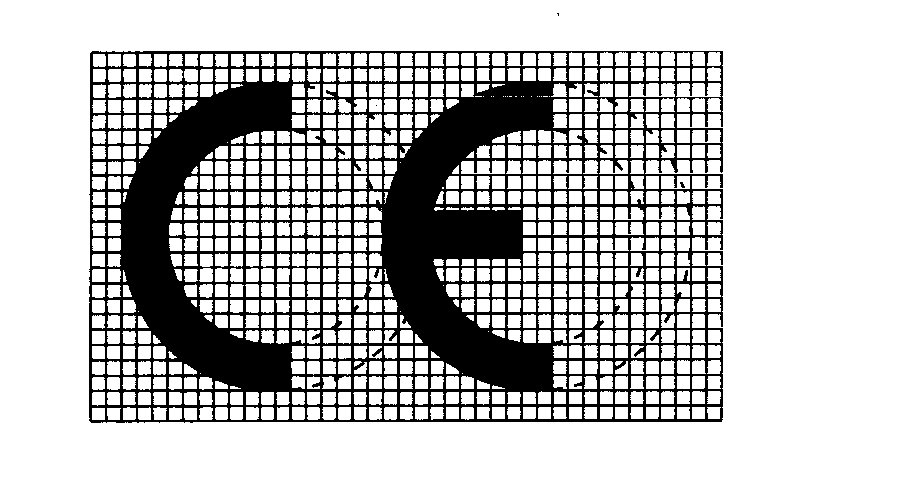
— La marcatura CE di conformità è costituita dalle iniziali «CE» secondo il simbolo grafico che segue:
—
01990L0385 — IT — 11.10.2007 — 003.007
Il presente testo è un semplice strumento di documentazione e non produce alcun effetto giuridico. Le istituzioni dell’Unione non assumono alcuna responsabilità per i suoi contenuti. Le versioni facenti fede degli atti pertinenti, compresi i loro preamboli, sono quelle pubblicate nella Gazzetta ufficiale dell’Unione europea e disponibili in EUR-Lex. Tali testi ufficiali sono direttamente accessibili attraverso i link inseriti nel presente documento
|
DIRETTIVA DEL CONSIGLIO del 20 giugno 1990 per il ravvicinamento delle legislazioni degli Stati membri relative ai dispositivi medici impiantabili attivi (GU L 189 dell'20.7.1990, pag. 17) |
Modificato da:
|
|
|
Gazzetta ufficiale |
||
|
n. |
pag. |
data |
||
|
L 169 |
1 |
12.7.1993 |
||
|
L 220 |
1 |
30.8.1993 |
||
|
REGOLAMENTO (CE) N. 1882/2003 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO del 29 settembre 2003 |
L 284 |
1 |
31.10.2003 |
|
|
L 247 |
21 |
21.9.2007 |
||
Rettificato da:
DIRETTIVA DEL CONSIGLIO
del 20 giugno 1990
per il ravvicinamento delle legislazioni degli Stati membri relative ai dispositivi medici impiantabili attivi
(90/385/CEE)
Articolo 1
1. La presente direttiva si applica ai dispositivi medici impiantabili attivi.
2. Ai fini della presente direttiva si applicano le definizioni seguenti:
a) «dispositivo medico»: qualunque strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compresi gli accessori tra cui il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo stesso, destinato dal fabbricante ad essere impiegato sull’uomo a fini di:
— diagnosi, prevenzione, controllo, trattamento o attenuazione di malattie,
— diagnosi, controllo, trattamento, attenuazione o compensazione di una ferita o di un handicap,
— studio, sostituzione o modifica dell’anatomia oppure di un processo fisiologico,
— controllo del concepimento,
che non eserciti nel o sul corpo umano l’azione principale cui è destinato con mezzi farmacologici, immunologici o mediante processi metabolici, ma la cui funzione possa essere coadiuvata da tali mezzi;
b) dispositivo medico attivo: qualsiasi dispositivo medico legato per il suo funzionamento a una fonte di energia elettrica o a qualsiasi altra fonte di energia diversa da quella prodotta direttamente dal corpo umano o dalla gravità;
c) dispositivo medico impiantabile attivo: qualsiasi dispositivo medico attivo destinato ad essere impiantato interamente o parzialmente mediante intervento chirurgico o medico nel corpo umano o mediante intervento medico in un orifizio naturale e destinato a restarvi dopo l'intervento;
d) «dispositivo su misura»: qualsiasi dispositivo fabbricato appositamente sulla base della prescrizione scritta di un medico debitamente qualificato che precisi, sotto la propria responsabilità, le caratteristiche specifiche di progettazione e destinato ad essere utilizzato solo per un determinato paziente.
I dispositivi fabbricati con metodi di produzione in serie che devono essere adattati per soddisfare un’esigenza specifica del medico o di un altro utilizzatore professionale non sono considerati dispositivi su misura;
e) «dispositivi per indagini cliniche»: qualsiasi dispositivo destinato ad essere utilizzato da un medico debitamente qualificato per lo svolgimento di indagini cliniche di cui all’allegato 7, punto 2.1, in un ambiente clinico umano adeguato.
Per l’esecuzione delle indagini cliniche, al medico debitamente qualificato è assimilata ogni altra persona la quale, in base alle qualifiche professionali, sia autorizzata a svolgere tali indagini;
f) «destinazione»: l’utilizzazione alla quale è destinato il dispositivo secondo le indicazioni fornite dal fabbricante sull’etichetta, nelle istruzioni per l’uso e/o nei materiali pubblicitari;
g) messa in servizio: messa a disposizione del corpo medico per l'impianto;
h) immissione in commercio: la prima messa a disposizione a titolo oneroso o gratuito di dispositivi, esclusi quelli destinati alle indagini cliniche, in vista della distribuzione e/o utilizzazione sul mercato comunitario, indipendentemente dal fatto che si tratti di dispositivi nuovi o rimessi a nuovo;
i) fabbricante: la persona fisica o giuridica responsabile della progettazione, della fabbricazione, dell'imballaggio e dell'etichettatura di un dispositivo in vista dell'immissione in commercio a proprio nome, indipendentemente dal fatto che queste operazioni siano eseguite da questa stessa persona o da un terzo per suo conto.
Gli obblighi della presente direttiva che si impongono al fabbricante valgono anche per la persona fisica o giuridica che compone, provvede all'imballaggio, tratta, rimette a nuovo e/o etichetta uno o più prodotti prefabbricati e/o assegna loro la destinazione di dispositivo in vista dell'immissione in commercio a proprio nome. Il presente comma non si applica alla persona la quale, senza essere il fabbricante ai sensi del primo comma, compone o adatta dispositivi già immessi in commercio in funzione della loro destinazione ad un singolo paziente.
j) «mandatario»: la persona fisica o giuridica stabilita nella Comunità che, dopo essere stata espressamente designata dal fabbricante, agisce e può essere interpellata dalle autorità e dagli organi della Comunità in vece del fabbricante per quanto riguarda gli obblighi che la presente direttiva impone a quest’ultimo;
k) «dati clinici»: le informazioni sulla sicurezza e/o sulle prestazioni ricavate dall’impiego di un dispositivo. I dati clinici provengono dalle seguenti fonti:
— indagini cliniche relative al dispositivo in questione, o
— indagini cliniche o altri studi pubblicati nella letteratura scientifica relativi a un dispositivo analogo di cui è dimostrabile l’equivalenza al dispositivo in questione, o
— relazioni pubblicate e/o non pubblicate su altre pratiche cliniche relative al dispositivo in questione o a un dispositivo analogo di cui è dimostrabile l’equivalenza al dispositivo in questione.
3. Quando un dispositivo medico impiantabile attivo è destinato a somministrare una sostanza definita «medicinale» ai sensi dell’articolo 1 della direttiva 2001/83/CE del Parlamento europeo e del Consiglio ( 1 ), tale dispositivo è disciplinato dalla presente direttiva, fatte salve le disposizioni della direttiva 2001/83/CE riguardanti il medicinale.
4. Quando un dispositivo medico impiantabile attivo incorpora come parte integrante una sostanza che, se utilizzata separatamente, può essere considerata un medicinale ai sensi dell’articolo 1 della direttiva 2001/83/CE e può avere effetti sul corpo umano con un’azione accessoria a quella del dispositivo, quest’ultimo deve essere valutato e autorizzato conformemente alla presente direttiva.
4 bis. Quando un dispositivo incorpora come parte integrante una sostanza, di seguito denominata «derivato del sangue umano», la quale, se utilizzata separatamente, può essere considerata un componente di un medicinale o un medicinale derivato dal sangue o dal plasma umano ai sensi dell’articolo 1 della direttiva 2001/83/CE e può avere effetti sul corpo umano con un’azione accessoria a quella del dispositivo, quest’ultimo è valutato e autorizzato in base alla presente direttiva.
5. La presente direttiva costituisce una direttiva specifica ai sensi dell’articolo 1, paragrafo 4, della direttiva 2004/108/CE ( 2 ).
6. La presente direttiva non si applica:
a) ai medicinali contemplati dalla direttiva 2001/83/CE. Nello stabilire se un determinato prodotto rientri nell’ambito di applicazione di tale direttiva oppure in quello della presente direttiva, si tiene conto in particolare del principale meccanismo d’azione del prodotto stesso;
b) al sangue umano, ai prodotti derivati dal sangue umano, al plasma o alle cellule ematiche di origine umana, né ai dispositivi che, al momento dell’immissione in commercio, contengono tali prodotti derivati da sangue, plasma o cellule, ad eccezione dei dispositivi di cui al paragrafo 4 bis;
c) a organi, tessuti o cellule di origine umana, né a prodotti comprendenti o derivati da tessuti o cellule di origine umana, ad eccezione dei dispositivi di cui al paragrafo 4 bis;
d) a organi, tessuti o cellule di origine animale, a meno che il dispositivo non sia fabbricato utilizzando tessuti animali resi non vitali o prodotti non vitali derivati da tessuti animali.
Articolo 2
Gli Stati membri adottano tutte le misure necessarie per garantire che i dispositivi siano immessi in commercio e/o messi in servizio solo se rispettano i requisiti stabiliti nella presente direttiva, qualora siano debitamente forniti, correttamente impiantati e/o installati, formino oggetto di adeguata manutenzione e siano usati secondo la loro destinazione.
Articolo 3
I dispositivi medici impiantabili attivi di cui all’articolo 1, paragrafo 2, lettere c), d) ed e), di seguito denominati «dispositivi», soddisfano i requisiti essenziali di cui all’allegato 1 che sono loro applicabili, tenendo conto della destinazione dei dispositivi in questione.
Laddove esista un rischio, i dispositivi che sono anche macchine ai sensi dell’articolo 2, lettera a), della direttiva 2006/42/CE del Parlamento europeo e del Consiglio, del 17 maggio 2006, relativa alle macchine ( 3 ), rispettano altresì i requisiti essenziali in materia di salute e sicurezza stabiliti nell’allegato I di tale direttiva, qualora detti requisiti essenziali in materia di salute e sicurezza siano più specifici dei requisiti essenziali stabiliti nell’allegato I della presente direttiva.
Articolo 4
1. Gli Stati membri non pongono ostacoli all’immissione sul mercato e alla messa in servizio nel loro territorio dei dispositivi conformi alle disposizioni della presente direttiva e recanti la marcatura CE di cui all’articolo 12, che indica che hanno formato oggetto della procedura di valutazione della conformità ai sensi dell’articolo 9.
2. Gli Stati membri non pongono ostacoli a che:
— i dispositivi destinati ad indagini cliniche possano essere messi a disposizione di medici debitamente qualificati o persone autorizzate a tale scopo se soddisfano le condizioni di cui all’articolo 10 e all’allegato 6,
— i dispositivi su misura possano essere immessi sul mercato e messi in servizio se soddisfano le condizioni di cui all’allegato 6 e sono accompagnati dalla dichiarazione prevista in detto allegato, la quale è messa a disposizione del paziente specificamente individuato.
Questi dispositivi non recano la marcatura CE.
3. Nelle fiere, esposizioni, dimostrazioni, ecc. gli Stati membri non pongono ostacoli a che siano presentati dispositivi non conformi alla presente direttiva, a condizione che sia apposta un’indicazione visibile da cui risulti chiaramente che gli stessi non possono essere commercializzati o messi in servizio prima che il fabbricante o il suo mandatario li abbiano resi conformi.
4. Gli Stati membri possono esigere che le indicazioni di cui all'allegato 1, punti 13, 14 e 15 siano, nella fase di messa in servizio di un dispositivo, redatte nelle loro lingue nazionali.
5.
a) Qualora i dispositivi siano disciplinati da altre direttive relative a differenti aspetti e che prevedono l'apposizione della marcatura CE, questa indica che i dispositivi si presumono soddisfare anche le prescrizioni di queste altre direttive.
b) Tuttavia, nel caso in cui una o più delle suddette direttive lascino al fabbricante la facoltà di scegliere il regime da applicare durante un periodo transitorio, la marcatura CE indica che i dispositivi soddisfano soltanto le disposizioni delle direttive applicate dal fabbricante. In tal caso, i riferimenti alle direttive applicate, pubblicati nella Gazzetta ufficiale delle Comunità europee, devono essere riportati nei documenti, nelle avvertenze o nei fogli di istruzione stabiliti dalle suddette direttive e che accompagnano tali dispositivi; tali documenti, avvertenze o fogli di istruzione devono essere accessibili senza che si debba distruggere l'imballaggio che assicura la sterilità del dispositivo.
Articolo 5
1. Gli Stati membri presumono conformi ai requisiti essenziali di cui all’articolo 3 i dispositivi che soddisfano le norme nazionali corrispondenti, adottate in applicazione delle norme armonizzate i cui numeri di riferimento sono stati pubblicati nella Gazzetta ufficiale dell’Unione europea; gli Stati membri pubblicano i numeri di riferimento di dette norme nazionali.
2. Ai fini della presente direttiva, il riferimento alle norme armonizzate comprende anche le monografie della Farmacopea europea, in particolare relative all’interazione tra medicinali e materiali impiegati in dispositivi contenenti detti medicinali, i cui riferimenti sono stati pubblicati nella Gazzetta ufficiale dell’Unione europea.
Articolo 6
1. Qualora uno Stato membro o la Commissione ritengano che le norme armonizzate di cui all'articolo 5 non soddisfano interamente i requisiti essenziali di cui all'articolo 3, la Commissione e lo Stato membro interessato sollevano la questione dinanzi al comitato permanente istituito dalla direttiva ►M4 98/34/CE ( 4 ) ◄ , precisandone i motivi. Il comitato formula immediatamente un parere.
Alla luce del parere di detto comitato la Commissione notifica agli Stati membri le misure da prendere per quanto concerne le norme e la pubblicazione di cui all'articolo 5.
2. La Commissione è assistita da un comitato permanente (di seguito denominato «comitato»).
3. Nei casi in cui è fatto riferimento al presente paragrafo, si applicano gli articoli 5 e 7 della decisione 1999/468/CE, tenendo conto delle disposizioni dell’articolo 8 della stessa.
Il termine di cui all’articolo 5, paragrafo 6, della decisione 1999/468/CE è fissato a tre mesi.
4. Nei casi in cui è fatto riferimento al presente paragrafo, si applicano l’articolo 5 bis, paragrafi da 1 a 4, e l’articolo 7 della decisione 1999/468/CE, tenendo conto delle disposizioni dell’articolo 8 della stessa.
5. Nei casi in cui è fatto riferimento al presente paragrafo, si applicano l’articolo 5 bis, paragrafi 1, 2, 4 e 6, e l’articolo 7 della decisione 1999/468/CE, tenendo conto delle disposizioni dell’articolo 8 della stessa.
Articolo 7
1. Qualora uno Stato membro constati che i dispositivi di cui all'articolo 1, paragrafo 2, lettere c) e d), correttamente messi in servizio e utilizzati conformemente alla loro destinazione rischiano di compromettere la salute e/o la sicurezza dei pazienti, degli operatori o, se del caso, dei terzi, esso prende tutti gli opportuni provvedimenti per ritirare tali dispositivi dal mercato, per vietarne o limitarne l'immissione sul mercato o la messa in servizio.
Lo Stato membro informa immediatamente la Commissione di questo provvedimento, precisando i motivi della sua decisione e, in particolare, se la non conformità alle disposizioni della presente direttiva sia dovuta:
a) alla mancata osservanza dei requisiti essenziali di cui all'articolo 3 nel caso in cui il dispositivo non corrisponda in tutto o in parte alle norme di cui all'articolo 5,
b) all'applicazione scorretta delle norme di cui all'articolo 5,
c) a carenze esistenti nelle norme stesse di cui all'articolo 5.
2. La Commissione procede nel minor tempo possibile a consultazioni con le parti interessate. Se dopo tali consultazioni essa ritiene:
— che il provvedimento è giustificato, essa ne informa immediatamente lo Stato membro che ha preso la misura e gli altri Stati membri. Qualora la decisione di cui al paragrafo 1 sia motivata da carenze esistenti nelle norme, la Commissione, dopo aver consultato le parti interessate, adisce il comitato di cui all'articolo 6, paragrafo 1, entro un termine di due mesi, se lo Stato membro che ha adottato il provvedimento intende mantenerlo in vigore, ed avvia la procedura prevista all'articolo 6, paragrafo 1;
— che il provvedimento è ingiustificato, essa ne informa immediatamente lo Stato membro che ha preso la misura nonché il fabbricante o il suo mandatario stabilito nella Comunità.
3. Qualora un dispositivo non conforme rechi la ►M2 marcatura CE ◄ , lo Stato membro competente prende le misure appropriate contro chiunque abbia apposto tale marchio e ne informa la Commissione e gli altri Stati membri.
4. La Commissione garantisce che gli Stati membri siano tenuti al corrente degli sviluppi e dell'esito di tale procedura.
Articolo 8
1. Gli Stati membri adottano le misure necessarie affinché i dati loro comunicati riguardanti gli incidenti di seguito elencati e inerenti ad un dispositivo siano registrati e valutati su base centralizzata:
a) qualsiasi malfunzionamento o alterazione delle caratteristiche e delle prestazioni di un dispositivo nonché qualsiasi inadeguatezza nell’etichettatura o nelle istruzioni per l’uso che possano essere o essere stati causa di decesso o grave peggioramento delle condizioni di salute di un paziente o di un utilizzatore;
b) qualsiasi motivo di ordine tecnico o medico connesso alle caratteristiche o alle prestazioni di un dispositivo, per le ragioni di cui alla lettera a), che comporti il ritiro sistematico dei dispositivi dello stesso tipo da parte del fabbricante.
2. Se uno Stato membro richiede che i medici o le istituzioni sanitarie informino le competenti autorità degli incidenti di cui al paragrafo 1, esso adotta le misure necessarie affinché il fabbricante del dispositivo in questione, o il suo mandatario, siano parimenti informati dell’incidente.
3. Dopo aver svolto una valutazione, se possibile assieme al fabbricante o al suo mandatario, gli Stati membri, fatto salvo l’articolo 7, informano immediatamente la Commissione e gli altri Stati membri delle misure adottate o previste per minimizzare il ripetersi degli incidenti di cui al paragrafo 1, comprese le informazioni sugli incidenti alla base.
4. Le misure necessarie per l’attuazione del presente articolo sono adottate secondo la procedura di regolamentazione di cui all’articolo 6, paragrafo 3.
Articolo 9
1. Per i dispositivi diversi dai dispositivi su misura e da quelli destinati ad indagini cliniche, il fabbricante, ai fini dell'apposizione della ►M2 marcatura CE ◄ , può scegliere tra:
a) svolgere la procedura relativa alla dichiarazione di conformità CE di cui all'allegato 2, oppure
b) seguire la procedura relativa alla certificazione CE di cui all'allegato 3 insieme con:
i) la procedura relativa alla verifica CE di cui all'allegato 4, oppure
ii) la procedura relativa alla dichiarazione CE di conformità al tipo di cui all'allegato 5.
2. Per i dispositivi su misura, il fabbricante deve presentare prima dell'immissione sul mercato di ciascun dispositivo la dichiarazione di cui all'allegato 6.
3. Le procedure previste agli allegati 3, 4 e 6 possono all'occorrenza essere svolte dal mandatario del fabbricante, stabilito nella Comunità.
4. I documenti e la corrispondenza riguardanti le procedure di cui ai paragrafi 1, 2 e 3 dovranno essere redatti in una lingua ufficiale dello Stato membro nel quale si svolgono le procedure stesse e/o in una lingua accettata dall'organismo notificato, di cui all'articolo 11.
5. Nel procedimento di valutazione della conformità del dispositivo, il fabbricante e/o l'organismo notificato tengono conto di tutti i risultati disponibili delle operazioni di valutazione e di verifica eventualmente svolte, secondo il disposto della presente direttiva, in una fase intermedia della fabbricazione.
6. Se il procedimento di valutazione della conformità presuppone l'intervento di un organismo notificato, il fabbricante, o il suo mandatario stabilito nella Comunità, può rivolgersi ad un organismo di sua scelta nell'ambito delle competenze per le quali l'organismo stesso è stato notificato.
7. L'organismo notificato può esigere, se debitamente giustificato, informazioni o dati necessari per stabilire e mantenere il certificato di conformità ai fini della procedura scelta.
8. Le decisioni adottate dagli organismi notificati a norma degli allegati 2, 3 e 5 hanno una validità massima di cinque anni e possono essere prorogate per periodi successivi di cinque anni al massimo, su richiesta presentata entro il termine convenuto nel contratto firmato dalle due parti.
9. In deroga ai paragrafi 1 e 2, le autorità competenti possono autorizzare su richiesta debitamente motivata, l'immissione in commercio e la messa in servizio, nel territorio dello Stato membro interessato, di singoli dispositivi per i quali le procedure di cui ai paragrafi 1 e 2 non sono state espletate ma il cui impiego è nell'interesse della protezione della salute.
10. Le misure intese a modificare elementi non essenziali della presente direttiva, anche integrandola, relative alle modalità attraverso le quali possono essere fornite, alla luce dei progressi tecnici e tenuto conto degli utilizzatori cui sono destinati i dispositivi in questione, le informazioni di cui all’allegato 1, punto 15, sono adottate secondo la procedura di regolamentazione con controllo di cui all’articolo 6, paragrafo 4.
Articolo 9 bis
1. Uno Stato membro presenta una domanda debitamente motivata alla Commissione affinché questa adotti le misure necessarie qualora lo Stato membro ritenga che:
— la conformità di un dispositivo o di una categoria di dispositivi debba essere stabilita, in deroga all’articolo 9, ricorrendo esclusivamente all’applicazione di una dedle procedure specifiche scelta tra quelle previste all’articolo 9,
— si renda necessaria una decisione sull’applicabilità ad un prodotto o a un gruppo di prodotti di una delle definizioni di cui all’articolo 1, paragrafo 2, lettere a), c), d) oppure e).
Qualora le misure siano ritenute necessarie ai sensi del primo trattino del presente paragrafo, esse sono adottate secondo la procedura di regolamentazione di cui all’articolo 6, paragrafo 3.
2. La Commissione informa gli Stati membri in merito alle misure adottate.
Articolo 10
1. Per i dispositivi destinati ad indagini cliniche, il fabbricante o il ►M4 ————— ◄ mandatario stabilito nella Comunità notifica, almeno sessanta giorni prima dell'inizio delle indagini, la dichiarazione di cui all'allegato 6 alle autorità competenti dello Stato membro nel quale si intende svolgere le indagini.
2. Il fabbricante può avviare le indagini cliniche in questione al termine di un periodo di sessanta giorni dalla notifica, a meno che le autorità competenti gli abbiano comunicato entro detto termine una decisione contraria, fondata su considerazioni di pubblica sanità e di ordine pubblico.
Gli Stati membri possono tuttavia autorizzare i fabbricanti ad iniziare le indagini cliniche in questione prima della scadenza dei sessanta giorni, purché il comitato etico interessato abbia espresso un parere favorevole in relazione al programma di indagini in questione, compresa la sua revisione del piano dell’indagine clinica.
2 bis L'autorizzazione di cui al paragrafo 2, secondo comma, può essere sottoposta al visto dell'autorità competente.
3. Gli Stati membri adottano, ove necessario, le misure adeguate per garantire la sanità pubblica e l’ordine pubblico. Se uno Stato membro rifiuta l’autorizzazione per un’indagine clinica o la sospende deve comunicare tale decisione e i pertinenti motivi a tutti gli Stati membri e alla Commissione. Qualora uno Stato membro abbia chiesto una modifica significativa o un’interruzione temporanea di un’indagine clinica esso informa gli Stati membri interessati in merito alle proprie azioni comunicandone i motivi.
4. Il fabbricante o il suo mandatario notifica alle competenti autorità degli Stati membri interessati la conclusione dell’indagine clinica, indicando i motivi in caso di conclusione anticipata. In caso di conclusione anticipata dell’indagine clinica per motivi di sicurezza tale notifica è comunicata a tutti gli Stati membri e alla Commissione. Il fabbricante o il suo mandatario tengono a disposizione delle autorità competenti la relazione di cui all’allegato 7, punto 2.3.7.
5. Le indagini cliniche sono svolte secondo le disposizioni dell’allegato 7. Le misure intese a modificare elementi non essenziali della presente direttiva relative alle disposizioni in materia di indagini cliniche di cui all’allegato 7 sono adottate secondo la procedura di regolamentazione con controllo di cui all’articolo 6, paragrafo 4.
Articolo 10 bis
1. Il fabbricante che immette in commercio dispositivi a nome proprio secondo la procedura di cui all’articolo 9, paragrafo 2, comunica l’indirizzo della sede e la descrizione dei dispositivi in questione alle autorità competenti dello Stato membro in cui ha la sede.
Gli Stati membri possono chiedere di essere informati in merito a tutti i dati che consentono di identificare i dispositivi, unitamente all’etichetta e alle istruzioni per l’uso, quando i dispositivi sono messi in servizio sul loro territorio.
2. Se non ha la sede in uno Stato membro, il fabbricante che immette in commercio un dispositivo a nome proprio designa un unico mandatario nell’Unione europea.
Per i dispositivi di cui al paragrafo 1, primo comma, il mandatario comunica all’autorità competente dello Stato membro nel quale ha sede tutti i dati di cui al paragrafo 1.
3. A richiesta, gli Stati membri informano gli altri Stati membri e la Commissione circa i dati di cui al paragrafo 1, primo comma, forniti dal fabbricante o dal mandatario.
Articolo 10 ter
1. I dati regolamentari di cui alla presente direttiva sono memorizzati in una banca dati europea accessibile alle autorità competenti in modo che queste ultime possano svolgere i propri compiti connessi alla presente direttiva in base a informazioni esaurienti.
La banca dati contiene i seguenti elementi:
a) i dati relativi ai certificati rilasciati, modificati, integrati, sospesi, ritirati o rifiutati secondo le procedure di cui agli allegati da 2 a 5;
b) i dati ottenuti in base alla procedura di vigilanza definita all’articolo 8;
c) i dati relativi alle indagini cliniche di cui all’articolo 10.
2. I dati sono trasmessi in un formato standard.
3. Le misure necessarie per l’attuazione dei paragrafi 1 e 2 del presente articolo, in particolare del paragrafo 1, lettera c), sono adottate secondo la procedura di regolamentazione di cui all’articolo 6, paragrafo 3.
Articolo 10 quater
Se, in relazione ad un dato prodotto o gruppo di prodotti, uno Stato membro ritiene che, per garantire la tutela della salute e della sicurezza e/o per assicurare il rispetto delle esigenze di sanità pubblica, detti prodotti dovrebbero essere ritirati dal mercato o che la loro immissione in commercio e la loro messa in servizio dovrebbero essere vietate, limitate o soggette a particolari requisiti, può adottare tutte le misure transitorie necessarie e giustificate.
Lo Stato membro informa allora la Commissione e tutti gli altri Stati membri in merito alle misure transitorie, indicando le ragioni della sua decisione.
La Commissione consulta, ove possibile, le parti interessate e gli Stati membri. La Commissione emette un parere, comunicando se le misure nazionali sono giustificate o meno. La Commissione informa tutti gli Stati membri e le parti interessate che sono state consultate.
Ove opportuno, le necessarie misure intese a modificare elementi non essenziali della presente direttiva, integrandola, in relazione al ritiro dal mercato, al divieto d’immissione in commercio o di messa in servizio di un dato prodotto o gruppo di prodotti ovvero a limitazioni o all’introduzione di requisiti particolari a tal fine sono adottate secondo la procedura di regolamentazione con controllo di cui all’articolo 6, paragrafo 4. Per imperativi motivi d’urgenza, la Commissione può avvalersi della procedura d’urgenza di cui all’articolo 6, paragrafo 5.
Articolo 11
1. Gli Stati membri notificano alla Commissione e agli altri Stati membri gli organismi da essi designati per espletare le procedure di cui all'articolo 9 nonché i compiti specifici per i quali tali organismi sono stati designati e i numeri di identificazione che sono stati loro attribuiti in precedenza dalla Commissione.
La Commissione pubblica nella Gazzetta ufficiale delle Comunità europee un elenco degli organismi notificati in cui figurano il loro numero di identificazione nonché i compiti per i quali essi sono stati notificati. Essa provvede all'aggiornamento di tale elenco.
2. Nel designare gli organismi gli Stati membri applicano i criteri minimi esposti nell'allegato 8. Si presume che gli organismi i quali soddisfano i criteri fissati nell'ambito delle pertinenti norme armonizzate soddisfino i criteri minimi pertinenti.
Ove opportuno, alla luce dei progressi tecnologici, le misure dettagliate necessarie per garantire un’applicazione coerente dei criteri di cui all’allegato 8 della presente direttiva per la designazione degli organismi da parte degli Stati membri sono adottate secondo la procedura di regolamentazione di cui all’articolo 6, paragrafo 3.
3. Lo Stato membro che ha notificato un organismo deve revocare questa notifica se constata che l'organismo non soddisfa più i criteri di cui al paragrafo 2. Esso ne informa immediatamente gli altri Stati membri e la Commissione.
4. L'organismo notificato ed il fabbricante o il suo ►M4 mandatario ◄ stabiliscono di comune accordo i termini per portare a termine le operazioni di valutazione e di verifica di cui agli allegati da 2 a 5.
5. L’organismo notificato fornisce alla propria autorità competente tutte le informazioni sui certificati rilasciati, modificati, integrati, sospesi, ritirati o rifiutati e agli altri organismi notificati ai sensi della presente direttiva quelle sui certificati sospesi, ritirati o rifiutati e, su richiesta, sui certificati rilasciati. L’organismo notificato mette inoltre a disposizione, su richiesta, tutte le informazioni supplementari pertinenti.
6. Qualora un organismo notificato constati che i requisiti pertinenti della presente direttiva non sono stati o non sono più soddisfatti dal fabbricante oppure che un certificato non avrebbe dovuto essere rilasciato, esso sospende, ritira o sottopone a limitazioni il certificato rilasciato, tenendo conto del principio di proporzionalità, a meno che la conformità a tali requisiti non sia assicurata mediante l’applicazione di appropriate misure correttive da parte del fabbricante.
In caso di sospensione, ritiro o limitazioni del certificato o nei casi in cui risulti necessario l’intervento dell’autorità competente, l’organismo notificato ne informa la propria autorità competente.
Lo Stato membro informa gli altri Stati membri e la Commissione.
7. L’organismo notificato fornisce, su richiesta, tutte le informazioni e i documenti pertinenti, compresi i documenti di bilancio, necessari allo Stato membro per verificare la conformità ai criteri di cui all’allegato 8.
Articolo 12
1. Ogni dispositivo, diverso dai dispositivi su misura e da quelli destinati ad indagini cliniche, ritenuto rispondente ai requisiti essenziali di cui all'articolo 3 deve formare oggetto di una marcatura CE di conformità.
2. La ►M2 marcatura CE ◄ di conformità, riprodotta nell'allegato 9, deve essere apposta in modo da risultare visibile, leggibile e indelebile sull'imballaggio che garantisce la sterilità, sull'eventuale imballaggio commerciale e sulle istruzioni per l'uso.
Essa deve essere seguita dal numero di identificazione dell'organismo notificato responsabile dell'attuazione delle procedure di cui agli allegati 2, 4 e 5.
3. È vietato apporre marcature che possano indurre in errore i terzi circa il significato e il simbolo grafico della marcatura CE. Sull'imballaggio o sul foglio di istruzioni che accompagna il dispositivo può essere apposto ogni altro marchio purché questo non limiti la visibilità e la leggibilità della marcatura CE.
Articolo 13
Fatto salvo l’articolo 7:
a) ogni constatazione da parte di uno Stato membro che il marchio CE è stato indebitamente apposto o manca in violazione della presente direttiva comporta per il fabbricante o il suo mandatario stabilito nella Comunità l’obbligo di far cessare l’infrazione alle condizioni stabilite dallo Stato membro stesso;
b) nel caso in cui persista la mancanza di conformità, lo Stato membro deve adottare tutte le misure atte a limitare o vietare l’immissione sul mercato del dispositivo in questione o a garantire il ritiro dal commercio secondo le procedure previste all’articolo 7.
Tali disposizioni si applicano anche quando il marchio CE è stato apposto in conformità delle procedure di cui alla presente direttiva, ma impropriamente, su prodotti che non sono contemplati dalla presente direttiva.
Articolo 14
Ogni decisione presa in applicazione della presente direttiva:
a) intesa a vietare o limitare l’immissione sul mercato o la messa in servizio di un dispositivo ovvero lo svolgimento di indagini cliniche;
o
b) intesa a ritirare dispositivi dal mercato,
è motivata in maniera precisa. Tale decisione è notificata all’interessato al più presto, indicandogli i mezzi di ricorso ammessi dal diritto vigente nello Stato membro in questione e i termini entro i quali gli stessi devono essere presentati.
Nel caso della decisione di cui al primo comma il fabbricante o il suo mandatario ►M4 ————— ◄ deve avere la possibilità di presentare preventivamente il proprio punto di vista, a meno che tale consultazione non sia resa impossibile dall'urgenza del provvedimento.
Articolo 15
1. Fatte salve le disposizioni e le prassi nazionali vigenti in materia di segreto medico, gli Stati membri provvedono a che tutte le parti interessate dall’applicazione della presente direttiva siano obbligate a mantenere riservate tutte le informazioni ottenute nell’espletamento delle loro funzioni.
Ciò lascia impregiudicati gli obblighi degli Stati membri e degli organismi notificati in materia di informazione reciproca e di diffusione degli avvisi di sicurezza nonché gli obblighi delle persone interessate a fornire informazioni ai sensi del diritto penale.
2. Le seguenti informazioni non sono considerate riservate:
a) informazioni sulla registrazione delle persone responsabili dell’immissione in commercio di dispositivi ai sensi dell’articolo 10 bis;
b) informazioni agli utilizzatori inviate dal fabbricante, dal mandatario o dal distributore in relazione a una misura prevista dall’articolo 8;
c) informazioni contenute in certificati rilasciati, modificati, integrati, sospesi o ritirati.
3. Le misure intese a modificare elementi non essenziali della presente direttiva, integrandola, relative alla determinazione delle condizioni in base alle quali possono essere rese pubbliche le informazioni diverse da quelle elencate al paragrafo 2, in particolare riguardanti l’obbligo dei fabbricanti di preparare e mettere a disposizione una sintesi delle informazioni e dei dati relativi al dispositivo, sono adottate secondo la procedura di regolamentazione con controllo di cui all’articolo 6, paragrafo 4.
Articolo 15 bis
Gli Stati membri adottano le misure idonee a garantire che le competenti autorità degli Stati membri cooperino tra loro e con la Commissione e si trasmettano reciprocamente le informazioni necessarie per far sì che la presente direttiva sia applicata in modo uniforme.
La Commissione assicura l’organizzazione di uno scambio di esperienze tra le autorità responsabili della vigilanza del mercato al fine di coordinare l’applicazione uniforme della presente direttiva.
Ferme restando le disposizioni della presente direttiva, la cooperazione può formare parte di iniziative sviluppate a livello internazionale.
Articolo 16
1. Gli Stati membri adottano e pubblicano anteriormente al 1o luglio 1992 le disposizioni legislative, regolamentari ed amministrative necessarie per conformarsi alla presente direttiva. Essi ne informano immediatamente la Commissione.
Essi applicano tali disposizioni a decorrere dal 1o gennaio 1993.
2. Gli Stati membri comunicano alla Commissione il testo delle disposizioni di diritto interno che essi adottano nel settore disciplinato dalla presente direttiva.
3. Gli Stati membri ammettono, fino al 31 dicembre 1994, l'immissione in commercio e la messa in servizio degli apparecchi conformi alle norme vigenti nel loro territorio al 31 dicembre 1992.
Articolo 17
Gli Stati membri sono destinatari della presente direttiva.
ALLEGATO 1
REQUISITI ESSENZIALI
I. REQUISITI GENERALI
|
1. |
I dispositivi devono essere progettati e fabbricati in modo tale che il loro impiego non comprometta lo stato clinico né la sicurezza dei pazienti, quando siano impiantati alle condizioni e per i fini previsti. Non devono presentare rischi per le persone che li impiantano né per eventuali terzi. |
|
2. |
I dispositivi devono fornire le prestazioni previste dal fabbricante: devono cioè essere progettati e fabbricati in modo da soddisfare una o più delle funzioni previste dall'articolo 1, paragrafo 2, lettera a), e ivi specificate. |
|
3. |
Le caratteristiche e le prestazioni di cui ai punti 1 e 2 non devono essere alterate in modo da compromettere lo stato clinico e la sicurezza dei pazienti e, se del caso, di terzi nel periodo di vita utile dei dispositivi prevista dal fabbricante, quando questi ultimi sono sottoposti alle sollecitazioni che possono verificarsi in condizioni normali di impiego. |
|
4. |
I dispositivi devono essere progettati, fabbricati e confezionati in modo che le loro caratteristiche e le loro prestazioni non siano alterate nelle condizioni di magazzinaggio e di trasporto previste dal fabbricante (temperatura, umidità, ecc.). |
|
5. |
Eventuali effetti collaterali e indesiderabili devono costituire rischi accettabili in rapporto alle prestazioni previste. |
|
5 bis. |
La dimostrazione della conformità con i requisiti essenziali deve comprendere una valutazione clinica a norma dell'allegato 7. |
II. REQUISITI RELATIVI ALLA PROGETTAZIONE E ALLA COSTRUZIONE
|
6. |
Le soluzioni scelte dal fabbricante nella progettazione e nella costruzione dei dispositivi devono attenersi ai principi di integrazione della sicurezza e tener conto dello stato della tecnica generalmente riconosciuto. |
|
7. |
I dispositivi impiantabili devono essere progettati, fabbricati e confezionati in imballaggi non riutilizzabili conformemente a procedure adeguate, in modo che siano sterili all'atto della loro immissione sul mercato e mantengano tale qualità, nelle condizioni di magazzinaggio e di trasporto previste dal fabbricante, fino all'apertura dell'imballaggio per l'impianto. |
|
8. |
I dispositivi devono essere progettati e fabbricati in modo da eliminare o ridurre al minimo per quanto possibile: — i rischi di lesioni connessi alle loro caratteristiche fisiche, anche dimensionali; — i rischi connessi con l'utilizzazione delle fonti di energia, facendo particolarmente attenzione, in caso di utilizzazione dell'elettricità, all'isolamento, alle correnti di dispersione e al riscaldamento dei dispositivi; — i rischi connessi con condizioni ambientali ragionevolmente prevedibili, in particolare quelli connessi con i campi magnetici, le influenze elettriche esterne, le scariche elettrostatiche, la pressione o le variazioni di pressione, l'accelerazione; — i rischi connessi ad interventi medici, in particolare quelli risultanti dall'impiego dei defibrillatori o delle apparecchiature chirurgiche ad alta frequenza; — rischi connessi alle radiazioni ionizzanti provenienti da sostanze radioattive che fanno parte del dispositivo, nel rispetto dei requisiti di protezione stabiliti dalla direttiva 96/29/Euratom del Consiglio, del 13 maggio 1996, che stabilisce le norme fondamentali di sicurezza relative alla protezione sanitaria della popolazione e dei lavoratori contro i pericoli derivanti dalle radiazioni ionizzanti ( 5 ), e dalla direttiva 97/43/Euratom del Consiglio, del 30 giugno 1997, riguardante la protezione sanitaria delle persone contro i pericoli delle radiazioni ionizzanti connesse a esposizioni mediche ( 6 ); — i rischi che possano verificarsi qualora la manutenzione o la taratura non siano possibili, connessi in particolare con: — — l'eccessivo aumento delle correnti di dispersione; — l'invecchiamento dei materiali utilizzati; — un eccessivo aumento del calore prodotto dal dispositivo; — un deterioramento della precisione di un qualsiasi meccanismo di misurazione o di controllo. |
|
9. |
I dispositivi devono essere progettati e fabbricati in modo tale da garantire le caratteristiche e le prestazioni di cui al punto I «Requisiti generali», con particolare riguardo per: — la scelta dei materiali utilizzati, in particolare per quanto riguarda gli aspetti della tossicità; — la reciproca compatibilità tra i materiali utilizzati e i tessuti, le cellule biologiche e i liquidi corporei, tenuto conto dell"impiego previsto per il dispositivo; — la compatibilità dei dispositivi con le sostanze che devono somministrare; — la qualità delle connessioni, in particolare sul piano della sicurezza; — l'affidabilità della fonte d'energia; — se del caso, un'adeguata tenuta alla penetrazione dei liquidi; — il buon funzionamento dei sistemi di comando, di programmazione e di controllo, compreso il software. ►M4 Per i dispositivi che incorporano un software o costituiscono in sé un software medico, il software è convalidato secondo lo stato dell'arte, tenendo conto dei principi del ciclo di vita dello sviluppo, della gestione dei rischi, della validazione e della verifica. ◄ |
|
10. |
Quando un dispositivo incorpora come parte integrante una sostanza che, se utilizzata separatamente, può essere considerata un medicinale ai sensi dell'articolo 1 della direttiva 2001/83/CE e può avere effetti sul corpo umano con un'azione accessoria a quella del dispositivo, è necessario verificare la qualità, la sicurezza e l'utilità della sostanza, applicando per analogia i metodi previsti dall'allegato I della direttiva 2001/83/CE. Nel caso di sostanze di cui al primo comma, l'organismo notificato, previa verifica dell'utilità della sostanza come parte del dispositivo medico e tenuto conto della destinazione del dispositivo, chiede a una delle autorità competenti designate dagli Stati membri o all'Agenzia europea per i medicinali (EMEA), che opera in particolare attraverso il suo comitato in conformità del regolamento (CE) n. 726/2004 ( 7 ), un parere scientifico sulla qualità e sulla sicurezza della sostanza, ivi compreso il profilo clinico rischi/benefici relativo all'incorporazione della sostanza nel dispositivo. Nell'esprimere il parere, l'autorità competente o l'EMEA tengono conto del processo di fabbricazione e dei dati relativi all'utilità dell'incorporazione della sostanza nel dispositivo come stabilito dall'organismo notificato. Quando un dispositivo incorpora, come parte integrante, un derivato del sangue umano, l'organismo notificato, previa verifica dell'utilità della sostanza come parte del dispositivo medico e tenuto conto della destinazione del dispositivo, chiede all'EMEA, che opera in particolare attraverso il suo comitato, un parere scientifico sulla qualità e sulla sicurezza della sostanza, ivi compreso il profilo clinico rischi/benefici dell'incorporazione del derivato del sangue umano nel dispositivo medico. Nell'esprimere il parere, l'EMEA tiene conto del processo di fabbricazione e dei dati relativi all'utilità dell'incorporazione della sostanza nel dispositivo, come stabilito dall'organismo notificato. Le modifiche apportate a una sostanza accessoria incorporata in un dispositivo, in particolare quelle connesse al processo di fabbricazione, sono comunicate all'organismo notificato, il quale consulta l'autorità per i medicinali competente (cioè quella che ha partecipato alla consultazione iniziale), per confermare il mantenimento della qualità e della sicurezza della sostanza accessoria. ►C1 L'autorità competente tiene conto dei dati relativi all'utilità dell'incorporazione della sostanza nel dispositivo come stabilito dall'organismo notificato, al fine di assicurarsi che le modifiche non abbiano alcuna ripercussione negativa sul profilo rischi/benefici definito, relativo all'inclusione della sostanza nel dispositivo medico. ◄ Allorché la pertinente autorità medica competente (ossia quella che ha partecipato alla consultazione iniziale) ha avuto un'informazioni sulla sostanza accessoria che potrebbe avere un impatto sul profilo rischi/benefici definito relativo all'aggiunta della sostanza al dispositivo, fornisce all'organismo notificato un parere in cui stabilisce se tale informazione abbia o meno un impatto sul profilo rischi/benefici definito relativo all'aggiunta di tale sostanza nel dispositivo. L'organismo notificato tiene conto del parere scientifico aggiornato riconsiderando la propria valutazione della procedura di valutazione di conformità. |
|
11. |
I dispositivi e, se del caso, i loro componenti devono essere identificati in modo da rendere possibile le azioni appropriate che si rivelassero necessarie a seguito della scoperta di un potenziale rischio connesso con i dispositivi e i loro componenti. |
|
12. |
I dispositivi devono recare un codice che permetta lidentificazione univoca del dispositivo stesso (segnatamente il tipo di dispositivo e l'anno di fabbricazione) e del fabbricante; il codice deve poter essere rilevato, se del caso, senza dover ricorrere ad un intervento chirurgico. |
|
13. |
Quando un dispositivo o i relativi accessori recano le istruzioni necessarie per il funzionamento del dispositivo o indicano parametri di funzionamento o di regolazione mediante un sistema di visualizzazione, tali informazioni devono poter essere comprese dall'operatore e, se del caso, dal paziente. |
|
14. |
Ogni dispositivo deve recare sull'imballaggio in modo leggibile e indelebile, eventualmente mediante codici generalmente riconosciuti, le seguenti indicazioni: 14.1. Sull'imballaggio che assicura la sterilità: — metodo di sterilizzazione; — indicazione che consenta di riconoscere detto imballaggio; — nome e indirizzo del fabbricante; — denominazione dell'apparecchio; — qualora si tratti di un dispositivo destinato ad indagini cliniche, l'indicazione «esclusivamente per indagini cliniche»; — qualora si tratti di un dispositivo su misura, l'indicazione «apparecchio su misura»; — indicazione che il dispositivo impiantabile è sterile; — indicazione del mese e dell'anno di fabbricazione; — indicazione della data limite di impianto del dispositivo in tutta sicurezza. 14.2. Sull'imballaggio commerciale: — nome e indirizzo del fabbricante e nome e indirizzo del mandatario qualora il fabbricante non abbia sede nella Comunità; — denominazione del dispositivo; — destinazione del dispositivo; — caratteristiche pertinenti per il suo impiego; — qualora si tratti di un dispositivo destinato a indagini cliniche, l'indicazione «esclusivamente per indagini cliniche»; — qualora si tratti di un dispositivo su misura, l'indicazione «dispositivo su misura»; — indicazione che il dispositivo impiantabile è sterile; — indicazione del mese e dell'anno di fabbricazione; — indicazione della data limite di impianto del dispositivo in tutta sicurezza; — condizioni per il trasporto e il magazzinaggio del dispositivo; — nel caso di un dispositivo ai sensi dell'articolo 1, paragrafo 4 bis, indicazione che il dispositivo incorpora un derivato del sangue umano. |
|
15. |
All'atto dell'immissione sul mercato, ogni dispositivo deve essere accompagnato da istruzioni comprendenti i seguenti elementi: — anno di autorizzazione dell"apposizione della ►M2 marcatura CE ◄ ; — indicazioni di cui ai punti 14.1 e 14.2, tranne quelle di cui all'ottavo e nono trattino; — prestazioni di cui al punto 2, nonché eventuali effetti secondari indesiderabili; — informazioni atte a consentire al medico di selezionare il dispositivo adeguato, nonché il software e gli accessori adeguati; — informazioni che costituiscono le avvertenze per l'uso e consentono al medico ed eventualmente al paziente di utilizzare correttamente il dispositivo, i suoi accessori e il software, nonché informazioni relative a natura, portata e intervalli dei controlli e delle prove di funzionamento ed eventualmente misure di manutenzione; — informazioni utili da seguire, se del caso, per evitare taluni rischi connessi con l'impianto del dispositivo; — informazioni relative ai rischi di mutue interferenze ( 8 ) connessi con la presenza del dispositivo in caso di indagini o trattamenti specifici; — istruzioni necessarie in caso di rottura dell'imballaggio che assicura la sterilità e, se del caso, indicazione dei metodi adeguati per la risterilizzazione; — se del caso, avviso che un dispositivo può essere riutilizzato solo se è stato ricondizionato sotto la responsabilità del fabbricante per essere conforme ai requisiti essenziali. Le istruzioni devono inoltre comprendere le indicazioni atte a consentire al medico di informare il paziente sulle controindicazioni e le precauzioni da prendere. Tali indicazioni riguardano in particolare: — le informazioni che consentono di determinare la durata di vita della fonte di energia, — le precauzioni da prendere in caso di variazioni di prestazione del dispositivo, — le precauzioni da prendere per quanto riguarda l'esposizione, in condizioni ambientali ragionevolmente prevedibili, a campi magnetici, alle influenze elettriche esterne, alle scariche elettrostatiche, alla pressione o a variazioni di pressione, all'accelerazione, ecc., — le informazioni adeguate relative ai medicinali che il dispositivo in questione deve somministrare, — data di emissione o dell'ultima versione delle istruzioni per l'uso. |
|
16. |
La conferma dell'osservanza dei requisiti inerenti alle caratteristiche e alle prestazioni, di cui al punto I «Requisiti generali», del dispositivo in condizioni normali di impiego, nonché la valutazione degli effetti secondari o indesiderabili devono basarsi su dati clinici accertati in conformità delle disposizioni dell'allegato 7. |
ALLEGATO 2
DICHIARAZIONE CE DI CONFORMITÀ
(Sistema completo di garanzia della qualità)
|
1. |
Il fabbricante applica il sistema qualità approvato per la progettazione, la fabbricazione, l'ispezione finale dei prodotti in questione come è specificato ai punti 3 e 4 ed è sottoposto al controllo CE come è specificato al punto 5. |
|
2. |
La dichiarazione di conformità è la procedura mediante la quale il fabbricante che ottemperi agli obblighi di cui al punto 1 garantisce e dichiara che i prodotti in questione soddisfano le pertinenti disposizioni della direttiva. Il fabbricante o il suo mandatario stabilito nella Comunità appone la marcatura CE in conformità dell'articolo 12 e fornisce una dichiarazione scritta di conformità. Detta dichiarazione riguarda uno o più dispositivi chiaramente individuati mediante il nome del prodotto, il relativo codice o altro riferimento non ambiguo e deve essere conservata dal fabbricante. La marcatura CE è accompagnata dal numero di identificazione dell'organismo notificato responsabile. |
|
3. |
Sistema qualità
|
|
4. |
Esame della progettazione del prodotto
|
|
5. |
Controllo
|
|
6. |
Disposizioni amministrative
▼M4 ————— |
|
7. |
Applicazione ai dispositivi di cui all'articolo 1, paragrafo 4 bis Al termine della fabbricazione di ogni lotto di dispositivi di cui all'articolo 1, paragrafo 4 bis, il fabbricante informa l'organismo notificato del rilascio di tale lotto di dispositivi e gli trasmette il certificato ufficiale di rilascio del lotto del derivato del sangue umano utilizzato in tale dispositivo, emesso da un laboratorio di Stato o da un laboratorio designato a tal fine da uno Stato membro, a norma dell'articolo 114, paragrafo 2, della direttiva 2001/83/CE. |
ALLEGATO 3
CERTIFICAZIONE CE
|
1. |
La certificazione CE è la procedura mediante la quale un organismo notificato constata e certifica che un esemplare rappresentativo della produzione in programma soddisfa le disposizioni pertinenti della direttiva. |
|
2. |
La domanda di certificazione CE è presentata dal fabbricante o dal suo mandatario stabiliti nella Comunità ad un organismo notificato. Tale richiesta deve comprendere: — nome ed indirizzo del fabbricante e, qualora la domanda sia presentata da un suo mandatario, anche il nome e l'indirizzo di quest'ultimo, — una dichiarazione scritta che specifichi che la domanda non è stata presentata a nessun altro organismo notificato, — la documentazione di cui al punto 3, necessaria per permettere di valutare la conformità dell'esemplare rappresentativo della produzione prevista, in appresso denominato «tipo», con i requisiti della direttiva. Il richiedente metterà a disposizione dell'organismo notificato un tipo. L'organismo notificato potrà richiederne, se necessario, altri esemplari. |
|
3. |
La documentazione deve illustrare chiaramente la progettazione, la fabbricazione e le prestazioni del prodotto. La documentazione deve contenere in particolare i seguenti elementi: — una descrizione generale del tipo, comprese le varianti previste, e la/le destinazione/destinazioni d'uso, — disegni di progettazione, metodi di fabbricazione previsti soprattutto in materia di sterilizzazione, schemi di componenti, sottoinsiemi, circuiti, ecc.; — le descrizioni e spiegazioni necessarie per comprendere detti disegni e schemi ed il funzionamento del prodotto; — un elenco delle norme di cui all'articolo 5, applicate integralmente o parzialmente, nonché le descrizioni delle soluzioni adottate perché siano soddisfatti i requisiti essenziali, qualora le norme di cui all'articolo 5 non siano state applicate; — i risultati dei calcoli di progettazione, dell'analisi dei rischi, delle indagini, delle prove tecniche svolte, ecc., — una dichiarazione che indichi se il dispositivo incorpora o meno, come parte integrante, una sostanza o un derivato del sangue umano di cui al punto 10 dell'allegato 1, nonché i dati relativi alle pertinenti prove svolte, necessarie a valutare la sicurezza, la qualità e l'utilità di tale sostanza o derivato del sangue umano, tenendo conto della destinazione del dispositivo, — la valutazione preclinica, — la valutazione clinica di cui all'allegato 7, — la bozza di istruzioni per l'uso. |
|
4. |
L'organismo notificato: 4.1. esamina e valuta la documentazione, controlla che il tipo sia stato fabbricato conformemente a quest'ultima e individua gli elementi che sono stati progettati in conformità delle disposizioni applicabili delle norme di cui all'articolo 5, nonché gli elementi la cui progettazione non si basa sulle appropriate disposizioni di dette norme; 4.2. effettua o fa effettuare i controlli del caso e le prove necessarie per verificare se le soluzioni adottate dal fabbricante rispondano alle esigenze essenziali della direttiva, qualora le norme di cui all'articolo 5 non siano state applicate; 4.3. effettua o fa effettuare i controlli appropriati e le prove necessarie per verificare se, qualora il fabbricante abbia deciso di applicare le norme pertinenti, queste siano state realmente applicate; 4.4. concorda con il richiedente la sede in cui devono svolgersi i controlli e le prove necessarie. |
|
5. |
Qualora il tipo sia conforme alle disposizioni della direttiva, l'organismo notificato rilascia al richiedente un certificato di esame CE di tipo. Il certificato reca il nome e l'indirizzo del fabbricante, le conclusioni del controllo, le condizioni di validità del certificato e i dati necessari per l'identificazione del tipo approvato. Le parti significative della documentazione sono allegate al certificato ed una copia viene conservata dall'organismo notificato. Nel caso dei dispositivi di cui all'allegato 1, punto 10, secondo comma, prima di prendere una decisione l'organismo notificato consulta, per quanto riguarda gli aspetti contemplati da tale punto, una delle competenti autorità designate dagli Stati membri a norma della direttiva 2001/83/CE o l'EMEA. Il parere dell'autorità nazionale competente o dell'EMEA è elaborato entro 210 giorni dal ricevimento della documentazione valida. Il parere scientifico dell'autorità nazionale competente o dell'EMEA deve essere inserito nella documentazione relativa al dispositivo. Nell'adottare la decisione, l'organismo notificato tiene in debita considerazione i pareri espressi nel contesto della consultazione. Esso provvede a informare l'organo competente interessato della sua decisione finale. Nel caso dei dispositivi di cui all'allegato 1, punto 10, terzo comma, il parere scientifico dell'EMEA dev'essere inserito nella documentazione concernente il dispositivo. Il parere è elaborato entro 210 giorni dal ricevimento di una valida documentazione. Nell'adottare la decisione, l'organismo notificato tiene in debita considerazione il parere dell'EMEA. L'organismo notificato non può rilasciare il certificato se il parere scientifico dell'EMEA è sfavorevole. Esso provvede ad informare l'EMEA della sua decisione finale. |
|
6. |
Il richiedente informa di eventuali modifiche apportate al prodotto approvato l'organismo notificato che ha rilasciato il certificato CE di esame di tipo. Qualora le modifiche apportate al prodotto approvato possano rimettere in discussione la conformità ai requisiti essenziali o alle condizioni previste di utilizzazione del prodotto, esse devono ottenere un'approvazione supplementare da parte dell'organismo notificato che ha rilasciato il certificato CE di esame. L'approvazione supplementare è rilasciata eventualmente come addendum al certificato CE iniziale di esame di tipo. |
|
7. |
Disposizioni amministrative 7.1. Ove richiesto, ciascun organismo notificato mette a disposizione degli altri organismi notificati e dell'autorità competente tutte le informazioni pertinenti riguardanti i certificati di esame CE di tipo e gli addenda rilasciati, rifiutati e ritirati. 7.2. Gli altri organismi notificati possono ottenere una copia dei certificati di esame CE di tipo e/o dei loro addenda. Gli allegati dei certificati sono tenuti a disposizione degli altri organismi notificati, dietro richiesta motivata, previa informazione del fabbricante. 7.3. Il fabbricante o il suo mandatario conserva, unitamente alla documentazione tecnica, una copia degli attestati di esame CE di tipo e dei loro documenti aggiuntivi per un periodo minimo di ►M4 quindici anni dalla fabbricazione dell'ultimo prodotto ◄ . ▼M4 ————— |
ALLEGATO 4
VERIFICA CE
|
1. |
La verifica CE è la procedura mediante la quale il fabbricante o il suo mandatario stabilito nella Comunità garantisce e dichiara che i prodotti sottoposti alle prescrizioni del paragrafo 3 sono conformi al tipo descritto nel certificato di esame «CE del tipo» e soddisfano i requisiti della direttiva che ad essi si applica. |
|
2. |
Il fabbricante adotta tutte le misure necessarie a che il processo di fabbricazione garantisca la conformità dei prodotti al tipo descritto nel certificato «CE del tipo» ed ai requisiti della presente direttiva che ad essi si applica. Il fabbricante o il suo mandatario stabilito nella Comunità appone la marcatura CE su ogni prodotto e fornisce una dichiarazione scritta di conformità. |
|
3. |
Prima dell'inizio dei processi di fabbricazione, il fabbricante deve redigere la documentazione che definisce tali processi soprattutto per quanto concerne la sterilizzazione, nonché le disposizioni prestabilite e sistematiche che saranno attuate per garantire l'omogeneità della produzione e la conformità dei prodotti al tipo descritto nel certificato d'esame CE del tipo e ai requisiti della direttiva ad essi applicabile. |
|
4. |
Il fabbricante si impegna ad instaurare ed aggiornare un ►M4 sistema di sorveglianza post-vendita comprendente le disposizioni di cui all'articolo 7 ◄ . Tale impegno comporta l'obbligo da parte del fabbricante di informare, se ne è a conoscenza, le autorità competenti degli incidenti seguenti: i) ogni alterazione delle caratteristiche e delle prestazioni, nonché ogni inadeguatezza del foglio di istruzioni di un dispositivo che potrebbe provocare o aver provocato la morte o un peggioramento delle condizioni di salute di un paziente; ii) ogni motivo di carattere tecnico o medico che abbia comportato il ritiro dal mercato di un dispositivo da parte del fabbricante. |
|
5. |
L'organismo notificato effettua gli esami e le prove atte a verificare la conformità del prodotto ai requisiti della presente direttiva, con controllo e prova su base statistica, come specificato al punto 6. Il fabbricante deve autorizzare l'organismo notificato a valutare l'efficacia delle misure adottate in applicazione del punto 3, se necessario mediante verifica sistematica. |
|
6. |
Verifica statistica
|
|
7. |
Applicazione ai dispositivi di cui all'articolo 1, paragrafo 4 bis Al termine della fabbricazione di ogni lotto di dispositivi di cui all'articolo 1, paragrafo 4 bis, il fabbricante informa l'organismo notificato del rilascio di tale lotto di dispositivi e gli trasmette il certificato ufficiale di rilascio del lotto del derivato del sangue umano utilizzato in tale dispositivo, emesso da un laboratorio di Stato o da un laboratorio designato a tal fine da uno Stato membro, a norma dell'articolo 114, paragrafo 2, della direttiva 2001/83/CE. |
ALLEGATO 5
DICHIARAZIONE CE DI CONFORMITÀ AL TIPO
(Garanzia della qualità della produzione)
|
1. |
Il fabbricante applica il sistema qualità approvato per la fabbricazione e l'ispezione finale dei prodotti in questione come previsto al punto 3, ed è sottoposto alla sorveglianza di cui al punto 4. |
|
2. |
Questa dichiarazione di conformità è l'elemento di procedura mediante il quale il fabbricante, che ottempera agli obblighi di cui al punto 2, garantisce e dichiara che i prodotti interessati sono conformi al tipo descritto nel certificato CE di esame del tipo e rispondono alle pertinenti disposizioni della direttiva. Il fabbricante o il suo mandatario stabilito nella Comunità appone la marcatura CE conformemente all'articolo 12 e redige una dichiarazione di conformità. Questa dichiarazione comprende uno o più ►M4 dispositivi fabbricati, chiaramente identificati con il nome del prodotto, il relativo codice o un altro riferimento non ambiguo, e deve essere conservata dal fabbricante ◄ . La marcatura CE è corredata dal numero di identificazione dell'organismo notificato responsabile. |
|
3. |
Sistema qualità
|
|
4. |
Sorveglianza
|
|
5. |
L'organismo notificato comunica agli altri organismi notificati le informazioni pertinenti riguardanti le approvazioni rilasciate, rifiutate o ritirate dei sistemi di qualità. |
|
6. |
Applicazione ai dispositivi di cui all'articolo 1, paragrafo 4 bis Al termine della fabbricazione di ogni lotto di dispositivi di cui all'articolo 1, paragrafo 4 bis, il fabbricante informa l'organismo notificato del rilascio di tale lotto di dispositivi e gli trasmette il certificato ufficiale di rilascio del lotto del derivato del sangue umano utilizzato in tale dispositivo, emesso da un laboratorio di Stato o da un laboratorio designato a tal fine da uno Stato membro, a norma dell'articolo 114, paragrafo 2, della direttiva 2001/83/CE. |
ALLEGATO 6
DICHIARAZIONE RELATIVA AI DISPOSITIVI CON DESTINAZIONI PARTICOLARI
|
1. |
Il fabbricante o il suo mandatario stabilito nella Comunità redige per i dispositivi su misura o per i dispositivi destinati a indagini cliniche la dichiarazione comprendente gli elementi specificati al punto 2. |
|
2. |
La dichiarazione comprende le seguenti indicazioni.
|
|
3. |
Il fabbricante si impegna a tenere a disposizione delle autorità nazionali competenti:
|
|
4. |
Le informazioni contenute nelle dichiarazioni previste dal presente allegato sono conservate per un periodo di almeno quindici anni a partire dalla data di fabbricazione dell'ultimo prodotto. |
|
5. |
Per quanto concerne i dispositivi su misura, il fabbricante si impegna a valutare e a documentare l'esperienza acquisita nella fase successiva alla produzione, anche sulla base delle disposizioni di cui all'allegato 7, nonché a predisporre i mezzi idonei all'applicazione degli interventi correttivi eventualmente necessari. Detto impegno deve comprendere l'obbligo per il fabbricante di informare le autorità competenti degli incidenti seguenti, non appena egli ne venga a conoscenza, e dei pertinenti interventi correttivi: i) qualsiasi disfunzione o deterioramento delle caratteristiche e/o delle prestazioni di un dispositivo, nonché qualsiasi carenza dell'etichettatura o delle istruzioni per l'uso di un dispositivo che possano causare o aver causato la morte o un grave peggioramento dello stato di salute di un paziente o di un utilizzatore; ii) le ragioni di ordine tecnico o medico connesse con le caratteristiche o le prestazioni di un dispositivo che determinino, per i motivi elencati al punto i), il ritiro sistematico da parte del fabbricante dei dispositivi appartenenti allo stesso tipo. |
ALLEGATO 7
VALUTAZIONE CLINICA
1. Disposizioni generali
|
1.1. |
Di regola la conferma del rispetto dei requisiti relativi alle caratteristiche e alle prestazioni specificate ai punti 1 e 2 dell'allegato 1 in condizioni normali di utilizzazione del dispositivo e la valutazione degli effetti collaterali e dell'accettabilità del rapporto rischi/benefici di cui al punto 5 dell'allegato 1 devono basarsi su dati clinici. La valutazione di tali dati, di seguito denominata «valutazione clinica», che tiene conto ove necessario delle eventuali norme armonizzate pertinenti, segue una procedura definita e metodologicamente valida fondata alternativamente su: 1.1.1. un'analisi critica della letteratura scientifica pertinente attualmente disponibile sui temi della sicurezza, delle prestazioni, delle caratteristiche di progettazione e della destinazione d'uso del dispositivo qualora: — sia dimostrata l'equivalenza tra il dispositivo in esame e il dispositivo cui si riferiscono i dati, e — i dati dimostrino adeguatamente la conformità ai requisiti essenziali pertinenti; 1.1.2. un'analisi critica di tutte le indagini cliniche condotte; 1.1.3. un'analisi critica dei dati clinici combinati di cui ai punti 1.1.1 e 1.1.2. |
|
1.2. |
Vengono condotte indagini cliniche, salvo che non sia debitamente giustificato fondarsi sui dati clinici esistenti. |
|
1.3. |
La valutazione clinica e il relativo esito sono documentati. La documentazione tecnica del dispositivo contiene tali documenti e/o i relativi riferimenti completi. |
|
1.4. |
La valutazione clinica e la relativa documentazione sono attivamente aggiornate con dati derivanti dalla sorveglianza post-vendita. Ove non si consideri necessario il follow-up clinico post-vendita nell'ambito del piano di sorveglianza post-vendita applicato al dispositivo, tale conclusione va debitamente giustificata e documentata. |
|
1.5. |
Qualora non si ritenga opportuna la dimostrazione della conformità ai requisiti essenziali in base ai dati clinici, occorre fornire un'idonea giustificazione di tale esclusione in base ai risultati della gestione del rischio, tenendo conto anche della specificità dell'interazione tra il dispositivo e il corpo, delle prestazioni cliniche attese e delle affermazioni del fabbricante. Va debitamente provata l'adeguatezza della dimostrazione della conformità ai requisiti essenziali che si fondi solo sulla valutazione delle prestazioni, sulle prove al banco e sulla valutazione preclinica. |
|
1.6. |
Tutti i dati devono rimanere riservati a meno che se ne ritenga essenziale la divulgazione. |
2. Indagini cliniche
2.1. Obiettivi
Gli obiettivi delle indagini cliniche sono:
— verificare che, nelle normali condizioni di utilizzazione, le prestazioni del dispositivo siano conformi a quelle di cui all'allegato 1, punto 2, e
— determinare gli eventuali effetti secondari indesiderati nelle normali condizioni di utilizzazione e valutare se essi comportano rischi accettabili rispetto alle prestazioni attese dal dispositivo.
2.2. Considerazioni etiche
L'indagine clinica deve essere effettuata conformemente alla dichiarazione di Helsinki approvata dalla 18a Conferenza medica mondiale svoltasi a Helsinki, Finlandia, nel 1964, emendata dalla 29a Conferenza medica mondiale svoltasi a Tokyo, Giappone, nel 1975, e dalla 35a Conferenza medica mondiale svoltasi a Venezia, Italia, nel 1983. È imperativo che tutte le considerazioni relative alla tutela dell'uomo tengano conto dello spirito della dichiarazione di Helsinki. Quanto sopra comprende ogni fase dell'indagine clinica, dalla prima riflessione sulla necessità e la giustificazione dello studio fino alla pubblicazione dei risultati.
2.3. Metodi
|
2.3.1. |
Le indagini cliniche vanno eseguite secondo un adeguato programma d'indagine conforme allo stato della scienza e della tecnica, definito in modo da confermare o respingere le affermazioni del fabbricante a proposito del dispositivo; tali indagini comportano un numero di osservazioni sufficienti per garantire la validità scientifica delle conclusioni. |
|
2.3.2. |
Le procedure utilizzate per svolgere le indagini devono essere congrue con il dispositivo in esame. |
|
2.3.3. |
Le indagini cliniche devono essere effettuate in circostanze equivalenti a quelle che si presenterebbero in condizioni normali di uso del dispositivo. |
|
2.3.4. |
Vanno esaminate tutte le caratteristiche pertinenti, comprese quelle attinenti alla sicurezza, alle prestazioni del dispositivo e agli effetti sul paziente. |
|
2.3.5. |
Tutti gli eventi avversi gravi devono essere registrati integralmente e immediatamente comunicati a tutte le autorità competenti degli Stati membri in cui è condotta l'indagine clinica. |
|
2.3.6. |
Le indagini vanno eseguite sotto la responsabilità di un ►M4 medico debitamente qualificato o persona autorizzata ◄ , in un ambiente adeguato. Il medico responsabile avrà accesso ai dati tecnici relativi al dispositivo. |
|
2.3.7. |
La relazione scritta, firmata dal medico responsabile, comprende una valutazione critica di tutti i dati ottenuti nel corso delle indagini cliniche. |
ALLEGATO 8
CRITERI MINIMI CHE DEVONO INTERVENIRE AI FINI DELLA DESIGNAZIONE DEGLI ORGANISMI DA NOTIFICARE
1. L'organismo, il suo direttore e il personale incaricato di eseguire le operazioni di valutazione e di verifica non possono essere né il progettista, né il fabbricante, né il fornitore, né l'istallatore dei dispositivi che essi controllano, né il mandatario di una di queste persone. Essi non possono intervenire né direttamente, né come mandatari nella progettazione, costruzione, commercializzazione o manutenzione di tali dispositivi. Ciò non esclude la possibilità di uno scambio di informazioni tecniche tra il fabbricante e l'organismo.
2. L'organismo e il personale incaricato del controllo debbono eseguire le operazioni di valutazione e di verifica con il massimo di integrità professionale e di competenza tecnica e devono inoltre essere liberi da qualsivoglia pressione e incentivo, soprattutto di ordine finanziario, che possa influenzare il loro giudizio o i risultati dei controlli, in particolare da pressioni che provengano da persone o gruppi di persone interessati ai risultati delle verifiche.
3. L'organismo deve poter assicurare l'insieme dei compiti che gli sono stati assegnati in uno degli allegati da 2 a 5 e per i quali è stato designato, sia che questi compiti siano svolti dal medesimo o sotto la sua responsabilità. Esso deve segnatamente disporre del personale e possedere i mezzi necessari, per eseguire in modo adeguato le operazioni tecniche e amministrative connesse con l'esecuzione delle valutazioni e delle verifiche; esso deve inoltre avere accesso al materiale necessario per le verifiche richieste.
4. Il personale incaricato dei controlli deve possedere:
— una buona formazione professionale per quanto riguarda il complesso di operazioni di valutazione e verifica per le quali l'organismo notificato è designato;
— una adeguata conoscenza delle norme relative ai controlli che effettua, nonché una sufficiente esperienza pratica di tali controlli;
— la capacità necessaria a compilare gli attestati, i verbali e le relazioni in cui sono riportati i risultati dei controlli effettuati.
5. Deve esser garantita l'indipendenza del personale incaricato del controllo. La retribuzione di ciascun agente non deve essere fissata in funzione del numero dei controlli eseguiti né dei risultati di tali controlli.
6. L'organismo deve sottoscrivere un contratto di assicurazione «responsabilità civile» a meno che detta responsabilità non sia coperta dallo Stato a norma del diritto nazionale o che i controlli non siano effettuati direttamente dallo Stato membro.
7. Il personale dell'organismo è legato dal segreto professionale per tutto quanto viene a sapere nell'esercizio delle sue funzioni (tranne che nei confronti delle autorità amministrative competenti dello Stato in cui esso esercita la propria attività) nell'ambito della presente direttiva o di qualsiasi disposizione di diritto interno concernente la sua applicazione.
ALLEGATO 9
MARCATURA CE DI CONFORMITÀ
— La marcatura CE di conformità è costituita dalle iniziali «CE» secondo il simbolo grafico che segue:
—
— in caso di riduzione o di ingrandimento della marcatura CE, devono essere rispettate le proporzioni indicate per il simbolo graduato di cui sopra;
— i diversi elementi della marcatura CE devono avere sostanzialmente la stessa dimensione verticale che non può essere inferiore a 5 mm.
— Per i dispositivi di piccole dimensioni si può derogare a detta dimensione minima.
( 1 ) GU L 311 del 28.11.2001, pag. 67. Direttiva modificata da ultimo dal regolamento (CE) n. 1901/2006 (GU L 378 del 27.12.2006, pag. 1).
( 2 ) Direttiva 2004/108/CE del Parlamento europeo e del Consiglio, del 15 dicembre 2004, concernente il ravvicinamento delle legislazioni degli Stati membri relative alla compatibilità elettromagnetica (GU L 390 del 31.12.2004, pag. 24).
( 3 ) GU L 157 del 9.6.2006, pag. 24.
( 4 ) Direttiva 98/34/CE del Parlamento europeo e del Consiglio, del 22 giugno 1998, che prevede una procedura d’informazione nel settore delle norme e delle regolamentazioni tecniche e delle regole relative ai servizi della società dell’informazione (GU L 204 del 21.7.1998, pag. 37). Direttiva modificata da ultimo dall’atto di adesione del 2003.
( 5 ) GU L 159 del 29.6.1996, pag. 1.
( 6 ) GU L 180 del 9.7.1997, pag. 22.
( 7 ) Regolamento (CE) n. 726/2004 del Parlamento europeo e del Consiglio, del 31 marzo 2004, che istituisce procedure comunitarie per l'autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario, e che istituisce l'agenzia europea per i medicinali (GU L 136 del 30.4.2004, pag. 1). Regolamento modificato da ultimo dal regolamento (CE) n. 1901/2006.
( 8 ) Per «rischi di mutue interferenze» si intendono le influenze negative sul dispositivo provocate da strumenti presenti al momento delle indagini o dei trattamenti e viceversa.