|
8)
|
Nella parte B sono aggiunti i seguenti capitoli:
"B.63 PROVA DI SCREENING DELLA TOSSICITÀ PER LA RIPRODUZIONE/LO SVILUPPO
INTRODUZIONE
|
1.
|
Il presente metodo di prova è equivalente alla linea guida n. 421 dell’OCSE (2016). Le linee guida dell’OCSE per le prove sulle sostanze chimiche sono periodicamente rivedute e aggiornate alla luce del progresso scientifico. La linea guida n. 421 sulla prova di screening è stata adottata inizialmente nel 1995, sulla base di un protocollo per una «prova preliminare di screening della tossicità per la riproduzione» di cui si è discusso nell’ambito di due riunioni di esperti, a Londra nel 1990 (1) e a Tokyo nel 1992 (2).
|
|
2.
|
Il presente metodo di prova è stato aggiornato mediante l’aggiunta di endpoint pertinenti per il rilevamento di interferenti endocrini, come seguito all’attività ad alta priorità avviata dall’OCSE nel 1998 volta a rivedere le linee guida esistenti e a elaborarne di nuove, per lo screening e la sperimentazione relativi a potenziali interferenti endocrini (3). La linea guida n. 407 dell’OCSE (studio della tossicità orale con somministrazione ripetuta di dosi per 28 giorni sui roditori, corrispondente al capitolo B.7 del presente allegato), ad esempio, è stata migliorata nel 2008 mediante l’aggiunta di parametri adatti per l’individuazione dell’attività endocrina delle sostanze in esame. La linea guida n. 421 è stata aggiornata con l’intento di includere gli endpoint pertinenti per il rilevamento di interferenti endocrini nelle linee guida in cui i periodi di esposizione coprono alcuni dei periodi sensibili dello sviluppo (i periodi precedenti o immediatamente successivi alla nascita).
|
|
3.
|
Gli ulteriori endpoint pertinenti per il rilevamento di interferenti endocrini selezionati, che fanno parte anche della linea guida n. 443 (studio esteso di tossicità per la riproduzione su una generazione, corrispondente al capitolo B.56 del presente allegato), sono stati aggiunti alla linea guida n. 421 sulla base di uno studio di fattibilità riguardante questioni scientifiche e tecniche relative alla loro inclusione, come pure gli eventuali adattamenti del disegno sperimentale necessari per la loro inclusione (4).
|
|
4.
|
Il presente metodo di prova è inteso a generare informazioni limitate concernenti gli effetti delle sostanze chimiche in esame sulla capacità riproduttiva maschile e femminile, quali la funzione delle gonadi, il comportamento in fase di accoppiamento, il concepimento, lo sviluppo dell’organismo concepito e il parto. Esso non rappresenta un’alternativa ai metodi di prova esistenti B.31, B.34, B.35 e B.56, né li sostituisce.
|
CONSIDERAZIONI INIZIALI
|
5.
|
Il presente metodo di prova di screening può essere utilizzato per fornire informazioni iniziali concernenti i possibili effetti sulla riproduzione e/o lo sviluppo, nella fase iniziale di valutazione delle proprietà tossicologiche delle sostanze chimiche o nella valutazione di sostanze potenzialmente pericolose. Esso può essere utilizzato anche nell’ambito di una serie di prove di screening iniziali per le sostanze chimiche esistenti per le quali le informazioni tossicologiche disponibili sono scarse o inesistenti, come studio volto a determinare gli intervalli di dosaggio per studi più approfonditi sulla riproduzione o sullo sviluppo o in altri casi in cui sia ritenuto opportuno. Durante lo svolgimento dello studio è opportuno seguire i principi guida e le considerazioni di cui al documento di orientamento dell’OCSE n. 19, Recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation (5).
|
|
6.
|
Il presente metodo di prova non fornisce informazioni complete su tutti gli aspetti della riproduzione e dello sviluppo. In particolare, esso offre solo mezzi limitati per individuare manifestazioni postnatali di un’esposizione prenatale o effetti eventualmente riconducibili a un’esposizione postnatale. Tenuto conto, tra le altre cose, del numero relativamente ridotto di animali nei gruppi di trattamento, della selettività degli endpoint e della breve durata dello studio, il presente metodo non fornisce prove sufficienti per trarre conclusioni definitive circa l’assenza di effetti. Inoltre, in assenza di dati desunti da altre prove di tossicità per la riproduzione/lo sviluppo, risultati positivi sono utili per una valutazione iniziale dei pericoli e permettono di prendere decisioni circa la necessità di procedere a ulteriori prove e i tempi per effettuarle.
|
|
7.
|
I risultati ottenuti per quanto riguarda i parametri endocrini devono essere interpretati alla luce del «Quadro concettuale dell’OCSE per le prove e la valutazione delle sostanze chimiche che alterano il sistema endocrino» (6). La linea guida migliorata dell’OCSE n. 421 fa parte del livello 4 di questo quadro concettuale come saggio in vivo in grado di fornire dati sugli effetti nocivi sugli endpoint pertinenti per il sistema endocrino. Un segnale endocrino potrebbe tuttavia non essere considerato di per sé una prova sufficiente ad attestare che la sostanza chimica in esame è un interferente endocrino.
|
|
8.
|
Il presente metodo di prova prevede la somministrazione orale della sostanza chimica da esaminare. Potrebbero essere necessarie modifiche in caso di utilizzo di altre vie di somministrazione.
|
|
9.
|
Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie laddove esista una disposizione normativa che obblighi a sottoporre a prova la miscela.
|
|
10.
|
L’appendice 1 contiene le definizioni dei termini utilizzati.
|
PRINCIPIO DELLA PROVA
|
11.
|
La sostanza chimica in esame viene somministrata in dosi graduate a diversi gruppi di maschi e femmine. Ai maschi le dosi devono essere somministrate per almeno quattro settimane e fino al prima della soppressione programmata, compreso tale giorno (tale periodo comprende un minimo di due settimane prima dell’accoppiamento, il tempo dell’accoppiamento e circa due settimane dopo l’accoppiamento). Tenuto conto della durata limitata del periodo di esposizione prima dell’accoppiamento nei maschi, la fertilità potrebbe non essere un particolare indicatore di sensibilità della tossicità testicolare. È pertanto essenziale un esame istologico particolareggiato delle prove. La combinazione di un periodo di esposizione di due settimane prima dell’accoppiamento (seguito da osservazioni sull’accoppiamento e la fertilità) e di un periodo di esposizione complessivo di almeno quattro settimane (seguito da un dettagliato esame istopatologico delle gonadi maschili) è considerata sufficiente a consentire l’individuazione della maggior parte degli effetti sulla fertilità maschile e sulla spermatogenesi.
|
|
12.
|
Alle femmine la sostanza va somministrata durante l’intero studio. Tale periodo comprende due settimane prima dell’accoppiamento (con l’obiettivo di coprire almeno due cicli estrali completi), l’intervallo variabile che precede il concepimento, la durata della gestazione e almeno tredici giorni dopo il parto, fino al giorno precedente alla soppressione programmata, compreso tale giorno.
|
|
13.
|
La durata dello studio, dopo l’acclimatazione e la valutazione del ciclo estrale precedente la somministrazione, dipende dal comportamento della femmina ed è di circa 63 giorni [almeno 14 giorni prima dell’accoppiamento, un massimo di 14 giorni per l’accoppiamento, 22 giorni di gestazione, 13 giorni di allattamento].
|
|
14.
|
Durante il periodo di somministrazione gli animali vengono esaminati attentamente e quotidianamente al fine di rilevare eventuali segni di tossicità. Gli animali deceduti o soppressi durante il periodo della prova vengono sottoposti a necroscopia; al termine della prova gli animali superstiti vengono soppressi e sottoposti a necroscopia.
|
DESCRIZIONE DEL METODO
Selezione della specie animale
|
15.
|
Il presente metodo di prova è destinato ad essere utilizzato con i ratti. Se i parametri specificati nel metodo di prova sono studiati in un’altra specie di roditori, occorre fornire una giustificazione dettagliata. Nel programma internazionale di validazione per l’individuazione degli interferenti endocrini nella linea guida n. 407 dell’OCSE (che corrisponde al capitolo B.7 del presente allegato) il ratto è stata l’unica specie animale utilizzata. Non vanno usati ceppi a bassa fecondità o con nota elevata incidenza di difetti dello sviluppo. Si devono utilizzare animali vergini sani, non sottoposti in precedenza a esperimenti. Gli animali utilizzati nella prova devono essere caratterizzati quanto alle specie, al ceppo, al sesso, al peso e all’età. All’inizio dello studio la variazione ponderale degli animali utilizzati deve essere minima e non superare il 20 % del peso medio di ciascun sesso. Per gli studi preliminari a studi a lungo termine o effettuati su un’intera generazione, è preferibile ricorrere in entrambi gli studi ad animali dello stesso ceppo e della stessa provenienza.
|
Condizioni di stabulazione e alimentazione
|
16.
|
Tutte le procedure devono attenersi agli standard locali in materia di cura degli animali da esperimento. La temperatura dello stabulario deve essere di 22 °C (± 3 °). L’umidità relativa deve raggiungere almeno il 30 % e preferibilmente non superare il 70 % (eccetto nel corso delle pulizie degli ambienti), ma occorre puntare a un valore del 50-60 %. L’illuminazione deve essere artificiale, con un fotoperiodo di 12 ore di luce e 12 ore di oscurità. Per quanto concerne l’alimentazione, si possono usare le diete convenzionali da laboratorio con una quantità illimitata di acqua da bere. La scelta della dieta può essere influenzata dalla necessità di garantire un’adeguata miscela della sostanza chimica in esame, se quest’ultima è somministrata con il cibo.
|
|
17.
|
Gli animali devono essere sistemati nelle gabbie in piccoli gruppi dello stesso sesso; possono essere sistemati in gabbie individuali se necessario per ragioni scientifiche. Ciascuna gabbia non deve ospitare più di cinque animali. L’accoppiamento va effettuato in gabbie adeguate allo scopo. Le femmine gravide devono essere tenute in gabbie individuali e rifornite di materiale per la costruzione della tana. Le femmine che allattano vanno poste in gabbie individuali insieme alla prole.
|
|
18.
|
L’alimentazione deve essere analizzata periodicamente per verificare la presenza di contaminanti. Un campione del mangime somministrato deve essere conservato fino al completamento della relazione.
|
Preparazione degli animali
|
19.
|
Gli animali adulti, sani e giovani vengono suddivisi a caso in gruppi di controllo e gruppi di trattamento. Le gabbie devono essere sistemate in modo da ridurre al minimo eventuali effetti dovuti alla loro collocazione. Gli animali vanno identificati inequivocabilmente e acclimatati alle condizioni di laboratorio per almeno cinque giorni prima dell’inizio dello studio.
|
Preparazione delle dosi
|
20.
|
Si raccomanda di somministrare la sostanza chimica in esame per via orale, a meno che non si considerino più appropriate altre vie di somministrazione. Se si sceglie la via orale, la sostanza chimica è generalmente somministrata mediante sonda gastrica; tuttavia, in alternativa, può essere somministrata con la dieta o l’acqua da bere.
|
|
21.
|
Ove necessario, la sostanza in esame è disciolta o sospesa in un mezzo disperdente adeguato. Si raccomanda di prendere anzitutto in considerazione, ove possibile, l’uso di una soluzione/sospensione acquosa, e in seconda battuta quello di una soluzione/emulsione in olio (ad esempio olio di semi di mais) e infine la possibile soluzione in altri mezzi disperdenti. Le caratteristiche tossiche dei mezzi disperdenti diversi dall’acqua devono essere note. È necessario determinare la stabilità e l’omogeneità della sostanza chimica in esame nel mezzo disperdente.
|
PROCEDURA
Numero e sesso degli animali
|
22.
|
Si raccomanda di costituire gruppi di almeno 10 maschi e 12-13 femmine. Nella fase pre-esposizione si procede alla valutazione della ciclicità estrale nelle femmine e gli animali che non presentano un ciclo tipico di 4-5 giorni non sono inclusi nello studio; si raccomanda pertanto di utilizzare un numero supplementare di femmine per ottenere 10 femmine per ciascun gruppo. Eccetto i casi in cui gli effetti tossici sono notevoli, si dovrebbero ottenere per ciascun gruppo almeno 8 femmine gravide, che generalmente è il numero minimo accettabile. Lo scopo è ottenere un numero di gravidanze e nidiate che consenta una valutazione significativa del potenziale della sostanza chimica in esame di influire negativamente sulla fertilità, sulla gravidanza, sul comportamento materno, sulla suzione, sulla crescita e sullo sviluppo della progenie F1, dal concepimento al tredicesimo giorno dopo il parto.
|
Dosaggio
|
23.
|
Si utilizzano generalmente almeno tre gruppi da trattare e un gruppo di controllo. I livelli di dosaggio possono essere basati sulle informazioni derivanti dalle prove di tossicità acuta o sui risultati di studi con dosi ripetute. Fatta eccezione per la somministrazione della sostanza chimica in esame, gli animali del gruppo di controllo devono essere trattati in modo identico agli esemplari dei gruppi sottoposti al trattamento. Se si usa un mezzo disperdente per la somministrazione della sostanza chimica in esame, al gruppo di controllo verrà somministrato il medesimo mezzo disperdente nel volume massimo utilizzato.
|
|
24.
|
I livelli di dosaggio devono essere selezionati tenendo conto di tutti i dati disponibili sulla tossicità e le caratteristiche (tossico-)cinetiche. Bisogna inoltre tener conto del fatto che potrebbero esserci differenze di sensibilità tra le femmine gravide e quelle non gravide. Il livello massimo di dosaggio deve essere tale da indurre effetti tossici senza cagionare la morte o sofferenze gravi. Deve essere inoltre definita una serie decrescente di livelli di dosaggio al fine di individuare un’eventuale correlazione dose-risposta e l’assenza di effetti avversi al dosaggio minimo (NOAEL, no-observed-adverse effects). In genere, per determinare i livelli decrescenti di dosaggio si consiglia un intervallo con un fattore compreso tra 2 e 4 e spesso è preferibile aggiungere un quarto gruppo di studio piuttosto che avere un intervallo eccessivamente lungo (ad esempio superiore a un fattore 10) fra un dosaggio e l’altro.
|
|
25.
|
Nel caso di tossicità generale osservata (ad es. riduzione del peso corporeo, effetti a livello epatico, cardiaco, polmonare o renale ecc.) o di altri cambiamenti che potrebbero non essere dovuti ad effetti tossici (ad es. diminuzione dell’assunzione di alimenti, dilatazione del fegato), gli effetti rilevati sugli endpoint endocrini devono essere interpretati con cautela.
|
Prova limite
|
26.
|
Se uno studio orale, effettuato secondo le procedure descritte per il presente studio, con un livello di dosaggio di almeno 1 000 mg/kg di peso corporeo/giorno o in caso di somministrazione con gli alimenti o l’acqua, a una concentrazione equivalente, non produce effetti tossici osservabili e se i dati relativi a sostanze di struttura analoga non indicano tossicità, si può considerare che non è necessario eseguire uno studio completo utilizzando diversi livelli di dosaggio. Si applica la prova limite, tranne quando l’esposizione umana indica la necessità di utilizzare un livello di dosaggio orale più elevato. Per altri tipi di somministrazione, ad esempio inalazione o applicazione cutanea, la concentrazione massima raggiungibile dipende in molti casi dalle proprietà fisico-chimiche delle sostanze chimiche da esaminare.
|
Somministrazione delle dosi
|
27.
|
Agli animali viene somministrata una dose giornaliera della sostanza chimica in esame sette giorni alla settimana. Se viene effettuata per via intragastrica, la somministrazione deve avvenire in dose singola mediante sonda gastrica o idonea cannula per intubazione. Il volume massimo di liquido che può essere somministrato in una sola volta dipende dalla taglia dell’animale. Il volume non deve superare 1 ml/100 g di peso corporeo, tranne nel caso delle soluzioni acquose che possono essere somministrate in quantità pari a 2 ml/100 g di peso corporeo. Salvo nel caso di sostanze chimiche irritanti o corrosive, i cui effetti di norma tendono a esacerbarsi con l’aumentare della concentrazione, la variabilità del volume somministrato deve essere ridotta al minimo adeguando la concentrazione, in modo da mantenere un volume costante per tutti i livelli di dosaggio.
|
|
28.
|
Per le sostanze chimiche somministrate con la dieta o l’acqua da bere è importante impedire che la quantità della sostanza chimica in esame interferisca con la normale alimentazione o il normale bilancio dei liquidi. Se la sostanza chimica in esame è somministrata con la dieta, si può utilizzare una concentrazione costante nella dieta (ppm) o un livello di dosaggio costante in funzione del peso corporeo di ciascun animale; la scelta di eventuali alternative va specificata. Nel caso di sostanze chimiche somministrate mediante sonda gastrica, la dose va somministrata ogni giorno all’incirca agli stessi orari e regolata almeno settimanalmente per mantenere un livello costante delle dosi rispetto al peso corporeo degli animali.
|
Protocollo sperimentale
|
29.
|
La somministrazione della sostanza a entrambi i sessi deve iniziare almeno due settimane prima dell’accoppiamento, dopo un periodo di acclimatazione di almeno cinque giorni e dopo avere osservato nelle femmine un ciclo estrale normale (durante un periodo di pre-trattamento di 2 settimane). Lo studio deve essere programmato in modo tale che la valutazione del ciclo estrale inizi subito dopo il raggiungimento della maturità sessuale completa da parte degli animali. Ciò può variare leggermente a seconda dei ceppi di ratti e dei laboratori, ad esempio 10 settimane per i ratti Sprague Dawley e circa 12 settimane per i ratti Wistar. Le femmine con prole devono essere soppresse il tredicesimo giorno dopo il parto o poco dopo. Il giorno della nascita (ossia quello in cui viene completato il parto) è il giorno 0 post-parto. Le femmine per le quali non vi sono prove di avvenuta copulazione sono soppresse 24-26 giorni dopo l’ultimo giorno del periodo di accoppiamento. La somministrazione della sostanza va continuata, in entrambi i sessi, durante il periodo di accoppiamento. Dopo tale periodo, la somministrazione deve proseguire per i maschi almeno fino al completamento del periodo di dosaggio totale minimo di 28 giorni. I maschi sono in seguito soppressi o, in alternativa, mantenuti in vita e continuano a ricevere la sostanza in esame ai fini di un eventuale secondo accoppiamento, ove ritenuto opportuno.
|
|
30.
|
La somministrazione giornaliera delle dosi alle femmine riproduttrici va continuata per tutta la gravidanza e almeno fino al tredicesimo giorno dopo il parto o al giorno prima della soppressione incluso. Per gli studi in cui la sostanza chimica in esame è somministrata tramite inalazione o per via cutanea, la somministrazione deve proseguire almeno fino al diciannovesimo giorno di gestazione incluso e deve essere iniziata di nuovo appena possibile e comunque entro il quarto giorno dopo il parto (PND 4).
|
|
31.
|
Un diagramma del calendario della prova è contenuto nell’appendice 2.
|
Procedura di accoppiamento
|
32.
|
Nel presente studio si utilizza normalmente il sistema di accoppiamento 1:1 (un maschio per una femmina). Possono essere previste eccezioni in caso di morte occasionale di esemplari maschi. Si deve mettere una femmina con lo stesso maschio fino a quando la copulazione è comprovata o sono trascorse due settimane. Ogni mattina le femmine devono essere esaminate per verificare la presenza di sperma o di tappi vaginali. Il giorno 0 della gravidanza è definito come il giorno in cui si riscontra la prova dell’avvenuta copulazione (presenza di un tappo vaginale o di sperma). Nel caso in cui l’accoppiamento non abbia successo, si può valutare la possibilità di far accoppiare le femmine con maschi di comprovata capacità riproduttiva dello stesso gruppo.
|
Dimensioni della nidiata
|
33.
|
Il quarto giorno dopo la nascita è possibile uniformare le dimensioni di ogni nidiata eliminando i piccoli in eccesso tramite selezione casuale, in modo da ottenere, nella misura del possibile, quattro o cinque piccoli per sesso per ciascuna nidiata, a seconda delle dimensioni normali della nidiata nei ceppi di ratti utilizzati. I campioni di sangue devono essere prelevati da due dei piccoli in eccedenza, raggruppati e utilizzati per determinare i livelli sierici di T4. L’eliminazione selettiva dei piccoli, ad esempio in base al peso corporeo o alla distanza anogenitale (AGD), non è opportuna. Ogni volta che il numero di piccoli maschi o femmine non permette di avere quattro o cinque animali di ciascun sesso per nidiata, è accettabile una regolazione parziale (ad esempio, sei maschi e quattro femmine). Se le dimensioni della nidiata scendono al di sotto della soglia di uniformazione (8 o 10 piccoli per ciascuna nidiata), nessun piccolo sarà eliminato. Qualora vi sia un solo piccolo al di sopra della soglia di uniformazione, esso sarà eliminato e utilizzato per prelevare il campione di sangue ai fini della determinazione dei livelli sierici di T4.
|
|
34.
|
Se le dimensioni della nidiata non sono uniformate, si sacrificano due piccoli per nidiata il quarto giorno dopo la nascita e si prelevano campioni di sangue per valutare la concentrazione sierica degli ormoni tiroidei. Se possibile, nel selezionare i due piccoli per nidiata da sacrificare, scegliere due femmine per tenere da parte i maschi per la valutazione della retrazione del capezzolo, tranne nel caso in cui l’eliminazione di questi piccoli non lasci alcuna femmina per le valutazioni da effettuare al termine. Non bisogna eliminare alcun piccolo se vi sono meno di 8 o 10 piccoli per nidiata (a seconda delle dimensioni normali della nidiata nei ceppi di ratti utilizzati). Qualora vi sia un solo piccolo in più rispetto alle dimensioni normali della nidiata, un solo piccolo sarà eliminato e utilizzato per prelevare il campione di sangue ai fini della determinazione dei livelli sierici di T4.
|
Osservazioni in vivo
Osservazioni cliniche
|
35.
|
Durante l’intero periodo della prova, le osservazioni cliniche generali vanno effettuate almeno una volta al giorno, con una frequenza maggiore se si constatano segni di tossicità. Le osservazioni devono essere effettuate preferibilmente ogni giorno alla stessa ora, tenendo conto del periodo probabile di massima intensità degli effetti dopo la somministrazione. Si devono registrare tutte le variazioni comportamentali pertinenti, i segni di parto difficile o prolungato e tutti i sintomi di tossicità, compresa la mortalità. Le registrazioni devono includere il momento dell’insorgenza, la gravità e la durata degli effetti tossici.
|
Peso corporeo e consumo di cibo/acqua
|
36.
|
I maschi e le femmine devono essere pesati il primo giorno della somministrazione, in seguito almeno settimanalmente, e al termine della somministrazione. Le femmine devono essere pesate durante la gravidanza i giorni 0, 7, 14 e 20 ed entro 24 ore dal parto (giorno 0 o 1 dopo il parto) e almeno il quarto e il tredicesimo giorno dopo il parto. Queste osservazioni vanno riportate singolarmente per ciascun animale adulto.
|
|
37.
|
Durante il periodo precedente l’accoppiamento e durante la gravidanza e l’allattamento, l’assunzione di cibo deve essere misurata almeno una volta alla settimana. La misurazione dell’assunzione di cibo durante l’accoppiamento è facoltativa. Se la sostanza chimica in esame è somministrata con acqua da bere, durante questi periodi va misurata anche l’assunzione di acqua.
|
Cicli estrali
|
38.
|
I cicli estrali devono essere monitorati prima dell’inizio del trattamento per selezionare per lo studio femmine aventi una ciclicità regolare (cfr. il paragrafo 22). Vanno monitorati anche gli strisci vaginali, ogni giorno dall’inizio del periodo di trattamento finché l’accoppiamento non risulti avvenuto. Se vi è il sospetto che effetti legati a forte stress all’inizio del trattamento possano alterare i cicli estrali, i laboratori possono esporre gli animali utilizzati nella prova per due settimane e quindi effettuare ogni giorno strisci vaginali per monitorare il ciclo estrale per almeno due settimane iniziando nel periodo precedente l’accoppiamento e proseguendo durante il periodo dell’accoppiamento finché quest’ultimo non risulti avvenuto. Durante il prelievo delle cellule vaginali/cervicali occorre prestare attenzione a non ledere la mucosa per evitare un’eventuale induzione di pseudogravidanza (7) (8).
|
Parametri relativi alla progenie
|
39.
|
La durata della gestazione deve essere registrata e calcolata dal giorno 0 di gravidanza. Ogni nidiata deve essere esaminata non appena possibile dopo il parto per stabilire il numero e il sesso dei piccoli, dei nati morti, dei nati vivi e degli esemplari più piccoli del normale (i piccoli di dimensioni molto inferiori ai corrispondenti piccoli di controllo) e la presenza di grosse anomalie.
|
|
40.
|
Si procede a contare i piccoli vivi e a identificarne il sesso e a pesare le nidiate entro 24 ore dal parto (giorno 0 o 1 dopo il parto) e almeno il quarto e il tredicesimo giorno dopo il parto. Oltre a quanto osservato al paragrafo 35, deve essere registrato qualsiasi comportamento anomalo della prole.
|
|
41.
|
Occorre misurare la distanza anogenitale (AGD) di ogni piccolo nello stesso giorno dalla nascita, tra il PND 0 e il PND 4. Il peso corporeo del piccolo va registrato il giorno in cui ne viene misurata l’AGD, che deve essere normalizzata in funzione della taglia, preferibilmente usando la radice cubica del peso corporeo (9). Va contato il numero di capezzoli/areole nei piccoli di sesso maschile il PND 12 o 13, come raccomandato nella linea guida n. 151 dell’OCSE (10).
|
Biochimica clinica
|
42.
|
Vanno prelevati campioni di sangue da un sito specifico in base al seguente programma:
|
—
|
da almeno due piccoli per nidiata il quarto giorno dopo la nascita, se il numero di piccoli lo consente (cfr. i paragrafi 33-34)
|
|
—
|
da tutte le madri e almeno due piccoli di 13 giorni per nidiata al termine dell’esperimento e
|
|
—
|
da tutti i maschi adulti, al termine dell’esposizione.
|
|
Tutti i campioni di sangue vanno conservati in condizioni adeguate. Si procede alla valutazione della concentrazione sierica per gli ormoni tiroidei (T4) nei campioni di sangue dei piccoli di 13 giorni e degli adulti maschi. Se del caso, si effettua un’ulteriore valutazione dell’ormone T4 nei campioni di sangue delle madri e dei piccoli di 4 giorni. Se opportuno, si possono misurare facoltativamente anche i livelli di altri ormoni. I prelievi di sangue dei piccoli possono essere raggruppati per nidiata ai fini dell’analisi degli ormoni tiroidei. Gli ormoni tiroidei (T4 e TSH) sono misurati di preferenza come «totale».
|
43.
|
I fattori seguenti possono influenzare la variabilità e le concentrazioni assolute delle analisi ormonali:
|
—
|
momento della soppressione, per via della variazione diurna delle concentrazioni ormonali;
|
|
—
|
metodi di soppressione, evitando di stressare inutilmente gli animali in quanto ciò potrebbe incidere sulle concentrazioni ormonali;
|
|
—
|
kit per le analisi ormonali che possono differire per le loro curve standard.
|
|
|
44.
|
I campioni di plasma destinati specificatamente all’analisi ormonale devono essere prelevati nelle stesse ore della giornata. I valori numerici ottenuti dalle analisi delle concentrazioni ormonali differiscono in funzione dei kit disponibili in commercio utilizzati.
|
Patologia
Necroscopia macroscopica
|
45.
|
Al momento della soppressione o del decesso durante lo studio va effettuato un esame macroscopico dei soggetti adulti alla ricerca di eventuali anomalie o alterazioni patologiche. Occorre prestare particolare attenzione agli organi dell’apparato riproduttivo e prendere nota del numero dei siti di impianto. Gli strisci vaginali devono essere esaminati al mattino il giorno della necroscopia per determinare lo stadio del ciclo estrale e consentire di stabilire correlazioni con l’istopatologia delle ovaie.
|
|
46.
|
Testicoli ed epididimi, come pure l’insieme composto dalla prostata e dalle vescicole seminali con le ghiandole della coagulazione, di tutti i maschi adulti vanno opportunamente liberati da eventuali tessuti aderenti e pesati umidi immediatamente dopo la dissezione, per evitare l’essiccamento. Inoltre, si possono pesare facoltativamente altri organi, come l’insieme del muscolo elevatore dell’ano e del muscolo bulbocavernoso, le ghiandole bulbouretrali e il glande nei maschi e le due ovaie (peso a umido) e l’utero (compresa la cervice) nelle femmine; se effettuate, queste misurazioni facoltative devono avvenire subito dopo la dissezione.
|
|
47.
|
I piccoli morti o soppressi il tredicesimo giorno dopo il parto, o poco dopo, devono essere sottoposti almeno a un attento esame esterno volto a individuare eventuali anomalie evidenti. Particolare attenzione deve essere dedicata all’apparato riproduttivo esterno, che va esaminato alla ricerca di eventuali alterazioni dello sviluppo. Il tredicesimo giorno bisogna conservare la tiroide di 1 piccolo maschio e di 1 piccolo femmina per nidiata.
|
|
48.
|
Le ovaie, i testicoli, gli organi sessuali accessori (utero e cervice, epididimi, prostata, vescicole seminali con ghiandole di coagulazione), la tiroide e tutti gli organi degli animali adulti che presentano lesioni macroscopiche devono essere conservati. La fissazione in formalina è sconsigliata per l’esame di routine di testicoli ed epididimi. Un metodo accettabile per questi tessuti è l’uso del liquido fissativo di Bouin o del liquido fissativo di Davidson modificato (11). La tunica albuginea può essere perforata, con un ago, con delicatezza e superficialmente in entrambi i poli dell’organo per consentire la rapida penetrazione del fissativo.
|
Esame istopatologico
|
49.
|
Va eseguito un esame istologico dettagliato di ovaie, testicoli ed epididimi (con particolare attenzione alle fasi della spermatogenesi e dell’istopatologia della struttura delle cellule interstiziali dei testicoli) degli animali del gruppo cui viene somministrata la dose più elevata e del gruppo di controllo. Gli altri organi conservati, compresa la tiroide dei piccoli e degli animali adulti, possono essere esaminati se necessario. Il peso della tiroide può essere stabilito dopo la fissazione. Anche in questo caso l’ablazione deve essere eseguita con cautela e solo previa fissazione per evitare di danneggiare i tessuti. L’eventuale danneggiamento dei tessuti infatti potrebbe compromettere l’analisi istopatologica. Si procede a questi esami anche sugli animali degli altri gruppi trattati se nel gruppo cui viene somministrata la dose più elevata si osservano alterazioni. Il documento di orientamento sull’istopatologia (11) fornisce informazioni supplementari sulla dissezione, la fissazione, l’asportazione e l’istopatologia dei tessuti endocrini.
|
DATI E RELAZIONE
Dati
|
50.
|
Devono essere forniti dati individuali su ciascun animale. Inoltre, tutti i dati vanno riassunti sotto forma di tabella, evidenziando per ciascun gruppo di studio il numero di animali all’inizio della prova, il numero di animali rinvenuti morti durante la prova o sottoposti a eutanasia, il momento di eventuali decessi o soppressioni, il numero di animali fertili, il numero di femmine gravide, il numero di animali che mostrano segni di tossicità, una descrizione dei segni di tossicità osservati, ivi compresi il momento dell’insorgenza, la durata e la gravità degli effetti tossici, i tipi di alterazioni istopatologiche, nonché tutti i dati di rilievo riguardanti le nidiate. Un formato tabulare di relazione sintetica che si è rivelato molto utile per la valutazione degli effetti sulla riproduzione/sullo sviluppo è contenuto nell’appendice 3.
|
|
51.
|
Tenuto conto della portata ridotta dello studio, le analisi statistiche sotto forma di prove intese ad accertare la «significatività» dei risultati hanno un valore limitato per molti endpoint, soprattutto quelli riproduttivi. Se si ricorre ad analisi statistiche, il metodo scelto deve essere adatto alla distribuzione della variabile esaminata e selezionato prima di iniziare lo studio. L’analisi statistica dell’AGD e della retrazione del capezzolo deve essere basata sui dati individuali dei piccoli, tenendo conto degli effetti sulle nidiate. Se opportuno, utilizzare la nidiata come unità di analisi. L’analisi statistica del peso corporeo dei piccoli deve essere basata sui dati individuali dei piccoli, tenendo conto delle dimensioni della nidiata. A causa delle dimensioni ridotte del gruppo, può essere utile anche rifarsi ad eventuali dati di controllo storici (ad esempio, per le dimensioni della nidiata) per facilitare l’interpretazione dei dati emersi dallo studio.
|
Valutazione dei risultati
|
52.
|
I risultati del presente studio di tossicità vanno valutati in base agli effetti osservati e ai risultati della necroscopia e dell’esame microscopico. La valutazione deve includere il rapporto fra la dose della sostanza chimica in esame e la presenza o l’assenza, l’incidenza e la gravità delle anomalie, comprese eventuali lesioni macroscopiche, gli organi bersaglio identificati, l’infertilità, le anomalie cliniche, la capacità riproduttiva, gli effetti sulla generazione successiva, le alterazioni del peso corporeo, gli effetti sulla mortalità ed eventuali altri effetti tossici.
|
|
53.
|
Tenuto conto del breve periodo di trattamento del maschio, l’istopatologia dei testicoli e degli epididimi deve essere considerata insieme ai dati sulla fertilità nel quadro della valutazione degli effetti sulla riproduzione del maschio. Può essere utile anche utilizzare eventuali dati di controllo storici sulla riproduzione/lo sviluppo (ad esempio per le dimensioni della nidiata, l’AGD, la retrazione del capezzolo, i livelli sierici di T4) per facilitare l’interpretazione dello studio.
|
|
54.
|
Ai fini del controllo di qualità, si suggerisce di raccogliere dati di controllo storici e di calcolare i coefficienti di variazione per i dati numerici, in particolare per i parametri legati all’individuazione degli interferenti endocrini. Questi dati possono essere utilizzati, a fini di confronto, in fase di valutazione degli studi effettivamente realizzati.
|
Relazione sull’esecuzione della prova
|
55.
|
La relazione sull’esecuzione della prova deve comprendere le seguenti informazioni:
|
|
Sostanza chimica in esame:
|
—
|
origine, numero di lotto, data limite per l’uso, se disponibili;
|
|
—
|
stabilità della sostanza chimica in esame, se nota;
|
|
|
|
Sostanza monocostituente:
|
—
|
aspetto fisico, idrosolubilità e, se del caso, ulteriori proprietà fisico-chimiche;
|
|
—
|
dati di identificazione chimica: denominazioni IUPAC o CAS, numero CAS, codice SMILES o InChI, formula strutturale, purezza, identità chimica delle impurezze, se del caso e se le condizioni pratiche lo consentono, ecc.;
|
|
|
|
Sostanza multicostituente, UVCB e miscele:
|
—
|
caratterizzate nella massima misura possibile con l’identità chimica (cfr. sopra), con la presenza quantitativa e con le proprietà fisico-chimiche pertinenti dei costituenti;
|
|
|
|
Mezzo disperdente (se del caso):
|
—
|
giustificazione per la scelta del mezzo disperdente utilizzato, se diverso dall’acqua;
|
|
|
|
Animali utilizzati nella prova:
|
—
|
specie/ceppo impiegati;
|
|
—
|
numero, età e sesso degli animali;
|
|
—
|
origine, condizioni di alloggio, dieta, ecc.;
|
|
—
|
peso di ciascun animale all’inizio della prova;
|
|
—
|
qualora non siano stati utilizzati ratti, spiegazione del motivo;
|
|
|
|
Condizioni sperimentali:
|
—
|
criteri di selezione delle dosi;
|
|
—
|
informazioni dettagliate sulla formulazione della sostanza chimica in esame/preparazione della dieta, sulle concentrazioni finali, sulla stabilità e sull’omogeneità del preparato;
|
|
—
|
modalità precise di somministrazione della sostanza chimica in esame;
|
|
—
|
se del caso, conversione della concentrazione della sostanza nella dieta o nell’acqua (ppm) in dose effettiva (mg/kg di peso corporeo/giorno);
|
|
—
|
dettagli sulla qualità del cibo e dell’acqua;
|
|
—
|
descrizione dettagliata del protocollo di randomizzazione utilizzato per selezionare gli eventuali piccoli da sopprimere;
|
|
|
|
Risultati:
|
—
|
peso corporeo/cambiamenti del peso corporeo;
|
|
—
|
assunzione di cibo ed eventualmente di acqua;
|
|
—
|
dati sulla risposta tossica per sesso e per dose, compresi i dati sulla fertilità, la gestazione ed eventuali altri sintomi di tossicità;
|
|
—
|
durata della gestazione,
|
|
—
|
effetti tossici o di altro tipo sulla riproduzione, sulla prole, sulla crescita postnatale, ecc.;
|
|
—
|
natura, gravità e durata dei segni clinici (sia reversibili che non reversibili);
|
|
—
|
numero di femmine adulte con ciclo estrale normale o anomalo e durata del ciclo;
|
|
—
|
numero di nati vivi e di perdite post-impianto;
|
|
—
|
dati relativi al peso corporeo dei piccoli;
|
|
—
|
AGD di tutti i piccoli (e peso corporeo il giorno della misurazione dell’AGD);
|
|
—
|
retrazione del capezzolo nei piccoli di sesso maschile;
|
|
—
|
livelli degli ormoni tiroidei, piccoli di 13 giorni e maschi adulti (e madri e piccoli di 4 giorni se sottoposti a misurazione);
|
|
—
|
numero di piccoli con anomalie evidenti, valutazione macroscopica degli organi genitali esterni, numero di esemplari più piccoli del normale;
|
|
—
|
momento del decesso durante lo studio o indicazione della sopravvivenza degli animali alla conclusione della prova;
|
|
—
|
numero di impianti e dimensioni e peso della nidiata al momento della registrazione;
|
|
—
|
peso corporeo al momento della soppressione e dati sul peso degli organi negli animali genitori;
|
|
—
|
risultati della necroscopia;
|
|
—
|
descrizione dettagliata dei risultati istopatologici;
|
|
—
|
dati sull’assorbimento (se disponibili);
|
|
—
|
elaborazione statistica dei risultati, se del caso.
|
|
Discussione dei risultati
Conclusioni
|
Interpretazione dei risultati
|
56.
|
Lo studio fornirà valutazioni della tossicità per la riproduzione/lo sviluppo associata alla somministrazione di dosi ripetute (cfr. i paragrafi 5 e 6). Esso può fornire indicazioni sulla necessità di condurre ulteriori indagini e fornisce orientamenti in merito alla progettazione di studi successivi. Si consulti il documento di orientamento n. 43 dell’OCSE per indicazioni sull’interpretazione dei risultati in merito alla riproduzione e allo sviluppo (12). Il documento di orientamento n. 106 dell’OCSE sulla valutazione istologica delle prove endocrine e sulla riproduzione nei roditori (11) fornisce informazioni sulla preparazione e la valutazione degli organi (endocrini) e degli strisci vaginali che possono essere utili per questa linea guida.
|
BIBLIOGRAFIA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Available Upon Request at Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L.(1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Organisation for Economic Cooperation and Development, Paris.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Organisation for Economic Cooperation and Development, Paris.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
Appendice 1
DEFINIZIONI (CFR. ANCHE LA LINEA GUIDA N. 150 DELL'OCSE (6))
Androgenicità: la capacità di una sostanza chimica di agire come un ormone androgenico naturale (ad es. il testosterone) in un mammifero.
Antiandrogenicità: la capacità di una sostanza chimica di inibire l'attività di un ormone androgenico naturale (ad es. il testosterone) in un mammifero.
Antiestrogenicità: la capacità di una sostanza chimica di inibire l'attività di un ormone estrogenico naturale (ad es. l'estradiolo 17ß) in un mammifero.
Attività antitiroidea: la capacità di una sostanza chimica di inibire l'attività di un ormone tiroideo naturale (ad es. T3) in un mammifero.
Sostanza chimica: una sostanza o una miscela.
Tossicità per lo sviluppo: la manifestazione della tossicità per la riproduzione, che risulta in disturbi prenatali, perinatali, postnatali, strutturali o funzionali nella progenie.
Dosaggio: termine generale che ricomprende la dose, la frequenza e la durata della somministrazione.
Dose: quantità di sostanza chimica somministrata. La dose è espressa come peso della sostanza chimica in esame per unità di peso corporeo dell'animale utilizzato nella prova per giorno (mg/kg peso corporeo/giorno) o come una concentrazione costante nella dieta.
Tossicità evidente: termine generale che designa i segnali evidenti di tossicità a seguito della somministrazione di una sostanza chimica. Questi segni devono essere sufficienti per consentire la valutazione dei pericoli ed essere tali che si possa prevedere che l'aumento della dose somministrata comporti la comparsa di segni di tossicità grave e probabilmente la mortalità.
Compromissione della fertilità: i disturbi delle funzioni o della capacità riproduttive maschili o femminili.
Tossicità materna: gli effetti nocivi sulle femmine gravide, che si verificano in modo specifico (effetto diretto) o non specifico (effetto indiretto).
NOAEL: l'abbreviazione di no-observed-adverse-effect level, ossia la dose più elevata alla quale non si osservano effetti avversi legati al trattamento.
Estrogenicità: la capacità di una sostanza chimica di agire come un ormone estrogenico naturale (ad es. l'estradiolo 17ß) in un mammifero.
Tossicità della riproduzione: gli effetti nocivi sulla progenie e/o la compromissione delle funzioni o della capacità riproduttive maschili e femminili.
Sostanza chimica in esame: qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova.
Attività tiroidea: la capacità di una sostanza chimica di agire come un ormone tiroideo naturale (ad es. T3) in un mammifero.
Validazione: processo scientifico destinato a caratterizzare i requisiti e i limiti operativi di un metodo di prova e a dimostrarne l'affidabilità e la pertinenza per un fine specifico.
Appendice 2
DIAGRAMMA DEL PROTOCOLLO SPERIMENTALE INDICANTE LA DURATA MASSIMA DELLO STUDIO, BASATO SU UN PERIODO DI ACCOPPIAMENTO COMPLETO DI 14 GIORNI

Appendice 3
RELAZIONE SINTETICA IN FORMATO TABULARE DEGLI EFFETTI SULLA RIPRODUZIONE/SULLO SVILUPPO
|
OSSERVAZIONI
|
VALORI
|
|
|
|
Dosaggio (unità)
|
0 (controllo)
|
...
|
...
|
...
|
...
|
|
Coppie formate (N)
|
|
|
|
|
|
|
Ciclo estrale (almeno durata media e frequenza dei cicli irregolari)
|
|
|
|
|
|
|
Femmine per le quali la copulazione risulta avvenuta (N)
|
|
|
|
|
|
|
Femmine gravide (N)
|
|
|
|
|
|
|
Giorni di concepimento 1 - 5 (N)
|
|
|
|
|
|
|
Giorni di concepimento 6 -...(
(21)
) (N)
|
|
|
|
|
|
|
Gravidanza ≤ 21 giorni (N)
|
|
|
|
|
|
|
Gravidanza = 22 giorni (N)
|
|
|
|
|
|
|
Gravidanza ≥ 23 giorni (N)
|
|
|
|
|
|
|
Madri che hanno partorito piccoli vivi (N)
|
|
|
|
|
|
|
Madri con piccoli vivi il quarto giorno dopo il parto (N)
|
|
|
|
|
|
|
Impianti/madre (media)
|
|
|
|
|
|
|
Piccoli vivi/madre alla nascita (media)
|
|
|
|
|
|
|
Piccoli vivi/madre al quarto giorno (media)
|
|
|
|
|
|
|
Rapporto numerico tra i sessi (m/f) alla nascita (media)
|
|
|
|
|
|
|
Rapporto numerico tra i sessi (m/f) al quarto giorno (media)
|
|
|
|
|
|
|
Peso della nidiata alla nascita (media)
|
|
|
|
|
|
|
Peso della nidiata al quarto giorno (media)
|
|
|
|
|
|
|
Peso dei piccoli alla nascita (media)
|
|
|
|
|
|
|
Peso dei piccoli al momento della misurazione dell'AGD (media maschi, media femmine)
|
|
|
|
|
|
|
AGD dei piccoli misurata lo stesso giorno dalla nascita, tra la nascita e il giorno 4 (media maschi, media femmine, prendere nota del PND)
|
|
|
|
|
|
|
Peso dei piccoli al quarto giorno (media)
|
|
|
|
|
|
|
Retrazione del capezzolo dei piccoli maschi al tredicesimo giorno (media)
|
|
|
|
|
|
|
Peso dei piccoli al tredicesimo giorno (media)
|
|
|
|
|
|
|
|
|
PICCOLI CON ANOMALIE
|
|
Madri con 0
|
|
|
|
|
|
|
Madri con 1
|
|
|
|
|
|
|
Madri con ≥ 2
|
|
|
|
|
|
|
|
|
PERDITA DELLA PROGENIE
|
|
|
|
Perdite prenatali/dopo l'impianto (impianti meno piccoli nati vivi)
|
|
Femmine con 0
|
|
|
|
|
|
|
Femmine con 1
|
|
|
|
|
|
|
Femmine con 2
|
|
|
|
|
|
|
Femmine con ≥ 3
|
|
|
|
|
|
|
|
|
Perdite postnatali (piccoli nati vivi meno piccoli vivi il tredicesimo giorno dopo il parto)
|
|
Femmine con 0
|
|
|
|
|
|
|
Femmine con 1
|
|
|
|
|
|
|
Femmine con 2
|
|
|
|
|
|
|
Femmine con ≥ 3
|
|
|
|
|
|
B.64 STUDIO DI TOSSICITÀ CON DOSE RIPETUTA COMBINATO CON LA PROVA DI SCREENING DELLA TOSSICITÀ PER LA RIPRODUZIONE/LO SVILUPPO
INTRODUZIONE
|
1.
|
Il presente metodo di prova è equivalente alla linea guida n. 422 dell’OCSE (2016). Le linee guida dell’OCSE per le prove sulle sostanze chimiche sono periodicamente rivedute e aggiornate alla luce del progresso scientifico. La linea guida n. 422 sulla prova di screening è stata adottata inizialmente nel 1996, sulla base di un protocollo per una «prova di screening della tossicità con dose ripetuta combinata con una prova di screening della tossicità per la riproduzione e lo sviluppo» di cui si è discusso nell’ambito di due riunioni di esperti, a Londra nel 1990 (1) e a Tokyo nel 1992 (2).
|
|
2.
|
Il presente metodo di prova consta di una parte in cui è effettuato uno screening della tossicità per la riproduzione/lo sviluppo, basata sull’esperienza acquisita negli Stati membri attraverso l’utilizzo del metodo originario con le sostanze chimiche esistenti a elevato volume di produzione e la conduzione di prove esplorative con sostanze utilizzate come controllo positivo (3) (4), e di una parte in cui viene esaminata la tossicità mediante prove con dosi ripetute, conformemente alla linea guida per le prove n. 407 dell’OCSE (studio della tossicità orale con somministrazione ripetuta di dosi per 28 giorni sui roditori, corrispondente al capitolo B.7 del presente allegato).
|
|
3.
|
Il presente metodo di prova è stato aggiornato mediante l’aggiunta di endpoint pertinenti per il rilevamento di interferenti endocrini, come seguito all’attività ad alta priorità avviata dall’OCSE nel 1998 volta a rivedere le linee guida esistenti e a elaborarne di nuove, per lo screening e la sperimentazione relativi a potenziali interferenti endocrini (5). In tale contesto, la linea guida n. 407 dell’OCSE (che corrisponde al capitolo B.7 del presente allegato) è stata migliorata nel 2008 mediante l’aggiunta di parametri adatti per l’individuazione dell’attività endocrina delle sostanze chimiche in esame. La linea guida n. 422 è stata aggiornata con l’intento di includere gli endpoint pertinenti per il rilevamento di interferenti endocrini nelle linee guida in cui i periodi di esposizione coprono alcuni dei periodi sensibili dello sviluppo (i periodi precedenti o immediatamente successivi alla nascita).
|
|
4.
|
Gli ulteriori endpoint pertinenti per il rilevamento di interferenti endocrini selezionati, che fanno parte anche della linea guida n. 443 (studio esteso di tossicità per la riproduzione su una generazione, corrispondente al capitolo B.56 del presente allegato), sono stati aggiunti alla linea guida n. 422 sulla base di uno studio di fattibilità riguardante questioni scientifiche e tecniche relative alla loro inclusione, come pure gli eventuali adattamenti del disegno sperimentale necessari per la loro inclusione (6).
|
|
5.
|
Il presente metodo di prova è inteso a generare informazioni limitate concernenti gli effetti delle sostanze chimiche in esame sulla capacità riproduttiva maschile e femminile, quali la funzione delle gonadi, il comportamento in fase di accoppiamento, il concepimento, lo sviluppo dell’organismo concepito e il parto. Esso non rappresenta un’alternativa ai metodi di prova esistenti B.31, B.34, B.35 e B.56, né li sostituisce.
|
CONSIDERAZIONI INIZIALI
|
6.
|
Nella valutazione e nell’esame delle caratteristiche tossiche di una sostanza chimica è possibile determinare la tossicità orale utilizzando dosi ripetute dopo aver ottenuto dati preliminari sulla tossicità mediante prove di tossicità acuta. Il presente studio fornisce informazioni sui rischi che l’esposizione ripetuta per un periodo di tempo relativamente limitato può comportare per la salute. Il metodo prevede uno studio di base della tossicità a dosi ripetute che può essere utilizzato per le sostanze chimiche per le quali uno studio di 90 giorni non si giustifica (ad esempio quando il volume di produzione non supera determinate quantità) o prima di uno studio a lungo termine. Durante lo svolgimento dello studio è opportuno seguire i principi guida e le considerazioni di cui al documento di orientamento dell’OCSE n. 19, Recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation (7).
|
|
7.
|
Esso comprende inoltre una prova di screening della tossicità per la riproduzione/lo sviluppo e, pertanto, può essere utilizzato anche per fornire informazioni iniziali sui possibili effetti sulla capacità riproduttiva maschile e femminile, quali la funzione delle gonadi, il comportamento in fase di accoppiamento, il concepimento, lo sviluppo dell’organismo concepito e il parto, nella fase iniziale di valutazione delle proprietà tossicologiche delle sostanze chimiche o nella valutazione di sostanze potenzialmente pericolose. Il presente metodo di prova non fornisce informazioni complete su tutti gli aspetti della riproduzione e dello sviluppo. In particolare, esso offre solo mezzi limitati per individuare manifestazioni postnatali di un’esposizione prenatale o effetti eventualmente riconducibili a un’esposizione postnatale. Tenuto conto (tra le altre cose) della selettività degli endpoint e della breve durata dello studio, il presente metodo non fornisce prove sufficienti per trarre conclusioni definitive circa l’assenza di effetti sulla riproduzione o lo sviluppo. Inoltre, in assenza di dati desunti da altre prove di tossicità per la riproduzione/lo sviluppo, risultati positivi sono utili per una valutazione iniziale dei pericoli e permettono di prendere decisioni circa la necessità di procedere a ulteriori prove e i tempi per effettuarle.
|
|
8.
|
I risultati ottenuti per quanto riguarda i parametri endocrini devono essere interpretati alla luce del «Quadro concettuale dell’OCSE per le prove e la valutazione delle sostanze chimiche che alterano il sistema endocrino» (8). La linea guida migliorata dell’OCSE n. 422 fa parte del livello 4 di questo quadro concettuale come saggio in vivo in grado di fornire dati sugli effetti nocivi sugli endpoint pertinenti per il sistema endocrino. Un segnale endocrino potrebbe tuttavia non essere considerato di per sé una prova sufficiente ad attestare che la sostanza chimica in esame è un interferente endocrino.
|
|
9.
|
Il presente metodo di prova attribuisce inoltre particolare importanza agli effetti neurologici in quanto parametro specifico di valutazione e comporta la necessità di un’accurata osservazione clinica degli animali per ottenere il maggior numero possibile di informazioni. Tale metodo è finalizzato all’individuazione di sostanze chimiche dotate di un potenziale neurotossico, che potranno successivamente richiedere indagini più approfondite al riguardo. Inoltre, il metodo può fornire anche un’indicazione di base degli effetti immunologici.
|
|
10.
|
In assenza di dati desunti da altri studi sulla tossicità sistemica, la tossicità per la riproduzione/lo sviluppo, la neurotossicità e/o l’immunotossicità, risultati positivi sono utili per una valutazione iniziale dei pericoli e permettono di prendere decisioni circa la necessità di procedere a ulteriori prove e i tempi per effettuarle. La prova può essere particolarmente utile nell’ambito delle serie di dati di informazione di monitoraggio dell’OCSE per la valutazione delle sostanze chimiche esistenti per le quali le informazioni tossicologiche sono scarse o inesistenti e può essere utilizzata come alternativa alla conduzione di due prove distinte, rispettivamente per la tossicità a dosi ripetute (linea guida n. 407 dell’OCSE, corrispondente al capitolo B.7 del presente allegato) e la tossicità per la riproduzione/lo sviluppo (linea guida n. 421 dell’OCSE, corrispondente al capitolo B.63 del presente allegato). Può essere utilizzata anche come studio di determinazione degli intervalli di dosaggio per studi più approfonditi sulla riproduzione o sullo sviluppo o in altri casi in cui sia ritenuto opportuno.
|
|
11.
|
In genere, si presuppone che vi siano differenze a livello di sensibilità tra le femmine gravide e quelle non gravide. Di conseguenza, rispetto alla conduzione di prove distinte, in questa prova combinata può essere più complicato determinare i livelli di dosaggio adeguati per valutare sia la tossicità sistemica generale sia la tossicità specifica per la riproduzione/lo sviluppo. Inoltre, rispetto alla conduzione di uno studio a dosi ripetute, l’interpretazione dei risultati della prova per quanto riguarda la tossicità sistemica generale può essere più difficile, soprattutto quando i parametri sierici e istopatologici non sono valutati contemporaneamente nello studio. A causa di queste complessità tecniche, per eseguire questa prova di screening combinata è richiesta molta esperienza nella conduzione di prove di tossicità. Dall’altro lato, oltre a coinvolgere un numero inferiore di animali, la prova combinata può consentire di distinguere meglio gli effetti diretti sulla riproduzione/lo sviluppo da quelli secondari rispetto ad altri effetti (sistemici).
|
|
12.
|
Il periodo di esposizione di questa prova è più lungo rispetto a quello di un convenzionale studio a dosi ripetute di 28 giorni. Tuttavia, il numero di animali di ciascun sesso per gruppo è inferiore a quello utilizzato nel caso di un convenzionale studio a dosi ripetute di 28 giorni condotto in aggiunta a una prova di screening della tossicità per la riproduzione/lo sviluppo.
|
|
13.
|
Il presente metodo di prova prevede la somministrazione orale della sostanza chimica da esaminare. Potrebbero essere necessarie modifiche in caso di utilizzo di altre vie di somministrazione.
|
|
14.
|
Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie laddove esista una disposizione normativa che obblighi a sottoporre a prova la miscela.
|
|
15.
|
L’appendice 1 contiene le definizioni dei termini utilizzati.
|
PRINCIPIO DELLA PROVA
|
16.
|
La sostanza chimica in esame viene somministrata in dosi graduate a diversi gruppi di maschi e femmine. Ai maschi le dosi devono essere somministrate per almeno quattro settimane e fino al giorno precedente alla soppressione programmata, compreso tale giorno (tale periodo comprende un minimo di due settimane prima dell’accoppiamento, il tempo dell’accoppiamento e circa due settimane dopo l’accoppiamento). Tenuto conto della breve durata del periodo di esposizione prima dell’accoppiamento nei maschi, la fertilità potrebbe non essere un particolare indicatore di sensibilità per la tossicità testicolare. È pertanto essenziale un esame istologico particolareggiato delle prove. La combinazione di un periodo di esposizione di due settimane prima dell’accoppiamento (seguito da osservazioni sull’accoppiamento e la fertilità) e di un periodo di esposizione complessivo di almeno quattro settimane (seguito da un dettagliato esame istopatologico delle gonadi maschili) è considerata sufficiente a consentire l’individuazione della maggior parte degli effetti sulla fertilità maschile e sulla spermatogenesi.
|
|
17.
|
Alle femmine la sostanza va somministrata durante l’intero studio. Tale periodo comprende due settimane prima dell’accoppiamento (con l’obiettivo di coprire almeno due cicli estrali completi), l’intervallo variabile che precede il concepimento, la durata della gestazione e almeno tredici giorni dopo il parto, fino al giorno precedente alla soppressione programmata, compreso tale giorno.
|
|
18.
|
La durata dello studio, dopo l’acclimatazione e la valutazione del ciclo estrale precedente la somministrazione, dipende dal comportamento della femmina ed è di circa 63 giorni [almeno 14 giorni prima dell’accoppiamento, un massimo di 14 giorni per l’accoppiamento, 22 giorni di gestazione, 13 giorni di allattamento].
|
|
19.
|
Durante il periodo di somministrazione gli animali vengono esaminati attentamente e quotidianamente al fine di rilevare eventuali segni di tossicità. Gli animali deceduti o soppressi durante l’esperimento vengono sottoposti a necroscopia; al termine della prova gli animali superstiti vengono soppressi e sottoposti a necroscopia.
|
DESCRIZIONE DEL METODO
Selezione della specie animale
|
20.
|
Il presente metodo di prova è destinato ad essere utilizzato con i ratti. Se i parametri specificati nella presente linea guida n. 422 sono studiati in un’altra specie di roditori occorre fornire una giustificazione dettagliata. Nel programma internazionale di validazione per l’individuazione degli interferenti endocrini nella linea guida n. 407 il ratto è stata l’unica specie animale utilizzata. Non vanno usati ceppi a bassa fecondità o con nota elevata incidenza di difetti dello sviluppo. Si devono utilizzare animali vergini sani, non sottoposti in precedenza a esperimenti. Gli animali utilizzati nella prova devono essere caratterizzati quanto alle specie, al ceppo, al sesso, al peso e all’età. All’inizio dello studio la variazione ponderale degli animali utilizzati deve essere minima e non superare il 20 % circa del peso medio di ciascun sesso. Per gli studi preliminari a studi a lungo termine o effettuati su un’intera generazione, è preferibile ricorrere in entrambi gli studi ad animali dello stesso ceppo e della stessa provenienza.
|
Condizioni di stabulazione e alimentazione
|
21.
|
Tutte le procedure devono attenersi agli standard locali in materia di cura degli animali da esperimento. La temperatura dello stabulario deve essere di 22 °C (± 3 °C). L’umidità relativa deve essere non inferiore al 30 % e, preferibilmente, non superiore al 70 %, tranne durante la pulizia dei locali. L’illuminazione deve essere artificiale, con un fotoperiodo di 12 ore di luce e 12 ore di oscurità. Per quanto concerne l’alimentazione, si possono usare le diete convenzionali da laboratorio con una quantità illimitata di acqua da bere. La scelta della dieta può essere influenzata dalla necessità di garantire un’adeguata miscela della sostanza chimica in esame, se quest’ultima è somministrata con il cibo.
|
|
22.
|
Gli animali devono essere sistemati nelle gabbie in piccoli gruppi dello stesso sesso; possono essere sistemati in gabbie individuali se necessario per ragioni scientifiche. Ciascuna gabbia non deve ospitare più di cinque animali. L’accoppiamento va effettuato in gabbie adeguate allo scopo. Le femmine gravide devono essere tenute in gabbie individuali e rifornite di materiale per la costruzione della tana. Le femmine che allattano vanno poste in gabbie individuali insieme alla prole.
|
|
23.
|
L’alimentazione deve essere analizzata periodicamente per verificare la presenza di contaminanti. Un campione del mangime somministrato deve essere conservato fino al completamento della relazione.
|
Preparazione degli animali
|
24.
|
Vengono scelti a caso soggetti giovani adulti destinati ai gruppi di trattamento e alle gabbie. Le gabbie devono essere sistemate in modo da ridurre al minimo eventuali effetti dovuti alla loro collocazione. Gli animali vanno identificati inequivocabilmente e acclimatati alle condizioni di laboratorio per almeno cinque giorni prima dell’inizio dello studio.
|
Preparazione delle dosi
|
25.
|
Si raccomanda di somministrare la sostanza chimica in esame per via orale, a meno che non si considerino più appropriate altre vie di somministrazione. Se si sceglie la via orale, la sostanza chimica è generalmente somministrata mediante sonda gastrica; tuttavia, in alternativa, può essere somministrata anche con la dieta o l’acqua da bere.
|
|
26.
|
Ove necessario, la sostanza in esame è disciolta o sospesa in un mezzo disperdente adeguato. Si raccomanda di prendere anzitutto in considerazione, ogni qualvolta possibile, l’uso di una soluzione/sospensione acquosa, e in seconda battuta quello di una soluzione/sospensione in olio (ad esempio olio di semi di mais) e infine la possibile soluzione in altri mezzi disperdenti. Le caratteristiche tossiche dei mezzi disperdenti non acquosi devono essere note. È necessario determinare la stabilità e l’omogeneità della sostanza chimica in esame nel mezzo disperdente.
|
PROCEDURA
Numero e sesso degli animali
|
27.
|
Si raccomanda di costituire gruppi di almeno 10 maschi e 12-13 femmine. Nella fase pre-esposizione si procede alla valutazione della ciclicità estrale nelle femmine e gli animali che non presentano un ciclo tipico di 4-5 giorni non sono inclusi nello studio; si raccomanda pertanto di utilizzare un numero supplementare di femmine per ottenere 10 femmine per ciascun gruppo. Eccetto i casi in cui gli effetti tossici sono notevoli, si dovrebbero ottenere per ciascun gruppo almeno 8 femmine gravide, che generalmente è il numero minimo accettabile. Lo scopo è ottenere un numero di gravidanze e nidiate che consenta una valutazione significativa del potenziale della sostanza chimica in esame di influire negativamente sulla fertilità, sulla gravidanza, sul comportamento materno, sulla suzione, sulla crescita e sullo sviluppo della progenie F1, dal concepimento al tredicesimo giorno dopo il parto. Qualora si preveda di sacrificare a intervalli intermedi alcuni animali, il numero va aumentato del numero di animali che si prevede di sacrificare prima del completamento dello studio. Si può considerare di includere un gruppo satellite supplementare di cinque animali (5 per sesso) nel gruppo di controllo e nel gruppo trattato con la dose più elevata al fine di monitorare la reversibilità, la persistenza o l’insorgenza ritardata di effetti tossici sistemici, per almeno 14 giorni dopo il trattamento. Gli animali del gruppo satellite non vengono fatti accoppiare e, di conseguenza, non sono utilizzati ai fini della valutazione della tossicità per la riproduzione/lo sviluppo.
|
Dosaggio
|
28.
|
Si utilizzano generalmente almeno tre gruppi da trattare e un gruppo di controllo. Per stabilire le dosi da utilizzare, in mancanza di dati adeguati relativi alla tossicità in generale si può effettuare uno studio preliminare di tipo range finding. Fatta eccezione per la somministrazione della sostanza chimica in esame, gli animali del gruppo di controllo devono essere trattati in modo identico agli esemplari dei gruppi sottoposti al trattamento. Se si usa un mezzo disperdente per la somministrazione della sostanza chimica in esame, al gruppo di controllo verrà somministrato il medesimo mezzo disperdente nel volume massimo utilizzato.
|
|
29.
|
I livelli di dosaggio devono essere selezionati tenendo conto di tutti i dati disponibili sulla tossicità e le caratteristiche (tossico-)cinetiche. Bisogna inoltre tener conto del fatto che potrebbero esserci differenze di sensibilità tra le femmine gravide e quelle non gravide. Il livello massimo di dosaggio deve essere tale da indurre effetti tossici senza cagionare la morte o sofferenze gravi. Sarà inoltre definita una serie decrescente di dosaggi al fine di individuare eventuali risposte a dosi determinate e dimostrare l’assenza di effetti avversi al dosaggio minimo. Per la determinazione dei livelli di dose decrescenti risulta spesso ottimale applicare un fattore di divisione compreso tra due e quattro; è comunque preferibile aggiungere un quarto gruppo di studio piuttosto che avere uno scarto eccessivo (ad esempio superiore a un fattore 10) tra un dosaggio e l’altro.
|
|
30.
|
Nel caso di tossicità generale osservata (ad es. riduzione del peso corporeo, effetti a livello epatico, cardiaco, polmonare o renale ecc.) o di altri cambiamenti che potrebbero non essere dovuti ad effetti tossici (ad es. diminuzione dell’assunzione di alimenti, dilatazione del fegato), gli effetti rilevati sugli endpoint endocrini devono essere interpretati con cautela.
|
Prova limite
|
31.
|
Qualora uno studio orale, effettuato in conformità con il metodo descritto, con un livello di dosaggio di almeno 1 000 mg/kg di peso corporeo/giorno o, in caso di somministrazione con gli alimenti o l’acqua, ad una concentrazione equivalente (in funzione del peso corporeo), non produca effetti tossici osservabili e se i dati relativi a sostanze di struttura analoga non indicano tossicità, si può ritenere non necessario uno studio completo con diversi dosaggi. La prova limite va effettuata, tranne quando i dati sull’esposizione umana indicano la necessità di utilizzare un livello di dosaggio più elevato. Per altri tipi di somministrazione, ad esempio per inalazione o applicazione cutanea, il livello massimo di esposizione realizzabile dipende in molti casi dalle proprietà fisico-chimiche delle sostanze chimiche da esaminare.
|
Somministrazione delle dosi
|
32.
|
Agli animali viene somministrata una dose giornaliera della sostanza chimica in esame sette giorni alla settimana. Se viene effettuata per via intragastrica, la somministrazione deve avvenire in dose singola mediante sonda gastrica o idonea cannula per intubazione. Il volume massimo di liquido che può essere somministrato in una sola volta dipende dalla taglia dell’animale. Il volume non deve superare 1 ml/100 g di peso corporeo, tranne nel caso delle soluzioni acquose che possono essere somministrate in quantità pari a 2 ml/100 g di peso corporeo. Salvo nel caso di sostanze chimiche irritanti o corrosive, i cui effetti di norma tendono a esacerbarsi con l’aumentare della concentrazione, la variabilità del volume somministrato deve essere ridotta al minimo adeguando la concentrazione, in modo da mantenere un volume costante per tutti i livelli di dosaggio.
|
|
33.
|
Per le sostanze chimiche somministrate con la dieta o l’acqua da bere è importante impedire che la quantità della sostanza chimica in esame interferisca con la normale alimentazione o il normale bilancio dei liquidi. Se la sostanza chimica in esame è somministrata con la dieta, si può utilizzare una concentrazione costante nella dieta (ppm) o un livello di dosaggio costante in funzione del peso corporeo di ciascun animale; la scelta di eventuali alternative va specificata. Nel caso di sostanze chimiche somministrate mediante sonda gastrica, la dose va somministrata ogni giorno all’incirca agli stessi orari e regolata almeno settimanalmente per mantenere un livello costante delle dosi rispetto al peso corporeo degli animali. Se lo studio combinato è preliminare a uno studio sulla tossicità a lungo termine o a uno studio effettuato su un’intera generazione, occorre utilizzare una dieta simile in entrambi gli studi.
|
Protocollo sperimentale
|
34.
|
La somministrazione della sostanza a entrambi i sessi deve iniziare due settimane prima dell’accoppiamento, dopo un periodo di acclimatazione di almeno cinque giorni e dopo avere osservato nelle femmine un ciclo estrale normale (durante un periodo di pre-trattamento di 2 settimane). Lo studio deve essere programmato in modo tale che la valutazione del ciclo estrale inizi subito dopo il raggiungimento della maturità sessuale completa da parte degli animali. Ciò può variare leggermente a seconda dei ceppi di ratti e dei laboratori, ad esempio 10 settimane per i ratti Sprague Dawley e circa 12 settimane per i ratti Wistar. Le femmine con prole devono essere soppresse il tredicesimo giorno dopo il parto o poco dopo. Per consentire il digiuno notturno delle madri prima del prelievo di sangue (se si preferisce tale opzione), non è necessario sopprimere le madri e la loro progenie lo stesso giorno. Il giorno della nascita (ossia quello in cui viene completato il parto) è il giorno 0 post-parto. Le femmine per le quali non vi sono prove di avvenuta copulazione sono soppresse 24-26 giorni dopo l’ultimo giorno del periodo di accoppiamento. La somministrazione della sostanza va continuata, in entrambi i sessi, durante il periodo di accoppiamento. Dopo tale periodo, la somministrazione deve proseguire per i maschi almeno fino al completamento del periodo di dosaggio totale minimo di 28 giorni. I maschi sono in seguito soppressi o mantenuti in vita e continuano a ricevere la sostanza in esame ai fini di un eventuale secondo accoppiamento, ove ritenuto opportuno.
|
|
35.
|
La somministrazione giornaliera delle dosi alle femmine riproduttrici va continuata per tutta la gravidanza e almeno fino al tredicesimo giorno dopo il parto o al giorno prima della soppressione incluso. Per gli studi in cui la sostanza chimica in esame è somministrata tramite inalazione o per via cutanea, la somministrazione deve proseguire almeno fino al diciannovesimo giorno di gestazione incluso e deve essere iniziata di nuovo appena possibile e comunque entro il quarto giorno dalla nascita (PND 4).
|
|
36.
|
Gli animali del gruppo satellite destinati al monitoraggio di follow-up, se inclusi, non vengono fatti accoppiare. Essi devono essere esaminati per almeno altri 14 giorni dopo la prima soppressione pianificata delle madri, senza alcun trattamento, al fine di individuare l’insorgenza tardiva, la persistenza o la scomparsa degli effetti tossici.
|
|
37.
|
Un diagramma del calendario della prova è contenuto nell’appendice 2.
|
Cicli estrali
|
38.
|
I cicli estrali devono essere monitorati prima dell’inizio del trattamento per selezionare per lo studio femmine aventi una ciclicità regolare (cfr. il paragrafo 27). Vanno monitorati anche gli strisci vaginali, ogni giorno dall’inizio del periodo di trattamento finché l’accoppiamento non risulti avvenuto. Se vi è il sospetto che effetti legati a forte stress all’inizio del trattamento possano alterare i cicli estrali, i laboratori possono esporre gli animali utilizzati nella prova per due settimane e quindi effettuare ogni giorno strisci vaginali per monitorare il ciclo estrale per almeno due settimane iniziando nel periodo precedente l’accoppiamento e proseguendo durante il periodo dell’accoppiamento finché quest’ultimo non risulti effettuato. Durante il prelievo delle cellule vaginali/cervicali occorre prestare attenzione a non ledere la mucosa per evitare un’eventuale induzione di pseudogravidanza (8) (9).
|
Procedura di accoppiamento
|
39.
|
Nel presente studio si utilizza normalmente il sistema di accoppiamento 1:1 (un maschio per una femmina). Possono essere previste eccezioni in caso di morte occasionale di esemplari maschi. Si deve mettere una femmina con lo stesso maschio fino a quando la copulazione è comprovata o sono trascorse due settimane. Ogni mattina le femmine devono essere esaminate per verificare la presenza di sperma o di tappi vaginali. Il giorno 0 della gravidanza è definito come il giorno in cui si riscontra la prova dell’avvenuta copulazione (presenza di un tappo vaginale o di sperma). Nel caso in cui l’accoppiamento non abbia successo, si può valutare la possibilità di far accoppiare le femmine con maschi di comprovata capacità riproduttiva dello stesso gruppo.
|
Dimensioni della nidiata
|
40.
|
Il quarto giorno dopo la nascita è possibile uniformare le dimensioni di ogni nidiata eliminando i piccoli in eccesso tramite selezione casuale, in modo da ottenere, nella misura del possibile, quattro o cinque piccoli per sesso per ciascuna nidiata, a seconda delle dimensioni normali della nidiata nei ceppi di ratti utilizzati. I campioni di sangue devono essere prelevati da due dei piccoli in eccedenza, raggruppati e utilizzati per determinare i livelli sierici di T4. L’eliminazione selettiva dei piccoli, ad esempio in base al peso corporeo o alla distanza anogenitale (AGD), non è opportuna. Ogni volta che il numero di piccoli maschi o femmine non permette di avere quattro o cinque animali di ciascun sesso per nidiata, è accettabile una regolazione parziale (ad esempio, sei maschi e quattro femmine). Se le dimensioni della nidiata scendono al di sotto della soglia di uniformazione (8 o 10 piccoli per ciascuna nidiata), nessun piccolo sarà eliminato. Qualora vi sia un solo piccolo al di sopra della soglia di uniformazione, esso sarà eliminato e utilizzato per prelevare il campione di sangue ai fini della determinazione dei livelli sierici di T4.
|
|
41.
|
Se le dimensioni della nidiata non sono uniformate, si sacrificano due piccoli per nidiata il quarto giorno dopo la nascita e si prelevano campioni di sangue per valutare la concentrazione sierica degli ormoni tiroidei. Se possibile, nel selezionare i due piccoli per nidiata da sacrificare, scegliere due femmine per tenere da parte i maschi per la valutazione della retrazione del capezzolo, tranne nel caso in cui l’eliminazione di questi piccoli non lasci alcuna femmina per le valutazioni da effettuare al termine. Non bisogna eliminare alcun piccolo se vi sono meno di 8 o 10 piccoli per nidiata (a seconda delle dimensioni normali della nidiata nei ceppi di ratti utilizzati). Qualora vi sia un solo piccolo in più rispetto alle dimensioni normali della nidiata, un solo piccolo sarà eliminato e utilizzato per prelevare il campione di sangue ai fini della determinazione dei livelli sierici di T4.
|
Osservazioni
|
42.
|
Le osservazioni cliniche generali devono essere effettuate almeno una volta al giorno, preferibilmente alla stessa ora e tenendo conto del periodo di massima intensità degli effetti previsti dopo la somministrazione. Vanno registrate le informazioni concernenti le condizioni di salute degli animali. Almeno due volte al giorno, tutti gli animali vengono esaminati al fine di determinare la morbilità e la mortalità.
|
|
43.
|
Una volta prima dell’esposizione iniziale (per consentire un confronto sullo stesso soggetto) e, successivamente, almeno una volta alla settimana tutti gli animali genitori vengono sottoposti ad osservazioni cliniche particolareggiate. A tale scopo gli animali vengono tolti dalle gabbie, collocati in un recinto standard ed esaminati di preferenza sempre alla stessa ora, ogni giorno. Occorre registrare con cura le osservazioni, preferibilmente usando sistemi di punteggio statistico definiti appositamente dal laboratorio che esegue la prova. Si avrà cura di ridurre al minimo le variazioni delle condizioni sperimentali; le osservazioni devono essere effettuate da persone che non sono a conoscenza del trattamento somministrato. Si terrà conto, tra l’altro, di tutte le alterazioni della cute, del pelo, degli occhi, delle mucose, della comparsa di secrezioni ed escrezioni e dell’attività neurovegetativa (per esempio lacrimazione, piloerezione, ampiezza pupillare, ritmo respiratorio insolito). Vanno inoltre registrati cambiamenti dell’andatura, della postura e della risposta alla manipolazione, nonché la presenza di movimenti clonici o tonici, stereotipie (ad esempio tolettatura eccessiva, continuo girare in cerchio), parto difficoltoso o prolungato o comportamenti insoliti (ad esempio automutilazione, marcia a ritroso) (10).
|
|
44.
|
Ad un certo punto durante lo studio, devono essere svolte valutazioni della reattività sensoriale a stimoli di vario tipo (ad esempio stimoli uditivi, visivi e propriocettivi) (8) (9) (11), della forza di presa (12) e dell’attività motoria (13) per cinque maschi e cinque femmine, selezionati da ciascun gruppo secondo un criterio di casualità. Ulteriori indicazioni sui procedimenti utilizzabili sono contenute nelle voci bibliografiche citate. Tuttavia possono essere applicate anche procedure alternative non indicate nella bibliografia. Nei maschi queste osservazioni funzionali devono essere effettuate verso la fine del periodo di esposizione, poco prima della soppressione programmata ma prima del prelievo dei campioni di sangue per gli esami ematologici o gli esami biochimici clinici (cfr. i paragrafi 53-56, compresa la nota a piè di pagina 1). La prova sulle femmine, che devono trovarsi in uno stato fisiologico simile durante queste prove funzionali, deve essere effettuata di preferenza una sola volta nell’ultima settimana di allattamento (ad esempio, il sesto-tredicesimo giorno di allattamento), poco prima della soppressione programmata. Nella misura del possibile, ridurre i tempi di separazione delle madri e dei piccoli.
|
|
45.
|
Le osservazioni funzionali effettuate una volta verso la fine dello studio possono essere evitate nel caso di uno studio preliminare ad un successivo studio subcronico (90 giorni) o ad un successivo studio a lungo termine. In questa eventualità, le osservazioni funzionali devono essere incluse nello studio complementare. D’altro canto, le informazioni ricavate dalle osservazioni funzionali nel corso dello studio a dosi ripetute possono essere utili nella determinazione dei livelli di dosaggio per un successivo studio subcronico o a lungo termine.
|
|
46.
|
Eccezionalmente è possibile omettere le osservazioni funzionali per i gruppi che evidenzino comunque segni di tossicità tali da interferire in modo significativo con l’esecuzione degli esami funzionali.
|
|
47.
|
La durata della gestazione deve essere registrata e calcolata dal giorno 0 di gravidanza. Ogni nidiata deve essere esaminata non appena possibile dopo il parto per stabilire il numero e il sesso dei piccoli, dei nati morti, dei nati vivi e degli esemplari più piccoli del normale (i piccoli molto più piccoli dei piccoli di controllo) e la presenza di grosse anomalie.
|
|
48.
|
Si procede a contare e identificare il sesso dei piccoli vivi e a pesare le nidiate entro 24 ore dal parto (giorno 0 o 1 dopo il parto) e almeno il quarto e il tredicesimo giorno dopo il parto. Oltre a quanto osservato per gli animali genitori (cfr. i paragrafi 43 e 44), deve essere registrato qualsiasi comportamento anomalo della prole.
|
|
49.
|
Occorre misurare la distanza anogenitale (AGD) di ogni piccolo nello stesso giorno dalla nascita, tra il PND 0 e il PND 4. Il peso corporeo del piccolo va registrato il giorno in cui ne viene misurata l’AGD, che deve essere normalizzata in funzione della taglia, preferibilmente usando la radice cubica del peso corporeo (14). Va contato il numero di capezzoli/areole nei piccoli di sesso maschile il PND 12 o 13, come raccomandato nella linea guida n. 151 dell’OCSE (15).
|
Peso corporeo e consumo di cibo/acqua
|
50.
|
I maschi e le femmine devono essere pesati il primo giorno della somministrazione, in seguito almeno settimanalmente, e al termine della somministrazione. Le femmine devono essere pesate durante la gravidanza i giorni 0, 7, 14 e 20 ed entro 24 ore dal parto (giorno 0 o 1 dopo il parto) e almeno il quarto e il tredicesimo giorno dopo il parto. Queste osservazioni vanno riportate singolarmente per ciascun animale adulto.
|
|
51.
|
Durante il periodo precedente l’accoppiamento e durante la gravidanza e l’allattamento, l’assunzione di cibo deve essere misurata almeno una volta alla settimana. La misurazione dell’assunzione di cibo durante l’accoppiamento è facoltativa. Se la sostanza chimica in esame è somministrata con acqua da bere, durante questi periodi va misurata anche l’assunzione di acqua.
|
Ematologia
|
52.
|
Una volta durante lo studio devono essere effettuati i seguenti esami ematologici su cinque maschi e cinque femmine, selezionati da ciascun gruppo secondo un criterio di casualità: ematocrito, concentrazioni di emoglobina, conteggio degli eritrociti, reticulociti, conteggio totale e differenziale dei leucociti, numero di placchette e misura del tempo e del potenziale di coagulazione. Se la sostanza in esame o i suoi metaboliti putativi hanno o possono avere proprietà ossidanti occorre effettuare altre analisi, relative tra l’altro alla concentrazione di metaemoglobine o ai corpi di Heinz.
|
|
53.
|
I campioni di sangue devono essere prelevati da un determinato sito. Durante il prelievo dei campioni le femmine devono trovarsi in uno stato fisiologico simile. Allo scopo di evitare le difficoltà pratiche connesse alla variabilità all’inizio della gestazione, i prelievi di sangue nelle femmine possono essere effettuati alla fine del periodo precedente l’accoppiamento in alternativa al prelievo dei campioni di sangue al momento della soppressione degli animali o nel periodo immediatamente precedente. Per i maschi, i campioni di sangue devono essere prelevati di preferenza al momento della soppressione degli animali o nel periodo immediatamente precedente. In alternativa, i prelievi di sangue nei maschi possono essere effettuati anche alla fine del periodo precedente l’accoppiamento se per le femmine è stato prescelto questo momento.
|
|
54.
|
I campioni devono essere conservati in condizioni adeguate.
|
Biochimica clinica
|
55.
|
Le determinazioni biochimiche cliniche per lo studio degli effetti tossici gravi sui tessuti e, specificamente, degli effetti su reni e fegato, vanno condotte su campioni di sangue prelevati dai cinque maschi e dalle cinque femmine di ciascun gruppo selezionati. Si raccomanda di lasciare gli animali a digiuno la notte precedente la raccolta dei campioni (22). Le analisi sul plasma o sul siero comprenderanno il sodio, il potassio, il glucosio, il colesterolo totale, l’urea, la creatinina, le proteine totali e l’albumina, almeno due enzimi indicatori degli effetti epatocellulari (come l’alanina aminotransferasi, l’aspartato aminotransferasi e la sorbitol deidrogenasi). Le determinazioni di altri enzimi (di origine epatica o di altro tipo) e della bilirubina possono talvolta fornire indicazioni utili.
|
|
56.
|
Vanno prelevati campioni di sangue da un sito specifico in base al seguente programma:
|
—
|
da almeno due piccoli per nidiata il quarto giorno dopo la nascita, se il numero di piccoli lo consente (cfr. i paragrafi 40-41)
|
|
—
|
da tutte le madri e almeno due piccoli di 13 giorni per nidiata al termine dell’esperimento e
|
|
—
|
da tutti i maschi adulti, al termine dell’esperimento.
|
Tutti i campioni di sangue vanno conservati in condizioni adeguate. Si procede alla valutazione della concentrazione sierica per gli ormoni tiroidei (T4) nei campioni di sangue dei piccoli di 13 giorni e degli adulti maschi. Se del caso, si effettua un’ulteriore valutazione dell’ormone T4 nei campioni di sangue delle madri e dei piccoli di 4 giorni. Se opportuno, si possono misurare facoltativamente anche i livelli di altri ormoni. I prelievi di sangue dei piccoli possono essere raggruppati per nidiata ai fini dell’analisi degli ormoni tiroidei. Gli ormoni tiroidei (T4 e TSH) sono misurati di preferenza come «totale».
|
|
57.
|
A titolo facoltativo, nel corso dell’ultima settimana dello studio si possono effettuare le seguenti analisi delle urine su campioni raccolti in momenti specifici: aspetto, volume, osmolalità o densità relativa, pH, proteine, glucosio e sangue/cellule ematiche.
|
|
58.
|
È inoltre necessario considerare la possibilità di condurre studi sui marker sierologici dei danni generici ai tessuti. Altre determinazioni dovranno essere eseguite qualora si abbia motivo di ritenere o di sospettare che le proprietà della sostanza chimica in esame possano alterare i profili metabolici riguardanti il calcio, il fosfato, i trigliceridi a digiuno e la glicemia a digiuno, gli ormoni specifici, la metemoglobina e la colinesterasi. Questi devono essere confermati caso per caso.
|
|
59.
|
I fattori seguenti possono influenzare la variabilità e le concentrazioni assolute delle analisi ormonali:
|
—
|
momento della soppressione, per via della variazione diurna delle concentrazioni ormonali;
|
|
—
|
metodi di soppressione, evitando di stressare inutilmente gli animali in quanto ciò potrebbe incidere sulle concentrazioni ormonali;
|
|
—
|
kit per le analisi ormonali che possono differire per le loro curve standard.
|
|
|
60.
|
I campioni di plasma destinati specificatamente all’analisi ormonale devono essere prelevati nelle stesse ore della giornata. I valori numerici ottenuti dalle analisi delle concentrazioni ormonali differiscono in funzione dei kit disponibili in commercio utilizzati.
|
|
61.
|
Se i dati di riferimento storici sono inadeguati, occorre tenere conto delle variabili ematologiche e di biochimica clinica prima di iniziare i dosaggi, di preferenza su un gruppo di animali diverso dal gruppo in esame. Per le femmine, i dati devono riguardare animali che allattano.
|
PATOLOGIA
Necroscopia macroscopica
|
62.
|
Tutti gli animali adulti utilizzati nello studio vanno sottoposti a un’autopsia macroscopica completa e dettagliata che comprenda un attento esame della superficie esterna del corpo, di tutti gli orifizi e delle cavità cranica, toracica e addominale e del loro contenuto. Occorre prestare particolare attenzione agli organi dell’apparato riproduttivo e prendere nota del numero dei siti di impianto. Gli strisci vaginali devono essere esaminati il giorno dell’autopsia per determinare lo stadio del ciclo estrale e consentire di stabilire correlazioni con l’istopatologia degli organi riproduttivi femminili.
|
|
63.
|
Testicoli ed epididimi, come pure l’insieme composto dalla prostata e dalle vescicole seminali con le ghiandole della coagulazione, di tutti i maschi adulti vanno opportunamente liberati da eventuali tessuti aderenti e pesati umidi immediatamente dopo la dissezione, per evitare l’essiccamento. Inoltre, si possono pesare facoltativamente altri organi, come l’insieme del muscolo elevatore dell’ano e del muscolo bulbocavernoso, le ghiandole bulbouretrali e il glande nei maschi e le due ovaie (peso a umido) e l’utero (compresa la cervice) nelle femmine; se effettuate, queste misurazioni facoltative devono avvenire subito dopo la dissezione. Le ovaie, i testicoli, gli epididimi, gli organi sessuali accessori e tutti gli organi degli animali adulti che presentano lesioni macroscopiche devono essere conservati.
|
|
64.
|
Per tutti i maschi e le femmine adulti e per un piccolo maschio e un piccolo femmina di tredici giorni di ciascuna nidiata, le ghiandole della tiroide devono essere conservate nel mezzo di fissazione più adatto per il successivo esame istopatologico che si intende effettuare. Il peso della tiroide può essere stabilito dopo la fissazione. Anche in questo caso l’ablazione deve essere eseguita con cautela e solo previa fissazione per evitare di danneggiare i tessuti. L’eventuale danneggiamento dei tessuti infatti potrebbe compromettere l’analisi istopatologica. I campioni di sangue devono essere prelevati da un determinato sito immediatamente prima o durante la soppressione degli animali e conservati in condizioni adeguate (cfr. il paragrafo 56).
|
|
65.
|
Inoltre, per almeno cinque maschi e femmine adulti, selezionati a caso da ciascun gruppo (tranne quelli trovati moribondi e/o sacrificati prima della conclusione dello studio), fegato, reni, ghiandole surrenali, timo, milza, cervello e cuore vanno opportunamente liberati da eventuali tessuti aderenti e pesati umidi immediatamente dopo la dissezione, per evitare l’essiccamento. I seguenti tessuti vanno conservati nel mezzo di fissazione più adatto, sia per il tipo di tessuto, sia per il successivo esame istopatologico che si intende effettuare: tutte le lesioni macroscopiche, cervello (porzioni rappresentative comprendenti cervello, cervelletto e ponte), midollo spinale, occhi, stomaco, intestino tenue e crasso (comprese le placche di Peyer), fegato, reni, ghiandole surrenali, milza, cuore, timo, trachea e polmoni (conservati con dilatazione mediante fissativo e poi immersione), gonadi (testicoli e ovaie), organi sessuali accessori (utero e collo dell’utero, epididimi, prostata, vescicole seminali con ghiandole della coagulazione), vagina, vescica e linfonodi [oltre al linfonodo più vicino un altro linfonodo, in funzione dell’esperienza del laboratorio (16)], nervo periferico (sciatico o tibiale) preferibilmente in prossimità del muscolo, muscolo e osso dello scheletro con il midollo osseo (una sezione o un preparato fresco di midollo osseo aspirato). Si raccomanda di fissare i testicoli mediante immersione in un fissativo di Bouin o di Davidson modificato (16) (17) (18); la fissazione in formalina è sconsigliata per questi tessuti. La tunica albuginea può essere perforata, con un ago, con delicatezza e superficialmente in entrambi i poli dell’organo per consentire la rapida penetrazione del fissativo. I risultati clinici e di altro tipo possono evidenziare la necessità di esaminare altri tessuti. Vanno inoltre conservati tutti gli organi considerati organi bersaglio in base alle proprietà note della sostanza in esame.
|
|
66.
|
I tessuti elencati qui di seguito possono apportare informazioni utili sugli effetti endocrini: gonadi (ovaie e testicoli), organi sessuali accessori (utero, collo dell’utero, epididimi, vescicole seminali con ghiandole di coagulazione, prostata dorso laterale e ventrale), vagina, ipofisi, ghiandola mammaria maschile e ghiandola surrenale. Non ci sono sufficienti riscontri di alterazioni nelle ghiandole mammarie maschili, ma questo parametro può essere molto sensibile alle sostanze con attività estrogenica. L’osservazione degli organi/tessuti non ripresi nel paragrafo 65 è facoltativa.
|
|
67.
|
I piccoli morti o soppressi il tredicesimo giorno dopo il parto, o poco dopo, devono essere sottoposti almeno a un attento esame esterno volto a individuare eventuali anomalie evidenti. Particolare attenzione deve essere dedicata all’apparato riproduttivo esterno, che va esaminato alla ricerca di eventuali alterazioni dello sviluppo.
|
Esame istopatologico
|
68.
|
Gli organi e i tessuti conservati di tutti gli animali selezionati del gruppo di controllo e del gruppo trattato con la dose più elevata vanno sottoposti a un esame istopatologico completo (con particolare attenzione alle fasi della spermatogenesi nelle gonadi maschili e dell’istopatologia della struttura delle cellule interstiziali dei testicoli). La ghiandola della tiroide dei piccoli e degli animali adulti restanti può essere esaminata se necessario. Si procede a questi esami anche sugli animali degli altri gruppi se nel gruppo cui viene somministrata la dose più elevata si osservano alterazioni correlate al trattamento. Il documento di orientamento sull’istopatologia (10) fornisce informazioni supplementari sulla dissezione, la fissazione, l’asportazione e l’istopatologia dei tessuti endocrini.
|
|
69.
|
Vanno esaminate tutte le lesioni macroscopiche. Allo scopo di contribuire alla determinazione dei NOAEL, devono essere esaminati gli organi bersaglio di altri gruppi-dose, soprattutto dei gruppi che presenterebbero un NOAEL.
|
|
70.
|
Quando si utilizza un gruppo satellite l’esame istopatologico va eseguito sui tessuti e sugli organi per i quali sono stati osservati effetti nei gruppi trattati.
|
DATI E RELAZIONE
Dati
|
71.
|
Devono essere forniti dati individuali su ciascun animale. Inoltre, tutti i dati vanno riassunti sotto forma di tabella, evidenziando per ciascun gruppo di studio il numero di animali all’inizio della prova, il numero di animali rinvenuti morti durante la prova o sottoposti a eutanasia, il momento di eventuali decessi o soppressioni, il numero di animali fertili, il numero di femmine gravide, il numero di animali che mostrano segni di tossicità, una descrizione dei segni di tossicità osservati, ivi compresi il momento dell’insorgenza, la durata e la gravità degli effetti tossici, i tipi di alterazioni istopatologiche, nonché tutti i dati di rilievo riguardanti le nidiate. Un formato tabulare di relazione sintetica che si è rivelato molto utile per la valutazione degli effetti sulla riproduzione/sullo sviluppo è contenuto nell’appendice 3.
|
|
72.
|
Se possibile, i risultati numerici devono essere valutati sulla base di un metodo statistico appropriato e comunemente accettato. Per i confronti degli effetti osservati nell’ambito di un intervallo di dosaggio si deve evitare il ricorso a prove t multiple. I metodi statistici devono essere selezionati durante la fase di progettazione dello studio. L’analisi statistica dell’AGD e della retrazione del capezzolo deve essere basata sui dati individuali dei piccoli, tenendo conto degli effetti sulle nidiate. Se opportuno, utilizzare la nidiata come unità di analisi. L’analisi statistica del peso corporeo dei piccoli deve essere basata sui dati individuali dei piccoli, tenendo conto delle dimensioni della nidiata. Tenuto conto della portata ridotta dello studio, le analisi statistiche sotto forma di prove intese ad accertare la «significatività» dei risultati hanno un valore limitato per molti endpoint, soprattutto quelli riproduttivi. Alcuni dei metodi più ampiamente utilizzati, soprattutto le prove parametriche per la valutazione della tendenza centrale, sono inadeguati. Se si ricorre ad analisi statistiche, il metodo scelto deve essere adatto alla distribuzione della variabile esaminata e selezionato prima dell’inizio dello studio.
|
Valutazione dei risultati
|
73.
|
I risultati del presente studio di tossicità vanno valutati in base agli effetti osservati e ai risultati della necroscopia e dell’esame microscopico. La valutazione deve includere il rapporto fra la dose della sostanza chimica in esame e la presenza o l’assenza, l’incidenza e la gravità delle anomalie, comprese eventuali lesioni macroscopiche, gli organi bersaglio identificati, l’infertilità, le anomalie cliniche, la capacità riproduttiva, gli effetti sulla generazione successiva, le alterazioni del peso corporeo, gli effetti sulla mortalità ed eventuali altri effetti tossici.
|
|
74.
|
Tenuto conto del breve periodo di trattamento del maschio, l’istopatologia dei testicoli e degli epididimi deve essere considerata insieme ai dati sulla fertilità nel quadro della valutazione degli effetti sulla riproduttività del maschio. Può essere utile anche utilizzare eventuali dati di controllo storici sulla riproduzione/lo sviluppo (ad esempio per le dimensioni della nidiata, l’AGD, la retrazione del capezzolo, i livelli sierici di T4) per facilitare l’interpretazione dello studio.
|
|
75.
|
Ai fini del controllo di qualità, si suggerisce di raccogliere dati di controllo storici e di calcolare i coefficienti di variazione per i dati numerici, in particolare per i parametri legati all’individuazione degli interferenti endocrini. Questi dati possono essere utilizzati, a fini di confronto, in fase di valutazione degli studi effettivamente realizzati.
|
Relazione sull’esecuzione della prova
|
76.
|
La relazione sull’esecuzione della prova deve comprendere le seguenti informazioni:
|
|
Sostanza chimica in esame:
|
—
|
origine, numero di lotto, data limite per l’uso, se disponibili;
|
|
—
|
stabilità della sostanza chimica in esame, se nota;
|
|
|
|
Sostanza monocostituente:
|
—
|
aspetto fisico, idrosolubilità e, se del caso, ulteriori proprietà fisico-chimiche;
|
|
—
|
dati di identificazione chimica: denominazioni IUPAC o CAS, numero CAS, codice SMILES o InChI, formula strutturale, purezza, identità chimica delle impurezze, se del caso e se le condizioni pratiche lo consentono, ecc.;
|
|
|
|
Sostanza multicostituente, UVCB e miscele:
|
—
|
caratterizzate nella massima misura possibile con l’identità chimica (cfr. sopra), con la presenza quantitativa e con le proprietà fisico-chimiche pertinenti dei costituenti
|
; |
|
|
Mezzo disperdente (se del caso):
|
—
|
giustificazione per la scelta del mezzo disperdente utilizzato, se diverso dall’acqua;
|
|
|
|
Animali utilizzati nella prova:
|
—
|
specie/ceppo impiegati;
|
|
—
|
numero, età e sesso degli animali;
|
|
—
|
origine, condizioni di alloggio, dieta, ecc.;
|
|
—
|
peso di ciascun animale all’inizio della prova;
|
|
—
|
qualora non siano stati utilizzati ratti, spiegazione del motivo;
|
|
|
|
Condizioni sperimentali:
|
—
|
criteri di selezione delle dosi;
|
|
—
|
informazioni dettagliate sulla formulazione della sostanza chimica in esame/incorporazione nella dieta, sulla concentrazione finale, sulla stabilità e sull’omogeneità del preparato;
|
|
—
|
modalità precise di somministrazione della sostanza chimica in esame;
|
|
—
|
se del caso, conversione della concentrazione della sostanza nella dieta o nell’acqua (ppm) in dose effettiva (mg/kg di peso corporeo/giorno);
|
|
—
|
dettagli sulla qualità del cibo e dell’acqua;
|
|
—
|
descrizione dettagliata del protocollo di randomizzazione utilizzato per selezionare gli eventuali piccoli da sopprimere;
|
|
|
|
Risultati:
|
—
|
peso corporeo/cambiamenti del peso corporeo;
|
|
—
|
assunzione di cibo ed eventualmente di acqua;
|
|
—
|
dati sulla risposta tossica per sesso e per dose, compresa la fertilità, la gestazione ed eventuali altri sintomi di
|
|
—
|
durata della gestazione, effetti tossici o di altro tipo sulla riproduzione, sulla prole, sulla crescita postnatale, ecc.;
|
|
—
|
natura, gravità e durata dei segni clinici (sia reversibili che non reversibili);
|
|
—
|
valutazione dell’attività sensoriale, della forza prensile e dell’attività motoria;
|
|
—
|
esami ematologici con i relativi valori basali;
|
|
—
|
esami biochimici clinici con i relativi valori basali;
|
|
—
|
numero di femmine adulte con ciclo estrale normale o anomalo e durata del ciclo;
|
|
—
|
numero di nati vivi e di perdite post-impianto;
|
|
—
|
numero di piccoli con anomalie evidenti; valutazione macroscopica degli organi genitali esterni, numero di esemplari più piccoli del normale;
|
|
—
|
momento del decesso durante lo studio o indicazione della sopravvivenza degli animali alla conclusione della prova;
|
|
—
|
numero di impianti e dimensioni e peso della nidiata al momento della registrazione;
|
|
—
|
dati relativi al peso corporeo dei piccoli;
|
|
—
|
AGD di tutti i piccoli (e peso corporeo il giorno della misurazione dell’AGD);
|
|
—
|
retrazione del capezzolo nei piccoli di sesso maschile;
|
|
—
|
livelli di ormoni tiroidei, piccoli di 13 giorni e maschi adulti (e madri e piccoli di 4 giorni se sottoposti a misurazione);
|
|
—
|
peso corporeo al momento della soppressione e dati sul peso degli organi negli animali genitori;
|
|
—
|
risultati della necroscopia;
|
|
—
|
descrizione dettagliata dei risultati istopatologici;
|
|
—
|
dati sull’assorbimento (se disponibili);
|
|
—
|
elaborazione statistica dei risultati, se del caso.
|
|
|
|
Discussione dei risultati
|
|
Interpretazione dei risultati
|
77.
|
Lo studio fornirà valutazioni della tossicità per la riproduzione/lo sviluppo associata alla somministrazione di dosi ripetute. In particolare, poiché lo studio verte sia sulla tossicità generale sia sugli endpoint di tossicità per la riproduzione/lo sviluppo, i risultati consentiranno di distinguere gli effetti sulla riproduzione o sullo sviluppo che si osservano in assenza di tossicità generale da quelli espressi solo a livelli che risultano tossici anche per gli animali genitori (cfr. i paragrafi 7-11). Esso può fornire indicazioni sulla necessità di condurre ulteriori indagini e orientamenti in merito alla progettazione di studi successivi. Si consulti il documento di orientamento n. 43 dell’OCSE per indicazioni sull’interpretazione dei risultati in merito alla riproduzione e allo sviluppo (19). Il documento di orientamento dell’OCSE n. 106 sulla valutazione istologica delle prove endocrine e sulla riproduzione nei roditori (16) fornisce informazioni sulla preparazione e la valutazione degli organi (endocrini) e degli strisci vaginali che possono essere utili per il presente metodo di prova.
|
BIBLIOGRAFIA
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Disponibile su richiesta presso l’Organizzazione per la cooperazione e lo sviluppo economico (OCSE), Parigi.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Disponibile su richiesta presso l’Organizzazione per la cooperazione e lo sviluppo economico (OCSE), Parigi.
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, Available upon request at Organisation for Economic Cooperation and Development, Paris
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). «Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights», Reproductive Toxicology, 13: 383-390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Organisation for Economic Cooperation and Development, Paris.
|
|
(16)
|
OECD (2009).Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Organisation for Economic Cooperation and Development, Paris.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Organisation for Economic Cooperation and Development, Paris.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Organisation for Economic Cooperation and Development, Paris.
|
Appendice 1
DEFINIZIONI (CFR. ANCHE LA LINEA GUIDA N. 150 DELL'OCSE (20))
Androgenicità la capacità di una sostanza chimica di agire come un ormone androgenico naturale (ad es. il testosterone) in un mammifero.
Antiandrogenicità la capacità di una sostanza chimica di inibire l'attività di un ormone androgenico naturale (ad es. il testosterone) in un mammifero.
Antiestrogenicità la capacità di una sostanza chimica di inibire l'attività di un ormone estrogenico naturale (ad es. l'estradiolo 17ß) in un mammifero.
Attività antitiroidea la capacità di una sostanza chimica di inibire l'attività di un ormone tiroideo naturale (ad es. T3) in un mammifero.
Sostanza chimica una sostanza o una miscela.
Tossicità per lo sviluppo la manifestazione della tossicità per la riproduzione, che risulta in disturbi prenatali, perinatali, postnatali, strutturali o funzionali nella progenie.
Dose quantità di sostanza chimica somministrata. La dose è espressa col peso della sostanza chimica in esame per unità di peso corporeo dell'animale utilizzato nella prova per giorno (mg/kg peso corporeo/giorno) o come una concentrazione costante nella dieta.
Dosaggio termine generale che ricomprende la dose, la frequenza e la durata della somministrazione.
Tossicità evidente termine generale che designa i segnali evidenti di tossicità a seguito della somministrazione di una sostanza chimica. Questi segni devono essere sufficienti per consentire la valutazione dei pericoli ed essere tali che si possa prevedere che l'aumento della dose somministrata comporti la comparsa di segni di tossicità grave e probabilmente la mortalità.
Compromissione della fertilità i disturbi delle funzioni o della capacità riproduttive maschili o femminili.
Tossicità materna gli effetti nocivi sulle femmine gravide, che si verificano in modo specifico (effetto diretto) o non specifico (effetto indiretto) e correlati allo stato di gravidanza.
NOAEL l'abbreviazione di no-observed-adverse-effect level, ossia la dose più elevata alla quale non si osservano effetti avversi legati al trattamento.
Estrogenicità la capacità di una sostanza chimica di agire come un ormone estrogenico naturale (ad es. l'estradiolo 17ß) in un mammifero.
Tossicità della riproduzione gli effetti nocivi sulla progenie e/o la compromissione delle funzioni o della capacità riproduttive maschili e femminili.
Sostanza chimica in esame qualsiasi sostanza o miscela saggiata seguendo il presente metodo di prova.
Attività tiroidea la capacità di una sostanza chimica di agire come un ormone tiroideo naturale (ad es. T3) in un mammifero.
Validazione processo scientifico destinato a caratterizzare i requisiti e i limiti operativi di un metodo di prova e a dimostrarne l'affidabilità e la pertinenza per un fine specifico.
Appendice 2
DIAGRAMMA DEL PROTOCOLLO SPERIMENTALE INDICANTE LA DURATA MASSIMA DELLO STUDIO, BASATO SU UN PERIODO DI ACCOPPIAMENTO COMPLETO DI 14 GIORNI
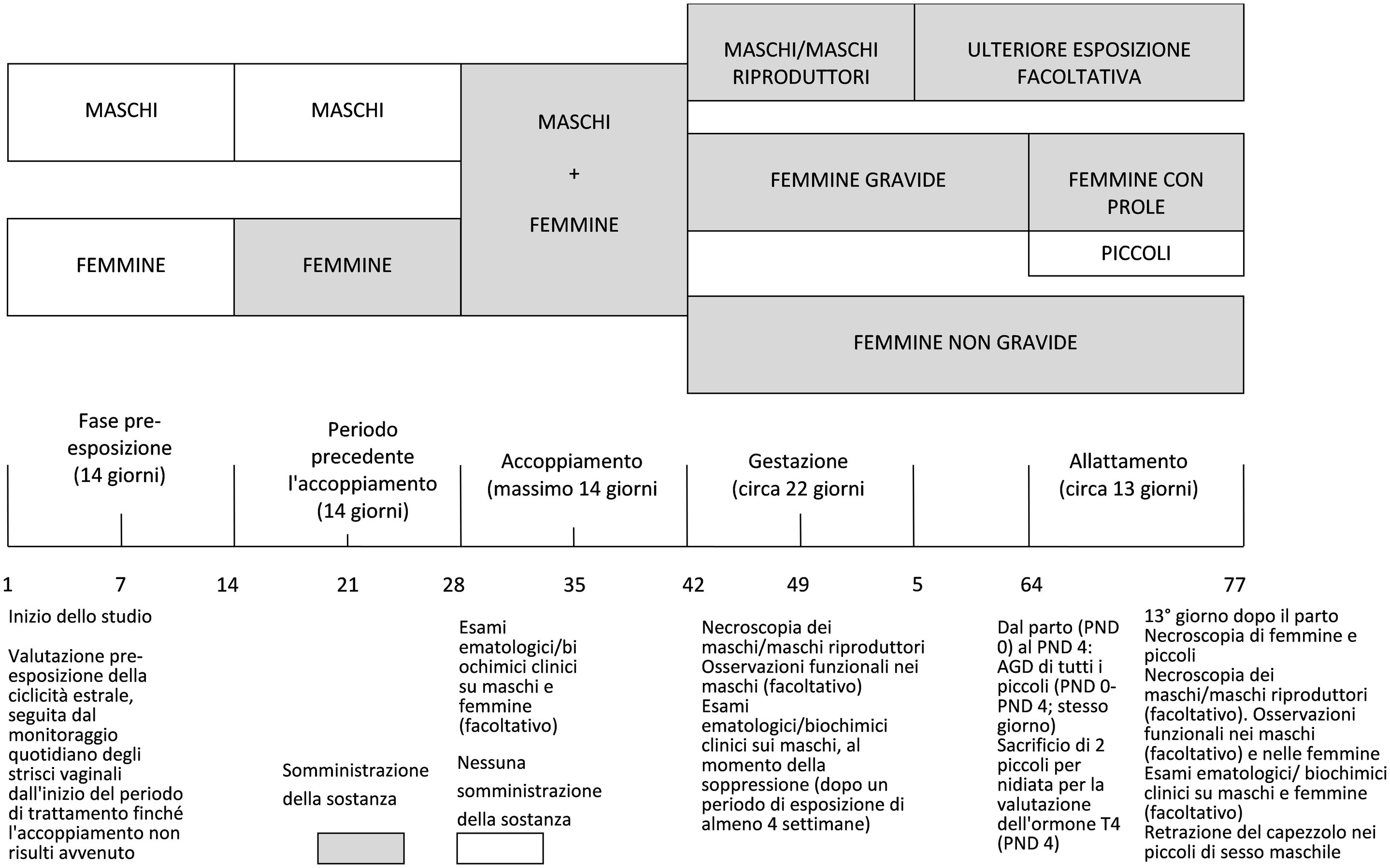
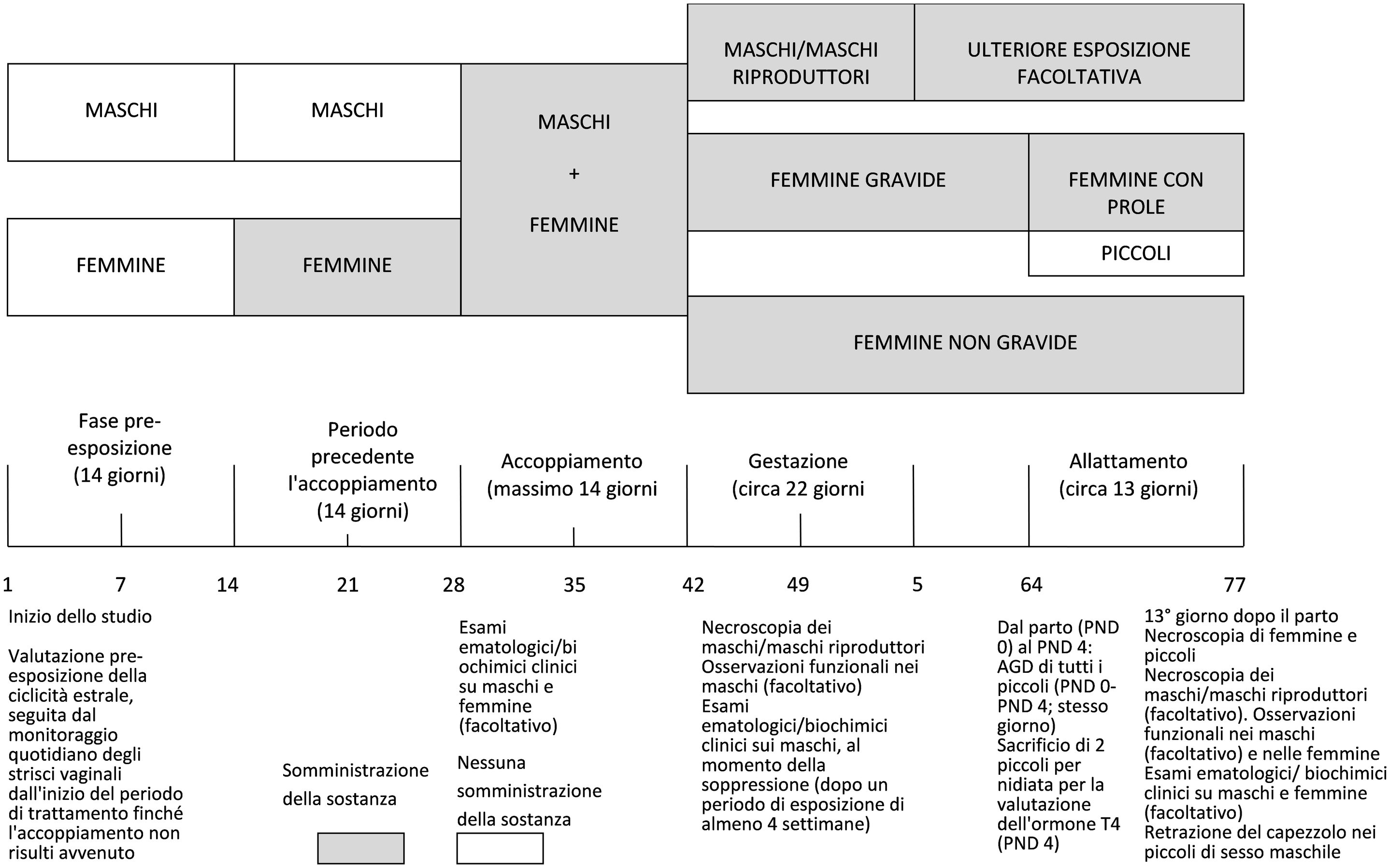
Appendice 3
RELAZIONE SINTETICA IN FORMATO TABULARE DEGLI EFFETTI SULLA RIPRODUZIONE/SULLO SVILUPPO
|
OSSERVAZIONI
|
VALORI
|
|
Dosaggio (unità)…….
|
0 (controllo)
|
…
|
…
|
…
|
…
|
|
Coppie formate (N)
|
|
|
|
|
|
|
Ciclo estrale (almeno durata media e frequenza dei cicli irregolari)
|
|
|
|
|
|
|
Femmine per le quali la copulazione risulta avvenuta (N)
|
|
|
|
|
|
|
Femmine gravide (N)
|
|
|
|
|
|
|
Giorni di concepimento 1 - 5 (N)
|
|
|
|
|
|
|
Giorni di concepimento 6 -…([1]
(23)
) (N)
|
|
|
|
|
|
|
Gravidanza≤21 giorni (N)
|
|
|
|
|
|
|
Gravidanza = 22 giorni (N)
|
|
|
|
|
|
|
Gravidanza ≥ 23 giorni (N)
|
|
|
|
|
|
|
Madri che hanno partorito piccoli vivi (N)
|
|
|
|
|
|
|
Madri con piccoli vivi il quarto giorno dopo il parto (N)
|
|
|
|
|
|
|
Impianti/madre (media)
|
|
|
|
|
|
|
Piccoli vivi/madre alla nascita (media)
|
|
|
|
|
|
|
Piccoli vivi/madre al quarto giorno (media)
|
|
|
|
|
|
|
Rapporto numerico tra i sessi (m/f) alla nascita (media)
|
|
|
|
|
|
|
Rapporto numerico tra i sessi (m/f) al quarto giorno (media)
|
|
|
|
|
|
|
Peso della nidiata alla nascita (media)
|
|
|
|
|
|
|
Peso della nidiata al quarto giorno (media)
|
|
|
|
|
|
|
Peso dei piccoli alla nascita (media)
|
|
|
|
|
|
|
Peso dei piccoli al momento della misurazione dell'AGD (media maschi, media femmine)
|
|
|
|
|
|
|
AGD dei piccoli misurata lo stesso giorno dalla nascita, tra la nascita e il giorno 4 (media maschi, media femmine, prendere nota del PND)
|
|
|
|
|
|
|
Peso dei piccoli al quarto giorno (media)
|
|
|
|
|
|
|
Peso dei piccoli al tredicesimo giorno (media)
|
|
|
|
|
|
|
Retrazione del capezzolo dei piccoli maschi al tredicesimo giorno (media)
|
|
|
|
|
|
|
PICCOLI CON ANOMALIE
|
|
Madri con 0
|
|
|
|
|
|
|
Madri con 1
|
|
|
|
|
|
|
Madri con ≥ 2
|
|
|
|
|
|
|
PERDITA DELLA PROGENIE
|
|
Perdite prenatali (impianti meno piccoli nati vivi)
|
|
Femmine con 0
|
|
|
|
|
|
|
Femmine con 1
|
|
|
|
|
|
|
Femmine con 2
|
|
|
|
|
|
|
Femmine con ≥ 3
|
|
|
|
|
|
|
Perdite postnatali (piccoli nati vivi meno piccoli vivi il tredicesimo giorno dopo il parto)
|
|
Femmine con 0
|
|
|
|
|
|
|
Femmine con 1
|
|
|
|
|
|
|
Femmine con 2
|
|
|
|
|
|
|
Femmine con ≥ 3
|
|
|
|
|
|
B.65 METODO DI PROVA IN VITRO CON MEMBRANA IMPERMEABILE PER LA CORROSIONE CUTANEA
INTRODUZIONE
|
1.
|
Il presente metodo di prova è equivalente alla linea guida OCSE. n. 435 (2015) Per corrosione cutanea si intende la produzione di lesioni irreversibili della pelle sotto forma di necrosi visibili nell’epidermide e nel derma, a seguito dell’applicazione di una sostanza chimica in esame, secondo la definizione del Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (sistema GHS dell’ONU) (1) e del regolamento (CE) n. 1272/2008 del Parlamento europeo e del Consiglio relativo alla classificazione, all’etichettatura e all’imballaggio delle sostanze e delle miscele (in appresso «regolamento CLP» (24)). Il presente metodo, equivalente alla linea guida aggiornata n. 435 dell’OCSE, costituisce un metodo di prova in vitro su membrana impermeabile che può essere utilizzato per individuare le sostanze chimiche corrosive. Il presente metodo di prova prevede l’utilizzo di una membrana artificiale destinata a reagire alle sostanze chimiche corrosive in modo analogo alla cute degli animali in situ.
|
|
2.
|
La corrosività cutanea è generalmente valutata applicando la sostanza chimica in esame sulla pelle di animali vivi e determinando l’ampiezza della lesione dei tessuti dopo un determinato lasso di tempo (2). Oltre al presente metodo di prova, per individuare le sostanze chimiche corrosive sono stati adottati alcuni metodi di prova in vitro come alternativa (3) (4) al metodo standard in vivo sui conigli (Capitolo B.4 del presente allegato, equivalente alla linea guida n. 404 dell’OCSE). La strategia di prova e valutazione su più livelli del sistema GHS dell’ONU per la stima e la classificazione della corrosività cutanea e le linee guida dell’OCSE sugli approcci integrati di prova e valutazione (Integrated Approaches to Testing and Assessment - IATA) per l’irritazione e la corrosione cutanea autorizzano l’utilizzo di procedure di prova in vitro validate e accettate di cui ai moduli 3 e 4 (1)(5). Le linee guida IATA descrivono diversi moduli che raggruppano le fonti di informazione e gli strumenti di analisi e i) forniscono orientamenti su come integrare e utilizzare i dati sperimentali e altri tipi di dati per valutare il potenziale di irritazione e di corrosione cutanea delle sostanze chimiche in esame e ii) propongono un approccio quando sono necessarie prove aggiuntive, anche quando si ottengono risultati negativi (5). Nell’ambito di questo approccio modulare, i risultati positivi dei metodi di prova in vitro possono essere utilizzati per classificare una sostanza chimica nella categoria «corrosivo» senza dover effettuare prove sugli animali, riducendo ed ottimizzando dunque l’utilizzo degli animali e evitando dolore e stress agli stessi.
|
|
3.
|
Sono stati realizzati studi di validazione del modello in vitro della membrana impermeabile reperibile in commercio sotto il nome Corrositex® (6)(7)(8), che hanno evidenziato una precisione del 79 % per quanto riguarda la previsione della corrosività cutanea (128/163), una sensibilità dell’85 % (76/89) e una specificità del 70 % (52/74) per una base di dati di 163 sostanze chimiche e miscele (7). Sulla base della sua validità riconosciuta, l’utilizzo di questo metodo di riferimento validato (validated reference method - VRM) è stato raccomandato nell’ambito di una strategia sperimentale su più livelli per valutare il rischio potenziale di corrosione cutanea generato dalle sostanze chimiche (5)(7). Prima di poter utilizzare a fini regolamentari un modello in vitro di membrana impermeabile per valutare la corrosione cutanea, occorre determinarne l’affidabilità, la pertinenza (accuratezza) e i limiti per l’uso proposto al fine di garantire che sia equivalente al VRM (9), conformemente agli standard di prestazione predefiniti (10). L’applicazione del sistema dell’OCSE di reciproca accettazione dei dati sarà garantito solo una volta che i metodi di prova proposti, nuovi o aggiornati e conformi agli standard di prestazione, sono stati esaminati e integrati nella corrispondente linea guida dell’OCSE. Attualmente la linea guida n. 435 dell’OCSE e il presente metodo di prova coprono un solo un metodo in vitro: il modello Corrositex® disponibile in commercio.
|
|
4.
|
Altri metodi per le prove sulla corrosività cutanea si basano sull’utilizzo di pelle umana ricostituita (linea guida n. 431 dell’OCSE) (3) e di pelle di ratto ricostituita (linea guida n. 430 dell’OCSE) (4). Il presente metodo di prova consente inoltre la sottocategorizzazione delle sostanze chimiche corrosive nelle tre sottocategorie di corrosività nell’ambito del sistema UN GHS e nei tre gruppi di imballaggio ONU per i trasporti per quanto concerne il rischio di corrosività. La presente linea guida è stata inizialmente adottata nel 2006 e aggiornata nel 2015 per tenere conto del documento di orientamento IATA e aggiornare l’elenco delle sostanze chimiche per la verifica della competenza tecnica.
|
DEFINIZIONI
|
5.
|
Le definizioni utilizzate sono riportate nell’appendice.
|
CONSIDERAZIONI INIZIALI E LIMITI
|
6.
|
La prova descritta nel presente metodo di prova consente di individuare le sostanze e le miscele chimiche corrosive e di classificarle in sottocategorie di sostanze corrosive conformemente al sistema GHS dell’ONU/ CLP (tabella 1). Questo metodo di prova può essere utilizzato anche per decidere in merito alla corrosività e alla non corrosività di classi specifiche di sostanze chimiche, ad esempio, gli acidi organici e minerali, i derivati acidi (25) e le basi in alcune prove di trasporto (7)(11)(12). Il presente metodo di prova descrive un protocollo generico simile ad un metodo di prova di riferimento validato (7). Il presente metodo di prova non fornisce informazioni adeguate sull’irritazione cutanea, ma è opportuno notare che il metodo di prova B.46 (equivalente alla linea guida n. 439 dell’OCSE) riguarda in modo specifico l’irritazione cutanea in vitro e gli effetti sulla salute (13). Per una valutazione completa degli effetti cutanei locali dopo una singola esposizione della pelle, si raccomanda di consultare il documento di orientamento dell’OCSE relativo agli approcci integrati in materia di prove e valutazioni (IATA) (5).
Tabella 1
La categoria e le sottocategorie di corrosività cutanea del sistema GHS dell’ONU (26)
|
Categoria di corrosività (categoria 1) (per le autorità che non utilizzano sottocategorie)
|
Possibili sottocategorie di corrosività (26) (per le autorità che utilizzano sottocategorie, ivi compreso il regolamento CLP)
|
Corrosivo per ≥ 1 animale su 3
|
|
Esposizione
|
Osservazioni
|
|
Corrosivo
|
Sottocategoria di corrosione 1A
|
≤ 3 minuti
|
≤ 1 ora
|
|
Sottocategoria di corrosione 1B
|
> 3 minuti / ≤ 1 ora
|
≤ 14 giorni
|
|
Sottocategoria di corrosione 1C
|
> 1 ora / ≤ 4 ore
|
≤ 14 giorni
|
|
|
7.
|
Un limite del metodo di riferimento validato (7) consiste nel fatto che, in base agli esiti del test di compatibilità iniziale, questo metodo non potrà essere applicato a numerose sostanze chimiche non corrosive e alcune sostanze chimiche corrosive (cfr. il paragrafo 13). Le sostanze acquose aventi un pH compreso tra 4,5 e 8,5 spesso non sono adattate alla prova; tuttavia, l’85 % delle sostanze chimiche testate in questa gamma di valori del pH è risultato non corrosivo per i test sugli animali (7). Il metodo in vitro della membrana impermeabile può essere utilizzato per testare solidi (solubili o insolubili in acqua), liquidi (acquosi o non acquosi) ed emulsioni. Tuttavia, le sostanze chimiche in esame che non causano un cambiamento rilevabile nella prova di compatibilità, ad esempio, un cambiamento di colore nel sistema di rilevamento chimico (Chemical Detection System - CDS) del metodo di prova di riferimento validato, non possono essere sottoposte a prova con il metodo della membrana impermeabile, rendendo necessario il ricorso ad altri metodi di prova.
|
PRINCIPIO DELLA PROVA
|
8.
|
Il sistema di prova si compone di due elementi: una biobarriera macromolecolare sintetica e un sistema di rilevamento chimico (CDS); in questo metodo di prova il CDS rileva i danni provocati dalle sostanze chimiche in esame corrosive sulla membrana impermeabile dopo l’applicazione della sostanza chimica in esame sulla superficie della membrana impermeabile macromolecolare sintetica (7), danni che probabilmente sono dovuti a uno o più dei meccanismi di corrosione simili a quelli che agiscono sulla pelle viva.
|
|
9.
|
Si può misurare la penetrazione della membrana impermeabile (o la sua permeazione) mediante una serie di procedure o CDS, in particolare un cambiamento di colore di un colorante indicatore di pH o di qualsiasi altra proprietà della soluzione di indicatore situata sotto la barriera.
|
|
10.
|
Occorre stabilire l’idoneità della membrana impermeabile, ossia la sua pertinenza e affidabilità, per l’uso previsto. Occorre quindi assicurarsi che le varie preparazioni garantiscano le proprietà di impermeabilità, ossia che siano in grado di mantenere una barriera contro le sostanze chimiche non corrosive, e siano atte a classificare le proprietà corrosive delle sostanze chimiche in varie sottocategorie di corrosività nell’ambito del sistema GHS dell’ONU (1). La classificazione assegnata si basa sul tempo impiegato dalla sostanza chimica per penetrare attraverso la membrana impermeabile fino alla soluzione di indicatore.
|
DIMOSTRAZIONE DELLA COMPETENZA DI LABORATORIO
|
11.
|
Prima di utilizzare sistematicamente il metodo della barriera impermeabile in vitro, i laboratori sono tenuti a dimostrare la loro competenza tecnica classificando correttamente le dodici sostanze di riferimento raccomandate nella tabella 2. Qualora una sostanza elencata non sia disponibile o nel caso in cui sia giustificato, può essere utilizzata un’altra sostanza per la quale sono disponibili dati di riferimento in vivo e in vitro (ad esempio scegliendo dalla lista delle sostanze chimiche di riferimento (10)), a condizione che siano applicati i medesimi criteri di selezione di cui alla tabella 1.
Tabella 2
Sostanze chimiche per la verifica della competenza tecnica
(27)
|
Sostanza (28)
|
Numero di registrazione CAS)
|
Classe chimica
|
Sottocategoria GHS ONU in vivo
(29)
|
Sottocategoria GHS ONU in vitro
(29)
|
|
Trifluoruro di boro diidrato
|
13319-75-0
|
Acidi inorganici
|
1A
|
1A
|
|
Acido nitrico
|
7697-37-2
|
Acidi inorganici
|
1A
|
1A
|
|
Pentacloruro di fosforo
|
10026-13-8
|
Precursori di acidi inorganici
|
1A
|
1A
|
|
Cloruro di valerile
|
638-29-9
|
Cloruri di acidi
|
1B
|
1B
|
|
Idrossido di sodio
|
1310-73-2
|
Basi inorganiche
|
1B
|
1B
|
|
1-(2-Amminoetil) piperazina
|
140-31-8
|
Ammine alifatiche
|
1B
|
1B
|
|
Cloruro di benzensolfonile
|
98-09-9
|
Cloruri di acidi
|
1C
|
1C
|
|
N,N-dimetil benzilammina
|
103-83-3
|
Aniline
|
1C
|
1C
|
|
Tetraetilene pentammina
|
112-57-2
|
Ammine alifatiche
|
1C
|
1C
|
|
Eugenolo
|
97-53-0
|
Fenoli
|
NC
|
NC
|
|
Acrilato di nonile
|
2664-55-3
|
Acrilati/metacrilati
|
NC
|
NC
|
|
Bicarbonato di sodio
|
144-55-8
|
Sali inorganici
|
NC
|
NC
|
|
PROCEDURA
|
12.
|
I paragrafi successivi contengono la descrizione dei componenti e delle procedure di un metodo di prova con membrana impermeabile artificiale per la valutazione della corrosività (7) (15) sulla base dell’attuale VRM, ossia il metodo commercialmente disponibile Corrositex®. La membrana impermeabile e le soluzioni di compatibilità e di indicatore nonché le soluzioni che consentono la categorizzazione possono essere prodotte, preparate o acquistate sul mercato, come nel caso del VRM Corrositex®. È disponibile un protocollo del metodo di prova su campione del metodo di prova di riferimento validato (7). Le prove devono essere effettuate a temperatura ambiente (17-25 °C) e i componenti devono soddisfare le condizioni indicate qui di seguito.
|
Prova di compatibilità della sostanza chimica in esame
|
13.
|
Prima di eseguire la prova con la membrana impermeabile, si effettua una prova di compatibilità per determinare se la sostanza in esame è rilevabile dal CDS. Se il CDS non rileva la sostanza chimica in esame, il metodo di prova con la membrana impermeabile non è adatto per valutare la potenziale corrosività di questa particolare sostanza chimica e deve essere utilizzato un metodo di prova diverso. Il CDS e le condizioni di esposizione utilizzati per la prova di compatibilità devono riprodurre l’esposizione subita nella successiva prova con la membrana impermeabile.
|
Prova di categoria di scala temporale della sostanza chimica in esame
|
14.
|
Se il metodo di prova lo consente, una sostanza chimica che è risultata idonea alla prova di compatibilità può essere oggetto di una prova di categoria di scala temporale, ossia uno screening che consente di distinguere tra acidi o basi deboli e forti. Ad esempio, nel metodo di prova di riferimento validato viene utilizzata una prova di classificazione di scala temporale per stabilire quale delle due scale temporali dovrebbe essere utilizzata a seconda che venga rilevata una riserva acida o una riserva alcalina significativa. Per determinare la corrosività e la sottocategoria GHS dell’ONU per la corrosività cutanea, si dovrebbero usare due diversi tempi di permeazione, in base alla riserva acida o alcalina della sostanza chimica in esame.
|
COMPONENTI DEL METODO DI PROVA DELLA MEMBRANA IMPERMEABILE
Membrana impermeabile
|
15.
|
La membrana impermeabile è costituita da due elementi: un gel acquoso macromolecolare proteico e una membrana di supporto permeabile. Il gel proteico dovrebbe essere impermeabile ai liquidi e ai solidi, ma può essere corroso e permeabilizzato. È opportuno conservare la membrana impermeabile pronta all’uso in condizioni prestabilite al fine di evitare il deterioramento del gel, ad esempio per via dell’essiccazione, di una crescita microbica, dello spostamento degli strati, dello screpolamento, che inficerebbero le sue prestazioni. Verrà stabilito un periodo di conservazione accettabile e le preparazioni di membrane impermeabili non saranno utilizzate dopo lo scadere di tale periodo.
|
|
16.
|
La membrana di supporto permeabile garantisce un supporto meccanico al gel proteico durante il processo di gelificazione e l’esposizione alla sostanza chimica in esame. La membrana di supporto dovrebbe impedire il cedimento e lo spostamento del gel ed essere facilmente permeabile a tutte le sostanze chimiche in esame.
|
|
17.
|
Il gel proteico costituito da proteine (ad esempio cheratina, collagene, o miscele di proteine, che formano una matrice di gel) funge da obiettivo per la sostanza chimica in esame. Il materiale proteico è posto sulla superficie della membrana di sostegno e, dopo la sua gelificazione, si colloca la membrana impermeabile sulla soluzione di indicatore. Il gel proteico dovrebbe essere di pari spessore e densità nel corso dell’intero processo, privo di bolle d’aria o difetti che potrebbero comprometterne l’integrità funzionale.
|
Sistema di rilevamento chimico (CDS)
|
18.
|
La soluzione d’indicatore, che è la stessa soluzione utilizzata per la prova di compatibilità, dovrebbe reagire alla presenza di una sostanza chimica in esame. Si può impiegare un colorante o una combinazione di coloranti indicatori di pH, ad esempio il rosso cresolo o il metilarancio, che cambia colore in presenza della sostanza chimica in esame. Il sistema di misurazione può essere visivo o elettronico.
|
|
19.
|
È opportuno valutare la pertinenza e l’affidabilità dei sistemi di rilevamento messi a punto per individuare il passaggio della sostanza chimica in esame attraverso la membrana impermeabile al fine di definire la gamma di sostanze chimiche che possono essere rilevate e i limiti quantitativi di rilevamento.
|
ESECUZIONE DELLA PROVA
Assemblaggio dei componenti del metodo di prova
|
20.
|
La membrana impermeabile è posta in una fiala (o tubo) contenente la soluzione d’indicatore in modo che la membrana di appoggio sia interamente a contatto con la soluzione d’indicatore e non si formino bolle d’aria. Occorre accertarsi che l’integrità della membrana sia preservata.
|
Applicazione della sostanza chimica in esame
|
21.
|
Una quantità adeguata della sostanza chimica in esame, ad esempio 500 μl di un liquido o 500 mg di un solido finemente polverizzato (7), viene accuratamente depositata sulla superficie superiore della membrana impermeabile e ripartita in modo uniforme. Per ciascuna sostanza chimica in esame e i controlli corrispondenti viene preparato un numero di repliche adeguato, ad esempio quattro (7) (cfr. i paragrafi da 23 a 25). Si prende nota dell’ora in cui viene applicata la sostanza chimica in esame alla membrana impermeabile. Per garantire la registrazione accurata dei periodi brevi di corrosione, le applicazioni della sostanza chimica in esame nelle fiale sono scaglionate.
|
Misurazione delle penetrazioni nella membrana impermeabile
|
22.
|
Ogni fiala è sottoposta a un monitoraggio adeguato, viene registrato il momento del primo cambiamento della soluzione d’indicatore, vale a dire la penetrazione della barriera, e viene determinato il tempo trascorso tra l’applicazione e la penetrazione della membrana impermeabile.
|
Controlli
|
23.
|
Nelle prove che prevedono l’utilizzo di un mezzo disperdente o di un solvente con la sostanza chimica di prova, questi devono essere compatibili con il sistema della membrana impermeabile, ossia non devono intaccare l’integrità di tale sistema né modificare la corrosività della sostanza chimica in esame. Se del caso, il controllo con solvente (o con il mezzo disperdente) dovrebbe essere testato in concomitanza per dimostrare la compatibilità del solvente con il sistema della barriera impermeabile.
|
|
24.
|
È opportuno inoltre testare contemporaneamente alla sostanza chimica di prova una sostanza chimica di controllo (corrosiva) la cui attività di corrosione è di grado medio, ad esempio 110 ± 15 mg di idrossido di sodio (sottocategoria di corrosività 1B del sistema GHS dell’ONU) (7) al fine di valutare se le prestazioni del sistema sono accettabili. Potrebbe essere utile includere un secondo controllo positivo della stessa classe chimica della sostanza chimica in esame per valutare il potenziale di corrosività relativo di una sostanza chimica in esame corrosiva. Occorre selezionare controlli positivi di media corrosività (sottocategoria 1B del sistema GHS dell’ONU) al fine di individuare i cambiamenti del tempo di penetrazione eventualmente troppo lunghi o troppo brevi rispetto al valore di riferimento stabilito, e che evidenziano pertanto un malfunzionamento del sistema di prova. A tal fine, le sostanze chimiche estremamente corrosive (sottocategoria 1A del sistema GHS dell’ONU) o non corrosive sono di utilità limitata. Una sostanza chimica corrosiva della sottocategoria 1B del sistema GHS dell’ONU consentirebbe di rilevare una durata di permeazione troppo breve o troppo lunga. Una sostanza chimica poco corrosiva (sottocategoria 1C del sistema GHS dell’ONU) potrebbe essere utilizzata come controllo positivo per misurare la capacità del metodo di prova di distinguere sistematicamente le sostanze chimiche poco corrosive da quelle non corrosive. A prescindere dall’approccio utilizzato, occorre stabilire un intervallo accettabile di risposta dei controlli positivi sulla base dell’intervallo storico delle durate di permeazione del o dei controlli positivi utilizzati, ad esempio la media di ± 2-3 deviazioni standard. In ogni studio, è opportuno stabilire il tempo di permeazione esatto del controllo positivo in modo da poter individuare le deviazioni che si situano al di fuori dell’intervallo accettabile.
|
|
25.
|
Contemporaneamente alla sostanza chimica in esame occorre testare anche un controllo negativo (non corrosivo), ad esempio acido citrico al 10 % e acido propionico al 6 % (7), per ottenere una misura di controllo di qualità supplementare a dimostrazione dell’integrità funzionale della membrana impermeabile.
|
Criteri di accettabilità dello studio
|
26.
|
Secondo i parametri di tempo stabiliti per ciascuna sottocategoria di corrosività (del sistema GHS delle Nazioni Unite), il tempo (in minuti) trascorso tra l’applicazione di una sostanza chimica in esame sulla membrana impermeabile e la penetrazione della membrana è utilizzata per prevedere la corrosività della sostanza chimica in esame. Affinché uno studio sia considerato accettabile, il controllo positivo concomitante dovrebbe dare il tempo di risposta di penetrazione previsto (ad esempio 8-16 minuti di tempo permeazione per l’idrossido di sodio se utilizzato come controllo positivo), il controllo negativo concomitante non dovrebbe essere corrosivo e il corrispondente controllo con solvente (se presente) non dovrebbe essere corrosivo né alterare il potenziale di corrosività della sostanza chimica in esame. Prima di utilizzare sistematicamente un metodo conforme al presente metodo di prova, i laboratori sono tenuti a dimostrare la loro competenza tecnica avvalendosi delle 12 sostanze raccomandate nella tabella 2. Per i nuovi metodi «strutturalmente analoghi» sviluppati nell’ambito di questo metodo di prova che sono strutturalmente e funzionalmente simili al metodo di riferimento validato (14), dovrebbero essere utilizzati gli standard di prestazione predefiniti per dimostrare l’affidabilità e l’accuratezza del nuovo metodo prima del suo utilizzo per le prove a fini regolamentari (10).
|
Interpretazione dei risultati e classificazione di corrosività della sostanza chimica in esame
|
27.
|
Il tempo (in minuti) trascorso tra l’applicazione della sostanza chimica in esame alla membrana impermeabile e la penetrazione della membrana è utilizzato per classificare la sostanza chimica in esame secondo le sottocategorie GHS dell’ONUI (1) e, se del caso, secondo i gruppi di imballaggio delle Nazioni Unite (16). I valori di tempo limite di ciascuna delle tre sottocategorie di corrosività sono definiti per ciascun metodo di prova proposto. Le decisioni finali sui tempi limite dovrebbero tenere conto dell’esigenza di minimizzare la sottoclassificazione del pericolo di corrosione (ad esempio, falsi negativi). Nella presente linea guida è opportuno utilizzare i tempi limite del metodo Corrositex® descritti nella tabella 3, in quanto si tratta dell’unico metodo di prova che ad oggi rispetta gli orientamenti della linea guida (7).
Tabella 3
Modello predittivo Corrositex®
|
Tempo medio di permeazione (min.)
|
Predizione UN GHS (32)
|
|
Sostanza chimica in esame di categoria 1 (30) (determinata dalla prova di categorizzazione del metodo)
|
Sostanza chimica in esame di categoria 2 (31) (determinata dalla prova di categorizzazione del metodo)
|
|
0-3 min.
|
0-3 min.
|
Corrosivo Sottocategoria 1A facoltativa
|
|
> 3 a 60 min.
|
> 3 a 30 min.
|
Corrosivo Sottocategoria 1B facoltativa
|
|
> 60 a 240 min.
|
> 30 a 60 min.
|
Corrosivo Sottocategoria 1C facoltativa
|
|
> 240 min.
|
> 60 min.
|
Sostanza chimica non corrosiva
|
|
DATI E RELAZIONE
Dati
|
28.
|
Il tempo (in minuti) trascorso tra l’applicazione della sostanza in esame e la penetrazione della barriera per la sostanza in esame e il o i controlli positivi devono essere riportati sotto forma di tabelle contenenti i risultati di ciascuna replica e come media ± di deviazione standard per ciascuna prova.
|
Relazione sull’esecuzione della prova
|
29.
|
La relazione sull’esecuzione della prova deve comprendere le seguenti informazioni.
|
|
Sostanze chimiche in esame e sostanze di controllo:
|
—
|
sostanza monocostituente: identificazione chimica, come denominazioni IUPAC o CAS, numero CAS, codice SMILES o InChI, formula strutturale, purezza, identità chimica delle impurezze se del caso e se le condizioni pratiche lo consentono, ecc.;
|
|
—
|
sostanza multicostituente, UVCB o miscela: caratterizzata, nella misura del possibile, attraverso l’identità chimica (cfr. sopra), le proporzioni quantitative e le proprietà fisico-chimiche pertinenti dei costituenti;
|
|
—
|
aspetto fisico, idrosolubilità e, se del caso, ulteriori proprietà fisico-chimiche pertinenti;
|
|
—
|
origine, numero del lotto se disponibile;
|
|
—
|
trattamento della sostanze chimica in esame/sostanza di controllo prima della prova, se applicabile (ad esempio, riscaldamento, frantumazione);
|
|
—
|
stabilità della sostanza chimica in esame, data limite di utilizzo, data della nuova analisi, se nota;
|
|
—
|
condizioni di conservazione.
|
|
|
|
Mezzo disperdente:
|
—
|
identificazione, concentrazione (ove pertinente), volume utilizzato;
|
|
—
|
motivazione della scelta del mezzo.
|
|
|
|
Modello della barriera impermeabile in vitro e protocollo utilizzati, ivi compresa la precisione e affidabilità dimostrate
|
|
|
Condizioni sperimentali:
|
—
|
descrizione dell’apparecchiatura e delle procedure di preparazione utilizzate;
|
|
—
|
fonte e composizione della membrana impermeabile in vitro utilizzata;
|
|
—
|
composizione e proprietà della soluzione d’indicatore;
|
|
—
|
quantità della sostanza chimica in esame e delle sostanze di controllo;
|
|
—
|
descrizione e giustificazione del test di categorizzazione della scala temporale;
|
|
—
|
metodo di applicazione;
|
|
—
|
descrizione dei criteri di valutazione e classificazione impiegati;
|
|
—
|
dimostrazione della competenza nell’esecuzione del metodo di prova prima del suo uso sistematico testando le sostanze chimiche per la verifica della competenza tecnica.
|
|
|
|
Risultati:
|
—
|
presentazione sotto forma di tabella dei dati grezzi individuali ottenuti da ciascun campione di prova e di controllo per ogni replica;
|
|
—
|
descrizioni di altri effetti osservati;
|
|
—
|
classificazione ottenuta con riferimento al modello predittivo/ai criteri decisionali utilizzati.
|
|
Discussione dei risultati
Conclusioni
|
BIBLIOGRAFIA
|
(1)
|
United Nations (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Genev, 2013. Disponibile qui: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Capitolo B.4 del presente allegato: Irritazione/corrosione cutanea acuta.
|
|
(3)
|
Capitolo B.40 bis del presente allegato, Corrosione cutanea in vitro: test su un modello di epidermide umana ricostituita (RHE)
|
|
(4)
|
Capitolo B.40 del presente allegato, Corrosione cutanea in vitro: Resistenza elettrica transcutanea (TER).
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Organizzazione per la cooperazione e lo sviluppo economici (OCSE)
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 2. Results and evaluation by the Management Team. Toxicology In Vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In Vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on Testing and Assessment (N. 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Organizzazione per la cooperazione e lo sviluppo economici (OCSE) Disponibile qui: http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (20 settembre 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
Capitolo B.46 del presente allegato, Irritazione cutanea in vitro: metodo di prova su un modello di epidermide umana ricostituita. ICCVAM (2004). ICCVAM Recommended Performance Standards for In Vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Disponibile qui: http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Disponibile qui: http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
United Nations (UN) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Disponibile qui: http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Appendice
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza" per indicare la percentuale di risultati corretti di un metodo di prova (9).
Sostanza chimica
: una sostanza o una miscela.
Sistema di rilevamento chimico (CDS)
: sistema di misurazione visivo o elettronico che si avvale di una soluzione di indicatore che reagisce alla presenza di una sostanza chimica in esame (ad esempio con la modifica di un colorante indicatore di pH o di una combinazione di questi coloranti) che registra un cambiamento di colore in risposta alla presenza della sostanza in esame o evidenzia altri tipi di reazioni chimiche o elettrochimiche.
Concordanza
: misura dell'efficacia del metodo di prova per i metodi che forniscono un risultato ordinabile in categorie; si tratta di un aspetto della pertinenza. Il termine è usato a volte come sinonimo di "accuratezza" ed è definito come la proporzione di tutte le sostanze chimiche in esame che sono correttamente classificate come positive o negative. La concordanza dipende strettamente dalla prevalenza di risultati positivi in tutti i tipi di sostanze chimiche in esame (9).
Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (GHS dell'ONU)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi standardizzati e livelli di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di precauzioni e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (1).
IATA
: Approcci integrati in materia di prove e valutazioni (Integrated Approaches to Testing and Assessment).
Miscela
: una miscela o una soluzione composta di due o più sostanze.
Sostanza monocostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno l'80 % (p/p).
Sostanza multicostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui più costituenti principali sono presenti in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente è che una miscela è ottenuta attraverso la miscelazione di due o più sostanze senza che avvenga una reazione chimica. Una sostanza multicostituente è il risultato di una reazione chimica.
NC
: Non corrosivo.
Standard di prestazione
: standard, basati su un metodo di riferimento validato, che consentono di valutare la comparabilità di un metodo proposto che è simile sotto il profilo meccanicistico e funzionale. Detti standard comprendono: i) gli elementi essenziali del metodo di prova; ii) un elenco minimo di sostanze chimiche di riferimento scelte tra le sostanze utilizzate per dimostrare le prestazioni accettabili del metodo di prova validato; e iii) in funzione dei risultati ottenuti con il metodo di riferimento validato, i livelli comparabili di affidabilità e accuratezza che il metodo proposto dovrebbe ottenere quando viene valutato utilizzando l'elenco minimo di sostanze di riferimento (9).
Pertinenza
: descrizione del rapporto tra la prova e l'effetto studiato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (9).
Affidabilità
: misura in cui l'esecuzione di un metodo di prova può essere riprodotta nel tempo all'interno dello stesso laboratorio o da laboratori diversi seguendo il medesimo protocollo. È valutata calcolando la riproducibilità intra-laboratorio e inter-laboratori (9).
Sensibilità
: proporzione di tutte le sostanze chimiche positive/attive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo di prova (9).
Corrosione cutanea in vivo
: il manifestarsi di lesioni irreversibili della pelle, vale a dire, necrosi visibile dell'epidermide e del derma, a seguito dell'applicazione della sostanza chimica di prova per non più di quattro ore. Gli effetti tipici della corrosione sono ulcere, sanguinamento, croste sanguinolente e, al termine di un periodo di osservazione di 14 giorni, depigmentazione cutanea dovuta all'effetto sbiancante, chiazze di alopecia e cicatrici. Per valutare le lesioni dubbie potrebbe essere necessario ricorrere a un esame istopatologico.
Specificità
: proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di un metodo di prova (9).
Sostanza
: un elemento chimico e i suoi composti, allo stato naturale od ottenuti per mezzo di un procedimento di fabbricazione, compresi gli additivi necessari a mantenerne la stabilità e le impurezze derivanti dal procedimento utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione.
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
UVCB
: sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici.
B.66 SAGGI DI TRANSATTIVAZIONE IN VITRO TRAMITE TRASFEZIONE STABILE PER L’INDIVIDUAZIONE DI SOSTANZE AGONISTE E ANTAGONISTE DEI RECETTORI ESTROGENICI
INTRODUZIONE GENERALE
Linea guida dell’OCSE per i metodi di prova basata su standard di prestazione
|
1.
|
Il presente metodo di prova è equivalente alla linea guida n. 455 (2016) dell’OCSE. La linea guida n. 455 è basata su standard di performance (PBTG) che descrive la metodologia della transattivazione in vitro tramite trasfezione stabile per l’individuazione delle sostanze agoniste e antagoniste dei recettori estrogenici. Comprende una serie di metodi di prova strutturalmente e funzionalmente simili per l’individuazione degli agonisti e degli antagonisti dei recettori estrogenici (ERα e/o ERβ) e dovrebbe agevolare lo sviluppo di nuovi metodi di prova simili o modificati, conformemente ai principi di validazione stabiliti nel documento di orientamento dell’OCSE intitolato «OECD Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment» (1). I seguenti metodi di prova di riferimento pienamente validati (appendice 2 e appendice 3) costituiscono la base per la presente linea guida:
|
—
|
il metodo di TA tramite trasfezione stabile (STTA) (2) che utilizza la linea cellulare (h) ERα-HeLa-9903; e
|
|
—
|
il metodo di VM7Luc ER TA (3) che utilizza la linea cellulare VM7Lu4E2 (33), che esprime prevalentemente hERα e, parzialmente, hERββ(4)(5).
|
Sono disponibili standard di prestazione (6) (7) per facilitare l’elaborazione e la validazione di metodi di prova simili finalizzati al medesimo endpoint di sicurezza e per consentire di aggiornare tempestivamente la PBTG 455 integrandola con nuovi protocolli simili. Ciò può avvenire, tuttavia, solo dopo che l’OCSE abbia esaminato e approvato tali nuovi protocolli, dichiarando che rispettano gli standard di prestazione. Qualsiasi metodo di prova incluso nella linea guida TG 455 può essere scelto e applicato ai fini della conformità con i requisiti nazionali in materia di prove di transattivazione dei recettori estrogenici (ER TA) nel quadro del sistema dell’OCSE di reciproca accettazione dei dati.
|
Contesto e principi relativi ai metodi di prova inclusi nella presente linea guida
|
2.
|
Nel 1998 l’OCSE ha avviato lavori a carattere altamente prioritario destinati a rivedere le linee guida esistenti e a elaborarne di nuove per lo screening e la sperimentazione relativi alle sostanze chimiche ritenute potenziali interferenti endocrini. Nel 2012 è stato rivisto il quadro concettuale dell’OCSE per la sperimentazione e la valutazione delle sostanze chimiche potenzialmente capaci di alterare il sistema endocrino. Le versioni originali e riviste del presente quadro concettuale figurano come allegati nel documento di orientamento dell’OCSE sulle linee guida standardizzate per le prove di valutazione delle sostanze chimiche che alterano il sistema endocrino (8). Il quadro concettuale comprende cinque livelli, ciascuno dei quali corrisponde a un diverso livello di complessità biologica. Le prove di ER TA descritte nella presente line guida corrispondono al livello 2, che comprende «saggi in vitro che forniscono dati su determinati meccanismi/vie di attivazione endocrina». La presente linea guida concerne i metodi di prova in vitro di transattivazione (TA) intesi a individuare gli agonisti e gli antagonisti dei recettori estrogenici.
|
|
3.
|
L’interazione degli estrogeni con gli ER può influenzare la trascrizione dei geni regolati dagli estrogeni, il che può portare all’induzione o all’inibizione di processi cellulari, compresi i meccanismi necessari alla proliferazione cellulare, allo sviluppo fetale normale e alla funzione riproduttiva (9) (10) (11). La perturbazione dei sistemi estrogenici normali può potenzialmente indurre effetti nocivi sul normale sviluppo (ontogenesi), sulla salute riproduttiva e sull’integrità del sistema riproduttivo.
|
|
4.
|
I metodi di prova TA in vitro sono basati sull’interazione diretta o indiretta tra le sostanze e uno specifico recettore che regola la trascrizione del prodotto di un gene reporter. Essi sono stati ampiamente utilizzati per valutare l’espressione genica regolata da recettori nucleari specifici, quali gli ER (12) (13) (14) (15) (16). Sono stati proposti per individuare l transattivazione estrogenica regolata da ER (17) (18) (19). Esistono almeno due principali sottotipi di ER nucleari - designati α e β - che sono codificati da geni distinti. Le proteine corrispondenti presentano funzioni biologiche differenti, così come diverse distribuzioni tessutali e affinità di legame con i ligandi (20) (21) (22) (23) (24) (25) (26). Gli ERα nucleari sono i mediatori della risposta estrogenica classica (27) (28) (29) (30); pertanto la maggior parte dei modelli attualmente in fase di sviluppo per misurare l’attivazione o l’inibizione degli ER sono specifici per l’ERα. I metodi di prova servono ad individuare le sostanze chimiche che attivano (o inibiscono) gli ER mediante legame del ligando con il recettore; il complesso recettore-ligando si lega successivamente a specifici elementi di risposta del DNA e transattiva un gene reporter, inducendo l’aumento dell’espressione cellulare di un marcatore proteico. Questi metodi di prova si basano su diverse risposte dei geni reporter. Nei sistemi basati sulla luciferasi, l’enzima luciferasi trasforma il suo substrato - la luciferina - in un prodotto bioluminescenti che può essere misurato quantitativamente con un luminometro. Tra gli altri esempi di marcatori/reporter comuni figurano la proteina fluorescente e il gene LacZ che codifica la β-galattosidasi, un enzima che può trasformare il substrato incolore X-gal (5-bromo-4-cloro-indolil-galattopiranoside) in un prodotto blu che può essere quantificato mediante spettrofotometro. Tali marcatori possono essere valutati in modo rapido e poco costoso grazie ai kit di analisi disponibili in commercio.
|
|
5.
|
Gli studi di validazione dei metodi di STTA e VM7Luc TA hanno dimostrato la loro pertinenza e affidabilità per i fini previsti (3)(4)(5)(30). Gli standard di prestazione per i metodi di ER TA basati sulla luminescenza che utilizzano linee cellulari di ghiandole mammarie sono riportati nella relazione dell’ICCVAM sulla valutazione del metodo di prova LUMI-CELL® ER (VM7Luc ER TA): An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals (3). Questi standard di prestazione sono stati modificati per renderli applicabili sia al metodo di STTA sia al metodo VM7Luc TA (2).
|
|
6.
|
Le definizioni e le abbreviazioni utilizzate nel presente metodo di prova figurano nell’appendice 1.
|
Portata e limiti dei metodi di prova di transattivazione
|
7.
|
I presenti metodi di prova sono proposti a fini di screening e di definizione delle priorità, ma possono fornire anche informazioni sui meccanismi d’azione che possono rivelarsi utili nel quadro di un approccio basato sulla forza probante dei dati. Si basano sulla transattivazione (TA) indotta dall’instaurazione di un legame chimico con gli ER in un sistema in vitro. Pertanto non è opportuno che i risultati ottenuti siano direttamente estrapolati e trasposti ai complessi meccanismi di segnalazione e regolazione che caratterizzano un sistema endocrino intatto in vivo.
|
|
8.
|
La TA mediata dagli ER è considerato uno dei meccanismi chiave di perturbazione endocrina, benché esistano altri meccanismi che possono indurre il medesimo effetto, tra i quali: i) le interazioni con altri recettori e sistemi enzimatici del sistema endocrino; ii) la sintesi ormonale; iii) l’attivazione e/o l’inattivazione metabolica degli ormoni; iv) la distribuzione ormonale nei tessuti bersaglio; e v) l’eliminazione degli ormoni dall’organismo. Nessuno dei metodi di prova oggetto della presente PBTG si basa sulle descritte modalità di azione.
|
|
9.
|
Il presente metodo di prova concerne la capacità delle sostanze chimiche di attivare (attività agonista) e anche di inibire (attività antagonista) la trascrizione regolata dagli ER. Alcune sostanze chimiche possono, a seconda del tipo cellulare utilizzato, presentare attività sia agonista sia antagonista, e sono note come «modulatori selettivi dei recettori estrogenici» (SERM). Si potrebbero considerare di sottoporre le sostanze chimiche che danno una risposta negativa con questi metodi di prova a prove di legame agli ER prima di concludere che tali sostanze in esame non si legano al recettore. Inoltre, il metodo di prova fornisce solo un indicatore probabile dell’attività della molecola madre, tenendo conto delle capacità metaboliche limitate dei sistemi cellulari in vitro. Considerando che la validazione ha avuto per oggetto solo sostanze singole, non esiste alcuna informazione circa l’applicabilità della prova alle miscele. Ad ogni modo, il metodo di prova è in ogni caso teoricamente applicabile alle prove sulle sostanze multicostituente, sulle UVCB e sulle miscele. Prima di applicare il presente metodo di prova a una sostanza multicostituente, un UVCB o una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie laddove esista una disposizione normativa che obblighi a sottoporre a prova la miscela.
|
|
10.
|
A fini di informazione, la tabella 1 fornisce i risultati delle prove agoniste delle 34 sostanze testate con entrambi i metodi di prova di riferimento pienamente validati descritti nel presente metodo di prova. Di queste sostanze, 26 sono definitivamente classificate come agoniste degli ER e 8 come negativi in base a relazioni pubblicate, comprese le prove in vitro di legame agli ER e di TA e/o le prove uterotrofiche (2)(3)(18)(31)(32)(33)(34). La tabella 2 fornisce i risultati delle prove antagoniste delle 15 sostanze testate con entrambi i metodi di prova di riferimento pienamente validati descritti nel presente metodo di prova. Di queste sostanze, 4 sono definitivamente/presumibilmente classificate come antagoniste degli ER e 10 come negative in base alle relazioni pubblicate, comprese le prove in vitro di legame agli ER e di TA (2)(3)(18)(31). Con riferimento ai dati sintetizzati nelle tabelle 1 e 2, i due metodi di prova di riferimento sono giunti ad una conclusione identica circa la classificazione di tutte le sostanze, tranne una (Mifepristone) per la prova antagonista, e ciascuna sostanza è stata correttamente classificata come agonista/antagonista degli ER o come negativa. Ulteriori informazioni su questo gruppo di sostanze chimiche così come sulle sostanze aggiuntive testate nei metodi di STTA e VM7Luc ER TA nel quadro degli studi di validazione sono fornite negli standard di prestazione per la prova di ER TA (6)(7), appendice 2 (tabelle 1, 2 e 3).
|
Tabella 1
Quadro d’insieme dei risultati dei metodi di prova di STTA e VM7Luc ER TA ottenuti per le sostanze testate in base ai due metodi agonisti e classificate come agoniste degli ER (Pos) o negative (Neg)
|
|
Sostanza
|
N. CAS
|
Metodo di STTA (35)
|
Metodo VM7Luc ER TA (36)
|
Fonte dei dati per la classificazione (38)
|
|
Attività di ER TA
|
PC10 (M)
|
PC50
(2) (M)
|
Attività di ER TA
|
EC50
(2), (37) (M)
|
Altri metodi di prova di ER TA (34)
|
Prove di legame agli ER
|
Prove uterotrofiche
|
|
1
|
17ß-estradiolo (1)
|
50-28-2
|
Pos.
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
Pos.
|
5,63 × 10-12
|
Pos. (227/227)
|
Pos.
|
Pos.
|
|
2
|
17α-estradiolo (1)
|
57-91-0
|
Pos.
|
7,24 × 10-11
|
6,44 × 10-10
|
Pos.
|
1,40 × 10-9
|
Pos. (11/11)
|
Pos.
|
Pos.
|
|
3
|
17α-etinilestradiolo (1)
|
57-63-6
|
Pos.
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
Pos.
|
7,31 × 10-12
|
Pos. (22/22)
|
Pos.
|
Pos.
|
|
4
|
17β-trenbolone
|
10161-33-8
|
Pos.
|
1,78 × 10-8
|
2,73 × 10-7
|
Pos.
|
4,20 × 10-8
|
Pos. (2/2)
|
NT
|
NT
|
|
5
|
19-nortestosterone (1)
|
434-22-0
|
Pos.
|
9,64 × 10-9
|
2,71 × 10-7
|
Pos.
|
1,80 × 10-6
|
Pos. (4/4)
|
Pos.
|
Pos.
|
|
6
|
4-cumilfenolo (1)
|
599-64-4
|
Pos.
|
1,49 × 10-7
|
1,60 × 10-6
|
Pos.
|
3,20 × 10-7
|
Pos. (5/5)
|
Pos.
|
NT
|
|
7
|
4-terz-ottilfenolo (1)
|
140-66-9
|
Pos.
|
1,85 × 10-9
|
7,37 × 10-8
|
Pos.
|
3,19 × 10-8
|
Pos. (21/24)
|
Pos.
|
Pos.
|
|
8
|
Apigenin (1)
|
520-36-5
|
Pos.
|
1,31 × 10-7
|
5,71 × 10-7
|
Pos.
|
1,60 × 10-6
|
Pos. (26/26)
|
Pos.
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (30/30)
|
Neg.
|
NT
|
|
10
|
Bisfenolo A (1)
|
80-05-7
|
Pos.
|
2,02 × 10-8
|
2,94 × 10-7
|
Pos.
|
5,33 × 10-7
|
Pos. (65/65)
|
Pos.
|
Pos.
|
|
11
|
Bisfenolo B (1)
|
77-40-7
|
Pos.
|
2,36 × 10-8
|
2,11 × 10-7
|
Pos.
|
1,95 × 10-7
|
Pos. (6/6)
|
Pos.
|
Pos.
|
|
12
|
Ftalato di butilbenzile (1)
|
85-68-7
|
Pos.
|
1,14 × 10-6
|
4,11 × 10-6
|
Pos.
|
1,98 × 10-6
|
Pos. (12/14)
|
Pos.
|
Neg.
|
|
13
|
Corticosterone (1)
|
50-22-6
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (6/6)
|
Neg.
|
NT
|
|
14
|
Cumestrolo (1)
|
479-13-0
|
Pos.
|
1,23 × 10-9
|
2,00 × 10-8
|
Pos.
|
1,32 × 10-7
|
Pos. (30/30)
|
Pos.
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
Pos.
|
1,76 × 10-8
|
1,51 × 10-7
|
Pos.
|
7,95 × 10-7
|
Pos. (39/39)
|
Pos.
|
Pos.
|
|
16
|
Dietilstilbestrolo (1)
|
56-53-1
|
Pos.
|
< 1,00 × 10-11
|
2,04 × 10-11
|
Pos.
|
3,34 × 10-11
|
Pos. (42/42)
|
Pos.
|
NT
|
|
17
|
Di-n-butilftalato
|
84-74-2
|
Pos.
|
4,09 × 10-6
|
|
Pos.
|
4,09 × 10-6
|
Pos. (6/11)
|
Pos.
|
Neg.
|
|
18
|
Etilparabene
|
120-47-8
|
Pos.
|
5,00 × 10-6
|
(no PC50)
|
Pos.
|
2,48 × 10-5
|
Pos.
|
|
NT
|
|
19
|
Estrone (1)
|
53-16-7
|
Pos.
|
3,02 × 10-11
|
5,88 × 10-10
|
Pos.
|
2,34 × 10-10
|
Pos. (26/28)
|
Pos.
|
Pos.
|
|
20
|
Genistein (1)
|
446-72-0
|
Pos.
|
2,24 × 10-9
|
2,45 × 10-8
|
Pos.
|
2,71 × 10-7
|
Pos. (100/102)
|
Pos.
|
Pos.
|
|
21
|
Aloperidolo
|
52-86-8
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (2/2)
|
Neg.
|
NT
|
|
22
|
Kaempferol (1)
|
520-18-3
|
Pos.
|
1,36 × 10-7
|
1,21 × 10-6
|
Pos.
|
3,99 × 10-6
|
Pos. (23/23)
|
Pos.
|
NT
|
|
23
|
Kepone (1)
|
143-50-0
|
Pos.
|
7,11 × 10-7
|
7,68 × 10-6
|
Pos.
|
4,91 × 10-7
|
Pos. (14/18)
|
Pos.
|
NT
|
|
24
|
Chetoconazolo
|
65277-42-1
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (2/2)
|
Neg.
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (8/8)
|
Neg.
|
NT
|
|
26
|
meso-esestrolo (1)
|
84-16-2
|
Pos.
|
< 1,00 × 10-11
|
2,75 × 10-11
|
Pos.
|
1,65 × 10-11
|
Pos. (4/4)
|
Pos.
|
NT
|
|
27
|
Metiltestosterone (1)
|
58-18-4
|
Pos.
|
1,73 × 10-7
|
4,11 × 10-6
|
Pos.
|
2,68 × 10-6
|
Pos. (5/6)
|
Pos.
|
NT
|
|
28
|
Morin
|
480-16-0
|
Pos.
|
5,43 × 10-7
|
4,16 × 10-6
|
Pos.
|
2,37 × 10-6
|
Pos. (2/2)
|
Pos.
|
NT
|
|
29
|
Noretinodrel (1)
|
68-23-5
|
Pos.
|
1,11 × 10-11
|
1,50 × 10-9
|
Pos.
|
9,39 × 10-10
|
Pos. (5/5)
|
Pos.
|
NT
|
|
30
|
p,p’-metossicolor (1)
|
72-43-5
|
Pos.
|
1,23 × 10-6
|
(no PC50) (2)
|
Pos.
|
1,92 × 10-6
|
Pos. (24/27)
|
Pos.
|
Pos.
|
|
31
|
Fenobarbital (1)
|
57-30-7
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (2/2)
|
Neg.
|
NT
|
|
32
|
Reserpina
|
50-55-5
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (4/4)
|
Neg.
|
NT
|
|
33
|
Spironolattone (1)
|
52-01-7
|
Neg.
|
—
|
—
|
Neg.
|
—
|
Neg. (4/4)
|
Neg.
|
NT
|
|
34
|
Testosterone
|
58-22-0
|
Pos.
|
2,82 × 10-8
|
9,78 × 10-6
|
Pos.
|
1,75 × 10-5
|
Pos. (5/10)
|
Pos.
|
NT
|
|
Abbreviazioni: N CAS = numero di registrazione CAS (Chemical Abstracts Service Registry Number). M = molare; EC50 = concentrazione efficace della sostanza in esame che induce una risposta pari alla metà della risposta massima; Neg.= negativo; Pos.= positivo; NT = non testato; PC10 (e PC50) = concentrazione della sostanza in esame per la quale la risposta è pari al 10 % (o 50 % per il valore PC50) dell’attività massima indotta dal controllo positivo (E2, 1 nm) in ciascuna piastra.
|
Tabella 2
Confronto dei risultati dei metodi di prova di STTA e VM7Luc ER TA ottenuti per le sostanze testate in base ai due metodi antagonisti e classificate come antagoniste degli ER (Pos.) o negative (Neg)
|
|
Sostanza (3)
|
N. CAS
|
Metodi di ER STTA (40)
|
Metodo VM7Luc ER TA (41)
|
Risposte attese ER STTA (43)
|
Consenso di classificazione ICCVAM (44)
|
Classe chimica nel sistema MeSH (45)
|
Classe di prodotto (46)
|
|
Attività di ER TA
|
IC50
(39) (M)
|
Attività di ER TA
|
IC50
(39), (42) (M)
|
|
1
|
4-idrossitamoxifen
|
68047-06-3
|
Pos.
|
3,97 × 10-9
|
Pos.
|
2,08 × 10-7
|
Pos. moderato
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
2
|
Dibenzo[a.h]antracene
|
53-70-3
|
Pos.
|
No IC50
|
Pos.
|
No IC50
|
Pos.
|
PP
|
Composto policiclico
|
Prodotto chimico di laboratorio, prodotto naturale
|
|
3
|
Mifepristone
|
84371-65-3
|
Pos.
|
5,61 × 10-6
|
Neg.
|
—
|
lievemente Pos.
|
Neg.
|
Steroide
|
Prodotto farmaceutico
|
|
4
|
Raloxifene HCl
|
82640-04-8
|
Pos.
|
7,86 × 10-10
|
Pos.
|
1,19 × 10-9
|
Pos. moderato
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
5
|
Tamoxifene
|
10540-29-1
|
Pos.
|
4,91 × 10-7
|
Pos.
|
8,17 × 10-7
|
Pos.
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
6
|
17β-estradiolo
|
50-28-2
|
Neg.
|
—
|
Neg.
|
—
|
PN
|
PN
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
7
|
Apigenin
|
520-36-5
|
Neg.
|
—
|
Neg.
|
—
|
Neg.
|
Neg.
|
Composto eterociclico
|
Colorante, prodotto naturale, intermediario farmaceutico
|
|
8
|
Atrazina
|
1912-24-9
|
Neg.
|
—
|
Neg.
|
—
|
Neg.
|
PN
|
Composto eterociclico
|
Erbicida
|
|
9
|
Di-n-butilftalato
|
84-74-2
|
Neg.
|
—
|
Neg.
|
—
|
Neg.
|
Neg.
|
Estere, acido ftalico
|
Ingrediente cosmetico, prodotto chimico industriale, plasticizzante
|
|
10
|
Fenarimol
|
60168-88-9
|
Neg.
|
—
|
Neg.
|
—
|
non testato
|
PN
|
Composto eterociclico, pirimidina
|
Fungicida
|
|
11
|
Flavone
|
525-82-6
|
Neg.
|
—
|
Neg.
|
—
|
PN
|
PN
|
Flavonoide, Composto eterociclico
|
Prodotto naturale, prodotto farmaceutico
|
|
12
|
Flutamide
|
13311-84-7
|
Neg.
|
—
|
Neg.
|
—
|
Neg.
|
PN
|
Ammide
|
Prodotto farmaceutico e veterinario
|
|
13
|
Genisteina
|
446-72-0
|
Neg.
|
—
|
Neg.
|
—
|
PN
|
Neg.
|
Flavonoide, Composto eterociclico
|
Prodotto naturale, prodotto farmaceutico
|
|
14
|
p-n-nonilfenolo
|
104-40-5
|
Neg.
|
—
|
Neg.
|
—
|
non testato
|
Neg.
|
Fenolo
|
Intermediario chimico
|
|
15
|
Resveratrolo
|
501-36-0
|
Neg.
|
—
|
Neg.
|
—
|
PN
|
Neg.
|
Idrocarburo (ciclico)
|
Prodotto naturale
|
|
Abbreviazioni: N CAS = numero di registrazione CAS (Chemical Abstracts Service Registry Number). M = molare; IC50 = concentrazione della sostanza in esame che induce la metà dell’inibizione massima; Neg.= negativo; PN = presunto negativo; Pos.= positivo; PP = presunto positivo.
|
COMPONENTI DEL METODO DI PROVA ER TA
Componenti essenziali del metodo di prova
|
11.
|
La presente linea guida si applica ai metodi che utilizzano un recettore ERα trasfettato in modo stabile o endogeno e un costrutto con gene reporter trasfettato in modo stabile e regolato da uno o più componenti di risposta agli estrogeni; altri recettori, quali ERβ, possono tuttavia essere presenti. I seguenti elementi sono essenziali ai fini del metodo di prova.
|
Controlli
|
12.
|
Sono descritte le ragioni che hanno portato a proporre gli standard di riferimento per la prova agonista e per la prova antagonista. I controlli inclusi simultaneamente nella prova (negativo, con solvente e positivo), come opportuno, servono ad indicare che il metodo di prova funziona alle condizioni sperimentali e forniscono gli elementi di comparazione tra esperimenti; essi rientrano generalmente tra i criteri di accettabilità di un determinato esperimento (1).
|
Procedure standard di controllo della qualità
|
13.
|
Le procedure standard di controllo della qualità vanno eseguite come descritto per ciascun protocollo al fine di garantire che la linea cellulare rimanga stabile durante i molteplici passaggi, rimanga esente da micoplasma (ossia priva di contaminazione batterica) e conservi la capacità di fornire le risposte mediate dagli ER nel tempo. Le linee cellulari devono essere ulteriormente controllate per verificarne l’identità esatta e la presenza di altri contaminanti (ad esempio funghi, lieviti e virus).
|
Dimostrazione della competenza del laboratorio
|
14.
|
Prima di testare sostanze chimiche sconosciute con uno qualsiasi dei metodi previsti dalla presente linea guida, ciascun laboratorio deve dimostrare la propria competenza ad eseguire il metodo di prova. Per dimostrare la competenza, ciascun laboratorio deve sottoporre a prova le 14 sostanze di prova a fini di competenza elencate nella tabella 3 per il saggio agonista e le 10 sostanze di prova a fini di competenza elencate nella tabella 4 per il saggio antagonista. La dimostrazione della competenza permette inoltre di confermare la reattività del sistema di prova. L’elenco delle sostanze da utilizzare per stabilire la competenza è un sottoinsieme delle sostanze di riferimento fornite negli standard di prestazione per i metodi di prova ER TA (6). Tali sostanze sono disponibili sul mercato, rappresentano le classi di sostanze chimiche comunemente associate all’attività agonista o antagonista degli ER, presentano una gamma di attività corrispondente alle affinità attese per gli agonisti/antagonisti degli ER (da debole a forte) e includono le sostanze negative. Le sostanze di prove a fini di competenza devono essere testate almeno due volte, in giorni diversi. La competenza è dimostrata quando ciascuna sostanza testata ai fini della competenza è classificata correttamente (risposta positiva/negativa). La prova ai fini della competenza è ripetuta da ciascun tecnico nell’ambito della formazione al presente metodo di prova. A seconda del tipo di cellula, alcune di queste sostanze di prova a fini di competenza possono comportarsi come SERM e presentare un’attività al tempo stesso agonista e antagonista. Tuttavia, nelle tabelle 3 e 4 le sostanze di prova a fini di competenza sono classificate in base alla loro attività conosciuta come prevalente, che sarà utilizzata per la valutazione della competenza a eseguire il metodo di prova.
|
|
15.
|
Per dimostrare la competenza e a fini di controllo qualitativo ciascun laboratorio costruisce banche di dati storici agonisti e antagonisti con i dati dello standard di riferimento (ossia, 17β-estradiolo e tamoxifene), delle sostanze di controllo positive e negative e del controllo con solvente (ad es. DMSO). Inizialmente, la base di dati è generata a partire da almeno 10 prove agoniste indipendenti (ad es. 17β-estradiolo) e 10 prove antagoniste indipendenti (ad es. tamoxifene). Occorrerà aggiungervi i risultati di ulteriori analisi condotte su tali standard di riferimento e controlli con solvente in modo da ampliare la base di dati al fine di garantirne la coerenza e le prestazioni del biosaggio da parte del laboratorio nel corso del tempo.
|
Tabella 3
Elenco delle (14) sostanze di prova a fini di competenza per la prova agonista
(54)
|
No
(53)
|
Sostanza
|
N. CAS
|
Risposta prevista (47)
|
Metodo STTA
|
Metodo VM7Luc ER TA
|
Classe chimica nel sistema MeSH (51)
|
Classe di prodotto (52)
|
|
PC10 (M) (48)
|
PC50 (M) (48)
|
Intervallo di concentrazione della prova (M)
|
VM7Luc EC50 (M) (49)
|
Conc. più alta per il test di range finding (M) (50)
|
|
14
|
Dietilstilbestrolo
|
56-53-1
|
Pos.
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14 – 10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico e veterinario
|
|
12
|
17α-estradiolo
|
57-91-0
|
Pos.
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11 – 10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
15
|
meso-esestrolo
|
84-16-2
|
Pos.
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11 – 10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Idrocarburo (ciclico), fenolo
|
Prodotto farmaceutico e veterinario
|
|
11
|
4-terz-ottilfenolo
|
140-66-9
|
Pos.
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11 – 10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenolo
|
Intermediario chimico
|
|
9
|
Genisteina
|
446-72-0
|
Pos.
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11 – 10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonoide, Composto eterociclico
|
Prodotto naturale, prodotto farmaceutico
|
|
6
|
Bisfenolo A
|
80-05-7
|
Pos.
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11 – 10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenolo
|
Intermediario chimico
|
|
2
|
Kaempferol
|
520-18-3
|
Pos.
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11 – 10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonoide, Composto eterociclico
|
Prodotto naturale
|
|
3
|
Ftalato di butilbenzile
|
85-68-7
|
Pos.
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11 – 10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Acido carbossilico, estere, acido ftalico
|
Plasticizzante, prodotto chimico industriale
|
|
4
|
p,p’- Metossicloro
|
72-43-5
|
Pos.
|
1,23 × 10-6
|
—
|
10-11 – 10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Idrocarburo (alogenato)
|
Pesticida, prodotto veterinario
|
|
1
|
Etilparabene
|
120-47-8
|
Pos.
|
5,00 × 10-6
|
—
|
10-11 – 10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Acido carbossilico, fenolo
|
Prodotto farmaceutico, conservante
|
|
17
|
Atrazina
|
1912-24-9
|
Neg.
|
—
|
—
|
10-10 – 10-4
|
—
|
4,64 × 10-4
|
Composto eterociclico
|
Erbicida
|
|
20
|
Spironolattone
|
52-01-7
|
Neg.
|
—
|
—
|
10-11 – 10-5
|
—
|
2,40 × 10-3
|
Lattone, steroide
|
Prodotto farmaceutico
|
|
21
|
Chetoconazolo
|
65277-42-1
|
Neg.
|
—
|
—
|
10-11 – 10-5
|
—
|
9,41 × 10-5
|
Composto eterociclico
|
Prodotto farmaceutico
|
|
22
|
Reserpina
|
50-55-5
|
Neg.
|
—
|
—
|
10-11 – 10-5
|
—
|
1,64 × 10-3
|
Composto eterociclico, indolo
|
Prodotto farmaceutico e veterinario
|
|
Abbreviazioni: N CAS = numero di registrazione CAS (Chemical Abstracts Service Registry Number). EC50 = concentrazione efficace della sostanza in esame che induce una risposta pari alla metà della risposta massima; Neg. = negativo; Pos. = positivo; PC10 (e PC50) = concentrazione della sostanza in esame per la quale la risposta è pari al 10 % (o 50 % per il valore PC50) dell’attività massima indotta dal controllo positivo (E2, 1 nm) in ciascuna piastra.
|
Tabella 4
Elenco delle (10) sostanze di prova di competenza per la prova antagonista
|
|
Sostanza (4)
|
N. CAS
|
Metodo ER STTA (55)
|
Metodo VM7Luc ER TA (56)
|
Risposte attese ER STTA (55)
|
ICCVAM (59) Consenso di classificazione nel sistema
|
MeSH (60) Classe chimica
|
Classe di prodotto (61)
|
|
Attività di ER TA
|
IC50 (M)
|
Intervallo di concentrazione della prova (M)
|
Attività di ER TA
|
IC50
(57) (M)
|
Conc. più alta per il test di range-finding (M) (58)
|
|
1
|
4-idrossitamoxifen
|
68047-06-3
|
Pos.
|
3,97 × 10-9
|
10-12 – 10-7
|
Pos.
|
2,08 × 10-7
|
2,58 × 10-4
|
Pos. moderato
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
2
|
Raloxifene HCl
|
82640-04-8
|
Pos.
|
7,86 × 10-10
|
10-12 – 10-7
|
Pos.
|
1,19 × 10-9
|
1,96 × 10-4
|
Pos. moderato
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
3
|
Tamoxifene
|
10540-29-1
|
Pos.
|
4,91 × 10-7
|
10-10 – 10-5
|
Pos.
|
8,17 × 10-7
|
2,69 × 10-4
|
Pos.
|
Pos.
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico
|
|
4
|
17β-estradiolo
|
50-28-2
|
Neg.
|
—
|
10-9 – 10-4
|
Neg.
|
—
|
3,67 × 10-3
|
Neg. (*4)
|
PN
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
5
|
Apigenin
|
520-36-5
|
Neg.
|
—
|
10-9 – 10-4
|
Neg.
|
—
|
3,70 × 10-4
|
Neg.
|
Neg.
|
Composto eterociclico
|
Colorante, prodotto naturale, intermediario farmaceutico
|
|
6
|
Di-n-butilftalato
|
84-74-2
|
Neg.
|
—
|
10-8 – 10-3
|
Neg.
|
—
|
3,59 × 10-3
|
Neg.
|
Neg.
|
Estere, acido ftalico
|
Ingrediente cosmetico, prodotto chimico industriale, plasticizzante
|
|
7
|
Flavone
|
525-82-6
|
Neg.
|
—
|
10-8 – 10-3
|
Neg.
|
—
|
4,50 × 10-4
|
Neg. (*4)
|
PN
|
Flavonoide, Composto eterociclico
|
Prodotto naturale, prodotto farmaceutico
|
|
8
|
Genisteina
|
446-72-0
|
Neg.
|
—
|
10-9 – 10-4
|
Neg.
|
—
|
3,70 × 10-4
|
Neg. (*4)
|
Neg.
|
Flavonoide, Composto eterociclico
|
Prodotto naturale, prodotto farmaceutico
|
|
9
|
p-n-nonilfenolo
|
104-40-5
|
Neg.
|
—
|
10-9 – 10-4
|
Neg.
|
—
|
4,54 × 10-4
|
non testato
|
Neg.
|
Fenolo
|
Intermediario chimico
|
|
10
|
Resveratrolo
|
501-36-0
|
Neg.
|
—
|
10-8 – 10-3
|
Neg.
|
—
|
4,38 × 10-4
|
Neg. (*4)
|
Neg.
|
Idrocarburo (ciclico)
|
Prodotto naturale
|
|
Abbreviazioni: N CAS = numero di registrazione CAS (Chemical Abstracts Service Registry Number). M = molare; IC50 = concentrazione della sostanza in esame che induce la metà dell’inibizione massima; Neg.= negativo; PN = presunto negativo; Pos.= positivo.
|
Criteri di accettabilità della batteria di prove
|
16.
|
L’accettazione o il rigetto di una batteria di prove si basa sulla valutazione dei risultati ottenuti per gli standard di riferimento e i controlli utilizzati per ciascun esperimento. I valori di PC50 (EC50) o IC50 per gli standard norme di riferimento devono soddisfare i criteri di accettabilità previsti per il metodo di prova selezionato (per il saggio STTA cfr. l’appendice 2, per il saggio VM7Luc ER TA cfr. l’appendice 3) e tutti i controlli positivi/negativi devono essere classificati in modo corretto per ciascun esperimento accettato. Un laboratorio dimostra la propria capacità di condurre un metodo di prova in modo omogeneo grazie alla creazione e al mantenimento di una banca di dati storici per gli standard di riferimento e per i controlli (cfr. il paragrafo 15). Le deviazioni standard (SD) o i coefficienti di variazione (CV) delle medie dei parametri di approssimazione delle curve degli standard di riferimento ottenuti dopo prove multiple possono essere utilizzati come misura di riproducibilità intra-laboratorio. Inoltre, devono essere rispettati i seguenti principi relativi ai criteri di accettabilità:
|
—
|
I dati dovrebbero essere sufficienti per valutare in modo quantitativo il tasso di attivazione (per la prova sugli agonisti) o di inibizione (per la prova sugli antagonisti) (efficacia e potenza).
|
|
—
|
L’attività media del marcatore ottenuto con la concentrazione di riferimento della sostanza estrogenica di riferimento è pari o superiore alla risposta minima specificata nei metodi di prova, con riferimento alla risposta del controllo con mezzo disperdente (con solvente) al fine di garantire un’adeguata sensibilità. Nel caso dei metodi di STTA e di TA ER VM7Luc, tali risultati corrispondono a quattro volte la risposta media del controllo con mezzo disperdente per ciascuna piastra.
|
|
—
|
Le concentrazioni testate devono rimanere inferiori al limite di solubilità delle sostanze chimiche in esame e non presentare citotossicità.
|
|
Analisi dei dati
|
17.
|
Per classificare una risposta come positiva o negativa, si applica la procedura di interpretazione dei dati definita per ciascun metodi di prova.
|
|
18.
|
Il rispetto dei criteri di accettabilità (paragrafo 16) comprova che la prova funziona correttamente, ma non garantisce che qualsiasi prova produrrà dati accurati. Replicare i risultati della prima prova è la migliore indicazione che sono stati generati dati accurati. Se due batterie di prove danno risultati riproducibili (ad esempio, se entrambe concludono che una sostanza chimica è positiva), non è necessario effettuarne una terza.
|
|
19.
|
due batterie di prove non forniscono risultati riproducibili (ad esempio, una sostanza chimica in esame è positiva in una e negativa nell’altra), o se è richiesto un maggiore grado di certezza per questo metodo di prova, dovrebbero essere condotte almeno tre batterie di prove indipendenti. In questo caso la classificazione si basa sui due risultati concordanti dei tre risultati ottenuti.
|
Criteri generali di interpretazione dei dati
|
20.
|
Attualmente non esiste alcun metodo universalmente concordato per l’interpretazione dei dati di una prova ER TA. Tuttavia, la valutazione qualitativa (ad es. positivo/negativo) e/o quantitativa (ad es. EC50, PC50, IC50), dell’attività mediata dagli hrER deve fondarsi su dati empirici e giudizi scientificamente validi. Se possibile, i risultati positivi devono essere caratterizzati sia dall’entità dell’effetto indotto rispetto al controllo con mezzo disperdente (solvente) o all’estrogeno di riferimento sia dalla concentrazione alla quale si produce l’effetto (ad es. EC50, PC50, RPCMax, IC50, ecc.).
|
Relazione sull’esecuzione della prova
|
21.
|
La relazione sull’esecuzione della prova deve includere le informazioni seguenti:
|
|
Metodo di prova:
|
—
|
metodo di prova utilizzato;
|
|
—
|
controlli/standard di riferimento/sostanza chimica in esame;
|
|
—
|
origine, numero di lotto, data limite d’uso, se disponibili;
|
|
—
|
stabilità della sostanza chimica in esame, se nota;
|
|
—
|
solubilità e stabilità della sostanza chimica in esame nel solvente, se note;
|
|
—
|
misurazione del pH, dell’osmolalità e del precipitato nel terreno di coltura a cui è stata aggiunta la sostanza chimica in esame, se del caso.
|
|
|
|
Sostanza monocostituente
|
—
|
aspetto fisico, idrosolubilità e ulteriori proprietà fisico-chimiche pertinenti;
|
|
—
|
dati di identificazione chimica: denominazioni IUPAC o CAS, numero CAS, codice SMILES o InChI, formula strutturale, identità chimica o impurezze, se del caso e se le condizioni pratiche lo consentono, ecc.
|
|
|
|
Sostanza multicostituente, UVCB e miscele
|
—
|
caratterizzazione, per quanto possibile, mediante l’identità chimica dei costituenti (vedi sopra), loro presenza quantitativa e loro proprietà fisico-chimiche pertinenti.
|
|
|
|
Solvente/mezzo disperdente
|
—
|
Caratterizzazione (natura, fornitore e lotto);
|
|
—
|
motivazione della scelta del solvente/mezzo disperdente;
|
|
—
|
solubilità e stabilità della sostanza in esame nel solvente/mezzo disperdente, se note.
|
|
|
|
Cellule
|
—
|
tipo e origine delle cellule;
|
—
|
L’ER è espresso in modo endogeno? In caso contrario, quale o quali recettori sono stati trasfettati?
|
|
—
|
costrutto o costrutti di geni reporter utilizzati (comprese le specie di origine);
|
|
—
|
metodo di selezione applicato per mantenere stabile una trasfezione(se del caso);
|
|
—
|
il metodo di trasfezione permette di ottenere linee stabili?
|
|
|
—
|
numero di passaggi in coltura delle cellule (dopo scongelamento);
|
|
—
|
numero di passaggi in coltura delle cellule al momento dello scongelamento;
|
|
—
|
metodi usati per la conservazione delle colture cellulari.
|
|
|
|
Condizioni sperimentali
|
—
|
descrizione dei metodi di valutazione della vitalità applicati;
|
|
—
|
composizione dei terreni di coltura, concentrazione di CO2;
|
|
—
|
concentrazione della sostanza chimica in esame;
|
|
—
|
volume del mezzo disperdente e della sostanza chimica in esame aggiunto;
|
|
—
|
temperatura di incubazione e umidità;
|
|
—
|
durata del trattamento;
|
|
—
|
densità cellulare all’inizio e durante il trattamento;
|
|
—
|
standard di riferimento positivi e negativi;
|
|
—
|
reagenti reporter (nome del prodotto, fornitore e lotto);
|
|
—
|
criteri in base ai quali i risultati delle prove sono considerati positivi, negativi o equivoci.
|
|
|
|
Verifica dell’accettabilità:
|
—
|
moltiplicazione dell’induzione in ciascuna piastra di prova e confronto con il minimo richiesto per il metodo di prova in base ai dati storici dei controlli;
|
|
—
|
valori reali per i criteri di accettabilità, ad es. log10EC50, log10PC50, logIC50 e pendenza di Hill per i controlli positivi e standard di riferimento simultaneamente inclusi nella prova.
|
|
|
|
Risultati
|
—
|
dati grezzi e normalizzati;
|
|
—
|
livello di induzione massimo;
|
|
—
|
dati relativi alla citotossicità;
|
|
—
|
se esiste, la più bassa concentrazione efficace (LEC);
|
|
—
|
valori RPCMax, PCMax, PC50, IC50 e/o EC50, come opportuno;
|
|
—
|
relazione concentrazione-risposta, se possibile;
|
|
—
|
eventuali analisi statistiche, assieme alla misura dell’errore e della confidenza (ad es. SEM, SD, CV o 95 % IC) e descrizione di come tali dati sono stati ottenuti.
|
|
|
|
Discussione dei risultati
|
|
BIBLIOGRAFIA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Organisation for Economic Cooperation and Development, Paris.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In Vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Organisation for Economic Cooperation and Development, Paris.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Organisation for Economic Cooperation and Development, Paris.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In Vitro and In Vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In Vitro and In Vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERαIn Vivo and In Vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In Vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Appendice 1
DEFINIZIONI E ABBREVIAZIONI
Criteri di accettabilità
: requisiti minimi di prestazione relativi ai controlli sperimentali e agli standard di riferimento. Tutti i criteri di accettabilità devono essere soddisfatti affinché un metodo di prova sia considerato valido.
Accuratezza (concordanza)
: grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza", per indicare la proporzione di risultati corretti di un metodo di prova (1).
Agonista
: sostanza che provoca una risposta, ad esempio una trascrizione, quando si lega a un recettore specifico.
Antagonista
: tipo di ligando di un recettore o tipo di sostanza chimica che non provoca, di per sé, una risposta biologica quando si lega a un recettore, ma blocca o attenua la risposta mediata dagli agonisti.
Attività antiestrogenica
: capacità di una sostanza chimica di inibire l'azione del 17β-estradiolo mediata dai recettori estrogenici.
Morfologia cellulare
: forma e aspetto delle cellule coltivate in monostrato in un unico pozzetto di una piastra di coltura tessutale. Le cellule che stanno per morire presentano spesso una morfologia anomala.
CF
: quadro concettuale dell'OCSE per la sperimentazione e la valutazione degli interferenti endocrini.
Trattamento al carbone-destrano
: trattamento del siero usato per la coltura cellulare. Spesso denominato stripping, questo trattamento con carbone rivestito di destrano permette di eliminare gli ormoni endogeni e le proteine che legano gli ormoni.
Sostanza chimica
: una sostanza o una miscela.
Citotossicità
: effetti nocivi per la struttura o le funzioni della cellula che possono finire per provocare la morte cellulare e che possono manifestarsi mediante una riduzione del numero di cellule presenti nel pozzetto alla fine del periodo di esposizione o una riduzione della capacità di misurare una funzione cellulare rispetto al concomitante controllo con mezzo disperdente.
CV
: coefficiente di variazione.
DCC-FBS
: (Dextran-coated charcoal treated fetal bovine serum) siero bovino fetale trattato al carbone rivestito di destrano.
DMEM
: (Dulbecco's Modification of Eagle's Medium) mezzo di Eagle modificato da Dulbecco.
DMSO
: dimetilsolfossido
E2
: 17β-estradiolo
EC50
: concentrazione efficace di una sostanza chimica in esame che produce un effetto pari alla metà dell'effetto massimo.
ED
: (Endocrine disruption) interferenza con il sistema endocrino.
hERα
: recettore estrogenico alfa umano
hERß
: recettore estrogenico beta umano
EFM
: terreno di coltura privo di estrogeni. Si tratta del mezzo di Eagle modificato da Dulbecco (DMEM) integrato da 4,5 % di FBS trattato con carbone-destrano, 1,9 % di L-glutammina e 0,9 % di Pen-Strep.
ER
: recettore estrogenico
ERE
: elemento di risposta agli estrogeni
Attività estrogenica
: capacità di una sostanza chimica di riprodurre la capacità del 17β-estradiolo di legarsi ai recettori estrogenici e di attivarli. L'attività estrogenica mediata dall'hERα può essere individuata mediante il presente metodo di prova.
ERTA
: transattivazione dei recettori estrogenici
FBS
: siero fetale bovino
HeLa
: Linea cellulare immortalizzata derivata da cellule umane prelevate da cervice uterina
HeLa9903
: subclone della linea cellulare HeLa trasfettato in modo stabile con un hERαα e un gene reporter della luciferasi
IC50
: concentrazione efficace di una sostanza chimica in esame che induce la metà dell'effetto massimo di inibizione.
ICCVAM
: Interagency Coordinating Committee on the Validation of Alternative Methods (comitato di coordinamento interagenzie per la validazione di metodi alternativi).
Riproducibilità inter-laboratori
: espressione della misura in cui laboratori qualificati diversi possano ottenere, seguendo lo stesso protocollo e testando le stesse sostanze di prova, risultati simili in termini qualitativi e quantitativi. La riproducibilità inter-laboratori è determinata nel corso dei processi di pre-validazione e validazione e indica la misura in cui un metodo di prova può essere trasferito con successo tra laboratori; è detta anche riproducibilità tra laboratori (1).
Riproducibilità intra-laboratorio
: espressione della misura in cui membri diversi del personale qualificato all'interno del medesimo laboratorio riescono a ottenere risultati simili da una prova effettuata seguendo un protocollo specifico in momenti diversi; è detta anche "riproducibilità all'interno dello stesso laboratorio" (1).
LEC
: la concentrazione minima della sostanza chimica in esame che produce una risposta (ossia la più bassa concentrazione della sostanza chimica in esame alla quale l'incremento dell'effetto indotto è statisticamente diverso da quello del concomitante controllo con mezzo disperdente).
Test strutturalmente analoghi
: espressione che indica un metodo di prova strutturalmente e funzionalmente analogo a un metodo di prova di riferimento validato e accettato. È sinonimo di "metodo di prova simile".
MT
: metallotioneina
MMTV
: (Mouse Mammary Tumor Virus) virus del tumore mammario dei topi
OHT
: 4-idrossitamoxifene
PBTG
: (Performance-Based Test Guideline) Linea guida basata su standard di prestazione
PC (controllo positivo)
: sostanza fortemente attiva, di preferenza il 17ß-estradiolo, inclusa in tutte le prove per contribuire a garantire il corretto funzionamento del metodo di prova.
PC10
: la concentrazione di una sostanza chimica in esame alla quale l'attività misurata nel corso di un metodo di prova è pari al 10 % dell'attività massima indotta dal controllo positivo (E2 a 1 nM per il metodo di STTA) in ciascuna piastra.
PC50
: la concentrazione di una sostanza chimica in esame alla quale l'attività misurata nel corso di un metodo di prova è pari al 50 % dell'attività massima indotta dal controllo positivo (E2 alla concentrazione di riferimento specificata nel metodo di prova) in ciascuna piastra.
PCMax
: la concentrazione di una sostanza chimica in esame che induce l'RPCMax
Standard di prestazione
: standard, basati su un metodo di prova validato, che consentono di valutare la comparabilità di un metodo di prova proposto che è simile sotto il profilo strutturale e funzionale. Comprendono: 1) gli elementi essenziali del metodo di prova; 2) un elenco minimo di sostanze chimiche di riferimento scelte tra le sostanze utilizzate per dimostrare le prestazioni accettabili del metodo di prova validato; e 3) in funzione dei risultati ottenuti con il metodo di prova validato, i livelli comparabili di accuratezza e affidabilità che il metodo proposto dovrebbe ottenere quando viene valutato utilizzando l'elenco minimo di sostanze chimiche di riferimento (1).
Sostanze di prova ai fini di competenza
: sottoinsieme delle sostanze di riferimento incluse negli standard di prestazione che possono essere utilizzate da un laboratorio per dimostrare la sua competenza tecnica ad effettuare un metodo di prova standardizzato. In generale, i criteri di selezione per tali sostanze includono la rappresentatività di tutta la gamma delle risposte, la loro disponibilità sul mercato e l'esistenza di dati di riferimento di elevata qualità a loro corredo.
Competenza
: la capacità di eseguire correttamente un metodo di prova, dimostrata prima di testare sostanze sconosciute.
Sostanza estrogenica di riferimento (controllo positivo, PC)
: 17β-estradiolo (E2, CAS 50-28-2).
Standard di riferimento
: sostanza di riferimento usata per dimostrare l'adeguatezza di un metodo di prova. Il 17β-estradiolo è lo standard di riferimento per i metodi di STTA e VM7Luc ER TA.
Metodi di prova di riferimento
: i metodi su cui si basa la linea guida PBTG 455.
Pertinenza
: descrizione del rapporto tra la prova e l'effetto studiato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (1).
Affidabilità
: misura in cui l'esecuzione di un metodo di prova può essere riprodotta nel tempo all'interno dello stesso laboratorio o da laboratori diversi seguendo il medesimo protocollo. È valutata calcolando la riproducibilità intra-laboratorio e inter-laboratori.
RLU
: unità relative di luce
RNA
: acido ribonucleico
RPCMax
: livello massimo di risposta indotto da una sostanza chimica in esame, espresso in percentuale della risposta indotta da 1 nM di E2 sulla stessa piastra
RPMI
: terreno di coltura RPMI 1 640 integrato da 0,9 % di Pen-Strep e 8,0 % di siero fetale bovino (FBS)
Batteria di prove
: singolo esperimento che valuta l'azione della sostanza chimica sull'aspetto biologico del metodo di prova. Ciascuna batteria di prove consiste in un esperimento completo eseguito su pozzetti in replicato, contemporaneamente e con cellule che provengono dalla medesima riserva di cellule.
Batteria di prove indipendente
: esperimento distinto e indipendente che valuta l'azione della sostanza chimica sull'aspetto biologico del metodo di prova, utilizzando cellule provenienti da una diversa riserva di cellule, sostanze chimiche diluite di recente, ed eseguito in giorni diversi oppure lo stesso giorno ma da personale diverso.
SD
: deviazione standard
Sensibilità
: proporzione di tutte le sostanze positive/attive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante da prendere in considerazione nel valutare la pertinenza di un metodo di prova (1).
Specificità
: proporzione di tutte le sostanze negative/inattive correttamente classificate dal metodo di prova. Misura l'accuratezza di un metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante da prendere in considerazione nel valutare la pertinenza di un metodo di prova (1).
Trasfezione stabile
: processo che consiste nel trasfettare DNA nelle cellule di coltura in modo tale che esso sia stabilmente integrato nel loro genoma, il che permette l'espressione stabile di geni trasfettati. Cloni di cellule trasfettate stabilmente sono quindi selezionati con marcatori stabili (ad esempio in funzione della loro resistenza al G418).
Saggio di STTA
: Stably Transfected Transactivation Assay, il saggio di transattivazione degli ERα basato sulla linea cellulare HeLa 9 903.
Studio
: l'intera gamma di lavori sperimentali eseguiti per valutare un'unica sostanza specifica mediante un metodo di prova specifico. Uno studio comprende tutte le fasi, incluse le prove di diluizione della sostanza chimica in esame nel mezzo di prova, la batteria di prove preliminari volte a determinare l'intervallo delle concentrazioni, tutte le necessarie batterie di prove complete, l'analisi dei dati, l'assicurazione della qualità, la valutazione della citotossicità, ecc. Il completamento di uno studio consente la classificazione dell'attività della sostanza chimica in esame rispetto al tipo di tossicità (ossia attivo, inattivo o non conclusivo) valutata con il metodo di prova e permette una stima dell'efficacia rispetto alla sostanza di riferimento positiva.
Sostanza
: nel contesto del REACH (62), per «sostanza» si intende un elemento chimico e i suoi composti, allo stato naturale od ottenuti per mezzo di un procedimento di fabbricazione, compresi gli additivi necessari a mantenerne la stabilità e le impurezze derivanti dal procedimento utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione. Una definizione molto simile è utilizzata nel contesto del GHS delle Nazioni Unite (1).
TA (transattivazione)
: avvio della sintesi di mRNA in risposta a un segnale chimico specifico, come il legame di un estrogeno al un recettori estrogenico.
Metodo di prova
: nel contesto del presente metodo di prova, un metodo di prova è una delle metodologie riconosciute valide per soddisfare i criteri di prestazione indicati. Le componenti del metodo di prova comprendono, ad esempio, la linea cellulare specifica con le condizioni di crescita associate, i terreni di coltura specifici in cui è condotta la prova, le condizioni di configurazione della piastra, la disposizione e le diluizioni delle sostanze chimiche in esame, nonché tutte le altre misure richieste per il controllo della qualità e le relative fasi di valutazione dei dati.
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Trascrizione
: sintesi dell'RNA messaggero
UVCB
: sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici
Metodo di prova validato
: saggio per il quale sono stati completati studi di validazione volti a determinare la pertinenza (compresa l'accuratezza) e l'affidabilità per uno scopo specifico. Va sottolineato che un metodo di prova validato potrebbe non avere una prestazione sufficiente in termini di accuratezza e affidabilità per essere ritenuto accettabile per lo scopo determinato (1).
Validazione
: il processo mediante il quale si stabilisce l'affidabilità e la pertinenza di uno specifico approccio, metodo, saggio, processo o di una specifica valutazione per un determinato scopo (1).
VC (controllo con mezzo disperdente)
: Il solvente utilizzato per dissolvere le sostanze chimiche in esame e di controllo è testato unicamente come mezzo disperdente, senza la sostanza chimica disciolta.
VM7
: cellule immortalizzate di adenocarcinoma che esprimono i recettori di estrogeni in modo endogeno.
VM7Luc4E2
: la linea cellulare VM7Lu4E2 è derivata da cellule VM7 immortalizzate di adenocarcinoma di origine umana capaci di esprimere i due tipi di recettori estrogenici (ERα e ERβ) in maniera endogena e che sono state trasfettate in modo stabile con il plasmide pGudLuc7.ERE. Questo plasmide contiene quattro copie di un oligonucleotide di sintesi contenente l'elemento di risposta agli estrogeni a monte del promotore del virus del tumore mammario del topo (MMTV) e il gene reporter della luciferasi di lucciola.
Controllo positivo debole
: sostanza debolmente attiva selezionata tra le sostanze chimiche di riferimento, inclusa in tutte le prove per contribuire a garantire il corretto funzionamento del metodo di prova.
Appendice 2
SAGGIO DI TRANSATTIVAZIONE MEDIANTE IL RECETTORE ESTROGENICO ALFA UMANO TRASFETTATO IN MODO STABILE PER L'INDIVIDUAZIONE DELL'ATTIVITÀ AGONISTA E ANTAGONISTA DELLE SOSTANZE CHIMICHE UTILIZZANDO LA LINEA CELLULARE HERΑ-HELA-9903
CONSIDERAZIONI INIZIALI E LIMITI (CFR. ANCHE INTRODUZIONE GENERALE)
|
1.
|
Il presente saggio di transattivazione (TA) utilizza la linea cellulare HERα-HeLa-9 903 per individuare l'attività estrogenica agonista mediata dal recettore estrogenico alfa umano (hERα). Lo studio di validazione del saggio di transattivazione mediante trasfezione stabile (STTA) condotto dall'Istituto giapponese di ricerca e valutazione delle sostanze chimiche (CERI), che utilizza la linea cellulare hERα-HeLa-9 903 per individuare l'attività estrogenica agonista e antagonista mediata dal recettore estrogenico alfa umano (hERα), ha dimostrato la pertinenza e l'affidabilità del saggio per lo scopo previsto (1).
|
|
2.
|
Il presente metodo di prova è concepito in modo specifico per individuare la transattivazione mediata dall'hERα misurando la chemiluminescenza come endpoint. Tuttavia, segnali di luminescenza non mediati dai recettori sono state riportati per concentrazioni di fitoestrogeni superiori a 1 μM, a motivo della sovrattivazione del gene reporter della luciferasi (2) (3). Mentre la curva dose-risposta indica che la reale attivazione del sistema ER avviene a concentrazioni inferiori, l'espressione della luciferasi ottenuta a concentrazioni elevate di fitoestrogeni o di composti simili sospettati di indurre una sovrattivazione del gene reporter della luciferasi mediante un meccanismo analogo a quello dei fitoestrogeni deve essere esaminata con attenzione nei sistemi di prova di ER TA trasfettati in modo stabile (appendice 1).
|
|
3.
|
Le sezioni "INTRODUZIONE GENERALE" e "COMPONENTI DEL METODO DI PROVA DI LEGAME ER TA" vanno lette prima di applicare il presente metodo di prova per fini regolamentari. Le definizioni e le abbreviazioni utilizzate nella presente linea guida figurano nell'appendice 2.1.
|
PRINCIPIO DEL METODO DI PROVA (CFR. ANCHE INTRODUZIONE GENERALE)
|
4.
|
Il metodo di prova è condotto per segnalare il legame di un ligando a un recettore estrogenico. Una volta stabilito tale legame, il complesso recettore-ligando trasloca verso il nucleo nel quale si fissa a elementi di risposta specifici del DNA e transattiva il gene reporter della luciferasi di lucciola, il che risulta in una maggiore espressione cellulare dell'enzima luciferasi. La luciferina è un substrato trasformato dall'enzima luciferasi in prodotto bioluminescente misurabile quantitativamente con un luminometro. L'attività della luciferasi può essere valutata in modo rapido e poco costoso grazie ai numerosi kit di analisi disponibili in commercio.
|
|
5.
|
Il sistema di prova utilizza la linea cellulare hERα-HeLa-9 903, derivata da un tumore della cervice uterina di origine umana, che comporta due costrutti trasfettati stabilmente: i) il costrutto di espressione dell'hERαα (che codifica il recettore umano a lunghezza intera), e ii) un costrutto del gene reporter della luciferasi di lucciola che porta cinque sequenze ripetute in tandem di un elemento di risposta agli estrogeni (ERE) della vitellogenina e indotto da un elemento promotore della metallotioneina (MT) del ratto con motivo TATA. È stato dimostrato che il gene MT TATA di ratto produce i migliori risultati ed è quindi comunemente utilizzato. Di conseguenza, la linea cellulare hERα-HeLa-9 903 permette di misurare la capacità di una sostanza chimica di indurre una transattivazione dell'espressione del gene della luciferasi mediata dall'hERα.
|
|
6.
|
Nel caso del saggio agonista degli ER, l'interpretazione dei dati poggia sul criterio seguente: il livello di risposta massimo indotto da una sostanza chimica in esame è superiore o uguale a una risposta agonista corrispondente al 10 % della risposta provocata da una concentrazione che induce una risposta massima del controllo positivo, ossia 1 nM di 17β-estradiolo (E2), cioè un valore PC10)? Nel caso del saggio antagonista, l'interpretazione dei dati poggia sul criterio seguente: la risposta mostra una riduzione di almeno il 30 % dell'attività rispetto alla risposta indotta dal controllo che produce il picco (25 pM di E2) senza citotossicità? L'analisi e l'interpretazione dei dati sono discusse in dettaglio nei paragrafi 34-48.
|
PROCEDURA
Linee cellulari
|
7.
|
Per il presente metodo di prova si utilizza la linea cellulare hERα-HeLa-9 903 trasfettata in modo stabile. La linea cellulare può essere ottenuta presso la banca cellulare JCRB (Japanese Collection of Research Bioresources) (63) previa sottoscrizione di un accordo di trasferimento di materiale (Material Transfer Agreement, MTA).
|
|
8.
|
Per il presente metodo di prova si utilizzano solo cellule esenti da micoplasmi. Il metodo migliore di individuazione sensibile di un'infezione da micoplasma è il RT-PCR (reazione a catena della polimerasi in tempo reale) (4) (5) (6).
|
Stabilità della linea cellulare
|
9.
|
Per monitorare la stabilità della linea cellulare, è opportuno utilizzare l'E2, il 17α-estradiolo, il 17-α-metiltestosterone e il corticosterone come standard di riferimento per la prova sugli agonisti; è inoltre opportuno che la curva concentrazione-risposta completa sia misurata sull'intero intervallo di concentrazioni di prova indicate nella tabella 1 almeno una volta nel corso della conduzione della prova e che i risultati siano conformi a quelli riportati nella tabella 1.
|
|
10.
|
Nel caso della prova sugli antagonisti, è opportuno misurare simultaneamente curve di concentrazione complete per due standard di riferimento, il tamoxifene e il flutamide, in ciascuna batteria di prove. Occorre controllare che la classificazione qualitativa sia correttamente individuata come positiva o negativa per le due sostanze chimiche.
|
Condizioni di coltura e di piastratura delle cellule
|
11.
|
Le cellule sono mantenute in un mezzo minimo essenziale di Eagle (EMEM) senza rosso fenolo, integrato da 60 mg/l di antibiotico (Kanamicina) e 10 % di siero bovino fetale trattato al carbone-destrano (DCC-FBS), in un incubatore con CO2 (5 % di CO2) a 37 ± 1 °C. Quando è raggiunta una confluenza del 75 -90 %, le cellule possono essere suddivise in colture secondarie di 10 ml contenenti 0,4 x 105 – 1 x 105 cellule/ml in piastre per colture cellulari di 100 mm di diametro. Le cellule sono messe in sospensione in FBS-EMEM al 10 % (equivalente a EMEM con DCC-FBS) e poi piastrate nei pozzetti di una micropiastra fino a una densità di 1 x 104 cellule/100 μl/pozzetto. Successivamente, le cellule sono preincubate in un incubatore con 5 % di CO2 a 37 ± 1 oC per 3 ore prima dell'esposizione alla sostanza chimica. Il materiale in plastica deve essere esente da qualsiasi attività estrogenica.
|
|
12.
|
Per preservare l'integrità della risposta, le cellule coltivate sono sottoposte a passaggio dallo stock congelato al mezzo di coltura condizionato; il numero di passaggi non deve superare 40. Per la linea cellulare hERα-HeLa-9 903, ciò avviene in meno di tre mesi. Tuttavia, la prestazione delle cellule può essere ridotta se le cellule sono coltivate in condizioni colturali inadeguate.
|
|
13.
|
Il DCC-FBS può essere preparato come descritto nell'appendice 2.2 o ottenuto da fonti commerciali.
|
Criteri di accettabilità
Standard di riferimento positivi e negativi per la prova sugli agonisti degli ER
|
14.
|
Prima e durante lo studio occorre verificare la reattività del sistema di prova usando le concentrazioni appropriate di un forte estrogeno E2, di un estrogeno debole (17α-estradiolo), di un agonista molto debole (17α-metiltestosterone) e di una sostanza che induce una risposta negativa (corticosterone). L'intervallo dei valori accettabili derivanti dallo studio di validazione (1) sono riportati nella tabella 1. Questi 4 standard di riferimento concomitanti sono inclusi in ciascun esperimento e i risultati devono rientrare entro i limiti accettabili stabiliti. In caso contrario, occorre determinare la causa del mancato rispetto dei criteri di accettabilità (ad esempio, manipolazione cellulare, la qualità e la concentrazione di siero e antibiotici) e la prova va ripetuta. Quando i criteri di accettabilità sono soddisfatti, è essenziale utilizzare in modo coerente i materiali di coltura cellulare per garantire una variabilità minima dei valori EC50, PC50 and PC10. I quattro standard di riferimento concomitanti, che vanno inclusi in ciascun esperimento (condotto nelle medesime condizioni, inclusi i materiali, il livello di passaggio delle cellule e i tecnici), garantiscono la sensibilità della prova nella misura in cui i valori PC10 dei tre standard di riferimento positivi rientrano nell'intervallo accettabile, così come i valori PC50 e EC50 laddove possono essere calcolati (cfr. tabella 1).
|
Tabella 1
Intervallo dei valori accettabili dei quattro standard di riferimento per la prova sugli agonisti degli ER
|
Nome
|
logPC50
|
logPC10
|
logEC50
|
Pendenza di Hill
|
Intervallo di prova
|
|
17β-estradiolo (E2) N. CAS: 50-28-2
|
-11,4~-10,1
|
<-11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-estradiolo N. CAS: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Corticosterone N. CAS: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-metiltestosterone N. CAS: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Standard di riferimento positivi e negativi per la prova sugli antagonisti degli ER
|
15.
|
Prima e durante lo studio occorre verificare la reattività del sistema di prova usando le concentrazioni appropriate di una sostanza positiva (tamoxifene) e di una sostanza negativa (flutamide). L'intervallo dei valori accettabili derivanti dallo studio di validazione (1) sono riportati nella tabella 2. Questi 2 standard di riferimento concomitanti sono inclusi in ciascun esperimento e i risultati devono permettere di classificarli correttamente, come indicato nei criteri. In caso contrario, occorre determinare la causa del mancato rispetto dei criteri (ad esempio, manipolazione cellulare, la qualità e la concentrazione di siero e antibiotici) e la prova va ripetuta. Occorre inoltre calcolare i valori IC50 per una sostanza positiva (tamoxifene) e i risultati devono rientrano nei limiti accettabili indicati. Quando i criteri di accettabilità sono soddisfatti, è essenziale utilizzare in modo coerente i materiali di coltura cellulare per garantire una variabilità minima dei valori IC50. I due standard di riferimento concomitanti, concomitanti, che vanno inclusi in ciascun esperimento (condotto nelle stesse condizioni, inclusi i materiali, il livello di passaggio delle cellule e i tecnici), possono garantire la sensibilità della prova (cfr. tabella 2).
|
Tabella 2
Criteri e intervallo dei valori accettabili dei due standard di riferimento per la prova sugli antagonisti degli ER
|
Nome
|
Criteri
|
LogIC50
|
Intervallo di prova
|
|
Tamoxifene N. CAS: 10 540 -29-1
|
positivo: Va calcolato il valore IC50
|
-5,942~-7,596
|
10-10~10-5M
|
|
Flutamide N. CAS: 13 311 -84-7
|
negativo: Non va calcolato il valore IC30
|
—
|
10-10 ~10-5M
|
Controllo positivo e controllo con mezzo disperdente
|
16.
|
Il controllo positivo (PC) per la prova sugli agonisti (1 nM di E2) e la prova sugli antagonisti (10 μM TAM) degli ER deve essere testato almeno in triplicato in ciascuna piastra. Il mezzo disperdente usato per sciogliere la sostanza chimica in esame deve essere testato come controllo con mezzo disperdente (VC) almeno in triplicato in ciascuna piastra. Oltre a questo VC, se il controllo positivo utilizza un mezzo disperdente diverso da quello della sostanza chimica in esame, questo altro mezzo disperdente deve essere testato almeno in triplicato sulla stessa piastra del controllo positivo.
|
Criteri di qualità per la prova sugli agonisti degli ER
|
17.
|
L'attività della luciferasi media del controllo positivo (1 nM di E2) deve essere almeno pari a 4 volte quella dell'attività media del VC su ciascuna piastra. La scelta di questo criterio poggia sull'attendibilità dei valori degli endpoint derivanti dallo studio di validazione (storicamente aumenti di induzione tra 4 e 30 volte).
|
|
18.
|
Per quanto riguarda il controllo della qualità della prova, l'aumento di induzione corrispondente al valore PC10 nel controllo positivo concomitante (1 nM di E2) deve essere superiore a 1 + 2 SD del valore iniziale di induzione (= 1) del controllo con mezzo disperdente concomitante. Ai fini della definizione delle priorità, il valore PC10 può essere utile per semplificare l'analisi dei dati richiesta rispetto a un'analisi statistica. Sebbene fornisca informazioni sulla significatività statistica, un'analisi statistica non costituisce un parametro quantitativo per quanto riguarda il potenziale basato sulle concentrazioni, ed è pertanto meno utile ai fini della definizione delle priorità.
|
Criteri di qualità per la prova sugli antagonisti degli ER
|
19.
|
L'attività della luciferasi media del controllo che produce il picco (25 nM di E2) deve essere almeno pari a 4 volte quella dell'attività media del VC per ciascuna piastra. La scelta di questo criterio poggia sull'attendibilità dei valori degli endpoint derivanti dallo studio di validazione.
|
|
20.
|
Per quanto riguarda il controllo di qualità del metodo di prova, l'attivazione trascrizionale relativa (RTA) di 1 nM di E2 deve superare il 100 %, l'RTA di 1μM di 4-idrossitamoxifene (OHT) deve essere inferiore a 40,6 % e l'RTA di 100 μM di digitonina (Dig) inferiore a 0 %.
Dimostrazione della competenza del laboratorio (cfr. il paragrafo 14 e le tabelle 3 e 4 nella sezione «COMPONENTI DEL METODO DI PROVA DI LEGAME ER TA» di questo metodo di prova).
|
Mezzo disperdente
|
21.
|
Il dimetilsolfossido (DMSO), o un solvente idoneo, è utilizzato come controllo con mezzo disperdente concomitante alla stessa concentrazione usata per i diversi controlli positivi e negativi e per le sostanze chimiche in esame. Le sostanze chimiche in esame vanno disciolte in un solvente capace di solubilizzarle che sia miscibile con il mezzo di coltura cellulare. L'acqua, l'etanolo (purezza tra il 95 % e il 100 %) e il DMSO sono mezzi disperdenti idonei. Se si usa il DMSO, il livello non deve superare lo 0,1 % (v/v). Per ogni mezzo disperdente va dimostrato che il volume massimo utilizzato non è citotossico e non interferisce con le prestazioni della prova.
|
Preparazione della sostanza chimica di prova
|
22.
|
In generale, le sostanze chimiche in esame vanno sciolte in DMSO o in altro solvente idoneo; tale preparazione è quindi frazionata in serie di soluzioni identiche diluite con lo stesso solvente in rapporto 1:10. Tali soluzioni sono destinate alla diluizione con i terreni di coltura.
|
Solubilità e citotossicità: considerazioni sulla prova preliminare di determinazione delle concentrazioni
|
23.
|
Occorre effettuare una prova preliminare per determinare l'adeguato intervallo (range-finding) delle concentrazioni della sostanza chimica da testare e verificare se la sostanza chimica in esame può presentare eventuali problemi di solubilità e di citotossicità. In un primo tempo le sostanze chimiche sono testate fino alla concentrazione massima di 1 μl/ml, 1 mg/ml o 1 mM (si scelga il valore più basso). Sulla base del grado di citotossicità o della mancanza di solubilità osservato nella prova preliminare, la prima batteria di prove definitiva deve testare la sostanza chimica a diluizioni in serie logaritmica, partendo dalla massima concentrazione accettabile (ad esempio 1 mM, 100 μM, 10 μM, ecc.); va riportata la presenza di intorbidimento o precipitato o citotossicità. Le prove sono ripetute una seconda e, se necessario, una terza volta adeguando le concentrazioni per caratterizzare meglio la curva concentrazione-risposta ed evitare le concentrazioni alle quali la sostanza chimica è risultata insolubile o eccessivamente citotossica.
|
|
24.
|
Per quanto riguarda gli agonisti e gli antagonisti degli ER, l'interpretazione dei dati tiene conto dell'influenza di livelli crescenti di citotossicità che possono alterare in modo significativo o eliminare la risposta sigmoide tipica. Vanno applicati metodi di prova della citotossicità in grado di fornire informazioni sulla vitalità cellulare dell'80 %, mediante un apposito test basato sull'esperienza del laboratorio.
|
|
25.
|
Se i risultati della prova di citotossicità indicano che la concentrazione della sostanza chimica in esame ha ridotto del 20 % o più il numero di cellule, occorre considerare citotossica tale concentrazione e escludere dalla valutazione tutte le concentrazioni pari o superiori a tale soglia.
|
Esposizione alla sostanza chimica di prova e configurazione delle piastre di prova
|
26.
|
La procedura per le diluizioni delle sostanze chimiche (fasi 1 e 2) e l'esposizione alle cellule (fase 3) possono essere eseguite come segue:
|
Fase-1: Diluire in serie ciascuna sostanza chimica in esame in DMSO, o in un solvente idoneo; ciascuna diluizione è versata nei pozzetti di una piastra da microtitolazione per ottenere le serie di concentrazioni finali, come stabilito precedentemente nella prova di determinazione dell'intervallo di concentrazioni (generalmente in una gamma che comprende, ad esempio, mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM, and 10 pM (10-3-10-11 M)] per le prove in triplicato.
Fase-2: diluizione della sostanza chimica: in primo luogo diluire 1,5 μl della sostanza chimica in esame nel solvente a un volume di 500 μl di mezzo di coltura.
Fase-3: esposizione delle cellule alla sostanza chimica: aggiungere 50 μl della diluizione nel mezzo di coltura (preparato nella fase 2) in un pozzetto di prova contenente 104 cellule/ 100 μl/pozzetto.
Il volume finale di mezzo di coltura raccomandato in ciascun pozzetto è di 150 μl. I campioni di prova e gli standard di riferimento possono essere ripartiti come indicato nella tabella 3 e nella tabella 4.
Tabella 3
Esempio di ripartizione delle concentrazioni degli standard di riferimento nella piastra di prova in una prova sugli agonisti degli ER
|
Riga
|
17α-metiltestosterone
|
Corticosterone
|
17α-estradiolo
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc 1 (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc 2 (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc 3 (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc 4 (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc 5 (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc 6 (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc 7 (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: controllo con mezzo disperdente (0,1 % di DMSO) PC: controllo positivo (1 nM di E2)
|
|
27.
|
Gli standard di riferimento (E2, 17-α-estradiolo, 17α-metiltestosterone e corticosterone) sono inclusi in ciascuna prova (tabella 3) Ciascuna piastra di prova (tabella 4) include anche pozzetti contenenti il controllo positivo, trattati con 1 nM di E2 che può provocare un'induzione massima di E2, e pozzetti contenenti il controllo con mezzo disperdente, trattati con il DMSO (o solvente idoneo) solo. Se sono utilizzate cellule provenienti da fonti differenti (ad esempio numero di passaggi diversi, lotti diversi, ecc.), gli standard di riferimento vanno testati per ciascuna fonte cellulare.
|
Tabella 4
VC: controllo con mezzo disperdente (0,1 % di DMSO) PC: controllo positivo (1 nM di E2)
|
Riga
|
Sost. in esame 1
|
Sost. in esame 2
|
Sost. in esame 3
|
Sost. in esame 4
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
conc 1 (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
conc 2 (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
conc 3 (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
conc 4 (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
conc 5 (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
conc 6 (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
conc 7 (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
Esempio di ripartizione delle concentrazioni delle sostanze chimiche in esame e degli standard di controllo nella piastra di prova in una prova sugli agonisti degli ER
|
Tabella 5
Esempio di ripartizione delle concentrazioni degli standard di riferimento nella piastra di prova in una prova sugli antagonisti degli ER
|
28.
|
Per valutare l'attività antagonista delle sostanze chimiche occorre addizionare i pozzetti di prova situati nelle righe da A a G con 25 pM di E2. Gli standard di riferimento (tamoxifene e flutamide) vanno testati in ciascuna batteria di prove. Ciascuna piastra di prova include anche pozzetti contenenti il controllo positivo (PC), trattati con 1 nM di E2 che possono fungere da controllo della qualità della linea cellulare hERα-HeLa-9 903, pozzetti contenenti il controllo con mezzo disperdente (VC) trattati con DMSO (o con solvente idoneo), pozzetti contenenti 0,1 % di DMSO, trattati con DMSO aggiunto alla concentrazione di E2 addizionata e corrispondente al «controllo addizionato», pozzetti trattati con una concentrazione finale di 1 μM OHT e pozzetti trattati con 100 μM di Dig (tabella 5). La piastra di prova successiva deve seguire la medesima disposizione sulla piastra, senza i pozzetti contenenti gli standard di riferimento (tabella 6). Se sono utilizzate cellule provenienti da fonti differenti (ad esempio numero di passaggi diversi, lotti diversi, ecc.), gli standard di riferimento vanno testati per ciascuna fonte cellulare.
|
Tabella 6
Esempio di ripartizione delle concentrazioni delle sostanze chimiche in esame e degli standard di controllo nella piastra di prova nella prova sugli antagonisti degli ER
|
29.
|
La mancanza di effetti di bordo dovrebbe essere confermata, a seconda dei casi, e qualora esista il sospetto che tali effetti possano prodursi, occorre modificare la disposizione sulla piastra in modo da evitare che ciò si verifichi. Ad esempio, può essere seguita una disposizione sulla piastra che escluda l'utilizzo dei pozzetti situati sul bordo.
|
|
30.
|
Dopo l'aggiunta delle sostanze chimiche, le piastre di prova sono collocate in un incubatore a 5 % di CO2 a 37 ± 1 oC per 20-24 ore, al fine di indurre la sintesi di prodotti di geni reporter.
|
|
31.
|
Quando si tratta di composti ad elevata volatilità, vanno applicate considerazioni particolari. In questi casi i pozzetti di controllo adiacenti possono generare falsi positivi e ciò va considerato alla luce dei valori di controllo storici e previsti. Nei pochi casi in cui la volatilità può destare preoccupazione, si raccomanda l'utilizzo di dispositivi di chiusura «ermetica» per le piastre in modo da isolare efficacemente i singoli pozzetti durante la sperimentazione.
|
|
32.
|
Per garantire l'indipendenza dei risultati occorre ripetere le prove definitive per la stessa sostanza chimica in giorni diversi.
|
Prova della luciferasi
|
33.
|
Per questa prova può essere utilizzato un reagente commerciale [ad esempio Steady-Glo® Luciferase Assay System (Promega, E2510, o equivalente)] o un sistema di prova della luciferasi standard (ad es. Promega, E1500, o equivalente), a condizione che i criteri di accettabilità siano soddisfatti. I reagenti di prova vanno selezionati in base alla sensibilità del luminometro utilizzato. Nel caso di un sistema di prova della luciferasi standard, va utilizzato un reagente per lisi cellulare (ad es. Promega, E1531, o equivalente) prima di aggiungere il substrato. Il reagente luciferasi deve essere applicato seguendo le istruzioni fornite dal fabbricante.
|
ANALISI DEI DATI
Prova sugli agonisti degli ER
|
34.
|
Nel caso di una prova sugli agonisti degli ER, l'attività trascrizionale relativa al controllo positivo (1 nM di E2) è ottenuta mediante analisi dei segnali luminescenti della medesima piastra conformemente alle seguenti fasi (sono anche accettabili altri processi matematici equivalenti):
|
Fase 1. Calcolare il valore medio corrispondente al VC.
Fase 2. Sottrarre il valore medio corrispondente al VC dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati.
Fase 3. Calcolare la media dei valori normalizzati per il PC.
Fase 4. Dividere il valore normalizzato ottenuto per ciascun pozzetto della piastra per la media dei valori normalizzati per il PC (PC = 100 %).
Il valore finale di ciascun pozzetto corrisponde all'attività trascrizionale relativa di tale pozzetto rispetto alla risposta corrispondente al PC.
Fase 5. Calcolare il valore medio dell'attività trascrizionale relativa per ciascun gruppo di concentrazione della sostanza chimica in esame. La risposta comporta due dimensioni: l'attività trascrizionale media (risposta) e la concentrazione che induce tale risposta (cfr. la sezione successiva).
Determinazione delle induzioni EC50, PC50 e PC10
|
35.
|
Il calcolo della EC50 si basa su una curva completa concentrazione-risposta, che non è sempre realizzabile o pratica a causa di eventuali limiti dell'intervallo delle concentrazioni di prova (ad esempio, in caso di problemi di citotossicità o di solubilità). Tuttavia, poiché la EC50 e il livello massimo di induzione (corrispondente al valore superiore dell'equazione di Hill) sono parametri informativi, essi devono essere indicati ove possibile. Il calcolo della EC50 e del livello massimo di induzione si effettua mediante software statistico adeguato (ad es. Graphpad Prism). Se l'equazione logistica di Hill si applica ai dati concentrazione-risposta, la EC50 va calcolata mediante la seguente equazione (7):
Y = base + (vertice - base) / (1 + 10 exp ((log EC50 - X) x pendenza di Hill)), dove:
X è il logaritmo della concentrazione; e
Y è la risposta, misurata tra la base (bottom) e il vertice (top) della curva sigmoide. La base è stabilita a zero nell'equazione logistica di Hill.
|
|
36.
|
I seguenti dati sono forniti per ciascuna sostanza chimica in esame:
l'RPCMax, che è il livello massimo di risposta indotto dalla sostanza chimica in esame, espresso in percentuale della risposta indotta da 1 nM di E2 sulla stessa piastra, nonché il PCMax (concentrazione corrispondente all'RPCMax); e
per le sostanze chimiche positive, le concentrazioni che corrispondono al PC10 e, se del caso, al PC50.
|
|
37.
|
Il valore PCx può essere calcolato mediante interpolazione tra 2 punti della curva X-Y, uno posto immediatamente sopra e l'altro immediatamente sotto al valore PCx cercato. Quando i punti che si trovano immediatamente al di sopra e al di sotto del valore PCx hanno, rispettivamente, le coordinate (a,b) e (c,d), il valore PCx può essere calcolato con la seguente equazione:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
La descrizione dei valori relativi al PC è rappresentata nel grafico 1.
Grafico 1
Esempio di deduzione dei valori relativi al PC. Il PC (1 nM di E2) è incluso in ciascuna piastra di prova
|
Prova sugli antagonisti degli ER
|
39.
|
Nel caso di una prova sugli antagonisti degli ER, l'attività trascrizionale relativa (RTA) al controllo spike-in (25 pM di E2) è ottenuta mediante analisi dei segnali luminescenti della stessa piastra conformemente alle seguenti fasi (sono anche accettabili altri processi matematici equivalenti):
Fase 1. Calcolare il valore medio corrispondente al VC.
Fase 2. Sottrarre il valore medio corrispondente al VC dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati. Fase 3. Calcolare la media dei valori normalizzati per il controllo spike-in.
Fase 4. Dividere il valore normalizzato ottenuto per ciascun pozzetto della piastra per la media dei valori normalizzati per il controllo spike-in (controllo spike-in = 100 %).
Il valore finale di ciascun pozzetto corrisponde all'attività trascrizionale relativa di tale pozzetto rispetto alla risposta del controllo spike-in.
Fase 5. Calcolare il valore medio dell'attività trascrizionale relativa per ciascun gruppo di concentrazione.
|
Determinazione delle induzioni IC30 e IC50
|
40.
|
Per le sostanze chimiche positive, sono fornite le concentrazioni che corrispondono al valore IC30 e, se del caso, all'IC50.
|
|
41.
|
Il valore ICx può essere calcolato mediante interpolazione tra 2 punti della curva X-Y, uno posto immediatamente sopra e l'altro immediatamente sotto al valore ICx cercato. Quando i punti che si trovano immediatamente al di sopra e al di sotto del valore ICx hanno, rispettivamente, le coordinate (c,d) e (a,b), il valore ICx può essere calcolato con la seguente equazione:
lin ICx = a-(b-(100-x)) (a-c) /(b-d)
Grafico 2
Esempio di deduzione dei valori relativi all'IC. Il controllo spike-in (25 pM di E2) è incluso in ciascuna piastra di prova
RTA: attività trascrizionale relativa
|
|
42.
|
I risultati dovrebbero basarsi su due (o tre) batterie di prove indipendenti. Se due batterie di prove forniscono risultati comparabili e pertanto riproducibili, non è necessario effettuare una terza batteria di prove. Per essere accettabili, i risultati devono:
|
—
|
soddisfare i criteri di accettabilità (cfr. i criteri di accettabilità, paragrafi da 14 a 20);
|
|
Criteri di interpretazione dei dati
Tabella 7
Criteri di interpretazione dei risultati positivi e negativi per il metodo di prova sugli agonisti degli ER
|
Positivo
|
Se il RPCMax ottenuto è pari o superiore al 10 % della risposta del controllo positivo in due batterie di prove su due oppure in almeno due batterie di prove su tre.
|
|
Negativo
|
Se il RPCMax ottenuto rimane inferiore al 10 % della risposta del controllo positivo in due batterie di prove su due oppure in almeno due batterie di prove su tre.
|
Tabella 8
Criteri di interpretazione dei risultati positivi e negativi per il metodo di prova sugli antagonisti degli ER
|
Positivo
|
Se il valore IC30 è calcolato in due batterie di prove su due oppure in almeno due batterie di prove su tre.
|
|
Negativo
|
Se non si può calcolare il valore IC30 in due batterie di prove su due oppure in almeno due batterie di prove su tre.
|
|
43.
|
I criteri di interpretazione dei dati sono indicati nelle tabelle 7 e 8. I risultati positivi saranno caratterizzati sia dall'ordine di grandezza dell'effetto sia dalla concentrazione corrispondente a tale effetto. L'espressione dei risultati sotto forma di concentrazione che induce una risposta pari al 50 % (PC50) o al 10 % (PC10) delle risposte ottenute con il PC nella prova sugli agonisti e un'inibizione del 50 % (IC50) o del 30 % (IC30) del valore del controllo spike-in nella prova sugli antagonisti permette di soddisfare entrambi questi obiettivi. Tuttavia, una sostanza chimica in esame è considerata positiva se la risposta massima indotta dalla sostanza chimica in esame (RPCMax) è uguale o superiore al 10 % della risposta del PC in due batterie di prove su due o in almeno due batterie di prove su tre, mentre una sostanza chimica in esame è considerata negativa se la RPCMax non raggiunge almeno il 10 % della risposta del controllo positivo in due batterie di prove su due o in almeno due batterie di prove su tre.
|
|
44.
|
I valori di PC10, PC50 e PCMax nella prova sugli agonisti degli ER e di IC30 e IC50 nella prova sugli antagonisti degli ER possono essere ottenuti mediante un foglio di calcolo disponibile con la Linea guida sul sito internet pubblico dell'OCSE (64).
|
|
45.
|
Due prove ripetute dovrebbero essere sufficienti per determinare i valori PC10 o PC50 e IC30 o IC50. Tuttavia, se il livello di riferimento dei dati nello stesso intervallo di concentrazioni mostra un coefficiente di variazione (VC; %) inaccettabilmente elevato, i dati possono essere considerati non affidabili e occorre individuare la fonte dell'elevata variabilità. Il VC dei dati grezzi triplicati (ossia i dati sull'intensità della luminescenza) corrispondenti ai punti di dati utilizzati per il calcolo del PC10 deve essere inferiore al 20 %.
|
|
46.
|
Il rispetto dei criteri di accettabilità indica che il sistema di prova funziona correttamente, ma non garantisce che qualsiasi prova produrrà dati accurati. Duplicare i risultati della prima batteria di prove è la migliore garanzia che i dati generati sono corretti.
|
|
47.
|
Nel caso della prova sugli agonisti degli ER, quando sono necessarie informazioni supplementari in aggiunta alle finalità di screening e di definizione delle priorità della presente linea guida concernente le sostanze chimiche in esame positive, in particolare le sostanze chimiche che vanno da PC10 a PC49, così come le sostanze chimiche di cui si sospetta che inducano una sovrastimolazione della luciferasi, una conferma che l'attività luciferasi osservata è dovuta esclusivamente alla risposta mediata da ERα può essere ottenuta utilizzando un antagonista ERα (cfr. l'appendice 2.1).
|
RELAZIONE SULL'ESECUZIONE DELLA PROVA
|
48.
|
Cfr. il paragrafo 20 della sezione "ELEMENTI DEL METODO DI PROVA DEL METODO ER TA").
|
BIBLIOGRAFIA
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1 459-1 469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4 252-4 263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
Appendice 2.1
FALSI POSITIVI: VALUTAZIONE DEI SEGNALI DI LUMINESCENZA NON MEDIATI DAI RECETTORI
|
1.
|
I falsi positivi nella prova sugli agonisti degli ER possono essere generati dall'attivazione del gene luciferasi non mediata da ER, o dall'attivazione diretta di un prodotto del gene o da una fluorescenza la cui origine è indeterminata. Tali effetti sono segnalati da una curva dose-risposta incompleta o insolita. Se tali effetti sono sospetti, va esaminato l'effetto di un antagonista dell'ER (ad esempio, 4-idrossitamoxifene [OHT] a una concentrazione non tossica) sulla risposta. L'antagonista puro ICI 1827 80 potrebbe non essere adatto a questo scopo perché una concentrazione sufficiente di ICI 1827 80 può diminuire il valore corrispondente al VC, alterando l'analisi dei dati.
|
|
2.
|
Per garantire la validità di questo approccio occorre testare nella stessa piastra gli elementi seguenti:
|
—
|
attività agonista della sostanza chimica sconosciuta in presenza / in assenza di 10 μM di OHT;
|
|
—
|
1 nM di E2 (in triplicato) come controllo positivo agonista;
|
|
—
|
1 nM di E2 + OHT (in triplicato).
|
|
Criteri di interpretazione dei dati
Nota: tutti i pozzetti devono contenere la stessa concentrazione di mezzo disperdente.
|
—
|
Se l'attività agonista della sostanza chimica sconosciuta NON è modificata dal trattamento con l'antagonista degli ER, il risultato è considerato «negativo».
|
|
—
|
Se l'attività agonista della sostanza chimica sconosciuta è completamente inibita, si applicano i criteri di interpretazione.
|
|
—
|
Se l'attività agonista alla concentrazione più bassa è pari o superiore alla risposta del PC10, la sostanza sconosciuta è inibita in misura uguale o superiore alla risposta del PC10. Calcolare la differenza tra le risposte relative ai pozzetti che hanno ricevuto l'antagonista degli ER e quelli che non l'hanno ricevuto; tale differenza è quindi considerata la vera risposta e va utilizzata per il calcolo dei parametri appropriati per consentire una decisione sulla classificazione della sostanza chimica.
|
Analisi dei dati
Verificare lo standard di prestazione.
Verificare il VC tra i pozzetti trattati alle medesime condizioni.
|
1.
|
Calcolare il valore medio corrispondente al VC;
|
|
2.
|
sottrarre il valore medio del VC dal valore ottenuto per ciascun pozzetto non trattato con OHT;
|
|
3.
|
calcolare il valore medio corrispondente a OHT;
|
|
4.
|
sottrarre il valore medio del VC dal valore ottenuto per ciascun pozzetto trattato con OHT;
|
|
5.
|
calcolare il valore medio ottenuto con il controllo positivo;
|
|
6.
|
calcolare l'attività trascrizionale relativa di tutti gli altri pozzetti rispetto al controllo positivo.
|
Appendice 2.2
PREPARAZIONE DEL SIERO TRATTATO CON CARBONE RIVESTITO DI DESTRANO (DCC)
|
1.
|
Il trattamento del siero con il carbone rivestito di destrano (DCC) è un metodo generico per eliminare i composti estrogenici dal siero aggiunto al terreno di coltura cellulare, al fine di escludere dalla risposta qualsiasi distorsione (bias) causata da residui estrogenici nel siero. Questa procedura consente di trattare 500 ml di siero fetale bovino (FBS).
|
Componenti
|
2.
|
Sono necessari i seguenti materiali e attrezzature:
|
Materiali
|
Cloruro di magnesio esaidrato (MgCl2· 6H2O)
|
|
1 M di soluzione tampone HEPES (pH 7,4)
|
|
Acqua ultra pura ottenuta per filtrazione
|
Attrezzature
Recipiente di vetro sterilizzato in autoclave (le dimensioni devono essere adattate alle necessità) Centrifuga classica da laboratorio (con temperatura regolabile a 4 °C)
Procedura
|
3.
|
La seguente procedura è adeguata per l'uso di provette per centrifuga da 50 ml:
[Giorno 1] Preparare una sospensione di carbone-destrano in 1 litro di acqua ultra pura contenente 1,5 mM di MgCl2, 0,25 M di saccarosio, 2,5 g di carbone, 0,25 g di destrano e 5 mM di HEPES e agitare alla temperatura di 4 °C tutta la notte.
[Giorno 2] Distribuire la sospensione nelle provette per centrifuga da 50 ml e centrifugare a 10 000 giri al minuto a 4 °C per 10 minuti. Rimuovere il supernatante e mettere da parte la metà del sedimento di carbone a 4 °C per utilizzarlo il Giorno 3. Sospendere l'altra metà del carbone con FBS che sarà stato scongelato lentamente per evitare la formazione di precipitato, quindi inattivarla termicamente a 56 °C per 30 minuti e trasferirla in un recipiente di vetro sterilizzato in autoclave, ad esempio un matraccio di Erlenmeyer. Agitare la sospensione moderatamente a 4 °C, per tutta la notte.
[Giorno 3] Distribuire la sospensione con FBS nelle provette per centrifuga da 50 ml e centrifugare a 10 000 giri al minuto a 4 °C per 10 minuti. Raccogliere il FBS e trasferirlo nel nuovo sedimento di carbone che è stato preparato e conservato il Giorno 2. Disperdere il sedimento di carbone nel FBS ed agitare delicatamente la sospensione in un recipiente in vetro sterilizzato, alla temperatura di 4 °C, per tutta la notte.
[Giorno 4] Distribuire la sospensione per una centrifugazione a 10 000 giri al minuto a 4 °C per 10 minuti; sterilizzare il supernatante per filtrazione con filtro sterile di 0,2 μm. Questo FBS trattato al DCC è conservato a -20 °C e può essere utilizzato per un anno al massimo.
|
Appendice 3
METODO DI PROVA DI TRANSATTIVAZIONE CHE UTILIZZA IL RECETTORE ESTROGENICO VM7LUC PER INDIVIDUARE GLI AGONISTI E GLI ANTAGONISTI DEGLI ER
CONSIDERAZIONI INIZIALI E LIMITI (CFR. ANCHE INTRODUZIONE GENERALE)
|
1.
|
Il presente metodo di prova utilizza la linea cellulare VM7Lu4E2 (65). È stato validato dal National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) e dal comitato di coordinamento inter-agenzie per la validazione dei metodi alternativi (ICCVAM) (1). Le linee cellulari VM7Luc esprimono, in modo endogeno, prevalentemente gli ERα e una minima quantità di ERβ (2) (3) (4).
|
|
2.
|
Il presente metodo di prova è applicabile a una vasta gamma di sostanze, a condizione che possano essere disciolte in dimetilsulfossido (DMSO; CASRN 67-68-5), non reagiscano con il DMSO né con il terreno di coltura cellulare e non siano citotossiche alle concentrazioni testate. Se non è possibile utilizzare il DMSO, si può usare un altro mezzo disperdente, come l'etanolo o l'acqua (cfr. il paragrafo 12). Le dimostrazioni del funzionamento del metodo di prova del VM7 Luc ER TA sugli (ant)agonisti indicano che i dati generati possono fornire informazioni sui meccanismi d'azione mediati dagli ER e possono essere presi in considerazione per determinare quali sostanze sottoporre a ulteriori prove in via prioritaria.
|
|
3.
|
Il presente metodo di prova è concepito in modo specifico per individuare la transattivazione mediata da hERαα e hERß utilizzando la chemiluminescenza come endpoint. I bio-saggi utilizzano diffusamente la chemiluminescenza, poiché la luminescenza ha un elevato rapporto segnale/rumore di fondo. Tuttavia, l'attività della luciferasi di lucciola può essere perturbata dalle sostanze che inibiscono l'enzima luciferasi, il che provoca un'inibizione manifesta o un aumento della luminescenza dovuta alla stabilizzazione della proteina. Inoltre, in alcune prove che utilizzano il gene reporter luciferasi attivato dagli ER, sono stati osservati segnali di luminescenza non mediati dai recettori per concentrazioni di fitoestrogeni superiori a 1 μM, a motivo della sovrattivazione del gene reporter della luciferasi (9) (11). Mentre la curva dose-risposta indica che la reale attivazione del sistema ER avviene a concentrazioni inferiori, l'espressione della luciferasi ottenuta a concentrazioni elevate di fitoestrogeni o di composti simili sospettati di indurre una sovrattivazione del gene reporter della luciferasi mediante un meccanismo analogo a quello dei fitoestrogeni deve essere esaminata con attenzione nei sistemi di prova di ER TA trasfettati in modo stabile.
|
|
4.
|
Le sezioni "INTRODUZIONE GENERALE" e "COMPONENTI DEL METODO DI PROVA ER TA" vanno lette prima di applicare il presente metodo di prova per fini regolamentari. Le definizioni e le abbreviazioni utilizzate nel presente metodo di prova figurano nell'appendice 1.
|
PRINCIPIO DEL METODO DI PROVA (CFR. ANCHE INTRODUZIONE GENERALE)
|
5.
|
Il metodo di prova serve ad indicare la formazione di un legame tra l'ER e il ligando, seguita dalla traslocazione del complesso recettore-ligando costituito verso il nucleo. Nel nucleo, il complesso recettore-ligando si lega a specifici elementi di risposta del DNA e transattiva il gene reporter (luc), che induce la produzione della luciferasi e la successiva emissione luminosa, che può essere quantificata con un luminometro. L'attività della luciferasi può essere valutata in modo rapido e poco costoso grazie ai numerosi kit di analisi disponibili in commercio. Il metodo di prova VM7Luc ER TA utilizza una linea cellulare resistente di adenocarcinoma del seno di origine umana che esprime gli ER, che è stata trasfettata in modo stabile con un costrutto del gene reporter della luciferasi di lucciola (luc) regolata da quattro elementi di risposta agli estrogeni inseriti a monte del promotore del virus delle neoplasie mammarie (MMTV) dei topi, al fine di individuare le sostanze che presentano un'attività estrogenica agonista o antagonista in vitro. Il promotore del MMTV mostra soltanto una debole reattività incrociata con altri ormoni steroidei e non steroidei (8). I criteri d'interpretazione dei dati sono descritti in dettaglio al paragrafo 41. In sintesi, una risposta positiva è caratterizzata da una curva concentrazione-risposta contenente almeno tre punti le cui barre di errore (media ± SD) non si sovrappongono, nonché da una variazione dell'ampiezza (unità di luce relativa normalizzata [RLU]) pari ad almeno il 20 % del valore massimo dello standard di riferimento (17β-estradiolo [E2; n. CAS 50-28-2] per la prova sugli agonisti, raloxifene HCl [Ral; n. CAS 84 449-90-1]/E2 per la prova sugli antagonisti).
|
PROCEDURA
Linea cellulare
|
6.
|
Per il presente metodo di prova si utilizza la linea cellulare VM7Luc4E2 trasfettata in modo stabile. Questa linea cellulare può essere ottenuta soltanto mediante sottoscrizione di un accordo di licenza tecnica rilasciato dall'Università di California, Davis, California, USA (66), e da Xenobiotic Detection Systems Inc., Durham, North Carolina, Stati Uniti (67).
|
Stabilità della linea cellulare
|
7.
|
Per preservare la stabilità e l'integrità della linea cellulare, le cellule coltivate sono sottoposte a più di un passaggio tra lo stock congelato e il mezzo di conservazione (cfr. il paragrafo 9). Le cellule non sono più coltivate oltre i 30 passaggi. Per la linea cellulare VM7Lu4E2, ci vorranno circa tre mesi per effettuare 30 passaggi.
|
Condizioni di coltura e di piastratura delle cellule
|
8.
|
Per garantire la qualità di tutti i materiali e metodi e mantenere l'integrità, la validità e la riproducibilità delle prove effettuate, occorre seguire le procedure indicate nel documento Guidance on Good Cell Culture Pract (5) (6).
|
|
9.
|
Le cellule VM7Lu4E2 sono conservate nel mezzo RPMI 1 640 integrato con 0,9 % di Pen-Strep e 8,0 % di siero fetale bovino (FBS) in un apposito incubatore per la coltura tessutale a 37 oC ± 1 oC, con un'umidità di 90 % ± 5 % e un'atmosfera contenente 5,0 % ± 1 % di CO2.
|
|
10.
|
Quando la confluenza raggiunge ~ 80 %, le cellule VM7Lu4E2 sono replicate e condizionate in un mezzo senza estrogeni per 48 ore. Esse sono disposte su piastre da 96 pozzetti per essere esposte alle sostanze chimiche in esame e per l'analisi dell'induzione estrogena dipendente dall'attività della luciferasi. Il mezzo senza estrogeni (EFM) contiene il mezzo di Eagle modificato da Dulbecco (DMEM) senza rosso fenolo, integrato da 4,5 % di FBS trattato con carbone-destrano, 1,9 % di L-glutammina e 0,9 % di Pen-Strep. Tutti i materiali in plastica devono essere esenti da attività estrogeniche [cfr. il protocollo dettagliato (7)].
|
Criteri di accettabilità
|
11.
|
L'accettazione o il rigetto di una prova si basa sulla valutazione dei risultati ottenuti con gli standard di riferimento e i controlli di ciascun esperimento condotto su una piastra a 96 pozzetti. Ciascuno standard di riferimento è testato a diverse concentrazioni e la prova prevede multiple repliche di ciascuna concentrazione di riferimento e di controllo. I risultati sono quindi confrontati con i controlli di qualità (QC) per questi parametri, che sono derivati dalle basi di dati storici per gli effetti agonisti e antagonisti generati da ciascun laboratorio durante la dimostrazione del livello di competenza. Le basi di dati storiche sono costantemente aggiornate con i valori degli standard di riferimento e dei controlli. Qualsiasi cambiamento nell'attrezzatura o nelle condizioni di laboratorio può richiedere lo sviluppo di basi di dati storiche aggiornate.
|
Prova dell'effetto agonista
Prova di determinazione dell'intervallo delle concentrazioni (prova di range finding)
|
•
|
Induzione: si misura l'effetto indotto sulla piastra dividendo il valore medio delle unità relative di luce (RLU) massime ottenute con lo standard di riferimento E2 per il valore medio di RLU del controllo DMSO. Si osserva generalmente un'induzione moltiplicata per cinque, ma ai fini dell'accettazione della prova l'induzione deve essere uguale o superiore a quattro volte.
|
|
•
|
Risultati del controllo DMSO: i valori di RLU del controllo con solvente devono situarsi entro 2,5 volte la deviazione standard del valore medio storico delle RLU del controllo con solvente.
|
|
•
|
Se uno di questi criteri non è soddisfatto, l'esperimento deve essere invalidato e ripetuto.
|
Prova completa
La prova è soggetta ai medesimi criteri di accettabilità applicabili alla prova di determinazione dell'intervallo di concentrazioni per gli effetti agonisti, ai quali si aggiungono:
|
•
|
i risultati dello standard di riferimento: la curva concentrazione-risposta dello standard di riferimento E2 corrisponde a un sigmoide e comporta almeno tre valori nella porzione lineare di detta curva.
|
|
•
|
i risultati del controllo positivo: i valori di RLU del controllo metossicloro devono essere superiori alla media di DMSO più tre volte la deviazione standard di tale media.
|
|
•
|
Se uno qualsiasi di questi criteri non è soddisfatto, l'esperimento deve essere invalidato e ripetuto.
|
Prova dell'effetto antagonista
Prova di determinazione dell'intervallo delle concentrazioni (prova di range finding)
|
•
|
Inibizione: si misura l'effetto inibitorio sulla piastra dividendo il valore medio delle unità relative di luce (RLU) massime ottenute con lo standard di riferimento Ral/E2 per il valore medio di RLU del controllo DMSO. Si osserva generalmente un'inibizione moltiplicata pari a cinque volte il valore del controllo, ma ai fini dell'accettazione della prova l'inibizione deve essere uguale o superiore a tre volte.
|
|
•
|
risultati del controllo E2: i valori di RLU del controllo E2 devono situarsi entro 2,5 volte la deviazione standard del valore medio storico delle RLU del controllo E2.
|
|
•
|
Risultati del controllo DMSO: i valori di RLU del controllo DMSO devono situarsi entro 2,5 volte la deviazione standard del valore medio storico delle RLU del controllo con solvente.
|
|
•
|
Se uno qualsiasi di questi criteri non è soddisfatto, l'esperimento deve essere invalidato e ripetuto.
|
Prova completa
La prova è soggetta ai medesimi criteri di accettabilità applicabili alla prova di definizione dell'intervallo per gli effetti antagonisti, ai quali si aggiungono:
|
•
|
i risultati dello standard di riferimento: la curva concentrazione-risposta dello standard di riferimento Ral/E2 corrisponde a un sigmoide e comporta almeno tre valori nella porzione lineare di detta curva.
|
|
•
|
i risultati del controllo positivo: i valori di RLU del controllo tamoxifene/E2 devono essere inferiori alla media del controllo E2 meno tre volte la deviazione standard di tale media.
|
|
•
|
Se uno qualsiasi di questi criteri non è soddisfatto, l'esperimento deve essere invalidato e ripetuto.
|
Standard di riferimento, controlli positivi e controlli con mezzo disperdente
Controllo con mezzo disperdente (prove per l'effetto agonista e antagonista)
|
12.
|
Il mezzo disperdente usato per sciogliere le sostanze chimiche in esame deve essere testato come controllo con mezzo disperdente. Il mezzo disperdente utilizzato durante la validazione della prova VM7Luc ER TA era l'1 % (v/v) di dimetilsolfossido (DMSO, n. CAS 67-68-5) (cfr. il paragrafo 24). Se si usa un mezzo disperdente diverso dal DMSO, tutti gli standard di riferimento, i controlli e le sostanze chimiche in esame devono, se del caso, essere testati nel medesimo mezzo disperdente.
|
Standard di riferimento (Range finding per l'effetto agonista)
|
13.
|
Lo standard di riferimento è l'E2 (n. CAS 50-28-2). Per la prova di range finding, lo standard di riferimento E2 è sottoposto a una serie di 4 diluizioni (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 e 2,87 x 10-12 M), con ciascuna concentrazione testata in due pozzetti.
|
Standard di riferimento (prova completa per l'effetto agonista)
|
14.
|
L'E2 per la prova completa è soggetto a una serie di diluizioni successive di 1:2 fino a ottenere 11 concentrazioni (che vanno da 3,67 x 10-10 a 3,59 x 10-13 M), con ciascuna concentrazione testata in due pozzetti.
|
Standard di riferimento (Range finding per l'effetto antagonista)
|
15.
|
Lo standard di riferimento è una combinazione di Ral (n. CAS 84 449-90-1) e E2 (n. CAS 50-28-2). Per la prova di range finding, la miscela Ral/E2 è sottoposta a una serie di diluizioni fino a ottenere tre concentrazioni di Ral (3,06 x 10-9, 7,67 x 10-10, e 1,92 x 10-10M) più una concentrazione costante di E2 (9,18 × 10-11 M) con ciascuna diluizione testata in due pozzetti.
|
Standard di riferimento (prova completa per l'effetto antagonista)
|
16.
|
Per la prova completa, la miscela Ral/E2 è sottoposta a una serie di diluizioni successive di 1:2 di Ral (che vanno da 2,45x 10-8 a 9,57 x 10-11M) più una concentrazione costante di E2 (9,18 × 10-11 M) per ottenere nove concentrazioni di Ral/E2, ciascuna testata in due pozzetti.
|
Controllo debolmente positivo (agonista)
|
17.
|
Il controllo debolmente positivo debole è una soluzione di 9,06 x 10-6 M p,p’-metossicloro (metossicloro; n. CAS 72-43-5) in EFM.
|
Controllo debolmente positivo (antagonista)
|
18.
|
Il controllo debolmente positivo è una soluzione di tamoxifene (n. CAS 10 540-29-1) a 3,36 x 10-6 M e di E2 a 9,18 × 10-11 M in EFM.
|
Controllo E2 (solo prova dell'effetto antagonista)
|
19.
|
Il controllo E2 è una soluzione di 9,18 × 10-11 M in EFM ed è usato come controllo negativo di riferimento.
|
Aumento dell'induzione (effetto agonista)
|
20.
|
L'induzione dell'attività della luciferasi nello standard di riferimento (E2) è misurata dividendo il valore medio di RLU più elevate ottenute per l'E2 con il valore medio di RLU corrispondente al controllo di DMSO; il risultato deve essere almeno quattro volte maggiore.
|
Aumento dell'inibizione (effetto antagonista)
|
21.
|
L'inibizione media dell'attività della luciferasi osservata nello standard di riferimento (Ral/E2) è misurata dividendo il valore medio di RLU più elevate ottenute per la miscela Ral/E2 con il valore medio di RLU corrispondente al controllo di DMSO; il risultato deve essere almeno tre volte maggiore.
Dimostrazione della competenza del laboratorio (cfr. il paragrafo 14 e le tabelle 3 e 4 nei « COMPONENTI DEL METODO DI PROVA ER TA» di questo metodo di prova).
|
Mezzo disperdente
|
22.
|
Le sostanze chimiche in esame vanno disciolte in un solvente capace di solubilizzare la sostanza chimica in esame e miscibile con il mezzo di coltura cellulare. L'acqua, l'etanolo (purezza tra il 95 % e il 100 %) e il DMSO sono mezzi disperdenti idonei. Se si usa il DMSO, il livello non deve superare l'1 % (v/v). Per ogni mezzo disperdente va dimostrato che il volume massimo utilizzato non è citotossico e non interferisce con le prestazioni del metodo di prova. Gli standard di riferimento e i controlli sono disciolti in un solvente del 100 % e poi diluiti fino a raggiungere le opportune concentrazioni in EFM.
|
Preparazione delle sostanze chimiche in esame
|
23.
|
Le sostanze chimiche in esame sono disciolte in 100 % di DMSO (o solvente idoneo) e poi diluite fino a raggiungere le opportune concentrazioni in EFM. Prima di essere disciolte e diluite, tutte le sostanze chimiche in esame sono portate a temperatura ambiente. Per ciascuna prova vanno preparate fresche soluzioni della sostanza chimica in esame. Non devono presentare né precipitato visibile né intorbidimento. Le soluzioni primarie degli standard di riferimento e dei controlli possono essere preparati in anticipo; ma le soluzioni e le diluizioni finali dello standard di riferimento, dei controlli e delle sostanze chimiche in esame devono essere preparate e utilizzate entro 24 ore prima di ciascuna prova.
|
Solubilità e citotossicità: Considerazioni ai fini della determinazione preliminare delle concentrazioni
|
24.
|
La prova preliminare di range finding consiste in sette diluizioni successive di rapporto 1:10 in duplicato. Inizialmente le sostanze chimiche in esame sono testate fino alla concentrazione massima di 1 mg/ml (~ 1 mM) per le prove per l'effetto agonista e 20 μg/ml (~ 10μ M) per le prove per l'effetto antagonista. Le prove di range finding permettono di determinare quanto segue:
|
|
—
|
le concentrazioni di partenza della sostanza chimica in esame da utilizzare durante le prove complete;
|
|
—
|
le diluizioni della sostanza chimica in esame da utilizzare durante le prove complete.
|
|
25.
|
La valutazione della vitalità/citotossicità cellulare è inclusa nei protocolli di prova per l'effetto agonista e antagonista (7), sia nelle prove di range finding sia nelle prove complete. La prova di citotossicità utilizzata per valutare la vitalità cellulare durante la validazione del metodo VM7Luc ER TA (1) era fondato su un metodo di osservazione visiva qualitativa per gradi; ma può essere utilizzato un metodo quantitativo per il medesimo scopo (cfr. il protocollo (7)). I dati relativi alle concentrazioni della sostanza chimica in esame che causano una riduzione della vitalità cellulare superiore al 20 % non possono essere utilizzati.
|
Esposizione alla sostanza chimica in esame e configurazione della piastra di prova
|
26.
|
Le cellule sono contate e depositate in piastre di coltura tessutale a 96 pozzetti (2 x 105 cellule per pozzetto) in EFM e incubate per 24 ore in modo da consentire alle cellule di aderire alla piastra. L'EFM è quindi eliminato e sostituito con le soluzioni di sostanze di riferimento e di sostanze chimiche in esame in EFM e la piastra è incubata per 19-24 ore. Particolare considerazione deve essere prestata alle sostanze altamente volatili in quanto possono generare falsi positivi quando si trovano accanto a pozzetti contenenti un controllo. In tali casi, si raccomanda l'utilizzo di dispositivi di chiusura «ermetica» per le piastre in modo da isolare efficacemente i singoli pozzetti durante la sperimentazione.
|
Prove di determinazione dell'intervallo delle concentrazioni (range finding)
|
27.
|
Per le prove di range finding si utilizzano tutti i pozzetti della piastra a 96 pozzetti per testare fino a sei sostanze chimiche in esame in sette concentrazioni diverse ottenute con diluizioni successive in rapporto 1:10, valutate in duplicato (cfr. Grafici 1 e 2).
|
|
—
|
La prova di range finding per l'effetto agonista utilizza quattro concentrazioni di E2 in duplicato come standard di riferimento e quattro repliche del controllo DMSO.
|
|
—
|
La prova di range finding per l'effetto antagonista utilizza tre concentrazioni della miscela Ral/E2 (in cui l'E2 rimane costante a 9,18 × 10-11 M) in duplicato, come standard di riferimento, e tre pozzetti assegnati rispettivamente ai controlli con E2 e DMSO.
|
Figura 1
Configurazione della piastra a 96 pozzetti per la prova di range finding per l'effetto agonista
Abbreviazioni: da E2-1 a E2-4 = concentrazioni dello standard di riferimento E2 (in ordine decrescente); da TC1-1 a TC1-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 1 (TC1); da TC2-1 a TC2-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 2 (TC2); da TC3-1 a TC3-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 3 (TC3); da TC4-1 a TC4-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 4 (TC4); da TC5-1 a TC5-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 5 (TC5); da TC6-1 a TC6-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 6 (TC6); VC = controllo con mezzo disperdente (DMSO a 1 % v/v EFM).
Figura 2
Configurazione della piastra a 96 pozzetti per la prova di range finding per l'effetto antagonista
Abbreviazioni: E2 = controllo E2; da Ral-1 a Ral-3 = concentrazioni dello standard di riferimento Raloxifene/E2 (in ordine decrescente); da TC1-1 a TC1-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 1 (TC1); da TC2-1 a TC2-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 2 (TC2); da TC3-1 a TC3-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 3 (TC3); da TC4-1 a TC4-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 4 (TC4); da TC5-1 a TC5-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 5 (TC5); da TC6-1 a TC6-7 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 6 (TC6); VC = controllo con mezzo disperdente (DMSO a 1 % v/v EFM).
Nota: Tutte le sostanze chimiche in esame sono sottoposte a prova in presenza di 9,18 × 10 -11 M di E2.
|
28.
|
Il volume finale di mezzo raccomandato in ciascun pozzetto è di 200 μl. Utilizzare solo piastre di prova in cui le cellule in tutti i pozzetti diano una vitalità pari o superiore all'80 %.
|
|
29.
|
La determinazione delle concentrazioni iniziali per le prove complete per l'effetto
agonista
è descritta dettagliatamente nel protocollo relativo a tale effetto (7). In sintesi, si utilizzano i seguenti criteri:
|
|
—
|
Se non vi sono punti sulla curva di concentrazione della sostanza chimica in esame che sono superiori al valore medio del controllo del DMSO più tre volte la sua deviazione standard, la prova completa va eseguita con una serie di 11 diluizioni successive con rapporto 1:2, a partire dalla concentrazione di saturazione della sostanza chimica in esame.
|
|
—
|
Se vi sono punti sulla curva di concentrazione della sostanza chimica in esame che sono superiori al valore medio del controllo DMSO più tre volte la sua deviazione standard, la prova completa utilizza una serie di 11 diluizioni successive a partire da una concentrazione superiore di 1 log alla concentrazione che induce le RLU adattate più elevate nella prova di range finding. Tale serie di 11 diluizioni è effettuata in base a rapporti di diluizione 1:2 o 1:5 in funzione dei seguenti criteri:
|
si raccomanda di utilizzare una serie di diluizioni con rapporto 1:2 se la gamma di concentrazioni risultante permette di osservare l'insieme delle risposte previste sulla base della curva concentrazione-risposta ottenuta nella prova di range finding. Altrimenti utilizzare una diluizione con rapporto 1:5.
|
|
|
—
|
Se una sostanza chimica in esame presenta una curva di concentrazione-risposta bifasica nella prova di range finding, entrambe le fasi vanno studiate nella prova completa.
|
|
30.
|
La determinazione delle concentrazioni iniziali per le prove complete per l'effetto
antagonista
è descritta dettagliatamente nel protocollo relativo a tale effetto (7). In sintesi, si utilizzano i seguenti criteri:
|
|
—
|
Se non vi sono punti sulla curva di concentrazione della sostanza chimica in esame che sono inferiori al valore medio del controllo di E2 meno tre volte la sua deviazione standard, la prova completa va eseguita con una serie di 11 diluizioni successive con rapporto 1:2, a partire dalla concentrazione di saturazione della sostanza chimica in esame.
|
|
—
|
Se vi sono punti sulla curva di concentrazione della sostanza chimica in esame che sono inferiori al valore medio del controllo di E2 meno tre volte la sua deviazione standard, la prova completa utilizza una serie di 11 diluizioni successive a partire da una delle concentrazioni seguenti:
|
—
|
la concentrazione che dà il valore più basso (di RLU adattate) nella prova di range finding;
|
|
—
|
la concentrazione massima solubile (cfr. protocollo per l'effetto antagonista (7), grafico 14-2);
|
|
—
|
la concentrazione minima che induce un effetto citotossico (cfr. protocollo per l'effetto antagonista (7), grafico 14-3 per un esempio in materia);
|
|
|
—
|
Tale serie di 11 diluizioni è effettuata in base a rapporti di diluizione 1:2 o 1:5 in funzione dei seguenti criteri:
|
si raccomanda di utilizzare una serie di diluizioni con rapporto 1:2 se la gamma di concentrazioni risultante permette di osservare l'insieme delle risposte previste sulla base della curva concentrazione-risposta ottenuta nella prova di range finding. Altrimenti usare una diluizione 1:5.
|
|
Prove complete
|
31.
|
Le prove complete consistono in una serie di 11 diluizioni successive (con rapporto 1:2 o 1:5 in funzione della concentrazione iniziale selezionata in base ai criteri pertinenti), con ciascuna concentrazione testata in tre pozzetti della piastra a 96 pozzetti (cfr. Figure 3 e 4).
|
|
—
|
La prova completa per l'effetto agonista utilizza 11 concentrazioni di E2 in duplicato come standard di riferimento. Su ciascuna piastra, quattro pozzetti sono utilizzati come repliche per il controllo di DMSO e quattro pozzetti per il controllo metossicloro (9,06 x 10 -6 M).
|
|
—
|
La prova di range finding per l'effetto antagonista utilizza nove concentrazioni della miscela Ral/E2 in duplicato, come standard di riferimento, quattro repliche del controllo di E2 a 9,18 x 10-11 M, quattro repliche del controllo DMSO e quattro repliche del tamoxifene a 3.36 x 10-6M.
|
Figura 3
Configurazione della piastra a 96 pozzetti per la prova completa per l'effetto agonista
Abbreviazioni: da TC1-1 a TC1-11 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 1; da TC2-1 a TC2-11 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 2; da E2-1 a E2-11 = concentrazioni dello standard di riferimento E2 (in ordine decrescente); Meth = controllo debolmente positivo p, p’-metossicloro; VC = controllo con mezzo disperdente DMSO (1 % v/v) in EFM
Figura 4
Configurazione della piastra a 96 pozzetti per la prova completa per l'effetto antagonista

Abbreviazioni: E2 = controllo E2; da Ral-1 a Ral-9 = concentrazioni dello standard di riferimento Raloxifene/E2 (in ordine decrescente); Tam = controllo debolmente positivo Tamoxifene/E2; da TC1-1 a TC1-11 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 1 (TC1); da TC2-1 a TC2-11 = concentrazioni (in ordine decrescente) della sostanza chimica in esame 2 (TC2); VC = controllo con mezzo disperdente (DMSO a 1 % v/v EFM).
Nota: Come osservato, tutti i pozzetti contenenti lo standard di riferimento e la sostanza chimica in esame mostrano la medesima concentrazione di E2 (9,18 x 10-11 M)
|
32.
|
Per garantire l'indipendenza dei risultati occorre ripetere le prove complete per la stessa sostanza chimica in giorni diversi. È opportuno svolgere almeno due prove complete. Se i risultati delle prove si contraddicono (ad esempio, una delle prove è positiva, l'altra negativa), oppure se una delle prove è inadeguata, va effettuata una terza prova supplementare.
|
Misurazione della luminescenza
|
33.
|
La luminescenza è misurata nell'intervallo da 300 a 650 nm, mediante un luminometro dotato di un sistema di iniezione e un software che controlla il volume di iniezione e l'intervallo di misurazione (7). L'emissione luminosa osservata per ciascun pozzetto è espressa come RLU per pozzetto.
|
ANALISI DEI DATI
Determinazione della EC50 e della IC50
|
34.
|
Il valore EC50 (concentrazione della sostanza chimica in esame corrispondente alla metà della risposta massima [effetto agonista]) e il valore IC50 (concentrazione della sostanza chimica in esame corrispondente alla metà dell'inibizione massima [effetto antagonista]) sono determinati a partire dai dati della curva concentrazione-risposta. Per le sostanze chimiche in esame positive a una o più concentrazioni, la concentrazione della sostanza chimica in esame che provoca la metà della risposta massima (IC50 o EC50) è calcolata utilizzando una funzione di Hill o un altro metodo appropriato. La funzione di Hill è un modello matematico logistico a quattro parametri che associa la concentrazione della sostanza chimica in esame a una risposta (solitamente secondo una funzione sigmoide), utilizzando la seguente equazione:
|
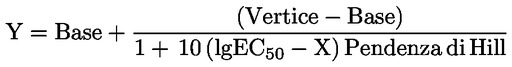
dove:
Y= risposta (in RLU);
X= logaritmo della concentrazione;
Base= risposta minima;
Vertice= risposta massima;
lg EC50 (o lg IC50)= logaritmo della concentrazione corrispondente alla risposta situata a metà strada tra la base e il vertice;
pendenza di Hill= pendenza (coefficiente angolare) della curva.
Il modello calcola i valori più vicini ai parametri vertice, base, pendenza di Hill e IC50 e EC50. Il calcolo dei valori EC50 e IC50 si effettua mediante software statistico adeguato (ad es. Graphpad PrismR).
Determinazione di valori anomali
|
35.
|
È possibile agevolare la qualità del giudizio statistico includendo (ma non limitato a) un test Q (cfr. i protocolli per gli effetti agonista e antagonista) (7) per determinare i pozzetti "inutilizzabili" che saranno esclusi dall'analisi dei dati.
|
|
36.
|
Per le repliche dello standard di riferimento E2 (campione composto da due pozzetti), il valore di RLU adattato di una replica a una data concentrazione di E2 è considerato un valore erratico se il suo valore è superiore o inferiore del 20 % al valore di RLU adattato per tale concentrazione nella banca dati storica.
|
Raccolta e adeguamento dei dati del luminometro per la prova di range finding
|
37.
|
I dati grezzi forniti dal luminometro sono trasferiti su un foglio di calcolo elaborato specificamente per il metodo di prova. Occorre determinare se vi siano punti con valori anomali che devono essere rimossi. (Cfr. Criteri di accettabilità della prova per i parametri ottenuti nelle analisi.) Sono effettuati i calcoli seguenti:
|
Effetto agonista
|
Fase 1
|
Calcolare il valore medio del controllo con mezzo disperdente DMSO (VC).
|
|
Fase 2
|
Sottrarre il valore medio del VC DMSO dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati.
|
|
Fase 3
|
Calcolare la moltiplicazione media dell'induzione osservata con lo standard di riferimento (E2).
|
|
Fase 4
|
Calcolare il valore medio della EC50 per la sostanza chimica in esame.
|
Effetto antagonista
|
Fase 1
|
Calcolare il valore medio di VC DMSO.
|
|
Fase 2
|
Sottrarre il valore medio del VC DMSO dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati.
|
|
Fase 3
|
Calcolare la moltiplicazione media dell'inibizione osservata con lo standard di riferimento (Ral/E2).
|
|
Fase 4
|
Calcolare il valore medio dello standard di riferimento E2.
|
|
Fase 5
|
Calcolare il valore medio della IC50 per le sostanze chimiche in esame.
|
Raccolta e adeguamento dei dati del luminometro per le prove complete
|
38.
|
I dati grezzi forniti dal luminometro sono trasferiti su un foglio di calcolo elaborato specificamente per il metodo di prova. Occorre determinare se vi siano punti con valori anomali che devono essere rimossi. (Cfr. Criteri di accettabilità della prova per i parametri ottenuti nelle analisi.) Sono effettuati i calcoli seguenti:
|
Effetto agonista
|
Fase 1
|
Calcolare il valore medio di VC DMSO.
|
|
Fase 2
|
Sottrarre il valore medio del VC DMSO dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati.
|
|
Fase 3
|
Calcolare la moltiplicazione media dell'induzione osservata con lo standard di riferimento (E2).
|
|
Fase 4
|
Calcolare il valore medio della EC50 per l'E2 e per la sostanza chimica in esame, rispettivamente.
|
|
Fase 5
|
Calcolare il valore medio in RLU adattate ottenuto con il metossicloro.
|
Effetto antagonista
|
Fase 1
|
Calcolare il valore medio di VC DMSO.
|
|
Fase 2
|
Sottrarre il valore medio del VC DMSO dal valore ottenuto per ciascun pozzetto al fine di normalizzare i dati.
|
|
Fase 3
|
Calcolare la moltiplicazione media dell'induzione osservata con lo standard di riferimento (Ral/E2).
|
|
Fase 4
|
Calcolare il valore medio della IC50 per la miscela Ral/E2 e per le sostanze chimiche in esame, rispettivamente.
|
|
Fase 5
|
Calcolare il valore medio in RLU adattato ottenuto con il tamoxifene.
|
|
Fase 6
|
Calcolare il valore medio dello standard di riferimento E2.
|
Criteri di interpretazione dei dati
|
39.
|
Il metodo VM7Luc ER TA è inteso come parte di un approccio basato sulla forza probante dei dati per contribuire a stabilire quali sostanze testare in via prioritaria mediante la prova in vivo dell'effetto di perturbazione del sistema endocrino. Una parte di tale procedura di definizione delle priorità consisterà nella classificazione della sostanza chimica in esame come positiva o negativa per l'attività agonista o antagonista sugli ER. I criteri di interpretazione dei risultati positivi e negativi utilizzati nell'ambito dello studio di validazione della prova VM7Luc ER TA sono descritti nella tabella 1.
|
Tabella 1
Criteri di interpretazione dei risultati positivi e negativi
|
|
|
ATTIVITÀ AGONISTA
|
|
Positivo
|
|
—
|
Tutte le sostanze chimiche in esame classificate come positive all'attività agonista sugli ER presentano una curva concentrazione-risposta che consiste in una linea di base, seguita da una pendenza positiva e che si conclude con un plateau o un picco. In alcuni casi possono essere definite solo due di queste caratteristiche (pendenza-linea di base o pendenza-picco).
|
|
—
|
La linea che definisce la pendenza positiva comprende almeno tre punti le cui barre di errore non si sovrappongono (media ± SD). I punti che costituiscono la linea di base sono esclusi, ma la parte lineare della curva può comprendere il picco o il primo punto del plateau.
|
|
—
|
Per essere classificata positiva occorre che la sostanza chimica induca una risposa la cui ampiezza - la differenza tra la linea di base e il picco - sia almeno superiore o uguale al 20 % del valore massimo indotto dallo standard di riferimento E2 (cioè almeno 2 000 RLU se il valore massimo indotto dallo standard di riferimento [E2] è adattato a 10 000 RLU).
|
|
—
|
Nella misura del possibile, calcolare il valore EC50 per ciascuna sostanza chimica positiva.
|
|
|
Negativo
|
Il valore medio adattato di RLU ottenuto per una data concentrazione è uguale o inferiore al valore medio di RLU del controllo DMSO più tre volte la deviazione standard di RLU del DMSO.
|
|
Equivoco
|
I dati che non possono essere interpretati come validi per stabilire una eventuale attività a causa di importanti limiti qualitativi o quantitativi sono considerati equivoci e non possono servire per determinare se la sostanza chimica in esame è positiva o negativa. In tal caso, occorre testare nuovamente le sostanze chimiche in esame.
|
|
|
|
ATTIVITÀ ANTAGONISTA
|
|
Positivo
|
|
—
|
I dati relativi alla sostanza chimica in esame producono una curva concentrazione-risposta che consiste in una linea di base, seguita da una retta con pendenza negativa.
|
|
—
|
La retta che definisce la pendenza negativa comprende almeno tre punti le cui barre di errore non si sovrappongono; i punti che costituiscono la linea di base sono esclusi, ma la parte lineare della curva può comprendere il picco o il primo punto del plateau.
|
|
—
|
Si osserva una riduzione di attività di almeno il 20 % rispetto alla risposta massima ottenuta con lo standard di riferimento Ral/E2 (cioè 8 000 RLU o meno se la risposta massima dello standard di riferimento [Ral/E2] è adattata a 10 000 RLU).
|
|
—
|
Le concentrazioni non citotossiche più elevate della sostanza chimica in esame sono inferiori o uguali a 1x10 -5M.
|
|
—
|
Nella misura del possibile, calcolare il valore IC50 per ciascuna sostanza chimica positiva.
|
|
|
Negativo
|
Tutti i punti di dati sono superiori al valore EC80 (80 % della risposta di E2 o 8 000 RLU) a concentrazioni inferiori a 1,0 x 10-5M.
|
|
Equivoco
|
I dati che non possono essere interpretati come validi per stabilire una eventuale attività a causa di importanti limiti qualitativi o quantitativi sono considerati equivoci e non possono servire per determinare se la sostanza chimica in esame è positiva o negativa. In tal caso, occorre testare nuovamente la sostanza chimica in esame.
|
|
40.
|
I risultati positivi saranno caratterizzati sia dall'ampiezza dell'effetto sia dalla concentrazione corrispondente a tale effetto, se possibile. I grafici 5 e 6 mostrano esempi di dati positivi, negativi e equivoci.
|
Grafico 5
Esempi di risultati positivi, negativi e equivoci per l'effetto agonista
La linea tratteggiata indica il 20 % della risposta di E2, cioè 2 000 RLU (valore adattato e normalizzato).
Grafico 6
Esempi di risultati positivi, negativi e equivoci per l'effetto antagonista
La linea tratteggiata indica l'80 % della risposta di Ral/E2, cioè 8 000 RLU (valore adattato e normalizzato).
La linea continua indica 1,00 x 10-5 M. Per essere considerata positiva, la risposta deve essere inferiore alla linea delle 8 000 RLU, e a concentrazioni inferiori a 1,00 x 10-5 M.
Le concentrazioni contrassegnate da un asterisco nel grafico del meso-esadestrolo indicano un punteggio di vitalità di «2» o più.
I risultati delle prove per il meso-esestrolo sono considerati equivoci poiché l'unica risposta inferiore a 8 000 RLU corrisponde a una concentrazione di 1,00 x 10-5 M.
|
41.
|
I calcoli dei valori EC50 e IC50 possono essere effettuati con la funzione di Hill a quattro parametri (cfr. i protocolli per gli effetti agonista e antagonista per maggiori dettagli (7)). Il rispetto dei criteri di accettabilità indica che il sistema funziona correttamente, ma non garantisce che qualsiasi prova produrrà dati accurati. Duplicare i risultati della prima batteria di prove è la migliore garanzia che i dati generati sono accurati (cfr. il paragrafo 19 della sezione "ELEMENTI DEL METODO DI PROVA DEL METODO ER TA").
|
RELAZIONE SULL'ESECUZIONE DELLA PROVA
|
42.
|
Cfr. il paragrafo 20 della sezione "ELEMENTI DEL METODO DI PROVA DEL METODO ER TA").
|
BIBLIOGRAFIA
|
(1)
|
ICCVAM: (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In Vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In Vitro Mol. Toxicol.,13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology,139(10):4 252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In Vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In Vitro & Molec. Toxicol. 13, 67-82.
|
Appendice 4
Metodo di prova di transattivazione mediante il recettore estrogenico alfa umano trasfettato in modo stabile per individuare l'attività agonista e antagonista delle sostanze chimiche utilizzando la linea cellulare erα calux
CONSIDERAZIONI INIZIALI E LIMITI (CFR. ANCHE INTRODUZIONE GENERALE)
|
1.
|
Il metodo di prova di transattivazione ERα CALUX utilizza la linea cellulare umana U2OS per individuare l'attività estrogenica agonista e antagonista mediata dal recettore estrogenico alfa umano (hERα). Lo studio di validazione del biosaggio che utilizza la linea cellulare ERα CALUX trasfettata in maniera stabile condotto da BioDetection Systems BV (Amsterdam, Paesi Bassi) ha dimostrato la pertinenza e l'affidabilità della prova ai fini previsti (1). La linea cellulare ERα CALUX esprime unicamente il recettore estrogenico ERα umano trasfettato in modo stabile (2) (3).
|
|
2.
|
Il presente metodo di prova è concepito in modo specifico per individuare la transattivazione mediata da hERα misurando la bioluminescenza come endpoint. La bioluminescenza è comunemente usata nei biosaggi a motivo dell'elevato rapporto segnale/rumore (4).
|
|
3.
|
È stato riportato che le concentrazioni di fitoestrogeni superiori a 1 μM inducono una sovrattivazione del gene reporter della luciferasi, che genera segnali di luminescenza non mediati dal recettore (5) (6) (7). Pertanto, le concentrazioni più elevate di fitoestrogeni o di altri composti simili che possono sovrattivare l'espressione della luciferasi devono essere esaminate con attenzione nelle prove di transattivazione che utilizzano il recettore estrogenico trasfettato in modo stabile (cfr. l'appendice 2).
|
|
4.
|
Le sezioni "INTRODUZIONE GENERALE" e "COMPONENTI DEL METODO DI PROVA ER TA" vanno lette prima di applicare il presente metodo di prova per fini regolamentari. Le definizioni e le abbreviazioni utilizzate nel presente metodo di prova figurano nell'appendice 1.
|
PRINCIPIO DEL METODO DI PROVA (CFR. ANCHE INTRODUZIONE GENERALE)
|
5.
|
Il biosaggio serve a valutare la formazione di un legame tra l'ER e il ligando, e la successiva traslocazione del complesso recettore-ligando costituitosi verso il nucleo. Nel nucleo, il complesso recettore-ligando si fissa a elementi di risposta specifici del DNA e transattiva il gene reporter della luciferasi di lucciola, inducendo una maggiore espressione cellulare dell'enzima luciferasi. Dopo l'aggiunta, il substrato della luciferasi (luciferina) viene trasformato in un prodotto bioluminescente. La luce prodotta può essere facilmente individuata e quantificata mediante luminometro.
|
|
6.
|
Il sistema di prova utilizza cellule ERα CALUX trasfettate in modo stabile. Le cellule ERα CALUX derivano dalla linea delle cellule umane U2OS osteoblastiche di osteosarcoma. Le cellule umane U2OS sono state trasfettate in modo stabile con 3xHRE-TATA-Luc e pSG5-neo-hERα, mediante il metodo di co-precipitazione del fosfato di calcio. La linea cellulare U2OS è stata selezionata come migliore candidato per fungere da linea cellulare reporter reattiva agli estrogeni (e ad altri ormoni steroidei), poiché presenta un'attività debole o nulla del recettore endogeno. L'assenza di recettori endogeni è stata valutata utilizzando esclusivamente plasmidi reporter della luciferasi, i quali non hanno manifestato alcuna attività quando sono stati aggiunti i ligandi del recettore. Inoltre, questa linea cellulare ha sostenuto forti risposte mediate dagli ormoni quando sono stati introdotti in modo transitorio i recettori apparentati (2) (3) (8).
|
|
7.
|
La sperimentazione di sostanze chimiche ad attività estrogenica o antiestrogenica mediante la linea cellulare ERα CALUX include una prova di preselezione e batterie di prove complete. Durante la prova di preselezione, sono determinati la solubilità, la citotossicità e l'intervallo di concentrazione affinato delle sostanze chimiche in esame ai fini delle prove complete. Durante le batterie di prove complete, gli intervalli di concentrazione affinati delle sostanze chimiche in esame sono testati nei biosaggi che utilizzano la linea cellulare ERα CALUX; successivamente, viene stabilita la classificazione delle sostanze chimiche in esame per la loro attività agonista o antagonista.
|
|
8.
|
I criteri d'interpretazione dei dati sono descritti in dettaglio al paragrafo 59. In sintesi, una sostanza chimica in esame è considerata positiva per l'attività agonista se almeno due concentrazioni consecutive della sostanza chimica in esame mostrano una risposta uguale o superiore al 10 % della risposta massima dello standard di riferimento 17β-estradiolo (PC10). Una sostanza chimica in esame è considerata positiva per l'attività antagonista se almeno due concentrazioni consecutive della sostanza chimica in esame mostrano una risposta pari o inferiore all'80 % della risposta massima dello standard di riferimento tamoxifene (PC80).
|
PROCEDURA
Linee cellulari
|
9.
|
Per il presente metodo di prova si utilizza la linea cellulare U2OS ERα CALUX trasfettata in modo stabile. La linea cellulare può essere ottenuta da BioDetection Systems BV, Amsterdam, Paesi Bassi, sottoscrivendo un accordo di licenza tecnica.
|
|
10.
|
Vanno utilizzate soltanto colture cellulari prive di micoplasma. I lotti di cellule utilizzati devono essere certificati negativi per la contaminazione da micoplasmi; in alternativa, prima dell'uso deve essere effettuato un test di individuazione di micoplasmi. La RT-PCR (reazione a catena della polimerasi in tempo reale) è utilizzata ai fini della individuazione sensibile di infezioni da micoplasma (4) (5) (9).
|
Stabilità della linea cellulare
|
11.
|
Per mantenerne la stabilità e l'integrità, le cellule CALUX vanno conservate in azoto liquido (-80 °C). Dopo lo scongelamento di cellule per avviare una nuova cultura, le cellule sono poste in coltura secondaria almeno due volte prima di essere utilizzate per la valutazione dell'attività estrogenica agonista e antagonista delle sostanze chimiche. Le cellule non sono più coltivate oltre i 30 passaggi.
|
|
12.
|
Per monitorare la stabilità della linea cellulare nel tempo, la reattività del sistema di prova agonista e antagonista deve essere verificata mediante la valutazione di EC50 o IC50 dello standard di riferimento. Occorre inoltre monitorare l'induzione relativa del campione di controllo positivo (PC) e del campione di controllo negativo (NC). I risultati devono essere conformi ai criteri di accettazione per la prova ERα CALUX per quanto concerne l'attività agonista (tabella 3C) o antagonista (tabella 4C). Gli standard di riferimento, i controlli positivi e quelli negativi sono indicati nella tabella 1 (attività agonista) e nella tabella 2 (attività antagonista).
|
Condizioni di piastratura e di coltura cellulare
|
13.
|
Le cellule U2OS sono coltivate nel terreno di coltura [DMEM/F12 (1:1) con rosso fenolo come indicatore del pH, integrato da siero fetale bovino (7,5 %), amminoacidi non essenziali (1 %), 10 unità/ml di penicillina, streptomicina e geneticina (G-418) come marcatore di selezione]. Le cellule sono collocate in un incubatore di CO2 (5 % di CO2) a 37 oC e al 100 % di umidità. Quando raggiungono una confluenza dell'85-95 %, le cellule sono poste in coltura secondaria oppure preparate per l'inseminazione in piastre da microtitolazione a 96 pozzetti. In questo secondo caso, le cellule sono rimesse in sospensione alla concentrazione di 1x105 cellule/ml nel mezzo di prova privo di estrogeni [DMEM/F12 (1:1) senza rosso fenolo, integrato da siero fetale bovino trattato con carbone-destrano (5 % v/v), amminoacidi non essenziali (1 % v/v), 10 unità/ml di penicillina e streptomicina] e collocate in piastre da microtitolazione a 96 pozzetti (100 μl di sospensione cellulare omogeneizzata). Le cellule sono quindi preincubate in un incubatore di CO2 (5 % CO2, 37 oC, 100 % umidità) per 24 ore prima dell'esposizione. I materiali in plastica devono essere privi di estrogeni.
|
Criteri di accettabilità
|
14.
|
Le attività agoniste e antagoniste della sostanza o delle sostanze in esame sono testate in serie. Una serie di prove è composta da un massimo di 6 piastre da microtitolazione. Ciascuna serie di prove contiene almeno 1 serie completa di diluizioni di uno standard di riferimento, di un campione di controllo positivo, di un campione di controllo negativo e di controlli contenenti solvente. Le figure 1 e 2 illustrano la configurazione della piastra per la serie di prove agoniste e antagoniste.
|
|
15.
|
Ciascuna diluizione degli standard di riferimento, delle sostanze chimiche in esame, di tutti i controlli con solvente e dei controlli positivi e negativi è analizzata in triplicato. Ciascuna delle analisi in triplicato deve soddisfare i requisiti indicati nelle tabelle 3A e 4A.
|
|
16.
|
Una serie completa di diluizioni dello standard di riferimento (17β-estradiolo per l'attività agonista; tamoxifene per l'attività antagonista) è misurata sulla prima piastra in ciascuna serie di prove. Per poter confrontare i risultati delle analisi delle restanti 5 piastre da microtitolazione con la prima piastra da microtitolazione contenente la curva completa concentrazione-risposta dello standard di riferimento, tutte le piastre devono contenere 3 campioni di controllo: il controllo con solvente, la concentrazione più elevata dello standard di riferimento testato e la concentrazione di valore EC50 (attività agonista) o IC50 (attività antagonista) approssimativa dello standard di riferimento. Il rapporto tra i campioni di controllo medi della prima piastra e le restanti 5 piastre deve essere conforme ai requisiti di cui alla tabella 3C (attività agonista) o alla tabella 4C (attività antagonista).
|
|
17.
|
Per ciascuna delle piastre da microtitolazione all'interno di una serie di prove è calcolato il fattore z (10). Il fattore z va calcolato utilizzando le risposte corrispondenti alla concentrazione più alta e più bassa dello standard di riferimento. Una piastra da microtitolazione è considerata valida se soddisfa i requisiti di cui alla tabella 3C (attività agonista) o alla tabella 4C (attività antagonista).
|
|
18.
|
Lo standard di riferimento deve dar luogo a una curva sigmoide di dose-risposta. Il valore EC50 o IC50 derivato dalla risposta della serie di diluizioni dello standard di riferimento deve soddisfare i requisiti indicati nella tabella 3C (attività agonista) o nella tabella 4C (attività antagonista).
|
|
19.
|
Ciascuna serie di prove deve contenere un campione di controllo positivo e un campione di controllo negativo. L'induzione relativa calcolata sia sulla base del campione di controllo positivo che di quello negativo deve soddisfare i requisiti indicati nella tabella 3C (attività agonista) o nella tabella 4C (attività antagonista).
|
|
20.
|
Durante tutte le misurazioni, il fattore di induzione della concentrazione più alta dello standard di riferimento è misurato dividendo la più elevata risposta media dello standard di riferimento 17β-estradiolo in unità di luce relative (RLU) per la risposta media del controllo con solvente in RLU. Questo fattore di induzione dovrebbe soddisfare i requisiti minimi relativi al fattore moltiplicativo di incremento dell'induzione come indicato nella tabella 3C (attività agonista) o nella tabella 4C (attività antagonista).
|
|
21.
|
Solo le piastre da microtitolazione che soddisfano tutti i criteri sopraindicati sono considerate valide e possono essere usate per valutare la risposta delle sostanze chimiche in esame.
|
|
22.
|
I criteri di accettazione sono applicabili sia alla prova di preselezione che alle batterie di prove complete.
|
Tabella 1
Concentrazioni di standard di riferimento, controllo positivo (PC) e controllo negativo (NC) per la prova CALUX di individuazione dell'attività agonista
|
|
Sostanza
|
Numero CAS
|
Intervallo di prova (M)
|
|
Standard di riferimento
|
17β-estradiolo
|
50-28-2
|
1*10-13 - 1*10-10
|
|
Controllo positivo (PC)
|
17α-metiltestosterone
|
58-18-4
|
3*10-6
|
|
Controllo negativo (NC)
|
corticosterone
|
50-22-6
|
1*10-8
|
Tabella 2
Concentrazioni di standard di riferimento, controllo positivo (PC) e controllo negativo (NC) per la prova CALUX di individuazione dell'attività antagonista
|
|
Sostanza
|
Numero CAS
|
Intervallo di prova (M)
|
|
Standard di riferimento
|
tamoxifene
|
10540-29-1
|
3*10-9 - 1*10-5
|
|
Controllo positivo (PC)
|
4-idrossitamoxifen
|
68047-06-3
|
1*10-9
|
|
Controllo negativo (NC)
|
resveratrolo
|
501-36-0
|
1*10-5
|
Tabella 3
Criteri di accettazione per la prova ERα CALUX di individuazione dell'attività agonista
|
A - campioni individuali su una piastra
|
Criterio
|
|
1
|
% massima di SD dei pozzetti in triplicato (per NC, PC, ciascuna diluizione della sostanza chimica in esame e lo standard di riferimento, tranne C0)
|
< 15 %
|
|
2
|
% massima di SD dei pozzetti in triplicato (per lo standard di riferimento e i controlli con solvente della sostanza chimica in esame (C0, SC))
|
< 30 %
|
|
3
|
Perdita massima di LDH, come misura della citotossicità.
|
< 120 %
|
|
B - all'interno di una singola piastra da microtitolazione
|
|
|
4
|
Rapporto tra il controllo con solvente dello standard di riferimento (C0; piastra 1) e il controllo con solvente della sostanza chimica in esame (SC; piastre da 2 a x)
|
0,5 - 2,0
|
|
5
|
Rapporto tra il valore approssimativo di EC50 e delle concentrazioni più elevate dello standard di riferimento sulla piastra 1 e il valore approssimativo di EC50 e delle concentrazioni più elevate dello standard di riferimento sulle piastre da 2 a x (C4, C8)
|
0,70 - 1,30
|
|
6
|
Fattore Z per ciascuna piastra
|
> 0,6
|
|
C - per una singola serie di analisi (tutte le piastre di una stessa serie)
|
|
|
7
|
Curva sigmoide dello standard di riferimento
|
Sì (17ß-estradiolo)
|
|
8
|
Intervallo di EC50 dello standard di riferimento 17ß-estradiolo
|
4*10-12 – 4*10-11 M
|
|
9
|
Fattore moltiplicatore minimo dell'incremento dell'induzione della concentrazione più elevata di 17ß-estradiolo, rispetto al controllo con solvente dello standard di riferimento.
|
5
|
|
10
|
Induzione relativa (%) PC.
|
> 30 %
|
|
11
|
Induzione relativa (%) NC.
|
< 10 %
|
Appr.: approssimativo; PC: controllo positivo; NC: controllo negativo: SC: controllo con solvente della sostanza chimica in esame; C0: controllo con solvente dello standard di riferimento; SD: deviazione standard; LDH: lattato deidrogenasi
Tabella 4
Criteri di accettazione per la prova ERα CALUX di individuazione dell'attività antagonista
|
A - campioni individuali su una piastra
|
Criterio
|
|
1
|
% massima di SD dei pozzetti in triplicato (per NC, PC, ciascuna diluizione della sostanza chimica in esame, lo standard di riferimento, il controllo con solvente (C0))
|
< 15 %
|
|
2
|
% massima di SD dei pozzetti in triplicato (per il controllo con mezzo disperdente (VC) e la concentrazione più elevata dello standard di riferimento (C8))
|
< 30 %
|
|
3
|
Perdita massima di LDH, come misura della citotossicità.
|
< 120 %
|
|
B - all'interno di una singola piastra da microtitolazione
|
|
|
4
|
Rapporto tra il controllo con solvente dello standard di riferimento (C0; piastra 1) e il controllo con solvente della sostanza chimica in esame (SC; piastre da 2 a x)
|
0,70 - 1,30
|
|
5
|
Rapporto tra il valore approssimativo di IC50 dello standard di riferimento sulla piastra 1 e il valore approssimativo di IC50 dello standard di riferimento sulle piastre da 2 a x (C4)
|
0,70 - 1,30
|
|
6
|
Rapporto tra le concentrazioni più elevate dello standard di riferimento sulla piastra 1 e le concentrazioni più elevate dello standard di riferimento sulle piastre da 2 a x (C8)
|
0,50 - 2,0
|
|
7
|
Fattore Z per ciascuna piastra
|
> 0,6
|
|
C - per una singola serie di analisi (tutte le piastre di una stessa serie)
|
|
|
8
|
Curva sigmoide dello standard di riferimento
|
Sì (Tamoxifene)
|
|
9
|
Intervallo di IC50 dello standard di riferimento (tamoxifene)
|
1*10-8 - 1*10-7 M
|
|
10
|
Fattore moltiplicatore minimo dell'incremento dell'induzione del controllo con solvente dello standard di riferimento, rispetto alla concentrazione più elevata di Tamoxifene.
|
2,5
|
|
11
|
Induzione relativa (%) PC.
|
< 70 %
|
|
12
|
Induzione relativa (%) NC.
|
> 85 %
|
Appr.: approssimativo; PC: controllo positivo; NC: controllo negativo: VC: controllo con mezzo disperdente (controllo con solvente senza concentrazione fissa dello standard di riferimento per l'attività agonista); SC: controllo con solvente della sostanza chimica in esame; C0: controllo con solvente dello standard di riferimento; SD: deviazione standard; LDH: lattato deidrogenasi
Controllo con solvente/mezzo disperdente, standard di riferimento, controlli positivi, controlli negativi
|
23.
|
I controlli con solvente/mezzo disperdente, standard di riferimento, controlli positivi, controlli negativi utilizzati sono identici sia per la prova di preselezione che per le prove complete. Inoltre, la concentrazione degli standard di riferimento, dei controlli positivi e dei controlli negativi deve essere identica.
|
Controllo con solvente
|
24.
|
Il solvente usato per sciogliere le sostanze chimiche in esame è testato come controllo con solvente. Il dimetilsolfossido (DMSO, 1 % (v/v); n. CAS 67-68-5) è stato utilizzato come mezzo disperdente durante la validazione del biosaggio ERα CALUX. Se si usa un solvente diverso dal DMSO, tutti gli standard di riferimento, i controlli e le sostanze chimiche in esame devono essere testati nel medesimo mezzo disperdente. Si osservi che, per gli studi di attività antagonista, il controllo con solvente contiene una concentrazione fissa dello standard di riferimento per l'attività agonista, il 17β-estradiolo (approssimativamente la concentrazione EC50). Per testare il solvente utilizzato per gli studi sull'attività antagonista, occorre preparare e testare un controllo con mezzo disperdente.
|
Controllo con mezzo disperdente (antagonismo)
|
25.
|
Per la sperimentazione sull'attività antagonista il mezzo di prova è integrato da una concentrazione fissa dello standard di riferimento per l'attività agonista il 17β-estradiolo (approssimativamente la concentrazione EC50). Per testare il solvente usato per sciogliere le sostanze chimiche in esame per l'attività antagonista, occorre preparare un mezzo che non contiene una concentrazione fissa dello standard di riferimento per l'attività agonista (17β-estradiolo). Questo campione di controllo è indicato come controllo con mezzo disperdente. Il dimetilsolfossido (DMSO, 1 % (v/v); n. CAS 67-68-5) è stato utilizzato come mezzo disperdente durante la validazione del biosaggio ERα CALUX. Se si usa un solvente diverso dal DMSO, tutti gli standard di riferimento, i controlli e le sostanze chimiche in esame devono essere testati nel medesimo mezzo disperdente.
|
Standard di riferimento
|
26.
|
Lo standard di riferimento per l'attività agonista è il 17β-estradiolo (tabella 1). Gli standard di riferimento includono una serie di diluizioni di otto concentrazioni di 17β-estradiolo (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Lo standard di riferimento per l'attività antagonista è il tamoxifene (tabella 2). Gli standard di riferimento includono una serie di diluizioni di otto concentrazioni di tamoxifene (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Ciascuna delle concentrazioni dello standard di riferimento per l'attività antagonista è co-incubata con una concentrazione fissa dello standard di riferimento agonista 17β-estradiolo (3*10-12 M).
|
Controllo positivo
|
28.
|
Il controllo positivo per gli studi di attività agonista è il 17-α-metiltestosterone (tabella 1).
|
|
29.
|
Il controllo positivo per gli studi di attività antagonista è il 4-idrossitamoxifene (tabella 2). Il controllo positivo per l'attività antagonista è co-incubato con una concentrazione fissa dello standard di riferimento agonista 17β-estradiolo (3*10-12 M).
|
Controllo negativo
|
30.
|
Il controllo negativo per gli studi di attività agonista è il corticosterone (tabella 1).
|
|
31.
|
Il controllo negativo per gli studi di attività antagonista è il resveratrolo (tabella 2). Il controllo negativo per l'attività antagonista è co-incubato con una concentrazione fissa dello standard di riferimento agonista 17β-estradiolo (3*10-12 M).
|
Dimostrazione della competenza del laboratorio (cfr. il paragrafo 14 e le tabelle 3 e 4 nei «COMPONENTI DEL METODO DI PROVA ER TA» di questo metodo di prova).
Mezzo disperdente
|
32.
|
Il solvente utilizzato per sciogliere le sostanze chimiche in esame devono solubilizzare completamente la sostanza chimica in esame e risultare miscibile con il mezzo cellulare. Il DMSO, l'acqua e l'etanolo (purezza 95-100 %) sono solventi appropriati. Se si usa il DMSO come solvente, la concentrazione massima di DMSO durante l'incubazione non deve superare 1 % (v/v). Prima dell'uso, il solvente deve essere testato per verificare l'assenza di citotossicità e di interferenza con le prestazioni delle prove.
|
Preparazione di standard di riferimento, controlli positivi, controlli negativi e sostanze chimiche in esame
|
33.
|
Gli standard di riferimento, i controlli positivi, i controlli negativi e le sostanze chimiche in esame sono dissolti in 100 % di DMSO (o un solvente appropriato). Diluizioni (in serie) appropriate sono quindi preparate nel medesimo solvente. Prima dello scioglimento, tutte le sostanze sono lasciate riposare fino a raggiungere la temperatura ambiente. Le soluzioni primarie, preparate recentemente, degli standard di riferimento, dei controlli positivi, dei controlli negativi e delle sostanze chimiche in esame non devono presentare precipitato né intorbidimento visibili. Le soluzioni primarie degli standard di riferimento e dei controlli possono essere preparate in anticipo. Le soluzioni primarie delle sostanze chimiche in esame sono preparate immediatamente prima di ciascun esperimento. Le diluizioni finali degli standard di riferimento, dei controlli positivi, dei controlli negativi e delle sostanze chimiche in esame devono essere preparate e utilizzate entro 24 ore prima di ciascuna prova.
|
Solubilità, citotossicità e intervallo delle concentrazioni
|
34.
|
Durante la prova di preselezione, si determina la solubilità delle sostanze chimiche in esame nel solvente scelto. È preparata una soluzione primaria di concentrazione massima di 0,1 M. Se tale concentrazione presenta problemi di solubilità, vanno preparate soluzioni primarie di concentrazione inferiore fino a solubilizzazione completa delle sostanze chimiche in esame. Durante la prova di preselezione, sono testate diluizioni successive a 1:10 della sostanza chimica in esame. La concentrazione massima delle prove per l'attività agonista e antagonista è 1 mM. A seguito della prova di preselezione, si ricava un adeguato intervallo di concentrazioni per le sostanze chimiche in esame che deve essere testato nel corso delle batterie di prove complete. Le diluizioni utilizzate per le prove complete devono essere 1x, 3x, 10x, 30x, 100x, 300x, 1000x e 3000x.
|
|
35.
|
Prove di citotossicità sono condotte nel quadro del protocollo sull'attività agonista e antagonista (11). Tali prove di citotossicità sono condotte sia durante la prova di preselezione che nelle batterie di prove complete. Il metodo utilizzato per valutare la citotossicità durante la validazione della prova ERα CALUX combina la prova di perdita di lattato deidrogenasi (LDH) con un'ispezione visiva qualitativa delle cellule (cfr. l'appendice 4.1) dopo esposizione alle sostanze chimiche in esame. È tuttavia possibile utilizzare altri metodi quantitativi per la determinazione della citotossicità [ad esempio il test colorimetrico al sale di tetrazolium (MTT) o la prova CALUX di citotossicità]. In generale, le concentrazioni della sostanza chimica in esame che mostrano una riduzione di oltre il 20 % della vitalità cellulare sono considerate citotossiche e non possono pertanto essere utilizzate per la valutazione dei dati. Per quanto riguarda la prova di perdita di DLH, la concentrazione della sostanza chimica in esame è considerata citotossica quando la perdita di LDH è superiore al 120 %.
|
Esposizione alle sostanze chimiche in esame e configurazione della piastra di prova
|
36.
|
Dopo tripsinazione di un matraccio di cellule confluenti in coltura, le cellule sono nuovamente messe in sospensione alla concentrazione di 1 x105 cellule/ml nel mezzo di prova privo di estrogeni. 100 μl di cellule rimesse in sospensione sono inserite nei pozzetti interni di una piastra da microtitolazione a 96 pozzetti. I pozzetti esterni sono riempiti con 200 μl di tampone fosfato salino (PBS) (cfr. figure 1 e 2). Le cellule piastrate sono pre-incubate in un incubatore a CO2 (5 % CO2, 37 oC, 100 % umidità) per 24 ore.
|
|
37.
|
Dopo la pre-incubazione, le piastre sono ispezionate visivamente per verificare la citotossicità (cfr. l'appendice 4.1), la contaminazione e la confluenza. Per la prova si usano esclusivamente le piastre che non mostrano citotossicità visiva né contaminazione e presentano una confluenza minima dell'85 %. Il mezzo prelevato dai pozzetti interni è accuratamente rimosso e sostituito da 200 μl di mezzo di prova privo di estrogeni contenente una serie di diluizioni appropriate degli standard di riferimento, delle sostanze chimiche in esame, dei controlli positivi, dei controlli negativi e dei controlli con solvente (tabella 5: studi dell'attività agonista; tabella 6: studi dell'attività antagonista). Tutti gli standard di riferimento, le sostanze chimiche in esame, i controlli positivi, i controlli negativi e i controlli con solvente sono testati in triplicato. La figura 1 illustra la configurazione della piastra per le prove sull'attività agonista. La figura 2 illustra la configurazione della piastra per le prove sull'attività antagonista. La configurazione della piastra per la prova di preselezione e le prove complete è identica. Per le prove sull'attività antagonista, tutti i pozzetti interni, esclusi i pozzetti dei controlli con mezzo disperdente (VC), contengono anche una concentrazione fissa dello standard di riferimento per l'attività agonista 17β-estradiolo (3*10-12 M). Si noti che a ciascuna piastra contenente la sostanza chimica in esame vanno aggiunti gli standard di riferimento C8 e C4.
|
|
38.
|
Dopo l'esposizione delle cellule a tutte le sostanze chimiche, le piastre da microtitolazione a 96 pozzetti sono incubate per altre 24 ore in un incubatore a CO2 (5 % di CO2, 37 oC, 100 % di umidità).
|
Figura 1
Configurazione delle piastre da microtitolazione a 96 pozzetti per la prova di preselezione e la valutazione dell'effetto agonista

C0 = solvente con standard di riferimento.
C(1-8) = serie di diluizioni (1-8, concentrazioni da deboli a elevate) dello standard di riferimento.
PC = controllo positivo.
NC = controllo negativo.
TCx-(1-8) = diluizioni (1-8, concentrazioni da deboli a elevate) della sostanza chimica in esame per la prova di preselezione e la valutazione dell'effetto agonista della sostanza chimica in esame x.
SC = controllo con solvente della sostanza chimica in esame (di preferenza lo stesso solvente utilizzato in C0, ma eventualmente da un altro lotto).
Caselle grigie: = pozzetti esterni, riempiti con 200 μl di PBS.
Figura 2
Configurazione delle piastre da microtitolazione a 96 pozzetti per la prova di preselezione antagonista e la valutazione dell'effetto antagonista

C0 = solvente con standard di riferimento.
C(1-8) = serie di diluizioni (1-8, concentrazioni da deboli a elevate) dello standard di riferimento.
NC = controllo negativo.
PC = controllo positivo.
TCx-(1-8) = diluizioni (1-8, concentrazioni da deboli a elevate) della sostanza chimica in esame per la prova di preselezione e la valutazione dell'effetto agonista della sostanza chimica in esame x.
SC = controllo con solvente della sostanza chimica in esame (di preferenza lo stesso solvente utilizzato in C0, ma eventualmente da un altro lotto).
VC = controllo con mezzo disperdente (controllo con solvente senza concentrazione fissa dello standard di riferimento per l'attività agonista 17β-estradiolo);
Caselle grigie: = pozzetti esterni, riempiti con 200 μl di PBS.
Nota: tutti i pozzetti interni, esclusi i pozzetti dei controlli con mezzo disperdente (VC), contengono anche una concentrazione fissa dello standard di riferimento per l'attività agonista 17β-estradiolo (3,0 * 10-12 M).
Misurazione della luminescenza
|
39.
|
La misurazione della luminescenza è descritta dettagliatamente nel protocollo di prova per l'attività agonista e antagonista (10). Il mezzo dei pozzetti è rimosso e le cellule devono essere sottoposte a lisi dopo 24 ore di incubazione per aprire la membrana cellulare e consentire la misurazione dell'attività della luciferasi.
|
|
40.
|
Per misurare la luminescenza, questa procedura richiede un luminometro dotato di 2 iniettori. La reazione della luciferasi viene avviata con l'iniezione della luciferina (substrato della luciferasi). La reazione è interrotta aggiungendo 0,2 M NaOH, per impedire il trasferimento di luminescenza da un pozzetto all'altro.
|
|
41.
|
La luce emessa da ciascun pozzetto è espressa in unità di luce relative (RLU) per pozzetto.
|
Prova di preselezione
|
42.
|
I risultati dell'analisi di preselezione sono usati per determinare un intervallo affinato di concentrazioni delle sostanze chimiche in esame per le prove complete. La valutazione dei risultati delle analisi di preselezione e la determinazione dell'intervallo di concentrazioni affinato delle sostanze chimiche in esame per le prove complete sono descritte in dettaglio nel protocollo di prova sull'attività agonista e antagonista (10). I paragrafi seguenti riassumono le procedure per determinare l'intervallo delle concentrazioni delle sostanze chimiche in esame per le prove sull'attività agonista e antagonista. Cfr. le tabelle 5 e 6 per orientamenti sulla concezione delle diluizioni in serie.
|
Selezione delle concentrazioni per la valutazione degli effetti agonisti
|
43.
|
Durante la prova di preselezione, le sostanze chimiche in esame vanno testate usando le serie di diluizioni indicate nelle tabelle 5 (attività agonista) e 6 (attività antagonista). Tutte le concentrazioni sono testate nei pozzetti in triplicato secondo la configurazione della piastra come indicato nella figura 1 (attività agonista) o 2 (attività antagonista).
|
|
44.
|
Solo i risultati delle analisi che soddisfano i criteri di accettabilità (tabella 3) sono considerati validi e possono essere usati per valutare la risposta delle sostanze chimiche in esame. Se una o più piastre da microtitolazione in una serie di analisi non soddisfano i criteri di accettazione, tali piastre vanno analizzate nuovamente. Se la prima piastra contenente la serie completa di diluizioni dello standard di riferimento non soddisfa i criteri di accettazione, occorre procedere ad una nuova analisi delle serie di prove complete (6 piastre).
|
|
45.
|
Occorre adeguare gli intervalli delle concentrazioni iniziali delle sostanze chimiche in esame e ripetere la prova di preselezione qualora:
|
—
|
si osservi citotossicità. La procedura di preselezione deve essere ripetuta con concentrazioni inferiori non citotossiche della sostanza chimica in esame.
|
|
—
|
la prova di preselezione della sostanza chimica in esame non mostra una curva dose-risposta completa poiché le concentrazioni testate generano un livello di induzione massima. La prova di preselezione deve essere ripetuta con concentrazioni inferiori della sostanza chimica in esame.
|
|
|
46.
|
Quando si osserva una relazione dose-risposta valida, occorre selezionare la (più bassa) concentrazione alla quale si osserva l'induzione massima che non genera citotossicità. La concentrazione più elevata della sostanza chimica in esame da testare nelle prove complete deve essere 3 volte la concentrazione selezionata.
|
|
47.
|
Una serie affinata completa di diluizioni della sostanza chimica in esame è preparata seguendo le fasi di diluizione indicate nella tabella 5, iniziando con la concentrazione più elevata come sopra indicato.
|
|
48.
|
La sostanza chimica in esame che non produce effetti agonisti è testata nelle batterie di prove complete, cominciando dalla concentrazione non citotossica più elevata individuata durante la preselezione.
|
Selezione delle concentrazioni per la valutazione degli effetti antagonisti
|
49.
|
Solo i risultati delle analisi che soddisfano i criteri di accettabilità (tabella 4) sono considerati validi e possono essere usati per valutare la risposta delle sostanze chimiche in esame. Se una o più piastre da microtitolazione in una serie di analisi non soddisfano i criteri di accettazione, tali piastre vanno analizzate nuovamente. Se la prima piastra contenente la serie completa di diluizioni dello standard di riferimento non soddisfa i criteri di accettazione, occorre procedere ad una nuova analisi delle serie di prove complete (6 piastre).
|
|
50.
|
Occorre adeguare gli intervalli delle concentrazioni iniziali delle sostanze chimiche in esame e ripetere la prova di preselezione qualora:
|
—
|
si osservi citotossicità. La procedura di preselezione deve essere ripetuta con concentrazioni inferiori non citotossiche della sostanza chimica in esame.
|
|
—
|
la prova di preselezione della sostanza chimica in esame non mostra una curva dose-risposta completa poiché le concentrazioni testate generano un livello di inibizione massima. La preselezione deve essere ripetuta con concentrazioni inferiori della sostanza chimica in esame.
|
|
|
51.
|
Quando si riscontra una relazione dose-risposta valida, occorre selezionare la (più bassa) concentrazione alla quale si osserva l'inibizione massima che non genera citotossicità. La concentrazione più elevata della sostanza chimica in esame da testare nelle prove complete deve essere 3 volte la concentrazione selezionata.
|
|
52.
|
Una serie affinata completa di diluizioni della sostanza chimica in esame è preparata seguendo le fasi di diluizione indicate nella tabella 6, iniziando con la concentrazione più elevata come sopra indicato.
|
|
53.
|
Le sostanze chimiche in esame che non producono effetti antagonisti sono testate nelle batterie di prove complete, cominciando dalla concentrazione non citotossica più elevata testata durante la preselezione.
|
Prove complete
|
54.
|
A seguito della selezione degli intervalli affinati di concentrazioni, le sostanze chimiche in esame sono testate in maniera esaustiva usando le serie di diluizioni indicate nelle tabelle 5 (attività agonista) e 6 (attività antagonista). Tutte le concentrazioni sono testate nei pozzetti in triplicato secondo la configurazione della piastra come indicato nella figura 1 (attività agonista) o 2 (attività antagonista).
|
|
55.
|
Solo i risultati delle analisi che soddisfano i criteri di accettabilità (tabella 3 e 4) sono considerati validi e possono essere usati per valutare la risposta delle sostanze chimiche in esame. Se una o più piastre da microtitolazione in una serie di analisi non soddisfano i criteri di accettazione, tali piastre vanno analizzate nuovamente. Se la prima piastra contenente la serie completa di diluizioni dello standard di riferimento non soddisfa i criteri di accettazione, occorre procedere ad una nuova analisi delle serie di prove complete (6 piastre).
|
Tabella 5
Concentrazione e diluizioni degli standard di riferimento, dei controlli e delle sostanze chimiche in esame utilizzati per le prove sull'attività agonista
|
Conc. (M) dello
|
Diluizione della TCx
|
Diluizione della TCx
|
Conc. (M)
|
|
standard di riferimento 17β-estradiolo
|
durante la prova di range finding
|
durante la prova completa
|
dei controlli
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx - sostanza chimica in esame x
PC - controllo positivo (17α-metiltestosterone)
NC - controllo negativo (corticosterone)
C0 - controllo con solvente dello standard di riferimento
SC - controllo con solvente della sostanza chimica in esame
|
Tabella 6
Concentrazione e diluizioni degli standard di riferimento, dei controlli e delle sostanze chimiche in esame utilizzati per la prova sugli antagonisti
|
Conc. (M) dello
|
Diluizione della TCx
|
Diluizione della TCx
|
Conc. (M)
|
|
standard di riferimento tamoxifene
|
durante la prova di range finding
|
durante la prova completa
|
dei controlli
|
|
C0
|
0
|
TCx-1
|
10000000 x
|
TCx-1
|
3000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1000000 x
|
TCx-2
|
1000 x
|
NC
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Conc. (M) dello standard di riferimento agonista
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
Conc. (M) dello
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-estradiolo
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx - sostanza chimica in esame x
PC - controllo positivo (4-idrossitamixefene)
NC - controllo negativo (resveratrolo)
C0 - controllo con solvente dello standard di riferimento
SC - controllo con solvente della sostanza chimica in esame
VC - controllo con mezzo disperdente (non contiene una concentrazione fissa dello standard di riferimento agonista 17β-estradiolo (3*10-12 M).
|
Raccolta e analisi dei dati
|
56.
|
A seguito della prova di preselezione e delle prove complete, i valori EC10, EC50, PC10, PC50 e l'induzione massima (TCxmax) di una sostanza chimica in esame sono determinati per la prova sull'attività agonista. Per la prova sull'attività antagonista, vanno calcolati i valori IC20, IC50, PC80, PC50 e l'induzione massima (TCxmin). I grafici 3 (attività agonista) e 4 (attività antagonista) illustrano questi parametri. I parametri richiesti sono calcolati in base all'induzione relativa di ciascuna sostanza chimica in esame [rispetto all'induzione massima dello standard di riferimento (= 100 %)]. Per la valutazione dei dati si utilizza la regressione non lineare (pendenza variabile, 4 parametri) applicando alla seguente equazione:
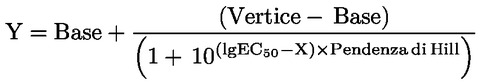
dove:
X = Log della dose o concentrazione
Y = Risposta (induzione relativa (%))
Vertice = Induzione massima (%)
Base = Induzione minima (%)
LogEC50 = Logaritmo della concentrazione corrispondente al 50 % della risposta massima
Pendenza di Hill = coefficiente della pendenza di Hill
|
|
57.
|
I dati grezzi del luminometro, espressi in unità di luce relative (RLU), sono trasferiti al foglio di calcolo contenente l'analisi dei dati destinata alla preselezione e alle batterie di prove complete. I dati grezzi devono soddisfare i criteri di accettazione indicati nella tabella 3A e nella tabella 3B (attività agonista) o 4A e 4B (attività antagonista). Se i dati grezzi soddisfano i criteri di accettazione, si eseguono le seguenti fasi di calcolo per determinare i parametri richiesti:
|
Attività agonista
|
—
|
Sottrarre le RLU medie del controllo con solvente dello standard di riferimento da ciascuno dei dati grezzi delle analisi relativi agli standard di riferimento.
|
|
—
|
Sottrarre le RLU medie del controllo con solvente della sostanza chimica in esame da ciascuno dei dati grezzi delle analisi relativi alle sostanze chimiche in esame.
|
|
—
|
Calcolare l'induzione relativa di ciascuna concentrazione dello standard di riferimento; fissare al 100 % l'induzione della concentrazione più elevata dello standard di riferimento.
|
|
—
|
Calcolare l'induzione relativa di ciascuna concentrazione della sostanza chimica in esame rispetto alla concentrazione massima dello standard di riferimento al 100 %.
|
|
—
|
Valutare i risultati dell'analisi seguendo una curva di regressione non lineare (pendenza variabile, 4 parametri).
|
|
—
|
Determinare i valori EC50 e EC10 dello standard di riferimento.
|
|
—
|
Determinare i valori EC50 e EC10 delle sostanze chimiche in esame.
|
|
—
|
Determinare l'induzione relativa massima della sostanza chimica in esame (TCmax).
|
|
—
|
Determinare i valori PC10 e PC50 delle sostanze chimiche in esame.
|
Per le sostanze chimiche in esame, non è sempre possibile ottenere una curva dose-risposta completa a motivo, ad esempio, di problemi di citotossicità o di solubilità. Pertanto i valori EC50, EC10 e PC50 non possono essere determinati. In tal caso, solo i valori PC10 e TCmax possono essere determinati.
Attività antagonista
|
—
|
Sottrarre le RLU medie della più elevata concentrazione dello standard di riferimento da ciascuno dei dati grezzi delle analisi relativi agli standard di riferimento.
|
|
—
|
Sottrarre le RLU medie della più elevata concentrazione dello standard di riferimento da ciascuno dei dati grezzi delle analisi relativi alle sostanze chimiche in esame.
|
|
—
|
Calcolare l'induzione relativa di ciascuna concentrazione dello standard di riferimento; fissare al 100 % l'induzione della concentrazione più bassa dello standard di riferimento.
|
|
—
|
Calcolare l'induzione relativa di ciascuna concentrazione della sostanza chimica in esame rispetto alla concentrazione più bassa dello standard di riferimento al 100 %.
|
|
—
|
Valutare i risultati dell'analisi seguendo una curva di regressione non lineare (pendenza variabile, 4 parametri).
|
|
—
|
Determinare i valori IC50 e IC20 dello standard di riferimento.
|
|
—
|
Determinare i valori IC50 e IC20 delle sostanze chimiche in esame.
|
|
—
|
Determinare l'induzione relativa minima della sostanza chimica in esame (TCmin).
|
|
—
|
Determinare i valori PC80 e PC50 delle sostanze chimiche in esame.
|
Grafico 3
Parametri determinati nella prova sull'effetto agonista

EC10 = concentrazione di una sostanza corrispondente al 10 % della sua risposta massima
EC50 = concentrazione di una sostanza corrispondente al 50 % della sua risposta massima
PC10 = concentrazione della sostanza chimica in esame equivalente alla EC10 dello standard di riferimento.
PC50 = concentrazione della sostanza chimica in esame equivalente alla EC50 dello standard di riferimento.
TCxmax = induzione relativa massima della sostanza chimica in esame.
Grafico 4
Parametri determinati nella prova sull'effetto antagonista

IC20 = concentrazione di una sostanza corrispondente all'80 % della sua risposta massima (20 % di inibizione)
IC50 = concentrazione di una sostanza corrispondente all'50 % della sua risposta massima (50 % di inibizione)
PC80 = concentrazione di una sostanza chimica in esame equivalente alla IC20 dello standard di riferimento.
PC50 = concentrazione di una sostanza chimica in esame equivalente alla IC50 dello standard di riferimento.
TCxmin = induzione relativa minima della sostanza chimica in esame.
Per le sostanze chimiche in esame, non è sempre possibile ottenere una curva dose-risposta completa a motivo, ad esempio, di problemi di citotossicità o di solubilità. Pertanto i valori IC50, IC20 e PC50 non possono essere determinati. In tal caso, solo i valori PC20 e TCmin possono essere determinati.
|
58.
|
I risultati dovrebbero basarsi su due (o tre) batterie di prove indipendenti. Se due batterie di prove forniscono risultati comparabili e pertanto riproducibili, non è necessario effettuare una terza batteria di prove. Per essere accettabili, i risultati devono:
|
—
|
soddisfare i criteri di accettabilità (cfr. i criteri di accettabilità, paragrafi da 14 a 22);
|
|
Criteri di interpretazione dei dati
|
59.
|
Per interpretare i dati e decidere se una sostanza chimica in esame è considerata positiva o negativa, si utilizzano i seguenti criteri:
|
Per ciascuna prova completa, una sostanza chimica è considerata positiva se:
|
1
|
Il valore TCmax è uguale o superiore al 10 % della risposta massima dello standard di riferimento (REF10).
|
|
2
|
Almeno 2 concentrazioni consecutive della sostanza chimica in esame sono uguali o superiori al REF10.
|
|
|
Per ciascuna prova completa, una sostanza chimica è considerata negativa se:
|
1
|
Il valore TCmax non è superiore al 10 % della risposta massima dello standard di riferimento (REF10).
|
|
2
|
Meno di 2 concentrazioni della sostanza chimica in esame sono uguali o superiori al REF10.
|
|
|
Per ciascuna prova completa, una sostanza chimica è considerata positiva se:
|
1
|
Il valore TCmin è uguale o inferiore all'80 % della risposta massima dello standard di riferimento (REF80 = 20 % di inibizione).
|
|
2
|
Almeno 2 concentrazioni consecutive della sostanza chimica in esame sono uguali o inferiori al REF80.
|
|
|
Per ciascuna prova completa, una sostanza chimica è considerata negativa se:
|
1
|
Il valore TCmin è superiore all'80 % della risposta massima dello standard di riferimento (REF80 = 20 % di inibizione).
|
|
2
|
Meno di 2 concentrazioni della sostanza chimica in esame sono uguali o inferiori al REF80.
|
|
|
|
60.
|
Per caratterizzare la potenza della reazione positiva di una sostanza chimica in esame, l'ampiezza dell'effetto (attività agonista: TCmax; attività antagonista: TCmin) e la concentrazione alla quale si verifica l'effetto (attività agonista: EC10, EC50, PC10, PC50; attività antagonista: IC20, IC50, PC80, PC50) sono riportate.
|
RELAZIONE SULL'ESECUZIONE DELLA PROVA
|
61.
|
Cfr. il paragrafo 20 della sezione "ELEMENTI DEL METODO DI PROVA DEL METODO ER TA").
|
BIBLIOGRAFIA
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay - transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Organisation for Economic Cooperation and Development, Paris
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P. (2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
Appendice 4.1:
ISPEZIONE VISIVA DELLA VITALITÀ CELLULARE
B.67 PROVE IN VITRO DI MUTAZIONE GENICA SU CELLULE DI MAMMIFERO UTILIZZANDO IL GENE TIMIDINA CHINASI
INTRODUZIONE
|
1.
|
Questo metodo di prova è equivalente alla linea guida dell’OCSE per le prove sulle sostanze chimiche n. 490 (2016). I metodi di prova vengono periodicamente riesaminati e rivisti alla luce dei progressi scientifici, delle esigenze in materia di regolamentazione, e di considerazioni relative al benessere degli animali. La prova sul linfoma di topo (MLA) e la prova TK6 con il locus timidina chinasi (TK) erano in origine contenute nel metodo di prova B.17. Successivamente il gruppo di lavoro degli esperti MLA dell’International Workshop on Genotoxicity Testing (IWGT) ha elaborato raccomandazioni armonizzate a livello internazionale per i criteri di accettazione delle prove e per l’interpretazione dei dati della prova MLA (1)(2)(3)(4)(5), che sono state inserite nel presente nuovo metodo di prova B.67. Il presente metodo di prova è elaborato per la prova MLA e, poiché usa anche il locus TK, anche per la prova TK6. La prova MLA è ampiamente usata a fini regolamentari, mentre la prova TK6 è stata usata molto meno frequentemente. Va osservato che, nonostante le similitudini fra gli endpoint, le due linee cellulari non sono intercambiabili e i programmi regolamentari possono esprimere validamente una preferenza per l’una o l’altra, a seconda di un particolare uso regolamentare. A titolo di esempio, la validazione della prova MLA ne ha dimostrato l’idoneità a rilevare non solo la mutazione genica, bensì anche la capacità di una sostanza chimica in esame di indurre danni strutturali a carico dei cromosomi. Il presente metodo di prova è parte integrante di una serie di metodi di prova sulla tossicologia genetica. Un documento contenente informazioni succinte sulle prove di tossicologia genetica dell’OCSE, comprensivo di un compendio delle modifiche recentemente apportate alla rispettiva linea guida è stato elaborato dall’OCSE (6).
|
|
2.
|
Scopo della prova in vitro di mutazione genica su cellule di mammifero è identificare mutazioni geniche indotte da sostanze chimiche. Le linee cellulari utilizzate in queste prove misurano le mutazioni "in avanti" dei geni reporter, in particolare il gene endogeno timidina chinasi (TK per le cellule umane e Tk per le cellule di roditori, denominate collettivamente TK in questo metodo di prova). Il presente metodo di prova è destinato a essere usato con due linee cellulari: le cellule di linfoma di topo L5178Y TK+/--3.7.2C (in generale denominate L5178Y) e le cellule linfoblastoidi umane TK6 (in generale denominate TK6). Sebbene le due linee cellulari differiscano a causa della loro origine, crescita cellulare, status del gene p53, ecc., le prove di mutazione genica TK possono essere effettuate in maniera simile su entrambi i tipi di cellule, come illustrato nel presente metodo di prova.
|
|
3.
|
La natura autosomica ed eterozigote del gene timidina chinasi consente di individuare colonie vitali le cui cellule sono carenti dell’enzima timidina chinasi in seguito a una mutazione da TK+/-
a TK-/-
. Tale carenza può risultare da fenomeni genetici che colpiscono il gene TK, compresi sia le mutazioni genetiche (mutazioni puntiformi, mutazioni per spostamento del sistema di lettura, piccole delezioni, ecc.) che i fenomeni cromosomici (ampie delezioni, riarrangiamenti cromosomici e ricombinazione mitotica). Questi ultimi fenomeni sono espressi come perdita di eterozigosi, ossia un cambiamento genetico comune dei geni soppressori dei tumori nell’oncogenesi umana. Teoricamente, nella prova MLA si può rilevare la perdita dell’intero cromosoma portante il gene TK che fa seguito al danneggiamento dell’elica e/o alla non disgiunzione mitotica. In effetti una combinazione di analisi citogenetica e molecolare mostra chiaramente che alcuni mutanti MLA TK sono il risultato di una non disgiunzione. Il peso dell’evidenza mostra tuttavia che le prove di mutazione genica TK non possono rilevare in modo affidabile gli aneugeni applicando i criteri standard di citotossicità (come descritto nel presente metodo di prova) e pertanto non è appropriato utilizzare tali metodi di prova per rilevare gli aneugeni (7) (8) (9).
|
|
4.
|
Nelle prove di mutazione genica TK, si generano due classi fenotipiche distinte di mutanti TK; i mutanti aventi una crescita normale che crescono allo stesso ritmo delle cellule eterozigoti TK e i mutanti aventi una crescita lenta che crescono con tempi di raddoppiamento prolungati. I mutanti a crescita normale e i mutanti a crescita lenta si riconoscono nella prova MLA come mutanti che producono colonie grandi e mutanti che producono colonie piccole e, nella prova TK6, come mutanti che producono colonie a comparsa precoce e mutanti che producono colonie a comparsa tardiva. La natura molecolare e citogenetica dei due tipi di mutanti (quelli che producono colonie grandi e quelli che producono colonie piccole nella prova MLA) è stata studiata in modo approfondito (8) (10) (11) (12) (13). La natura molecolare e citogenetica dei due tipi di mutanti TK6 a comparsa precoce e a comparsa tardiva è stata anch’essa ampiamente studiata (14) (15) (16) (17). I mutanti a crescita lenta di entrambi i tipi di cellule hanno subito danni genetici che comportano uno o più geni che si presumono responsabili di regolare la crescita presso il locus TK, il che determina tempi di raddoppiamento prolungati e la formazione di piccole colonie o a comparsa tardiva (18). La presenza di mutanti a crescita lenta è stata messa in relazione con sostanze chimiche che causano rilevanti cambiamenti strutturali a livello cromosomico. Le cellule i cui danni non riguardano il o i geni che si presumono responsabili di regolare la crescita presso il locus TK crescono a una velocità analoga a quella delle cellule parentali e diventano mutanti a crescita normale. L’induzione di mutanti principalmente a crescita normale è associata a sostanze chimiche che fungono principalmente da mutageni puntiformi. È quindi fondamentale conteggiare sia i mutanti a crescita lenta che quelli a crescita normale, al fine di recuperare tutti i mutanti e ottenere informazioni sul o sui tipi di danni (mutageni o clastogeni) indotti dalla sostanza chimica in esame (10) (12) (18) (19).
|
|
5.
|
Il metodo di prova è organizzato in modo da fornire informazioni generali applicabili a entrambe le prove MLA e TK6 nonché orientamenti specializzati per le singole prove.
|
|
6.
|
Le definizioni usate sono riportate nell’appendice 1.
|
CONSIDERAZIONI INIZIALI E LIMITI
|
7.
|
Le prove in vitro richiedono in generale l’uso di una fonte esogena di attivazione metabolica. Il sistema esogeno di attivazione metabolica non simula perfettamente le condizioni in vivo.
|
|
8.
|
Occorre adoperarsi per evitare condizioni che porterebbero a falsi risultati positivi (vale a dire potenziali interazioni con il sistema di prova), non causati da un’interazione fra la sostanza chimica in esame e il materiale genetico della cellula; tali condizioni includono modifiche del pH o dell’osmolalità, un’interazione con i componenti del terreno di coltura (20) (21) o una eccessiva citotossicità (22)(23)(24). Per le prove MLA e TK6 è considerata eccessiva una citotossicità che superi i massimi livelli di citotossicità raccomandati definiti al paragrafo 28. Va inoltre osservato che le sostanze chimiche in esame analoghe della timidina o che si comportano come gli analoghi della timidina possono aumentare la frequenza dei mutanti mediante crescita selettiva dei mutanti spontanei di fondo durante il trattamento delle cellule e richiedono metodi di prova supplementari ai fini di un’adeguata valutazione (25).
|
|
9.
|
Possono essere necessari adattamenti specifici di questo metodo di prova, non descritti in questa sede, per i nanomateriali di sintesi.
|
|
10.
|
Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tale verifica non è necessaria se la miscela viene sottoposta a prova in ottemperanza a un obbligo regolamentare.
|
|
11.
|
Le cellule mutanti carenti di attività enzimatica della timidina chinasi a causa della mutazione TK
+/- —> TK
-/- sono resistenti agli effetti citostatici dell’analogo pirimidinico trifluorotimidina (TFT). Le cellule capaci di produrre TK sono sensibili alla TFT, che provoca l’inibizione del metabolismo cellulare e arresta la divisione cellulare. Di conseguenza, le cellule mutanti sono in grado di proliferare in presenza di TFT e di formare colonie, mentre le cellule contenenti l’enzima TK non lo sono.
|
PRINCIPIO DELLA PROVA
|
12.
|
Le cellule in sospensione sono esposte alla sostanza in esame, con e senza fonte esogena di attivazione metabolica (cfr. il paragrafo 19), per un tempo adeguato (cfr. il paragrafo 33), poi si allestiscono sub-culture per determinare la citotossicità e permettere l’espressione fenotipica prima della selezione dei mutanti. La citotossicità è determinata dalla crescita relativa totale (RTG - cfr. il paragrafo 25) per la prova MLA e dal tasso di sopravvivenza relativa (RS - cfr. il paragrafo 26) per la prova TK6. Le colture trattate sono mantenute nel terreno di coltura per un periodo sufficiente, specifico per ciascun tipo cellulare (cfr. il paragrafo 37), per permettere l’espressione fenotipica ottimale delle mutazioni indotte. Secondo l’espressione fenotipica, la frequenza dei mutanti è determinata mediante inseminazione di un numero noto di cellule in terreno contenente l’agente selettivo, per individuare le colonie mutanti, e in terreno non contenente l’agente selettivo, per determinare l’efficacia di clonazione (vitalità). Dopo un adeguato periodo di incubazione le colonie sono contate. La frequenza dei mutanti è calcolata sulla base del numero di colonie di mutanti corretto per l’efficienza di clonazione al momento della selezione dei mutanti.
|
DESCRIZIONE DEL METODO
Preparazioni
Cellule
|
13.
|
Per la prova MLA: poiché il metodo di prova MLA è stato elaborato e caratterizzato dal ricorso alla sottolinea TK
+/- -3.7.2C di cellule L5178Y, per la presente prova si deve utilizzare questa sottolinea specifica. La linea cellulare L5178Y è stata derivata da un linfoma timico indotto da metilcolantrene in un topo DBA-2 (26). Clive e colleghi hanno trattato le cellule L5178Y (indicate da Clive come TK+/+ -3) con etilmetan sulfonato e hanno isolato un clone TK-/- (indicato come TK-/- -3.7), utilizzando bromodeossiuridina come agente selettivo. A partire dal clone TK-/- sono stati isolati e caratterizzati per essere utilizzati nella prova MLA un clone spontaneo TK+/- (indicato come TK+/- -3.7.2.) e un sottoclone (indicato come TK+/--3.7.2C) (27). Il cariotipo della linea cellulare è stato pubblicato (28)(29)(30)(31). Il numero modale dei cromosomi è 40. Vi è un cromosoma metacentrico (t12;13) che deve essere conteggiato come un cromosoma. Nel topo il locus TK è situato all’estremità distale del cromosoma 11. La linea cellulare L5178Y TK
+/- -3.7.2C presenta mutazioni in entrambi gli alleli p53 e produce la proteina mutante p53 (32) (33). Si presume che la capacità di individuare danni su ampia scala sia riconducibile allo status p53 della linea cellulare TK+/--3.7.2C (17).
|
|
14.
|
Per la prova TK6: si tratta di una linea cellulare linfoblastoide umana. La linea cellulare parentale consiste nella linea cellulare trasformata dal virus di Epstein-Barr WI-L2, in origine derivata da un soggetto maschio di cinque anni affetto da sferocitosi ereditaria. Il primo clone isolato, HH4, è stato mutagenizzato con ICR191 e si è generata una linea cellulare eterozigote TK, TK6 (34). Le cellule TK6 sono quasi diploidi e il cariotipo rappresentativo è 47, XY, 13+, t(14; 20), t(3; 21) (35). Nell’uomo il locus TK è situato sul braccio lungo del cromosoma 17. La prova TK6 è una linea cellulare competente per il gene p53, in quanto presenta una sequenza p53 di tipo casuale su entrambi gli alleli ed esprime unicamente la proteina p53 di tipo casuale (36).
|
|
15.
|
In entrambe le prove MLA e TK6, quando si costituisce o si ricostituisce una popolazione di cellule, si raccomanda al laboratorio che esegue la prova di garantire l’assenza di contaminazione da Mycoplasma, di procedere al cariotipaggio delle cellule o colorare i cromosomi che presentano il locus TK e di controllare i tempi di raddoppiamento della popolazione. Occorre stabilire la durata normale del ciclo cellulare delle linee cellulari utilizzate nel laboratorio di prova, che deve essere coerente con le caratteristiche cellulari pubblicate (16) (19) (37). Tale popolazione madre va conservata a -150 °C o a temperatura inferiore e va utilizzata per preparare tutti gli stock cellulari di lavoro.
|
|
16.
|
Prima di costituire un ampio numero di stock di lavoro crioconservati o appena prima di utilizzarli in un esperimento, la coltura può dover essere ripulita delle cellule mutanti preesistenti [salvo nel caso in cui la frequenza dei mutanti (MF) nel solvente di controllo si trovi già entro l’intervallo accettabile, cfr. tabella 2 per la prova MLA)]. Questo avviene utilizzando metotressato (aminopterina) per scartare le cellule carenti in TK e aggiungendo alla coltura timidina, ipoxantina e glicina (L5178Y) o 2’-deossicitidina (TK6) al fine di garantire la crescita ottimale delle cellule competenti per la TK (19)(38)(39) (e (40) per la prova TK6). Ai paragrafi (19)(31)(37)(39)(41) figurano le indicazioni generali relative alle buone prassi per il mantenimento delle colture cellulari nonché raccomandazioni specifiche relative alle cellule L5178Y e TK6. Per i laboratori che necessitano di popolazioni cellulari madri per avviare la prova MLA o TK6 o che devono ottenere nuove popolazioni madri, è disponibile una banca di cellule qualificata (37).
|
Terreni e condizioni di coltura
|
17.
|
Per entrambe le prove, le colture vanno mantenute in terreni di coltura e condizioni di incubazione (ad es. recipienti di coltura, atmosfera umidificata al 5 % di CO2, temperatura di incubazione di 37 °C) adeguati. Le colture cellulari sono sempre mantenute in condizioni che garantiscano una crescita in fase esponenziale. È particolarmente importante che i terreni e le condizioni di coltura siano scelti in modo da garantire la crescita ottimale delle cellule durante il periodo di espressione e di clonazione delle cellule mutanti e non mutanti. Nelle prove MLA e TK6 è altresì importante che le condizioni di coltura garantiscano una crescita ottimale dei mutanti TK delle grandi colonie/a comparsa precoce come delle piccole colonie/a comparsa tardiva. Ai paragrafi (19)(31)(38)(39)(40)(42) sono reperibili maggiori particolari relativi alla coltura, compresa l’esigenza di inattivare termicamente il siero di cavallo se si utilizza il terreno di coltura RPMI nella selezione dei mutanti.
|
Preparazione delle colture
|
18.
|
Le cellule provengono da colture primarie, sono inseminate in un terreno di coltura ad una densità tale che le colture in sospensione proseguiranno la loro crescita esponenziale durante le fasi di trattamento ed espressione.
|
Attivazione metabolica
|
19.
|
Se si utilizzano cellule L5178Y e TK6 in quanto prive di un’adeguata capacità di attivazione metabolica endogena, si deve ricorrere a sistemi di attivazione metabolica esogeni. Il sistema più comunemente usato, raccomandato in tutti i casi salvo alternativa motivata, è una frazione post-mitocondriale integrata di cofattore (S9), prelevata dal fegato di roditori (in generale ratti) trattati con induttori enzimatici come Aroclor 1254 (43) (44) (45) o con una combinazione di fenobarbitone e β-naftoflavone (46) (47) (48) (49) (50) (51). Quest’ultima combinazione è conforme alla convenzione di Stoccolma sugli inquinanti organici persistenti (52) e ha dimostrato di essere tanto efficace quanto l’Aroclor 1254 nell’indurre ossidasi a funzione mista (45) (46) (47) (48) (49). La frazione S9 viene in generale usata a concentrazioni comprese tra 1-2 % (v/v) ma può aumentare fino al 10 % (v/v) nel terreno di coltura finale. La scelta del tipo e della concentrazione del sistema di attivazione metabolica esogena o dell’induttore metabolico usato può dipendere dalla classe delle sostanze chimiche in esame.
|
Preparazioni della sostanza chimica in esame
|
20.
|
Le sostanze chimiche in esame solide devono essere preparate in adeguati solventi e, se necessario, diluite prima del trattamento delle cellule (cfr. il paragrafo 21). Le sostanze chimiche liquide in esame possono essere aggiunte direttamente alla coltura e/o diluite prima del trattamento del sistema di prova. Le sostanze chimiche in esame gassose o volatili devono essere sottoposte alla prova modificando adeguatamente i protocolli standard (trattamento in recipienti di coltura ermetici) (53) (54) (55). Devono essere utilizzati preparati della sostanza chimica approntati immediatamente prima del trattamento, salvo qualora siano disponibili dati sulla sua stabilità che dimostrino che la conservazione è accettabile.
|
CONDIZIONI DI PROVA
Solventi
|
21.
|
La scelta del solvente deve favorire l’ottimizzazione della solubilità della sostanza chimica in esame, senza nuocere alla conduzione del metodo di prova (ad esempio influenzando la crescita cellulare), compromettere l’integrità della sostanza chimica in esame, reagire con recipienti di coltura o pregiudicare il sistema di attivazione metabolica. Si raccomanda di prendere in considerazione in primo luogo, se possibile, l’uso di un solvente acquoso (o terreno di coltura). Solventi il cui uso è consolidato sono l’acqua e il dimetilsolfossido. In generale è opportuno che i solventi organici non superino l’1 % (v/v) e quelli acquosi (soluzione fisiologica o acqua) il 10 % (v/v) nel terreno di coltura finale. L’uso di solventi poco noti (ad esempio, etanolo o acetone) è ammesso purché suffragato da dati che ne provino la compatibilità con le sostanze chimiche in esame e l’assenza di tossicità genetica alla concentrazione utilizzata. In assenza di tali dati, è importante includere controlli non trattati (cfr. l’appendice 1, definizioni) per dimostrare che il solvente scelto non induce effetti nocivi o mutageni.
|
MISURAZIONE DELLA CITOTOSSICITÀ E SCELTA DELLE CONCENTRAZIONI DI TRATTAMENTO
|
22.
|
Nel determinare la concentrazione più elevata della sostanza chimica in esame, occorre evitare le concentrazioni che hanno la capacità di produrre falsi risultati positivi, come quelle che causano eccessiva citotossicità (cfr. il paragrafo 28), precipitazione nel terreno di coltura (cfr. il paragrafo 29) o variazioni marcate del pH od osmolalità (cfr. il paragrafo 8). Se la sostanza chimica in esame provoca un’alterazione marcata del pH del terreno al momento della sua aggiunta, è possibile adeguare il pH mediante l’azione tampone del terreno di trattamento finale in modo da evitare falsi risultati positivi e da mantenere condizioni di coltura appropriate.
|
|
23.
|
La scelta della concentrazione si basa sulla citotossicità e su altre considerazioni (cfr. i paragrafi 27-30). Mentre la valutazione della citotossicità in una prova iniziale può essere utile per definire meglio le concentrazioni da utilizzare nell’esperimento principale, non è necessario condurre una prova iniziale. Anche se si effettua una valutazione iniziale della citotossicità, nella prova principale è ancora richiesta la misurazione della citotossicità di ciascuna coltura. Se si effettuano esperimenti per determinare gli intervalli di dose, essi dovrebbero interessare un’ampia gamma di concentrazioni e possono essere conclusi al giorno 1 dopo il trattamento o proseguiti attraverso l’espressione del giorno 2 e la selezione dei mutanti (se risulta che le concentrazioni utilizzate sono appropriate).
|
|
24.
|
La citotossicità va determinata per ciascuna coltura di prova e di controllo: i metodi di prova per le prove MLA (2) e TK6 (15) sono definiti da prassi concordate a livello internazionale.
|
|
25.
|
Per entrambe le versioni (gel di agarosio e microtitolazione) della prova MLA: la citotossicità va valutata mediante la crescita relativa totale (RTG), in origine definita da Clive e Spector nel 1975 (2). Questa misurazione include la crescita in sospensione relativa (RSG: coltura di prova e coltura di controllo con solvente) durante il trattamento delle cellule, il tempo di espressione e l’efficienza relativa di clonazione (RCE: coltura di prova e coltura di controllo con solvente) al momento della selezione dei mutanti (2). Va osservato che la RSG include le eventuali perdite di cellule che si verificano nella coltura di prova durante il trattamento (cfr. l’appendice 2 per la formula).
|
|
26.
|
Per la prova TK6: la citotossicità va valutata per mezzo del tasso di sopravvivenza relativa (RS), ossia l’efficienza di clonazione delle cellule piastrate subito dopo il trattamento, adeguata per le eventuali perdite di cellule durante il trattamento, in base alla conta cellulare, rispetto ai controlli negativi (assegnata una sopravvivenza del 100 %) (cfr. l’appendice 2 per la formula).
|
|
27.
|
Si usino almeno quattro concentrazioni di prova (senza contare i controlli positivi e i controlli con solvente) che soddisfano i criteri di accettabilità (citotossicità adeguata, numero di cellule, ecc.). Sebbene sia consigliabile l’utilizzo di colture doppie, per ciascuna concentrazione di prova si possono utilizzare colture replicate o uniche. I risultati ottenuti con colture replicate con una determinata concentrazione devono essere riportati separatamente ma possono essere aggregati per l’analisi dei dati (55). Per le sostanze chimiche in esame la cui citotossicità è bassa o nulla, sono in generale adeguati intervalli di concentrazione di un fattore di circa 2 a 3. In caso di citotossicità, le concentrazioni di prova selezionate devono coprire l’intervallo di citotossicità a partire dai valori che producono citotossicità come descritto al paragrafo 28 e che include le concentrazioni alle quali si riscontra una citotossicità modesta o nulla. Molte sostanze chimiche in esame presentano curve di concentrazione-risposta a forte pendenza e, per coprire l’intero intervallo di citotossicità o per valutare la concentrazione-risposta in dettaglio, può essere necessario utilizzare concentrazioni più ravvicinate e più di quattro concentrazioni, in particolare nei casi in cui è necessario ripetere l’esperimento (cfr. il paragrafo 70). L’utilizzo di più di quattro concentrazioni può essere particolarmente importante quando si utilizzano colture uniche.
|
|
28.
|
Se la concentrazione massima è basata sulla citotossicità, la concentrazione massima deve mirare a realizzare un tasso di RTG compreso fra 20 e 10 % per la prova MLA e un tasso di RS fra 20 e 10 % per la prova TK6 (paragrafo 67).
|
|
29.
|
Per le sostanze chimiche in esame scarsamente solubili che non sono citotossiche a concentrazioni inferiori alla concentrazione insolubile più bassa, la concentrazione più elevata analizzata dovrebbe presentare una torbidità o un precipitato visibile ad occhio nudo o con un microscopio invertito alla fine del trattamento con la sostanza chimica in esame. Anche se la citotossicità si verifica a concentrazioni superiori a quella minima insolubile, è indicato effettuare la prova a una sola concentrazione che produce torbidità o un precipitato visibile, perché il precipitato può falsare gli effetti. Poiché le prove MLA e TK6 utilizzano colture in sospensione, che produce un precipitato, occorre assicurarsi in particolare che quest’ultimo non interferisca con la conduzione della sperimentazione. Può essere utile anche valutare la solubilità nel terreno di coltura prima della prova.
|
|
30.
|
Se non si osserva una citotossicità limitante o si rileva la presenza di un precipitato, la concentrazione massima di prova dovrebbe essere pari a 10 mM, 2 mg/ml or 2 μl/ml, (si scelga il valore più basso) (57) (58). Quando la composizione della sostanza chimica in esame non è definita, ad esempio nel caso di sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici (ossia sostanze di composizione sconosciuta o variabile, UVCB), prodotti estratti dall’ambiente, ecc., può essere necessario aumentare la concentrazione massima (ad es. 5 mg/ml) in assenza di citotossicità sufficiente, per aumentare la concentrazione di ciascun componente. Va tuttavia rilevato che tali requisiti possono essere diversi per i prodotti farmaceutici per uso umano (59).
|
Controlli
|
31.
|
Per ciascuna condizione di prova si effettuano anche controlli negativi concomitanti (cfr. il paragrafo 21), consistenti nel solo solvente nel terreno di trattamento; i controlli vanno trattati come le colture di trattamento.
|
|
32.
|
I controlli positivi concomitanti sono necessari per dimostrare la capacità del laboratorio di individuare mutageni alle condizioni del protocollo di prova, l’efficacia del sistema di attivazione metabolica esogena, se del caso, nonché un’adeguata rilevazione dei mutanti TK, sia in piccole colonie/a comparsa tardiva, sia in grandi colonie/a comparsa precoce. Esempi di controlli positivi sono indicati nella tabella 1 in appresso. Sostanze alternative possono essere usate per i controlli positivi, se necessario. Poiché le prove di genotossicità in vitro su cellule di mammifero sono sufficientemente standardizzate per i trattamenti concomitanti di breve durata (3-4 ore) effettuati con e senza attivazione metabolica per la stessa durata di trattamento, l’uso dei controlli positivi può essere limitato a un mutageno che richiede attivazione metabolica. In questo caso, questa sola risposta in un controllo positivo dimostrerà sia l’attività del sistema di attivazione metabolica che la capacità di risposta del sistema di prova. Se utilizzato, il trattamento a lungo termine (ossia 24 ore senza S9) richiede tuttavia l’esecuzione di un proprio controllo positivo, in quanto la durata del trattamento differisce da quella della prova con attivazione metabolica. Ciascun controllo positivo va utilizzato con una o più concentrazioni che in generale danno luogo a un aumento riproducibile e individuabile rispetto al valore di fondo per dimostrare la sensibilità del sistema sperimentale e la risposta non può essere compromessa da una citotossicità superiore ai limiti stabiliti nel presente metodo di prova (cfr. il paragrafo 28).
|
Tabella 1
Sostanze chimiche di riferimento raccomandate per la valutazione della competenza dei laboratori e per la selezione dei controlli positivi
|
Categoria
|
Sostanza
|
N. CAS
|
|
1. Mutageni attivi senza attivazione metabolica
|
|
Metansolfonato di metile
|
66-27-3
|
|
Mitomicina C
|
50-07-7
|
|
4-Nitrochinolina-N–ossido
|
56-57-5
|
|
2. Mutageni che richiedono attivazione metabolica
|
|
Benzo(a)pirene
|
50-32-8
|
|
Ciclofosfamide (monoidrato)
|
50-18-0 (6055-19-2)
|
|
7,12-Dimetilbenzantracene
|
57-97-6
|
|
3-Metilcolantrene
|
56-49-5
|
PROCEDURA
Trattamento con la sostanza chimica in esame
|
33.
|
Le cellule in proliferazione sono trattate con la sostanza chimica in esame in presenza e in assenza di un sistema di attivazione metabolica. L’esposizione deve protrarsi per un periodo adeguato (si considerano di norma adeguate 3-4 ore). Va tuttavia rilevato che tali requisiti possono essere diversi per i prodotti farmaceutici per uso umano (59). Per la prova MLA, nei casi in cui il trattamento di breve durata produca risultati negativi e in assenza di informazioni che indichino l’esigenza di un trattamento più lungo [ad es. analoghi nucleosidici, sostanze chimiche scarsamente solubili, (5) (59)], va presa in considerazione la conduzione della prova con un trattamento più lungo, ossia 24 ore senza S9.
|
|
34.
|
Il numero minimo di cellule utilizzate per ciascuna coltura di prova (di controllo e trattate) in ogni fase della prova è basata sulla frequenza dei mutanti spontanei. Un orientamento generale è trattare e preparare sub-colture di un numero sufficiente di cellule per mantenere almeno 10 ma idealmente 100 mutanti spontanei in ciascuna coltura sperimentale in tutte le fasi della prova (trattamento, espressione fenotipica e selezione dei mutanti) (56).
|
|
35.
|
Per la prova MLA la frequenza accettabile dei mutanti spontanei raccomandata è compresa fra 35-140 × 10-6 (gel di agarosio) e 50-170 × 10-6 (microtitolazione) (cfr. tabella 2). Per ottenere almeno 10 e idealmente 100 mutanti spontanei che sopravvivano al trattamento per ciascuna coltura di prova, è necessario trattare almeno 6 × 106 cellule. Trattare questo numero di cellule e mantenere un numero di cellule sufficiente durante l’espressione e la clonazione per la selezione dei mutanti fornisce un numero sufficiente di mutanti spontanei (10 o più) in tutte le fasi della prova, anche per le colture trattate a concentrazioni con il 90 % di citotossicità (misurata da un RTG del 10 %) (19)(38)(39).
|
|
36.
|
Per la prova TK6, la frequenza dei mutanti spontanei di norma è compresa fra 2 e 10 × 10-6. Per ottenere almeno 10 mutanti spontanei che sopravvivano al trattamento per ciascuna coltura, è necessario trattare almeno 20 × 106 cellule. Trattare un tale numero di cellule fornisce un numero sufficiente di mutanti spontanei (10 o più) anche per le colture trattate a concentrazioni che causano il 90 % di citotossicità durante il trattamento (RS 10 %). Inoltre durante il periodo di espressione per la selezione dei mutanti va coltivato e piastrato un numero sufficiente di cellule (60).
|
Tempo dell’espressione fenotipica e misurazione della citotossicità e della frequenza dei mutanti
|
37.
|
Alla fine del periodo di trattamento le cellule sono coltivate per un tempo definito per consentire l’espressione fenotipica quasi ottimale delle mutazioni neoindotte; specifica di ciascuna linea cellulare. Per la prova MLA, il periodo di espressione fenotipica è di 2 giorni. Per la prova TK6, il periodo di espressione fenotipica è di 3-4 giorni. Se si utilizza un periodo di trattamento di 24 ore, il periodo di espressione inizia alla fine del trattamento.
|
|
38.
|
Durante il periodo di espressione fenotipica le cellule sono conteggiate con cadenza giornaliera. Per la prova MLA le conte giornaliere delle cellule sono utilizzate per calcolare la crescita in sospensione quotidiana (SG). In seguito al periodo di espressione di 2 giorni, le cellule sono sospese nel terreno di coltura con e senza agente selettivo per determinare rispettivamente il numero dei mutanti (piastre di selezione) e l’efficienza di clonazione (piastre di vitalità). Per la prova MLA esistono due metodi ugualmente accettabili per clonare la selezione dei mutanti: il primo si avvale del gel di agarosio e l’altro utilizza un terreno di coltura liquido in piastre a 96 pozzetti (19) (38) (39). La clonazione nella prova TK6 è condotta utilizzando terreni di coltura liquidi e piastre a 96 pozzetti (16).
|
|
39.
|
La trifluorotimidina (TFT) è l’unico agente selettivo raccomandato per i mutanti TK (61).
|
|
40.
|
Per la prova MLA, le piastre di agarosio e le piastre di microtitolazione sono sottoposte a conta dopo un’incubazione di 10-12 giorni. Per la prova TK6, le colonie nelle piastre di microtitolazione sono conteggiate dopo 10-14 giorni per i mutanti a comparsa precoce. Al fine di recuperare i mutanti TK6 a crescita lenta (comparsa tardiva), è necessario rialimentare le cellule con terreno di coltura e TFT, previa conta dei mutanti a comparsa precoce e quindi incubare le piastre per 7-10 giorni supplementari (62). Cfr. i paragrafi 42 e 44 per una discussione relativa all’enumerazione dei mutanti TK a crescita lenta e normale.
|
|
41.
|
Nell’appendice 2 sono riportati i calcoli appropriati per le due prove, compresi i due metodi (gel di agarosio e microtitolazione) per la prova MLA. Per il metodo del gel di agarosio della prova MLA, si conteggiano le colonie e si adegua il numero di mutanti per l’efficienza di clonazione per calcolare una MF. Per la versione a microtitolazione delle prove MLA e TK6, l’efficienza di clonazione di entrambe le piastre di selezione ed efficienza di clonazione è determinata secondo la distribuzione di Poisson (63). La MF è calcolata a partire da queste due efficienze di clonazione.
|
Caratterizzazione della colonia mutante
|
42.
|
Per la prova MLA, se la sostanza chimica in esame è positiva (cfr. i paragrafi 62 e 63), la caratterizzazione per dimensione o crescita della stessa deve essere effettuata su almeno una delle colture trattate (in generale quella con la concentrazione positiva più alta) nonché sui controlli negativi e positivi. Se la sostanza chimica in esame è negativa (cfr. il paragrafo 64), la caratterizzazione della colonia mutante va effettuata sui controlli negativi e positivi. Per il metodo della microtitolazione della prova MLA, una piccola colonia di mutanti è definita come quella che copre meno del 25 % del diametro del pozzetto e una grande colonia di mutanti come quella che copre oltre il 25 % del diametro del pozzetto. Per la versione con gel di agarosio, si utilizza un contatore di colonie automatico per conteggiare le colonie di mutanti e determinare la dimensione della colonia. Gli approcci alla determinazione della dimensione della colonia sono illustrati in dettaglio nella letteratura scientifica (19)(38)(40). La caratterizzazione della colonia nei controlli negativi e positivi è necessaria per dimostrare che gli studi sono stati adeguatamente condotti.
|
|
43.
|
La sostanza chimica in esame non può essere determinata come negativa se i mutanti di entrambe le colonie, grandi e piccole, non sono adeguatamente rilevati nel controllo positivo. La caratterizzazione della colonia può essere utilizzata per ottenere informazioni generali relative alla capacità della sostanza chimica in esame di indurre mutazioni puntiformi e/o fenomeni cromosomici (paragrafo 4).
|
|
44.
|
TK6: i mutanti a crescita normale e lenta si differenziano per il tempo di incubazione (cfr. il paragrafo 40). Per la prova TK6 in generale si conteggiano sia i mutanti a comparsa precoce, sia quelli a comparsa tardiva, per tutte le colture, compresi i controlli negativi e positivi. La caratterizzazione della colonia nei controlli negativi e positivi è necessaria per dimostrare che gli studi sono stati adeguatamente condotti. La sostanza chimica in esame non può essere determinata come negativa se i mutanti di entrambe le colonie a comparsa precoce e tardiva non sono adeguatamente rilevati nel controllo positivo. La caratterizzazione della colonia può essere utilizzata per ottenere informazioni generali relative alla capacità della sostanza chimica in esame di indurre mutazioni puntiformi e/o fenomeni cromosomici (paragrafo 4).
|
Competenza del laboratorio
|
45.
|
Al fine di dimostrare una sufficiente esperienza con un metodo di prova prima di utilizzarlo regolarmente, il laboratorio deve aver effettuato una serie di esperienze con sostanze di riferimento positive che agiscono mediante meccanismi differenti (almeno una con attivazione metabolica e una senza attivazione metabolica, con sostanze selezionate dalle sostanze chimiche elencate nella tabella 1) e con vari controlli negativi (comprese colture non trattate e diversi solventi/mezzi disperdenti). Le risposte dei controlli positivi e negativi devono essere coerenti con la letteratura scientifica. Questo requisito non si applica ai laboratori che hanno già maturato un’esperienza, vale a dire che dispongono di una banca di dati storici quale definita ai paragrafi 47-50. Per la prova MLA, i valori ottenuti per i controlli negativi e positivi devono essere coerenti con le raccomandazioni dell’International Workshop on Genotoxicity Testing (IWGT) (cfr. tabella 2).
|
|
46.
|
Una selezione di sostanze chimiche utilizzate come sostanze di controllo positivo (cfr. tabella 1) deve essere verificata con trattamenti di breve e lunga durata (se si utilizzano trattamenti lunghi) in assenza attivazione metabolica, e anche con trattamenti brevi in presenza di attivazione metabolica, allo scopo di dimostrare che il laboratorio dispone della competenza necessaria per individuare le sostanze chimiche mutagene, determinare l’efficacia del sistema di attivazione metabolica e dimostrare l’adeguatezza delle condizioni di crescita delle cellule durante il trattamento, l’espressione fenotipica e la selezione dei mutanti nonché dei metodi di conteggio. Occorrerà definire un intervallo di concentrazione delle sostanze selezionate che consenta di ottenere aumenti riproducibili e correlati alla concentrazione rispetto ai valori di fondo allo scopo di dimostrare la sensibilità e l’intervallo dinamico del sistema di prova.
|
Dati storici di controllo
|
47.
|
Il laboratorio deve stabilire:
|
—
|
un intervallo e una distribuzione dei controlli positivi storici;
|
|
—
|
un intervallo e una distribuzione dei controlli negativi storici (non trattati, con solvente).
|
|
|
48.
|
Al momento della prima acquisizione di dati per stabilire la distribuzione dei controlli negativi storici, i dati dei controlli negativi concomitanti devono essere coerenti con i dati pubblicati. Con l’aumento dei dati sperimentali aggiunti alla distribuzione dei controlli, i controlli negativi concomitanti dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % di tale distribuzione (64) (65).
|
|
49.
|
La banca dati dei controlli negativi storici del laboratorio deve inizialmente essere costituita con un minimo di 10 esperimenti ma preferibilmente con almeno 20 esperimenti svolti in condizioni sperimentali analoghe. I laboratori devono utilizzare metodi di controllo della qualità, quali diagrammi di controllo (ad esempio carte C o carte X medio (65)), per rilevare la variabilità dei loro dati di controllo positivi e negativi e dimostrare che la metodologia è "sotto controllo" nel laboratorio (66). Ulteriori dettagli e raccomandazioni su come sviluppare e utilizzare i dati storici sono reperibili nella letteratura scientifica (64).
|
|
50.
|
I dati dei controlli negativi consistono nelle frequenze di mutanti da un’unica coltura o preferibilmente da colture replicate, come descritto al paragrafo 27. I controlli negativi concomitanti dovrebbero idealmente situarsi entro i limiti di tale controllo al 95 % della distribuzione della banca dati dei controlli negativi storici del laboratorio. Se i dati dei controlli negativi si situano al di fuori del limite di controllo al 95 %, la loro inclusione nella distribuzione dei controlli storici può essere accettata a condizione che tali dati non siano valori erratici estremi, che sia provato che il sistema di prova è "sotto controllo" (cfr. il paragrafo 49) e che non vi sono stati problemi tecnici o errori umani.
|
|
51.
|
Qualsiasi modifica del protocollo sperimentale deve essere valutata in termini di coerenza dei dati con le banche dati storiche esistenti del laboratorio. Eventuali notevoli incoerenze devono tradursi nella creazione di una nuova banca dati storica sui controlli.
|
DATI E RELAZIONE
Presentazione dei risultati
|
52.
|
La presentazione dei dati per entrambe le prove MLA e TK6 deve includere, sia per le colture trattate che per quelle di control4lo, i dati necessari ai fini del calcolo della citotossicità (rispettivamente RTG o RS) e le frequenze dei mutanti, come indicato oltre.
|
|
53.
|
Per la prova MLA, per ciascuna coltura si indicano i dati relativi ai tassi RSG, RTG, l’efficienza di clonazione al momento della selezione dei mutanti e il numero di colonie di mutanti (gel di agarosio) o il numero di pozzetti vuoti (microtitolazione). La MF deve essere espressa come il numero delle cellule mutanti per milione di cellule sopravvissute. Se la risposta è positiva, per almeno una concentrazione della sostanza chimica in esame (in generale la concentrazione positiva più elevata) si indicano le MF delle colonie piccole e grandi (e/o la percentuale della MF totale) nonché i controlli negativi e positivi. In caso di risposta negativa, si indicano le MF delle colonie piccole e grandi per i controlli nativi e positivi.
|
|
54.
|
Per la prova TK6, per il tasso di RS si indicano i dati relativi a ciascuna coltura, l’efficienza di clonazione al momento della selezione dei mutanti e il numero di pozzetti vuoti per i mutanti a comparsa precoce e quelli a comparsa tardiva. La MF va espressa come numero di cellule mutanti per numero di cellule sopravvissute e include la MF totale nonché la MF (e/o la percentuale della MF totale) dei mutanti a comparsa precoce e tardiva.
|
Criteri di accettabilità
|
55.
|
Per entrambe le prove MLA e TK6, i seguenti criteri devono essere soddisfatti prima di determinare i risultati complessivi di una specifica sostanza chimica in esame:
|
—
|
sono state applicate due condizioni sperimentali (trattamento di breve durata con e senza attivazione metabolica, cfr. il paragrafo 33), salvo nel caso in cui una sia abbia prodotto risultati positivi.
|
|
—
|
Deve essere possibile analizzare un numero adeguato di cellule e concentrazioni (cfr. i paragrafi 27, 34-36).
|
|
—
|
I criteri di selezione della concentrazione massima sono coerenti con quelli descritti ai paragrafi 28-30.
|
|
Criteri di accettabilità per i controlli positivi e negativi
|
56.
|
L’analisi del gruppo di lavoro di esperti MLA dell’IWGT condotta su un’ingente mole di dati MLA ha permesso di raggiungere il consenso internazionale relativamente ai criteri specifici di accettabilità della prova MLA (1)(2)(3)(4)(5). Il presente metodo di prova fornisce pertanto raccomandazioni specifiche per determinare l’accettabilità dei controlli negativi e positivi nonché per valutare i risultati della singola sostanza nella prova MLA. La prova TK6 dispone di una banca dati molto più ristretta e non è stata sottoposta alla valutazione di un gruppo di lavoro.
|
|
57.
|
Per la prova MLA, in ciascuna sperimentazione si valuta se il controllo non trattato/con solvente soddisfa i criteri di accettazione del gruppo di lavoro MLA dell’IWGT ((4) e tabella 2, oltre) relativamente a: 1) MF (si osservi che i valori MF ritenuti accettabili dall’IWGT sono diversi per le versioni con gel di agarosio e microtitolazione della prova MLA), 2) l’efficienza di clonazione (CE) al momento della selezione dei mutanti e 3) la crescita in sospensione (SG) per il controllo con solvente (cfr. l’appendice 2 per le formule).
Tabella 2
Criteri di accettabilità per la prova MLA
|
Parametro
|
Metodo del gel di agarosio
|
Metodo della microtitolazione
|
|
Frequenza dei mutanti
|
35 – 140 × 10-6
|
50 – 170 × 10-6
|
|
Efficienza di clonazione
|
65 – 120 %
|
65 – 120 %
|
|
Crescita in sospensione
|
Fattore 8 – 32 (trattamento di 3-4 ore) Fattore 32 – 180 (trattamento di 24 ore, se effettuato)
|
Fattore 8 – 32 (trattamento di 3-4 ore) Fattore 32 – 180 (trattamento di 24 ore, se effettuato)
|
|
|
58.
|
Per la prova MLA, ciascuna prova deve anche valutare se il o i controlli positivi soddisfano almeno uno dei due criteri di accettazione elaborati dal gruppo di lavoro dell’IWGT:
|
—
|
il controllo positivo deve dimostrare un incremento assoluto della MF totale, ossia un incremento superiore ai mutanti spontanei di fondo [MF indotta, IMF] di almeno 300 × 10-6. Almeno il 40 % dell’IMF deve rispecchiarsi nella MF della piccola colonia.
|
|
—
|
Il controllo positivo presenta un incremento di MF nella piccola colonia di almeno 150 × 10-6 rispetto a quanto osservato nel controllo concomitante non trattato/con solvente (IMF della piccola colonia pari a 150 × 10-6).
|
|
|
58.
|
Per una prova TK6, la prova è ritenuta accettabile se il controllo negativo concomitante è considerato accettabile per essere aggiunto nella banca dati sui controlli negativi storici di laboratorio come descritto ai paragrafi 48-49. Inoltre, i controlli positivi concomitanti (cfr. il paragrafo 32) devono produrre risposte compatibili con quelle generate nella banca dati dei controlli positivi storici e produrre un aumento statisticamente significativo rispetto al controllo negativo concomitante.
|
|
60.
|
Per entrambe le prove il limite superiore della citotossicità osservata nella coltura di controllo positiva deve essere uguale a quello delle colture sperimentali, ossia, il tasso RTG/RS non deve essere inferiore al 10 %. È sufficiente utilizzare una concentrazione unica (o una delle concentrazioni delle colture di controllo positivo se si utilizza più di una concentrazione) per dimostrare che i criteri di accettazione del controllo positivo sono soddisfatti. Inoltre, la MF del controllo positivo deve situarsi nell’intervallo accettabile stabilito per il laboratorio.
|
Analisi e interpretazione dei risultati
|
61.
|
Per la prova MLA, il gruppo di lavoro Mouse Lymphoma Expert dell’IWGT ha condotto un importante lavoro sulla rilevanza biologica e i criteri di risposta positiva (4). Pertanto il presente metodo di prova fornisce raccomandazioni specifiche per interpretare i risultati della prova MLA sulla sostanza chimica (cfr. i paragrafi 62-64). La prova TK6 dispone di una banca dati molto più ristretta e non è stata sottoposta alla valutazione di un gruppo di lavoro. Le raccomandazioni per interpretare i dati della prova TK6 sono quindi fornite in termini più generali (cfr. i paragrafi 65-66). Le raccomandazioni supplementari si applicano a entrambe le prove (cfr. i paragrafi 67-71).
|
MLA
|
62.
|
Si raccomanda di adottare un approccio volto a definire le risposte positive e negative per assicurare che l’incremento della MF sia biologicamente rilevante. Anziché l’analisi statistica utilizzata in generale per altre prove, esso si fonda sull’uso di una frequenza dei mutanti indotta (ossia un incremento della MF superiore al controllo concomitante) predefinita, designata come "fattore di valutazione globale" (Global Evaluation Factor, GEF) e basata sull’analisi della distribuzione dei dati relativi alla MF del controllo negativo ottenuti dai laboratori partecipanti (4). Per la versione con gel di agarosio della prova MLA, il GEF è 90 × 10-6 e per la versione con microtitolazione della prova MLA il GEF è 126 × 10-6.
|
|
63.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente positiva se, in una qualsiasi delle condizioni sperimentali esaminate (cfr. il paragrafo 33), l’incremento della MF superiore al fondo concomitante è superiore al valore GEF e l’incremento è correlato alla concentrazione (ad es. mediante un test di tendenza). La sostanza chimica in esame è quindi ritenuta in grado di indurre mutazioni nel sistema di prova.
|
|
64.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente negativa se, in una qualsiasi delle condizioni sperimentali esaminate (cfr. il paragrafo 33), non risulta una risposta correlata alla concentrazione o se l’incremento della MF non è superiore al valore GEF. La sostanza chimica in esame è quindi ritenuta non in grado di indurre mutazioni nel sistema di prova.
|
TK6
|
65.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente positiva se, in una qualsiasi delle condizioni sperimentali esaminate (cfr. il paragrafo 33):
|
—
|
almeno una delle concentrazioni di prova presenta un aumento statisticamente significativo rispetto al controllo negativo concomitante;
|
|
—
|
un metodo adeguato di analisi della tendenza evidenzia che l’aumento è collegato alla dose (cfr. il paragrafo 33);
|
|
—
|
vi sono risultati che non rientrano nella distribuzione dei dati dei controlli negativi storici (ad esempio limite di controllo al 95 % di una distribuzione di Poisson; cfr. il paragrafo 48).
|
Se tutti i criteri sono soddisfatti, la sostanza chimica in esame è ritenuta in grado di indurre mutazioni nel sistema di prova. Raccomandazioni relative ai metodi statistici più appropriati figurano nella letteratura scientifica (66) (67).
|
|
66.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata chiaramente negativa se, in tutte le condizioni sperimentali esaminate (cfr. il paragrafo 33):
|
—
|
nessuna delle concentrazioni sperimentali presenta un incremento statisticamente significativo rispetto al concomitante controllo negativo;
|
|
—
|
un test di tendenza appropriato dimostra che non vi è aumento correlato alla concentrazione;
|
|
—
|
tutti i risultati si situano entro la distribuzione dei dati di controllo negativi storici (ad es. limite del controllo al 95 % in base alla distribuzione di Poisson, cfr. il paragrafo 48).
|
La sostanza chimica in esame è quindi ritenuta non in grado di indurre mutazioni nel sistema di prova.
|
Per entrambe le prove MLA e TK6:
|
67.
|
se la concentrazione massima è basata sulla citotossicità, la concentrazione più elevata dovrebbe mirare a realizzare un tasso di RTG/RS compreso fra 20 e 10 %. Opinione prevalente è che occorre prestare un’attenzione particolare nell’interpretare i risultati positivi ottenuti solo a un tasso di RTG/RS compreso fra il 20 e il 10 % e che un risultato non sarebbe considerato positivo se l’incremento della MF si verificasse solo a un tasso di RTG/RS del 10 % o inferiore (se valutato) (2)(59).
|
|
68.
|
In talune circostanze le informazioni supplementari possono contribuire a determinare se una sostanza chimica in esame non sia mutagena nel caso in cui non vi siano colture che presentano un tasso di RTG/RS compreso fra il 10 e il 20 %. Tali situazioni sono delineate come segue. 1) Non vi sono prove di mutagenicità (ad es. nessuna risposta alla dose, nessuna frequenza dei mutanti superiore a quelle osservate nei controlli negativi concomitanti o negli intervalli storici di fondo, ecc.) in una serie di dati puntuali del tasso di RTG/RS compresi fra il 100 e il 20 % con almeno un dato puntuale del tasso di RTG/RS compreso fra il 20 e il 25 %. 2) Non vi sono prove di mutagenicità (ad es. nessuna risposta alla dose, nessuna frequenza dei mutanti superiore a quelle osservate nei controlli negativi concomitanti o negli intervalli storici di fondo, ecc.) in una serie di dati puntuali del tasso di RTG/RS situati fra il 100 e il 25 % e si registra anche un dato puntuale del tasso di RTG/RS negativo lievemente inferiore al 10 %. In entrambe le situazioni si può concludere che la sostanza in esame è negativa.
|
|
69.
|
Non è necessario verificare una risposta palesemente positiva o negativa.
|
|
70.
|
Se la risposta non è chiaramente negativa o positiva, come descritto in precedenza, e/o al fine di contribuire a stabilire la rilevanza biologica di un risultato, i dati devono venire sottoposti al giudizio di esperti e/o a indagini più approfondite. Può essere utile ripetere un esperimento modificandone eventualmente le condizioni (ad esempio, intervallazione delle concentrazioni per aumentare la probabilità di raggiungere i dati puntuali situati nell’intervallo del tasso di RTG/RS compreso fra il 10 e il 20 %, altre condizioni di attivazione metabolica (ossia concentrazione S9 o di origine S9) e durata del trattamento].
|
|
71.
|
In rari casi, anche dopo ulteriori analisi, la serie di dati non consente di valutare i risultati come positivi o negativi. Pertanto la risposta della sostanza chimica in esame va considerata equivoca (interpretata come altrettanto positiva o negativa).
|
RELAZIONE SULL’ESECUZIONE DELLA PROVA
|
72.
|
La relazione sull’esecuzione della prova comprende le informazioni riportate di seguito.
|
|
Sostanza chimica in esame:
|
—
|
origine, numero di lotto e data di scadenza, se disponibile;
|
|
—
|
stabilità della sostanza chimica in esame, se nota;
|
|
—
|
solubilità e stabilità della sostanza in esame nel solvente, se note;
|
|
—
|
misurazione del pH, dell’osmolalità e del precipitato nel terreno di coltura al quale è stata aggiunta la sostanza chimica in esame, se del caso.
|
|
|
Sostanza monocostituente:
|
—
|
aspetto fisico, idrosolubilità e, se del caso, ulteriori proprietà fisico-chimiche;
|
|
—
|
identificazione chimica, come la denominazione IUPAC o CAS, il numero CAS, il codice SMILES o InChI, la formula strutturale, la purezza, l’identità chimica delle impurezze, se del caso e se le condizioni pratiche lo consentono, ecc.
|
|
|
|
Sostanza multicostituente, UVCB e miscele:
|
—
|
caratterizzate nella massima misura possibile con l’identità chimica (vedasi sopra), con la presenza quantitativa e con le proprietà fisico-chimiche pertinenti dei costituenti.
|
|
|
|
|
Solvente:
|
—
|
giustificazione della scelta del solvente;
|
|
—
|
percentuale di solvente nel terreno di coltura finale.
|
|
|
|
Cellule:
|
|
Per le colture madri di laboratorio:
|
—
|
tipo e provenienza delle cellule nonché dati storici del laboratorio che esegue la prova;
|
|
—
|
caratteristiche del cariotipo e/o numero modale di cromosomi;
|
|
—
|
metodi usati per il mantenimento delle colture cellulari;
|
|
—
|
tempi di raddoppiamento delle cellule.
|
|
|
|
|
Condizioni sperimentali:
|
—
|
criteri di selezione delle concentrazioni e del numero di colture cellulari: ad esempio i dati relativi alla citotossicità e ai limiti di solubilità;
|
|
—
|
composizione dei terreni di coltura, concentrazione di CO2, livello di umidità;
|
|
—
|
concentrazione della sostanza chimica in esame espressa come concentrazione finale nel terreno di coltura (ad esempio μg o mg/ml o mM di terreno di coltura);
|
|
—
|
concentrazione (e/o volume) del solvente e della sostanza chimica in esame aggiunti nel terreno di coltura;
|
|
—
|
temperatura di incubazione;
|
|
—
|
durata del trattamento;
|
|
—
|
densità delle cellule durante il trattamento;
|
|
—
|
tipo e composizione del sistema di attivazione metabolica (fonte di S9, metodo di preparazione della miscela S9, concentrazione o volume della miscela S9 e di S9 nel terreno di coltura finale, controlli di qualità S9);
|
|
—
|
sostanze di controllo positive e negative, concentrazioni finali per ciascuna condizione di trattamento;
|
|
—
|
durata del periodo di espressione (con numero di cellule inseminate, sub-colture e protocolli di alimentazione, se del caso);
|
|
—
|
identità e concentrazione dell’agente selettivo;
|
|
—
|
per la prova MLA, si indica la versione utilizzata (gel di agarosio o microtitolazione)
|
|
—
|
criteri di accettabilità delle prove;
|
|
—
|
metodi usati per contare il numero di cellule vitali e mutanti;
|
|
—
|
metodi utilizzati per la misurazione della citotossicità;
|
|
—
|
eventuali informazioni supplementari pertinenti per la citotossicità e metodo utilizzato;
|
|
—
|
durata dei tempi di incubazione dopo la piastratura;
|
|
—
|
definizione delle colonie considerate, per dimensione e tipo (compresi i criteri in base a cui le colonie sono considerate "piccole" o "grandi", se del caso).
|
|
—
|
criteri in base ai quali i risultati sono considerati positivi, negativi o ambigui;
|
|
—
|
metodi utilizzati per determinare il pH, l’osmolalità e la precipitazione, se del caso.
|
|
|
|
Risultati:
|
—
|
numero di cellule trattate e numero di cellule sottoposte a sub-coltura per ciascuna coltura;
|
|
—
|
parametri di tossicità (tasso di RTG per MLA e di RS per TK6);
|
|
—
|
segni di precipitazione e momento della determinazione;
|
|
—
|
numero di cellule piastrate nel terreno di coltura selettivo e in quello non selettivo;
|
|
—
|
numero di colonie nel terreno di coltura non selettivo e numero di colonie resistenti in quello selettivo e relative frequenze dei mutanti;
|
|
—
|
dimensione delle colonie per i controlli negativi e positivi e, se la sostanza chimica in esame è positiva, almeno una concentrazione nonché le relative frequenze dei mutanti;
|
|
—
|
relazione concentrazione-risposta, se possibile;
|
|
—
|
dati sui controlli concomitanti negativi (solvente) e positivi (concentrazioni e solventi);
|
|
—
|
dati sui controlli storici negativi (solvente) e positivi (concentrazioni e solventi), con intervalli, medie e deviazioni standard; numero di prove su cui si basano i controlli storici;
|
|
—
|
analisi statistiche (per le colture singole e le repliche raggruppate se del caso), e gli eventuali valori-p; e per la prova MLA, la valutazione GEF.
|
|
|
|
Discussione dei risultati
|
|
BIBLIOGRAFIA
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Paris.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O’Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In Vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In Vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In Vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411–416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In Vitro Methods: Yan Z and Caldwell(Eds), 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In Vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In Vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In Vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In Vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In Vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Consultabile al seguente indirizzo: [https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In Vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Organisation for Economic Cooperation and Development, Paris.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
Appendice 1
DEFINIZIONI
Aneugeno
: detto di qualsiasi sostanza chimica o processo che, interagendo con le componenti del ciclo mitotico e meiotico di divisione cellulare, determina aneuploidia nelle cellule o negli organismi.
Aneuploidia
: qualsiasi deviazione dal normale numero diploide (o aploide) di cromosomi da parte di uno o più cromosomi, ma non dell'intero corredo di cromosomi (poliploidia).
Mutageni che provocano la sostituzione di coppie di basi
: sostanze che provocano la sostituzione di una o più coppie di basi del DNA.
Sostanza chimica
: una sostanza o una miscela.
Efficienza di clonazione
: percentuale di cellule piastrate a bassa densità in grado di crescere in una colonia che può essere contata.
Clastogeno
: qualsiasi sostanza chimica o processo in grado di provocare aberrazioni cromosomiche strutturali in popolazioni di cellule o organismi.
Citotossicità
: per le prove di cui al presente metodo di prova, la citotossicità è identificata come riduzione della crescita relativa totale (RTG) o della sopravvivenza relativa (RS), rispettivamente per le prove MLA e TK6.
Mutazione "in avanti"
: una mutazione genica in cui dal tipo parentale alla forma mutante si ha alterazione o perdita dell'attività enzimatica o della funzione della proteina codificata.
Mutageni che provocano la mutazione della fase di lettura
: sostanze chimiche che provocano l'inserzione o la delezione di una o più coppie di basi nella molecola di DNA.
Genotossico
: termine generico che comprende tutti i tipi di danno a carico del DNA o dei cromosomi, tra cui rottura del DNA, cancellazioni, riarrangiamenti, mutazioni, aberrazioni cromosomiche e aneuploidia. Non tutti i tipi di effetti genotossici determinano alterazioni cromosomiche o danni permanenti ai cromosomi.
Ricombinazione mitotica
: durante la mitosi, ricombinazione fra cromatidi omologhi risultanti nell'eventuale induzione di rotture del doppio filamento del DNA o in una perdita di eterozigosi.
Mutageno
: un fattore in grado di provocare mutazioni ereditarie della o delle sequenze di coppie di basi del DNA nei geni o della struttura dei cromosomi (aberrazioni cromosomiche).
Frequenza dei mutanti (MF)
: numero di cellule mutanti diviso per il numero di cellule vitali.
Tempo di espressione fenotipica
: il tempo trascorso dal trattamento durante il quale l'alterazione genetica si fissa nel genoma e tutti i prodotti genici preesistenti scompaiono fino all'alterazione del tratto fenotipico.
Tasso di sopravvivenza relativa (RS)
: il tasso di RS è utilizzato come misura della citotossicità del trattamento nella prova TK6. Si tratta dell'efficienza di clonazione (CE) delle cellule piastrate subito dopo il trattamento, adeguata per le eventuali perdite di cellule durante il trattamento, rispetto all'efficienza di clonazione nei controlli negativi.
Crescita in sospensione relativa (RSG)
: per la prova MLA, la crescita totale relativa di due giorni in sospensione della coltura sperimentale rispetto alla crescita totale di due giorni in sospensione del controllo negativo/con solvente (Clive e Spector, 1975). Il tasso di RSG include la crescita relativa della coltura sperimentale rispetto al controllo negativo/con solvente durante il periodo di trattamento.
Crescita relativa totale (RTG)
: il tasso di RTG è utilizzato come misura della citotossicità del trattamento nella prova MLA. È una misura della crescita relativa (del controllo con mezzo disperdente) delle colture sperimentali durante le fasi di trattamento, di espressione di due giorni e di clonazione della selezione dei mutanti della prova. Il tasso di RSG di ciascuna coltura sperimentale è moltiplicato per l'efficienza di clonazione relativa della coltura sperimentale al momento della selezione dei mutanti ed espressa in relazione all'efficienza di clonazione del controllo negativo/con solvente (Clive and Spector, 1975).
Frazioni S9 di fegato
: supernatante dell'omogenato di fegato centrifugato a 9 000 g (estratto di fegato crudo).
Miscela di frazione S9
: miscela di frazione S9 di fegato e dei cofattori necessari all'attività degli enzimi metabolici.
Crescita in sospensione (SG)
: fattore di incremento del numero di cellule durante le fasi di trattamento ed espressione della prova MLA. Il valore di SG è calcolato moltiplicando l'aumento del giorno 1 per l'aumento del giorno 2 nel trattamento di breve durata (3-4 ore). Se si applica un trattamento di 24 ore, il valore di SG è pari all'aumento durante le 24 ore di trattamento moltiplicato per l'aumento dei giorni di espressione 1 e 2.
Controllo con solvente
: termine generico che designa le colture di controllo che ricevono unicamente il solvente utilizzato per dissolvere la sostanza chimica in esame.
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Controllo non trattato
: i controlli non trattati sono colture non sottoposte a trattamento (che non ricevono alcuna sostanza chimica in esame né solvente) ma preparate in modo identico alle culture esposte alla sostanza chimica in esame.
Appendice 2
FORMULE
Citotossicità
Per entrambe le versioni (gel di agarosio e microtitolazione) della prova MLA
La citotossicità è definita come la crescita relativa totale (RTG) che comprende la crescita in sospensione relativa (RSG) durante il periodo di espressione di 2 giorni e l'efficienza di clonazione relativa (RCE) ottenuta al momento della selezione dei mutanti. I tassi di RTG, RSG e RCE sono tutti espressi come percentuali.
Calcolo del tasso di RSG: la crescita in sospensione 1 (SG1) corrisponde al tasso di crescita fra il giorno 0 e il giorno 1 (concentrazione delle cellule il giorno 1 / concentrazione delle cellule il giorno 0) e la crescita in sospensione 2 (SG2) corrisponde al tasso di crescita fra il giorno 1 e il giorno 2 (concentrazione delle cellule il giorno 2 / concentrazione delle cellule il giorno 1). Il tasso di RSG corrisponde al valore totale di SG (SG1 x SG2) della coltura trattata rispetto al controllo non trattato/con solvente. Ossia: RSG = [SG1(test) x SG2(test)] / [SG1(control) x SG2(control)] Il valore di SG1 va calcolato a partire dalla concentrazione delle cellule iniziale utilizzata all'inizio del trattamento delle stesse. Questo spiega un'eventuale citotossicità differenziale che si verifica nella o nelle colture sperimentali durante il trattamento delle cellule.
Il tasso di RCE corrisponde all'efficienza di clonazione relativa della coltura sperimentale rispetto all'efficienza di clonazione relativa del controllo non trattato/con solvente ottenuta al momento della selezione dei mutanti.
Crescita relativa totale (RTG):
RTG=RSG x RCE
TK6
Tasso di sopravvivenza relativa (RS)
La citotossicità è valutata mediante il tasso di sopravvivenza relativa, ossia l'efficienza di clonazione (CE) delle cellule piastrate subito dopo il trattamento, adeguata per le eventuali perdite di cellule durante il trattamento, rispetto all'efficienza di clonazione nei controlli negativi (cui è assegnata una sopravvivenza del 100 %). L'adeguamento per la perdita di cellule durante il trattamento è calcolato come:

Il tasso di RS per una coltura trattata con una sostanza chimica in esame è calcolato come:

Frequenza dei mutanti per entrambe le prove MLA e TK6
La frequenza dei mutanti (MF) è l'efficienza di clonazione delle colonie mutanti nel terreno di coltura selettivo (CEM) adattato per l'efficienza di clonazione nel terreno non selettivo al momento della selezione (CEV). Ossia MF=CEM/CEV. Il calcolo di queste due efficienze di clonazione è descritto oltre per i metodi di clonazione con gel di agarosio e microtitolazione.
MLA, versione gel di agarosio
: nella versione della prova MLA con gel di agarosio, il numero di colonie sulla piastra di selezione dei mutanti (CM) e il numero di colonie presenti su quella non selezionata o destinata a misurare l'efficienza di clonazione (conta della vitalità) (CV) sono ottenuti mediante conteggio diretto dei cloni. Se si piastrano 600 cellule ai fini dell'efficienza di clonazione (CE), le piastre utilizzate per la selezione dei mutanti (CEM) e le piastre non selezionate o destinate a misurare l'efficienza di clonazione (conta della vitalità) (CEV) e che per la selezione dei mutanti si usano 3 x 106 cellule,
CEM = CM / (3 x 106) = (CM / 3) x 10-6
CEV = CV / 600
MLA e TK6, versione a microtitolazione:
nella versione a microtitolazione della prova MLA, i valori CM e CV sono determinati come il prodotto del numero totale di pozzetti (TW) e il numero probabile di colonie per pozzetto (P) sulle piastre di microtitolazione.
CM = PM x TWM
CV = PV x TWV
Dal termine zero della distribuzione di Poisson (Furth et al., 1981), P è dato da
P = - ln (EW / TW)
Dove EW è il numero di pozzetti vuoti e TW è il numero totale di pozzetti. Quindi,
CEM = CM / TM = (PM x TWM) / TM
CEV = CV / TV = (PV x TWV) / TV
Per la versione a microtitolazione della prova MLA, le frequenze dei mutanti delle colonie piccole e grandi si calcolano in modo identico, utilizzando il pertinente numero di pozzetti vuoti per le colonie piccole e grandi.
Per la prova TK6 le frequenze di mutanti delle colonie piccole e grandi sono basate sulla comparsa precoce e tardiva dei mutanti.
B.68 METODO DI PROVA DI EPOSIZIONE IN VITRO DI BREVE DURATA PER L’IDENTIFICAZIONE DI i) SOSTANZE CHIMICHE CHE INDUCONO GRAVI LESIONI OCULARI E ii) SOSTANZE CHIMICHE CHE NON RICHIEDONO CLASSIFICAZIONE PER IRRITAZIONE OCULARE O GRAVI LESIONI OCULARI
INTRODUZIONE
|
1.
|
Il presente metodo di prova è equivalente alla linea guida dell’OCSE per le prove sulle sostanze chimiche n. 491 (2017). L’esposizione di breve durata (STE, Short Time Exposure) è un metodo di prova in vitro che può essere utilizzato, in determinate circostanze e con specifiche limitazioni, per la classificazione delle sostanze chimiche (sostanze e miscele) come sostanze pericolose che inducono gravi lesioni oculari e come sostanze che non richiedono classificazione per irritazione oculare o gravi lesioni oculari, secondo la definizione del Sistema mondiale armonizzato di classificazione e di etichettatura delle sostanze chimiche delle Nazioni Unite (Globally Harmonized System of Classification and Labelling of Chemicals (GHS) (1) e del regolamento (CE) n. 1272/2008 relativo alla classificazione, all’etichettatura e all’imballaggio delle sostanze e delle miscele (CLP, Classification, Labelling, Packaging) (68).
|
|
2.
|
Da molti anni il potenziale di pericolo che una sostanza chimica rappresenta per gli occhi è valutato usando principalmente la prova in vivo sugli occhi del coniglio (metodo B.5 (8), equivalente alla linea guida dell’OCSE n. 405). È generalmente riconosciuto che nel prossimo futuro non sarà disponibile alcuna prova alternativa in vitro che da sola possa sostituire completamente la prova oculare in vivo sul coniglio per prevedere tutte le risposte rientranti nella gamma delle lesioni oculari gravi/irritazione oculare indotte dalle diverse classi chimiche. Non è tuttavia da escludere che la prova oculare sul coniglio (2) possa essere sostituita da combinazioni strategiche di metodi alternativi nell’ambito di una strategia sperimentale (sequenziale). L’approccio "dall’alto" è indicato quando, in base alle informazioni esistenti, si prevede che la sostanza chimica abbia un elevato potenziale di irritazione o induca gravi lesioni oculari, mentre l’approccio "dal basso" è appropriato per le sperimentazioni in cui, in base alle informazioni esistenti, si può prevedere che la sostanza chimica non causi un’irritazione oculare tale da richiedere la classificazione. Sebbene considerato inadeguato a sostituire completamente la prova oculare in vivo sul coniglio, il metodo STE può però inserirsi validamente in una strategia di prove in sequenza per la classificazione e l’etichettatura a fini regolamentari, come l’approccio "dall’alto"/"dal basso", per individuare senza ulteriore sperimentazione: i) le sostanze chimiche che inducono gravi lesioni oculari (categoria 1 del GHS dell’ONU/CLP), e ii) le sostanze chimiche (escluse quelle altamente volatili e tutte quelle solide diverse dai tensioattivi) che non richiedono classificazione per irritazione oculare o gravi lesioni oculari ("senza categoria" del GHS dell’ONU/CLP) (1)(2). Tuttavia, la sostanza chimica che con il metodo STE non risulta causare gravi lesioni oculari (categoria 1 del GHS dell’ONU/CLP) né rientrare in alcuna classificazione ("senza categoria" del GHS dell’ONU/CLP) deve essere sottoposta a ulteriore sperimentazione per stabilire la classificazione definitiva. Occorre inoltre consultare le autorità di regolamentazione competenti prima di utilizzare il metodo STE in un approccio "dal basso" nell’ambito di sistemi di classificazione diversi dal sistema GHS dell’ONU/CLP. La scelta del metodo di prova più adatto e l’uso del presente metodo devono essere valutati nel contesto del documento di orientamento dell’OCSE Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation (14).
|
|
3.
|
Scopo del presente metodo di prova è descrivere le procedure impiegate per valutare il pericolo potenziale che la sostanza chimica in esame presenta per l’occhio, sulla base della capacità della sostanza di indurre citotossicità in una prova di esposizione di breve durata. L’effetto citotossico delle sostanze chimiche sulle cellule epiteliali corneali è un meccanismo d’azione importante che provoca lesioni dell’epitelio corneale e irritazione oculare. La vitalità cellulare nel metodo di prova STE è valutata tramite misurazione quantitativa dei cristalli di formazan blu estratti dalle cellule, che sono prodotti dalle cellule vive per conversione enzimatica del colorante vitale MTT [3-(bromuro di 4,5-dimetiltiazol-2-ile)-2,5-difeniltetrazolio], noto anche come, bromuro di tiazolil blu tetrazolio (3). La vitalità cellulare osservata è confrontata con il controllo con solvente (vitalità relativa) e usata per stimare il pericolo potenziale che la sostanza chimica in esame presenta per l’occhio. La sostanza chimica in esame è classificata nella categoria 1 del GHS dell’ONU/CLP quando, sia al 5 % sia allo 0,05 % di concentrazione, la vitalità cellulare risulta inferiore o pari (≤) al 70 %. È invece considerata "senza categoria" nel GHS dell’ONU/CLP quando, sia al 5 % sia allo 0,05 % di concentrazione, la vitalità cellulare risulta superiore (>) al 70 %.
|
|
4.
|
Nel presente metodo di prova, il termine "sostanza chimica in esame" fa riferimento alla sostanza oggetto della prova a prescindere dall’applicabilità del metodo STE alla sperimentazione sulle sostanze e/o miscele. Le definizioni sono riportate nell’appendice.
|
CONSIDERAZIONI INIZIALI E LIMITI
|
5.
|
Il presente metodo di prova si basa su un protocollo elaborato da Kao Corporation (4), che è stato oggetto di due diversi studi di validazione: l’uno a cura del comitato di validazione della società giapponese per le alternative alla sperimentazione animale (JSAAE, Japanese Society for Alternative to Animal Experiments) (5), l’altro a cura del Centro giapponese per la validazione di metodi alternativi (JACVAM, Japanese Center for the Validation of Alternative Methods) (6). Una revisione inter pares è stata condotta da NICEATM/ICCVAM sulla base dei rapporti degli studi di validazione e dei Background Review Documents sul metodo di prova (7).
|
|
6.
|
I dati ottenuti per le 125 sostanze chimiche (sostanze e miscele) testate con il metodo STE per individuare quelle che inducono gravi lesioni oculari (categoria 1 del GHS dell’ONU/CLP) (1) presentano un’accuratezza complessiva dell’83 % (104/125), una percentuale di falsi positivi dell’1 % (1/86) e una percentuale di falsi negativi del 51 % (20/39), rispetto alla prova in vivo sugli occhi del coniglio (7). La percentuale di falsi negativi ottenuta non è preoccupante in questo contesto perché tutte le sostanze chimiche in esame che producono una vitalità cellulare ≤ 70 % alla concentrazione del 5 % e > 70 % alla concentrazione dello 0,05 % saranno successivamente testate con altri metodi in vitro debitamente validati o, come ultima ratio, mediante la prova oculare in vivo sul coniglio, in funzione dei requisiti regolamentari e in conformità della strategia sperimentale sequenziale e degli approcci basati sul peso dell’evidenza attualmente raccomandati (1) (8). Le prove sono state eseguite prevalentemente su sostanze monocostituente, sebbene esista anche una quantità limitata di dati relativi alle prove sulle miscele. Il metodo di prova è in ogni caso tecnicamente applicabile alle prove sulle sostanze multicostituente e sulle miscele. Tuttavia, prima di testare una miscela con questo metodo di prova per ottenere dati a fini regolamentari, è opportuno chiedersi se, e in caso affermativo perché, i dati ottenuti possono essere ritenuti idonei per i fini regolamentari perseguiti. Tali considerazioni non sono necessarie laddove esista una disposizione normativa che obblighi a sottoporre a prova la miscela. Quando è stato usato per individuare le sostanze chimiche classificate nella categoria 1 del GHS dell’ONU/CLP, il metodo di prova STE non ha mostrato alcun altro limite particolare. I ricercatori potrebbero considerare di usare questo metodo per le sostanze chimiche, accettando una vitalità cellulare ≤ 70 % alle due concentrazioni del 5 % e dello 0,05 % come indice di risposta che induce gravi lesioni oculari, tali da giustificare la classificazione della sostanza nella categoria 1 del GHS dell’ONU/CLP senza ulteriore sperimentazione.
|
|
7.
|
I dati ottenuti per le 130 sostanze chimiche (sostanze e miscele) sottoposte al metodo STE per individuare quelle che non richiedono classificazione per irritazione oculare o gravi lesioni oculari ("senza categoria" del GHS dell’ONU/CLP) presentano un’accuratezza complessiva dell’85 % (110/130), una percentuale di falsi negativi del 12 % (9/73) e una percentuale di falsi positivi del 19 % (11/57), rispetto ai dati ottenuti con la prova in vivo sugli occhi del coniglio (7). Se si escludono dai dati quelli relativi alle sostanze altamente volatili e a quelle solide diverse dai tensioattivi, l’accuratezza complessiva del metodo è pari al 90 % (92/102), la percentuale di falsi negativi al 2 % (1/54) e i falsi positivi al 19 % (9/48) (7). Pertanto i limiti potenziali del metodo di prova STE quando usato per individuare le sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari ("senza categoria" del GHS dell’ONU/CLP) consistono in un’alta percentuale di falsi negativi per i) le sostanze chimiche altamente volatili con una pressione di vapore superiore a 6 kPa e ii) le sostanze chimiche solide (sostanze e miscele) diverse dai tensioattivi e le miscele composte solo da tensioattivi. Queste sostanze chimiche sono escluse dall’ambito di applicabilità del metodo di prova STE (7).
|
|
8.
|
Oltre alle sostanze chimiche di cui i paragrafi 6 e 7, i dati generati dal metodo di prova STE contengono anche dati interni relativi a 40 miscele che, rispetto alla prova oculare di Draize in vivo, presentano un’accuratezza dell’88 % (35/40), una percentuale di falsi positivi del 50 % (5/10) e assenza di falsi negativi (0/30) per fare previsioni sulle miscele che non richiedono classificazione secondo i sistemi GHS dell’ONU/CLP (9). Il metodo STE può pertanto essere applicato in un approccio "dal basso" per individuare le miscele "senza categoria" secondo il GHS dell’ONU/CLP, ad eccezione delle miscele solide diverse da quelle composte solo da tensioattivi, come estensione del suo limite di applicabilità alle sostanze solide. Inoltre le miscele contenenti sostanze con una pressione di vapore superiore a 6 kPa devono essere valutate attentamente per evitare previsioni ottimistiche, e vanno giustificate caso per caso.
|
|
9.
|
Il metodo STE non può essere usato per identificare le sostanze chimiche appartenenti alla categoria 2 del GHS dell’ONU/CLP o alle categorie 2A (irritazione oculare) o 2B (irritazione oculare moderata) del GHS dell’ONU, a causa dell’ingente numero di sostanze chimiche di categoria 1 del GHS dell’ONU sottovalutate e considerate di categoria 2, 2A o 2B e di sostanze chimiche "senza categoria" nel GHS dell’ONU/CLP sopravvalutate e considerate di categoria 2, 2A o 2B (7). A tal fine possono essere necessarie ulteriori prove con un altro metodo adeguato.
|
|
10.
|
Il metodo STE è adatto per le sostanze chimiche in esame disciolte o in sospensione uniforme per almeno 5 minuti in una soluzione salina fisiologica, in dimetilsulfossido (DMSO) al 5 % in soluzione salina, o in olio minerale. Il metodo STE non è adatto per le sostanze insolubili o che non possono essere in sospensione uniforme per almeno 5 minuti in una soluzione salina fisiologica, in dimetilsulfossido (DMSO) al 5 % in soluzione salina, o in olio minerale. È possibile usare olio minerale nel metodo STE perché l’esposizione è di breve durata. Questo metodo è quindi adatto per prevedere il pericolo potenziale che le sostanze chimiche insolubili in esame (ad esempio, alcoli grassi a catena lunga o chetoni) presentano per l’occhio, a condizione che siano miscibili in almeno uno dei tre solventi proposti sopra (4).
|
|
11.
|
Il termine "sostanza chimica in esame" utilizzato nel presente metodo di prova designa l’oggetto della prova (69), a prescindere dall’applicabilità del metodo STE alla sperimentazione su sostanze e/o miscele.
|
PRINCIPIO DELLA PROVA
|
12.
|
Il metodo STE è un saggio di citotossicità in vitro eseguito su un monostrato confluente di cellule di cornea di coniglio dello Statens Seruminstitut (SIRC, Statens Seruminstitut Rabbit Cornea), coltivate su micropiastre in policarbonato a 96 pozzetti (4). Dopo 5 minuti di esposizione alla sostanza in esame si determina quantitativamente la citotossicità misurando, mediante il test dell’MTT (4), la vitalità relativa delle cellule SIRC. Una diminuzione della vitalità cellulare è usata per predire gli effetti nocivi che causano lesioni oculari.
|
|
13.
|
È stato segnalato che l’80 % di una soluzione instillata nell’occhio del coniglio è escreta attraverso il sacco congiuntivale nei tre o quattro minuti successivi all’applicazione, mentre più dell’80 % di una soluzione instillata nell’occhio umano è escreta nell’arco di uno o due minuti (10). Il metodo STE è inteso ad avvicinare questi tempi di esposizione e si avvale della citotossicità come endpoint per valutare le lesioni subite dalle cellule SIRC dopo cinque minuti di esposizione alla sostanza in esame.
|
DIMOSTRAZIONE DELLA COMPETENZA DI LABORATORIO
|
14.
|
Prima di utilizzare il metodo STE qui descritto, i laboratori devono dimostrare le loro competenze tecniche classificando correttamente le undici sostanze raccomandate nella tabella 1. Tali sostanze sono state selezionate quali rappresentative dell’intera gamma di risposte corrispondenti a lesioni oculari gravi o irritazione oculare, sulla base dei risultati di prove in vivo sugli occhi del coniglio (linea guida n. 405) e del sistema di classificazione GHS dell’ONU (1). Altri criteri di selezione sono stati la disponibilità in commercio delle sostanze, la disponibilità di dati di riferimento in vivo di elevata qualità e la disponibilità di dati in vitro di elevata qualità ottenuti con il metodo STE (3). È possibile utilizzare un’altra sostanza per la quale esistano adeguati dati di riferimento in vivo e in vitro se una delle sostanze elencate non è disponibile o se il ricorso ad un’altra sostanza si giustifica, purché si applichino gli stessi criteri descritti nel presente metodo.
Tabella 1
Elenco delle sostanze chimiche per la verifica della competenza tecnica
|
Sostanza
|
N. CAS
|
Classe chimica (70)
|
Stato fisico
|
Categoria GHS ONU/CLP in vivo
(71)
|
Solvente nella prova STE
|
Categoria GHS ONU/CLP STE
|
|
Cloruro di benzalconio (10 %, acquoso)
|
8001-54-5
|
Composto onio
|
Liquido
|
Categoria 1
|
Soluzione salina
|
Categoria 1
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Etere
|
Liquido
|
Categoria 1
|
Soluzione salina
|
Categoria 1
|
|
Acid Red 92
|
18472-87-2
|
Composto eterociclico; composto del bromo; composto del cloro
|
Solido
|
Categoria 1
|
Soluzione salina
|
Categoria 1
|
|
Idrossido di sodio
|
1310-73-2
|
Alcali; sostanza chimica inorganica
|
Solido
|
Categoria 1 (72)
|
Soluzione salina
|
Categoria 1
|
|
Butirolattone
|
96-48-0
|
Lattone, composto eterociclico
|
Liquido
|
Categoria 2A (Categoria 2 del CLP)
|
Soluzione salina
|
Non è possibile fare previsioni
|
|
1-ottanolo
|
111-87-5
|
Alcol
|
Liquido
|
Categoria 2A/B (73) (Categoria 2 del CLP)
|
Oli minerali
|
Non è possibile fare previsioni
|
|
Ciclopentanolo
|
96-41-3
|
alcol; idrocarburo, ciclico
|
Liquido
|
Categoria 2A/B (74) (Categoria 2 del CLP)
|
Soluzione salina
|
Non è possibile fare previsioni
|
|
2-Etossietil acetato
|
111-15-9
|
alcol; etere
|
Liquido
|
Senza categoria
|
Soluzione salina
|
Senza categoria
|
|
Dodecano
|
112-40-3
|
Idrocarburo, aciclico
|
Liquido
|
Senza categoria
|
Oli minerali
|
Senza categoria
|
|
Metilisobutilchetone
|
108-10-1
|
Chetone
|
Liquido
|
Senza categoria
|
Oli minerali
|
Senza categoria
|
|
Solfato di 1,1-dimetilguanidina
|
598-65-2
|
Ammidina; composto di solfo
|
Solido
|
Senza categoria
|
Soluzione salina
|
Senza categoria
|
|
Abbreviazioni: N. CAS (Chemical Abstracts Service Registry Number) = Numero CAS (numero di registrazione nell’inventario europeo delle sostanze chimiche).
|
|
PROCEDURA
Preparazione del monostrato cellulare
|
15.
|
Per l’esecuzione della prova con il metodo STE si utilizza la linea cellulare di cornea di coniglio SIRC. Si raccomanda che le cellule SIRC provengano da una banca di cellule riconosciuta, quale la American Type Culture Collection CCL60.
|
|
16.
|
Le cellule SIRC sono coltivate a 37 °C in atmosfera umidificata al 5 % di CO2, in una fiasca di coltura contenente un mezzo di coltura costituito di mezzo essenziale minimo di Eagle (MEM, Minimum Essential Medium) con aggiunta di siero fetale bovino (FBS, Fetal Bovine Serum) al 10 %, 2 mM di L-glutammina, 50–100 unità/ml di penicillina e 50-100 μg/ml di streptomicina. Le cellule che sono diventate confluenti nella fiasca di coltura devono essere separate per mezzo di una soluzione di tripsina-acido tetraetilendiaminoacetico, con o senza l’ausilio di un raschiatore. Le cellule sono moltiplicate in una fiasca da coltura (ad esempio, 2 o 3 passaggi) prima di essere utilizzate nelle prove di routine e non possono essere sottoposte a più di 25 passaggi dopo lo scongelamento.
|
|
17.
|
Le cellule pronte ad essere utilizzate nella prova STE sono preparate alla densità adeguata e inoculate in piastre a 96 pozzetti. La densità di inoculazione raccomandata è di 6,0 × 103 cellule per pozzetto quando le cellule sono utilizzate quattro giorni dopo l’inoculazione, o 3,0 × 103 cellule per pozzetto quando sono utilizzate cinque giorni dopo l’inoculazione, per un volume di coltura di 200 μl. Le cellule utilizzate per la prova STE che sono inoculate in un mezzo di coltura alla densità adeguata raggiungeranno una confluenza superiore all’80 % al momento della prova, ossia quattro o cinque giorni dopo l’inoculazione.
|
Applicazione della sostanza chimica in esame e delle sostanze di controllo
|
18.
|
Il solvente da privilegiare per disciogliere o sospendere la sostanza chimica in esame è la soluzione salina fisiologica. Se la sostanza chimica in esame si dimostra poco solubile, insolubile o non in grado di essere in sospensione uniforme per almeno cinque minuti nella soluzione salina, si opta per il DMSO al 5 % (n. CAS 67-68-5) in soluzione salina. Se la sostanza chimica in esame non può essere disciolta o messa in sospensione uniforme per almeno cinque minuti nella soluzione salina fisiologica né nel DMSO al 5 % in soluzione salina, il terzo solvente possibile è l’olio minerale (n. CAS 8042-47-5).
|
|
19.
|
Le sostanze chimiche sono disciolte o sospese uniformemente a una concentrazione del 5 % (p/p) nel solvente prescelto e ulteriormente diluite con una serie di diluizioni di fattore 10 fino alle concentrazioni dello 0,5 % e dello 0,05 %. Ogni sostanza chimica deve essere testata alle due concentrazioni (5 % e 0,05 %). Le cellule coltivate sulla piastra a 96 pozzetti sono esposte a 200 μl/pozzetto di soluzione (o sospensione) della sostanza chimica in esame a 5 % o 0,05 % per cinque minuti a temperatura ambiente. Le sostanze chimiche in esame (sostanze monocostituente o sostanze o miscele multicostituente) sono considerate pure e diluite o sospese secondo il presente metodo, indipendentemente dal loro grado di purezza.
|
|
20.
|
Il mezzo di coltura descritto al paragrafo 16 è utilizzato come controllo in ogni piastra di ogni ripetizione. Inoltre, le cellule devono anche essere esposte, per ogni piastra di ogni ripetizione, a campioni di controllo con solvente. È assodato che i solventi indicati al paragrafo 18 non hanno effetti nocivi sulla vitalità delle cellule SIRC.
|
|
21.
|
Nel metodo di prova STE si deve utilizzare laurilsolfato di sodio (SLS) allo 0,01 % come controllo positivo in ogni piastra di ogni ripetizione. Per calcolare la vitalità cellulare del controllo positivo ogni piastra di ogni ripetizione deve includere anche un controllo del solvente con soluzione salina.
|
|
22.
|
Un bianco è necessario per determinare la compensazione della densità ottica, da eseguirsi in pozzetti contenenti solo tampone fosfato in soluzione salina, senza calcio, magnesio (PBS-), né cellule.
|
|
23.
|
Ogni campione (sostanza chimica in esame a 5 % e 0,05 %, controllo con mezzo, controllo con solvente e controllo positivo) deve essere sottoposto a prova con tre repliche per ogni ripetizione, esponendo le cellule a 200 μl della sostanza chimica adeguata, in esame o di controllo, per cinque minuti a temperatura ambiente.
|
|
24.
|
Le sostanze chimiche di riferimento sono utili per valutare il potenziale di irritazione oculare di sostanze chimiche sconosciute appartenenti a una determinata classe di sostanze o prodotti chimici o per valutare il potenziale di irritazione relativo di una sostanza irritante per gli occhi nell’ambito di una serie specifica di risposte d’irritazione.
|
Misurazione della vitalità cellulare
|
25.
|
Dopo l’esposizione lavare le cellule due volte con 200 μl di PBS e aggiungere 200 μl di soluzione MTT (0,5 mg MTT/ml di mezzo di coltura). Dopo due ore di reazione in un incubatore (37 °C, 5 % CO2) decantare la soluzione MTT, estrarre l’MTT formazan aggiungendo 200 μl di 0,04 N di isopropanolo-acido cloridrico per 60 minuti al buio a temperatura ambiente e misurare l’assorbanza della soluzione MTT formazan a 570 nm con un lettore di piastre. Si verifica interferenza tra la sostanza chimica in esame e l’MTT (dovuta a coloranti o a riduttori diretti dell’MTT) solo se una quantità significativa della sostanza chimica in esame permane nel sistema sperimentale dopo il lavaggio che segue l’esposizione, come nel caso della cornea umana ricostruita in 3D o dei tessuti dell’epidermide umana ricostruiti, ma non nelle colture cellulari in 2D utilizzate nel metodo di prova STE.
|
Interpretazione dei risultati e modello predittivo
|
26.
|
I valori della densità ottica (OD, Optical Density) ottenuti per ciascuna sostanza in esame sono poi utilizzati per calcolare la vitalità cellulare rispetto al controllo con solvente, il cui valore è fissato al 100 %. La vitalità cellulare relativa è espressa in percentuale ed è ottenuta dividendo la densità ottica della sostanza chimica in esame per la densità ottica del controllo con solvente dopo aver sottratto la densità ottica del bianco da ciascun valore.

Analogamente, la vitalità cellulare relativa di ogni controllo con solvente è espressa in percentuale ed è ottenuta dividendo la densità ottica di ogni controllo con solvente per la densità ottica del controllo con mezzo, dopo aver sottratto la densità ottica del bianco da ciascun valore.
|
|
27.
|
Si devono eseguire tre ripetizioni indipendenti, contenenti ciascuna tre repliche (ossia n = 9). Si usa la media aritmetica dei tre pozzetti per ogni sostanza chimica in esame e ogni controllo con solvente in ogni ripetizione indipendente per calcolare la media aritmetica della vitalità cellulare relativa. La media aritmetica finale della vitalità cellulare è calcolata a partire dalle tre ripetizioni indipendenti.
|
|
28.
|
Di seguito sono indicati i valori limite della vitalità cellulare per identificare le sostanze chimiche in esame in grado di indurre gravi lesioni oculari (categoria 1 del GHS dell’ONU/CLP) e le sostanze chimiche in esame che non richiedono classificazione per l’irritazione oculare o per gravi lesioni oculari ("senza categoria" del GHS dell’ONU/CLP).
|
Tabella 2
Modello predittivo del metodo di prova STE
|
Vitalità cellulare
|
Classificazione GHS dell’ONU/CLP
|
Applicabilità
|
|
Al 5 %
|
Allo 0,05 %
|
|
> 70 %
|
> 70 %
|
Senza categoria
|
Sostanze e miscele, tranne: i) sostanze altamente volatili con una pressione di vapore superiore a 6 kPa (75) e ii) sostanze chimiche solide (sostanze e miscele) diverse dai tensioattivi e miscele composte solo da tensioattivi.
|
|
≤ 70 %
|
> 70 %
|
Non è possibile fare previsioni
|
Non pertinente
|
|
≤ 70 %
|
≤ 70 %
|
Categoria 1
|
Sostanze e miscele (76)
|
Criteri di accettabilità
|
29.
|
I risultati delle prove sono ritenuti accettabili se sono soddisfatti tutti i criteri seguenti:
|
a)
|
la densità ottica del controllo con mezzo (esposto al mezzo di coltura) è pari o superiore a 0,3 dopo sottrazione della densità ottica del bianco;
|
|
b)
|
la vitalità del controllo con solvente è pari o superiore all’80 % rispetto a quella del controllo con mezzo. Se ad ogni ripetizione si usano controlli con più di un solvente, ogni controllo deve dimostrare una vitalità cellulare superiore all’80 % per poter qualificare le sostanze chimiche saggiate con questi solventi.
|
|
c)
|
La vitalità cellulare risultante dal controllo positivo (0,01 % SLS) si deve situare nell’intervallo tra due deviazioni standard della media storica. I limiti superiore e inferiore di accettabilità per il controllo positivo vanno aggiornati con frequenza, ossia a cadenza trimestrale oppure ogniqualvolta si esegua una prova accettabile nei laboratori in cui le prove sono eseguite sporadicamente (ossia, meno di una volta al mese). Se il laboratorio non porta a termine un numero sufficiente di esperimenti per stabilire una distribuzione dei controlli positivi statisticamente solida, è ammesso l’uso dei limiti superiore e inferiore di accettabilità stabiliti al momento della messa a punto del metodo, ossia tra il 21,1 % e il 62,3 % secondo i dati storici del laboratorio, e la distribuzione interna è determinata durante le prime prove di routine;
|
|
d)
|
la deviazione standard della vitalità cellulare finale risultante da tre ripetizioni indipendenti deve essere inferiore al 15 % per entrambe le concentrazioni, al 5 % e allo 0,05 %, della sostanza chimica in esame.
Se uno o più criteri non sono soddisfatti i risultati devono essere scartati e devono essere condotte altre tre ripetizioni indipendenti.
|
|
DATI E RELAZIONE
Dati
|
30.
|
I dati da riferire sono quelli ottenuti per ogni pozzetto (ad esempio, i valori della vitalità cellulare) di ogni ripetizione, così come la media globale, la deviazione standard e la classificazione.
|
Relazione sull’esecuzione della prova
|
31.
|
I seguenti dati devono figurare nella relazione sull’esecuzione della prova.
|
|
Sostanze chimiche in esame e di controllo
|
—
|
Sostanza monocostituente: identificazione della sostanza chimica, mediante denominazioni quali IUPAC o CAS, il numero CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
—
|
Sostanza multicostituente, UVCB e miscela: caratterizzazione, nella misura del possibile, ad esempio attraverso l’identità chimica (cfr. sopra), la purezza, le proporzioni quantitative e le proprietà fisico-chimiche pertinenti (cfr. sopra) dei costituenti, secondo i dati disponibili.
|
|
—
|
Stato fisico, volatilità, pH, LogP, peso molecolare, classe chimica e altre proprietà fisico-chimiche pertinenti per la realizzazione dello studio, secondo i dati disponibili.
|
|
—
|
Purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico ecc.
|
|
—
|
Trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione).
|
|
—
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili.
|
|
|
|
Condizioni e procedure del metodo di prova
|
—
|
Nome e indirizzo dello sponsor, dell’infrastruttura utilizzata per la prova e del responsabile dello studio.
|
|
—
|
Descrizione del metodo di prova.
|
|
—
|
Linea cellulare utilizzata e sua provenienza, numero di passaggi e livello di confluenza delle cellule utilizzate per la prova.
|
|
—
|
Dettagli della procedura di prova.
|
|
—
|
Numero di ripetizioni e di repliche.
|
|
—
|
Concentrazioni della sostanza chimica in esame (se diverse da quelle raccomandate).
|
|
—
|
Giustificazione della scelta del solvente per ciascuna sostanza chimica in esame.
|
|
—
|
Durata dell’esposizione alla sostanza chimica in esame (se diversa da quella raccomandata).
|
|
—
|
Descrizione delle eventuali modifiche del protocollo sperimentale.
|
|
—
|
Descrizione dei criteri di valutazione e di decisione.
|
|
—
|
Riferimenti alla media e alla deviazione standard storiche dei controlli positivi.
|
|
—
|
Dimostrazione della competenza del laboratorio nell’applicazione del metodo di prova (ad esempio, nel saggio delle sostanze di riferimento) o dimostrazione della riproducibilità delle prestazioni del metodo di prova nel tempo.
|
|
|
|
Risultati
|
—
|
Per ciascuna sostanza chimica in esame e di controllo, e per ciascuna concentrazione testata, presentare in una tabella i valori della densità ottica per ogni pozzetto, la media aritmetica dei valori della densità ottica per ogni ripetizione indipendente, la percentuale della vitalità cellulare per ogni ripetizione indipendente e la media aritmetica finale della percentuale della vitalità cellulare e della deviazione standard per le tre ripetizioni.
|
|
—
|
Risultati del controllo con mezzo, del controllo con solvente e del controllo positivo comprovanti che lo studio soddisfa i criteri di accettabilità.
|
|
—
|
Descrizione di altri effetti osservati.
|
|
—
|
Classificazione generale stabilita con riferimento al modello predittivo/ai criteri di decisione applicati.
|
|
|
|
Discussione dei risultati
|
|
BIBLIOGRAFIA
|
(1)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Disponibile all’indirizzo: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In Vitro Eye Irritation Test Using SIRC Cells. Toxicol. In Vitro 22,760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In Vitro 25,796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In Vitro 27, pp.1 855-1 869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Disponibile all’indirizzo: [http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
Capitolo B.5 del presente allegato, Irritazione/corrosione oculare acuta.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1 648-1 653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In Vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442–449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Organisation for Economic Cooperation and Development, Paris.
|
Appendice
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza" per indicare la percentuale di risultati corretti di un metodo di prova (13).
Sostanza di riferimento
: la sostanza usata come standard di confronto rispetto alla sostanza chimica in esame. La sostanza di riferimento ha le seguenti proprietà: i) origine o origini coerenti e affidabili; ii) analogia strutturale e funzionale alla classe delle sostanze in esame; iii) caratteristiche fisiche/chimiche note; iv) dati di supporto relativi agli effetti noti; v) efficacia nota nell'ambito della risposta auspicata.
Approccio "dal basso"
: l'approccio graduale applicato nel caso di una sostanza chimica che si ritiene non debba essere classificata in termini di irritazione oculare o gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che non richiedono classificazione (esito negativo) rispetto ad altre sostanze chimiche (esito positivo).
Sostanza chimica
: sostanza o miscela.
Irritazione oculare
: la produzione di alterazioni nell'occhio in seguito all'applicazione della sostanza chimica in esame sulla superficie anteriore dell'occhio, totalmente reversibili entro 21 giorni dall'applicazione. Sinonimo di "effetti reversibili sugli occhi" e "categoria 2 del GHS dell'ONU".
Percentuale di falsi negativi
: la percentuale di tutte le sostanze chimiche positive falsamente identificate come negative da un metodo di prova. È un indicatore dell'efficienza del metodo di prova.
Percentuale di falsi positivi
: la percentuale di tutte le sostanze chimiche negative falsamente identificate come positive da un metodo di prova. È un indicatore dell'efficienza del metodo di prova.
Pericolo
: la proprietà intrinseca di un agente o di una situazione che ha il potenziale di causare effetti nocivi se un organismo, un sistema o una (sotto)popolazione vi sono esposti.
Controllo con mezzo
: la replica non trattata che contiene tutti i componenti di un sistema di prova. Il campione subisce il medesimo procedimento dei campioni trattati con la sostanza chimica in esame e degli altri campioni di controllo per determinare se il solvente interagisce con il sistema di prova.
Miscela
: la miscela o soluzione composta di due o più sostanze.
Sostanza monocostituente
: la sostanza, definita in base alla sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno l'80 % (p/p).
MTT
: bromuro di 3-(4,5-dimetiltiazol-2-ile)-2,5-difeniltetrazolio; tiazolil blu tetrazolio bromuro.
Sostanza multicostituente
: la sostanza, definita in base alla sua composizione quantitativa, in cui più di un costituente principale è presente in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). La sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente risiede nel fatto che la miscela è ricavata mischiando due o più sostanze senza che avvenga una reazione chimica, mentre la sostanza multicostituente è il risultato di una reazione chimica.
OD
: densità ottica.
Controllo positivo
: la replica contenente tutti i componenti di un sistema di prova, trattata con una sostanza che notoriamente induce una risposta positiva. Per poter valutare la variabilità nel tempo della risposta dei controlli positivi, l'entità della risposta positiva non deve essere eccessiva.
Pertinenza
: descrizione del rapporto tra la prova e l'effetto studiato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (10).
Affidabilità
: la misura in cui il metodo di prova può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna al laboratorio e la ripetibilità fra i laboratori (13).
Sensibilità
: la percentuale di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l'accuratezza del metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza del metodo di prova (10).
Gravi lesioni oculari
: la produzione di danni ai tessuti oculari o indebolimento grave della vista in seguito all'applicazione della sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Sinonimo di "effetti irreversibili sugli occhi" e "categoria 1 del GHS dell'ONU".
Controllo con solvente/mezzo disperdente
: il campione non trattato che contiene tutti i componenti del sistema di prova, compreso il solvente o il mezzo disperdente, e subisce il medesimo procedimento dei campioni trattati con la sostanza chimica in esame e degli altri campioni di controllo per stabilire la risposta di base nei campioni trattati con la sostanza chimica in esame disciolta nello stesso solvente o mezzo disperdente. Nelle prove con controlli paralleli con mezzo, questo campione dimostra anche se il solvente o il mezzo disperdente può interagire con il sistema sperimentale.
Specificità
: la percentuale di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l'accuratezza del metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza del metodo (13).
Sostanza
: l'elemento chimico e i suoi composti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a mantenerne la stabilità e le impurità derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione.
Tensioattivo (o surfattante)
: la sostanza chimica, come un detergente, in grado di ridurre la tensione superficiale di un liquido consentendo quindi di formare schiuma o penetrare solidi; è noto anche come agente umettante.
Sostanza chimica in esame
: la sostanza o miscela testata applicando il presente metodo di prova.
Strategia di prova in sequenza
: la strategia graduale di prova in cui sono vagliate, secondo un ordine specificato, tutte le informazioni disponibili sulla sostanza chimica in esame, seguendo in ciascuna fase un approccio basato sul peso dell'evidenza, al fine di stabilire se vi sono informazioni sufficienti per prendere una decisione di classificazione del pericolo prima di procedere alla fase successiva. Se è possibile assegnare il potenziale di irritazione della sostanza chimica in esame in base alle informazioni disponibili, non è necessario svolgere altre prove. Se non è possibile assegnare il potenziale di irritazione della sostanza chimica in esame in base alle informazioni disponibili, si esegue una procedura sperimentale graduale in sequenza sugli animali fino a stabilire una classificazione inequivocabile.
Approccio "dall'alto"
: l'approccio graduale applicato nel caso di una sostanza chimica in esame sospettata di causare gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che inducono gravi lesioni oculari (esito positivo) rispetto ad altre sostanze chimiche (esito negativo).
Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (GHS dell'ONU)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi e livelli standardizzati di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di prudenza e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (1).
Categoria 1 del GHS dell'ONU
: cfr. "Gravi lesioni oculari".
Categoria 2 del GHS dell'ONU
: cfr. "Irritazione oculare".
Senza categoria del GHS dell'ONU/CLP
: la sostanza chimica che non rientra nella categoria 1 o 2 del GHS dell'ONU/CLP (o categoria 2A o 2B del GHS dell'ONU).
UVCB (Substance of Unknown or Variable Composition, Complex Reaction Products or Biological Materials)
: sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici.
B.69 METODO DI PROVA SU MODELLO DI EPITELIO CORNEALE UMANO RICOSTITUITO (RhCE) PER L’IDENTIFICAZIONE DELLE SOSTANZE CHIMICHE CHE NON RICHIEDONO CLASSIFICAZIONE NÉ ETICHETTATURA PER IRRITAZIONE OCULARE O GRAVI LESIONI OCULARI
INTRODUZIONE
|
1.
|
Il presente metodo di prova è equivalente alla linea guida dell’OCSE per le prove sulle sostanze chimiche n. 492 (2017). Per «grave lesione oculare» s’intende la lesione dei tessuti oculari o la degradazione grave della vista causate dall’applicazione della sostanza chimica in esame sulla superficie anteriore dell’occhio, non completamente reversibile entro 21 giorni dall’applicazione, secondo la definizione del Sistema mondiale armonizzato di classificazione e di etichettatura delle sostanze chimiche delle Nazioni Unite (GHS, Globally Harmonized System of Classification and Labelling of Chemicals) (1) e del regolamento (CE) n. 1272/2008 relativo alla classificazione, all’etichettatura e all’imballaggio delle sostanze e delle miscele (CLP, Classification, Labelling, Packaging) (77). Sempre secondo il GHS dell’ONU e il CLP, per «irritazione oculare» s’intende la produzione di alterazioni nell’occhio a seguito dell’applicazione della sostanza chimica in esame sulla superficie anteriore dell’occhio, totalmente reversibili entro 21 giorni dall’applicazione. Le sostanze chimiche in esame che inducono gravi lesioni oculari sono classificate nella categoria 1 del GHS dell’ONU e del CLP, mentre quelle che inducono irritazione oculare sono classificate nella categoria 2 del GHS dell’ONU e del CLP. Le sostanze chimiche in esame non classificate in termini di irritazione oculare o gravi lesioni oculari sono definite come non rispondenti ai criteri di classificazione delle categorie 1 o 2 (2A o 2B) del GHS dell’ONU e del CLP, vale a dire sono considerate «senza categoria» nei suddetti sistemi.
|
|
2.
|
Tradizionalmente, la valutazione delle lesioni oculari gravi o dell’irritazione oculare implicava l’uso di animali da esperimento (metodo di prova B.5) (2). La scelta del metodo di prova più adatto e l’uso del presente metodo devono essere valutati nel contesto del documento di orientamento dell’OCSE Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation (39).
|
|
3.
|
Il presente metodo descrive una procedura in vitro che permette di identificare le sostanze chimiche (sostanze e miscele) che non richiedono classificazione né etichettatura per l’irritazione oculare o per gravi lesioni oculari in conformità dei sistemi GHS dell’ONU e CLP. Utilizza un modello di epitelio corneale umano ricostituito (Reconstructed human Cornea-Like Epithelium, RhCE) che riproduce fedelmente le proprietà istologiche, morfologiche biochimiche e fisiologiche dell’epitelio corneale umano. Altri quattro metodi di prova in vitro sono stati validati, considerati scientificamente validi e adottati come metodi prova B.47 (3), B.48 (4), B.61 (5) e B.68 (6) per studiare l’endpoint per la salute umana delle gravi lesioni oculari/dell’irritazione oculare.
|
|
4.
|
Il presente metodo contempla due prove validate che usano modelli di RhCE disponibili in commercio. La prova d’irritazione oculare EpiOcular™ Eye Irritation Test («EpiOcular™ EIT») e la prova d’irritazione oculare su epitelio corneale umano SkinEthic™ Human Corneal Epithelium Human Corneal Epithelium Eye Irritation Test («SkinEthic™ HCE EIT») sono state usate in studi di validazione (7)(8)(9)(10)(11)(12)(13) per valutare l’irritazione oculare/le gravi lesioni oculari. Queste due prove utilizzano come sistema sperimentale modelli tessutali di RhCE disponibili in commercio, di seguito denominati «metodi di riferimento validati» (Validated Reference Method, VRM), nella fattispecie VRM1 e VRM2. In base a questi studi di validazione e alla revisione inter pares indipendente (9)(12), è stato concluso che le prove d’irritazione oculare EpiOcular™ EIT e SkinEthic™ HCE EIT possono identificare correttamente le sostanze chimiche (sostanze e miscele) che non richiedono classificazione né etichettatura per l’irritazione oculare o per gravi lesioni oculari in conformità del sistema GHS dell’ONU, e sono state raccomandate per tale finalità in quando riconosciute scientificamente valide (13).
|
|
5.
|
È generalmente riconosciuto che nel prossimo futuro non sarà disponibile alcun metodo di prova in vitro che da solo possa sostituire completamente la prova oculare di Draize in vivo (2) (14) per prevedere tutte le risposte rientranti nella gamma delle lesioni oculari gravi/irritazione oculare indotte dalle diverse classi chimiche. Non è tuttavia da escludere che la prova oculare di Draize possa essere completamente sostituita da combinazioni strategiche di metodi alternativi nell’ambito di strategie di prova (sequenziali), come l’approccio «dal basso»/«dall’alto» (15). L’approccio «dal basso» (15) è indicato per i casi in cui, in base alle informazioni esistenti, la sostanza chimica non dovrebbe causare un’irritazione oculare tale da richiedere la classificazione, mentre l’approccio «dall’alto» (15) è appropriato nei casi in cui, in base alle informazioni esistenti, si prevede che la sostanza chimica induca gravi lesioni oculari. Le prove d’irritazione oculare EpiOcular™ EIT e SkinEthic™ HCE EIT sono raccomandate per identificare, senza ulteriore sperimentazione, le sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari secondo il sistema GHS dell’ONU/CLP («senza categoria»), nell’ambito di una strategia di sperimentazione quale l’approccio «dal basso»/«dall’alto» suggerito da Scott et al., ad esempio come prima fase di un approccio «dal basso» o come una delle ultime fasi di un approccio «dall’alto» (15). Tuttavia, le prove d’irritazione oculare EpiOcular™ EIT e SkinEthic™ HCE EIT non sono concepite per stabilire se una sostanza chimica rientra nella categoria 1 (gravi lesioni oculari) o nella categoria 2 (irritazione oculare) del GHS dell’ONU/CLP. Tale distinzione dovrà essere effettuata in un’altra fase di una strategia di prove in sequenza (15). La sostanza chimica in esame che con le prove d’irritazione oculare EpiOcular™ EIT e SkinEthic™ HCE EIT risulti causare irritazione oculare/gravi lesioni oculari dovrà essere sottoposta a ulteriori prove (in vitro e/o in vivo) per stabilirne la classificazione definitiva (nella categoria 1, 2 o «senza categoria» del GHS dell’ONU/CLP, utilizzando, ad esempio, i metodi di prova B.47, B.48, B.61 o B.68.
|
|
6.
|
Scopo del presente metodo di prova è descrivere la procedura impiegata per valutare il pericolo potenziale che la sostanza chimica in esame presenta per l’occhio, sulla base della capacità della sostanza di indurre citotossicità in un modello tessutale di RhCE, misurata con il test dell’MTT (16) (cfr. il paragrafo 21). La vitalità del tessuto RhCE dopo l’esposizione alla sostanza in esame è determinata confrontandola con quella dei tessuti trattati con la sostanza scelta per il controllo negativo (% di vitalità), e successivamente utilizzata per prevedere il pericolo potenziale che la sostanza chimica in esame presenta per l’occhio.
|
|
7.
|
Sono disponibili standard di prestazione (17) per semplificare la validazione delle prove in vitro, nuove o modificate, che usano RhCE e che sono simili alle prove d’irritazione oculare EpiOcular™ EIT e SkinEthic™ HCE EIT, in conformità dei principi stabiliti nel documento di orientamento dell’OCSE n. 34 (18), e per modificare rapidamente la linea guida dell’OCSE n. 492 per potervele includere. L’accettazione reciproca dei dati conformemente all’accordo OCSE sarà garantita solo per le prove validate in base agli standard di prestazione, se tali prove sono state esaminate e integrate nella corrispondente linea guida dell’OCSE.
|
DEFINIZIONI
|
8.
|
Le definizioni figurano nell’appendice 1.
|
CONSIDERAZIONI INIZIALI E LIMITI
|
9.
|
Il presente metodo si basa su modelli di tessuto tridimensionali di RhCE disponibili in commercio e prodotti a partire da cheratinociti primari dell’epidermide umana (EpiOcular™ OCL-200) o da cellule epiteliali corneali umane immortalizzate (SkinEthic™ HCE/S). I costrutti tessutali di RhCE EpiOcular™ OCL-200 e SkinEthic™ HCE/S sono simili alla struttura tridimensionale dell’epitelio corneale in vivo e sono prodotti a partire da cellule della specie d’interesse (19)(20). Inoltre le prove misurano direttamente la citotossicità dovuta alla penetrazione transcorneale della sostanza chimica e alla produzione di lesioni cellulari e tessutali; in base alla risposta citotossica si determina quindi la risposta globale di grave lesione oculare/irritazione oculare in vivo. Le lesioni cellulari possono essere indotte da varie modalità d’azione (cfr. il paragrafo 20), ma la citotossicità svolge un ruolo meccanicistico importante, se non primario, nel determinare la reazione complessiva risultante in grave lesione oculare/irritazione oculare dopo esposizione a una sostanza chimica, che si manifesta in vivo principalmente mediante opacità della cornea, infiammazione dell’iride (irite), rossore e/o chemosi della congiuntiva, indipendentemente dai processi fisico chimici delle lesioni tessutali.
|
|
10.
|
Ai fini dello studio di validazione su cui si fonda il presente metodo di prova è stata testata una vasta gamma di sostanze chimiche, che rappresentano svariati tipi e classi chimiche, pesi molecolari, LogP, strutture chimiche ecc. La banca dati di validazione della prova d’irritazione oculare EpiOcular™ EIT conteneva complessivamente 113 sostanze chimiche che, secondo un’analisi dell’OCSE svolta con l’ausilio del sofware QSAR, rappresentavano 95 gruppi organici funzionali diversi (8). La maggior parte di queste sostanze erano monocostituente, ma lo studio includeva anche molte sostanze multicostituente (tra cui 3 omopolimeri, 5 copolimeri e 10 quasi polimeri). Per quanto riguarda lo stato fisico e le categorie del GHS dell’ONU/CLP, le 113 sostanze chimiche testate erano così distribuite: 13 liquidi di categoria 1, 15 solidi di categoria 1, 6 liquidi di categoria 2A, 10 solidi di categoria 2A, 7 liquidi di categoria 2B, 7 solidi di categoria 2B, 27 liquidi senza categoria e 28 solidi senza categoria (8). La banca dati di validazione della prova d’irritazione oculare SkinEthic™ HCE EIT conteneva complessivamente 200 sostanze chimiche, che rappresentavano 165 gruppi organici funzionali diversi (8)(10)(11). La maggior parte di queste sostanze chimiche erano monocostituente, ma lo studio includeva anche molte sostanze multicostituente (tra cui 10 omopolimeri). Per quanto riguarda lo stato fisico e le categorie del GHS dell’ONU/CLP, le 200 sostanze chimiche testate erano così distribuite: 27 liquidi di categoria 1, 24 solidi di categoria 1, 19 liquidi di categoria 2A, 10 solidi di categoria 2A, 9 liquidi di categoria 2B, 8 solidi di categoria 2B, 50 liquidi senza categoria e 53 solidi senza categoria (10) (11).
|
|
11.
|
Il presente metodo di prova è applicabile a sostanze e miscele solide, liquide, semisolide e sotto forma di cere. I liquidi possono essere acquosi o non acquosi, i solidi possono essere solubili o insolubili in acqua. Quando possibile, prima dell’applicazione i solidi devono essere frantumati in polvere fine; non sono necessarie altre forme di pretrattamento del campione. I gas e gli aerosol non sono stati valutati in alcuno studio di validazione. Benché sia ipotizzabile che possano essere testati utilizzando la tecnologia RhCE, l’attuale metodo di prova non permette la sperimentazione su gas e aerosol.
|
|
12.
|
Le sostanze chimiche in esame che assorbono la luce alla stessa lunghezza d’onda dell’MTT formazan (naturalmente o dopo il trattamento) e quelle che possono ridurre direttamente il colorante vitale MTT (in MTT formazan) potrebbero interferire con le misurazioni della vitalità tessutale, perciò occorre utilizzare controlli adattati per correggere la prova in funzione di queste interferenze. Il tipo di controlli adattati necessari dipenderà dal tipo d’interferenza prodotta dalla sostanza chimica in esame e dalla procedura utilizzata per quantificare l’MTT formazan (cfr. i paragrafi 36-42).
|
|
13.
|
I risultati ottenuti dagli studi di pre-validazione (21) (22) e di validazione (8) (10) (11) hanno dimostrato che entrambe le prove d’irritazione oculare, EpiOcular™ EIT e SkinEthic™ HCE EIT, possono essere condotte da laboratori senza alcuna specifica esperienza su dette prove, e che sono riproducibili a livello intralaboratorio e interlaboratori. Da questi studi risulta che, in base ai dati relativi a 113 sostanze chimiche, il livello atteso di riproducibilità della prova d’irritazione oculare EpiOcular™ EIT per quanto riguarda la concordanza delle previsioni è dell’ordine del 95 % a livello intralaboratorio e del 93 % a livello interlaboratori. Il livello atteso di riproducibilità della prova d’irritazione oculare SkinEthic™ HCE EIT per quanto riguarda la concordanza delle previsioni, in base ai dati relativi a 120 sostanze chimiche è dell’ordine del 92 % a livello intralaboratorio e del 95 % a livello interlaboratori.
|
|
14.
|
La prova d’irritazione oculare EpiOcular™ EIT può essere usata per identificare le sostanze chimiche che non richiedono una classificazione per irritazione oculare o per gravi lesioni oculari secondo il sistema di classificazione GHS dell’ONU/CLP. Considerando i dati ottenuti nello studio di validazione (8), la prova d’irritazione oculare EpiOcular™ EIT ha un’accuratezza globale dell’80 % (su 112 sostanze chimiche), una sensibilità del 96 % (su 57 sostanze chimiche), un tasso di falsi negativi del 4 % (su 57 sostanze chimiche), una specificità del 63 % (su 55 sostanze chimiche) e un tasso di falsi positivi del 37 % (su 55 sostanze chimiche), rispetto ai dati di riferimento ottenuti con la prova in vivo sugli occhi del coniglio (metodo di prova B.5) (2)(14) classificati secondo il GHS dell’ONU/CLP. In uno studio in cui sono state testate 97 formulazioni di sostanze agrochimiche liquide, la prova d’irritazione oculare EpiOcular™ EIT ha dimostrato prestazioni simili a quelle dello studio di validazione per questo tipo di miscele (23). Le 97 formulazioni erano così distribuite: 21 nella categoria 1, 19 nella categoria 2A, 14 nella categoria 2B e 43 senza categoria, secondo il GHS dell’ONU e sulla base dei dati di riferimento ottenuti con la prova in vivo sugli occhi del coniglio (metodo di prova B.5) (2)(14). Si è ottenuto un’accuratezza globale dell’82 % (su 97 formulazioni), una sensibilità del 91 % (su 54 formulazioni), un tasso di falsi positivi del 9 % (su 54 formulazioni), una specificità del 72 % (su 43 formulazioni) e un tasso di falsi positivi del 28 % (su 43 formulazioni) (23).
|
|
15.
|
La prova d’irritazione oculare SkinEthic™ HCE EIT può essere usata per identificare le sostanze chimiche che non richiedono una classificazione per irritazione oculare o per gravi lesioni oculari secondo il sistema di classificazione GHS dell’ONU/CLP. Considerando i dati ottenuti nello studio di validazione (10)(11), la prova d’irritazione oculare SkinEthic™ HCE EIT ha un’accuratezza globale dell’84 % (su 200 sostanze chimiche), una sensibilità del 95 % (su 97 sostanze chimiche), un tasso di falsi negativi del 5 % (su 97 sostanze chimiche), una specificità del 72 % (su 103 sostanze chimiche) e un tasso di falsi positivi del 28 % (su 103 sostanze chimiche), rispetto ai dati di riferimento ottenuti con la prova in vivo sugli occhi del coniglio (metodo di prova B.5) (2)(14) classificati secondo il GHS dell’ONU/CLP.
|
|
16.
|
Le percentuali di falsi negativi ottenuti con entrambe le prove RhCE, su sostanze o miscele, rientrano nel 12 % di probabilità generale che le sostanze chimiche siano classificate nella categoria 2 oppure «senza categoria» in base al sistema GHS dell’ONU e della classificazione CLP a seguito di una prova oculare in vivo di Draize, in prove ripetute a causa della variabilità intrinseca al metodo durante l’esecuzione della prova (24). Le percentuali di falsi negativi ottenuti con entrambe le prove RhCE, su sostanze o miscele, non sono preoccupanti in questo contesto perché tutte le sostanze chimiche in esame che producono una vitalità tessutale pari o inferiore alle soglie definite (cfr. il paragrafo 44) richiederanno ulteriore sperimentazione con altri metodi di prova in vitro, o, come ultima ratio, sul coniglio, in funzione dei requisiti regolamentari e in conformità della strategia sperimentale sequenziale in un approccio basato sul peso dell’evidenza. Questi metodi di prova possono essere usati per tutti i tipi di sostanze chimiche in cui è accettabile un risultato negativo al fine di non classificare una sostanza chimica per irritazione oculare o gravi lesioni oculari («senza categoria» secondo il sistema GHS dell’ONU/CLP). Occorre consultare le autorità di regolamentazione competenti prima di utilizzare i metodi EpiOcular™ EIT e SkinEthic™ HCE EIT nell’ambito di sistemi di classificazione diversi dal sistema GHS dell’ONU/CLP.
|
|
17.
|
Un limite di questo metodo di prova è che non permette di distinguere le sostanze chimiche che producono irritazione cutanea o effetti reversibili sugli occhi (categoria 2) da quelle che determinano gravi lesioni oculari o effetti irreversibili sugli occhi (categoria 1 del sistema GHS dell’ONU/CLP), né le sostanze che producono irritazione oculare (categoria facoltativa 2A) da quelle moderatamente irritanti per gli occhi (categoria facoltativa 2B), come definite nella classificazione GHS dell’ONU (1). Per operare tale distinzione, occorre procedere a ulteriore sperimentazione con metodi di prova in vitro.
|
|
18.
|
Il termine «sostanza chimica in esame» utilizzato nel presente metodo di prova designa l’oggetto della prova (78), a prescindere dall’applicabilità del metodo RhCE alle prove di sostanze e/o miscele.
|
PRINCIPIO DELLA PROVA
|
19.
|
La sostanza in esame viene applicata localmente ad almeno due modelli tridimensionali di RhCE e viene misurata la vitalità del tessuto dopo il periodo di esposizione e di incubazione post-trattamento. Il tessuto RhCE è un tessuto ricostituito da cheratinociti epiteliali primari di origine umana o da cellule epiteliali corneali immortalizzate di origine umana, che sono stati coltivati per più giorni fino a formare un epitelio stratificato, squamoso, altamente differenziato e morfologicamente simile all’epitelio corneale umano. Il costrutto tessutale RhCE EpiOcular™ EIT consiste di almeno tre strati di cellule vitali e di una superficie non cheratinizzata, con una struttura simile a quella della cornea in vivo. Il costrutto tessutale RhCE SkinEthic™ HCE consiste di almeno 4 strati di cellule vitali, incluse cellule basali colonnari, cellule amplificatrici transitorie e cellule squamose superficiali simili a quelle che si trovano nel normale epitelio corneale umano (20) (26).
|
|
20.
|
Gravi danni oculari/irritazioni oculari indotti da una sostanza chimica che si manifestano in vivo, principalmente mediante opacità corneale, infiammazione dell’iride (irite) e rossore e/o chemosi congiuntivale, sono il risultato di una cascata di eventi che iniziano con la penetrazione della sostanza chimica nella cornea e/o nella congiuntiva e producono lesioni cellulari. Le lesioni cellulari possono essere generate mediante varie modalità d’azione, tra cui: la lisi della membrana cellulare (solventi organici o di agenti tensioattivi); la coagulazione di macromolecole (proteine, in particolare mediante solventi organici, tensioattivi, acidi e alcali); saponificazione dei lipidi (alcali, ad esempio); e alchilazione o altre interazioni covalenti con macromolecole (ad es. mediante prodotti sbiancanti, alchilanti e perossidi) (15) (27) (28). Tuttavia, è stato dimostrato che la citotossicità svolge un ruolo meccanicistico importante, se non primario, nel determinare la reazione complessiva risultante in grave lesione oculare/irritazione oculare dopo esposizione a una sostanza chimica, indipendentemente dai processi fisico-chimici che sottendono alle lesioni tessutali (29)(30). Inoltre, il potenziale di una sostanza chimica di causare gravi lesioni oculari/irritazione oculare è determinato principalmente mediante l’entità del danno iniziale (31), che è correlato al grado di morte cellulare (29) e all’entità delle successive reazioni e delle eventuali conseguenze (32). Pertanto, le sostanze con un leggero effetto irritante interessano in genere solo l’epitelio corneale superficiale, le sostanze moderatamente irritanti ledono principalmente l’epitelio e lo stroma superficiale, mentre le sostanze che causano gravi irritazioni danneggiano l’epitelio, lo stroma profondo e talvolta l’endotelio della cornea (30) (33). La misurazione della vitalità di un costrutto tessutale RhCE dopo esposizione topica a una sostanza chimica allo scopo di individuare le sostanze che non richiedono classificazione per irritazione oculare o gravi lesioni oculari («senza categoria» del GHS dell’ONU/CLP) si basano sull’assunto che tutte le sostanze che producono gravi lesioni oculari o irritazione oculare inducono citotossicità nell’epitelio corneale e/o nella congiuntiva.
|
|
21.
|
La vitalità del tessuto RhCE è tradizionalmente misurata tramite conversione enzimatica del colorante vitale MTT [bromuro di 3-(4,5-dimetiltiazol-2-ile)-2,5-difeniltetrazolio; bromuro di tiazolil blu tetrazolio; numero CAS 298-93-1] da parte delle cellule vitali del tessuto in un sale, il blu MTT di formazan, misurato quantitativamente dopo l’estrazione dai tessuti (16). Le sostanze chimiche che non richiedono classificazione né etichettatura in base al sistema GHS dell’ONU/CLP («senza categoria») sono identificate come quelle che non provocano una diminuzione della vitalità dei tessuti al di sotto di una determinata soglia (ossia, vitalità del tessuto > 60 %, nelle prove EpiOcular™ EIT e SkinEthic™ HCE EITL (79), o > 50 %, nella prova SkinEthic™ HCE EITS (80)) (cfr. il paragrafo 44).
|
DIMOSTRAZIONE DELLA COMPETENZA DI LABORATORIO
|
22.
|
Prima di utilizzare come test di routine le prove su RhCE a fini regolamentari, i laboratori sono tenuti a dimostrare la loro competenza tecnica classificando correttamente le 15 sostanze per la verifica della competenza elencate nella tabella 1. Tali sostanze chimiche sono state selezionate sulla base delle sostanze chimiche utilizzate negli studi di validazione dei VRM (8) (10) (11). La selezione comprende, nei limiti del possibile, le sostanze chimiche che: i) si presentano in diversi stati fisici; ii) rappresentano la gamma completa di reazioni in vivo di irritazione oculare/grave lesione oculare in base ai risultati di alta qualità ottenuti con la prova in vivo sugli occhi dei conigli (metodo di prova B.5) (2)(14) e del sistema di classificazione UN GHS (ossia le categorie 1, 2A, 2B, o «senza categoria») (1) e del sistema di classificazione CLP (ossia le categorie 1, 2 o «senza categoria»); iii) coprono i diversi criteri di classificazione in vivo (24) (25); iv) sono rappresentative delle classi chimiche usate nello studio di validazione (8)(10)(11); v) rappresentano una grande varietà di gruppi funzionali organici (8) (10) (11); vi) hanno una struttura chimica ben definita (8) (10) (11); vii) sono colorate e/o riduttori diretti dell’MTT; viii) hanno prodotto risultati riproducibili nel corso della validazione dei metodi di prova sul RhCE; ix) sono state correttamente classificate dai metodi di prova sul RhCE durante la loro validazione; x) coprono l’intera gamma di risposte in vitro in base a dati di alta qualità provenienti da metodi di prova sul RhCE (vitalità 0-100 %); xi) sono disponibili in commercio; e xii) non sono eccessivamente costose da acquisire e/o smaltire. Nelle situazioni in cui una delle sostanze elencate non sia disponibile o non possa essere utilizzata per altri motivi giustificati, può essere utilizzata un’altra sostanza chimica che soddisfi i criteri sopra descritti, ad es. selezionata tra le sostanze chimiche utilizzate nella validazione del VRM. Una tale modifica deve tuttavia essere giustificata.
Tabella 1
Elenco delle sostanze chimiche per la verifica della competenza tecnica
|
Denominazione chimica
|
N. CAS
|
Gruppo organico funzionale (81)
|
Stato fisico
|
Prestazione del VRM1 (%) (82)
|
Prestazione del VRM2 (%) (83)
|
Predizione del VRM
|
Riduttore dell’MTT
|
Interferenza di colori
|
|
Categoria in vivo 1 (84)
|
|
|
Metil tioglicolato
|
2365-48-2
|
Estere di acido carbossilico tioalcol
|
L
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Non è possibile fare previsioni
|
Sì (elevato)
|
No
|
|
Acrilato di idrossietile
|
818-61-1
|
Acrilato; Alcol
|
L
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Non è possibile fare previsioni
|
No
|
No
|
|
2,5-Dimetil-2,5-esandiolo
|
110-03-2
|
Alcol
|
S
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Non è possibile fare previsioni
|
No
|
No
|
|
Ossalato di sodio
|
62-76-0
|
Acido carbossilico
|
S
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Non è possibile fare previsioni
|
No
|
No
|
|
Categoria in vivo 2A (84)
|
|
|
di-D-gluconato, composto di N,N″-bis(4-clorofenil)-3,12-diimino- 2,4,11,13-tetraazatetradecane-diimidamide (20 %, acquoso) (86)
|
18472-51-0
|
Alogenuro aromatico eterociclico; alogenuro di arile; gruppo diidrossile; guanidina
|
L
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Non è possibile fare previsioni
|
No
|
Sì (debole)
|
|
Benzoato di sodio
|
532-32-1
|
Arile; Acido carbossilico
|
S
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Non è possibile fare previsioni
|
No
|
No
|
|
Categoria in vivo 2B (84)
|
|
|
Dietil-toluammide
|
134-62-3
|
Benzammide
|
L
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Non è possibile fare previsioni
|
No
|
No
|
|
2,2-dimetil-3-metilenbiciclo[2.2.1]eptano
|
79-92-5
|
Alcano, ramificato con carbonio terziario; Alchene; Bicicloeptano; composti carbociclici ad anello reticolati cicloalcani
|
S
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Non è possibile fare previsioni
|
No
|
No
|
|
In Vivo Senza categoria (84)
|
|
|
1-Etil-3-metilimidazolio etilsolfato
|
342573-75-5
|
Alcossi; Sale di ammonio; Arile; imidazolo; solfato
|
L
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Senza cat.
|
No
|
No
|
|
Etere dicaprilico
|
629-82-3
|
Alcossi; etere
|
L
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Senza cat.
|
No
|
No
|
|
Piperonil butossido
|
51-03-6
|
Alcossi; benzodiossolo; benzile; etere
|
L
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Senza cat.
|
No
|
No
|
|
Polietilenglicole (PRG-40); Olio di ricino idrogenato
|
61788-85-0
|
acile; alcol; allile; etere
|
Viscoso
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Senza cat.
|
No
|
No
|
|
1-(4-Clorofenil)-3-(3,4-diclorofenil)urea
|
101-20-2
|
Alogenuro aromatico eterociclico; alogenuro di arile; derivati dell’urea
|
S
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Senza cat.
|
No
|
No
|
|
2,2′-metilene-bis-6-(2H-benzotriazolo-2-il)-4-(1,1,3,3-tetrametilbutil)fenolo
|
103597-45-1
|
Alcano, ramificato con carbonio quaternario; Composto carbociclico aromatico; eterocicli saturi policiclici; precursori di composti di quinone; terz-butile
|
S
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Senza cat.
|
No
|
No
|
|
Tetrafluoroborato di potassio
|
14075-53-7
|
Sale inorganico
|
S
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Senza cat.
|
No
|
No
|
|
Abbreviazioni:
N. CAS (Chemical Abstracts Service Registry Number) = Numero CAS (numero di registrazione nell’inventario europeo delle sostanze chimiche). GHS dell’ONU = Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (1); VRM1 = «metodo di riferimento validato», EpiOcular™ EIT; VRM2 = «metodo di riferimento validato», SkinEthic™ HCE EIT; Interferenza di colori = interferenza di colere con la misurazione dell’assorbanza standard (densità ottica - OD) dell’MTT formazan.
|
|
|
23.
|
Nell’ambito della dimostrazione della competenza tecnica, si raccomanda agli utilizzatori di verificare le proprietà di barriera dei tessuti dopo averli ricevuti, in conformità delle specifiche del produttore del modello di tessuto RhCE (cfr. i paragrafi 25, 27 e 30). Tale verifica è particolarmente importante se i tessuti vengono trasportati per lunghe distanze/per viaggi lunghi. Quando una prova è consolidata e la sua correttezza d’impiego è stata dimostrata, non è più necessario effettuare la verifica in maniera sistematica. Tuttavia, se una prova viene utilizzata correntemente, si raccomanda di continuare a verificare le proprietà di barriera a intervalli regolari.
|
PROCEDURA
|
24.
|
Le prove che attualmente rientrano nel presente metodo di prova sono le prove scientificamente valide EpiOcular™ EIT e SkinEthic™ HCE EIT (9)(12)(13), denominate «metodo di riferimento validato» (rispettivamente VRM1 e VRM2). Le procedure operative standard per i metodi di prova sul RhCE sono disponibili e dovrebbero essere applicate quando si eseguono questi metodi di prova in laboratorio (34) (35). Nei paragrafi che seguono e nell’appendice 2 è fornita una descrizione dei principali elementi e le procedure dei metodi di prova sul RhCE.
|
COMPONENTI DEI METODI DI PROVA SUL RHCE
Condizioni generali
|
25.
|
Cellule derivate da cellule umane devono essere utilizzate per ricostituire un modello tridimensionale di tessuto epiteliale della cornea, che deve essere composto di cellule progressivamente stratificate ma non cheratinizzate. Il costrutto di tessuto RhCE è preparato in piastre di coltura con una membrana sintetica porosa attraverso la quale i nutrienti possono raggiungere le cellule. Il modello di epitelio corneale ricostituito è formato da diversi strati di cellule epiteliali vitali e non cheratinizzate. Il costrutto di RhCE presenta superficie epiteliale a diretto contatto con l’aria, in modo che l’esposizione locale diretta alle sostanze chimiche avvenga in modo simile a come avverrebbe l’esposizione di un epitelio corneale in vivo. Il costrutto di tessuto RhCE deve formare una barriera funzionale solida capace di resistere alla penetrazione rapida delle sostanze di riferimento citotossiche, ad esempio, sodio dodecil solfato (SDS) o Triton X-100. La funzione di barriera deve essere dimostrata e può essere valutata determinando il tempo di esposizione necessario per ridurre la vitalità tessutale del 50 % (ET50) a seguito dell’applicazione di una sostanza di riferimento a una concentrazione fissa predeterminata (ad es. 100 μl allo 0,3 % (v/v) Triton X-100), oppure la concentrazione alla quale una sostanza di riferimento riduce la vitalità dei tessuti del 50 % (IC50) dopo un tempo di esposizione fisso (ad es. 30 minuti di trattamento con 50 μl di SDS) (cfr. il paragrafo 30). Le proprietà di contenimento del costrutto tessutale RhCE devono essere tali da impedire che la sostanza chimica in esame possa passare attorno ai tessuti vitali, il che inciderebbe negativamente sulla qualità della modellizzazione dell’esposizione corneale. Le cellule di origine umana utilizzate nel modello di RhCE devono essere esenti da contaminazione da batteri, virus, micoplasmi o funghi. Il fornitore deve verificare la sterilità del costrutto di tessuto e l’assenza di contaminazione da funghi o batteri.
|
Condizioni funzionali
Vitalità
|
26.
|
La prova utilizzata per la determinazione della vitalità è il test dell’MTT (16). Le cellule vitali del costrutto tessutale RhCE riducono il colorante vitale MTT in un precipitato di formazan blu, che è successivamente estratto dal tessuto con solvente (isopropanolo o simile). L’estratto di formazan può essere quantificato mediante misurazione standard dell’assorbanza (densità ottica, OD) o analisi con spettrofotometria HPLC/CLUP (36). La densità ottica (OD) del solo solvente di estrazione dovrebbe essere sufficientemente bassa, ossia OD < 0,1. Gli utilizzatori del modello RhCE devono accertarsi che ciascun lotto del costrutto di tessuto RhCE impiegato soddisfi i criteri definiti per il controllo negativo. Gli intervalli di accettabilità dei valori OD del controllo negativo per i VRM sono indicati nella tabella 2. Gli utilizzatori della spettrofotometria HPLC/CLUP dovrebbero utilizzare gli intervalli di valori OD del controllo negativo di cui alla tabella 2, come criteri di accettabilità per il controllo negativo. Occorre documentare nella relazione sull’esecuzione della prova che i tessuti trattati con la sostanza di controllo negativo sono stabili in coltura (ossia presentano misurazioni di vitalità tessutale simili) nel corso del periodo di esposizione. Una procedura analoga dovrebbe essere seguita dal produttore di un tessuto nell’ambito del controllo di qualità dei lotti di tessuto, ma in questo caso i criteri di accettabilità sono diversi da quelli specificati nella tabella 2. Lo sviluppatore/il fornitore del costrutto tessutale RhCE deve stabilire un intervallo di accettabilità (limite superiore e inferiore) per i valori OD del controllo negativo (alle condizioni del metodo di prova per il controllo di qualità).
|
Tabella 2
Intervalli di accettabilità dei valori OD del controllo negativo (per gli utilizzatore della prova)
|
Prova
|
Limite inferiore di accettabilità
|
Limite superiore di accettabilità
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (protocolli per le sostanze sia solide che liquide)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (protocolli per le sostanze sia solide che liquide)
|
> 1,0
|
≤ 2,5
|
Funzione di barriera
|
27.
|
Il costrutto tessutale RhCE deve essere sufficientemente spesso e robusto da resistere alla penetrazione rapida delle sostanze di riferimento citotossiche, valutata dai fattori ET50 (Triton X-100) o IC50 (SDS) (tabella 3). La funzione di barriera di ciascun lotto del modello del RhCE è dimostrata dallo sviluppatore/fornitore del tessuto RhCE al momento della fornitura dei tessuti all’utilizzatore finale (cfr. il paragrafo 30).
|
Morfologia
|
28.
|
L’esame istologico del RhCE dovrebbe evidenziare una struttura simile a quella dell’epitelio della cornea umana (compresi almeno 3 strati di cellule epiteliali vitali e una superficie non cheratinizzata). Per i VRM, la morfologia adeguata è stata dimostrata dallo sviluppatore/fornitore e non è dunque necessario dimostrare nuovamente ogni volta che il metodo di prova viene eseguito su un nuovo lotto.
|
Riproducibilità
|
29.
|
I risultati del controllo positivo (PC) e dei controlli negativi (NC) del metodo devono mostrare riproducibilità nel tempo.
|
Controllo di qualità (QC)
|
30.
|
Il costrutto tessutale RhCE deve essere utilizzato soltanto se lo sviluppatore/fornitore ha dimostrato che ogni lotto del costrutto di tessuto RhCE utilizzato rispetta determinati criteri di fabbricazione, i più rilevanti dei quali sono quelli relativi alla vitalità (cfr. il paragrafo 26) e alla funzione di barriera (cfr. il paragrafo 27). Lo sviluppatore/il fornitore del costrutto tessutale RhCE deve stabilire un intervallo di accettabilità (limite superiore e inferiore) per le funzioni di barriera, misurate mediante ET50 o IC50 (cfr. i paragrafi 25 e 26). L’intervallo di accettabilità per i valori ET50 e IC50 scelti come criteri per il controllo di qualità del lotto dallo sviluppatore/fornitore del costrutto tessutale RhCE (utilizzato nei VRM), è riportato nella tabella 3. Lo sviluppatore/fornitore del costrutto tessutale RhCE deve fornire dati che dimostrino la conformità con tutti i criteri di produzione agli utilizzatori del presente metodo di prova, in maniera che possano includere tali informazioni nella relazione sull’esecuzione della prova. Solo i risultati ottenuti con tessuti che soddisfano tutti i criteri di fabbricazione definiti possono essere accettati per una previsione affidabile sulle sostanze chimiche che non richiedono classificazione né etichettatura per irritazione oculare o gravi lesioni oculari secondo il sistema GHS dell’ONU/CLP.
|
Tabella 3
Criteri di controllo della qualità dei lotti
|
Prova
|
Limite inferiore di accettabilità
|
Limite superiore di accettabilità
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl di Triton X-100 allo 0,3 % (v/v))
|
ET50 = 12,2 min
|
ET50 = 37,5 min
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 minuti di trattamento con 50 μl SDS)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
Applicazione della sostanza chimica in esame e delle sostanze di controllo
|
31.
|
Per ogni sostanza chimica in esame e per ciascuna sostanza di controllo in ciascuna batteria di prove devono essere utilizzate almeno due repliche di tessuto. Si seguono due diversi protocolli di trattamento per le sostanze chimiche in esame, uno per le sostanze liquide e uno per le sostanze solide (34) (35). In entrambi i metodi e i protocolli, la superficie del costrutto tessutale deve essere umidificata con tampone fosfato salino di Dulbecco senza sale di calcio e magnesio (DPBS privo di Ca2+/Mg2+-free DPBS), prima di essere esposta alla sostanza chimica in esame, in modo da riprodurre il più fedelmente possibile l’umidità dell’occhio umano. Il trattamento dei tessuti inizia con l’esposizione alla sostanza o sostanze chimiche in esame e alle sostanze di controllo. Nei due protocolli di entrambi i VRM occorre applicare una quantità della sostanza in esame o di controllo sufficiente per coprire uniformemente la superficie epiteliale, evitando tuttavia di utilizzare una dose infinita (cfr. i paragrafi 32 e 33) (appendice 2).
|
|
32.
|
Le sostanze chimiche in esame che possono essere applicate con pipetta a 37 °C o a temperature inferiori (con l’ausilio di una pipetta a stantuffo, se necessario) sono trattate come liquidi nei VRM. In caso contrario, devono essere trattate come solidi (cfr. il paragrafo 33). Nei VRM, la sostanza chimica in esame è applicata uniformemente sulla superficie del tessuto (cioè un’applicazione di almeno 60 μl/cm2) (cfr. l’appendice 2 (33) (34)). Si dovrebbero evitare, per quanto possibile, effetti di capillarità (effetti di tensione superficiale) che possano essere osservati a motivo dei limitati volumi applicati agli inserti (sulla superficie tessutale), al fine di garantire il corretto dosaggio sui tessuti. I tessuti trattati con sostanze chimiche liquide sono incubati per 30 minuti in condizioni di coltura [37±2 °C, 5±1 % CO2, ≥ 95 % di umidità relativa (UR)]. Al termine del periodo di esposizione, la sostanza in esame liquida e le sostanze di controllo vengono rimosse con cura dalla superficie del tessuto mediante risciacquo abbondante con DPBS privo di Ca2+/Mg2+ a temperatura ambiente. Questa fase di risciacquo è seguita da un’immersione post-esposizione in un nuovo mezzo di coltura a temperatura ambiente (per eliminare eventuale sostanza in esame assorbita dal tessuto) per un periodo prestabilito che varia in funzione del VRM utilizzato. Solo nel caso del VRM1, l’incubazione post-trattamento in nuovo mezzo di coltura a condizioni di coltura standard, è applicata prima di effettuare il test dell’MTT [cfr. l’appendice 2 (34) (35)].
|
|
33.
|
Le sostanze chimiche in esame che non possono essere applicate con pipetta a 37 °C o a temperature inferiori sono trattate come solidi nei VRM. La quantità applicata deve essere sufficiente a coprire completamente la superficie di tessuto, corrispondente cioè a un’applicazione di almeno 60 mg/cm2 (appendice 2). Quando possibile, i solidi dovrebbero essere testati sotto forma di polvere fine. I tessuti trattati con le sostanze chimiche in esame solide devono essere incubati per un periodo predefinito (in funzione del VRM utilizzato) in condizioni di coltura standard [cfr. l’appendice 2 (34) (35)]. Al termine del periodo di esposizione, la sostanza in esame solida e le sostanze di controllo vengono rimosse con cura dalla superficie del tessuto mediante risciacquo abbondante con DPBS privo di Ca2+/Mg2+ a temperatura ambiente. Questa fase di risciacquo è seguita da un’immersione post-esposizione in un nuovo mezzo di coltura a temperatura ambiente (per eliminare eventuale sostanza in esame assorbita dal tessuto) per un periodo prestabilito che varia in funzione del VRM utilizzato, e dall’incubazione post-esposizione in nuovo mezzo di coltura a condizioni di coltura standard, prima di effettuare il test dell’MTT [cfr. l’appendice 2 (34) (35)].
|
|
34.
|
Controlli negativi e positivi concomitanti sono inclusi in ciascuna batteria di prove per dimostrare che la vitalità (determinata con il controllo negativo) e la sensibilità (determinata con il controllo positivo) dei tessuti rientrano in un definito intervallo storico di accettabilità. Il controllo negativo permette anche di stabilire il valore di riferimento (vitalità tessutale al 100 %) da cui è calcolata la vitalità relativa (in percentuale) dei tessuti trattati con la sostanza chimica in esame (%Vitalitàtest). La sostanza raccomandata per il controllo positivo da utilizzare nei VRM è l’acetato di metile puro (N. CAS 79-20-9, disponibile in commercio presso, ad es., Sigma-Aldrich, Cat. n. 45997; liquido). Le sostanze raccomandate per il controllo negativo da utilizzare nel VRM1 e nel VRM2 sono, rispettivamente, l’H2O ultrapura e il DPBS privo di Ca2+/Mg2+. Queste sostanze sono utilizzate come controlli in studi di pre-validazione e validazione dei VRM, e sono quelle per le quali esistono più dati storici. L’uso di adeguate sostanze alternative per i controlli positivi e negativi deve essere debitamente giustificato scientificamente. I controlli positivi e negativi devono essere testati secondo il medesimo o i medesimi protocolli utilizzati per le sostanze chimiche in esame incluse nella batteria di prove (cioè, per liquidi e solidi). Questa applicazione è seguita da un trattamento mediante esposizione, da un risciacquo, da un’immersione post-trattamento, e da un’incubazione post-trattamento se del caso, come descritto per le batterie di controllo contestuali alle sostanze chimiche in esame liquide (cfr. il paragrafo 32) o le batterie di controllo contestuali alle sostanze chimiche in esame solide (cfr. il paragrafo 33), prima di effettuare il test dell’MTT (cfr. il paragrafo 35) (34) (35). Una singola serie di controlli negativi e positivi è sufficiente per tutte le sostanze chimiche in esame nello stesso stato fisico (liquido o solido).
|
Misurazione della vitalità dei tessuti
|
35.
|
Il test dell’MTT è un metodo quantitativo standardizzato (16) che dovrebbe essere utilizzato per misurare la vitalità dei tessuti nell’ambito del presente metodo di prova. È compatibile con l’utilizzo di un costrutto tessutale tridimensionale. Il test dell’MTT è eseguito immediatamente dopo il periodo di incubazione post-trattamento. Nei VRM, l’RhCE è immerso in 0,3 ml di soluzione MTT a 1 mg/ml per 180 ± 15 minuti in condizioni di coltura standard. Il colorante vitale MTT è ridotto a un precipitato di MTT formazan dalle cellule vitali del costrutto tessutale RhCE. Il precipitato di formazan blu è successivamente estratto dal tessuto mediante un adeguato volume di solvente (isopropanolo o simile) (34)(35). Per tessuti esposti a sostanze chimiche in esame liquide, l’estrazione è effettuata da strati sia inferiori sia superiori dei tessuti, mentre per i tessuti esposti alle sostanze in esame solide e a liquidi colorati, l’estrazione è effettuata soltanto dallo strato inferiore del tessuto (per ridurre al minimo qualsiasi potenziale rischio di contaminazione della soluzione di isopropanolo usata per l’estrazione con un eventuale residuo della sostanza chimica in esame presente nel tessuto). Anche nel caso di tessuti esposti a sostanze chimiche liquide difficili da risciacquare, l’estrazione può essere effettuata soltanto dallo strato inferiore di tessuto. Le sostanze di controllo positive e negative testate in parallelo sono trattate allo stesso modo delle sostanze chimiche in esame. L’MTT formazan estratto è quantificato mediante misurazione dell’assorbanza standard (DO) a 570 nm con un filtro passa-banda di larghezza massima di ± 30 nm, oppure mediante spettrofotometria HPLC/CLUP (cfr. il paragrafo 42) (11) (36).
|
|
36.
|
Le proprietà ottiche della sostanza chimica in esame o la sua azione chimica sull’MTT potrebbero interferire con la misurazione dell’MTT formazan, portando a una stima errata della vitalità dei tessuti. Le sostanze chimiche in esame possono interferire con il test dell’MTT mediante riduzione diretta dell’MTT in blu di formazan, e/o mediante interferenza dei colori se la sostanza in esame assorbe, naturalmente o a seguito di trattamento, nello stesso spettro di OD del formazan (cioè, circa 570 nm). Vanno effettuate verifiche preliminari prima della prova, al fine di individuare le sostanze che sono potenziali riduttori diretti dell’MTT e/o che causano interferenze nei colori, e sono preparati controlli supplementari per individuare le interferenze che possono essere causate da tali sostanze chimiche e correggere la prova di conseguenza (cfr. i paragrafi 37-41). Ciò è particolarmente importante quando la sostanza in esame non è stata rimossa completamente dal modello di RhCE mediante risciacquo o quando penetra nel modello di epitelio corneale ed è, quindi, presente nel costrutto tessutale RhCE al momento dell’esecuzione del test dell’MTT. Per le sostanze chimiche in esame che assorbono la luce nello stesso spettro dell’MTT formazan (naturalmente o a seguito di trattamento), e che sono incompatibili con la misurazione dell’assorbanza (OD) del formazan a motivo di un’eccessiva interferenza (assorbanza che raggiunge 570 ± 30 nm), può essere effettuata una procedura analitica mediante spettrofotometria HPLC/CLUP per misurare la presenza di formazan (cfr. i paragrafi 41 e 42) (11) (36). Per una descrizione dettagliata di come individuare e correggere la riduzione diretta dell’MTT e le interferenze da parte degli agenti coloranti consultare le procedure operative standard dei VRM (34)(35). Un diagramma di flusso illustrativo e orientativo della procedura per identificare e trattare le sostanze chimiche in esame riduttrici dirette dell’MTT e/o che provocano un’interferenza di colori nei VRM1 e VRM2 figura, rispettivamente, nelle appendici 3 e 4.
|
|
37.
|
Per individuare eventuali interferenze dovute alle sostanze chimiche in esame che assorbono nello stesso spettro del formazan (naturalmente o a seguito di trattamento) e stabilire se sono necessari controlli aggiuntivi, la sostanza chimica in esame è addizionata di acqua e/o isopropanolo e incubata per un periodo adeguato a temperatura ambiente (cfr. l’appendice 2) (34) (35). Se la sostanza chimica in esame in acqua e/o isopropanolo assorbe sufficiente luce ad una lunghezza d’onda di 570 ± 20 nm per VRM1 (cfr. l’appendice 3), o se si ottiene una soluzione colorata mescolando la sostanza chimica in esame con acqua per il VRM2 (cfr. l’appendice 4), si ritiene che la sostanza chimica in esame interferisca con la misurazione dell’assorbanza (DO) del formazan; in tal caso devono essere preparati controlli colorati supplementari oppure, in alternativa, si procede all’analisi mediante spettrofotometria HPLC/CLUP, nel qual caso non sono necessari controlli supplementari (cfr. i paragrafi 41 e 42 e le appendici 3 e 4) (34) (35). Quando si effettuano le misurazioni dell’assorbanza standard (OD), ciascuna sostanza chimica in esame che interferisce con la prova è applicata su almeno due repliche di tessuti vitali, che sono sottoposte all’intera procedura sperimentale ma sono incubate nel mezzo anziché in soluzione MTT durante la fase di incubazione con MTT, al fine di generare un controllo di colore non specifico in tessuti vivi (NSCliving) (34) (35). Il controllo NSCliving deve essere testato in parallelo alla prova sulla sostanza chimica in esame colorata e, in caso di prova multipla, deve essere effettuato un controllo NSCliving indipendente per ciascuna prova (di ciascuna batteria di prove) per tener conto della variabilità biologica intrinseca dei tessuti vivi. La vitalità reale dei tessuti è calcolata come segue: la percentuale di vitalità dei tessuti ottenuta nei tessuti vivi, esposti a sostanze chimiche che causano interferenze e incubate con soluzione MTT (%Vitalitàtest) meno la percentuale di colore non specifico ottenuta nei tessuti vivi esposti a sostanze chimiche che causano interferenze e incubate in mezzo privo di MTT, in parallelo alla prova da correggere (%NSCliving), cioè la vitalità reale dei tessuti = [%Vitalitàtest] - [%NSCliving].
|
|
38.
|
Per individuare i riduttori diretti dell’MTT, occorre aggiungere ciascuna sostanza chimica in esame a una soluzione di MTT appena preparata. Una quantità adeguata della sostanza chimica in esame è aggiunta a una soluzione MTT e la miscela è incubata per circa 3 ore in condizioni di coltura standard (cfr. le appendici 3 e 4) (34) (35). Se la miscela di MTT e della sostanza chimica in esame (o la sospensione nel caso di sostanze chimiche insolubili) diventa blu/viola, si ritiene che la sostanza chimica in esame è un riduttore diretto dell’MTT e sono necessarie ulteriori verifiche funzionali su costrutto di tessuto RhCE non vitale, indipendentemente dalla scelta di misurare l’assorbanza (OD) o di procedere ad analisi mediante spettrofotometria HPLC/CLUP. Tale ulteriore verifica funzionale è eseguita su tessuti uccisi che presentano solo un’attività metabolica residuale, ma assorbono e trattengono la sostanza chimica in esame in proporzioni simili ai tessuti vitali. In VRM1, i tessuti uccisi sono preparati mediante esposizione a basse temperature («uccisi per congelamento»). In VRM2, i tessuti uccisi sono preparati mediante un lungo periodo di incubazione (almeno 24 ± 1 ore) in acqua, seguito da conservazione a bassa temperatura («uccisi in acqua»). Ciascuna sostanza chimica in esame che riduce l’MTT è applicata su almeno due repliche di tessuti uccisi, che sono sottoposte all’intera procedura sperimentale per generare un controllo di riduzione non specifico dell’MTT (NSMTT) (34) (35). È sufficiente un solo controllo NSMTT per ciascuna sostanza chimica in esame, a prescindere dal numero di prove/batterie di prove indipendenti eseguite. La vitalità reale dei tessuti è calcolata come segue: la percentuale di vitalità dei tessuti ottenuta nei tessuti vivi, esposti al riduttore dell’MTT (%Vitalitàtest) meno la percentuale del riduttore dell’MTT ottenuta nei tessuti uccisi esposti al medesimo riduttore dell’MTT e calcolati in proporzione del controllo negativo testato, in parallelo alla prova da correggere (%NSC), cioè la vitalità reale dei tessuti = [%Vitalitàtest] - [%NSC].
|
|
39.
|
Nel caso delle sostanze chimiche che sono state individuate quali sostanze in grado di causare interferenze di colori (cfr. il paragrafo 37) e la riduzione diretta dell’MTT (cfr. il paragrafo 38), si rende necessario un terzo gruppo di controlli quando si esegue la misurazione dell’assorbanza standard (OD), in aggiunta ai controlli NSMTT e NSCliving descritti nei paragrafi precedenti. In generale, questo caso si verifica per le sostanze chimiche di colore scuro che assorbono la luce nello spettro di 570 ± 30 nm (blu, viola e nero) in quanto il loro colore intrinseco impedisce di valutare il loro potenziale di riduzione diretta dell’MTT descritta al paragrafo 38. Ciò rende automaticamente obbligatori i controlli NSMTT in parallelo ai controlli NSCliving. Le sostanze chimiche in esame richiedono controlli sia NSMTT che NSCliving possono essere assorbiti e trattenuti sia dai tessuti vivi che da quelli uccisi. Pertanto, in tal caso l’uso del controllo NSMTT può permettere di correggere la prova non solo in funzione del potenziale di riduzione diretta dell’MTT della sostanza chimica in esame, ma anche dell’interferenza di colore derivante dall’assorbimento e dal trattenimento della sostanza chimica in esame da parte dei tessuti uccisi. Ciò può portare a una doppia correzione dell’interferenza di colori, dato che il controllo NSCliving consente già di tener conto dell’interferenza di colori dovuta all’assorbimento e al trattenimento della sostanza chimica in esame da parte dei tessuti vivi. Per evitare una doppia correzione dell’interferenza di colori, deve essere effettuato un terzo controllo per colore non specifico nei tessuti uccisi (NSCkilled) (cfr. le appendici 3 e 4) (34) (35). In tale controllo supplementare, ciascuna sostanza chimica in esame è applicata su almeno due repliche di tessuti uccisi, che sono sottoposte all’intera procedura sperimentale ma sono incubate nel mezzo anziché in soluzione MTT durante la fase di incubazione con MTT. È sufficiente un solo controllo NSCkilled per ciascuna sostanza chimica in esame, a prescindere dal numero di prove/batterie di prove indipendenti eseguite, ma dovrebbe essere condotto in parallelo al controllo NSMTT e sul medesimo lotto di tessuti. La vitalità reale dei tessuti è calcolata come segue: la percentuale di vitalità dei tessuti ottenuta nei tessuti vivi esposti alla sostanza chimica in esame (%Vitalitàtest) meno %NSMTT meno %NSCliving
più la percentuale di colore non specifico ottenuta nei tessuti uccisi esposti alla sostanza in esame che causa un’interferenza e incubati nel mezzo senza MTT, calcolata in proporzione del controllo negativo testato, in parallelo alla prova da correggere (%NSCkilled), cioè la vitalità reale dei tessuti = [%Vitalitàtest] - [%NSMTT] - [%NSCliving] + [%NSCkilled].
|
|
40.
|
È importante notare che la riduzione non specifica dell’MTT e le interferenze non specifiche di colore possono aumentare l’OD (quando si eseguono le misurazioni di assorbanza standard) dell’estratto di tessuto al di sopra dell’intervallo di linearità dello spettrofotometro e che una riduzione non specifica dell’MTT può anche ampliare l’area di picco del formazan (quando si eseguono misurazioni mediante spettrofotometria HPLC/CLUP) dell’estratto tessutale al di sopra dell’intervallo di linearità dello spettrofotometro. È quindi importante che ciascun laboratorio determini l’intervallo di linearità del proprio spettrofotometro (per l’OD/area di picco) ad esempio mediante formazan, (n. CAS 57360-69-7), disponibile in commercio presso la società Sigma-Aldrich (Cat. n. M2003), prima di procedere alle prove sulle sostanze chimiche a fini regolamentari.
|
|
41.
|
La misurazione dell’assorbanza standard (OD) mediante spettrofotometro risulta appropriata per valutare le sostanze chimiche che agiscono da riduttori diretti dell’MTT e causano interferenze di colori, quando l’interferenza osservata non è troppo forte rispetto alla misurazione dell’MTT formazan (cioè la densità ottica degli estratti di tessuto ottenuta per la sostanza chimica in esame senza correzione per tener conto della riduzione diretta dell’MTT e/o dell’interferenza di colori rientrano nell’intervallo di linearità dello spettrofotometro). Tuttavia, i risultati relativi alle sostanze chimiche in esame che producono %NSMTT e/o %NSCliving ≥ 60 % (in VRM1, e in VRM2 per il protocollo sui liquidi) o 50 % (VRM2 per il protocollo dei solidi) del controllo negativo devono essere utilizzati con cautela, in quanto tali soglie corrispondono a quelle stabilite nei VRM per distinguere tra sostanze chimiche rientranti in una categoria e quelle «senza categoria» (cfr. il paragrafo 44). Per contro, l’assorbanza standard (OD) non può essere misurata quando l’interferenza con la misurazione di MTT formazan è troppo forte (cioè se l’OD non corretta negli estratti di tessuto testati si situano al di fuori dell’intervallo di linearità dello spettrofotometro). Le sostanze chimiche in esame colorate o quelle che si colorano a contatto con l’acqua o l’isopropanolo, che interferiscano troppo con la misurazione dell’assorbanza standard (OD) dell’MTT formazan possono comunque essere valutate mediante spettrofotometria HPLC/CLUP (cfr. le appendici 3 e 4), il quanto il sistema HPLC/CLUP permette di separare l’MTT formazan dalla sostanza chimica prima della quantificazione (36). Per questo motivo, i controlli NSCliving o NSCkilled non sono mai necessari per l’analisi con spettrofotometria HPLC/CLUP, indipendentemente dalla sostanza chimica da testare. I controlli NSMTT sono tuttavia necessari se si prevede che la sostanza chimica in esame sia un riduttore diretto dell’MTT (seguendo la procedura descritta al paragrafo 38). I controlli NSMTT sono necessari anche per le sostanze chimiche in esame colorate (naturalmente o a contatto con l’acqua) il cui colore impedisce di valutare il loro potenziale di riduzione diretta dell’MTT, come descritto nel paragrafo 38. Quando si ricorre all’analisi con spettrofotometria HPLC/CLUP per misurare l’MTT formazan, la vitalità del tessuto è calcolata come percentuale dell’area di picco di MTT formazan ottenuta con tessuti vivi esposti alla sostanza chimica in esame rispetto all’area di picco dell’MTT formazan ottenuta con il controllo negativo concomitante. Per le sostanze chimiche in esame in grado di ridurre direttamente l’MTT, la vitalità reale dei tessuti è calcolata come segue: %Vitalitàtest
meno %NSMTT, come descritto nell’ultima frase del paragrafo 38. Infine, va osservato che i riduttori dell’MTT diretti o i riduttori dell’MTT diretti che causano anche interferenze di colore, trattenuti nei tessuti dopo il trattamento e la cui capacità di riduzione MTT è tale da portare a valori dell’OD (utilizzando misurazioni dell’OD standard) o ad aree di picco (utilizzando la spettrofotometria HPLC/CLUP) degli estratti di tessuto in esame che si situano al di fuori dell’intervallo di linearità dello spettrofotometro, non possono essere valutati con i metodi di prova sul RhCE; si ritiene tuttavia che ciò avvenga solo in rare occasioni.
|
|
42.
|
La procedura di misurazione mediante spettrofotometria HPLC/CLUP può essere utilizzata con tutti i tipi di sostanze chimiche in esame (colorate, non colorate, riduttori di MTT e non riduttori di MTT) ai fini della misurazione dell’MTT formazan (11) (36). Poiché esistono vari sistemi di spettrofotometria HPLC/CLUP, non è ipotizzabile che tutti gli utilizzatori riusciranno a riprodurre identiche condizioni per le attrezzature utilizzate. Per questo motivo, devono essere fornite prove dell’efficacia dell’apparecchiatura di spettrofotometria HPLC/CLUP prima di utilizzarla per quantificare l’MTT formazan negli estratti di tessuto, e devono essere rispettati i criteri di accettabilità per una serie di parametri standard di qualificazione sulla base dei parametri descritti nelle raccomandazioni della Food and Drug Administration statunitense per la validazione dei metodi di bio-analisi (36) (38). Questi parametri di base e loro criteri di accettazione figurano nell’appendice 5. Una volta che i criteri di accettabilità definiti nell’appendice 5 sono soddisfatti, l’apparecchiatura di spettrofotometria HPLC/CLUP è considerata qualificata e può essere utilizzata per misurare l’MTT formazan alle condizioni sperimentali descritte nel presente metodo di prova.
|
Criteri di accettabilità
|
43.
|
Per ciascuna batteria di prove che utilizza lotti di tessuto RhCE che soddisfa i criteri del controllo di qualità (cfr. il paragrafo 30), i tessuti trattati con la sostanza di controllo negativo devono mostrare una densità ottica che rifletta la qualità dei tessuti sia dopo le fasi di spedizione e ricezione, sia durante l’applicazione di tutti i protocolli e devono situarsi entro i limiti storici descritti nella tabella 2 (cfr. il paragrafo 26). Analogamente, i tessuti trattati con la sostanza di controllo positivo (acetato di metile) devono presentare una vitalità tessutale < 50 % rispetto al controllo negativo nel VRM1 per il protocollo di liquidi o solidi, e ≤ 30 % (protocollo per i liquidi) o ≤ 20 % (protocollo per i solidi) rispetto al controllo negativo nel VRM2; tali valori riflettono la capacità dei tessuti di reagire a una sostanza chimica in esame irritante alle condizioni del metodo di prova (34)(35). La variabilità tra le repliche di tessuto per le sostanze chimiche in esame e le sostanze di controllo deve rientrare nei limiti accettabili (cioè, la differenza di vitalità tra due repliche di tessuti dovrebbe essere inferiore al 20 % oppure la deviazione standard tra tre repliche di tessuto non dovrebbe essere superiore al 18 %). Se uno dei controlli (positivo o negativo) in una batteria di prove si situa al di fuori degli intervalli accettabili, la batteria di prove deve essere considerata «non valida» e deve essere ripetuta. Se la variabilità tra le repliche di tessuto di una sostanza chimica si situa al di fuori dell’intervallo di valori accettabile, la prova deve essere considerata «non valida» e la sostanza chimica in esame deve essere nuovamente testata.
|
Interpretazione dei risultati e modello predittivo
|
44.
|
I valori di OD dei controlli/aree di picco ottenuti con le repliche degli estratti di tessuto per ciascuna sostanza chimica in esame sono utilizzati per calcolare la percentuale media di vitalità dei tessuti (media tra le repliche di tessuto) rispetto alla vitalità del controllo negativo (fissata al 100 %). La tabella 4 riporta i valori limite della percentuale della vitalità tessutale per identificare le sostanze chimiche in esame che non richiedono classificazione per l’irritazione oculare o per gravi lesioni oculari («senza categoria» del GHS dell’ONU/CLP). Pertanto i risultati sono interpretati come segue:
|
—
|
La sostanza chimica in esame è considerata sostanza che non richiede classificazione né etichettatura secondo il rientrano in nessuna categoria («senza categoria» secondo il sistema GHS dell’ONU/CLP) se la percentuale media di vitalità tessutale dopo l’esposizione e l’incubazione post-esposizione è superiore (>) al valore limite stabilito per la percentuale di vitalità del tessuto, come riportato nella tabella 4. In questo caso, non occorre procedere a prove supplementari con altri metodi di prova.
|
|
—
|
Per contro, nessuna predizione è possibile se la percentuale media di vitalità tessutale dopo l’esposizione e l’incubazione post-esposizione è inferiore o uguale (≤) al valore limite stabilito per la percentuale di vitalità del tessuto, come riportato nella tabella 4. In questo caso, è necessario effettuare prove supplementari con altri metodi di prova, giacché i metodi di prova sul RhCE producono un numero significativo di falsi positivi (cfr. i paragrafi 14 e 15) e non permettono di distinguere tra le categorie 1 e 2 del GHS dell’ONU e CLP (cfr. il paragrafo 17).
|
Tabella 4
Modelli predittivi secondo la classificazione del sistema GHS dell’ONU e del regolamento CLP
|
VRM
|
Senza categoria
|
Non è possibile fare previsioni
|
|
VRM 1 - EpiOcular™ EIT (per entrambi i protocolli)
|
Vitalità tessutale media > 60 %
|
Vitalità tessutale media ≤ 60 %
|
|
VRM 2 - SkinEthic™ HCE EIT (per il protocollo dei liquidi)
|
Vitalità tessutale media > 60 %
|
Vitalità tessutale media ≤ 60 %
|
|
VRM2 - SkinEthic™ HCE EIT (per il protocollo dei solidi)
|
Vitalità tessutale media > 50 %
|
Vitalità tessutale media ≤ 50 %
|
|
|
45.
|
Una singola prova costituita da almeno due repliche di tessuto dovrebbe essere sufficiente per una sostanza chimica in esame, se la classificazione che ne risulta è inequivocabile. Tuttavia, in caso di risultati inconclusivi, tra cui misurazioni discordanti delle repliche e/o una percentuale media della vitalità dei tessuti pari a 60±5 % (VRM1, e VRM2 per il protocollo dei liquidi) o 50±5 % (VRM2 per il protocollo dei solidi), si dovrebbe considerare l’opportunità di eseguire una seconda prova, oltre che una terza nell’eventualità di risultati discordanti tra le prime due prove.
|
|
46.
|
È anche possibile utilizzare valori soglia di vitalità dei tessuti diversi per distinguere le sostanze chimiche che rientrano o non rientrano in una data categoria in casi particolari di miscele, se necessario e giustificato, in modo da migliorare le prestazioni complessive del metodo di prova per questi tipi di miscele (cfr. il paragrafo 14). Le sostanze chimiche di riferimento sono utili per valutare il potenziale di grave lesione oculare/irritazione oculare di sostanze chimiche o miscele sconosciute o per valutare il potenziale di irritazione oculare relativo di una sostanza chimica appartenente a una determinata classe di sostanze o prodotti chimici nell’ambito di una specifica gamma di risposte positive.
|
DATI E RELAZIONE
Dati
|
47.
|
Per ciascuna prova, i dati ottenuti da singole repliche dei tessuti (ad esempio, i valori OD/aree di picco dell’MTT formazan e le percentuali di vitalità tessutale calcolate per ogni sostanza chimica in esame e per ogni controllo, compresa la previsione conclusiva del metodo di prova sul RhCE) sono presentati sotto forma di tabella per ciascuna sostanza chimica in esame, compresi i dati delle prove ripetute, se del caso. Inoltre, devono essere riportate la percentuale media della vitalità del tessuto e la differenza di vitalità tra due repliche di tessuto (se n = 2 repliche di tessuto) o la deviazione standard (se n ≥ 3 repliche di tessuto) per ogni sostanza chimica in esame e controllo. Eventuali interferenze con il test dell’MTT osservate per una sostanza chimica in esame a motivo di una riduzione diretta dell’MTT e/o di interferenza nei colori devono essere indicate per ciascuna sostanza chimica in esame.
|
Relazione sull’esecuzione della prova
|
48.
|
La relazione sull’esecuzione della prova comprende le informazioni riportate di seguito.
Sostanza chimica in esame
|
|
Sostanza monocostituente:
|
—
|
identificazione della sostanza chimica, mediante denominazioni quali IUPAC o CAS, il numero CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
—
|
stato fisico, volatilità, pH, LogP, peso molecolare, classe chimica e altre proprietà fisico-chimiche pertinenti per la realizzazione dello studio, secondo i dati disponibili;
|
|
—
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
—
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
—
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili.
|
|
|
|
Sostanza multicostituente, UVCB e miscela:
|
—
|
caratterizzazione, nella misura del possibile, ad esempio attraverso l’identità chimica (cfr. sopra), la purezza, le proporzioni quantitative e le proprietà fisico-chimiche pertinenti (cfr. sopra) dei costituenti, secondo i dati disponibili;
|
|
—
|
stato fisico e altre proprietà fisico-chimiche pertinenti per la realizzazione dello studio, secondo i dati disponibili;
|
|
—
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
—
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
—
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili.
|
|
Sostanze di controllo positive e negative
|
—
|
identificazione della sostanza chimica, mediante denominazioni quali IUPAC o CAS, il numero CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
—
|
stato fisico, volatilità, peso molecolare, classe chimica e altre proprietà fisico-chimiche pertinenti per la realizzazione dello studio, secondo i dati disponibili;
|
|
—
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
—
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
—
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
—
|
giustificazione dell’utilizzo di un controllo negativo diverso dall’acqua (H2O) ultrapura o il DPBS privo di Ca 2+/Mg2+, se applicabile;
|
|
—
|
giustificazione dell’utilizzo di un controllo positivo diverso dall’acetato di metile puro, se applicabile;
|
|
—
|
riferimento ai dati storici relativi ai controlli positivi e negativi che dimostrano la conformità ai criteri di accettabilità.
|
Informazioni relative allo sponsor e al laboratorio sperimentale
|
—
|
Nome e indirizzo dello sponsor, del centro di prova e del responsabile dello studio.
|
|
—
|
Costrutto tessutale RhCE e protocollo applicato (spiegazione dei motivi delle scelte operate, se del caso)
|
Condizioni del metodo di prova
|
—
|
Il costrutto tessutale RhCE utilizzato, compreso numero di lotto;
|
|
—
|
lunghezza d’onda e larghezza di banda (se del caso) utilizzati per la quantificazione dell’MTT formazan, e intervallo di linearità degli apparecchi di misurazione (ad es. spettrofotometro);
|
|
—
|
descrizione del metodo applicato per quantificare l’MTT formazan;
|
|
—
|
descrizione del sistema di spettrofotometria-HPLC/CLUP utilizzato, se del caso;
|
|
—
|
informazioni complete sul modello specifico tessuto RhCE utilizzato e sulla sua efficienza. Esse dovrebbero includere (elenco non esaustivo):
|
i)
|
vitalità all’atto del controllo di qualità (fornitore);
|
|
ii)
|
vitalità alle condizioni del metodo di prova (utilizzatore);
|
|
iii)
|
funzione di barriera durante il controllo della qualità;
|
|
iv)
|
morfologia, se informazioni disponibili;
|
|
v)
|
riproducibilità e predittività;
|
|
vi)
|
altri controlli di qualità (QC) del costrutto tessutale RhCE, in base alle informazioni disponibili;
|
|
|
—
|
riferimenti a dati storici del costrutto tessutale RhCE, compresi (elenco non esaustivo): accettabilità dei dati di QC in relazione ai dati storici del lotto;
|
|
—
|
dichiarazione di dimostrazione della competenza tecnica del laboratorio nell’applicazione del metodo di prova, prima della sua applicazione sistematica, sottoponendo a prova le sostanze di riferimento;
|
Criteri di accettabilità per la prova e le batterie di prove
|
—
|
medie e intervalli di valori accettabili per i controlli positivi e negativi, basati su dati storici;
|
|
—
|
variabilità accettabile tra le repliche di tessuto per i controlli positivi e negativi;
|
|
—
|
variabilità accettabile tra le repliche di tessuto per la sostanza chimica in esame;
|
Procedura di prova
|
—
|
dettagli della procedura di prova utilizzata;
|
|
—
|
dosi delle sostanze chimiche in esame e delle sostanze di controllo utilizzate;
|
|
—
|
durata e temperatura dei periodi di esposizione, di immersione post-esposizione e di incubazione post-esposizione (se del caso);
|
|
—
|
descrizione delle eventuali modifiche apportate al protocollo sperimentale;
|
|
—
|
indicazione dei controlli usati per riduttori dell’MTT diretti e/o sostanze chimiche in esame coloranti, se del caso;
|
|
—
|
numero di repliche dei tessuti utilizzati per la sostanza chimica in esame e i controlli (controllo positivo, controllo negativo, NSMTT, NSCliving and NSCkilled, se del caso).
|
Risultati
|
—
|
tabella dei dati per ciascuna sostanza chimica in esame e sostanza di controllo per ciascuna batteria di prove (compresi gli esperimenti ripetuti, se del caso) e per la misurazione di ciascuna replica, compresi i valori OD o l’area di picco dell’MTT formazan, la percentuale di vitalità dei tessuti, la percentuale media della vitalità dei tessuti, la differenza o la deviazione standard tra le repliche di tessuti e la previsione finale;
|
|
—
|
se del caso, i risultati dei controlli effettuati per riduttori diretti dell’MTT e/o delle sostanze chimiche in esame colorate, compresi i valori OD o l’area di picco dell’MTT formazan, %NSMTT, %NSCliving, %NSCkilled, la differenza o la deviazione standard tra le repliche di tessuti, la percentuale finale corretta di vitalità dei tessuti e la previsione finale;
|
|
—
|
risultati ottenuti per la o le sostanze chimiche in esame e le sostanze di controllo in relazione ai criteri di accettabilità stabiliti per la prova e le batterie di prove;
|
|
—
|
descrizione di altri effetti osservati, ad esempio la colorazione dei tessuti da parte di una sostanza chimica in esame colorata.
|
Discussione dei risultati
Conclusione
|
BIBLIOGRAFIA
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: United Nations. Disponibile all’indirizzo: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
Capitolo B.5 del presente allegato, Irritazione/corrosione oculare acuta.
|
|
(3)
|
Capitolo B.47 del presente allegato, Metodo di prova dell’opacità e della permeabilità della cornea nei bovini per l’identificazione di i) sostanze chimiche che inducono gravi lesioni oculari e ii) sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari.
|
|
(4)
|
Capitolo B.48 del presente allegato, Metodo di prova sull’occhio isolato dei polli (Isolated Chicken Eye — ICE) per l’identificazione di i) sostanze chimiche che inducono gravi lesioni oculari e ii) sostanze chimiche che non richiedono classificazione.
|
|
(5)
|
Capitolo B.61 del presente allegato, Metodo di prova di diffusione della fluoresceina per l’individuazione di sostanze corrosive e gravemente irritanti per gli occhi
|
|
(6)
|
Capitolo B.68 del presente allegato, Metodo di prova di esposizione in vitro di breve durata per l’identificazione di i) sostanze chimiche che inducono gravi lesioni oculari e ii) sostanze chimiche che non richiedono classificazione per irritazione oculare o gravi lesioni oculari.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2010, 261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28 125 EN; doi:10.2787/41680. Disponibile all’indirizzo: http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28 173 EN; doi: 10.2787/043697. Disponibile all’indirizzo: http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In Vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In Vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28 175 EN; doi: 10.2787/390390. Disponibile all’indirizzo: http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in Preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In Vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In Vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Organisation for Economic Cooperation and Development, Paris.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In Vitro 27, 1 476-1 488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In Vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In Vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In Vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922–936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In Vitro 15, 115–130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2 610–2 625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41–54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Disponibile all’indirizzo: [https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Disponibile all’indirizzo: https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. May 2001. Disponibile all’indirizzo: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Organisation for Economic Cooperation and Development, Paris.
|
Appendice 1
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza" per indicare la percentuale di risultati corretti di un metodo di prova (18).
Sostanza chimica di riferimento
: sostanza chimica usata come standard di confronto rispetto a una sostanza chimica in esame. Una sostanza di riferimento dovrebbe presentare le seguenti proprietà: i) fonte o fonti coerenti e affidabili per la sua identificazione e caratterizzazione; ii) analogia strutturale, funzionale e/o chimica o analogia alla classe di prodotto della sostanza o delle sostanze in esame; iii) caratteristiche fisiche/chimiche note; iv) dati di supporto relativi agli effetti noti; e v) potenza nota nell'intervallo della reazione auspicata.
Approccio "dal basso"
: approccio graduale applicato a una sostanza chimica in esame che si presume non debba essere classificata né etichettata per irritazione oculare o grave lesione oculare, la cui prima fase consiste nel distinguere le sostanze chimiche che non richiedono classificazione né etichettatura (esito negativo) dalle altre sostanze chimiche (esito positivo).
Sostanza chimica
: sostanza o miscela.
Concordanza
: si veda "Accuratezza".
Cornea
: parte trasparente frontale del bulbo oculare che copre l'iride e la pupilla e consente il passaggio della luce verso l'interno.
CV
: coefficiente di variazione
Dev
: Deviazione
EIT
: (Eye Irritation Test) test di irritazione oculare.
EURL ECVAM
: (European Union Reference Laboratory for Alternatives to Animal Testing) Laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale.
Irritazione oculare
: produzione di alterazioni nell'occhio in seguito all'applicazione di una sostanza chimica in esame sulla superficie anteriore dell'occhio, totalmente reversibili entro 21 giorni dall'applicazione. Sinonimo di "effetti reversibili sugli occhi" e di "categoria 2 del GHS dell'ONU".
ET50
: tempo di esposizione necessario per ridurre la vitalità dei tessuti del 50 % dopo l'applicazione di una sostanza chimica di riferimento ad una concentrazione fissa predeterminata.
Percentuale di falsi negativi
: percentuale di tutte le sostanze positive erroneamente identificate come negative da un metodo di prova. È un indicatore dell'efficienza del metodo di prova.
Percentuale di falsi positivi
: percentuale di tutte le sostanze negative erroneamente identificate come positive da un metodo di prova. È un indicatore dell'efficienza del metodo di prova.
Pericolo
: la proprietà intrinseca di un agente o di una situazione che ha il potenziale di causare effetti nocivi se un organismo, un sistema o una (sotto)popolazione vi sono esposti.
HCE
: epitelio corneale umano di SkinEthic™.
HPLC
: cromatografia liquida ad alta prestazione.
IC50
: concentrazione alla quale una sostanza chimica di riferimento riduce la vitalità dei tessuti del 50 % dopo un tempo di esposizione fisso (ad es. 30 minuti di trattamento con SDS).
Dose infinita
: quantità di sostanza chimica in esame applicata al costrutto tessutale RhCE che supera la quantità necessaria per coprire in maniera completa e uniforme la superficie epiteliale.
Effetti irreversibili sugli occhi
: cfr. "Gravi lesioni oculari".
LLOQ
: limite inferiore di quantificazione.
LogP
: logaritmo del coefficiente di ripartizione ottanolo/acqua.
Miscela
: la miscela o soluzione composta di due o più sostanze.
Sostanza monocostituente
: la sostanza, definita in base alla sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno l'80 % (p/p).
Sostanza multicostituente
: la sostanza, definita in base alla sua composizione quantitativa, in cui più di un costituente principale è presente in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). La sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente risiede nel fatto che la miscela è ricavata mischiando due o più sostanze senza che avvenga una reazione chimica, mentre la sostanza multicostituente è il risultato di una reazione chimica.
MTT
: bromuro di 3-(4,5-dimetiltiazol-2-ile)-2,5-difeniltetrazolio; tiazolil blu tetrazolio bromuro.
Controllo negativo
: campione contenente tutti i componenti di un sistema di prova, trattato con una sostanza che notoriamente induce una risposta positiva nel sistema di prova. Il campione subisce il medesimo procedimento dei campioni trattati con la sostanza chimica in esame e degli altri campioni di controllo ed è utilizzato per determinare la vitalità dei tessuti al 100 %.
Non classificata
: sostanza chimica non classificata in termini di irritazione oculare (categoria GHS dell'ONU/CLP 2, categoria GHS dell'ONU 2A o 2B) o gravi lesioni oculari (categoria GHS dell'ONU/CLP 1). Termine intercambiabile con "Senza categoria GHS dell'ONU".
NSCkilled
: colore non specifico nei tessuti morti.
NSCliving
: colore non specifico nei tessuti vivi.
NSMTT
: riduzione non specifica di MTT.
OD
: densità ottica.
Standard di prestazione
: standard basati su un metodo di riferimento validato e riconosciuto come scientificamente valido, che consentono di valutare la comparabilità di un metodo proposto simile sotto il profilo strutturale e funzionale. Detti standard comprendono: i) i componenti essenziali del metodo di prova; ii) un elenco minimo di sostanze di riferimento scelte tra le sostanze utilizzate per dimostrare l'accettabilità delle prestazioni del metodo di riferimento validato; e iii) in funzione dei risultati ottenuti con il metodo di riferimento validato, i livelli comparabili di accuratezza e affidabilità che il metodo proposto dovrebbe ottenere quando viene valutato utilizzando l'elenco minimo di sostanze di riferimento (18).
Controllo positivo
: campione contenente tutti i componenti di un sistema di prova, trattato con una sostanza che notoriamente induce una risposta positiva nel sistema di prova. Il campione subisce il medesimo procedimento dei campioni trattati con la sostanza chimica in esame e degli altri campioni di controllo. Per poter valutare la variabilità nel tempo della risposta dei controlli positivi, l'entità della risposta positiva non deve essere eccessiva.
Pertinenza
: descrizione del rapporto tra la prova e l'effetto studiato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (18).
Affidabilità
: la misura in cui il metodo di prova può essere riprodotto nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna al laboratorio e la ripetibilità fra i laboratori (18).
Prova di sostituzione
: una prova progettata per sostituire un metodo di prova applicato correntemente e accettato per l'individuazione dei pericoli e/o la valutazione dei rischi, e che è stata determinata per fornire una protezione equivalente o maggiore della salute dell'uomo o degli animali oppure dell'ambiente, se del caso, rispetto alla prova accettata, per tutte le possibili situazioni sperimentali e le sostanze di prova (18).
Riproducibilità
: concordanza dei risultati ottenuti dall'esecuzione di prove ripetute sulla stessa sostanza chimica in esame in applicazione dello stesso protocollo sperimentale (cfr. Affidabilità) (18).
Effetti reversibili sugli occhi
: cfr. "Irritazione oculare".
RhCE
: (Reconstructed human Cornea-like Epithelium) modello di epitelio corneale umano ricostituito.
Batteria di prove
: una batteria di prove consiste nel testare una o più sostanze chimiche in esame simultaneamente a un controllo negativo e a un controllo positivo.
SD
: deviazione standard.
Sensibilità
: percentuale di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l'accuratezza del metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza del metodo di prova (18).
Gravi lesioni oculari
: la produzione di danni ai tessuti oculari o indebolimento grave della vista in seguito all'applicazione della sostanza chimica in esame sulla parte anteriore dell'occhio, non completamente reversibile entro 21 giorni dall'applicazione. Sinonimo di "effetti irreversibili sugli occhi" e di "categoria 1 del GHS dell'ONU/CLP".
Procedure operative standard
: Procedure scritte formali che descrivono in dettaglio le modalità di esecuzione in laboratorio di specifiche operazioni di routine e nel quadro di una specifica prova. Rientrano nelle buone pratiche di laboratorio.
Specificità
: percentuale di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l'accuratezza del metodo di prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza del metodo (18).
Sostanza
: un elemento chimico e i suoi composti, allo stato naturale od ottenuti per mezzo di un procedimento di produzione, compresi gli additivi necessari a mantenerne la stabilità e le impurezze derivanti dal procedimento utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione.
Prova
: Una singola sostanza chimica in esame sottoposta a prova in parallelo in almeno due repliche tessutali, come definito nella corrispondente procedura operativa standard.
Vitalità tessutale
: parametro che misura l'attività totale di una popolazione di cellule in un tessuto ricostituito in termini della loro capacità di ridurre il colorante vitale MTT), che, in funzione del parametro misurato e del tipo di disegno sperimentale utilizzato, corrisponde al numero totale e/o alla vitalità delle cellule vive.
Approccio "dall'alto"
: approccio graduale applicato nel caso di una sostanza chimica in esame sospettata di causare gravi lesioni oculari, che inizia con la determinazione delle sostanze chimiche che inducono gravi lesioni oculari (esito positivo) rispetto ad altre sostanze chimiche (esito negativo).
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Strategia di prova in sequenza
: strategia sperimentale in tappe progressive, che applica i metodi sperimentali in maniera sequenziale; tutte le informazioni disponibili sulla sostanza chimica in esame sono vagliate in ciascuna fase, seguendo un approccio basato sul peso dell'evidenza, al fine di stabilire se vi sono informazioni sufficienti per prendere una decisione di classificazione del pericolo, prima di procedere alla fase successiva della strategia sperimentale. Se è possibile assegnare il potenziale di pericolo/la potenza della sostanza chimica in esame in base alle informazioni disponibili ad una determinata fase, non è necessario svolgere altre prove (18).
ULOQ
: limite superiore di quantificazione.
Sistema mondiale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (GHS dell'ONU)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi e livelli standardizzati di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di prudenza e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (1).
Categoria 1 del GHS dell'ONU/CLP
: cfr. "Gravi lesioni oculari".
Categoria 2 del GHS dell'ONU/CLP
: cfr. "Irritazione oculare".
Senza categoria del GHS dell'ONU/CLP
: sostanza chimica che non rientra nella categoria 1 o 2 del GHS dell'ONU/CLP (o categoria 2A o 2B del GHS dell'ONU). Sostituibile con "Non classificata".
UPLC
: cromatografia liquida a ultra alta prestazione.
UVCB (Substance of Unknown or Variable Composition, Complex Reaction Products or Biological Materials)
: sostanze di composizione sconosciuta o variabile, prodotti di una reazione complessa o materiali biologici.
Metodo di prova valido
: metodo di prova la cui pertinenza e affidabilità sono ritenute soddisfacenti per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in termini assoluti ma soltanto in relazione a un obiettivo definito (18).
Metodo di prova validato
: metodo di prova in base al quale sono stati completati studi di validazione per determinare la pertinenza (compresa l'accuratezza) e l'affidabilità per un fine specifico. Va sottolineato che un metodo di prova validato potrebbe non avere un rendimento sufficiente in termini di valori di accuratezza e affidabilità ritenuti accettabili per il raggiungimento dell'obiettivo prefissato (18).
VRM
: Metodo di riferimento validato.
VRM1
: Con VRM1 (Metodo di riferimento validato 1) si intende il metodo EpiOcular™ EIT.
VRM2
: Con VRM2 (Metodo di riferimento validato 2) si intende il metodo SkinEthic™ HCE EIT.
Peso dell'evidenza
: processo che consiste nel tener conto dei punti di forza e di debolezza di informazioni diverse per conseguire e supportare una data conclusione relativa al potenziale di pericolo di una sostanza chimica in esame.
Appendice 2
Principali Componenti dei Metodi di Prova su Rhce Validati ai Fini dell'Identificazione Delle Sostanze Chimiche che non Richiedono Classificazione né Etichettatura per Irritazione Oculare o Gravi Lesioni Oculari
|
Componenti della prova
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protocolli
|
Liquidi
(pipettabile a ≤ 37 ± 1 oC per 15 minuti)
|
Solidi
(non pipettabile)
|
Liquidi e viscosi
(pipettabile)
|
Solidi
(non pipettabile)
|
|
Superficie del modello
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
Numero di repliche tessutali
|
Almeno 2
|
Almeno 2
|
Almeno 2
|
Almeno 2
|
|
Verifica preventiva di interferenze colore
|
50 μl + 1 ml H2O per 60 min a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR (sostanze chimiche in esame non colorate), o 50 μl + 2 ml di isopropanolo mescolato per 2-3 ore a T ambiente (sostanze chimiche in esame colorate)
|
 Se la densità ottica della sostanza chimica in esame a 570 ± 20 nm, dopo sottrazione della OD per l'isopropanolo o acqua è > 0,08 (corrispondente a circa il 5 % della OD media del controllo negativo), devono essere eseguiti adeguati controlli su organismi viventi. Se la densità ottica della sostanza chimica in esame a 570 ± 20 nm, dopo sottrazione della OD per l'isopropanolo o acqua è > 0,08 (corrispondente a circa il 5 % della OD media del controllo negativo), devono essere eseguiti adeguati controlli su organismi viventi.
|
|
50 mg + 1 ml H2O per 60 min a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR (sostanze chimiche in esame non colorate)
e/o
50 mg + 2 ml di isopropanolo mescolato per 2-3 ore a T ambiente (sostanze chimiche in esame colorate e non colorate)
|
 Se la densità ottica della sostanza chimica in esame a 570 ± 20 nm, dopo sottrazione della OD per l'isopropanolo o acqua è > 0,08 (corrispondente a circa il 5 % della OD media del controllo negativo), devono essere eseguiti adeguati controlli su organismi viventi. Se la densità ottica della sostanza chimica in esame a 570 ± 20 nm, dopo sottrazione della OD per l'isopropanolo o acqua è > 0,08 (corrispondente a circa il 5 % della OD media del controllo negativo), devono essere eseguiti adeguati controlli su organismi viventi.
|
|
10 μl + 90 μl H2O mescolati per 30 ± 2 min a T ambiente (TA, 18-28 oC)
|
 se la sostanza chimica in esame è colorata, devono essere eseguiti adeguati controlli su organismi viventi. se la sostanza chimica in esame è colorata, devono essere eseguiti adeguati controlli su organismi viventi.
|
|
10 mg + 90 μl H2O mescolati per 30 ± 2 min a T ambiente
|
 se la sostanza chimica in esame è colorata, devono essere eseguiti adeguati controlli su organismi viventi. se la sostanza chimica in esame è colorata, devono essere eseguiti adeguati controlli su organismi viventi.
|
|
|
Verifica preventiva di riduzione diretta dell'MTT
|
50 μl + 1 ml MTT 1 mg/ml di soluzione per 180 ± 15 min a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
 Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi per congelamento Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi per congelamento
(come controllo negativo si usa 50 μl di acqua distillata sterile in una soluzione MTT)
|
|
50 mg + 1 ml di soluzione MTT 1 mg/ml per 180 ± 15 min a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
 Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi per congelamento Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi per congelamento
(come controllo negativo si usa 50 μl di acqua distillata sterile in una soluzione MTT)
|
|
30 μl + 300 μl di soluzione MTT 1 mg/ml per 180 ± 15 min a 37±2oC, 5±1 % CO2, ≥95 % UR
|
 Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi in acqua Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi in acqua
(come controllo negativo si usa 30 μl di acqua distillata sterile in una soluzione MTT)
|
|
30 mg + 300 μl MTT di soluzione MTT 1 mg/ml per 180 ± 15 min a 37±2oC, 5±1 % CO2, ≥95 % UR
|
 Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi in acqua Se la soluzione vira al blu/viola, devono essere eseguiti adeguati controlli su tessuti uccisi in acqua
(come controllo negativo si usa 30 μl di acqua distillata sterile in una soluzione MTT)
|
|
|
Pretrattamento
|
20 μl di DPBS privo di Ca2+/Mg2+
per 30 ± 2 min a 37±2oC, 5±1 % CO2, ≥95 % UR, al riparo dalla luce.
|
20 μl di DPBS privo di Ca2+/Mg2+
per 30 ± 2 min a 37±2oC, 5±1 % CO2, ≥95 % UR, al riparo dalla luce.
|
-
|
-
|
|
Dosi e trattamento
|
50 μl (83.3 μl/cm2)
|
50 mg (83,3 mg/cm2), misurati con strumenti calibrati (ad es. un cucchiaio raso è calibrato per contenere 50 mg di cloruro di sodio).
|
10 μl di DPBS privo di Ca2+/Mg2+ + 30 ± 2 μl (60 μl/cm2)
Per sostanze viscose, usare maglie di nylon
|
30 μl di DPBS privo di Ca2+/Mg2+ + 30 ± 2 mg (60 mg/cm2)
|
|
Tempo e temperatura di esposizione
|
30 min (± 2 min)
nel mezzo di coltura
a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
6 ore (± 0,25 h)
nel mezzo di coltura
a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
30 min (± 2 min)
nel mezzo di coltura
a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
4 ore (± 0,1 h)
nel mezzo di coltura
a 37 ± 2 oC, 5 ± 1 % CO2, ≥ 95 % UR
|
|
Risciacquo a temperatura ambiente
|
3 volte in 100 ml di DPBS privo di Ca2+/Mg2+
|
3 volte in 100 ml di DPBS privo di Ca2+/Mg2+
|
20 ml di DPBS privo di Ca2+/Mg2+
|
25 ml di DPBS privo di Ca2+/Mg2+
|
|
Immersione post-esposizione
|
12 min (± 2 min) a T amb. nel mezzo di coltura
|
25 min (± 2 min) a T amb. nel mezzo di coltura
|
30 min (± 2 min) a 37oC, 5 % CO2, 95 % UR nel mezzo di coltura
|
30 min (± 2 min) a T amb. nel mezzo di coltura
|
|
Incubazione post-esposizione
|
120 min (± 15 min) nel mezzo di coltura a 37±2oC, 5±1 % CO2, ≥95 % UR
|
18 h (± 0,25 h) nel mezzo di coltura a 37±2oC, 5±1 % CO2, ≥95 % UR
|
nessuna
|
18 h (± 0,5 h) nel mezzo di coltura a 37±2oC, 5±1 % CO2, ≥95 % UR
|
|
Controllo negativo
|
50 μl H2O
Testato in parallelo
|
50 μl H2O
Testato in parallelo
|
30 + 2μl di DPBS privo di Ca2+/Mg2+
Testato in parallelo
|
30 + 2μl di DPBS privo di Ca2+/Mg2+
Testato in parallelo
|
|
Controllo positivo
|
50 μl di acetato di metile
Testato in parallelo
|
50 μl di acetato di metile
Testato in parallelo
|
30 + 2μl di acetato di metile
Testato in parallelo
|
30 + 2μl di acetato di metile
Testato in parallelo
|
|
Soluzione di MTT
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
Tempo e temperatura dell'incubazione di MTT
|
180 min (± 15 min) a 37±2oC, 5±1 % CO2, ≥95 % UR
|
180 min (± 15 min) a 37±2oC, 5±1 % CO2, ≥95 % UR
|
180 min (± 15 min) a 37±2oC, 5±1 % CO2, ≥95 % UR
|
180 min (± 15 min) a 37±2oC, 5±1 % CO2, ≥95 % UR
|
|
Solvente di estrazione
|
2 ml di isopropanolo
(estratto della parte superiore e inferiore dell'inserto bucando il tessuto)
|
2 ml di isopropanolo
(estratto della parte superiore dell'inserto bucando il tessuto)
|
1,5 ml di isopropanolo
(estratto della parte superiore e inferiore dell'inserto)
|
1,5 ml di isopropanolo
(estratto della parte superiore dell'inserto)
|
|
Tempo e temperatura di estrazione
|
2-3 ore agitando (~120 giri/min.) a T amb. o per una notte a 4-10 oC
|
2-3 ore agitando (~120 giri/min.) a T amb. o per una notte a 4-10 oC
|
4 ore agitando (~120 giri/min.) a T amb. o almeno per una notte senza agitare a 4-10 oC
|
Almeno 2 h agitando (~120 giri/min.) a T amb.
|
|
Lettura della densità ottica
|
570 nm (550 - 590 nm)
senza filtro di riferimento
|
570 nm (550-590 nm)
senza filtro di riferimento
|
570 nm (540 - 600 nm)
senza filtro di riferimento
|
570 nm (540 - 600 nm)
senza filtro di riferimento
|
|
Controllo di qualità dei tessuti
|
Trattamento con 100 μl di Triton X-100 allo 0,3 % (v/v)
12,2 min ≤ ET50 ≤ 37,5 min
|
Trattamento con 100 μl di Triton X-100 allo 0,3 % (v/v)
12,2 min ≤ ET50 ≤ 37,5 min
|
Trattamento di 30 min con SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
Trattamento di 30 min con SDS (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Criteri di accettabilità
|
|
1.
|
La OD media delle repliche tessutali trattate con il controllo negativo deve essere > 0,8 e < 2,5
|
|
2.
|
La vitalità media delle repliche tessutali esposte per 30 minuti con il controllo positivo, espresso in % del controllo negativo, deve essere < 50 %
|
|
3.
|
La differenza della vitalità tra due repliche tessutali non deve superare il 20 %.
|
|
|
1.
|
La OD media delle repliche tessutali trattate con il controllo negativo deve essere > 0,8 e < 2,5
|
|
2.
|
La vitalità media delle repliche tessutali esposte per 6 ore con il controllo positivo, espresso in % del controllo negativo, deve essere < 50 %
|
|
3.
|
La differenza della vitalità tra due repliche tessutali non deve superare il 20 %.
|
|
|
1.
|
La OD media delle repliche dei tessuti trattate con il controllo negativo deve essere > 1,0 e < 2,5
|
|
2.
|
La vitalità media delle repliche tessutali esposte per 30 minuti con il controllo positivo, espresso in % del controllo negativo, deve essere ≤ 30 %
|
|
3.
|
La differenza della vitalità tra due repliche tessutali non deve superare il 20 %.
|
|
|
1.
|
La OD media delle repliche dei tessuti trattate con il controllo negativo deve essere > 1,0 e < 2,5
|
|
2.
|
La vitalità media delle repliche tessutali esposte per 4 ore con il controllo positivo, espresso in % del controllo negativo, deve essere ≤ 20 %
|
|
3.
|
La differenza della vitalità tra due repliche tessutali non deve superare il 20 %.
|
|
Appendice 3
DIAGRAMMA DI FLUSSO ILLUSTRATIVO E ORIENTATIVO DELLA PROCEDURA PER IDENTIFICARE E TRATTARE LE SOSTANZE CHIMICHE IN ESAME RIDUTTRICI DIRETTE DELL'MTT E/O CHE PROVOCANO UN'INTERFERENZA DI COLORI, SULLA BASE DELLA PROCEDURA OPERATIVA STANDARD DEL VRM1
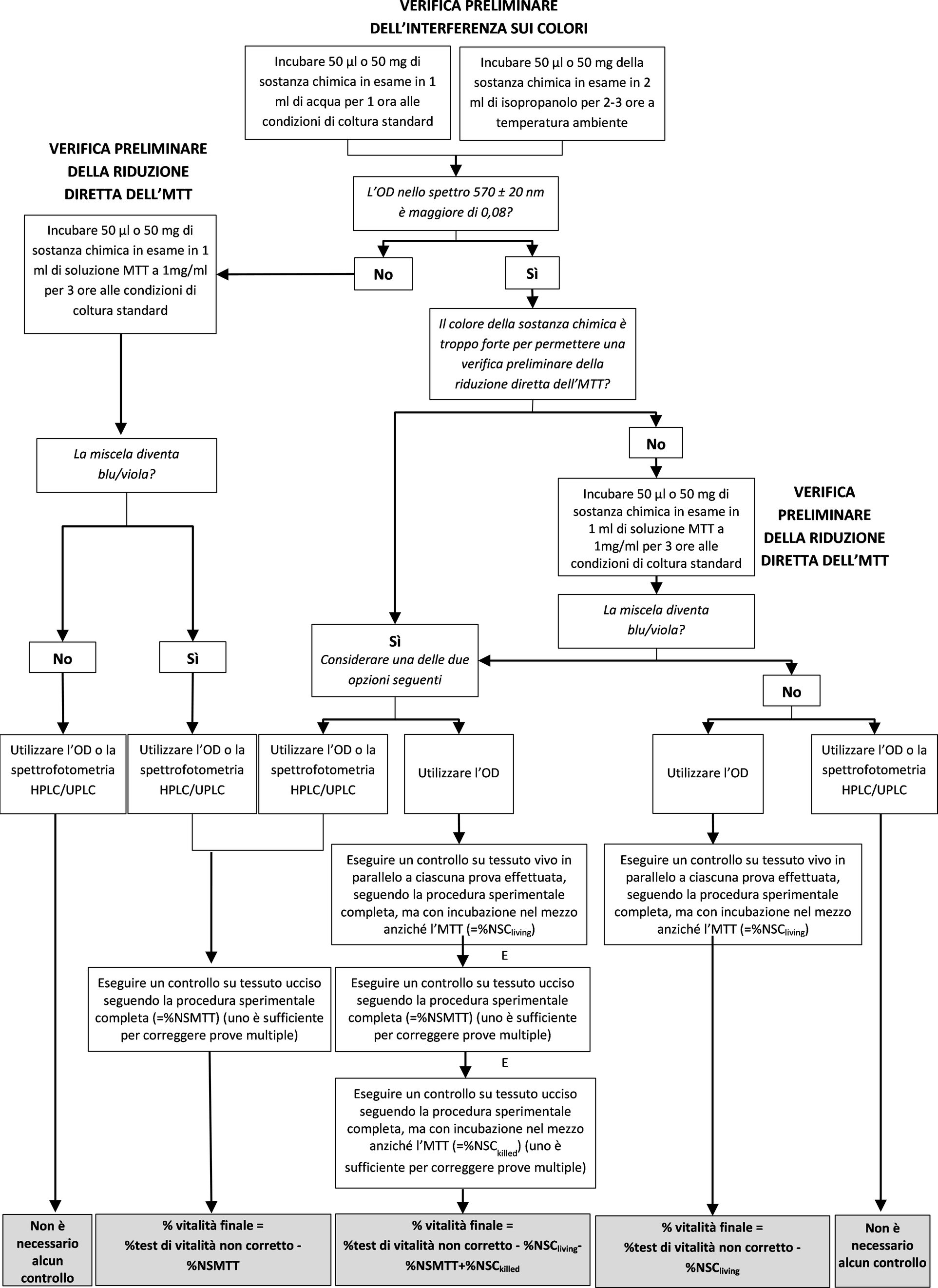
Appendice 4
DIAGRAMMA DI FLUSSO ILLUSTRATIVO E ORIENTATIVO DELLA PROCEDURA PER IDENTIFICARE E TRATTARE LE SOSTANZE CHIMICHE IN ESAME RIDUTTRICI DIRETTE DELL'MTT E/O CHE PROVOCANO UN'INTERFERENZA DI COLORI, SULLA BASE DELLA PROCEDURA OPERATIVA STANDARD DEL VRM2
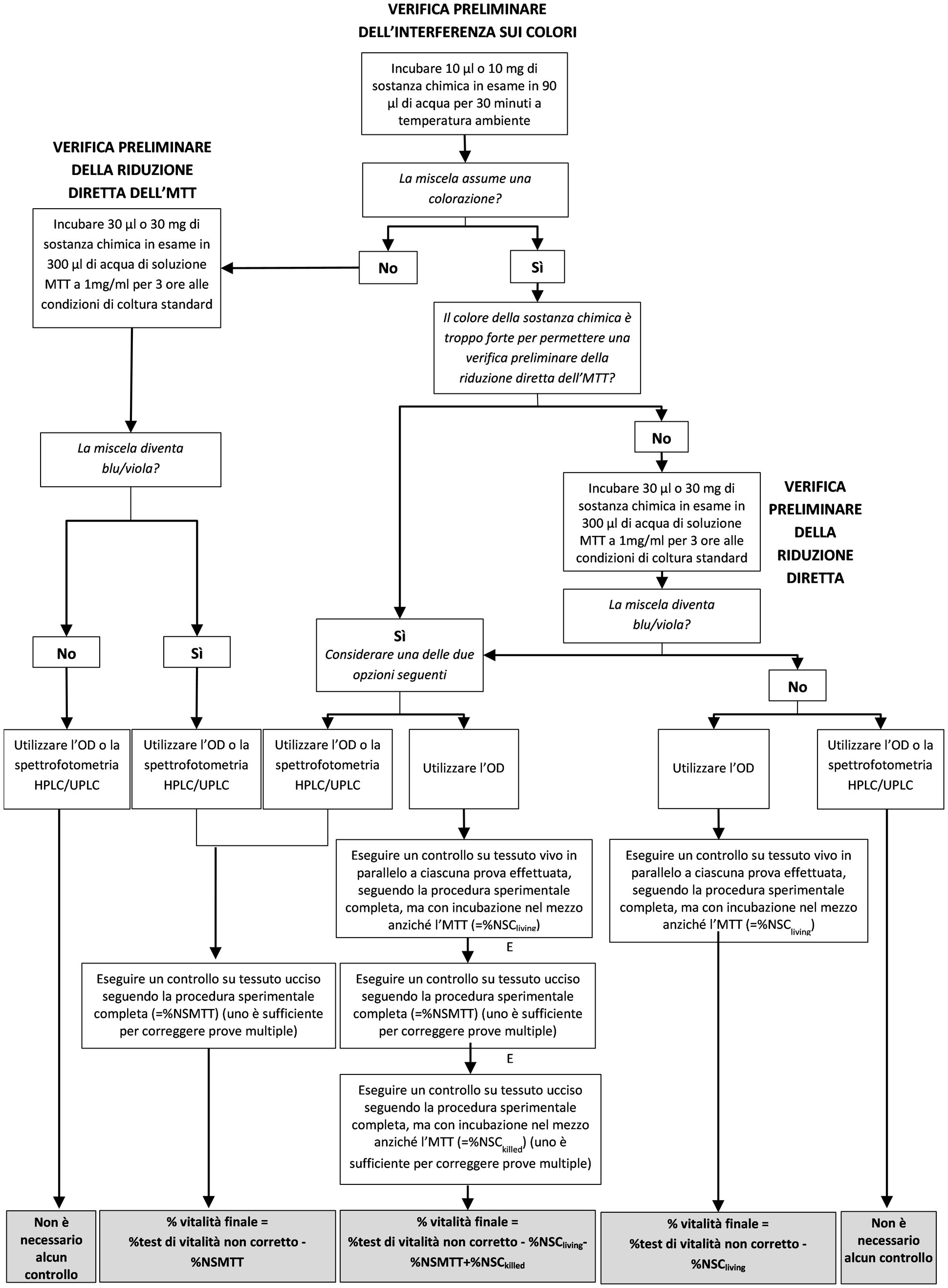
Appendice 5
PRINCIPALI PARAMETRI E CRITERI DI ACCETTAZIONE DELLA PROVA DI EFFICACIA DI UN SISTEMA DI SPETTROMETRIA HPLC/CLUP PER LA MISURAZIONE DI UN ESTRATTO DI MTT FORMAZAN ESTRATTO DA COSTRUTTI TESSUALI DI RHCE
|
Parametro
|
Protocollo derivato dagli orientamenti della FDA (36)(38)
|
Criteri di accettabilità
|
|
Selettività
|
Analisi dell'isopropanolo, del bianco vivente (estratto dal costrutto tessutale di RhCE vivente senza trattamento in isopropanolo), bianco ucciso (estratto dal costrutto tessutale di RhCE ucciso senza trattamento in isopropanolo) e di un colorante (blu di metilene, ad esempio)
|
Areainterferenza ≤ 20 % della AreaLLOQ
(88)
|
|
Precisione
|
Controlli della qualità (ossia, MTT formazan a 1,6 μg/ml, 16 μg/ml e 160 μg/ml) in isopropanolo (n=5)
|
CV ≤ 15 % o ≤ 20 % per il LLOQ
|
|
Accuratezza
|
Controlli della qualità in isopropanolo (n=5)
|
% scarto ≤ 15 % o ≤ 20 % per il LLOQ
|
|
Effetto matrice
|
Controlli della qualità nel bianco vivente (n=5)
|
85 % ≤ % effetto matrice ≤ 115 %
|
|
Effetto residuo
|
Analisi dell'isopropanolo dopo un ULOQ (89) standard
|
Areainterferenza ≤ 20 % della AreaLLOQ
|
|
Riproducibilità (nello stesso giorno)
|
3 curve di calibrazione indipendenti (sulla base di 6 consecutive diluizioni di 1/3 del MTT formazan in isopropanolo, a partire da ULSQ, cioè 200 μg/ml);
Controlli della qualità in isopropanolo (n=5)
|
Curve di calibrazione: % scarto ≤ 15 % o ≤ 20 % per il LLOQ
Controlli della qualità: % scarto ≤ 15 % e CV ≤ 15 %
|
|
Riproducibilità (in giorni diversi)
|
Giorno 1: 1 curva di taratura e controlli della qualità in isopropanolo (n = 3)
Giorno 2: 1 curva di taratura e controlli della qualità in isopropanolo (n = 3)
Giorno 3: 1 curva di taratura e controlli della qualità in isopropanolo (n = 3)
|
|
Stabilità a breve termine di MTT formazan in un estratto tessutale di RHCE
|
Controlli della qualità nel bianco vivente (n = 3) analizzato il giorno della preparazione, dopo conservazione per 24 ore a temperatura ambiente
|
% scarto ≤ 15 %
|
|
Stabilità a lungo termine dell'MTT formazan in un estratto tessutale di RHCE, se necessario
|
Controlli della qualità nel bianco vivente (n = 3) analizzato il giorno della preparazione, dopo diversi giorni di conservazione a -20 °C
|
% scarto ≤ 15 %
|
B.70 SAGGIO IN VITRO CHE UTILIZZA IL RECETTORE ESTROGENICO RICOMBINANTE UMANO (hrER) PER INDIVIDUARE LE SOSTANZE CHIMICHE CHE PRESENTANO UN’AFFINITÀ DI LEGAME CON I RECETTORI DI ESTROGENI
INTRODUZIONE GENERALE
Linea guida dell’OCSE per i metodi di prova basata su standard di prestazione
|
1.
|
Il presente metodo di prova è equivalente alla linea guida n. 493 (2015) dell’OCSE. La linea guida n. 493 è basata su standard di prestazione (performance-based test guideline - PBTG) che descrive la metodologia applicabile ai metodi di prova in vitro che si avvalgono del recettore estrogenico ricombinante umano per individuare le sostanze che presentano affinità di legame con i recettori di estrogeni (prove di legame all’hrER). Comprende due metodi di prova strutturalmente e funzionalmente simili volti ad individuare i ligandi dei recettori di estrogeni (ERα) e mira a facilitare l’elaborazione di nuovi metodi di prova simili o modificati, conformemente ai principi di validazione illustrati nel documento di orientamento dell’OCSE Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment (1). La presente PBTG si basa sui seguenti protocolli di riferimento (cfr. l’appendice 2 e appendice 3) completamente validati:
|
—
|
il saggio di Freyberg-Wilson (FW): metodo di prova in vitro di legame ai recettori di estrogeni (ER) che utilizza un ERα ricombinante umano a lunghezza intera (2); e
|
|
—
|
il saggio del CERI (Chemical Evaluation and Research Institute): metodo di prova in vitro di legame ai recettori di estrogeni (ER) che utilizza la proteina che costituisce il dominio di legame del ligando di un ER ricombinante umano (2).
|
Sono disponibili standard di prestazione (PS) (3) per facilitare l’elaborazione e la validazione di metodi di prova simili finalizzati al medesimo endpoint di sicurezza e per consentire di aggiornare tempestivamente la PBTG 493 integrandola con nuovi protocolli simili. Ciò può avvenire, tuttavia, solo dopo che l’OCSE abbia esaminato e approvato tali nuovi protocolli, dichiarando che rispettano gli standard di prestazione. Qualsiasi metodo di prova incluso nella linea guida TG 493 può essere scelto e applicato ai fini della conformità con i requisiti nazionali in materia di prove di legame ai recettori estrogenici nel quadro del sistema dell’OCSE di reciproca accettazione dei dati.
|
Contesto e principi relativi ai metodi di prova inclusi nella presente linea guida
|
2.
|
Nel 1998 l’OCSE ha avviato lavori a carattere altamente prioritario destinati a rivedere le linee guida esistenti, e a elaborarne di nuove, per lo screening e la sperimentazione relativi alle sostanze chimiche ritenute potenziali interferenti endocrini. Nel 2012 è stato rivisto il quadro concettuale dell’OCSE per la sperimentazione e la valutazione delle sostanze chimiche potenzialmente capaci di alterare il sistema endocrino. Le versioni originali e riviste del quadro concettuale figurano come allegati nel documento Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (4). Il quadro concettuale comprende cinque livelli, ciascuno dei quali corrisponde a un diverso livello di complessità biologica. Le prove di legame agli ER descritte nel presente metodo di prova sono di livello 2, che comprende «saggi in vitro che forniscono dati su determinati meccanismi/vie di attivazione endocrina». Il presente metodo di prova si applica alle prove in vitro di legame ai recettori concepite per individuare i ligandi del recettore estrogenico alfa umano (ERα).
|
|
3.
|
La pertinenza delle prove in vitro di legame agli ER sotto il profilo delle funzioni biologiche è stata chiaramente dimostrata. Le prove di legame agli ER sono concepite per individuare le sostanze chimiche potenzialmente capaci di interferire con i meccanismi d’azione dell’ormone estrogeno, e sono state ampiamente utilizzate nel corso degli ultimi due decenni per caratterizzare la distribuzione tessutale degli ER e per individuare gli agonisti/antagonisti degli ER. Queste prove si basano sull’interazione ligando-recettore, che costituisce la fase iniziale del percorso di segnalazione degli estrogeni, essenziale per la funzione riproduttiva in tutti i vertebrati.
|
|
4.
|
L’interazione degli estrogeni con gli ER può influenzare la trascrizione dei geni controllati dagli estrogeni e indurre effetti non genomici, che possono portare all’induzione o all’inibizione di processi cellulari, compresi quelli necessari alla proliferazione cellulare, allo sviluppo fetale normale e alla funzione riproduttiva (5) (6) (7). La perturbazione dei sistemi estrogenici normali può potenzialmente indurre effetti negativi sul normale sviluppo (ontogenesi), sulla salute riproduttiva e sull’integrità del sistema riproduttivo. Una errata segnalazione degli ER può comportare, ad esempio, un maggiore rischio di cancro ormonedipendente, una ridotta fertilità e alterazioni della crescita e dello sviluppo del feto (8).
|
|
5.
|
Le prove di legame in vitro sono basate sull’interazione diretta tra una sostanza e il sito specifico di legame recettore-ligando che regola la trascrizione genica. La prova di legame al recettore estrogenico alfa ricombinante umano (hrERα) consiste principalmente nel misurare la capacità di un ligando radiomarcato ([3H]-17β-estradiolo) di legarsi agli ER in presenza di concentrazioni crescenti di una sostanza chimica in esame (denominata «competitore»). Le sostanze chimiche in esame che presentano un’alta affinità di legame agli ER competono con il ligando radiomarcato a una concentrazione inferiore rispetto alle sostanze chimiche che hanno una più debole affinità per il recettore. Il presente metodo di prova consta di due componenti principali: una prova di legame a saturazione per caratterizzare i parametri di interazione recettore-ligando e documentare la specificità dei legami agli ER, seguita da una prova di legame competitivo per determinare in che misura una sostanza chimica in esame compete con un ligando radiomarcato per legarsi agli ER.
|
|
6.
|
Gli studi di validazione dei saggi di legame del CERI e di FW hanno dimostrato la loro pertinenza e affidabilità per i fini previsti (2).
|
|
7.
|
Le definizioni e le abbreviazioni utilizzate nel presente metodo di prova figurano nell’appendice 1.
|
Portata e limiti delle prove di legame ai recettori
|
8.
|
I presenti metodi di prova sono proposti a fini di screening e di definizione delle priorità, ma possono fornire anche informazioni sull’evento molecolare scatenante (MIE), utile nel quadro di un approccio basato sulla forza probante dei dati. Essi studiano il legame chimico con il dominio di legame di un ligando all’ERα in un sistema in vitro. Pertanto non è opportuno che i risultati siano direttamente estrapolati e trasposti ai complessi meccanismi di segnalazione e regolazione che caratterizzano un sistema endocrino intatto in vivo.
|
|
9.
|
Il legame del ligando naturale 17β-estradiolo agli ER è la prima fase di una serie di eventi molecolari che attiva la trascrizione di geni bersaglio e, in ultima analisi, determina un cambiamento fisiologico (9). Pertanto il legame al dominio di legame del ligando all’ERα è considerato uno dei meccanismi chiave di perturbazione endocrina mediata dagli ER, benché esistano altri meccanismi capaci di indurre una perturbazione endocrina, tra i quali: i) le interazioni con i siti dell’ERα diversi dai siti di legame del ligando, ii) le interazioni con altri recettori pertinenti per la segnalazione estrogenica, gli ERβ e gli ER accoppiati alla proteina G ed altri recettori e sistemi enzimatici del sistema endocrino; iii) la sintesi ormonale; iv) l’attivazione metabolica e/o l’inattivazione ormonale; v) la distribuzione di ormoni nei tessuti bersaglio; e vi) l’eliminazione degli ormoni dall’organismo. Nessuna delle prove oggetto del presente metodo di prova tratta i descritti meccanismi di azione.
|
|
10.
|
Il presente metodo di prova studia la capacità delle sostanze di legarsi all’ERα umano e non distingue tra agonisti o antagonisti dell’ERα. Il metodo di prova non riguarda nemmeno gli eventi che avvengono più a valle, quali la trascrizione genica o i cambiamenti fisiologici. Considerando che durante la validazione sono state utilizzate solo singole sostanze monocostituente, non esistono risultati utili ai fini dell’applicabilità alle miscele. Cionondimeno, il metodo di prova è teoricamente applicabile alle prove sulle sostanze multicostituente e sulle miscele. Prima di applicare il presente metodo di prova a una miscela per generare dati ai fini regolamentari previsti, si deve considerare se, e in caso affermativo, perché, esso possa fornire risultati adeguati a tale scopo. Tali considerazioni non sono necessarie in presenza di un obbligo normativo di prova sulla miscela.
|
|
11.
|
I sistemi recettoriali cell-free non hanno alcuna capacità metabolica intrinseca e non sono stati validati in combinazione con sistemi enzimatici metabolici. Tuttavia, potrebbe essere possibile incorporare l’attività metabolica nella progettazione dello studio, a condizione che siano effettuati ulteriori lavori di validazione.
|
|
12.
|
Le sostanze chimiche capaci di indurre la denaturazione della proteina (ossia la proteina che costituisce il recettore), come i tensioattivi o le sostanze chimiche che possono modificare il pH della soluzione tampone, non possono essere sottoposte alla prova oppure possono essere testate solo alle concentrazioni alle quali non si verificano tali interazioni. Negli altri casi, l’intervallo delle concentrazioni di prova di una sostanza chimica è limitato dalla sua solubilità nella soluzione tampone della prova.
|
|
13.
|
A fini di informazione, la tabella 1 fornisce i risultati delle prove relative alle 24 sostanze testate con i due metodi di prova pienamente validati descritti nella presente linea guida. Di queste sostanze, 17 sono classificate come ligandi degli ER e 6 come non ligandi sulla base di relazioni pubblicate, segnatamente le prove in vitro per l’attivazione trascrizionale e/o la prova uterotrofica (9) (10) (11) (13) (14) (15). Con riferimento ai dati sintetizzati nella tabella 1, i due metodi di prova sono giunti ad una conclusione quasi identica circa la classificazione di tutte le sostanze fino a 10-4 M, e ciascuna sostanza è stata correttamente classificata come ligando o non ligando degli ER. Ulteriori informazioni su questo gruppo di sostanze nonché sulle sostanze aggiuntive testate nelle prove di legame agli ER durante gli studi di validazione sono fornite negli standard di prestazione per la prova di legame agli hrER (3) nell’appendice 2 (tabelle 1, 2 e 3).
Tabella 1
Classificazione delle sostanze come ligandi o non ligandi degli ER in esito alle prove di legame agli hrER in base ai metodi FW e CERI, e confronto con la risposta prevista
|
|
Nome della sostanza
|
Numero CAS
|
Risposta prevista
|
Saggio di FW
|
Saggio del CERI
|
Classe chimica nel sistema MeSH
|
Classe di prodotto
|
|
|
Intervallo di concentrazione (M)
|
Classificazione
|
Intervallo di concentrazione (M)
|
Classificazione
|
|
1
|
17β-estradiolo
|
50-28-2
|
Ligando
|
1×10-11 – 1×10-6
|
Ligando
|
1×10-11 – 1×10-6
|
Ligando
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
2
|
Noretinodrel
|
68-23-5
|
Ligando
|
3×10-9 – 30×10-4
|
Ligando
|
3×10-9 – 30×10-4
|
Ligando
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
3
|
Noretindrone
|
68-22-4
|
Ligando
|
3×10-9 – 30×10-4
|
Ligando
|
3×10-9 – 30×10-4
|
Ligando
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
4
|
Di-n-butilftalato
|
84-74-2
|
Non ligando (*5)
|
1×10-10 – 1×10-4
|
Non-ligando (*6)
(1)
|
1×10-10 – 1×10-4
|
Non-ligando (*6)
(1)
|
Idrocarburo (ciclico), estere
|
Plasticizzante, intermediario chimico
|
|
5
|
DES
|
56-53-1
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (ciclico), fenolo
|
Prodotto farmaceutico e veterinario
|
|
6
|
17α-etinilestradiolo
|
57-63-6
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
7
|
Meso-esestrolo
|
84-16-2
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (ciclico), fenolo
|
Prodotto farmaceutico e veterinario
|
|
8
|
Genisteina
|
446-72-0
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (eterociclico), flavonoide
|
Prodotto naturale
|
|
9
|
Equol
|
531-95-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Fitoestrogeni Metabolite
|
Prodotto naturale
|
|
10
|
Parabene (n-butil-4-idrossibenzoato) di butile
|
94-26-8
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Parabene
|
Conservante
|
|
11
|
Nonilfenolo (miscela)
|
84852-15-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Alchilfenolo
|
Composto intermedio
|
|
12
|
o,p’-DDT
|
789-02-6
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Organoclorurato
|
Insetticida
|
|
13
|
Corticosterone
|
50-22-6
|
Non ligando (*5)
|
1×10-10 – 1×10-4
|
Non ligando
|
1×10-10 – 1×10-4
|
Non-ligando
|
Steroide
|
Prodotto naturale
|
|
14
|
Zearalenone
|
17924-92-4
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (eterociclico), lattone
|
Prodotto naturale
|
|
15
|
Tamoxifen
|
10540-29-1
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico e veterinario
|
|
16
|
5α-diidrotestosterone
|
521-18-6
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Steroide, non fenolico
|
Prodotto naturale
|
|
17
|
Bisfenolo A
|
80-05-7
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Fenolo
|
Intermediario chimico
|
|
18
|
4-n-eptilfenolo
|
1987-50-4
|
Ligando
|
1×10-10 – 1×10-3
|
Ambiguo (5)
|
1×10-10 – 1×10-3
|
Ligando
|
Alchilfenolo
|
Intermediario
|
|
19
|
Kepone (Clordecone)
|
143-50-0
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Idrocarburo (alogenato)
|
Pesticida
|
|
20
|
Benzo(a)antracene
|
56-55-3
|
Non-ligando
|
1×10-10 – 1×10-3
|
Non-ligando (6)
|
1×10-10 – 1×10-3
|
Non-ligando (6)
|
Idrocarburo aromatico
|
Intermediario
|
|
21
|
Enterolattone
|
78473-71-9
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
1×10-10 – 1×10-3
|
Ligando
|
Fitoestrogeno
|
Prodotto naturale
|
|
22
|
Progesterone
|
57-83-0
|
Non ligando (*5)
|
1×10-10 – 1×10-4
|
Non-ligando
|
1×10-10 – 1×10-4
|
Non-ligando
|
Steroide
|
Prodotto naturale
|
|
23
|
Ottiltrietossisilano
|
2943-75-1
|
Non ligando
|
1×10-10 – 1×10-3
|
Non-ligando
|
1×10-10 – 1×10-3
|
Non-ligando
|
Silano
|
Modificatore di superficie
|
|
24
|
Atrazina
|
1912-24-9
|
Non ligando (*5)
|
1×10-10 – 1×10-4
|
Non-ligando
|
1×10-10 – 1×10-4
|
Non-ligando
|
Composto eterociclico
|
Erbicida
|
|
COMPONENTI DEL METODO DI PROVA DI LEGAME ALL’hrER
Componenti essenziali del metodo di prova
|
14.
|
Il presente metodo di prova si applica alle prove che utilizzano un recettore estrogenico e un ligando di tale recettore che presenti un’affinità sufficientemente forte. Tale ligando può fungere da marcatore/tracciante della prova ed essere spiazzato dalla sostanza chimica in esame man mano che le concentrazioni aumentano. Le prove di legame comprendono i seguenti due componenti principali: 1) legame a saturazione; e 2) legame competitivo. La prova di legame a saturazione è utilizzata per confermare la specificità e l’attività delle preparazioni di recettori, mentre per valutare la capacità della sostanza chimica in esame di legarsi all’hrER si ricorre ad una prova di legame competitivo.
|
Controlli
|
15.
|
Occorre spiegare in dettaglio il motivo della scelta della sostanza estrogenica di riferimento e delle sostanze di controllo concomitanti. I controlli concomitanti - che consistono, come opportuno, in un controllo con solvente (mezzo disperdente), un controllo positivo (ligando degli ER; affinità forte e debole), un controllo negativo (non ligando) - servono ad attestare il corretto funzionamento del metodo di prova nelle condizioni sperimentali e forniscono elementi di confronto tra esperimenti; essi rientrano generalmente tra i criteri di accettabilità di un determinato esperimento (1). Per ciascuna batteria di prove, una medesima piastra deve essere utilizzata per stabilire le curve di tutte le concentrazioni della sostanza estrogenica di riferimento e dei controlli (ossia, ligando debole e non ligando). Tutte le altre piastre devono contenere: 1) una concentrazione elevata (che spiazza quasi completamente il ligando radiomarcato) e una concentrazione media (corrispondente circa all’IC50) di E2 e del ligando debole in triplicato; 2) controlli con solvente e ligandi non specifici, ciascuno in triplicato.
|
Procedure standard di controllo della qualità
|
16.
|
Le procedure standard di controllo della qualità vanno applicate come descritto per ciascuna prova al fine di garantire che i recettori siano attivi, le concentrazioni chimiche siano corrette, l’intervallo di tolleranza rimanga stabile nel corso delle multiple replicazioni della prova e che la prova mantenga la capacità di fornire nel tempo le previste risposte relative ai legami agli ER.
|
Dimostrazione della competenza del laboratorio
|
17.
|
Prima di testare sostanze chimiche sconosciute secondo il presente metodo di prova, ciascun laboratorio deve dimostrare la propria competenza nell’uso della prova effettuando prove di legame a saturazione per confermare la specificità e l’attività della preparazione dei recettori, nonché prove di legame competitivo con la sostanza estrogenica di riferimento e i controlli (ligando debole e non ligando). Ciascun laboratorio dovrebbe costituire una banca dati storica contenente i risultati ottenuti per la sostanza estrogenica di riferimento e i controlli in esito a 3-5 esperimenti indipendenti condotti in giorni diversi. Tali esperimenti costituiranno la base per la sostanza estrogenica di riferimento e i controlli storici del laboratorio e saranno utilizzati come valutazione parziale dell’accettabilità della prova per le batterie di prove future.
|
|
18.
|
La reattività del sistema di prova sarà confermata anche testando le sostanze da utilizzare per stabilire la competenza del laboratorio elencate nella tabella 2. L’elenco delle sostanze da utilizzare per stabilire la competenza del laboratorio è un sottoinsieme delle sostanze di riferimento indicate negli standard di prestazione per le prove di legame agli ER (3). Tali sostanze sono disponibili sul mercato, rappresentano le classi di sostanze chimiche comunemente associate all’attività di legame agli ER, presentano un idoneo intervallo dell’attività biologica (potency) attesa per i ligandi agli ER (da forte a debole) oppure l’assenza di legame (ossia, negativi). Per ciascuna sostanza da utilizzare ai fini della competenza del laboratorio, le concentrazioni testate devono coprire gli intervalli di valori indicati nella tabella 2. Per ogni sostanza occorre eseguire almeno tre esperimenti e i risultati devono essere conformi alla prevista attività chimica. Ciascun esperimento va condotto in modo indipendente (ossia con nuove diluizioni del recettore, delle sostanze chimiche e del reagente), con tre repliche per ciascuna concentrazione. La competenza è dimostrata quando ciascuna sostanza testata ai fini della competenza è classificata correttamente (risposta positiva/negativa). La prova ai fini della competenza è effettuata da ciascun tecnico nell’ambito della formazione al presente metodo di prova.
Tabella 2
Elenco di controlli e di sostanze testate ai fini della competenza per i metodi di prova di legame competitivo all’hrER
(90)
|
N.
|
Nome della sostanza
|
Numero CAS (91)
|
Risposta prevista (92)
(93)
|
Intervallo delle concentrazioni di prova (M)
|
Classe chimica nel sistema MeSH (94)
|
Classe di prodotto (95)
|
|
Controlli (sostanza estrogenica di riferimento, ligando debole, non ligando)
|
|
1
|
17-εστραδιολο
|
50-28-2
|
Ligando
|
1×10-11 – 1×10-6
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
2
|
Noretinodrel (o) Noretindrone
|
68-23-5 (o) 68-22-4
|
Ligando
|
3×10-9 – 30×10-6
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
3
|
Ottiltrietossisilano
|
2943-75-1
|
Non ligando
|
1×10-10 – 1×10-3
|
Silano
|
Modificatore di superficie
|
|
Sostanze di prova ai fini di competenza (95)
|
|
4
|
Dietilstilbestrolo
|
56-53-1
|
Ligando
|
1×10-11 – 1×10-6
|
Idrocarburo (ciclico), fenolo
|
Prodotto farmaceutico e veterinario
|
|
5
|
17α-etinilestradiolo
|
57-63-6
|
Ligando
|
1×10-11 – 1×10-6
|
Steroide
|
Prodotto farmaceutico e veterinario
|
|
6
|
meso-esestrolo
|
84-16-2
|
Ligando
|
1×10-11 – 1×10-6
|
Idrocarburo (ciclico), fenolo
|
Prodotto farmaceutico e veterinario
|
|
7
|
Tamoxifen
|
10540-29-1
|
Ligando
|
1×10-11 – 1×10-6
|
Idrocarburo (ciclico)
|
Prodotto farmaceutico e veterinario
|
|
8
|
Genisteina
|
446-72-0
|
Ligando
|
1×10-10 – 1×10-3
|
Composto eterociclico, flavonoide
|
Prodotto naturale
|
|
9
|
Bisfenolo A
|
80-05-7
|
Ligando
|
1×10-10 – 1×10-3
|
Fenolo
|
Intermediario chimico
|
|
10
|
Zearalonone
|
17924-92-4
|
Ligando
|
1×10-11 – 1×10-3
|
Composto eterociclico, lattone
|
Prodotto naturale
|
|
11
|
Butilparabene
|
94-26-8
|
Ligando
|
1×10-11 – 1×10-3
|
Acido carbossilico, fenolo
|
Conservante
|
|
12
|
Atrazina
|
1912-24-9
|
Non ligando
|
1×10-11 – 1×10-6
|
Composto eterociclico
|
Erbicida
|
|
13
|
Di-n-butilftalato (DBP) (96)
|
84-74-2
|
Non ligando (97)
|
1×10-10 – 1×10-4
|
Idrocarburo (ciclico), estere
|
Plasticizzante, intermediario chimico
|
|
14
|
Corticosterone
|
50-22-6
|
Non ligando
|
1×10-11 – 1×10-4
|
Steroide
|
Prodotto naturale
|
|
Determinazione della solubilità e dell’intervallo delle concentrazioni delle sostanze chimiche in esame
|
19.
|
Occorre effettuare una prova preliminare per determinare il limite di solubilità di ciascuna sostanza chimica in esame e per identificare l’intervallo delle concentrazioni appropriato da utilizzare durante l’esecuzione della prova. Il limite di solubilità di ciascuna sostanza chimica in esame deve essere inizialmente determinato nel solvente e ulteriormente confermato in condizioni sperimentali. La concentrazione finale testata nella prova non deve superare 1 mM. La prova per determinare l’intervallo delle concentrazioni consiste in un controllo con solvente più una serie logaritmica di otto diluizioni, partendo dalla concentrazione massima accettabile (ad es. 1 mM o meno, secondo il limite di solubilità), prendendo nota dell’eventuale presenza di torbidità o precipitato. Le concentrazioni nella seconda e nella terza prova devono essere opportunamente approssimate in modo da caratterizzare meglio la curva concentrazione-risposta.
|
Criteri di accettabilità della batteria di prove
|
20.
|
L’accettazione o il rigetto di una batteria di prove si basa sulla valutazione dei risultati ottenuti per la sostanza estrogenica di riferimento e i controlli utilizzati per ciascun esperimento. Innanzitutto, per la piastra 1, le curve concentrazione-risposta complete ottenute per i controlli di riferimento di ciascun esperimento devono corrispondere alle misure della prestazione basate sui parametri di approssimazione delle curve (ad es. IC50 e pendenza di Hill) che derivano dai risultati dei saggi del CERI e di FW, rispettivamente (appendice 2 e 3), e dai dati storici relativi al controllo di cui dispone il laboratorio che ha condotto la prova. Per ogni esperimento devono essere correttamente classificati tutti i controlli (sostanza estrogenica di riferimento, ligando e non ligando). In secondo luogo, i controlli di tutte le piastre successive devono essere valutati per verificarne la coerenza con la piastra 1. È opportuno usare un intervallo di concentrazioni della sostanza chimica in esame sufficiente per definire chiaramente il vertice della curva di legame competitivo. La variabilità fra le repliche a ciascuna concentrazione della sostanza chimica in esame, così come tra le tre batterie di test indipendenti deve essere ragionevole e giustificabile sotto il profilo scientifico. Un laboratorio dimostra la propria capacità di ripetere un metodo di prova in modo omogeneo sviluppando e mantenendo una banca dati dei risultati storici ottenuti per la sostanza estrogenica di riferimento e per i controlli. Le deviazioni standard (SD) o i coefficienti di variazione (CV) delle medie dei parametri di approssimazione delle curve della sostanza estrogenica di riferimento e dei controlli di ligando debole ottenuti dopo multipli esperimenti possono essere utilizzati come misura di riproducibilità intralaboratorio. Si deve ricorrere al giudizio professionale di un esperto per analizzare i risultati dei controlli della piastra per ciascuna batteria di prove e per ciascuna sostanza chimica in esame.
Inoltre, devono essere rispettati i seguenti principi relativi ai criteri di accettabilità:
|
—
|
i dati devono essere sufficienti per una valutazione quantitativa del legame agli ER;
|
|
—
|
le concentrazioni testate devono rimanere nell’ambito dell’intervallo di solubilità della sostanza chimica in esame.
|
|
Analisi di dati
|
21.
|
La procedura di analisi definita per i risultati delle prove di legame a saturazione e di legame competitivo deve essere conforme ai principi fondamentali di caratterizzazione delle interazioni recettore-ligando. I dati relativi al legame a saturazione sono generalmente analizzati applicando un modello di regressione non lineare che tiene conto del legame totale e non specifico. Può essere necessario eseguire una correzione per la perdita di ligando (ad es. Swillens, 1995 (19)) per determinare la capacità massima di legame Bmax e la costante di legame Kd. I dati delle prove di legame competitivo sono generalmente trasformati (ad es. percentuale di legame specifico, logaritmo della concentrazione della sostanza chimica in esame). Le stime del log (IC50) di ciascuna sostanza chimica in esame sono determinate utilizzando un idoneo software di approssimazione delle curve con regressione non lineare basato su un’equazione di Hill a quattro parametri. Dopo un’analisi iniziale, si determinano i parametri di approssimazione della curva e quindi si verifica visivamente se i dati relativi al legame corrispondono alla curva di legame competitivo ottenuta. In alcuni casi possono essere necessarie ulteriori analisi per ottenere un’approssimazione ottimale della curva (ad esempio, restringendo il vertice e/o la base della curva o applicando la regola del 10 % (cfr. l’appendice 4 e riferimento 2, sezione III.A.2).
|
|
22.
|
Il rispetto dei criteri di accettabilità (paragrafo 20) indica che il sistema di prova funziona correttamente, ma non garantisce che qualsiasi prova produrrà dati accurati. La replicazione dei risultati corretti della prima batteria di prove è la migliore garanzia che i dati generati sono accurati.
|
Criteri generali di interpretazione dei dati
|
23.
|
Attualmente non esiste alcun metodo universalmente concordato per l’interpretazione dei dati di una prova di legame agli ER. Tuttavia, la valutazione qualitativa (ad es. ligando/non ligando) e/o quantitativa (ad es. log IC50, affinità di legame relativa (RBA), ecc.) dell’attività mediata dagli hrER deve fondarsi su basi empirici e giudizi scientificamente validi.
|
Relazione sull’esecuzione della prova
|
24.
|
La relazione sull’esecuzione della prova deve comprendere le informazioni seguenti.
|
|
Metodo di prova:
|
—
|
metodo di prova utilizzato;
|
|
|
|
Controllo/riferimento/sostanza chimica in esame
|
—
|
origine, numero di lotto, data limite d’uso, se disponibili;
|
|
—
|
stabilità della sostanza chimica in esame, se nota.
|
|
—
|
solubilità e stabilità della sostanza chimica in esame nel solvente, se note;
|
|
—
|
misurazione del pH, dell’osmolalità e del precipitato nel terreno di coltura al quale è stata aggiunta la sostanza chimica in esame, se del caso.
|
|
|
|
Sostanza monocostituente:
|
—
|
aspetto fisico, idrosolubilità e ulteriori proprietà fisico-chimiche pertinenti;
|
|
—
|
dati di identificazione chimica: denominazioni IUPAC o CAS, numero CAS, codice SMILES o InChI, formula strutturale, identità chimica o impurezze, se del caso e se le condizioni pratiche lo consentono, ecc.
|
|
|
|
Sostanza multicostituente, UVCB e miscele:
|
—
|
caratterizzate, per quanto possibile mediante l’identità chimica (cfr. sopra), la presenza quantitativa e le proprietà fisico-chimiche pertinenti dei costituenti.
|
|
|
|
Solvente/mezzo disperdente:
|
—
|
Caratterizzazione (natura, fornitore e lotto);
|
|
—
|
motivazione della scelta del solvente/mezzo disperdente;
|
|
—
|
solubilità e stabilità della sostanza in esame nel solvente/mezzo disperdente, se note.
|
|
|
|
Recettori:
|
—
|
origine dei recettori (fornitore, n. di riferimento, lotto, tipo di recettore, concentrazione dei recettori attivi indicata dal fornitore, certificazione del fornitore);
|
|
—
|
caratterizzazione dei recettori (compresi i risultati delle prove di legame a saturazione): Kd, Bmax,
|
|
—
|
stoccaggio dei recettori.
|
|
—
|
fornitore, numero di riferimento, lotto, attività specifica.
|
|
|
|
Condizioni sperimentali:
|
—
|
limiti di solubilità in condizioni sperimentali;
|
|
—
|
composizione della soluzione tamponata della prova di legame;
|
|
—
|
concentrazione di recettore;
|
|
—
|
concentrazione di tracciante (ossia, ligando radiomarcato);
|
|
—
|
Concentrazione di sostanza chimica in esame;
|
|
—
|
percentuale di mezzo disperdente nella prova finale;
|
|
—
|
temperatura e tempo di incubazione;
|
|
—
|
metodo di separazione dei ligandi legati/liberi;
|
|
—
|
controlli/sostanze di riferimento positivi e negativi;
|
|
—
|
criteri in base ai quali i risultati sono considerati positivi, negativi o equivoci.
|
|
|
|
Verifica dell’accettabilità:
|
—
|
valori reali di IC50 e pendenza di Hill per controlli positivi e le sostanze di riferimento inclusi nella prova di legame competitivo.
|
|
|
|
Risultati:
|
—
|
dati grezzi, dati relativi ai ligandi legati/liberi;
|
|
—
|
verifica della denaturazione, se del caso;
|
|
—
|
se esiste, la più bassa concentrazione efficace (LEC);
|
|
—
|
valori RBA e/o IC50, come opportuno;
|
|
—
|
relazione concentrazione-risposta, se possibile;
|
|
—
|
eventuali analisi statistiche, corredate della misurazione dell’errore e della confidenza (ad es. SEM, SD, CV o IC 95 %) e di una descrizione di come sono stati ottenuti tali valori.
|
|
|
|
Discussione dei risultati:
|
—
|
applicazione della regola del 10 %.
|
|
Conclusione
|
BIBLIOGRAFIA
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Organisation for Economic Cooperation and Development, Paris.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Organisation for Economic Cooperation and Development, Paris.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In Vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In Vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In Vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In Vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In Vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Organisation for Economic Cooperation and Development, Paris.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
Appendice 1
Definizioni e Abbreviazioni
Regola del 10 %
: opzione che permette di escludere dei punti di dati dalle analisi quando la percentuale di legame specifico media del [3H]-17β-estradiolo per tutte le repliche di una concentrazione supera del 10 % o più la media corrispondente a una concentrazione inferiore (cfr. l'appendice 4).
Criteri di accettabilità
: requisiti minimi di prestazione relativi ai controlli sperimentali e agli standard di riferimento. Tutti i criteri di accettabilità devono essere soddisfatti affinché un metodo di prova sia considerato valido.
Accuratezza (concordanza)
: grado di concordanza tra i risultati ottenuti con il metodo di prova e i valori di riferimento accettati. Misura l'efficienza del metodo di prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza" per indicare la proporzione dei risultati corretti di un metodo di prova (1).
CF
: quadro concettuale dell'OCSE per la sperimentazione e la valutazione degli interferenti endocrini.
Sostanza chimica
: una sostanza o una miscela.
CV
: coefficiente di variazione.
E2
: 17β-estradiolo
ED: (Endocrine disruption)
: interferenza con il sistema endocrino
hERα
: recettore estrogenico alfa umano
ER
: recettore estrogenico
Attività estrogenica
: capacità di una sostanza chimica di mimare la capacità del 17β-estradiolo di legarsi ai recettori di estrogeni. Il legame con l'hERα può essere individuato con il presente metodo di prova.
IC50
: concentrazione efficace di una sostanza chimica in esame che induce la metà dell'effetto massimo di inibizione.
ICCVAM
: Interagency Coordinating Committee on the Validation of Alternative Methods (comitato di coordinamento interagenzie per la validazione di metodi alternativi).
Riproducibilità inter-laboratori
: espressione della misura in cui laboratori qualificati diversi possano ottenere, seguendo lo stesso protocollo e testando le stesse sostanze di prova, risultati simili in termini qualitativi e quantitativi. La riproducibilità inter-laboratori è determinata nel corso dei processi di pre-validazione e validazione e indica la misura in cui un metodo di prova può essere trasferito con successo tra laboratori; è detta anche riproducibilità tra laboratori (1).
Riproducibilità intra-laboratorio
: espressione della misura in cui membri diversi del personale qualificato all'interno del medesimo laboratorio riescono a ottenere risultati simili da una prova effettuata seguendo un protocollo specifico in momenti diversi; è detta anche "riproducibilità all'interno dello stesso laboratorio" (1).
LEC
: la concentrazione minima della sostanza chimica in esame che produce una risposta (ossia la più bassa concentrazione della sostanza chimica in esame alla quale l'incremento dell'effetto indotto è statisticamente diverso da quello del concomitante controllo con mezzo disperdente).
Test strutturalmente analoghi
: espressione che indica un metodo di prova strutturalmente e funzionalmente analogo a un metodo di prova di riferimento validato e accettato. È sinonimo di "metodo di prova simile".
PBTG
: (Performance-Based Test Guideline) Linea guida basata su standard di prestazione
Standard di prestazione
: standard, basati su un metodo di riferimento validato, che consentono di valutare la comparabilità di un metodo proposto che è simile sotto il profilo strutturale e funzionale. Comprendono: 1) gli elementi essenziali del metodo di prova; 2) un elenco minimo di sostanze chimiche di riferimento scelte tra le sostanze utilizzate per dimostrare le prestazioni accettabili del metodo di prova validato; e 3) in funzione dei risultati ottenuti con il metodo di prova validato, i livelli comparabili di accuratezza e affidabilità che il metodo proposto dovrebbe ottenere quando viene valutato utilizzando l'elenco minimo di sostanze chimiche di riferimento (1).
Sostanze di prova ai fini di competenza
: sottoinsieme delle sostanze di riferimento incluse negli standard di prestazione che possono essere utilizzate dai laboratori per dimostrare la competenza tecnica ad effettuare un metodo di prova standardizzato. In generale, i criteri di selezione per tali sostanze includono la rappresentatività di tutta la gamma delle risposte, la loro disponibilità sul mercato e l'esistenza di dati di riferimento di elevata qualità a loro corredo.
Competenza
: la capacità di eseguire correttamente un metodo di prova, dimostrata prima di testare sostanze sconosciute.
Sostanza estrogenica di riferimento
: 17ß-estradiolo (E2, CAS 50-28-2).
Metodi di prova di riferimento
: i saggi su cui si basa la linea guida PBTG 493.
RBA
: Relative Binding Affinity. Affinità di legame relativa. La RBA di una sostanza è espressa in percentuale del log(IC50) della sostanza in rapporto al log(IC50) del 17β-estradiolo.
Pertinenza
: descrizione del rapporto tra la prova e l'effetto studiato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (1).
Affidabilità
: misura in cui l'esecuzione di un metodo di prova può essere riprodotta nel tempo all'interno dello stesso laboratorio o da laboratori diversi seguendo il medesimo protocollo. È valutata calcolando la riproducibilità intra-laboratorio e inter-laboratori.
SD
: deviazione standard
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Metodo di prova validato
: saggio per il quale sono stati completati studi di validazione volti a determinare la pertinenza (compresa l'accuratezza) e l'affidabilità per uno scopo specifico. Va sottolineato che un metodo di prova validato potrebbe non avere una prestazione sufficiente in termini di accuratezza e affidabilità per essere ritenuto accettabile per lo scopo determinato (1).
Validazione
: il processo mediante il quale si stabilisce l'affidabilità e la pertinenza di uno specifico approccio, metodo, saggio, processo o di una specifica valutazione per un determinato scopo (1).
Appendice 2
Saggio di Freyberg-Wilson (fw) Relativo Alle Prove in Vitro di Legame a Saturazione e di Legame Competitivo ai Recettori Estrogenici (erα) che Utilizza l'Intera Lunghezza di un erα Ricombinante
CONSIDERAZIONI INIZIALI E LIMITI (CFR. ANCHE INTRODUZIONE GENERALE)
|
1.
|
Il presente metodo di prova in vitro di legame a saturazione e di legame competitivo ai recettori estrogenici (ERα) utilizza l'intera lunghezza del recettore estrogenico alfa umano ERαα(hrERα), prodotto e isolato a partire da cellule di insetti infettati da baculovirus. Il protocollo, sviluppato da Freyberger e Wilson, è stato sottoposto a uno studio internazionale di validazione realizzato da diversi laboratori (2) che ha dimostrato la pertinenza e l'affidabilità di questo metodo di prova per gli scopi previsti.
|
|
2.
|
Il presente metodo di prova costituisce una procedura di screening per individuare le sostanze che possono legarsi all'intera lunghezza dell'hrERα; è utilizzato per determinare la capacità di una sostanza chimica in esame di competere con il 17β-estradiolo nel legarsi all'hrERα. I risultati sotto il profilo quantitativo possono comprendere il valore IC50 (la concentrazione della sostanza chimica in esame necessaria per spiazzare la metà del [3H]-17β-estradiolo legato all'hrERα) e le affinità di legame relative delle sostanze chimiche in esame per l'hrERα rispetto al 17β-estradiolo. Ai fini dello screening chimico, i risultati accettabili sotto il profilo qualitativo possono comprendere la classificazione delle sostanze chimiche in esame come ligandi o non ligandi dell'hrERα, oppure generanti una risposta equivoca, in funzione dei criteri descritti per le curve di legame.
|
|
3.
|
Atteso che il metodo di prova utilizza un ligando radioattivo, il laboratorio deve richiedere una licenza per trattare materiali radioattivi. Tutte le procedure che implicano radioisotopi e sostanze chimiche pericolose devono essere conformi ai regolamenti e alle procedure stabiliti dalla legislazione nazionale.
|
|
4.
|
Le sezioni «INTRODUZIONE GENERALE» e «COMPONENTI DEL METODO DI PROVA DI LEGAME ALL'hrER» vanno lette prima di applicare il presente metodo di prova per fini regolamentari. Le definizioni e le abbreviazioni utilizzate nella presente linea guida figurano nell'appendice 1.
|
PRINCIPI DEL METODO DI PROVA (CFR. ANCHE INTRODUZIONE GENERALE)
|
5.
|
La prova di legame al recettore hrERα misura la capacità di un ligando radiomarcato ([3H]-17β-estradiolo) di legarsi agli ER in presenza di concentrazioni crescenti di una sostanza chimica in esame (denominata «competitore»). Le sostanze chimiche in esame che presentano un'alta affinità di legame agli ER competono con il ligando radiomarcato a una concentrazione inferiore rispetto alle sostanze chimiche che hanno una più debole affinità per il recettore.
|
|
6.
|
Il presente metodo di prova consta di due componenti principali: un esperimento di legame a saturazione per caratterizzare i parametri di interazione recettore-ligando, seguito da un esperimento di legame competitivo per determinare in che misura una sostanza chimica in esame compete con un ligando radiomarcato per legarsi agli ER.
|
|
7.
|
Scopo dell'esperimento di legame a saturazione è caratterizzare il numero e l'affinità di legame dei recettori di un particolare lotto in vista dell'esperimento di legame competitivo. L'esperimento di legame a saturazione misura, in condizioni di equilibrio, l'affinità di una concentrazione fissa di recettori degli estrogeni rispetto al loro ligando naturale (rappresentata dalla costante di dissociazione, Kd) e la concentrazione di siti recettori attivi (Bmax).
|
|
8.
|
L'esperimento di legame competitivo misura l'affinità di una sostanza a legarsi agli ER in competizione con il [3H]-17β-estradiolo. L'affinità è quantificata dalla concentrazione della sostanza in esame che, in condizioni di equilibrio, inibisce il 50 % del legame specifico del [3H]-17β-estradiolo (definita «concentrazione che induce il 50 % di inibizione» o «IC50»). Essa si può esprimere anche come l'affinità di legame relativa (RBA, calcolata in rapporto all'IC50 di estradiolo misurata separatamente nell'ambito della stessa prova). L'esperimento di legame competitivo misura il legame del [3H]-17β-estradiolo a una concentrazione fissa in presenza di un ampio intervallo (otto ordini di grandezza) di concentrazioni della sostanza chimica in esame. I dati sono quindi approssimati, ove possibile, a una forma dell'equazione di Hill (Hill, 1910) che descrive lo spiazzamento del ligando radiomarcato da parte di un ligando competitore su un sito unico. L'ampiezza dello spiazzamento dell'estradiolo radiomarcato, in condizioni di equilibrio, è utilizzata per caratterizzare la sostanza chimica in esame come ligando, non ligando o generante una risposta equivoca.
|
PROCEDURA
Dimostrazione dell'accettabilità della prestazione della proteina hrERα
|
9.
|
Prima di effettuare le prove di routine relative al legame competitivo e al legame a saturazione, si deve verificare che ciascun lotto di hrERα stia funzionando correttamente nel laboratorio in cui sarà utilizzato. Un processo in due fasi è applicato per dimostrare tale prestazione. Le due fasi in questione consistono in:
|
—
|
una prova di legame a saturazione [3H]-17β-estradiolo per dimostrare la specificità e la saturazione dell'hrERα. Un'analisi di regressione non lineare dei dati ottenuti (ad es. BioSoft; McPherson, 1985; Motulsky, 1995) e il grafico di Scatchard così ottenuto documenteranno l'affinità di legame all'hrERα del [3H]-17β-estradiolo (Kd) e il numero di recettori attivi (Bmax) per ciascun lotto di hrERα.
|
|
—
|
una prova di legame competitivo utilizzando le sostanze di controllo (sostanza estrogenica di riferimento 17β-estradiolo), un ligando debole (ad esempio, noretinodrel o noretindrone) e un non ligando (ottiltrietossisilano, OTES). Ciascun laboratorio è tenuto a creare una banca dati storica per documentare che l'IC50 e gli altri valori pertinenti per la sostanza estrogenica di riferimento e un ligando debole rimangono coerenti tra una prova e l'altra e tra i diversi lotti di hrERα. I parametri delle curve di legame competitivo per le sostanze di controllo devono rientrare nei limiti dell'intervallo di confidenza del 95 % (cfr. la tabella 1) sviluppati utilizzando i dati dei laboratori che hanno partecipato allo studio di validazione della prova (2).
|
|
Tabella 1
Criteri di prestazione sviluppati per la sostanza estrogenica di riferimento e il ligando debole nella prova di legame all'hrER di FW
|
Sostanza
|
Parametro
|
Media (7)
|
Deviazione standard (n)
|
Intervallo di confidenza al 95 % (8)
|
|
Limite inferiore
|
Limite superiore
|
|
17β-estradiolo
|
Vertice (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Base (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Pendenza di Hill
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrel
|
Vertice (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Base (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Pendenza di Hill
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindrone (9)
|
Vertice (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Base (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Pendenza di Hill
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50 (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
Dimostrazione delle competenze del laboratorio
|
10.
|
Cfr. i paragrafi 17 e 18 e la Tabella 2 nella sezione «ELEMENTI DEL METODO DI PROVA DI LEGAME ALL'hrER» del presente metodo di prova. Ciascuna prova (di legame a saturazione e di legame competitivo) deve consistere in tre batterie indipendenti di prove (ossia con nuove diluizioni di recettore, sostanze chimiche e reagente) condotte in giorni differenti, e ciascuna batteria di prove deve comportare tre repliche.
|
Determinazione della concentrazione di recettore (hrERα)
|
11.
|
La concentrazione di recettori attivi varia leggermente in funzione del lotto e delle condizioni di conservazione. Per questo motivo, occorre determinare la concentrazione di recettori attivi del lotto ricevuto dal fornitore. Questa fase permette di ottenere la concentrazione esatta di recettori attivi al momento della prova.
|
|
12.
|
Alle stesse condizioni della prova di legame competitivo (ossia, [3H]-estradiolo a 1 nM), le concentrazioni nominali di 0,25, 0,5 0,75 e 1 nM di recettore sono incubate in assenza (legame totale) e in presenza (legame non specifico) di estradiolo non marcato a 1 μM. Il legame specifico, calcolato come differenza tra il legame totale e il legame non specifico, è rappresentato graficamente in funzione della concentrazione nominale di recettore. La concentrazione di recettore alla quale il legame specifico corrisponde al 20 % del ligando radiomarcato aggiunto permette di dedurre la concentrazione nominale del recettore; quest'ultima è utilizzata per esperimenti di legame a saturazione e di legame competitivo. Frequentemente, tale condizione è soddisfatta da una concentrazione finale di hrER di 0,5 nM.
|
|
13.
|
Se il criterio del 20 % fallisce ripetutamente, occorre verificare l'impianto sperimentale per individuare eventuali errori. Il mancato raggiungimento del criterio del 20 % può indicare che il lotto di recettori ricombinanti contiene pochissimi siti attivi; occorre quindi considerare il ricorso a un altro lotto di recettori.
|
Prova a saturazione
|
14.
|
Sono valutate otto concentrazioni crescenti di [3H]-17β-estradiolo in triplicato, rispettando le tre condizioni seguenti (cfr. tabella 2):
|
—
|
In assenza di 17β-estradiolo non marcato e in presenza di ER. Questa condizione permette di determinare il legame totale misurando la radioattività nei pozzetti che contengono soltanto [3H]-17β-estradiolo.
|
|
—
|
In presenza di una concentrazione di 17β-estradiolo non marcato 1 000 volte superiore a quella di 17β-estradiolo radiomarcato e in presenza di ER. Questa condizione è intesa a saturare i siti di legame attivi con 17β-estradiolo non marcato e a determinare il legame non specifico misurando la radioattività presente nei pozzetti. Si considera che l'estradiolo caldo (radiomarcato) eventualmente rimanente capace di legarsi al recettore si leghi a un sito non specifico, in quanto l'estradiolo freddo (non marcato) deve essere in concentrazione talmente elevata da legarsi a tutti i siti specifici disponibili sul recettore.
|
|
—
|
In assenza di 17β-estradiolo non marcato e in assenza di ER (determinazione della radioattività totale).
|
|
Preparazione di soluzioni di [3H]-17β-estradiolo e di 17β-estradiolo non marcato
|
15.
|
Le diluizioni di [3H]-17β-estradiolo sono preparate aggiungendo il tampone di prova a 12 nM di soluzione madre di [3H]-17β-estradiolo per ottenere concentrazioni che inizialmente vanno da 0,12 nM a 12 nM. Aggiungendo 40 μl di tali diluizioni ai rispettivi pozzetti di prova di una piastra da microtitolazione a 96 pozzetti (per un volume finale di 160 μl), si otterranno le concentrazioni finali della prova, che vanno da 0,03 a 3,0 nM. La preparazione del tampone di prova, della soluzione madre di [3H]-17β-estradiolo e delle diluizioni e la determinazione delle concentrazioni sono descritte in dettaglio nel protocollo di FW (2).
|
|
16.
|
Le diluizioni delle soluzioni etanoliche di 17β-estradiolo sono preparate aggiungendo il tampone di prova in modo da ottenere otto concentrazioni crescenti che inizialmente vanno da 0,06 μM a 6 μM. Aggiungendo 80 μl di tali soluzioni ai rispettivi pozzetti di prova di una piastra da microtitolazione a 96 pozzetti (in un volume finale di 160 μl), si otterranno le concentrazioni finali, che vanno da 0,03 μM a 3 μM. La concentrazione finale di 17β-estradiolo non marcato in ciascun pozzetto di prova di legame non specifico deve essere 1 000 volte superiore alla concentrazione del [3H]-17β- estradiolo radiomarcato. La preparazione di diluizioni di 17β-estradiolo non marcato è descritta in dettaglio nel protocollo di FW (2).
|
|
17.
|
Per la prova, va utilizzata la concentrazione nominale del recettore che produce un legame specifico di 20 ± 5 % (cfr. i paragrafi 12 e 13). La soluzione di hrERα deve essere preparata immediatamente prima dell'uso.
|
|
18.
|
Le piastre da microtitolazione a 96 pozzetti sono preparate come illustrato nella tabella 2, con 3 repliche per concentrazione. L'appendice 2.2 contiene un esempio di piastra indicante la concentrazione e la distribuzione dei volumi di [3H]-17β-estradiolo, di 17β-estradiolo non marcato, della soluzione tamponata e del recettore.
|
Tabella 2
Configurazione della piastra da microtitolazione nella prova di legame a saturazione
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Legame totale (solvente)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E
2
+ ER
+ 0,10 μM E
2
|
Legame non specifico
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E
2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2: [3H]-17β-estradiolo
ER: recettore degli estrogeni
E2: 17β-estradiolo non marcato
|
|
19.
|
Le piastre da microtitolazione per la prova sono incubate tra 2 e 8 oC per 16-20 ore e sottoposte a rotazione durante il periodo di incubazione.
|
Misurazione del [3H]-17β-estradiolo legato all'hrERα
|
20.
|
Per separare il [3H]-17β-estradiolo legato all'hrERα dal [3H]-17β-estradiolo libero, si aggiungono 80 μl di sospensione fredda di DCC in ciascun pozzetto, quindi le piastre da microtitolazione sono agitate per 10 minuti e infine centrifugate a circa 2 500 giri al minuto. Per ridurre al minimo la dissociazione del [3H]-17β-estradiolo dall'hrERα durante questo processo, è estremamente importante che le soluzioni tamponate e i pozzetti di prova siano mantenuti tra 2 e 8 oC e che ciascuna fase sia eseguita rapidamente. Un agitatore per le piastre da microtitolazione è necessario per un trattamento efficace e rapido delle piastre.
|
|
21.
|
Prelevare quindi 50 μl del supernatante contenente il [3H]-17β-estradiolo legato all'hrERα con estrema cura, in modo da evitare qualsiasi contaminazione dei pozzetti per contatto con il DCC, e collocarli in una seconda piastra da microtitolazione.
|
|
22.
|
Aggiungere 200 μl di liquido di scintillazione, in grado di convertire l'energia cinetica delle emissioni nucleari in energia luminosa, in ciascun pozzetto (da A1 a B12 e da D1 a E12). I pozzetti G1-H12 (identificati come disintegrazioni al minuto - dpm - totali) rappresentano le diluizioni successive del [3H]-17β-estradiolo (40 μl) da versare direttamente nel liquido di scintillazione nei pozzetti della piastra di misurazione, come indicato nella tabella 3; questi pozzetti contengono quindi solo 200 μl di liquido di scintillazione e la diluizione appropriata di [3H]-17β-estradiolo. Queste misurazioni indicano la quantità di [3H]-17β-estradiolo (espressa in dpm) aggiunta a ciascuna serie di pozzetti per il legame totale e il legame non specifico.
|
Tabella 3
Configurazione della piastra da microtitolazione nella prova di legame a saturazione, misurazione della radioattività
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3H] E2 + ER
|
0,06 nM [3H] E2 + ER
|
0,08 nM [3H] E2 + ER
|
0,10 nM [3 H] E2 + ER
|
Legame totale (solvente)
|
|
B
|
0,30 nM [3H] E2 + ER
|
0,60 nM [3H] E2 + ER
|
1,0 nM [3H] E2 + ER
|
3,0 nM [3H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3H] E2 + ER + 0,03 μM E2
|
0,06 nM [3H] E2 + ER + 0,06 μM E2
|
0,08 nM [3H] E2 + ER + 0,08 μM E2
|
0,10 nM [3H] E
2
+ ER
+ 0,10 μM E
2
|
Legame non specifico
|
|
E
|
0,30 nM [3H] E2 + ER + 0,30 μM E2
|
0,60 nM [3H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E
2
|
3,0 nM [3H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [3H] E
2 (totale in dpm)
|
0,06 nM [3H] E
2
|
0,08 nM [3H] E
2
|
0,10 nM [3H] E
2
|
(totale in dpm) (*7)
|
|
H
|
0,30 nM [3H] E2
|
0,60 nM [3H] E2
|
1,0 nM [3H] E2
|
3,0 nM [3H] E2
|
|
[3H] E2: [3H]-17β-estradiolo
ER: recettore degli estrogeni
E2: 17β-estradiolo non marcato
dpm: disintegrazioni al minuto.
|
|
23.
|
La misurazione della scintillazione inizia almeno due ore dopo l'aggiunta, e dura 40 minuti per pozzetto. Per la determinazione delle disintegrazioni al minuto si usa un contatore a scintillazione per piastre da microtitolazione, applicando una correzione dell'attenuazione. In alternativa, se non si dispone di un contatore a scintillazione, i campioni possono essere misurati mediante un contatore convenzionale. In tali condizioni può essere presa in considerazione una riduzione del tempo di misurazione.
|
Prova di legame competitivo
|
24.
|
La prova di legame competitivo misura i legami di [3H]-17β-estradiolo in concentrazione fissa in presenza di concentrazioni crescenti della sostanza chimica in esame. Per ciascuna concentrazione si dovranno utilizzare tre repliche concomitanti nella stessa batteria di prove. Inoltre, per ogni sostanza chimica sottoposta a prova devono essere effettuate tre batterie di prove non concomitanti. La prova richiede una o più piastre da microtitolazione a 96 pozzetti.
|
Controlli
|
25.
|
Durante l'esecuzione della prova vanno inclusi in ciascun esperimento il solvente e i controlli concomitanti (ossia, la sostanza estrogenica di riferimento, ligando debole e non ligandi). Per ciascuna batteria di prove, una medesima piastra deve essere utilizzata per stabilire le curve di tutte le concentrazioni della sostanza estrogenica di riferimento e dei controlli (ossia, ligando debole e non ligando). Tutte le altre piastre devono contenere: i) una concentrazione elevata (massimo spiazzamento) e media (corrispondente circa alla IC50) di E2 e un ligando debole in triplicato; ii) il controllo con solvente e ligandi non specifici, ciascuno almeno in triplicato. Le procedure per la preparazione della soluzione tamponata, dei controlli, del [3H]-17β-estradiolo, dell'hrERα e delle soluzioni della sostanza chimica in esame sono descritte nel riferimento 2 (allegato K, cfr. protocollo di FW).
|
Controllo con solvente:
|
26.
|
Il controllo trattato solo con il solvente conferma che il solvente non interagisce con il sistema di prova e permette di misurare il legame totale. Di preferenza si utilizzerà l'etanolo come solvente. In alternativa, se la concentrazione massima della sostanza chimica in esame non è solubile in etanolo, si può usare il DMSO. Alla fine della prova, la concentrazione di etanolo finale o DMSO, se utilizzato, sarà pari all'1,5 % nei pozzetti di prova e non dovrà superare il 2 %.
|
Controllo con soluzione tamponata:
|
27.
|
Il controllo con soluzione tamponata contiene tutti gli altri elementi della prova, tranne il solvente e la sostanza chimica in esame. I risultati del controllo con soluzione tamponata sono confrontati con il controllo con solvente per verificare che il solvente utilizzato non interferisca con il sistema di prova.
|
Ligando forte (sostanza estrogenica di riferimento)
|
28.
|
Il 17β-estradiolo (CAS 50-28-2) è il ligando endogeno che presenta una forte affinità di legame all'ER di tipo alfa. Si rappresenti una curva standard utilizzando il 17β-estradiolo non marcato per ciascuna prova di legame competitivo all'hrERα, al fine di valutare la variabilità delle prove condotte nel corso del tempo nello stesso laboratorio. Otto soluzioni di 17β-estradiolo non marcato sono preparate nell'etanolo, con concentrazioni nei pozzetti di prova che variano da 100 nM a 10 pM (-7[logM] a -11[logM]), distanziate come segue: (-7[logM], -8[logM], -8,5[logM], -9[logM], - 9,5[logM], -10[logM], -11[logM]). La concentrazione più elevata di 17β-estradiolo non marcato (1 μM) serve anche da indicatore di legame non specifico. Tale concentrazione è distinta dall'indicazione «NSB» nella tabella 4, sebbene faccia parte anche della curva standard.
|
Ligando debole
|
29.
|
Per dimostrare la sensibilità di ciascun esperimento e consentire una valutazione della variabilità delle prove condotte nel corso del tempo, si deve includere un ligando debole (noretinodrel (CAS68-23-5) o noretindrone (CAS 68-22-4). Otto soluzioni di ligando debole sono preparate nell'etanolo, con concentrazioni nei pozzetti di prova che variano da 3 nM a 30 pM (-8,5[logM] a -4,5[logM]), distanziate come segue: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM],-7,5[logM], -8,5[logM].
|
Non ligando
|
30.
|
Si utilizzi l'ottiltrietossisilano (OTES, CAS 2 943-75-1) come controllo negativo (non ligando). Esso garantisce che la prova, così come è condotta, permetterà di individuare le sostanze chimiche in esame che non si legano all'hrERα. Otto soluzioni di non ligando debole sono preparate nell'etanolo, con concentrazioni nei pozzetti di prova che variano da 0,1 nM a 1 000 μM (-10[logM] a -3[logM]), distanziate con incrementi logaritmici. Lo ftalato di di-n-butile (DBP) può essere utilizzato come controllo non ligando alternativo. È stato dimostrato che la sua solubilità massima è -4[logM].
|
Concentrazione di hrERα
|
31.
|
Occorre utilizzare la quantità di recettore che produce un legame specifico di 20 ± 5 % del ligando radiomarcato a 1 nM (cfr. i paragrafi 12 e 13 dell'allegato 2). La soluzione di hrERα deve essere preparata immediatamente prima dell'uso.
|
[3H]-17β-estradiolo
|
32.
|
La concentrazione di [3H]-17β-estradiolo nei pozzetti di prova deve essere di 1,0 nM.
|
Sostanze chimiche in esame
|
33.
|
Occorre effettuare una prova preliminare per determinare il limite di solubilità di ciascuna sostanza chimica in esame e per identificare l'intervallo delle concentrazioni appropriato da utilizzare durante l'esecuzione del protocollo sperimentale. Il limite di solubilità di ciascuna sostanza chimica in esame deve essere inizialmente determinato nel solvente e ulteriormente confermato in condizioni sperimentali. La concentrazione finale della prova non dovrebbe superare 1 mM. La prova per determinare l'intervallo delle concentrazioni consiste in un controllo con solvente più una serie logaritmica di 8 diluizioni della concentrazione massima accettabile (ad es. 1 mM o meno, secondo il limite di solubilità). Si noterà l'apparizione di torbidità o precipitato (cfr. anche paragrafo 35). La sostanza chimica testata è analizzata mediante le curve ottenute per la serie logaritmica di otto concentrazioni stabilite dalla prova di determinazione dell'intervallo (range-finding test) condotta precedentemente. Le concentrazioni nella seconda e nella terza prova devono essere opportunamente approssimate in modo da caratterizzare meglio la curva concentrazione-risposta.
|
|
34.
|
Le diluizioni della sostanza chimica in esame vanno preparate nel solvente appropriato (cfr. il paragrafo 26 dell'appendice 2). Se la concentrazione più elevata della sostanza chimica in esame non è solubile in etanolo o in DMSO e se aggiungendo più solvente la concentrazione del solvente nei pozzetti a fine prova risulta superiore al limite accettabile, la concentrazione più elevata può essere ridotta alla concentrazione immediatamente inferiore. In tal caso, è possibile aggiungere una concentrazione supplementare all'inizio della serie di concentrazioni. Le altre concentrazioni della serie rimangono invariate.
|
|
35.
|
I pozzetti di prova sono monitorati attentamente durante l'aggiunta delle soluzioni della sostanza chimica in esame, in quanto tale aggiunta può provocare un precipitato. I dati relativi a tutti i pozzetti che contengono precipitato vanno esclusi dall'approssimazione della curva e la ragione di esclusione dei dati va riportata.
|
|
36.
|
Se esistono informazioni precedenti provenienti da altre fonti che forniscono dati sul log(IC50) di una sostanza chimica in esame, può essere opportuno ripartire geometricamente le diluzioni (ossia, 0,5 unità logaritmiche attorno al log(IC50) atteso. Il risultato finale deve corrispondere a un intervallo di concentrazioni sufficientemente ripartite ai due lati del log(IC50), compresi il vertice (top) e la base (bottom) della curva di legame, al fine di caratterizzarla in modo adeguato.
|
Configurazione della piastra di prova
|
37.
|
Le piastre da microtitolazione comprendono serie di sei repliche incubate, distinte con codici corrispondenti al controllo con solvente, alla concentrazione più elevata di sostanza estrogenica di riferimento che serve anche come indicatore di legame non specifico (NSB), e al controllo della soluzione tamponata, nonché le incubazioni in triplicato codificate per ciascuna delle otto concentrazioni del controllo non ligando (ottiltrietossisilano), le sette concentrazioni inferiori della sostanza estrogenica di riferimento, le otto concentrazioni del ligando debole e le otto concentrazioni di ciascuna sostanza chimica in esame (TC). Un esempio di configurazione della piastra di prova per ottenere curve complete a partire da tutte le concentrazioni della sostanza estrogenica di riferimento e i controlli è riportato nella tabella 4. Piastre da microtitolazione aggiuntive sono utilizzate per le sostanze chimiche in esame e includono: 1) una concentrazione elevata (massimo spiazzamento) e una concentrazione media (circa IC50) di E2 e del ligando debole in triplicato; 2) controlli con solvente e ligandi non specifici, ciascuno in sei repliche (tabella 5). Nell'appendice 2.3 è riportato un esempio di configurazione della piastra da microtitolazione per la prova di legame competitivo di tre sostanze sconosciute. Le concentrazioni indicate nelle tabelle 4 e 5 sono le concentrazioni finali della prova. La concentrazione massima di E2 deve essere pari a 1 × 10-7 M mentre, per il ligando debole, è utilizzata la concentrazione più elevata della piastra 1. La concentrazione IC50 è determinata dal laboratorio a partire della sua banca dei dati storici. Si prevede che questo valore sarà analogo a quello osservato negli studi di validazione (cfr. tabella 1).
|
Tabella 4
Configurazione della piastra da microtitolazione per la prova di legame competitivo, curve concentrazione-risposta complete per la sostanza estrogenica di riferimento e i controlli (piastra 1)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (Solo solvente)
|
TB (Solo solvente)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Bianco (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Bianco (E2 caldo) (*9)
|
Bianco (E2 caldo) (*9)
|
Controllo con tampone
|
Controllo con tampone
|
|
In questo esempio, il ligando debole è noretinodrel (NE)
|
Tabella 5
Configurazione della piastra da microtitolazione per la prova di legame competitivo, curve concentrazione-risposta complete per le sostanze chimiche in esame e i controlli della piastra
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (Solo solvente)
|
TB (Solo solvente)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50)
|
NE (1×10-4,5)
|
E2 (IC50)
|
E2 (1×10-7)
|
|
In questo esempio, il ligando debole è noretinodrel (NE)
|
Completamento della prova di legame competitivo
|
38.
|
Come indicato nella tabella 6, ai pozzetti vanno aggiunti 80 μl di controllo con solvente, di controllo con soluzione tamponata, di sostanza estrogenica di riferimento, di ligando debole, di non ligando, e di sostanza chimica in esame preparata in soluzione tamponata. Aggiungere a ciascun pozzetto 40 μl di soluzione di [3H]-17β-estradiolo a 4 nM. Dopo leggera rotazione per 10-15 minuti tra 2o e 8 oC, aggiungere 40 μl di soluzione di hrERα in ciascun pozzetto. Le piastre da microtitolazione per la prova sono poste su un rotatore e incubate a 2-8 oC per 16-20 ore.
|
Tabella 6
Volume dei costituenti della prova di legame competitivo all'hrER, piastre da microtitolazione
|
Volume (μl)
|
Costituente
|
|
80
|
17β-estradiolo non marcato, noretinodrel, OTES, sostanze chimiche in esame, solvente o soluzione tamponata
|
|
40
|
Soluzione di [3H]-17β-estradiolo a 4 nM
|
|
40
|
Soluzione di hrERα, concentrazione determinata dalla prova preliminare
|
|
160
|
Volume totale in ciascun pozzetto di prova
|
|
39.
|
Il [3H]-17β-estradiolo legato all'hrERα è separato dal [3H]-17β-estradiolo libero aggiungendo 80 μl di sospensione fredda di DCC in ogni pozzetto, e quindi quantificato seguendo il metodo descritto nei paragrafi da 20 a 23 per la prova di legame a saturazione.
|
|
40.
|
I pozzetti H1-H6 (indicati come «bianco (E2 caldo)» nella tabella 4) forniscono le disintegrazioni al minuto del [3H]-estradiolo in 40 μl. Le aliquote di 40 μl sono versate direttamente nel liquido di scintillazione nei pozzetti da H1 a H6.
|
Criteri di accettabilità
Prova di legame a saturazione
|
41.
|
La curva di legame specifica deve raggiungere un plateau man mano che si utilizzano concentrazioni crescenti di [3H]-17β-estradiolo, il che indica la saturazione degli hrERα con il ligando.
|
|
42.
|
Il ligando specifico di [3H]-17β-estradiolo a 1 nM deve corrispondere ad una radioattività pari a 15-25 % della media della radioattività totale misurata in tutte le batteria di prove. Sono ammissibili lievi escursioni al di fuori di tale intervallo, ma se ciò avviene costantemente o se una particolare batteria di prove dà risultati significativamente al di fuori di tale intervallo, occorre adattare la concentrazione delle proteine e ripetere la prova di legame a saturazione.
|
|
43.
|
I dati devono generare un grafico di Scatchard lineare.
|
|
44.
|
Il legame non specifico non deve essere eccessivo: il valore è generalmente inferiore al 35 % del legame totale. Tuttavia, il rapporto può occasionalmente eccedere tale limite quando si misurano disintegrazioni per minuto molto deboli per la concentrazione testata più bassa di 17β-estradiolo radiomarcato.
|
Prova di legame competitivo
|
45.
|
Crescenti concentrazioni di 17β-estradiolo dovrebbero spiazzare il [3H]-17β-estradiolo dal recettore secondo un modello di legame competitivo in un unico sito.
|
|
46.
|
Il valore IC50 per la sostanza estrogenica di riferimento (17β-estradiolo) deve essere approssimativamente uguale alla concentrazione molare di [3H]-17β-estradiolo più la Kd stabilita per la prova di legame a saturazione.
|
|
47.
|
Il legame specifico totale deve situarsi costantemente nell'intervallo di 20 ± 5 % quando la concentrazione media della radioattività totale aggiunta a ciascun pozzetto è di 1 nM in tutte le batterie di prove. Sono ammissibili lievi escursioni al di fuori di tale intervallo, ma se ciò avviene costantemente o se una particolare batteria di prove dà risultati significativamente al di fuori di tale intervallo, occorre adattare la concentrazione delle proteine.
|
|
48.
|
Il solvente non deve alterare la sensibilità o la riproducibilità della prova. I risultati del controllo con solvente (pozzetti TB) sono confrontati con il controllo con soluzione tamponata per verificare che il solvente utilizzato non interagisca con il sistema di prova. Se il solvente non influisce sulla prova, i risultati del TB e del controllo con soluzione tamponata devono essere comparabili.
|
|
49.
|
Il non ligando non dovrebbe spiazzare più del 25 % di [3H]-17β-estradiolo dall'hrERα durante la prova a concentrazioni che vanno fino a 10-3 M (OTES) o 10-4 M (DBP).
|
|
50.
|
Sono stati definiti criteri di prestazione per la sostanza estrogenica di riferimento e per i due ligandi deboli (ad esempio, noretinodrel, noretindrone) utilizzando i dati dello studio di validazione del saggio di legame all'hrER di FW (allegato N del riferimento 2). Vengono forniti intervalli di confidenza al 95 % per la media (n) ± SD per tutte le batterie di controllo effettuate dai laboratori che partecipano allo studio di validazione. Gli intervalli di confidenza al 95 % sono stati calcolati per i parametri di approssimazione della curva (ossia, il vertice e la base della curva, la pendenza di Hill, logIC50) per la sostanza estrogenica di riferimento e i ligandi deboli e per il log10RBA dei ligandi deboli rispetto alla sostanza estrogenica di riferimento; tali intervalli sono forniti come criteri di prestazione per i controlli positivi. La tabella 1 fornisce gli intervalli attesi per i parametri di approssimazione della curva che possono essere utilizzati come criteri di prestazione. In pratica, l'intervallo di IC50 può variare leggermente in funzione della Kd della preparazione dei recettori e della concentrazione del ligando.
|
|
51.
|
Non sono stati sviluppati criteri di prestazione per i parametri di approssimazione delle curve corrispondenti alle sostanze chimiche in esame poiché esiste una grande diversità di sostanze chimiche che possono essere potenzialmente oggetto di prova così come una grande variazione nelle affinità e risultati potenziali corrispondenti (ad es. approssimazione completa, parziale o nulla delle curve). Tuttavia, è necessario ricorrere al giudizio professionale di un esperto per analizzare i risultati di ciascuna batteria di prove su una sostanza chimica in esame. Occorre usare un intervallo di concentrazioni della sostanza chimica in esame sufficiente per definire chiaramente il vertice della curva di legame competitivo (ad es. 90-100 % di legame). La variabilità fra le repliche di ciascuna concentrazione della sostanza chimica in esame, nonché tra le tre batterie di prove non concomitanti deve essere ragionevole e giustificabile sotto il profilo scientifico. I controlli di ciascuna batteria di prove sulla sostanza in esame devono avvicinarsi alle misure di prestazione riportate per questo saggio di FW ed essere coerenti con i dati storici ottenuti per i controlli da ciascun laboratorio.
|
ANALISI DEI DATI
Prova di legame a saturazione
|
52.
|
La prova misura il legame totale e il legame non specifico. Tali valori permettono di calcolare il legame specifico indotto da concentrazioni crescenti di [3H]-17β-estradiolo in condizioni di equilibrio, sottraendo il legame non specifico dal legame totale. La curva che rappresenta il legame specifico in funzione della concentrazione di [3H]-17β-estradiolo deve raggiungere un plateau per il legame specifico massimo, che indica la saturazione degli hrERα con il [3H]-17β-estradiolo. Inoltre, l'analisi dei dati documenta il legame del [3H]-17β-estradiolo a un unico sito di legame ad alta affinità. La curva di legame a saturazione deve indicare il legame non specifico, il legame totale e il legame specifico. Una regressione non lineare consente di approfondire l'analisi dei dati (ad es. BioSoft; McPherson, 1985; Motulsky, 1995), e i risultati finali sono presentati sotto forma di grafico di Scatchard.
|
|
53.
|
L'analisi dei dati deve determinare la Bmax e la Kd sulla base dei soli dati di legame totale, nell'ipotesi che il legame non specifico sia lineare, a meno che venga fornita una giustificazione dell'utilizzo di un altro metodo. Inoltre, si determina la migliore approssimazione delle curve mediante una solida analisi di regressione; in caso contrario se ne deve fornire la motivazione. Va indicato il metodo scelto per questa solida analisi di regressione. Una correzione relativa alla perdita di ligando (ad es. con il metodo di Swillens, 1995) va sempre applicata per determinare la Bmax e la Kd a partire dai dati di legame a saturazione.
|
Prova di legame competitivo
|
54.
|
Tracciare la curva di legame competitivo come legame specifico di [3H]-17β-estradiolo in funzione della concentrazione (in unità log10) del competitore. La concentrazione della sostanza chimica in esame che inibisce il 50 % del legame massimo specifico di [3H]-17β-estradiolo corrisponde al valore IC50.
|
|
55.
|
Le stime dei valori di log(IC50) per i controlli positivi (ad es. sostanza estrogenica di riferimento e ligando debole) sono determinate utilizzando un software di approssimazione delle curve con regressione non lineare appropriata che si basa su un'equazione di Hill a quattro parametri (ad es. BioSoft; McPherson, 1985; Motulsky, 1995). Tali approssimazioni sono effettuate senza imporre restringimenti al vertice e alla base della curva, alla pendenza e al log(IC50). Si determina la migliore approssimazione delle curve mediante una solida analisi di regressione; in caso contrario se ne deve fornire la motivazione. Non va applicata la correzione relativa alla perdita di ligandi. In seguito all'analisi iniziale, ciascuna curva di legame deve essere rivista per garantire la migliore approssimazione al modello. L'affinità di legame relativa (RBA) di un ligando debole è espressa in percentuale corrispondente al rapporto tra log(IC50) del ligando debole e il log(IC50) del 17β-estradiolo. I risultati dei controlli positivi e del controllo non ligando devono essere valutati in base alle misurazioni della prestazione della prova indicate nei paragrafi 45-50 della presente appendice 2.
|
|
56.
|
I dati relativi a tutte le sostanze chimiche in esame devono essere analizzati mediante un approccio graduale per garantire un'adeguata classificazione dei dati e un'adeguata classificazione di ciascuna curva di legame competitivo. Si raccomanda che ciascuna batteria di prove per una sostanza chimica in esame sia inizialmente sottoposta a un'analisi normalizzata dei dati, identica a quella utilizzata per la sostanza estrogenica di riferimento e i controlli con ligando debole (cfr. il paragrafo 55). Una volta completata l'analisi, si procede a un esame tecnico dei parametri di approssimazione della curva e a un esame visivo per determinare in che modo i dati corrispondono alla curva di legame competitivo ottenuta per ciascuna batteria di prove. Tale esame tecnico si basa su tre osservazioni che indicano che la prova e le analisi sono state condotte correttamente: una diminuzione della percentuale di [3H]-17β-estradiolo collegata ai siti specifici in funzione della concentrazione; una variabilità debole tra le repliche tecniche di ciascuna concentrazione della sostanza chimica in esame; la coerenza dei parametri di approssimazione tra le tre batterie di prove.
|
Interpretazione dei dati
|
57.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata un ligando dell'hrERα se la curva di legame può essere approssimata e se il punto più basso della curva di risposta ottenuta per l'intervallo dei dati corrisponde a un legame inferiore al 50 % (Grafico 1).
|
|
58.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata non ligando dell'hrERα se:
|
—
|
la sua curva di legame può essere approssimata e se il punto più basso sulla curva di risposta approssimata ottenuta per l'intervallo di dati corrisponde a un legame superiore al 75 %, oppure
|
|
—
|
la sua curva di legame non può essere approssimata e se la media delle percentuali di legame non livellate di tutti i gruppi di concentrazione è superiore al 75 %.
|
|
|
59.
|
Le sostanze chimiche in esame sono considerate equivoche se non è soddisfatta nessuna delle condizioni di cui sopra (ad esempio il punto più basso della curva di risposta approssimata si situa tra 76 e 51 %).
|
Tabella 7
Criteri di classificazione di una sostanza chimica in esame basata sulla sua curva di legame competitivo
|
Classificazione
|
Criteri
|
|
Ligandoa
|
È possibile approssimare la curva di legame.
Il punto più basso della curva di risposta ottenuto per l'intervallo di dati corrisponde a un legame inferiore a 50 %.
|
|
Non-ligandob
|
Se è possibile approssimare la curva di legame,
il punto più basso della curva di risposta approssimata ottenuto per l'intervallo di dati corrisponde a un legame superiore a 75 %.
Se non è possibile approssimare la curva di legame,
sla media delle percentuali di legame non livellate di tutti i gruppi di concentrazione della prova è superiore a 75 %.
|
|
Equivococ
|
Una prova valutabile che non può esser classificata come ligando né non ligando.
(ad esempio il punto più basso della curva di risposta approssimata si situa tra 76 e 51 %).
|
Grafico 1
Esempi di classificazione della sostanza chimica in esame in base ad una curva di legame competitivo.
|
60.
|
Le multiple prove condotte all'interno di un laboratorio su una sostanza chimica in esame sono combinate attribuendo valori numerici a ciascuna batteria di prove e calcolando la media di tali valori, come indicato nella tabella 8. I risultati combinati delle batterie di prove di ciascun laboratorio sono confrontati con la classificazione prevista per ciascuna sostanza chimica in esame.
|
Tabella 8
Metodo di classificazione della sostanza chimica in esame mediante multiple batterie di prove condotte all'interno di un laboratorio
|
Attribuzione di un valore a ciascuna batteria di prove:
|
|
|
Classificazione
|
Valore numerico
|
|
Ligando
|
2
|
|
Equivoco
|
1
|
|
Non ligando
|
0
|
|
Classificazione sulla base della media dei valori numerici di tutte le batterie di prove:
|
|
|
Classificazione
|
Valore numerico
|
|
Ligando
|
Media ≥ 1,5
|
|
Equivoco
|
0,5 ≤ media < 1,5
|
|
Non ligando
|
Media < 0,5
|
RELAZIONE SULL'ESECUZIONE DELLA PROVA
|
61.
|
Cfr. il paragrafo 24 nella sezione «ELEMENTI DEL METODO DI PROVA DI LEGAME ALL'hrER» del presente metodo di prova. 3
|
Appendice 2.1
ELENCO DEI TERMINI
[3H]E2
: 17β-estradiolo radiomarcato con trizio
DCC
: Dextran-coated charcoal (carbone rivestito di destrano)
E2
: 17β-estradiolo non marcato (inerte)
Soluzione di prova tamponata
: soluzione composta da 10 mM di tris, 10 mg/ml di albumina serica bovina, 2 mM di DTT, 10 % di glicerolo, 0,2 mM di leupeptina, con pH 7,5
hrERα
: recettore estrogenico alfa ricombinante umano
Replica
: Uno dei multipli pozzetti che contengono lo stesso contenuto alle stesse concentrazioni e che sono testati contemporaneamente in un'unica batteria di prove. Nel presente protocollo, ogni concentrazione della sostanza chimica in esame è testata in triplicato; ciò significa che tre repliche di ciascuna concentrazione della sostanza chimica in esame sono testate simultaneamente.
Batteria di prove
: serie completa di prove simultanee nei pozzetti di una piastra da microtitolazione che fornisce tutte le informazioni necessarie per caratterizzare l'affinità di legame all'hrERα di una sostanza chimica in esame (ossia, quantità totale di [3H]-17β-estradiolo aggiunta in un pozzetto, il legame massimo all'hrERα di [3H]-17β-estradiolo, il legame non specifico e il legame totale per diverse concentrazioni della sostanza chimica in esame. Una batteria di prove può consistere anche in un unico pozzetto di prova (replica) per concentrazione, ma poiché il presente protocollo richiede prove in triplicato, una batteria di prove consiste di tre pozzetti di prova per concentrazione. Inoltre, il presente protocollo impone di realizzare tre batterie di prove indipendenti (non simultanee) per sostanza chimica.
Appendice 2.2
SAGGIO TIPICO DI SATURAZIONE CON [3H]-17Β-ESTRADIOLO IN TRIPLICATO
|
Saggio tipico di saturazione con [3H]-17β-estradiolo in triplicato
|
|
Posizione
|
Replica
|
Codice per tipo di pozzetto
|
Concentrazione iniziale di E2 caldo (nM)
|
Volume di E2 caldo (μl)
|
Concentrazione finale di E2 caldo (nM)
|
Concentrazione iniziale di E2 freddo (μM)
|
Volume di E2 freddo (μl)
|
Concentrazione finale di E2 freddo (μM)
|
Volume di soluzione tamponata (μl)
|
Volume del recettore (μl)
|
Volume totale nei pozzetti
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
caldo
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
caldo
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
caldo
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
caldo
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
caldo
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
caldo
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
caldo
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
caldo
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
caldo
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
caldo
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
caldo
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
caldo
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
caldo
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
caldo
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
caldo
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
caldo
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
caldo
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
caldo
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
caldo
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
caldo
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
caldo
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
caldo
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
caldo
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
caldo
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
N.B. I pozzetti "caldi" sono vuoti durante l'incubazione. I 40 μl che sono aggiunti successivamente servono soltanto per il conteggio per scintillazione.
|
Appendice 2.3
CONFIGURAZIONE DEI POZZETTI PER LA BATTERIA DI PROVE DI LEGAME COMPETITIVO
|
Piastra
|
Posizione
|
Replica
|
Tipo di pozzetto
|
Codice per pozzetto
|
Codice della concentrazione
|
Concentrazione iniziale del competitore (M)
|
Volume di hrER (μl)
|
Volume di soluzione tamponata (μl)
|
Volume di tracciante (E2 caldo) (μl)
|
Volume della piastra di diluizione (μl)
|
Volume finale (μl)
|
Concentrazione finale del competitore (M)
|
|
S
|
A1
|
1
|
legame totale
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A2
|
2
|
legame totale
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A3
|
3
|
legame totale
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A4
|
1
|
legame totale
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A5
|
2
|
legame totale
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A6
|
3
|
legame totale
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
S
|
A7
|
1
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A8
|
2
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A9
|
3
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A10
|
1
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A11
|
2
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
A12
|
3
|
E2 freddo (elevato)
|
NSB
|
S0
|
2.00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
B1
|
1
|
E2 freddo
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B2
|
2
|
E2 freddo
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B3
|
3
|
E2 freddo
|
S
|
S1
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
B4
|
1
|
E2 freddo
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B5
|
2
|
E2 freddo
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B6
|
3
|
E2 freddo
|
S
|
S2
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
B7
|
1
|
E2 freddo
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B8
|
2
|
E2 freddo
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B9
|
3
|
E2 freddo
|
S
|
S3
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
B10
|
1
|
E2 freddo
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B11
|
2
|
E2 freddo
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
B12
|
3
|
E2 freddo
|
S
|
S4
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
C1
|
1
|
E2 freddo
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C2
|
2
|
E2 freddo
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C3
|
3
|
E2 freddo
|
S
|
S5
|
6,00E-10
|
40
|
—
|
40
|
80
|
160
|
3,0E-10
|
|
S
|
C4
|
1
|
E2 freddo
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C5
|
2
|
E2 freddo
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C6
|
3
|
E2 freddo
|
S
|
S6
|
2,00E-10
|
40
|
—
|
40
|
80
|
160
|
1,0E-10
|
|
S
|
C7
|
1
|
E2 freddo
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C8
|
2
|
E2 freddo
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C9
|
3
|
E2 freddo
|
S
|
S7
|
2,00E-11
|
40
|
—
|
40
|
80
|
160
|
1,0E-11
|
|
S
|
C10
|
1
|
bianco
|
bianco
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C11
|
2
|
bianco
|
bianco
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
C12
|
3
|
bianco
|
bianco
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
S
|
D1
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D2
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D3
|
1
|
Noretinodrel
|
NE
|
WP1
|
6,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D5
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D6
|
1
|
Noretinodrel
|
NE
|
WP2
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
1,0E-05
|
|
S
|
D7
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D8
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D9
|
1
|
Noretinodrel
|
NE
|
WP3
|
6,00E-06
|
40
|
—
|
40
|
80
|
160
|
3,0E-06
|
|
S
|
D10
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D11
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
D12
|
1
|
Noretinodrel
|
NE
|
WP4
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
E1
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E2
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E3
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-07
|
40
|
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP
|
2,00E-07
|
40
|
|
40
|
80
|
160
|
1,0E-07
|
|
S
|
E7
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E8
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E9
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-08
|
40
|
—
|
40
|
80
|
160
|
3,0E-08
|
|
S
|
E10
|
1
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E11
|
2
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
E12
|
3
|
Noretinodrel
|
NE
|
WP
|
6,00E-09
|
40
|
—
|
40
|
80
|
160
|
3,0E-09
|
|
S
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00E-03
|
40
|
—
|
40
|
80
|
160
|
1,0E-03
|
|
S
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00E-04
|
40
|
—
|
40
|
80
|
160
|
1,0E-04
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00E-05
|
40
|
—
|
40
|
80
|
160
|
3,0E-05
|
|
S
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
S
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00E-07
|
40
|
—
|
40
|
80
|
160
|
3,0E-07
|
|
S
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00E-08
|
40
|
—
|
40
|
80
|
160
|
1,0E-08
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00E-09
|
40
|
—
|
40
|
80
|
160
|
1,0E-09
|
|
S
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00E-10
|
40
|
—
|
40
|
—
|
160
|
1,0E-10
|
|
S
|
H1
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H2
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H3
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H4
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H5
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H6
|
1
|
caldo
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
S
|
H7
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H8
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H9
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H10
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H11
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
S
|
H12
|
1
|
Ctrl con tampone
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
N.B. I pozzetti "caldi" sono vuoti durante l'incubazione. I 40 μl che sono aggiunti successivamente servono soltanto per il conteggio per scintillazione.
|
Configurazione dei pozzetti per la batteria di prove di legame competitivo
|
|
Piastra
|
Posizione
|
Replica
|
Tipo di pozzetto
|
Codice del pozzetto
|
Codice della concentrazione
|
Concentrazione iniziale del competitore (M)
|
Volume di hrER (μl)
|
Volume di soluzione tamponata (μl)
|
Volume di tracciante (E2 caldo) (μl)
|
Volume della piastra di diluizione μl)
|
Volume finale (μl)
|
Concentrazione finale del competitore (M)
|
|
P1
|
A1
|
1
|
legame totale
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
legame totale
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
legame totale
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
legame totale
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
legame totale
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
legame totale
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A8
|
2
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A9
|
3
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A10
|
1
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A11
|
2
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
A12
|
3
|
E2 freddo (elevato)
|
NSB
|
S0
|
2,00E-06
|
40
|
—
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B1
|
1
|
Sost. in esame 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B2
|
2
|
Sost. in esame 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B3
|
3
|
Sost. in esame 1
|
TC1
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
B4
|
1
|
Sost. in esame 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B5
|
2
|
Sost. in esame 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B6
|
3
|
Sost. in esame 1
|
TC1
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
B7
|
1
|
Sost. in esame 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B8
|
2
|
Sost. in esame 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B9
|
3
|
Sost. in esame 1
|
TC1
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
B10
|
1
|
Sost. in esame 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B11
|
2
|
Sost. in esame 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
B12
|
3
|
Sost. in esame 1
|
TC1
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
C1
|
1
|
Sost. in esame 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C2
|
2
|
Sost. in esame 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C3
|
3
|
Sost. in esame 1
|
TC1
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
C4
|
1
|
Sost. in esame 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C5
|
2
|
Sost. in esame 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C6
|
3
|
Sost. in esame 1
|
TC1
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
C7
|
1
|
Sost. in esame 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C8
|
2
|
Sost. in esame 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C9
|
3
|
Sost. in esame 1
|
TC1
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
C10
|
1
|
Sost. in esame 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C11
|
2
|
Sost. in esame 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
C12
|
3
|
Sost. in esame 1
|
TC1
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
D1
|
1
|
Sost. in esame 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D2
|
2
|
Sost. in esame 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D3
|
3
|
Sost. in esame 2
|
TC2
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
D4
|
1
|
Sost. in esame 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D5
|
2
|
Sost. in esame 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D6
|
3
|
Sost. in esame 2
|
TC2
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
D7
|
1
|
Sost. in esame 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D8
|
2
|
Sost. in esame 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D9
|
3
|
Sost. in esame 2
|
TC2
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
D10
|
1
|
Sost. in esame 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D11
|
2
|
Sost. in esame 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
D12
|
3
|
Sost. in esame 2
|
TC2
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
E1
|
1
|
Sost. in esame 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E2
|
2
|
Sost. in esame 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E3
|
3
|
Sost. in esame 2
|
TC2
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
E4
|
1
|
Sost. in esame 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E5
|
2
|
Sost. in esame 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E6
|
3
|
Sost. in esame 2
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
E7
|
1
|
Sost. in esame 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E8
|
2
|
Sost. in esame 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E9
|
3
|
Sost. in esame 2
|
TC2
|
7
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
E10
|
1
|
Sost. in esame 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E11
|
2
|
Sost. in esame 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
E12
|
3
|
Sost. in esame 2
|
TC2
|
8
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
F1
|
1
|
Sost. in esame 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F2
|
2
|
Sost. in esame 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F3
|
3
|
Sost. in esame 3
|
TC3
|
1
|
2.00E-03
|
40
|
0
|
40
|
80
|
160
|
1.0E-03
|
|
P1
|
F4
|
1
|
Sost. in esame 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F5
|
2
|
Sost. in esame 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F6
|
3
|
Sost. in esame 3
|
TC3
|
2
|
2.00E-04
|
40
|
0
|
40
|
80
|
160
|
1.0E-04
|
|
P1
|
F7
|
1
|
Sost. in esame 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F8
|
2
|
Sost. in esame 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F9
|
3
|
Sost. in esame 3
|
TC3
|
3
|
2,00E-05
|
40
|
0
|
40
|
80
|
160
|
1,0E-05
|
|
P1
|
F10
|
1
|
Sost. in esame 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F11
|
2
|
Sost. in esame 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
F12
|
3
|
Sost. in esame 3
|
TC3
|
4
|
2,00E-06
|
40
|
0
|
40
|
80
|
160
|
1,0E-06
|
|
P1
|
G1
|
1
|
Sost. in esame 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G2
|
2
|
Sost. in esame 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G3
|
3
|
Sost. in esame 3
|
TC3
|
5
|
2,00E-07
|
40
|
0
|
40
|
80
|
160
|
1,0E-07
|
|
P1
|
G4
|
1
|
Sost. in esame 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G5
|
2
|
Sost. in esame 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G6
|
3
|
Sost. in esame 3
|
TC3
|
6
|
2,00E-08
|
40
|
0
|
40
|
80
|
160
|
1,0E-08
|
|
P1
|
G7
|
1
|
Sost. in esame 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G8
|
2
|
Sost. in esame 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G9
|
3
|
Sost. in esame 3
|
TC3
|
7
|
2,00E-09
|
40
|
0
|
40
|
80
|
160
|
1,0E-09
|
|
P1
|
G10
|
1
|
Sost. in esame 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G11
|
2
|
Sost. in esame 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
G12
|
3
|
Sost. in esame 3
|
TC3
|
8
|
2,00E-10
|
40
|
0
|
40
|
80
|
160
|
1,0E-10
|
|
P1
|
H1
|
1
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
Noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
Noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
Noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
Noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
E2 freddo S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
E2 freddo S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
E2 freddo S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
E2 freddo S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
E2 freddo S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
E2 freddo S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
Appendice 3
METODO DI PROVA IN VITRO DI LEGAME AL RECETTORE ESTROGENICO (ER) BASATO SULLA PROTEINA DEL DOMINIO DI LEGAME DEL LIGANDO DELL'ERΑ RICOMBINANTE UMANO SECONDO IL METODO DEL CERI (CHEMICAL EVALUATION AND RESEARCH INSTITUTE)
CONSIDERAZIONI INIZIALI E LIMITI (CFR. ANCHE INTRODUZIONE GENERALE)
|
1.
|
Questo metodo di prova in vitro di legame a saturazione e di legame competitivo al recettore estrogenico (ERα) utilizza il dominio di legame del ligando (LBD) del recettore estrogenico α umano (hrERα). Questo costrutto proteico è stato realizzato dal Chemical Evaluation and Research Institute (CERI, Giappone) ed esiste sotto forma di proteina di fusione glutatione-S-transferasi (GST), espressa in E. coli. Il protocollo sviluppato dal CERI è stato sottoposto a uno studio internazionale di validazione realizzato da diversi laboratori (2) che ha dimostrato la pertinenza e l'affidabilità di questo metodo di prova per gli scopi previsti.
|
|
2.
|
Il presente metodo di prova costituisce una procedura di screening intesa ad individuare le sostanze che possono legarsi all'hrERα. Esso permette di determinare la capacità di una sostanza chimica in esame di competere con il 17β-estradiolo nel formare legami con il dominio di legame del ligando dell'hrERα. I risultati sotto il profilo quantitativo possono comprendere il valore IC50 (la concentrazione della sostanza chimica in esame necessaria per spiazzare la metà del [3H]-17β-estradiolo legato all'hrERα) e le affinità di legame relative delle sostanze chimiche in esame per l'hrERα rispetto al 17β-estradiolo. Ai fini dello screening chimico, i risultati accettabili sotto il profilo qualitativo possono comprendere la classificazione delle sostanze chimiche in esame come ligandi o non ligandi dell'hrERα, oppure generanti una risposta equivoca, in funzione dei criteri descritti per le curve di legame.
|
|
3.
|
Atteso che il metodo di prova utilizza un ligando radioattivo, il laboratorio deve richiedere una licenza per trattare materiali radioattivi. Tutte le procedure che implicano radioisotopi e sostanze chimiche pericolose devono essere conformi ai regolamenti e alle procedure stabiliti dalla legislazione nazionale.
|
|
4.
|
Le sezioni "INTRODUZIONE GENERALE" e "COMPONENTI DEL METODO DI PROVA DI LEGAME ALL'hrER" vanno lette prima di applicare il presente metodo di prova per fini regolamentari. Le definizioni e le abbreviazioni utilizzate nella presente linea guida figurano nell'appendice 1.
|
PRINCIPI DEL METODO DI PROVA (CFR. ANCHE INTRODUZIONE GENERALE)
|
5.
|
La prova di legame (hrERα) consiste principalmente nel misurare la capacità di un ligando radiomarcato ([3H]-17β-estradiolo) a legarsi agli ER in presenza di concentrazioni crescenti di una sostanza chimica in esame (denominata "competitore"). Le sostanze chimiche in esame che presentano un'alta affinità di legame agli ER competono con il ligando radiomarcato a una concentrazione inferiore rispetto alle sostanze chimiche che hanno una più debole affinità per il recettore.
|
|
6.
|
Il presente metodo di prova consta di due componenti principali: un esperimento di legame a saturazione per caratterizzare i parametri di interazione recettore-ligando, seguito da un esperimento di legame competitivo per determinare in che misura una sostanza chimica in esame compete con un ligando radiomarcato per legarsi agli ER.
|
|
7.
|
Scopo dell'esperimento di legame a saturazione è caratterizzare il numero e l'affinità di legame dei recettori di un particolare lotto in vista dell'esperimento di legame competitivo. L'esperimento di legame a saturazione misura, in condizioni di equilibrio, l'affinità di una concentrazione fissa di recettori degli estrogeni rispetto al loro ligando naturale (rappresentata dalla costante di dissociazione, Kd) e la concentrazione di siti recettori attivi (Bmax).
|
|
8.
|
L'esperimento di legame competitivo misura l'affinità di una sostanza a legarsi agli ER in competizione con il [3H]-17β-estradiolo. L'affinità è quantificata dalla concentrazione della sostanza in esame che, in condizioni di equilibrio, inibisce il 50 % del legame specifico del [3H]-17β-estradiolo (definita «concentrazione che induce un'inibizione del 50 %» o «IC50»). Essa si può esprimere anche come l'affinità di legame relativa (RBA, calcolata in rapporto all'IC50 di estradiolo misurata separatamente nell'ambito della stessa batteria di prove). L'esperimento di legame competitivo misura il legame di [3H]-17β-estradiolo a una concentrazione fissa in presenza di un ampio intervallo (otto ordini di grandezza) di concentrazioni della sostanza chimica in esame. I dati sono quindi approssimati, ove possibile, a una forma dell'equazione di Hill (Hill, 1910) che descrive lo spiazzamento del ligando radiomarcato da parte di un ligando competitore su un sito unico. L'ampiezza dello spiazzamento dell'estradiolo radiomarcato, in condizioni di equilibrio, è utilizzata per caratterizzare la sostanza chimica in esame come ligando, non ligando o generante una risposta equivoca.
|
PROCEDURA
Dimostrazione dell'accettabilità della prestazione della proteina hrERα
|
9.
|
Prima di effettuare le prove di routine relative al legame competitivo e al legame a saturazione, si deve verificare che ciascun lotto di hrERα stia funzionando correttamente nel laboratorio in cui sarà utilizzato. Un processo in due fasi è applicato per dimostrare tale prestazione. Le due fasi in questione consistono in:
|
—
|
una prova di legame a saturazione [3H]-17β-estradiolo per dimostrare la specificità e la saturazione dell'hrERα. Un'analisi di regressione non lineare dei dati ottenuti (ad es. BioSoft; McPherson, 1985; Motulsky, 1995) e il grafico di Scatchard così ottenuto documenteranno l'affinità di legame del [3H]-17β-estradiolo (Kd) e il numero di recettori (Bmax) per un determinato lotto di hrERα.
|
|
—
|
una prova di legame competitivo utilizzando le sostanze di controllo (sostanza estrogenica di riferimento 17β-estradiolo), un ligando debole (ad esempio, noretinodrel o noretindrone) e un non ligando (ottiltrietossisilano, OTES). Ciascun laboratorio è tenuto a creare una banca di dati storici per documentare che l'IC50 e i valori pertinenti per la sostanza estrogenica di riferimento e un ligando debole rimangono coerenti tra una prova e l'altra e tra i diversi lotti di hrERα. Inoltre, i parametri delle curve di legame competitivo per le sostanze di controllo dovrebbero rientrare nei limiti dell'intervallo di confidenza del 95 % (cfr. la tabella 1) sviluppati utilizzando i dati dei laboratori che hanno partecipato allo studio di validazione della prova (2).
|
Tabella 1
Criteri di prestazione definiti per la sostanza estrogenica di riferimento e il ligando debole nella prova di legame all'hrER del CERI.
|
Sostanza
|
Parametro
|
Media (10)
|
Deviazione standard (n)
|
Intervallo di confidenza al 95 % (11)
|
|
Limite inferiore
|
Limite superiore
|
|
17β-estradiolo
|
Vertice
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Base
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Pendenza di Hill
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretinodrel
|
Vertice
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Base
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Pendenza di Hill
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindrone (12)
|
Vertice
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Base
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Pendenza di Hill
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
Dimostrazione delle competenze del laboratorio
|
10.
|
Cfr. i paragrafi 17 e 18 e la Tabella 2 nella sezione "ELEMENTI DEL METODO DI PROVA DI LEGAME ALL'hrER" del presente metodo di prova. Ciascuna prova (di legame a saturazione e di legame competitivo) deve consistere in tre batterie indipendenti di prove (ossia con nuove diluizioni di recettore, sostanze chimiche e reagente) condotte in giorni differenti, e ciascuna batteria di prove deve comportare tre repliche.
|
Determinazione della concentrazione di recettore (hrERα)
|
11.
|
La concentrazione di recettori attivi varia leggermente in funzione del lotto e delle condizioni di conservazione. Per questo motivo, occorre determinare la concentrazione di recettori attivi del lotto ricevuto dal fornitore. Questa fase permette di ottenere la concentrazione esatta di recettori attivi al momento della prova.
|
|
12.
|
Alle stesse condizioni della prova di legame competitivo (ossia, 0,5 nM [3H]-estradiolo), le concentrazioni nominali di 0,1, 0,2 0,4 e 0,6 nM di recettore sono incubate in assenza (legame totale) e in presenza (legame non specifico) di estradiolo non marcato a 1 μM. Il legame specifico, calcolato come differenza tra il legame totale e il legame non specifico, è rappresentato graficamente in funzione della concentrazione nominale di recettore. La concentrazione di recettore che fornisce valori di legame specifici corrispondenti al 40 % del ligando radiomarcato aggiunto è correlata alla corrispondente concentrazione di recettore; quest'ultima è utilizzata per esperimenti di legame a saturazione e di legame competitivo. Frequentemente, tale condizione è soddisfatta da una concentrazione finale di hrER di 0,2 nM.
|
|
13.
|
Se il criterio del 40 % fallisce ripetutamente, occorre verificare l'impianto sperimentale per individuare eventuali errori. Il mancato raggiungimento del criterio del 40 % può indicare che il lotto di recettori ricombinanti contiene pochissimi siti attivi; occorre quindi considerare il ricorso a un altro lotto di recettori.
|
Prova a saturazione
|
14.
|
Sono valutate otto concentrazioni crescenti di [3H]-17β-estradiolo in triplicato, rispettando le tre condizioni seguenti (cfr. tabella 2):
|
a.
|
In assenza di 17β-estradiolo non marcato e in presenza di ER. Questa condizione permette di determinare il legame totale misurando la radioattività nei pozzetti che contengono soltanto [3H]-17β-estradiolo.
|
|
b.
|
In presenza di una concentrazione di 17β-estradiolo non marcato 2000 volte superiore a quella di 17β-estradiolo marcato e in presenza di ER. Questa condizione è intesa a saturare i siti di legame attivi con 17β-estradiolo non marcato e a determinare il legame non specifico misurando la radioattività presente nei pozzetti. Si considera che l'estradiolo caldo (radiomarcato) eventualmente rimanente capace di legarsi al recettore si leghi a un sito non specifico, in quanto l'estradiolo freddo (non marcato) deve essere in concentrazione talmente elevata da legarsi a tutti i siti specifici disponibili sul recettore.
|
|
c.
|
In assenza di 17β-estradiolo non marcato e in assenza di ER (determinazione della radioattività totale).
|
Preparazione delle soluzioni di [3H]-17β-estradiolo, di 17β-estradiolo non marcato e di hrERα.
|
15.
|
Preparare una soluzione di 40 nM di [3H]-17β-estradiolo a partire da una soluzione madre di 1 μM di [3H]-17β-estradiolo nel DMSO, aggiungendo il DMSO (per preparare 200 nM) e la soluzione tamponata di prova a temperatura ambiente (prepararne 40 nM). Tale soluzione di 40 nM è quindi diluita con la soluzione tamponata a temperatura ambiente per preparare la serie di diluizioni di [3H]-17β-estradiolo, che vanno da 0,313 nM a 40 nM (conformemente alla colonna 12 della tabella 2). Le concentrazioni di prova finali, da 0,0313 a 4,0 nM, si ottengono aggiungendo 10 μl di tali soluzioni nei rispettivi pozzetti di prova di una piastra da microtitolazione a 96 pozzetti (cfr. tabelle 2 e 3). La preparazione della soluzione tamponata, la caratterizzazione della soluzione madre di [3H]-17β-estradiolo a partire dalla sua attività specifica, la preparazione delle diluizioni e la determinazione delle concentrazioni sono descritte in dettaglio nel protocollo del CERI (2).
|
|
16.
|
Le diluizioni delle soluzioni di 17β-estradiolo non marcato sono preparate aggiungendo la soluzione tamponata alla soluzione madre di 17β-estradiolo a 1 nM in modo da ottenere otto concentrazioni crescenti che inizialmente vanno da 0,625 μM a 80 μM. Le concentrazioni di prova finali, da 0,0625 a 8 μM, si ottengono aggiungendo 10 μl di tali soluzioni nei rispettivi pozzetti di prova di una piastra da microtitolazione a 96 pozzetti che serve per misurare il legame non specifico (cfr. tabelle 2 e 3). La preparazione di diluizioni di 17β-estradiolo non marcato è descritto in dettaglio nel protocollo del CERI (2).
|
|
17.
|
Occorre utilizzare la concentrazione di recettore che produce un legame specifico di 40 ± 10 % (cfr. i paragrafi 12 e 13). La soluzione di hrERα è preparata con soluzione di prova tamponata ghiacciata immediatamente prima della prova, cioè quando sono stati preparati tutti i pozzetti che servono per determinare il legame totale, il legame non specifico e quelli che contengono solo il ligando caldo.
|
|
18.
|
Le piastre da microtitolazione a 96 pozzetti sono preparate come illustrato nella tabella 2, con 3 repliche per concentrazione di [3H]-17β-estradiolo. La ripartizione dei volumi di [3H]-17β-estradiolo, di 17β-estradiolo non marcato, di soluzione tampone e di recettore figurano nella tabella 3.
Tabella 2
Configurazione della piastra da microtitolazione nella prova di legame a saturazione
|
|
1 (*10)
|
2 (*10)
|
3 (*10)
|
4 (*10)
|
5 (*10)
|
6 (*10)
|
7 (*10)
|
8 (*10)
|
9 (*10)
|
10
|
11 (*11)
|
12 (*11)
|
|
Per misurare il TB
|
Per misurare il NSB
|
Per misurare il ligando caldo solo
|
|
Diluizioni di E2 non marcate per le colonne 4-6 della piastra
|
Diluizioni di [3H]E2 non marcate per le colonne 1-9 della piastra
|
|
A
|
0,0313 nM [3H]E2+ ER
|
0,0313 nM [3H]E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H]E2+ ER
|
0,0625 nM [3H]E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H]E2+ ER
|
0,125 nM [3H]E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H]E2+ ER
|
0,250 nM [3H]E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [3H] E2+ ER
|
0,50 nM [3H]E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H]E2+ ER
|
1,00 nM [3H]E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H]E2+ ER
|
2,00 nM [3H]E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H]E2+ ER
|
4,00 nM [3H]E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
:
|
legame totale.
|
|
NSB
|
:
|
legame non specifico.
|
|
[3H] E2
|
:
|
[3H]-17β-estradiolo
|
|
E2
|
:
|
17β-estradiolo non marcato
|
|
Tabella 3
Volumi di reagente nella piastra da microtitolazione nella prova di legame a saturazione
|
Numero di colonna
|
1
|
2
|
3
|
4
|
5
|
6
|
7 (*12)
|
8 (*12)
|
9 (*12)
|
|
Fasi della preparazione
|
Pozzetti TB
|
Pozzetti NSB
|
Solo ligando caldo
|
|
Volume dei componenti da versare nei pozzetti di prova, e ordine dell'aggiunta
|
Soluzione tampone
|
60 μl
|
50 μl
|
90 μl
|
|
E2 non marcato della colonna 11 nella tabella 2
|
–
|
10 μl
|
–
|
|
[3H]E2 dalla colonna 12 nella tabella 2
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
–
|
|
Volume di reazione totale
|
100 μl
|
100 μl
|
100 μl
|
|
Incubazione
|
DOPO 2 ORE DI REAZIONE PER INCUBAZIONE
|
Quantificazione della radioattività immediatamente dopo la preparazione. Nessuna incubazione
|
|
Aggiunta di DCC a 0,4 %
|
Sì
|
Sì
|
No
|
|
Volume di DCC a 0,4 %
|
100 μl
|
100 μl
|
–
|
|
Filtrazione
|
Sì
|
Sì
|
No
|
|
MISURAZIONE DELLE DPM
|
|
Volume di quantificazione aggiunto al cocktail di scintillazione
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
Le piastre da microtitolazione per la determinazione del legame totale e del legame non specifico devono essere poste in incubazione per due ore a temperatura ambiente (da 22 oC a 28 oC).
|
|
Misurazione del [3H]-17β-estradiolo legato all'hrERα
|
20.
|
Dopo le due ore di incubazione, il [3H]-17β-estradiolo legato al hrERα è preparato dal [3H]-17β-estradiolo libero con l'aggiunta di 100 μl di una sospensione ghiacciata di DCC a 0,4 % in ciascun pozzetto. Mettere le piastre in ghiaccio per 10 minuti e filtrare la miscela di reazione e la sospensione di DCC mediante filtro per piastra di microtitolazione al fine di rimuovere il DCC. Aggiungere quindi 100 μl del filtrato al liquido di scintillazione nelle fiale per LSC per determinare le disintegrazioni al minuto (DPM) di ciascuna fiala mediante conteggio per scintillazione liquida.
|
|
21.
|
In alternativa, se non è disponibile un filtro per piastra da microtitolazione, la rimozione del DCC può essere effettuata mediante centrifugazione. Un volume di 50 μl del supernatante contenente il [3H]-17β-estradiolo legato all'hrERα è quindi prelevato con estrema cura, in modo da evitare qualsiasi contaminazione dei pozzetti per contatto con il DCC, ed è quindi sottoposto a conteggio per scintillazione.
|
|
22.
|
Il ligando caldo solo permette di determinare le disintegrazioni al minuto (dpm) del [3H]-17β-estradiolo aggiunto ai pozzetti di prova. La radioattività è quantificata immediatamente dopo la preparazione. Questi pozzetti non sono incubati e on sono trattati con la sospensione di DCC, e il loro contenuto è trasferito direttamente nel liquido di scintillazione. Queste misurazioni indicano la quantità di [3H]-17β-estradiolo (espressa in dpm) aggiunta a ciascuna serie di pozzetti per il legame totale e il legame non specifico.
|
Prova di legame competitivo
|
23.
|
La prova di legame competitivo misura i legami di [3H]-17β-estradiolo in concentrazione fissa in presenza di concentrazioni crescenti della sostanza chimica in esame. Per ciascuna concentrazione si dovranno utilizzare tre repliche concomitanti nella stessa batteria di prove. Inoltre, per ogni sostanza chimica sottoposta a prova devono essere effettuate tre batterie di prove non concomitanti. La prova richiede una o più piastre da microtitolazione a 96 pozzetti.
|
Controlli
|
24.
|
Durante l'esecuzione della prova vanno inclusi in ciascun esperimento il solvente e i controlli concomitanti (ossia, la sostanza estrogenica di riferimento, ligando debole e non ligandi). Per ciascuna batteria di prove, una medesima piastra deve essere utilizzata per stabilire le curve di tutte le concentrazioni della sostanza estrogenica di riferimento e dei controlli (ossia, ligando debole e non ligando). Tutte le altre piastre devono contenere: i) una concentrazione elevata (massimo spiazzamento, ossia circa la piena sostituzione del ligando radiomarcato) e media (corrispondente circa alla IC50) di E2 e un ligando debole in triplicato; 2) i controlli con solvente e ligandi non specifici, ciascuno in triplicato. Le procedure per la preparazione della soluzione tamponata, del [3H]-17β-estradiolo, dell'hrERα e delle soluzioni della sostanza chimica in esame sono descritte in dettaglio nel protocollo del CERI (2).
|
Controllo con solvente:
|
25.
|
Il controllo trattato solo con il solvente conferma che il solvente non interagisce con il sistema di prova e permette di misurare il legame totale (TB). Di preferenza si utilizzerà il DMSO come solvente. In alternativa, se la concentrazione massima della sostanza chimica in esame non è solubile in DMSO, si può usare l'etanolo. La concentrazione del DMSO nei pozzetti a fine prova deve essere pari al 2,05 %, ma potrebbe essere aumentata fino al 2,5 % se la sostanza chimica in esame non è sufficientemente solubile. Le concentrazioni di DMSO superiori al 2,5 % non devono essere utilizzate perché concentrazioni più elevate di solvente interferiscono con la prova. Per le sostanze chimiche in esame che non sono solubili nel DMSO, ma solubili in etanolo, è possibile utilizzare fino al 2 % di etanolo nella prova senza che si generi interferenza.
|
Controllo con soluzione tamponata:
|
26.
|
Il controllo con soluzione tamponata contiene tutti gli altri elementi della prova, tranne il solvente e la sostanza chimica in esame. I risultati del controllo con soluzione tamponata sono confrontati con il controllo con solvente per verificare che il solvente utilizzato non interferisca con il sistema di prova.
|
Ligando forte (sostanza estrogenica di riferimento)
|
27.
|
Il 17β-estradiolo (CAS 50-28-2) è il ligando endogeno che presenta una forte affinità di legame all'ER di tipo alfa. Si rappresenti una curva standard utilizzando il 17β-estradiolo non marcato per ciascuna prova di legame competitivo all'hrERα, al fine di valutare la variabilità delle prove condotte nel corso del tempo nello stesso laboratorio. Otto soluzioni di 17β-estradiolo non marcato sono preparate nel DMSO e nella soluzione tamponata, al fine di ottenere le seguenti concentrazioni finali nei pozzetti di prova: 10–6, 10–7, 10–8, 10–8.5, 10–9, 10–9.5, 10–10, 10–11 M. La concentrazione più alta di 17β–estradiolo non marcato (1 μM) funge da indicatore di legame non specifico. Tale concentrazione è distinta dall'indicazione «NSB» nella tabella 4, sebbene faccia parte anche della curva standard.
|
Ligando debole
|
28.
|
Per dimostrare la sensibilità di ogni esperimento e consentire una valutazione della variabilità delle prove condotte nel corso del tempo, viene incluso un ligando debole (noretinodrel (CAS68-23-5) o, in alternativa, noretindrone (CAS 68-22-4). Otto soluzioni di ligando debole sono preparate nel DMSO e nella soluzione tamponata, al fine di ottenere le seguenti concentrazioni finali nei pozzetti di prova: 10–4.5, 10–5.5, 10–6, 10–6.5, 10–7, 10–7.5, 10–8 and 10–9 M.
|
Non ligando
|
29.
|
Si utilizzerà l'ottiltrietossisilano (OTES, CAS 2943-75-1) come controllo negativo (non ligando). Ciò consente di verificare che la conduzione della prova individuerà le sostanze chimiche in esame che non si legano all'hrERα. Otto soluzioni di non ligando sono preparate nel DMSO e nella soluzione tamponata, al fine di ottenere le seguenti concentrazioni finali nei pozzetti di prova: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Si può utilizzare anche lo ftalato di di–n–butile (DBP, CAS 84-72-2) come non ligando alternativo, ma limitando la concentrazione massima analizzata a 10–4M. Infatti, è stato dimostrato che la solubilità massima del DBP nella prova è pari a 10–4 M.
|
Concentrazione di hrERα
|
30.
|
Occorre utilizzare la quantità di recettore che produce un legame specifico di 40 ± 10 % (cfr. i paragrafi 12 e 13 dell'allegato 3). La soluzione di hrERα è preparata immediatamente prima della prova mediante diluizione dell'hrERα funzionale in soluzione di prova tamponata ghiacciata.
|
[3H]-17β-estradiolo
|
31.
|
La concentrazione finale di [3H]-17β-estradiolo nei pozzetti di prova deve essere di 0,5 nM.
|
Sostanze chimiche in esame
|
32.
|
Occorre effettuare una prova preliminare per determinare il limite di solubilità di ciascuna sostanza chimica in esame e per identificare l'intervallo delle concentrazioni appropriato da utilizzare durante l'esecuzione del protocollo sperimentale. Il limite di solubilità di ciascuna sostanza chimica in esame deve essere inizialmente determinato nel solvente e quindi ulteriormente confermato alle condizioni sperimentali. La concentrazione finale utilizzata nella prova non deve essere superiore a 1 mM. La prova per determinare l'intervallo delle concentrazioni (range finding test) consiste in un controllo con solvente più una serie logaritmica di 8 diluizioni della concentrazione massima (ad es. 1 mM o meno, secondo il limite di solubilità). Si noterà l'apparizione di torbidità o precipitato (cfr. anche paragrafo 35 dell'appendice 3). Una volta determinato l'intervallo di concentrazioni per la prova, una sostanza chimica in esame deve essere testata utilizzando 8 concentrazioni in serie logaritmica spaziate opportunamente, come definito nella precedente prova d'individuazione dell'intervallo. Se necessario, le concentrazioni testate nella seconda e nella terza prova devono essere ulteriormente adattate in modo da caratterizzare meglio la curva concentrazione-risposta.
|
|
33.
|
Le diluizioni della sostanza chimica in esame vanno preparate nel solvente appropriato (cfr. il paragrafo 25 dell'appendice 3). Se la concentrazione più elevata della sostanza chimica in esame non è solubile in DMSO o in etanolo e se aggiungendo più solvente la concentrazione del solvente nei pozzetti al termine della prova risulta superiore al limite accettabile, la concentrazione più elevata può essere ridotta alla concentrazione immediatamente inferiore. In tal caso, è possibile aggiungere una concentrazione supplementare all'inizio della serie di concentrazioni. Le altre concentrazioni della serie rimangono invariate.
|
|
34.
|
I pozzetti di prova sono monitorati attentamente durante l'aggiunta delle soluzioni della sostanza chimica in esame, in quanto tale aggiunta può provocare un precipitato. I dati relativi a tutti i pozzetti che contengono precipitato vanno esclusi dall'approssimazione della curva e la ragione di esclusione dei dati va riportata.
|
|
35.
|
Se esistono informazioni precedenti provenienti da altre fonti che forniscono dati sul log(IC50) di una sostanza chimica in esame, può essere opportuno ripartire geometricamente le diluzioni in modo più ravvicinato attorno al log(IC50) atteso (ossia, 0,5 unità logaritmiche). I risultati finali devono corrispondere a un intervallo di concentrazioni sufficientemente ripartite ai due lati del log(IC50), compresi il vertice (top) e la base (bottom) della curva di legame, al fine di caratterizzarla in modo adeguato.
|
Configurazione della piastra di prova
|
36.
|
Le piastre di microtitolazione sono preparate usando serie di sei repliche incubate distinte per il controllo con solvente, alla concentrazione più elevata della sostanza estrogenica di riferimento (E2) che serve anche come indicatore di legame non specifico (NSB), il controllo della soluzione tamponata, incubazioni in triplicato per ciascuna delle otto concentrazioni del controllo non ligando (ottiltrietossisilano), le sette concentrazioni inferiori della sostanza estrogenica di riferimento (E2), le otto concentrazioni del ligando debole (noretinodrel o noretindrone) e le otto concentrazioni di ciascuna sostanza chimica in esame (TC). Un esempio di configurazione delle piastre per ottenere curve complete a partire da tutte le concentrazioni della sostanza estrogenica di riferimento e i controlli è riportato nella tabella 4. Si utilizzano piastre di microtitolazione aggiuntive per la sostanza chimica in esame contenenti: i) una concentrazione elevata (massimo spiazzamento) e media (circa IC50) di E2 e del ligando debole in triplicato; ii) il controllo con solvente (come legame totale) e legame non specifico, ciascuno in sei replicati (tabella 5). Nell'appendice 3.3 è riportato un esempio di configurazione della piastra da microtitolazione per la prova di legame competitivo di tre sostanze sconosciute. Le concentrazioni indicate nel foglio di lavoro, così come nelle tabelle 4 e 5, si riferiscono alle concentrazioni finali utilizzate in ciascun pozzetto di prova. La concentrazione massima di E2 deve essere pari a 1 × 10-7 M mentre, per il ligando debole, è utilizzata la concentrazione più elevata della piastra 1. La concentrazione IC50 è determinata dal laboratorio a partire della sua banca dei dati storici. Si prevede che questo valore sarà analogo a quello osservato negli studi di validazione (cfr. tabella 1).
|
Tabella 4
Configurazione della piastra da microtitolazione per la prova di legame competitivo (98)
(99), curve concentrazione-risposta complete per la sostanza estrogenica di riferimento e i controlli (piastra 1).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Controllo con soluzione tamponata e controllo positivo (E2)
|
Controllo debolmente positivo (Noretinodrel)
|
Controllo negativo (OTES)
|
TB e NSB
|
|
A
|
Bianco (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (controllo con solvente) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Controllo con tampone
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Bianco (E2 caldo) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
Tabella 5
Configurazione della piastra da microtitolazione per la prova di legame competitivo, piastre aggiuntive per le sostanze chimiche in esame e i controlli della piastra.
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
Sostanza chimica in esame-1 (TC-1)
|
Sostanza chimica in esame-2 (TC-2)
|
Sostanza chimica in esame-3 (TC-3)
|
Controlli
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10
-7
M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10
–4.5
M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (Controllo con solvente)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
In questo esempio, il ligando debole è noretinodrel (NE)
|
Completamento della prova di legame competitivo
|
37.
|
Tutti i pozzetti, ad eccezione di quelli che servono a determinare il legame totale e i pozzetti per i bianchi (con ligando caldo), come indicato nella tabella 6, ricevono 50 μl di soluzione tamponata, quindi mescolata con 10 μl di controllo con solvente, di sostanza estrogenica di riferimento (E2), di ligando debole, di non ligando, e delle sostanze chimiche in esame, rispettivamente, e di 10 μl di una soluzione di [3H]-17β-estradiolo a 5 nM. Infine, si aggiungono ai pozzetti di ciascuna piastra 30μl di soluzione di recettore ghiacciata e si mescola delicatamente. La soluzione di hrERα è l'ultimo reattivo aggiunto. Le piastre di prova da microtitolazione sono incubate a temperatura ambiente (da 22 oC a 28 oC) per 2 ore.
Tabella 6
Volume dei costituenti della prova di legame competitivo all'hrER, piastre da microtitolazione
|
Fasi della preparazione
|
Pozzetti diversi da TB
|
Pozzetti TB
|
Bianco (E2 caldo)
|
|
Volume dei componenti da versare nei pozzetti di prova, e ordine dell'aggiunta
|
Soluzione tamponata a temperatura ambiente
|
50 μl
|
60 μl
|
90 μl
|
|
E2 non marcato, ligando debole, non ligando, solvente e sostanze chimiche in esame (*16)
|
10 μl
|
–
|
–
|
|
[3H]-17β-estradiolo in quantità sufficiente per una concentrazione finale di 0,5 nM (ossia 5 nM)
|
10 μl
|
10 μl
|
10 μl
|
|
Concentrazione di hrERα come determinata (cfr. i paragrafi 12-13)
|
30 μl
|
30 μl
|
-
|
|
Volume totale in ciascun pozzetto di prova
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Il [3H]-17β-estradiolo legato all'hrERα è separato dal [3H]-17β-estradiolo libero aggiungendo 100 μl di sospensione fredda di DCC in ogni pozzetto, e quindi quantificato seguendo il metodo descritto nei paragrafi da 21 a 23 dell'appendice 3 per la prova di legame a saturazione.
|
|
39.
|
I pozzetti G10-G12 e H10-H12 (identificati come «bianco (E2 caldo)» nella tabella 4) forniscono le disintegrazioni al minuto del [3 H]-estradiolo in 10 μl. Le aliquote di 10 μl sono versate direttamente nel liquido di scintillazione.
|
Criteri di accettabilità
Prova di legame a saturazione
|
40.
|
La curva di legame specifica deve raggiungere un plateau man mano che si utilizzano concentrazioni crescenti di [3H]-17β-estradiolo, il che indica la saturazione degli hrERα con il ligando.
|
|
41.
|
Il legame specifico di [3H] -17β-estradiolo a 0,5 nM deve corrispondere ad un intervallo accettabile di 30-50 % della media della radioattività in tutte le batteria di prove. Sono ammissibili lievi escursioni al di fuori di tale intervallo, ma se ciò avviene costantemente o se una particolare batteria di prove dà risultati significativamente al di fuori di tale intervallo, occorre adattare la concentrazione delle proteine e ripetere la prova di legame a saturazione.
|
|
42.
|
I dati devono generare un grafico di Scatchard lineare.
|
|
43.
|
Il legame non specifico non deve essere eccessivo: il valore è generalmente inferiore al 35 % del legame totale. Tuttavia, il rapporto può occasionalmente eccedere tale limite quando si misurano disintegrazioni al minuto molto deboli per la concentrazione più bassa di 17β-estradiolo radiomarcato testata.
|
Prova di legame competitivo
|
44.
|
Crescenti concentrazioni di 17β-estradiolo dovrebbero spiazzare il [3H]-17β-estradiolo dal recettore secondo un modello di legame competitivo in un unico sito.
|
|
45.
|
Il valore IC50 per la sostanza estrogenica di riferimento (17β-estradiolo) deve essere approssimativamente uguale alla concentrazione molare di [3H]-17β-estradiolo più la Kd stabilita per la prova di legame a saturazione.
|
|
46.
|
Il legame specifico totale deve situarsi costantemente nell'intervallo di 40 ± 10 % quando la concentrazione media della radioattività totale aggiunta a ciascun pozzetto è di 0,5 nM in tutte le batterie di prove. Sono ammissibili lievi escursioni al di fuori di tale intervallo, ma se ciò avviene costantemente o se una particolare batteria di prove dà risultati significativamente al di fuori di tale intervallo, occorre adattare la concentrazione delle proteine.
|
|
47.
|
Il solvente non deve alterare la sensibilità o la riproducibilità della prova. I risultati del controllo con solvente (pozzetti TB) sono confrontati con il controllo con soluzione tamponata per verificare che il solvente utilizzato non interagisca con il sistema di prova. Se il solvente non influisce sulla prova, i risultati del TB e del controllo con soluzione tamponata devono essere comparabili.
|
|
48.
|
Il non ligando non dovrebbe spiazzare più del 25 % di [3H]-17β-estradiolo dall'hrERα durante la prova fino a 10–3 M (OTES) o 10–4 M (DBP).
|
|
49.
|
Sono stati definiti criteri di prestazione per la sostanza estrogenica di riferimento e per i due ligandi deboli (ad esempio, noretinodrel, noretindrone) utilizzando i dati dello studio di validazione della prova di legame all'hrER del CERI (allegato N del riferimento 2). Vengono forniti intervalli di confidenza al 95 % per la media ± SD (n) per tutti i cicli di controllo effettuati da quattro laboratori che partecipano allo studio di validazione. Gli intervalli di confidenza al 95 % sono stati calcolati per i parametri di approssimazione della curva (ossia, il vertice e la base della curva, la pendenza di Hill, il logIC50) per la sostanza estrogenica di riferimento e i ligandi deboli e per il log10 RBA dei ligandi deboli rispetto alla sostanza estrogenica di riferimento. La tabella 1 fornisce gli intervalli attesi per i parametri di approssimazione della curva che possono essere utilizzati come criteri di prestazione. In pratica, l'intervallo della IC50 può variare leggermente in funzione della Kd, derivata sperimentalmente, della preparazione dei recettori e della concentrazione del ligando utilizzata per la prova.
|
|
50.
|
Non sono stati sviluppati criteri di prestazione per i parametri di approssimazione della curva per le sostanze chimiche in esame poiché esiste una grande diversità delle sostanze chimiche che possono essere potenzialmente oggetto di prova così come una grande variazione nelle affinità e risultati potenziali corrispondenti (ad es. approssimazione completa, parziale o nulla delle curve). Tuttavia, è necessario ricorrere al giudizio professionale di un esperto per analizzare i risultati di ciascuna batteria di prove su una sostanza chimica in esame. Occorre usare un intervallo di concentrazioni della sostanza chimica in esame sufficiente per definire chiaramente il vertice della curva di legame competitivo (ad es. 90-100 % di legame). La variabilità fra le repliche di ciascuna concentrazione della sostanza chimica in esame, nonché tra le tre batterie di prove non concomitanti deve essere ragionevole e giustificabile sotto il profilo scientifico. I controlli di ciascuna batteria di prove sulla sostanza in esame devono avvicinarsi alle misure di prestazione riportate per questo saggio del CERI ed essere coerenti con i dati storici di controllo di ciascun rispettivo laboratorio.
|
ANALISI DEI DATI
Prova di legame a saturazione
|
51.
|
La prova misura il legame totale e il legame non specifico. Tali valori permettono di calcolare il legame specifico indotto da concentrazioni crescenti di [3H]-17β-estradiolo in condizioni di equilibrio, sottraendo il legame non specifico dal legame totale. La curva che rappresenta il legame specifico in funzione della concentrazione di [3H]-17β-estradiolo deve raggiungere un plateau per il legame specifico massimo, che indica la saturazione degli hrERα con il [3H]-17β-estradiolo. Inoltre, l'analisi dei dati documenta il legame del [3H]-17β-estradiolo a un unico sito di legame ad alta affinità. La curva di legame a saturazione deve indicare il legame non specifico, il legame totale e il legame specifico. Una regressione non lineare consente di approfondire l'analisi dei dati (ad es. BioSoft; McPherson, 1985; Motulsky, 1995), e i risultati finali sono presentati sotto forma di grafico di Scatchard.
|
|
52.
|
L'analisi dei dati deve determinare la Bmax e la Kd sulla base dei soli dati di legame totale, nell'ipotesi che il legame non specifico sia lineare, a meno che venga fornita una giustificazione dell'utilizzo di un altro metodo. Inoltre, si determina la migliore approssimazione delle curve mediante una solida analisi di regressione; in caso contrario se ne deve fornire la motivazione. Va indicato il metodo scelto per questa solida analisi di regressione. Una correzione relativa alla perdita di ligando (ad es. con il metodo di Swillens, 1995) va sempre applicata per determinare la Bmax e la Kd a partire dai dati di legame a saturazione.
|
Prova di legame competitivo
|
53.
|
La curva di legame competitivo presenta il legame specifico del [3H]-17β-estradiolo in funzione della concentrazione (in log10) del competitore. La concentrazione della sostanza chimica in esame che inibisce il 50 % del legame massimo specifico di [3H]-17β-estradiolo corrisponde al valore IC50.
|
|
54.
|
Le stime dei valori di log(IC50) per i controlli positivi (ad es. sostanza estrogenica di riferimento e ligando debole) sono determinate utilizzando un software di approssimazione delle curve con regressione non lineare appropriata che si basa su un'equazione di Hill a quattro parametri (ad es. BioSoft; McPherson, 1985; Motulsky, 1995). Tali approssimazioni sono effettuate senza imporre restringimenti al vertice e alla base della curva, alla pendenza e al log(IC50). Si determina la migliore approssimazione delle curve mediante una solida analisi di regressione; in caso contrario se ne deve fornire la motivazione. Non va applicata la correzione relativa alla perdita di ligandi. In seguito all'analisi iniziale, ciascuna curva di legame deve essere rivista per garantire la migliore approssimazione al modello. L'affinità di legame relativa (RBA) di un ligando debole è espressa in percentuale corrispondente al rapporto tra log(IC50) del ligando debole e il log(IC50) del 17β-estradiolo. I risultati dei controlli positivi e del controllo non ligando devono essere valutati in base alle misurazioni della prestazione della prova indicate nei paragrafi 44-49 dell'appendice 3.
|
|
55.
|
I dati relativi a tutte le sostanze chimiche in esame devono essere analizzati mediante un approccio graduale per garantire un'adeguata classificazione dei dati e un'adeguata classificazione di ciascuna curva di legame competitivo. Si raccomanda che, all'inizio, ciascuna batteria di prove per una sostanza chimica in esame sia sottoposta a un'analisi normalizzata dei dati, identica a quella utilizzata per la sostanza estrogenica di riferimento e i controlli con ligando debole (cfr. il paragrafo 54 della presente appendice 3). Una volta completata l'analisi, si procede a un esame tecnico dei parametri di approssimazione della curva e a un esame visivo per determinare in che modo i dati corrispondono alla curva di legame competitivo ottenuta per ciascuna batteria di prove. Tale esame tecnico si basa su tre osservazioni che indicano che la prova e gli esami sono stati condotti correttamente: una diminuzione della percentuale di [3H]-17β-estradiolo collegata ai siti specifici in funzione della concentrazione; una variabilità debole tra le repliche tecniche di ciascuna concentrazione chimica della sostanza chimica in esame; la coerenza dei parametri di approssimazione tra le tre batterie di prove.
|
Interpretazione dei dati
|
56.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata un ligando dell'hrERα se la curva di legame può essere approssimata e se il punto più basso della curva di risposta ottenuta per l'intervallo dei dati corrisponde a un legame inferiore al 50 % (Grafico 1).
|
|
57.
|
A condizione che siano soddisfatti tutti i criteri di accettabilità, la sostanza chimica in esame è considerata non ligando dell'hrERα se:
|
—
|
la sua curva di legame può essere approssimata e se il punto più basso sulla curva di risposta approssimata ottenuta per l'intervallo di dati corrisponde a un legame superiore al 75 %, oppure
|
|
—
|
la sua curva di legame non può essere approssimata e se la media delle percentuali di legame non livellate di tutti i gruppi di concentrazione è superiore al 75 %.
|
|
|
58.
|
Le sostanze chimiche in esame sono considerate equivoche se non è soddisfatta nessuna delle condizioni di cui sopra (ad esempio il punto più basso della curva di risposta approssimata si situa tra 76 e 51 %).
|
Tabella 7
Criteri di classificazione di una sostanza chimica in esame basata sulla sua curva di legame competitivo
|
Classificazione
|
Criteri
|
|
Ligandoa
|
È possibile approssimare la curva di legame.
Il punto più basso della curva di risposta ottenuto per l'intervallo di dati corrisponde a un legame inferiore a 50 %.
|
|
Non-ligandob
|
Se è possibile approssimare la curva di legame, il
punto più basso della curva di risposta approssimata ottenuto per l'intervallo di dati corrisponde a un legame superiore a 75 %.
Se non è possibile approssimare la curva di legame,
la media delle percentuali di legame non livellate di tutti i gruppi di concentrazione della prova è superiore a 75 %.
|
|
Equivococ
|
Una prova valutabile che non può esser classificata come ligando né non ligando.
(ad esempio il punto più basso della curva di risposta approssimata si situa tra 76 e 51 %).
|
Grafico 1
Esempi di classificazione della sostanza chimica in esame in base ad una curva di legame competitivo

|
59.
|
Le multiple prove condotte all'interno di un laboratorio su una sostanza chimica in esame sono combinate attribuendo valori numerici a ciascuna batteria di prove e calcolando la media di tali valori, come indicato nella tabella 8. I risultati combinati delle batterie di prove di ciascun laboratorio sono confrontati con la classificazione prevista per ciascuna sostanza chimica in esame.
|
Tabella 8
Metodo di classificazione della sostanza chimica in esame mediante multiple batterie di prove condotte all'interno di un laboratorio.
|
Attribuzione di un valore a ciascuna batteria di prove:
|
|
Classificazione
|
Valore numerico
|
|
Ligando
|
2
|
|
Equivoco
|
1
|
|
Non ligando
|
0
|
|
Classificazione sulla base della media dei valori numerici di tutte le batterie di prove:
|
|
Classificazione
|
Valore numerico
|
|
Ligando
|
Media ≥ 1,5
|
|
Equivoco
|
0,5 ≤ media < 1,5
|
|
Non ligando
|
Media < 0,5
|
RELAZIONE SULL'ESECUZIONE DELLA PROVA
|
60.
|
Cfr. il paragrafo 24 nella sezione "ELEMENTI DEL METODO DI PROVA DI LEGAME ALL'hrER" del presente metodo di prova.
|
Appendice 3.1
Elenco dei Termini
[3H]E2
: 17β-estradiolo radiomarcato con trizio
DCC
: Dextran-coated charcoal (carbone rivestito di destrano)
E2
: 17β-estradiolo non marcato (inerte)
Soluzione di prova tamponata
: soluzione di 10 mM di Tris-HCl con pH 7,4, contenente 1 mM di EDTA, 1mM di EGTA, 1 mM di NaVO3, 10 % di glicerolo, 0,2 mM di leupeptina, 1 mM di ditiotreitolo e 10 mg/ml di albumina serica bovina.
hrERα
: recettore estrogenico alfa ricombinante umano (dominio di legame del ligando)
Replica
: Uno dei multipli pozzetti che contengono lo stesso contenuto alle stesse concentrazioni e che sono testati contemporaneamente in un'unica batteria di prove. Nel presente protocollo, ogni concentrazione della sostanza chimica in esame è testata in triplicato; ciò significa che tre repliche di ciascuna concentrazione della sostanza chimica in esame sono testate simultaneamente.
Batteria di prove
: serie completa di prove simultanee nei pozzetti di una piastra da microtitolazione che fornisce tutte le informazioni necessarie per caratterizzare l'affinità di legame all'hrERα di una sostanza chimica in esame (ossia, quantità totale di [3H]-17β-estradiolo aggiunta in un pozzetto, il legame massimo all'hrERα di [3H]-17β-estradiolo, il legame non specifico e il legame totale per diverse concentrazioni della sostanza chimica in esame. Una batteria di prove può consistere anche in un unico pozzetto di prova (replica) per concentrazione, ma poiché il presente protocollo richiede prove in triplicato, una batteria di prove consiste di tre pozzetti di prova per concentrazione. Inoltre, il presente protocollo impone di realizzare tre batterie di prove indipendenti (non simultanee) per sostanza chimica.
Appendice 3.2
CONFIGURAZIONE DEI POZZETTI PER LA BATTERIA DI PROVE DI LEGAME COMPETITIVO
|
Piastra
|
Posizione
|
Replica
|
Tipo di pozzetto
|
Codice del pozzetto
|
Codice della concentrazione
|
Concentrazione iniziale del competitore (M)
|
Volume di soluzione madre di hrER (μl)
|
Volume di soluzione tamponata (μl)
|
Volume di tracciante (E2 caldo) (μl)
|
Volume della piastra di diluizione (μl)
|
Volume finale (μl)
|
Concentrazione finale del competitore (M)
|
|
S
|
A1
|
1
|
Bianco
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A2
|
2
|
Bianco
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
A3
|
3
|
Bianco
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
S
|
B1
|
1
|
E2 freddo
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B2
|
2
|
E2 freddo
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
B3
|
3
|
E2 freddo
|
S
|
S1
|
1,00E-10
|
30
|
50
|
10
|
10
|
100
|
1,0E-11
|
|
S
|
C1
|
1
|
E2 freddo
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C2
|
2
|
E2 freddo
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
C3
|
3
|
E2 freddo
|
S
|
S2
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
D1
|
1
|
E2 freddo
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D2
|
2
|
E2 freddo
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
D3
|
3
|
E2 freddo
|
S
|
S3
|
3,16E-09
|
30
|
50
|
10
|
10
|
100
|
3,2E-10
|
|
S
|
E1
|
1
|
E2 freddo
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E2
|
2
|
E2 freddo
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
E3
|
3
|
E2 freddo
|
S
|
S4
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
F1
|
1
|
E2 freddo
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F2
|
2
|
E2 freddo
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
F3
|
3
|
E2 freddo
|
S
|
S5
|
3,16E-08
|
30
|
50
|
10
|
10
|
100
|
3,2E-09
|
|
S
|
G1
|
1
|
E2 freddo
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G2
|
2
|
E2 freddo
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
G3
|
3
|
E2 freddo
|
S
|
S6
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
H1
|
1
|
E2 freddo
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H2
|
2
|
E2 freddo
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
H3
|
3
|
E2 freddo
|
S
|
S7
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
A4
|
1
|
Noretinodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A5
|
2
|
Noretinodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
A6
|
3
|
Noretinodrel
|
NE
|
WP1
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B4
|
1
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B5
|
2
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
B6
|
3
|
Noretinodrel
|
NE
|
WP2
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C4
|
1
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C5
|
2
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
C6
|
3
|
Noretinodrel
|
NE
|
WP3
|
3,16E-07
|
30
|
50
|
10
|
10
|
100
|
3,2E-08
|
|
S
|
D4
|
1
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D5
|
2
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D6
|
3
|
Noretinodrel
|
NE
|
WP4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E4
|
1
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E5
|
2
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
E6
|
3
|
Noretinodrel
|
NE
|
WP5
|
3,16E-06
|
30
|
50
|
10
|
10
|
100
|
3,2E-07
|
|
S
|
F4
|
1
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F5
|
2
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F6
|
3
|
Noretinodrel
|
NE
|
WP6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
G4
|
1
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G5
|
2
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
G6
|
3
|
Noretinodrel
|
NE
|
WP7
|
3,16E-05
|
30
|
50
|
10
|
10
|
100
|
3,2E-06
|
|
S
|
H4
|
1
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H5
|
2
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
H6
|
3
|
Noretinodrel
|
NE
|
WP8
|
3,16E-04
|
30
|
50
|
10
|
10
|
100
|
3,2E-05
|
|
S
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
S
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
S
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
S
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
S
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
S
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
S
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
S
|
A10
|
1
|
legame totale
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A11
|
2
|
legame totale
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
A12
|
3
|
legame totale
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B10
|
4
|
legame totale
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B11
|
5
|
legame totale
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
B12
|
6
|
legame totale
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
S
|
C10
|
1
|
E2 freddo (elevato)
|
NSB
|
S1
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C11
|
2
|
E2 freddo (elevato)
|
NSB
|
S2
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
C12
|
3
|
E2 freddo (elevato)
|
NSB
|
S3
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D10
|
4
|
E2 freddo (elevato)
|
NSB
|
S4
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D11
|
5
|
E2 freddo (elevato)
|
NSB
|
S5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
D12
|
6
|
E2 freddo (elevato)
|
NSB
|
S6
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
S
|
E10
|
1
|
Ctrl con tampone
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E11
|
2
|
Ctrl con tampone
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
E12
|
3
|
Ctrl con tampone
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F10
|
4
|
Ctrl con tampone
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F11
|
5
|
Ctrl con tampone
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
F12
|
6
|
Ctrl con tampone
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
S
|
G10 (*17)
|
1
|
Bianco (E2 caldo)
|
caldo
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G11 (*17)
|
2
|
Bianco (E2 caldo)
|
caldo
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
G12 (*17)
|
3
|
Bianco (E2 caldo)
|
caldo
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H10 (*17)
|
4
|
Bianco (E2 caldo)
|
caldo
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H11 (*17)
|
5
|
Bianco (E2 caldo)
|
caldo
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
S
|
H12
|
6
|
Bianco (E2 caldo)
|
caldo
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
Sconosciuto 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A2
|
2
|
Sconosciuto 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A3
|
3
|
Sconosciuto 1
|
U1
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B1
|
1
|
Sconosciuto 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B2
|
2
|
Sconosciuto 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B3
|
3
|
Sconosciuto 1
|
U1
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C1
|
1
|
Sconosciuto 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C2
|
2
|
Sconosciuto 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C3
|
3
|
Sconosciuto 1
|
U1
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D1
|
1
|
Sconosciuto 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D2
|
2
|
Sconosciuto 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D3
|
3
|
Sconosciuto 1
|
U1
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E1
|
1
|
Sconosciuto 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E2
|
2
|
Sconosciuto 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E3
|
3
|
Sconosciuto 1
|
U1
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F1
|
1
|
Sconosciuto 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F2
|
2
|
Sconosciuto 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F3
|
3
|
Sconosciuto 1
|
U1
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G1
|
1
|
Sconosciuto 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G2
|
2
|
Sconosciuto 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G3
|
3
|
Sconosciuto 1
|
U1
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H1
|
1
|
Sconosciuto 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H2
|
2
|
Sconosciuto 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H3
|
3
|
Sconosciuto 1
|
U1
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A4
|
1
|
Sconosciuto 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A5
|
2
|
Sconosciuto 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A6
|
3
|
Sconosciuto 2
|
U2
|
1
|
1,00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B4
|
1
|
Sconosciuto 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B5
|
2
|
Sconosciuto 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B6
|
3
|
Sconosciuto 2
|
U2
|
2
|
1,00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C4
|
1
|
Sconosciuto 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C5
|
2
|
Sconosciuto 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C6
|
3
|
Sconosciuto 2
|
U2
|
3
|
1,00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D4
|
1
|
Sconosciuto 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D5
|
2
|
Sconosciuto 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D6
|
3
|
Sconosciuto 2
|
U2
|
4
|
1,00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E4
|
1
|
Sconosciuto 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E5
|
2
|
Sconosciuto 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E6
|
3
|
Sconosciuto 2
|
U2
|
5
|
1,00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F4
|
1
|
Sconosciuto 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F5
|
2
|
Sconosciuto 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F6
|
3
|
Sconosciuto 2
|
U2
|
6
|
1,00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G4
|
1
|
Sconosciuto 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G5
|
2
|
Sconosciuto 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
G6
|
3
|
Sconosciuto 2
|
U2
|
7
|
1,00E-03
|
30
|
50
|
10
|
10
|
100
|
1,0E-04
|
|
P1
|
H4
|
1
|
Sconosciuto 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H5
|
2
|
Sconosciuto 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
H6
|
3
|
Sconosciuto 2
|
U2
|
8
|
1,00E-02
|
30
|
50
|
10
|
10
|
100
|
1,0E-03
|
|
P1
|
A7
|
1
|
Sconosciuto 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A8
|
2
|
Sconosciuto 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
A9
|
3
|
Sconosciuto 3
|
U3
|
1
|
1.00E-09
|
30
|
50
|
10
|
10
|
100
|
1,0E-10
|
|
P1
|
B7
|
1
|
Sconosciuto 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B8
|
2
|
Sconosciuto 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
B9
|
3
|
Sconosciuto 3
|
U3
|
2
|
1.00E-08
|
30
|
50
|
10
|
10
|
100
|
1,0E-09
|
|
P1
|
C7
|
1
|
Sconosciuto 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C8
|
2
|
Sconosciuto 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
C9
|
3
|
Sconosciuto 3
|
U3
|
3
|
1.00E-07
|
30
|
50
|
10
|
10
|
100
|
1,0E-08
|
|
P1
|
D7
|
1
|
Sconosciuto 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D8
|
2
|
Sconosciuto 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
D9
|
3
|
Sconosciuto 3
|
U3
|
4
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1,0E-07
|
|
P1
|
E7
|
1
|
Sconosciuto 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E8
|
2
|
Sconosciuto 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E9
|
3
|
Sconosciuto 3
|
U3
|
5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F7
|
1
|
Sconosciuto 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F8
|
2
|
Sconosciuto 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
F9
|
3
|
Sconosciuto 3
|
U3
|
6
|
1.00E-04
|
30
|
50
|
10
|
10
|
100
|
1,0E-05
|
|
P1
|
G7
|
1
|
Sconosciuto 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G8
|
2
|
Sconosciuto 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
G9
|
3
|
Sconosciuto 3
|
U3
|
7
|
1.00E-03
|
30
|
50
|
10
|
10
|
100
|
1.0E-04
|
|
P1
|
H7
|
1
|
Sconosciuto 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H8
|
2
|
Sconosciuto 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
H9
|
3
|
Sconosciuto 3
|
U3
|
8
|
1.00E-02
|
30
|
50
|
10
|
10
|
100
|
1.0E-03
|
|
P1
|
A10
|
1
|
Controllo E2 (max)
|
S
|
E2max1
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A11
|
2
|
Controllo E2 (max)
|
S
|
E2max2
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
A12
|
3
|
Controllo E2 (max)
|
S
|
E2max3
|
1.00E-06
|
30
|
50
|
10
|
10
|
100
|
1.00E-07
|
|
P1
|
B10
|
1
|
Controllo E2 (IC50)
|
S
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
Controllo E2 (IC50)
|
S
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
Controllo E2 (IC50)
|
S
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
Controllo NE (max)
|
S
|
Nemax1
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C11
|
2
|
Controllo NE (max)
|
S
|
Nemax2
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
C12
|
3
|
Controllo NE (max)
|
S
|
Nemax3
|
1.00E-3,5
|
30
|
50
|
10
|
10
|
100
|
1.00E-4,5
|
|
P1
|
D10
|
1
|
Controllo NE (IC50)
|
S
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
Controllo NE (IC50)
|
S
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
Controllo NE (IC50)
|
S
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
E2 freddo (elevato)
|
NSB
|
S1
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E11
|
2
|
E2 freddo (elevato)
|
NSB
|
S2
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
E12
|
3
|
E2 freddo (elevato)
|
NSB
|
S3
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F10
|
4
|
E2 freddo (elevato)
|
NSB
|
S4
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F11
|
5
|
E2 freddo (elevato)
|
NSB
|
S5
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
F12
|
6
|
E2 freddo (elevato)
|
NSB
|
S6
|
1.00E-05
|
30
|
50
|
10
|
10
|
100
|
1,0E-06
|
|
P1
|
G10
|
1
|
legame totale
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
legame totale
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
legame totale
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
legame totale
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
legame totale
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
legame totale
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
Appendice 4
CONSIDERAZIONI RELATIVE ALL'ANALISI DEI DATI DELLA PROVA DI LEGAME COMPETITIVO ALL'HRER
|
1.
|
La prova di legame competitivo all'hrERα misura i legami del [3H]-17β-estradiolo in concentrazione fissa in presenza di concentrazioni crescenti della sostanza chimica in esame. La curva di legame competitivo presenta il legame specifico del [3H]-17β-estradiolo in funzione della concentrazione (in log10) del competitore. La concentrazione della sostanza chimica in esame che inibisce il 50 % del legame massimo specifico del [3H]-17β-estradiolo corrisponde al valore IC50.
|
Analisi dei dati per la sostanza estrogenica di riferimento e il ligando debole (1)
|
2.
|
I dati delle batterie di prove sui controlli sono trasformati (in percentuale di legame specifico del [3H]-17β-estradiolo e logaritmo della concentrazione della sostanza chimica di controllo) per le ulteriori analisi. Le stime dei valori di log(IC50) per i controlli positivi (ad es. sostanza estrogenica di riferimento e ligando debole) sono determinate utilizzando un idoneo software di approssimazione delle curve con regressione non lineare che si basa su un'equazione di Hill a quattro parametri (ad es. BioSoft; GraphPad Prism) (2). Tali approssimazioni possono generalmente essere effettuate senza imporre limiti al vertice e alla base della curva, alla pendenza e al log(IC50). Si determina la migliore approssimazione delle curve mediante una solida analisi di regressione; in caso contrario se ne deve fornire la motivazione. Va indicato il metodo scelto per questa solida analisi di regressione. I saggi di FW o del CERI sul legame all'hrER non richiedono alcuna correzione relativa alla perdita di ligandi, che però può essere presa in considerazione se necessario. In seguito all'analisi iniziale, ciascuna curva di legame deve essere rivista per garantire la conformità al modello. L'affinità di legame relativa (RBA) di un ligando debole può essere calcolata come percentuale del log (IC50) per il ligando debole relativa al log (IC50) per il 17β-estradiolo. I risultati ottenuti per i controlli positivi e il controllo non ligando devono essere valutati mediante misure di prestazione della prova e criteri di accettabilità descritti nel presente metodo di prova (paragrafo 20), appendice 2 (saggio di FW, paragrafi 41-51) e appendice 3 (saggio del CERI, paragrafi 41-51). Esempi di 3 batterie di prove per la sostanza estrogenica di riferimento e per il ligando debole figurano nel Grafico 1.
|
Grafico 1
Esempi di curve di legame competitivo per la sostanza estrogenica di riferimento e il ligando debole di controllo
Analisi dei dati per le sostanze chimiche in esame
|
3.
|
I dati relativi a tutte le sostanze chimiche in esame devono essere analizzati mediante un approccio graduale per garantire un'adeguata classificazione dei dati e un'adeguata classificazione di ciascuna curva di legame competitivo. Ciascuna batteria di prove per una sostanza chimica in esame è inizialmente sottoposta a un'analisi normalizzata dei dati, identica a quella utilizzata per la sostanza estrogenica di riferimento e i controlli con ligando debole. Una volta completata l'analisi, si procede a un esame tecnico dei parametri di approssimazione della curva e a un esame visivo per determinare in che modo i dati corrispondono alla curva di legame competitivo ottenuta per ciascuna batteria di prove. Tale esame tecnico si basa su tre osservazioni che indicano che la prova e gli esami sono stati condotti correttamente: una diminuzione della percentuale di [3H]-17β-estradiolo collegata ai siti specifici in funzione della concentrazione; una variabilità debole tra le repliche tecniche di ciascuna concentrazione chimica della sostanza chimica in esame; la coerenza dei parametri di approssimazione tra le tre batterie di prove. È opportuno fare ricorso al giudizio professionale di un esperto al riesame dei risultati di ciascuna batteria di prove per la sostanza chimica in esame, e i dati utilizzati per classificare ciascuna sostanza chimica in esame come ligando o non ligando devono essere giustificabili sotto il profilo scientifico.
|
|
4.
|
Occasionalmente alcuni dati possono richiedere un'ulteriore attenzione al fine di analizzare e interpretare in modo adeguato i dati relativi al legame all'hrER. Studi precedenti hanno rivelato casi in cui l'analisi e l'interpretazione di dati relativi al legame competitivo ai recettori possono essere complicate da una rimonta della percentuale del legame specifico per le concentrazioni di prova più elevate (Grafico 2). Si tratta di un problema ben noto che è stato riscontrato durante l'esecuzione dei protocolli in diverse prove di legame competitivo al recettore (3). In questi casi si osserva che la risposta è dipendente dalla concentrazione alle concentrazioni inferiori, ma quando la concentrazione della sostanza chimica in esame si avvicina al limite di solubilità, il [3H]-17β-estradiolo cessa di essere spiazzato dalla sostanza chimica in esame. In questi casi, i dati relativi alle concentrazioni più elevate indicano che è stato raggiunto il limite biologico della prova. Ad esempio, questo fenomeno è spesso associato a insolubilità chimica e a precipitazione a forte concentrazione; potrebbe anche essere un riflesso del superamento della capacità del DCC di catturare il ligando radiomarcato libero durante il processo di separazione, alle concentrazioni chimiche più elevate. Conservare tali punti durante l'approssimazione dei dati del legame competitivo a una curva sigmoide può a volte portare a una classificazione errata del potenziale di legame all'ER di una sostanza chimica in esame (Grafico 2). Per evitare tale situazione, i protocolli per i saggi di FW e del CERI includono un'opzione per escludere punti di dati dalle analisi quando la media delle repliche per la percentuale di legame specifico del [3H]-17β-estradiolo supera del 10 % o più la media del legame osservato a una concentrazione inferiore (ciò è spesso denominato "regola del 10 %"). Questa regola può essere utilizzata una sola volta per una determinata curva e devono essere disponibili dati relativi ad almeno 6 concentrazioni affinché la curva possa essere correttamente classificata.
|
Grafico 2
Esempi di curve di legame competitivo con e senza applicazione della regola del 10 %.
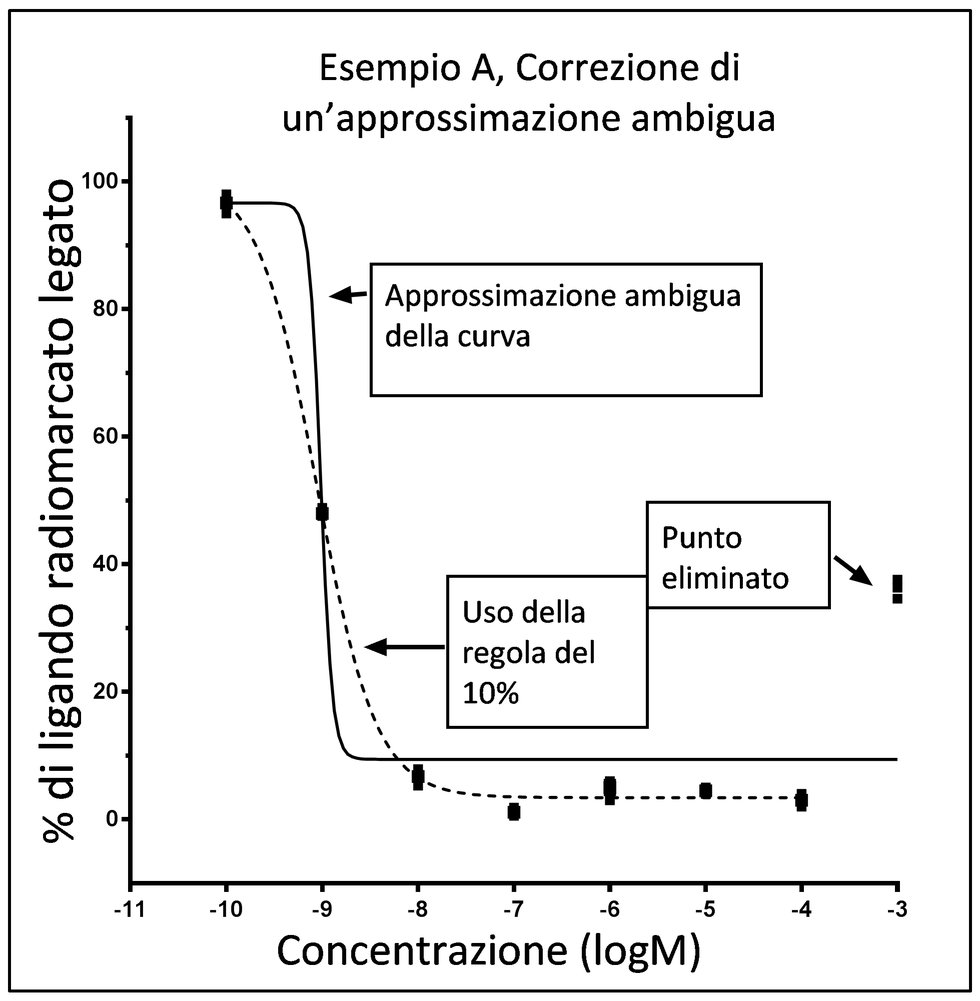
|
5.
|
Occorre considerare attentamente se è appropriato applicare la regola del 10 % per correggere tali curve; la regola va riservata alle sostanze chimiche per le quali esistono forti indicazioni che siano dei ligandi dell'hrER. Nel corso della sperimentazione effettuata per lo studio di validazione del saggio di FW di legame all'hrER è stato osservato che la regola del 10 % può, a volte, avere conseguenze indesiderate e impreviste. Infatti, le sostanze chimiche che non interagivano con il recettore (ossia, non ligandi) mostravano spesso una variabilità superiore al 10 % nell'insieme delle concentrazioni testate, quando il legame del ligando radiomarcato era vicino al 100 %. Se il valore di legame più basso corrisponde a una concentrazione debole, l'applicazione della regola del 10 % potrebbe comportare l'eliminazione dall'analisi dei dati di tutte le concentrazioni superiori, anche se tali concentrazioni potrebbero essere utili per stabilire che la sostanza chimica è un non ligando. Il grafico 3 mostra alcuni esempi in cui il ricorso alla regola del 10 % non è appropriato.
|
Grafico 3
Esempi di curve di legame competitivo in cui non è appropriato applicare la regola del 10 %
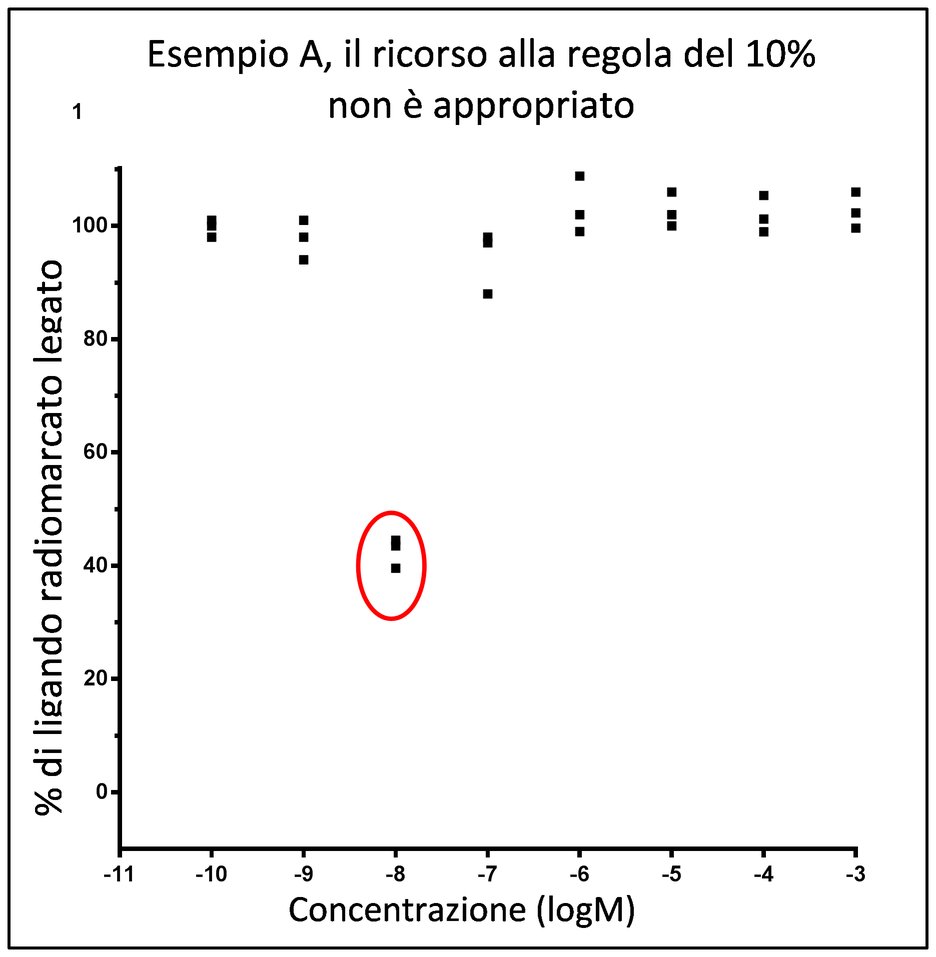
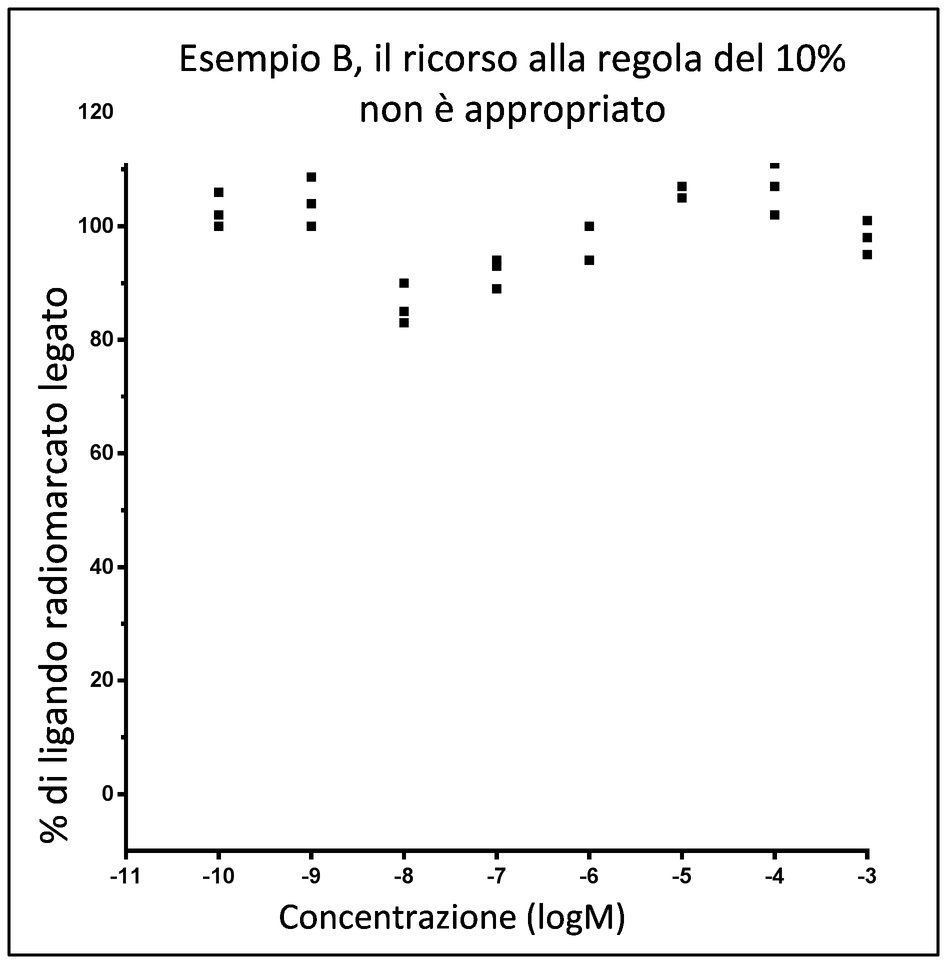
BIBLIOGRAFIA
|
(1)
|
OECD (2015). Guidance Document on Fish Gonadal Histopathology. Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Organisation for Economic Cooperation and Development, Paris.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71 PROVE DI SENSIBILIZZAZIONE CUTANEA IN VITRO RIGUARDANTI L’EVENTO CHIAVE NELL’ATTIVAZIONE DI CELLULE DENDRITICHE NEL MECCANISMO D’AZIONE DEGLI EFFETTI AVVERSI (AOP) PER LA SENSIBILIZZAZIONE CUTANEA
INTRODUZIONE GENERALE
Metodo di prova basato sull’evento chiave nell’attivazione di cellule dendritiche
|
1.
|
Per sensibilizzante cutaneo si intende una sostanza che provoca una reazione allergica a contatto con la pelle, secondo la definizione del Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS) (1) e del regolamento (CE) n. 1272/2008 dell’Unione europea relativo alla classificazione, all’etichettatura e all’imballaggio delle sostanze e delle miscele (CLP) (100). Vi è consenso generale circa le principali fasi del processo biologico di sensibilizzazione cutanea. Le attuali conoscenze relative ai meccanismi chimici e biologici associati alla sensibilizzazione cutanea sono state riassunte nel concetto del meccanismo d’azione degli effetti avversi (AOP, Adverse Outcome Pathway) nell’ambito del programma AOP dell’OCSE (2), che va dall’evento molecolare scatenante fino agli effetti avversi per la salute (dermatite allergica da contatto), passando attraverso le fasi intermedie. In questo caso l’evento molecolare scatenante (cioè il primo evento chiave) è il legame covalente tra sostanze chimiche elettrofile e i centri nucleofili nelle proteine della pelle. Il secondo evento chiave nell’AOP avviene a livello di cheratinociti e comprende risposte infiammatorie e variazioni di espressione genica, associate a specifiche vie di segnalazione intercellulare come le vie dipendenti dall’elemento di risposta antiossidante/elettrofilo (ARE, Antioxidant Response Element). Il terzo evento chiave è l’attivazione di cellule dendritiche (DC), generalmente valutata attraverso l’espressione di specifici marcatori di superficie cellulare, chemochine e citochine. Il quarto evento chiave è l’attivazione e proliferazione dei linfociti T, valutata indirettamente con il test sui linfonodi locali (LLNA) su topi (3).
|
|
2.
|
Il presente metodo di prova è equivalente alla linea guida dell’OCSE per le prove sulle sostanze chimiche n. 442E (2017). Esso descrive i saggi in vitro riguardanti i meccanismi descritti nell’ambito dell’evento chiave nell’attivazione di cellule dendritiche dell’AOP per la sensibilizzazione cutanea (2). Il metodo di prova comprende prove da utilizzare per distinguere i sensibilizzanti dai non sensibilizzanti cutanei, secondo la definizione del sistema UN GHS e della classificazione CLP.
|
|
Nel presente metodo di prova sono descritte le prove seguenti:
|
—
|
test di attivazione della linea cellulare umana - (h-CLAT)
|
|
—
|
test di attivazione della linea cellulare U937 - (U-SENS™)
|
|
—
|
test con il gene reporter dell’interleuchina-8 (metodo di prova IL-8 Luc).
|
|
|
|
3.
|
Le prove facenti parte del presente metodo di prova e la corrispondente linea guida dell’OCSE possono differire per quanto riguarda la procedura utilizzata per generare i dati e i risultati misurati, ma possono essere indifferentemente utilizzate per rispondere alle esigenze dei paesi in materia di risultati sperimentali riguardanti l’evento chiave nell’attivazione di cellule dendritiche nell’AOP di sensibilizzazione cutanea, beneficiando nel contempo del sistema di accettazione reciproca dei dati nel quadro dell’OCSE.
|
Fondamenti e principi delle prove comprese nel metodo di prova basato sull’evento chiave
|
4.
|
Tipicamente, la valutazione della sensibilizzazione cutanea è effettuata su cavie. I metodi classici che utilizzano cavie - il test di massimizzazione su cavie (Guinea Pig Maximisation Test, GPMT) di Magnusson e Kligman e il test di Buehler (metodo di prova B.6) (4) - valutano sia le fasi di induzione che quelle di reazione della sensibilizzazione cutanea. Sono inoltre utilizzati test sui topi - il test LLNA (metodo di prova B.42) (3) e le sue due varianti non radioattive, LLNA: DA (metodo di prova B.50) (5) e LLNA: BrdU-ELISA (metodo di prova B.51) (6) - che riguardano esclusivamente la reazione di induzione, garantendo un vantaggio rispetto ai test su cavie per quanto riguarda il benessere degli animali e la possibilità di ottenere una misurazione obiettiva della fase di induzione della sensibilizzazione cutanea.
|
|
5.
|
Per contribuire alla valutazione del potenziale di sensibilizzazione cutanea delle sostanze chimiche sono stati recentemente adottati metodi di prova in chemico e in vitro di tipo meccanicistico riguardanti il primo evento chiave [metodo di prova B.59; saggio di reattività peptidica diretta (7)] e il secondo evento chiave [metodo di prova B.60; metodo di prova della luciferasi ARE-Nrf2 (8)] dell’AOP di sensibilizzazione cutanea.
|
|
6.
|
Le prove descritte nel presente metodo di prova consentono di quantificare la variazione di espressione dei marcatori di superficie cellulare associati al processo di attivazione di monociti e DC in seguito all’esposizione a sensibilizzanti (ad es. CD54, CD86) o le variazioni di espressione di IL-8, una citochina associata all’attivazione di DC. È stato segnalato che i sensibilizzanti cutanei inducono l’espressione di marcatori di membrana cellulare associati all’attivazione di DC (2), quali CD40, CD54, CD80, CD83 e CD86, nonché di citochine proinfiammatorie quali IL-1β e TNF-α e di varie chemochine, tra cui IL-8 (CXCL8) e CCL3 (9) (10) (11) (12).
|
|
7.
|
Tuttavia, poiché l’attivazione di DC costituisce soltanto uno degli eventi chiave dell’AOP di sensibilizzazione cutanea (2) (13), le informazioni ricavate da prove che misurano unicamente i marcatori dell’attivazione di DC possono non essere sufficienti per stabilire se una sostanza chimica presenta o no un potenziale di sensibilizzazione cutanea. Pertanto i dati ricavati dalle prove descritte nel presente metodo di prova possono contribuire a distinguere tra sensibilizzanti (categoria 1 del GHS dell’ONU/CLP) e non sensibilizzati cutanei se utilizzati nell’ambito di approcci integrati in materia di prove e valutazioni (IATA), in combinazione con altre informazioni complementari ricavate ad esempio da saggi in vitro che prendano in esame altri eventi chiave dell’AOP di sensibilizzazione cutanea nonché da metodi non sperimentali, compresi i metodi read-across utilizzati con sostanze chimiche analoghe (13). Esistono in letteratura esempi di utilizzo di dati ricavati da queste prove nell’ambito di approcci definiti, vale a dire approcci standardizzati per quanto riguarda le fonti di informazione utilizzate e la procedura applicata ai dati per formulare previsioni (13), che possono costituire elementi utili nell’ambito di un approccio IATA.
|
|
8.
|
Le prove descritte nel presente metodo di prova non possono essere utilizzate da sole, né per la classificazione dei sensibilizzanti cutanei nelle sottocategorie 1A e 1B del sistema UN GHS/CLP, per le autorità che applicano queste due sottocategorie facoltative, né per prevedere la potenza di sensibilizzazione in sede di valutazione della sicurezza. Tuttavia, in funzione del quadro normativo applicabile, i risultati positivi ottenuti con questi metodi possono essere usati isolatamente per classificare una sostanza chimica nella categoria 1 del GHS dell’ONU/CLP.
|
|
9.
|
Il termine «sostanza chimica in esame» utilizzato nel presente metodo di prova designa l’oggetto della prova (101) e non si riferisce all’applicabilità delle prove per testare sostanze monocostituente, sostanze multicostituente e/o miscele. Attualmente disponiamo di informazioni limitate quanto all’applicabilità delle prove a sostanze multicostituente e a miscele (14) (15). Le prove sono comunque tecnicamente applicabili alle prove su sostanze multicostituente e miscele. Tuttavia, prima di applicare questo metodo di prova a una miscela per ottenere dati a fini regolamentari, è opportuno chiedersi se, e in caso affermativo perché, i dati ottenuti possono essere ritenuti idonei per i fini regolamentari previsti (102). Tali considerazioni non sono necessarie in presenza di un obbligo normativo di prova sulla miscela. Inoltre, quando si testano sostanze multicostituente o miscele, occorre prestare attenzione alle possibili interferenze dei costituenti citotossici con le risposte osservate.
|
BIBLIOGRAFIA
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Consultabile al seguente indirizzo: https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Consultabile al seguente indirizzo: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
Capitolo B.42 del presente allegato: Test sui linfonodi locali (LLNA).
|
|
(4)
|
Capitolo B.6 del presente allegato: Sensibilizzazione cutanea.
|
|
(5)
|
Capitolo B.50 del presente allegato: Sensibilizzazione cutanea: test sui linfonodi locali (LLNA): DA.
|
|
(6)
|
Capitolo B.51 del presente allegato: Sensibilizzazione cutanea: test sui linfonodi locali (LLNA): BrdU-ELISA.
|
|
(7)
|
Capitolo B.59 del presente allegato: Sensibilizzazione cutanea in chemico: saggio di reattività peptidica diretta (DPRA).
|
|
(8)
|
Capitolo B.60 del presente allegato: Sensibilizzazione cutanea in vitro: metodo di prova della luciferasi ARE-Nrf2.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
Appendice 1
Sensibilizzazione Cutanea in vitro: Test di Attivazione Della Linea Cellulare Umana (h-clat)
CONSIDERAZIONI INIZIALI E LIMITI
|
1.
|
Il metodo h-CLAT consente di quantificare le variazioni di espressione dei marcatori di superficie cellulare associati al processo di attivazione di monociti e cellule dendritiche (DC) (cioè CD86 e CD54) nella linea cellulare di leucemia monocitica umana THP-1 in seguito all'esposizione a sensibilizzanti (1)(2). I livelli misurati di espressione dei marcatori di superficie cellulare CD86 e CD54 sono quindi utilizzati per contribuire a distinguere tra sensibilizzanti e non sensibilizzanti cutanei.
|
|
2.
|
Il metodo h-CLAT è stato oggetto di uno studio di validazione in uno dei laboratori di riferimento dell'Unione europea per le alternative alla sperimentazione animale (European Union Reference Laboratory for Alternatives to Animal Testing - EURL ECVAM) e di una successiva revisione indipendente inter pares sotto la guida del comitato scientifico consultivo dello stesso laboratorio (ESAC). Tenuto conto di tutti i dati disponibili e dei pareri formulati dalle autorità di regolamentazione e dai portatori di interessi, l'EURL ECVAM ha raccomandato l'uso del metodo h-CLAT (3) nell'ambito di una metodologia integrata di tipo IATA per contribuire a distinguere tra sensibilizzanti e non sensibilizzanti cutanei a fini di classificazione ed etichettatura del pericolo. Nella letteratura scientifica figurano esempi dell'uso dei dati h-CLAT in combinazione con altre informazioni (4)(5)(6)(7)(8)(9)(10)(11).
|
|
3.
|
È stato dimostrato che il metodo h-CLAT può essere applicato in laboratori con esperienza nel campo delle tecniche di coltura cellulare e dell'analisi mediante citometria a flusso. Il livello di riproducibilità atteso delle predizioni è dell'ordine dell'80 % a livello intralaboratorio e a livello interlaboratori (3)(12). I risultati dello studio di validazione (13) e di altri studi pubblicati (14) indicano globalmente che, rispetto ai risultati ottenuti con il metodo LLNA, l'accuratezza nella distinzione tra sensibilizzanti (categoria 1 del GHS dell'ONU/CLP) e non sensibilizzanti cutanei è pari all'85 % (N = 142) per una sensibilità del 93 % (94/101) e una specificità del 66 % (27/41) [sulla base di una nuova analisi dell'EURL ECVAM (12) che ha tenuto conto di tutti i dati esistenti ma non dei risultati negativi per le sostanze chimiche con un Log Kow superiore a 3,5, come descritto al paragrafo 4]. È probabile che i falsi negativi nelle predizioni formulate con il metodo h-CLAT riguardino piuttosto le sostanze chimiche con una potenza di sensibilizzazione cutanea da bassa a moderata (sottocategoria 1B del GHS dell'ONU/CLP) rispetto alle sostanze chimiche con una potenza di sensibilizzazione cutanea elevata (sottocategoria 1A del GHS dell'ONU/CLP) (4)(13)(15). Queste informazioni, nel loro insieme, indicano l'utilità del metodo h-CLAT per l'identificazione dei rischi di sensibilizzazione cutanea. Tuttavia, i valori di accuratezza forniti in questa sede per il metodo h-CLAT utilizzato isolatamente sono puramente indicativi, in quanto la prova dovrebbe essere considerata in combinazione con altre fonti di informazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei paragrafi 7 e 8 dell'Introduzione generale. Inoltre, nel valutare i metodi di studio della sensibilizzazione cutanea che non utilizzano la sperimentazione animale si deve tenere presente che l'LLNA, come pure altre prove che utilizzano la sperimentazione animale, può non riflettere pienamente la situazione per l'uomo.
|
|
4.
|
I dati attualmente disponibili indicano che il metodo h-CLAT è applicabile alle sostanze chimiche in esame relative a diversi gruppi funzionali organici, meccanismi di reazione, potenze di sensibilizzazione cutanea (determinate negli studi in vivo) e proprietà fisico-chimiche (3)(14)(15). Il metodo h-CLAT è applicabile alle sostanze chimiche solubili o che formano una dispersione stabile (colloide o sospensione in cui la sostanza chimica in esame non si deposita né si separa dal solvente/mezzo disperdente formando più fasi) in un solvente/disperdente idoneo (cfr. il paragrafo 14). Le sostanze chimiche con un Log Kow superiore a 3,5 tendono a produrre risultati falsi negativi (14). Pertanto non si deve tener conto dei risultati negativi ottenuti da sostanze chimiche con un Log Kow superiore a 3,5. Tuttavia, i risultati positivi ottenuti da sostanze chimiche che presentano un Log Kow superiore a 3,5 possono comunque essere utilizzati nel processo di identificazione della sostanza chimica come sensibilizzante cutaneo. Inoltre, a causa della capacità metabolica limitata della linea cellulare utilizzata (16) e a causa delle condizioni sperimentali, anche i proapteni (sostanze che richiedono un'attivazione enzimatica, ad esempio mediante enzimi P450) e i preapteni (sostanze attivate mediante ossidazione), in particolare quelli a ossidazione lenta, possono dare risultati negativi con il metodo h-CLAT (15). Le sostanze chimiche fluorescenti possono essere valutate con il metodo h-CLAT (17); tuttavia le sostanze chimiche fortemente fluorescenti che emettono alla stessa lunghezza d'onda dell'isotiocianato di fluorescina (FITC) o dello ioduro di propidio (PI) interferiscono con la rilevazione mediante citometria a flusso e pertanto non possono essere correttamente valutate mediante anticorpi coniugati con FITC o PI. In questo caso si può fare ricorso, rispettivamente, ad altri anticorpi marcati con fluorocromo o ad altri marcatori di citotossicità, purché si possa dimostrare, ad esempio testando le sostanze di riferimento indicate nell'appendice 1-2, che essi producono risultati simili agli anticorpi marcati con FITC (cfr. il paragrafo 24) o PI (cfr. il paragrafo 18). Alla luce di quanto precede, i risultati negativi andranno interpretati nel contesto dei limiti indicati e in combinazione con altre fonti di informazione nell'ambito di un approccio IATA. Nel caso in cui si dimostri che il metodo di prova h-CLAT non è applicabile ad altre categorie specifiche di sostanze chimiche in esame, è opportuno evitare di utilizzarlo per tali categorie.
|
|
5.
|
Come già precisato, il metodo h-CLAT aiuta a distinguere tra sensibilizzanti e non sensibilizzanti cutanei. Tuttavia, esso può contribuire anche alla valutazione della potenza di sensibilizzazione cutanea (4)(5)(9) se utilizzato nell'ambito di approcci integrati quali IATA. Sono tuttavia necessarie ulteriori ricerche, di preferenza basate su dati relativi all'uomo, per determinare in che modo i risultati del metodo h-CLAT possano contribuire alla valutazione della potenza di sensibilizzazione.
|
|
6.
|
Le definizioni figurano nell'appendice 1.1.
|
PRINCIPIO DELLA PROVA
|
7.
|
Il metodo h-CLAT è una prova in vitro che consente di quantificare le variazioni di espressione dei marcatori di superficie cellulare (CD86 e CD54) in una linea cellulare di leucemia monocitica umana (cellule THP-1) dopo 24 ore di esposizione alla sostanza chimica in esame. Tali molecole superficiali sono marcatori tipici dell'attivazione dei monociti THP-1 e possono mimare l'attivazione di DC, che svolge un ruolo cruciale nel priming dei linfociti T. Le variazioni di espressione dei marcatori di superficie sono misurate mediante citometria a flusso dopo colorazione cellulare con anticorpi marcati con fluorocromo. In parallelo è inoltre effettuata una misurazione della citotossicità per stabilire se la sovraespressione del marcatore di superficie avviene in presenza di concentrazioni sub-citotossiche. L'intensità di fluorescenza relativa dei marcatori di superficie rispetto al controllo con solvente/disperdente è calcolata e utilizzata in un modello predittivo (cfr. il paragrafo 26) ai fini della distinzione tra sensibilizzanti e non sensibilizzanti cutanei.
|
DIMOSTRAZIONE DELLA COMPETENZA TECNICA
|
8.
|
Prima di utilizzare come test di routine la prova descritta nella presente appendice relativa al metodo di prova B.71, i laboratori devono dimostrare le loro competenze tecniche utilizzando le 10 sostanze di riferimento elencate nell'appendice 1.2. Inoltre gli utilizzatori del metodo devono mantenere una banca dati storica contenente i dati ottenuti con i controlli di reattività (cfr. il paragrafo 11) nonché con i controlli positivi e con solvente/disperdente (cfr. i paragrafi 20-22) e utilizzare tali dati per confermare la persistenza nel tempo della riproducibilità della prova nel loro laboratorio.
|
PROCEDURA
|
9.
|
La presente prova si basa sul protocollo h-CLAT n. 158 del Servizio dati sui metodi alternativi alla sperimentazione animale (DataBase service on ALternative Methods to animal experimentation - DB-ALM) (18) utilizzato per lo studio di validazione coordinato dal laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale (EURL ECVAM). Si raccomanda di utilizzare questo protocollo nell'applicazione e nell'impiego del metodo h-CLAT in laboratorio. Di seguito è fornita una descrizione degli elementi e delle procedure principali del metodo h-CLAT, che comprende due fasi: la prova di determinazione della dose e la misurazione dell'espressione di CD86/CD54.
|
Preparazione delle cellule
|
10.
|
Per l'esecuzione del metodo h-CLAT si utilizza la linea cellulare di leucemia monocitica umana (THP-1). Si raccomanda che le cellule (TIB-202™) provengano da una banca di cellule riconosciuta, quale la American Type Culture Collection.
|
|
11.
|
Le cellule THP-1 sono coltivate a 37oC in atmosfera umidificata al 5 % di CO2, su mezzo di coltura RPMI-1640 integrato da 10 % di siero fetale bovino (FBS), 0,05 mM di 2-mercaptoetanolo, 100 unità/ml di penicillina e 100 μg/ml di streptomicina. L'uso di penicillina e streptomicina nel mezzo di coltura può essere evitato. In tal caso, tuttavia, gli utilizzatori devono verificare che l'assenza di antibiotici nel mezzo di coltura non incida sui risultati, ad esempio testando le sostanze di riferimento indicate nell'appendice 1.2. In ogni caso, per ridurre al minimo il rischio di contaminazione si dovranno seguire buone prassi di coltura cellulare, a prescindere dalla presenza o no di antibiotici nel mezzo di coltura. Le cellule THP-1 sono regolarmente inoculate ogni 2-3 giorni a una densità compresa tra 0,1 e 0,2 × 106 cellule/ml e vanno mantenute a densità comprese tra 0,1 e 1,0 × 106 cellule/ml. Prima di essere utilizzate per la prova, le cellule devono essere qualificate mediante un controllo di reattività da realizzarsi due settimane dopo lo scongelamento mediante 2,4-dinitroclorobenzene (DNCB) (n. CAS 97-00-7, purezza ≥ 99 %) e solfato di nichel (NiSO4) (n. CAS 10101-97-0, purezza ≥ 99 %) come controlli positivi e acido lattico (LA) (n. CAS 50-21-5, purezza ≥ 85 %) come controllo negativo. Sia il DNCB che il NiSO4 devono produrre una risposta positiva per entrambi i marcatori di superficie cellulare CD86 e CD54; LA deve produrre una risposta negativa per entrambi i marcatori di superficie cellulare CD86 e CD54. Possono essere utilizzate per la prova soltanto le cellule che superano il controllo di reattività. Le cellule possono essere moltiplicate fino a due mesi dopo lo scongelamento senza superare 30 passaggi. Il controllo di reattività va effettuato secondo le procedure descritte ai paragrafi 20-24.
|
|
12.
|
Per la prova, le cellule THP-1 sono inoculate a una densità di 0,1 × 106 cellule/ml o di 0,2 × 106 cellule/ml e precoltivate in fiasche di coltura per 72 o 48 ore rispettivamente. È importante che la densità cellulare nella fiasca di coltura subito dopo il periodo di pre-coltura sia quanto più possibile costante in ogni esperimento (grazie al ricorso a una delle due condizioni di pre-coltura descritte sopra). Infatti, la densità cellulare nella fiasca di coltura subito dopo la pre-coltura può influire sull'espressione di CD86/CD54 indotta da allergeni (19). Il giorno della prova le cellule raccolte dalla fiasca di coltura sono rimesse in sospensione in un nuovo mezzo di coltura a una densità di 2 × 106 cellule/ml. Quindi le cellule sono ripartite su una piastra a fondo piatto a 24 pozzetti (500 μl, 1 × 106 cellule/pozzetto) o su una piastra a fondo piatto a 96 pozzetti (80 μl, 1,6 × 105 cellule/pozzetto).
|
Prova di determinazione della dose
|
13.
|
Una prova di determinazione della dose è realizzata per determinare il valore CV75, vale a dire la concentrazione della sostanza chimica in esame che produce una vitalità cellulare (CV) del 75 % rispetto al controllo con solvente/disperdente. Il valore CV75 è utilizzato per determinare la concentrazione delle sostanze chimiche in esame per la misurazione dell'espressione di CD86/CD54 (cfr. i paragrafi 20-24).
|
Preparazione delle sostanze chimiche in esame e delle sostanze di controllo
|
14.
|
Le sostanze chimiche in esame e le sostanze di controllo vengono preparate il giorno della prova. Nel metodo h-CLAT le sostanze chimiche in esame sono disciolte o disperse stabilmente (cfr. anche il paragrafo 4) utilizzando di preferenza, come solvente/disperdente, una soluzione salina o un mezzo o, come seconda opzione se la sostanza chimica in esame non è solubile o non forma una dispersione stabile nei due solventi/mezzi disperdenti precedenti, dimetilsulfossido (DMSO, purezza ≥ 99 %), fino a raggiungere una concentrazione finale di 100 mg/ml (nella soluzione salina o nel mezzo) o di 500 mg/ml (nel DMSO). È possibile utilizzare solventi/mezzi disperdenti diversi da quelli descritti sopra fornendo una sufficiente giustificazione scientifica. Si deve tenere conto della stabilità della sostanza chimica in esame nel solvente/disperdente finale.
|
|
15.
|
A partire dalle soluzioni madre delle sostanze chimiche in esame (100 mg/ml nella soluzione salina o nel mezzo o 500 mg/ml nel DMSO) si procede alle seguenti diluizioni:
|
—
|
se il solvente/disperdente è una soluzione salina o un mezzo: vengono preparate otto soluzioni madre (otto concentrazioni) mediante diluizioni doppie in serie realizzate con l'opportuno solvente/disperdente. Queste soluzioni madre sono poi nuovamente diluite di un fattore 50 nel mezzo di coltura (soluzioni di lavoro). Se la concentrazione finale massima di 1 000 μg/ml nella piastra non è tossica occorre determinare nuovamente la concentrazione massima mediante un altro test di citotossicità. La concentrazione finale nella piastra non deve superare 5 000 μg/ml per le sostanze chimiche in esame disciolte o disperse stabilmente in una soluzione salina o un mezzo.
|
|
—
|
Se il solvente/disperdente è il DMSO: vengono preparate otto soluzioni madre (otto concentrazioni) mediante diluizioni doppie in serie realizzate con l'opportuno solvente/disperdente. Queste soluzioni madre sono poi nuovamente diluite di un fattore 250 nel mezzo di coltura (soluzioni di lavoro). La concentrazione finale nella piastra non deve superare 1 000 μg/ml, anche se tale concentrazione non è tossica.
|
Le soluzioni di lavoro sono infine utilizzate per l'esposizione aggiungendo un volume uguale di soluzione di lavoro al volume della sospensione di cellule THP-1 nella piastra (cfr. anche il paragrafo 17) per ottenere un'ulteriore diluizione doppia (generalmente, le concentrazioni finali nella piastra vanno da 7,81 a 1 000 μg/ml).
|
|
16.
|
Il controllo con solvente/disperdente utilizzato nel metodo h-CLAT è il mezzo di coltura [per le sostanze chimiche in esame solubilizzate o disperse stabilmente (cfr. il paragrafo 4) nel mezzo o nella soluzione salina] o il DMSO (per le sostanze chimiche in esame solubilizzate o disperse stabilmente nel DMSO) ed è testato a una concentrazione finale unica nella piastra di 0,2 %. Esso è sottoposto alla stessa diluizione descritta per le soluzioni di lavoro al paragrafo 15.
|
Applicazione delle sostanze chimiche in esame e delle sostanze di controllo
|
17.
|
Il mezzo di coltura o le soluzioni di lavoro descritti ai paragrafi 15 e 16 sono mescolati nella proporzione 1:1 (v/v) con le sospensioni cellulari preparate nella piastra a fondo piatto a 24 o 96 pozzetti (cfr. il paragrafo 12). Le piastre trattate sono quindi messe in incubazione per 24 ± 0,5 ore a una temperatura di 37oC con il 5 % di CO2. Si avrà cura di evitare l'evaporazione delle sostanze chimiche volatili in esame nonché la contaminazione incrociata tra i pozzetti da parte delle sostanze stesse, ad esempio sigillando la piastra prima dell'incubazione con le sostanze chimiche in esame (20).
|
Colorazione con ioduro di propidio (PI)
|
18.
|
Dopo 24 ± 0,5 ore di esposizione le cellule sono trasferite in provette e raccolte mediante centrifugazione. I supernatanti vengono scartati e le cellule rimanenti sono rimesse in sospensione in 200 μl (in caso di piastra a 96 pozzetti) o 600 μl (in caso di piastra a 24 pozzetti) di tampone fosfato isotonico contenente lo 0,1 % di seroalbumina bovina (tampone di colorazione). 200 μl di sospensione cellulare sono trasferiti in una piastra a fondo rotondo a 96 pozzetti (in caso di piastra a 96 pozzetti) o in una microprovetta (in caso di piastra a 24 pozzetti) e lavati due volte con 200 μl (in caso di piastra a 96 pozzetti) o 600 μl (in caso di piastra a 24 pozzetti) di tampone di colorazione. Infine, le cellule sono rimesse in sospensione nel tampone di colorazione (ad es. 400 μl) ed è aggiunta una soluzione di PI (ad es. 20 μl) (per ottenere, ad esempio, una concentrazione finale di PI di 0,625 μg/ml). È possibile utilizzare altri marcatori di citotossicità, tra cui la 7-aminoactinomicina D (7-AAD) e il blu di tripano, purché si dimostri che tali coloranti alternativi producono risultati simili al PI, ad esempio testando le sostanze di riferimento di cui all'appendice 1.2.
|
Misurazione della citotossicità mediante citometria a flusso e stima del valore CV75
|
19.
|
L'assorbimento di PI è analizzato mediante citometria a flusso sul canale di acquisizione FL-3. Vengono complessivamente raccolte 10 000 cellule vive (negative al PI). La vitalità cellulare può essere calcolata dal programma di analisi del citometro mediante l'equazione riportata di seguito. Se la vitalità cellulare è bassa si dovrebbero raccogliere fino a 30 000 cellule, comprese le cellule morte. Un'opzione alternativa consiste nell'acquisire i dati per un minuto dall'inizio dell'analisi.

Il valore CV75 (cfr. il paragrafo 13), cioè una concentrazione che produce il 75 % di sopravvivenza di cellule THP-1 (25 % di citotossicità), è calcolato per interpolazione log-lineare mediante la seguente equazione:
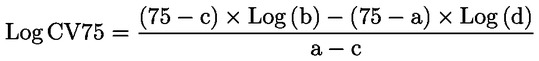
in cui:
a è il valore minimo di vitalità cellulare superiore a 75 %
c è il valore massimo di vitalità cellulare inferiore a 75 %
b e d sono le concentrazioni che producono rispettivamente i valori di vitalità cellulare a e c
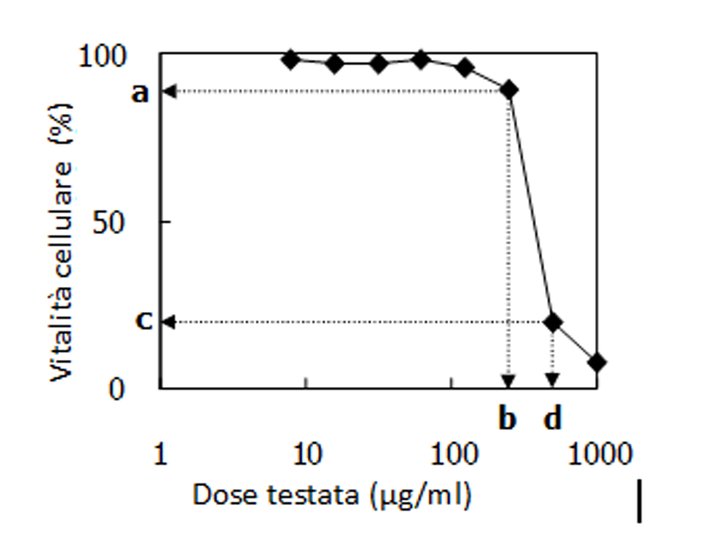
È possibile utilizzare altri metodi per calcolare il valore CV75, purché sia provato che ciò non incide sui risultati (ad es. testando le sostanze di riferimento).
|
Misurazione dell'espressione di CD86/CD54
Preparazione delle sostanze chimiche in esame e delle sostanze di controllo
|
20.
|
È utilizzato il solvente/disperdente appropriato (soluzione salina, mezzo o DMSO; cfr. il paragrafo 14) per dissolvere le sostanze chimiche in esame o creare una dispersione stabile. Le sostanze chimiche in esame sono dapprima diluite a una concentrazione pari a 100 volte (soluzione salina o mezzo) o 500 volte (DMSO) il valore CV75 × 1,2 determinato nella prova di determinazione della dose (cfr. il paragrafo 19). Se non è possibile determinare il valore CV75 (cioè se la tossicità osservata nella prova di determinazione della dose non è sufficiente), si utilizzerà come concentrazione inziale la concentrazione solubile o in dispersione stabile più elevata della sostanza chimica in esame preparata con ciascun solvente/disperdente. Si osservi che la concentrazione finale nella piastra non deve superare 5 000 μg/ml (in caso di soluzione salina o mezzo) o 1 000 μg/ml (in caso di DMSO). Vengono quindi effettuate diluizioni in serie di un fattore 1,2 utilizzando il solvente/disperdente corrispondente per ottenere le soluzioni madre [otto concentrazioni comprese tra 100×1,2 × CV75 e 100×0,335 × CV75 (soluzione salina o mezzo) o tra 500×1,2 × CV75 e 500×0,335 × CV75 (DMSO)] che saranno testate con il metodo h-CLAT (per un esempio di schema di dosaggio si veda il protocollo DB-ALM n. 158). Le soluzioni madre sono quindi ulteriormente diluite di un fattore 50 (soluzione salina o mezzo) o 250 (DMSO) nel mezzo di coltura (soluzioni di lavoro). Queste soluzioni di lavoro sono infine utilizzate per l'esposizione, dopo una diluizione finale di fattore 2 nella piastra. Se i risultati non soddisfano i criteri di accettabilità descritti ai paragrafi 29 e 30 per la vitalità cellulare, la prova di determinazione della dose può essere ripetuta per calcolare un valore CV75 più preciso. Si osservi che per la misurazione dell'espressione di CD86/CD54 possono essere utilizzate unicamente piastre a 24 pozzetti.
|
|
21.
|
Il controllo con solvente/disperdente è preparato come descritto al paragrafo 16. Il controllo positivo utilizzato nel metodo h-CLAT è il DNCB (cfr. il paragrafo 11), di cui si preparano soluzioni madre in DMSO che sono poi diluite come descritto per le soluzioni madre al paragrafo 20. Il DNCB deve essere utilizzato come controllo positivo per la misurazione dell'espressione di CD86/CD54 a una concentrazione finale unica nella piastra (generalmente 4,0 μg/ml). Per ottenere una concentrazione di 4,0 μg/ml di DNCB nella piastra si prepara una soluzione madre di 2 mg/ml di DNCB in DMSO e la si diluisce ulteriormente di un fattore 250 nel mezzo di coltura fino ad ottenere una soluzione di lavoro di 8 μg/ml. In alternativa è possibile utilizzare, come concentrazione del controllo positivo, il valore CV75 del DNCB determinato in ciascun centro di prova. Altri controlli positivi adeguati possono essere utilizzati in caso di disponibilità di dati storici da cui ricavare criteri di accettabilità comparabili per la batteria di prove. Per i controlli positivi la concentrazione finale nella piastra non deve superare 5 000 μg/ml (in caso di soluzione salina o mezzo) o 1 000 μg/ml (in caso di DMSO). I criteri di accettabilità della batteria di prove sono identici a quelli descritti per la sostanza chimica in esame (cfr. il paragrafo 29), ad eccezione dell'ultimo criterio di accettabilità in quanto il controllo positivo è testato a una concentrazione unica.
|
Applicazione delle sostanze chimiche in esame e delle sostanze di controllo
|
22.
|
Per ogni sostanza chimica in esame e sostanza di controllo è necessario un esperimento al fine di ottenere una predizione. Ogni esperimento consiste in almeno due batterie di prove indipendenti per la misurazione dell'espressione di CD86/CD54 (cfr. i paragrafi 26-28). Le batterie di prove indipendenti hanno luogo in giorni diversi, o nello stesso giorno a condizione che, per ciascuna batteria di prove: a) siano preparate soluzioni madre e soluzioni di lavoro nuove e indipendenti della sostanza chimica in esame e delle soluzioni di anticorpi e b) siano utilizzate cellule raccolte in modo indipendente (cioè provenienti da matracci diversi); tuttavia, le cellule possono provenire dallo stesso passaggio. Le sostanze chimiche in esame e le sostanze di controllo preparate come soluzioni di lavoro (500 μl) sono miscelate con 500 μl di cellule in sospensione (1x106 cellule) in proporzione di 1:1 e le cellule sono messe in incubazione per 24 ± 0,5 ore come descritto ai paragrafi 20 e 21. Per ogni prova è sufficiente un'unica replica per ciascuna concentrazione della sostanza chimica in esame e della sostanza di controllo, in quanto la predizione è ricavata da almeno due batterie di prove indipendenti.
|
Colorazione cellulare e analisi
|
23.
|
Dopo 24 ± 0,5 ore di esposizione le cellule sono trasferite dalla piastra a 24 pozzetti in provette, raccolte per centrifugazione e successivamente lavate due volte con un 1 ml di tampone di colorazione (se necessario possono essere eseguite ulteriori tappe di lavaggio). Dopo il lavaggio le cellule vengono bloccate con 600 μl di soluzione bloccante [tampone di colorazione contenente lo 0,01 % (p/v) di globulina (frazione di Cohn II, III, umana; SIGMA, #G2388-10G o equivalente)] e messe in incubazione a 4oC per 15 minuti. Una volta bloccate le cellule vengono ripartite in tre aliquote da 180 μl su una piastra a fondo rotondo a 96 pozzetti o in una microprovetta.
|
|
24.
|
Dopo la centrifugazione le cellule sono sottoposte a colorazione con 50 μl di anticorpi marcati con FITC anti-CD86, anti-CD54 o di anticorpi murini IgG1 (isotipo) a 4oC per 30 minuti. Gli anticorpi descritti nel protocollo h-CLAT n. 158 della banca dati DB-ALM (18) devono essere diluiti in un tampone di colorazione in proporzione di 3:25 v/v [per CD86 (BD-PharMingen, #555657; Clone: Fun-1)] o di 3:50 v/v [per CD54 (DAKO, #F7143; Clone: 6.5B5) e IgG1 (DAKO, #X0927)]. Tali fattori di diluizione degli anticorpi sono stati definiti dagli sviluppatori del metodo di prova come i fattori che offrono il migliore rapporto segnale-disturbo. Sulla base dell'esperienza degli sviluppatori del metodo di prova, l'intensità di fluorescenza dei vari lotti di anticorpi è generalmente costante. Tuttavia gli utilizzatori possono decidere di effettuare la titolazione degli anticorpi nelle loro condizioni di laboratorio per definire le concentrazioni più adatte all'uso. È possibile avvalersi di altri anticorpi marcati con fluorocromo anti-CD86 e/o anti-CD54 purché si possa dimostrare che essi producono risultati simili agli anticorpi coniugati con FITC, ad esempio testando le sostanze di riferimento di cui all'appendice 1.2. Va osservato che un cambiamento di clone o di fornitore di anticorpi quale descritto nel protocollo h-CLAT n. 158 della banca dati DB-ALM (18) può incidere sui risultati. Dopo essere state lavate due o più volte con 150 μl di tampone di colorazione, le cellule sono rimesse in sospensione in un campione di colorazione (ad es. 400 μl) cui è aggiunta la soluzione di PI (ad es. 20 μl per ottenere una concentrazione finale di 0,625 μg/ml) o di un altro marcatore di tossicità (cfr. il paragrafo 18). I livelli di espressione di CD86 e CD54 e la vitalità cellulare sono analizzati mediante citometria a flusso.
|
DATI E RELAZIONI
Valutazione dei dati
|
25.
|
Il livello di espressione di CD86 e CD54 è analizzato mediante citometria a flusso sul canale di acquisizione FL-1. Sulla base della media geometrica dell'intensità di fluorescenza (MFI), l'intensità di fluorescenza relativa (RFI) di CD86 e CD54 per le cellule di controllo positivo (ctrl) e per quelle trattate con la sostanza chimica è calcolata mediante la seguente equazione:

È inoltre calcolata la vitalità cellulare delle cellule di controllo isotipo (ctrl) [colorate con anticorpi murini IgG1 (isotipo)] mediante l'equazione descritta al paragrafo 19.
|
Modello predittivo
|
26.
|
Per la misurazione dell'espressione di CD86/CD54 ciascuna sostanza chimica è testata in almeno due prove indipendenti per ottenere una predizione unica (POSITIVA o NEGATIVA). Una predizione h-CLAT è considerata POSITIVA se almeno una delle condizioni elencate di seguito è soddisfatta in 2 prove indipendenti su 2 o in almeno 2 prove indipendenti su 3; in caso contrario, la predizione h-CLAT è considerata NEGATIVA (figura 1):
|
—
|
l'RFI di CD86 è pari o superiore a 150 % in tutte le concentrazioni testate (con una vitalità cellulare ≥ 50 %);
|
|
—
|
l'RFI di CD54 è pari o superiore a 200 % in tutte le concentrazioni testate (con una vitalità cellulare ≥ 50 %).
|
|
|
27.
|
Sulla base di quanto precede, se le prime due batterie di prove sono entrambe positive per CD86 e/o sono entrambe positive per CD54, la predizione h-CLAT è considerata POSITIVA e non è necessario eseguire una terza batteria di prove. Analogamente, se le prime due batterie di prove sono negative per entrambi i marcatori, la predizione h-CLAT è considerata NEGATIVA (tenuto conto di quanto disposto al paragrafo 30) e non è necessario eseguire una terza batteria di prove. Tuttavia, se i risultati delle prime due batterie di prove differiscono per almeno uno dei marcatori (CD54 o CD86), è necessario effettuare una terza batteria di prove e la predizione finale sarà basata sul risultato ottenuto nella maggioranza delle tre batterie di prove individuali (cioè 2 su 3). A questo proposito va osservato che se vengono svolte due batterie di prove indipendenti e una di queste è positiva soltanto per CD86 (di seguito, P1) e l'altra è positiva soltanto per CD54 (di seguito, P2), è necessario procedere a una terza batteria di prove. Se questa terza batteria di prove è negativa per entrambi i marcatori (di seguito, N), la predizione h-CLAT è considerata NEGATIVA. D'altro canto, se la terza batteria di prove è positiva per uno dei marcatori (P1 o P2) o per entrambi i marcatori (di seguito, P12), la predizione h-CLAT è considerata POSITIVA.
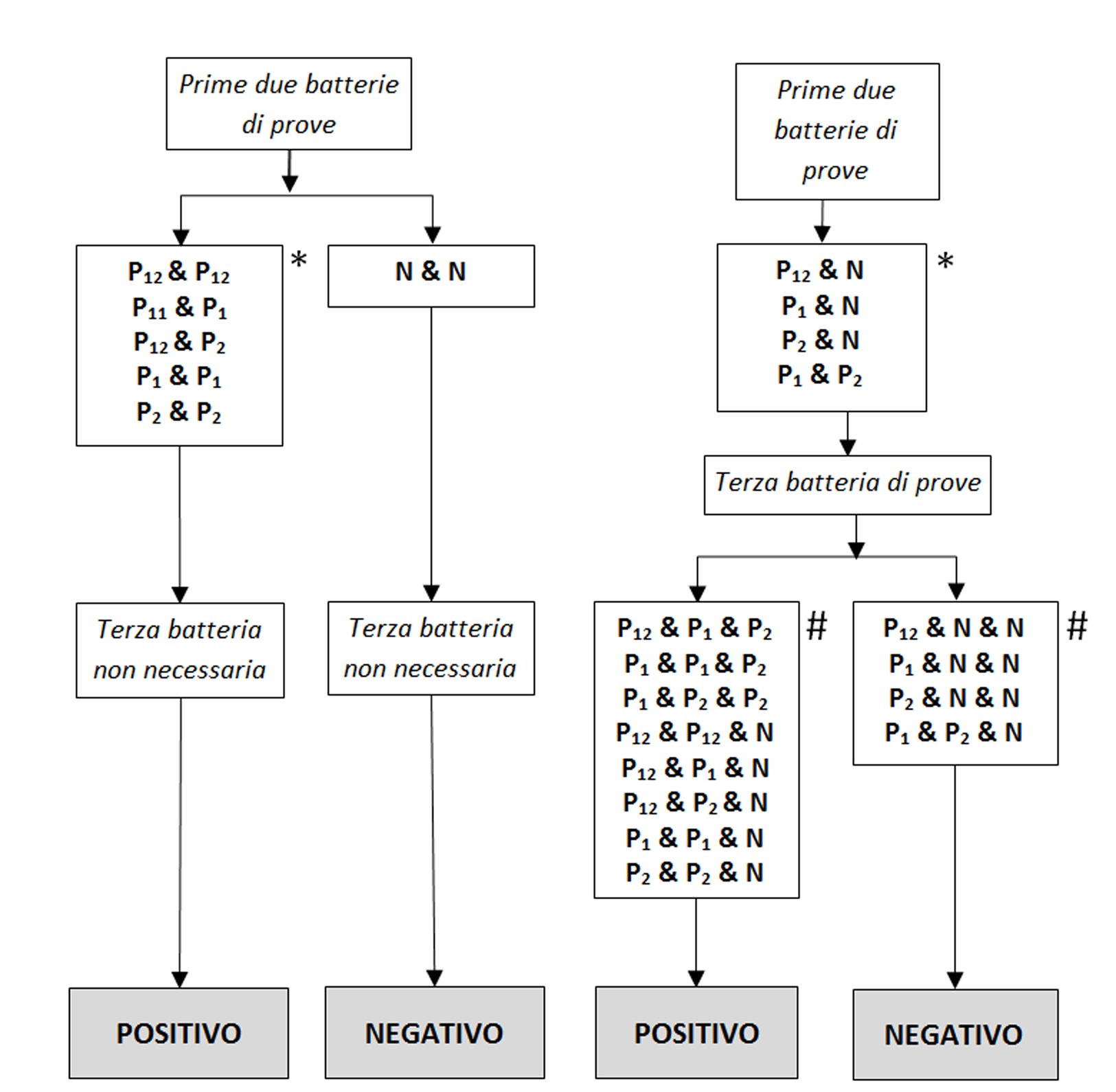
Figura 1: Modello predittivo utilizzato nel metodo di prova h-CLAT. Una predizione h-CLAT va considerata nell'ambito di un approccio IATA e in conformità alle disposizioni dei paragrafi 7 e 8 dell'Introduzione generale.
P1: prova positiva soltanto per CD86; P2; prova positiva soltanto per CD54; P12: prova positiva sia per CD86 che per CD54; N: prova non positiva per CD86 né per CD54;
* I riquadri mostrano le combinazioni pertinenti di risultati delle prime due batterie di prove, a prescindere dall'ordine in cui possono essere ottenuti.
# I riquadri mostrano le combinazioni pertinenti di risultati delle prime tre batterie di prove sulla base dei risultati ottenuti nelle prime due batterie di prove (riportati nel riquadro che precede), ma non rispecchiano l'ordine in cui possono essere ottenuti.
|
|
28.
|
Per le sostanze chimiche in esame per le quali il metodo h-CLAT ha fornito una predizione POSITIVA possono essere eventualmente determinati due valori di concentrazione efficace (EC) (EC150 per CD86 e EC200 per CD54), vale a dire la concentrazione alla quale le sostanze chimiche in esame producono un'RFI di 150 o 200. Tali valori EC potrebbero contribuire alla valutazione della potenza di sensibilizzazione cutanea (9) se utilizzati nell'ambito di approcci integrati quali IATA (4) (5) (6) (7) (8). Essi possono essere calcolati mediante le seguenti equazioni:
EC 150 (per CD86) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (per CD86) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
in cui
Aconc è la concentrazione più bassa in μg/ml con RFI < 150 (CD86) o 200 (CD54)
Bconc è la concentrazione più elevata in μg/ml con RFI > 150 (CD86) o 200 (CD54)
ARFI è l'RFI alla concentrazione più bassa in μg/ml con RFI < 150 (CD86) o 200 (CD54)
BRFI è l'RFI alla concentrazione più elevata in μg/ml con RFI > 150 (CD86) o 200 (CD54)
Per determinare con maggior precisione i valori EC150 e EC200 possono essere necessarie tre prove indipendenti di misurazione dell'espressione di CD86/CD54. I valori finali EC150 e EC200 sono quindi determinati come mediana delle EC calcolate sulla base delle tre batterie di prove indipendenti. Se solo due delle tre batterie di prove indipendenti soddisfano i criteri di positività (cfr. i paragrafi 26-27) viene adottato il valore EC150 o EC200 più elevato tra i due valori calcolati.
|
Criteri di accettabilità
|
29.
|
I seguenti criteri di accettabilità devono essere soddisfatti quando si utilizza il metodo di prova h-CLAT (22) (27).
|
—
|
I valori di vitalità cellulare dei controlli con mezzo e con solvente/disperdente devono essere superiori a 90 %.
|
|
—
|
Nel controllo con solvente/disperdente i valori RFI di CD86 e CD54 non devono superare i criteri di positività (CD86 RFI ≥ 150 % e CD54 RFI ≥ 200 %). I valori RFI del controllo con solvente/disperdente sono calcolati mediante la formula descritta al paragrafo 25 [sostituendo «MFI della sostanza chimica» con «MFI del solvente/disperdente» e «MFI del solvente/disperdentē» con «MFI del controllo (mezzo)»].
|
|
—
|
Per entrambi i controlli (mezzo e solvente/disperdente) il rapporto MFI CD86-controllo isotipo e MFI CD54-controllo isotipo deve essere > 105 %.
|
|
—
|
Nel controllo positivo (DNCB) i valori RFI di CD86 e CD54 devono soddisfare i criteri di positività (CD86 RFI ≥ 150 e CD54 RFI ≥ 200) e la vitalità cellulare deve essere superiore a 50 %.
|
|
—
|
Per la sostanza chimica in esame la vitalità cellulare deve essere superiore a 50 % in almeno quattro concentrazioni testate in ciascuna batteria di prove.
|
|
|
30.
|
Un risultato negativo è accettabile soltanto per le sostanze chimiche in esame che presentano una vitalità cellulare inferiore a 90 % alla massima concentrazione testata (cioè 1,2 × CV75 in base allo schema di diluizione in serie descritto al paragrafo 20). Se la vitalità cellulare a 1,2 × CV75 è pari o superiore a 90 % il risultato negativo non deve essere preso in considerazione. In tal caso si raccomanda di cercare di migliorare la scelta delle dosi ripetendo la determinazione del valore CV75. Va osservato che ove si utilizzi una concentrazione di 5 000 μg/ml in soluzione salina (o mezzo o altri solventi/disperdenti), una concentrazione di 1 000 μg/ml in DMSO o la concentrazione massima di solubilità come concentrazione massima di prova di una sostanza chimica in esame, un risultato negativo è accettabile anche in presenza di una vitalità cellulare superiore a 90 %.
|
Relazione sulla prova
|
31.
|
La relazione sulla prova comprende le informazioni riportate di seguito.
|
Sostanza chimica in esame
|
Sostanza monocostituente:
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, Log Kow, idrosolubilità, solubilità nel DMSO, peso molecolare e proprietà fisico-chimiche pertinenti aggiuntive, a seconda dei dati disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
Sostanza multicostituente, UVCB o miscela:
|
caratterizzazione, nella misura del possibile, ad esempio attraverso l'identità chimica (cfr. sopra), la purezza, le proporzioni quantitative e le proprietà fisico-chimiche pertinenti (cfr. sopra) dei costituenti, secondo i dati disponibili;
|
|
aspetto fisico, idrosolubilità, solubilità nel DMSO e proprietà fisico-chimiche pertinenti aggiuntive, a seconda dei dati disponibili;
|
|
peso molecolare o peso molecolare apparente nel caso di miscele/polimeri di composizione nota o altre informazioni pertinenti per la realizzazione dello studio;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
|
Controlli
|
Controllo positivo
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, Log Kow, idrosolubilità, solubilità nel DMSO, peso molecolare e proprietà fisico-chimiche pertinenti aggiuntive, se del caso e a seconda dei dati disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
riferimento ai dati storici relativi ai controlli positivi che dimostrano la conformità ai criteri di accettabilità, se del caso.
|
|
|
|
Controllo negativo e controllo con solvente/disperdente
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
aspetto fisico, peso molecolare e altre proprietà fisico-chimiche pertinenti, nel caso siano utilizzati solventi/mezzi disperdenti diversi da quelli menzionati nelle linee guida e se disponibili;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
Condizioni della prova
|
nome e indirizzo dello sponsor, dell'infrastruttura utilizzata per la prova e del responsabile dello studio;
|
|
descrizione della prova utilizzata
|
|
linea cellulare utilizzata, condizioni di conservazione e provenienza (ad esempio, l'infrastruttura dalla quale provengono le cellule);
|
|
tipo di citometria a flusso utilizzato (ad es. modello), comprese le impostazioni dello strumento, globulina, anticorpi e marcatore di citotossicità utilizzati;
|
|
procedura utilizzata per dimostrare la competenza del laboratorio nell'esecuzione della prova mediante sostanze di riferimento o nel dimostrare la riproducibilità della prova nel tempo, ad esempio dati storici dei controlli e/o dei controlli di reattività.
|
|
|
Criteri di accettabilità della prova:
|
vitalità cellulare, valori MFI e RFI ottenuti con il controllo con solvente/disperdente rispetto agli intervalli di accettabilità;
|
|
vitalità cellulare e valori RFI ottenuti con il controllo positivo rispetto agli intervalli di accettabilità;
|
|
vitalità cellulare di tutte le concentrazioni testate della sostanza chimica in esame.
|
|
|
Procedura
|
numero di prove realizzate;
|
|
concentrazioni della sostanza chimica in esame, applicazione e tempo di esposizione (se diverso da quello raccomandato);
|
|
descrizione dei criteri di valutazione e di decisione impiegati;
|
|
descrizione di qualsiasi modifica della procedura sperimentale.
|
|
|
Risultati
|
presentazione dei dati in formato tabulare, ivi compreso CV75 (se del caso), MFI geometrica individuale, RFI, valori di vitalità cellulare, valori EC150/EC200 (se del caso) ottenuti per la sostanza chimica in esame e per il controllo positivo in ciascuna batteria di prove, e indicazione della classificazione della sostanza chimica in esame secondo il modello predittivo;
|
|
descrizione di eventuali altre osservazioni pertinenti, se del caso.
|
|
|
Discussione dei risultati
|
discussione dei risultati ottenuti con il metodo di prova h-CLAT;
|
|
esame dei risultati della prova nel quadro di un approccio di tipo IATA, qualora siano disponibili altre informazioni pertinenti.
|
|
|
BIBLIOGRAFIA
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. Consultabile al seguente indirizzo: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). Consultabile al seguente indirizzo: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report consultabile al seguente indirizzo: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. Consultabile al seguente indirizzo: http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Parigi, Francia, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Consultabile al seguente indirizzo: http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Consultabile al seguente indirizzo: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
Appendice 1.1
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con la prova e i valori di riferimento comunemente accettati. Misura l'efficienza della prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di "concordanza", per indicare la proporzione di risultati corretti di una prova (21).
AOP (Adverse Outcome Pathway, meccanismo d'azione degli effetti avversi)
: sequenza di eventi che, a partire dalla struttura chimica di una sostanza chimica bersaglio o di un gruppo di sostanze chimiche simili, attraverso l'evento molecolare scatenante, produce un effetto avverso in vivo (22).
Sostanza chimica
: una sostanza o una miscela.
CV75
: concentrazione stimata che presenta una vitalità cellulare del 75 %.
EC150
: concentrazioni che producono RFI di 150 per l'espressione di CD86.
EC200
: concentrazioni che producono RFI di 200 per l'espressione di CD54.
Citometria a flusso
: tecnica citometrica in cui le cellule sospese in un fluido passano velocemente e singolarmente attraverso un fascio luminoso di eccitazione, producendo schemi di diffusione della luce che sono caratteristici delle cellule e dei loro componenti; le cellule sono spesso marcate con marcatori fluorescenti di modo che la luce sia dapprima assorbita e quindi emessa a un'altra frequenza.
Pericolo
: proprietà intrinseca di un agente o di una situazione che ha il potenziale di causare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti.
IATA (Integrated Approaches to Testing and Assessment, approcci integrati in materia di prove e valutazioni)
: approccio strutturato utilizzato per l'identificazione del pericolo (potenziale), la caratterizzazione del pericolo (potenza) e/o la valutazione della sicurezza (potenziale/potenza ed esposizione) di una sostanza chimica o di un gruppo di sostanze chimiche, che integra in modo strategico e ponderato tutti i dati pertinenti per orientare una decisione di tipo regolamentare concernente il pericolo potenziale e/o il rischio e/o la necessità di effettuare altre prove mirate e, pertanto, limitate allo stretto necessario.
Controllo con mezzo
: una replica non trattata che contiene tutti i componenti di un sistema di prova. Questo campione subisce il medesimo procedimento dei campioni trattati con la sostanza chimica in esame e di altri campioni di controllo al fine di determinare se il solvente/disperdente interagisce con il sistema di prova.
Miscela
: una miscela o una soluzione composta di due o più sostanze.
Sostanza monocostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno 80 % (p/p).
Sostanza multicostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui più costituenti principali sono presenti in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente è che una miscela è ottenuta attraverso la miscelazione di due o più sostanze senza che avvenga una reazione chimica. Una sostanza multicostituente è il risultato di una reazione chimica.
Controllo positivo
: replica contenente tutti i componenti di un sistema di prova, trattata con una sostanza che notoriamente induce una risposta positiva. Perché sia possibile valutare la variabilità nel tempo della risposta del controllo positivo, l'intensità di tale risposta non dovrebbe essere eccessiva.
Preapteni
: sostanze chimiche attivate tramite trasformazione abiotica.
Proapteni
: sostanze chimiche che necessitano di una bioattivazione enzimatica per dispiegare il loro potenziale di sensibilizzazione cutanea.
Intensità di fluorescenza relativa (RFI)
: valore relativo della media geometrica dell'intensità di fluorescenza (MFI) delle cellule esposte alla sostanza chimica in esame confrontato con l'MFI di cellule trattate con solvente/disperdente.
Pertinenza
: descrizione del rapporto tra la prova e l'effetto ricercato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l'effetto biologico di interesse. La pertinenza comprende una valutazione dell'accuratezza (concordanza) di una prova (21).
Affidabilità
: misura in cui una prova può essere riprodotta nel tempo all'interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna ai laboratori e la ripetibilità fra i laboratori (21).
Batteria di prove
: una batteria di prove consiste nel testare una o più sostanze chimiche in esame simultaneamente a un controllo con solvente/disperdente e a un controllo positivo.
Sensibilità
: proporzione di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l'accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (21).
Tampone di colorazione
: tampone fosfato salino contenente 0,1 % di albumina di siero bovino.
Controllo con solvente/disperdente
: campione non trattato contenente tutti i componenti di un sistema di prova, esclusa la sostanza chimica in esame, ma incluso il solvente/disperdente utilizzato. È utilizzato per stabilire la risposta di base per i campioni trattati con la sostanza chimica in esame disciolta o in dispersione stabile nello stesso solvente/disperdente. Se testato in concomitanza con un controllo con mezzo, questo campione dimostra anche se il solvente/disperdente interagisce con il sistema di prova.
Specificità
: proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l'accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (21).
Sostanza
: un elemento chimico e i suoi composti, allo stato naturale od ottenuti per mezzo di un procedimento di produzione, compresi gli additivi necessari a mantenerne la stabilità e le impurità derivanti dal procedimento utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione.
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi e livelli standardizzati di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di prudenza e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell'ambiente (23).
UVCB
: sostanze la cui composizione non è conosciuta o è variabile, prodotti di una reazione complessa o materiali origine biologica.
Metodo di prova valido
: metodo di prova la cui pertinenza e affidabilità sono ritenute soddisfacenti per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in assoluto ma solo in relazione a un determinato scopo (21).
Appendice 1.2
SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA
Prima di utilizzare come test di routine la prova descritta nella presente appendice per il metodo di prova B.71, i laboratori sono tenuti a dimostrare la loro competenza tecnica ottenendo correttamente la predizione attesa con il metodo h-CLAT per le 10 sostanze raccomandate nella tabella 1 e ottenendo valori CV75, EC150 e EC200 che rientrino nel rispettivo intervallo di riferimento per almeno 8 delle 10 sostanze di riferimento. Tali sostanze sono state selezionate per rappresentare la gamma di risposte per quanto riguarda i pericoli di sensibilizzazione cutanea. Altri criteri di selezione riguardavano la disponibilità in commercio delle sostanze e la disponibilità di dati di riferimento in vivo e di dati in vitro di elevata qualità ottenuti con il metodo h-CLAT. Sono inoltre disponibili dati di riferimento pubblicati per il metodo h-CLAT (3) (14).
Tabella 1
Sostanze raccomandate per la verifica della competenza tecnica con il metodo h-CLAT
|
Sostanze di prova per la verifica della competenza
|
N. CAS
|
Stato fisico
|
Predizione in vivo
(103)
|
Intervallo di riferimento CV75 in μg/ml (104)
|
Risultati h-CLAT per CD86 (intervallo di riferimento EC150 in μg/ml) (104)
|
Risultati h-CLAT per CD54 (intervallo di riferimento EC200 in μg/ml) (104)
|
|
2,4-Dinitroclorobenzene
|
97-00-7
|
Solido
|
Sensibilizzante (estremamente elevato)
|
2-12
|
Positivo (0,5-10)
|
Positivo (0,5-15)
|
|
4-Fenilendiammina
|
106-50-3
|
Solido
|
Sensibilizzante (elevato)
|
5-95
|
Positivo (<40)
|
Negativo (>1,5) (105)
|
|
Solfato di nichel
|
10101-97-0
|
Solido
|
Sensibilizzante (moderato)
|
30-500
|
Positivo (<100)
|
Positivo (10-100)
|
|
2-Mercaptobenzotiazolo
|
149-30-4
|
Solido
|
Sensibilizzante (moderato)
|
30-400
|
Negativo (>10) (105)
|
Positivo (10-140)
|
|
R(+)-Limonene
|
5989-27-5
|
Liquido
|
Sensibilizzante (debole)
|
>20
|
Negativo (>5) (105)
|
Positivo (<250)
|
|
Imidazolidinil urea
|
39236-46-9
|
Solido
|
Sensibilizzante (debole)
|
25-100
|
Positivo (20-90)
|
Positivo (20-75)
|
|
Isopropanolo
|
67-63-0
|
Liquido
|
Non sensibilizzante
|
>5 000
|
Negativo (>5 000 )
|
Negativo (>5 000 )
|
|
Glicerolo
|
56-81-5
|
Liquido
|
Non sensibilizzante
|
>5 000
|
Negativo (>5 000 )
|
Negativo (>5 000 )
|
|
Acido lattico
|
50-21-5
|
Liquido
|
Non sensibilizzante
|
1500-5 000
|
Negativo (>5 000 )
|
Negativo (>5 000 )
|
|
Acido 4-amminobenzoico
|
150-13-0
|
Solido
|
Non sensibilizzante
|
>1 000
|
Negativo (>1 000 )
|
Negativo (>1 000 )
|
|
Abbreviazioni: N. CAS (<Chemical Abstracts Service Registry Number) = Numero CAS (numero di registrazione nell'inventario europeo delle sostanze chimiche).
|
Appendice 2
SENSIBILIZZAZIONE CUTANEA IN VITRO: TEST DI ATTIVAZIONE DELLA LINEA CELLULARE U937 - (U-SENS™)
CONSIDERAZIONI INIZIALI E LIMITI
|
1.
|
Il metodo U-SENS™ consente di quantificare la variazione di espressione di un marcatore di superficie cellulare associato al processo di attivazione di monociti e cellule dendritiche (DC) (cioè CD86) nella linea cellulare di linfoma istiocitico umano U937 in seguito all'esposizione a sensibilizzanti (1). I livelli misurati di espressione del marcatore di superficie cellulare CD86 nella linea cellulare U937 sono quindi utilizzati per contribuire a distinguere tra sensibilizzanti e non sensibilizzanti cutanei.
|
|
2.
|
Il metodo U-SENS™ è stato oggetto di uno studio di validazione (2) coordinato da L'Oreal e di una successiva revisione indipendente inter pares effettuata sotto la guida del comitato scientifico consultivo (ESAC) del laboratorio di riferimento dell'Unione europea per le alternative alla sperimentazione animale (European Union Reference Laboratory for Alternatives to Animal Testing - EURL ECVAM) (3). Tenuto conto di tutti i dati disponibili e dei pareri formulati dalle autorità di regolamentazione e dai portatori di interessi, l'EURL ECVAM ha raccomandato l'uso del metodo U-SENS™ (4) nell'ambito di una metodologia integrata di tipo IATA per contribuire a distinguere tra sensibilizzanti e non sensibilizzanti cutanei a fini di classificazione ed etichettatura del pericolo. Nel suo documento orientativo sull'elaborazione di relazioni riguardanti approcci strutturati di integrazione dei dati e uso di fonti individuali di informazioni nell'ambito dell'approccio IATA per la sensibilizzazione cutanea, l'OCSE analizza una serie di studi di caso e descrive differenti strategie di prova e modelli predittivi. Uno degli approcci definiti si basa sul metodo di prova U-SENS (5). Esistono inoltre in letteratura (4)(5)(7) esempi dell'uso dei dati U-SENS™ in combinazione con altre informazioni, compresi dati storici e dati umani validi preesistenti (6).
|
|
3.
|
È stato dimostrato che il metodo U-SENS™ può essere applicato in laboratori con esperienza nel campo delle tecniche di coltura cellulare e dell'analisi mediante citometria a flusso. Il livello di riproducibilità atteso delle predizioni è dell'ordine del 90 % a livello intralaboratorio e dell'84 % a livello interlaboratori (8). I risultati dello studio di validazione (8) e di altri studi pubblicati (1) indicano globalmente che, rispetto ai risultati ottenuti con l'LLNA, l'accuratezza nella distinzione tra sensibilizzanti cutanei (categoria 1 del GHS dell'ONU/CLP) e non sensibilizzanti è pari all'86 % (N=166) per una sensibilità del 91 % (118/129) e una specificità del 65 % (24/37). Rispetto ai risultati ottenuti nell'uomo, l'accuratezza nella distinzione tra sensibilizzanti cutanei (categoria 1 del GHS dell'ONU/CLP) e non sensibilizzanti è pari al 77 % (N=101) per una sensibilità del 100 % (58/58) e una specificità del 47 % (20/43). Rispetto all'LLNA, è più probabile che i falsi negativi nelle predizioni formulate con il metodo U-SENS™ riguardino sostanze chimiche con una potenza di sensibilizzazione cutanea da bassa a moderata (sottocategoria 1B del GHS dell'ONU/CLP) piuttosto che sostanze chimiche con una potenza di sensibilizzazione cutanea elevata (sottocategoria 1A del GHS dell'ONU/CLP) (1)(8)(9). L'insieme di queste informazioni indica l'utilità del metodo di prova U-SENS™ come elemento in grado di contribuire all'identificazione dei pericoli di sensibilizzazione cutanea. Tuttavia, i valori di accuratezza forniti in questa sede per il metodo U-SENS™ utilizzato isolatamente sono puramente indicativi, in quanto la prova dovrebbe essere considerata in combinazione con altre fonti di informazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei paragrafi 7 e 8 dell'Introduzione generale. Inoltre, nel valutare i metodi di studio della sensibilizzazione cutanea che non utilizzano la sperimentazione animale si deve tenere presente che l'LLNA, come pure altre prove che utilizzano la sperimentazione animale, può non riflettere pienamente la situazione per l'uomo.
|
|
4.
|
I dati attualmente disponibili mostrano che il metodo di prova U-SENS™ è applicabile a sostanze chimiche in esame (compresi ingredienti di cosmetici quali conservanti, tensioattivi, sostanze attive, coloranti) che coprono diversi gruppi funzionali organici, proprietà fisico-chimiche, potenze di sensibilizzazione cutanea (determinate nell'ambito di studi in in vivo) e l'insieme dei meccanismi di reazione notoriamente associati alla sensibilizzazione cutanea (accettore di Michael, sintesi di una base di Schiff, agente acilante, sostituzione nucleofila bimolecolare [SN2], o sostituzione nucleofila aromatica [SNAr]) (1)(8)(9)(10). Il metodo di prova U-SENS™ è applicabile alle sostanze chimiche solubili o che formano una dispersione stabile (colloide o sospensione in cui la sostanza chimica in esame non si deposita né si separa dal solvente/mezzo disperdente formando più fasi) in un solvente/disperdente idoneo (cfr. il paragrafo 13). Le sostanze chimiche della banca dati classificate come preapteni (sostanze attivate mediante ossidazione) o proapteni (sostanze che richiedono un'attivazione enzimatica, ad esempio mediante enzimi P450) sono state correttamente identificate con il metodo U-SENS™ (1) (10). Le sostanze chimiche in grado di provocare la rottura della membrana possono dar luogo a falsi positivi a causa di un aumento non specifico dell'espressione di CD86. In effetti, 3 dei 7 falsi positivi in relazione alla classificazione di riferimento in vivo erano tensioattivi (1). Pertanto risultati positivi con tensioattivi vanno considerati con cautela, mentre risultati negativi con tensioattivi possono essere utilizzati nel processo di identificazione della sostanza chimica in esame come non sensibilizzante. Le sostanze chimiche fluorescenti possono essere valutate con il metodo U-SENS™ (1); tuttavia, le sostanze chimiche fortemente fluorescenti che emettono alla stessa lunghezza d'onda dell'isotiocianato di fluorescina (FITC) o dello ioduro di propidio (PI) interferiscono con la rilevazione mediante citometria a flusso e pertanto non possono essere correttamente valutate mediante anticorpi coniugati con FITC (rischio di falso negativo) o PI (vitalità non misurabile). In questo caso si può fare ricorso, rispettivamente, ad altri anticorpi marcati con fluorocromo o ad altri marcatori di citotossicità, purché si possa dimostrare, ad esempio testando le sostanze di riferimento indicate nell'appendice 2.2, che essi producono risultati simili agli anticorpi marcati con FITC o PI (cfr. il paragrafo 18). Alla luce di quanto precede, i risultati positivi con tensioattivi e i risultati negativi con sostanze chimiche fortemente fluorescenti andranno interpretati nel contesto dei limiti indicati e in combinazione con altre fonti di informazione nell'ambito di un approccio IATA. Nel caso in cui si dimostri che il metodo di prova U-SENS™ non è applicabile ad altre categorie specifiche di sostanze chimiche in esame, è opportuno evitare di utilizzarlo per tali categorie.
|
|
5.
|
Come già precisato, il metodo U-SENS™ aiuta a distinguere tra sensibilizzanti e non sensibilizzanti cutanei. Tuttavia, esso può contribuire anche alla valutazione della potenza di sensibilizzazione cutanea se utilizzato nell'ambito di approcci integrati quali IATA. Sono però necessarie ulteriori ricerche, di preferenza basate su dati relativi all'uomo, per determinare in che modo i risultati del metodo U-SENS™ possano contribuire alla valutazione della potenza di sensibilizzazione.
|
|
6.
|
Le definizioni figurano nell'appendice 2.1.
|
Principio Della Prova
|
7.
|
Il metodo U-SENS™ è una prova in vitro che consente di quantificare le variazioni di espressione del marcatore di superficie cellulare CD86 nella linea cellulare di linfoma istiocitico umano (cellule U937) dopo 45 ± 3 ore di esposizione alla sostanza chimica in esame. Il marcatore di superficie CD86 è un marcatore tipico dell'attivazione delle cellule U937. Si tratta di una molecola di costimolazione capace di stimolare l'attivazione monocitaria, che svolge un ruolo cruciale nel priming dei linfociti T. Le variazioni di espressione del marcatore di superficie cellulare CD86 sono misurate mediante citometria a flusso dopo colorazione cellulare, generalmente effettuata con anticorpi marcati con isotiocianato di fluorescina (FITC). In parallelo è inoltre effettuata una misurazione della citotossicità (ad esempio mediante PI) per stabilire se la sovraespressione del marcatore di superficie cellulare CD86 avviene in presenza di concentrazioni sub-citotossiche. L'indice di stimolazione (S.I.) del marcatore di superficie cellulare CD86 rispetto al controllo con solvente/disperdente è calcolato e utilizzato in un modello predittivo (cfr. il paragrafo 19) ai fini della distinzione tra sensibilizzanti e non sensibilizzanti cutanei.
|
Dimostrazione Della Competenza Tecnica
|
8.
|
Prima di utilizzare come test di routine la prova descritta nella presente appendice relativa al metodo di prova B.71, i laboratori devono dimostrare le loro competenze tecniche utilizzando le 10 sostanze di riferimento elencate nell'appendice 2.2, conformemente alle buone pratiche per i metodi in vitro (11). Inoltre gli utilizzatori del metodo devono mantenere una banca dati storica contenente i dati ottenuti con i controlli di reattività (cfr. il paragrafo 11) nonché con i controlli positivi e con solvente/disperdente (cfr. i paragrafi 15-16) e utilizzare tali dati per confermare la persistenza nel tempo della riproducibilità della prova nel loro laboratorio.
|
Procedura
|
9.
|
La presente prova si basa sul protocollo U-SENS™ n. 183 del Servizio dati sui metodi alternativi alla sperimentazione animale (DataBase service on ALternative Methods to animal experimentation - DB-ALM) (12). Nell'applicare e utilizzare la prova U-SENS™ in laboratorio vanno seguite le procedure operative standard (SOP). È possibile avvalersi di un sistema automatizzato per l'esecuzione della prova U-SENS™ purché si dimostri, ad esempio testando le sostanze di riferimento di cui all'appendice 2.2, che tale sistema produce risultati simili. Di seguito è fornita una descrizione dei principali elementi e procedure del metodo U-SENS™.
|
Preparazione delle cellule
|
10.
|
Per l'esecuzione del metodo U-SENS™ si utilizza la linea cellulare di linfoma istiocitico umano (cellule U937) (13). Le cellule (clone CRL1593.2) devono provenire da una banca di cellule riconosciuta, quale la American Type Culture Collection.
|
|
11.
|
Le cellule U937 sono coltivate a 37°C in atmosfera umidificata al 5 % di CO2, su mezzo di coltura RPMI-1 640 integrato da 10 % di siero fetale di vitello (FCS), 2 mM di L-glutammina, 100 unità/ml di penicillina e 100 μg/ml di streptomicina (mezzo completo). Le cellule U937 sono regolarmente messe in subcultura ogni 2-3 giorni a una densità di 1,5 o 3 × 105 cellule/ml rispettivamente. La densità cellulare non deve superare 2 × 106 cellule/ml e la vitalità cellulare misurata con esclusione del blu di tripano deve essere ≥ 90 % (questo non si applica al primo passaggio successivo allo scongelamento). Prima di essere utilizzato per la prova, ogni lotto di cellule, FCS o anticorpi deve essere qualificato mediante un controllo di reattività da realizzarsi almeno una settimana dopo lo scongelamento mediante acido picrilsulfonico (acido 2,4,6-trinitrobenzensulfonico: TNBS) (n. CAS 2 508-19-2, purezza ≥ 99 %) come controllo positivo e acido lattico (LA) (n. CAS 50-21-5, purezza ≥ 85 %) come controllo negativo. Per il controllo di reattività devono essere testate sei concentrazioni finali per ciascuno dei due controlli (TNBS: 1, 12,5, 25, 50, 75, 100μg/ml e LA: 1, 10, 20, 50, 100, 200μg/ml). Il TNBS solubilizzato nel mezzo completo deve dar luogo a una risposta di CD86 positiva e correlata alla concentrazione (ad es. quando una concentrazione positiva, S.I. CD86 ≥ 150, è seguita da una concentrazione con S.I. CD86 crescente) e l'acido lattico solubilizzato nel mezzo completo deve dar luogo a una risposta negativa di CD86 (cfr. il paragrafo 21). Possono essere utilizzati per la prova soltanto i lotti di cellule che superano per due volte il controllo di reattività. Le cellule possono essere moltiplicate fino a sette settimane dopo lo scongelamento senza superare 21 passaggi. Il controllo di reattività va effettuato secondo le procedure descritte ai paragrafi 18-22.
|
|
12.
|
Per la prova, le cellule U937 sono inoculate a una densità di 3 × 105 cellule/ml o di 6 × 105 cellule/ml e precoltivate in fiasche di coltura per 2 giorni o 1 giorno rispettivamente. Possono essere utilizzate condizioni di precoltura diverse da quelle descritte sopra purché si forniscano sufficienti motivazioni scientifiche e si dimostri, ad esempio testando le sostanze di riferimento di cui all'appendice 2.2, che tali condizioni alternative producono risultati simili. Il giorno della prova le cellule raccolte dalla fiasca di coltura sono rimesse in sospensione in un nuovo mezzo di coltura a una densità di 5 × 105 cellule/ml. Quindi le cellule sono ripartite su una piastra a fondo piatto a 96 pozzetti con 100 μl (densità cellulare finale di 0,5 × 105 cellule/pozzetto).
|
Preparazione delle sostanze chimiche in esame e delle sostanze di controllo
|
13.
|
La valutazione della solubilità è realizzata prima dell'esecuzione della prova. A questo scopo le sostanze chimiche in esame sono dissolte o disperse stabilmente a una concentrazione di 50 mg/ml utilizzando di preferenza come solvente il mezzo completo o, se la sostanza chimica in esame non è solubile nel mezzo completo, dimetilsulfossido (DMSO, purezza ≥ 99 %) come seconda scelta di solvente/disperdente. Per la prova, la sostanza chimica in esame è disciolta fino a una concentrazione finale di 0,4 mg/ml nel mezzo completo, se la sostanza è solubile in tale solvente/disperdente. Se è solubile unicamente in DMSO, la sostanza chimica è disciolta a una concentrazione di 50 mg/ml. È possibile utilizzare solventi/mezzi disperdenti diversi da quelli descritti sopra fornendo una sufficiente giustificazione scientifica. Si deve tenere conto della stabilità della sostanza chimica in esame nel solvente/disperdente finale.
|
|
14.
|
Le sostanze chimiche in esame e le sostanze di controllo vengono preparate il giorno della prova. Poiché non viene effettuata una prova di determinazione della dose, per la prima batteria di prove vengono testate 6 concentrazioni finali (1, 10, 20, 50, 100 e 200 μg/ml) nel corrispondente solvente/disperdente, vale a dire mezzo completo o DMSO a 0,4 % nel mezzo. Per le batterie di prove successive, a partire dalle soluzioni delle sostanze chimiche in esame alla concentrazione di 0,4 mg/ml nel mezzo completo o 50 mg/ml in DMSO vengono preparate almeno 4 soluzioni di lavoro (cioè almeno 4 concentrazioni) con il solvente/disperdente corrispondente. Le soluzioni di lavoro sono infine utilizzate per il trattamento aggiungendo un volume uguale di sospensione di cellule U937 (cfr. il paragrafo 11 supra) al volume della soluzione di lavoro nella piastra per ottenere un'ulteriore diluizione di fattore 2 (12). Le concentrazioni (almeno 4) per eventuali batterie di prove supplementari sono scelte sulla base dei risultati individuali di tutte le batterie di prove precedenti (8). Le concentrazioni finali utilizzabili sono 1, 2, 3, 4, 5, 7,5, 10, 12.5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 e 200 μg/ml. La concentrazione finale massima è 200 μg/ml. Se si ottiene un valore positivo per CD86 a 1 μg/ml, si valuta 0,1 μg/ml per trovare la concentrazione della sostanza chimica in esame che non supera la soglia di positività per CD86. Per ogni batteria di prove si calcola il valore EC150 (concentrazione alla quale la sostanza chimica raggiunge la soglia di positività di 150 % per CD86 - cfr. il paragrafo 19) se per CD86 si osserva una risposta positiva correlata alla concentrazione. Se la sostanza chimica in esame induce una risposta positiva per CD86 non correlata alla concentrazione, il calcolo del valore EC150 potrebbe non essere pertinente, come descritto nel protocollo U-SENS™ n. 183 del DB-ALM (12). Per ogni batteria di prove si calcola il valore CV70 (concentrazione alla quale la sostanza chimica raggiunge la soglia di citotossicità di 70 % - cfr. il paragrafo 19) ogniqualvolta possibile. Per valutare l'effetto della risposta correlata alla concentrazione dell'aumento di CD86, tutte le concentrazioni scelte tra quelle utilizzabili devono essere uniformemente ripartite tra EC150 (o la massima concentrazione non citotossica negativa per CD86) e CV70 (o la massima concentrazione consentita, vale a dire 200 μg/ml). Devono essere testate almeno 4 concentrazioni per batteria di prove, di cui almeno 2 comuni alla o alle batterie di prove precedenti, a fini di raffronto.
|
|
15.
|
Il controllo con solvente/disperdente utilizzato nel metodo U-SENS™ è il mezzo completo (per le sostanze chimiche in esame solubilizzate o disperse stabilmente nel mezzo completo) (cfr. il paragrafo 4) o DMSO a 0,4 % nel mezzo completo (per le sostanze chimiche in esame solubilizzate o disperse stabilmente in DMSO).
|
|
16.
|
Il controllo positivo utilizzato nel metodo U-SENS™ è il TNBS (cfr. il paragrafo 11), preparato in mezzo completo. Il TNBS deve essere utilizzato come controllo positivo per la misurazione dell'espressione di CD86 a una concentrazione finale unica nella piastra (50 μg/ml) che produce una vitalità cellulare > 70 %. Per ottenere una concentrazione di 50 μg/ml di TNBS nella piastra si prepara una soluzione madre di TNBS a 1 M (cioè 293 mg/ml) in mezzo completo e la si diluisce ulteriormente di un fattore 2 930 nel mezzo completo fino ad ottenere una soluzione di lavoro di 100 μg/ml. L'acido lattico (LA, CAS 50-21-5) è utilizzato come controllo negativo a 200 μg/ml, solubilizzato in mezzo completo (a partire da una soluzione madre di 0,4 mg/ml). Per ogni piastra di ciascuna batteria di prove si preparano tre repliche di controllo con mezzo completo non trattato, controllo con solvente/disperdente, controlli negativo e positivo (12). Altri controlli positivi adeguati possono essere utilizzati in caso di disponibilità di dati storici da cui ricavare criteri di accettabilità comparabili per la batteria di prove. I criteri di accettabilità della batteria di prove sono identici a quelli descritti per la sostanza chimica in esame (cfr. il paragrafo 12).
|
Applicazione delle sostanze chimiche in esame e delle sostanze di controllo
|
17.
|
Il controllo con solvente/disperdente o le soluzioni di lavoro descritte ai paragrafi 14-16 sono mescolati nella proporzione 1:1 (v/v) con le sospensioni cellulari preparate nella piastra a fondo piatto a 96 pozzetti (cfr. il paragrafo 12). Le piastre trattate sono quindi messe in incubazione per 45 ± 3 ore a una temperatura di 37 °C con 5 % di CO2. Prima dell'incubazione le piastre sono sigillate con una membrana semipermeabile al fine di evitare l'evaporazione di sostanze chimiche volatili e la contaminazione incrociata tra le cellule trattate con le sostanze chimiche in esame (12).
|
Colorazione cellulare
|
18.
|
Dopo 45 ± 3 ore di esposizione le cellule sono trasferite in una piastra per microtitolazione con fondo a V e raccolte mediante centrifugazione. L'interferenza di solubilità è definita dalla presenza di cristalli o gocce visibili al microscopio 45 ± 3 ore dopo il trattamento (prima della colorazione cellulare). I supernatanti vengono scartati e le cellule rimanenti sono lavate una volta con 100 μl di tampone fosfato salino (PBS) ghiacciato contenente 5 % di siero fetale di vitello (tampone di colorazione). Dopo la centrifugazione le cellule sono rimesse in sospensione in 100 μl di tampone di colorazione e colorate con 5 μl (cioè 0,25 μg) di anticorpi anti-CD86 o anticorpi murini IgG1 (isotipo) marcati con FITC a 4°C per 30 minuti al riparo dalla luce. Devono essere utilizzati gli anticorpi descritti nel protocollo U-SENS™ n. 183 del DB-ALM (12) (per CD86: BD-PharMingen #5556 57 Clone: Fun-1, o Caltag/Invitrogen # MHCD8601 Clone: BU63; e per IgG1: BD-PharMingen #5557 48, o Caltag/Invitrogen # GM4992). Sulla base dell'esperienza degli sviluppatori del metodo di prova, l'intensità di fluorescenza dei vari lotti di anticorpi è generalmente costante. Per la prova possono essere usati altri cloni o fornitori di anticorpi che abbiano superato il controllo di reattività (cfr. il paragrafo 11). Tuttavia gli utilizzatori possono decidere di effettuare la titolazione degli anticorpi nelle loro condizioni di laboratorio per definire la concentrazione più adatta all'uso. È possibile avvalersi di altri sistemi di rilevazione, quali anticorpi marcati con fluorocromo anti-CD86, purché si possa dimostrare che essi producono risultati simili agli anticorpi coniugati con FITC, ad esempio testando le sostanze di riferimento di cui all'appendice 2.2. Dopo due lavaggi con 100 μl di tampone di colorazione e un lavaggio con 100 μl di PBS ghiacciato, le cellule sono rimesse in sospensione in PBS ghiacciato (ad es. 125 μl per i campioni analizzati manualmente provetta per provetta o 50 μl se si utilizza un autocampionatore) ed è aggiunta una soluzione di PI (concentrazione finale di 3 μg/ml). È possibile utilizzare altri marcatori di citotossicità, tra cui la 7-aminoactinomicina D (7-AAD) e il blu di tripano, purché si dimostri che tali coloranti alternativi producono risultati simili al PI, ad esempio testando le sostanze di riferimento di cui all'appendice 2.2.
|
Analisi mediante citometria a flusso
|
19.
|
Il livello di espressione di CD86 e la vitalità cellulare sono analizzati mediante citometria a flusso. Le cellule sono rappresentate su un grafico a punti in scala logaritmica in base alla taglia (FSC) e alla granularità (SSC) per identificare chiaramente la popolazione in un primo gate R1 ed eliminare i residui. L'obiettivo è acquisire un totale di 10 000 cellule nel gate R1 per ogni pozzetto. Le cellule appartenenti allo stesso gate R1 sono rappresentate su un grafico a punti FL3 o FL4/SSC. Le cellule vitali sono identificate tracciando un secondo gate R2 di selezione della popolazione cellulare negativa per lo ioduro di propidio (canale FL3 o FL4). La vitalità cellulare può essere calcolata dal programma di analisi del citometro mediante l'equazione riportata di seguito. Se la vitalità cellulare è bassa possono essere raccolte fino a 20 000 cellule, comprese le cellule morte. Un'opzione alternativa consiste nell'acquisire i dati per un minuto dall'inizio dell'analisi.
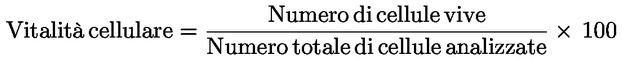
Si misura quindi la percentuale di cellule FL1-positive tra le cellule vitali comprese nel gate R2 (all'interno di R1). L'espressione di CD86 sulla superficie cellulare è analizzata mediante un grafico a punti FL1/SSC limitato alle cellule vitali (R2).
Per i pozzetti contenenti mezzo completo/IgG1 il marcatore di analisi è fissato in prossimità della popolazione principale in modo che i controlli con mezzo completo presentino un valore IgG1 compreso tra 0,6 e 0,9 %.
L'interferenza di colore è definita come scostamento della dispersione corrispondente agli IgG1 marcati con FITC (media geometrica di S.I. IgG1 FL1 ≥ 150 %).
L'indice di stimolazione (S.I.) di CD86 per le cellule di controllo (non trattate o in DMSO a 0,4 %) e per le cellule trattate con la sostanza chimica è calcolato mediante la seguente equazione:.
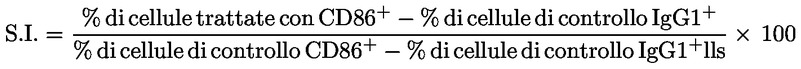
% di cellule di controllo non trattate IgG1+: percentuale di cellule IgG1 FL1-positive che superano il marcatore di analisi (intervallo di accettabilità ≥ 0,6 % e < 1,5 %, cfr. il paragrafo 22) tra le cellule vitali non trattate.
% di cellule di controllo IgG1+/cellule trattate CD86+: percentuale di cellule IgG1/CD86 FL1-positive misurata senza spostare il marcatore di analisi, tra le cellule vitali di controllo/cellule trattate.
|
Dati E Relazioni
Valutazione dei dati
|
20.
|
I seguenti parametri sono calcolati con il metodo U-SENS™: il valore CV70, vale a dire la concentrazione che produce il 70 % di sopravvivenza di cellule U937 (30 % di citotossicità), e il valore EC150, vale a dire la concentrazione alla quale le sostanze chimiche in esame producono un indice di stimolazione (S.I.) di CD86 di 150 %.
Il valore CV70 è calcolato per interpolazione log-lineare mediante la seguente equazione:
CV70 = C1 + [(V1 - 70) / (V1 – V2) * (C2 – C1)]
in cui:
V1 è il valore minimo di vitalità cellulare superiore a 70 %
V2 il valore massimo di vitalità cellulare inferiore a 70 %
C1 e C2 sono le concentrazioni che producono rispettivamente i valori di vitalità cellulare V1 e V2.

È possibile utilizzare altri metodi per calcolare il valore CV70, purché sia provato che ciò non incide sui risultati (ad es. testando le sostanze di riferimento).
Il valore EC150 è calcolato per interpolazione log-lineare mediante la seguente equazione:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
in cui:
C1 è la concentrazione più elevata in μg/ml con S.I. CD86 < 150 % (S.I. 1)
C2 è la concentrazione più bassa in μg/ml con S.I. CD86 ≥ 150 % (S.I. 2)
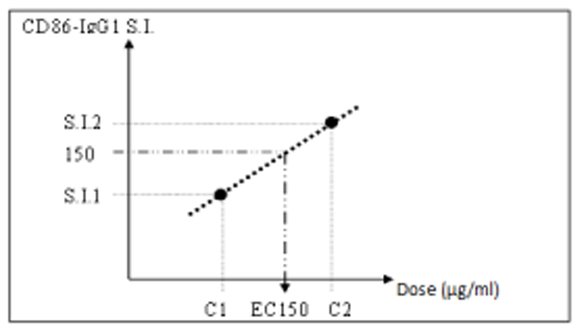
I valori EC150 e CV70 sono calcolati
|
—
|
per ogni batteria di prove: i valori individuali EC150 e CV70 sono utilizzati per valutare l'effetto della risposta correlata alla concentrazione dell'aumento di CD86 (cfr. il paragrafo 14),
|
|
—
|
il valore CV70 globale è determinato in funzione della vitalità media (12),
|
|
—
|
il valore EC150 globale di una sostanza chimica in esame la cui predizione è risultata POSITIVA con il metodo U-SENS™ è determinato in funzione dei valori medi di S.I. di CD86 (cfr. il paragrafo 21) (12).
|
|
Modello predittivo
|
21.
|
Per la misurazione dell'espressione di CD86 ciascuna sostanza chimica è testata in almeno quattro concentrazioni e in almeno due batterie di prove indipendenti (realizzate in giorni diversi) per ottenere una predizione unica (POSITIVA o NEGATIVA).
|
—
|
La conclusione individuale di una batteria di prove U-SENS™ è considerata negativa (di seguito, N) se l'S.I. di CD86 è inferiore a 150 % a tutte le concentrazioni non citotossiche (vitalità cellulare ≥ 70 %) e se non è stata osservata alcuna interferenza (citotossicità, solubilità: cfr. il paragrafo 18 o colore: cfr. il paragrafo 19, a prescindere dalle concentrazioni non citotossiche a cui è osservata l'interferenza). In tutti gli altri casi: se l'S.I. di CD86 è superiore o uguale a 150 % e/o si osservano interferenze, la conclusione individuale di una batteria di prove U-SENS™ è considerata positiva (di seguito, P).
|
|
—
|
Una predizione U-SENS™ è considerata NEGATIVA se almeno due batterie di prove indipendenti sono negative (N) (figura 1). Se le prime due batterie di prove sono negative (N), la predizione U-SENS™ è considerata NEGATIVA e non è necessario eseguire una terza batteria di prove.
|
|
—
|
Una predizione U-SENS™ è considerata POSITIVA se almeno due batterie di prove indipendenti sono positive (P) (figura 1). Se le prime due batterie di prove sono positive (P), la predizione U-SENS™ è considerata POSITIVA e non è necessario eseguire una terza batteria di prove.
|
|
—
|
Poiché non viene effettuata una prova di determinazione della dose, si ha un'eccezione se, nella prima batteria di prove, l'S.I. di CD86 è superiore o uguale a 150 % unicamente alla massima concentrazione citotossica. La batteria di prove è considerata NON CONCLUSIVA (NC) e si testano altre concentrazioni (tra la massima concentrazione non citotossica e la minima concentrazione citotossica - cfr. il paragrafo 20) in ulteriori batterie di prove. Se la batteria di prove risulta NC si svolgono almeno due batterie di prove supplementari; una quarta batteria di prove è svolta in caso di discordanza tra le batterie di prove 2 e 3 (N e/o P in qualunque combinazione) (figura 1). Le batterie di prove successive saranno considerate positive anche nel caso in cui soltanto una delle concentrazioni non citotossiche dia luogo a un valore di CD86 uguale o superiore a 150 %, dal momento che i parametri di concentrazione sono stati specificamente adattati alla sostanza chimica in esame. La predizione finale sarà basata sul risultato ottenuto nella maggioranza delle tre o quattro batterie di prove individuali (cioè 2 su 3 o 2 su 4) (figura 1).
|
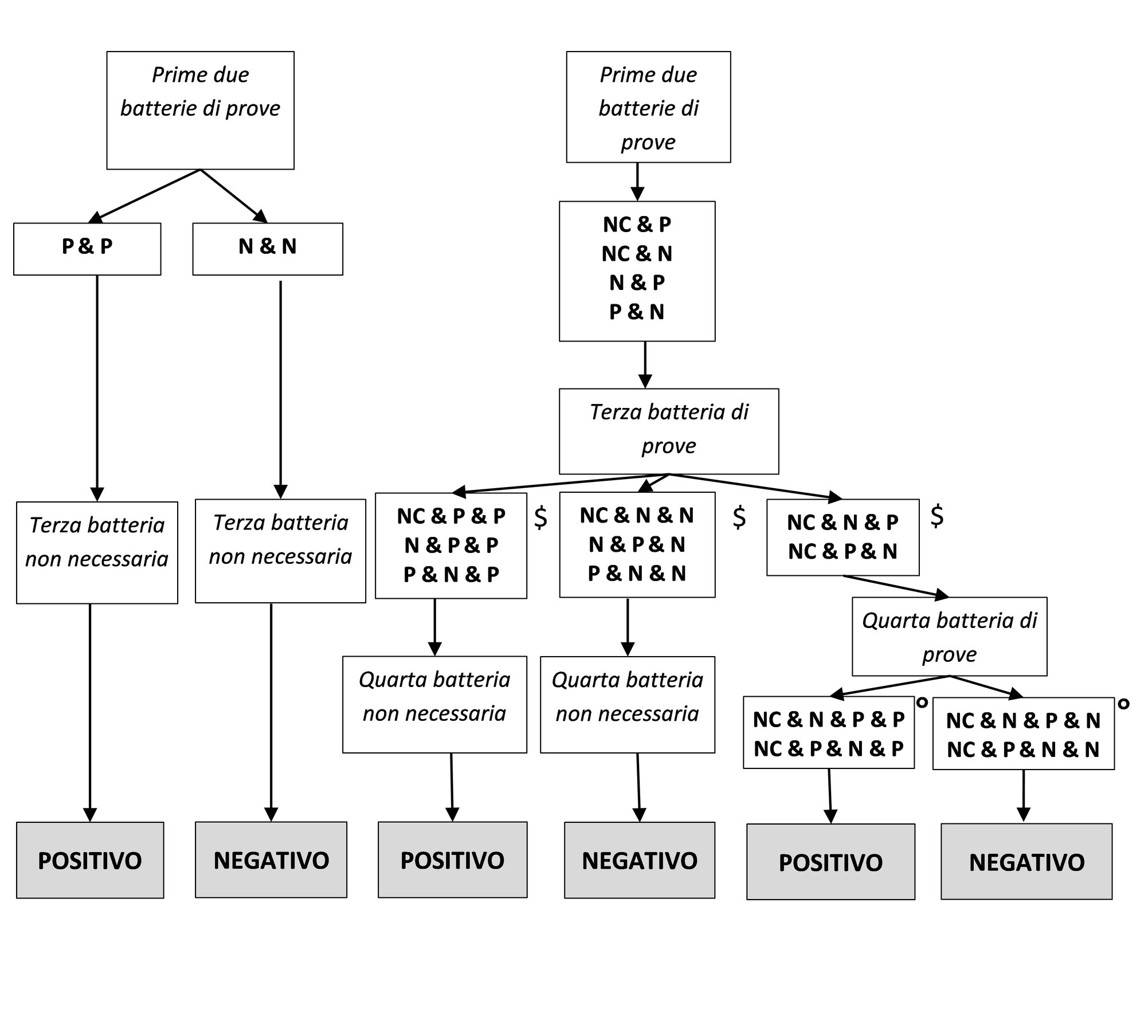
Figura 1: Modello predittivo utilizzato nel metodo U-SENS™. Una previsione U-SENS™ va considerata nell'ambito di un approccio IATA e in conformità alle disposizioni del paragrafo 4 e del paragrafi 7, 8 e 9 dell'Introduzione generale.
N: batteria di prove senza risultato positivo per CD86 né interferenza;
P: batteria di prove con risultato positivo per CD86 e/o interferenza/interferenze;
NC: non conclusiva. Prima batteria di prove non conclusiva se CD86 è positivo unicamente alla massima concentrazione non citotossica;
#: un risultato individuale non conclusivo (NC) attribuito unicamente alla prima batteria di prove comporta automaticamente la necessità di una terza batteria di prove per raggiungere una maggioranza di risultati positivi (P) o negativi (N) in almeno 2 batterie di prove indipendenti su 3.
$: i riquadri mostrano le combinazioni pertinenti di risultati delle prime tre batterie di prove sulla base dei risultati ottenuti nelle prime due batterie di prove (riportati nel riquadro che precede).
o: i riquadri mostrano le combinazioni pertinenti di risultati delle prime quattro batterie di prove sulla base dei risultati ottenuti nelle prime tre batterie di prove (riportati nel riquadro che precede).
|
Criteri di accettabilità
|
22.
|
I seguenti criteri di accettabilità devono essere soddisfatti quando si utilizza il metodo di prova U-SENS™ (12).
|
—
|
Al termine di un periodo di esposizione di 45 ± 3 ore la vitalità media delle tre repliche di cellule U937 non trattate deve essere > 90 % e non deve essere osservato alcuno scostamento dell'espressione di CD86. L'espressione basale di CD86 nelle cellule U937 non trattate deve essere compresa tra ≥ 2 % e ≤ 25 %.
|
|
—
|
Se il solvente utilizzato è il DMSO, la sua validità come controllo con mezzo disperdente è valutata calcolando l'S.I. del DMSO rispetto a quello delle cellule non trattate, e la vitalità media delle tre repliche di cellule deve essere > 90 %. Il controllo con mezzo disperdente DMSO è valido se l'S.I. medio per CD86 nelle tre repliche è inferiore a 250 % dell'S.I. medio di CD86 nelle tre repliche di cellule U937 non trattate.
|
|
—
|
Le batterie di prove sono considerate valide se almeno due dei tre valori IgG1 delle cellule U937 non trattate sono compresi tra ≥ 0,6 % e < 1,5 %.
|
|
—
|
Il controllo negativo testato in parallelo (acido lattico) è considerato valido se almeno due delle tre repliche sono negative (S.I. CD86 < 150 %) e non citotossiche (vitalità cellulare ≥ 70 %).
|
|
—
|
Il controllo positivo (TNBS) è considerato valido se almeno due delle tre repliche sono positive (S.I. CD86 ≥ 150 %) e non citotossiche (vitalità cellulare ≥ 70 %).
|
|
Relazione sulla prova
|
23.
|
La relazione sulla prova comprende le informazioni riportate di seguito.
|
Sostanza chimica in esame
Sostanza monocostituente:
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, solubilità nel mezzo completo, solubilità nel DMSO, peso molecolare e proprietà fisico-chimiche pertinenti aggiuntive, a seconda dei dati disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
Sostanza multicostituente, UVCB o miscela:
|
caratterizzazione, nella misura del possibile, ad esempio attraverso l'identità chimica (cfr. sopra), la purezza, le proporzioni quantitative e le proprietà fisico-chimiche pertinenti (cfr. sopra) dei costituenti, secondo i dati disponibili;
|
|
aspetto fisico, solubilità nel mezzo completo, solubilità nel DMSO e proprietà fisico-chimiche pertinenti aggiuntive, a seconda dei dati disponibili;
|
|
peso molecolare o peso molecolare apparente nel caso di miscele/polimeri di composizione nota o altre informazioni pertinenti per la realizzazione dello studio;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
Controlli
Controllo positivo
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, solubilità nel DMSO, peso molecolare e proprietà fisico-chimiche pertinenti aggiuntive, se del caso e a seconda dei dati disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
riferimento ai dati storici relativi ai controlli positivi che dimostrano la conformità ai criteri di accettabilità, se del caso.
|
Controllo negativo e controllo con solvente/disperdente
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
aspetto fisico, peso molecolare e altre proprietà fisico-chimiche pertinenti, nel caso siano utilizzati solventi/mezzi disperdenti diversi da quelli menzionati nelle linee guida e se disponibili;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
Condizioni di prova
|
nome e indirizzo dello sponsor, dell'infrastruttura utilizzata per la prova e del responsabile dello studio;
|
|
descrizione del sistema di prova utilizzato;
|
|
linea cellulare utilizzata, condizioni di conservazione e provenienza (ad esempio, l'infrastruttura dalla quale provengono le cellule);
|
|
tipo di citometria a flusso utilizzato (ad es. modello), comprese le impostazioni dello strumento, anticorpi e marcatore di citotossicità utilizzati;
|
|
procedura utilizzata per dimostrare la competenza del laboratorio nell'esecuzione della prova mediante sostanze di riferimento o nel dimostrare la riproducibilità della prova nel tempo, ad esempio dati storici dei controlli e/o dei controlli di reattività.
|
|
|
Criteri di accettabilità della prova
|
vitalità cellulare e valori S.I. CD86 ottenuti con il controllo con solvente/disperdente rispetto agli intervalli di accettabilità;
|
|
vitalità cellulare e valori S.I. ottenuti con il controllo positivo rispetto agli intervalli di accettabilità;
|
|
vitalità cellulare di tutte le concentrazioni testate della sostanza chimica in esame.
|
|
|
Procedura
|
numero di prove realizzate;
|
|
concentrazioni della sostanza chimica in esame, applicazione e tempo di esposizione (se diverso da quello raccomandato);
|
|
descrizione dei criteri di valutazione e di decisione impiegati;
|
|
descrizione di qualsiasi modifica della procedura sperimentale.
|
|
|
Risultati
|
presentazione dei dati in formato tabulare, ivi compreso CV70 (se del caso), S.I., valori di vitalità cellulare, valori EC150 (se del caso) ottenuti per la sostanza chimica in esame e per il controllo positivo in ciascuna batteria di prove, e indicazione della classificazione della sostanza chimica in esame secondo il modello predittivo;
|
|
descrizione di eventuali altre osservazioni pertinenti, se del caso.
|
|
|
Discussione dei risultati
|
discussione dei risultati ottenuti con il metodo di prova U-SENS™;
|
|
esame dei risultati della prova nel quadro di un approccio di tipo IATA, qualora siano disponibili altre informazioni pertinenti.
|
|
Conclusioni
|
BIBLIOGRAFIA
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. Consultabile al seguente indirizzo: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Consultabile al seguente indirizzo: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Consultabile al seguente indirizzo: https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: [ http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. Consultabile al seguente indirizzo: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. Consultabile al seguente indirizzo: http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Consultabile al seguente indirizzo: https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
Appendice 2.1
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con la prova e i valori di riferimento comunemente accettati. Misura l’efficienza della prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di «concordanza», per indicare la proporzione di risultati corretti di una prova (14).
AOP (Adverse Outcome Pathway, meccanismo d’azione degli effetti avversi)
: sequenza di eventi che, a partire dalla struttura chimica di una sostanza chimica bersaglio o di un gruppo di sostanze chimiche simili, attraverso l’evento molecolare scatenante, produce un effetto avverso in vivo (15).
Risposta di CD86 indotta dalla concentrazione
: si ha dipendenza dalla concentrazione (o risposta indotta dalla concentrazione) quando una concentrazione che induce una risposta positiva (I.S. CD86 ≥ 150) è seguita da una concentrazione con un I.S. CD86 crescente.
Sostanza chimica
: una sostanza o una miscela.
CV70
: concentrazione stimata che presenta una vitalità cellulare del 70 %.
Deriva
: i) il valore corretto %CD86+ della replica 3 del controllo non trattato è inferiore a 50 % della media corretta del valore %CD86+ delle repliche 1 e 2 del controllo non trattato; e ii) il valore corretto %CD86+ della replica 3 del controllo negativo è inferiore a 50 % della media corretta del valore %CD86+ delle repliche 1 e 2 del controllo negativo.
EC150
: concentrazioni stimate che producono un I.S. di 150 % di espressione di CD86.
Citometria a flusso
: tecnica citometrica in cui le cellule sospese in un fluido passano velocemente e singolarmente attraverso un fascio luminoso di eccitazione, producendo schemi di diffusione della luce che sono caratteristici delle cellule e dei loro componenti; le cellule sono spesso marcate con marcatori fluorescenti di modo che la luce sia dapprima assorbita e quindi emessa a un’altra frequenza.
Pericolo
: proprietà intrinseca di un agente o di una situazione che ha il potenziale di causare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti.
IATA (Integrated Approaches to Testing and Assessment, approcci integrati in materia di prove e valutazioni)
: approccio strutturato utilizzato per l’identificazione del pericolo (potenziale), la caratterizzazione del pericolo (potenza) e/o la valutazione della sicurezza (potenziale/potenza ed esposizione) di una sostanza chimica o di un gruppo di sostanze chimiche, che integra in modo strategico e ponderato tutti i dati pertinenti per orientare una decisione di tipo regolamentare concernente il pericolo potenziale e/o il rischio e/o la necessità di effettuare altre prove mirate e, pertanto, limitate allo stretto necessario.
Miscela
: una miscela o una soluzione composta di due o più sostanze.
Sostanza monocostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno 80 % (p/p).
Sostanza multicostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui più costituenti principali sono presenti in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente è che una miscela è ottenuta attraverso la miscelazione di due o più sostanze senza che avvenga una reazione chimica. Una sostanza multicostituente è il risultato di una reazione chimica.
Controllo positivo
: replica contenente tutti i componenti di un sistema di prova, trattata con una sostanza che notoriamente induce una risposta positiva. Perché sia possibile valutare la variabilità nel tempo della risposta del controllo positivo, l’intensità di tale risposta non dovrebbe essere eccessiva.
Preapteni
: sostanze chimiche che diventano sensibilizzanti tramite trasformazione abiotica, ad es. l’ossidazione.
Proapteni
: sostanze chimiche che necessitano di una bioattivazione enzimatica per dispiegare il loro potenziale di sensibilizzazione cutanea.
Pertinenza
: descrizione del rapporto tra la prova e l’effetto ricercato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l’effetto biologico di interesse. La pertinenza comprende una valutazione dell’accuratezza (concordanza) di una prova (14).
Affidabilità
: misura in cui una prova può essere riprodotta nel tempo all’interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna ai laboratori e la ripetibilità fra i laboratori (14).
Batteria di prove
: una batteria di prove consiste nel testare una o più sostanze chimiche in esame simultaneamente a un controllo con solvente/disperdente e a un controllo positivo.
Sensibilità
: proporzione di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l’accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (14).
S.I.
: indice di stimolazione. Valore relativo della media geometrica dell’intensità di fluorescenza (MFI) delle cellule esposte alla sostanza chimica in esame confrontato con l’MFI di cellule trattate con solvente/disperdente.
Controllo con solvente/disperdente
: campione non trattato contenente tutti i componenti di un sistema di prova, esclusa la sostanza chimica in esame, ma incluso il solvente/disperdente utilizzato. È utilizzato per stabilire la risposta di base per i campioni trattati con la sostanza chimica in esame disciolta o in dispersione stabile nello stesso solvente/disperdente. Se testato in concomitanza con un controllo con mezzo, questo campione dimostra anche se il solvente/disperdente interagisce con il sistema di prova.
Specificità
: proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l’accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (14).
Tampone di colorazione
: tampone fosfato salino contenente 5 % di siero fetale di vitello.
Sostanza
: un elemento chimico e i suoi composti, allo stato naturale od ottenuti per mezzo di un procedimento di produzione, compresi gli additivi necessari a mantenerne la stabilità e le impurità derivanti dal procedimento utilizzato, ma esclusi i solventi che possono essere separati senza compromettere la stabilità della sostanza o modificarne la composizione.
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi e livelli standardizzati di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di prudenza e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell’ambiente (16).
UVCB
: sostanze la cui composizione non è conosciuta o è variabile, prodotti di una reazione complessa o materiali origine biologica.
Metodo di prova valido
: metodo di prova la cui pertinenza e affidabilità sono ritenute soddisfacenti per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in assoluto ma solo in relazione a un determinato scopo (14).
Appendice 2.2
SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA
Prima di utilizzare come test di routine la prova descritta nella presente appendice per il metodo di prova B.71, i laboratori sono tenuti a dimostrare la loro competenza tecnica ottenendo correttamente la predizione attesa con il metodo U-SENS™ per le 10 sostanze raccomandate nella tabella 1 e ottenendo valori CV70 e EC150 che rientrino nel rispettivo intervallo di riferimento per almeno 8 delle 10 sostanze di riferimento. Tali sostanze sono state selezionate per rappresentare la gamma di risposte per quanto riguarda i pericoli di sensibilizzazione cutanea. Altri criteri di selezione riguardavano la disponibilità in commercio delle sostanze e la disponibilità di dati di riferimento in vivo e di dati in vitro di elevata qualità ottenuti con il metodo U-SENS™. Sono inoltre disponibili dati di riferimento pubblicati per il metodo U-SENS™ (1) (8).
Tabella 1
Sostanze raccomandate per la verifica della competenza tecnica con il metodo di prova U-SENS™
|
Sostanze di prova per la verifica della competenza
|
N. CAS
|
Stato fisico
|
Predizione in vivo (106)
|
U-SENS Solvente/disperdente
|
U-SENS Intervallo di riferimento CV70 in μg/ml (107)
|
U-SENS Intervallo di riferimento EC150 in μg/ml) (107)
|
|
4-Fenilendiammina
|
106-50-3
|
Solido
|
Sensibilizzante (elevato)
|
Mezzo completo (108)
|
<30
|
Positivo (≤10)
|
|
Acido picrilsulfonico
|
2508-19-2
|
Liquido
|
Sensibilizzante (elevato)
|
Mezzo completo
|
>50
|
Positivo (≤50)
|
|
Maleato di dietile
|
141-05-9
|
Liquido
|
Sensibilizzante (moderato)
|
DMSO
|
10-100
|
Positivo (≤20)
|
|
Resorcinolo
|
108-46-3
|
Solido
|
Sensibilizzante (moderato)
|
Mezzo completo
|
>100
|
Positivo (≤50)
|
|
Alcol cinnamico
|
104-54-1
|
Solido
|
Sensibilizzante (debole)
|
DMSO
|
>100
|
Positivo (10-100)
|
|
4-Allilanisolo
|
140-67-0
|
Liquido
|
Sensibilizzante (debole)
|
DMSO
|
>100
|
Positivo (<200)
|
|
Saccarina
|
81-07-2
|
Solido
|
Non sensibilizzante
|
DMSO
|
>200
|
Negativo (>200)
|
|
Glicerolo
|
56-81-5
|
Liquido
|
Non sensibilizzante
|
Mezzo completo
|
>200
|
Negativo (>200)
|
|
Acido lattico
|
50-21-5
|
Liquido
|
Non sensibilizzante
|
Mezzo completo
|
>200
|
Negativo (>200)
|
|
Acido salicilico
|
69-72-7
|
Solido
|
Non sensibilizzante
|
DMSO
|
>200
|
Negativo (>200)
|
|
Abbreviazioni: N.CAS (Chemical Abstracts Service Registry Number) = Numero CAS (numero di registrazione nell’inventario europeo delle sostanze chimiche).
|
Appendice 3
SENSIBILIZZAZIONE CUTANEA IN VITRO: METODO DI PROVA IL-8 LUC
CONSIDERAZIONI INIZIALI E LIMITI
|
1.
|
Contrariamente ai metodi di prova che analizzano l'espressione di marcatori di superficie cellulare, il metodo di prova IL8-Luc quantifica le variazioni dell'espressione di IL-8, una citotossina associata all'attivazione di cellule dendritiche (DC). Nella linea cellulare reporter IL-8 derivata dalla linea cellulare THP-1 (THP-G8, stabilita dalla linea cellulare di leucemia monocitica acuta umana THP-1), l'espressione di IL-8 è misurata in seguito all'esposizione a sensibilizzanti (1). Il livello di espressione della luciferasi è quindi utilizzato per contribuire a distinguere tra sensibilizzanti e non sensibilizzanti cutanei.
|
|
2.
|
Il metodo di prova IL-8 Luc è stato oggetto di uno studio di validazione (2) condotto dal Centro giapponese di validazione di metodi alternativi (JaCVAM), dal ministero dell'Economia, del commercio e dell'industria (METI) e dalla Società giapponese per i metodi alternativi alla sperimentazione animale (JSAAE), cui ha fatto seguito una revisione indipendente inter pares (3) sotto la guida del JaCVAM e del ministero della Salute, del lavoro e della sicurezza sociale (MHLW) con il sostegno della Cooperazione internazionale relativa ai metodi alternativi alla sperimentazione animale (ICATM). Tenuto conto di tutti i dati disponibili e dei pareri formulati dalle autorità di regolamentazione e dai portatori di interessi, si ritiene che il metodo di prova IL-8 Luc possa efficacemente contribuire, nell'ambito di una metodologia integrata di tipo IATA, a distinguere tra sensibilizzanti e non sensibilizzanti cutanei a fini di classificazione ed etichettatura del pericolo. In letteratura figurano esempi dell'uso dei dati del metodo di prova IL-8 Luc in combinazione con altre informazioni (4) (5) (6).
|
|
3.
|
È stato dimostrato che il metodo di prova IL-8 Luc può essere applicato in laboratori con esperienza di coltura cellulare e misurazione della luciferasi. Il livello di riproducibilità era dell'ordine dell'87,7 % all'interno dello stesso laboratorio e dell'87,5 % fra laboratori diversi (2). I dati ottenuti nello studio di valutazione (2) e in altri studi pubblicati (1) (6) indicano che, rispetto all'LLNA, il metodo di prova IL-8 Luc ha permesso di ottenere una predizione positiva o negativa per 118 sostanze chimiche su 143 e ha fornito risultati non conclusivi per 25 sostanze; l'accuratezza del metodo IL-8 Luc nella distinzione tra sensibilizzanti (categoria 1 del GHS dell'ONU/CLP) e non sensibilizzanti cutanei («senza categoria» del GHS dell'ONU/CLP) è dell'86 % (101/118), con una sensibilità del 96 % (92/96) e una specificità del 41 % (9/22). Se non si tiene conto delle sostanze chimiche che esulano dall'ambito di applicabilità descritto di seguito (paragrafo 5), il metodo di prova IL-8 Luc ha permesso di classificare 113 sostanze chimiche su 136 come positive o negative e 23 sostanze come non conclusive; l'accuratezza del metodo IL-8 Luc è dell'ordine dell'89 % (101/113), con una sensibilità del 96 % (92/96) e una specificità del 53 % (9/17). Se ci si avvale dei dati relativi all'uomo citati in Urbisch et al. (7), il metodo di prova IL-8 Luc ha permesso di classificare 76 sostanze chimiche su 90 come positive o negative e 14 sostanze come non conclusive; l'accuratezza dell'IL-8 Luc è dell'ordine dell'80 % (61/76), con una sensibilità del 93 % (54/58) e una specificità del 39 % (7/18). Se non si tiene conto delle sostanze chimiche che esulano dall'ambito di applicabilità, il metodo di prova IL-8 Luc ha permesso di classificare 71 sostanze chimiche su 84 come positive o negative e 13 sostanze come non conclusive; l'accuratezza dell'IL-8 Luc è dell'ordine dell'86 % (61/71), con una sensibilità del 93 % (54/58) e una specificità del 54 % (7/13). È probabile che i falsi negativi nelle predizioni formulate con il metodo di prova IL-8 Luc riguardino piuttosto le sostanze chimiche con una potenza di sensibilizzazione cutanea da bassa a moderata (sottocategoria 1B del GHS dell'ONU/CLP) rispetto alle sostanze chimiche con una potenza elevata (sottocategoria 1A del GHS dell'ONU/CLP) (6). L'insieme di queste informazioni indica che l'IL-8 Luc può efficacemente contribuire all'identificazione del pericolo di sensibilizzazione cutanea. I valori di accuratezza forniti per il metodo di prova IL-8 Luc utilizzato isolatamente sono puramente indicativi, in quanto la prova dovrebbe essere considerata in combinazione con altre fonti di informazione nell'ambito di un approccio IATA e in conformità alle disposizioni dei paragrafi 7 e 8 dell'Introduzione generale. Inoltre, nel valutare le prove di sensibilizzazione cutanea che non utilizzano la sperimentazione animale si deve tenere presente che l'LLNA, come pure altre prove che utilizzano la sperimentazione animale, può non riflettere pienamente la situazione per l'uomo.
|
|
4.
|
I dati attualmente disponibili indicano che il metodo di prova IL-8 Luc è applicabile alle sostanze chimiche in esame relative a diversi gruppi funzionali organici, meccanismi di reazione, potenze di sensibilizzazione cutanea (determinate negli studi in vivo) e proprietà fisico-chimiche (2)(6).
|
|
5.
|
Nonostante utilizzi il solvente X-VIVOTM 15, il metodo di prova IL-8 Luc ha permesso di valutare correttamente sostanze chimiche con un Log Kow > 3,5 e sostanze con un grado di idrosolubilità di circa 100 μg/ml calcolato con il programma EPI SuiteTM e la sua efficienza nell'individuare sensibilizzanti scarsamente idrosolubili è superiore a quella del metodo di prova IL-8 Luc realizzato con il solvente dimetilsulfossido (DMSO) (2). Tuttavia, i risultati negativi ottenuti con sostanze chimiche in esame che non vengono disciolte a 20 mg/ml possono dar luogo a falsi negativi, in quanto le sostanze non sono solubili in X-VIVOTM 15. Per tali sostanze chimiche, quindi, non si devono prendere in considerazione risultati negativi. Nell'ambito dello studio di validazione si è osservato un tasso elevato di falsi negativi per le anidridi. Inoltre, a causa della capacità metabolica limitata della linea cellulare utilizzata (8) e delle condizioni sperimentali, i proapteni (sostanze che richiedono un'attivazione enzimatica) e i preapteni (sostanze attivate mediante ossidazione) possono dare risultati negativi nella prova. Tuttavia, anche se i risultati negativi ottenuti con potenziali pre/proapteni devono essere interpretati con cautela, va osservato che il metodo di prova IL-8 Luc ha permesso di identificare correttamente 11 preapteni su 11, 6 proapteni su 6 e 6 pre/proapteni su 8 nella serie di dati dell'IL-8 Luc (2). L'esame globale recentemente effettuato su tre metodi di prova che non comportano l'impiego di animali (DPRA, KeratinoSens™ e h-CLAT) per l'individuazione di preapteni e proapteni (9) e il fatto che le cellule THP-G8 utilizzate nel metodo di prova IL-8 Luc sono una linea cellulare derivata dalla linea THP-1 che è utilizzata nell'h-CLAT, consentono di concludere che l'IL-8 Luc, in combinazione con altri metodi, può inoltre contribuire a migliorare la sensibilità delle prove senza l'impiego di animali nell'individuazione di pre- e proapteni. I tensioattivi testati fino ad oggi hanno dato risultati (falsi) positivi a prescindere dal tipo (cationici, anionici o non-ionici). Infine, le sostanze chimiche che interferiscono con la luciferasi possono alterarne l'attività/misurazione, provocando un'inibizione apparente o una maggiore luminescenza (10). Ad esempio, è stato segnalato che concentrazioni di fitoestrogeni superiori a 1 μM interferirebbero con i segnali di luminescenza in altri metodi di prova con geni reporter basati sulla luciferasi a causa della sovrattivazione del gene reporter della luciferasi. Di conseguenza, è necessario esaminare attentamente l'espressione della luciferasi ottenuta in presenza di concentrazioni elevate di fitoestrogeni o di composti sospettati di indurre un'attivazione del gene reporter della luciferasi comparabile a quella causata dai fitoestrogeni (11). Sulla base di quanto precede, i tensioattivi, le anidridi e le sostanze chimiche che interferiscono con la luciferasi non rientrano nel campo di applicabilità del presente metodo di prova. Nel caso in cui si dimostri che il metodo di prova IL-8 Luc non è applicabile ad altre categorie specifiche di sostanze chimiche in esame, è opportuno evitare di utilizzare tale protocollo per tali categorie.
|
|
6.
|
Come già precisato, il metodo di prova IL-8 Luc aiuta a distinguere tra sensibilizzanti e non sensibilizzanti cutanei. Sono necessarie ulteriori ricerche, di preferenza basate su dati relativi all'uomo, per determinare se i risultati del protocollo IL-8 Luc possano contribuire, insieme ad altre fonti di informazione, alla valutazione della potenza di sensibilizzazione.
|
|
7.
|
Le definizioni figurano nell'appendice 3.1.
|
PRINCIPIO DELLA PROVA
|
8.
|
Il metodo di prova IL-8 Luc si avvale di una linea cellulare di leucemia monocitica umana THP-1 proveniente dall'American Type Culture Collection (Manassas, VA, USA). A partire da questa linea cellulare il dipartimento di dermatologia della facoltà di medicina dell'università di Tohoku ha sviluppato la linea THP-G8, una linea cellulare reporter IL-8 derivata dalla THP-1 che contiene i geni della luciferasi arancione (Stable Luciferase Orange, SLO) e della luciferasi rossa (Stable Luciferase Red, SLR) sotto il controllo dei promotori dell'IL-8 e della gliceraldeide-3-fosfato deidrogenasi (GAPDH) rispettivamente (1). Questo sistema consente di quantificare l'induzione del gene della luciferasi rilevando la luminescenza emessa da substrati di luciferasi che producono una luminescenza soddisfacente come indicatore dell'attività di IL-8 e della GAPDH nelle cellule in seguito all'esposizione a sostanze sensibilizzanti.
|
|
9.
|
Il sistema di prova bicromatico comprende una luciferasi che emette luce arancione (SLO; λmax = 580 nm) (12) per l'espressione genica del promotore IL-8 e una luciferasi che emette luce rossa (SLR; λmax = 630 nm) (13) per l'espressione genica del promotore del controllo interno, GAPDH. Le due luciferasi emettono colori diversi quando reagiscono con la d-luciferina di lucciola e la loro luminescenza è misurata simultaneamente in una reazione a fase singola dividendo la luce emessa dalla miscela di prova mediante un filtro ottico (14) (appendice 3.2).
|
|
10.
|
Le cellule THP-G8 sono trattate per 16 ore con la sostanza chimica in esame, quindi si misura l'attività della luciferasi SLO (SLO-LA), indice dell'attività del promotore IL-8, e l'attività della luciferasi SLR (SLR-LA), indice dell'attività del promotore GAPDH. Per facilitare la comprensione delle abbreviazioni, SLO-LA e SLR-LA sono indicate rispettivamente come IL8LA e GAPLA. Nella tabella 1 figura una descrizione dei termini associati all'attività della luciferasi nel metodo di prova IL-8 Luc. I valori misurati sono utilizzati per calcolare l'IL8LA normalizzata (nIL8LA), che è il rapporto IL8LA/GAPLA, l'induzione di nIL8LA (Ind-IL8LA), che è il rapporto tra la media aritmetica dei quattro valori misurati di nIL8LA delle cellule THP-G8 trattate con una sostanza chimica in esame e i valori di nIL8LA delle cellule THP-G8 non trattate, e l'inibizione di GAPLA (Inh-GAPLA), che è il rapporto tra la media aritmetica dei quattro valori misurati di GAPLA delle cellule THP-G8 trattate con una sostanza chimica in esame e i valori di GAPLA delle cellule THP-G8 non trattate, e che è utilizzata come indicatore di citotossicità.
Tabella 1
Descrizione dei termini associati all'attività della luciferasi nel metodo di prova IL-8 Luc.
|
Abbreviazioni
|
Definizione
|
|
GAPLA
|
Attività della luciferasi SLR indicativa dell'attività del promotore GAPDH
|
|
IL8LA
|
Attività della luciferasi SLO indicativa dell'attività del promotore IL-8
|
|
nIL8LA
|
IL8LA / GAPLA
|
|
Ind-IL8LA
|
nIL8LA delle cellule THP-G8 trattate con le sostanze chimiche / nIL8LA delle cellule non trattate
|
|
Inh-GAPLA
|
GAPLA delle cellule THP-G8 trattate con le sostanze chimiche /GAPLA delle cellule non trattate
|
|
CV05
|
La concentrazione minima della sostanza chimica alla quale Inh-GAPLA è < 0,05.
|
|
|
11.
|
Sono disponibili standard di prestazione (15) che facilitano la validazione di metodi di prova della luciferasi IL-8 in vitro modificati simili al metodo di prova IL-8 Luc e permettono di modificare rapidamente la linea guida dell'OCSE 442E per poterveli inserire. L'accettazione reciproca dei dati nel quadro dell'OCSE sarà garantita solo per i metodi di prova validati in base agli standard di prestazione, se tali metodi di prova sono stati esaminati e integrati dall'OCSE nella linea guida 442E.
|
DIMOSTRAZIONE DELLA COMPETENZA
|
12.
|
Prima di utilizzare come test di routine la prova descritta nella presente appendice relativa al metodo di prova B.71, i laboratori devono dimostrare le loro competenze tecniche utilizzando le 10 sostanze di riferimento elencate nell'appendice 3.3, conformemente alle buone pratiche per i metodi in vitro (17). Inoltre gli utilizzatori del metodo devono mantenere una banca dati storica contenente i dati ottenuti con i controlli di reattività (cfr. il paragrafo 15) nonché con i controlli positivi e con solvente/disperdente (cfr. i paragrafi 21-24) e utilizzare tali dati per confermare la persistenza nel tempo della riproducibilità della prova nel loro laboratorio.
|
PROCEDURA
|
13.
|
La procedura operativa standard per il metodo di prova IL-8 Luc è disponibile e dovrebbe essere applicata quando si utilizza questo metodo di prova (18). I laboratori che intendono applicare questo metodo di prova possono ottenere la linea cellulare ricombinante THP-G8 dal laboratorio GPC Lab. Co. Ltd., Tottori, Giappone, previa sottoscrizione di un accordo di trasferimento di materiale (Material Transfer Agreement, MTA) secondo le condizioni riportate nel modello dell'OCSE. Nei paragrafi che seguono è fornita una descrizione dei principali elementi e procedure del metodo di prova.
|
Preparazione delle cellule
|
14.
|
Per l'esecuzione del metodo di prova IL-8 Luc è opportuno utilizzare la linea cellulare THP-G8 del laboratorio GPC Lab. Co. Ltd., Tottori, Giappone, (cfr. i paragrafi 8 e 13). Al ricevimento, le cellule sono moltiplicate (2-4 passaggi) al fine di costituire uno stock omogeneo da conservare in stato di congelamento. Le cellule di questo stock possono essere moltiplicate fino a un massimo di 12 passaggi o per un massimo di 6 settimane. Il mezzo di coltura utilizzato per la propagazione cellulare è l'RPMI-1640 contenente 10 % di siero fetale bovino (FBS), una soluzione antibiotica/antimicotica (100 U/ml di penicillina G, 100 μg/ml di streptomicina e 0,25 μg/ml di amfotericina B in una soluzione salina allo 0,85 %) (ad es. GIBCO Cat#15240-062), 0,15 μg/ml di puromicina (ad es. CAS:58-58-2) e 300 μg/ml di G418 (ad es. CAS:108321-42-2).
|
|
15.
|
Prima di essere utilizzate per la prova, le cellule devono essere qualificate mediante un controllo di reattività da realizzarsi dopo 1-2 settimane o 2-4 passaggi dallo scongelamento mediante 4-nitrobenzilbromuro (4-NBB) (CAS:100-11-8, purezza ≥ 99 %) come controllo positivo e acido lattico (LA) (CAS:50-21-5, purezza ≥ 85 %) come controllo negativo. Il 4-NBB deve dar luogo a una risposta positiva per Ind-IL8LA (≥ 1,4) e l'acido lattico deve dar luogo a una risposta negativa per Ind-IL8LA (< 1,4). Per la prova vengono utilizzate unicamente le cellule che superano il controllo di reattività. Il controllo va effettuato secondo le procedure descritte ai paragrafi 22-24.
|
|
16.
|
Per la prova, le cellule THP-G8 sono inoculate a una densità compresa tra 2 e 5 × 105 cellule/ml e precoltivate in fiasche di coltura per una durata compresa tra 48 e 96 ore. Il giorno della prova le cellule raccolte dalla fiasca di coltura sono lavate con RPMI-1640 contenente 10 % di FBS senza antibiotici, poi sono rimesse in sospensione a una densità di 1 × 106 cellule/ml con RPMI-1640 contenente 10 % di FBS senza antibiotici. Quindi le cellule sono ripartite su una piastra nera a fondo piatto a 96 pozzetti (ad es. Costar Cat#3603) con 50 μl (5 × 104 cellule/pozzetto).
|
Preparazione della sostanza chimica in esame e delle sostanze di controllo
|
17.
|
La sostanza chimica in esame e le sostanze di controllo vengono preparate il giorno della prova. Per il metodo di prova IL-8 Luc, le sostanze chimiche in esame vengono disciolte in X-VIVOTM 15, un mezzo senza siero disponibile in commercio (Lonza, 04-418Q), fino a una concentrazione finale di 20 mg/ml. Il mezzo X-VIVOTM 15 è addizionato a 20 mg di sostanza chimica in esame (a prescindere dalla solubilità della sostanza chimica) in una provetta da microcentrifuga fino a raggiungere un volume di 1 ml e successivamente agitato vigorosamente su vortex e posto su un rotore a una velocità massima di 8 rpm per 30 minuti a una temperatura ambiente di circa 20oC. Inoltre, se le sostanze chimiche solide continuano a essere insolubili, la provetta è sonicata fino a dissoluzione completa della sostanza o fino all'ottenimento di una dispersione stabile. Se le sostanze chimiche in esame sono solubili in X-VIVOTM 15, la soluzione è diluita di un fattore 5 con X-VIVOTM 15 e utilizzata come soluzione madre della sostanza chimica in X-VIVOTM 15 (4 mg/ml). Se le sostanze chimiche in esame non sono solubili in X-VIVOTM 15, la miscela è nuovamente agitata su rotore per almeno 30 minuti e successivamente centrifugata a 15000 rpm (≈ 20000 g) per 5 minuti; il supernatante così ottenuto è utilizzato come soluzione madre della sostanza chimica in esame in X-VIVOTM 15. Qualora si utilizzino altri solventi, quali DMSO, acqua o mezzo di coltura, è necessario fornire un'adeguata motivazione scientifica. L'appendice 3.5 illustra la procedura dettagliata per la diluizione delle sostanze chimiche. Le soluzioni in X-VIVOTM 15 descritte ai paragrafi 18-23 sono mescolate nella proporzione 1:1 (v/v) con le sospensioni cellulari preparate nella piastra nera a fondo piatto a 96 pozzetti (cfr. il paragrafo 16).
|
|
18.
|
La prima batteria di prove è intesa a determinare la concentrazione citotossica e a valutare il potenziale di sensibilizzazione cutanea delle sostanze chimiche. Avvalendosi di X-VIVOTM 15, si effettuano diluizioni in serie di fattore 2 delle soluzioni madre delle sostanze chimiche in esame in X-VIVOTM 15 (cfr. l'appendice 3.5) mediante un blocco a 96 pozzetti (ad es. Costar Cat#EW-01729-03). Quindi 50 μl/pozzetto di soluzione diluita sono addizionati a 50 μl di sospensione cellulare in una piastra nera a fondo piatto da 96 pozzetti. Pertanto, per le sostanze chimiche in esame solubili in X-VIVO TM 15, le concentrazioni finali della sostanza chimica vanno da 0,002 a 2 mg/ml (appendice 3.5). Per le sostanze chimiche in esame non solubili in X-VIVO TM 15 a 20 mg/ml vengono determinati unicamente fattori di diluizione compresi tra 2 e 210, nonostante le concentrazioni finali effettive delle sostanze chimiche rimangano incerte e dipendano dalla concentrazione di saturazione delle sostanze stesse nella soluzione madre di X-VIVOTM 15.
|
|
19.
|
Nelle batterie di prove successive (seconda, terza e quarta replica), la soluzione madre di X-VIVOTM 15 è preparata a una concentrazione 4 volte superiore alla concentrazione di vitalità cellulare 05 (CV05; la concentrazione minima alla quale Inh-GAPLA è < 0,05) osservata nel primo esperimento. Se il valore Inh-GAPLA non scende al di sotto di 0,05 alla concentrazione massima utilizzata nella prima prova, la soluzione madre X-VIVOTM 15 è preparata alla concentrazione massima della prima batteria di prove. La concentrazione CV05 è calcolata dividendo la concentrazione della soluzione madre della prima batteria di prove per il fattore di diluizione di CV05 (X) necessario per diluire la soluzione madre fino a raggiungere il valore CV05 (cfr. l'appendice 3.5). Per le sostanze chimiche in esame non solubili in X-VIVO a 20 mg/ml, il valore CV05 è determinato dalla concentrazione della soluzione madre × 1/X. Per le batterie di prove da 2 a 4 viene preparata una seconda soluzione madre a una concentrazione di 4 × CV05 (appendice 3.5).
|
|
20.
|
Diluizioni in serie di fattore 1,5 delle seconde soluzioni madre di X-VIVOTM 15 vengono effettuate utilizzando un blocco a 96 pozzetti. Quindi 50 μl/pozzetto di soluzione diluita sono addizionati a 50 μl di sospensione cellulare nei pozzetti di una piastra nera a fondo piatto da 96 pozzetti. Ciascuna concentrazione della sostanza chimica in esame deve essere testata in 4 pozzetti. I campioni sono successivamente miscelati su un agitatore di piastre e messi in incubazione per 16 ore a 37 oC e 5 % CO2, quindi si misura l'attività della luciferasi come descritto nel prosieguo.
|
|
21.
|
Il controllo con solvente è la miscela di 50 μl/pozzetto di X-VIVOTM 15 e 50 μl/pozzetto di sospensione cellulare in RPMI-1640 contenente 10 % di FBS.
|
|
22.
|
Il controllo positivo raccomandato è il 4-NBB. A 20 mg di 4-NBB preparati in una provetta da microcentrifuga da 1,5-ml si aggiunge X-VIVOTM 15 fino a raggiungere 1 ml. La provetta è agitata vigorosamente su vortex e posta su un rotore a una velocità massima di 8 rpm per almeno 30 minuti. Dopo centrifugazione a 20000 g per 5 minuti, il supernatante è diluito di un fattore 4 con X-VIVOTM 15 e 500 μl del supernatante diluito sono trasferiti in un blocco a 96 pozzetti. Il supernatante diluito è ulteriormente diluito con X-VIVOTM 15 di un fattore 2 e 4 e 50 μl della soluzione sono addizionati a 50 μl di sospensione cellulare THP-G8 nei pozzetti di una piastra nera a fondo piatto a 96 pozzetti (appendice 3.6). Ciascuna concentrazione del controllo positivo deve essere testata in 4 pozzetti. La piastra è agitata su un agitatore e messa in incubazione in un incubatore a CO2 per 16 ore (37 oC, 5 % CO2), quindi si misura l'attività della luciferasi come descritto al paragrafo 29.
|
|
23.
|
Il controllo negativo raccomandato è l'acido lattico. A 20 mg di acido lattico preparati in una provetta da microcentrifuga da 1,5-ml si aggiunge X-VIVOTM 15 fino a raggiungere 1 ml (20 mg/ ml). Si diluiscono 20 mg/ml di soluzione di acido lattico di un fattore 5 con X-VIVOTM 15 (4 mg/ml); 500 μl di questa soluzione di acido lattico a 4 mg/ml vengono poi trasferiti in un pozzetto di un blocco a 96 pozzetti. Questa soluzione viene diluita di un fattore 2 con X-VIVOTM 15, quindi nuovamente diluita di un fattore 2 per ottenere soluzioni a 2 mg/ml e 1 mg/ml. 50 μl di queste 3 soluzioni e del controllo con disperdente (X-VIVOTM 15) sono addizionati a 50 μl di sospensione cellulare THP-G8 nei pozzetti di una piastra nera a fondo piatto a 96 pozzetti. Ciascuna concentrazione del controllo negativo è testata in 4 pozzetti. La piastra è agitata su un agitatore e messa in incubazione in un incubatore a CO2 per 16 ore (37 oC, 5 % CO2), quindi si misura l'attività della luciferasi come descritto al paragrafo 29.
|
|
24.
|
Altri controlli positivi o negativi adeguati possono essere utilizzati in caso di disponibilità di dati storici da cui ricavare criteri di accettabilità comparabili per la prova.
|
|
25.
|
Si avrà cura di evitare l'evaporazione delle sostanze chimiche volatili in esame nonché la contaminazione incrociata tra i pozzetti da parte delle sostanze stesse, ad esempio sigillando la piastra prima dell'incubazione con le sostanze chimiche in esame.
|
|
26.
|
Per le sostanze chimiche in esame e il controllo con solvente occorrono da 2 a 4 batterie di prove per ottenere una predizione negativa o positiva (cfr. la tabella 2). Ogni batteria di prove è eseguita in un giorno diverso con una nuova soluzione madre delle sostanze chimiche in esame in X-VIVOTM 15 e cellule raccolte in modo indipendente. Le cellule possono provenire dallo stesso passaggio.
|
Misurazioni dell'attività della luciferasi
|
27.
|
La luminescenza è misurata con un luminometro per micropiastre a 96 pozzetti dotato di filtri ottici, ad es. delle serie Phelios (ATTO, Tokyo, Giappone), Tristan 941 (Berthold, Bad Wildbad, Germania) o ARVO (PerkinElmer, Waltham, MA, USA). Il luminometro deve essere calibrato per ogni prova al fine di garantire la riproducibilità (19). Per la calibrazione esistono luciferasi ricombinanti a emissione arancione e rossa.
|
|
28.
|
In ciascun pozzetto della piastra contenente la sospensione cellulare trattata con o senza sostanza chimica si trasferiscono 100 μl di reagente preriscaldato Tripluc® Luciferase (Tripluc). La piastra è agitata per 10 minuti a una temperatura ambiente di circa 20 oC e successivamente collocata nel luminometro per misurare l'attività della luciferasi. La bioluminescenza è misurata per 3 secondi senza filtro ottico (F0) e per altri 3 secondi con filtro ottico (F1). L'eventuale ricorso a impostazioni alternative, ad esempio per adeguarsi al modello di luminometro utilizzato, deve essere opportunamente giustificato.
|
|
29.
|
Per ogni concentrazione i parametri sono calcolati a partire dai valori misurati, ad esempio IL8LA, GAPLA, nIL8LA, Ind-IL8LA, Inh-GAPLA, la media ± deviazione standard (SD) di IL8LA, la media ± SD di GAPLA, la media ± SD di nIL8LA, la media ± SD di Ind-IL8LA, la media ± SD di Inh-GAPLA e l'intervallo di confidenza del 95 % per Ind-IL8LA. Le definizioni dei parametri utilizzati nel presente paragrafo sono riportate rispettivamente nelle appendici 3.1 e 3.4.
|
|
30.
|
Prima di procedere alla misurazione, nelle prove con reporter policromatici si effettua generalmente una discriminazione cromatica mediante rivelatori (luminometro e lettore di piastre) dotati di filtri ottici, quali filtri taglia banda (passa-alto o passa-basso) o filtri passa-banda. I coefficienti di trasmissione dei filtri per ciascun colore del segnale di bioluminescenza devono essere calibrati prima della prova, come descritto nell'appendice 3.2.
|
DATI E RELAZIONI
Valutazione dei dati
|
31.
|
I criteri da rispettare in ogni batteria di prove per formulare una decisione positiva o negativa sono i seguenti:
|
—
|
una predizione formulata con il metodo di prova IL-8 Luc è considerata positiva se una sostanza chimica in esame presenta un valore Ind-IL8LA ≥ 1,4 e il limite inferiore dell'intervallo di confidenza del 95 % di Ind-IL8LA ≥ 1,0
|
|
—
|
una predizione formulata con il metodo di prova IL-8 Luc è considerata negativa se una sostanza chimica in esame presenta un valore Ind-IL8LA < 1,4 e il limite inferiore dell'intervallo di confidenza del 95 % di Ind-IL8LA < 1,0
|
|
Modello predittivo
|
32.
|
Le sostanze chimiche in esame che producono due risultati positivi nella 1a, 2a, 3a o 4a batteria di prove sono considerate positive, quelle che producono tre risultati negativi nella 1a, 2a, 3a o 4a batteria di prove sono considerate presumibilmente negative (tabella 2). Tra le sostanze chimiche presumibilmente negative, le sostanze disciolte a 20 mg/ml di X-VIVOTM 15 sono considerate negative, mentre le sostanze non disciolte a 20 mg/ml di X-VIVOTM 15 non sono prese in considerazione (figura 1).
Tabella 2
Criteri di identificazione delle sostanze positive e presumibilmente negative
|
1a batteria di prove
|
2a batteria di prove
|
3a batteria di prove
|
4a batteria di prove
|
Predizione finale
|
|
Positiva
|
Positiva
|
–
|
–
|
Positiva
|
|
Negativa
|
Positiva
|
–
|
Positiva
|
|
Negativa
|
Positiva
|
Positiva
|
|
Negativa
|
Presumibilmente negativa
|
|
Negativa
|
Positiva
|
Positiva
|
–
|
Positiva
|
|
Negativa
|
Positiva
|
Positiva
|
|
Negativa
|
Presumibilmente negativa
|
|
Negativa
|
Positiva
|
Positiva
|
Positiva
|
|
Negativa
|
Presumibilmente negativa
|
|
Negativa
|
–
|
Presumibilmente negativa
|
|
Figura 1
Modello predittivo per la decisione finale

Criteri di accettabilità
|
33.
|
Quando si utilizza il metodo di prova IL-8 Luc devono essere soddisfatti i criteri di accettabilità elencati di seguito:
|
—
|
il valore Ind-IL8LA deve essere superiore a 5,0 ad almeno una concentrazione del controllo positivo, 4-NBB, in ciascuna batteria di prove;
|
|
—
|
il valore Ind-IL8LA deve essere inferiore a 1,4 ad ogni concentrazione del controllo negativo, l'acido lattico, in ciascuna batteria di prove;
|
|
—
|
i dati ottenuti da piastre in cui il valore GAPLA dei pozzetti di controllo contenenti cellule e Tripluc, ma non sostanze chimiche, è inferiore a 5 volte il valore del pozzetto che contiene unicamente il mezzo di prova (50 μl/pozzetto di RPMI-1640 contenente 10 % di FBS e 50 μl/pozzetto di X-VIVOTM 15) devono essere scartati;
|
|
—
|
i dati ottenuti da piastre in cui il valore Inh-GAPLA di tutte le concentrazioni delle sostanze chimiche in esame o delle sostanze di controllo è inferiore a 0,05 devono essere scartati. In questo caso occorre ripetere la prima prova in modo che la concentrazione finale massima della prova ripetuta corrisponda alla concentrazione finale minima della prova precedente.
|
|
Relazione sulla prova
|
34.
|
La relazione sulla prova comprende le informazioni riportate di seguito.
|
Sostanze chimiche in esame
Sostanza monocostituente:
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, idrosolubilità, peso molecolare, altre proprietà fisico-chimiche pertinenti, se disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
solubilità in X-VIVOTM 15. Per le sostanze chimiche insolubili in X-VIVOTM 15, se si osserva precipitazione o flottazione dopo la centrifugazione;
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
se non è stato utilizzato X-VIVOTM 15, motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
Sostanza multicostituente, UVCB o miscela:
|
caratterizzazione, nella misura del possibile, ad esempio attraverso l'identità chimica (cfr. sopra), purezza, proporzioni quantitative e proprietà fisico-chimiche pertinenti (cfr. sopra) dei costituenti, secondo i dati disponibili;
|
|
aspetto fisico, idrosolubilità, altre proprietà fisico-chimiche pertinenti, se disponibili;
|
|
peso molecolare o peso molecolare apparente nel caso di miscele/polimeri di composizione nota o altre informazioni pertinenti per la realizzazione dello studio;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
solubilità in X-VIVOTM 15. Per le sostanze chimiche insolubili in X-VIVOTM 15, se si osserva precipitazione o flottazione dopo la centrifugazione;
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
se non è stato utilizzato X-VIVOTM 15, motivazione della scelta del solvente/disperdente per ciascuna sostanza chimica in esame.
|
|
|
Controlli
Controllo positivo:
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS, il codice SMILES o InChI, la formula di struttura e/o altri identificatori;
|
|
aspetto fisico, idrosolubilità, peso molecolare e proprietà fisico-chimiche pertinenti aggiuntive, se del caso e a seconda dei dati disponibili;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
trattamento prima della prova, se del caso (ad esempio riscaldamento, frantumazione);
|
|
concentrazione/i testata/e;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
riferimento ai dati storici relativi ai controlli positivi che dimostrano la conformità ai criteri di accettabilità, se del caso.
|
Controllo negativo:
|
identificazione della sostanza chimica: denominazioni quali i nomi IUPAC o CAS, i numeri CAS e/o altri identificatori;
|
|
purezza, identità chimica delle impurezze, se del caso e se fattibile dal punto di vista pratico, ecc.;
|
|
aspetto fisico, peso molecolare e altre proprietà fisico-chimiche pertinenti, nel caso siano utilizzati controlli negativi diversi da quelli menzionati nelle linee guida e se disponibili;
|
|
condizioni di conservazione e stabilità, a seconda dei dati disponibili;
|
|
motivazione della scelta del solvente per ciascuna sostanza chimica in esame.
|
|
|
Condizioni della prova
|
nome e indirizzo dello sponsor, dell'infrastruttura utilizzata per la prova e del responsabile dello studio;
|
|
descrizione del sistema di prova utilizzato;
|
|
linea cellulare utilizzata, condizioni di conservazione e provenienza (ad esempio, l'infrastruttura dalla quale provengono le cellule);
|
|
numero di lotto e origine dell'FBS, nome del fornitore, numero di lotto della piastra nera a fondo piatto a 96 pozzetti e numero di lotto del reagente Tripluc;
|
|
numero di passaggi e densità cellulare utilizzata per la prova;
|
|
metodo di conteggio delle cellule utilizzato per l'inoculazione prima della prova e misure prese per assicurare una distribuzione omogenea del numero di cellule;
|
|
luminometro utilizzato (ad esempio il modello), compresi le impostazioni dello strumento, il substrato di luciferasi utilizzato e la dimostrazione della qualità delle misurazioni della luminescenza sulla base della prova descritta nell'appendice 3.2;
|
|
procedura utilizzata per dimostrare la competenza del laboratorio nell'esecuzione della prova (ad esempio sottoponendo la prova a sostanze di riferimento) o per dimostrare la riproducibilità della prova nel tempo.
|
|
|
Procedura
|
numero di repliche e di batterie di prove eseguite;
|
|
concentrazioni della sostanza chimica in esame, modalità di applicazione e tempo di esposizione (se diversi da quelli raccomandati);
|
|
descrizione dei criteri di valutazione e di decisione impiegati;
|
|
descrizione dei criteri di accettabilità dello studio;
|
|
descrizione di qualsiasi modifica della procedura sperimentale.
|
|
|
Risultati
|
misurazioni di IL8LA e GAPLA;
|
|
calcoli per nIL8LA, Ind-IL8LA e Inh-GAPLA;
|
|
intervallo di confidenza del 95 % di Ind-IL8LA;
|
|
grafico raffigurante le curve dose-risposta per l'induzione dell'attività della luciferasi e la vitalità;
|
|
descrizione di eventuali altre osservazioni pertinenti, se del caso.
|
|
|
Discussione dei risultati
|
discussione dei risultati ottenuti con il metodo di prova IL-8 Luc;
|
|
esame dei risultati della prova nel quadro di un approccio di tipo IATA, qualora siano disponibili altre informazioni pertinenti.
|
|
Conclusione
|
BIBLIOGRAFIA
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). In attesa di pubblicazione - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OCSE, Parigi, Francia
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OCSE, Parigi, Francia.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Organisation for Economic Cooperation and Development, Paris. Consultabile al seguente indirizzo: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, consultabile al seguente indirizzo: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OCSE, Parigi, Francia.
|
|
(21)
|
Nazioni Unite (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1.Consultabile al seguente indirizzo: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
Appendice 3.1
DEFINIZIONI
Accuratezza
: grado di concordanza tra i risultati ottenuti con la prova e i valori di riferimento comunemente accettati. Misura l’efficienza della prova e costituisce un aspetto della pertinenza. Il termine è spesso utilizzato come sinonimo di «concordanza», per indicare la proporzione di risultati corretti di una prova (16).
AOP (Adverse Outcome Pathway, meccanismo d’azione degli effetti avversi)
: sequenza di eventi che, a partire dalla struttura chimica di una sostanza chimica bersaglio o di un gruppo di sostanze chimiche simili, attraverso l’evento molecolare scatenante, produce un effetto avverso in vivo (20).
Sostanza chimica
: una sostanza o una miscela.
CV05
: vitalità cellulare 05, cioè la concentrazione minima alla quale le sostanze chimiche producono un valore Inh-GAPLA inferiore a 0,05.
FInSLO-LA a: bbreviazione utilizzata nella relazione di validazione e in studi precedenti concernenti il metodo di prova IL-8 Luc per fare riferimento a Ind IL8LA. Cfr. la definizione di Ind-IL8LA.
GAPLA
: attività di luciferasi che emette Stable Luciferase Red (SLR) (λmax = 630 nm), regolata dal promotore GAPDH, che dimostra la vitalità cellulare e il numero di cellule vitali.
Pericolo
: proprietà intrinseca di un agente o di una situazione che ha il potenziale di causare effetti nocivi se un organismo, un sistema o una (sotto-)popolazione vi sono esposti.
IATA (Integrated Approaches to Testing and Assessment, approcci integrati in materia di prove e valutazioni)
: approccio strutturato utilizzato per l’identificazione del pericolo (potenziale), la caratterizzazione del pericolo (potenza) e/o la valutazione della sicurezza (potenziale/potenza ed esposizione) di una sostanza chimica o di un gruppo di sostanze chimiche, che integra in modo strategico e ponderato tutti i dati pertinenti per orientare una decisione di tipo regolamentare concernente il pericolo potenziale e/o il rischio e/o la necessità di effettuare altre prove mirate e, pertanto, limitate allo stretto necessario.
II-SLR-LA
: abbreviazione utilizzata nella relazione di validazione e in studi precedenti concernenti il metodo di prova IL-8 Luc per fare riferimento a Inh-GAPLA. Cfr. la definizione di Inh-GAPLA.
IL-8 (Interleuchina-8)
: citochina derivata da cellule endoteliali, fibroblasti, cheratinociti, macrofagi e monociti che provoca la chemotassi di neutrofili e dei linfociti T.
IL8LA
: attività della luciferasi che emette Stable Luciferase Orange (SLO) (λmax = 580 nm), regolata dal promotore IL-8.
Ind-IL8LA
: variazione di induzione di nIL8LA. Si calcola dividendo il valore nIL8LA delle cellule THP-G8 trattate con le sostanze chimiche per il valore nIL8LA delle cellule THP-G8 non stimolate e rappresenta l’induzione del promotore IL-8 da parte delle sostanze chimiche.
Inh-GAPLA
: inibizione di GAPLA. Si calcola dividendo il valore GAPLA delle cellule THP-G8 trattate con sostanze chimiche per il valore GAPLA delle cellule THP-G8 non trattate e rappresenta la citotossicità delle sostanze chimiche.
Soglia minima di induzione (MIT)
: la concentrazione minima alla quale la sostanza chimica soddisfa il criterio di positività.
Miscela
: una miscela o una soluzione composta di due o più sostanze.
Sostanza monocostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui un costituente principale è presente in percentuale pari ad almeno 80 % (p/p).
Sostanza multicostituente
: sostanza, definita attraverso la sua composizione quantitativa, in cui più di un costituente principale è presente in concentrazione ≥ 10 % (p/p) e < 80 % (p/p). Una sostanza multicostituente è il risultato di un processo di fabbricazione. La differenza tra miscela e sostanza multicostituente è che una miscela è ottenuta attraverso la miscelazione di due o più sostanze senza che avvenga una reazione chimica. Una sostanza multicostituente è il risultato di una reazione chimica.
nIL8LA
: l’attività della luciferasi SLO che rispecchia l’attività del promotore IL 8 (IL8LA) normalizzata dall’attività della luciferasi SLR che rispecchia l’attività del promotore GAPDH (GAPLA). Rappresenta l’attività del promotore IL-8 tenendo in considerazione la vitalità cellulare o il numero delle cellule.
nSLO-LA
: abbreviazione utilizzata nella relazione di validazione e in studi precedenti concernenti il metodo di prova IL-8 Luc per fare riferimento a nIL8LA. Cfr. la definizione di nIL8LA.
Controllo positivo
: replica contenente tutti i componenti di un sistema di prova, trattata con una sostanza che notoriamente induce una risposta positiva. Perché sia possibile valutare la variabilità nel tempo della risposta del controllo positivo, l’intensità di tale risposta non dovrebbe essere eccessiva.
Preapteni
: sostanze chimiche che diventano sensibilizzanti tramite trasformazione abiotica.
Proapteni
: sostanze chimiche che necessitano di un’attivazione enzimatica per dispiegare il loro potenziale di sensibilizzazione cutanea.
Pertinenza
: descrizione del rapporto tra la prova e l’effetto ricercato; indica se la prova è significativa e utile per uno scopo specifico. È il grado con cui la prova misura o prevede correttamente l’effetto biologico di interesse. La pertinenza comprende una valutazione dell’accuratezza (concordanza) di una prova (16).
Affidabilità
: misura in cui una prova può essere riprodotta nel tempo all’interno dello stesso laboratorio o da laboratori diversi utilizzando il medesimo protocollo. È valutata calcolando la riproducibilità interna ai laboratori e la ripetibilità fra i laboratori (16).
Batteria di prove
: una batteria di prove consiste nel testare una o più sostanze chimiche in esame simultaneamente a un controllo con solvente/disperdente e a un controllo positivo.
Sensibilità
: proporzione di tutte le sostanze chimiche positive/attive correttamente classificate dalla prova. Misura l’accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (16).
SLO-LA
: abbreviazione utilizzata nella relazione di validazione e in studi precedenti concernenti il metodo di prova IL-8 Luc per fare riferimento a IL8LA. Cfr. la definizione di IL8LA.
SLR-LA
: abbreviazione utilizzata nella relazione di validazione e in studi precedenti concernenti il metodo di prova IL-8 Luc per fare riferimento a GAPLA. Cfr. la definizione di GAPLA.
Controllo con solvente/disperdente
: campione non trattato contenente tutti i componenti di un sistema di prova, esclusa la sostanza chimica in esame, ma incluso il solvente/disperdente utilizzato. È utilizzato per stabilire la risposta di base per i campioni trattati con la sostanza chimica in esame disciolta o in dispersione stabile nello stesso solvente/disperdente. Se testato in concomitanza con un controllo con mezzo, questo campione dimostra anche se il solvente/disperdente interagisce con il sistema di prova.
Specificità
: proporzione di tutte le sostanze chimiche negative/inattive correttamente classificate dalla prova. Misura l’accuratezza di una prova che produce risultati ordinabili in categorie ed è un elemento importante per valutare la pertinenza di una prova (16).
Sostanza
: elemento chimico e suoi composti allo stato naturale o ottenuti mediante un processo di produzione, compresi gli additivi necessari a conservare la stabilità del prodotto e le impurezze derivanti dal processo utilizzato, ma esclusi i solventi che possono essere separati senza ripercussioni sulla stabilità della sostanza o modifiche della sua composizione.
Tensioattivo
: denominato anche surfattante, è una sostanza, come un detergente, in grado di ridurre la tensione superficiale di un liquido consentendo quindi di formare schiuma o di penetrare solidi; è noto anche come agente umettante. (TG n. 437)
Sostanza chimica in esame
: qualsiasi sostanza o miscela testata applicando il presente metodo di prova.
THP-G8
: linea cellulare reporter IL-8 utilizzata nel metodo di prova IL-8 Luc. La linea cellulare umana macrofagica THP-1 è stata trasfettata con i geni della luciferasi SLO e SLR sotto il controllo dei promotori IL-8 e GAPDH, rispettivamente.
Sistema globale armonizzato di classificazione ed etichettatura delle sostanze chimiche delle Nazioni Unite (UN GHS)
: sistema di classificazione delle sostanze chimiche (sostanze e miscele) secondo tipi e livelli standardizzati di rischio fisico, sanitario e ambientale, che elabora i relativi elementi di comunicazione, quali pittogrammi, avvertenze, indicazioni di pericolo, consigli di prudenza e schede informative di sicurezza, per trasmettere informazioni sugli effetti avversi di dette sostanze a tutela delle persone (compresi datori di lavoro, lavoratori, trasportatori, consumatori e personale di pronto intervento) e dell’ambiente (21).
UVCB
: sostanze la cui composizione non è conosciuta o è variabile, prodotti di una reazione complessa o materiali origine biologica.
Metodo di prova valido
: metodo di prova la cui pertinenza e affidabilità sono ritenute soddisfacenti per uno scopo specifico e che si fonda su principi scientificamente provati. Un metodo di prova non è mai valido in assoluto ma solo in relazione a un determinato scopo.
Appendice 3.2
PRINCIPIO DI MISURAZIONE DELL’ATTIVITÀ DELLA LUCIFERASI E DETERMINAZIONE DEI COEFFICIENTI DI TRASMISSIONE DEI FILTRI OTTICI PER SLO E SLR
Il sistema di prova MultiReporter - Tripluc - può essere utilizzato con un luminometro per micropiastre dotato di un sistema di rilevazione policromatico con filtro ottico [ad es. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)]. Il filtro ottico utilizzato per la misurazione è un filtro passa-alto o passa-basso di 600–620 nm o un filtro passa-banda di 600–700 nm.
Misurazione di luciferasi bicromatiche con un filtro ottico
L’esempio che segue è realizzato con un apparecchio AB-2350 (ATTO). Questo luminometro è dotato di un filtro passa-alto di 600 nm (R60 HOYA Co., 600 nm LP, filtro 1) che consente di separare la luminescenza SLO (λmax = 580 nm) dalla luminescenza SLR (λmax = 630 nm).
Per determinare i coefficienti di trasmissione del filtro 600 nm LP si utilizzano in primo luogo enzimi luciferasi SLO e SLR purificati per i) misurare l’intensità di bioluminescenza di SLO e SLR senza filtro (F0), ii) misurare l’intensità di bioluminescenza di SLO e SLR che attraversa il filtro 600 nm LP (filtro 1), e iii) calcolare i coefficienti di trasmissione del filtro 600 nm LP per SLO e SLR indicati di seguito.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Se l’intensità di SLO e SLR nel campione di prova è definita rispettivamente come O e R, i) l’intensità della luce senza filtro (sistema interamente ottico) F0 e ii) l’intensità della luce trasmessa attraverso il filtro 600 nm LP (filtro 1) F1 sono descritte come segue.
F0 = O + R
F1 = κOR60 x O + κRR60 x R
Tali formule possono essere esplicitate come segue:

Quindi, a partire dai fattori di trasmittanza calcolati (κOR60 e κRR60) e dai valori F0 e F1 misurati, è possibile calcolare i valori O e R come segue:

Materiali e metodi per la determinazione del fattore di trasmittanza
|
(1)
|
Reagenti
Enzimi luciferasi purificati:
|
enzima SLO purificato liofilizzato
|
|
enzima SLR purificato liofilizzato
|
|
(nello studio di validazione gli enzimi sono stati ottenuti dal laboratorio GPC Lab. Co. Ltd., Tottori, Giappone, con una linea cellulare THP-G8)
|
Reagente della prova:
|
Tripluc® Luciferase (ad esempio da TOYOBO Cat#MRA-301)
|
Mezzo: per la prova della luciferasi (30 ml, conservato a 2 - 8 °C)
|
|
Reagente
|
Concentrazione
|
Concentrazione finale nel mezzo
|
Volume necessario
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
(2)
|
Preparazione della soluzione enzimatica:
Sciogliere l’enzima luciferasi purificato liofilizzato in una provetta aggiungendo 200 μl di 10 ~ 100 mM Tris/HCl o Hepes/HCl (pH 7,5 ~ 8,0) con aggiunta di 10 % (p/v) di glicerolo, suddividere la soluzione enzimatica in aliquote da 10 μl in provette monouso da 1,5 ml e riporre in congelatore a -80 °C. La soluzione enzimatica congelata può essere utilizzata per un massimo di 6 mesi. Per utilizzare la soluzione, aggiungere 1 ml di mezzo della prova di luciferasi (RPMI-1640 con 10 % di FBS) a ciascuna provetta contenente la soluzione enzimatica (soluzione enzimatica diluita) e conservare le provette su ghiaccio per evitare la disattivazione.
|
|
(3)
|
Misurazione della bioluminescenza
Scongelare il reagente della prova di luciferasi Tripluc® (Tripluc) e conservarlo a temperatura ambiente a bagnomaria o all’aria. Accendere il luminometro 30 minuti prima di iniziare la misurazione per consentire la stabilizzazione del fotomoltiplicatore. Trasferire 100 μl di soluzione enzimatica diluita in una piastra nera a fondo piatto a 96 pozzetti (campione di riferimento SLO in #B1, #B2, #B3, campione di riferimento SLR in #D1, #D2, #D3). Quindi, per mezzo di una pipetta automatica, trasferire 100 μl di Tripluc preriscaldato in ciascun pozzetto della piastra contenente la soluzione enzimatica diluita. Agitare la piastra per 10 minuti a temperatura ambiente (25 °C circa) su un agitatore per piastre. Asportare eventuali bolle dalle soluzioni contenute nei pozzetti. Collocare la piastra nel luminometro per misurare l’attività della luciferasi. La bioluminescenza è misurata per 3 secondi senza filtro ottico (F0) e per altri 3 secondi con filtro ottico (F1).
Il coefficiente di trasmissione del filtro ottico è stato calcolato come segue:
coefficiente di trasmissione (SLO (κOR60))= (#B1 di F1+ #B2 di F1+ #B3 di F1) / (#B1 di F0+ #B2 di F0+ #B3 of F0)
coefficiente di trasmissione (SLR (κRR60))= (#D1 di F1+ #D2 di F1+ #D3 di F1) / (#D1 di F0+ #D2 di F0+ #D3 of F0)
I fattori di trasmittanza calcolati sono utilizzati per tutte le misurazioni realizzate con lo stesso luminometro.
|
Controllo di qualità dell’attrezzatura
Devono essere utilizzate le procedure descritte nel protocollo IL-8 Luc (18).
Appendice 3.3
SOSTANZE CHIMICHE PER LA VERIFICA DELLA COMPETENZA TECNICA
Prima di utilizzare come test di routine la prova descritta nella presente appendice per il metodo di prova B.71, i laboratori sono tenuti a dimostrare la loro competenza tecnica ottenendo la predizione attesa con il metodo di prova IL-8 Luc per le 9 sostanze raccomandate nella tabella 1 e ottenendo valori che rientrino nel rispettivo intervallo di riferimento per almeno 8 delle 9 sostanze di riferimento (selezionate in modo da rappresentare la gamma delle risposte corrispondenti ai pericoli di sensibilizzazione cutanea). Altri criteri di selezione riguardavano la disponibilità in commercio delle sostanze e la disponibilità di dati di riferimento in vivo e di dati in vitro di elevata qualità ottenuti con il metodo di prova IL-8 Luc. Sono inoltre disponibili dati di riferimento pubblicati per il metodo di prova IL-8 Luc (6) (1).
Tabella 1
Sostanze raccomandate per la verifica della competenza tecnica con il metodo di prova IL-8 Luc
|
Sostanze di prova per la verifica della competenza
|
N. CAS
|
Stato
|
Solubilità in X-VIVO15 a 20 mg/ml
|
Predizione in vivo
(109)
|
Predizione IL-8 Luc (110)
|
Intervallo di riferimento (μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-Dinitroclorobenzene
|
97-00-7
|
Solido
|
Insolubile
|
Sensibilizzante (estremamente elevato)
|
Positivo
|
2,3-3,9
|
0,5-2,3
|
|
Formaldeide
|
50-00-0
|
Liquido
|
Solubile
|
Sensibilizzante (elevato)
|
Positivo
|
9-30
|
4-9
|
|
2-Mercaptobenzotiazolo
|
149-30-4
|
Solido
|
Insolubile
|
Sensibilizzante(moderato)
|
Positivo
|
250-290
|
60-250
|
|
Etilendiammina
|
107-15-3
|
Liquido
|
Solubile
|
Sensibilizzante (moderato)
|
Positivo
|
500-700
|
0,1-0,4
|
|
Etilene glicol dimetacrilato
|
97-90-5
|
Liquido
|
Insolubile
|
Sensibilizzante (debole)
|
Positivo
|
>2000
|
0,04-0,1
|
|
4-Allilanisolo (estragolo)
|
140-67-0
|
Liquido
|
Insolubile
|
Sensibilizzante (debole)
|
Positivo
|
>2000
|
0,01-0,07
|
|
Solfato di streptomicina
|
3810-74-0
|
Solido
|
Solubile
|
Non sensibilizzante
|
Negativo
|
>2000
|
>2000
|
|
Glicerolo
|
56-81-5
|
Liquido
|
Solubile
|
Non sensibilizzante
|
Negativo
|
>2000
|
>2000
|
|
Isopropanolo
|
67-63-0
|
Liquido
|
Solubile
|
Non sensibilizzante
|
Negativo
|
>2000
|
>2000
|
|
Abbreviazioni: N. CAS (Chemical Abstracts Service Registry Number) = Numero CAS (numero di registrazione nell'inventario europeo delle sostanze chimiche).
|
Appendice 3.4
INDICI E CRITERI DI VALUTAZIONE
nIL8LA (nSLO-LA)
La j-esima ripetizione (j = 1 - 4) della i-esima concentrazione (i = 0 - 11) è misurata per IL8LA (SLO-LA) e GAPLA (SLR-LA) rispettivamente. Il valore IL8LA normalizzato, detto nIL8LA (nSLO-LA), è definito come segue:
nIL8LAij = IL8LAij/GAPLAij
Si tratta dell’unità di misura di base per questo metodo di prova.
Ind-IL8LA (FInSLO-LA)
L’aumento del valore medio nIL8LA (nSLO-LA) per la ripetizione alla i-esima concentrazione rispetto alla concentrazione 0, Ind-IL8LA, è la misura principale di questo metodo di prova. Tale rapporto è espresso dalla formula seguente:

Il laboratorio principale ha proposto che un valore di 1,4 corrisponda a un risultato positivo per la sostanza chimica in esame. Tale valore è basato sullo studio dei dati storici del laboratorio principale. L’équipe che si occupa della gestione dei dati ha utilizzato tale valore in tutte le fasi dello studio di validazione. Come risulta dall’equazione, il risultato principale, Ind-IL8LA, è il rapporto di 2 medie aritmetiche.
Intervallo di confidenza del 95 % (CI 95 %)
La precisione della misura di tale risultato principale è stimata mediante l’intervallo di confidenza del 95 % (CI 95 %) basato sul rapporto. Il limite inferiore di CI 95 % ≥ 1 indica che il valore nIL8LA alla i-esima concentrazione è significativamente più elevato del valore ottenuto con il controllo con solvente. L’intervallo di confidenza CI 95 % può essere ricavato in diversi modi. Nel presente studio è stato utilizzato il metodo noto come teorema di Fieller, secondo cui l’intervallo di confidenza del 95 % è calcolato mediante la formula seguente:
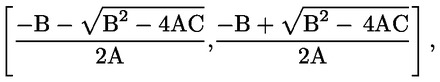
in cui
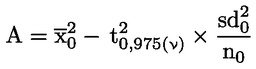 ; ;
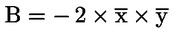 ; ;
 , ,
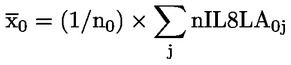 , ,
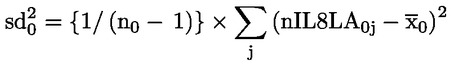 , ,
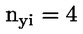 , ,
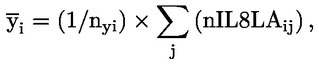
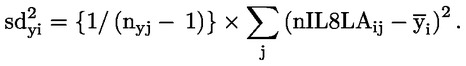
t0,975(ν) è il 97,5° percentile della distribuzione t centrale con il valore ν del grado di libertà, in cui
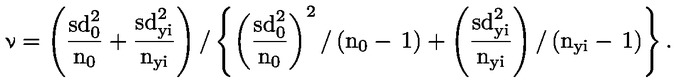
Inh-GAPLA (II-SLR-LA)
Il valore Inh-GAPLA è il rapporto del valore GAPLA medio (SLR-LA) per la ripetizione della i-esima concentrazione rispetto al valore ottenuto con il controllo con solvente, ed è espresso alla seguente formula:
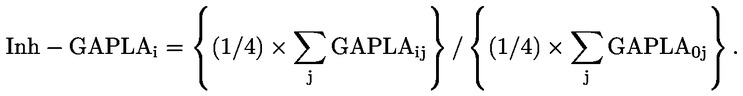
Essendo GAPLA il denominatore di nIL8LA, un valore estremamente basso determina una grande variazione di nIL8LA. Pertanto i valori Ind-IL8LA con un valore Inh-GAPLA estremamente basso (inferiore a 0,05) vanno considerati di scarsa precisione.
Appendice 3.5
SCHEMA DEI METODI DI DISSOLUZIONE DELLE SOSTANZE CHIMICHE PER IL METODO DI PROVA IL-8 LUC
|
(a)
|
Per le sostanze chimiche disciolte in X-VIVOTM 15 a 20 mg/ml
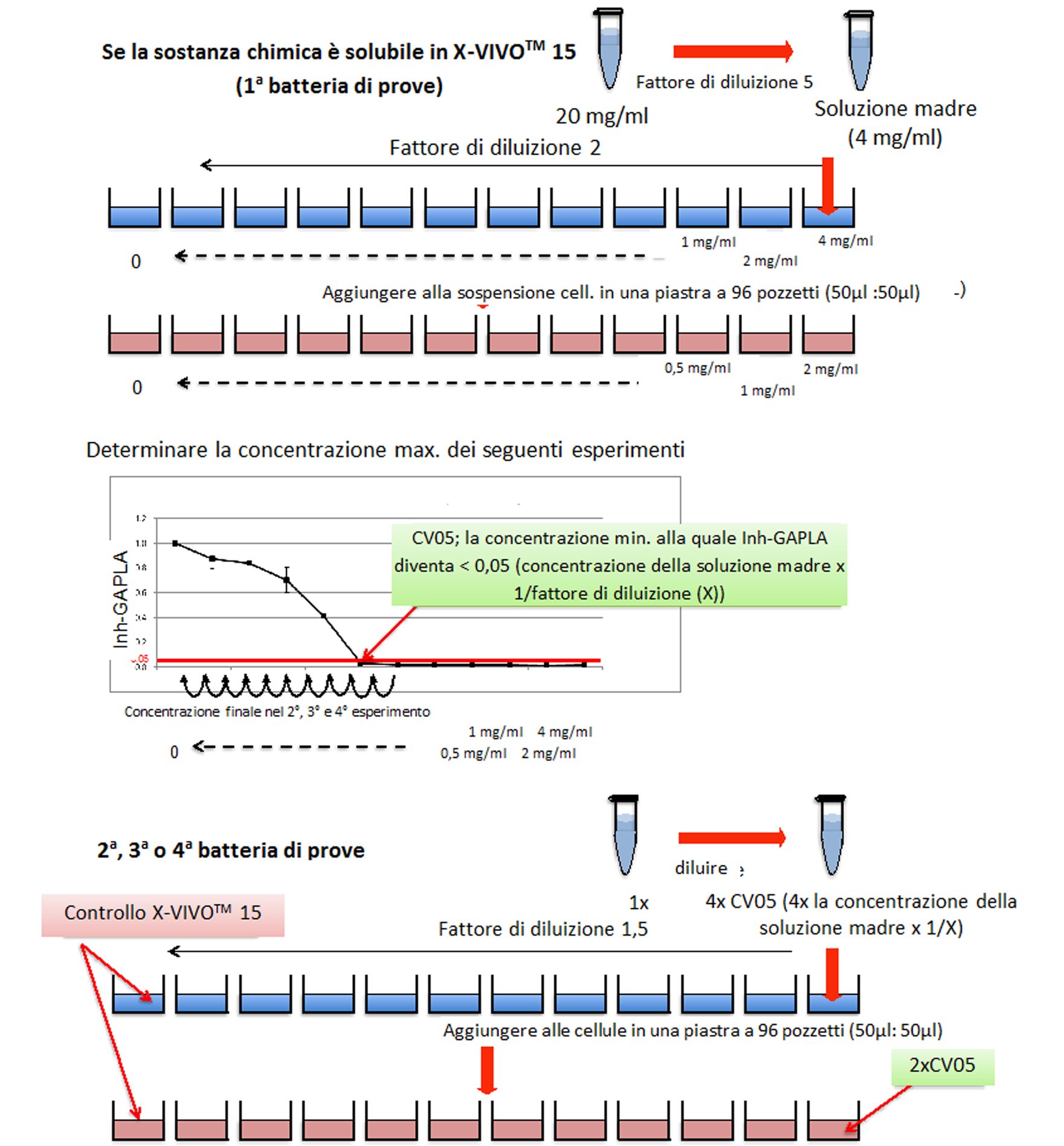
|
|
(b)
|
Per le sostanze chimiche insolubili in X-VIVOTM 15 a 20 mg/ml
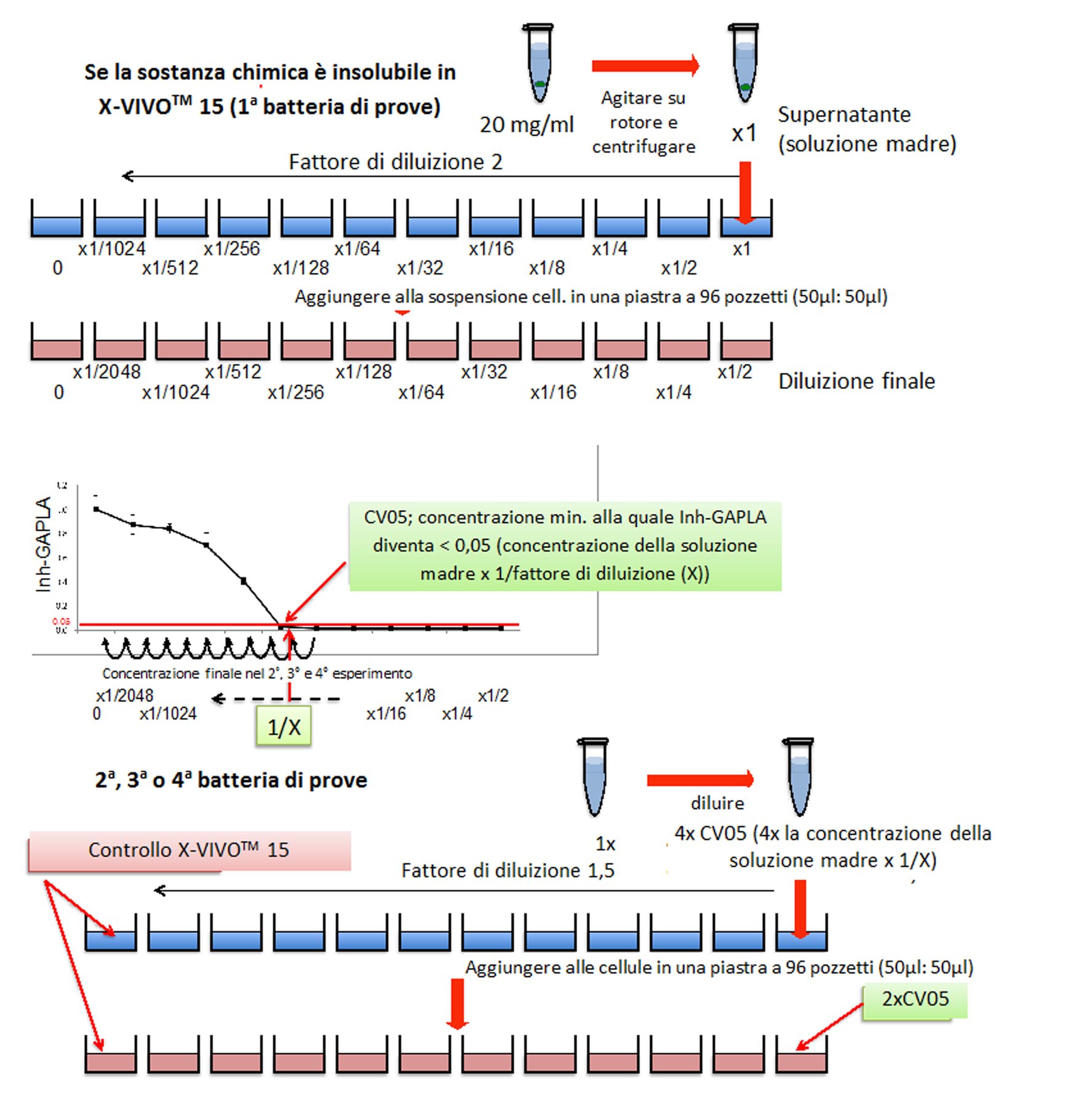
|
Appendice 3.6
SCHEMA DEL METODO DI DISSOLUZIONE DEL 4-NBB PER IL CONTROLLO POSITIVO PER IL METODO DI PROVA IL-8 LUC
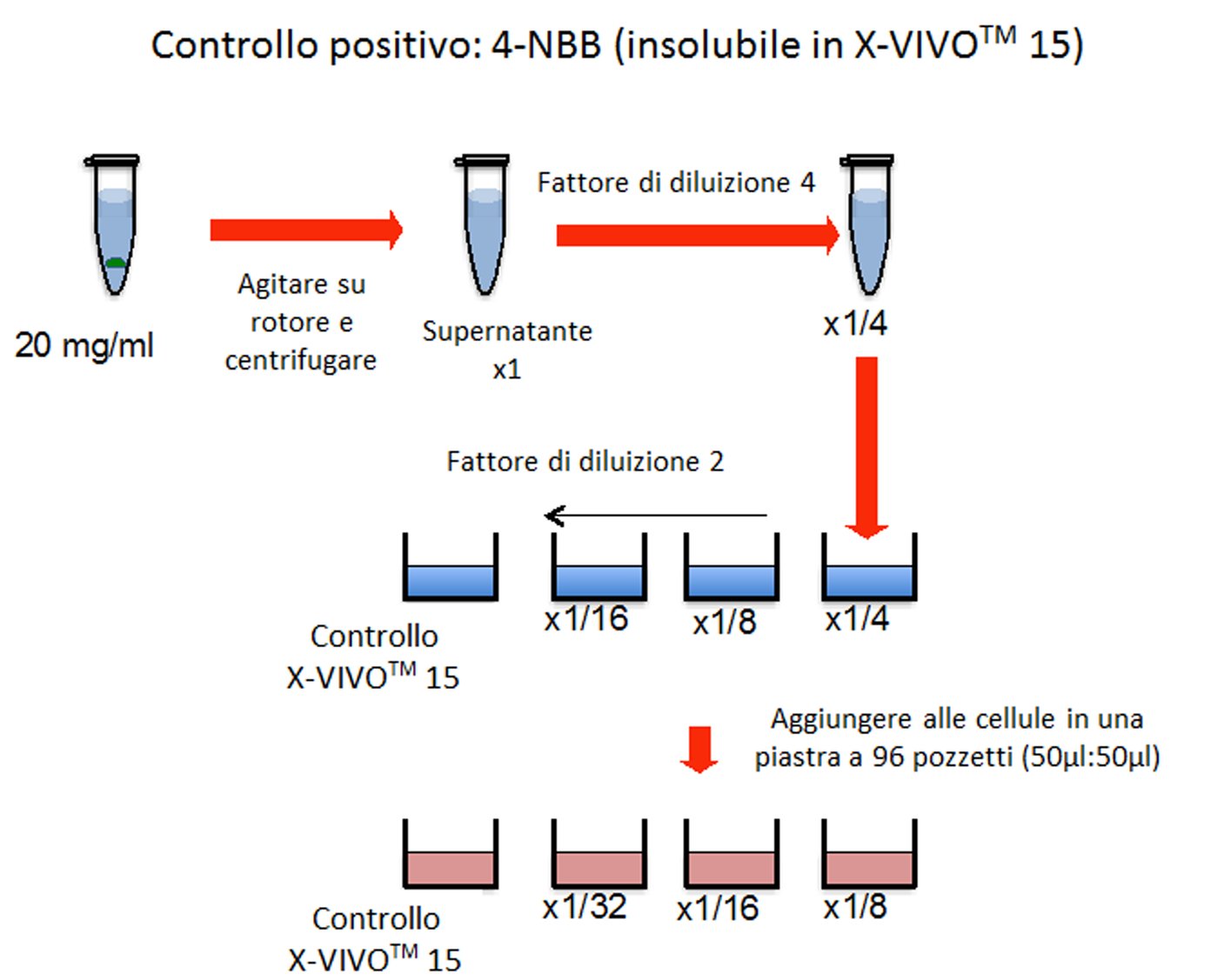
" |