|
3.
|
A melléklet a következő fejezetekkel egészül ki:
„B.49
IN VITRO CELLULÁRIS MIKRONUKLEUSZ-VIZSGÁLAT EMLŐSÖKÖN
BEVEZETÉS
|
1.
|
Az in vitro mikronukleusz-vizsgálat (MNvit vizsgálat) interfázisban lévő sejtek citoplazmájában mikronukleuszok (MN) kimutatására szolgáló genotoxicitási vizsgálat. A mikronukleuszok acentrikus (vagyis centromérával nem rendelkező) kromoszómafragmentumokból származhatnak, vagy olyan ép kromoszómákból, amelyek a sejtosztódás anafázisa során nem képesek a sejtpólusokra vándorolni. A vizsgálattal klasztogén és aneugén kémiai anyagok (anyagok és keverékek) (1) (2) aktivitása mutatható ki osztódó sejteken, a vizsgált anyaggal történő expozíció során vagy azt követően. Ez a vizsgálati módszer egyaránt lehetővé teszi az aktin-polimerizációt gátló citokalazin-B-t (citoB) tartalmazó és nélkülöző protokollok használatát. A citoB célzott mitózist megelőző hozzáadása az egy mitotikus cikluson átesett sejtek esetében lehetővé teszi a mikronukleuszok gyakoriságának azonosítását és szelektív analízisét, mivel ezek a sejtek kétmagvúak (3) (4). Ez a vizsgálati módszer a citokinézis-gátlást nem alkalmazó protokollok használatát is lehetővé teszi, feltéve, hogy a vizsgált sejtpopuláció mitózisa bizonyítható.
|
|
2.
|
Amellett, hogy MNvit vizsgálat alkalmazható a mikronukleuszokat indukáló vegyi anyagok (anyagok vagy keverékek) azonosítására, a citokinézis-gátlás alkalmazása, a kinetochorok immunkémiai jelölése, illetve a centroméra/teloméra próbák hibridizációja (fluoreszcens in situ hibridizáció (FISH)) arról is információkat nyújthat, hogy milyen mechanizmussal történt a kromoszómasérülés és a mikronukleusz-képződés (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16). A jelölést és a hibridizációs technikákat akkor lehet használni, ha fokozódik a mikronukleusz-képződés, és a vizsgáló személy szeretné megállapítani, hogy ez a fokozódás klasztogén és/vagy aneugén hatásra jött-e létre.
|
|
3.
|
A mikronukleuszok olyan károsodást jeleznek, amely átkerült az utódsejtekbe, míg a metafázisban lévő sejtekben talált kromoszómaaberrációk nem feltétlenül öröklődnek. Mivel az interfázisos sejtben lévő mikronukleuszok viszonylag objektívan vizsgálhatók, a laboratóriumi személyzetnek csupán azt kell meghatároznia, hogy a sejtek átestek-e osztódáson, és hogy hány sejt tartalmaz mikronukleuszt. Ennek következtében a preparátumok viszonylag gyorsan értékelhetők, és a vizsgálat automatizálható. Ez a gyakorlatban kezelésenként több száz helyett több ezer sejt értékelését teszi lehetővé, növelve a vizsgálat erejét. Végül megállapítható, hogy mivel a mikronukleuszok lemaradó kromoszómákból keletkezhetnek, lehetséges olyan aneuploidiát indukáló anyagok azonosítása, amelyeket a hagyományos kromoszómaaberrációs vizsgálatokkal, például az OECD 473. vizsgálati iránymutatása (e melléklet B.10. fejezete) szerint nehéz vizsgálni (17). Az MNvit vizsgálat ugyanakkor speciális technikák, például a 2. bekezdésben leírt FISH alkalmazása nélkül nem teszi lehetővé a poliploidiát okozó és a klasztogén hatású vegyi anyagok elkülönítését.
|
|
4.
|
Az MNvit vizsgálat in vitro módszer, amelyet jellemzően humán és rágcsáló eredetű tenyésztett sejteken alkalmaznak. Mivel aneugén és klasztogén hatású anyagok egyaránt kimutathatóak, széleskörű alapot biztosít a kromoszómakárosító hatás in vitro vizsgálatához.
|
|
5.
|
Az MNvit vizsgálat számos különféle sejttípus esetében, valamint citoB jelenlétében és hiányában is megbízható és hatékony. Az MNvit vizsgálat hitelességét különböző rágcsálósejtvonalak (CHO, V79, CHL/IU és L5178Y) és humán limfociták alkalmazásával nyert nagy mennyiségű adat támasztja alá (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31). Ezek közé tartoznak különösen a Société Française de Toxicologie Génétique (SFTG) által koordinált nemzetközi hitelesítési tanulmányok (18) (19) (20) (21) (22) és a genotoxicitás vizsgálatával foglalkozó nemzetközi műhely (International Workshop on Genotoxicity Testing) jelentései (4) (16). A rendelkezésre álló adatokat az Európai Bizottság Alternatív Módszerek Validálásával Foglalkozó Európai Központja (ECVAM) is újraértékelte egy, a bizonyítékok súlyát mérlegelő retrospektív hitelesítési vizsgálatban, és a vizsgálati módszert az ECVAM tudományos tanácsadó bizottsága (ESAC) tudományosan hitelesített módszerként jóváhagyta (32) (33) (34). A humán TK6 limfoblasztoid sejtvonal (35), HepG2 sejtek (36) (37) és szíriai aranyhörcsög-embrió elsődleges sejtjeinek (38) felhasználását is leírták, bár ezeket hitelesítési vizsgálatok során nem használják.
|
FOGALOMMEGHATÁROZÁSOK
|
6.
|
Az alkalmazott fogalmak meghatározása az 1. függelékben található.
|
KIINDULÁSI MEGFONTOLÁSOK
|
7.
|
Az in vitro végrehajtott vizsgálatok rendszerint szükségessé teszik a metabolikus aktiválás valamilyen exogén forrásának használatát, kivéve, ha a sejtek metabolizálják a vizsgált anyagokat. A külső metabolikus aktivációs rendszer nem utánozza teljesen az in vivo körülményeket. Ügyelni kell az olyan körülmények elkerülésére, amelyek műhibaként a belső mutagenitást nem tükröző pozitív eredményekhez vezetnének, és amelyek olyan tényezőkből adódhatnak, mint például a pH vagy az ozmolalitás jelentős változásai vagy a citotoxicitás magas szintje (39) (40) (41). Amennyiben a vizsgált vegyi anyag a hozzáadásakor változást idéz elő a közeg pH-jában, akkor a pH-t – lehetőleg a törzsoldat pufferolásával – úgy kell beállítani, hogy a térfogatok minden vizsgált koncentráció és minden kontroll esetében azonosak maradjanak.
|
|
8.
|
A mikronukleuszok indukálásának vizsgálatához elengedhetetlen, hogy a kezelt és a nem kezelt tenyészetekben egyaránt mitózis menjen végbe. A mikronukleuszok számolásához az a leginformatívabb szakasz, amikor a sejt a vizsgált anyaggal végzett kezelés során vagy után egy mitózison esett át.
|
A VIZSGÁLAT ELVE
|
9.
|
A humán vagy emlős eredetű sejttenyészeteket metabolikus aktiváció exogén forrásának jelenlétében és anélkül egyaránt kiteszik a vizsgált anyag hatásának, kivéve, ha megfelelő metabolizáló képességgel rendelkező sejteket használnak. A párhuzamos oldószeres/vivőanyagos (VC) és pozitív kontrollanyagok (PC) minden vizsgálathoz hozzátartoznak.
|
|
10.
|
A vizsgált anyaggal végzett expozíció során vagy után a sejteket elegendő ideig kell tenyészteni ahhoz, hogy kromoszóma- vagy orsókárosodás jöhessen létre, és ez az interfázisban lévő sejtekben mikronukleuszok képződéséhez vezessen. Az aneuploidia indukálásához a vizsgált anyagnak a mitózis során a szokott módon jelen kell lennie. A mikronukleuszok jelenlétét a begyűjtött és megfestett, interfázisban lévő sejtekben vizsgálják. Ideális esetben a mikronukleuszokat csak azokban a sejtekben kell megszámolni, amelyek a vizsgált anyaggal történt expozíció során vagy – ha van ilyen – az expozíció utáni időszakban estek át mitózison. A citokinezis-gátlóval kezelt tenyészetekben ezt úgy érjük el, hogy csak a kétmagvú sejteket értékeljük. Citokinezis-gátló hiányában fontos annak bizonyítása, hogy a vizsgált sejtek valószínűleg sejtosztódáson estek át a vizsgált anyaggal végzett expozíció során vagy után. Minden protokoll esetében fontos bizonyítani, hogy a sejtproliferáció a kontroll és a kezelt tenyészetekben egyaránt végbement, és a mikronukleuszok számolásához használt tenyészetekben (vagy a párhuzamos tenyészetekben) meg kell állapítani a vizsgált anyag által kiváltott citotoxicitás vagy citosztázis mértékét.
|
A VIZSGÁLAT LEÍRÁSA
Előkészületek
|
11.
|
Humán perifériás vérből származó tenyésztett primer limfociták (5) (19) (42) (43) és bizonyos rágcsáló sejtvonalak, például a CHO, V79, CHL/IU és L5178Y sejtek alkalmazhatók (18) (19) (20) (21) (22) (25) (26) (27) (28) (30). Egyéb sejtvonalak és sejttípusok alkalmazását a vizsgálat során mutatott teljesítményük alapján, az Elfogadhatósági kritériumok című részben leírtak szerint kell indokolni. Mivel a mikronukleusz-képződés alapgyakorisága befolyásolja a vizsgálat érzékenységét, ezért alacsony, stabil mikronukleusz-képződési gyakorisággal rendelkező sejttípusok alkalmazása javasolt.
|
|
12.
|
A limfociták kinyeréséhez a humán perifériás vért fiatal (körülbelül 18-35 éves), egészséges, nem dohányzó személytől kell venni, akiről nem ismert, hogy az utóbbi időben genotoxikus vegyi anyagoknak vagy sugárhatásnak lett volna kitéve. Amennyiben több donortól származó sejteket együtt használnak fel, akkor a donorok számát meg kell adni. A mikronukleusz-képződés gyakorisága az életkor előrehaladtával fokozódik, és ez a tendencia a nőknél erősebb, mint a férfiaknál (44), így ezt a donorsejtek gyűjtésre történő kiválasztása során figyelembe kell venni.
|
Tenyésztőközegek és tenyésztési körülmények
|
13.
|
A tenyészetek fenntartásához megfelelő tenyésztőközeget és inkubálási körülményeket (tenyésztőedényeket, CO2-koncentrációt, hőmérsékletet és páratartalmat) kell biztosítani. Az igazolt sejtvonalaknál és törzseknél rutinszerűen ellenőrizni kell a modális kromoszómaszám stabilitását és a mikoplazma-fertőzés hiányát, és amennyiben a modális kromoszómaszám megváltozott, nem szabad felhasználni a tenyészetet. A vizsgálólaboratóriumban a tenyésztéshez alkalmazott normál sejtciklus idejének ismertnek kell lennie. Amennyiben citokinezis-gátlási módszert alkalmaznak, akkor a citokinezis-gátló anyag koncentrációját az adott sejttípusnak megfelelően optimalizálni kell, és bizonyítottnak kell lennie, hogy az adott koncentráció az értékeléshez megfelelő számú kétmagvú sejtet eredményez.
|
Tenyészetek készítése
|
14.
|
Igazolt sejtvonalak és törzsek: a törzstenyészetből származó sejteket széleszteni kell, majd tápközegben, olyan denzitásban kell tenyészteni, hogy az egy rétegben növő tenyészetek (monolayerek) ne váljanak egybefüggővé, és a szuszpenziós tenyészetek ne érjenek el túlzott denzitást a begyűjtés előtt, majd 37 °C hőmérsékleten inkubálni kell.
|
|
15.
|
Limfociták: a vizsgált anyaggal és a citoB-vel való expozíció előtt véralvadásgátlóval (például heparinnal) kezelt teljes vért vagy izolált limfocitákat tenyésztünk valamilyen mitogén anyag, például fitohemagglutinin (PHA) jelenlétében.
|
Metabolikus aktiváció
|
16.
|
Megfelelő endogén metabolikus kapacitással nem rendelkező sejtek használata esetén exogén metabolizáló rendszereket kell alkalmazni. A leggyakrabban használt rendszer egy kofaktorral kiegészített posztmitokondriális frakció (S9), amelyet enziminduktor szerekkel, például Aroclor 1254-gyel (45) (46) vagy fenobarbiton és β-naftoflavon kombinációjával (46) (47) (48) (49) kezelt rágcsálók májából állítanak elő. Ez utóbbi kombináció nem sérti a környezetben tartósan megmaradó szerves szennyező anyagokra vonatkozó Stockholmi Egyezményt (50) és a környezetben tartósan megmaradó szerves szennyező anyagokról szóló 850/2004/EK rendeletet (66), és a vegyes funkciójú oxidázok indukciója terén ugyanolyan hatékonynak bizonyult, mint az Aroclor 1254 (46) (47) (48) (49). Az S9 frakciót a végleges vizsgálati tenyésztőközegben általában az 1–10 % (v/v) közötti koncentrációtartományban alkalmazzák. A metabolikus aktivációs rendszer jellemzői függhetnek a vizsgált kémiai anyag csoportjától, és bizonyos esetekben megfelelő lehet több S9 koncentráció alkalmazása.
|
|
17.
|
A humán vagy rágcsáló eredetű aktiváló enzimeket termelő, géntechnológiával előállított sejtvonalak kiküszöbölhetik az exogén metabolikus aktivációs rendszer alkalmazásának szükségességét, és alkalmazhatók vizsgálathoz. Ilyen esetekben az alkalmazott sejtvonalra vonatkozó döntésnek – például a vegyes funkciójú oxidázoknak a vizsgált anyag metabolizmusában betöltött szerepe (51), valamint az ismert klasztogén és aneugén hatású anyagokra adott válaszreakciójuk alapján – tudományosan megalapozottnak kell lennie (lásd az Elfogadhatósági kritériumokra vonatkozó külön részt). Szem előtt kell tartani, hogy a vizsgált anyagot a termelt vegyes funkciójú oxidáz(ok) nem metabolizálhatják, ellenkező esetben ugyanis a negatív eredmények nem jelentenék azt, hogy a vizsgált anyag nem indukálja mikronukleuszok képződését.
|
A vizsgált anyag előkészítése
|
18.
|
A szilárd halmazállapotú vegyi anyagokat a sejtek kezelése előtt oldószerben vagy vivőanyagban kell feloldani, és szükség esetén hígítani kell. A folyékony anyagokat közvetlenül is lehet a vizsgálati rendszerhez adagolni és/vagy a kezelés előtt hígíthatók. Gáznemű vagy illékony vegyi anyagok a szabványos protokoll megfelelő módosításával vizsgálhatók, ilyen lehet például, hogy az anyagok kezelését légmentesen lezárt edényekben kell végezni (52) (53). A vizsgált anyagból friss készítményeket kell alkalmazni, kivéve, ha a stabilitási adatok a tárolás elfogadhatóságát bizonyítják.
|
Vizsgálati körülmények
Oldószerek/vivőanyagok
|
19.
|
Az oldószer/vivőanyag nem léphet reakcióba a vizsgált anyaggal, és nem befolyásolhatja a sejtek túlélőképességét vagy az S9 aktivitásának fennmaradását az alkalmazott koncentrációban. Ha nem jól bevált oldószert/vivőanyagot (például víz, sejttenyésztőközeg, dimetil-szulfoxid) alkalmaznak, akkor a választott szer alkalmazását a vizsgált anyaggal való kompatibilitást és a genotoxicitás hiányát mutató adatokkal kell alátámasztani. Amennyiben lehetséges, először valamilyen vizes oldószer/vivőanyag használatát kell megfontolni.
|
CitoB alkalmazása citokinezis-gátlóként
|
20.
|
Az MNvit vizsgálat teljesítményét illetően az egyik legfőbb szempont annak biztosítása, hogy az értékelt sejtek a kezelés során vagy – ha volt ilyen – a kezelés utáni inkubációs periódusban mitózison essenek át. A citokinezis gátlására a legelterjedtebben alkalmazott szer a citoB, mivel gátolja az aktin kapcsolódását, ezzel megakadályozza az utódsejtek mitózis utáni szétválását, ami kétmagvú sejtek képződéséhez vezet (5) (54) (55). A mikronukleuszok számolása ezért azokra a sejtekre korlátozható, amelyek a kezelés során vagy utána átestek a mitózison. A vizsgált anyag által a sejtproliferáció kinetikájára gyakorolt hatás párhuzamosan mérhető. A citoB akkor alkalmazandó citokinezis-gátlóként, ha humán limfocitákat alkalmazunk, mivel a sejtciklusidők adott tenyészeten belül és donorok között változóak lesznek, és nem az összes limfocita fog reagálni a PHA-ra. Egyéb módszereket alkalmazunk, ha azért vizsgáljuk a sejtvonalakat, hogy meghatározzuk, osztódtak-e az értékelt sejtek; ezek alább szerepelnek (lásd a 26. bekezdést).
|
|
21.
|
A citoB megfelelő koncentrációját az egyes sejttípusokra vonatkozóan a laboratóriumnak kell meghatározni annak érdekében, hogy az oldószerrel/vivőanyaggal kezelt kontroll tenyészetekben megfelelő gyakorisággal alakuljanak ki kétmagvú sejtek. A citoB megfelelő koncentrációja általában 3–6 μg/ml.
|
A sejtproliferáció és a citotoxicitás mérése, valamint az expozíciós koncentráció kiválasztása
|
22.
|
A vizsgált anyag legmagasabb vizsgálandó koncentrációjának meghatározásakor kerülni kell azokat a koncentrációkat, amelyek mellett álpozitív válaszreakciók alakulhatnak ki, ilyenek például a rendkívüli citotoxicitást kiváltó, a tenyésztőközegben kicsapódást okozó vagy a pH-ban, illetve az ozmolalitásban jelentős változásokat okozó koncentrációk (39) (40) (41).
|
|
23.
|
Mérni kell a sejtproliferációt, és ezáltal meg kell győződni arról, hogy a kezelt sejtek a vizsgálat során átestek a mitózison, és hogy a kezeléseket megfelelő szintű citotoxicitás mellett végezzük (lásd a 29. bekezdést). A citotoxicitást a metabolikus aktivációt igénylő sejtekben meg kell határozni metabolikus aktiváció mellett és anélkül is, a sejtszám relatív növekedése (RICC) vagy a relatív populációduplázódás (RPD) alapján (lásd a 2. függelék képleteit), kivéve, ha citoB-t alkalmazunk. CitoB alkalmazása esetén a citotoxicitás meghatározható a replikációs index (RI) segítségével (a képletet lásd a 2. függelékben).
|
|
24.
|
A tenyészetek citoB-vel történő kezelése, valamint a tenyészetben található egymagvú, kétmagvú és többmagvú sejtek relatív gyakoriságainak mérése pontos módszer a kezelés sejtproliferációt előidéző és citotoxikus vagy citosztatikus hatásának mennyiségi meghatározására (5), valamint biztosítja, hogy csak a kezelés során vagy után osztódott sejtek kerüljenek értékelésre.
|
|
25.
|
A citoB-vel végzett vizsgálatokban a citosztázis/citotoxicitás mennyiségi meghatározása a citokinezis-gátlási proliferációs indexből (CBPI) (5) (26) (56) végezhető, vagy tenyészetenként legalább 500 sejt RI-jéből (a képleteket lásd a 2. függelékben) vezethető le. Amennyiben a sejtproliferáció értékeléséhez citoB-t alkalmazunk, a CBPI-t vagy az RI-t tenyészetenként legalább 500 sejtből kell meghatározni. Többek között ezek a mérések alkalmazhatók a citotoxicitásnak a kezelt tenyészetekben és a kontrolltenyészetekben kapott értékek összehasonlításával történő becslésére. A citotoxicitás egyéb markereinek (például egybefüggőség, sejtszám, apoptózis, nekrózis, metafázis számolása) értékelése szintén hasznos információkat szolgáltathat.
|
|
26.
|
A citoB nélkül végzett vizsgálatokban igazolni kell azt, hogy a tenyészetben értékelt sejtek a vizsgált anyaggal végzett kezelés alatt vagy az után átestek az osztódáson, ellenkező esetében álnegatív lehet a válaszreakció. Annak biztosítására, hogy az osztódott sejtek kerüljenek értékelésre, alkalmazható például a bromodezoxiuridin (BrdU) beépítése és azt követő kimutatása az osztódott sejtek azonosítása érdekében (57), a klónképződés, amikor immortalizált sejtvonalat kezelnek és értékelnek in situ tárgylemezen, mikroszkóp használatával (proliferációs index, [PI]) (25) (26) (27) (28), illetve a relatív populációduplázódás (RPD), a sejtszám relatív növekedése (RICC) vagy egyéb bizonyított igazolt módszerek (16) (56) (58) (59) (a képleteket lásd a 2. függelékben). A citotoxicitás vagy citosztázis egyéb markereinek (például egybefüggőség, sejtszám, apoptózis, nekrózis, metafázis számolása) értékelése szintén hasznos információkat szolgáltathat.
|
|
27.
|
Legalább három elemezhető vizsgálati koncentrációt kell értékelni. Ennek érdekében szükséges lehet nagyobb számú, egymáshoz közeli koncentrációkkal végezni a kísérletet, és a mikronukleusz-képződést a megfelelő tartományba eső citotoxicitást eredményező koncentrációk alkalmazásával elemezni. Egy másik lehetséges stratégia szerint előzetes citotoxicitási vizsgálatot kell végezni és ezzel leszűkíteni a tartományt a végleges vizsgálathoz.
|
|
28.
|
A legmagasabb koncentrációval 55 ± 5 %-os citotoxicitás előidézését kell megcélozni. Nagyobb koncentrációk a citotoxicitás másodlagos hatásaként kromoszómakárosodást idézhetnek elő (60). Amennyiben citotoxicitás lép fel, a kiválasztott koncentrációknak le kell fedni az 55 ± 5 %-os citotoxicitástól kezdve a kisfokú citotoxicitáson át a citotoxicitást nem okozó koncentrációk tartományát.
|
|
29.
|
Amennyiben citotoxicitás vagy kicsapódás nem figyelhető meg, akkor a legmagasabb vizsgált koncentrációnak 0,01 M-nak, 5 mg/ml-nek vagy 5 μl/ml-nek kell megfelelnie, attól függően, hogy melyik a legalacsonyabb érték. A vizsgálathoz kiválasztott koncentrációk között általában nem lehet 10-nél nagyobb távolság. Olyan vizsgált anyagok esetében, amelyek meredek koncentráció-válasz görbével rendelkeznek, szükséges lehet kisebb különbségeket hagyni a vizsgált anyag koncentrációi között, hogy a közepes és alacsony toxicitási tartományban lévő tenyészetek is értékelésre kerüljenek.
|
|
30.
|
Amennyiben az oldhatóság korlátozó tényező, akkor a maximális koncentrációnak – ha nem korlátozza citotoxicitás – azt a legalacsonyabb koncentrációt kell választani, amely mellett csak minimális precipitátumok láthatók a tenyészetekben, feltéve, hogy nem zavarják az értékelést. A precipitáció értékelését például fénymikroszkópos módszerrel kell végezni, figyelembe véve azokat a precipitátumokat, amelyek a tenyésztés során tartósan megmaradtak vagy ekkor (a kezelés végéig) jelentek meg.
|
Kontrollok
|
31.
|
Minden kísérletben egyidejűleg pozitív és oldószeres/vivőanyagos kontrollokat is vizsgálni kell, metabolikus aktiválással és anélkül is.
|
|
32.
|
A pozitív kontrollok azért szükségesek, hogy igazoljuk az alkalmazott sejtek és a vizsgálati protokoll alkalmasságát a klasztogén és aneugén hatású anyagok azonosítására, valamint az S9 készítmény metabolikus képességének megerősítésére. A pozitív kontrollnak a mikronukleusz-képződés ismert induktorát olyan koncentrációban kell tartalmaznia, amely a háttérértékekben várhatóan kismértékű, de reprodukálható növekedést okoz, és bizonyítja a vizsgálati rendszer érzékenységét. A pozitív kontrollok koncentrációját úgy kell kiválasztani, hogy a hatások egyértelműek legyenek, de a kiértékelő számára ne árulkodjanak azonnal a kódolt tárgylemezek tartalmáról.
|
|
33.
|
A metabolikus kompetencia és a vizsgálati rendszer klasztogén anyagok kimutatására való képességének igazolására egyaránt metabolikus aktivációt igénylő klasztogén anyagokat (például ciklofoszfamid; benzo[a]pirén) kell alkalmazni. Indokolt esetben más pozitív kontroll is alkalmazható. Mivel a metabolikus aktivációt igénylő egyes pozitív kontrollok bizonyos kezelési körülmények mellett, illetve bizonyos sejtvonalakban exogén metabolikus aktiváció nélkül is aktívak lehetnek, ezért a metabolikus aktiváció szükségességét, valamint az S9 preparátum aktivitását a kiválasztott sejtvonalban és a kiválasztott koncentrációk mellett kell vizsgálni.
|
|
34.
|
Jelenleg nem ismert olyan aneugén hatású anyag, amely genotoxikus aktivitásához metabolikus aktivációt igényelne (16). Az aneugén aktivitás vizsgálata során alkalmazandó jelenleg elfogadott pozitív kontroll például a kolhicin és a vinblasztin. Más vegyi anyagok is alkalmazhatók, amennyiben a mikronukleuszok képződését kizárólag, vagy elsődlegesen aneugén hatásuk révén indukálják. Annak elkerülésére, hogy metabolikus aktiváció nélkül két pozitív kontrollt (a klasztogén és az aneugén hatás vizsgálatára) kelljen alkalmazni, az aneugenitás vizsgálata során alkalmazott kontroll S9 nélkül szolgálhat pozitív kontrollként, a klasztogén hatás vizsgálatához alkalmazott kontroll pedig használható az alkalmazott metabolikus aktiváló rendszer megfelelőségének vizsgálatára. Az S9 preparátumot nem igénylő sejtekben a klasztogén és az aneugén hatás vizsgálatához egyaránt szükséges pozitív kontrollt alkalmazni. A javasolt pozitív kontrollokat a 3. függelék tartalmazza.
|
|
35.
|
Megfontolható a vegyi anyag csoportjához tartozó pozitív kontroll használata, amennyiben a megfelelő vegyi anyagok rendelkezésre állnak. Az alkalmazott pozitív kontrolloknak meg kell felelniük a sejttípusnak és az aktiválás körülményeinek.
|
|
36.
|
Minden begyűjtési időpontban oldószeres/vivőanyagos kontrollokat kell alkalmazni. Ezenfelül kezeletlen negatív kontrollt (amelyben nincs oldószer/vivőanyag) szintén alkalmazni kell, kivéve, ha rendelkezésre állnak olyan publikált vagy a laboratóriumban nyert régebbi kontrolladatok, amelyek igazolják, hogy a választott oldószer az alkalmazott koncentrációban nem idéz elő genotoxikus vagy egyéb ártalmas hatásokat.
|
VIZSGÁLATI ELJÁRÁS
A kezelés ütemterve
|
37.
|
A sejtciklus meghatározott fázisában ható aneugén, illetve klasztogén anyagok azonosítási valószínűségének maximalizálása érdekében fontos, hogy a sejtciklusok valamennyi fázisa során megfelelő számú sejtet kezeljünk a vizsgált anyaggal. A kezelés ütemterve a sejtvonalak és primer sejttenyészetek esetében ezért valamelyest elérthet a limfocitákétól, amelyek mitogén stimulációt igényelnek ahhoz, hogy megkezdődjön a sejtciklus; ennek tárgyalása a 41–43. bekezdésekben szerepel (16).
|
|
38.
|
Az elméleti megfontolások a publikált adatokkal együtt (18) azt mutatják, hogy a legtöbb aneugén és klasztogén anyagot rövid, 3–6 órás kezelési időszak alkalmazásával – S9 jelenlétében vagy hiányában – lehet kimutatni, amelyet a vizsgált anyag eltávolítása és 1,5–2,0 sejtciklusnyi szaporodási időszak követ (6). A sejteket a kezelés kezdetétől vagy végétől számítva a normál sejtciklus (vagyis a kezeletlen sejtek sejtciklusa) körülbelül 1,5–2,0-szeresének megfelelő idő elteltével kell begyűjteni (lásd az 1. táblázatot). A begyűjtési vagy visszanyerési idők meghosszabbíthatók, amennyiben ismert vagy feltételezhető, hogy a vizsgált anyag befolyásolja a sejtciklus idejét (például nukleozidanalógok vizsgálata során).
|
|
39.
|
Az S9 preparátumok tenyésztett emlőssejtek esetében tapasztalható potenciális citotoxicitása miatt a normál sejtciklus-idő 1,5–2,0-szeresének megfelelő meghosszabbított expozíciós kezelést kizárólag S9 hiányában végeznek. Meghosszabbított kezelés esetén lehetőség kínálkozik a sejtek vizsgált vegyi anyaggal történő kezelésének citoB hiányában vagy jelenlétében történő végzésére. Ezeknek a választási lehetőségeknek azokban a helyzetekben van jelentőségük, amikor problémát jelenthet a vizsgált anyag és a citoB közötti lehetséges kölcsönhatás.
|
|
40.
|
A sejtek kezelésének javasolt ütemtervét az 1. táblázat mutatja be. Ezek az általános kezelési ütemtervek a vizsgált anyag stabilitásától és reaktivitásától, illetve az alkalmazott sejtek konkrét szaporodási jellemzőitől függően módosíthatóak. Minden kezelésnek az exponenciális szaporodás időszakában kell kezdődnie és véget érnie. Ezeket az ütemterveket a következő, 41–47. bekezdések mutatják be részletesebben.
1. táblázat
A sejtek kezelésének és begyűjtésének ideje az MNvit vizsgálat esetében
|
CitoB-vel kezelt limfociták, primer sejtek és sejtvonalak
|
+ S9
|
A sejteket 3–6 órán át S9 jelenlétében kezeljük;
eltávolítjuk az S9-et és a kezelési közeget;
friss közeget és citoB-t adunk hozzá;
a normál sejtciklus 1,5–2,0-szeresének elteltével begyűjtjük a sejteket.
|
|
– S9
Rövid expozíció
|
A sejteket 3–6 órán át kezeljük;
eltávolítjuk a kezelési közeget;
friss közeget és citoB-t adunk hozzá;
a normál sejtciklus 1,5–2,0-szeresének elteltével begyűjtjük a sejteket.
|
|
– S9
Meghoszszabbított expozíció
|
»A«lehetőség: A sejteket a normál sejtciklus 1,5–2-szeresének megfelelő ideig citoB jelenlétében kezeljük;
az expozíciós idő végén begyűjtjük a sejteket.
»B«lehetőség: A sejteket a normál sejtciklus 1,5–2-szeresének megfelelő ideig kezeljük;
eltávolítjuk a vizsgált anyagot;
friss közeget és citoB-t adunk hozzá;
a normál sejtciklus 1,5–2,0-szeresének elteltével begyűjtjük a sejteket.
|
|
CitoB nélkül kezelt sejtvonalak
(Megegyezik a fentebb ismertetett kezelési ütemtervvel, azzal a kivétellel, hogy citoB hozzáadására nem kerül sor)
|
|
CitoB-vel kezelt limfociták, primer sejtek és sejtvonalak
|
41.
|
Limfociták esetében a leghatékonyabb módszer, ha a vizsgált anyaggal történő expozíciót a PHA-stimuláció után 44–48 órával kezdjük, amikorra a ciklusszinkronizáció már eltűnik (5). Az első vizsgálat során a sejteket 3–6 órán át, S9 hiányában és jelenlétében kezeljük a vizsgált anyaggal. Eltávolítjuk a kezelési közeget, citoB-t tartalmazó friss közeggel pótoljuk, majd a normál sejtciklus 1,5–2,0-szeresének megfelelő idő elteltével begyűjtjük a sejteket.
|
|
42.
|
Amennyiben a rövid (3–6 órás) kezeléssel végzett mindkét első vizsgálat negatív vagy kétes eredményű, akkor meghosszabbított expozíciós idővel, S9 nélkül újabb kezelést végzünk. Kétféle kezelési lehetőség áll rendelkezésre, amelyek mindegyike egyformán elfogadott. Stimulált limfociták esetében, amelyeknél az exponenciális szaporodási fázis a stimulációt követő 96. órától csökkenhet, megfelelőbb lehet az »A« lehetőség szerint eljárni. Ezenfelül a »B« lehetőség esetében a sejttenyészeteknek a végleges mintavétel időpontjáig nem szabad egybefüggővé válniuk.
|
—
|
»A« lehetőség: A sejteket a normál sejtciklus 1,5–2,0-szeresének megfelelő ideig kezeljük a vizsgált anyaggal, majd a kezelési idő végén begyűjtjük.
|
|
—
|
»B« lehetőség: A sejteket a normál sejtciklus 1,5–2,0-szeresének megfelelő ideig kezeljük a vizsgált anyaggal. Eltávolítjuk a kezelési közeget és friss közeggel pótoljuk, majd a normál sejtciklus 1,5–2,0-szeresének megfelelő további időszak elteltével begyűjtjük a sejteket.
|
|
|
43.
|
A primer sejteket és sejtvonalakat a limfocitákhoz hasonló módon kell kezelni, azzal a kivétellel, hogy a PHA-val 44–48 órán át végzett stimuláció nem szükséges. A limfocitáktól eltérő egyéb sejtek esetében az expozíciót úgy kell végezni, hogy a vizsgálat befejezésének időpontjában a sejtek még a szaporodás logaritmikus fázisában legyenek.
|
CitoB nélkül kezelt sejtvonalak
|
44.
|
A sejteket 3–6 órán át, S9 jelenlétében és hiányában kell kezelni. Eltávolítjuk a kezelési közeget és friss közeggel pótoljuk, majd a normál sejtciklus 1,5–2,0-szeresének megfelelő idő elteltével begyűjtjük a sejteket.
|
|
45.
|
Amennyiben a rövid (3–6 órás) kezeléssel végzett mindkét első vizsgálat negatív vagy kétes eredményű, akkor meghosszabbított expozíciós idővel újabb kezelést végzünk (S9 nélkül). Kétféle kezelési lehetőség áll rendelkezésre, amelyek mindegyike egyformán elfogadott:
|
—
|
»A« lehetőség: A sejteket a normál sejtciklus 1,5–2,0-szeresének megfelelő ideig kezeljük a vizsgált anyaggal, majd a kezelési idő végén begyűjtjük.
|
|
—
|
»B« lehetőség: A sejteket a normál sejtciklus 1,5–2,0-szeresének megfelelő ideig kezeljük a vizsgált anyaggal. Eltávolítjuk a kezelési közeget és friss közeggel pótoljuk, majd a normál sejtciklus 1,5–2,0-szeresének megfelelő további időszak elteltével begyűjtjük a sejteket.
|
|
|
46.
|
Az egy rétegben növő tenyészetekben (monolayerek) a 3-6 órás kezelés végén mitotikus sejtek (ismertetőjegyük, hogy alakjuk kerek, és leválnak a felszínről) lehetnek jelen. Mivel ezek a mitotikus sejtek könnyen leválnak, a vizsgált anyagot tartalmazó közeg eltávolításakor elveszhetnek. Ügyelni kell arra, hogy a tenyészetek átmosásakor összegyűjtsük ezeket a sejteket, és visszahelyezzük a tenyészetbe, ezáltal a begyűjtéskor ne veszítsünk el mitózisban lévő és a mikronuleusz-képződés kockázatának kitett sejteket.
|
A tenyészetek száma
|
47.
|
A vizsgált anyag minden koncentrációjához, valamint a vivőanyagos/oldószeres és negatív kontroll tenyészetekhez két párhuzamos tenyészetet kell alkalmazni. Amennyiben a párhuzamos tenyészetek között a korábbi laboratóriumi adatok alapján csak minimális eltérések igazolhatók, elfogadható lehet egy tenyészet alkalmazása. Egy tenyészet alkalmazása esetén javasolt nagyobb számú koncentrációkat vizsgálni.
|
A sejtek begyűjtése és a metszet elkészítése
|
48.
|
A begyűjtést és a feldolgozást minden tenyészetnél külön kell végezni. A sejtek előkészítéséhez hozzátartozhat a hipotóniás kezelés, ez a lépés azonban nem szükséges, ha a sejtek megfelelő szétterítése egyéb módon is elérhető. A metszetkészítés során különböző technikák alkalmazhatók, amennyiben ezek az értékeléshez jó minőségű sejtpreparátumokat eredményeznek. A sejt citoplazmáját meg kell tartani, hogy ki lehessen mutatni a mikronukleuszokat és (a citokinezis-gátlási módszer esetén) megbízhatóan azonosítani lehessen a kétmagvú sejteket.
|
|
49.
|
A metszetek különböző módszerekkel festhetők meg, például Giemsa vagy fluoreszkáló DNS-specifikus festékekkel (59). DNS-specifikus festék (például akridin narancs (61) vagy Hoechst 33258 plusz pironin-Y (62)) használatával kiküszöbölhetők a nem DNS-specifikus festékkel járó bizonyos műhibák. A mikronukleuszok tartalmának (kromoszóma/kromoszómafragmentum) azonosítására antikinetokor antitestek, pancentromérás DNS-próbákkal végzett FISH vagy pancentroméra-specifikus primerekkel végzett in situ jelölés, illetve megfelelő DNS-kontrasztfestés alkalmazható, amennyiben a mikronukleuszok keletkezésének mechanizmusára vonatkozó információkra van szükség (15)(16). A klasztogén és aneugén hatású anyagok megkülönböztetésére egyéb, korábban hatékonynak bizonyult módszerek is alkalmazhatók.
|
Elemzés
|
50.
|
A mikroszkópos elemzés előtt az összes metszetet, köztük az oldószeres/vivőanyagos metszeteket és a kontrollokat is egymástól függetlenül kell kódolni. A kódolt minták hitelesített, automatikus flow citometriával vagy képelemzési rendszerrel is elemezhetők.
|
|
51.
|
A citoB-vel kezelt tenyészetekben a mikronukleusz-képződés gyakoriságát koncentrációnként legalább 2 000 kétmagvú sejt értékelésével (tenyészeteként legalább 1 000 kétmagvú sejt; két tenyészet koncentrációként) kell vizsgálni. Ha csak egy tenyészetet alkalmazunk, akkor az adott tenyészetből koncentrációként legalább 2 000 kétmagvú sejtet kell értékelni. Ha az egyes koncentrációkban tenyészetenként 1 000-nél vagy – egy tenyészet alkalmazása esetén – 2 000-nél jelentősen kevesebb kétmagvú sejt áll rendelkezésre, és nem mutatható ki a mikronukleuszok számának jelentős növekedése, akkor vizsgálatot szükség szerint több sejt vagy kevésbé toxikus koncentrációk alkalmazásával meg kell ismételni. Ügyelni kell arra, hogy nem szabad olyan kétmagvú sejteket értékelni, amelyek szabálytalan alakúak vagy amelyeknél nagyobb méretű a két mag, továbbá nem szabad összetéveszteni a rosszul szétterített sejteket a többmagvú sejtekkel. A kettőnél több fő magot tartalmazó sejteknél nem szabad vizsgálni a mikronukleuszokat, mivel a ezekben a sejtekben magasabb lehet a mikronukleusz-képződés háttérgyakorisága (63) (64). Egymagvú sejtek értékelése elfogadható, amennyiben a vizsgált anyagról bebizonyosodott, hogy interferál a citoB aktivitásával.
|
|
52.
|
A citoB nélkül vizsgált sejtvonalakban a mikronukleusz-képződés gyakoriságát koncentrációnként legalább 2 000 sejt értékelésével (tenyészetenként legalább 1 000 sejt; két tenyészet koncentrációként) kell vizsgálni. Ha koncentrációnként csak egy tenyészetet alkalmazunk, akkor az adott tenyészetből legalább 2 000 sejtet kell értékelni.
|
|
53.
|
Amennyiben citoB-t alkalmazunk, a sejtproliferáció értékelése érdekében tenyészetenként legalább 500 sejtből meg kell határozni a CBPI-t vagy az RI-t (lásd 2. függelék). Amennyiben a kezeléseket citoB hiányában végzik, elengedhetetlen annak bizonyítása, hogy az értékelt sejtek osztódtak, amint ezt a 24–27. bekezdések is tárgyalták.
|
Elfogadhatósági kritériumok
|
54.
|
A vizsgálati módszerben leírt MNvit tesztet alkalmazni kívánó laboratóriumnak igazolnia kell, hogy metabolikus aktivációval és anélkül, megbízatóan és pontosan ki tudja mutatni az ismert aneugén és klasztogén hatású anyagokat, valamint az ismert negatív vegyi anyagokat a 3. függelékben felsorolt referenciaanyagok alkalmazásával. Amennyiben a vizsgálatot citoB használata nélkül végzik, akkor a vizsgálati módszer helyes elvégzésének alátámasztására a laboratóriumnak igazolnia kell, hogy a mikronukleusz-képződés vizsgálatához értékelt sejtek egy magosztódáson átestek.
|
|
55.
|
Referenciaanyagként történő alkalmazásra a 3. függelékben szereplő vegyi anyagok javasoltak. Helyettesítő vagy további vegyi anyagok is alkalmazhatók, ha ezek ismert hatásúak, és ugyanolyan hatásmechanizmussal indukálják a mikronukleusz-képződést, továbbá igazoltan relevánsak az MNvit eljárással vizsgálandó vegyi anyagok szempontjából. Ezt olyan hitelesítési vizsgálat elvégzésével lehet indokolni, amelyhez az anyagok széles körét alkalmazzák vagy a vizsgált vegyi anyag kémiai csoportja vagy a vizsgált károsodás mechanizmusa alapján egy szűkebb körre összpontosítanak.
|
|
56.
|
Az oldószeres/vivőanyagos kontrollnak és a kezeletlen tenyészeteknek reprodukálhatóan alacsony és következetes mikronukleusz-képződési gyakoriságot kell produkálniuk (a 11. bekezdésben azonosított sejttípusok esetében általában 5–25 mikronukleusz/1 000 sejt). Az egyéb sejttípusok esetében a válaszreakciók eltérő tartományokba eshetnek, amelyeket a sejtek MNvit vizsgálathoz történő alkalmazásának hitelesítésekor meg kell határozni. A historikus kontrolltartományok megállapításához a negatív kontroll, az oldószer és a pozitív kontroll adatait kell felhasználni. A vizsgálat során párhuzamosan értékelt negatív kontrollok/pozitív kontrollok megfelelőségének megítéléséhez ezeket az értékeket kell felhasználni.
|
|
57.
|
Amennyiben a vizsgálati protokollon kisebb változtatásokat terveznek (például manuális értékelés helyett automatikus; új sejttípus használata), akkor ahhoz, hogy a módosított protokollt használatra elfogadhatónak lehessen tekinteni, a változtatás után igazolni kell a vizsgálat hatékonyságát. A hatékonyság igazolása magába foglalja annak bizonyítását, hogy a kromoszómatörés, valamint a szám feletti vagy hiányzó kromoszómák kimutathatók, és a vizsgálandó kémiai anyagok adott csoportjával vagy anyagok széles körével a vizsgálat megfelelő pozitív vagy negatív eredményeket ad.
|
ADATOK ÉS JELENTÉS
Eredmények kezelése
|
58.
|
Amennyiben a citokinezis-gátlási technikát alkalmazzuk, a mikronuleusz-képződés indukciójának értékelése során (a mikronukleuszok sejtenkénti számától függetlenül) csak a mikronukleuszokkal rendelkező kétmagvú sejtek gyakoriságát használjuk fel. Az egy, két vagy több mikronukleusszal rendelkező sejtek pontozása hasznos információval szolgálhat, de nem kötelező.
|
|
59.
|
Egyidejűleg az összes kezelt, illetve oldószeres/vivőanyagos kontroll tenyészet esetében meg kell határozni a citotoxicitás és/vagy a citosztázis paramétereit is (58). Amennyiben a citokinezis-gátlási módszert alkalmazzuk, a kezelt és kontrolltenyészetek esetében a sejtcikus késésének mutatójaként ki kell számolni a CBPI-t vagy az RI-t. CitoB nélkül végzett vizsgálatoknál az RPD-t, az RICC-t vagy a PI-t kell alkalmazni (lásd a 2. függeléket).
|
|
60.
|
Meg kell adni a tenyészetre vonatkozó egyedi adatokat. Ezenkívül táblázatos formában az összes adatot össze kell foglalni.
|
|
61.
|
Az MNvit vizsgálatban a mikronukleusz-képződést indukáló vegyi anyagok azért válthatnak ki ilyen hatást, mert kromoszómatörést, kromoszómavesztést vagy a kettő együttesét idézik elő. Annak vizsgálatára, hogy a mikronukleusz-képződés indukciója klasztogén és/vagy aneugén hatás eredménye-e, antikinetokor antitestekkel, centroméraspecifikus in situ próbákkal vagy egyéb módszerekkel végzett további elemzések alkalmazhatók.
|
Az eredmények értékelése és értelmezése
|
62.
|
Egyértelműen pozitív vagy negatív eredmény esetén nincs előírva további vizsgálattal végzendő igazolás. A kétes eredmények tisztázhatók – a vizsgálat vak jellegének fenntartása érdekében – az összes tenyészetből vett újabb 1 000 sejt vizsgálatával. Ha ezzel a módszerrel nem lehet egyértelmű eredményre jutni, akkor további vizsgálatokat kell végezni. A nyomonkövetési kísérletek során mérlegelendő a vizsgálati paraméterek módosítása a vizsgálati paraméterek tartományának szükség szerinti növelésével vagy csökkentésével. A módosítható vizsgálati paraméterek közé tartozik a vizsgált koncentrációk különbsége, a sejtek begyűjtésének időzítése és/vagy a metabolikus aktiváció körülményei.
|
|
63.
|
A pozitív eredmény kimondásának számos feltétele van, például a mikronukleuszokat tartalmazó sejtek számában bekövetkezett koncentrációfüggő vagy statisztikailag szignifikáns növekedés. Elsőként mérlegelendő az eredmények biológiai relevanciája. A válaszreakció biológiai jelentőségének értékelésekor támpontot adhat annak mérlegelése, hogy a megfigyelt értékek a kontrollokkal kapott régebbi eredmények tartományába esnek-e. A vizsgálati eredmények értékelése során segítségként megfelelő statisztikai módszerek alkalmazhatók (65). A statisztikai próbák eredményeit azonban a dózis-válasz összefüggés figyelembevételével kell értékelni. A reprodukálhatóságot és a régebbi adatokat szintén figyelembe kell venni.
|
|
64.
|
Bár a legtöbb kísérlet egyértelmű pozitív vagy negatív eredményeket fog hozni, bizonyos esetekben előfordul, hogy a rendelkezésre álló adatok alapján nem lehet a vizsgált anyag aktivitásával kapcsolatban egyértelműen állást foglalni. Előfordul, hogy az eredmények továbbra is többféleképpen magyarázhatók vagy megkérdőjelezhetők maradnak függetlenül attól, hogy hány alkalommal ismételik meg a kísérletet.
|
|
65.
|
Az MNvit vizsgálattal kapott pozitív eredmény azt jelzi, hogy a vizsgált anyag tenyésztett emlőssejtekben kromoszómatörést vagy kromoszómavesztést indukál. A negatív eredmények azt jelzik, hogy az alkalmazott vizsgálati körülmények között a vizsgált anyag tenyésztett emlőssejtekben nem idéz elő kromoszómatöréseket és/vagy szám feletti kromoszómákat vagy kromoszómahiányt.
|
Vizsgálati jelentés
|
66.
|
A vizsgálati jegyzőkönyvnek legalább a következő információkat kell tartalmaznia, amennyiben azok a vizsgálat lefolytatása szempontjából relevánsak:
|
|
Vizsgált vegyi anyag:
|
—
|
azonosítási adatok, valamint a CAS nyilvántartási szám és EU-szám,
|
|
—
|
fizikai jelleg és tisztaság,
|
|
—
|
a vizsgálat elvégzése szempontjából releváns fizikai-kémiai tulajdonságok,
|
|
—
|
a vizsgált vegyi anyag reaktivitása az oldószerrel/vivőanyaggal vagy a sejttenyészethez alkalmazott tápközeggel,
|
|
|
|
Oldószer/vivőanyag:
|
—
|
az oldószer/vivőanyag kiválasztásának indoklása,
|
|
—
|
a vizsgált anyag oldhatósága és stabilitása az oldószerben/vivőanyagban,
|
|
|
|
Sejtek:
|
—
|
az alkalmazott sejtek típusa és eredete,
|
|
—
|
az alkalmazott sejtek alkalmassága,
|
|
—
|
adott esetben a mikoplazma-fertőzöttség hiánya,
|
|
—
|
a sejtciklus hosszára, a duplázódási időre, illetve a proliferációs indexre vonatkozó információk,
|
|
—
|
limfociták alkalmazása esetén adott esetben a véradók neme, életkora és száma,
|
|
—
|
limfociták alkalmazása esetén meg kell adni, hogy teljes vér vagy szeparált limfociták expozíciója történt-e,
|
|
—
|
adott esetben az átoltások száma,
|
|
—
|
adott esetben a sejttenyészetek fenntartására alkalmazott módszerek,
|
|
—
|
modális kromoszómaszám,
|
|
—
|
a normál (negatív kontroll) sejtciklusidő,
|
|
|
|
Vizsgálati körülmények:
|
—
|
amennyiben alkalmaznak citokinezis-gátló anyagot, annak megnevezése (például citoB), illetve koncentrációja és a sejtek expozíciójának időtartama,
|
|
—
|
a koncentrációk kiválasztásának és a tenyészetek számának indoklása, beleértve a citotoxicitási adatokat és az oldhatóságra vonatkozó korlátokat, ha rendelkezésre állnak,
|
|
—
|
közegek összetétele, adott esetben a CO2–koncentráció,
|
|
—
|
a vizsgált anyag koncentrációi,
|
|
—
|
a hozzáadott vivőanyag és vizsgált anyag koncentrációja (és/vagy térfogata),
|
|
—
|
inkubációs hőmérséklet és idő,
|
|
—
|
a kezelés utáni begyűjtés időpontja,
|
|
—
|
adott esetben sejtsűrűség a leoltáskor,
|
|
—
|
a metabolikus aktiváló rendszer típusa és összetétele, ideértve az elfogadhatósági kritériumokat,
|
|
—
|
pozitív kontrollanyagok és negatív kontrollok,
|
|
—
|
az alkalmazott metszetkészítési módszer és festési technika,
|
|
—
|
a mikronukleusz azonosításának kritériumai,
|
|
—
|
a vizsgált sejtek száma,
|
|
—
|
a citotoxicitás mérésére alkalmazott módszerek,
|
|
—
|
a citotoxicitás szempontjából lényeges kiegészítő információk,
|
|
—
|
a pozitív, negatív és kétes vizsgálati eredmény kritériumai,
|
|
—
|
az alkalmazott statisztikai elemzési módszer(ek),
|
|
—
|
szükség esetén annak meghatározására alkalmazott módszerek, hogy a mikronukleuszok teljes kromoszómákat vagy csak kromoszómatöredékeket tartalmaznak,
|
|
|
|
Eredmények:
|
—
|
a citokinezis-gátlási módszer esetében az alkalmazott citotoxicitás mérése, például CBPI vagy RI meghatározásával; citokinezis-gátlási módszerek alkalmazása nélkül az RICC, RPD vagy PI meghatározása; szükség esetén egyéb megfigyelések, például a sejtek egybefüggősége, apoptózis, nekrózis, metafázis számolás, kétmagvú sejtek előfordulási gyakorisága,
|
|
—
|
a kezelési közeg pH-jára és ozmolalitására vonatkozó adatok, amennyiben meghatározásra kerültek,
|
|
—
|
az elemzéshez elfogadható sejtek meghatározása,
|
|
—
|
az egymagvú, kétmagvú és többmagvú sejtek megoszlása, amennyiben citokinezis-gátlási módszert alkalmazunk,
|
|
—
|
a mikronukleuszokkal rendelkező sejtek száma minden kezelt és kontrolltenyészetre megadva, valamint adott esetben annak meghatározása, hogy kétmagvú vagy egymagvú sejtben találhatók-e,
|
|
—
|
amennyiben lehetséges, a koncentráció-válasz összefüggés,
|
|
—
|
a párhuzamos negatív (oldószeres/vivőanyagos) kontroll és pozitív kontroll kémiai adatai,
|
|
—
|
a negatív (oldószeres/vivőanyagos) kontroll és pozitív kontroll historikus kémiai adatai a tartományokkal, átlagokkal, szórással és konfidenciaintervallummal (például 95 %) együtt,
|
|
—
|
statisztikai elemzés; p-értékek, ha meghatározásra kerültek.
|
|
|
SZAKIRODALOM
|
(1)
|
Kirsch-Volders, M. (1997), Towards a validation of the micronucleus test. Mutation Res., 392, 1-4.
|
|
(2)
|
Parry, J.M. and Sors, A. (1993), The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project, Mutation Res., 287, 3-15.
|
|
(3)
|
Fenech, M. and Morley, A.A. (1985), Solutions to the kinetic problem in the micronucleus assay, Cytobios., 43, 233-246.
|
|
(4)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr, Lorge, E., Norppa, H., Surralles, J., von der Hude, W. and Wakata, A. (2000), Report from the In Vitro Micronucleus Assay Working Group, Environ. Mol. Mutagen., 35, 167-172.
|
|
(5)
|
Fenech, M. (2007), Cytokinesis-block micronucleus cytome assay, Nature Protocols, 2(5), 1084-1104.
|
|
(6)
|
Fenech, M. and Morley, A.A. (1986), Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation, Mutation Res., 161, 193-198.
|
|
(7)
|
Eastmond, D.A. and Tucker, J.D. (1989), Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody, Environ. Mol. Mutagen., 13, 34-43.
|
|
(8)
|
Eastmond, D.A. and Pinkel, D. (1990), Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probes, Mutation Res., 234, 9-20.
|
|
(9)
|
Miller, B.M., Zitzelsberger, H.F., Weier, H.U. and Adler, I.D. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesis, 6, 297-302.
|
|
(10)
|
Farooqi, Z., Darroudi, F. and Natarajan, A.T. (1993), The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes, Mutagenesis, 8, 329-334.
|
|
(11)
|
Migliore, L., Bocciardi, R., Macri, C. and Lo Jacono, F. (1993), Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe, Mutation Res., 319, 205-213.
|
|
(12)
|
Norppa, H., Renzi, L. and Lindholm, C. (1993), Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization, Mutagenesis, 8, 519-525.
|
|
(13)
|
Eastmond, D.A, Rupa, D.S. and Hasegawa, L.S. (1994), Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes, Mutation Res., 322, 9-20.
|
|
(14)
|
Marshall, R.R., Murphy, M., Kirkland, D.J. and Bentley, K.S. (1996), Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy, Mutation Res., 372, 233-245.
|
|
(15)
|
Zijno, P., Leopardi, F., Marcon, R. and Crebelli, R. (1996), Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes, Mutation Res., 372, 211-219.
|
|
(16)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate Jr., M., Lorge, E., Norppa, H., Surrallés, J., von der Hude, W. and Wakata, A. (2003), Report from the in vitro micronucleus assay working group. Mutation Res., 540, 153-163.
|
|
(17)
|
OECD (1997), In Vitro Mammalian Chromosome Aberration Test, Test Guideline No. 473, OECD Guidelines for Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [www.oecd.org/env/testguidelines]
|
|
(18)
|
Lorge, E., Thybaud, V., Aardema, M.J., Oliver, J., Wakata, A., Lorenzon G. and Marzin, D. (2006), SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study, Mutation Res., 607, 13-36.
|
|
(19)
|
Clare, G., Lorenzon, G., Akhurst, L.C., Marzin, D., van Delft, J., Montero, R., Botta, A., Bertens, A., Cinelli, S., Thybaud, V. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes, Mutation Res., 607, 37-60.
|
|
(20)
|
Aardema, M.J., Snyder, R.D., Spicer, C., Divi, K., Morita, T., Mauthe, R.J., Gibson, D.P., Soelter, S., Curry, P.T., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells, Mutation Res., 607, 61-87.
|
|
(21)
|
Wakata, A., Matsuoka, A., Yamakage, K., Yoshida, J., Kubo, K., Kobayashi, K., Senjyu, N., Itoh, S., Miyajima, H., Hamada, S., Nishida, S., Araki, H., Yamamura, E., Matsui, A., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells, Mutation Res., 607, 88-124.
|
|
(22)
|
Oliver, J., Meunier, J.-R., Awogi, T., Elhajouji, A., Ouldelhkim, M.-C., Bichet, N., Thybaud, V., Lorenzon, G., Marzin, D. and Lorge, E. (2006), SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells, Mutation Res., 607, 125-152.
|
|
(23)
|
Albertini, S., Miller, B., Chetelat, A.A. and Locher, F. (1997), Detailed data on in vitro MNT and in vitro CA: industrial experience, Mutation Res., 392, 187-208.
|
|
(24)
|
Miller, B., Albertini, S., Locher, F., Thybaud, V. and Lorge, E. (1997), Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience, Mutation Res., 392, 45-59.
|
|
(25)
|
Miller, B., Potter-Locher, F., Seelbach, A., Stopper, H., Utesch, D. and Madle, S. (1998), Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft fur Umwelt-Mutations-forschung, Mutation Res., 410, 81-116.
|
|
(26)
|
Kalweit, S., Utesch, U., von der Hude, W. and Madle, S. (1999), Chemically induced micronucleus formation in V79 cells – comparison of three different test approaches, Mutation Res. 439, 183-190.
|
|
(27)
|
Kersten, B., Zhang, J., Brendler Schwaab, S.Y., Kasper, P. and Müller, L. (1999), The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity, Mutation Res. 445, 55-71.
|
|
(28)
|
von der Hude, W., Kalweit, S., Engelhardt, G., McKiernan, S., Kasper, P., Slacik-Erben, R., Miltenburger, H.G., Honarvar, N., Fahrig, R., Gorlitz, B., Albertini, S., Kirchner, S., Utesch, D., Potter-Locher, F., Stopper, H. and Madle, S. (2000), In vitro micronucleus assay with Chinese hamster V79 cells – results of a collaborative study with in situ exposure to 26 chemical substances, Mutation Res., 468, 137-163.
|
|
(29)
|
Garriott, M.L., Phelps, J.B. and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 517, 123-134.
|
|
(30)
|
Matsushima, T., Hayashi, M., Matsuoka, A., Ishidate, M. Jr., Miura, K.F., Shimizu, H., Suzuki, Y., Morimoto, K., Ogura, H., Mure, K., Koshi, K. and Sofuni, T. (1999), Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU), Mutagenesis, 14, 569-580.
|
|
(31)
|
Elhajouji, A., and Lorge, E. (2006), Special Issue: SFTG International collaborative study on in vitro micronucleus test, Mutation Res., 607, 1-152.
|
|
(32)
|
ECVAM (2006), Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. ESAC 25th meeting, 16-17 November, 2006. Elektronikus formátumban: [http://ecvam.jrc.it/index.htm]
|
|
(33)
|
ESAC (2006), ECVAM Scientific Advisory Committee (ESAC) Peer Review, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel. Elektronikus formátumban: [http://ecvam.jrc.it/index.htm]
|
|
(34)
|
Corvi, R., Albertini, S., Hartung, T., Hoffmann, S., Maurici, D., Pfuhler, S, van Benthem, J., Vanparys P. (2008), ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT), Mutagenesis, 23, 271-283.
|
|
(35)
|
Zhang, L.S., Honma, M., Hayashi, M., Suzuki, T., Matsuoka, A. and Sofuni, T. (1995), A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test, Mutation Res., 347, 105-115.
|
|
(36)
|
Ehrlich, V., Darroudi, F., Uhl, M., Steinkellner, S., Zsivkovits, M. and Knasmeuller, S. (2002), Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells, Mutagenesis, 17, 257-260.
|
|
(37)
|
Knasmüller, S., Mersch-Sundermann, V., Kevekordes, S., Darroudi, F., Huber, W.W., Hoelzl, C., Bichler, J. and Majer, B.J. (2004), Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge, Toxicol., 198, 315-328.
|
|
(38)
|
Gibson, D.P., Brauninger, R., Shaffi, H.S., Kerckaert, G.A., LeBoeuf, R.A., Isfort, R.J. and Aardema, M.J. (1997), Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals, Mutation Res., 392, 61-70.
|
|
(39)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M. Jr., Brusick, D., Ashby, J. and Myhr, B.C. (1991), International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9, Mutation Res., 257, 147-205.
|
|
(40)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992), Clastogenicity of low pH to various cultured mammalian cells, Mutation Res., 268, 297-305.
|
|
(41)
|
Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environ. Mutagen., 8, 789-886.
|
|
(42)
|
Fenech, M. and Morley, A.A. (1985), Measurement of micronuclei in lymphocytes, Mutation Res., 147, 29-36.
|
|
(43)
|
Fenech, M. (1997), The advantages and disadvantages of cytokinesis-blood micronucleus method, Mutation Res., 392, 11-18.
|
|
(44)
|
Bonassi, S., Fenech, M., Lando, C., Lin, Y.P., Ceppi, M., Chang, W.P., Holland, N., Kirsch-Volders, M., Zeiger, E., Ban, S., Barale, R., Bigatti, M.P., Bolognesi, C., Jia, C., Di Giorgio, M., Ferguson, L.R., Fucic, A., Lima, O.G., Hrelia, P., Krishnaja, A.P., Lee, T.K., Migliore, L., Mikhalevich, L., Mirkova, E., Mosesso, P., Muller, W.U., Odagiri, Y., Scarffi, M.R., Szabova, E., Vorobtsova, I., Vral, A. and Zijno, A. (2001), HUman MicroNucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei, Environ. Mol. Mutagen.
37, 31-45.
|
|
(45)
|
Maron, D.M. and Ames, B.N. (1983), Revised methods for the Salmonella mutagenicity test, Mutation Res., 113, 173-215.
|
|
(46)
|
Ong, T.-m., Mukhtar, M., Wolf, C.R. and Zeiger, E. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, J. Environ. Pathol. Toxicol., 4, 55-65.
|
|
(47)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992), Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, 7, 175-177.
|
|
(48)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976), A safe substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, In: de Serres, F.J., Fouts, J. R., Bend, J.R. and Philpot, R.M. (eds), In Vitro Metabolic Activation in Mutagenesis Testing, Elsevier, North-Holland, 85-88.
|
|
(49)
|
Johnson, T.E., Umbenhauer, D.R. and Galloway, S.M. (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environ. Mol. Mutagen., 28, 51-59.
|
|
(50)
|
UNEP (2001), Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP). Elektronikus formátumban: [http://www.pops.int/]
|
|
(51)
|
Doherty, A.T., Ellard, S., Parry, E.M. and Parry, J.M. (1996), An investigation into the activation and deactivation of chlorinated hydrocarbons to genotoxins in metabolically competent human cells, Mutagenesis, 11, 247-274.
|
|
(52)
|
Krahn, D.F., Barsky, F.C. and McCooey, K.T. (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, In: Tice, R.R., Costa, D.L. and Schaich, K.M. (eds), Genotoxic Effects of Airborne Agents. New York, Plenum, pp. 91-103.
|
|
(53)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983), Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay, Environ. Mutagenesis 5, 795-801.
|
|
(54)
|
Fenech, M. (1993), The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations, Mutation Res., 285, 35-44.
|
|
(55)
|
Phelps, J.B., Garriott, M.L., and Hoffman, W.P. (2002), A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity, Mutation Res., 521, 103-112.
|
|
(56)
|
Kirsch-Volders, M., Sofuni, T., Aardema, M., Albertini, S., Eastmond, D., Fenech, M., Ishidate, M. Jr., Kirchner, S., Lorge, E., Morita, T., Norppa, H., Surralles, J., Vanhauwaert, A. and Wakata, A. (2004), Corrigendum to »Report from the in vitro micronucleus assay working group«, Mutation Res., 564, 97-100.
|
|
(57)
|
Pincu, M., Bass, D. and Norman, A. (1984), An improved micronuclear assay in lymphocytes, Mutation Res., 139, 61-65.
|
|
(58)
|
Lorge, E., Hayashi, M., Albertini, S. and Kirkland, D. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Res., 655, 1-3.
|
|
(59)
|
Surralles, J., Xamena, N., Creus, A., Catalan, J., Norppa, H. and Marcos, R. (1995), Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures, Mutation Res., 341, 169-184.
|
|
(60)
|
Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environ. Molec. Mutagenesis 35, 191-201.
|
|
(61)
|
Hayashi, M., Sofuni, T., and Ishidate, M. Jr. (1983), An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test, Mutation Res., 120, 241-247.
|
|
(62)
|
MacGregor, J. T., Wehr, C. M., and Langlois, R. G. (1983), A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y, Mutation Res., 120, 269-275.
|
|
(63)
|
Hayashi, M., Sofuni, T. and Ishidate, M. Jr. (1983), An application of acridine orange fluorescent staining to the micronucleus test, Mutation Res., 120, 241-247.
|
|
(64)
|
Fenech, M., Chang, W.P., Kirsch-Volders, M., Holland, N., Bonassi, S. and Zeiger, E. (2003), HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures, Mutation Res., 534, 65-75.
|
|
(65)
|
Hoffman, W.P., Garriott, M.L. and Lee, C. (2003), In vitro micronucleus test, In: Encyclopedia of Biopharmaceutical Statistics, Second edition. S. Chow (ed.), Marcel Dekker, Inc. New York, NY, 463-467.
|
|
(66)
|
Az Európai Parlament és a Tanács 850/2004/EK rendelete (2004. április 29.) a környezetben tartósan megmaradó szerves szennyező anyagokról és a 79/117/EGK irányelv módosításáról, HL L 229., 2004.4.30., 5. o.
|
1. függelék
Fogalommeghatározások
Aneugén: olyan anyag vagy folyamat, amely a mitózissal vagy meiózissal történő sejtosztódási ciklus elemeivel kölcsönhatásba lépve a sejtek vagy organizmusok aneuploidiáját eredményezi.
Aneuploidia: bármilyen eltérés a normális diploid (vagy haploid) kromoszómaszámtól, amely lehet egy vagy több szám feletti vagy hiányzó kromoszóma, nem tartozik viszont ide a teljes kromoszómakészlet többszöröződése (poliploidia).
Apoptózis: programozott sejthalál olyan lépések során keresztül, amelyek a sejt membránhoz kötött részecskékre történő szétesését eredményezik; a részecskék ezután fagocitózissal és endocitózissal kerülnek eltávolításra.
Sejtproliferáció: a sejtek számának növekedése mitotikus sejtosztódással.
Centroméra: A kromoszóma DNS-ének azon része, ahol a két kromatida összeér, és amelyhez a két kinetokor oldalirányban kapcsolódik.
Klasztogén: bármilyen anyag vagy folyamat, amely a sejtek vagy organizmusok populációjában szerkezeti kromoszómakárosodásokat okoz.
Citokinezis: a mitózist közvetlenül követő sejtosztódási folyamat, amelynek eredményeként két utódsejt képződik egy-egy sejtmaggal.
Citokinezis-gátlási proliferációs index (CBPI): a kezelt populációban tapasztalható másodszor osztódó sejtek aránya a kezeletlen kontrollhoz képest (a képletet lásd a 2. függelékben).
Citosztázis: a sejtszaporodás gátlása (a képletet lásd a 2. függelékben).
Citotoxicitás: a sejt szerkezetét vagy működését károsító hatások, amelyek végső soron sejthalált idéznek elő.
Genotoxikus: általános kifejezés, amely magába foglalja a DNS- vagy kromoszómakárosodás valamennyi típusát, köztük a töréseket, adduktok képződését, átrendeződéseket, mutációkat, kromoszómaaberrációkat és aneuploidiát. A genotoxikus hatás nem minden típusa eredményez mutációkat vagy stabil kromoszómakárosodást.
Interfázisban lévő sejtek: azok a sejtek, amelyek nincsenek a mitózis fázisában.
Kinetokor: protein tartalmú struktúra, amely a kromoszóma centroméra részén épül fel, és amelyhez az orsórostok a sejtosztódás során kötődnek, lehetővé téve a leánykromoszómák szabályos mozgását az utódsejtek pólusaihoz.
Mikronukleuszok: kis magvak, amelyek a sejtmagtól külön, azon felül találhatók a sejtekben, és a mitózis vagy meiózis telofázisa során, lemaradó kromoszómatöredékek vagy teljes kromoszómák révén képződnek.
Mitózis: a sejtmag osztódása, amely általában profázisra, prometafázisra, metafázisra, anafázisra és telofázisra osztható fel.
Mitotikus index: a metafázisban lévő sejtek aránya osztva a sejtpopulációban megfigyelt sejtek összes számával; az adott populációban a sejtproliferáció fokát jelzi.
Mutagén: örökletes elváltozást idéz elő a génekben lévő DNS-bázispár szekvenciában vagy szekvenciákban vagy a kromoszómák szerkezetében (kromoszómaaberrációk).
Nondiszjunkció: a kromatidpárok nem válnak szét, és különülnek el a kifejlődő utódsejtekbe, rendellenes kromoszómaszámmal rendelkező utódsejteket eredményezve.
Poliploidia: ellentétben a csak egyetlen kromoszómát vagy bizonyos kromoszómákat érintő számbeli rendellenességekkel (aneuploidia), poliploidia esetében a teljes kromoszómakészlet(ek) számbeli rendellenessége áll fenn.
Proliferációs index (PI): a citotoxicitás mérésére használható módszer olyan esetekben, amikor nem alkalmaznak citoB-t (a képletet lásd a 2. függelékben).
A sejtszám relatív növekedése (RICC): a citotoxicitás mérésére használható módszer olyan esetekben, amikor nem alkalmaznak citoB-t (a képletet lásd a 2. függelékben).
Relatív populációduplázódás (RPD): a citotoxicitás mérésére használható módszer olyan esetekben, amikor nem alkalmaznak citoB-t (a képletet lásd a 2. függelékben).
Replikációs index (RI): a kezelt tenyészetben tapasztalható sejtciklusok aránya a kezeletlen kontrollhoz képest, az expozíciós időszakban és a helyreállás időszakában (a képletet lásd a 2. függelékben).
Vizsgált vegyi anyag (más néven vizsgált anyag): Bármely anyag vagy keverék, amelyet ezzel a vizsgálati módszerrel vizsgálnak.
2. függelék
A citotoxicitás értékelésére szolgáló képletek
|
1.
|
CitoB alkalmazása esetén a citotoxicitás értékelését a citokinezis-gátlási proliferációs index (CBPI) vagy replikációs index (RI) alapján kell végezni (16) (58). A CBPI a citoB-expozíció időszaka alatt megmutatja a sejtenkénti sejtciklusok átlagos számát, így alkalmazható a sejtproliferáció kiszámítására. Az RI a kezelt tenyészetekben a sejtmagok viszonylagos számát mutatja meg a kontroll tenyészetekhez képest, így alkalmazható a %-os citosztázis kiszámítására:
%-os citosztázis = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)}
valamint:
|
T
|
=
|
a vizsgált vegyi anyaggal kezelt tenyészet
|
|
C
|
=
|
vivőanyagos kontrolltenyészet
|
ahol:

Tehát az 1-es CBPI (az összes sejt egymagvú) 100 %-os citosztázisnak felel meg.
Citosztázis = 100 – RI

|
T
|
=
|
kezelt tenyészetek
|
|
C
|
=
|
kontrolltenyészetek
|
|
|
2.
|
Tehát az 53 %-os RI azt jelenti, hogy a kontroll tenyészetben kétmagvú és többmagvú sejtekké osztódott sejtek számához képest a kezelt tenyészetben ennek csak 53 %-a osztódott, vagyis a citosztázis 47 %-os.
|
|
3.
|
Amennyiben citoB-t nem alkalmaznak, a citotoxicitás értékelésére a sejtszám relatív növekedése (RICC) vagy a relatív populációduplázódás (RPD) ajánlott (58), mivel mindkét módszer figyelembe veszi az osztódott sejtpopuláció arányát.


ahol:
Populációduplázódás = [log (kezelés utáni sejtszám ÷ kiindulási sejtszám)] ÷ log 2
|
|
4.
|
Tehát az 53 %-os RICC vagy RPD 47 %-os citotoxicitást/citosztázist jelez.
|
|
5.
|
Proliferációs index (PI) alkalmazásával a citotoxicitás az 1 sejtből (cl1), 2 sejtből (cl2), 3-4 sejtből (cl4) és 5-8 sejtből (cl8) álló telepek megszámlálásával határozható meg.
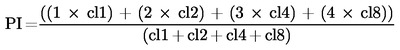
|
|
6.
|
A PI-t a citotoxicitás értékes és megbízható paramétereként alkalmazzák a citoB nélkül in situ tenyésztett sejtvonalak esetében is (25) (26) (27) (28).
|
3. függelék
A teljesítmény értékeléséhez ajánlott referenciaanyagok
(16)
|
Kategória
|
Vegyi anyag
|
CAS-szám
|
EU-szám
|
| 1. Metabolikus aktiváció nélkül is aktív klasztogének
|
|
|
Citozin-arabinozid
|
147-94-4
|
205-705-9
|
|
|
Mitomicin C
|
50-07-7
|
200-008-6
|
| 2. Metabolikus aktivációt igénylő klasztogének
|
|
|
Benzo(a)pirén
|
50-32-8
|
200-028-5
|
|
|
Ciklofoszfamid
|
50-18-0
|
200-015-4
|
| 3. Aneugén anyagok
|
|
|
Kolhicin
|
64-86-8
|
200-598-5
|
|
|
Vinblasztin
|
143-67-9
|
205-606-0
|
| 4. Negatív anyagok
|
|
|
Di-(2-etilhexil)-ftalát
|
117-81-7
|
204-211-0
|
|
|
Nalidixsav
|
389-08-2
|
206-864-7
|
|
|
Pirén
|
129-00-0
|
204-927-3
|
|
|
Nátrium-klorid
|
7647-14-5
|
231-598-3
|
B.50. BŐRSZENZIBILIZÁCIÓ: LOKÁLIS NYIROKCSOMÓ-VIZSGÁLATI MÓDSZER (LLNA): DA
BEVEZETÉS
|
1.
|
Az OECD vegyi anyagok vizsgálatára vonatkozó iránymutatásait és az uniós vizsgálati módszereket időnként felülvizsgálják a tudományos fejlődés, a változó szabályozási igények és állatjóléti megfontolások fényében. A bőrszenzitizáció egereknél történő meghatározására szolgáló első vizsgálati módszer (B.42.), a lokális nyirokcsomó-vizsgálat (LLNA; az OECD 429. vizsgálati iránymutatása) átdolgozásra került (1). Az LLNA hitelesítésének részleteit és az ezzel kapcsolatos munka áttekintését publikálták (2)(3)(4)(5)(6)(7)(8)(9). Az LLNA során a limfociták proliferációjának mérése radioizotópos timidin vagy jód alkalmazásával történik, ezért amennyiben a radioaktív anyagok beszerzése, használata vagy ártalmatlanítása problémát jelent, a vizsgálat csak korlátozottan alkalmazható. Az LLNA: DA (a Daicel Chemical Industries, Ltd. fejlesztése) az LLNA nem radioaktív változata, amely az adenozin-trifoszfát (ATP) tartalom mennyiségi meghatározását biolumineszcencia útján végzi, amely alkalmazható a limfocitaproliferáció indikátoraként. Az LLNA: DA vizsgálati módszert egy szakértőkből álló nemzetközi értékelő testület hitelesítette, vizsgálta felül és ajánlotta olyan módszerként, amely a bőrszenzitizációt okozó, illetve nem okozó vegyi anyagok meghatározására bizonyos megkötésekkel hasznosnak tekinthető (10) (11) (12) (13). A vizsgálati módszer célja a vegyi anyagok (anyagok és keverékek) bőrszenzibilizáló hatásának állatoknál történő felmérése. E melléklet B.6. fejezete és az OECD 406. vizsgálati iránymutatása tengerimalac-vizsgálatokat alkalmaz, nevezetesen a tengerimalac-maximizációs módszert és Bühler-vizsgálatot (14). Az LLNA (e melléklet B.42. fejezete; az OECD 429. vizsgálati iránymutatása) és a két nem radioaktív módosított eljárás, az LLNA: DA (e melléklet B.50. fejezete; az OECD 442. A. vizsgálati iránymutatása) és az LLNA: BrdU-ELISA (e melléklet B.51. fejezete; az OECD 442. B. vizsgálati iránymutatása) egyaránt előnyösebb a B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában (14) szereplő tengerimalac-vizsgálatnál abból a szempontból, hogy kevesebb állat használatát igényli és kíméletesebb.
|
|
2.
|
Az LLNA-hoz hasonlóan az LLNA: DA is a bőrszenzibilizáció indukciós fázisát vizsgálja, és a dózis-válasz értékelésére alkalmas mennyiségi adatokat szolgáltat. Emellett azáltal, hogy a bőrszenzibilizáló anyagokat a DNS radioaktív jelölése nélkül lehet kimutatni, a munka során kiküszöbölhető a radioaktivitásnak való kitettség, valamint a hulladék ártalmatlanításának problémája. Ez viszont az egerek nagyobb arányú használatát jelentheti a bőrszenzibilizáló anyagok kimutatására, ami a bőrszenzibilizáló képesség vizsgálata során tovább csökkenthetné a tengerimalacok használatát (pl. B.6. fejezet; az OECD 406. vizsgálati iránymutatása) (14).
|
FOGALOMMEGHATÁROZÁSOK
|
3.
|
Az alkalmazott fogalmak meghatározása az 1. függelékben található.
|
KIINDULÁSI MEGFONTOLÁSOK ÉS KORLÁTOK
|
4.
|
Az LLNA: DA egy módosított LLNA módszer, amely bizonyos korlátozások mellett a potenciális bőrszenzibilizáló vegyi anyagok azonosítására szolgál. Ez nem feltétlenül jelenti azt, hogy az LLNA vagy a tengerimalac-vizsgálat (lásd B.6. fejezet, az OECD 406. vizsgálati iránymutatása) (14) helyett minden esetben az LLNA: DA-t kell alkalmazni, inkább azt, hogy ez a vizsgálati módszer ugyanolyan hasznos, és olyan alternatív módszerként alkalmazható, amely esetében a pozitív és negatív eredmények általában már nem igényelnek további megerősítést (10) (11). A vizsgálat elvégzése előtt a vizsgáló laboratóriumnak a vizsgált anyagról rendelkezésre álló összes adatot figyelembe kell vennie. Ezen információk közé tartozik a vizsgált anyag azonosítása, kémiai szerkezete, fizikai-kémiai tulajdonságai, valamint a vizsgált anyaggal korábban végzett in vitro vagy in vivo toxicitási vizsgálatok eredményei és a szerkezetileg rokon vegyi anyagok toxikológiai adatai is. Mindezen információk figyelembevételével dönthető el, hogy az LLNA: DA módszer alkalmazható-e a vizsgált anyag esetében (tekintve, hogy bizonyos vegyianyag-típusoknál inkompatibilitás áll fenn [lásd az 5. bekezdést]), és milyen dózist célszerű választani a vizsgálatokhoz.
|
|
5.
|
Az LLNA: DA in vivo módszer, következésképpen nem küszöbölhető ki vele az állatok felhasználása az allergiás kontakt szenzibilizáló hatás vizsgálata során. Ugyanakkor a tengerimalac-vizsgálatokhoz képest (B.6. fejezet; az OECD 406. vizsgálati iránymutatása) lehetővé teszi az állatok ilyen célú felhasználásának csökkentését (14). Továbbá az állatok kontakt szenzibilizációs vizsgálatok során történő felhasználásának módját tekintve az LLNA: DA jelentősen kíméletesebb (kevesebb fájdalmat és szenvedést okoz), mivel a B.6. fejezet és az OECD 406. vizsgálati iránymutatásával ellentétben az LLNA: DA esetében nincs szükség indukált bőr-hiperszenzitivitási reakciók előidézésére. A B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában foglalt módszerrel (14) szembeni előnyei ellenére az LLNA: DA módszernek vannak bizonyos korlátai, amelyek szükségessé tehetik a B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában foglaltak alkalmazását (pl. bizonyos fémek vizsgálata, álpozitív eredmények bizonyos bőrirritáló anyagokkal [például bizonyos felületaktív anyagokkal] (6) [1, illetve e melléklet B.42. fejezete], vagy a vizsgált anyag oldhatósága). Továbbá az olyan funkcionális csoportokat tartalmazó anyagcsoportok, illetve anyagok, amelyek potenciális zavaró tényezőként szerepelnek (16) szükségessé tehetik a tengerimalac-vizsgálatok (vagyis a B.6. fejezet; az OECD 406. vizsgálati iránymutatása (14)) használatát. Az LLNA esetében megállapított korlátozásokat (1, illetve e melléklet B.42. fejezete) az LLNA: DA módszerre is ajánlott alkalmazni (10). Ezenkívül az LLNA: DA módszer nem feltétlenül alkalmas olyan anyagok vizsgálatára, amelyek befolyásolják az ATP szintjét (például ATP-gátlóként ható anyagok) vagy az intracelluláris ATP pontos mérését (például az ATP-t bontó enzimek jelenléte vagy extracelluláris ATP jelenléte a nyirokcsomóban). E meghatározott hiányosságokon kívül, az LLNA: DA bármilyen anyag vizsgálatára megfelelő, kivéve, ha ezek az anyagok olyan tulajdonságokkal rendelkeznek, amelyek befolyásolják az LLNA: DA pontosságát. Emellett abban az esetben, ha a stimulációs indexre (SI) kapott érték 1,8 és 2,5 közé esik figyelembe kell venni a határértéken lévő pozitív eredmények lehetőségét is (lásd a 31–32. bekezdést). Ez egy legalább 1,8 értékű stimulációs indexszel rendelkező, 44 anyagot magában foglaló hitelesítési adatbázison alapul (lásd a 6. bekezdést), amely esetében az LLNA: DA módszer helyesen azonosította mind a 32, az LLNA módszer szerint szenzibilizáló anyagot, de hibásan azonosított három anyagot az LLNA módszerrel nem szenzibilizálóként meghatározott, 1,8 és 2,5 közötti SI-értékkel (vagyis határértéken lévő pozitív) rendelkező 12 anyagok közül (10). Mivel azonban az SI-értékek beállításához és a vizsgálat prediktív jellemzőinek kiszámításához ugyanazt az adatállományt alkalmazták, a megadott eredmények túlbecsülhetik a valós prediktív jellemzőket.
|
A VIZSGÁLATI MÓDSZER ELVE
|
6.
|
Az LLNA: DA módszer hátterében az az alapelv áll, hogy a szenzibilizáló anyagok elsődleges limfocitaproliferációt indukálnak a vizsgált anyag alkalmazási területének nyirokelvezetéséről gondoskodó nyirokcsomókban. Ez a proliferáció arányos az alkalmazott allergén dózisával és erősségével, valamint egyszerű módszert biztosít a szenzibilizáció kvantitatív méréséhez. A proliferációt úgy mérik, hogy a vizsgálati csoportokban mért átlagos proliferációt összehasonlítják a vivőanyaggal kezelt csoportban mért proliferációval. Meghatározzák az egyes kezelt csoportokban és a párhuzamosan vivőanyaggal kezelt kontrollcsoportokban mért átlagos proliferáció arányát, amelyet stimulációs indexnek (SI) neveznek, és amelynek legalább ≥ 1,8-nek kell lennie ahhoz, hogy indokolt legyen a vizsgált anyag potenciális bőrszenzibilizáló anyagként történő további vizsgálata. Az itt leírt eljárások az ATP tartalom biolumineszcenciával történő mérésén alapulnak (az ATP-ről ismert, hogy korrelál az élő sejtek számával) (17), amely az elvezető aurikuláris nyirokcsomókban lévő proliferáló sejtek számának növekedését jelzi (18) (19). A biolumineszcens módszer során alkalmazott luciferáz enzim ATP-ből és luciferinből fény képződését katalizálja a következő reakció lezajlásával:
ATP + luciferin + O
2
 Oxiluciferin + AMP + PPi
+ CO
2 + fény
Oxiluciferin + AMP + PPi
+ CO
2 + fény
A kibocsátott fény intenzitása lineáris összefüggést mutat az ATP koncentrációjával, és luminométerrel mérhető. A luciferin-luciferáz vizsgálat széles körében alkalmazott, érzékeny módszer az ATP mennyiségi meghatározására (20).
|
A VIZSGÁLAT LEÍRÁSA
Az állatfaj kiválasztása
|
7.
|
Ehhez a vizsgálathoz az egér a választandó faj. A LLNA: DA módszerhez a hitelesítési vizsgálatokat kizárólag a CBA/J törzzsel végezték, amely ezért preferált törzsnek tekintendő (12) (13). Fiatal felnőtt nullipara és nem vemhes nőstény egereket kell használni. A vizsgálat elején az állatok korának 8-12 hétnek kell lennie, az állatok testtömege között az eltérésnek minimálisnak kell lennie, és a testtömeg nem haladhatja meg az átlagos testtömeg 20 %-át. Ha elegendő adat áll rendelkezésre arra nézve, hogy az LLNA: DA módszerrel kapott válaszreakcióban nincsenek jelentős törzs- és/vagy ivarspecifikus különbségek, akkor más törzsek, illetve hím állatok is alkalmazhatók.
|
Az állatok tartásának és etetésének körülményei
|
8.
|
Az egereket csoportosan kell tartani (21), kivéve, ha az egyedenkénti tartásra megfelelő tudományos indok merül fel. A kísérleti állatok tartásául szolgáló helyiség hőmérsékletének 22 °C ± 3 °C-nak kell lennie. Noha a helyiség relatív páratartalmának legalább 30 %-nak kell lennie, és – a takarítás időtartamától eltekintve – lehetőség szerint nem haladhatja meg a 70 %-ot, a célértéknek 50–60 % között kell lennie. A világítás legyen mesterséges, 12 órás világos, 12 órás sötét periódusok váltakozásával. Az etetéshez a szokásos laboratóriumi takarmány alkalmazható korlátlan mennyiségű ivóvíz biztosítása mellett.
|
Az állatok előkészítése
|
9.
|
Az állatokat véletlenszerűen választják ki, majd egyedi azonosítóval látják el (viszont az állat fülén semmiféle azonosító nem alkalmazható), és a kezelés megkezdése előtt legalább 5 napig a ketrecükben tartják őket, hogy hozzászokhassanak a laboratóriumi körülményekhez. A kezelés megkezdése előtt minden állatot megvizsgálnak, hogy meggyőződjenek arról, nincs rajtuk látható bőrsérülés.
|
A dózisok előkészítése
|
10.
|
A szilárd halmazállapotú vegyi anyagokat az egér fülén történő alkalmazás előtt oldószerben/vivőanyagban kell feloldani vagy szuszpendálni, és szükség esetén hígítani kell. A folyadék halmazállapotú vegyi anyagokat hígítatlanul vagy az adagolás előtt hígított formában lehet alkalmazni. A nem oldható vegyi anyagokat, például az orvosi eszközökben általánosan előforduló anyagokat megfelelő oldószerben történő agresszív extrakciónak kell kitenni, hogy a vizsgálat során az egér fülén való alkalmazás előtt az összes kivonható összetevőt feltárjuk. A vizsgált anyagokat naponta kell elkészíteni, kivéve, ha stabilitási adatok igazolják, hogy a tárolás elfogadható.
|
A megbízhatóság ellenőrzése
|
11.
|
A vizsgálat megfelelő teljesítményének megfelelő és reprodukálható szenzitivitással adott válaszreakciókkal történő igazolására pozitív kontrollanyagokat (PK) alkalmaznak olyan szenzibilizáló tesztanyagként, amelynél a válaszreakció mértéke pontosan meghatározott. Pozitív kontroll párhuzamos vizsgálata ajánlott, mert ez igazolja, hogy a laboratórium megfelelő szakértelemmel végzi az egyes vizsgálatokat, és lehetővé teszi a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság és összehasonlíthatóság értékelését. Néhány szabályozó hatóság szintén előírja a pozitív kontroll minden vizsgálat során történő alkalmazását, ezért az LLNA: DA módszer elvégzése előtt ajánlott egyeztetni az illetékes hatóságokkal. Ennek megfelelően javasolt a pozitív kontroll rutinszerű párhuzamos vizsgálata, hogy elkerülhetőek legyenek az olyan további állatkísérletek, amelyeket a pozitív kontrollt csak időszakosan alkalmazó laboratórium az előírásoknak való megfelelés érdekében végezne (lásd a 12. bekezdést). A pozitív kontrollnak az LLNA: DA módszerrel olyan expozíciós szint mellett kell pozitív válaszreakciót adnia, amely a negatív kontrollcsoporthoz (NK) képest várhatóan legalább 1,8-del növeli a stimulációs indexet (SI). A pozitív kontroll dózisát úgy kell megválasztani, hogy ne okozzon túlzott bőrirritációt vagy szisztémás toxicitást, és az indukció reprodukálható, de ne túl nagymértékű legyen (például SI > 10 már túlzottnak tekinthető). Az előnyben részesítendő pozitív kontrollanyagok a 25 %-os hexil-cinnemaldehid (Chemical Abstracts Service [CAS] szám: 101-86-0) és a 25 %-os eugenol (CAS-szám: 97-53-0) aceton:olívaolaj (4:1, v/v) arányú keverékében. Adódhatnak olyan körülmények, amelyek fennállásakor, megfelelő indoklás esetén a fenti kritériumoknak megfelelő más pozitív kontrollanyagok is alkalmazhatók.
|
|
12.
|
Bár ajánlott a pozitív kontrollcsoport párhuzamos vizsgálata, lehetnek azonban olyan helyzetek, amikor a pozitív kontroll időszakos (például legalább 6 havonta végzett) vizsgálata elfogadható lehet az LLNA: DA vizsgálatot rendszeresen (vagyis legalább havonta egyszeri gyakorisággal) végző laboratóriumok esetében, amelyek rendelkeznek pozitív kontrollok igazolt, historikus adatbázisával, és ezzel bizonyítható, hogy a laboratórium a pozitív kontrollokkal képes reprodukálható és pontos eredményeket előállítani. Az LLNA: DA módszerben való jártasság alátámasztható a pozitív kontrollal megfelelő időszakon belül (kevesebb mint egy éve) végzett legalább 10 független vizsgálat során kapott következetesen pozitív eredményekkel.
|
|
13.
|
Pozitív kontrollcsoport párhuzamos vizsgálata minden alkalommal szükséges, valahányszor az LLNA: DA vizsgálati módszerében változtatást hajtanak végre (például a képzett személyzet, a vizsgálati eljárás során használt anyagok és/vagy reagensek, a vizsgálati eszközök, a kísérleti állatok beszerzési forrásának megváltoztatása), és ezeket a változásokat dokumentálni kell a laboratóriumi jelentésekben. Figyelembe kell venni, hogy ezek a változtatások mennyiben befolyásolják a korábban létrehozott historikus adatbázis megfelelőségét, és el kell dönteni, hogy szükséges-e új historikus adatbázis felállítása a pozitív kontroll eredményei következetességének dokumentálása érdekében.
|
|
14.
|
A vizsgálatot végző személyeknek tisztában kell lenniük azzal, hogy amennyiben a párhuzamos vizsgálat helyett a pozitív kontrollok időszakos vizsgálata mellett döntenek, ez hatással lehet a pozitív kontrollok időszakos vizsgálatai közötti időszakokban, párhuzamos pozitív kontroll vizsgálata nélkül előállított negatív vizsgálati eredmények megfelelőségére és elfogadhatóságára. Ha például a pozitív kontrollal végzett időszakos vizsgálat álnegatív eredményt adott, akkor megkérdőjelezhetők azok a negatív eredmények, amelyek az utolsó elfogadható eredményű és az elfogadhatatlan eredményű időszakos pozitív kontroll vizsgálat között eltelt időszakban keletkeztek. Az ilyen következmények jelentőségét körültekintően mérlegelni kell annak eldöntésekor, hogy pozitív kontrollokat párhuzamosan vagy csak időszakosan vizsgáljanak-e. Szintén megfontolandó a kevesebb állat használata a párhuzamos pozitív kontrollcsoportban, amennyiben ez tudományosan indokolt, és a laboratóriumra specifikus historikus adatok alapján a laboratórium igazolja, hogy megfelelő a kevesebb egér használata (22).
|
|
15.
|
Bár a pozitív kontrollt olyan vivőanyagban kell vizsgálni, amelyről ismert, hogy következetes válaszreakciót vált ki (például aceton:olívaolaj; 4:1, v/v arányú keveréke), lehetnek olyan szabályozási helyzetek, amikor nem szokványos vivőanyagokban (klinikailag/kémiailag releváns készítmény) történő vizsgálat elvégzésére is szükség van (23). Amennyiben a párhuzamos pozitív kontrollt a vizsgált anyagétól eltérő vivőanyagban vizsgálják, akkor a párhuzamos pozitív kontrollal együtt egy külön vivőanyagos kontrollt is vizsgálni kell.
|
|
16.
|
Azokban az estekben, amikor egy bizonyos vegyi csoportba tartozó anyagokat vagy a válaszreakciók tartományát értékelik, szintén hasznos lehet a referenciaanyagok alkalmazása annak igazolására, hogy a vizsgálati módszer megfelelően működik az ilyen típusú anyagok bőrszenzibilizáló hatásának kimutatására. A megfelelő referenciaanyagoknak a következő tulajdonságokkal kell rendelkezniük:
|
—
|
szerkezeti és funkcionális hasonlóság a vizsgált anyag csoportjával,
|
|
—
|
ismert fizikai/kémiai tulajdonságok,
|
|
—
|
az LLNA: DA módszerrel kapott alátámasztó adatok,
|
|
—
|
egyéb állatmodellekből és/vagy emberből származó alátámasztó adatok.
|
|
VIZSGÁLATI ELJÁRÁS
Az állatok száma és a dózisszintek
|
17.
|
Minden dóziscsoportban legalább négy állatot kell használni, és a vizsgált anyagból legalább három különböző koncentrációt, továbbá párhuzamos negatív kontrollcsoportot, amelyet csak a vizsgált anyaghoz használt vivőanyaggal kezelnek, valamint egy pozitív kontroll csoportot kell alkalmazni (pozitív kontroll párhuzamos vagy a közelmúltban végzett vizsgálata a laboratórium szabályzatának megfelelően, figyelembe véve a 11–15. bekezdésben foglaltakat). Megfontolandó a pozitív kontroll többféle dózisának vizsgálata, különösen, ha a pozitív kontroll vizsgálatát időszakosan végzik. A kontrollcsoportban lévő állatokkal ugyanúgy kell bánni és dolgozni, mint a kezelt csoportokban lévőkkel, azzal a kivétellel, hogy az állatokat nem kezelik a vizsgált anyaggal.
|
|
18.
|
A dózist és a vivőanyagot a (2) és (24) számú hivatkozásokban megadott ajánlások alapján kell kiválasztani. Az egymás utáni dózisok kiválasztása általában megfelelő koncentrációsorból, például 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % stb. történik. Az alkalmazott koncentrációsort megfelelő tudományos megfontolások alapján kell kiválasztani. A három egymás utáni koncentráció kiválasztásakor figyelembe kell venni a vizsgált anyagról esetlegesen rendelkezésre álló valamennyi toxikológiai információt (például akut toxicitás és bőrirritáció), valamint szerkezeti és fizikai-kémiai adatot, hogy a legmagasabb koncentrációval a lehető legnagyobb expozíciót idézzük elő, amely még nem okoz szisztémás toxicitást és/vagy túlzott mértékű lokális bőrirritációt (24) (25). Ilyen jellegű információk hiányában szükséges lehet a megelőző előszűrő vizsgálat (lásd 21–24. bekezdés).
|
|
19.
|
A vivőanyagnak nem szabad megzavarnia vagy befolyásolnia a vizsgálat eredményét, és aszerint kell kiválasztani, hogy a vizsgált anyag oldhatósága a lehető legnagyobb koncentráció elérése érdekében maximális legyen, ugyanakkor a vizsgált anyag felvitelére alkalmas oldatot/szuszpenziót kapjunk. A javasolt vivőanyagok az aceton:olívaolaj (4:1 v/v) keveréke, N,N-dimetilformamid, metil-etil-keton, propilén-glikol és dimetil-szulfoxid (6), de megfelelő tudományos indoklás esetén más vivőanyagok is alkalmazhatók. Bizonyos esetekben további kontrollként szükséges lehet valamely klinikai szempontból releváns oldószer használata, vagy annak a kereskedelmi formulának az alkalmazása, amelyben a vizsgált anyagot forgalomba hozzák. Különösen vigyázni kell arra, hogy megfelelő szolubilizáló szerek (pl. 1 % Pluronic® L92) alkalmazásával a hidrofil anyagokat olyan vivőanyag-rendszerbe építsük be, amely benedvesíti a bőrt, de nem folyik le azonnal. A teljesen vizes alapú vivőanyagokat tehát kerülni kell.
|
|
20.
|
A nyirokcsomók egyedenként történő feldolgozása lehetővé teszi az egyedek közötti variabilitás, valamint a vizsgált anyaggal és a vivőanyaggal kezelt csoport eredményeinek statisztikai összehasonlítását (lásd a 33. bekezdést). Emellett csak akkor lehet értékelni, hogy lehetséges-e csökkenteni a pozitív kontrollcsoportban lévő egerek számát, ha az egyedi állatokra vonatkozóan gyűjtünk adatokat (22). Ezen túlmenően, egyes szabályozó hatóságok is megkövetelik az egyedi állatokon alapuló adatgyűjtést. Az egyedi állatokon alapuló adatgyűjtés rendszeresítése állatjóléti előnyökkel jár azáltal, hogy elkerülhető a vizsgálatok megismétlése abban az esetben, amikor a vizsgált anyagról eredetileg csak egyféleképpen gyűjtötték az adatokat (pl. összevont állatkísérletes adatok), míg a szabályozó hatóság később eltérő követelményeket állít fel (pl. egyedi állatokon alapuló adatok).
|
Előszűrő vizsgálat
|
21.
|
A legmagasabb vizsgálandó dózis meghatározását lehetővé tevő információ hiányában (lásd a 18. bekezdést) az LLNA: DA módszerhez alkalmazandó megfelelő dózisszint megállapítása érdekében előszűrő vizsgálatot kell végezni. Az előszűrő vizsgálat célja, hogy támpontot adjon az LLNA: DA vizsgálat során alkalmazandó maximális dózis kiválasztásához, amennyiben a szisztémás toxicitást (lásd a 24. bekezdést) és/vagy nagyfokú bőrirritációt (lásd a 23. bekezdést) okozó koncentrációra vonatkozó információk nem állnak rendelkezésre. Folyadékok esetében a legmagasabb vizsgált dózisnak 100 %-os koncentrációnak kell lennie, szilárd anyagok vagy szuszpenziók esetében pedig a lehetséges maximális koncentrációnak.
|
|
22.
|
Az előszűrő vizsgálatot a fő LLNA: DA vizsgálathoz hasonló körülmények között kell végezni, eltekintve attól, hogy nem kell a nyirokcsomó proliferációt értékelni, valamint dóziscsoportonként kevesebb állat használható. Dóziscsoportonként egy vagy két állat javasolt. Az egereket a szisztémás toxicitás, illetve az alkalmazás helyén fellépő helyi irritáció jeleinek szempontjából naponta megfigyelik. A testtömeget a vizsgálat előtt és az állat exterminálása előtt is (8. nap) feljegyzik. A bőrpírt az egerek mindkét fülén vizsgálják és pontozzák az 1. táblázat (25) szerint. A fülvastagság értékeit egy vastagságmérővel (például digitális mikrométer, vagy Peacock Dial vastagságmérő) állapítják meg az 1. napon (az adagolás előtt), a 3. napon (hozzávetőleg 48 órával az első dózis után), valamint a 7. napon (az exterminálás előtt 24 órával) és a 8. napon. A 8. napon a fülvastagságot ezen kívül a fülből lyukasztással eltávolított szövet tömegének meghatározásával is meg lehet állapítani, amit az állat exterminálása után kell elvégezni. Nagyfokú helyi irritációnak minősül a legalább ≥ 3-as pontszámú bőrpír és/vagy legalább ≥ 25 %-os fülvastagság a vizsgálat bármely napján (26) (27). A fő LLNA: DA vizsgálathoz azt a dózist kell maximális dózisként választani, amely az előszűrés során alkalmazott koncentrációsorban (lásd a 18. bekezdést) a szisztémás toxicitást és/vagy nagyfokú helyi bőrirritációt okozó koncentrációnál eggyel alacsonyabb.
1. táblázat
Bőrpír (erythema) pontszámai
|
Megfigyelés
|
Pontszám
|
|
Nincs erythema
|
0
|
|
Nagyon enyhe erythema (alig észlelhető)
|
1
|
|
Jól körülírt erythema
|
2
|
|
Közepes-súlyos erythema
|
3
|
|
Súlyos (céklavörös elszíneződés), pörkképződéig terjedő erythema, amely lehetetlenné teszi az erythema besorolását
|
4
|
|
|
23.
|
A 25 %-os fülvastagság-növekedésen felül (26) (27), a bőrirritáló anyagok LLNA módszerrel végzett kimutatásához a kezelt állatok fülvastagságának kontrollcsoporthoz viszonyított statisztikailag szignifikáns növekedését is használják (28) (29) (30) (31) (32) (33) (34). Noha 25 % alatti fülvastagság esetén előfordulhat statisztikailag szignifikáns növekedés, a nagyfokú irritációval azonban ezt különösen nem hozták összefüggésbe (30) (31) (32) (33) (34).
|
|
24.
|
Az integrált értékelés részeként alkalmazva a következő klinikai tünetek jelezhetnek szisztémás toxicitást (35), és ezáltal kijelölhetik a fő LLNA: DA vizsgálat során alkalmazandó maximális dózist: az idegrendszer működésében bekövetkező változások (például piloerekció, ataxia, tremorok és görcsrohamok); a viselkedésben bekövetkező változások (például agresszivitás, a tisztálkodási tevékenységekben bekövetkező változás, az aktivitási szint jelentős változása); a légzési mintázat változása (például a légvételek gyakoriságában és az intenzitásában bekövetkező változás, úgymint dyspnoe, zihálás és szörtyzörejek), valamint változások az táplálék- és vízfogyasztásban. Továbbá az értékelés során figyelembe kell venni a letargia és/vagy reakciókészség hiányának jeleit, valamint az enyhénél és pillanatnyinál fokozottabb fájdalom vagy szenvedés bármilyen klinikai tünetét, az > 5 %-nál nagyobb arányú testtömeg-csökkenést az 1. naptól a 8. napig tartó időszakban, valamint a mortalitást. Az elhullásközeli állapotban lévõ vagy súlyos fájdalmak és szenvedés jeleit mutató állatokat humánus módon exterminálni kell (36).
|
A fő vizsgálat kísérleti ütemterve
|
25.
|
A vizsgálat kísérleti ütemterve a következő:
— 1. nap: Külön meg kell határozni, és fel kell jegyezni mindegyik állat testtömegét és az esetleges klinikai megfigyeléseket. Vigyük fel 1 % nátrium-lauril-szulfát (SLS) vizes oldatát mindkét fül dorzális oldalára egy SLS-oldatba mártott kefével, és négy-öt húzással be kell fedni a fülek teljes dorzális felszínét. Az SLS-tal végzett kezelés után egy órával vigyünk fel a vizsgált anyag megfelelő hígításából 25 μl-t, valamint a vivőanyagot önmagában, illetve pozitív kontrollt (pozitív kontroll párhuzamos vagy a közelmúltban végzett vizsgálata a laboratórium szabályzatának megfelelően, figyelembe véve a 11–15. bekezdésekben foglaltakat) a fülek dorzális oldalára.
— 2., 3. és 7. nap: Az 1. napnak megfelelően ismételjük meg az SLS 1 %-os vizes oldatával végzett előkezelést, valamint a vizsgált anyag felviteli eljárását.
— 4., 5. és 6. nap: Nincs kezelés.
— 8. nap: Fel kell jegyezni minden állat testtömegét és minden klinikai megfigyelést. Az állatokat az alkalmazás megkezdése után körülbelül 24–30 órával a 7. napon humánus módon extermináljuk. Mindegyik egér füléből vágjuk ki a kezelt terület nyirokelvezetéséről gondoskodó füli nyirokcsomókat, és tegyük be azokat állatonként külön-külön foszfáttal pufferelt sóoldatba (PBS). A nyirokcsomók azonosításának és preparálásának részletei és az ehhez kapcsolódó ábrák az (22) hivatkozásban találhatók. A helyi bőrreakció fő vizsgálatban történő további monitorozásához olyan további paramétereket lehet belevenni a vizsgálati protokollba, mint például a bőrpír pontozása vagy a fülvastagság mérése (akár vastagságmérővel, akár elölés utáni füldarabsúly-meghatározással).
|
A sejtszuszpenziók elkészítése
|
26.
|
Mindegyik egér esetében el kell készíteni a két oldalról kimetszett nyirokcsomó-sejtek (LNC) különálló sejtekből álló szuszpenzióját; ehhez a nyirokcsomót két üveg tárgylemez közé kell helyezni, és a nyirokcsomók összetöréséhez enyhe nyomást kell rá gyakorolni. Miután meggyőződtünk róla, hogy a szövet vékony rétegben szétterjedt, válasszuk szét a két tárgylemezt. Szuszpendáljuk a két tárgylemezen található szövetet PBS-ben; ehhez a tárgylemezeket Petri-csésze fölött ferdén tartva öblítsük le PBS-oldattal, eközben sejtkaparóval kaparjuk le a szövetet a tárgylemezről. A negatív kontrollként szolgáló állatok nyirokcsomói kisméretűek, ezért az SI-értékre gyakorolt mesterséges hatások elkerülése érdekében fontos az elővigyázatos kezelés. A két tárgylemez leöblítéséhez összesen 1 ml térfogatú PBS-oldatot kell használni. A Petri-csészében lévő LNC-szuszpenziót a sejtkaparóval enyhén homogenizálni kell. Ezután az LNC-szuszpenzióból mikropipettával 20 μl-nyi adagot fel kell szívni, ügyelve arra, hogy szemmel látható membránt ne szívjunk fel, majd el kell keverni 1,98 ml PBS-oldattal, hogy 2 ml-nyi mintát kapjunk. Ezután ugyanilyen eljárással újabb 2 ml-es mintát kell készíteni, hogy minden állat esetében két minta álljon rendelkezésre.
|
A sejtproliferáció meghatározása (a limfociták ATP-tartalmának mérése)
|
27.
|
A nyirokcsomók ATP-tartalmában bekövetkezett növekedést a luciferin/luciferáz módszerrel ATP-mérési készlet segítségével határozzuk meg, amely a biolumineszcenciát relatív lumineszcencia egységekben (RLU) méri. A vizsgálat idejének az állat exterminálásától az ATP-tartalom méréséig mindegyik állat esetében azonosnak, körülbelül 30 percesnek kell lennie, ugyanis az ATP tartalom az állat exterminálása után idővel feltételezhetően fokozatosan csökken (12). Tehát az eljárások sorát az aurikuláris nyirokcsomók kimetszésétől az ATP méréséig, az előre meghatározott, mindegyik állat esetében azonos ütemterv szerint 20 percen belül el kell végezni. Az ATP-lumineszcenciát mindegyik 2 ml-es mintában mérni kell, hogy minden állat esetében összesen két ATP mérési eredmény álljon rendelkezésre. Ezután meghatározzuk az átlagos ATP-lumeneszcenciát, és a további számításokhoz ezt alkalmazzuk (lásd a 30. bekezdést).
|
MEGFIGYELÉSEK
Klinikai megfigyelések
|
28.
|
Naponta legalább egyszer minden egérnél meg kell vizsgálni az alkalmazás helyén kialakuló lokális vagy szisztémás toxicitás esetleges tüneteit. Minden megfigyelést szisztematikusan fel kell jegyezni minden egyes egér esetében, egyedi adatsor felvételével. A monitoring tervnek tartalmaznia kell azon kritériumokat, amelyek alapján azonnal azonosíthatóak az eutanáziát igénylő, szisztémás toxicitást, kiterjedt helyi bőrirritációt vagy bőrkorróziót mutató egerek (36).
|
Testtömeg
|
29.
|
Ahogyan az a 25. bekezdésben szerepel, az egyes állatok testtömegét a vizsgálat kezdetén, valamint a humánusan történő exterminálás tervezett időpontjában kell megmérni.
|
AZ EREDMÉNYEK KISZÁMÍTÁSA
|
30.
|
Az egyes kezelési csoportokra vonatkozó eredményeket átlagos stimulációs indexben (SI) fejezzük ki. Az SI kiszámítása úgy történik, hogy a vizsgált anyaggal kezelt egyes csoportokban és a pozitív kontrollcsoportban kapott állatonkénti átlagos RLU-értéket el kell osztani az oldószerrel/vivőanyaggal kezelt kontrollcsoport átlagos állatonkénti RLU-értékével. A vivőanyaggal kezelt kontrollcsoportok átlagos SI-értéke tehát 1.
|
|
31.
|
A válaszreakciót akkor lehet pozitívan értékelni, ha a stimulációs index ≥ 1,8 (10). Amikor azonban azt kell meghatározni, hogy egy határértéken lévő eredmény (vagyis amikor az SI-érték 1,8 és 2,5 között van) pozitívnak tekintendő-e, akkor a dózis-válasz összefüggés erőssége, a statisztikai szignifikancia és az oldószerrel/vivőanyaggal, valamint a pozitív kontrollal kapott válaszreakciók következetessége is felhasználható (2) (3) (37).
|
|
32.
|
Az 1,8 és 2,5 közötti SI-vel járó, határértéken lévő pozitív válaszreakciók esetében a felhasználók dönthetnek úgy, hogy az eredmények pozitivitásának megerősítéséhez további információkat – például a dózis-válasz összefüggést, szisztémás toxicitás vagy nagyfokú irritáció jelét, illetve adott esetben a statisztikai szignifikanciát az SI értékekkel együtt – kívánnak figyelembe venni (10). Figyelembe kell venni a vizsgált anyag különféle tulajdonságait is, köztük azt, hogy mutat-e szerkezeti rokonságot ismert bőrszenzibilizáló anyagokkal, okoz-e nagyfokú bőrirritációt egereknél, illetve a megfigyelt dózis-válasz összefüggés jellegét. Ezek, illetve további megfontolások részletesebb tárgyalását lásd a (4) hivatkozásban.
|
|
33.
|
Az egyedi állatokon alapuló adatgyűjtés lehetővé teszi a dózis-válasz összefüggés adatokban tükröződő fennállásának és mértékének statisztikai elemzését. Bármely statisztikai elemzés tartalmazhatja a dózis-válasz összefüggés értékelését, valamint a vizsgálati csoportok megfelelően korrigált összehasonlításait (például a dóziscsoportok páronkénti összehasonlítása a párhuzamos oldószeres/vivőanyagos kontrollcsoportokkal). A statisztikai elemzésekben a dózis-válasz tendenciák értékelésénél szerepelhet például lineáris regresszió vagy William-féle próba, a páronkénti összehasonlításoknál pedig Dunnett-féle próba. A megfelelő statisztikai elemzési módszer kiválasztásakor a vizsgálónak figyelemmel kell lennie a lehetséges varianciaegyenlőtlenségekre és más kapcsolódó problémákra, amelyek miatt az adatok transzformációjára vagy nem paraméteres statisztikai elemzésre lehet szükség. A vizsgálónak minden esetben el kell végeznie az SI-k kiszámítását és statisztikai elemzéseket bizonyos adatpontokkal (az úgynevezett »kiugró adatokkal«) és azok nélkül.
|
ADATOK ÉS JELENTÉS
Adatok
|
34.
|
Az adatokat táblázatos formában kell összefoglalni, feltüntetve az egyedi állatok RLU-értékeit, a csoport átlagos RLU/állat értékét, a hozzá tartozó hibamutatóval (például SD, SEM), valamint az egyes dóziscsoportokban kapott átlagos stimulációs indexeket (SI) összehasonlítva a párhuzamos oldószeres/vivőanyagos kontrollcsoport értékeivel.
|
Vizsgálati jelentés
|
35.
|
A vizsgálati jelentésnek a következő információkat kell tartalmaznia:
|
|
Vizsgált anyagok és kontrollanyagok:
|
—
|
azonosító adatok (pl. adott esetben a CAS-szám és az EU-szám; eredet; tisztaság; ismert szennyezők; tételszám),
|
|
—
|
fizikai megjelenés és fizikai-kémiai tulajdonságok (pl. illékonyság, stabilitás, oldhatóság),
|
|
—
|
elegy esetén annak összetétele és az egyes komponensek egymáshoz viszonyított aránya,
|
|
|
|
Oldószer/vivőanyag:
|
—
|
azonosító adatok (adott esetben tisztaság; koncentráció; alkalmazott térfogat),
|
|
—
|
a vivőanyag kiválasztásának indoklása,
|
|
|
|
Kísérleti állatok:
|
—
|
CBA egértörzs származása,
|
|
—
|
az állatok mikrobiológiai státusa, amennyiben ismert,
|
|
—
|
az állatok száma és életkora,
|
|
—
|
az állatok származása, tartásának körülményei, takarmánya stb.,
|
|
|
|
Vizsgálati körülmények:
|
—
|
az ATP készlet származása, tételszáma, a gyártó minőségbiztosítási/minőség-ellenőrzési adatai,
|
|
—
|
a vizsgált anyagból előállított készítménnyel és annak alkalmazásával kapcsolatos adatok,
|
|
—
|
a dózisszintek megválasztásának indoklása (ezen belül adott esetben az előszűrési vizsgálatok eredményei),
|
|
—
|
a vivőanyag és a vizsgált anyag alkalmazott koncentrációja, valamint a vizsgált anyagból alkalmazott teljes mennyiség,
|
|
—
|
a táplálék és a víz minőségére vonatkozó adatok (ezen belül a takarmány típusa/eredete, a víz eredete),
|
|
—
|
a kezelés adatai és a mintavételek ütemezése,
|
|
—
|
a toxicitás mérésére alkalmazott módszerek,
|
|
—
|
a pozitív és negatív vizsgálati eredmény kimondásának kritériumai,
|
|
—
|
a protokolltól való esetleges eltérések adatai, és annak kifejtése, hogy az eltérés hogyan érintheti vizsgálati tervet és az eredményeket,
|
|
|
|
A megbízhatóság ellenőrzése:
|
—
|
a legutóbbi megbízhatósági ellenőrzés eredményeinek összefoglalása az alkalmazott anyag, koncentráció és vivőanyag adataival együtt,
|
|
—
|
a vizsgálólaboratórium párhuzamosan és/vagy korábban vizsgált pozitív és párhuzamosan vizsgált negatív (oldószer/vivőanyag) kontrollokra vonatkozó adatai,
|
|
—
|
ha nem vizsgáltak párhuzamosan pozitív kontrollt, a laboratóriumban legutóbb végzett időszakos pozitív kontroll dátuma és laboratóriumi jelentése, valamint pozitív kontroll vizsgálatának korábbi adatait részletező jelentés, amely indokolja, hogy miért nem végzik a laboratóriumban pozitív kontroll párhuzamos vizsgálatát,
|
|
|
|
Eredmények:
|
—
|
az egyes állatok testtömege a kezelés kezdetén és az exterminálás tervezett időpontjában, illetve az átlagos és vonatkozó hibamutatók (pl. SD, SEM) kezelési csoportonként,
|
|
—
|
a toxicitási tünetek, ezen belül az alkalmazás helyén esetleg kialakuló bőrirritáció megjelenésének időpontja és időbeli alakulása minden egyes állatra vonatkozóan,
|
|
—
|
az állat exterminálásának időpontja és az ATP mérésének időpontja az egyes állatok esetében,
|
|
—
|
az állatonkénti RLU-értékek és dóziscsoportonkénti SI-értékek táblázata,
|
|
—
|
átlagos és kapcsolódó hibamutatók (pl. SD, SEM) az állatonkénti RLU-értékek esetében, az egyes kezelés csoportokban, és a kiugró értékek elemzése az egyes kezelési csoportokban,
|
|
—
|
a kiszámított SI és a variabilitás megfelelő mutatója, amely az egyedek közti variabilitást a vizsgált anyaggal kezelt csoportban és a kontrollcsoportokban egyaránt figyelembe veszi,
|
|
—
|
dózis-válasz összefüggés,
|
|
—
|
adott esetben statisztikai elemzések,
|
|
|
|
Az eredmények tárgyalása:
|
—
|
rövid kommentár az eredményekhez, a dózis-válasz elemzéshez és adott esetben a statisztikai elemzésekhez, és következtetés levonása arra vonatkozóan, hogy a vizsgált anyagot bőrszenzibilizáló hatásúnak kell-e tekinteni vagy sem.
|
|
|
SZAKIRODALOM
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999-1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985-997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-333.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf]
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258-273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274-286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol. 34, 249-257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd., based on ATP content test method protocol (LLNA:DA). NIH Publication No. 10-7551A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm]
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
|
|
(12)
|
Idehara, K., Yamagishi, G., Yamashita, K. and Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a non-radio isotopic endpoint. J. Pharmacol. Toxicol. Meth., 58, 1-10.
|
|
(13)
|
Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. and Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11-26.
|
|
(14)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(15)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904.
|
|
(16)
|
Basketter, D., Ball, N., Cagen, S., Carrillo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96.
|
|
(17)
|
Crouch, S.P., Kozlowski, R., Slater, K.J. and Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81-88.
|
|
(18)
|
Ishizaka, A., Tono-oka, T. and Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127-132.
|
|
(19)
|
Dexter, S.J., Cámara, M., Davies, M. and Shakesheff, K.M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27-34.
|
|
(20)
|
Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346-370.
|
|
(21)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(22)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09-7357, Research Triangle Park, NC: National Institute of Environmental Health Science. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf]
|
|
(23)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89.
|
|
(24)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31.
|
|
(25)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(26)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(27)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(28)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195-206.
|
|
(29)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94.
|
|
(30)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256.
|
|
(31)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491-506.
|
|
(32)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter-laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68.
|
|
(33)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728.
|
|
(34)
|
Patterson, R.M., Noga, E. and Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028.
|
|
(35)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm]
|
|
(36)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(37)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Health, 53 563-79.
|
|
(38)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
1. függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság: A vizsgálati módszerrel kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálati módszer teljesítményének mutatója, valamint a relevanciájának egyik megítélési szempontja. E kifejezést gyakran használják az »egyezés« megfelelőjeként, amely egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát fejezi ki (38).
Referenciaanyag: A vizsgált anyaggal való összehasonlítás céljából etalonként használt szenzibilizáló vagy nem szenzibilizáló anyag. A referenciaanyaggal szemben a következő elvárások fogalmazhatók meg: i. állandó és megbízható beszerzési forrásból vagy forrásokból kell származnia; ii. a vizsgált anyagok csoportjához hasonló szerkezettel és funkcionalitással kell rendelkeznie; iii. ismert fizikai-kémiai tulajdonságokkal kell rendelkeznie; iv. rendelkezésre kell állniuk az anyag ismert hatásait alátámasztó adatoknak; és v. a kívánt hatástartományban ismert hatásúnak kell lennie.
Álnegatív eredmény: Valamely vizsgálati módszerrel tévesen negatívnak vagy inaktívnak minősített anyag, amely valójában pozitív vagy aktív.
Álpozitív: Valamely vizsgálattal tévesen pozitívnak vagy aktívnak minősített anyag, amely valójában negatív vagy inaktív.
Veszély: Káros egészségügyi vagy ökológiai hatások előidézésének lehetősége. A káros hatás csak akkor jelenik meg, ha kellő szintű az expozíció.
Laboratóriumok közötti reprodukálhatóság: Annak mértéke, hogy a különböző minősített laboratóriumok azonos protokoll és azonos vizsgált anyagok alkalmazásával mennyire képesek minőségileg és mennyiségileg azonos eredményeket produkálni. A laboratóriumok közötti reprodukálhatóság az előhitelesítési és hitelesítési folyamat során kerül meghatározásra, és annak mértékét jelzi, hogy az adott vizsgálat mennyire sikeresen vihető át laboratóriumok között; más néven laboratóriumok közötti megismételhetőség (38).
Laboratóriumon belüli reprodukálhatóság: Annak meghatározása, hogy az ugyanazon laboratóriumban dolgozó képzett személyek meghatározott protokoll különböző időpontokban történő alkalmazásával mennyire sikeresen tudják reprodukálni az eredményeket. Más néven laboratóriumon belüli megismételhetőség (38).
Kiugró érték: A kiugró érték olyan megfigyelés, amely szembetűnően eltér az egy populációból vett véletlenszerű minta egyéb értékeitől.
Minőségbiztosítás: Irányítási folyamat, amelynek során a laboratóriumi vizsgálati szabványok és követelmények betartását, a jegyzőkönyvek vezetését, valamint az adattovábbítás pontosságát a vizsgálatot végző személyektől független személyek ellenőrzik.
Megbízhatóság: Azonos protokoll szerint végzett vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti reprodukálhatóságának mérésére szolgál. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg (38).
Bőrszenzibilizáció: Immunológiai folyamat, amely akkor jelentkezik, ha egy érzékeny egyén lokálisan érintkezik valamely indukáló hatású allergén vegyi anyaggal, amely esetlegesen kontakt szenzibilizáció kialakulásához vezető immunválaszt vált ki a bőrben.
Stimulációs index (SI): Adott vizsgált anyag bőrszenzibilizációt okozó képességének meghatározására kiszámított érték, amely az anyaggal kezelt csoportban és a párhuzamosan vivőanyaggal kezelt kontrollcsoportban tapasztalt proliferáció aránya.
Vizsgált anyag (más néven vizsgált vegyi anyag): Bármely anyag vagy keverék, amelyet ezzel a vizsgálati módszerrel vizsgálnak.
B.51. BŐRSZENZIBILIZÁCIÓ: LOKÁLIS NYIROKCSOMÓ-VIZSGÁLATI MÓDSZER (LLNA): BRDU-ELISA
BEVEZETÉS
|
1.
|
Az OECD vegyi anyagok vizsgálatára vonatkozó iránymutatásait és az uniós vizsgálati módszereket időnként felülvizsgálják a tudományos fejlődés, a változó szabályozási igények és állatjóléti megfontolások fényében. A bőrszenzibilizáció egereknél történő meghatározására szolgáló első vizsgálati módszer (B.42.), a lokális nyirokcsomó-vizsgálat (LLNA; az OECD 429. vizsgálati iránymutatása) átdolgozásra került (e melléklet 1. és B.42. fejezete). Az LLNA hitelesítésének részleteit és az ezzel kapcsolatos munka áttekintését publikálták (2) (3) (4) (5) (6) (7) (8) (9). Az LLNA során a limfociták proliferációjának mérése radioizotópos timidin vagy jód alkalmazásával történik, ezért amennyiben a radioaktív anyagok beszerzése, használata vagy ártalmatlanítása problémát jelent, a vizsgálat csak korlátozottan alkalmazható. Az LLNA: BrdU-ELISA (enzimhez kapcsolt immunszorbens vizsgálat) az LLNA vizsgálati módszer nem radioaktív változata, amely radioaktívan nem jelölt 5-bromo-2-dezoxiuridin (BrdU) (Chemical Abstracts Service [CAS] szám: 59-14-3) felhasználásával, ELISA alapú vizsgálati rendszerben méri a limfocitaproliferációt. Az LLNA: BrdU-ELISA-t vizsgálati módszert egy szakértőkből álló független nemzetközi értékelő testület hitelesítette, vizsgálta felül és ajánlotta olyan módszerként, amely a bőrszenzibilizációt okozó, illetve nem okozó vegyi anyagok meghatározására bizonyos megkötésekkel hasznosnak tekinthető (10) (11) (12). A vizsgálati módszer célja a vegyi anyagok (anyagok és keverékek) bőrszenzibilizáló hatásának állatoknál történő felmérése. E melléklet B.6. fejezete és az OECD 406. vizsgálati iránymutatása tengerimalac-vizsgálatokat alkalmaz, nevezetesen a tengerimalac-maximizációs módszert és Bühler-vizsgálatot (13). Az LLNA (e melléklet B.42. fejezete; az OECD 429. vizsgálati iránymutatása) és a két nem radioaktív módosított eljárás, az LLNA: BrdU-ELISA (e melléklet B.51. fejezete; az OECD 442. B. vizsgálati iránymutatása) és az LLNA: DA (e melléklet B.50. fejezete; az OECD 442. A. vizsgálati iránymutatása) egyaránt előnyösebb a B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában (13) szereplő tengerimalac-vizsgálatnál abból a szempontból, hogy kevesebb állat használatát igényli és kíméletesebben.
|
|
2.
|
Az LLNA-hoz hasonlóan az LLNA: BrdU-ELISA vizsgálat is a bőrszenzibilizáció indukciós fázisát vizsgálja, és a dózis-válasz értékelésére alkalmas mennyiségi adatokat szolgáltat. Emellett azáltal, hogy a bőrszenzibilizáló anyagokat a DNS radioaktív jelölése nélkül lehet kimutatni, a munka során kiküszöbölhető a radioaktivitásnak való kitettség, valamint a hulladék ártalmatlanításának problémája. Ez viszont az egerek nagyobb arányú használatát jelentheti a bőrszenzibilizáló anyagok kimutatására, ami a bőrszenzibilizáló képesség vizsgálata során tovább csökkenthetné a tengerimalacok használatát (pl. B.6. fejezet; az OECD 406. vizsgálati irányelve) (13).
|
FOGALOMMEGHATÁROZÁSOK
|
3.
|
Az alkalmazott fogalmak meghatározása az 1. függelékben található.
|
KIINDULÁSI MEGFONTOLÁSOK ÉS KORLÁTOK
|
4.
|
Az LLNA: BrdU-ELISA egy módosított LLNA (helyi nyirokcsomó-vizsgálati) módszer, amely bizonyos korlátozások mellett a bőrön túlérzékenységi reakciót kiváltó vegyi anyagok azonosítására szolgál. Ez nem feltétlenül jelenti azt, hogy az LLNA vagy a tengerimalac-vizsgálat (lásd B.6. fejezet, az OECD 406. vizsgálati iránymutatása) (13) helyett minden esetben az LLNA: BrdU-ELISA-t kell alkalmazni, inkább azt, hogy ez a vizsgálati módszer ugyanolyan hasznos, és olyan alternatív módszerként alkalmazható, amely esetében a pozitív és negatív eredmények általában már nem igényelnek további megerősítést (10) (11). A vizsgálat elvégzése előtt a vizsgáló laboratóriumnak a vizsgált anyagról rendelkezésre álló összes adatot figyelembe kell vennie. Ezen információk közé tartozik a vizsgált anyag azonosítása, kémiai szerkezete, fizikai-kémiai tulajdonságai, valamint a vizsgált anyaggal korábban végzett in vitro vagy in vivo toxicitási vizsgálatok eredményei és a szerkezetileg rokon vegyi anyagok toxikológiai adatai is. Mindezen információk figyelembevételével dönthető el, hogy az LLNA: BrdU-ELISA módszer alkalmazható-e a vizsgált anyag esetében (tekintve, hogy bizonyos vegyianyag-típusoknál inkompatibilitás áll fenn [lásd az 5. bekezdést]), és milyen dózist célszerű választani a vizsgálatokhoz.
|
|
5.
|
Az LLNA: BrdU-ELISA in vivo módszer, következésképpen nem küszöbölhető ki vele az állatok felhasználása az allergiás kontakt szenzibilizáló hatás vizsgálata során. Ugyanakkor a tengerimalac-vizsgálatokhoz képest (B.6. fejezet; az OECD 406. vizsgálati iránymutatása) lehetővé teszi az állatok ilyen célú felhasználásának csökkentését (13). Továbbá az állatok kontakt szenzibilizációs vizsgálatok során történő felhasználásának módját tekintve az LLNA: BrdU-ELISA jelentősen kíméletesebb, mivel a B.6. fejezetben és a 406-os OECD vizsgálati irányelvben foglaltakkal ellentétben az LLNA: BrdU-ELISA esetében nincs szükség indukált bőr-hiperszenzitivitási reakciók előidézésére. Továbbá az LLNA: BrdU-ELISA során a tengerimalac-maximizációs vizsgálattal ellentétben nincs szükség adjuváns alkalmazására sem (e melléklet B.6. fejezete, 13). Az LLNA: BrdU-ELISA alkalmazásával tehát csökkenthető az állatok szenvedése. A B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában (13) foglalt módszerrel szembeni előnyei ellenére az LLNA: BrdU-ELISA módszernek vannak bizonyos korlátai, amelyek szükségessé tehetik a B.6. fejezetben és az OECD 406. vizsgálati iránymutatásában foglaltak alkalmazását (pl. bizonyos fémek vizsgálata, álpozitív eredmények bizonyos bőrirritáló anyagokkal [például bizonyos felületaktív anyagokkal] (6) [1, illetve e melléklet B.42. fejezete], vagy a vizsgált anyag oldhatósága). Továbbá az olyan funkcionális csoportokat tartalmazó anyagcsoportok, illetve anyagok, amelyek potenciális zavaró tényezőként szerepelnek (15) szükségessé tehetik a tengerimalac-vizsgálatok (vagyis a B.6. fejezet; az OECD 406. vizsgálati iránymutatása (13)) használatát. Az LLNA esetében megállapított korlátozásokat (1, illetve e melléklet B.42. fejezete) az LLNA: BrdU-ELISA módszerre is javasolt alkalmazni (10). E meghatározott hiányosságokon kívül, az LLNA: BrdU-ELISA bármilyen vegyi anyag vizsgálatára megfelelő, kivéve, ha ezek a vegyi anyagok olyan tulajdonságokkal rendelkeznek, amelyek befolyásolják az LLNA: BrdU-ELISA pontosságát. Emellett abban az esetben, ha a stimulációs indexre (SI) kapott érték 1,6 és 1,9 közé esik (lásd a 31–32. bekezdést) figyelembe kell venni a határértéken lévő pozitív eredmények lehetőségét. Ez egy legalább 1,6 értékű stimulációs indexszel rendelkező, 43 anyagot magában foglaló hitelesítési adatbázison alapul (lásd a 6. bekezdést), a mely esetében az LLNA: BrdU-ELISA módszer helyesen azonosította mind a 32, az LLNA módszer szerint szenzibilizáló anyagot, de hibásan azonosított két anyagot az LLNA módszerrel nem szenzibilizálóként meghatározott, 1,6 és 1,9 közötti SI-értékkel (vagyis határértéken lévő pozitív értékkel) rendelkező 11 anyag közül (10). Mivel azonban az SI-értékek beállításához és a vizsgálat prediktív jellemzőinek kiszámításához ugyanazt az adatállományt alkalmazták, a megadott eredmények túlbecsülhetik a valós prediktív jellemzőket.
|
A VIZSGÁLATI MÓDSZER ELVE
|
6.
|
Az LLNA: BrdU-ELISA módszer hátterében az az alapelv áll, hogy a szenzibilizáló anyagok elsődleges limfocitaproliferációt indukálnak a vizsgált anyag alkalmazási területének nyirokelvezetéséről gondoskodó nyirokcsomókban. Ez a proliferáció arányos az alkalmazott allergén dózisával és erősségével, valamint egyszerű módszert biztosít a szenzibilizáció kvantitatív méréséhez. A proliferációt úgy mérik, hogy az egyes vizsgálati csoportokban mért átlagos proliferációt összehasonlítják a vivőanyaggal kezelt kontrollcsoportban (VK) mért átlagos proliferációval. Meghatározzák az egyes kezelt csoportokban és a párhuzamosan vivőanyaggal kezelt kontrollcsoportokban mért átlagos proliferáció arányát, amelyet stimulációs indexnek (SI) neveznek, és amelynek legalább ≥ 1,6-nek kell lennie ahhoz, hogy indokolt legyen a vizsgált anyag potenciális bőrszenzibilizáló anyagként történő további vizsgálata. Az itt leírt eljárások a BrdU tartalom mérésén alapulnak, amely jelzi az elvezető aurikuláris nyirokcsomókban lévő proliferáló sejtek számának növekedését. A BrdU egy timidinanalóg, amely hasonlóan beépül a proliferáló sejtek DNS-ébe. A BrdU beépülését ELISA-val mérjük, amely peroxidázzal is jelölt BrdU-specifikus antitestet alkalmaz. A szubsztrát hozzáadáskor a peroxidáz reakcióba lép a szubsztráttal, és színes termék képződik, amelynek mennyisége meghatározott abszorbancia mellett mikrotiterlemez-leolvasó segítségével mérhető.
|
A VIZSGÁLAT LEÍRÁSA
Az állatfaj kiválasztása
|
7.
|
Ehhez a vizsgálathoz az egér a választandó faj. A LLNA: BrdU-ELISA módszerhez a hitelesítési vizsgálatokat kizárólag a CBA/JN törzzsel végezték, amely ezért preferált törzsnek tekintendő (10) (12). Fiatal felnőtt nullipara és nem vemhes nőstény egereket kell használni. A vizsgálat elején az állatok korának 8-12 hétnek kell lennie, az állatok testtömege között az eltérésnek minimálisnak kell lennie, és a testtömeg nem haladhatja meg az átlagos testtömeg 20 %-át. Ha elegendő adat áll rendelkezésre arra nézve, hogy az LLNA:BrdU-ELISA módszerrel kapott válaszreakcióban nincsenek jelentős törzs- és/vagy ivarspecifikus különbségek, akkor más törzsek, illetve hím állatok is alkalmazhatók.
|
Az állatok tartásának és etetésének körülményei
|
8.
|
Az egereket csoportosan kell tartani (16), kivéve, ha az egyedenkénti tartásra megfelelő tudományos indok merül fel. A kísérleti állatok tartásául szolgáló helyiség hőmérsékletének 22 °C ± 3 °C-nak kell lennie. Noha a helyiség relatív páratartalmának legalább 30 %-nak kell lennie, és – a takarítás időtartamától eltekintve – lehetőség szerint nem haladhatja meg a 70 %-ot, a célértéknek 50–60 % között kell lennie. A világítás legyen mesterséges, 12 órás világos, 12 órás sötét periódusok váltakozásával. Az etetéshez a szokásos laboratóriumi takarmány alkalmazható korlátlan mennyiségű ivóvíz biztosítása mellett.
|
Az állatok előkészítése
|
9.
|
Az állatokat véletlenszerűen választják ki, majd egyedi azonosítóval látják el (viszont az állat fülén semmiféle azonosító nem alkalmazható), és a kezelés megkezdése előtt legalább 5 napig a ketrecükben tartják őket, hogy hozzászokhassanak a laboratóriumi körülményekhez. A kezelés megkezdése előtt minden állatot megvizsgálnak, hogy meggyőződjenek arról, nincs rajtuk látható bőrsérülés.
|
A dózisok előkészítése
|
10.
|
A szilárd halmazállapotú vegyi anyagokat az egér fülén történő alkalmazás előtt oldószerben/vivőanyagban kell feloldani vagy szuszpendálni, és szükség esetén hígítani kell. A folyadék halmazállapotú vegyi anyagokat hígítatlanul vagy az adagolás előtt hígított formában lehet alkalmazni. A nem oldható vegyi anyagokat, például az orvosi eszközökben általánosan előforduló anyagokat megfelelő oldószerben történő agresszív extrakciónak kell kitenni, hogy a vizsgálat során az egér fülén való alkalmazás előtt az összes kivonható összetevőt feltárjuk. A vizsgált anyagokat naponta kell elkészíteni, kivéve, ha stabilitási adatok igazolják, hogy a tárolás elfogadható.
|
A megbízhatóság ellenőrzése
|
11.
|
A vizsgálat megfelelő teljesítményének megfelelő és reprodukálható szenzitivitással adott válaszreakciókkal történő igazolására pozitív kontrollanyagokat (PK) alkalmaznak; ezek olyan szenzibilizáló tesztanyagok, amelyek esetében a válaszreakció mértéke pontosan meghatározott. Pozitív kontroll párhuzamos vizsgálata ajánlott, mivel ez igazolja, hogy a laboratórium megfelelő szakértelemmel végzi az egyes vizsgálatokat, és lehetővé teszi a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság és összehasonlíthatóság értékelését. Néhány szabályozó hatóság szintén előírja a pozitív kontroll minden vizsgálat során történő alkalmazását, ezért az LLNA: BrdU-ELISA módszer elvégzése előtt ajánlott egyeztetni az illetékes hatóságokkal. Ennek megfelelően javasolt a pozitív kontroll rutinszerű párhuzamos vizsgálata, hogy elkerülhetőek legyenek az olyan további állatkísérletek, amelyeket a pozitív kontrollt csak időszakosan alkalmazó laboratórium az előírásoknak való megfelelés érdekében végezne (lásd 12. bekezdés). A pozitív kontrollnak az LLNA: BrdU-ELISA módszerrel olyan expozíciós szint mellett kell pozitív válaszreakciót adnia, amely a negatív kontrollcsoporthoz (NK) képest várhatóan legalább 1,6-del növeli a stimulációs indexet (SI). A pozitív kontroll dózisát úgy kell megválasztani, hogy ne okozzon túlzott bőrirritációt vagy szisztémás toxicitást, és az indukció reprodukálható, de ne túl nagymértékű legyen (például SI > 14 már túlzottnak tekinthető). Az előnyben részesítendő pozitív kontrollanyagok a 25 %-os hexil-cinnemaldehid (CAS-szám: 101-86-0) és a 25 %-os eugenol (CAS-szám: 97-53-0) aceton:olívaolaj (4:1, v/v) arányú keverékében. Adódhatnak olyan körülmények, amelyek fennállásakor, megfelelő indoklás esetén a fenti kritériumoknak megfelelő más pozitív kontrollanyagok is alkalmazhatók.
|
|
12.
|
Bár ajánlott a pozitív kontrollcsoport párhuzamos vizsgálata, lehetnek azonban olyan helyzetek, amikor a pozitív kontroll időszakos (például legalább 6 havonta végzett) vizsgálata elfogadható lehet az olyan, az LLNA: BrdU-ELISA vizsgálatot rendszeresen (vagyis legalább havonta egyszeri gyakorisággal) végző laboratóriumok esetében, amelyek rendelkeznek a pozitív kontrollok igazolt, historikus adatbázisával, és ezzel bizonyítható, hogy a laboratórium a pozitív kontrollokkal képes reprodukálható és pontos eredményeket előállítani. Az LLNA: BrdU-ELISA módszerben való jártasság alátámasztható a pozitív kontrollal megfelelő időszakon belül (kevesebb mint egy éve) végzett legalább 10 független vizsgálat során kapott következetesen pozitív eredményekkel.
|
|
13.
|
Pozitív kontrollcsoport párhuzamos vizsgálata minden alkalommal szükséges, valahányszor az LLNA: BrdU-ELISA vizsgálati módszerében változtatást hajtanak végre (például a képzett személyzet, a vizsgálati eljárás során használt anyagok és/vagy reagensek, a vizsgálati eszközök, a kísérleti állatok beszerzési forrásának megváltoztatása), és ezeket a változásokat dokumentálni kell a laboratóriumi jelentésekben. Figyelembe kell venni, hogy ezek a változtatások mennyiben befolyásolják a korábban létrehozott historikus adatbázis megfelelőségét, és el kell dönteni, hogy szükséges-e új historikus adatbázis felállítása a pozitív kontroll eredményei következetességének dokumentálása érdekében.
|
|
14.
|
A vizsgálatot végző személyeknek tisztában kell lenniük azzal, hogy amennyiben a párhuzamos vizsgálat helyett a pozitív kontrollok időszakos vizsgálata mellett döntenek, ez hatással lehet a pozitív kontrollok időszakos vizsgálatai közötti időszakokban, párhuzamos pozitív kontroll vizsgálata nélkül előállított negatív vizsgálati eredmények megfelelőségére és elfogadhatóságára. Ha például a pozitív kontrollal végzett időszakos vizsgálat álnegatív eredményt adott, akkor megkérdőjelezhetők azok a negatív eredmények, amelyek az utolsó elfogadható eredményű és az elfogadhatatlan eredményű időszakos pozitív kontroll vizsgálat között eltelt időszakban keletkeztek. Az ilyen következmények jelentőségét körültekintően mérlegelni kell annak eldöntésekor, hogy pozitív kontrollokat párhuzamosan vagy csak időszakosan vizsgáljanak-e. Szintén megfontolandó a kevesebb állat használata a párhuzamos pozitív kontrollcsoportban, amennyiben ez tudományosan indokolt, és a laboratóriumra specifikus historikus adatok alapján a laboratórium igazolja, hogy megfelelő a kevesebb egér használata (17).
|
|
15.
|
Bár a pozitív kontrollt olyan vivőanyagban kell vizsgálni, amelyről ismert, hogy következetes válaszreakciót vált ki (például aceton:olívaolaj; 4:1, v/v arányú keveréke), lehetnek olyan szabályozási helyzetek, amikor nem szokványos vivőanyagokban (klinikailag/kémiailag releváns készítmény) történő vizsgálat elvégzésére is szükség van (18). Amennyiben a párhuzamos pozitív kontrollt a vizsgált anyagétól eltérő vivőanyagban vizsgálják, akkor a párhuzamos pozitív kontrollal együtt egy külön vivőanyagos kontrollt is vizsgálni kell.
|
|
16.
|
Azokban az estekben, amikor egy bizonyos vegyi csoportba tartozó vizsgált anyagokat vagy a válaszreakciók tartományát értékelik, szintén hasznos lehet a referenciaanyagok alkalmazása annak igazolására, hogy a vizsgálati módszer megfelelően működik az ilyen típusú vizsgált anyagok bőrszenzibilizáló hatásának kimutatására. A megfelelő referenciaanyagoknak a következő tulajdonságokkal kell rendelkezniük:
|
—
|
szerkezeti és funkcionális hasonlóság a vizsgált anyag csoportjával,
|
|
—
|
ismert fizikai/kémiai tulajdonságok,
|
|
—
|
az LLNA: BrdU-ELISA módszerrel kapott alátámasztó adatok,
|
|
—
|
egyéb állatmodellekből és/vagy emberből származó alátámasztó adatok.
|
|
VIZSGÁLATI ELJÁRÁS
Az állatok száma és a dózisszintek
|
17.
|
Minden dóziscsoportban legalább négy állatot kell használni, és a vizsgált anyagból legalább három különböző koncentrációt, továbbá párhuzamos negatív kontrollcsoportot, amelyet csak a vizsgált anyaghoz használt vivőanyaggal kezelnek, valamint egy pozitív kontroll csoportot kell alkalmazni (pozitív kontroll párhuzamos vagy a közelmúltban végzett vizsgálata a laboratórium szabályzatának megfelelően, figyelembe véve a 11–15. bekezdésben foglaltakat). Megfontolandó a pozitív kontroll többféle dózisának vizsgálata, különösen, ha a pozitív kontroll vizsgálatát időszakosan végzik. A kontrollcsoportban lévő állatokkal ugyanúgy kell bánni és dolgozni, mint a kezelt csoportokban lévőkkel, azzal a kivétellel, hogy az állatokat nem kezelik a vizsgált anyaggal.
|
|
18.
|
A dózist és a vivőanyagot a 2. és 19. számú hivatkozásokban megadott ajánlások alapján kell kiválasztani. Az egymás utáni dózisok kiválasztása általában megfelelő koncentrációsorból, például 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % stb. történik. Az alkalmazott koncentrációsort megfelelő tudományos megfontolások alapján kell kiválasztani. A három egymás utáni koncentráció kiválasztásakor figyelembe kell venni a vizsgált anyagról esetlegesen rendelkezésre álló valamennyi toxikológiai információt (például akut toxicitás és bőrirritáció), valamint szerkezeti és fizikokémiai adatot, hogy a legmagasabb koncentrációval a lehető legnagyobb expozíciót idézzük elő, amely még nem okoz szisztémás toxicitást és/vagy túlzott mértékű lokális bőrirritációt (19) (20, illetve e melléklet B.4. fejezete). Ilyen jellegű információk hiányában szükséges lehet a megelőző előszűrő vizsgálat (lásd a 21–24. bekezdést).
|
|
19.
|
A vivőanyagnak nem szabad megzavarnia vagy befolyásolnia a vizsgálat eredményét, és aszerint kell kiválasztani, hogy a vizsgált anyag oldhatósága a lehető legnagyobb koncentráció elérése érdekében maximális legyen, ugyanakkor a vizsgált anyag felvitelére alkalmas oldatot/szuszpenziót kapjunk. A javasolt vivőanyagok az aceton:olívaolaj (4:1 v/v) keveréke, N,N-dimetilformamid, metil-etil-keton, propilén-glikol és dimetil-szulfoxid (6), de megfelelő tudományos indoklás esetén más vivőanyagok is alkalmazhatók. Bizonyos esetekben további kontrollként szükséges lehet valamely klinikai szempontból releváns oldószer használata, vagy annak a kereskedelmi formulának az alkalmazása, amelyben a vizsgált anyagot forgalomba hozzák. Különösen vigyázni kell arra, hogy megfelelő szolubilizáló szerek (pl. 1 % Pluronic® L92) alkalmazásával a hidrofil vizsgált anyagokat olyan vivőanyagrendszerbe építsük be, amely benedvesíti a bőrt, de nem folyik le azonnal. A teljesen vizes alapú vivőanyagokat tehát kerülni kell.
|
|
20.
|
A nyirokcsomók egyedenként történő feldolgozása lehetővé teszi az egyedek közötti variabilitás, valamint a vizsgált anyaggal és a vivőanyaggal kezelt csoport eredményeinek statisztikai összehasonlítását (lásd a 33. bekezdést). Emellett csak akkor lehet értékelni, hogy lehetséges-e csökkenteni a pozitív kontrollcsoportban lévő egerek számát, ha az egyedi állatokra vonatkozóan gyűjtünk adatokat (17). Ezen túlmenően, egyes szabályozó hatóságok is megkövetelik az egyedi állatokon alapuló adatgyűjtést. Az egyedi állatokon alapuló adatgyűjtés rendszeresítése állatjóléti előnyökkel jár azáltal, hogy elkerülhető a vizsgálatok megismétlése abban az esetben, amikor a vizsgált anyagról eredetileg csak egyféleképpen gyűjtötték az adatokat (pl. összevont állatkísérletes adatok), míg a szabályozó hatóság később eltérő követelményeket állít fel (pl. egyedi állatokon alapuló adatok).
|
Előszűrő vizsgálat
|
21.
|
A legmagasabb vizsgálandó dózis meghatározását lehetővé tevő információ hiányában (lásd a 18. bekezdést) az LLNA: BrdU-ELISA módszerhez alkalmazandó megfelelő dózisszint megállapítása érdekében előszűrő vizsgálatot kell végezni. Az előszűrő vizsgálat célja, hogy támpontot adjon az LLNA: BrdU-ELISA vizsgálat során alkalmazandó maximális dózis kiválasztásához, amennyiben a szisztémás toxicitást (lásd a 24. bekezdést) és/vagy nagyfokú bőrirritációt (lásd a 23. bekezdést) okozó koncentrációra vonatkozó információk nem állnak rendelkezésre. Folyadékok esetében a legmagasabb vizsgált dózisnak 100 %-os koncentrációnak kell lennie, szilárd anyagok vagy szuszpenziók esetében pedig a lehetséges maximális koncentrációnak.
|
|
22.
|
Az előszűrő vizsgálatot a fő LLNA: BrdU-ELISA vizsgálathoz hasonló körülmények között kell végezni, eltekintve attól, hogy nem kell a nyirokcsomó-proliferációt értékelni, valamint dóziscsoportonként kevesebb állat használható. Dóziscsoportonként egy vagy két állat javasolt. Az egereket a szisztémás toxicitás, illetve az alkalmazás helyén fellépő helyi irritáció jeleinek szempontjából naponta megfigyelik. A testtömeget a vizsgálat előtt és az állat exterminálása előtt is (6. nap) feljegyzik. A bőrpírt az egerek mindkét fülén vizsgálják és pontozzák az 1. táblázat (20, illetve e melléklet B.4. fejezete) szerint. A fülvastagság értékeit egy vastagságmérővel (például digitális mikrométer, vagy Peacock Dial vastagságmérő) állapítják meg az 1. napon (az adagolás előtt), a 3. napon (hozzávetőleg 48 órával az első dózis után), valamint a 6. napon. A 6. napon a fülvastagságot ezen kívül a fülből lyukasztással eltávolított szövet tömegének meghatározásával is meg lehet állapítani, amit az állat exterminálása után kell elvégezni. Nagyfokú helyi irritációnak minősül a legalább ≥ 3-as pontszámú bőrpír és/vagy legalább ≥ 25 %-os fülvastagság a vizsgálat bármely napján (21) (22). A fő LLNA: BrdU-ELISA vizsgálathoz azt a dózist kell maximális dózisként választani, amely az előszűrés során alkalmazott koncentrációsorban (lásd a 18. bekezdést) a szisztémás toxicitást és/vagy nagyfokú helyi bőrirritációt okozó koncentrációnál eggyel alacsonyabb.
1. táblázat
Bőrpír (erythema) pontszámai
|
Megfigyelés
|
Pontszám
|
|
Nincs erythema
|
0
|
|
Nagyon enyhe erythema (alig észlelhető)
|
1
|
|
Jól körülírt erythema
|
2
|
|
Közepes-súlyos erythema
|
3
|
|
Súlyos (céklavörös elszíneződés), pörkképződéig terjedő erythema, amely lehetetlenné teszi az erythema besorolását
|
4
|
|
|
23.
|
A 25 %-os fülvastagság-növekedésen felül (21) (22), a bőrirritáló anyagok LLNA módszerrel végzett kimutatásához a kezelt állatok fülvastagságának kontrollcsoporthoz viszonyított statisztikailag szignifikáns növekedését is használják (22) (23) (24) (25) (26) (27) (28). Noha 25 % alatti fülvastagság esetén előfordulhat statisztikailag szignifikáns növekedés, a nagyfokú irritációval azonban ezt különösen nem hozták összefüggésbe (25) (26) (27) (28) (29).
|
|
24.
|
Az integrált értékelés részeként alkalmazva a következő klinikai tünetek jelezhetnek szisztémás toxicitást (30), és ezáltal kijelölhetik a fő LLNA:BrdU-ELISA vizsgálat során alkalmazandó maximális dózist: az idegrendszer működésében bekövetkező változások (például piloerekció, ataxia, tremorok és görcsrohamok); a viselkedésben bekövetkező változások (például agresszivitás, a tisztálkodási tevékenységekben bekövetkező változás, az aktivitási szint jelentős változása); a légzési mintázat változása (például a légvételek gyakoriságában és az intenzitásában bekövetkező változás, úgymint dyspnoe, zihálás és szörtyzörejek), valamint változások az táplálék- és vízfogyasztásban. Az értékelés során emellett figyelembe kell venni a letargia és/vagy reakciókészség hiányának jeleit, valamint az enyhénél és pillanatnyinál fokozottabb fájdalom vagy szenvedés bármilyen klinikai tünetét, az > 5 %-nál nagyobb arányú testtömegcsökkenést az 1. naptól a 6. napig tartó időszakban, valamint a mortalitást. Az elhullásközeli állapotban lévõ vagy súlyos fájdalmak és szenvedés jeleit mutató állatokat humánus módon exterminálni kell (31).
|
A fő vizsgálat kísérleti ütemterve
|
25.
|
A vizsgálat kísérleti ütemterve a következő:
— 1. nap: Külön meg kell határozni, és fel kell jegyezni mindegyik állat testtömegét és az esetleges klinikai megfigyeléseket. Vigyünk fel 25 μl térfogatú, megfelelő hígítású vizsgált anyagot, valamint a vivőanyagot önmagában vagy pozitív kontrollt (pozitív kontroll párhuzamos vagy a közelmúltban végzett vizsgálata a laboratórium szabályzatának megfelelően, figyelembe véve a 11–15. bekezdésekben foglaltakat) a fülek dorzális oldalára.
— 2. és 3. nap: Ismételjük meg az 1. napon elvégzett felviteli eljárást.
— 4. nap: Nincs kezelés.
— 5. nap: Fecskendezzünk be 0,5 mL (5 mg/egér) BrdU (10 mg/ml) oldatot intraperitoneálisan.
— 6. nap: Fel kell jegyezni minden állat testtömegét és minden klinikai megfigyelést. A BrdU befecskendezése után körülbelül 24 órával (24 h) extermináljuk az állatokat. Mindegyik egér füléből vágjuk ki a kezelt terület nyirokelvezetéséről gondoskodó füli nyirokcsomókat, és tegyük be azokat állatonként külön-külön foszfáttal pufferelt sóoldatba (PBS). A nyirokcsomók azonosításának és preparálásának részletei és az ehhez kapcsolódó ábrák a (17) hivatkozásban találhatók. A helyi bőrreakció fő vizsgálatban történő további monitorozásához olyan további paramétereket lehet belevenni a vizsgálati protokollba, mint például a bőrpír pontozása vagy a fülvastagság mérése (akár vastagságmérővel, akár elölés utáni füldarabsúly-meghatározással).
|
A sejtszuszpenziók elkészítése
|
26.
|
Az egyes állatokból két oldalról vett nyirokcsomósejtekből (LNC) egysejtszuszpenziót kell készíteni a sejtek mechanikus úton történő óvatos szétválasztásával, amely végezhető 200 mikrométeres lyukméretű rozsdamentes acélból készült fémhálóval vagy egyéb elfogadható módszerrel (pl. a nyirokcsomók egyszer használatos műanyag mozsárban történő összetörése, majd 70-es nejlonhálón való átpréselése). Az LNC szuszpenzió elkészítése a vizsgálat szempontjából döntő fontosságú, ezért a módszert a vizsgálatot végző valamennyi munkatársnak előre el kell sajátítani. A negatív kontrollként szolgáló állatok nyirokcsomói kisméretűek, ezért az SI-értékre gyakorolt mesterséges hatások elkerülése érdekében fontos az elővigyázatos kezelés. Az LNC szuszpenzió céltérfogatát minden esetben a meghatározott optimalizált térfogathoz kell igazítani (körülbelül 15 ml). Az optimális térfogat meghatározása a negatív kontrollcsoport 0,1–0,2 közötti átlagos abszorbanciájának elérésén alapul.
|
A sejtproliferáció meghatározása (a BrdU limfociták DNS-ébe való beépülésének mérése)
|
27.
|
A BrdU mérése a kereskedelemben kapható ELISA készlet (például Roche Applied Science, Mannheim, Németország, katalógusszám: 11 647 229 001) használatával történik. A mérés lényege, hogy az LNC-szuszpenzióból három adagban 100 μl-t kell egy lapos aljú mikrotiterlemez mélyedéseibe adagolni. Az LNC fixálása és denaturálása után mindegyik mélyedésbe anti-BrdU-antitestet kell adni, majd várni kell, hogy a reakció kialakuljon. Ezután az anti-BrdU antitestet mosással el kell távolítani, majd hozzá kell adni a szubsztrátoldatot, és várni kell, hogy kialakuljon a kromogén termék. Ekkor 370 nm-en 492 nm-es referencia-hullámhossz mellett mérni kell az abszorbanciát. A vizsgálati körülményeket minden esetben optimalizálni kell (lásd a 26. bekezdést).
|
MEGFIGYELÉSEK
Klinikai megfigyelések
|
28.
|
Naponta legalább egyszer minden egérnél meg kell vizsgálni az alkalmazás helyén kialakuló lokális vagy szisztémás toxicitás esetleges tüneteit. Minden megfigyelést szisztematikusan fel kell jegyezni minden egyes egér esetében, egyedi adatsor felvételével. A monitoring tervnek tartalmaznia kell azon kritériumokat, amelyek alapján azonnal azonosíthatóak az eutanáziát igénylő, szisztémás toxicitást, kiterjedt helyi bőrirritációt vagy bőrkorróziót mutató egerek (31).
|
Testtömeg
|
29.
|
Ahogyan az a 25. bekezdésben szerepel, az egyes állatok testtömegét a vizsgálat kezdetén, valamint a humánusan történő exterminálás tervezett időpontjában kell megmérni.
|
AZ EREDMÉNYEK KISZÁMÍTÁSA
|
30.
|
Az egyes kezelési csoportokra vonatkozó eredményeket átlagos stimulációs indexben (SI) fejezzük ki. Az SI kiszámítása úgy történik, hogy a vizsgált anyaggal kezelt egyes csoportokban és a pozitív kontrollcsoportban kapott állatonkénti átlagos BrdU jelölési indexet el kell osztani az oldószerrel/vivőanyaggal kezelt kontrollcsoport átlagos BrdU jelölési indexével. A vivőanyaggal kezelt kontrollcsoportok átlagos SI-értéke tehát 1.
A BrdU jelölési index meghatározása:
BrdU jelölési index = (ABSem – ABS vakem) – (ABSref – ABS vakref)
em = kibocsátott hullámhossz; ref = referencia hullámhossz.
|
|
31.
|
A válaszreakciót akkor lehet pozitívan értékelni, ha a stimulációs index ≥ 1,6 (10). Amikor azonban azt kell meghatározni, hogy egy határértéken lévő eredmény (vagyis amikor az SI-érték 1,6 és 1,9 között van) pozitívnak tekintendő-e, akkor a dózis-válasz összefüggés erőssége, a statisztikai szignifikancia és az oldószerrel/vivőanyaggal, valamint a pozitív kontrollal kapott válaszreakciók következetessége is felhasználható (3) (6) (32).
|
|
32.
|
Az 1,6 és 1,9 közötti SI-vel járó, határértéken lévő pozitív válaszreakciók esetében a felhasználók dönthetnek úgy, hogy az eredmények pozitivitásának megerősítéséhez további információkat – például a dózis-válasz összefüggést, szisztémás toxicitás vagy nagyfokú irritáció jelét, illetve adott esetben a statisztikai szignifikanciát az SI értékekkel együtt – kívánnak figyelembe venni (10). Figyelembe kell venni a vizsgált anyag különféle tulajdonságait is, köztük azt, hogy mutat-e szerkezeti rokonságot ismert bőrszenzibilizáló anyagokkal, okoz-e nagyfokú bőrirritációt egereknél, illetve a megfigyelt dózis-válasz összefüggés jellegét. Ezek, illetve további megfontolások részletesebb tárgyalását lásd a (4) hivatkozásban.
|
|
33.
|
Az egyedi állatokon alapuló adatgyűjtés lehetővé teszi a dózis-válasz összefüggés adatokban tükröződő fennállásának és mértékének statisztikai elemzését. Bármely statisztikai elemzés tartalmazhatja a dózis-válasz összefüggés értékelését, valamint a vizsgálati csoportok megfelelően korrigált összehasonlításait (például a dóziscsoportok páronkénti összehasonlítása a párhuzamos oldószeres/vivőanyagos kontrollcsoportokkal). A statisztikai elemzésekben a dózis-válasz tendenciák értékelésénél szerepelhet például lineáris regresszió vagy William-féle próba, a páronkénti összehasonlításoknál pedig Dunnett-féle próba. A megfelelő statisztikai elemzési módszer kiválasztásakor a vizsgálónak figyelemmel kell lennie a lehetséges varianciaegyenlőtlenségekre és más kapcsolódó problémákra, amelyek miatt az adatok transzformációjára vagy nem paraméteres statisztikai elemzésre lehet szükség. A vizsgálónak minden esetben el kell végeznie az SI-k kiszámítását és statisztikai elemzéseket bizonyos adatpontokkal (az úgynevezett »kiugró adatokkal«) és azok nélkül.
|
ADATOK ÉS JELENTÉS
Adatok
|
34.
|
Az adatokat táblázatos formában kell összefoglalni, feltüntetve az egyedi állatok BrdU jelölési indexének értékeit, a csoport átlagos BrdU jelölés/állat értékét, a hozzá tartozó hibamutatóval (például SD, SEM), valamint az egyes dóziscsoportokban kapott átlagos stimulációs indexeket (SI) összehasonlítva a párhuzamos oldószeres/vivőanyagos kontrollcsoportéval.
|
Vizsgálati jelentés
|
35.
|
A vizsgálati jelentésnek a következő információkat kell tartalmaznia:
|
|
Vizsgált anyagok és kontrollanyagok:
|
—
|
azonosító adatok (pl. adott esetben a CAS-szám és az EU-szám; eredet; tisztaság; ismert szennyezők; tételszám),
|
|
—
|
fizikai megjelenés és fizikai-kémiai tulajdonságok (pl. illékonyság, stabilitás, oldhatóság),
|
|
—
|
elegy esetén annak összetétele és az egyes komponensek egymáshoz viszonyított aránya,
|
|
|
|
Oldószer/vivőanyag:
|
—
|
azonosító adatok (adott esetben tisztaság; koncentráció; alkalmazott térfogat),
|
|
—
|
a vivőanyag kiválasztásának indoklása,
|
|
|
|
Kísérleti állatok:
|
—
|
CBA egértörzs származása,
|
|
—
|
az állatok mikrobiológiai státusa, amennyiben ismert,
|
|
—
|
az állatok száma és életkora,
|
|
—
|
az állatok származása, tartásának körülményei, takarmánya stb.,
|
|
|
|
Vizsgálati körülmények:
|
—
|
az ELISA készlet esetében a származás, a tételszám, a gyártó minőségbiztosítási/minőség-ellenőrzési adatai (antitest szenzitivitása és specificitása, illetve a kimutatási határ),
|
|
—
|
a vizsgált anyagból előállított készítménnyel és annak alkalmazásával kapcsolatos adatok,
|
|
—
|
a dózisszintek megválasztásának indoklása (ezen belül adott esetben az előszűrési vizsgálatok eredményei),
|
|
—
|
a vivőanyag és a vizsgált anyag alkalmazott koncentrációja, valamint a vizsgált anyagból alkalmazott teljes mennyiség,
|
|
—
|
a táplálék és a víz minőségére vonatkozó adatok (ezen belül a takarmány típusa/eredete, a víz eredete),
|
|
—
|
a kezelés adatai és a mintavételek ütemezése,
|
|
—
|
a toxicitás mérésére alkalmazott módszerek,
|
|
—
|
a pozitív és negatív vizsgálati eredmény kimondásának kritériumai,
|
|
—
|
a protokolltól való esetleges eltérések adatai, és annak kifejtése, hogy az eltérés hogyan érintheti vizsgálati tervet és az eredményeket,
|
|
|
|
A megbízhatóság ellenőrzése:
|
—
|
a legutóbbi megbízhatósági ellenőrzés eredményeinek összefoglalása az alkalmazott anyag, koncentráció és vivőanyag adataival együtt,
|
|
—
|
a vizsgálólaboratórium párhuzamosan és/vagy korábban vizsgált pozitív és párhuzamosan vizsgált negatív (oldószer/vivőanyag) kontrollokra vonatkozó adatai,
|
|
—
|
ha nem vizsgáltak párhuzamosan pozitív kontrollt, a laboratóriumban legutóbb végzett időszakos pozitív kontroll dátuma és laboratóriumi jelentése, valamint pozitív kontroll vizsgálatának korábbi adatait részletező jelentés, amely indokolja, hogy miért nem végzik a laboratóriumban pozitív kontroll párhuzamos vizsgálatát,
|
|
|
|
Eredmények:
|
—
|
az egyes állatok testtömege a kezelés kezdetén és a humánusan történő exterminálás tervezett időpontjában, illetve az átlagos és vonatkozó hibamutatók (pl. SD, SEM) kezelési csoportonként,
|
|
—
|
a toxicitási tünetek, ezen belül az alkalmazás helyén esetleg kialakuló bőrirritáció megjelenésének időpontja és időbeli alakulása minden egyes állatra vonatkozóan, amennyiben volt ilyen,
|
|
—
|
az állatonkénti BrdU jelölési indexek és kezelési csoportonkénti SI értékek táblázata,
|
|
—
|
átlagos és kapcsolódó hibamutatók (pl. SD, SEM) az állatonkénti BrdU jelölési index esetében, az egyes kezelés csoportokban, és a kiugró értékek elemzése az egyes kezelési csoportokban,
|
|
—
|
a kiszámított SI és a variabilitás megfelelő mutatója, amely az egyedek közti variabilitást a vizsgált anyaggal kezelt csoportban és a kontrollcsoportokban egyaránt figyelembe veszi,
|
|
—
|
dózis-válasz összefüggés,
|
|
—
|
adott esetben statisztikai elemzések,
|
|
|
|
Az eredmények tárgyalása:
|
—
|
rövid kommentár az eredményekhez, a dózis-válasz elemzéshez és adott esetben a statisztikai elemzésekhez, és következtetés levonása arra vonatkozóan, hogy a vizsgált anyagot bőrszenzibilizáló hatásúnak kell-e tekinteni vagy sem.
|
|
|
SZAKIRODALOM
|
(1)
|
OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(2)
|
Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999-1002.
|
|
(3)
|
Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985-997.
|
|
(4)
|
Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-33.
|
|
(5)
|
Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59.
|
|
(6)
|
ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program Center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf]
|
|
(7)
|
Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258-273.
|
|
(8)
|
Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274-286.
|
|
(9)
|
Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249-257.
|
|
(10)
|
ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA:BrdU-ELISA). NIH Publication No. 10-7552A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm]
|
|
(11)
|
ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf]
|
|
(12)
|
Takeyoshi, M., Iida, K., Shiraishi, K. and Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by non-radioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129-134.
|
|
(13)
|
OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(14)
|
Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904.
|
|
(15)
|
Basketter, D., Ball, N., Cagen, S., Carrilo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96.
|
|
(16)
|
ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
|
|
(17)
|
ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09-7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban:
[http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna-ps/LLNAPerfStds.pdf]
|
|
(18)
|
McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89.
|
|
(19)
|
Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31.
|
|
(20)
|
OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(21)
|
Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
|
|
(22)
|
ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
|
|
(23)
|
Hayes, B.B., Gerber, P.C., Griffey, S.S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195-206.
|
|
(24)
|
Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemical-induced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94.
|
|
(25)
|
Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256.
|
|
(26)
|
Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491-506.
|
|
(27)
|
Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68.
|
|
(28)
|
Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728.
|
|
(29)
|
Patterson, R.M., Noga, E. and Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028.
|
|
(30)
|
ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Elektronikus formátumban: [http://iccvam.niehs.nih.gov/methods/acutetox/Tox_workshop.htm].
|
|
(31)
|
OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Elektronikus formátumban: [http://www.oecd.org/env/testguidelines]
|
|
(32)
|
Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.l Health, 53, 563-79.
|
|
(33)
|
OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Available at: [http://www.oecd.org/env/testguidelines]
|
1. függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság: A vizsgálati módszerrel kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálati módszer teljesítményének mutatója, valamint a relevanciájának egyik megítélési szempontja. E kifejezést gyakran használják az »egyezés« megfelelőjeként, amely egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát fejezi ki (33).
Referenciaanyag: A vizsgált anyaggal való összehasonlítás céljából etalonként használt szenzibilizáló vagy nem szenzibilizáló anyag. A referenciaanyaggal szemben a következő elvárások fogalmazhatók meg: i. állandó és megbízható beszerzési forrásból vagy forrásokból kell származnia; ii. a vizsgált anyagok csoportjához hasonló szerkezettel és funkcionális jellemzőkkel kell rendelkeznie; iii. ismert fizikai-kémiai tulajdonságokkal kell rendelkeznie; iv. rendelkezésre kell állniuk az anyag ismert hatásait alátámasztó adatoknak; és v. a kívánt válaszreakciók tartományában ismert hatásúnak kell lennie.
Álnegatív eredmény: Valamely vizsgálati módszerrel tévesen negatívnak vagy inaktívnak minősített vizsgált anyag, amely valójában pozitív vagy aktív (33).
Álpozitív: Valamely vizsgálattal tévesen pozitívnak vagy aktívnak minősített vizsgált anyag, amely valójában negatív vagy inaktív (33).
Veszély: Káros egészségügyi vagy ökológiai hatások előidézésének lehetősége. A káros hatás csak akkor jelenik meg, ha kellő szintű az expozíció.
Laboratóriumok közötti reprodukálhatóság: Annak mértéke, hogy a különböző minősített laboratóriumok azonos protokoll és azonos vizsgált anyag alkalmazásával mennyire képesek minőségileg és mennyiségileg azonos eredményeket produkálni. A laboratóriumok közötti reprodukálhatóság az előhitelesítési és hitelesítési folyamat során kerül meghatározásra, és annak mértékét jelzi, hogy az adott vizsgálat mennyire sikeresen vihető át laboratóriumok között; más néven laboratóriumok közötti megismételhetőség (33).
Laboratóriumon belüli reprodukálhatóság: Annak meghatározása, hogy az ugyanazon laboratóriumban dolgozó képzett személyek meghatározott protokoll különböző időpontokban történő alkalmazásával mennyire sikeresen tudják reprodukálni az eredményeket. Más néven laboratóriumon belüli megismételhetőség (33).
Kiugró érték: A kiugró érték olyan megfigyelés, amely szembetűnően eltér az egy populációból vett véletlenszerű minta egyéb értékeitől.
Minőségbiztosítás: Irányítási folyamat, amelynek során a laboratóriumi vizsgálati szabványok és követelmények betartását, a jegyzőkönyvek vezetését, valamint az adattovábbítás pontosságát a vizsgálatot végző személyektől független személyek ellenőrzik.
Megbízhatóság: Azonos protokoll szerint végzett vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti reprodukálhatóságának mérésére szolgál. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg (33).
Bőrszenzibilizáció: Immunológiai folyamat, amely akkor jelentkezik, ha egy érzékeny egyén lokálisan érintkezik valamely indukáló hatású allergén vegyi anyaggal, amely esetlegesen kontakt szenzibilizáció kialakulásához vezető immunválaszt vált ki a bőrben.
Stimulációs index (SI): Adott vizsgált anyag bőrszenzibilizációt okozó képességének meghatározására kiszámított érték, amely az anyaggal kezelt csoportban és a párhuzamosan vivőanyaggal kezelt kontrollcsoportban tapasztalt proliferáció aránya.
Vizsgált anyag (más néven vizsgált vegyi anyag): Bármilyen anyag vagy keverék, amelyet ezzel a vizsgálati módszerrel vizsgálnak.
” |
