ISSN 1725-5090
doi:10.3000/17255090.L_2009.220.hun
Az Európai Unió
Hivatalos Lapja
L 220

Magyar nyelvű kiadás
Jogszabályok
52. évfolyam
2009. augusztus 24.
|
ISSN 1725-5090 doi:10.3000/17255090.L_2009.220.hun |
||
|
Az Európai Unió Hivatalos Lapja |
L 220 |
|
|
|
||
|
Magyar nyelvű kiadás |
Jogszabályok |
52. évfolyam |
|
Tartalom |
|
I Az EK-Szerződés/Euratom-Szerződés alapján elfogadott jogi aktusok, amelyek közzététele kötelező |
Oldal |
|
|
|
RENDELETEK |
|
|
|
* |
A Bizottság 761/2009/EK rendelete (2009. július 23.) a vegyi anyagok regisztrálásáról, értékeléséről, engedélyezéséről és korlátozásáról (REACH) szóló 1907/2006/EK európai parlamenti és a tanácsi rendelet értelmében alkalmazandó vizsgálati módszerek megállapításáról szóló 440/2008/EK bizottsági rendeletnek a műszaki fejlődéshez való hozzáigazítása céljából történő módosításáról ( 1 ) |
|
|
|
|
|
(1) EGT-vonatkozású szöveg |
|
HU |
Azok a jogi aktusok, amelyek címe normál szedéssel jelenik meg, a mezőgazdasági ügyek napi intézésére vonatkoznak, és rendszerint csak korlátozott ideig maradnak hatályban. Valamennyi más jogszabály címét vastagon szedik, és előtte csillag szerepel. |
I Az EK-Szerződés/Euratom-Szerződés alapján elfogadott jogi aktusok, amelyek közzététele kötelező
RENDELETEK
|
24.8.2009 |
HU |
Az Európai Unió Hivatalos Lapja |
L 220/1 |
A BIZOTTSÁG 761/2009/EK RENDELETE
(2009. július 23.)
a vegyi anyagok regisztrálásáról, értékeléséről, engedélyezéséről és korlátozásáról (REACH) szóló 1907/2006/EK európai parlamenti és a tanácsi rendelet értelmében alkalmazandó vizsgálati módszerek megállapításáról szóló 440/2008/EK bizottsági rendeletnek a műszaki fejlődéshez való hozzáigazítása céljából történő módosításáról
(EGT-vonatkozású szöveg)
AZ EURÓPAI KÖZÖSSÉGEK BIZOTTSÁGA,
tekintettel az Európai Közösséget létrehozó szerződésre,
tekintettel a vegyi anyagok regisztrálásáról, értékeléséről, engedélyezéséről és korlátozásáról (REACH), az Európai Vegyianyag-ügynökség létrehozásáról, az 1999/45/EK irányelv módosításáról, valamint a 793/93/EGK tanácsi rendelet, az 1488/94/EK bizottsági rendelet, a 76/769/EGK tanácsi irányelv, a 91/155/EGK, a 93/67/EGK, a 93/105/EK és a 2000/21/EK bizottsági irányelv hatályon kívül helyezéséről szóló, 2006. december 18-i 1907/2006/EK európai parlamenti és tanácsi rendeletre (1) és különösen annak 13. cikke (3) bekezdésére,
mivel:
|
(1) |
A Bizottság 440/2008/EK rendelete (2) tartalmazza az 1907/2006/EK rendelet céljából alkalmazandó anyagok fizikai-kémiai tulajdonságainak, toxicitásának és ökotoxicitásának meghatározására szolgáló vizsgálati módszereket. |
|
(2) |
Szükséges a Bizottság 440/2008/EK rendeletét aktualizálni bizonyos vizsgálati módszerekben végrehajtott változtatásokkal és az OECD által elfogadott bizonyos új vizsgálati módszerekkel történő kiegészítéssel. A javaslatról egyeztetés történt az érintettekkel. Az említett módosítások a tudományos és műszaki fejlődéshez igazítják a kérdéses módszereket. |
|
(3) |
Felül kell vizsgálni a gőznyomásra vonatkozó rendelkezéseket, hogy azok az új effúziós módszerre is kiterjedjenek. |
|
(4) |
A rostátmérő hosszal súlyozott mértani középértékének méréséhez új módszer felvételére van szükség. |
|
(5) |
Célszerű a 440/2008/EK rendeletet mindenekelőtt egy új in vitro bőrirritációs módszerrel aktualizálni annak érdekében, hogy a kísérleti és egyéb tudományos célokra felhasznált állatok védelmére vonatkozó tagállami törvényi, rendeleti és közigazgatási rendelkezések közelítéséről szóló, 1986. november 24-i 86/609/EGK tanácsi irányelvnek megfelelően a kísérleti célokra használt állatok számában csökkenést lehessen elérni (3). Bár az OECD-n belül még folyamatban van az egyeztetés egy in vitro bőrirritációs vizsgálati módszer tervezetével kapcsolatban, a helyzet kivételes jellege indokolja, hogy e rendeletben a B.46. módszer is helyet kapjon. Amint az OECD-n belül létrejön a megegyezés, illetve ha bármilyen időközben felmerülő információ azt szükségessé teszi, a B.46. módszert haladéktalanul felül kell vizsgálni. |
|
(6) |
Az algásodásgátlási vizsgálatra vonatkozó rendelkezéseket szükséges felülvizsgálni további fajok felvétele és a veszélyértékelésre, valamint a vegyianyag-besorolásra vonatkozó követelmények teljesítése érdekében. |
|
(7) |
Szükség van a biológiai lebonthatóság szimulációs vizsgálatán alapuló, a felszíni vízben zajló aerob mineralizáció mérésére szolgáló új módszer felvételére, valamint a Lemna nemzetségre gyakorolt toxicitás megállapítására szolgáló növekedésgátlási vizsgálaton alapuló új módszer felvételére. |
|
(8) |
A 440/2008/EK rendeletet ennek megfelelően módosítani kell. |
|
(9) |
Az e rendeletben előírt intézkedések összhangban vannak az 1907/2006/EK rendelet 133. cikkével létrehozott bizottság véleményével, |
ELGOGADTA EZT A RENDELETET:
1. cikk
A 440/2008/EK rendelet melléklete a következőképpen módosul:
|
1. |
Az A. rész a következőképpen módosul:
|
|
2. |
A B. rész a következőképpen módosul: A rész az e rendelet III. mellékletében megállapított B.46. fejezettel egészül ki. |
|
3. |
A C. rész a következőképpen módosul:
|
2. cikk
Ez a rendelet az Európai Unió Hivatalos Lapjában történő kihirdetését követő harmadik napon lép hatályba.
Ez a rendelet teljes egészében kötelező és közvetlenül alkalmazandó valamennyi tagállamban.
Kelt Brüsszelben, 2009. július 23-án.
a Bizottság részéről
Stavros DIMAS
a Bizottság tagja
(1) HL L 396., 2006.12.30., 1. o.
(2) HL L 142., 2008.5.31., 1. o.
(3) HL L 358., 1986.12.18., 1. o.
I. MELLÉKLET
|
A.4. |
GŐZNYOMÁS |
1. MÓDSZER
Ez a módszer megfelel az OECD TG 104 (2004) jelű módszernek.
1.1. BEVEZETÉS
Az A.4. (1) módszer jelen átdolgozott változata tartalmaz egy újabb módszert: „Effúziós módszer: izoterm termogravimetria”, amit nagyon kis gőznyomású (10–10 Pa-ig) anyagokra dolgoztak ki. Az eljárásokra vonatkozó igények, különösen a kis gőznyomású anyagok gőznyomásának mérése iránti igények alapján a módszer más eljárásai is átértékelésre kerültek további alkalmazási tartományokat illetően.
Termodinamikailag egyensúlyban lévő tiszta anyagok gőznyomása csak a hőmérséklet függvénye. Az alapelvek ismertetése megtalálható az irodalomban (2)(3).
Nincs egyetlen mérési eljárás a gőznyomás mérésére a 10–10 Pa alattitól a 105 Pa-ig terjedő tartomány egészére. Ez a módszer nyolc különböző gőznyomásmérési módszert foglal magában, melyek különböző gőznyomás-tartományokra alkalmazhatóak. Az 1. táblázat a különböző módszereket hasonlítja össze az alkalmazásuk és a mérési tartományok szerint. A módszereket csak olyan anyagok esetén lehet alkalmazni, amelyek nem bomlanak a mérés körülményei között. Azokban az esetekben, amikor kísérleti módszerek eljárástechnikai okok miatt nem alkalmazhatók, a gőznyomás becsléssel is megállapítható, és a függelék tartalmaz egy ajánlott becslési módszert.
1.2. DEFINÍCIÓK ÉS MÉRTÉKEGYSÉGEK
Egy anyag gőznyomása definíció szerint a szilárd vagy folyékony anyag felett kialakult, telített gőz nyomása.
A nyomás SI mértékegységét, a pascalt (Pa) kell használni. Korábban alkalmazott más mértékegységek, és átváltási tényezőik:
|
1 torr |
= |
1 Hgmm |
= |
1,333 × 102 Pa |
|
1 atmoszféra |
= |
1,013 × 105 Pa |
|
|
|
1 bar |
= |
105 Pa |
|
|
Az SI szerinti hőmérséklet-mértékegység a kelvin (K). A Celsius fok átváltása kelvin fokra a következő egyenlet alapján történik:
T = t + 273,15
ahol T a kelvinben mért, más néven termodinamikai hőmérséklet, t pedig a hőmérséklet Celsius fokban.
1. táblázat
|
Mérési módszer |
Anyag |
becsült ismételhetőség |
becsült reprodukálhatóság |
ajánlott tartomány |
|
|
szilárd |
folyékony |
||||
|
Dinamikus módszer |
kis olvadáspont |
Igen |
25 %-ig 1–5 % |
25 %-ig 1–5 % |
103 Pa–2 × 103 Pa 2 × 103 Pa–105 Pa |
|
Statikus eljárás |
Igen |
Igen |
5–10 % |
5–10 % |
10 Pa–105 Pa 10–2 Pa–105 Pa (1) |
|
Izoteniszkópos módszer |
Igen |
Igen |
5–10 % |
5–10 % |
102 Pa–105 Pa |
|
Effúziós módszer: gőznyomás-egyensúly |
Igen |
Igen |
5–20 % |
50 %-ig |
10–3–1 Pa |
|
Effúziós módszer: Knudsen-cella |
Igen |
Igen |
10–30 % |
— |
10–10–1 P |
|
Effúziós módszer: izoterm termogravimetria |
Igen |
Igen |
5–30 % |
50 %-ig |
10–10–1 Pa |
|
Gáztelítéses eljárás |
Igen |
Igen |
10–30 % |
50 %-ig |
10–10–103 Pa |
|
Rotoros eljárás |
Igen |
Igen |
10–20 % |
— |
10–4–0,5 Pa |
1.3. A MÉRÉS ELVE
A gőznyomás meghatározása általában különböző hőmérsékleteken történik. Véges hőmérséklettartományban a tiszta anyag gőznyomásának logaritmusa lineáris függvénye a termodinamikai hőmérséklet reciprokának, az egyszerűsített Clapeyron-Clausius egyenlet szerint:

ahol:
|
p |
= |
gőznyomás, pascal (Pa) |
|
ΔHv |
= |
párolgási entalpia, J mól–1 |
|
R |
= |
egyetemes gázállandó, 8,314 J mól–1 K–1 |
|
T |
= |
hőmérséklet, kelvin fok (K) |
1.4. REFERENCIAANYAGOK
Referenciaanyagokat nem szükséges használni. Ezek elsődlegesen a módszer alkalmasságának rendszeres ellenőrzésére szolgálnak, illetve lehetővé teszik a különböző módszerekkel kapott eredmények összehasonlítását.
1.5. AZ ELJÁRÁS LEÍRÁSA
1.5.1. Dinamikus eljárás (Cottrell-módszer)
1.5.1.1. Elv
Az anyag gőznyomását a forráspontnak különböző meghatározott nyomásokon (kb. 103 és 105 Pa közötti tartományban) történő mérésével határozzuk meg. Ez az eljárás ajánlott a forráspont meghatározására is. Erre a célra egészen 600 K-ig használható. A folyadékok forráspontja 3–4 cm-rel a folyadék felszíne alatt közelítőleg 0,1 oC-kal nagyobb, mint a felszínén, a folyadékoszlop hidrosztatikai nyomása miatt. A Cottrell-módszernél (4) a hőmérő a folyadék felszíne fölötti gőztérben van, és a forrásban lévő folyadék a forrás szivattyúzó hatásának köszönhetően folyamatosan elborítja a hőmérő érzékelőjét. Ekkor egy vékony folyadékréteg fedi be a hőmérő érzékelőgömbjét, amely egyensúlyt tart a gőzzel a környezeti nyomáson. A hőmérő így a valódi forráspontot mutatja, a túlhevítés, vagy a hidrosztatikai nyomásból eredő hiba nélkül. Az eredeti Cottrell-szivattyú az 1. ábrán látható. Az A jelű lombik tartalmazza a forrásban lévő folyadékot. Egy platinaszál (B) van a lombik fenekébe forrasztva, biztosítva az egyenletes forrást. A C jelű csonk egy kondenzátorral van összekötve, a D jelű köpeny pedig megakadályozza, hogy a hideg kondenzátum elérje a hőmérőt (E). Amikor a folyadék forr a lombikban, a tölcsér által felfogott buborékok és folyadék a szivattyú két ágán (F) keresztül ráfolyik a hőmérő gömbjére.
|
1. ábra
|
2 ábra
|
Cottrell-szivattyú (4)
|
A: |
Hőelem |
|
B: |
Vákuumpuffer |
|
C: |
Nyomásmérő |
|
D: |
Vákuum |
|
E: |
Mérőpont |
|
F: |
Fűtőelem, körülbelül 150 W-os |
1.5.1.2. Készülék
A Cottrell-elv alapján működő, nagyon pontos készülék látható a 2. ábrán. Ez egy lombikból áll, amelynek az alsó részén történik a forralás, a középső részén van egy hűtő, és a felső részén van egy kivezetés és egy zárófedél. A Cottrell-szivattyú a forrástérben van elhelyezve, amit egy elektromos fűtőbetét hevít. A hőmérsékletet a zárófedélen keresztül bevezetett tokozott hőelem, vagy pedig ellenállás-hőmérő méri. A kivezetés a nyomásszabályozó rendszerbe van bekötve. Az utóbbi egy vákuumszivattyúból, egy puffertérből, egy nitrogénadagolással működő nyomásszabályozóból, és egy nyomásmérőből áll.
1.5.1.3. Eljárás
Az anyagot a forrástérbe helyezzük. Nem porszerű szilárd anyagok problémát jelenthetnek, de ezek néha kiküszöbölhetőek a hűtőköpeny fűtésével. A készüléket a fedéllel lezárjuk, és az anyagot gáztalanítjuk. Habzó anyag nem mérhető ezzel a módszerrel.
Ezután beállítjuk a legkisebb kívánt nyomást és bekapcsoljuk a fűtést. Ugyanekkor a hőmérsékletérzékelőt rákötjük egy regisztrálóműszerre.
Az egyensúlyt akkor értük el, amikor állandó nyomás mellett a regisztrálóműszer állandó forrási hőmérsékletet mutat. Külön figyelmet kell fordítani arra, hogy elkerüljük a megfutást a forralás alatt. Fontos továbbá, hogy teljes kondenzáció történjen a hűtőben. Amikor kis olvadáspontú szilárd anyag gőznyomását határozzuk meg, figyelmet kell fordítani a hűtő esetleges eltömődésének elkerülésére is.
Az egyensúlyi pont regisztrálása után állítsuk be a következő kívánt nyomást. A folyamatot egészen a 105 Pa eléréséig folytatjuk (összesen kb. 5–10 mérési pont). Ellenőrzésként ismételjük meg az eljárást visszafelé, úgy, hogy a nyomásértékek csökkennek.
1.5.2. Statikus eljárás
1.5.2.1. Elv
A statikus eljárásban (5), a gőznyomást egy adott hőmérsékleten termodinamikai egyensúlyban határozzuk meg. Ez az eljárás olyan anyagokhoz, többkomponensű folyadékokhoz és szilárd anyagokhoz használható, melyek gőznyomástartománya 10–1–105 Pa tartományban van, valamint, megfelelő gondossággal, használható az 1–10 Pa tartományban is.
1.5.2.2. Készülék
A készülék egy állandó hőmérsékletű (pontosság: ±0,2 K) fürdőből, egy vákuumvezetékhez kapcsolt mintatartályból, egy nyomásmérőből, és egy nyomásszabályozó rendszerből áll. A mintatartály (3a. ábra) egy szelepen és egy nyomáskülönbség-mérőn (U-csöves manométer alkalmas folyadékkal) keresztül kapcsolódik a vákuumvezetékhez, ez utóbbi a nulla nyomáskülönbség jelzésére szolgál. A nyomástartománytól és a mérendő anyag kémiai viselkedésétől függően higany, szilikonok és ftalátok használhatók az U-csöves manométerben. Környezetvédelmi szempontok miatt azonban a higany használata lehetőség szerint kerülendő. A mérendő anyag nem oldódhat észlelhető mértékben az U-csöves manométerben lévő folyadékban, és nem léphet vele reakcióba. Az U-csöves manométer helyett használható nyomásmérő (3b. ábra). Az U-csöves manométerben higany a légköri nyomástól a 102 Pa-ig terjedő tartományban használható, míg a szilikonfolyadékok és ftalátok 102 Pa alatt is használhatók, egészen 10 Pa nyomásig. Léteznek más nyomásmérők is, amelyeket lehet 102 Pa alatt használni, és a fűthető membrános kapacitív nyomásmérők használhatók még 10–1 Pa alatt is. A hőmérséklet mérése a mintát tartalmazó edény külső falán vagy magában az edényben történik.
1.5.2.3. Eljárás
A 3a. ábrán látható készülék használata esetén a mérés megkezdése előtt az U-csöves manométert megtöltjük a kiválasztott folyadékkal, amelyet a mérések megkezdése előtt melegítéssel gáztalanítani kell. Helyezzük be a mérendő anyagot a készülékbe és gáztalanítsuk csökkentett hőmérsékleten. Többkomponensű minta esetében olyan kis hőmérsékletet kell alkalmazni, amelyen a mérendő anyag összetétele még nem változik meg. Az egyensúly beállását keveréssel gyorsíthatjuk. A minta folyékony nitrogénnel vagy szárazjéggel hűthető, de vigyázni kell, hogy elkerüljük a levegőben lévő pára, vagy a szivattyúfolyadék lecsapódását A mintatartó edény fölött lévő szelepet kinyitva néhány perces szivattyúzással eltávolítjuk a levegőt. Ha szükséges, a gáztalanítási műveltet néhányszor megismételjük.
|
3a. ábra
|
3b. ábra
|
Amikor a szelep zárva van és hevítjük az anyagot, a gőznyomás nő. Ez megváltoztatja a folyadék egyensúlyát az U-csőben. Ellensúlyozandó ezt a hatást, nitrogént vagy levegőt engedünk a rendszerbe addig, amíg a nyomáskülönbség-mérő ismét nullát nem mutat. A kiegyenlítéshez szükséges nyomás értéke leolvasható a manométerről, vagy egy nagyobb pontosságú műszerről. Ez a nyomás felel meg az anyag gőznyomásának az adott hőmérsékleten. Ha a 3b. ábrán látható készüléket használjuk, akkor a gőznyomás közvetlenül leolvasható.
A gőznyomás meghatározása megfelelő kis hőmérsékletintervallumonként történik (összesen kb. 5–10 mérési pont) egészen a kívánt legnagyobb hőmérsékletig.
A kis hőmérsékletértékeket eredményező méréseket ellenőrzés céljából meg kell ismételni. Ha a megismételt mérés eredménye nem esik egybe a növekvő hőmérsékletre kapott görbével, az a következő két eset valamelyikére vezethető vissza:
|
i. |
a minta még mindig tartalmaz levegőt (pl. nagy viszkozitású anyagok esetében) vagy kis forráspontú anyagokat, melyek felszabadulnak a melegítés alatt; |
|
ii. |
az anyagnál kémiai reakció lép fel a vizsgált hőmérséklettartományban (pl. bomlás, polimerizáció). |
1.5.3. Izoteniszkópos eljárás
1.5.3.1. Elv
Az izoteniszkóp (6) a statikus eljárás elvén alapul. A módszernél az anyagot egy állandó hőmérsékleten tartott gömbbe helyezzük, és csatlakoztatunk egy manométert és egy vákuumszivattyút. Az anyagnál illékonyabb szennyeződések csökkentett nyomás mellett gáztalanítással eltávolíthatók. A minta gőznyomását adott hőmérsékleteken ismert nyomású semleges gázzal egyensúlyba hozzuk. Az izoteniszkópot egyes folyékony szénhidrogének mérésére fejlesztették ki, de alkalmas szilárd anyagok vizsgálatára is. A módszer általában nem alkalmazható többkomponensű rendszerek esetében. Nem illékony szennyeződéseket tartalmazó minták esetében az eredményekben csak kisebb hibák jelentkeznek. Az ajánlott tartomány 102–105 Pa.
1.5.3.2. Készülék
A mérőkészülékre a 4. ábrán látható példa. Teljes leírás az ASTM D 2879–86 szabványban (6) található.
1.5.3.3. Eljárás
Folyadékok esetén magát az anyagot használjuk töltőfolyadékként az U-csöves manométerben. Az izoteniszkópba elegendő folyadékot töltünk ahhoz, hogy a gömb és a manométer rövidebb szára feltöltődjön. Az izoteniszkópot csatlakoztatjuk egy vákuumrendszerhez, kiszivattyúzzuk a levegőt majd megtöltjük nitrogénnel. A kiszivattyúzást és a rendszer átöblítését kétszer megismételjük, hogy eltávolítsuk a maradék oxigént. A megtöltött izoteniszkópot vízszintes helyzetbe állítjuk, hogy a minta vékony rétegben szétterüljön a mintatartó gömbben és a manométerben. A rendszer nyomását 133 Pa-ra csökkentjük, és a mintát óvatosan melegítjük, amíg éppen forrni nem kezd (az oldott gázok eltávolítása). Ezután az izoteniszkópot úgy helyezzük el, hogy a minta visszatérjen a gömbbe és feltöltse a manométer rövid szárát. A nyomást 133 Pa-on kell tartani. A mintatartó gömb elnyújtott csúcsát kis lánggal hevítjük úgy, hogy a mintából felszabaduló gőz elegendően kiterjedjen ahhoz, hogy kiszorítsa a minta egy részét a mintatartó gömb felső részéből és a manométer szárából a manométerbe, előállítva így egy gőzzel töltött, nitrogénmentes teret. Ezután az izoteniszkópot állandó hőmérsékletű fürdőbe merítjük, és a nitrogén nyomását beállítjuk úgy, hogy egyenlő legyen a mintáéval. Egyensúlyban a nitrogén nyomása megegyezik az anyag gőznyomásával.
4. ábra

Szilárd anyag esetén a hőmérséklet- és nyomástartománytól függően a manométer töltőfolyadékaként szilikonfolyadékok, vagy ftalátok használhatók. A gáztalanított manométerfolyadékot az izoteniszkóp hosszú szárán kialakított öblösebb részbe töltjük be. Ezután a vizsgálandó szilárd anyagot behelyezzük a mintatartó gömbbe, és melegítéssel gáztalanítjuk. Ezt követően az izoteniszkópot megdöntjük, hogy a manométerfolyadék befolyjon az U-csőbe.
1.5.4. Effúziós eljárás: gőznyomás-egyensúly (7)
1.5.4.1. Elv
A vizsgálati anyagból vett mintát légüres búra alatt egy kis kemencében hevítjük. A kemence fedelén ismert átmérőjű kis lyukak vannak. Az anyag gőze kilép az egyik lyukon és egy nagyon érzékeny mérleg serpenyőjébe jut, ami szintén a légüres búra alatt van. Néhány konstrukcióban a mérleg serpenyője egy hűtődobozban van, ami hővezetéssel kivonja a hőt a kültérbe, és a serpenyőt hősugárzás révén hűti, hogy a gőz kondenzálódjon rajta. A kilépő gőz nyomatéka erőként hat a mérlegre. A gőznyomást kétféleképpen kaphatjuk meg: közvetlenül a mérleg serpenyőjére ható erőből, vagy pedig a párolgási sebességből, a Hertz-Knudsen egyenlet segítségével (2):
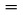
ahol:
|
G |
= |
párolgási sebesség (kg s–1 m–2) |
|
M |
= |
moláris tömeg (g mol–1) |
|
T |
= |
hőmérséklet (K) |
|
R |
= |
egyetemes gázállandó (J mol–1 K–1) |
|
p |
= |
gőznyomás (Pa) |
Az ajánlott tartomány 10–3–1 Pa.
1.5.4.2. Készülék
A készülék működési elve az 5. ábrán látható.
5. ábra

|
A: |
Alaplemez |
F: |
Hűtődoboz és hűtőrúd |
|
B: |
Mozgó tekercses berendezés |
G: |
Párologtató kemence |
|
C: |
Búra |
H: |
Dewar-edény folyékony nitrogénnel |
|
D: |
Mérleg serpenyővel |
I: |
Minta hőmérsékletének mérése |
|
E: |
Vákuummérő műszer |
J: |
Mérendő anyag |
1.5.5. Effúziós eljárás: Knudsen-cella
1.5.5.1. Elv
Az eljárás a vizsgált anyag Knudsen-cellából (8) mikronyíláson ultravákuumban egységnyi idő alatt gőz formában kilépő tömegének becslésén alapul. A kilépő gőz tömegét meghatározhatjuk a cella tömegvesztéséből, vagy úgy, hogy a gőzt kis hőmérsékleten kondenzáltatjuk, és az elpárolgott anyag mennyiségét kromatográfiásan meghatározzuk. A gőznyomást a Hertz-Knudsen összefüggésből számolhatjuk ki (lásd 1.5.4.1.), a készülék paramétereitől függő korrekciós tényezőkkel (9). Az ajánlott tartomány 10–10–1 Pa (10)(11)(12)(13)(14).
1.5.5.2. Készülék
A készülék működési elve a 6.ábrán látható.
6. ábra

|
1: |
Vákuumcsatlakozás |
7: |
Menetes fedél |
|
2: |
Platina ellenállás-hőmérő számára, vagy hőmérsékletmérésre és -szabályozásra szolgáló furatok |
8: |
Szárnyas anyák |
|
3: |
Vákuumtartály fedele |
9: |
Csavarok |
|
4: |
O-gyűrű |
10: |
Effúziós cella saválló acélból |
|
5: |
Alumínium vákuumtartály |
11: |
Fűtőbetét |
|
6: |
Eszköz az effúziós cellák be- és kihelyezéséhez |
|
|
1.5.6. Effúziós eljárás: izoterm termogravimetria
1.5.6.1. Elv
A módszer a vizsgált anyag nagyobb hőmérsékleten és környezeti nyomáson fellépő megnövelt párolgási sebességeinek termogravimetriás módszerrel történő meghatározásán alapul (10)(15)(16)(17)(18)(19)(20). A párolgási sebességeket (vT) úgy kapjuk, hogy a kiválasztott anyagot lassan áramló inert gáznak tesszük ki, és megfigyeljük az adott T izoterm hőmérsékleten (kelvin) megfelelő időegységek alatt bekövetkező tömegveszteséget. A gőznyomást (pT) a vT értékekből számítjuk ki, a gőznyomás logaritmusa és a párolgási sebesség logaritmusa közötti lineáris összefüggés segítségével. Ha szükséges, a (log pT)-(1/T) görbét regressziós analízissel extrapolálhatjuk 20 °C-ra és 25 °C-ra. Az eljárás alkalmas nagyon kicsi, 10–10 Pa (10–12 mbar) gőznyomású anyagok méréséhez is, de csak nagyon nagy tisztaságú (majdnem 100 %-os) anyagok esetén, elkerülendő a mért tömegvesztés hibás értelmezését.
1.5.6.2. Készülék
Az kísérleti készülék működési elve a 7. ábrán látható.
7. ábra

A mintatartó tányér szabályozott hőmérsékletű kamrában függ egy mikromérlegen, és száraz nitrogén gáz áramlik át rajta, amely elszállítja a mérendő anyagból elpárolgó molekulákat. A kamrából kilépő gázáramot egy abszorpciós egység tisztítja.
1.5.6.3. Eljárás
A mérendő anyagot homogén rétegben szétterítjük egy érdesített felületű üvegtányéron. Szilárd anyagok esetén a tányért egyenletesen benedvesítjük az anyag megfelelő oldószerben képzett oldatával, és inert gázzal megszárítjuk. A méréshez a tányért, rajta a mintával, felfüggesztjük a termogravimetriás analizátorban, és folyamatosan mérjük a tömegvesztését az idő függvényében.
Az adott hőmérséklethez tartozó párolgási sebességet (vT) a mintatartó tányér Δm tömegvesztéséből számítjuk a következő képlettel:
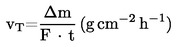
Ahol az F a mérendő anyag felszíne, szokásosan a mintatányér felszíne, a t pedig a Δm tömegvesztéshez tartozó időtartam.
A gőznyomás (pT) a vT párolgási sebesség függvényeként számolható:
Log pT = C + D log vT,
ahol a C és D állandók, melyek az adott kísérleti elrendezésre jellemzőek és a használt mérőkamra átmérőjétől és a gáz áramlási sebességétől függenek. Ezeket az állandókat egyszer kell meghatározni, ismert gőznyomású vegyületek mérésével és a kapott (log pT)-(log v) függvények regresszióelemzésével (11)(21)(22).
A gőznyomás (pT)és a hőmérséklet T (kelvin) közötti összefüggést a következő egyenlet adja meg:
Log pT = A + B 1/T
ahol A és B a (log pT)-(1/T) függvény regresszióelemzésével kapott állandók. Az egyenlet segítségével a gőznyomás extrapolációval átszámolható bármely más hőmérsékletre.
1.5.7. Gáztelítéses eljárás (23)
1.5.7.1. Elv
Semleges gáz ismert sebességgel, szobahőmérsékleten áramlik a mintaanyag felett vagy azon keresztül, elég lassan ahhoz, hogy megtörténjen a telítődés. A gázfázis telítettségének elérése kritikus fontosságú. A gáz által szállított anyagot felfogjuk, általában abszorbenssel, és ezt a felfogott anyagmennyiséget határozzuk meg. A gőz felfogása és az azt követő analízis alternatívájaként a szállított anyag mennyiségi meghatározására folyamatos analitikai technikák is használhatók, mint például gázkromatográfia. A gőznyomás annak a feltételezésnek az alapján számítható ki, hogy érvényes az ideálisgáz-törvény, és hogy a gázkeverék össznyomása megegyezik az azt alkotó gázok parciális nyomásának összegével. A mintaanyag parciális nyomása, azaz a gőznyomás az ismert össz-gáztérfogatból és a gáz által szállított anyag tömegéből számítható ki.
A gáztelítéses eljárás szilárd, vagy folyékony anyagok mérésére használható. Egészen 10–10 Pa gőznyomásig alkalmazható (10)(11)(12)(13)(14). Az eljárás 103 Pa gőznyomás alatt a legmegbízhatóbb. Általában 103 Pa felett a gőznyomás értéke túlbecsült, valószínűleg aeroszol-képződés miatt. Mivel a gőznyomás-mérés szobahőmérsékleten történik, nincs szükség nagy hőmérsékletről történő extrapolálásra – amely gyakran komoly hibákat okoz – és így az elkerülhető.
1.5.7.2. Készülék
Az eljárás állandó hőmérsékletű készülék használatát igényli. A 8. ábrán lévő rajzon látható készülékben három-három mintatartó van folyékony, illetve szilárd mintákhoz, ami lehetővé teszi egy szilárd vagy folyékony minta hármas elemzését. A hőmérsékletszabályozás pontossága ±0,5 °C, vagy jobb.
8. ábra

Inert vivőgázként általában nitrogént használunk, de esetenként más gáz lehet szükséges (24). A vivőgáznak száraznak kell lennie. A gázáram 6 részáramra van osztva, amelyeket tűszelep szabályoz (kb. 0,79 mm-es szelepnyílás), és 3,8 mm belső átmérőjű rézcsöveken keresztül jut be az edénybe. A hőmérséklet kiegyenlítődése után a gáz átáramlik a mintán és az abszorbensen, majd kilép a készülékből.
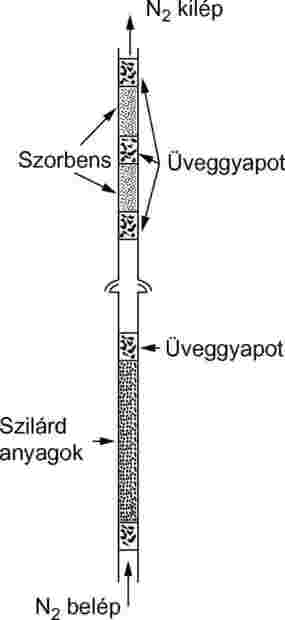
Szilárd mintákat 5 mm-es belső átmérőjű üvegcsőbe, üveggyapot-dugók közé töltünk (9. ábra). A 10. ábrán egy folyadékminta-tartó és abszorpciós rendszer látható. Folyadékok gőznyomás-mérésének legjobban reprodukálható módszere az, hogy a folyadékot üveggyöngyre vagy inert abszorbens anyagra (pl. szilika) visszük fel, és a mintatartót ezekkel a gyöngyökkel töltjük meg. Más megoldásként a vivőgáz durva üvegszemcséken áramlik keresztül, és átbuborékol a vizsgált anyag folyadékoszlopán.
|
9. ábra
|
10. ábra
|
Az abszorbensrendszer egy elülső és egy hátsó abszorbensszakaszból áll. Nagyon kis gőznyomáson az abszorbens csak csekély mennyiséget tart vissza és a minta és az abszorbens között lévő üveggyapoton és üvegcsövön lejátszódó abszorpció komoly probléma lehet.
Egy másik hatékony módja az elpárolgott anyag felfogásának a szárazjéggel hűtött folyadékcsapda. Nem okoz ellennyomást a telítő oszlopon, és a felfogott anyag teljes mennyisége is könnyen visszanyerhető.
1.5.7.3. Eljárás
A kilépő vivőgáz áramlási sebességét szobahőmérsékleten határozzuk meg. Az áramlási sebességet gyakran kell ellenőrizni a kísérlet folyamán, biztosítandó, hogy pontos érték álljon rendelkezésre a vivőgáz össztérfogatára vonatkozóan. Célszerű folyamatos ellenőrzést végezni tömegáram-mérővel. A gázfázis telítése meglehetősen hosszú tartózkodási időt igényel, így a gázáramlási sebességek meglehetősen kicsik lesznek (25).
A kísérlet végén mind az elülső, mind a hátsó abszorbensszakaszt külön-külön elemezzük. A vegyületet mindegyik szakaszban oldószer hozzáadással deszorbeáljuk. A kapott oldatokat kvantitatív elemzésnek vetjük alá az egyes szakaszokból deszorbeált mennyiségek meghatározására. A vizsgált anyag jellege szabja meg az analitikai módszer kiválasztását (illetve az abszorbens és a deszorpcióhoz használt oldószer kiválasztását is). A deszorpció hatékonyságát úgy határozzuk meg, hogy ismert mennyiségű mintát juttatunk az abszorbensre, majd deszorbeáljuk és meghatározzuk a visszanyert anyagmennyiséget. Fontos ellenőrizni a deszorpció hatékonyságát a kísérletben használt, vagy ahhoz közeli mintakoncentrációkkal.
Három különböző gázáramlási sebességet kell használni, biztosítandó, hogy a vivőgáz telítődjön a minta gőzével. Ha a számított gőznyomás-értékek azt mutatják, hogy függetlenek az áramlási sebességtől, akkor a gázt telítettnek tételezzük fel.
A gőznyomás a következő egyenletből számítható ki:
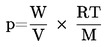
ahol:
|
p |
= |
gőznyomás (Pa) |
|
W |
= |
elpárolgott minta tömege (g) |
|
V |
= |
a telített gáz térfogata (m3) |
|
R |
= |
egyetemes gázállandó, 8,314 (J mol–1 K–1) |
|
T |
= |
hőmérséklet (K) |
|
M |
= |
a vizsgált anyag moláris tömege (g mol–1) |
A mért térfogatot korrigálni kell az áramlásmérő és a telítő közötti nyomás- és hőmérsékletkülönbségekkel.
1.5.8. Rotoros eljárás
1.5.8.1. Elv
Ez az eljárás rotoros viszkozitásmérőt használ, amelyben a mérőelem egy mágneses térben lebegő, kis acélgolyó, amelyet forgó mágneses mező forgat (26)(27)(28). A forgási sebességét felvevőtekercsek mérik. Amikor a golyó elért egy adott fordulatszámot (rendszerint kb. 400 fordulat/sec), kikapcsoljuk a forgatómágneseket és a gázban fellépő súrlódás következtében a golyó lassulni kezd. A forgási sebesség csökkenését az idő függvényében mérjük. A gőznyomást az acélgolyó nyomásfüggő lassulásából számíthatjuk. Az ajánlott tartomány 10–4–0,5 Pa.
1.5.8.2. Készülék
A kísérleti készülék sematikus ábrája a 11. ábrán látható. A mérőfej egy 0,1 °C pontossággal szabályozott állandó hőmérsékletű szekrényben van. A mintatartó egy külön szekrényben van, és ennek is 0,1 °C-on belül szabályozzuk a hőmérsékletét. A készülék minden más részén a hőmérsékletnek ennél nagyobbnak kell lennie, hogy elkerüljük a kondenzációt. Az egész készülék egy nagy vákuumot biztosító rendszerrel van összekapcsolva.
11. ábra

2. ADATOK ÉS DOKUMENTÁCIÓ
2.1. ADATOK
A fenti ELJÁRÁSOK bármelyikével kapott gőznyomást legalább két hőmérsékleten kell meghatározni. Három, vagy ennél több pont ajánlott 0–50 °C tartományban, hogy ellenőrizzük a gőznyomás-görbe linearitását. Az effúziós módszer (Knudsen-cella és izoterm termogravimetria) és a gáztelítéses módszer esetén a 0–50 °C mérési hőmérséklettartomány helyett 120–150 °C tartomány ajánlott.
2.2. MÉRÉSI JEGYZŐKÖNYV
A mérési jegyzőkönyvnek tartalmaznia kell a következő információkat:
|
— |
alkalmazott módszer, |
|
— |
a minta pontos leírása (megnevezés és szennyezők), és adott esetben az előzetes tisztítási műveletek, |
|
— |
legalább két gőznyomás- és hőmérsékletérték (de három vagy több ajánlott) a 0–50 °C (vagy 120–150 °C) tartományban, |
|
— |
legalább egy mérésnek 25 °C-on vagy az alatt kell történnie, ha a választott módszerrel ez technikailag lehetséges, |
|
— |
minden eredeti adat, |
|
— |
a (log p)–(1/T) görbe, |
|
— |
a gőznyomás becslése 20 vagy 25 °C-ra. |
Ha átalakulás lép fel (halmazállapot-változás, bomlás), a következő információkat fel kell tüntetni jegyzőkönyvben:
|
— |
a változás jellege, |
|
— |
az a hőmérséklet, ahol a változás bekövetkezett légköri nyomáson, |
|
— |
gőznyomás 10 és 20c°C-kal az átalakulási hőmérséklet alatt és 10 és 20 °C-kal e hőmérséklet felett (kivéve, ha az átalakulás szilárd állapotból gáz állapotba történik). |
Minden, az eredmények értelmezése szempontjából fontos információt és észrevételt fel kell tüntetni a jegyzőkönyvben, különösen a minta szennyeződéseire és az anyag fizikai állapotára vonatkozókat.
3. IRODALOM
|
(1) |
Official Journal of the European Communities L 383 A, 26–47 (1992). |
|
(2) |
Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Neindre, B., and Vodar, B., Eds., Butterworths, London. |
|
(3) |
Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Vol. I, Part I. Chapter IX, Interscience Publ., New York. |
|
(4) |
Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York. |
|
(5) |
NF T 20–048 AFNOR (September 1985). Chemical products for industrial use – Determination of vapour pressure of solids and liquids within a range from 10–1 to 105 Pa – Static method. |
|
(6) |
ASTM D 2879–86, Standard test method for vapour pressure – temperature relationship and initial decomposition temperature of liquids by isoteniscope. |
|
(7) |
NF T 20–047 AFNOR (September 1985). Chemical products for industrial use –Determination of vapour pressure of solids and liquids within range from 10–3 to 1 Pa – Vapour pressure balance method. |
|
(8) |
Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. |
|
(9) |
Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, 1173. |
|
(10) |
Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, 521–532. |
|
(11) |
Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000) |
|
(12) |
Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), 22–28 |
|
(13) |
Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), 269–278. |
|
(14) |
Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twenty-nine halogenated dibenzo-p-dioxins, Thermochimia Acta 112 Issue 1 (1987), 117–122. |
|
(15) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973) 137–147. |
|
(16) |
Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974) 393–400. |
|
(17) |
Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982) 161–168. |
|
(18) |
Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based on Evaporation Rate; Pesticide Science 45 (1995) 27–31. |
|
(19) |
Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science, 53 (1998) 300–310. |
|
(20) |
Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998) 1512–20. |
|
(21) |
Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range – 25 °C to 150 °C. |
|
(22) |
Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002) |
|
(23) |
40 CFR, 796. (1993). pp 148–153, Office of the Federal Register, Washington DC |
|
(24) |
Rordorf B.F. (1985). Thermochimica Acta 85, 435. |
|
(25) |
Westcott et al. (1981). Environ. Sci. Technol. 15, 1375. |
|
(26) |
Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), 2440. |
|
(27) |
Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), 642. |
|
(28) |
Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), 1715. |
(1) Kapacitív manometer használatakor.
Függelék
Becslési eljárás
BEVEZETÉS
A gőznyomásra becsült értéket használhatunk a következő esetekben:
|
— |
annak eldöntésére, hogy melyik kísérleti módszer a megfelelő, |
|
— |
becsült érték vagy határérték megadására, amikor a kísérleti módszer technikai okok miatt nem kivitelezhető. |
BECSLÉSI MÓDSZER
Folyadékok és szilárd anyagok gőznyomása megbecsülhető a módosított Watson-korrelációval (a). Az egyetlen szükséges kísérleti adat a normál forráspont. Az eljárás a 10–5 Pa–105 Pa tartományban alkalmazható.
Részletes információk találhatók a „Handbook of Chemical Property Estimation Methods” című kézikönyvben (b). Lásd még: OECD Environmental Monograph No. 67 (c).
SZÁMÍTÁSI ELJÁRÁS
A gőznyomás a következők szerint számolható:

ahol:
|
T |
= |
a kérdéses hőmérséklet |
|
Tb |
= |
a normál forráspont |
|
Pvp |
= |
gőznyomás a T hőmérsékleten |
|
ΔHvb |
= |
párolgási entalpia |
|
ΔZb |
= |
kompresszibilitási tényező (becsült: 0,97) |
|
m |
= |
tapasztalati tényező, a vizsgált hőmérsékleten fennálló fizikai állapottól függ |
Továbbá,

ahol KF egy tapasztalati tényező az anyag polaritásának figyelembevétele céljából. Néhány vegyülettípus KF tényezője megtalálható az irodalomban (b).
Elég gyakran olyan adatok állnak rendelkezésre, ahol a forráspont kisebb nyomásra van megadva. Ebben az esetben a gőznyomást a következők szerint számítjuk:

ahol T1 a kisebb P1 nyomáson mért forráspont.
DOKUMENTÁCIÓ
A becslési módszer használata esetén a jegyzőkönyvnek tartalmaznia kell a számítás átfogó dokumentációját.
IRODALOM
|
(a) |
Watson, K.M. (1943). Ind. Eng. Chem, 35, 398. |
|
(b) |
Lyman, W.J., Reehl, W.F., Rosenblatt, D.H. (1982). Handbook of Chemical Property Estimation Methods, McGraw-Hill. |
|
(c) |
OECD Environmental Monograph No.67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). |
II. MELLÉKLET
|
A.22. |
ROSTOK HOSSZAL SÚLYOZOTT ÁTLAGOS GEOMETRIAI ÁTMÉRŐJE |
1. MÓDSZER
1.1. BEVEZETÉS
Ez a módszer a mesterséges ásványi rostok (MMMF) hosszal súlyozott átlagos geometriai átmérőjének (LWGMD) mérési módszerét írja le. Mivel a minta sokaság hosszal súlyozott átlagos geometriai átmérője 95 %-os valószínűséggel a 95 %-os konfidenciaszinteken belül lesz (LWGMD ± két standard hiba), a jelentett érték (a vizsgálati érték) a minta alsó 95 %-os konfidenciahatára lesz (azaz LWGMD – 2 két standard hiba). A módszer egy HSE ipari eljárástervezet frissítésén alapul (1994. június), melyet az ECFIA és a HSE Chester tartott megbeszélésén fogadott el, 1993. szeptember 26-án, és egy második laboratóriumközi vizsgálat részére készült, illetve abból dolgozták ki (1, 2). Ez a mérési módszer az ömlesztett anyagok vagy a mesterséges ásványi szálakat, beleértve a refraktor kerámia rostokat (RCF), mesterséges üvegszálakat (MMVF), kristályos és polikristályos szálakat tartalmazó termékek rostátmérőjének jellemzésére használható.
A hosszal történő súlyozás az anyagok mintavételezésekor vagy mozgatásakor a hosszú rostok törése által okozott átmérő eloszlásra gyakorolt hatást egyenlíti ki. Az MMMF átmérők méreteloszlásának méréséhez geometriai statisztikát (geometriai átlag) alkalmaznak, mert ezeknek az átmérőknek a méreteloszlása rendszerint a logaritmiko-normális eloszláshoz közelít.
A hossz és az átmérő mérése egyaránt unalmas és időigényes, de csak ha azokat a rostokat mérik, melyek érintik a SEM látómező egy végtelenül vékony vonalát, egy adott rost kiválasztásának lehetősége arányos annak hosszával. Mivel a hosszal súlyozott számításokban ez figyelemmel van a hosszra, az egyetlen megkívánt mérés az átmérő mérése, és a LWGMD-2SE a leírtak szerint számítható.
1.2. FOGALOMMEGHATÁROZÁSOK
Részecske: Olyan tárgy, melynél a hossznak a szélességhez viszonyított aránya kisebb mint 3:1.
Rost: Olyan tárgy, melynél a hossznak a szélességhez viszonyított aránya (méretarány) legalább 3:1.
1.3. ALKALMAZÁSI TERÜLET ÉS KORLÁTOK
A módszer a 0,5 µm és 6 µm közti átlagátmérőjű átmérő eloszlás vizsgálatára született. A nagyobb átmérők kisebb SEM nagyítással vizsgálhatóak, de a módszer növekvő mértékben korlátozóvá válik a finomabb rosteloszlás esetében és 0,5 µm alatti átlagátmérő esetében TEM (transzmissziós elektronmikroszkóp) mérés javasolt.
1.4. A VIZSGÁLATI MÓDSZER ELVE
A rostpaplanból vagy a szabad ömlesztett rostból több reprezentatív magmintát vesznek. Az ömlesztett rostok hosszát aprítási eljárással csökkentik és a reprezentatív almintát vízben oszlatják el. Az aliquotokat kivonatolják és 0,2 µm pórusméretű, polikarbonát szűrőn átszűrik, majd előkészítik a pásztázó elektronmikroszkóp (SEM) technikájú vizsgálatra. A rostátmérőket 10 000-szeres vagy nagyobb képernyőnagyítással mérik (1), egyenes metszés módszer alkalmazásával, az átlagátmérő torzítatlan becslése végett. A kisebb 95 % konfidenciaintervallumot (egyoldalas vizsgálaton alapulva) számítják, és az eredmény az anyag átlag geometriai rostátmérőjének legkisebb becsült értéke.
1.5. A VIZSGÁLATI MÓDSZER LEÍRÁSA
1.5.1. Biztonság/óvintézkedések
A lebegő rostoknak történő személyi kitettséget minimalizálni kell, és a száraz rostok mozgatásakor füstszekrényt vagy kesztyűs manipulátort kell használni. A módszerek hatékonyságának meghatározására időszakos személyi kitettség monitoringot kell végezni. Mesterséges ásványi rostok mozgatásakor a bőrirritációk csökkentésére és a kereszt-szennyeződés megelőzése érdekében eldobható kesztyűt kell viselni.
1.5.2. Készülék / berendezés
|
— |
Préselő és sajtoló (10 MPa előállítására képes). |
|
— |
0,2 µm pórusméretű polikarbonát kapilláris pórus szűrők (25 mm átmérő). |
|
— |
5 µm pórusméretű cellulózészter membránszűrő alátétszűrőnek. |
|
— |
Üveg szűrőberendezés (vagy eldobható szűrőrendszerek) 25 mm átmérőjű szűrőkhöz (pl. Millipore üveg mikroanalízis készlet, XX10 025 00 típusú). |
|
— |
Mikroorganizmusok eltávolítására 0,2 µm pórusátmérőjű szűrőn átszűrt, frissen desztillált víz. |
|
— |
Katódbevonó arany vagy arany/palládium anyaggal. |
|
— |
Pásztázó elektronmikroszkóp, mely 10 nm felbontású és 10 000-szeres nagyításra képes. |
|
— |
Vegyes: spatulák, 24-es típusú szikepenge, csipeszek, SEM tárgylemezek, szén ragasztó vagy szén tapadó szalag, ezüst vezető. |
|
— |
Ultrahangos szonda vagy asztali ultrahangos fürdő. |
|
— |
Fúró mintavevő vagy dugófúró, a mesterséges ásványi szál paplanból történő magmintavételre. |
1.5.3. A vizsgálat kivitelezése
1.5.3.1. Mintavétel
Paplanok és lapok esetében egy 25 mm-es magfúróval vagy dugófúróval vesznek mintákat a keresztmetszetből. Ezek a paplanok egy rövid hosszoldalán, annak szélességében egyenlő távolságra kell, hogy elhelyezkedjenek, vagy pedig véletlenszerűen kell venni őket, amennyiben elegendő hosszú hosszdarabok áll rendelkezésre. Ugyanezt a berendezést szabad rostokból történő véletlenszerű minták vételére is lehet használni. Lehetőség szerint hat mintát kell venni, az ömlesztett anyag térbeli eltéréseinek tükrözése céljából.
A hat magmintát 50 mm átmérőjű sajtolóban, 10 MPa-on kell összetörtni. Az anyagot egy spatulával kell összekeverni, majd 10 MPa-on újra összenyomni. Ezután az anyag kikerül a sajtolóból és lezárt üvegben kerül tárolásra.
1.5.3.2. A minta előkészítése
Ha szükséges, a szerves kötőanyag eltávolítható a rostnak körülbelül egy órára 450 °C-os kemencébe történő helyezésével.
A mintát kúp alakúra kell formálni, majd negyedelve szétosztani (ezt porkamrában kell végezni).
Spatula segítségével adjuk a minta egy kis mennyiségét (< 0,5 g) 100 ml frissen desztillált, 0,2 µm-es membránszűrőn átszűrt vízhez (ha kielégítőnek bizonyulnak, más, alternatív ultratiszta víz forrásokat is lehet használni). 100 W teljesítményen működtetett, kavitációra beállított ultrahangos szondával alaposan oszlassuk el. (Ha nem áll rendelkezésre szonda, a következő módszert alkalmazzuk: ismételten rázzuk és invertáljuk 30 másodpercig; asztali ultrahangos fürdőben ultrahangozzuk öt percig; majd ismételten rázzuk és invertáljuk további 30 másodpercig).
Rögtön a rost szétoszlatását követően vegyünk ki több aliquotot (pl. három darab, 3, 6 és 10 ml aliquot) egy széles szájú pipetta segítségével (2–5 ml kapacitás).
Mindegyik aliquotot vákuumszűrőzzük át egy 0,2 µm polikarbonát szűrőn, melyet egy 5 µm pórusú MEC alátét szűrővel egészít ki, egy 25 mm-es hengeres tartályos, üvegszűrős tölcsérrel. Körülbelül 5 ml szűrt desztillált vizet kell a tölcsérbe helyezni, és az aliquotot lassan át kell pipettázni át a vízbe, a pipetta csúcsát a meniszkusz alatt tartva. A pipettázást követően a pipettát és a tartályt alaposan át kell öblíteni, mivel a vékony rostok hajlamosak inkább a felületen maradni.
Óvatosan távolítsuk el a szűrőt és válasszuk el az alátét szűrőtől, mielőtt egy tartályba helyeznénk szárítani.
24-es típusú szikével a szűrt üledékből vágjunk egy negyed vagy fél szűrőrészt, ide-oda mozgó mozdulatokkal. A kivágott részt óvatosan helyezzük a SEM tárgylemezre, szén ragasztószalaggal vagy szénragasztóval rögzítve. A szűrő széleinek és a tárgylemez villamos érintkezésének javítása érdekében legalább három helyen ezüst vezetőt kell alkalmazni. Amikor a ragasztó/ezüst vezető száraz, szórással vonja be az üledék felületét körülbelül 50 nm arany/palládium bevonattal.
1.5.3.3. SEM kalibrálása és működtetése
1.5.3.3.1. Kalibrálás
A SEM kalibrálását legalább hetente egyszer ellenőrizni kell (ideálisan naponta egyszer), hitelesített kalibrációs ráccsal. A kalibrációt össze kell hasonlítani egy hitelesített standarddal és ha a mért érték (SEM) nincs a hitelesített érték ±2 %-án belül, a SEM kalibrációt be kell állítani és újra kell ellenőrizni.
A SEM-nek legalább látható 0,2 µm átmérő felbontással kell rendelkeznie, valós minta mátrixot alkalmazva, 2 000-szeres nagyításon.
1.5.3.3.2. Működtetés
A SEM-et 10 000-szeres nagyításon kell működtetni (2), olyan körülményeket alkalmazva, melyek jó felbontást és elfogadható képet adnak lassú, például 5 másodperc per kép letapogatási sebességnél. Bár a különböző SEM-ek üzemeltetési követelményei eltérhetnek, általánosságban a legjobban látható és legjobb felbontás érdekében, viszonylag alacsony atomsúlyú anyagokkal 5–10 keV gyorsítófeszültséget kell alkalmazni kis fénypont mérettel és rövid működési távolsággal. Mivel lineáris metszést végzünk, 0o-os dőlést kell alkalmazni a minimális újrafókuszáláshoz, vagy, ha az SEM-nek van eucentrikus állapota, az eucentrikus működési távolságot kell használni. Kisebb nagyítás alkalmazható, ha az anyag nem tartalmaz kis (átmérőjű) rostokat, és a rostok átmérője nagy (> 5 µm).
1.5.3.4. Méretezés
1.5.3.4.1. Kisebb nagyítású vizsgálat a minta értékelésére
A mintát először kis nagyítással kell vizsgálni összetapadt nagy rostok jeleit keresve, és a rostok sűrűségét vizsgálva. Túlzott tapadás esetén javasolt új minta készítése.
A statisztikai pontosság érdekében fontos egy minimum számú rost mérése, és kívánatos a magas rostsűrűség, mivel az üres mezők vizsgálata időigényes és nem segíti az elemzést. Ugyanakkor, ha a szűrő túltöltött, nehézkes az összes mérhető rost mérése, mert a kis rostokat eltakarhatják a nagyobbak, ezért ezek esetleg figyelmen kívül maradnak.
Az LWGMD túlbecslése annak eredménye lehet, ha a lineáris átlón a rostsűrűség meghaladja millimétereként a 150 rostot. Másrészről az alacsony rostkoncentrációk növelik az elemzés idejét, és gyakran költséghatékonyabb az optimálishoz közeli rostsűrűségű minta készítése, mint az alacsony koncentrációjú szűrők számolgatásához ragaszkodás. Az optimális rostsűrűség esetén 5 0000-szeres nagyításnál egy látómezőben átlag egy vagy két megszámlálható rost található. Ugyanakkor az optimum sűrűség függ a rostok méretétől (átmérő), ezért fontos, hogy a műveletet végző személy szakszerűen döntsön arról, hogy a rostsűrűség optimálishoz közeli-e vagy sem.
1.5.3.4.2. A rostátmérők hosszal történő súlyozása
Csak azokat a rostokat kell számolni, melyek a SEM képernyőjén lévő (végtelenül) vékony vonalat érintik (vagy keresztezik). Ebből az okból kifolyólag a képernyő közepén egy vízszintes (vagy függőleges) vonal húzódik.
Alternatívaként a képernyő közepén egyetlen pont található, és a szűrőn keresztül egy irányban zajlik a folyamatos pásztázás. Minden olyan rost átmérője, melynek méretaránya nagyobb mint 3:1, és érinti, vagy keresztezi ezt a pontot, mérésre és rögzítésre kerül.
1.5.3.4.3. Rostok méretezése
Minimum 300 rost mérése javasolt. Minden rostot csak egyszer, a képen lévő vonallal vagy ponttal kapott metszéspontban mérünk (vagy ha a rost végei takarva vannak, a metszésponthoz közel). Ha nem szokványos keresztmetszetű rostok fordulnak elő, a rost átlag átmérőjét képviselő mérést kell alkalmazni. Gondosan kell eljárni a végek meghatározásakor, illetve a rost végei közötti legrövidebb távolság mérésekor. A méretezés végezhető on-line vagy pedig a tárolt képekkel vagy fotókkal off-line. Félautomatikus képi mérési rendszerek – melyek az adatokat közvetlenül egy táblázatba töltik le – használata ajánlott, mivel ezek időt takarítanak meg, kiküszöbölik az elírási hibákat és a számítások automatikusak.
A hosszú rostok végeit kis nagyítással kell ellenőrizni, meggyőződve arról, hogy azok nem göndörödnek vissza a mérési látómezőbe és csak egyszer kerülnek mérésre.
2. ADATOK
2.1. EREDMÉNYEK KEZELÉSE
A rostátmérők rendszerint nem mutatnak normális eloszlást. Ugyanakkor a logtranszformáció elvégzésével a normálishoz közelítő eloszlást nyerhetünk.
Kiszámítjuk az n rostátmérők (D) természetes e alapú logaritmusa (lnD) számtani átlagát (mean lnD) és standard eltérését (SDlnD):
|
|
(1) |
|
|
(2) |
A standard hiba (SElnD) meghatározásához a standard eltérést elosztjuk a mérések számának (n) négyzetgyökével.
|
|
(3) |
Az átlagból kivonjuk a standard hiba kétszeresét, és kiszámítjuk az érték (mínusz két standardhiba) exponenciálisát, így megkapjuk a geometriai átlag mínusz két standard hiba értéket.
|
|
(4) |
3. JELENTÉS
VIZSGÁLATI JELENTÉS
A vizsgálati jelentésnek legalább a következő információt kell tartalmaznia:
|
— |
Az LWGMD-2SE értéke. |
|
— |
Bármilyen eltérés és különösen azok, melyek hatással lehetnek az eredmények precízségére vagy pontosságára, megfelelő indoklással. |
4. HIVATKOZÁSOK
|
1. |
B. Tylee SOP MF 240. Health and Safety Executive. February 1999. |
|
2. |
G. Burdett and G. Revell. Development of a standard method to measure the length-weigthed geometric mean fibre diameter: Results of the Second inter-laboratory exchange. IR/L/MF/94/07. Project R42.75 HPD. Health and Safety Executive. Research and Laboratory Services Division. 1994. |
(1) Ez a nagyítási érték 3 µm rostokhoz javasolt, a 6 µm rostok esetében 5 000-szeres nagyítás megfelelőbb lehet.
(2) A 3 μm rostok esetében lásd az előző jegyzetet.
III. MELLÉKLET
|
B.46. |
IN VITRO BŐRIRRITÁCIÓ: REKONSTRUÁLT EMBERI FELHÁMMODELLEN VÉGZETT VIZSGÁLAT |
1. MÓDSZER
1.1. BEVEZETÉS
A bőrirritáció a vizsgálandó anyag alkalmazását követően 4 órán belül megjelenő, visszafordítható bőrkárosodás (az ENSZ vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszerének [GHS] meghatározása szerint) (1). E vizsgálati módszer a vizsgálati stratégián belül önálló helyettesítő vizsgálatként olyan in vitro eljárást garantál, amely a tájékoztatási követelményektől függően lehetővé teszi az egyes anyagok által okozott bőrirritáció meghatározását az adatok bizonyító erejének mérlegelésével (2).
A bőrirritáció vizsgálata jellemzően állatkísérletek alkalmazásával jár. (Lásd a B.4 módszert)(3). Állatjólléti szempontokat érvényesítve a B.4 módszer lehetővé teszi a bőrkorróziónak/bőrirritációnak alépcsőzetes vizsgálati stratégia részét képező validált in vitro és ex vivo módszerek alkalmazásával történő meghatározását, megkímélve ezzel az állatokat a fájdalomtól és szenvedéstől. A B.4 lépcsőzetes vizsgálati stratégia maró hatással foglalkozó része esetében három hasznos in vitro vizsgálati módszer, illetve vizsgálati iránymutatás ismert: a B.40, B.40A és a TG 435 (4, 5, 6).
E vizsgálati módszer rekonstruált emberi epidermisz-modelleken alapul, amelyek általános felépítésükben (sejtforrásként, szövet- és szövetfelépítés-mintaként emberi felhám keratinsejteket alkalmaznak) élethűen imitálják az emberi bőr felső rétegének, azaz a felhámnak (epidermisznek) a biokémiai és fiziológiai tulajdonságait. Az e vizsgálati módszer alapján leírt eljárás lehetővé teszi az irritatív hatású anyagok veszélyességének besorolását az ENSZ által javasolt GHS szerinti 2. kategóriának megfelelően. E vizsgálati módszer teljesítményszabvány-készletet is magában foglal a hasonló és módosított rekonstruált emberi epidermisz alapú vizsgálati módszerek értékeléséhez (7).
Két olyan in vitro vizsgálati módszerhez készültek elővalidálási, optimalizálási és validálási tanulmányok (8, 9, 10, 11, 12, 13, 14, 15, 16, 17), amelyek rekonstruált emberi epidermisz-modelleket alkalmaznak és a kereskedelmi forgalomban EpiSkin™ és EpiDerm™ néven kaphatók. Ezen tanulmányok az R38 besorolást veszik alapul. A 25. hivatkozás a GHS-szempontú újraszámítás bizonyos aspektusait tárgyalja. Az EpiSkin™ módszerrel (1. validált referencia-módszer) működési szempontból egyenértékű módszerek javasoltak önálló helyettesítő vizsgálati módszerként a GHS szerinti 2. kategóriába tartozó irritatív hatású anyagok besorolása érdekében nyulakon végzett in vivo vizsgálathoz. Az EpiDerm™ módszerrel (a 2. validált referencia-módszer) egyenértékű módszerek alkalmazása a GHS szerinti 2. kategóriába tartozó irritatív hatású anyagok besorolására csak szűrővizsgálati módszerként, vagy az adatok bizonyító erejének mérlegelésével alépcsőzetes vizsgálati stratégia részeként javasolt. Mielőtt a javasolt in vitro rekonstruált emberi felhámmodellen végzett bőrirritációs vizsgálatot szabályozási célokból el lehetne végezni, meg kell határozni a javasolt alkalmazáshoz a vizsgálat megbízhatóságát, alkalmazhatóságát (pontosságát) és korlátait annak érdekében, hogy az 1. referencia-módszerrel való összehasonlíthatóság biztosított legyen az e vizsgálati módszerben előírt teljesítményszabványoknak megfelelően (függelék).
Két másik in vitro rekonstruált emberi felhámmodellen végzett vizsgálati módszer került hitelesítésére e vizsgálati módszerben előírt követelményeknek megfelelően és mutat hasonló eredményeket, mint az 1. hitelesített referencia-módszer (18). Ezek a módosított EpiDerm™ vizsgálati módszer (módosított 2. referencia-módszer) és a SkinEthic RHE™ vizsgálati módszer (1. „me-too” módszer).
1.2. FOGALOMMEGHATÁROZÁSOK
E vizsgálati módszerben a következő fogalommeghatározások szerepelnek:
Pontosság: egy vizsgálati eredmény és az elfogadott referenciaérték közötti egyezés mértéke. A vizsgálati módszer teljesítményének mérésére szolgál, és az alkalmazhatóság egyik szempontja. Gyakran használják e kifejezést az „egyezés” megfelelőjeként, amely jelzi egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát.
Tételenkénti kontrollanyag: mérőanyag-mennyiség, amely a szövet közepes mértékű sejtéletképesség-reakcióját váltja ki.
Sejt-életképesség: valamely sejtpopuláció teljes aktivitását mérő paraméter (például a sejt- mitokondriális dehidrogenázok MTT ([3–(4,5-Dimetiltiazol-2il)-2,5-difeniltetrazólium bromid, Tiazolil kék] vitális festékredukáló képessége) amely – a mért végponttól és a használt vizsgálati tervtől függően – korrelációt mutat a sejtek számával és/vagy életképességével.
ET50 : a sejt életképességének 50 %-kal való csökkentéséhez szükséges expozíciós idő a markeranyag meghatározott koncentrációban történő alkalmazásakor, lásd még az IC50-et.
Hamis negatív arány: egy vizsgálati módszer során az összes tévesen negatívnak minősített pozitív anyag aránya. Ez a vizsgálati módszer teljesítményének egyik mutatója.
Hamis pozitív arány: az összes tévesen pozitívnak minősített negatív (inaktív) anyag aránya. Ez a vizsgálati módszer teljesítményének egyik mutatója.
Végtelen dózis: a vizsgált anyag bőrre adagolt azon mennyisége, amely meghaladja a bőr felszínének teljes és egyenletes befedéséhez szükséges mennyiséget.
GHS (A vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszere): anyagok és keverékek szabványosított fizikai, egészségi és környezeti kockázatok szerinti besorolására, valamint azok megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javasolt rendszer, az anyagok káros hatásairól szóló információk továbbítására az emberek (köztük a munkaadók, munkavállalók, fuvarozók, fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében (1), amelyet az Európai Unióban az 1272/2008/EK rendelet juttat érvényre.
IC50 : a markeranyag azon koncentrációja, amelynél a szövetek életképessége 50 %-kal visszaesik(IC50) egy meghatározott expozíciós idő elteltével, lásd még az ET50-et.
Teljesítményszabvány: hitelesített referencia-módszeren alapuló szabványok, amelyek a javasolt, végrehajtás és funkcionalitás szempontjából hasonló vizsgálati módszer összehasonlíthatósági értékelésének alapjául szolgálnak. Ide tartoznak I. a szükséges vizsgálati módszer elemei II. a hitelesített referencia-módszer elfogadható működésének igazolására alkalmazott anyagok közül kiválasztásra került referenciaanyagok minimális listája, valamint III. a pontosságnak és megbízhatóságnak azon összehasonlítható szintjei, amelyeket a javasolt vizsgálati módszernek a referenciaanyagok minimális listájával történő értékelésekor el kell érnie.
Megbízhatóság: egy vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti reprodukálhatóságának leírására szolgál ugyanazon eljárás alkalmazása mellett. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg.
Érzékenység: az összes olyan pozitív/aktív anyagnak az aránya, amelyet a vizsgálat helyesen sorolt be. A vizsgálati módszer pontosságának olyan mérése, amely kategorikus eredményre vezet és fontos a vizsgálati módszer alkalmazhatóságának megítélésében.
Specificitás: az összes olyan negatív/inaktív anyag aránya, amelyet a vizsgálat helyesen sorolt be. A vizsgálati módszer pontosságának olyan mérése, amely kategorikus eredményre vezet és fontos a vizsgálati módszer alkalmazhatóságának megítélésében.
Bőrirritáció: a vizsgálandó anyag alkalmazását követően 4 órán belül megjelenő, visszafordítható bőrkárosodás. A bőrirritáció helyileg jelentkező, nem immunogén reakció, amely röviddel a stimuláció után megjelenik (24). Fő jellemzője, hogy gyulladásos reakciókkal járó visszafordítható folyamat a gyulladásos folyamatokra jellemző legtöbb klinikai tünettel együtt (bőrpír, ödéma, viszketés és fájdalom).
1.3. ALKALMAZÁSI KÖR ÉS KORLÁTOZÁSOK
Az e vizsgálati módszerhez tartozó rekonstruált emberi felhámvizsgálatok egyik korlátját az jelenti, hogy a bőrirritálónak minősülő anyagokat csak az ENSZ GHS szerinti 2. kategóriába sorolják be. Mivel az anyagoknak az ENSZ által javasolt GHS meghatározása szerinti opcionális 3. kategóriába történő besorolását nem teszik lehetővé, az összes többi anyag besorolás nélkül (kategória nélküli) marad. A szabályozási igényektől és a „me-too” vizsgálatok esetleges jövőbeli új végpontokkal történő kiegészítésétől, javításától, illetve fejlesztésétől függően szükség lehet e vizsgálati módszer felülvizsgálatára.
E vizsgálati módszer lehetővé teszi az egykomponensű irritatív hatású anyagok veszélyességének besorolását (19), de nem nyújt megfelelő információt a bőrkorrózióra nézve. A gázok és aeroszolok nem vizsgálhatók, amíg a keverékek vonatkozásában nem történt meg a módszer validálási célú értékelése.
1.4. A VIZSGÁLAT ELVE
A vizsgált anyagot helyileg egy olyan normál emberi felhám-keratinsejteket tartalmazó, háromdimenziós emberi felhámmodellre viszik fel, amelyeket úgy tenyésztettek ki, hogy többrétegű, rendkívül differenciált emberi felhámmodellt alkossanak. Ez szervezett bazális, tüskés, és granuláris sejtrétegekből, valamint a sejtek közötti lamellás lipidrétegeket tartalmazó többrétegű stratum corneumból (szarurétegből) áll az in vivo mintáknak megfelelő elrendezésben.
A rekonstruált emberi felhámmodellen végzett vizsgálat elve azon alapul, hogy az irritatív hatású anyagok képesek diffúzióval áthatolni a stratum corneumon és citotoxikusak az alatta levő sejtrétegek sejtjeire. A sejtéletképességet a MTT [3–(4,5-Dimetiltiazol-2il)-2,5-difeniltetrazólium bromid, Tiazolil kék; EINECS-szám: 206–069–5, CAS-szám: 298–93–1)] vitális festék kék formazán sóvá történő dehidrogenáz átalakulásával mérik, amelynek mennyiségi mérésére a szövetekből történő extrakció után kerül sor (20). Az irritatív hatású anyagokat azon képességük alapján azonosítják, hogy a sejtéletképességet meghatározott határérték alá csökkentik (azaz 50 % alá, az ENSZ által javasolt GHS szerinti 2. kategóriájú irritatív anyagok esetében). A határérték feletti sejtéletképességet produkáló anyagokat nem sorolják be (azaz 50 % felett nincsenek kategóriába sorolva).
A rekonstruált emberi felhámmodell-rendszereket szilárd, folyékony, félszilárd anyagokon és viaszokon végzett vizsgálatoknál lehet alkalmazni. A folyadékokban lehet víz, illetve lehetnek vízmentesek, a szilárd anyagok lehetnek vízben oldódóak, illetve vízben nem oldódóak. Amennyiben lehetséges, a szilárd anyagokat finom por alakban kell vizsgálni. Mivel a rekonstruált emberi felhámmodellen végzett vizsgálati rendszerek hitelesítésébe a kémiai osztályok széles spektrumát képviselő 58 gondosan kiválasztott anyagot vontak be, ezért e módszerek várhatóan a különféle kémiai osztályokban általánosan alkalmazhatóak lesznek (16). A hitelesítés során 13, a GHS szerint 2. kategóriájú irritatív anyagot vizsgáltak. Meg kell említeni, hogy a hitelesítés nem terjedt ki a nem maró hatású savakra, lúgokra, sókra és egyéb szervetlen anyagokra, illetve nem, vagy csak korlátozott mértékben érintette a szerves irritatív anyagok bizonyos ismert osztályait (pl. hidrogén-peroxidok, fenolok, felületaktív anyagok).
1.5. AZ ALKALMASSÁG BIZONYÍTÁSA
E vizsgálati módszer alapján hitelesített módszer rutinszerű alkalmazását megelőzően a laboratóriumok célszerűnek tarthatják megbizonyosodni műszaki alkalmasságukról az 1. táblázatban javasolt tíz anyag felhasználásával. E vizsgálati módszer alapján az ENSZ által javasolt GHS szerinti 3. kategória nem minősül kategóriának. Az olyan új, hasonló (me-too) vizsgálati módszerek esetében, amelyek e vizsgálati módszer alapján kerülnek kifejlesztésre és amelyek szerkezetileg és funkcionálisan hasonlóak a validált referencia-módszerekhez, illetve a hitelesített módszerek módosításai esetében a szabványos tesztelésre való használatukat megelőzően az új vizsgálati módszer összehasonlítható megbízhatóságának és pontosságának bizonyítására e vizsgálati módszer Függelékében leírt teljesítményszabványokat kell használni.
1. táblázat
A függelékben felsorolt referenciaanyagok részhalmazát képező anyagok
|
Anyag |
CAS-szám |
In vivo eredmény |
Halmazállapot |
GHS szerinti kategória |
|
naftalin ecetsav |
86–87–3 |
0 |
Sz |
kat. nélkül |
|
izopropanol |
67–63–0 |
0,3 |
F |
kat. nélkül |
|
metil sztearát |
112–61–8 |
1 |
Sz |
kat. nélkül |
|
heptil butirát |
5870–93–9 |
1,7 |
F |
Opcionális 3. kat. |
|
Hexil-szalicilát |
6259–76–3 |
2 |
F |
Opcionális 3. kat. |
|
ciklámen aldehid |
103–95–7 |
2,3 |
F |
2. kat. |
|
1- brómhexán |
111–25–1 |
2,7 |
F |
2. kat. |
|
butil metakrilát |
97–88–1 |
3 |
F |
2. kat. |
|
1-metil-3-fenil-1-piperazin |
5271–27–2 |
3,3 |
Sz |
2. kat. |
|
Heptanal |
111–71–7 |
4 |
F |
2. kat. |
1.6. A VIZSGÁLATI MÓDSZER LEÍRÁSA
Az alábbiakban a rekonstruált emberi felhámmodellen végzett, bőrirritációt elemző vizsgálat leírására kerül sor. A rekonstruált emberi felhámmodell felépíthető, elkészíthető vagy kereskedelmi forgalomban kapható (pl. EpiSkin™, EpiDerm™ és SkinEthic RHE™). Az EpiSkin™, EpiDerm™ és SkinEthic RHE™-vel használható szabvány vizsgálati eljárások elérhetők a [http://ecvam.jrc.ec.europa.eu] címen (21, 22, 23). A vizsgálatot az alábbiak szerint kell végrehajtani:
1.6.1. Rekonstruált emberi felhámmodell összetevői
1.6.1.1. A modellre vonatkozó általános feltételek
Az epitélium felépítéséhez normál emberi keratinsejteket kell használni. Élő epitélsejtek több rétegének (bazális, tüskés, és granuláris sejtréteg) kell jelen lenni a funkcionális stratum corneum alatt. A stratum corneumnak többrétegűnek kell lennie, és megfelelő lipidszelvénnyel kell rendelkeznie, hogy nagy ellenálló képességgel funkcionális barriert képezzen a citotoxikus markeranyagok, pl. a nátrium-dodecil-szulfát (SDS) vagy a Triton X-100 gyors áthatolása ellen. A barrierfunkció értékelése vagy a marker anyag azon koncentrációjának meghatározásával történik, amely a szövet életképességét 50 %-kal csökkenti (IC50) egy meghatározott expozíciós idő elteltével, vagy a markeranyag meghatározott koncentrációban történő alkalmazásakor a sejt életképességének 50 %-kal való csökkentéséhez szükséges expozíciós idő (ET50) meghatározásával. A modell visszaszorító tulajdonságainak meg kell akadályozni, hogy az anyag áthatoljon a stratum corneum körül az élő szövetbe. A vizsgált vegyszerek stratum corneum körül történő áthatolása a bőr expozíciójának gyenge modellezését eredményezi. A bőrmodellnek baktérium-, vírus-, mikoplazma- és gombafertőzéstől mentesnek kell lennie.
1.6.1.2. Funkcionális modellfeltételek
1.6.1.2.1. Életképesség
Az életképesség terjedelmének meghatározását lehetőleg az MTT segítségével kell végezni (20). A negatív kontrollanyaggal (NC) kezelt szövetből kivont (szolibilizált) festékanyag optikai sűrűségének (OD) legalább 20-szor nagyobbnak kell lennie az extrahálásra használt oldószer OD-jénél. Dokumentálni kell, hogy az NC-vel kezelt kontrollszövet a vizsgálati expozíciós időszak alatt a tenyészetben állandó (hasonló életképesség-mérést nyújt).
1.6.1.2.2. Barrierfunkció
A stratum corneumnak és lipidszelvényeinek megfelelő ellenálló képességgel kell rendelkezni a citotoxikus markeranyagok, pl. a nátrium-dodecil-szulfát (SDS) vagy a Triton X-100 gyors áthatolása ellen, az IC50 vagy ET50 alapján végzett becslések szerint.
1.6.1.2.3. Morfológia
A rekonstruált bőr/epidermisz szövettani vizsgálatát olyan megfelelően képzett személyzetnek kell végezni, akik emberi bőr/epidermisz-szerű struktúrákon (beleértve a többrétegű stratum corneumot is) végzik a bizonyító vizsgálatot.
1.6.1.2.4. Reprodukálhatóság
A specifikus modellt alkalmazó vizsgálati eredményeknek idővel reprodukálhatóknak kell lenniük, lehetőleg a megfelelő tételenkénti (összehasonlító) kontrollanyaga szerint. (Lásd a Függeléket).
1.6.1.2.5. A modell minőségellenőrzése (QC)
Minden felhasználásra kerülő felhámmodell-csoportnak meghatározott előállítási kritériumoknak kell megfelelnie, amelyek között az életképességre (1.6.1.2.1. bekezdés) és a barrierfunkcióra (1.6.1.2.2. bekezdés) vonatkozóak a leglényegesebbek. A bőrmodell szállítója (illetve saját előállítású modell esetén a vizsgálatot végző személy) köteles elfogadhatósági tartományt (felső és alsó határértéket) megállapítani az IC50 vagy a ET50 számára. A szövetek átvétele után a laboratóriumnak ellenőriznie kell a szövetek barrier tulajdonságait. Az irritatív hatások megbízható előrejelzéséhez kizárólag a megfelelő szövetek által produkált eredményeket szabad elfogadni. A hitelesített referencia-módszer elfogadhatósági tartományai például a következők.
2. táblázat
Példák a minőségellenőrzési tételkibocsátás kritériumaira
|
|
Alsó elfogadási határ |
Elfogadási tartomány középértéke |
Felső elfogadási határ |
|
1. Hitelesített referencia-módszer (18 órás nátrium-dodecil-szulfátos (SDS) kezelés) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
2. Hitelesített referencia-módszer (1 % Triton X100) |
ET50 = 4,8 óra |
ET50 = 6,7 óra |
ET50 = 8,7 óra |
1.6.1.3. A vizsgálat alkalmazása és a kontrollanyagok
Valamennyi kezeléshez és kontrollhoz megfelelő számú szövetpéldányt (szakaszonként legalább három példányt) kell használni. Folyékony és szilárd anyagok esetén a bőrön egyenletesen elosztva elegendő vizsgált anyagot használjunk, azaz minimum 25 μL/cm2 vagy 25 mg/cm2-ot, elkerülve ugyanakkor a végtelen dózis használatát (lásd az 1.2. fogalommeghatározást). Szilárd anyagok alkalmazása előtt az epidermisz felületét deionizált vagy desztillált vízzel nedvesítsük be a bőrrel való jó érintkezés biztosítására. A szilárd anyagokat, amikor csak lehetséges finom por alakban kell vizsgálni. Az expozíciós idő lejártával a vizsgált anyagot megfelelő pufferrel vagy 0,9 %-os NaCl-dal óvatosan le kell mosni a bőrről. A felhasznált rekonstruált emberi felhámtól függően az expozíciós idő 15 és 60 perc között, az inkubációs hőmérséklet pedig 20 és 37 °C között változhat. A három módszerre vonatkozó részleteket lásd a szabványműveleti előírásokban (Standard Operating Procedures) (21, 22, 23).
Minden vizsgálathoz egyidejűleg negatív (NC) és pozitív (PC) kontrollértékeket kell használni annak bemutatására, hogy a sejtek életképessége, a barrierfunkció és a kapott szövetérzékenység (PC) a meghatározott történeti elfogadási tartományon belül van. A javasolt pozitív kontrollanyag a nátrium-dodecil-szulfát (SDS) 5 %-os vizes oldata. A javasolt negatív kontrollanyagok a víz, vagy a sós foszfátpuffer (PBS).
1.6.1.4. A sejtek életképességének mérése
A vizsgálati eljárás legfontosabb eleme, hogy az életképesség mérését ne közvetlenül a vizsgált anyagok hatásának kitett időszak után végezzük, hanem a friss közegben átöblített szövetek kezelés utáni, megfelelő hosszúságú inkubációs időszakát követően. Ezen időszak lehetővé teszi egyrészt az enyhe irritatív hatások alóli felépülést, másrészt az egyértelmű citotoxikus hatások megjelenését. A vizsgálat optimalizálásának szakaszában (9, 10, 11, 12, 13) egy 42-órás kezelés utáni inkubációs időszak bizonyult optimálisnak és következésképpen ezt alkalmazták a referencia vizsgálati módszerek validálása során.
Az MTT átalakulásának vizsgálata egy olyan kvantitatív validált módszer, amelyet a sejtek életképességének mérésekor kell alkalmazni. Ez összeegyeztethető egy háromdimenziós szövetkonstrukcióban történő használattal. A bőrmintát 3 órára egy megfelelő koncentrációjú MTT-oldatba (pl. 0,3–1 mg/ml) teszik. A kicsapódott kék formazán terméket aztán oldószer (pld. izopropanol, savas izopropanol) használatával eltávolítják a szövetből, és egy maximum ±30 nm sávszűrő alkalmazásával 570 nm hullámhosszon az OD-meghatározása útján megmérik a formazán koncentrációját.
A vizsgált anyag optikai tulajdonságai, vagy az MTT-re gyakorolt vegyi hatása megzavarhatja a vizsgálatot, ami az életképesség téves megállapításához vezet (mivel a vizsgált anyag akadályozhatja, megváltoztathatja, vagy akár okozhatja is az elszíneződést). Ez akkor következhet be, amikor egy adott vizsgált anyagot öblítéssel nem távolítottak el teljesen a bőrről, vagy amikor az áthatol az epidermiszen. Ha a vizsgált anyag közvetlenül az MTT-re hat, természetes színezéket tartalmaz, vagy a szövetkezelés során színeződik el, további ellenőrzéseket kell alkalmazni annak megállapítására, hogy a vizsgált anyag befolyásolja-e az életképesség mérésének módját, illetve el kell végezni a megfelelő korrekciókat. Kérjük, az MTT-csökkenésvizsgálat részletes leírásáért forduljon a validált referencia-módszerekhez készült vizsgálati módszer jegyzőkönyvhöz (21, 22, 23). Az e befolyásoló tényezőknek köszönhetően jelentkező nem specifikus szín (NSC) nem haladhatja meg az NC 30 %-át (a korrekciók esetében). Ha az NSC > 30 %, a vizsgált anyag a vizsgálattal összeférhetetlennek minősül.
1.6.1.5. A vizsgálat elfogadhatósági kritériumai
Minden érvényes tételt alkalmazó vizsgálat esetében (lásd az 1.6.1.2.5. bekezdést), az NC-vel kezelt szöveteknek olyan OD-t kell mutatniuk, amely tükrözi a szövetek minőségét az összes szállítási és átvételi lépés, valamint az összes irritációs eljárási folyamat után. A kontrollok OD értékei nem lehetnek kisebbek a megállapított történeti alsó határértékeknél. Hasonlóképpen a PC-vel, azaz nátrium-dodecil-szulfát (SDS) 5 %-os vizes oldatával kezelt szöveteknek, tükrözniük kell szövetek által megtartott érzékenységet, valamint az irritatív hatású anyaggal szembeni reagálóképességüket minden vizsgálati körülmény esetében (pl. az életképesség ≤ 40 % az 1. validált referencia-módszer esetén, és ≤ 20 % a 2. validált referencia-módszer esetén). Meg kell határozni a szövetpéldányok közötti, ezzel kapcsolatos eltérés megfelelő mértékét (pld. szórás alkalmazása esetén 18 %-nál nem lehet nagyobb).
2. ADATOK
2.1. ADATOK
Minden kezelés esetében, az egyes párhuzamos vizsgálati minták adatait (pl. minden vizsgált anyagra vonatkozó OD-értéket és a százalékban meghatározott sejtéletképesség-értékeket, a besorolásokat is beleértve) táblázatba foglaltan kell megadni, ideértve adott esetben a megismételt kísérletek során kapott adatokat. Továbbá meg kell adni az átlagértékeket és azokhoz viszonyított ± szórást. Az MTT reagenssel és a színezett vizsgált anyagokkal tapasztalt kölcsönhatásokat minden vizsgált anyag esetében fel kell jegyezni.
2.2. AZ EREDMÉNYEK ÉRTELMEZÉSE
Minden vizsgálati mintánál a kapott OD-értékek felhasználhatók az NC-hez viszonyított életképesség százalékos kiszámításához, amely 100 %-ra van beállítva. Egyértelműen meg kell határozni, és írásban rögzíteni kell az irritatív hatású vizsgált anyagokat a besorolás nélküli vizsgált anyagoktól megkülönböztető, százalékban megállapított sejt-életképesség határértéket, valamint azon bizonyítottan megfelelő statisztikai eljárás(oka)t, amelyeket az eredmények értékelésénél és az irritatív hatású anyagok azonosításánál alkalmaznak. A validált referenciamódszerekhez kapcsolódó becsült irritációs hatás határértékei az alábbiak:
A vizsgált anyag bőrirritáló hatásúnak minősül az ENSZ által javasolt GHS szerinti 2. kategória szerint:
|
i. |
ha a vizsgált anyagoknak kitett időszak és a kezelést követő inkubációs időszak után a szövetek életképessége kisebb, vagy egyenlő, mint (≤) 50 %; |
A vizsgált anyagot nem tekintik egyik kategóriába tartozónak sem:
|
ii. |
ha a vizsgált anyagoknak kitett időszak és a kezelést követő inkubációs időszak után a szövetek életképessége nagyobb mint (>) 50 %. |
3. JEGYZŐKÖNYVEZÉS
3.1. VIZSGÁLATI JEGYZŐKÖNYV
A vizsgálati jegyzőkönyvnek a következő információkat kell tartalmaznia:
Vizsgálati és kontrollanyagok:
|
— |
a vegyszer(ek) neve, mint IUPAC- vagy CAS-név és CAS-szám, ha ismert, |
|
— |
az anyag tisztasága és összetétele (tömegszázalékban), |
|
— |
a vizsgálat elvégzése szempontjából lényeges fizikai-kémiai tulajdonságok (pl. fizikai állapot, stabilitás és illékonyság, pH, vízben való oldhatóság, ha ismert), |
|
— |
a vizsgálati/kontrollanyagok kezelése a vizsgálatot megelőzően, ha történt ilyen (pl. melegítés, őrlés), |
|
— |
tárolási feltételek. |
A használt bőrmodell és protokoll indokolása.
Vizsgálati körülmények:
|
— |
a felhasznált sejtrendszer, |
|
— |
a sejt életképességének mérésére használt eszköz (pl. spektrofotométer) és sávszűrő bemérésére vonatkozó kalibrálási információk, |
|
— |
a használt specifikus bőrmodellre vonatkozó teljes háttérinformáció, beleértve annak teljesítményét. Ez magában foglalja, de nem korlátozódik a következőkre:
|
|
— |
a használt vizsgálati eljárás részletei, |
|
— |
a használt vizsgálati dózisok, a vizsgált anyag hatásának kitett időszak és a kezelés utáni inkubációs időszak hossza, |
|
— |
a vizsgálati eljárás bármilyen változtatásának leírása, |
|
— |
hivatkozás a modell történeti adataira. Ez magában foglalja, de nem korlátozódik a következőkre:
|
|
— |
A használt értékelési ismérvek leírása, beleértve az előrejelző modell számára a határérték(ek) kiválasztásának indoklását. |
Eredmények:
|
— |
az egyedi vizsgálati minták adatainak táblázatba foglalása, |
|
— |
a megfigyelt egyéb jelenségek leírása. |
Az eredmények értékelése.
Következtetés.
4. HIVATKOZÁSOK
|
1. |
United Nations (UN) (2007). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Second revised edition, UN New York and Geneva, 2007. Available at: http://www.unece.org/trans/danger/publi/ghs/ghs_rev02/02files_e.html. |
|
2. |
REACH: Guidance on Information Requirements and Chemical Safety Assessment. Available at: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1232447649. |
|
3. |
B.4. Vizsgálati módszer: AKUT TOXICITÁS; BŐRIRRITÁCIÓ/BŐRKORRÓZIÓS HATÁS. |
|
4. |
B.40. Vizsgálati módszer: IN VITRO BŐRKORRÓZIÓ: TRANSZKUTÁN ELEKTROMOS REZISZTENCIA VIZSGÁLAT (TER). |
|
5. |
B.40.A. IN VITRO BŐRKORRÓZIÓ: EMBERI BŐRMODELLEN VÉGZETT VIZSGÁLAT. |
|
6. |
OECD (2006). Test Guideline 435. OECD Guideline for the Testing of Chemicals. In Vitro Membrane Barrier Test Method. Adopted July 19, 2006. Available at: http://www.oecd.org/document/22/0,2340,en_2649_34377_1916054_1_1_1_1,00.html. |
|
7. |
ECVAM (2009) Performance Standards for applying human skin models to in vitro skin irritation. Available under Download Study Documents, at: http://ecvam.jrc.ec.europa.eu. |
|
8. |
Fentem, J.H., Briggs, D., Chesné, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Roguet, R., van de Sandt, J.J.M. & Botham, P. (2001). A prevalidation study on in vitro tests for acute skin irritation. Results and evaluation by the Management Team. Toxicology in Vitro 15, 57–93. |
|
9. |
Portes, P., Grandidier, M.H., Cohen, C. & Roguet, R.(2002). Refinement of the EPISKIN protocol for the assessment of acute skin irritation of chemicals: follow-up to the ECVAM prevalidation study. Toxicology in Vitro 16, 765–770. |
|
10. |
Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B. & Spielmann, H. (2004). Optimisation of the EpiDerm test protocol for the upcoming ECVAM validation study on in vitro skin irritation tests. ALTEX 21, 107–114. |
|
11. |
Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D. & Spielmann H. (2005) The EpiDerm Test Protocol fort the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests – An Assessment of the Performance of the Optimised Test. ATLA 33, 351–367. |
|
12. |
Cotovio, J., Grandidier, M.–H., Portes, P., Roguet, R. & G. Rubinsteen. (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model within the Framework of the ECVAM Validation Process. ATLA 33, 329–249. |
|
13. |
Zuang, V., Balls, M., Botham, P.A., Coquette, A., Corsini, E., Curren, R.D., Elliot, G.R., Fentem, J.H., Heylings, J.R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J.J.M., Wiemann, C. & Worth, A.(2002). Follow-up to the ECVAM prevalidation study on in vitro tests for acute skin irritation. ECVAM Skin Irritation Task Force Report 2. ATLA 30,109–129. |
|
14. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559–601. |
|
15. |
Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-α. 135 pp. + annexes. Available under Download Study Documents, at: http://ecvam.jrc.ec.europa.eu. |
|
16. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603–619. |
|
17. |
J. Cotovio, M.–H. Grandidier, D. Lelièvre, R. Roguet, E. Tinois-Tessonneaud, J. Leclaire (2007). In vitro acute skin irritancy of chemicals using the validated EPISKIN model in a tiered strategy -Results and performances with 184 cosmetic ingredients, AATEX, Special Issue-proceedings from WC6. Vol.14, 351–358. |
|
18. |
ESAC statement on updated EpiDerm and similar SkinEthic assays, 2008. november 5. |
|
19. |
(2006) EK. A vegyi anyagok regisztrálásáról, értékeléséről, engedélyezéséről és korlátozásáról (REACH), az Európai Vegyianyag-ügynökség létrehozásáról, az 1999/45/EK irányelv módosításáról, valamint a 793/93/EGK tanácsi rendelet, az 1488/94/EK bizottsági rendelet, a 76/769/EGK tanácsi irányelv, a 91/155/EGK, a 93/67/EGK, a 93/105/EK és a 2000/21/EK bizottsági irányelv hatályon kívül helyezéséről szóló, 2006. december 18-i 1907/2006/EK európai parlamenti és tanácsi rendelet. Az Európai Unió Hivatalos Lapja L396/1, 2006.12.30., OPOCE, Luxembourg. |
|
20. |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55–63. |
|
21. |
EpiSkin™ SOP, Version 1.6 (January 2005). Validation of the EpiSkin Skin Irritation Test – 42 Hours Assay For The Prediction of Acute Skin Irritation of Chemicals. Available under Download Study Documents, at: http://ecvam.jrc.ec.europa.eu. |
|
22. |
EpiDerm™ SOP, Version 5.0 (October 2004). Draft Standard Operating Procedure. In Vitro Skin Irritation Test: Human Skin Model. Model: EpiDerm™- 200. Available under Download Study Documents, at: http://ecvam.jrc.ec.europa.eu. |
|
23. |
SkinEthic RHE™ SOP. Will be available under Download Study Documents, at: http://ecvam.jrc.ec.europa.eu. |
|
24. |
Harvell, J.D., Lamminstausta, K, Maibach H.I. (1995) Irritant contact dermatitis IN: Guin J.D. Practical Contact Dermatitis Mc Graw-Hill New York, pp 7–18 |
|
25. |
Griesinger C, Barroso J & Zuang V: ECVAM background document on the recent adaptations of the ECVAM performance standards for in vitro skin irritation testing in the context of the drafting process of an EU test method and an OECD draft test guideline. Ispra, 2008. november 13. |
Függelék
A javasolt in vitro rekonstruált emberi felhámmodellen végzett bőrirritációs vizsgálat teljesítményjellemzőinek értékelése
BEVEZETÉS
E vizsgálati módszer alapján alkalmazásra javasolt eljárásokat a megbízhatóságuk és pontosságuk meghatározása érdekében a Draize-féle irritációs pontértékek teljes tartományát képviselő anyagok felhasználásával értékelni kell. A húsz javasolt referenciaanyag (2. táblázat) felhasználásával történő értékelés során a javasolt eljárás által elért megbízhatósági és pontossági értékeknek és az 1. hitelesített referencia-módszer ugyanezen értékeinek (3. táblázat) összehasonlíthatóknak kell lenniük (1). A II. és III. pontban kerülnek meghatározásra az elérni kívánt pontossági és megbízhatósági szabványértékek. A javasolt vizsgálati módszer és az 1. hitelesített referencia-módszer megbízhatóságának és működésének (érzékenység, specificitás, hamis negatív arányok, hamis pozitív arányok és pontosság) összeegyeztethetősége érdekében bevonták az összes kémiai osztályt képviselő besorolás nélküli, valamint (az ENSZ által javasolt GHS szerinti 2. kategóriába) besorolt anyagot. Új anyagok vizsgálatára történő alkalmazását megelőzően a vizsgálati módszer megbízhatóságát, valamint az ENSZ által javasolt GHS szerinti 2. kategóriába tartozó irritatív hatású anyagok helyes azonosítására való képességét bizonyítani kell.
TELJESÍTMÉNYSZABVÁNYOK
A teljesítményszabványok a következő három alkotóelemből állnak: I. a vizsgálati módszer alapvető összetevői, II. referenciaanyagok és III. a pontosság és megbízhatóság meghatározott értékei (2). E teljesítményszabványok az ECVAM bőrirritáció-validálási tanulmány elkészítése után meghatározásra került teljesítményszabványokon alapulnak (3).
I. A vizsgálati módszer alapvető összetevői
A modellre vonatkozó általános feltételek
Az epitélium felépítéséhez normál emberi keratinsejteket kell használni. Élő epitélsejtek több rétegének (bazális, tüskés, és granuláris sejtrétegnek) kell jelen lenni a funkcionális stratum corneum alatt. A stratum corneumnak többrétegűnek kell lennie, és megfelelő lipidszelvénnyel kell rendelkeznie, hogy nagy ellenálló képességgel funkcionális barriert képezzen a citotoxikus markeranyagok, pl. a nátrium-dodecil-szulfát (SDS) vagy a Triton X-100 gyors áthatolása ellen. A barrierfunkció értékelését vagy a markeranyag azon koncentrációjának meghatározásával lehet végezni, amelynél a szövetek életképessége 50 %-kal visszaesik(IC50) egy meghatározott expozíciós idő elteltével, vagy a sejt életképességének 50 %-kal való csökkentéséhez szükséges expozíciós idő meghatározásának segítségével a markeranyag meghatározott koncentrációban történő alkalmazásakor. A modell visszaszorító tulajdonságainak meg kell akadályozni, hogy az anyag áthatoljon a stratum corneum körül az élő szövetbe. A vizsgált vegyszerek stratum corneum körül történő áthatolása a bőr expozíciójának gyenge modellezését eredményezné. A bőrmodellnek baktérium-, vírus-, mikoplazma- és gombafertőzéstől mentesnek kell lennie.
Funkcionális modellfeltételek
Életképesség
Az életképesség terjedelmének meghatározását lehetőleg az MTT segítségével kell végezni (4). A negatív kontrollszövetből (NC) kivont (szolibilizált) festékanyag optikai sűrűségének (OD) legalább 20-szor nagyobbnak kell lennie az extrahálásra használt oldószer OD-jénél. Dokumentálni kell, hogy az NC-vel kezelt kontrollszövet a vizsgálati expozíciós időszak alatt a tenyészetben állandó (hasonló életképesség-mérést nyújt).
Barrierfunkció
A stratum corneumnak és lipidszelvényeinek megfelelő ellenálló képességgel kell rendelkezni a citotoxikus markeranyagok, pl. a nátrium-dodecil-szulfát (SDS) vagy a Triton X-100 gyors áthatolása ellen, az IC50 vagy ET50 alapján végzett becslések szerint.
Morfológia
A rekonstruált bőr/epidermisz szövettani vizsgálatát olyan megfelelően képzett személyzetnek kell végezni, akik emberi bőr/epidermisz-szerű struktúrákon (beleértve a többrétegű stratum corneumot is) végzik a bizonyító vizsgálatot.
Reprodukálhatóság
A specifikus modellt alkalmazó vizsgálati eredményeknek idővel reprodukálhatóknak kell lenniük, lehetőleg a megfelelő tételenkénti kontroll (összehasonlító) anyag szerint. (Lásd a fogalommeghatározásokat az 1.2. szakaszban).
A modell minőségellenőrzése (QC)
Minden felhasználásra kerülő felhámmodell csoportnak meghatározott előállítási kritériumoknak kell megfelelni, amelyek között az életképességre (4. bekezdés) és a barrierfunkcióra (5. bekezdés) vonatkozóak a leglényegesebbek. A bőrmodell szállítója (illetve saját előállítású modell esetén a vizsgálatot végző személy) köteles elfogadhatósági tartományt (felső és alsó határértéket) megállapítani az IC50 vagy a ET50 számára. A szövetek átvétele után a laboratóriumnak ellenőriznie kell a szövetek barrier tulajdonságait. Az irritatív hatások megbízható előrejelzéséhez kizárólag a megfelelő szövetek által produkált eredményeket szabad elfogadni. A hitelesített referencia-módszer elfogadhatósági tartományai például a következők.
1. táblázat
Példák a minőség-ellenőrzési tételkibocsátás kritériumaira
|
|
Alsó elfogadási határ |
Elfogadási tartomány középértéke |
Felső elfogadási határ |
|
1. Hitelesített referencia-módszer (18 órás nátrium-dodecil-szulfátos (SDS) kezelés) |
IC50 = 1,0 mg/ml |
IC50 = 2,32 mg/ml |
IC50 = 3,0 mg/ml |
|
2. Hitelesített referencia-módszer (1 % Triton X100) |
ET50 = 4,8 óra |
ET50 = 6,7 óra |
ET50 = 8,7 óra |
II. Referenciaanyagok
Referenciaanyagokat kell alkalmazni annak megállapítására, hogy a javasolt, rekonstruált emberi felhámmodellen végzett és a hitelesített referencia-módszerekhez szerkezetileg és funkcionálisan bizonyítottan hasonló, vagy egy hitelesített referencia-módszer kisebb módosítását képviselő új in vitro vizsgálati módszer megbízhatósága és pontossága összeegyeztethető eredményeket mutat-e az 1. hitelesített referencia-módszerével (1). A 2. táblázatban felsorolt 20 referenciaanyag között megtalálhatók különböző vizsgált kémiai osztályokba tartozó, valamint az ENSZ által javasolt GHS szerinti 2. kategóriába tartózó anyagok is. E listán a felsorolt anyagok között tíz az ENSZ által javasolt GHS szerinti 2. kategóriába tartózó anyag, három az ENSZ által javasolt GHS meghatározás szerinti opcionális 3. kategóriába tartozó anyag, és hét besorolás nélküli anyag található. E vizsgálati módszer alapján az opcionális 3. kategória nem minősül kategóriának. E referenciaanyagok képezik azon anyagok minimális számát, amelyeket fel kell használni a javasolt, rekonstruált emberi felhámmodellen végzett bőrirritációs vizsgálati módszer pontosságának és megbízhatóságának értékelésekor. Olyan helyzetekben, amikor a felsorolt anyagok nem állnak rendelkezésre, megfelelő in vivo referenciaadatokkal rendelkező egyéb anyagok is felhasználhatók. Szükség esetén a javasolt vizsgálati módszer pontosságának további értékeléséhez a referenciaanyagok minimális listája kiegészíthető más kémiai osztályba tartozó és megfelelő in vivo referenciaadatokkal rendelkező további anyagokkal.
2. táblázat
A rekonstruált emberi felhám (epidermisz) bőrirritációs modelleknél a pontossági és megbízhatósági értékek meghatározásához használt referenciaanyagok
|
Anyagok (1) |
CAS-szám |
EINECS-szám |
halmazállapot |
In vivo eredmény |
GHS in vitro kat. |
GHS in vivo kat. |
|
1-bróm-4-klórbután |
6940-78-9 |
230-089-3 |
F |
0 |
2. kat. |
Kat. nélkül |
|
Dietilftalát |
84-66-2 |
201-550-6 |
F |
0 |
Kat. nélkül |
Kat. nélkül |
|
Naftalin ecetsav |
86-87-3 |
201-705-8 |
Sz |
0 |
Kat. nélkül |
Kat. nélkül |
|
Allil fenoxi-acetát |
7493-74-5 |
231-335-2 |
F |
0,3 |
Kat. nélkül |
Kat. nélkül |
|
Izopropanol |
67-63-0 |
200-661-7 |
F |
0,3 |
Kat. nélkül |
Kat. nélkül |
|
4-metil-thio-benzaldehid |
3446-89-7 |
222-365-7 |
F |
1 |
2. kat. |
Kat. nélkül |
|
Metil sztearát |
112-61-8 |
203-990-4 |
Sz |
1 |
Kat. nélkül |
Kat. nélkül |
|
Heptil butirát |
5870-93-9 |
227-526-5 |
F |
1,7 |
Kat. nélkül |
Opcionális 3. Kat. |
|
Hexil-szalicilát |
6259-76-3 |
228-408-6 |
F |
2 |
Kat. nélkül |
Opcionális 3. kat. |
|
tri-izobutil foszfát |
126-71-6 |
204-798-3 |
F |
2 |
2. kat. |
Opcionális 3. kat. |
|
1-dekanol |
112-30-1 |
203-956-9 |
F |
2,3 |
2. kat. |
2. kat. |
|
Ciklámen aldehid |
103-95-7 |
203-161-7 |
F |
2,3 |
2. kat. |
2. kat. |
|
1-brómhexán |
111-25-1 |
203-850-2 |
F |
2,7 |
2. kat. |
2. kat. |
|
2-klórmetil-3,5-dimetil-4-metoxipiridin hidroklorid |
86604-75-3 |
434-680-9 |
Sz |
2,7 |
2. kat. |
2. kat. |
|
A-terpineol |
98-55-5 |
202-680-6 |
F |
2,7 |
2. kat. |
2. kat. |
|
di-n-propil diszulfid |
629-19-6 |
211-079-8 |
F |
3 |
Kat. nélkül |
2. kat. |
|
Butil metakrilát |
97-88-1 |
202-615-1 |
F |
3 |
2. kat. |
2. kat. |
|
Benzoltiol, 5-(1,1-dimetil-etil)-2-metil |
7340-90-1 |
438-520-9 |
F |
3,3 |
2. kat. |
2. kat. |
|
1-metil-3-fenil-1-piperazin |
5271-27-2 |
431-180-2 |
Sz |
3,3 |
2. kat. |
2. kat. |
|
heptanal |
111-71-7 |
203-898-4 |
F |
4 |
2. kat. |
2. kat. |
A 2. táblázatban felsorolt anyagok az ECVAM nemzetközi bőrirritáció-validálási tanulmányban alkalmazott 58 anyag reprezentatív megoszlását mutatják (1). Kiválasztásuk a következő ismérvek alapján történt:
|
— |
kereskedelmi forgalomban kapható anyagok, |
|
— |
a Draize-féle irritációs pontértékek teljes tartományát reprezentáló anyagok (a nem irritatív hatásútól az erősen irritatív hatásúig), |
|
— |
jól meghatározott kémiai szerkezettel rendelkeznek, |
|
— |
reprezentatív módon jellemzik a hitelesített módszer reprodukálhatóságát és előre jelezhetőségét az ECVAM validálási tanulmányban meghatározottak szerint, |
|
— |
reprezentálják a validálási eljárásban felhasznált kémiai működést, |
|
— |
nem kapcsolódik hozzájuk fokozottan toxikus jelleg (pl. rákkeltő vagy toxikus hatás a reprodukciós rendszerre) és tiltó jellegű ártalmatlanítási költségek sem. |
III. A pontosság és megbízhatóság meghatározott értékei
A javasolt vizsgálati módszer és az 1. hitelesített referencia-módszer működésének (érzékenység, specificitás, hamis negatív arányok, hamis pozitív arányok, és pontosság) összeegyeztethetőknek kell lenniük (3. táblázat); azaz az érzékenységnek nagyobbnak vagy egyenlőnek kell lennie, mint (≥) 80 %, a specificitásnak nagyobbnak vagy egyenlőnek kell lennie, mint (≥) 70 %, és a pontosságnak nagyobbnak vagy egyenlőnek kell lennie (≥) 75 %-nál. A húsz anyagra a vizsgálatokban résztvevő különböző laboratóriumokban kapott összes besorolást alkalmazni kell a működés számításánál. Minden egyes anyag besorolását minden egyes laboratóriumban az életképesség átlagértékének felhasználásával kell meghatározni a különböző szakaszok (minimum három érvényes szakasz) végrehajtása során.
3. táblázat
Az 1. hitelesített referencia-módszer működése (2)
|
Vizsgálati módszer |
Anyagok száma |
Érzékenység |
Specificitás |
Hamis negatív arány |
Hamis pozitív arány |
Pontosság |
|
1. hitelesített referencia-módszer (3) |
58 |
87,2 % (4) |
71,1 % (5) |
12,8 % |
29,9 % |
74,7 % |
|
1. hitelesített referencia-módszer (3) |
20 |
90 % |
73,3 % |
10 % |
26,7 % |
81,7 % |
A javasolt vizsgálati módszer és a hitelesített referencia-módszerek megbízhatóságának összeegyeztethetőknek kell lenniük.
Laboratóriumon belüli reprodukálhatóság
A laboratóriumon belüli eltérések vizsgálata esetén a húsz referenciaanyag egyetlen laboratóriumban történő, különböző, egymástól független vizsgálata során kapott besorolási értékeknek (2. kategória/kategória nélküli) 90 %-nál nagyobb, vagy azzal egyenlő mértékű egyezést kell mutatni.
Laboratóriumok közötti reprodukálhatóság
A laboratóriumok közötti reprodukálhatóság vizsgálata nem szükséges, amennyiben a javasolt vizsgálati módszer csak egyetlen laboratóriumon belül kerül alkalmazásra. A laboratóriumok között átadásra kerülő módszerek esetében a húsz referenciaanyag lehetőleg minimum három laboratóriumban történő, különböző, egymástól független vizsgálata során kapott besorolási értékeknek (2. kategória/kategória nélküli) 80 %-nál nagyobb, vagy azzal egyenlő mértékű egyezést kell mutatni.
HIVATKOZÁSOK
|
1. |
Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovió, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007) The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test. ATLA 35, 559–601. |
|
2. |
OECD (2005) Guidance Document No. 34 on the validation and international acceptance of new or updated test methods for hazard assessment. OECD, Paris. |
|
3. |
ECVAM (2007) Performance Standards for applying human skin models to in vitro skin irritation. Available under Download Study Documents, at http://ecvam.jrc.ec.europa.eu. Accessed on 27.10.2008. |
|
4. |
Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, 55–63. |
|
5. |
Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, 603–619. |
(1) A húsz referenciaanyag abból az 58 anyagból került reprezentatív kiválasztásra, amelyeket eredetileg az 1. referenciamódszer (EpiSkin™) validálásához használtak fel. A vizsgált anyagok teljes listája és kiválasztási szempontjai rendelkezésre állnak (5).
(2) A 3. táblázat az 1. hitelesített referencia-módszer működését az irritatív hatású anyagok (az ENSZ által javasolt GHS szerinti 2. kategória), valamint a besorolásra nem került anyagok (kategória nélkül, ideértve az opcionális 3. kategóriát is) helyes azonosítására való képessége alapján ismerteti az 58, illetve a 20 referenciaanyag esetére (2. táblázat).
(3) EpiSkin™
(4) 13 GHS szerinti 2. kat. irritatív anyag alapján.
(5) 45 GHS szerinti 3. kat. irritatív vagy a GHS szerint be nem sorolt vegyi anyag alapján.
IV. MELLÉKLET
|
C.3. |
SZAPORODÁSGÁTLÁSI VIZSGÁLAT, ÉDESVÍZI ALGA ÉS CIANOBAKTÉRIUM |
1. MÓDSZER
A módszer egyenértékű az OECD TG 201 (2006) (1) jelű módszerrel.
1.1. BEVEZETÉS
A tudományos haladás fényében a vizsgálati módszerek időről időre felülvizsgálatra és korszerűsítésre kerülnek. Szükség volt a C.3 jelű vizsgálati módszer átdolgozására, hogy kiterjedjen további fajokra is, és hogy megfelelhessen a vegyületekkel szemben támasztott kockázatértékelési és besorolási követelményeknek. A módszer átdolgozása a kiterjedt gyakorlati tapasztalatok, az algákra ható toxicitás vizsgálatának területén végbement tudományos fejlődés és a módszer eredeti elfogadása óta folyamatban lévő széles körű hatósági alkalmazás tapasztalatai alapján történt.
1.2. DEFINÍCIÓK
E vizsgálati módszerben az alábbi definíciók és rövidítések használatosak:
„Élőanyag”: egy populációban jelen lévő élő anyag száraz tömege egy adott mennyiségben kifejezve; pl. mg alga/liter vizsgálati oldat. Általában az „élőanyag” tömegként van definiálva, de ebben a vizsgálatban a szó „tömeg/térfogat”-ot jelent. Szintén ebben a vizsgálatban az élőanyag tömege helyett jellemzően sejtszám, fluoreszcencia stb. mérése történik, és az „élőanyag” kifejezés használata így ezekre a helyettesítő mérőszámokra is utal.
„Variációs együttható (CV)”: egy paraméter változékonyságának dimenzió nélküli mérőszáma; definíció szerint a szórás és az átlag aránya. Ezt százalékos formában is ki lehet fejezni. A párhuzamos kontrolltenyészetekben az átlagos fajlagos szaporodási sebesség átlagos variációs együtthatóját az alábbiak szerint lehet kiszámítani:
|
1. |
Ki kell számítani az átlagos fajlagos szaporodási sebesség %-os variációs együtthatóját a naponkénti/szakaszonkénti szaporodási sebességekből a megfelelő párhuzamos tenyészetekre. |
|
2. |
Ki kell számítani az 1. pontban kiszámított összes érték átlagát, ami megadja a párhuzamos kontrolltenyészetekben a naponkénti/szakaszonkénti átlagos fajlagos szaporodási sebesség átlagos variációs együtthatóját. |
„ECx ”: a vizsgálati oldatban oldott vizsgált anyag olyan koncentrációja, ami meghatározott expozíciós idő alatt (ha eltér a teljes vagy szokásos kísérleti időtartamtól, akkor pontosan meg kell adni) a vizsgált organizmus x %-os (pl. 50 %) szaporodásgátlását eredményezi. A szaporodási sebességből (r), illetve a szaporulatból (y) levezetett EC-értékek egyértelmű jelölésére az „ErC”, illetve az „EyC” használatos.
„Tápoldat”: az a teljes mesterséges tenyésztő közeg, amelyben a vizsgált algák a vizsgált anyag jelenlétében szaporodnak. A vizsgált anyag feloldása általában a kísérleti oldattal történik.
„Szaporodási sebesség (átlagos fajlagos szaporodási sebesség)”: az élőanyag expozíciós idő alatti növekedésének logaritmusa.
„Észlelhető hatást okozó legkisebb koncentráció (LOEC)”: az a legkisebb mért koncentráció, amelyben az anyagnak a kontrolltenyészethez képest észlelhető, statisztikailag szignifikáns (p < 0,05) szaporodásgátló hatása van egy adott expozíciós idő alatt. Ugyanakkor minden, az LOEC-nél nagyobb koncentrációnak az LOEC-nél kapottal azonos, vagy nagyobb gátló hatást kell kifejtenie. Ha ez a két feltétel nem teljesül, az LOEC (és ennélfogva az NOEC) megválasztását teljes körűen meg kell indokolni.
„Észlelhető hatást még nem okozó koncentráció (NOEC)”: közvetlenül az LOEC alatti koncentráció.
„Hatásváltozó”: a toxikus hatás becslésére használt változó, amely különböző számítási módokkal az élőanyag leírására szolgáló bármelyik mért paraméterből levezethető. Ebben a módszerben a hatásváltozók a szaporodási sebesség, illetve a szaporulat, amelyek közvetlenül az élőanyag mérésével, vagy pedig az említett helyettesítő mérőszámok valamelyikének mérésével kaphatók meg.
„Fajlagos szaporodási sebesség”: egy mérési paraméter (ebben a vizsgálati módszerben az élőanyag mennyiségének változása) természetes alapú logaritmusainak különbségéből és a változáshoz tartozó időtartamból képzett hányadosként definiált hatásváltozó.
„Szaporulat”: az élőanyag vizsgálat alatti mennyiségi növekedésének a kifejezésére használt mérési változó értéke; az expozíciós idő végén mért változóérték mínusz az expozíciós idő kezdetén mért változóérték.
1.3. A VIZSGÁLAT ALKALMAZHATÓSÁGA
Ez a vizsgálati módszer legegyszerűbben olyan vízoldható anyagokra használható, amelyek a vizsgálati körülmények között valószínűleg a vízben maradnak. Az illékony, erősen adszorbeálódó, színes, vízben rosszul oldódó, esetleg a tápoldatban lévő tápanyagok vagy ásványi anyagok felvételére hatással lévő anyagok esetében szükség lehet a leírt módszer bizonyos módosítására (pl. zárt rendszer, a vizsgálathoz használt eszközök kondicionálása). Néhány megfelelő módosításhoz útmutatás található az irodalomban (2), (3) és (4).
1.4. A VIZSGÁLAT ELVE
A vizsgálat célja egy adott anyag által az édesvízi mikroalgákra és/vagy cianobaktériumokra gyakorolt hatások meghatározása. A vizsgált anyag exponenciálisan szaporodó organizmusokra hat egyszeres (batch) tenyészetekben, szokásosan 72 órán keresztül. A viszonylag rövid vizsgálati időtartam ellenére több generációra kiterjedő hatások vizsgálhatók.
A rendszerben fellépő hatás az, hogy csökken a szaporodás a vizsgált anyag különböző koncentrációinak kitett algatenyészetekben (kísérleti egységek). A hatás értékelése a koncentráció függvényében történik, összehasonlítva a szaporodást a párhuzamos, nem kezelt kontrolltenyészetekben észlelt szaporodással. A rendszerben a toxikus hatások teljes kifejlődéséhez (optimális érzékenység) a tenyészetek számára korlátlan exponenciális szaporodást kell biztosítani – megfelelő tápkörülmények és folyamatos világítás mellett – elegendően hosszú ideig ahhoz, hogy mérni lehessen a fajlagos szaporodási sebesség csökkenését.
A szaporodás és szaporodásgátlás mennyiségi meghatározásához mérni kell az alga élőanyag-tömegét az idő függvényében. Az élőanyag mennyisége definíció szerint száraz tömeg/térfogat, pl. mg alga/liter tápoldat. A száraz tömeg mérése azonban bonyolult, ezért helyettesítő paraméterek használatosak. Ezek közül a sejtszám a leggyakrabban használt paraméter. További helyettesítő paraméterek lehetnek a sejttérfogat, a fluoreszcencia, az optikai sűrűség stb. A mért helyettesítő paraméterek és az élőanyag-tömeg közötti átszámítási tényezőt ismerni kell.
A kísérlet végpontja a szaporodás gátlása, az élőanyag-tömeg expozíciós idő alatti növekedésének logaritmikusaként kifejezve (átlagos fajlagos szaporodási sebesség). A kísérleti oldatsorozattal mért átlagos fajlagos szaporodási sebességekből meg kell határozni azt a koncentrációt, amely a szaporodási sebességet adott x %-os (pl. 50 %-os) mértékben gátolja, és azt ErCx-ként (pl. ErC50) kell kifejezni.
E módszernek az EU szabályozási keretén belül történő alkalmazásához az eredményeket a 2.2 pontban leírt okokból kifolyólag az átlagos fajlagos szaporodási sebesség alapján kell kiszámítani. Ebben a vizsgálati módszerben használt másik hatásváltozó a szaporulat, amely szükséges lehet néhány országban az adott hatósági előírások teljesítéséhez. A szaporulat definíció szerint az expozíciós idő végén és az expozíciós idő kezdetén meglévő élőanyag tömegének különbsége. A kísérleti oldatsorozatban mért szaporulatból meghatározásra kerül az a koncentráció, amely a szaporulatot adott x %-os (pl. 50 %-os) mértékben gátolja, és azt EyCx-ként (pl. EyC50) kell kifejezni.
Emellett statisztikai módszerekkel meghatározható az észlelhető hatást okozó legkisebb koncentráció (LOEC) és az észlelhető hatást még nem okozó koncentráció (NOEC).
1.5. INFORMÁCIÓK A VIZSGÁLT ANYAGRÓL
A kísérleti körülmények kialakításához hasznosak lehetnek egyes információk a vizsgált anyagról, mint pl. szerkezeti képlet, tisztaság, fényérzékenység, stabilitás a kísérleti körülmények között, fényabszorpciós tulajdonságok, pKa és az átalakulási vizsgálatok eredményei, beleértve a biológiai lebonthatóságot vízben.
A vízben való oldhatóságot, az oktanol/víz megoszlási hányadost (Pow) és a vizsgált anyag gőznyomását ismerni kell, valamint rendelkezésre kell állnia egy, a kísérleti oldatban jelen lévő anyag mennyiségi meghatározására alkalmas validált módszernek, dokumentált visszanyerési hatásfokkal és kimutatási határral.
1.6. REFERENCIAANYAG
A vizsgálati eljárás ellenőrzéseként a kísérlet elvégezhető referenciaanyaggal (referenciaanyagokkal), például a nemzetközi körvizsgálatban (4) használt 3,5-diklór-fenollal. A kálium-bikromát szintén használható referenciaanyagként, zöldalgák esetében. Tanácsos legalább kétszer egy évben kísérletet végezni a referenciaanyaggal.
1.7. A VIZSGÁLAT ÉRVÉNYESSÉGE
A vizsgálat érvényességéhez a következő kritériumoknak kell teljesülniük:
|
— |
A kontrolltenyészetekben a élőanyag mennyiségének exponenciálisan kell növekednie, legalább 16-szorosára a 72-órás kísérleti idő alatt. Ez 0,92 nap–1-es fajlagos szaporodási sebességnek felel meg. A leggyakrabban használt fajoknál a szaporodási sebesség rendszerint jelentősen nagyobb ennél (lásd 1. függelék). Ez a kritérium az 1. függelékben felsoroltaknál lassabban szaporodó fajok alkalmazása esetében nem feltétlenül teljesül. Ebben az esetben a kísérleti időt meg kell hosszabbítani, hogy legalább 16-szoros szaporodást kapjunk a kontrolltenyészetekben úgy, hogy a szaporodás mindvégig exponenciális marad a kísérlet ideje alatt. A kísérlet ideje lerövidíthető (de legalább 48 órának kell lennie), hogy a korlátlan exponenciális szaporodás a kísérlet alatt végig fennálljon, feltéve hogy megmarad a legalább 16-szoros szaporodás. |
|
— |
A szakaszonkénti (0–1., 1–2. és 2–3. nap a 72 órás kísérletnél) fajlagos szaporodási sebesség átlagos variációs együtthatója a kontrolltenyészeteknél (lásd 1.2. pont, „variációs együttható”) nem haladhatja meg a 35 %-ot. A szakaszonkénti fajlagos szaporodási sebesség kiszámítására vonatkozóan lásd a 2.2.1. pont második bekezdését. Ez a kritérium a párhuzamos kontrolltenyészetekre számított variációs együttható átlagértékére vonatkozik. |
|
— |
Az átlagos fajlagos szaporodási sebesség variációs együtthatója a kísérleti idő alatt a párhuzamos kontrolltenyészetekben semmikor sem haladhatja meg a 7 %-ot a Pseudokirchneriella subcapitata fajjal és Desmodesmus subspicatus fajjal folytatott vizsgálatokban. A többi, kevésbé gyakran vizsgált faj esetében ez az érték nem lehet nagyobb 10 %-nál. |
1.8. A MÓDSZER LEÍRÁSA
1.8.1. Felszerelés
A tápoldatokkal érintkezésbe kerülő kísérleti eszközöknek és más felszereléseknek teljes egészében üvegből, vagy kémiailag semleges más anyagból kell készülniük. Alaposan át kell mosni őket, biztosítandó, hogy szerves vagy szervetlen szennyezések ne befolyásolhassák az algák szaporodását, vagy a tápoldat összetételét.
A kísérleti eszközök általában olyan méretű üveglombikok, amelyek a vizsgálat során végzett mérésekhez megfelelő térfogatú tenyészetet képesek befogadni, és biztosítják a kívánt mértékű CO2-átadást a környezeti levegőből (lásd az 1.8.9. pont második bekezdését). Figyelni kell arra, hogy a folyadéktérfogat elegendő legyen az analitikai meghatározások elvégzéséhez (lásd az 1.8.11. pont ötödik bekezdését).
Ezentúl szükséges néhány, vagy az összes az alábbi eszközök közül:
|
— |
Tenyésztőberendezés: tenyésztőszekrény vagy -fülke ajánlott, amelyekben a választott inkubációs hőmérsékletet ±2 °C pontossággal tartható. |
|
— |
Fénymérő berendezés: fontos megjegyezni, hogy a fényerő mérési módszere, és főleg az érzékelő (kollektor) típusa hatással lesz a mért értékre. A mérésekhez gömb alakú (4 π) érzékelő (amely a mérési sík alatti és feletti bármely szögből érkező közvetlen és visszavert fényre reagál), vagy félgömb alakú (2 π) érzékelő (amely a mérési sík feletti bármely szögből érkező fényre reagál) használata preferált. |
|
— |
Az alga élőanyag-tömegének meghatározására alkalmas berendezés: az ilyen élőanyag mérésére a leggyakrabban használt helyettesítő módszer, a sejtszámlálás elvégezhető elektronikus részecskeszámláló, Bürker-kamrával ellátott mikroszkóp, vagy áramlásos citométer segítségével. Más helyettesítő paraméterek mérhetők áramlásos citométerrel, fluoriméterrel, spektrofotométerrel vagy koloriméterrel. Célszerű kiszámítani a sejtszám és a száraz tömeg közötti átszámítási tényezőt. Spektrofotométer használata esetén szükséges lehet legalább 4 cm fényúttal rendelkező küvetták használatára, hogy a módszer kis élőanyag-koncentrációknál is használható méréseket szolgáltasson. |
1.8.2. Vizsgált organizmusok
A nem kitapadó mikroalgák és cianobaktériumok több faja használható. Az 1. függelékben felsorolt törzsekről már bizonyították, hogy megfelelően használhatók ennek a vizsgálati módszernek az eljárásai során.
Más faj használata esetén a törzset és/vagy az eredetet dokumentálni kell. Igazolni kell, hogy a vizsgálathoz kiválasztott alga exponenciális szaporodása a kísérlet ideje alatt mindvégig fennmarad az adott körülmények között.
1.8.3. Tápoldat
Két választható tápoldat, az OECD-tápoldat és az AAP-tápoldat ajánlott. Ezeknek a tápoldatoknak az összetétele a 2. függelékben látható. Figyelembe kell venni, hogy a két tápoldat kiindulási pH-értéke és pufferkapacitása (a pH-növekedés szabályozása) eltérő. Ennek megfelelően az alkalmazott tápoldattól függően a vizsgálati eredmények különbözőek lehetnek, kiváltképp disszociáló anyagok vizsgálata során.
Bizonyos célokhoz (pl. fémek és kelátképző szerek vizsgálatakor, vagy ha a kísérlet különböző pH-értékeken történik) a tápoldat módosítására lehet szükség. A módosított tápoldat használatát részletesen ismertetni és indokolni kell (3)(4).
1.8.4. Kiindulási élőanyag-koncentráció
A kiindulási élőanyagnak minden vizsgált tenyészetben ugyanannyinak és megfelelően kevésnek kell lennie, hogy exponenciálisan növekedhessen végig az expozíciós idő alatt, a tápanyagok elfogyásának veszélye nélkül. A kiindulási élőanyag mennyisége nem lépheti túl a 0,5 mg száraz tömeg/l értéket. Az alábbi kiindulási sejtkoncentrációk ajánlottak:
|
Pseudokirchneriella subcapitata: |
5·103 – 104 |
sejt/ml |
|
Desmodesmus subspicatus |
2–5 x 103 |
sejt/ml |
|
Navicula pelliculosa |
104 |
sejt/ml |
|
Anabaena flos-aquae |
104 |
sejt/ml |
|
Synechococcus leopoliensis |
5·104 – 105 |
sejt/ml |
1.8.5. A vizsgált anyag -koncentrációi
Az a koncentrációtartomány, amelyben a hatások valószínűleg fellépnek, koncentrációtartomány-meghatározási vizsgálatok eredményei alapján állapítható meg. A végleges meghatározó vizsgálathoz legalább öt, 3,2-nél nem nagyobb kvóciensű mértani sorozat szerinti koncentrációt kell kiválasztani. Olyan anyagok esetében, ahol a koncentráció-hatás görbe lapos, nagyobb kvóciens használata lehet indokolt. A koncentrációsorozatnak lehetőleg az algaszaporodási sebesség 5–75 %-os gátlását okozó tartományt kell lefednie.
1.8.6. Párhuzamos tenyészetek és kontrolltenyészetek
A vizsgálatot úgy kell megtervezni, hogy minden vizsgált koncentrációra három párhuzamos tenyészetet tartalmazzon. Ha az NOEC meghatározása nem követelmény, a vizsgálat ettől eltérő lehet úgy, hogy a koncentrációk száma nagyobb lesz, az egy koncentrációhoz tartozó párhuzamos tenyészetek száma pedig kisebb. A párhuzamos kontrolltenyészetek számának legalább háromnak kell lennie, és ideálisan kétszer annyinak, mint az egyes vizsgált koncentrációkhoz tartozó párhuzamos tenyészetek száma.
Külön oldatsorozatot lehet készíteni a vizsgált anyag koncentrációinak analitikai meghatározásához (lásd az 1.8.11. pont negyedik és hatodik bekezdését).
Ha a vizsgált anyag oldhatóvá tételéhez oldószert kell használni, akkor a vizsgálatnak további kontrolltenyészeteket is tartalmaznia kell, amelyekben az oldószernek ugyanabban a koncentrációban kell jelen lennie, mint a vizsgált tenyészetekben.
1.8.7. Az oltótenyészet elkészítése
A tápoldattal 2–4 nappal a kísérlet kezdete előtt oltótenyészetet kell készíteni, hogy a vizsgált algák hozzászokjanak a kísérleti körülményekhez, és hogy exponenciális szaporodási fázisban legyenek a kísérleti oldatok beoltásakor. Az alga élőanyag-mennyiségét úgy kell beállítani, hogy a kísérlet kezdetéig az exponenciális szaporodás érvényesüljön az oltótenyészetben. Az oltótenyészetet a kísérleti tenyészetekkel megegyező körülmények között kell inkubálni. Mérni kell az élőanyag mennyiségének növekedését az oltótenyészetben, ellenőrizendő, hogy a vizsgált törzsnek az adott tenyésztési körülmények közötti szaporodása a megfelelő tartományon belül marad-e. Az algatenyésztési eljárás egy példája a 3. függelékben található. Elkerülendő a kísérlet alatt az egyidejű sejtosztódást, szükség lehet esetleg az oltótenyészet egy második szaporítási lépésére is.
1.8.8. A kísérleti oldatok elkészítése
Minden kísérleti oldatnak azonos koncentrációban kell tartalmaznia a tápoldatot és a vizsgált algák kiindulási élőanyag-tömegét. A kiválasztott koncentrációjú kísérleti oldatokat rendszerint a vizsgált anyag törzsoldatának, a tápoldatnak és az oltótenyészetnek az összekeverésével kell előállítani. A törzsoldatokat általában a vizsgált anyagnak a kísérleti oldattal való feloldásával kell elkészíteni.
Oldószerek (pl. aceton, tercier butilalkohol és dimetil-formamid) használhatók hordozóanyagként a vízben rosszul oldódó anyagoknak a kísérleti oldatba juttatásához (2)(3). Az oldószer-koncentráció nem haladhatja meg a 100 µl/l–t, és az oldószert a kísérletsorozatban használt minden tenyészethez (beleértve a kontrolltenyészeteket is) azonos koncentrációban hozzá kell adni.
1.8.9. Inkubáció
A kísérleti lombikokat légáteresztő dugóval kell lezárni. A lombikokat rázás után a tenyésztőberendezésbe kell helyezni. A kísérlet folyamán az algáknak lebegniük kell, és elő kell segíteni a CO2-átadást. Ebből a célból folyamatos rázást vagy keverést kell alkalmazni. A tenyészeteket 21–24 °C tartományon belüli hőmérsékleten kell tartani, ±2 °C-os pontossággal szabályozva. Az 1. függelékben nem szereplő fajok, pl. a trópusi fajok esetében nagyobb hőmérséklet lehet megfelelő, feltéve hogy az érvényességi kritériumok teljesülnek. Ajánlott a lombikok véletlenszerű elrendezése majd naponkénti áthelyezése az inkubátorban.
A kísérlet során a kontrolloldatban a pH nem nőhet 1,5 egységnél többel. Fémek, és a kísérlet pH-ja körüli értéken részlegesen disszociáló vegyületek esetében szükséges lehet a pH-eltolódás korlátozása a reprodukálható és jól definiált eredmények érdekében. A 0,5 pH-egységnél kisebb eltolódás biztosítása technikailag kivitelezhető és a környezeti levegőből az oldatba történő CO2-bevitel megfelelő sebességének biztosításával, például a rázás intenzitásának növelésével, elérhető. Másik lehetőség a CO2-igény csökkentése a kiindulási élőanyag mennyiségének vagy a kísérlet idejének csökkentésével.
A felületnek, ahol a tenyészetek inkubálása történik, folytonos egyenletes fluoreszcens megvilágítást (pl. „hideg-fehér” vagy „nappali” fény) kell kapnia. Az alga- és cianobaktériumtörzsek fényigénye eltérő. A fényerőt úgy kell megválasztani, hogy az megfeleljen a vizsgált organizmusnak. A javasolt zöldalga-fajokra a kísérleti oldatok szintjén a fényerőt a 60–120 µE·m–2·s–1 tartományból kell kiválasztani, a megfelelő érzékelővel a fotoszintetikusan hatékony, 400–700 nm-es hullámhossz-tartományban mérve. Néhány faj, főként az Anabaena flos-aquae jól növekszik kisebb fényerő mellett, és a nagyobb fényerő károsíthatja. Ezekre a fajokra a 40–60 µE·m–2·s–1 tartományba eső átlagos fényerőt kell választani. (A luxban kalibrált fénymérő berendezéseknél a hideg fehér fényre megadott 4 440–8 880 luxos tartomány felel meg az ajánlott 60–120 µE·m–2·s–1-es fényerőnek). A fényerő az inkubációs területen ±15 %-nál jobban nem térhet el az átlagtól.
1.8.10. A kísérlet időtartama
A kísérlet hossza szokásos esetben 72 óra. Használható azonban rövidebb vagy hosszabb kísérleti időtartam is, feltéve, hogy az 1.7. pontban előírt összes érvényességi kritérium teljesül.
1.8.11. Mérések és analitikai meghatározások
Az alga élőanyag-mennyiségét a kísérlet ideje alatt minden lombikban legalább naponta meg kell határozni. Ha a mérések a kísérleti oldatból pipettával vett kis térfogatú mintákkal történnek, ezeket a mintákat nem kell visszajuttatni a lombikba.
Az élőanyag mennyiségének mérése mikroszkópos manuális sejtszámlálással vagy elektronikus részecskeszámlálással (sejtszámlálás és/vagy élőanyag térfogata) történik. Alternatív eljárások, pl. áramlásos citometria, in vitro vagy in vivo klorofillfluoreszcencia (6)(7), vagy optikai sűrűség mérése is használható, feltéve, ha igazolni lehet az élőanyag-tömeggel fennálló kielégítő korrelációt az élőanyag-tömegnek a kísérletben előforduló tartományában.
Az oldatok pH-ját a kísérlet kezdetén és végén meg kell mérni.
Amennyiben az alkalmazott koncentrációtartományban a vizsgált anyag meghatározásához rendelkezésre áll analitikai módszer, akkor a kísérleti oldatokat elemezni kell a kiindulási koncentrációk ellenőrzése, illetve annak ellenőrzése céljából, hogy a kísérletben használandó koncentrációk mindvégig fennállnak-e a kísérlet során.
Elegendő lehet a vizsgált anyag egy alsó és felső koncentráció-határértékének, valamint a várható EC50 érték körüli koncentrációjának a mérése a kísérlet kezdetén és végén, ha valószínű, hogy a kísérlet során a koncentráció kevesebb mint 20 %-kal tér el a névleges értéktől. Ha nem valószínű, hogy a koncentrációk a névleges érték 80–120 %-án belül maradnak, ajánlott az összes kísérleti koncentráció mérése a kísérlet kezdetén és végén. Illékony, instabil és erősen adszorbeálódó anyagoknál további mintavétel ajánlott 24 óránként végzendő elemzéshez a kísérlet folyamán mindvégig, hogy az anyagveszteség még jobban meghatározható legyen. Ilyen anyagoknál további párhuzamos tenyészetek szükségesek. A vizsgált anyag koncentrációjának meghatározását minden esetben elegendő csak egyetlen párhuzamos tenyészet lombikjából elvégezni az egyes kísérleti koncentrációkra (vagy a párhuzamos tenyészetek egyesítésével készített mintából).
A kifejezetten a vizsgálat folyamán előálló kísérleti koncentrációk méréséhez készített oldatokat a kísérlethez használtakkal megegyező módon kell kezelni, azaz ezeket is be kell oltatni algával és a kísérleti körülményekkel azonos körülmények között inkubálni kell. Ha az oldott anyag koncentrációját kell mérni, szükséges lehet az algák eltávolítása az oldatból. Az elválasztást lehetőleg az algák kiülepedéséhez elegendő, de kis fordulatszámú centrifugálással kell végezni.
Ha egyértelmű, hogy a vizsgált anyag koncentrációja kielégítő módon a névleges koncentráció vagy a mért kezdeti koncentráció ±20 %-án belül marad a kísérlet során, akkor az eredmények értékelése alapulhat a névleges értéken vagy a mért kezdeti koncentráció értékén. Ha a névleges vagy a mért kezdeti koncentrációtól való eltérés nagyobb mint ±20 %, akkor az eredmények értékelésének alapja a kísérlet alatti koncentrációk mértani átlaga, vagy a vizsgált anyag koncentrációjának csökkenését leíró modell (3)(8).
Az algaszaporodás gátlásának vizsgálata dinamikusabb vizsgálati rendszer, mint a legtöbb hasonló, a vízi toxicitást mérő rövid időtartamú vizsgálat. Ennek következtében a tényleges kísérleti koncentrációkat esetleg nehezen lehet meghatározni, főleg kis koncentrációk esetén, és ha a vizsgált anyag abszorbeálódik. Ilyen esetekben az, ha a vizsgált anyag a növekvő élőanyagban való adszorpciója miatti eltűnik az oldatból, nem jelenti azt, hogy eltávozott a vizsgált rendszerből. A kísérlet eredményeinek elemzése során ellenőrizni kell, hogy a vizsgált anyag koncentrációjának a vizsgálat során történő csökkenése együtt jár-e a szaporodásgátlás csökkenésével. Ha ez a helyzet áll fenn, mérlegelni lehet egy olyan megfelelő modell alkalmazását, amely leírja a vizsgált anyag koncentrációjának csökkenését (8). Ha ez nem áll fenn, akkor az eredmények elemzését megfelelő lehet a kiindulási (névleges vagy mért) koncentrációkra alapozni.
1.8.12. További vizsgálatok
Az oltótenyészet normális és egészséges fenotípusának ellenőrzésére, illetve a kísérlet végén az algák esetleges abnormális fenotípusának (ami lehet a vizsgált anyag hatása) megállapítására mikroszkópos vizsgálatokat célszerű végezni.
1.8.13. Határérték-vizsgálat
Bizonyos körülmények között, például amikor egy előzetes vizsgálat azt jelzi, hogy a vizsgált anyagnak nincs toxikus hatása 100 mg·l–1 koncentrációig, vagy a kísérleti oldatban való oldhatósága határáig (amelyik a kettő közül kisebb), határérték-vizsgálat végezhető, amely egy kontrollcsoport és egy kezelt csoport (100 mg·l–1-es vagy az oldhatósági határral megegyező koncentrációval) szaporodási viselkedésének összehasonlítását jelenti. Határozottan ajánlott ezt a kísérletben használt koncentráció mérésével alátámasztani. A határérték-vizsgálatra érvényes az összes korábban leírt kísérleti körülmény és érvényességi kritérium, kivéve, hogy a kezelt párhuzamos tenyészetek számának legalább hatnak kell lennie. A kontroll- és a kezelt csoport hatásváltozóit az átlagok összevetésére használható statisztikai analízissel (pl. Student-féle t-próba) lehet elemezni. Ha a két csoport szórásnégyzetei nem egyenlőek, akkor egyenlőtlen szórásnégyzetekre korrigált t-próbát kell végezni.
1.8.14. Elváltozás erősen színezett anyagok esetén
A besugárzást (fényerő) az e vizsgálati módszerben meghatározott legfelsőbb tartományban, vagyis 120µE m–2 s–1 vagy e fölött kell elvégezni.
A fényutat a kísérleti oldatok mennyiségének a csökkentésével (5–25 ml tartományban) le kell rövidíteni.
Megfelelő keverési eljárással (például mérsékelt rázással) kell biztosítani, hogy a vizsgált algatenyészet felszíne gyakran legyen kitéve erős sugárzásnak.
2. ADATOK
2.1. A SZAPORODÁSI GÖRBÉK SZERKESZTÉSE
A kísérleti lombikokban lévő élőanyag mennyiségét ki lehet fejezni a mérésre használt helyettesítő paraméter (pl. sejtszám, fluoreszcencia) mértékegységében is.
Táblázatba kell foglalni a vizsgált tenyészetekben és a kontrolltenyészetekben a becsült élőanyag-koncentrációkat, a vizsgált anyag koncentrációival és a legalább egész órákra kerekített mérési időpontokkal együtt, és ezek alapján kell megszerkeszteni a szaporodási görbéket. Ebben az első szakaszban használható akár logaritmikus, akár lineáris skála, a logaritmikus skála azonban kötelező, és általában jobban megjeleníti a kísérleti időszak alatt a szaporodási viselkedésben bekövetkező eltéréseket. Megjegyzendő, hogy logaritmikus ábrázolás esetén az exponenciális szaporodás egyenes vonalat ad, és hogy a vonal meredeksége adja meg a fajlagos szaporodási sebességet.
A görbék segítségével meg kell vizsgálni, hogy a kontrolltenyészetek a vizsgálat során mindvégig exponenciálisan szaporodtak-e, a várt sebességgel. Kritikusan meg kell vizsgálni minden adatpontot és a görbék lefutását, valamint ellenőrizni kell az adatsorokat és eljárásokat a lehetséges hibák tekintetében. Főleg azokat az adatok igényelnek ellenőrzést, melyeknél az eltérés valószínűleg szisztematikus hibából ered. Ha nyilvánvaló, hogy eljárási hiba történt, illetve ennek nagy a valószínűsége, akkor az adott adatpontot érvénytelenként meg kell jelölni, és ki kell zárni a további statisztikai elemzésből. (Ha a két vagy három párhuzamos tenyészetből az egyik lombik algakoncentrációja nulla, az azt jelezheti, hogy a lombik nem megfelelően lett beoltva, vagy megtisztítva). Az érvénytelenként elvetett adatpontoknál az elvetést egyértelműen indokolni kell a mérési jegyzőkönyvben. Csak eljárási hiba (ritka) lehet elfogadható indok, egyszerű pontatlanság nem. Az érvénytelen adatpontok azonosítására szolgáló statisztikai módszerek csak korlátozottan használhatóak az ilyen típusú problémákra, és nem helyettesítik a szakértői megítélést. Az érvénytelen adatpontokat (ilyenként megjelölve) célszerű megtartani az adatpontok között, feltüntetve azokat a későbbi grafikus vagy táblázatos megjelenítések során.
2.2. HATÁSVÁLTOZÓK
A vizsgálat célja a vizsgált anyag által az alga szaporodására gyakorolt hatás meghatározása. Ez a vizsgálati módszer két hatásváltozót ír le, mivel a tagországoknak különböző preferenciáik és szabályozási igényeik vannak. Ahhoz, hogy a vizsgálati eredmények minden tagországban elfogadhatóak legyenek, a hatásokat ki kell értékelni az alább leírt mindkét hatásváltozóval (a) és b)).
|
a) |
Átlagos fajlagos szaporodási sebesség: ez a hatásváltozó az élőanyag tömegének a kísérlet alatt bekövetkező logaritmikus növekedése alapján számítható, egy napra vetítve. |
|
b) |
Szaporulat: ez a hatásváltozó a kísérlet végén mérhető élőanyag-tömeg mínusz a kiindulási élőanyag-tömeg. |
E módszernek az EU szabályozási keretén belül történő alkalmazásához az eredményeket az alább leírt okokból kifolyólag az átlagos fajlagos szaporodási sebesség alapján kell kiszámítani. Meg kell jegyezni, hogy e két hatásváltozóval számított toxicitásértékek nem hasonlíthatóak össze, és a vizsgálati eredmények felhasználása során a kettő közötti különbségnek tudatában kell lenni. Az átlagos fajlagos szaporodási sebességen alapuló ECx értékek (ErCx) a két megközelítés matematikai alapjának következtében általában nagyobbak a szaporulaton alapuló eredményeknél (EyCx), ha e vizsgálati módszer kísérleti feltételei teljesülnek. Ez nem a két hatásváltozó érzékenysége közötti eltérésként értelmezendő, egyszerűen az értékek matematikailag eltérőek. Az átlagos fajlagos szaporodási sebesség koncepciója az algák korlátozás nélküli tenyészetekben történő általános exponenciális szaporodási sémáján alapszik, ahol a toxicitás mértéke a szaporodási sebességre gyakorolt hatások alapján becsülhető, függetlenül a kontrolltenyészet fajlagos szaporodási sebességének abszolút értékétől, a koncentráció-hatás görbe meredekségétől, vagy a kísérlet időtartamától. Ezzel szemben a „szaporulat” hatásváltozón alapuló eredmények függenek az említett többi változótól. Az EyCx függ az egyes vizsgálatokban használt algafajok fajlagos szaporodási sebességétől és a legnagyobb fajlagos szaporodási sebességétől, ami a fajok, sőt még különböző algatörzsek között is eltérő lehet. Ez a hatásváltozó nem használható az algafajok, de még a különböző törzsek mérgező anyagokkal szembeni érzékenységének összehasonlítására sem. Noha tudományos szempontból a toxicitás becsléséhez az átlagos fajlagos szaporodási sebesség használata preferált, e vizsgálati módszer a szaporulaton alapuló toxicitásbecsléseket is tartalmazza, hogy kielégítse a néhány országban érvényben lévő hatósági előírásokat.
2.2.1. Átlagos szaporodási sebesség
Az adott időszakra vonatkozó átlagos fajlagos szaporodási sebesség az egyes kontrolltenyészetekre és kezelt tenyészetekre az élőanyag mennyiségének logaritmikus növekedéseként számítható ki a következő egyenletből:
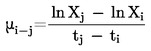 (nap–1)
(nap–1)
ahol:
|
μi–j |
az átlagos fajlagos szaporodási sebesség i-től j időpontig |
|
X i |
az élőanyag mennyisége i időpontban |
|
X j |
az élőanyag mennyisége j időpontban. |
Minden kezelt és kontrollcsoportra ki kell számítani egy átlagot a szaporodási sebességre, valamint meg kell becsülni a szórásnégyzetet.
Az átlagos fajlagos szaporodási sebességet ki kell számolni a teljes kísérleti időszakra (általában 0–3. nap), kezdeti értékként a névlegesen beoltott élőanyagot, nem pedig a mért kezdeti értéket használva, mert ilyen módon általában nagyobb pontosság érhető el. Ha az élőanyag mérésére használt mérőeszköz lehetővé teszi az oltó élőanyag kis mennyiségének megfelelően pontos meghatározását (pl. áramlásos citométer), akkor használható a mért kezdeti élőanyag-koncentráció. A szakaszonkénti szaporodási sebességet is meg kell állapítani a vizsgálat minden egyes napjára vonatkozóan (0–1., 1–2. és 2–3. nap), ugyanúgy számítva, mint a fajlagos szaporodási sebességet, és meg kell vizsgálni, hogy a kontrolltenyészet szaporodási sebessége állandó marad-e (lásd érvényességi kritériumok, 1.7. pont). Az első napon kapott, a teljes átlagos fajlagos szaporodási sebességnél szignifikánsan kisebb fajlagos szaporodási sebesség késési (lag) fázist jelez. Míg a késési (lag) fázis az előtenyészet megfelelő adagolásával minimalizálható és gyakorlatilag kiküszöbölhető a kontrolltenyészetekben, addig a vizsgált anyagnak kitett tenyészetekben a késési (lag) fázis a kezdeti toxicitás-stressz utáni helyreállást, vagy pedig a vizsgált anyagnak az expozíció elején történő elveszése (beleértve az élőanyagba történő szorpciót) miatti kisebb gátló hatást jelezhet. Ebből következően a szakaszonkénti szaporodási sebesség használható a vizsgált anyagnak a kísérlet során mutatott hatásainak az értékeléséhez. A szakaszonkénti szaporodási sebesség és az átlagos szaporodási sebesség közötti lényeges különbségek az állandó exponenciális szaporodástól való eltérést jelzik, és azt, hogy a szaporodási görbék alapos vizsgálata indokolt.
A szaporodási sebesség százalékos gátlása az egyes kezelt párhuzamos tenyészetekre vonatkozóan az alábbi egyenletből számítható ki:

ahol:
|
%Ir: |
átlagos szaporodási sebesség százalékos gátlása |
|
µC: |
átlagos szaporodási sebesség átlaga (μ) a kontrollcsoportban |
|
µT: |
átlagos szaporodási sebesség átlaga a kezelt párhuzamos tenyészetben. |
Ha a kísérleti oldatok oldószer felhasználásával készülnek, oldószer nélküli kontrolltenyészetek helyett oldószeres kontrolltenyészeteket kell használni a százalékos gátlás számításakor.
2.2.2. Szaporulat
A szaporulatot az egyes kontrolltenyészetekre és kezelt tenyészetekre úgy kell kiszámítani, hogy a kísérlet végén mért élőanyag-tömegből kivonjuk a kiindulási élőanyag-tömeget. Minden kísérleti koncentrációra és kontrolltenyészetre vonatkozóan ki kell számítani a szaporulatra egy átlagot, valamint meg kell becsülni a szórásnégyzetet. Az egyes kezelt párhuzamos tenyészetekre a szaporulatra vonatkozó százalékos gátlás (% Iy) az alábbiak szerint számítható ki:

ahol:
|
% Iy: |
százalékos szaporulatgátlás |
|
YC: |
a szaporulat átlaga a kontrollcsoportban |
|
YT: |
a szaporulat értéke a kezelt párhuzamos tenyészetekben. |
2.3. A KONCENTRÁCIÓ-HATÁS GÖRBE SZERKESZTÉSE
A százalékos szaporodásgátlást ábrázolni kell a vizsgált anyag koncentrációjának (logaritmus) függvényében, és alaposan meg kell vizsgálni a görbét, figyelmen kívül hagyva azokat az adatpontokat, amelyek az első szakaszban érvénytelenként lettek megjelölve. Egy egyenest kell illeszteni az adatpontokra kézzel vagy számítógépes interpolációval, ami egy első benyomást ad a koncentráció-hatás összefüggésről, majd ezután kifinomultabb módszert, lehetőleg számítógépes statisztikai módszert kell használni. Az adatok tervezett felhasználásának, minőségének (pontosságának) és mennyiségének, valamint a rendelkezésre álló adatelemző eszközök függvényében lehet úgy dönteni (és néha jól meg lehet indokolni), hogy ezen a szinten befejeződik az adatelemzés és a meghatározó értékek, azaz az EC50 és az EC10 (és/vagy EC20) egyszerűen leolvashatók a kézzel illesztett görbéről (lásd még alább a stimulációs hatásokról szóló pontot). A következők indokolhatják a statisztikai módszer mellőzését:
|
— |
Az adatok nem megfelelőek ahhoz, hogy számítógépes módszerrel további, még megbízhatóbb eredmény szülessen, mint a szakértői megítéléssel – ilyen esetekben néhány számítógépes program nem is képes megbízható megoldást adni (az iteráció nem konvergál stb.). |
|
— |
Stimulációs szaporodási viselkedés nem kezelhető pontosan a rendelkezésre álló számítógépes programokkal (lásd alább). |
2.4. STATISZTIKAI ELJÁRÁSOK
A cél kvantitatív koncentráció-hatás összefüggés előállítása regresszióanalízissel. Használható súlyozott lineáris regresszió, a hatásadatok például probit, logit vagy Weibull egységekbe történő lineáris transzformációja után (9), de célszerű előnyben részesíteni a nem lineáris regressziós technikákat, amelyek jobban kezelik az elkerülhetetlen adatszabálytalanságokat és az egyenletes eloszlástól való eltéréseket. Akár a nulla gátlás, akár a teljes gátlás közelében a transzformáció megsokszorozhat bizonyos szabálytalanságokat, ami hibás elemzést eredményezhet (9). Meg kell jegyezni, hogy a transzformációval kapott probit, logit vagy Weibull egységeket használó szabványos elemzési módszerek kvantált adatok (pl. halálozás vagy túlélés) használatára vannak tervezve, és a szaporodási adatokkal vagy az élőanyagra vonatkozó adatokkal való alkalmazásukhoz módosítani kell őket. Az ECx értékek folytonos adatokból történő meghatározására szolgáló speciális eljárásokat lásd az irodalomban, (10), (11) és (12). A nem lineáris regresszió használatáról a 4. függelékben található részletesebb információ.
Az ECx értékek pontbecsléseinek kiszámításához a koncentráció-hatás összefüggést kell használni minden elemzendő hatásváltozóra. Lehetőség szerint minden ilyen becsléshez meg kell határozni a 95 %-os megbízhatósághoz tartozó határokat. A hatásváltozók regressziós modellhez való illeszkedésének jóságát grafikusan vagy statisztikai módszerrel kell megállapítani. A regresszióanalízist az egyedi párhuzamos tenyészetekkel kapott adatokkal kell elvégezni, nem pedig a kezelt csoportok átlagaival. Ha azonban a nem lineáris görbeillesztés bonyolult vagy nem sikerül az adatok túl nagy szórása miatt, a problémát meg lehet kerülni a csoportátlagok regresszióanalízésével, ami egy gyakorlati mód a feltehető érvénytelen adatpontok hatásának csökkentésére. Ennek a lehetőségnek a használatát a szokásos eljárástól való eltérésként fel kell tüntetni a mérési jegyzőkönyvben, hivatkozva arra, hogy a görbeillesztések az egyedi párhuzamos tenyészeteknél kapott adatokkal nem adtak jó eredményt.
Az EC50 becsült értékeit és a megbízhatósági határokat visszatevéses mintavétellel („bootstrap” módszer) együtt alkalmazott lineáris interpolációval (13) is megkaphatjuk, ha a rendelkezésre álló regressziós modellek/módszerek nem használhatók a kapott adatokkal.
Az LOEC és ennélfogva az NOEC becsléséhez is, illetve a vizsgált anyag által a szaporodási sebességre gyakorolt hatásnak a becsléséhez szórásnégyzet-elemzési (ANOVA) eljárások segítségével össze kell hasonlítani a kezelt csoport átlagait. Ezután az egyes koncentrációk átlagát össze kell hasonlítani a kontrolltenyészetek átlagával megfelelő többszörös adat-összehasonlító vagy trendelemző módszer segítségével. A Dunnett- vagy a Williams-féle módszer használható lehet erre a célra (14)(15)(16)(17)(18). Meg kell állapítani, hogy fennáll-e a szórásnégyzetnek az ANOVA módszerben feltételezett homogenitása. Ez történhet grafikusan vagy formális próbával (18). A Levene- és Bartlett-féle próbák megfelelőek ehhez. Ha a szórásnégyzet homogenitásának követelménye nem teljesül, az néha az adatok logaritmikus transzformációjával korrigálható. Ha a szórásnégyzet rendkívül heterogén és transzformációval nem korrigálható, akkor fontolóra kell venni az olyan módszerekkel végzett elemzést, mint például a redukáló Jonkheere trendpróbák (step-down Jonkheere trend tests). Az NOEC meghatározásához további útmutatás is található az irodalomban (12).
Az újabb keletű tudományos eredmények az NOEC-koncepció elhagyásának ajánlásához és az ECx regresszión alapuló pontbecsléssel történő meghatározásával való helyettesítéséhez vezettek. Ehhez az algavizsgálathoz még nem állapítottak meg megfelelő x értéket. A 10–20 %-os tartomány azonban megfelelőnek tűnik (a választott hatásváltozótól függően), és célszerű mind az EC10-et, mind az EC20-at megadni a mérési jegyzőkönyvben.
2.5. SZAPORODÁSSTIMULÁCIÓ
Kis koncentrációk mellett néha szaporodásstimuláció (negatív gátlás) figyelhető meg. Ezt vagy a hormesis (kétfázisú reagálás) jelensége („toxikus anyag által kiváltott stimuláció”), vagy a vizsgált anyaggal együtt az alkalmazott ásványi sókat tartalmazó tápoldatba került szaporodásserkentő tényezők okozhatják. Megjegyzendő, hogy a szervetlen tápanyagok hozzáadásának nem lehet semmilyen közvetlen hatása, mert a kísérleti oldatnak a kísérlet folyamán végig feleslegben kell tartalmaznia a tápanyagot. Kis dózisú stimuláció gyakran figyelmen kívül hagyható az EC50 kiszámítása során, hacsak mértéke nem rendkívül nagy. Ha azonban rendkívül nagy mértékű, vagy pedig kis x értékre kell ECx-et számítani, akkor speciális eljárásokra lehet szükség. Lehetőség szerint kerülni kell a stimulációs hatások törlését az adatelemzésből, és ha a rendelkezésre álló görbeillesztő szoftver nem képes kezelni a kisebb mértékű stimulációkat, akkor visszatevéses mintavétellel („bootstrap” módszer) együtt alkalmazott lineáris interpoláció használható. Ha a stimuláció rendkívül nagy, akkor mérlegelni lehet hormesismodell alkalmazását (19).
2.6. NEM A TOXIKUS ANYAG ÁLTAL KIVÁLTOTT SZAPORODÁSGÁTLÁS
A fényt abszorbeáló anyagok a szaporodási sebesség csökkenését okozhatják, hiszen az árnyékolás csökkenti a kapott fény mennyiségét. Az ilyen jellegű fizikai hatásokat a kísérleti körülmények módosításával el kell választani a toxikus hatásoktól, és az előbbit külön kell dokumentálni. Útmutatás az irodalomban található, (2) és (3).
3. DOKUMENTÁCIÓ
3.1. MÉRÉSI JEGYZŐKÖNYV
A mérési jegyzőkönyvnek az alábbiakat kell tartalmaznia:
Vizsgált anyag:
|
— |
fizikai jelleg és a vonatkozó fizikai és kémiai tulajdonságok, beleértve a vízoldhatósági határértéket is, |
|
— |
kémiai azonosítás, beleértve a tisztaságot is. |
Vizsgált fajok:
|
— |
a törzs, a szállító vagy az eredet, és az alkalmazott tenyésztési körülmények. |
Kísérleti körülmények:
|
— |
a kísérlet kezdetének időpontja és a kísérlet időtartama, |
|
— |
a kísérlet kivitelezésének leírása: kísérleti eszközök, a tenyészetek mennyisége, az élőanyag sűrűsége a vizsgálat kezdetén, |
|
— |
a tápoldat összetétele, |
|
— |
kísérleti koncentrációk és párhuzamos tenyészetek (pl. párhuzamos tenyészetek száma, kísérleti koncentrációk száma és az alkalmazott mértani sor), |
|
— |
a kísérleti oldatok készítésének leírása, beleértve az oldószerek használatát stb., |
|
— |
tenyésztőkészülék, |
|
— |
fényerő és -minőség (fényforrás, homogenitás), |
|
— |
hőmérséklet, |
|
— |
kísérleti koncentrációk: a névleges kísérleti koncentrációk és a lombikokban lévő vizsgált anyag koncentrációjának meghatározására szolgáló analízisek eredménye. A vizsgálati mátrixra vonatkozóan a mérési jegyzőkönyvben fel kell tüntetni a módszer visszanyerési hatékonyságát és a mennyiségi meghatározás határértékét, |
|
— |
minden eltérés ettől a vizsgálati módszertől, |
|
— |
az élőanyag tömegének meghatározási módszere, valamint a mért paraméter és a száraz tömeg közötti korreláció bizonyítéka. |
Eredmények:
|
— |
pH-értékek minden kezelés kezdetén és végén, |
|
— |
az élőanyag tömege minden lombikban és minden mérési pontban, valamint az élőanyag mérésének módszere, |
|
— |
szaporodási görbék (az élőanyag mennyiségének ábrázolása az idő függvényében), |
|
— |
számított hatásváltozók minden kezelt párhuzamos tenyészetre, átlagokkal és variációs együtthatókkal a párhuzamos tenyészetekre, |
|
— |
a koncentráció-hatás összefüggés grafikus ábrázolása, |
|
— |
toxicitásbecslés a hatásváltozókra, pl. EC50, EC10, EC20 és a kapcsolódó megbízhatósági intervallumok. Az LOEC és az NOEC, ha ezek kiszámítása megtörtént, és a meghatározásukhoz használt statisztikai módszer, |
|
— |
az ANOVA módszer használata esetén a kimutatható hatás mértéke (pl. a legkisebb szignifikáns különbség), |
|
— |
bármilyen észlelt szaporodásstimuláció a kezelt rendszerekben, |
|
— |
bármilyen más észlelt hatás, például az algák morfológiai változása, |
|
— |
az eredmények tárgyalása, beleértve az ettől a vizsgálati módszertől való eltérés eredményeképpen a vizsgálat kimenetelére gyakorolt hatásokat. |
4. IRODALOM
|
(1) |
OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test |
|
(2) |
ISO 1998: Water quality – Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442 |
|
(3) |
OECD 2000: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment, no. 23. |
|
(4) |
ISO 1998: Water quality – Sampling – Part 16: General Guidance for Biotesting. ISO 5667–16. |
|
(5) |
ISO 1993: Water quality – Algal growth inhibition test. ISO 8692 |
|
(6) |
Mayer, P., Cuhel, R. and Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31: 2525–2531. |
|
(7) |
Slovacey, R.E. and Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), pp.919–925 |
|
(8) |
Simpson, S.L., Roland, M.G.E., Stauber, J.L. and Batley, G.E. (2003) Effect of declining toxicant concentrations on algal bioassay endpoints. Environ. Toxicol. Chem 22, 2073–2079. |
|
(9) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713–718. |
|
(10) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157–167. |
|
(11) |
Bruce, R.D., and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11:1485–1494. |
|
(12) |
OECD (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(13) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05–88. USEPA, Duluth, MN. |
|
(14) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50: 1096–1121 |
|
(15) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482–491. |
|
(16) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27: 103–117. |
|
(17) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28: 510–531. |
|
(18) |
Draper, N.R. and Smith, H. (1981). Applied Regression Analysis, second edition. Wiley, New York. |
|
(19) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93–96. |
1. függelék
A vizsgálathoz bizonyítottan alkalmas törzsek
Zöldalgák
|
— |
Pseudokirchneriella subcapitata, (régebbi elnevezéssel Selenastrum capricornutum), ATCC 22662, CCAP 278/4, 61.81 SAG |
|
— |
Desmodesmus subspicatus (régebbi elnevezéssel Scenedesmus subspicatus) 86.81 SAG |
Kovamoszatok
|
— |
Navicula pelliculosa, UTEX 664 |
Cianobaktériumok
|
— |
Anabaena flos-aquae, UTEX 1444, ATCC 29413, CCAP 1403/13A |
|
— |
Synechococcus leopoliensis, UTEX 625, CCAP 1405/1 |
A törzsek eredete
Az ajánlott törzsek egyfajú algatenyészetként beszerezhetőek az alábbi törzsgyűjteményekből (ábécésorrendben):
|
|
|
|
|
|
|
|
|
|
|
|
A javasolt fajok fenotípusa és jellemzői
|
|
P. subcapitata |
D. subspicatus |
N. pelliculosa |
A. flos-aquae |
S. leopoliensis |
|
Fenotípus |
Hajlott, csavart egyedi sejtek |
Ovális, főként egyedi sejtek |
Pálcák |
Ovális sejtekből álló fonalak |
Pálcák |
|
Méret (hossz × szélesség) µm |
8–14 × 2–3 |
7–15 × 3–12 |
7,1 × 3,7 |
4,5 × 3 |
6 × 1 |
|
Sejttérfogat (µm3/sejt) |
40–60 (1) |
60–80 (1) |
40–50 (1) |
30–40 (1) |
2,5 (2) |
|
Sejt száraz tömege (mg/sejt) |
2–3 × 10–8 |
3–4 × 10–8 |
3–4 × 10–8 |
1–2 × 10–8 |
2–3 × 10–9 |
|
Szaporodási sebesség (3) (nap–1) |
1,5–1,7 |
1,2–1,5 |
1,4 |
1,1–1,4 |
2,0–2,4 |
Egyedi ajánlások a vizsgálathoz ajánlott fajok tenyésztésére és kezelésére
Pseudokirchneriella subcapitata és Desmodesmus subspicatus
Ezeket a zöldalgákat általában könnyű fenntartani különféle tápoldatokban. A megfelelő tápoldatról a tenyészetgyűjteményektől szerezhető be információ. A sejtek általában különállóak, és a sejtsűrűség könnyen mérhető elektronikus részecskeszámlálóval vagy mikroszkóppal.
Anabaena flos-aquae
Törzstenyészet fenntartására számos tápoldat használható. Különösen fontos, hogy megújításkor elkerüljük az egyszeres (batch) tenyészet túlfutását az exponenciális fázison, mert azután már nehéz helyreállítani a tenyészetet.
Az Anabaena flos-aquae fonaltelepekben fejlődik. Ezek mérete a tenyésztési körülményekkel változik. Mikroszkópos vagy elektronikus részecskeszámlálóval végzett élőanyag-meghatározáshoz fontos lehet ezeknek a telepeknek a feltörése.
Részminták ultrahangos kezelése használható a láncok feltörésére, hogy csökkenjen a sejtszámlálás változékonysága. A fonalak rövidebb szakaszokra töréséhez szükségesnél hosszabb ultrahangos kezelés roncsolhatja a sejteket. Az ultrahangos kezelés intenzitásának és hosszának minden kezelésnél azonosnak kell lennie.
A változékonyság kompenzálásának elősegítése céljából elegendően nagy számú mezőben kell a számlálást végezni a hemocitométerben (legalább 400 sejt). Ez növeli a mikroszkópos sűrűségmeghatározás megbízhatóságát.
Elektronikus részecskeszámláló használható az Anabaena teljes sejttérfogatának meghatározásához, a sejtfonalak óvatos ultrahangos széttörése után. Az ultrahang energiáját úgy kell beállítani, hogy elkerüljük a sejtek roncsolását.
Örvénykeverő segítségével vagy hasonló alkalmas módszerrel biztosítani kell, hogy a lombikok beoltására alkalmazott algaszuszpenzió megfelelően el legyen keverve és homogén legyen.
A lombikokat kb. 150/perc fordulatszámú körpályás, vagy lengőmozgású rázóasztalra kell helyezni. Más megoldásként szakaszosan keverés is használható az Anabaena összetapadási hajlamának csökkentésére. Ha összetapadás történik, ügyelni kell arra, hogy az élőanyag-mérésekhez szükséges minták reprezentatívak legyenek. Az összetapadt algák szétválasztásához erőteljes keverés lehet szükséges a mintavétel előtt.
Synechococcus leopoliensis
A törzstenyészet fenntartására különféle tápoldatok használhatók. A megfelelő tápoldatról a tenyészetgyűjteményektől szerezhető be információ.
A Synechococcus leopoliensis különálló pálca alakú sejtek formájában szaporodik. A sejtek nagyon kicsik, ami megnehezíti az élőanyag-meghatározáshoz szükséges mikroszkópos sejtszámlálást. Jól használhatók a kb. 1 µm méretű részecskék számlására alkalmas elektronikus részecskeszámlálók. In vitro fluorometriai mérések szintén alkalmazhatóak.
Navicula pelliculosa
A törzstenyészet fenntartására különféle tápoldatok használhatók. A megfelelő tápoldatról a tenyészetgyűjteményektől szerezhető be információ. Figyelni kell arra, hogy a tápoldatban lennie kell szilikátnak.
A Navicula pelliculosa bizonyos körülmények között telepeket hozhat létre. A lipidtermelés következtében az algasejtek néha hajlamosak rétegekben összegyűlni a felszínen. Ilyen körülmények között az élőanyag mennyiségének meghatározásához leveendő minták esetében speciális intézkedések szükségesek ahhoz, hogy a minták reprezentatívak legyenek. Erőteljes rázás, pl. örvénykeverő használata lehet szükséges.
(1) Elektronikus részecskeszámlálóval mérve
(2) A méretből számítva
(3) Leggyakrabban tapasztalt szaporodási sebesség OECD-tápoldatban kb. 70 µE·m–2·s–1 fényintenzitásnál és 21 °C-on.
2. függelék
Tápoldatok
A következő két tápoldat egyike használható:
OECD-tápoldat: Az OECD TG 201 jelű eredeti tápoldat, megfelel az ISO 8692-nek is.
Az USA Környezetvédelmi Hatóságának (EPA) AAP-tápoldata, megfelel az ASTM-nek is.
Ezeknek a tápoldatoknak az elkészítése során laboratóriumi reagenseket vagy analitikai tisztaságú vegyszereket, és ionmentesített vizet kell használni.
Az AAP-tápoldat (USA EPA) és OECD TG 201 jelű tápoldat összetétele.
|
Komponens |
EPA |
OECD |
||
|
|
mg/l |
mM |
mg/l |
mM |
|
NaHCO3 |
15,0 |
0,179 |
50,0 |
0,595 |
|
NaNO3 |
25,5 |
0,300 |
|
|
|
NH4Cl |
|
|
15,0 |
0,280 |
|
MgCl2·6(H2O) |
12,16 |
0,0598 |
12,0 |
0,0590 |
|
CaCl2·2(H2O) |
4,41 |
0,0300 |
18,0 |
0,122 |
|
MgSO4·7(H2O) |
14,6 |
0,0592 |
15,0 |
0,0609 |
|
K2HPO4 |
1,044 |
0,00599 |
|
|
|
KH2PO4 |
|
|
1,60 |
0,00919 |
|
FeCl3·6(H2O) |
0,160 |
0,000591 |
0,0640 |
0,000237 |
|
Na2EDTA·2(H2O) |
0,300 |
0,000806 |
0,100 |
0,000269 (1) |
|
H3BO3 |
0,186 |
0,00300 |
0,185 |
0,00299 |
|
MnCl2·4(H2O) |
0,415 |
0,00201 |
0,415 |
0,00210 |
|
ZnCl2 |
0,00327 |
0,000024 |
0,00300 |
0,0000220 |
|
CoCl2·6(H2O) |
0,00143 |
0,000006 |
0,00150 |
0,00000630 |
|
Na2MoO4·2(H2O) |
0,00726 |
0,000030 |
0,00700 |
0,0000289 |
|
CuCl2.2(H2O) |
0,000012 |
0,00000007 |
0,00001 |
0,00000006 |
|
pH |
7,5 |
8,1 |
||
A Navicula pelliculosa kovamoszattal végzett vizsgálat során mindkét tápoldathoz Na2SiO3·9H20-t kell adni úgy, hogy a koncentráció 1,4 mg Si/l legyen.
A tápoldat pH-ját akkor kell mérni, amikor egyensúly van az oldat karbonáttartalma és a környezeti levegőben lévő CO2 parciális nyomása között. Közelítő összefüggés a 25 °C-on mért pH és a moláris bikarbonátkoncentráció között:
pHeq = 11,30 + log [HCO3]
15 mg NaHCO3/l-rel pHeq = 7,5 (EPA-tápoldat), és 50 mg NaHCO3/l-rel pHeq = 8,1 (OECD-tápoldat).
A kísérleti oldatok elemi összetétele
|
Kémiai elem |
EPA |
OECD |
|
|
mg/l |
mg/l |
|
C |
2,144 |
7,148 |
|
N |
4,202 |
3,927 |
|
P |
0,186 |
0,285 |
|
K |
0,469 |
0,459 |
|
Na |
11,044 |
13,704 |
|
Ca |
1,202 |
4,905 |
|
Mg |
2,909 |
2,913 |
|
Fe |
0,033 |
0,017 |
|
Mn |
0,115 |
0,115 |
Az OECD-tápoldat elkészítése
|
Tápanyagok |
Koncentráció a törzsoldatban |
|
1. törzsoldat: makrotápanyagok |
|
|
NH4Cl MgCl2·6H2O CaCl2·2H2O MgSO4·7H2O KH2PO4 |
1,5 g·l–1 1,2 g·l–1 1,8 g·l–1 1,5 g·l–1 0,16 g·l–1 |
|
2. törzsoldat: vas |
|
|
FeCl3·6H2O Na2EDTA·2H2O |
64 mg·l–1 100 mg·l–1 |
|
3. törzsoldat: nyomelemek |
|
|
H3BO3 MnCl2·4H2O ZnCl2 CoCl2·6H2O CuCl2·2H2O Na2MoO4·2H2O |
185 mg·l–1 415 mg·l–1 3 mg·l–1 1,5 mg·l–1 0,01 mg·l–1 7 mg·l–1 |
|
4. törzsoldat: bikarbonát |
|
|
NaHCO3 |
50 g·l–1 |
|
Na2SiO3·9H2O |
|
A törzsoldatokat membránszűréssel (átlagos pórusátmérő 0,2 µm) vagy autoklávban (120 °C, 15 perc) sterilizálni kell. Az oldatokat 4 °C-on, sötét helyen kell tárolni.
A 2. és 4. törzsoldatokat nem szabad autoklávban kezelni, hanem membránszűréssel kell őket sterilizálni.
Készítsük el a tápoldatot az 1–4. törzsoldatokból vett megfelelő térfogatok és víz összekeverésével:
Adjunk kb. 500 ml sterilizált vízhez:
|
— |
10 ml-t az 1. törzsoldatból, |
|
— |
1 ml-t az 2. törzsoldatból, |
|
— |
1 ml-t az 3. törzsoldatból, |
|
— |
1 ml-t az 4. törzsoldatból. |
Töltsük fel az oldatot 1 000 ml-re sterilizált vízzel.
Várjuk meg, hogy a tápoldat egyensúlyba kerüljön a környezetben lévő CO2-vel, szükség esetén buborékoltassunk át az oldaton steril, szűrt levegőt néhány órán keresztül.
Az AAP-tápoldat elkészítése
|
A1.1. |
Adjunk az A1.2.1–A1.2.7. pontokban felsorolt törzsoldatok mindegyikéből 1–1 ml-t kb. 900 ml ionmentesített vagy desztillált vízhez, majd hígítsuk fel 1 literre. |
|
A1.2. |
A makrotápanyagok törzsoldatainak készítéséhez oldjuk fel az alábbiakat 500 ml ionmentesített vagy desztillált vízben. Az A1.2.1., A1.2.2., A1.2.3. és A1.2.4. pontokban szereplő reagensek egyesíthetők egy törzsoldatba. |
|
A1.2.1. |
NaNO3 –12,750 g. |
|
A1.2.2. |
MgCl2 ·6H2O–6,082 g. |
|
A1.2.3. |
CaCl2 ·2H2O–2,205 g. |
|
A1.2.4. |
Mikrotápanyagok törzsoldata– (lásd A1.3.). |
|
A1.2.5. |
MgSO4·7H2O–7,350 g. |
|
A1.2.6. |
K2HPO4 –0,522 g. |
|
A1.2.7. |
NaHCO3 –7,500 g. |
|
A1.2.8. |
Na2SiO3·9H2O Lásd az A1.1. megjegyzést. A1.1. megjegyzés: Csak kovamoszatfajokhoz alkalmazandó. Beadható közvetlenül (202,4 mg) vagy törzsoldat formájában úgy, hogy 20 mg/l Si legyen a tápoldatban a végkoncentráció. |
|
A1.3. |
A mikrotápanyagok törzsoldatának elkészítéséhez oldjuk fel az alábbiakat 500 ml ionmentesített vagy desztillált vízben: |
|
A1.3.1. |
H3BO3 –92,760 mg. |
|
A1.3.2. |
MnCl2·4H2O– 207,690 mg. |
|
A1.3.3. |
ZnCl2 –1,635 mg. |
|
A1.3.4. |
FeCl3·6H2O–79,880 mg. |
|
A1.3.5. |
CoCl2·6H2O–0,714 mg. |
|
A1.3.6. |
Na2MoO4·2H2O–3,630 mg. |
|
A1.3.7. |
CuCl2·2H2O–0,006 mg. |
|
A1.3.8. |
Na2EDTA·2H2O– 150,000 mg. [Dinátrium(etilén-dinitrilo)tetraacetát]. |
|
A1.3.9. |
Na2SeO4·5H2O–0,005 mg. Lásd az A1.2. megjegyzést. A1.2. megjegyzés: Csak kovamoszatfajok törzstenyészetének tápoldatában alkalmazandó. |
|
A1.4. |
Állítsuk be a pH-t 7,5 ± 0,1 értékre 0,1 N vagy 1,0 N NaOH-dal vagy HCl-dal. |
|
A1.5. |
Szűrjük a tápközeget steril edénybe, vagy 0,22 μm-os membránszűrőn ha részecskeszámlálót használunk, vagy 0,45 μm-os szűrőn ha nem részecskeszámlálót használunk. |
|
A1.6. |
Felhasználásig tároljuk a tápoldatot sötét helyen, kb. 4 °C-on. |
(1) Az EDTA/vas mólarány kissé nagyobb 1-nél. Ez megakadályozza a vas kicsapódását, és ugyanakkor a kelátképződés a nehézfém-ionokkal a lehető legkisebb lesz.
3. függelék
Példa algatenyésztési eljárására
Általános megjegyzések
A következő eljáráson alapuló tenyésztés célja algatenyészetek előállítása toxicitásvizsgálatokhoz.
Az algatenyészetek bakteriális fertőződésének elkerülése érdekében megfelelő módszereket kell használni. Tiszta (axenikus) tenyészetek kívánatosak lehetnek, de egyfajú algatenyészeteket kell létrehozni és felhasználni.
Minden beavatkozást steril körülmények között kell végezni, elkerülendő a baktériumokkal, vagy más algákkal történő szennyeződést.
Eszközök és anyagok
Lásd „Vizsgálati módszer: Felszerelés”.
Eljárások algatenyészetek előállításához
Tápoldatok készítése:
A tápoldat minden tápsóját tömény törzsoldatként készítjük, és sötét hideg helyen tároljuk. Ezeket az oldatokat szűréssel vagy autoklávban sterilizálni kell.
A tápoldat a megfelelő mennyiségű törzsoldat desztillált vízhez való hozzáadásával készül, ügyelve arra, hogy ne történjen befertőződés. Szilárd táptalajhoz 0,8 % agart kell adni.
Törzstenyészet:
A törzstenyészetek kicsi algatenyészetek, melyeket rendszeresen friss tápoldatba juttatunk, hogy kiindulási anyagként szolgáljanak a vizsgálathoz. Ha a tenyészeteket nem használjuk rendszeresen, agarral töltött ferde kémcsövekben szélesztjük őket. Ezeket a tenyészeteket legalább kéthavonta egyszer átvisszük friss táptalajra.
A törzstenyészetek kitenyésztése a megfelelő táptalajt (térfogat kb. 100 ml) tartalmazó Erlenmeyer-lombikokban történik. Ha az algákat 20 °C-on, folyamatos megvilágítás mellett inkubáljuk, heti átoltás szükséges.
„Öreg” tenyészetből olyan mennyiségét kell átvinni steril pipettával a friss táptalajt tartalmazó lombikba, hogy a gyorsan szaporodó fajok esetén a kezdeti koncentráció 100-szor kisebb legyen, mint az „öreg” tenyészetben.
A faj szaporodási sebessége a szaporodási görbéből határozható meg. Ha ez ismert, akkor megbecsülhető az a sűrűség, amelynek elérésekor a tenyészetből át kell vinni az új tápoldatba. Ezt azelőtt kell elvégezni, mielőtt a tenyészet eléri a pusztulási fázist.
Előtenyészet:
Az előtenyészet célja, hogy a vizsgálathoz használt tenyészet beoltásához megfelelő mennyiségű algát adjon. Az előtenyészetet a kísérleti körülmények között inkubáljuk és akkor használjuk fel, amikor még exponenciálisan szaporodó fázisban van, szokásosan 2–4 napos inkubációs időszakot követően. A deformált vagy abnormális sejteket tartalmazó algatenyészeteket ki kell selejtezni.
4. függelék
Adatelemzés nem-lineáris regresszióval
Általános szempontok
Az algaszaporodási és más mikrobiológiai szaporodási vizsgálatokban a hatás – az élőanyag növekedése – természeténél fogva folyamatos, azaz metrikus változó – folyamatsebesség, ha a szaporodási sebességet használjuk, illetve annak idő szerinti integrálja, ha az élőanyag tömegét választjuk. Mindkettő olyan nem kezelt, párhuzamos kontrollmintákban fellépő, megfelelő hatás átlagához van viszonyítva, amelyek maximálisan reagáltak az adott körülményekre – az algás vizsgálatoknál ezek közül meghatározó tényező a fény és a hőmérséklet. A rendszer statisztikai eloszlású, azaz homogén, és az élőanyag kontiniumnak tekinthető, nem pedig önálló sejteknek. Ilyen rendszereknél a jellemző hatás szórásnégyzet-eloszlása kizárólag a kísérleti tényezőktől (amelyeket jellemzően a log/normál vagy a normál hibaeloszlás ír le) függ. Ezzel szemben, a jellemző biológiai kísérletek kvantált adatokkal szolgálnak, amelyeknél az önálló organizmusok tűréséről (ami jellemzően binomiális eloszlású) gyakran feltételezik, hogy az a meghatározó komponens a szórásnégyzetben. A kontrollmintában fellépő hatások ekkor nullával, vagy a háttérszinttel egyenlőek.
Egyszerű esetben a normált vagy relatív válasz, r, monoton csökken 1-től (nulla gátlás) 0-ig (100-százalékos gátlás). Megjegyzendő, hogy minden hatással együtt jár egy belső hiba, és hogy a megjelenő negatív gátlások csak véletlen hiba eredményeként számolhatók.
Regresszióanalízis
Modellek
A regresszióanalízis célja, hogy egy matematikai regressziós függvény, Y = f (C), vagy még gyakrabban F (Z), ahol Z = log C, formájában mennyiségileg leírja a koncentráció-hatás görbét. A függvény inverze, a C = f–1 (Y), segítségével kiszámíthatók az ECx értékek, köztük az EC50, EC10 és EC20, valamint ezekre a 95 %-os megbízhatósághoz tartozó határok. Több egyszerű matematikai függvény bizonyítottan jól leírja az algákkal végzett szaporodásgátlási vizsgálatokban kapott koncentráció-hatás összefüggéseket. Ilyen függvény például a logisztikai egyenlet, az aszimmetrikus Weibull-egyenlet, és a log/normál eloszlási függvény, amelyek mind szigmoid görbék, melyek aszimptotikusan közelítenek 1-hez, ha C → 0-hoz, illetve 0-hoz, ha C → a végtelenhez.
Újabban ajánlott a folytonos küszöbérték-függvényre alapuló modellek (mint pl. a Kooijman-modell a „populációnövekedés gátlására”, Kooijman és mtsai, 1996) használata, akár az aszimptotikus modellek alternatívájaként. Ez a modell feltételezi, hogy egy adott küszöbérték, EC0+, alatt a koncentrációknak nincs hatása, és ennek a küszöbértéknek a meghatározása a koncentráció-hatás görbe egy, a kezdőpontban nem differenciálható egyszerű folyamatos függvény segítségével történő extrapolációjával és a koncentrációtengely metszésével történik.
Megjegyzendő, hogy az analízis lehet a legkisebb négyzetek összegének módszere (állandó szórásnégyzetet feltételezve), vagy a súlyozott legkisebb négyzetek módszere, ha a szórásnégyzet heterogenitása kompenzálva van.
Eljárás
Az eljárás a következő: választani kell egy megfelelő függvényt, az Y = f (C)-t, és nem lineáris regresszióval rá kell illeszteni az adatpontokra. Lehetőség szerint az egyes lombikokból származó méréseket használjuk, ne pedig a párhuzamos minták átlagait, hogy a lehető legtöbb információt nyerjük ki az adatokból. Ha viszont a szórásnégyzet értéke nagy, akkor a gyakorlati tapasztalatok azt mutatják, hogy a párhuzamos minták átlagai szilárdabb matematikai becslést eredményezhetnek (amelyet csak kevéssé befolyásolnak az adatokban meglévő véletlen hibák), mint a minden egyes megtartott adatpont használata.
Az illesztett görbe és a mért adatok megszerkesztése után meg kell vizsgálni, hogy a görbe illeszkedése megfelelő-e. A különbségek elemzése különösen hasznos eszköz lehet erre a célra. Ha a koncentráció-hatás pontokra való illesztéshez választott függvény nem írja le jól a teljes görbét, vagy annak egy adott, alapvetően fontos részét – mint például a kis koncentrációknál jelentkező hatásokat – akkor egy másik illesztési megoldást kell választani, a szimmetrikus helyett például aszimmetrikus görbét, mint pl. a Weibull-függvényt. Negatív gátlás esetén probléma lehet például a logaritmikus/normál eloszlási függvénnyel, ekkor szintén egy másik regressziófüggvényt kell használni. Az ilyen negatív értékek nullával vagy kis pozitív számmal történő helyettesítése nem ajánlott, mert torzítja a hibaeloszlást. Célravezetőbb lehet másik illesztést használni az érintett szakaszokra – például arra, ahol a szaporodásgátlás kis mértékű –, egy adott EClow x érték meghatározása céljából. Az illesztési egyenletből ki kell számítani (inverz számítással, C = f–1 (Y)) az ECx jellemző pontértékeit, és a mérési jegyzőkönyvben meg kell adni legalább az EC50-re és egy vagy két EClow x-re kapott értékeket. A gyakorlati tapasztalat azt mutatja, hogy az algás vizsgálatok pontossága általában elfogadhatóan pontos becslést tesz lehetővé 10 %-os gátlási szinten, ha van elég adatpont – kivéve, ha zavaró tényezőként stimuláció fordul elő kis koncentrációknál. Az EC20-becslés pontossága gyakran lényegesen jobb, mint az EC10-é, mert az EC20 rendszerint a koncentráció-hatás görbe középső, közelítőleg lineáris szakaszára esik. A szaporodásstimuláció miatt néha nehéz az EC10-et értelmezni. Összefoglalva, noha az EC10 általában kielégítő pontossággal meghatározható, mindig ajánlott az EC20 feltüntetése is a mérési jegyzőkönyvben.
Súlyozó tényezők
A tapasztalati szórásnégyzet általában nem állandó, és rendszerint tartalmaz egy proporcionális tagot, ezért előnyős rutinszerűen súlyozott regressziót végezni. Az ilyen analízishez használt súlyozó tényezők általában fordítottan arányosak a szórásnégyzettel:
Wi = 1/Var(ri)
Sok regresszióelemző programban lehetőség van a súlyozott regresszióanalízisre úgy, hogy a súlyozó tényezők egy táblázatban vannak felsorolva. Kényelmi szempontból a súlyozó tényezőket célszerű normálni, megszorozva őket n/Σ wi -vel (ahol n az adatpontok száma), hogy az összegük egyenlő legyen 1-gyel.
A hatásváltozók normálása
A kontrollmintával kapott hatások átlagával történő normálás elvi problémákat vet fel és meglehetősen bonyolult szórásnégyzet-szerkezethez vezethet. Ha a szaporodásgátlás százalékos meghatározásához a kapott hatásértékeket elosztjuk a kontrollmintával kapott hatások átlagával, további hibát viszünk be, amit a kontrollminta átlagának hibája okoz. Ha ez a hiba nem elhanyagolhatóan kicsi, akkor a regresszióhoz és a megbízhatósági határokhoz használt súlyozó tényezőket korrigálni kell a kontrollminták kovarianciájával (17). Megjegyzendő, hogy nagyon fontos a kontrollmintákkal kapott hatásértékek becsült átlagának a pontossága, hogy a lehető legkisebb legyen a relatív hatás általános szórásnégyzete. Ez a szórásnégyzet a következő:
az i alsó index az i-dik koncentrációkat, a 0 alsó index pedig a kontrollmintákat jelöli
Yi = Relatív hatás = ri/r0 = 1 – I = f (Ci)
szórásnégyzettel:
Var (Yi) = Var (ri/r0) ≅ (∂Yi / ∂ ri)2·Var(ri) + (∂ Yi/ ∂ r0)2·Var (r0)
és mivel
(∂ Yi/ ∂ ri) = 1/r0 and (∂ Yi / ∂ r0) = ri/r0 2
normál eloszlású adatoknál és mi és m0 párhuzamos mintákkal:
Var(ri) = σ2/mi
a relatív hatás, Yi, teljes szórásnégyzete így a következő:
Var(Yi) = σ2/(r0 2 mi) + ri 2·σ2/r0 4 m0
A kontrollminták átlagának hibája fordítottan arányos az átlagolt párhuzamos kontrollminták számának négyzetgyökével, és néha indokolt lehet régebbi adatokat is felhasználni és így nagy mértékben csökkenteni a hibát. Más megoldásként mellőzhető az adatok normálása és az illesztést az abszolút hatásértékekre kell elvégezni, beleértve a kontrollminta hatásadatait is, de ekkor újabb paraméterként be kell vezetni a kontrollminta hatásértékét, amelyre nem lineáris regresszióval el kell végezni az illesztést. A szokásos 2-paraméteres regresszióegyenlet esetén ehhez a módszerhez 3 paraméter illesztése szükséges, és így több adatpontot igényel, mint az előre beállított kontrollminta-hatás segítségével normált adatokon végzett nem lineáris regresszió.
Inverz megbízhatósági intervallumok
A nem lineáris regresszió megbízhatósági intervallumainak inverz becsléssel történő kiszámítása meglehetősen bonyolult és a szokásos statisztikai számítógépes programcsomagokban ez a lehetőség nem is áll rendelkezésre. A körülbelüli megbízhatósági határok megkaphatók szokásos nem lineáris regressziós programmal, a paraméterek megváltoztatásával (Bruce and Versteeg, 1992), ami a matematikai egyenletnek az átírását jelenti a kívánt pontbecslésekkel, pl. az EC10-zel és az EC50-nel mint megbecsülendő paraméterekkel. (Legyen a függvény I = f (α, β, koncentráció) és a definiáló egyenletek, f (α, β, EC10) = 0,1 és f (α, β, EC50) = 0,5 felhasználásával helyettesítsük be az f (α, β, koncentráció) függvényt egy ekvivalens g (EC10, EC50, koncentráció) függvénnyel.
Közvetlenebb számítás (Andersen és mtsai, 1998) végezhető az eredeti egyenlet megtartásával és az ri és r0 átlagok körüli Taylor-sorfejtés felhasználásával.
Újabban a visszatevéses mintavételes (bootstrap) módszerek váltak népszerűvé. Ezek a módszerek mért adatokat, és véletlenszám-generátor által irányított gyakori mintavételt használnak a tapasztalati szórásnégyzet eloszlásának becslésére.
Irodalom
Kooijman, S.A.L.M.; Hanstveit, A.O.; Nyholm, N. (1996): No-effect concentrations in algal growth inhibition tests. Water Research, 30, 1625–1632.
Bruce, R.D. and Versteeg,, D.J.(1992) A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem.11, 1485–1494.
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A. & Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics, 3, 405–420.
V. MELLÉKLET
|
C.25. |
AEROB MINERALIZÁCIÓ FELSZÍNI VIZEKBEN – VIZSGÁLAT A BIOLÓGIAI LEBONTÁS SZIMULÁLÁSÁRA |
1. MÓDSZER
Ez a módszer megegyezik az OECD TG 309 (2004) (1) jelű módszerrel.
1.1. BEVEZETÉS
A vizsgálat célja az aerob természetes vízben, kis koncentrációban jelenlévő vizsgálati anyag biológiai lebontása időtartamának meghatározása, és a megfigyelések reakciókinetikai kifejezések szerinti számszerűsítése. Maga a szimuláció egy laboratóriumi, rázó-lombikos egymenetes vizsgálat, amelynek a segítségével természetes felszíni vizek (édes-, brakk- vagy tengervíz) mintáiban jelen lévő szerves anyagok aerob biológiai lebontásának sebessége megadható. A módszer alapja az ISO/DIS 14592–1 (2), de magában hordozza a C.23 és a C.24 (3)(4) módszerek egyes elemeit is. Lehetőség van arra, hogy hosszú vizsgálati idő esetén az egymenetes működtetést félfolyamatos működtetésre változtassuk a vizsgálati mikrokozmosz romlásának megelőzése céljából. A szimulációs vizsgálat alapvető célja a felszíni vízben jelen lévő vizsgálni kívánt anyag mineralizációjának meghatározása, ahol a mineralizáció a lebontás kinetikája kifejézésének az alapját képezi. Emellett azonban a vizsgálat másodlagos célja, hogy információt szolgáltasson az elsődleges lebontásról és a keletkező fő transzformációs termékekről. Ez utóbbiak azonosítása, és amennyiben lehetséges, koncentrációjuk számszerűsítése a nagyon lassan mineralizálódó anyagok (pl. ha a teljes maradvány 14C izotóp felezési ideje meghaladja a 60 napot) esetében különösen fontos. Az analitikai megkötésekből következően a fő transzformációs termék azonosításához és mennyiségi meghatározásához a vizsgálati anyagot jellemzően magasabb koncentrációban (pl. > 100 μg/l) kell alkalmazni.
Ebben a vizsgálatban alacsony koncentráció alatt olyan koncentrációértéket értünk (pl. kisebb mint 1–100 μg/l), amely elég alacsony ahhoz, hogy biztosítsa azt, hogy a vizsgálatban kapott biológiai lebontási kinetika jól tükrözze a környezetben várható kinetikát. A vizsgálathoz használt természetes vízben található biológiailag lebontható szén szubsztrátok teljes mennyiségéhez képest a vizsgálati anyag csak kis koncentrációban van jelen, és csak másodlagos szubsztrátként fog szolgálni. Ez maga után vonja, hogy a várható biológiai lebontási kinetika elsőrendű (nem-növekedési kinetika), és hogy a vizsgálati anyag lebontása kometabolizmussal történhet. A kinetika elsőrendűségéből következik, hogy a lebontás sebessége (mg/l/nap) arányos a szubsztrát koncentrációjával, amelynek értéke az időben folyamatosan csökken. A valódi elsőrendű kinetikában a specifikus lebontási sebességi állandó, a k, független az időtől és a koncentrációtól. Vagyis k értéke nem változik jelentősen egy kísérlet során, és a különböző kísérletekben éppen alkalmazott vizsgálati anyag koncentrációjának hatására sem. Lényegében a specifikus lebontási sebességi állandó értéke megegyezik a koncentráció időegységenkénti relatív változásával: k = (1/C) · (dC/dt). Bár rendesen az előírt kritériumok teljesülése mellett az elsőrendű kinetika várható, lehetnek bizonyos körülmények, amelyek között más kinetikák megfelelőbbek. Az elsőrendű kinetikától való eltérést tapasztalhatunk, ha tömegátviteli jelenség, például a diffúziósebesség határozza meg a biotranszformáció sebességét, és nem a biológiai reakció sebessége. Ezek az adatok azonban majdnem mindig leírhatóak pszeudo-elsőrendű kinetikával, ha bevezetünk egy koncentráció-függő sebességi állandót is.
A kísérletek megtervezését és a kapott eredmények értelmezését megkönnyíti, ha a vizsgálat megkezdése előtt megfelelő információk (pl. szabványos szűrővizsgálatokból) állnak rendelkezésre a nagy koncentrációjú vizsgálati anyag biológiai lebonthatóságáról, valamint abiotikus lebontásáról, a transzformációs termékekről és vonatkozó fizikokémiai tulajdonságaikról. A teljes biológiai lebontás mértékének meghatározását a 14C-jelölt vizsgálati anyag alkalmazása, és annak a vizsgálat végén kapott fázismegoszlása teszi lehetővé. Jelöletlen vizsgálati anyag alkalmazása esetén a teljes biológiai lebontás mértékének megállapítása csak akkor lehetséges, ha a vizsgálati anyag koncentrációja magas és a fő transzformációs termékek ismertek.
1.2. FOGALOMMEGHATÁROZÁSOK
Elsődleges biológiai lebontás: Egy kémiai anyagban a mikroorganizmusok által létrehozott szerkezeti változás (transzformáció), amely a kémiai azonosság elvesztésével jár.
Funkcionális biológiai lebontás: Egy kémiai anyagban a mikroorganizmusok által létrehozott szerkezeti változás (transzformáció), amely a kiindulási anyag egy specifikus tulajdonságának elvesztésével jár.
Teljes aerob biológiai lebontás: Egy kémiai anyag oxigén jelenlétében történő, mikroorganizmusok általi lebontása szén-dioxiddá, vízzé és a jelen lévő bármely más elem ásványi sóivá (mineralizáció), valamint új biomassza és szerves mikrobiális bioszintézis-termékek létrehozása.
Mineralizáció: Egy vegyi vagy szerves anyag oxigén jelenlétében történő, mikroorganizmusok általi lebontása szén-dioxiddá, vízzé és a jelen lévő bármely más elem ásványi sóivá.
Lappangási fázis: A vizsgálat kezdetétől addig eltelt idő, amíg a lebontást végző mikrobák adaptációja megtörténik, és a vegyi anyag vagy a szerves anyag biológiai lebontásának mértéke a detektálható szint fölé emelkedik (pl. a legmagasabb elméleti biológiai lebontás 10 %-a, vagy ennél kisebb érték a mérési módszer pontosságától függően)
A legmagasabb biológiai lebontási szint: A vizsgált vegyi vagy szerves anyag biológiai lebontásának mértéke százalékban, amely felett további biológiai lebontás a vizsgálat folyamán már nem történik.
Elsődleges szubsztrát: A természetes szén- és energiaforrások összessége, amely a mikrobiális biomassza növekedésére és fenntartására fordítódik.
Másodlagos szubsztrát: Egy olyan, kis koncentrációban jelen lévő szubsztrátkomponens, amelynek a lebontása csak elhanyagolhatóan kis szén- és energiaforrást jelent az érintett mikroorganizmus számára, a fő komponensek (elsődleges szubsztrát) lebontásával szolgáltatott szén- és energiaforráshoz viszonyítva.
Lebontási sebességi állandó: Egy elsőrendű, vagy pszeudo-elsőrendű kinetikus sebességi állandó, k (nap–1), ami a lebontási folyamatok sebességét jelzi. Egymenetes kísérletben k a lappangási fázis vége után nyert lebontási görbe kezdeti szakaszából következtethető.
Felezési idő, t1/2 (nap): Az elsőrendű reakció sebességének leírására használt kifejezés. A koncentráció felére csökkenéséhez szükséges időintervallum. A felezési idő és a lebontási sebességi állandó a t1/2 = ln2/k egyenlet szerint aránylik egymáshoz.
Lebontásos felezési idő, DT50 (nap): A biológiai lebontási vizsgálat eredményének számszerűsítésére használt kifejezés. Az a lappangási fázist is tartalmazó időintervallum, amely az 50 %-os biológiai lebontási érték eléréséhez szükséges.
Mennyiségi meghatározás határa (LOD) és a mérhetőségi határ (LOQ): A mennyiségi meghatározás határa (LOD) az adott anyagnak az a koncentrációja, amely alatt az anyag egyértelmű azonosításához szükséges jel nem különböztethető meg az analitika háttérzajától. A mérhetőségi határ (LOQ) az adott anyagnak az a koncentrációja, amely alatt a koncentráció nem határozható meg elfogadható pontossággal.
Oldott szerves széntartalom (DOC): A vízminta szerves széntartalmának az a része, amely nem távolítható el meghatározott fázis szétválasztással (pl. 15 percig tartó 40 000 ms–2-es centrifugálással vagy 0,2 –0,45 μm pórusátmérőjű membránon keresztül történő szűréssel).
Teljes szerves 14C aktivitás (TOA): A szerves széntartalomhoz tartozó teljes 14C aktivitás.
Oldott szerves 14C aktivitás (DOA): Az oldott szerves széntartalomhoz tartozó teljes 14C aktivitás.
Szemcsés szerves 14C aktivitás (POA): A szemcsés szerves széntartalomhoz tartozó teljes 14C aktivitás.
1.3. A VIZSGÁLAT ALKALMAZHATÓSÁGA
Ez a szimulációs vizsgálat kis koncentrációban alkalmazott nem illékony, vagy kis mértékben illékony szerves anyagok vizsgálatára alkalmas. A légkör felé nyitott (pl. vattadugóval lezárt) lombikok alkalmazása mellett a gyakorlatban az 1 Pa·m3/mol-nál (kb. 10–5 atm·m3/mol) kisebb Henry-állandójú anyagok tekinthetők nem illékonynak. A gőztérrel rendelkező zárt lombikok esetében a kis mértékben illékony anyagok (Henry-állandójuk < 100 Pa·m3/mol vagy < 10–3 atm·m3/mol) is vizsgálhatóak anélkül, hogy anyagveszteség lépne fel. A megfelelő óvintézkedések elhagyása esetén a 14C-jelölt anyag elvesztése léphet fel, amikor a CO2 eltávozik a rendszerből. Ilyen esetekben szükséges lehet a CO2 visszatartása egy belső lúg abszorbenssel, vagy egy külső abszorbens rendszerrel (direkt 14CO2 meghatározás; lásd 3. melléklet). A biológiai lebontási kinetika meghatározásához a vizsgálati anyag koncentrációjának kisebbnek kell lennie a vízben való oldhatósági határánál. Meg kell jegyezni azonban, hogy a vízben való oldhatóság szakirodalmi értékei jelentősen magasabbak lehetnek, mint a vizsgálati anyag természetes vízben való oldhatósága. Lehetőség van arra is, hogy a vízben különösen gyengén oldódó vizsgálati anyagok oldhatóságát megállapítsuk a vizsgált természetes vizek alkalmazásával.
A biológiai lebontást szimuláló módszer használható mind durva részecskéktől mentes víz („nyílt vízi vizsgálat”), mind pedig olyan zavaros felszíni vizek esetében, amelyek pl. a víz/üledék határfelülethez közel fordulhatnak elő („lebegő-üledék vizsgálat”).
1.4. A VIZSGÁLAT ALAPELVE
A vizsgálatot egy adagban hajtják végre, vagy csak felszíni vízben inkubálva a vizsgálati anyagot („nyílt vízi vizsgálat”), vagy 0,01–1 g/l száraztömegű lebegő szilárd anyaggal/üledékkel kiegészített felszíni vízben inkubálva a vizsgálati anyagot (lebegő üledék vizsgálat). Ez utóbbi módszer szimulálja a lebegő szilárd anyagot vagy felkavart üledéket tartalmazó víztestet. A lebegő szilárd anyag/üledék ilyen koncentráció-intervallumának alsó tartománya jellemző a legtöbb felszíni vízre. A vizsgálati lombikok inkubálása aerob körülmények között, rázatva, környezeti hőmérsékleten, sötétben történik. A lebontásos kinetika meghatározásához legalább két különböző vizsgálatianyag-koncentráció alkalmazása szükséges. A koncentrációknak ötös-tizes szorzófaktorral kell eltérniük egymástól, és a környezetben várható koncentrációtartományt kell mutatniuk. A vizsgálati anyag maximális koncentrációja nem lépheti túl a 100 µg/l–t, de 10 µg/l alatti, vagy még kisebb tesztkoncentrációkat részesítenek előnyben annak biztosításához, hogy a biológiai lebontás valóban elsőrendű kinetikát kövessen. A minimális koncentráció nem lehet több 10 µg/l-nél, de az 1–2 μg/l, vagy az 1 μg/l-nél kisebb a legelőnyösebb. Normál esetben az ilyen alacsony koncentrációk megfelelő analízise a kereskedelmi forgalomból beszerezhető 14C-jelzett anyagokkal elvégezhető. Az analitikai módszer korlátjai miatt gyakran lehetetlen a vizsgálati anyag koncentrációjának kívánt pontosságú mérése abban az esetben, ha a vizsgálati anyag bemérési koncentrációja ≤ 100 µg/l (lásd az 1.7.2. pont második bekezdését). A fő transzformációs termék azonosításához és mennyiségi meghatározásához, illetve ha nem áll rendelkezésre alacsony mennyiségi meghatározási határral bíró specifikus analitikai módszer, a vizsgálati anyag magas koncentrációban is alkalmazható (> 100 µg/l és néha > 1 mg/l). Ilyen esetekben a kapott eredmények nem használhatóak az elsőrendű lebontási sebességi állandó és a felezési idő számításához, hiszen a lebontás valószínűleg nem elsőrendű kinetikát követ.
A lebontás nyomon követése megfelelő időpontokban mért maradó 14C koncentráció mérésével, vagy specifikus kémiai vizsgálat használata esetén a vizsgálati anyag maradvány koncentrációjának mérésével történik. A molekula legstabilabb részének 14C-jelölése biztosítja a teljes mineralizáció értékének meghatározását, míg a molekula kevésbé stabil részének jelölése, valamint a specifikus vizsgálat használata teszi lehetővé az elsődleges biológiai lebontás mértékének becslését. Mindamellett a legstabilabb rész nem feltétlenül foglalja magában a molekula vonatkozó funkciós csoportját (ami egy speciális tulajdonsággal, pl. toxicitással, bioakkumulációval stb. lehet kapcsolatos). Ebben az esetben célszerű lehet olyan vizsgálati anyag alkalmazása, amely a funkcionális részen 14C-jelölt, hogy az adott tulajdonság megszűnése követhetővé váljon.
1.5. INFORMÁECIÓK A VIZSGÁLATI ANYAGRÓL
Radioaktívan jelölt és nem jelölt anyagok egyaránt használhatóak a vizsgálat során. Ajánlott a 14C-jelöléses technika. A jelölésnek normál esetben a molekula legstabilabb részét kell érintenie (lásd az 1.4. szakaszt is). Egynél több aromás gyűrűt tartalmazó anyagok esetében célszerű minden gyűrű egy vagy több szénatomjának 14C-jelölése. Emellett javasolt a kapcsolódó, könnyen lebontható csoportok mindkét oldalán egy vagy több szénatom jelölése is. A vizsgálati anyag kémiai és/vagy radiokémiai tisztaságának > 95 %-nak kell lennie. Radioaktívan jelölt anyagok esetében javasolt a kb. 50 μCi/mg (1,85 MBq), vagy még nagyobb specifikus aktivitás, ami megkönnyíti a 14C mérést a kis kezdeti koncentrációjú vizsgálatokban. A vizsgálati anyagokról az alábbi információkkal kell rendelkezni:
|
— |
vízben való oldhatóság [A.6. módszer], |
|
— |
szerves oldószer(ek)ben való oldhatóság (oldószerrel együtt alkalmazott, vagy vízben rosszul oldódó anyagok), |
|
— |
disszociációs állandó (pKa), amennyiben az anyag hajlamos a protonálódásra, illetve deprotonálódásra [OECD TG 112] (5), |
|
— |
gőznyomás [A.4 módszer] és Henry-állandó, |
|
— |
kémiai stabilitás vízben és sötétben (hidrolízis) [C.7. módszer]. |
Amennyiben vízben rosszul oldódó anyagot vizsgálunk tengervízben, hasznos tudni a kisózási állandót (vagy „Setschenow-állandót”) Ks, amely a log (S/S’) = Ks Cm egyenletből fejezhető ki, ahol S és S’ az anyag oldhatósága édes-, illetve tengervízben és Cm a moláris sókoncentráció értéke.
Ha a vizsgálatokat „lebegő üledék vizsgálatként” végezzük el, az alábbi információkkal kell rendelkezni:
|
— |
n-oktanol/víz megoszlási együttható [A.8. módszer], |
|
— |
adszorpciós együttható [C.18. módszer]. |
További fontos adatok lehetnek:
|
— |
a vizsgálati anyag környezeti koncentrációja, ha ez ismert, vagy becsült, |
|
— |
a vizsgálati anyag mikroorganizmusokra gyakorolt toxicitása [C.11. módszer], |
|
— |
gyors és/vagy inherens lebonthatóságáról [C.4 A–F, C.12, C.9, OECD TG 302 (5) módszerek], |
|
— |
aerob vagy anaerob lebonthatóság a talaj és üledék/víz transzformációs kísérletekben [C.23., C.24. módszer]. |
1.6. REFERENCIAANYAG
Referenciaanyagként célszerű egy aerob körülmények között könnyen lebontható anyagot (pl. anilin, vagy nátrium-benzoát) használni. Az anilin és nátrium-benzoát lebontásának várható időtartama rendszerint kevesebb mint 2 hét. A referenciaanyag használatával megbizonyosodhatunk arról, hogy a mikrobiális aktivitás a tesztvízben bizonyos határokon belül mozog, azaz, hogy a vízminta tartalmazz aktív mikrobiális populációt.
1.7. MINŐSÉGI KRITÉRIUMOK
1.7.1. Visszanyerés (recovery)
Közvetlenül a vizsgálati anyag hozzáadása után minden egyes kiindulási vizsgálati koncentrációt 14C aktivitásméréssel, vagy nem jelölt anyag esetén kémiai analízissel ellenőrizni kell legalább ellenmintában. Ez információval szolgál az analitikai módszer alkalmazhatóságáról és ismételhetőségéről, valamint a vizsgálati anyag eloszlásának homogenitásáról. A későbbi adatkiértékelések során célszerű a kezdeti 14C aktivitás, vagy vizsgálatianyag-koncentráció adattal számolni a névleges koncentráció helyett, hiszen a szorpció miatti veszteségek és az adagolási hibák ezáltal kompenzálhatóak. 14C-jelölt vizsgálati anyag esetében a kísérlet végén történő visszanyerés értékét tömegkiegyenlítéssel adják meg (lásd 1.8.9.4. pont utolsó bekezdése). Ideális esetben a radioaktívan jelölt izotóp tömegkiegyenlítése 90 és 110 % közötti tartományban mozog, míg a nem jelölt vizsgálati anyag esetében a pontos analitikának 70–110 % közötti kezdeti visszanyerést kell eredményeznie. Ezeket a számokat mint elérendő céltartományokat, és nem mint a vizsgálat elfogadhatósági kritériumait kell kezelni. Az analízis megbízhatóságát választhatóan a kiindulásinál kisebb vizsgálatianyag-koncentrációra, vagy a fő transzformációs termékekre is meg lehet határozni.
1.7.2. Az analitikai módszer ismételhetősége és érzékenysége
A vizsgálati anyag és a transzformációs termékek mennyiségi meghatározásához az analitikai módszer ismételhetőségét (a kiindulási kivonás hatékonyságát is beleértve), amennyiben szükséges, a felszíni vízminta 5 független kivonatának analízisével kell ellenőrizni.
Az analitikai módszernek a vizsgálati anyagra és a transzformációs termékekre vonatkozó mennyiségi meghatározási határa (LOD) a tesztrendszerben alkalmazott kiindulási mennyiség legalább 1 %-a legyen. A mérhetőségi határnak (LOQ) az alkalmazott koncentráció 10 %-ával egyenlőnek, vagy annál kevesebbnek kell lennie. Számos szerves anyag és transzformációs termékeik kémiai analízise gyakran megkívánja a vizsgálati anyag viszonylag magas koncentrációjának (azaz > 100 μg/l) alkalmazását.
1.8. A VIZSGÁLATI MÓDSZER LEÍRÁSA
1.8.1. Eszközök
A vizsgálatok megfelelő térfogatú (pl. 0,5 vagy 1,0 literes), szilikon-, vagy gumidugóval lezárt kúp, vagy henger alakú lombikokban, illetve CO2-biztos tetővel (pl. butilgumi-szeptum) ellátott szérumlombikokban végezhetőek. A vizsgálat megvalósításának másik módja, ha nagyszámú lombikot használunk és minden mintavételi időpontban legalább kétszer feldolgozzuk a lombikok teljes tartalmát (lásd az 1.8.9.1. pont utolsó bekezdését). Radioaktívan nem jelölt, nem illékony vizsgálati anyagok esetében gázbiztos dugók vagy fedelek nem szükségesek, a kívülről történő beszennyeződéstől megóvó laza vattadugók megfelelőek (lásd az 1.8.9.1. pont második bekezdését). A kis mértékben illékony anyagok tesztelését biométer-típusú rendszerekben végezhetjük, a vízfázis óvatos kevertetése mellett. A felhasználásukat megelőzően hővel vagy autoklávozással a laboreszközök sterilizálhatóak, így a bakteriális beszennyeződésük kizárható. Ezek mellett az alábbi általános laboreszközök kerülnek felhasználásra:
|
— |
rázógép vagy mágneses keverő a tesztlombikok folyamatos kevertetéséhez, |
|
— |
centrifuga, |
|
— |
pH mérő, |
|
— |
turbiditásmérő a zavarosság mértékének meghatározására, |
|
— |
hevítőkemence vagy mikrohullámú sütő a száraz tömeg meghatározására, |
|
— |
membránszűrő apparátus, |
|
— |
autokláv vagy hőlégsterilizáló az üvegeszközök sterilizálásához, |
|
— |
14C-jelölt anyagokkal végzett munkához szükséges felszerelés, |
|
— |
műszer a 14C-activitás megkötött CO2-t tartalmazó oldatmintákból, vagy szükség esetén üledékmintákból történő mennyiségi meghatározásához, |
|
— |
specifikus kémiai analízis alkalmazása esetén a vizsgálati anyag (és a referenciaanyag) méréséhez szükséges analitikai berendezés (pl. gázkromatográf, nagynyomású folyadékkromatográf). |
1.8.2. A vizsgálati anyag törzsoldatai
A vizsgálat- és referenciaanyag törzsoldatainak elkészítéséhez ioncserélt vizet használunk (lásd az 1.8.7. pont első bekezdését). Az ioncserélt víznek a mikroorganizmusokra toxikus anyagoktól mentesnek kell lennie, és oldott szerves széntartalma (DOC) nem lehet több 1 mg/l-nél (6).
1.8.3. A felszíni vízminta begyűjtése és szállítása
A felszíni vízminta vételére szolgáló mintavételi helyet mindenkor a vizsgálat céljainak megfelelően kell kiválasztani. A mintavételi hely kiválasztásakor figyelembe kell venni a mezőgazdasági, az ipari és a háztartási eredetű korábbi hatásokat. Amennyiben ismeretes, hogy az elmúlt négy évben a vízbázis a vizsgálati anyaggal, vagy annak szerkezeti analógjaival szennyeződött, akkor tesztvíz vételére nem használható, hacsak a vizsgálatot végzőnek nem kifejezetten az a célja, hogy korábban szennyezett területen vizsgálja a lebontási sebességét. A víz pH-ját és hőmérsékletét a mintavételi helyszínen kell megmérni. Továbbá fel kell jegyezni a mintavétel mélységét és a vízminta állagát is (szín és zavarosság) (lásd 3. pont). Az aerob körülmények jelenlétének bizonyításához – hacsak ez már a területen végzett előző kísérletek és azok eredményei alapján, valamint a külső megjelenés alapján nyilvánvalóan el nem dönthető – a víz és az üledék felszíni rétege oxigén-koncentrációjának és/vagy redoxpotenciáljának mérése is szükséges. A felszíni vizet alaposan kitisztított edényben szállítsuk. A szállítás során a minta hőmérséklete jelentősen nem haladhatja meg a vizsgálat hőmérsékletét. Ha a szállítás 2–3 óránál hosszabb időt vesz igénybe, a mintát ajánlatos 4 °C-ra hűteni. A vízminta fagyasztása tilos.
1.8.4. A felszíni vízminta tárolása és előkészítése
A vizsgálatot célszerű a mintavételt követő egy napon belül elvégezni. Ha szükséges a minta tárolása, a tárolás időtartamát minimalizálni kell, és semmilyen körülmények között sem szabad 4 hétnél tovább tárolni. A felhasználásig a vízmintát levegőztetés mellett tartsuk 4 °C-on. A felhasználás előtt távolítsuk el a durva részecskéket (pl. kb. 100 μm pórusméretű nylon szűrőn vagy durva szűrőpapíron történő átszűréssel, vagy ülepítéssel).
1.8.5. Üledékkel kiegészített vízminta előkészítése (választható)
A lebegőüledékes iszap vizsgálathoz (a durva részecskéktől az 1.8.4. pontban leírtak szerint szűréssel megszabadított) természetes vízmintát tartalmazó lombikokba juttassunk felszíni üledéket, hogy szuszpenziót kapjunk. A lebegő szilárd anyag koncentrációjának 0,01 és 1 g/l közöttinek kell lennie. A felszíni üledéknek a vízminta kivételi helyéről kell származnia. A vízbázis adott környezetétől függően a felszíni üledék lehet nagy szervesszén-tartalmú (2,5–7,5 %) és finom szerkezetű, vagy kis szervesszén-tartalmú (0,5–2,5 %) és durva szerkezetű (3). A felszíni üledék előkészítésekor átlátszó műanyagcső segítségével emeljünk ki több üledékdarabot, a mintavétel után azonnal vágjuk le a felső aerob réteget (a felszíntől max. 5 mm-es mélységig), és egyesítsük ezeket. Az így kapott üledéket nagy légterű edényben kell szállítani, hogy az üledék aerob körülmények között maradjon (ha a szállítási idő 2–3 óránál több, hűtsük 4 °C-ra). Az üledékmintából 10-szeres mennyiségű vízzel készítsünk szuszpenziót, és a felhasználásig levegőztetés mellett 4 °C-on tartsuk. Ha szükséges az üledék tárolása, a tárolás időtartamát minimalizálni kell, és semmilyen körülmények között sem szabad 4 hétnél tovább tárolni.
1.8.6. Félfolyamatos eljárás (opcionális)
Ha hosszú várakozási időnek kell eltelnie azelőtt, hogy a vizsgálati anyag szignifikáns mértékű lebontását lehetne mérni, akkor hosszabb inkubációra (több hónap) lehet szükség. Ha ez az anyag korábbi vizsgálatai alapján ismert, akkor a vizsgálat félfolyamatos eljárással kezdhető, ami lehetővé teszi a tesztvíz vagy szuszpenzió egy részének időről időre történő kicserélését (lásd 2. melléklet). Másik megoldásként a szokásos egymenetes vizsgálat átalakítható félfolyamatos vizsgálattá, ha a vizsgálati anyagnál az egymenetes eljárással körülbelül 60 nap alatt sem tapasztalunk lebontást (lásd az 1.8.8.3. pont második bekezdése).
1.8.7. A vizsgálati anyag (vagy referenciaanyag) bejuttatása a rendszerbe
A vízben jól oldódó (> 1 mg/l) és kis illékonyságú (Henry-állandó < 1 Pa·m3/mól vagy < 10–5 atm·m3/mól) anyagokból ioncserélt vízzel (lásd 1.8.2. pont) törzsoldat készíthető. Ebből megfelelő mennyiséget a tesztedényekbe juttatva beállíthatjuk a kívánt koncentrációkat. A hozzáadott törzsoldat térfogatát a gyakorlatilag lehetséges minimum szinten kell tartani (ha lehetséges, a végső folyadéktérfogat 10 %-a alatt). Egy másik lehetséges módszer a vizsgálati anyag feloldása nagyobb térfogatú tesztvízben, ami a szerves oldószer használatának alternatívájaként tekinthető.
Ha ez elkerülhetetlen, a vízben rosszul oldódó nem illékony anyagok törzsoldatait illékony szerves oldószer felhasználásával is előállíthatjuk, de a tesztrendszerbe juttatott oldószer mennyisége nem lépheti túl az 1 tf %-ot, és nem lehet káros hatása a mikrobás aktivitásra, vagy a vizsgálati anyag stabilitására a vizes oldatban. Az oldószert annyira ki kell űzni az oldatból, hogy lehetőleg csak nagyon kis mennyiségben maradjon, ami már jelentős mértékben nem emeli a tesztvíz vagy szuszpenzió DOC koncentrációját. Ezt anyagspecifikus analízissel, vagy ha lehetséges DOC analízissel kell ellenőrizni (6). Figyelmet kell fordítani arra, hogy a bejuttatott oldószer mennyiségét az abszolút szükséges mennyiségre korlátozzuk, és biztosítani kell, hogy a vizsgálati anyag feloldódhasson a tesztvíz végleges térfogatában. A vizsgálati anyagnak a reakcióedényekbe történő bejuttatására szolgáló más módszereket a (7) és (8) referencia tartalmazza. Abban az esetben, ha a vizsgálati anyaghoz szerves oldószert adunk, tesztvizes (más adalék nélküli) oldószer-kontrollt és referencia szubsztrát-kontrollt is alkalmazni kell. Ez utóbbit hasonlóan kezeljük, mint a vivőoldószerben oldott vizsgálati anyaggal kiegészített aktív tesztedényeket. Az oldószer-kontrollok célja, hogy megvizsgáljuk a szolvens által okozott, a mikrobapopulációra gyakorolt lehetséges gátló hatást, amit a referenciaanyag lebontási kinetikája jelez előre.
1.8.8. Tesztkörülmények
1.8.8.1. Teszthőmérséklet
Az inkubációnak sötétben vagy szórt fényben, kontrollált hőmérsékleten (± 2 °C) kell történnie, amely lehet terepi hőmérséklet vagy 20–25 °C-os szobahőmérséklet. A terepi hőmérséklet a minta mintavételkor mért tényleges hőmérséklete, vagy a mintavételi hely átlagos terepi hőmérséklete.
1.8.8.2. Kevertetés
A kevertetés a folyamatos rázatás vagy keverés révén gondoskodik a részecskék és a mikrobák szuszpenzióban tartásáról. A kevertetés elősegíti a gáztérből az oxigén bejutását a folyadékfázisba, ezáltal az aerob körülmények megfelelő módon fenntarthatóak. Helyezzük a lombikokat a rázógépbe (kb. 100 rpm-es kevertetés) vagy használjunk mágneses keverőt. A kevertetésnek folyamatosnak kell lennie. Mindazonáltal a rázatásnak vagy a keverésnek a homogén szuszpenzió fenntartása mellett a lehető legenyhébbnek kell lennie.
1.8.8.3. A vizsgálat időtartama
A vizsgálat időtartama általános esetben nem lépi túl a 60 napot, hacsak a félfolyamatos eljárás szerint a vizsgálati szuszpenziót időről időre nem cseréljük (lásd a 1.8.6. pontot és a 2. mellékletet). Az egymenetes vizsgálati periódus is kiterjeszthető azonban maximum 90 napra, ha a vizsgálati anyag lebontása az első 60 napon belül megkezdődött. A lebontás rendszeres időközönkénti nyomon követése a visszamaradó 14C aktivitás, vagy a kibocsátott 14CO2 (lásd 1.8.9.4. pont) mérésével és/vagy a kémiai analízissel (lásd 1.8.9.5. pont) történik. A lebontási folyamat értékelhetőségéhez az inkubációs időnek megfelelően hosszúnak kell lennie. A lebontás mértékének lehetőleg 50 % felett kell lennie, lassan bomló anyagoknál a lebontás mértékének megfelelőnek (normális esetben 20 % feletti lebontás) kell lennie ahhoz, hogy biztosítsa a kinetikus lebontásos sebességi állandó becsülhetőségét.
A vizsgálati rendszerben a pH-t és az oxigénkoncentrációt rendszeresen mérni kell, hacsak az ugyanarról a területről származó víz- és üledékmintákkal végzett hasonló vizsgálatokból leszűrhető tapasztalatok ezt szükségtelenné nem teszik. Bizonyos körülmények között a vízben vagy üledékben sokkal nagyobb koncentrációban jelen lévő elsődleges szubsztrátok metabolizmusa elegendő CO2-termelődést és oxigénfogyást eredményez ahhoz, hogy a vizsgálat során a kísérleti körülmények jelentősen megváltozzanak.
1.8.9. Kísérleti eljárás
1.8.9.1. A lombikok előkészítése nyílt vízi vizsgálathoz
Juttassunk megfelelő mennyiségű tesztvizet a tesztlombikokba, a lombikokat térfogatuk kb. harmadáig töltve, de legalább kb. 100 ml tesztvizet töltve beléjük. Több lombik használata esetén (ami lehetővé teszi a lombik teljes tartalmának feldolgozását minden mintavételi időpontban) is kb. 100 ml a kívánt tesztvíz-mennyiség, mivel a kis mintatérfogatok hatással lehetnek a lappangási fázis hosszára. A vizsgálati anyag törzsoldatból történő bejuttatása az 1.8.2. és 1.8.7. pontokban leírtak szerint történik. Hogy a lebontási kinetika meghatározása és a kinetikus lebontásos sebességi állandó számíthatóvá váljon, a vizsgálati anyagot legalább két, egymástól ötös-tizes szorzófaktorral eltérő koncentrációban kell használni. Mindegyik választott koncentrációnak 100 µg/l-nél kisebbnek kell lennie, lehetőleg a < 1–10 µg/l tartományban.
Zárjuk le a lombikokat a levegő és CO2 számára áthatolhatatlan dugókkal vagy fedelekkel. 14C izotóppal nem jelölt nem-illékony vizsgálati anyagok esetében elegendő laza vattadugókat használni a légkörből érkező szennyezések elkerülésére (lásd 1.8.1. pont), feltéve hogy az összes fő transzformációs termék ismerten nem illékony, és indirekt CO2 meghatározást (lásd 3. melléklet) alkalmazunk.
Inkubáljuk a lombikokat a választott hőmérsékleten (lásd 1.8.8.1. pont). Különítsünk el mintákat a kémiai analízishez vagy a 14C méréshez a vizsgálat kezdetén (azaz mielőtt a biológiai lebontás megindulna; 1.7.1. pont), majd végig a vizsgálat folyamán megfelelő időközönként. A mintavétel történhet úgy, hogy minden párhuzamosból kis mennyiségű mintát (pl. 5 ml-es alikvótok) veszünk, vagy minden egyes mintavételi időpontban feldolgozhatjuk teljes lombikok tartalmát. A vizsgálati anyag mineralizációját meghatározhatjuk indirekt és direkt módon is (lásd 3. melléklet). A sebességi állandó megbízható becsléséhez általában legalább öt mintavételi pont szükséges a lebontási fázisban (azaz a lappangási fázist követően), hacsak nem igazolható, hogy három mérési pont elégséges a gyorsan bomló anyagok esetében. Lassan bomló anyagoknál a lebontásos fázisban több mérés is könnyen kivitelezhető, ennek következtében a k számításához több adatpontot használhatunk. Mivel a biológiai lebontás sebessége eltérő, egy fix mintavételi ütemterv nem határozható meg, bár ha a lebontás lassú heti egy mintavétel elvégzése ajánlatos. Ha a vizsgálati anyag lebontása gyors, az első három napban napi egy mintát, majd ezt követően két-háromnaponta egy mintát kell venni. Bizonyos körülmények között (például gyorsan hidrolizáló anyagok esetében) óránkénti mintavételre is szükség lehet. A vizsgálat megkezdése előtt ajánlott elővizsgálatok lefolytatása a mintavételi intervallumok meghatározására. Ha a mintákat további specifikus analízisnek is alá kell vetni, akkor tanácsos több mintát venni, majd az analizálandókat a kísérlet végén, a visszafelé mérés stratégiáját alkalmazva (azaz az utoljára levett mintákat analizáljuk először) kiválasztani (a minták tárolás közbeni stabilitására vonatkozó útmutatót lásd az 1.8.9.5. pont második bekezdésében).
1.8.9.2. A lombikok és minták száma
Állítsunk be megfelelő számú tesztlombikot, hogy rendelkezésre álljanak a követezők:
|
— |
minden egyes vizsgálati anyag-koncentrációhoz legalább két párhuzamos (lehetőleg legalább 3), vagy minden egyes koncentrációhoz számos tesztlombik, ha minden mintavételkor teljes lombikok tartalmának feldolgozása történik (FT-vel jelölve), |
|
— |
tesztlombikok az tömegkiegyenlítés-kalkulációhoz legalább két párhuzamosban minden egyes tesztkoncentrációhoz (FM-mel jelölve), |
|
— |
vizsgálati anyag nélküli vak kontroll; legalább egy, csak tesztvizet tartalmazó tesztlombik (FB-mel jelölve), |
|
— |
referenciakontroll; a referenciaanyagot (pl. anilin vagy nátrium-benzoát) tartalmazó tesztlombikok két párhuzamosban (FC-vel jelölve). A referenciakontroll célja a mikrobiális aktivitás minimumának ellenőrzése. Ha lehetséges, radioaktívan jelölt referenciaanyag is használható, még akkor is, ha a vizsgálati anyag lebontását kémiai analízissel követjük, |
|
— |
steril kontroll; egy-két sterilizált tesztvizet tartalmazó lombik az abiotikus lebontás, vagy a vizsgálati anyag más, nem biológiai eredetű elvesztésének vizsgálatához (FS-sel jelölve). A biológiai aktivitás leállítható a tesztvíz autoklávozásával (121 °C; 20 perc), mérgező anyag hozzáadásával (pl. 10–20 g/l nátrium-azid (NaN3), 100 mg/l higany-klorid (HgCl2), vagy 100 mg/l formalin), vagy gammasugárzással. HgCl2 használata esetén ezeket mint veszélyes hulladékot kell ártalmatlanítani. Nagy mennyiségben alkalmazott vizes üledékeknél a steril körülményeket nem könnyű kialakítani, ebben az esetben ismételt (pl. háromszori) autoklávozás javasolt. Figyelembe kell venni, hogy az autoklávozás hatására az üledék szorpciós tulajdonságai megváltozhatnak, |
|
— |
tesztvizet, referenciaanyagot és tesztvizet tartalmazó oldószer kontrollok; azonos mennyiségű oldószerrel és a vizsgálati anyag alkalmazásával megegyező módon előállított lombikok két párhuzamosban. Ennek célja az oldószer esetleges gátló hatásának megállapítása a referenciaanyag lebontásának meghatározásán keresztül. |
A vizsgálat megtervezése során a vizsgálatot kivitelező személynek fontolóra kell vennie, hogy számára inkább a nagyobb számú kísérleti ismétlés, vagy ezzel szemben a nagyobb számú mintavétel a fontosabb. A szükséges lombikok pontos száma a lebontás mérésére szolgáló módszertől függ (lásd az 1.8.9.1. pont harmadik bekezdését; az 1.8.9.4. pontot és a 3. mellékletet).
Minden mintavételi időpontban minden lombikból két mintát (pl. 5ml-es alikvótokat) kell venni. Ha számos lombik használatos, hogy teljes lombikok tartalmát fel lehessen dolgozni, akkor minden mintavételi időpontban legalább két lombikot kell feláldozni (lásd az 1.8.9.1. pont első bekezdését).
1.8.9.3. Lombikok előkészítése lebegő üledék vizsgálathoz [választható]
Ha szükséges, juttassunk megfelelő mennyiségű tesztvizet és üledéket a tesztedényekbe (lásd 1.8.5. pont). A lombikoknak a lebegő üledék vizsgálathoz történő előkészítése megegyezik a nyílt vízi vizsgálatnál leírtakkal (lásd 1.8.9.1. és 1.8.9.2. pontok). Használjunk inkább szérumpalackokat vagy hasonló alakú lombikokat. Helyezzük a lezárt lombikokat vízszintesen a rázógépbe. A nem 14C-jelölt, nem-illékony anyagokat tartalmazó nyitott lombikokat nyilvánvalóan függőlegesen kell elhelyezni, ebben az esetben mágneses keverés, vagy üveggel bevont mágnesrudak használata ajánlott. Ha szükséges, a megfelelő aerob körülmények eléréséhez levegőztessük a reakcióedényeket.
1.8.9.4. Radiokémai mérések
A képződő 14CO2 mérhető indirekt, vagy direkt módon (lásd 3. melléklet). Az indirekt 14CO2 meghatározás a tesztvíz vagy szuszpenzió kezdeti 14CO2 aktivitásának, és a mintavétel alkalmával a minta pH 2–3-ra történő lesavanyítása és a CO2 kiűzése után mért teljes maradó aktivitásnak a különbsége alapján történik. Így a szervetlen széntartalom is távozik, és a maradó aktivitás a szerves anyagtól származik. Ha a vizsgálati anyag átalakulása során illékony fő transzformációs termék képződik, az indirekt 14CO2 meghatározás nem használható (lásd 3. melléklet). Amennyiben lehetséges, a 14CO2 képződését minden egyes mintavételi pontban legalább egy lombikban direkt módon kell mérni (lásd 3. melléklet). Ez a módszer lehetővé teszi mind az tömegkiegyenlítés, mind a biológiai lebontási folyamat nyomon követését, de használata korlátozott a zárt lombikos vizsgálatok esetében.
Ha a vizsgálat során a termelődő 14CO2-t direkt módon mérjük, erre a célra a vizsgálat kezdetekor több lombikot kell beállítani. Ha a vizsgálati anyagból illékony fő transzformációs termék képződik, ez a módszer az ajánlott. A többi tesztlombik tartalmát minden mérési pontban pH 2–3-ra savanyítjuk, és a 14CO2-t belső vagy külső abszorbenssel kötjük meg (lásd 3. melléklet).
A 14C-jelölt vizsgálati anyag és a fő transzformációs termék koncentrációját esetleg radiokromatográfiásan (pl. vékonyréteg-kromatográfia, RAD-TLC), vagy radiokémiai detekcióval, HPLC (nagynyomású folyadékkromatográfia) segítségével is meghatározhatjuk.
Opcionálisan a maradó radioaktivitás (lásd 1. melléklet), a vizsgálati anyag és a transzformációs termék fázismegoszlását is meghatározhatjuk.
A vizsgálat végén a tömegkiegyenlítést direkt 14CO2 méréssel kell megállapítani, olyan független tesztlombikokból, amelyekből a vizsgálat folyamán nem vettünk mintákat (lásd 3. melléklet).
1.8.9.5. Specifikus kémiai analízis
Ha lehetőség van érzékeny, specifikus analitikai módszer használatára, az elsődleges biológiai lebontás mértéke az izotópos jelölés helyett a vizsgálati anyag teljes maradó koncentrációjának mérésével állapítható meg. Amennyiben jelölt vizsgálati anyagot használunk (a teljes mineralizáció mérésére), a specifikus kémiai analízis ezzel párhuzamosan elvégezhető, hasznos többlet-információval szolgál, és ellenőrizhetjük vele a módszert. A specifikus kémiai analízis a vizsgálati anyag lebontása során keletkező transzformációs termék mérésére is használható, és ez ajánlott is olyan anyagok esetében, amelyek mineralizációs felezési ideje 60 napnál hosszabb. A vizsgálati anyag és a transzformációs termék koncentrációját minden mintavételi pontban mérni kell és ki kell értékelni (koncentrációban vagy százalékos formában). Általánosságban az alkalmazott koncentráció 10 %-a ≤, bármely mintavételi időpontban kimutatható transzformációs termékeket azonosítani kell, hacsak nincs más értelmű ésszerű indoklás. A kísérlet során folyamatosan emelkedő koncentrációban jelen lévő transzformációs termékek azonosítását is meg kell fontolni, még akkor is, ha ezek nem érik el a fenti határt, mivel ez a lebontással szembeni ellenállóképességet jelezhet. Ha a vizsgálati anyag gyors abiotikus lebontása lehetségesnek tűnik (pl. hidrolízis), akkor fontolóra kell venni a transzformációs termékek analízisét a steril kontrollokban. A transzformációs termékek mennyiségi és minőségi meghatározását az adott esetben kell eldönteni, és az indoklásokkal együtt a kiértékelésben közzétenni. A szerves oldószerrel végzett extrakciós technikákat a megfelelő analitikai módszer előírásai alapján kell elvégezni.
Minden mintát 2 és 4 °C között, légmentesen kell tárolni, ha az analízis 24 órán belül megtörténik (ajánlott). Hosszabb tárolásnál a mintákat –18 °C alá kell fagyasztani, vagy kémiai módszerrel tartósítani kell. Utóbbi módszerhez a savanyítás nem javasolt, hiszen ezek a minták instabilak lehetnek. Ha a mintákat 24 órán belül nem analizálják és hosszabb tárolásra kerülnek, akkor egy tárolási stabilitás-vizsgálatot kell kapcsolni a kísérlethez, hogy a vizsgált vegyületek kémiai stabilitása –18 °C alatt vagy tartósított állapotban demonstrálható legyen. Oldószeres extrakciós vagy szilárd fázisú extrakciós (SPE) lépést is magában foglaló analitikai módszereknél az extrakciót azonnal a mintavétel után, vagy a fagyasztott minta maximum 24 óráig tartó tárolása után el kell végezni.
Az analitikai módszer érzékenységének függvényében a pont 16. sorában jelzetteknél nagyobb mintatérfogatok lehetnek szükségesek. A vizsgálat könnyedén kivitelezhető 2–3 literes lombikokban egy literes teszttérfogatokkal, amely lehetővé teszi kb. 100 ml minta levételét.
2. ADATOK ÉS DOKUMENTÁLÁSUK
2.1. AZ EREDMÉNYEK KEZELÉSE
2.1.1. Az adatok ábrázolása
Kerekítsük a mintavételi időpontokat egész órákra (hacsak az anyag lényeges mértékű lebontása nem percek kérdése), de ne egész napokra. Ábrázoljuk a vizsgálati anyag becsült maradó aktivitását (14C-jelölt anyagok esetében), vagy maradó koncentrációját (nem jelölt anyagok esetében) az idő függvényében mind lineáris, mind fél-logaritmikus skálán (lásd 1a., 1b. ábra). Ha lebontás játszódik le, hasonlítsuk össze az FT lombikok eredményeit az FS lombikokéval. Ha a vizsgálati anyagos lombikok (FT) eredményeinek átlaga kevesebb mint 10 %-kal tér el a steril lombikokétól (FS), akkor megállapítható, hogy a tapasztalt lebontás alapvetően abiotikus. Amennyiben az FS lombikokban a lebontás kisebb mértékű, az értékekkel korrigáljuk az FT lombikoknál kapott értékeket (kivonással), hogy becsülni tudjuk a biológiai lebontás mértékét. Ha a fő transzformációs termékek választható analízisét elvégeztük, akkor ezek keletkezését és fogyását is ábrázolni kell a vizsgálati anyag fogyása mellett.
Határozzuk meg a lappangási fázis tL hosszát a lebontási görbéből (fél-logaritmikus ábrázolás) úgy, hogy extrapoláljuk ennek lineáris részét a zéró lebontásig, vagy úgy, hogy meghatározzuk a 10 %-os lebontáshoz szükséges időt (lásd 1a., 1b. ábra). A fél-logaritmikus ábrából becsüljük meg az elsőrendű sebességi állandót (k) és standard hibáját az idő függvényében felvett ln (maradó 14C aktivitás vagy vizsgálati anyag-koncentráció) lineáris regressziójából. Főleg a 14C méréseknél csak a görbének lappangási fázis vége utáni kezdeti lineáris szakaszát használjuk, és a kiválasztásnál a kisszámú, reprezentatív adatokat részesítsük előnyben a nagyobb számú, de bizonytalan adatokkal szemben. A bizonytalanság tartalmazza a mért maradó 14C aktivitások javasolt direkt felhasználásából eredő hibákat (lásd lent). Ha a lebontás kétfázisú lefutást követ, néha fontos lehet két különböző sebességi állandó számítása. Ebből a célból a lebontási görbe két különböző fázisa definiálható. Az ugyanabból a lombikból vett minták esetében a párhuzamosokban minden egyes lombikra számítani kell a sebességi állandót (k) és a felezési időt (t½ = ln2/k); vagy ha a lombik teljes tartalmát analizáljuk (lásd az 1.8.9.2. pont utolsó bekezdése), akkor az eredmények átlagából. Amikor az első módszert használjuk, a sebességi állandót és a felezési időt megadjuk a párhuzamosok minden egyes lombikjára, és ezek átlagát is a standard hibával. Magas koncentrációjú vizsgálati anyag esetében a lebontási görbe jelentősen eltérhet az egyenestől (fél-logaritmikus ábrázolás) és az elsőrendű kinetika érvénytelen lehet. Ennek következtében a felezési idő megadásának nincs értelme. Egy limitált adattartományra azonban használható pszeudo-elsőrendű kinetika, és a lebontási felezési idő DT50 (az 50 %-os lebontáshoz szükséges idő) becsülhető. Ugyanakkor fontos szem előtt tartani, hogy a lebontás időtartama a kiválasztott adattartományon túl a DT50 használatával nem határozható meg, mivel ez az érték csupán ennek az adatsornak a sajátja. A statisztikai számításokat és a görbeillesztést könnyítő segédeszközök könnyen hozzáférhetőek, és az ilyen típusú szoftverek alkalmazása ajánlott is.
Specifikus kémiai analízis elvégzése esetén határozzuk meg a lebontási állandókat és felezési időket az elsődleges lebontásra, a teljes mineralizációra leírtak szerint (lásd fent). Ha az elsődleges lebontás a meghatározó folyamat, néha a teljes lebontási időtartam pontjai felhasználhatóak. Ennek oka az, hogy szemben a 14C aktivitásméréssel, ezek a mérések direktek.
Ha 14C-jelölt anyagot használunk, a tömegkiegyenlítést a bemért kezdeti koncentráció százalékában kell kifejezni, legalább a vizsgálat végeztével.
2.1.2. Maradó aktivitás
Amikor a 14C-jelzett szerves anyag lebontódik, a 14C nagy része 14CO2-dá alakul, míg a másik része a biomassza növekedésére és/vagy extracelluláris metabolitok szintézisére használódik fel. Ennek következtében az anyag biológiai lebontásának maximális szintje nem eredményezi széntartalmának 100 %-os átalakulását 14CO2-dá. A 14C bioszintézis-termékekbe épül be, majd ezt követően, a másodlagos mineralizációnak köszönhetően lassan, 14CO2 formában felszabadul. Ezen okok miatt a maradó 14C aktivitás (a CO2 kiűzése után) vagy az idő függvényében felvett termelődő 14CO2 ábrázolásakor a lebontás befejeződése után egy „lecsengési fázist” fogunk látni. Ez bonyolultabbá teszi az adatok kinetikai interpretációját, ezért csupán a görbe kezdeti szakaszát (a lappangási fázis befejeződése után és a kb. 50 %-os lebontási szint elérése előtt) használjuk a lebontási sebességi állandó számításához. Ha a vizsgálati anyag lebomlik, a teljes maradó szerves 14C aktivitás mindig nagyobb, mint a maradó intakt vizsgálati anyaghoz kapcsolható 14C aktivitás. Ha a vizsgálati anyag elsőrendű reakció szerint bomlik, és egy állandó α mennyisége CO2-dá mineralizálódik, a 14C csökkenés görbéjének (teljes szerves 14C az idő függvényében) kezdeti meredeksége α-szorosa lesz a vizsgálati anyag koncentrációjának (vagy egészen pontosan a vizsgálati anyag 14C-gyel jelölt részének) megfelelő görbe meredekségének. A teljes szerves 14C aktivitás korrigálatlan mért értékeit használva a számított lebontási sebességi állandó ennek következtében csak óvatos becslés lesz. Különféle egyszerűsítő feltételeken alapuló, a vizsgálati anyag koncentrációját a mért radiokémiai aktivitásokból becslő módszer a szakirodalomból ismert (2)(9)(10)(11). Az ilyen eljárások a legkönnyebben a gyorsan bomló anyagokra alkalmazhatóak.
2.2. AZ EREDMÉNYEK INTERPRETÁCIÓJA
Amennyiben k függetlennek bizonyul az alkalmazott koncentrációtól (azaz a k számított értéke közel azonos a különböző vizsgálati anyag-koncentrációk esetén) megállapítható, hogy az elsőrendű sebességi állandó reprezentatív az adott körülményekre, azaz a vizsgálati anyagra, a vízmintára és a teszthőmérsékletre. Hogy az eredmények milyen mértékben általánosíthatóak és vetíthetőek ki más rendszerekre, csak szakértői vélemények alapján határozható meg. Amennyiben a vizsgálati anyagot magas koncentrációban alkalmazzuk, és így a lebontás nem elsőrendű kinetikát követ, az adatok nem használhatóak fel elsőrendű lebontási sebességi állandó és a megfelelő felezési idő megadásához. Az ilyen adatok azonban még mindig használhatóak a teljes mineralizáció mértékének becslésére és/vagy a transzformációs termékek kimutatására és mennyiségi meghatározására.
Amennyiben a biológiai lebontáson kívüli egyéb, a vizsgálati anyag mennyiségének csökkenésével járó folyamatok (hidrolízis vagy volatilizáció) sebessége ismert, ezek értékét a teljes koncentráció csökkenési sebességéből a vizsgálat során kivonva egy közelítő biológiai lebontási sebességet kaphatunk. A hidrolízis-adatokhoz a steril kontrollból vagy nagyobb koncentrációjú párhuzamos vizsgálati anyag-kísérletből juthatunk.
A 14CO2 indirekt és direkt meghatározása (lásd 1.8.9.4. pont és 3. melléklet) csupán a vizsgálati anyag CO2-dá történő mineralizációja mértékének mérésére használható. A 14C-jelölt vizsgálati anyag koncentrációját és a fő transzformációs termék (lásd 1.8.9.4. pont harmadik bekezdését) keletkezését radiokromatográfiával (RAD-TLC) vagy HPLC-vel analizálhatjuk. A felezési idő direkt meghatározásához szükséges, hogy ne legyen jelen fő transzformációs termék (definíció szerint az alkalmazott vizsgálati anyag 10 %-a ≤). Ha a definíció szerint ilyen anyag jelen van, az adatok részletes kiértékelésére van szükség. Ez jelenthet megismételt vizsgálatot, és/vagy a transzformációs termék azonosítását (lásd 1.8.9.5. pont első bekezdése), hacsak a termék sorsa a gyakorlatban egyértelműen meg nem állapítható (pl. a lebontási útvonal ismerete). Mivel a vizsgálati anyag CO2-dá alakult hányada változó (erősen függ a vizsgálati anyag és más hozzáférhető szubsztrátok koncentrációjától, a tesztkörülményektől és a mikrobiális populációtól) ez a vizsgálat nem alkalmas a teljes biológiai lebontás közvetlen számítására, amit pl. a DOC fogyási vizsgálatban láthatunk, de az eredmények hasonlóak a respirometriás vizsgálatból kaphatóakkal. A mineralizáció mértéke így a teljes biológiai lebontás minimális szintjénél kisebb, vagy azzal egyenlő lesz. Hogy még teljesebb képet kapjunk a teljes biológiai lebontásról (mineralizáció és biomasszába történő beépülés), a 14C fázismegoszlásának analízisét a vizsgálat végén el kell végezni (lásd 1. melléklet). A 14C a diszpergált fázisban a bakteriális biomasszába beépült 14C-ből és a szerves részecskékhez tapadt 14C-ből fog állni.
2.3. A VIZSGÁLAT VALIDITÁSA
Amennyiben a referenciaanyag nem bomlik le a várt időtartam alatt (anilinre és nátrium-benzoátra általában kevesebb mint két hét), a vizsgálat validitása kétségbe vonható és további ellenőrzése szükséges, vagy a vizsgálatot új vízmintával meg kell ismételni. A módszer egy ISO-körméréses tesztsorozatában, amelyben Európa-szerte hét laboratórium vett részt, az anilinre alkalmas lebontási sebességi állandó a 0,3–1,7 nap–1-es tartományban mozgott, 20 oC-on 0,8 nap–1-es átlaggal és ±0,4 nap–1 standard hibával (t½ = 0,9 nap). A vizsgált vízmintákra vonatkozóan milliliterenként 103–104 CFU-nak (telepképző egység) megfelelő bakteriális biomassza-tartalmat írtak le. A lebontási sebességek az ásványi anyagban dúsabb közép-európai vizekben nagyobbak voltak, mint a skandináv oligotróf vizekben, amit a különböző ásványianyag-ellátottság, vagy a kémiai anyagokkal való előzetes kitettség okozhat.
A kísérlet végén a teljes visszanyerésnek (anyagmérleg) 90 és 110 % között kell lennie radioaktívan jelölt anyagoknál, míg nem jelölt anyagok esetében a kezdeti visszanyerésnek (recovery) a kísérlet kezdetén 70 és 110 % között kell lennie. Az említett tartományok azonban csak megcélzandó értékek, és nem használhatók a vizsgálat elfogadhatósági kritériumaiként.
3. VIZSGÁLATI JEGYZŐKÖNYV
A vizsgálat típusának (azaz nyílt vízi vagy lebegő üledék vizsgálat) egyértelműen meg kell jelennie a vizsgálat jegyzőkönyvében, amelynek az alábbi információt is tartalmaznia kell:
Vizsgálati anyag és referenciaanyag(ok):
|
— |
közkeletű nevek, kémiai elnevezések (ajánlottak a IUPAC és/vagy CAS elnevezések), CAS számok, szerkezeti képletek (ha az alkalmazott anyag radioaktívan jelölt a 14C pozíciójának megjelölésével) és a vizsgálati anyag és referenciaanyag fontos fizikokémiai tulajdonságai (lásd 1.5. és 1.6. pont), |
|
— |
a transzformációs termék azonosításához és mennyiségi meghatározásához használt standardok kémiai elnevezései, CAS számai, szerkezeti képletei (ha az alkalmazott anyag radioaktívan jelölt, a 14C pozíciójának megjelölésével), fontos fizikokémiai tulajdonságai, |
|
— |
a vizsgálati anyag és referenciaanyag tisztasága (szennyezései), |
|
— |
a jelölt vegyület radiokémiai tisztasága és specifikus aktivitása (ahol szükséges). |
Felszíni víz:
A levett vízmintáról az alábbi minimális információkat közre kell adni:
|
— |
a mintavételi hely fekvése és leírása, ha lehetséges, szennyezettségi előtörténete, |
|
— |
a mintavétel időpontja, |
|
— |
ásványi anyagok (totál N, ammónium, nitrit, nitrát, totál P, oldott ortofoszfát), |
|
— |
mintavételi mélység, |
|
— |
a minta állaga (pl. szín és zavarosság), |
|
— |
DOC és TOC, |
|
— |
BOD, |
|
— |
hőmérséklet és pH a mintavétel helyén és idején, |
|
— |
oxigén vagy redoxpotenciál (csak abban az esetben kötelező, ha az aerob körülmények megléte nem nyilvánvaló), |
|
— |
ionerősség vagy konduktivitás (tengervíz és brakk víz esetében), |
|
— |
lebegő szilárd anyagok (zavaros minta esetén), |
|
— |
egyéb, esetleg fontos információk a mintavétel időpontjában a mintavétel helyszínéről (pl. a folyók, vagy tengeráramlatok aktuális vagy múltbéli adatai, közeli fő kiömlések és ezek típusa, időjárási körülmények a mintavétel időpontját megelőzően), |
és választhatóan:
|
— |
mikrobiális biomassza (pl. akridinnarancs direkt sejtszám vagy telepképző egység), |
|
— |
szervetlen szén, |
|
— |
klorofill-a koncentráció, mint az alga- biomassza speciális mérése. |
Ezen túlmenően lebegő üledék vizsgálat végzése esetén az alábbi információkat kell rendelkezésre bocsátani az üledékről:
|
— |
az üledék mintavételi mélysége, |
|
— |
az üledék állaga (úgymint színes, saras, iszapos, vagy homokos), |
|
— |
textúra (pl. durva homok, finom homok, iszap és agyag %-ban), |
|
— |
a lebegő szilárd anyag száraztömege g/l-ben, TOC koncentráció vagy égetés során fellépő tömegcsökkenés mint a szervesanyag-tartalom mérőszáma, |
|
— |
pH, |
|
— |
oxigén vagy redoxpotenciál (csak abban az esetben kötelező, ha az aerob körülmények megléte nem nyilvánvaló). |
Tesztkörülmények:
|
— |
a mintavétel és a laboratóriumi felhasználás között eltelt idő, mintatárolás és a minta előkezelése, a vizsgálatok kivitelezésének időpontja, |
|
— |
az alkalmazott vizsgálati anyag mennyisége, tesztkoncentráció és referenciaanyag, |
|
— |
a vizsgálati anyag alkalmazásának módszere, oldószer használata, |
|
— |
a használt felszíni víz és üledék (ha használjuk) térfogata, és az egyes időközönként analízisre levett minták térfogata, |
|
— |
az alkalmazott tesztrendszer leírása, |
ha a kivitelezés nem sötétben folyik, a szórt fényre vonatkozó információ;
|
— |
információ a steril kontrollok beállításának módszereiről (pl. hőmérséklet, az autoklávozások ideje és száma), |
|
— |
inkubációs hőmérséklet, |
|
— |
információ az analitikai technikákról, a radiokémiai mérési módszerekről, a tömegkiegyenlítés ellenőrzésének és a fázismegoszlás mérésének (amennyiben használjuk) módszeréről, |
|
— |
a párhuzamosok száma. |
Eredmények:
|
— |
a visszanyerés százaléka (lásd 1.7.1. pont), |
|
— |
az alkalmazott analitikai módszer ismételhetősége és érzékenysége, beleértve a mennyiségi meghatározási határt (LOD) és a mérhetőségi határt (LOQ) (lásd 1.7.2. pont), |
|
— |
az összes mért adat (beleértve a mintavételi időpontokat) és számított érték táblázatos formában, a lebontási görbe minden egyes tesztkoncentrációra minden párhuzamosban, a logaritmikus ábrázolás meredekségének korrelációs koefficiense, a becsült lappangási fázis és az elsőrendű vagy pszeudo-elsőrendű sebességi állandó (ha lehet), és a megfelelő lebontási felezési idő (vagy féléletidő, t50), |
|
— |
fontos értékek, mint az egyedi mintákból kapott eredmények átlagai, pl. a lappangási fázis hossza, lebontási sebességi állandó, lebontási felezési idő (vagy t50), |
|
— |
a rendszer nem adaptálódottként, vagy adaptálódottként történő kategorizálása a lebontási görbe lefutásából, vagy a tesztkoncentráció esetleges hatásából, |
|
— |
a végső anyagmérleg-ellenőrzés eredményei és a fázismegoszlás-mérések eredményei (ha vannak), |
|
— |
a mineralizálódott 14C aránya és specifikus kémiai analízis használata esetén az elsődleges biológiai lebontás végső szintje, |
|
— |
az alkalmazott és fő transzformációs termékek azonosítása, moláris koncentrációja és százalékos aránya (lásd az 1.8.9.5. pont első bekezdése), ahol szükséges, |
|
— |
a transzformáció feltételezett lebontási útja, ahol szükséges, |
|
— |
az eredmények összegzése. |
4. IRODALOM
|
1. |
OECD TG 309 (2004) Aerobic Mineralisation in surface water – Simulation Biodegradation Test |
|
2. |
ISO/DIS 14592–1 (1999) Water quality – Evaluation of the aerobic biodegradability of organic compounds at low concentrations – Part 1: Shake flask batch test with surface water or surface water/sediment suspensions. |
|
3. |
Testing Method C.23. Aerobic and anaerobic transformation in soil. |
|
4. |
Testing Method C.24. Aerobic and anaerobic transformation in aquatic sediments. |
|
5. |
OECD (1993). Guidelines for the Testing of Chemicals. OECD, Paris. |
|
6. |
ISO 8245 (1999). Water quality – Guidelines on the determination of total organic carbon (TOC) and dissolved organic carbon (DOC). |
|
7. |
ISO 10634 (1995). Water quality – Guidance for the preparation and treatment of poorly water-soluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium. |
|
8. |
OECD draft (2000). Guidance Document on aquatic toxicity testing of difficult substances and mixtures. Environmental Health and Safety Publications. Series on Testing and Assessment. No 22 (to be published summer 2000). |
|
9. |
Simkins, S. and Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol.47, 394–401. |
|
10. |
Ingerslev, F. and N. Nyholm. (2000). Shake-flask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, 274–283. |
|
11. |
ISO/CD 14592–1 (1999). Ring test report: Water Quality – Evaluation of the aerobic biodegradability of organic compounds at low concentrations part 1 – report of 1998/1999 ring-test. Shake flask batch test with surface water or surface water/sediment suspensions. |
1. melléklet
A 14C Fázismegoszlása
Az eljárás ellenőrzéséhez a maradó teljes szerves 14C aktivitás (TOA) rutinvizsgálatait ki kell egészíteni a tömegkiegyenlítés-mérésekkel, amelyek magukban foglalják a képződő, abszorbenssel megkötött 14CO2 direkt meghatározását is (lásd 3. melléklet). A pozitív 14CO2 termelődés önmagában közvetlen bizonyíték a biológiai lebontásra, szemben az abiotikus lebontással, vagy más anyagvesztési mechanizmussal, mint a volatilizáció és a szorpció. A biológiai lebontási viselkedést jellemző hasznos többletinformáció nyerhető a részecskék membránszűréssel vagy centrifugálással történő eltávolítását követően, az oldat fázis (oldott szerves 14C aktivitás, DOA) és a szuszpenzió fázis (szuszpendált szerves 14C aktivitás, POA) közötti TOA megoszlásának méréséből. A POA a mikrobiális biomasszára és más részecskékre kitapadt vizsgálati anyagból, és e mellett a vizsgálati anyag széntartalmának azon részéből áll, amely a szaporodó sejtek anyagának szintézisére fordítódik, és így beépül a szuszpendált biomassza frakcióba. Az oldott 14C szerves anyag képződése DOA-ként mérhető a biológiai lebontás végén (a degradáció-idő görbe telítési szakasza).
Számítsuk ki a maradó 14C fázismegoszlását a kiválasztott, 0,22–0,45 μm-es szűrőkön átszűrt mintákban. A szűrő anyaga nem adszorbeálhat jelentősebb mennyiségű vizsgálati anyagot (a polikarbonát filterek megfelelőek lehetnek). Ha a vizsgálati anyag szorpciója a filteren túl nagy mértékű (a kísérletet megelőzően tesztelni kell), használatát mellőzzük, helyette nagysebességű centrifugálás (2 000 g; 10 perc) használható.
A szűrlettel vagy a centrifugált mintával járjunk el a 3. melléklet szűretlen mintákra leírt módszere szerint. Oldjuk fel a membránfiltert a megfelelő szcintillációs folyadékban, és mérjük a beütésszámot a szokott módon, a kioltás korrigálására normálisan a külső standard-arány módszert, vagy mintaoxidálást használva. Ha centrifugálást használtunk, reszuszpendáljuk a részecskéket tartalmazó fázist 1–2 ml desztillált vízben, és juttassuk szcintillációs üvegcsébe. Ezt követően mossuk még kétszer, 1–1 ml desztillált vízzel, és a mosóvizet is juttassuk az üvegcsébe. Ha szükséges, a folyadékszcintillációs méréshez a szuszpenzió gélbe is ágyazható.
2. melléklet
Félfolyamatos eljárás
A nehezen bomló anyagok megfelelő lebontásának eléréséhez meghosszabbított, több hónapos inkubáció is szükséges lehet. A tesztidőszak hossza normál esetben nem haladhatja meg a 60 napot, hacsak az eredeti vízminta jellemzőinek fenntartása nem a tesztszuszpenzió lecserélésével történik. Mindamellett a tesztperiódust ki lehet terjeszteni maximum 90 napra a tesztszuszpenzió lecserélése nélkül is, ha a vizsgálati anyag lebontása az első 60 napon belül megkezdődött.
Hosszú idejű inkubáció során a mikrobiális populáció diverzitása különböző anyagvesztési mechanizmusok, valamint a vízminta alapvető ásványisó-tartalmának és elsődleges széntartalmú szubsztrátjainak kimerülése miatt lecsökkenhet. A lassan bomló anyagok esetében a lebontási sebesség pontos meghatározásához ezért a félfolyamatos vizsgálat alkalmazása ajánlott. Ha az előzetes tapasztalatok alapján a vizsgálati anyag 20 %-os lebontásának eléréséhez egy három hónapos inkubáció várható, a vizsgálatot a félfolyamatos eljárással kell elősegíteni. A másik megoldás az lehet, hogy ha a vizsgálati anyag lebontása egymenetes körülmények között körülbelül 60 nap alatt nem történik meg, akkor a normál egymenetes vizsgálatot a félfolyamatos vizsgálatra váltjuk. A félfolyamatos eljárás is leállítható és egymenetes kísérletként folytatható, ha jellemző lebontás történik (pl. > 20 %).
A félfolyamatos vizsgálatban a vizsgálati szuszpenzió térfogatának körülbelül harmadát kéthetente a kiindulási koncentrációra beállított, vizsgálati anyaggal ellátott, frissen vett vízzel cseréljük le. Ha a választható lebegő üledék vizsgálatot hajtjuk végre, akkor üledéket adunk ugyanígy cserevízhez, a kezdeti koncentrációt (0,01 és 1 g/l között) hozva létre. A lebegő üledék vizsgálat kivitelezése során fontos, hogy a rendszert teljesen felszuszpendáljuk a vízcseréhez is, valamint hogy a tartózkodási időnek azonosnak kell lennie a szilárd anyagokra és a vízre, mivel egyébként a kívánt hasonlóságot a homogén vizes rendszerekkel elveszthetjük. Ezen okok miatt a félfolyamatos eljárás alkalmazása során a lebegő üledék kezdeti koncentrációjának a megadott intervallum alsó tartományába kell esnie.
A vizsgálati anyag előírt hozzáadásába beleértendő, hogy a vizsgálati anyag kezdeti koncentrációját a tesztszuszpenzió részleges megújításával nem lépjük túl, ennélfogva az adaptáció jelensége, ami a vizsgálati anyag magas koncentrációinál gyakran fellép, kiküszöbölődik. Mivel az eljárás mind re-inokulációval, mind a kimerülő ásványi anyagok és elsődleges szubsztrátok pótlásával jár, a vizsgálat hossza alapjában véve korlátlanul megnyújtható. Fontos megjegyezni, hogy félfolyamatos eljárás alkalmazása esetén a vizsgálati anyag maradó koncentrációját korrigálni kell az egyes cserék során hozzáadott és eltávolított vizsgálati anyag-mennyiséggel. A teljes és az oldott vizsgálati anyag-koncentráció a csekély mértékben adszorbeálódó vegyületek esetében felcserélhető egymással. A szorpció az adott körülmények között (0,1–1 g szilárd anyag/l) a log Kow < 3 értékű (neutrális, lipofil vegyületekre érvényes) anyagokra vonatkozóan elhanyagolható (< 5 %). Ezt a következő számítási példán keresztül mutatjuk be. 0,1 g/l szilárd anyag durván 10 mg szénnek felel meg literenként (szénfrakció, fC = 0,01). Ha feltételezzük, hogy:
Log Kow (vizsgálati anyag) = 3
Koc = 0,42 × Kow
Megoszlási koefficiens, Kd = fC × Koc
akkor a teljes koncentráció oldott frakciója (C-víz (Cw)/C-teljes (Ct):
Cw/Ct = 1/(1 + Kd × SS) = 1(1 + Koc × fC × SS) = 1/(1 + 0,42 × 103 × 0,01 × 0,1 × 10–3) = 0,999
3. melléklet
A 14CO2 meghatározása
Indirekt 14CO2 meghatározás
Rutinmérésekhez az indirekt módszer általában a legkevésbé időigényes és legpontosabb módszer, ha a vizsgálati anyag nem illékony és nem alakul át illékony transzformációs termékekké. Egyszerűen juttassuk a szűretlen mintákat, pl. 5 ml térfogatú szcintillációs üvegcsékbe. Kezdetben a megfelelő aktivitás a mintákban 5 000 dpm–10 000 dpm (80–170 Bq), és a minimum kezdeti aktivitás kb. 1 000 dpm. Az egy-két csepp tömény H3PO4-val vagy HCl-val pH 2–3-ra történő savanyítás után a CO2-t ki kell űzni a rendszerből. Ez kb. fél-egy órán át tartó levegő-átbuborékoltatással történhet. Más megoldásként az üvegcsék intenzíven rázathatók 1–2 óráig (például mikrotányéros rázón), vagy óvatosabb rázatás mellett egy éjszakán keresztül. A CO2 kiűzésének hatásfokát ellenőrizni kell (a levegőztetés vagy a rázatási periódus meghosszabbításával). Ezután a vizes minták beütésszámának meghatározására alkalmas szcintillációs folyadékot adunk a rendszerbe, majd a minta vortexes homogenizálása után a radioaktivitás folyadék szcintillációs méréssel, a vizsgálat vakok (FB) háttéraktivitásának kivonásával meghatározható. Hacsak a tesztvíz nem nagyon színes, vagy nem tartalmaz nagy koncentrációban részecskéket, a minták rendes körülmények között egységes kioltást mutatnak, és elegendő külső standard alkalmazásával kioltási korrekciót végezni. Ha a tesztvíz nagyon színes, a kioltási korrekciót belső standard hozzáadása révén szükséges kalkulálni. Ha részecske-koncentráció magas, nem mindig lehetséges homogén oldathoz vagy gélhez jutni, vagy nagy lehet a kioltás eltérése a minták között. Ilyen esetekben az alábbiakban bemutatott tesztzagy-mérési módszert kell alkalmazni. Ha a vizsgálat mint lebegő üledék vizsgálatot végezzük el, a 14CO2 mérést indirekt módon kell elvégezni egy homogén 10 ml-es tesztvíz/szuszpenzió mintából, ahol a fázisokat megfelelő sebességű (pl. 40 000 m/s2-en 15 percig) centrifugálással szeparáltuk. Ezután a vizes fázissal a fentiek szerint járunk el. A 14C aktivitás mérése a lebegő részecskéket tartalmazó fázisból (POA) úgy történik, hogy az üledéket kevés vízben reszuszpendáljuk, majd szcintillációs üvegcsékbe juttatjuk és szcintillációs folyadékot adunk hozzá, hogy gél jöjjön létre (erre a célra speciális szcintillációs folyadékok szerezhetőek be). A részecskék természetétől (pl. szervesanyag-tartalmuk) függően megvalósítható lehet a minták szövet-szolubilizálóval történő, egy éjszakán keresztüli emésztése, majd a szcintillációs folyadék hozzáadását megelőző vortexes homogenizálása. Más megoldásként a POA oxigénfeleslegben, mintaoxidálóval történő hevítés segítségével is meghatározható. A mérés során mindig használni kell belső standardokat, valamint szükséges lehet kioltási korrekciókat végrehajtani belső standard hozzáadásával, minden egyes egyedi mintára vonatkozóan.
Direkt 14CO2 meghatározás
Ha a képződő 14CO2-t közvetlenül mérjük, akkor a vizsgálat kezdetekor több lombikot kell beállítani, majd begyűjtéskor a vzsgálati lombikokat pH 2–3 közé kell savanyítani minden egyes mérési pontnál, és a 14CO2-t fel kell fogni egy (a vizsgálat kezdetekor minden egyes vizsgálati lombikba behelyezett) belső vagy külső abszorberrel. Az abszorbeáló anyag lehet lúg (pl. 1 N NaOH oldat, vagy NaOH golyó), etanolamin vagy egy etanolamin-alapú, kereskedelmi forgalomból beszerezhető abszorber. A 14CO2 direkt méréséhez a lombikokat le kell zárni pl. butilgumi szeptummal.
1a. ábra
Példa az adatok grafikus ábrázolására (maradó aktivitás az idő függvényében)
1b. ábra
Példa az adatok fél-logaritmikus ábrázolására (ln maradó aktivitás az idő függvényében)

VI. MELLÉKLET
|
C.26. |
LEMNA SP. NÖVEKEDÉSGÁTLÁSI VIZSGÁLATA |
1. MÓDSZER
A módszer egyenértékű az OECD TG 221 (2006) jelű vizsgálatával (1). Az európai uniós hatóságok széles körben egyetértenek abban, hogy az erősen színezett anyagok esetében a Lemna-vizsgálat megfelelő módon helyettesíti az algákon végzett vizsgálatot (2)(3).
1.1. BEVEZETÉS
Ez a vizsgálati módszer különböző anyagok által a Lemna (békalencse) nemzetséghez tartozó édesvízi növényekre kifejtett toxikus hatás meghatározására szolgál. A módszer meglévő iránymutatásokon (4)(5)(6)(7)(8)(9) alapul, de azoktól több kulcsfontosságú kérdés tekintetében, a legfrissebb kutatási eredmények és szakmai egyeztetések figyelembevétele érdekében eltér. A javasolt módszer validálása nemzetközi körvizsgálattal (10) történt.
A vizsgálati módszer Lemna gibba és Lemna minor felhasználásával végzendő toxicitási vizsgálatot ír le; mindkét fajon számos vizsgálatot végeztek, és a fent említett szabványosított vizsgálatok is ezekre vonatkoznak. A Lemna nemzetség rendszertana nehezen leírható, amit tovább bonyolít a létező fenotípusok nagy száma. Bár a Lemna nemzetségben a genetikai eltérések a toxikus anyagokkal szembeni viselkedés változatosságát eredményezhetik, erről a változóról egyelőre kevés adat áll rendelkezésre ahhoz, hogy az ismertetendő eljárás keretében konkrét klón használatát ajánlhassunk. Megjegyzendő, hogy a vizsgálat nem steril körülmények között zajlik, de az eljárás egyes szakaszaiban erőfeszítéseket kell tenni annak érdekében, hogy más élő szervezetek zavaró hatása a lehető legkisebb legyen.
Az alábbiakban részletesen ismertetjük a vizsgálati oldat cseréjével végzett (félstatikus és átfolyásos) és a vizsgálati oldat cseréje nélkül végzett (statikus) vizsgálatokat. A vizsgálat céljától és a jogszabályi követelményektől függően a félstatikus és az átfolyásos vizsgálat alkalmazását például az oldatból gyorsan eltávozó (illékony, fény hatására lebomló, kicsapódó vagy biológiailag lebomló) anyagok esetén érdemes megfontolni. További iránymutatást a (11) irodalom tartalmaz.
1.2. FOGALOMMEGHATÁROZÁSOK
Az eljárás alkalmazásában:
Élőanyag-tömeg: a populációban lévő élő anyag száraz tömege. A vizsgálat során az élőanyag-tömeg helyett jellemzően helyettesítő mennyiségeket, például levélszámot vagy levélterületet mérünk, és ezekre a helyettesítő mennyiségekre is „élőanyag-tömegként” hivatkozunk.
Klorózis: a levélszövet sárgulása.
Klón: egy adott egyedből ivartalan szaporodással létrejövő szervezet vagy sejt. Az azonos klónhoz tartozó egyedek tehát genetikai szempontból azonosak.
Telep: egymáshoz kapcsolódó anya- és leánylevelek (általában 2–4) összessége. Néha növénynek is nevezik.
ECx : a vizsgálati oldatban feloldott vizsgált anyag azon koncentrációja, amely adott expozíciós idő alatt a Lemna növekedésének x %-os (például 50 %-os) csökkenését okozza (az expozíciós időt külön meg kell adni, ha eltér a vizsgálat rendes vagy teljes időtartamától). Hogy egyértelmű legyen, hogy az EC értékét a növekedési sebességből vagy a kihozatalból nyertük-e, növekedési sebesség esetén „ErC”-t, kihozatal esetén „EyC”-t írunk, majd ezt követően megadjuk a mérési változót, például: „ErC (levélszám)”.
Átfolyásos vizsgálat: olyan vizsgálat, amelynek során a vizsgálati oldatot folyamatosan cseréljük.
Levél: a békalencse növény egyetlen különálló, „levélszerű” struktúrája. Ez a szaporodásra képes legkisebb egység (egyed).
Púposság: púpos vagy duzzadt megjelenést mutató levelek.
Növekedés: a vizsgálat ideje alatt a mérési változóban (például a levélszámban, a száraz tömegben, a nedves tömegben vagy a levélterületben) bekövetkező növekedés.
Növekedési sebesség (átlagos fajlagos növekedési sebesség): az élőanyag-tömeg logaritmikus növekedése az expozíciós idő alatt.
Észlelhető hatást okozó legkisebb koncentráció (LOEC): az a legkisebb vizsgált koncentráció, amely mellett adott expozíciós idő alatt az anyag statisztikailag szignifikáns mértékben (p < 0,05) csökkenti a növekedést a kontrolltenyészethez képest. Az LOEC felett ugyanakkor minden koncentrációnak legalább akkora káros hatást kell eredményeznie, mint amelyet az LOEC okoz. Ha ez a két feltétel nem elégíthető ki, az LOEC (és így az NOEC) megválasztását részletesen indokolni kell.
Mérési változók: minden olyan változó, amelyet a vizsgálat végpontjának egy vagy több különböző hatásváltozó segítségével történő kifejezése érdekében mérünk. Ebben az eljárásban mérési változó a levélszám, a levélterület, a nedves tömeg és a száraz tömeg.
Monokultúra: egyetlen növényfajból álló tenyészet.
Nekrózis: elhalt (például fehér vagy vízzel átitatott) levélszövet.
Észlelhető hatást még nem okozó koncentráció (NOEC): az a vizsgált koncentráció, amely közvetlenül az LOEC alatt helyezkedik el.
Fenotípus: az élőlény észlelhető, génjei és a környezet kölcsönhatása által meghatározott jellemzői.
Hatásváltozók: a toxikus hatás becslésére szolgáló, az élőanyag-tömeget leíró mérési változókból különböző számítási módszerekkel származtatott változók. Ebben az eljárásban hatásváltozó a növekedési sebesség és a kihozatal, amelyeket a levélszámból, a levélterületből, a nedves tömegből és a száraz tömegből mint mérési változókból származtatunk.
Félstatikus (oldatcserés) vizsgálat: olyan vizsgálat, amelynek során a vizsgálati oldatot bizonyos időközönként lecseréljük.
Statikus vizsgálat: olyan vizsgálat, amelynek során a vizsgálati oldatot nem cseréljük le.
A vizsgálat végpontja: az az általános jellemző, amelyre a vizsgálat irányul, és amely a vizsgált vegyület hatására a kontrolltenyészethez képest megváltozik. Ebben az eljárásban a vizsgálat végpontja a növekedésgátlás, amely különböző, egy vagy több mérési változóból származtatott hatásváltozók segítségével fejezhető ki.
Vizsgálati oldat: az a teljes, szintetikus tápoldat, amelyben a vizsgált növények a vizsgált anyag jelenlétében növekednek. A vizsgált anyag általában oldott formában van jelen a vizsgálati oldatban.
Kihozatal: az élőanyag-tömeget leíró mérési változó értéke az expozíciós idő végén, mínusz ugyanezen változó értéke az expozíciós idő kezdetén.
1.3. A VIZSGÁLAT ELVE
Exponenciálisan növekedő, a Lemna nemzetséghez tartozó növénytenyészeteket monokultúraként hét napig növekedni hagyunk a vizsgált anyag különböző koncentrációinak jelenlétében. A vizsgálat célja: a vizsgálat időtartama alatt a vizsgált anyag vegetatív növekedésre gyakorolt hatásának számszerű kifejezése alkalmasan megválasztott mérési változók értékelése alapján. Elsődleges mérési változóként a levélszám szolgál. Emellett legalább még egy mérési változót (a levelek összterületét, a száraz tömeget vagy a nedves tömeget) is mérni kell, mert bizonyos anyagok más mérési változókat sokkal inkább befolyásolnak, mint a levélszámot. A vizsgált anyag hatásainak számszerű kifejezése érdekében a vizsgálati oldatban bekövetkező növekedést összehasonlítjuk a kontrolltenyészetben bekövetkező növekedéssel, és meghatározzuk azt az ECx (például EC50) koncentrációt, amely egy adott x %-os (például 50 %-os) növekedésgátlást idéz elő.
A vizsgálat végpontjaként a növekedésgátlás szolgál, amelyet a mérési változóban az expozíciós idő alatt bekövetkező logaritmikus növekedéssel (az átlagos fajlagos növekedési sebességgel) fejezünk ki. A vizsgálati oldatokra egyenként kiszámított átlagos fajlagos növekedési sebességek alapján meghatározzuk azt az ErCx (például ErC50) koncentrációt, amely egy adott x %-os (például 50 %-os) növekedésgátlást idéz elő.
Az eljárás másik hatásváltozója a kihozatal, amelyre bizonyos országok esetében a jogszabályi követelmények teljesítéséhez lehet szükség. A kihozatal a mérési változóknak az expozíciós idő végén érvényes értéke és az expozíciós idő kezdetén érvényes értéke közötti különbséget jelenti. A vizsgálati oldatokra egyenként kiszámított kihozatalok alapján meghatározzuk azt az EyCx (például EyC50) koncentrációt, amely egy adott x %-os (például 50 %-os) növekedésgátlást idéz elő.
Statisztikai eljárással meghatározható továbbá az észlelhető hatást okozó legkisebb koncentráció (LOEC) és az észlelhető hatást még nem okozó koncentráció (NOEC) értéke is.
1.4. A VIZSGÁLT ANYAG ADATAI
Rendelkezésre kell állnia egy olyan analitikai eljárásnak, amellyel a vizsgálati oldatban lévő anyag mennyisége kellő pontossággal meghatározható.
A vizsgálati körülmények rögzítése érdekében célszerű lehet tájékozódni többek között a vizsgált anyag szerkezeti képletéről, tisztaságáról, vízben való oldhatóságáról, vízben és fény hatására tanúsított stabilitásáról, pKa és Kow értékéről, gőznyomásáról és biológiai lebonthatóságáról. A vízben való oldhatóságból és a gőznyomásból meghatározható a Henry-állandó, amely megmutatja, mennyire kell arra számítani, hogy a vizsgálat során a vizsgált anyag jelentős mennyiségben eltávozik. Ennek alapján eldönthető, kell-e külön gondoskodni az így adódó veszteségek ellensúlyozásáról. Ha a vizsgált anyag oldhatóságára és stabilitására vonatkozó információk bizonytalanok, célszerű ezeket a vizsgálat körülményei, azaz a vizsgálati oldat, hőmérséklet és fényviszonyok mellett meghatározni.
Ha különösen fontos a vizsgálati oldat pH-értékének szabályozása, például könnyen hidrolizálódó fémek vagy egyéb anyagok vizsgálata esetén, akkor ajánlatos a vizsgálati oldatot pufferrel kiegészíteni (lásd az 1.7.4. szakasz első bekezdését). A (11) irodalom további iránymutatást ad olyan anyagok vizsgálatához, amelyek fizikai-kémiai jellegzetességeiknél fogva nehezen vizsgálhatók.
1.5. REFERENCIAANYAG
A vizsgálati eljárás referenciaanyag(ok), mint például a nemzetközi körvizsgálatban (10) alkalmazott 3,5-diklór-fenol segítségével ellenőrizhető. Referenciaanyaggal legalább évente kétszer (ha ennél ritkábban végzünk vizsgálatot, akkor a vizsgált anyag toxicitásának meghatározásával párhuzamosan mindig) ajánlott vizsgálatot végezni.
1.6. A VIZSGÁLAT ÉRVÉNYESSÉGE
A vizsgálat akkor érvényes, ha a kontrolltenyészetben a levélszám megkettőződési ideje 2,5 napnál (60 óránál) kisebb, ami hét nap alatt körülbelül hétszeres növekedést, illetve 0,275 1/nap átlagos fajlagos növekedési sebességet jelent. A jelen módszerben ismertetett vizsgálati oldat és vizsgálati körülmények alkalmazása esetén ez a feltétel statikus vizsgálattal kielégíthető (8). Várható, hogy a feltétel félstatikus és átfolyásos vizsgálat esetén is kielégíthető. A megkettőződési idő számítását a 2.1. szakasz ismerteti.
1.7. A MÓDSZER LEÍRÁSA
1.7.1. Eszközök
Minden, a vizsgálati oldattal érintkező eszköz üvegből vagy más, kémiailag semleges anyagból legyen. A tenyésztés és a vizsgálat céljára felhasznált üvegeszközök sterilek legyenek, és meg kell őket tisztítani minden olyan kémiai szennyeződéstől, amely a vizsgálati oldatba bemosódhatna. A vizsgálati edények legyenek kellően szélesek ahhoz, hogy a kontrolltenyészetekben a különböző telepek levelei a vizsgálat végéig átfedésmentesen növekedhessenek. Nem jelent problémát, ha a gyökér az edény alját éri, de ajánlatos legalább 20 mm mélységű és legalább 100 ml térfogatú vizsgálati edényeket alkalmazni. Ha ezek a feltételek teljesülnek, a vizsgálati edények megválasztása nem kritikus. A korábbiakban a kellő méretű üveg főzőpoharak, kristályosító tégelyek és üveg Petri-csészék egyaránt alkalmasnak bizonyultak. A vizsgálati edényeket a párolgás és a véletlen szennyeződések megakadályozása érdekében le kell fedni oly módon, hogy a levegő cserélődhessen. A vizsgálati edény, különösen pedig a fedő lehetőleg ne keltsen árnyékot vagy okozza a fény spektrális jellemzőinek módosulását.
A tenyészeteket és a vizsgálati edényeket ne tartsuk együtt. A legjobb megoldás, ha külön környezeti növesztőkamrákat, inkubátorokat vagy helyiségeket veszünk igénybe. A fényviszonyok és a hőmérsékletek szabályozhatók legyenek, és állandó szinten kell őket tartani (lásd az 1.7.8. szakaszt).
1.7.2. Vizsgált szervezet
A vizsgálat során Lemna gibba vagy Lemna minor egyedeket kell használni. A toxicitási vizsgálatokhoz korábban már felhasznált békalencsefajok rövid leírását az 1. függelék adja meg. A növényanyag törzsgyűjteményből, más laboratóriumból vagy a terepről nyerhető. A terepről származó növényanyag tenyészetét felhasználás előtt legalább nyolc hétig a vizsgálat során alkalmazandóval megegyező tápoldatban kell tartani. A tenyészet kialakításához a növényanyagot nyilvánvaló szennyeződéstől mentes helyszínről kell begyűjteni. A más laboratóriumból vagy törzsgyűjteményből származó növényanyagot legalább három hétig kell hasonló körülmények között tartani. A mérési jegyzőkönyvben mindig rögzíteni kell a növényanyag eredetét és a vizsgálathoz felhasznált fajt és klónt (ha ismert).
Olyan monokultúrát használjunk, amelyet más élő szervezetek, például algák vagy egysejtűek szemmel érzékelhető módon nem szennyeznek. Az egészséges L. minor növények kettő–öt levélből állnak, míg a L. gibba egészséges telepei akár hét levelet is tartalmazhatnak.
A vizsgálat során felhasznált növények minősége és egységessége jelentősen befolyásolja a vizsgálat eredményeit, ezért megválasztásuk során kellő gondossággal kell eljárni. Fiatal, gyorsan növekvő, látható sérüléstől és fakulástól (klorózistól) mentes egyedeket kell felhasználni. A jó minőségű tenyészetet az jellemzi, hogy nagy arányban tartalmaz legalább két levélből álló telepeket. A sok egyedülálló levél kedvezőtlen környezeti körülményekre, például tápanyaghiányra utal, ezért ilyen tenyészetekből vett egyedeket nem szabad a vizsgálatban felhasználni.
1.7.3. Tenyésztés
Az átoltás gyakoriságának csökkentése érdekében (például ha egy ideig előreláthatóan nem végzünk Lemna-vizsgálatokat) a tenyészet csökkentett fényviszonyok mellett és hőmérsékleten (4–10 °C) tartható. A tenyésztés körülményeit részletesen a 2. függelék ismerteti. Ha olyan nyilvánvaló jeleket figyelünk meg, amelyek algák vagy más élő szervezetek okozta szennyeződésre utalnak, akkor a Lemna-levelek egy részét felületi sterilizálásnak kell alávetni, majd másik oldatba kell áthelyezni (lásd a 2. függeléket), a fennmaradó szennyezett tenyészetet pedig hulladékként el kell távolítani.
A vizsgálatok előtt legalább hét nappal elegendő mennyiségű telepet csíramentesen friss, steril oldatba kell áthelyezni, majd hét–tíz napon át a vizsgálatnak megfelelő körülmények között tartani.
1.7.4. Vizsgálati oldat
Lemna minor és Lemna gibba esetén eltérő oldatot ajánlott alkalmazni az alábbiak szerint. Kellő körültekintéssel meg kell fontolni pH-puffer alkalmazását a vizsgálati oldatban (MOPS (4-morfolinpropánszulfonsav, CAS-szám: 1132–61–2; EINECS-szám: 214–478–5 a L. minor és NaHCO3 a L. gibba vizsgálati oldatában), ha gyanítható, hogy az oldat reakcióba léphet a vizsgált anyaggal és befolyásolhatja a toxikus hatást. Az érvényességi kritériumok betartása mellett Steinberg-féle tápoldat (12) is alkalmazható.
A L. minor tenyésztése és vizsgálata során a svéd szabvány (SIS) szerinti Lemna-tápoldat módosított változatát ajánlott alkalmazni. E tápoldat összetételét a 3. függelék ismerteti.
A L. gibba tenyésztése és vizsgálata során a 3. függelék szerinti 20X–AAP tápoldatot ajánlott alkalmazni.
A 3. függelék szerinti Steinberg-féle tápoldat ugyancsak alkalmas L. minor esetén, és az érvényességi kritériumok betartása mellett L. gibba esetén is alkalmazható.
1.7.5. Vizsgálati oldatok
A vizsgálati oldatokat általában törzsoldat hígításával állítjuk elő. A vizsgált anyag törzsoldatait általában úgy készítjük, hogy az anyagot tápoldatban feloldjuk.
A vizsgált anyag legnagyobb vizsgálati koncentrációja általában ne haladja meg az anyag vizsgálati körülmények között érvényes vízben való oldhatóságát. Megjegyzendő azonban, hogy a Lemna nemzetség egyedei a víz felszínén úsznak, ezért olyan anyagok hatásának is ki lehetnek téve, amelyek jellemzően a víz és a levegő határfelületén fordulnak elő (például vízben nehezen oldódó vagy víztaszító anyagok, felületaktív anyagok). Ilyenkor az expozíció nem a vizsgálati oldatban lévő anyag következtében áll elő, és a vizsgálati koncentráció a vizsgált anyag jellemzőitől függően meghaladhatja a vízben való oldhatóságot. Vízben kismértékben oldódó vizsgált anyag esetén szükséges lehet szerves oldó- vagy diszpergálószer alkalmazásával nagy koncentrációjú törzsoldatot vagy -diszperziót készíteni annak érdekében, hogy könnyebb legyen a vizsgálati anyagból a pontos mennyiséget a vizsgálati oldathoz adagolni, és könnyebben végbemenjen az oldódás vagy a diszperzióképződés. Ilyen anyag alkalmazását lehetőség szerint kerülni kell. A segédoldószer vagy -diszpergálószer alkalmazása nem járhat fitotoxikus hatással. A széles körben alkalmazott oldószerek közül például az aceton vagy a dimetil-formamid 100 μl/l koncentrációig nem okoz fitotoxikus hatást. Az esetleg alkalmazott oldó- vagy diszpergálószer végső koncentrációja – amelyet a mérési jegyzőkönyvben rögzíteni kell – a lehető legkisebb legyen (legfeljebb 100 μl/l), és az oldó- vagy diszpergálószert a vizsgálathoz és a kontrolltenyészet esetében azonos koncentrációban kell alkalmazni. A (11) irodalom bővebb iránymutatást ad a diszpergálószerekre vonatkozóan.
1.7.6. Vizsgálati csoportok és kontrollcsoportok
A vizsgált anyag Lemna-fajokra gyakorolt toxikus hatásának előzetes ismerete alapján, amely származhat például koncentrációtartomány-meghatározási vizsgálatból, megválaszthatók az alkalmas vizsgálati koncentrációk. A végleges toxicitási vizsgálatnak általában öt, egymással mértani sorozatot alkotó vizsgálati koncentrációra kell kiterjednie. A koncentrációk sorozatának hányadosa általában ne legyen 3,2-nél nagyobb, de lapos koncentráció–hatás görbe esetén ennél nagyobb érték lehet indokolt. Ötnél kevesebb koncentráció alkalmazását indokolni kell. Minden koncentrációval legalább három párhuzamos tenyészetet kell megvizsgálni.
A vizsgálati koncentrációk tartományának kijelölése során (akár a koncentrációtartomány-meghatározási vizsgálatot, akár a végleges vizsgálatot megelőzően) a következőket célszerű megfontolni.
|
— |
ECx meghatározása esetén a kellő konfidenciaszint biztosítása érdekében a vizsgálati koncentrációknak közre kell fogniuk az ECx értékét. Ha például az EC50 értéket kívánjuk meghatározni, akkor a legnagyobb vizsgálati koncentrációnak nagyobbnak kell lennie EC50-nél. Ha az EC50 kívül esik a vizsgálati koncentrációk tartományán, akkor a kiadódó konfidenciaintervallumok szélesek lesznek, és előfordulhat, hogy nem végezhető el a modell statisztikai illesztése. |
|
— |
LOEC és/vagy NOEC meghatározása esetén a legkisebb vizsgálati koncentráció legyen elegendően kicsiny ahhoz, hogy a növekedési sebesség ne legyen jelentősen kisebb, mint a kontrollcsoportban. A legnagyobb koncentráció ugyanakkor legyen elegendően nagy ahhoz, hogy a növekedési sebesség jelentősen kisebb legyen, mint a kontrollcsoportban. Ha ez nem teljesül, a vizsgálatot más koncentrációtartománnyal meg kell ismételni (kivéve akkor, ha a legnagyobb koncentráció megegyezik az oldhatósági határral vagy az előírt maximális határkoncentrációval, például 100 mg/l-rel). |
Az egyes vizsgálatok mellett kontrolltenyészeteket is kell vizsgálni a vizsgálati edényekével megegyező táptalaj, levél- és telepszám, környezeti körülmények és eljárások alkalmazásával, de a vizsgált anyag nélkül. Segédoldószer vagy -diszpergálószer alkalmazása esetén egy további kontrolltenyészet is szükséges, amelyben az oldó- vagy diszpergálószer azonos koncentrációban van jelen, mint a vizsgálati edényekben. Legalább a koncentrációnként alkalmazott edényszámmal megegyező, ideális esetben kétszer annyi párhuzamos kontrolltenyészetet (illetőleg szükség szerint oldószeres edényt) kell alkalmazni.
Ha nem kívánjuk meghatározni az NOEC értékét, a vizsgálati terv módosítható oly módon, hogy több koncentrációt, de koncentrációnként kevesebb párhuzamos tenyészetet tartalmazzon. A párhuzamos kontrolltenyészetek száma azonban ilyenkor is legalább három legyen.
1.7.7. Expozíció
Kettő–négy látható levélből álló telepeket a kiindulási tenyészetből csíramentesen a kísérleti edényekbe helyezünk, véletlenszerűen elosztva. A kísérleti edényekbe egyenként összesen 9–12 levél kerüljön. Minden kísérleti edényben azonos számú levél és telep legyen. A módszer alkalmazása és a körvizsgálat során szerzett tapasztalatok azt mutatják, hogy a készítmények közötti, a növekedési sebesség alapján meghatározott növekedésgátlás 4–7 %-os értékének (a kihozatal alapján meghatározott növekedésgátlás 10–15 %-os értékének) megfelelő nagyságú növekedéskülönbség kimutatásához elegendő, ha készítményenként három párhuzamos tenyészetet és edényenként 9–12 kiindulási levelet alkalmazunk (10).
Az inkubátorban a kísérleti edényeket véletlenszerű elrendezésben kell elhelyezni annak érdekében, hogy a fényerősség és a hőmérséklet térbeli változatossága minél kisebb hatást gyakoroljon a vizsgálatokra. A megfigyelések végzésekor (vagy gyakrabban) az edényeket véletlenszerűen vagy blokkonkénti kiosztási terv alapján át kell rendezni.
Ha az előzetesen elvégzett stabilitásvizsgálatok azt jelzik, hogy a vizsgált anyag koncentrációja a vizsgálat időtartama (7 nap) alatt nem tartható fenn (azaz a mért koncentráció a kezdeti mért koncentráció 80 %-a alá csökken), akkor félstatikus vizsgálatot ajánlatos végezni. Ennek keretében a telepeket a vizsgálat során legalább két alkalommal (a 3. és az 5. napon) friss vizsgálati és kontrolloldatokba kell helyezni. A friss oldatba helyezés gyakorisága a vizsgált anyag stabilitásától függ; nagymértékben instabil vagy illékony anyagok esetén a közel állandó koncentráció fenntartása gyakoribb oldatcserét igényelhet. Bizonyos esetekben átfolyásos vizsgálatot célszerű végezni (11)(13).
Ez az eljárás nem tartalmazza a levélre történő felvitellel (permetezéssel) megvalósuló expozíciót; erről lásd a (14) irodalmat.
1.7.8. Inkubációs körülmények
Folyamatos meleg- vagy hidegfehér fluoreszcens megvilágítást alkalmazzunk oly módon, hogy a fotoszintetikusan aktív sugárzásban (400–700 nm), a fényforrástól a Lemna-levelekkel megegyező távolságban lévő pontokban mért fényintenzitás a 85–135 μE/m2s tartományban előzetesen felvett értéknek feleljen meg (ez 6 500–10 000 lux értéket jelent). A vizsgálat területén a fényintenzitás az előzetesen felvett értéktől legfeljebb ±15 %-kal térhet el. A fényerősség érzékelésére és mérésére szolgáló módszer, különösen pedig a fényérzékelő befolyásolja a mért értéket. A gömbérzékelők (amelyek a mérési sík alatt és felett tetszőleges irányból érkező fény mérésére alkalmasak) és a „koszinuszos” érzékelők (amelyek csak a mérési sík felett tetszőleges irányból érkező fény mérésére alkalmasak) előnyben részesítendők az egytengelyű érzékelőkkel szemben, mert a jelen módszerben előírt többpontos fényforrás esetén nagyobb értékeket szolgáltatnak.
A vizsgálati edényekben a hőmérséklet 24 ± 2 °C legyen. A vizsgálat során a kontrolltenyészet tápoldatának pH-értéke nem növekedhet 1,5-nél nagyobb mértékben. Az 1,5-nél nagyobb eltérés ugyanakkor nem teszi érvénytelenné a vizsgálatot, ha kimutatható, hogy az érvényességi feltételek teljesülnek. A pH-érték eltolódása egyes különleges esetekben, például instabil anyag vagy fém vizsgálata esetén fokozott körültekintést tesz szükségessé. További iránymutatást a (11) irodalom tartalmaz.
1.7.9. A vizsgálat időtartama
A vizsgálat a növények vizsgálati edényekbe helyezése után 7 nappal fejeződik be.
1.7.10. Mérések és analitikai vizsgálatok
A vizsgálat kezdetén meg kell számlálni a vizsgálati edényekben lévő leveleket, és az értéket fel kell jegyezni a mérési jegyzőkönyvben; a számlálás során ügyelni kell arra, hogy a kiálló, vizuálisan elkülönülő leveleket vegyük figyelembe. A szabályszerűnek vagy rendellenesnek tűnő levelek számát a vizsgálat kezdetén, az expozíció időtartama alatt legalább háromnaponta (azaz a hét nap során legalább kétszer), valamint a vizsgálat végén kell meghatározni. Fel kell jegyezni a növények fejlődésében beálló változásokat, például a levélméret vagy a megjelenés változásait, a nekrózisra, a klorózisra vagy a púposságra utaló jeleket, a telepek felbomlását vagy az úszóképesség megszűnését, a gyökerek hosszának vagy megjelenésének megváltozását. Fel kell továbbá jegyezni a vizsgálati oldat jellegzetességeit (például oldatlan anyag jelenléte, algaképződés a vizsgálati edényben) is.
A vizsgálat során a levélszám mellett a vizsgált anyagnak a következő mérési változók közül legalább egy továbbira gyakorolt hatását is meg kell határozni:
|
i. |
a levelek összterülete; |
|
ii. |
száraz tömeg; |
|
iii. |
nedves tömeg. |
A levelek összterülete azért előnyös mérési változó, mert a vizsgálat kezdetén, folyamán és végén minden vizsgálati és kontrolltenyészetre külön-külön meghatározható. A száraz és a nedves tömeget a vizsgálat elején a vizsgálatban alkalmazandó kiindulási tenyészet szempontjából reprezentatív mintán, majd a vizsgálat végén az egyes vizsgálati és kontrolltenyészetek növényanyagán kell meghatározni. Ha a levélterületet nem mérjük, a száraz tömeg mérését előnyben kell részesíteni a nedves tömeg mérésével szemben.
A levelek összterülete, a száraz tömeg és a nedves tömeg a következőképpen határozható meg:
|
i. |
A levelek összterülete. A telepekben előforduló levelek összterülete meghatározható képelemzés segítségével. Videokamerával (például az edényt világító testre helyezve) rögzíthetők a vizsgálati edény és a növények körvonalai, majd a kiadódó kép digitalizálható. Ismert területű síkidomokhoz hasonlítva meghatározható a vizsgálati edényekben lévő levelek összterülete. Kellő gondossággal ki kell küszöbölni a vizsgálati edény pereme miatti zavarás hatását. Másik, munkaigényesebb lehetőség, ha a vizsgálati edényeket és a növényeket lefényképezzük, a kiadódó körvonalak mentén kivágjuk az alakokat, majd levélterület-elemzővel vagy milliméterpapíron meghatározzuk a levélterületet. Egyéb technikák (például a papírból kivágott terület egységnyi területű idomokhoz hasonlítása a tömegek összevetése alapján) ugyancsak megoldást jelenthetnek. |
|
ii. |
Száraz tömeg. A telepeket összegyűjtjük a vizsgálati edényekből, desztillált vagy ionmentesített vízzel leöblítjük őket, leitatjuk a felesleges vizet, és a telepeket 60 °C-on tömegállandóságig hevítjük. A kapcsolódó gyökereket nem távolítjuk el. A száraz tömeget legalább 0,1 mg pontossággal kell kifejezni. |
|
iii. |
Nedves tömeg. A telepeket előzetesen megmért tömegű, kerek aljukon kis (1 mm-es) lyukakkal ellátott polisztirol (vagy más kémiailag semleges anyagból készült) csövekbe helyezzük. A csöveket ezt követően 3 000 1/perc fordulatszámon, szobahőmérsékleten 10 percig centrifugáljuk. Megmérjük a most már szárított telepeket tartalmazó csövek tömegét, és a nedves tömeget az üres csövek tömegének levonásával nyerjük. |
1.7.10.1. A mérések és az analitikai vizsgálatok gyakorisága
Statikus vizsgálat esetén a vizsgálat elején és végén megmérjük az egyes készítmények pH-értékét. Félstatikus vizsgálat esetén a pH-értéket minden oldatcsere alkalmával meghatározzuk mind a friss, mind az elhasznált oldaton.
A fényintenzitást a növesztőkamrában, az inkubátorban vagy a vizsgálati helyiségben a fényforrástól a Lemna-levelekkel megegyező távolságban elhelyezkedő pontokban mérjük. Fényintenzitást a vizsgálat során legalább egyszer kell mérni. A növesztőkamrában, az inkubátorban vagy a vizsgálati helyiségben azonos körülmények között külön erre a célra tartott edényben legalább naponta mérjük a tápoldat hőmérsékletét.
A vizsgálat során alkalmas időközönként meg kell határozni a vizsgált anyag koncentrációját. Statikus vizsgálat esetén a koncentrációt legalább a vizsgálat elején és végén kell meghatározni.
Félstatikus vizsgálat esetén, ha a vizsgált anyag koncentrációja várhatóan nem marad a névleges koncentráció ± 20 %-os környezetében, akkor minden friss vizsgálati oldatot elkészítésekor, továbbá minden lecserélt oldatot analitikai vizsgálatnak kell alávetni (lásd az 1.7.7. szakasz harmadik bekezdését). Olyan vizsgálatok esetén ugyanakkor, amelyekben a vizsgált anyag mért kezdeti koncentrációja ugyan nem a névleges koncentráció ± 20 %-os környezetében van, de adatokkal kellően bizonyítható, hogy a kezdeti koncentrációk megismételhetők és stabilak (azaz a kezdeti koncentráció 80–120 %-os környezetében vannak), akkor az analitikai vizsgálatokat elegendő csak a legkisebb és a legnagyobb vizsgálati koncentráción elvégezni. Az oldatcsere előtt a vizsgált anyag koncentrációját minden esetben elegendő vizsgálati koncentrációnként egy párhuzamos oldaton (vagy az edények párhuzamos tenyészetenként egyesített tartalmán) elvégezni.
Átfolyásos vizsgálat esetén a félstatikus vizsgálathoz hasonló rendben, így a vizsgálat elején, felénél és végén kell mintákat venni, de ilyenkor nincs értelme az „elhasznált” oldat vizsgálatának. Átfolyásos vizsgálat esetén naponta ellenőrizni kell az oldószer és a vizsgált anyag vagy a vizsgált anyag törzsoldata adagolási sebességét.
Ha kimutatható, hogy a vizsgált anyag koncentrációját a vizsgálat időtartama alatt sikerült a névleges vagy a mért kezdeti koncentráció ± 20 %-os környezetében tartani, akkor az eredmények a névleges vagy mért kezdeti értékek alapján elemezhetők. Ha a névleges vagy a mért kezdeti koncentrációtól való eltérés meghaladja a ± 20 %-ot, akkor az eredményeket az expozíció idején érvényes koncentrációértékek mértani középértéke vagy a vizsgált anyag koncentrációjának csökkenését leíró modellek alapján kell elemezni (11).
1.7.11. Határérték-vizsgálat
Egyes esetekben, például ha előzetesen elvégzett vizsgálatok azt jelzik, hogy a vizsgált anyag 100 mg/l koncentrációig vagy az adott vizsgálati oldatban való oldhatósági határig (a kettő közül a kisebbikig) nem okoz toxikus hatást, akkor egy kontrollcsoport és egyetlen (100 mg/l koncentrációnak vagy az adott vizsgálati oldatban való oldhatósági határhoz tartozó koncentrációnak megfelelő) expozíciónak kitett csoport összehasonlításával úgynevezett határérték-vizsgálat végezhető. A vizsgálat során fokozottan ajánlatos elemezni az expozíció koncentrációját. A határérték-vizsgálat során biztosítani kell valamennyi fent leírt vizsgálati körülmény fennállását és érvényességi feltétel teljesülését azzal az eltéréssel, hogy kétszer annyi párhuzamos készítményt kell vizsgálni. A kontrollcsoportban és az expozíciónak kitett csoportban tapasztalt növekedés olyan statisztikai próbával (például a Student-féle t-próbával) elemezhető, amely a középértékek összehasonlításán alapul.
2. ADATOK ÉS JEGYZŐKÖNYVEZÉS
2.1. MEGKETTŐZŐDÉSI IDŐ
A vizsgálat során a levélszám megkettőződéséhez szükséges Td idő meghatározásához és az erre vonatkozó érvényességi feltétel (lásd az 1.6. szakaszt) ellenőrzéséhez a kontrolltenyészetek adataira a következő összefüggést kell alkalmazni:
Td = ln 2/μ,
ahol μ a 2.2.1. szakasz első és második bekezdése szerint meghatározott átlagos fajlagos növekedési sebesség.
2.2. HATÁSVÁLTOZÓK
A vizsgálat célja annak meghatározása, milyen hatást gyakorol a vizsgált anyag a Lemna-telepek vegetatív növekedésére. Ez a vizsgálati módszer két hatásváltozót tartalmaz, mert a tagállamok igényei és a tagállamokban érvényes szabályozás eltérő. Hogy a vizsgálati eredmények valamennyi tagállamban elfogadhatók legyenek, a hatást mindkét alábbi hatásváltozó alapján meg kell határozni:
|
a) |
átlagos fajlagos növekedési sebesség: ezt a hatásváltozót a levélszám logaritmusának, valamint egy további mért paraméter (a levelek összterülete, a száraz tömeg vagy a nedves tömeg) logaritmusának napi változása alapján számítjuk a kontrolltenyészetek és az egyes expozíciónak kitett tenyészetek figyelembevételével. Ezt a mennyiséget relatív növekedési sebességnek is nevezik (15); |
|
b) |
kihozatal: ezt a hatásváltozót a levélszám, valamint egy további mért paraméter (a levelek összterülete, a száraz tömeg vagy a nedves tömeg) értékének napi változása alapján számítjuk a kontrolltenyészetek és az egyes expozíciónak kitett tenyészetek figyelembevételével. |
Megjegyzendő, hogy a két hatásváltozó alapján számított toxicitásértékek nem hasonlíthatók egymáshoz, és a vizsgálatok eredményeinek felhasználása során erre a különbségre tekintettel kell lenni. A jelen vizsgálati módszerben előírt vizsgálati körülmények betartása mellett a két számítási eljárás matematikai háttere következtében az átlagos fajlagos növekedési sebességen alapuló ECx értékek (ErCx) általában nagyobbra adódnak, mint a kihozatalon alapuló ECx értékek (EyCx). Ezt az eltérést nem szabad úgy értelmezni, hogy egyik hatásváltozó érzékenyebb lenne a másiknál; egyszerűen arról van szó, hogy a két érték matematikailag mást jelent. Az átlagos fajlagos növekedési sebesség a békalencse korlátozatlan tenyészetekben tanúsított általános exponenciális növekedési mintáján alapul, és a toxicitást a növekedési sebességre kifejtett hatás alapján, a kontrolltenyészetekben tapasztalható növekedési sebesség abszolút nagyságától, a koncentráció–hatás görbe meredekségétől és a vizsgálat időtartamától függetlenül becsli. A kihozatalból származtatott eredmények ezzel szemben mindezektől a változóktól is függnek. Az EyCx függ a békalencse egyes vizsgálatokban felhasznált egyedeinek fajlagos növekedési sebességétől és a legnagyobb fajlagos növekedési sebességtől, amely fajonként, sőt klónonként is eltérő lehet. Ez a hatásváltozó nem alkalmas az egyes békalencsefajok vagy klónok toxikus hatással szembeni érzékenységében fennálló különbségek megítélésére. Bár tudományos szempontból a toxikus hatást indokoltabb az átlagos fajlagos növekedési sebességből meghatározni, ez a vizsgálati módszer a kihozatalon alapuló toxicitásszámítást is tartalmazza, mert egyes országok hatályos jogszabályi követelményei ezt is szükségessé teszik.
A toxicitást a levélszám és egy további mérési változó (a levelek összterülete, a száraz tömeg vagy a nedves tömeg) alapján is meg kell határozni, mert bizonyos anyagok egy másik mérési változót sokkal erősebben befolyásolhatnak, mint a levélszámot. Ez a hatás nem volna kimutatható, ha csak a levélszámból indulnánk ki.
A levélszám, valamint a többi feljegyzett mérési változó (a levelek összterülete, a száraz tömeg vagy a nedves tömeg) értékét az egyes mérések alkalmával a koncentrációértékek szerint táblázatba kell rendezni. A további adatelemzés, azaz az LOEC, az NOEC vagy az ECx meghatározása során a párhuzamos tenyészetek külön-külön vett adataiból, nem pedig az egyes csoportok középértékéből kell kiindulni.
2.2.1. Átlagos fajlagos növekedési sebesség
Az adott időszakhoz tartozó átlagos fajlagos növekedési sebességet a növekedést leíró változók (levélszám, valamint egy további mérési változó: a levelek összterülete, száraz tömeg vagy nedves tömeg) logaritmusának növekedéseként számítjuk a vizsgálati és a kontrolltenyészetek minden egyes példányára külön-külön, a következő összefüggés szerint:

ahol:
|
– |
μi–j: |
az átlagos fajlagos növekedési sebesség az „i”-től a „j” időpillanatig |
|
– |
Ni: |
a mérési változó értéke a vizsgálati vagy a kontrolltenyészetben az „i” időpillanatban |
|
– |
Nj: |
a mérési változó értéke a vizsgálati vagy a kontrolltenyészetben a „j” időpillanatban |
|
– |
t: |
az „i”-től „j”-ig terjedő időtartam. |
A vizsgálati és a kontrollcsoportok mindegyikére meg kell határozni a növekedési sebesség középértékét és tapasztalati szórásnégyzetét.
Az átlagos fajlagos növekedési sebességet a vizsgálat teljes időtartamára (tehát úgy, hogy a fenti összefüggésben „i” a vizsgálat kezdetét, „j” a vizsgálat végét jelöli) kell meghatározni. Minden egyes vizsgálati koncentrációra és kontrollcsoportra meg kell határozni az átlagos fajlagos növekedési sebesség középértékét és tapasztalati szórásnégyzetét. Emellett (például a növekedési görbék logaritmikus transzformáltjának szemügyre vételével) becslést adunk a növekedési sebesség szakaszonkénti értékére is annak érdekében, hogy megítélhető legyen a vizsgált anyag által az expozíciós időszak folyamán kifejtett hatás. Ha a szakaszonkénti növekedési sebesség és az átlagos növekedési sebesség között nagy különbséget tapasztalunk, az azt jelenti, hogy a növekedés mintája erősen eltér az állandó kitevőjű exponenciális növekedéstől és különösen ajánlott a növekedési görbék részletes vizsgálata. Ilyenkor a biztonság javára járunk el, ha az összehasonlítást a legnagyobb gátlás időszakában a vizsgált tenyészetekben és az ugyanebben az időszakban a kontrolltenyészetekben tapasztalt fajlagos növekedési sebesség között végezzük.
A növekedési sebesség Ir százalékos gátlását ezután koncentrációnként (készítménycsoportonként) a következő összefüggésből kell meghatározni:

ahol:
|
– |
% Ir: |
az átlagos fajlagos növekedési sebesség százalékos gátlása |
|
– |
μC: |
a μ középértéke a kontrollcsoportban |
|
– |
μT: |
a μ középértéke a vizsgált csoportban. |
2.2.2. Kihozatal
A kihozatalra gyakorolt hatást vizsgálati edényenként, két mérési változó: a levélszám és egy további mérési változó (a levelek összterülete, a száraz tömeg vagy a nedves tömeg) vizsgálat kezdetén és vizsgálat végén mért értékéből számítjuk. A száraz tömeg és a nedves tömeg esetében a kezdeti élőanyag-tömeget a vizsgálati edények tartalmának összeállításához (lásd az 1.7.3. szakasz második bekezdését) használt tételből származó levelekből vett mintán határozzuk meg. Minden egyes vizsgálati koncentráció és kontrollcsoport esetén kiszámítjuk a kihozatal középértékét és tapasztalati szórásnégyzetét. Az átlagos százalékos kihozatalgátlás (% Iy) készítménycsoportonként határozható meg a következő összefüggésből:
ahol:
|
– |
% Iy: |
a kihozatal százalékos csökkenése |
|
– |
bC: |
az élőanyag-tömeg végső értéke és kiindulási értéke közötti különbség a kontrollcsoportban |
|
– |
bT: |
az élőanyag-tömeg végső értéke és kiindulási értéke közötti különbség a vizsgált csoportban. |
2.2.3. A koncentráció–hatás görbék elkészítése
El kell készíteni a hatásváltozóhoz tartozó átlagos százalékos gátlás értékét (a 2.2.1., illetve a 2.2.2. szakasz utolsó bekezdésében meghatározott Ir vagy Iy értéket) a vizsgálati koncentráció logaritmusa függvényében ábrázoló koncentráció–hatás görbéket.
2.2.4. Az ECx becslése
Az ECx (például EC50) becslését mind az átlagos fajlagos növekedési sebesség (ErCx), mind a kihozatal (EyCx) alapján el kell végezni, amelyeket viszont a levélszámból és egy további mérési változóból (levelek összterülete, száraz tömeg vagy nedves tömeg) egyaránt ki kell számítani. Ez azért szükséges, mert egyes vizsgálati anyagok a levélszámra és a második mérési változóra eltérő hatást gyakorolnak. Ennek megfelelően tehát minden x gátlási szinthez négy ECx toxicitási paramétert állítunk elő: ErCx (levélszám); ErCx (levelek összterülete, száraz tömeg vagy nedves tömeg); EyCx (levélszám); és EyCx (levelek összterülete, száraz tömeg vagy nedves tömeg).
2.3. STATISZTIKAI SZÁMÍTÁSOK
A statisztikai számítások célja, hogy regressziószámítással kvantitatív koncentráció–hatás összefüggést állítsunk elő. Lehetőség van arra, hogy a hatásadatok – például probit-, logit- vagy Weibull-modell (16) szerinti – linearizációs transzformálását követően súlyozott lineáris regressziót végezzünk, de előnyösebb inkább nemlineáris regressziót alkalmazni, mert ez jobban kezeli az adatok elkerülhetetlen irregularitását és a sima eloszlástól való eltérést. A nulla és a teljes gátlás környezetében az irregularitást a transzformáció felnagyíthatja, ami a számítás szempontjából előnytelen (16). Megjegyzendő, hogy a probit-, logit- vagy Weibull-transzformáltat alkalmazó szokásos számítási módszerek elsősorban kvantált (például elhalálozás kontra túlélés) adatok feldolgozására alkalmasak, így módosítást igényelnek, ha a növekedési sebességre vagy a kihozatalra kívánjuk őket alkalmazni. Az ECx-értékek folytonos adatokból való számítására alkalmas módszereket a (17), (18) és (19) irodalom ismertet.
Az egyes hatásváltozók elemzéséhez a koncentráció–válasz összefüggésből állítsuk elő az ECx egyes pontokban becsült értékét. Ha lehetséges, meg kell határozni az egyes becslések 95 %-os konfidenciahatárát. A hatásadatok regressziós modellhez illeszkedésének jóságát grafikus vagy statisztikai módszerrel kell megállapítani. A regressziószámítást az egyes vizsgálati edényeken nyert értékekkel, nem pedig a vizsgálati csoportok középértékével kell elvégezni.
Ha a rendelkezésre álló regressziós modellek/módszerek nem alkalmasak a konkrét adatok elemzésére, az EC50-re vonatkozó becsléseket és a konfidenciahatárokat „bootstrap” technikával együtt alkalmazott lineáris interpolációval (20) is előállíthatjuk.
Az LOEC, és így az NOEC becsült értékének előállítása érdekében varianciaanalízis (ANOVA) segítségével összevetjük az egyes készítményekhez tartozó középértékeket. Az egyes koncentrációkhoz tartozó középértékeket ezután alkalmasan megválasztott többszörös összehasonlító módszerrel vagy trendpróbával a kontrollcsoporton nyert középértékhez hasonlítjuk. Hasznos lehet a Dunnett- és a Williams-próba alkalmazása (21)(22)(23)(24). Lényeges ellenőrizni, hogy fennáll-e az ANOVA-nak a szórásnégyzetek homogenitására vonatkozó feltevése. Ezt az ellenőrzést grafikusan vagy formális próbával (25) végezhetjük. Erre a célra a Levene- és a Bartlett-próba alkalmas. A szórásnégyzetek homogenitására vonatkozó feltevés nem teljesülése gyakran kiküszöbölhető az adatok logaritmikus transzformálásával. Ha a szórásnégyzetek heterogenitása túlságosan szélsőségesnek bizonyul, és transzformációval nem javítható, akkor célszerű megfontolni például a lépésenkénti Jonkheere-trendpróba alkalmazását. Az NOEC meghatározásához további útmutatást ad a (19) irodalom.
A legújabb kutatási eredmények alapján az az ajánlás fogalmazható meg, hogy az NOEC kiszámítása helyett célszerűbb az ECx értékét becsülni pontonkénti regresszióval. A jelen Lemna-vizsgálathoz egyelőre nem áll rendelkezésre alkalmas x érték, bár a 10 és 20 % közötti tartomány megfelelőnek látszik (a választott hatásváltozótól függően), és ajánlott mind az EC10, mind az EC20 értéket jegyzőkönyvbe foglalni.
3. JEGYZŐKÖNYVEZÉS
3.1. VIZSGÁLATI JEGYZŐKÖNYV
A vizsgálati jegyzőkönyv tartalmazza a következő információkat:
Vizsgált anyag:
|
— |
fizikai jelleg és fizikokémiai tulajdonságok, vízben való oldhatósági határ, |
|
— |
kémiai azonosító adatok (pl. CAS-szám), tisztaság. |
Vizsgált faj:
|
— |
tudományos név, klón (ha ismert) és eredet. |
Vizsgálati körülmények:
|
— |
alkalmazott vizsgálati eljárás (statikus, félstatikus vagy átfolyásos), |
|
— |
a vizsgálat megkezdésének időpontja és időtartama, |
|
— |
vizsgálati oldat, |
|
— |
a kísérleti terv ismertetése: vizsgálati edények és fedők, oldattérfogatok, telep- és levélszámok vizsgálati edényenként a vizsgálat megkezdésekor, |
|
— |
vizsgált koncentrációk (szükség szerint névleges és mért értékek) és a párhuzamos tenyészetek száma koncentrációnként, |
|
— |
a törzs- és a vizsgálati oldatok elkészítésének módszere, az esetleg alkalmazott oldó- vagy diszpergálószer, |
|
— |
hőmérséklet a vizsgálat folyamán, |
|
— |
fényforrás, a fény intenzitása és homogenitása, |
|
— |
a vizsgálati és a kontrolloldatok pH-értékei, |
|
— |
a vizsgált anyag koncentrációi és az analízis módszere a megfelelő minőség-ellenőrzési adatokkal együtt (az érvényesség vizsgálata, a szórás vagy az analízis konfidenciahatára), |
|
— |
a levélszám és a további mérési változók (száraz tömeg, nedves tömeg vagy levélterület) meghatározásának módszere, |
|
— |
a jelen vizsgálati módszertől való esetleges eltérések. |
Eredmények:
|
— |
nyers mérési adatok: levélszám és a további mérési változók minden egyes vizsgálati és kontrolltenyészetben minden adatfelvételkor és analízis alkalmával, |
|
— |
az egyes mérési változók középértékei és szórásai, |
|
— |
növekedési görbék minden egyes koncentrációra (logaritmikusan transzformált mérési változókkal ajánlott megadni, lásd a 2.2.1. szakasz második bekezdését), |
|
— |
megkettőződési idő, illetve növekedési sebesség a kontrolltenyészetben, a levélszám alapján, |
|
— |
hatásváltozók számított értéke minden párhuzamos tenyészetre, továbbá vizsgálati csoportonként a középérték és a szórásnégyzet, |
|
— |
a koncentráció–hatás összefüggés grafikus formában, |
|
— |
a hatásváltozók toxikus végpontjának becsült értékei, például EC50, EC10, EC20 és a hozzájuk tartozó konfidenciaintervallumok. Ha kiszámítottuk, az LOEC, illetve az NOEC értéke, továbbá a számításhoz alkalmazott statisztikai módszer, |
|
— |
ANOVA alkalmazása esetén a hatás kimutatásának mérethatára (például a legkisebb szignifikáns különbség), |
|
— |
a vizsgálatok során észlelt esetleges növekedésserkentés, |
|
— |
fitotoxicitásra utaló esetleges látható jelek, valamint a vizsgálati oldatokkal kapcsolatos észrevételek, |
|
— |
az eredmények szöveges összefoglalása és értékelése, ezen belül a jelen vizsgálati módszertől való eltérések vizsgálati eredményekre gyakorolt hatásának elemzése. |
4. IRODALOM
|
(1) |
OECD TG 221 (2006) Lemna Sp. Growth Inhibition Test |
|
(2) |
A Lemna-vizsgálat színezett anyagokra való alkalmazását az EU Manual of Decisions (2006. július) 13.5.3. szakasza tárgyalja: http://ecb.jrc.ec.europa.eu/new-chemicals |
|
(3) |
Guidance on information requirements and chemical safety assessment – Chapter R.7b: Endpoint specific guidance; Table 7.8.3 Summary of difficult substance testing issues: http://guidance.echa.europa.eu/docs/guidance_document/information_requirements_en.htm?time=1234958685#A |
|
(4) |
ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415–91 (Reapproved 1998). pp. 733–742. In, Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA. |
|
(5) |
USEPA – United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft”. EPA 712-C-96–156. 8pp. |
|
(6) |
AFNOR – Association Française de Normalisation. (1996). XP T 90–337: Détermination de l’inhibition de la croissance de Lemna minor. 10pp. |
|
(7) |
SSI – Svéd Szabványügyi Intézet. (1995). Vízminőség. A növekedésgátlás meghatározása (7-d) Lemna minor, békalencse. SS 02 82 13. 15pp. (svéd nyelven). |
|
(8) |
Environment Canada. (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37–120 pp |
|
(9) |
Environment Canada. (1993) Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series No. 145. |
|
(10) |
Sims I., Whitehouse P., and Lacey R. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc – Environment Agency. |
|
(11) |
OECD (2000). Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publications, Series on Testing and Assessment No.23. |
|
(12) |
ISO DIS 20079. Water Quality – Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) – Duckweed Growth Inhibition Test. |
|
(13) |
Walbridge C. T. (1977). A flow-through testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory – Duluth, Minnesota 55804. US EPA Report No. EPA-600/3–77 108. September 1977. |
|
(14) |
Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia, 118/119, 353–359. |
|
(15) |
Huebert, D.B. and Shay J.M. (1993) Considerations in the assessment of toxicity using duckweeds. Environmental Toxicology and Chemistry, 12, 481–483. |
|
(16) |
Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, 713–718. |
|
(17) |
Nyholm, N. Sørensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, 157–167. |
|
(18) |
Bruce R.D. and Versteeg D.J. (1992) A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry, 11, 1485–1494. |
|
(19) |
OECD. (2004). Guidance Document on Statistical Analysis of Ecotoxicity Data. |
|
(20) |
Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment Center Technical Report 05–88. USEPA, Duluth, MN. |
|
(21) |
Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc., 50, 1096–1121. |
|
(22) |
Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics, 20, 482–491. |
|
(23) |
Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics, 27: 103–117. |
|
(24) |
Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics, 28: 510–531. |
|
(25) |
Brain P. and Cousens R. (1989). An equation to describe dose-responses where there is stimulation of growth at low doses. Weed Research, 29, 93–96. |
1. függelék
A Lemna nemzetség leírása
A Lemna nemzetséghez tartozó, békalencseként közismert vízinövények a Lemnaceae családba tartoznak, amelynek négy nemzetsége számos világszerte elterjedt fajt felölel. Különböző megjelenésüket és rendszertanukat a szakirodalom kimerítően ismerteti (1)(2). A Lemna gibba és a L. minor a mérsékelt éghajlati övezet jellegzetes képviselői; a toxicitási vizsgálatokban többnyire ezeket alkalmazzák. Mindkét fajnak úszó vagy alámerülő, korong alakú levelei és nagyon vékony, az egyes levelek alsó felszínének közepéről eredő gyökerei vannak. A Lemna-fajok többnyire nem virágzanak, a szaporodás érdekében a növények vegetatív úton új leveleket hoznak létre (3). Az idősebb növényekhez képest a fiatalabbak általában halványabbak, rövidebb gyökereik vannak és két-három, különböző méretű levélből állnak. A Lemna-növények kis mérete, egyszerű struktúrája, ivartalan szaporodása és rövid generációs ideje a nemzetség fajait különösen alkalmassá teszi laboratóriumi vizsgálatokra (4)(5).
Mivel az egyes fajok előreláthatóan eltérő mértékben lesznek érzékenyek a különböző behatásokra, az érzékenységet kizárólag fajon belül szabad összehasonlítani.
Példák a vizsgálatokban korábban már alkalmazott Lemna-fajokra, fajonként szakirodalmi hivatkozásokkal
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major: Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. phys. Chem., 29: 935–941.
Lemna minor: United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft”. EPA 712-C-96–156. 8pp.
Association Française de Normalisation (AFNOR). (1996). XP T 90–337: Détermination de l'inhibition de la croissance de Lemna minor. 10pp.
Swedish Standards Institute (SIS). (1995). Vízminőség. A növekedésgátlás meghatározása (7-d) Lemna minor, békalencse. SS 02 82 13. 15pp. (svéd nyelven).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415–91 (Reapproved 1998). pp. 733–742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., „Public draft”. EPA 712-C-96–156. 8pp.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol., 10:1959–1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation and depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf., 5:87–96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem., 12:481–483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicity and synergism to floating aquatic weeds. Verh.-Int. Ver. Limnol., 19:2102–2111.
A Lemna-fajok beszerzési forrásai
|
University of Toronto Culture Collection of Algae and Cyanobacteria |
|
Department of Botany, University of Toronto |
|
Toronto, Ontario, Canada, M5S 3 B2 |
|
Tel. +1-416-978-3641 |
|
Fax.+1-416-978-5878 |
|
e-mail: jacreman@botany.utoronto.ca |
|
http://www.botany.utoronto.ca/utcc |
|
North Carolina State University |
|
Forestry Dept |
|
Duckweed Culture Collection |
|
Campus Box 8002 |
|
Raleigh, NC 27695-8002 |
|
United States |
|
phone 001 (919) 515-7572 |
|
astomp@unity.ncsu.edu |
|
Institute of Applied Environmental Research (ITM) Stockholm University |
|
SE-106 91 STOCKHOLM |
|
SWEDEN |
|
Tel. +46 8 674 7240 |
|
Fax +46 8 674 7636 |
|
Federal Environmental Agency (UBA) |
|
FG III 3.4 |
|
Schichauweg 58 |
|
12307 Berlin |
|
Germany |
|
e-mail: lemna@uba.de |
|
http://www.umweltbundesamt.de/contact.htm |
Irodalom
|
(1) |
Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental literature. The Botanical Review, 27:221–287. |
|
(2) |
Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland. |
|
(3) |
Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82–991150–0–0. The Agricultural Research Council of Norway, University of Oslo. |
|
(4) |
Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B, 11:1–14. |
|
(5) |
Wang, W. (1990). Literature review on duckweed toxicity testing. Environmental Research, 52:7–22. |
2. függelék
A törzstenyészetek tartása
A törzstenyészetek alacsony hőmérsékleteken (4–10 °C) hosszabb ideig átoltás nélkül is fenntarthatók. A Lemna tápoldata megegyezhet a vizsgálatok során alkalmazottal, de a törzstenyészetek más, tápanyagokban gazdag oldatban is tarthatók.
Időről időre néhány fiatal, világoszöld növényt csíramentes eljárással friss tápoldatot tartalmazó növesztőedénybe helyezünk. A javasolt hűvösebb körülmények között elegendő lehet akár háromhavonta is átoltást végezni.
Kémiailag tiszta (savval lemosott) és steril üveg növesztőedényeket alkalmazzunk, és a kezelés során biztosítsunk csíramentes körülményeket. A törzskultúra (például algával vagy gombákkal történő) fertőződése esetén meg kell kísérelni a szennyező organizmusok eltávolítását. Algák és a legtöbb más szennyező organizmus esetén ez felületi fertőtlenítéssel valósítható meg. A fertőződött növényanyagból mintát veszünk, és a gyökereket levágjuk. A növényeket ezután tiszta vízben intenzíven megrázzuk, majd 30 másodperc és 5 perc közötti időtartamra 0,5 térfogatszázalékos nátrium-hipoklorit-oldatba merítjük. A növényanyagot ezután steril vízben átöblítjük, majd több részletben friss tápoldatot tartalmazó növesztőedényekbe helyezzük. A kezelés eredményeképpen számos levél elhal, különösen hosszabb expozíciós idő esetén, de a fennmaradók közül néhány általában fertőzésmentes lesz. Ez a tenyészet a későbbiekben újabbak leoltására használható fel.
3. függelék
Tápoldatok
L. minor és L. gibba esetén eltérő tápoldatot ajánlott alkalmazni. L. minor esetén a svéd szabvány (SIS) szerinti tápoldat módosított változatát, míg L. gibba esetén a 20X–AAP tápoldatot javasoljuk. Az alábbiakban megadjuk minkét tápoldat összetételét. A tápoldatok elkészítése során reagens vagy analitikai tisztaságú vegyületeket és ionmentesített vizet kell alkalmazni.
Svéd szabvány (SIS) szerinti Lemna-tápoldat
|
— |
Az I–V. törzsoldatot autoklávozással (120 °C, 15 perc) vagy membránszűréssel (körülbelül 0,2 μm pórusméret) sterilizáljuk. |
|
— |
A VI. (és szükség esetén a VII.) törzsoldatot kizárólag membránszűréssel sterilizáljuk, autoklávozás nem alkalmazható. |
|
— |
A steril törzsoldatokat hideg, sötét helyen tároljuk. Az I–V. törzsoldatot hat hónap elteltével ki kell önteni, míg a VI. (és szükség esetén a VII.) törzsoldat eltarthatósági ideje egy hónap.
|
||||||||||||||||||||||||||||||||||||||||||||||||||
|
— |
Egy liter SIS szerinti tápoldat elkészítéséhez 900 ml ionmentesített vízhez a következőket adagoljuk:
Megjegyzés: A VII. törzsoldatot (MOPS puffer) bizonyos vizsgált anyagok esetében kell alkalmazni (lásd az 1.4. szakasz utolsó bekezdését). |
|
— |
A pH-értéket 0,1 vagy 1 mol HCl vagy NaOH hozzáadásával 6,5 ± 0,2 értékre állítjuk, majd a térfogatot ionmentesített víz hozzáadásával egy literre kiegészítjük. |
20X–AAP tápoldat
A törzsoldatokat steril desztillált vagy ionmentesített vízzel készítjük.
A steril törzsoldatokat hideg, sötét helyen tároljuk. Ilyen körülmények között eltarthatósági idejük legalább 6–8 hét.
A 20X–AAP tápoldat előállításához öt, tápanyagokat tartalmazó törzsoldatot (A1., A2., A3., B és C) készítünk reagens minőségű vegyületek felhasználásával. A tápoldat előállításához kb. 850 ml ionmentesített vízhez 20 ml-t adunk minden törzsoldatból. A pH-értéket 0,1 vagy 1 mol HCl vagy NaOH hozzáadásával 7,5 ± 0,1 értékre állítjuk, majd a térfogatot ionmentesített víz hozzáadásával egy literre kiegészítjük. Ezután a tápoldatot egy kb. 0,2 μm pórusméretű membránszűrőn át steril edénybe szűrjük.
A vizsgálatok céljára szánt tápoldatot egy-két nappal a felhasználás előtt kell elkészíteni annak érdekében, hogy pH-értékük stabilizálódhasson. A tápoldat pH-értékét felhasználás előtt ellenőrizni kell, és szükség esetén 0,1 vagy 1 mol HCl vagy NaOH hozzáadásával a fentieknek megfelelően újra be kell állítani.
|
Törzsoldat száma |
Anyag |
Koncentráció a törzsoldatban (g/l) (1) |
Koncentráció a kész tápoldatban (mg/l) (1) |
Kész tápoldat |
|
|
Kémiai elem |
Koncentráció (mg/l) (1) |
||||
|
A1. |
NaNO3 MgCl2·6H2O CaCl2·2H2O |
26 12 4,4 |
510 240 90 |
Na; N Mg Ca |
190; 84 58,08 24,04 |
|
A2. |
MgSO4·7H2O |
15 |
290 |
S |
38,22 |
|
A3. |
K2HPO4·3H2O |
1,4 |
30 |
K; P |
9,4; 3,7 |
|
B. |
H3BO3 MnCl2·4H2O FeCl3·6H2O Na2EDTA·2H2O ZnCl2 CoCl2·6H2O Na2MoO4·2H2O CuCl2·2H2O |
0,19 0,42 0,16 0,30 3,3 mg/l 1,4 mg/l 7,3 mg/l 0,012 mg/l |
3,7 8,3 3,2 6,0 66 μg/l 29 μg/l 145 μg/l 0,24 μg/l |
B Mn Fe – Zn Co Mo Cu |
0,65 2,3 0,66 – 31 μg/l 7,1 μg/l 58 μg/l 0,080 μg/l |
|
C. |
NaHCO3 |
15 |
300 |
Na; C |
220; 43 |
Lábjegyzet: Az elméletileg szükséges hidrogén-karbonát-koncentráció (amely mellett nincs szükség a pH-érték jelentős mértékű utánállítására) 15 mg/l, nem 300 mg/l. A 20X–AAP tápoldattal végzett korábbi vizsgálatok és a jelen módszerhez kapcsolódó körvizsgálat azonban egyaránt 300 mg/l-en alapult. [I. Sims, P. Whitehouse and R. Lacey. (1999) The OECD Lemna Growth Inhibition Test. Development and Ring-testing of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc – Environment Agency.]
STEINBERG-féle tápoldat (az ISO 20079 szerint)
Koncentrációk és törzsoldatok
|
— |
A módosított Steinberg-féle tápoldatot az ISO 20079 kizárólag Lemna minor esetére írja elő (mert a szabvány csak Lemna minor alkalmazását engedélyezi), de korábbi vizsgálatok Lemna gibba esetén is kedvező eredményekre vezettek. |
|
— |
A tápoldat készítése során reagens vagy analitikai tisztaságú anyagokat és ionmentesített vizet kell alkalmazni. |
|
— |
A tápoldatot elkészíthetjük törzsoldatokból vagy a tízszeres töménységű oldatból, amely maximális oldatkoncentrációt tesz lehetővé kicsapódás nélkül. |
1. táblázat
pH-stabilizált STEINBERG-féle tápoldat (Altenburger szerint módosítva)
|
Anyag |
Tápoldat |
||
|
Makroelemek |
móltömeg |
mg/l |
mmol/l |
|
KNO3 |
101,12 |
350,00 |
3,46 |
|
Ca(NO3)2 · 4H2O |
236,15 |
295,00 |
1,25 |
|
KH2PO4 |
136,09 |
90,00 |
0,66 |
|
K2HPO4 |
174,18 |
12,60 |
0,072 |
|
MgSO4 · 7H2O |
246,37 |
100,00 |
0,41 |
|
Mikroelemek |
móltömeg |
µg/l |
µmol/l |
|
H3BO3 |
61,83 |
120,00 |
1,94 |
|
ZnSO4 · 7H2O |
287,43 |
180,00 |
0,63 |
|
Na2MoO4 · 2H2O |
241,92 |
44,00 |
0,18 |
|
MnCl2 · 4H2O |
197,84 |
180,00 |
0,91 |
|
FeCl3 · 6H2O |
270,21 |
760,00 |
2,81 |
|
Dinátrium-EDTA-dihidrát |
372,24 |
1 500,00 |
4,03 |
2. táblázat
Törzsoldatok (makroelemek)
|
g/l |
||
|
1. törzsoldat: |
|||
|
KNO3 |
17,50 |
||
|
KH2PO4 |
4,5 |
||
|
K2HPO4 |
0,63 |
||
|
2. törzsoldat: |
|||
|
MgSO4 · 7H2O |
5,00 |
||
|
3. törzsoldat: |
|||
|
Ca(NO3)2 · 4H2O |
14,75 |
||
3. táblázat
Törzsoldatok (mikroelemek)
|
mg/l |
||
|
4. törzsoldat: |
|||
|
H3BO3 |
120,0 |
||
|
5. törzsoldat: |
|||
|
ZnSO4 · 7H2O |
180,0 |
||
|
6. törzsoldat: |
|||
|
Na2MoO4 · 2H2O |
44,0 |
||
|
7. törzsoldat: |
|||
|
MnCl2 · 4H2O |
180,0 |
||
|
8. törzsoldat: |
|||
|
FeCl3 · 6H2O |
760,00 |
||
|
Dinátrium-EDTA-dihidrát |
1 500,00 |
||
|
— |
A 2. és 3., illetve külön a 4–7. törzsoldat egyesíthető (a kívánt koncentrációk figyelembevételével). |
|
— |
A hosszabb eltarthatóság érdekében kezeljük a törzsoldatokat autoklávban 121 °C-on 20 percig, vagy végezzünk membránszűréses (0,2 µm) sterilizálást. A 8. törzsoldat esetében fokozottan ajánlott membránszűréses (0,2 µm) sterilizálást végezni. |
A végleges koncentrációjú (módosított) STEINBERG-féle tápoldat elkészítése
|
— |
A kicsapódás elkerülése érdekében mintegy 900 ml ionmentesített vízhez adjunk az 1., 2. és 3. törzsoldatból (2. táblázat) egyaránt 20 ml-t. |
|
— |
Az oldathoz adjunk a 4., 5., 6., 7. és 8. törzsoldatból (3. táblázat) egyaránt 1,0 ml-t. |
|
— |
A pH-érték 5,5 ± 0,2 legyen (ezt az értéket minél kevesebb NaOH-oldat vagy HCl hozzáadásával állítsuk be). |
|
— |
Víz hozzáadásával egészítsük ki az oldat térfogatát 1 000 ml-re. |
|
— |
Ha a törzsoldatok sterilizáltak és megfelelő minőségű vizet alkalmazunk, akkor további sterilizálás nem szükséges. Ha a végleges tápoldatot sterilizáljuk, a 8. törzsoldatot az autoklávozás (121 °C-on 20 percig) után kell hozzáadni. |
A 10-szeres töménységű (módosított) STEINBERG-féle tápoldat elkészítése ideiglenes tárolásra
|
— |
A kicsapódás elkerülése érdekében mintegy 30 ml vízhez adjunk az 1., 2. és 3. törzsoldatból (2. táblázat) egyaránt 20 ml-t. |
|
— |
Az oldathoz adjunk a 4., 5., 6., 7. és 8. törzsoldatból (3. táblázat) egyaránt 1,0 ml-t. Víz hozzáadásával egészítsük ki az oldat térfogatát 1 000 ml-re. |
|
— |
Ha a törzsoldatok sterilizáltak és megfelelő minőségű vizet alkalmazunk, akkor további sterilizálás nem szükséges. Ha a végleges tápoldatot sterilizáljuk, a 8. törzsoldatot az autoklávozás (121 °C-on 20 percig) után kell hozzáadni. |
|
— |
A (végleges koncentrációjú) tápoldat pH-értéke 5,5 ± 0,2 legyen. |
(1) Eltérő előírás hiányában.