

EUR-Lex Access to European Union law
This document is an excerpt from the EUR-Lex website
Document 32015R1833
Commission Implementing Regulation (EU) 2015/1833 of 12 October 2015 amending Regulation (EEC) No 2568/91 on the characteristics of olive oil and olive-residue oil and on the relevant methods of analysis
Regolamento di esecuzione (UE) 2015/1833 della Commissione, del 12 ottobre 2015, che modifica il regolamento (CEE) n. 2568/91 relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi a essi attinenti
Regolamento di esecuzione (UE) 2015/1833 della Commissione, del 12 ottobre 2015, che modifica il regolamento (CEE) n. 2568/91 relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi a essi attinenti
OJ L 266, 13.10.2015, p. 29–52
(BG, ES, CS, DA, DE, ET, EL, EN, FR, HR, IT, LV, LT, HU, MT, NL, PL, PT, RO, SK, SL, FI, SV)
 No longer in force, Date of end of validity: 23/11/2022; abrog. impl. da 32022R2104
No longer in force, Date of end of validity: 23/11/2022; abrog. impl. da 32022R2104
|
13.10.2015 |
IT |
Gazzetta ufficiale dell'Unione europea |
L 266/29 |
REGOLAMENTO DI ESECUZIONE (UE) 2015/1833 DELLA COMMISSIONE
del 12 ottobre 2015
che modifica il regolamento (CEE) n. 2568/91 relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi a essi attinenti
LA COMMISSIONE EUROPEA,
visto il trattato sul funzionamento dell'Unione europea,
visto il regolamento (UE) n. 1308/2013 del Parlamento europeo e del Consiglio, del 17 dicembre 2013, recante organizzazione comune dei mercati dei prodotti agricoli e che abroga i regolamenti (CEE) n. 922/72, (CEE) n. 234/79, (CE) n. 1037/2001 e (CE) n. 1234/2007 del Consiglio (1), in particolare l'articolo 91, primo comma, lettera d), e secondo comma,
considerando quanto segue:
|
(1) |
Il regolamento (CEE) n. 2568/91 della Commissione (2) definisce le caratteristiche fisico-chimiche e organolettiche degli oli di oliva e degli oli di sansa di oliva e stabilisce i metodi di valutazione di tali caratteristiche. Detti metodi sono regolarmente aggiornati in base al parere degli esperti di chimica e conformemente all'operato svolto nell'ambito del Consiglio oleicolo internazionale (COI). |
|
(2) |
Per garantire l'applicazione a livello dell'Unione delle più recenti norme internazionali stabilite dal COI, è opportuno aggiornare taluni metodi di analisi stabiliti nel regolamento (CEE) n. 2568/91. |
|
(3) |
Alla luce dell'esperienza acquisita risulta che il metodo per rilevare la presenza di oli vegetali estranei negli oli di oliva può produrre falsi positivi. È pertanto opportuno eliminare i riferimenti a tale metodo. |
|
(4) |
È opportuno modificare di conseguenza il regolamento (CEE) n. 2568/91. |
|
(5) |
Le misure di cui al presente regolamento sono conformi al parere del comitato per l'organizzazione comune dei mercati agricoli, |
HA ADOTTATO IL PRESENTE REGOLAMENTO:
Articolo 1
Il regolamento (CEE) n. 2568/91 è così modificato:
|
1) |
all'articolo 2, il paragrafo 1 è così modificato:
|
|
2) |
il sommario degli allegati è così modificato:
|
|
3) |
nell'allegato I ter, l'appendice 1 è modificata in conformità dell'allegato I del presente regolamento; |
|
4) |
l'allegato V è modificato in conformità dell'allegato II del presente regolamento; |
|
5) |
l'allegato IX è sostituito dal testo che figura nell'allegato III del presente regolamento; |
|
6) |
gli allegati X A e X B sono sostituiti dal testo figurante nell'allegato IV del presente regolamento; |
|
7) |
l'allegato XII è modificato in conformità dell'allegato V del presente regolamento; |
|
8) |
l'allegato XIX è modificato in conformità dell'allegato VI del presente regolamento; |
|
9) |
l'allegato XX bis è soppresso. |
Articolo 2
Il presente regolamento entra in vigore il terzo giorno successivo alla pubblicazione nella Gazzetta ufficiale dell'Unione europea.
Il presente regolamento è obbligatorio in tutti i suoi elementi e direttamente applicabile in ciascuno degli Stati membri.
Fatto a Bruxelles, il 12 ottobre 2015
Per la Commissione
Il presidente
Jean-Claude JUNCKER
(1) GU L 347 del 20.12.2013, pag. 671.
(2) Regolamento (CEE) n. 2568/91 della Commissione, dell'11 luglio 1991, relativo alle caratteristiche degli oli d'oliva e degli oli di sansa d'oliva nonché ai metodi ad essi attinenti (GU L 248 del 5.9.1991, pag. 1).
ALLEGATO I
Nell'allegato I ter dell'appendice 1 del regolamento (CEE) n. 2568/91, la tavola di corrispondenza è così modificata:
|
1) |
le righe corrispondenti agli isomeri trans degli acidi grassi e alla composizione di acidi grassi sono sostituite dalle seguenti:
|
|
2) |
la riga corrispondente agli alcoli alifatici è sostituita dalla seguente:
|
ALLEGATO II
Nell'allegato V del regolamento (CEE) n. 2568/91, il punto 6.2 è sostituito dal seguente:
|
«6.2. |
Si calcola il contenuto percentuale di ogni singolo sterolo dal rapporto fra l'area del picco corrispondente e la somma delle aree dei picchi degli steroli:
in cui:
|
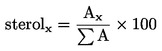
ALLEGATO III
«ALLEGATO IX
ANALISI SPETTROFOTOMETRICA NELL'ULTRAVIOLETTO
PREMESSA
L'analisi spettrofotometrica nell'ultravioletto può fornire informazioni sulla qualità di una sostanza grassa, sul suo stato di conservazione e sulle modificazioni indotte da processi tecnologici. L'assorbimento alle lunghezze d'onda specificate nel metodo è dovuto alla presenza di sistemi dienici e trienici coniugati derivanti da processi di ossidazione e/o da pratiche di raffinazione. I valori di tale assorbimento sono espressi come estinzione specifica (estinzione di una soluzione della sostanza grassa all'1 % (p/v) nel solvente prescritto, in una cuvetta di 10 mm) convenzionalmente indicata con K (detto anche “coefficiente di estinzione”).
(estinzione di una soluzione della sostanza grassa all'1 % (p/v) nel solvente prescritto, in una cuvetta di 10 mm) convenzionalmente indicata con K (detto anche “coefficiente di estinzione”).
1. OGGETTO
Il presente allegato descrive il procedimento per l'esecuzione dell'analisi spettrofotometrica nell'ultravioletto dell'olio d'oliva.
2. PRINCIPIO DEL METODO
Un campione viene disciolto nel solvente stabilito e l'assorbanza della soluzione è misurata alle lunghezze d'onda prescritte, in riferimento al solvente puro.
Si determinano i valori dell'estinzione specifica alle lunghezze d'onda di 232 e 268 nm nell'isoottano o di 232 e 270 nm nel cicloesano, per una concentrazione dell'1 % p/v in una cuvetta di 10 mm.
3. APPARECCHIATURA
3.1. Spettrofotometro per misurazioni a lunghezze d'onda dell'ultravioletto (da 220 a 360 nm), con possibilità di lettura per ogni unità nanometrica. Si raccomanda l'esecuzione di controlli regolari in relazione all'accuratezza e alla riproducibilità delle scale di lunghezza d'onda e di assorbanza, nonché alla luce parassita.
3.1.1. Scala delle lunghezza d'onda: questo controllo può essere effettuato mediante materiale di riferimento costituito da un filtro di vetro ottico contenente ossido di olmio o una soluzione di ossido di olmio (sigillata o no), che presenta bande di assorbanza separate. I materiali di riferimento sono destinati alla verifica e alla taratura della scala delle lunghezze d'onda di spettrofotometri UV-visibile con ampiezza di banda spettrale nominale uguale o inferiore a 5 nm. Le misurazioni sono effettuate rispetto a una prova in bianco all'aria su un intervallo di lunghezze d'onda compreso tra 640 e 240 nm, secondo le istruzioni accluse ai materiali di riferimento. Ad ogni alterazione della larghezza della fessura (slit) si esegue una correzione della linea di base con un percorso ottico libero. Le lunghezze d'onda della norma sono elencate nel certificato del materiale di riferimento.
3.1.2. Scala di assorbanza: tale controllo può essere eseguito utilizzando materiali di riferimento sigillati reperibili in commercio, costituiti da soluzioni acide di dicromato di potassio a determinate concentrazioni e con valori certificati di assorbanza a λmax (quattro soluzioni di dicromato di potassio in acido perclorico, sigillate in quattro cuvette UV in quarzo utilizzate per misurare la linearità e l'accuratezza fotometrica di riferimento nell'ultravioletto). Le soluzioni di dicromato di potassio sono misurate rispetto a una prova in bianco dell'acido utilizzato, previa correzione della linea di base, conformemente alle istruzioni accluse al materiale di riferimento. I valori di assorbanza sono elencati nel certificato del materiale di riferimento.
Per verificare la risposta della fotocellula e del fotomoltiplicatore è inoltre possibile procedere come segue: pesare 0,2000 g di potassio cromato puro per spettrofotometria e scioglierlo in una soluzione di idrossido di potassio 0,05 N in un matraccio tarato da 1 000 ml e portare a volume. Prelevare esattamente 25 ml della soluzione ottenuta, trasferirla in un matraccio tarato da 500 ml e portare a volume con la stessa soluzione di idrossido di potassio.
Misurare l'estinzione della soluzione così ottenuta a 275 nm, utilizzando come riferimento la soluzione di idrossido di potassio. L'estinzione misurata con cuvetta da 1 cm deve essere di 0,200 ± 0,005.
3.2. Cuvette in quarzo rettangolari, con coperchio, adatte per misurazioni a lunghezze d'onda dell'ultravioletto (da 220 a 360 nm), con un percorso ottico di 10 mm. Riempite di acqua o con altro solvente idoneo, le cuvette non devono presentare fra loro differenze superiori a 0,01 unità di estinzione.
3.3. Matracci volumetrici tarati da 25 ml, classe A.
3.4. Bilancia analitica, idonea a fornire una lettura con l'approssimazione di 0,0001 g.
4. REAGENTI
Salvo indicazione contraria, durante l'analisi utilizzare soltanto reagenti di qualità analitica riconosciuta e acqua distillata o demineralizzata o acqua di purezza equivalente.
Solvente: Isoottano (2,2,4 trimetilpentano) per misurazioni a 232 nm e 268 nm o cicloesano per misurazioni a 232 nm e 270 nm, con assorbanza inferiore a 0,12 a 232 nm e a 0,05 a 270 nm rispetto all'acqua distillata, misurata in una cuvetta di 10 mm.
5. PROCEDIMENTO
5.1. Il campione deve essere perfettamente omogeneo ed esente da impurezze sospese. In caso contrario deve essere filtrato su carta alla temperatura di circa 30 °C.
5.2. Del campione così preparato si versano circa 0,25 g (con un'approssimazione di 1 mg) in un matraccio tarato da 25 ml, si porta a volume con il solvente prescritto e si omogeneizza. La soluzione risultante deve essere perfettamente limpida. Qualora si riscontri opalescenza o torbidità si filtra rapidamente con carta.
NOTA: in generale, è sufficiente una massa di 0,25-0,30 g per misurare l'assorbenza di oli di oliva vergini ed extra vergini a 268 nm e 270 nm. Per misurazioni a 232 nm occorrono di norma 0,05 g di campione; in tal caso si preparano generalmente due soluzioni distinte. Per misurare l'assorbanza di oli di sansa d'oliva, oli d'oliva raffinati e oli d'oliva adulterati, che presentano un'assorbanza più elevata, è generalmente sufficiente un campione di quantità inferiore (ad esempio 0,1 g).
5.3. Se necessario, si corregge la linea di base (220-290 nm) con solvente in entrambe le cuvette di quarzo (campione e riferimento), quindi si riempie con la soluzione di prova la cuvetta di quarzo del campione e si misurano le estinzioni a 232, 268 o 270 nm rispetto al solvente utilizzato come riferimento.
I valori di estinzione registrati devono essere compresi tra 0,1 e 0,8 o nell'intervallo di linearità dello spettrofotometro (che deve essere verificato). In caso contrario è necessario ripetere le misurazioni utilizzando soluzioni più concentrate o più diluite, a seconda del caso.
5.4. Dopo aver misurato l'assorbanza a 268 o 270 nm, si misura l'assorbanza a λmax, λmax + 4 e λmax – 4. Questi valori di assorbanza sono utilizzati per determinare la variazione dell'estinzione specifica (ΔΚ).
NOTA: il valore di λmax è fissato a 268 nm per l'isoottano utilizzato come solvente e a 270 nm per il cicloesano.
6. ESPRESSIONE DEI RISULTATI
6.1. Si riportano le estinzioni specifiche (coefficienti di estinzione) alle varie lunghezze d'onda calcolate come segue:

in cui:
|
Kλ |
= |
estinzione specifica alla lunghezza d'onda λ; |
|
Eλ |
= |
estinzione misurata alla lunghezza d'onda λ; |
|
c |
= |
concentrazione della soluzione in g/100 ml; |
|
s |
= |
spessore della cuvetta di quarzo in cm; |
il valore è espresso con due cifre decimali.
6.2. Variazione dell'estinzione specifica (ΔΚ)
La variazione del valore assoluto dell'estinzione (ΔΚ) è data da:
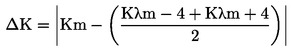
in cui Km è l'estinzione specifica alla lunghezza d'onda di massimo assorbimento a 270 nm e 268 nm, a seconda del solvente utilizzato.
Il valore è espresso con due cifre decimali.»
ALLEGATO IV
ALLEGATO X
DETERMINAZIONE DEGLI ESTERI METILICI DEGLI ACIDI GRASSI MEDIANTE GASCROMATOGRAFIA
1. OGGETTO
Il presente allegato fornisce orientamenti per la determinazione mediante gascromatografia degli acidi grassi liberi e legati nei grassi e negli oli vegetali dopo la loro conversione in esteri metilici di acidi grassi (FAME).
Gli acidi grassi legati dei triacilgliceroli (TAG) e, a seconda del metodo di esterificazione, gli acidi grassi liberi (FFA), sono convertiti in esteri metilici di acidi grassi (FAME), determinati mediante gascromatografia con colonna capillare.
Il metodo descritto nel presente allegato consente di determinare gli esteri metilici degli acidi grassi da C12 a C24, compresi quelli saturi, monoinsaturi cis e trans e polinsaturi cis e trans.
2. PRINCIPIO
La gascromatografia (GC) è utilizzata per l'analisi quantitativa dei FAME. I FAME sono preparati conformemente alla parte A. Essi sono successivamente iniettati e vaporizzati nell'iniettore. La separazione dei FAME viene eseguita su colonne analitiche di polarità e lunghezza specifiche. Un rivelatore a ionizzazione di fiamma (FID) è utilizzato per il rilevamento dei FAME. Le condizioni di analisi figurano nella parte B.
L'idrogeno o l'elio possono essere utilizzati come gas vettore (fase mobile) nella gascromatografia dei FAME con FID. L'idrogeno accelera la separazione e produce picchi più marcati. La fase stazionaria consiste in un microscopico strato di una sottile pellicola liquida su una superficie solida inerte di silice fusa.
Mentre passano attraverso la colonna capillare i composti volatili in esame interagiscono con la fase stazionaria che riveste la superficie interna della colonna. A causa di tale differenza di interazione dei diversi composti, la loro eluizione avviene in un momento diverso, detto “tempo di ritenzione” del composto per un determinato insieme di parametri analitici. La comparazione dei tempi di ritenzione serve a identificare i diversi composti.
PARTE A
PREPARAZIONE DEGLI ESTERI METILICI DI ACIDI GRASSI DA OLIO DI OLIVA E DA OLIO DI SANSA DI OLIVA
1. OGGETTO
La presente parte illustra la preparazione degli esteri metilici degli acidi grassi e include metodi di preparazione degli esteri metilici di acidi grassi da oli di oliva e di oli di sansa di oliva.
2. CAMPO DI APPLICAZIONE
La preparazione degli esteri metilici di acidi grassi da oli di oliva e oli di sansa di oliva avviene mediante transesterificazione con soluzione metanolica di idrossido di potassio a temperatura ambiente. La necessità di depurare il campione prima della transesterificazione dipende dal tenore di acidi grassi liberi del campione e dal parametro analitico da determinare; il metodo può essere scelto secondo la seguente tabella:
|
Categoria di olio |
Metodo |
||||||
|
Olio di oliva vergine di acidità ≤ 2,0 %, |
|
||||||
|
Olio di oliva raffinato |
|||||||
|
Olio di oliva composto da oli d'oliva raffinati e da oli d'oliva vergini |
|||||||
|
Olio di sansa di oliva raffinato |
|||||||
|
Olio di sansa di oliva |
|||||||
|
Olio di oliva vergine con acidità > 2,0 % Olio di sansa di oliva greggio |
|
3. METODOLOGIA
3.1. Transesterificazione in soluzione metanolica di idrossido di potassio a temperatura ambiente
3.1.1. Principio
Gli esteri metilici si formano per transesterificazione in una soluzione metanolica di idrossido di potassio come fase intermedia prima della saponificazione.
3.1.2. Reagenti
3.1.2.1. Metanolo con tenore di acqua non superiore allo 0,5 % (m/m).
3.1.2.2. Esano per cromatografia.
3.1.2.3. Eptano per cromatografia.
3.1.2.4. Etere dietilico, stabilizzato per analisi.
3.1.2.5. Acetone per cromatografia.
3.1.2.6. Solvente di eluizione per la purificazione dell'olio mediante cromatografia su colonna/estrazione su fase solida (SPE); miscela di esano/etere dietilico in proporzioni 87:13(v/v).
3.1.2.7. Idrossido di potassio, soluzione metanolica di circa 2M: sciogliere 11,2 g di idrossido di potassio in 100 ml di metanolo.
3.1.2.8. Cartucce di gel di silice, 1 g (6 ml), per l'estrazione in fase solida (SPE).
3.1.3. Apparecchiatura
3.1.3.1. Provette con tappo a vite (volume 5 ml) munito di guarnizione in PTFE.
3.1.3.2. Pipette graduate o automatiche da 2 ml e 0,2 ml.
3.1.4. Purificazione dei campioni di olio
All'occorrenza i campioni verranno purificati facendo passare l'olio su una cartuccia di gel di silice per estrazione in fase solida. Inserire una cartuccia di gel di silice (3.1.2.8) in un apparecchio di eluizione sotto vuoto e lavare con 6 ml di esano (3.1.2.2); effettuare il lavaggio senza vuoto. Quindi immettere nella colonna una soluzione d'olio (0,12 g circa) in 0,5 ml di esano (3.1.2.2). Far scendere la soluzione per eluizione con 10 ml di esano/etere dietilico (87:13 v/v) (3.1.2.6). Omogeneizzare tutti gli eluati e dividerli in due volumi simili. Fare evaporare un'aliquota fino ad essiccamento in un evaporatore rotante, a pressione ridotta e temperatura ambiente. Dissolvere il residuo in 1 ml di eptano: si ottiene una soluzione pronta per l'analisi degli acidi grassi mediante GC. Far evaporare la seconda aliquota e dissolvere il residuo in 1 ml di acetone per l'analisi dei trigliceridi mediante HPLC, se necessario.
3.1.5. Procedura
Pesare circa 0,1 g del campione di olio in una provetta da 5 ml con tappo a vite (3.1.3.1). Aggiungere 2 ml di eptano e mescolare (3.1.2.2). Aggiungere 0,2 ml di soluzione metanolica di idrossido di potassio (3.1.2.7), chiudere con il tappo munito di guarnizione in PTFE, stringere bene il tappo e agitare energicamente per 30 secondi. Lasciare depositare finché la parte superiore della soluzione diventa chiara. Far decantare lo strato superiore che contiene gli esteri metilici. La soluzione di eptano ottenuta è pronta per essere iniettata nel gascromatografo. Si consiglia di conservare la soluzione in frigorifero fino al momento dell'analisi gascromatografica. Non si consiglia di conservare la soluzione per un periodo superiore alle 12 ore.
PARTE B
ANALISI DEGLI ESTERI METILICI DEGLI ACIDI GRASSI MEDIANTE GASCROMATOGRAFIA
1. OGGETTO
La presente parte fornisce un orientamento generale per l'applicazione della gascromatografia con colonne capillari ai fini della determinazione della composizione qualitativa e quantitativa di una miscela di esteri metilici degli acidi grassi ottenuta con il metodo specificato nella parte A.
La presente parte non è applicabile agli acidi grassi polimerizzati.
2. REAGENTI
2.1. Gas vettore
Gas inerte (elio o idrogeno) completamente essiccato ed avente un tenore di ossigeno inferiore a 10 mg/kg.
|
Nota 1: |
L'idrogeno può raddoppiare la velocità dell'analisi, ma è pericoloso. Sono disponibili dispositivi di sicurezza. |
2.2. Gas ausiliari
2.2.1. Idrogeno (purezza ≥ 99,9 %), esente da impurezze organiche.
2.2.2. Aria od ossigeno, esente da impurezze organiche.
2.2.3. Azoto (purezza > 99 %).
2.3. Standard di riferimento
Miscela di esteri metilici di acidi grassi puri oppure esteri metilici di un grasso di composizione nota, di preferenza analogo a quello della sostanza grassa da analizzare. Gli isomeri cis e trans di esteri metilici ottadecadienoici, ottadecenoici e ottadecatrienoici sono utili per l'identificazione degli isomeri trans degli acidi insaturi.
Si deve prestare attenzione a prevenire l'ossidazione degli acidi grassi polinsaturi.
3. APPARECCHIATURA
Le istruzioni precisano che deve essere usata la consueta apparecchiatura per gascromatografia, facendo uso di colonne capillari e di un rivelatore a ionizzazione di fiamma.
3.1. Gascromatografo
Il gascromatografo deve comprendere i seguenti elementi.
3.1.1. Sistema d'iniezione
Utilizzare un sistema d'iniezione con colonne capillari, nel qual caso il sistema di iniezione deve essere appositamente progettato per l'utilizzo delle colonne. L'iniezione può essere del tipo split o splitless (iniezione on-column).
3.1.2. Forno
Il forno deve poter scaldare la colonna capillare ad almeno 260 °C e mantenere la temperatura desiderata con l'approssimazione di ± 0,1 °C. Quest'ultimo requisito è particolarmente importante se si usa una provetta in silice fusa.
L'utilizzo di un apparecchio dotato di un programmatore di temperatura è raccomandato in tutti i casi e in particolare per gli acidi grassi con meno di 16 atomi di carbonio.
3.1.3. Colonna capillare
3.1.3.1. Tubo costituito di materiale inerte rispetto alle sostanze da analizzare (di solito vetro o silice fusa). Il diametro interno deve essere compreso tra 0,20 mm e 0,32 mm. La superficie interna deve esser sottoposta a un opportuno trattamento (ad es. preparazione della superficie, inattivazione) prima di ricevere la pellicola della fase stazionaria. Una lunghezza di 60 m è sufficiente per gli acidi grassi e per gli isomeri cis e trans degli acidi grassi.
3.1.3.2. Fase stazionaria, polisilossano polare (cianopropilsilicone). Le colonne a fase legata (a legami reticolari) sono adeguate.
|
Nota 2: |
Vi è il rischio che i polisilossani polari creino difficoltà nell'identificazione e separazione dell'acido linolenico e degli acidi a C20. |
Lo spessore delle pellicole deve essere sottile, ad esempio 0,1 μm-0,2 μm.
3.1.3.3. Montaggio e condizionamento della colonna
Osservare le normali precauzioni necessarie per il montaggio delle colonne capillari, ovvero sistemazione della colonna nel forno (supporto), scelta e collegamento dei giunti (a tenuta stagna), sistemazione delle estremità della colonna nell'iniettore e nel rivelatore (riduzione degli spazi morti). Far fluire attraverso la colonna un flusso di gas vettore [ad es. 0,3 bar (30 kPa) per una colonna di 25 m di lunghezza e di 0,3 mm di diametro interno].
Condizionare la colonna programmando la temperatura del forno a 3 °C/min dalla temperatura ambiente a una temperatura di 10 °C inferiore al limite di decomposizione della fase stazionaria. Mantenere il forno a questa temperatura per 1 h fino a stabilizzazione della linea di base. Riportarla a 180 °C in modo da lavorare in condizioni isotermiche.
|
Nota 3: |
Sono disponibili in commercio adeguate colonne precondizionate. |
3.1.4. Rivelatore a ionizzazione di fiamma e convertitore-amplificatore
3.2. Siringa
La siringa deve avere una capacità massima di 10 μl ed essere graduata in divisioni di 0,1 μl.
3.3. Sistema di acquisizione dei dati
Sistema di acquisizione dei dati collegato online con i rilevatori e utilizzato con un software adeguato per l'integrazione e la normalizzazione dei picchi.
4. PROCEDURA
Le operazioni descritte dal paragrafo 4.1 al paragrafo 4.3 riguardano l'utilizzo di un rivelatore a ionizzazione di fiamma.
4.1. Condizioni di prova
4.1.1. Selezione delle condizioni operative ottimali per colonne capillari
In considerazione delle caratteristiche di efficienza e di permeabilità delle colonne capillari, la separazione dei costituenti e la durata dell'analisi sono ampiamente dipendenti dal flusso del gas vettore nella colonna. Sarà pertanto necessario ottimizzare le condizioni operative adeguando questo parametro (o semplicemente la perdita di carico in testa alla colonna), a seconda che si voglia migliorare la separazione o accelerare l'analisi.
Le seguenti condizioni si sono rivelate adatte per la separazione dei FAME (da C4 a C26). Esempi di cromatogrammi figurano nell'appendice B:
|
Temperatura dell'iniettore: |
250 °C |
|
Temperatura del rivelatore: |
250 °C |
|
Temperatura del forno: |
da 165 °C (8 min) a 210 °C a 2 °C/min |
|
Gas vettore idrogeno: pressione in testa alla colonna: |
179 kPa |
|
Flusso totale: |
154,0 ml/min; |
|
Rapporto di partizione (split ratio): |
1:100 |
|
Volume di iniezione: |
1 μl |
4.1.2. Determinazione della risoluzione (cfr. appendice A)
Calcolare la risoluzione, R, di due picchi vicini I e II, utilizzando la seguente formula:
R = 2 × ((dr(II) – d r(I))/(ω(I) + ω(II))) or R = 2 × ((tr(II) – t r(I))/(ω(I) + ω(II))) (USP) (United States Pharmacopeia),
Oppure
R = 1,18 × ((tr(II) – tr(I))/(ω0,5(I) + ω0,5(II))) (EP, BP, JP, DAB), (JP (Japanese Pharmacopeia), EP (Pharmacopée Européenne), (BP (British Pharmacopeia))
in cui:
|
|
d r(I) è la distanza di ritenzione del picco I; |
|
|
d r(II) è la distanza di ritenzione del picco II; |
|
|
t r(I) è il tempo di ritenzione del picco I; |
|
|
t r(II) è il tempo di ritenzione del picco II; |
|
|
ω(I) è la larghezza della base del picco I; |
|
|
ω(II) è la larghezza della base del picco II; |
|
|
ω0,5 è la larghezza del picco dello specifico composto, a metà altezza del picco; |
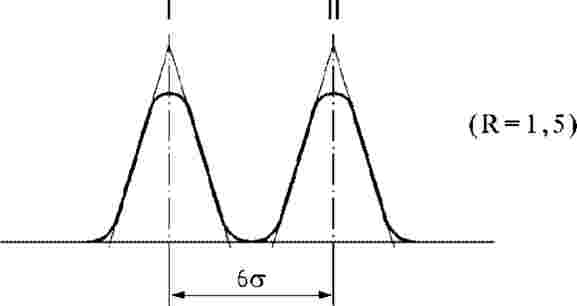
Se ω(I) ≈ ω(II), calcolare R applicando le seguenti formule:
R = (dr(II) – d r(I))/ω = (dr(II) – d r(I))/4σ
in cui:
σ è la deviazione standard (cfr. appendice A, figura 1).
Se la distanza dr tra due picchi d r(II) – d r(I) è uguale a 4σ, il fattore di risoluzione R = 1.
Se due picchi non sono completamente separati, le tangenti ai punti di flesso dei due picchi si intersecano al punto C. Per separare completamente i due picchi, la distanza tra i due picchi deve essere pari a:
d r(II) – d r(I) = 6 σ da cui R = 1,5 (cfr. appendice A, figura 3).
5. ESPRESSIONE DEI RISULTATI
5.1. Analisi qualitativa
Identificare i picchi dell'estere metilico del campione dal cromatogramma nell'appendice B, figura 1, se necessario per interpolazione, o dal raffronto con quelli delle miscele degli esteri metilici di riferimento (come indicato al punto 2.3).
5.2. Analisi quantitativa
5.2.1. Determinazione della composizione
Calcolare la frazione di massa wi dei singoli esteri metilici di acidi grassi, espressa come percentuale in massa degli esteri metilici, come segue:
5.2.2. Metodo di calcolo
5.2.2.1. Caso generale
Calcolare il contenuto di un dato componente i, espresso come percentuale in massa degli esteri metilici, determinando la percentuale rappresentata dal rapporto tra l'area del picco corrispondente e la somma delle aree di tutti i picchi, usando la formula seguente:
wi = (Ai/ΣA) × 100
in cui:
|
|
Ai è l'area del picco del singolo estere metilico di acidi grassi i; |
|
|
ΣA è la somma delle aree dei picchi di tutti i singoli esteri metilici degli acidi grassi. |
I risultati sono espressi con due cifre decimali.
|
Nota 4: |
Per oli e grassi, la frazione in massa degli esteri metilici di acidi grassi è pari alla frazione di massa dei triacilgliceroli in grammi per 100 g. Per i casi in cui tale ipotesi non è ammessa, cfr. 5.2.2.2. |
5.2.2.2. Uso di fattori correttivi
In taluni casi, ad esempio in presenza di acidi grassi con meno di otto atomi di carbonio oppure di acidi con gruppi secondari, le aree devono essere corrette con specifici fattori di correzione (Fci). Questi fattori sono determinati per ogni singolo strumento. A tal fine vanno utilizzati materiali di riferimento appropriati con una composizione in acidi grassi certificata nel corrispondente intervallo.
|
Nota 5: |
Questi fattori correttivi non sono identici ai fattori di correzione teorici FID, riportati nell'appendice A, poiché comprendono anche le prestazioni del sistema di iniezione ecc. Tuttavia, in caso di differenze maggiori, l'intero sistema dovrebbe essere controllato per verificarne le prestazioni. |
Per questa miscela di riferimento, la percentuale in massa del FAME i è data dalla formula:
wi = (mi /Σm) × 100
in cui:
|
|
m i è la massa del FAME i nella miscela di riferimento; |
|
|
Σm è il totale delle masse dei vari componenti come i FAME della miscela di riferimento. |
Dal cromatogramma della miscela di riferimento calcolare la percentuale in area del FAME i come segue:
wi = (Ai/ΣA) × 100
in cui:
|
|
Ai è l'area del FAME i nella miscela di riferimento; |
|
|
ΣA è la somma delle aree di tutti i FAME della miscela di riferimento. |
Il fattore di correzione Fc è quindi
Fc = (mi × ΣA)/(Ai/Σm)
Per il campione, la percentuale in massa di ciascun FAME i è:
wi = (Fi × Ai)/Σ (Fi × Ai)
I risultati sono espressi con due cifre decimali.
|
Nota 6: |
Il valore calcolato corrisponde alla percentuale in massa dei singoli acidi grassi calcolata come triacilgliceroli per 100 g di grassi. |
5.2.2.3. Uso di uno standard interno
In alcune analisi (ad es. quando non tutti gli acidi grassi sono quantificati e quando sono presenti acidi con 4 e 6 atomi di carbonio accanto ad acidi con 16 e 18 atomi di carbonio, oppure quando è necessario determinare il quantitativo assoluto di un acido grasso in un campione) è necessario ricorrere a uno standard interno. Vengono spesso usati acidi grassi con 5, 15 o 17 atomi di carbonio. Deve essere determinato l'eventuale fattore di correzione per lo standard interno.
La percentuale in massa del componente i, espressa in esteri metilici, è data dalla formula:
wi = (mIS × Fi × Ai)/(m × FIS × AIS)
in cui:
|
|
A i è l'area del FAME i; |
|
|
A IS è l'area dello standard interno; |
|
|
F i è il fattore di correzione dell'acido grasso i, espressa come FAME; |
|
|
F IS è il fattore di correzione dello standard interno; |
|
|
m è la massa in milligrammi della quantità di sostanza prelevata per l'analisi, |
|
|
m IS è la massa in milligrammi dello standard interno. |
I risultati sono espressi con due cifre decimali.
6. RELAZIONE SULLA PROVA
La relazione sulla prova deve specificare i metodi usati per la preparazione degli esteri metilici e per l'analisi gascromatografica. Essa deve citare altresì tutti i dettagli operativi non specificati nel presente metodo standard oppure considerati facoltativi, nonché i particolari di qualsiasi evento che possa avere influenzato i risultati.
La relazione sulla prova deve comprendere tutti i dati necessari per la completa identificazione del campione.
7. PRECISIONE DEL METODO
7.1. Risultati del test interlaboratorio
Maggiori dettagli di un test interlaboratorio per verificare la precisione del metodo sono stabiliti nell'allegato C della norma COI/T.20/Doc. n. 33. I valori tratti da questa prova interlaboratorio potrebbero non essere applicabili a intervalli di concentrazioni e a matrici diverse da quelle date.
7.2. Ripetibilità
La differenza assoluta tra i risultati di due prove indipendenti, eseguite dallo stesso operatore entro un breve lasso di tempo, utilizzando la stessa attrezzatura e lo stesso metodo sul medesimo materiale di prova, nello stesso laboratorio, eccede in non più del 5 % dei casi il valore di ripetibilità (r) riportato nelle tabelle 1-14 dell'allegato C della norma COI/T.20/Doc. n. 33.
7.3. Riproducibilità
La differenza assoluta tra i risultati di due prove, eseguite da diversi operatori utilizzando lo stesso metodo su materiale di prova identico in laboratori diversi con diverse attrezzature, eccede in non più del 5 % dei casi il valore di riproducibilità (R) riportato nelle tabelle 1-14 dell'allegato C della norma COI/T.20/Doc. n. 33.
Appendice A
Figura 1

Con larghezza di ω0,5 a metà altezza del triangolo (ABC) e larghezza b a metà altezza del triangolo (NPM).
|
Figura 2 |
Figura 3 |
|
|
|

Appendice B
Figura 1
Profilo gascromatografico di un olio di sansa di oliva, ottenuto con il metodo della metilazione a freddo
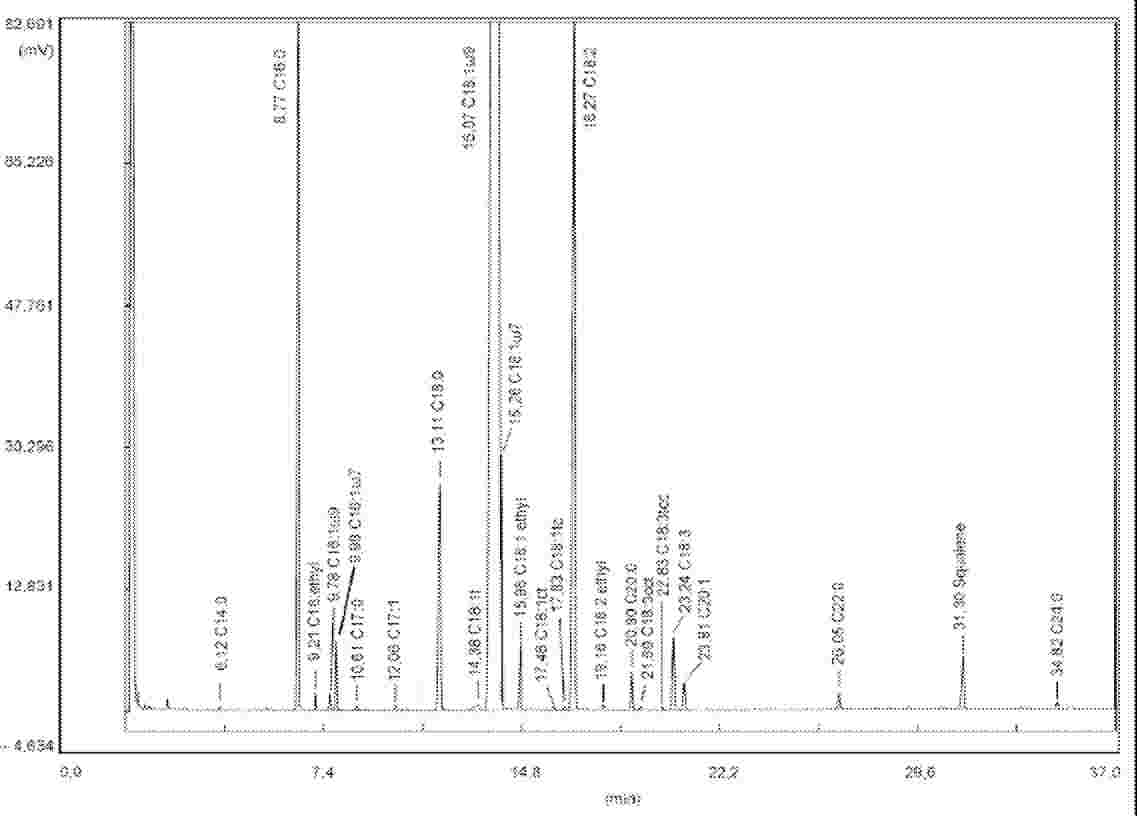
I picchi cromatografici corrispondono agli esteri metilici ed etilici, salvo altre indicazioni.
ALLEGATO V
L'allegato XII del regolamento (CEE) n. 2568/91 è così modificato:
|
1) |
il punto 1 è sostituito dal seguente testo: «1. OGGETTO E CAMPO D'APPLICAZIONE Il presente metodo internazionale stabilisce una procedura che consente di valutare le caratteristiche organolettiche degli oli di oliva vergini ai sensi dell'allegato VII, parte VIII, punto 1, del regolamento (UE) n. 1308/2013 del Parlamento europeo e del Consiglio (1) e di classificarli in base a tali caratteristiche. Il metodo contiene inoltre indicazioni per un'etichettatura facoltativa. Il metodo descritto è applicabile soltanto agli oli di oliva vergini e alla loro classificazione o etichettatura in funzione dell'intensità dei difetti percepiti e del flavor fruttato, determinata da un gruppo di assaggiatori selezionati, addestrati e controllati, costituito in panel. Le norme COI citate nel presente allegato si intendono nell'ultima versione disponibile. (1) Regolamento (UE) n. 1308/2013 del Parlamento europeo e del Consiglio, del 17 dicembre 2013, recante organizzazione comune dei mercati dei prodotti agricoli e che abroga i regolamenti (CEE) n. 922/72, (CEE) n. 234/79, (CE) n. 1037/2001 e (CE) n. 1234/2007 del Consiglio (GU L 347 del 20.12.2013, pag. 671).»" |
|
2) |
i punti 3.2, 3.3 e 3.4 sono sostituiti dai seguenti: «3.1.1. Altri attributi negativi
3.2. Attributi positivi
3.3. Terminologia facoltativa ai fini dell'etichettatura Su richiesta, il capo panel può certificare che gli oli valutati corrispondono alle definizioni e agli intervalli relativi agli aggettivi di seguito elencati, in funzione dell'intensità e della percezione degli attributi. Attributi positivi (fruttato, amaro e piccante): in funzione dell'intensità della percezione:
|
|
3) |
al punto 7, dopo il punto 7.1 è inserito il seguente punto: «7.1.1. Vicecapo panel Per motivi giustificati, il capo panel può essere sostituito nei suoi compiti concernenti la realizzazione delle prove da un vicecapo. Il vicecapo deve possedere tutte le competenze necessarie per la funzione di capo panel.» |
|
4) |
il punto 7.2 è sostituito dal seguente testo: «7.2. Assaggiatori Le persone che intervengono come assaggiatori nelle prove organolettiche di oli di oliva devono farlo a titolo volontario. Si raccomanda pertanto di richiedere ai candidati la presentazione di una domanda scritta. I candidati sono selezionati, addestrati ed esaminati dal capo panel in base alla capacità di distinguere campioni simili; occorre ricordare che la precisione dell'assaggiatore migliora con l'addestramento. L'assaggiatore deve comportarsi come un vero osservatore sensoriale e riferire esclusivamente le sensazioni percepite, senza tener conto dei gusti personali. Svolgerà il suo lavoro in silenzio, con animo disteso e senza fretta, prestando la massima attenzione al campione che sta analizzando. Per ciascuna prova occorrono da 8 a 12 assaggiatori. È bene prevedere alcuni assaggiatori di riserva, per supplire a eventuali assenze.» |
|
5) |
il punto 9.3 è sostituito dal seguente testo: «9.3. Uso dei dati da parte del capo panel Il capo panel raccoglie le schede di profilo compilate dagli assaggiatori e controlla le intensità assegnate ai diversi attributi; se constata un'anomalia, chiede all'assaggiatore di rivedere la sua scheda di profilo e, se necessario, di ripetere la prova. Il capo panel introduce i dati della valutazione di ogni componente del panel in un programma informatico come quello previsto dalla norma COI/T.20/Doc. n. 15 e procede a calcolare statisticamente i risultati dell'analisi, basandosi sul calcolo della mediana. V. punto 9.4 e Appendice del presente Allegato. L'inserimento dei dati per un campione va effettuato servendosi di una matrice composta di 9 colonne corrispondenti ai 9 attributi sensoriali e di 9 righe corrispondenti agli n componenti del panel impiegati. Quando un difetto percepito è riportato alla voce “altri” da almeno il 50 % del panel, il capo panel deve procedere al calcolo della mediana del difetto in questione e alla corrispondente classificazione. Il valore del coefficiente di variazione robusto che definisce la classificazione (difetto con l'intensità più alta e attributo fruttato) deve essere inferiore o pari al 20 %. Altrimenti, il capo panel deve ripetere la valutazione del campione in questione in una seduta di assaggio distinta. Se tale situazione si verifica frequentemente, si raccomanda al capo panel di fornire agli assaggiatori un ulteriore addestramento specifico (COI/T.20/Doc. n. 14, § 5) e di controllare le prestazioni dell'assaggiatore avvalendosi dell'indice di ripetibilità e di deviazione (COI/T.20/Doc. n. 14, § 6).» |
|
6) |
il punto 9.4 è sostituito dal seguente testo: «9.4. Classificazione dell'olio di oliva L'olio è classificato nelle categorie sotto riportate in funzione della mediana dei difetti e della mediana dell'attributo fruttato. Per mediana dei difetti si intende la mediana del difetto percepito con l'intensità più alta. La mediana dei difetti e la mediana del fruttato sono espresse con una sola cifra decimale. La classificazione dell'olio avviene confrontando il valore della mediana dei difetti e della mediana del fruttato con gli intervalli di riferimento indicati di seguito. Poiché i limiti di questi intervalli sono stati stabiliti tenendo conto del margine di errore del metodo, sono considerati assoluti. I programmi informatici consentono di visualizzare la classificazione su una tabella di dati statistici o un grafico.
Per le analisi eseguite al fini del controllo di conformità, si effettua una unica prova. Nel caso di analisi contraddittorie il capo panel deve provvedere a far realizzare l'analisi in doppio in sessioni distinte; la mediana degli attributi sarà calcolata sulla base della totalità dei dati della scheda di profilo per entrambe le prove.» |
|
7) |
la figura 1 è sostituita dalla seguente: «Figura 1 SCHEDA DI PROFILO DELL'OLIO DI OLIVA VERGINE
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
ALLEGATO VI
L'allegato XIX del regolamento (CEE) n. 2568/91 è così modificato:
|
1) |
il titolo è sostituito dal seguente: «DETERMINAZIONE DEL CONTENUTO DI ALCOLI ALIFATICI E TRITERPENICI MEDIANTE GASCROMATOGRAFIA CON COLONNA CAPILLARE» |
|
2) |
il punto 1 è sostituito dal seguente testo: «1. OGGETTO Il presente allegato descrive un procedimento per la determinazione del contenuto di alcoli alifatici e triterpenici negli oli e nelle sostanze grasse.» |
|
3) |
il punto 4.11 è sostituito dal seguente testo:
|
|
4) |
i punti 5.2.5 e 5.2.6 sono sostituiti dai seguenti:
|
|
5) |
il punto 5.4.4 è sostituito dal seguente testo: «5.4.4. Identificazione dei picchi. L'identificazione dei singoli picchi viene effettuata in base ai tempi di ritenzione e per paragone con miscele di TMSE degli alcoli, analizzate nelle medesime condizioni. Nelle figure 2 e 3 dell'appendice sono riportati esempi di cromatogramma della frazione alcolica di un olio di oliva raffinato.» |
|
6) |
l'appendice è sostituita dalla seguente: «Appendice Esempio di separazione cromatografica su strato sottile ed esempi di cromatogramma Figura 1 Cromatografia su strato sottile della frazione insaponificabile dall'olio di oliva eluito in esano/etere etilico (65/35) 
Figura 2 Cromatogramma della frazione alcolica di un olio di oliva raffinato 
Figura 3 Alcoli alifatici e triterpenici di un olio di oliva raffinato e un olio di oliva di seconda centrifugazione 
|