti sono quelli che rappresentano almeno ≥ 10 % dei residui totali nei tessuti del pesce, quelli che rappresentano ≥ 5 % in due tempi consecutivi di campionamento, che aumentano i livelli durante la fase di assorbimento, infine, quelli di rilevanza tossicologica. Se il BCF per l'intero pesce in termini di residui totali con marcatura radioattiva è ≥ 500, può essere consigliabile, anche fortemente raccomandato nel caso di talune categorie di sostanze chimiche come i pesticidi, individuare e quantificare i principali metaboliti. La quantificazione di tali metaboliti può essere richiesta da alcune autorità di regolamentazione. Se si identificano e quantificano prodotti di degradazione che rappresentano ≥ 10 % dei residui totali con marcatura radioattiva nei tessuti del pesce, si raccomanda di identificarli e quantificarli anche nell'acqua di prova. Se ciò non fosse possibile, va spiegato nella relazione. La concentrazione della sostanza in esame viene di solito determinata su ciascun pesce pesato. Se ciò non è possibile, si possono raggruppare i campioni in occasione di ciascun campionamento, ma, poiché tale operazione limita il trattamento statistico dei risultati, è pertanto opportuno includere un numero sufficiente di pesci nella prova per tener conto del raggruppamento, della procedura statistica e della potenza desiderati. I riferimenti (27) e (28) possono essere utilizzati per l'introduzione alle pertinenti procedure di raggruppamento dei campioni. Il BCF va espresso in modo normalizzato in funzione di un pesce con un contenuto di grassi del 5 % (in relazione al peso umido) in aggiunta al BCF ottenuto direttamente dalla prova (cfr. paragrafo 21), a meno che non si possa dimostrare che la sostanza in esame non si accumula principalmente nei grassi Occorre, se possibile, determinare il contenuto lipidico dei pesci a ciascun campionamento, di preferenza a partire dallo stesso estratto prodotto per l'analisi della sostanza in esame, perché i lipidi devono spesso venire rimossi dall'estratto prima di poterlo analizzare per via cromatografica. Tuttavia, l'analisi delle sostanze in esame richiede spesso specifiche procedure di estrazione che potrebbero essere in contraddizione con i metodi di prova per la determinazione dei lipidi. In questo caso (finché non siano disponibili idonei metodi strumentali non distruttivi), si raccomanda di impiegare una strategia differente per determinare il contenuto di lipidi del pesce (cfr. paragrafo 56). Si devono usare metodi adatti per la determinazione del contenuto lipidico (20). Si raccomanda la tecnica di estrazione cloroformio/metanolo (29) come metodo standard (30), ma anche il metodo Smedes (31) può essere utilizzato in alternativa. Quest'ultimo metodo è caratterizzato da efficienza di estrazione, alta precisione, uso di solventi organici meno tossici e facilità di esecuzione. Altri metodi di precisione comparabili ai metodi raccomandati potrebbero essere utilizzati a condizione di giustificarne adeguatamente la scelta. È importante fornire informazioni dettagliate sulla metodologia applicata. Misurazione della crescita dei pesci All'inizio della prova, pesare individualmente e misurare la lunghezza di 5-10 pesci dello stock. Può trattarsi degli stessi pesci utilizzati per l'analisi dei lipidi (cfr. paragrafo 56). Misurare il peso e la lunghezza dei pesci appartenenti ai gruppi di controllo e di prova utilizzati per ogni campionamento prima di procedere all'analisi chimica o dei lipidi. La misurazione di questi pesci campionati può essere usata per calcolare il peso e la lunghezza dei pesci rimasti nelle vasche di prova e di controllo (cfr. paragrafo 45). DATI E RELAZIONE Trattamento dei risultati La curva di assorbimento della sostanza in esame è ottenuta riportando graficamente la sua concentrazione nel/sul pesce (o tessuti specificati) durante la fase di assorbimento in funzione del tempo su scale aritmetiche. Se la curva ha raggiunto un andamento costante, cioè è diventata approssimativamente asintotica rispetto all'asse del tempo, il fattore di bioconcentrazione allo stato stazionario (BCFSS) è calcolato secondo la formula seguente:
Lo sviluppo di C f può essere influenzato dalla crescita dei pesci (cfr. paragrafi 72 e 73). La concentrazione media di esposizione (Cw ) è influenzata da variazioni nel tempo. Si può prevedere che una concentrazione media ponderata nel tempo sia più pertinente e precisa per studi di bioaccumulo, anche se la variazione si mantiene all'interno del pertinente intervallo di validità (cfr. paragrafo 24). La media ponderata per il tempo della concentrazione in acqua può essere calcolata seguendo le istruzioni di cui all'appendice 5, sezione 1. Il fattore di bioconcentrazione cinetico (BCFk ) è determinato come il rapporto k1/k2, le due costanti cinetiche di primo ordine. Le costanti cinetiche k1 e k2 e il BCFk possono essere ottenuti adattando simultaneamente la fase di assorbimento e quella di depurazione. Un'altra soluzione consiste nel determinare k1 e k2 in sequenza (cfr. l'appendice 5 per una descrizione e un raffronto di questi metodi). La costante cinetica di depurazione (k2) può essere corretta dall'effetto di diluizione dovuta alla crescita (cfr. paragrafi 72 e 73). Se è evidente che la curva di assorbimento e/o depurazione non è di primo ordine, bisogna allora impiegare modelli più complessi (cfr. bibliografia dell'appendice 5) con l'assistenza di un esperto in biostatistica e/o farmacocinetica. Dati relativi al peso/alla lunghezza dei pesci Il peso umido e la lunghezza totale di ogni esemplare, a ciascun tempo di campionamento, sono riportati separatamente per i gruppi di prova e di controllo durante le fasi di assorbimento e di depurazione (compreso lo stock ittico all'inizio del periodo di assorbimento). Per ogni singolo pesce, il peso e la lunghezza sono collegati alla concentrazione chimica analizzata, ad esempio dotando ciascun pesce di un codice d'identificazione univoco. Il peso è il parametro più indicato per la misurazione della crescita al fine di correggere i valori del BCF cinetico dall'effetto di diluizione dovuta alla crescita. Il paragrafo 73 e l'appendice 5 presentano il metodo utilizzato per la correzione dall'effetto di diluizione dovuta alla crescita. Correzione dall'effetto di diluizione dovuta alla crescita e normalizzazione dei lipidi La crescita del pesce durante la fase di depurazione può ridurre le concentrazioni della sostanza chimica misurate nel pesce, con la conseguenza che, nel complesso, la costante cinetica di depurazione (k2) è di fatto maggiore rispetto a quanto si otterrebbe dai soli processi di depurazione (ad esempio respirazione, metabolismo, egestione). I fattori di bioconcentrazione cinetici sono corretti dall'effetto di diluizione dovuta alla crescita. Il BCFSS è influenzato anche dalla crescita, ma non esiste una procedura di correzione concordata in materia In caso di forte crescita, deve essere calcolato anche il BCFk, corretto dall'effetto della crescita (BCFkg), perché può risultare più pertinente del fattore di bioconcentrazione. Il contenuto di liquidi nel pesce sottoposto a trattamento (fortemente associato al bioaccumulo delle sostanze idrofobe) può variare sufficientemente in pratica perché sia necessaria la normalizzazione sulla base di un tasso di lipidi predefinito (5 % del peso umido) al fine di ottenere significativi fattori di bioconcentrazione cinetico e allo stato stazionario — a meno che non sia possibile dimostrare che la sostanza non si accumula principalmente nei grassi (ad esempio alcune sostanze perfluorurate possono legarsi alle proteine). Le equazioni ed esempi per questi calcoli figurano nell'appendice 5. Per correggere un BCF cinetico dall'effetto di diluizione dovuta alla crescita, la costante cinetica di depurazione deve essere corretta per la crescita. La costante cinetica di depurazione corretta per la crescita (k 2g) è calcolata sottraendo il tasso di crescita costante (k g, ricavato dai dati relativi al peso misurato) dalla complessiva costante cinetica di depurazione (k 2). Il fattore di bioconcentrazione cinetico corretto per la crescita è calcolato dividendo la costante cinetica di assorbimento (k 1), la costante cinetica di depurazione corretta per la crescita (k 2g) (cfr. appendice 5). In alcuni casi questo metodo è inadeguato. Ad esempio, per sostanze a depurazione molto lenta saggiate in pesci in rapida crescita, il k2g derivato può essere molto piccolo e quindi l'errore nelle due costanti cinetiche utilizzate per determinarlo è cruciale, e in alcuni casi le stime di kg possono essere superiori a k 2. Un approccio alternativo che evita la necessità di correzione dall'effetto di diluizione dovuto alla crescita comporta l'utilizzo dei dati di depurazione relativi alla massa della sostanza in esame per pesce (pesci interi), invece dei dati (concentrazione) relativi alla massa normale della sostanza in esame per unità di massa del pesce. Tale calcolo è facilmente ottenibile dato che le prove eseguite conformemente al presente metodo di prova dovrebbero collegare le concentrazioni registrate di tessuto al peso dei singoli pesci. La semplice procedura da seguire è descritta nell'appendice 5. Si noti che k 2 deve essere comunque registrato, anche se si applica questo metodo alternativo. I fattori di bioconcentrazione cinetico e allo stato stazionario vanno registrati anche rispetto ad un contenuto standard di lipidi del pesce (5 % p/p), a meno che si possa sostenere che la sostanza in esame non si accumula principalmente nei grassi. I dati relativi alla concentrazione nel pesce, o il BCF, sono normalizzati in base al rapporto tra il 5 % e l'effettivo contenuto (individuale) di lipidi (in % di peso umido) (cfr. appendice 5). Se l'analisi chimica e l'analisi dei lipidi sono state condotte sullo stesso pesce, si devono utilizzare i dati normalizzati relativi ai lipidi del singolo pesce ai fini del calcolo del BCF normalizzato in funzione dei lipidi. In alternativa, se il tasso di crescita è simile nei gruppi esposti e dei gruppi di controllo, si può utilizzare soltanto il contenuto lipidico dei pesci di controllo per la correzione dei lipidi (cfr. paragrafo 56). Un metodo di calcolo del BCF normalizzato è descritto nell'appendice 5. Interpretazione dei risultati Se le concentrazioni misurate delle soluzioni di prova sono prossime al limite di rilevazione del metodo analitico, i risultati devono essere interpretati con cautela. Per escludere effetti tossici, in linea di principio la crescita media sia del gruppo di prova sia del gruppo di controllo non dovrebbe essere significativamente diversa. Le costanti del tasso di crescita o le curve di crescita dei due gruppi vanno confrontate mediante appropriata procedura (52). Curve di assorbimento e depurazione chiaramente definite sono un'indicazione della buona qualità dei dati di bioconcentrazione. Per le costanti cinetiche, il risultato del test della bontà di adattamento χ2 dovrebbe mostrare un buon adattamento (ossia una bassa percentuale di errore nelle misurazioni (32)) per il modello di bioaccumulo, affinché le costanti cinetiche possano essere considerate attendibili (cfr. appendice 5). Se si utilizza più di una concentrazione di prova, la variazione nelle costanti di assorbimento/depurazione tra le concentrazioni di prova deve essere minore del 20 % (53). In caso contrario, ciò potrebbe indicare una dipendenza dalla concentrazione. Se si osservano differenze significative nelle velocità di assorbimento/depurazione tra le concentrazioni di prova applicate, registrarle e fornire una possibile spiegazione. In generale, un intervallo di confidenza al 95 % del BCF negli studi ben impostati è vicino a ± 20 % del BCF ottenuto. Se si saggiano due o più concentrazioni, i risultati di entrambe o tutte le concentrazioni sono utilizzati per esaminare la coerenza dei risultati e per mettere in evidenza un'eventuale dipendenza dalla concentrazione. Se si utilizza una sola concentrazione per ridurre il numero di animali e/o le risorse impiegate, occorre giustificare tale scelta. Il BCFSS è incerto se il BCFk è significativamente più grande del BCFSS, poiché ciò può indicare che lo stato stazionario non è stato raggiunto o che la diluizione dovuta alla crescita e i processi di perdita non sono stati presi in considerazione. Nei casi in cui il BCFSS è molto più elevato del BCFk, è necessario verificare la presenza di possibili errori e calcolare nuovamente le costanti cinetiche di assorbimento e depurazione. Una diversa procedura di adattamento potrebbe migliorare la stima del BCFk (cfr. appendice 5). Relazione sulla prova Oltre alle informazioni di cui al paragrafo 3, la relazione comprende i dati seguenti:
C.13 — II Prova ridotta di esposizione dei pesci attraverso l'ambiente acquatico INTRODUZIONE La crescente esperienza acquisita nella conduzione e nell'interpretazione della prova completa, sia in laboratorio sia da parte degli organismi di regolamentazione, dimostra che, con poche eccezioni, è applicabile la cinetica di primo ordine per calcolare le costanti cinetiche di assorbimento e depurazione. Pertanto, è possibile stimare le costanti cinetiche di assorbimento e depurazione con un minimo di punti di campionamento e ottenere il BCF cinetico. Lo scopo iniziale di esaminare disegni sperimentali alternativi per lo studio del BCF consisteva nello sviluppare una prova ridotta da applicare in una fase di prova intermedia al fine di confutare o confermare le stime del BCF basate su valori Kow e QSAR, ed eliminare in tal modo la necessità di eseguire uno studio completo per molte sostanze, e anche per minimizzare i costi e il numero di animali utilizzati, riducendo i campionamenti e le sequenze analitiche effettuate. Pur seguendo il principale disegno del precedente metodo di prova per consentire l'integrazione dei risultati della sperimentazione con i dati esistenti relativi al BCF e per facilitare l'esecuzione di prove e interpretazione dei dati, l'obiettivo consisteva nel fornire stime del BCF sufficientemente accurate e precise per valutare i rischi e adottare le decisioni pertinenti. Valgono molte delle considerazioni fatte per la prova completa, ad esempio i criteri di validità (cfr. paragrafo 24) e quelli per porre fine ad una prova se l'assorbimento riscontrato è trascurabile al termine della fase di assorbimento (cfr. paragrafi 16 e 38). Le sostanze che si prestano a questo disegno sperimentale ridotto devono essere pertinenti per il settore generale per il quale il presente metodo di prova è stato elaborato, vale a dire le sostanze organiche non polari (cfr. paragrafo 49). Qualora risulti che la sostanza in esame si comporta diversamente (se ad esempio presenta una chiara deviazione dalla cinetica di primo ordine), è opportuno effettuare, a fini regolamentari, una prova completa. In genere, la prova ridotta non si esegue su un periodo sperimentale più breve rispetto alla prova standard del BCF, ma richiede un ridotto campionamento dei pesci (cfr. l'appendice 6 per una spiegazione). Tuttavia, il periodo di depurazione può essere ridotto per le sostanze a depurazione rapida in modo da evitare che le concentrazioni nel pesce scendano al di sotto del limite di rilevazione/quantificazione prima della fine della prova. Una prova ridotta di esposizione attraverso l'ambiente acquatico con un'unica concentrazione può servire a determinare se è necessario condurre una verifica completa, e se i dati utilizzati per calcolare le costanti cinetiche e il BCF sono affidabili (cfr. paragrafo 93), la prova completa può essere omessa a condizione che il BCF ottenuto sia lontano dai valori regolamentari che destano preoccupazione. In alcuni casi può essere utile eseguire la prova con più di una concentrazione di prova, come prova preliminare per determinare se le stime del fattore di bioconcentrazione (BCF) per una sostanza chimica sono dipendenti dalla concentrazione. Se sulla base della prova ridotta le stime del BCF risultano dipendenti dalla concentrazione, è necessario eseguire una prova completa. Se, invece, sulla base di una prova ridotta, le stime del BCF risultano indipendenti dalla concentrazione, ma i risultati non sono considerati definitivi, qualsiasi ulteriore prova completa può essere effettuata con un'unica concentrazione, riducendo in tal modo il numero degli animali utilizzati rispetto a una prova completa con due (o più) concentrazioni. Le sostanze potenzialmente ammissibili alla prova ridotta devono:
PROGRAMMA DI CAMPIONAMENTO PER GLI STUDI ESEGUITI IN BASE AL DISEGNO SPERIMENTALE RIDOTTO Campionamento del pesce Il campionamento dei pesci è ridotto a quattro tempi di campionamento:
Campionamento dell'acqua Nella prova ridotta, l'acqua viene prelevata come nella prova completa (cfr. paragrafo 54) o almeno cinque volte ad intervalli regolari durante la fase di assorbimento e una volta alla settimana durante la fase di depurazione. Modifiche del disegno sperimentale In funzione delle proprietà della sostanza in esame, della validità delle previsioni QSAR e delle specifiche finalità dello studio, possono essere prese in considerazione alcune modifiche nel disegno sperimentale.
Calcoli Il motivo di questa impostazione è che il fattore di bioconcentrazione in una prova completa può essere determinato come fattore di bioconcentrazione allo stato stazionario (BCFSS) calcolando il rapporto tra la concentrazione della sostanza in esame nei tessuti del pesce e la concentrazione della sostanza in esame nell'acqua, o calcolando il fattore di bioconcentrazione cinetico (BCFK), vale a dire il rapporto tra la costante cinetica di assorbimento k1 e la costante cinetica di depurazione k2. Il BCFK sarebbe valido anche se una concentrazione di una sostanza allo stato stazionario non è raggiunta durante l'assorbimento, a condizione che l'assorbimento e la depurazione seguano essenzialmente processi cinetici di primo ordine. Almeno due punti sono necessari per stimare le costanti cinetiche di assorbimento e di depurazione, alla fine della fase di assorbimento (ossia all'inizio della fase di depurazione) e l'altro alla fine della fase di depurazione (o in uno stadio avanzato della fase di depurazione). Il punto di campionamento intermedio è raccomandato per controllare le cinetiche di assorbimento e di depurazione (57). Per i calcoli, cfr. le appendici 5 e 6. Interpretazione dei risultati Per valutare la validità e il valore informativo della prova, occorre verificare che il periodo di depurazione sia superiore a un tempo di dimezzamento. Analogamente, si confronta il BCFKm (BCF cinetico ottenuto da una prova ridotta) con il valore del BCFSS ridotto (che corrisponde al BCFSS calcolato alla fine della fase di assorbimento, supponendo che sia stato raggiunto lo stato stazionario. Ciò può essere solo presunto, in quanto il numero di punti di campionamento non è sufficiente a dimostrarlo). Se il BCFKm < BCFSS ridotto, quest'ultimo sarà il valore da privilegiare. Se BCFKm è inferiore al 70 % del BCFSS ridotto, i risultati non sono validi, e occorre eseguire una prova completa. Se la prova ridotta dà un BCFKm che si colloca nell'intervallo di qualsiasi valore che desta preoccupazione in base alla regolamentazione, dovrà essere eseguita una prova completa. Se il risultato è lontano da qualunque valore regolamentare preoccupante (nettamente superiore o inferiore), una prova completa può non essere necessaria, o si potrà effettuare uno studio completo con un'unica concentrazione se richiesto dal pertinente quadro normativo. Se al termine di una prova ridotta con un'unica concentrazione risulta necessario eseguire una prova completa, questa potrà essere eseguita con una seconda concentrazione. Se i risultati corrispondono, si potrà fare a meno di una prova completa con una concentrazione diversa, poiché la bioconcentrazione della sostanza non dovrebbe dipendere dalla concentrazione. Se la prova ridotta è stata eseguita con due concentrazioni e i risultati non mostrano una dipendenza dalla concentrazione, si può eseguire la prova completa con un'unica concentrazione (cfr. paragrafo 87) Relazione sulla prova La relazione della prova ridotta comprende tutte le informazioni richieste per la prova completa (cfr. paragrafo 81), a eccezione di quelle che non è possibile ottenere (vale a dire la curva indicante il rapporto tempo/stato stazionario e tempo/fattore di bioconcentrazione allo stato stazionario; per quest'ultimo dovrà essere indicato, invece, il BCFss ridotto). Occorrerà altresì precisare le ragioni per le quali è stato deciso di condurre una sperimentazione ridotta, e indicare il BCFKm risultante. C.13 — III Prova di bioaccumulo nei pesci con esposizione per via alimentare INTRODUZIONE Il metodo descritto nella presente sezione si applica alle sostanze che non si prestano a una prova mediante l'esposizione in ambiente acquatico (ad esempio perché non è possibile mantenere concentrazioni stabili e misurabili in acqua o il carico corporeo non può essere raggiunto in 60 giorni di esposizione; cfr. le sezioni precedenti per il metodo di esposizione in ambiente acquatico). Va rilevato tuttavia che il risultato da questa prova sarà un fattore di biomagnificazione per via alimentare (BMF) e non un fattore di bioconcentrazione (BCF) (58). Nel maggio 2001 un nuovo metodo per le prove di bioaccumulo di sostanze organiche scarsamente solubili in acqua è stato presentato alla conferenza SETAC Europe tenutasi a Madrid (36). Questo lavoro si basa su una serie di studi di bioaccumulo riportati nella letteratura che utilizzano un metodo di misurazione con una dieta addizionata [cfr. ad esempio (37)]. Nei primi mesi del 2004 un progetto di protocollo (38) volto a misurare il potenziale di bioaccumulo delle sostanze organiche scarsamente solubili in acqua alle quali non era applicabile il metodo di bioconcentrazione mediante l'esposizione attraverso l'ambiente acquatico, insieme a un documento di riferimento (39), è stato sottoposto al gruppo di lavoro PBT dell'UE. Tra le ragioni addotte per applicare tale metodo, è stato indicato che l'esposizione potenziale nell'ambiente a tali sostanze scarsamente solubili (log Kow > 5) avviene probabilmente in larga misura per via alimentare [cfr. (40) (41) (42) (43) (44)]. Per questa ragione, le prove con esposizione per via alimentare sono indicate in taluni regolamenti relativi alle sostanze chimiche (59). Va rilevato, tuttavia, che il metodo qui descritto evita accuratamente l'esposizione attraverso l'ambiente acquatico e, di conseguenza, un BMF ottenuto da tale metodo di prova non è direttamente comparabile con un BMF ottenuto con uno studio sul campo (che consente una combinazione di esposizione in ambiente acquatico e per via alimentare). Questa sezione si basa sul presente protocollo (38) e presenta una nuova metodologia che non figurava nella versione precedente del metodo C.13. Questa prova alternativa consente di esaminare direttamente l'esposizione per via alimentare, in condizioni controllate di laboratorio. Prima di procedere a tale esame, occorre fare riferimento ai paragrafi da 1 a 14 del presente metodo di prova per comprendere le circostanze nelle quali l'esperimento con esposizione per via alimentare è preferibile all'esame con esposizione in ambiente acquatico. Tali paragrafi contengono anche informazioni sulle sostanze, e occorre prenderne conoscenza prima di eseguire la prova. L'utilizzo di sostanze radiomarcate può essere considerato per gli stessi motivi addotti per il metodo di esposizione in ambiente acquatico (cfr. paragrafi 6 e 65). Il metodo di esposizione per via alimentare può servire ad analizzare più sostanze in un'unica prova, se sono soddisfatti determinati criteri, esaminati in dettaglio nel paragrafo 112. Per maggiore semplicità, il metodo qui presentato descrive la prova eseguita con una sola sostanza in esame. La prova con esposizione per via alimentare è simile alla prova con esposizione attraverso l'ambiente acquatico sotto molto aspetti ad eccezione, ovviamente, della modalità di esposizione. Pertanto il metodo qui proposto si sovrappone in molti punti al metodo di esposizione in ambiente acquatico di cui alla sezione precedente. Per quanto possibile sono stati introdotti rinvii ai paragrafi pertinenti della sezione precedente, ma per motivi di leggibilità e di comprensione alcune ripetizioni risultano inevitabili. PRINCIPIO DELLA PROVA Si possono utilizzare condizioni a flusso continuo o semistatiche (cfr. paragrafo 4). Le prove a flusso continuo sono preferibili per limitare l'esposizione potenziale della sostanza in esame attraverso l'ambiente acquatico a seguito di desorbimento degli alimenti addizionati o feci. La prova consta di due fasi: assorbimento (sostanza in esame-mangimi addizionati) e depurazione (mangimi «puliti», non trattati) (cfr. paragrafo 16). Durante la fase di assorbimento, un gruppo di pesci soggetto a esposizione è alimentato quotidianamente con mangimi commerciali specifici per specie ittiche di composizione nota, addizionati con la sostanza in esame. Idealmente, i pesci dovrebbero consumare tutti i prodotti alimentari offerti (cfr. paragrafo 141). Durante la fase di depurazione, i pesci sono quindi alimentati con il medesimo mangime commerciale “puro” (non trattato). Con il metodo di esposizione attraverso l'ambiente acquatico, è possibile, se necessario, utilizzare più di un gruppo variando la concentrazione della sostanza in esame, ma per la maggior parte delle sostanze organiche fortemente idrofobe è sufficiente un solo gruppo di trattamento (cfr. paragrafi 49 e 107). In condizioni semistatiche il pesce è trasferito in un nuovo ambiente e/o una nuova vasca di trattamento alla fine della fase di assorbimento (nel caso in cui il terreno o l'apparecchiatura utilizzata durante la fase di assorbimento siano stati contaminati dalla sostanza in esame mediante lisciviazione). Le concentrazioni della sostanza in esame nel pesce sono misurate in entrambe le fasi della prova. Oltre al gruppo di pesci alimentati con una dieta addizionata (il gruppo di trattamento), un gruppo di pesci di controllo viene mantenuto in condizioni identiche e con il medesimo regime alimentare, tranne il fatto che il mangime non è addizionato della sostanza in esame. Il gruppo di controllo permette di quantificare la concentrazione della sostanza in esame nei pesci non esposti e serve da termine di riferimento qualora si osservassero effetti nocivi imputabili al trattamento nel gruppo o gruppi di trattamento (60). Ciò consente inoltre il confronto dei tassi di crescita costanti tra i gruppi per accertare che siano state consumate medesime quantità di alimentazione (occorre anche tenere conto delle qualità organolettiche potenzialmente diverse dei mangimi per spiegare la differenza tra le costanti cinetiche di crescita; Cfr. paragrafo 138). È importante che durante le fasi di assorbimento e di depurazione, i regimi alimentari dei gruppi di trattamento e di controllo siano equivalenti sotto il profilo nutrizionale. Una fase di assorbimento che dura tra 7 e 14 giorni è generalmente sufficiente, sulla base dell'esperienza maturata dagli sperimentatori che hanno messo a punto tali metodi di prova (38) (39). Tale durata dovrebbe consentire di minimizzare i costi dell'esperimento, garantendo un'esposizione sufficiente per la maggior parte delle sostanze. Tuttavia, in alcuni casi può essere più opportuno prolungare la fase di assorbimento (cfr. paragrafo 127). Durante la fase di assorbimento, la concentrazione della sostanza nei pesci può non raggiungere lo stato stazionario, pertanto il trattamento dei dati e i risultati ottenuti con tale metodo si basano in generale su una analisi cinetica dei residui tessutali. (Nota: si possono applicare anche in questo caso le equazioni per calcolare il tempo di raggiungimento dello stato stazionano utilizzate nella prova con esposizione in ambiente acquatico (cfr. appendice 5). La fase di depurazione inizia dal momento in cui si somministrano ai pesci mangimi non addizionati; essa dura solitamente fino a 28 giorni, o qualora ciò richieda tempi più brevi, fino a quando la sostanza in esame non sia più quantificabile nel pesce intero. La fase di depurazione può essere abbreviata o prolungata oltre i 28 giorni, secondo le variazioni nel tempo delle concentrazioni chimiche misurate e delle dimensioni dei pesci. Questo metodo consente di determinare il tempo di dimezzamento specifico della sostanza (t1/2, in base alla costante cinetica di depurazione, k2), l'efficienza di assimilazione (assorbimento intestinale; a), il fattore di biomagnificazione alimentare cinetico (BMFK), il fattore di biomagnificazione cinetico per via alimentare corretto per la crescita (BMFKg) e il fattore di biomagnificazione cinetico per via alimentare corretto per il tenore lipidico (BMFKL) (e/o il fattore di biomagnificazione cinetico per via alimentare corretto per la crescita e per i grassi, BMFKgL) per la sostanza in esame nel pesce (61). Per quanto riguarda il metodo di esposizione in ambiente acquatico, l'aumento di peso del pesce durante l'esperimento provocherà la diluizione della sostanza in esame. Conseguentemente, il BMF (cinetico) risulterà sottostimato se non corretto per la crescita (cfr. paragrafi 162 e 163). Inoltre, se si ritiene che lo stato stazionario sia stato raggiunto nella fase di assorbimento, si può calcolare il BMF indicativo allo stato stazionario. Diversi metodi consentono di calcolare un fattore di bioconcentrazione cinetico (BCFK) a partire dai dati ottenuti nello studio per via alimentare [ad esempio (44) (45) (46) (47) (48)]. I pro e i contro di questi approcci sono analizzati nell'appendice 8. La prova è stata concepita in primo luogo per le sostanze organiche non polari scarsamente solubili in acqua che seguono essenzialmente cinetiche di assorbimento e di depurazione di primo ordine nei pesci. Quando la sostanza in esame non segue cinetiche di assorbimento e di depurazione di primo ordine, occorre impiegare modelli più complessi (cfr. bibliografia dell'appendice 5) con l'assistenza di un esperto in biostatistica e/o farmacocinetica. Di norma, si determina il BMF utilizzando l'analisi della sostanza in esame per il pesce intero (peso umido). Se pertinenti in relazione agli obiettivi dello studio, è possibile prelevare determinati tessuti (ad esempio muscolo, fegato) se il pesce è diviso in parti commestibili e non commestibili (cfr. paragrafo 21). Inoltre, la rimozione e l'analisi separata del tubo gastroenterico possono servire a determinare il contributo alle concentrazioni in pesci interi a diversi tempi di campionamento, alla fine della fase di assorbimento e all'inizio della fase di depurazione, o nel quadro di un approccio basato sul bilancio di massa. Il tenore lipidico dei pesci (interi) campionati è misurato per garantire che le concentrazioni siano corrette per i grassi, tenendo conto del tenore lipidico sia contenuto nei mangimi sia presente nei pesci (cfr. paragrafi 56 e 57 e appendice 7). Ogni pesce campionato viene pesato e il peso viene registrato e collegato alla concentrazione chimica analizzata per tale esemplare (ad esempio attribuendo un identificatore unico a ciascun pesce campionato), per calcolare la crescita del pesce in fase di prova. Nella misura del possibile, occorre misurare la lunghezza totale del pesce (62). I dati relativi al peso sono necessari anche per stimare il fattore di bioconcentrazione (BCF) utilizzando i dati della fase di depurazione dalla prova con esposizione per via alimentare. INFORMAZIONI SULLA SOSTANZA IN ESAME Occorre disporre delle informazioni sulla sostanza in esame descritte ai paragrafi 3 e 22. Un metodo per analizzare le concentrazioni della sostanza in esame nell'acqua non è in genere necessario; i metodi da applicare devono presentare una sensibilità appropriata per misurare le concentrazioni nei mangimi per pesce e nei tessuti del pesce. Il metodo può essere impiegato per valutare più di una sostanza durante un'unica prova. Tuttavia, le sostanze in esame dovranno essere compatibili tra loro, di modo che non interagiscano o cambino la loro identità chimica dopo l'addizione in un mangime per pesci. L'obiettivo è che i risultati misurati per ogni sostanza testata congiuntamente non differiscano notevolmente dai risultati che sarebbero stati ottenuti con prove individuali per ciascuna sostanza in esame. Una valutazione analitica preliminare dovrebbe stabilire che ciascuna sostanza può essere isolata da un campione di pesce o di mangime addizionato di diverse sostanze, con i) livelli elevati di recupero (> 85 % del valore nominale) e ii) la sensibilità necessaria al corretto svolgimento della prova. La quantità totale delle sostanze testate congiuntamente dovrebbe essere inferiore alla concentrazione combinata che potrebbe causare effetti tossici (cfr. paragrafo 51). Il disegno sperimentale dovrebbe inoltre tener conto dei potenziali effetti nocivi nei pesci e delle possibili interazioni (effetti metabolici) associati all'esame congiunto di più sostanze. Occorre evitare di esaminare contemporaneamente le sostanze ionizzabili. In termini di esposizione, il metodo si presta anche a miscele complesse (cfr. paragrafo 13, anche se i limiti dell'analisi saranno gli stessi con qualsiasi metodo). VALIDITÀ DELLA PROVA La validità della prova è subordinata all'osservanza delle seguenti condizioni (cfr. paragrafo 24):
SOSTANZE DI RIFERIMENTO Se un laboratorio non ha effettuato la prova in precedenza o se ha apportato modifiche significative (cambio di ceppo o di fornitore di pesci, specie ittica diversa, cambiamento significativo delle dimensioni o dell'alimentazione dei pesci, o del metodo di arricchimento, ecc.), si consiglia di verificare le competenze tecniche disponibili, utilizzando una sostanza di riferimento. La sostanza di riferimento viene usata principalmente per stabilire se la tecnica di arricchimento dell'alimentazione consente di garantire l'omogeneità e la biodisponibilità massime delle sostanze di prova. Ad esempio, la sostanza di riferimento per le sostanze idrofobe non polari è l'esaclorobenzene (HCB), ma a causa delle proprietà pericolose dell'HCB dovrebbero essere considerate altre sostanze per le quali sono disponibili dati affidabili sull'assorbimento e sulla biomagnificazione (63). Se utilizzata, informazioni generali sulla sostanza di riferimento devono figurare nella relazione sulla prova, in particolare il nome, la purezza, il numero CAS, la struttura, la tossicità (se disponibile), come per le sostanze in esame (cfr. paragrafi 3 e 22). DESCRIZIONE DEL METODO Apparecchiatura Il materiale e le apparecchiature vanno utilizzati come descritto per la prova di esposizione in ambiente acquatico (cfr. paragrafo 26). Occorre utilizzare un sistema di rinnovo a flusso continuo o statico che fornisca un volume di acqua di diluizione sufficiente alle vasche sperimentali. Le portate devono essere registrate. Acqua L'acqua di prova è utilizzata come descritto per la prova di esposizione in ambiente acquatico (cfr. paragrafi da 27 a 29). Il mezzo di prova deve presentare le caratteristiche richieste e la sua qualità va mantenuta costante per tutta la durata della prova. Il contenuto naturale di particolato e il carbonio organico totale deve essere il più basso possibile (≤ 5 mg/l di particolato; ≤ 2 mg/l di carbonio organico totale) prima dell'inizio della prova. Il carbonio organico totale è misurato solo prima della prova al momento della caratterizzazione dell'acqua utilizzata per la prova (cfr. paragrafo 53). Regime alimentare Si raccomanda l'uso di mangime per pesci disponibile in commercio (con granuli galleggianti e/o a discesa lenta), caratterizzato almeno in termini di proteine e di materie grasse. I granuli presentano una dimensione uniforme per accrescere l'efficienza dell'esposizione per via alimentare; in questo modo, i pesci mangeranno di più, poiché non si limiteranno a mangiare solo i pezzi più grossi, tralasciando quelli piccoli. È inoltre necessario che la dimensione dei granuli sia adeguata alla dimensione dei pesci all'inizio della prova (diametro di circa 0,6-0,85 mm per i pesci di lunghezza compresa tra 3 e 7 cm, e di 0,85-1,2 mm per i pesci di lunghezza compresa tra 6 e 12 cm). È possibile uniformare la dimensione dei granuli in funzione dello sviluppo dei pesci all'inizio della fase di depurazione. L'appendice 7 contiene un esempio di un'adeguata composizione degli alimenti per uso commerciale. Nello sviluppo del presente metodo sono stati utilizzati in genere regimi alimentari con un tenore lipidico compreso fra 15 e 20 % (peso umido). È possibile che mangimi per pesci con un tenore di grassi così elevato non siano disponibili in determinate regioni. In tal caso, la prova può essere effettuata con un tenore di grassi inferiore e, se necessario, il tasso di somministrazione va adeguato in modo da mantenere lo stato di salute dei pesci (sulla base di una prova preliminare). Il tenore totale di lipidi dell'alimentazione del gruppo trattato e del gruppo di controllo è misurato e registrato prima dell'inizio della prova e al termine della fase di assorbimento. La relazione sulla prova deve presentare le informazioni dettagliate indicate dal fornitore di mangimi commerciali relativamente all'analisi dei nutrienti, del tenore di umidità, fibre e ceneri e, se possibile, dei minerali e dei residui di pesticidi (ad es. inquinanti prioritari “standard”). Al momento di addizionare il mangime con la sostanza in esame, occorre garantire, per quanto possibile, l'omogeneità dei mangimi utilizzati durante la prova. La concentrazione della sostanza in esame nella dieta del gruppo trattato è selezionata in funzione della sensibilità della tecnica di analisi, della tossicità della sostanza in esame (NOEC se nota) e dei dati fisico-chimici pertinenti. Se utilizzata, è opportuno incorporare la sostanza di riferimento ad una concentrazione di circa il 10 % di quella della sostanza in esame (o comunque quanto più bassa possibile), in funzione della sensibilità dell'analisi (ad esempio per l'esaclorobenzene, una concentrazione nei mangimi di 1-100 μg/g è stata giudicata accettabile; cfr. (47) per maggiori informazioni sull'efficienza di assimilazione dell'HCB). La sostanza in esame può essere aggiunta ai mangimi per pesci in modi diversi, secondo le caratteristiche fisiche e la solubilità (cfr. appendice 7 per maggiori informazioni sui metodi di arricchimento):
In alcuni casi, ad esempio nelle prove con sostanze meno idrofobe con maggiore probabilità di deassorbimento dagli alimenti, potrebbe essere necessario rivestire i granuli con una piccola quantità di olio di pesce/mais (cfr. paragrafo 142). In tal caso, anche il mangime di controllo va trattato in modo analogo e il mangime così preparato va utilizzato ai fini della misurazione del tenore di grassi. Se del caso, i risultati sulla sostanza di riferimento devono essere comparabili con i dati delle prove descritte in letteratura e condotte in condizioni analoghe con una razione alimentare comparabile (cfr. paragrafo 45), e parametri specifici alla sostanza di riferimento devono corrispondere ai pertinenti criteri di cui al paragrafo 113 (3°, 4° e 5° punto). Se un olio o un solvente è utilizzato come disperdente per la sostanza in esame, si mescoli un quantitativo equivalente di tale disperdente (escludendo la sostanza in esame) al mangime di controllo al fine di mantenere l'equivalenza con il mangime addizionato. È importante che durante le fasi di assorbimento e di depurazione i gruppi trattati e di controllo ricevano un'alimentazione equivalente. La dieta arricchita è conservata in condizioni che mantengono la stabilità della sostanza in esame nel mix di mangimi (ad es. mediante refrigerazione) e tali condizioni vanno registrate. Selezione della specie ittica Tale prova può essere effettuata con le specie ittiche indicate per l'esposizione in ambiente acquatico (cfr. paragrafo 32 e appendice 3). Prima della pubblicazione del presente metodo di prova, gli studi sul bioaccumulo mediante regime alimentare con sostanze organiche utilizzavano sistematicamente la trota iridea (Oncorhynchus mykiss), la carpa (Cyprinus carpio) e il ciprinide Fathead minnow (Pimephales promelas). È opportuno che le specie esaminate abbiano un comportamento alimentare consistente in un rapido consumo della razione somministrata in modo da ridurre al minimo l'influenza potenziale di qualsiasi fattore che incide sulla concentrazione della sostanza in esame nell'alimento (ad esempio, lisciviazione in acqua e possibile esposizione all'ambiente acquatico). La lunghezza e il peso dei pesci utilizzati sono compresi nei limiti raccomandati (cfr. appendice 3). I pesci non devono essere troppo piccoli da ostacolare le analisi sui singoli esemplari. L'esame delle specie in un periodo di rapido accrescimento può rendere difficile l'interpretazione dei dati, e gli elevati tassi di crescita possono influenzare il calcolo dell'efficienza di assimilazione (64). Mantenimento dei pesci I criteri relativi all'acclimatazione, alla mortalità e ad eventuali malattie sono gli stessi del metodo di esposizione in ambiente acquatico (cfr. paragrafi da 33 a 35). ESECUZIONE DELLA PROVA Documento di lavoro e test di definizione dell'intervallo di concentrazioni Un'analisi preliminare è necessaria per dimostrare la possibilità di isolare la sostanza nei mangimi addizionati o nei tessuti dei pesci. Non è sempre necessario eseguire una prova di determinazione dell'intervallo di concentrazione (range-finding test) per decidere la concentrazione adeguata della sostanza chimica nel mangime. Al fine di dimostrare che non si osserva alcun effetto nocivo, di valutare le qualità organolettiche dei mangimi addizionati, di determinare la sensibilità del metodo di analisi nei tessuti dei pesci e dei mangimi e di definire la razione alimentare e i tempi di campionamento adeguati durante la fase di depurazione, ecc., è possibile — ma non obbligatorio — procedere a prove di alimentazione preliminari. Uno studio di osservazione preliminare può essere utile per stimare il numero di pesci necessari per il campionamento durante la fase di depurazione. Ciò può ridurre notevolmente il numero di pesci utilizzati, soprattutto per le sostanze in esame che sono particolarmente sensibili al metabolismo. Condizioni di esposizione Durata della fase di assorbimento Una fase di assorbimento di 7-14 giorni è normalmente sufficiente. In questa fase a un gruppo di pesci viene somministrato il mangime di controllo e a un altro gruppo il mangime trattato. La razione alimentare che viene loro somministrata ogni giorno dipende dalla specie utilizzata e dalle condizioni sperimentali; sarà, ad esempio, tra l'1 e il 2 % del peso del pesce (peso umido) nel caso della trota iridea. La frequenza di alimentazione va definita in modo da evitare una rapida crescita e un aumento significativo del tenore di grassi. Se necessario, è possibile prolungare la fase di assorbimento in funzione degli insegnamenti tratti da studi precedenti o di informazioni note sull'assorbimento o sulla depurazione della sostanza in esame (o analoga) nei pesci. L'inizio della prova è stabilito al momento della prima somministrazione della dieta addizionata. Un giorno di prova è calcolato dal momento della somministrazione di una razione alimentare fino a poco prima della razione successiva (ad esempio, un'ora prima). Pertanto, nella fase di assorbimento, il primo giorno di prova inizia con la prima somministrazione della dieta addizionata e termina subito prima della seconda razione addizionata. In pratica, la fase di assorbimento termina subito prima (ad esempio un'ora prima) della prima somministrazione di cibo non addizionato della sostanza in esame, dato che il pesce continua a digerire gli alimenti addizionati e ad assorbire la sostanza in esame nel corso delle successive 24 ore. È importante assicurare che il carico corporeo della sostanza in esame sia sufficientemente elevato (non tossico) per quanto riguarda il metodo di analisi, affinché si possa misurare un calo di almeno un ordine di grandezza durante la fase di depurazione. In taluni casi si può prolungare la fase di assorbimento (fino a 28 giorni) effettuando campionamenti supplementari per approfondire le conoscenze sulla cinetica di assorbimento. Durante l'assorbimento, la concentrazione nel pesce può non raggiungere lo stato stazionario. Per stimare il tempo occorrente per raggiungere lo stato stazionario, e avere così un'indicazione della probabile durata necessaria per ottenere concentrazioni significative nel pesce, è possibile applicare le equazioni indicate per la prova di esposizione in ambiente acquatico (cfr. appendice 5). In alcuni casi potrebbe essere noto in anticipo che una durata di assorbimento di 7-14 giorni della sostanza chimica nel pesce non è sufficiente perché la concentrazione nei mangimi consenta di raggiungere una concentrazione nel pesce sufficientemente elevata per analizzare una diminuzione di almeno un ordine di grandezza durante la depurazione, a causa della scarsa sensibilità del metodo di analisi o di un'efficienza di assimilazione troppo bassa. In tal caso, può essere utile protrarre la fase iniziale di alimentazione oltre i 14 giorni o, soprattutto nel caso di sostanze ad elevata metabolizzazione, prevedere una concentrazione maggiore nella dieta. Occorre tuttavia mantenere il carico corporeo durante l'assorbimento al di sotto della concentrazione senza effetti osservati (NOEC) cronica (stimata) nei tessuti del pesce (cfr. paragrafo 138). Durata della fase di depurazione Di norma, la depurazione dura fino a 28 giorni ed inizia con la prima somministrazione ai pesci del gruppo di prova di mangimi puri, non addizionati, dopo la fase di assorbimento. La depurazione inizia con la prima razione non addizionata anziché dall'ultima razione addizionata, poiché il pesce continua a digerire gli alimenti e ad assorbire la sostanza in esame nel corso delle successive 24 ore, come indicato al paragrafo 126. Pertanto, il primo campione della fase di depurazione è prelevato immediatamente prima della seconda razione non addizionata. Tale periodo di depurazione è inteso a individuare le sostanze con un tempo di dimezzamento di 14 giorni, corrispondente alle caratteristiche delle sostanze bioaccumulabili, una prova della durata di 28 giorni comprende quindi due tempi di dimezzamento di tali sostanze (65). Con le sostanze fortemente bioaccumulabili, può essere utile estendere la fase di depurazione (se indicato dall'esame preliminare). Se una sostanza è eliminata molto lentamente al punto che non si possa determinare un tempo di dimezzamento esatto durante la fase di depurazione, le informazioni ottenute possono comunque essere sufficienti ai fini della valutazione e indicare un livello elevato di bioaccumulo. Al contrario, se la sostanza è eliminata molto rapidamente al punto che non si può ottenere né alcuna concentrazione affidabile al tempo 0 (concentrazione alla fine della fase di assorbimento o all'inizio della fase di depurazione, C 0,d) né k 2, si può procedere a una stima prudente di k 2 (cfr. appendice 7). Se le prime analisi dei pesci campionati (7 o 14 giorni) indicano che la sostanza in esame è stata eliminata al di sotto dei livelli di rilevabilità prima della fine del periodo completo di 28 giorni, si può annullare il campionamento successivo e interrompere la prova. In alcuni casi non si constata alcun assorbimento misurabile della sostanza in esame alla fine del periodo di assorbimento (o con il secondo campione nella fase di depurazione). Se si può dimostrare che i) i criteri di validità di cui al paragrafo 113 sono soddisfatti, e che ii) tale scarso assorbimento non è dovuto ad un difetto della prova (ad esempio, durata di assorbimento insufficiente, tecnica di arricchimento inadeguata con scarsa biodisponibilità, sensibilità insufficiente del metodo d'analisi, mancato consumo dell'alimento da parte dei pesci, ecc.), è possibile porre fine allo studio senza dover ripetere la prova con una durata di assorbimento più lunga. Se lo studio preliminare indica che ciò possa essere il caso, può essere consigliabile, se possibile, analizzare le feci per esaminare la sostanza in esame non digerita secondo un approccio basato sul bilancio di massa. Numero di pesci sperimentali Come per la prova di esposizione attraverso l'ambiente acquatico, occorre scegliere pesci di peso e lunghezza simili e il più piccolo di essi non deve avere un peso inferiore a due terzi del pesce più grande (cfr. paragrafi da 40 a 42). Il numero totale di pesci utilizzati per lo studio va stabilito in funzione del programma di campionamento (almeno un prelievo alla fine della fase di assorbimento e quattro o sei prelievi durante la fase di depurazione, secondo la durata di ciascuna fase). Occorre inoltre tenere conto della sensibilità della tecnica di analisi, la concentrazione che può essere raggiunta alla fine della fase di assorbimento (secondo le informazioni disponibili ex ante) e la durata della fase di depurazione (se le informazioni disponibili in precedenza ne consentono una stima). Occorre prelevare ogni volta tra cinque e dieci pesci e quantificarne la crescita (peso e lunghezza totale) prima dell'analisi chimica o del contenuto lipidico. A causa della variabilità inevitabile delle dimensioni, della crescita e della fisiologia dei pesci e della quantità probabilmente variabile di cibo ingerita da ciascuno, è necessario prelevare per ciascun tempo di campionamento almeno cinque pesci di prova e cinque esemplari del gruppo di controllo per accertare in modo adeguato la concentrazione media e la relativa variabilità. La variabilità dei parametri nei pesci può incrementare la variabilità incontrollata complessiva della prova più di quanto dipenda dalla variabilità intrinseca dei metodi di analisi applicati, il che giustifica l'utilizzo, in alcuni casi, di un massimo di dieci esemplari per campionamento. Tuttavia, se i livelli di fondo delle concentrazioni della sostanza in esame nei pesci di controllo non sono misurabili all'inizio della fase di depurazione, l'analisi chimica di due o tre pesci di controllo nell'ultimo periodo di campionamento può essere sufficiente solo se si continua a campionare gli esemplari rimanenti del gruppo di controllo in funzione del loro peso e lunghezza totale (in modo da prelevare ogni volta lo stesso numero nei gruppi di prova e di controllo per tener conto della loro crescita). I pesci sono conservati, pesati individualmente (anche se successivamente si rende necessario combinare i risultati dei prelievi) e misurati (lunghezza totale). Pertanto, una prova tipo che prevede una fase di depurazione di 28 giorni e cinque prelievi richiederà un totale di 59-120 pesci nel gruppo di prova e tra 50 e 110 pesci nel gruppo di controllo, supponendo che la tecnica di analisi della sostanza consenta di analizzare il contenuto di grassi su uno stesso pesce. Se non è possibile analizzare la sostanza chimica e il tenore di lipidi sullo stesso pesce e non è nemmeno possibile utilizzare un pesce di controllo per analizzarne il contenuto di grassi (cfr. paragrafo 56), si devono aggiungere 15 esemplari (tre dallo stock di pesci all'inizio della prova, tre rispettivamente dal gruppo di controllo e dal gruppo di prova all'inizio della fase di depurazione e tre rispettivamente dal gruppo di controllo e dal gruppo di prova alla fine dell'esperimento). Un programma di campionamento con il numero dei pesci utilizzati è riportato nell'appendice 4. Carico Usare rapporti acqua/pesce altrettanto elevati di quelli utilizzati nel metodo di esposizione in ambiente acquatico (cfr. paragrafi 43 e 44). Sebbene i tassi di carico pesci/acqua non influiscano in alcun modo sulle concentrazioni di esposizione durante la prova, si raccomanda di applicare un tasso di carico compreso tra 0,1 e 1,0 g di pesce (peso umido) per litro d'acqua per giorno in modo da mantenere le concentrazioni di ossigeno disciolto e ridurre al minimo lo stress per gli organismi testati. Dieta e somministrazione durante la prova Durante il periodo di acclimatazione, i pesci ricevono un'alimentazione appropriata (cfr. paragrafo 117). Se la prova è eseguita con un sistema a flusso continuo, è opportuno interrompere il flusso al momento della somministrazione del cibo. Durante la prova, l'alimentazione del gruppo di prova deve essere conforme ai criteri descritti in precedenza (cfr. paragrafi da 116 a 121). Oltre alle caratteristiche specifiche della sostanza in esame, della sensibilità del metodo di analisi, della concentrazione prevista nell'alimentazione in funzione delle condizioni ambientali e dei livelli di tossicità cronica/carico corporeo, occorre tener conto, nel definire la concentrazione di prova della sostanza, delle qualità organolettiche dei mangimi (di modo che i pesci non evitino di assumerli). La concentrazione nominale della sostanza in esame va indicata nella relazione sulla prova. In base all'esperienza, le concentrazioni comprese tra 1 e 1 000 μg/g offrono un intervallo operativo adeguato per valutare le sostanze che non presentano alcun meccanismo tossico specifico. Per quanto riguarda le sostanze che agiscono con un meccanismo non specifico, i livelli nei residui tissutali non dovrebbero essere superiori a 5 mol/g di grassi, altrimenti rischiano di provocare effetti cronici (19) (48) (50) (66). Per le altre sostanze, è opportuno assicurarsi che nessun effetto nocivo derivi dall'esposizione cumulata (cfr. paragrafo 127). Ciò vale in particolar modo se più sostanze sono testate congiuntamente (cfr. paragrafo 112). La quantità adeguata della sostanza in esame può essere addizionata ai mangimi per pesci in tre modi (cfr. paragrafo 119 e appendice 7). I metodi e le procedure di arricchimento utilizzati vanno specificati nella relazione sulla prova. I pesci di controllo sono alimentati con mangimi non addizionati; nella fase di assorbimento, tali mangimi devono contenere la medesima quantità di olio eventualmente utilizzato come disperdente nel mangime addizionato oppure la stessa quantità di solvente “puro”, se il solvente è stato utilizzato come disperdente nella preparazione del mangime addizionato da somministrare al gruppo di prova. È necessario analizzare almeno tre campioni della concentrazione della sostanza in esame nei mangimi, trattati e non trattati, prima dell'inizio e alla fine della fase di assorbimento. Dopo l'esposizione al regime addizionato (fase di assorbimento) i pesci (entrambi i gruppi) ricevono mangimi non trattati (fase di depurazione). I pesci ricevano una razione alimentare determinata (a seconda della specie; circa 1-2 % del peso umido al giorno nel caso della trota iridata). L'esatta razione alimentare va determinata in modo da evitare una rapida crescita dei pesci e un forte aumento del tenore di grassi. È opportuno specificare nella relazione le dosi esatte somministrate per tutta la durata della prova. La prima razione si basa sul peso dello stock di pesce misurato appena prima dell'inizio della prova. La quantità di mangime deve essere adattata in funzione del peso umido dei pesci prelevati ai differenti tempi di campionamento previsti, per tener conto della loro crescita nel corso dell'esperimento. Il peso e la lunghezza dei pesci nelle vasche sperimentali e di controllo possono essere stimati in base al peso e alla lunghezza totale dei pesci di prova utilizzati in occasione di ogni campionamento; non pesare né misurare i pesci rimasti nelle vasche sperimentali e di controllo. È importante mantenere la medesima razione alimentare durante l'intero esperimento. Occorre osservare l'alimentazione dei pesci per assicurarsi che i pesci consumino chiaramente tutto il mangime presentato in modo da garantire che i tassi di ingestione utilizzati nei calcoli siano corretti. Si considererà l'opportunità di eseguire esperimenti preliminari di somministrazione o di tener conto di esperienze precedenti nella scelta di un regime alimentare che garantisca che la razione giornaliera somministrata in un'unica soluzione viene totalmente consumata. Se una parte del mangime rimane sistematicamente non consumato, può essere opportuno ripartire la razione su un ulteriore periodo di somministrazione in ciascun giorno di esperimento (ad es. la stessa quantità giornaliera ma somministrata in due volte anziché una). Se ciò è necessario, la seconda somministrazione deve avvenire in un momento preciso, da stabilirsi in modo che intercorra il massimo tempo possibile prima del campionamento del pesce (ad esempio, la seconda razione viene somministrata nella prima metà del giorno di esperimento). Sebbene in generale i pesci consumino rapidamente il cibo somministrato, è importante garantire che la sostanza in esame rimanga adsorbita negli alimenti. Occorre pertanto fare in modo che la sostanza in esame non si disperda in acqua, il che esporrebbe i pesci a concentrazioni della sostanza in esame nell'ambiente acquatico in aggiunta alla via alimentare. A tal fine, è opportuno eliminare il cibo non consumato (e le feci) dalle vasche sperimentali e di controllo entro un'ora o, di preferenza, entro 30 minuti dalla somministrazione. Inoltre, si può utilizzare un sistema di depurazione continua dell'acqua mediante un filtro a carbone, che adsorbe qualsiasi contaminante disciolto. I sistemi a flusso continuo possono contribuire a evacuare rapidamente le particelle alimentari e le sostanze sciolte (67). Talvolta, una leggera modifica della tecnica di arricchimento degli alimenti può contribuire ad attenuare il problema (cfr. paragrafo 119). Illuminazione e temperatura Per quanto riguarda il metodo di esposizione in ambiente acquatico (cfr. paragrafo 48) si raccomanda un fotoperiodo di 12-16 ore e una temperatura (± 2 °C) adeguata alla specie utilizzata (cfr. appendice 3). Tipo e caratteristiche dell'illuminazione devono essere noti e documentati. Controlli Occorre utilizzare un gruppo di controllo, alimentato con lo stesso mangime del gruppo di prova che però non contiene la sostanza in esame. Se per addizionare gli alimenti del gruppo di prova è stato utilizzato un olio o un solvente come disperdente, occorre trattare il cibo del gruppo di controllo esattamente allo stesso modo, ma senza l'aggiunta della sostanza in esame affinché l'alimentazione dei gruppi di prova e di controllo siano equivalenti (cfr. paragrafi 121 e 139). Frequenza delle misurazioni della qualità dell'acqua Le condizioni descritte per la prova di esposizione in ambiente acquatico si applicano anche in questo caso, ad eccezione del fatto che il carbonio organico totale è misurato solo prima della prova, al momento della caratterizzazione dell'acqua utilizzata per la prova (cfr. paragrafo 53). Campionamento e analisi dei pesci e dell'alimentazione Analisi dei campioni alimentari Almeno tre campioni di cibo addizionato e non addizionato sono analizzati, per determinare la concentrazione della sostanza in esame e il contenuto di lipidi, almeno prima dell'inizio e alla fine della fase di assorbimento. I metodi di analisi e le procedure applicate per garantire l'omogeneità del regime alimentare vanno specificati nella relazione sulla prova. Occorre analizzare la sostanza in esame nei campioni conformemente alla metodologia definita e convalidata. Si procede a uno studio per stabilire il limite di quantificazione, la percentuale di isolamento, le interferenze e la variabilità dell'analisi nella prevista matrice del campione. Se viene testata una sostanza radiomarcata, si dovranno fare considerazioni analoghe a quelle relative al metodo di esposizione in ambiente acquatico, nelle quali l'analisi degli alimenti sostituisce l'analisi dell'acqua (cfr. paragrafo 65). Analisi dei pesci Ad ogni campionamento si prelevano tra 5 e 10 esemplari in ciascuno dei gruppi di controllo e di prova (in alcuni casi il numero dei pesci di controllo può essere ridotto; cfr. paragrafo 134). I campionamenti devono avvenire nello stesso momento in ciascun giorno di sperimentazione (in funzione del momento di somministrazione alimentare), e dovrebbe essere stabiliti in modo da ridurre la probabilità che il cibo rimanga nell'intestino durante la fase di assorbimento e l'inizio della fase di depurazione al fine di evitare di falsare le concentrazioni totali della sostanza in esame (cioè i pesci campionati vanno rimossi alla fine di un giorno sperimentale, tenendo presente che un giorno di sperimentazione inizia al momento della somministrazione di cibo e termina al momento della successiva alimentazione, circa 24 ore più tardi. La fase di depurazione inizia con la prima razione di cibo «non addizionato» (cfr. paragrafo 128). Il primo campione della fase di depurazione (prelevato immediatamente prima della seconda razione non addizionata) è importante in quanto serve a calcolare la concentrazione al tempo 0, mediante estrapolazione al giorno prima (C 0,d, la concentrazione nel pesce alla fine dell'assorbimento/all'inizio della depurazione). In alternativa, si possono prelevare e analizzare separatamente il tratto gastrointestinale del pesce alla fine dell'assorbimento e nei giorni 1 e 3 della fase di depurazione. Per ciascun tempo di campionamento occorre prelevare i pesci dalle vasche sperimentali e di controllo e trattarli allo stesso modo descritto per il metodo di esposizione in ambiente acquatico (cfr. paragrafi da 61 a 63). Misurare le concentrazioni della sostanza in esame nel pesce (peso umido) almeno alla fine della fase di assorbimento e durante la fase di depurazione nei gruppi di controllo e di prova. Durante la fase di depurazione, si raccomandano 4-6 tempi di campionamento (ad esempio nei giorni 1, 3, 7, 14 e 28). In alternativa, è possibile includere un tempo di campionamento supplementare dopo 1-3 giorni di assorbimento al fine dell'efficienza di assimilazione dalla fase lineare di assorbimento per i pesci mentre è ancora vicina all'inizio del periodo di esposizione. Esistono due principali deviazioni rispetto al programma: i) se si utilizza una proroga della fase di assorbimento ai fini dello studio della cinetica di assorbimento, ci saranno ulteriori tempi di campionamento durante la fase di assorbimento e di conseguenza dovrà essere incluso un numero maggiore di pesci (cfr. paragrafo 126); ii) se allo studio è stato posto termine alla fine della fase di assorbimento, a motivo della mancanza di assorbimento misurabile (cfr. paragrafo 131). Gli esemplari di pesci campionati sono pesati (e la loro lunghezza totale misurata) per determinare le costanti cinetiche di crescita. Possono essere misurate anche le concentrazioni della sostanza in specifici tessuti dei pesci (parti commestibili e non commestibili) alla fine della fase di assorbimento e in selezionati punti della fase di depurazione. Se viene testata una sostanza radiomarcata, si dovranno fare considerazioni analoghe a quelle relative al metodo di esposizione in ambiente acquatico, nelle quali l'analisi degli alimenti sostituisce l'analisi dell'acqua (cfr. paragrafo 65). In caso di utilizzo periodico di una sostanza di riferimento (cfr. paragrafo 25), le concentrazioni vanno misurate di preferenza nel gruppo di prova alla fine dell'assorbimento e in tutti i tempi della depurazione specificati per la sostanza in esame (pesce intero); le concentrazioni nel gruppo di controllo vanno analizzate solo alla fine dell'assorbimento (pesce intero). In alcune circostanze (ad esempio quando le tecniche di analisi della sostanza in esame e della sostanza di riferimento sono incompatibili al punto che è necessario l'utilizzo di pesci supplementari per rispettare il programma di campionamento), si potrà applicare un metodo alternativo per ridurre al minimo il numero di esemplari supplementari necessari. Le concentrazioni della sostanza di riferimento sono misurate durante la fase di depurazione solo nei giorni 1, 3 e in due altri tempi di campionamento selezionati in modo da ottenere stime affidabili della concentrazione al tempo 0 (C 0,d) e del valore k2 per la sostanza di riferimento. Se possibile il contenuto lipidico di ciascun pesce dovrebbe essere determinato ad ogni campionamento, o almeno all'inizio e alla fine della fase di assorbimento e alla fine della fase di depurazione. (cfr. paragrafi 56 e 67). In funzione del metodo analitico utilizzato (cfr. paragrafo 67 e appendice 4), è possibile utilizzare gli stessi pesci per determinare il tenore di grassi e la concentrazione della sostanza in esame. Ciò è preferibile in quanto consente di ridurre al minimo il numero di esemplari necessari. Tuttavia, quando ciò non è possibile, occorre applicare il metodo descritto per il metodo di esposizione in ambiente acquatico (cfr. paragrafo 56 per le diverse opzioni di misurazione del tenore di grassi). Il metodo applicato per quantificare il tenore lipidico va registrato nella relazione sulla prova. Qualità del metodo analitico Sono necessarie prove preliminari per verificare la specificità, precisione e riproducibilità della tecnica di analisi specifica per la sostanza e l'isolamento della sostanza in esame nell'alimentazione e nel pesce. Misurazione della crescita dei pesci All'inizio della prova, occorre pesare e misurare un campione di pesce proveniente dallo stock ittico. I pesci sono prelevati poco prima (un'ora prima) della somministrazione del mangime addizionato e assegnati al giorno di prova 0. Il numero di animali sottoposti a campionamento deve essere uguale o superiore a quello dei pesci prelevati successivamente durante la prova. Alcuni di questi possono essere gli stessi pesci utilizzati per l'analisi dei lipidi eseguita prima dell'inizio della fase di assorbimento (cfr. paragrafo 153). In occasione di ciascun campionamento, i pesci sono pesati e misurati. Per ciascun esemplare, il peso e la lunghezza sono collegati alla concentrazione chimica analizzata (e eventualmente al tenore di grassi), ad esempio assegnando un codice di identificazione unico a ciascun pesce campionato. Tali misure possono servire a stimare il peso (e la lunghezza dei pesci rimasti nelle vasche di prova e di controllo. Valutazione della prova Le osservazioni relative alla mortalità devono essere registrate ogni giorno. Vanno effettuate ulteriori osservazioni per accertare eventuali effetti nocivi, ad esempio un comportamento anomalo o una pigmentazione, che devono essere registrati. I pesci sono considerati morti in assenza di movimenti respiratori o di reazione ad un leggero stimolo meccanico. Ogni esemplare morto o visibilmente morente dovrà essere rimosso. RELAZIONE SULLA PROVA E RISULTATI Trattamento dei risultati I risultati delle prove sono utilizzati per calcolare la costante cinetica di depurazione (k2 ) in funzione del peso umido totale del pesce. La costante cinetica di crescita, kg , basata sull'incremento medio del peso del pesce è calcolata e utilizzata per produrre la costante cinetica di depurazione corretta per la crescita, k2g, se del caso. Si dovrebbe inoltre registrare l'efficienza di assimilazione (a; assunzione intestinale), il fattore di biomagnificazione cinetico (BMFK) (se necessario corretto per la crescita, BMFKg), il suo valore corretto per i lipidi (BMFKL o BMFKgL, se corretto dall'effetto di diluizione dovuto alla crescita) e il regime alimentare. Inoltre, qualora sia possibile stimare il tempo necessario per raggiungere lo stato stazionario durante la fase di assorbimento (ad esempio 95 % dello stato stazionario o t 95 = 3,0/k 2), si deve includere una stima del BMF allo stato stazionario (BMFSS) (cfr. paragrafi 105 e 106 e appendice 5) se il valore t95 indica che lo stato stazionario è raggiunto. Occorre applicare al BMFSS la stessa correzione per i lipidi applicata al BMF cinetico (BMFK) per ottenere un valore corretto per il tenore di grassi, BMFSS (non esiste alcuna procedura concordata per correggere un BMF allo stato stazionario dall'effetto di diluizione dovuto alla crescita). Le formule e gli esempi di calcolo sono illustrati nell'appendice 7. Diversi metodi consentono di calcolare un fattore di bioconcentrazione cinetico (BCFK) a partire dai dati ottenuti nello studio per via alimentare. Essi sono riportati nell'appendice 8. Peso e lunghezza dei pesci Il peso umido e la lunghezza di ciascun pesce, a ciascun tempo di campionamento, sono presentati separatamente per i gruppi di prova e i gruppi di controllo durante la fase di assorbimento [(stock ittico all'inizio del periodo di assorbimento; gruppo di controllo e gruppo di prova alla fine del periodo di assorbimento e, se eseguite, la fase iniziale (ad esempio giorni 1-3 della fase di assorbimento) e la fase di depurazione (ad esempio giorni 1, 2, 4, 7, 14, 28, per il gruppo di prova e il gruppo di controllo)]. Il peso è la misura preferita per la crescita ai fini della correzione dall'effetto di diluizione dovuto alla crescita. I paragrafi 162 e 163 e l'appendice 5 presentano il metodo o i metodi utilizzati per la correzione. Concentrazione della sostanza in esame nel pesce Le misurazioni dei residui della sostanza in esame effettuate su ogni singolo pesce (o su campioni raggruppati se le singole misurazioni non sono possibili), espresse in termini di concentrazioni del peso umido (w/w), vengono presentate in tabelle, separatamente per ciascun punto di campionamento, per i gruppi di controllo e di prova. Se è stata effettuata un'analisi lipidica su ciascuno dei pesci prelevati, le concentrazioni individuali corrette per il tenore di grassi, espresse in tenore lipidico del pesce umido (w/w lipid), possono essere calcolate e registrate.
Velocità di depurazione e fattore di biomagnificazione Per calcolare il fattore di biomagnificazione, occorre innanzitutto ottenere l'efficienza di assimilazione (assorbimento della sostanza in esame per via intestinale, α). A tal fine, è opportuno applicare l'equazione A7.1 dell'appendice 7 che richiede la conoscenza della concentrazione nel pesce ottenuta al giorno 0 della fase di depurazione (C 0,d), la costante cinetica di depurazione (globale) (k2), la concentrazione negli alimenti (C food), la costante cinetica di ingestione (I) e la durata della fase di assorbimento (t). La pendenza e l'intercetta della relazione lineare tra il logaritmo naturale della concentrazione e il tempo di depurazione sono registrate come la costante cinetica di depurazione complessiva (k2 = pendenza) e la concentrazione al tempo 0 (C 0,d = eintercept), come indicato in precedenza. Occorre verificare la plausibilità biologica dei valori ottenuti (ad esempio, l'efficienza di assimilazione sotto forma di percentuale non è superiore a 1). (I) è calcolata dividendo la massa degli alimenti per il peso del pesce alimentato ogni giorno (se alimentato al 2 % del suo peso (I) è pari a 0,02). Tuttavia può rendersi necessario adeguare il regime alimentare utilizzato nei calcoli in funzione della crescita degli esemplari (utilizzando la costante cinetica di crescita per stimare il peso del pesce ad ogni campionamento durante la fase di assorbimento; cfr. appendice 7). Nei casi in cui non è possibile ottenere k2 e C 0,d perché, ad esempio, le concentrazioni sono scese al di sotto della soglia di rilevamento al momento del secondo campione di depurazione, si può procedere ad una stima prudente di k2 e si può calcolare un “limite superiore” di BMFk (cfr. appendice 7) Una volta ottenuta l'efficienza di assimilazione (α) si può calcolare il fattore di biomagnificazione moltiplicando α per la costante cinetica di ingestione (I) e dividendola per la costante cinetica di depurazione (globale) (k2). Il fattore di biomagnificazione corretto dall'effetto di diluizione dovuto alla crescita è calcolato nello stesso modo, ma utilizzando la costante cinetica di depurazione corretta per la crescita (k 2g; cfr. paragrafi 162 e 163). In alternativa, è possibile stimare l'efficienza di assimilazione se sono stati analizzati i tessuti dei pesci prelevati nel corso della fase iniziale, lineare dell'assorbimento (cfr. paragrafo 151 e appendice 7). Tale valore rappresenta una stima indipendente dell'efficienza di assimilazione per un organismo praticamente non esposto (vale a dire il pesce prelevato all'inizio della fase di assorbimento). L'efficienza di assimilazione stimata sulla base dei dati della fase di depurazione è solitamente utilizzata per ottenere il BMF. Correzione per il tenore di grassi e correzione dalla diluizione dovuta alla crescita La crescita dei pesci durante la fase di depurazione può ridurre le concentrazioni chimiche misurate nel pesce, aumentando la costante cinetica di depurazione complessiva, k2, più di quanto provocherebbero i soli processi di depurazione (es.: metabolismo, egestione) (cfr. paragrafo 72). Il contenuto di liquidi nel pesce di prova (fortemente associato al bioaccumulo delle sostanze chimiche idrofobe) e il contenuto di lipidi degli alimenti possono variare in pratica sufficientemente per richiederne la correzione al fine di ottenere fattori di biomagnificazione significativi. Il fattore di biomagnificazione è corretto dall'effetto di diluizione dovuto alla crescita (come il BCF cinetico nel metodo di esposizione in ambiente acquatico) e corretto per il tenore lipidico degli alimenti in funzione di quello del pesce (il fattore di correzione per i lipidi). Le appendici 5 e 7 presentano le equazioni e forniscono esempi di calcoli di questo tipo. Per correggere la diluizione della crescita, si deve calcolare la costante cinetica di depurazione corretta per la crescita (k 2g) (cfr. appendice 5 per le formule). La costante cinetica di depurazione corretta (k 2g) è utilizzata per calcolare il fattore di biomagnificazione corretto per la crescita, come indicato al paragrafo 73. In alcuni casi, tale metodo non è possibile. Un altro metodo che evita la necessità di una correzione della diluizione dovuto alla crescita consiste nell'utilizzare la massa della sostanza in esame per pesce (intero) della depurazione anziché la normale massa della sostanza in esame per unità di massa del pesce (concentrazione). Ciò è facilmente calcolabile, poiché le prove che rispettano il presente metodo devono collegare le concentrazioni registrate nei tessuti al peso del singolo pesce. L'appendice 5 illustra la procedura di calcolo semplice. Si noti che il ricorso a tale metodo alternativo non esonera dalla valutazione e dalla registrazione del valore di k2. Per correggere per il tenore lipidico degli alimenti e del pesce quando l'analisi lipidica non è stata effettuata su tutti i pesci campionati, si calcolano le parti grasse medie (del pesce umido) nel pesce e nei mangimi (68). Il fattore di correzione per il tenore di grassi (Lc ) è calcolato dividendo la parte grassa media del pesce per la parte grassa media di alimenti. Il fattore di biomagnificazione, corretto per la crescita, è diviso per il fattore di correzione per il tenore di grassi al fine di calcolare il fattore di biomagnificazione corretto per i lipidi. Se l'analisi chimica e l'analisi dei lipidi sono state condotte sullo stesso pesce allo stesso tempo di campionamento è opportuno utilizzare questi dati corretti per i lipidi sui tessuti dei singoli pesci al fine di calcolare direttamente un BMF corretto per i lipidi [cfr. (37)]. La curva della concentrazione corretta in funzione dei lipidi dà C 0,d e k2. Si può procedere ad un'analisi matematica utilizzando le equazioni nell'appendice 7, ma l'efficienza di assimilazione è calcolata utilizzando la costante cinetica di ingestione normalizzata per il tenore lipidico (I lipid) e la concentrazione nell'alimentazione in funzione dei lipidi (C food-lipid). I parametri corretti per il tenore di grassi sono poi impiegati in modo analogo per calcolare il BMF (per calcolare il BMFKgL corretto per il tenore di grassi e per la crescita, si applica anche la correzione della costante cinetica di crescita alla parte grassa anziché al peso del pesce umido). Interpretazione dei risultati La crescita media del gruppo di prova e del gruppo di controllo non dovrebbe in linea di principio differire molto per escludere effetti tossici. Le costanti cinetiche di crescita o le curve di crescita dei due gruppi vanno confrontate mediante una procedura adeguata (69). Relazione sulla prova Dopo il completamento dello studio, occorre redigere una relazione finale contenente le informazioni sulla sostanza in esame, la specie utilizzata e le condizioni di prova di cui al paragrafo 81 (per il metodo di esposizione in ambiente acquatico). Inoltre, la relazione deve includere anche le seguenti informazioni:
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI E UNITÀ DI MISURA
BIBLIOGRAFIA
Appendice 2 ALCUNE CARATTERISTICHE CHIMICHE DI UN'ACQUA DI DILUIZIONE ACCETTABILE
Appendice 3 SPECIE ITTICHE RACCOMANDATE PER LA PROVA
Varie specie di estuario e marine sono meno utilizzate, ad esempio:
I pesci d'acqua dolce suelencati sono facilmente allevabili e/o sono largamente disponibili per tutto l'anno, mentre la disponibilità delle specie marine e di estuario è parzialmente confinata ai rispettivi paesi. Possono riprodursi e venire allevati sia in stabilimenti di acquicoltura sia in laboratorio, in condizioni di controllo delle malattie e dei parassiti, in modo che gli animali di prova siano sani e geneticamente controllati. Questi pesci sono disponibili in molte parti del mondo. BIBLIOGRAFIA
Appendice 4 PROGRAMMA DI CAMPIONAMENTO PER LE PROVE DI ESPOSIZIONE PER VIA ALIMENTARE IN AMBIENTE ACQUATICO 1. Esempio teorico di un programma di campionamento per una prova di bioconcentrazione con esposizione esclusivamente in ambiente acquatico su una sostanza con log ko/w = 4.
2. Esempio teorico di un programma di campionamento per prova di bioaccumulo della sostanza per via alimentare con fasi di assorbimento e di depurazione, rispettivamente, di 10 e 42 giorni.
Nota relativa alla durata delle fasi e ai tempi di campionamento: la fase di assorbimento inizia con la prima razione di mangimi addizionati. Il primo giorno di prova inizia con la prima somministrazione di cibo e termina subito prima della successiva, 24 ore più tardi. Il primo campionamento (1 nella tabella) dovrebbe avvenire poco prima della somministrazione di cibo (un'ora prima, ad esempio). Idealmente, si dovrebbe ogni volta prelevare i pesci immediatamente prima della razione del giorno successivo (circa 23 ore dopo l'ultima razione). La fase di assorbimento termina subito prima della somministrazione di mangimi non addizionati, quando comincia la fase di depurazione (è probabile che i pesci del gruppo di prova stiano ancora digerendo gli alimenti addizionati nelle 24 ore dall'ultima somministrazione di mangime addizionato). Ciò significa che l'ultimo prelievo della fase di assorbimento avviene poco prima della somministrazione del mangime non addizionato, e il primo campionamento della fase di depurazione è eseguito circa 23 ore dopo la prima razione di mangimi non addizionati. Appendice 5 CALCOLI GENERALI
1. INTRODUZIONE Il modello generale di bioaccumulo in ambiente acquatico nei pesci può essere descritto in termini di processo di assorbimento e di perdita, ignorando l'assunzione alimentare. L'equazione differenziale (dC f/dt) che descrive il tasso di variazione della concentrazione nel pesce (mg·kg– 1·giorno– 1) è data da (1):
Dove
Per quanto riguarda sostanze bioaccumulabili, si può prevedere che un valore medio ponderato nel tempo (TWA) sia la concentrazione di esposizione nell'acqua (Cw ) all'interno dell'intervallo di fluttuazione autorizzato (cfr. paragrafo 24). Si raccomanda di calcolare una media ponderata rispetto al tempo della concentrazione in acqua secondo le istruzioni fornite nell'appendice 6 del metodo di prova C.20 (2). Si noti che il logaritmo naturale della concentrazione in acqua è adeguato quando si prevede un declino esponenziale tra i periodi di rinnovo, ad esempio in condizioni di prova semistatiche. Con un sistema dinamico, la trasformazione in logaritmo naturale delle concentrazioni di esposizione non è sempre necessaria. Se si ottiene una media ponderata nel tempo della concentrazione in acqua, è opportuno registrarla e utilizzarla per i calcoli successivi. In una prova BCF standard eseguita sui pesci, l'assorbimento e la depurazione possono essere descritti in termini di due processi cinetici di primo ordine.
Allo stato stazionario, in un'ipotesi di crescita e metabolismo trascurabili (i valori per kg e km non sono distinguibili da zero), la cinetica di assorbimento è uguale alla cinetica di depurazione, e quindi combinando le equazioni A5.2 e A5.3 si ottiene la seguente relazione:
Dove
Il rapporto k1/k2 è noto come fattore di bioconcentrazione cinetico (BCFk ) e dovrebbe essere pari al valore del fattore di bioconcentrazione (BCF) allo stato stazionario (BCFSS) ottenuto dal rapporto tra la concentrazione allo stato stazionario nel pesce e la concentrazione allo stato stazionario in acqua, ma variazioni sono possibili se lo stato stazionario è incerto o se correzioni per la crescita sono state applicate al BCF. Tuttavia, una volta che k 1 e k 2 sono costanti, non è necessario che venga raggiunto lo stato stazionario per ottenere un BCFk. Sulla base di queste equazioni di primo ordine, la presente appendice 5 riporta le operazioni generali di calcolo necessarie per i due metodi di bioaccumulo, con esposizione in ambiente acquatico e esposizione per via alimentare. Tuttavia, le sezioni 5, 6 e 8 sono rilevanti solo per il metodo di esposizione in ambiente acquatico, ma sono state qui riportate come “tecniche generali”. I metodi sequenziali (sezioni 4 e 5) e simultaneo (sezione 6) permettono di calcolare le costanti di assorbimento e di depurazione che servono a ottenere i BCF cinetici. Il metodo sequenziale per calcolare k2 (sezione 4) è importante per l'esposizione per via alimentare, in quanto necessario per calcolare l'efficienza di assimilazione e il BMF. L'appendice 7 illustra in dettaglio i calcoli specifici per il metodo di esposizione per via alimentare. 2. PREVISIONE DELLA DURATA DELLA FASE DI ASSORBIMENTO Prima di eseguire la prova, si può ricavare una stima di k2 e quindi di una data percentuale del tempo occorrente per arrivare allo stato stazionario da relazioni empiriche tra k2 e il coefficiente di ripartizione n-ottanolo/acqua (Kow) o tra k1 e il BCF. Si dovrebbe essere, tuttavia, consapevoli che le equazioni in questa sezione sono applicabili unicamente quando l'assorbimento e la depurazione seguano una cinetica di primo ordine. Se ciò non è evidentemente il caso, si raccomanda di chiedere il parere di un esperto in biostatistica e/o farmacocinetica, per stabilire se le previsioni della durata della fase di assorbimento sono auspicabili. Una stima di k2 (giorni-1) si può ottenere con diversi metodi. Ad esempio, le seguenti relazioni empiriche possono essere utilizzate in primo luogo: (87)
oppure
W = media del peso del pesce trattato (in grammi di pesce umido) alla fine dell'assorbimento/inizio di depurazione (88) Per altre relazioni associate, cfr. (6). Può essere utile impiegare modelli più complessi per ricavare una stima di k2 se, ad esempio, è probabile che si verifichi un metabolismo significativo (7) (8). Tuttavia, a causa della maggiore complessità del modello, si provvederà a prestare particolare attenzione all'interpretazione delle previsioni. Pertanto, la presenza di gruppi nitrici potrebbe indicare un metabolismo rapido, ma non è sempre questo il caso. Pertanto, l'utente deve ponderare i risultati con metodi predittivi sulla base della struttura chimica e ogni altra informazione pertinente (ad esempio studi preliminari) nella programmazione di uno studio. Il tempo necessario per raggiungere una certa percentuale dello stato stazionario si può ricavare, applicando la stima k2, dall'equazione cinetica generale che descrive l'assorbimento e la depurazione (cinetica di primo ordine), nell'ipotesi di crescita e metabolismo trascurabili. Se si verifica una crescita sostanziale nel corso dello studio, le stime indicate di seguito non saranno attendibili. In tale caso, è meglio utilizzare il valore k2g corretto per la crescita come descritto più avanti (cfr. sezione 7 della presente appendice):
o, se C è costante:
Approssimandosi allo stato stazionario, (t — ∞), l'equazione A5.10 può essere ridotta a (cfr. (9) (10)]:
oppure
BCF × C w è pertanto un'approssimazione della concentrazione nel pesce allo stato stazionario (C f-SS). [Nota: Lo stesso approccio può essere usato per calcolare il BMF allo stato stazionario durante una prova con esposizione per via alimentare. In tal caso, il valore BCF è sostituito dal BMF, e la C w dal C food, la concentrazione negli alimenti, nelle equazioni di cui sopra.] L'equazione A5.10 può essere riformulata come segue:
oppure
Applicando l'equazione A5.14, il tempo necessario per raggiungere una certa percentuale dello stato stazionario si può prevedere quando k2 sia stato pre-stimato mediante l'equazione A5.5 o A5.6. A titolo indicativo, la durata statisticamente ottimale della fase di assorbimento per ottenere dati statisticamente accettabili (BCFK ) è il periodo necessario perché la curva del logaritmo della concentrazione della sostanza in esame nel pesce in funzione del tempo raggiunga almeno il 50 % dello stato stazionario (0.69/k2), e non più del 95 % dello stato stazionario (3.0/k2) (11). Se l'accumulo supera il 95 % dello stato stazionario, il calcolo del BCFSS diventa possibile. Il tempo necessario per raggiungere l'80 % dello stato stazionario si ottiene da (equazione A5.14):
oppure
Analogamente il tempo necessario per raggiungere il 95 % dello stato stazionario si ottiene da:
A titolo di esempio, la durata della fase di assorbimento (cioè il tempo necessario all'ottenimento di una certa percentuale dello stato stazionario, ad esempio t 80 o t 95) di una sostanza in esame con log Ko/w = 4 raggiungerebbe (utilizzando le equazioni A5.5, A5.16 e A5.17): logk 2 = 1,47 – 0,414 · 4 k2 = 0.652 giorni-1
oppure In alternativa, si può utilizzare l'espressione:
per calcolare il tempo affinché uno stato stazionario efficace ((t eSS ) possa essere raggiunto (12). Per una sostanza in esame con log Kow = 4 ciò comporta, infatti: teSS = 6,54 · 10 – 3 · 104 + 55,31 = 121 hours 3. PREVISIONE DELLA DURATA DELLA FASE DI DEPURAZIONE Una previsione del tempo necessario per ridurre il carico corporeo ad una certa percentuale della concentrazione iniziale si può ricavare anch'essa dall'equazione generale che descrive l'assorbimento e la depurazione (presupponendo una cinetica di primo ordine (cfr. equazione A5.9 (1) (13). Per la fase di depurazione, si assume che C w (o C food per la prova di esposizione per via alimentare) sia pari a zero. L'equazione si può ridurre a:
oppure
dove C f,0 è la concentrazione all'inizio del periodo di depurazione. 50 per cento di depurazione verrà allora raggiunta al tempo (t 50):
oppure
Analogamente il 95 % di depurazione verrà raggiunto a:
Se si usa un assorbimento dell'80 % per il primo periodo (1.6/k2) e una perdita del 95 % nella fase di depurazione (3.0/k2), la fase di depurazione sarà pari a circa il doppio della durata della fase di assorbimento. Occorre osservare che le stime si basano sull'ipotesi secondo cui il processo di assorbimento e di depurazione segue una cinetica di primo ordine. Se tali procedimenti non rispondono manifestamente a una cinetica di primo ordine, tali stime non sono valide. 4. METODO SEQUENZIALE: DETERMINAZIONE DELLA COSTANTE CINETICA DI DEPURAZIONE (PERDITA) K2 Si è ipotizzato che la maggior parte dei dati riguardanti la bioconcentrazione fossero “ragionevolmente” ben descritti mediante un semplice modello a due compartimenti e due parametri, come indicato dalla curva rettilinea che approssima i punti che rappresentano la concentrazione nel pesce (su un grafico logaritmico), durante la fase di depurazione.
Si noti che le deviazioni dalla linea retta possono indicare uno schema di depurazione più complesso di una cinetica di primo ordine. Il metodo grafico può essere applicato per risolvere i tipi di depurazione che si discostano dalla cinetica di primo ordine. Per calcolare k2 per multipli punti (di campionamento) nel tempo, effettuare una regressione lineare del logaritmo naturale della concentrazione in funzione del tempo. Il coefficiente angolare della linea di regressione è una stima di k2, la costante cinetica di depurazione (89) Dall'ordinata, la concentrazione media nel pesce all'inizio della fase di depurazione (C0, D; che è pari alla media delle concentrazioni nel pesce alla fine della fase di assorbimento), può essere facilmente calcolata (compresi i margini di errore) (89):
Per calcolare k2 quando sono disponibili solo due punti di (campionamento) nel tempo (come nella prova ridotta), sostituire le due concentrazioni medie nell'equazione:
Dove ln (C f1) e ln (C f2) sono i logaritmi naturali delle concentrazioni rispettivamente ai tempi t1 e t2 e t2 e t1 sono i momenti in cui i due campioni sono stati raccolti in relazione all'inizio della depurazione (90). 5. METODO SEQUENZIALE: DETERMINAZIONE DELLA COSTANTE CINETICA DI ASSORBIMENTO K1 (SOLO NEL METODO DI ESPOSIZIONE IN AMBIENTE ACQUATICO) Per trovare un valore di k1 data una serie di valori sequenziali della concentrazione in funzione del tempo per la fase di assorbimento, occorre utilizzare un programma informatico che corrisponda al seguente modello:
Quando k2 è dato dal precedente calcolo, C f(t) e C w(t) sono le concentrazioni nel pesce e nell'acqua, rispettivamente, al tempo t. Per calcolare k1 quando sono disponibili solo due punti di (campionamento) nel tempo (come nella prova ridotta), si utilizza la formula seguente:
Quando k2 è dato dal precedente calcolo, C f è la concentrazione nel pesce all'inizio della fase di depurazione e Cw è la concentrazione media in acqua durante la fase di assorbimento (91). Per valutare la bontà di adattamento, si può procedere ad un'ispezione visiva delle pendenze k1 e k2 rispetto ai valori misurati ai tempi di campionamento riportati nel grafico. Se risulta che il metodo sequenziale ha fornito una scarsa stima di k1, è opportuno applicare il metodo simultaneo per calcolare k1 e k2 (cfr. sezione 6 in appresso). Ancora in questo caso, per valutare la bontà di adattamento, è necessario paragonare visivamente le pendenze ottenute con i valori misurati contenuti nel grafico. Se l'adattamento non è ancora soddisfacente, ciò può significare che la cinetica di primo ordine non si applica e che occorre impiegare modelli più complessi. 6. METODO DI CALCOLO SIMULTANEO DELLE COSTANTI CINETICHE DI ASSORBIMENTO E DEPURAZIONE (PERDITA) (SOLO METODO DI ESPOSIZIONE IN AMBIENTE ACQUATICO) È possibile utilizzare i programmi informatici per calcolare i valori di k1 e k2 data una serie di valori sequenziali della concentrazione in funzione del tempo e il modello:
dove
Tale approccio fornisce direttamente errori standard per le stime di k1 e k2. Se k1/k2 è sostituita dal BCF (cfr. equazione A5.4) nelle equazioni A5.25 e A5.26, è possibile stimare l'errore standard e l'intervallo di confidenza del 95 % del fattore di bioconcentrazione (BCF). Ciò è particolarmente utile per comparare le stime diverse derivanti dalla trasformazione dei dati. La variabile dipendente (concentrazione nel pesce) può essere adattata con o senza trasformazione in logaritmo naturale, e si può valutare l'incertezza circa il BCF ottenuto. A causa della forte correlazione tra i due parametri, k1 e k2, se sono stati stimati simultaneamente, si consiglia di calcolare anzitutto k2 dai risultati della depurazione (cfr. sopra). Nella maggior parte dei casi, k2 può essere stimato a partire dalla curva di depurazione con una precisione relativamente elevata. Successivamente, k1 può essere calcolato a partire dai dati di assorbimento con una regressione non lineare (92). Si raccomanda di trasformare i dati nello stesso modo in caso di adattamento sequenziale. Per valutare la bontà di adattamento, si può procedere ad un'ispezione visiva delle pendenze ottenute riportando su un grafico i dati misurati al tempo di campionamento. Se risulta che tale metodo fornisce una stima scarsa di k1, occorre applicare il metodo alternativo per calcolare k1 e k2. Anche in questo caso, per valutare la bontà di adattamento, si dovrebbe comparare visivamente il modello adeguato ai dati misurati contenuti nel grafico, e le stime dei parametri per k1, k2 e il BCF ottenuto nonché i loro errori standard e/o gli intervalli di fiducia devono essere confrontati tra diversi tipi di adattamento. Se la bontà di adattamento non è ancora soddisfacente, ciò può significare che la cinetica di primo ordine non si applica e che occorre impiegare modelli più complessi. Una delle complicazioni più comuni è la crescita del pesce durante la prova. 7. CORREZIONE DELL'EFFETTO DI DILUIZIONE DOVUTO ALLA CRESCITA PER IL BCF CINETICO E IL BMF La presente sezione descrive un metodo standard per la correzione dovuta alla crescita dei pesci durante la prova (il cosiddetto “effetto di diluizione dovuto alla crescita”) che vale solo quando si applica una cinetica di primo ordine. Se non si applica la cinetica di primo ordine, si consiglia di rivolgersi ad un esperto di biostatistica per correggere i dati dell'effetto di diluizione dovuto alla crescita o di utilizzare l'approccio basato sulla massa descritti di seguito. In alcuni casi, tale metodo di correzione dell'effetto di diluizione della crescita manca di precisione o non funziona (ad esempio, per sostanze testate che si eliminano molto lentamente nei pesci in rapida crescita, la costante cinetica di depurazione corretta per la crescita, k2g, può essere molto modesta, e quindi l'errore nelle due costanti cinetiche usate per calcolarlo è cruciale, e in alcuni casi il valore kg stimato può essere superiore a k2). È possibile utilizzare un altro metodo (approccio di massa), che evita di apportare eventuali correzioni e opera anche in assenza di una cinetica di primo ordine. Questo metodo è presentato alla fine della presente sezione. Metodo di correzione della crescita mediante sottrazione della costante cinetica di crescita In base alla metodologia standard, i pesi e le lunghezze individuali sono convertiti in logaritmi naturali e ln (peso) o ln (1/ peso) sono riportati graficamente in funzione del tempo (giorno) in due grafici distinti per i gruppi di controlli e il gruppo di prova. Si procede analogamente con i dati ottenuti separatamente per le fasi di assorbimento e di depurazione. In generale, per correggere gli effetti di diluizione dovuto alla crescita, è più opportuno fare riferimento al peso dell'insieme dello studio per ottenere la costante cinetica di crescita (kg), ma differenze statisticamente significative tra le costanti cinetiche di crescita sia per la fase di assorbimento sia per la fase di depurazione possono far ritenere opportuno usare la costante cinetica della fase di depurazione. I tassi di crescita globali osservati in studi con esposizione in ambiente acquatico per i gruppi di controllo e di prova possono essere utilizzati per accertare eventuali effetti correlati al trattamento. Una correlazione dei minimi quadrati lineari è calcolata per ln (peso del pesce) in funzione del tempo (giorno) (ln (1/ peso) in funzione del tempo per ciascun gruppo (gruppi di prova e di controllo, dati individuali, medie non giornaliere) per l'intero studio, le fasi di assorbimento e di depurazione applicando i metodi statistici standard. Le variazioni delle pendenze delle linee sono calcolate e utilizzate per valutare la significatività statistica (p = 0,05) della differenza tra le pendenze (costanti cinetiche di crescita) utilizzando il test t (o di ANOVA se sono testate più di una concentrazione). Si preferisce generalmente utilizzare i dati sul peso per le correzioni per la crescita. Le lunghezze, trattate allo stesso modo, possono essere usate per valutare gli effetti del trattamento su gruppi di controllo e di prova. Se non si rileva alcuna differenza statisticamente significativa nell'analisi dei dati sul peso, si possono raggruppare i dati dei gruppi di controllo e di prova e calcolare la costante cinetica di crescita del pesce globale per lo studio (kg) vale a dire la pendenza globale della correlazione lineare. Se si osservano differenze statisticamente significative, si registrano separatamente le costanti cinetiche di crescita per ciascun gruppo di pesci e/o ciascuna fase della prova. La costante cinetica per ciascun gruppo trattato viene utilizzata per le correzioni dell'effetto di diluizione dovuto alla crescita in questo gruppo. Se vi sono differenze statistiche tra le costanti cinetiche di assorbimento e di depurazione, si usano le costanti cinetiche ricavate dalla fase di depurazione. La costante cinetica di crescita calcolata (kg espresso in giorni-1) può essere dedotta dalla costante cinetica di depurazione complessiva (k2) per garantire la costante cinetica di depurazione, k2g..
Suddividendo la costante cinetica di assorbimento per la costante cinetica di depurazione corretta per la crescita per ottenere il fattore di bioconcentrazione cinetico corretto per l'effetto di diluizione dovuto alla crescita, denominato BCFK g (o BMFKg).
La costante cinetica di crescita ottenuta per una prova con esposizione per via alimentare è utilizzata nell'equazione A7.5 per calcolare il BMFKg corretto per la crescita (cfr. appendice 7). Metodo di correzione della crescita basata sulla massa Un'alternativa al suddetto “Metodo di correzione della crescita mediante sottrazione della costante cinetica di crescita”, che consente di evitare la necessità di correggere per la crescita, può essere utilizzata nel modo seguente. Si tratta fondamentalmente di utilizzare dati sulla depurazione in funzione della massa per pesce intero piuttosto che in funzione della concentrazione. Convertire le concentrazioni riscontrate nei tessuti in fase di depurazione (massa della sostanza in esame/unità di massa del pesce) in massa della sostanza in esame/pesce: mettere in corrispondenza, sotto forma di tabella, le concentrazioni e il peso di ciascun pesce (ad esempio utilizzando un foglio di calcolo elettronico) e moltiplicando ciascuna concentrazione per il totale del peso del pesce in modo che la misura fornisca una serie di massa della sostanza in esame/pesce in tutte le sessioni di campionamento della fase di depurazione. Tracciare il logaritmo naturale ottenuto con i dati della massa della sostanza chimica in funzione del tempo (fase di depurazione) come si farebbe normalmente. Per il metodo di esposizione in ambiente acquatico, calcolare la costante cinetica di assorbimento come di consueto (cfr. sezioni 4 e 6; il valore k2“normale” è utilizzato nelle equazioni di interpolazione della curva di k1) e detrarre la costante cinetica di depurazione da tali dati. Poiché il valore ottenuto per la costante cinetica di depurazione è indipendente dalla crescita, in quanto è stato ottenuto in base alla massa per pesce intero, va indicato come k2g e non k2. 8. NORMALIZZAZIONE DEI LIPIDI AL 5 % DEL TENORE DI GRASSI (SOLO METODO DI ESPOSIZIONE IN AMBIENTE ACQUATICO) I risultati del BCF (cinetico e allo stato stazionario) delle prove con esposizione in ambiente acquatico sono registrati anche in funzione del contenuto lipidico standard del 5 % del peso umido dei pesci, tranne quando si può provare che la sostanza in esame non si accumula nei grassi (ad esempio alcune sostanze perfluorurate possono legarsi alle proteine). Occorre convertire le concentrazioni nei pesci, o il BCF, in un tenore di lipidi del 5 % rispetto al peso umido. Se i pesci sono stati utilizzati per misurare le concentrazioni della sostanza e il contenuto lipidico in tutti i tempi di campionamento, è opportuno correggere le concentrazioni misurate individualmente in funzione del contenuto lipidico dei pesci.
Dove
Se non è stata eseguita un'analisi dei lipidi in tutti i pesci campionati, si utilizza un valore medio dei lipidi per normalizzare il BCF. Per quanto riguarda il fattore di bioconcentrazione (BCF) allo stato stazionario, occorre utilizzare il valore medio registrato alla fine della fase di assorbimento nel gruppo trattato. Per quanto concerne la normalizzazione del BCF cinetico vi possono essere casi in cui è giustificato un approccio diverso, ad esempio se il tenore di lipidi cambia notevolmente durante la fase di assorbimento e di depurazione. Tuttavia, si consiglia di prevedere un regime alimentare che consenta di ridurre al minimo qualunque cambiamento significativo del tenore di grassi.
Dove
BIBLIOGRAFIA
Appendice 6 EQUAZIONI PER LA PROVA DI ESPOSIZIONE IN AMBIENTE ACQUATICO: DISEGNO SPERIMENTALE DELLE PROVE RIDOTTE I motivi che giustificano siffatta impostazione consistono nel fatto che il fattore di bioconcentrazione in una prova completa può essere determinato come fattore di bioconcentrazione allo stato stazionario (BCFSS) calcolando il rapporto tra la concentrazione della sostanza in esame nei tessuti del pesce e la concentrazione della sostanza in esame nell'acqua, o calcolando il fattore di bioconcentrazione cinetico (BCFk ), come il rapporto tra la costante cinetica di assorbimento k1 alla costante cinetica di depurazione k2. Il BCFk è valido anche se una concentrazione allo stato stazionario di una sostanza chimica non è raggiunta durante l'assorbimento, a condizione che l'assorbimento e la depurazione agiscano approssimativamente secondo processi cinetici di primo ordine. Se si misura la concentrazione della sostanza nei tessuti (C f1) alla fine dell'esposizione (t1) e si misura la concentrazione nei tessuti (C f2) nuovamente dopo un certo tempo (t2), si può stimare la costante cinetica di depurazione (k2) con l'equazione A5.22 dell'appendice 5. La costante cinetica di assorbimento, k1, può essere determinata in modo algebrico con l'equazione A5.23 dell'appendice 5 (dove C f è pari a C f1 e t è pari a t1 ) (1). Il fattore di bioconcentrazione cinetico per il disegno sperimentale della prova ridotta (designato come BCFK m per distinguerlo dai fattori di bioconcentrazione cinetica determinati con altri metodi) è:
È opportuno correggere le concentrazioni o i risultati dell'effetto di diluizione dovuto alla crescita e normalizzare rispetto a un contenuto di grassi nel pesce del 5 % (cfr. appendice 5). Il BCFSS minimizzato corrisponde al BCF calcolato alla fine della fase di assorbimento, supponendo che sia raggiunto lo stato stazionario. Ciò può essere solo presunto, in quanto il numero dei punti di campionamento non è sufficiente per dimostrarlo.
Dove C f-minSS = concentrazione nel pesce allo stato stazionario assunto alla fine dell'assorbimento (mg kg– 1 di peso umido). C w-minSS = concentrazione in acqua allo stato stazionario presunto alla fine dell'assorbimento (mg kg– 1).BIBLIOGRAFIA
Appendice 7 EQUAZIONE PER LA PROVA CON ESPOSIZIONE PER VIA ALIMENTARE
1. ESEMPIO DI QUANTITÀ DI COMPONENTI DI UN ADEGUATO MANGIME COMMERCIALE PER PESCI
2. ESEMPI DI TECNICHE DI ADDIZIONAMENTO DELL'ALIMENTO Aspetti generali L'alimentazione per il gruppo di controllo è preparata esattamente allo stesso modo dei mangimi addizionati, ma senza l'aggiunta della sostanza in esame. Per conoscere le concentrazioni nei mangimi trattati, occorre estrarre tre campioni di alimenti trattati con un idoneo un metodo di estrazione e misurare la radioattività e la concentrazione della sostanza in esame negli estratti. La possibilità di isolare la sostanza in esame (> 85 %) e la modesta variazione tra i campioni (tre concentrazioni della sostanza misurata su campioni prelevati all'inizio della prova non devono variare di oltre ± 15 % rispetto al valore medio) devono essere dimostrate. Durante la prova con esposizione per via alimentare, è opportuno raccogliere tre campioni di alimenti al giorno 0 e alla fine della fase di assorbimento per determinare la concentrazione della sostanza in esame negli alimenti. Preparazione di mangimi per pesci con un materiale sperimentale liquido (puro) Stabilire una concentrazione nominale di prova nel regime alimentare dei pesci, ad esempio 500 μg di sostanza in esame/g di mangimi. La quantità appropriata (in funzione della massa molare o radioattività specifica) della sostanza in esame pura è addizionata ad una massa nota di un mangime per pesci in un barattolo di vetro o un evaporatore rotante. La massa di cibo deve essere sufficiente per tutta la durata della fase di assorbimento (tenere conto della necessità di aumentare le razioni a motivo della crescita dei pesci). I mangimi per pesci e la sostanza in esame vanno mescolati lentamente nel corso della notte (ad esempio mediante un miscelatore “roto-rack” o, in caso di utilizzo di un evaporatore rotante, a rotazione). La dieta addizionata è conservata in condizioni che mantengano la stabilità della sostanza in esame all'interno della miscela (mediante refrigerazione) fino all'utilizzo. Preparazione di mangimi per pesci con un disperdente (olio di pesce/mais) Le sostanze solide devono essere triturate in polvere fine in un mortaio. Le sostanze liquide possono essere aggiunte direttamente all'olio di mais o di pesce. La sostanza sotto esame viene disciolta in una quantità nota di olio di mais o di pesce (per esempio da 5-15 ml). L'olio è trasferito quantitativamente trasferito mediante un evaporatore di tipo rotativo di dimensioni adatte. Il recipiente utilizzato per preparare i dosaggi di olio dovrebbe essere sciacquato con due piccole aliquote di olio e queste aggiunte all'evaporatore per assicurare che sia trasferita tutta la sostanza in esame disciolta. Per garantire la completa dissoluzione o dispersione nell'olio (o se più di una sostanza in esame viene utilizzata nello studio), è aggiunto un micro-miscelatore, il recipiente tappato e la miscela agitata rapidamente durante la notte. Una quantità adeguata di mangimi per pesci (solitamente sotto forma di pellet) per la prova è aggiunta all'evaporatore e i contenuti dello stesso vengono mescolati in modo omogeneo facendo ruotare continuamente l'evaporatore di vetro per almeno 30 minuti, ma preferibilmente durante la notte. In seguito, il mangime addizionato è conservato in modo appropriato (ad.es. refrigerato) per garantire la stabilità della sostanza in esame nel mangime fino all'utilizzo. Preparazione di mangimi per pesci con un solvente organico Una quantità adeguata della sostanza in esame (per massa molare o radioattività specifica) sufficiente per raggiungere l'obiettivo di dose è disciolta in un solvente organico idoneo (ad esempio, cicloesano o acetone; 10-40 ml, ma un maggiore volume se necessario in funzione della quantità di mangime da addizionare). Miscelare un'aliquota di questa soluzione o la sua totalità (riferita alla porzione addizionata) con un'adeguata massa di mangime per pesci, sufficiente perché la prova ottenga il necessario livello di dose nominale. Il mangime/sostanza in esame può essere miscelato in vasca in acciaio inossidabile e il mangime addizionato recentemente deve rimanere nella vasca in una cappa di laboratorio agitata per due giorni (occasionalmente) per consentire l'eccesso di solvente supplementare, oppure essere mescolato in un evaporatore rotante a bulbo con rotazione continua. L'eccesso di solvente può essere rimosso mediante un getto di aria o azoto, se necessario. Fare attenzione a che la sostanza in esame non cristallizzi man mano che il solvente viene rimosso. La dieta addizionata deve essere conservata in condizioni di refrigerazione (ad esempio) che mantengono la stabilità della sostanza in esame nel mix di mangime fino all'utilizzo. 3. CALCOLO DELL'EFFICIENZA DI ASSIMILAZIONE E DEL FATTORE DI BIOMAGNIFICAZIONE Per calcolare l'efficienza di assimilazione occorre innanzitutto stimare la costante cinetica di depurazione globale come indicato nella sezione 4 dell'appendice 5 (con il metodo sequenziale, cioè con una regressione lineare standard) con concentrazioni medie misurate nei campioni della fase di depurazione. La costante della razione alimentare, I, e la durata di assorbimento, t, sono parametri noti dello studio. Cfood, la concentrazione media misurata della sostanza in esame negli alimenti, è una variabile misurata nel corso dello studio. C 0,d la concentrazione della sostanza in esame nel pesce alla fine della fase di assorbimento, è comunemente calcolata dall'intercetta della rappresentazione grafica del logaritmo naturale della concentrazione in funzione del giorno di depurazione. L'efficienza di assimilazione della sostanza in esame (a, assorbimento della sostanza in esame nell'intestino) è calcolata come segue:
Dove C0, D = concentrazione nel pesce ottenuta al giorno 0 della fase di depurazione (mg kg-1);k2 = costante cinetica di depurazione (non corretta per la crescita) globale (giorno-1), calcolato in base alle equazioni di cui all'appendice 5, sezione 3; I = costante cinetica di ingestione (g di cibo g– 1 di pesce giorno– 1); Cfood= concentrazione nel cibo (mg kg-1 di cibo); t= durata del periodo di somministrazione (giorno) Tuttavia può rendersi necessario adeguare alla crescita dei pesci la razione alimentare, I, utilizzato nei calcoli per ottenere un'efficienza di assimilazione (a) più precisa. In una prova in cui i pesci crescono in misura significativa durante la fase di assorbimento (durante la quale non si corregge la quantità di cibo per mantenere il tasso di somministrazione stabilito), il tasso effettivo di somministrazione alimentare man mano che la fase di assorbimento progredisce diventerà inferiore a quello stabilito, con un conseguente valore della “effettiva” efficienza di assimilazione superiore. (Questo aspetto non è rilevante per i calcoli complessivi del BMF, poiché i termini I si annullano tra le equazioni A7.1 e A7.4). Il tasso medio di somministrazione corretto dell'effetto di diluizione dovuto alla crescita, Ig, può essere ottenuto in vari modi, ma un metodo semplice e rigoroso consiste nell'utilizzare la costante cinetica di crescita (kg) nota per stimare il peso dei pesci testati in determinati periodi della fase di assorbimento:
Dove W f(t)= peso medio dei pesci al giorno t della fase di assorbimento Wf,0= media del peso dei pesci all'inizio dell'esperimento In questo modo (almeno), si può stimare il peso medio dei pesci nell'ultimo giorno di esposizione (W f,end-of-uptake). Poiché il tasso di somministrazione alimentare è stato stabilito in base a W f,0, l'effettivo tasso per ogni giorno di assorbimento può essere calcolato utilizzando i valori relativi al peso. Il tasso di somministrazione alimentare corretto per la crescita, Ig (g di cibo g-1 di pesce giorno-1), da utilizzare in sostituzione di I in caso di rapida crescita durante la fase di assorbimento, può essere calcolata come
Una volta ottenuta l'efficienza di assimilazione, si può calcolare il BMF moltiplicandolo per la costante del tasso di alimentazione I (o Ig, se quest'ultimo è stato utilizzato per calcolare α) e dividendo il prodotto per la costante cinetica di depurazione globale k2:
Il fattore di biomagnificazione corretto per la crescita è calcolato secondo le stesse modalità, impiegando la costante cinetica di depurazione corretta per la crescita (ottenuta come indicato nella sezione 7 dell'appendice 5). Ancora una volta, se Ig è servito a calcolare α, è opportuno utilizzarlo anche qui invece di I:
dove: α = efficienza di assimilazione (assorbimento della sostanza in esame per via intestinale).k2 = costante cinetica di depurazione (non corretta per la crescita) globale (giorno-1), calcolata con le equazioni di cui all'appendice 5, sezione 3; k2g= costante cinetica di depurazione corretta per la crescita (giorno-1); I = costante cinetica di ingestione (g di cibo g-1 di pesce giorno-1); Il tempo di dimezzamento corretto per la crescita (t1/2) è calcolato come segue:
È possibile stimare anche l'efficienza di assimilazione della sostanza chimica con il cibo se sono determinati residui nei tessuti durante la fase lineare della fase di assorbimento (tra i giorni 1 e 3). In questo caso occorre analizzare l'efficienza di assimilazione della sostanza (α) come segue:
Dove C fish (t) = concentrazione della sostanza in esame nel pesce di prova al tempo t (mg kg-1 di peso umido). 4. CORREZIONE DEL TENORE DI GRASSI Se il tenore di lipidi è stato misurato negli stessi pesci sottoposti all'analisi chimica in tutti i tempi di campionamento, si consiglia di correggere le concentrazioni individuali in funzione dei lipidi e di tracciare il logaritmo naturale della concentrazione, corretto per il tenore di grassi in funzione della depurazione (giorno) per ottenere C 0,d e k 2. L'efficienza di assimilazione (equazione A7.1) può esser calcolata sulla base dei lipidi utilizzando C food sulla base dei lipidi (cioè C food è moltiplicato per la frazione media di grassi negli alimenti). Calcoli successivi con le equazioni A7.4 e A7.5 daranno il BMF corretto per il tenore lipidico (e dell'effetto di diluizione dovuto alla crescita) direttamente. In caso contrario, le frazioni medie di grassi (peso umido) nel pesce e nei mangimi sono ottenute per entrambi i gruppi, di controllo e di prova (per quanto riguarda il cibo e i pesci del gruppo di controllo, ciò si ottiene generalmente dai dati misurati all'inizio e alla fine dell'esposizione; per il gruppo di trattamento, si ottiene generalmente dai dati misurati solo alla fine dell'esposizione). In alcuni studi, il contenuto lipidico dei pesci può aumentare sensibilmente; in tal caso è preferibile utilizzare una concentrazione media dei lipidi nel pesce testato calcolata sulla base dei valori misurati alla fine dell'esposizione e alla fine della depurazione. In generale, i dati dal solo gruppo di prova dovrebbero essere utilizzati per ottenere entrambe le frazioni di grassi. Il fattore di correzione del tenore di grassi (Lc) è calcolato come segue:
In cui L fish and L food sono le frazioni medie di grassi rispettivamente nel pesce e nei mangimi. I Il fattore di correzione del tenore di grassi è usato per il calcolo del fattore di biomagnificazione corretto per il tenore lipidico (BMFL):
5. VALUTAZIONE DELLE DIFFERENZE TRA LA CONCENTRAZIONE MISURATA AL TEMPO 0 (C 0,M) E LA CONCENTRAZIONE CALCOLATA PER IL TEMPO 0 (C0,D) Occorre confrontare la concentrazione misurata al tempo 0 (C 0,m) e la concentrazione calcolata per il tempo 0 (C 0,d). Se sono molto simili, si può ritenere adeguato il modello di primo ordine utilizzato per ottenere i parametri di depurazione. In alcuni studi, si osserverà una differenza sostanziale tra il valore ottenuto per il tempo 0, C 0,d, e la concentrazione media misurata al tempo 0, C 0,m (cfr. ultimo trattino del paragrafo 159). Se C0, D è nettamente inferiore a C 0, C 0,m (C 0,d << C 0,m), tale differenza può indicare la presenza, nell'intestino dei pesci, di mangimi addizionati non digeriti. Per averne la prova, si possono analizzare separatamente gli intestini escissi se, alla fine della fase di assorbimento, sono stati prelevati e conservati esemplari supplementari di pesce. In caso contrario, qualora risulti da un test di outlier statisticamente valido applicato alla regressione lineare della fase di depurazione che il primo punto di campionamento della fase di depurazione è erroneamente elevato, può essere utile proseguire la regressione lineare per ottenere k2, ma omettendo la concentrazione al primo tempo della depurazione. In tal caso, se l'incertezza nella regressione lineare appare fortemente diminuita e appare evidente che il processo di depurazione abbia seguito approssimativamente una cinetica di primo ordine, si possono utilizzare i valori C 0,d e k 2 ottenuti con il calcolo dell'efficienza di assimilazione. Ciò deve essere debitamente giustificato nella relazione di prova. È inoltre possibile che la fase di depurazione non abbia seguito una cinetica di primo ordine. Se tale ipotesi è probabile (cioè i dati trasformati con logaritmo naturale sembrano seguire una curva rispetto alla retta di regressione lineare), è poco probabile che i calcoli di k2 e C 0,d siano validi, e si raccomanda di ricorrere al consiglio di un esperto in biostatistica. Se C 0,d è nettamente superiore al valore misurato (C 0,d > > C 0,dm) ciò potrebbe significare: che la sostanza è stata eliminata in tempi rapidi (il tempo di campionamento si avvicina al limite di quantificazione del metodo analitico molto all'inizio della fase di depurazione, cfr. la sezione 6); che il processo di depurazione non segue una cinetica di primo ordine; che la regressione lineare per ottenere k2 e C 0,d è errata; o che si è verificato un problema con le concentrazioni misurate nel corso dello studio in alcuni punti di campionamento. È quindi necessario esaminare il tracciato della regressione lineare per identificare i campioni al limite o in prossimità del limite di quantificazione, i valori anomali e una curva manifesta (che suggerirebbe che la depurazione non ha seguito una cinetica di primo ordine) e evidenziarne chiaramente i risultati nella relazione di prova. Ogni successiva rivalutazione della regressione lineare per migliorare le stime dovrà essere descritta e giustificata. Se si registra una deviazione significativa della cinetica di primo ordine, è poco probabile che i calcoli di k2 e C 0,d siano validi, e si raccomanda di ricorrere al consiglio di un esperto in biostatistica. 6. ORIENTAMENTI PER TESTARE SOSTANZE CHE SONO ELIMINATE MOLTO RAPIDAMENTE Come indicato al paragrafo 129, alcune sostanze possono avere un'eliminazione talmente rapida da non poter ricavare una concentrazione affidabile al tempo 0, C 0,d né k2 perché la sostanza non è più effettivamente misurata (concentrazioni sono al limite della quantificazione) già molto presto nel corso della fase di depurazione (a partire dal secondo campionamento). Tale situazione, rilevata in occasione della prova inter-laboratori eseguita con il benzo [a] pirene, è stata riportata nella relazione di validazione del presente metodo di prova. In questo caso non è possibile proseguire la regressione lineare in modo attendibile, in quanto risulterebbe probabilmente una stima eccessivamente elevata di C 0,d, con un'efficienza di assimilazione che apparirebbe nettamente superiore a 1. In tal caso, si può procedere a una stima prudente di k2 e si può calcolare un “limite superiore” di BMF. Utilizzando i dati dei tempi della fase di depurazione in cui è stata misurata la concentrazione, fino alla prima concentrazione non rilevata (ivi compresa) (concentrazione al limite della quantificazione) una regressione lineare (basata sulle concentrazioni trasformate in logaritmi naturali in funzione del tempo) fornirà una stima di k2. Ciò richiederà spesso solo due tempi di misurazione (ad esempio i giorni di campionamento 1 e 2 della fase di depurazione) e k2 potrà poi essere stimato con l'equazione A5.22 presentata nell'appendice 5. Tale stima di k2 può servire a calcolare l'efficienza di assimilazione con l'equazione A7.1, sostituendo al valore C 0,d nella formula la concentrazione misurata al tempo 0 (C 0,m) qualora la stima di C 0,d sia nettamente superiore a quanto il test avrebbe consentito di ottenere. Se C 0,m non era misurabile, utilizzare il limite di rilevamento analitico nei tessuti dei pesci. Se, in alcuni casi, ciò dà un valore α > 1 allora si presume che l'efficienza di assimilazione pari a 1 rappresenti il “caso peggiore”. Il valore massimo di BMFK può essere stimato con l'equazione A7.4 e dovrà essere indicato come un valore “molto inferiore a” (<<). Ad esempio, uno studio condotto con un tasso di alimentazione del 3 % e un tempo di dimezzamento di depurazione inferiore a 3 giorni, e il “caso peggiore” di α = 1, il BMFK rischia di essere inferiore a circa 0,13. Dato lo scopo di questa stima e il fatto che i valori avranno carattere prudenziale, non è necessario correggerli degli effetti di diluizione dovuto alla crescita e per il contenuto di lipidi nel pesce o nel cibo. Appendice 8 METODI PER STIMARE I BCF PROVVISORI SULLA BASE DEI DATI RACCOLTI NELLO STUDIO CON ESPOSIZIONE PER VIA ALIMENTARE Il metodo di esposizione per via alimentare è presentato nel presente metodo di prova per testare il bioaccumulo delle sostanze che non possono essere testate con il metodo di esposizione attraverso l'ambiente acquatico. Il metodo di esposizione attraverso l'ambiente acquatico dà un fattore di bioconcentrazione, mentre quello per via alimentare fornisce direttamente informazioni sul potenziale di biomagnificazione degli alimenti. In diversi regimi di sicurezza dei prodotti chimici sono necessarie informazioni sulla bioconcentrazione acquatica (ad esempio per la valutazione dei rischi o il sistema mondiale armonizzato di classificazione). È pertanto necessario utilizzare i dati ottenuti da uno studio di esposizione per via alimentare per calcolare un fattore di bioconcentrazione comparabile alle prove effettuate secondo il metodo di esposizione attraverso l'ambiente acquatico (94). Questa sezione esamina diversi approcci in tal senso, pur riconoscendo i limiti intrinseci a tali stime. Lo studio per via alimentare misura la depurazione per ottenere la costante cinetica di depurazione, k2. Se si può stimare la costante cinetica di assorbimento con i dati disponibili per la situazione in cui il pesce è stato esposto alla sostanza in esame attraverso l'ambiente acquatico, si potrà calcolare un BCF cinetico. La stima di una costante cinetica di assorbimento per l'esposizione attraverso l'ambiente acquatico alla sostanza in esame si basa su numerose ipotesi, che contribuiscono tutte all'incertezza dei risultati. Inoltre, tale questo approccio per stimare un BCF presuppone che la velocità complessiva di depurazione (compresi i fattori rilevanti, quali la ripartizione nel corpo e i processi di depurazione individuali) sia indipendente dalla tecnica di esposizione utilizzata per produrre un carico corporeo della sostanza in esame. Le principali ipotesi inerenti a tale approccio di stima possono essere riassunte come segue: La depurazione a seguito di assunzione alimentare è la stessa della depurazione a seguito di esposizione attraverso l'ambiente acquatico a una determinata sostanza; L'assorbimento attraverso l'ambiente acquatico segue una cinetica di primo ordine; A seconda del metodo utilizzato per stimare l'assorbimento:
La banca dati utilizzata per sviluppare il metodo di stima dell'assorbimento è rappresentativa della sostanza in esame. Numerose pubblicazioni in letteratura riportano le equazioni che mettono in correlazione l'assorbimento di acqua nei pesci attraverso le branchie e il coefficiente di ripartizione ottanolo/acqua di una sostanza, il peso del pesce (1) (2) (3) (4), il volume e/o il contenuto lipidico, la permeabilità/diffusione delle membrane (5) (6), il volume di ventilazione dei pesci (7) e mediante un approccio fugacità/ bilancio di massa (8) (9) (10). Una valutazione dettagliata di tali approcci in questo contesto figura in Crookes & Brooke (11). Una pubblicazione di Barber (12), che si è adoperato per modellizzare il bioaccumulo associato all'assunzione alimentare, si rivela utile in questo contesto, poiché comprende i contributi di modelli di cinetica di assorbimento attraverso le branchie. Anche una sezione del documento di riferimento sul protocollo alimentare del 2004 (13) è dedicata a questo aspetto. Per la maggior parte, tali modelli sembrano elaborati a partire da banche dati limitate. Per quanto riguarda i modelli per i quali sono disponibili informazioni dettagliate sulle banche dati utilizzate per la loro elaborazione, sembra che i tipi di sostanze utilizzate presentino spesso una struttura simile o rientrino nella stessa classe (in termini di funzionalità, ad esempio i composti organoclorurati). Ciò aumenta l'incertezza di utilizzare un modello al fine di prevedere la costante cinetica di assorbimento per un tipo di sostanza diverso, in aggiunta a considerazioni specifiche alla prova quali le specie sperimentali, la temperature, ecc. Una panoramica delle tecniche disponibili (11) mostra che nessuna metodologia è “più corretta” delle altre. Occorre pertanto giustificare chiaramente la scelta del modello utilizzato. Quando sono disponibili diversi metodi la cui applicazione può essere giustificata, è prudente presentare più di una stima di k1 (e quindi del BCF) o un intervallo di valori di k1 (e del BCF) secondo i diversi possibili metodi di stima dell'assorbimento. Tuttavia, date le differenze tra i tipi di modello e le banche dati utilizzate per svilupparli, non sarebbe opportuno adottare una media delle stime ottenute con questi diversi metodi. Alcuni ricercatori ipotizzano che tali stime del fattore di bioconcentrazione (BCF) richiedano una correzione della biodisponibilità per tener conto dell'adsorbimento della sostanza rispetto al carbonio organico disciolto (DOC) in un ambiente acquatico, affinché la stima corrisponda ai risultati degli studi di esposizione in ambiente acquatico [ad esempio (13) (14)]. Tale correzione non è però necessariamente adeguata a causa dei bassi livelli di COD richiesti in uno studio con esposizione in ambiente acquatico per una stima nel “caso peggiore” (ossia il rapporto tra la sostanza biodisponibile e la sostanza misurata in soluzione). Con le sostanze estremamente idrofobe, l'assorbimento attraverso le branchie può essere limitato dal tasso di diffusione passivo in prossimità della superficie delle branchie; in tal caso, è possibile che la correzione tenga conto di questo effetto e non di quello che si voleva correggere. È consigliabile concentrarsi sui metodi che richiedono dati di base facilmente disponibili sulle sostanze esaminate con il metodo di esposizione per via alimentare qui descritto (vale a dire il log Ko/w, e il peso dei pesci). Altri metodi che richiedono dati di base più complessi sono applicabili, ma potrebbero richiedere ulteriori misurazioni nel corso della prova o una conoscenza approfondita della sostanza in esame o sulle specie sperimentali che potrebbero essere difficili da ottenere. Inoltre, la scelta del modello può essere influenzato dal livello di validazione e dal campo di applicabilità (cfr. (11) per una panoramica e un raffronto dei diversi metodi). Occorre tenere presente che la risultante stima di k1 o il BCF stimato, sono valori incerti e potrebbero richiedere l'applicazione del “peso dell'evidenza” assieme al BMF ottenuto e ai parametri relativi alla sostanza (ad es. dimensione molecolare) per avere una visione d'insieme del potenziale di bioaccumulo di una sostanza. L'interpretazione e l'uso di tali parametri possono variare in funzione del quadro normativo. BIBLIOGRAFIA
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(17) |
Nella parte C, il capitolo C.20 è sostituito dal seguente: «C.20 Prova di riproduzione con Daphnia magna INTRODUZIONE Il presente metodo di prova è equivalente alla linea guida dell'OCSE per le prove sulle sostanze chimiche n. 211 (2012). Le linee guida dell'OCSE per le prove dei prodotti chimici sono periodicamente rivedute e aggiornate alla luce del progresso scientifico. La linea guida n. 211 sulla prova di riproduzione deriva dalla linea guida n. 202, parte II, Prova di riproduzione con Daphnia sp. (1984). È generalmente riconosciuto che i dati derivanti da prove effettuate secondo la linea guida n. 202 possono essere variabili. Per questa ragione sono stati intrapresi notevoli sforzi per identificare i motivi alla base di questa variabilità, con l'obiettivo di produrre un metodo di prova migliore. La linea guida n. 211 si basa sui risultati di tali attività di ricerca, su prove interlaboratorio e su studi di validazione effettuati nel 1992 (1), 1994 (2) e 2008 (3). Le principali differenze tra la versione iniziale (linea guida n. 202, 1984) e la seconda versione (linea guida n. 211, 1998) della linea guida sulla prova di riproduzione sono le seguenti:
L'appendice 1 contiene le definizioni dei termini utilizzati. PRINCIPIO DELLA PROVA Il principale obiettivo della prova è valutare l'effetto delle sostanze chimiche sulla capacità riproduttiva di Daphnia magna. A tal fine giovani femmine di Daphnia (animali riproduttori), di età inferiore alle 24 ore al momento dell'inizio della prova, vengono esposte alla sostanza chimica in esame aggiunta all'acqua a un intervallo di concentrazioni diverse. La durata della prova è di 21 giorni. Alla fine della prova, viene verificato il numero totale di piccoli vivi prodotti. La capacità riproduttiva degli animali parentali può essere espressa in altri modi (ad esempio, con il numero dei piccoli vivi prodotti giornalmente da ciascun animale a partire dal primo giorno di comparsa della prole) ma questi dati dovrebbero essere riportati solo ad integrazione del numero totale di piccoli vivi prodotti alla fine del test. A causa della particolare concezione della prova semistatica rispetto ad altri metodi di prova sulla riproduzione di invertebrati, è altresì possibile contare il numero di piccoli vivi prodotti da ciascun animale riproduttore. In questo modo, contrariamente ad altri metodi di prova sulla riproduzione di invertebrati, è possibile escludere dalla valutazione i dati corrispondenti alla prole di un animale riproduttore che muoia accidentalmente e/o casualmente durante il periodo di prova. Di conseguenza, in presenza di mortalità parentale nelle repliche esposte si deve stabilire se essa segue un modello concentrazione-risposta, ad esempio se vi è una significativa regressione della risposta rispetto alla concentrazione della sostanza chimica in esame con una pendenza positiva (è possibile utilizzare una prova statistica quale il test di Cochran-Armitage per le tendenze). Se la mortalità non segue un modello concentrazione-risposta, le repliche che presentano mortalità parentale vanno escluse dall'analisi del risultato della prova. Se la mortalità segue un modello concentrazione-risposta, la mortalità parentale è assimilata a un effetto della sostanza chimica in esame e le repliche non vanno escluse dall'analisi. Se l'animale riproduttore si rivela essere un maschio, o se muore durante la prova sia accidentalmente, per incuria o incidente, sia casualmente a causa di un incidente che non trova spiegazione e che non è collegato all'effetto della sostanza chimica in esame, questa replica viene esclusa dall'analisi (per saperne di più, cfr. paragrafo 51). L'eventuale effetto tossico della sostanza chimica in esame sul tasso di riproduzione è misurato da valori espressi come ECx, eseguendo, per regressione non lineare, un aggiustamento dei dati a un modello adeguato allo scopo di calcolare la concentrazione che causerebbe x % di riduzione del tasso riproduttivo, o in alternativa dal valore NOEC/LOEC (4). Le concentrazioni di prova devono comprendere le più basse concentrazioni con effetto utilizzate (ad esempio EC10), il che significa che questo valore è calcolato per interpolazione e non per estrapolazione. Vanno riportati anche la sopravvivenza degli animali riproduttori e il tempo intercorso fino alla produzione della prima schiusa. È possibile esaminare altri effetti della sostanza chimica su parametri quali la crescita (per es. la lunghezza) e, possibilmente, il tasso intrinseco di aumento della popolazione (cfr. paragrafo 44). INFORMAZIONI SULLA SOSTANZA CHIMICA IN ESAME I risultati di un test di tossicità acuta (cfr. capitolo C.2 del presente allegato: Saggio di immobilizzazione acuta in Daphnia sp.) effettuato su Daphnia magna possono essere utili per selezionare l'adeguato intervallo di concentrazioni di prova da utilizzare nei test sulla riproduzione. È necessario conoscere la solubilità in acqua e la pressione di vapore della sostanza chimica in esame e deve essere disponibile un metodo analitico affidabile per quantificare la sostanza chimica nelle soluzioni di prova con un'efficienza di recupero e un limite di rilevamento noti. Le informazioni sulla sostanza chimica in esame che possono essere utili per stabilire le condizioni di prova comprendono: formula strutturale, purezza della sostanza, fotostabilità, e stabilità nelle condizioni di esecuzione della prova, pKa, Pow e risultati di una prova di pronta biodegradabilità [cfr. capitoli C.4 (Determinazione della pronta biodegradabilità), C.29 (pronta biodegradabilità — CO 2 in recipienti ermetici) del presente allegato]. VALIDITÀ DELLA PROVA Perché la prova sia valida, nei gruppi di controllo devono essere soddisfatti i seguenti criteri di prestazione:
Nota: Lo stesso criterio di validità (20 %) può essere utilizzato per la mortalità parentale accidentale e casuale nei controlli nonché in ciascuna delle concentrazioni di prova. DESCRIZIONE DEL METODO Apparecchiature I recipienti e le altre apparecchiature destinate a entrare in contatto con le soluzioni di prova devono essere interamente di vetro o di altro materiale chimicamente inerte. Di norma si utilizzano beaker di vetro come recipienti di prova. Inoltre sono necessarie alcune o tutte le seguenti apparecchiature:
Organismo sperimentale La specie da utilizzare nella prova è la Daphnia magna Straus (95). Di preferenza il clone va identificato tramite determinazione del genotipo. La ricerca (1) ha dimostrato che le prestazioni riproduttive del Clone A (proveniente dall'IRCHA, in Francia) (5), allevato nelle condizioni descritte nel presente metodo di prova, soddisfa costantemente il criterio di validità di una media di ≥ 60 di piccoli vivi per animale riproduttore sopravvissuto. Sono comunque accettabili altri cloni, purché si dimostri che la coltura di Daphnia soddisfa i criteri di validità della prova. All'inizio della prova gli animali devono avere meno di 24 ore di vita e non devono provenire dalla prima nidiata. Devono provenire da una popolazione sana (senza segni di stress quali un alto tasso di mortalità, presenza di maschi e formazione di efippi, ritardo nella produzione della prima nidiata, decolorazione ecc.). Gli animali riproduttori vanno mantenuti in condizioni di coltura (luce, temperatura, mezzo, alimentazione e numero di animali per unità di volume) simili a quelle che verranno utilizzate nella prova. Se il mezzo di coltura per la Dafnia da usare nella prova è diverso da quello utilizzato di routine per la coltura di Dafnia, è buona prassi prevedere un periodo di acclimatazione prima del test, normalmente di tre settimane (cioè una generazione), per evitare di sottoporre a stress gli animali destinati alla riproduzione. Mezzo di prova Si raccomanda di usare per questa prova un mezzo completamente definito: ciò può evitare l'uso di additivi (ad esempio alghe, estratto di terra), che sono difficili da caratterizzare, e dunque aumentare la possibilità di standardizzazione fra vari laboratori. I mezzi M4 (6) e M7 di Elendt (cfr. appendice 2) si sono rivelati adatti a questo scopo. Sono comunque accettabili altri mezzi [es. (7) (8)], purché si dimostri che le prestazioni della coltura di Daphnia soddisfano i criteri di validità della prova. Se si impiegano mezzi contenenti additivi non ben definiti, occorre descriverli in dettaglio aggiungendo nella relazione informazioni sulla loro composizione, con particolare riferimento al contenuto di carbonio, che potrebbe influire sulla dieta. Si raccomanda di determinare il carbonio organico totale (TOC) e/o la domanda chimica di ossigeno (COD) della preparazione madre dell'additivo organico e di effettuare una stima del contributo dato al TOC/COD del mezzo di prova. Si raccomanda inoltre che i livelli di TOC nel mezzo (cioè prima dell'aggiunta delle alghe) siano inferiori a 2 mg/l (9). Quando si testano sostanze chimiche contenenti metalli è importante tener conto del fatto che le proprietà del mezzo di prova (ad esempio la durezza e la capacità di chelazione) possono influire sulla tossicità della sostanza chimica in esame. Per questo motivo è consigliabile utilizzare un mezzo la cui composizione sia completamente conosciuta. Attualmente, però, gli unici mezzi di questo tipo noti per essere adatti alla coltura a lungo termine di Daphnia magna sono l'M4 e l'M7 di Elendt. Entrambi i mezzi contengono l'agente chelante EDTA. La ricerca ha dimostrato (2) che la «tossicità apparente» del cadmio è generalmente inferiore quando il test sulla riproduzione viene eseguito nei mezzi M4 e M7 invece che in mezzi non contenenti EDTA. I mezzi M4 e M7 non sono pertanto raccomandati per testare sostanze chimiche contenenti metalli; occorre inoltre evitare anche altri mezzi che contengono agenti chelanti. Per le sostanze chimiche contenenti metalli può essere consigliabile utilizzare un mezzo alternativo come ad esempio l'acqua dolce dura ricostituita secondo le indicazioni dell'ASTM (9), che non contiene EDTA. Questa combinazione di acqua dolce dura ricostituita ed estratto di alghe marine è adatta alla coltura a lungo termine (10) di Daphnia magna (2). La concentrazione dell'ossigeno disciolto deve essere superiore a 3 mg/l, all'inizio e durante la prova. Il pH deve collocarsi nell'intervallo 6-9 senza di norma variare di oltre 1,5 unità nell'ambito di una prova. Si raccomanda una durezza superiore a 140 mg/l (come CaCO3). Le prove eseguite con un valore pari o superiore a questo livello hanno dimostrato che le prestazioni riproduttive sono conformi ai criteri di validità (11) (12). Soluzioni di prova Le soluzioni di prova alle concentrazioni prescelte vanno in genere preparate per diluizione di una soluzione madre. Se possibile, le soluzioni madre devono essere preparate preferibilmente senza l'impiego di eventuali solventi o disperdenti, mediante miscelazione o agitazione della sostanza chimica in esame nel mezzo di prova utilizzando mezzi meccanici (es. per agitazione, mescolatura, ultrasuoni) o altri metodi appropriati. È preferibile esporre i sistemi di prova alle concentrazioni della sostanza chimica da utilizzare nello studio per tutto il tempo necessario a dimostrare il mantenimento della stabilità delle concentrazioni di esposizione prima dell'introduzione degli organismi di prova. Se una sostanza chimica in esame è difficile da sciogliere in acqua, vanno applicate le procedure descritte nel documento di orientamento dell'OCSE per la manipolazione di sostanze “difficili” (13). Andrebbe evitato l'uso di solventi o disperdenti, ma in alcuni casi può rendersi necessario utilizzarli per ottenere una soluzione madre di adeguata concentrazione per il dosaggio. Occorre allestire, oltre alle concentrazioni di prova, un campione di controllo con l'acqua di diluizione con adeguate repliche e, se ciò si rivelasse inevitabile, un controllo con solvente con adeguate repliche. Per la prova vanno utilizzati solo i solventi o disperdenti che hanno dimostrato avere effetti minimi o inesistenti sulla variabile di risposta. In (13) sono indicati esempi di solventi (ad esempio l'acetone, l'etanolo, il metanolo, la dimetilformammide e il glicole trietilenico) e di disperdenti adatti (ad esempio il Cremophor RH 40, la metilcellulosa 0,01 % e l'HCO-40). Se si utilizza un solvente, o un disperdente, la sua concentrazione finale non deve superare 0,1 ml/l (13) e deve essere identica in tutti i recipienti sperimentali, salvo che per il campione di controllo con acqua di diluizione. Tuttavia, occorre compiere ogni sforzo per mantenere la concentrazione di solvente al minimo. PROCEDURA Condizioni di esposizione Durata La durata del test è di 21 giorni. Carico Gli esemplari riproduttori vengono mantenuti individualmente, uno per ciascun recipiente di prova, solitamente con 50-100 ml di mezzo per ogni recipiente (per la Daphnia magna sono possibili volumi più piccoli, specialmente per dafnidi più piccoli, ad esempio Ceriodaphnia dubia), a meno che sia necessario allestire una prova a flusso continuo. Talvolta, per soddisfare i requisiti della procedura analitica usata per determinare la concentrazione della sostanza chimica in esame, occorre utilizzare un volume maggiore, sebbene sia consentito il raggruppamento delle repliche per l'analisi chimica. Nel caso si utilizzino volumi superiori a 100 ml, potrebbe essere necessario aumentare la razione fornita alla Daphnia per assicurare un'adeguata disponibilità del cibo e il rispetto dei criteri di validità. Animali da esperimento Per le prove semistatiche occorrono almeno 10 animali, mantenuti singolarmente, per ogni concentrazione di prova e almeno 10 animali, mantenuti singolarmente, nella serie di controllo. E stato dimostrato che per le prove a flusso continuo è adeguato utilizzare 40 animali suddivisi in quattro gruppi di 10 per ciascuna concentrazione di prova (1). È possibile utilizzare un numero inferiore di organismi sperimentali, ma si raccomanda comunque di utilizzare un minimo di 20 animali per concentrazione, divisi in due o più repliche con un numero uguale di animali (ad esempio quattro repliche con cinque dafnidi ciascuna). Si noti che per le prove nelle quali gli animali sono mantenuti in gruppi, non sarà possibile escludere nessuna prole dall'analisi statistica in caso di mortalità parentale casuale/accidentale in fase di riproduzione già avviata, e pertanto in questi casi la capacità riproduttiva va espressa come numero totale di piccoli vivi prodotti da ciascun animale riproduttore presente all'inizio della prova. Occorre randomizzare l'assegnazione dei trattamenti ai recipienti di prova e tutte le successive manipolazioni. In caso contrario si potrebbero verificare “bias” che potrebbero essere interpretate come un effetto della concentrazione. In particolare, se le unità sperimentali vengono manipolate in ordine di trattamento o di concentrazione, alcuni effetti collegati al tempo (ad esempio la stanchezza dell'operatore o un altro errore) possono produrre effetti maggiori alle concentrazioni più alte. Inoltre, se si ritiene che i risultati della prova possano essere influenzati da condizioni sperimentali iniziali o ambientali (ad esempio la posizione nel laboratorio), bisogna considerare la possibilità di porre termine alla prova. Alimentazione Nelle prove semistatiche è preferibile nutrire gli animali ogni giorno, e comunque almeno tre volte alla settimana (in concomitanza con la sostituzione del mezzo). Occorre tenere conto dell'eventuale diluizione delle concentrazioni di esposizione dovuta all'aggiunta del prodotto alimentare, evitandola, per quanto possibile, grazie a sospensioni ben concentrate di alghe. Se non si osserva questo modello (per esempio nelle prove a flusso continuo) occorre segnalarlo nella relazione. Durante la prova la dieta degli animali riproduttori deve consistere preferibilmente in alghe unicellulari vive di una o più delle seguenti specie: Chlorella sp., Pseudokirchneriella subcapitata (in precedenza Selenastrum capricornutum) e Desmodesmus subspicatus (in precedenza Scenedesmus subspicatus). La dieta deve essere basata sulla quantità di carbonio organico (C) fornita a ciascun animale riproduttore. Ricerche (14) hanno dimostrato che per ottenere il numero di piccoli vivi di Daphnia magna necessari per soddisfare i criteri di validità sono sufficienti razioni comprese fra 0,1 e 0,2 mg C/Daphnia/die. È possibile somministrare la razione in maniera costante per tutto il periodo della prova oppure una razione inferiore all'inizio della prova della prova ed una razione più alta durante la prova, tenendo conto della crescita degli animali riproduttori. In questo caso la razione deve comunque restare sempre nell'intervallo raccomandato di 0,1 — 0,2 mg C/Daphnia/die. Se per comodità (visto che la misurazione del tenore di carbonio richiede molto tempo) si utilizzano altri parametri di misurazione, quali la conta delle cellule algali o l'assorbanza della luce, per somministrare la razione necessaria, ogni laboratorio deve elaborare un proprio nomogramma che correli il parametro di misurazione scelto al contenuto di carbonio della coltura di alghe (cfr. appendice 3 per l'elaborazione del nomogramma). I nomogrammi vanno controllati almeno una volta all'anno e con maggiore frequenza in caso di modifica delle condizioni di coltura delle alghe. È stato dimostrato che per il contenuto di carbonio l'assorbanza della luce è un indicatore migliore che non il numero di cellule (15). Occorre alimentare le Daphnia con una sospensione concentrata di alghe per ridurre al minimo il volume del mezzo di coltura algale trasferito nei recipienti di prova. La concentrazione delle alghe può essere ottenuta per centrifugazione e successiva risospensione nel mezzo di coltura delle Daphnia. Luce 16 ore di luce a un'intensità non superiore a 15 – 20 μE · m– 2 · s– 1 misurata sulla superficie dell'acqua del recipiente. Per gli strumenti per la misurazione della luce calibrati in lux, un intervallo equivalente di 1 000-1 500 lux per la luce bianca fredda corrisponde da vicino all'intensità raccomandata della luce, cioè 15 – 20 μE · m-2 · s – 1. Temperatura La temperatura del mezzo di prova si colloca nell'intervallo 18-22 °C. Tuttavia, per ogni test la temperatura non deve variare quotidianamente, se possibile, di più di 2 °C all'interno dell'intervallo indicato (ossia, deve mantenersi nei seguenti intervalli: 18-20, 19-21 o 20-22 °C) Per controllare la temperatura si può utilizzare un recipiente di prova aggiuntivo. Aerazione I recipienti di prova non vanno aerati durante la prova. Disegno sperimentale Prova di determinazione dell'intervallo delle concentrazioni Ove necessario, si effettua una prova per determinare l'intervallo delle concentrazioni (range finding test): ad esempio, cinque concentrazioni della sostanza chimica in esame e due repliche per ogni gruppo di trattamento e di controllo. Ulteriori informazioni sulla tossicità acuta per Daphnia e/o per altri organismi acquatici, derivanti da prove con sostanze chimiche simili o tratte dalla letteratura specializzata, possono anche essere utili per decidere l'intervallo di concentrazioni da sottoporre alla prova di determinazione degli intervalli delle concentrazioni. La durata della prova di determinazione degli intervalli delle concentrazioni è 21 giorni o una durata sufficiente a prevedere in modo attendibile i livelli degli effetti. Al termine della prova viene valutata la capacità riproduttiva della Daphnia. Vanno registrati il numero di animali riproduttori e la comparsa di prole. Prova definitiva In genere occorrono almeno cinque concentrazioni di prova, soffermandosi sulle concentrazioni efficaci (ad esempio ECx) e disposte in una serie geometrica con un fattore di separazione preferibilmente non superiore a 3,2. Vanno usate un numero adeguato di repliche per ciascuna concentrazione di prova (cfr. paragrafi 24-25). L'uso di un numero di concentrazioni inferiore a cinque va giustificato. Le sostanze chimiche non vanno provate al di sopra del loro limite di solubilità nel mezzo di prova. Prima di condurre l'esperimento si consiglia di prendere in considerazione la potenza statistica del disegno di prova e l'uso di metodi statistici adeguati (4). Nel definire l'intervallo delle concentrazioni è necessario tenere conto dei seguenti elementi:
Se non si osservano effetti alla concentrazione massima durante la prova di determinazione degli intervalli delle concentrazioni (ad esempio a 10 mg/l), o quando è altamente probabile che la sostanza chimica in esame sia di tossicità scarsa o nulla basandosi sulla tossicità nulla rilevata per altri organismi, e/o quando tali organismi la assorbono poco o per nulla, la prova di riproduzione può essere eseguita come una prova limite, con una concentrazione di prova, ad esempio, di 10 mg/l e un controllo. Occorre utilizzare dieci repliche sia per i gruppi di trattamento sia per i gruppi di controllo. Se la prova limite richiede un sistema a flusso continuo, può essere adeguato utilizzare meno repliche. Una prova limite fornirà l'occasione di dimostrare che non esiste un effetto statisticamente significativo alla concentrazione limite, ma se invece vengono registrati degli effetti sarà in genere necessario procedere a una prova completa. Controlli In aggiunta alle concentrazioni con la sostanza chimica in esame, dovrebbe essere allestita una serie di controllo con il mezzo di prova ed eventualmente una serie di controllo contenente il solvente o il disperdente. La concentrazione dell'eventuale solvente o disperdente deve essere identica a quella usata nei recipienti contenenti la sostanza chimica in esame. Anche per i controlli occorre usare un numero adeguato di repliche (cfr. paragrafi 23-24). Generalmente, in un test ben condotto, il coefficiente di variazione del numero medio di piccoli vivi prodotti da ciascun animale riproduttore nel o nei controlli dovrebbe essere ≤ 25 %; il coefficiente di variazione va riportato per le prove dove gli animali riproduttori sono mantenuti individualmente. Rinnovo del mezzo di prova La frequenza con cui il mezzo viene rinnovato dipende dalla stabilità della sostanza chimica in esame, ma dovrebbe essere almeno tre volte alla settimana. Se da prove preliminari sulla stabilità (cfr. paragrafo 7) la concentrazione della sostanza chimica in esame non risulta stabile (è cioè al di fuori dell'intervallo 80-120 % della concentrazione nominale o al di sotto dell'80 % della concentrazione iniziale misurata) durante il periodo massimo di rinnovo (tre giorni), si consiglia di rinnovare il mezzo con maggiore frequenza oppure di eseguire una prova a flusso continuo. Quando si rinnova il mezzo nelle prove semistatiche si prepara una seconda serie di recipienti dove vengono trasferiti gli animali riproduttori mediante, ad esempio, una pipetta di vetro di diametro adeguato. Il volume del mezzo trasferito con le Daphnia dovrebbe essere il più piccolo possibile. Osservazioni I risultati delle osservazioni fatte durante il test vanno registrati su apposite schede di raccolta dei dati (cfr. appendici 4 e 5). Dovendo fornire i dati di altre misurazioni (cfr. paragrafo 44) possono essere richieste ulteriori osservazioni. Prole La prole prodotta da ciascun animale riproduttore dovrebbe essere di preferenza tolta dal recipiente e contata ogni giorno a partire dalla comparsa della prima schiusa, per impedire che consumi cibo destinato all'animale riproduttore. Ai fini del metodo di prova qui descritto è necessario contare solo il numero di piccoli vivi, ma è consigliabile registrare anche la presenza di uova abortite o piccoli morti. Mortalità La mortalità fra gli animali riproduttori va rilevata di preferenza quotidianamente, o almeno ad ogni conta dei piccoli. Altri parametri Sebbene questo metodo di prova sia fondamentalmente inteso a valutare gli effetti sulla capacità riproduttiva, è possibile che altri effetti siano sufficientemente quantificati da permettere un'analisi statistica. È possibile registrare la capacità riproduttiva per animale riproduttore superstite, ossia il numero di piccoli vivi prodotti durante la prova per riproduttore superstite. Ciò può essere confrontato con la principale variabile di risposta (la capacità riproduttiva all'inizio della prova, per animale riproduttore che non sia vittima di morte casuale o accidentale durante la prova). In presenza di mortalità parentale nelle repliche esposte si deve stabilire se essa segue un modello concentrazione-risposta, ad esempio se vi è una significativa regressione della risposta rispetto alla concentrazione della sostanza chimica in esame con una pendenza positiva (è possibile utilizzare una prova statistica quale il test di Cochran-Armitage per le tendenze). Se la mortalità non segue un modello concentrazione-risposta, le repliche che presentano mortalità parentale vanno escluse dall'analisi del risultato della prova. Se la mortalità segue un modello concentrazione-risposta, la mortalità parentale è assimilata a un effetto della sostanza chimica in esame e le repliche non vanno escluse dall'analisi del risultato della prova. La misura della crescita è particolarmente auspicabile in quanto fornisce informazioni su possibili effetti subletali, che potrebbero essere utili e affiancarsi alla sola misura della riproduzione; si raccomanda di misurare la lunghezza degli animali riproduttori (lunghezza del corpo esclusa la spina posteriore del carapace) alla fine del test. Altri parametri che possono essere misurati o calcolati sono: il periodo intercorso fino alla produzione della prima schiusa (e delle schiuse successive), il numero e le dimensioni delle schiuse per animale, il numero delle schiuse abortite, la presenza di maschi neonati (OCSE, 2008) o efippi e possibilmente il tasso intrinseco di aumento della popolazione (cfr. appendice 1 per la definizione e l'appendice 7 per l'identificazione del sesso dei neonati). Frequenza delle determinazioni e delle misurazioni analitiche Concentrazione dell'ossigeno, temperatura, durezza e pH dovrebbero essere misurati almeno una volta alla settimana, nei mezzi freschi (dopo il rinnovo) e vecchi (prima del rinnovo), nel/i controllo/i e nella concentrazione massima della sostanza chimica in esame. Durante la prova le concentrazioni della sostanza chimica in esame vanno determinate a intervalli regolari. Nelle prove semistatiche in cui si prevede che la concentrazione della sostanza chimica in esame resti intorno a ± 20 % del valore nominale (e cioè entro l'intervallo dell'80-120 %; cfr. paragrafi 6, 7 e 39), si raccomanda di analizzare almeno le concentrazioni minima e massima di prova subito dopo la loro preparazione e in occasione di un rinnovo del mezzo nel corso della prima settimana del test (le analisi vanno effettuate su un campione della stessa soluzione, appena preparata e al momento di rinnovarla). Queste determinazioni vanno poi ripetute a intervalli almeno settimanali. Per le prove in cui non si prevede che la concentrazione della sostanza in esame resti entro ± 20 % del valore nominale, è necessario analizzare tutte le concentrazioni di prova, appena preparate e al momento di rinnovarle. Tuttavia, per le prove in cui la concentrazione iniziale misurata della sostanza chimica in esame non è entro ± 20 % del valore nominale, ma si può fornire prova che le concentrazioni iniziali sono ripetibili e stabili (cioè entro un range dell'80-120 % delle concentrazioni iniziali), nella seconda e terza settimana della prova le determinazioni chimiche possono limitarsi alle concentrazioni massima e minima. In tutti i casi la determinazione delle concentrazioni della sostanza chimica in esame prima del rinnovo può limitarsi a un unico recipiente per ciascuna concentrazione. Per le prove a flusso continuo è appropriato l'uso di un regime di campionamento simile a quello descritto per le prove semistatiche (sebbene in questo caso non ci sia la misurazione delle soluzioni “vecchie”). Può essere però consigliabile aumentare il numero di campionamenti durante la prima settimana (ad esempio, tre serie di misurazioni) per accertare la stabilità delle concentrazioni di prova. In questi tipi di prova dovrebbe essere controllato quotidianamente il tasso di flusso del diluente e della sostanza chimica in esame. Potendo dimostrare che durante l'intera prova la concentrazione della sostanza chimica in esame in soluzione è stata mantenuta in modo soddisfacente entro ± 20 % della concentrazione nominale o della concentrazione iniziale misurata, i risultati possono essere basati sui valori nominali o sui valori iniziali misurati. Se la deviazione dalla concentrazione iniziale nominale o misurata è maggiore del ± 20 %, i risultati vanno espressi in termini di media ponderata nel tempo (v. orientamenti per il calcolo nell'appendice 6). DATI E RELAZIONI Trattamento dei risultati La presente prova è intesa a determinare gli effetti della sostanza chimica in esame sulla capacità riproduttiva. Occorre calcolare il numero complessivo di piccoli vivi per animale riproduttore per ciascun recipiente di prova (replica). Inoltre, la capacità riproduttiva può essere calcolata sulla base della produzione di piccoli vivi per animale riproduttore superstite. Tuttavia, la variabile di risposta ecologicamente più rilevante corrisponde al numero totale di piccoli vivi prodotti da ciascun animale riproduttore che non sia vittima di morte accidentale (96) o casuale (97) durante la prova. Se l'animale riproduttore muore accidentalmente o casualmente durante il test, o si rivela essere un maschio, la replica viene esclusa dall'analisi. L'analisi si baserà quindi su un numero ridotto di repliche. In presenza di mortalità parentale nelle repliche esposte si deve stabilire se essa segue un modello concentrazione-risposta, ad esempio se vi è una significativa regressione della risposta rispetto alla concentrazione della sostanza chimica in esame con una pendenza positiva (è possibile utilizzare una prova statistica quale il test di Cochran-Armitage per le tendenze). Se la mortalità non segue un modello concentrazione-risposta, le repliche che presentano mortalità parentale vanno escluse dall'analisi del risultato della prova. Se la mortalità segue un modello concentrazione-risposta, la mortalità parentale è assimilata a un effetto della sostanza chimica in esame e le repliche non vanno escluse dall'analisi del risultato della prova. In sintesi, quando gli effetti sono espressi come LOEC e NOEC o ECx si raccomanda di calcolare gli effetti sulla riproduzione mediante l'uso delle due variabili di risposta succitate, vale a dire
e di utilizzare in seguito come risultato finale il più basso valore LOEC e NOEC o ECx calcolato utilizzando queste due variabili di risposta. Prima di ricorrere all'analisi statistica, ad esempio tramite analisi ANOVA o il confronto dei gruppi trattati e dei gruppi di controllo con i test di Student (test t), di Dunnett, di Williams o di Jonckheere-Terpstra (test di tendenza regressiva), si raccomanda di considerare la trasformazione dei dati se ciò è necessario per soddisfare i requisiti del particolare test statistico. Come alternativa non parametrica, si possono prendere in considerazione i test di Dunn o Mann-Whitney. Per le medie di ciascuna concentrazione sono calcolati intervalli di confidenza al 95 %. Il numero di animali riproduttori sopravvissuti nei controlli non trattati è un criterio di validità che deve essere documentato e registrato. Anche tutti gli altri effetti negativi, ad esempio comportamenti anomali e risultati tossicologici significativi, devono essere registrati nella relazione finale. ECx I valori ECx, e i relativi limiti di confidenza superiori e inferiori, vanno calcolati utilizzando metodi statistici appropriati (funzione logistica o di Weibull, metodo semplificato di Spearman-Karber o semplice interpolazione). Per calcolare la EC10, la EC50 o qualsiasi altra ECx, occorre sottoporre la serie completa di dati a un'analisi di regressione. NOEC/LOEC Se l'analisi statistica intende determinare la NOEC/LOEC occorre fare uso di metodi statistici appropriati in base al Documento n. 54 dell'OCSE “Current Approaches in the Statistical Analysis of Ecotoxicity Data: a Guidance to Application” (4): In generale, gli effetti nocivi della sostanza chimica in esame rispetto al controllo sono analizzati applicando un'ipotesi unilaterale con p ≤ 0,05. La distribuzione normale dei dati e l'omogeneità della varianza possono ad esempio essere analizzate mediante il test di Shapiro-Wilk o il test di Levene (p ≤ 0,05), rispettivamente. Possono essere eseguiti un'analisi della varianza ANOVA a un fattore e successivi test multi-comparativi. I test multi-comparativi (ad esempio, test di Dunnett) o i test di tendenza regressiva (ad esempio, test di Williams o di Jonckheere-Terpstra) possono essere utilizzati ai fini del calcolo di eventuali differenze significative (p ≤0,05) tra i controlli e le varie concentrazioni della sostanza chimica in esame [(per scegliere la prova raccomandata si consulti il documento n. 54 dell'OCSE (4)]. Altrimenti, per determinare la LOEC e la NOEC si possono utilizzare metodi non parametrici (test U di Bonferroni secondo il test di tendenza di Holm o di Jonckheere-Terpstra). Prova limite Se è stata svolta una prova limite (confronto tra il controllo e un unico trattamento) e se sono rispettati i presupposti necessari per le procedure delle prove parametriche (normalità, omogeneità), è possibile valutare le risposte metriche mediante il test di Student (t-test). In caso contrario, si può ricorrere a un test t per varianze disuguali (es.: test di Welch) o a un test non parametrico, come il test di Wilcoxon-Mann-Whithey. Per determinare significative divergenze tra i controlli (campione di controllo e controllo con solvente o disperdente), le repliche di ciascun controllo possono essere testate come descritto per la prova limite. Se le prove non rilevano alcuna differenza significativa, è possibile raggruppare assieme tutte le repliche di controllo e di controllo con solvente. In caso contrario, occorre confrontare tutti i trattamenti con il controllo con solvente. Relazione sulla prova La relazione sulla prova include le informazioni indicate di seguito.
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Ai fini del presente metodo di prova si applicano le seguenti definizioni: Mortalità accidentale : mortalità non collegata a sostanze chimiche e causata da un evento accidentale (cioè da una causa conosciuta). Sostanza chimica : una sostanza o una miscela. ECx : concentrazione della sostanza chimica in esame disciolta in acqua che provoca una percentuale x di riduzione della capacità riproduttiva della Daphnia magna entro un determinato periodo di esposizione. Mortalità casuale : mortalità non collegata a sostanze chimiche e priva di causa conosciuta. Tasso intrinseco di aumento della popolazione : misura della crescita della popolazione che integra la capacità riproduttiva e la mortalità specifica in base all'età (1) (2) (3). Nelle popolazioni in equilibrio dinamico è uguale a zero. Per le popolazioni in crescita è positivo, mentre per quelle in diminuzione è negativo. Ovviamente, quest'ultimo tasso non è sostenibile e, alla fine, porta all'estinzione. Limite di rilevazione : concentrazione minima che può essere individuata ma non quantificata. Limite di determinazione : concentrazione minima misurabile quantitativamente. Minima concentrazione con effetti significativi (Lowest Observed Effect Concentration — LOEC) : la concentrazione più bassa sottoposta a prova alla quale si osserva che la sostanza chimica produce un effetto statisticamente significativo sulla riproduzione e sulla mortalità parentale (con p < 0,05) rispetto ai controlli, entro un periodo di esposizione definito. Tuttavia, tutte le concentrazioni di prova superiori alla LOEC devono avere un effetto dannoso uguale o superiore a quelli osservati alla LOEC. Se non è possibile soddisfare queste due condizioni, è necessario fornire una spiegazione esauriente sulle modalità di scelta della LOEC (e di conseguenza della NOEC). Mortalità : si considerano morti gli animali che, entro 15 secondi dopo lieve agitazione del contenitore usato per la prova, restano immobili, cioè non sono in grado di nuotare, o nei quali non si osserva alcun movimento delle appendici o della parte posteriore dell'addome. (Nel caso si usi un'altra definizione, essa deve essere indicata insieme al relativo riferimento bibliografico). Massima concentrazione senza effetti significativi (No Observed Effect Concentration — NOEC) : concentrazione di prova immediatamente inferiore alla LOEC che, se confrontata con i controlli, non ha un effetto statisticamente significativo (con p < 0,05), entro un periodo di esposizione definito. Prole : piccoli di Daphnia generati dagli animali riproduttori nel corso della prova. Animali riproduttori : femmine di Daphnia presenti all'inizio della prova la cui capacità riproduttiva è oggetto dello studio. Capacità riproduttiva : numero di piccoli vivi prodotti da animali riproduttori entro il periodo di prova. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata applicando il presente metodo di prova. BIBLIOGRAFIA
Appendice 2 PREPARAZIONE DEI MEZZI M7 E M4 DI ELENDT TOTALMENTE DEFINITI Acclimatazione ai mezzi M7 e M4 di Elendt Alcuni laboratori hanno avuto difficoltà nel trasferire direttamente le Daphnia nei mezzi M4 (1) e M7. Qualche risultato soddisfacente è stato invece ottenuto con un'acclimatazione graduale, cioè trasferendo le Daphnia dal proprio mezzo ad un mezzo Elendt al 30 %, poi al 60 % e infine ad un mezzo Elendt al 100 %. I periodi di acclimatazione possono avere anche una durata di un mese. Preparazione Oligoelementi Inizialmente si preparano soluzioni madre distinte (I) dei singoli oligoelementi in acqua di adeguata purezza, ad esempio deionizzata, distillata o sottoposta a osmosi inversa. Da queste diverse soluzioni madre (I) si prepara una seconda soluzione madre singola (II) che contiene tutti gli oligoelementi (soluzione combinata), e cioè:
Mezzi M4 e M7 I mezzi di coltura M4 ed M7 sono preparati usando la soluzione madre II, i macronutrienti e le vitamine, nel modo seguente:
La soluzione madre combinata di vitamine si conserva congelata in piccole aliquote. Le vitamine si aggiungono al mezzo di coltura poco prima dell'uso.
Appendice 3 ANALISI DEL CARBONIO ORGANICO TOTALE (TOC) E PRODUZIONE DI UN NOMOGRAMMA PER IL CONTENUTO DI TOC DELL'ALIMENTO ALGALE È riconosciuto che il contenuto di carbonio dell'alimento algale non viene di norma misurato direttamente, bensì mediante correlazioni (cioè nomogrammi) con misure sostitutive quali il numero di cellule algali o l'assorbanza della luce. Il TOC dovrebbe essere misurato per ossidazione ad alta temperatura piuttosto che mediante UV o metodi con persolfati. (Per suggerimenti cfr.: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB). Per la produzione del nomogramma, le alghe vanno separate dal mezzo di crescita mediante centrifugazione, seguita da risospensione in acqua distillata. Occorre misurare il parametro surrogato e la concentrazione del TOC in ciascun campione in triplicato. Vanno analizzati i bianchi di acqua distillata e la loro concentrazione di TOC viene dedotta dalla concentrazione del TOC nel campione di alghe. I nomogrammi devono essere lineari nell'intervallo richiesto di concentrazioni del carbonio. Di seguito sono riportati alcuni esempi.
Chlorella vulgaris var. viridis (CCAP 211/12). Regressione di mg/l di peso secco su mg C/1. Dati da sospensioni concentrate di cellule in coltura in semicontinuo, risospese in acqua distillata. Asse x: mg C/1 di alimento algale concentrato Asse y: mg/1 peso secco di alimento algale concentrato Coefficiente correttore -0,980 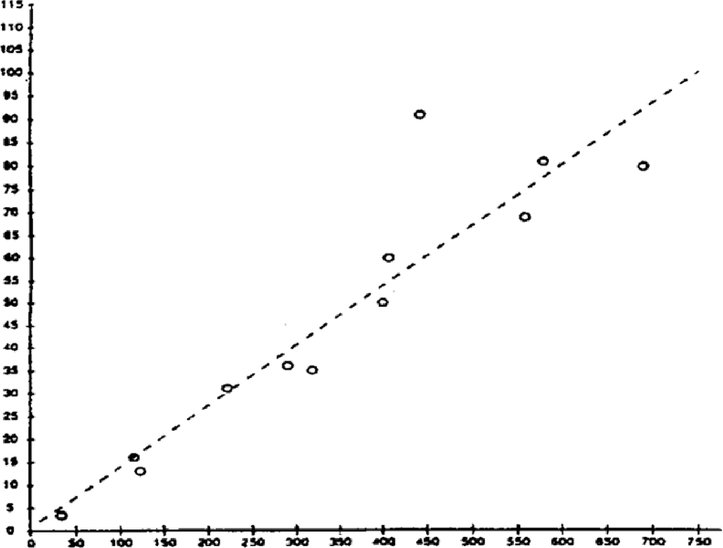
Chlorella vulgaris var. viridis (CCAP 211/12). Regressione del numero di cellule su mg C/1. Dati da sospensioni concentrate di cellule in coltura in semicontinuo, risospese in acqua distillata. Asse x: mg C/1 di alimento algale concentrato Asse y: n. di cellule/1 di alimento algale concentrato Coefficiente correttore -0,926 
Chlorella vulgaris var. viridis (CCAP 211/12). Regressione dell'assorbanza su mg C/1 (1 cm di cammino ottico). Dati da sospensioni concentrate di cellule in coltura in semicontinuo, risospese in acqua distillata. Asse x: mg C/1 di alimento algale concentrato Asse y: Assorbanza a 440 nm di una diluizione 1/10 di alimento algale concentrato Coefficiente correttore – 0,998 Appendice 4 ESEMPIO DI SCHEDA PER LA RACCOLTA DI DATI SUL RINNOVO DEL MEZZO, IL MONITORAGGIO FISICO/CHIMICO, L'ALIMENTAZIONE, LA RIPRODUZIONE DELLE DAPHNIA E LA MORTALITÀ PARENTALE
Appendice 5 ESEMPIO DI SCHEDA PER LA REGISTRAZIONE DEI RISULTATI DELLE ANALISI CHIMICHE (a) Concentrazioni misurate
(b) Concentrazioni misurate come percentuale del valore nominale
Appendice 6 CALCOLO DI UNA MEDIA PONDERATA NEL TEMPO Media ponderata nel tempo Dato che la concentrazione della sostanza chimica in esame può diminuire nel periodo fra i rinnovi del mezzo è necessario considerare quale concentrazione vada scelta come rappresentativa dell'intervallo di concentrazioni a cui sono state esposte le Daphnia riproduttrici. La selezione deve basarsi su considerazioni biologiche oltre che statistiche. Per esempio, se si ritiene che la riproduzione venga influenzata soprattutto dalla concentrazione picco, si deve utilizzare la concentrazione massima. Se invece si ritiene più importante l'effetto accumulato o a più lungo termine della sostanza chimica tossica, allora risulta più pertinente una concentrazione media. In questo caso una media adeguata è la concentrazione media ponderata nel tempo, in quanto tiene conto della variazione della concentrazione istantanea nel corso del tempo. Figura 1 Esempio di media ponderata in funzione tempo 
La Figura 1 mostra un esempio di test (semplificato) della durata di sette giorni con rinnovo del mezzo nei giorni 0, 2 e 4.
La media ponderata nel tempo viene calcolata in modo che l'area ad essa sottostante sia uguale all'area sotto la curva della concentrazione. Il calcolo per l'esempio in figura è illustrato nella tabella 1. Tabella 1 Calcolo di una media ponderata nel tempo
Area è l'area sotto la curva esponenziale per ciascun periodo di rinnovamento. Viene calcolata nel modo seguente:
La media ponderata nel tempo (media ponderata/t) è l'Area totale divisa per i Giorni totali. Ovviamente per la prova di riproduzione con Daphnia occorre prolungare la tabella fino a coprire 21 giorni. È chiaro che quando le osservazioni vengono effettuate solo all'inizio e alla fine di ciascun periodo di rinnovamento non è possibile confermare che il processo di decadimento è effettivamente esponenziale. Una curva diversa produrrebbe un calcolo diverso per l'Area. Tuttavia, è tuttavia plausibile che un processo di decadimento sia esponenziale e questa è probabilmente la curva migliore da usare in assenza di altre informazioni. Occorre però procedere con cautela se l'analisi chimica non rileva alcuna sostanza alla fine del periodo di rinnovo. A meno che non sia possibile stimare la rapidità con cui la sostanza chimica è scomparsa dalla soluzione, è impossibile ottenere un'area sotto la curva che sia realistica, e pertanto è impossibile ottenere una ragionevole media ponderata nel tempo. Appendice 7 ORIENTAMENTI PER L'IDENTIFICAZIONE DEL SESSO DEI NEONATI La produzione di neonati di sesso maschile può avvenire modificando le condizioni ambientali, ad esempio abbreviando i fotoperiodi, modificando la temperatura, riducendo la concentrazione degli alimenti e aumentando la densità demografica (Hobaek e Larson, 1990; Kleiven et al., 1992). La produzione di neonati di sesso maschile è anche una risposta nota ad alcuni regolatori della crescita degli insetti (Oda et al., 2005). In condizioni in cui agenti chimici stressanti inducono una diminuzione della prole da femmine partenogenetiche, si può prevedere un incremento del numero dei maschi (OCSE, 2008). Sulla base delle informazioni disponibili non è possibile prevedere quale parametro, sia esso il rapporto numerico tra i sessi o la riproduzione, sarà quello più sensibile; tuttavia, vi sono indicazioni (cfr. “relazione di validazione”, parte 1) secondo cui questo aumento del numero dei maschi sarebbe meno sensibile della diminuzione dei piccoli vivi. Dato che l'obiettivo primario del presente metodo di prova è valutare il numero di piccoli vivi prodotti, l'osservazione della comparsa di maschi va considerata come facoltativa. Se in uno studio si decide di valutare questo endpoint facoltativo, occorre allora introdurre un ulteriore criterio di validità della prova utilizzando non oltre il 5 % di maschi nei controlli. Il modo più veloce e semplice per differenziare il sesso delle Daphnia consiste nell'utilizzare le loro caratteristiche fenotipiche, in quanto maschi e femmine sono geneticamente identici e il sesso è determinato dalle condizioni ambientali. I maschi e le femmine sono diversi per la lunghezza e la morfologia delle prime antenne, più lunghe nei maschi che nelle femmine (fig. 1). Tale differenza è riconoscibile subito dopo la nascita, sebbene lo sviluppo di caratteristiche sessuali secondarie avvenga con la crescita (cfr., ad esempio, fig. 2 in Olmstead e LeBlanc, 2000). Per osservare le caratteristiche sessuali morfologiche, i neonati prodotti da ciascun animale sperimentale devono essere trasferiti tramite una pipetta e inseriti in una capsula Petri con mezzo di prova. Il mezzo di prova è mantenuto al minimo per limitare il movimento degli animali. È possibile osservare le prime antenne con un microscopio stereoscopico (× 10-60). Fig. 1 Esemplari di Daphna magna di 24 ore: maschio (sinistra) e femmina (destra). I maschi si distinguono dalle femmine per la lunghezza e la morfologia delle prime antenne, evidenziate nelle cerchiature (Tatarazako et al., 2004). 
RIFERIMENTI BIBLIOGRAFICI Hobaek A e Larson P. 1990. Sex determination in Daphnia magna. Ecology 71: 2255-2268. Kleiven O.T., Larsson P., Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65, 197-206. Oda S., Tatarazako N, Watanabe H., Morita M., e Iguchi T. 2005. Production of male neonates in Daphnia magna (Cladocera, Crustacea) exposed to juvenile hormones and their analogs. Chemosphere 61:1168-1174. OECD, 2008. Validation report for an enhancement of OECD TG 211 Daphnia magna reproduction test. OECD Series on Testing and Assessment, Number 88. Organisation for Economic Co-operation and Development, Paris. Olmstead, A.W., LeBlanc, G.A., 2000. Effects of endocrine-active chemicals on the development characteristics of Daphnia magna. Environmental Toxicology and Chemistry 19:2107-2113. Tatarazako, N., Oda, S., Abe, R., Morita M. and Iguchi T., 2004. Development of a screening method for endocrine disruptors in crustaceans using Daphnia magna (Cladocera, Crustacea). Environmental Science 17, 439-449. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(18) |
Nella parte C, capitolo C.29, il paragrafo 66 è sostituito da:
|
|
(19) |
nella parte C sono aggiunti i seguenti capitoli: «C. 47 Prova di tossicità sui pesci nei primi stadi di vita INTRODUZIONE
PRINCIPIO DELLA PROVA
INFORMAZIONI SULLA SOSTANZA CHIMICA IN ESAME
VALIDITÀ DELLA PROVA
DESCRIZIONE DEL METODO Vasche di prova
Scelta delle specie
Pesci riproduttori
Manipolazione delle uova fecondate, degli embrioni e delle larve
Acqua
Soluzioni di prova
PROCEDURA Condizioni di esposizione Durata
Carico
Illuminazione e temperatura
Alimentazione
Concentrazioni di prova
Controlli
Frequenza delle misurazioni e delle determinazioni analitiche
Osservazioni 26. Stadio embrionale : verificare il più accuratamente possibile lo stadio embrionale all'inizio dell'esposizione alla sostanza chimica in esame. A tal fine si può utilizzare un campione rappresentativo di uova adeguatamente conservate e pulite. 27. Schiusa e sopravvivenza : l'osservazione della schiusa e della sopravvivenza si esegue almeno una volta al giorno e se ne registrano i numeri. Si contano e si scartano le uova che all'inizio della fase di sviluppo embrionale (il primo o il secondo giorno di prova, ad esempio) presentano muffe. Gli embrioni, le larve e gli individui giovani morti vanno rimossi non appena individuati in quanto possono decomporsi rapidamente ed essere fatti a pezzi dagli altri pesci. Nel rimuovere gli individui morti occorre fare estrema attenzione a non danneggiare le uova/larve adiacenti. I segni di decesso dipendono dalla specie e dallo stadio di vita. Ad esempio:
28. Aspetto anomalo : con una frequenza congrua alla durata della prova e alla natura dell'anomalia, si registra il numero di larve o pesci giovani che presentano anomalie morfologiche. La presenza di larve e pesci giovani anomali è un fenomeno naturale che, per alcune specie, può essere dell'ordine di svariati punti percentuali nel/i controllo/i. Se le malformazioni e i comportamenti anomali associati sono considerati gravi al punto da causare grandi e irreversibili sofferenze, l'organismo colpito può essere ritirato dalla prova. Gli animali in queste condizioni sono soppressi in modo incruento e considerati casi di mortalità nella successiva analisi dei dati. Uno sviluppo embrionale normale per la maggior parte delle specie raccomandate nel presente metodo di prova è descritto in letteratura (9) (10) (11) (12). 29. Comportamento anomalo : con una frequenza congrua alla durata della prova (ad esempio, una volta al giorno per le specie tropicali) si registrano le anomalie osservate, quali iperventilazione, nuoto scoordinato, immobilità o comportamento alimentare atipici. L'eventuale osservazione di tali effetti, per quanto difficili da quantificare, può facilitare l'interpretazione dei dati sulla mortalità. 30. Peso : alla fine della prova si pesano i pesci sopravvissuti, almeno per replica (registrando il numero di pesci presenti nella replica e il peso medio per pesce); è preferibile utilizzare il peso umido (previa asciugatura su carta assorbente), ma si può anche indicare nella relazione il peso secco (13). 31. Lunghezza : alla fine della prova si misura ogni pesce. Si raccomanda di misurarne la lunghezza totale, ma in caso di marcescenza della pinna caudale o erosione delle pinne è possibile utilizzare la lunghezza standard. Si applica lo stesso metodo per tutti i pesci utilizzati nella prova. I pesci possono essere misurati, ad esempio, con un calibro, una macchina fotografica digitale, o un micrometro oculare graduato. Le lunghezze minime tipiche sono indicate nell'appendice 2. DATI E RELAZIONE Trattamento dei risultati
Relazione sulla prova
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI
Appendice 2 CONDIZIONI SPERIMENTALI, DURATA E CRITERI DI SOPRAVVIVENZA PER LE SPECIE RACCOMANDATE
Appendice 3 ORIENTAMENTI PER L'ALIMENTAZIONE E LA MANIPOLAZIONE DEI PESCI RIPRODUTTORI E DEI PESCI UTILIZZATI NELLE PROVE, DELLE SPECIE RACCOMANDATE
Appendice 4 ALCUNE CARATTERISTICHE CHIMICHE DI UN'ACQUA DI DILUIZIONE ACCETTABILE
Appendice 5 ORIENTAMENTI PER L'ANALISI STATISTICA CON CUI DETERMINARE LA NOEC Orientamenti generali Ogni replica costituisce un'unità di analisi. Pertanto, nel caso delle misurazioni continue, come le dimensioni, si calcola la media o la mediana di ogni replica e i valori ottenuti costituiscono i dati da analizzare. Occorre dimostrare la potenza dei test utilizzati, di preferenza facendo riferimento a una base adeguata di dati storici del laboratorio. Per ogni endpoint si indica il test statistico da utilizzare e la variazione (minima) delle dimensioni dei pesci rilevabile con una potenza statistica del 75-80 %. Le basi di dati disponibili al momento dell'elaborazione del presente metodo di prova definiscono la potenza ottenibile con i metodi statistici raccomandati. Il laboratorio deve dimostrarsi in grado di soddisfare il requisito della potenza effettuando un'analisi della potenza o dimostrando che il coefficiente di variazione (CV) di ogni risposta non supera il 90o percentile dei coefficienti di variazione utilizzati per elaborare il metodo di prova, indicati nella tabella 1. Se si dispone unicamente delle medie e delle mediane delle repliche, il coefficiente di variazione intra repliche può essere ignorato. Tabella 1 90° percentile dei coefficienti di variazione per le specie d'acqua dolce selezionate
Per quasi tutti i test statistici utilizzati per valutare gli studi tossicologici condotti in laboratorio, i confronti di interesse riguardano i gruppi esposti rispetto al controllo. Non è, pertanto, giustificato esigere un test F ANOVA statisticamente significativo prima di utilizzare il test di Dunnett o il test di Williams, né un test statisticamente significativo di Kruskal-Wallis prima di realizzare il test di Jonckheere-Terpstra, di Mann-Whitney, o di Dunn (Hochberg e Tamhane 1987, Hsu 1996, Dunnett 1955, 1964, Williams 1971, 1972, 1975, 1977, Robertson et al. 1988, Jonckheere 1954, Dunn 1964). Il test di Dunnett consente confronti multipli e l'uso di un test F come filtro ne pregiudica i tassi di falsi positivi e falsi negativi. Analogamente, i test regressivi di Williams e di Jonckheere-Terpstra con un livello di significatività di 0,05 in ogni fase mantengono un tasso complessivo di falsi positivi del 5 % e questo tasso, così come la potenza dei test, sono pregiudicati dall'utilizzo del test F o del test di Kruskal-Wallis come filtro. Il test di Mann-Whitney e il test di Dunn devono essere corretti ai fini dei confronti multipli, e si consiglia la correzione di Bonferroni-Holm. In OCSE (2006) si trova un'ampia bibliografia e un'analisi approfondita della maggior parte delle raccomandazioni sui test di ipotesi e sulla verifica dei presupposti sottostanti. Trattamento dei controlli quando si utilizza un solvente Se si utilizza un solvente è necessario prevedere un controllo dell'acqua di diluizione e un controllo del solvente. Si confronta la risposta ottenuta per il primo con la risposta ottenuta per il secondo e, se non presentano differenze statisticamente significative, i due controlli vengono combinati nell'analisi statistica. In caso contrario, si utilizza il controllo del solvente per determinare la NOEC o per stimare l'ECx, ma non il controllo dell'acqua di diluizione. Si veda la restrizione enunciata nei criteri di validità (paragrafo 7). Per quanto riguarda la lunghezza, il peso, la proporzione di uova schiuse, larve morte o larve anomale e il primo o l'ultimo giorno della schiusa o dell'affioramento, si utilizza un test T o un test di Mann-Whitney per confrontare il controllo dell'acqua di diluizione e il controllo del solvente al livello di significatività dello 0,05, ignorando tutti i gruppi esposti. I risultati di questi test devono figurare nella relazione. Misurazioni morfologiche (lunghezza e peso) La lunghezza e il peso dei pesci possono seguire una distribuzione normale o log normale. In entrambi i casi i valori medi per replica tendono a una distribuzione normale, come previsto dal teorema del limite centrale e corroborato dai risultati di oltre un centinaio di studi condotti nei primi stadi di vita di tre specie di acqua dolce. In alternativa, se i dati o le basi di dati storici indicano una distribuzione log normale dei valori relativi alle dimensioni dei pesci, si può calcolare il logaritmo della media dei valori corrispondenti ai pesci che formano ogni replica e i dati da analizzare saranno gli antilogaritmi di tali logaritmi di medie per replica. Si valuta la compatibilità dei dati con una distribuzione normale e con una varianza omogenea. A tal fine, si utilizzano i residui di un modello ANOVA in cui la concentrazione sia l'unica variabile esplicativa. Si può ricorrere a rappresentazioni sotto forma di grafico a dispersione, istogrammi e grafici a ramo e foglia; oppure si può utilizzare un test formale, come il test di Shapiro-Wilk o il test di Anderson-Darling. La compatibilità con una varianza omogenea può essere valutata tramite un esame visivo dello stesso grafico a dispersione o, formalmente, per mezzo del test di Levene. La verifica della normalità e dell'omogeneità della varianza è necessaria solo per i test parametrici (per esempio, Williams o Dunnett). Occorre prestare attenzione a eventuali valori anomali e alle conseguenti implicazioni sull'analisi. È possibile ricorrere al test dei valori anomali di Tukey e all'esame visivo dei summenzionati grafici dei residui. Si rammenta che l'osservazione corrisponde alla replica intera, pertanto l'esclusione di valori anomali dall'analisi necessita attenta ponderazione. I test statistici che si basano sulle caratteristiche del disegno sperimentale e sulle aspettative biologiche sono test di tendenza regressivi, come il test di Williams e il test di Jonckheere-Terpstra. Questi test presuppongono una relazione concentrazione-risposta monotona, per cui occorre valutare la compatibilità dei dati con tale presupposto; la valutazione può essere effettuata visivamente osservando il grafico a dispersione delle medie per replica in funzione della concentrazione di prova. Sarà utile sovrapporre al grafico a dispersione un grafico lineare a tratti che colleghi le concentrazioni medie ponderate per la dimensione del campione della replica; un diagramma lineare che si discosta molto da un tracciato monotono può indicare la necessità di applicare test non parametrici. In alternativa si possono utilizzare test formali. Un test formale semplice consiste nel calcolare i contrasti lineari e quadratici delle medie della concentrazione: se il contrasto quadratico è significativo e il contrasto lineare non lo è, può esserci un problema di monotonia, da rivalutare sulla base dei suddetti grafici; se la normalità e l'omogeneità della varianza pongono dubbi, i suddetti contrasti possono essere costruiti a partire dalla trasformazione dei dati in ranghi. Si può ricorrere a metodi alternativi, come il test di monotonia di Bartholomew, che però aggiungono complessità. Figura 2 Diagramma per la determinazione della NOEC in base alle misurazioni morfologiche (lunghezza e peso) 
A meno che i dati non siano compatibili con i requisiti di questi test, si determina la NOEC per applicazione regressiva del test di Williams o del test di Jonckheere-Terpstra. In OCSE (2006) si trovano informazioni dettagliate sull'applicazione di questi metodi. Se i dati non sono compatibili con i requisiti di un test di tendenza regressivo, si può utilizzare il test di Dunnett o il test di Tamhane-Dunnett (T3), entrambi i quali permettono confronti multipli. Questi test presuppongono una distribuzione normale e, nel caso del test di Dunnett, omogeneità della varianza. Se queste condizioni non sono soddisfatte, si può ricorrere al test non parametrico di Dunn. OCSE (2006) fornisce maggiori informazioni su tutti questi test. La figura 2 illustra schematicamente come scegliere il test adatto. Schiusa delle uova e sopravvivenza delle larve I dati da analizzare sono la proporzione di uova che si schiudono o di larve che sopravvivono in ogni replica. Si valutano tali proporzioni in termini di varianza extrabinomiale, che è frequente ma non universale in questi due casi. Il diagramma nella figura 3 serve a orientare la scelta del test adatto, a corredo del testo esplicativo. Due tipi di test sono normalmente utilizzati: il test C (α) di Tarone (Tarone, 1979), e i test Chi-quadrato, ciascuno applicato separatamente ad ogni concentrazione di prova. Se si osserva una varianza extrabinomiale, anche solo in una concentrazione di prova, occorre utilizzare metodi che la contemplino. Formula 1 test C(α) di Tarone (Tarone 1979)
dove ̂ è la proporzione media per una data concentrazione, m è il numero delle repliche (vasche), nj è il numero degli individui presenti nella replica j, e xj è il numero degli individui che rispondono in detta replica, ossia i casi di non schiusa o di decesso. Questo test si applica separatamente a ciascuna concentrazione. Può essere considerato un test Chi-quadrato corretto, ma nelle simulazioni di bassa potenza si è dimostrato più potente di un Chi-quadrato. Figura 3 Diagramma per determinare la NOEC in base alla schiusa delle uova e dalla mortalità delle larve 
Se non vi sono indizi significativi di varianza extrabinomiale, si può utilizzare il test regressivo di Cochran-Armitage; poiché questo test ignora le repliche, si raccomanda che, nel caso vi siano indizi significativi di varianza extrabinomiale, si applichi la correzione di Rao-Scott (RSCA), che tiene conto delle repliche, delle loro dimensioni e della varianza extrabinomiale. Altre possibilità sono i test regressivi di Williams e di Jonckheere-Terpstra, così come il test di Dunnett, illustrati nella sezione relativa alle misurazioni morfologiche. Questi test possono essere applicati indipendentemente dall'esistenza di varianza extrabinomiale, ma la loro potenza è minore (Agresti 2002, Morgan 1992, Rao e Scott 1992, 1999, Fung et al. 1994, 1996). Primo o ultimo giorno di schiusa o affioramento La risposta da analizzare è un numero intero, che corrisponde al giorno della prova in cui l'osservazione indicata è effettuata in una determinata replica. L'intervallo di valori è generalmente molto limitato e sono frequenti proporzioni elevate di valori collegati, ad esempio, il primo giorno di schiusa è identico in tutte le repliche del controllo e, eventualmente, per la concentrazione più bassa o per le due concentrazioni più basse. I test parametrici, come quelli di Williams e Dunnett, non sono adatti all'analisi di tali dati. A meno che non vi siano indizi di netta non monotonia, il test regressivo di Jonckheere-Terpstra è molto potente nel rilevare gli effetti delle sostanze chimiche. In caso contrario si può utilizzare il test di Dunn. Larve con anomalie La risposta da analizzare è il numero di larve nelle quali è stata rilevata un'anomalia di qualsiasi genere. Questa risposta ha spesso un'incidenza bassa e presenta gli stessi problemi del primo giorno di schiusa, così come, talvolta, una relazione concentrazione-risposta talvolta erratica. Se dai dati si evince una funzione approssimativamente monotona della concentrazione, il test regressivo di Jonckheere-Terpstra è potente nel rilevamento degli effetti; in caso contrario si può utilizzare il test di Dunn. BIBLIOGRAFIA Agresti, A. (2002); Categorical Data Analysis, second edition, Wiley, Hoboken. Dunnett C. W. (1955); A multiple comparison procedure for comparing several treatments with a control, J. American Statistical Association 50, 1096-1121. Dunn O. J. (1964 ); Multiple Comparisons Using Rank Sums, Technometrics 6, 241-252. Dunnett C. W. (1964); New tables for multiple comparisons with a control, Biometrics 20, 482-491. Fung, K.Y., D. Krewski, J.N.K. Rao, A.J. Scott (1994); Tests for Trend in Developmental Toxicity Experiments with Correlated Binary Data, Risk Analysis 14, 639-648. Fung, K.Y, D. Krewski, R.T. Smythe (1996); A comparison of tests for trend with historical controls in carcinogen bioassay, Canadian Journal of Statistics 24, 431-454. Hochberg, Y. and A. C. Tamhane (1987); Multiple Comparison Procedures, Wiley, New York. Hsu, J.C. (1996); Multiple Comparisons: Theory and Methods; Chapman and Hall/CRC Press, Boca Raton. Jonckheere A. R. (1954); A distribution-free k-sample test against ordered alternatives, Biometrika 41, 133. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. OECD (2006). Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series on Testing and Assessment, No. 54. Organisation for Economic Co-operation and Development, OECD, Paris.. Rao J.N.K. and Scott A.J. (1992) — A simple method for the analysis of clustered binary data, Biometrics 48, 577-585. Rao J.N.K. and Scott A.J. (1999) — A simple method for analyzing overdispersion in clustered Poisson data, Statistics in Medicine 18, 1373-1385. Robertson, T., Wright F.T. and Dykstra R.L. (1988); Order restricted statistical inference, Wiley. Tarone, R.E. (1979); Testing the goodness of fit of the Binomial distribution, Biometrika 66, 585-590. Williams D.A. (1971); A test for differences between treatment means when several dose levels are compared with a zero dose control, Biometrics 27, 103-117. Williams D.A. (1972); The comparison of several dose levels with a zero dose control, Biometrics 28, 519-531. Williams D. A. (1975); The Analysis of Binary Responses from Toxicological Experiments Involving Reproduction and Teratotlogy, Biometrics 31, 949-952. Williams D.A. (1977); Some inference procedures for monotonically ordered normal means, Biometrika 64, 9-14. Appendice 6 ORIENTAMENTI PER L'ANALISI STATISTICA DELLE STIME PER REGRESSIONE Orientamenti generali Le osservazioni alle quali adattare il modello sono le medie (della lunghezza e del peso) per replica o le proporzioni (della schiusa delle uova e della mortalità larvale) per replica (OCSE 2006). Si raccomanda, in generale, una regressione ponderata, utilizzando come fattore di ponderazione le dimensioni del campione della replica. Sono possibili altri metodi di ponderazione, ad esempio utilizzando come fattore di ponderazione la risposta media prevista o una combinazione di questa e delle dimensioni del campione della replica. Non si raccomanda la ponderazione per l'inverso della varianza dei campioni a ciascuna concentrazione (Bunke et al. 1999, Seber e Wild, 2003, Motulsky e Christopoulos 2004, Huet et al. 2003). Qualsiasi trasformazione delle risposte prima dell'analisi deve mantenere l'indipendenza delle osservazioni; inoltre l'ECx e i limiti del relativo intervallo di confidenza devono essere espressi nelle unità di misura originali e non in unità trasformate. Per esempio, una variazione del 20 % del logaritmo della lunghezza non equivale a una variazione del 20 % della lunghezza (Lyles et.al 2008, Draper e Smith 1999). Il diagramma della figura 4 schematizza il procedimento per la stima dell'ECx, a corredo della spiegazione dettagliata fornita nel testo. Figura 4 Diagramma per la stima dell'ECx in funzione delle medie della lunghezza, del peso o della proporzione di schiusa delle uova o mortalità larvale per replica (per i dettagli si veda il testo) 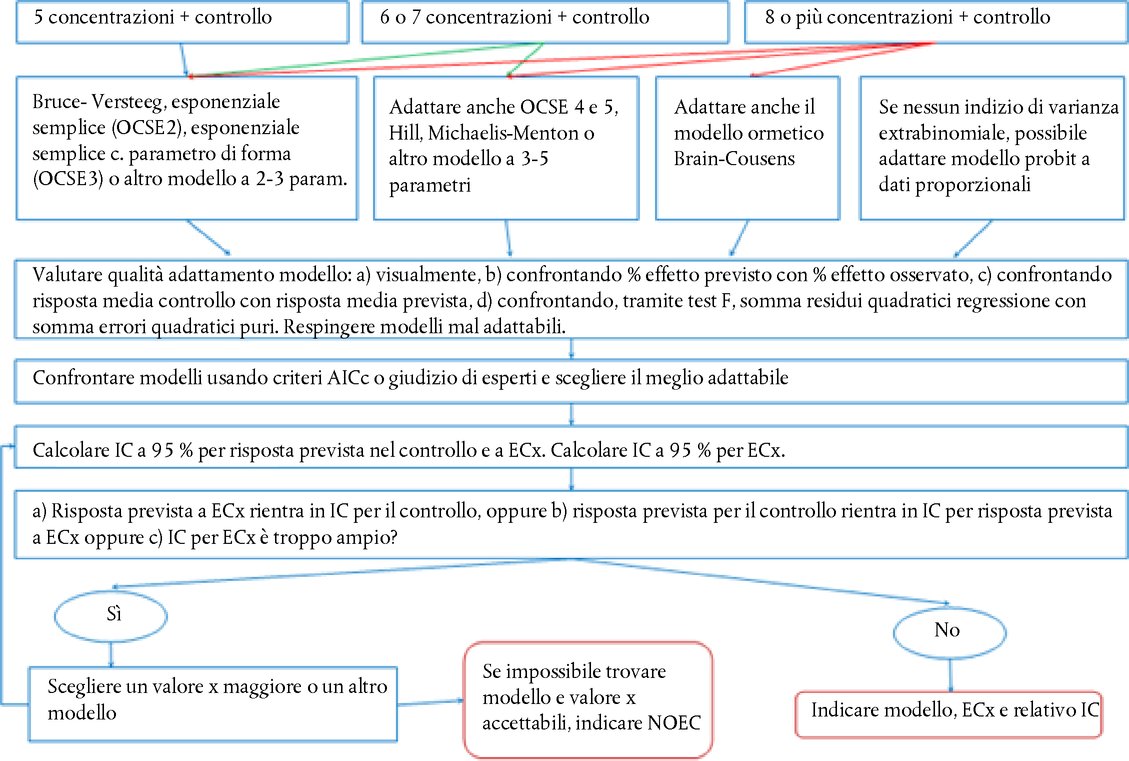
Considerazioni sulla schiusa delle uova e sulla mortalità larvale Per la schiusa delle uova e la mortalità larvale è in genere preferibile adattare un modello decrescente, a meno che non si intenda adattare un modello probit nel seguente modo: si adatta il modello alla proporzione di uova non schiuse o di larve morte; la ragione per cui si procede così è che l'ECx si riferisce alla concentrazione alla quale si verifica una variazione dell'x % rispetto alla risposta media del controllo. Se il 5 % delle uova del controllo non si schiudono e il modello traduce la mancata schiusa, l'EC20 corrisponderà alla concentrazione alla quale si verifica una variazione del 20 % della proporzione del 5 % di uova non schiuse nel controllo, ossia una variazione pari a 0,2 x 0,05 = 0,01; si tratta di un aumento di 1 punto percentuale, che innalza al 6 % la proporzione di uova non schiuse. Una variazione così piccola non può essere stimata in modo significativo sulla base dei dati disponibili e non ha rilevanza biologica. Se invece il modello traduce la proporzione di uova schiuse, la proporzione corrispondente al controllo sarà, in questo esempio, del 95 % e una riduzione del 20 % rispetto alla media del controllo equivarrà a una variazione di 0,95 x 0,2 = 0,18, ossia una variazione della riuscita della schiusa dal 95 % al 77 % (= 95-18); la concentrazione che produce questo effetto può così essere stimata e presumibilmente è di maggiore interesse. Il problema non si pone con le misurazioni morfologiche, sebbene gli effetti negativi in tal caso corrispondano in genere a una diminuzione di peso o lunghezza. Modelli applicabili alla morfologia (lunghezza o peso) e alla riuscita della schiusa o alla sopravvivenza larvale. Ad eccezione del modello ormetico Brain-Cousens, tutti i seguenti modelli sono descritti e raccomandati in OCSE (2006). I modelli OCSE da 2 a 5 sono discussi per esperienze di ecotossicità anche in Slob (2002). Esistono, naturalmente, molti altri modelli che potrebbero essere utili: Bunke, et al. (1999) ne enumera una serie che non sono qui citati e abbondano i riferimenti ad altri. I modelli elencati di seguito, ampiamente utilizzati, sono considerati particolarmente adatti per esperienze di ecotossicità. Con 5 concentrazioni della sostanza di prova più un controllo
Con 6 o più concentrazioni della sostanza di prova più un controllo
In presenza di ormesi comprovata visivamente (improbabile in caso di schiusa delle uova o di sopravvivenza larvale, ma talvolta registrata nelle osservazioni morfologiche)
Modelli alternativi per la mancata schiusa delle uova e la mortalità larvale
Qualità dell'adattamento di un modello
Confronto dei modelli
Qualità della stima dell'ECx L'intervallo di confidenza (IC) dell'ECx non deve essere troppo ampio. Occorre vagliare statisticamente l'ampiezza massima che può avere l'intervallo di confidenza affinché l'ECx sia utile. Dalle simulazioni dell'adattamento dei modelli di regressione ai dati della schiusa delle uova e ai dati morfologici emerge che l'ampiezza di circa il 75 % degli intervalli di confidenza dell'ECx (x = 10, 20 o 30) non include più di due concentrazioni di prova. Questo dato fornisce un orientamento generale di quanto è accettabile e realizzabile. Molti autori sostengono che è necessario riportare nella relazione gli intervalli di confidenza di tutti i parametri del modello e che parametri con intervalli ampi sono indice d'inaccettabilità del modello (Ott e Longnecker 2008, Alvord e Rossio 1993, Motulsky e Christopoulos 2004, Lyles et al. 2008, Seber and Wild 2003, Bunke et al. 1999, Environment Canada 2005). L'intervallo di confidenza corrispondente all'ECx (o a qualsiasi altro parametro del modello) non deve includere il valore zero (Motulsky and Christopoulos 2004). Si tratta dell'equivalente, nella regressione, della differenza minima con significatività spesso citata negli approcci sperimentali basati sulla verifica delle ipotesi (ad esempio, Wang et al 2000). Corrisponde anche al fatto che l'intervallo di confidenza delle risposte medie alla LOEC non contiene la media del controllo. È importante riflettere sulla plausibilità scientifica delle stime dei parametri, ad esempio, se l'intervallo di confidenza corrispondente a y0 è ±20 % non sarà plausibile alcuna stima dell'EC10. Se il modello prevede un effetto del 20 % alla concentrazione C e l'effetto massimo osservato a questa concentrazione e a concentrazioni inferiori è 10 %, l'EC20 non è plausibile (Motulsky e Christopoulos 2004, Wang et al. 2000, Environment Canada 2005). L'ECx non è estrapolabile al di fuori dell'intervallo delle concentrazioni positive (Draper and Smith 1999; OCSE, 2006). Ad esempio, un orientamento generale potrebbe essere che l'ECx non sia inferiore di oltre il 25 % circa alla concentrazione più bassa saggiata, né superiore nella stessa misura a quella più alta. BIBLIOGRAFIA Alvord, W.G., Rossio, J.L. (1993); Determining confidence limits for drug potency in immunoassay, Journal of Immunological Methods 157, 155-163. Brain P. and Cousens R. (1989); An equation to describe dose responses where there is stimulation of growth at low doses. Weed res. 29: 93-96. Bunke, O., Droge, B. and Polzehl, J. (1999). Model selection, transformations and variance estimation in nonlinear regression. Statistics 33, 197-240. Collett, D. (2002); Modelling Binary Data, second edition, Chapman and Hall, London. Collett, D. (2003); Modelling Survival Data in Medical Research, second edition, Chapman and Hall, London. Draper, N.R. and Smith, H. (1999); Applied Regression Analysis, third edition. New York: John Wiley & Sons. Environment Canada (2005); Guidance Document on Statistical Methods for Environmental Toxicity Tests, Report EPS 1/RM/46 Huet, S., A. Bouvier, M.-A. Poursat, E. Jolivet (2003); Statistical Tools for Nonlinear Regression: A Practical Guide with S-PLUS and R Examples, Springer Series in Statistics, New York. Lyles, R. H., C. Poindexter, A. Evans, M. Brown, and C.R. Cooper (2008); Nonlinear Model-Based Estimates of IC50 for Studies Involving Continuous Therapeutic Dose-Response Data, Contemp Clin Trials. 2008 November; 29(6): 878–886. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. Motulsky, H., A. Christopoulos (2004); Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, USA. O'Hara Hines, R. J. and J. F. Lawless (1993); Modelling Overdispersion in Toxicological Mortality Data Grouped over Time, Biometrics Vol. 49, pp. 107-121 OECD (2006); Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Series Testing and Assessment, No. 54, Organisation for Economic Co-operation and Development, OECD, Paris. Ott, R.L., M.T. Longnecker, An Introduction to Statistical Methods and Data Analysis, sixth edition, 2008, Brooks-Cole, Belmont, CA Ratkowsky, D.A. (1993); Principles of nonlinear regression, Journal of Industrial Microbiology 12, 195-199. Seber, G.A.F., C.J. Wild, Nonlinear Regression, Wiley, 2003 Slob W. (2002); Dose-response modelling of continuous endpoints. Toxicol. Sci., 66, 298-312 Wang, Q., D.L. Denton, and R. Shukla (2000); Applications and Statistical Properties Of Minimum Significant Difference-Based Criterion Testing In a Toxicity Testing Program, Environmental Toxicology and Chemistry, Vol. 19, pp. 113–117, 2000. C.48 Saggio di tossicità a breve termine sulla riproduzione di pesci INTRODUZIONE
CONSIDERAZIONI INIZIALI E LIMITI
PRINCIPIO DELLA PROVA
CRITERI DI ACCETTAZIONE DELLA PROVA
DESCRIZIONE DEL METODO Apparecchiature
Acqua
Soluzioni di prova
Mantenimento dei pesci
Pre-esposizione e selezione dei pesci
DISEGNO SPERIMENTALE
Selezione delle concentrazioni sperimentali
PROCEDURA Selezione e pesatura dei pesci da sottoporre alla prova
Condizioni di esposizione Durata
Alimentazione
Illuminazione e temperatura
Frequenza delle determinazioni e delle misurazioni analitiche
Osservazioni
Sopravvivenza
Comportamento e aspetto
Fecondità
Soppressione incruenta
Osservazione dei caratteri sessuali secondari
Vitellogenina (VTG)
Esame istopatologico delle gonadi
DATI E RELAZIONE Valutazione delle risposte dei biomarcatori mediante l'analisi della varianza (ANOVA)
Elaborazione di relazioni sui risultati della prova
ORIENTAMENTI PER L'INTERPRETAZIONE E L'ACCETTAZIONE DEI RISULTATI
BIBLIOGRAFIA
Appendice 1 ABBREVIAZIONI E DEFINIZIONI Sostanza chimica : sostanza o miscela CV : coefficiente di variazione ELISA : prova di immunoassorbimento enzimatico HPG (Hypothalamic-pituitary-gonadal) Axis : asse ipotalamo-ipofisi-gonadi Tasso di carico : peso a umido dei pesci per volume di acqua MTC : concentrazione massima tollerata, che rappresenta circa il 10 % della LC50 Densità della popolazione : numero di pesci per volume di acqua Sostanza chimica in esame : qualsiasi sostanza o miscela testata applicando il presente metodo di prova. VTG : la vitellogenina è una lipo-glico-fosfo-proteina precursore delle proteine del tuorlo normalmente prodotta dalle femmine sessualmente attive di tutte le specie ovipare Appendice 2 CONDIZIONI SPERIMENTALI PER LO SCREENING DEL SISTEMA ENDOCRINO DEI PESCI
Appendice 3 ALCUNE CARATTERISTICHE CHIMICHE DI UN'ACQUA DI DILUIZIONE ACCETTABILE
Appendice 4A SUBSTRATO DI RIPRODUZIONE PER DANIO RERIO (DANIO ZEBRATO) Piattaforma di riproduzione : piatto per strumenti in vetro, ad esempio di dimensioni 22 × 15 × 5,5 (larghezza × L × h), coperto da una griglia amovibile di acciaio inossidabile (con maglie di 2 mm di larghezza). La griglia di copertura è posta ad un'altezza inferiore al bordo del piatto. 
Il substrato di riproduzione è fissato sulla griglia, creando una struttura in cui i pesci possono muoversi. Allo scopo sono adatte, ad esempio, piante artificiali per acquario di plastica verde (NB: bisogna tener presente il possibile adsorbimento della sostanza chimica in esame sulla materia plastica). La plastica è lisciviata in un volume sufficiente di acqua calda e per un tempo sufficiente affinché nessuna sostanza sia rilasciata nell'acqua di prova. Se si utilizzano materiali in vetro, bisogna assicurare che i pesci non si feriscano o siano impediti nei movimenti durante le attività più vigorose. La distanza tra il piatto e la parete della vasca deve essere di almeno 3 cm affinché la riproduzione non avvenga al di fuori del piatto. Le uova depositate sul piatto passano attraverso la griglia e possono essere prelevate 45-60 minuti dopo l'inizio dell'illuminazione. Le uova traslucide non aderiscono e possono facilmente essere contate alla luce trasversale. In presenza di cinque femmine per vasca, il numero di uova deposte può essere considerato basso se inferiore o pari a 20 al giorno, medio se compreso tra 20 e 100, e alto se superiore a 100. Il piatto di riproduzione è rimosso, le uova raccolte e la piattaforma di riproduzione reintrodotta nella vasca sperimentale, il più tardi possibile in serata o di prima mattina. La reintroduzione della piattaforma deve avvenire entro un'ora al massimo, perché altrimenti il segnale del substrato di riproduzione può indurre singoli accoppiamenti e una riproduzione al di fuori dei tempi controllati. Se la situazione richiede una successiva introduzione della piattaforma di riproduzione, occorre attendere almeno nove ore dopo l'inizio dell'illuminazione. A quest'ora tarda della giornata, la deposizione delle uova non è più indotta. Appendice 4B SUBSTRATO DI RIPRODUZIONE PER LA SPECIE PIMEPHALES PROMELAS Due o tre piastre e piatti di riproduzione combinati in plastica/ceramica/vetro o acciaio inossidabile sono collocati in ciascuna vasca sperimentale (ad esempio un pezzo di grondaia di forma semicircolare grigia di 80 mm di lunghezza posto su una piastrella di metallo dotata di bordi rialzati, lunga 130 mm) (v. foto). È dimostrato che le piastrelle in PVC o in ceramica opportunamente trattate possono costituire idonei substrati di riproduzione (Thorpe et al, 2007). Si raccomanda l'uso di piastrelle abrase per migliorarne l'aderenza. Il piatto è inoltre munito di uno schermo di protezione per impedire che i pesci abbiano accesso alle uova depositate sul fondo a meno che sia stato dimostrato che le uova aderiscono efficacemente al substrato di riproduzione utilizzato. 
La base è progettata per contenere tutte le uova che non aderiscono alla superficie della piastrella e che ricadrebbero quindi sul fondo della vasca (o le uova depositate direttamente sulla base di plastica piatta). Tutti i substrati di riproduzione sono lisciviati per almeno 12 ore in acqua di diluizione prima dell'uso. Thorpe KL, Benstead R, Hutchinson TH, Tyler CR, 2007. An optimised experimental test procedure for measuring chemical effects on reproduction in the fathead minnow, Pimephales promelas. Aquatic Toxicology, 81, 90–98. Appendice 5A VALUTAZIONE DEI CARATTERI SESSUALI SECONDARI DI PIMEPHALES PROMELAS AI FINI DELL'INDIVIDUAZIONE DI DETERMINATE SOSTANZE ATTIVE A LIVELLO ENDOCRINO Sintesi Negli esemplari adulti della specie Pimephales promelas, le caratteristiche fisiche che possono assumere rilevanza ai fini della sperimentazione sugli interferenti endocrini sono le seguenti: colore della livrea (chiaro/scuro), motivi di colorazione (presenza o assenza di bande verticali), forma del corpo (forma della testa e della regione toracica, distensione addominale) e caratteri sessuali secondari specifici della specie (numero e dimensioni dei tubercoli nuziali, dimensioni del cuscinetto dorsale e dell'ovopositore). I tubercoli nuziali sono situati sulla testa (cuscinetto dorsale) nei maschi adulti riproduttori, e sono generalmente disposti in modo bilaterale e simmetrico (Jensen et al. 2001). Le femmine delle vasche di controllo e i giovani esemplari maschi e femmine non mostrano alcuno sviluppo di tubercoli (Jensen et al. 2001). È possibile individuare fino a otto singoli tubercoli attorno agli occhi e tra le narici degli esemplari maschi. I tubercoli più grandi e numerosi formano due linee parallele situate immediatamente al di sotto delle narici e sopra la bocca. In molti pesci si riscontrano gruppi di tubercoli sotto la mascella inferiore; vicino alla bocca se ne trova generalmente solo una coppia mentre la parte ventrale può comprendere fino a quattro tubercoli. Il numero di tubercoli raramente supera i 30 (range di 18-28; Jensen et al. 2001). I tubercoli più numerosi presentano un'unica struttura di forma tondeggiante, di altezza approssimativamente pari al raggio. Nella maggior parte dei maschi riproduttori, alcuni tubercoli sono talmente estesi e prominenti che è impossibile distinguerli gli uni dagli altri. Alcuni tipi di interferenti endocrini possono provocare l'insorgere abnorme di caratteri sessuali secondari nel sesso opposto; ad esempio, gli antagonisti dei recettori degli androgeni, quali il 17α-metiltestosterone o il 17β-trenbolone, possono provocare l'insorgere di tubercoli nuziali nei ciprinidi femmina (Smith 1974; Ankley et al. 2001; 2003), mentre gli antagonisti dei ricettori degli estrogeni possono ridurre il numero o la dimensione dei tubercoli nuzionali nei maschi (Miles-Richardson et al. 1999; Harries et al. 2000). Segue una descrizione della caratterizzazione dei tubercoli nuziali in Pimephales promelas (ciprinidi), basata sul protocollo sperimentale utilizzato dal laboratorio dell'Agenzia per la protezione dell'ambiente degli Stati Uniti (Duluth, MN). I prodotti e/o le attrezzature specifiche possono essere sostituiti da materiali comparabili disponibili. L'utilizzazione di una lente di ingrandimento munita di illuminazione o microscopio binoculare da dissezione con illuminazione (3x) consente un'osservazione ottimale. Si osserverà il pesce in posizione dorsale, con la parte anteriore davanti (testa verso l'osservatore).
Conteggio e classificazione dei tubercoli Sono state individuate sei aree specifiche per la valutazione della presenza e dello sviluppo di tubercoli in Pimephales promelas (ciprinidi) adulti. È stato creato un modello per definire la localizzazione dei tubercoli e la quantità di tubercoli presenti (cfr. parte finale della presente appendice). Va registrato il numero di tubercoli che possono essere classificati come segue in funzione delle loro dimensioni: 0-nullo; 1-presente; 2-esteso e 3-prominente per ciascun organismo (figura 1). Classe 0-: assenza di qualsiasi tubercolo. Classe 1- tubercolo presente: un tubercolo che ha un unico punto di altezza quasi uguale al raggio. Classe 2-tubercolo esteso: individuato dal tessuto somigliante ad un asterisco, di solito con un'ampia base radiale con solchi che partono dal centro. L'altezza dei tubercoli è spesso più frastagliata ma può talvolta essere leggermente arrotondata. Classe 3-tubercolo prominente: generalmente abbastanza ampio, di forma arrotondata, con una struttura meno ben definita. Questi tubercoli si agglomerano talvolta fino a formare un'unica massa lungo una zona o più zone (B, C e D, si veda la descrizione qui sotto). Il colore e la forma sono simili alla classe 2, ma sono talvolta piuttosto indeterminati. Questo sistema di classificazione permette generalmente di ottenere un risultato globale di <50 in un maschio normale di controllo che presenta un numero di tubercoli compreso tra 18 e 20 (Jensen et al. 2001). Figura 1 
Alcuni pesci possono presentare più tubercoli di quelli risultanti dalle caselle del modello per una particolare zona. In tal caso, codici supplementari possono essere indicati all'interno, a destra o a sinistra della casella. Il modello non deve pertanto necessariamente presentarsi simmetrico. Un'altra tecnica per indicare i tubercoli in coppia o riuniti verticalmente lungo il piano orizzontale della bocca potrebbe consistere nell'indicare codici a 2 cifre in un'unica casella. Aree di localizzazione:
BIBLIOGRAFIA
Appendice 5B VALUTAZIONE DEI CARATTERI SESSUALI SECONDARI NEL MEDAKA AL FINE DI INVIDIDUARE ALCUNE SOSTANZE CHIMICHE CON ATTIVITÀ ENDOCRINA Di seguito è descritta la misurazione dei processi papillari (*10), che costituiscono i caratteri sessuali secondari nel medaka (Oryzias latipes).
Misurazione
Figura 1. Differenze sessuali in base a forma e dimensioni della pinna anale. A, maschio; B, femmina. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Figura 2. A, Processi che avvengono sui segmenti congiunti del raggio della pinna anale. J.P. (joint plate), segmento congiunto; A.S., spazio assiale; P., processo. B, estremità distale della pinna anale. Gli attinotrichi (Act.) si trovano sulle punte. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Figura 3. Fotografia del corpo del pesce che mostra la linea di taglio quando la gonade è fissata in una soluzione diversa dalla formalina tamponata al 10 %. In tal caso, il resto del corpo è tagliato fra la regione anteriore della pinna anale e l'ano mediante rasoio (linea rossa). La testa è riposta nella soluzione fissativa per conservare la gonade, e la coda in formalina tamponata al 10 %. 
Appendice 6 PROCEDURE RACCOMANDATE PER I PRELIEVI EFFETTUATI AI FINI DELL'ANALISI DELLA VTG Si avrà cura di evitare la contaminazione incrociata tra i campioni di VTG dei maschi e delle femmine. Procedura 1A: Ciprinidi della specie Pimephales promelas, prelievo di sangue dall'arteria/vena caudale Dopo anestesia, il peduncolo caudale è parzialmente reciso con un bisturi ed è prelevato un campione di sangue dall'arteria/vena caudale mediante un tubo capillare per microematocrito eparinato. Dopo il prelievo di sangue, il plasma viene rapidamente separato tramite centrifugazione a temperatura ambiente per 3 minuti a 15 000 g (o a una temperatura di 4 °C per 10 minuti a 15 000 g). A seguito della centrifugazione, si può determinare la percentuale di ematocrito. Il plasma viene successivamente ritirato dal tubo microematocrito e immagazzinato in un tubo da centrifuga con 0,13 unità di aprotinina (un inibitore di proteasi) a – 80 °C fino alla misurazione della VTG. Secondo la dimensione del Ciprinide (che dipende dal sesso), i volumi di plasma prelevabili sono generalmente di 5-60 ml per individuo (Jensen et al. 2001). Procedura 1 B: Ciprinidi della specie Pimephales promelas, prelievo di sangue mediante puntura cardiaca In alternativa, è possibile effettuare un prelievo di sangue con puntura cardiaca mediante siringa eparinizzata (1 000 unità di eparina per ml). Il sangue è trasferito in provette Eppendorf (mantenute nel ghiaccio) e quindi centrifugato (5 minuti a 7 000 g a temperatura ambiente). Il plasma va trasferito in provette Eppendorf pulite (in varie porzioni se il volume di plasma lo consente) e successivamente congelato rapidamente a – 80 °C fino all'analisi (Panter et al., 1998). Procedura 2A: Escissione del fegato in Oryzias latipes (medaka) Rimozione dei pesci oggetto della prova dalla vasca sperimentale
Escissione del fegato
Dopo l'escissione del fegato, la carcassa è pronta per l'esame istologico delle gonadi e la misurazione dei caratteri sessuali secondari. Campioni Conservare i campioni di fegato prelevati dai pesci oggetto di prova ad una temperatura di ≤ – 70 °C se non utilizzati per il pre-trattamento subito dopo l'escissione. Figura 1 Praticare con le forbici un taglio nella parte anteriore delle pinne pettorali. 
Figura 2 Tagliare la linea mediana dell'addome con forbici da un punto situato a circa 2 mm dal cranio fino all'ano. 
Figura 3 Allargare le pareti addominali con pinzette per esporre il fegato e gli altri organi interni (in alternativa le pareti addominali possono essere pinzate lateralmente). La freccia indica il fegato. 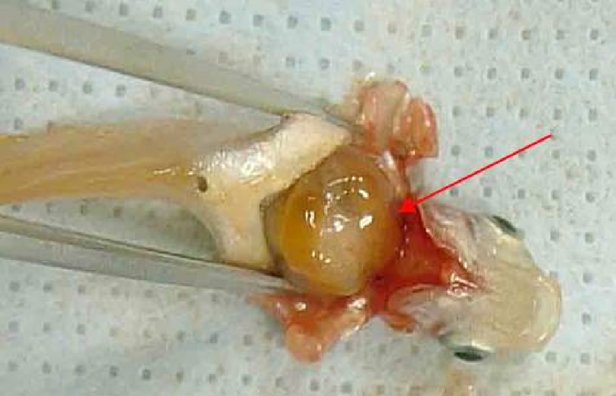
Figura 4 Il fegato è dissezionato e rimosso mediante le pinzette. 
Figura 5 Gli intestini sono rimossi delicatamente con le pinzette. 
Figura 6 Le due estremità degli intestini e gli attacchi del mesenterio sono separati con le forbici. 
Figura 7 (femmina) La procedura è identica per le femmine. 
Figura 8 Procedura completata. 
Procedura 2B: Pre-trattamento del fegato per l'analisi della vitellogenina in Oryzias latipes (medaka) Ritirare la bottiglia contenente il tampone di omogeneizzazione dal kit ELISA e raffreddarla con ghiaccio tritato (temperatura della soluzione: ≤ 4 °C). Se si usa un tampone di omogeneizzazione proveniente da un kit EnBio ELISA, scongelare la soluzione a temperatura ambiente e quindi raffreddare la bottiglia con ghiaccio tritato. Calcolare il volume di tampone di omogeneizzazione per il fegato in base al peso di quest'ultimo (aggiungere 50 μl di tampone di omogeneizzazione per mg di fegato). Per esempio: se il fegato pesa 4,5 mg, il volume del tampone di omogeneizzazione per il fegato è di 225 μl. Stilare un elenco dei volumi di tampone di omogeneizzazione per tutti i fegati. Preparazione del fegato per il pre-trattamento
Svolgimento del pre-trattamento 1) Aggiunta del tampone di omogenato Verificare nell'elenco il volume di tampone di omogeneizzazione da utilizzare per un determinato campione di fegato e aggiustare la micropipetta (intervallo dei volumi: 100-1 000 μl) al volume adeguato. Attaccare un puntale pulito alla micropipetta. Rimuovere il tampone di omogeneizzazione dalla bottiglia del reagente e aggiungere il tampone nella microprovetta da 1,5 ml contenente il fegato. Aggiungere il tampone di omogeneizzazione in tutte le microprovette da 1,5 ml contenenti i campioni di fegato seguendo la procedura sopra descritta. Non è necessario sostituire il puntale della micropipetta con uno nuovo, a meno che non sia contaminato o si sospetti possa esserlo. 2) Omogeneizzazione del fegato
3) Centrifugazione dell'omogenato epatico in sospensione
4) Raccolta del supernatante
Conservazione degli esemplari Conservare le microprovette da 0,5 ml contenenti il supernatante dell'omogenato epatico a una temperatura ≤ – 70 °C fino all'esecuzione dell'analisi ELISA. Procedura 3A: Prelievo di campioni ematici dall'arteria/vena caudale nel danio zebrato Immediatamente dopo l'anestesia sezionare trasversalmente il peduncolo caudale ed eseguire un prelievo ematico dall'arteria/vena caudale mediante tubo capillare microematocrito eparinato. I volumi di sangue raccolti variano tra 5 e 15 microlitri in funzione della dimensione del pesce. Un volume equivalente di tampone di aprotinina [6 microgrammi/ml di soluzione salina tamponata al fosfato (PBS)] è aggiunto al tubo capillare e il plasma è separato dal sangue mediante centrifugazione (5 minuti a 600 g). Il plasma è raccolto in provette e conservato a – 20 °C fino alla misurazione della VTG o di altre proteine di interesse. Procedura 3B: Prelievo di sangue mediante puntura cardiaca nel danio zebrato Per evitare la coagulazione del sangue e la degradazione della proteina, i campioni sono prelevati in una soluzione salina tamponata al fosfato (PBS) contenente eparina (1 000 unità/ml) e aprotinina, inibitore di proteasi (2 TIU/ml). Come ingredienti del tampone, si raccomanda di utilizzare eparina, sale ammonico e aprotinina liofilizzata. Per il prelievo ematico, si raccomanda di utilizzare una siringa (1 ml) con ago fisso sottile (Braun Omnikan-F, ad esempio). La siringa va preriempita con il tampone (circa 100 microlitri) per eluire completamente i piccoli volumi sanguigni da ciascun pesce. I prelievi di sangue avvengono mediante puntura cardiaca. Inizialmente il pesce viene anestetizzato con MS-222 (100 mg/l). Un'anestesia adeguata consente di distinguere il battito cardiaco del danio zebrato. Durante la puntura cardiaca, mantenere una pressione leggera sullo stantuffo della siringa. I volumi sanguigni che possono essere raccolti variano tra 20 e 40 microlitri. Dopo la puntura cardiaca, la miscela sangue/tampone è versata nella provetta. Il plasma è separato dal sangue mediante centrifugazione (20 minuti a 5 000 g) e dovrebbe essere conservato a – 80 °C fino al momento dell'analisi. Procedura 3C POS: omogeneizzazione della testa e della coda nel danio zebrato
Appendice7 CAMPIONI FORTIFICATI DI VITELLOGENINA E STANDARD DI RIFERIMENTO INTER-PROVA Ogni giorno in cui sono effettuate prove sulla VTG, è analizzato un campione fortificato in applicazione di uno standard di riferimento inter-prova. La VTG utilizzata per preparare lo standard di riferimento inter-prova proverrà da un lotto diverso da quello utilizzato per preparare gli standard di calibrazione per la prova in corso. Per preparare il campione fortificato, si aggiunge una quantità nota di standard inter-prova ad un campione di plasma di esemplare maschio di controllo. Il campione sarà ulteriormente fortificato fino a raggiungere una concentrazione di VTG da 10 a 100 volte superiore alla concentrazione di VTG prevista nei maschi di controllo. Il campione di plasma di esemplare maschio di controllo da fortificare può provenire da un unico esemplare o da più esemplari. Un sottocampione del plasma di un esemplare maschio di controllo non fortificato sarà analizzato in almeno due pozzetti in duplicato. Anche il campione fortificato va analizzato in almeno due pozzetti in duplicato. Al fine di determinare la concentrazione prevista, la quantità media di VTG di entrambi i campioni non fortificati di plasma di maschio di controllo viene aggiunta alla quantità calcolata di VTG aggiunta per arricchire i campioni. Il rapporto tra la concentrazione prevista e la concentrazione misurata dovrebbe essere rilevato assieme ai risultati dei singoli test effettuati lo stesso giorno. Appendice 8 DIAGRAMMA DECISIONALE PER L'ANALISI STATISTICA 
C.49 Prova di tossicità acuta sugli embrioni di pesci INTRODUZIONE
PRINCIPIO DELLA PROVA
CONSIDERAZIONI INIZIALI
VALIDITÀ DELLA PROVA
DESCRIZIONE DEL METODO
Strumentazione
Recipienti di prova
Acqua e condizioni sperimentali
Soluzioni di prova
Mantenimento dei pesci riproduttori
Prove di competenza
Produzione delle uova
Differenziazione delle uova
PROCEDURA Condizioni di esposizione
Concentrazioni di prova
Controlli
Inizio dell'esposizione e durata della prova
Distribuzione delle uova nelle piastre a 24 pozzetti
Osservazioni
Misurazioni analitiche
PROVA LIMITE
DATI E RELAZIONE Trattamento dei risultati
Relazione di prova
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Endpoint apicale : indicatore di effetti a livello della popolazione. Blastula : formazione cellulare intorno al polo animale che ricopre una determinata parte del vitello. Sostanza chimica : una sostanza o una miscela. Epibolia : proliferazione massiccia di cellule prevalentemente epidermiche nella fase di gastrulazione dell'embrione e loro spostamento dal lato dorsale verso quello ventrale mediante il quale gli strati cellulari entodermici si invaginano e il vitello si ritrova all'interno dell'embrione. Prova a flusso continuo : prova nella quale le soluzioni saggiate scorrono nel sistema sperimentale con un flusso continuo durante il periodo di esposizione. Controllo interno della piastra : controllo interno costituito, in una piastra a 24 pozzetti, da 4 pozzetti riempiti di acqua di diluizione al fine di individuare la contaminazione potenziale delle piastre causata dal fabbricante o dal ricercatore durante la procedura e ricercare eventuali effetti che possano influire sull'esito della prova (ad esempio, gradiente di temperatura). IUPAC : International Union of Pure and Applied Chemistry — Unione internazionale di chimica pura e applicata. Acqua di mantenimento : acqua in cui si allevano i pesci adulti. Concentrazione letale mediana (LC50) : concentrazione della sostanza chimica in esame ritenuta letale per il 50 % degli organismi esposti nell'arco temporale della prova. Prova con rinnovo semistatico : prova con un regolare rinnovo delle soluzioni di prova a cadenza prestabilita (ad esempio, ogni 24 ore). SMILES : Simplified Molecular Input Line Entry Specification (notazione semplificata lineare delle molecole). Somite : negli embrioni dei vertebrati, gruppo di cellule mesodermiche posto ai lati del tubo neurale da cui si svilupperanno il derma (dermatomo), i muscoli scheletrici (miotomo) e le vertebre (sclerotomo). Prova statica : prova durante la quale le soluzioni di prova restano sempre inalterate. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata secondo il presente metodo di prova. UVCB : sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici. Appendice 2 MANTENIMENTO, RIPRODUZIONE E CONDIZIONI TIPO PER LE PROVE DI TOSSICITÀ ACUTA SUGLI EMBRIONI DI DANIO ZEBRATO
Appendice 3 SVILUPPO NORMALE DEL DANIO ZEBRATO A 26 °C 


Appendice 4 Figura 1 Allestimento delle piastre a 24 pozzetti 
Figura 2 Schema della procedura sperimentale di tossicità acuta sugli embrioni di Danio zebrato (da sinistra a destra): produzione delle uova, raccolta delle uova, pre-esposizione subito dopo la fecondazione in recipienti di vetro, selezione delle uova fecondate con un microscopio rovesciato o un binoculare e distribuzione delle uova fecondate nelle piastre a 24 pozzetti preparate con le rispettive concentrazioni di prova/controlli, n = numero di uova necessarie per ogni concentrazione di prova/controllo (in questo caso 20), hpf = ore trascorse dalla fecondazione 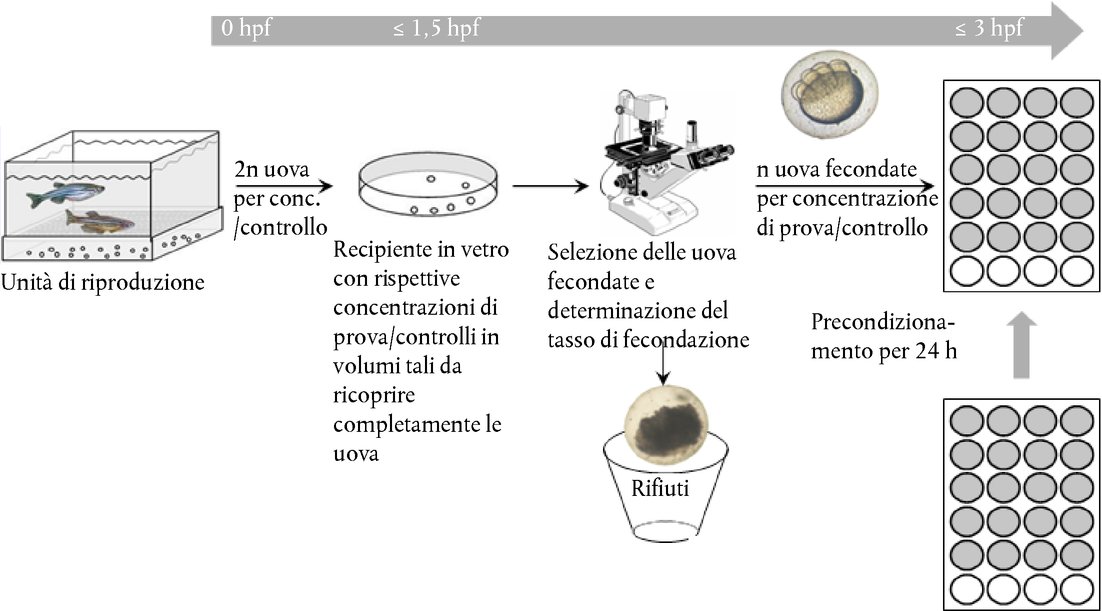
Appendice 5 ATLANTE DEGLI ENDPOINT LETALI PER LA PROVA DI TOSSICITÀ ACUTA SUGLI EMBRIONI DI DANIO ZEBRATO I seguenti endpoint apicali indicano tossicità acuta e, di conseguenza, il decesso degli embrioni: coagulazione dell'embrione, mancato distacco dell'abbozzo caudale, mancata formazione dei somiti e assenza di battito cardiaco. Per illustrarli sono state selezionate le seguenti micrografie. Figura 1 Coagulazione dell'embrione: 
con illuminazione a campo chiaro si distingue una serie di inclusioni opache negli embrioni coagulati di danio zebrato. Figura 2 Mancata formazione dei somiti: 
malgrado il ritardo di sviluppo di circa 10 ore, l'embrione di danio zebrato di 24 ore in (a) presenta somiti ben sviluppati (→), mentre l'embrione in (b) non presenta alcun segno di formazione di somiti (→). Si osserva la netta formazione di somiti (→) nell'embrione di danio zebrato di 48 ore in (c), nonostante un edema pronunciato del sacco vitellino (*), mentre l'embrione di danio zebrato di 96 ore in (d) non presenta alcun segno di formazione di somiti (→). Si noti anche in (d) la deviazione della colonna vertebrale (scoliosi) e l'edema pericardico (*). Figura 3 Vista laterale del mancato distacco dell'abbozzo caudale 
Figura 4 Assenza di battito cardiaco 
L'assenza di battito cardiaco è, per ovvie ragioni, difficile da illustrare in una micrografia. La non convulsione del cuore (doppia freccia) indica l'assenza di battito cardiaco. L'immobilità delle cellule ematiche, ad esempio nell'aorta addominale (→ nel riquadro) non è un indicatore dell'assenza di battito cardiaco. Si osservi anche la mancata formazione dei somiti in questo embrione (*, aspetto omogeneo anziché segmentale dei tessuti muscolari). Il tempo di osservazione per la registrazione dell'assenza di battito cardiaco deve essere di almeno un minuto con un ingrandimento minimo di 80×. C.50 Prova di tossicità su Myriophyllum spicatum in un sistema di prova senza sedimento INTRODUZIONE
PRINCIPIO DELLA PROVA
INFORMAZIONI SULLA SOSTANZA CHIMICA IN ESAME
VALIDITÀ DELLA PROVA
SOSTANZE CHIMICHE DI RIFERIMENTO
DESCRIZIONE DEL METODO Apparecchiature
Organismo sperimentale
Colture
Mezzo di prova
Soluzioni di prova
Gruppi sperimentali e gruppi di controllo
Esposizione
Condizioni di prova
Durata
Misure e determinazioni analitiche
Frequenza delle misurazioni e determinazioni analitiche
Prova limite
DATI E RELAZIONI Variabili di risposta
Tasso di crescita specifico medio
Rendimento
Tempo di raddoppio
Tracciato delle curve concentrazione-risposta
Stima del valore ECx
Procedure statistiche
Relazioni
BIBLIOGRAFIA
Appendice 1 DEFINIZIONI Biomassa : peso fresco e/o secco della materia vivente presente in una popolazione. Nella presente prova la biomassa comprende il germoglio principale, tutti i rami laterali e tutte le radici. Sostanza chimica : una sostanza o una miscela. Clorosi : il cambiamento di colore di un organismo di prova, in particolare dei germogli, dal verde a un colore tendente al giallo. ECx : concentrazione della sostanza chimica in esame disciolta nel mezzo di prova che determina una riduzione dell'x % (per esempio, 50 %) della crescita di Myriophyllum spicatum entro un periodo di esposizione definito (che deve essere esplicitato se diverso dalla durata totale o normale della prova). Per indicare in modo inequivoco se il valore EC si riferisce al tasso di crescita o al rendimento si utilizzano le abbreviazioni “ErC” per il tasso di crescita e “EyC” per il rendimento, seguite dalla variabile di misurazione utilizzata, ad esempio ErC (lunghezza del germoglio principale). Crescita : aumento della variabile di misurazione, ad esempio la lunghezza del germoglio principale, la lunghezza totale dei rami laterali, la lunghezza totale dei germogli, la lunghezza totale delle radici, il peso fresco, il peso secco o il numero di verticilli, nel corso del periodo di prova. Tasso di crescita : (tasso di crescita specifico medio): aumento logaritmico della variabile di misurazione durante il periodo di esposizione. Nota: La risposta relativa al tasso di crescita è indipendente dalla durata della prova a condizione che gli organismi di controllo non esposti siano soggetti a un andamento di crescita esponenziale. Concentrazione minima a cui si osserva un effetto statisticamente significativo (LOEC — Lowest Observed Effect Concentration ): la concentrazione più bassa saggiata di una sostanza alla quale si osserva un effetto di riduzione statisticamente significativo della crescita (p < 0,05) rispetto al controllo, nell'arco di un periodo di esposizione definito. Tutte le concentrazioni di prova superiori alla LOEC, tuttavia, devono avere un effetto dannoso uguale o superiore a quello osservato per la LOEC. Quando queste due condizioni non possono essere soddisfatte occorre fornire una spiegazione dettagliata per spiegare come è stata scelta la LOEC (e di conseguenza la NOEC). Variabile di misurazione : qualsiasi tipo di variabile che viene misurata per esprimere l'endpoint della prova utilizzando una o più variabili di risposta. Nella presente prova le variabili di misurazione consistono nella lunghezza del germoglio principale, nella lunghezza totale dei rami laterali, nella lunghezza totale dei germogli, nella lunghezza totale delle radici, nel peso fresco, nel peso secco e nel numero di verticilli. Monocultura : coltura con una sola specie vegetale. Necrosi : tessuto morto (ossia di aspetto bianco o marrone scuro) dell'organismo di prova. Concentrazione senza effetti osservati (NOEC — No Observed Effect Concentration ) : concentrazione di prova immediatamente inferiore alla LOEC. Variabile di risposta : la variabile per la stima della tossicità derivata da qualsiasi parametro misurato che descrive la biomassa mediante metodi diversi di calcolo. Nel presente metodo di prova, il tasso e il rendimento di crescita sono variabili di risposta derivate dalle variabili di misurazione come la lunghezza del germoglio principale, la lunghezza totale dei germogli, il peso fresco, il peso secco o il numero di verticilli. Prova semistatica (con rinnovo) : prova in cui la soluzione di prova è periodicamente sostituita a determinati intervalli durante la prova. Prova statistica : prova senza rinnovo della soluzione di prova durante la prova. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Endpoint della prova : indica il fattore generale che sarà modificato, rispetto al controllo, dalla sostanza chimica in esame. Nel presente metodo di prova l'endpoint è l'inibizione della crescita che può essere espressa da più variabili di risposta dedotte da una o più variabili di misurazione. Mezzo di prova : mezzo di crescita sintetico completo in cui le piante sperimentali crescono quando sono esposte alla sostanza chimica in esame. Quest'ultimo è di norma disciolto nel mezzo di prova. UVCB : una sostanza di composizione sconosciuta o variabile, il prodotto di una reazione complessa o materiale biologico. Rendimento : valore di una variabile di misurazione che esprime la differenza tra la biomassa al termine del periodo di esposizione e il valore della stessa variabile all'inizio del periodo di esposizione. Nota: quando l'andamento della crescita è esponenziale, le variabili di risposta basate sul rendimento diminuiscono con l'aumento della durata della prova. Appendice 2 MEZZO DI PROVA DI ANDREWS MODIFICATO PER COLTURE MADRE E PRE-COLTURE Il mezzo di prova di Andrews modificato necessario per le pre-colture e le colture madre è preparato partendo da cinque soluzioni madre nutritive elaborate separatamente, cui va aggiunto un 3 % di saccarosio. Tabella 1 omposizione della soluzione nutritiva di Andrews': (Designazione ASTM: E 1913-04)
Le soluzioni madre possono essere conservate in frigorifero per 6 mesi (a una temperatura compresa tra i 5 e i 10 °C). Solo la soluzione madre n. 5 ha una durata di conservazione inferiore (due mesi). Tabella 2 Produzione della soluzione madre n. 3.1 che serve per la preparazione della soluzione madre n. 3.
Una volta ottenuta la soluzione madre n. 3.1 (tabella 2), è necessario congelarla (a una temperatura di almeno – 18 °C) in aliquote di circa 11 ml. Queste porzioni congelate hanno una durata di conservazione di cinque anni. Per la preparazione della soluzione madre 3, scongelare la soluzione 3.1, versarne 10 ml in un matraccio tarato da 1 litro e aggiungere dell'acqua distillata ultrapura fino all'apposito segno sul matraccio. Per ottenere un mezzo di prova di Andrews modificato, versare circa 2 500 ml di acqua distillata ultrapura in un matraccio tarato a 5 l. Aggiungere 50 ml di ciascuna soluzione madre, riempire il 90 % del matraccio con acqua distillata ultrapura e portare a un pH di 5,8. In seguito, aggiungere 150 g di saccarosio disciolto (3 % per 5 l); successivamente riempire il matraccio con acqua distillata ultrapura fino all'apposito segno. Infine, versare la soluzione nutritiva in matracci Schott da 1 l e sottoporre a trattamento in autoclave a 121 °C per 20 minuti. La soluzione nutritiva così ottenuta può essere mantenuta in stato sterile in un refrigeratore (a 5-10 °C) per tre mesi. Mezzo di prova di Andrews per prove di tossicità senza sedimento Per ottenere le soluzioni di prova, si parte dalle cinque soluzioni madre nutritive già menzionate nelle tabelle 1 e 2 e si prepara un mezzo di prova di Andrews non modificato con concentrazione decuplicata, con un'aggiunta di saccarosio pari al 30 %. Per fare ciò è necessario versare circa 100 ml di acqua distillata ultrapura in un matraccio tarato a 1 l. Aggiungere 100 ml di ciascuna delle succitate soluzioni madre, raggiungere un pH di 5,8. In seguito, aggiungere il 30 % di saccarosio disciolto (300 g per 1 000 ml); successivamente riempire il matraccio con acqua distillata ultrapura fino all'apposito segno. Infine, versare la soluzione nutritiva in matracci Schott da 0,5 l e porre in autoclave a 121 °C per 20 minuti. La soluzione nutritiva modificata e concentrata così ottenuta può essere mantenuta in stato sterile in un refrigeratore (a 5-10 °C) per tre mesi. Appendice 3 MANTENIMENTO DI UNA COLTURA MADRE Nella presente appendice 3 si descrive la coltura madre Myriophyllum spicatum L (103), una specie di piante acquatiche sommerse della classe delle dicotiledoni, del genere delle millefoglie d'acqua. Tra giugno e agosto produce dei fiori non molto appariscenti di color rosa-bianco che emergono dallo specchio d'acqua. Le radici delle piante sono ancorate al suolo con un sistema di rizomi robusti. Queste piante crescono nell'intero emisfero boreale in acque stagnanti eutrofiche ma non inquinate e con un tenore di calcio piuttosto elevato, con un substrato fangoso. Il Myriophyllum spicatum predilige l'acqua dolce, ma cresce anche in acque salmastre. Per creare una coltura madre in un sistema senza sedimento a condizioni di laboratorio è necessario ricorrere a piante sterili. Tali piante possono essere reperite dal laboratorio ecotossicologico dell'Ufficio federale tedesco per l'ambiente (Deutsches Umweltbundesamt). In alternativa gli organismi di prova possono essere preparati usando piante non sterili conformemente alla designazione ASTM E 1913-04. Il seguente estratto dalla guida generale ASTM descrive la procedura necessaria per ottenere una coltura di Myriophyllum sibiricum reperiti in natura: “Se si opta per la raccolta di piante non sterili in natura, si raccomanda di raccogliere i turioni in autunno. Inserire i turioni in un acquario da 20 l che contiene 5 cm di sedimento sterile coperto da sabbia di silicea ad esempio da Turface® e 18 l di acqua di reazione. Aerare l'acquario e mantenerlo a una temperatura di 15 °C con un flusso tra i 200 e i 300 μmol m– 2 s– 1 per 16 ore al giorno. La coltura di piante nell'acquario può essere mantenuta come fonte di riserva di piante nel caso in cui le colture di piante sterili fossero distrutte da malfunzionamenti meccanici nella camera di crescita o per altre ragioni. Le piante cresciute nell'acquario non sono sterili e le colture sterili non possono essere conservate in un sistema di coltura in batch. Al fine di sterilizzare la coltura, le piante sono rimosse dall'acquario e sciacquate con acqua corrente deionizzata per circa 0,5 h. In condizioni asettiche in una camera con flusso d'aria laminare, le piante sono disinfettate per meno di 20 minuti (fino a quando i tessuti della maggior parte delle piante non sono sbiancati e rimane verde solo l'apice in crescita) in una soluzione di ipoclorito di sodio al 3 % (peso/volume) contenente lo 0,01 % di un tensioattivo idoneo. Agitare il disinfettante e il materiale vegetale. I segmenti che presentano diversi nodi sono trasferiti in tubi di coltura sterili che contengono 45 ml di mezzo di prova di Andrews sterile modificato e sono chiusi con tappi standard. In ogni camera di prova va inserito un solo segmento vegetale. Per far sì che i recipienti di prova siano ben chiusi, la sigillazione avviene con pellicola da laboratorio. Una volta stabilita la coltura stabile, i segmenti vegetali che presentano diversi nodi vanno trasferiti nelle nuove camere di prova che contengono mezzo nutritivo liquido fresco preparato ogni dieci-dodici giorni. Come dimostrato con la creazione di colture su piastra di agar, le piante devono essere sterili e rimanere tali per otto settimane prima che si possa iniziare la prova.” Poiché il mezzo di prova di Andrews modificato contiene saccarosio (che stimola la crescita di funghi e batteri), tutti i materiali, le soluzioni e la creazione di colture vanno tenuti in condizioni sterili. Tutti i liquidi e tutta l'attrezzatura sono sterilizzati prima dell'uso. La sterilizzazione avviene tramite un trattamento termico ad aria calda (210 °C) dalla durata di 4 ore o tramite un trattamento in autoclave di 20 minuti a 121 °C. Inoltre, tutti i matracci, le piastre, le ciotole, ecc. e le altre attrezzature sono sottoposti a trattamento a fiamma su un piano di lavoro sterilizzato immediatamente prima dell'uso. Le colture madre possono essere conservate a bassa illuminazione e temperatura (50 μE m-2 s-1, 20 ± 2 °C) per lunghi periodi senza che occorra ristabilirle. Il mezzo di crescita del Myriophyllum può essere identico a quello utilizzato per le prove, ma è possibile utilizzare anche altri mezzi ricchi di nutrienti per le colture madre. I segmenti vegetali sono distribuiti in maniera axenica in diversi matracci Erlenmeyer da 500 ml e/o matracci Fernbach da 2 000 ml, con un contenuto per ciascun matraccio di rispettivamente 450 ml o 1 000 ml di mezzo di prova di Andrews modificato. In seguito i matracci sono chiusi in maniera axenica con tappi di cellulosa. Oltre a ciò è assolutamente necessario sottoporre a trattamento a fiamma su un piano di lavoro sterilizzato immediatamente prima dell'uso. In funzione del numero e delle dimensioni, le piante vanno trasferite in una soluzione nutritiva fresca circa ogni tre settimane. Per questa coltura rinnovata è possibile usare apici e segmenti della parte centrale del fusto. Il numero e la dimensione delle piante (o dei segmenti vegetali) dipendono dalla quantità di piante necessaria. Ad esempio, è possibile trasferire cinque segmenti di germoglio nel matraccio Fernbach e tre segmenti di germoglio in un matraccio Erlenmeyer, ciascuno con una lunghezza di 5 cm. Scartare tutte le parti che presentano radici, fioriture, componenti morte o altri elementi evidenti. Figura 1 sezionamento delle piante per la coltura madre e la pre-coltura dopo 3 settimane di coltivazione. 
La coltivazione delle piante avviene in matracci Erlenmeyer da 500 ml e in flaconi Fernbach da 2 000 ml in un incubatore di raffreddamento a 20 (± 2) °C con illuminazione costante a 100-150 μE m– 2 s– 1 o 6 000-9 000 Lux (emessa dalla camera di illuminazione con una temperatura di colore corrispondente a una “luce bianca calda”). Figura 2 Coltivazione di piante in un incubatore di raffreddamento in camera illuminata. 
Occorre utilizzare recipienti di coltura in vetro sterili e chimicamente puliti (lavati con acido) e manipolare il materiale secondo tecniche asettiche. In caso di contaminazione della coltura madre, ad es. da alghe, funghi e/o batteri, per rinnovarla va preparata una nuova coltura o coltura madre proveniente da un altro laboratorio. Appendice 4 MANTENIMENTO DI UNA PRE-COLTURA E PREPARAZIONE DI UN ORGANISMO DI PROVA Per ottenere una pre-coltura, tagliare i germogli della coltura madre in segmenti, ciascuno con due verticilli. Inserire questi segmenti nei matracci Fernbach riempiti con mezzo di prova di Andrews modificato (con 3 % di saccarosio). Ciascun matraccio può contenere fino a 50 segmenti di germoglio. Tuttavia è necessario far sì che i segmenti siano vitali e non presentino nessuna radice, né rami laterali, né le loro gemme (cfr. la figura 1 dell'appendice 3). La pre-coltura dura da 14 a 21 giorni in condizioni sterili in una camera ambientale con fasi alternate buio/luce di 16:8 ore. L'intensità luminosa sarà compresa tra 100 e 150 μE m– 2 s– 1. La temperatura nei recipienti di prova deve essere mantenuta a 23 (± 2) °C. Poiché il mezzo di prova di Andrews modificato contiene saccarosio (che stimola la crescita di alghe, funghi e batteri), sia la preparazione delle soluzioni della sostanza chimica in esame, sia la creazione di colture vanno condotte in condizioni sterili. Tutti i liquidi e tutta l'attrezzatura sono sterilizzati prima dell'uso. La sterilizzazione avviene tramite un trattamento termico ad aria calda (210 °C) dalla durata di 4 ore o tramite trattamento in autoclave di 20 minuti a 121 °C. Inoltre, tutti i matracci, le piastre, le ciotole, ecc. e le altre attrezzature sono sottoposti a trattamento a fiamma su un piano di lavoro sterilizzato immediatamente prima dell'uso. I germogli sono rimossi in maniera axenica dai matracci che contengono le pre-colture, avendo cura di scegliere, nei limiti del possibile, materiale omogeneo. Ciascuna sperimentazione richiede almeno 60 organismi sperimentali (con otto concentrazioni chimiche di prova). Per la prova è necessario servirsi di rami laterali freschi di pre-colture, accorciare a 2,5 dalla base (misurazione con righello) e trasferirli in un becher che contiene mezzo di prova di Andrews modificato. I rami laterali freschi possono essere usati per il test di tossicità di Myriophyllum spicatum in un sistema senza sedimento. Figura 2 Taglio delle piante della pre-coltura per il test di tossicità su Myriophyllum spicatum in un sistema senza sedimento 
C.51 Prova di tossicità su Myriophyllum spicatum in un sistema di prova acqua-sedimento INTRODUZIONE
PRINCIPIO DELLA PROVA
INFORMAZIONI SULLA SOSTANZA CHIMICA IN ESAME
VALIDITÀ DELLA PROVA
SOSTANZE CHIMICHE DI RIFERIMENTO
DESCRIZIONE DEL METODO Apparecchiatura di prova
Organismo sperimentale
Coltivazione dell'organismo di prova:
Sedimento
Mezzo di prova
Disegno sperimentale
Concentrazioni della sostanza chimica in esame e gruppi di controllo
Prova limite
Soluzioni di prova
PROCEDURA DI PROVA
Fase di impianto
Selezione di materiale vegetale uniforme
Esposizione tramite la fase acquatica
Esposizione tramite il sedimento
Mantenimento del livello dell'acqua nel corso della prova
Condizioni di prova
Durata della prova
Misure e determinazioni analitiche
Frequenza delle misurazioni e determinazioni analitiche
Misurazioni analitiche della sostanza chimica in esame
VALUTAZIONE DEI DATI
Variabili di risposta
Tasso di crescita specifico medio
Rendimento
Tracciato delle curve concentrazione-risposta
Stima del valore ECx
Procedure statistiche
RELAZIONI
BIBLIOGRAFIA
Appendice 1 COMPOSIZIONE DEL MEZZO DI PROVA DI SMART E BARKO
Appendice 2 DEFINIZIONI Biomassa : peso fresco e/o secco della materia vivente presente in una popolazione. Nella presente prova la biomassa comprende il germoglio principale, tutti i rami laterali e tutte le radici. Sostanza chimica : una sostanza o una miscela. Clorosi : il cambiamento di colore di un organismo di prova, in particolare dei germogli, dal verde a un colore tendente al giallo. ECx : concentrazione della sostanza chimica in esame disciolta nel mezzo di prova che determina una riduzione dell'x % (per esempio, 50 %) della crescita di Myriophyllum spicatum entro un periodo di esposizione definito (che deve essere esplicitato se diverso dalla durata totale o normale della prova). Per indicare in modo inequivoco se il valore EC si riferisce al tasso di crescita o al rendimento si utilizzano le abbreviazioni “ErC” per il tasso di crescita e “EyC” per il rendimento, seguite dalla variabile di misurazione utilizzata, ad esempio ErC (lunghezza del germoglio principale). Crescita : aumento della variabile di misurazione, ad esempio la lunghezza del germoglio principale, la lunghezza totale dei rami laterali, la lunghezza totale dei germogli, la lunghezza totale delle radici, il peso fresco, il peso secco o il numero di verticilli, nel corso del periodo di prova. Tasso di crescita : (tasso di crescita specifico medio): aumento logaritmico della variabile di misurazione durante il periodo di esposizione. Nota: La risposta relativa al tasso di crescita è indipendente dalla durata della prova a condizione che gli organismi di controllo non esposti siano soggetti a un andamento di crescita esponenziale. Concentrazione minima a cui si osserva un effetto statisticamente significativo (LOEC — Lowest Observed Effect Concentration): la concentrazione più bassa saggiata di una sostanza alla quale si osserva un effetto di riduzione statisticamente significativo della crescita (p < 0,05) rispetto al controllo, nell'arco di un periodo di esposizione definito. Tutte le concentrazioni di prova superiori alla LOEC, tuttavia, devono avere un effetto dannoso uguale o superiore a quello osservato per la LOEC. Tutte le concentrazioni di prova superiori alla LOEC, tuttavia, devono avere un effetto dannoso uguale o superiore a quello osservato per la LOEC. Quando queste due condizioni non possono essere soddisfatte occorre fornire una spiegazione dettagliata per spiegare come è stata scelta la LOEC (e di conseguenza la NOEC). Variabile di misurazione : qualsiasi tipo di variabile che viene misurata per esprimere l'endpoint della prova utilizzando una o più variabili di risposta. Nella presente prova le variabili di misurazione consistono nella lunghezza del germoglio principale, nella lunghezza totale dei rami laterali, nella lunghezza totale dei germogli, nella lunghezza totale delle radici, nel peso fresco, nel peso secco e nel numero di verticilli. Monocultura : coltura con una sola specie vegetale. Necrosi : tessuto morto (ossia di aspetto bianco o marrone scuro) dell'organismo di prova. Concentrazione senza effetti osservati (NOEC — No Observed Effect Concentration ) : concentrazione di prova immediatamente inferiore alla LOEC. Variabile di risposta : la variabile per la stima della tossicità derivata da qualsiasi parametro misurato che descrive la biomassa mediante metodi diversi di calcolo. Nel presente metodo di prova, il tasso e il rendimento di crescita sono variabili di risposta derivate dalle variabili di misurazione come la lunghezza del germoglio principale, la lunghezza totale dei germogli, il peso fresco, il peso secco o il numero di verticilli. Prova semistatica (con rinnovo) : prova in cui la soluzione di prova è periodicamente sostituita a determinati intervalli durante la prova. Prova statistica : prova senza rinnovo della soluzione di prova durante la prova. Sostanza chimica in esame : qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova. Endpoint della prova : indica il fattore generale che sarà modificato, rispetto al controllo, dalla sostanza chimica in esame. Nel presente metodo di prova l'endpoint è l'inibizione della crescita che può essere espressa da più variabili di risposta dedotte da una o più variabili di misurazione. Mezzo di prova : mezzo di crescita sintetico completo in cui le piante sperimentali crescono quando sono esposte alla sostanza chimica in esame. Quest'ultimo è di norma disciolto nel mezzo di prova. UVCB : una sostanza di composizione sconosciuta o variabile, il prodotto di una reazione complessa o materiale biologico. Rendimento : valore di una variabile di misurazione che esprime la differenza tra la biomassa al termine del periodo di esposizione e il valore della stessa variabile all'inizio del periodo di esposizione. Nota: quando l'andamento della crescita è esponenziale, le variabili di risposta basate sul rendimento diminuiscono con l'aumento della durata della prova. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(1) Non è disponibile alcun valore per la temperatura di 20 °C, ma si può ritenere che la variabilità dei risultati della misurazione sia superiore alle variazioni dovute alla temperatura
(2) Regolamento (CE) n. 1907/2006 del Parlamento europeo e del Consiglio, del 18 dicembre 2006, concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH), che istituisce un'Agenzia europea per le sostanze chimiche, che modifica la direttiva 1999/45/CE e che abroga il regolamento (CEE) n. 793/93 del Consiglio e il regolamento (CE) n. 1488/94 della Commissione, nonché la direttiva 76/769/CEE del Consiglio e le direttive della Commissione 91/155/CEE, 93/67/CEE, 93/105/CE e 2000/21/CE (GU L 304 del 22.11.2007, pag. 1).
(3) The US Interagency Coordinating Committee on the Validation of Alternative Methods.
(*1) Indicare la zona di opacità corneale
(4) Per l'applicazione di una strategia sperimentale integrata per l'irritazione oculare nell'ambito del RACH, si veda anche la linea guida ECHA sugli obblighi di informazione e la valutazione della sicurezza chimica, capitolo R.7a: “Endpoint specific guidance”, http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
(5) Gli statistici che applicano una metodologia di modellizzazione quale l'utilizzo dei modelli lineari generalizzati (GLM) possono effettuare l'analisi in modo diverso ma comparabile, senza tuttavia ricavare necessariamente la tradizionale tabella ANOVA che risale a concetti algoritmici del calcolo statistico elaborati in epoca preinformatica.
(6) Gli statistici che applicano una metodologia di modellizzazione quale l'utilizzo dei modelli lineari generalizzati (GLM) possono effettuare l'analisi in modo diverso ma comparabile, senza tuttavia ricavare necessariamente la tradizionale tabella ANOVA che risale a concetti algoritmici del calcolo statistico elaborati in epoca preinformatica.
(7) Regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio, del 16 dicembre 2008, relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006 (GU L 353 del 31.12.2008, pag. 1).
(8) Ciascuna sostanza chimica in esame è stata assegnata a classi chimiche definite in base a un sistema di classificazione standard, basato sul sistema di classificazione della National Library of Medicine Medical Subject Headings (MeSH)(disponibile online al sito www.nlm.nih.gov/mesh).
(9) In base ai risultati del test in vivo sugli occhi dei conigli (OCSE TG 405) (17) e sul sistema di classificazione UN GHS (4).
(10) La classificazione nelle categorie 2A o 2B dipende dall'interpretazione del criterio UN GHS inteso a distinguere fra queste due categorie, ossia 1 su 3 oppure 2 su 3 animali presentano al giorno 7 gli effetti necessari per determinare una classificazione nella categoria 2A. Lo studio in vivo ha incluso 3 animali. Tutti gli endpoint, tranne il rossore della congiuntiva in un animale, sono regrediti a un punteggio pari a zero entro il giorno 7 o prima. L'unico animale che non aveva recuperato interamente entro il giorno 7 presentava un punteggio di rossore della congiuntiva pari a 1 (al giorno 7), interamente regredito al giorno 10.
(11) Le dimensioni fornite si riferiscono a un supporto corneale utilizzato per bovini di età compresa fra 12 e 60 mesi. Se l'esame è svolto su animali fra i 6 e i 12 mesi di età, il supporto deve essere realizzato in modo tale che ciascuna camera abbia una capacità di 4 ml e che ciascuna camera interna presenti un diametro di 1,5 cm e una profondità di 2,2 cm. Per ogni nuovo supporto corneale è necessario che fra l'area della cornea esposta e il volume della camera posteriore sia mantenuto lo stesso rapporto del supporto corneale tradizionale. Ciò è necessario per garantire la corretta determinazione dei valori di permeabilità per il calcolo del valore IVIS con la formula proposta.
(12) Regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio, del 16 dicembre 2008, relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006 (GU L 353 del 31.12.2008, pag. 1).
(*2) Massimo punteggio medio osservato a qualsiasi scadenza temporale
(*3) Massimo punteggio medio osservato a qualsiasi scadenza temporale (basato sui punteggi di opacità definiti nella tabella 1).
(*4) In base ai punteggi definiti nella tabella 2.
(*5) Combinazioni meno probabili.
(13) Ciascuna sostanza di prova è stata assegnata a classi chimiche definite in base a un sistema di classificazione standard, basato sul sistema di classificazione della National Library of Medicine Medical Subject Headings (MeSH) (disponibile online al sito http//www.nlm.nih.gov/mesh).
(14) Basata sui risultati del test sugli occhi dei conigli in vivo (OECD TG 405) e sul sistema di classificazione UN GHS (4)(6).
(15) In base ai risultati ICE descritti nella tabella 6.
(16) Combinazione dei punteggi ICE diversi da quelli descritti nella tabella 6 per identificare l'assenza di categoria o la categoria 1 del sistema UN GHS (cfr. tabella 6)
(17) La classificazione nelle categorie 2A o 2B dipende dall'interpretazione del criterio UN GHS inteso a distinguere fra queste due categorie, ossia 1 su 3 oppure 2 su 3 animali presentano al giorno 7 gli effetti necessari per determinare una classificazione nella categoria 2A. Lo studio in vivo ha incluso 3 animali. Tutti gli endpoint, tranne il rossore della congiuntiva in un animale, sono regrediti a un punteggio pari a zero entro il giorno 7 o prima. L'unico animale che non aveva recuperato interamente entro il giorno 7 presentava un punteggio di rossore della congiuntiva pari a 1 (al giorno 7), interamente regredito al giorno 10.
(18) Regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio, del 16 dicembre 2008, relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006, GU L 353 del 31.12.2008, pag. 1.
(19) In tutto il presente documento con “media” si intende la media aritmetica.
(20) Le cifre corrispondono a valori soglia ottenuti per elaborazione statistica e non fanno riferimento alla precisione della misurazione.
(21) Una previsione DPRA deve essere presa in considerazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei punti 9 e 12.
(22) Le cifre corrispondono a valori soglia ottenuti per elaborazione statistica e non fanno riferimento alla precisione della misurazione.
(23) Una previsione DPRA deve essere presa in considerazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei punti 9 e 12.
(24) Le previsioni in vivo del pericolo (e la potenza sensibilizzante) sono basate su dati LLNA (19). La potenza in vivo è determinata utilizzando i criteri proposti da ECETOC (23).
(25) Una previsione DPRA deve essere presa in considerazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei punti 9 e 11.
(26) Intervalli determinati sulla base di almeno 10 valori di deplezione stabiliti da 6 laboratori indipendenti.
(27) Regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio, del 16 dicembre 2008, relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006 (GU L 353 del 31.12.2008, pag. 1).
(28) La predizione del pericolo (e della potenza) in vivo si basa sui dati dell'LLNA (13). La potenza in vivo è determinata mediante criteri proposti da ECETOC (24).
(29) Una predizione KeratinoSensTM deve essere considerata nel quadro di un approccio di tipo IATA e conformemente alle disposizioni dei paragrafi 9 e 11 del presente metodo di prova.
(30) Sulla base dei dati storici osservati (12).
(31) Regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio, del 16 dicembre 2008, relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006 (GU L 353 del 31.12.2008, pag. 1).
(32) Ciascuna sostanza chimica in esame è stata assegnata a classi chimiche definite in base a un sistema di classificazione standard, basato sul sistema di classificazione della National Library of Medicine Medical Subject Headings (MeSH) (disponibile sul sito http//www.nlm.nih.gov/mesh).
(33) Basata sui risultati della prova sugli occhi dei conigli in vivo (OECD TG 405), sul sistema di classificazione GHS dell'ONU e sul regolamento CLP.
(34) Basata sui risultati FL (protocollo INVITTOX n. 71 (6)).
(35) Gli statistici che applicano una metodologia di modellizzazione quale l'utilizzo dei modelli lineari generalizzati (GLM) possono effettuare l'analisi in modo diverso ma comparabile, senza tuttavia ricavare necessariamente la tradizionale tabella ANOVA che risale a concetti algoritmici del calcolo statistico elaborati in epoca preinformatica.
(36) Cfr. appendice 1, Definizioni e unità di misura.
(37) Talvolta indicati con P OW; determinati con il metodo del dibattimento in pallone nel metodo di prova A.8 (3), con il metodo HPLC nel metodo di prova A.24 (4) e con il metodo dell'agitazione lenta nel metodo di prova A.23 (5). La tecnica del generatore a colonna è talvolta utilizzata per la determinazione di log K OW. Un numero limitato di studi che utilizzano questa tecnica è disponibile, essenzialmente per le dibenzo-diossine e i difenili clorurati (e.g. Li and Doucette, 1993) (3). Per le sostanze ionizzabili, il log K OW dovrebbe fare riferimento alla forma non ionizzata.
(38) Cfr. appendice 1, Definizioni e unità di misura.
(39) CPT: cromatografia su strato sottile; HPLC: cromatografia liquida ad alta pressione; GC: cromatografia in fase gassosa.
(40) In alcuni quadri normativi le analisi dei metaboliti possono essere obbligatorie qualora siano soddisfatte talune condizioni (cfr. paragrafo 65).
(41) In generale, le concentrazioni misurate nell'acqua durante la fase di assorbimento dovrebbero essere di almeno un ordine di grandezza sopra il limite di quantificazione in modo che più di un tempo di dimezzamento del carico corporeo può essere misurato nella fase di depurazione dello studio.
(42) Cfr. appendice 1, Definizioni e unità di misura.
(43) Per la maggior parte delle sostanze in esame, idealmente non si dovrebbe procedere ad alcun rilevamento nell'acqua di controllo. Le concentrazioni di fondo dovrebbero applicarsi solo ai materiali presenti in natura (ad esempio metalli) e alle sostanze largamente diffuse nell'ambiente.
(44) Qualora sia evidente che non avviene una reazione di primo ordine, si devono impiegare modelli più complessi (cfr. bibliografia dell'appendice 5) con l'assistenza di un esperto di biostatistica.
(45) L'assorbimento può essere limitato da una bassa concentrazione di esposizione a causa della scarsa solubilità in acqua nella prova di bioconcentrazione, mentre concentrazioni di esposizione molto maggiori possono essere ottenute nella prova con esposizione per via alimentare.
(46) Per le sostanze multicomponenti, le sostanze UVCB e le miscele, si deve prendere in considerazione la solubilità in acqua di ogni componente al fine di determinare le opportune concentrazioni di esposizione.
(47) Il TOC include il carbonio organico del particolato e il carbonio organico disciolto, TOC = POC + DOC.
(48) Pur non essendo generalmente raccomandati, se si utilizzano solventi o agenti solubilizzanti, il carbonio organico proveniente da tali agenti deve essere aggiunto al carbonio organico della sostanza in esame per valutare la concentrazione del carbonio organico nel recipiente di prova.
(49) Se il tenore di lipidi e la sostanza in esame non sono analizzati nello stesso pesce, i pesci devono almeno avere peso analogo ed essere (se del caso) dello stesso sesso.
(50) Questa alternativa è valida solo se i pesci in tutti i gruppi di prova sono mantenute in gruppi con dimensioni analoghe, i pesci sono rimossi secondo lo stesso modello e alimentati allo stesso modo. Ciò garantisce che una crescita analoga dei pesci in tutti i gruppi di prova, se la concentrazione è inferiore all'intervallo di tossicità. Se il tasso di crescita è simile, anche il contenuto lipidico dovrebbe essere simile. Una crescita diversa nel gruppo di controllo può indicare un effetto della sostanza e vanificherebbe lo studio.
(51) Oltre al peso, va registrata la lunghezza totale perché il confronto tra le entità di aumento della lunghezza durante la prova è un buon indicatore per stabilire se si sia verificato un effetto negativo.
(52) Può essere effettuato un test t sulle costanti del tasso di crescita, per verificare se la crescita varia tra gruppi di controllo e di prova, o un test F nel caso di analisi della varianza. Se necessario, un test F o del rapporto di verosimiglianza può essere utilizzato come ausilio per la scelta del modello di crescita (monografia 54 dell'OCSE (32).
(53) Tali percentuali presuppongono che i metodi analitici sono affidabili e che il tempo di dimezzamento è < 14 giorni. Se i metodi d'analisi sono meno affidabili o il dimezzamento è (molto) maggiore questi numeri saranno più elevati.
CI: Intervallo di confidenza (se possibile stimarlo)
SD: Deviazione standard (se possibile stimarla)
(56) La prova ridotta può infatti essere utilizzata per dimostrare un metabolismo rapido, quando è noto che un metabolismo rapido è probabile.
(57) Se sono misurati solo due dati, è possibile ottenere stime degli intervalli di confidenza per il BCFKm mediante una tecnica di bootstrap. Quando sono disponibili anche valori intermedi è possibile calcolare anche gli intervalli di confidenza per il BCFKm come nella prova completa.
(58) Cfr. appendice 1, Definizioni e unità di misura.
(59) Ai fini dell'applicazione del regolamento (CE) n. 1907/2006 concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH) (GU L 396 del 30.12.2006, pag. 1), la questione è affrontata nella “Guida alle prescrizioni in materia di informazione e alla valutazione della sicurezza chimica”, capitolo R.7c, R.7.10.3.1, pp 13; R.7.10.4.1, pp 31-32; e figura R7.10-2, pp 50.
(60) Per la maggior parte delle sostanze in esame idealmente non si dovrebbe individuare alcunché nell'acqua di controllo. Le concentrazioni naturali sono pertinenti solo per i materiali presenti in natura (alcuni metalli) e per le sostanze presenti ovunque nell'ambiente.
(61) Il BMF è definito come il rapporto tra la concentrazione di una sostanza in un organismo e la concentrazione della sostanza nell'alimentazione di tale organismo allo stato stazionario, i grassi sono presi in considerazione correggendo i valori ottenuti in funzione dei lipidi presenti nell'organismo e nei mangimi, da cui il termine più preciso di “correzione”. Tale approccio differisce dalla “normalizzazione” rispetto a un determinato tenore in lipidi dell'organismo, come avviene nella prova di bioconcentrazione mediante l'esposizione in ambiente acquatico.
(62) La lunghezza totale deve essere registrata durante la prova, poiché costituisce un buon indicatore degli eventuali effetti nocivi osservati.
(63) L'HCB figura negli elenchi degli allegati A e C della convenzione di Stoccolma, nonché negli allegati I e III del regolamento (CE) n. 850/2004 relativo agli inquinanti organici persistenti (GU L 158 del 30.4.2004, pag. 7).
(64) In caso di rapida crescita durante la fase di assorbimento, la razione alimentare reale scenderà al di sotto di quella definita all'inizio dell'esposizione.
(65) Nel quadro di uno studio con esposizione attraverso l'ambiente acquatico, un tempo di dimezzamento di 14 giorni corrisponde a un fattore di bioconcentrazione di circa 10 000 l/kg utilizzando pesci di 1 g con un tasso di assorbimento di circa 500 l/kg/d [secondo l'equazione di Sijm et al. (46)].
(66) Atteso che le concentrazioni interne reali possono essere determinate solo al termine della prova, è necessario stimare la concentrazione interna prevista (ad esempio tramite il BMF previsto e la concentrazione nel mangime; cfr. l'appendice 5 (A5.8).
(67) La presenza della sostanza nel mezzo di prova attraverso le feci del pesce o a seguito di lisciviazione del cibo non può essere del tutto evitata. È possibile ad esempio misurare la concentrazione della sostanza presente nell'acqua alla fine della fase di assorbimento, soprattutto quando si utilizza un sistema semistatico, per stabilire se si è verificata un'esposizione in ambiente acquatico.
(68) Questo metodo è specifico per lo studio basato sull'alimentazione e differisce dalla procedura seguita per l'esposizione in ambiente acquatico. Per tale motivo è stato usato il termine “correzione” anziché “normalizzazione” onde evitare confusione — cfr. la nota n. 34.
(69) Si può eseguire un test t per le costanti cinetiche di crescita, per verificare se la crescita tra i gruppi di controllo e di prova varia, o un test F in caso dell'analisi della varianza. Se necessario, si può utilizzare un test F o test del rapporto di verosimiglianza per facilitare la scelta del modello di crescita adeguato [monografia OCSE n. 54 (32)].
(70) Tecnica di analisi degli alimenti per verificare il tenore di proteine, lipidi, in cellulosa e ceneri. Tali informazioni sono di solito disponibili presso il fornitore.
CI: intervallo di confidenza (se possibile stimarlo)
SD: Deviazione standard (se possibile stimarla)
(73) Meyer et al. (1)
(74) Durante la prova stessa, è preferibile utilizzare il peso per misurare le derivazioni delle costanti cinetiche di crescita. Si riconosce tuttavia che la lunghezza è una misura più pratica se i pesci devono essere selezionati a vista all'inizio dell'esperimento (all'interno della popolazione dello stock ittico).
(75) Tale intervallo di lunghezze è riportato nel documento Testing Methods for New Chemical Substances ecc., basato sulla legislazione relativa al controllo delle sostanze chimiche del Giappone (CSCL).
(76) I valori tra parentesi corrispondono al numero di campioni (acqua, pesce) da prelevare se si esegue un campionamento supplementare.
(77) La stima preliminare di k2 di una sostanza con log Kow = 4.0 è di 0.652 giorni– 1. La durata totale dell'esperimento è impostata su 3 × t SS = 3 × 4.6, cioè 14 giorni. Per la stima di t SS cfr. appendice 5.
(78) Prelevare un campione di acqua dopo che è stato versato l'equivalente del volume di almeno tre recipienti di prova.
(79) Questi pesci sono prelevati dallo stock ittico.
(80) Se è necessaria una maggiore precisione su studi di metabolismo che richiedono un maggior numero di pesci, questi devono essere campionati specificamente alla fine delle fasi di assorbimento e di depurazione (cfr. paragrafo 40).
(81) Almeno altri 3 pesci potranno essere necessari per analizzare il contenuto di grassi se non è possibile utilizzare i pesci prelevati per misurare le concentrazioni della sostanza in esame all'inizio della prova, alla fine della fase di assorbimento e alla fine della fase di depurazione. Nota dovrebbe essere possibile in molti casi utilizzare solo i 3 pesci di controllo (cfr. paragrafo 56).
(82) Tre campioni di alimenti sia dei gruppi di controllo sia dei gruppi di prova sono analizzati per misurare le concentrazioni della sostanza in esame e il contenuto di lipidi.
(83) I pesci vengono prelevati a fini di campionamento dallo stock il più tardi possibile prima dell'inizio dello studio; almeno tre esemplari di tale stock sono prelevati all'inizio della prova per misurare il tenore di grassi.
(84) Il campionamento (facoltativo) all'inizio della fase di assorbimento fornisce i dati necessari per calcolare l'assimilazione della sostanza in esame consumata per via alimentare, che si può paragonare all' efficienza di assimilazione calcolata a partire dai dati ottenuti nel corso della fase di depurazione.
(85) Si possono prelevare cinque pesci supplementari per l'analisi dei tessuti specifici.
(86) Almeno tre esemplari supplementari potranno essere necessari per analizzare il contenuto di grassi se non è possibile utilizzare i pesci prelevati per misurare le concentrazioni della sostanza in esame all'inizio della prova, alla fine della fase di assorbimento e alla fine della fase di depurazione. È opportuno precisare che dovrebbe essere possibile in molti casi utilizzare solo i 3 pesci di controllo (cfr. paragrafi 56 e 153).
(87) Come per ogni relazione empirica, occorre verificare che la sostanza rientri nel campo di applicabilità della relazione.
(88) Il peso del pesce alla fine della fase di assorbimento può essere stimato sulla base dei dati di uno studio precedente o conoscenze sulla specie in esame, di cui è noto il probabile aumento delle dimensioni in base al peso all'inizio della prova abituale e per una durata di assorbimento abituale (ad es. 28 giorni).
(89) Nella maggior parte dei programmi che consentono una regressione lineare, sono forniti anche gli errori standard e l'intervallo di confidenza (IC) delle stime, ad esempio in Microsoft Excel utilizzando i dati dello strumento di analisi Pack.
(90) Contrariamente al metodo di regressione lineare, questa formula non darà generalmente un errore standard di k2.
(91) In contrasto con una procedura di approssimazione, questo metodo lineare di solito non generare un errore standard o intervallo di confidenza per la stima di k1.
(92) Occorre tener conto del fatto che l'incertezza nella stima di k2 non è utilizzata adeguatamente nel modello di bioaccumulo quando è essenzialmente considerata come costante continua quando si adatta k1 con il metodo sequenziale. L'incertezza circa il BCF ottenuto sarà, perciò, differente a seconda che si applica il metodo di adattamento simultaneo o sequenziale.
(93) In alcune regioni, potrebbe essere possibile ottenere solo mangimi per pesco con un tenore lipidico molto inferiore a tale massimale. In tal caso, occorre effettuare la prova con il tenore di grassi inferiore nei mangimi e adeguare la razione alimentare in modo da mantenere i pesci in buona salute. È preferibile non aumentare artificialmente il tenore lipidico dei mangimi aggiungendo troppo di olio.
(94) In natura, il meccanismo che porta alla maggiore esposizione in ambiente acquatico è probabilmente l'ingestione di sostanze estremamente idrofobe; pertanto, un BCF stimato non è necessariamente rappresentativo del potenziale di bioaccumulo di tali sostanze.
(95) È possibile usare altre specie di Dafnidi, purché soddisfino adeguatamente i criteri di validità (il criterio di validità relativo alla capacità riproduttiva nei controlli deve essere pertinente per tutte le specie). Se sono utilizzate altre specie di Dafnidi, occorre identificarle chiaramente e motivare la scelta.
(96) Mortalità accidentale: mortalità non legata a sostanze chimiche e causata da un evento accidentale (della quale, cioè, si conosce la causa).
(97) Mortalità casuale: mortalità non legata a sostanze chimiche e della quale non si conosce la causa.
(*6) Indicare quale recipiente è stato usato per l'esperimento
(*7) Registrare le schiuse abortite inserendo la sigla “AB” (aborted broods) nella casella corrispondente.
(*8) Registrare la mortalità di ogni animale riproduttore (usando l'iniziale “M”) nella casella corrispondente.
(98) Il valore medio minimo tipico della lunghezza totale non è un criterio di validità ma le medie inferiori al valore indicato devono essere esaminate attentamente per valutare la sensibilità della prova. I valori medi minimi della lunghezza totale sono determinati sulla base di una selezione dei dati disponibili.
(99) Il particolare ceppo di trota iridea studiato può richiedere l'uso di altre temperature. I pesci riproduttori devono essere mantenuti alla stessa temperatura delle uova. Le uova che provengono da un fornitore commerciale richiedono un breve periodo d'adattamento (1-2 ore) alla temperatura alla quale si svolgerà la prova.
(100) Durante la settimana successiva alla schiusa le larve sono tenute al buio, tranne quando sono esaminate; per tutto il resto della prova si mantiene un'illuminazione fioca (fotoperiodo compreso tra 12 e 16 ore) (4).
(101) Indipendentemente dalle condizioni sperimentali, l'illuminazione prescelta deve essere costante.
(102) La deviazione ammessa in tutte le prove è di ± 20/00.
(*9) Cibo fornito ad libitum. Per evitare l'accumulo di rifiuti il cibo non consumato e gli escrementi sono eliminati, quando necessario.
|
AC |
= |
artemie congelate; adulti di Artemia sp |
|
NA |
= |
naupli di artemia; appena schiusi |
|
NA48 |
= |
naupli di artemia; di 48 ore di età |
(1) le larve provviste di sacco vitellino non hanno bisogno di essere alimentate
(2) filtrati da coltura mista
(3) granuli da processo di fermentazione
(*10) I processi papillari sono generalmente presenti soltanto nei maschi adulti e si situano tra il secondo e il settimo/ottavo raggio della pinna contando a partire dall'estremità posteriore della pinna anale (figure 1 e 2). Tuttavia è raro che i processi siano presenti sul primo raggio della pinna contando a partire dall'estremità posteriore della pinna anale. La procedura operativa standard (POS) consente di misurare i processi presenti sul primo raggio della pinna (il numero del raggio si conta a partire dall'estremità posteriore della pinna anale nella presente POS).
(*11) Tampone di omogeneizzazione:
|
— |
50 mM Tris-HCl pH 7,4; 1 % cocktail di inibitori di proteasi (Sigma)): 12 ml Tris-HCl pH 7,4 + 120 μl cocktail di inibitori di proteasi. |
|
— |
TRIS: TRIS-ULTRA PURE (ICN) e.g. da Bie & Berntsen, Danimarca. |
|
— |
Cocktail di inibitori di proteasi: da Sigma (per i tessuti di mammiferi) n. del prodotto P 8340. |
NOTA: Il tampone di omogeneizzazione va utilizzato il giorno stesso in cui è preparato. Mantenere nel ghiaccio durante l'uso.
(103) Carl von Linné (* 23 maggio, 1707 a Råshult/Älmhult; † 10 gennaio 1778 a Uppsala).
(*12) acqua demineralizzata (vale a dire distillata o deionizzata)







 , pari a 0,095 mm. È opportuno richiamare l'attenzione degli sperimentatori sul fatto che i microscopi con lampade a fessura possono produrre misurazioni dello spessore della cornea diverse a seconda delle diverse larghezze su cui è impostata la fessura.
, pari a 0,095 mm. È opportuno richiamare l'attenzione degli sperimentatori sul fatto che i microscopi con lampade a fessura possono produrre misurazioni dello spessore della cornea diverse a seconda delle diverse larghezze su cui è impostata la fessura.