|
26.2.2009
|
IT
|
Gazzetta ufficiale dell’Unione europea
|
L 54/1
|
REGOLAMENTO (CE) N. 152/2009 DELLA COMMISSIONE
del 27 gennaio 2009
che fissa i metodi di campionamento e d'analisi per i controlli ufficiali degli alimenti per gli animali
(Testo rilevante ai fini del SEE)
LA COMMISSIONE DELLE COMUNITÀ EUROPEE ,
visto il trattato che istituisce la Comunità europea,
visto il regolamento (CE) n. 882/2004 del Parlamento europeo e del Consiglio, del 29 aprile 2004, relativo ai controlli ufficiali intesi a verificare la conformità alla normativa in materia di mangimi e di alimenti e alle norme sulla salute e sul benessere degli animali (1), in particolare l’articolo 11, paragrafo 4, lettere a), b) e c),
considerando quanto segue:
|
(1)
|
Ai fini dell'applicazione della direttiva 70/373/CEE sono stati adottati e rimangono in vigore, conformemente all'articolo 61, paragrafo 2, del regolamento (CE) n. 882/2004, i seguenti atti normativi:
|
—
|
prima direttiva 71/250/CEE della Commissione, del 15 giugno 1971, che fissa i metodi d'analisi comunitari per controlli ufficiali degli alimenti per gli animali (2),
|
|
—
|
seconda direttiva 71/393/CEE della Commissione, del 18 novembre 1971, che fissa i metodi d'analisi comunitari per i controlli ufficiali degli alimenti per gli animali (3),
|
|
—
|
terza direttiva 72/199/CEE della Commissione, del 27 aprile 1972, che fissa i metodi di analisi comunitari per i controlli degli alimenti per gli animali (4),
|
|
—
|
quarta direttiva 73/46/CEE della Commissione, del 5 dicembre 1972, che fissa i metodi d'analisi comunitari per i controlli ufficiali degli alimenti per gli animali (5),
|
|
—
|
prima direttiva 76/371/CEE della Commissione, del 1o marzo 1976, che fissa i modi comunitari di prelevamento dei campioni per il controllo ufficiale degli alimenti per gli animali (6),
|
|
—
|
settima direttiva 76/372/CEE della Commissione, del 1o marzo 1976, che fissa i metodi d'analisi comunitari per i controlli ufficiali degli alimenti per gli animali (7),
|
|
—
|
ottava direttiva 78/633/CEE della Commissione, del 15 giugno 1978, che fissa i metodi d'analisi comunitari per il controllo ufficiale degli alimenti per gli animali (8),
|
|
—
|
nona direttiva 81/715/CEE della Commissione, del 31 luglio 1981, che fissa i metodi d'analisi comunitari per il controllo ufficiale degli alimenti per animali (9),
|
|
—
|
decima direttiva 84/425/CEE della Commissione, del 25 luglio 1984, che fissa i metodi d'analisi comunitari per il controllo ufficiale degli alimenti per animali (10),
|
|
—
|
direttiva 86/174/CEE della Commissione, del 9 aprile 1986, che fissa il metodo di calcolo del valore energetico degli alimenti composti destinati al pollame (11),
|
|
—
|
undicesima direttiva 93/70/CEE della Commissione, del 28 luglio 1993, che fissa i metodi d'analisi comunitari per il controllo degli alimenti per animali (12),
|
|
—
|
dodicesima direttiva 93/117/CE della Commissione, del 17 dicembre 1993, che fissa i metodi d'analisi comunitari per il controllo ufficiale degli alimenti per animali (13),
|
|
—
|
direttiva 98/64/CE della Commissione, del 3 settembre 1998, che fissa i metodi di analisi comunitari per la determinazione degli amminoacidi, delle materie grasse grezze e dell'olaquindox negli alimenti per animali e che modifica la direttiva 71/393/CEE (14),
|
|
—
|
direttiva 1999/27/CE della Commissione, del 20 aprile 1999, che fissa i metodi di analisi comunitari per la determinazione dell'amprolium, del diclazuril e del carbadox negli alimenti per animali, che modifica le direttive 71/250/CEE e 73/46/CEE e che revoca la direttiva 74/203/CEE (15),
|
|
—
|
direttiva 1999/76/CE della Commissione, del 23 luglio 1999, che fissa i metodi di analisi comunitari per la determinazione del lasalocid sodico negli alimenti per animali (16),
|
|
—
|
direttiva 2000/45/CE della Commissione, del 6 luglio 2000, che fissa i metodi di analisi comunitari per la determinazione della vitamina A, della vitamina E e del triptofano negli alimenti per animali (17),
|
|
—
|
direttiva 2002/70/CE della Commissione, del 26 luglio 2002, che stabilisce i requisiti per la determinazione dei livelli di diossine e PCB diossina-simili nei mangimi (18),
|
|
—
|
direttiva 2003/126/CE della Commissione, del 23 dicembre 2003, che stabilisce il metodo analitico per la determinazione dei costituenti di origine animale nell'ambito del controllo ufficiale degli alimenti per animali (19).
|
|
|
(2)
|
Dal momento che la direttiva 70/373/CEE è stata sostituita dal regolamento (CE) n. 882/2004 è opportuno sostituire i provvedimenti attuativi di tale direttiva con un regolamento unico. Al contempo vanno adeguate le metodiche tenendo conto degli sviluppi delle conoscenze in campo scientifico e tecnologico. I metodi non più validi per i fini previsti vanno soppressi. È previsto l'aggiornamento a tempo debito delle disposizioni relative al campionamento al fine di tener conto degli ultimi sviluppi nel campo della produzione, dell'immagazzinamento, del trasporto e della commercializzazione degli alimenti per animali, ma è opportuno, per il momento, mantenere le disposizioni vigenti in materia.
|
|
(3)
|
Occorre di conseguenza abrogare le direttive 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE e 2003/126/CE.
|
|
(4)
|
Le misure di cui al presente regolamento sono conformi al parere del comitato permanente per la catena alimentare e la salute degli animali,
|
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
Il prelievo dei campioni destinati al controllo ufficiale degli alimenti per animali, per quanto concerne la determinazione dei costituenti, degli additivi e delle sostanze indesiderabili, eccettuati i residui di pesticidi e i microorganismi, è effettuato secondo i metodi descritti nell'allegato I.
Articolo 2
La preparazione dei campioni per l'analisi e l'espressione dei risultati sono conformi ai metodi indicati nell'allegato II.
Articolo 3
Le analisi per i controlli ufficiali degli alimenti per animali sono effettuate secondo i metodi indicati nell'allegato III (Metodi di analisi per il controllo della composizione delle materie prime per alimenti per animali e degli alimenti composti), nell'allegato IV (Metodi di analisi per il controllo del contenuto di additivi autorizzati negli alimenti per animali), nell'allegato V (Metodi di analisi per il controllo della presenza di sostanze indesiderabili negli alimenti per animali) e nell'allegato VI (Metodi di analisi per la determinazione dei costituenti di origine animale nell'ambito del controllo ufficiale degli alimenti per animali).
Articolo 4
Il valore energetico degli alimenti composti destinati al pollame è calcolato conformemente al metodo descritto nell'allegato VII.
Articolo 5
I metodi di analisi per il controllo della presenza illecita di additivi il cui uso non è più autorizzato negli alimenti per animali di cui all'allegato VIII sono applicati a fini di conferma.
Articolo 6
Le direttive 71/250/CEE, 71/393/CEE, 72/199/CEE, 73/46/CEE, 76/371/CEE, 76/372/CEE, 78/633/CEE, 81/715/CEE, 84/425/CEE, 86/174/CEE, 93/70/CEE, 93/117/CE, 98/64/CE, 1999/27/CE, 1999/76/CE, 2000/45/CE, 2002/70/CE e 2003/126/CE sono abrogate.
I riferimenti alle direttive abrogate s'intendono fatti al presente regolamento e vanno letti secondo le tavole di concordanza che figurano nell'allegato IX.
Articolo 7
Il presente regolamento entra in vigore il ventesimo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Esso si applica a decorrere dal 26 agosto 2009.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 27 gennaio 2009.
Per la Commissione
Androulla VASSILIOU
Membro della Commissione
(1) GU L 165 del 30.4.2004, pag. 1; rettifica nella GU L 191 del 28.5.2004, pag. 1.
(2) GU L 155 del 12.7.1971, pag. 13.
(3) GU L 279 del 20.12.1971, pag. 7.
(4) GU L 123 del 29.5.1972, pag. 6.
(5) GU L 83 del 30.3.1973, pag. 21.
(6) GU L 102 del 15.4.1976, pag. 1.
(7) GU L 102 del 15.4.1976, pag. 8.
(8) GU L 206 del 29.7.1978, pag. 43.
(9) GU L 257 del 10.9.1981, pag. 38.
(10) GU L 238 del 6.9.1984, pag. 34.
(11) GU L 130 del 16.5.1986, pag. 53.
(12) GU L 234 del 17.9.1993, pag. 17.
(13) GU L 329 del 30.12.1993, pag. 54.
(14) GU L 257 del 19.9.1998, pag. 14.
(15) GU L 118 del 6.5.1999, pag. 36.
(16) GU L 207 del 6.8.1999, pag. 13.
(17) GU L 174 del 13.7.2000, pag. 32.
(18) GU L 209 del 6.8.2002, pag. 15.
(19) GU L 339 del 24.12.2003, pag. 78.
ALLEGATO I
METODI DI CAMPIONAMENTO
1. FINALITÀ E CAMPO D'APPLICAZIONE
I campioni destinati al controllo ufficiale degli alimenti per animali sono prelevati secondo le modalità sottoindicate. Tali campioni sono da considerarsi rappresentativi delle partite campionate.
2. PERSONALE ADDETTO AL CAMPIONAMENTO
I campioni sono prelevati da personale autorizzato, appositamente designato dagli Stati membri.
3. DEFINIZIONI
Partita da campionare: quantità di prodotto costituente un'unità e avente caratteristiche presunte uniformi.
Campione elementare: quantità prelevata da un punto della partita campionata.
Campione globale: insieme di campioni elementari prelevati da una stessa partita campionata.
Campione ridotto: parte rappresentativa del campione globale, ottenuta mediante riduzione di quest'ultimo.
Campione finale: parte del campione ridotto o del campione globale omogeneizzato.
4. APPARECCHIATURA
|
4.1.
|
Gli strumenti necessari per il prelievo dei campioni devono essere costruiti con materiali che non contaminino i prodotti da campionare e possono essere ufficialmente approvati dagli Stati membri.
|
4.2. Strumenti raccomandati per il prelievo di campioni di alimenti solidi per animali
4.2.1. Campionamento manuale
|
4.2.1.1.
|
Pala a fondo piatto e a bordi laterali verticali
|
|
4.2.1.2.
|
Sonda a lungo setto o a partizioni. Le dimensioni della sonda devono essere adeguate alle caratteristiche della partita (profondità del recipiente, misure del sacco, ecc.) e alla dimensione delle particelle costituenti l'alimento.
|
4.2.2. Campionamento meccanico
Per il prelievo di campioni di alimenti in flusso possono essere utilizzati dispositivi meccanici autorizzati.
4.2.3. Divisore
Per i prelievi di campioni elementari nonché per la preparazione di campioni ridotti e di campioni finali possono essere utilizzati strumenti che servono a dividere i campioni in parti approssimativamente uguali.
5. REQUISITI QUANTITATIVI
|
5.A.
|
Per il controllo delle sostanze o dei prodotti ripartiti in modo uniforme negli alimenti per animali
|
|
5.A.1.
|
Partita da campionare
L'entità della partita da campionare deve essere tale da consentire il prelievo di campioni in ogni sua parte.
|
|
5.A.2.
|
Campioni elementari
|
|
5.A.2.1.
|
Alimenti alla rinfusa:
|
Numero minimo di campioni elementari:
|
|
5.A.2.1.1.
|
partite di peso non superiore a 2,5 t
|
sette
|
|
5.A.2.1.2.
|
partite di peso superiore a 2,5 t
|
√ di 20 volte il numero di tonnellate costituenti la partita da campionare (1), con un massimo di 40 campioni elementari
|
|
5.A.2.2.
|
Alimenti in confezioni:
|
Numero minimo di confezioni da campionare (2):
|
|
5.A.2.2.1.
|
Confezioni di contenuto superiore a 1 kg:
|
|
5.A.2.2.1.1.
|
partite costituite da 1-4 confezioni
|
tutte le confezioni
|
|
5.A.2.2.1.2.
|
partite costituite da 5-16 confezioni
|
quattro
|
|
5.A.2.2.1.3.
|
partite costituite da oltre 16 confezioni
|
√ del numero di confezioni costituenti la partita da campionare (1), per un massimo di 20 confezioni
|
|
5.A.2.2.2.
|
Confezioni di contenuto pari o inferiore a 1 kg
|
quattro
|
|
5.A.2.3.
|
Alimenti liquidi o semiliquidi:
|
Numero minimo di recipienti da campionare (2):
|
|
5.A.2.3.1.
|
Recipienti di contenuto superiore a 1 litro:
|
|
5.A.2.3.1.1.
|
partite costituite da 1-4 recipienti
|
tutti i recipienti
|
|
5.A.2.3.1.2.
|
partite costituite da 5-16 recipienti
|
quattro
|
|
5.A.2.3.1.3.
|
partite costituite da oltre 16 recipienti
|
√ del numero di recipienti costituenti la partita da campionare (1), per un massimo di 20 recipienti
|
|
5.A.2.3.2.
|
Recipienti di contenuto pari o inferiore a un litro
|
quattro
|
|
5.A.2.4.
|
Alimenti minerali formellati o mattonelle di sali minerali:
|
Numero minimo di formellati o mattonelle da campionare (2):
un formellato o una mattonella per partita di 25 unità, per un massimo di quattro formellati o mattonelle
|
|
5.A.3.
|
Campione globale
È richiesto un solo campione globale per partita. La massa totale dei campioni elementari destinati a costituire il campione globale non è inferiore ai seguenti quantitativi:
|
|
5.A.3.1.
|
Alimenti alla rinfusa
|
4 kg
|
|
5.A.3.2.
|
Alimenti in confezioni:
|
|
|
5.A.3.2.1.
|
confezioni di contenuto superiore a 1 kg
|
4 kg
|
|
5.A.3.2.2.
|
confezioni di contenuto pari o inferiore a 1 kg
|
peso del contenuto di quattro confezioni d'origine
|
|
5.A.3.3.
|
Alimenti liquidi o semiliquidi:
|
|
|
5.A.3.3.1.
|
recipienti di contenuto superiore a un litro
|
4 litri
|
|
5.A.3.3.2.
|
recipienti di contenuto pari o inferiore a un litro
|
volume del contenuto di quattro recipienti d'origine
|
|
5.A.3.4.
|
Alimenti minerali in formellati o mattonelle di sali minerali:
|
|
5.A.3.4.1.
|
di peso unitario superiore a 1 kg
|
4 kg
|
|
5.A.3.4.2.
|
di peso unitario pari o inferiore a 1 kg
|
peso di quattro formellati o mattonelle d'origine
|
|
5.A.4.
|
Campioni finali
Dopo riduzione, se necessaria, si ottengono dal campione globale campioni finali. È richiesta l'analisi di almeno un campione finale. La massa del campione finale destinato all'analisi non deve essere inferiore ai seguenti quantitativi:
|
|
|
Alimenti solidi
|
500 g
|
|
|
Alimenti liquidi o semiliquidi:
|
500 ml
|
|
5.B.
|
Per il controllo delle sostanze o dei prodotti indesiderabili che possono essere distribuiti in modo non uniforme negli alimenti come le aflatossine, l'ergotina di segale, il ricino, la crotalaria nelle materie prime per alimenti per animali (3)
|
|
5.B.1.
|
Partita da campionare: cfr. punto 5.A.1.
|
|
5.B.2.
|
Campioni elementari
|
|
5.B.2.1.
|
Alimenti alla rinfusa: cfr. punto 5.A.2.1.
|
|
5.B.2.2.
|
Alimenti in confezioni:
|
Numero minimo di confezioni da campionare:
|
|
5.B.2.2.1.
|
partite costituite da 1-4 confezioni
|
tutte le confezioni
|
|
5.B.2.2.2.
|
partite costituite da 5-16 confezioni
|
quattro
|
|
5.B.2.2.3.
|
partite costituite da oltre 16 confezioni
|
√ del numero di confezioni costituenti la partita da campionare (1) con un massimo di 40 confezioni
|
|
5.B.3.
|
Campione globale
Il numero di campioni globali varia secondo la dimensione della partita. Il numero minimo di campioni globali per partita è indicato di seguito. Il peso totale dei campioni elementari destinati a costituire il campione globale non deve risultare inferiore a 4 kg
|
|
5.B.3.1.
|
Alimenti alla rinfusa
|
|
|
|
Peso della partita in tonnellate:
|
Numero minimo di campioni globali per partita:
|
|
|
fino a 1
|
1
|
|
|
più di 1 e fino a 10
|
2
|
|
|
più di 10 e fino a 40
|
3
|
|
|
più di 40
|
4
|
|
5.B.3.2.
|
Alimenti confezionati
|
|
|
|
Quantità di alimenti confezionati costituenti la partita da campionare in numero di confezioni:
|
Numero minimo di campioni globali per partita:
|
|
|
da 1 a 16
|
1
|
|
|
da 17 a 200
|
2
|
|
|
da 201 a 800
|
3
|
|
|
più di 800
|
4
|
|
5.B.4.
|
Campioni finali
Dopo riduzione, se necessaria, si ottengono dal campione globale campioni finali. Per ciascun campione globale è richiesta l'analisi di almeno un campione finale. Il peso del campione finale destinato all'analisi non deve essere inferiore a 500 g.
|
6. ISTRUZIONI RELATIVE AI PRELIEVI, ALLA FORMAZIONE E AL CONDIZIONAMENTO DEI CAMPIONI
6.1. Indicazioni generali
Prelevare e formare i campioni quanto più rapidamente possibile prendendo le precauzioni necessarie per evitare qualsiasi alterazione o contaminazione del prodotto. Le superfici, i recipienti e gli strumenti impiegati devono essere puliti e asciutti.
6.2. Campioni elementari
6.2.A Destinati al controllo delle sostanze o dei prodotti ripartiti in modo uniforme nell'alimento
I campioni elementari vanno prelevati a caso dall'insieme della partita da campionare e devono risultare d'entità approssimativamente uguale.
6.2.A.1. Alimenti alla rinfusa
Dividere simbolicamente la partita da campionare in un numero di parti approssimativamente uguali. Scegliere a caso un numero di parti corrispondente al numero di campioni elementari di cui al punto 5.A.2 e prelevare almeno un campione da ciascuna parte.
Eventualmente, procedere al campionamento al momento della messa in movimento della partita (carico o scarico).
6.2.A.2. Alimenti in confezioni
Prelevare con una sonda o pala una parte del contenuto di ciascuna confezione da campionare secondo quanto indicato al punto 5.A.2. Eventualmente vuotare separatamente le confezioni. Se necessario, schiacciare i grumi separatamente per ciascun campione globale (togliendoli eventualmente dalla massa e riunendo quindi il tutto).
6.2.A.3. Alimenti liquidi o semiliquidi omogenei o omogeneizzabili
Dal numero prescritto di recipienti da campionare selezionati secondo quanto indicato al punto 5.A.2, prelevare una parte del contenuto di ciascun recipiente, se necessario, dopo omogeneizzazione.
I campioni elementari possono eventualmente essere prelevati al momento del travaso del prodotto.
6.2.A.4. Alimenti liquidi o semiliquidi non omogeneizzabili
Dal numero prescritto di recipienti da campionare selezionati secondo quanto indicato al punto 5.A.2, prelevare i campioni a diversi livelli.
I campioni possono essere prelevati anche al momento del travaso del prodotto, dopo eliminazione delle prime frazioni.
In entrambi i casi, il volume totale dei prelievi non deve essere inferiore a 10 litri.
6.2.A.5. Alimenti minerali formellati o mattonelle di sali minerali
Dal numero prescritto di formellati o mattonelle da campionare selezionati secondo quanto indicato al punto 5.A.2, prelevare una parte da ciascuno di essi.
6.2.B. Destinati al controllo delle sostanze o dei prodotti indesiderabili che possono essere distribuiti in modo non uniforme negli alimenti per animali come le aflatossine, l'ergotina di segale, il ricino, la crotalaria presenti nelle materie prime per alimenti per animali
Dividere idealmente la partita in un numero di parti approssimativamente uguali corrispondente a quello dei campioni globali di cui al punto 5.B.3. Se tale numero è superiore a 1, ripartire il numero totale dei campioni elementari di cui al punto 5.B.2 in modo approssimativamente uguale tra le diverse parti. Prelevare quindi quantità approssimativamente uguali (4) in modo che la massa totale dei campioni relativi a ciascuna parte non sia inferiore al quantitativo minimo di 4 kg necessario per ciascun campione globale. Non riunire i campioni elementari provenienti da parti diverse.
6.3. Formazione dei campioni globali
6.3.A. Destinati al controllo delle sostanze o dei prodotti ripartiti in modo uniforme negli alimenti per animali
Riunire i campioni elementari per costituire un solo campione globale.
6.3.B. Destinati al controllo delle sostanze o dei prodotti indesiderabili che possono essere distribuiti in modo non uniforme negli alimenti come le aflatossine, l'ergotina di segale, il ricino, la crotalaria nelle materie presenti prime per alimenti per animali
Riunire i campioni elementari prelevati da ciascuna frazione della partita per ottenere il numero di campioni globali previsti al punto 5.B.3, avendo cura di annotare la provenienza di ciascun campione globale.
6.4. Formazione dei campioni finali
Mescolare con cura ciascun campione globale per ottenere un campione omogeneo (5). Se necessario ridurre, a tal fine, il campione globale a 2 kg o a due litri (campione ridotto), con l'aiuto eventualmente di un divisore meccanico o con il metodo della suddivisione in quarti.
Formare, quindi, almeno tre campioni finali di massa o di volume approssimativamente uguale e rispondenti ai requisiti quantitativi di cui ai punti 5.A.4 o 5.B.4. Introdurre ciascun campione in un recipiente idoneo. Prendere tutte le precauzioni del caso per evitare qualsiasi modifica alla composizione del campione o qualsiasi contaminazione o alterazione fortuita durante il trasporto o l'immagazzinaggio.
6.5. Condizionamento dei campioni finali
Sigillare ed etichettare i recipienti o le confezioni (l'etichetta deve essere incorporata nel sigillo) in modo che non possano essere aperti senza violare il sigillo.
7. VERBALI DEL CAMPIONAMENTO
Per ogni operazione di campionamento va redatto un verbale che permetta di identificare in modo univoco la partita campionata.
8. DESTINAZIONE DEI CAMPIONI
Per ciascuna partita trasmettere il più rapidamente possibile almeno un campione finale a uno dei laboratori incaricati dell'analisi con le indicazioni necessarie all'analisi stessa.
(1) Se il risultato è un numero decimale si arrotonda al numero intero superiore.
(2) Per confezioni o recipienti di contenuto pari o inferiore a 1 kg o un litro, nonché per formellati o mattonelle di sali minerali di peso unitario non superiore a 1 kg, il campione elementare è dato dal contenuto di una confezione o di un recipiente d'origine, di un formellato o di una mattonella.
(3) Per il controllo delle aflatossine, dell'ergotina di segale, del ricino, della crotalaria negli alimenti completi e complementari si applicano i metodi di cui al punto 5.A.
(4) Nel caso degli alimenti in confezioni, prelevare con una sonda o pala una parte del contenuto delle confezioni da campionare, eventualmente dopo averle vuotate separatamente.
(5) Se necessario, schiacciare i grumi separatamente per ciascun campione globale (togliendoli eventualmente dalla massa e riunendo quindi il tutto).
ALLEGATO II
DISPOSIZIONI GENERALI RELATIVE AI METODI DI ANALISI DEGLI ALIMENTI PER ANIMALI
A. PREPARAZIONE DEI CAMPIONI PER LE ANALISI
1. Finalità
Le modalità qui di seguito descritte riguardano la preparazione per l'analisi dei campioni finali, trasmessi ai laboratori di controllo dopo essere stati prelevati conformemente alle diposizioni di cui all'allegato I.
La preparazione di questi campioni deve assicurare che le aliquote di sostanza previste dai metodi di analisi siano omogenee e rappresentative dei campioni finali.
2. Precauzioni necessarie
Le modalità di preparazione dei campioni sono scelte in funzione dei metodi di analisi impiegati. È pertanto di fondamentale importanza assicurare la coerenza delle modalità prescelte con il metodo di analisi applicato.
Effettuare tutte le operazioni in modo da evitare, nei limiti del possibile, di contaminare il campione o di modificarne le composizione.
Effettuare le triturazioni, le miscelazioni e le setacciature con la massima rapidità, esponendo al minimo il campione all'aria e alla luce. Evitare l'impiego di mulini o trituratori suscettibili di riscaldare eccessivamente il campione.
Per gli alimenti particolarmente sensibili al calore si raccomanda la triturazione manuale. Assicurarsi altresì che l'apparecchiatura in se stessa non costituisca una fonte di contaminazione da oligoelementi.
Se il campione non può essere preparato senza variazione sensibile del contenuto di umidità, quest'ultimo va determinato prima e dopo la preparazione, secondo il metodo di dosaggio dell'umidità previsto nell'allegato III, parte A.
3. Procedimento
Suddividere il campione in adeguati sottocampioni da analizzare e a cui fare riferimento, utilizzando metodi di frazionamento appropriati quali le palate alternate o un ripartitore fisso o rotante. Il metodo del cono e della quartatura è sconsigliato in quanto può dar luogo a sottocampioni con un elevato errore di ripartizione. Conservare il campione di riferimento in un recipiente appropriato pulito e asciutto, a chiusura ermetica, e preparare i sottocampioni da analizzare di peso non inferiore a 100 g secondo le modalità indicate di seguito.
3.1. Alimenti che possono essere macinati direttamente
Salvo diversa indicazione specifica nei metodi di analisi, setacciare la totalità del campione su un setaccio a maglie quadrate di un millimetro di lato (conforme alla raccomandazione ISO R565) eventualmente dopo averlo frantumato. Evitare ogni frantumazione superflua.
Mescolare il campione setacciato e raccoglierlo in un recipiente appropriato pulito e asciutto, provvisto di chiusura ermetica. Mescolare di nuovo, immediatamente prima di prelevare la quantità da analizzare.
3.2. Alimenti che possono essere macinati dopo essiccazione
Salvo diversa indicazione specifica nei metodi di analisi, essiccare il campione, in modo da portarne il contenuto di umidità ad un livello compreso tra l'8 e il 12 %, applicando il procedimento di preessiccazione indicato al punto 4.3 del metodo di dosaggio dell'umidità menzionato nell'allegato III, parte A. Procedere quindi come indicato al punto 3.1.
3.3. Alimenti liquidi o semiliquidi
Raccogliere il campione in un recipiente appropriato pulito e asciutto, provvisto di chiusura ermetica. Mescolarlo accuratamente, immediatamente prima di prelevare la quantità da analizzare.
3.4. Altri alimenti
Se il campione non può essere preparato secondo uno dei procedimenti di cui sopra, applicare qualsiasi altro procedimento di preparazione che consenta di ottenere quantità da analizzare omogenee e rappresentative dei campioni finali.
4. Conservazione e immagazzinamento dei campioni
Conservare i campioni a una temperatura tale da non alterare la loro composizione. Per i campioni destinati all'analisi di vitamine o di sostanze particolarmente sensibili alla luce, utilizzare recipienti in vetro scuro.
B. DISPOSIZIONI CONCERNENTI I REATTIVI E L'APPARECCHIATURA DA UTILIZZARE NEI METODI DI ANALISI
|
1.
|
Tutti i reattivi, in mancanza di altre indicazioni, devono essere puri per analisi (p.a.). Per l'analisi degli oligoelementi, la purezza dei reattivi deve essere controllata con una prova in bianco. A seconda del risultato ottenuto, può rendersi necessaria una purificazione supplementare dei reattivi.
|
|
2.
|
Per le operazioni di dissoluzione, diluizione, risciacquo o lavaggio, menzionate nei metodi di analisi senza indicazioni riguardo alla natura del solvente o del diluente, deve essere utilizzata acqua. Di norma, l'acqua deve essere demineralizzata o distillata. In casi particolari, indicati nei metodi di analisi, deve essere sottoposta a procedimenti specifici di purificazione.
|
|
3.
|
Tenuto conto dell'abituale equipaggiamento dei laboratori di controllo, l'apparecchiatura descritta nei metodi di analisi si limita agli strumenti e agli apparecchi speciali o rispondenti a prescrizioni d'uso specifiche. Detto materiale deve essere pulito, soprattutto per le determinazioni di quantità minime di sostanze.
|
C. APPLICAZIONE DEI METODI DI ANALISI ED ESPRESSIONE DEI RISULTATI
1. Procedura di estrazione
Diversi metodi determinano una procedura di estrazione specifica. In linea di massima, è possibile applicare procedure di estrazione diverse da quella indicata nel metodo, a condizione che abbiano dimostrato un'efficienza di estrazione, per la matrice analizzata, equivalente a quella della procedura indicata nel metodo.
2. Procedura di purificazione
Diversi metodi determinano una procedura di purificazione specifica. In linea di massima, è possibile applicare procedure di purificazione diverse da quella indicata nel metodo, a condizione che abbiano dimostrato di produrre risultati, per la matrice analizzata, equivalenti a quelli prodotti dalla procedura indicata nel metodo.
3. Comunicazione del metodo di analisi applicato
Per determinare le sostanze presenti negli alimenti per animali si applica, di norma, un solo metodo d'analisi. Qualora siano previsti più metodi, nel bollettino d'analisi deve essere specificato il metodo applicato dal laboratorio di controllo.
4. Numero di determinazioni
Il risultato indicato nel bollettino d'analisi è il valore medio ottenuto in base ad almeno due determinazioni, effettuate su aliquote distinte di sostanza, la cui ripetibilità sia soddisfacente.
Tuttavia, qualora si analizzino sostanze indesiderabili, se il risultato della prima determinazione risulta nettamente inferiore (> 50 %) al valore della specifica oggetto di controllo, non è necessario procedere a ulteriori determinazioni, a condizione che si applichino le procedure appropriate in materia di qualità.
Se, nel controllare il titolo dichiarato di una sostanza o di un dato ingrediente, da una prima determinazione questo risulta essere esatto (ossia, lo scarto tra il risultato dell'analisi e il titolo dichiarato rientra in un margine di variazione accettabile), non è necessario ripetere la determinazione, purché siano rispettati gli opportuni controlli di qualità.
In alcuni casi il margine di variazione accettabile è definito da uno specifico atto legislativo, ad esempio la direttiva 79/373/CEE del Consiglio (1).
5. Comunicazione dei risultati dell'analisi
Il risultato deve essere espresso secondo le indicazioni fornite nel metodo di analisi con un numero appropriato di cifre significative e, ove necessario, corretto in funzione del contenuto di umidità del campione finale prima della sua preparazione.
6. Incertezza delle misurazioni e tasso di recupero in caso di analisi di sostanze indesiderabili
Per quanto riguarda le sostanze indesiderabili ai sensi della direttiva 2002/32/CE, comprese le diossine e i PCB di tipo diossina, un prodotto destinato all'alimentazione animale è considerato non conforme al tasso massimo fissato quando il risultato d'analisi è giudicato superiore al tasso massimo, tenuto conto dell'incertezza di misura estesa e della correzione per il recupero. Al fine di valutare la conformità, si utilizzano la concentrazione risultante dall'analisi, corretta per il fattore di recupero, e l'incertezza di misura estesa sottratta. Tale procedura è applicabile unicamente nei casi in cui il metodo d'analisi consenta la stima dell'incertezza di misura e della correzione per il fattore di recupero (non è possibile, ad esempio, in caso di analisi microscopica).
Il risultato dell'analisi è riportato come segue (quando il metodo d'analisi consente di valutare l'incertezza di misura e il tasso di recupero):
|
a)
|
corretto per il recupero, indicando il tasso di recupero. La correzione non è necessaria nel caso in cui il tasso di recupero sia compreso tra 90 e 110 %;
|
|
b)
|
nella forma «x +/- U», dove x è il risultato dell'analisi e U l'incertezza di misura estesa, calcolata per mezzo di un fattore di copertura 2 che dà un livello di affidabilità del 95 % circa.
|
Tuttavia, qualora il risultato dell'analisi risulti molto inferiore (> 50 %) al valore della specifica oggetto di controllo, e a condizione che si rispettino le procedure appropriate in materia di qualità e l'analisi serva unicamente a verificare la conformità alle norme giuridiche pertinenti, il risultato d'analisi può essere presentato senza correzioni per il recupero e, in questo caso, il tasso di recupero e l'incertezza della misura possono essere omessi.
(1) GU L 86 del 6.4.1979, pag. 30.
ALLEGATO III
METODI DI ANALISI PER IL CONTROLLO DELLA COMPOSIZIONE DELLE MATERIE PRIME PER ALIMENTI PER ANIMALI E DEGLI ALIMENTI COMPOSTI
A. DETERMINAZIONE DELL'UMIDITÀ
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di umidità degli alimenti per animali. Nel caso degli alimenti contenenti sostanze volatili, ad esempio acidi organici, va osservato che oltre al contenuto di umidità si rileva anche un'importante presenza di materie volatili.
Il metodo non riguarda l'analisi dei prodotti lattieri in quanto materie prime per alimenti per animali, delle sostanze minerali e miscele essenzialmente composte di sostanze minerali, l'analisi dei grassi e oli vegetali, né quella dei semi e frutti oleaginosi.
2. Principio
Il campione è sottoposto a essiccazione in condizioni ben definite, varianti in funzione della natura dell'alimento. La perdita di peso è determinata per pesata. È necessario procedere a una preessiccazione quando si tratta di alimenti solidi aventi un elevato contenuto di umidità.
3. Apparecchiatura
|
3.1.
|
Mulino da laboratorio costruito in materiale non assorbente l'umidità, facile a pulirsi, che permetta una macinazione rapida e uniforme senza provocare un sensibile riscaldamento, eviti al massimo il contatto con l'aria in ambiente esterno e risponda ai requisiti di cui ai punti 4.1.1 e 4.1.2 (ad esempio micromacinatori a martelli o a raffreddamento ad acqua, mulini a coni smontabili, macinatori a movimento lento o a dischi dentati).
|
|
3.2.
|
Bilancia analitica (precisione 1 mg).
|
|
3.3.
|
Recipienti asciutti, di vetro o di metallo inossidabile, provvisti di coperchi a chiusura ermetica; superficie utile che permetta di ottenere una ripartizione della quantità di prodotto su cui si opera dell'ordine di 0,3 g/cm2.
|
|
3.4.
|
Stufa isotermica (± 2 °C) a riscaldamento elettrico, adeguatamente ventilata, capace di assicurare una regolazione rapida della temperatura (1).
|
|
3.5.
|
Stufa a vuoto a riscaldamento elettrico regolabile munita di pompa a olio e di un dispositivo per l'introduzione di aria calda disidratata o di un agente disidratante (ad esempio ossido di calcio).
|
|
3.6.
|
Essiccatore a piastra in metallo o in porcellana, spessa, perforata, contenente un efficace disidratante.
|
4. Procedimento
|
Nota:
|
Le operazioni descritte in questo capitolo debbono essere effettuate immediatamente dopo l'apertura degli imballaggi contenenti i campioni. Le analisi vanno eseguite almeno due volte.
|
4.1. Preparazione
4.1.1. Alimenti per animali diversi da quelli menzionati ai punti 4.1.2 e 4.1.3
Prelevare almeno 50 g di campione. Se necessario macinare o dividere in particelle di grandezza appropriata in modo da evitare ogni variazione del contenuto di umidità (cfr. punto 6).
4.1.2. Cereali e semole
Prelevare almeno 50 g di campione. Macinare in particelle di cui almeno la metà passi per un setaccio a maglie di 0,5 mm e non lasci più del 10 % di residuo su un setaccio a maglie rotonde di 1 mm di diametro.
4.1.3. Alimenti liquidi o pastosi, alimenti costituiti essenzialmente da sostanze grasse
Prelevare almeno 25 g di campione, pesati con l'approssimazione di 10 mg, e aggiungere una quantità appropriata di sabbia anidra, pesare con l'approssimazione di 10 mg e mescolare sino a ottenere un prodotto omogeneo.
4.2. Essiccazione
4.2.1. Alimenti per animali diversi da quelli menzionati ai punti 4.2.2 e 4.2.3
Tarare, con la precisione di 1 mg, un recipiente (3.3), provvisto di coperchio. Pesare nel recipiente tarato, con l'approssimazione di 1 mg, circa 5 g di prodotto e ripartirli uniformemente. Porre il recipiente, senza coperchio, nella stufa preriscaldata a 103 °C. Per evitare che la temperatura della stufa scenda troppo, introdurre il recipiente nel più breve tempo possibile. Lasciare essiccare per quattro ore a partire dal momento in cui la stufa ha nuovamente raggiunto la temperatura di 103 oC. Rimettere il coperchio sul recipiente, estrarre quest'ultimo dalla stufa, lasciare raffreddare per 30-45 minuti nell'essiccatore (3.6) e pesare con l'approssimazione di 1 mg.
Nel caso di alimenti costituiti essenzialmente da sostanze grasse, lasciare essiccare per altri 30 minuti nella stufa a 130 oC. La differenza tra le due pesate non deve superare lo 0,1 % di umidità.
4.2.2. Cereali, farine, semole e semolini
Tarare, con la precisione di 0,5 mg, un recipiente (3.3), provvisto di coperchio. Pesare nel recipiente tarato, con l'approssimazione di 1 mg, 5 g circa di prodotto macinato e ripartirlo uniformemente. Porre il recipiente, senza coperchio, nella stufa preriscaldata a 130 °C. Per evitare che la temperatura della stufa scenda troppo, introdurre il recipiente nel più breve tempo possibile. Lasciare essiccare per due ore a partire dal momento in cui la stufa ha nuovamente raggiunto la temperatura di 130 oC. Rimettere il coperchio sul recipiente, estrarre quest'ultimo dalla stufa, lasciare raffreddare per 30-45 minuti nell'essiccatore (3.6) e pesare con l'approssimazione di 1 mg.
|
4.2.3.
|
Alimenti composti contenenti più del 4 % di saccarosio o lattosio: materie prime per mangimi quali carrube, prodotti a base di cereali idrolizzati, germi di malto, fettucce di barbabietola, «solubili» di pesce e zuccheri; alimenti composti contenenti più del 25 % di sali minerali con acqua di cristallizzazione.
Tarare, con la precisione di 0,5 mg, un recipiente (3.3), provvisto di coperchio. Pesare nel recipiente tarato, con l'approssimazione di 1 mg, circa 5 g di prodotto e ripartirli uniformemente. Porre il recipiente, privo del coperchio, nella stufa a vuoto (3.5) preriscaldata a una temperatura di 80-85 oC. Per evitare che la temperatura della stufa scenda troppo, introdurre il recipiente nel più breve tempo possibile.
Portare la pressione a 100 Torr e lasciare essiccare a questa pressione per quattro ore in corrente d'aria secca e calda, o mediante un disidratante (300 g circa per 20 campioni). In quest'ultimo caso interrompere la connessione con la pompa a vuoto quando è raggiunta la pressione prescritta. Misurare la durata dell'essiccazione a partire dal momento in cui la stufa ha nuovamente raggiunto la temperatura di 80-85 oC. Riportare poi con precauzione la stufa alla pressione atmosferica. Aprire la stufa, coprire immediatamente il recipiente con il coperchio, estrarlo dalla stufa, lasciarlo raffreddare per 30-45 minuti nell'essiccatore (3.6) e pesare con l'approssimazione di 1 mg. Essiccare per altri 30 minuti nella stufa a vuoto, alla temperatura di 80-85 oC, e pesare di nuovo. La differenza tra le due pesate non deve superare lo 0,1 % di umidità.
|
4.3. Preessiccazione
4.3.1. Alimenti per animali diversi da quelli menzionati al punto 4.3.2
Gli alimenti solidi, il cui contenuto di umidità è elevato e rende difficile la macinazione, devono essere preessiccati nel modo indicato di seguito.
Pesare, con l'approssimazione di 10 mg, 50 g di prodotto non macinato (un grossolano spezzettamento può essere effettuato, se necessario, nel caso di alimenti compressi o agglomerati) in un recipiente appropriato (ad esempio, una piastra di alluminio di 20 × 12 cm con bordo di 0,5 cm). Lasciar essiccare in una stufa alla temperatura di 60-70 oC, sino a che il contenuto di umidità sia portato a un valore compreso tra l'8 e il 12 %. Togliere dalla stufa, lasciar raffreddare il prodotto non coperto nel laboratorio per un'ora e pesare con l'approssimazione di 10 mg. Macinare subito dopo come indicato al punto 4.1.1 ed effettuare l'essiccazione come indicato ai punti 4.2.1 o 4.2.3 secondo la natura dell'alimento.
4.3.2. Cereali
I cereali in granella aventi una percentuale di umidità superiore al 17 % debbono essere preessiccati nel modo indicato di seguito.
Pesare, con l'approssimazione di 10 mg, 50 g di cereali in granella non moliti in un recipiente appropriato (ad esempio una piastra di alluminio di 20 × 12 cm con bordo di 0,5 cm). Lasciare essiccare in una stufa per 5-7 minuti alla temperatura di 130 oC. Togliere dalla stufa, lasciar raffreddare il prodotto non coperto nel laboratorio per due ore e pesare con l'approssimazione di 10 mg. Molire subito dopo come indicato al punto 4.1.2 ed effettuare l'essiccazione come indicato al punto 4.2.2.
5. Calcolo dei risultati
Il contenuto di umidità (X), espresso in percentuale del campione, è dato dalle seguenti formule:
5.1. Essiccazione senza preessiccazione
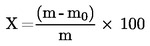
dove
|
m
|
=
|
peso iniziale, in grammi, della quantità di prodotto sottoposta all'analisi,
|
|
m0
|
=
|
peso, in grammi, del campione essiccato.
|
5.2. Essiccazione con preessiccazione

dove
|
m
|
=
|
peso iniziale, in grammi, della quantità di prodotto sottoposta all'analisi,
|
|
m1
|
=
|
peso, in grammi, della quantità di prodotto sottoposta all'analisi, dopo la preessiccazione,
|
|
m2
|
=
|
peso, in grammi, della quantità di prodotto sottoposta all'analisi, dopo la macinazione o frantumazione,
|
|
m0
|
=
|
peso, in grammi, del campione essiccato.
|
5.3. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non supera lo 0,2 % di umidità (valore assoluto).
6. Osservazione
Qualora sia necessario procedere alla macinazione e questa comporti una variazione nel contenuto di umidità del prodotto, i risultati dell'analisi dei componenti dell'alimento devono essere corretti in funzione del contenuto di umidità del campione iniziale.
B. DETERMINAZIONE DELL'UMIDITA NEI GRASSI E NEGLI OLI ANIMALI E VEGETALI
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di umidità (acqua ed altre materie volatili) dei grassi e degli oli animali e vegetali.
2. Principio
Il campione viene sottoposto ad essiccazione a 103 oC fino a cessazione della diminuzione di massa (la perdita di peso tra due pesate consecutive deve essere inferiore o pari a 1 mg). La perdita di peso è determinata per pesata.
3. Apparecchiatura
|
3.1.
|
Recipiente a fondo piano, di materiale resistente alla corrosione, diametro da 8 a 9 cm, altezza di 3 cm circa.
|
|
3.2.
|
Termometro con bulbo rinforzato e camera di espansione all'estremità superiore, tarato da circa 80 oC ad almeno 110 oC , lunghezza di 10 cm circa.
|
|
3.3.
|
Bagno di sabbia o piastra elettrica riscaldante.
|
|
3.4.
|
Essiccatore, contenente un efficace disidratante.
|
|
3.5.
|
Bilancia per analisi.
|
4. Procedimento
Pesare, con l'approssimazione di 1 mg, 20 g circa del campione omogeneizzato nel recipiente (3.1), essiccato e tarato, contenente il termometro (3.2). Riscaldare sul bagno di sabbia o sulla piastra riscaldante (3.3), agitando continuamente con il termometro, in modo da far salire la temperatura a 90 oC in circa 7 minuti.
Ridurre l'intensità del riscaldamento secondo la frequenza con la quale le bolle risalgono dal fondo del recipiente. La temperatura non deve superare i 105 oC. Continuare ad agitare raschiando il fondo del recipiente sino a quando non si formano più bolle.
Per assicurare l'eliminazione completa dell'umidità, riscaldare più volte a 103 ± 2 oC, raffreddando a 93 oC tra i riscaldamenti successivi. Lasciare quindi raffreddare nell'essiccatore (3.4) sino a temperatura ambiente e pesare. Ripetere l'operazione fino a quando la perdita di peso tra due pesate consecutive non supera i 2 mg.
|
Nota
|
Un aumento del peso del campione dopo i ripetuti riscaldamenti indica un'ossidazione del grasso. In questo caso, calcolare il risultato basandosi sulla pesata effettuata immediatamente prima che il peso abbia incominciato ad aumentare.
|
5. Calcolo dei risultati
Il contenuto di umidità, espresso in percentuale del campione, è dato dalla seguente formula:

dove
|
m
|
=
|
peso, in grammi, della quantità di prodotto sottoposta all'analisi,
|
|
m1
|
=
|
peso, in grammi, del recipiente con il suo contenuto prima del riscaldamento,
|
|
m2
|
=
|
peso, in grammi, del recipiente con il suo contenuto dopo il riscaldamento.
|
I risultati inferiori allo 0,05 % debbono recare la dicitura «meno di 0,05 %».
Ripetibilità
La differenza tra i risultati di due determinazioni parallele, effettuate sullo stesso campione, non deve oltrepassare lo 0,05 % in valore assoluto.
C. DETERMINAZIONE DEL CONTENUTO DI PROTEINE GREGGE
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto in proteine gregge degli alimenti per animali a partire dal contenuto di azoto, dosato secondo il metodo di Kjeldahl.
2. Principio
Il campione viene mineralizzato in acido solforico in presenza di un catalizzatore. La soluzione acida è alcalinizzata con una soluzione d'idrossido di sodio. L'ammoniaca viene isolata per distillazione e raccolta in una quantità determinata di acido solforico, il cui eccesso è titolato con una soluzione standard d'idrossido di sodio.
In alternativa, l'ammoniaca liberatasi viene distillata in un eccesso di soluzione di acido borico e successivamente titolata con una soluzione di acido cloridrico o acido solforico.
3. Reattivi
|
3.1.
|
Solfato di potassio.
|
|
3.2.
|
Catalizzatore: ossido rameico (II) CuO o solfato di rame (II) pentaidrato CuSO4 5H2 O.
|
|
3.4.
|
Acido solforico, ρ20 = 1,84 g/ml.
|
|
3.5.
|
Acido solforico, soluzione volumetrica standard, c(H2SO4) = 0,25 mol/l.
|
|
3.6.
|
Acido solforico, soluzione volumetrica, c(H2SO4) = 0,10 mol/l.
|
|
3.7.
|
Acido solforico, soluzione volumetrica standard, c(H2SO4) = 0,05 mol/l.
|
|
3.8.
|
Indicatore rosso di metile: sciogliere 300 mg di rosso di metile in 100 ml di etanolo, σ = 95-96 % (v/v).
|
|
3.9.
|
Soluzione d'idrossido di sodio (è possibile usare quello di purezza tecnica), β = 40 g/100 ml (m/v: 40 %).
|
|
3.10.
|
Idrossido di sodio, soluzione standard volumetrica, c(NaOH) = 0,25 mol/l.
|
|
3.11.
|
Idrossido di sodio, soluzione volumetrica standard, c(NaOH) = 0,10 mol/l.
|
|
3.12.
|
Pietra pomice in granulati, lavata con acido cloridrico e calcinata.
|
|
3.13.
|
Acetanilide (p.f. = 114 oC, contenuto N = 10,36 %).
|
|
3.14.
|
Saccarosio (esente da azoto).
|
|
3.15.
|
Acido borico (H3BO3).
|
|
3.16.
|
Soluzione d'indicatore rosso di metile: sciogliere 100 mg di rosso di metile in 100 ml di etanolo o metanolo.
|
|
3.17.
|
Soluzione di verde di bromocresolo: sciogliere 100 mg di verde di bromocresolo in 100 ml di etanolo o metanolo.
|
|
3.18.
|
Soluzione di acido borico (da 10 g/l a 40 g/l a seconda dell'apparecchiatura utilizzata)
Quando è applicato il metodo di determinazione colorimetrica del punto finale vanno aggiunti alle soluzioni di acido borico gli indicatori rosso di metile e bromocresolo. Nella preparazione di 1 litro di soluzione di acido borico, prima di portare a volume, aggiungere 7 ml di soluzione d'indicatore rosso metile (3.16) e 10 ml di soluzione di verde di bromocresolo (3.17).
A seconda dell'acqua utilizzata, il pH della soluzione di acido borico può variare da una partita all'altra. Per ottenere una prova in bianco positiva è spesso necessario un trattamento con una piccola quantità di alcali.
|
Nota.:
|
L'aggiunta di circa 3-4 ml di NaOH (3.11) a 1 litro di soluzione di acido borico a 10 g/l dà in genere buoni risultati. Conservare la soluzione a temperatura ambiente e proteggerla dalla luce e da sorgenti di fumi di ammoniaca.
|
|
|
3.19.
|
Acido cloridrico, soluzione volumetrica standard, c(HCl) = 0,10 mol/l.
|
Nota.:
|
Si possono utilizzare altre concentrazioni di soluzioni volumetriche (3.5, 3.6, 3.7, 3.10, 3.11 e 3.19) a condizione di apportare le necessarie correzioni nei calcoli. Le concentrazioni sono espresse sempre in cifre a quattro decimali.
|
|
4. Apparecchiatura
Apparecchi per mineralizzazione, distillazione e titolazione secondo il metodo di Kjeldahl.
5. Procedimento
5.1. Mineralizzazione
Pesare, con l'approssimazione di 1 mg, 1 g del campione e introdurlo nel pallone dell'apparecchio di mineralizzazione. Aggiungere 15 g di solfato di potassio (3.1), una quantità appropriata di catalizzatore (3.2) [da 0,3 g a 0,4 g di ossido rameico, oppure da 0,9 g a 1,2 g di solfato di rame (II) pentaidrato], 25 ml di acido solforico (3.4) e, se necessario, frammenti di pietra pomice (3.12). Mescolare.
Riscaldare il pallone prima moderatamente, agitando di tanto in tanto, se necessario, fino a carbonizzazione della massa e a scomparsa della schiuma; quindi scaldare più intensamente sino a ebollizione regolare del liquido. Il riscaldamento è adeguato se l'acido bollendo si condensa sulle pareti del pallone. Evitare che le pareti si surriscaldino e che particelle organiche vi aderiscano.
Quando la soluzione diventa limpida e di colore verde pallido prolungare l'ebollizione ancora per due ore. Lasciare quindi raffreddare.
5.2. Distillazione
Aggiungere con precauzione un quantitativo d'acqua sufficiente a sciogliere completamente i solfati. Lasciar raffreddare. Aggiungere quindi, se necessario, qualche granulo di zinco (3.3). Procedere secondo il punto 5.2.1 o 5.2.2.
5.2.1. Distillazione in acido solforico
Introdurre nel matraccio collettore dell'apparecchio di distillazione 25 ml misurati esattamente di acido solforico (3.5 o 3.7, a seconda del presunto contenuto di azoto) e qualche goccia di indicatore al rosso di metile (3.8).
Collegare il pallone di mineralizzazione al refrigerante dell'apparecchio di distillazione e immergere l'estremità di quest'ultimo per almeno 1 cm nel liquido del matraccio collettore (cfr. osservazione al punto 8.3). Versare lentamente nel pallone 100 ml di soluzione di idrossido di sodio (3.9) senza provocare una perdita di ammoniaca (cfr. osservazione al punto 8.1). Scaldare il pallone fino a distillazione completa dell'ammoniaca.
5.2.2. Distillazione in acido borico
Quando la titolazione del contenuto di ammoniaca del distillato è realizzata manualmente, si applica il metodo indicato di seguito. Quando l'unità di distillazione è interamente automatizzata, anche per quanto riguarda la titolazione del tenore di ammoniaca del distillato, seguire le istruzioni d'uso di tale unità fornite dal fabbricante.
Collocare un matraccio collettore contenente 25-30 ml della soluzione di acido borico (3.18) alla bocca d'uscita del refrigerante in modo che il tubo di evacuazione si trovi al di sotto della superficie dell'eccesso di soluzione di acido borico. Regolare l'unità di distillazione per ottenere 50 ml di soluzione di idrossido di sodio (3.9). Far funzionare l'unità di distillazione conformemente alle istruzioni fornite dal fabbricante ed eliminare l'ammoniaca liberatasi per distillazione aggiungendo la soluzione di idrossido di sodio. Raccogliere il distillato nella soluzione di acido borico. La quantità di distillato (tempo di distillazione in corrente di vapore) dipende dal tenore di azoto nel campione. Seguire le indicazioni fornite dal fabbricante.
|
Nota:
|
In un'unità di distillazione semiautomatica l'aggiunta di eccesso di idrossido di sodio e la distillazione in corrente di vapore avvengono automaticamente.
|
5.3. Titolazione
Procedere conformemente al punto 5.3.1 o 5.3.2.
5.3.1. Acido solforico
Titolare l'eccesso di acido solforico nel matraccio collettore per mezzo di una soluzione d'idrossido di sodio (3.10 o 3.11) secondo la concentrazione dell'acido solforico usato, sino a raggiungere il punto finale.
5.3.2. Acido borico
Titolare il contenuto del matraccio collettore con la soluzione volumetrica standard di acido cloridrico (3.19) o con la soluzione volumetrica standard di acido solforico (3.6) per mezzo di una buretta e leggere la quantità di soluzione titolata utilizzata.
Quando è applicato il metodo di determinazione colorimetrica del punto finale, si raggiunge il punto finale all'apparire della prima traccia di colore rosa nel contenuto. Valutare il contenuto della buretta con l'approssimazione di 0,05 ml. La visualizzazione del punto finale può essere facilitata per mezzo di un rivelatore fotometrico.
È possibile farlo automaticamente per mezzo di un'unità di distillazione in corrente di vapore a titolazione automatica.
Seguire le istruzioni del fabbricante relative al funzionamento dell'unità di distillazione o del distillatore titolatore, a seconda dei casi.
|
Nota:
|
Quando è utilizzato un sistema di titolazione automatica, con soluzione di acido borico all'1 % (3.18), la titolazione inizia non appena avviene la distillazione.
In caso di utilizzo di un'unità di distillazione interamente automatica, anche la titolazione automatica dell'ammoniaca può essere eseguita applicando il metodo di determinazione del punto finale per mezzo di un sistema di titolazione potenziometrica del pH.
In questo caso si ricorre ad un titolatore automatico con pH-metro, tarato in maniera appropriata entro una gamma di valori compresa tra pH 4 e pH 7 conformemente ai normali metodi di taratura del pH in laboratorio.
Il punto finale in pH della titolazione è raggiunto a pH 4,6, che corrisponde al punto di inflessione (o di flesso) della curva di titolazione.
|
5.4. Prova in bianco
Per confermare che i reattivi sono esenti da azoto, effettuare una prova in bianco (mineralizzazione, distillazione e titolazione) sostituendo il campione con 1 g di saccarosio (3.14).
6. Calcolo dei risultati
I calcoli sono effettuati conformemente ai punti 6.1 o 6.2.
6.1. Calcolo per la titolazione conformemente al punto 5.3.1
Calcolare il tenore di proteine gregge, espresso in percentuale di peso, con la formula seguente:
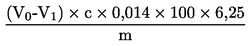
dove
|
V0
|
=
|
volume (ml) di NaOH (3.10 o 3.11) usato nella prova in bianco;
|
|
V1
|
=
|
volume (ml) di NaOH (3.10 o 3.11) usato nella titolazione del campione;
|
|
c
|
=
|
concentrazione (mol/l) dell'idrossido di sodio (3.10 o 3.11);
|
|
m
|
=
|
peso (g) del campione.
|
6.2. Calcolo per la titolazione conformemente al punto 5.3.2
6.2.1. Titolazione con acido cloridrico
Calcolare il tenore di proteine gregge, espresso in percentuale di peso, con la formula seguente:
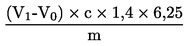
dove
|
m
|
=
|
peso (g) della quantità di sostanza sottoposta all'analisi;
|
|
c
|
=
|
concentrazione (mol/l) della soluzione volumetrica standard di acido cloridrico (3.19);
|
|
V0
|
=
|
volume (ml) di acido cloridrico usato nella prova in bianco;
|
|
V1
|
=
|
volume (ml) di acido cloridrico usato per la quantità di sostanza sottoposta all'analisi.
|
6.2.2. Titolazione con acido solforico
Calcolare il tenore di proteine gregge, espresso in percentuale di peso, con la formula seguente:

dove
|
m
|
=
|
peso (g) della quantità di sostanza sottoposta all'analisi;
|
|
c
|
=
|
concentrazione (mol/l) della soluzione volumetrica standard di acido solforico (3.6);
|
|
V0
|
=
|
volume (ml) di acido solforico usato per la prova in bianco;
|
|
V1
|
=
|
volume (ml) di acido solforico (3.6) usato per la quantità di sostanza sottoposta all'analisi.
|
7. Verifica del metodo
7.1. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
lo 0,2 % in valore assoluto per i contenuti di proteine gregge inferiori al 20 %;
|
|
—
|
l'1,0 % rispetto al valore più elevato, per i contenuti di proteine gregge compresi fra il 20 e il 40 %;
|
|
—
|
lo 0,4 % in valore assoluto per i contenuti di proteine gregge superiori al 40 %.
|
7.2. Precisione
Eseguire l'analisi (mineralizzazione, distillazione e titolazione) su 1,5-2,0 g di acetanilide (3.13) in presenza di 1 g di saccarosio (3.14); 1 g di acetanilide consuma 14,80 ml di acido solforico (3.5). Il recupero deve essere almeno del 99 %.
8. Osservazioni
|
8.1.
|
L'apparecchiatura può essere di tipo manuale, semiautomatico o automatico. Se l'apparecchio richiede un travasamento tra mineralizzazione e distillazione, il travasamento deve essere effettuato senza perdite. Se il pallone dell'apparecchio di distillazione non è provvisto di un imbuto separatore, aggiungere la soluzione d'idrossido di sodio immediatamente prima di collegare il pallone al refrigerante, lasciando colare lentamente il liquido lungo le pareti.
|
|
8.2.
|
Se il prodotto mineralizzato si solidifica, ricominciare la determinazione usando un quantitativo di acido solforico (3.4) superiore a quello indicato sopra.
|
|
8.3.
|
Per i prodotti poveri di sostanze azotate, il volume di acido solforico (3.7) da introdurre nel matraccio collettore può essere ridotto, se necessario, a 10 o 15 ml e portato a 25 ml con acqua.
|
|
8.4.
|
Per analisi ordinarie possono essere applicati metodi alternativi per determinare le proteine gregge, ma il metodo di Kjeldahl descritto in questa parte C costituisce il metodo di riferimento. L'equivalenza tra i risultati ottenuti con il metodo alternativo (ad esempio, il metodo Dumas) e quelli del metodo di riferimento va dimostrata per ciascuna matrice singolarmente. Dato che i risultati ottenuti con un metodo diverso, anche dopo verifica dell'equivalenza, possono deviare leggermente da quelli ottenuti con il metodo di riferimento, è necessario indicare nella relazione di analisi il metodo utilizzato per la determinazione delle proteine gregge.
|
D. DETERMINAZIONE DELL'UREA
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di urea negli alimenti per animali.
2. Principio
Il campione è messo in sospensione nell'acqua in presenza di un chiarificante. La sospensione è filtrata. Il contenuto di urea del filtrato è determinato, dopo aggiunta di 4-dimetilamminobenzaldeide (4-DMAB), misurando la densità ottica alla lunghezza d'onda di 420 nm.
3. Reattivi
|
3.1.
|
Soluzione di 4-dimetilamminobenzaldeide: sciogliere 1,6 g di 4-DMAB in 100 ml di etanolo al 96 % e aggiungere 10 ml di acido cloridrico (ρ20 1,19 g/ml). Questo reattivo si conserva soltanto per due settimane.
|
|
3.2.
|
Soluzione di Carrez I: sciogliere in acqua 21,9 g di acetato di zinco, Zn(CH3COO)2 2H2O e 3 g di acido acetico glaciale. Portare a 100 ml con acqua.
|
|
3.3.
|
Soluzione di Carrez II: sciogliere nell'acqua 10,6 g di ferrocianuro di potassio, K4 Fe (CN)6 3H2O. Portare a 100 ml con acqua.
|
|
3.4.
|
Carbone attivo non assorbente urea (eseguire un controllo).
|
|
3.5.
|
Urea, soluzione allo 0,1 % (p/v).
|
4. Apparecchiatura
|
4.1.
|
Agitatore rotativo a capovolgimento: circa 35-40 giri per minuto.
|
|
4.2.
|
Provette: 160 × 16 mm, a tappo smerigliato.
|
5. Procedimento
5.1. Analisi del campione
Pesare, con l'approssimazione di 1 mg, 2 g del campione ed introdurli con 1 g di carbone attivo (3.4) in un matraccio tarato da 500 ml. Aggiungere 400 ml di acqua e 5 ml di soluzione di Carrez I (3.2), agitare per circa 30 secondi e aggiungere 5 ml di soluzione di Carrez II (3.3). Mescolare per mezz'ora mediante l'agitatore rotativo a capovolgimento. Portare a volume con acqua, agitare e filtrare.
Prelevare 5 ml di filtrati limpidi ed incolori, introdurli nei tubi da prova con tappo smerigliato, aggiungere 5 ml di soluzione di 4-DMAB (3.1) e mescolare. Porre i tubi in bagnomaria a 20 oC (+/- 4 oC). Dopo 15 minuti, misurare la densità ottica della soluzione del campione con lo spettrofotometro a 420 nm rispetto alla soluzione della prova in bianco dei reattivi.
5.2. Curva di taratura
Prelevare volumi di 1, 2, 4, 5 e 10 ml della soluzione di urea (3.5), introdurli in matracci tarati da 100 ml e portare a volume con acqua. Prelevare 5 ml di ciascuna soluzione aggiungendo ogni volta 5 ml di soluzione di 4-DMAB (3.1), mescolare e misurare la densità ottica, come indicato dianzi, rispetto ad una soluzione testimone contenente 5 ml di 4-DMAB e 5 ml d'acqua, esente da urea. Tracciare la curva di taratura.
6. Calcolo dei risultati
Determinare la quantità d'urea, nella parte di campione sottoposta all'analisi, servendosi della curva di taratura.
Esprimere il risultato in percentuale del campione.
7. Osservazioni
|
7.1.
|
Per contenuti di urea superiori al 3 %, ridurre la quantità di campione da sottoporre all'analisi a 1 g o diluire la soluzione originale, per non avere più di 50 mg d'urea in 500 ml.
|
|
7.2.
|
Per bassi tenori di urea, aumentare la quantità di campione da sottoporre all'analisi fino a quando il filtrato resta limpido ed incolore.
|
|
7.3.
|
Quando il prodotto contiene composti azotati semplici, ad esempio amminoacidi, si effettua la misurazione della densità ottica a 435 nm.
|
E. DETERMINAZIONE DELLE BASI AZOTATE VOLATILI
I. PER MICRODIFFUSIONE
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di basi azotate volatili, espresse in ammoniaca, degli alimenti per gli animali.
2. Principio
Il campione viene estratto con acqua, la soluzione viene chiarificata e filtrata. Le basi azotate volatili sono spostate per mezzo di una soluzione di carbonato di potassio per microdiffusione, raccolte in una soluzione di acido borico e titolate con acido solforico.
3. Reattivi
|
3.1.
|
Soluzione al 20 % (p/v) di acido tricloroacetico.
|
|
3.2.
|
Indicatore: sciogliere 33 mg di verde di bromocresolo e 65 mg di rosso di metile in 100 ml d'etanolo al 95-96 % (v/v).
|
|
3.3.
|
Soluzione di acido borico: in un pallone tarato da 1 litro, sciogliere 10 g d'acido borico in 200 ml d'etanolo al 95-96 % (v/v) e 700 ml d'acqua. Aggiungere 10 ml di indicatore (3.2). Mescolare e aggiustare, se necessario, il colore della soluzione al rosso chiaro mediante aggiunta di una soluzione d'idrossido di sodio. 1 ml di questa soluzione consente di fissare al massimo 300 μg di ammoniaca.
|
|
3.4.
|
Soluzione satura di carbonato di potassio: sciogliere 100 g di carbonato di potassio in 100 ml d'acqua portata all'ebollizione. Lasciar raffreddare e filtrare.
|
|
3.5.
|
Acido solforico 0,01 mol/l.
|
4. Apparecchiatura
|
4.1.
|
Agitatore rotativo a capovolgimento: circa 35-40 giri al minuto
|
|
4.2.
|
Celle di Conway (cfr. figura), in vetro o in materia plastica.
|
|
4.3.
|
Microburette, graduate a 1/100 ml.
|
5. Procedimento
Pesare, con l'approssimazione di 1 mg, 10 g di sostanza e porli in un pallone tarato da 200 ml con 100 ml d'acqua. Mescolare o agitare il pallone, nell'agitatore rotativo, per 30 minuti. Aggiungere 50 ml della soluzione d'acido tricloroacetico (3.1), portare a volume con acqua, agitare vigorosamente e filtrare su un filtro a pieghe.
Prelevare con una pipetta 1 ml della soluzione di acido borico (3.3) nell'incavo centrale della cella Conway e, nella corona della cella, 1 ml del filtrato ottenuto dal campione. Coprire parzialmente con il coperchio ingrassato. Lasciar scolare rapidamente nella corona periferica 1 ml della soluzione satura di carbonato di potassio (3.4) e chiudere ermeticamente il coperchio. Agitare con precauzione la cella facendola rotare su un piano orizzontale in modo da assicurare la miscelazione dei due reattivi. Mantenere l'apparecchio per almeno quattro ore alla temperatura ambiente o per un'ora a 40 oC.
Titolare le basi volatili nella soluzione d'acido borico con la soluzione di acido solforico (3.5) per mezzo di una microburetta (4.3).
Effettuare una prova in bianco applicando lo stesso procedimento, in assenza del campione da analizzare.
6. Calcolo dei risultati
1 ml di H2SO40,01 mol/l corrisponde a 0,34 mg di ammoniaca.
Esprimere il risultato in percentuale del campione.
Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non supera:
|
—
|
il 10 %, in valore relativo, per i contenuti di ammoniaca inferiori a 1,0 %,
|
|
—
|
lo 0,1 %, in valore assoluto, per i contenuti di ammoniaca uguali o superiori all'1,0 %.
|
7. Osservazione
Se il contenuto di ammoniaca del campione è superiore allo 0,6 %, diluire il filtrato iniziale.
CONWAY CELL
Scale 1/1

II. PER DISTILLAZIONE
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di basi azotate volatili, espresse in ammoniaca, delle farine di pesce non contenenti, praticamente, urea. Il metodo è applicabile unicamente a contenuti di ammoniaca inferiori a 0,25 %.
2. Principio
Il campione viene estratto con acqua, la soluzione è chiarificata e filtrata. Le basi azotate volatili sono portate/spostate all'ebollizione per aggiunta di ossido di magnesio e raccolte in una quantità determinata d'acido solforico il cui eccesso viene titolato di ritorno con una soluzione d'idrossido di sodio.
3. Reattivi
|
3.1.
|
Soluzione al 20 % (p/v) di acido tricloroacetico.
|
|
3.3.
|
Emulsione antischiuma (ad esempio, silicone).
|
|
3.4.
|
Acido solforico 0,05 mol/l.
|
|
3.5.
|
Soluzione di idrossido di sodio (0,1 mol/l).
|
|
3.6.
|
Soluzione allo 0,3 % (v/v) di rosso di metile in etanolo al 95-96 % (v/v).
|
4. Apparecchiatura
|
4.1.
|
Agitatore rotativo a capovolgimento: circa 35-40 giri al minuto.
|
|
4.2.
|
Apparecchio per distillazione del tipo Kjeldahl.
|
5. Procedimento
Pesare, con l'approssimazione di 1 mg, 10 g di sostanza e introdurli in un pallone tarato da 200 ml con 100 ml d'acqua. Mescolare o agitare il pallone, nell'agitatore rotativo, per 30 minuti. Aggiungere 50 ml della soluzione d'acido tricloroacetico (3.1), portare a volume con acqua, agitare vigorosamente e filtrare su un filtro a pieghe.
Prelevare una quantità di filtrato limpido in funzione del presunto contenuto di basi azotate volatili (100 ml sono generalmente sufficienti). Diluire a 200 ml e aggiungere 2 g di ossido di magnesio (3.2) e qualche goccia di emulsione antischiuma (3.3). La soluzione deve risultare alcalina alla cartina tornasole, altrimenti aumentare la quantità di ossido di magnesio (3.2). Procedere conformemente ai punti 5.2 e 5.3 del metodo di analisi per la determinazione del contenuto di proteine gregge (parte C del presente allegato).
Effettuare una prova in bianco applicando lo stesso procedimento, in assenza del campione da analizzare.
6. Calcolo dei risultati
1 ml di H2SO4 0,05 mol/l corrisponde a 1,7 mg di ammoniaca.
Esprimere il risultato in percentuale del campione.
Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non supera, in valore relativo, il 10 % di ammoniaca.
F. DETERMINAZIONE DEGLI AMMINOACIDI (TRIPTOFANO ESCLUSO)
1. Finalità e campo d'applicazione
Il metodo serve per determinare gli amminoacidi liberi (sia di sintesi che naturali) e totali (legati a peptidi e liberi) negli alimenti per animali mediante un analizzatore di amminoacidi. Il metodo è applicabile ai seguenti amminoacidi: cist(e)ina, metionina, lisina, treonina, alanina, arginina, acido aspartico, acido glutammico, glicina, istidina, isoleucina, leucina, fenilalanina, prolina, serina, tirosina e valina.
Il metodo non distingue gli amminoacidi dai loro sali e non può distinguere la forma D degli amminoacidi dalla forma L; esso non è adatto per la determinazione del triptofano né degli analoghi idrossilati degli amminoacidi.
2. Principio
2.1. Amminoacidi liberi
Gli amminoacidi liberi sono estratti con acido cloridrico diluito. Le macromolecole azotate coestratte vengono fatte precipitare con acido solfosalicilico e vengono rimosse per filtrazione. La soluzione filtrata viene portata a pH 2,20. Gli amminoacidi sono separati mediante cromatografia a scambio ionico e determinati per reazione con la ninidrina e con rivelazione fotometrica a 570 nm.
2.2. Amminoacidi totali
La scelta della procedura dipende dagli amminoacidi oggetto dell'analisi. Cist(e)ina e metionina devono essere ossidate ad acido cisteico e al metionina sulfone prima dell'idrolisi. La tirosina deve essere determinata in idrolizzati di campioni non ossidati. Tutti gli altri amminoacidi elencati al punto 1 possono essere determinati sia nel campione ossidato sia in quello non ossidato.
L'ossidazione viene eseguita a 0 oC con una miscela di acido performico e di fenolo. L'eccesso del reagente di ossidazione viene decomposto con metabisolfito di sodio. Il campione ossidato o non ossidato è idrolizzato con acido cloridrico (3.20) per 23 ore. L'idrolizzato è regolato a pH 2,20. Gli amminoacidi sono separati mediante cromatografia a scambio ionico e determinati per reazione con la ninidrina e con rivelazione fotometrica a 570 nm (440 nm per la prolina).
3. Reattivi
Usare acqua bidistillata o di qualità equivalente (conduttività < 10 μS).
|
3.1.
|
Perossido d'idrogeno, p (p/p) = 30 %.
|
|
3.2.
|
Acido formico, p (p/p) = 98-100 %.
|
|
3.4.
|
Metabisolfito di sodio.
|
|
3.6.
|
Acido 5-solfosalicilico diidrato.
|
|
3.7.
|
Acido cloridrico, densità circa 1,18 g/ml.
|
|
3.8.
|
Citrato trisodico diidrato.
|
|
3.9.
|
2,2'-Tiodietanolo (tiodiglicole).
|
|
3.12.
|
Etere di petrolio, intervallo di ebollizione: 40-60 oC.
|
|
3.13.
|
Norleucina o altro composto adatto ad essere utilizzato come standard interno.
|
|
3.14.
|
Azoto gassoso (< 10 ppm di ossigeno).
|
|
3.16.1.
|
Sostanze di riferimento elencate al punto 1. Composti puri non contenenti acqua di cristallizzazione. Essiccare sotto vuoto su P2O5 o H2SO4 per una settimana prima dell'uso.
|
|
3.16.3.
|
Metionina sulfone.
|
|
3.17.
|
Soluzione di idrossido di sodio, c = 7,5 mol/l:
sciogliere 300 g di NaOH (3.5) in acqua e portare a 1 litro.
|
|
3.18.
|
Soluzione di idrossido di sodio, c = 1 mol/l:
sciogliere 40 g di NaOH (3.5) in acqua e portare a 1 litro.
|
|
3.19.
|
Soluzione di acido formico-fenolo:
miscelare 889 g di acido formico (3.2) con 111 g d'acqua e aggiungere 4,73 g di fenolo (3.3).
|
|
3.20.
|
Miscela di idrolisi, c = HCl 6 mol/l contenente 1 g di fenolo:
aggiungere 1 g di fenolo (3.3) a 492 ml di HCl (3.7) e portare a 1 litro con acqua.
|
|
3.21.
|
Miscela di estrazione, c = HCl 0,1 mol/l contenente 2 % di tiodiglicole: introdurre 8,2 ml di HCl (3.7) in circa 900 ml d'acqua e miscelare, aggiungere 20 ml di tiodiglicolo (3.9) e portare a 1 litro con acqua (non miscelare direttamente le sostanze di cui ai punti 3.7 e 3.9).
|
|
3.22.
|
Acido 5-solfosalicilico: β = 6 %:
sciogliere 60 g di acido 5-solfosalicilico (3.6) in acqua e portare a 1 litro con acqua.
|
|
3.23.
|
Miscela di ossidazione (acido performico-fenolo):
miscelare 0,5 ml di perossido di idrogeno (3.1) con 4,5 ml di soluzione di acido formico e fenolo (3.19) in un piccolo becher. Tenere in incubazione a 20-30 oC per un'ora per ottenere acido performico; lasciare raffreddare su un bagno di acqua gelata (15 min) prima di aggiungere il campione.
Attenzione: evitare il contatto con la pelle e indossare indumenti di protezione.
|
|
3.24.
|
Tampone citrato, c = Na+ 0,2 mol/l, pH 2,20:
sciogliere 19,61 g di citrato di sodio (3.8), 5 ml di tiodiglicole (3.9), 1 g di fenolo (3.3) e 16,50 ml di HCl (3.7) in cira 800 ml d'acqua. Portare il pH a 2,20. Portare a 1 litro con acqua.
|
|
3.25.
|
Tamponi di eluizione preparati conformemente alle prescrizioni relative all'analizzatore (4.9).
|
|
3.26.
|
Reattivo alla ninidrina preparato conformemente alle prescrizioni relative all'analizzatore (4.9).
|
|
3.27.
|
Soluzioni standard di amminoacidi. Conservare queste soluzioni a temperatura inferiore a 5 oC.
|
|
3.27.1.
|
Soluzione madre standard di amminoacidi (3.16.1).
c = 2,5 μmol/ml di ciascuno in acido cloridrico.
Reperibile in commercio.
|
|
3.27.2.
|
Soluzione madre standard di acido cisteico e metionina sulfone, c = 1,25 μmol/ml.
Sciogliere 0,2115 g di acido cisteico (3.16.2) e 0,2265 g di metionina sulfone (3.16.3) in tampone citrato (3.24) in un matraccio tarato da 1 litro e portare a volume con tampone citrato. Conservare a temperatura inferiore a 5 oC al massimo per 12 mesi. Questa soluzione non viene utilizzata se la soluzione madre standard (3.27.1) contiene acido cisteico e metionina sulfone.
|
|
3.27.3.
|
Soluzione madre standard, ad esempio norleucina, c = 20 μmol/ml.
Sciogliere 0,6560 g di norleucina (3.13) in tampone citrato (3.24) in un matraccio tarato e portare a 250 ml con tampone citrato. Conservare a temperatura inferiore a 5 oC al massimo per 6 mesi.
|
|
3.27.4.
|
Soluzione di taratura degli amminoacidi standard da usarsi con idrolizzati, c = 5 nmol/50 μl di acido cisteico e metionina sulfone e c = 10 nmol/50 μl di altri amminoacidi. Sciogliere 2,2 g di cloruro di sodio (3.10) in un becher da 100 ml con 30 ml di tampone citrato (3.24). Aggiungere 4,00 ml di soluzione madre standard di amminoacidi (3.27.1), 4,00 ml di soluzione madre standard di acido cisteico e metionina sulfone (3.27.2) e, se del caso, 0,50 ml di soluzione madre standard dello standard interno (3.27.3). Portare il pH a 2,20 con idrossido di sodio (3.18).
Trasferire quantitativamente in un matraccio tarato da 50 ml, portare a volume con tampone citrato (3.24) e miscelare.
Conservare a temperatura inferiore a 5 oC al massimo per 3 mesi.
Cfr. anche le osservazioni al punto 9.1.
|
|
3.27.5.
|
Soluzione di taratura degli amminoacidi standard da usarsi con idrolizzati preparati secondo quanto prescritto al punto 5.3.3.1 e destinati all'uso con estratti (5.2). Preparare la soluzione di taratura secondo quanto indicato al punto 3.27.4, ma senza cloruro di sodio.
Conservare a temperatura inferiore a 5 oC al massimo per 3 mesi.
|
4. Apparecchiatura
|
4.1.
|
Pallone a fondo arrotondato da 100 o 250 ml provvisto di refrigerante a ricadere.
|
|
4.2.
|
Flacone di vetro borosilicatico da 100 ml con tappo a vite e guarnizione di gomma/teflon (ad esempio Duran, Schott) utilizzabile in stufa.
|
|
4.3.
|
Stufa a ventilazione forzata con regolazione della temperatura avente una precisione superiore a ± 2 oC.
|
|
4.4.
|
pH-metro (a tre cifre decimali).
|
|
4.5.
|
Filtro a membrana, da 0,22 μm.
|
|
4.7.
|
Evaporatore rotante sotto vuoto.
|
|
4.8.
|
Agitatore meccanico o mescolatore magnetico.
|
|
4.9.
|
Analizzatore di amminoacidi o apparecchiatura HPLC provvista di colonna a scambio ionico, dispositivo per ninidrina, derivatizzatore post-colonna e rivelatore fotometrico.
La colonna deve essere riempita di resine polistireniche sulfonate in grado di separare gli amminoacidi uno dall'altro e da altri prodotti reattivi alla ninidrina. Il flusso del tampone e del reattivo di ninidrina è garantito da pompe il cui grado di stabilità di portata è ±0,5 % nell'intervallo tra l'analisi della soluzione di taratura e l'analisi del campione.
Con alcuni analizzatori di amminoacidi si possono utilizzare metodi di idrolisi in cui l'idrolizzato presenta una concentrazione di sodio di c = 0,8 mol/l e contiene tutto l'acido formico residuo dalla fase di ossidazione. Altri apparecchi non consentono una separazione soddisfacente di certi amminoacidi se l'idrolizzato contiene troppo acido formico e/o presenta elevate concentrazioni di ioni sodio. In questo caso il volume dell'acido è ridotto a circa 5 ml mediante evaporazione dopo l'idrolisi e prima della regolazione del pH. L'evaporazione è eseguita sotto vuoto a temperatura non superiore a 40 oC.
|
5. Procedimento
5.1. Preparazione del campione
Il campione viene triturato in modo che possa passare attraverso un vaglio da 0,5 mm. I campioni molto umidi devono essere essiccati all'aria ad una temperatura non superiore a 50 oC o liofilizzati prima di essere triturati. I campioni ad alto tenore di grassi sono estratti con etere di petrolio (3.12) prima di essere triturati.
5.2. Determinazione degli amminoacidi liberi negli alimenti per animali e nelle premiscele
Pesare con l'approssimazione di 0,2 mg una quantità appropriata (1-5 g) del campione preparato (5.1) in una beuta e aggiungere 100,0 ml di miscela di estrazione (3.2.1). Agitare la miscela per 60 minuti con un agitatore o meccanico o miscelatore magnetico (4.8). Lasciar depositare il sedimento e pipettare 10,0 ml della soluzione surnatante in un becher da 100 ml.
Aggiungere 5,0 ml di soluzione di acido solfosalicilico (3.22) sempre agitando e continuare ad agitare con agitatore magnetico per 5 minuti. Filtrare o centrifugare il surnatante per rimuovere eventuali precipitati. Introdurre 10,0 ml della soluzione risultante in un becher da 100 ml e portare il pH a 2,20 con una soluzione di idrossido di sodio (3.18), trasferire in un matraccio tarato di volume appropriato utilizzando tampone citrato (3.24) e portare a volume con la soluzione tampone (3.24).
Se si usa uno standard interno, aggiungere 1,00 ml di standard interno (3.27.3) ogni 100 ml di soluzione finale e portare a volume con la soluzione tampone (3.24).
Procedere alla fase della cromatografia secondo quanto indicato al punto 5.4.
Se gli estratti non vengono cromatografati lo stesso giorno, conservarli a temperatura inferiore a 5 oC.
5.3. Determinazione degli amminoacidi totali
5.3.1. Ossidazione
Pesare, con l'approssimazione di 0,2 mg, da 0,1 a 1 g del campione preparato (5.1) in:
|
—
|
un pallone a fondo arrotondato da 100 ml (4.1) per l'idrolisi in sistema aperto (5.3.2.3), o
|
|
—
|
un pallone a fondo arrotondato da 250 ml (4.1) se è richiesta una bassa concentrazione di sodio, o
|
|
—
|
un flacone da 100 ml dotato di tappo a vite (4.2) per l'idrolisi in sistema chiuso (5.3.2.4).
|
La porzione di campione pesata deve avere un contenuto d'azoto di circa 10 mg e un contenuto di umidità non superiore a 100 mg.
Introdurre il pallone o il flacone in un bagno di acqua e ghiaccio e raffreddare a 0 oC; aggiungere 5 ml di miscela di ossidazione (3.23) e miscelare con una spatola di vetro con un estremità ricurva. Sigillare il pallone o flacone contenente la spatola con una pellicola impermeabile all'aria, introdurre il bagno di acqua e ghiaccio contenente il contenitore sigillato in un frigorifero a 0 oC e lasciarvelo per 16 ore. Dopo 16 ore togliere dal frigorifero e decomporre l'eccesso di reagente di ossidazione mediante l'aggiunta di 0,84 g di bisolfito di sodio (3.4).
Procedere come indicato al punto 5.3.2.1.
5.3.2. Idrolisi
5.3.2.1.
Idrolisi dei campioni ossidati
Aggiungere al campione ossidato, preparato conformemente al punto 5.3.1, 25 ml di miscela di idrolisi (3.20) avendo cura di risciacquare eventuali residui di campione che aderiscono alle pareti del recipiente e alla spatola.
Procedere come indicato al punto 5.3.2.3 o 5.3.2.4, a seconda del metodo di idrolisi utilizzato.
5.3.2.2.
Idrolisi dei campioni non ossidati
Pesare in un pallone a fondo arrotondato da 100 ml o 250 ml (4.1) o in un flacone da 100 ml con tappo a vite (4.2) (con l'approssimazione di 0,2 mg) una quantità da 0,1 a 1 g del campione preparato (5.1). La porzione di campione pesata deve avere un contenuto di azoto di circa 10 mg. Aggiungere con cautela 25 ml di miscela di idrolisi (3.20) e miscelare con il campione. Procedere come indicato al punto 5.3.2.3 o 5.3.2.4.
5.3.2.3.
Idrolisi, sistema aperto
Aggiungere 3 biglie di vetro alla miscela (preparata conformemente al punto 5.3.2.1 o 5.3.2.2) contenuta nel pallone e far bollire a ricadere per 23 ore a ebollizione continua. Al completamento dell'idrolisi, risciacquare il refrigerante con 5 ml di tampone citrato (3.24). Togliere il pallone e farlo raffreddare in un bagno di ghiaccio.
Procedere come indicato al punto 5.3.3.
5.3.2.4.
Idrolisi, sistema chiuso
Introdurre il flacone contenente la miscela, preparata conformemente al punto 5.3.2.1 o 5.3.2.2, in un forno (4.3) a 110 oC. Durante la prima ora, allo scopo di evitare un accumulo di pressione in conseguenza dello sviluppo di sostanze gassose e di evitare un'esplosione, porre il tappo a vite sopra al recipiente senza chiuderlo. Dopo un'ora, chiudere il recipiente con il tappo e lasciarlo nel forno (4.3) per 23 ore. Al completamento dell'idrolisi, togliere il flacone dal forno, aprire con cautela il tappo del flacone e introdurre il flacone in un bagno di acqua e ghiaccio. Lasciar raffreddare.
Secondo la procedura usata per la regolazione del pH (5.3.3), trasferire quantitativamente il contenuto del flacone in un becher da 250 ml o in un pallone a fondo tondo da 250 ml utilizzando tampone citrato (3.24).
Procedere come indicato al punto 5.3.3.
5.3.3. Regolazione del pH
Procedere, conformemente al punto 5.3.3.1 o 5.3.3.2, alla regolazione del pH, in funzione della tolleranza al sodio dell'analizzatore di amminoacidi (4.9).
5.3.3.1.
Per sistemi cromatografici (4.9) che richiedono una bassa concentrazione di sodio.
Quando si impiegano analizzatori di amminoacidi che richiedono una bassa concentrazione di sodio (quando il volume dell'acido deve essere ridotto), è consigliabile utilizzare una soluzione madre standard interna (3.27.3).
In questo caso aggiungere 2,00 ml della soluzione standard interna (3.27.3) all'idrolizzato prima dell'evaporazione.
Aggiungere 2 gocce di 1-ottanolo (3.15) all'idrolizzato ottenuto conformemente alla procedura di cui al punto 5.3.2.3 o 5.3.2.4.
Utilizzando un evaporatore rotante (4.7), ridurre il volume a 5-10 ml sotto vuoto a 40 oC. Se il volume viene ridotto accidentalmente a meno di 5 ml, scartare l'idrolizzato e ricominciare l'analisi.
Portare il pH a 2,20 con soluzione di idrossido di sodio (3.18) e procedere conformemente al punto 5.3.4.
5.3.3.2.
Per tutti gli altri analizzatori di amminoacidi (4.9)
Neutralizzare parzialmente gli idrolizzati ottenuti conformemente al punto 5.3.2.3 o 5.3.2.4, aggiungendovi con cautela, sempre agitando, 17 ml di soluzione di idrossido di sodio (3.17), facendo attenzione che la temperatura rimanga al di sotto di 40 oC.
Portare il pH a 2,20 a temperatura ambiente utilizzando la soluzione di idrossido di sodio di cui al punto 3.17 e, infine, una soluzione di idrossido di sodio conforme al punto 3.18. Procedere conformemente al punto 5.3.4.
5.3.4. Soluzione del campione per cromatografia
Trasferire quantitativamente l'idrolizzato portato a pH 2,20 (5.3.3.1 o 5.3.3.2) con tampone citrato (3.24) in un matraccio tarato da 200 ml e portare a volume con tampone (3.24).
Se non è stato ancora utilizzato uno standard interno, aggiungerne 2,00 ml (3.27.3) e portare a volume con tampone citrato (3.24). Miscelare accuratamente.
Procedere alla fase della cromatografia (5.4).
Se le soluzioni del campione non vengono cromatografate nello stesso giorno, conservarle a temperatura inferiore a 5 oC.
5.4. Cromatografia
Prima della cromatografia, portare l'estratto (5.2) o l'idrolizzato (5.3.4) a temperature ambiente. Agitare la miscela e filtrarne una quantità appropriata attraverso un filtro a membrana da 0,22 μm (4.5). La soluzione limpida risultante viene sottoposta a cromatografia a scambio ionico utilizzando un analizzatore di amminoacidi (4.9).
L'iniezione può venire eseguita manualmente o automaticamente. È importante iniettare sempre la stessa quantità (±0,5 %) di soluzione nella colonna per l'analisi degli standard e dei campioni, salvo quando si usa uno standard interno; inoltre i rapporti sodio/amminoacidi nello standard e nelle soluzioni del campione devono essere il più possibile simili.
La frequenza delle iniezioni della soluzione di taratura dipende dalla stabilità del reattivo ninidrina e del sistema analitico. La soluzione standard o del campione è diluita con tampone citrato (3.24) in misura tale che l'area del picco della soluzione standard sia compresa Fra il 30 % e il 200 % dell'area del picco dell'amminoacido nel campione.
La cromatografia degli amminoacidi varierà leggermente secondo il tipo di analizzatore impiegato e la resina usata. Il sistema scelto deve essere in grado di separare gli amminoacidi uno dall'altro e dalle sostanze reattive alla ninidrina. Nel corso dell'operazione, il sistema cromatografico deve dare una risposta lineare alle variazioni delle quantità di amminoacidi introdotti in colonna.
Durante la fase di cromatografia, quando si analizza una soluzione equimolare (degli amminoacidi sottoposti a determinazione), si applicano i rapporti di altezza valle/picco citati più avanti. La soluzione equimolare deve contenere almeno il 30 % del carico massimo di ciascun amminoacido che può essere determinato con precisione tramite il sistema di analisi degli amminoacidi (4.9).
Per la separazione treonina-serina, il rapporto di altezza valle/picco del più basso dei due amminoacidi che si sovrappongono sul cromatogramma non deve superare 2/10 [se la determinazione viene effettuata solo su cist(e)ina, metionina, treonina e lisina, una insufficiente separazione di picchi adiacenti influirà sfavorevolmente sulla determinazione]. Per tutti gli altri amminoacidi la separazione deve essere maggiore di 1/10.
Il sistema deve poter separare la lisina da «artefatti di lisina» e dall'ornitina.
6. Calcolo dei risultati
Determinare le aree del picco del campione e della soluzione standard per ogni singolo amminoacido e calcolare la quantità (X) in g di amminoacido per kg di campione secondo la seguente formula:
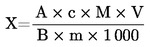
Se si usa uno standard interno moltiplicare per: 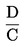
|
A
|
=
|
area del picco dell'idrolizzato o dell'estratto
|
|
B
|
=
|
area del picco della soluzione standard di taratura
|
|
C
|
=
|
area del picco dello standard interno nell'idrolizzato o nell'estratto
|
|
D
|
=
|
area del picco dello standard interno, soluzione standard di taratura
|
|
M
|
=
|
peso molecolare dell'amminoacido determinato
|
|
c
|
=
|
concentrazione dello standard in μmol/ml
|
|
m
|
=
|
peso del campione in g (corretto al peso originale se essiccato o sgrassato)
|
|
V
|
=
|
ml totali di idrolizzato (5.3.4) o ml del volume di diluizione totale calcolati dell'estratto (6.1).
|
La cistina e la cisteina sono determinate entrambe sotto forma di acido cisteico negli idrolizzati di campione ossidato, ma calcolate come cistina (C6H12N2O4S2, M 240,30 g/mol) utilizzando un peso molecolare di 120,15 g/mol (= 0,5 × 240,30 g/mol).
La metionina è determinata come metionina sulfone negli idrolizzati del campione ossidato, ma calcolata come metionina utilizzando il peso molecolare della metionina (149,21 g/mol).
La metionina libera aggiunta viene determinata dopo estrazione sotto forma di metionina; per il calcolo si usa lo stesso valore M.
|
6.1.
|
Il volume della diluizione totale degli estratti (F) per la determinazione degli amminoacidi liberi (5.2) è calcolato come segue:

|
V
|
=
|
volume dell'estratto finale.
|
|
7. Valutazione del metodo
Il metodo è stato testato nel 1990, nel corso di un confronto internazionale tra laboratori sulla base di quattro diversi alimenti per animali (alimento composto per suini, alimento composto per polli da ingrasso, concentrato proteico, premiscela). Le medie e le deviazioni standard ottenute dopo l'eliminazione dei risultati anomali figurano nelle seguenti tabelle:
Medie in g/kg
|
Materiale di riferimento
|
Amminoacido
|
|
Treonina
|
Cist(e)ina
|
Metionina
|
Lisina
|
|
Alimento composto per suini
|
6,94
n = 15
|
3,01
n = 17
|
3,27
n = 17
|
9,55
n = 13
|
|
Alimento composto per polli da ingrasso
|
9,31
n = 16
|
3,92
n = 18
|
5,08
n = 18
|
13,93
n = 16
|
|
Concentrato proteico
|
22,32
n = 16
|
5,06
n = 17
|
12,01
n = 17
|
47,74
n = 15
|
|
Premiscela
|
58,42
n = 16
|
—
|
90,21
n = 16
|
98,03
n = 16
|
|
n
|
=
|
numero di laboratori partecipanti.
|
|
7.1. Ripetibilità
La ripetibilità (espressa come «deviazione standard entro il laboratorio») del confronto tra laboratori è presentata nelle seguenti tabelle:
Deviazione standard entro il laboratorio (Sr) in g/kg
|
Materiale di riferimento
|
Amminoacido
|
|
Treonina
|
Cist(e)ina
|
Metionina
|
Lisina
|
|
Alimento composto per suini
|
0,13
n = 15
|
0,10
n = 17
|
0,11
n = 17
|
0,26
n = 13
|
|
Alimento composto per polli da ingrasso
|
0,20
n = 16
|
0,11
n = 18
|
0,16
n = 18
|
0,28
n = 16
|
|
Concentrato proteico
|
0,48
n = 16
|
0,13
n = 17
|
0,27
n = 17
|
0,99
n = 15
|
|
Premiscela
|
1,30
n = 16
|
—
|
2,19
n = 16
|
2,06
n = 16
|
|
n
|
=
|
numero di laboratori partecipanti.
|
|
Coefficiente di variabilità (%) per la deviazione standard entro il laboratorio (Sr)
|
Materiale di riferimento
|
Amminoacido
|
|
Treonina
|
Cist(e)ina
|
Metionina
|
Lisina
|
|
Alimento composto per suini
|
1,9
n = 15
|
3,3
n = 17
|
3,4
n = 17
|
2,8
n = 13
|
|
Alimento composto per polli da ingrasso
|
2,1
n = 16
|
2,8
n = 18
|
3,1
n = 18
|
2,1
n = 16
|
|
Concentrato proteico
|
2,7
n = 16
|
2,6
n = 17
|
2,2
n = 17
|
2,4
n = 15
|
|
Premiscela
|
2,2
n = 16
|
—
|
2,4
n = 16
|
2,1
n = 16
|
|
n
|
=
|
numero di laboratori partecipanti.
|
|
7.2. Riproducibilità
I risultati relativi alla deviazione standard tra laboratori ottenuti dal confronto tra laboratori figurano nelle seguenti tabelle:
Deviazione standard tra laboratori (Sr) in g/kg
|
Materiale di riferimento:
|
Amminoacido
|
|
Treonina
|
Cist(e)ina
|
Metionina
|
Lisina
|
|
Alimento composto per suini
|
0,28
n = 15
|
0,30
n = 17
|
0,23
n = 17
|
0,30
n = 13
|
|
Alimento composto per polli da ingrasso
|
0,48
n = 16
|
0,34
n = 18
|
0,55
n = 18
|
0,75
n = 16
|
|
Concentrato proteico
|
0,85
n = 16
|
0,62
n = 17
|
1,57
n = 17
|
1,24
n = 15
|
|
Premiscela
|
2,49
n = 16
|
—
|
6,20
n = 16
|
6,62
n = 16
|
|
n
|
=
|
numero di laboratori partecipanti.
|
|
Coefficiente di variabilità (%) per la deviazione standard tra laboratori (Sr)
|
Materiale di riferimento:
|
Amminoacido
|
|
Treonina
|
Cist(e)ina
|
Metionina
|
Lisina
|
|
Alimento composto per suini
|
4,1
n = 15
|
9,9
n = 17
|
7,0
n = 17
|
3,2
n = 13
|
|
Alimento composto per polli
|
5,2
n = 16
|
8,8
n = 18
|
10,9
n = 18
|
5,4
n = 16
|
|
Concentrato proteico
|
3,8
n = 16
|
12,3
n = 17
|
13,0
n = 17
|
3,0
n = 15
|
|
Premiscela
|
4,3
n = 16
|
—
|
6,9
n = 16
|
6,7
n = 16
|
|
n
|
=
|
numero di laboratori partecipanti.
|
|
8. Utilizzo di materiali di riferimento
La corretta applicazione del metodo può essere verificata ripetendo le misurazioni dei materiali di riferimento certificati, se disponibili. Si raccomanda di effettuare la taratura con una soluzione di taratura certificata di amminoacidi.
9. Osservazioni
|
9.1.
|
A causa delle differenze tra gli analizzatori di amminoacidi, le concentrazioni finali delle soluzioni di taratura degli amminoacidi standard (cfr. punti 3.27.4 e 3.27.5) e dell'idrolizzato (cfr. punto 5.3.4) sono considerate valori indicativi.
Per tutti gli amminoacidi va verificato il range della risposta lineare dell'apparecchiatura.
La soluzione standard è diluita con tampone citrato per ottenere aree dei picchi al centro del range.
|
|
9.2.
|
Se si fa uso di apparecchi per cromatografia liquida ad alta prestazione per analizzare gli idrolizzati, le condizioni sperimentali devono essere ottimizzate conformemente alle raccomandazioni del fabbricante.
|
|
9.3.
|
L'applicazione del metodo agli alimenti per animali contenenti cloruro in misura superiore all'1 % (concentrato, alimenti a base di minerali, alimenti integrativi) potrebbe comportare una sottostima della metionina; va previsto pertanto un trattamento speciale.
|
G. DETERMINAZIONE DEL TRIPTOFANO
1. Finalità e campo d'applicazione
Il metodo serve per la determinazione del triptofano totale e libero negli alimenti per animali. Esso non distingue la forma D dalla forma L.
2. Principio
Per la determinazione del triptofano totale, il campione è idrolizzato in ambiente alcalino con una soluzione satura di idrossido di bario e riscaldato a 110 oC per 20 ore. A idrolisi avvenuta, viene aggiunto lo standard interno.
Per la determinazione del triptofano libero, il campione è estratto in ambiente moderatamente acido in presenza dello standard interno.
Il triptofano e lo standard interno nell'idrolizzato e nell'estratto sono determinati per HPLC per mezzo di un rivelatore a fluorescenza.
3. Reattivi
|
3.1.
|
Usare acqua bidistillata o di qualità equivalente (conduttività < 10 μS/cm).
|
|
3.2.
|
Sostanza di riferimento (standard): triptofano (purezza/contenuto ≥ 99 %) essiccato sotto vuoto su pentossido di fosforo.
|
|
3.3.
|
Standard interno: α-metil-triptofano (purezza/contenuto ≥ 99 %), essiccato sotto vuoto su pentossido di fosforo.
|
|
3.4.
|
Idrossido di bario ottaidrato (prestare attenzione a non esporre eccessivamente il Ba(OH)2.8 H2O all'aria, per evitare la formazione di BaCO3, che potrebbe disturbare la determinazione) (cfr. osservazione al punto 9.3).
|
|
3.6.
|
Acido ortofosforico, p (p/p) = 85 %.
|
|
3.7.
|
Acido cloridico, ρ20 1,19 g/ml.
|
|
3.8.
|
Metanolo, di qualità HPLC.
|
|
3.9.
|
Etere di petrolio, punto di ebollizione tra 40 e 60 oC.
|
|
3.10.
|
Soluzione di idrossido di sodio, c = 1 mol/l:
sciogliere 40,0 g di NaOH (3.5) in acqua e portare a 1 litro con acqua (3.1).
|
|
3.11.
|
Acido cloridrico, c = 6 mol/l:
prelevare 492 ml di HCl (3.7) e portare a 1 litro con acqua.
|
|
3.12.
|
Acido cloridrico, c = 1 mol/l:
prelevare 82 ml di HCl (3.7) e portare a 1 litro con acqua.
|
|
3.13.
|
Acido cloridrico, c = 0,1 mol/l:
prelevare 8,2 ml di HCl (3.7) e portare a 1 litro con acqua.
|
|
3.14.
|
Acido ortofosforico, c = 0,5 mol/l:
prelevare 34 ml di acido ortofosforico (3.6) e portare a 1 litro con acqua (3.1).
|
|
3.15.
|
Soluzione concentrata di triptofano (3.2), c = 2,50 μmol/ml:
in un matraccio tarato da 500 ml sciogliere 0,2553 g di triptofano (3.2) in acido cloridrico (3.13) e portare a volume con acido cloridrico (3.13). Conservare a - 18 oC al massimo per quattro settimane.
|
|
3.16.
|
Soluzione concentrata di standard interno, c = 2,50 μmol/ml:
in un matraccio tarato da 500 ml sciogliere 0,2728 g di α-metil-triptofano (3.3) in acido cloridrico (3.13) e portare a volume con acido cloridrico (3.13). Conservare a - 18 oC al massimo per quattro settimane.
|
|
3.17.
|
Soluzione madre di taratura di triptofano e di standard interno:
prelevare 2,00 ml di soluzione concentrata di triptofano (3.15) e 2,00 ml di soluzione concentrata di standard interno (α-metil-triptofano) (3.16). Diluire con acqua (3.1) e metanolo (3.8) a circa lo stesso volume e a circa la stessa concentrazione di metanolo (10-30 %) dell'idrolizzato finito.
Questa soluzione deve essere preparata al momento, prima dell'uso.
Proteggere dalla luce solare diretta durante la preparazione.
|
|
3.19.
|
1,1,1-tricloro-2-metil-2-propanolo.
|
|
3.20.
|
Etanolammina p (p/p) > 98 %.
|
|
3.21.
|
Soluzione di 1 g di 1,1,1-tricloro-2-metil-2-propanolo (3.19) in 100 ml di metanolo (3.8).
|
|
3.22.
|
Fase mobile per HPLC: 3,00 g di acido acetico (3.18) + 900 ml di acqua (3.1) +50,0 ml di soluzione (3.21) di 1,1,1-tricloro-2-metil-2-propanolo (3.19) in metanolo (3.8) (1 g/100 ml); regolare il pH a 5,00 utilizzando etanolammina (3.20); portare a 1 000 ml con acqua (3.1).
|
4. Apparecchiatura
|
4.1.
|
Apparecchiatura HPLC con rilevatore spettrofluorimetrico.
|
|
4.2.
|
Colonna per cromatografia liquida, 125 mm × 4 mm, C18, particelle di riempimento 3 μm, o equivalente.
|
|
4.4.
|
Matraccio di polipropilene, capacità 125 ml, a collo largo e coperchio a vite.
|
|
4.5.
|
Filtro a membrana, da 0,45 μm.
|
|
4.6.
|
Autoclave, 110 (± 2) oC, 1,4 (±0,1) bar.
|
|
4.7.
|
Agitatore meccanico o mescolatore magnetico.
|
5. Procedimento
5.1. Preparazione dei campioni
Il campione viene triturato in modo che possa passare attraverso un vaglio da 0,5 mm. I campioni molto umidi devono essere essiccati all'aria a una temperatura non superiore a 50 oC o liofilizzati prima di essere triturati. I campioni ad alto tenore di grassi sono estratti con etere di petrolio (3.9) prima di essere triturati.
5.2. Determinazione del triptofano libero (estratto)
In una beuta pesare (con l'approssimazione di 1 mg) una quantità adeguata (1-5 g) del campione preparato (5.1); aggiungere 100,0 ml di acido cloridrico (3.13) e 5,00 ml di soluzione concentrata di standard interno (3.16). Agitare o miscelare per 60 minuti con un agitatore meccanico o magnetico (4.7). Lasciare che si depositi il sedimento e pipettare 10,0 ml della soluzione surnatante in un becher. Aggiungere 5 ml di acido orto-fosforico (3.14). Portare il pH al valore 3 utilizzando idrossido di sodio (3.10). Aggiungere sufficiente metanolo (3.8) per ottenere una concentrazione compresa tra il 10 e il 30 % di metanolo nel volume finale. Trasferire in un matraccio tarato di volume adeguato e diluire con acqua sino al volume necessario per la cromatografia [approssimativamente lo stesso volume della soluzione madre di taratura (3.17)].
Filtrare alcuni ml della soluzione con un filtro a membrana da 0,45 μm (4.5) prima dell'iniezione nella colonna HPLC. Procedere alla fase della cromatografia secondo quanto indicato al punto 5.4.
Proteggere la soluzione madre e gli estratti dalla luce solare diretta. Se non è possibile analizzare gli estratti nello stesso giorno, questi devono essere conservati a 5 oC al massimo per 3 giorni.
5.3. Determinazione del triptofano totale (idrolizzato).
Pesare (con l'approssimazione di 0,2 mg) da 0,1 a 1 g del campione preparato (5.1) nel matraccio di polipropilene (4.4). La porzione di campione pesata ha un contenuto di azoto di circa 10 mg. Aggiungere 8,4 g di idrossido di bario (ottaidrato) (3.4) e 10 ml di acqua. Miscelare con un miscelatore Vortex (4.8) o con agitatore magnetico (4.7). Lasciare il magnete rivestito di teflon nella miscela. Sciacquare le pareti del recipiente con 4 ml di acqua. Porre il coperchio a vite e chiudere il matraccio senza stringere. Trasferire in una autoclave (4.6) con acqua bollente e vapore per 30-60 minuti. Chiudere l'autoclave e metterla in funzione a 110 (± 2) oC per 20 ore.
Prima di aprire l'autoclave, ridurre la temperatura poco al di sotto di 100 oC. Per evitare la cristallizzazione del Ba(OH)2 8H2O, aggiungere alla miscela calda 30 ml di acqua a temperatura ambiente. Scuotere o agitare non violentemente. Aggiungere 2,00 ml di soluzione concentrata di standard interno (α-metil-triptofano) (3.16). Lasciare raffreddare i recipienti in un bagno di acqua e ghiaccio per 15 minuti.
Aggiungere quindi 5 ml di acido orto-fosforico (3.14). Tenere il recipiente nel bagno di refrigerazione e neutralizzare con HCl (3.11) sempre agitando; regolare quindi il pH a 3,0 utilizzando HCl (3.12). Aggiungere una quantità sufficiente di metanolo per ottenere una concentrazione compresa tra 10 e 30 % di metanolo nel volume finale. Trasferire in un matraccio tarato di volume adeguato e diluire con acqua sino al volume necessario per la cromatografia (ad esempio, 100 ml). L'aggiunta di metanolo non provoca precipitazione.
Filtrare alcuni ml della soluzione con un filtro a membrana da 0,45 μm (4.5) prima dell'iniezione nella colonna HPLC. Procedere alla fase della cromatografia secondo quanto indicato al punto 5.4.
Proteggere la soluzione madre e gli idrolizzati dalla luce solare diretta. Se non è possibile analizzare gli idrolizzati il giorno stesso, questi devono essere conservati a 5 oC al massimo per 3 giorni.
5.4. Determinazione HPLC
Le seguenti condizioni di eluizione isocratica sono proposte a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti (cfr. anche osservazioni 9.1 e 9.2):
|
Colonna per cromatografia liquida (4.2):
|
125 mm × 4 mm, C18, particelle di riempimento 3 μm o equivalente.
|
|
Temperatura della colonna:
|
temperatura ambiente
|
|
Fase mobile (3.22):
|
3,00 g di acido acetico (3.18) + 900 ml di acqua (3.1) +50,0 ml di soluzione (3.21) di 1,1,1-trichloro-2-metil-2-propanolo (3.19) in metanolo (3.8) (1 g/100 ml); regolare il pH a 5,00 per mezzo di etanolammina (3.20); portare a 1 000 ml con acqua (3.1)
|
|
Velocità di efflusso:
|
1 ml/min
|
|
Durata totale dell'eluizione:
|
circa 34 min
|
|
Lunghezza d'onda di rivelazione:
|
eccitazione: 280 nm, emissione: 356 nm
|
|
Volume di iniezione:
|
20 μl
|
6. Calcolo dei risultati
Calcolare la quantità di triptofano (X), espressa in g, per 100 g di campione secondo la seguente formula:

|
A
|
=
|
area del picco dello standard interno, soluzione madre di taratura (3.17)
|
|
B
|
=
|
area del picco del triptofano, estratto (5.2) o idrolizzato (5.3)
|
|
V1
|
=
|
volume in ml (2 ml) di soluzione concentrata di triptofano (3.15) aggiunta alla soluzione di taratura (3.17)
|
|
c
|
=
|
concentrazione in μmol/ml (= 2,50) di soluzione concentrata di triptofano (3.15) aggiunta alla soluzione di taratura (3.17)
|
|
V2
|
=
|
volume in ml della soluzione concentrata di standard interno (3.16) aggiunta all'estrazione (5.2) (= 5,00 ml) o all'idrolizzato (5.3) (= 2,00 ml)
|
|
C
|
=
|
area del picco dello standard interno, dell'estratto (5.2) o dell'idrolizzato (5.3)
|
|
D
|
=
|
area del picco del triptofano, soluzione madre di taratura (3.17)
|
|
V3
|
=
|
volume in ml (= 2,00 ml) della soluzione concentrata dello standard interno (3.16) aggiunto alla soluzione madre di taratura (3.17)
|
|
m
|
=
|
peso del campione in g (corretto al peso iniziale se essiccato e/o sgrassato)
|
|
M
|
=
|
peso molecolare del triptofano (= 204,23 g/mol)
|
7. Ripetibilità
La differenza tra i risultati di due determinazioni, effettuate in parallelo sullo stesso campione, non deve superare il 10 % del valore ottenuto più elevato.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio collaborativo a livello comunitario (quarta intercomparazione) nel corso del quale dodici laboratori hanno analizzato tre campioni al fine di convalidare il metodo di idrolisi. Ogni campione è stato analizzato cinque volte. I risultati sono stati i seguenti:
|
|
Campione 1
Alimento per suini
|
Campione 2
Alimento per suini con aggiunta di L-triptofano
|
Campione 3
Alimento concentrato per suini
|
|
L
|
12
|
12
|
12
|
|
n
|
50
|
55
|
50
|
|
Media [g/kg]
|
2,42
|
3,40
|
4,22
|
|
sr [g/kg]
|
0,05
|
0,05
|
0,08
|
|
r [g/kg]
|
0,14
|
0,14
|
0,22
|
|
CVr [%]
|
1,9
|
1,6
|
1,9
|
|
SR [g/kg]
|
0,15
|
0,20
|
0,09
|
|
R [g/kg]
|
0,42
|
0,56
|
0,25
|
|
CVR [%]
|
6,3
|
6,0
|
2,2
|
|
L
|
=
|
numero di laboratori che hanno trasmesso risultati
|
|
n
|
=
|
numero di risultati individuali considerati una volta eliminati gli outlier (valori aberranti identificati in base al test di Cochran-Dixon)
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
r
|
=
|
ripetibilità
|
|
R
|
=
|
riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, in %
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, in %.
|
È stato organizzato un altro studio collaborativo a livello comunitario (terza intercomparazione), in cui tredici laboratori hanno analizzato due campioni al fine di convalidare il metodo di estrazione del triptofano libero. Ogni campione è stato analizzato cinque volte. I risultati sono stati i seguenti:
|
|
Campione 4
Miscela di frumento e soia
|
Campione 5
Miscela di frumento e soia (= campione 4) con aggiunta di triptofano (0,457g/kg)
|
|
L
|
12
|
12
|
|
n
|
55
|
60
|
|
Media [g/kg]
|
0,391
|
0,931
|
|
sr [g/kg]
|
0,005
|
0,012
|
|
r [g/kg]
|
0,014
|
0,034
|
|
CVr [%]
|
1,34
|
1,34
|
|
SR [g/kg]
|
0,018
|
0,048
|
|
R [g/kg]
|
0,050
|
0,134
|
|
CVR [%]
|
4,71
|
5,11
|
|
L
|
=
|
numero di laboratori che hanno trasmesso risultati
|
|
n
|
=
|
numero di risultati individuali considerati una volta eliminati gli outlier (valori aberranti identificati in base al test di Cochran-Dixon)
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
r
|
=
|
ripetibilità
|
|
R
|
=
|
riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, in %
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, in %.
|
È stato effettuato un altro studio di intercomparazione comunitario in cui sono stati analizzati quattro campioni da 7 laboratori ai fini della certificazione del triptofano per idrolisi. Ogni campione è stato analizzato cinque volte. I risultati sono stati i seguenti:
|
|
Campione 1
Alimento composto per suini
(CRM 117)
|
Campione 2
Farina di pesce a basso tenore di grassi
(CRM 118)
|
Campione 3
Farina di soia
(CRM 119)
|
Campione 4
Latte scremato in polvere
(CRM 120)
|
|
L
|
7
|
7
|
7
|
7
|
|
n
|
25
|
30
|
30
|
30
|
|
Media [g/kg]
|
2,064
|
8,801
|
6,882
|
5,236
|
|
sr [g/kg]
|
0,021
|
0,101
|
0,089
|
0,040
|
|
r [g/kg]
|
0,059
|
0,283
|
0,249
|
0,112
|
|
CVr [ %]
|
1,04
|
1,15
|
1,30
|
0,76
|
|
SR [g/kg]
|
0,031
|
0,413
|
0,283
|
0,221
|
|
R [g/kg]
|
0,087
|
1,156
|
0,792
|
0,619
|
|
CVR [ %]
|
1,48
|
4,69
|
4,11
|
4,22
|
|
L
|
=
|
numero di laboratori che hanno trasmesso risultati
|
|
n
|
=
|
numero di risultati individuali considerati una volta eliminati gli outlier (valori aberranti identificati in base al test di Cochran-Dixon)
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
r
|
=
|
ripetibilità
|
|
R
|
=
|
riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, in %
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, in %.
|
9. Osservazioni
|
9.1.
|
Speciali condizioni cromatografiche consentono una migliore separazione tra triptofano e α-metil-triptofano.
Eluizione isocratica seguita da pulitura della colonna a gradiente:
|
Colonna per cromatografia liquida:
|
125 mm × 4 mm, C18, particelle di riempimento 5 μm, o equivalente
|
|
Temperatura della colonna:
|
32 oC
|
|
Fase mobile:
|
A: 0,01 mol/l KH2PO4/metanolo, 95 + 5 (V + V).
B: metanolo
|
|
Programma del gradiente:
|
0 min
|
100 % A
|
0 % B
|
|
|
15 min
|
100 % A
|
0 % B
|
|
|
17 min
|
60 % A
|
40 % B
|
|
|
19 min
|
60 % A
|
40 % B
|
|
|
21 min
|
100 % A
|
0 % B
|
|
|
33 min
|
100 % A
|
0 % B
|
|
Velocità di efflusso:
|
1,2 ml/min
|
|
Durata totale dell'eluizione:
|
circa 33 min.
|
|
|
9.2.
|
La cromatografia varierà a seconda del tipo di HPLC e di materiale di riempimento utilizzati. Il sistema scelto deve permettere una completa separazione dei picchi del triptofano e dello standard interno. Inoltre è importante che i prodotti di degradazione siano nettamente separati dal triptofano e dallo standard interno. Vanno fatti passare idrolizzati senza standard interno in modo da verificare l'assenza di impurità sulla linea di base sotto lo standard interno. È importante che il tempo di eluizione sia sufficientemente lungo per consentire l'eluizione di tutti i prodotti di degradazione; altrimenti gli ultimi picchi di eluizione possono interferire con le operazioni cromatografiche successive.
Nell'intervallo operativo, il sistema cromatografico dà una risposta lineare. Tale risposta è misurata a una concentrazione costante (concentrazione normale) dello standard interno e a concentrazioni variabili di triptofano. È importante che le altezze dei picchi del triptofano e dello standard interno si situino nell'intervallo lineare del sistema HPLC/fluorescenza. Se il picco o i picchi del triptofano e/o dello standard interno sono troppo bassi o troppo alti, l'analisi viene ripetuta con altre dimensioni del campione e/o un volume finale modificato.
|
|
9.3.
|
Idrossido di bario
Con il tempo, l'idrossido di bario si scioglie con maggiore difficoltà. Ciò causa una soluzione torbida per la determinazione HPLC, che può produrre bassi valori per il triptofano.
|
H. DETERMINAZIONE DEGLI OLI E GRASSI GREGGI
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di oli e grassi greggi negli alimenti per gli animali, ma non riguarda l'analisi dei semi e frutti oleaginosi.
L'applicazione dei due procedimenti sotto descritti dipende dalla natura e dalla composizione dell'alimento, nonché dalla ragione per cui si esegue l'analisi.
1.1. Procedimento A — Oli e grassi greggi estraibili direttamente
Il metodo è applicabile alle materie prime per alimenti per animali di origine vegetale, fatta eccezione per quelle alle quali si applica il procedimento B.
1.2. Procedimento B — Oli e grassi greggi totali
Il metodo è applicabile alle materie prime per alimenti per animali di origine animale e a tutti gli alimenti composti. Esso deve essere usato per tutte le materie prime dai quali gli oli e i grassi non possono essere completamente estratti senza idrolisi preliminare, ad esempio i glutini, i lieviti, le proteine di patata e i prodotti che sono stati sottoposti a procedimenti quali l'estrusione, la fioccatura ed il riscaldamento.
1.3. Interpretazione dei risultati
Se il valore ottenuto con il procedimento B è più elevato di quello ottenuto con il procedimento A, si considera valore reale il risultato ottenuto con il procedimento B.
2. Principio
2.1. Procedimento A
Il campione è estratto con etere di petrolio. Il solvente è eliminato per distillazione e il residuo è essiccato e pesato.
2.2. Procedimento B
Il campione è trattato a caldo con acido cloridrico. La miscela è raffreddata e filtrata. Dopo essere stato lavato ed essiccato, il residuo è sottoposto all'analisi secondo il procedimento A.
3. Reattivi
|
3.1.
|
Etere di petrolio, intervallo di ebollizione: 40-60 oC. L'indice di bromo deve essere inferiore a 1 e il residuo all'evaporazione inferiore a 2 mg/100 ml.
|
|
3.2.
|
Solfato di sodio, anidro.
|
|
3.3.
|
Acido cloridrico, c = 3 mol/l.
|
|
3.4.
|
Coadiuvante di filtrazione, ad esempio farina fossile, Hyflo-supercel.
|
4. Apparecchiatura
|
4.1.
|
Estrattore. Se l'apparecchio è munito di un sifone (apparecchio di Soxhlet), la portata del riflusso è regolata in modo da ottenere almeno 10 cicli l'ora; se si tratta di un apparecchio senza sifone, il liquido rifluisce in quantità pari a circa 10 ml al minuto.
|
|
4.2.
|
Ditali da estrazione, esenti da sostanze solubili nell'etere di petrolio, la cui porosità sia compatibile con le esigenze di cui al punto 4.1.
|
|
4.3.
|
Stufa per essiccazione sia a vuoto a 75 ± 3 oC o a pressione atmosferica a 100 ± 3 oC.
|
5. Procedimento
5.1. Procedimento A (cfr. punto 8.1)
Pesare, con l'approssimazione di 1 mg, 5 g del campione; introdurli in un ditale da estrazione (4.2) e coprire con un tampone di cotone sgrassato.
Porre il ditale in un estrattore (4.1) ed estrarre per 6 ore con etere di petrolio (3.1). Raccogliere l'estratto in un pallone asciutto e tarato, contenente frammenti di pietra pomice (2).
Eliminare il solvente per distillazione. Essiccare il residuo introducendo il pallone in una stufa per essiccazione (4.3), lasciandovelo per un'ora e mezza. Lasciar raffreddare in essiccatore e pesare. Essiccare una seconda volta per 30 minuti, onde assicurarsi che il peso della sostanza oleosa o grassa rimanga costante (la perdita di peso tra due pesate consecutive deve essere pari o inferiore a 1 mg).
5.2. Procedimento B
Pesare, con l'approssimazione di 1 mg, 2,5 g del campione (cfr. punto 8.2); introdurli in un becher da 400 ml o in una beuta da 300 ml e aggiungere 100 ml di acido cloridrico (3.3) e qualche granulo di pietra pomice. Ricoprire il becher con un vetro da orologio o applicare sulla beuta un refrigerante a ricadere. Portare la miscela a lenta ebollizione su piccola fiamma o su piastra riscaldante e lasciarvela per un'ora. Evitare che la sostanza aderisca alle pareti del recipiente.
Raffreddare e aggiungere una quantità sufficiente di un coadiuvante di filtrazione (3.4) per evitare qualsiasi perdita di sostanza grassa durante la filtrazione stessa. Filtrare su un doppio filtro di carta bagnato, esente da materie grasse. Lavare il residuo con acqua fredda fino a reazione neutra del filtrato. Verificare che il filtrato non contenga sostanza grasse. La presenza di queste nel filtrato indica che, prima dell'idrolisi, deve essere effettuata un'estrazione del campione con etere di petrolio, secondo il procedimento A.
Porre il doppio filtro con il residuo su un vetro da orologio ed essiccare per un'ora e mezza nella stufa a pressione atmosferica (4.3) a 100 ± 3 oC.
Introdurre il doppio filtro contenente il residuo secco in un ditale da estrazione (4.2) e coprire con un tampone di cotone sgrassato. Porre il ditale in un estrattore (4.1) e operare come indicato al punto 5.1, secondo e terzo capoverso.
6. Espressione del risultato
Esprimere il risultato della pesata del residuo in percentuale del campione.
7. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione dallo stesso analista non supera:
|
—
|
lo 0,2 %, in valore assoluto, per contenuti di materie oleose e grasse gregge inferiori al 5 %,
|
|
—
|
il 4,0 % del valore ottenuto più elevato, per contenuti compresi fra 5 e 10 %,
|
|
—
|
lo 0,4 %, in valore assoluto, per contenuti superiori al 10 %.
|
8. Osservazioni
|
8.1.
|
Per i prodotti a elevato tenore di sostanze oleose e grasse, difficili da macinare o non appropriati per il prelevamento di una piccola quantità omogenea, procedere nel modo che segue.
Pesare, con l'approssimazione di 1 mg, 20 g di campione e mescolarli con 10 g o più di solfato di sodio anidro (3.2). Procedere all'estrazione con etere di petrolio (3.1) come indicato al punto 5.1. Portare l'estratto ottenuto al volume di 500 ml con etere di petrolio (3.1) e mescolare. Introdurre 50 ml della soluzione in un palloncino asciutto, contenente qualche granulo di pietra pomice, e tarato. Eliminare il solvente per distillazione, essiccare e proseguire come indicato al punto 5.1, ultimo capoverso.
Eliminare il solvente dal residuo d'estrazione che si trova nel ditale, macinare il residuo alla finezza di 1 mm, porlo nuovamente nel ditale (non aggiungere solfato di sodio) e proseguire come indicato al punto 5.1, secondo e terzo capoverso.
Il contenuto di materie oleose e grasse gregge, espresso in percentuale del campione, è dato dalla seguente formula:
(10m1 + m2) × 5
dove
|
m1
|
=
|
peso, in grammi, del residuo della prima estrazione (parte aliquota dell'estratto),
|
|
m2
|
=
|
peso, in grammi, del residuo della seconda estrazione.
|
|
|
8.2.
|
La quantità di sostanza sottoposta all'analisi nel caso di prodotti poveri di materie oleose e grasse può essere portata a 5 g.
|
|
8.3.
|
Gli alimenti per animali da compagnia, contenenti un elevato tenore d'acqua, possono rendere necessaria l'aggiunta di solfato di sodio anidro prima dell'idrolisi e dell'estrazione secondo il procedimento B.
|
|
8.4.
|
Nel procedimento descritto al punto 5.2 potrebbe rivelarsi più efficace usare acqua calda, invece che fredda, per lavare il residuo dopo la filtrazione.
|
|
8.5.
|
Per alcuni alimenti per animali il tempo di essiccazione (1 h 30) deve essere prolungato; tuttavia è evitato un tempo di essiccazione eccessivo, che potrebbe dare scarsi risultati. È possibile usare altresì un forno a microonde.
|
|
8.6.
|
Se il tenore di sostanza oleosa o grassa è superiore al 15 %, nel procedimento A si raccomanda di effettuare una estrazione preliminare prima dell'idrolisi e nel procedimento B una seconda estrazione. Ciò può dipendere in parte dalla natura degli alimenti per animali e da quella della sostanza oleosa o grassa contenuta in tali alimenti.
|
I. DETERMINAZIONE DELLA CELLULOSA GREZZA
1. Finalità e campo d'applicazione
Il metodo consente di determinare negli alimenti per animali le materie organiche esenti da grasso, insolubili in ambiente acido e in ambiente alcalino, convenzionalmente designate con il nome di cellulosa grezza.
2. Principio
Il campione, eventualmente sgrassato, è trattato successivamente con soluzioni bollenti di acido solforico e di idrossido di potassio di determinate concentrazioni. Il residuo è separato per filtrazione su vetro sinterizzato, lavato, essiccato, pesato e incenerito a 475-500 oC. La perdita di peso conseguente all'incenerimento corrisponde alla cellulosa grezza della quantità di prodotto sottoposta all'analisi.
3. Reattivi
|
3.1.
|
Acido solforico, c = 0,13 mol/l.
|
|
3.2.
|
Agente antischiuma (ad esempio, n-ottanolo).
|
|
3.3.
|
Coadiuvante della filtrazione (Celite 545 o equivalente), riscaldato a 500 oC per quattro ore (8.6).
|
|
3.5.
|
Etere di petrolio, con punto di ebollizione compreso fra 40 oC e 60 oC.
|
|
3.6.
|
Acido cloridrico, c = 0,5 mol/l.
|
|
3.7.
|
Soluzione di idrossido di potassio, c = 0,23 mol/l.
|
4. Apparecchiatura
|
4.1.
|
L'unità riscaldante per la mineralizzazione con soluzioni di acido solforico e di idrossido di potassio, dotata di supporto per crogiolo filtrante (4.2) e di un tubo di uscita con valvola di svuotamento e a vuoto, eventualmente con aria compressa. Prima dell'uso quotidiano preriscaldare per cinque minuti l'unità con acqua bollente.
|
|
4.2.
|
Crogiolo filtrante in vetro con filtro in vetro sinterizzato, dimensione dei pori: 40-90 μm. Prima della messa in uso, riscaldare a 500 oC per alcuni minuti e raffreddare (8.6).
|
|
4.3.
|
Cilindro da almeno 270 ml con refrigerante a ricadere, adatto all'ebollizione.
|
|
4.4.
|
Stufa per essiccazione con termostato.
|
|
4.5.
|
Forno a muffola con termostato.
|
|
4.6.
|
Unità per estrazione consistente in una piastra di supporto per il crogiolo filtrante (4.2) e in un tubo di scarico con valvola di svuotamento e a vuoto.
|
|
4.7.
|
Anelli di giunzione per assemblare l'unità riscaldante (4.1), il crogiolo (4.2) e il cilindro (4.3) e per collegare l'unità di estrazione a freddo (4.6) e il crogiolo.
|
5. Procedimento
Pesare, con l'approssimazione di 1 mg, 1 g del campione preparato e introdurlo nel crogiolo (4.2) (cfr. osservazioni ai punti 8.1, 8.2 e 8.3) e aggiungere 1 g di coadiuvante di filtrazione (3.3).
Assemblare l'unità riscaldante (4.1) e il crogiolo filtrante (4.2), quindi applicare il cilindro (4.3) al crogiolo. Versare 150 ml di acido solforico bollente (3.1) nel cilindro e crogiolo assemblati e, se necessario, aggiungere qualche goccia di agente antischiuma (3.2).
Portare il liquido all'ebollizione in 5 ± 2 minuti e lasciar bollire vivacemente per esattamente 30 minuti.
Posizionare la valvola verso il tubo di scarico (4.1) e, sotto vuoto, filtrare l'acido solforico attraverso il crogiolo filtrante e sciacquare il residuo tre volte con 30 ml di acqua bollente ciascuna, controllando che il residuo sia filtrato a secco dopo ogni risciacquo.
Richiudere la valvola di svuotamento e versare 150 ml di soluzione di idrossido di potassio bollente (3.7) nel cilindro e nel crogiolo assemblati e aggiungere qualche goccia di antischiuma (3.2). Portare il liquido all'ebollizione entro 5 ± 2 minuti e lasciar bollire vivacemente per esattamente 30 minuti. Filtrare e ripetere la procedura di risciacquo applicata all'acido solforico.
Dopo il risciacquo finale e l'essiccazione, staccare il crogiolo e il suo contenuto e ricollegarlo all'unità di estrazione a freddo (4.6). Applicare il tubo vuoto e risciacquare il residuo nel crogiolo tre volte con porzioni di 25 ml di acetone ciascuna (3.4) assicurandosi che il residuo sia filtrato a secco dopo ogni lavaggio.
Essiccare il crogiolo nel forno a 130 oC fino a cessazione della diminuzione di massa. Dopo ogni essiccazione lasciar raffreddare nell'essiccatore e pesare rapidamente. Introdurre quindi il crogiolo nel forno a muffola e incenerire fino a peso costante (la perdita di peso tra due pesate consecutive deve essere pari o inferiore a 2 mg) ad una temperature compresa tra 450 e 500 oC per almeno 30 minuti.
Dopo ciascun riscaldamento, lasciar raffreddare in un primo momento nel forno, poi nell'essiccatore e pesare.
Procedere ad una prova in bianco in assenza del campione. La perdita di peso conseguente all'incenerimento non deve essere superiore a 4 mg.
6. Calcolo dei risultati
Il contenuto di cellulosa grezza, in percentuale del campione, è dato dalla seguente formula:
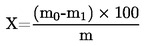
dove
|
m
|
=
|
peso del campione, in grammi;
|
|
m0
|
=
|
perdita di peso conseguente all'incenerimento nel corso della determinazione, in grammi;
|
|
m1
|
=
|
perdita di peso conseguente all'incenerimento, durante la prova in bianco, in grammi.
|
7. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
lo 0,6 % in valore assoluto, per i contenuti di cellulosa grezza inferiori al 10 %,
|
|
—
|
il 6 % rispetto al valore più elevato, per i contenuti di cellulosa grezza uguali o superiori al 10 %.
|
8. Osservazioni
|
8.1.
|
Gli alimenti per animali che contengono più del 10 % di sostanze grasse gregge debbono essere sgrassati prima dell'analisi con etere di petrolio (3.5). Collegare il crogiolo filtrante (4.2) e il suo contenuto all'unità di estrazione a freddo (4.6), applicare il vuoto e sciacquare il residuo tre volte con volumi di 30 ml di etere di petrolio ciascuna, assicurandosi che il residuo sia secco. Collegare il crogiolo e il suo contenuto all'unità riscaldante (4.1) e proseguire come indicato al punto 5.
|
|
8.2.
|
Gli alimenti per animali contenenti materie grasse che non possono essere estratte direttamente con etere di petrolio (3.5) vanno sgrassate una prima volta come descritto al punto 8.1 e una seconda volta dopo ebollizione con l'acido. Dopo l'ebollizione con acido e successivi lavaggi, collegare il crogiolo e il suo contenuto all'unità di estrazione a freddo (4.6), lavare tre volte con 30 ml di acetone e rilavare altre tre volte con 30 ml di etere di petrolio. Filtrare sotto vuoto fino a completa essiccazione e continuare l'analisi secondo le indicazioni fornite al punto 5, iniziando con il trattamento con idrossido di potassio.
|
|
8.3.
|
Se l'alimento contiene più del 5 % di carbonati, espressi in carbonato di calcio, collegare il crogiolo (4.2) con il campione pesato all'unità riscaldante (4.1). Lavare il campione tre volte con 30 ml di acido cloridrico (3.6). Dopo ogni aggiunta lasciare riposare il campione per circa un minuto prima di filtrarlo. Risciacquare una volta con 30 ml di acqua e proseguire come descritto al punto 5.
|
|
8.4.
|
Se l'apparecchio utilizzato è composto da più crogioli collegati alla stessa unità riscaldante, non eseguire due prove sullo stesso campione nella stessa serie.
|
|
8.5.
|
Se, dopo la bollitura, il filtraggio delle soluzioni acida e basica risulta difficile, iniettare aria compressa nel tubo di scarico dell'unità riscaldante e continuare a filtrare.
|
|
8.6.
|
La temperatura d'incenerimento non supera i 500 oC in modo da prolungare la durata dei crogioli filtranti in vetro. Vanno evitati possibili effetti da choc termico eccessivo durante i cicli di riscaldamento e di raffreddamento.
|
J. DETERMINAZIONE DEGLI ZUCCHERI
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto in zuccheri riduttori ed in zuccheri totali dopo inversione, espressi in glucosio e, se del caso, in saccarosio, per conversione mediante fattore 0,95. Il metodo è applicabile agli alimenti composti. Particolari modalità sono previste per altri alimenti. All'occorrenza si doserà separatamente il lattosio e se ne terrà conto nel calcolo dei risultati.
2. Principio
Gli zuccheri presenti sono sciolti nell'etanolo diluito; la soluzione è chiarificata con reattivi di Carrez I e II. Dopo l'eliminazione dell'etanolo, le determinazioni sono effettuate, prima e dopo l'inversione, secondo il metodo di Luff-Schoorl.
3. Reattivi
|
3.1.
|
Soluzione di etanolo al 40 % (v/v), densità: 0,948 g/ml a 20 oC, neutralizzato alla fenolftaleina.
|
|
3.2.
|
Soluzione di Carrez I: sciogliere in acqua 21,9 g di acetato di zinco Zn (CH3COO)2 2H2O e 3 g di acido acetico glaciale. Portare a 100 ml con acqua.
|
|
3.3.
|
Soluzione di Carrez II: sciogliere nell'acqua 10,6 g di ferrocianuro di potassio K4Fe (CN)6 3H2O. Portare a 100 ml con acqua.
|
|
3.4.
|
Soluzione allo 0,1 % (p/v) di metilarancio.
|
|
3.5.
|
Acido cloridrico, 4 mol/l.
|
|
3.6.
|
Acido cloridrico, 0,1 mol/l.
|
|
3.7.
|
Soluzione di idrossido di sodio, 0,1 mol/l.
|
|
3.8.
|
Reattivo di Luff-Schoorl:
aggiungere, agitando prudentemente, la soluzione d'acido citrico (3.8.2) alla soluzione di carbonato di sodio (3.8.3). Aggiungere quindi la soluzione di solfato di rame (3.8.1) e portare al volume di 1 litro con acqua. Lasciar riposare una notte e filtrare.
Controllare la concentrazione del reattivo così ottenuto (Cu 0,05 mol/l; Na2 CO3 1 mol/l), cfr. punto 5.4, ultimo capoverso. Il pH della soluzione è circa 9,4.
|
|
3.8.1.
|
Soluzione di solfato di rame: sciogliere 25 g di solfato di rame, Cu SO4 5H2O, esente da ferro, in 100 ml d'acqua.
|
|
3.8.2.
|
Soluzione di acido citrico: sciogliere 50 g di acido citrico, C6H8O7 H2O, in 50 ml d'acqua.
|
|
3.8.3.
|
Soluzione di carbonato di sodio: sciogliere 143,8 g di carbonato di sodio anidro in circa 300 ml d'acqua calda. Lasciar raffreddare.
|
|
3.9.
|
Soluzione di tiosolfato di sodio, 0,1 mol/l.
|
|
3.10.
|
Soluzione d'amido: aggiungere una miscela di 5 g di amido solubile in 30 ml d'acqua a 1 litro d'acqua bollente. Far bollire per 3 minuti, lasciar raffreddare, aggiungere eventualmente, come conservante, 10 mg di ioduro di mercurio.
|
|
3.11.
|
Acido solforico 3 mol/l.
|
|
3.12.
|
Soluzione al 30 % (p/v) di ioduro di potassio.
|
|
3.13.
|
Granuli di pietra pomice bolliti nell'acido cloridrico, lavati in acqua ed essiccati.
|
|
3.14.
|
3-metilbutano-1-olo.
|
4. Apparecchiatura
Agitatore rotativo a capovolgimento: circa 35-40 giri al minuto.
5. Procedimento
5.1. Estrazione del campione
Introdurre 2,5 g di sostanza, pesata con l'approssimazione di 1 mg, in un pallone tarato da 250 ml. Aggiungere 200 ml di etanolo (3.1) e far girare il pallone per un’ora nell'apparecchio rotativo. Aggiungere 5 ml di soluzione di Carrez I (3.2) e agitare per circa 30 secondi. Aggiungere quindi 5 ml di soluzione di Carrez II (3.3) e agitare nuovamente per un minuto. Portare a volume con etanolo (3.1), omogeneizzare e filtrare. Prelevare 200 ml del filtrato ed evaporare circa la metà del volume allo scopo di eliminare la maggior parte di etanolo. Trasferire quantitativamente il residuo d'evaporazione mediante acqua calda in un pallone tarato da 200 ml, raffreddare, portare a volume con acqua, omogeneizzare e filtrare, se necessario. Questa soluzione sarà utilizzata per la determinazione degli zuccheri riduttori e, dopo inversione, per la determinazione degli zuccheri totali.
5.2. Determinazione degli zuccheri riduttori
Con una pipetta prelevare un volume di soluzione non superiore a 25 ml e contenente meno di 60 mg di zuccheri riduttori, espressi in glucosio. Se necessario, portare a 25 ml con acqua distillata e determinare il contenuto in zuccheri riduttori secondo il metodo di Luff-Schoorl. Il risultato è espresso in percentuale di glucosio presente nel campione.
5.3. Determinazione degli zuccheri totali dopo inversione
Con una pipetta prelevare 50 ml della soluzione e introdurli in un pallone tarato da 100 ml. Aggiungere qualche goccia di soluzione di metilarancio (3.4), poi, con molta attenzione, e sempre agitando, aggiungere acido cloridrico (3.5) fino a ottenere un viraggio nettamente al rosso. Aggiungere 15 ml di acido cloridrico (3.6), immergere il pallone per 30 minuti in bagnomaria a forte ebollizione e lasciarvelo per 30 minuti. Raffreddare rapidamente alla temperatura di circa 20 oC e aggiungere 15 ml di soluzione di idrossido di sodio (3.7). Portare al volume di 100 ml con acqua ed omogeneizzare. Prelevare un volume non superiore a 25 ml e contenente meno di 60 mg di zuccheri riduttori, espressi in glucosio. Se necessario, portare a 25 ml con acqua distillata e determinare il contenuto in zuccheri riduttori secondo il metodo di Luff-Schoorl. Esprimere il risultato in percentuale di glucosio, o se del caso, di saccarosio moltiplicando per il fattore 0,95.
5.4. Titolazione secondo il metodo di Luff-Schoorl
Con una pipetta prelevare 25 ml del reattivo di Luff-Schoorl (3.8) e introdurlo in un erlenmeyer da 300 ml; aggiungere 25 ml esatti della soluzione zuccherina chiarificata. Dopo aver aggiunto due granuli di pietra pomice (3.13) riscaldare, agitando a mano, su fiamma libera di altezza media e portare il liquido a ebollizione in circa 2 minuti. Collocare immediatamente l'erlenmeyer su una tela metallica, provvista di uno schermo d'amianto munito di un foro del diametro di circa 6 cm, sulla quale è stata preventivamente accesa una fiamma. Questa è regolata in modo tale che venga riscaldato soltanto il fondo dell'erlenmeyer. Adattare sull'erlenmeyer un refrigerante a riflusso. A decorrere da questo momento far bollire per 10 minuti esatti. Raffreddare immediatamente in acqua fredda e dopo circa 5 minuti titolare nel seguente modo:
Aggiungere 10 ml di soluzione di ioduro di potassio (3.12) e, subito dopo, con molta attenzione (a causa del rischio di formazione di abbondante schiuma), 25 ml di acido solforico (3.11). Titolare quindi con la soluzione di tiosolfato di sodio (3.9) sino all'apparire di una colorazione giallo opaco, aggiungere l'indicatore all'amido (3.10) e terminare la titolazione.
Effettuare la stessa titolazione su di una miscela, esattamente misurata, di 25 ml di reattivo di Luff-Schoorl (3.8) e 25 ml d'acqua, dopo aver aggiunto 10 ml di soluzione di ioduro di potassio (3.12) e 25 ml di acido solforico (3.11) senza portare a ebollizione.
6. Calcolo dei risultati
Stabilire, a mezzo della tabella, la quantità (in mg) di glucosio corrispondente alla differenza tra i risultati delle due titolazioni, espresse in mg di tiosolfato di sodio 0,1 mol/l. Esprimere il risultato in percentuale del campione.
7. Procedimenti speciali
|
7.1.
|
Per gli alimenti molto ricchi di melasso e altri alimenti poco omogenei, pesare 20 g e introdurli con 500 ml d'acqua in un pallone tarato da 1 1itro. Far girare nell'apparecchio rotativo per un'ora. Chiarificare con i reattivi di Carrez I (3.2) e Carrez II (3.3) come descritto al punto 5.1 utilizzando, tuttavia, una quantità quattro volte più elevata di ciascun reattivo. Portare a volume con etanolo all'80 % (v/v).
Omogeneizzare e filtrare. Eliminare l'etanolo come descritto al punto 5.1. In assenza di amido destrinizzato, portare a volume con acqua distillata.
|
|
7.2.
|
Per i melassi e le materie prime per alimenti, ricchi in zuccheri e praticamente esenti da amido (carrube, fettucce essiccate di barbabietole, ecc.), pesare 5 g, introdurli in un pallone tarato da 250 ml, aggiungere 200 ml di acqua distillata e far girare nell'apparecchio rotativo, per un'ora o più, se necessario. Chiarificare con i reattivi di Carrez I (3.2) e Carrez II (3.3), come descritto al punto 5.1. Portare a volume con acqua, omogeneizzare e filtrare. Per la determinazione degli zuccheri totali, procedere come descritto al punto 5.3.
|
8. Osservazioni
|
8.1.
|
È consigliabile aggiungere circa 1 ml di 3-metilbutan-l-olo (3.14) (senza tener conto del volume), prima dell'ebollizione con il reattivo di Luff-Schoorl, onde evitare la formazione di schiuma.
|
|
8.2.
|
La differenza tra il contenuto di zuccheri totali dopo l'inversione, espressi in glucosio, e il contenuto di zuccheri riduttori, sempre espressi in glucosio, moltiplicata per il fattore 0,95, dà il contenuto in percentuale di saccarosio.
|
|
8.3.
|
La determinazione del contenuto di zuccheri riduttori, a esclusione del lattosio, può avvenire in due modi.
|
|
8.3.1.
|
Per un calcolo approssimativo si moltiplica per 0,675 il contenuto di lattosio stabilito con un metodo d'analisi diverso e si detrae il risultato ottenuto dal contenuto di zuccheri riduttori.
|
|
8.3.2.
|
Per un calcolo esatto degli zuccheri riduttori, fatta eccezione del lattosio, è necessario partire dallo stesso campione per le due determinazioni finali. Una delle analisi è effettuata su una parte della soluzione ottenuta conformemente al procedimento di cui al punto 5.1, l'altra su una parte della soluzione ottenuta durante la determinazione del lattosio, secondo il metodo previsto a tal fine (dopo la fermentazione degli altri tipi di zuccheri e la chiarificazione).
In entrambi i casi, la quantità di zucchero presente è determinata secondo il metodo di Luff-Schoorl e calcolata in mg di glucosio. Uno dei valori è detratto dall'altro e la differenza è espressa in percentuale del campione.
Esempio
Le due quantità prelevate corrispondono, per ciascuna analisi, a un campione di 250 mg.
Nel primo caso si consumano 17 ml di una soluzione di tiosolfato di sodio di 0,1 mol/l, corrispondenti a 44,2 mg di glucosio, nel secondo 11 ml, corrispondenti a 27,6 mg di glucosio.
La differenza è di 16,6 mg di glucosio.
Il contenuto in zuccheri riduttori (eccettuato il lattosio), calcolato in glucosio, è pertanto di:

|
Tabella dei valori per 25 ml di reattivo di Luff-Schoorl
ml di Na2 S2 O3 0,1 mol/l, 2 minuti di riscaldamento, 10 minuti di ebollizione
|
Na2 S2 O3
0,1 mol/l
|
Glucosio, fruttosio, zuccheri invertiti
C6 H12 O6
|
Lattosio
C12 H22 O11
|
Maltosio
C12 H22 O11
|
Na2 S2 O3
0,1 mol/l
|
|
ml
|
mg
|
differenza
|
mg
|
differenza
|
mg
|
differenza
|
ml
|
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
2,4
4,8
7,2
9,7
12,2
14,7
17,2
19,8
22,4
25,0
27,6
30,3
33,0
35,7
38,5
41,3
44,2
47,1
50,0
53,0
56,0
59,1
62,2
|
2,4
2,4
2,5
2,5
2,5
2,5
2,6
2,6
2,6
2,6
2,7
2,7
2,7
2,8
2,8
2,9
2,9
2,9
3,0
3,0
3,1
3,1
|
3,6
7,3
11,0
14,7
18,4
22,1
25,8
29,5
33,2
37,0
40,8
44,6
48,4
52,2
56,0
59,9
63,8
67,7
71,7
75,7
79,8
83,9
88,0
|
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,8
3,8
3,8
3,8
3,8
3,8
3,9
3,9
3,9
4,0
4,0
4,1
4,1
4,1
|
3,9
7,8
11,7
15,6
19,6
23,5
27,5
31,5
35,5
39,5
43,5
47,5
51,6
55,7
59,8
63,9
68,0
72,2
76,5
80,9
85,4
90,0
94,6
|
3,9
3,9
3,9
4,0
3,9
4,0
4,0
4,0
4,0
4,0
4,0
4,1
4,1
4,1
4,1
4,1
4,2
4,3
4,4
4,5
4,6
4,6
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
K. DETERMINAZIONE DEL LATTOSIO
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di lattosio negli alimenti per animali che ne contengono più dello 0,5 %.
2. Principio
Gli zuccheri sono disciolti nell'acqua. La soluzione è sottoposta a fermentazione col lievito Saccharomyces cerevisiae che lascia intatto il lattosio. Dopo chiarificazione e filtrazione il contenuto in lattosio del filtrato è determinato con il metodo di Luff-Schoorl.
3. Reattivi
|
3.1.
|
Sospensione di Saccharomyces cerevisiae: porre in sospensione 25 g di lievito fresco in 100 ml di acqua. La sospensione si conserva in frigorifero al massimo per una settimana.
|
|
3.2.
|
Soluzione di Carrez I: sciogliere in acqua 21,9 g di acetato di zinco, Zn (CH3 COO)2 2H2O e 3 g di acido acetico glaciale. Portare a 100 ml con acqua.
|
|
3.3.
|
Soluzione di Carrez II: sciogliere in acqua 10,6 g di ferrocianuro di potassio, K4 Fe (CN)6 3H2O. Portare a 100 ml con acqua.
|
|
3.4.
|
Reattivo di Luff-Schoorl:
versare, agitando sempre lentamente, la soluzione di acido citrico (3.4.2) nella soluzione di carbonato di sodio (3.4.3). Aggiungere la soluzione di solfato di rame (3.4.1) e portare al volume di 1 litro con acqua. Lasciar riposare una notte e filtrare. Controllare la concentrazione del reattivo così ottenuto (Cu 0,05 mol/l; Na2 CO3 1 mol/l). Il pH della soluzione è circa 9,4.
|
|
3.4.1.
|
Soluzione di solfato di rame: sciogliere 25 g di solfato di rame, Cu SO4 5H2O, esente da ferro, in 100 ml d'acqua.
|
|
3.4.2.
|
Soluzione di acido citrico: sciogliere 50 g di acido citrico, C6H8O7·H2O, in 50 ml d'acqua.
|
|
3.4.3.
|
Soluzione di carbonato di sodio: sciogliere 143,8 g di carbonato di sodio anidro in circa 300 ml d'acqua calda. Lasciar raffreddare.
|
|
3.5.
|
Granuli di pietra pomice bolliti nell'acido cloridrico, lavati in acqua ed essiccati.
|
|
3.6.
|
Soluzione al 30 % (p/v) di ioduro di potassio.
|
|
3.7.
|
Acido solforico (3 mol/l).
|
|
3.8.
|
Soluzione di tiosolfato di sodio (0,1 mol/l).
|
|
3.9.
|
Soluzione d'amido: aggiungere una miscela di 5 g d'amido solubile in 30 ml d'acqua a 1 litro d'acqua bollente. Far bollire per 3 minuti, lasciar raffreddare e aggiungere eventualmente, come conservante, 10 mg di ioduro di mercurio.
|
4. Apparecchiatura
Bagnomaria con termostato regolato a 38-40 oC.
5. Procedimento
Pesare, con l'approssimazione di 1 mg, 1 g del campione e introdurlo in un pallone tarato da 100 ml. Aggiungere 25-30 ml d'acqua. Porre il pallone per 30 minuti in un bagnomaria bollente e lasciarlo poi raffreddare a circa 35 oC. Aggiungere 5 ml di sospensione di lievito (3.1) e mescolare. Lasciar riposare il pallone per 2 ore in un bagnomaria, a una temperatura di 38-40 oC. Raffreddare quindi sino a circa 20 oC.
Aggiungere 2,5 ml di soluzione di Carrez I (3.2) e agitare per 30 secondi; aggiungere poi 2,5 ml di soluzione di Carrez II (3.3) e agitare nuovamente per 30 secondi. Portare a 100 ml con acqua, mescolare e filtrare. Prelevare con la pipetta un quantitativo di filtrato non eccedente i 25 ml e contenente preferibilmente da 40 a 80 mg di lattosio; porlo quindi in un erlenmeyer da 300 ml. Se necessario portare a 25 ml con acqua.
Procedere nello stesso modo a una prova in bianco con 5 ml di sospensione di lievito (3.1). Determinare il contenuto di lattosio secondo il metodo Luff-Schoorl come segue: aggiungere 25 ml esatti del reattivo di Luff-Schoorl (3.4) e due granuli di pietra pomice (3.5). Riscaldare agitando a mano su una fiamma libera di media altezza e portare il liquido all'ebollizione in circa 2 minuti. Trasferire immediatamente l'erlenmeyer su una tela metallica, provvista di uno schermo d'amianto munito di un foro del diametro di circa 6 cm, sulla quale è stata preventivamente accesa una fiamma. Questa è regolata in modo tale che venga riscaldato soltanto il fondo dell'erlenmeyer. Adattare sull'erlenmeyer un refrigerante a riflusso. A decorrere da questo momento far bollire per 10 minuti esatti. Raffreddare immediatamente in acqua fredda e dopo circa 5 minuti titolare nel seguente modo:
aggiungere 10 ml di soluzione di ioduro di potassio (3.6) e, subito dopo, con molta attenzione (a causa del rischio di formazione di abbondante schiuma), 25 ml di acido solforico (3.7). Titolare quindi con la soluzione di tiosolfato di sodio (3.8) sino all'apparire di una colorazione giallo opaco, aggiungere l'indicatore all'amido (3.10) e terminare la titolazione.
Effettuare la stessa titolazione su di una miscela, esattamente misurata, di 25 ml di reattivo di Luff-Schoorl (3.4) e 25 ml d'acqua, dopo aver aggiunto 10 ml di soluzione di ioduro di potassio (3.6) e 25 ml di acido solforico (3.7) senza portare a ebollizione.
6. Calcolo dei risultati
Stabilire, a mezzo della tabella allegata, la quantità di lattosio in mg corrispondente alla differenza tra i risultati delle due titolazioni, espressi in ml di tiosolfato di sodio 0,1 mol/l.
Esprimere il risultato in percentuale del lattosio anidro del campione.
7. Osservazione
Per i prodotti contenenti più del 40 % di zuccheri fermentescibili, utilizzare più di 5 ml di sospensione di lievito (3.1).
Tabella dei valori per 25 ml di reattivo di Luff-Schoorl
ml di Na2 S2 O3 0,1 mol/l, 2 minuti di riscaldamento, 10 minuti di ebollizione
|
Na2 S2 O3
0,1 mol/l
|
Glucosio, fruttosio, zuccheri invertiti
C6 H12 O6
|
Lattosio
C12 H22 O11
|
Maltosio
C12 H22 O11
|
Na2 S2 O3
0,1 mol/l
|
|
ml
|
mg
|
differenza
|
mg
|
differenza
|
mg
|
differenza
|
ml
|
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
2,4
4,8
7,2
9,7
12,2
14,7
17,2
19,8
22,4
25,0
27,6
30,3
33,0
35,7
38,5
41,3
44,2
47,1
50,0
53,0
56,0
59,1
62,2
|
2,4
2,4
2,5
2,5
2,5
2,5
2,6
2,6
2,6
2,6
2,7
2,7
2,7
2,8
2,8
2,9
2,9
2,9
3,0
3,0
3,1
3,1
|
3,6
7,3
11,0
14,7
18,4
22,1
25,8
29,5
33,2
37,0
40,8
44,6
48,4
52,2
56,0
59,9
63,8
67,7
71,7
75,7
79,8
83,9
88,0
|
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,7
3,8
3,8
3,8
3,8
3,8
3,8
3,9
3,9
3,9
4,0
4,0
4,1
4,1
4,1
|
3,9
7,8
11,7
15,6
19,6
23,5
27,5
31,5
35,5
39,5
43,5
47,5
51,6
55,7
59,8
63,9
68,0
72,2
76,5
80,9
85,4
90,0
94,6
|
3,9
3,9
3,9
4,0
3,9
4,0
4,0
4,0
4,0
4,0
4,0
4,1
4,1
4,1
4,1
4,1
4,2
4,3
4,4
4,5
4,6
4,6
|
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
L. DETERMINAZIONE DELL'AMIDO
METODO POLARIMETRICO
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto in amido e in prodotti di degradazione dell'amido ad alto peso molecolare negli alimenti per animali, al fine di controllarne la conformità al valore energetico dichiarato (disposizioni di cui all'allegato VII) e alla direttiva 96/25/CE del Consiglio (3).
2. Principio
Il metodo prevede una duplice determinazione. Nella prima, il campione è trattato con acido cloridrico diluito. Dopo chiarificazione e filtrazione si misura per polarimetria il potere rotatorio della soluzione.
Nella seconda, il campione viene estratto con etanolo al 40 %. Dopo acidificazione del filtrato con acido cloridrico, chiarificazione e filtrazione, si misura il potere rotatorio come nella prima determinazione.
La differenza tra le due misure, moltiplicata per un fattore noto, dà il contenuto in amido del campione.
3. Reattivi
|
3.1.
|
Acido cloridrico, soluzione al 25 % (p/p),densità: 1,126 g/ml.
|
|
3.2.
|
Acido cloridrico, soluzione all'1,13 % (p/v).
La concentrazione deve essere verificata per titolazione mediante una soluzione di idrossido di sodio 0,1 mol/l in presenza di rosso di metile allo 0,1 % (p/v) in etanolo al 94 % (v/v). Per la neutralizzazione di 10 ml, sono necessari 30,94 ml di NaOH 0,1 mol/l.
|
|
3.3.
|
Soluzione di Carrez I: sciogliere in acqua 21,9 g di acetato di zinco Zn(CH3COO)2 2H2O e 3 g di acido acetico glaciale. Portare a 100 ml con acqua.
|
|
3.4.
|
Soluzione di Carrez II: sciogliere nell'acqua 10,6 g di ferrocianuro di potassio, K4Fe(CN)6 3H2O. Portare a 100 ml con acqua.
|
|
3.5.
|
Etanolo, soluzione al 40 % (v/v), densità: 0,948 g/ml a 20 oC.
|
4. Apparecchiatura
|
4.1.
|
Erlenmeyer da 250 ml a cono normalizzato con collo smerigliato, con refrigerante a ricadere.
|
|
4.2.
|
Polarimetro o saccarimetro.
|
5. Procedimento
5.1. Preparazione del campione
Macinare il campione in modo che passi tutto attraverso un setaccio a maglie rotonde di 0,5 mm di diametro.
5.2. Determinazione del potere rotatorio totale (P o S) (cfr. osservazione 7.1)
Pesare, con l'approssimazione di 1 mg, 2,5 g del campione macinato e introdurli in un pallone tarato da 100 ml. Aggiungere 25 ml di acido cloridrico (3.2), agitare per ottenere una buona ripartizione della sostanza e aggiungere altri 25 ml di acido cloridrico (3.2). Immergere il pallone in bagnomaria bollente, agitando energicamente e regolarmente per i primi tre minuti allo scopo di evitare la formazione di grumi. La quantità d'acqua del bagnomaria deve essere sufficiente per mantenere l'ebollizione quando vi è immerso il pallone. Quest'ultimo non può essere tolto dal bagnomaria durante l'agitazione. Dopo 15 minuti esatti togliere il pallone dal bagnomaria, aggiungere 30 ml d'acqua fredda e raffreddare immediatamente sino a 20 °C.
Aggiungere 5 ml di soluzione di Carrez I (3.3) ed agitare per circa 30 secondi. Aggiungere quindi 5 ml di soluzione di Carrez II (3.4) e agitare nuovamente per circa 30 secondi. Portare a volume con acqua, mescolare e filtrare. Se il filtrato non è perfettamente limpido (caso poco frequente), ripetere l'analisi usando una maggiore quantità di soluzione di Carrez I e II, ad esempio 10 ml.
Misurare quindi il potere rotatorio della soluzione, in un tubo da 200 mm, con un polarimetro od un saccarimetro.
5.3. Determinazione del potere rotatorio (P' o S') delle sostanze solubili in etanolo al 40 %
Pesare, con l'approssimazione di 1 mg, 5 g del campione, introdurli in un pallone tarato da 100 ml e aggiungere circa 80 ml di etanolo (3.5) (cfr. osservazione 7.2). Lasciar riposare il pallone per un'ora a temperatura ambiente; durante questo intervallo agitarlo energicamente sei volte in modo che la sostanza sia ben mescolata con l'etanolo. Portare quindi a volume con etanolo (3.5), mescolare e filtrare.
Pipettare 50 ml del filtrato (corrispondenti a 2,5 g del campione) in una beuta da 250 ml, aggiungere 2,1 ml di acido cloridrico (3.1) e agitare energicamente. Applicare un refrigerante a ricadere sulla beuta e immergerla in bagnomaria bollente. Dopo 15 minuti esatti ritirare la beuta dal bagno, travasarne il contenuto in un pallone tarato da 100 ml, sciacquando con un po' di acqua fredda, e raffreddare sino a 20 oC.
Chiarificare quindi con le soluzioni di Carrez I (3.3) e II (3.4), portare a volume con acqua, mescolare, filtrare e misurare il potere rotatorio come indicato al punto 5.2, secondo e terzo capoverso.
6. Calcolo dei risultati
Il contenuto di amido (in %) è calcolato come segue:
6.1. Misurazioni effettuate con il polarimetro

|
P
|
=
|
potere rotatorio totale in gradi d'arco
|
|
P'
|
=
|
potere rotatorio in gradi d'arco delle sostanze solubili nell'etanolo al 40 % (v/v)
|
|

|
=
|
potere rotatorio specifico dell'amido puro. I valori numerici D comunemente accettati per tale fattore sono i seguenti
|
+ 185,9o:
|
amido di riso
|
|
+ 185,7o:
|
fecola di patate
|
|
+ 184,6o:
|
amido di mais
|
|
+ 182,7o:
|
amido di frumento
|
|
+ 181,5o:
|
amido d'orzo
|
|
+ 181,3o:
|
amido d'avena
|
|
+ 184,0o:
|
altri tipi di amido e miscele di amido negli alimenti composti
|
|
6.2. Misurazioni effettuate con il saccarimetro

|
S
|
=
|
potere rotatorio totale in gradi saccarimetrici
|
|
S'
|
=
|
potere rotatorio in gradi saccarimetrici delle sostanze solubili nell'etanolo al 40 % (v/v)
|
|
N
|
=
|
peso (in g) del saccarosio in 100 ml di acqua che dà un potere rotatorio di 100 gradi saccarimetrici misurati con un tubo di 200 mm
16,29 g per i saccarimetri francesi
26,00 g per i saccarimetri tedeschi
20,00 g per i saccarimetri misti.
|
|

|
=
|
potere rotatorio specifico dell'amido puro (cfr. 6.1)
|
6.3. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare lo 0,4 in valore assoluto, per i contenuti di amido inferiori al 40 %, e l'1 % in valore relativo, per i contenuti di amido uguali o superiori al 40 %.
7. Osservazioni
|
7.1.
|
Se il campione contiene più del 6 % di carbonati, espressi in carbonato di calcio, prima di determinare il potere rotatorio totale essi vanno distrutti per trattamento con l'esatta quantità necessaria di acido solforico diluito.
|
|
7.2.
|
Nel caso di prodotti ad alto contenuto di lattosio, quali il siero di latte in polvere o il latte scremato in polvere, dopo aver aggiunto 80 ml di etanolo (3.5) procedere come segue. Applicare un refrigerante a ricadere sulla beuta e immergerla per 30 minuti in bagnomaria a 50 oC. Lasciare poi raffreddare e continuare l'analisi come indicato al punto 5.3.
|
|
7.3.
|
Quando la determinazione del contenuto di amido viene effettuata col metodo polarimetrico, le materie prime elencate di seguito, se presenti negli alimenti per animali in quantità significative, possono dar luogo a interferenze capaci di condurre a risultati inesatti:
|
—
|
prodotti della barbabietola (da zucchero), come polpa di barbabietola (da zucchero), melasse di barbabietola (da zucchero), polpa di barbabietola (da zucchero) melassata, borlanda di barbabietola (da zucchero), zucchero di barbabietola,
|
|
—
|
semi di lino; panello di lino; farina di estrazione di lino,
|
|
—
|
semi di colza; panello di colza; farina di estrazione di colza; corteccia di colza,
|
|
—
|
semi di girasole; farina di estrazione di girasole; farina di estrazione di girasole, parzialmente decorticato,
|
|
—
|
panello di copra; farina di estrazione di copra,
|
|
—
|
prodotti ricchi di inulina (ad esempio fettucce e farina di topinambur),
|
|
M. DETERMINAZIONE DELLE CENERI GREZZE
1. Finalità e campo d'applicazione
Il metodo consente di determinare il tenore di ceneri grezze negli alimenti per gli animali.
2. Principio
Il campione è incenerito alla temperatura di 550 oC; il residuo viene pesato.
3. Reattivi
Nitrato d'ammonio, soluzione al 20 % (p/v).
4. Apparecchiatura
|
4.1.
|
Piastra riscaldante.
|
|
4.2.
|
Forno elettrico a muffola, dotato di termostato.
|
|
4.3.
|
Crogioli da incenerimento in quarzo, porcellana o platino, rettangolari (60 × 40 × 25 mm) o circolari (diametro di 60-75 mm, altezza da 20 a 40 mm).
|
5. Procedimento
Pesare, con l'approssimazione di 1 mg, 5 g circa (o 2,5 g per i prodotti aventi tendenza a rigonfiare) del campione in un crogiolo per incenerimento preventivamente riscaldato a 550 °C, raffreddato e tarato. Porre il crogiolo sopra la piastra riscaldante e scaldare progressivamente fino a carbonizzazione della sostanza. Incenerire conformemente ai punti 5.1 o 5.2.
|
5.1.
|
Introdurre quindi il crogiolo nel forno a muffola regolato a 550 oC. Mantenere a tale temperatura fino a ottenere ceneri di colore bianco, grigio chiaro o rossastro, apparentemente esenti da particelle carboniose. Porre il crogiolo in un essiccatore, lasciare raffreddare e pesare immediatamente.
|
|
5.2.
|
Introdurre il crogiolo nel forno a muffola regolato a 550 oC. Incenerire per 3 ore. Porre il crogiolo in un essiccatore, lasciare raffreddare e pesare immediatamente. Incenerire nuovamente per 30 minuti, onde assicurarsi che il peso delle ceneri rimanga costante (la perdita di peso tra due pesate consecutive deve essere pari o inferiore a 1 mg).
|
6. Calcolo dei risultati
Calcolare il peso del residuo deducendone la tara.
Esprimere il risultato in percentuale del campione.
7. Osservazioni
|
7.1.
|
I prodotti di difficile incenerimento vanno sottoposti a un primo incenerimento di almeno 3 ore, lasciati raffreddare e addizionati di alcune gocce di una soluzione al 20 % di nitrato d'ammonio o acqua (con cautela, per evitare la proiezione o l'aggregazione delle ceneri). Proseguire la calcinazione dopo essiccazione nel forno. Ripetere eventualmente l'operazione fino a completo incenerimento.
|
|
7.2.
|
Per i prodotti che resistono al trattamento di cui al punto 7.1, operare come segue: dopo un incenerimento di 3 ore trasferire le ceneri in acqua calda e filtrare su un piccolo filtro senza ceneri. Incenerire il filtro e il suo contenuto nello stesso crogiolo. Aggiungere il filtrato nel crogiolo raffreddato, portare a secco, incenerire e pesare.
|
|
7.3.
|
Nel caso degli oli e dei grassi, pesare esattamente una quantità di prodotto di 25 g in un crogiolo di capacità appropriata. Carbonizzare accendendo il prodotto per mezzo di una miccia di carta da filtro senza ceneri. Dopo combustione, umettare con la quantità minima d'acqua. Essiccare e incenerire come indicato al punto 5.
|
N. DETERMINAZIONE DELLE CENERI INSOLUBILI IN ACIDO CLORIDRICO
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di sostanze minerali insolubili nell'acido cloridrico presenti negli alimenti per gli animali. Si può procedere in due modi in funzione della natura del campione.
|
1.1.
|
Metodo A: applicabile alle materie prime di origine organica per alimenti per animali ed alla maggior parte degli alimenti composti.
|
|
1.2.
|
Metodo B: applicabile ai composti e alle miscele minerali oltre che agli alimenti composti il cui contenuto di sostanze insolubili in acido cloridrico, determinato secondo il metodo A, è superiore all'1 %.
|
2. Principio
|
2.1.
|
Metodo A: il campione è incenerito e le ceneri sono trattate all'ebollizione con acido cloridrico e il residuo insolubile è filtrato e pesato.
|
|
2.2.
|
Metodo B: il campione è trattato con acido cloridrico. La soluzione è filtrata, il residuo incenerito e le ceneri ottenute vengono trattate come nel metodo A.
|
3. Reattivi
|
3.1.
|
Acido cloridrico, 3 mol/l.
|
|
3.2.
|
Acido tricloroacetico, soluzione al 20 % (p/v).
|
|
3.3.
|
Acido tricloroacetico, soluzione all'1 % (p/v).
|
4. Apparecchiatura
|
4.1.
|
Piastra riscaldante.
|
|
4.2.
|
Forno elettrico a muffola, dotato di termostato.
|
|
4.3.
|
Crogioli da incenerimento in quarzo, porcellana o platino, rettangolari (60 × 40 × 25 mm) o circolari (diametro di 60-75 mm, altezza da 20 a 40 mm).
|
5. Procedimento
5.1. Metodo A
Incenerire la sostanza da analizzare secondo il procedimento previsto per la determinazione delle ceneri grezze. Possono essere utilizzate anche le ceneri ottenute in tale determinazione.
Trasferire le ceneri in un becher da 250-400 ml con 75 ml d'acido cloridrico (3.1). Portare lentamente a ebollizione e lasciare bollire adagio per 15 minuti. Filtrare la soluzione calda su filtro di carta senza ceneri e lavare il residuo con acqua calda sino a scomparsa della reazione acida. Essiccare il filtro contenente il residuo e incenerire in un crogiolo tarato a temperatura non inferiore a 550 oC e non superiore a 700 oC. Raffreddare in un essiccatore e pesare.
5.2. Metodo B
Pesare, con l'approssimazione di 1 mg, 5 g di campione e introdurli in un becher da 250 a 400 ml. Aggiungere successivamente 25 ml d'acqua e 25 ml d'acido cloridrico (3.1), agitare sino a cessazione dell'effervescenza. Aggiungere altri 50 ml d'acido cloridrico (3.1). Attendere la fine di nuova effervescenza e porre il becher in un bagno di acqua bollente e tenervelo per la durata di 30 minuti o più, se necessario, al fine di idrolizzare completamente l'amido eventualmente presente. Filtrare a caldo su un filtro senza ceneri e lavare il filtro mediante 50 ml di acqua calda (cfr. osservazione 7). Porre il filtro contenente il residuo in un crogiolo da incenerimento, essiccare e incenerire a temperatura non inferiore a 550 oC e non superiore a 700 oC. Trasferire infine le ceneri in un becher della capacità di 250-400 ml mediante 75 ml di acido cloridrico (3.1) e continuare come descritto al punto 5.1, secondo capoverso.
6. Calcolo dei risultati
Calcolare il peso del residuo deducendone la tara. Esprimere il risultato in percentuale del campione.
7. Osservazione
Se la filtrazione si rivela difficile, ricominciare la determinazione, sostituendo i 50 ml di acido cloridrico (3.1) con 50 ml di acido tricloroacetico al 20 % (3.2) lavando il filtro mediante una soluzione calda di acido tricloroacetico all'1 % (3.3).
O. DETERMINAZIONE DEI CARBONATI
1. Finalità e campo d'applicazione
Il metodo consente la determinazione dei carbonati, convenzionalmente espressi in carbonato di calcio, nella maggior parte degli alimenti per animali.
Tuttavia, in alcuni casi (ad esempio con il carbonato di ferro) bisogna impiegare un metodo particolare.
2. Principio
I carbonati sono decomposti in acido cloridrico; l'anidride carbonica liberata è raccolta in un tubo tarato e il suo volume è comparato a quello sprigionato, nelle medesime condizioni, da una quantità nota di carbonato di calcio.
3. Reattivi
|
3.1.
|
Acido cloridrico, densità 1,10 g/ml.
|
|
3.2.
|
Carbonato di calcio.
|
|
3.3.
|
Acido solforico 0,05 mol/litro circa, colorato con rosso di metile.
|
4. Apparecchiatura
Apparecchio di Scheibler-Dietrich (cfr. schema) o apparecchio equivalente.
5. Procedimento
Secondo il contenuto di carbonati del campione, pesare una quantità di prodotto come indicato di seguito:
|
—
|
0,5 g per i prodotti contenenti da 50 a 100 % di carbonati, espressi in carbonato di calcio,
|
|
—
|
1 g per i prodotti contenenti da 40 a 50 % di carbonati, espressi in carbonato di calcio,
|
|
—
|
da 2 a 3 g in tutti gli altri casi.
|
Introdurre la quantità di sostanza da analizzare nello speciale recipiente (4) dell'apparecchio, provvisto di un piccolo tubo in materiale infrangibile, contenente 10 ml di acido cloridrico (3.1), e collegare il recipiente all'apparecchio. Girare il rubinetto a tre vie (5), in modo che il tubo (1) comunichi con l'esterno. Per mezzo del tubo mobile (2), che è riempito di acido solforico colorato (3.3) ed è collegato al tubo tarato (1), portare il livello del liquido alla gradazione zero. Girare il rubinetto (5) in modo da far comunicare i tubi (1) e (3) e verificare se il livello è a zero.
Versare lentamente l'acido cloridrico (3.1) sulla sostanza inclinando il recipiente (4). Pareggiare la pressione abbassando il tubo (2). Agitare il recipiente (4) fino a cessazione completa dell'emissione di anidride carbonica.
Ristabilire la pressione riportando il liquido allo stesso livello nei tubi (1) e (2). Eseguire la lettura dopo qualche minuto quando il volume gassoso è divenuto costante.
Effettuare nelle stesse condizioni una prova comparativa su 0,5 g di carbonato di calcio (3.2).
6. Calcolo dei risultati
Il contenuto di carbonati, espressi in carbonato di calcio, è dato dalla seguente formula:

dove
|
X
|
=
|
% (p/p) di carbonati presenti nel campione, espressi come carbonati di calcio
|
|
V
|
=
|
ml di CO2 liberati dalla quantità di sostanza sottoposta all'analisi
|
|
V1
|
=
|
ml di CO2 liberati da 0,5 g di CaCO3
|
|
m
|
=
|
peso (in g) della sostanza sottoposta ad analisi.
|
7. Osservazioni
|
7.1.
|
Quando la quantità di sostanza sottoposta all'analisi è superiore a 2 g, introdurre innanzitutto 15 ml di acqua distillata nel recipiente (4) e mescolare prima di cominciare la prova. Usare lo stesso volume d'acqua per la prova comparativa.
|
|
7.2.
|
Quando si utilizza un apparecchio di volume diverso da quello di Scheibler-Dietrich, apportare le conseguenti variazioni alla quantità di sostanza su cui si opera, alla quantità di carbonato di calcio nel saggio-tipo, nonché al calcolo dei risultati.
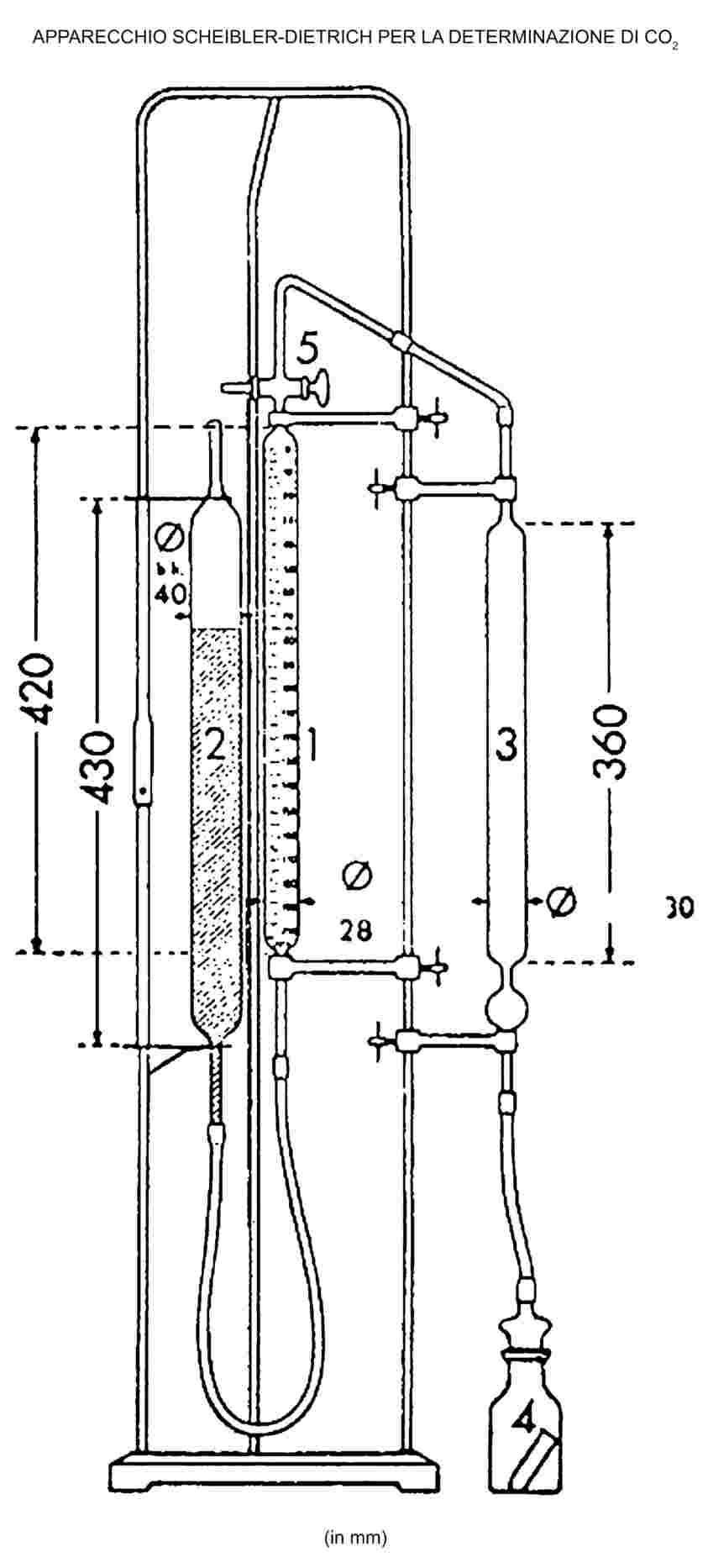
|
P. DETERMINAZIONE DEL FOSFORO TOTALE
METODO FOTOMETRICO
1. Finalità e campo d'applicazione
Il metodo consente la determinazione del contenuto di fosforo totale negli alimenti per gli animali. Esso è particolarmente indicato per l'analisi dei prodotti poveri di fosforo. In certi casi (prodotti ricchi di fosforo) può essere applicato un metodo gravimetrico.
2. Principio
Il campione viene mineralizzato, per via secca (per gli alimenti organici) o per via umida (per i composti minerali e gli alimenti liquidi) e trasferito in soluzione acida. La soluzione viene trattata con il reattivo vanado-molibdico. La densità ottica della soluzione gialla così formatasi, viene misurata allo spettrofotometro a 430 nm.
3. Reattivi
|
3.1.
|
Carbonato di calcio.
|
|
3.2.
|
Acido cloridico, ρ20 = 1,10 g/ml (6 mol/l circa).
|
|
3.3.
|
Acido nitrico, ρ20 = 1,045 g/ml.
|
|
3.4.
|
Acido nitrico, ρ20 = 1,38-1,42 g/ml.
|
|
3.5.
|
Acido solforico, ρ20 = 1,84 g/ml.
|
|
3.6.
|
Reattivo vanado-molibdico: mescolare, in un pallone tarato da 1 litro, 200 ml di soluzione di eptamolibdato d'ammonio (3.6.1) con 200 ml di soluzione di monovanadato d'ammonio (3.6.2) e 134 ml di acido nitrico (3.4). Portare a volume con acqua.
|
|
3.6.1.
|
Soluzione di eptamolibdato d'ammonio: sciogliere in acqua calda 100 g di eptamolibdato d'ammonio (NH4) 6Mo7O24.4H2O. Aggiungere 10 ml di ammoniaca (densità 0,91 g/ml) e portare a 1 litro con acqua.
|
|
3.6.2.
|
Soluzione di monovanadato di ammonio: sciogliere 2,35 g di monovanadato di ammonio NH4VO3 in 400 ml di acqua calda. Aggiungere lentamente sempre agitando 20 ml di acido nitrico diluito [7 ml di HNO3 (3.4) + 13 ml di H2O] e portare a 1 litro con acqua.
|
|
3.7.
|
Soluzione tipo, contenente 1 mg di fosforo per ml: sciogliere in acqua 4,387 g di potassio diidrogeno fosfato KH2PO4. Portare a 1 litro con acqua.
|
4. Apparecchiatura
|
4.1.
|
Crogioli da incenerimento di quarzo, porcellana o platino.
|
|
4.2.
|
Forno elettrico a muffola munito di termostato regolato a 550 oC.
|
|
4.3.
|
Matraccio di Kjeldahl da 250 ml.
|
|
4.4.
|
Palloni tarati e pipette di precisione.
|
|
4.6.
|
Tubi da saggio a tappo smerigliato, diametro circa 16 mm, a smerigliatura normalizzata 14,5 mm; capacità: 25-30 ml.
|
5. Procedimento
5.1. Preparazione della soluzione
Secondo la natura del campione preparare una soluzione come indicato al punto 5.1.1 o 5.1.2.
5.1.1. Procedura abituale
Pesare, con l'approssimazione di 1 mg, 1 g o più del campione. Introdurre la quantità da analizzare in un matraccio di Kjeldahl, aggiungere 20 ml di acido solforico (3.5), agitare per imbibire d'acido tutta la sostanza e per evitare che essa aderisca alle pareti del pallone; riscaldarla e farla bollire per 10 minuti. Lasciar raffreddare leggermente, aggiungere 2 ml di acido nitrico (3.4), riscaldare lentamente, lasciare ancora raffreddare leggermente, aggiungere dell'altro acido nitrico (3.4) e portare il tutto nuovamente a ebollizione. Ripetere le operazioni fino a ottenere una soluzione incolore. Far raffreddare, aggiungere un po' d'acqua, travasare il liquido in un pallone tarato da 500 ml lavando il pallone con acqua calda. Lasciar raffreddare, portare a volume con acqua, mescolare e filtrare.
5.1.2. Campioni contenenti sostanze organiche ed esenti da diidrogenofosfati di calcio e magnesio
Pesare, con l'approssimazione di 1 mg, 2,5 g circa di campione e introdurli in un crogiolo da incenerimento. Miscelare il campione da analizzare sino ad amalgamarlo con 1 g di carbonato di calcio (3.1). Incenerire nel forno a 550 oC fino a ottenere ceneri bianche o grigie (una piccola quantità di carbone non disturba). Trasferire le ceneri in un becher da 250 ml. Aggiungere 20 ml d'acqua e acido cloridrico (3.2) sino a cessazione dell'effervescenza. Aggiungere altri 10 ml di acido cloridrico (3.2). Porre il becher su un bagno di sabbia e evaporare a secco per insolubilizzare la silice. Riprendere il residuo con 10 ml di acido nitrico (3.3) e far bollire per 5 minuti sul bagno di sabbia o sulla piastra riscaldante senza arrivare a secco. Travasare il liquido in un pallone tarato da 500 ml lavando il becher, a più riprese, con acqua calda. Lasciar raffreddare, portare a volume con acqua, mescolare e filtrare.
5.2. Sviluppo della colorazione e misura della densità ottica
Diluire una parte del filtrato ottenuto conformemente a quanto indicato al punto 5.1.1 o 5.1.2 in modo da ottenere una concentrazione di fosforo in quantità non superiore a 40 μg/ml. Introdurre in un tubo da saggio (4.6) 10 ml di questa soluzione ed aggiungervi 10 ml del reattivo vanado-molibdico (3.6). Mescolare e lasciar riposare almeno 10 minuti alla temperatura di 20 oC. Misurare la densità ottica allo spettrofotometro a 430 nm per comparazione con una soluzione ottenuta per aggiunta di 10 ml di reattivo vanado-molibdico (3.6) a 10 ml di acqua.
5.3. Curva di taratura
Preparare, a partire dalla soluzione tipo (3.7), soluzioni contenenti rispettivamente 5, 10, 20, 30 e 40 μg di fosforo per ml. Prelevare 10 ml da ciascuna soluzione e aggiungervi 10 ml di reattivo vanado-molibdico. Mescolare e lasciar riposare almeno 10 minuti alla temperatura di 20 oC. Misurarne la densità ottica come indicato al punto 5.2. Tracciare la curva di taratura portando in ordinata i valori di densità ottica ed in ascissa le quantità corrispondenti di fosforo. Per concentrazioni comprese tra 0 e 40 μg/ml, la curva sarà lineare.
6. Calcolo dei risultati
Determinare la quantità di fosforo nella sostanza sottoposta all'analisi riferendosi alla curva di taratura.
Esprimere il risultato in percentuale del campione.
Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non supera:
|
—
|
il 3 %, rispetto al valore più elevato, per contenuti di fosforo inferiori al 5 %,
|
|
—
|
lo 0,15 %, in valore assoluto, per i contenuti in fosforo uguali o superiori al 5 %.
|
Q. DETERMINAZIONE DEL CLORO DEI CLORURI
1. Finalità e campo d'applicazione
Il metodo consente di determinare il cloro dei cloruri solubili in acqua, convenzionalmente espresso in cloruro di sodio. Esso è applicabile a tutti gli alimenti per gli animali.
2. Principio
I cloruri sono disciolti in acqua. Se il prodotto contiene sostanze organiche, si procede ad una chiarificazione. La soluzione è leggermente acidificata con acido nitrico e i cloruri sono precipitati sotto forma di cloruro d'argento a mezzo di una soluzione di nitrato d'argento. L'eccesso di nitrato d'argento è titolato mediante una soluzione di tiocianato di ammonio, secondo il metodo Volhard.
3. Reattivi
|
3.1.
|
Soluzione di tiocianato di ammonio 0,1 mol/l.
|
|
3.2.
|
Soluzione di nitrato d'argento 0,1 mol/l.
|
|
3.3.
|
Soluzione satura di bis(solfato)di ammonio e ferro (NH4)Fe(SO4)2.
|
|
3.4.
|
Acido nitrico, densità: 1,38 g/ml.
|
|
3.7.
|
Soluzione di Carrez I: sciogliere in acqua 21,9 g di acetato di zinco Zn (CH3COO)2·2H2O e 3 g di acido acetico glaciale. Portare a 100 ml con acqua.
|
|
3.8.
|
Soluzione di Carrez II: sciogliere in acqua 10,6 g di ferrocianuro di potassio, K4Fe(CN)6·3H2O. Portare a 100 ml con acqua.
|
|
3.9.
|
Carbone attivo, esente da cloruri e non assorbente cloro.
|
4. Apparecchiatura
Agitatore rotativo a capovolgimento: circa 35-40 giri al minuto.
5. Procedimento
5.1. Preparazione della soluzione
Secondo la natura del campione preparare una soluzione come indicato ai punti 5.1.1, 5.1.2 o 5.1.3.
Effettuare in parallelo una prova in bianco, senza il campione da analizzare.
5.1.1. Campioni esenti da sostanze organiche
Pesare, con l'approssimazione di 1 mg, un campione di peso non superiore a 10 g e contenente al massimo 3 g di cloro sotto forma di cloruri. Introdurlo in un matraccio tarato da 500 ml con 400 ml di acqua a 20 oC circa. Agitare per 30 minuti con l'agitatore rotativo, portare a volume, mescolare e filtrare.
5.1.2. Campioni contenenti materie organiche, a eccezione dei prodotti indicati al punto 5.1.3
Pesare con l'approssimazione di 1 mg un quantitativo di campione di circa 5 g e introdurlo con un grammo di carbone attivo in un matraccio tarato da 500 ml. Aggiungere 400 ml di acqua a circa 20 oC e 5 ml di soluzione di Carrez I (3.7), agitare per 30 secondi e aggiungere 5 ml di soluzione di Carrez II (3.8). Miscelare per 30 minuti nell'agitatore rotativo, portare a volume, mescolare e filtrare.
5.1.3. Alimenti cotti, panelli e farina di lino, prodotti ricchi di farina di lino e altri prodotti ricchi di mucillaggini o di sostanze colloidali (ad esempio amido destrinizzato)
Preparare la soluzione come indicato al punto 5.1.2 ma senza filtrare. Dopo decantazione (se necessario centrifugare) prelevare 100 ml del liquido surnatante e introdurli in un pallone tarato da 200 ml. Mescolare con acetone (3.6) e portare a volume con tale solvente, miscelare e filtrare.
5.2. Titolazione
Porre in un erlenmeyer, con la pipetta, da 25 a 100 ml del filtrato (a seconda del contenuto presunto di cloro) ottenuto secondo quanto indicato ai punti 5.1.1, 5.1.2 o 5.1.3. L'aliquota non deve contenere più di 150 mg di cloro (Cl). Diluire, se necessario, con acqua fino a ottenere una soluzione di 50 ml, aggiungere 5 ml di acido nitrico (3.4), 20 ml di soluzione satura di bis(solfato) di ammonio e ferro (3.3) e 2 gocce di soluzione di tiocianato di ammonio (3.1) proveniente dalla buretta riempita a questo scopo fino al tratto zero. Far gocciolare in seguito da una buretta la soluzione di nitrato d'argento (3.2) fino ad averne un eccesso di 5 ml. Aggiungere 5 ml di etere dietilico (3.5) e agitare energicamente per far coagulare il precipitato. Titolare l'eccesso di nitrato d'argento con la soluzione di tiocianato di ammonio (3.1) fino a quando il viraggio al rosso-bruno persiste per 1 minuto.
6. Calcolo dei risultati
La quantità di cloro (X), espressa in cloruro di sodio, è data dalla formula seguente:

dove
|
V1
|
=
|
ml di soluzione di nitrato d'argento 0,1 mol/l aggiunti
|
|
V2
|
=
|
ml di soluzione di tiocianato di ammonio 0,1 mol/l utilizzati per la titolazione
|
|
m
|
=
|
peso del campione.
|
Qualora la prova in bianco indichi un consumo di soluzione di nitrato d'argento di 0,1 mol/l detrarre questo valore dal volume (V1 - V2).
7. Osservazioni
|
7.1.
|
La titolazione può essere realizzata anche con il metodo potenziometrico.
|
|
7.2.
|
Per i prodotti molto ricchi in sostanze grasse, procedere a uno sgrassaggio preliminare con etere dietilico o etere di petrolio.
|
|
7.3.
|
Per le farine di pesce, la titolazione può essere effettuata secondo il metodo di Mohr.
|
(1) Per l'essiccazione dei cereali nonché delle farine, semole e semolini, la stufa deve avere una capacità calorifica tale che, preventivamente regolata alla temperatura di 131 oC, possa raggiungere nuovamente tale temperatura in meno di 45 minuti, dopo avervi introdotto il numero massimo di campioni da essiccare simultaneamente. La stufa deve avere una ventilazione tale che, essiccando per due ore tutti i campioni di frumento tenero che essa può contenere, i risultati presentino una differenza inferiore allo 0,15 % rispetto ai risultati ottenuti con quattro ore di essiccazione.
(2) Sostituire i frammenti di pietra con alcune palline di vetro, quando si debbano eseguire ulteriori esami qualitativi sulla sostanza oleosa o grassa.
(3) GU L 125 del 23.5.1996, pag. 35.
ALLEGATO IV
METODI DI ANALISI PER IL CONTROLLO DEL CONTENUTO DI ADDITIVI AUTORIZZATI NEGLI ALIMENTI PER ANIMALI
A. DETERMINAZIONE DELLA VITAMINA A
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di vitamina A (retinolo) negli alimenti per animali e nelle premiscele. La vitamina A comprende l'alcole retinilico tutto trans e i suoi isomeri cis che vengono determinati con tale metodo. Il contenuto di vitamina A è espresso in unità internazionali (UI) per kg. Una UI corrisponde all'attività di 0,300 μg di alcole di vitamina A tutto trans oppure 0,344 μg di acetato di vitamina A tutto trans oppure 0,550 μg di palmitato di vitamina A tutto trans.
Il limite di quantificazione è di 2 000 UI di vitamina A/kg.
2. Principio
Il campione viene idrolizzato con una soluzione etanolica di idrossido di potassio e la vitamina A viene estratta in etere di petrolio. Il solvente è rimosso per evaporazione e il residuo sciolto in metanolo e, se necessario, diluito alla concentrazione richiesta. Il contenuto di vitamina A è determinato per cromatografia liquida ad alta prestazione a fase inversa (RP-HPLC) utilizzando un rivelatore UV o a fluorescenza. I parametri cromatografici sono scelti in modo che non vi sia separazione tra l'alcole di vitamina A tutto trans e i suoi isomeri cis.
3. Reattivi
|
3.2.
|
Etere di petrolio, intervallo di ebollizione: 40-60 oC
|
|
3.4.
|
Soluzione di idrossido di potassio, c = 50 g/100 ml
|
|
3.5.
|
Soluzione di ascorbato di sodio, c = 10 g/100 ml (cfr. osservazione al punto 7.7)
|
|
3.6.
|
Solfuro di sodio, Na2S x H2O (x = 7-9)
|
|
3.6.1.
|
Soluzione di solfuro di sodio, c = 0,5 mol/l in glicerolo, β = 120 g/l (per x = 9) (cfr. osservazione al punto 7.8)
|
|
3.7.
|
Soluzione di fenolftaleina, c = 2 g/100 ml in etanolo (3.1)
|
|
3.9.
|
Fase mobile per HPLC: miscela di metanolo (3.3) e acqua, ad esempio 980 + 20 (v + v). Il rapporto esatto sarà determinato dalle caratteristiche della colonna utilizzata.
|
|
3.10.
|
Azoto, senza ossigeno
|
|
3.11.
|
Acetato di vitamina A tutto trans, purissimo, di attività certificata, ad esempio 2,80 × 106 UI/g
|
|
3.11.1.
|
Soluzione madre di acetato di vitamina A tutto trans: in un matraccio tarato da 100 ml pesare, con l'approssimazione di 0,1 mg, 50 mg di acetato di vitamina A (3.11). Sciogliere in 2-propanolo (3.8) e portare a volume con lo stesso solvente. La concentrazione nominale di questa soluzione è 1 400 UI di vitamina A/ml. Il contenuto esatto deve essere determinato conformemente al punto 5.6.3.1.
|
|
3.12.
|
Palmitato di vitamina A tutto trans, purissimo, di attività certificata, ad esempio 1,80 × 106UI/g
|
|
3.12.1.
|
Soluzione madre di palmitato di vitamina A tutto trans: in un matraccio tarato da 100 ml pesare, con l'approssimazione di 0,1 mg, 80 mg di palmitato di vitamina A (3.12). Sciogliere in 2-propanolo (3.8) e portare a volume con lo stesso solvente. La concentrazione nominale di questa soluzione è 1 400 UI di vitamina A/ml. Il contenuto esatto deve essere determinato conformemente al punto 5.6.3.2.
|
|
3.13.
|
2,6-di-tert-butil-4-metilfenolo (BHT) (cfr. osservazioni al punto 7.5).
|
4. Apparecchiatura
|
4.1.
|
Evaporatore rotante sotto vuoto
|
|
4.2.
|
Vetreria in vetro ambra
|
|
4.2.1.
|
Matracci a fondo piatto o beute, da 500 ml, con base di vetro smerigliato
|
|
4.2.2.
|
Matracci tarati con tappi di vetro smerigliato, a collo piccolo, da 10, 25, 100 e 500 ml
|
|
4.2.3.
|
Imbuti separatori, conici, da 1 000 ml, con tappi di vetro smerigliato
|
|
4.2.4.
|
Matracci a pera, da 250 ml, con base di vetro smerigliato
|
|
4.3.
|
Refrigerante Allihn, lunghezza tubo di raffreddamento 300 mm, con giunzione di vetro smerigliato e con adattatore per tubo di alimentazione gas
|
|
4.4.
|
Carta filtro pieghettata per separazione di fase; diametro 185 mm (ad esempio Schleicher & Schuell 597 HY 1/2)
|
|
4.5.
|
Apparecchiatura HPLC, con dispositivo di iniezione
|
|
4.5.1.
|
Colonna per cromatografia liquida, 250 mm × 4 mm, C18, particelle di riempimento 5 o 10 μm, o equivalente (criterio di prestazione: un unico picco per tutti gli isomeri di retinolo in condizioni HPLC)
|
|
4.5.2.
|
Rivelatore UV o a fluorescenza, con regolazione lunghezza d'onda variabile
|
|
4.6.
|
Spettrofotometro con celle al quarzo da 10 mm
|
|
4.7.
|
Bagnomaria con agitatore magnetico
|
|
4.8.
|
Il dispositivo di estrazione (cfr. figura 1) è costituito da:
|
|
4.8.1.
|
un cilindro di vetro della capacità di 1 litro, con collo e tappo di vetro smerigliato
|
|
4.8.2.
|
un insert di vetro smerigliato con braccio laterale e tubo regolabile che attraversa la parte centrale. Questo tubo ha l'estremità inferiore a U e un beccuccio all'estremità opposta, in modo che lo strato liquido superiore nel cilindro possa essere trasferito in un imbuto separatore.
|
5. Procedimento
|
Nota:
|
La vitamina A è sensibile alla luce ultravioletta e all'ossidazione. Tutte le operazioni sono effettuate in assenza di luce (utilizzando vetreria di vetro ambra o vetreria protetta con foglio di alluminio) e di ossigeno (eliminato con un getto di azoto). Durante l'estrazione, l'aria al di sopra del liquido è sostituita con azoto (evitare una pressione eccessiva alzando ogni tanto il tappo).
|
5.1. Preparazione del campione
Frantumare il campione affinché passi attraverso un vaglio da 1 mm, evitando la produzione di calore. La frantumazione deve essere effettuata immediatamente prima della pesatura e della saponificazione, altrimenti si possono verificare perdite di vitamina A.
5.2. Saponificazione
A seconda del contenuto di vitamina A, pesare, con l'approssimazione di 1 mg, da 2 a 25 g di campione in un matraccio da 500 ml a fondo piatto o conico (4.2.1). Aggiungere, rimestando ogni volta, 130 ml di etanolo (3.1), circa 100 mg di BHT (3.13), 2 ml di soluzione di ascorbato di sodio (3.5) e 2 ml di soluzione di solfuro di sodio (3.6). Adattare un refrigerante (4.3) al matraccio e immergere quest'ultimo in un bagno d'acqua con agitatore magnetico (4.7). Portare a ebollizione e lasciar rifluire per 5 minuti. Aggiungere quindi 25 ml di soluzione di idrossido di potassio (3.4) attraverso il refrigerante (4.3) e lasciar rifluire per altri 25 minuti, sempre agitando sotto una debole corrente di azoto. Risciacquare il refrigerante con circa 20 ml di acqua e lasciare raffreddare il contenuto del matraccio a temperatura ambiente.
5.3. Estrazione
Trasferire quantitativamente per decantazione la soluzione di saponificazione risciacquando con un volume totale di 250 ml di acqua in un imbuto separatore da 1 000 ml (4.2.3) o nel dispositivo di estrazione (4.8). Risciacquare successivamente il matraccio di saponificazione con 25 ml di etanolo (3.1) e 100 ml di etere di petrolio (3.2) e trasferire il liquido di lavaggio nell'imbuto separatore o nel dispositivo di estrazione. Il rapporto di acqua ed etanolo nelle soluzioni combinate deve essere di circa 2:1. Agitare energicamente per 2 minuti e lasciare riposare per 2 minuti.
5.3.1. Estrazione con imbuto separatore (4.2.3)
Quando gli strati sono separati (cfr. osservazione al punto 7.3) trasferire lo strato di etere di petrolio in un altro separatore (4.2.3). Ripetere questa estrazione due volte con 100 ml di etere di petrolio (3.2), poi ancora due volte con 50 ml di etere di petrolio (3.2).
Lavare due volte gli estratti combinati nell'imbuto separatore rimestando delicatamente (per evitare la formazione di emulsioni) con volumi di 100 ml di acqua e quindi agitando ripetutamente con altri volumi di 100 ml di acqua, finché l'acqua risulti incolore con l'aggiunta di soluzione di fenolftaleina (3.7) (in genere sono sufficienti quattro successivi lavaggi). Filtrare l'estratto lavato attraverso un filtro asciutto per separazione di fase (4.4) per rimuovere eventuale acqua residua e trasferire in un matraccio tarato da 500 ml (4.2.2). Risciacquare l'imbuto separatore ed il filtro con 50 ml di etere di petrolio (3.2), portare a volume con etere di petrolio (3.2) e mescolare bene.
5.3.2. Estrazione con apparecchiatura per estrazione (4.8)
Quando gli strati si sono separati (cfr. osservazione al punto 7.3) sostituire il tappo del cilindro di vetro (4.8.1) con l'insert di vetro smerigliato (4.8.2) e disporre l'estremità inferiore a U del tubo regolabile in modo che risulti appena al di sopra del livello dell'interfaccia. Esercitando una pressione con getto d'azoto nel braccio laterale, trasferire lo strato superiore di etere di petrolio in un imbuto separatore da 1 000 ml (4.2.3). Aggiungere 100 ml di etere di petrolio (3.2) nel cilindro di vetro, tappare e scuotere bene. Lasciare che gli strati si separino e trasferire lo strato superiore nell'imbuto separatore come prima. Ripetere la procedura di estrazione con altri 100 ml di etere di petrolio (3.2), e altre due volte con frazioni di 50 ml di etere di petrolio (3.2); aggiungere gli strati di etere di petrolio nell'imbuto separatore.
Lavare gli estratti di etere di petrolio combinati come descritto al punto 5.3.1 e procedere come ivi indicato.
5.4. Preparazione della soluzione del campione per HPLC
Pipettare un'aliquota della soluzione di etere di petrolio (risultante dalla procedura di cui ai punti 5.3.1 o 5.3.2) in un matraccio a pera da 250 ml (4.2.4). Far evaporare il solvente quasi interamente nell'evaporatore rotante (4.1), a pressione ridotta, a una temperatura del bagno non superiore a 40 oC. Ripristinare la pressione atmosferica facendovi fluire azoto (3.10) e togliere il matraccio dall'evaporatore. Togliere il solvente rimanente con un flusso di azoto (3.10) e disciogliere immediatamente il residuo in un volume noto (10-100 ml) di metanolo (3.3) (la concentrazione di vitamina A deve essere dell'ordine di 5 UI/ml-30 UI/ml).
5.5. Determinazione HPLC
La vitamina A è separata su una colonna C18 a fase inversa (4.5.1) e la concentrazione è misurata mediante rivelatore UV (325 nm) o a fluorescenza (eccitazione: 325 nm, emissione: 475 nm) (4.5.2).
Iniettare un'aliquota (ad esempio 20 μl) della soluzione di metanolo ottenuta conformemente al punto 5.4 ed eluire con la fase mobile (3.9). Calcolare l'altezza (area) media dei picchi di diverse iniezioni della stessa soluzione del campione e le altezze medie (aree) dei picchi di diverse iniezioni di soluzioni di taratura (5.6.2).
Condizioni HPLC
Le seguenti condizioni sono proposte a titolo indicativo; è possibile operare in condizioni diverse purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida (4.5.1):
|
250 mm × 4 mm, C18, particelle di riempimento 5 o 10 μm, o equivalente
|
|
Fase mobile (3.9):
|
miscela di metanolo (3.3) e acqua, ad esempio, 980 + 20 (v + v).
|
|
Velocità di efflusso:
|
1-2 ml/min
|
|
Rivelatore (4.5.2):
|
rivelatore UV (325 nm) o rivelatore a fluorescenza
(eccitazione: 325 nm/emissione: 475 nm)
|
5.6. Taratura
5.6.1. Preparazione delle soluzioni madre di lavoro
Pipettare 20 ml della soluzione madre di acetato di vitamina A (3.11.1) o 20 ml della soluzione madre di palmitato di vitamina A (3.12.1) in un matraccio a fondo piatto o in una beuta da 500 ml (4.2.1) e idrolizzare come indicato al punto 5.2, ma senza aggiunta di BHT. Successivamente, estrarre con etere di petrolio (3.2) secondo le indicazioni di cui al punto 5.3 e portare a 500 ml con etere di petrolio (3.2). Fare evaporare 100 ml di questo estratto nell'evaporatore rotante (cfr. punto 5.4) sin quasi all'essiccazione, togliere il solvente residuo con una corrente di azoto (3.10) e ridisciogliere il residuo in 10,0 ml di metanolo (3.3). La concentrazione nominale di questa soluzione è 560 UI di vitamina A/ml. Il contenuto esatto deve essere determinato conformemente al punto 5.6.3.3. La soluzione madre di lavoro deve essere preparata al momento, poco prima dell'uso.
Pipettare 2,0 ml di questa soluzione madre di lavoro in un matraccio tarato da 20 ml, portare a volume con metanolo (3.3) e mescolare. La concentrazione nominale di questa soluzione madre di lavoro diluita è 56 UI di vitamina A/ml.
5.6.2. Preparazione delle soluzioni di taratura e della curva di taratura
In una serie di matracci tarati da 20 ml trasferire 1,0 ml, 2,0 ml, 5,0 ml e 10,0 ml di soluzione madre di lavoro diluita; portare a volume con metanolo (3.3) e mescolare. Le concentrazioni nominali di queste soluzioni sono 2,8, 5,6, 14,0 e 28,0 UI di vitamina A/ml.
Iniettare più volte 20 μl di ogni soluzione di taratura e determinare le altezze medie dei picchi (aree). Sulla base delle altezze medie (aree) dei picchi tracciare una curva di taratura tenendo presente i risultati del controllo UV (5.6.3.3).
5.6.3. Standardizzazione UV delle soluzioni madre
5.6.3.1.
Soluzione madre di acetato di vitamina A
Pipettare 2,0 ml della soluzione madre di acetato di vitamina A (3.11.1) in un matraccio tarato da 50 ml (4.2.2) e portare a volume con 2-propanolo (3.8). La concentrazione nominale di questa soluzione è di 56 UI di vitamina A/ml. Pipettare 3,0 ml di questa soluzione di acetato di vitamina A diluita in un matraccio tarato da 25 ml e portare a volume con 2-propanolo (3.8). La concentrazione nominale di questa soluzione è di 6,72 UI di vitamina A/ml. Misurare lo spettro UV di questa soluzione su 2-propanolo (3.8) nello spettrofotometro (4.6) tra 300 nm e 400 nm. Il massimo di estinzione deve situarsi tra 325 nm e 327 nm.
Calcolo del contenuto di vitamina A:
UI di vitamina A/ml = E326 × 19,0
( per l'acetato di vitamina A = 1 530 a 326 nm in 2-propanolo)
per l'acetato di vitamina A = 1 530 a 326 nm in 2-propanolo)
5.6.3.2.
Soluzione madre di palmitato di vitamina A
Pipettare 2,0 ml della soluzione madre di palmitato di vitamina A (3.12.1) in un matraccio tarato da 50 ml (4.2.2) e portare a volume con 2-propanolo (3.8). La concentrazione nominale di questa soluzione è di 56 UI di vitamina A/ml. Pipettare 3,0 ml di questa soluzione di palmitato di vitamina A diluita in un matraccio tarato da 25 ml e portare a volume con 2-propanolo (3.8). La concentrazione nominale di questa soluzione è di 6,72 UI di vitamina A/ml. Misurare lo spettro UV di questa soluzione su 2-propanolo (3.8) nello spettrofotometro (4.6) tra 300 nm e 400 nm. Il massimo di estinzione deve situarsi tra 325 nm e 327 nm.
Calcolo del contenuto di vitamina A:
UI di vitamina A/ml = E326 × 19,0
( per il palmitato di vitamina A = 957 a 326 nm in 2-propanolo)
per il palmitato di vitamina A = 957 a 326 nm in 2-propanolo)
5.6.3.3.
Soluzione standard di lavoro di vitamina A
Pipettare 3,0 ml della soluzione standard di lavoro di vitamina A non diluita, preparata secondo quanto indicato al punto 5.6.1, in un matraccio tarato da 50 ml (4.2.2) e portare a volume con 2-propanolo (3.8). Pipettare 5,0 ml di questa soluzione in un matraccio tarato da 25 ml e portare a volume con 2-propanolo (3.8). La concentrazione nominale di questa soluzione è di 6,72 UI di vitamina A/ml. Misurare lo spettro UV di questa soluzione su 2-propanolo (3.8) nello spettrofotometro (4.6) tra 300 nm e 400 nm. Il massimo di estinzione deve situarsi tra 325 nm e 327 nm.
Calcolo del contenuto di vitamina A:
UI di vitamina A/ml = E325 × 18,3
( per l'alcole di vitamina A = 1 821 a 325 nm in 2-propanolo)
per l'alcole di vitamina A = 1 821 a 325 nm in 2-propanolo)
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in UI/ml in base all'altezza (area) media dei picchi della vitamina A, per riferimento alla curva di taratura (5.6.2).
Il contenuto w di vitamina A (in UI/kg) del campione è dato dalla seguente formula:
 [UI/kg]
[UI/kg]
dove
|
c
|
=
|
concentrazione di vitamina A nella soluzione del campione (5.4), in UI/ml
|
|
V1
|
=
|
volume della soluzione del campione (5.4), in ml
|
|
V2
|
=
|
volume dell'aliquota prelevata come indicato al punto 5.4, in ml
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Osservazioni
|
7.1.
|
Per i campioni con scarsa concentrazione di vitamina A può essere utile combinare gli estratti di etere di petrolio delle due saponificazioni (quantità pesata: 25 g) con una soluzione del campione, per la determinazione HPLC.
|
|
7.2.
|
Il campione prelevato per l'analisi non contiene più di 2 g di sostanze grasse.
|
|
7.3.
|
Se non ha luogo la separazione di fase, aggiungere circa 10 ml di etanolo (3.1) affinché l'emulsione si rompa.
|
|
7.4.
|
Con olio di fegato di merluzzo ed altri grassi puri, il tempo di saponificazione è portato a 45-60 minuti.
|
|
7.5.
|
In sostituzione del BHT si può utilizzare idrochinone.
|
|
7.6.
|
Utilizzando una colonna a fase normale, è possibile separare gli isomeri del retinolo. In questo caso, tuttavia, a fini di calcolo vanno sommate le altezze (aree) dei picchi degli isomeri tutto trans e cis.
|
|
7.7.
|
In sostituzione della soluzione di ascorbato di sodio si possono utilizzare circa 150 mg di acido ascorbico.
|
|
7.8.
|
La soluzione di solfuro di sodio può essere sostituita da circa 50 mg di EDTA.
|
|
7.9.
|
In caso di analisi della vitamina A negli alimenti d'allattamento, va prestata particolare attenzione
|
—
|
alla saponificazione (5.2): a causa della quantità di grasso presente nel campione, potrebbe rivelarsi necessario aumentare la soluzione di idrossido di potassio (3.4),
|
|
—
|
all'estrazione (5.3): a causa della presenza di emulsioni, potrebbe essere necessario adeguare il rapporto 2:1 acqua/etanolo.
|
Per verificare se il metodo di analisi applicato consente di ottenere risultati affidabili relativi a questa matrice specifica (alimenti d'allattamento), è effettuata una prova di recupero su un campione supplementare. Se il tasso di recupero è inferiore all'80 %, il risultato dell'analisi va corretto per il fattore di recupero.
|
8. Ripetibilità
La differenza fra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare il 15 % del risultato più elevato.
9. Risultati di uno studio effettuato in cooperazione tra vari laboratori
(1)
|
|
Premiscela
|
Alimento premiscelato
|
Concentrato minerale
|
Concentrato proteico
|
Alimento per suinetti
|
|
L
|
13
|
12
|
13
|
12
|
13
|
|
n
|
48
|
45
|
47
|
46
|
49
|
|
media [UI/kg]
|
17,02 × 106
|
1,21 × 106
|
537 100
|
151 800
|
18 070
|
|
sr [UI/kg]
|
0,51 × 106
|
0,039 × 106
|
22 080
|
12 280
|
682
|
|
r [UI/kg]
|
1,43 × 106
|
0,109 × 106
|
61 824
|
34 384
|
1 910
|
|
CVr [%]
|
3,0
|
3,5
|
4,1
|
8,1
|
3,8
|
|
sR [UI/kg]
|
1,36 × 106
|
0,069 × 106
|
46 300
|
23 060
|
3 614
|
|
R [UI/kg]
|
3,81 × 106
|
0,193 × 106
|
129 640
|
64 568
|
10 119
|
|
CVR [%]
|
8,0
|
6,2
|
8,6
|
15
|
20
|
|
L
|
=
|
numero di laboratori
|
|
n
|
=
|
numero di valori singoli
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
sR
|
=
|
deviazione standard della riproducibilità
|
|
r
|
=
|
ripetibilità
|
|
R
|
=
|
riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità
|

B. DETERMINAZIONE DELLA VITAMINA E
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di vitamina E negli alimenti per animali e nelle premiscele. Il contenuto di vitamina E è espresso in mg di acetato di DL-α-tocoferolo per kg. 1 mg di acetato di DL-α-tocoferolo corrisponde a 0,91 mg di DL-α-tocoferolo (vitamina E).
Il limite di quantificazione è di 2 mg di vitamina E/kg. Tale limite è ottenibile unicamente mediante rivelatore a fluorescenza. Utilizzando un rivelatore UV il limite di quantificazione è di 10 mg/kg.
2. Principio
Il campione è idrolizzato con una soluzione etanolica di idrossido di potassio e la vitamina E è estratta in etere di petrolio. Il solvente viene rimosso per evaporazione e il residuo è disciolto in metanolo e, se necessario, diluito alla concentrazione richiesta. Il contenuto di vitamina E viene determinato per cromatografia liquida ad alta prestazione a fase inversa (RP-HPLC) utilizzando un rivelatore UV o a fluorescenza.
3. Reattivi
|
3.2.
|
Etere di petrolio, intervallo di ebollizione: 40-60 oC
|
|
3.4.
|
Soluzione di idrossido di potassio, c = 50 g/100 ml
|
|
3.5.
|
Soluzione di ascorbato di sodio, c = 10 g/100 ml (cfr. osservazione al punto 7.7)
|
|
3.6.
|
Solfuro di sodio, Na2S × H2O (x = 7-9)
|
|
3.6.1.
|
Soluzione di solfuro di sodio, c = 0,5 mol/l in glicerolo, β = 120 g/l (per x = 9) (cfr. osservazione al punto 7.8)
|
|
3.7.
|
Soluzione di fenolftaleina, c = 2 g/100 ml in etanolo (3.1)
|
|
3.8.
|
Fase mobile per HPLC: miscela di metanolo (3.3) e acqua, ad esempio 980 + 20 (v + v). Il rapporto esatto sarà determinato dalle caratteristiche della colonna utilizzata.
|
|
3.9.
|
Azoto, senza ossigeno
|
|
3.10.
|
Acetato di DL-α-tocoferolo, purissimo, di attività certificata
|
|
3.10.1.
|
Soluzione madre di acetato di DL-α-tocoferolo: in un matraccio tarato da 100 ml, pesare, con l'approssimazione di 0,1 mg, 100 mg di acetato di DL-α-tocoferolo (3.10). Sciogliere in etanolo (3.1) e portare a volume con lo stesso solvente. 1 ml di questa soluzione contiene 1 mg di acetato di DL-α-tocoferolo. (per il controllo UV, cfr. punto 5.6.1.3; per la stabilizzazione cfr. osservazioni al punto 7.4).
|
|
3.11.
|
DL-α-tocoferolo, purissimo, di attività certificata
|
|
3.11.1.
|
Soluzione madre di acetato di DL-α-tocoferolo in un matraccio tarato da 100 ml, pesare, con l'approssimazione di 0,1 mg, 100 mg di DL-α-tocoferolo (3.11). Sciogliere in etanolo (3.1) e portare a volume con lo stesso solvente. 1 ml di questa soluzione contiene 1 mg di DL-α-tocoferolo. (per il controllo UV, cfr. punto 5.6.2.3; per la stabilizzazione cfr. osservazioni al punto 7.4).
|
|
3.12.
|
2,6-di-tert-butil-4-metilfenolo (BHT) (cfr. osservazione al punto 7.5).
|
4. Apparecchiatura
|
4.1.
|
Evaporatore rotante a film
|
|
4.2.
|
Vetreria in vetro ambra
|
|
4.2.1.
|
Matracci a fondo piatto o beute, da 500 ml, con base di vetro smerigliato
|
|
4.2.2.
|
Matracci tarati con tappi di vetro smerigliato, a collo piccolo, da 10, 25, 100 e 500 ml
|
|
4.2.3.
|
Imbuti separatori, conici, da 1 000 ml, con tappi di vetro smerigliato
|
|
4.2.4.
|
Matracci a pera, da 250 ml, con base di vetro smerigliato
|
|
4.3.
|
Refrigerante Allihn, lunghezza tubo di raffreddamento 300 mm, con giunzione di vetro smerigliato, con adattatore per tubo di alimentazione gas
|
|
4.4.
|
Carta filtro pieghettata per separazione di fase; diametro 185 mm (ad esempio Schleicher & Schuell 597 HY 1/2)
|
|
4.5.
|
Apparecchiatura HPLC, con dispositivo di iniezione
|
|
4.5.1.
|
Colonna per cromatografia liquida, 250 mm × 4 mm, C18, particelle di riempimento 5 o 10 μm, o equivalente
|
|
4.5.2.
|
Rivelatore UV o a fluorescenza, con regolazione lunghezza d'onda variabile
|
|
4.6.
|
Spettrofotometro con celle al quarzo da 10 mm
|
|
4.7.
|
Bagnomaria con agitatore magnetico
|
|
4.8.
|
Il dispositivo di estrazione (cfr. figura 1) è costituito da:
|
|
4.8.1.
|
un cilindro di vetro della capacità di 1 litro, con collo e tappo di vetro smerigliato
|
|
4.8.2.
|
un insert di vetro smerigliato con braccio laterale e tubo regolabile che attraversa la parte centrale. Tale tubo ha l'estremità inferiore a U e un beccuccio all'estremità opposta, in modo che lo strato liquido superiore nel cilindro possa essere trasferito in un imbuto separatore.
|
5. Procedimento
|
Nota:
|
La vitamina E è sensibile alla luce ultravioletta e all'ossidazione. Tutte le operazioni sono effettuate in assenza di luce (utilizzando vetreria di vetro ambra o vetreria protetta con foglio di alluminio) e di ossigeno (eliminato con un getto di azoto). Durante l'estrazione, l'aria al di sopra del liquido è sostituita con azoto (evitare una pressione eccessiva alzando ogni tanto il tappo).
|
5.1. Preparazione del campione
Frantumare il campione affinché passi attraverso un vaglio da 1 mm, evitando la produzione di calore. La frantumazione deve essere effettuata immediatamente prima della pesatura e della saponificazione, altrimenti si possono verificare perdite di vitamina E.
5.2. Saponificazione
A seconda del contenuto di vitamina E, pesare, con l'approssimazione di 0,01 g, da 2 g a 25 g di campione in un matraccio da 500 ml a fondo piatto o conico (4.2.1). Aggiungere successivamente, sempre rimestando, 130 ml di etanolo (3.1), circa 100 mg di BHT (3.13), 2 ml di soluzione di ascorbato di sodio (3.5) e 2 ml di soluzione di solfuro di sodio (3.6). Adattare un refrigerante (4.3) al matraccio e immergere quest'ultimo in un bagno d'acqua con agitatore magnetico (4.7). Portare a ebollizione e lasciare rifluire per 5 minuti. Aggiungere 25 ml di soluzione di idrossido di potassio (3.4) attraverso il refrigerante (4.3) e lasciare rifluire per altri 25 minuti, sempre agitando sotto una debole corrente di azoto. Risciacquare il refrigerante con circa 20 ml di acqua e lasciare raffreddare il contenuto del matraccio a temperatura ambiente.
5.3. Estrazione
Trasferire quantitativamente per decantazione la soluzione di saponificazione risciacquando con un volume totale di 250 ml di acqua in un imbuto separatore da 1 000 ml (4.2.3) o nel dispositivo di estrazione (4.8). Risciacquare successivamente il matraccio di saponificazione con 25 ml di etanolo (3.1) e 100 ml di etere di petrolio (3.2) e trasferire il liquido di lavaggio nell'imbuto separatore o nel dispositivo di estrazione. Il rapporto di acqua ed etanolo nelle soluzioni combinate deve essere di circa 2:1. Agitare energicamente per 2 minuti e lasciare riposare per 2 minuti.
5.3.1. Estrazione con imbuto separatore (4.2.3)
Quando gli strati sono separati (cfr. osservazione al punto 7.3) trasferire lo strato di etere di petrolio in un altro separatore (4.2.3). Ripetere questa estrazione due volte con 100 ml di etere di petrolio (3.2), poi ancora due volte con 50 ml di etere di petrolio (3.2).
Lavare due volte gli estratti combinati nell'imbuto separatore rimestando delicatamente (per evitare la formazione di emulsioni) con volumi di 100 ml di acqua e quindi agitando ripetutamente con altri volumi di 100 ml di acqua finché l'acqua risulti incolore con l'aggiunta di soluzione di fenolftaleina (3.7) (in genere sono sufficienti quattro successivi lavaggi). Filtrare l'estratto lavato attraverso un filtro asciutto per separazione di fase (4.4) per rimuovere eventuale acqua residua e trasferire in un matraccio tarato da 500 ml (4.2.2). Risciacquare l'imbuto separatore e il filtro con 50 ml di etere di petrolio (3.2), portare a volume con etere di petrolio (3.2) e mescolare bene.
5.3.2. Estrazione con apparecchiatura per estrazione (4.8)
Quando gli strati si sono separati (cfr. osservazione al punto 7.3) sostituire il tappo del cilindro di vetro (4.8.1) con l'insert di vetro smerigliato (4.8.2) e disporre la terminazione inferiore a U del tubo adattabile in modo che risulti appena al di sopra del livello dell'interfaccia. Esercitando una pressione con getto d'azoto nel braccio laterale, trasferire lo strato superiore di etere di petrolio in un imbuto separatore da 1 000 ml (4.2.3). Aggiungere 100 ml di etere di petrolio (3.2) nel cilindro di vetro, tappare e scuotere bene. Lasciare che gli strati si separino e trasferire lo strato superiore nell'imbuto separatore come prima. Ripetere la procedura di estrazione con altri 100 ml di etere di petrolio (3.2), poi ancora due volte con 50 ml di etere di petrolio (3.2); aggiungere gli strati di etere di petrolio nell'imbuto separatore.
Lavare gli estratti di etere di petrolio combinati come descritto al punto 5.3.1 e procedere come ivi indicato.
5.4. Preparazione della soluzione del campione per HPLC
Pipettare un'aliquota della soluzione di etere di petrolio (risultante dalla procedura di cui ai punti 5.3.1 o 5.3.2) in un matraccio a pera da 250 ml (4.2.4). Far evaporare il solvente sin quasi all'essiccazione nell'evaporatore rotante (4.1), a pressione ridotta, ad una temperatura del bagno non superiore a 40 oC. Ripristinare la pressione atmosferica facendovi fluire azoto (3.9) e togliere il matraccio dall'evaporatore. Togliere il solvente rimanente con un flusso di azoto (3.9) e disciogliere il residuo immediatamente in un volume noto (10-100 ml) di metanolo (3.3) (la concentrazione di DL-α-tocoferolo deve essere dell'ordine di 5 μg/ml a 30 μg/ml).
5.5. Determinazione HPLC
La vitamina E viene separata su una colonna C18 a fase inversa (4.5.1) e la concentrazione è misurata mediante rivelatore a fluorescenza (eccitazione: 295 nm, emissione: 330 nm) (4.5.2) o un rivelatore UV (292 nm) (4.5.2).
Iniettare un'aliquota (ad esempio 20 μl) della soluzione di metanolo ottenuta conformemente al punto 5.4 ed eluire con la fase mobile (3.8). Calcolare le altezze medie (aree) dei picchi di diverse iniezioni della stessa soluzione del campione e le altezze medie dei picchi (aree) di diverse iniezioni delle soluzioni di taratura (5.6.2).
Condizioni HPLC
Le seguenti condizioni vengono proposte a titolo indicativo; è possibile operare in condizioni diverse purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida (4.5.1):
|
250 mm × 4 mm, C18, particelle di riempimento 5 μm o 10 μm o equivalente
|
|
Fase mobile (3.8):
|
miscela metanolo (3.3) e acqua, ad esempio, 980 + 20 (v + v).
|
|
Velocità di efflusso:
|
1-2 ml/min
|
|
Rivelatore (4.5.2):
|
rivelatore a fluorescenza
(eccitazione: 295 nm/emissione: 330 nm) o rivelatore UV (292 nm)
|
5.6. Taratura (acetato di DL-α-tocoferolo o DL-α-tocoferolo)
5.6.1. Standard di acetato di DL-α-tocoferolo
5.6.1.1.
Preparazione della soluzione standard di lavoro
Pipettare 25 ml della soluzione madre di acetato di DL-α-tocoferolo (3.10.1) in un matraccio a fondo piatto o in una beuta da 500 ml (4.2.1) e idrolizzare come indicato al punto 5.2. Successivamente, estrarre con etere di petrolio (3.2) secondo quanto indicato al punto 5.3 e portare a 500 ml con etere di petrolio. Far evaporare 25 ml di questo estratto nell'evaporatore (cfr. punto 5.4) sin quasi all'essiccazione, togliere il solvente residuo con una corrente di azoto (3.9) e ridisciogliere il residuo in 25,0 ml di metanolo (3.3). La concentrazione nominale di questa soluzione è di 45,5 μg di DL-α-tocoferolo/ml, equivalente a 50 μg di acetato di DL-α-tocoferolo/ml. La soluzione standard di lavoro deve essere preparata al momento, poco prima dell'uso.
5.6.1.2.
Preparazione delle soluzioni di taratura e della curva di taratura
In una serie di matracci tarati da 20 ml trasferire 1,0, 2,0, 4,0 e 10,0 ml della soluzione standard di lavoro, portare a volume con metanolo (3.3) e miscelare. Le concentrazioni nominali di queste soluzioni sono 2,5, 5,0, 10,0 e 25,0 μg/ml di acetato di DL-α-tocoferolo e cioè 2,28, 4,55, 9,10 e 22,8 μg/ml di DL-α-tocoferolo.
Iniettare più volte 20 μl di ogni soluzione di taratura e determinare le altezze medie (aree) dei picchi. Sulla base delle altezze medie (aree) dei picchi tracciare una curva di taratura.
5.6.1.3.
Standardizzazione UV della soluzione madre di acetato di DL-α-tocoferolo (3.10.1)
Diluire 5,0 ml della soluzione madre di acetato di DL-α-tocoferolo (3.10.1) sino a 25,0 ml con etanolo e misurare lo spettro UV di questa soluzione su etanolo (3.1), nello spettrofotometro (4.6), tra 250 e 320 nm.
Il massimo di assorbimento si situa a 284 nm:
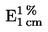 = 43,6 a 284 nm in etanolo
= 43,6 a 284 nm in etanolo
A questa diluizione, si deve ottenere un valore di estinzione compreso tra 0,84 e 0,88.
5.6.2. Standard di DL-α-tocoferolo
5.6.2.1.
Preparazione della soluzione standard di lavoro
Trasferire con pipetta 2 ml della soluzione madre di DL-α-tocoferolo (3.11.1) in un matraccio tarato da 50 ml, sciogliere in metanolo (3.3) e portare a volume con metanolo. La concentrazione nominale di questa soluzione è di 40 μg di DL-α-tocoferolo/ml, equivalente a 44,0 μg di acetato di DL-α-tocoferolo/ml. La soluzione standard di lavoro deve essere preparata al momento, poco prima dell'uso.
5.6.2.2.
Preparazione delle soluzioni di taratura e della curva di taratura
In una serie di matracci tarati da 20 ml trasferire 1,0, 2,0, 4,0 e 10,0 ml della soluzione standard di lavoro, portare a volume con metanolo (3.3) e miscelare. Le concentrazioni nominali di queste soluzioni sono 2,0, 4,0, 8,0 e 20,0 μg/ml di DL-α-tocoferolo e cioè 2,20, 4,40, 8,79 e 22,0 μg/ml di acetato di DL-α-tocoferolo.
Iniettare più volte 20 μl di ogni soluzione di taratura e determinare le altezze medie dei picchi (aree). Sulla base delle altezze medie (aree) dei picchi tracciare una curva di taratura.
5.6.2.3.
Standardizzazione UV della soluzione madre di DL-α-tocoferolo (3.11.1)
Diluire 2,0 ml della soluzione madre di DL-α-tocoferolo (3.11.1) sino a 25,0 ml con etanolo e misurare lo spettro UV di questa soluzione su etanolo (3.1) nello spettrofotometro (4.6) tra 250 nm e 320 nm. Il massimo di assorbimento si situa a 292 nm:
 = 75,8 a 292 nm in etanolo
= 75,8 a 292 nm in etanolo
A questa diluizione si dovrebbe ottenere un valore di estinzione di 0,6.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml (calcolata come acetato di α-tocoferolo) in base all'altezza (area) media dei picchi della vitamina E, per riferimento alla curva di taratura (5.6.1.2 o 5.6.2.2).
Il contenuto w di vitamina E (in mg/kg) del campione è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
c
|
=
|
concentrazione di vitamina E (come α-tocoferolo) nella soluzione del campione (5.4), in μg/ml
|
|
V1
|
=
|
volume della soluzione del campione (5.4), in ml
|
|
V2
|
=
|
volume dell'aliquota prelevata come indicato al punto 5.4, in ml
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in g.
|
7. Osservazioni
|
7.1.
|
Per i campioni con debole concentrazione di vitamina E può essere utile combinare gli estratti di etere di petrolio delle due saponificazioni (quantità pesata: 25 g) con una soluzione del campione, per la determinazione HPLC.
|
|
7.2.
|
Il peso del campione prelevato per l'analisi non contiene più di 2 g di sostanze grasse.
|
|
7.3.
|
Se non ha luogo la separazione di fase, aggiungere circa 10 ml di etanolo (3.1) affinché l'emulsione si rompa.
|
|
7.4.
|
Dopo la misurazione spettrofotometrica dell'acetato di DL-α-tocoferolo o della soluzione di DL-α-tocoferolo secondo il punto 5.6.1.3 o, rispettivamente, 5.6.2.3, aggiungere circa 10 mg di BHT (3.12) alla soluzione (3.10.1 o 3.10.2) e conservare questa soluzione in frigorifero (tempo massimo di conservazione: 4 settimane).
|
|
7.5.
|
In sostituzione del BHT si può utilizzare idrochinone.
|
|
7.6.
|
Utilizzando una colonna a fase normale, è possibile separare i tocoferoli α, β, γ e δ.
|
|
7.7.
|
La soluzione di ascorbato di sodio può essere sostituita da circa 150 mg di acido ascorbico.
|
|
7.8.
|
La soluzione di solfuro di sodio può essere sostituita da circa 50 mg di EDTA.
|
|
7.9.
|
L'acetato di vitamina E viene idrolizzato molto velocemente in condizioni alcaline ed è pertanto molto sensibile all'ossidazione, in particolare in presenza di oligoelementi quali il ferro e il rame. Nel caso della determinazione della vitamina E nelle premiscele a livelli superiori a 5 000 mg/kg può prodursi una degradazione della vitamina E. Si raccomanda pertanto a titolo di conferma l'applicazione di un metodo HPLC che includa la mineralizzazione enzimatica della formulazione della vitamina E senza la fase della saponificazione alcalina.
|
8. Ripetibilità
La differenza fra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare il 15 % del risultato più elevato.
9. Risultati di uno studio effettuato in cooperazione tra vari laboratori
(2)
|
|
Premiscela
|
Alimento premiscelato
|
Concentrato minerale
|
Concentrato proteico
|
Alimento per suinetti
|
|
L
|
12
|
12
|
12
|
12
|
12
|
|
n
|
48
|
48
|
48
|
48
|
48
|
|
media [mg/kg]
|
17 380
|
1 187
|
926
|
315
|
61,3
|
|
sr [mg/kg]
|
384
|
45,3
|
25,2
|
13,0
|
2,3
|
|
r [mg/kg]
|
1 075
|
126,8
|
70,6
|
36,4
|
6,4
|
|
CVr [%]
|
2,2
|
3,8
|
2,7
|
4,1
|
3,8
|
|
sR mg/kg]
|
830
|
65,0
|
55,5
|
18,9
|
7,8
|
|
R [mg/kg]
|
2 324
|
182,0
|
155,4
|
52,9
|
21,8
|
|
CVR [%]
|
4,8
|
5,5
|
6,0
|
6,0
|
12,7
|
|
L
|
=
|
numero di laboratori
|
|
n
|
=
|
numero di valori singoli
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
sR
|
=
|
deviazione standard della riproducibilità
|
|
r
|
=
|
ripetibilità
|
|
R
|
=
|
riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità
|
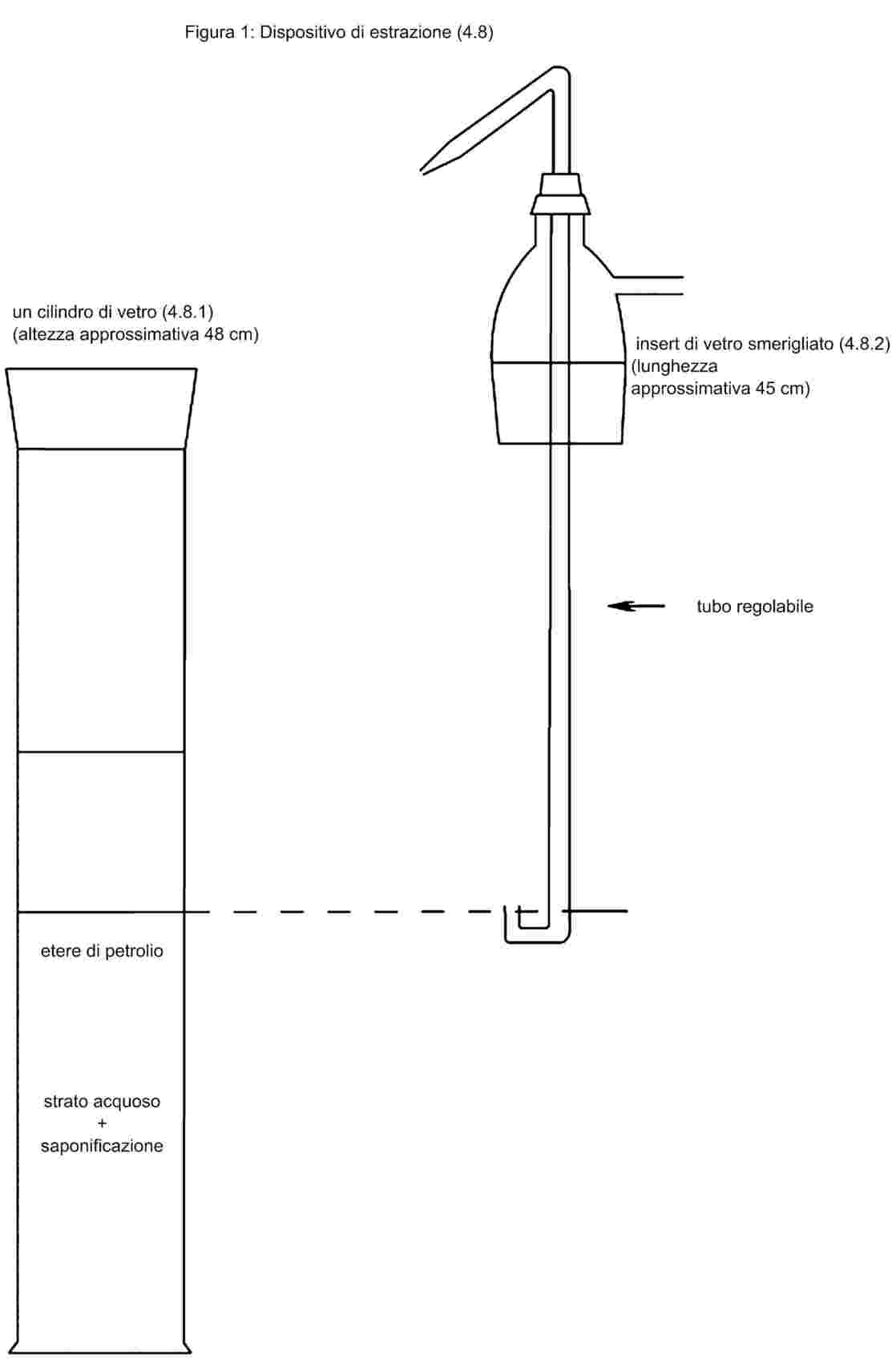
C. DETERMINAZIONE DEGLI OLIGOELEMENTI FERRO, RAME, MANGANESE E ZINCO
1. Finalità e campo d'applicazione
Questo metodo consente di determinare gli oligoelementi ferro, rame, manganese e zinco contenuti negli alimenti per animali. Le soglie di quantificazione dei suddetti elementi sono:
|
—
|
manganese (Mn): 20 mg/kg
|
2. Principio
Il campione viene disciolto in acido cloridrico dopo distruzione delle sostanze organiche eventualmente presenti. Gli elementi ferro, rame, manganese e zinco sono determinati, dopo opportuna diluizione, mediante spettrometria di assorbimento atomico.
3. Reattivi
Osservazioni preliminari
L'acqua utilizzata per la preparazione dei reattivi e delle soluzioni per l'analisi deve essere esente da cationi da determinare, preparata o per doppia distillazione dell'acqua in un apparecchio in borosilicato o in quarzo o per doppio passaggio su resine a scambio ionico.
I reattivi debbono essere di purezza almeno «analitica». L'assenza dell'elemento da determinare deve essere controllata mediante una prova in bianco. Se necessario, i reattivi dovranno essere ulteriormente purificati.
In luogo delle soluzioni standard appresso elencate è possibile impiegare soluzioni standard commerciali purché fornite di garanzia e controllate prima dell'uso.
|
3.1.
|
Acido cloridrico (d:1,19 g/ml).
|
|
3.2.
|
Acido cloridrico (6 mol/l).
|
|
3.3.
|
Acido cloridrico (0,5 mol/l).
|
|
3.4.
|
Acido fluoridrico al 38-40 % (v/v), con un contenuto di ferro (Fe) inferiore a 1 mg/l e un residuo d'evaporazione (espresso in solfati) inferiore a 10 mg/1.
|
|
3.5.
|
Acido solforico (d: 1,84 g/ml).
|
|
3.6.
|
Perossido di idrogeno a 100 volumi circa di ossigeno (30 % in peso).
|
|
3.7.
|
Soluzione standard di ferro (1 000 μg Fe/ml) preparata come segue o soluzione equivalente in commercio: sciogliere 1 g di fil di ferro in 200 ml di acido cloridrico 6 mol/l (3.2), aggiungere 16 ml di perossido d'idrogeno (3.6) e portare a 1 litro con acqua.
|
|
3.7.1.
|
Soluzione standard di ferro (100 μg Fe/ml): diluire un volume della soluzione standard (3.7) con 9 volumi d'acqua.
|
|
3.8.
|
Soluzione standard di rame (1 000 μg Cu/ml) preparata come segue o soluzione equivalente in commercio:
|
—
|
sciogliere 1 g di rame in polvere in 25 ml di acido cloridrico 6 mol/l (3.2), aggiungere 5 ml di perossido d'idrogeno (3.6) e portare a 1 litro con acqua.
|
|
|
3.8.1.
|
Soluzione standard di rame (10 μg Cu/ml): diluire un volume della soluzione standard (3.8) con 9 volumi d'acqua, poi ridiluire una parte della soluzione così ottenuta con 9 volumi di acqua.
|
|
3.9.
|
Soluzione standard di manganese (1 000 μg Mn/ml) preparata come segue o soluzione equivalente in commercio:
|
—
|
sciogliere 1 g di manganese in polvere in 25 ml di acido cloridrico 6 mol/l (3.2) e portare a 1 litro con acqua.
|
|
|
3.9.1.
|
Soluzione standard di rame (10 μg Mn/ml): diluire un volume della soluzione standard (3.9) con 9 volumi d'acqua, poi ridiluire una parte della soluzione così ottenuta con 9 volumi di acqua.
|
|
3.10.
|
Soluzione standard di zinco (1 000 μg Zn/ml) preparata come segue o soluzione equivalente in commercio:
|
—
|
sciogliere 1 g di zinco in lamine o in fogli in 25 ml di acido cloridrico 6 mol/l (3.2) e portare a 1 litro con acqua.
|
|
|
3.10.1.
|
Soluzione standard di zinco (10 μg Zn/ml): diluire un volume della soluzione standard (3.10) con 9 volumi d'acqua, poi ridiluire una parte della soluzione così ottenuta con 9 volumi di acqua.
|
|
3.11.
|
Soluzione di cloruro di lantanio: sciogliere 12 g di ossido di lantanio in 150 ml d'acqua, aggiungere 100 ml di acido cloridrico 6 mol/l (3.2) e portare a 1 litro con acqua.
|
4. Apparecchiatura
|
4.1.
|
Muffola a temperatura regolabile e preferibilmente controllata.
|
|
4.2.
|
Vetreria resistente in borosilicato; si raccomanda di riservarla esclusivamente alla determinazione degli oligoelementi.
|
|
4.3.
|
Spettrofotometro di assorbimento atomico, conforme ai requisiti del metodo per quanto riguarda la sensibilità e la precisione nell'intervallo delle misurazioni richieste.
|
5. Procedimento
(3)
5.1. Campioni contenenti sostanze organiche
5.1.1. Incenerimento e preparazione della soluzione da analizzare
(4)
|
5.1.1.1.
|
Porre 5-10 g di campione, pesato con l'approssimazione di 0,2 mg, in un crogiolo di quarzo o di platino [cfr. nota b)], essiccare in stufa a 105 oC e introdurre il crogiolo nel forno a muffola freddo (4.1). Chiudere il forno [cfr. nota c)] e innalzare progressivamente la temperatura fino a raggiungere 450-475 oC in 90 minuti circa. Mantenere questa temperatura per 4-16 ore (ad esempio tutta una notte) in modo da eliminare la materia carboniosa; aprire poi il forno e lasciar raffreddare [cfr. nota d)].
Umettare le ceneri con acqua e trasferirle in un becher da 250 ml. Lavare bene il crogiolo con complessivi 5 ml circa di acido cloridrico (3.1) e travasare lentamente e accuratamente quest'ultimo nel becher (si può avere una reazione violenta per formazione di CO2). Aggiungere goccia a goccia dell'acido cloridrico (3.1), agitando finché l'effervescenza sia terminata. Evaporare a secco agitando ogni tanto con un bacchetta di vetro.
Aggiungere al residuo 15 ml di acido cloridrico 6 mol/l (3.2) e successivamente circa 120 ml d'acqua. Rimescolare con la bacchetta di vetro, che va lasciata nel becher, e coprire quest'ultimo con un vetro di orologio. Portare il liquido a leggera ebollizione e mantenerla finché non si vedono più dissolversi le ceneri. Filtrare su carta filtro senza ceneri e raccogliere il filtrato in un matraccio tarato da 250 ml. Lavare il becher e il filtro prima con 5 ml di acido cloridrico 6 mol/l caldo (3.2), poi, per due volte, con acqua bollente. Portare a volume con acqua (la concentrazione dell'acido cloridrico è 0,5 mol/l).
|
|
5.1.1.2.
|
Se il residuo nel filtro è nero (presenza di carbone), rimetterlo nel forno a muffola e incenerire di nuovo a 450-475 oC. Questo incenerimento, che richiede solo alcune ore (da 3 a 5 ore), è completo allorché le ceneri appaiono bianche o quasi. Sciogliere di nuovo il residuo con circa 2 ml di acido cloridrico (3.1), evaporare a secco e aggiungere 5 ml di acido cloridrico 6 mol/l (3.2). Riscaldare la soluzione e filtrarla nel matraccio tarato e portare a volume con acqua (la concentrazione dell'acido cloridrico è 0,5 mol/l circa).
Note:
|
a)
|
Nella determinazione degli oligoelementi è importante vigilare sui rischi di contaminazione, in particolare quella da zinco, rame e ferro. L'apparecchiatura impiegata nella preparazione dei campioni deve quindi essere esente da tali metalli.
Per ridurre i rischi generali di contaminazione, lavorare in ambiente esente da polvere con apparecchiature scrupolosamente pulite e vetreria accuratamente lavata. La determinazione dello zinco è particolarmente sensibile a molti tipi di contaminazione, ad esempio quella dovuta alla vetreria, ai reattivi, alla polvere, ecc.
|
|
b)
|
Calcolare il peso del campione da incenerire in funzione del contenuto approssimativo di oligoelementi dell'alimento per animali in esame e della sensibilità dello spettrofotometro adoperato. Per taluni alimenti per animali a basso tenore di oligoelementi può essere necessario partire da un campione del peso di 10-20 g e limitare a soli 100 ml il volume della soluzione finale.
|
|
c)
|
L'incenerimento deve essere effettuato in forno chiuso, senza insufflazione d'aria o di ossigeno.
|
|
d)
|
La temperatura indicata dal pirometro non deve superare i 475 oC.
|
|
5.1.2. Determinazione spettrofotometrica
5.1.2.1.
Preparazione delle soluzioni di taratura
Per ciascun oligoelemento da dosare, preparare una serie di soluzioni di taratura a partire dalle soluzioni standard di lavoro di cui ai punti 3.7.1, 3.8.1, 3.9.1 e 3.10.1, in modo che la concentrazione dell'acido cloridrico in ciascuna di esse sia all'incirca 0,5 mol/l, e, nel caso del ferro, del manganese e dello zinco, la concentrazione del cloruro di lantanio corrisponda allo 0,1 % di La (p/v).
Le concentrazioni prescelte per gli oligoelementi devono trovarsi nell'intervallo di sensibilità dello spettrofotometro utilizzato. Le tabelle di seguito riportano, a titolo di esempio, alcuni tipi di composizione delle soluzioni di taratura; nondimeno a seconda del tipo e della sensibilità dello spettrofotometro impiegato potrebbe essere necessario scegliere altre concentrazioni.
Ferro
|
μg Fe/ml
|
0
|
0,5
|
1
|
2
|
3
|
4
|
5
|
|
ml di soluzione standard di lavoro (3.7.1) (1 ml = 100 μg Fe)
|
0
|
0,5
|
1
|
2
|
3
|
4
|
5
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml di soluzione di cloruro di lantanio (3.11); portare a 100 ml con acqua
|
Rame
|
μg Cu/ml
|
0
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
1,0
|
|
ml di soluzione standard di lavoro (3.7.1) (1 ml = 10 μg Cu)
|
0
|
1
|
2
|
4
|
6
|
8
|
10
|
|
ml HCl (3.2)
|
8
|
8
|
8
|
8
|
8
|
8
|
8
|
Manganese
|
μg Mn/ml
|
0
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
1,0
|
|
ml di soluzione standard di lavoro (3.7.1) (1 ml = 10 μg Mn)
|
0
|
1
|
2
|
4
|
6
|
8
|
10
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml di soluzione di cloruro di lantanio (3.11); portare a 100 ml con acqua
|
Zinco
|
μg Zn/ml
|
0
|
0,05
|
0,1
|
0,2
|
0,4
|
0,6
|
0,8
|
|
ml di soluzione standard di lavoro (3.7.1) (1 ml = 10 μg Zn)
|
0
|
0,5
|
1
|
2
|
4
|
6
|
8
|
|
ml HCl (3.2)
|
7
|
7
|
7
|
7
|
7
|
7
|
7
|
|
+ 10 ml di soluzione di cloruro di lantanio (3.11); portare a 100 ml con acqua
|
5.1.2.2.
Preparazione della soluzione da sottoporre ad analisi
Per la determinazione del rame, la soluzione preparata secondo quanto indicato al punto 5.1.1 può in linea generale essere utilizzata direttamente. Se è necessario portare la sua concentrazione nell'intervallo delle concentrazioni delle soluzioni di taratura, una sua aliquota può essere pipettata in un pallone tarato da 100 ml e portata a volume con acido cloridrico 0,5 mol/l (3.3).
Per la determinazione del ferro, del manganese e dello zinco, pipettare un'aliquota della soluzione preparata secondo quanto indicato al punto 5.1.1 in un pallone tarato da 100 ml; aggiungere 10 ml di soluzione di cloruro di lantanio (3.11) e portare a volume con acido cloridrico 0,5 mol/l (3.3) (cfr. anche punto 8, «Osservazione»).
5.1.2.3.
Prova in bianco
Effettuare una prova in bianco comprendente tutte le operazioni prescritte nel procedimento, escludendo la presenza del campione. La soluzione di taratura «0» non deve essere utilizzata come «bianco».
5.1.2.4.
Misurazione dell'assorbimento atomico
Misurare l'assorbimento delle soluzioni di taratura e della soluzione da analizzare, impiegando una fiamma ossidante aria-acetilene, alle lunghezze d'onda seguenti:
Ripetere ogni misurazione quattro volte.
5.2. Alimenti a base di minerali
In assenza di materiali organici, l'incenerimento preliminare è superfluo. Operare come descritto al punto 5.1.1.1, a partire dal secondo capoverso. L'evaporazione in presenza di acido fluoridrico può essere tralasciata.
6. Calcolo dei risultati
Tramite la curva di taratura calcolare la concentrazione degli oligoelementi nella soluzione in esame ed esprimere il risultato in mg d'oligoelemento per kg di campione (ppm).
7. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione dallo stesso analista non supera:
|
—
|
5 mg/kg, in valore assoluto, per i tenori di oligoelementi fino a 50 mg/kg,
|
|
—
|
il 10 % del risultato più elevato per i tenori superiori a 50 e fino a 100 mg/kg,
|
|
—
|
10 mg/kg, in valore assoluto, per i tenori superiori a 100 e fino a 200 mg/kg,
|
|
—
|
il 5 % del risultato più elevato per i tenori superiori a 200 mg/kg.
|
8. Osservazione
La presenza di notevoli quantità di fosfati può interferire nella determinazione del ferro, del manganese e dello zinco. Questa interferenza deve essere corretta aggiungendo soluzione di cloruro di lantanio (3.11). Tuttavia, se nel campione il rapporto di peso Ca + Mg/P risulta > 2, non è necessario aggiungere soluzione di cloruro di lantanio (3.11) alla soluzione da analizzare e alle soluzioni di taratura.
D. DETERMINAZIONE DELL'ALOFUGINONE
DL-trans-7-bromo-6-cloro-3-[3-(3-idrossi-2-piperidil)acetonil]chinazolin-4(3H)-one bromidrato
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di alofuginone negli alimenti per animali. Il limite di quantificazione è di 1 mg/kg.
2. Principio
Dopo essere stato trattato con acqua calda, l'alofuginone è estratto come base libera in acetato di etile e successivamente ripartito come cloridrato in soluzione acquosa acida. L'estratto è purificato mediante cromatografia a scambio ionico. Il tenore di alofuginone è determinato mediante cromatografia liquida ad alta prestazione (HPLC) a fase inversa, usando un rivelatore UV.
3. Reattivi
|
3.1.
|
Acetonitrile, di qualità HPLC.
|
|
3.2.
|
Resina amberlite XAD-2
|
|
3.5.
|
Acido acetico glaciale
|
|
3.6.
|
Alofuginone standard (DL-trans-7-bromo-6-cloro-3-[3-(3-idrossi-2-piperidil)acetonil]-chinazolin-4-(3H)-one bromidrato, E 764)
|
|
3.6.1.
|
Soluzione madre di alofuginone, 100 μg/ml
In un matraccio tarato da 500 ml pesare, con l'approssimazione di 0,1 mg, 50 mg di alofuginone (3.6), sciogliere in una soluzione tampone di acetato ammonico (3.18), portare a volume con la soluzione tampone e mescolare. Questa soluzione è stabile per tre settimane a 5 oC se conservata al buio.
|
|
3.6.2.
|
Soluzioni di taratura
In una serie di palloni graduati da 100 ml trasferire 1,0, 2,0, 3,0, 4,0 e 6,0 ml della soluzione madre standard (3.6.1). Portare a volume con la fase mobile (3.21) e agitare. Queste soluzioni hanno concentrazioni rispettivamente di 1,0, 2,0, 3,0, 4,0 e 6,0 μg/ml di alofuginone. Devono essere preparate al momento, poco prima dell'uso.
|
|
3.7.
|
Acido cloridico, (ρ20 circa 1,16 g/ml).
|
|
3.13.
|
EDTA (acido etilendiamminotetracetico, sale bisodico)
|
|
3.14.
|
Acqua, di qualità HPLC
|
|
3.15.
|
Soluzione di carbonato di sodio, c = 10 g/100 ml
|
|
3.16.
|
Soluzione di carbonato di sodio saturata con cloruro di sodio, c =5 g/100 ml
Sciogliere 50 g di carbonato di sodio (3.11) in acqua, portare a 1 litro e aggiungere cloruro di sodio (3.12) fino a saturazione.
|
|
3.17.
|
Acido cloridrico circa 0,1 mol/l.
Diluire 10 ml di HCI (3.7) con acqua e portare a 1 litro.
|
|
3.18.
|
Soluzione tampone di acetato di ammonio, circa 0,25 mol/l
Sciogliere 19,3 g di acetato di ammonio (3.3) e 30 ml di acido acetico (3.5) in acqua (3.14) e portare a 1 litro.
|
|
3.19.
|
Preparazione della resina amberlite XAD-2
Sciacquare un quantitativo adatto di amberlite (3.2) con acqua fino a eliminazione completa di tutti gli ioni cloruro, eliminazione che viene verificata eseguendo una prova al nitrato di argento (3.20) sulla fase acquosa che è stata messa da parte. Sciacquare quindi la resina con 50 ml di metanolo (3.8), eliminare il metanolo e conservare la resina in metanolo fresco.
|
|
3.20.
|
Soluzione di nitrato di argento, circa 0,1 mol/l
Sciogliere 0,17 g di nitrato di argento (3.9) in 10 ml di acqua.
|
|
3.21.
|
Fase mobile per HPLC
Mescolare 500 ml di acetonitrile (3.1), 300 ml di soluzione tampone di acetato ammonico (3.18) e 1 200 ml di acqua (3.14). Regolare il pH a 4,3 con acido acetico (3.5). Filtrare la soluzione attraverso un filtro di 0,22 μm (4.8) e degassarla (ad esempio sottoponendola per 10 minuti a trattamento con ultrasuoni). Questa soluzione è stabile per un mese se conservata al buio in un contenitore chiuso.
|
4. Apparecchiatura
|
4.4.
|
Apparecchiatura HPLC a rivelatore UV, a lunghezza d'onda variabile, oppure rivelatore a serie di diodi.
|
|
4.4.1.
|
Colonna per cromatografia liquida, 300 mm × 4 mm, C18, con riempimento 10 μm, o colonna equivalente
|
|
4.5.
|
Colonna di vetro (300 mm × 10 mm) con filtro in vetro sinterizzato e di rubinetto di arresto
|
|
4.6.
|
Filtri in fibra di vetro, diametro 150 mm
|
|
4.7.
|
Filtri a membrana, da 0,45 μm.
|
|
4.8.
|
Filtri a membrana, da 0,22 μm.
|
5. Procedimento
|
Nota:
|
L'alofuginone in quanto base libera è instabile in soluzioni alcaline e di acetato di etile. Non resta in acetato di etile per oltre 30 minuti.
|
5.1. Indicazioni generali
|
5.1.1.
|
Analizzare un alimento di riferimento (bianco), per accertare l'assenza di alofuginone o di altre sostanze che possono interferire.
|
|
5.1.2.
|
Eseguire una prova di recupero analizzando il bianco addizionato di una quantità di alofuginone analoga a quella presente nel campione. Per ottenere una concentrazione di 3 mg/kg, aggiungere 300 μl della soluzione madre standard (3.6.1) a 10 g del bianco, mescolare e attendere 10 minuti prima di procedere all'estrazione (5.2).
|
Nota:
|
Ai fini del presente metodo, il bianco ha una composizione simile a quella del campione e all'analisi l'alofuginone non risulta presente.
|
|
5.2. Estrazione
Pesare, con l'approssimazione di 0,1 g, 10 g del campione preparato in una provetta da centrifuga da 200 ml, aggiungere 0,5 g di ascorbato sodico (3.10), 0,5 g di EDTA (3.13) e 20 ml di acqua e agitare. Immergere la provetta in bagnomaria (80 oC) per 5 minuti. Dopo aver fatto raffreddare a temperatura ambiente, aggiungere 20 ml di soluzione di carbonato di sodio (3.15) e agitare. Aggiungere immediatamente 100 ml di acetato di etile (3.4) e agitare vigorosamente a mano per 15 secondi. Introdurre quindi la provetta nel bagno ultrasonico (4.1) per 3 minuti e allentare il tappo. Centrifugare per 2 minuti e decantare la fase di acetato etilico attraverso un filtro in fibra di vetro (4.6) in un imbuto separatore da 500 ml. Ripetere l'estrazione del campione con un secondo volume di 100 ml di acetato di etile. Lavare gli estratti combinati, per un minuto, con 50 ml di soluzione di carbonato di sodio saturata con cloruro di sodio (3.16) ed eliminare lo strato acquoso.
Estrarre lo strato organico, per un minuto, con 50 ml di acido cloridrico (3.17). Raccogliere lo strato acido inferiore in un imbuto separatore da 250 ml. Riestrarre lo strato organico per 1,5 minuti con altri 50 ml di acido cloridrico e aggiungere al primo estratto. Lavare gli estratti acidi combinati agitando per 10 secondi con 10 ml di acetato di etile (3.4).
Trasferire quantitativamente lo strato acquoso in un pallone a fondo arrotondato da 250 ml ed eliminare la fase organica. Far evaporare dalla soluzione acida tutto l'acetato di etile restante mediante un evaporatore rotante (4.2). La temperatura del bagnomaria non deve superare i 40 oC. Con un vuoto di circa 25 mbar tutto l'acetato di etile residuo sarà rimosso entro 5 minuti a 38 oC.
5.3. Purificazione
5.3.1. Preparazione della colonna di amberlite
Per ciascun estratto di campione viene preparata una colonna XAD-2. Mediante metanolo (3.8) trasferire in una colonna di vetro (4.5) 10 g di amberlite preparata (3.19). Aggiungere un piccolo tampone di lana di vetro alla sommità del letto della resina. Lasciar colare il metanolo dalla colonna e lavare la resina con 100 ml d'acqua, fermando il flusso quando il liquido raggiunge la sommità del letto della resina. Attendere che la colonna si stabilizzi per 10 minuti prima di utilizzarla. Evitare comunque che la colonna si essicchi.
5.3.2. Purificazione del campione
Trasferire quantitativamente l'estratto (5.2) sulla sommità della colonna di amberlite preparata (5.3.1) ed eluire, scartando l'eluato. La velocità di eluizione non deve superare 20 ml/min. Risciacquare il pallone con 20 ml di acido cloridrico (3.17) e usare quest'ultimo liquido per lavare la colonna di resina. Eliminare eventuali residui di soluzione acida insufflando aria. Eliminare i liquidi di lavaggio. Aggiungere 100 ml di metanolo (3.8) alla colonna e lasciar eluire per 5-10 minuti, raccogliendo l'eluato in un pallone da 250 ml. Attendere che il metanolo residuo si stabilizzi per 10 minuti con la resina e continuare l'eluizione con un flusso non superiore a 20 ml/min, raccogliendo l'eluato nello stesso pallone. Evaporare il metanolo sull'evaporatore rotante (4.2); la temperatura del bagnomaria non deve superare i 40 oC. Trasferire quantitativamente il residuo in un pallone tarato da 10 ml applicando la fase mobile (3.21). Portare a volume con la fase mobile e agitare. Filtrare una parte attraverso un filtro a membrana (4.7). Riservare questa soluzione per la determinazione mediante HPLC (5.4).
5.4. Determinazione HPLC
5.4.1. Parametri
I parametri qui riportati sono di riferimento. È possibile operare in condizioni diverse, purché si ottengano risultati equivalenti.
Colonna per cromatografia liquida (4.4.1)
Fase mobile per HPLC (3.21)
Velocità di efflusso: 1,5-2 ml/min
Lunghezza d'onda di rivelazione: 243 nm
Volume iniettato: 40-100 μl.
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.6.2) contenente 3,0 μg/ml fino a ottenimento di altezze (aree) del picco e tempi di ritenzione costanti.
5.4.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.6.2) e misurare le altezze (aree) dei picchi per ciascuna concentrazione. Tracciare la curva di taratura riportando in ordinate le altezze medie o le aree medie dei picchi delle soluzioni di taratura e in ascisse le corrispondenti concentrazioni in μg/ml.
5.4.3. Soluzione del campione
Iniettare più volte l'estratto di campione (5.3.2) utilizzando lo stesso volume usato per le soluzioni di taratura e determinare l'altezza (area) media dei picchi dell'alofuginone.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi dell'alofuginone, per riferimento alla curva di taratura (5.4.2).
Il contenuto di alofuginone w (mg/kg) del campione è dato dalla formula seguente:

dove
|
c
|
=
|
concentrazione di alofuginone nella soluzione del campione, in μg/ml
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure usando un rivelatore a serie di diodi in cui vengono confrontati gli spettri dell'estratto di campione e della soluzione di taratura (3.6.2) contenente 6,0 μg/ml.
7.1.1. Co-cromatografia
Un estratto di campione viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.6.2). Il quantitativo di alofuginone addizionato deve essere analogo a quello stimato di alofuginone rilevato nell'estratto di campione.
Deve aumentare soltanto l'altezza del picco dell'alofuginone, tenuto conto sia della quantità addizionata che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza massima, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda di assorbimento massimo degli spettri del campione e dello standard, registrata all'apice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in genere di circa 2 nm;
|
|
b)
|
tra 225 e 300 nm, gli spettri del campione e dello standard registrati all'apice del picco sul cromatogramma non debbono essere diversi per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è rispettato quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra i due spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
tra 225 e 300 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dello spettro all'apice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni parallele effettuate sullo stesso campione non deve superare 0,5 mg/kg per contenuti di alofuginone sino a un massimo di 3 mg/kg.
7.3. Recupero
Per quanto riguarda il campione in bianco addizionato il recupero non è inferiore all'80 %.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio collaborativo (5) nel corso del quale otto laboratori hanno analizzato tre campioni.
Risultati
|
|
Per il campione A
(bianco)
Al ricevimento
|
Per il campione B (farina)
|
Per il campione C (granulare)
|
|
|
|
Al ricevimento
|
Dopo due mesi
|
Al ricevimento
|
Dopo due mesi
|
|
media [mg/kg]
|
NR
|
2,80
|
2,42
|
2,89
|
2,45
|
|
SR [mg/kg]
|
—
|
0,45
|
0,43
|
0,40
|
0,42
|
|
CVR [%]
|
—
|
16
|
18
|
14
|
17
|
|
Rec. [%]
|
|
86
|
74
|
88
|
75
|
|
NR
|
=
|
non riscontrata
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità (%)
|
|
Rec. [%]
|
=
|
recupero (%).
|
E. DETERMINAZIONE DELLA ROBENIDINA
Cloridrato di 1,3-bis[(4-clorobenzilidene) amino]guanidina
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di robenidina negli alimenti per animali. Il limite di quantificazione è di 5 mg/kg.
2. Principio
Il campione è estratto con metanolo acidificato. L'estratto è essiccato e una parte aliquota è purificata in una colonna di ossido di alluminio. La robenidina, eluita dalla colonna con metanolo, è concentrata e portata volume adeguato con la fase mobile. Il tenore di robenidina è determinato mediante cromatografia liquida ad alta prestazione (HPLC) a fase inversa, utilizzando un rivelatore UV.
3. Reattivi
|
3.2.
|
Metanolo acidificato
Travasare 4,0 ml di acido cloridrico (ρ20 = 1,18 g/ml) in un pallone tarato da 500 ml, portare a volume con metanolo (3.1) e agitare. Questa soluzione è preparata al momento, poco prima dell'uso.
|
|
3.3.
|
Acetonitrile, di qualità HPLC.
|
|
3.4.
|
Setacci molecolari
Sferette di tipo 3A, 8-12 mesh (1,6-2,5 mm, alluminosilicato cristallino, diametro dei pori 0,3 mm).
|
|
3.5.
|
Allumina acida, grado di attività I per cromatografia su colonna
Trasferire 100 g di allumina acida in un contenitore adatto e aggiungere 2,0 ml di acqua. Tappare e agitare per circa 20 minuti. Conservare in un contenitore ben chiuso.
|
|
3.6.
|
Soluzione di potassio diidrogeno fosfato, c = 0,025 mol/l
Sciogliere 3,40 g di potassio diidrogeno fosfato in acqua (qualità HPLC) in un matraccio tarato da 1 000 ml, portare a volume e agitare.
|
|
3.7.
|
Soluzione di fosfato di sodio bibasico, c = 0,025 mol/l
Sciogliere 3,55 g di fosfato di sodio bibasico anidro (o 4,45 g di diidrato, o 8,95 g di dodecaidrato) in acqua (di qualità HPLC), in un matraccio tarato da 1 litro, portare a volume e agitare.
|
|
3.8.
|
Fase mobile per HPLC
Miscelare i seguenti reattivi:
|
|
650 ml di acetonitrile (3.3),
|
|
|
250 ml di acqua (di qualità HPLC),
|
|
|
50 ml di soluzione di potassio diidrogeno fosfato (3.6),
|
|
|
50 ml di soluzione di fosfato di sodio bibasico (3.7).
|
Filtrare la soluzione attraverso un filtro di 0,22 μm (4.6) e degassarla (ad esempio, sottoponendola per 10 minuti a trattamento con ultrasuoni).
|
|
3.9.
|
Sostanza di riferimento (standard)
Robenidina pura: 1,3-bis [(4-clorobenziliden) amino] guanidina cloridrato.
|
|
3.9.1.
|
Robenidina, soluzione madre standard: 300 μg/ml
Pesare, con l'approssimazione di 0,1 mg, 30 mg di robenidina standard (3.9). Sciogliere con metanolo acidificato (3.2) in un matraccio tarato da 100 ml, portare a volume con lo stesso solvente e agitare. Avvolgere il matraccio in un foglio di alluminio e conservarlo al buio.
|
|
3.9.2.
|
Soluzione standard intermedia di robenidina: 12 μg/ml
Travasare 10,0 ml della soluzione madre standard (3.9.1) in un matraccio tarato da 250 ml, portare a volume con la fase mobile (3.8) e agitare. Avvolgere il matraccio in un foglio di alluminio e conservarlo al buio.
|
|
3.9.3.
|
Soluzioni di taratura
In una serie di matracci tarati da 50 ml travasare 5,0, 10,0, 15,0, 20,0 e 25,0 ml della soluzione standard intermedia (3.9.2). Portare a volume con la fase mobile (3.8) e agitare. Queste soluzioni corrispondono rispettivamente a 1,2, 2,4, 3,6, 4,8 e 6,0 μg/ml di robenidina. Devono essere preparate al momento, poco prima dell'uso.
|
|
3.10.
|
Acqua di qualità HPLC
|
4. Apparecchiatura
|
4.1.
|
Colonna di vetro
Colonna realizzata in vetro ambra, dotata di rubinetto e serbatoio di circa 150 ml di capacità, diametro interno 10-15 mm, lunghezza 250 mm.
|
|
4.2.
|
Agitatore meccanico o magnetico
|
|
4.3.
|
Evaporatore rotante a film
|
|
4.4.
|
Apparecchiatura per HPLC con rivelatore a ultravioletti, a lunghezza d'onda variabile, oppure con rivelatore a serie di diodi, funzionante nell'intervallo di 250-400 nm.
|
|
4.4.1.
|
Colonna per cromatografia liquida: 300 mm × 4 mm, C18, particelle di riempimento 10 μm, o equivalente
|
|
4.5.
|
Carta da filtro in fibra di vetro (Whatman GF/A o equivalente)
|
|
4.6.
|
Filtri a membrana, da 0,22 μm.
|
|
4.7.
|
Filtri a membrana, da 0,45 μm.
|
5. Procedimento
|
Nota:
|
La robenidina è sensibile alla luce. Si usa vetreria ambrata in tutte le operazioni.
|
5.1. Indicazioni generali
|
5.1.1.
|
Analizzare un alimento di riferimento (bianco) per accertare l'assenza di robenidina o di altre sostanze che possono interferire.
|
|
5.1.2.
|
Procedere a una prova di recupero, analizzando un campione dell'alimento bianco (5.1.1) addizionato di una quantità di robenidina simile a quella presente nel campione. Per ottenere una concentrazione di 60 mg/kg, trasferire 3,0 ml della soluzione madre standard (3.9.1) in una beuta da 250 ml. Far evaporare la soluzione fino a circa 0,5 ml in corrente d'azoto. Aggiungere 15 g dell'alimento bianco, mescolare per 10 minuti prima di procedere all'estrazione (5.2).
|
Nota:
|
Ai fini del presente metodo, l'alimento bianco ha una composizione simile a quella del campione e all'analisi la robenidina non deve risultare presente.
|
|
5.2. Estrazione
Pesare, con l'approssimazione di 0,01 g, 15 g circa del campione preparato, trasferirli in una beuta da 250 ml, aggiungere 100,0 ml di metanolo acidificato (3.2), tappare e agitare per un'ora con l'agitatore (4.2). Filtrare la soluzione attraverso un filtro in fibra di vetro (4.5) e raccogliere tutto il filtrato in una beuta da 150 ml. Aggiungere 7,5 g di setacci molecolari (3.4), tappare e agitare per cinque minuti. Filtrare immediatamente attraverso un filtro in fibra di vetro. Conservare questa soluzione per la fase di purificazione (5.3).
5.3. Purificazione
5.3.1. Preparazione della colonna di ossido di alluminio
Introdurre un piccolo tampone di lana di vetro all'estremità inferiore della colonna (4.1) e comprimerlo con un'asticella di vetro. Pesare e trasferire nella colonna 11,0 g di allumina preparata (3.5). Nel corso di questa operazione far sì che l'esposizione all'atmosfera sia ridotta al minimo. Battere delicatamente l'estremità inferiore della colonna per lasciar depositare l'allumina.
5.3.2. Purificazione del campione
Trasferire nella colonna con una pipetta 5,0 ml dell'estratto del campione preparato secondo quanto indicato al punto 5.2. Posizionare la punta della pipetta contro la parete della colonna e lasciando che l'allumina assorba la soluzione. Eluire la robenidina con 100 ml di metanolo (3.1) alla velocità di 2-3 ml/minuto e raccogliere l'eluato in un pallone a fondo arrotondato da 250 ml. Evaporare a secco la soluzione di metanolo, a pressione ridotta, e a 40 oC mediante evaporatore rotante (4.3). Ridisciogliere il residuo in 3-4 ml di fase mobile (3.8) e travasarlo quantitativamente in un pallone tarato da 10 ml. Lavare il pallone con più volumi di 1-2 ml di fase mobile e travasare questi liquidi di lavaggio nel pallone tarato. Portare a volume con lo stesso solvente e agitare. Filtrare un'aliquota attraverso un filtro a membrana di 0,45 μm (4.7). Conservare questa soluzione per la determinazione HPLC (5.4).
5.4. Determinazione HPLC
5.4.1. Parametri
I parametri qui riportati sono di riferimento; è possibile tuttavia operare in condizioni diverse, purché si ottengano risultati equivalenti:
Colonna per cromatografia liquida (4.4.1.)
Fase mobile per HPLC (3.8),
Velocità di efflusso: 1,5-2 ml/minuto
Lunghezza d'onda di rivelazione: 317 nm
Volume di iniezione: 20-50 μl.
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.9.3) contenente 3,6 μg/ml sino ad ottenimento di altezze (aree) del picco e tempi di ritenzione costanti.
5.4.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.9.3) e misurare le altezze (aree) dei picchi per ciascuna concentrazione. Tracciare una curva di taratura riportando le altezze medie dei picchi o le aree medie delle soluzioni di taratura sulle ordinate e le corrispondenti concentrazioni in μg/ml sulle ascisse.
5.4.3. Soluzione del campione
Iniettare più volte l'estratto del campione (5.3.2) utilizzando lo stesso volume usato per le soluzioni di taratura e determinare l'altezza (area) media dei picchi della robenidina.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi della robenidina, per riferimento alla curva di taratura (5.4.2).
Il contenuto di robenidina, w (mg/kg) del campione è dato dalla seguente formula:

dove
|
c
|
=
|
concentrazione di robenidina nella soluzione del campione, in μg/ml,
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione e della soluzione di taratura (3.9.3) contenente 6 μg/ml.
7.1.1. Co-cromatografia
Un estratto del campione viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.9.3). Il quantitativo di robenidina addizionato deve essere analogo a quello stimato di robenidina rilevato nell'estratto del campione.
Deve aumentare soltanto l'altezza del picco della robenidina, tenuto conto sia della quantità di robenidina addizionata che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza massima, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda di assorbimento massimo degli spettri del campione e dello standard, registrata all'apice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in generale di 2 nm circa;
|
|
b)
|
tra 225 e 300 nm, gli spettri del campione e dello standard registrati all'apice del picco sul cromatogramma non devono essere diversi per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
tra 250 e 400 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dello spettro all'apice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare il 10 % del risultato più elevato per contenuti di robenidina superiori a 15 mg/kg.
7.3. Recupero
Per il campione in bianco addizionato il recupero non è inferiore all'85 %.
8. Risultati di uno studio collaborativo
La Comunità europea ha organizzato uno studio collaborativo nel corso del quale dodici laboratori hanno analizzato quattro campioni di alimenti per volatili da cortile e per conigli, in forma di farina o di granulare. Ogni campione è stato analizzato due volte. I risultati dello studio figurano nella seguente tabella:
|
|
Volatili
|
Conigli
|
|
|
Farina
|
Granulare
|
Farina
|
Granulare
|
|
media [mg/kg]
|
27,00
|
27,99
|
43,6
|
40,1
|
|
sr [mg/kg]
|
1,46
|
1,26
|
1,44
|
1,66
|
|
Cvr [%]
|
5,4
|
4,5
|
3,3
|
4,1
|
|
SR [mg/kg]
|
4,36
|
3,36
|
4,61
|
3,91
|
|
CVR [%]
|
16,1
|
12,0
|
10,6
|
9,7
|
|
Recupero [%]
|
90,0
|
93,3
|
87,2
|
80,2
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, %
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, %
|
F. DETERMINAZIONE DEL DICLAZURIL
(+)-4-clorofenil[2,6-dicloro-4-(2,3,4,5-tetraidro-3,5-diosso-1,2,4-triazina-2-il)fenil]acetonitrile
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di diclazuril negli alimenti per animali e nelle premiscele. Il limite di rilevazione è di 0,1 mg/kg; il limite di quantificazione è di 0,5 mg/kg.
2. Principio
Dopo aggiunta di uno standard interno, il campione viene estratto con metanolo acidificato. Nel caso degli alimenti per animali, una parte aliquota dell'estratto viene purificata su una cartuccia C18 per estrazione in fase solida. Il diclazuril viene eluito dalla cartuccia con una miscela di metanolo acidificato e acqua. Dopo evaporazione, il residuo è disciolto in una miscela DMF/acqua. Nelle premiscele, l'estratto viene evaporato e il residuo disciolto in una miscela DMF/acqua. Il contenuto di diclazuril è determinato per cromatografia liquida ad alta prestazione (HPLC), a gradiente ternario e in fase inversa, utilizzando un rivelatore UV.
3. Reattivi
|
3.1.
|
Acqua, di qualità HPLC.
|
|
3.3.
|
Solfidrato di tetrabutilammonio (TBHS)
|
|
3.4.
|
Acetonitrile, di qualità HPLC
|
|
3.5.
|
Metanolo, di qualità HPLC
|
|
3.6.
|
N, N-dimetilformammide (DMF)
|
|
3.7.
|
Acido cloridrico, ρ20 = 1,19 g/ml
|
|
3.8.
|
Sostanza di riferimento (standard): diclazuril II-24: (+)-4-clorofenil[2,6-dicloro-4-(2,3,4,5-tetraidro-3,5-diosso-1,2,4-triazina-2-il)fenil]aceto nitrile, di purezza garantita, E771.
|
|
3.8.1.
|
Soluzione madre standard di diclazuril, 500 μg/m l
In un matraccio tarato da 50 ml, pesare, con l'approssimazione di 0,1 mg, 25 mg di diclazuril standard (3.8). Sciogliere in DMF (3.6), portare a volume con DMF (3.6) e mescolare. Avvolgere il matraccio in un foglio d'alluminio (o impiegare un matraccio in vetro scuro) e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.8.2.
|
Soluzione madre standard di diclazuril, 50 μg/m l
Trasferire 5,00 ml della soluzione madre (3.8.l) in un matraccio tarato da 50 ml, portare a volume con DMF (3.6) e mescolare. Avvolgere il matraccio in un foglio d'alluminio (o utilizzare un matraccio in vetro scuro) e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.9.
|
Standard interno: 2,6 dicloro-α-(4-clorofenil)-4-[4,5-diidro-3,5-diosso-l,2,4-triazina-2(3H)-il] α-metilbenzenebenzene-acetonitrile
|
|
3.9.1.
|
Soluzione madre dello standard interno di diclazuril, 500 μg/m l
In un matraccio tarato da 50 ml, pesare, con l'approssimazione di 0,1 mg, 25 mg dello standard interno (3.9). Sciogliere in DMF (3.6), portare a volume con DMF (3.6) e mescolare. Avvolgere il matraccio in un foglio d'alluminio (o utilizzare un matraccio in vetro scuro) e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.9.2.
|
Soluzione dello standard interno di diclazuril, 50 μg/m l
Trasferire 5,00 ml della soluzione madre dello standard interno (3.9.l) in un matraccio tarato da 50 ml, portare a volume con DMF (3.6) e mescolare. Avvolgere il matraccio in un foglio d'alluminio (o utilizzare un matraccio in vetro scuro) e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.9.3.
|
Soluzione dello standard interno per le premiscele, p/1 000 mg/ml
(p = contenuto nominale di diclazuril nella premiscela, in mg/kg)
In un matraccio tarato da 100 ml, pesare, con l'approssimazione di 0,1 mg, p/10 mg dello standard interno, sciogliere in DMF (3.6) in bagno ultrasonico (4.6), portare a volume con DMF e mescolare. Avvolgere il matraccio in un foglio d'alluminio (o utilizzare un matraccio in vetro scuro) e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.10.
|
Soluzione di taratura, 2 μg/ml.
In un matraccio tarato da 50 ml, pipettare 2,00 ml di soluzione standard di diclazuril (3.8.2) e 2,00 ml di soluzione dello standard interno (3.9.2). Aggiungere 16 ml di DMF (3.6), portare a volume con acqua e mescolare. Questa soluzione deve essere preparata al momento, prima dell'uso.
|
|
3.11.
|
Cartuccia C18 per estrazione in fase solida, ad esempio Bond Elut, misura: 1 cm., peso sorbente: 100 mg.
|
|
3.12.
|
Solvente di estrazione: metanolo acidificato.
Pipettare 5,0 ml di acido cloridrico (3.7) in 1 000 ml di metanolo (3.5) e mescolare.
|
|
3.13.
|
Fase mobile per HPLC
|
|
3.13.1.
|
Eluente A: soluzione di acetato di ammonio — solfidrato di tetrabutilammonio.
Sciogliere 5 g di acetato d'ammonio (3.2) e 3,4 g di TBHS (3.3) in 1 000 ml d'acqua (3.l) e mescolare.
|
|
3.13.2.
|
Eluente B: acetonitrile (3.4).
|
|
3.13.3.
|
Eluente C: metanolo (3.5).
|
4. Apparecchiatura
|
4.2.
|
Apparecchiatura per HPLC a gradiente ternario
|
|
4.2.1.
|
Colonna per cromatografia liquida, 100 mm × 4,6 mm, con riempimento in Hypersil ODS da 3 μm, o equivalente
|
|
4.2.2.
|
Rivelatore UV a lunghezza d'onda variabile o rivelatore a serie di diodi
|
|
4.3.
|
Evaporatore rotante a film
|
|
4.4.
|
Filtro a membrana, da 0,45 μm
|
5. Procedimento
5.1. Indicazioni generali
5.1.1. Alimento bianco
Analizzare un alimento di riferimento (bianco), per accertare l'assenza di diclazuril o di altre sostanze che possono interferire. Il bianco ha una composizione simile a quella del campione e all'analisi il diclazuril o sostanze capaci di interferire non risultano presenti.
5.1.2. Prova di recupero
Effettuare una prova di recupero analizzando il bianco addizionato di una quantità di diclazuril analoga a quella presente nel campione. Per ottenere una concentrazione di 1 mg/kg, aggiungere 0,1 ml della soluzione madre (3.8.1) a 50 g di bianco, mescolare accuratamente e lasciar riposare per 10 minuti, agitando nuovamente varie volte prima di procedere all'estrazione (5.2).
Se non fosse disponibile un bianco di tipo simile a quello del campione (cfr. punto 5.1.1), la prova di recupero può essere eseguita col metodo di addizione della sostanza di riferimento. In questo caso, un'aliquota del campione da analizzare viene addizionata di una quantità nota di diclazuril, analoga a quella già presente. Quest'aliquota viene analizzata insieme a una del campione non addizionato e il recupero può essere calcolato per differenza.
5.2. Estrazione
5.2.1. Alimenti per animali
Pesare, con l'approssimazione di 0,01 g, 50 g circa del campione. Trasferire in una beuta da 500 ml, aggiungere 1,00 ml di soluzione dello standard interno (3.9.2) e 200 ml di solvente di estrazione (3.12), poi tappare la beuta. Mantenere sotto agitazione la miscela per una notte, nell'agitatore (4.l). Lasciare depositare per 10 minuti. Trasferire una parte aliquota da 20 ml del surnatante in un contenitore adatto in vetro e diluire con 20 ml d'acqua. Trasferire questa soluzione in una cartuccia di estrazione (3.11) e filtrare sotto vuoto (4.5). Lavare la cartuccia con 25 ml di una miscela di solvente di estrazione (3.12) e d'acqua, 65 + 35 (V + V). Eliminare le frazioni raccolte ed eluire i composti con 25 ml di una miscela di solvente di estrazione (3.12) e d'acqua, 80 + 20 (V + V). Evaporare questa frazione a 60 oC nell'evaporatore rotante (4.3) fino a quando comincia a seccare. Sciogliere il residuo con 1,0 ml di DMF (3.6), aggiungere 1,5 ml d'acqua (3.1) e mescolare. Filtrare con filtro a membrana (4.4). Procedere alla determinazione HPLC (5.3).
5.2.2. Premiscele
Pesare, con l'approssimazione di 0,001 g, 1 g circa del campione. Trasferire in una beuta da 500 ml, aggiungere 1,00 ml di soluzione dello standard interno (3.9.3) e 200 ml di solvente di estrazione (3.12), poi tappare la beuta. Agitare per una notte sull'agitatore (4.1). Lasciare depositare per 10 minuti. Trasferire un'aliquota da 10 000/p ml (p = contenuto nominale di diclazuril nella premiscela, espresso in mg/kg) del surnatante in un matraccio a fondo arrotondato di dimensioni adatte. Evaporare nell'evaporatore rotante (4.3), sotto pressione ridotta e a 60 oC, fino a quando il composto comincia a seccare. Sciogliere il residuo con 10,0 ml DMF (3.6), aggiungere 15,0 ml d'acqua (3.1) e mescolare. Procedere alla determinazione HPLC (5.3).
5.3. Determinazione HPLC
5.3.1. Parametri
Le seguenti condizioni vengono proposte a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida(4.2.1):
|
100 mm × 4,6 mm, con riempimento in Hypersil ODS da 3 μm, o equivalente
|
|
|
Fase mobile:
|
Eluente A (3.13.1):
|
soluzione acquosa di acetato di ammonio e di solfidrato di tetrabutilammonio
|
|
|
Eluente B (3.13.2):
|
acetonitrile
|
|
|
Eluente C (3.13.3):
|
metanolo
|
|
Metodo di eluzione:
|
|
—
|
condizioni iniziali: A + B + C = 60 + 20 + 20 (V + V + V)
|
|
—
|
dopo 10 minuti, eluizione a gradiente per 30 minuti fino a: A + B + C = 45 + 20 + 35 (V + V + V).
|
Risciacquare con B per 10 minuti.
|
|
Velocità di efflusso:
|
1,5-2 ml/min
|
|
|
Volume di iniezione:
|
20 μl
|
|
|
Lunghezza d'onda di rivelazione:
|
280 nm
|
|
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.10), contenente 2 μg/ml, fino a ottenimento di altezze (aree) del picco e tempi di ritenzione costanti.
5.3.2. Soluzione di taratura
Iniettare più volte 20 μl della soluzione di taratura (3.10) e determinare l'altezza (area) media dei picchi del diclazuril e dello standard interno.
5.3.3. Soluzione del campione
Iniettare più volte 20 μl della soluzione del campione (5.2.1 o 5.2.2) e determinare l'altezza (area) media dei picchi del diclazuril e dello standard interno.
6. Calcolo dei risultati
6.1. Alimenti per animali
Il contenuto w di diclazuril nel campione, espresso in mg/kg, è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
hd,s
|
=
|
altezza (area) del picco del diclazuril nella soluzione del campione (5.2.1)
|
|
hi,s
|
=
|
altezza (area) del picco dello standard interno nella soluzione del campione (5.2.1)
|
|
hd,c
|
=
|
altezza (area) del picco del diclazuril nella soluzione di taratura (3.10)
|
|
hi,c
|
=
|
altezza (area) del picco dello standard interno nella soluzione di taratura (3.10)
|
|
cd,c
|
=
|
concentrazione di diclazuril nella soluzione di taratura, in μg/ml (3.10)
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi
|
|
V
|
=
|
volume dell'estratto del campione conformemente al punto 5.2.1 (ossia 2,5 ml)
|
6.2. Premiscele
Il contenuto w di diclazuril nel campione, espresso in mg/kg, è dato dalla seguente formula:
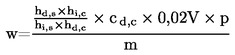 [mg/kg]
[mg/kg]
dove
|
hd,c
|
=
|
altezza (area) del picco del diclazuril nella soluzione di taratura (3.10)
|
|
hi,c
|
=
|
altezza (area) del picco dello standard interno nella soluzione di taratura (3.10)
|
|
hd,s
|
=
|
altezza (area) del picco del diclazuril nella soluzione del campione (5.2.2)
|
|
hi,s
|
=
|
altezza (area) del picco dello standard interno nella soluzione del campione (5.2.2)
|
|
cd,c
|
=
|
concentrazione di diclazuril nella soluzione di taratura, in μg/ml (3.10)
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi
|
|
V
|
=
|
volume dell'estratto del campione conformemente al punto 5.2.2 (ossia 25 ml)
|
|
p
|
=
|
contenuto nominale di diclazuril nella premiscela, in mg/kg
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione (5.2.1 o 5.2.2) e della soluzione di taratura (3.10).
7.1.1. Co-cromatografia
A un estratto del campione (5.2.1 o 5.2.2) viene addizionato un quantitativo adeguato della soluzione di taratura (3.10). Il quantitativo di diclazuril addizionato deve essere analogo a quello di diclazuril rilevato nell'estratto del campione.
Devono aumentare soltanto le altezze del picco del diclazuril e dello standard interno, tenuto conto dei quantitativi aggiunti e della diluizione dell'estratto. L'ampiezza del picco, a metà altezza, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale del picco del diclazuril o dello standard interno del campione non addizionato.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda dell'assorbimento massimo degli spettri del campione e dello standard, registrata al vertice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi essa è in genere di 2 nm circa;
|
|
b)
|
fra 210 e 320 nm, gli spettri del campione e dello standard registrati al vertice del picco sul cromatogramma non devono essere diversi per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi massimi e quando in tutti i punti osservati lo scarto fra gli spettri non supera il 15 % dell'assorbanza dello standard dell'analita;
|
|
c)
|
fra 230 e 320 nm, gli spettri della curva ascendente, dell'apice e della curva discendente del picco prodotta dall'estratto del campione non devono essere diversi gli uni dagli altri per le parti dello spettro situate fra il 10 % e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto fra gli spettri non supera il 15 % dell'assorbanza dello spettro all'apice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
il 30 % rispetto al valore più elevato, per i contenuti di diclazuril compresi fra 0,5 e 2,5 mg/kg,
|
|
—
|
0,75 mg/kg, per i contenuti di diclazuril compresi fra 2,5 e 5 mg/kg,
|
|
—
|
il 15 % del valore più elevato, per i contenuti di diclazuril superiori a 5 mg/kg.
|
7.3. Recupero
Per un campione addizionato (bianco), il recupero non è inferiore all'80 %.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio collaborativo nel corso del quale undici laboratori hanno analizzato cinque campioni costituiti da due premiscele: una mescolata con una matrice organica (O 100) e l'altra con una matrice inorganica (A 100). Il contenuto teorico era di 100 mg di diclazuril/kg. I tre alimenti misti per volatili da cortile sono stati preparati da tre diversi produttori (NL) (L1/Z1/K1). Il contenuto teorico era di 1 mg di diclazuril/kg. Ai laboratori è stato chiesto di analizzare ciascun campione uno sola volta o in duplicato. (Per ulteriori informazioni consultare il Journal of AOAC International, Volume 77, n. 6, 1994, pagg. 1359-1361). I risultati dello studio figurano nella seguente tabella:
|
|
Campione 1
A 100
|
Campione 2
O 100
|
Campione 3
L1
|
Campione 4
Z1
|
Campione 5
K1
|
|
L
|
11
|
11
|
11
|
11
|
6
|
|
n
|
19
|
18
|
19
|
19
|
12
|
|
Media
|
100,8
|
103,5
|
0,89
|
1,15
|
0,89
|
|
Sr (mg/kg)
|
5,88
|
7,64
|
0,15
|
0,02
|
0,03
|
|
CVr (%)
|
5,83
|
7,38
|
17,32
|
1,92
|
3,34
|
|
SR (mg/kg)
|
7,59
|
7,64
|
0,17
|
0,11
|
0,12
|
|
CVR (%)
|
7,53
|
7,38
|
18,61
|
9,67
|
13,65
|
|
contenuto nominale (mg/kg]
|
100
|
100
|
1
|
1
|
1
|
|
L
|
=
|
numero di laboratori
|
|
n
|
=
|
numero di valori singoli
|
|
Sr
|
=
|
deviazione standard della ripetibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità
|
9. Osservazioni
Deve essere dimostrato preliminarmente che il diclazuril dà una risposta lineare per l'insieme delle concentrazioni misurate.
G. DETERMINAZIONE DEL LASALOCID SODICO
Sale sodico di un acido monocarbossilico polietere prodotto da Streptomyces lasaliensis
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di lasalocid sodico negli alimenti per animali e nelle premiscele. Il limite di rilevazione è di 5 mg/kg, il limite di quantificazione è di 10 mg/kg.
2. Principio
Il lasalocid sodico viene estratto dal campione in metanolo acidificato e viene determinato per cromatografia in fase liquida ad alta prestazione (HPLC) a fase inversa, utilizzando un rivelatore spettrofluorimetrico.
3. Reattivi
|
3.1.
|
Potassio diidrogeno fosfato (KH2PO4)
|
|
3.2.
|
Acido ortofosforico, p (p/p) = 85 %
|
|
3.3.
|
Soluzione di acido ortofosforico, c = 20 %
Diluire con acqua 23,5 ml di acido ortofosforico (3.2) e portare a 100 ml.
|
|
3.4.
|
6-metil-2-eptilammina (1,5-dimetilesilammina), p (p/p) = 99 %
|
|
3.5.
|
Metanolo, di qualità HPLC
|
|
3.6.
|
Acido cloridrico, densità: 1,19 g/ml.
|
|
3.7.
|
Soluzione tampone fosfato, c = 0,01 mol/l
In 500 ml di acqua (3.11) sciogliere 1,36 g di KH2PO4 (3.1), aggiungere 3,5 ml di acido ortofosforico (3.2) e 10,0 ml di 6-metil-2-eptilammina (3.4). Portare il pH a 4,0 con soluzione di acido ortofosforico (3.3) e portare a 1 000 ml con acqua (3.11).
|
|
3.8.
|
Metanolo acidificato
Travasare 5,0 ml di acido cloridrico (3.6) in un matraccio tarato da 1 000 ml, portare a volume con metanolo (3.5) e mescolare. Questa soluzione deve essere preparata al momento, prima dell'uso.
|
|
3.9.
|
Fase mobile per HPLC, soluzione tampone fosfato e metanolo 5 + 95 (V + V)
Mescolare 5 ml di soluzione tampone fosfato (3.7) con 95 ml di metanolo (3.5)
|
|
3.10.
|
Sostanza standard lasalocid sodico, di purezza garantita, C34H53O8Na (sale sodico di un acido monocarbossilico polietere prodotto da Streptomyces lasaliensis), E763
|
|
3.10.1.
|
Soluzione madre standard di lasalocid sodico, 500 μg/ml
In un matraccio tarato da 100 ml, pesare, con l'approssimazione di 0,1 mg, 50 mg di lasalocid sodico (3.10), sciogliere in metanolo acidificato (3.8), portare a volume con lo stesso solvente e mescolare. Questa soluzione deve essere preparata al momento, poco prima dell'uso.
|
|
3.10.2.
|
Soluzione standard intermedia di lasalocid sodico, 50 μg/ml
In un matraccio tarato da 100 ml, pipettare 10,0 ml di soluzione madre standard (3.10.1), portare a volume con metanolo acidificato (3.8) e mescolare. Questa soluzione deve essere preparata al momento, prima dell'uso.
|
|
3.10.3.
|
Soluzioni di taratura
In una serie di matracci tarati da 50 ml trasferire 1,0 ml, 2,0 ml, 4,0 ml, 5,0 ml e 10,0 ml di soluzione madre intermedia (3.10.2). Portare a volume con metanolo acidificato (3.8) e mescolare. Queste soluzioni corrispondono rispettivamente a 1,0, 2,0, 4,0, 5,0 e 10,0 g di lasalocid sodico per ml e vanno preparate al momento, prima dell'uso.
|
|
3.11.
|
Acqua, di qualità HPLC
|
4. Apparecchiatura
|
4.1.
|
Bagno ultrasonico (o bagnomaria con agitatore), con sistema di termoregolazione
|
|
4.2.
|
Filtri a membrana, 0,45 μm.
|
|
4.3.
|
Apparecchiatura HPLC con sistema di iniezione adeguato per volumi da 20 μl.
|
|
4.3.1.
|
Colonna per cromatografia liquida, 125 mm × 4 mm, C18, particelle di riempimento 5 μm, o equivalente
|
|
4.3.2.
|
Spettrofluorimetro con regolazione a lunghezza d'onda variabile (eccitazione e emissione)
|
5. Procedimento
5.1. Indicazioni generali
5.1.1. Alimento «bianco»
Per eseguire la prova di recupero (5.1.2), analizzare un alimento di riferimento (bianco), per accertare l'assenza di lasalocid sodico o di altre sostanze che possono interferire. Il campione bianco ha una composizione simile a quella del campione e all'analisi il lasalocid sodico o sostanze capaci di interferire non risultano presenti.
5.1.2. Prova di recupero
Effettuare una prova di recupero analizzando il bianco addizionato di una quantità di lasalocid sodico analoga a quella presente nel campione. Per ottenere una concentrazione di 100 mg/kg, trasferire 10,0 ml della soluzione madre di riserva (3.10.1) in una beuta da 250 ml e far evaporare la soluzione a circa 0,5 ml. Aggiungere 50 g del bianco, mescolare il tutto e attendere per 10 minuti, agitando più volte, prima di procedere all'estrazione (5.2).
Se non fosse disponibile un bianco di tipo simile a quello del campione (cfr. punto 5.1.1), la prova di recupero può essere eseguita col metodo di addizione della sostanza di riferimento. In questo caso, un'aliquota del campione da analizzare viene addizionata di una quantità nota di lasalocid sodico, analoga a quella già presente. Quest'aliquota viene analizzata insieme a una del campione non addizionato, e il recupero può essere calcolato per differenza.
5.2. Estrazione
5.2.1. Alimenti per animali
In una beuta da 250 ml con tappo, pesare, con l'approssimazione di 0,01 g, da 5 a 10 g del campione. Aggiungere con pipetta 100,0 ml di metanolo acidificato (3.8). Chiudere senza stringere e scuotere per disperdere. Collocare la beuta in un bagno ultrasonico (4.1) a circa 40 oC per 20 minuti; quindi toglierla e lasciarla raffreddare a temperatura ambiente. Lasciare riposare per circa un'ora finché la sostanza in sospensione si sia depositata; filtrare quindi un'aliquota con filtro a membrana da 0,45 μm (4.2) in un recipiente adeguato. Procedere alla determinazione HPLC (5.3).
5.2.2. Premiscele
In un matraccio tarato pesare, con l'approssimazione di 0,001 g, circa 2 g di premiscela non macinata. Aggiungere 100,0 ml di metanolo acidificato (3.8) e scuotere per disperdere. Collocare la beuta e il suo contenuto in un bagno ultrasonico (4.1) a circa 40 oC per 20 minuti; quindi toglierla e lasciarla raffreddare a temperatura ambiente. Diluire e portare a volume con metanolo acidificato (3.8) e mescolare accuratamente. Lasciare riposare per un'ora finché la sostanza in sospensione si sia depositata; filtrare quindi un'aliquota con filtro a membrana da 0,45 μm (4.2). Diluire un volume adeguato di filtrato chiaro con metanolo acidificato (3.8) ottenendo così una soluzione di prova finale contenente circa 4 μg/ml di lasalocid sodico. Procedere alla determinazione HPLC (5.3).
5.3. Determinazione HPLC
5.3.1. Parametri
Le seguenti condizioni vengono proposte a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida:
|
125 mm × 4 mm, colonna a fase inversa (4.3.1) C18, particelle di riempimento 5 μm, o equivalente
|
|
Fase mobile (3.9):
|
miscela di soluzione tampone fosfato (3.7) e metanolo (3.5), 5 + 95 (V + V)
|
|
Velocità di efflusso:
|
1,2 ml/min
|
|
Lunghezze d'onda di rivelazione:
|
|
|
eccitazione:
|
310 nm
|
|
emissione:
|
419 nm
|
|
Volume da iniezione:
|
20 μl
|
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.10.3) contenente 4,0 μg/ml, fino a ottenimento di altezze (aree) del picco e tempi di ritenzione costanti.
5.3.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.10.3) e determinare le altezze (aree) medie dei picchi per ciascuna concentrazione. Tracciare la curva di taratura, riportando in ordinata le altezze (aree) medie dei picchi e in ascissa le corrispondenti concentrazioni, espresse in μg/ml.
5.3.3. Soluzione del campione
Iniettare più volte gli estratti dei campioni di cui ai punti 5.2.1 o 5.2.2, utilizzando lo stesso volume di quello prelevato per le soluzioni di taratura e determinare le altezze (aree) medie dei picchi del lasalocid sodico.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi del lasalocid sodico ottenuta per iniezione (5.3.3), per riferimento alla curva di taratura.
6.1. Alimenti per animali
Il contenuto w di lasalocid sodico nel campione, espresso in mg/kg, è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
c
|
=
|
concentrazione di lasalocid sodico nella soluzione del campione (5.2.1), in μg/ml
|
|
V1
|
=
|
volume dell'estratto del campione, espresso in ml, conformemente al punto 5.2.1 (ossia 100 ml)
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
6.2. Premiscele
Il contenuto w di lasalocid sodico nel campione, espresso in mg/kg, è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
c
|
=
|
concentrazione di lasalocid sodico nella soluzione del campione (5.2.2), in μg/ml
|
|
V2
|
=
|
volume dell'estratto del campione, espresso in ml, conformemente al punto 5.2.2 (ossia 250 ml)
|
|
f
|
=
|
fattore di diluizione conformemente al punto 5.2.2
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
I metodi basati sulla spettrofluorimetria sono meno soggetti a interferenza di quelli in cui si fa ricorso alla rivelazione UV. L'identità dell'analita può essere confermata mediante co-cromatografia.
7.1.1. Co-cromatografia
Un estratto del campione (5.2.1 o 5.2.2) viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.10.3). Il quantitativo di lasalocid sodico aggiunto deve essere analogo a quello di lasalocid sodico rilevato nell'estratto del campione. Deve aumentare soltanto l'altezza del picco del lasalocid sodico, tenuto conto della quantità di lasalocid aggiunto e della diluizione dell'estratto. L'ampiezza del picco, a metà altezza, deve corrispondere, con uno scartamento massimo del 10 %, all'ampiezza originale prodotta dall'estratto del campione non addizionato.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
il 15 % del valore più elevato, per i contenuti di lasalocid sodico compresi fra 30 e 100 mg/kg,
|
|
—
|
15 mg/kg per contenuti di lasalocid sodico compresi fra 100 e 200 mg/kg,
|
|
—
|
il 7,5 % del valore più elevato per i contenuti di lasalocid sodico superiori a 200 mg/kg.
|
7.3. Recupero
Per il campione addizionato (bianco) di alimento, il recupero non è inferiore all'80 %. Per i campioni addizionati di premiscela, il recupero non è inferiore al 90 %.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio collaborativo (6) nel corso del quale dodici laboratori hanno analizzato due 2 premiscele (campioni 1 e 2) e cinque alimenti per animali (campioni 3-7). Ciascun campione è stato analizzato due volte. I risultati figurano nella tabella seguente:
|
|
Campione 1
Premiscela per polli
|
Campione 2
Premiscela per tacchini
|
Campione 3
Granulare per tacchini
|
Campione 4
Briciolame per polli
|
Campione 5
Mangime per tacchini
|
Campione 6
Mangime per polli A
|
Campione 7
Mangime per polli B
|
|
L
|
12
|
12
|
12
|
12
|
12
|
12
|
12
|
|
n
|
23
|
23
|
23
|
23
|
23
|
23
|
23
|
|
media [mg/kg]
|
5 050
|
16 200
|
76,5
|
78,4
|
92,9
|
48,3
|
32,6
|
|
sr [mg/kg]
|
107
|
408
|
1,71
|
2,23
|
2,27
|
1,93
|
1,75
|
|
CVr [%]
|
2,12
|
2,52
|
2,24
|
2,84
|
2,44
|
4,00
|
5,37
|
|
sR [mg/kg]
|
286
|
883
|
3,85
|
7,32
|
5,29
|
3,47
|
3,49
|
|
CVR [%]
|
5,66
|
5,45
|
5,03
|
9,34
|
5,69
|
7,18
|
10,70
|
|
Contenuto nominale [mg/kg]
|
5 000 (7)
|
16 000 (7)
|
80 (7)
|
105 (7)
|
120 (7)
|
50 (8)
|
35 (8)
|
|
L
|
=
|
numero di laboratori
|
|
n
|
=
|
numero di valori singoli
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
sR
|
=
|
deviazione standard della riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, %
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, %
|
(1) Studio effettuato dal gruppo di lavoro Alimenti per animali del Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(2) Studio effettuato dal gruppo di lavoro Alimenti per animali del Verband Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
(3) Possono essere utilizzati altri metodi di mineralizzazione purché abbiano dimostrato di dare risultati simili (ad esempio la mineralizzazione in pressione a microonde).
(4) Nel foraggio verde (fresco o essiccato) possono essere presenti notevoli quantità di silice vegetale, che è capace di trattenere gli oligoelementi e che va quindi eliminata. Ai campioni di tali prodotti va pertanto applicato il procedimento seguente. Eseguire le operazioni descritte al punto 5.1.1.1, fino alla filtrazione. Lavare per due volte con acqua bollente la carta da filtro contenente il residuo insolubile e porla in un crogiolo di quarzo o di platino (4.3). Calcinare in muffola (4.1) a temperatura inferiore ai 550 oC, fino a scomparsa completa delle sostanze carboniose. Lasciar raffreddare, aggiungere qualche goccia d'acqua, poi 10-15 ml di acido fluoridrico (3.4), quindi evaporare a secco a 150 oC circa. Se nel residuo rimane ancora della silice, scioglierla in pochi ml di acido fluoridrico (3.4) ed evaporare a secco. Aggiungere 5 gocce di acido solforico (3.5) e riscaldare fino a scomparsa dei fumi bianchi. Addizionare 5 ml di acido cloridrico 6 mol/l (3.2) e 30 ml circa d'acqua; riscaldare, filtrare la soluzione in un matraccio tarato da 250 ml e portare a volume con acqua (la concentrazione dell'acido cloridrico è di 0,5 mol/l circa). Procedere quindi nella determinazione a partire dal punto 5.1.3.
(5) The Analyst 108, 1983: pagg. 1252-1256.
(6) Analyst, 1995, 120, 2175-2180.
(7) contenuto dichiarato dal produttore
(8) alimento preparato in laboratorio
ALLEGATO V
METODI DI ANALISI PER IL CONTROLLO DELLA PRESENZA DI SOSTANZE INDESIDERABILI NEGLI ALIMENTI PER ANIMALI
A. DETERMINAZIONE DEL GOSSIPOLO LIBERO E TOTALE
1. Finalità e campo d'applicazione
Il metodo consente di determinare i livelli di gossipolo libero, di gossipolo totale e di sostanze chimicamente affini nei semi, nelle farine e nei panelli di cotone, nonché negli alimenti composti che contengono tali materie prime. Il limite inferiore di determinabilità del metodo è di 20 mg/kg.
2. Principio
Il gossipolo viene estratto, in presenza di 3-amino-1-propanolo, con una miscela di isopropanolo-esano nel caso della determinazione del gossipolo libero e con la dimetilformamide nel caso della determinazione del gossipolo totale. Il gossipolo è trasformato mediante anilina in gossipolo-dianilina, la cui densità ottica è misurata a 440 nm.
3. Reattivi
|
3.1.
|
Miscela di isopropanolo-esano: mescolare 60 parti in volume di isopropanolo con 40 parti in volume di esano normale.
|
|
3.2.
|
Solvente A: porre in un pallone tarato da 1 litro, 500 ml circa della miscela isopropanolo-esano (3.1), 2 ml di 3-amino-1-propanolo, 8 ml di acido acetico glaciale e 50 ml d'acqua. Portare a volume con miscela di isopropanolo-esano (3.1). Questo reattivo è stabile per una settimana.
|
|
3.3.
|
Solvente B: pipettare in un pallone tarato da 100 ml, 2 ml di 3-amino-1-propanolo e 10 ml di acido acetico glaciale. Raffreddare a temperatura ambiente e portare a volume con N,N-dimetilformamide. Questo reattivo è stabile per una settimana.
|
|
3.4.
|
Anilina: se la densità ottica della prova in bianco supera il valore 0,022, l'anilina deve essere distillata su polvere di zinco eliminando la prima e l'ultima frazione pari al 10 % del distillato. Posta in bottiglia chiusa di vetro bruno ed in frigorifero questo reattivo si conserva per parecchi mesi.
|
|
3.5.
|
Soluzione tipo A di gossipolo: porre in un pallone tarato da 250 ml, 27,9 mg d'acetato di gossipolo. Sciogliere e portare a volume con il solvente A (3.2). Pipettare 5,0 ml di questa soluzione in un matraccio tarato da 250 ml e portare a volume con il solvente A. La concentrazione di gossipolo in questa soluzione è 0,02 mg/ml. Lasciar riposare per un'ora a temperatura ambiente prima dell'uso.
|
|
3.6.
|
Soluzione tipo B di gossipolo: porre in un pallone tarato da 50 ml, 27,9 mg d'acetato di gossipolo, sciogliere e portare a volume con il solvente B (3.3). La concentrazione di gossipolo in questa soluzione è di 0,5 mg/ml.
Le soluzioni tipo A e B di gossipolo restano stabili per 24 ore se sono conservate al buio.
|
4. Apparecchiatura
|
4.1.
|
Agitatore rotativo a capovolgimento: circa 35 giri al minuto.
|
5. Procedimento
5.1. Quantità di sostanza da sottoporre all'analisi
La quantità di sostanza da sottoporre all'analisi è proporzionale al presunto contenuto di gossipolo nel campione. È preferibile operare su una piccola quantità di sostanza e su una parte aliquota del filtrato relativamente importante, in modo da ottenere una quantità di gossipolo sufficiente per effettuare una misura spettofotometrica precisa. Per la determinazione del gossipolo libero nei semi, nelle farine e nei panelli di cotone, la quantità di sostanza da sottoporre all'analisi non supera 1 g; per gli alimenti composti potrà arrivare sino a 5 g. Una parte aliquota di 10 ml di filtrato è sufficiente nella maggioranza dei casi; essa conterrà da 50 a 100 μg di gossipolo. Per la determinazione del gossipolo totale, la quantità di sostanza da sottoporre ad analisi varierà da 0,5 a 5 g in modo che una parte aliquota di 2 ml di filtrato contenga da 40 a 200 μg di gossipolo.
Effettuare l'analisi a una temperatura ambiente di circa 20 oC.
5.2. Determinazione del gossipolo libero
Porre la quantità di sostanza da sottoporre all'analisi in un pallone da 250 ml a collo smerigliato il cui fondo è ricoperto di polvere di vetro. Aggiungere, con una pipetta, 50 ml del solvente A (3.2), tappare il pallone e porlo per un'ora nel mescolatore rotativo a capovolgimento. Filtrare su un filtro asciutto e raccogliere il filtrato in un palloncino a collo smerigliato. Nel corso della filtrazione ricoprire l'imbuto con un vetro da orologio.
Introdurre con una pipetta in due palloni tarati da 25 ml (A e B), parti aliquote identiche di filtrato contenenti da 50 a 100 μg di gossipolo. Completare eventualmente a 10 ml con il solvente A (3.2). Portare quindi a volume il contenuto del pallone (A) con la miscela di isopropanolo-esano (3.1). Questa soluzione sarà utilizzata come soluzione di riferimento per la misura della soluzione del campione.
Pipettare 10 ml del solvente A (3.2) in altri due palloni tarati da 25 ml (C e D). Portare quindi a volume il contenuto del pallone (C) con la miscela di isopropanolo-esano (3.1). Questa soluzione sarà utilizzata come soluzione di riferimento per la misura della soluzione della prova in bianco.
Aggiungere 2 ml d'anilina (3.4) nei palloni (D) e (B). Riscaldare per 30 minuti su bagno d'acqua bollente per far sviluppare la colorazione. Raffreddare a temperatura ambiente, portare a volume con la miscela di isopropanolo-esano (3.1), mescolare e lasciar riposare per un'ora.
Determinare allo spettrofotometro a 440 nm, in vaschette di vetro da 1 cm, la densità ottica della soluzione della prova in bianco (D) confrontandola con la soluzione di riferimento (C) e la densità ottica della soluzione del campione (B) confrontandola con la soluzione di riferimento (A).
Sottrarre la densità ottica della soluzione della prova in bianco da quella della soluzione del campione (= densità ottica corretta). Calcolare a partire da questo valore il contenuto in gossipolo libero come indicato al punto 6.
5.3. Determinazione del gossipolo totale
Introdurre la quantità di sostanza da sottoporre all'analisi, contenente da 1 a 5 mg di gossipolo, in un pallone tarato da 50 ml e aggiungere 10 ml di solvente B (3.3). Preparare simultaneamente una prova in bianco, introducendo 10 ml di solvente B (3.3) in un altro pallone tarato da 50 ml. Riscaldare i due palloni per 30 minuti su un bagno d'acqua bollente. Raffreddare a temperatura ambiente e portare a volume il contenuto di ciascun pallone con la miscela di isopropanolo-esano (3.1). Mescolare e lasciar depositare per 10-15 minuti, quindi filtrare e raccogliere i filtrati in palloni a tappo smerigliato.
Pipettare 2 ml del filtrato del campione in due palloni tarati da 25 ml e 2 ml del filtrato della prova in bianco in due altri palloni da 25 ml. Prendere un pallone di ciascuna serie e completare i rispettivi contenuti a 25 ml con la miscela di isopropanolo-esano (3.1). Queste soluzioni saranno utilizzate come soluzione di riferimento.
Aggiungere 2 ml d'anilina (3.4) negli altri due palloni. Riscaldare per 30 minuti su bagno d'acqua bollente per far sviluppare la colorazione. Raffreddare a temperatura ambiente, portare a 25 ml con miscela di isopropanolo-esano (3.1), mescolare e lasciar riposare per un'ora.
Determinare la densità ottica come indicato al punto 5.2 per il gossipolo libero. Calcolare a partire da questo valore il contenuto in gossipolo totale come indicato al punto 6.
6. Calcolo dei risultati
Il calcolo dei risultati può essere fatto sia in base alla densità ottica specifica (6.1), sia per riferimento a una curva di taratura (6.2).
6.1. In base alla densità ottica specifica
Nelle condizioni descritte le densità ottiche specifiche sono le seguenti:
|
Gossipolo libero
|

|
|
Gossipolo totale
|

|
Il contenuto di gossipolo libero o totale nel campione è dato dalla formula seguente:

dove
|
E
|
=
|
densità ottica corretta, determinata come indicato al punto 5.2
|
|
p
|
=
|
quantità in g di sostanza analizzata;
|
|
a
|
=
|
parte aliquota del filtrato espressa in ml.
|
6.2. In base a una curva di taratura
6.2.1. Gossipolo libero
Predisporre 2 serie di 5 palloni tarati da 25 ml. In ogni serie pipettare rispettivamente 2,0, 4,0, 6,0, 8,0 e 10,0 ml della soluzione tipo A di gossipolo (3.5). Completare i volumi a 10 ml con il solvente A (3.2). Completare ogni serie con un pallone tarato da 25 ml contenente soltanto 10 ml del solvente A (3.2) (prova in bianco).
Portare a 25 ml il volume dei palloni della prima serie (compreso quello per la prova in bianco) con la miscela isopropanolo-esano (3.1) (serie di riferimento).
Aggiungere 2 ml d'anilina (3.4) in ciascuno dei palloni della seconda serie (compreso quello per la prova in bianco). Riscaldare per 30 minuti su bagno d'acqua bollente per far sviluppare la colorazione. Raffreddare a temperatura ambiente, portare a volume con la miscela isopropanolo-esano (3.1), mescolare e lasciar riposare per un'ora (serie tipo).
Determinare la densità ottica di ogni soluzione della serie tipo confrontandola con la soluzione corrispondente della serie di riferimento, alle condizioni indicate al punto 5.2. Tracciare la curva di taratura portando in ordinata i valori di densità ottica e in ascissa le quantità corrispondenti di gossipolo (in μg).
6.2.2. Gossipolo totale
Predisporre 6 palloni tarati da 50 ml. Porre nel primo 10 ml del solvente B (3.3) e negli altri rispettivamente 2,0, 4,0, 6,0, 8,0 e 10,0 ml della soluzione tipo B di gossipolo (3.6). Completare il contenuto di ogni pallone a 10 ml con il solvente B (3.3). Riscaldare per 30 minuti su bagno d'acqua bollente. Raffreddare a temperatura ambiente, portare a volume con la miscela isopropanolo-esano (3.1) e mescolare.
Introdurre 2,0 ml di queste soluzioni in due serie di 6 palloni tarati da 25 ml. Completare a 25 ml il contenuto dei palloni della prima serie con la miscela isopropanolo-esano (3.1) (serie di riferimento).
Aggiungere 2 ml d'anilina (3.4) in ciascuno dei palloni della seconda serie. Riscaldare per 30 minuti su bagno d'acqua bollente. Raffreddare a temperatura ambiente, portare a volume con la miscela isopropanolo-esano (3.1), mescolare e lasciar riposare per un'ora (serie tipo).
Determinare la densità ottica di ogni soluzione della serie tipo confrontandola con la soluzione corrispondente della serie di riferimento, alle condizioni indicate al punto 5.2. Tracciare la curva di taratura portando in ordinata i valori di densità ottica e in ascissa le quantità corrispondenti di gossipolo (in μg).
6.3. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
il 15 % in valore relativo rispetto al valore più elevato per i contenuti di gossipolo inferiori a 500 ppm,
|
|
—
|
75 ppm in valore assoluto per i contenuti compresi tra 500 e 750 ppm,
|
|
—
|
il 10 % in valore relativo rispetto al valore più elevato per i contenuti superiori a 750 ppm.
|
B. DETERMINAZIONE DEI LIVELLI DI DIOSSINE (PCDD/PCDF) E DI PCB DIOSSINA-SIMILI
I. METODI DI CAMPIONAMENTO E INTERPRETAZIONE DEI RISULTATI D'ANALISI
1. Finalità e campo d'applicazione
I campioni destinati al controllo ufficiale dei livelli di diossine [policlorodibenzodiossine (PCDF) e di policlorodibenzofurani (PCDF)] e di difenili policlorurati diossina-simili (PCB) (1) negli alimenti per animali sono prelevati secondo le disposizioni di cui all'allegato I. Vanno applicate le prescrizioni quantitative per il controllo delle sostanze o dei prodotti ripartiti in modo uniforme negli alimenti per animali di cui al punto 5.A dell'allegato I. I campioni globali così ottenuti sono considerati rappresentativi delle partite o sottopartite da cui sono prelevati. In base ai livelli determinati nei campioni di laboratorio, si accerta il rispetto o meno dei livelli massimi fissati nella direttiva 2002/32/CE del Parlamento europeo e del Consiglio (2).
2. Conformità della partita o sottopartita alle specifiche
La partita si considera accettata quando il risultato di una singola analisi non supera il livello massimo corrispondente fissato nella direttiva 2002/32/CE, tenuto conto dell'approssimazione della misurazione.
La partita non è conforme al livello massimo stabilito dalla direttiva 2002/32/CE se il risultato analitico con il limite superiore (3), confermato da una doppia analisi (4), supera il livello massimo oltre ogni ragionevole dubbio tenendo conto dell'incertezza della misura.
Dell'incertezza della misurazione si può tener conto in uno dei seguenti modi:
|
—
|
calcolando l’incertezza estesa, utilizzando un fattore di copertura di 2 corrispondente ad un livello di affidabilità del 95 % circa. Una partita non è conforme se il valore misurato meno U (incertezza estesa) supera il livello massimo. Qualora le diossine e i PCB diossina-simili siano oggetto di determinazioni separate, per la somma di entrambi si utilizza la somma delle stime dell'incertezza estesa dei risultati d'analisi ottenuti separatamente per le diossine e i PCB diossina-simili;
|
|
—
|
stabilendo il limite di decisione (CCα) conformemente alle disposizioni della decisione 2002/657/CE della Commissione (5) (punto 3.1.2.5 dell'allegato — nel caso di sostanze con un limite stabilito consentito). Una partita non è conforme se il valore misurato è pari o superiore al CCα.
|
Le presenti norme di interpretazione si applicano ai risultati d'analisi ottenuti dal campione destinato al controllo ufficiale. Resta salva la facoltà degli Stati membri di applicare norme nazionali ad analisi effettuate a fini di difesa o arbitrato.
II. PREPARAZIONE DEI CAMPIONI E PRESCRIZIONI PER I METODI D'ANALISI IMPIEGATI NEL CONTROLLO UFFICIALE DEI LIVELLI DI DIOSSINE (PCDD/PCDF) E DI PCB DIOSSINA-SIMILI
1. Finalità e campo d'applicazione
Queste prescrizioni si applicano all'analisi delle materie prime per mangimi e degli alimenti per animali volta a determinare la presenza di diossine [policlorodibenzodiossine (PCDD) e policlorodibenzofurani (PCDF)] e difenili policlorurati diossina-simili (PCB).
Il controllo della presenza di diossine negli alimenti per animali può essere effettuato mediante una strategia che preveda un metodo di screening al fine di selezionare i campioni i cui contenuti di diossine e di PCB diossina-simili eccedano il livello considerato o siano inferiori a quest'ultimo di un valore che non superi il 25 %. Occorre poi determinare e/o confermare la concentrazione di diossine nei campioni che presentano livelli significativi tramite un metodo di conferma.
L'obiettivo dei metodi di screening è rilevare la presenza di diossine e PCB diossina-simili al livello considerato. Essi sono dotati di una grande capacità di trattamento di campioni, il che consente di passare al vaglio un elevato numero di campioni per ricercare quelli che potrebbero rivelarsi positivi. Inoltre, sono destinati specificamente a evitare falsi risultati negativi.
I metodi di conferma forniscono informazioni complete o complementari che consentono di individuare e quantificare in maniera inequivocabile le diossine e i PCB diossina-simili al livello considerato.
2. Contesto
Dal momento che i campioni prelevati dall'ambiente e i campioni biologici (inclusi quelli di materie prime per mangimi e di alimenti per animali) contengono in generale miscele complesse di diversi congeneri di diossine, per agevolare la valutazione dei rischi è stato elaborato il concetto di «fattori di tossicità equivalente» (TEF). I TEF consentono di esprimere concentrazioni di miscele di PCDD e PCDF sostituiti alle posizioni 2,3,7,8 e alcune forme di PCB non-orto e mono-orto clorosostituiti aventi proprietà simili a quelle delle diossine in equivalenti tossici (TEQ) di 2,3,7,8-TCDD. Le concentrazioni delle singole sostanze in un dato campione sono dapprima moltiplicate per il corrispondente TEF e poi sommate per ottenere la concentrazione totale dei composti diossina-simili espressa in TEQ.
Unicamente ai fini del presente regolamento, il limite specifico accettato di quantificazione di un congenere è la concentrazione di un analita nell'estratto di un campione che produca una risposta strumentale a due ioni differenti, da controllare con un rapporto S/R (segnale/rumore) di 3:1 per il segnale meno sensibile e il rispetto dei requisiti di base, quali, ad esempio, il tempo di ritenzione e il rapporto isotopico, secondo la procedura di determinazione descritta nel metodo EPA 1613, revisione B.
3. Prescrizioni in materia di garanzia della qualità nella preparazione dei campioni
Si applicano le disposizioni generali sulla preparazione dei campioni destinati all'analisi indicate nell'allegato II.
Occorre inoltre che siano rispettate le seguenti prescrizioni:
|
—
|
i campioni devono essere conservati e trasportati in appositi contenitori di vetro, alluminio, polipropilene o polietilene, dopo avere rimosso ogni traccia di polvere di carta dal contenitore. Gli strumenti in vetro vengono risciacquati con solventi sottoposti a un controllo volto a determinare la presenza di diossine,
|
|
—
|
occorre effettuare un'analisi in bianco, ovvero effettuare l'intera procedura analitica, in assenza del campione,
|
|
—
|
il peso dell'estratto deve essere tale da rispondere ai requisiti relativi alla sensibilità.
|
4. Prescrizioni relative ai laboratori
|
—
|
I laboratori devono dimostrare la validità del metodo nell'intervallo di tolleranza del livello considerato, ad esempio a livelli di 0,5 volte, 1 volta e 2 volte il livello considerato, con un coefficiente di variazione accettabile per analisi ripetute. Per ulteriori informazioni sui criteri di validità, cfr. il punto 5.
|
|
—
|
Il limite di quantificazione per un metodo di conferma non deve essere superiore a un quinto del livello considerato, per garantire coefficienti di variazione accettabili nell'intervallo summenzionato.
|
|
—
|
Vanno effettuati regolarmente controlli in bianco ed esperimenti o analisi sui campioni di controllo con l'aggiunta di indicatori (di preferenza, se disponibile, materiale di riferimento certificato), quali misure interne di garanzia della qualità.
|
|
—
|
Il modo migliore per i laboratori di dimostrare la loro capacità di effettuare analisi specifiche consiste nel partecipare con successo a studi condotti in collaborazione con altri riguardanti la valutazione della competenza dei laboratori. Tuttavia, il buon esito della partecipazione a studi interlaboratori, ad esempio su campioni di suolo o di acque residue, non basta a dimostrare che il laboratorio è altrettanto competente a trattare campioni di prodotti alimentari o alimenti per animali, caratterizzati da livelli di contaminazione inferiori. È pertanto requisito imprescindibile la partecipazione regolare a studi interlaboratori sulla determinazione di contenuti di diossina e di PCB diossina-simili in matrici di prodotti alimentari e/o alimenti per animali corrispondenti.
|
|
—
|
I laboratori sono accreditati da un organismo riconosciuto in conformità con la Guida ISO 58, che certifichi l'applicazione delle procedure di garanzia della qualità relativamente ai metodi d'analisi. I laboratori sono accreditati in base alla norma ISO/IEC/17025.
|
5. Prescrizioni relative alle procedure d'analisi per le diossine e i PCB diossina-simili
Requisiti di base relativi alla validità delle procedure d'analisi
|
—
|
Elevata sensibilità e limiti di rilevabilità bassi. Per quanto concerne le PCDD e i PCDF, le quantità rilevabili devono essere dell'ordine del picogrammo di TEQ (10-12 g), data l'estrema tossicità di alcuni di questi composti. È noto che i PCB si manifestano in contenuti più elevati rispetto alle PCDD e ai PCDF. Per quanto concerne la maggior parte dei congeneri del gruppo dei PCB, è sufficiente una sensibilità dell'ordine del nanogrammo (10-9 g). Tuttavia, per determinare i congeneri più tossici di PCB diossina-simili (in particolare i congeneri non-orto sostituiti) bisogna arrivare alla stessa sensibilità delle PCDD e dei PCDF.
|
|
—
|
Alta selettività (specificità). Occorre distinguere le PCDD, i PCDF e i PCB diossina-simili da una moltitudine di altri composti che, estratti simultaneamente dal campione e suscettibili d'interferire, sono presenti in concentrazioni di molto superiori a quelle degli analiti da rilevare. Per quanto concerne i metodi di gascromatografia e/o spettrometria di massa (GC/MS), è necessario distinguere tra diversi congeneri, in particolare tra quelli tossici (ad esempio i diciassette PCDD e PCDF sostituiti alle posizioni 2,3,7,8 e i PCB diossina-simili) e altri congeneri. Mediante biotest dovrebbe essere possibile determinare selettivamente i valori di TEQ, in quanto somma di PCDD, PCDF e PCB diossina-simili.
|
|
—
|
Estrema accuratezza (esattezza e precisione). La determinazione fornisce una stima valida e affidabile della concentrazione reale presente in un campione. È necessario porre estrema cura (accuratezza della misurazione: grado di concordanza tra il risultato della misurazione e il valore reale del misurando o attribuito a esso) per evitare che i risultati dell'analisi di un campione siano respinti a causa della scarsa affidabilità della stima dei TEQ. L'accuratezza è espressa come esattezza (differenza tra il valore medio misurato per un analita in un materiale certificato e il suo valore certificato, espressa in percentuale di tale valore) e precisione (deviazione standard relativa RSDR calcolata in base a risultati ottenuti in condizioni di riproducibilità).
|
I metodi di screening possono comprendere biotest e metodi GC/MS, mentre i metodi di conferma sono costituiti dalla cromatografia in fase gassosa ad alta risoluzione e dalla spettrometria di massa ad alta risoluzione (HRGC/HRMS).
Per il valore totale in TEQ vanno osservati i seguenti criteri:
|
|
Metodi di screening
|
Metodi di conferma
|
|
Percentuale di falsi negativi
|
< 1 %
|
|
|
Esattezza
|
|
da - 20 % a + 20 %
|
|
Precisione RSDR
|
< 30 %
|
< 15 %
|
6. Prescrizioni specifiche relative ai metodi d'analisi GC/MS con finalità di screening o di conferma
|
—
|
Per convalidare la procedura d'analisi, occorre aggiungere all'inizio dell'analisi, ad esempio prima dell'estrazione, standard interni di PCDD/F clorosostituiti alle posizioni 2,3,7,8 e marcati con 13C e standard interni di PCB diossina-simili marcati con 13C. Va aggiunto almeno un congenere per ciascun gruppo omologo di PCDD/F da tetra a octaclorati e almeno un congenere per ciascun gruppo omologo di PCB diossina-simile (in alternativa, è possibile aggiungere almeno un congenere per ciascuna funzione di registrazione di ioni selezionati tramite spettrometria di massa utilizzata per il controllo di PCDD/F e PCB diossina-simili). Si consiglia vivamente, soprattutto per i metodi di conferma, di utilizzare l'insieme dei diciassette standard interni di PCDD/F sostituiti alle posizioni 2,3,7,8 marcati con 13C, nonché la totalità dei dodici standard interni di PCB diossina-simili marcati con 13C.
|
|
—
|
Sono inoltre determinati i fattori di risposta relativa per quei congeneri ai quali non è stato aggiunto alcun analogo marcato con 13C, utilizzando soluzioni di taratura adeguate.
|
|
—
|
Per gli alimenti per animali d'origine vegetale e per gli alimenti per animali d'origine animale con un contenuto di grassi inferiore al 10 %, è obbligatoria l'aggiunta di standard interni prima dell'estrazione. Per i prodotti d'origine animale con un contenuto di grasso superiore al 10 %, gli standard interni possono essere aggiunti o prima dell'estrazione o dopo l'estrazione del grasso. L'efficacia dell'estrazione è adeguatamente convalidata, a seconda della fase in cui sono stati introdotti gli standard interni e del modo in cui i risultati sono riportati (sulla base del prodotto o dei grassi).
|
|
—
|
Prima dell'analisi GC/MS, occorre aggiungere 1 o 2 standard di recupero (surrogato).
|
|
—
|
È necessario effettuare un controllo del recupero. Per quanto riguarda i metodi di conferma, i tassi di recupero dei singoli standard interni devono essere compresi tra il 60 % e il 120 %. Tassi di recupero inferiori o superiori per singoli congeneri, in particolare per alcune dibenzodiossine e alcuni dibenzofurani epta e octaclorati, sono accettabili, purché il loro contributo al valore TEQ non superi il 10 % del valore totale TEQ (tenendo conto della somma di PCDD/F e di PCB diossina-simili). Per quanto concerne i metodi di screening, i recuperi devono essere compresi tra il 30 % e il 140 %.
|
|
—
|
Separare le diossine dai composti clorurati interferenti, quali i PCB non diossina-simili e gli eteri clorurati di difenile, ricorrendo ad adeguate tecniche cromatografiche (di preferenza tramite una colonna di florisil, d'allumina e/o di carbone).
|
|
—
|
È sufficiente la separazione gascromatografica degli isomeri (< 25 % da picco a picco tra 1,2,3,4,7,8-HxCDF e 1,2,3,6,7,8-HxCDF).
|
|
—
|
Per la determinazione, si fa riferimento al metodo EPA Method 1613, revisione B: «Tetra- through Octachlorinated Dioxins and Furans by Isotope Dilution HRGC-HRMS», o ad un altro metodo con criteri di rendimento equivalenti.
|
|
—
|
La differenza tra il livello massimo e il livello minimo non deve essere superiore al 20 % per gli alimenti per animali con una contaminazione da diossina pari o superiore al livello massimo. Per gli alimenti per animali che presentano livelli di contaminazione nettamente inferiori al livello massimo, la differenza può essere dell'ordine del 25-40 %.
|
7. Metodi analitici di screening
7.1. Introduzione
Il metodo di screening consente di applicare vari approcci analitici: un approccio puramente di screening e un approccio quantitativo.
Approccio di screening
La risposta dei campioni è confrontata con quella di un campione di riferimento al livello considerato. I campioni la cui risposta è inferiore a quella del campione di riferimento sono considerati negativi, mentre quelli con risposta superiore sono ritenuti positivi. Prescrizioni:
|
—
|
in ogni serie di prove un campione di riferimento e uno bianco devono essere estratti e analizzati allo stesso momento e alle medesime condizioni. La risposta del campione di riferimento deve essere decisamente superiore a quella del campione in bianco,
|
|
—
|
includere campioni di riferimento supplementari con concentrazione pari a 0,5 volte e 2 volte il livello considerato, per dimostrare l'efficacia del test nell'intervallo considerato per il controllo del livello considerato,
|
|
—
|
qualora si analizzino altre matrici, occorre dimostrare la validità dei campioni di riferimento, utilizzando di preferenza campioni il cui livello di TEQ, stabilito tramite HRGC/HRMS, sia simile a quello del campione di riferimento o di un bianco addizionato per raggiungere tale livello,
|
|
—
|
dato che nei biotest non si possono utilizzare standard interni, i test di ripetibilità sono estremamente importanti per ottenere informazioni sulla deviazione standard nell'ambito di una serie di prove. Il coefficiente di variazione deve essere inferiore al 30 %,
|
|
—
|
per quanto concerne i biotest, definire quali sono i composti-bersaglio, le potenziali interferenze e il valore massimo tollerato per il bianco.
|
Approccio quantitativo
L'approccio quantitativo comprende obbligatoriamente una serie di diluizioni tipo, un processo di purificazione e di misurazione doppio o triplo, nonché analisi in bianco e controlli del recupero. Il risultato può essere espresso in TEQ, dando per scontato che i composti responsabili del segnale soddisfino il principio TEQ. A tal fine, si può impiegare la TCDD (o una miscela-tipo di diossine/furani/PCB diossina-simili) per elaborare una curva di taratura che consenta di calcolare il livello di TEQ nell'estratto e, di conseguenza, nel campione. Tale risultato è poi corretto con il livello di TEQ calcolato per un campione in bianco (per tenere conto di impurezze derivanti dai solventi e dalle sostanze chimiche utilizzate) e per il recupero (quest'ultima quantità è calcolata a partire dal livello di TEQ in un campione di controllo qualità la cui concentrazione è analoga a quella del livello massimo considerato). È fondamentale osservare che una parte della perdita apparente del recupero può essere dovuta agli effetti della matrice e/o alle differenze tra i valori dei TEF nei biotest e i valori dei TEF ufficiali stabiliti dall'OMS.
7.2. Prescrizioni relative ai metodi d'analisi utilizzati per lo screening
|
—
|
Lo screening può essere effettuato tramite metodi d'analisi GC/MS e biotest. Ai metodi GC/MS si applicano le prescrizioni stabilite al punto 6. Prescrizioni specifiche sono stabilite al punto 7.3 per i biotest cellulari, e al punto 7.4 per i biotest realizzati con kit.
|
|
—
|
Sono necessarie informazioni sul numero di risultati falsi positivi e falsi negativi di un'ampia serie di campioni al di sopra e al di sotto del livello massimo o della soglia d'azione, raffrontati al valore di TEQ determinato tramite metodo analitico di conferma. La percentuale reale di falsi negativi deve essere inferiore all'1 %. Affinché il ricorso al metodo di screening risulti vantaggioso, la percentuale di campioni falsi positivi è sufficientemente bassa.
|
|
—
|
I risultati positivi devono essere sempre convalidati tramite un metodo analitico di conferma (HRGC/HRMS). Inoltre, i campioni corrispondenti a una vasta gamma di TEQ sono confermati tramite HRGC/HRMS (circa 2-10 % dei campioni negativi). Sono forniti dati sulla corrispondenza tra i risultati dei biotest e quelli della HRGC/HRMS.
|
7.3. Prescrizioni specifiche relative ai biotest cellulari
|
—
|
Quando si effettua un biotest, si deve utilizzare in ogni prova una serie di concentrazioni di riferimento di TCDD o una miscela di diossine/furani (curva di risposta con un R2 > 0,95 per una dose completa). Tuttavia, ai fini dello screening, nell'analisi dei campioni a bassa concentrazione è possibile utilizzare una curva più dettagliata nei livelli bassi.
|
|
—
|
Per i risultati del biotest in un intervallo di tempo costante, si usa una concentrazione di riferimento di TCDD (circa 3 volte il limite di quantificazione) su un modulo di controllo della qualità. In alternativa, ci si può basare anche sulla risposta relativa di un campione di riferimento paragonata a una curva di taratura di TCDD, dato che la risposta delle cellule può dipendere da molteplici fattori.
|
|
—
|
Compilare e verificare i grafici del controllo della qualità (QC) per ogni tipo di materiale di riferimento allo scopo di garantire che il risultato sia conforme agli orientamenti indicati.
|
|
—
|
L'induzione della diluizione utilizzata per il campione deve situarsi nella parte lineare della curva di risposta, in particolare per i calcoli quantitativi. I campioni situati al di sopra di detta parte lineare devono essere diluiti e sottoposti a una nuova analisi. Occorre pertanto analizzare 3 o più diluizioni alla volta.
|
|
—
|
La deviazione standard percentuale non è superiore al 15 % quando si effettua una misurazione tripla per ogni diluizione del campione, né superiore al 30 % per tre esperimenti indipendenti.
|
|
—
|
È possibile scegliere come limite di determinazione un valore equivalente a 3 volte la deviazione standard della soluzione di solvente in bianco o della risposta di fondo. Un altro metodo consiste nell'applicare una risposta che sia superiore alla risposta di fondo sulla curva di taratura del giorno (fattore d'induzione equivalente a 5 volte il solvente in bianco). È possibile scegliere come limite di quantificazione un valore 5-6 volte superiore alla deviazione standard del solvente in bianco o della risposta di fondo oppure utilizzare una concentrazione che corrisponda ad una risposta nettamente superiore alla risposta di fondo sulla curva di taratura del giorno(fattore d'induzione 10 volte superiore al solvente in bianco).
|
7.4. Prescrizioni specifiche relative ai biotest effettuati con kit
|
—
|
Occorre assicurarsi che i biotest effettuati con kit dimostrino una sensibilità e un'affidabilità sufficienti per poter essere applicati agli alimenti per animali.
|
|
—
|
Occorre seguire le istruzioni del fabbricante relative alla preparazione dei campioni e alle analisi.
|
|
—
|
Non si utilizza il kit oltre la data di scadenza indicata.
|
|
—
|
Non si utilizzano materiali o componenti previsti per altri kit.
|
|
—
|
I kit sono conservati e utilizzati alle temperature di conservazione e di impiego indicate.
|
|
—
|
Il limite di rilevazione per gli immunodosaggi si ottiene sommando la media e un valore pari a 3 volte la deviazione standard, basandosi su una serie di dieci analisi del bianco, e dividendo questa somma per il valore della pendenza nell'equazione di regressione lineare.
|
|
—
|
Si utilizzano standard di riferimento per le prove di laboratorio al fine di garantire che la risposta allo standard rientri in un intervallo di valori accettabile.
|
8. Comunicazione dei risultati
A condizione che il metodo d'analisi impiegato lo consenta, i risultati dell'analisi comprendono i tenori dei singoli congeneri di PCDD/F e PCB e i risultati analitici devono essere indicati come limite inferiore, superiore e intermedio, onde fornire la maggior quantità di dati possibile e consentire così d'interpretare i risultati in base alle prescrizioni specifiche.
La relazione menziona inoltre il contenuto lipidico del campione e il metodo impiegato per l'estrazione del grasso.
I tassi di recupero dei singoli standard interni devono essere forniti se si situano al di fuori dell'intervallo menzionato al punto 6 o qualora eccedano il livello massimo. In tutti gli altri casi vanno forniti su richiesta.
Occorre indicare anche l'incertezza della misura poiché tale parametro va preso in considerazione nel determinare la conformità di un campione. I risultati analitici sono pertanto indicati utilizzando la formula «x +/- U», dove x è il risultato dell'analisi e U l'incertezza di misura estesa, calcolata per mezzo di un fattore di copertura 2 che dà un livello di affidabilità del 95 % circa. Qualora le diossine e i PCB diossina-simili siano oggetto di determinazioni separate, per la somma di entrambi si utilizza la somma delle stime dell'incertezza di misura estesa di risultati di analisi separate delle diossine e dei PCB diossina-simili.
Qualora venga presa in considerazione l'incertezza della misura applicando il CCα (quale descritto nell'allegato I, capo 2, di questa parte B), va indicato tale parametro.
(1) Tabella TEF («fattori di tossicità equivalente») per diossine, furani e PCB diossina-simili
|
Congenere
|
Valore TEF
|
Congenere
|
Valore TEF
|
|
Dibenzo-p-diossine («PCDD»)
|
|
PCB diossina-simili:
|
|
|
2,3,7,8-TCDD
|
1
|
|
|
|
1,2,3,7,8-PeCDD
|
1
|
Non-orto PCB
|
|
|
1,2,3,4,7,8-HxCDD
|
0,1
|
PCB 77
|
0,0001
|
|
1,2,3,6,7,8-HxCDD
|
0,1
|
PCB 81
|
0,0001
|
|
1,2,3,7,8,9-HxCDD
|
0,1
|
PCB 126
|
0,1
|
|
1,2,3,4,6,7,8-HpCDD
|
0,01
|
PCB 169
|
0,01
|
|
OCDD
|
0,0001
|
Mono-orto PCBs
|
|
|
|
|
PCB 105
|
0,0001
|
|
Dibenzofurani («PCDF»)
|
|
PCB 114
|
0,0005
|
|
2,3,7,8-TCDF
|
0,1
|
PCB 118
|
0,0001
|
|
1,2,3,7,8-PeCDF
|
0,05
|
PCB 123
|
0,0001
|
|
2,3,4,7,8-PeCDF
|
0,5
|
PCB 156
|
0,0005
|
|
1,2,3,4,7,8-HxCDF
|
0,1
|
PCB 157
|
0,0005
|
|
1,2,3,6,7,8-HxCDF
|
0,1
|
PCB 167
|
0,00001
|
|
1,2,3,7,8,9-HxCDF
|
0,1
|
PCB 189
|
0,0001
|
|
2,3,4,6,7,8-HxCDF
|
0,1
|
|
|
|
1,2,3,4,6,7,8-HpCDF
|
0,01
|
|
|
|
1,2,3,4,7,8,9-HpCDF
|
0,01
|
|
|
|
OCDF
|
0,0001
|
|
|
|
Abbreviazioni: «T» = tetra; «Pe» = penta; «Hx» = esa; «Hp» = epta; «O» = octa; «CDD» = clorodibenzodiossina; «CDF» = clorodibenzofurano; «CB» = clorobifenile.
|
(2) GU L 140 del 30.5.2002, pag. 10.
(3) Per il calcolo del «limite superiore», si suppone che il contributo all'equivalente tossico (TEQ) di ogni congenere non quantificato sia uguale alla soglia di quantificazione.
Per il calcolo del «limite inferiore», si suppone che il contributo al TE di ogni congenere non quantificato sia uguale a zero.
Per il calcolo del «valore intermedio», si suppone che il contributo al TE di ogni congenere non quantificato sia uguale alla metà della soglia di quantificazione.
(4) La doppia analisi è necessaria per escludere la possibilità di una contaminazione incrociata interna o di scambio accidentale dei campioni. La prima analisi, che tiene conto dell’incertezza della misura, è utilizzata per verificare la conformità.
Qualora l'analisi avvenga nell’ambito di un incidente di contaminazione da diossina, è possibile omettere la conferma mediante doppia analisi se i campioni selezionati per l'analisi possono essere associati, grazie alla tracciabilità, a tale incidente.
(5) GU L 221 del 17.8.2002, pag. 8.
ALLEGATO VI
METODI D'ANALISI PER LA DETERMINAZIONE DEI COSTITUENTI DI ORIGINE ANIMALE NELL'AMBITO DEL CONTROLLO UFFICIALE DEGLI ALIMENTI PER ANIMALI
Condizioni per la rilevazione microscopica, l'identificazione o la stima dei costituenti di origine animale negli alimenti per animali
1. Finalità e campo d'applicazione
Queste condizioni si applicano all'individuazione, mediante esame microscopico, dei costituenti di origine animale (definiti come prodotti della trasformazione di carcasse e parti di carcasse di mammiferi, volatili da cortile e pesci) presenti negli alimenti per animali che ha luogo nell'ambito del programma di controllo coordinato nel settore dell'alimentazione animale in conformità del regolamento (CE) n. 882/2004 del Parlamento europeo e del Consiglio (1). A condizione che i metodi descritti nel presente allegato siano utilizzati in tutte le analisi ufficiali, può essere effettuata una seconda analisi con metodi diversi o alternativi, al fine di migliorare l'individuazione di alcuni tipi di costituenti di origine animale o per determinarne con maggior precisione l'origine. Inoltre, può essere utilizzata una variante del protocollo per esaminare alcuni tipi di costituenti specifici di origine animale come, ad esempio, il plasma o le ossa con sevo (cfr. anche il punto 9), sempre che tali analisi siano effettuate in aggiunta a quelle previste dal programma di controllo coordinato.
2. Sensibilità
A seconda della natura dei costituenti di origine animale, è possibile individuare negli alimenti per animali quantità anche minime (< 0,1 %).
3. Principio
Ai fini dell'individuazione si utilizza un campione rappresentativo, prelevato secondo le disposizioni di cui all'allegato I, e adeguatamente preparato. Il protocollo che segue serve al trattamento di alimenti a basso tenore di umidità. Gli alimenti con un tenore di umidità superiore al 14 % sono essiccati (condensati) prima del trattamento. Alimenti per animali o materie prime speciali per tali alimenti (ad esempio grassi e oli) necessitano di un trattamento particolare (cfr. punto 9). I costituenti di origine animale sono individuati sulla base di caratteristiche tipiche, identificabili al microscopio (ad esempio fibre muscolari o altre particelle di carne, cartilagine, ossa, corna, peli, setole, sangue, piume, gusci d'uovo, lische, scaglie). L'individuazione va fatta sia sulla frazione granulometrica (6.1) sia sul sedimento concentrato (6.2) del campione.
4. Reattivi
4.1. Agenti di rivestimento
|
4.1.1.
|
Cloralio idrato (acquoso, 60 % in p/v)
|
|
4.1.2.
|
Liscivia (NaOH a 2,5 % in p/v o KOH a 2,5 % in p/v) per le frazioni granulometriche
|
|
4.1.3.
|
Olio di paraffina o glicerolo (viscosità: 68-81) per le osservazioni al microscopio del sedimento
|
4.2. Agenti di risciacquo
4.3. Agenti concentratori
|
4.3.1.
|
Tetracloroetilene (densità 1,62)
|
4.4. Reagenti di mordenzatura
|
4.4.1.
|
Soluzione iodata/di ioduro di potassio (sciogliere 2 g di ioduro di potassio in 100 ml di acqua e aggiungere 1 g di iodio agitando ripetutamente)
|
|
4.4.2.
|
Rosso alizarina (diluire 2,5 ml di acido idroclorico 1M in 100 ml di acqua, aggiungere 200 mg di rosso alizarina alla soluzione)
|
|
4.4.3.
|
Reagente cisteina (2 g di acetato di piombo, 10 g di NaOH/100 ml H2O)
|
|
4.4.4.
|
Soluzione iodata/di ioduro di potassio (sciolta in etanolo al 70 %)
|
4.5. Reagente sbiancante
|
4.5.1
|
Soluzione di ipoclorito di sodio in commercio (9,6 % cloro attivo)
|
5. Apparecchiatura e accessori
|
5.1.
|
Bilancia analitica (precisione a 0,01 g, salvo per il sedimento concentrato: 0,001 g)
|
|
5.2.
|
Strumento per frantumazione (frantumatore o mortaio speciale per alimenti per animali il cui contenuto di grassi al momento dell'analisi è superiore al 15 %).
|
|
5.3.
|
Setaccio a maglie quadrate di 0,50 mm al massimo
|
|
5.4.
|
Imbuto separatore o coppa di decantazione a fondo conico
|
|
5.5.
|
Stereomicroscopio (ingrandimento minimo di 40 volte)
|
|
5.6.
|
Microscopio composto (ingrandimento minimo di 400 volte), in trasparenza o a luce polarizzata
|
|
5.7.
|
Normale vetreria da laboratorio
Tutta l'apparecchiatura è perfettamente pulita. Gli imbuti separatori e la vetreria non devono essere lavati a mano. I setacci vanno puliti usando una spazzola a setole rigide.
|
6. Procedimento
Il mangime granulare può essere pre-setacciato se le due frazioni sono analizzate come campione separato.
Si trattano almeno 50 g del campione [macinati con cura usando gli strumenti adatti per ottenere la struttura desiderata (5.2)]. Dal materiale macinato si prelevano due parti rappresentative, una per la frazione granulometrica (almeno 5 g) (6.1) e l'altra per il sedimento concentrato (almeno 5 g) (6.2). A fini di individuazione si può procedere anche alla colorazione mediante reagenti di mordenzatura (6.3).
Allo scopo di indicare la natura delle proteine animali e l'origine delle particelle si può ricorrere al supporto fornito da un sistema qual è ARIES o a campioni di riferimento.
6.1. Individuazione di costituenti di origine animale nelle frazioni granulometriche
Setacciare almeno 5 g del campione attraverso il setaccio (5.3) in due frazioni.
La frazione o le frazioni granulometriche a particelle grandi (o una parte rappresentativa della frazione) vengono versate in sottile strato su un supporto adatto e analizzate sistematicamente con lo stereomicroscopio (5.5) a vari ingrandimenti per individuare i costituenti di origine animale.
Vetrini con la frazione granulometrica delle particelle più sottili sono analizzate sistematicamente al microscopio composto (5.6) a vari ingrandimenti per individuare i costituenti di origine animale.
6.2. Identificazione di costituenti di origine animale nel sedimento concentrato
Trasferire almeno 5 g (precisione 0,01 g) del campione in un imbuto separatore o in una coppa di decantazione a fondo conico e trattarli con almeno 50 ml di tetracloroetilene (4.3.1). Agitare o mescolare ripetutamente la miscela così ottenuta.
|
—
|
Se si usa un imbuto separatore chiuso lasciar riposare il sedimento per un tempo sufficiente (almeno 3 minuti) prima di separare il sedimento. Agitare ripetutamente e lasciare riposare di nuovo per almeno 3 minuti. Il sedimento si separa ancora una volta.
|
|
—
|
Se si usa una coppa aperta, il sedimento deve riposare per almeno 5 minuti prima di separarlo.
|
Il residuo totale viene fatto asciugare e successivamente pesato (precisione 0,001 g). La pesatura è necessaria unicamente nel caso in cui sia richiesta una valutazione. Se il sedimento è composto da diverse particelle più grandi può essere setacciato in due frazioni (5.3). Il residuo secco è esaminato allo stereomicroscopio (5.5) e al microscopio composto (5.6) per individuare costituenti a base di ossa.
6.3. Utilizzo di agenti di rivestimento e di reagenti di mordenzatura
L'individuazione al microscopio di costituenti di origine animale può essere facilitata con l'uso di speciali agenti di rivestimento o reagenti di mordenzatura.
Cloralio idrato (4.1.1): scaldando con cautela, le strutture cellulari sono più chiaramente visibili grazie alla gelatinizzazione dei grani di amido e all'evacuazione delle cellule indesiderabili.
Liscivia (4.1.2.): sia l'idrossido di sodio che l'idrossido di potassio chiarificano le materie prime dell'alimento, facilitando così l'individuazione di fibre muscolari, peli o altre strutture cheratiniche.
Olio di paraffina e glicerolo (4.1.3.): i costituenti a base di ossa possono essere individuati facilmente in questo agente di rivestimento in quanto la maggior parte delle lacune si riempie d'aria e si presenta quindi sotto forma di buchi neri di circa 5-15 μm.
Soluzione di iodio/di ioduro di potassio (4.4.1): utilizzato per il rilevamento dell'amido (colore blu-violetto) e delle proteine (colore giallo-arancio). Le soluzioni possono essere diluite se necessario.
Soluzione di rosso alizarina (4.4.2.): colorazione rosso/rosa di ossa, lische e scaglie. Prima di essiccarlo (cfr. punto 6.2), trasferire il sedimento totale in un tubo di vetro e risciacquarlo due volte con circa 5 ml di alcool (4.2.1.) (utilizzare ogni volta un miscelatore, lasciare riposare per circa un minuto ed eliminare il solvente). Prima di utilizzare questo reagente di mordenzatura il sedimento è sbiancato aggiungendo almeno 1 ml di soluzione di ipoclorito di sodio (4.5.1). Lasciare reagire per almeno 10 minuti. Riempire il tubo con acqua, lasciar riposare la miscela per 2-3 minuti ed eliminare l'acqua e le particelle in sospensione. Risciacquato ancora due volte il residuo con circa 10 ml di acqua (usare un miscelatore, lasciare riposare e versare via l'acqua ogni volta). Aggiungere da due a dieci gocce o più della soluzione rosso alizarina (a seconda della quantità di residuo). Agitare la miscela e lasciare reagire per qualche secondo. Risciacquare due volte il sedimento colorato con circa 5 ml di alcool (4.2.1) e poi con acetone (4.2.2) (usare un miscelatore, lasciare riposare per circa un minuto ed eliminare il solvente). Il residuo è ora pronto per essere essiccato.
Reagente cisteina (4.4.3.): scaldando con cautela, i costituenti che contengono cisteina (peli, piume, ecc.) prendono una colorazione nera/marrone.
6.4. Analisi di alimenti per animali che possono contenere farina di pesce
Osservare al microscopio composto almeno un vetrino preparato con la frazione setacciata fine e con la frazione di sedimento fine (cfr. punti 6.1 e 6.2).
Se l'etichetta indica che tra gli ingredienti figura la farina di pesce o se nel corso di un primo esame si sospetta o si accerta la presenza di farina di pesce, vanno esaminati almeno altri due vetrini della frazione setacciata fine del campione originale e della frazione del residuo totale.
7. Calcolo e valutazione
Gli Stati membri assicurano che quando viene effettuata un'analisi ufficiale siano applicate le procedure descritte in questo punto allo scopo di stimare il contenuto (e non solo la presenza) di costituenti di origine animale.
Il calcolo può essere effettuato solo se i costituenti di origine animale contengono frammenti ossei.
Nei preparati al microscopio si possono distinguere frammenti di ossa delle specie terrestri a sangue caldo (ad esempio mammiferi ed uccelli) dai diversi tipi di ossa di pesce grazie alle lacune tipiche. La percentuale di costituenti di origine animale nel campione è valutata prendendo in considerazione:
|
—
|
la percentuale stimata (peso %) di frammenti ossei nel sedimento concentrato, e
|
|
—
|
la percentuale di osso (peso %) nei costituenti di origine animale.
|
La stima deve essere basata su almeno tre (se possibile) preparati e almeno cinque campi per preparato. Negli alimenti per animali composti il sedimento concentrato non contiene in genere solo frammenti di ossa di animali terrestri e di lische di pesce, ma anche altre particelle dal peso specifico elevato, come ad esempio minerali, sabbia, frammenti di minerali lignificati, ecc.
7.1. Valore stimato della percentuale di frammenti ossei
% di frammenti ossei di animali terrestri = (S × c)/W
% di frammenti di lische e scaglie = (S × d)/W
|
[S =
|
peso del sedimento (mg), c = fattore di correzione ( %) per la percentuale stimata di ossa di animali terrestri nel sedimento, d = fattore di correzione ( %) per la percentuale stimata di frammenti di ossa e scaglie di pesce nel sedimento, W = peso del campione di materiale utilizzato per la sedimentazione (mg)].
|
7.2. Valore stimato dei costituenti di origine animale
La percentuale di ossa nei prodotti di origine animale può variare in modo notevole. (La percentuale di osso è del 50-60 % nel caso di farine d'ossa ed è dell'ordine del 20-30 % nel caso di farine di carne; nelle farine di pesce il tenore di ossa e di scaglie varia in funzione della categoria e dell'origine della farina di pesce, ma è normalmente compresa tra il 10 e il 20 %).
Se si conosce il tipo di farina animale contenuta nel campione, è possibile procedere alla stima.
Contenuto stimato dei costituenti derivati da prodotti a base di animali terrestri ( %) = (S × c)/(W × f) × 100
Contenuto stimato di costituenti derivati da prodotti a base di pesce ( %) = (S × d)/(W × f) × 100
|
[S =
|
peso del sedimento (mg), c = fattore di correzione ( %) per la percentuale stimata di costituenti derivati da ossa di animali terrestri nel sedimento, d = fattore di correzione ( %) per la percentuale stimata di frammenti di ossa e scaglie di pesce nel sedimento, f = fattore di correzione per la percentuale di ossa nei costituenti di origine animale presenti nel campione esaminato, W = peso del campione di materiale utilizzato per la sedimentazione (mg)].
|
8. Espressione del risultato dell'esame
La relazione contiene almeno informazioni sulla presenza di costituenti derivati da animali terrestri e da farina di pesce. I diversi casi sono presentati nella maniera seguente:
|
8.1.
|
Per quanto riguarda la presenza di costituenti derivati da animali terrestri:
|
—
|
l'esame microscopico non ha permesso di rilevare la presenza di costituenti derivati da animali terrestri nel campione esaminato,
oppure
|
|
—
|
l'esame microscopico ha permesso di rilevare la presenza di costituenti derivati da animali terrestri nel campione esaminato.
|
|
|
8.2.
|
Per quanto riguarda la presenza di farina di pesce:
|
—
|
l'esame microscopico non ha permesso di rilevare la presenza di costituenti derivati da pesce nel campione esaminato,
oppure
|
|
—
|
l'esame microscopico ha permesso di rilevare la presenza di costituenti derivati da pesce nel campione esaminato.
|
Nel caso in cui si sia riscontrata la presenza di costituenti derivati da pesce o da animali terrestri, la relazione sui risultati dell'esame può, se necessario, indicare una stima della quantità di costituenti rilevati (x %, < 0,1 %, 0,1-0,5 %, 0,5-5 % o > 5 %), e ulteriori indicazioni riguardo al tipo di animali terrestri, se possibile, e di costituenti di origine animale individuati (fibre muscolari, cartilagini, ossa, corna, peli, setole, piume, sangue, gusci d'uovo, scaglie e lische).
Nel caso in cui sia accertata la quantità d'ingredienti di origine animale va indicato il fattore di correzione f.
Qualora si sia riscontrata la presenza di costituenti derivati da ossa di animali terrestri la relazione deve riportare la seguente indicazione:
«Non si può escludere la possibilità che i costituenti sopra descritti provengano da mammiferi.»
Tale precisazione è superflua qualora sia stato accertato che i frammenti ossei di animali terrestri derivano da pollame o da mammiferi.
|
9. Protocollo facoltativo per l'analisi di grasso o olio
Per l'analisi di grasso o olio può essere applicato il seguente protocollo.
|
—
|
Se il grasso è solido, si scalda, ad esempio in un forno a microonde, finché non diventa liquido.
|
|
—
|
Con un contagocce trasferire 40 ml di grasso dal fondo del campione a un tubo di centrifugazione.
|
|
—
|
Centrifugare per 10 minuti a 4 000 giri al minuto.
|
|
—
|
Se il grasso è solido dopo la centrifugazione, scaldarlo nuovamente nel forno finché non ridiventa liquido. Ripetere la centrifugazione per 5 minuti a 4 000 giri al minuto.
|
|
—
|
Con un cucchiaino o una spatola trasferire metà delle impurità sedimentate in una piccola scatola di Petri o un vetrino microscopico per l'individuazione al microscopio di un possibile contenuto di costituenti di origine animale (fibre di carne, piume, frammenti d'osso, ecc.). Si raccomanda l'uso di un agente di rivestimento come l'olio di paraffina o il glicerolo.
|
|
—
|
L'altra metà di impurità è utilizzata per la sedimentazione come descritto al punto 6.2.
|
(1) GU L 165 del 30.4.2004, pag. 1; rettifica nella GU L 191 del 28.5.2004, pag. 1.
ALLEGATO VII
METODO DI CALCOLO DEL VALORE ENERGETICO DEGLI ALIMENTI DESTINATI AL POLLAME
1. Metodo di calcolo ed espressione del valore energetico
Il valore energetico degli alimenti composti destinati ai volatili da cortile è calcolato secondo la formula che segue, in base alle percentuali di alcuni componenti analitici degli alimenti; il valore è espresso in megajoule (MJ) di energia metabolizzabile (ME), corretta in azoto, per chilogrammo di alimento composto:
MJ/kg di ME = 0,1551 × % proteina greggia +0,3431 × % sostanze grasse gregge +0,1669 × % amido +0,1301 × % zuccheri totali (espressi in saccarosio).
2. Tolleranze applicabili ai valori dichiarati
Se, a seguito dei controlli ufficiali si constata una differenza (in più o in meno) fra il risultato del controllo del valore energetico dell'alimento e il valore energetico dichiarato, è applicata una tolleranza minima di 0,4 MJ/kg di EM.
3. Espressione dei risultati
Previa applicazione della formula suindicata, il risultato ottenuto è approssimato al primo decimale.
4. Modalità di prelievo dei campioni e metodi di analisi
Il prelievo del campione dell'alimento composto e la determinazione dei tenori dei componenti analitici impiegati nel metodo di calcolo devono essere effettuati rispettivamente secondo i modi di campionamento e i metodi di analisi comunitari per il controllo ufficiale degli alimenti per animali.
Vanno applicati:
|
—
|
per la determinazione delle sostanze grasse gregge: la procedura B del metodo per la determinazione delle sostanze grasse gregge, di cui all'allegato III, parte H,
|
|
—
|
per la determinazione dell'amido: il metodo polarimetrico, di cui all'allegato III, parte L.
|
ALLEGATO VIII
METODI DI ANALISI PER IL CONTROLLO DELLA PRESENZA ILLECITA NEGLI ALIMENTI PER ANIMALI DI ADDITIVI NON PIÙ AUTORIZZATI
Osservazioni importanti:Possono essere applicati metodi di analisi più sensibili di quelli citati nel presente allegato per individuare la presenza illecita di additivi il cui uso negli alimenti per animali non è più autorizzato.I metodi di analisi citati nel presente allegato sono utilizzati a fini di conferma.
A. DETERMINAZIONE DEL METILBENZOQUATO
(7-benzilossi-6-butil-3-metossicarbonil-4-chinolone)
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di metilbenzoquato negli alimenti per animali. Il limite di quantificazione è di 1 mg/kg.
2. Principio
Il metilbenzoquato viene estratto dal campione con una soluzione metanolica di acido metansolfonico. L'estratto è purificato con diclorometano, mediante cromatografia a scambio ionico e poi nuovamente con diclorometano. Il tenore del metilbenzoquato è determinato mediante cromatografia liquida ad alta prestazione (HPLC) in fase inversa utilizzando un rivelatore UV.
3. Reattivi
|
3.2.
|
Metanolo, di qualità HPLC
|
|
3.3.
|
Fase mobile per HPLC
miscela di metanolo (3.2) e acqua (di qualità HPLC) 75 + 25 (v + v).
Filtrare la soluzione attraverso un filtro di 0,22 μm (4.5) e degassarla (ad esempio sottoponendola per 10 minuti a trattamento con ultrasuoni).
|
|
3.4.
|
Soluzione di acido metansolfonico, c = 2 %
Diluire 20,0 ml di acido metansolfonico e portarlo a 1 000 ml con metanolo (3.2).
|
|
3.5.
|
Soluzione di acido cloridrico, c = 10 %.
Prelevare 100 ml d'acido cloridrico (ρ20 1,18 g/ml) e portare a 1 000 ml con acqua.
|
|
3.6.
|
Resina scambiatrice di cationi Amberlite CG-120 (Na), 100-200 mesh
La resina viene pretrattata prima dell'uso. Sospendere 100 g di resina con 500 ml di soluzione di acido cloridrico (3.5) e portare ad ebollizione su piastra calda continuando ad agitare. Lasciare raffreddare e decantare l'acido. Filtrare attraverso carta da filtro sotto vuoto. Lavare due volte la resina con 500 ml di acqua e poi con 250 ml di metanolo (3.2). Risciacquare una seconda volta la resina con 250 ml di metanolo ed essiccarla insufflando aria attraverso il panello del filtro. Conservare la resina essiccata in una bottiglia chiusa con tappo.
|
|
3.7.
|
Sostanza di riferimento (standard): metilbenzoquato puro (7-benzilossi-6-butil-3-metossicarbonil-4-chinolone).
|
|
3.7.1.
|
Soluzione madre standard di metilbenzoquato, 500 μg/ml
Pesare, con l'approssimazione di 0,1 mg, 50 mg di sostanza standard (3.7), scioglierli in soluzione di acido metansolfonico (3.4) in un pallone tarato da 100 ml, portare a volume e miscelare.
|
|
3.7.2.
|
Soluzione standard intermedia di metilbenzoquato, 50 μg/ml
Trasferire 5,0 ml della soluzione madre standard di metilbenzoquato (3.7.1) in un pallone tarato da 50 ml, portare a volume con metanolo (3.2) e miscelare.
|
|
3.7.3.
|
Soluzioni di taratura
In una serie di palloni tarati da 25 ml, trasferire 1,0, 2,0, 3,0, 4,0 e 5,0 ml della soluzione standard intermedia di metilbenzoquato (3.7.2). Portare a volume con la fase mobile (3.3) e miscelare. Queste soluzioni presentano rispettivamente concentrazioni di 2,0, 4,0, 6,0, 8,0 e 10,0 μg/ml di metilbenzoquato. Esse devono essere preparate al momento, poco prima dell'uso.
|
4. Apparecchiatura
|
4.1.
|
Agitatore da laboratorio
|
|
4.2.
|
Evaporatore rotante a film
|
|
4.3.
|
Colonna di vetro (250 mm × 15 mm) dotata di rubinetto e serbatoio della capacità di circa 200 ml
|
|
4.4.
|
Apparecchiatura HPLC a rivelatore UV, a lunghezza d'onda variabile, oppure rivelatore a serie di diodi.
|
|
4.4.1.
|
Colonna per cromatografia liquida: 300 mm × 4 mm, C18, particelle di riempimento 10 μm, o equivalente
|
|
4.5.
|
Filtri a membrana, da 0,22 μm.
|
|
4.6.
|
Filtri a membrana, da 0,45 μm.
|
5. Procedimento
5.1. Indicazioni generali
|
5.1.1.
|
Analizzare un campione di alimento bianco, per accertare l'assenza del metilbenzoquato o di altre sostanze che possono interferire.
|
|
5.1.2.
|
Procedere a una prova di recupero, analizzando un campione in bianco dell'alimento addizionato con una quantità di metilbenzoquato simile a quella presente nel campione. Per ottenere una concentrazione di 15 mg/kg, aggiungere 600 μl della soluzione madre standard (3.6.1) a 20 g dell'alimento bianco, mescolare e attendere 10 minuti prima di procedere all'estrazione (5.2).
Nota: Ai fini del presente metodo, l'alimento bianco ha una composizione simile a quella del campione e all'analisi il metilbenzoquato non deve risultare presente.
|
5.2. Estrazione
Pesare, con l'approssimazione di 0,01 g, circa 20 g del campione preparato e trasferirli in una beuta da 250 ml. Aggiungere 100,0 ml di soluzione di acido metansolfonico (3.4) e agitare in agitatore meccanico (4.1) per 30 minuti. Filtrare la soluzione attraverso carta da filtro e conservare il filtrato per la fase di separazione liquido-liquido (5.3).
5.3. Separazione liquido-liquido
In un imbuto separatore da 500 ml contenente 100 ml di soluzione di acido cloridrico (3.5), trasferire 25,0 ml del filtrato ottenuto come descritto al punto (5.2). Introdurre nell'imbuto 100 ml di diclorometano (3.1) e agitare per 1 minuto. Lasciare separare gli strati e versare lo strato inferiore (diclorometano) in un pallone da 500 ml. Ripetere l'estrazione della fase acquosa con altri due volumi di 40 ml di diclorometano e riunirli con il primo estratto nel pallone a fondo arrotondato. Evaporare a secchezza nell'evaporatore rotante (4.2) l'estratto di diclorometano a 40 oC a pressione ridotta. Sciogliere il residuo in 20-25 ml di metanolo (3.2), tappare il pallone e conservare tutto l'estratto per la cromatografia a scambio ionico (5.4).
5.4. Cromatografia a scambio ionico
5.4.1. Preparazione della colonna a scambio cationico
Inserire un tappo di lana di vetro nell'estremità inferiore di una colonna di vetro (4.3). Preparare una sospensione di 5,0 g della resina a scambio cationico trattata (3.6) con 50 ml di acido cloridrico (3.5), versare nella colonna di vetro e far decantare. Scaricare l'eccesso di acido fino a poco sopra la superficie della resina e lavare la colonna con acqua fino a quando l'effluente è neutro al tornasole. Trasferire 50 ml di metanolo (3.2) nella colonna e drenare fino alla superficie della resina.
5.4.2. Cromatografia su colonna
Mediante una pipetta, trasferire accuratamente l'estratto ottenuto come descritto al punto 5.3 nella colonna. Risciacquare il pallone con due volumi di 5-10 ml di metanolo (3.2) e trasferire questi liquidi di lavaggio nella colonna. Scaricare l'estratto fino alla superficie della resina e lavare la colonna con 50 ml di metanolo facendo attenzione che lai defluiscano ad una velocità pari o inferiore a 5 ml al minuto. Eliminare tutto il filtrato. Eluire il metilbenzoquato dalla colonna utilizzando 150 ml di soluzione di acico metansolfonico (3.4) e raccogliere l'eluato della colonna in una beuta da 250 ml.
5.5. Separazione liquido-liquido
Trasferire l'eluato ottenuto come descritto al punto 5.4.2 in un imbuto separatore da 1 litro. Risciacquare la beuta con 5-10 ml di metanolo (3.2) e aggiungere il liquido di risciacquo al contenuto dell'imbuto separatore. Aggiungere 300 ml di soluzione di acido cloridrico (3.5) e 130 ml di diclorometano (3.1). Agitare per 1 minuto e lasciar separare le fasi. Versare lo strato inferiore (diclorometano) in un matraccio a fondo arrotondato da 500 ml. Ripetere l'estrazione della fase acquosa con altri due volumi di 70 ml di diclorometano e aggiungere questi estratti al primo nel matraccio.
In evaporatore rotante evaporare a secchezza (4.2) l'estratto di diclorometano a 40 oC a pressione ridotta. Sciogliere il residuo contenuto nel pallone con circa 5 ml di metanolo (3.2) e trasferire quantitativamente questa soluzione in un matraccio tarato da 10 ml. Risciacquare il pallone con altri due volumi di 1-2 ml di metanolo e trasferire anche questi nel matraccio tarato. Portare a volume con il metanolo e miscelare. Un'aliquota viene filtrata attraverso un filtro a membrana (4.6). Conservare questa soluzione per la determinazione mediante HPLC (5.6).
5.6. Determinazione HPLC
5.6.1. Parametri
I parametri seguenti sono proposti a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti:
|
—
|
Colonna per cromatografia liquida (4.4.1.)
|
|
—
|
Fase mobile per HPLC: miscela metanolo-acqua (3.3)
|
|
—
|
Velocità di effllusso: 1-1,5 ml/minuto
|
|
—
|
Lunghezza d'onda di rivelazione: 265 nm
|
|
—
|
Volume d'iniezione: 20-50 μl.
|
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.7.3) contenente 4 μg/ml fino a ottenimento di altezze o aree del picco e tempi di ritenzione costanti.
5.6.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.7.3) e misurare le altezze (aree) del picco per ciascuna concentrazione. Tracciare una curva di taratura riportando in ordinate l'altezza media o l'area media dei picchi delle soluzioni di taratura e in ascisse le corrispondenti concentrazioni in μg/ml.
5.6.3. Soluzione del campione
Iniettare più volte l'estratto del campione (5.5) utilizzando lo stesso volume usato per le soluzioni di taratura e determinare l'altezza (area) media dei picchi del metilbenzoquato.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in g/ml in base all'altezza (area) media dei picchi del metilbenzoquato, per riferimento alla curva di taratura (5.6.2).
Il contenuto di metilbenzoquato w (mg/kg) del campione è dato dalla formula seguente:

dove
|
c
|
=
|
concentrazione del metilbenzoquato nella soluzione del campione in g/ml
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione e della soluzione di taratura (3.7.3) contenente 10 μg/ml.
7.1.1. Co-cromatografia
Un estratto del campione viene «rinforzato» mediante un quantitativo adeguato della soluzione standard intermedia (3.7.2). Il quantitativo di metilbenzoquato addizionato deve essere analogo a quello stimato di metilbenzoquato rilevato nell'estratto del campione.
Deve aumentare soltanto l'altezza del picco del metilbenzoquato, tenuto conto sia della quantità di metilbenzoquato aggiunta che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza massima, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda di assorbimento massimo degli spettri del campione e dello standard, registrata all'apice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in genere di circa 2 nm;
|
|
b)
|
fra 225 e 300 nm, gli spettri del campione e dello standard registrati all'apice del picco sul cromatogramma non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra i due spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
tra 220 e 350 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto fra gli spettri non supera il 15 % dell'assorbanza dello spettro al vertice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare: il 10 % del valore ottenuto più elevato per contenuti di metilbenzoquato compresi tra 4 e 20 mg/kg.
7.3. Recupero
Per il campione bianco addizionato, il recupero non è inferiore al 90 %.
8. Risultati di uno studio collaborativo
Cinque campioni sono stati analizzati da 10 laboratori. Ogni campione è stato analizzato due volte.
|
|
In bianco
|
Farina 1
|
Granulare 1
|
Farina 2
|
Granulare 2
|
|
media [mg/kg]
|
NR
|
4,50
|
4,50
|
8,90
|
8,70
|
|
sr [mg/kg]
|
—
|
0,30
|
0,20
|
0,60
|
0,50
|
|
CVr [ %]
|
—
|
6,70
|
4,40
|
6,70
|
5,70
|
|
sR [mg/kg]
|
—
|
0,40
|
0,50
|
0,90
|
1,00
|
|
CVR [ %]
|
—
|
8,90
|
11,10
|
10,10
|
11,50
|
|
Recupero [ %]
|
—
|
92,00
|
93,00
|
92,00
|
89,00
|
|
NR
|
=
|
non riscontrata
|
|
sr
|
=
|
deviazione standard della ripetibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità, %
|
|
sR
|
=
|
deviazione standard della riproducibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità, %
|
B. DETERMINAZIONE DELL'OLAQUINDOX
2-[N-2'-(idrossietil) carbamoil]-3-metilchinoxalin-N1,N4-biossido
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di olaquindox negli alimenti per animali. Il limite di quantificazione è di 5 mg/kg.
2. Principio
Il campione viene estratto con una miscela acqua/metanolo. Il tenore di olaquindox è determinato mediante cromatografia liquida ad alta prestazione (HPLC) a fase inversa, utilizzando un rivelatore UV.
3. Reattivi
|
3.2.
|
Metanolo, di qualità HPLC
|
|
3.3.
|
Acqua, di qualità HPLC
|
|
3.4.
|
Fase mobile per HPLC
Miscela acqua (3.3)-metanolo (3.2), 900 + + 100 (v + v)
|
|
3.5.
|
Sostanza di riferimento (standard): olaquindox puro: 2-[N-2'-(idrossietil)carbamoil]-3-metilchinossalina-N1,N4-biossido, E 851.
|
|
3.5.1.
|
Soluzione madre standard di olaquindox, 250 μg/ml
Pesare, con l'approssimazione di 0,1 mg, 50 mg di olaquindox (3.5) in un matraccio tarato da 200 ml ed aggiungere circa 190 ml di acqua. Immergere quindi il matraccio per 20 minuti in un bagno ultrasonico (4.1). Dopo il trattamento ultrasonico raffreddare la soluzione a temperatura ambiente, portare a volume con acqua e mescolare. Avvolgere il matraccio in un foglio di alluminio e conservare in frigorifero. La soluzione va preparata di fresco ogni mese.
|
|
3.5.2.
|
Soluzione standard intermedia di olaquindox, 25 μg/ml.
Travasare 10,0 ml di soluzione madre standard (3.5.1) in un matraccio tarato da 100 ml, portare a volume con la fase mobile (3.4) ed agitare. Avvolgere il matraccio in un foglio di alluminio e conservare in frigorifero. La soluzione va preparata di fresco quotidianamente.
|
|
3.5.3.
|
Soluzioni di taratura
In una serie di matracci tarati da 50 ml trasferire 1,0, 2,0, 5,0, 10,0, 15,0 e 20,0 ml della soluzione standard intermedia (3.5.2). Portare a volume con la fase mobile (3.4) e miscelare. Avvolgere il pallone in foglio di alluminio. Queste soluzioni corrispondono rispettivamente a 0,5, 1,0, 2,5, 5,0, 7,5 e 10,0 μg/ml di olaquindox.
Queste soluzioni vanno preparate di fresco quotidianamente.
|
4. Apparecchiatura
|
4.3.
|
Apparecchiatura HPLC con rivelatore UV, a lunghezza d'onda variabile, oppure con rivelatore a serie diodi
|
|
4.3.1.
|
Colonna per cromatografia liquida, da 250 mm × 4 mm, C18, particelle di riempimento 10 μm, o equivalente
|
|
4.4.
|
Filtri a membrana, da 0,45 μm.
|
5. Procedimento
|
Nota:
|
L'olaquindox è fotosensibile. Effettuare tutte le operazioni in luce soffusa, oppure usare vetreria scura.
|
5.1. Indicazioni generali
|
5.1.1.
|
Analizzare un alimento di riferimento (bianco), per accertare l'assenza di olaquindox o di altre sostanze che possono interferire.
|
|
5.1.2.
|
Effettuare una prova di recupero analizzando il bianco addizionato di una quantità di olaquindox analoga a quella presente nel campione. Per ottenere una concentrazione di 50 mg/kg, trasferire 10,0 ml della soluzione madre standard (3.5.1) in una beuta da 250 ml e far evaporare la soluzione a circa 0,5 ml. Aggiungere 50 g del bianco, mescolare il tutto e attendere 10 minuti, agitando più volte, prima di procedere all'estrazione (5.2).
|
Nota:
|
Ai fini del presente metodo, il bianco ha una composizione simile a quella del campione e all'analisi l'olaquindox non deve risultare presente.
|
|
5.2. Estrazione
Pesare, con l'approssimazione di 0,01 g, 50 g circa del campione. Trasferire in una beuta da 1 000 ml, aggiungere 100 ml di metanolo (3.1) e immergere per 5 minuti la beuta in un bagno ultrasonico (4.1). Aggiungere 410 ml di acqua e lasciare nel bagno ultrasonico per altri 15 minuti. Togliere la beuta dal bagno, agitare per 30 minuti mediante l'agitatore (4.2) e filtrare attraverso un filtro a pieghe. Trasferire 10,0 ml del filtrato in un matraccio tarato da 20 ml, portare a volume con acqua e agitare. Filtrare un'aliquota attraverso un filtro a membrana (4.4). (cfr. osservazione al punto 9). Procedere alla determinazione HPLC (5.3).
5.3. Determinazione HPLC
5.3.1. Parametri:
Le seguenti condizioni vengono proposte a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti.
|
Colonna analitica (4.3.1)
|
|
|
Fase mobile (3.4):
|
miscela acqua (3.3) — metanolo (3.2) 900 + 100 (v + v)
|
|
Velocità di efflusso:
|
1,5-2 ml/min
|
|
Lunghezza d'onda di rivelazione:
|
380 nm
|
|
Volume di iniezione:
|
20 μl-100 μl
|
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.5.3) contenente 2,5 μg/ml, fino a ottenimento di altezze del picco e tempi di ritenzione costanti.
5.3.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.5.3) e determinare le altezze (le aree) medie dei picchi per ciascuna concentrazione. Tracciare una curva di taratura indicando le altezze (aree) medie dei picchi delle soluzioni di taratura sulle ordinate e le concentrazioni corrispondenti in μg/ml nelle ascisse.
5.3.3. Soluzione del campione
Iniettare più volte l'estratto del campione (5.2) utilizzando lo stesso volume usato per le soluzioni di taratura e determinare l'altezza (area) media dei picchi dell'olaquindox.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi dell'olaquindox, per riferimento alla curva di taratura (5.3.2).
Il tenore w di olaquindox, espresso in mg/kg del campione, è dato dalla seguente formula:

dove
|
c
|
=
|
concentrazione di olaquindox nell'estratto del campione (5.2) espressa in μg/ml
|
|
m
|
=
|
peso della sostanza da analizzare in g (5.2).
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia, oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione (5.2) e della soluzione di taratura (3.7.3) contenente 5,0 μg/ml.
7.1.1. Co-cromatografia
Un estratto del campione (5.2) viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.5.3). Il quantitativo di olaquindox aggiunto deve essere simile a quello di olaquindox rilevato nell'estratto del campione.
Deve aumentare soltanto l'altezza del picco dell'olaquindox, tenuto conto sia della quantità aggiunta che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale del picco dell'olaquindox nell'estratto del campione non addizionato.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda di assorbimento massimo degli spettri del campione e dello standard, registrata all'apice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in genere di circa 2 nm;
|
|
b)
|
fra 220 e 400 nm, gli spettri del campione e dello standard registrati all'apice del picco sul cromatogramma non devono essere diversi per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra i due spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
fra 220 e 400 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dello spettro al vertice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza tra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare il 15 % del risultato più elevato per contenuti di olaquindox compresi tra 10 e 200 mg/kg.
7.3. Recupero
Per il campione bianco addizionato, il recupero non è inferiore al 90 %.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio comunitario in cooperazione tra vari laboratori nel corso del quale tredici laboratori hanno analizzato quattro campioni di alimenti per suinetti, ivi compreso un alimento bianco. I risultati dello studio figurano nella tabella seguente:
|
|
Campione 1
|
Campione 2
|
Campione 3
|
Campione 4
|
|
L
|
13
|
10
|
11
|
11
|
|
n
|
40
|
40
|
44
|
44
|
|
media [mg/kg]
|
—
|
14,6
|
48,0
|
95,4
|
|
Sr [mg/kg]
|
—
|
0,82
|
2,05
|
6,36
|
|
SR [mg/kg]
|
—
|
1,62
|
4,28
|
8,42
|
|
CVr [ %]
|
—
|
5,6
|
4,3
|
6,7
|
|
CVR [ %]
|
—
|
11,1
|
8,9
|
8,8
|
|
Contenuto nominale
|
|
|
|
|
|
[mg/kg]
|
—
|
15
|
50
|
100
|
|
recupero %
|
—
|
97,3
|
96,0
|
95,4
|
|
L
|
=
|
numero di laboratori
|
|
n
|
=
|
numero di valori singoli
|
|
Sr
|
=
|
deviazione standard della ripetibilità
|
|
SR
|
=
|
deviazione standard della riproducibilità
|
|
CVr
|
=
|
coefficiente di variazione della ripetibilità
|
|
CVR
|
=
|
coefficiente di variazione della riproducibilità
|
9. Osservazione
Anche se il metodo non è stato convalidato per alimenti contenenti quantità superiori a 100 mg/kg di olaquindox, è possibile ottenere risultati soddisfacenti pesando una quantità di campione più piccola e/o diluendo l'estratto (5.2) fino a ottenere una concentrazione entro il campo della curva di taratura.
C. DETERMINAZIONE DELL'AMPROLIUM
Cloridrato di cloruro di 1-[(4-ammino-2-propil-5 pirimidinil)metil]-2-picolinio
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di amprolium negli alimenti per animali e nelle premiscele. Il limite di rilevazione è di 1 mg/kg, la soglia di quantificazione è di 5 mg/kg.
2. Principio
Il campione è estratto con una miscela metanolo/acqua. Dopo diluizione con la fase mobile e filtrazione per membrana, il contenuto di amprolium è determinato per cromatografia in fase liquida ad alta prestazione (HPLC) a scambio cationico, utilizzando un rivelatore UV.
3. Reattivi
|
3.2.
|
Acetonitrile, di qualità HPLC
|
|
3.3.
|
Acqua, di qualità HPLC
|
|
3.4.
|
Soluzione di fosfato monosodico, c = 0,1 mol/1
In un matraccio tarato da 1 000 ml, sciogliere 13,80 g di fosfato monosodico monoidrato in acqua (3.3), portare a volume con acqua (3.3) e mescolare.
|
|
3.5.
|
Soluzione di perclorato sodico, c = 1,6 mol/1
In un matraccio tarato da 1 000 ml, sciogliere 224,74 g di perclorato sodico monoidrato in acqua (3.3), portare a volume con acqua (3.3) e mescolare.
|
|
3.6.
|
Fase mobile per HPLC (cfr. osservazione 9.l)
Miscela di acetonitrile (3.2), soluzione di fosfato monosodico (3.4) e soluzione di perclorato sodico (3.5), 450+450+100 (v+v+v). Prima dell'impiego, filtrare attraverso filtro a membrana da 0,22 μm (4.3) e degassare la soluzione [ad esempio nel bagno ultrasonico (4.4), per almeno 15 minuti].
|
|
3.7.
|
Sostanza di riferimento (standard): cloridrato di cloruro di 1-[(4-ammino-2-propilpirimidin5-il)metil]-2-metil-piridinio (amprolium, E 750), di purezza garantita (cfr. punto 9.2).
|
|
3.7.1.
|
Soluzione madre standard di amprolium, 500 μg/ml
In un matraccio tarato da 100 ml, pesare, con l'approssimazione di 0,1 mg, 50 mg di amprolium (3.7); sciogliere in 80 ml di metanolo (3.l) e immergere per 10 minuti il matraccio in bagno ultrasonico (4.4). Dopo il trattamento a ultrasuoni raffreddare la soluzione a temperatura ambiente, portare a volume con acqua e mescolare. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.7.2.
|
Soluzione standard intermedia di amprolium, 50 μg/ml.
In un matraccio tarato da 50 ml, pipettare 5,0 ml della soluzione madre standard (3.7.l), portare a volume col solvente di estrazione (3.8) e mescolare. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.7.3.
|
Soluzioni di taratura
Trasferire 0,5, 1,0 e 2,0 ml della soluzione standard intermedia (3.7.2) in una serie di matracci tarati da 50 ml. Portare a volume con la fase mobile (3.6) e miscelare. Queste soluzioni corrispondono rispettivamente a 0,5, 1,0 e 2,0 μg/ml di amprolium. Esse vanno preparate al momento, prima dell'uso.
|
|
3.8.
|
Solvente di estrazione
Miscela metanolo (3.l)-acqua 2+1 (v+v)
|
4. Apparecchiatura
|
4.1.
|
Apparecchiatura HPLC con sistema di iniezione adeguato per volumi da 100 μl.
|
|
4.1.1.
|
Colonna per cromatografia liquida, 125 mm x 4 mm, a scambio cationico (riempimento: Nucleosil 10 SA, 5 o 10 μm) o equivalente.
|
|
4.1.2.
|
Rivelatore UV a lunghezza d'onda variabile o rivelatore a serie di diodi
|
|
4.2.
|
Filtro a membrana in PTFE, da 0,45 μm.
|
|
4.3.
|
Filtro a membrana, da 0,22 μm
|
|
4.5.
|
Agitatore meccanico o magnetico
|
5. Procedimento
5.1. Indicazioni generali
5.1.1. Alimento «bianco»
Per poter procedere alla prova di recupero (5.l.2) analizzare un alimento di riferimento (bianco), per accertare l'assenza di amprolium o di altre sostanze che possono interferire. L'alimento bianco ha una composizione simile a quella del campione e all'analisi l'amprolium o sostanze capaci di interferire non devono risultare presenti.
5.1.2 Prova di recupero
Effettuare una prova di recupero analizzando il bianco addizionato di una quantità di amprolium simile a quella presente nel campione. Per ottenere una concentrazione di 100 mg/kg, trasferire 10,0 ml della soluzione madre standard (3.7.1) in una beuta da 250 ml e far evaporare la soluzione a circa 0,5 ml. Aggiungere 50 g del bianco, mescolare il tutto e attendere per 10 minuti, agitando più volte, prima di procedere all'estrazione (5.2).
Se non fosse disponibile un bianco di tipo simile a quello del campione (cfr. punto 5.1.1), la prova di recupero può essere eseguita col metodo di addizione della sostanza di riferimento. In questo caso, un'aliquota del campione da analizzare viene addizionata di una quantità nota di amprolium, analoga a quella già presente. Quest'aliquota viene analizzata insieme a una del campione non addizionato e il recupero può essere calcolato per differenza.
5.2. Estrazione
5.2.1. Premiscele (contenuto < 1 % di amprolium) e alimenti per animali
A seconda del contenuto di amprolium, pesare, con l'approssimazione di 0,01 g, da 5 g a 40 g di campione in una beuta da 500 ml a fondo conico e aggiungere 200 ml di solvente di estrazione (3.8). Collocare la beuta nel bagno ultrasonico (4.4) e mantenervela per 15 minuti. Togliere la beuta dal bagno, agitare per un'ora mediante l'agitatore o col mescolatore magnetico (4.5). Diluire una parte di estratto con la fase mobile (3.6) sino ad ottenere un tenore di amprolium di 0,5-2 μg/ml e mescolare (cfr. osservazione 9.3). Filtrare 5-10 ml di questa soluzione diluita attraverso un filtro a membrana (4.2). Procedere alla determinazione HPLC (5.3).
5.2.2. Premiscele (contenuto ≥ 1 % di amprolium)
A seconda del contenuto di amprolium, pesare, con l'approssimazione di 0,001 g, da 1 a 4 g di premiscela in una beuta da 500 ml e aggiungere 200 ml di solvente di estrazione (3.8). Collocare la beuta nel bagno ultrasonico (4.4) e mantenervela per 15 minuti. Togliere la beuta dal bagno, agitare per un'ora mediante l'agitatore o col mescolatore magnetico (4.5). Diluire una parte di estratto con la fase mobile (3.6) sino ad ottenere un contenuto di amprolium di 0,5-2 μg/ml e mescolare. Filtrare 5-10 ml di questa soluzione diluita attraverso un filtro a membrana (4.2). Procedere alla determinazione HPLC (5.3).
5.3. Determinazione HPLC
5.3.1. Parametri:
Le seguenti condizioni vengono proposte a titolo indicativo; è possibile operare in condizioni diverse, purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida (4.1.1):
|
125 mm × 4 mm, a scambio cationico (riempimento: Nucleosil 10 SA, 5 o 10 μm, o equivalente)
|
|
Fase mobile (3.6):
|
miscela di acetonitrile (3.2), soluzione di fosfato monosodico (3.4) e soluzione di perclorato sodico (3.5), 450+450+100 (v+v+v).
|
|
Velocità di efflusso:
|
0,7-1 ml/min
|
|
Lunghezza d'onda di rivelazione:
|
264 nm
|
|
Volume d'iniezione:
|
100 μl
|
Verificare la stabilità del sistema cromatografico iniettando varie volte la soluzione di taratura (3.7.3) contenente 1,0 μg/ml, fino a ottenimento di altezze del picco e tempi di ritenzione costanti.
5.3.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.7.3) e determinare le altezze (aree) medie del picco per ciascuna concentrazione. Tracciare una curva di taratura indicando le altezze (aree) medie del picco delle soluzioni di taratura sulle ordinate e le concentrazioni corrispondenti in μg/ml nelle ascisse.
5.3.3. Soluzione del campione
Iniettare più volte l'estratto del campione (5.2), utilizzando lo stesso volume di quello prelevato per le soluzioni di taratura e determinare le altezze (aree) medie del picco dell'amprolium.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi dell'amprolium, per riferimento alla curva di taratura (5.3.2).
Il contenuto w di amprolium nel campione, espresso in mg/kg, è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
V
|
=
|
volume del solvente di estrazione (3.8) espresso in ml conformemente al punto 5.2 (ossia 200 ml)
|
|
c
|
=
|
concentrazione di amprolium nell'estratto del campione (5.2) espresso in μg/m
|
|
f
|
=
|
fattore di diluizione conformemente al punto 5.2.2
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione (5.2) e della soluzione di taratura (3.7.3) contenente 2,0 μg/ml.
7.1.1. Co-cromatografia
Un estratto del campione (5.2) viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.7.3). Il quantitativo di amprolium aggiunto deve essere simile a quello di amprolium rilevato nell'estratto del campione.
Deve aumentare soltanto l'altezza del picco dell'amprolium, tenuto conto sia del quantitativo aggiunto che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza, deve corrispondere, con uno scostamento massimo del 10 %, all'ampiezza originale del picco dell'amprolium nell'estratto del campione non addizionato.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda dell'assorbimento massimo degli spettri del campione e dello standard, registrata al vertice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in genere di circa 2 nm;
|
|
b)
|
fra 210 e 320 nm, gli spettri del campione e dello standard registrati al vertice del picco sul cromatogramma non devono essere diversi per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto fra gli spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
fra 210 e 320 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per le parti dello spettro situate fra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto fra gli spettri non supera il 15 % dell'assorbanza dello spettro al vertice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
La differenza fra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare:
|
—
|
il 15 % del valore più elevato, per i contenuti di amprolium compresi fra 25 mg/kg e 500 mg/kg,
|
|
—
|
75 mg/kg per i contenuti di amprolium compresi fra 500 mg/kg e 1 000 mg/kg,
|
|
—
|
il 7,5 % del valore più elevato, per i contenuti di amprolium superiori a 1 000 mg/kg.
|
7.3. Recupero
Per un campione addizionato (bianco), il recupero non è inferiore al 90 %.
8. Risultati di uno studio collaborativo
E' stato organizzato uno studio in cooperazione tra vari laboratori nel corso del quale sono stati analizzati tre alimenti per pollame (campioni 1-3), un alimento minerale (campione 4) e una premiscela (campione 5). I risultati dello studio figurano nella seguente tabella:
|
|
Campione 1 (bianco)
|
Campione 2
|
Campione 3
|
Campione 4
|
Campione 5
|
|
L
|
14
|
14
|
14
|
14
|
15
|
|
n
|
56
|
56
|
56
|
56
|
60
|
|
media [mg/kg]
|
—
|
45,5
|
188
|
5 129
|
25 140
|
|
sr [mg/kg]
|
—
|
2,26
|
3,57
|
178
|
550
|
|
CVr [ %]
|
—
|
4,95
|
1,90
|
3,46
|
2,20
|
|
sR [mg/kg]
|
—
|
2,95
|
11,8
|
266
|
760
|
|
CVR [ %]
|
—
|
6,47
|
6,27
|
5,19
|
3,00
|
|
contenuto nominale [mg/kg]
|
—
|
50
|
200
|
5 000
|
25 000
|
|
L:
|
numero di laboratori
|
|
n:
|
numero di valori singoli
|
|
sr:
|
deviazione standard della ripetibilità
|
|
CVr:
|
coefficiente di variazione della ripetibilità
|
|
sR:
|
deviazione standard della riproducibilità
|
|
CVR:
|
coefficiente di variazione della riproducibilità
|
9. Osservazioni
|
9.1.
|
Se il campione contiene tiamina, il relativo picco compare nel cromatogramma poco prima di quello dell'amprolium. Secondo questo metodo, l'amprolium e la tiamina debbono essere separati. Se l'amprolium e la tiamina non vengono separati dalla colonna (4.1.1) impiegata con questo metodo, sostituire con metanolo fino al 50 % della porzione di acetonitrile della fase mobile (3.6).
|
|
9.2.
|
Secondo la farmacopea britannica, lo spettro di una soluzione di amprolium (c = 0,02 mol/l) in acido cloridrico (c = 0,1 mol/l) presenta valori massimi a 246 nm e 262 nm. L'assorbanza è di 0,84 a 246 nm e di 0,80 a 262 nm.
|
|
9.3.
|
L'estratto deve essere sempre diluito con la fase mobile, poiché altrimenti il tempo di ritenzione del picco dell'amprolium potrebbe spostarsi significativamente per effetto delle variazioni della forza ionica.
|
D. DETERMINAZIONE DEL CARBADOX
Metil-3-(2-chinossalinilinmetilene) carbazato-N1,N4-diossido
1. Finalità e campo d'applicazione
Il metodo consente di determinare il contenuto di carbadox negli alimenti per animali, nelle premiscele e nei preparati. Il limite di rilevazione è di 1 mg/kg, la soglia di quantificazione è di 5 mg/kg.
2. Principio
Il campione viene equilibrato con acqua ed estratto con metanolo-acetonitrile. Nel caso degli alimenti per animali, un'aliquota dell'estratto filtrato è sottoposta a purificazione su una colonna di allumina. Per le premiscele e i preparati, un'aliquota dell'estratto filtrato è diluita con acqua, metanolo e aceto nitrile, fino a ottenere una concentrazione adeguata. Il contenuto di carbadox è determinato mediante cromatografia liquida ad alta prestazione (HPLC) in fase inversa, usando un rivelatore UV.
3. Reattivi
|
3.2.
|
Acetonitrile, di qualità HPLC
|
|
3.3.
|
Acido acetico, w = 100 %
|
|
3.4.
|
Ossido d'alluminio: neutro, grado di attività I
|
|
3.5.
|
Metanolo-acetonitrile 1 + 1 (v + v)
Miscelare 500 ml di metanolo (3.l) con 500 ml di acetonitrile (3.2).
|
|
3.6.
|
Acido acetico, σ = 10 %
Diluire con acqua 10 ml di acido acetico (3.3) e portare a 100 ml.
|
|
3.8.
|
Acqua, di qualità HPLC
|
|
3.9.
|
Soluzione tampone di acetato, c = 0,01 mol/1, pH = 6,0
Sciogliere 0,82 g di acetato di sodio (3.7) in 700 ml d'acqua (3.8) e regolare il pH a 6,0 con acido acetico (3.6). Trasferire in un matraccio tarato da 1000 ml, portare a volume con acqua (3.8) e mescolare.
|
|
3.10.
|
Fase mobile per HPLC
Mescolare 825 ml di soluzione tampone di acetato (3.9) con 175 ml di acetonitrile (3.2).
Filtrare la soluzione attraverso un filtro da 0,22 μm (4.5) e degassarla (ad esempio sottoponendola per 10 minuti a trattamento con ultrasuoni).
|
|
3.11.
|
Sostanza di riferimento (standard):
Carbadox allo stato puro: Metil-3-(2-chinossalinilinmetilene) carbazato-N1,N4-diossido, E 850
|
|
3.11.1.
|
Soluzione madre standard di carbadox, 100 μg/ml (cfr. nota 5. Procedimento):
In un matraccio tarato da 250 ml, pesare, con l'approssimazione di 0,1 mg, 25 mg di carbadox standard. Sciogliere la sostanza in metanolo-acetonitrile (3.5) sottoponendola a trattamento con ultrasuoni (4.7). Dopo il trattamento con ultrasuoni, raffreddare a temperatura ambiente, portare a volume con metanolo-acetonitrile (3.5) e mescolare. Avvolgere il matraccio in un foglio d'alluminio o usare vetreria scura e conservare in frigorifero. A temperatura non superiore a 4 oC la soluzione si mantiene stabile per un mese.
|
|
3.11.2.
|
Soluzioni di taratura
Trasferire 2,0, 5,0, 10,0 e 20,0 ml della soluzione madre standard (3.11.1) in una serie di matracci tarati da 100 ml. Aggiungere 30 ml d'acqua, portare a volume con metanolo-acetonitrile (3.5) e mescolare. Avvolgere i matracci in foglio d'alluminio. Queste soluzioni corrispondono rispettivamente a 2,0, 5,0, 10,0 e 20,0 μg/ml di carbadox.
Le soluzioni di taratura devono essere preparate al momento, poco prima dell'uso.
|
Nota:
|
per la determinazione del carbadox negli alimenti per animali contenenti meno di 10 mg/kg si dovranno preparare soluzioni di taratura a concentrazioni inferiori a 2,0 μg/ml.
|
|
|
3.12.
|
Miscela acqua -[metanolo-acetonitrile] (3.5), 300 + 700 (v + v).
Mescolare 300 ml d'acqua con 700 ml della miscela di metanolo-acetonitrile (3.5).
|
4. Apparecchiatura
|
4.1.
|
Agitatore da laboratorio o mescolatore magnetico
|
|
4.2.
|
Carta da filtro in fibra di vetro (Whatman GF/A o equivalente)
|
|
4.3.
|
Colonna in vetro (lunghezza: 300-400 mm, diametro interno: 10 mm circa), con setto in vetro sinterizzato e valvola di scarico.
|
Nota
|
:
|
Si può anche impiegare una colonna di vetro provvista di rubinetto oppure una colonna di vetro a estremità affusolata. In questo caso, si deve inserire un piccolo tampone di lana di vetro all'estremità inferiore e pressarlo verso il basso con una bacchetta di vetro.
|
|
|
4.4.
|
Apparecchiatura HPLC con sistema di iniezione adeguato per volumi da 20 μl.
|
|
4.4.1.
|
Colonna per cromatografia liquida: 300 mm × 4 mm, C18, particelle di riempimento 10 μm, o equivalente
|
|
4.4.2.
|
Rivelatore UV con regolazione variabile della lunghezza d'onda o rivelatore a serie di diodi, funzionante nell'intervallo di 225-400 nm.
|
|
4.5.
|
Filtro a membrana, da 0,22 μm.
|
|
4.6.
|
Filtro a membrana, da 0,45 μm.
|
5. Procedimento
|
Nota
|
:
|
Il carbadox è fotosensibile. Effettuare tutte le operazioni in luce soffusa oppure usare vetreria scura o avvolta in foglio d'alluminio.
|
5.1. Indicazioni generali
5.1.1. Alimento«bianco»
Per poter procedere alla prova di recupero (5.l.2) analizzare un alimento di riferimento (bianco), per accertare l'assenza di carbadox o di altre sostanze che possono interferire. Il bianco ha una composizione simile a quella del campione e all'analisi il carbadox o sostanze capaci di interferire non devono risultare presenti.
5.1.2. Prova di recupero
Effettuare una prova di recupero analizzando il bianco (5.1.1) addizionato di una quantità di carbadox simile a quella presente nel campione. Per ottenere una concentrazione di 50 mg/kg, introdurre 5,0 ml della soluzione madre standard (3.11.1) in una beuta da 200 ml. Far evaporare la soluzione a 0,5 ml circa, in corrente di azoto. Aggiungere 10 g dell'alimento bianco, mescolare per 10 minuti prima di procedere all'estrazione (5.2).
Se non fosse disponibile un bianco di tipo simile a quello del campione (cfr. punto 5.1.1), la prova di recupero può essere eseguita col metodo di addizione della sostanza di riferimento. In questo caso, un'aliquota del campione da analizzare viene addizionata di una quantità nota di carbadox, analoga a quella già presente. Quest'aliquota viene analizzata insieme a una del campione non addizionato e il recupero può essere calcolato per differenza.
5.2. Estrazione
5.2.1. Alimenti per animali
Pesare, con l'approssimazione di 0,01 g, 10 g del campione e trasferirli in una beuta da 200 ml. Aggiungere 15,0 ml di acqua, mescolare ed equilibrare per 5 minuti. Aggiungere 35,0 ml di metanolo-acetonitrile (3.5), tappare e agitare per 30 minuti sull'agitatore o col mescolatore magnetico (4.l). Filtrare la soluzione attraverso carta da filtro in fibra di vetro (4.2). Conservare questa soluzione per la fase di purificazione (5.3).
5.2.2. Premiscele (0,1-2,0 %)
Pesare, con l'approssimazione di 0,001 g, 1 g del campione non macinato e trasferirlo in una beuta da 200 ml. Aggiungere 15,0 ml di acqua, mescolare ed equilibrare per 5 minuti. Aggiungere 35,0 ml di metanolo-acetonitrile (3.5), tappare ed agitare per 30 minuti sull'agitatore o col mescolatore magnetico (4.l). Filtrare la soluzione attraverso carta da filtro in fibra di vetro (4.2).
Pipettare un'aliquota del filtrato in un matraccio tarato da 50 ml. Aggiungere 15,0 ml d'acqua, portare a volume con metanolo-acetonitrile (3.5) e mescolare. La concentrazione di carbadox nella soluzione finale è di circa 10 g/ml. Filtrare un'aliquota attraverso un filtro a membrana da 0,45 μm (4.6).
Procedere alla determinazione HPLC (5.4).
5.2.3. Preparati (> 2 %)
Pesare, con l'approssimazione di 0,001 g, 0,2 g del campione non macinato e trasferirli in una beuta da 250 ml. Aggiungere 45,0 ml d'acqua, mescolare ed equilibrare per 5 minuti. Aggiungere 105,0 ml di metanolo-acetonitrile (3.5), coprire e omogeneizzare. Sottoporre il campione a ultrasuoni (4.7) per 15 minuti, poi scuotere o agitare per 15 minuti (4.1). Filtrare la soluzione attraverso carta da filtro in fibra di vetro (4.2).
Diluire un'aliquota del filtrato con la miscela acqua-metanolo-acetonitrile (3.12) fino ad ottenere una concentrazione finale di carbadox dell'ordine di 10-15 μg/ml (per un preparato al 10 %, il fattore di diluizione è 10). Filtrare un'aliquota attraverso un filtro da 0,45 μm (4.6).
Procedere alla determinazione HPLC (5.4).
5.3. Purificazione
5.3.1. Preparazione della colonna di ossido di alluminio
Pesare 4 g di ossido di alluminio (3.4) e trasferire nella colonna di vetro (4.3).
5.3.2. Purificazione del campione
Far passare 15 ml dell'estratto filtrato (5.2.1) per la colonna di ossido di alluminio ed eliminare i primi 2 ml di eluato. Raccogliere i successivi 5 ml e filtrare un'aliquota attraverso un filtro da 0,45 μm (4.6).
Procedere alla determinazione HPLC (5.4).
5.4. Determinazione HPLC
5.4.1. Parametri
Le seguenti condizioni sono proposte a titolo indicativo; è possibile operare in condizioni diverse purché si ottengano risultati equivalenti.
|
Colonna per cromatografia liquida (4.4.1):
|
300 mm × 4 mm, C18, particelle di riempimento 10 μm o equivalente
|
|
Fase mobile (3.10):
|
Miscela di soluzione tampone di acetato (3.9) e acetonitrile (3.2), 825 + 175 (v + v)
|
|
Velocità di efflusso:
|
1,5-2 ml/min
|
|
Lunghezza d'onda di rivelazione:
|
365 nm
|
|
Volume di iniezione:
|
20 μl
|
Verificare la stabilità del sistema cromatografico iniettando più volte la soluzione di taratura (3.11.2) contenente 5,0 μg/ml, fino a ottenimento di altezze del picco e tempi di ritenzione costanti.
5.4.2. Curva di taratura
Iniettare più volte ciascuna soluzione di taratura (3.11.2) e determinare le altezze (aree) del picco per ciascuna concentrazione. Tracciare una curva di taratura riportando in ordinata le altezze (aree) medie del picco della soluzione di taratura e in ascissa le corrispondenti concentrazioni, espresse in μg/ml.
5.4.3. Soluzione del campione
Iniettare più volte l'estratto del campione [(5.3.2) per gli alimenti, (5.2.2) per le premiscele e (5.2.3) per i preparati], e determinare l'altezza (area) media dei picchi del carbadox.
6. Calcolo dei risultati
Determinare la concentrazione della soluzione del campione in μg/ml in base all'altezza (area) media dei picchi del carbadox, per riferimento alla curva di taratura (5.4.2).
6.1. Alimenti per animali
Il contenuto w (mg/kg) di carbadox nel campione è dato dalla seguente formula:
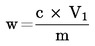 [mg/kg]
[mg/kg]
dove
|
c
|
=
|
concentrazione di carbadox nell'estratto del campione (5.2) espresso in μg/ml
|
|
V1
|
=
|
volume dell'estratto espresso in ml (ossia 50 ml)
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
6.2. Premiscele e preparati
Il contenuto w (mg/kg) di carbadox nel campione è dato dalla seguente formula:
 [mg/kg]
[mg/kg]
dove
|
c
|
=
|
concentrazione di carbadox nell'estratto del campione (5.2.2 o 5.2.3) espresso in μg/ml
|
|
V2
|
=
|
volume dell'estratto espresso in ml (ossia 50 ml per le premiscele;) 150 ml per i preparati)
|
|
f
|
=
|
fattore di diluizione conformemente ai punti 5.2.2 (premiscele) e 5.2.3 (preparati)
|
|
m
|
=
|
peso della quantità di sostanza da analizzare, in grammi.
|
7. Convalida dei risultati
7.1. Identità
L'identità dell'analita può essere confermata mediante co-cromatografia oppure mediante un rivelatore a serie di diodi che permetta di confrontare gli spettri dell'estratto del campione e della soluzione di taratura (3.11.2) contenente 10,0 μg/ml.
7.1.1. Co-cromatografia
Un estratto del campione viene «rinforzato» mediante aggiunta di un quantitativo adeguato della soluzione di taratura (3.11.2). Il quantitativo di carbadox aggiunto deve essere simile a quello stimato rilevato nell'estratto del campione.
Deve aumentare soltanto l'altezza del picco del carbadox tenuto conto sia del quantitativo di carbadox aggiunto che della diluizione dell'estratto. L'ampiezza del picco, a metà della sua altezza massima, deve corrispondere, con uno scostamento massimo del 10 %, dall'ampiezza originale.
7.1.2. Rivelazione a serie di diodi
I risultati sono valutati in base ai seguenti criteri:
|
a)
|
la lunghezza d'onda dell'assorbimento massimo degli spettri del campione e dello standard, registrata al vertice del picco sul cromatogramma, deve essere la stessa entro un margine determinato dal potere di risoluzione del sistema di rivelazione. Per la rivelazione a serie di diodi, tale margine è in genere di circa 2 nm;
|
|
b)
|
fra 225 e 400 nm, gli spettri del campione e dello standard registrati all'apice del picco cromatografico non devono differire tra loro per le parti dello spettro situate fra il 10 % e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dell'analita standard;
|
|
c)
|
fra 225 e 400 nm, gli spettri relativi all'estratto del campione, registrati nel tratto ascendente, all'apice e nel tratto discendente del picco cromatografico, non devono differire tra loro per quelle parti dello spettro situate tra il 10 e il 100 % dell'assorbanza relativa. Questo criterio è soddisfatto quando sono presenti gli stessi valori massimi e quando in tutti i punti osservati lo scarto tra gli spettri non supera il 15 % dell'assorbanza dello spettro all'apice del picco.
|
Se uno di questi criteri non è soddisfatto, la presenza dell'analita non è confermata.
7.2. Ripetibilità
Per contenuti di 10 mg/kg e superiori, la differenza fra i risultati di due determinazioni effettuate in parallelo sullo stesso campione non deve superare il 15 % rispetto al risultato più elevato.
7.3. Recupero
Per un campione addizionato (bianco), il recupero non è inferiore al 90 %.
8. Risultati di uno studio collaborativo
È stato organizzato uno studio in cooperazione tra vari laboratori nel corso del quale otto laboratori hanno analizzato sei alimenti per animali, quattro premiscele e tre preparati. Ogni campione è stato analizzato due volte. (Per maggiori informazioni consultare: Journal of the AOAC, Volume 71, 1998, pag. 484-490). I risultati (ad esclusione di quelli fuori campo) sono mostrati di seguito:
Tabella 1.
Risultati dello studio collaborativo sugli alimenti per animali
|
|
Campione 1
|
Campione 2
|
Campione 3
|
Campione 4
|
Campione 5
|
Campione 6
|
|
L
|
8
|
8
|
8
|
8
|
8
|
8
|
|
n
|
15
|
14
|
15
|
15
|
15
|
15
|
|
media (mg/kg)
|
50,0
|
47,6
|
48,2
|
49,7
|
46,9
|
49,7
|
|
Sr (mg/kg)
|
2,90
|
2,69
|
1,38
|
1,55
|
1,52
|
2,12
|
|
CVr ( %)
|
5,8
|
5,6
|
2,9
|
3,1
|
3,2
|
4,3
|
|
SR (mg/kg)
|
3,92
|
4,13
|
2,23
|
2,58
|
2,26
|
2,44
|
|
CVR ( %)
|
7,8
|
8,7
|
4,6
|
5,2
|
4,8
|
4,9
|
|
contenuto nominale (mg/kg)
|
50,0
|
50,0
|
50,0
|
50,0
|
50,0
|
50,0
|
Tabella 2.
Risultati dello studio collaborativo sulle premiscele e sui preparati
|
|
Premiscele
|
Preparati
|
|
A
|
B
|
C
|
D
|
A
|
B
|
C
|
|
L
|
7
|
7
|
7
|
7
|
8
|
8
|
8
|
|
n
|
14
|
14
|
14
|
14
|
16
|
16
|
16
|
|
media (g/kg)
|
8,89
|
9,29
|
9,21
|
8,76
|
94,6
|
98,1
|
104
|
|
Sr (g/kg)
|
0,37
|
0,28
|
0,28
|
0,44
|
4,1
|
5,1
|
7,7
|
|
CVr ( %)
|
4,2
|
3,0
|
3,0
|
5,0
|
4,3
|
5,2
|
7,4
|
|
SR (g/kg)
|
0,37
|
0,28
|
0,40
|
0,55
|
5,4
|
6,4
|
7,7
|
|
CVR ( %)
|
4,2
|
3,0
|
4,3
|
6,3
|
5,7
|
6,5
|
7,4
|
|
contenuto nominale (g/kg)
|
10,0
|
10,0
|
10,0
|
10,0
|
100
|
100
|
100
|
|
L:
|
numero di laboratori
|
|
n:
|
numero di valori singoli
|
|
Sr:
|
deviazione standard della ripetibilità
|
|
CVr:
|
coefficiente di variazione della ripetibilità
|
|
SR:
|
deviazione standard della riproducibilità
|
|
CVR:
|
coefficiente di variazione della riproducibilità
|
ALLEGATO IX
TAVOLE DI CONCORDANZA DI CUI ALL'ARTICOLO 6
1. Direttiva 71/250/CEE
|
Direttiva 71/250/CEE
|
Il presente regolamento
|
|
Articolo 1, primo comma
|
Articolo 3
|
|
Articolo 1, secondo comma
|
Articolo 2
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato, parte 1
|
Allegato II
|
|
Allegato, parte 2
|
—
|
|
Allegato, parte 3
|
—
|
|
Allegato, parte 4
|
Allegato III, parte O
|
|
Allegato, parte 5
|
Allegato III, parte M
|
|
Allegato, parte 6
|
Allegato III, parte N
|
|
Allegato, parte 7
|
Allegato III, parte Q
|
|
Allegato, parte 9
|
Allegato III, parte K
|
|
Allegato, parte 10
|
—
|
|
Allegato, parte 11
|
—
|
|
Allegato, parte 12
|
Allegato III, parte J
|
|
Allegato, parte 14
|
Allegato III, parte D
|
|
Allegato, parte 16
|
—
|
2. Direttiva 71/393/CEE
|
Direttiva 71/393/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato, parte I
|
Allegato III, parte A
|
|
Allegato, parte II
|
Allegato III, parte E
|
|
Allegato, parte III
|
Allegato III, parte P
|
|
Allegato, parte IV
|
Allegato III, parte H
|
3. Direttiva 72/199/CEE
|
Direttiva 72/199/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Allegato I, parte 1
|
Allegato III, parte L
|
|
Allegato I, parte 2
|
Allegato III, parte C
|
|
Allegato I, parte 3
|
—
|
|
Allegato I, parte 4
|
—
|
|
Allegato I, parte 5
|
Allegato V, parte A
|
|
Allegato II
|
—
|
4. Direttiva 73/46/CEE
|
Direttiva 73/46/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Allegato I, parte 1
|
Allegato III, parte B
|
|
Allegato I, parte 2
|
—
|
|
Allegato I, parte 3
|
Allegato III, parte I
|
5. Direttiva 76/371/CEE
|
Direttiva 76/371/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 1
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
Allegato I
|
6. Direttiva 76/372/CEE
|
Direttiva 76/372/CEE
|
Il presente regolamento
|
|
Articolo 1
|
—
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
—
|
7. Direttiva 78/633/CEE
|
Direttiva 78/633/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato, parte 1
|
—
|
|
Allegato, parte 2
|
—
|
|
Allegato, parte 3
|
Allegato IV, parte C
|
8. Direttiva 81/715/CEE
|
Direttiva 81/715/CEE
|
Il presente regolamento
|
|
Articolo 1
|
—
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
—
|
9. Direttiva 84/425/CEE
|
Direttiva 84/425/CEE
|
Il presente regolamento
|
|
Articolo 1
|
—
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
—
|
10. Direttiva 86/174/CEE
|
Direttiva 86/174/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 4
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
Allegato VII
|
11. Direttiva 93/70/CEE
|
Direttiva 93/70/CEE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato
|
Allegato IV, parte D
|
12. Direttiva 93/117/CE
|
Direttiva 93/117/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articoli 3 e 5
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Allegato, parte 1
|
Allegato IV, parte E
|
|
Allegato, parte 2
|
Allegato VIII, parte A
|
13. Direttiva 98/64/CE
|
Direttiva 98/64/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articoli 3 e 5
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Allegato, parte A
|
Allegato III, parte F
|
|
Allegato, parte C
|
Allegato VIII, parte B
|
14. Direttiva 1999/27/CE
|
Direttiva 1999/27/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articoli 3 e 5
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Articolo 5
|
—
|
|
Articolo 6
|
—
|
|
Articolo 7
|
—
|
|
Allegato, parte A
|
Allegato VIII, parte C
|
|
Allegato, parte B
|
Allegato IV, parte F
|
|
Allegato, parte C
|
Allegato VIII, parte D
|
15. Direttiva 1999/76/CE
|
Direttiva 1999/76/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Allegato
|
Allegato IV, parte G
|
16. Direttiva 2000/45/CE
|
Direttiva 2000/45/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Allegato, parte A
|
Allegato IV, parte A
|
|
Allegato, parte B
|
Allegato IV, parte B
|
|
Allegato, parte C
|
Allegato III, parte G
|
17. Direttiva 2002/70/CE
|
Direttiva 2002/70/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 1
|
|
Articolo 2
|
Articoli 2 e 3
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Articolo 5
|
—
|
|
Allegato I
|
Allegato I e allegato V, parte B (I)
|
|
Allegato II
|
Allegato II e allegato V, parte B (II)
|
18. Direttiva 2003/126/CE
|
Direttiva 2003/126/CE
|
Il presente regolamento
|
|
Articolo 1
|
Articolo 3
|
|
Articolo 2
|
—
|
|
Articolo 3
|
—
|
|
Articolo 4
|
—
|
|
Articolo 5
|
—
|
|
Articolo 6
|
—
|
|
Allegato
|
Allegato VI
|