ISSN 1725-258X
Gazzetta ufficiale
dell'Unione europea
L 39

Edizione in lingua italiana
Legislazione
52° anno
10 febbraio 2009
|
ISSN 1725-258X |
||
|
Gazzetta ufficiale dell'Unione europea |
L 39 |
|
|
|
||
|
Edizione in lingua italiana |
Legislazione |
52° anno |
|
|
|
II Atti adottati a norma dei trattati CE/Euratom la cui pubblicazione non è obbligatoria |
|
|
|
|
DECISIONI |
|
|
|
|
Commissione |
|
|
|
|
2009/108/CE |
|
|
|
* |
Decisione della Commissione, del 3 febbraio 2009, che modifica la decisione 2002/364/CE relativa alle specifiche tecniche comuni per i dispositivi medico-diagnostici in vitro [notificata con il numero C(2009) 565] ( 1 ) |
|
|
|
||
|
|
* |
|
|
|
|
|
(1) Testo rilevante ai fini del SEE |
|
IT |
Gli atti i cui titoli sono stampati in caratteri chiari appartengono alla gestione corrente. Essi sono adottati nel quadro della politica agricola ed hanno generalmente una durata di validità limitata. I titoli degli altri atti sono stampati in grassetto e preceduti da un asterisco. |
I Atti adottati a norma dei trattati CE/Euratom la cui pubblicazione è obbligatoria
REGOLAMENTI
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/1 |
REGOLAMENTO (CE) N. 116/2009 DEL CONSIGLIO
del 18 dicembre 2008
relativo all'esportazione di beni culturali
(Versione codificata)
IL CONSIGLIO DELL’UNIONE EUROPEA,
visto il trattato che istituisce la Comunità europea, in particolare l'articolo 133,
vista la proposta della Commissione,
considerando quanto segue:
|
(1) |
Il regolamento (CEE) n. 3911/92 del Consiglio, del 9 dicembre 1992, relativo all'esportazione di beni culturali (1), è stato modificato in modo sostanziale e a più riprese (2). A fini di razionalità e chiarezza occorre provvedere alla codificazione di tale regolamento. |
|
(2) |
Ai fini del mantenimento del mercato interno è necessario adottare una normativa per gli scambi con i paesi terzi, la quale assicuri la protezione dei beni culturali. |
|
(3) |
Sembra necessario prendere misure in particolare per garantire che le esportazioni di beni culturali siano sottoposte a controlli uniformi alle frontiere esterne della Comunità. |
|
(4) |
Un siffatto sistema dovrebbe prevedere l'obbligo di presentare una licenza rilasciata dallo Stato membro competente, prima dell'esportazione dei beni culturali contemplati dal presente regolamento. Ciò richiede una precisa definizione del campo di applicazione di dette misure e delle loro modalità di attuazione. La realizzazione del sistema dovrebbe presentare la massima semplicità ed efficacia. |
|
(5) |
Le misure necessarie per l'attuazione del presente regolamento dovrebbero essere adottate secondo la decisione 1999/468/CE del Consiglio, del 28 giugno 1999, recante modalità per l'esercizio delle competenze di esecuzione conferite alla Commissione (3). |
|
(6) |
Data la notevole esperienza acquisita dalle autorità degli Stati membri nell'applicare il regolamento (CE) n. 515/97 del Consiglio, del 13 marzo 1997, relativo alla mutua assistenza tra le autorità amministrative degli Stati membri e alla collaborazione tra queste e la Commissione per assicurare la corretta applicazione delle normative doganale e agricola (4), detto regolamento dovrebbe essere applicato nel presente settore. |
|
(7) |
L'allegato I del presente regolamento ha lo scopo di definire le categorie di beni culturali che dovrebbero formare oggetto di particolare protezione negli scambi con i paesi terzi, ferma restando la libertà degli Stati membri di definire i beni da considerare patrimonio nazionale ai sensi dell'articolo 30 del trattato, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
Definizione
Fatti salvi i poteri degli Stati membri ai sensi dell'articolo 30 del trattato, per «beni culturali» s'intendono, ai fini del presente regolamento, i beni elencati nell'allegato I.
Articolo 2
Licenza di esportazione
1. L'esportazione di beni culturali al di fuori del territorio della Comunità è subordinata alla presentazione di una licenza di esportazione.
2. La licenza di esportazione è rilasciata, su richiesta dell'interessato:
|
a) |
da un'autorità competente dello Stato membro sul cui territorio si trovava lecitamente e definitivamente il bene culturale alla data del 1o gennaio 1993; |
|
b) |
oppure, dopo la suddetta data, da un'autorità competente dello Stato membro sul cui territorio il bene culturale si trova dopo essere stato lecitamente e definitivamente spedito da un altro Stato membro o dopo essere stato importato da un paese terzo o reimportato da un paese terzo in seguito a una spedizione lecita da uno Stato membro verso detto paese terzo. |
Tuttavia, fermo restando il paragrafo 4, lo Stato membro competente conformemente al primo comma, lettera a) o lettera b), può non richiedere licenze di esportazione per i beni culturali elencati nell'allegato I, categoria A.1, primo e secondo trattino, qualora detti beni abbiano un interesse archeologico o scientifico limitato e purché non provengano direttamente da scavi, scoperte o siti archeologici in uno Stato membro e la loro presenza sul mercato sia lecita.
La licenza di esportazione può essere negata, ai sensi del presente regolamento, qualora i beni culturali in questione siano contemplati da una legislazione che tutela il patrimonio nazionale avente valore artistico, storico o archeologico nello Stato membro di cui trattasi.
Se necessario, l'autorità di cui al primo comma, lettera b), prende contatto con le autorità competenti dello Stato membro da cui il bene culturale proviene, in particolare le autorità competenti ai sensi della direttiva 93/7/CEE del Consiglio, del 15 marzo 1993, relativa alla restituzione dei beni culturali usciti illecitamente dal territorio di uno Stato membro (5).
3. La licenza di esportazione è valida in tutta la Comunità.
4. Fatte salve le disposizioni dei paragrafi da 1 a 3, l'esportazione diretta dal territorio doganale della Comunità di beni del patrimonio nazionale di valore artistico, storico o archeologico che non rientrano nella definizione di beni culturali ai sensi del presente regolamento è soggetta alla normativa nazionale dello Stato membro di esportazione.
Articolo 3
Autorità competenti
1. Gli Stati membri comunicano alla Commissione l'elenco delle autorità competenti per il rilascio delle licenze di esportazione di beni culturali.
2. La Commissione pubblica l'elenco di queste autorità, nonché le eventuali modifiche dello stesso, nella Gazzetta ufficiale dell'Unione europea, serie C.
Articolo 4
Presentazione della licenza
La licenza di esportazione è presentata, a sostegno della dichiarazione di esportazione, al momento dell'espletamento delle formalità doganali di esportazione, presso l'ufficio doganale competente per accettare tale dichiarazione.
Articolo 5
Restrizione del numero degli uffici doganali competenti
1. Gli Stati membri possono limitare il numero degli uffici doganali competenti per espletare le formalità di esportazione di beni culturali.
2. Quando si avvalgono della possibilità di cui al paragrafo 1, gli Stati membri comunicano alla Commissione l'elenco degli uffici doganali debitamente abilitati.
La Commissione pubblica tali informazioni nella Gazzetta ufficiale dell'Unione europea, serie C.
Articolo 6
Cooperazione amministrativa
Ai fini dell'attuazione del presente regolamento, si applicano mutatis mutandis le disposizioni del regolamento (CE) n. 515/97, in particolare quelle relative alla riservatezza delle informazioni.
Oltre a cooperare ai sensi del primo comma, gli Stati membri fanno tutto il necessario per stabilire, sul piano dei loro rapporti reciproci, una cooperazione tra le autorità doganali e le autorità competenti di cui all'articolo 4 della direttiva 93/7/CEE.
Articolo 7
Misure di attuazione
Le misure necessarie all'attuazione del presente regolamento, in particolare quelle relative al formulario da utilizzare (ad esempio, il modello e le caratteristiche tecniche), sono adottate secondo la procedura di cui all'articolo 8, paragrafo 2.
Articolo 8
Comitato
1. La Commissione è assistita da un comitato.
2. Nei casi in cui è fatto riferimento al presente paragrafo, si applicano gli articoli 3 e 7 della decisione 1999/468/CE.
Articolo 9
Sanzioni
Gli Stati membri determinano le sanzioni da irrogare in caso di violazione delle norme del presente regolamento e adottano ogni provvedimento necessario per assicurare l’applicazione delle sanzioni stesse. Le sanzioni devono essere efficaci, proporzionate e dissuasive.
Articolo 10
Relazione
1. Ogni Stato membro informa la Commissione delle misure che prende per l'esecuzione del presente regolamento.
La Commissione comunica tali informazioni agli altri Stati membri.
2. Ogni tre anni, la Commissione trasmette al Parlamento europeo, al Consiglio e al Comitato economico e sociale europeo una relazione sull'attuazione del presente regolamento.
Il Consiglio, su proposta della Commissione, procede ogni tre anni a esaminare e se del caso a rivalutare gli importi indicati nell'allegato I, per tener conto degli indicatori economici e monetari nella Comunità.
Articolo 11
Abrogazione
Il regolamento (CEE) n. 3911/92, come modificato dai regolamenti elencati all'allegato II, è abrogato.
I riferimenti al regolamento abrogato si intendono fatti al presente regolamento e si leggono secondo la tavola di concordanza dell’allegato III.
Articolo 12
Entrata in vigore
Il presente regolamento entra in vigore il ventesimo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, addì 18 dicembre 2008.
Per il Consiglio
Il presidente
M. BARNIER
(1) GU L 395 del 31.12.1992, pag. 1.
(2) Cfr. allegato II.
(3) GU L 184 del 17.7.1999, pag. 23.
(4) GU L 82 del 22.3.1997, pag. 1.
(5) GU L 74 del 27.3.1993, pag. 74.
ALLEGATO I
Categorie di beni culturali di cui all'articolo 1
|
1. |
Reperti archeologici aventi più di 100 anni, provenienti da: |
|
||
|
9705 00 00 |
|||
|
9706 00 00 |
|||
|
||||
|
2. |
Elementi costituenti parte integrante di monumenti artistici, storici o religiosi e provenienti dallo smembramento dei monumenti stessi, aventi più di 100 anni |
9705 00 00 9706 00 00 |
||
|
3. |
Quadri e pitture diversi da quelli appartenenti alla categoria 4 o 5, fatti interamente a mano, su qualsiasi supporto e con qualsiasi materiale (1) |
9701 |
||
|
4. |
Acquerelli, guazzi e pastelli eseguiti interamente a mano, su qualsiasi supporto (1) |
9701 |
||
|
5. |
Mosaici, diversi da quelli delle categorie 1 o 2, realizzati interamente a mano, con qualsiasi materia, e disegni fatti interamente a mano su qualsiasi supporto e con qualsiasi materia (1) |
6914 9701 |
||
|
6. |
Incisioni, stampe, serigrafie e litografie originali e relative matrici, nonché manifesti originali (1) |
Capitolo 49 9702 00 00 8442 50 99 |
||
|
7. |
Opere originali dell'arte statuaria o dell'arte scultoria e copie ottenute con il medesimo procedimento dell'originale (1), diverse da quelle della categoria 1 |
9703 00 00 |
||
|
8. |
Fotografie, film e relativi negativi (1) |
3704 3705 3706 4911 91 80 |
||
|
9. |
Incunaboli e manoscritti, comprese le carte geografiche e gli spartiti musicali, isolati o in collezione (1) |
9702 00 00 9706 00 00 4901 10 00 4901 99 00 4904 00 00 4905 91 00 4905 99 00 4906 00 00 |
||
|
10. |
Libri aventi più di 100 anni, isolati o in collezione |
9705 00 00 9706 00 00 |
||
|
11. |
Carte geografiche stampate aventi più di 200 anni |
9706 00 00 |
||
|
12. |
Archivi di qualsiasi natura e supporto, comprendenti elementi aventi più di 50 anni |
3704 3705 3706 4901 4906 9705 00 00 9706 00 00 |
||
|
13. |
|
9705 00 00 |
||
|
9705 00 00 |
|||
|
14. |
Mezzi di trasporto aventi più di 75 anni |
9705 00 00 Capitoli 86-89 |
||
|
15. |
Altri oggetti d'antiquariato non contemplati dalle categorie da A.1 a A.14 |
|
||
|
|
|||
|
giocattoli, giochi |
Capitolo 95 |
|||
|
vetrerie |
7013 |
|||
|
articoli di oreficeria |
7114 |
|||
|
mobili e oggetti d'arredamento |
Capitolo 94 |
|||
|
strumenti ottici, fotografici o cinematografici |
Capitolo 90 |
|||
|
strumenti musicali |
Capitolo 92 |
|||
|
orologi |
Capitolo 91 |
|||
|
opere in legno |
Capitolo 44 |
|||
|
vasellame |
Capitolo 69 |
|||
|
arazzi |
5805 00 00 |
|||
|
tappeti |
Capitolo 57 |
|||
|
carte da parati |
4814 |
|||
|
armi |
Capitolo 93 |
|||
|
9706 00 00 |
I beni culturali rientranti nelle categorie da A.1 a A.15 sono disciplinati dal presente regolamento soltanto se il loro valore è pari o superiore ai valori di cui al punto B.
B. Valori applicabili a talune categorie di cui al punto A (in EUR)
Valori:
|
|
qualunque ne sia il valore
|
|
|
15 000
|
|
|
30 000
|
|
|
50 000
|
|
|
150 000
|
Il rispetto delle condizioni relative ai valori deve essere accertato al momento della presentazione della domanda di licenza di esportazione. Il valore è quello del bene culturale nello Stato membro di cui all'articolo 2, paragrafo 2, del regolamento.
Per gli Stati membri che non adottano l'euro, i valori espressi in euro nell'allegato I sono convertiti e espressi nelle monete nazionali al tasso di cambio del 31 dicembre 2001 pubblicato nella Gazzetta ufficiale delle Comunità europee. Tale controvalore nelle monete nazionali è rivisto ogni due anni dal 31 dicembre 2001 in poi. Il calcolo del controvalore si basa sulla media del valore quotidiano di tali monete, espresso in euro, relativo al periodo di ventiquattro mesi terminante l'ultimo giorno del mese di agosto che precede la revisione avente effetto dal 31 dicembre. Questo metodo di calcolo è riesaminato, su proposta della Commissione, dal comitato consultivo dei beni culturali, in linea di principio due anni dopo la prima applicazione. Per ogni revisione i valori espressi in euro e i loro controvalori in moneta nazionale sono periodicamente pubblicati nella Gazzetta ufficiale dell’Unione europea nei primi giorni del mese di novembre precedente la data da cui ha effetto la revisione.
(1) Aventi più di 50 anni e non appartenenti all'autore.
(2) Quali definite dalla Corte di giustizia nella sentenza n. 252/84: «Gli oggetti da collezione ai sensi della voce 97.05 della TDC sono quelli che possiedono le qualità richieste per far parte di una collezione, cioè gli oggetti relativamente rari, che non sono normalmente usati secondo la loro destinazione originaria, che formano oggetto di transazioni speciali al di fuori del mercato abituale degli analoghi oggetti di uso comune e hanno un valore elevato.»
ALLEGATO II
Regolamento abrogato e sue modificazioni successive
|
Regolamento (CEE) n. 3911/92 del Consiglio |
|
|
Regolamento (CE) n. 2469/96 del Consiglio |
|
|
Regolamento (CE) n. 974/2001 del Consiglio |
|
|
Regolamento (CE) n. 806/2003 del Consiglio |
limitatamente all’allegato I, punto 2 |
ALLEGATO III
TAVOLA DI CONCORDANZA
|
Regolamento (CEE) n. 3911/92 |
Presente regolamento |
|
Articolo 1 |
Articolo 1 |
|
Articolo 2, paragrafo 1 |
Articolo 2, paragrafo 1 |
|
Articolo 2, paragrafo 2, primo comma, alinea |
Articolo 2, paragrafo 2, primo comma, alinea |
|
Articolo 2, paragrafo 2, primo comma, primo trattino |
Articolo 2, paragrafo 2, primo comma, lettera a) |
|
Articolo 2, paragrafo 2, primo comma, secondo trattino |
Articolo 2, paragrafo 2, primo comma, lettera b) |
|
Articolo 2, paragrafo 2, secondo comma |
Articolo 2, paragrafo 2, secondo comma |
|
Articolo 2, paragrafo 2, terzo comma |
Articolo 2, paragrafo 2, terzo comma |
|
Articolo 2, paragrafo 2, quarto comma |
Articolo 2, paragrafo 2, quarto comma |
|
Articolo 2, paragrafo 3 |
Articolo 2, paragrafo 3 |
|
Articolo 2, paragrafo 4 |
Articolo 2, paragrafo 4 |
|
Articoli da 3 a 9 |
Articoli da 3 a 9 |
|
Articolo 10, primo comma |
Articolo 10, paragrafo 1, primo comma |
|
Articolo 10, secondo comma |
Articolo 10, paragrafo 1, secondo comma |
|
Articolo 10, terzo comma |
Articolo 10, paragrafo 2, primo comma |
|
Articolo 10, quarto comma |
— |
|
Articolo 10, quinto comma |
Articolo 10, paragrafo 2, secondo comma |
|
— |
Articolo 11 |
|
Articolo 11 |
Articolo 12 |
|
Allegato, punti A.1, A.2 e A.3 |
Allegato I, punti A.1, A.2 e A.3 |
|
Allegato, punto A.3 bis |
Allegato I, punto A.4 |
|
Allegato, punto A.4 |
Allegato I, punto A.5 |
|
Allegato, punto A.5 |
Allegato I, punto A.6 |
|
Allegato, punto A.6 |
Allegato I, punto A.7 |
|
Allegato, punto A.7 |
Allegato I, punto A.8 |
|
Allegato, punto A.8 |
Allegato I, punto A.9 |
|
Allegato, punto A.9 |
Allegato I, punto A.10 |
|
Allegato, punto A.10 |
Allegato I, punto A.11 |
|
Allegato, punto A.11 |
Allegato I, punto A.12 |
|
Allegato, punto A.12 |
Allegato I, punto A.13 |
|
Allegato, punto A.13 |
Allegato I, punto A.14 |
|
Allegato, punto A.14 |
Allegato I, punto A.15 |
|
Allegato, punto B |
Allegato I, punto B |
|
— |
Allegato II |
|
— |
Allegato III |
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/8 |
REGOLAMENTO (CE) N. 117/2009 DELLA COMMISSIONE
del 9 febbraio 2009
recante fissazione dei valori forfettari all’importazione ai fini della determinazione del prezzo di entrata di taluni ortofrutticoli
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
visto il regolamento (CE) n. 1234/2007 del Consiglio, del 22 ottobre 2007, recante organizzazione comune dei mercati agricoli e disposizioni specifiche per taluni prodotti agricoli (regolamento unico OCM) (1),
visto il regolamento (CE) n. 1580/2007 della Commissione, del 21 dicembre 2007, recante modalità di applicazione dei regolamenti (CE) n. 2200/96, (CE) n. 2201/96 e (CE) n. 1182/2007 nel settore degli ortofrutticoli (2), in particolare l’articolo 138, paragrafo 1,
considerando quanto segue:
Il regolamento (CE) n. 1580/2007 prevede, in applicazione dei risultati dei negoziati commerciali multilaterali dell’Uruguay round, i criteri per la fissazione da parte della Commissione dei valori forfettari all’importazione dai paesi terzi, per i prodotti e i periodi indicati nell’allegato XV, parte A, del medesimo regolamento,
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
I valori forfettari all’importazione di cui all’articolo 138 del regolamento (CE) n. 1580/2007 sono quelli fissati nell’allegato del presente regolamento.
Articolo 2
Il presente regolamento entra in vigore il 10 febbraio 2009.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 9 febbraio 2009.
Per la Commissione
Jean-Luc DEMARTY
Direttore generale dell'Agricoltura e dello sviluppo rurale
(1) GU L 299 del 16.11.2007, pag. 1.
(2) GU L 350 del 31.12.2007, pag. 1.
ALLEGATO
Valori forfettari all’importazione ai fini della determinazione del prezzo di entrata di taluni ortofrutticoli
|
(EUR/100 kg) |
||
|
Codice NC |
Codice paesi terzi (1) |
Valore forfettario all'importazione |
|
0702 00 00 |
IL |
111,0 |
|
JO |
68,6 |
|
|
MA |
45,0 |
|
|
TN |
134,4 |
|
|
TR |
89,8 |
|
|
ZZ |
89,8 |
|
|
0707 00 05 |
JO |
155,5 |
|
MA |
134,2 |
|
|
TR |
151,1 |
|
|
ZZ |
146,9 |
|
|
0709 90 70 |
MA |
116,3 |
|
TR |
117,2 |
|
|
ZZ |
116,8 |
|
|
0709 90 80 |
EG |
126,4 |
|
ZZ |
126,4 |
|
|
0805 10 20 |
EG |
47,5 |
|
IL |
54,0 |
|
|
MA |
59,3 |
|
|
TN |
40,6 |
|
|
TR |
65,8 |
|
|
ZA |
44,9 |
|
|
ZZ |
52,0 |
|
|
0805 20 10 |
IL |
152,1 |
|
MA |
100,5 |
|
|
TR |
52,0 |
|
|
ZZ |
101,5 |
|
|
0805 20 30, 0805 20 50, 0805 20 70, 0805 20 90 |
CN |
72,2 |
|
IL |
87,2 |
|
|
JM |
101,6 |
|
|
MA |
158,6 |
|
|
PK |
40,0 |
|
|
TR |
62,7 |
|
|
ZZ |
87,1 |
|
|
0805 50 10 |
EG |
64,1 |
|
MA |
67,1 |
|
|
TR |
53,5 |
|
|
ZZ |
61,6 |
|
|
0808 10 80 |
AR |
91,9 |
|
CA |
90,4 |
|
|
CL |
67,8 |
|
|
CN |
82,1 |
|
|
MK |
32,6 |
|
|
US |
114,6 |
|
|
ZZ |
79,9 |
|
|
0808 20 50 |
AR |
107,7 |
|
CL |
73,7 |
|
|
CN |
58,5 |
|
|
US |
108,5 |
|
|
ZA |
104,3 |
|
|
ZZ |
90,5 |
|
(1) Nomenclatura dei paesi stabilita dal regolamento (CE) n. 1833/2006 della Commissione (GU L 354 del 14.12.2006, pag. 19). Il codice «ZZ» rappresenta le «altre origini».
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/10 |
REGOLAMENTO (CE) N. 118/2009 DELLA COMMISSIONE
del 9 febbraio 2009
recante modifica dei prezzi rappresentativi e dei dazi addizionali all'importazione per taluni prodotti del settore dello zucchero, fissati dal regolamento (CE) n. 945/2008, per la campagna 2008/2009
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
visto il regolamento (CE) n. 1234/2007, del Consiglio, del 22 ottobre 2007, recante organizzazione comune dei mercati agricoli e disposizioni specifiche per taluni prodotti agricoli (regolamento unico OCM) (1),
visto il regolamento (CE) n. 951/2006 della Commissione, del 30 giugno 2006, recante modalità di applicazione del regolamento (CE) n. 318/2006 del Consiglio per quanto riguarda gli scambi di prodotti del settore dello zucchero con i paesi terzi (2), in particolare l'articolo 36, paragrafo 2, secondo comma, seconda frase,
considerando quanto segue:
|
(1) |
Gli importi dei prezzi rappresentativi e dei dazi addizionali applicabili all'importazione di zucchero bianco, di zucchero greggio e di taluni sciroppi per la campagna 2008/2009 sono stati fissati dal regolamento (CE) n. 945/2008 della Commissione (3). Tali prezzi e dazi sono stati modificati da ultimo dal regolamento (CE) n. 100/2009 della Commissione (4). |
|
(2) |
Alla luce dei dati attualmente in possesso della Commissione risulta necessario modificare gli importi in vigore, in conformità delle norme e delle modalità previste dal regolamento (CE) n. 951/2006, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
I prezzi rappresentativi e i dazi addizionali applicabili all'importazione dei prodotti contemplati dall'articolo 36 del regolamento (CE) n. 951/2006, fissati dal regolamento (CE) n. 945/2008 per la campagna 2008/2009, sono modificati e figurano nell'allegato del presente regolamento.
Articolo 2
Il presente regolamento entra in vigore il 10 febbraio 2009.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 9 febbraio 2009.
Per la Commissione
Jean-Luc DEMARTY
Direttore generale dell'Agricoltura e dello sviluppo rurale
(1) GU L 299 del 16.11.2007, pag. 1.
(2) GU L 178 dell'1.7.2006, pag. 24.
(3) GU L 258 del 26.9.2008, pag. 56.
(4) GU L 34 del 4.2.2009, pag. 3.
ALLEGATO
Importi modificati dei prezzi rappresentativi e dei dazi addizionali all'importazione per lo zucchero bianco, lo zucchero greggio e i prodotti del codice NC 1702 90 95 applicabili a partire dal 10 febbraio 2009
|
(EUR) |
||
|
Codice NC |
Importo del prezzo rappresentativo per 100 kg netti di prodotto |
Importo del dazio addizionale per 100 kg netti di prodotto |
|
1701 11 10 (1) |
25,95 |
3,50 |
|
1701 11 90 (1) |
25,95 |
8,56 |
|
1701 12 10 (1) |
25,95 |
3,37 |
|
1701 12 90 (1) |
25,95 |
8,13 |
|
1701 91 00 (2) |
29,84 |
10,31 |
|
1701 99 10 (2) |
29,84 |
5,79 |
|
1701 99 90 (2) |
29,84 |
5,79 |
|
1702 90 95 (3) |
0,30 |
0,35 |
(1) Importo fissato per la qualità tipo definita nell'allegato IV, punto III, del regolamento (CE) n. 1234/2007.
(2) Importo fissato per la qualità tipo definita nell'allegato IV, punto II, del regolamento (CE) n. 1234/2007.
(3) Importo fissato per 1 % di tenore di saccarosio.
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/12 |
REGOLAMENTO (CE) N. 119/2009 DELLA COMMISSIONE
del 9 febbraio 2009
che stabilisce un elenco di paesi terzi, o di parti di essi, nonché i requisiti di certificazione veterinaria ai fini dell’importazione nella Comunità, o del transito sul suo territorio, della carne dei leporidi selvatici, di alcuni mammiferi terrestri selvatici e dei conigli d’allevamento
(Testo rilevante ai fini del SEE)
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
vista la direttiva 2002/99/CE del Consiglio, del 16 dicembre 2002, che stabilisce norme di polizia sanitaria per la produzione, la trasformazione, la distribuzione e l’introduzione di prodotti di origine animale destinati al consumo umano (1), in particolare l’articolo 8, paragrafo 1, primo comma, l’articolo 9, paragrafo 2, punto b), e l’articolo 9, paragrafo 4, punti b) e c),
visto il regolamento (CE) n. 852/2004 del Parlamento europeo e del Consiglio, del 29 aprile 2004, sull’igiene dei prodotti alimentari (2), e in particolare l’articolo 12,
visto il regolamento (CE) n. 853/2004 del Parlamento europeo e del Consiglio, del 29 aprile 2004, che stabilisce norme specifiche in materia di igiene per gli alimenti di origine animale (3), e in particolare l’articolo 9,
visto il regolamento (CE) n. 854/2004 del Parlamento europeo e del Consiglio, del 29 aprile 2004, che stabilisce norme specifiche per l’organizzazione di controlli ufficiali sui prodotti di origine animale destinati al consumo umano (4), in particolare l’articolo 11, paragrafo 1 e l’articolo 14, paragrafo 4,
visto il regolamento (CE) n. 882/2004 del Parlamento europeo e del Consiglio, del 29 aprile 2004, relativo ai controlli ufficiali intesi a verificare la conformità alla normativa in materia di mangimi e di alimenti e alle norme sulla salute e sul benessere degli animali (5), in particolare l’articolo 48, paragrafo 1,
considerando quanto segue:
|
(1) |
La decisione 2000/585/CE della Commissione (6), del 7 settembre 2000, stabilisce l’elenco dei paesi terzi da cui gli Stati membri possono autorizzare l’importazione di carni di coniglio e di alcune carni di selvaggina in libertà e di selvaggina d’allevamento e definisce le condizioni sanitarie e di polizia sanitaria nonché la certificazione veterinaria relative a tali importazioni. |
|
(2) |
Per motivi di coerenza della legislazione comunitaria, la normativa comunitaria sulle importazioni della carne di leporidi selvatici, di alcuni mammiferi terrestri selvatici e dei conigli d’allevamento deve tener conto dei requisiti di polizia sanitaria fissati nei regolamenti (CE) n. 852/2004, (CE) n. 853/2004, (CE) n. 854/2004 e (CE) n. 882/2004. |
|
(3) |
Le misure previste dal presente regolamento non pregiudicano le norme d’attuazione del regolamento (CE) n. 338/97 del Consiglio, del 9 dicembre 1996, relativo alla protezione di specie della flora e della fauna selvatiche mediante il controllo del loro commercio (7). |
|
(4) |
Al fine di armonizzare le condizioni comunitarie che disciplinano le importazioni nella Comunità delle merci interessate, al fine di renderle più trasparenti e di semplificare le procedure legislative necessarie a modificarle, occorre che quelle condizioni siano definite nei rispettivi modelli dei certificati veterinari descritti nel presente regolamento. |
|
(5) |
I certificati veterinari necessari all’importazione nella Comunità, al transito e allo stoccaggio durante il transito sul territorio comunitario, della carne dei leporidi selvatici, di alcuni mammiferi terrestri selvatici e dei conigli d’allevamento devono conformarsi ai rispettivi modelli standard descritti nell’allegato I della decisione 2007/240/CE della Commissione, del 16 aprile 2007, che istituisce nuovi certificati veterinari per l’introduzione nella Comunità di animali vivi, sperma, embrioni, ovuli e prodotti d’origine animale nell’ambito delle decisioni 79/542/CEE, 92/260/CEE, 93/195/CEE, 93/196/CEE, 93/197/CEE, 95/328/CE, 96/333/CE, 96/539/CE, 96/540/CE, 2000/572/CE, 2000/585/CE, 2000/666/CE, 2002/613/CE, 2003/56/CE, 2003/779/CE, 2003/804/CE, 2003/858/CE, 2003/863/CE, 2003/881/CE, 2004/407/CE, 2004/438/CE, 2004/595/CE, 2004/639/CE e 2006/168/CE (8). |
|
(6) |
I modelli dei certificati veterinari, descritti nel presente regolamento, necessari all’importazione nella Comunità, al transito e allo stoccaggio durante il transito sul territorio comunitario, della carne dei leporidi selvatici, di alcuni mammiferi terrestri selvatici e dei conigli d’allevamento devono anche essere compatibili con il sistema TRACES, come previsto dalla decisione 2004/292/CE della Commissione, del 30 marzo 2004, relativa all’applicazione del sistema TRACES (9). |
|
(7) |
L’elenco dei paesi terzi o di parti di essi, di cui all’allegato II della decisione 79/542/CEE del Consiglio (10), deve essere usato per le importazioni nella Comunità, o il transito sul suo territorio, della carne di leporidi selvatici e di conigli d’allevamento. L’elenco dei paesi è anche necessario per importare nella Comunità, o far transitare sul suo territorio, la carne di mammiferi terrestri selvatici diversi dagli ungulati e dai leporidi. |
|
(8) |
È opportuno prevedere condizioni specifiche per il transito attraverso la Comunità di partite da e per la Russia, data la situazione geografica di Kaliningrad che riguarda solo la Lettonia, la Lituania e la Polonia. |
|
(9) |
Per evitare perturbazioni degli scambi, occorre autorizzare l’uso dei certificati veterinari rilasciati in conformità della decisione 2000/585/CE per un periodo transitorio. |
|
(10) |
Per motivi di chiarezza della legislazione comunitaria, occorre abrogare la decisione 2000/585/CE e sostituirla con il presente regolamento. |
|
(11) |
Le misure di cui al presente regolamento sono conformi al parere del Comitato permanente per la catena alimentare e la salute degli animali, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
Oggetto e campo di applicazione
1. Il presente regolamento stabilisce:
|
a) |
un elenco di paesi terzi, o di parti di essi, dai quali possono essere importate nella Comunità, o fatte transitare sul suo territorio, le seguenti merci:
|
|
b) |
i requisiti della certificazione veterinaria per le merci elencate ai punti i), ii) e iii) (di seguito, «le merci»). |
2. Senza pregiudicare la restrizione di cui all’articolo 5, paragrafo 2, ai fini del presente regolamento, «transito» copre lo stoccaggio durante il transito [e la messa in deposito come stabilito dall’articolo 12, paragrafo 4, e dall’articolo 13 della direttiva 97/78/CE del Consiglio (11)].
3. Il presente regolamento si applica lasciando impregiudicati:
|
i) |
requisiti specifici di certificazione previsti da accordi comunitari con paesi terzi; |
|
ii) |
le regole di certificazione contenute nelle norme di attuazione del regolamento (CE) n. 338/97 relativo alla protezione di specie della flora e della fauna selvatiche mediante il controllo del loro commercio. |
Articolo 2
Definizione
Ai fini del presente regolamento, «leporidi selvatici» indica conigli selvatici e lepri.
Articolo 3
Elenchi di paesi terzi o di parti di essi dai quali le merci possono essere importate nella Comunità o fatte transitare sul suo territorio
Le merci possono essere importate nella Comunità o transitare sul suo territorio solo da paesi terzi, o da parti di essi, che figurino nell’elenco dell’allegato I, parte 1, o in esso citati.
Articolo 4
Certificazione veterinaria
1. Le merci importate nella Comunità saranno accompagnate da un certificato veterinario conforme al modello descritto nell’allegato II per la merce interessata, compilato in conformità delle note di cui all’allegato I, parte 4.
2. Le merci in transito sul territorio comunitario saranno accompagnate da un certificato conforme al modello di cui all’allegato III.
3. La conformità alle garanzie complementari, richieste per un determinato Stato membro o parte di esso nelle colonne 4, 6 e 8 della tabella di cui all’allegato I, parte 1, e descritte nell’allegato I, parte 3, verrà attestata compilando, nel certificato veterinario, la relativa sezione della merce interessata.
4. È consentito l’uso della certificazione elettronica e di altri sistemi concordati, armonizzati a livello comunitario.
Articolo 5
Deroga per il transito attraverso la Lettonia, la Lituania e la Polonia
1. In deroga all’articolo 4, paragrafo 2, il transito di partite da e per la Russia, diretto o attraverso un altro paese terzo, per strada o ferrovia tra i posti d’ispezione frontalieri in Lettonia, Lituania e Polonia elencati all’allegato della decisione 2001/881/CE della Commissione (12), è consentito a condizione che:
|
a) |
il veterinario ufficiale in servizio presso il posto d’ispezione frontaliero d’ingresso abbia sigillato la partita con un sigillo numerato progressivamente; |
|
b) |
il veterinario ufficiale in servizio presso il posto d’ispezione frontaliero d’ingresso, abbia timbrato con la dicitura «Solo per il transito verso la Russia attraverso la CE» ogni pagina dei documenti, di cui all’articolo 7 della direttiva 97/78/CE, che accompagnano la partita; |
|
c) |
siano soddisfatti i requisiti procedurali di cui all’articolo 11 della direttiva 97/78/CE; |
|
d) |
il veterinario ufficiale in servizio presso il posto d’ispezione frontaliero d’ingresso abbia certificato sul documento veterinario comune di entrata l’ammissibilità della partita al transito. |
2. In ottemperanza all’articolo 12, paragrafo 4, o all’articolo 13 della direttiva 97/78/CE, le partite di cui al paragrafo 1 non possono essere scaricate o stoccate nel territorio comunitario.
3. L’autorità competente effettua controlli regolari volti a garantire che il numero delle partite di cui al paragrafo 1 e i quantitativi corrispondenti dei prodotti in uscita dal territorio comunitario corrispondano al numero di partite e ai quantitativi in entrata.
Articolo 6
Abrogazione
La decisione 2000/585/CE è abrogata.
I riferimenti alle decisioni abrogate si intendono fatti al presente regolamento e vanno letti secondo la tavola di concordanza di cui all’allegato IV.
Articolo 7
Disposizioni transitorie
Le merci oggetto dei relativi certificati veterinari rilasciati in conformità della decisione 2000/585/CE possono essere importate nella Comunità o transitare sul suo territorio fino al 30 giugno 2009.
Articolo 8
Entrata in vigore e applicabilità
Il presente regolamento entra in vigore il ventesimo giorno successivo a quello della sua pubblicazione nella Gazzetta ufficiale dell’Unione europea.
Esso si applica a decorrere dal 1o giugno 2009.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 9 febbraio 2009.
Per la Commissione
Androulla VASSILIOU
Membro della Commissione
(1) GU L 18 del 23.1.2003, pag. 11.
(2) GU L 139 del 30.4.2004, pag. 1; rettifica nella GU L 226 del 25.6.2004, pag. 3.
(3) GU L 139 del 30.4.2004, pag. 55; rettifica nella GU L 226 del 25.6.2004, pag. 22.
(4) GU L 139 del 30.4.2004, pag. 206; rettifica nella GU L 226 del 25.6.2004, pag. 83.
(5) GU L 165 del 30.4.2004, pag. 1; rettifica nella GU L 191 del 28.5.2004, pag. 1.
(6) GU L 251 del 6.10.2000, pag. 1.
(7) GU L 61 del 3.3.1997, pag. 1.
(8) GU L 104 del 21.4.2007, pag. 37.
(9) GU L 94 del 31.3.2004, pag. 63.
(10) GU L 146 del 14.6.1979, pag. 15.
(11) GU L 24 del 30.1.1998, pag. 9.
(12) GU L 326 dell’11.12.2001, pag. 44.
ALLEGATO I
CARNE DI LEPORIDI SELVATICI, DI ALCUNI MAMMIFERI TERRESTRI SELVATICI E DI CONIGLI D’ALLEVAMENTO
PARTE 1
Elenco dei paesi terzi, di parti di essi e delle garanzie complementari
|
Paese |
Codice del territorio |
Leporidi |
Mammiferi terrestri selvatici diversi dagli ungulati e dai leporidi |
||||||||||
|
Selvatici |
Conigli d’allevamento |
||||||||||||
|
MC |
AG |
MC |
AG |
MC |
AG |
||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
||||||
|
Australia |
AU |
WL |
|
RM |
|
WM |
|
||||||
|
Canada |
CA |
WL |
|
RM |
|
WM |
|
||||||
|
Groenlandia |
GL |
WL |
|
RM |
|
WM |
|
||||||
|
Nuova Zelanda |
NZ |
WL |
|
RM |
|
WM |
|
||||||
|
Russia |
RU |
WL |
|
RM |
|
WM |
|
||||||
|
Qualsiasi altro paese terzo, o parte di esso, elencato nelle colonne 1 e 3 della tabella di cui alla decisione 79/542/CEE del Consiglio, allegato II, parte 1. |
WL |
|
RM |
|
|
|
|||||||
|
|||||||||||||
PARTE 2
Modello dei certificati veterinari
Modello/i:
|
«WL» |
: |
modello di certificato veterinario per le carni dei leporidi selvatici (conigli e lepri) |
|
«WM» |
: |
modello di certificato veterinario per la carne dei mammiferi terrestri selvatici diversi dagli ungulati e dai leporidi |
|
«RM» |
: |
modello di certificato veterinario per la carne dei conigli d’allevamento |
PARTE 3
Garanzie complementari
PARTE 4
Note per la certificazione veterinaria
|
a) |
I certificati veterinari basati sui modelli di cui al presente allegato, parte 2, e sullo schema del modello corrispondente alla merce interessata sono rilasciati dal paese terzo esportatore o da una parte di esso. Essi devono contenere, nell’ordine indicato dal modello, le attestazioni richieste per qualsiasi paese terzo ed eventualmente i requisiti sanitari complementari necessari per il paese terzo esportatore o per una parte di esso. L’originale del certificato veterinario indicherà anche eventuali garanzie complementari richieste dallo Stato membro dell’UE di destinazione per la merce interessata. |
|
b) |
Per ogni partita di merce interessata, esportata verso la stessa destinazione da un territorio che figuri nel presente allegato, parte 1, colonna 2, e trasportata nello stesso vagone ferroviario, autocarro, aereo o nave, occorre presentare un certificato singolo separato. |
|
c) |
L’originale dei certificati consiste in un foglio singolo, stampato su entrambi i lati; se occorrono più pagine, esso dovrà essere tale che l’insieme delle pagine formi un tutto unico non separabile. |
|
d) |
Il certificato va redatto almeno in una lingua ufficiale dello Stato membro in cui avviene l’ispezione al posto di frontiera e in una lingua ufficiale dello Stato membro di destinazione. Tali Stati membri possono tuttavia consentire l’uso di una lingua comunitaria diversa dalla propria, accompagnata se necessario da una traduzione ufficiale. |
|
e) |
Se, al fine di identificare i vari elementi che compongono la partita, si allegano al certificato delle pagine supplementari, anche queste ultime vanno considerate parte integrante dell’originale del certificato se su ciascuna di esse figurano la firma e il timbro del veterinario ufficiale che rilascia la certificazione. |
|
f) |
Se il certificato, comprendente pagine supplementari di cui alla nota e), si compone di più pagine, ciascuna pagina deve recare, in basso, una numerazione del tipo «–x(page number) di y(numero totale delle pagine)–» e, in alto, il numero di codice del certificato, attribuito dall’autorità competente. |
|
g) |
L’originale del certificato va compilato e firmato da un veterinario ufficiale non prima delle 24 ore che precedono il carico della partita destinata a essere importata nella Comunità, a meno che la legislazione comunitaria non specifichi altrimenti. A tal fine l’autorità competente del paese esportatore garantisce l’applicazione di criteri di certificazione equivalenti a quelli previsti dalla direttiva 96/93/CE del Consiglio (1). Il colore della firma deve essere diverso da quello del testo stampato. La stessa norma si applica ai timbri diversi da quelli a secco. |
|
h) |
L’originale del certificato deve accompagnare la spedizione fino al posto d’ispezione d’ingresso del confine della Comunità europea. |
ALLEGATO II
MODELLI DI CERTIFICATI VETERINARI DESTINATI ALL’IMPORTAZIONE NELLA COMUNITÀ EUROPEA DELLE CARNI DI LEPORIDI SELVATICI, DI ALCUNI MAMMIFERI TERRESTRI SELVATICI E DI CONIGLI D’ALLEVAMENTO
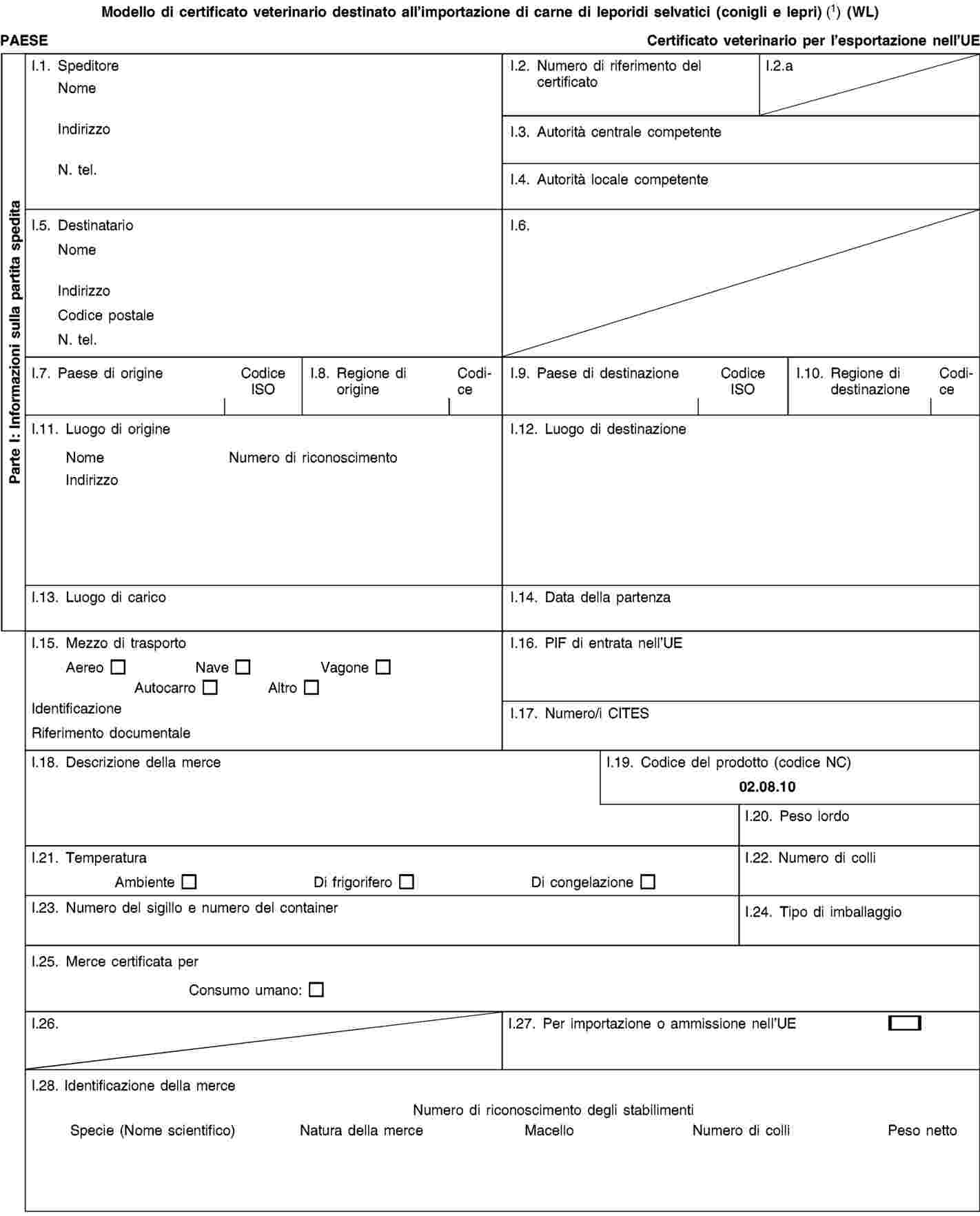





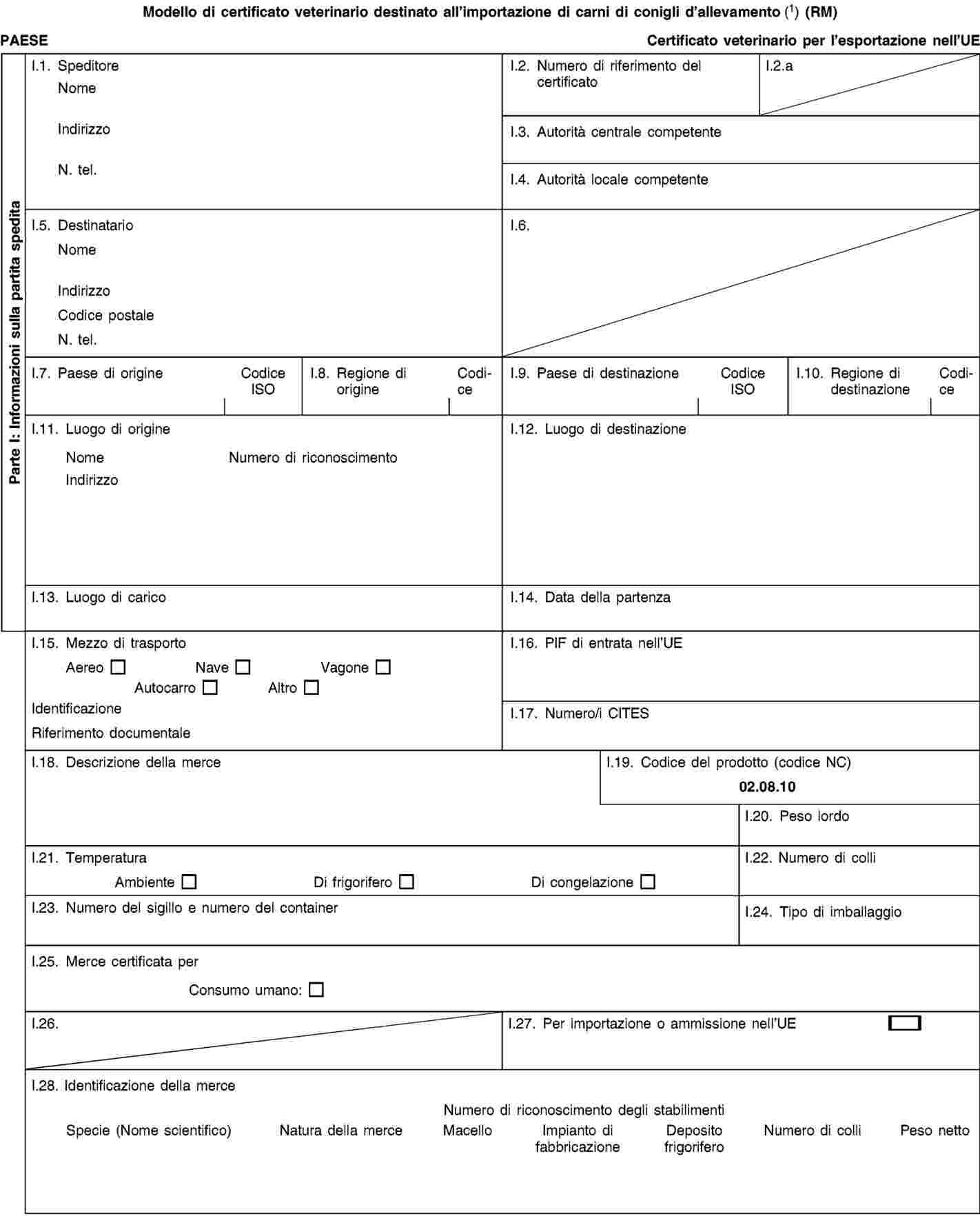


ALLEGATO III
(di cui all’articolo 4, paragrafo 2)
Modello di certificato veterinario destinato al transito/stoccaggio di carni di leporidi selvatici, di conigli d’allevamento e di mammiferi terrestri selvatici diversi dagli ungulati


ALLEGATO IV
(di cui all’articolo 6)
Tabella di concordanza
|
Decisione 2000/585/CE |
Presente regolamento |
|
Articolo 2 |
Articolo 1 |
|
— |
Articolo 2 |
|
Articolo 2 bis, lettera a) |
Articolo 3 |
|
Articolo 2 bis, lettere b), c) e d) |
Articolo 4 |
|
Articolo 2 ter |
Articolo 5 |
|
Articolo 4, paragrafo 1 |
Articolo 6 |
|
Articolo 4, paragrafo 2 |
Articolo 7 |
|
Articolo 3 |
Articolo 8 |
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/29 |
REGOLAMENTO (CE) N. 120/2009 DELLA COMMISSIONE
del 9 febbraio 2009
recante modifica del regolamento (CEE) n. 574/72 del Consiglio che stabilisce le modalità di applicazione del regolamento (CEE) n. 1408/71 relativo all'applicazione dei regimi di sicurezza sociale ai lavoratori subordinati e ai loro familiari che si spostano all'interno della Comunità
(Testo rilevante ai fini del SEE)
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
visto il regolamento (CEE) n. 574/72 del Consiglio, del 21 marzo 1972, che stabilisce le modalità di applicazione del regolamento (CEE) n. 1408/71 relativo all'applicazione dei regimi di sicurezza sociale ai lavoratori subordinati e ai loro familiari che si spostano all'interno della Comunità (1), in particolare l'articolo 122,
considerando quanto segue:
|
(1) |
Alcuni Stati membri o le loro autorità competenti hanno richiesto che siano apportate modifiche agli allegati del regolamento (CEE) n. 574/72. |
|
(2) |
Le modifiche proposte derivano da decisioni adottate dagli Stati membri interessati o dalle loro autorità competenti che designano le autorità incaricate di vigilare affinché la legislazione sulla sicurezza sociale sia attuata conformemente al diritto comunitario. |
|
(3) |
Le convenzioni bilaterali stipulate in vista dell'applicazione delle disposizioni del regolamento (CEE) n. 574/72 sono elencate nell'allegato 5 di tale regolamento. |
|
(4) |
È stato ottenuto il parere unanime della Commissione amministrativa per la Sicurezza sociale dei lavoratori migranti, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
Gli allegati da 2 a 5 del regolamento (CEE) n. 574/72 sono modificati conformemente all'allegato del presente regolamento.
Articolo 2
Il presente regolamento entra in vigore il ventesimo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 9 febbraio 2009.
Per la Commissione
Vladimír ŠPIDLA
Membro della Commissione
(1) GU L 74 del 27.3.1972, pag. 1.
ALLEGATO
Gli allegati da 2 a 5 del regolamento (CEE) n. 574/72 sono modificati come segue.
|
1. |
L'allegato 2 è modificato come segue.
|
|
2. |
L'allegato 3 è modificato come segue.
|
|
3. |
L'allegato 4 è modificato come segue.
|
|
4. |
L'allegato 5 è modificato come segue.
|
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/33 |
REGOLAMENTO (CE) N. 121/2009 DELLA COMMISSIONE
del 9 febbraio 2009
che fissa l'importo supplementare da versare in Bulgaria per le pesche destinate alla trasformazione nell'ambito della campagna di commercializzazione 2007-2008 conformemente al regolamento (CE) n. 679/2007
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
visto il trattato di adesione della Bulgaria e della Romania,
visto l'atto di adesione della Bulgaria e della Romania,
visto il regolamento (CE) n. 679/2007 della Commissione, del 18 giugno 2007, recante fissazione, per la campagna di commercializzazione 2007-2008, dell'importo dell'aiuto per le pesche destinate alla trasformazione (1), in particolare l'articolo 2, paragrafo 1,
considerando quanto segue:
|
(1) |
In applicazione dell'articolo 39, paragrafo 2, del regolamento (CE) n. 1535/2003 della Commissione, del 29 agosto 2003, recante modalità di applicazione del regolamento (CE) n. 2201/96 del Consiglio per quanto riguarda il regime di aiuti nel settore dei prodotti trasformati a base di ortofrutticoli (2), la Bulgaria ha notificato alla Commissione che 119,46 tonnellate di pesche avevano beneficiato di un aiuto alla trasformazione nel quadro del suddetto regime per la campagna 2007-2008. Il limite di trasformazione fissato per questo Stato membro nell'allegato III del regolamento (CE) n. 2201/96 del Consiglio (3) non è perciò stato superato. Un importo supplementare di 11,92 EUR per tonnellata dovrà di conseguenza essere versato per i suddetti quantitativi. |
|
(2) |
Per la campagna di commercializzazione 2007-2008, i produttori della Romania non hanno presentato domande di aiuto per le pesche destinate alla trasformazione. In tale Stato membro non è pertanto necessario versare alcun importo supplementare per la suddetta campagna, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
L'importo supplementare di cui all'articolo 2, paragrafo 1, del regolamento (CE) n. 679/2007, pari a 11,92 EUR per tonnellata di pesche destinate alla trasformazione, è versato in Bulgaria successivamente alla campagna di commercializzazione 2007-2008.
Articolo 2
Il presente regolamento entra in vigore il terzo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 9 febbraio 2009.
Per la Commissione
Mariann FISCHER BOEL
Membro della Commissione
(1) GU L 157 del 19.6.2007, pag. 12.
(2) GU L 218 del 30.8.2003, pag. 14.
(3) GU L 297 del 21.11.1996, pag. 29.
II Atti adottati a norma dei trattati CE/Euratom la cui pubblicazione non è obbligatoria
DECISIONI
Commissione
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/34 |
DECISIONE DELLA COMMISSIONE
del 3 febbraio 2009
che modifica la decisione 2002/364/CE relativa alle specifiche tecniche comuni per i dispositivi medico-diagnostici in vitro
[notificata con il numero C(2009) 565]
(Testo rilevante ai fini del SEE)
(2009/108/CE)
LA COMMISSIONE DELLE COMUNITÀ EUROPEE,
visto il trattato che istituisce la Comunità europea,
vista la direttiva 98/79/CE del Parlamento europeo e del Consiglio, del 27 ottobre 1998, relativa ai dispositivi medico-diagnostici in vitro (1), in particolare l’articolo 5, paragrafo 3, secondo comma,
considerando quanto segue:
|
(1) |
La decisione 2002/364/CE della Commissione (2) descrive le specifiche tecniche comuni dei dispositivi medico-diagnostici in vitro. |
|
(2) |
Nell’interesse della sanità pubblica, e al fine di tener conto dei progressi tecnici, soprattutto quelli realizzati nel campo delle prestazioni e della sensibilità analitica dei dispositivi, è opportuno rivedere le specifiche tecniche comuni descritte nella decisione 2002/364/CE. |
|
(3) |
Occorre affinare la definizione di prova rapida perché risulti più precisa. Per motivi di chiarezza, occorre aggiungere ulteriori definizioni. |
|
(4) |
Per allineare le specifiche tecniche comuni alle attuali pratiche scientifiche e tecniche è necessario aggiornare una serie di riferimenti scientifici e tecnici. |
|
(5) |
Occorre chiarire i requisiti d’analisi degli screening sull’HIV. Per garantire che le specifiche tecniche comuni riflettano adeguatamente i criteri di prestazione delle odierne tecnologie, è necessario aggiungere requisiti relativi alle prove combinate anticorpo/antigene dell’HIV e ulteriori specifiche sui requisiti del campione di talune prove. |
|
(6) |
L’allegato della decisione 2002/364/CE deve pertanto essere modificato di conseguenza e, a fini di chiarezza, sostituito. |
|
(7) |
Occorre concedere ai produttori, che abbiano già dispositivi sul mercato, un periodo di transizione per adeguarsi alle nuove specifiche tecniche comuni. D’altra parte, nell’interesse della sanità pubblica, i produttori che lo desiderino devono poter applicare le nuove specifiche tecniche comuni prima della scadenza del periodo di transizione. |
|
(8) |
Le misure di cui alla presente decisione sono conformi al parere del comitato istituito a norma dell’articolo 6, paragrafo 2, della direttiva 90/385/CEE del Consiglio (3), |
HA ADOTTATO LA PRESENTE DECISIONE:
Articolo 1
L’allegato della decisione 2002/364/CE è sostituito dall’allegato della presente decisione.
Articolo 2
La presente decisione si applica dal 1o dicembre 2010 ai dispositivi immessi per la prima volta sul mercato prima del 1o dicembre 2009.
Essa si applica a tutti gli altri dispositivi dal 1o dicembre 2009.
Gli Stati membri permetteranno tuttavia ai produttori di applicare i requisiti di cui all’allegato prima delle date specificate nei due commi precedenti.
Articolo 3
Gli Stati membri sono destinatari della presente decisione.
Fatto a Bruxelles, il 3 febbraio 2009.
Per la Commissione
Günter VERHEUGEN
Vicepresidente
(1) GU L 331 del 7.12.1998, pag. 1.
(2) GU L 131 del 16.5.2002, pag. 17.
(3) GU L 189 del 20.7.1990, pag. 17.
ALLEGATO
«ALLEGATO
SPECIFICHE TECNICHE COMUNI (CTS) PER I DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO
1. CAMPO DI APPLICAZIONE
Le specifiche tecniche comuni descritte nel presente allegato vanno applicate nell’ambito delle finalità di cui all’elenco A dell’allegato II della direttiva 98/79/CE.
2. DEFINIZIONI E TERMINI
Sensibilità (diagnostica)
La probabilità che il dispositivo fornisca un risultato positivo in presenza del marcatore bersaglio.
Vero positivo
Un campione noto come positivo per il marcatore bersaglio e correttamente classificato dal dispositivo.
Falso negativo
Un campione noto come positivo per il marcatore bersaglio e classificato erroneamente dal dispositivo.
Specificità (diagnostica)
La probabilità che il dispositivo fornisca un risultato negativo in assenza del marcatore bersaglio.
Falso positivo
Un campione noto come negativo per il marcatore bersaglio e classificato erroneamente dal dispositivo.
Vero negativo
Un campione noto come negativo per il marcatore bersaglio e classificato correttamente dal dispositivo.
Sensibilità analitica
La sensibilità analitica può essere espressa come il limite di rivelazione, cioè la quantità minima di marcatore bersaglio che può essere esattamente rivelata.
Specificità analitica
La specificità analitica indica la capacità del metodo di determinare il solo marcatore bersaglio.
Tecniche per l’amplificazione degli acidi nucleici (Nucleic acid amplification techniques — NAT)
La sigla “NAT” è usata nelle prove destinate a rivelare o a quantificare gli acidi nucleici mediante l’amplificazione di una serie bersaglio, l’amplificazione di un segnale o l’ibridazione.
Test rapido
“Test rapidi” indica dispositivi medico-diagnostici in vitro qualitativi o semiquantitativi, usati separatamente o in una piccola serie, comprendenti procedure non automatizzate e destinati a fornire un risultato in tempi rapidi.
Robustezza
La robustezza di una procedura analitica indica la capacità di quest’ultima di non essere influenzata da piccole ma volute variazioni dei parametri di metodo e indicazione anche l’affidabilità della procedura analitica nell’uso normale.
Tasso globale d’errore del sistema
Il tasso globale d’errore del sistema è la frequenza dei fallimenti quando l’intero processo è eseguito come prescritto dal fabbricante.
Test di conferma
“Test di conferma” indica una prova usata per confermare un risultato reattivo di un test di screening.
Prova di tipizzazione virale
“Prova di tipizzazione virale” indica una prova di tipizzazione con campioni positivi già noti, non utilizzati per la diagnosi primaria dell’infezione o per lo screening.
Sieroconversione dei campioni di HIV
“Sieroconversione dei campioni di HIV” indica:
|
— |
antigene p24 e/o HIV RNA positivo, e |
|
— |
riconosciuto da tutte le prove di screening dell’anticorpo, e |
|
— |
risultato positivo o indeterminato nei test di conferma. |
Sieroconversione precoce dei campioni di HIV
“Sieroconversione precoce dei campioni di HIV” indica:
|
— |
antigene p24 e/o HIV RNA positivo, e |
|
— |
non riconosciuto da tutte le prove di screening dell’anticorpo, e |
|
— |
risultato indeterminato o negativo nei test di conferma. |
3. SPECIFICHE TECNICHE COMUNI (CTS) DEI PRODOTTI ELENCATI ALL’ALLEGATO II, ELENCO A, DELLA DIRETTIVA 98/79/CE.
3.1. CTS per la valutazione delle prestazioni dei reagenti e dei prodotti reagenti per la rilevazione, la conferma e la quantificazione in campioni umani dei marcatori di infezione da HIV (HIV 1 e HIV 2), HTLV I e II ed epatite B, C e D
Principi generali
3.1.1. I dispositivi atti a identificare infezioni virali, immessi sul mercato come test di “screening” e/o come test diagnostici devono rispondere agli stessi requisiti di sensibilità e specificità (cfr. tabella 1). Cfr. inoltre il principio 3.1.11 per i test di screening.
3.1.2. I dispositivi che il fabbricante ha destinato all’analisi di liquidi biologici diversi dal siero o dal plasma, come l’urina, la saliva, ecc. devono soddisfare gli stessi requisiti di sensibilità e specificità delle CTS per le prove sul siero o sul plasma. La valutazione delle prestazioni va effettuata su campioni degli stessi soggetti in entrambi i test da approvare e in un’analisi del rispettivo siero o plasma.
3.1.3. I dispositivi che il fabbricante ha destinato all’autodiagnosi, cioè ad essere utilizzati a domicilio, devono soddisfare gli stessi requisiti di sensibilità e specificità delle CTS dei corrispondenti dispositivi per uso professionale. Le parti pertinenti della valutazione delle prestazioni vanno eseguite (o ripetute) da degli utenti “profani” al fine di convalidare il funzionamento del dispositivo e le istruzioni per l’uso.
3.1.4. Tutte le valutazioni delle prestazioni vanno effettuate per confronto diretto con un dispositivo già in uso, allineato ai più recenti aggiornamenti. Se il dispositivo usato per il confronto è sul mercato al momento della valutazione delle prestazioni, esso dovrà recare il marchio CE.
3.1.5. Se, durante la valutazione si individuano risultati di test discordanti, tali discordanze vanno, per quanto possibile, risolte; ad esempio:
|
— |
effettuando test complementari sul campione discordante, |
|
— |
ricorrendo ad altri metodi o ad altri marcatori, |
|
— |
riesaminando lo stato clinico e la diagnosi del paziente, nonché |
|
— |
sottoponendo a test campioni successivi. |
3.1.6. Le valutazioni delle prestazioni sono effettuate su una popolazione equivalente alla popolazione europea.
3.1.7. I campioni positivi usati nella valutazione delle prestazioni sono selezionati in modo da riflettere stadi diversi della malattia, diversi modelli anticorpali, diversi genotipi e sottotipi, mutanti, ecc.
3.1.8. La sensibilità con i campioni veri positivi e di sieroconversione va valutata come segue.
|
3.1.8.1. |
La sensibilità dei test diagnostici durante la sieroconversione deve corrispondere agli standard più aggiornati. Che l’ulteriore analisi degli stessi pannelli o dei pannelli supplementari della sieroconversione sia effettuato dall’organismo notificato o dal produttore, i risultati devono confermare i dati iniziali di valutazione delle prestazioni (cfr. tabella 1). I pannelli della sieroconversione dovrebbero iniziare con uno o più test sanguigni negativi e gli intervalli tra i test dovrebbero essere brevi. |
|
3.1.8.2. |
Riguardo ai dispositivi usati per gli screening del sangue (esclusi i test HBsAg e anti-HBc), tutti i campioni veri positivi devono risultare positivi al test effettuato con il dispositivo cui va apposto il marchio CE (tabella 1). Per i test HBsAg e anti-HBc il nuovo dispositivo deve avere una prestazione globale almeno equivalente a quella del dispositivo già in uso (cfr. punto 3.1.4). |
|
3.1.8.3. |
Riguardo ai test HIV:
|
3.1.9. L’analisi delle prestazioni dei test di screening deve comprendere 25 campioni di siero e/o plasma fresco (se disponibile per le infezioni rare) “dello stesso giorno” (≤ 1 giorno dopo il prelievo).
3.1.10. I campioni negativi usati in una valutazione delle prestazioni sono definiti in modo da rappresentare le popolazioni bersaglio a cui il test è destinato, ad esempio donatori di sangue, pazienti ricoverati, donne in gravidanza, ecc.
3.1.11. Per valutare le prestazioni dei test di screening (tabella 1), le popolazioni di donatori di sangue esaminate devono provenire da almeno due centri trasfusionali e consistere in donazioni di sangue consecutive, non selezionate al fine di escludere donatori alla prima donazione.
3.1.12. A meno che non sia diversamente specificato nelle tabelle allegate, i dispositivi devono presentare una specificità pari ad almeno il 99,5 % per le donazioni di sangue. La specificità è calcolata in base alla frequenza dei risultati ripetutamente reattivi (cioè “falsi positivi”) tra i donatori di sangue negativi per il relativo marcatore.
3.1.13. Nell’ambito della valutazione delle prestazioni, occorre esaminare i dispositivi per stabilire l’effetto di potenziali sostanze interferenti. Le potenziali sostanze interferenti da valutare dipenderanno in parte dalla composizione del reagente e dalla configurazione dell’analisi. Le potenziali sostanze interferenti vanno identificate nell’ambito dell’analisi obbligatoria dei rischi, requisito essenziale di ogni nuovo dispositivo, ma può includere, ad esempio:
|
— |
campioni rappresentanti infezioni “affini”, |
|
— |
campioni provenienti da donne multipare (donne che hanno avuto più di una gravidanza) o da pazienti positivi per il fattore reumatoide, |
|
— |
per gli antigeni ricombinanti, anticorpi umani contro i componenti del sistema di espressione, ad esempio anti-E. coli o anti-lievito. |
3.1.14. Per i dispositivi destinati dal fabbricante ad essere usati con il siero e il plasma, la valutazione delle prestazioni deve dimostrare l’equivalenza tra siero e plasma. Ciò va dimostrato per almeno 50 donazioni (25 positive e 25 negative).
3.1.15. Per i dispositivi destinati dal fabbricante ad essere usati con il plasma, la valutazione delle prestazioni deve verificare le prestazioni del dispositivo utilizzando tutti i coagulanti indicati dal fabbricante per l’uso del dispositivo. Ciò va dimostrato per almeno 50 donazioni (25 positive e 25 negative).
3.1.16. Nel quadro dell’analisi obbligatoria dei rischi, il tasso globale d’errore del sistema che porti a risultati falsi negativi va stabilito in base a test ripetuti su campioni a bassa positività.
3.1.17. Se un nuovo dispositivo medico-diagnostico in vitro di cui all’allegato II, elenco A, non è specialmente coperto dalle specifiche tecniche comuni (CTS), si ricorrerà a CTS per un dispositivo affine. Si possono individuare dispositivi affini per vari motivi, ad esempio per lo stesso uso, o per uno simile, o per rischi analoghi.
3.2. Requisiti supplementari per i test combinati anticorpo/antigene dell’HIV
3.2.1. I test combinati anticorpo/antigene dell’HIV destinati alla detezione degli anticorpi anti-HIV e dell’antigene p24, destinati a individuare anche il solo antigene p24, devono essere conformi alle tabelle 1 e 5, compresi i criteri di sensibilità analitica per l’antigene p24.
3.2.2. I test combinati anticorpo/antigene dell’HIV destinati alla detezione degli anticorpi anti-HIV e dell’antigene p24, non destinati a individuare anche il solo antigene p24, devono essere conformi alle tabelle 1 e 5, esclusi i criteri di sensibilità analitica per l’antigene p24.
3.3. Requisiti supplementari per le tecniche di amplificazione dell’acido nucleico (NAT)
I criteri per la valutazione delle prestazioni dei test NAT si trovano nella tabella 2.
3.3.1. Per i test di amplificazione di una sequenza bersaglio, il controllo di funzionalità di ogni campione da provare (controllo interno) deve corrispondere agli standard più aggiornati. Se possibile, occorre effettuare tale controllo nel corso dell’intero processo: estrazione, amplificazione/ibridazione, rilevazione.
3.3.2. La sensibilità analitica o il limite di rilevazione dei test NAT va espresso da un valore limite positivo del 95 %. Questa è la concentrazione dell’analita in cui il 95 % dei test sono positivi dopo diluizioni in serie di un materiale di riferimento internazionale, come potrebbe essere un materiale di riferimento standard dell’OMS o un altro su di esso calibrato.
3.3.3. La rilevazione del genotipo va dimostrata con un’adeguata convalida della concezione del primer o della sonda e con test effettuati su campioni di genotipi caratterizzati.
3.3.4. I risultati dei NAT quantitativi saranno conformi a standard internazionali o a materiali di riferimento calibrati, se disponibili, e andranno espressi nelle unità internazionali usate nello specifico campo di applicazione.
3.3.5. I test NAT possono essere usati per individuare il virus in campioni anticorpo-negativi, cioè campioni precedenti la sieroconversione. I virus di complessi immuni possono comportarsi diversamente rispetto ai virus liberi, per esempio nella fase di centrifugazione. Negli studi di robustezza è perciò importante che siano compresi campioni anticorpo-negativi (“pre-sieroconversione”).
3.3.6. Per indagare potenziali reazioni incrociate, durante gli esami di robustezza vanno effettuati almeno 5 test alternando campioni ad alta positività e negatività. I campioni ad alta positività devono includere campioni con titoli virali naturalmente elevati.
3.3.7. Il tasso totale di insuccesso del sistema che conduce ai risultati falsi negativi va determinato provando campioni debolmente positivi. I campioni debolmente positivi devono contenere una concentrazione di virus pari a 3 volte la concentrazione virale positiva al 95 %.
3.4. CTS per il controllo del rilascio, da parte del fabbricante, di reagenti e di prodotti reagenti per la rilevazione, la conferma e la quantificazione in campioni umani dei marcatori di infezione da HIV (HIV 1 e HIV 2), HTLV I e HTLV II ed epatite B, C e D (solo test immunologici)
3.4.1. I criteri per il controllo del rilascio da parte del fabbricante devono garantire che ogni lotto identifichi in modo coerente i relativi antigeni, epitopi e anticorpi.
3.4.2. Il controllo da parte del fabbricante del rilascio dei lotti per test di screening comprende almeno 100 campioni negativi per l’analita corrispondente.
3.5. CTS per valutare le prestazioni di reagenti e prodotti reagenti al fine di determinare gli antigeni dei seguenti gruppi sanguigni: sistema del gruppo sanguigno ABO: ABO1 (A), ABO2 (B), ABO3 (A,B); sistema Rh: RH1 (D), RH2 (C), RH3 (E), RH4 (c), RH5 (e); sistema Kell: KEL1 (K)
Nella tabella 9 sono elencati i criteri per valutare le prestazioni di reagenti e prodotti reagenti al fine di determinare gli antigeni dei gruppi sanguigni: sistema del gruppo sanguigno ABO: ABO01 (A), ABO2 (B), ABO3 (A,B); sistema Rh: RH1 (D), RH2 (C), RH3 (E), RH4 (c), RH5 (e); sistema Kell: KEL1 (K)
3.5.1. Tutte le valutazioni delle prestazioni vanno effettuate per confronto diretto con un dispositivo già in uso, allineato ai più recenti aggiornamenti. Se il dispositivo usato per il confronto è sul mercato al momento della valutazione delle prestazioni, esso dovrà recare il marchio CE.
3.5.2. Se, durante la valutazione si individuano risultati di test discordanti, tali discordanze vanno, per quanto possibile, risolte; ad esempio:
|
— |
effettuando test complementari sul campione discordante, |
|
— |
utilizzando altri metodi. |
3.5.3. Le valutazioni delle prestazioni sono effettuate su una popolazione equivalente alla popolazione europea.
3.5.4. I campioni positivi utilizzati per la valutazione delle prestazioni sono selezionati in modo da riflettere l’espressione di antigeni varianti e deboli.
3.5.5. Nell’ambito della valutazione delle prestazioni, occorre esaminare i dispositivi per stabilire l’effetto di potenziali sostanze interferenti. Le potenziali sostanze interferenti da valutare dipenderanno in parte dalla composizione del reagente e dalla configurazione dell’analisi. Tali sostanze sono identificate nel quadro dell’analisi dei rischi prevista dai requisiti essenziali per ogni nuovo dispositivo.
3.5.6. Per i dispositivi destinati dal fabbricante ad essere usati con il plasma, la valutazione delle prestazioni deve verificare le prestazioni del dispositivo utilizzando tutti i coagulanti indicati dal fabbricante per l’uso del dispositivo. La dimostrazione deve essere effettuata per almeno 50 donazioni.
3.6. CTS per il controllo del rilascio, da parte del fabbricante, di reagenti e prodotti reagenti al fine di determinare gli antigeni dei seguenti gruppi sanguigni: sistema del gruppo sanguigno ABO: ABO1 (A), ABO2 (B), ABO3 (A,B); sistema Rh: RH1 (D), RH2 (C), RH3 (E), RH4 (c), RH5 (e); sistema Kell: KEL1 (K)
3.6.1. I criteri per il controllo del rilascio da parte del fabbricante devono garantire che ogni lotto identifichi in modo coerente i relativi antigeni, epitopi e anticorpi.
3.6.2. La tabella 10 elenca i requisiti per il controllo del rilascio dei lotti da parte del fabbricante.
Tabella 1
Test di screening: anti-HIV 1 e 2, anti-HTLV I e II, anti-HCV, HBsAg, anti-HBc
|
|
|
anti-HIV 1 e 2 |
anti-HTLV I e II |
anti-HCV |
HBsAg |
anti-HBc |
|
Sensibilità diagnostica |
Campioni positivi |
400 HIV 1 100 HIV 2 compresi 40 sottotipi non-B, tutti i sottotipi HIV 1 disponibili dovrebbero essere rappresentati da almeno 3 campioni per sottotipo |
300 HTLV I 100 HTLV II |
400 (campioni positivi) Compresi i campioni da stadi d’infezione diversi e che riflettono diversi modelli anticorpali. Genotipo 1-4: > 20 campioni per genotipo (compresi sottotipi non-A di genotipo 4); 5: > 5 campioni 6: se disponibili. |
400 tenendo conto dei sottotipi |
400 compresa la valutazione di altri marcatori HBV |
|
Pannelli di sieroconversione |
20 pannelli 10 pannelli supplementari (presso l’organismo notificato o il fabbricante) |
Da definire quando disponibile |
20 pannelli 10 pannelli supplementari (presso l’organismo notificato o il fabbricante) |
20 pannelli 10 pannelli supplementari (presso l’organismo notificato o il fabbricante) |
Da definire quando disponibile |
|
|
Sensibilità analitica |
Norme |
|
|
|
0,130 IU/ml (seconda norma internazionale per HBsAg, sottotipo adw2, genotipo A, codice NIBSC: 00/588) |
|
|
Specificità |
Donatori non selezionati (compresi i donatori per la prima volta) |
5 000 |
5 000 |
5 000 |
5 000 |
5 000 |
|
Pazienti ospedalizzati |
200 |
200 |
200 |
200 |
200 |
|
|
Campioni di sangue a possibile reazione incrociata(RF+, virus affini, donne incinte, ecc.) |
100 |
100 |
100 |
100 |
100 |
Tabella 2
Test NAT per HIV1, HCV, HBV, HTLV I e II (test qualitativi e quantitativi; senza tipizzazione molecolare)
|
HIV1 |
HCV |
HBV |
HTLV I e II |
Criteri di accettazione |
|||||
|
NAT |
qualitativi |
quantitativi |
qualitativi |
quantitativi |
qualitativi |
quantitativi |
qualitativi |
quantitativi |
|
|
come per i test quantitativi HIV |
come per i test quantitativi HIV |
come per i test quantitativi HIV |
|||||||
|
Sensibilità Limite di rilevazione Rilevazione di sensibilità analitica (IU/ml; definita da norme OMS o su materiali di riferimento calibrati) |
Secondo gli orientamenti di convalida della PE (1): varie diluizioni successive in concentrazione limite; analisi statistica (ad esempio analisi Probit) sulla base di almeno 24 replicati; calcolo del valore limite al 95 % |
Limite di rilevazione come per i test qualitativi; limite di quantificazione: diluizioni (mezzo-log10 o meno) di preparazioni calibrate di riferimento, definizione del limite di quantificazione inferiore e superiore, precisione, accuratezza, campo di misurazione “lineare”, “campo dinamico”. Dimostrare la riproducibilità a diversi livelli di concentrazione |
Secondo gli orientamenti di convalida della PE (1): varie diluizioni successive in concentrazione limite; analisi statistica (ad esempio analisi Probit) sulla base di almeno 24 replicati; calcolo del valore limite al 95 % |
|
Secondo gli orientamenti di convalida della PE (1): varie diluizioni successive in concentrazione limite; analisi statistica (ad esempio analisi Probit) sulla base di almeno 24 replicati; calcolo del valore limite al 95 % |
|
Secondo gli orientamenti di convalida della PE (1): varie diluizioni successive in concentrazione limite; analisi statistica (ad esempio analisi Probit) sulla base di almeno 24 replicati; calcolo del valore limite al 95 % |
|
|
|
Efficacia della rilevazione/quantificazione del genotipo/sottotipo |
Almeno 10 campioni per sottotipo (se disponibili) |
Serie di diluizioni di tutti i genotipi/sottotipi pertinenti, preferibilmente dei materiali di riferimento, se disponibili |
Almeno 10 campioni per sottotipo (se disponibili) |
|
Se sono disponibili materiali di riferimento calibrati del genotipo |
|
Se sono disponibili materiali di riferimento calibrati del genotipo |
|
|
|
Supernatanti di coltura cellulare (possono sostituire i sottotipi rari di HIV 1) |
Possono essere usati trascrizioni o plasmidi quantificati da metodi appropriati. |
|
|
|
|
|
|
|
|
|
Secondo gli orientamento di convalida della PE (1) se esistono materiali di riferimento calibrati per sottotipo; la trascrizione in vitro è accettabile |
|
Secondo gli orientamento di convalida della PE (1) se esistono materiali di riferimento calibrati per sottotipo; la trascrizione in vitro è accettabile |
|
Secondo gli orientamento di convalida della PE (1) se esistono materiali di riferimento calibrati per sottotipo; la trascrizione in vitro è accettabile |
|
Secondo gli orientamento di convalida della PE (1) se esistono materiali di riferimento calibrati per sottotipo; la trascrizione in vitro è accettabile |
|
|
|
|
Campioni negativi di specificità diagnostica |
500 donatori di sangue |
100 donatori di sangue |
500 donatori di sangue |
|
500 donatori di sangue |
|
500 donazioni singole di sangue |
|
|
|
Marcatori con possibile reazione incrociata |
Dimostrando che la concezione del test è adeguata (ad esempio comparando le sequenze) e/o provando almeno 10 campioni positivi di retrovirus umani (ad esempio HTLV) |
Come per i test qualitativi |
Secondo la concezione del test e/o provando almeno 10 campioni positivi di flavivirus (ad esempio HGV, YFV) |
|
Secondo la concezione del test e/o provando almeno 10 campioni positivi al virus DNA |
|
Secondo la concezione del test e/o provando almeno 10 campioni positivi di retrovirus umani (ad esempio HGV) |
|
|
|
Robustezza |
|
Come per i test qualitativi |
|
|
|
|
|
|
|
|
Contaminazione incrociata |
Almeno 5 serie, alternando campioni fortemente positivi (noti per apparire naturalmente) e negativi |
|
Almeno 5 serie, alternando campioni fortemente positivi (noti per apparire naturalmente) e negativi |
|
Almeno 5 serie, alternando campioni fortemente positivi (noti per apparire naturalmente) e negativi |
|
Almeno 5 serie, alternando campioni fortemente positivi (noti per apparire naturalmente) e negativi |
|
|
|
Inibizione |
Controllo interno preferibilmente per l’intera procedura NAT |
|
Controllo interno preferibilmente per l’intera procedura NAT |
|
Controllo interno preferibilmente per l’intera procedura NAT |
|
Controllo interno preferibilmente per l’intera procedura NAT |
|
|
|
Tasso totale di insuccesso del sistema che conduce a risultati falsi |
Almeno 100 campioni infettati dal virus con 3 × la concentrazione virale positiva del 95 % |
|
Almeno 100 campioni infettati dal virus con 3 × la concentrazione virale positiva del 95 % |
|
Almeno 100 campioni infettati dal virus con 3 × la concentrazione virale positiva del 95 % |
|
Almeno 100 campioni infettati dal virus con 3 × la concentrazione virale positiva del 95 % |
|
99 % di test positivi |
Tabella 3
Test rapidi anti-HIV 1 e 2, anti-HCV, HBsAg, anti-HBc, anti-HTLV I e II
|
|
|
anti-HIV1 e 2 |
anti-HCV |
HBsAg |
anti-HBc |
anti-HTLV I e II |
Criteri di accettazione |
|
Sensibilità diagnostica |
Campioni positivi |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
|
Pannelli di sieroconversione |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
Criteri identici a quelli dei test di screening |
|
|
Specificità diagnostica |
Campioni negativi |
1 000 donazioni di sangue |
1 000 donazioni di sangue |
1 000 donazioni di sangue |
1 000 donazioni di sangue |
1 000 donazioni di sangue |
≥ 99 % (anti-HBc: ≥ 96 %) |
|
200 campioni clinici |
200 campioni clinici |
200 campioni clinici |
200 campioni clinici |
200 campioni clinici |
|||
|
200 campioni da donne incinte |
200 campioni da donne incinte |
200 campioni da donne incinte |
|
200 campioni da donne incinte |
|||
|
100 campioni potenzialmente interferenti |
100 campioni potenzialmente interferenti |
100 campioni potenzialmente interferenti |
100 campioni potenzialmente interferenti |
100 campioni potenzialmente interferenti |
Tabella 4
Test di conferma e/o supplementari per anti-HIV 1 e 2, anti-HTLV I e II, anti-HCV, HBsAg
|
|
|
Test di conferma anti-HIV |
Test di conferma anti-HTLV |
Test supplementare HCV |
Test di conferma HBsAg |
Criteri di accettazione |
|
Sensibilità diagnostica |
Campioni positivi |
200 HIV 1 e 100 HIV 2 |
200 HTLV I e 100 HTLV II |
300 HCV (campioni positivi) |
300 HBsAg |
Identificazione corretta come positivo (o indeterminato), non come negativo |
|
Compresi i campioni da stadi d’infezione diversi e che riflettono diversi modelli anticorpali. |
|
Compresi i campioni da stadi d’infezione diversi e che riflettono diversi modelli anticorpali. Genotipi 1-4: > 20 campioni per genotipo (compresi sottotipi non-A di genotipo 4); 5: > 5 campioni 6: se disponibili. |
Compresi i campioni da stadi d’infezione diversi 20 campioni “altamente positivi” (> 26 IU/ml); 20 campioni vicini al valore limite |
|
||
|
Pannelli di sieroconversione |
15 pannelli di sieroconversione/pannelli a basso titolo |
|
15 pannelli di sieroconversione/pannelli a basso titolo |
15 pannelli di sieroconversione/pannelli a basso titolo |
|
|
|
Sensibilità analitica |
Norme |
|
|
|
Seconda norma internazionale per l’HBsAg, sottotipo adw2, genotipo A, codice NIBSC: 00/588 |
|
|
Specificità diagnostica |
Campioni negativi |
200 donazioni di sangue |
200 donazioni di sangue |
200 donazioni di sangue |
10 falsi positivi se disponibili dalla valutazione delle prestazioni del test di screening (2) |
Nessun risultato falso positivo/ (2) nessuna neutralizzazione |
|
200 campioni clinici che comprendano donne incinte |
200 campioni clinici che comprendano donne incinte |
200 campioni clinici che comprendano donne incinte |
|
|
||
|
50 campioni potenzialmente interferenti, comprendenti campioni con risultati indeterminati in altri test di conferma |
50 campioni potenzialmente interferenti, comprendenti campioni con risultati indeterminati in altri test di conferma |
50 campioni potenzialmente interferenti, comprendenti campioni con risultati indeterminati in altri test supplementari |
50 campioni potenzialmente interferenti |
|
Tabella 5
Antigene dell’HIV 1
|
|
Test dell’antigene dell’HIV 1 |
Criteri di accettazione |
|
|
Sensibilità diagnostica |
Campioni positivi |
50 positivi all’antigene dell’HIV 1 50 supernatanti di coltura cellulare comprendenti diversi sottotipi di HIV 1 e HIV 2 |
Identificazione corretta (dopo neutralizzazione) |
|
Pannelli di sieroconversione |
20 pannelli di sieroconversione/pannelli a basso titolo |
|
|
|
Sensibilità analitica |
Norme |
Antigene p24 all’HIV 1, primo reagente di riferimento internazionale, codice NIBSC: 90/636 |
≤ 2 IU/ml |
|
Specificità diagnostica |
|
200 donazioni di sangue 200 campioni clinici 50 campioni potenzialmente interferenti |
≥ 99,5 % dopo neutralizzazione |
Tabella 6
Test di sierotipizzazione e genotipizzazione: HCV
|
|
Test di sierotipizzazione e genotipizzazione dell’HCV |
Criteri di accettazione |
|
|
Sensibilità diagnostica |
Campioni positivi |
200 (campioni positivi) Compresi i campioni da stadi d’infezione diversi e che riflettono diversi modelli anticorpali. Genotipi 1-4: > 20 campioni per genotipo (compresi sottotipi non-A di genotipo 4); 5: > 5 campioni; 6: se disponibili |
≥ 95 % di concordanza tra sierotipizzazione e genotipizzazione ≥ 95 % di concordanza tra genotipizzazione e sequenziamento |
|
Specificità diagnostica |
Campioni negativi |
100 |
|
Tabella 7
Marcatori dell’HBV: anti-HBs, anti -HBc IgM, anti-HBe, HBeAg
|
|
anti-HBs |
anti-HBc IgM |
anti-HBe |
HBeAg |
Criteri di accettazione |
|
|
Sensibilità diagnostica |
Campioni positivi |
100 soggetti vaccinati |
200 |
200 |
200 |
≥ 98 % |
|
100 soggetti infettati per via naturale |
Compresi i campioni da stadi d’infezione diversi (acuti/cronici, ecc.) I criteri di accettazione vanno applicati solo a campioni provenienti da stadi d’infezione acuti |
Compresi i campioni da stadi d’infezione diversi (acuti/cronici, ecc.) |
Compresi i campioni da stadi d’infezione diversi (acuti/cronici, ecc.) |
|||
|
Pannelli di sieroconversione |
10 pannelli di campioni sequenziali o sieroconversioni anti-HBs |
Se disponibili |
|
|
|
|
|
Sensibilità analitica |
Norme |
Primo preparato di riferimento internazionale OMS del 1977; NIBSC, Regno Unito |
|
|
Antigene di riferimento 82 per HBe; PEI, Germania |
Anti-HBs: < 10 mIU/ml |
|
Specificità diagnostica |
Campioni negativi |
500 |
500 donazioni di sangue |
200 donazioni di sangue |
200 donazioni di sangue |
≥ 98 % |
|
Compresi campioni clinici 50 campioni potenzialmente interferenti |
200 campioni clinici 50 campioni potenzialmente interferenti |
200 campioni clinici 50 campioni potenzialmente interferenti |
200 campioni clinici 50 campioni potenzialmente interferenti |
|||
Tabella 8
Marcatori HDV: anti-HDV, anti-HDV IgM, antigene Delta
|
|
anti-HDV |
anti-HDV IgM |
Antigene Delta |
Criteri di accettazione |
|
|
Sensibilità diagnostica |
Campioni positivi |
100 |
50 |
10 |
≥ 98 % |
|
Indicazione di marcatori HBV |
Indicazione di marcatori HBV |
Indicazione di marcatori HBV |
|||
|
Specificità diagnostica |
Campioni negativi |
200 |
200 |
200 |
≥ 98 % |
|
Compresi campioni clinici |
Compresi campioni clinici |
Compresi campioni clinici |
|||
|
50 campioni potenzialmente interferenti |
50 campioni potenzialmente interferenti |
50 campioni potenzialmente interferenti |
|||
Tabella 9
Antigeni del gruppo sanguigno nei sistemi dei gruppi sanguigni ABO, Rh e Kell
|
|
1 |
2 |
3 |
|
Specificità |
Numero di prove per metodo raccomandato |
Numero totale di campioni da provare per il lancio di un prodotto |
Numero totale di campioni da provare per una nuova formulazione o per l’uso di reagenti ben caratterizzati |
|
Anti-ABO1 (anti-A), anti-ABO2 (anti-B), anti-ABO3 (anti-A,B) |
500 |
3 000 |
1 000 |
|
Anti-RH1 (anti-D) |
500 |
3 000 |
1 000 |
|
Anti-RH2 (anti-C), anti-RH4 (anti-c), anti-RH3 (anti-E) |
100 |
1 000 |
200 |
|
Anti-RH5 (anti-e) |
100 |
500 |
200 |
|
Anti-KEL1 (anti-K) |
100 |
500 |
200 |
Criteri di accettazione:
Tutti i reagenti di cui sopra devono dare risultati di prova comparabili a quelli dei reagenti già in uso e avere prestazioni accettabili riguardo alla reattività prevista del dispositivo. Per i reagenti già in uso, di cui siano stati modificati o estesi l’uso o l’applicazione, vanno effettuate ulteriori prove in conformità ai requisiti descritti alla colonna 1 (cfr. sopra).
La valutazione delle prestazioni dei reagenti anti-D comprenderà prove su una serie di campioni di RH1 (D) debole e di RH1 (D) parziale, a seconda dell’uso previsto del prodotto.
Caratteristiche:
|
Campioni clinici |
: |
10 % della popolazione studiata |
|
Campioni neonatali |
: |
> 2 % della popolazione studiata |
|
Campioni ABO |
: |
> 40 % A, B positivi |
|
“D debole” |
: |
> 2 % di RH1 (D) positivi |
Tabella 10
Criteri di rilascio dei lotti per reagenti e prodotti reagenti destinati a determinare antigeni del gruppo sanguigno nei sistemi dei gruppi sanguigni ABO, Rh e Kell
Requisiti di valutazione della specificità per ogni reagente
1. Reagenti per test
|
Reagenti del gruppo sanguigno |
Numero minimo di cellule di controllo da esaminare |
|||||||
|
|
Reazioni positive |
|
Reazioni negative |
|||||
|
|
A1 |
A2B |
Ax |
|
|
B |
0 |
|
|
Anti-ABO1 (anti-A) |
2 |
2 |
2 (3) |
|
2 |
2 |
|
|
|
|
B |
A1B |
|
|
A1 |
0 |
|
|
|
Anti-ABO2 (anti-B) |
2 |
2 |
|
|
2 |
2 |
|
|
|
|
A1 |
A2 |
Ax |
B |
0 |
|
|
|
|
Anti-ABO3 (anti-A,B) |
2 |
2 |
2 |
2 |
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R1r |
R2r |
D debole |
|
r’r |
r’’r |
rr |
|
|
Anti-RH1 (anti-D) |
2 |
2 |
2 (3) |
|
1 |
1 |
1 |
|
|
|
R1R2 |
R1r |
r’r |
|
R2R2 |
r’’r |
rr |
|
|
Anti-RH2 (anti-C) |
2 |
1 |
1 |
|
1 |
1 |
1 |
|
|
|
R1R2 |
R1r |
r’r |
|
R1R1 |
|
|
|
|
Anti-RH4 (anti-c) |
1 |
2 |
1 |
|
3 |
|
|
|
|
|
R1R2 |
R2r |
r’’r |
|
R1R1 |
r’r |
rr |
|
|
Anti-RH 3 (anti-E) |
2 |
1 |
1 |
|
1 |
1 |
1 |
|
|
|
R1R2 |
R2r |
r’’r |
|
R2R2 |
|
|
|
|
Anti-RH5 (anti-e) |
2 |
1 |
1 |
|
3 |
|
|
|
|
|
Kk |
|
|
|
kk |
|
|
|
|
Anti-KEL1 (anti-K) |
4 |
|
|
|
3 |
|
|
|
Criteri di accettazione:
Per ogni lotto di reagenti i risultati devono essere inequivocabilmente positivi o negativi per tutte le tecniche raccomandate, conformemente ai risultati ottenuti con i dati della valutazione delle prestazioni.
2. Materiali di controllo (globuli rossi)
Il fenotipo dei globuli rossi utilizzati per il controllo dei reagenti per la tipizzazione dei gruppi sanguigni sopraelencati deve essere confermato utilizzando un dispositivo riconosciuto.»
(1) Orientamento della Farmacopea europea.
Note: I criteri di accettazione per il “tasso totale di insuccesso del sistema che conduce a risultati falsi” è di 99 test positivi su 100.
Per i test quantitativi NAT verrà effettuato uno studio su almeno 100 campioni positivi che riflettano le condizioni abituali degli utenti (ad esempio nessuna preselezione dei campioni). Parallelamente, dovranno essere generati risultati comparativi con un altro sistema di test NAT.
Per i test NAT qualitativi verrà effettuato uno studio sulla sensibilità diagnostica utilizzando almeno 10 pannelli di sieroconversione. Parallelamente, dovranno essere generati risultati comparativi con un altro sistema di test NAT.
(2) Criteri di accettazione: nessuna neutralizzazione per test di conferma HBsAg.
(3) Solo mediante tecniche raccomandate, se viene indicata la reattività a tali antigeni.
Nota: I reagenti policlonali devono essere provati su un pannello di cellule più ampio per confermare la specificità ed escludere la presenza di anticorpi di contaminazione indesiderati.
|
10.2.2009 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 39/s3 |
NOTA PER IL LETTORE
Le istituzioni hanno deciso di non fare più apparire nei loro testi la menzione dell'ultima modifica degli atti citati.
Salvo indicazione contraria, nei testi qui pubblicati il riferimento è fatto agli atti nella loro versione in vigore.