|
8.
|
A B. rész a következő fejezetekkel egészül ki:
„B.63 REPRODUKCIÓS/FEJLŐDÉSI TOXICITÁSI SZŰRŐVIZSGÁLAT
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 421. vizsgálati iránymutatásában (2016) leírt módszerrel. A vegyi anyagok vizsgálatának OECD-iránymutatásait időszakonként felülvizsgálják a tudományos fejlődés fényében. A szűrővizsgálatról szóló eredeti 421. irányelvet 1995-ben fogadták el egy „előzetes reprodukciós toxicitási szűrővizsgálat” protokollja alapján, amelyet két, 1990-ben Londonban (1) és 1992-ben Tokióban (2) megrendezett szakértői találkozón vitattak meg.
|
|
2.
|
Az OECD által 1998-ban indított, a meglévő vizsgálati iránymutatások felülvizsgálatára, valamint az endokrin rendszert esetlegesen károsító anyagok kiszűrésére és vizsgálatára vonatkozó új vizsgálati iránymutatások kidolgozására irányuló, kiemelt fontosságú tevékenység (3) folyományaként e vizsgálati módszert frissítették az endokrin rendszert károsító anyagok tekintetében releváns végpontokkal. Az OECD 407. iránymutatását (Rágcsálókon végzett 28 napos, ismételt dózisú orális toxicitási vizsgálat, e melléklet B.7. fejezete) például 2008-ban továbbfejlesztették olyan paraméterek bevezetésével, amelyek alkalmasak a vizsgálati vegyi anyagok endokrin rendszerre gyakorolt hatásának kimutatására. A 421. vizsgálati iránymutatást abból a célból frissítették, hogy azokba a szűrővizsgálatokat érintő vizsgálati iránymutatásokba, amelyek expozíciós időszakai lefedik a fejlődés egyes érzékeny szakaszait (prenatális vagy korai posztnatális időszakokat), bekerüljenek bizonyos, az endokrin rendszert károsító anyagok tekintetében releváns végpontok.
|
|
3.
|
Az endokrin rendszert károsító anyagok kiválasztott további releváns végpontjait, amelyek részét képezik a 443. vizsgálati iránymutatásnak is (Kibővített egygenerációs reprodukciós toxicitási vizsgálat, e melléklet B.56. fejezete), belefoglalták a 421. vizsgálati iránymutatásba azon megvalósíthatósági tanulmány alapján, amely a belefoglalásukkal kapcsolatos tudományos és szakmai kérdésekkel, valamint a vizsgálati terv belefoglalásukhoz szükséges lehetséges módosításaival foglalkozott (4).
|
|
4.
|
E vizsgálati módszer célja, hogy korlátozott információkat szolgáltasson a vizsgálati vegyi anyagoknak a hím és női szaporodási teljesítményre, azon belül az ivarmirigyek működésére, a párzási viselkedésre, a fogantatásra, a konceptus fejlődésére és az ellésre gyakorolt hatásairól. A módszer nem képezi a meglévő B.31., B.34., B.35. és B.56. vizsgálati módszer alternatíváját, és nem helyettesíti azokat.
|
ALAPVETŐ MEGFONTOLÁSOK
|
5.
|
Ez a szűrővizsgálati módszer arra használható, hogy kiindulási pontként szolgáló információkat nyújtson a szaporodásra és/vagy a fejlődésre gyakorolt hatásokról, akár a vegyi anyagokra jellemző toxikológiai tulajdonságok felmérésének korai szakaszában, akár az aggodalomra okot adó vegyi anyagok esetében. Használható emellett olyan létező vegyi anyagok kezdeti szűrővizsgálatainak részeként, amelyekről nem vagy csak csekély mértékben állnak rendelkezésre toxikológiai adatok, továbbá szélesebb körű reprodukciós/fejlődési vizsgálatok dózistartomány-kereső vizsgálataként és egyéb releváns esetekben. A vizsgálatok elvégzése során a klinikai tüneteknek, mint a biztonsági értékeléshez használt kísérleti állatok humánus végpontjainak felismeréséről, értékeléséről és használatáról szóló 19. OECD-iránymutatásban (5) ismertetett vezérelveket és szempontokat kell követni.
|
|
6.
|
Ez a vizsgálati módszer nem nyújt teljes körű tájékoztatást a szaporodás és a fejlődés minden vonatkozásáról. Alkalmazhatósága különösen a prenatális expozíció posztnatális tüneteinek, valamint a posztnatális expozíció során előidézett hatások kimutatása tekintetében korlátozott. A dóziscsoportokat alkotó állatok viszonylag csekély száma, a végpontok szelektivitása, valamint a vizsgálat rövid időtartama (és egyéb okok) miatt ez a módszer nem szolgáltat bizonyítékot annak végleges megállapítására, hogy az adott vegyi anyag nem gyakorol hatást. Ezen túlmenően, más reprodukciós/fejlődési toxicitási vizsgálatokból nyert adatok hiányában a pozitív eredmények a veszély kezdeti értékelésére használhatók, illetve hozzájárulnak a további vizsgálatok szükségességére és időzítésére vonatkozó döntésekhez.
|
|
7.
|
Az endokrin rendszerrel kapcsolatos paraméterek alkalmazásával kapott eredményeket az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére vonatkozó fogalmi keretének összefüggésében kell értelmezni (6). Ebben a fogalmi keretben az OECD továbbfejlesztett 421. vizsgálati iránymutatása a 4. szinten helyezkedik el, mint olyan in vivo vizsgálat, amely az endokrin rendszer releváns végpontjaira gyakorolt káros hatásokról szolgáltat adatokat. Egy endokrin jel önmagában azonban még nem tekinthető elegendő bizonyítéknak arra, hogy a vizsgálati vegyi anyag az endokrin rendszert károsító anyag lenne.
|
|
8.
|
Ez a vizsgálati módszer a vizsgálati vegyi anyag orális adagolását feltételezi. Más beadási mód alkalmazása esetén szükség lehet a módszer kiigazítására.
|
|
9.
|
A vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény.
|
|
10.
|
A használt fogalmak meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLAT ELVE
|
11.
|
A vizsgálati vegyi anyagot fokozatosan növekvő dózisokban kell adagolni hím és nőstény állatok több csoportjának. A hímeket legalább négy hétig kell kezelni, egészen a tervezett elpusztításukat megelőző napig (ez az időtartam magában foglalja a pároztatást megelőző minimum két hetet, a párzási időszakot, valamint a pároztatást követő mintegy két hetes időszakot). Tekintettel a hímek pároztatást megelőző rövid kezelési időszakára, előfordulhat, hogy a termékenység nem kifejezetten érzékeny mutatója a heretoxicitásnak. Ezért a heréken mindenképpen részletes szövettani vizsgálatot kell végezni. A pároztatást megelőző kéthetes kezelési időszak és az azt követően a párzással/termékenységgel kapcsolatban tett megfigyelések, az összesen legalább négy hétig tartó kezelés, majd a hím ivarmirigyek részletes kórszövettani vizsgálata összességében elegendőnek bizonyulnak ahhoz, hogy kimutatható legyen a hím termékenységre és spermatogenezisre gyakorolt hatások többsége.
|
|
12.
|
A nőstényeket a vizsgálat teljes időszaka alatt kezelni kell. Ez magában foglalja a párzást megelőző két hetes időszakot (amelynek célja, hogy lefedjen legalább két teljes ivari ciklust), a fogantatáshoz szükséges változó időtartamot, a vemhesség időszakát, az ellést követő legalább tizenhárom napot és a tervezett elpusztítást megelőző napig tartó további kezelést.
|
|
13.
|
Az akklimatizációt és az ivari ciklus kezelés előtti felmérését követően megkezdett vizsgálat időtartama a nőstények teljesítményétől függően megközelítőleg 63 nap [legalább 14 napos pároztatás előtti időszak, (legfeljebb) 14 napos párzási időszak, 22 napos vemhesség és 13 napos szoptatás].
|
|
14.
|
Az adagolási időszakban az állatokat a toxicitás jeleinek észlelése érdekében minden nap gondosan megfigyelik. A vizsgálati időszak során elpusztult vagy elpusztított állatokat felboncolják, továbbá a vizsgálat befejezése után az életben maradt állatokat is elpusztítják és felboncolják.
|
A MÓDSZER LEÍRÁSA
Az állatfaj kiválasztása
|
15.
|
Ezt a vizsgálati módszert patkányok használatára tervezték. Ha az e vizsgálati módszerben meghatározott paramétereket más rágcsálófajon vizsgálják, a döntést részletesen indokolni kell. Az OECD (e melléklet B.7. fejezetének megfelelő) 407. vizsgálati iránymutatása alapján végzett nemzetközi validációs programban a patkány volt az endokrin rendszert károsító anyagok kimutatására használt egyetlen faj. Nem szabad olyan törzseket alkalmazni, amelyek kevéssé termékenyek, vagy amelyekben magas a fejlődési defektusok előfordulási gyakorisága. Korábban vizsgálatban még részt nem vett, egészséges szűz állatokat kell használni. A kísérleti állatokat faj, törzs, nem, súly és kor szerint kell jellemezni. A vizsgálat kezdetén az állatok tömege közötti eltérésnek minimálisnak kell lennie és nem haladhatja meg az ivaronkénti átlag ± 20 %-át. Ha a vizsgálatot egy hosszú távú vagy teljes generáción ál tartó vizsgálat előzetes kísérleteként végzik, ajánlatos mindkét vizsgálathoz ugyanabból a törzsből való, azonos eredetű állatokat használni.
|
Az állatok tartása és etetése
|
16.
|
Minden eljárásnak meg kell felelnie a laboratóriumi állatgondozásra vonatkozó helyi szabványoknak. A kísérleti állatok tartására szolgáló helyiség hőmérsékletének 22°C-nak (±3°C) kell lennie. Noha a helyiség relatív páratartalmának legalább 30 %-nak kell lennie, és az – a takarítás időtartamától eltekintve – lehetőség szerint nem haladhatja meg a 70 %-ot, a célértéknek 50–60 % között kell lennie. A világítás legyen mesterséges, 12 órás világos és 12 órás sötét periódusok váltsák egymást. Az etetéshez standard laboratóriumi takarmány alkalmazható, korlátlan mennyiségű ivóvíz biztosítása mellett. A takarmány megválasztását a vizsgálati vegyi anyaggal való megfelelő keveredés igénye is befolyásolhatja, ha a vizsgálati vegyi anyagot a takarmánnyal adják be.
|
|
17.
|
Az állatok azonos nemű egyedekből álló kisebb csoportokban tartandók; ha tudományos szempontból indokolt, az állatok egyedileg is elhelyezhetők. Ketrecben való csoportos elhelyezés esetén egy ketrecben legfeljebb öt állat tartható. A pároztatást erre a célra alkalmas ketrecekben kell végezni. A vemhes nőstényeket egyedileg kell elhelyezni, és el kell látni az alom kialakításához használt (fészeképítő) anyaggal. A szoptató nőstények és utódaik számára külön ketrecet kell biztosítani.
|
|
18.
|
Rendszeresen ellenőrizni kell, hogy a takarmány nem tartalmaz-e szennyező anyagokat. A takarmányból egy mintát a jelentés véglegesítéséig meg kell őrizni.
|
Az állatok előkészítése
|
19.
|
Egészséges, fiatal, ivarérett állatokat osztunk véletlenszerűen a kontroll- és kezelt csoportokba. A ketreceket úgy kell elrendezni, hogy minimálisak legyenek a ketrec elhelyezéséből adódó esetleges hatások. Az állatokat az egyedi azonosíthatóság céljából megjelölik, és a vizsgálat megkezdése előtt legalább öt napig a ketrecükben tartják a laboratóriumi körülményekhez való szoktatás céljából.
|
A dózisok előkészítése
|
20.
|
A vizsgálati vegyi anyagot ajánlatos orálisan adagolni, kivéve ha más beadási módot tartanak megfelelőbbnek. Orális adagolás esetén a vizsgálati vegyi anyag beadása általában gyomorszondával történik; a vizsgálati vegyi anyagok azonban beadhatók a takarmánnyal vagy az ivóvízzel is.
|
|
21.
|
A vizsgálati vegyi anyagot szükség esetén alkalmas vivőanyagban feloldják vagy szuszpendálják. Lehetőség szerint elsősorban vizes oldatot/szuszpenziót ajánlatos alkalmazni, másodsorban olajos (például kukoricacsíra-olajban elkészített) oldatot vagy emulziót, és csak harmadsorban más vivőanyagokban elkészített oldatot. A víztől eltérő vivőanyagok esetében ismerni kell a vivőanyag toxikológiai tulajdonságait. Meg kell határozni továbbá azt is, hogy a vizsgálati vegyi anyag mennyire stabil és homogén az adott vivőanyagban.
|
ELJÁRÁS
Az állatok száma és neme
|
22.
|
Az egyes csoportok kiindulási létszámát ajánlatos 10 hím és 12–13 nőstény egyedben meghatározni. Az expozíciót megelőzően felmérik a nőstények ivari ciklusát, és a szokásos 4–5 naptól eltérő időtartamú ciklussal rendelkező állatokat nem használják a vizsgálatban; ezért célszerű néhány plusz nőstényről gondoskodni, hogy végül maradjon csoportonként 10 nőstény. Ha nem mutatkoznak jelentős toxikus tünetek, ezzel a létszámmal várhatóan csoportonként legalább 8 vemhes nőstény biztosítható, ami a vemhes nőstények csoportonkénti elfogadható számának alsó határértéke. A cél az, hogy megfelelő mennyiségű vemhesség és utód jöjjön létre ahhoz, hogy érdemileg fel lehessen mérni a vizsgálati vegyi anyag hatását a termékenységre, a vemhességre, az anyai és a szopási viselkedésre, valamint az F1 utódnemzedék növekedésére és fejlődésére a fogantatástól az ellést követő 13. napig.
|
Adagolás
|
23.
|
Általánosan 3 kezelt csoportot és egy kontrollcsoportot alkalmaznak. A dózisok alapulhatnak az akut toxicitási vizsgálatok adatain vagy az ismételt dózisú vizsgálatok eredményein. A kontrollcsoportba tartozó állatokat a vizsgálati vegyi anyag beadásától eltekintve ugyanolyan módon kell kezelni, mint a vizsgálati csoportokban lévőket. Ha a vizsgálati vegyi anyag beadásához vivőanyagot alkalmaznak, a kontrollcsoportnak a vivőanyagból a felhasznált legnagyobb mennyiséget kell kapnia.
|
|
24.
|
A dózisok kiválasztásánál figyelembe veszik a toxicitásra és (toxiko)kinetikára vonatkozóan rendelkezésre álló adatokat. Emellett ügyelni kell arra is, hogy érzékenység tekintetében különbségek lehetnek a vemhes és nem vemhes állatok között. A legmagasabb dózist úgy kell kiválasztani, hogy az toxikus tüneteket keltsen, de ne okozzon halált vagy súlyos szenvedést a kísérleti állatoknak. Ezt követően úgy kell csökkenő sorrendben kiválasztani a kisebb dózisokat, hogy meg lehessen határozni a dózissal összefüggő válaszokat, illetve a legkisebb dózisnál a nem észlelhető káros hatások szintjét (NOAEL). A csökkenő dózisok beállításához gyakran optimális a 2–4-szeres intervallumok alkalmazása, a dózisok közötti nagyon nagy intervallumok (például 10-nél magasabb szorzófaktor) alkalmazásával szemben pedig gyakran előnyben részesítendő egy negyedik vizsgálati csoport beiktatása.
|
|
25.
|
Ha általános toxicitást (például testsúlycsökkenést, a májra, a szívre, a tüdőre vagy a vesére kifejtett hatásokat stb.) vagy egyéb, nem feltétlenül toxikus reakcióra utaló változásokat (például csökkent táplálékfogyasztást, májmegnagyobbodást) figyelnek meg, az endokrin rendszer szempontjából érzékeny végpontok tekintetében megfigyelt hatások értelmezésekor óvatosan kell eljárni.
|
Határérték-vizsgálat
|
26.
|
Ha egy egyetlen, legalább 1 000 mg/testtömeg-kg/nap orális dózis felhasználásával végzett, vagy táplálékkal vagy ivóvízzel történő beadás esetén a táplálékban vagy ivóvízben ezzel egyenértékű százalékos arány alkalmazásával történő és az itt ismertetett eljárások szerint lefolytatott vizsgálat nem idéz elő észlelhető toxikus hatást, és ha a szerkezet tekintetében rokon anyagokra vonatkozó adatok alapján nem is várható a toxicitás bekövetkezése, a teljes, több dózist átfogó tanulmány elvégzése szükségtelennek tekinthető. Határérték-vizsgálatot kell minden esetben alkalmazni, kivéve, ha a humán expozíció magasabb orális dózis alkalmazását teszi szükségessé. Más típusú beadási módok, így például inhalálás vagy dermális alkalmazás esetén a vizsgálati vegyi anyag fizikai-kémiai tulajdonságai gyakran előre jelezhetik a lehetséges koncentráció maximumát.
|
A dózisok beadása
|
27.
|
Az állatoknak a vizsgálati vegyi anyagot heti hét napon át, naponta adagolják. Ha a vizsgálati vegyi anyagot szondán át adagolják, lehetőleg egyetlen adagban kell gyomorszondán vagy megfelelő intubációs kanülön át beadni. Az egy alkalommal beadható folyadék maximális mennyisége a kísérleti állat méretétől függ. A térfogat nem haladhatja meg az 1 ml/100 g testtömegarányt, kivéve vizes oldatok esetében, amikor 2 ml/100 g testtömegarány is alkalmazható. A magasabb koncentrációértékek esetén rendszerint fokozottabb hatást kiváltó irritatív vagy korrozív vizsgálati vegyi anyagok kivételével a koncentráció megfelelő beállításával a minimálisra kell csökkenteni a vizsgálati vegyi anyag mennyiségének változásait, hogy a mennyiség valamennyi dózisnál konstans legyen.
|
|
28.
|
A takarmánnyal vagy ivóvízzel adagolt vizsgálati vegyi anyagok esetében fontos annak biztosítása, hogy a vizsgálati vegyi anyag beadott mennyisége ne zavarja meg a szokásos táplálékfelvételt vagy a vízháztartás egyensúlyát. Ha a vizsgálati vegyi anyagot a táplálékkal együtt adják be, vagy állandó koncentrációban (ppm) keverik be, vagy az állat testsúlyától függő, állandó nagyságú dózisban adagolják; a kiválasztott alternatívát meg kell nevezni. A gyomorszondával bejuttatott vizsgálati vegyi anyagok esetében a dózist mindig a nap hasonló időszakában kell beadni, és legalább hetente újra be kell állítani ahhoz, hogy az állat testtömegéhez viszonyítva állandó értéken lehessen tartani.
|
A kísérleti program
|
29.
|
Az adagolást mindkét nem esetében legalább 2 héttel a pároztatás előtt el kell kezdeni, miután az állatok legalább öt napon keresztül akklimatizálódtak, és (egy kéthetes kezelés előtti időszak során) ellenőrizték, hogy a nőstények ivari ciklusa megfelel-e a normál ciklusidőnek. A vizsgálatot úgy kell időzíteni, hogy az ivari ciklus értékelése röviddel a teljes ivarérettség elérését követően megkezdődjön. Ez az egyes laboratóriumokban használt különféle patkánytörzsek függvényében némileg változó, a Sprague-Dawley törzsbe tartozó patkányok esetében például 10 hetes korukban, a Wistar-törzsbe tartozók esetében pedig 12 hetes korukban következik be. Az utódokkal rendelkező anyaállatokat az ellést követő 13. napon vagy röviddel azután el kell pusztítani. A születés napja (vagyis amikor lezárul az ellés) a post-partum 0. napja. Azokat a nőstényeket, amelyek semmi jelét nem mutatják annak, hogy párosodtak volna, a párzási időszak utolsó napját követő 24–26 nappal el kell pusztítani. Az adagolást a párzási időszakban sem szabad abbahagyni egyik nem esetében sem. A hímeket a párzási időszakot követően továbbra is kezelni kell, legalább a minimális, 28 napos adagolási időszak végéig. Ezt követően leölik, vagy ha úgy ítélik megfelelőnek, megtartják azokat, és egy esetleges második pároztatás céljából tovább folytatják kezelésüket.
|
|
30.
|
Az anyaállatok napi szintű kezelését folytatni kell a vemhesség teljes időszakán át egészen a post-partum 13. napjáig vagy az elpusztításukat megelőző napig. Azon vizsgálatoknál, amelyek keretében a vizsgálati vegyi anyagot inhalálás vagy dermális alkalmazás útján adják be, a kezelést folytatni kell legalább a vemhesség 19. napjáig, majd a lehető leghamarabb, de legkésőbb a 4. posztnatális napon újra kell kezdeni.
|
|
31.
|
A kísérleti ütemterv ábráját a 2. függelék tartalmazza.
|
A pároztatási eljárás
|
32.
|
Rendes esetben e vizsgálat során 1:1 (egy hím-egy nőstény) pároztatást kell alkalmazni. Ettől abban az esetben lehet eltekinteni, ha a hímek közül elpusztul valamelyik. A nőstényt egy adott hímmel kell összerakni, és addig kell együtt hagyni azokat, amíg meg nem győződtek a párzás megtörténtéről, vagy 2 hét el nem telik. Minden reggel meg kell vizsgálni, hogy a nőstényben észlelhető-e sperma vagy a hüvelydugó. A vemhesség 0. napja definíció szerint az a nap, amikor a párosodás tényét megerősítették (hüvelydugó vagy sperma figyelhető meg a nőstényben). Ha a pároztatás sikertelen volt, fontolóra lehet venni a nőstények újrapároztatását egy azonos csoportbeli és már sikeresen pároztatott hímmel.
|
Az alom mérete
|
33.
|
Az ellés utáni negyedik napon az egyes almok egyedszáma a létszám fölötti kölykök véletlenszerű eltávolításával módosítható, hogy almonként az eredmény – amennyire lehetséges – a használt patkánytörzsre jellemző ivaronkénti négy vagy öt utód legyen. Két létszám feletti kölyöktől vérmintát kell venni, egybe kell gyűjteni és fel kell használni a T4 szérumszintek meghatározásához. A kölykök szelektív, például a testtömeg vagy az anogenitális távolság (AGD) alapján történő eltávolítása nem elfogadható. Ha a hím és nőstény utódok száma nem teszi lehetővé a nemenként négy vagy öt azonos nemű utódot tartalmazó almok kialakítását, részleges kiegyenlítés is elfogadható (például hat hím és négy nőstény). Amennyiben az alom egyedszáma a selejtezési célérték (almonként 8 vagy 10 utód) alá csökkenne, nem kell eltávolítani kölyköket. Ha az alom mérete csupán eggyel haladja meg a selejtezési célértéket, csak egy kölyköt távolítanak el, és attól vesznek vért a T4 szérumszintek esetleges értékelésekhez.
|
|
34.
|
Ha nem igazítják ki az alom egyedszámát, az ellést követő 4. napon almonként két kölyköt elpusztítanak, és vérmintát vesznek tőlük a pajzsmirigyhormonok szérumkoncentrációjának méréséhez. Lehetőség szerint az almonkénti két kölyök nőstény legyen, hogy maradjanak hímek az emlőbimbó-visszamaradás megállapításához, kivéve abban az esetben, ha a nőstény utódok eltávolítása miatt nem maradna nőstény a vizsgálat végi értékeléshez. Amennyiben az egyedszám az almonkénti 8 vagy 10 utód alá esne (a használt patkánytörzsre jellemző szokásos alommérettől függően), nem kell eltávolítani kölyköket. Ha az alom egyedszáma csupán eggyel haladja meg az alom szokásos méretét, csak egy kölyköt távolítanak el, és attól vesznek vért a T4 szérumszintek esetleges értékelésekhez.
|
Megfigyelések élő állatban
Klinikai megfigyelések
|
35.
|
A vizsgálat teljes időtartama alatt általános klinikai megfigyeléseket legalább naponta kell végezni, vagy gyakrabban, ha toxicitás jelei figyelhetők meg. A megfigyeléseket lehetőleg mindig a nap azonos időszakában/időszakaiban kell végezni, figyelembe véve azt is, hogy a várt hatások a dózis beadása után mikor érik el a maximumukat. Feljegyzést kell készíteni a lényeges magatartászavarokról, az elhúzódó vagy nehéz ellés jeleiről, valamint a toxikus hatásra utaló minden egyéb tünetről, beleértve a mortalitást is. A feljegyzéseknek tartalmazniuk kell a toxicitási tünetek fellépésének időpontját, mértékét és időtartamát.
|
Testtömeg és táplálék-/vízfogyasztás
|
36.
|
A hímek és nőstények testsúlyát meg kell mérni az adagolás első napján, majd ezt követően legalább hetente és a vizsgálat végén meg kell ismételni a mérést. A nőstényeket a vemhesség alatt a 0., 7., 14. és 20. napon kell megmérni, továbbá az ellést követő 24 órán belül (a post-partum 0. vagy 1. napján) és legalább a post-partum 4. és 13. napján. A megfigyeléseket minden egyes felnőtt állat esetében külön-külön kell rögzíteni.
|
|
37.
|
A pároztatás előtt, valamint a vemhesség és a szoptatás ideje alatt a takarmányfogyasztást legalább hetente kell mérni. A párzási időszak során nem kell feltétlenül mérni a takarmányfogyasztást. Ha a vizsgálati vegyi anyagot az ivóvízzel adagolják, ezen időszakok során a vízfogyasztást is mérni kell.
|
Ivari ciklus
|
38.
|
A kezelés megkezdése előtt ellenőrizni kell az ivari ciklust, hogy szabályos ciklussal rendelkező nőstények kerüljenek kiválasztásra a vizsgálathoz (lásd a 22. pontot). Emellett napi szinten kell vizsgálni a hüvelyi kenetet is, a kezelési időszak kezdetétől mindaddig, amíg be nem bizonyosodik, hogy megtörtént a párzás. Ha felmerül a lehetősége annak, hogy az adagolás megkezdése olyan akut stresszhatást idéz elő, amely módosíthatja az ivari ciklust, a laboratóriumok eljárhatnak úgy is, hogy két hétig kezelik a vizsgált állatokat, majd a pároztatás előtti időszakban legalább két hétig naponta hüvelyi kenetet vesznek az ivari ciklus ellenőrzéséhez, amelyet a párzási időszak során is folyamatosan figyelemmel kísérnek egészen a párzás megtörténtéig. A hüvelyi/méhnyaki sejtvétel során Ügyelni kell arra, hogy meg ne sérüljön a nyálkahártya, mert az álvemhességet idézhet elő (7) (8).
|
Az utódok paraméterei
|
39.
|
A vemhesség nulladik napjától számítandó terhességi (gesztációs) időszakról feljegyzést kell készíteni. Az ellés után minden almot a lehető leghamarabb meg kell vizsgálni a kölykök számának és ivarának, a halvaszületések, élveszületések és fejletlen egyedek (a megfelelő kontrollcsoportba tartozó kölyköknél jelentősen kisebb utódok) számának, valamint a súlyos rendellenességek jelenlétének megállapításához.
|
|
40.
|
Az élő kölyköket meg kell számolni, nemüket meg kell állapítani, és az almok egyedeinek súlyát meg kell mérni a születést követő 24 órán belül (a post-partum 0. vagy 1. napján) és legalább a post-partum 4. és 13. napján. A 35. pontban említett megfigyelések mellett az utódok minden rendellenes viselkedéséről be kell számolni.
|
|
41.
|
Az anogenitális távolságot (AGD) minden utódon legalább egy alkalommal meg kell mérni a 0. és 4. posztnatális nap között. A kölykök testtömegét azon a napon kell megmérni, amikor az AGD-t is mérik, és az AGD-t normálni kell a kölykök méretére, ami lehetőleg a testtömeg köbgyöke (9). Az OECD 151. vizsgálati iránymutatásának (10) ajánlása szerint a hím utódok emlőbimbóinak/emlőbimbóudvarainak számát meg kell számolni a 12. vagy 13. posztnatális napon.
|
Klinikai biokémiai vizsgálatok
|
42.
|
Vérmintákat egy meghatározott helyről kell venni, az alábbi ütemezés szerint:
|
—
|
almonként legalább két kölyöktől a születést követő 4. napon, amennyiben azt az utódok száma lehetővé teszi (lásd a 33–34. pontot),
|
|
—
|
az összes anyaállattól és almonként legalább két kölyöktől a vizsgálat végét jelentő 13. napon, és
|
|
—
|
az összes felnőtt hímtől a vizsgálat végén,
|
|
A vérmintákat megfelelő körülmények között tárolják. A 13 napos utódoktól és a felnőtt hímektől vett vérminták alapján értékelik a pajzsmirigyhormon (T4) szérumszinteket. Szükség esetén a T4 további értékeléséhez felhasználhatók az anyaállatoktól és a 4 napos utódoktól vett vérminták. Ha a vizsgálat szempontjából lényeges, más hormonok is mérhetők. A pajzsmirigyhormon analízisekhez almonként egyesíteni lehet a kölyköktől vett vérmintákat. A pajzsmirigyhormonokra (T4 és TSH) vonatkozó mérésekhez lehetőleg a teljes vérmintát kell használni.
|
43.
|
A hormonok meghatározásának variabilitását és az azzal kapott abszolút koncentrációt az alábbi tényezők befolyásolhatják:
|
—
|
az elpusztítás időpontja, tekintettel a hormonkoncentrációk nap folyamán tapasztalható ingadozására,
|
|
—
|
az állatot érő indokolatlan stressz elkerülése érdekében alkalmazott elpusztítási mód, amely hatással lehet a hormonkoncentrációkra,
|
|
—
|
a hormonmeghatározásokhoz alkalmazott, standard görbéjük tekintetében esetlegesen eltérő vizsgálati készletek.
|
|
|
44.
|
A kifejezetten hormon-meghatározási célra szánt plazmamintákat a nap hasonló időpontjában kell gyűjteni. A kereskedelmi fogalomban kapható különböző vizsgálati készletek eltérő számszerű értékeket adnak a hormonkoncentrációk elemzése során.
|
Patológia
Makroszkópos boncolás
|
45.
|
A vizsgálat során bekövetkező elpusztítás vagy elhullás időpontjában makroszkóposan meg kell vizsgálni a felnőtt állatokban az esetleges rendellenességeket vagy patológiás elváltozásokat. Külön figyelmet kell szentelni a szaporítószerveknek. A beágyazódási helyek számát fel kell jegyezni. A boncolás napjának reggelén meg kell vizsgálni a hüvelyi kenetet az ivari ciklus aktuális szakaszának meghatározásához, hogy lehetővé váljon a korreláció felállítása a petefészkek kórszövettanával.
|
|
46.
|
Az összes hím felnőtt állat heréit és mellékheréit, prosztatáját és ondóhólyagjait a koaguláló mirigyekkel egy egységként meg kell tisztítani minden rátapadt szövettől, és a kiszáradás megelőzése érdekében a boncolást követően a lehető legrövidebb időn belül meg kell mérni nedves tömegüket. Ezen túlmenően további szervtömegmérések végezhetők hímeknél a végbélemelő és a bulbocavernosus izomra, a Cowper-mirigyekre és a glans penisre, nőstényeknél a páros petefészekre (nedves tömeg) és a méhre (valamint a méhnyakra) kiterjedőleg; ha ezeket a szerveket is bevonják a mérésbe, tömegüket a boncolás után lehető leghamarabb meg kell határozni.
|
|
47.
|
Az elhullott és a post-partum 13. napján vagy röviddel azután elpusztított utódokat legalább külsőleg alaposan meg kell vizsgálni, hogy láthatók-e rajtuk súlyos rendellenességek. Különös figyelmet kell szentelni a külső ivarszervek vizsgálatának, és ezen belül a rendellenes fejlődés jeleinek. A 13. napon almonként 1 hím és 1 nőstény utód pajzsmirigyét meg kell őrizni.
|
|
48.
|
Ezenfelül meg kell őrizni az összes felnőtt állat petefészkeit, heréit, a járulékos ivarszerveket (a méhet és a méhnyakat, a mellékheréket, a prosztatát és az ondóhólyagokat a koaguláló mirigyekkel), a pajzsmirigyet, valamint a makroszkópos vizsgálat során léziókat mutató szerveket. A herék és mellékherék rutinvizsgálatához a formalinos fixálás használata nem ajánlott. E szövetek elfogadható vizsgálati módszere a Bouin-féle vagy a módosított Davidson-féle fixáló használata (11). A tunica albugineán a fixálószer gyors bejutása érdekében tűvel óvatosan sekély szúrást lehet ejteni a szerv mindkét végén.
|
Kórszövettan
|
49.
|
Részletes szövettani vizsgálatot kell végrehajtani a legmagasabb dózissal kezelt csoportba és a kontrollcsoportba tartozó állatok petefészkein, heréin és mellékheréin (különös figyelmet fordítva a spermatogenezis szakaszaira és a here intersticiális sejtjeinek felépítésére). Amennyiben szükséges, vizsgálni lehet az utódoktól és a felnőtt állatoktól származó egyéb megőrzött szerveket, például a pajzsmirigyet. A pajzsmirigy súlya a fixálást követően is meghatározható. A megtisztítás a szövetkárosodás elkerülése érdekében ez esetben is igen óvatosan, kizárólag a fixálást követően végzendő el. A szövetkárosodás veszélyeztetheti a kórszövettani elemzést. Ha a legmagasabb dózissal kezelt csoport egyedei esetében elváltozások figyelhetők meg, a vizsgálatokat a többi, más dózissal kezelt csoport egyedeire is ki kell terjeszteni. A kórszövettani iránymutatás (11) további információkat tartalmaz az endokrin szövetek boncolásával, fixálásával, metszeteinek elkészítésével és kórszövettani vizsgálatával kapcsolatban.
|
ADATOK ÉS JELENTÉS
Adatok
|
50.
|
Az állatokra vonatkozóan egyedi adatsorokat kell felvenni. Emellett minden adatot táblázatos formában is össze kell foglalni, amelyben minden egyes vizsgálati csoport esetében fel kell tüntetni az állatok számát a vizsgálat kezdetén, a vizsgálat során elpusztult vagy humánus okok miatt exterminált állatok számát, az elhullások és a humánus okok miatti elpusztítások időpontját, a termékeny állatok számát, a vemhes nőstények számát, a toxicitás jeleit mutató állatok számát, a megfigyelt toxicitási tünetek leírását, ezen belül bármely toxikus tünet megjelenésének időpontját, valamint ezek időtartamát és súlyosságát, a kórszövettani elváltozások típusait és az almokra vonatkozó minden idevágó adatot. A 3. mellékletben közzéteszünk egy, a reprodukciós/fejlődési hatások értékelése során nagyon hasznosnak bizonyult táblázatos formájú, összefoglaló jelentést.
|
|
51.
|
A vizsgálat korlátozott terjedelme miatt a „szignifikancia” felmérésére szolgáló vizsgálatok formájában végzett statisztikai elemzések számos végpont, különösen a reprodukciós végpontok szempontjából csekély jelentőséggel bírnak. Amennyiben statisztikai elemzésekre támaszkodnak, még a vizsgálat megkezdése előtt olyan módszert kell választani, amelyik megfelel a vizsgált változók eloszlásának. Az anogenitális távolság és az emlőbimbó-visszamaradás statisztikai elemzésének az utódokra vonatkozó egyedi adatokon kell alapulnia, ugyanakkor figyelembe kell vinni az alom által gyakorolt hatásokat. Ahol megfelelő, az elemzés egysége az alom. A kölykök testsúlyára vonatkozó statisztikai elemzésnek a kölykök egyedi adatain kell alapulnia, ugyanakkor figyelembe kell vinni az alom által gyakorolt hatásokat. A csoportok alacsony egyedszáma miatt az esetleg rendelkezésre álló (pl. az alomméretre vonatkozó) korábbi kontrolladatok szintén hasznos segítséget nyújthatnak a vizsgálat értelmezéséhez.
|
Az eredmények értékelése
|
52.
|
E toxicitási vizsgálat eredményeit a megfigyelt hatások, valamint a boncolási és mikroszkópos eredmények szempontjából kell értékelni. Az értékelés során ki kell térni a vizsgálati vegyi anyag dózisa és a rendellenességek, ezen belül a súlyos léziók, az azonosított célszervek, a terméketlenség, a klinikai rendellenességek, a csökkent reproduktív teljesítmény és egyedszám, a testtömegváltozások, a mortalitásra gyakorolt hatások és egyéb toxikus hatások megléte vagy hiánya, illetve gyakorisága és súlyossága közötti összefüggésekre.
|
|
53.
|
A hímek rövid kezelési időszaka miatt a hímekre gyakorolt reprodukciós hatások felmérése során figyelembe kell venni a herék és mellékherék kórszövettanát, valamint a termékenységre vonatkozó adatokat. A szaporodásra/fejlődésre (pl. alomméretre, anogenitális távolságra, emlőbimbó-visszamaradásra, T4 szérumszintekre) vonatkozólag rendelkezésre álló történeti kontrolladatok szintén hasznos segítséget nyújthatnak a vizsgálat értelmezéséhez.
|
|
54.
|
A minőség-ellenőrzéshez javasolt történeti kontrolladatokat gyűjteni, a számszerű adatokhoz pedig kiszámítani a variációs együtthatót, különösen az endokrin rendszert károsító anyagok kimutatásával összefüggő paraméterek tekintetében. Ezek az adatok felhasználhatók összehasonlítási célokra a tényleges vizsgálatok kiértékelése során.
|
Vizsgálati jelentés
|
55.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálati vegyi anyag:
|
—
|
eredet, gyártási szám, felhasználási határidő, ha rendelkezésre áll;
|
|
—
|
a vizsgálati vegyi anyag stabilitása, ha ismert;
|
|
|
|
Egy összetevőből álló anyag:
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján
|
|
|
|
Több összetevőből álló anyag, UVCB-k és keverékek:
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai révén jellemezve.
|
|
|
|
Vivőanyag (ha van):
|
—
|
a vivőanyag megválasztásának indokolása, ha az nem víz.
|
|
|
|
Kísérleti állatok:
|
—
|
az állatok száma, kora és neme;
|
|
—
|
az állatok származása, tartási körülmények, takarmány stb.;
|
|
—
|
az egyes állatok testtömege a vizsgálat kezdetekor;
|
|
—
|
patkánytól eltérő faj alkalmazásának indokolása.
|
|
|
|
Vizsgálati körülmények:
|
—
|
a dózisok kiválasztásának indokolása;
|
|
—
|
a vizsgálati vegyi anyag formulázásával/a takarmány előkészítésével, az elért koncentrációval, a készítmény stabilitásával és homogenitásával kapcsolatos adatok;
|
|
—
|
a vizsgálati vegyi anyag beadására vonatkozó részletek;
|
|
—
|
átszámítás a vizsgálati vegyi anyagnak a takarmányban/ivóvízben meglévő koncentrációjáról (ppm) a tényleges dózisra (mg/kg testsúly/nap), ha ez alkalmazható;
|
|
—
|
a táplálék és víz minőségére vonatkozó részletek;
|
|
—
|
a selejtezés során a kölykök kiválasztására használat randomizálási eljárás részletes leírása, ha sor került selejtezésre.
|
|
|
|
Eredmények:
|
—
|
testsúly és annak változásai;
|
|
—
|
táplálék- és vízfogyasztásra vonatkozó adatok, ha rendelkezésre állnak;
|
|
—
|
a toxikus reakciók adatai nemenként és dózisonként, beleértve a termékenységet, a gesztációs időt és a toxicitás egyéb tüneteit is;
|
|
—
|
a vemhesség időtartama;
|
|
—
|
a szaporodásra, az utódokra, a posztnatális növekedésre stb. gyakorolt toxikus vagy egyéb hatások;
|
|
—
|
a klinikai megfigyelések jellege, súlyossága és időtartama (a változások visszafordíthatók-e vagy sem);
|
|
—
|
normális vagy abnormális ivari ciklust mutató felnőtt nőstények száma és a ciklus időtartama;
|
|
—
|
az élveszületések száma és a beágyazódás utáni veszteség;
|
|
—
|
az összes utód anogenitális távolsága (és az AGD-mérés napján rögzített testsúlyuk);
|
|
—
|
emlőbimbó-visszamaradás a hím utódoknál;
|
|
—
|
pajzsmirigyhormonok szintje 13 napos kölyköknél és felnőtt hímeknél (és ha mérték, az anyaállatoknál és 4 napos kölyköknél);
|
|
—
|
a szemmel jól látható rendellenességeket mutató utódok száma, a külső nemi szervek makroszkopikus értékelése, a fejletlen utódok száma;
|
|
—
|
a vizsgálat során bekövetkezett elhullás ideje, illetve hogy az állatok megélték-e a vizsgálat végét;
|
|
—
|
a beágyazódások száma, az alomméret és az almokban lévő egyedek méréskor rögzített testsúlya;
|
|
—
|
a testtömeg az elpusztításkor, valamint a szülők szervtömegadatai;
|
|
—
|
a kórszövettani leletek részletes ismertetése;
|
|
—
|
felszívódási adatok (ha vannak);
|
|
—
|
adott esetben az eredmények statisztikai feldolgozása.
|
|
Az eredmények értékelése.
Következtetések
|
Az eredmények értelmezése
|
56.
|
Ez a vizsgálat az ismételt dózisok útján történő kezeléssel összefüggésben kimutatható reprodukciós/fejlődési toxicitás értékelésére szolgál (lásd az 5. és a 6. pontot). Jelezheti a további vizsgálatok szükségességét, és iránymutatást nyújt a későbbi vizsgálatok kialakításához. A szaporodási és fejlődési eredmények értelmezésével kapcsolatban az OECD 43. iránymutatását kell alkalmazni (12). A rágcsálókon végzett endokrin és reprodukciós vizsgálatok szövettani értékeléséről szóló 106. OECD-iránymutatás (11) olyan információkkal szolgál az (endokrin) szervek és a hüvelyi kenet előkészítéséről és értékeléséről, amelyek segítséget nyújthatnak e vizsgálati iránymutatáshoz.
|
SZAKIRODALOM
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Available upon request at Organisation for Economic and Cooperation and Development, Paris.
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Kérésre elérhető a Gazdasági Együttműködési és Fejlesztési Szervezetnél, Párizs.
|
|
(3)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998. Kérésre elérhető a Gazdasági Együttműködési és Fejlesztési Szervezetnél, Párizs.
|
|
(4)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(5)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations. Series on Testing and Assessment, (No 19), Organisation for Economic Cooperation and Development, Paris.
|
|
(6)
|
OECD (2011). Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment(No 150), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(7)
|
Goldman, J.M., Murr A.S., Buckalew A.R., Ferrell J.M. and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(8)
|
Sadleir R.M.F.S (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(9)
|
Gallavan R.H. Jr, Holson J.F., Stump D.G., Knapp J.F. and Reynolds V.L. (1999). Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights, Reproductive Toxicology, 13: 383-390.
|
|
(10)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(11)
|
OECD (2009). Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No106), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(12)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
1. függelék
FOGALOMMEGHATÁROZÁSOK (LÁSD MÉG: OECD 150. IRÁNYMUTATÁSA (6))
Androgenitás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében természetes androgén hormonokhoz (például a tesztoszteronhoz) hasonló hatást fejtsen ki.
Antiandrogenitás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes androgén hormonok (például a tesztoszteron) hatását gátolja.
Antiösztrogenitás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes ösztrogén hormonok (például a 17ß-ösztradiol) hatását gátolja.
Antitiroid hatás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes pajzsmirigyhormon (például a T3) hatását gátolja.
Vegyi anyag: anyag vagy keverék.
Fejlődési toxicitás: reprodukciós toxicitás kifejeződése az utódokban pre-, peri-, posztnatális, szerkezeti vagy funkcionális rendellenességek formájában.
Adagolás: általános fogalom, amely a dózist, valamint az adagolás gyakoriságát és időtartamát foglalja magában.
Dózis: a vizsgálati vegyi anyag beadott mennyisége. A dózist a vizsgálati vegyi anyag súlyának és a kísérleti állat egységnyi testsúlyának napi hányadosaként (például mg/testsúlykilogramm/nap), vagy a takarmányban lévő állandó koncentrációként fejezik ki.
Nyilvánvaló toxicitás: általános fogalom, amely a toxicitás vizsgálati vegyi anyag beadását követően kialakuló egyértelmű tüneteire vonatkozik. E tüneteknek elegendőnek kell lenniük a veszély értékeléséhez, továbbá olyan szintet kell képviselniük, amelynél a beadott dózis növelése várhatóan a toxicitás súlyos jeleinek kialakulásához és valószínű mortalitáshoz vezet.
Termékenységkárosodás: a hím vagy női szaporodási funkciók vagy képesség terén jelentkező rendellenességeket jelenti.
Anyai toxicitás: a vemhes nőstényeket érő káros hatások, amelyek történhetnek specifikusan (közvetlen hatás) vagy nem specifikusan (közvetett hatás).
NOAEL: a „no-observed-adverse-effect level” (nem észlelhető káros hatásszint) rövidítése. Ez a legmagasabb dózis, amelynél még nem figyelhetők meg a kezelés okozta káros hatások.
Ösztrogenitás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes ösztrogén hormonokhoz (például a 17ß-ösztradiolhoz) hasonló hatást fejtsen ki.
Reprodukciós toxicitás: az utódoknál jelentkező káros hatásokat és/vagy a hím vagy női szaporodási funkciók vagy képesség csökkenését jelenti.
Vizsgálati vegyi anyag: az e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Tiroid hatás: egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes pajzsmirigyhormonhoz (például a T3-hoz) hasonló hatást gyakoroljon.
Validáció: tudományos folyamat, amelynek célja egy adott vizsgálati módszer működési követelményeinek és korlátjainak jellemzése, valamint megbízhatóságának és egy adott célhoz kapcsolódó relevanciájának igazolása.
2. függelék
A KÍSÉRLETI ÜTEMTERV ÁBRÁJA, AMELY JELZI A VIZSGÁLAT TELJES 14 NAPOS PÁRZÁSI IDŐSZAKON ALAPULÓ MAXIMÁLIS IDŐTARTAMÁT

3. függelék
TÁBLÁZATOS ÖSSZEFOGLALÓ JELENTÉS A SZAPORODÁSRA/FEJLŐDÉSRE GYAKOROLT HATÁSOKRÓL
|
MEGFIGYELÉSEK
|
ÉRTÉKEK
|
|
|
|
Adagolás (egységek)
|
0 (kontroll)
|
...
|
...
|
...
|
...
|
|
Kezdő párok (Sz)
|
|
|
|
|
|
|
Ivari ciklus (legalább az átlagos időtartam és az irreguláris ciklusok gyakorisága)
|
|
|
|
|
|
|
Bizonyítottan párosított nőstények (Sz)
|
|
|
|
|
|
|
Vemhességet elért nőstények (Sz)
|
|
|
|
|
|
|
Fogantatás napja 1–5. (Sz)
|
|
|
|
|
|
|
Fogantatás napja 6–...(
(21)
) (Sz)
|
|
|
|
|
|
|
Vemhesség ≤ 21 nap (Sz)
|
|
|
|
|
|
|
Vemhesség = 22 nap (Sz)
|
|
|
|
|
|
|
Vemhesség ≥ 23 nap (Sz)
|
|
|
|
|
|
|
Élve született utódokkal rendelkező anyaállatok (Sz)
|
|
|
|
|
|
|
Post-partum 4. napján élő utódokkal rendelkező anyaállatok (Sz)
|
|
|
|
|
|
|
Beágyazódások/anyaállat (átlag)
|
|
|
|
|
|
|
Élő utódok/anyaállat az ellésnél (átlag)
|
|
|
|
|
|
|
Élő utódok/anyaállat a 4. napon (átlag)
|
|
|
|
|
|
|
Ivararány (h/n) az ellésénél (átlag)
|
|
|
|
|
|
|
Ivararány (h/n) a 4. napon (átlag)
|
|
|
|
|
|
|
Alom egyedsúlya az ellésnél (átlag)
|
|
|
|
|
|
|
Alom egyedsúlya a 4. napon (átlag)
|
|
|
|
|
|
|
Utódok súlya születéskor (átlag)
|
|
|
|
|
|
|
Utódok súlya az AGD-mérés időpontjában (hímek átlaga, nőstények átlaga)
|
|
|
|
|
|
|
Utódok AGD-értéke ugyanazon a posztnatális napon, születés – 4. nap (hímek átlaga, nőstények átlaga, posztnatális nap feljegyzése)
|
|
|
|
|
|
|
Utódok súlya a 4. napon (átlag)
|
|
|
|
|
|
|
Hím utódok emlőbimbó-visszamaradása a 13. napon (átlag)
|
|
|
|
|
|
|
Utódok súlya a 13. napon (átlag)
|
|
|
|
|
|
|
|
|
RENDELLENES UTÓDOK
|
|
Anyaállatok 0-val
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok ≥ 2-vel
|
|
|
|
|
|
|
|
|
UTÓDOK ELVESZTÉSE
|
|
|
|
Prenatális/poszt-implantációs időszak (beágyazódások mínusz élveszületések száma)
|
|
Anyaállatok 0-vel
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok 2-vel
|
|
|
|
|
|
|
Anyaállatok ≥ 3-mal
|
|
|
|
|
|
|
|
|
Posztnatális időszak (élveszületések mínusz a 13. posztnatális napon élők száma)
|
|
Anyaállatok 0-vel
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok 2-vel
|
|
|
|
|
|
|
Anyaállatok ≥ 3-mal
|
|
|
|
|
|
B.64. KOMBINÁLT ISMÉTELT DÓZISÚ TOXICITÁSI VIZSGÁLAT ÉS REPRODUKCIÓS/FEJLŐDÉSI TOXICITÁSI SZŰRŐVIZSGÁLAT
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 422. vizsgálati iránymutatásában (2016) leírt módszerrel. A vegyi anyagok vizsgálatának OECD-iránymutatásait időszakonként felülvizsgálják a tudományos fejlődés fényében. A szűrővizsgálatról szóló eredeti 422. vizsgálati iránymutatást 1996-ban fogadták el a „kombinált ismételt dózisú és reprodukciós/fejlődési szűrővizsgálat” protokollja alapján, amelyet két, 1990-ben Londonban (1) és 1992-ben Tokióban (2) megrendezett szakértői találkozón vitattak meg.
|
|
2.
|
Ez a vizsgálati módszer két vizsgálatot ötvöz, egyfelől egy reprodukciós/fejlődési toxicitási szűrővizsgálatot, amely részben a már létező és nagy mennyiségben előállított vegyi anyagokra vonatkozó eredeti módszer alkalmazása során a tagállamokban szerzett tapasztalatokon, részben a pozitív kontrollanyagokkal végzett kísérletekben szerzett ismereteken alapul (3) (4), másfelől egy ismételt dózisú toxicitási vizsgálatot, amely összhangban áll az OECD 407. vizsgálati iránymutatásával (Rágcsálókon végzett 28 napos, ismételt dózisú orális toxicitási tanulmány, e melléklet B.7. fejezete).
|
|
3.
|
Az OECD által 1998-ban indított, a meglévő vizsgálati iránymutatások felülvizsgálatára, valamint az endokrin rendszert esetlegesen károsító anyagok kiszűrésére és vizsgálatára vonatkozó új vizsgálati iránymutatások kidolgozására irányuló, kiemelt fontosságú tevékenység (5) folyományaként e vizsgálati módszert frissítették az endokrin rendszert károsító anyagok tekintetében releváns végpontokkal. Ebben az összefüggésben az (e melléklet B.7. fejezetének megfelelő) 407. vizsgálati iránymutatást 2008-ban továbbfejlesztették olyan paraméterekkel, amelyek alkalmasak a vizsgálati vegyi anyagok endokrin rendszerre gyakorolt hatásának kimutatására. A 422. vizsgálati iránymutatást abból a célból frissítették, hogy azokba a szűrővizsgálatokat érintő vizsgálati iránymutatásokba, amelyek expozíciós időszakai lefedik a fejlődés egyes érzékeny szakaszait (prenatális vagy korai posztnatális időszakokat), bekerüljenek bizonyos, az endokrin rendszert károsító anyagok tekintetében releváns végpontok.
|
|
4.
|
Az endokrin rendszert károsító anyagok tekintetében releváns kiválasztott végpontokat, amelyek részét képezik a 443. vizsgálati iránymutatásnak is (Kibővített egygenerációs reprodukciós toxicitási vizsgálat, amely e melléklet B.56. fejezetének felel meg), belefoglalták a 422. vizsgálati iránymutatásba azon megvalósíthatósági tanulmány alapján, amely a belefoglalásukkal kapcsolatos tudományos és szakmai kérdésekkel, valamint a vizsgálati terv belefoglalásukhoz szükséges lehetséges módosításaival foglalkozott (6).
|
|
5.
|
E vizsgálati módszer célja, hogy korlátozott információkat szolgáltasson a vizsgálati vegyi anyagoknak a hím és női szaporodási teljesítményre, azon belül az ivarmirigyek működésére, a párzási viselkedésre, a fogantatásra, a konceptus fejlődésére és az ellésre gyakorolt hatásairól. A módszer nem képezi a meglévő B.31., B.34., B.35. és B.56. vizsgálati módszer alternatíváját, és nem helyettesíti azokat.
|
ALAPVETŐ MEGFONTOLÁSOK
|
6.
|
Egy vizsgálati vegyi anyag toxikus jellemzőinek értékelése és felmérése során az ismételt dózisú orális toxicitást azután lehet meghatározni, hogy az akut vizsgálatok kezdeti eredményei már rendelkezésre állnak. Ez a vizsgálat ismereteket szolgáltat azokról a lehetséges egészségügyi veszélyekről, amelyek egy viszonylag korlátozott időszakon keresztül ismétlődő expozíció következtében alakulhatnak ki. A módszer egy alapvető, ismételt dózisú toxicitási vizsgálatot foglal magában, amely a 90 napos vizsgálatot nem igénylő vegyi anyagok esetén (például abban az esetben, ha az előállított mennyiség nem halad meg bizonyos határértékeket), vagy egy hosszú távú vizsgálat előzetes vizsgálataként alkalmazható. A vizsgálatok elvégzése során a klinikai tüneteknek, mint a biztonsági értékeléshez használt kísérleti állatok humánus végpontjainak felismeréséről, értékeléséről és használatáról szóló 19. OECD-iránymutatásban (7) ismertetett vezérelveket és szempontokat kell követni.
|
|
7.
|
A módszer magában foglal továbbá egy reprodukciós/fejlődési toxicitási vizsgálatot, ezért használható arra is, hogy kiindulási pontként szolgáló információkat nyújtson a hím és női szaporodási teljesítményre, azon belül az ivarmirigyek működésére, a párzási viselkedésre, a fogantatásra, a konceptus fejlődésére és az ellésre gyakorolt lehetséges hatásokról, akár a vegyi anyagokra jellemző toxikológiai tulajdonságok felmérésének korai szakaszában, akár az érintett vegyi anyagok esetében. Ez a vizsgálati módszer nem nyújt teljes körű tájékoztatást a szaporodás és a fejlődés minden vonatkozásáról. Alkalmazhatósága különösen a prenatális expozíció posztnatális tüneteinek, valamint a posztnatális expozíció során esetlegesen előidézett hatásoknak a kimutatása tekintetében korlátozott. A végpontok szelektivitása, a vizsgálat rövid időtartama (és egyéb okok) miatt ez a módszer nem szolgáltat bizonyítékot annak végleges megállapítására, hogy az adott vegyi anyag nem gyakorol hatást a szaporodásra/fejlődésre. Ezen túlmenően, más reprodukciós/fejlődési toxicitási vizsgálatokból nyert adatok hiányában a pozitív eredmények a veszély kezdeti értékelésére használhatók, illetve hozzájárulnak a további vizsgálatok szükségességére és időzítésére vonatkozó döntésekhez.
|
|
8.
|
Az endokrin rendszerrel kapcsolatos paraméterek alkalmazásával kapott eredményeket az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére vonatkozó fogalmi keretének összefüggésében kell értelmezni (8). Ebben a fogalmi keretben az OECD továbbfejlesztett 422. vizsgálati iránymutatása a 4. szinten helyezkedik el, mint olyan in vivo vizsgálat, amely az endokrin rendszer releváns végpontjaira gyakorolt káros hatásokról szolgáltat adatokat. Egy endokrin jel önmagában azonban még nem tekinthető elegendő bizonyítéknak arra, hogy a vizsgálati vegyi anyag az endokrin rendszert károsító anyag lenne.
|
|
9.
|
A vizsgálati módszer emellett hangsúlyt fektet a neurológiai hatásokra mint különleges végpontra, valamint az állatok gondos klinikai megfigyelésének jelentőségére a lehető legtöbb információ megszerzése érdekében. A módszer segítségével azonosíthatók a neurotoxikus hatással rendelkező vegyi anyagok, ami aztán további alaposabb, ilyen irányú kutatást indokolhat. Mindezek mellett a módszer alapinformációkkal szolgálhat az immunológiai hatásokkal kapcsolatban is.
|
|
10.
|
Más szisztémás toxicitási, reprodukciós/fejlődési toxicitási, neurotoxicitási és/vagy immunotoxicitási vizsgálatokból nyert adatok hiányában a pozitív eredmények a veszély kezdeti értékelésére használhatók, illetve hozzájárulnak a további vizsgálatok szükségességére és időzítésére vonatkozó döntésekhez. A vizsgálat különösen hasznosnak bizonyulhat, ha az OECD szűrővizsgálati információs adatkészletének (SIDS) részeként alkalmazzák olyan létező vegyi anyagok értékelésére, amelyekről nem vagy csak csekély mértékben állnak rendelkezésre toxikológiai adatok, továbbá helyettesíthet két különálló vizsgálatot: az OECD 407. vizsgálati iránymutatásával egyenértékű (e melléklet B.7. fejezetében ismertetett) ismételt dózisú toxicitási vizsgálatot és az OECD 421. vizsgálati iránymutatásával egyenértékű (e melléklet B.63. fejezetében ismertetett) reprodukciós/fejlődési toxicitási vizsgálatot. Használható ezenkívül szélesebb körű reprodukciós/fejlődési vizsgálatok dózistartomány-kereső vizsgálataként és egyéb releváns esetekben.
|
|
11.
|
Általában feltételezhető, hogy érzékenység tekintetében különbségek vannak a vemhes és nem vemhes állatok között. Következésképpen a két vizsgálat külön-külön történő elvégzéséhez képest e kombinált vizsgálat keretében bonyolultabb lehet meghatározni olyan dózisokat, amelyek egyaránt alkalmasak az általános szisztémás toxicitás és a specifikus reprodukciós/fejlődési toxicitás felmérésére. Emellett előfordulhat, hogy az önállóan végzett ismételt dózisú vizsgálathoz viszonyítva a vizsgálati eredmények nehezebben értelmezhetők az általános szisztémás toxicitás szempontjából, különösen akkor, ha a vizsgálat során nem egy időben értékelik a szérumra és a kórszövettanra vonatkozó paramétereket. A kombinált szűrővizsgálat összetettebb jellege miatt annak végrehajtása jelentős tapasztalatot kíván a toxicitási vizsgálatok terén. xxxMásfelől viszont – amellett, hogy kevesebb számú kísérleti állat részvételét igényli – a kombinált vizsgálat hatékonyabb eszközt nyújthat a szaporodásra/fejlődésre gyakorolt közvetlen hatásoknak a másodlagos, egyéb (szisztémás) hatásoktól való elkülönítéséhez.
|
|
12.
|
Ebben a vizsgálatban az adagolási időszak hosszabb az ismételt dózisú vizsgálat szokásos 28 napos időtartamánál. Ugyanakkor csoportonként mindkét nemből kevesebb állat szükséges a vizsgálathoz ahhoz képest, mint amikor a reprodukciós/fejlődési toxicitási vizsgálat mellett végezik el a hagyományos 28 napos, ismételt dózisú vizsgálatot.
|
|
13.
|
Ez a vizsgálati módszer a vizsgálati vegyi anyag orális adagolását feltételezi. Más beadási mód alkalmazása esetén szükség lehet a módszer kiigazítására.
|
|
14.
|
A vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény.
|
|
15.
|
A használt fogalmak meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLAT ELVE
|
16.
|
A vizsgálati vegyi anyagot fokozatosan növekvő dózisokban kell adagolni hím és nőstény állatok több csoportjának. A hímeket legalább négy hétig kell kezelni, egészen a tervezett leölésüket megelőző napig (ez az időtartam magában foglalja a pároztatást megelőző minimum két hetet, a párzási időszakot, valamint a pároztatást követő mintegy két hetes időszakot). Tekintettel a hímek pároztatást megelőző rövid kezelési időszakára, előfordulhat, hogy a termékenység nem kifejezetten érzékeny mutatója a heretoxicitásnak. Ezért a heréken mindenképpen részletes szövettani vizsgálatot kell végezni. A pároztatást megelőző kéthetes kezelési időszak és az azt követően a párzással/termékenységgel kapcsolatban tett megfigyelések, az összesen legalább négy hétig tartó kezelés, majd a hím ivarmirigyek részletes kórszövettani vizsgálata összességében elegendőnek bizonyulnak ahhoz, hogy kimutatható legyen a hím termékenységre és spermatogenezisre gyakorolt hatások többsége.
|
|
17.
|
A nőstényeket a vizsgálat teljes időszaka alatt kezelni kell. Ez magában foglalja a párzást megelőző két hetes időszakot (amelynek célja, hogy lefedjen legalább két teljes ivari ciklust), a fogantatáshoz szükséges változó időtartamot, a vemhesség időszakát, az ellést követő legalább tizenhárom napot és a tervezett elpusztítást megelőző napig tartó további kezelést.
|
|
18.
|
Az akklimatizációt és az ivari ciklus kezelés előtti felmérését követően megkezdett vizsgálat időtartama a nőstények teljesítményétől függően megközelítőleg 63 nap [legalább 14 napos pároztatás előtti időszak, (legfeljebb) 14 napos párzási időszak, 22 napos vemhesség és 13 napos szoptatás].
|
|
19.
|
Az adagolási időszakban az állatokat a toxicitás jeleinek észlelése érdekében minden nap gondosan megfigyelik. A vizsgálat során elpusztult, illetve leölt állatokat felboncolják, illetőleg a vizsgálat befejezése után a túlélő állatokat is leölik és felboncolják.
|
A MÓDSZER LEÍRÁSA
Az állatfaj kiválasztása
|
20.
|
Ezt a vizsgálati módszert patkányok használatára tervezték. Ha ebben a 422. vizsgálati iránymutatásban meghatározott paramétereket más rágcsálófajon vizsgálják, a döntést részletesen indokolni kell. A 407. vizsgálati iránymutatás alapján végzett nemzetközi validációs programban a patkány volt az endokrin rendszert károsító anyagok kimutatására használt egyedüli faj. Nem szabad olyan törzseket alkalmazni, amelyek kevéssé termékenyek vagy amelyekben magas a fejlődési defektusok előfordulási gyakorisága. Korábban vizsgálatban még részt nem vett, egészséges szűz állatokat kell használni. A kísérleti állatokat faj, törzs, nem, súly és kor szerint kell jellemezni. A vizsgálat kezdetén az állatok tömege közötti eltérésnek minimálisnak kell lennie és nem haladhatja meg az ivaronkénti átlag ± 20 %-át. Ha a vizsgálatot egy hosszú távú vagy teljes generáción ál tartó vizsgálat előzetes kísérleteként végzik, ajánlatos mindkét vizsgálathoz ugyanabból a törzsből való, azonos eredetű állatokat használni.
|
Az állatok tartása és etetése
|
21.
|
Minden eljárásnak meg kell felelnie a laboratóriumi állatgondozásra vonatkozó helyi szabványoknak. A kísérleti állatok tartására szolgáló helyiség hőmérsékletének 22°C-nak (±3 °) kell lennie. A relatív páratartalomnak legalább 30 %-nak kell lennie, és a takarítás időtartamától eltekintve lehetőleg ne haladja meg a 70 %-ot. A világítás legyen mesterséges, 12 órás világos és 12 órás sötét periódusok váltsák egymást. Az etetéshez standard laboratóriumi takarmány alkalmazható, korlátlan mennyiségű ivóvíz biztosítása mellett. A takarmány megválasztását a vizsgálati vegyi anyaggal való megfelelő keveredés igénye is befolyásolhatja, ha a vizsgálati vegyi anyagot a takarmánnyal adják be.
|
|
22.
|
Az állatok azonos nemű egyedekből álló kisebb csoportokban tartandók; ha tudományos szempontból indokolt, az állatok egyedileg is elhelyezhetők. Ketrecben való csoportos elhelyezés esetén egy ketrecben legfeljebb öt állat tartható. A pároztatást erre a célra alkalmas ketrecekben kell végezni. A vemhes nőstényeket egyedileg kell elhelyezni, és el kell látni az alom kialakításához használt (fészeképítő) anyaggal. A szoptató nőstények és utódaik számára külön ketrecet kell biztosítani.
|
|
23.
|
Rendszeresen ellenőrizni kell, hogy a takarmány nem tartalmaz-e szennyező anyagokat. A takarmányból egy mintát a jelentés véglegesítéséig meg kell őrizni.
|
Az állatok előkészítése
|
24.
|
Az egészséges, fiatal, ivarérett állatokat véletlenszerű kiválasztás útján kell a kezelési csoportokba és ketrecekbe osztani. A ketreceket úgy kell elrendezni, hogy minimálisak legyenek a ketrec elhelyezéséből adódó esetleges hatások. Az állatokat az egyedi azonosíthatóság céljából megjelölik, és a vizsgálat megkezdése előtt legalább öt napig a ketrecükben tartják a laboratóriumi körülményekhez való szoktatás céljából.
|
A dózisok előkészítése
|
25.
|
A vizsgálati vegyi anyagot ajánlatos orálisan adagolni, kivéve ha más beadási módot tartanak megfelelőbbnek. Orális adagolás esetén a vizsgálati vegyi anyag beadása általában gyomorszondával történik; a vizsgálati vegyi anyagok azonban beadhatók a takarmánnyal vagy az ivóvízzel is.
|
|
26.
|
A vizsgálati vegyi anyagot szükség esetén alkalmas vivőanyagban feloldják vagy szuszpendálják. Lehetőség szerint elsősorban vizes oldatot/emulziót ajánlatos alkalmazni, másodsorban olajos (például kukoricacsíra-olajban elkészített) oldatot vagy emulziót, és csak harmadsorban más vivőanyagokban elkészített oldatot. A víztől eltérő vivőanyagok esetében ismerni kell a vivőanyag toxikológiai tulajdonságait. Meg kell határozni továbbá azt is, hogy a vizsgálati vegyi anyag mennyire stabil és homogén az adott vivőanyagban.
|
ELJÁRÁS
Az állatok száma és neme
|
27.
|
Az egyes csoportok kiindulási létszámát ajánlatos 10 hím és 12–13 nőstény egyedben meghatározni. Az expozíciót megelőzően felmérik a nőstények ivari ciklusát, és a szokásos 4–5 naptól eltérő időtartamú ciklussal rendelkező állatokat nem használják a vizsgálatban; ezért célszerű néhány plusz nőstényről gondoskodni, hogy végül maradjon csoportonként 10 nőstény. Ha nem mutatkoznak jelentős toxikus tünetek, ezzel a létszámmal várhatóan csoportonként legalább 8 vemhes nőstény biztosítható, ami a vemhes nőstények csoportonkénti elfogadható számának alsó határértéke. A cél az, hogy megfelelő mennyiségű vemhesség és utód jöjjön létre ahhoz, hogy érdemileg fel lehessen mérni a vizsgálati vegyi anyag hatását a termékenységre, a vemhességre, az anyai és a szopási viselkedésre, valamint az F1 utódnemzedék növekedésére és fejlődésére a fogantatástól az ellést követő 13. napig. Ha időközi leölést terveznek, a csoport létszámát a kísérlet során leölni szándékozott állatok számával meg kell növelni. Meg kell fontolni egy további, ivaronként öt állatból álló kísérő csoport alkalmazását a kontrollcsoportban és a legnagyobb adaggal kezelt csoportban, amelyen a kezelés időszakát követően legalább 14 napig megfigyelhető a szisztémás toxikus hatás visszafordíthatósága, tartóssága vagy késleltetett kialakulása. A kísérő csoportba tartozó állatok nem vesznek részt a pároztatásban, ezért nem használhatók a reprodukciós/fejlődési toxicitás értékelésére.
|
Adagolás
|
28.
|
Általánosan 3 kezelt csoportot és egy kontrollcsoportot alkalmaznak. Ha nem állnak rendelkezésre megfelelő általános toxikológiai adatok, az alkalmazandó dózisok meghatározása céljából tartománybehatároló vizsgálat végezhető (azonos törzsből való, azonos eredetű állatokon). A kontrollcsoportba tartozó állatokat a vizsgálati vegyi anyag beadásától eltekintve ugyanolyan módon kell kezelni, mint a vizsgálati csoportokban lévőket. Ha a vizsgálati vegyi anyag beadásához vivőanyagot alkalmaznak, a kontrollcsoportnak a vivőanyagból a felhasznált legnagyobb mennyiséget kell kapnia.
|
|
29.
|
A dózisok kiválasztásánál figyelembe veszik a toxicitásra és (toxiko)kinetikára vonatkozóan rendelkezésre álló adatokat. Emellett ügyelni kell arra is, hogy érzékenység tekintetében különbségek lehetnek a vemhes és nem vemhes állatok között. A legmagasabb dózist úgy kell kiválasztani, hogy az toxikus tüneteket keltsen, de ne okozzon halált vagy nyilvánvaló szenvedést a kísérleti állatoknak. Ezután az adagokat fokozatosan csökkentik, azzal a céllal, hogy kimutassák az adagolási szinttől függő hatásokat, illetve a legalacsonyabb adagolási szinten a káros hatások hiányát. Sokszor bizonyul optimálisnak a kettő–négyszeres intervallum alkalmazása, a nagyon nagy intervallumok (pl. több mint 10-szeres szorzófaktor) alkalmazása helyett pedig gyakran célszerű egy negyedik vizsgálati csoportot beiktatni.
|
|
30.
|
Ha általános toxicitást (például testsúlycsökkenést, a májra, a szívre, a tüdőre vagy a vesére kifejtett hatásokat stb.) vagy egyéb, nem feltétlenül toxikus reakcióra utaló változásokat (például csökkent táplálékfogyasztást, májmegnagyobbodást) figyelnek meg, az endokrin rendszer szempontjából érzékeny végpontok tekintetében megfigyelt hatások értelmezésekor óvatosan kell eljárni.
|
Határérték-vizsgálat
|
31.
|
Ha egy egyetlen, legalább 1 000 mg/testtömeg-kg/nap orális dózis felhasználásával végzett, vagy táplálékkal vagy ivóvízzel történő beadás esetén a táplálékban vagy ivóvízben ezzel egyenértékű (a testsúly alapján végzett számításokon alapuló) százalékos arány alkalmazásával történő és az itt ismertetett eljárások szerint lefolytatott vizsgálat nem idéz elő észlelhető toxikus hatást, és ha a szerkezet tekintetében rokon anyagokra vonatkozó adatok alapján nem is várható a toxicitás bekövetkezése, a teljes, több dózist átfogó tanulmány elvégzése szükségtelennek tekinthető. A határérték-vizsgálat alkalmazandó, kivéve, ha az emberi expozíció ennél nagyobb adagolási szint használatát indokolja. Más típusú beadási módok, így például inhalálás vagy dermális alkalmazás esetén a vizsgálati vegyi anyag fizikai-kémiai tulajdonságai gyakran előre jelezhetik az expozíció lehetséges maximumát.
|
A dózisok beadása
|
32.
|
Az állatoknak a vizsgálati vegyi anyagot heti hét napon át, naponta adagolják. Ha a vizsgálati vegyi anyagot szondán át adagolják, lehetőleg egyetlen adagban kell gyomorszondán vagy megfelelő intubációs kanülön át beadni. Az egy alkalommal beadható folyadék maximális mennyisége a kísérleti állat méretétől függ. A térfogat nem haladhatja meg az 1 ml/100 g testtömegarányt, kivéve vizes oldatok esetében, amikor 2 ml/100 g testtömegarány is alkalmazható. A magasabb koncentrációértékek esetén rendszerint fokozottabb hatást kiváltó irritatív vagy korrozív vizsgálati vegyi anyagok kivételével a koncentráció megfelelő beállításával a minimálisra kell csökkenteni a vizsgálati vegyi anyag mennyiségének változásait, hogy a mennyiség valamennyi dózisnál konstans legyen.
|
|
33.
|
A takarmánnyal vagy ivóvízzel adagolt vizsgálati vegyi anyagok esetében fontos annak biztosítása, hogy a vizsgálati vegyi anyag beadott mennyisége ne zavarja meg a szokásos táplálékfelvételt vagy a vízháztartás egyensúlyát. Ha a vizsgálati vegyi anyagot a táplálékkal együtt adják be, vagy állandó koncentrációban (ppm) keverik be, vagy az állat testsúlyától függő, állandó nagyságú dózisban adagolják; a kiválasztott alternatívát meg kell nevezni. A gyomorszondával bejuttatott vizsgálati vegyi anyagok esetében a dózist mindig a nap hasonló időszakában kell beadni, és legalább hetente újra be kell állítani ahhoz, hogy az állat testtömegéhez viszonyítva állandó értéken lehessen tartani. Ha a kombinált vizsgálatot egy hosszan tartó vagy teljes generáción át tartó toxicitási vizsgálat előkísérleteként végzik, a két vizsgálatban azonos étrendet alkalmaznak.
|
A kísérleti program
|
34.
|
Az adagolást mindkét ivar esetében 2 héttel a pároztatás előtt el kell kezdeni, miután az állatok legalább öt napon keresztül akklimatizálódtak, és (egy kéthetes kezelés előtti időszak során) ellenőrizték, hogy a nőstények ivari ciklusa megfelel-e a normál ciklusidőnek. A vizsgálatot úgy kell időzíteni, hogy az ivari ciklus értékelése röviddel a teljes ivarérettség elérését követően megkezdődjön. Ez az egyes laboratóriumokban használt különféle patkánytörzsek függvényében némileg változó, a Sprague-Dawley törzsbe tartozó patkányok esetében például 10 hetes korukban, a Wistar-törzsbe tartozók esetében pedig 12 hetes korukban következik be. Az utódokkal rendelkező anyaállatokat az ellést követő 13. napon vagy röviddel azután el kell pusztítani. Annak érdekében, hogy a vérvétel előtti éjszaka koplaljanak az anyaállatok (ha ez a kiválasztott módszer), nem feltétlenül szükséges ugyanazon a napon elpusztítani az anyaállatot és utódait. A születés napja (vagyis amikor lezárul az ellés) a post-partum 0. napja. Azokat a nőstényeket, amelyek semmi jelét nem mutatják annak, hogy párosodtak volna, a párzási időszak utolsó napját követő 24–26 nappal el kell pusztítani. Az adagolást a párzási időszakban sem szabad abbahagyni egyik nem esetében sem. A hímeket a párzási időszakot követően továbbra is kezelni kell, legalább a minimális, 28 napos adagolási időszak végéig. Ezt követően leölik, vagy ha úgy ítélik megfelelőnek, megtartják azokat, és egy esetleges második pároztatás céljából tovább folytatják kezelésüket.
|
|
35.
|
Az anyaállatok napi szintű kezelését folytatni kell a vemhesség teljes időszakán át egészen a post-partum 13. napjáig vagy az elpusztításukat megelőző napig. Azon vizsgálatoknál, amelyek keretében a vizsgálati vegyi anyagot inhalálás vagy dermális alkalmazás útján adják be, a kezelést folytatni kell legalább a vemhesség 19. napjáig, majd a lehető leghamarabb, de legkésőbb a 4. posztnatális napon (PNN) újra kell kezdeni.
|
|
36.
|
Amennyiben vizsgálatot követő megfigyelés céljából létrehoztak kísérő csoportot, a csoportba tartozó állatok nem vesznek részt a pároztatásban. Ezeket az állatokat az anyaállatok első tervezett leölését követően még legalább 14 napig tartják kezelés nélkül, hogy megfigyeljék a toxikus hatások késleltetett előfordulását, fennmaradását vagy visszafordíthatóságát.
|
|
37.
|
A kísérleti ütemterv ábráját a 2. függelék tartalmazza.
|
Ivari ciklus
|
38.
|
A kezelés megkezdése előtt ellenőrizni kell az ivari ciklust, hogy szabályos ciklussal rendelkező nőstények kerüljenek kiválasztásra a vizsgálathoz (lásd a 27. pontot). Emellett napi szinten kell vizsgálni a hüvelyi kenetet is, a kezelési időszak kezdetétől mindaddig, amíg be nem bizonyosodik, hogy megtörtént a párzás. Ha felmerül a lehetősége annak, hogy az adagolás megkezdése olyan akut stresszhatást idéz elő, amely módosíthatja az ivari ciklust, a laboratóriumok eljárhatnak úgy is, hogy két hétig kezelik a vizsgálati állatokat, majd a pároztatás előtti időszakban legalább két hétig naponta hüvelyi kenetet vesznek az ivari ciklus ellenőrzéséhez, amelyet a párzási időszak során is folyamatosan figyelemmel kísérnek egészen a párzás megtörténtéig. A hüvelyi/méhnyaki sejtvétel során ügyelni kell arra, hogy meg ne sérüljön a nyálkahártya, mert az álvemhességet idézhet elő (8) (9).
|
A pároztatási eljárás
|
39.
|
Rendes esetben e vizsgálat során 1:1 (egy hím-egy nőstény) pároztatást kell alkalmazni. Ettől abban az esetben lehet eltekinteni, ha a hímek közül elpusztul valamelyik. A nőstényt egy adott hímmel kell összerakni, és addig kell együtt hagyni azokat, amíg meg nem győződtek a párzás megtörténtéről, vagy 2 hét el nem telik. Minden reggel meg kell vizsgálni, hogy a nőstényben észlelhető-e sperma vagy a hüvelydugó. A vemhesség 0. napja definíció szerint az a nap, amikor a párosodás tényét megerősítették (hüvelydugó vagy sperma figyelhető meg a nőstényben). Ha a pároztatás sikertelen volt, fontolóra lehet venni a nőstények újrapároztatását egy azonos csoportbeli és már sikeresen pároztatott hímmel.
|
Az alom mérete
|
40.
|
Az ellés utáni negyedik napon az egyes almok egyedszáma a létszám fölötti kölykök véletlenszerű eltávolításával módosítható, hogy almonként az eredmény – amennyire lehetséges – a használt patkánytörzsre jellemző ivaronkénti négy vagy öt utód legyen. Két létszám feletti kölyöktől vérmintát kell venni, egybe kell gyűjteni és fel kell használni a T4 szérumszintek meghatározásához. A kölykök szelektív, például a testtömeg vagy az anogenitális távolság (AGD) alapján történő eltávolítása nem elfogadható. Ha a hím és nőstény utódok száma nem teszi lehetővé a nemenként négy vagy öt azonos nemű utódot tartalmazó almok kialakítását, részleges kiegyenlítés is elfogadható (például hat hím és négy nőstény). Amennyiben az alom egyedszáma a selejtezési célérték (almonként 8 vagy 10 utód) alá csökkenne, nem kell eltávolítani kölyköket. Ha az alom mérete csupán eggyel haladja meg a selejtezési célértéket, csak egy kölyköt távolítanak el, és attól vesznek vért a T4 szérumszintek esetleges értékelésekhez.
|
|
41.
|
Ha nem igazítják ki az alom egyedszámát, az ellést követő 4. napon almonként két kölyköt elpusztítanak, és vérmintát vesznek tőlük a pajzsmirigyhormonok szérumkoncentrációjának méréséhez. Lehetőség szerint az almonkénti két kölyök nőstény legyen, hogy maradjanak hímek az emlőbimbó-visszamaradás megállapításához, kivéve abban az esetben, ha a nőstény utódok eltávolítása miatt nem maradna nőstény a vizsgálat végi értékeléshez. Amennyiben az egyedszám az almonkénti 8 vagy 10 utód alá esne (a használt patkánytörzsre jellemző szokásos alommérettől függően), nem kell eltávolítani kölyköket. Ha az alom egyedszáma csupán eggyel haladja meg az alom szokásos méretét, csak egy kölyköt távolítanak el, és attól vesznek vért a T4 szérumszintek esetleges értékelésekhez.
|
Megfigyelések
|
42.
|
Naponta legalább egyszer, lehetőleg minden nap ugyanazon időpont(ok)ban általános klinikai megfigyelést kell végezni, figyelembe véve az adagolást követően várható hatások érvényesülésének csúcsidőszakát. Fel kell jegyezni a kísérleti állatok klinikai állapotát. Naponta legalább kétszer meg kell vizsgálni az összes állatot morbiditás és mortalitás szempontjából.
|
|
43.
|
Az első expozíció előtt egyszer (az egyedek közötti összehasonlítás érdekében), azt követően pedig legalább hetente egyszer az összes szülőt részletes klinikai vizsgálatoknak kell alávetni. E megfigyeléseket azokon a ketreceken kívül, ahol egyébként tartják őket, lehetőleg egy szabványos karámban, minden alkalommal a nap hasonló időszakában végzik el. A megfigyeléseket gondosan feljegyzik; lehetőleg a vizsgálatot végző laboratórium által meghatározott pontozásos rendszer segítségével. Törekedni kell arra, hogy minimálisak legyenek a kísérleti körülmények közötti eltérések, és hogy a megfigyeléseket lehetőleg olyan megfigyelők végezzék, akik nem tudnak a kezelésről. A feljegyzett észleléseknek ki kell terjedniük többek között a bőr, a szőrzet, a szemek és a nyálkahártyák állapota, a váladékok és kiválasztott anyagok előfordulása, valamint az autonóm aktivitás (például könnyezés, szőrzet felborzolódása, pupillaméret, szokatlan légzési ritmusok) tekintetében megfigyelhető változásokra. A járás, a testtartás és a kézbevételre adott reakció megváltozását, valamint a klónusos vagy tónusos görcsök, sztereotip viselkedés (például túlzott mosdás, megkergülés), az elhúzódó vagy nehéz ellés, illetve bármilyen bizarr viselkedésmód (például öncsonkítás, hátrafelé járás) megfigyelt eseteit is fel kell jegyezni (10).
|
|
44.
|
A vizsgálat során egy alkalommal minden csoportból véletlenszerűen kiválasztott öt hímen és öt nőstényen meg kell figyelni a különféle (pl. hallószervi, vizuális, proprioceptív) ingerekre adott szenzoros reaktivitást (8) (9) (11), meg kell mérni a fogóerőt (12) és értékelni kell a motoros aktivitást (13). A követendő eljárásokkal kapcsolatban további részletek a vonatkozó szakirodalomban találhatók. Ettől függetlenül a hivatkozottaktól eltérő alternatív eljárások is alkalmazhatók. A hímek esetében ezeket a funkcionalitási megfigyeléseket az adagolási időszak vége felé, röviddel tervezett leölésük előtt, de még a hematológiai és klinikai kémiai célú vérvételt megelőzően kell elvégezni (lásd az 53–56. pontot és az 1. lábjegyzetet). A nőstényeknek fiziológiai szempontból hasonló állapotban kell lenniük a funkcionalitási vizsgálatok során, ezért célszerű vizsgálatukat röviddel tervezett leölésük előtt, a szoptatás utolsó hetében elvégezni (pl. a 6–13. szoptatási napon). Törekedni kell arra, hogy az anyaállatok és kölykeik a lehető legrövidebb ideig legyenek elválasztva egymástól.
|
|
45.
|
A vizsgálat vége felé esedékes, egyszeri funkcionalitási megfigyelések elhagyhatók, ha a vizsgálatot egy azt követő (90 napos) szubkrónikus vagy hosszú távú vizsgálat előzetes tanulmányaként végzik. Ebben az esetben a funkcionalitási megfigyeléseket az utóbbi vizsgálat részeként kell elvégezni. Másrészt az ismételt dózisú vizsgálat során végzett funkcionalitási megfigyelések adatainak megléte segítheti az azt követő szubkrónikus vagy hosszú távú vizsgálat során a dózisok kiválasztását.
|
|
46.
|
Kivételes esetekben a funkcionalitási megfigyelések elhagyhatók azon csoportoknál is, amelyek egyébként olyan mértékű toxicitási tüneteket mutatnak, hogy az jelentős mértékben megzavarná a funkcionalitási vizsgálat eredményeit.
|
|
47.
|
A vemhesség nulladik napjától számítandó terhességi (gesztációs) időszakról feljegyzést kell készíteni. Az ellés után minden almot a lehető leghamarabb meg kell vizsgálni a kölykök számának és ivarának, a halvaszületések, élveszületések és fejletlen egyedek (a megfelelő kontrollcsoportba tartozó kölyköknél jelentősen kisebb utódok) számának, valamint a súlyos rendellenességek jelenlétének megállapításához.
|
|
48.
|
Az élő kölyköket meg kell számolni, nemüket meg kell állapítani, és az almok egyedeinek súlyát meg kell mérni a születést követő 24 órán belül (a post-partum 0. vagy 1. napján) és legalább a post-partum 4. és 13. napján. A szülőkön végzett (43. és 44. pontban említett) megfigyelések mellett az utódok minden rendellenes viselkedéséről be kell számolni.
|
|
49.
|
Az anogenitális távolságot (AGD) minden utódon legalább egy alkalommal meg kell mérni a 0. és 4. posztnatális nap között. A kölykök testtömegét azon a napon kell megmérni, amikor az AGD-t is mérik, és az AGD-t normálni kell a kölykök méretére, ami lehetőleg a testtömeg köbgyöke (14). Az OECD 151. vizsgálati iránymutatásának (15) ajánlása szerint a hím utódok emlőbimbóinak/emlőbimbóudvarainak számát meg kell számolni a 12. vagy 13. posztnatális napon.
|
Testtömeg és táplálék-/vízfogyasztás
|
50.
|
A hímek és nőstények testsúlyát meg kell mérni az adagolás első napján, majd ezt követően legalább hetente és a vizsgálat végén meg kell ismételni a mérést. A nőstényeket a vemhesség alatt a 0., 7., 14. és 20. napon kell megmérni, továbbá az ellést követő 24 órán belül (a post-partum 0. vagy 1. napján) és legalább a post-partum 4. és 13. napján. A megfigyeléseket minden egyes felnőtt állat esetében külön-külön kell rögzíteni.
|
|
51.
|
A pároztatás előtt, valamint a vemhesség és a szoptatás ideje alatt a takarmányfogyasztást legalább hetente kell mérni. A párzási időszak során nem kell feltétlenül mérni a takarmányfogyasztást. Ha a vizsgálati vegyi anyagot az ivóvízzel adagolják, ezen időszakok során a vízfogyasztást is mérni kell.
|
Hematológia
|
52.
|
A vizsgálat során egy alkalommal minden csoportból véletlenszerűen kiválasztott öt hímen és öt nőstényen el kell végezni az alábbi hematológiai vizsgálatokat: hematokrit, hemoglobin koncentrációk, vörösvértestszám, retikulocitaszám, leukocitaszám (teljes és differenciált vérkép), trombocitaszám és a véralvadási idő/képesség mérése. Ha a vizsgálati vegyi anyagról vagy annak vélt metabolitjairól ismert vagy feltételezhető, hogy oxidáló tulajdonságokkal rendelkeznek, az említett vizsgálatokon kívül többek között a methemoglobin-koncentrációt és a Heinz-testek előfordulását is meg kell határozni.
|
|
53.
|
A vérmintákat egy megnevezett helyről kell levenni. A nőstényeknek fiziológiai szempontból hasonló állapotban kell lenniük a vérvétel során. A vemhesség kialakulásának eltérő időpontjából adódó gyakorlati nehézségek elkerülése érdekében a nőstényektől a pároztatás előtti időszak végén is vehető vérminta ahelyett, hogy közvetlenül az állatok elaltatása előtt vagy az elaltatási folyamat részeként vennék le a mintát. A hímektől viszont célszerű közvetlenül az állatok végleges elaltatása előtt vagy az elaltatási folyamat részeként vérmintát venni. Alternatív megoldásként a hímektől is vehető vér a pároztatás előtti időszak végén, amennyiben a nőstények esetén is ez az időpont került kiválasztásra.
|
|
54.
|
A vérmintákat megfelelő feltételek mellett kell tárolni.
|
Klinikai biokémiai vizsgálatok
|
55.
|
A szövetekben jelentkező fontosabb toxikus hatások és különösen a vesékre és a májra gyakorolt hatás vizsgálata érdekében a klinikai biokémiai értékek meghatározását az egyes csoportokból kiválasztott öt hím és öt nőstény vérmintáján végzik el. A vérvétel előtti éjszaka ajánlatos az állatokat koplaltatni (22). A vérplazma- és vérszérum-meghatározások során vizsgálandó elemek: nátrium, kálium, vércukor, összkoleszterin, karbamid, kreatinin, összprotein és albumin, továbbá legalább két, a májsejti hatásokat jelző enzim (pl. alanin-aminotranszferáz, aszpartát-aminotranszferáz és szorbitol dehidrogenáz) és epesavak. Bizonyos körülmények között hasznos információk nyerhetők további (májból vagy máshonnan származó) enzimek, valamint a bilirubin méréséből.
|
|
56.
|
Vérmintákat egy meghatározott helyről kell venni, az alábbi ütemezés szerint:
|
—
|
almonként legalább két kölyöktől a születést követő 4. napon, amennyiben azt az utódok száma lehetővé teszi (lásd a 40–41. pontot),
|
|
—
|
az összes anyaállattól és almonként legalább két kölyöktől a vizsgálat végét jelentő 13. napon, és
|
|
—
|
az összes felnőtt hímtől a vizsgálat végén.
|
A vérmintákat megfelelő körülmények között tárolják. A 13 napos utódoktól és a felnőtt hímektől vett vérminták alapján értékelik a pajzsmirigyhormon (T4) szérumszinteket. Szükség esetén a T4 további értékeléséhez felhasználhatók az anyaállatoktól és a 4 napos utódoktól vett vérminták. Ha a vizsgálat szempontjából lényeges, más hormonok is mérhetők. A pajzsmirigyhormon analízisekhez almonként egyesíteni lehet a kölyköktől vett vérmintákat. A pajzsmirigyhormonokra (T4 és TSH) vonatkozó mérésekhez lehetőleg a teljes vérmintát kell használni.
|
|
57.
|
Kiegészítésként, a vizsgálat utolsó hetében, amikor a vizeletet előre meghatározott időpontokban gyűjtik, az alábbi vizeletminta-vizsgálatokat lehet elvégezni az egyes csoportokból véletlenszerűen kiválasztott öt hím tekintetében: megjelenés, mennyiség, ozmolalitás vagy fajsúly, pH-érték, fehérjék, glukóz és vér/vérsejtek.
|
|
58.
|
Ezeken túlmenően megfontolható az általános szöveti roncsolódás szérummarkereinek vizsgálata. Más vizsgálatokat is elvégeznek, ha a vizsgálati vegyi anyagnak ismert vagy feltételezhető hatása van a kapcsolódó anyagcsere-folyamatokra; ilyen pl. a kalcium, a foszfát, éhgyomorra mért triglicerid és glukóz, egyes hormonok, a methemoglobin és a kolinészteráz mérése. Ezeket eseti alapon kell meghatározni.
|
|
59.
|
A hormonok meghatározásának variabilitását és az azzal kapott abszolút koncentrációt az alábbi tényezők befolyásolhatják:
|
—
|
az elpusztítás időpontja, tekintettel a hormonkoncentrációk nap folyamán tapasztalható ingadozására,
|
|
—
|
az állatot érő indokolatlan stressz elkerülése érdekében alkalmazott elpusztítási mód, amely hatással lehet a hormonkoncentrációkra,
|
|
—
|
a hormonmeghatározásokhoz alkalmazott, standard görbéjük tekintetében esetlegesen eltérő vizsgálati készletek.
|
|
|
60.
|
A kifejezetten hormon-meghatározási célra szánt plazmamintákat a nap hasonló időpontjában kell gyűjteni. A kereskedelmi fogalomban kapható különböző vizsgálati készletek eltérő számszerű értékeket adnak a hormonkoncentrációk elemzése során.
|
|
61.
|
Ha a rendelkezésre álló kiinduló adatok nem bizonyulnak elegendőnek, megfontolandó a hematológiai és klinikai biokémiai változók meghatározása az adagolás kezdete előtt, lehetőség szerint az állatok egy, a kísérleti csoportokba nem tartozó csoportjánál. Nőstények esetében az adatok szoptató anyaállatokról kell származzanak.
|
PATOLÓGIA
Makroszkópos boncolás
|
62.
|
A vizsgálatokba bevont összes ivarérett állatot teljes körű, beható autopsziának kell alávetni, ideértve a következők alapos vizsgálatát: a test külső felszíne, az összes testnyílás, a koponya, a mellüreg, a hasüreg és ezek tartalma. Külön figyelmet kell szentelni a szaporítószerveknek. A beágyazódási helyek számát fel kell jegyezni. A boncolás napján meg kell vizsgálni a hüvelyi kenetet az ivari ciklus aktuális szakaszának meghatározásához, hogy lehetővé váljon a korreláció felállítása a női szaporítószervek kórszövettanával.
|
|
63.
|
Az összes hím felnőtt állat heréit és mellékheréit, prosztatáját és ondóhólyagjait a koaguláló mirigyekkel egy egységként meg kell tisztítani minden rátapadt szövettől, és a kiszáradás megelőzése érdekében a boncolást követően a lehető legrövidebb időn belül meg kell mérni nedves tömegüket. Ezen túlmenően további szervtömegmérések végezhetők hímeknél a végbélemelő és a bulbocavernosus izomra, a Cowper-mirigyekre és a glans penisre, nőstényeknél a páros petefészekre (nedves tömeg) és a méhre (valamint a méhnyakra) kiterjedőleg; ha ezeket a szerveket is bevonják a mérésbe, tömegüket a boncolás után lehető leghamarabb meg kell határozni. Meg kell őrizni az összes felnőtt állat petefészkeit, heréit, mellékheréit, a járulékos ivarszerveket, valamint a makroszkópikus léziókat mutató szerveket.
|
|
64.
|
Ezenfelül a későbbi tervezett kórszövettani vizsgálatok szempontjából leginkább megfelelő fixálószerben meg kell őrizni valamennyi felnőtt hím és nőstény, továbbá minden alomból egy hím és egy nőstény 13 napos kölyök pajzsmirigyét. A pajzsmirigy súlya a fixálást követően is meghatározható. A megtisztítás a szövetkárosodás elkerülése érdekében ez esetben is igen óvatosan, kizárólag a fixálást követően végzendő el. A szövetkárosodás veszélyeztetheti a kórszövettani elemzést. A vérmintákat egy megnevezett helyről kell venni, közvetlenül az állatok végleges elaltatása előtt vagy aközben, és azokat megfelelő körülmények között kell tárolni (lásd a 56. pontot).
|
|
65.
|
Ezen túlmenően (a haldokló és/vagy a vizsgálat vége előtt elaltatott egyedeken kívül) minden csoportból véletlenszerűen kiválasztott legalább öt felnőtt hím és nőstény máját, veséit, mellékveséit, csecsemőmirigyét, lépét, agyát és szívét megtisztítják minden rátapadt szövettől, és a kiszáradás megelőzése érdekében a boncolást követő lehető legrövidebb időn belül megmérik nedves tömegüket. Az alábbi testszöveteket konzerválni kell a szövetek típusa és a szándékolt későbbi kórszövettani vizsgálatok szempontjából legmegfelelőbb fixálószerben: szövetet, amelyen van makroszkopikus lézió, az agyat (jellemző részeket, mint pl. a nagyagy, a kisagy, a nyúltagyi híd), a gerincvelőt, a szemet, a gyomrot, a vékony- és vastagbelet (beleértve a Payer-plakkot), a májat, a veséket, a mellékveséket, a lépet, a szívet, a csecsemőmirigyet, a légcsövet és a tüdőket (utóbbit először megtöltik fixálószerrel, és úgy merítik alá), az ivarmirigyeket (heréket és petefészkeket), a járulékos nemi szerveket (a méhet és a méhnyakat, a mellékheréket, a prosztatát és az ondóhólyagokat a koaguláló mirigyekkel), a hüvelyt, a húgyhólyagot, a nyirokcsomókat (a legközelebbi elvezető nyirokcsomó mellett a laboratórium tapasztalata alapján másik nyirokcsomót is vételezni kell (16)), a perifériás idegeket (csípő vagy sípcsont környéki), lehetőleg az izomzathoz közeli helyről véve, a vázizomzatot és csontot a csontvelővel együtt (metszetet és/vagy egy, friss csontvelőből felszívott mintát). A herék fixálását javasolt Bouin- vagy módosított Davidson-féle fixálószerbe merítéssel végezni (16) (17) (18); e szövetek esetében formalinos fixálás használata nem ajánlott. A tunica albugineán a fixálószer gyors bejutása érdekében tűvel óvatosan sekély szúrást lehet ejteni a szerv mindkét végén. A klinikai és más jellegű megfigyelések alapján további testszövetek vizsgálata is szükséges lehet. A fentieken túl meg kell őrizni minden olyan más szervet is, amely a vizsgálati vegyi anyag ismert tulajdonságai alapján valószínűleg célszervnek tekinthető.
|
|
66.
|
Az alábbi szövetek értékes információkat nyújthatnak az endokrin rendszerre gyakorolt hatásokról: ivarmirigyek (petefészkek és herék), járulékos ivarszervek (méh és méhnyak, mellékherék, ondóhólyagok a koaguláló mirigyekkel, a prosztata dorzolaterális és ventrális része), hüvely, agyalapi mirigy, hím emlőmirigy és mellékvese. A hím emlőmirigyek elváltozásairól nem áll rendelkezésre kellő dokumentáció, azonban ez a paraméter igen érzékeny lehet az ösztrogénhatású anyagokra. A 65. pontban nem szereplő szervek/szövetek megfigyelése nem kötelező.
|
|
67.
|
Az elhullott és a post-partum 13. napján vagy röviddel azután elpusztított utódokat legalább külsőleg alaposan meg kell vizsgálni, hogy láthatók-e rajtuk súlyos rendellenességek. Különös figyelmet kell szentelni a külső ivarszervek vizsgálatának, és ezen belül a rendellenes fejlődés jeleinek.
|
Kórszövettan
|
68.
|
A kontrollcsoport és a magas dózissal kezelt csoport kiválasztott egyedeinek tartósított szerveit és szöveteit teljes kórszövettani vizsgálatnak vetik alá (különös figyelmet fordítva a hím ivarmirigyek spermatogenezisének szakaszaira és a here intersticiális sejtfelépítésének kórszövettanára). Szükség esetén vizsgálni lehet az utódoktól és a fennmaradó felnőtt állatoktól származó pajzsmirigyet. Ha a magas dózissal kezelt csoport egyedei esetében a kezelésnek tulajdonítható elváltozások figyelhetők meg, e vizsgálatokat ki kell terjeszteni más dózissal kezelt csoportok egyedeire is. A kórszövettani iránymutatás (10) további információkat tartalmaz az endokrin szövetek boncolásával, fixálásával, metszeteinek elkészítésével és kórszövettani vizsgálatával kapcsolatban.
|
|
69.
|
Minden makroszkopikus léziót meg kell vizsgálni. A NOAEL értelmezéséhez segítséget nyújthat más dózissal kezelt csoportokból származó célszervek vizsgálata, különösen azokból, amelyekben a kezelés állítólag nem járt káros hatással.
|
|
70.
|
Kísérő csoport használata esetén azon szerveken és szöveteken kell kórszövettani vizsgálatot végezni, amelyeknél a kezelt csoportok egyedei esetében elváltozásokat lehetett kimutatni.
|
ADATOK ÉS JELENTÉS
Adatok
|
71.
|
Az állatokra vonatkozóan egyedi adatsorokat kell felvenni. Emellett minden adatot táblázatos formában is össze kell foglalni, amelyben minden egyes vizsgálati csoport esetében fel kell tüntetni az állatok számát a vizsgálat kezdetén, a vizsgálat során elpusztult vagy humánus okok miatt elaltatott állatok számát, az elhullások és az eutanáziák időpontját, a termékeny állatok számát, a vemhes nőstények számát, a toxicitás jeleit mutató állatok számát, a megfigyelt toxicitási tünetek leírását, ezen belül bármely toxikus tünet megjelenésének időpontját, valamint ezek időtartamát és súlyosságát, a kórszövettani elváltozások típusait és az almokra vonatkozó minden idevágó adatot. A 3. mellékletben közzéteszünk egy, a reprodukciós/fejlődési hatások értékelése során nagyon hasznosnak bizonyult, táblázatos formájú összefoglaló jelentést.
|
|
72.
|
A számszerű eredményeket lehetőség szerint megfelelő és általánosan elfogadott statisztikai módszer alkalmazásával kell kiértékelni. A hatás dózistartomány mentén történő összehasonlításaival kiküszöbölhető a több t-próba alkalmazása. A statisztikai módszereket a vizsgálat tervezésekor kell megválasztani. Az anogenitális távolság és az emlőbimbó-visszamaradás statisztikai elemzésének az utódokra vonatkozó egyedi adatokon kell alapulnia, ugyanakkor figyelembe kell vinni az alom által gyakorolt hatásokat. Ahol megfelelő, az elemzés egysége az alom. A kölykök testsúlyára vonatkozó statisztikai elemzésnek a kölykök egyedi adatain kell alapulnia, ugyanakkor figyelembe kell vinni az alom által gyakorolt hatásokat. A vizsgálat korlátozott terjedelme miatt a „szignifikancia” felmérésére szolgáló vizsgálatok formájában végzett statisztikai elemzések számos végpont, különösen a reprodukciós végpontok szempontjából csekély jelentőséggel bírnak. A legelterjedtebb módszerek némelyike – különösen a központi érték mérésére szolgáló paraméteres próbák – nem megfelelők. Amennyiben statisztikai elemzésekre támaszkodnak, még a vizsgálat megkezdése előtt olyan módszert kell választani, amelyik megfelel a vizsgált változók eloszlásának.
|
Az eredmények értékelése
|
73.
|
E toxicitási vizsgálat eredményeit a megfigyelt hatások, valamint a boncolási és mikroszkópos eredmények szempontjából kell értékelni. Az értékelés során ki kell térni a vizsgálati vegyi anyag dózisa és a rendellenességek, ezen belül a súlyos léziók, az azonosított célszervek, a terméketlenség, a klinikai rendellenességek, a csökkent reproduktív teljesítmény és egyedszám, a testtömegváltozások, a mortalitásra gyakorolt hatások és egyéb toxikus hatások megléte vagy hiánya, illetve gyakorisága és súlyossága közötti összefüggésekre.
|
|
74.
|
A hímek rövid kezelési időszaka miatt a hímekre gyakorolt reprodukciós hatások felmérése során figyelembe kell venni a herék és mellékherék kórszövettanát, valamint a termékenységre vonatkozó adatokat. A szaporodásra/fejlődésre (pl. alomméretre, anogenitális távolságra, emlőbimbó-visszamaradásra, T4 szérumszintekre) vonatkozólag rendelkezésre álló korábbi kontrolladatok szintén hasznos segítséget nyújthatnak a vizsgálat értelmezéséhez.
|
|
75.
|
A minőség-ellenőrzéshez javasolt történeti kontrolladatokat gyűjteni, a számszerű adatokhoz pedig kiszámítani a variációs együtthatót, különösen az endokrin rendszert károsító anyagok kimutatásával összefüggő paraméterek tekintetében. Ezek az adatok felhasználhatók összehasonlítási célokra a tényleges vizsgálatok kiértékelése során.
|
Vizsgálati jelentés
|
76.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálati vegyi anyag:
|
—
|
eredet, gyártási szám, felhasználási határidő, ha rendelkezésre áll;
|
|
—
|
a vizsgálati vegyi anyag stabilitása, ha ismert;
|
|
|
|
Egy összetevőből álló anyag:
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján
|
|
|
|
Több összetevőből álló anyag, UVCB-k és keverékek:
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai révén jellemezve
|
. |
|
|
Vivőanyag (ha van):
|
—
|
a vivőanyag kiválasztásának indokolása, ha az nem víz.
|
|
|
|
Kísérleti állatok:
|
—
|
az állatok száma, kora és neme;
|
|
—
|
az állatok származása, tartási körülmények, takarmány stb.;
|
|
—
|
az egyes állatok testtömege a vizsgálat kezdetekor;
|
|
—
|
patkánytól eltérő faj alkalmazásának indokolása.
|
|
|
|
Vizsgálati körülmények:
|
—
|
a dózisok kiválasztásának indokolása;
|
|
—
|
a vizsgálati vegyi anyag formulázásával/a takarmány előkészítésével, az elért koncentrációval, a készítmény stabilitásával és homogenitásával kapcsolatos adatok;
|
|
—
|
a vizsgálati vegyi anyag beadására vonatkozó részletek;
|
|
—
|
átszámítás a vizsgálati vegyi anyagnak a takarmányban/ivóvízben meglévő koncentrációjáról (ppm) a tényleges dózisra (mg/kg testsúly/nap), ha ez alkalmazható;
|
|
—
|
a táplálék és víz minőségére vonatkozó részletek;
|
|
—
|
a selejtezés során a kölykök kiválasztására használat randomizálási eljárás részletes leírása, ha sor került selejtezésre.
|
|
|
|
Eredmények:
|
—
|
testsúly és annak változásai;
|
|
—
|
táplálék- és vízfogyasztásra vonatkozó adatok, ha alkalmazható;
|
|
—
|
a toxikus reakciók adatai nemenként és dózisonként, beleértve a termékenységet, a gesztációs időt és a toxicitás egyéb
|
|
—
|
a vemhesség időtartama; a szaporodásra, az utódokra, a posztnatális növekedésre stb. gyakorolt toxikus vagy egyéb hatások;
|
|
—
|
a klinikai megfigyelések jellege, súlyossága és időtartama (a változások visszafordíthatók-e vagy sem);
|
|
—
|
az érzékszervi aktivitás, a szorítási erősség és a motoros aktivitás értékelése;
|
|
—
|
hematológiai vizsgálatok eredményei, a megfelelő viszonyítási értékekkel;
|
|
—
|
a klinikai biokémiai vizsgálatok eredményei, a megfelelő viszonyítási értékekkel;
|
|
—
|
normális vagy abnormális ivari ciklust mutató felnőtt nőstények száma és a ciklus időtartama;
|
|
—
|
az élveszületések száma és a beágyazódás utáni veszteség;
|
|
—
|
a szemmel látható rendellenességeket mutató kölykök száma; a külső nemi szervek makroszkopikus értékelése, a fejletlen utódok száma;
|
|
—
|
a vizsgálat során bekövetkezett elhullás ideje, illetve hogy az állatok megélték-e a vizsgálat végét;
|
|
—
|
a beágyazódások száma, az alomméret és az almokban lévő egyedek méréskor rögzített testsúlya;
|
|
—
|
az összes utód anogenitális távolsága (és az AGD-mérés napján rögzített testsúlyuk);
|
|
—
|
emlőbimbó-visszamaradás a hím utódoknál;
|
|
—
|
pajzsmirigyhormonok szintje 13 napos kölyköknél és felnőtt hímeknél (az anyaállatoknál és 4 napos kölyköknél, ha mérték);
|
|
—
|
a testtömeg az elpusztításkor, valamint a szülők szervtömegadatai;
|
|
—
|
a kórszövettani leletek részletes ismertetése;
|
|
—
|
felszívódási adatok (ha vannak);
|
|
—
|
adott esetben az eredmények statisztikai feldolgozása.
|
|
|
|
Az eredmények értékelése.
|
|
Az eredmények értelmezése
|
77.
|
Ez a vizsgálat az ismételt dózisok útján történő kezeléssel összefüggésben kimutatható reprodukciós/fejlődési toxicitás értékelésére szolgál. Különösen mivel mind az általános toxicitási, mind a reprodukciós/fejlődési végpontokra hangsúlyt fektetnek, a vizsgálat eredményei lehetővé teszik az általános toxicitás hiányában is fellépő reprodukciós/fejlődési hatások megkülönböztetését azoktól, amelyek csak a szülőkre is toxikus szintek mellett fejeződnek ki (lásd a 7–11. pontot). Jelezheti a további vizsgálatok szükségességét, és iránymutatást nyújthat a későbbi vizsgálatok kialakításához. A szaporodási és fejlődési eredmények értelmezésével kapcsolatban az OECD 43. iránymutatását kell alkalmazni (19). A rágcsálókon végzett endokrin és reprodukciós vizsgálatok szövettani értékeléséről szóló 106. OECD-iránymutatás (16) olyan információkkal szolgál az (endokrin) szervek és a hüvelyi kenet előkészítéséről és értékeléséről, amelyek segítséget nyújthatnak e vizsgálati módszerhez.
|
SZAKIRODALOM
|
(1)
|
OECD (1990). Room Document No 1 for the 14th Joint Meeting of the Chemicals Group and Management Committee. Kérésre elérhető a Gazdasági Együttműködési és Fejlesztési Szervezetnél, Párizs
|
|
(2)
|
OECD (1992). Chairman’s Report of the ad hoc Expert Meeting on Reproductive Toxicity Screening Methods, Tokyo, 27th-29th October, 1992. Kérésre elérhető a Gazdasági Együttműködési és Fejlesztési Szervezetnél, Párizs
|
|
(3)
|
Mitsumori K., Kodama Y., Uchida O., Takada K., Saito M. Naito K., Tanaka S., Kurokawa Y., Usami, M., Kawashima K., Yasuhara K., Toyoda K., Onodera H., Furukawa F., Takahashi M. and Hayashi Y. (1994). Confirmation Study, Using Nitro-Benzene, of the Combined Repeat Dose and Reproductive/ Developmental Toxicity Test Protocol Proposed by the Organization for Economic Cooperation and Development (OECD). J. Toxicol, Sci., 19, 141-149.
|
|
(4)
|
Tanaka S., Kawashima K., Naito K., Usami M., Nakadate M., Imaida K., Takahashi M., Hayashi Y., Kurokawa Y. and Tobe M. (1992). Combined Repeat Dose and Reproductive/Developmental Toxicity Screening Test (OECD): Familiarization Using Cyclophosphamide. Fundam. Appl. Toxicol., 18, 89-95.
|
|
(5)
|
OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11th March 1998, kérésre elérhető a Gazdasági Együttműködési és Fejlesztési Szervezetnél, Párizs
|
|
(6)
|
OECD (2015). Feasibility Study for Minor Enhancements of TG 421/422 with ED Relevant Endpoints. Environment, Health and Safety Publications, Series on Testing and Assessment (No 217), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(7)
|
OECD (2000). Guidance Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluations, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 19), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(8)
|
Goldman J.M., Murr A.S., Buckalew A.R., Ferrell J.M.and Cooper R.L. (2007). The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies, Birth Defects Research, Part B, 80 (2), 84-97.
|
|
(9)
|
Sadleir R.M.F.S. (1979). Cycles and Seasons, in Auston C.R. and Short R.V. (Eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York.
|
|
(10)
|
IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document (No 60).
|
|
(11)
|
Moser V.C., McDaniel K.M. and Phillips P.M. (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol., 108, 267-283.
|
|
(12)
|
Meyer O.A., Tilson H.A., Byrd W.C. and Riley M.T. (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rats and Mice. Neurobehav. Toxicol., 1, 233-236.
|
|
(13)
|
Crofton K.M., Howard J.L., Moser V.C., Gill M.W., Reiter L.W., Tilson H.A., MacPhail R.C. (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13, 599-609.
|
|
(14)
|
Gallavan R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds. (1999). „Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights”, Reproductive Toxicology, 13: 383–390.
|
|
(15)
|
OECD (2013). Guidance Document in Support of the Test Guideline on the Extended One Generation Reproductive Toxicity Study. Environment, Health and Safety Publications, Series on Testing and Assessment (No 151). Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(16)
|
OECD (2009).Guidance Document for Histologic Evaluation of Endocrine and Reproductive Tests in Rodents. Environment, Health and Safety Publications, Series on Testing and Assessment (No. 106) Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(17)
|
Hess RA and Moore BJ. (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (Eds.). Academic Press: San Diego, CA, pp. 52-85.
|
|
(18)
|
Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM. (2002). Fixation of Testes and Eyes Using a Modified Davidson’s Fluid: Comparison with Bouin’s Fluid and Conventional Davidson’s fluid. Toxicol. Pathol. 30, 524-533.
|
|
(19)
|
OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 43), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(20)
|
OECD (2011), Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption (No 150), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
1. függelék
FOGALOMMEGHATÁROZÁSOK (LÁSD MÉG AZ OECD 150. IRÁNYMUTATÁSÁT (20))
Androgenitás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében természetes androgén hormonokhoz (például a tesztoszteronhoz) hasonló hatást fejtsen ki.
Antiandrogenitás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes androgén hormonok (például a tesztoszteron) hatását gátolja.
Antiösztrogenitás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes ösztrogén hormonok (például a 17ß-ösztradiol) hatását gátolja.
Antitiroid hatás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes pajzsmirigyhormon (például a T3) hatását gátolja.
Vegyi anyag anyag vagy keverék.
Fejlődési toxicitás reprodukciós toxicitás kifejeződése az utódokban pre-, peri-, posztnatális, szerkezeti vagy funkcionális rendellenességek formájában.
Dózis a vizsgálati vegyi anyag beadott mennyisége. A dózist a vizsgálati vegyi anyag súlyának és a kísérleti állat egységnyi testsúlyának napi hányadosaként (például mg/testsúlykilogramm/nap), vagy a takarmányban lévő állandó koncentrációként fejezik ki.
Adagolás általános fogalom, amely a dózist, valamint az adagolás gyakoriságát és időtartamát foglalja magában.
Nyilvánvaló toxicitás általános fogalom, amely a toxicitás vizsgálati vegyi anyag beadását követően kialakuló egyértelmű tüneteire vonatkozik. E tüneteknek elegendőnek kell lenniük a veszély értékeléséhez, továbbá olyan szintet kell képviselniük, amelynél a beadott dózis növelése várhatóan a toxicitás súlyos jeleinek kialakulásához és valószínű mortalitáshoz vezet.
Termékenységkárosodás a hím vagy női szaporodási funkciók vagy képesség terén jelentkező rendellenességeket jelenti.
Anyai toxicitás a vemhes nőstényeket érő, a vemhességhez kapcsolható káros hatások, amelyek történhetnek specifikusan (közvetlen hatás) vagy nem specifikusan (közvetett hatás).
NOAEL a „no-observed-adverse-effect level” (nem észlelhető káros hatásszint) rövidítése. Ez a legmagasabb dózis, amelynél még nem figyelhetők meg a kezelés okozta káros hatások.
Ösztrogenitás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes ösztrogén hormonokhoz (például a 17ß-ösztradiolhoz) hasonló hatást fejtsen ki.
Reprodukciós toxicitás az utódoknál jelentkező káros hatásokat és/vagy a hím vagy női szaporodási funkciók vagy képesség csökkenését jelenti.
Vizsgálati vegyi anyag: az e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Tiroid hatás egy vegyi anyag azon képessége, hogy emlősállatok szervezetében a természetes pajzsmirigyhormonhoz (például a T3-hoz) hasonló hatást gyakoroljon.
Validáció tudományos folyamat, amelynek célja egy adott vizsgálati módszer működési követelményeinek és korlátjainak jellemzése, valamint megbízhatóságának és egy adott célhoz kapcsolódó relevanciájának igazolása.
2. függelék
A KÍSÉRLETI ÜTEMTERV ÁBRÁJA, AMELY JELZI A VIZSGÁLAT TELJES 14 NAPOS PÁRZÁSI IDŐSZAKON ALAPULÓ MAXIMÁLIS IDŐTARTAMÁT


3. függelék
TÁBLÁZATOS ÖSSZEFOGLALÓ JELENTÉS A SZAPORODÁSRA/FEJLŐDÉSRE GYAKOROLT HATÁSOKRÓL
|
MEGFIGYELÉSEK
|
ÉRTÉKEK
|
|
Adagolás (egységek).......
|
0 (kontroll)
|
...
|
...
|
...
|
...
|
|
Kezdő párok (Sz)
|
|
|
|
|
|
|
Ivari ciklus (legalább az átlagos időtartam és az irreguláris ciklusok gyakorisága)
|
|
|
|
|
|
|
Bizonyítottan párosított nőstények (Sz)
|
|
|
|
|
|
|
Vemhességet elért nőstények (Sz)
|
|
|
|
|
|
|
Fogantatás napja 1–5. (Sz)
|
|
|
|
|
|
|
Fogantatás napja 6–... (23) (Sz)
|
|
|
|
|
|
|
Vemhesség ≤ 21 nap (Sz)
|
|
|
|
|
|
|
Vemhesség = 22 nap (Sz)
|
|
|
|
|
|
|
Vemhesség ≥ 23 nap (Sz)
|
|
|
|
|
|
|
Élve született utódokkal rendelkező anyaállatok (Sz)
|
|
|
|
|
|
|
Post-partum 4. napján élő utódokkal rendelkező anyaállatok (Sz)
|
|
|
|
|
|
|
Beágyazódások/anyaállat (átlag)
|
|
|
|
|
|
|
Élő utódok/anyaállat az ellésnél (átlag)
|
|
|
|
|
|
|
Élő utódok/anyaállat a 4. napon (átlag)
|
|
|
|
|
|
|
Ivararány (h/n) az ellésénél (átlag)
|
|
|
|
|
|
|
Ivararány (h/n) a 4. napon (átlag)
|
|
|
|
|
|
|
Alom egyedsúlya az ellésnél (átlag)
|
|
|
|
|
|
|
Alom egyedsúlya a 4. napon (átlag)
|
|
|
|
|
|
|
Utódok súlya születéskor (átlag)
|
|
|
|
|
|
|
Utódok súlya az AGD-mérés időpontjában (hímek átlaga, nőstények átlaga)
|
|
|
|
|
|
|
Utódok AGD-értéke ugyanazon a posztnatális napon, születés utáni 4. napon (hímek átlaga, nőstények átlaga, posztnatális nap feljegyzése)
|
|
|
|
|
|
|
Utódok súlya a 4. napon (átlag)
|
|
|
|
|
|
|
Utódok súlya a 13. napon (átlag)
|
|
|
|
|
|
|
Hím utódok emlőbimbó-visszamaradása a 13. napon (átlag)
|
|
|
|
|
|
|
RENDELLENES UTÓDOK
|
|
Anyaállatok 0-val
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok ≥ 2-vel
|
|
|
|
|
|
|
UTÓDOK ELVESZTÉSE
|
|
Prenatális időszak (beágyazódások mínusz élveszületések száma)
|
|
Anyaállatok 0-vel
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok 2-vel
|
|
|
|
|
|
|
Anyaállatok ≥ 3-mal
|
|
|
|
|
|
|
Posztnatális időszak (élveszületések mínusz a 13. posztnatális napon élők száma)
|
|
Anyaállatok 0-vel
|
|
|
|
|
|
|
Anyaállatok 1-gyel
|
|
|
|
|
|
|
Anyaállatok 2-vel
|
|
|
|
|
|
|
Anyaállatok ≥ 3-mal
|
|
|
|
|
|
B.65.
IN VITRO BŐRKORRÓZIÓS MEMBRÁNBARRIER-VIZSGÁLATI MÓDSZER
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 435. vizsgálati iránymutatásában (2015) leírt módszerrel. A bőrkorrózió – az Egyesült Nemzetek Szervezete (ENSZ) által kidolgozott, vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszerében (GHS) (1), valamint az Európai Unió (EU) anyagok és keverékek osztályozásáról, címkézéséről és csomagolásáról szóló 1272/2008/EK rendeletében (CLP-rendelet) (24) meghatározottak szerint – valamely vizsgálati vegyi anyag alkalmazását követően megjelenő, visszafordíthatatlan bőrkárosodás, amely a felhámon át az irhára is átterjedő látható szövetelhalást okoz. Ez a vizsgálati módszer, amely egyenértékű az OECD 435. átdolgozott vizsgálati iránymutatásával, egy olyan in vitro membránbarrier-vizsgálatot foglal magában, amellyel azonosíthatók a korrozív vegyi anyagok. A vizsgálati módszer egy olyan mesterséges membránt alkalmaz, amely az állatbőrhöz hasonló módon reagál a korrozív vegyi anyagokra in situ.
|
|
2.
|
A hagyományos bőrkorróziós vizsgálatoknál a vizsgálati vegyi anyagot élő állatok bőrére viszik fel, majd egy meghatározott idő elteltével felmérik a szövetkárosodás mértékét (2). E vizsgálati módszeren túl elfogadtak néhány más in vitro vizsgálati módszert is a korrozív vegyi anyagok azonosítására használt (az OECD 404. vizsgálati iránymutatásával egyenértékű és e melléklet B.4. fejezetében ismertetett), standardnak számító, nyulakon végzett in vivo bőrkorróziós eljárás (2) alternatívájaként (3) (4). Az ENSZ GHS többszintes vizsgálati és értékelési stratégiát határoz meg a bőrkorrózió felméréséhez és osztályozásához, a bőrirritáció/bőrkorrózió vonatkozásában végzett vizsgálatok és értékelések integrált megközelítéseiről (IATA) szóló OECD-iránymutatás 3. és 4. modulja (1) (5) pedig validált és elfogadott in vitro vizsgálati módszerek használatát ajánlja. Az integrált vizsgálati és értékelési megközelítés (IATA) információforrásokat és adatelemző eszközöket csoportosító több modult ismertet, továbbá i. iránymutatást nyújt arra nézve, hogy miként integrálhatók és használhatók az ismert vizsgálati és nem vizsgálati adatok a vegyi anyagok potenciális bőrirritációs és bőrkorróziós hatásainak felmérésére, és ii. javaslatot tesz egy, a további vizsgálatok szükségessége esetén alkalmazható megközelítésre, amely kiterjed a negatív vizsgálati eredmények esetére is (5). E moduláris megközelítés révén az in vitro vizsgálati módszerek pozitív eredményei alapján megállapítható a vegyi anyagok korrozivitása anélkül, hogy állatkísérletekre lenne szükség, ezáltal visszaszorul és finomodik az állatok kísérleti felhasználása, és elkerülhető mindaz a fájdalom és szorongás, amely előfordulhatott volna, ha állatokkal végzik a kísérletet.
|
|
3.
|
A kereskedelmi forgalomban Corrositex® (6) (7) (8) néven kapható in vitro membránbarrier-modellen végzett validálási vizsgálatok kimutatták, hogy a modell egy 163 anyagból és keverékből álló adatbázis (7) tekintetében összességében 79 %-os (128/163) pontossággal, 85 %-os (76/89) érzékenységgel és 70 %-os (52/74) specificitással jelezi előre a bőrkorróziót. A modellt – elismert hitelessége alapján – validált referenciamódszerként arra ajánlják, hogy egy többszintű vizsgálati stratégia részeként alkalmazzák a vegyi anyagok által esetlegesen okozott bőrkorróziós veszély felmérésére (5)(7). Mielőtt egy in vitro bőrkorróziós membránbarrier-modell szabályozási célú alkalmazásra kerülne, a validált referenciamódszerhez (9) való hasonlóságának biztosítása érdekében az előre meghatározott teljesítményszabványok (10) követelményeinek megfelelően meg kell állapítani a javasolt felhasználása tekintetében fennálló megbízhatóságát, relevanciáját (pontosságát) és korlátait. Az adatok OECD-megállapodás szerinti kölcsönös elfogadása csak azután garantált, hogy a javasolt új vagy átdolgozott módszert a teljesítményszabványok szerint felülvizsgálták és belefoglalták az OECD megfelelő vizsgálati iránymutatásába. Jelenleg egyetlen in vitro módszer tartozik az OECD 435. vizsgálati iránymutatása alá, és ez a vizsgálati módszer a kereskedelmi forgalomban beszerezhető Corrositex® modell.
|
|
4.
|
A bőrkorrózió felmérésére szolgáló egyéb vizsgálati módszerek a rekonstruált humán bőr (az OECD 431. vizsgálati iránymutatása) (3) és az izolált patkánybőr (az OECD 430. vizsgálati iránymutatása) (4) használatán alapulnak. Emellett e vizsgálati iránymutatás lehetővé teszi azt, hogy a korrozív vegyi anyagokat alkategóriába sorolják az ENSZ GHS három korrozivitási alkategóriája és az ENSZ korróziós veszélyt jelentő anyagok szállítására vonatkozó három csomagolási csoportja szerint. Az eredetileg 2006-ben elfogadott vizsgálati iránymutatást 2015-ben átdolgozták, hogy figyelembe vegyék az IATA-iránymutatást és frissítsék a jártassági tesztanyagok jegyzékét.
|
FOGALOMMEGHATÁROZÁSOK
|
5.
|
Az alkalmazott fogalmak meghatározása a függelékben található.
|
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
6.
|
Az e vizsgálati módszer keretében leírt vizsgálat lehetővé teszi a korrozív vizsgálati vegyi anyagok azonosítását és ezen anyagok ENSZ GHS/CLP-rendelet szerinti alkategorizálását (1. táblázat). Emellett a vizsgálati módszer alkalmas arra, hogy a szállításukkal összefüggő vizsgálati célokból kifolyólag bizonyos kémiai anyagosztályok, pl. szerves és szervetlen savak, savszármazékok (25) és lúgok tekintetében megállapítsák a korrozivitást, illetve annak hiányát (7) (11) (12). Ez a vizsgálati módszer a validált vizsgálati referenciamódszerhez hasonló általános eljárást ír le (7). Ez a vizsgálati módszer ugyan nem biztosít elegendő információt a bőrirritáció felméréséhez, de (az OECD 439. vizsgálati iránymutatásával egyenértékű) B.46. vizsgálati módszer célja kifejezetten az, hogy a bőrirritációs egészségügyi hatásokat in vitro határozza meg (13). Egyetlen bőrexpozíció után a bőrt ért helyi hatások teljes kiértékeléséhez a vizsgálatok és értékelések integrált megközelítéseiről szóló OECD-iránymutatást kell tanulmányozni (5).
1. táblázat
Az ENSZ GHS szerinti bőrkorróziós kategóriák és alkategóriák
(26)
|
Korrozív kategória (1. kategória) (alkategóriákat nem használó hatóságoknak)
|
Alkalmazható korrozív alkategóriák (26) (alkategóriákat, pl. a CLP-rendeletet használó hatóságoknak)
|
Korrozív 3 állat közül legalább 1 esetében
|
|
Expozíció
|
Megfigyelés
|
|
Korrozív
|
1A korrozivitási alkategória
|
≤ 3 perc
|
≤ 1 óra
|
|
1B korrozivitási alkategória
|
> 3 perc / ≤ 1 óra
|
≤ 14 nap
|
|
1C korrozivitási alkategória
|
> 1 óra / ≤ 4 óra
|
≤ 14 nap
|
|
|
7.
|
A validált referenciamódszer (7) korlátja, hogy az előzetes kompatibilitási vizsgálat (lásd a 13. pontot) eredményei alapján számos nem korrozív vegyi anyagra és néhány korrozív vegyi anyagra nem alkalmazható a vizsgálat. A 4,5 és 8,5 közötti pH-értékkel rendelkező vízbázisú vegyi anyagok gyakran nem alkalmasak a vizsgálatra; ugyanakkor az e pH-tartományban vizsgált vegyi anyagok 85 %-a nem bizonyult korrozívnak az állatkísérletek során (7). Az in vitro membránbarrier módszer alkalmazható (vízben oldható és oldhatatlan) szilárd anyagok, (vizes és nem vízbázisú) folyadékok, valamint emulziók vizsgálatára. Azonban azokat a vizsgálati vegyi anyagokat, amelyek a kompatibilitási vizsgálat során nem idéznek elő kimutatható változást (vagyis színváltozást a validált vizsgálati referenciamódszer vegyi anyagok észlelésére szolgáló rendszerében (Chemical Detection System, CDS)), a membránbarrier módszertől eltérő vizsgálati módszerekkel kell vizsgálni.
|
A VIZSGÁLAT ELVE
|
8.
|
A vizsgálati rendszer két összetevőből áll: egy szintetikus makromolekuláris biobarrierből és egy kémiai érzékelő rendszerből (CDS); ez a vizsgálati módszer a kémiai érzékelő rendszeren keresztül – feltételezhetően az élő bőrre ható korróziós mechanizmussal/mechanizmusokkal megegyező módon – detektálja a membránbarrier korrozív vizsgálati vegyi anyagok okozta károsodását, miután a vizsgálati vegyi anyagot a szintetikus makromolekuláris membránbarrier felszínén alkalmazták (7).
|
|
9.
|
A membránbarrieren való áthatolás (vagy áttörés) számos eljárással vagy a kémiai érzékelő rendszerrel mérhető, többek között azáltal, hogy a pH-indikátor festék színe vagy az indikátoroldat bizonyos más tulajdonságai megváltoznak a barrier alatt.
|
|
10.
|
Meg kell állapítani a membránbarrier hitelességét, vagyis azt, hogy tervezett használata szempontjából releváns és megbízható. Ez magában foglalja annak biztosítását, hogy a különböző preparátumok barriertulajdonságai állandók, például képesek barrierként működni a nem korrozív vegyi anyagokkal szemben, valamint képesek a vegyi anyagok korrozív tulajdonságainak osztályozására az ENSZ GHS különböző korrozivitási alkategóriái szerint (1). A besorolás alapja az az idő, amely alatt egy adott vegyi anyag a membránbarrieren áthatolva az indikátoroldatba jut.
|
A JÁRTASSÁG BIZONYÍTÁSA
|
11.
|
Az e vizsgálati módszer alá tartozó in vitro membránbarrier módszer rutinszerű alkalmazása előtt a laboratóriumoknak a 2. táblázatban felsorolt tizenkét jártassági tesztanyag megfelelő besorolásával igazolniuk kell szakmai jártasságukat. Ha a jegyzékben szereplő valamely anyag nem áll rendelkezésre, illetve ha indokolható, megfelelő in vivo és in vitro referenciaadatokkal rendelkező más – például a referencia-vegyianyagok jegyzékében feltüntetett (10) – anyag is használható, amennyiben a kiválasztási kritériumok megegyeznek az 1. táblázatban foglaltakkal.
2. táblázat
Jártassági tesztanyagok
(27)
|
Anyag (28)
|
CAS-szám
|
Kémiai osztály
|
In vivo ENSZ GHS alkategória (29)
|
In vitro ENSZ GHS alkategória (29)
|
|
Bór-trifluorid-dihidrát
|
13319-75-0
|
Szervetlen savak
|
1A
|
1A
|
|
Salétromsav
|
7697-37-2
|
Szervetlen savak
|
1A
|
1A
|
|
Foszfor-pentaklorid
|
10026-13-8
|
Szervetlen savak prekurzorai
|
1A
|
1A
|
|
Valeril-klorid
|
638-29-9
|
Savkloridok
|
1B
|
1B
|
|
Nátrium-hidroxid
|
1310-73-2
|
Szervetlen lúgok
|
1B
|
1B
|
|
1-(2-aminoetil) piperazin
|
140-31-8
|
Alifás aminok
|
1B
|
1B
|
|
Benzolszulfonil-klorid
|
98-09-9
|
Savkloridok
|
1C
|
1C
|
|
N,N-dimetil-benzilamin
|
103-83-3
|
Anilinok
|
1C
|
1C
|
|
Tetraetilén-pentamin
|
112-57-2
|
Alifás aminok
|
1C
|
1C
|
|
Eugenol
|
97-53-0
|
Fenolok
|
NK
|
NK
|
|
Nonil-akrilát
|
2664-55-3
|
Akrilátok/metakrilátok
|
NK
|
NK
|
|
Nátrium-bikarbonát
|
144-55-8
|
Szervetlen sók
|
NK
|
NK
|
|
ELJÁRÁS
|
12.
|
Az alábbi pontok egy, a korrózió felmérésére szolgáló mesterséges membránbarrier vizsgálati módszer (7) (15) összetevőit és eljárásait ismertetik, amelynek alapja a jelenlegi validált referenciamódszer, vagyis a kereskedelmi forgalomban kapható Corrositex®. A membránbarrier, valamint a kompatibilitást meghatározó/indikátor- és kategorizáló oldatok összeállíthatók, elkészíthetők vagy kereskedelmi forgalomban beszerezhetők, mint a Corrositex® validált referenciamódszer. A validált vizsgálati referenciamódszerhez rendelkezésre áll a vizsgálati módszer mintaprotokollja (7). A vizsgálatot szobahőmérsékleten (17–25 °C) kell elvégezni, az összetevőknek pedig meg kell felelniük az alábbi feltételeknek.
|
A vizsgálati vegyi anyag kompatibilitási tesztje
|
13.
|
A membránbarrier-vizsgálat előtt egy kompatibilitási vizsgálat során meg kell határozni, hogy a kémiai érzékelő rendszer képes-e detektálni a vizsgálati vegyi anyagot. Amennyiben a kémiai érzékelő rendszer nem mutatja ki a vizsgálati vegyi anyagot, a membránbarrieres vizsgálati módszer nem alkalmas az adott vizsgálati vegyi anyag potenciális korróziós hatásának értékelésére, ezért mást vizsgálati módszert kell alkalmazni. A kompatibilitási vizsgálat során olyan kémiai érzékelő rendszert és expozíciós körülményeket kell alkalmazni, amelyek megegyeznek a későbbi membránbarrier-vizsgálat expozíciójával.
|
A vizsgálati vegyi anyag időskálán alapuló kategorizálási vizsgálata
|
14.
|
Amennyiben az a vizsgálati módszer szempontjából releváns, a kompatibilitási vizsgálat során alkalmasnak nyilvánított vizsgálati vegyi anyagokon el kell végezni egy időskálán alapuló kategorizálási vizsgálatot, vagyis egy olyan szűrővizsgálatot, amely megkülönbözteti a gyenge és az erős savakat és a bázisokat. A validált vizsgálati referenciamódszerben például egy időskálán alapuló kategorizálási vizsgálat során határozzák meg, hogy a két időskála közül melyiket használják, annak alapján, hogy kimutattak-e jelentős sav- vagy lúgtartalékot. A bőrkorrozivitás és az ENSZ GHS szerinti korrozivitási alkategória meghatározásához az adott vizsgálati vegyi anyag sav- vagy lúgtartaléka szerint két különböző, áthatolási időn alapuló időskála áll rendelkezésre.
|
A MEMBRÁNBARRIERES VIZSGÁLATI MÓDSZER ÖSSZETEVŐI
Membránbarrier
|
15.
|
A membránbarrier két összetevőből áll: egy fehérjetartalmú makromolekuláris vízbázisú gélből és egy áteresztő membránból. A fehérjetartalmú gél nem ereszti át a folyadékokat és a szilárd anyagokat, ugyanakkor korrózió útján áteresztővé tehető. A teljesen összeállított membránbarriert előre meghatározott körülmények között kell tárolni, amelyek bizonyítottan kizárják a gél minőségromlását, pl. kiszáradását, a mikrobák elszaporodását, elmozdulását, megrepedezését, ami csökkentené teljesítményét. Meg kell határozni az elfogadható tárolási időt, és annak lejártát követően nem szabad felhasználni a membránbarrier-preparátumokat.
|
|
16.
|
Az áteresztő membrán fizikai alátámasztást biztosít a fehérjetartalmú gélnek a gélesedési folyamat és a vizsgálati vegyi anyaggal való kezelés során. A membránnak meg kell akadályoznia a gél beesesét és elcsúszását, továbbá átjárhatónak kell lennie minden vizsgálati vegyi anyag számára.
|
|
17.
|
A fehérjetartalmú gél, amely gélmátrixot képező fehérjékből, például keratinból, kollagénből vagy fehérjekeverékekből tevődik össze, a vizsgálati vegyi anyag célpreparátuma. A fehérjetartalmú anyagot felviszik a membrán felületére, és megvárják, amíg gélesedik, majd ezt követően a membránbarriert az indikátoroldat fölé helyezik. A fehérjetartalmú gél teljes felületének egyforma vastagságúnak és sűrűségűnek kell lennie, továbbá nem tartalmazhat légbuborékokat és egyéb olyan hibákat, amelyek károsíthatják funkciójának épségét.
|
A kémiai érzékelő rendszer
|
18.
|
Az indikátoroldatnak, amely megegyezik a kompatibilitási vizsgálat során alkalmazott oldattal, reagálnia kell a vizsgálati vegyi anyag jelenlétére. Olyan pH-indikátor festéket vagy festékkombinációt (pl. krezolvörös és metilnarancs) kell használni, amely színváltozással reagál a vizsgálati vegyi anyag jelenlétére. A mérési rendszer lehet vizuális vagy elektronikus.
|
|
19.
|
A vizsgálati vegyi anyagok barriermembránon keresztüli áthaladásának észlelésére kifejlesztett detektorrendszerek alkalmasságát és megbízhatóságát értékelni kell, hogy igazolni lehessen az általuk észlelhető vegyi anyagok körét és az észlelés mennyiségi korlátait.
|
A VIZSGÁLAT KIÉRTÉKELÉSE
A vizsgálati módszer alkotóelemeinek összeállítása
|
20.
|
A membránbarriert az indikátoroldatot tartalmazó ampullába (vagy csőbe) helyezik oly módon, hogy a membrán teljes felületén érintkezzen az indikátoroldattal és ne alakuljanak ki légbuborékok. Ügyelni kell arra, hogy a barrier épsége fennmaradjon.
|
A vizsgálati vegyi anyag alkalmazása
|
21.
|
A vizsgálati vegyi anyagot megfelelő, azaz például folyadék esetében 500 μl, finomra porított szilárd anyag esetében 500 mg (7) mennyiségben óvatosan fel kell vinni a membránbarrier felső felszínére, majd egyenletesen szét kell oszlatni a felületen. Minden vizsgálati vegyi anyag és az annak megfelelő kontrollok esetében megfelelő számú, például négy (7) replikátumot kell készíteni (lásd a 23–25. pontot). A vizsgálati vegyi anyag membránbarrierre történő felvitelének időpontját rögzíteni kell. Annak érdekében, hogy a rövid hatóidejű korróziót is pontosan lehessen rögzíteni, a vizsgálati vegyi anyagot szakaszosan kell a replikátum csövekbe felvinni.
|
A membránbarrieren való áthatolás mérése
|
22.
|
Minden csövet gondosan megfigyelnek, és feljegyzik az indikátoroldat első elváltozásának, vagyis a barrieren történő áthatolásnak az időpontját, majd meghatározzák a felvitel és a membránbarrieren való áthatolás között eltelt időt.
|
Kontrollok
|
23.
|
Azokban a vizsgálatokban, amelyekben a vizsgálati vegyi anyagot vivőanyaggal vagy oldószerrel együtt alkalmazzák, a vivőanyagnak vagy az oldószernek kompatibilisnek kell lennie a membránbarrier rendszerrel, azaz nem lehet hatással sem a membránbarrier rendszer épségére, sem a vizsgálati vegyi anyag korrozivitására. Adott esetben a vizsgálati vegyi anyaggal párhuzamosan kontrollvizsgálatot kell végezni az oldószerrel (vagy a vivőanyaggal) annak igazolására, hogy az kompatibilis a membránbarrier rendszerrel.
|
|
24.
|
A vizsgálati vegyi anyaggal egyidejűleg egy közepesen korrozív anyaggal, például 110 ± 15 mg nátrium-hidroxiddal (amely az ENSZ GHS szerinti 1B alkategóriába tartozik) (7) pozitív (korrozív) kontrollt kell végezni a vizsgálati rendszer által nyújtott teljesítmény elfogadhatóságának értékelésre. A korrozív vizsgálati vegyi anyagok relatív korróziós potenciáljának felméréséhez hasznosnak bizonyulhat egy, a vizsgálati vegyi anyaggal azonos kémiai osztályba tartozó anyaggal végzett második pozitív kontroll. A pozitív kontrollanyagként közepesen korrozív (pl. az ENSZ GHS szerinti 1B alkategóriába tartozó) anyagot kell választani, hogy észlelni lehessen, ha az áthatolási idő elfogadhatatlanul hosszú vagy rövid a meghatározott referenciaértékhez képest, ami azt jelzi, hogy a vizsgálati rendszer nem működik megfelelően. E tekintetben a rendkívül korrozív (az ENSZ GHS szerinti 1A alkategóriába tartozó) vagy a nem korrozív hatású vegyi anyagok használhatósága korlátozott. Ugyanakkor az ENSZ GHS szerinti 1B alkategóriába tartozó korrozív vegyi anyagok lehetővé teszik a túl gyors vagy túl lassú áthatolási idő észlelését. Az enyhén korrozív (az ENSZ GHS szerinti 1C alkategóriába tartozó) anyagok használhatók pozitív kontrollként annak mérésére, hogy a vizsgálati módszer képes-e megbízhatóan különbséget tenni az enyhén korrozív és a nem korrozív hatású vegyi anyagok között. Az alkalmazott megközelítéstől függetlenül a használt pozitív kontrollanyag(ok) áthatolási idejének dokumentált tartománya alapján meg kell határozni a pozitív kontroll válaszreakciójának elfogadható tartományát, úgy mint az átlagérték ± 2–3 standard deviációt. Minden vizsgálat során meg kell állapítani a pozitív kontrollanyag pontos áthatolási idejét, hogy észlelni lehessen az elfogadható tartománytól való eltéréseket.
|
|
25.
|
Negatív kontrollként egy negatív (nem korrozív) kontrollanyagot – például 10 % citromsav, 6 % propionsav (7) – is vizsgálni kell a vizsgálati vegyi anyaggal egyidejűleg, amely egy másik minőség-ellenőrzési mérésként igazolja a membránbarrier épségét.
|
A vizsgálat elfogadhatósági kritériumai
|
26.
|
Az egyes ENSZ GHS szerinti korrozivitási alkategóriákra meghatározott időparaméterek alapján a vizsgálati vegyi anyag membránbarrierre történő felvitele és a barrieren való áthatolása között eltelt (percben mért) időt használják a vizsgálati vegyi anyag korrozivitásának előrejelzésére. Ahhoz, hogy egy vizsgálatot elfogadhatónak minősítsenek, a párhuzamos pozitív kontrollnak a várt áthatolási reakcióidőt kell biztosítania (ami például a nátrium-hidroxid pozitív kontrollként való alkalmazása esetén 8–16 perces áthatolási időt jelent), a párhuzamos negatív kontrollanyag nem lehet korrozív, és – amennyiben sor került rá – a párhuzamos oldószeres kontroll nem lehet korrozív és nem módosíthatja a vizsgálati vegyi anyag korróziós potenciálját. Az e vizsgálati módszer alá tartozó módszer rutinszerű alkalmazását megelőzően a laboratóriumoknak a 2. táblázatban javasolt tizenkét anyag felhasználásával igazolniuk kell szakmai jártasságukat. Az e vizsgálati módszer keretében kidolgozott, a validált referenciamódszerhez (14) strukturálisan és funkcionálisan hasonló, úgynevezett „me-too” vizsgálati módszerek szabályozási célú alkalmazása előtt az előre meghatározott teljesítményszabványok használatával igazolni kell az új módszer megbízhatóságát és pontosságát (10).
|
Az eredmények értelmezése és a vizsgálati vegyi anyagok korrozivitási osztályba sorolása
|
27.
|
A vizsgálati vegyi anyag membránbarrierre történő felvitele és a barrieren való áthatolása között eltelt (percben mért) időt használják a vizsgálati vegyi anyag – ENSZ GHS szerinti korrozivitási alkategóriák (1) és adott esetben az ENSZ csomagolási csoportjai (16) szerinti – osztályozására. Az egyes javasolt vizsgálati módszerekre nézve meghatározzák a három korrozivitási alkategóriát elválasztó időértékeket. A kategóriákat elválasztó időértékekre vonatkozó végső döntés során törekedni kell arra, hogy minimalizálják a korrozív veszély alulkategorizálását (vagyis az álnegatív eredményeket). E vizsgálati iránymutatás keretén belül a Corrositex® 3. táblázatban feltüntetett időértékeit kell figyelembe venni, mivel jelenleg ez az egyetlen vizsgálati módszer tartozik a vizsgálati iránymutatáshoz (7).
3. táblázat
Corrositex® előrejelzési modell
|
Átlagos áthatolási idő (percben)
|
ENSZ GHS előrejelzés (32)
|
|
1. kategóriába sorolt vizsgálati vegyi anyagok (30) (a módszer kategorizálási vizsgálata alapján)
|
2. kategóriába sorolt vizsgálati vegyi anyagok (31) (a módszer kategorizálási vizsgálata alapján)
|
|
0–3 perc
|
0–3 perc
|
Korrozív opcionális 1A alkategória
|
|
> 3–60 perc
|
> 3–30 perc
|
Korrozív opcionális 1B alkategória
|
|
> 60–240 perc
|
> 30–60 perc
|
Korrozív opcionális 1C alkategória
|
|
> 240 perc
|
> 60 perc
|
Nem korrozív
|
|
ADATOK ÉS JELENTÉS
Adatok
|
28.
|
Minden kísérlet esetében táblázatba kell foglalni a vizsgálati vegyi anyag és a pozitív kontrollanyag(ok) felvitele és a barrieren való áthatolás között eltelt (percben mért) időt az egyes ismétlések adataként, az átlagérték ± a szórás megadásával.
|
Vizsgálati jelentés
|
29.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálati vegyi anyag és kontrollként szolgáló anyagok:
|
—
|
Egy összetevőből álló anyag: kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján;
|
|
—
|
több összetevőből álló anyagok, UVCB-k és keverékek: amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai jellemzik;
|
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
eredet, gyártási szám, ha ismert;
|
|
—
|
a vizsgálati vegyi anyag/kontrollanyag kezelése a vizsgálatot megelőzően, ha történt ilyen (pl. melegítés, őrlés);
|
|
—
|
a vizsgálati vegyi anyag stabilitása, felhasználási határideje vagy ismételt elemzésének napja, ha ismert;
|
|
|
|
Vivőanyag:
|
—
|
név, (adott esetben) koncentráció, alkalmazott térfogat;
|
|
—
|
a vivőanyag megválasztásának indokolása.
|
|
|
|
Az alkalmazott in vitro membránbarrier-modell és protokoll, ideértve az igazolt pontosságot és megbízhatóságot
|
|
|
Vizsgálati körülmények:
|
—
|
az alkalmazott műszer és az elkészítési eljárások leírása;
|
|
—
|
az alkalmazott in vitro membránbarrier eredete és összetétele;
|
|
—
|
az indikátoroldat összetétele és jellemzői;
|
|
—
|
a vizsgálati vegyi anyag és a kontrollanyagok mennyisége;
|
|
—
|
az időskálán alapuló kategorizálási vizsgálat leírása és indokolása;
|
|
—
|
megfigyelési időszakok;
|
|
—
|
az alkalmazott értékelési és osztályozási kritériumok leírása;
|
|
—
|
a vizsgálati módszer végrehajtásában való jártasság bizonyítása a módszer rutinszerű használata előtt, a jártassági tesztanyagok vizsgálata révén.
|
|
|
|
Eredmények:
|
—
|
az egyes replikátumok egyedi vizsgálatából és kontrollmintáiból nyert egyedi nyers adatok táblázatba foglalása;
|
|
—
|
a megfigyelt egyéb hatások leírása;
|
|
—
|
az alkalmazott előrejelzési modellel/döntési kritériumokkal összhangban kialakított osztályozás.
|
|
Az eredmények értékelése
Következtetések
|
SZAKIRODALOM
|
(1)
|
Egyesült Nemzetek Szervezete (ENSZ) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Fifth Revised Edition, UN New York and Geneva, 2013. Elérhető a következő címen:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
E melléklet B.4., Akut bőrirritáció/bőrkorrózió fejezete.
|
|
(3)
|
E melléklet B.40bis., In vitro bőrkorrózió: rekonstruált emberi felhámmodellen végzett vizsgálati módszer című fejezete.
|
|
(4)
|
E melléklet B.40., In vitro bőrkorrózió: transzkután elektromos rezisztencia (TER) vizsgálat című fejezete.
|
|
(5)
|
OECD (2015). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203). Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(6)
|
Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhutter, H.-G. and Liebsch, M. (1998). The ECVAM International Validation Study on In vitro Tests for Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxicology In vitro 12, 483-524.
|
|
(7)
|
ICCVAM (1999). Corrositex®. An In vitro Test Method for Assessing Dermal Corrosivity Potential of Chemicals. The Results of an Independent Peer Review Evaluation Coordinated by ICCVAM, NTP and NICEATM. NIEHS, NIH Publication (No 99-4495.)
|
|
(8)
|
Gordon V.C., Harvell J.D. and Maibach H.I. (1994). Dermal Corrosion, the Corrositex® System: A DOT Accepted Method to Predict Corrosivity Potential of Test Materials. In vitro Skin Toxicology-Irritation, Phototoxicity, Sensitization. Alternative Methods in Toxicology 10, 37-45.
|
|
(9)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications. Series on testing and Assessment (No 34).
|
|
(10)
|
OECD (2014). Performance Standards for the Assessment of Proposed Similar or Modified In vitro Membrane Barrier Test Method for Skin Corrosion in Relation to TG 435. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen:http://www.oecd.org/chemicalsafety/testing/PerfStand-TG430-June14.pdf.
|
|
(11)
|
ECVAM (2001). Statement on the Application of the CORROSITEX® Assay for Skin Corrosivity Testing. 15th Meeting of ECVAM Scientific Advisory Committee (ESAC), Ispra, Italy. ATLA 29, 96-97.
|
|
(12)
|
U.S. DOT (2002). Exemption DOT-E-10904 (Fifth Revision). (September 20, 2002). Washington, D.C., U.S. DOT.
|
|
(13)
|
E melléklet B.46., In vitro bőrirritáció: Rekonstruált emberi felhámmodellen végzett vizsgálati módszer című fejezete. ICCVAM (2004). ICCVAM Recommended Performance Standards for In vitro Test Methods for Skin Corrosion. NIEHS, NIH Publication No 04-4510. Elérhető a következő címen:http://www.ntp.niehs.nih.gov/iccvam/docs/dermal_docs/ps/ps044510.pdf.
|
|
(14)
|
U.S. EPA (1996). Method 1120, Dermal Corrosion. Elérhető a következő címen:http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/1120.pdf.
|
|
(15)
|
Egyesült Nemzetek Szervezete (ENSZ) (2013). UN Recommendations on the Transport of Dangerous Goods, Model Regulations, 18th Revised Edition (Part, Chapter 2.8), UN, 2013. Elérhető a következő címen:http://www.unece.org/fileadmin/DAM/trans/danger/publi/unrec/rev18/English/Rev18_Volume1_Part2.pdf.
|
Függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság
: a vizsgálati módszerrel kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálati módszer teljesítményének mutatója, valamint a relevanciájának egyik megítélési szempontja. E kifejezést gyakran használják az „egyezés” megfelelőjeként, amely egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát fejezi ki (9).
Vegyi anyag
: anyag vagy keverék.
Kémiai érzékelő rendszer
: vizuális vagy elektronikus mérési rendszer indikátoroldattal, amely reagál a vizsgálati vegyi anyagok jelenlétére, például azáltal, hogy módosul a vizsgálati vegyi anyag jelenlétére színváltozással reagáló pH-indikátor festék vagy festékkombináció, illetve más típusú kémiai vagy elektrokémiai reakciók által.
Egyezés
: a kategorikus eredményt adó vizsgálati módszerek teljesítményének mutatója, valamint a relevanciájának egyik megítélési szempontja. A kifejezést esetenként használják a pontosság megfelelőjeként, és úgy határozzák meg, mint a helyesen pozitívként vagy negatívként besorolt összes vizsgálati vegyi anyag aránya. Az egyezés nagy mértékben függ a vizsgálat tárgyát képező vizsgálati vegyi anyag típusaiban előforduló pozitív eredmények gyakoriságától (9).
GHS (a vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszere)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (1).
IATA
: integrált vizsgálati és értékelési megközelítés.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő legalább 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
NK
: nem korrozív.
Teljesítményszabványok
: hitelesített referenciamódszeren alapuló szabványok, amelyek a javasolt, végrehajtás és funkcionalitás szempontjából hasonló vizsgálati módszer összehasonlíthatósági értékelésének alapjául szolgálnak. Ide tartoznak: i. a vizsgálati módszer alapvető összetevői; ii. a referencia-vegyianyagok minimális jegyzéke, amely a validált vizsgálati módszer által nyújtott teljesítmény elfogadhatóságának igazolására használt vegyi anyagokból került összeállításra; és iii. a validált vizsgálati módszer eredményei alapján meghatározott, azokhoz hasonló megbízhatósági és pontossági szint, amelyet a javasolt vizsgálati módszernek teljesítenie kell a referencia-vegyianyagok minimális jegyzéke szerinti értékelése során (9).
Relevancia
: a vizsgálati módszer és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálati módszer mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálati módszer pontosságát (az eredmények egyezését) figyelembe kell venni (9).
Megbízhatóság
: a vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg (9).
Érzékenység
: az összes olyan pozitív/aktív vegyi anyag aránya, amelyet a vizsgálati módszer helyesen sorolt be. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (9).
In vivo bőrkorrózió
: a vizsgálati vegyi anyag alkalmazását követően négy órán belül megjelenő, visszafordíthatatlan bőrkárosodás, azaz a felhámon át az irhára is átterjedő látható szövetelhalás. A korróziós tünetek jellemzően fekély, vérzés, véres hegesedés és – 14 napos megfigyelés végén – a bőr kifehéredése miatt elszíneződés, teljes szőrhullásos területek és hegek. A kérdéses lézió értékeléséhez figyelembe kell venni a kórszövettant.
Specificitás
: a vizsgálati módszerrel helyesen besorolt összes negatív/inaktív vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (9).
Anyag
: olyan természetes állapotban előforduló vagy gyártási eljárásból származó kémiai elem és vegyületei, amely a stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott eljárásból származó szennyező anyagokat is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
UVCB
: ismeretlen vagy változó összetételű anyagok, valamint komplex reakciótermékek vagy biológiai anyagok.
B.66. STABILAN TRANSZFEKTÁLT TRANSZAKTIVÁCIÓS IN VITRO VIZSGÁLATOK ÖSZTROGÉNRECEPTOROK AGONISTÁINAK ÉS ANTAGONISTÁINAK ÉSZLELÉSÉRE
ÁLTALÁNOS BEVEZETÉS
Az OECD teljesítményalapú vizsgálati iránymutatása
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 455. vizsgálati iránymutatásában (2016) leírt módszerrel. A 455. teljesítményalapú vizsgálati iránymutatás (PBTG) az ösztrogénreceptorok agonistáinak és antagonistáinak észlelésére szolgáló stabilan transzfektált transzaktivációs in vitro vizsgálatok (ER TA vizsgálatok) módszertanát ismerteti. Az iránymutatás több mechanikusan és funkcionálisan hasonló vizsgálati módszert tartalmaz az ösztrogénreceptor (vagyis az ERα és/vagy ERβ ösztrogénreceptor) agonistáinak és antagonistáinak azonosítására, és a veszély értékelésére szolgáló új és átdolgozott vizsgálati módszerek validálásáról és nemzetközi elfogadásáról szóló OECD-irányelvben (1) megfogalmazott validálási elvekkel összhangban megkönnyíti az új – hasonló vagy módosított – vizsgálati módszerek kidolgozását. A teljesítményalapú vizsgálati iránymutatás alapjául szolgáló, teljes körűen validált (a 2. és a 3. függelékben ismertetett) vizsgálati referenciamódszerek a következők:
|
—
|
a stabilan transzfektált transzaktivációs (STTA) vizsgálat (2), amely (h) ERα-HeLa-9903 sejtvonalat alkalmaz; valamint
|
|
—
|
a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat (3), amely VM7Luc4E2 sejtvonalat (33) alkalmaz, ami elsősorban a humán ösztrogénreceptor α-t, kisebb mértékben a humán ösztrogénreceptor β-t fejezi ki (4) (5).
|
Az ugyanezt a veszélyességi végpontot célzó hasonló vizsgálatok kidolgozásához és validálásához rendelkezésre állnak és alkalmazandók teljesítményszabványok (6) (7). A teljesítményszabványok lehetővé teszik a 455. teljesítményalapú vizsgálati iránymutatás időben történő módosítását, hogy az új, hasonló vizsgálatokat be lehessen építeni az átdolgozott iránymutatásba; hasonló vizsgálatok azonban csak azután foglalhatók bele az iránymutatásba, hogy az OECD felülvizsgálta és jóváhagyta a teljesítményszabványok alkalmazását. A 455. vizsgálati iránymutatás alá tartozó vizsgálatok egységesen felhasználhatók az OECD-tagállamok ösztrogénreceptor transzaktivációs vizsgálatok eredményeire vonatkozó követelményeinek kialakításához, ugyanakkor biztosítják az adatok kölcsönös elfogadásának előnyeit.
|
A vizsgálati módszerhez tartozó vizsgálatok háttere és elvei
|
2.
|
Az OECD 1998-ban kiemelt fontosságú tevékenységet indított az endokrin rendszert esetlegesen károsító vegyi anyagok kiszűrésére és vizsgálatára vonatkozó meglévő vizsgálati iránymutatások felülvizsgálata, valamint új vizsgálati iránymutatások kidolgozása érdekében. Az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére szolgáló fogalmi keretét 2012-ben átdolgozták. Az elvi keretrendszer eredeti és átdolgozott változatát mellékletként csatolták az endokrin rendszert károsító vegyi anyagok értékelésére vonatkozó szabványosított vizsgálati iránymutatásokról szóló OECD-iránymutatáshoz (8). Az elvi keretrendszer öt szintet határoz meg, amelyek mindegyike a biológiai komplexitás egy különálló szintjének felel meg. Az e vizsgálati módszer keretében ismertetett ösztrogénreceptor (ER) transzaktivációs (TA) vizsgálatok a 2. szinten helyezkednek el, amely magában foglalja „a kiválasztott endokrin mechanizmus(ok)ról/útvonal(ak)ról adatokat szolgáltató in vitro vizsgálatokat”. Ez a vizsgálati módszer in vitro transzaktivációs (TA) vizsgálatokra alkalmas, amelyek célja az ösztrogénreceptor (ER) agonistáinak és antagonistáinak azonosítása.
|
|
3.
|
Az ösztrogének ösztrogénreceptorokkal való kölcsönhatása befolyásolhatja az ösztrogén kontrollálta gének átírását (transzkripcióját), ami sejtes folyamatok indukciójához vagy gátlásához vezethet, beleértve azokat, amelyek szükségesek a sejtosztódáshoz, a normál magzati fejlődéshez és a szaporodási funkciókhoz (9) (10) (11). A normál ösztrogénrendszerek zavara károsan befolyásolhatja a normál egyedfejlődést (ontogenezist), a reprodukciós egészséget és a reproduktív rendszer integritását.
|
|
4.
|
Az in vitro transzaktivációs vizsgálatokban a receptor és ligandumja közötti direkt vagy indirekt kölcsönhatások egy riportergén bekapcsolását (transzkripcióját) szabályozzák. Az ilyen vizsgálatokat széles körben használják specifikus sejtmagi receptorok, például ösztrogénreceptorok által szabályozott génkifejeződés értékelésére (12) (13) (14) (15) (16). Javasolt felhasználási területük az ösztrogénreceptorok által szabályozott ösztrogénhatású transzaktiváció észlelése (17) (18) (19). A sejtmagi ösztrogénreceptoroknak legalább két fő altípusa létezik, az α és a β, amelyeket különböző gének kódolnak. A megfelelő fehérjéknek eltérő biológiai funkciói vannak, eltérő a szöveti jelenlétük és ligandumkötő affinitásuk (20) (21) (22) (23) (24) (25) (26). A sejtmagi ösztrogénreceptor α a klasszikus ösztrogénválaszt közvetíti (27) (28) (29) (30), ezért az ösztrogénreceptorok aktiválásának és gátlásának mérésére kifejlesztett modellek többsége az ösztrogénreceptor α-ra specifikus. A vizsgálatokat arra használják, hogy azonosítsanak olyan vegyi anyagokat, amelyek aktiválják (vagy gátolják) az ösztrogénreceptorokat a ligandumkötésben, a kötés révén létrejött receptor-ligandum komplex specifikusan DNS válaszelemekhez kötődik és transzaktivál egy riportergént, megnövelve ezáltal a markerfehérje celluláris kifejeződését. E vizsgálatokban különféle riportergének alkalmazhatók. A luciferáz alapú rendszerekben a luciferáz enzim a luciferin szubsztrátot átalakítja, miközben biolumineszcens fény keletkezik, amely luminométerrel számszerűen mérhető. Gyakori riporterre példák lehetnek még a fluoreszcens fehérjék génjei és a lacZ gén, ez utóbbi a β-galaktozidázt kódolja, egy olyan enzimet, amely képes a színtelen X-gal szubsztrátot (5-bromo-4-kloro-indolil-galaktopiranozid) kék termékké alakítani, amelynek mennyisége spektrofotométerrel mérhető. Ezek a riporterek kereskedelmi forgalomban kapható vizsgálati készletekkel gyorsan és költséghatékonyan értékelhetők.
|
|
5.
|
a stabilan transzfektált transzaktivációs és a VM7Luc transzaktivációs vizsgálatok validálási tanulmányai igazolták, hogy e vizsgálatok tervezett felhasználásuk tekintetében relevánsak és megbízhatóak (3) (4) (5) (30). A lumineszcencián alapuló, mellrák sejtvonalat alkalmazó ösztrogénreceptor transzaktivációs vizsgálatok teljesítményszabványait az ICCVAM belefoglalta a „LUMI-CELL® ösztrogénreceptor (VM7Luc ösztrogénreceptor transzaktivációs) vizsgálati módszer: a vegyi anyagok humán ösztrogénreceptorokra kifejtett agonista és antagonista aktivitásának azonosítására szolgáló in vitro vizsgálat” című (3), vizsgálati módszert értékelő jelentésébe. Ezeket a teljesítményszabványokat módosították, hogy alkalmazhatók legyenek mind a stabilan transzfektált transzaktivációs, mind a VM7Luc transzaktivációs vizsgálatra (2).
|
|
6.
|
A vizsgálati módszerben használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A transzaktivációs vizsgálatok alkalmazási területe és korlátai
|
7.
|
E vizsgálatokat szűrési és rangsorolási célra ajánlják, ugyanakkor a mechanizmusra vonatkozólag is képesek adatokat szolgáltatni, amelyek felhasználhatók egy bizonyítékok súlyozásán (WoE) alapuló megközelítésben. A vizsgálatok tárgya egy vegyi anyag ösztrogénreceptorokhoz való kötődése által előidézett transzaktiváció in vitro rendszerben. Ezért az eredményekből nem lehet közvetlenül következtetni az intakt in vivo endokrin rendszerben megvalósuló összetett jelátvitelre és szabályozásra.
|
|
8.
|
Habár az ösztrogénreceptorok által kiváltott transzaktivációt az endokrin zavarok (ED) hátterében álló egyik fő mechanizmusként tartják számon, más mechanizmusok is okozhatnak endokrin zavarokat, többek között i. az endokrin rendszeren belüli más receptorokkal és enzimrendszerekkel való interakciók, ii. hormonszintézis, iii. a hormonok metabolikus aktiválása és/vagy inaktiválása, iv. hormonok elszállítása a célszövetekhez és v. hormonok kiürülése a szervezetből. Ezekkel a hatásmechanizmusokkal a tárgyalt vizsgálati módszerhez kapcsolódó vizsgálatok egyike sem foglalkozik.
|
|
9.
|
E vizsgálati módszer célja, hogy felmérje a vegyi anyagok ösztrogénreceptor-dependens transzkripció aktiválására (vagyis agonistaként való viselkedésre) és gátlására (vagyis antagonistaként való viselkedésre) való képességét. Egyes vegyi anyagok a sejttípustól függően agonistaként és antagonistaként is viselkedhetnek, ezek az úgynevezett szelektív ösztrogénreceptor modulátorok (SERM). Az e vizsgálatokban negatívnak minősített vegyi anyagok tovább vizsgálhatók egy ösztrogénreceptorhoz kötődési tesztben, mielőtt megállapításra kerülne, hogy nem kötődnek a receptorhoz. Tekintettel az in vitro sejtrendszerek korlátozott metabolikus képességére, a vizsgálatok valószínűleg csak a kiindulási molekula hatásáról szolgáltatnak információt. Mivel a validálás során csak egykomponensű anyagokat használtak, a vizsgálat keverékekre való alkalmazhatósága nem vetődött fel. Mindazonáltal a vizsgálati módszer elvileg alkalmazható több összetevőből álló anyagok, UVCB-k és keverékek vizsgálatára. A vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, több összetevőből álló anyagokon, UVCB-ken és keveréken történő alkalmazása előtt meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény.
|
|
10.
|
Az 1. táblázat tájékoztatási céllal ismerteti az e vizsgálati módszer által leírt mindkét teljes körűen validált vizsgálati referenciamódszerben tesztelt 34 anyagra vonatkozó agonista vizsgálati eredményeket. A szóban forgó anyagok közül 26-ot egyértelmű ösztrogénreceptor agonistaként határoztak meg, a többi 8-at negatívnak minősítették a közzétett jelentések, többek között az in vitro ösztrogénreceptor-kötődési és transzaktivációs vizsgálatok és/vagy az uterotrofikus vizsgálat alapján (2) (3) (18) (31) (32) (33) (34). A 2. táblázat pedig ismerteti az e vizsgálati módszer által leírt mindkét teljes körűen validált vizsgálati referenciamódszerben vizsgált 15 anyagra vonatkozó antagonista vizsgálati eredményeket. Ezen anyagok közül 4-et egyértelmű/feltételezett ösztrogénreceptor antagonistaként határoztak meg, míg 10-et negatívnak minősítettek a közzétett jelentések, többek között az in vitro ösztrogénreceptor-kötődési és transzaktivációs vizsgálatok alapján (2) (3) (18) (31). Az 1. és 2. táblázatban összefoglalt adatok vonatkozásában 100 %-os egyezés mutatható ki a két vizsgálati referenciamódszer között valamennyi anyag besorolása tekintetében (az antagonista vizsgálatban szereplő mifepriszton kivételével), és minden anyagot helyesen osztályoztak ösztrogénreceptor agonistaként, ösztrogénreceptor antagonistaként vagy negatívként. E vegyianyag-csoportról, valamint a validálási tanulmányok során a stabilan transzfektált transzaktivációs és a VM7Luc ösztrogénreceptor transzaktivációs vizsgálatban tesztelt egyéb vegyi anyagokról az ösztrogénreceptor transzaktivációs vizsgálat teljesítményszabványai nyújtanak bővebb tájékoztatást (6) (7), 2. függelék (1., 2., és 3. táblázat).
|
1. táblázat
A stabilan transzfektált transzaktivációs és a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat eredményeinek áttekintése azon anyagok vonatkozásában, amelyeket mindkét agonista vizsgálatban teszteltek, és ösztrogénreceptor agonistaként (POZ) vagy negatívként (NEG) azonosítottak
|
|
Anyag
|
CAS-szám
|
STTA-vizsgálat (35)
|
VM7Luc ER TA vizsgálat (36)
|
Osztályozás alapját képező adatforrás (38)
|
|
ER TA aktivitás
|
PC10 érték (M)
|
PC50 érték (2) (M)
|
ER TA aktivitás
|
EC50 érték (2), (37) (M)
|
Egyéb ER TA-k (34)
|
ER- kötés
|
Uterotrofikus
|
|
1
|
17ß-ösztradiol (1)
|
50-28-2
|
POZ
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POZ
|
5,63 × 10-12
|
POZ (227/227)
|
POZ
|
POZ
|
|
2
|
17α-ösztradiol (1)
|
57-91-0
|
POZ
|
7,24 × 10-11
|
6,44 × 10-10
|
POZ
|
1,40 × 10-9
|
POZ (11/11)
|
POZ
|
POZ
|
|
3
|
17α-etinilösztradiol (1)
|
57-63-6
|
POZ
|
< 1,00 × 10-11
|
< 1,00 × 10-11
|
POZ
|
7,31 × 10-12
|
POZ (22/22)
|
POZ
|
POZ
|
|
4
|
17β-trenbolon
|
10161-33-8
|
POZ
|
1,78 × 10-8
|
2,73 × 10-7
|
POZ
|
4,20 × 10-8
|
POZ (2/2)
|
NT
|
NT
|
|
5
|
19-nortesztoszteron (1)
|
434-22-0
|
POZ
|
9,64 × 10-9
|
2,71 × 10-7
|
POZ
|
1,80 × 10-6
|
POZ (4/4)
|
POZ
|
POZ
|
|
6
|
4-kumolfenol (1)
|
599-64-4
|
POZ
|
1,49 × 10-7
|
1,60 × 10-6
|
POZ
|
3,20 × 10-7
|
POZ (5/5)
|
POZ
|
NT
|
|
7
|
4-terc-oktilfenol (1)
|
140-66-9
|
POZ
|
1,85 × 10-9
|
7,37 × 10-8
|
POZ
|
3,19 × 10-8
|
POZ (21/24)
|
POZ
|
POZ
|
|
8
|
Apigenin (1)
|
520-36-5
|
POZ
|
1,31 × 10-7
|
5,71 × 10-7
|
POZ
|
1,60 × 10-6
|
POZ (26/26)
|
POZ
|
NT
|
|
9
|
Atrazin (1)
|
1912-24-9
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (30/30)
|
NEG
|
NT
|
|
10
|
Biszfenol-A (1)
|
80-05-7
|
POZ
|
2,02 × 10-8
|
2,94 × 10-7
|
POZ
|
5,33 × 10-7
|
POZ (65/65)
|
POZ
|
POZ
|
|
11
|
Biszfenol-B (1)
|
77-40-7
|
POZ
|
2,36 × 10-8
|
2,11 × 10-7
|
POZ
|
1,95 × 10-7
|
POZ (6/6)
|
POZ
|
POZ
|
|
12
|
Butilbenzil-ftalát (1)
|
85-68-7
|
POZ
|
1,14 × 10-6
|
4,11 × 10-6
|
POZ
|
1,98 × 10-6
|
POZ (12/14)
|
POZ
|
NEG
|
|
13
|
Kortikoszteron (1)
|
50-22-6
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (6/6)
|
NEG
|
NT
|
|
14
|
Kumesztrol (1)
|
479-13-0
|
POZ
|
1,23 × 10-9
|
2,00 × 10-8
|
POZ
|
1,32 × 10-7
|
POZ (30/30)
|
POZ
|
NT
|
|
15
|
Daidzein (1)
|
486-66-8
|
POZ
|
1,76 × 10-8
|
1,51 × 10-7
|
POZ
|
7,95 × 10-7
|
POZ (39/39)
|
POZ
|
POZ
|
|
16
|
Dietilstilbösztrol (1)
|
56-53-1
|
POZ
|
< 1,00 × 10-11
|
2,04 × 10-11
|
POZ
|
3,34 × 10-11
|
POZ (42/42)
|
POZ
|
NT
|
|
17
|
Di-n-butil-ftalát
|
84-74-2
|
POZ
|
4,09 × 10-6
|
|
POZ
|
4,09 × 10-6
|
POZ (6/11)
|
POZ
|
NEG
|
|
18
|
Etilparabén
|
120-47-8
|
POZ
|
5,00 × 10-6
|
(nincs PC50)
|
POZ
|
2,48 × 10-5
|
POZ
|
|
NT
|
|
19
|
Esztron (1)
|
53-16-7
|
POZ
|
3,02 × 10-11
|
5,88 × 10-10
|
POZ
|
2,34 × 10-10
|
POZ (26/28)
|
POZ
|
POZ
|
|
20
|
Geniszten (1)
|
446-72-0
|
POZ
|
2,24 × 10-9
|
2,45 × 10-8
|
POZ
|
2,71 × 10-7
|
POZ (100/102)
|
POZ
|
POZ
|
|
21
|
Haloperidol
|
52-86-8
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
22
|
Kempferol (1)
|
520-18-3
|
POZ
|
1,36 × 10-7
|
1,21 × 10-6
|
POZ
|
3,99 × 10-6
|
POZ (23/23)
|
POZ
|
NT
|
|
23
|
Kepone (1)
|
143-50-0
|
POZ
|
7,11 × 10-7
|
7,68 × 10-6
|
POZ
|
4,91 × 10-7
|
POZ (14/18)
|
POZ
|
NT
|
|
24
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
25
|
Linuron (1)
|
330-55-2
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (8/8)
|
NEG
|
NT
|
|
26
|
meso-Hexestrol (1)
|
84-16-2
|
POZ
|
< 1,00 × 10-11
|
2,75 × 10-11
|
POZ
|
1,65 × 10-11
|
POZ (4/4)
|
POZ
|
NT
|
|
27
|
Metil-tesztoszteron (1)
|
58-18-4
|
POZ
|
1,73 × 10-7
|
4,11 × 10-6
|
POZ
|
2,68 × 10-6
|
POZ (5/6)
|
POZ
|
NT
|
|
28
|
Morin
|
480-16-0
|
POZ
|
5,43 × 10-7
|
4,16 × 10-6
|
POZ
|
2,37 × 10-6
|
POZ (2/2)
|
POZ
|
NT
|
|
29
|
Noretinodrel (1)
|
68-23-5
|
POZ
|
1,11 × 10-11
|
1,50 × 10-9
|
POZ
|
9,39 × 10-10
|
POZ (5/5)
|
POZ
|
NT
|
|
30
|
p,p’-Metoxiklór (1)
|
72-43-5
|
POZ
|
1,23 × 10-6
|
(nincs PC50) (2)
|
POZ
|
1,92 × 10-6
|
POZ (24/27)
|
POZ
|
POZ
|
|
31
|
Fenobarbitál (1)
|
57-30-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (2/2)
|
NEG
|
NT
|
|
32
|
Reszerpin
|
50-55-5
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
33
|
Spironolakton (1)
|
52-01-7
|
NEG
|
—
|
—
|
NEG
|
—
|
NEG (4/4)
|
NEG
|
NT
|
|
34
|
Tesztoszteron
|
58-22-0
|
POZ
|
2,82 × 10-8
|
9,78 × 10-6
|
POZ
|
1,75 × 10-5
|
POZ (5/10)
|
POZ
|
NT
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám; M = moláris; EC50 = a vizsgálati anyag maximális tényleges hatásának felét kiváltó koncentráció; NEG = negatív; POZ = pozitív; NT = nem tesztelt; PC10 (és PC50) = a vizsgálati anyagnak az a koncentrációja, amelynél a kiváltott válasz a pozitív kontroll (E2, 1 nm) egyes lemezeken indukált válaszának 10 %-a (illetve PC50 esetében 50 %-a).
|
2. táblázat
A stabilan transzfektált transzaktivációs és a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat eredményeinek összevetése azon anyagok vonatkozásában, amelyeket mindkét antagonista vizsgálatban teszteltek, és ösztrogénreceptor antagonistaként (POZ) vagy negatívként (NEG) azonosítottak
|
|
Anyag (3)
|
CAS-szám
|
ER STTA-vizsgálat (40)
|
VM7Luc ER TA vizsgálat (41)
|
ER STTA vizsgált anyagok hatása (43)
|
ICCVAM (44) konszenzuson alapuló besorolás
|
MeSH (45) Kémiai osztály
|
Termékosztály (46)
|
|
ER TA aktivitás
|
IC50 érték (39) (M)
|
ER TA aktivitás
|
IC50 érték (39), (42) (M)
|
|
1
|
4-hidroxitamoxifén
|
68047-06-3
|
POZ
|
3,97 × 10-9
|
POZ
|
2,08 × 10-7
|
mérsékelten POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
2
|
Dibenzo[a,h]antracén
|
53-70-3
|
POZ
|
Nincs IC50
|
POZ
|
Nincs IC50
|
POZ
|
FP
|
Policiklikus vegyület
|
Laboratóriumi vegyi anyag, természetes termék
|
|
3
|
Mifepriszton
|
84371-65-3
|
POZ
|
5,61 × 10-6
|
NEG
|
—
|
enyhén POZ
|
NEG
|
Szteroid
|
Gyógyszer
|
|
4
|
Raloxifen-hidroklorid
|
82640-04-8
|
POZ
|
7,86 × 10-10
|
POZ
|
1,19 × 10-9
|
mérsékelten POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
5
|
Tamoxifén
|
10540-29-1
|
POZ
|
4,91 × 10-7
|
POZ
|
8,17 × 10-7
|
POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
6
|
17β-ösztradiol
|
50-28-2
|
NEG
|
—
|
NEG
|
—
|
FN
|
FN
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
7
|
Apigenin
|
520-36-5
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Heterociklikus vegyület
|
Festék, természetes termék, gyógyszerészeti köztes termék
|
|
8
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
NEG
|
—
|
NEG
|
FN
|
Heterociklikus vegyület
|
Gyomirtó szer
|
|
9
|
Di-n-butil-ftalát
|
84-74-2
|
NEG
|
—
|
NEG
|
—
|
NEG
|
NEG
|
Észter, ftálsav
|
Kozmetikai összetevő, ipari vegyi anyag, lágyítószer
|
|
10
|
Fenarimol
|
60168-88-9
|
NEG
|
—
|
NEG
|
—
|
nem vizsgált
|
FN
|
Heterociklikus vegyület, pirimidin
|
Gombaölő szer
|
|
11
|
Flavon
|
525-82-6
|
NEG
|
—
|
NEG
|
—
|
FN
|
FN
|
Flavonid, heterociklikus vegyület
|
Természetes termék, gyógyszer
|
|
12
|
Flutamid
|
13311-84-7
|
NEG
|
—
|
NEG
|
—
|
NEG
|
FN
|
Amid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
13
|
Geniszten
|
446-72-0
|
NEG
|
—
|
NEG
|
—
|
FN
|
NEG
|
Flavonid, heterociklikus vegyület
|
Természetes termék, gyógyszer
|
|
14
|
p-n-nonilfenol
|
104-40-5
|
NEG
|
—
|
NEG
|
—
|
nem vizsgált
|
NEG
|
Fenol
|
Köztes vegyi anyag
|
|
15
|
Rezveratrol
|
501-36-0
|
NEG
|
—
|
NEG
|
—
|
FN
|
NEG
|
Szénhidrogén (ciklikus)
|
Természetes termék
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám; M = moláris; IC50 = a vizsgálati anyag maximális gátló hatásának felét kiváltó koncentráció; NEG = negatív; FN = feltételezhetően negatív; POZ = pozitív; FP = feltételezhetően pozitív.
|
AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI
A vizsgálat alapvető összetevői
|
11.
|
Ez a vizsgálati módszer olyan vizsgálatokra alkalmazható, amelyek stabilan transzfektált vagy endogén ösztrogénreceptor α-t, valamint egy stabilan transzfektált, egy vagy több ösztrogén válaszelemmel szabályozott riportergén konstrukciót használnak, amelyben azonban más receptorok, például ösztrogénreceptor β is jelen lehet. Ezek a vizsgálat alapvető összetevői.
|
Kontrollok
|
12.
|
Meg kell határozni az egyes agonista és antagonista vizsgálatokhoz javasolt, párhuzamosan tesztelt referenciaszabvány alapját. A párhuzamos kontrollok (negatív, pozitív és oldószeres) arra szolgálnak, hogy igazolják a vizsgálat tesztkörülmények közötti megfelelő működését, és megteremtsék az alapját az egyes kísérletek összevetésének; általában részét képezik a szóban forgó kísérlet elfogadhatósági kritériumainak (1).
|
Standard minőség-ellenőrzési eljárások
|
13.
|
Az egyes vizsgálatokhoz meghatározott standard minőség-ellenőrzési eljárásokat kell végezni annak biztosítása érdekében, hogy a sejtvonalak több passzálást követően is stabilak maradjanak, mikoplazmától (vagyis bakteriális fertőzéstől) mentesek legyenek, és később is megismételhetőek legyenek az ösztrogénreceptor által közvetített várt válaszreakciók. Emellett ellenőrizni kell a sejtvonalak megfelelő azonosságát, valamint az egyéb fertőző ágensek (pl. gombák, élesztő és vírusok) hiányát.
|
A laboratórium jártasságának igazolása
|
14.
|
Mielőtt ismeretlen vegyi anyagokat tesztelnének az e vizsgálati módszerhez kapcsolódó bármely vizsgálattal, a laboratóriumoknak igazolniuk kell az adott vizsgálat alkalmazásában szerzett jártasságukat. A jártasság igazolásához minden laboratóriumnak tesztelnie kell az agonista vizsgálat 3. táblázatban feltüntetett 14 jártassági tesztanyagát és az antagonista vizsgálat 4. táblázatban szereplő 10 jártassági tesztanyagát. E jártassági vizsgálatok bizonyítják a vizsgálati rendszer reagálóképességét is. A jártassági tesztanyagok jegyzéke az ösztrogénreceptor transzaktivációs vizsgálatok teljesítményszabványaiban (6) megadott referenciaanyagok egy csoportjából tevődik össze. Ezek az anyagok kereskedelmi forgalomban beszerezhetők, olyan kémiai osztályokat képviselnek, amelyeknél gyakori az ösztrogénreceptor agonista vagy antagonista aktivitás, az ösztrogénreceptor agonistáktól és antagonistáktól elvárt potenciál megfelelő tartományát képviselik (erőstől a gyengéig) és vannak közöttük negatív besorolású anyagok is. A jártassági tesztanyagok vizsgálatát legalább kétszer, különböző napokon meg kell ismételni. A jártasság az egyes jártassági tesztanyagok megfelelő (pozitív/negatív) osztályba sorolásával igazolható. A vizsgálat elsajátítása során a jártassági vizsgálatot minden technikusnak meg kell ismételnie. Sejttípustól függően e jártassági tesztanyagok némelyike szelektív ösztrogénreceptor modulátorként viselkedhet, vagyis kifejthet agonista és antagonista aktivitást is. Azonban a jártassági tesztanyagok 3. és 4. táblázatban ismertetett besorolása ismert domináns aktivitásuk alapján történt, azt kell figyelembe venni a jártassági értékelés során.
|
|
15.
|
A vizsgálat teljesítményének igazolása és minőség-ellenőrzés céljából minden laboratóriumnak referenciaszabványok (pl. 17β-ösztradiol és tamoxifén), kontrollként szolgáló pozitív és negatív vegyi anyagok, valamint oldószeres kontrollanyagok (pl. DMSO) adataiból álló agonista és antagonista adatbázisokat kell létrehozniuk. Az induló adatbázist legalább 10 független agonista (pl. 17β-ösztradiol) és 10 független antagonista (pl. tamoxifén) vizsgálatmenet alapján kell létrehozni. Az adatbázist ki kell bővíteni e referenciaszabványok és oldószeres kontrollanyagok későbbi elemzéseiből származó eredményekkel, hogy az idő múlásával is biztosítani lehessen a biológiai vizsgálat konzisztenciáját és teljesítményét.
|
3. táblázat
Az agonista vizsgálathoz használatos jártassági tesztanyagok (14) jegyzéke
(47)
|
Szám (54)
|
Anyag
|
CAS-szám
|
Várt válasz (48)
|
STTA-vizsgálat
|
VM7Luc ER TA vizsgálat
|
MeSH kémiai osztály (52)
|
Termékosztály (53)
|
|
PC10 érték (M) (49)
|
PC50 érték (M) (49)
|
Vizsgálati koncentrációtartomány (M)
|
VM7Luc EC50 érték (M) (50)
|
Legmagasabb konc. dózisbehatároláshoz (M) (51)
|
|
14
|
Dietilstilbösztrol
|
56-53-1
|
POZ
|
< 1,00 × 10-11
|
2,04 × 10-11
|
10-14–10-8
|
3,34 × 10-11
|
3,73 × 10-4
|
Szénhidrogén (ciklikus)
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
12
|
17α-ösztradiol
|
57-91-0
|
POZ
|
4,27 × 10-11
|
6,44 × 10-10
|
10-11–10-5
|
1,40 × 10-9
|
3,67 × 10-3
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
15
|
meso-Hexestrol
|
84-16-2
|
POZ
|
< 1,00 × 10-11
|
2,75 × 10-11
|
10-11–10-5
|
1,65 × 10-11
|
3,70 × 10-3
|
Szénhidrogén (ciklikus), fenol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
11
|
4-terc-oktilfenol
|
140-66-9
|
POZ
|
1,85 × 10-9
|
7,37 × 10-8
|
10-11–10-5
|
3,19 × 10-8
|
4,85 × 10-3
|
Fenol
|
Köztes vegyi anyag
|
|
9
|
Geniszten
|
446-72-0
|
POZ
|
2,24 × 10-9
|
2,45 × 10-8
|
10-11–10-5
|
2,71 × 10-7
|
3,70 × 10-4
|
Flavonid, heterociklikus vegyület
|
Természetes termék, gyógyszer
|
|
6
|
Biszfenol-A
|
80-05-7
|
POZ
|
2,02 × 10-8
|
2,94 × 10-7
|
10-11–10-5
|
5,33 × 10-7
|
4,38 × 10-3
|
Fenol
|
Köztes vegyi anyag
|
|
2
|
Kempferol
|
520-18-3
|
POZ
|
1,36 × 10-7
|
1,21 × 10-6
|
10-11–10-5
|
3,99 × 10-6
|
3,49 × 10-3
|
Flavonid, heterociklikus vegyület
|
Természetes termék
|
|
3
|
Butilbenzil-ftalát
|
85-68-7
|
POZ
|
1,14 × 10-6
|
4,11 × 10-6
|
10-11–10-5
|
1,98 × 10-6
|
3,20 × 10-4
|
Karbonsav, észter, ftálsav
|
Lágyítószer, ipari vegyi anyag
|
|
4
|
p,p’-Methoxyklór
|
72-43-5
|
POZ
|
1,23 × 10-6
|
—
|
10-11–10-5
|
1,92 × 10-6
|
2,89 × 10-3
|
Szénhidrogén (halogénezett)
|
Növényvédő szer, állatgyógyászati hatóanyag
|
|
1
|
Etilparabén
|
120-47-8
|
POZ
|
5,00 × 10-6
|
—
|
10-11–10-5
|
2,48 × 10-5
|
6,02 × 10-3
|
Karbonsav, fenol
|
Gyógyszer, konzerváló szer
|
|
17
|
Atrazin
|
1912-24-9
|
NEG
|
—
|
—
|
10-10–10-4
|
—
|
4,64 × 10-4
|
Heterociklikus vegyület
|
Gyomirtó szer
|
|
20
|
Spironolakton
|
52-01-7
|
NEG
|
—
|
—
|
10-11–10-5
|
—
|
2,40 × 10-3
|
Lakton, szteroid
|
Gyógyszer
|
|
21
|
Ketokonazol
|
65277-42-1
|
NEG
|
—
|
—
|
10-11–10-5
|
—
|
9,41 × 10-5
|
Heterociklikus vegyület
|
Gyógyszer
|
|
22
|
Reszerpin
|
50-55-5
|
NEG
|
—
|
—
|
10-11–10-5
|
—
|
1,64 × 10-3
|
Heterociklikus vegyület, indol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám; EC50 = a vizsgálati anyag maximális tényleges hatásának felét kiváltó koncentráció; NEG = negatív; POZ = pozitív; PC10 (és PC50) = a vizsgálati anyagnak az a koncentrációja, amelynél a kiváltott válasz a pozitív kontroll (E2, 1 nm) egyes lemezeken indukált válaszának 10 %-a (illetve PC50 esetében 50 %-a).
|
4. táblázat
Az antagonista vizsgálathoz használatos jártassági tesztanyagok (10) jegyzéke
|
|
Anyag (4)
|
CAS-szám
|
ER STTA-vizsgálat (55)
|
VM7Luc ER TA vizsgálat (56)
|
ER STTA (55) vizsgált anyagok hatása
|
ICCVAM (59) konszenzuson alapuló besorolás
|
MeSH (60) Kémiai osztály
|
Termékosztály (61)
|
|
ER TA aktivitás
|
IC50 (M)
|
Vizsgálati koncentrációtartomány (M)
|
ER TA aktivitás
|
IC50
(57) (M)
|
Legmagasabb konc. dózisbehatároláshoz (M) (58)
|
|
1
|
4-hidroxitamoxifén
|
68047-06-3
|
POZ
|
3,97 × 10-9
|
10-12 –10-7
|
POZ
|
2,08 × 10-7
|
2,58 × 10-4
|
mérsékelten POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
2
|
Raloxifen-hidroklorid
|
82640-04-8
|
POZ
|
7,86 × 10-10
|
10-12 –10-7
|
POZ
|
1,19 × 10-9
|
1,96 × 10-4
|
mérsékelten POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
3
|
Tamoxifén
|
10540-29-1
|
POZ
|
4,91 × 10-7
|
10-10 –10-5
|
POZ
|
8,17 × 10-7
|
2,69 × 10-4
|
POZ
|
POZ
|
Szénhidrogén (ciklikus)
|
Gyógyszer
|
|
4
|
17β-ösztradiol
|
50-28-2
|
NEG
|
—
|
10-9 –10-4
|
NEG
|
—
|
3,67 × 10-3
|
negatívnak kell lennie (*4)
|
FN
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
5
|
Apigenin
|
520-36-5
|
NEG
|
—
|
10-9 –10-4
|
NEG
|
—
|
3,70 × 10-4
|
NEG
|
NEG
|
Heterociklikus vegyület
|
Festék, természetes termék, gyógyszerészeti köztes termék
|
|
6
|
Di-n-butil-ftalát
|
84-74-2
|
NEG
|
—
|
10-8 –10-3
|
NEG
|
—
|
3,59 × 10-3
|
NEG
|
NEG
|
Észter, ftálsav
|
Kozmetikai összetevő, ipari vegyi anyag, lágyítószer
|
|
7
|
Flavon
|
525-82-6
|
NEG
|
—
|
10-8 –10-3
|
NEG
|
—
|
4,50 × 10-4
|
negatívnak kell lennie (*4)
|
FN
|
Flavonid, heterociklikus vegyület
|
Természetes termék, gyógyszer
|
|
8
|
Geniszten
|
446-72-0
|
NEG
|
—
|
10-9 –10-4
|
NEG
|
—
|
3,70 × 10-4
|
negatívnak kell lennie (*4)
|
NEG
|
Flavonid, heterociklikus vegyület
|
Természetes termék, gyógyszer
|
|
9
|
p-n-nonilfenol
|
104-40-5
|
NEG
|
—
|
10-9 –10-4
|
NEG
|
—
|
4,54 × 10-4
|
nem vizsgált
|
NEG
|
Fenol
|
Köztes vegyi anyag
|
|
10
|
Rezveratrol
|
501-36-0
|
NEG
|
—
|
10-8 –10-3
|
NEG
|
—
|
4,38 × 10-4
|
negatívnak kell lennie (*4)
|
NEG
|
Szénhidrogén (ciklikus)
|
Természetes termék
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám; M = moláris; IC50 = a vizsgálati anyag maximális gátló hatásának felét kiváltó koncentráció; NEG = negatív; FN = feltételezhetően negatív; POZ = pozitív.
|
A vizsgálatmenet elfogadhatósági kritériumai
|
16.
|
A vizsgálatmenetek elfogadására vagy elvetésére az egyes kísérletekben alkalmazott referenciaszabványok és kontrollanyagok eredményeinek értékelése alapján kerül sor. A referenciaszabványok PC50 (EC50) vagy IC50 értékének meg kell felelnie a választott vizsgálat elfogadhatósági kritériumainak (ezeket a stabilan transzfektált transzaktivációs vizsgálat esetében a 2. függelék, a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat esetében a 3. függelék tartalmazza), és minden elfogadott kísérletben megfelelően kell besorolni az összes pozitív/negatív kontrollanyagot. A vizsgálat következetes végrehajtásának képességét bizonyítani kell azáltal, hogy a laboratóriumok adatbázist hoznak létre és tartanak fent a referenciaszabványok és kontrollanyagok adataiból (lásd a 15. pontot). A laboratóriumon belüli reprodukálhatóság mérésére használhatók a referenciaszabványok átlagához illesztett, több kísérletből származó paraméterekből álló görbe standard deviációi (SD) vagy variációs koefficiensei (CV). Ezenkívül érvényesülniük kell az elfogadhatósági kritériumokra vonatkozó alábbi elveknek is:
|
—
|
Elegendő mennyiségű adattal kell rendelkezni ahhoz, hogy számszerűen értékelni lehessen az ösztrogénreceptor aktivitást (agonista vizsgálat esetén) vagy gátlást (antagonista vizsgálat esetén) (vagyis a hatékonyságot és a potenciált).
|
|
—
|
A megfelelő érzékenység biztosítása érdekében a referenciaösztrogén referenciakoncentrációjánál kifejtett átlagos riporter aktivitásnak – a vivőanyagos (oldószeres) kontroll esetén tapasztalt aktivitáshoz képest – el kell érnie legalább a vizsgálatokban meghatározott minimumszintet. A stabilan transzfektált transzaktivációs és a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat esetén ez a minimumszint az egyes lemezeken vizsgált vivőanyagos kontrollok átlagának négyszerese.
|
|
—
|
A vizsgált koncentrációknak a vizsgálati vegyi anyagok oldhatósági tartományán belül kell maradniuk és nem okozhatnak citotoxicitást.
|
|
Az adatok elemzése
|
17.
|
A válaszok pozitív vagy negatív kategóriába sorolásakor az egyes vizsgálatokhoz meghatározott adatértelmezési eljárásra kell támaszkodni.
|
|
18.
|
Az elfogadhatósági kritériumok (16. pont) teljesítése jelzi a vizsgálat megfelelő működését, de nem garantálja az egyes vizsgálatmenetek adatainak pontosságát. Az adatok pontosságát az jelzi a legjobban, ha az első vizsgálatmenet eredményei megismételhetők. Amennyiben két vizsgálatmenet bizonyította az eredmény reprodukálhatóságát (például egy adott vizsgálati vegyi anyag mindkét vizsgálatmenetben pozitívnak bizonyult), akkor harmadik vizsgálatmenetre nincs szükség.
|
|
19.
|
Ha két vizsgálatmenet nem bizonyítja az eredmény reprodukálhatóságát (például egy vizsgálati vegyi anyag az egyik vizsgálatmenetben pozitívnak, a másikban negatívnak minősült), vagy magasabb fokú bizonyosságot kell szerezni a vizsgálat kimeneteléről, legalább három egymástól független vizsgálatmenetet kell lefolytatni. Ebben az esetben az osztályozás alapja a három vizsgálatmenet során kialakult két megegyező eredmény lesz.
|
Az adatok értelmezésének általános kritériumai
|
20.
|
Jelenleg nincs általánosan elfogadott módszer az ösztrogénreceptor transzaktivációs adatok értelmezésére. Ugyanakkor az ösztrogénreceptor által közvetített aktivitás kvalitatív (pl. pozitív/negatív) és/vagy kvantitatív (pl. EC50, PC50, IC50) értékelésének empirikus adatokon és megalapozott tudományos véleményen kell nyugodnia. A pozitív eredményeket lehetőség szerint jellemezni kell mind a vivőanyagos (oldószeres) kontrollhoz vagy referenciaösztrogénhez képest előidézett hatás mértékével, mind a hatást kiváltó koncentrációval (pl. EC50, PC50, RPCMax, IC50 stb.).
|
Vizsgálati jelentés
|
21.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálat:
|
—
|
az alkalmazott vizsgálat;
|
|
—
|
kontroll/referenciaszabvány/vizsgálati vegyi anyag;
|
|
—
|
eredet, gyártási szám, felhasználási határidő, ha rendelkezésre áll;
|
|
—
|
A vizsgálati vegyi anyag stabilitása, ha ismert;
|
|
—
|
A vizsgálati vegyi anyag oldhatósága és stabilitása az oldószerben, ha ismert;
|
|
—
|
adott esetben a pH-érték, az ozmolalitás és a kicsapódás mérése abban a tápfolyadékban, amelyhez a vizsgálati vegyi anyagot hozzáadták.
|
|
|
|
Egy összetevőből álló anyag:
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján
|
|
|
|
Több összetevőből álló anyag, UVCB-k és keverékek:
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai révén jellemezve.
|
|
|
|
Oldószer/vivőanyag:
|
—
|
jellemzés (az anyag természete, forgalmazója és gyártási száma);
|
|
—
|
az oldószer/vivőanyag kiválasztásának indokolása;
|
|
—
|
A vizsgálati vegyi anyag oldhatósága és stabilitása az oldószerben/vivőanyagban, ha ismert.
|
|
|
|
Sejtek:
|
—
|
A sejtek típusa és eredete;
|
—
|
endogén módon fejeződött ki az ösztrogénreceptor? Ha nem, mely receptor(oka)t transzfektálták?;
|
|
—
|
az alkalmazott riporterkonstrukció(k) (ideértve a forrást);
|
|
—
|
transzfektálási módszer;
|
|
—
|
A stabil transzfekció fenntartására alkalmazott szelekció (ahol van);
|
|
—
|
A transzfektálási módszer releváns a stabil sejtvonalak szempontjából?
|
|
|
—
|
sejtpasszálások száma (felolvasztástól);
|
|
—
|
passzált sejtek száma felolvasztáskor;
|
|
—
|
A sejttenyészetek fenntartásának módszerei.
|
|
|
|
Vizsgálati körülmények:
|
—
|
az életképesség felmérésére alkalmazott módszer leírása;
|
|
—
|
A közeg összetétele, CO2-koncentráció;
|
|
—
|
A vizsgálati vegyi anyag koncentrációi;
|
|
—
|
A hozzáadott vivőanyag és vizsgálati vegyi anyag mennyisége;
|
|
—
|
inkubációs hőmérséklet és páratartalom;
|
|
—
|
sejtsűrűség a kezelés elején és annak időtartama alatt;
|
|
—
|
pozitív és negatív referenciaszabványok;
|
|
—
|
riporter reagensek (a termék neve, forgalmazója és gyártási száma);
|
|
—
|
azok a kritériumok, amelyek meghatározzák, hogy a vizsgálatmenetek pozitívnak, negatívnak vagy többféleképpen értelmezhetőnek tekintendők-e.
|
|
|
|
Elfogadhatóság ellenőrzése:
|
—
|
az egyes vizsgálati lemezek indukciós faktorai, illetve hogy az indukciós faktorok a dokumentált kontrollok alapján megfelelnek-e a vizsgálathoz szükséges minimumnak;
|
|
—
|
az elfogadhatósági kritériumok tényleges értékei, pl. a párhuzamos pozitív kontrollok/referenciaszabványok log10EC50, log10PC50, logIC50 és Hill-meredekségi értékei.
|
|
|
|
Eredmények:
|
—
|
nyers és normalizált adatok;
|
|
—
|
az indukciós faktor maximális szintje;
|
|
—
|
ha létezik, a legkisebb hatásos koncentráció (LEC);
|
|
—
|
adott esetben az RPCMax, PCMax, PC50, IC50 és/vagy EC50 értékek;
|
|
—
|
amennyiben lehetséges, a koncentráció-válasz összefüggés;
|
|
—
|
statisztikai elemzések, ha végeztek ilyeneket, hiba- és megbízhatósági számításokkal (pl. SEM, SD, CV vagy 95 % CI), és annak leírása, hogy milyen módon kapták ezeket az értékeket.
|
|
|
SZAKIRODALOM
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34.), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(2)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(3)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method, an In vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(4)
|
Pujol P. et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer. Res., 58(23): p. 5367-73.
|
|
(5)
|
Rogers J.M. and Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In vitro and Molecular Toxicology: Journal of Basic and Applied Research, 13(1): p. 67-82.
|
|
(6)
|
OECD (2012). Performance Standards For Stably Transfected Transactivation In vitro Assay to Detect Estrogen Receptor Agonists (for TG 455). Environment, Health and Safety Publications, Series on Testing and Assessment (No 173.), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(7)
|
OECD (2015). Performance Standards For Stably Transfected Transactivation In vitro Assay to Detect Estrogen Receptor Antagonists. Environment, Health and Safety Publications, Series on Testing and Assessment (No 174.), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(8)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environment, Health and Safety Publications, Series on Testing and Assessment (No 150.), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(9)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept. Climacteric, 5 Suppl 2: p. 20- 6.
|
|
(10)
|
Welboren W.J. et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and how are they Regulated? Endocr. Relat. Cancer, 16(4): p. 1073-89.
|
|
(11)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63- 6.
|
|
(12)
|
Jefferson W.N., et al. (2002). Assessing Estrogenic Activity of Phytochemicals Using Transcriptional Activation and Immature Mouse Uterotrophic Responses, Journal of Chromatography B, 777(1-2): p. 179-189.
|
|
(13)
|
Sonneveld E. et al. (2006). Comparison of In vitro and In vivo Screening Models for Androgenic and Estrogenic Activities, Toxicol. Sci., 89(1): p. 173-187.
|
|
(14)
|
Takeyoshi M. et al. (2002). The Efficacy of Endocrine Disruptor Screening Tests in Detecting Anti- Estrogenic Effects Downstream of Receptor-Ligand Interactions, Toxicology Letters, 126(2): p. 91- 98.
|
|
(15)
|
Combes R.D. (2000). Endocrine Disruptors: a Critical Review of In vitro and In vivo Testing Strategies for Assessing their Toxic Hazard to Humans, ATLA Alternatives to Laboratory Animals,28(1): p. 81-118.
|
|
(16)
|
Escande A. et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol,71(10): p. 1459-69.
|
|
(17)
|
Gray L.E. Jr. (1998). Tiered Screening and Testing Strategy for Xenoestrogens and Antiandrogens, Toxicol. Lett, 102-103, 677-680.
|
|
(18)
|
EDSTAC (1998). Endocrine Disruptor Screening and Testing Advisory Committee (EDSTAC) Final Report.
|
|
(19)
|
ICCVAM (2003). ICCVAM Evaluation of In vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(20)
|
Gustafsson J.Ö. (1999). Estrogen Receptor ß - A New Dimension in Estrogen Mechanism of Action, Journal of Endocrinology, 163(3): p. 379-383.
|
|
(21)
|
Ogawa S. et al. (1998). The Complete Primary Structure of Human Estrogen Receptor ß (hERß) and its Heterodimerization with ERα In vivo and In vitro, Biochemical and Biophysical Research Communications, 243(1): p. 122-126.
|
|
(22)
|
Enmark E. et al. (1997). Human Estrogen Receptor ß-Gene Structure, Chromosomal Localization, and Expression Pattern, Journal of Clinical Endocrinology and Metabolism,82(12): p. 4258-4265.
|
|
(23)
|
Ball L.J. et al. (2009). Cell Type- and Estrogen Receptor-Subtype Specific Regulation of Selective Estrogen Receptor Modulator Regulatory Elements, Molecular and Cellular Endocrinology, 299(2): p. 204-211.
|
|
(24)
|
Barkhem T. et al. (1998). Differential Response of Estrogen Receptor Alpha and Estrogen Receptor Beta to Partial Estrogen Agonists/Antagonists, Mol. Pharmacol, 54(1): p. 105-12.
|
|
(25)
|
Deroo B.J. and Buensuceso A.V. (2010). Minireview: Estrogen Receptor-ß: Mechanistic Insights from Recent Studies, Molecular Endocrinology, 24(9): p. 1703-1714.
|
|
(26)
|
Harris D.M. et al. (2005). Phytoestrogens Induce Differential Estrogen Receptor Alpha- or Beta- Mediated Responses in Transfected Breast Cancer Cells, Experimental Biology and Medicine, 230(8): p. 558-568.
|
|
(27)
|
Anderson J.N. Clark J.H. and Peck E.J.Jr. (1972). The Relationship Between Nuclear Receptor- Estrogen Binding and Uterotrophic Responses, Biochemical and Biophysical Research Communications, 48(6): p. 1460-1468.
|
|
(28)
|
Toft D. (1972). The Interaction of Uterine Estrogen Receptors with DNA, Journal of Steroid Biochemistry, 3(3): p. 515-522.
|
|
(29)
|
Gorski J. et al. (1968), Hormone Receptors: Studies on the Interaction of Estrogen with the Uterus, Recent Progress in Hormone Research, 24: p. 45-80.
|
|
(30)
|
Jensen E.V. et al. (1967), Estrogen-Receptor Interactions in Target Tissues, Archives d’Anatomie Microscopique et de Morphologie Experimentale, 56(3):p. 547-569.
|
|
(31)
|
ICCVAM (2002). Background Review Document: Estrogen Receptor Transcriptional Activation (TA) Assay. Appendix D, Substances Tested in the ER TA Assay, NIH Publication Report (No 03-4505.).
|
|
(32)
|
Kanno J. et al. (2001). The OECD Program to Validate the Rat Uterotrophic Bioassay to Screen Compounds for In vivo Estrogenic Responses: Phase 1, Environ. Health Persp., 109:785-94.
|
|
(33)
|
Kanno J. et al. (2003). The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two Dose -Response Studies, Environ. Health Persp., 111:1530-1549.
|
|
(34)
|
Kanno J. et al. (2003), The OECD Program to Validate the Rat Uterotrophic Bioassay: Phase Two – Coded Single-Dose Studies, Environ. Health Persp., 111:1550-1558.
|
|
(35)
|
Geisinger et al. (1989) Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(36)
|
Baldwin et al. (1998) BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(37)
|
Li, Y. et al. (2014) Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(38)
|
Rogers, J.M. and Denison, M.S. (2000) Recombinant cell bioassays for endocrine disruptors: development of a stably transfected human ovarian cell line for the detection of estrogenic and anti-estrogenic chemicals, In vitro & Molec. Toxicol. 13, 67-82.
|
1. függelék
FOGALOMMEGHATÁROZÁSOK ÉS RÖVIDÍTÉSEK
Elfogadhatósági kritériumok
: a kísérleti kontrollok és referenciaszabványok megfelelőségére vonatkozó minimális előírások. Ahhoz, hogy egy kísérlet érvényesnek minősüljön, meg kell felelni valamennyi elfogadhatósági kritériumnak.
Pontosság (egyezőség)
: a vizsgálati eredmények és az elfogadott referenciaértékek közötti egyezés eltérése. A vizsgálat megfelelőségének mutatója, és relevanciájának egyik szempontja. E kifejezést gyakran használják az „egyezőség” szinonimájaként, amely arra utal, hogy egy adott vizsgálat milyen arányban szolgáltat helyes eredményeket (1).
Agonista
: olyan anyag, amely egy specifikus receptorhoz kötődve valamilyen választ, pl. transzkripciót vált ki.
Antagonista
: a receptor ligandumok vagy vegyi anyagok azon típusa, amely egy receptorhoz kötődve nem vált ki biológiai válaszreakciót, hanem gátolja vagy tompítja az agonisták által indukált válaszokat.
Antiösztrogén aktivitás
: egy vegyi anyag azon képessége, hogy gátolja az ösztrogénreceptorok által közvetített 17β-ösztradiol hatását.
Sejtmorfológia
: egy szövettenyészetet tartalmazó lemez egyetlen lyukában lévő egyrétegű sejtek alakja és megjelenési formája. Az elhaló sejtek gyakran a normálistól eltérő sejtmorfológiával rendelkeznek.
CF
: Az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére szolgáló fogalmi kerete.
Aktív szén/dextrán kezelés
: a sejtkultúrához használt szérum kezelési módja. Az aktív szénnel/dextránnal végzett kezelés (szaknyelven „stripping-eljárás”) során eltávolítják az endogén hormonokat és hormonmegkötő fehérjéket.
Vegyi anyag
: anyag vagy keverék.
Citotoxicitás
: a sejtstruktúrát vagy a sejtfunkciót érő káros hatások, amelyek végső soron sejtpusztuláshoz vezethetnek, kimutatásához összevetik a lyukban növő sejtek számában az expozíciós időszak végére bekövetkezett csökkenést vagy a mérhető sejtfunkciók csökkenését a párhuzamos vivőanyagos kontrollal.
CV
: variációs koefficiens.
DCC-FBS
: dextránbevonatú aktív szénnel kezelt magzati borjú szérum.
DMEM
: Dulbecco-féle módosított Eagle-tápoldat.
DMSO
: dimetil-szulfoxid.
E2
: 17β-ösztradiol
EC50
: a vizsgálati vegyi anyag maximális hatásának felét kiváltó koncentráció.
ED
: az endokrin rendszer károsítása.
hERα
: humán ösztrogénreceptor alfa.
hERß
: humán ösztrogénreceptor béta.
EFM
: ösztrogénmentes közeg. A Dulbecco-féle módosított Eagle-tápoldat (DMEM) kiegészítve 4,5 % dextránbevonatú aktív szénnel kezelt magzati borjú szérummal, 1,9 % L-glutaminnal és 0,9 % penicillinnel és sztreptomicinnel.
ER
: ösztrogénreceptor.
ERE
: ösztrogén válaszelem.
Ösztrogénhatású aktivitás
: egy vegyi anyag azon képessége, hogy a 17β-ösztradiolhoz hasonlóan ösztrogénreceptorokhoz kötődjön és aktiválja azokat. Ezzel a vizsgálati módszerrel a humán ösztrogénreceptor alfa által közvetített ösztrogénhatású aktivitás észlelhető.
ER TA
: ösztrogénreceptor transzaktiváció.
FBS
: magzati borjú szérum.
HeLa
: humán méhnyakból származó halhatatlan sejtvonal.
HeLa9903
: egy hERαα-val és egy luciferáz riportergénnel stabilan transzfektált HeLa-sejt szubklónja.
IC
50
: egy gátló vizsgálati vegyi anyag maximális tényleges hatásának felét kiváltó koncentráció.
ICCVAM
: alternatív módszerek validálásával foglalkozó ügynökségközi koordinációs bizottság.
Laboratóriumok közötti reprodukálhatóság
: annak a mutatója, hogy különböző minősített laboratóriumok milyen mértékben képesek ugyanazon protokoll alkalmazásával és ugyanazon anyagok vizsgálatával hasonló kvalitatív és kvantitatív eredményeket elérni. A laboratóriumok közötti reprodukálhatóság az elővalidálási és validálási folyamat során kerül meghatározásra, és annak mértékét jelzi, hogy az adott vizsgálat mennyire sikeresen vihető át laboratóriumok között; más néven laboratóriumok közötti megismételhetőség (1).
Laboratóriumon belüli reprodukálhatóság
: annak meghatározása, hogy egy adott laboratóriumon belül dolgozó szakemberek milyen mértékben képesek egy meghatározott protokoll alapján különböző időpontokban sikeresen megismételni az eredményeket. Más néven „laboratóriumon belüli megismételhetőség” (1).
LEC
: a legkisebb hatásos koncentráció a vizsgálati vegyi anyag azon legalacsonyabb koncentrációja, amely választ vált ki (vagyis a vizsgálati vegyi anyag legkisebb koncentrációja, amelynél az indukciós faktor statisztikailag eltér a párhuzamos vivőanyagos kontrolltól).
Me-too vizsgálat
: olyan vizsgálatok köznapi elnevezése, amelyek strukturálisan és funkcionálisan hasonlóak egy validált és elfogadott, referenciaként szolgáló vizsgálati módszerhez. A hasonló vizsgálati módszer szinonimája.
MT
: metallotionein.
MMTV
: egér emlőtumor vírus.
OHT
: 4-hidroxitamoxifén.
PBTG
: teljesítményalapú vizsgálati iránymutatás.
PC (pozitív kontroll)
: erős hatást kifejtő anyag, lehetőség szerint 17β-ösztradiol, amelyet belefoglalnak minden vizsgálatba, hogy biztosítsák a vizsgálat megfelelő működését.
PC
10
: a vizsgálati vegyi anyagnak az a koncentrációja, amelynél az agonista vizsgálat során mért aktivitás a pozitív kontroll (a stabilan transzfektált transzaktivációs vizsgálatnál 1nM E2) egyes lemezeken indukált maximális aktivitásának 10 %-a.
PC
50
: a vizsgálati vegyi anyagnak az a koncentrációja, amelynél az agonista vizsgálat során mért aktivitás a pozitív kontroll (E2 a vizsgálati módszerben megadott referenciakoncentrációban) egyes lemezeken indukált maximális aktivitásának 50 %-a.
PC
Max
: a vizsgálati vegyi anyag RPCMax hatást kiváltó koncentrációja.
Teljesítményszabványok
: validált vizsgálaton alapuló szabványok, amelyek a javasolt, végrehajtás és funkcionalitás szempontjából hasonló vizsgálat összehasonlíthatósági értékelésének alapjául szolgálnak. Ide tartoznak: (1) a vizsgálat alapvető összetevői; (2) a referencia-vegyianyagok minimális jegyzéke, amely a validált vizsgálati módszer elfogadható teljesítményének igazolására használt vegyi anyagokból került összeállításra; és (3) a validált vizsgálati módszer eredményei alapján meghatározott, hasonló megbízhatósági és pontossági szintek, amelyeket a javasolt vizsgálatnak teljesítenie kell a minimális jegyzék referencia-vegyianyagainak felhasználásával történő értékelése során (1).
Jártassági tesztanyagok
: a teljesítményszabványokban megadott referenciaanyagokon belüli alcsoport, amelyet a laboratóriumok felhasználhatnak a szabványosított vizsgálati módszerben való szakmai jártasságuk igazolására. Ezeket az anyagokat általában az alapján választják ki, hogy összességükben lefedik a válaszreakciók lehetséges tartományát, kereskedelmi forgalomban kaphatók és rendelkezésre állnak rájuk vonatkozó minőségi referenciaadatok.
Jártasság
: annak igazolása, hogy a laboratórium képes megfelelően lefolytatni egy adott vizsgálatot, mielőtt ismeretlen anyagokat tesztelne.
Referenciaösztrogén (pozitív kontroll, PC)
: 17β-ösztradiol (E2, CAS-száma: 50-28-2).
Referenciaszabvány
: egy adott vizsgálat megfelelőségének bizonyítására használt referenciaanyag. A stabilan transzfektált transzaktivációs és a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat referenciaszabványa a 17β-ösztradiol.
Vizsgálati referenciamódszerek
: A 455. teljesítményalapú vizsgálati iránymutatás alapját képező vizsgálatok.
Relevancia
: valamely vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálat pontosságát (az eredmények egyezését) figyelembe kell venni (1).
Megbízhatóság
: a vizsgálat laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg.
RLU
: relatív fényegység.
RNS
: ribonukleinsav.
RPC
Max
: valamely vizsgálati vegyi anyag által előidézett maximális szintű válasz, amelyet ugyanazon lemezen vizsgált 1 nM mennyiségű E2 által kiváltott válasz százalékában adnak meg.
RPMI
: RPMI-1 640 tápoldat kiegészítve 0,9 % penicillinnel és sztreptomicinnel, valamint 8,0 % magzati borjú szérummal (FBS).
Vizsgálatmenet
: egyszeri kísérlet, amelynek során értékelik a vizsgálat biológiai eredményére gyakorolt kémiai hatást. Minden vizsgálatmenet egy teljes kísérletnek felel meg, amelyet közös sejtkészletből származó, ugyanabban az időben replikátum lyukakba kiszélesztett sejteken végeznek el.
Független vizsgálatmenet
: más sejtkészletből származó sejtekkel, frissen hígított vegyi anyagokkal, különböző napokon vagy ugyanazon a napon más szakemberek által végzett különálló, független kísérlet, amelynek során értékelik a vizsgálat biológiai eredményére gyakorolt kémiai hatást.
SD
: standard deviáció.
Érzékenység
: az összes olyan pozitív/aktív anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálat pontosságának mutatója, valamint fontos szempont a vizsgálat relevanciájának megítélésében (1).
Specificitás
: az összes olyan negatív/aktív anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálat pontosságának mutatója, valamint fontos szempont a vizsgálat relevanciájának megítélésében (1).
Stabil transzfekció
: amikor a DNS-t olyan módon transzfektálják a tenyésztett sejtekbe, hogy az stabilan integrálódik a sejtek genomjába, lehetővé téve ezáltal a transzfektált gének stabil kifejeződését. A stabilan transzfektált sejtek klónjainak szelektálását stabil markerekkel végzik (pl. G418-rezisztencia alapján).
STTA-vizsgálat
: stabilan transzfektált transzaktivációs vizsgálat, a HeLa 9 903 sejtvonalat alkalmazó ösztrogénreceptor alfa transzkripciós aktivációs vizsgálat.
Tanulmány
: egyetlen, konkrét anyag meghatározott vizsgálattal való értékeléséhez végzett teljes körű kísérleti munka. A tanulmány magában foglal minden lépést, egyebek mellett a vizsgálati anyag vizsgálati közegben végzett hígítási tesztjeit, az előzetes dózistartomány-kereső vizsgálatmeneteket, az összes szükséges teljes körű vizsgálatmenetet, az adatok elemzését, a minőség-ellenőrzést és a toxicitás felmérését. A tanulmány lezárásakor lehetővé válik a vizsgálati vegyi anyag toxicitási célpontra kifejtett és az alkalmazott vizsgált által értékelt hatásának besorolása (pl. aktív, inaktív vagy nem meggyőző), valamint a pozitív referencia-vegyianyaghoz viszonyított potenciáljának megbecslése.
Anyag
: a REACH-rendelet (62) értelmében az anyag természetes állapotban előforduló vagy gyártási folyamatból származó kémiai elem és vegyületei, amely az anyag stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott folyamatból származó szennyező anyagot is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető. Az ENSZ GHS (1) egy ehhez nagyon hasonló meghatározást alkalmaz.
TA (transzaktiváció)
: az mRNS szintézis elindítása egy specifikus kémiai jel, például az ösztrogén ösztrogénreceptorhoz való kötődésének hatására.
Vizsgálat
: e vizsgálati módszer keretében a vizsgálat azon hitelesnek elfogadott módszertanok egyike, amelyek eleget tesznek a meghatározott teljesítési kritériumoknak. A vizsgálat összetevői közé tartozik például a specifikus sejtvonal a hozzá kapcsolódó tenyésztési körülményekkel, a vizsgálat elvégzéshez használt specifikus tápoldat, a lemez elrendezésének körülményei, a vizsgálati vegyi anyag elrendezése és hígítása, továbbá bármely más szükséges minőség-ellenőrzési intézkedés és a kapcsolódó adatkiértékelési lépések.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Transzkripció
: mRNS szintézis.
UVCB
: ismeretlen vagy változó összetételű vegyi anyagok, komplex reakciótermékek vagy biológiai anyagok.
Validált vizsgálati módszer
: olyan vizsgálat, amelynek az adott céllal kapcsolatos relevanciája (pontosságát is ideértve) és megbízhatósága validálási vizsgálatok alapján megállapítást nyert. Fontos megjegyezni, hogy a validált vizsgálati módszer a javasolt felhasználási célra nem feltétlenül kínál megfelelő pontosságú és megbízhatóságú elfogadható eljárást (1).
Validálás
: az az eljárás, amelynek révén megállapítják egy bizonyos megközelítés, módszer, vizsgálat, eljárás vagy értékelés adott célra való megbízhatóságát és relevanciáját (1).
VC (Vivőanyagos kontroll)
: a vizsgálati és a kontrollként szolgáló vegyi anyagok feloldására használt oldószert önmagában, az oldott vegyi anyag nélkül, vivőanyagként vizsgálják.
VM7
: immortalizált adenokarcinoma sejtvonal, amely endogénen fejezi ki az ösztrogénreceptort.
VM7Luc4E2
: a VM7Luc4E2 sejtvonal VM7 immortalizált humán adenokarcinoma sejtekből származik, amelyek endogén módon kifejezik az ösztrogénreceptor mindkét formáját (ERα és ERβ), és ezt a sejtvonalat stabilan transzfektálták a pGudLuc7 plazmiddal. Ez a plazmid az egér emlőtumor vírus (MMTV) promóter előtt négy szintetikus oligonukleotid kópián hordozza az ösztrogén válaszelemet, és a szentjánosbogár luciferáz gén expresszióját szabályozza.
Gyenge pozitív kontroll
: a referencia-vegyianyagok jegyzékéből kiválasztott gyenge hatást kifejtő anyag, amelyet belefoglalnak minden vizsgálatba, hogy biztosítsák a vizsgálat megfelelő működését.
2. függelék
STABILAN TRANSZFEKTÁLT HUMÁN ÖSZTROGÉNRECEPTOR Α TRANSZAKTIVÁCIÓS VIZSGÁLAT VEGYI ANYAGOK ÖSZTROGÉNHATÁSÚ AGONISTA ÉS ANTAGONISTA AKTIVITÁSÁNAK ÉSZLELÉSÉRE HERΑ-HELA-9903 SEJTVONALBAN
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
1.
|
Ez a transzaktivációs (TA) vizsgálat hERα-HeLa-9903 sejtvonalat használ a humán ösztrogénreceptor alfa (hERα) által közvetített ösztrogénhatású agonista aktivitás észlelésére. A stabilan transzfektált transzaktivációs (STTA) vizsgálat validálási tanulmányát a japán Chemicals Evaluation and Research Institute (CERI) végezte el, hERα-HeLa-9903 sejtvonalat használva a humán ösztrogénreceptor alfa (hERα) által közvetített ösztrogénhatású agonista és antagonista aktivitás észlelésére, amely tanulmány bizonyította, hogy a vizsgálat tervezett felhasználása tekintetében releváns és megbízható (1).
|
|
2.
|
A vizsgálat kifejezetten azt a célt szolgálja, hogy a kémiai lumineszcencia mint végpont mérése révén észlelje a hERα által közvetített transzaktivációt. Azonban beszámoltak arról, hogy az 1 μM-nél magasabb fitoösztrogén-koncentrációnál a luciferáz riportergén túlzott aktiválása miatt nem receptorok által közvetített lumineszcencia jelek észlelhetők (2) (3). Bár a dózis-válasz görbe arra utal, hogy az ER-rendszer ténylegesen alacsonyabb koncentrációknál aktiválódik, a magas fitoösztrogén-koncentráció mellett vagy feltehetőleg a luciferáz riportergént a fitoösztrogénhez hasonlóan túlzottan aktiváló vegyületek magas koncentrációja mellett kapott luciferáz-kifejeződést körültekintően meg kell vizsgálni stabilan transzfektált ösztrogénreceptor transzaktivációs vizsgálati rendszerekben (1. függelék).
|
|
3.
|
E vizsgálat szabályozási célra való alkalmazása előtt el kell olvasni az „ÁLTALÁNOS BEVEZETÉS” és „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részt. Az ebben a vizsgálati iránymutatásban használt fogalmak és rövidítések meghatározását a 2.1. függelék tartalmazza.
|
A VIZSGÁLAT ELVE (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
4.
|
A vizsgálat az ösztrogénreceptor ligandumhoz kötődésének jelzésére szolgál. A ligandum kötődését követően a receptor-ligandum komplex transzlokálódik a sejtmagba, ahol specifikus DNS válaszelemekhez kötődve transzaktivál egy szentjánosbogár luciferáz riportergént, ami megnöveli a luciferáz enzim celluláris kifejeződését. A luciferin egy szubsztrát, amit a luciferáz enzim átalakít és közben biolumineszcens fény keletkezik, amely luminométerrel számszerűen mérhető. A luciferáz-aktivitás számos kereskedelmi forgalomban kapható vizsgálati készlettel gyorsan és költséghatékonyan értékelhető.
|
|
5.
|
A vizsgálati rendszer humán méhnyakdaganatból származó hERα-HeLa-9903 sejtvonalat használ, két stabilan inzertált konstrukcióval: i. a hERα expressziós konstrukcióval (amely a teljes humán ösztrogénreceptort kódolja), és ii. egy szentjánosbogár luciferáz riporterkonstrukcióval, amely egy vitellogenin ösztrogén válaszelem ötször ismétlődő szakaszát tartalmazza hozzákapcsolva egy egér metallotionein promóter TATA-boxához. Az egér metallotionein promóter TATA-box génkonstrukció biztosítja a legjobb teljesítményt, ezért használata általánosan elterjedt. Következésképpen a hERα-HeLa-9903 sejtvonal képes mérni egy adott vizsgálati vegyi anyag azon képességét, hogy beindítja a humán ösztrogénreceptor alfa által irányított luciferáz gén kifejeződést.
|
|
6.
|
ösztrogénreceptor agonista vizsgálat esetén az adatok értelmezése azon alapul, hogy a vizsgálati vegyi anyag által előidézett maximális szintű válasz megegyezik-e vagy meghaladja-e a pozitív kontrollanyagként használt 17β-ösztradiol (E2) indukáláshoz alkalmazott maximális (1 nM) koncentrációja által keltett agonista válasz 10 %-át (vagyis a PC10 értékét). ösztrogénreceptor antagonista vizsgálat esetén az adatok értelmezése azon alapul, hogy a válasz előidézi-e a kiegészítő kontroll (25 pM E2) által kiváltott válaszhoz képest az aktivitás legalább 30 %-os csökkenését, citotoxicitás nélkül. Az adatok elemzésének és értelmezésének részleteivel a 34–48. pont foglalkozik.
|
ELJÁRÁS
Sejtvonalak
|
7.
|
A vizsgálathoz stabilan transzfektált hERα-HeLa-9903 sejtvonalat kell használni. A sejtvonal egy anyagátadási megállapodás aláírását követően beszerezhető a japán Collection of Research Bioresources (JCRB) sejtbankból (63).
|
|
8.
|
A vizsgálat során kizárólag mikoplazmamentesnek nyilvánított sejtek használhatók. A mikoplazma-fertőzés érzékeny detektálására az RT-PCR (valós idejű polimeráz láncreakció) módszer alkalmazandó (4) (5) (6).
|
A sejtvonal stabilitása
|
9.
|
A sejtvonal stabilitásának nyomon követéséhez az agonista vizsgálat során referenciaszabványként E2-t, 17α-ösztradiolt, 17α-metiltesztoszteront és kortikoszteront kell használni, emellett a vizsgálat minden elvégzése során legalább egy alkalommal fel kell venni egy teljes koncentráció-válasz görbét az 1. táblázatban szereplő koncentrációtartományban, és az eredményeknek meg kell felelniük az 1. táblázatban feltüntetett eredményeknek.
|
|
10.
|
Antagonista vizsgálat során minden vizsgálatmenetben párhuzamosan meg kell határozni két referenciaszabvány, a tamoxifén és a flutamid teljes koncentrációgörbéjét. Ellenőrizni kell továbbá, hogy e két vegyi anyagot a kvalitatív osztályozás során helyesen sorolták-e pozitív, illetve negatív kategóriába.
|
Sejttenyésztés és a leültetés körülményei
|
11.
|
A sejteket fenolvörösmentes, 60 mg/l kanamicin antibiotikummal és 10 %-os dextránbevonatú aktív szénnel kezelt magzati borjú szérummal (DCC-FBS) kiegészített Eagle-féle minimum esszenciális tápoldatban (Eagle’s Minimum Essential Medium, EMEM), CO2 inkubátorban (5 % CO2), 37 ±1 °C-on kell fenntartani. 75–90 %-os konfluencia elérését követően a sejteket tovább kell oltani egy 100 mm-es sejttenyésztő edénybe, 10 ml térfogatban 0,4 × 105 –1 × 105 sejt/ml sűrűségben. 10 %-os FBS-EMEM (vagyis DCC-FBS-sel kiegészített EMEM) sejtszuszpenziót kell készíteni, majd a sejteket egy mikrotiterlemez lyukaiba 1 × 104 sejt/(100 μl × lyuk) sűrűségben ki kell széleszteni. A vegyi anyaggal való kezelést megelőzően a sejteket 5 %-os CO2-tartalmú inkubátorba n 37° ±1° C-on 3 órán át preinkubálni kell. A műanyag edénynek nem lehet ösztrogénhatású aktivitása.
|
|
12.
|
A válasz integritása érdekében a fagyasztott sejtkultúra sejtjein legalább két, de legfeljebb 40 passzálást kell végrehajtani a kondicionált tápoldatban. A hERα-HeLa-9903 sejtvonal esetében ez három hónapnál kevesebb időt vesz igénybe. A nem megfelelő tenyésztési körülmények között növő sejtek hatékonysága csökkenhet.
|
|
13.
|
A DCC-FBS elkészíthető a 2.2. függelékben leírtak szerint, de kereskedelmi forrásból is beszerezhető.
|
Elfogadhatósági kritériumok
Az ösztrogénreceptor agonista vizsgálathoz használandó pozitív és a negatív referenciaszabványok
|
14.
|
A tanulmány előtt és alatt a vizsgálati rendszer reakcióképességét ellenőrizni kell egy erős ösztrogén (E2), egy gyenge ösztrogén (17α-ösztradiol) és egy nagyon gyenge agonista (17α-metiltesztoszteron) megfelelő koncentrációkban való alkalmazásával, valamint egy negatív anyaggal (kortikoszteron). A validálási tanulmányból (1) származó elfogadható értéktartományokat a 1. táblázat ismerteti. E 4 párhuzamosan vizsgálandó referenciaszabványt bele kell foglalni minden kísérletbe, vizsgálatuk eredményének pedig az elfogadható határértékeken belül kell lennie. Ellenkező esetben meg kell határozni, hogy mi okból nem teljesültek az elfogadhatósági kritériumok (pl. a sejtek kezelése, a szérum és az antibiotikum minősége és koncentrációja), a vizsgálatot pedig meg kell ismételni. Az elfogadhatósági kritériumok teljesülése esetén az EC50, a PC50 és a PC10 értékek minimális szórása érdekében törekedni kell a sejttenyésztésben részt vevő anyagok következetes használatára. A négy párhuzamosan vizsgált referenciaszabvány – amelyeket minden kísérletben vizsgálni kell (azonos körülmények között, ideértve az anyagokat, a sejtek passzálási számát és a technikusokat is) – biztosíthatja a vizsgálat érzékenységét, mivel a három pozitív referenciaszabvány PC10 értékének az elfogadhatósági tartományba kell esnie, csakúgy mint PC50 és EC50 értékeknek, ha meghatározható (lásd az 1. táblázatot).
|
1. táblázat
Az ösztrogénreceptor agonista vizsgálathoz használandó négy referenciaszabvány elfogadhatósági tartománya
|
Elnevezés
|
logPC50
|
logPC10
|
logEC50
|
Hill-meredekség
|
Vizsgálati tartomány
|
|
17β-ösztradiol (E2) CAS-szám: 50-28-2
|
-11,4~-10,1
|
< -11
|
-11,3~-10,1
|
0,7~1,5
|
10-14~10-8M
|
|
17α-ösztradiol CAS-szám: 57-91-0
|
-9,6~-8,1
|
-10,7~-9,3
|
-9,6~-8,4
|
0,9~2,0
|
10-12~10-6M
|
|
Kortikoszteron CAS-szám: 50-22-6
|
—
|
—
|
—
|
—
|
10-10~10-4M
|
|
17α-metiltesztoszteron CAS-szám: 58-18-4
|
-6,0~-5,1
|
-8,0~-6,2
|
—
|
—
|
10-11~10-5M
|
Az ösztrogénreceptor antagonista vizsgálathoz használandó pozitív és negatív referenciaszabványok
|
15.
|
A tanulmány előtt és alatt a vizsgálati rendszer reakcióképességét egy pozitív anyag (tamoxifén) és egy negatív anyag (flutamid) megfelelő koncentrációkban való alkalmazásával ellenőrizni kell. A validálási tanulmányból (1) származó elfogadható értéktartományokat a 2. táblázat ismerteti. E két párhuzamosan vizsgálandó referenciaszabványt bele kell foglalni minden kísérletbe, vizsgálatuk eredményét pedig a kritériumoknak megfelelően kell értékelni. Ha az eredmény nem elfogadható, meg kell határozni, hogy mi okból nem teljesültek a kritériumok (pl. a sejtek kezelése, a szérum és az antibiotikum minősége és koncentrációja), a vizsgálatot pedig meg kell ismételni. Emellett meg kell határozni a pozitív anyag (tamoxifén) IC50 értékét, és az eredménynek az elfogadható határértékeken belül kell lennie. Az elfogadhatósági kritériumok teljesülése esetén az IC50 értékének minimális szórása érdekében mindenképpen biztosítani kell a sejttenyésztésben részt vevő anyagok következetes használatát. A két párhuzamosan vizsgált referenciaszabvány – amelyeket minden kísérletben vizsgálni kell (azonos körülmények között, ideértve az anyagokat, a sejtek passzálási számát és a technikusokat) – képes biztosítani a vizsgálat érzékenységét (lásd a 2. táblázatot).
|
2. táblázat
Az ösztrogénreceptor antagonista vizsgálathoz használandó két referenciaszabvány kritériuma és elfogadhatósági tartománya
|
Elnevezés
|
Kritériumok
|
LogIC50
|
Vizsgálati tartomány
|
|
Tamoxifén CAS-szám: 10540-29-1
|
Pozitív: meg kell határozni az IC50 értéket
|
-5,942 ~ -7,596
|
10-10 ~ 10-5 M
|
|
FlutamidCAS-szám: 13311-84-7
|
Negatív: nem kell meghatározni az IC30 értéket
|
—
|
10-10 ~ 10-5 M
|
Pozitív és vivőanyagos kontrollok
|
16.
|
A pozitív kontrollt az ösztrogénreceptor agonista vizsgálat esetén (1 nM mennyiségű E2) és az ösztrogénreceptor antagonista vizsgálat esetén (10 μM mennyiségű TAM) lemezenként legalább három ismétlésben kell végrehajtani. A vizsgálati vegyi anyag feloldására használt vivőanyagot vivőanyagos kontroll keretében lemezenként legalább három ismétlésben kell vizsgálni. Amennyiben a pozitív kontroll során a vizsgálati vegyi anyagétól eltérő vivőanyagot alkalmaznak, e vivőanyagos kontroll mellett egy másik vivőanyagos kontrollt kell végezni legalább három ismétlésben, a pozitív kontrollal azonos lemezen.
|
Az ösztrogénreceptor agonista vizsgálat minőségi kritériumai
|
17.
|
A pozitív kontroll (1 nM E2) során tapasztalt átlagos luciferáz-aktivitás minden lemezen legalább a vivőanyagos kontroll átlagértékének négyszerese legyen. E kritérium meghatározása a validálási tanulmány végpontértékeinek megbízhatóságán alapul (a dokumentált értékek négyszeres és harmincszoros között helyezkednek el).
|
|
18.
|
A vizsgálat minőség-ellenőrzése szempontjából a párhuzamos pozitív kontroll (1 nM E2) PC10 értékének megfelelő indukciós faktor nagyobbnak kell lennie, mint a párhuzamos vivőanyagos kontroll indukciós faktora (=1) plusz a standard deviáció kétszerese (1+2SD). Rangsorolási célból a PC10 érték a statisztikai elemzéshez képest leegyszerűsítheti a szükséges adatelemzést. Bár a statisztikai elemzés információt nyújt a szignifikanciáról, nem szolgáltat a koncentráció alapján megállapított potenciálhoz hasonló kvantitatív paramétert, ezért rangsorolás céljára kevésbé alkalmas.
|
Az ösztrogénreceptor antagonista vizsgálat minőségi kritériumai
|
19.
|
A kiegészítő kontroll (25 pM mennyiségű E2) luciferáz-aktivitásának átlaga lemezenként legalább négyszerese legyen a vivőanyagos kontroll átlagértékének. E kritérium meghatározása a validálási tanulmány végpontértékeinek megbízhatóságán alapul.
|
|
20.
|
A vizsgálat minőség-ellenőrzése szempontjából az 1 nM mennyiségű E2 relatív transzkripciós aktiválásának (RTA) meg kell haladnia a 100 %-ot, az 1 μM mennyiségű 4-hidroxitamoxifén (OHT) relatív transzkripciós aktiválás értékének kisebbnek kell lennie 40,6 %-nál, a 100 μM digitonin (Dig) relatív transzkripciós aktiválás értéke pedig nem haladhatja meg a 0 %-ot.
A laboratórium jártasságának igazolása (lásd a vizsgálati módszer „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részének 14. pontját, valamint 3. és 4. táblázatát).
|
Vivőanyag
|
21.
|
Párhuzamos vivőanyagos kontrollként dimetil-szulfoxidot (DMSO) vagy más megfelelő oldószert kell használni, azonos koncentrációban a különböző pozitív és negatív kontrollok és a vizsgálati vegyi anyag tesztelése során. A vizsgálati vegyi anyagokat olyan oldószerben kell feloldani, amelyik szolubilizálja az adott vizsgálati vegyi anyagot és elegyedik a sejtek tápoldatával. A megfelelő vivőanyagok közé tartozik a víz, az etanol (95–100 %-os tisztaságban) és a dimetil-szulfoxid. Dimetil-szulfoxid használata esetén az anyag mennyisége nem haladhatja meg a 0,1 térfogatszázalékot. Minden vivőanyag esetében igazolni kell, hogy maximális alkalmazott térfogatuk nem fejt ki citotoxikus hatást és nem befolyásolja a vizsgálat teljesítményét.
|
Vizsgálati vegyi anyagok előkészítése
|
22.
|
Általában a vizsgálati vegyi anyagokat dimetil-szulfoxidban vagy más alkalmas oldószerben fel kell oldani, és ugyanazzal az oldószerrel sorozatban 1:10 arányban hígítani kell, elkészítve az oldatokat a tápoldattal való hígításhoz.
|
Oldhatóság és citotoxicitás: a dózisbehatárolás szempontjai
|
23.
|
Elővizsgálatot kell végezni a vizsgálandó vegyi anyag megfelelő koncentrációtartományának meghatározására, illetve annak megállapítására, hogy a vizsgálati vegyi anyaggal kapcsolatban felmerül-e oldhatósági és citotoxicitási probléma. A vegyi anyagokat először a legmagasabb, vagyis 1 μl/ml, 1 mg/ml vagy 1 mM koncentrációig vizsgálják, melyek közül a legalacsonyabb a mérvadó. Az elővizsgálat során megfigyelt citotoxicitási szint vagy oldhatatlanság alapján az első végleges vizsgálatmenetben a legmagasabb elfogadható koncentrációtól (pl. 1 mM, 100 μM, 10 μM stb.) induló logaritmikus hígítási oldatsorozatban kell vizsgálni a vegyi anyagot, és fel kell jegyezni az esetlegesen tapasztalt zavarosságot, kicsapódást vagy citotoxicitást. A második, illetve ha szükséges a harmadik vizsgálatmenet koncentrációértékeit megfelelően ki kell igazítani, hogy jó koncentráció-válasz görbe alakuljon ki, és ki lehessen zárni azokat a koncentrációkat, amelyek oldhatatlannak bizonyultak vagy túlzott citotoxicitást idéztek elő.
|
|
24.
|
Az ER-agonisták és ER-antagonisták esetében a magas szintű citotoxicitás szignifikánsan módosíthatja vagy megakadályozhatja a tipikusan szigmoid választ, ezért figyelembe kell venni az adatok értelmezésénél. Olyan citotoxicitási vizsgálati módszereket kell alkalmazni, amelyek információt nyújtanak a 80 %-os sejtéletképességről, a megfelelő vizsgálatot a laboratórium tapasztalata alapján kell kiválasztani.
|
|
25.
|
Amennyiben a citotoxicitási vizsgálat eredményei azt jelzik, hogy a vizsgálati vegyi anyag koncentrációja legalább 20 %-kal csökkenti a sejtszámot, ezt a koncentrációt citotoxikusnak kell tekinteni, és a citotoxikus vagy afeletti koncentrációkat ki kell zárni az értékelésből.
|
Vegyi anyaggal történő kezelés és a vizsgálati lemez elrendezése
|
26.
|
A vegyi anyag hígításának elkészítése (1. és 2. lépés) és a sejtek kezelése (3. lépés) az alábbiak szerint is elvégezhető:
|
1. lépés: Az egyes vizsgálati vegyi anyagokkal DMSO-dal vagy megfelelő oldószerrel hígítási sorozatot kell készíteni, és a három ismétléses elrendezésben kell elhelyezni a mikrotiterlemez lyukaiban az előkísérletben megállapított dózistartománynak megfelelő végső koncentrációsorozat szerint (egy tipikus oldatsorozat például az 1 mM, 100 μM, 10 μM, 1μM, 100 nM, 10 nM, 1 nM, 100 pM és 10 pM (10-3–10-11 M)).
2. lépés: Vegyi anyag hígítása: az oldószerben először 1,5 μl mennyiségű vizsgálati vegyi anyagot oldjunk fel a tápoldat 500 μl-jében.
3. lépés: A sejtek kezelése a vegyi anyaggal: a (2. lépésben elkészített) tápoldattal hígított 50 μl mennyiségű anyagot adjuk hozzá a lyukanként és 100 μl-enként 104 sejtet tartalmazó vizsgálati lyukakhoz.
Az egyes lyukakba helyezendő tápoldat végleges ajánlott térfogata 150 μl. A vizsgálati minták és a referenciaszabványok elosztása történhet a 3. és 4. táblázatban foglaltak szerint.
3. táblázat
Példa az ösztrogénreceptor agonista vizsgálathoz használt vizsgálati lemezen tesztelt referenciaszabványok koncentrációértékeire
|
Sor
|
17α-metiltesztoszteron
|
Kortikoszteron
|
17α-ösztradiol
|
E2
|
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
1. konc. (10 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
2. konc. (1 μM)
|
→
|
→
|
10 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
3. konc. (100 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
4. konc. (10 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
5. konc. (1 nM)
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
6. konc. (100 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
7. konc. (10 pM)
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Vivőanyagos kontroll (0,1 % DMSO); PC: Pozitív kontroll (1 nM E2).
|
|
27.
|
A referenciaszabványokat (E2, 17α-ösztradiol, 17α-metiltesztoszteron és kortikoszteron) minden vizsgálatmenetben tesztelni kell (3. táblázat)vvMinden vizsgálati lemezen legyenek pozitív kontrollként szolgáló, a maximális indukciót előidéző 1 nM mennyiségű E2-vel kezelt lyukak, és kizárólag DMSO-dal (vagy megfelelő oldószerrel) kezelt, vivőanyagos kontrollként szolgáló lyukak (4. táblázat). Amennyiben egy adott kísérlet során különböző forrásból származó (pl. eltérő passzálásból, más tételből származó stb.) sejteket alkalmaznak, a referenciaszabványokat minden egyes sejtforráshoz külön vizsgálni kell.
|
4. táblázat
Példa az ösztrogénreceptor agonista vizsgálathoz használt vizsgálati lemez elrendezésére a vizsgálati és kontrollanyagok különböző koncentrációjánál
|
Sor
|
1. vizsgálati vegyi anyag
|
2. vizsgálati vegyi anyag
|
3. vizsgálati vegyi anyag
|
4. vizsgálati vegyi anyag
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
1. konc. (10 μM)
|
→
|
→
|
1 mM
|
→
|
→
|
1 μM
|
→
|
→
|
10 nM
|
→
|
→
|
|
B
|
2. konc. (1 μM)
|
→
|
→
|
100 μM
|
→
|
→
|
100 nM
|
→
|
→
|
1 nM
|
→
|
→
|
|
C
|
3. konc. (100 nM)
|
→
|
→
|
10 μM
|
→
|
→
|
10 nM
|
→
|
→
|
100 pM
|
→
|
→
|
|
D
|
4. konc. (10 nM)
|
→
|
→
|
1 μM
|
→
|
→
|
1 nM
|
→
|
→
|
10 pM
|
→
|
→
|
|
E
|
5. konc. (1 nM)
|
→
|
→
|
100 nM
|
→
|
→
|
100 pM
|
→
|
→
|
1 pM
|
→
|
→
|
|
F
|
6. konc. (100 pM)
|
→
|
→
|
10 nM
|
→
|
→
|
10 pM
|
→
|
→
|
0,1 pM
|
→
|
→
|
|
G
|
7. konc. (10 pM)
|
→
|
→
|
1 nM
|
→
|
→
|
1 pM
|
→
|
→
|
0,01 pM
|
→
|
→
|
|
H
|
VC
|
→
|
→
|
→
|
→
|
→
|
PC
|
→
|
→
|
→
|
→
|
→
|
|
VC: Vivőanyagos kontroll (0,1 % DMSO); PC: Pozitív kontroll (1 nM E2).
|
5. táblázat
Példa az ösztrogénreceptor antagonista vizsgálathoz használt vizsgálati lemez elrendezésére a referenciaszabványok különböző koncentrációjánál

|
28.
|
A vegyi anyagok antagonista aktivitásának értékeléséhez az A–G sor vizsgálati lyukaihoz hozzá kell adni 25 pM E2-t. A referenciaszabványokat (tamoxifén és flutamid) minden vizsgálatmenetben tesztelni kell. Minden vizsgálati lemezen legyenek pozitív kontrollként szolgáló, a hERα-HeLa-9903 sejtvonal minőség-ellenőrzésére használható 1 nM E2-vel kezelt lyukak, DMSO-dal (vagy megfelelő oldószerrel) kezelt, vivőanyagos kontrollként szolgáló lyukak, 0,1 % DMSO-dal kezelt, „kiegészítő kontrollként” szolgáló lyukak, amelyek DMSO mellett E2-t is tartalmaznak, 1 μM végső koncentrációjú 4-hidroxitamoxifennel kezelt lyukak, valamint 100 μM digitonionnal kezelt lyukak (5. táblázat). A további vizsgálati lemezeken a referenciaszabványokat tartalmazó lyukak kivételével ugyanezt az elrendezést kell alkalmazni (6. táblázat). Amennyiben egy adott kísérlet során különböző forrásból származó (pl. eltérő passzálásból, más tételből származó stb.) sejteket alkalmaznak, a referenciaszabványokat minden egyes sejtforráshoz külön vizsgálni kell.
|
6. táblázat
Példa az ösztrogénreceptor antagonista vizsgálathoz használt vizsgálati lemez elrendezésére a vizsgálati és kontrollanyagok különböző koncentrációjánál

|
29.
|
Adott esetben meg kell bizonyosodni arról, hogy nem érvényesül perem-hatás, ha gyaníthatóan fennáll peremhatás, annak kiküszöbölése érdekében módosítani kell a lemez elrendezését. Például olyan elrendezés alkalmazható, amelyen a széleken lévő lyukak üresen maradnak.
|
|
30.
|
A vegyi anyagok hozzáadását követően a vizsgálati lemezeket 5 %-os CO2-tartalmú inkubátorba n 37° ±1° C-on 20–24 órán át inkubálni kell, hogy a riportergén termékei kifejeződjenek.
|
|
31.
|
Rendkívül illékony vegyületek esetén egyedi szempontokat is figyelembe kell venni. Ilyenkor ugyanis a kontrollanyagot tartalmazó közeli lyukak álpozitív eredményt hozhatnak, amit a várt és az ismert kontrollértékek fényében kell értékelni. Abban a néhány esetben, amikor az illékonyság gondot okozhat, „zárófólia” használatával hatékonyan izolálhatók az egyes lyukak a vizsgálat során, ezért ilyen esetekben zárófólia alkalmazása ajánlott.
|
|
32.
|
A vizsgálatok függetlenségének biztosítása érdekében az azonos vegyi anyaggal végzett végleges vizsgálatokat különböző napokon kell megismételni.
|
Luciferáz vizsgálat
|
33.
|
A vizsgálathoz kereskedelmi forgalomban beszerezhető luciferáz vizsgálati reagens [pl. Steady-Glo® luciferáz vizsgálati rendszer (Promega, E2510 vagy ezekkel egyenértékű rendszer)] vagy standard luciferáz vizsgálati rendszer (pl. Promega, E1500 vagy ezekkel egyenértékű rendszer) is használható, amennyiben teljesülnek az elfogadhatósági kritériumok. A vizsgálati reagenseket a használt luminométer érzékenysége alapján kell kiválasztani. Standard luciferáz vizsgálati rendszer alkalmazása esetén a szubsztrátum hozzáadása előtt sejtkultúrát lizáló reagenst (pl. Promega, E1531 vagy ezekkel egyenértékű reagenst) kell használni. A luciferáz reagenst a gyártó utasításai szerint kell alkalmazni.
|
AZ ADATOK ELEMZÉSE
Ösztrogénreceptor agonista vizsgálat
|
34.
|
Az ösztrogénreceptor agonista vizsgálat esetében a pozitív kontrollhoz (1 nM E2) viszonyított, relatív transzkripciós aktivitás meghatározásához egy adott lemez lumineszcencia jelei az alábbi lépések végrehajtásával elemezhetők (ezzel egyenértékű más matematikai eljárások is elfogadhatók):
|
1. lépés Számítsuk ki a VC átlagértékét.
2. lépés Az adatok normalizálásához a VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
3. lépés Számítsuk ki a normalizált PC átlagértékét.
4. lépés Osszuk el a lemez egyes lyukainak normalizált értékét a PC normalizált átlagértékével (PC=100 %).
Az egyes lyukak végső értéke adja meg az adott lyuk PC-válaszhoz viszonyított, relatív transzkripciós aktivitását.
5. lépés Számítsuk ki a vizsgálati vegyi anyag egyes koncentrációcsoportjaira jellemző relatív transzkripciós aktivitás átlagértékét. A válasznak két vetülete van: az átlagos transzkripciós aktivitás (válasz) és az adott koncentráció, amelynél bekövetkezik a válasz (lásd a következő részt).
Az EC50, PC50 és PC10 indukció szempontjai
|
35.
|
Az EC50 meghatározásához teljes koncentráció-válasz görbére van szükség, ennek létrehozása azonban a vizsgált koncentrációtartomány korlátaiból fakadóan (például citotoxicitás vagy oldhatósági problémák miatt) nem mindig megvalósítható vagy nem mindig praktikus. Mivel azonban az EC50 és az indukció maximális szintje (ami a Hill-egyenlet felső értékének felel meg) lényeges információt hordozó paraméterek, lehetőség szerint bele kell foglalni őket a jelentésbe. Az EC50 és az indukció maximális szintjének meghatározásához megfelelő statisztikai szoftvert kell használni (ilyen például a Graphpad Prism statisztikai szoftver). Amennyiben a koncentráció-válasz adatokra alkalmazható a Hill-féle logisztikai egyenlet, az EC50 értékét az alábbi egyenlettel kell kiszámítani (7):
Y=görbe alja + (görbe teteje-alja) / (1+10 exp. ((log EC50 –X) × Hill-meredekség)), ahol:
az × a koncentráció logaritmusa; valamint
az Y a válasz, az Y a szigmoid görbe aljánál kezdődik és a tetejéig halad. A Hill-féle logisztikai egyenletben a görbe aljához rögzítik a nullpontot.
|
|
36.
|
Az egyes vizsgálati vegyi anyagok tekintetében az alábbi adatokat kell biztosítani:
az RPCMax értéket, amely valamely vizsgálati vegyi anyag által előidézett maximális szintű válasz, amelyet ugyanazon lemezen vizsgált 1 nM mennyiségű E2 által kiváltott válasz százalékában adnak meg, továbbá a PCMax értéket (az RPCMax választ kiváltó koncentráció); valamint
pozitív vegyi anyagok esetében azokat a koncentrációkat, amelyeknél PC10, illetve adott esetben PC50 hatás figyelhető meg.
|
|
37.
|
A PCx értéket interpolációval lehet számítani az X-Y koordinátatengely 2 pontja között, az egyik pontnak közvetlenül a PCx érték felett, a másiknak közvetlenül alatta kell elhelyezkednie. Amennyiben a közvetlenül a PCx érték felett elhelyezkedő adatpont koordinátája (a,b), az alatta elhelyezkedő adatponté pedig (c,d), akkor a PCx érték az alábbi egyenlet alapján határozható meg:
log[PCx] = log[c]+(x-d)/(d-b)
|
|
38.
|
A PC-értékek leírását az alábbi 1. ábra ismerteti.
1. ábra
Példa a PC-értékek meghatározására. A PC (1 nM mennyiségű E2) minden vizsgálati lemezen megtalálható

|
Ösztrogénreceptor antagonista vizsgálat
|
39.
|
Az ösztrogénreceptor antagonista vizsgálat esetében a kiegészítő kontrollhoz (25 pM E2) viszonyított, relatív transzkripciós aktivitás meghatározásához egy adott lemez lumineszcencia jelei az alábbi lépések végrehajtásával elemezhetők (ezzel egyenértékű más matematikai eljárások is elfogadhatók):
1. lépés: Számítsuk ki a VC átlagértékét.
2. lépés: Az adatok normalizálásához a VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
3. lépés: Számítsuk ki a normalizált kiegészítő kontroll átlagértékét.
4. lépés: Osszuk el a lemez egyes lyukainak normalizált értékét a normalizált kiegészítő kontroll átlagértékével (kiegészítő kontroll=100 %).
Az egyes lyukak végső értéke adja meg az adott lyuknak a kiegészítő kontroll válaszához viszonyított, relatív transzkripciós aktivitását.
5. lépés: Számítsuk ki az egyes kezelések során megfigyelt relatív transzkripciós aktivitás átlagértékét.
|
Az IC30 és IC50 indukció szempontjai
|
40.
|
A pozitív vegyi anyagok esetében meg kell adni azokat a koncentrációkat, amelyeknél IC30, illetve adott esetben IC50 hatás figyelhető meg.
|
|
41.
|
Az ICx értéket interpolációval lehet kiszámítani az X-Y koordinátatengely 2 pontja között, az egyik pontnak közvetlenül az ICx érték felett, a másiknak közvetlenül alatta kell elhelyezkednie. Amennyiben a közvetlenül az ICx érték felett elhelyezkedő adatpont koordinátája (c,d), az alatta elhelyezkedő adatponté pedig (a,b), akkor az ICx érték az alábbi egyenlet alapján határozható meg:
lin ICx = a-(b-(100-x)) (a-c)/(b-d)
2. ábra
Példa az IC-értékek meghatározására A kiegészítő kontroll (25 pM E2) minden vizsgálati lemezen megtalálható
RTA: relatív transzkripciós aktivitás
|
|
42.
|
Az eredményeknek két (vagy három) független vizsgálatmeneten kell alapulniuk. Amennyiben két vizsgálatmenet összevethető, vagyis reprodukálható eredményeket ad, akkor harmadik vizsgálatmenetre nincs szükség. Az eredmények akkor elfogadhatók, ha:
|
—
|
teljesülnek az elfogadhatósági kritériumok (e kritériumokat lásd a 14–20. pontban),
|
|
Az adatok értelmezésének kritériumai
7. táblázat
Pozitív és negatív besorolás feltételei az ösztrogénreceptor agonista vizsgálatoknál
|
Pozitív
|
Amikor az RPCMax érték két vagy három vizsgálatmenetből kettőben megegyezik a pozitív kontroll által előidézett válasz 10 %-ával, vagy meghaladja azt.
|
|
Negatív
|
Amikor az RPCMax érték két vagy három vizsgálatmenetből kettőben nem éri el legalább a pozitív kontroll által előidézett válasz 10 %-át.
|
8. táblázat
Pozitív és negatív besorolás feltételei az ösztrogénreceptor antagonista vizsgálatoknál
|
Pozitív
|
Amikor két vagy három vizsgálatmenetből kettőben számítható IC30 érték.
|
|
Negatív
|
Amikor két vagy három vizsgálatmenetből kettőben nem számítható IC30 érték.
|
|
43.
|
Az adatok értelmezésének kritériumait a 7. és a 8. táblázat tartalmazza. A pozitív eredményeket jellemezni kell mind a kiváltott hatás mértékével, mind a hatást előidéző koncentrációval. E két cél egyszerre teljesíthető azáltal, ha az eredményeket abban a koncentrációban fejezik ki, amelynél a PC-értékek 50 %-át (PC50) vagy 10 %-át (PC10) elérték az agonista vizsgálatban, és a kiegészítő kontroll értékének 50 %-a (IC50) vagy 30 %-a (IC30) gátlódott az antagonista vizsgálatban. Egy adott vizsgálati vegyi anyag pozitívnak minősül, ha a vizsgálati vegyi anyag által előidézett maximális válasz (RPCMax) két vagy három vizsgálatmenetből kettőben megegyezik a pozitív kontroll által előidézett válasz 10 %-ával, vagy meghaladja azt, ugyanakkor negatívnak tekintendő, ha az RPCMax értéke két vagy három vizsgálatmenetből kettőben nem éri el legalább a pozitív kontroll által előidézett válasz 10 %-át.
|
|
44.
|
Az ösztrogénreceptor agonista vizsgálat PC10, PC50 és PCMax értékeinek, valamint az ösztrogénreceptor antagonista vizsgálat IC30 és IC50 értékeinek kiszámításához igénybe lehet venni a vizsgálati iránymutatáshoz tartozó és az OECD nyilvános weboldalán (64) rendelkezésre álló táblázatot.
|
|
45.
|
A PC10 vagy PC50 és az IC30 vagy IC50 értékek legalább két alkalommal történő meghatározása általában elegendőnek bizonyul. Ha azonban egy adott koncentrációtartományba tartozó adatoknál az alapvonal szórása elfogadhatatlanul magas variációs koefficiensű (CV; %), megkérdőjelezhető az adatok megbízhatósága, és be kell azonosítani a nagy szórás okát. A PC10 érték meghatározásához használt adatpontok nyers adatainak (vagyis a lumineszcencia intenzitására vonatkozó adatoknak) három ismétlésből számolt variációs koefficiense nem érheti el a 20 %-ot.
|
|
46.
|
Az elfogadhatósági kritériumok teljesítése jelzi a vizsgálati rendszer megfelelő működését, de nem garantálja az egyes vizsgálatmenetek adatainak pontosságát. Az adatok pontosságát az biztosítja a legjobban, ha az első vizsgálatmenet eredményei megismételhetők.
|
|
47.
|
Azon ösztrogénreceptor agonista vizsgálatok esetében, amelyeknél a pozitív vizsgálati vegyi anyagok – különösen a PC10–PC49 értékkel rendelkező és a gyaníthatóan túlzott luciferáz-aktivitást okozó vegyi anyagok – tekintetében több információra van szükség, mint amit e szűrési és rangsorolási célú vizsgálati iránymutatás biztosít, egy ERα antagonista használatával megerősíthető, hogy a megfigyelt luciferáz-aktivitást kizárólag az ERα idézi elő (lásd a 2.1. függeléket).
|
VIZSGÁLATI JELENTÉS
|
48.
|
Lásd „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” című 20. pontot.
|
SZAKIRODALOM
|
(1)
|
OECD (2015). Report of the Inter-Laboratory Validation for Stably Transfected Transactivation Assay to detect Estrogenic and Anti-estrogenic Activity. Environment, Health and Safety Publications, Series on Testing and Assessment (No 225), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(2)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71, 1459-1469.
|
|
(3)
|
Kuiper G.G., et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinol., 139, 4252-4263.
|
|
(4)
|
Spaepen M., et al. (1992). Detection of Bacterial and Mycoplasma Contamination in Cell Cultures by Polymerase Chain Reaction, FEMS Microbiol. Lett., 78(1), 89-94.
|
|
(5)
|
Kobayashi H., et al. (1995). Rapid Detection of Mycoplasma Contamination in Cell Cultures by Enzymatic Detection of Polymerase Chain Reaction (PCR) Products, J. Vet. Med. Sci., 57(4), 769- 71.
|
|
(6)
|
Dussurget O. and Roulland-Dussoix D. (1994). Rapid, Sensitive PCR-Based Detection of Mycoplasmas in Simulated Samples of Animal Sera, Appl. Environ. Microbiol., 60(3), 953-9.
|
|
(7)
|
De Lean A., Munson P.J. and Rodbard D. (1978). Simultaneous Analysis of Families of Sigmoidal Curves: Application to Bioassay, Radioligand Assay, and Physiological Dose-Response Curves, Am. J. Physiol., 235, E97-El02.
|
2.1. függelék
ÁLPOZITÍV EREDMÉNYEK: A NEM RECEPTOR ÁLTAL KÖZVETÍTETT LUMINESZCENCIA JELEK ÉRTÉKELÉSE
|
1.
|
Az ösztrogénreceptor agonista vizsgálat álpozitív eredményei keletkezhetnek abból, hogy a luciferáz gént nem az ösztrogénreceptor által közvetített aktiválás indukálta, a géntermék közvetlenül aktiválódott, illetve a vizsgálattól független fluoreszcencia lépett fel. Az ilyen jellegű hatásokra a dózis-válasz görbe hiányosságából vagy szokatlan formájából lehet következtetni. Amennyiben gyaníthatóan ilyen hatások játszanak közre, meg kell vizsgálni egy ösztrogénreceptor antagonista (pl. nem toxikus koncentrációban alkalmazott 4-hidroxitamoxifén (OHT)) válaszra gyakorolt hatását. A tisztán antagonista ICI 1827 80 nem feltétlenül alkalmas erre a célra, mivel az ICI 1827 80 megfelelő koncentrációja csökkentheti a VC értékét, ami befolyásolja az adatelemzést.
|
|
2.
|
E megközelítés hitelességének biztosítása érdekében az alábbiakat kell vizsgálni ugyanazon a lemezen:
|
—
|
az ismeretlen vegyi anyag agonista hatású aktivitása 10 μM OHT-vel és anélkül vizsgálva,
|
|
—
|
agonista pozitív kontrollként 1 nM E2 (három ismétlés),
|
|
—
|
1 nM E2 + OHT (három ismétlés).
|
|
Az adatok értelmezésének kritériumai
Megjegyzés: Valamennyi lyukat a vivőanyag azonos koncentrációjával kell kezelni.
|
—
|
Amennyiben az ismeretlen vegyi anyag agonista hatású aktivitását NEM befolyásolja az ER-antagonistával való kezelése, akkor „negatívnak” minősítendő.
|
|
—
|
Ha az ismeretlen vegyi anyag agonista hatású aktivitását teljesen gátolta a kezelés, akkor a döntési kritériumokat kell alkalmazni.
|
|
—
|
Amennyiben a legalacsonyabb koncentrációnál az agonista hatású aktivitás megegyezik a PC10 válasszal vagy meghaladja azt, az ismeretlen vegyi anyag gátlása eléri vagy meghaladja a PC10 választ. Meg kell határozni a kezeletlen és az ER-antagonistával kezelt lyukak válaszreakciója közötti különbséget, és ez a különbség tekintendő valós válasznak, ez alapján kell kiszámítani a kategóriába sorolási döntést lehetővé tévő, megfelelő paramétereket.
|
Az adatok elemzése
Ellenőrizzük a teljesítményszabványokat.
Ellenőrizzük az azonos feltételekkel kezelt lyukak közötti variációs koefficienst.
|
1.
|
Számítsuk ki a VC átlagát.
|
|
2.
|
A VC átlagát vonjuk ki az egyes OHT-vel nem kezelt lyukaknál kapott értékből.
|
|
3.
|
Számítsuk ki az OHT átlagát.
|
|
4.
|
A VC átlagát vonjuk ki az egyes OHT-vel kezelt lyukaknál kapott értékből.
|
|
5.
|
Számítsuk ki a PC átlagát.
|
|
6.
|
Számítsuk ki a többi lyuk pozitív kontrollhoz viszonyított, relatív transzkripciós aktivitását.
|
2.2. függelék
A DEXTRÁNBEVONATÚ AKTÍV SZÉNNEL (DCC) KEZELT SZÉRUM ELKÉSZÍTÉSE
|
1.
|
A szérum dextránbevonatú aktív szénnel (DCC) történő kezelése egy általánosan használt módszer arra, hogy az ösztrogénhatású vegyületeket eltávolítsák a tápoldatot kiegészítő szérumból, biztosítva ezáltal, hogy a szérumban lévő maradék ösztrogének ne torzítsák el a válasz. Ezen eljárás keretében 500 ml magzati borjú szérumot (FBS) kezelnek.
|
Alkotóelemek
|
2.
|
Az alábbi anyagokra és eszközökre lesz szükség:
|
Anyagok
|
Magnézium-klorid-hexahidrát (MgCl2×6H2O)
|
|
1 M HEPES pufferoldat (pH-értéke 7,4)
|
|
Szűrőrendszerrel előállított ultra nagytisztaságú víz
|
Eszközök
Autokláv üvegedény (mérete szükség szerint módosítandó), általános laboratóriumi centrifuga (amely beállítható 4 oC-os hőmérsékletre)
Az eljárás
|
3.
|
Az alábbi eljárást 50 ml-es centrifugacsövek használatához alakítottuk:
[1. nap] Készítsük el a dextránbevonatú aktív szén szuszpenziót, ehhez szükséges 1 l ultra nagytisztaságú víz, ami 1,5 mM MgCl2-t, 0,25 M szacharózt, 2,5 g aktív szenet, 0,25 g dextránt és 5 mM HEPES puffert tartalmaz, majd 4 oC-on kevertessük egy éjszakán át.
[2. nap] Adagoljuk a szuszpenziót 50 ml-es centrifugacsövekbe, és 10 000 rpm sebességgel 4 oC-on centrifugáljuk 10 percig. Távolítsuk el a felülúszót, és az aktív szén üledék felét tároljuk 4 oC-on, ez a 3. napon kerül felhasználásra. Az aktív szén üledék másik felét szuszpendáljuk FBS-sel, amelyet előzőleg a kicsapódások elkerülése érdekében lassan felolvasztottunk és 56 oC-on 30 percig inaktiváltunk, majd helyezzük a szuszpenziót autoklávozott üvegedénybe, például Erlenmeyer-lombikba. Óvatosan kevertessük a szuszpenziót 4 oC-on egy éjszakán keresztül.
[3. nap] Adagoljuk az FBS-t tartalmazó szuszpenziót centrifugacsövekbe, és 10 000 rpm sebességgel 4 oC-on centrifugáljuk 10 percig. Gyűjtsük össze az FBS-t, és adjuk hozzá a második napon készített és eltárolt aktív szén üledéket. Szuszpendáljuk fel az aktív szén üledéket, majd a szuszpenziót egy autoklávozott üvegedényben óvatosan kevertessük 4 oC-on egy éjszakán keresztül.
[4. nap] Adagoljuk ki a szuszpenziót, és 10 000 rpm sebességgel 4 oC-on centrifugáljuk 10 percig, majd egy 0,2 μm steril szűrőn való átszűréssel sterilizáljuk a felülúszót. Ez a dextránbevonatú aktív szénnel kezelt magzati borjú szérum -20 oC-on tárolandó, és egy évig felhasználható.
|
3. Függelék
ÖSZTROGÉNRECEPTOR AGONISTÁK ÉS ANTAGONISTÁK ÉSZLELÉSÉRE SZOLGÁLÓ VM7LUC ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
1.
|
Ez a vizsgálat VM7Luc4E2 sejtvonalak (65) használatára épül. A vizsgálatot az Országos Toxikológiai Program Alternatív Toxikológiai Módszerek Értékelésével Foglalkozó Ügynökségközi Központja (NICEATM) és az alternatív módszerek validálásával foglalkozó ügynökségközi koordinációs bizottság (ICCVAM) validálta (1). A VM7Luc sejtvonalak elsősorban endogén ERα, kisebb mértékben endogén ERβ receptorokat fejeznek ki (2) (3) (4).
|
|
2.
|
Ez a vizsgálat anyagok széles skálájára alkalmazható, feltéve, ha oldódnak dimetil-szulfoxidban (DMSO; CAS-szám: 67-68-5), nem lépnek reakcióba a DMSO-dal vagy a sejttenyésztő közeggel, és a vizsgált koncentrációkban nem citotoxikusak. Amennyiben DMSO nem használható, más vivőanyag, például etanol vagy víz is alkalmazható (lásd a 12. pontot). A VM7Luc ösztrogénreceptor transzaktivációs (ant)agonista vizsgálat bizonyított hatékonysága arra enged következtetni, hogy a vizsgálat során nyert adatok információt nyújthatnak az ER-közvetített hatásmechanizmusokról, és alkalmasak az anyagok további vizsgálatra való rangsorolására.
|
|
3.
|
Ezt a vizsgálatot kifejezetten arra a célra tervezték, hogy a kémiai lumineszcencia mint végpont mérése révén észlelje a hERα és a hERß által közvetített transzaktivációt. A kémiai lumineszcenciát széles körben alkalmazzák a biológiai vizsgálatok során, mert a lumineszcencia nagy érzékenységgel mérhető a háttérzajhoz képest. A sejtalapú vizsgálatokban azonban a szentjánosbogár luciferáz aktivitását megzavarhatják olyan anyagok, amelyek gátlólag hatnak a luciferáz enzimre, és ezáltal egyaránt előidézhetnek látszólagos gátlást és fehérjestabilizációból fakadó erősebb lumineszcenciát. Emellett néhány luciferáz alapú ER riportergén vizsgálatban beszámoltak arról, hogy az 1 μM-nél magasabb fitoösztrogén-koncentrációnál a luciferáz riportergén túlzott aktiválása miatt nem receptorok által közvetített lumineszcencia jelek észlelhetők. Bár a dózis-válasz görbe arra utal, hogy az ER-rendszer ténylegesen alacsonyabb koncentrációknál aktiválódik, a magas fitoösztrogén-koncentráció mellett vagy feltehetőleg a luciferáz riportergént a fitoösztrogénhez hasonlóan túlzottan aktiváló vegyületek magas koncentrációja mellett kapott luciferáz-kifejeződést körültekintően kell vizsgálni a stabilan transzfektált ösztrogénreceptor transzaktivációs vizsgálati rendszerekben.
|
|
4.
|
E vizsgálat szabályozási célra való alkalmazása előtt el kell olvasni az „ÁLTALÁNOS BEVEZETÉS” és „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részt. A vizsgálati módszerben használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLAT ELVE (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
5.
|
Ez a vizsgálat arra szolgál, hogy jelezze az ösztrogénreceptor ligandumhoz kötődését, amely ezt követően mint receptor-ligandum komplex transzlokálódik a sejtmagba. A receptor-ligandum komplex a sejtmagban a specifikus DNS válaszelemekhez kötődve transzaktiválja a riportergént (luc), aminek eredményeként luciferáz termelődik, majd fény szabadul fel, amelynek mennyisége luminométerrel meghatározható. A luciferáz-aktivitás számos kereskedelmi forgalomban kapható készlettel gyorsan és költséghatékonyan értékelhető. A VM7Luc ösztrogénreceptor transzaktiváció során egy ösztrogénreceptor-reszponzív humán emlő adenokarcinoma sejtvonalat, a VM7-et használják, amelyet egy szentjánosbogár luciferáz riporterkonstrukcióval stabilan transzfektáltak, a riporter gén az egér emlőtumor vírus (MMTV) promóter előtt található négy ösztrogén válaszelem szabályzása alatt áll, így lehet észlelni in vitro az ösztrogénreceptor agonista vagy antagonista hatást kiváltó anyagokat. Ez az MMTV-promóter más szteroid és nem szteroid hormonokra csak csekély mértékben aktiválódik (8). Az adatok értelmezésének kritériumait részletesen a 41. pont ismerteti. Röviden: a pozitív válasz olyan koncentráció-válasz görbe alapján ismerhető fel, amely legalább három pontot tartalmaz, amelyeknek a szórása nem fed át (átlag ± SD), valamint a (normalizált relatív fényegységben [RLU] mért) válasz amplitúdója eléri a referenciaszabvány maximális értékének legalább 20 %-át (a referenciaszabvány a 17β-ösztradiol [E2; CAS-szám: 50-28-2] az agonista vizsgálat esetén, a raloxifen-hidroklorid [Ral; CAS-szám: 84 449-90-1]/E2 az antagonista vizsgálat esetén).
|
ELJÁRÁS
Sejtvonal
|
6.
|
A vizsgálathoz stabilan transzfektált VM7Luc4E2 sejtvonalat kell használni. A sejtvonal jelenleg kizárólag szakmai licencszerződés birtokában szerezhető be a Kaliforniai Egyetemtől, Davis, Kalifornia, USA (66) és a Xenobiotic Detection Systems Inc. vállalattól, Durham, Észak-Karolina, USA (67).
|
A sejtvonal stabilitása
|
7.
|
A sejtvonal stabilitásának és integritásának megőrzése érdekében a fagyasztott sejtkultúrából származó sejteket legalább két passzálásig megfelelő tápoldatban kell növeszteni (lásd a 9. pontot). A sejteket legfeljebb 30 alkalommal szabad passzálni. A VM7Luc4E2 sejtvonal esetében a 30 passzálás mintegy három hónapot vesz igénybe.
|
Sejttenyésztés és a leültetés körülményei
|
8.
|
Az összes anyag és módszer minőségének biztosítása érdekében az Iránymutatás a megfelelő sejttenyésztési gyakorlathoz (5) (6) című dokumentumban meghatározott eljárások szerint kell eljárni, hogy garantálni lehessen az elvégzett tevékenységek integritását, hitelességét és megismételhetőségét.
|
|
9.
|
A VM7Luc4E2 sejteket 0,9 % penicillinnel és sztreptomicinnel, valamint 8,0 % magzati borjú szérummal (FBS) kiegészített RPMI 1 640 tápoldatban tartják fenn, egy erre a célra alkalmas szövettenyésztő inkubátorban 37 oC-on (± 1 oC), 90 %-os (± 5 %) páratartalom és 5,0 %-os (± 1 %) CO2-tartalom mellett.
|
|
10.
|
Mintegy 80 %-os konfluencia elérését követően a VM7Luc4E2 sejteket szubkultúrákat hoznak létre és a leültetés előtt egy ösztrogénmentes környezetben 48 órán át kondicionálják, majd a sejteket leültetik egy 96 lyukú lemezre, ahol a vizsgálati vegyi anyaggal kezelik és elemzik a luciferáz-aktivitás ösztrogénfüggő indukcióját. Az ösztrogénmentes tápoldat (EFM) egy fenolvörösmentes Dulbecco-féle módosított Eagle-tápoldat (DMEM), kiegészítve 4,5 % dextránbevonatú aktív szénnel kezelt magzati borjú szérummal (FBS), 1,9 % L-glutaminnal, valamint 0,9 % penicillinnel és sztreptomicinnel. A műanyag edényeken nem történhet ösztrogénhatású aktivitás [lásd a részletes protokollt (7)].
|
Elfogadhatósági kritériumok
|
11.
|
Valamely vizsgálat elfogadására vagy elvetésére az egyes 96 lyukú lemezeken végzett kísérletekben alkalmazott referenciaszabványok és kontrollok eredményeinek értékelése alapján kerül sor. Minden referenciaszabványt több koncentrációban kell tesztelni, és minden referenciaszabvány- és kontrollanyag-koncentrációt több mintán kell vizsgálni. Az eredményeket össze kell vetni az e paraméterek minőség-ellenőrzésére szolgáló adatokkal, amelyek a laboratórium által a jártasság bizonyítása során létrehozott agonista és antagonista adatbázisokból származnak. Ezeket az adatbázisokat a referenciaszabványokra és a kontrollanyagokra kapott értékekkel folyamatosan frissítik. A felszerelés vagy a laboratóriumi körülmények változása esetén új adatbázisok létrehozására lehet szükség.
|
Agonista vizsgálat
Dózistartomány- kereső vizsgálat
|
•
|
Indukció: A lemezen tapasztalt aktivitás indukciós faktorának méréséhez el kell osztani az E2 referenciaszabvány átlagos legmagasabb relatív fényegységét (RLU) a DMSO-kontroll átlagos RLU-értékével. Általában ötszörös indukció figyelhető meg, de a legalább négyszeres indukció már elfogadható.
|
|
•
|
DMSO-kontroll eredményei: Az oldószeres kontroll RLU-értékeinek nem szabad meghaladniuk a dokumentált oldószeres kontrollok átlagos RLU-értékére jellemző standard deviáció 2,5-szeresét.
|
|
•
|
Azt a kísérletet, amelyben e két elfogadhatósági kritérium bármelyike nem teljesül, figyelmen kívül kell hagyni és meg kell ismételni.
|
Teljes körű vizsgálat
Az agonista dózistartomány-kereső vizsgálat elfogadhatósági kritériumai mellett az alábbiakat foglalja magában:
|
•
|
Referenciaszabvány eredményei: Az E2 referenciaszabvány koncentráció-válasz görbéje legyen szigmoid formájú és tartalmazzon legalább három értéket a koncentráció-válasz görbe lineáris szakaszán.
|
|
•
|
Pozitív kontroll eredményei: A metoxiklórral végzett kontroll RLU-értékei haladják meg a DMSO átlagától való standard deviáció háromszorosával növelt DMSO-átlagot.
|
|
•
|
Azt a kísérletet, amelyben valamely elfogadhatósági kritérium nem teljesül, figyelmen kívül kell hagyni és meg kell ismételni.
|
Antagonista vizsgálat
Dózistartomány- kereső vizsgálat
|
•
|
Aktivitás csökkenése: A lemezen tapasztalt aktivitás csökkenésének méréséhez el kell osztani a Ral/E2 referenciaszabvány átlagos legmagasabb RLU-értékét a DMSO-kontroll átlagos RLU-értékével. Általában ötszörös csökkenés figyelhető meg, de a legalább háromszoros csökkenés már elfogadható.
|
|
•
|
E2-kontroll eredményei: Az E2-kontroll RLU-értékeinek nem szabad meghaladniuk a dokumentált E2-kontrollok átlagos RLU-értékére jellemző standard deviáció 2,5-szeresét.
|
|
•
|
DMSO-kontroll eredményei: A DMSO-kontroll RLU-értékeinek nem szabad meghaladniuk a dokumentált oldószeres kontrollok átlagos RLU-értékére jellemző standard deviáció 2,5-szeresét.
|
|
•
|
Azt a kísérletet, amelyben valamely elfogadhatósági kritérium nem teljesül, figyelmen kívül hagyják és megismétlik.
|
Teljes körű vizsgálat
Az antagonista dózistartomány-kereső vizsgálat elfogadhatósági kritériumai mellett az alábbiakat foglalja magában:
|
•
|
Referenciaszabvány eredményei: A Ral/E2 referenciaszabvány koncentráció-válasz görbéje legyen szigmoid formájú és tartalmazzon legalább három értéket a koncentráció-válasz görbe lineáris szakaszán.
|
|
•
|
Pozitív kontroll eredményei: A tamoxifénnel/E2-vel végzett kontroll RLU-értékei nem haladhatják meg az E2-kontroll átlagától való standard deviáció háromszorosával csökkentett E2-átlagot.
|
|
•
|
Azt a kísérletet, amelyben valamely elfogadhatósági kritérium nem teljesül, figyelmen kívül hagyják és megismétlik.
|
Referenciaszabványok, pozitív és vivőanyagos kontrollok
Vivőanyagos kontroll (agonista és antagonista vizsgálat)
|
12.
|
A vizsgálati vegyi anyagok feloldására használt vivőanyagot vivőanyagos kontroll keretében vizsgálni kell. A VM7Luc ösztrogénreceptor transzaktivációs vizsgálat validálása során használt vivőanyag 1 térfogatszázaléknyi dimetil-szulfoxid (DMSO, CAS-szám: 67-68-5) (lásd a 24. pontot). DMSO-tól eltérő vivőanyag használata esetén valamennyi referenciaszabványt, kontrollanyagot és vizsgálati vegyi anyagot ugyanabban a vivőanyagban kell tesztelni, ha alkalmazandó.
|
Referenciaszabvány (agonista dózisbehatárolás)
|
13.
|
A referenciaszabvány az E2 (CAS-szám: 50-28-2). A dózistartomány-kereső vizsgálatban a referenciaszabvány az E2 négy elemből álló koncentrációsorozata (1,84 x 10-10, 4,59 x 10-11, 1,15 x 10-11 és 2,87 x 10-12 M), minden egyes koncentrációértéket replikátumban kell vizsgálni.
|
Referenciaszabvány (agonista teljes körű vizsgálat)
|
14.
|
A teljes körű vizsgálatban az E2 1:2 arányban hígított 11 elemes (3,67 x 10-10 és 3,59 x 10-13 M között terjedő) koncentrációsorozatból áll, a koncentrációértékeket replikátumban vizsgálják.
|
Referenciaszabvány (antagonista dózisbehatárolás)
|
15.
|
A referenciaszabvány a Ral (CAS-szám: 84 449-90-1) és az E2 (CAS-szám: 50-28-2) kombinációja. A dózistartomány-kereső vizsgálatban a Ral/E2 a Ral három elemből álló hígított koncentrációsorozatából (3,06 x 10-9, 7,67 x 10-10 és 1,92 x 10-10 M) és az E2 fix koncentrációjából (9,18 × 10-11 M) tevődik össze, minden koncentrációértéket replikátumban vizsgálnak.
|
Referenciaszabvány (antagonista teljes körű vizsgálat)
|
16.
|
A teljes körű vizsgálatban a Ral/E2 a Ral 1:2 arányban hígított (2,45 x 10-8 és 9,57 x 10-11 M között terjedő) koncentrációsorozatából és az E2 fix koncentrációjából (9,18 × 10-11 M) tevődik össze, a Ral/E2 kilenc koncentrációértékének mindegyikét replikátumban vizsgálják.
|
Gyenge pozitív kontroll (agonista)
|
17.
|
A gyenge pozitív kontroll az ösztrogénmentes közegben vizsgált 9,06 x 10-6 M p,p’-metoxiklór (metoxiklór; CAS-száma: 72-43-5).
|
Gyenge pozitív kontroll (antagonista)
|
18.
|
A gyenge pozitív kontroll az ösztrogénmentes közegben, 9,18 × 10-11 M E2 jelenlétében vizsgált 3,36 x 10-6 M tamoxifén (CAS-száma: 10 540-29-1).
|
E2-kontroll (csak antagonista vizsgálat esetén)
|
19.
|
Az alapvonal negatív kontrollként szolgáló E2-kontroll az ösztrogénmentes közegben vizsgált 9,18 × 10-11 M E2.
|
Indukciós faktor (agonista)
|
20.
|
A referenciaszabvány (E2) által előidézett luciferáz-aktivitás méréséhez el kell osztani az E2 referenciaszabvány átlagos legmagasabb RLU-értékét a DMSO-kontroll átlagos RLU-értékével; az eredménynek négyszeresnél nagyobb indukciót kell jeleznie.
|
Indukciós faktor (antagonista)
|
21.
|
A referenciaszabvány (Ral/E2) által előidézett átlagos luciferáz-aktivitás méréséhez el kell osztani a Ral/E2 referenciaszabvány átlagos legmagasabb RLU-értékét a DMSO-kontroll átlagos RLU-értékével, és az eredménynek háromszorosnál nagyobb csökkenést kell mutatnia.
A laboratórium jártasságának igazolása (lásd a vizsgálati módszer „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részének 14. pontját, valamint 3. és 4. táblázatát).
|
Vivőanyag
|
22.
|
A vizsgálati vegyi anyagokat olyan oldószerben kell feloldani, amelyik szolubilizálja a vizsgálati vegyi anyagot és elegyedik a tápoldattal. A megfelelő vivőanyagok közé tartozik a víz, az etanol (95–100 %-os tisztaságban) és a dimetil-szulfoxid. Dimetil-szulfoxid használata esetén az anyag mennyisége nem haladhatja meg az 1 térfogatszázalékot. Minden vivőanyag esetében igazolni kell, hogy maximális alkalmazott térfogatuk nem fejt ki citotoxikus hatást és nem zavarja a vizsgálat teljesítményét. A referenciaszabványokat és kontrollanyagokat 100 %-os oldószerben feloldják, majd ösztrogénmentes közegben a megfelelő koncentrációkra hígítják.
|
Vizsgálati vegyi anyagok előkészítése
|
23.
|
A vizsgálati vegyi anyagokat 100 %-os DMSO-ban (vagy megfelelő oldószerben) feloldják, majd ösztrogénmentes közegben a megfelelő koncentrációkra hígítják. Feloldás és hígítás előtt meg kell várni, hogy minden vizsgálati vegyi anyag szobahőmérsékletű legyen. A vizsgálati vegyi anyag oldatait minden kísérlethez frissen kell elkészíteni. Az oldatban nem lehet szemmel látható kicsapódás és az oldat nem lehet zavaros. A referenciaszabványok és kontrollanyagok nagy tételben is előkészíthetők; a referenciaszabványok és kontrollanyagok végleges oldatait, valamint a vizsgálati vegyi anyagokat minden egyes kísérlethez frissen kell készíteni, és az elkészítéstől számított 24 órán belül fel kell használni.
|
Oldhatóság és citotoxicitás: a dózisbehatárolás szempontjai
|
24.
|
A dózistartomány-kereső vizsgálat hét koncentrációban és duplikátumban vizsgált 1: 10 arányú oldatsorozatból áll. A vizsgálati vegyi anyagokat először a legmagasabb koncentrációig vizsgálják, ami az agonista vizsgálat esetében 1 mg/ml (~1 mM), az antagonista vizsgálat esetében pedig 20 μg/ml (~10 μM). A dózistartomány-kereső kísérletek az alábbiak meghatározására szolgálnak:
|
|
—
|
a vizsgálati vegyi anyag teljes körű vizsgálat során alkalmazandó induló koncentrációértékei;
|
|
—
|
a vizsgálati vegyi anyag teljes körű vizsgálat során alkalmazandó (1:2 vagy 1:5 arányú) hígításai.
|
|
25.
|
Az agonista és antagonista vizsgálati protokollokba belefoglalták a sejtéletképesség/citotoxicitás mérését (7), ami részét képezi mind a dózistartomány-kereső, mind a teljes körű vizsgálatnak. A VM7Luc ösztrogénreceptor transzaktivációs vizsgálat validálása során (1) a sejtéletképesség megállapítása céljából egy lépcsőzetes kvalitatív vizuális megfigyelési módszerrel mérték fel a citotoxicitást; a citotoxicitás meghatározásához azonban kvantitatív módszert is lehet alkalmazni (lásd a protokollt (7)). Az életképességet több mint 20 %-kal csökkentő vegyianyag-koncentrációkra vonatkozó adatok nem használhatók.
|
Vegyi anyaggal történő kezelés és a vizsgálati lemez elrendezése
|
26.
|
A sejteket megszámlálják és ösztrogénmentes tápoldatban (lyukanként 2 x 105 sejt sűrűségben) 96 lyukú szövettenyésztő lemezre szélesztik ki, majd 24 órán át inkubálják, hogy a sejtek kitapadjanak a lemezre. Ezt követően az ösztrogénmentes tápoldatot eltávolítják, és a sejteket ösztrogénmentes tápoldatba adagolt vizsgálati és referencia vegyi anyagokkal 19–24 órán keresztül inkubálják. Rendkívül illékony anyagok esetén egyedi szempontokat is figyelembe kell venni, mivel a kontrollanyagot tartalmazó közeli lyukak álpozitív eredményhez vezethetnek. Ilyen esetekben a „zárófólia” használata hatékonyan izolálhatja az egyes lyukakat a vizsgálat során, ezért alkalmazása ajánlott.
|
Dózistartomány-kereső vizsgálatok
|
27.
|
A dózistartomány-kereső vizsgálat során a 96 lyukú lemez összes lyukának felhasználásával akár hat vizsgálati vegyi anyag is tesztelhető hét koncentrációban, duplikátumban, 1:10 arányú hígítási sorozatban (lásd az 1. és a 2. ábrát).
|
|
—
|
Az agonista dózistartomány-kereső vizsgálat során referenciaszabványként az E2 négy koncentrációját mérik duplikátumban, és négy lyukat tartanak fent a DMSO-kontroll számára.
|
|
—
|
Az antagonista dózistartomány-kereső vizsgálat során pedig referenciaszabványként a Ral/E2 három koncentrációját mérik 9,18 × 10-11 M E2-vel duplikátumban, és három-három lyukat tartanak fent az E2 és a DMSO-kontroll számára.
|
1. ábra
A 96 lyukú lemez elrendezése agonista dózistartomány-kereső vizsgálathoz

Rövidítések: E2-1 és E2-4 között = az E2 referenciaszabvány koncentrációi (magastól az alacsony felé haladva); TC1-1 és TC1-7 között = az 1. vizsgálati vegyi anyag (TC1) koncentrációi (magastól az alacsony felé haladva); TC2-1 és TC2-7 között = a 2. vizsgálati vegyi anyag (TC2) koncentrációi (magastól az alacsony felé haladva); TC3-1 és TC3-7 között = a 3. vizsgálati vegyi anyag (TC3) koncentrációi (magastól az alacsony felé haladva); TC4-1 és TC4-7 között = a 4. vizsgálati vegyi anyag (TC4) koncentrációi (magastól az alacsony felé haladva); TC5-1 és TC5-7 között = az 5. vizsgálati vegyi anyag (TC5) koncentrációi (magastól az alacsony felé haladva); TC6-1 és TC6-7 között = a 6. vizsgálati vegyi anyag (TC6) koncentrációi (magastól az alacsony felé haladva); VC = vivőanyagos kontroll (DMSO [1 térfogatszázalék EFM-ben]).
2. ábra
A 96 lyukú lemez elrendezése antagonista dózistartomány-kereső vizsgálathoz

Rövidítések: E2 = E2-kontroll; Ral-1 és Ral-3 között = a raloxifen/E2 referenciaszabvány koncentrációi (magastól az alacsony felé haladva); TC1-1 és TC1-7 között = az 1. vizsgálati vegyi anyag (TC1) koncentrációi (magastól az alacsony felé haladva); TC2-1 és TC2-7 között = a 2. vizsgálati vegyi anyag (TC2) koncentrációi (magastól az alacsony felé haladva); TC3-1 és TC3-7 között = a 3. vizsgálati vegyi anyag (TC3) koncentrációi (magastól az alacsony felé haladva); TC4-1 és TC4-7 között = a 4. vizsgálati vegyi anyag (TC4) koncentrációi (magastól az alacsony felé haladva); TC5-1 és TC5-7 között = az 5. vizsgálati vegyi anyag (TC5) koncentrációi (magastól az alacsony felé haladva); TC6-1 és TC6-7 között = a 6. vizsgálati vegyi anyag (TC6) koncentrációi (magastól az alacsony felé haladva); VC = vivőanyagos kontroll (DMSO [1 térfogatszázalék EFM-ben]).
Megjegyzés: Minden vizsgálati vegyi anyagot 9,18 × 10-11 M E2 jelenlétében kell vizsgálni.
|
28.
|
Az egyes lyukakba helyezendő tápoldat végleges ajánlott térfogata 200 μl. Csak olyan vizsgálati lemezek használhatók, amelyek valamennyi lyukában a sejtek életképessége legalább 80 %.
|
|
29.
|
A teljes körű
agonista
vizsgálat induló koncentrációértékeinek meghatározását részletesen ismerteti az agonista protokoll (7). Röviden az alábbi kritériumoknak kell megfelelni:
|
|
—
|
Amennyiben a vizsgálati vegyi anyag koncentrációgörbéjén nincs olyan pont, amelyik meghaladná a DMSO-kontroll átlagától való standard deviáció háromszorosával növelt átlagot, a teljes körű vizsgálatot 1:2 arányban hígított, 11 elemből álló koncentrációsorozattal kell végrehajtani, a maximális oldható koncentrációval kezdve.
|
|
—
|
Amennyiben a vizsgálati vegyi anyag koncentrációgörbéjén vannak olyan pontok, amelyek meghaladják a DMSO-kontroll átlagától való standard deviáció háromszorosával növelt átlagot, a teljes körű vizsgálat 11 koncentrációjú hígítási rendszerének induló koncentrációértéke egy loggal magasabb legyen annál a koncentrációnál, amely a dózisbehatárolás során a legmagasabb kiigazított RLU-értéket adta. A 11 koncentrációjú hígítási rendszer alapja az 1:2 vagy 1:5 arányú hígítás, az alábbi kritériumok alapján:
|
Az 1:2 arányban hígított, 11 elemből álló koncentrációsorozat akkor alkalmazandó, ha az eredményül kapott koncentrációtartomány – a dózistartomány-kereső vizsgálat koncentráció-válasz görbéje alapján – magában foglalja a válaszok teljes tartományát. Ellenkező esetben 1:5 arányú hígítást kell alkalmazni.
|
|
|
—
|
Amennyiben valamely vizsgálati vegyi anyag dózistartomány-kereső vizsgálata kétfázisú koncentráció-válasz görbét eredményez, a teljes körű vizsgálat során mindkét fázist figyelembe kell venni.
|
|
30.
|
A teljes körű
antagonista
vizsgálat induló koncentrációértékeinek meghatározását részletesen ismerteti az antagonista protokoll (7). Röviden az alábbi kritériumoknak kell megfelelni:
|
|
—
|
Amennyiben a vizsgálati vegyi anyag koncentrációgörbéjén nincs olyan pont, ami kisebb mint az E2-kontroll átlagától való standard deviáció háromszorosával csökkentett átlag, a teljes körű vizsgálatot 1:2 arányban hígított, 11 elemből álló koncentrációsorozattal kell végrehajtani, a maximális oldható koncentrációval kezdve.
|
|
—
|
Amennyiben a vizsgálati vegyi anyag koncentrációgörbéjén vannak olyan pontok, amelyek nem haladják meg az E2-kontroll átlagától való standard deviáció háromszorosával csökkentett átlagot, a teljes körű vizsgálat 11 koncentrációjú hígítási rendszerének induló koncentrációértéke az alábbiak egyike kell legyen:
|
—
|
a dózisbehatárolás során a legalacsonyabb kiigazított RLU-értéket adó koncentráció;
|
|
—
|
a maximális oldható koncentráció (lásd az antagonista protokoll (7) 14-2. ábráját);
|
|
—
|
a legalacsonyabb citotoxikus koncentráció (vonatkozó példához lásd az antagonista protokoll (7) 14-3. ábráját).
|
|
|
—
|
A 11 koncentrációjú hígítási rendszer alapja az 1:2 vagy 1: 5 arányú hígítás, az alábbi kritériumok alapján:
|
Az 1:2 arányban hígított, 11 elemből álló koncentrációsorozat akkor alkalmazandó, ha az eredményül kapott koncentrációtartomány – a dózistartomány-kereső vizsgálat koncentráció-válasz görbéje alapján – magában foglalja a válaszok teljes tartományát. Ellenkező esetben 1:5 arányú hígítást kell alkalmazni.
|
|
Teljes körű tesztelés
|
31.
|
A teljes körű vizsgálat egy 11 elemes koncentrációsorozatból áll (1:2 vagy 1:5 arányú hígítás alkalmazandó a teljes körű vizsgálat induló koncentrációértékének meghatározási kritériumai alapján), és minden koncentrációértéket három ismétlésben mérnek egy 96 lyukú lemezen (lásd a 3. és 4. ábrát).
|
|
—
|
Az agonista teljes körű vizsgálat során referenciaszabványként az E2 11 koncentrációját mérik replikátumban. Mindegyik lemezen négy replikátum lyukat tartanak fent a DMSO-kontroll és négy replikátum lyukat a metoxiklórral végzett kontroll (9,06 x 10-6 M) számára.
|
|
—
|
Az antagonista teljes körű vizsgálat során referenciaszabványként a Ral/E2 kilenc koncentrációját mérik 9,18 × 10-11 M E2-vel duplikátumban, emellett négy replikátum lyukat tartanak fent a 9,18 x 10-11 M E2-kontroll, négy replikátum lyukat a DMSO-kontroll és négy replikátum lyukat a 3,36 x 10-6 M tamoxifén kontroll számára.
|
3. ábra
A 96 lyukú lemez elrendezése agonista teljes körű vizsgálathoz

Rövidítések: TC1-1 és TC1-11 között = a 1. vizsgálati vegyi anyag koncentrációi (magastól az alacsony felé haladva); TC2-1 és TC2-11 között = a 2. vizsgálati vegyi anyag koncentrációi (magastól az alacsony felé haladva); E2-1 és E2-11 között = az E2 referenciaszabvány koncentrációi (magastól az alacsony felé haladva); Met = p,p’-metoxiklórral végzett gyenge pozitív kontroll; VC = 1 térfogatszázalék DMSO-dal EFM-ben végzett vivőanyagos kontroll.
4. ábra
A 96 lyukú lemez elrendezése antagonista teljes körű vizsgálathoz
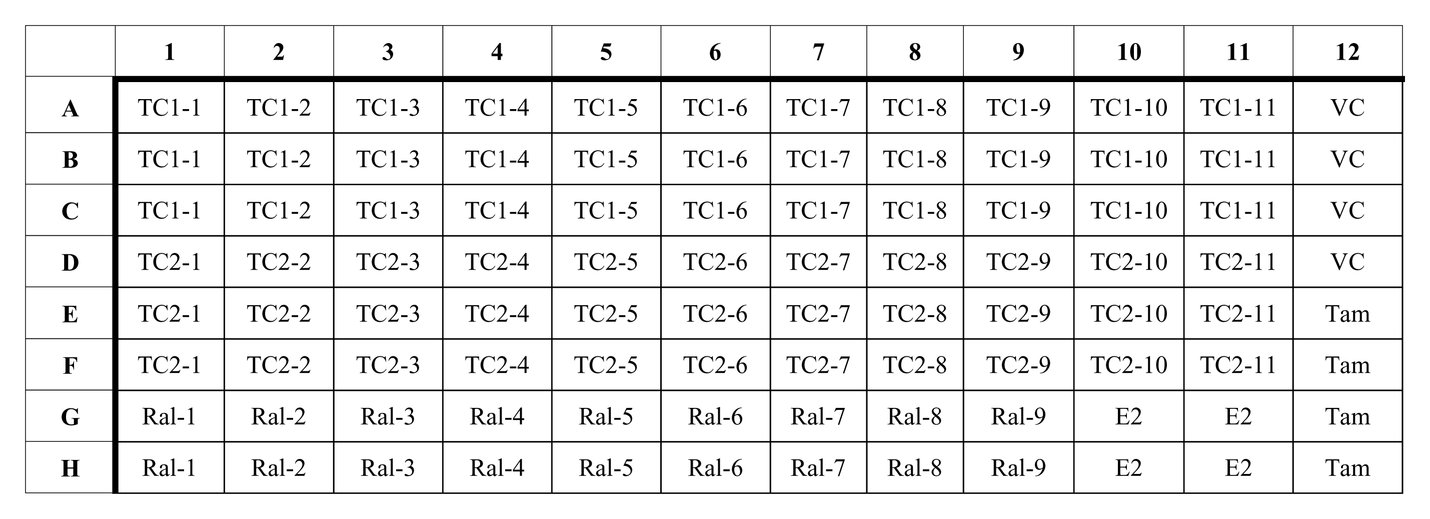
Rövidítések: E2 = E2-kontroll; Ral-1 és Ral-9 között = a raloxifen/E2 referenciaszabvány koncentrációi (magastól az alacsony felé haladva); Tam = tamoxifénnel/E2-vel végzett gyenge pozitív kontroll; TC1-1 és TC1-11 között = az 1. vizsgálati vegyi anyag (TC1) koncentrációi (magastól az alacsony felé haladva); TC2-1 és TC2-11 között = a 2. vizsgálati vegyi anyag (TC2) koncentrációi (magastól az alacsony felé haladva); VC = vivőanyagos kontroll (DMSO [1 térfogatszázalék EFM-ben]).
Megjegyzés: Amint az már említésre került, minden referencia és vizsgálati anyagot tartalmazó lyuk fix koncentrációban (9,18 x 10-11 M) E2-t tartalmaz.
|
32.
|
A vizsgálatok függetlenségének biztosítása érdekében az azonos vegyi anyaggal végzett teljes körű vizsgálatokat különböző napokon kell megismételni. Legalább két teljes körű vizsgálatot kell végezni. Ha az egyik vizsgálat eredménye ellentmond a másikénak (pl. az egyik pozitív, a másik negatív), vagy ha az egyik vizsgálat nem megfelelő eredményre vezetett, egy további, harmadik vizsgálatra van szükség.
|
A lumineszcencia mérése
|
33.
|
A lumineszcencia 300 és 650 nm közötti tartományban mérhető, a mérés egy injektor luminométer és egy szoftver segítségével történik, ez utóbbi szabályozza az injektált térfogatot és a mérési időközt (7). Az egyes lyukak által kibocsátott fényt lyukankénti RLU-ban fejezik ki.
|
AZ ADATOK ELEMZÉSE
Az EC50 /IC50 érték meghatározása
|
34.
|
Az EC50 érték (a vizsgálati vegyi anyag maximális hatásának felét kiváltó koncentráció [agonisták esetében]) és az IC50 érték (a vizsgálati vegyi anyag maximális gátló hatásának felét kiváltó koncentráció [antagonisták esetében]) a koncentráció-válasz adatok alapján kerül meghatározásra. A legalább egy koncentrációban pozitívnak bizonyult vizsgálati vegyi anyagoknál a vizsgálati vegyi anyag félmaximális választ kiváltó koncentrációját (IC50 vagy EC50) Hill-féle funkcióanalízissel vagy annak megfelelő alternatívájával számítják ki. A Hill-féle funkcióanalízis egy négy paraméteres logisztikai-matematikai modell, amely az alábbi egyenlet révén összekapcsolja a vizsgálati vegyi anyag koncentrációját a válasszal (ami általában egy szigmoid görbe formáját veszi fel): Y = Görbe alja +
(Görbe teteje - Görbe alja) 1 + 10(lgC
50 – x)Hill-meredekség
|
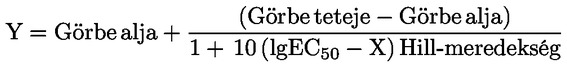
ahol:
Y= a válasz (vagyis az RLU-értékek);
X= a koncentráció logaritmusa;
Görbe alja= a minimális válasz;
Görbe teteje= a maximális válasz;
lg EC50 (vagy lg IC50)= az X logaritmusa, a görbe teteje és alja közötti válasz;
Hill-meredekség= a görbe meredeksége.
A modell kikalkulálja a felső, alsó, meredekségi, IC50 és EC50 paraméterek legjobb mérőszámait. Az EC50 és IC50 értékek meghatározásához megfelelő statisztikai szoftvert kell használni (ilyen például a Graphpad PrismR statisztikai szoftver).
Kiugró értékek meghatározása
|
35.
|
A statisztikai eredmények megfelelő értékelését megkönnyíti, ha a vizsgálat során (egyebek mellett) elvégeznek egy Q-tesztet (az adatelemzésből kizárandó „használhatatlan” lyukak azonosításáról lásd az agonista és antagonista protokollt (7)).
|
|
36.
|
Az E2 referenciaszabvány (két mintájú) replikátumai esetében az E2 bármely koncentrációjú replikátumának kiigazított RLU-értéke akkor tekinthető kiugrónak, ha az adott érték 20 %-kal alatta vagy felette van az adatbázisban az adott koncentrációra vonatkozó kiigazított RLU-értéknek.
|
Luminométerrel mért adatok begyűjtése és kiigazítása dózistartomány-kereső vizsgálathoz
|
37.
|
A luminométer által szolgáltatott nyers adatokat át kell vezetni a vizsgálat céljára létrehozott táblázatsablonba. Meg kell határozni, hogy vannak-e a vizsgálatból kizárandó kiugró adatok. (Lásd az elemzés során meghatározott paraméterekre vonatkozó elfogadhatósági kritériumokat.) Az alábbi számításokat kell elvégezni:
|
Agonista vizsgálat
|
1. lépés
|
Számítsuk ki a DMSO vivőanyagos kontroll (VC) átlagértékét.
|
|
2. lépés
|
Az adatok normalizálásához a DMSO VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
|
|
3. lépés
|
Számítsuk ki a referenciaszabvány (E2) átlagos indukciós faktorát.
|
|
4. lépés
|
Számítsuk ki a vizsgálati vegyi anyagok átlagos EC50 értékét.
|
Antagonista vizsgálat
|
1. lépés
|
Számítsuk ki a DMSO VC átlagértékét.
|
|
2. lépés
|
Az adatok normalizálásához a DMSO VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
|
|
3. lépés
|
Számítsuk ki a referenciaszabvány (Ral/E2) átlagos redukciós faktorát.
|
|
4. lépés
|
Számítsuk ki az E2 referenciaszabvány átlagértékét.
|
|
5. lépés
|
Számítsuk ki a vizsgálati vegyi anyagok átlagos IC50 értékét.
|
A teljes körű vizsgálat során a luminométerrel mért adatok begyűjtése és kiigazítása
|
38.
|
A luminométer által szolgáltatott nyers adatokat át kell vezetni a vizsgálat céljára létrehozott táblázatsablonba. Meg kell határozni, hogy vannak-e a vizsgálatból kizárandó kiugró adatok. (Lásd az elemzés során meghatározott paraméterekre vonatkozó elfogadhatósági kritériumokat.) Az alábbi számításokat végzik el:
|
Agonista vizsgálat
|
1. lépés
|
Számítsuk ki a DMSO VC átlagértékét.
|
|
2. lépés
|
Az adatok normalizálásához a DMSO VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
|
|
3. lépés
|
Számítsuk ki a referenciaszabvány (E2) átlagos indukciós faktorát.
|
|
4. lépés
|
Számítsuk ki az E2 és a vizsgálati vegyi anyagok átlagos EC50 értékét.
|
|
5. lépés
|
Számítsuk ki a metoxiklór átlagos kiigazított RLU-értékét.
|
Antagonista vizsgálat
|
1. lépés
|
Számítsuk ki a DMSO VC átlagértékét.
|
|
2. lépés
|
Az adatok normalizálásához a DMSO VC átlagértékét vonjuk ki az egyes lyukaknál kapott értékből.
|
|
3. lépés
|
Számítsuk ki a referenciaszabvány (Ral/E2) átlagos indukciós faktorát.
|
|
4. lépés
|
Számítsuk ki a Ral/E2 és a vizsgálati vegyi anyagok átlagos IC50 értékét.
|
|
5. lépés
|
Számítsuk ki a tamoxifén átlagos kiigazított RLU-értékét.
|
|
6. lépés
|
Számítsuk ki az E2 referenciaszabvány átlagértékét.
|
Az adatok értelmezésének kritériumai
|
39.
|
A VM7Luc ösztrogénreceptor transzaktivációs vizsgálat célja, hogy egy bizonyítékok súlyozásán (WoE) alapuló megközelítés részeként segítsen rangsorolni az anyagokat az endokrin zavarokat mérő in vivo vizsgálatokhoz. E rangsorolási folyamat egyik lépése az, amikor a vizsgálati vegyi anyagokat ösztrogénreceptor agonista vagy ösztrogénreceptor antagonista aktivitás szempontjából pozitívként vagy negatívként azonosítják. Az 1. táblázat ismerteti azokat a döntési kritériumokat, amelyek alapján a VM7Luc ösztrogénreceptor transzaktivációs vizsgálat validálási tanulmánya során pozitívnak vagy negatívnak minősítették az anyagokat.
|
1. táblázat
Pozitív és negatív döntési kritériumok
|
|
|
AGONISTA AKTIVITÁS
|
|
Pozitív
|
|
—
|
Az ösztrogénreceptor agonista aktivitás szempontjából pozitívnak minősített minden vizsgálati vegyi anyag koncentráció-válasz görbéjén lennie kell egy alapvonalnak, amelyet egy pozitív meredekség követ, amely egy platóban vagy csúcspontban zárul. Bizonyos esetekben e három jellemző közül csak kettő határozható meg (alapvonal-meredekség vagy meredekség-csúcspont).
|
|
—
|
A pozitív meredekséget meghatározó vonalnak legalább három olyan pontot kell tartalmaznia, amelyeknek a szórása nem fed át (átlag ± SD). Az alapvonalat alkotó pontok nem számítanak, de a görbe lineáris szakasza tartalmazhatja a csúcspontot vagy a plató első pontját.
|
|
—
|
A pozitív besoroláshoz szükséges továbbá, hogy a válasz amplitúdója (vagyis az alapvonal és a csúcspont közötti különbség) elérje legalább az E2 referenciaszabvány maximális értékének 20 %-át (vagyis legalább 2 000 RLU-t, amikor az [E2] referenciaszabvány kiigazított maximális válaszértéke 10 000 RLU).
|
|
—
|
Amennyiben megoldható, meg kell határozni minden pozitív vizsgálati vegyi anyag EC50 értékét.
|
|
|
Negatív
|
Egy adott koncentráció átlagos kiigazított RLU-értéke megegyezik azzal az értékkel, vagy alatta marad annak az értéknek, amely a DMSO-kontroll átlagos RLU-értéke plusz a DMSO RLU-értékétől való standard deviáció háromszorosa.
|
|
Nem megfelelő
|
Azok az adatok nem tekinthetők hitelesnek, amelyek jelentős kvalitatív vagy kvantitatív korlátok miatt az aktivitás meglétét vagy hiányát mutatják, és ezért nem megfelelőnek kell minősíteni azokat, és nem használhatók fel a vizsgálati vegyi anyag pozitívként vagy negatívként való besorolásához. Ilyenkor a vegyi anyagokat újbóli vizsgálatnak kell alávetni.
|
|
|
|
ANTAGONISTA AKTIVITÁS
|
|
Pozitív
|
|
—
|
A vizsgálati anyag adatai olyan koncentráció-válasz görbét rajzolnak ki, amelyik egy platóból induló negatív meredekség követ.
|
|
—
|
A negatív meredekséget meghatározó vonal legalább három pontot tartalmaz egymást nem fedő hibasávokkal; az alapvonalat alkotó pontok nem számítanak, de a görbe lineáris szakasza tartalmazhatja a plató első pontját.
|
|
—
|
A Ral/E2 referenciaszabvány maximális értékéhez képest legalább 20 %-ot kell csökkennie az aktivitásnak (vagyis legfeljebb 8 000 RLU lehet, amikor a [Ral/E2] referenciaszabvány kiigazított maximális válaszértéke 10 000 RLU).
|
|
—
|
A vizsgálati vegyi anyag legmagasabb nem toxikus koncentrációja legfeljebb 1 x 10-5 M lehet.
|
|
—
|
Amennyiben megoldható, meg kell határozni minden pozitív vizsgálati vegyi anyag IC50 értékét.
|
|
|
Negatív
|
Minden 1,0 x 10-5 M-nél kisebb koncentrációnak megfelelő adatpont az EC80 érték felett helyezkedik el (ami megegyezik az E2 válasz 80 %-ával vagy 8 000 RLU-val).
|
|
Nem megfelelő
|
Azok az adatok nem tekinthetők hitelesnek, amelyek jelentős kvalitatív vagy kvantitatív korlátok miatt az aktivitás meglétét vagy hiányát mutatják, és ezért nem megfelelőnek kell minősíteni azokat, és nem használhatók fel a vizsgálati vegyi anyag pozitívként vagy negatívként való besorolásához. Ilyenkor a vegyi anyagot újbóli vizsgálatnak kell alávetni.
|
|
40.
|
A pozitív eredményeket – ha lehetséges – jellemezni kell mind a kiváltott hatás mértékével, mind a hatást előidéző koncentrációval. A pozitív, negatív és nem megfelelő adatokra az 5. és a 6. ábra szolgáltat példákat.
|
5. ábra
Agonista példák: pozitív, negatív és nem megfelelő adatok

A szaggatott vonal az E2 válasz 20 %-át jelzi, ami 2 000 kiigazított és normalizált RLU-nak felel meg.
6. ábra
Antagonista példák: pozitív, negatív és nem megfelelő adatok

A szaggatott vonal a Ral/E2 válasz 80 %-át jelzi, ami 8 000 kiigazított és normalizált RLU-nak felel meg.
Az egybefüggő vonal az 1,00 x 10-5 M koncentrációt jelzi. Ahhoz, hogy egy választ pozitívnak lehessen tekinteni, alatta kell maradnia a 8 000 RLU-t jelző vonalon, és a koncentrációja nem haladhatja meg az 1,00 x 10-5 M értéket.
A meso-hexestrol grafikonon csillaggal jelzett koncentrációk „2” vagy annál nagyobb életképességi pontszámot jeleznek.
A meso-hexestrol vizsgálat eredményei nem megfelelőnek tekintendők, mert az egyetlen 8 000 RLU alatti válasz 1,00 x 10-5 M koncentrációnál fordul elő.
|
41.
|
Az EC50 és az IC50 érték kiszámítható a négy paraméteres Hill-féle funkcióanalízis alkalmazásával (részleteket lásd az agonista és az antagonista protokollban (7)). Az elfogadhatósági kritériumok teljesítése jelzi a rendszer megfelelő működését, de nem garantálja az egyes vizsgálatmenetek adatainak pontosságát. Az adatok pontosságát az biztosítja a legjobban, ha az első vizsgálatmenet eredményei megismételhetők (lásd „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” című 19. pontot).
|
VIZSGÁLATI JELENTÉS
|
42.
|
Lásd „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” című 20. pontot.
|
SZAKIRODALOM
|
(1)
|
ICCVAM. (2011). ICCVAM Test Method Evaluation Report on the LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In vitro Method for Identifying ER Agonists and Antagonists, National Institute of Environmental Health Sciences: Research Triangle Park, NC.
|
|
(2)
|
Monje P., Boland R. (2001). Subcellular Distribution of Native Estrogen Receptor α and β Isoforms in Rabbit Uterus and Ovary, J. Cell Biochem., 82(3): 467-479.
|
|
(3)
|
Pujol P., et al. (1998). Differential Expression of Estrogen Receptor-Alpha and -Beta Messenger RNAs as a Potential Marker of Ovarian Carcinogenesis, Cancer Res., 58(23): 5 367-5 373.
|
|
(4)
|
Weihua Z., et al. (2000). Estrogen Receptor (ER) β, a Modulator of ERα in the Uterus, Proceedings of the National Academy of Sciences of the United States of America 97(11): 936-5 941.
|
|
(5)
|
Balls M., et al. (2006). The Importance of Good Cell Culture Practice (GCCP), ALTEX, 23(Suppl): p. 270-273.
|
|
(6)
|
Coecke S., et al. (2005). Guidance on Good Cell Culture Practice: a Report of the Second ECVAM Task Force on Good Cell Culture Practice, Alternatives to Laboratory Animals, 33: p. 261-287.
|
|
(7)
|
ICCVAM (2011). ICCVAM Test Method Evaluation Report, The LUMI-CELL® ER (BG1Luc ER TA) Test Method: An In vitro Assay for Identifying Human Estrogen Receptor Agonist and Antagonist Activity of Chemicals, NIH Publication No 11-7 850.
|
|
(8)
|
Rogers J.M., Denison M.S. (2000). Recombinant Cell Bioassays for Endocrine Disruptors: Development of a Stably Transfected Human Ovarian Cell Line for the Detection of Estrogenic and Anti-Estrogenic Chemicals, In vitro Mol. Toxicol., 13(1):67-82.
|
|
(9)
|
Escande A., et al. (2006). Evaluation of Ligand Selectivity Using Reporter Cell Lines Stably Expressing Estrogen Receptor Alpha or Beta, Biochem. Pharmacol., 71(10):1 459-69.
|
|
(10)
|
Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology,17(6):646-57.
|
|
(11)
|
Kuiper G.G, et al. (1998). Interaction of Estrogenic Chemicals and Phytoestrogens with Estrogen Receptor Beta, Endocrinology, 139(10):4 252-63.
|
|
(12)
|
Geisinger, et al. (1989). Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors, Cancer 63, 280-288.
|
|
(13)
|
Baldwin, et al. (1998). BG-1 ovarian cell line: an alternative model for examining estrogen-dependent growth in vitro, In vitro Cell. Dev. Biol. – Animal, 34, 649-654.
|
|
(14)
|
Li, Y., et al. (2014). Research resource: STR DNA profile and gene expression comparisons of human BG-1 cells and a BG-1/MCF-7 clonal variant, Mol. Endo. 28, 2072-2081.
|
|
(15)
|
Rogers, J.M. and Denison, M.S. (2000). Recombinant cell bioassays for endocrine disruptors:development of a stably transfected human ovarian cell line for the detection of estrogenicand anti-estrogenic chemicals, In vitro & Molec. Toxicol. 13, 67-82.
|
4. Függelék
A VEGYI ANYAGOK ÖSZTROGÉNHATÁSÚ AGONISTA ÉS ANTAGONISTA AKTIVITÁSÁNAK ERΑ CALUX SEJTVONALLAL TÖRTÉNŐ ÉSZLELÉSÉRE SZOLGÁLÓ, STABILAN TRANSZFEKTÁLT HUMÁN ÖSZTROGÉNRECEPTOR ALFA TRANSZAKTIVÁCIÓS VIZSGÁLAT
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
1.
|
Az ösztrogénreceptor α CALUX transzaktivációs vizsgálat humán U2OS sejtvonalat használ a humán ösztrogénreceptor alfa (hERα) által közvetített ösztrogénhatású agonista és antagonista aktivitás észlelésére. A stabil transzfekciós ERα CALUX biológiai vizsgálat validálási tanulmányát a BioDetection Systems BV (Amszterdam, Hollandia) végezte, amely tanulmány bizonyította, hogy a vizsgálat tervezett felhasználása tekintetében releváns és megbízható (1). Az ERα CALUX sejtvonal kizárólag stabilan transzfektált humán ERα-t fejez ki (2) (3).
|
|
2.
|
A vizsgálat kifejezetten azt a célt szolgálja, hogy a biolumineszcencia mint végpont mérése révén észlelje a hERα által közvetített transzaktivációt. A biolumineszcenciát általánosan alkalmazzák a biológiai vizsgálatok során, a magas jel/háttér arány miatt (4).
|
|
3.
|
Az 1 μM-nél magasabb fitoösztrogén-koncentrációnál a luciferáz riportergén túlzott aktiválásáról számoltak be, ami nem receptorok által közvetített lumineszcenciához vezet (5) (6) (7). Ezért az ennél magasabb fitoösztrogén-koncentrációértékeket vagy a luciferáz-kifejeződést túlzottan aktiváló más hasonló vegyületeket körültekintően kell vizsgálni a stabilan transzfektált ösztrogénreceptor transzaktivációs vizsgálatok során (lásd a 2. függeléket).
|
|
4.
|
E vizsgálat szabályozási célra való alkalmazása előtt el kell olvasni az „ÁLTALÁNOS BEVEZETÉS” és „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részt. A vizsgálati módszerben használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLAT ELVE (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
5.
|
Ez a biológiai vizsgálat arra szolgál, hogy értékelje az ösztrogénreceptor ligandumhoz kötődését, és a receptor-ligandum komplex ezt követő transzlokációját a sejtmagba. A sejtmagban a receptor-ligandum komplex specifikus DNS válaszelemekhez kötődve transzaktivál egy szentjánosbogár luciferáz riportergént, ami megnöveli a luciferáz enzim celluláris kifejeződését. A luciferázhoz luciferin szubsztrátumot adagolnak, ezt követően a luciferin biolumineszcens termékké alakul. A felszabaduló fény könnyen észlelhető, mennyisége luminométer segítségével meghatározható.
|
|
6.
|
A vizsgálati rendszer stabilan transzfektált ERα CALUX sejteket alkalmaz. Az ERα CALUX sejtek a humán oszteoblasztás oszteoszarkóma U2OS sejtvonalból származnak. A humán U2OS sejteket a kalcium-foszfát transzfekciós módszerrel stabilan transzfektálták a 3xHRE-TATA-Luc és pSG5-neo-hERα konstrukciókkal. Az U2OS sejtvonal bizonyult a legjobb jelöltnek arra, hogy ösztrogén-reszponzív (és egyéb szteroidhormon-reszponzív) riporter sejtvonalként szolgáljon, ami azon a megfigyelésen alapul, hogy az U2OS sejtvonal csekély szintű endogén riporter aktivitást mutatott vagy egyáltalán nem mutatott endogén riporter aktivitást. Az endogén receptorok hiányát csak luciferáz riporter plazmidokkal mérték, amelyek a riporter ligandumok hozzáadását követően nem mutattak aktivitást. Emellett ez a sejtvonal erős hormon közvetítésű válaszokat adott hasonló receptorok tranziens transzfekciója esetén (2) (3) (8).
|
|
7.
|
Amikor az ERα CALUX sejtvonallal vizsgálják a vegyi anyagok ösztrogénhatású vagy antiösztrogén-hatású aktivitását, szükség van elővizsgálatra és teljes körű vizsgálatmenetekre. Az elővizsgálat során állapítják meg a vizsgálati vegyi anyagok oldhatóságát, citotoxicitását és pontosított koncentrációtartományát a teljes körű vizsgálatokhoz. A teljes körű vizsgálatmenetekben az ERα CALUX biológiai vizsgálat során tesztelik a vizsgálati vegyi anyagok pontosított koncentrációtartományát, majd agonista vagy antagonista aktivitás szempontjából elvégzik besorolásukat.
|
|
8.
|
Az adatok értelmezésének kritériumait részletesen a 59. pont ismerteti. Röviden: a vizsgálati vegyi anyag akkor tekinthető pozitív agonistának, ha az anyag legalább két egymást követő koncentrációértéke olyan választ ad, amely megegyezik a referenciaszabványként használt 17β-ösztradiol maximális válaszának 10 %-ával (PC10), illetve meghaladja azt. A vizsgálati vegyi anyag akkor minősíthető pozitív antagonistának, ha az anyag legalább két egymást követő koncentrációértéke olyan választ ad, amely megegyezik a referenciaszabványként használt tamoxifén maximális válaszának 80 %-ával (PC80), illetve alacsonyabb annál.
|
ELJÁRÁS
Sejtvonalak
|
9.
|
A vizsgálathoz stabilan transzfektált U2OS ERα CALUX sejtvonalat kell használni. A sejtvonal szakmai licencszerződés birtokában szerezhető be a BioDetection Systems BV vállalattól (Amszterdam, Hollandia).
|
|
10.
|
Csak mikoplazmamentes sejtkultúrák használhatók. A felhasznált sejttételeknek vagy igazoltan mikoplazmafertőzés-mentesnek kell lenniük, vagy használat előtt el kell végezni rajtuk egy mikoplazma vizsgálatot. A mikoplazma-fertőzés detektálására az RT-PCR (valós idejű polimeráz láncreakció) alkalmazandó (9).
|
A sejtvonal stabilitása
|
11.
|
A CALUX sejtek stabilitásának és integritásának megőrzése érdekében a sejteket folyékony nitrogénben (-80 0C-on) kell tárolni. Miután felolvasztották a sejteket, hogy új tenyészetet hozzanak létre, a sejteket legalább kétszer át kell ültetni, mielőtt felhasználnák azokat vegyi anyagok ösztrogénhatású agonista és antagonista aktivitásának értékelésére. A sejteket legfeljebb 30 alkalommal szabad passzálni.
|
|
12.
|
Annak nyomon követése érdekében, hogy a sejtvonal az idő múlásával is megőrzi stabilitását, a referenciaszabvány EC50 vagy IC50 értékének mérésével ellenőrizni kell az agonista és antagonista vizsgálati rendszer válaszadási képességét. Emellett ellenőrizni kell a pozitív kontrollminta (PC) és a negatív kontrollminta (NC) relatív indukciós képességét. Az eredményeknek meg kell felelniük az ERα CALUX biológiai vizsgálat agonista (3C táblázat) vagy antagonista (4C táblázat) tesztjére vonatkozó elfogadhatósági kritériumoknak. Az agonista, illetve az antagonista vizsgálat referenciaszabványát, pozitív és negatív kontrollanyagát az 1. táblázat, illetve a 2. táblázat ismerteti.
|
Sejttenyésztés és a leültetés körülményei
|
13.
|
Az U2OS sejteket pH-indikátor fenolvöröst tartalmazó, továbbá magzati borjú szérummal (7,5 %), nem esszenciális aminosavakkal (1 %), 10 egység/ml penicillinnel és sztreptomicinnel, valamint szelekciós markerként szolgáló geneticinnel (G-418) kiegészített tápoldatban (DMEM/F12 (1: 1)) kell tenyészteni. A sejteket 5 %-os CO2-tartalmú inkubátorba kell helyezni, 37° C-os hőmérséklet és 100 %-os páratartalom biztosítása mellett. 85–95 %-os konfluencia elérését követően a sejteket vagy tovább kell oltani, vagy elő kell készíteni a 96 lyukú mikrotiterlemezekbe történő leoltáshoz. Ez utóbbi esetben a sejteket egy ösztrogénmentes DMEM/F12 (1: 1) tápoldatban 1 x 105 sejt/ml sűrűségben felszuszpendálják és a homogenizált sejtszuszpenziót 100 μl-ként kiszélesztik a 96 lyukú mikrotiterlemezek lyukaiba, a tápoldat itt fenolvörösmentes, tartalmaz 5 térfogatszázalék dextránbevonatú aktív szénnel kezelt magzati borjú szérumot, 1 térfogatszázalék nem esszenciális aminosavakat és 10 egység/ml penicillin és sztreptomicint). A kezelést megelőzően a sejteket CO2 inkubátorban (5 % CO2, 37 0C, 100 % páratartalom) 24 órán keresztül preinkubálni kell. A műanyag edények legyenek ösztrogénmentesek.
|
Elfogadhatósági kritériumok
|
14.
|
A vizsgálati vegyi anyag(ok) agonista és antagonista aktivitását egy vizsgálatsorozaton keresztül mérik fel. Egy vizsgálatsorozat legfeljebb 6 mikrotiterlemezből áll. Minden vizsgálatsorozat magában foglalja a referenciaszabvány egy teljes oldatsorozatát, egy pozitív kontrollmintát, egy negatív kontrollmintát és az oldószeres kontrollokat. Az 1. és a 2. ábra ismerteti az agonista és az antagonista vizsgálatsorozatokhoz használt lemez elrendezését.
|
|
15.
|
A referenciaszabványok, a vizsgálati vegyi anyagok, az oldószeres kontrollanyagok, valamint a pozitív és negatív kontrollanyagok minden egyes oldatát triplikátumban kell vizsgálni. Mindhárom ismétlés eredményeinek meg kell felelniük a 3A és 4A táblázatban szereplő követelményeknek.
|
|
16.
|
Minden vizsgálatsorozat első lemezén megmérik a referenciaszabvány (agonista vizsgálat esetén a 17β-ösztradiol; antagonista vizsgálat esetén a tamoxifén) teljes oldatsorozatát. Annak érdekében, hogy a másik 5 mikrotiterlemez elemzésének eredményeit össze lehessen vetni a referenciaszabvány teljes koncentráció-válasz görbéjét tartalmazó első mikrotiterlemezzel, minden lemeznek 3 kontrollmintát kell tartalmaznia: az oldószeres kontrollt, a vizsgált referenciaszabvány legmagasabb koncentrációját és a referenciaszabvány körülbelüli EC50 (agonista) vagy IC50 (antagonista) koncentrációját. Az első lemez és a másik 5 lemez kontrollmintái átlagának aránya meg kell feleljen a 3C táblázatban (agonista vizsgálat) vagy a 4C táblázatban (antagonista vizsgálat) feltüntetett követelményeknek.
|
|
17.
|
Meg kell határozni a vizsgálatsorozatán belüli egyes mikrotiterlemezek z-tényezőjét (10). A z-tényezőt a referenciaszabvány legmagasabb és legalacsonyabb koncentrációjánál kapott válasz alapján kell kiszámítani. A mikrotiterlemezek eredményei akkor tekinthetők hitelesnek, ha eleget tesznek a 3C táblázatban (agonista vizsgálat) vagy a 4C táblázatban (antagonista vizsgálat) feltüntetett követelményeknek.
|
|
18.
|
A referenciaszabványnak szigmoid formájú dózis-válasz görbét kell eredményeznie. A referenciaszabvány oldatsorozatának válaszai alapján meghatározott EC50 vagy IC50 értéknek meg kell felelnie a 3C táblázatban (agonista vizsgálat) vagy a 4C táblázatban (antagonista vizsgálat) szereplő követelményeknek.
|
|
19.
|
Minden vizsgálatsorozatnak tartalmaznia kell egy pozitív és egy negatív kontrollmintát. Mind a pozitív, mind a negatív kontrollminta kalkulált relatív indukciós hatásának eleget kell tennie a 3C táblázatban (agonista vizsgálat) vagy a 4C táblázatban (antagonista vizsgálat) feltüntetett követelményeknek.
|
|
20.
|
A referenciaszabvány legmagasabb koncentrációjának indukciós faktorát minden mérés során úgy határozzák meg, hogy elosztják a 17β-ösztradiol referenciaszabvány átlagos legmagasabb relatív fényegység (RLU) válaszát a referencia oldószerrel végzett kontroll átlagos RLU-válaszával. Az indukciós faktornak eleget kell tennie a 3C táblázatban (agonista vizsgálat) vagy a 4C táblázatban (antagonista vizsgálat) feltüntetett, indukciós faktorra vonatkozó követelményeknek.
|
|
21.
|
Kizárólag a fenti elfogadhatósági kritériumoknak megfelelő mikrotiterlemezek tekinthetők hitelesnek, és használhatók fel a vizsgálati vegyi anyagok által adott válasz értékelésére.
|
|
22.
|
Az elfogadhatósági kritériumok egyaránt vonatkoznak az elővizsgálatra és a teljes körű vizsgálatmenetekre.
|
1. táblázat
Az agonista CALUX biológiai vizsgálathoz használt referenciaszabvány, pozitív kontrollanyag (PC) és negatív kontrollanyag (NC) koncentrációi
|
|
Anyag
|
CAS-szám
|
Vizsgálati tartomány (M)
|
|
Referenciaszabvány
|
17β-ösztradiol
|
50-28-2
|
1*10-13–1*10-10
|
|
Pozitív kontroll (PC)
|
17α-metiltesztoszteron
|
58-18-4
|
3*10-6
|
|
Negatív kontroll (NC)
|
kortikoszteron
|
50-22-6
|
1*10-8
|
2. táblázat
Az antagonista CALUX biológiai vizsgálathoz használt referenciaszabvány, pozitív kontrollanyag (PC) és negatív kontrollanyag (NC) koncentrációi
|
|
Anyag
|
CAS-szám
|
Vizsgálati tartomány (M)
|
|
Referenciaszabvány
|
tamoxifén
|
10540-29-1
|
3*10-9–1*10-5
|
|
Pozitív kontroll (PC)
|
4-hidroxitamoxifén
|
68047-06-3
|
1*10-9
|
|
Negatív kontroll (NC)
|
rezveratrol
|
501-36-0
|
1*10-5
|
3. táblázat
Az agonista ERα CALUX biológiai vizsgálat elfogadhatósági kritériumai
|
A – egy lemezen lévő egyes minták
|
Kritérium
|
|
1
|
A három ismétlésben vizsgált lyukak maximális SD-ja %-ban (a NC, a PC, a vizsgálati vegyi anyag és a referenciaszabvány minden oldata tekintetében, kivéve C0)
|
< 15 %
|
|
2
|
A három ismétlésben vizsgált lyukak maximális SD-ja %-ban (a referenciaszabvány és a vizsgálati vegyi anyag oldószeres kontrolljai tekintetében (C0, SC))
|
< 30 %
|
|
3
|
Az LDH maximális csökkenése, ami a citotoxicitás mérőszáma.
|
< 120 %
|
|
B – egy mikrotiterlemezen belül
|
|
|
4
|
A referenciaszabvány oldószeres kontrolljának (C0; 1. lemez) és a vizsgálati vegyi anyag oldószeres kontrolljának (SC; 2–x. lemez) aránya
|
0,5–2,0
|
|
5
|
A kb. EC50 és a referenciaszabvány legmagasabb koncentrációjának aránya az 1. lemezen, valamint a kb. EC50 és a referenciaszabvány legmagasabb koncentrációjának aránya a 2–x. lemezen (C4, C8)
|
0,70–1,30
|
|
6
|
Az egyes lemezek z-tényezője
|
> 0,6
|
|
C – egy analízissorozaton belül (egy sorozat összes lemeze)
|
|
|
7
|
A referenciaszabvány görbéje szigmoid formájú
|
Igen (17ß-ösztradiol)
|
|
8
|
A 17ß-ösztradiol referenciaszabvány EC50 tartománya
|
4*10-12–4*10-11 M
|
|
9
|
A legmagasabb 17β-ösztradiol koncentráció minimális indukciós faktora, a referenciaszabvány oldószeres kontrolljához képest.
|
5
|
|
10
|
PC relatív indukciója (%)
|
> 30 %
|
|
11
|
NC relatív indukciója (%)
|
< 10 %
|
Kb.: körülbelüli; PC: pozitív kontroll; NC: negatív kontroll; SC: a vizsgálati vegyi anyag oldószeres kontrollja; C0: a referenciaszabvány oldószeres kontrollja; SD: standard deviáció; LDH: laktát-dehidrogenáz.
4. táblázat
Az antagonista ERα CALUX biológiai vizsgálat elfogadhatósági kritériumai
|
A – egy lemezen lévő egyes minták
|
Kritérium
|
|
1
|
A három ismétlésben vizsgált lyukak maximális SD-ja %-ban (az NC, a PC, a vizsgálati vegyi anyag és a referenciaszabvány minden oldata, valamint az oldószeres kontroll (C0) tekintetében)
|
< 15 %
|
|
2
|
A három ismétlésben vizsgált lyukak maximális SD-ja %-ban (a vivőanyag kontroll (VC) és a referenciaszabvány legmagasabb koncentrációja (C8) tekintetében)
|
< 30 %
|
|
3
|
Az LDH maximális csökkenése, ami a citotoxicitás mérőszáma.
|
< 120 %
|
|
B – egy mikrotiterlemezen belül
|
|
|
4
|
A referenciaszabvány oldószeres kontrolljának (C0; 1. lemez) és a vizsgálati vegyi anyag oldószeres kontrolljának (SC; 2–x. lemez) aránya
|
0,70–1,30
|
|
5
|
Az 1. lemez referenciaszabvány-koncentrációinak kb. IC50 értékének és a 2–x. lemez referenciaszabvány-koncentrációinak kb. IC50 értékének (C4) aránya
|
0,70–1,30
|
|
6
|
Az 1. lemez legmagasabb referenciaszabvány-koncentrációinak és a 2–x. lemez legmagasabb referenciaszabvány-koncentrációinak (C8) aránya
|
0,50–2,0
|
|
7
|
Az egyes lemezek z-tényezője
|
> 0,6
|
|
C – egy analízissorozaton belül (egy sorozat összes lemeze)
|
|
|
8
|
A referenciaszabvány görbéje szigmoid formájú
|
Igen (tamoxifén)
|
|
9
|
A referenciaszabvány (tamoxifén) IC50 tartománya
|
1*10-8–1*10-7 M
|
|
10
|
A referenciaszabvány oldószeres kontrolljának minimális indukciós faktora, a tamoxifén legmagasabb koncentrációjához képest.
|
2,5
|
|
11
|
PC relatív indukciója (%)
|
< 70 %
|
|
12
|
NC relatív indukciója (%)
|
> 85 %
|
Kb.: körülbelüli; PC: pozitív kontroll; NC: negatív kontroll; VC: vivőanyagos kontroll (az agonista referenciaszabvány fix koncentrációja nélküli oldószeres kontroll); SC: a vizsgálati vegyi anyag oldószeres kontrollja; C0: a referenciaszabvány oldószeres kontrollja; SD: standard deviáció; LDH: laktát-dehidrogenáz.
Oldószeres/vivőanyagos kontroll, referenciaszabványok, pozitív kontrollok, negatív kontrollok
|
23.
|
Mind az elővizsgálatban, mind a teljes körű vizsgálatmenetekben ugyanazt a kontrollként szolgáló oldószert/vivőanyagot, referenciaszabványokat, pozitív és negatív kontrollanyagokat kell használni. Emellett változatlanul kell hagyni a referenciaszabványok, pozitív és negatív kontrollanyagok koncentrációértékeit is.
|
Oldószeres kontroll
|
24.
|
A vizsgálati vegyi anyagok feloldására használt oldószert oldószeres kontroll keretében vizsgálni kell. Az ERα CALUX biológiai vizsgálat validálása során vivőanyagként dimetil-szulfoxidot alkalmaztak (DMSO, 1 térfogatszázalékban, CAS-száma: 67-68-5). DMSO-tól eltérő oldószer használata esetén valamennyi referenciaszabványt, kontrollanyagot és vizsgálati vegyi anyagot ugyanabban a vivőanyagban kell tesztelni. Az antagonista tanulmányok oldószeres kontrollja fix koncentrációban (körülbelül EC50 koncentrációban) tartalmazza az agonista referenciaszabványt, a 17β-ösztradiolt. Az antagonista tanulmányokhoz használt oldószer teszteléséhez vivőanyagos kontrollt kell készíteni és tesztelni.
|
Vivőanyagos kontroll (antagonista vizsgálat)
|
25.
|
Az antagonista hatás vizsgálatához a vizsgálati tápoldatot az agonista referenciaszabvány, a 17β-ösztradiol fix koncentrációjával (körülbelül EC50 koncentrációval) egészítik ki. Az antagonista hatásuk felmérése céljából vizsgált vizsgálati vegyi anyagok feloldásához alkalmazott oldószer teszteléséhez az agonista referenciaszabványként használt 17β-ösztradiol fix koncentrációjától mentes vizsgálati közeget kell készíteni. Ez a kontrollminta az úgynevezett vivőanyagos kontroll. Az ERα CALUX biológiai vizsgálat validálása során vivőanyagként dimetil-szulfoxidot alkalmaztak (DMSO, 1 térfogatszázalékban, CAS-száma: 67-68-5). DMSO-tól eltérő oldószer használata esetén valamennyi referenciaszabványt, kontrollanyagot és vizsgálati vegyi anyagot ugyanabban a vivőanyagban kell tesztelni.
|
Referenciaszabványok
|
26.
|
Az agonista referenciaszabvány a 17β-ösztradiol (1. táblázat). A referenciaszabványok a 17β-ösztradiol nyolc koncentrációjából álló oldatsorozatot foglalnak magunkban (1*10-13, 3*10-13, 1*10-12, 3*10-12, 6*10-12, 1*10-11, 3*10-11, 1*10-10 M).
|
|
27.
|
Az antagonista referenciaszabvány a tamoxifén (2. táblázat). A referenciaszabványok egy, a tamoxifén nyolc koncentrációjából álló oldatsorozatot foglalnak magunkban (3*10-9, 1*10-8, 3*10-8, 1*10-7, 3*10-7, 1*10-6, 3*10-6, 1*10-5 M). Az antagonista referenciaszabvány minden egyes koncentrációját együtt inkubálják az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációjával (3*10-12 M).
|
Pozitív kontroll
|
28.
|
Az agonista tanulmányok pozitív kontrollanyaga a 17α-metiltesztoszteron (1. táblázat).
|
|
29.
|
Az antagonista tanulmányok pozitív kontrollanyaga a 4-hidroxitamoxifén (2. táblázat). Az antagonista pozitív kontrollanyagot együtt inkubálják az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációjával (3*10-12 M).
|
Negatív kontroll
|
30.
|
Az agonista tanulmányok negatív kontrollanyaga a kortikoszteron (1. táblázat).
|
|
31.
|
Az antagonista tanulmányok negatív kontrollanyaga a rezveratrol (2. táblázat). Az antagonista negatív kontrollanyagot együtt inkubálják az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációjával (3*10-12 M).
|
A laboratórium jártasságának igazolása (lásd a vizsgálati módszer „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” részének 14. pontját, valamint 3. és 4. táblázatát).
Vivőanyag
|
32.
|
A vizsgálati vegyi anyagok feloldásához olyan oldószert kell használni, amelyik teljesen szolubilizálja a vizsgálati vegyi anyagot és elegyedik a sejtek tápoldatával. A megfelelő oldószerek közé tartozik a DMSO, a víz és az etanol (95–100 %-os tisztaságban). Amennyiben DMSO-t használnak oldószerként, az inkubálás alatt a DMSO maximális koncentrációja nem haladhatja meg az 1 térfogatszázalékot. Az oldószert alkalmazása előtt tesztelni kell, hogy nem okoz-e citotoxicitást, és nem zavarja-e a vizsgálatok teljesítményét.
|
A referenciaszabványok, a pozitív és negatív kontrollanyagok, valamint a vizsgálati vegyi anyagok előkészítése
|
33.
|
A referenciaszabványokat, a pozitív és negatív kontrollanyagokat, valamint a vizsgálati vegyi anyagokat 100 %-os DMSO-ban (vagy más megfelelő oldószerben) kell feloldani. A megfelelő oldatokat (oldatsorozatot) ugyanabban az oldószerben kell elkészíteni. Feloldás előtt meg kell várni, hogy minden anyag szobahőmérsékletű legyen. A referenciaszabványok, a pozitív és negatív kontrollanyagok, valamint a vizsgálati vegyi anyagok frissen készített törzsoldataiban nem lehet szemmel látható kicsapódás és az oldatok nem lehet zavarosak. A referenciaszabványok és kontrollanyagok nagy tételben is előkészíthetők. A vizsgálati vegyi anyagok törzsoldatait minden kísérlet előtt frissen kell elkészíteni. A referenciaszabványok, a pozitív és negatív kontrollanyagok, valamint a vizsgálati vegyi anyagok végső oldatait minden egyes kísérlethez frissen kell készíteni, és az elkészítéstől számított 24 órán belül fel kell használni.
|
Oldhatóság, citotoxicitás és dózisbehatárolás
|
34.
|
Az elővizsgálat során meghatározzák a vizsgálati vegyi anyagok választott oldószerben való oldhatóságát. Elkészítik a maximális, 0,1 M törzsoldat-koncentrációt. Amennyiben ennél a koncentrációnál oldhatósági problémák jelentkeznek, alacsonyabb koncentrációjú törzsoldatokat kell készíteni mindaddig, amíg a vizsgálati vegyi anyagok teljesen oldhatónak nem bizonyulnak. Az elővizsgálat során a vizsgálati vegyi anyagok 1: 10 arányú oldatsorozatát tesztelik. Az agonista és antagonista vizsgálat maximális vizsgált koncentrációja az 1 mM. Az elővizsgálat megfelelően pontosítja a vizsgálati vegyi anyagok koncentrációtartományát, a teljes körű vizsgálatmenetek során ezt a tartományt kell vizsgálni. A teljes körű vizsgálat során a következő hígítási arányokat kell alkalmazni: 1x-es, 3x-os, 10x-es, 30x-os, 100x-os, 300x-os, 1000x-es és 3000x-es.
|
|
35.
|
A citotoxicitás vizsgálata egyaránt része az agonista és az antagonista vizsgálati protokollnak (11). Mind az elővizsgálat, mind a teljes körű vizsgálatmenetek tartalmaznak citotoxicitási vizsgálatot. Az ERα CALUX biológiai vizsgálat validálása során a citotoxicitás felmérésére a laktát-dehidrogenáz (LDH) szivárgását vizsgáló tesztet használták, és emellett kvalitatív vizuális módszerrel (lásd a 4.1. függeléket) megfigyelték a vizsgálati vegyi anyagokkal kezelt sejteket. Ugyanakkor a citotoxicitás meghatározásához használhatók más kvantitatív módszerek is (pl. a tetrazólium alapú kolorimetriás (MTT) vizsgálat vagy a CALUX citotoxicitási biológiai vizsgálat). Általában véve a vizsgálati vegyi anyagok azon koncentrációi, amelyek a sejtéletképesség több mint 20 %-os visszaesését okozzák, citotoxikusnak tekintendők, így az adatok értékeléséhez nem használhatók. Az LDH szivárgását vizsgáló teszt esetében a vizsgálati vegyi anyag koncentrációja akkor minősül citotoxikusnak, ha az LDH-szivárgás aránya meghaladja a 120 %-ot.
|
Vegyi anyaggal történő kezelés és a vizsgálati lemez elrendezése
|
36.
|
A megfelelő sűrűséget elérő sejtkultúra sejtjeit tripszin hozzáadással elválasztják a sejttenyésztő edény aljától, majd a sejteket ösztrogénmentes vizsgálati tápoldatban 1 x 105 sejt/ml sűrűségre állítják be. Az újraszuszpendált sejteket 100 μl mennyiségben kiszélesztik a 96 lyukú mikrotiterlemez belső lyukaiba. A lemezen a szélső sorok lyukait 200 μl foszfátpuffert tartalmazó sóoldattal (PBS) töltik meg (lásd az 1. és 2. ábrát). A leültetett sejteket 24 órán keresztül CO2 inkubátorban preinkubálják (5 % CO2, 37 °C, 100 % páratartalom mellett).
|
|
37.
|
A preinkubációt követően a lemezeken megfigyelik a citotoxicitás (lásd a 4.1. függeléket), a fertőzés és a konfluencia szabad szemmel látható jeleit. A vizsgálathoz kizárólag olyan lemezek használhatók fel, amelyeken nem látható citotoxicitás és fertőzés, a konfluencia mértéke pedig eléri a 85 %-ot. A belső lyukakban lévő sejtekről óvatosan lecserélik a tápoldatot, és 200 μl ösztrogénmentes vizsgálati tápoldattal töltik fel, amely tartalmazza a referenciaszabványok, a vizsgálati vegyi anyagok, a pozitív és negatív kontrollanyagok és az oldószerek megfelelő oldatsorozatát (5. táblázat: agonista tanulmányok; 6. táblázat: antagonista tanulmányok). A referenciaszabványokat, a vizsgálati vegyi anyagokat, a pozitív és a negatív kontrollanyagokat, valamint az oldószereket három ismétlésben kell vizsgálni. Az 1. ábra ismerteti az agonista vizsgálati lemez elrendezését. A 2. ábra ismerteti az antagonista vizsgálati lemez elrendezését. Az elővizsgálathoz és a teljes körű vizsgálathoz alkalmazott lemezek elrendezése azonos. Az antagonista vizsgálat során minden belső lyuk – a vivőanyagot (VC) tartalmazó lyukakat kivéve – tartalmazza az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációját is (3*10-12 M). C8-as és C4-es koncentrációjú referenciaszabványnak szerepelnie kell minden vizsgálati vegyi anyagot tesztelő lemezen.
|
|
38.
|
Miután minden vegyi anyaggal kezelték a sejteket, a 96 lyukú mikrotiterlemezeket ismét 24 órán keresztül CO2 inkubátorban inkubálják (5 % CO2, 37 °C, 100 % páratartalom mellett).
|
1. ábra
A 96 lyukú mikrotiterlemezek elrendezése agonista hatás elővizsgálathoz és értékeléséhez

C0 = a referenciaszabvány oldószere.
C(1–8) = a referenciaszabvány oldatsorozata (1–8, az alacsonytól a magas koncentráció felé).
PC = pozitív kontroll.
NK = negatív kontroll.
TCx-(1–8) = vizsgálati vegyi anyag oldatai (1–8, az alacsonytól a magas koncentráció felé) az x. vizsgálati vegyi anyag elővizsgálatához és agonista hatásának felméréséhez.
SC = a vizsgálati vegyi anyag oldószeres kontrollja (optimális esetben azonos a C0-val, de lehetőleg másik tételből származzon).
Szürke cellák: = szélső lyukak, 200 μl PBS-sel feltöltve.
2. ábra
A 96 lyukú mikrotiterlemezek elrendezése antagonista hatás elővizsgálathoz és értékeléséhez

C0 = a referenciaszabvány oldószere.
C(1–8) = a referenciaszabvány oldatsorozata (1–8, az alacsonytól a magas koncentráció felé).
NK = negatív kontroll.
PC = pozitív kontroll.
TCx-(1–8) = vizsgálati vegyi anyag oldatai (1–8, az alacsonytól a magas koncentráció felé) az x. vizsgálati vegyi anyag elővizsgálatához és agonista hatásának felméréséhez.
SC = a vizsgálati vegyi anyag oldószeres kontrollja (optimális esetben azonos a C0-val, de lehetőleg másik tételből származzon).
VC = vivőanyagos kontroll (oldószeres kontroll, az agonista referenciaszabvány, a 17β-ösztradiol fix koncentrációja nélkül).
Szürke cellák: = szélső lyukak, 200 μl PBS-sel feltöltve.
Megjegyzés: minden belső lyuk – a vivőanyagot (VC) tartalmazó lyukakat kivéve – magában foglalja az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációját is (3,0*10-12 M).
A lumineszcencia mérése
|
39.
|
A lumineszcencia mérésének részletes leírása megtalálható az agonista és az antagonista vizsgálati protokollban (10). A lyukakban lévő tápoldatot el kell távolítani, majd a sejteket 24 órás inkubálást követően lizálni kell, hogy feltárják a sejtmembránt, és mérhetővé váljon a luciferáz-aktivitás.
|
|
40.
|
A lumineszcencia méréséhez ebben az eljárásban 2 injektorral felszerelt luminométert kell használni. A luciferáz reakció megkezdéséhez beinjektálják a luciferin szubsztrátot. A reakciót 0,2 M NaOH hozzáadásával állítják le. A reakciót azért kell leállítani, hogy ne terjedjen át a lumineszcencia az egyik lyukból a másikba.
|
|
41.
|
Az egyes lyukak által kibocsátott fényt lyukankénti relatív fényegységekben (RLU-ban) fejezik ki.
|
Elővizsgálat
|
42.
|
Az elővizsgálat elemzésének eredményei alapján határozzák meg a vizsgálati vegyi anyagok teljes körű vizsgálatban alkalmazandó pontosított koncentrációtartományát. Az elővizsgálat eredményeinek értékeléséről és a vizsgálati vegyi anyagok teljes körű vizsgálatban alkalmazandó pontosított koncentrációtartományáról az agonista és az antagonista vizsgálati protokoll nyújt részletes tájékoztatást (10). Az alábbiak csak röviden összefoglalják azt az eljárást, amelynek alapján meghatározható az agonista és az antagonista vizsgálatban használt vizsgálati vegyi anyagok koncentrációtartománya. Az oldatsorozat elemeinek meghatározásához az 5. és a 6. táblázat nyújt iránymutatást.
|
Az agonista hatások felmérésére szolgáló koncentrációértékek kiválasztása
|
43.
|
Az elővizsgálat során a vizsgálati vegyi anyagokat az 5. táblázatban (agonista vizsgálat) és a 6. táblázatban (antagonista vizsgálat) jelzett oldatsorozatban kell vizsgálni. Minden koncentrációértéket három-három lyukban kell vizsgálni, az 1. ábrán (agonista vizsgálat) és a 2. ábrán (antagonista vizsgálat) szereplő lemezelrendezésben.
|
|
44.
|
Kizárólag az analízis elfogadhatósági kritériumoknak (3. táblázat) megfelelő eredményei tekinthetők hitelesnek, és használhatók fel a vizsgálati vegyi anyagok válaszának értékelésére. Amennyiben az elemzéssorozat egy vagy több mikrotiterlemeze nem felel meg az elfogadhatósági kritériumoknak, az adott mikrotiterlemezeket újbóli elemzésnek kell alávetni. Ha a referenciaszabvány teljes sorozatát tartalmazó első lemez nem teljesíti az elfogadhatósági kritériumokat, ismételten el kell végezni az egész vizsgálatsorozat (mind a 6 lemez) elemzését.
|
|
45.
|
A vizsgálati vegyi anyagok kezdeti koncentrációtartományait ki kell igazítani és az elővizsgálatot meg kell ismételni, ha:
|
—
|
citotoxicitást figyeltek meg. Ilyenkor az elővizsgálatot a vizsgálati vegyi anyag alacsonyabb, nem citotoxikus koncentrációival kell megismételni;
|
|
—
|
a vizsgálati vegyi anyag elővizsgálata nem biztosított teljes dózis-válasz görbét, mert a vizsgált koncentrációértékek maximális indukciót idéztek elő. Ilyenkor az elővizsgálatot a vizsgálati vegyi anyag alacsonyabb koncentrációival kell megismételni.
|
|
|
46.
|
Érvényes dózisfüggő válasz esetén azt a (legalacsonyabb) koncentrációt kell kiválasztani, amely maximális indukciót hoz létre, ugyanakkor nem okoz citotoxicitást. A vizsgálati vegyi anyag teljes körű vizsgálatmenetekben tesztelendő legnagyobb koncentrációja e kiválasztott koncentráció háromszorosa legyen.
|
|
47.
|
A vizsgálati vegyi anyag pontosított teljes oldatsorozatát az 5. táblázatban szereplő hígítási lépések szerint kell elkészíteni, kiindulva a fenti eljárás alapján meghatározott legnagyobb koncentrációtól.
|
|
48.
|
Azokat a vizsgálati vegyi anyagokat, amelyek nem váltanak ki agonista hatást, az elővizsgálat során azonosított legnagyobb nem toxikus koncentrációtól kezdve kell vizsgálni a teljes körű vizsgálatban.
|
Az antagonista hatások felmérésére szolgáló koncentrációértékek kiválasztása
|
49.
|
Kizárólag az analízis elfogadhatósági kritériumoknak (4. táblázat) megfelelő eredményei tekinthetők hitelesnek, és használhatók fel a vizsgálati vegyi anyagok válaszának értékelésére. Amennyiben az elemzéssorozat egy vagy több mikrotiterlemeze nem felel meg az elfogadhatósági kritériumoknak, az adott mikrotiterlemezeket újbóli elemzésnek kell alávetni. Ha a referenciaszabvány teljes sorozatát tartalmazó első lemez nem teljesíti az elfogadhatósági kritériumokat, ismételten el kell végezni az egész vizsgálatsorozat (mind a 6 lemez) elemzését.
|
|
50.
|
A vizsgálati vegyi anyagok kezdeti koncentrációtartományait ki kell igazítani és az elővizsgálatot meg kell ismételni, ha:
|
—
|
citotoxicitást figyeltek meg. Ilyenkor az elővizsgálatot a vizsgálati vegyi anyag alacsonyabb, nem citotoxikus koncentrációival kell megismételni;
|
|
—
|
a vizsgálati vegyi anyag elővizsgálata nem biztosított teljes dózis-válasz görbét, mert a vizsgált koncentrációértékek maximális gátlást idéztek elő. Ilyenkor az elővizsgálatot a vizsgálati vegyi anyag alacsonyabb koncentrációival kell megismételni.
|
|
|
51.
|
Érvényes dózisfüggő válasz esetén azt a (legalacsonyabb) koncentrációt kell kiválasztani, amely maximális gátlást hoz létre, ugyanakkor nem okoz citotoxicitást. A vizsgálati vegyi anyag teljes körű vizsgálatmenetekben tesztelendő legnagyobb koncentrációja e kiválasztott koncentráció háromszorosa legyen.
|
|
52.
|
A vizsgálati vegyi anyag pontosított teljes oldatsorozatát a 6. táblázatban szereplő hígítási lépések szerint kell elkészíteni, kiindulva a fenti eljárás alapján meghatározott legnagyobb koncentrációtól.
|
|
53.
|
Azokat a vizsgálati vegyi anyagokat, amelyek nem váltanak ki antagonista hatást, az elővizsgálat során tesztelt legnagyobb nem toxikus koncentrációtól kezdve kell vizsgálni a teljes körű vizsgálatban.
|
Teljes körű vizsgálatmenetek
|
54.
|
A pontosított koncentrációtartományok meghatározását követően a teljes körű vizsgálatmenetek során a vizsgálati vegyi anyagokat az 5. táblázatban (agonista vizsgálat) és a 6. táblázatban (antagonista vizsgálat) jelzett oldatsorozatban kell vizsgálni. Minden koncentrációértéket három-három lyukban kell vizsgálni, az 1. ábrán (agonista vizsgálat) és a 2. ábrán (antagonista vizsgálat) szereplő lemezelrendezésben.
|
|
55.
|
Kizárólag az analízis elfogadhatósági kritériumoknak (3. és 4. táblázat) megfelelő eredményei tekinthetők hitelesnek, és használhatók fel a vizsgálati vegyi anyagok válaszának értékelésére. Amennyiben az elemzéssorozat egy vagy több mikrotiterlemeze nem felel meg az elfogadhatósági kritériumoknak, az adott mikrotiterlemezeket újbóli elemzésnek kell alávetni. Ha a referenciaszabvány teljes sorozatát tartalmazó első lemez nem teljesíti az elfogadhatósági kritériumokat, ismételten el kell végezni az egész vizsgálatsorozat (mind a 6 lemez) elemzését.
|
5. táblázat
Az agonista vizsgálathoz használt referenciaanyagok, kontrollanyagok és vizsgálati vegyi anyagok koncentrációi és oldatai
|
Referencia 17β-ösztradiol
|
TCx – elővizsgálat
|
TCx – teljes körű vizsgálatmenet
|
Kontrollok
|
|
konc. (M)
|
oldatai
|
oldatai
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3000 x
|
PC
|
3*10-6
|
|
C1
|
1*10-13
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1000 x
|
NK
|
1*10-8
|
|
C2
|
3*10-13
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
1*10-12
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
3*10-12
|
TCx-5
|
1000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
6*10-12
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
|
|
|
C6
|
1*10-11
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
|
|
|
C7
|
3*10-11
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
|
|
|
C8
|
1*10-10
|
|
|
|
|
|
|
|
TCx –x. vizsgálati vegyi anyag
PC –pozitív kontroll (17α-metiltesztoszteron)
NK –negatív kontroll (kortikoszteron)
C0 –a referenciaszabvány oldószeres kontrollja
SC –a vizsgálati vegyi anyag oldószeres kontrollja
|
6. táblázat
Az antagonista vizsgálathoz használt referenciaanyagok, kontrollanyagok és vizsgálati vegyi anyagok koncentrációi és oldatai
|
Referencia tamoxifén
|
TCx – elővizsgálat
|
TCx – teljes körű vizsgálatmenet
|
Kontrollok
|
|
konc. (M)
|
oldatai
|
oldatai
|
konc. (M)
|
|
C0
|
0
|
TCx-1
|
10 000 000 x
|
TCx-1
|
3000 x
|
PC
|
1*10-9
|
|
C1
|
3*10-9
|
TCx-2
|
1 000 000 x
|
TCx-2
|
1000 x
|
NK
|
1*10-5
|
|
C2
|
1*10-8
|
TCx-3
|
100 000 x
|
TCx-3
|
300 x
|
C0
|
0
|
|
C3
|
3*10-8
|
TCx-4
|
10 000 x
|
TCx-4
|
100 x
|
SC
|
0
|
|
C4
|
1*10-7
|
TCx-5
|
1 000 x
|
TCx-5
|
30 x
|
|
|
|
C5
|
3*10-7
|
TCx-6
|
100 x
|
TCx-6
|
10 x
|
Hozzáadott agonista
|
|
C6
|
1*10-6
|
TCx-7
|
10 x
|
TCx-7
|
3 x
|
konc. (M)
|
|
C7
|
3*10-6
|
TCx-8
|
1 x
|
TCx-8
|
1 x
|
17β-ösztradiol
|
3*10-12
|
|
C8
|
1*10-5
|
|
|
|
|
|
|
|
TCx –x. vizsgálati vegyi anyag
PC –pozitív kontroll (4-hidroxitamoxifén)
NK –negatív kontroll (rezveratrol)
C0 –a referenciaszabvány oldószeres kontrollja
SC –a vizsgálati vegyi anyag oldószeres kontrollja
VC –vivőanyagos kontroll (nem tartalmazza az agonista referenciaszabványként szolgáló 17β-ösztradiol fix koncentrációját (3,0*10-12 M))
|
Adatgyűjtés és adatelemzés
|
56.
|
Az elővizsgálat és a teljes körű vizsgálatmenetek lefolytatását követően meg kell határozni az agonista vizsgálat EC10, EC50, PC10 és PC50 értéket, valamint a maximális indukciót (TCxmax). Antagonista vizsgálat esetében az IC20, IC50, PC80 és PC50 értéket, valamint a maximális indukciót (TCxmax) kell kiszámolni. E paraméterek grafikus ábrázolása látható a 3. ábrán (agonista vizsgálat) és 4. ábrán (antagonista vizsgálat). A szükséges paraméterek kiszámítása az egyes vizsgálati vegyi anyagok relatív (a 100 %-nak vett referenciaszabvány maximális indukciójához viszonyított) indukcióján alapul. Az adatok értékeléséhez nem lineáris regressziót (változó meredekség 4 paraméter alapján) kell alkalmazni, az alábbi egyenlet szerint:

ahol:
X = a dózis vagy a koncentráció logaritmusa
Y = válasz (relatív indukció (%))
Görbe teteje = maximális indukció (%)
Görbe alja = minimális indukció (%)
LogEC50 = annak a koncentrációnak a logaritmusa, amely a maximális válasz 50 %-át idézi elő
Hill-meredekség = meredekségi tényező vagy Hill-meredekség
|
|
57.
|
A luminométer által szolgáltatott, relatív fényegységekben (RLU) kifejezett nyers adatokat át kell vezetni az elővizsgálat és a teljes körű vizsgálatmenetek adatelemzése céljára létrehozott táblázatsablonba. A nyers adatoknak meg kell felelniük a 3A és 3B táblázatban (agonista vizsgálat) vagy a 4A és 4B táblázatban (antagonista vizsgálat) feltüntetett elfogadhatósági kritériumoknak. Amennyiben a nyers adatok megfelelnek az elfogadhatósági kritériumoknak, az alábbi kalkulációs lépéseken keresztül meg kell határozni a szükséges paramétereket:
|
Agonista vizsgálat
|
—
|
Az egyes referenciaszabványok nyers adatainak elemzéséből származó eredményből vonjuk ki a referenciaszabvány oldószeres kontrolljának átlagos RLU-értékét.
|
|
—
|
Az egyes vizsgálati vegyi anyagok nyers adatainak elemzéséből származó eredményből vonjuk ki a vizsgálati vegyi anyag oldószeres kontrolljának átlagos RLU-értékét.
|
|
—
|
Számítsuk ki a referenciaszabvány egyes koncentrációértékeinek relatív indukcióját. Határozzuk meg 100 %-ként a referenciaszabvány legmagasabb koncentrációjának indukcióját.
|
|
—
|
Számítsuk ki a vizsgálati vegyi anyagok egyes koncentrációinak a referenciaszabvány 100 %-nak vett legnagyobb koncentrációjához viszonyított, relatív indukcióját.
|
|
—
|
Értékeljük az analízis eredményeit nem lineáris regresszió technikájával (változó meredekség, 4 paraméter).
|
|
—
|
Határozzuk meg a referenciaszabvány EC50 és EC10 értékét.
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok EC50 és EC10 értékét.
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok maximális relatív indukcióját (TCmax).
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok PC10 és PC50 értékét.
|
A vizsgálati vegyi anyagok esetében citotoxicitás vagy oldhatósági problémák miatt nem mindig hozható létre teljes dózis-válasz görbe. Ekkor nem lehet meghatározni az EC50, EC10 és PC50 értékét. Ilyen esetben csak a PC10 és TCmax értéke határozható meg.
Antagonista vizsgálat
|
—
|
Az egyes referenciaszabványok nyers adatainak elemzéséből származó eredményből vonjuk ki a referenciaszabvány legnagyobb koncentrációjának átlagos RLU-értékét.
|
|
—
|
Az egyes vizsgálati vegyi anyagok nyers adatainak elemzéséből származó eredményből vonjuk ki a referenciaszabvány legnagyobb koncentrációjának átlagos RLU-értékét.
|
|
—
|
Számítsuk ki a referenciaszabvány egyes koncentrációértékeinek relatív indukcióját. Határozzuk meg 100 %-ként a referenciaszabvány legalacsonyabb koncentrációjának indukcióját.
|
|
—
|
Számítsuk ki a vizsgálati vegyi anyagok egyes koncentrációinak a referenciaszabvány 100 %-nak vett legalacsonyabb koncentrációjához viszonyított, relatív indukcióját.
|
|
—
|
Értékeljük az analízis eredményeit nem lineáris regresszió technikájával (változó meredekség, 4 paraméter).
|
|
—
|
Határozzuk meg a referenciaszabvány IC50 és IC20 értékét.
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok IC50 és IC20 értékét.
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok minimális relatív indukcióját (TCmin).
|
|
—
|
Határozzuk meg a vizsgálati vegyi anyagok PC80 és PC50 értékét.
|
3. ábra
Az agonista vizsgálatban meghatározott paraméterek áttekintése
EC10 = egy anyag azon koncentrációja, amely a maximális válaszának 10 %-át váltja ki.
EC50 = egy anyag azon koncentrációja, amely a maximális válaszának 50 %-át váltja ki.
PC10 = egy vizsgálati vegyi anyag azon koncentrációja, amely a referenciaszabvány EC10 értékével megegyező választ vált ki.
PC50 = egy vizsgálati vegyi anyag azon koncentrációja, amely a referenciaszabvány EC50 értékével megegyező választ vált ki.
TCxmax = a vizsgálati vegyi anyag maximális relatív indukciója.
4. ábra
Az antagonista vizsgálatban meghatározott paraméterek áttekintése

IC20 = egy anyag azon koncentrációja, amely a maximális válaszának 80 %-át váltja ki (20 %-os gátlást idéz elő).
IC50 = egy anyag azon koncentrációja, amely a maximális válaszának 50 %-át váltja ki (50 %-os gátlást idéz elő).
PC80 = egy vizsgálati vegyi anyag azon koncentrációja, amely a referenciaszabvány IC20 értékével megegyező választ vált ki.
PC50 = egy vizsgálati vegyi anyag azon koncentrációja, amely a referenciaszabvány IC50 értékével megegyező választ vált ki.
TCxmin = a vizsgálati vegyi anyag minimális relatív indukciója.
A vizsgálati vegyi anyagok esetében citotoxicitás vagy oldhatósági problémák miatt nem mindig hozható létre teljes dózis-válasz görbe. Ekkor nem lehet meghatározni az IC50, IC20 és PC50 értékét. Ilyen esetben csak a PC20 és TCmin értéke határozható meg.
|
58.
|
Az eredményeknek két (vagy három) független vizsgálatmeneten kell alapulniuk. Amennyiben két vizsgálatmenet összevethető, vagyis reprodukálható eredményeket ad, akkor harmadik vizsgálatmenetre nincs szükség. Az eredmények akkor elfogadhatók, ha:
|
—
|
teljesülnek az elfogadhatósági kritériumok (e kritériumokat lásd a 14–22. pontban),
|
|
Az adatok értelmezésének kritériumai
|
59.
|
Az adatok értelmezéséhez és annak eldöntéséhez, hogy egy adott vizsgálati vegyi anyag pozitív vagy negatív besorolást kapjon, az alábbi kritériumokat kell figyelembe venni:
|
Minden teljes körű vizsgálatmenet tekintetében valamely vizsgálati vegyi anyag az alábbi esetben tekinthető pozitívnak:
|
1.
|
A TCmax megegyezik a referenciaszabvány maximális válaszának 10 %-ával (REF10), vagy meghaladja azt.
|
|
2.
|
A vizsgálati vegyi anyag legalább 2 egymást követő koncentrációja megegyezik az REF10 értékével, vagy meghaladja azt.
|
|
|
Minden teljes körű vizsgálatmenet tekintetében valamely vizsgálati vegyi anyag az alábbi esetben tekinthető negatívnak:
|
1.
|
A TCmax nem haladja meg a referenciaszabvány maximális válaszának 10 %-át (REF10).
|
|
2.
|
A vizsgálati vegyi anyag kevesebb mint két koncentrációja egyezik meg az REF10 értékével, vagy haladja meg azt.
|
|
|
Minden teljes körű vizsgálatmenet tekintetében valamely vizsgálati vegyi anyag az alábbi esetben tekinthető pozitívnak:
|
1.
|
A TCmin megegyezik a referenciaszabvány maximális válaszának 80 %-ával (REF80 = 20 %-os gátlás), vagy alacsonyabb annál.
|
|
2.
|
A vizsgálati vegyi anyag legalább 2 egymást követő koncentrációja megegyezik az REF80 értékével, vagy alacsonyabb annál.
|
|
|
Minden teljes körű vizsgálatmenet tekintetében valamely vizsgálati vegyi anyag az alábbi esetben tekinthető negatívnak:
|
1.
|
A TCmin meghaladja a referenciaszabvány maximális válaszának 80 %-át (REF80 = 20 % gátlás).
|
|
2.
|
A vizsgálati vegyi anyag legfeljebb egy koncentrációja egyezik meg az REF80 értékével, vagy alacsonyabb annál.
|
|
|
|
60.
|
Egy adott vizsgálati vegyi anyag pozitív válasza által előidézett hatás jellemzése érdekében a jelentésbe egyaránt bele kell foglalni a kiváltott hatás mértékét (agonista vizsgálatnál: TCmax; antagonista vizsgálatnál: TCmin) és a hatást előidéző koncentrációt (agonista vizsgálatnál: EC10, EC50, PC10, PC50; antagonista vizsgálatnál: IC20, IC50, PC80, PC50).
|
VIZSGÁLATI JELENTÉS
|
61.
|
Lásd „AZ ÖSZTROGÉNRECEPTOR TRANSZAKTIVÁCIÓS VIZSGÁLAT ÖSSZETEVŐI” című 20. pontot.
|
SZAKIRODALOM
|
(1)
|
OECD (2016). Draft Validation report of the (anti-) ERα CALUX bioassay – transactivation bioassay for the detection of compounds with (anti)estrogenic potential. Environmental Health and Safety Publications, Series on Testing and Assessment (No 240). Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs
|
|
(2)
|
Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. (2005). Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol Sci. 83(1), 136-148.
|
|
(3)
|
Quaedackers ME, van den Brink CE, Wissink S, Schreurs RHMM, Gustafsson JA, van der Saag PT, and van der Burg B. (2001). 4-Hydroxytamoxifen trans-represses nuclear factor-kB Activity in human osteoblastic U2OS cells through estrogen receptor (ER)α and not through ERβ. Endocrinology 142(3), 1156-1166.
|
|
(4)
|
Thorne N, Inglese J and Auld DS. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology, Chemistry and Biology17(6):646-57.
|
|
(5)
|
Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V and Balaguer P.(2006). Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem. Pharmacol., 71, 1459-1469.
|
|
(6)
|
Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA. (1998). Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinol., 139, 4252-4263.
|
|
(7)
|
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. (2010). Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism. J. Steroid. Biochem. Mol. Biol., 122, 204–211.
|
|
(8)
|
Sonneveld E, Riteco JAC, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, and van der Burg B. (2006). Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci., 89(1), 173–187.
|
|
(9)
|
Kobayashi H, Yamamoto K, Eguchi M, Kubo M, Nakagami S, Wakisaka S, Kaizuka M and Ishii H. (1995). Rapid detection of mycoplasma contamination in cell cultures by enzymatic detection of polymerase chain reaction (PCR) products. J. Vet. Med. Sci., 57(4), 769-771.
|
|
(10)
|
Zhang J-H, Chung TDY, and Oldenburg KR. (1999). A simple statistical parameter for use in evaluation and validation of high throuphut screening assays. J. Biomol. Scr., 4, 67-73
|
|
(11)
|
Besselink H, Middelhof I, and Felzel, E. (2014). Transactivation assay for the detection of compounds with (anti)estrogenic potential using ERα CALUX cells. BioDetection Systems BV (BDS). Amsterdam, the Netherlands.
|
4.1. függelék:
A SEJTÉLETKÉPESSÉG VIZUÁLIS MEGFIGYELÉSE
B.67. EMLŐSSEJTEKEN TIMIDIN-KINÁZ GÉNNEL VÉGZETT IN VITRO GÉNMUTÁCIÓS VIZSGÁLATOK
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 490. vizsgálati iránymutatásával (2016). A vizsgálati módszereket a tudományos fejlődés, a szabályozási igények és az állatok kíméletének figyelembevételével rendszeresen felülvizsgálják és átdolgozzák. A timidin-kináz (TK) lókuszt alkalmazó egérlimfóma-vizsgálat (MLA) és TK6 vizsgálat eredetileg a B.17. vizsgálati módszer részét képezte. Később azonban a genotoxicitás vizsgálatával foglalkozó nemzetközi műhely (IWGT) MLA szakértői munkacsoportja az egérlimfóma-vizsgálat elfogadhatósági kritériumaira és adatainak értelmezésére vonatkozólag nemzetközi harmonizált ajánlásokat dolgozott ki (1) (2) (3) (4) (5), ezeket az ajánlásokat foglalja magában ez az új, B.67. vizsgálati módszer. Ez a vizsgálati módszer az egérlimfóma-vizsgálathoz készült, és mivel timidin-kináz lókuszt alkalmaz, a TK6 vizsgálatra is érvényes. Az egérlimfóma-vizsgálat ot széles körben alkalmazzák szabályozási célból, a TK6 vizsgálatot viszont sokkal ritkábban. Megjegyzendő, hogy a végpontok hasonlósága ellenére a két sejtvonal nem helyettesíthető egymással, ezért a szabályzási programok adott szabályozási célra jogosan részesítik előnyben az egyiket a másikkal szemben. Az MLA validálása például bizonyította, hogy a vizsgálat nem csupán a génmutáció észlelésére alkalmas, hanem a szerkezeti kromoszómakárosodást okozó vizsgálati vegyi anyagok kiszűrésére is. E vizsgálati módszer a genetikai toxikológiai vizsgálati módszerek sorozatának részét képezi. Az OECD kidolgozott egy, a genetikai toxikológiai vizsgálatokról tömör tájékoztatást nyújtó, az OECD genetikai toxikológiára vonatkozó vizsgálati iránymutatásainak közelmúltbeli változásait áttekintő dokumentumot (6).
|
|
2.
|
Az emlőssejteken végzett in vitro génmutációs vizsgálatok célja a vegyi anyagok által előidézett génmutációk észlelése. Az ezekben a vizsgálatokban használt sejtvonalak forward (előremutató) mutációkat mérnek riportergénekben, konkrétan az endogén timidin-kináz génben (TK emberi sejtekben és Tk rágcsálók sejtekben, e vizsgálati módszer keretében együttes megnevezésük TK). Ezt a vizsgálati módszert két sejtvonal használatára tervezték: az egyik az L5178Y TK+/--3.7.2C egérlimfóma-sejtvonal (általános nevén az L5178Y), a másik a TK6-os humán limfoblasztoid sejtvonal (elterjedt nevén a TK6). Bár a két sejtvonal eredetét, sejtnövekedését, p53 státuszát stb. tekintve különbözik egymástól, a TK génmutációs vizsgálatok hasonló módon végezhetők el mindkét sejttípusban, a vizsgálati módszerben leírtak szerint.
|
|
3.
|
A timidin-kináz gén autoszomális és heterozigóta jellege lehetővé teszi, hogy a TK+/- > TK-/- mutációt követően észlelni lehessen a timidin-kináz enzimmel nem rendelkező sejtekből álló életképes telepeket. Az enzimhiányt okozhatják a TK gént érintő genetikai események, amelyek lehetnek génmutációk (pontmutációk, a leolvasási keret eltolódása, kisebb deléciók stb.) és kromoszomális események (nagy deléciók, kromoszóma-átrendeződések és mitotikus rekombináció). Ez utóbbi események a heterozigótaság elvesztését okozhatják, ami a humán tumorgenezisben szerepet játszó tumorszupresszor gének gyakori genetikai változása. Az egérlimfóma-vizsgálat elvileg képes detektálni a TK gént hordozó teljes kromoszóma magorsókárosodásból és/vagy mitotikus nondiszjunkcióból fakadó elvesztését. A citogenetikus és a molekuláris analízis együttes alkalmazása egyértelműen mutatja, hogy az egérlimfóma-vizsgálat timidin-kináz mutánsainak egy része nondiszjunkcióval jött létre. A bizonyítékok súlyozása azonban arra utal, hogy a TK génmutációs vizsgálatok a standard (e vizsgálati módszerben ismertetett) citotoxicitási kritériumok alkalmazásával nem képesek megbízhatóan kimutatni aneugéneket, ezért ezek a vizsgálatok aneugének észlelésére nem megfelelők (7) (8) (9).
|
|
4.
|
A TK génmutációs vizsgálatokban a timidin-kináz mutánsok két eltérő fenotípus csoportba oszthatók: a normális növekedési sebességű mutánsok, amelyek a heterozigóta timidin-kináz sejtekkel megegyező ütemben növekednek, és a lassan növő mutánsok, amelyeknek hosszabb a generációs ideje. A normális növekedési sebességű és a lassan növő mutánsokat az egérlimfóma-vizsgálatban nagy és kis telepmutánsokként, a TK6 vizsgálatban pedig gyorsan és lassan megjelenő telepmutánsokként tartják számon. Az egérlimfóma-vizsgálat nagy és kis telepmutánsainak molekuláris és citogenetikus természetét már részletesen feltárták (8) (10) (11) (12) (13). A TK6 vizsgálat gyorsan és lassan megjelenő mutánsainak molekuláris és citogenetikus természetét is széles körű vizsgálatnak vetették alá (14) (15) (16) (17). Mindkét sejttípus lassan növő mutánsait genetikus károsodás érte, amely a TK lókusz közelében elhelyezkedő, feltételezetten növekedést szabályozó gén(eke)t érinti, és amely kiváltja a megduplázódási idő elhúzódását, azaz a lassan megjelenő vagy kis telepek létrejöttét (18). A lassan növő mutánsok megjelenését alapvető szerkezeti kromoszóma-rendellenességeket előidéző vegyi anyagokkal hozták összefüggésbe. Azok a sejtek, amelyeknél a mutáció nem érinti a TK lókusz közelében elhelyezkedő, feltételezhetően növekedést szabályozó gén(eke)t, a szülői sejtekhez hasonló ütemben nőnek és normális sebességgel növő mutánsokká válnak. A kezdetben normális növekedési sebességű mutánsoknál a vegyi anyagok elsődlegesen pontmutációkat okozó mutagének. Ebből következően mind a lassan növő, mind a normális növekedési sebességű mutánsokat meg kell számolni ahhoz, hogy számba vehessék az összes mutánst, és támpontokat lehessen szerezni arra nézve, hogy a vizsgálati vegyi anyag milyen típusú károsodás(oka)t idéz elő (mutagén vs. klasztogén) (10) (12) (18) (19).
|
|
5.
|
A vizsgálati módszert úgy építették fel, hogy egyaránt biztosítson mindkét vizsgálatra (egérlimfóma-vizsgálat és TK6) érvényes általános információkat, és egyedi iránymutatást az egyes vizsgálatokhoz.
|
|
6.
|
Az alkalmazott fogalommeghatározások az 1. függelékben találhatók.
|
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
7.
|
Az in vitro végrehajtott vizsgálatok rendszerint külső forrásból származó metabolikus aktiváció alkalmazását teszik szükségessé. Az exogén metabolikus aktivációs rendszer nem utánozza teljesen az in vivo körülményeket.
|
|
8.
|
Ügyelni kell arra, hogy ne álljanak elő olyan körülmények, amelyek álpozitív – tehát nem a vizsgálati vegyi anyag és a sejt genetikus anyagának kölcsönhatásából származó – eredményhez (hanem a vizsgálati rendszerrel való esetleges kölcsönhatáshoz) vezethetnének; ilyen körülmény többek között a pH-érték vagy az ozmolalitás változása, a tápfolyadék összetevőivel való kölcsönhatás (20) (21) vagy a túlzott mértékű citotoxicitás (22) (23) (24). Az egérlimfóma-vizsgálat és a TK6 vizsgálat szempontjából a 28. pontban meghatározott, ajánlott citotoxicitási felső határértékeket meghaladó citotoxicitás minősül túlzott mértékűnek. Emellett a timidin bázisanalóg, vagy timidin bázisanalógként viselkedő vizsgálati vegyi anyagok növelhetik a mutánsok gyakoriságát azáltal, hogy a sejtkezelés alatt elősegítik a spontán háttérmutánsok szelektív növekedését, így a megfelelő értékelés biztosítása érdekében további vizsgálati módszerek használatát teszik szükségessé (25).
|
|
9.
|
A mesterséges nanoanyagok esetében e vizsgálati módszer egyedi adaptálására lehet szükség, amelyet azonban ez a vizsgálati módszer nem ismertet.
|
|
10.
|
E vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény.
|
|
11.
|
Azok a mutáns sejtek, amelyek a TK+/- > TK
-/- mutáció miatt nem mutatnak timidinkináz-enzimaktivitást, rezisztensek a pirimidin bázisanalóg trifluoro-timidin (TFT) citotoxikus hatásaival szemben. A TK proficiens sejtek érzékenyek a TFT-re, ami a celluláris metabolizmus gátlását okozza, és megállítja a további sejtosztódást. Így tehát a mutáns sejtek TFT jelenlétében képesek sejtburjánzásra és látható telepek létrehozására, míg a timidin-kinázt tartalmazó sejtek nem képesek erre.
|
A VIZSGÁLAT ELVE
|
12.
|
Szuszpenzióban növő sejteket megfelelő időtartamig (lásd a 33. pontot) külső forrásból származó metabolikus aktivációval és anélkül (lásd a 19. pontot) kezelik a vizsgálati vegyi anyaggal, majd a sejtkultúrát átültetik, hogy meghatározzák a citotoxicitást és lehetővé tegyék a fenotípus expressziót a mutáns kiválasztása előtt. A citotoxicitást az egérlimfóma-vizsgálat esetében a relatív teljes növekedés (RTG – lásd a 25. pontot), a TK6 esetében pedig a relatív túlélés (RS – lásd a 26. pontot) alapján határozzák meg. A kezelt tenyészeteket tápoldatban tartják, az egyes sejttípusokra jellemző megfelelő ideig (lásd a 37. pontot), hogy lehetővé váljon az indukált mutációk közel optimális fenotípus expressziója. A fenotípus expressziót követően a mutációs gyakoriságot úgy határozzák meg, hogy leültetnek ismert számú sejtet a szelekciós ágenst tartalmazó tápoldatba a mutáns telepek kimutatására, valamint a szelekciós ágenst nem tartalmazó tápoldatba a kolóniaképző képesség (életképesség) meghatározására. Megfelelő inkubációs idő után megszámolják a telepeket. A mutációs gyakoriság kiszámításának alapja a mutáns telepek száma, kiigazítva a mutáns kiválasztásának idején mért kolóniaképző képességgel.
|
A MÓDSZER LEÍRÁSA
Előkészületek
Sejtek
|
13.
|
Az egérlimfóma-vizsgálat esetén: Mivel az egérlimfóma-vizsgálat kidolgozását és jellemzését az L5178Y sejtek TK
+/- -3.7.2C alvonalára alapozták, az egérlimfóma-vizsgálathoz ezt az alvonalat kell használni. Az L5178Y sejtvonal a DBA/2 egér vonalból származó, metilkolantrénnel indukált csecsemőmirigy limfómából származik (26). Clive és munkatársai az L5178Y (Clive-féle jelöléssel TK+/+ -3) sejteket etil-metán-szulfonáttal kezelték, majd bróm-dezoxiuridint szelekciós ágensként használva izolálták a TK-/- (Clive-féle jelöléssel TK-/- -3.7) klónt. A TK-/- klónból egy spontán keletkezett TK+/- klónt (Clive-féle jelöléssel TK+/- -3.7.2.) és egy alklónt (Clive-féle jelöléssel TK+/--3.7.2C) izoláltak és jellemeztek az egérlimfóma-vizsgálathoz (27). A sejtvonal kariotípusát közzétették (28) (29) (30) (31). Modális kromoszómaszáma 40. A sejtvonal tartalmaz egy metacentrikus kromoszómát (t12;13), amely egy kromoszómának tekintendő. Az egér timidin-kináz lókusza a 11. kromoszóma távoli végén helyezkedik el. Az L5178Y TK
+/- -3.7.2C sejtvonal mindkét p53 allélján mutáció figyelhető meg, és mutáns p53 fehérjét hoz létre (32) (33). Valószínűleg a TK+/--3.7.2C sejtvonal p53 státusza felelős azért, hogy a vizsgálat képes észlelni a nagymérvű károsodásokat (17).
|
|
14.
|
A TK6 esetében A TK6 egy humán limfoblasztoid sejtvonal. A kiinduló sejtvonal egy Epstein-Barr vírussal transzformált sejtvonal, a WI-L2, amely eredetileg egy örökletes szferocitózisban szenvedő 5 éves fiútól származik. A sejtvonal első izolált klónját, a HH4-et ICR191-gyel mutagenizálták, majd létrehozták a heterozigóta timidin-kináz sejtvonalat, a TK6-ot (34). A TK6 sejtek szinte diploid állapotúak, kromoszómakészletük jellemzői a következők: 47, XY, 13+, t(14; 20), t(3; 21) (35). A humán timidin-kináz lókusza a 17. kromoszóma hosszú karján helyezkedik el. A TK6 p53-kompetens sejtvonal, mivel mindkét allélon vad típusú p53-szekvenciával rendelkezik, és csak vad típusú p53 fehérjét fejez ki (36).
|
|
15.
|
Mind az egér-limfóma, mind a TK6 vizsgálatra érvényes, hogy az induló sejtkészlet létrehozása vagy lefagyasztása előtt ajánlatos a laboratóriumnak meggyőződnie a sejtek mikoplazmafertőzés-mentességéről, meghatároznia a sejtek kariotípusát vagy megfestenie azokat a kromoszómákat, amelyek a timidin-kináz lókuszt hordozzák és ellenőriznie a sejtpopuláció megduplázódási idejét. A laboratóriumban használt sejteknél meg kell állapítani a normál sejtciklus-időtartamot, és annak összhangban kell lennie a publikált jellemzőkkel (16) (19) (37). A mester törzskészletet -150 °C-on vagy az alatti hőmérsékleten kell tárolni, és minden munkakészletet ebből kell elkészíteni.
|
|
16.
|
Nagy mennyiségű fagyasztott munkakészlet létrehozását megelőzően, vagy közvetlenül a munkakészletek kísérleti felhasználása előtt szükség lehet arra, hogy a tenyészetet megtisztítsák a jelen lévő mutáns sejtektől [kivéve, ha az oldószeres kontroll mutációs gyakorisága (MF) már az elfogadható tartományon belül van – lásd az egérlimfóma-vizsgálat 2. táblázatát)]. Ehhez metotrexáttal (aminopterinnel) szelektálják a timidinkináz-hiányos sejteket, majd timidint, hipoxantint és glicint (L5178Y) vagy 2'-deoxicitidint (TK6) adagolnak a sejtkultúrához, hogy biztosítsák a TK-kompetens sejtek (19) (38) (39) és a TK6 (40) optimális növekedését. A sejttenyészetek fenntartásához általános gyakorlati tanácsok, valamint konkrétan az L5178Y és a TK6 sejtekre vonatkozó tanácsok a (19) (31) (37) (39) (41) szakirodalmi hivatkozásban találhatók. Azoknak a laboratóriumoknak, amelyek mester törzskészletet igényelnek az egérlimfóma- vagy a TK6 vizsgálat elvégzéséhez, illetve új törzskészletet kívánnak beszerezni, rendelkezésére áll egy megfelelően jellemzett sejteket tartalmazó törzsgyűjtemény (37).
|
Tápfolyadék és tenyésztési körülmények
|
17.
|
Mindkét vizsgálatban megfelelő tápfolyadékot és inkubációs feltételeket kell biztosítani a tenyészetek fenntartásához (pl. tenyésztőedények, 5 %-os CO2-koncentrációjú, vízgőzzel telített levegő és 37 °C-os inkubációs hőmérséklet). A sejttenyészeteket mindig olyan körülmények között kell tartani, amelyek biztosítják az exponenciális növekedési szakaszt. Különösen fontos, hogy a tápfolyadékot és a tenyésztési körülményeket úgy válasszák meg, hogy azok biztosítsák az optimális növekedési feltételeket az expressziós időszak alatt, és mind a mutáns, mind a vad típusú sejtvonalak kolóniaképző készsége optimális legyen. Az egérlimfóma- és a TK6 vizsgálatban egyaránt fontos az is, hogy a tenyésztési körülmények biztosítsák mind a nagy telepmutánsok/gyorsan megjelenő timidin kináz mutánsok, mind a kis telepmutánsok/lassan megjelenő timidin kináz mutánsok optimális fejlődését. A sejttenyésztés további részleteiről – egyebek mellett a lószérum megfelelő iválásának szükségességéről, ha a mutánsok kiválasztása során RPMI tápfolyadékot használnak – a (19) (31) (38) (39) (40) (42) szakirodalmi hivatkozás nyújt tájékoztatást.
|
A tenyészetek előkészítése
|
18.
|
A sejteket tárolt törzskultúrákból indítva tápfolyadékba oltják le olyan sejtszámmal, hogy a szuszpenzióban növő sejtek a teljes kezelési és expressziós időszakban exponenciálisan növekedjenek.
|
Metabolikus aktiválás
|
19.
|
L5178Y és TK6 sejtek használata esetén exogén metabolikus aktivációs rendszereket kell alkalmazni, mivel ezek a sejtek nem rendelkeznek megfelelő endogén metabolikus képességgel. Amennyiben az ettől való eltérés nem indokolt, az alapértelmezetten ajánlott, legáltalánosabban használt rendszer a rágcsálók (általában patkányok) enziminduktorral (ilyen például az Aroclor 1254 (43) (44) (45) vagy fenobarbitál és ß-naftoflavon kombinációjával (46) (47) (48) (49) (50) (51)) kezelt májából preparált, kofaktor-kiegészítésű posztmitokondriális frakció (S9). Ez utóbbi kombináció nem ütközik a környezetben tartósan megmaradó szerves szennyező anyagokról szóló stockholmi egyezménnyel (52), és kiderült róla, hogy olyan hatékony, mint az Aroclor 1254 a vegyes funkciójú oxidázok indukálásához (45) (46) (47) (48) (49). Az S9 frakció jellemzően 1–2 térfogatszázalék koncentrációban használatos, de a végső vizsgálati tápfolyadékban mennyisége 10 térfogatszázalékra növelhető. Az alkalmazott exogén metabolikus aktivációs rendszer vagy metabolikus induktor típusának és koncentrációjának megválasztását befolyásolhatja a vizsgálati vegyi anyagok osztálya.
|
A vizsgálati vegyi anyagok előkészítése
|
20.
|
A szilárd halmazállapotú vizsgálati vegyi anyagokat a sejtek kezelése előtt megfelelő oldószerrel elő kell kezelni, és szükség esetén hígítani kell (lásd a 21. pontot). A folyékony halmazállapotú vizsgálati vegyi anyagok közvetlenül hozzáadhatók a vizsgálati rendszerhez és/vagy hígíthatók a vizsgálati rendszer kezelése előtt. A gáz halmazállapotú vagy illékony vizsgálati vegyi anyagokat a standard protokollok megfelelő módosításával, például légmentesen lezárt tenyésztőedényekben történő kezeléssel kell vizsgálni (53) (54) (55). A vizsgálati vegyi anyagot tartalmazó készítményeket közvetlenül a kezelés előtt kell előállítani, kivéve, ha a stabilitási adatok a tárolhatóságot bizonyítják.
|
VIZSGÁLATI KÖRÜLMÉNYEK
Oldószerek
|
21.
|
Az oldószert oly módon kell kiválasztani, hogy optimális oldhatóságot biztosítson a vizsgálati vegyi anyagnak, ugyanakkor ne befolyásolja hátrányosan a vizsgálat lebonyolítását, vagyis ne változtassa meg a sejtek növekedését, ne befolyásolja a vizsgálati vegyi anyag integritását, ne lépjen reakcióba a tenyésztőedénnyel, és ne károsítsa a metabolikus aktivációs rendszert. Amennyiben lehetséges, először valamilyen vizes oldószer (vagy tápfolyadék) használatát érdemes megfontolni. Jól bevált oldószer a víz vagy a dimetil-szulfoxid. A végleges kezelési közegben a szerves oldószerek koncentrációja általában ne haladja meg az 1 térfogatszázalékot, a vizes oldószereké (só oldat vagy víz) pedig a 10 térfogatszázalékot. A jól bevált szerves oldószerektől eltérő oldószerek (például etanol vagy aceton) használatakor a vizsgálati vegyi anyaggal és a vizsgálati rendszerrel való összeegyeztethetőségükre utaló adatokkal kell alátámasztani használatukat, és azt, hogy a használt koncentrációnál nem genotoxikusak. Alátámasztó adatok hiányában szükséges nem kezelt kontrollok alkalmazása (lásd a fogalommeghatározásokat az 1. függelékben) annak bizonyítására, hogy a kiválasztott oldószer nem vált ki káros vagy mutagén hatást.
|
A CITOTOXICITÁS MÉRÉSE ÉS A KEZELÉS SORÁN ALKALMAZOTT KONCENTRÁCIÓSZINTEK KIVÁLASZTÁSA
|
22.
|
A vizsgálati vegyi anyag legmagasabb koncentrációjának meghatározása során kerülendő az olyan koncentráció, amely hamis pozitív válaszreakciókat válthat ki, ilyen a túlzott citotoxicitást okozó koncentráció (lásd a 28. pontot), a tápfolyadékban történő kicsapódást okozó koncentráció (lásd a 29. pontot), illetve a pH vagy az ozmolalitás jelentős változását okozó koncentráció (lásd a 8. pontot). Ha a vizsgálati vegyi anyag a hozzáadásakor jelentős változást okoz a tápfolyadék pH-jában, a pH a kezelésre szolgáló végleges tápfolyadék pufferelésével kiigazítható, hogy elkerülhetők legyenek a hamis pozitív eredmények és megfelelő tenyésztési feltételeket lehessen fenntartani.
|
|
23.
|
A koncentráció kiválasztása a citotoxicitás és egyéb megfontolások alapján történik (lásd a 27–30. pontot). Bár a fő vizsgálatban alkalmazandó koncentrációk jobb meghatározásához egy előzetes vizsgálatban érdemes meghatározni a citotoxicitást, az előzetes vizsgálat nem követelmény. Még ha sor is kerül a citotoxicitás előzetes meghatározására, a fő vizsgálat során minden kultúrára el kell végezni a citotoxicitás mérését. Amennyiben koncentrációtartomány-behatároló kísérletet végeznek, a kísérletnek le kell fednie a koncentrációk széles körét, a kísérlet pedig lezárható a kezelést követő 1. napon, vagy folytatható a 2. napon történő expresszióig és a mutánsok kiválasztásáig (ha bebizonyosodik, hogy a használt koncentrációértékek megfelelők).
|
|
24.
|
A citotoxicitást minden vizsgálati és kontrollként szolgáló sejtkultúra esetében meg kell határozni: az egérlimfóma- (2) és a TK6 (15) vizsgálatban alkalmazott módszert a nemzetközileg elfogadott gyakorlat alapján dolgozták ki.
|
|
25.
|
Az egérlimfóma-vizsgálat esetében mind az agarlemezen, mind a mikrotiterlemezen való tenyésztés során a citotoxicitást a relatív teljes növekedés (RTG) alapján kell értékelni, amelyet eredetileg Clive és Spector határozott meg 1975-ben (2). Ez a mérés magában foglalja a sejtek kezelése során tapasztalt relatív szuszpenziós növekedést (RSG: a vizsgálati kultúra összevetése az oldószeres kontrollal), az expressziós időt és a mutánsok kiválasztásának idején megfigyelt relatív kolóniaképző képességet (RCE: a vizsgálati kultúra összevetése az oldószeres kontrollal) (2). Megjegyzendő, hogy a relatív szuszpenziós növekedés számol a vizsgálati kultúrában a kezelés alatt bekövetkezett sejtpusztulással (a képletet lásd a 2. függelékben).
|
|
26.
|
A TK6 esetében a citotoxicitást a relatív túlélés alapján kell értékelni, ami a közvetlenül a kezelést követően leültetett sejtek kolóniaképző képessége, kiigazítva a kezelés során végbement sejtpusztulással, majd a kapott sejtszám összevetve a negatív kontrollal (amelynek túlélési aránya 100 %-nak tekintendő) (a képletet lásd a 2. mellékletben).
|
|
27.
|
Legalább négy, az elfogadhatósági kritériumokat (megfelelő citotoxicitás, sejtszám stb.) teljesítő kísérleti koncentráció eredményét kell értékelni (az oldószeres és a pozitív kontrollokon túl). Bár két párhuzamos tenyészet használata ajánlott, az egyes vizsgált koncentrációknál párhuzamos vagy szimpla kezelt tenyészetek használhatók. A párhuzamos tenyészetekben adott koncentráció mellett kapott eredményeket külön kell dokumentálni, az adatelemzéshez azonban összevonhatók (55). Az alacsony citotoxicitást vagy a citotoxicitás hiányát mutató vizsgálati vegyi anyagok esetében rendszerint megközelítőleg kétszeres-háromszoros koncentrációs intervallumok helyénvalóak. Citotoxicitás esetén olyan koncentrációkat kell választani, amelyek lefedik a 28. pontban ismertetett, citotoxicitást okozó koncentrációtól azokig a koncentrációkig terjedő tartományt, ahol mérsékelt vagy alacsony a citotoxicitás, illetve nem jelentkezik citotoxicitás. Számos vizsgálati vegyi anyagnak meredek a koncentráció-válasz görbéje, és annak érdekében, hogy a citotoxicitás teljes tartományát le lehessen fedni, vagy részletesen meg lehessen vizsgálni a koncentráció-válasz összefüggést, szükség lehet egymáshoz közelebbi koncentrációk és négynél több koncentráció használatára, különösen olyan helyzetekben, ahol megismételt kísérlet szükséges (lásd a 70. pontot). Négynél több koncentráció alkalmazásának különösen egyedi tenyészet alkalmazása esetében lehet jelentősége.
|
|
28.
|
Ha a legnagyobb koncentráció a citotoxicitáson alapul, a legmagasabb koncentrációval 10 és 20 % közötti relatív teljes növekedést kell megcélozni az egérlimfóma-vizsgálat , és 10 és 20 % közötti relatív túlélést a TK6 vizsgálat esetén (67. pont).
|
|
29.
|
A legalacsonyabb oldhatatlan koncentrációnál alacsonyabb koncentrációk mellett nem citotoxikus, nehezen oldható vizsgálati vegyi anyagok esetében a legmagasabb analizált koncentrációnak a vizsgálati vegyi anyaggal történő kezelés végén szemmel vagy inverz mikroszkóp segítségével látható zavarosságot vagy kicsapódást kell okoznia. Még abban az esetben is, ha citotoxicitás a legalacsonyabb oldhatatlan koncentráció felett jön létre, ajánlatos egyetlen egy, zavarosságot vagy látható kicsapódást okozó koncentrációt vizsgálni, mivel a kicsapódás fals hatásokat eredményezhet. Mivel az egérlimfóma- és a TK6 vizsgálat szuszpenzióban növő kultúrákat használ, különösen ügyelni kell annak biztosítására, hogy a kicsapódás ne zavarja a vizsgálat lebonyolítását. Hasznos lehet emellett a tápfolyadékban való oldhatóság meghatározása a kísérlet előtt.
|
|
30.
|
Ha nem figyelhető meg kicsapódás vagy korlátozó citotoxicitás, a legmagasabb vizsgálati koncentrációnak 10 mM-nek, 2 mg/ml-nek vagy 2 μl/ml-nek kell megfelelnie, melyek közül a legalacsonyabb a mérvadó (57) (58). Ha a vizsgálati vegyi anyag nem meghatározott összetételű, például ismeretlen vagy változó összetételű anyag, komplex reakciótermék vagy biológiai anyag [vagyis úgynevezett ismeretlen vagy változó összetételű vegyi anyag (UVCB)], környezeti extraktum stb., előfordulhat, hogy kellő citotoxicitás hiányában a legmagasabb koncentrációnak magasabbnak (például 5 mg/ml-nek) kell lennie ahhoz, hogy növekedjen az egyes összetevők koncentrációja. Megjegyzendő azonban, hogy ezek a követelmények eltérőek lehetnek a humán gyógyszerek esetében (59).
|
Kontrollok
|
31.
|
Minden kísérleti körülménynél párhuzamos negatív kontrollokat kell használni (lásd a 21. pontot), amelyeknél csak oldószert alkalmaznak a kezelt közegben, és a kezelésük ugyanolyan módon történik, mint a kezelt tenyészeteké.
|
|
32.
|
Párhuzamos pozitív kontrollok szükségesek annak igazolására, hogy a laboratórium képes az alkalmazott vizsgálati protokoll feltételei mellett mutagének azonosítására, hogy (adott esetben) az exogén metabolikus aktivációs rendszer eredményes, továbbá hogy megfelelően detektálhatók mind a kis telepmutánsok/lassan megjelenő TK6-mutánsok, mind a nagy telepmutánsok/gyorsan megjelenő TK6-mutánsok. Pozitív kontrollokra vonatkozó példák az alábbi 1. táblázatban találhatók. Indokolt esetben pozitív kontrollként alternatív anyagok használhatók. Mivel az emlőssejtek genetikai toxicitás szempontjából metabolikus aktiválással és anélkül végzett in vitro vizsgálatai a párhuzamosan lefolytatott, azonos időtartamú, rövid távú (3–4 órás) kezelések esetében kellően szabványosítottak, a pozitív kontrollok használata a metabolikus aktiválást igénylő mutagénekre korlátozódhat. Ebben az esetben egyetlen pozitív kontroll válaszreakciója a metabolikus aktiváló rendszer aktivitását és a vizsgálati rendszer reagálóképességét is bizonyítja. Hosszú távú (vagyis 24 órás, S9 nélküli) kezelés esetén azonban saját pozitív kontrollt kell végezni, mivel a kezelés időtartama eltér a metabolikus aktiválás alkalmazása mellett végzett vizsgálatétól. Mindegyik pozitív kontrollt egy vagy több olyan koncentrációban kell alkalmazni, amely a háttérértékekhez viszonyítva reprodukálható és kimutatható növekedést okoz, hogy bizonyítani lehessen a vizsgálati rendszer érzékenységét, és a választ nem veszélyeztetheti a vizsgálati módszerben meghatározott határértékeket meghaladó citotoxicitás (lásd a 28. pontot).
|
1. táblázat
A laboratóriumok jártasságának értékeléséhez és a pozitív kontrollok kiválasztásához ajánlott referenciaanyagok
|
Kategória
|
Anyag
|
CAS-szám
|
|
1. Metabolikus aktiválás nélkül is aktív mutagének
|
|
Metil-metánszulfonát
|
66-27-3
|
|
Mitomicin-C
|
50-07-7
|
|
4-Nitro-kinolin-N-oxid
|
56-57-5
|
|
2. Metabolikus aktiválást igénylő mutagének
|
|
Benzo(a)pirén
|
50-32-8
|
|
Ciklofoszfamid monohidrát
|
50-18-0 (6055-19-2)
|
|
7,12-Dimetil-benz[a]antracén
|
57-97-6
|
|
3-metilkolantrén
|
56-49-5
|
ELJÁRÁS
Kezelés a vizsgálati vegyi anyaggal
|
33.
|
A proliferáló sejteket metabolikus aktivációs rendszer jelenlétében és hiányában kezelik a vizsgálati vegyi anyaggal. Az expozíciónak megfelelő időtartamig kell tartania (ez rendszerint 3–4 óra). Megjegyzendő azonban, hogy ezek a követelmények eltérőek lehetnek a humán gyógyszerek esetében (59). Amennyiben az egérlimfóma-vizsgálatban a rövid távú kezelés negatív eredményre vezet, és léteznek a hosszabb kísérlet szükségességét alátámasztó adatok [pl. nukleozidanalógok, nehezen oldható vegyi anyagok esetében (5) (59)], fontolóra kell venni a vizsgálat hosszabb – vagyis 24 órás, S9 kiegészítők nélküli – kezeléssel járó elvégzését.
|
|
34.
|
Az egyes vizsgálati (kontroll és kezelt) tenyészetekhez a vizsgálat bármely szakaszában alkalmazott minimális sejtszámnak a spontán mutációs gyakoriságon kell alapulnia. Az általános iránymutatás szerint minden kísérleti tenyészetben elegendő sejtet kell kezelni és passzálni ahhoz, hogy a vizsgálat minden szakaszában (kezelés, fenotípusos expresszió és mutánsok kiválasztása) előforduljon legalább 10, ideális esetben azonban 100 spontán mutáció (56).
|
|
35.
|
Az egérlimfóma-vizsgálatban az ajánlott elfogadható spontán mutációs gyakoriság 35-140 × 10-6 (agarlemezen) és 50-170 × 10-6 (mikrotiterlemezen) között van (lásd a 2. táblázatot). Ahhoz, hogy minden vizsgálati sejtkultúrában legyen legalább 10, de ideális esetben 100, a kezelést túlélő spontán mutáns, legalább 6 × 106 sejt kezelése szükséges. Ilyen mennyiségű sejt kezelése, valamint a kifejeződés és a mutánsok kiválasztását lehetővé tévő kolóniaképzés során a megfelelő sejtszám fenntartása a kísérlet minden szakaszában garantálja a spontán mutánsok megfelelő számát (10 vagy több), még a 90 %-os citotoxicitást (vagyis 10 %-os relatív teljes növekedést) okozó koncentrációkkal kezelt tenyészetek esetében is (19) (38) (39).
|
|
36.
|
A TK6 vizsgálatban a spontán mutációs gyakoriság rendszerint 2 és 10 × 10-6 közé esik. Ahhoz, hogy minden sejtkultúrában legyen legalább 10, a kezelést túlélő spontán mutáns, legalább 20 × 106 sejt kezelése szükséges. Ilyen mennyiségű sejt kezelése még a 90 %-os citotoxicitást (10 %-os relatív túlélést) kiváltó koncentrációkkal kezelt tenyészetek esetében is megfelelő számú (10 vagy több) spontán mutánst biztosít. Emellett az expressziós időszak során elegendő számú sejtet kell tenyészteni, majd kiszéleszteni a mutánsok kiválasztásához (60).
|
A fenotípusos expresszió ideje, valamint a citotoxicitás és a mutációs gyakoriság mérése
|
37.
|
A kezelési időszak végén a sejteket meghatározott – az egyes sejtvonalakra jellemző – ideig fenntartják, hogy lehetővé tegyék az újonnan indukált mutánsok fenotípusának közel optimális expresszióját. Az egérlimfóma-vizsgálatban a fenotípusos expressziós időszak 2 nap. A TK6 vizsgálatban a fenotípusos expressziós időszak 3–4 nap. 24 órás kezelési időszak esetén az expressziós időszak a kezelés végét követően veszi kezdetét.
|
|
38.
|
A fenotípusos expressziós időszakban a sejteket naponta megszámlálják. Az egérlimfóma-vizsgálatban a naponta mért sejtszám alapján számítják ki a napi szuszpenziós növekedést (SG). A 2 napos expressziós időszakot követően a sejteket szuszpendálják szelekciós ágenst tartalmazó, illetve szelekciós ágens nélküli tápfolyadékban, hogy meghatározzák a mutánsok számát (a szelekciós lemezeken) és a kolóniaképző képességet (az életképességet mérő lemezeken). Az egérlimfóma-vizsgálatban két egyenértékű módszer létezik a mutánsok kiválasztását célzó kolóniaképzésre: az egyik lágyagar, a másik 96 lyukú lemezbe helyezett tápfolyadék használatán alapul (19) (38) (39). A TK6 vizsgálatban a kolóniaképzés 96 lyukú lemezbe helyezett tápfolyadékban zajlik (16).
|
|
39.
|
A trifluor-timidin (TFT) a timidin kináz mutánsok egyetlen ajánlott szelekciós ágense (61).
|
|
40.
|
Az egérlimfóma-vizsgálatban az agarlemezeken és a mikrotiterlemezeken 10–12 napi inkubálást követően végzik el a számlálást. A TK6 vizsgálatban a mikrotiterlemezeken fejlődő telepeken 10–14 nap elteltével számlálják meg a gyorsan megjelenő mutánsokat. A lassan növő (lassan megjelenő) TK6-mutánsok kinyeréséhez a gyorsan megjelenő mutánsok számba vételét követően a sejtekhez ismét tápfolyadékot és TFT-t kell adni, majd a lemezeket további 7–10 napon keresztül inkubálni kell (62). A lassú és normál fejlődésű timidin kináz mutánsok megszámlálására vonatkozó szempontokat lásd a 42–44. pontban.
|
|
41.
|
A két vizsgálat, azon belül az egérlimfóma-vizsgálat két módszerének (agarlemez és mikrotiterlemez) megfelelő számlálási módját a 2. függelék tartalmazza. Az egérlimfóma-vizsgálat agarlemezes módszerénél a mutációs gyakoriság meghatározásához megszámlálják a telepeket, és a mutáns telepek számát kiigazítják a kolóniaképző képességgel. Az egérlimfóma-vizsgálat mikrotiterlemezes módszerénél és a TK6 vizsgálatnál a kolóniaképző képességet mind a mutánsok kiválasztására, mind a kolóniaképző képesség mérésére szolgáló lemezeken Poisson-elosztással állapítják meg (63). A mutációs gyakoriságot a két lemezen tapasztalt kolóniaképző készség alapján határozzák meg.
|
Mutáns telepek jellemzése
|
42.
|
Amennyiben az egérlimfóma-vizsgálatban a vizsgálati vegyi anyag pozitív eredményt ad (lásd a 62–63. pontot), a telep méretének vagy növekedésének meghatározásával jellemezni kell a telepeket a vizsgált tenyészetek közül legalább egynél (általában a legnagyobb elfogadható pozitív koncentrációnál), valamint a negatív és a pozitív kontrolloknál. Ha a vizsgálati vegyi anyag negatívnak bizonyul (lásd a 64. pontot), a mutáns telepek jellemzését a negatív és a pozitív kontrollokon kell elvégezni. Az egérlimfóma-vizsgálat mikrotiterlemezes módszere esetén a kis telepmutánsok azok, amelyek a lyuk átmérőjének kevesebb mint 25 %-át fedik be, a nagy telepmutánsok pedig azok, amelyek a lyuk átmérőjének több mint 25 %-át fedik be. Az agarlemezes módszerben automatikus telepszámlálással határozzák meg a mutáns telepek számát és méretét. A telepméret mérésének módjairól a szakirodalom nyújt részletes tájékoztatást (19) (38) (40). A negatív és a pozitív kontrollok esetén a telepek jellemzései arra szolgálnak, hogy igazolják a vizsgálatok megfelelő végrehajtását.
|
|
43.
|
A vizsgálati vegyi anyag nem minősíthető negatívnak, ha sem a nagy, sem a kis telepmutánsokat nem észlelik megfelelően a pozitív kontrollban. A telepek jellemzése általános tájékoztatást nyújthat arról, hogy a vizsgálati vegyi anyag képes-e pontmutációkat és/vagy kromoszomális eseményeket előidézni (4. pont).
|
|
44.
|
TK6: a normális növekedési sebességű és a lassan növő mutánsok elkülönítésének alapja az eltérő inkubációs idő (lásd a 40. pontot). A TK6 vizsgálatban általában minden tenyészetnél – így a negatív és a pozitív kontrolloknál is – számba veszik mind a gyorsan, mind a lassan megjelenő mutánsokat. A negatív és a pozitív kontroll telepjellemzései arra szolgálnak, hogy igazolják a vizsgálatok megfelelő végrehajtását. A vizsgálati vegyi anyag nem minősíthető negatívnak, ha sem a gyorsan, sem a lassan megjelenő mutánsokat nem észleli megfelelően a pozitív kontroll. A telepek jellemzése általános tájékoztatást nyújthat arról, hogy a vizsgálati vegyi anyag képes-e pontmutációkat és/vagy kromoszomális eseményeket előidézni (4. pont).
|
A laboratórium jártassága
|
45.
|
A laboratóriumoknak a vizsgálat rutinszerű alkalmazása előtti elegendő tapasztalat igazolása érdekében kísérletsorozatot kell végrehajtaniuk referenciaként szolgáló, különböző mechanizmussal ható pozitív anyagokkal (legalább egy metabolikus aktiválással és legalább egy metabolikus aktiválás nélkül ható, az 1. táblázatban felsoroltak közül kiválasztott anyaggal), és különböző negatív kontrollokkal (nem kezelt tenyészetek és különféle oldószerek/vivőanyagok használatával). E pozitív és negatív kontrollok válaszreakcióinak összhangban kell lenniük a szakirodalommal. Ez nem vonatkozik azokra a laboratóriumokra, amelyek tapasztalattal rendelkeznek, azaz amelyeknek a rendelkezésére áll a 47–50. pontban meghatározott, dokumentált adatbázis. Az egérlimfóma-vizsgálatban a pozitív és a negatív kontrollból nyert adatoknak összhangban kell lenniük a genotoxicitás vizsgálatával foglalkozó nemzetközi műhely ajánlásaival (lásd a 2. táblázatot).
|
|
46.
|
A pozitív kontrollként kiválasztott anyagokat (lásd az 1. táblázatot) meg kell vizsgálni metabolikus aktiválás nélkül rövid és hosszú kezelési idővel (ha végeznek hosszú időtartalmú kezelést) és metabolikus aktiválást alkalmazó rövid kezelési idővel is, a laboratórium mutagén hatású vegyi anyagok kimutatásában és a metabolikus aktivációs rendszer hatékonyságának meghatározásában való jártasságának bizonyítására, valamint be kell mutatni, hogy a laboratórium megfelelően végzi a kezelés alatt a sejttenyésztést, a fenotípusos expressziót és a mutánsok kiválasztását, továbbá, hogy a kiértékelési eljárások is megfelelőek. A vizsgálati rendszer érzékenységének és dinamikus tartományának bizonyítása érdekében a kiválasztott anyagok koncentrációtartományát úgy kell megválasztani, hogy reprodukálható és koncentrációval összefüggő növekedést biztosítson a háttérértékekhez viszonyítva.
|
Történeti kontrolladatok
|
47.
|
A laboratóriumnak meg kell állapítania a következőket:
|
—
|
a pozitív történeti kontrollok tartománya és eloszlása,
|
|
—
|
a negatív (nem kezelt, oldószeres) történeti kontrollok tartománya és eloszlása.
|
|
|
48.
|
A negatív történeti kontrollok dokumentált eloszlásához kapcsolódó első adatgyűjtés során a párhuzamos negatív kontrolloknak összhangban kell lenniük a negatív kontrollokra vonatkozóan közzétett adatokkal. Amint több kísérleti adat adódik a kontrollok eloszlásához, a párhuzamos negatív kontrolloknak ideális esetben az eloszlás 95 %-os ellenőrzési határértékén belül kell lenniük (64) (65).
|
|
49.
|
A laboratórium negatív történeti kontrollokat tartalmazó adatbázisát első lépésként legalább 10 kísérlettel kell kiépíteni, de az adatbázisnak lehetőség szerint legalább 20, összehasonlítható kísérleti körülmények között végzett kísérletet kell tartalmaznia. A laboratóriumoknak minőségellenőrzési módszereket, például ellenőrzési diagramokat (C-diagramokat vagy X-bar diagramokat (65)) kell alkalmazniuk annak meghatározására, hogy a pozitív és a negatív kontrollokra vonatkozó adataik mennyire változóak, valamint annak bizonyítására, hogy náluk a módszertan „ellenőrzés alatt” áll (66). A dokumentált adatok gyűjtésére és használatára vonatkozó további részletek és ajánlások megtalálhatók a szakirodalomban (64).
|
|
50.
|
A negatív kontrollokra vonatkozó adatoknak tartalmazniuk kell a szimpla tenyészetből vagy lehetőség szerint a párhuzamos tenyészetekből származó mutációs gyakoriságot, ahogyan az a 27. pontban szerepel. A párhuzamos negatív kontrolloknak ideális esetben a laboratórium negatív történeti kontrollokat tartalmazó adatbázisa szerinti eloszlás 95 %-os ellenőrzési határértékén belül kell lenniük. Amennyiben a negatív kontrollokra vonatkozó adatok a 95 %-os ellenőrzési határértéken kívül esnek, akkor számíthatók be a kontrollok dokumentált eloszlásába, ha ezek az adatok nem szélsőségesen kiugró értékek, továbbá bizonyíték van arra, hogy a vizsgálati rendszer „ellenőrzés alatt áll” (lásd a 49. pontot), valamint arra, hogy nem áll fenn szakmai vagy emberi mulasztás.
|
|
51.
|
A kísérleti protokoll bármely módosítását meg kell vizsgálni abból a szempontból, hogy az adatok összhangban vannak-e a laboratórium meglévő történeti kontrolladatbázisaival. Jelentősebb következetlenségek fennállása esetén új történeti kontrolladatbázist kell létrehozni.
|
ADATOK ÉS JELENTÉS
Az eredmények értékelése
|
52.
|
Az adatok ismertetésének az egérlimfóma és a TK6 vizsgálat esetében is ki kell terjednie a kezelt és a kontrollként szolgáló tenyészetekre, a citotoxicitás (relatív teljes növekedés vagy relatív túlélés) kiszámításához szükséges adatokra és a mutációs gyakoriságra, az alábbiakban meghatározottak szerint.
|
|
53.
|
Az egérlimfóma-vizsgálatban az egyes tenyészetekre vonatkozólag meg kell adni a relatív szuszpenziós növekedést, a relatív teljes növekedést, a mutánsok kiválasztásának idején mért kolóniaképző képességet és a mutáns telepek számát (agarlemez használata esetén), illetve az üres lyukak számát (mikrotiterlemez használata esetén). A mutációs gyakoriságot a mutáns sejtek száma és 1 millió túlélő sejt hányadosaként kell kifejezni. Pozitív válasz esetén meg kell adni a kis és a nagy telepek mutációs gyakoriságát (és/vagy a teljes mutációs gyakoriság arányát) a vizsgálati vegyi anyag legalább egy koncentrációjánál (általában a legnagyobb pozitív koncentrációnál), valamint a negatív és a pozitív kontrolloknál. Negatív válasz esetén a negatív és a pozitív kontroll kis és nagy telepeinek mutációs gyakoriságát kell megadni.
|
|
54.
|
A TK6 vizsgálatban az egyes tenyészetekre vonatkozólag meg kell adni a relatív túlélést, a mutánsok kiválasztásának idején mért kolóniaképző képességet, valamint a gyorsan és a lassan megjelenő mutánsoknál megfigyelt üres lyukak számát. A mutációs gyakoriságot a mutáns sejtek számának és a túlélő sejtek számának hányadosaként kell meghatározni, továbbá meg kell adni a teljes mutációs gyakoriságot, valamint a gyorsan és a lassan megjelenő mutánsok mutációs gyakoriságát (és/vagy a teljes mutációs gyakoriság arányát).
|
Elfogadhatósági kritériumok
|
55.
|
Egy adott vizsgálati vegyi anyag összesített eredményeinek meghatározása előtt mind az egérlimfóma, mind a TK6 vizsgálatban teljesíteni kell az alábbi kritériumokat:
|
—
|
Két kísérleti körülmény (rövid időtartamú kezelés metabolikus aktiválással és metabolikus aktiválás nélkül – lásd a 33. pontot) vizsgálatára került sor, kivéve, ha az egyik pozitív eredményeket hozott.
|
|
—
|
Megfelelő számú sejt és koncentrációérték elemezhető (lásd a 27. és a 34–36. pontot).
|
|
—
|
A legmagasabb koncentráció kiválasztására vonatkozó kritériumok összhangban vannak a 28–30. pontban ismertetett legmagasabb koncentrációkkal.
|
|
Negatív és pozitív kontrollok elfogadhatósági kritériumai
|
56.
|
Miután az IWGT mlA szakértői munkacsoportja az egérlimfóma-vizsgálat ból származó adatok széles körét elemezte, nemzetközi konszenzus jött létre az egérlimfóma-vizsgálat ra vonatkozó konkrét elfogadhatósági kritériumokról (1) (2) (3) (4) (5). Ezért ez a vizsgálati módszer konkrét ajánlásokat fogalmaz meg a negatív és a pozitív kontrollok elfogadhatóságára, valamint az egérlimfóma-vizsgálatban tesztelt anyagok eredményeinek kiértékelésére vonatkozólag. A TK6 vizsgálat jelentősen kisebb adatbázissal rendelkezik, amelyet nem értékelt munkacsoport.
|
|
57.
|
Az egérlimfóma-vizsgálat minden kísérletét értékelni kell abból a szempontból, hogy a kezeletlen/oldószeres kontroll teljesíti-e az IWGT mlA munkacsoportja által felállított elfogadhatósági kritériumokat ((4) és az alábbi 2. táblázat) a következő mutatók tekintetében: (1) mutációs gyakoriság (az IWGT eltérő mutációs gyakoriságot minősített elfogadhatónak az egérlimfóma-vizsgálat agarlemezes és mikrotiterlemezes vizsgálatánál), (2) a mutánsok kiválasztásának idején mért kolóniaképző képesség (CE) és (3) az oldószeres kontroll során tapasztalt szuszpenziós növekedés (SG) (a képleteket lásd a 2. függelékben).
2. táblázat
Az egérlimfóma-vizsgálat elfogadhatósági kritériumai
|
Paraméter
|
Lágyagar módszer
|
Mikrotiter módszer
|
|
Mutációs gyakoriság
|
35–140 × 10-6
|
50–170 × 10-6
|
|
Kolóniaképző képesség
|
65–120 %
|
65–120 %
|
|
Szuszpenziós növekedés
|
8–32-szeres (3–4 órás kezelés) 32–180-szoros (24 órás kezelés, ha végeztek ilyet)
|
8–32-szeres (3–4 órás kezelés) 32–180-szoros (24 órás kezelés, ha végeztek ilyet)
|
|
|
58.
|
Az egérlimfóma-vizsgálatban minden kísérletet értékelni kell abból a szempontból is, hogy a pozitív kontroll(ok) teljesíti(k)-e az IWGT munkacsoport által kidolgozott két alábbi elfogadhatósági kritérium legalább egyikét:
|
—
|
A pozitív kontrollnak elő kell idéznie a teljes mutációs gyakoriság abszolút növekedését, vagyis a spontán háttér mutációs gyakoriságot legalább 300 × 10-6 mértékben meghaladó növekedést kell kiváltania [indukált MF (IMF)]. A kis kolóniáknál megfigyelt mutációs gyakoriságnak legalább 40 %-ban indukált mutációs gyakoriságnak kell lennie.
|
|
—
|
A pozitív kontrollnál a kis kolóniák mutációs gyakorisága legalább 150 × 10-6 mértékben haladja meg a párhuzamos kezeletlen/oldószeres kontrollnál tapasztalt mutációs gyakoriságot (azaz a kis kolóniák indukált mutációs gyakorisága 150 × 10-6).
|
|
|
59.
|
A TK6 vizsgálatban akkor elfogadhatók a kísérletek, amikor a párhuzamos negatív kontroll a 48–49. pont szerint belefoglalható a laboratórium negatív történeti kontrollokat tartalmazó adatbázisába. Ezenfelül a párhuzamos pozitív kontrolloknak (lásd a 32. pontot) a pozitív történeti kontrollokat tartalmazó adatbázisban generált válaszokkal összeegyeztethető válaszreakciókat, valamint a párhuzamos negatív kontrollhoz viszonyítva statisztikailag szignifikáns növekedést kell előidézniük.
|
|
60.
|
Mindkét vizsgálat esetében a pozitív kontrolltenyészetnél megfigyelt citotoxicitás felső határának meg kell egyeznie a vizsgált kultúráknál tapasztalt citotoxicitás felső határával. Ez azt jelenti, hogy a relatív teljes növekedés/relatív túlélés nem csökkenhet 10 % alá. Elegendő egyetlen koncentrációt (több koncentrációérték használata esetén a pozitív kontrolltenyészetek egyik koncentrációját) vizsgálni annak igazolására, hogy a pozitív kontroll elfogadhatósági kritériumai teljesülnek. Emellett szükséges az is, hogy a pozitív kontroll mutációs gyakorisága a laboratórium által meghatározott elfogadhatósági tartományon belül legyen.
|
Az eredmények értékelése és értelmezése
|
61.
|
Az IWGT egérlimfóma szakértői munkacsoportja jelentős munka keretében megállapította az egérlimfóma-vizsgálat pozitív válaszának biológiai relevanciáját és kritériumait (4). Ezért ez a vizsgálati módszer konkrét ajánlásokat fogalmaz meg az egérlimfóma-vizsgálatban tesztelt vizsgálati vegyi anyagok eredményeinek kiértékelésére vonatkozólag (lásd a 62–64. pontot). A TK6 vizsgálat jelentősen kisebb adatbázissal rendelkezik, amelyet nem értékelt munkacsoport. Ebből adódóan a TK6 vizsgálatból származó adatok értelmezésére vonatkozó ajánlások általánosabban fogalmaznak (lásd a 65–66. pontot). Mindkét vizsgálatra nézve megfogalmaztak kiegészítő ajánlásokat (lásd a 67–71. pontot).
|
EGÉRLIMFÓMA-VIZSGÁLAT
|
62.
|
Ajánlatos egységes megközelítést alkalmazni a pozitív és negatív válaszok meghatározására, mert ez biztosítja a megnövekedett mutációs gyakoriság biológiai relevanciáját. A más vizsgálatoknál általánosan alkalmazott statisztikai elemzés helyett az egérlimfóma-vizsgálat egy előre meghatározott indukált mutációs gyakoriságot (vagyis a mutációs gyakoriság párhuzamos kontrollal összevetett növekedését) veszi alapul, az úgynevezett globális értékelési faktort (GEF), amely a részt vevő laboratóriumok által elvégzett negatív kontrollokból nyert mutációs gyakorisági adatok eloszlásának elemzése alapján került meghatározásra (4). A GEF az egérlimfóma-vizsgálat agarlemezes változata esetén 90 × 10-6, mikrotiterlemezes változata esetén pedig 126 × 10-6.
|
|
63.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag egyértelműen pozitívnak minősül, ha a vizsgált kísérleti körülmények bármelyikében (lásd a 33. pontot) a mutációs gyakoriság párhuzamos háttérértékhez viszonyított növekedése meghaladja a GEF-et, és a növekedés (pl. egy trendpróbával) koncentrációfüggést mutat. Ezt követően úgy tekintendő, hogy a vizsgálati vegyi anyag képes mutációt előidézni ebben a vizsgálati rendszerben.
|
|
64.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag egyértelműen negatívnak minősül, ha a vizsgált kísérleti körülmények (lásd a 33. pontot) egyikében sem figyeltek meg koncentrációfüggő választ, illetve ha növekedett is a mutációs gyakoriság, a növekedés mértéke nem haladja meg a GEF-et. Ezt követően úgy tekintendő, hogy a vizsgálati vegyi anyag ebben a vizsgálati rendszerben nem képes mutációt előidézni.
|
TK6
|
65.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag egyértelműen pozitívnak minősül, ha a vizsgált kísérleti körülmények bármelyikében (lásd a 33. pontot):
|
—
|
legalább az egyik vizsgálati koncentráció statisztikailag szignifikáns növekedést mutat a párhuzamos negatív kontrollhoz viszonyítva,
|
|
—
|
a megfelelő trendpróbával történő értékelés arra utal, hogy a növekedés koncentrációfüggő (lásd a 33. pontot),
|
|
—
|
ezen eredmények bármelyike a negatív történeti kontrolladatok eloszlásán (például Poisson-eloszlású 95 %-os ellenőrzési határértéken) kívül esik; lásd a 48. pontot).
|
Mindezen kritériumok teljesülése esetén úgy tekintendő, hogy a vizsgálati vegyi anyag képes mutációt előidézni ebben a vizsgálati rendszerben. A legmegfelelőbb statisztikai módszerekre vonatkozó ajánlások megtalálhatók a szakirodalomban (66) (67).
|
|
66.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag egyértelműen negatívnak minősül, ha valamennyi vizsgált kísérleti körülmény mellett (lásd a 33. pontot):
|
—
|
a vizsgálati koncentrációk egyike sem mutat statisztikailag szignifikáns növekedést a párhuzamos negatív kontrollhoz viszonyítva,
|
|
—
|
a megfelelő trendpróbával végzett értékelés nem mutat koncentrációfüggő növekedést,
|
|
—
|
valamennyi eredmény a negatív történeti kontrolladatok eloszlásán (például Poisson-eloszlású 95 %-os ellenőrzési határértéken) belül van; lásd a 48. pontot).
|
Ezt követően úgy tekintendő, hogy a vizsgálati vegyi anyag ebben a vizsgálati rendszerben nem képes mutációt előidézni.
|
Az egérlimfóma- és a TK6 vizsgálatra egyaránt vonatkozó szempontok:
|
67.
|
Ha a legnagyobb koncentráció a citotoxicitáson alapul, a legmagasabb koncentrációval 10 és 20 % közötti relatív teljes növekedést/relatív túlélést kell megcélozni. Megegyezés szerint körültekintően kell eljárni a 10 és 20 % közötti relatív teljes növekedésnél/relatív túlélésnél jelentkező pozitív eredmények értékelése során, és az eredmények nem tekinthetők pozitívnak, ha a mutációs gyakoriság növekedése csak 10 %-os vagy az alatti relatív teljes növekedésnél/relatív túlélésnél tapasztalható (ha ez értékelt) (2) (59).
|
|
68.
|
Bizonyos körülmények között további információk segítséget nyújthatnak egy adott vizsgálati vegyi anyag nem mutagén jellegének meghatározásához, amikor egyik tenyészetnél sem mutatható ki 10–20 %-os relatív teljes növekedés/relatív túlélés közé eső relatív teljes növekedés. Az alábbi helyzetek tartoznak ide: (1) A 100 % és 20 % közötti relatív teljes növekedési/relatív túlélési tartományba eső adatpontoknál nincsenek mutagenitásra utaló jelek (pl. nincs dózisfüggő válasz, a mutációs gyakoriság nem haladja meg a párhuzamos negatív kontrollét vagy a dokumentált háttérérték-tartományt stb.), és legalább egy adatpont a 20 % és 25 % közötti relatív teljes növekedési/relatív túlélési tartományba esik. (2) A 100 % és 25 % közötti relatív teljes növekedési/relatív túlélési tartományba eső adatpontoknál nincsenek mutagenitásra utaló jelek (pl. nincs dózisfüggő válasz, a mutációs gyakoriság nem haladja meg a párhuzamos negatív kontrollét vagy a dokumentált háttérérték-tartományt stb.), és egy negatív adatpont kissé a 10 %-os relatív teljes növekedés/relatív túlélés alatt helyezkedik el. A vizsgálati vegyi anyag mindkét helyzetben negatívnak minősíthető.
|
|
69.
|
Az egyértelműen pozitív vagy negatív válasz igazolása nem követelmény.
|
|
70.
|
Olyan esetekben, amikor a válaszreakció – a fentiekben leírtak szerint – nem egyértelműen negatív vagy pozitív, illetve egy adott eredmény biológiai relevanciája megállapításának alátámasztása érdekében az adatokat szakértői vélemény és/vagy további vizsgálatok alapján kell értékelni. Hasznos lehet egy megismételt kísérlet lehetőleg módosított kísérleti körülmények melletti elvégzése [pl. a koncentrációk felosztásának módosításával megnövelni a valószínűségét annak, hogy az adatpontok a 10–20 %-os relatív teljes növekedési/relatív túlélési tartományba esnek, más metabolikus aktiválási feltételek (S9 koncentrációjának vagy forrásának megváltoztatása) vagy más kezelési időtartam alkalmazása).
|
|
71.
|
Ritka esetekben az adatkészlet – még további vizsgálatok után is – eleve kizárja a pozitív vagy negatív eredményekre vonatkozó következtetést. Ezért a vizsgálati vegyi anyag válaszreakcióját kétértelműnek kell minősíteni (amit úgy kell értelmezni, hogy egyenlő valószínűséggel tekinthető pozitívnak és negatívnak).
|
VIZSGÁLATI JELENTÉS
|
72.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálati vegyi anyag:
|
—
|
eredet, gyártási szám, felhasználási határidő, ha rendelkezésre áll;
|
|
—
|
a vizsgálati vegyi anyag stabilitása, ha ismert;
|
|
—
|
a vizsgálati vegyi anyag oldhatósága és stabilitása az oldószerben, ha ismert;
|
|
—
|
adott esetben a pH-érték, az ozmolalitás és a kicsapódás mérése abban a tápfolyadékban, amelyhez a vizsgálati vegyi anyagot hozzáadták.
|
|
|
Egy összetevőből álló anyag:
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján
|
|
|
|
Több összetevőből álló anyag, UVCB-k és keverékek:
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai révén jellemezve.
|
|
|
|
|
Oldószer:
|
—
|
az oldószer kiválasztásának indokolása;
|
|
—
|
az oldószernek a végleges tápfolyadékon belüli aránya.
|
|
|
|
Sejtek:
|
|
Laboratóriumi törzstenyészetek esetén:
|
—
|
a sejtek típusa és eredete, valamint a vizsgálólaboratóriumban rendelkezésre álló dokumentált adataik;
|
|
—
|
kariotípus jellemzői és/vagy modális kromoszómaszám;
|
|
—
|
a sejttenyészetek fenntartásának módszerei.
|
|
—
|
sejt megduplázódási ideje.
|
|
|
|
|
Vizsgálati körülmények:
|
—
|
a koncentrációk kiválasztásának és a sejttenyészetek számának indokolása; beleértve például a citotoxicitási adatokat és az oldhatóságra vonatkozó korlátokat;
|
|
—
|
a közeg összetétele, CO2-koncentráció, páratartalom;
|
|
—
|
a vizsgálati vegyi anyag koncentrációja a tápfolyadékon belüli végső koncentrációjaként kifejezve (például μg vagy mg/a tápfolyadék ml-e vagy mM-ja);
|
|
—
|
a tápfolyadékhoz adott oldószer és vizsgálati vegyi anyag koncentrációja (és/vagy térfogata);
|
|
—
|
inkubációs hőmérséklet;
|
|
—
|
sejtsűrűség a kezelés időtartama alatt;
|
|
—
|
a metabolikus aktivációs rendszer típusa és összetétele (az S9 eredete, az S9 keverék elkészítési módszerei, az S9 keverék és az S9 koncentrációja vagy térfogata a végleges tápfolyadékban, az S9 minőségellenőrzése);
|
|
—
|
pozitív és negatív kontrollként szolgáló anyagok, végső koncentráció az egyes kezelési körülmények esetén;
|
|
—
|
az expressziós időszak hossza (beleértve a leültetett sejtek számát, a szubkultúrákat és a tápfolyadékok cseréjét, ha szükséges);
|
|
—
|
a szelekciós ágens neve és koncentrációja;
|
|
—
|
egérlimfóma-vizsgálat esetén jelezni kell az alkalmazott vizsgálat típusát (agarlemezes vagy mikrotiterlemezes);
|
|
—
|
a vizsgálatok elfogadhatóságának kritériumai;
|
|
—
|
az életképes és mutáns sejtek számának megállapítására használt módszerek;
|
|
—
|
a citotoxicitás mérésére alkalmazott módszerek;
|
|
—
|
a citotoxicitás és az alkalmazott módszer szempontjából lényeges kiegészítő információk;
|
|
—
|
a leültetést követően végzett inkubálás időtartama;
|
|
—
|
azon telepek meghatározása, amelyek méretét és típusát figyelembe vették (beleértve a „kis” és „nagy” telepekre vonatkozó kritériumokat is);
|
|
—
|
azok a kritériumok, amelyek meghatározzák, hogy a vizsgálatok pozitívnak, negatívnak vagy többféleképpen értelmezhetőnek tekintendők-e;
|
|
—
|
adott esetben a pH-érték, az ozmolalitás és a kicsapódás meghatározására alkalmazott módszerek.
|
|
|
|
Eredmények:
|
—
|
az egyes tenyészetek esetében kezelt sejtek és átültetett sejtek száma;
|
|
—
|
citotoxicitási paraméterek (egérlimfóma-vizsgálat esetében a relatív teljes növekedés, a TK6 esetében a relatív túlélés);
|
|
—
|
a kicsapódás jelei és a meghatározás időpontja;
|
|
—
|
a szelektív és nem szelektív folyadékba leültetett sejtek száma;
|
|
—
|
a telepek száma a nem szelektív közegben és a rezisztens telepek száma a szelektív közegben, valamint az ezeknél tapasztalható mutációs gyakoriság;
|
|
—
|
a negatív és pozitív kontrollok telepeinek mérete, és ha a vizsgálati vegyi anyag pozitív, legalább egy koncentráció és a hozzá kapcsolódó mutációs gyakoriság;
|
|
—
|
amennyiben lehetséges, a koncentráció-válasz összefüggés;
|
|
—
|
a párhuzamos negatív (oldószeres) és pozitív kontrollokra vonatkozó adatok (koncentrációk és oldószerek);
|
|
—
|
történeti negatív (oldószeres) és pozitív kontroll adatok (koncentrációk és oldószerek), tartományokkal, átlagértékekkel és standard eltérésekkel; a dokumentált kontrolladatok alapjául szolgáló vizsgálatok száma;
|
|
—
|
statisztikai elemzések (az egyes tenyészetek és adott esetben összevont párhuzamos tenyészetek tekintetében), valamint p-értékek, ha meghatározásra kerültek; és az egérlimfóma-vizsgálat esetében a GEF értékelése.
|
|
|
SZAKIRODALOM
|
(1)
|
Moore, M.M., Honma, M. Clements, J. (Rapporteur), Awogi, T., Bolcsfoldi, G., Cole, J., Gollapudi, B., Harrington-Brock, K., Mitchell, A., Muster, W., Myhr, B., O'Donovan, M., Ouldelhkim, M-C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2000). Mouse Lymphoma Thymidine Kinase Locus (TK) Gene Mutation Assay: International Workshop on Genotoxicity Test Procedures (IWGTP) Workgroup Report, Environ. Mol. Mutagen., 35 (3): 185-190.
|
|
(2)
|
Moore, M.M., Honma, M., Clements, J., Harrington-Brock, K., Awogi, T., Bolcsfoldi, G., Cifone, M., Collard, D., Fellows, M., Flanders, K., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Kraycer, J., McEnaney, S., Muster, W., Myhr, B., O’Donovan, M., Oliver, Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Shimada, H. and Stankowski, L.F. Jr. (2002). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: Follow-Up International Workshop on Genotoxicity Test Procedures, New Orleans, Louisiana, (April 2000), Environ. Mol. Mutagen., 40 (4): 292-299.
|
|
(3)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Cifone, M., Delongchamp, R., Fellows, M., Gollapudi, B., Jenkinson, P., Kirby, P., Kirchner, S., Muster, W., Myhr, B., O’Donovan, M., Ouldelhkim, M-C., Pant, K., Preston, R., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A., Wakuri, S. and Yoshimura, I. (2003). Mouse Lymphoma Thymidine Kinase Locus Gene Mutation Assay: International Workshop (Plymouth, UK) on Genotoxicity Test Procedures Workgroup Report, Mutation Res., 540: 127-140.
|
|
(4)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Delongchamp, R., Durward, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Lloyd, M., Majeska, J., Myhr, B., O’Donovan, M., Omori, T., Riach, C., San, R., Stankowski, L.F. Jr., Thakur, A.K., Van Goethem, F., Wakuri, S. and Yoshimura, I. (2006). Mouse Lymphoma Thymidine Kinase Gene Mutation Assay: Follow-Up Meeting of the International Workshop on Genotoxicity Tests – Aberdeen, Scotland, 2003 – Assay Acceptance Criteria, Positive Controls, and Data Evaluation, Environ. Mol. Mutagen., 47 (1): 1-5.
|
|
(5)
|
Moore, M.M., Honma, M., Clements, J., Bolcsfoldi, G., Burlinson, B., Cifone, M., Clarke, J., Clay, P., Doppalapudi, R., Fellows, M., Gollapudi, B., Hou, S., Jenkinson, P., Muster, W., Pant, K., Kidd, D.A., Lorge, E., Lloyd, M., Myhr, B., O’Donovan, M., Riach, C., Stankowski, L.F. Jr., Thakur A.K. and Van Goethem, F. (2007). Mouse Lymphoma Thymidine Kinase Mutation Assay: Meeting of the International Workshop on Genotoxicity Testing, San Francisco, 2005, Recommendations for 24-h Treatment, Mutation. Res., 627 (1): 36-40.
|
|
(6)
|
OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No 234, OECD, Párizs.
|
|
(7)
|
Fellows M.D., Luker, T., Cooper, A. and O'Donovan, M.R. (2012). Unusual Structure-Genotoxicity Relationship in Mouse Lymphoma Cells Observed with a Series of Kinase Inhibitors. Mutation, Res., 746 (1): 21-28.
|
|
(8)
|
Honma, M., Momose, M., Sakamoto, H., Sofuni, T. and Hayashi, M. (2001). Spindol Poisons Induce Allelic Loss in Mouse Lymphoma Cells Through Mitotic Non-Disjunction. Mutation Res., 493 (1-2): 101-114.
|
|
(9)
|
Wang, J., Sawyer, J.R., Chen, L., Chen, T., Honma, M., Mei, N. and Moore, M.M. (2009). The Mouse Lymphoma Assay Detects Recombination, Deletion, and Aneuploidy, Toxicol. Sci.., 109 (1): 96-105.
|
|
(10)
|
Applegate, M.L., Moore, M.M., Broder, C.B., Burrell, A., and Hozier, J.C. (1990). Molecular Dissection of Mutations at the Heterozygous Thymidine Kinase Locus in Mouse Lymphoma Cells. Proc. National. Academy. Sci. USA, 87 (1): 51-55.
|
|
(11)
|
Hozier, J., Sawyer, J., Moore, M., Howard, B. and Clive, D. (1981). Cytogenetic Analysis of the L5178Y/TK+/- Leads to TK-/- Mouse Lymphoma Mutagenesis Assay System, Mutation Res., 84 (1): 169-181.
|
|
(12)
|
Hozier, J., Sawyer, J., Clive, D. and Moore, M.M. (1985). Chromosome 11 Aberrations in Small Colony L5178Y TK-/- Mutants Early in their Clonal History, Mutation Res., 147 (5): 237-242.
|
|
(13)
|
Moore, M.M., Clive, D., Hozier, J.C., Howard, B.E., Batson, A.G., Turner, N.T. and Sawyer, J. (1985). Analysis of Trifluorothymidine-Resistant (TFTr) Mutants of L5178Y/TK+/- Mouse Lymphoma Cells. Mutation Res., 151 (1): 161-174.
|
|
(14)
|
Liber H.L., Call K.M. and Little J.B. (1987). Molecular and Biochemical Analyses of Spontaneous and X-Ray-Induced Mutants in Human Lymphoblastoid Cells. Mutation Res., 178 (1): 143-153.
|
|
(15)
|
Li C.Y., Yandell D.W. and Little J.B. (1992). Molecular Mechanisms of Spontaneous and Induced Loss of Heterozygosity in Human Cells In vitro. Somat. Cell Mol. Genet., 18 (1): 77-87.
|
|
(16)
|
Honma M., Hayashi M. and Sofuni T. (1997). Cytotoxic and Mutagenic Responses to X-Rays and Chemical Mutagens in Normal and P53-Mutated Human Lymphoblastoid Cells. Mutation. Res., 374 (1): 89-98.
|
|
(17)
|
Honma, M., Momose, M., Tanabe, H., Sakamoto, H., Yu, Y., Little, J.B., Sofuni, T. and Hayashi, M. (2000). Requirement of Wild-Type P53 Protein for Maintenance of Chromosomal Integrity. Mol. Carcinogen., 28 (4): 203-14.
|
|
(18)
|
Amundson S.A. and Liber H.L. (1992). A Comparison of Induced Mutation at Homologous Alleles of the TK Locus in Human Cells. II. Molecular Analysis of Mutants. Mutation Res., 267 (1): 89-95.
|
|
(19)
|
Schisler M.R., Moore M.M. and Gollapudi B.B. (2013). In vitro Mouse Lymphoma (L5178Y TK+/- -3.7.2C) Forward Mutation Assay. In Protocols in Genotoxicity Assessment A. Dhawan and M. Bajpayee (Eds.), Springer Protocols, Humana Press: 27-50.
|
|
(20)
|
Long, L.H., Kirkland, D., Whitwell, J. and Halliwell, B. (2007). Different Cytotoxic and Clastogenic Effects of Epigallocatechin Gallate in Various Cell-Culture Media Due to Variable Rates of its Oxidation in the Culture Medium, Mutation Res., 634 (1-2): 177-183.
|
|
(21)
|
Nesslany, F., Simar-Meintieres, S., Watzinger, M., Talahari, I. and Marzin, D. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environ. Mol. Mutagen., 49 (6): 439-452.
|
|
(22)
|
Brusick D. (1986). Genotoxic Effects in Cultured Mammalian Cells Produced by Low pH Treatment Conditions and Increased Ion Concentrations. Environ. Mutagen., 8 (6): 879-886.
|
|
(23)
|
Morita, T., Nagaki, T., Fukuda, I. and Okumura, K. (1992). Clastogenicity of Low pH to Various Cultured Mammalian Cells. Mutation Res., 268 (2): 297-305.
|
|
(24)
|
Scott, D., Galloway, S.M., Marshall, R.R., Ishidate, M.Jr, Brusick, D., Ashby, J. and Myhr, B.C. (1991). Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9. Mutation Res., 257: 147-204.
|
|
(25)
|
Wang J., Heflich R.H. and Moore M.M. (2007). A Method to Distinguish Between the De Novo Induction of Thymidine Kinase Mutants and the Selection of Pre-Existing Thymidine Kinase Mutants in the Mouse Lymphoma Assay. Mutation Res., 626 (1-2): 185-190.
|
|
(26)
|
Fischer, G.A. (1958). Studies on the Culture of Leukemic Cells In vitro. Ann. N.Y. Academy Sci., 76: 673-680.
|
|
(27)
|
Clive, D., Johnson, K.O., Spector, J.F.S., Batson, A.G. and Brown, M.M.M. (1979). Validation and Characterization of the L5178Y/TK+/– Mouse Lymphoma Mutagen Assay System. Mutation Res., 59(1): 61-108.
|
|
(28)
|
Sawyer, J., Moore, M.M., Clive, D. and Hozier, J. (1985). Cytogenetic Characterization of the L5178Y TK+/- 3.7.2C Mouse Lymphoma Cell Line, Mutation Res., 147 (5): 243-253.
|
|
(29)
|
Sawyer J.R., Moore M.M. and Hozier J.C. (1989). High-Resolution Cytogenetic Characterization of the L5178Y TK+/- Mouse Lymphoma Cell Line, Mutation Res., 214 (2): 181-193.
|
|
(30)
|
Sawyer, J.R., Binz, R.L., Wang, J. and Moore, M.M. (2006). Multicolor Spectral Karyotyping of the L5178Y TK+/--3.7.2C Mouse Lymphoma Cell Line, Environ. Mol. Mutagen., 47 (2): 127-131.
|
|
(31)
|
Fellows, M.D., McDermott, A., Clare, K.R., Doherty, A. and Aardema, M.J. (2014). The Spectral Karyotype of L5178Y TK+/- Mouse Lymphoma Cells Clone 3.7.2C and Factors Affecting Mutant Frequency at the Thymidine Kinase (TK) Locus in the Microtitre Mouse Lymphoma Assay, Environ. Mol. Mutagen., 55 (1): 35-42.
|
|
(32)
|
Storer, R.D., Jraynak, A.R., McKelvey, T.W., Elia, M.C., Goodrow, T.L. and DeLuca, J.G. (1997). The Mouse Lymphoma L5178Y TK+/- Cell Line is Heterozygous for a Codon 170 Mutation in the P53 Tumor Suppressor Gene. Mutation. Res., 373 (2): 157-165.
|
|
(33)
|
Clark L.S., Harrington-Brock, K., Wang, J., Sargent, L., Lowry, D., Reynolds, S.H. and Moore, M.M. (2004). Loss of P53 Heterozygosity is not Responsible for the Small Colony Thymidine Kinase Mutant Phenotype in L5178Y Mouse Lymphoma Cells. Mutagen., 19 (4): 263-268.
|
|
(34)
|
Skopek T.R., Liber, H.L., Penman, B.W. and Thilly, W.G. (1978). Isolation of a Human Lymphoblastoid Line Heterozygous at the Thymidine Kinase Locus: Possibility for a Rapid Human Cell Mutation Assay. Biochem. Biophys. Res. Commun., 84 (2): 411-416.
|
|
(35)
|
Honma M. (2005). Generation of Loss of Heterozygosity and its Dependency on P53 Status in Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 45 (2-3): 162-176.
|
|
(36)
|
Xia, F., Wang, X., Wang, Y.H., Tsang, N.M., Yandell, D.W., Kelsey, K.T. and Liber, H.L. (1995). Altered P53 Status Correlates with Differences in Sensitivity to Radiation-Induced Mutation and Apoptosis in Two Closely Related Human Lymphoblast Lines. Cancer. Res., 55 (1): 12-15.
|
|
(37)
|
Lorge, E., M. Moore, J. Clements, M. O Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, A. Sutter, D.J. Kirkland (2015). Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. (Manuscript in preparation).
|
|
(38)
|
Lloyd M. and Kidd D. (2012). The Mouse Lymphoma Assay. Springer Protocols: Methods in Molecular Biology 817, Genetic Toxicology Principles and Methods, ed. Parry and Parry, Humana Press. ISBN, 978-1-61779-420-9, 35-54.
|
|
(39)
|
Mei N., Guo X. and Moore M.M. (2014). Methods for Using the Mouse Lymphoma Assay to Screen for Chemical Mutagenicity and Photo-Mutagenicity. In: Optimization in Drug Discover: In vitro Methods: Yan Z and Caldwell(Eds) , 2nd Edition, GW; Humana Press, Totowa, NJ.
|
|
(40)
|
Liber H.L. and Thilly W.G. (1982). Mutation Assay at the Thymidine Kinase Locus in Diploidhuman Lymphoblasts. Mutation Res., 94 (2): 467-485.
|
|
(41)
|
Coecke, S., Balls, M., Bowe, G., Davis, J., Gstraunthaler, G., Hartung, T., Hay, R., Merten, OW., Price, A., Schechtman, L., Stacey, G. and Stokes, W. (2005). Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice. ATLA, 33 (3): 261-287.
|
|
(42)
|
Moore M.M. and Howard B.E. (1982). Quantitation of Small Colony Trifluorothymidine-Resistant Mutants of L5178Y/TK+/- Mouse Lymphoma Cells in RPMI-1640 Medium, Mutation Res., 104 (4-5): 287-294.
|
|
(43)
|
Ames B.N., McCann J. and Yamasaki E. (1975). Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test. Mutation Res., 31 (6): 347-364.
|
|
(44)
|
Maron D.M. and Ames B.N. (1983). Revised Methods for the Salmonella Mutagenicity Test. Mutation Res., 113 (3-4): 173-215.
|
|
(45)
|
Natarajan, A.T., Tates, A.D, Van Buul, P.P.W., Meijers, M. and De Vogel, N. (1976). Cytogenetic Effects of Mutagens/Carcinogens After Activation in a Microsomal System In vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes. Mutation Res., 37 (1): 83-90.
|
|
(46)
|
Matsuoka A., Hayashi M. and Ishidate M. Jr. (1979). Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix In vitro. Mutation Res., 66 (3): 277-290.
|
|
(47)
|
Ong T.M., et al. (1980). Differential Effects of Cytochrome P450-Inducers on Promutagen Activation Capabilities and Enzymatic Activities of S-9 from Rat Liver, J. Environ. Pathol. Toxicol., 4 (1): 55-65
|
|
(48)
|
Elliott, B.M., Combes, R.D., Elcombe, C.R., Gatehouse, D.G., Gibson, G.G., Mackay, J.M. and Wolf, R.C. (1992). Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-Induced S9 in In vitro Genotoxicity Assays. Mutagen., 7 (3): 175-177.
|
|
(49)
|
Matsushima, T., Sawamura, M., Hara, K. and Sugimura, T. (1976). A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems. In: In vitro Metabolic Activation in Mutagenesis Testing. de Serres F.J., et al. (Eds, Elsevier, North-Holland, pp. 85-88.
|
|
(50)
|
Galloway S.M., et al. (1994). Report from Working Group on In vitro Tests for Chromosomal Aberrations. Mutation Res., 312 (3): 241-261.
|
|
(51)
|
Johnson T.E., Umbenhauer D.R. and Galloway S.M. (1996). Human Liver S-9 Metabolic Activation: Proficiency in Cytogenetic Assays and Comparison with Phenobarbital/Beta-Naphthoflavone or Aroclor 1254 Induced Rat S-9, Environ. Mol. Mutagen., 28 (1): 51-59.
|
|
(52)
|
UNEP (2001). Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP).
|
|
(53)
|
Krahn D.F., Barsky F.C. and McCooey K.T. (1982). CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids. In: Genotoxic Effects of Airborne Agents Tice R.R., Costa D.L.and Schaich K.M. (Eds.). New York, Plenum, pp. 91-103.
|
|
(54)
|
Zamora, P.O., Benson, J.M., Li, A.P. and Brooks, A.L. (1983). Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay. Environ. Mutagen., 5 (6): 795-801.
|
|
(55)
|
Asakura M., Sasaki T., Sugiyama T., Arito H., Fukushima, S. and Matsushima, T. (2008). An Improved System for Exposure of Cultured Mammalian Cells to Gaseous Compounds in the Chromosomal Aberration Assay. Mutation Res., 652 (2): 122-130.
|
|
(56)
|
Arlett C.F., et al. (1989). Mammalian Cell Gene Mutation Assays Based upon Colony Formation. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Ed.), CambridgeUniversity Press, pp. 66-101.
|
|
(57)
|
Morita T., Honma M. and Morikawa K. (2012). Effect of Reducing the Top Concentration Used in the In vitro Chromosomal Aberration Test in CHL Cells on the Evaluation of Industrial Chemical Genotoxicity. Mutation Res., 741 (1-2): 32-56.
|
|
(58)
|
Brookmire L., Chen J.J. and Levy D.D. (2013). Evaluation of the Highest Concentrations Used in the In vitro Chromosome Aberrations Assay. Environ. Mol. Mutagen., 54 (1): 36-43.
|
|
(59)
|
USFDA (2012). International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Elérhető a következő címen:[https://www.federalregister.gov/a/2012-13774].
|
|
(60)
|
Honma M. and Hayashi M. (2011). Comparison of In vitro Micronucleus and Gene Mutation Assay Results for P53-Competent Versus P53-Deficient Human Lymphoblastoid Cells. Environ. Mol. Mutagen., 52 (5): 373-384.
|
|
(61)
|
Moore-Brown, M.M., Clive, D., Howard, B.E., Batson, A.G. and Johnson, K.O. (1981). The Utilization of Trifluorothymidine (TFT) to Select for Thymidine Kinase-Deficient (TK-/-) Mutants from L5178Y/TK+/- Mouse Lymphoma Cells, Mutation Res., 85 (5): 363-378.
|
|
(62)
|
Liber H.L., Yandell D.W. and Little J.B. (1989). A Comparison of Mutation Induction at the TK and HRPT Loci in Human Lymphoblastoid Cells; Quantitative Differences are Due to an Additional Class of Mutations at the Autosomal TK locus. Mutation Res., 216 (1): 9-17.
|
|
(63)
|
Furth E.E., Thilly, W.G., Penman, B.W., Liber, H.L. and Rand, W.M. (1981). Quantitative Assay for Mutation in Diploid Human Lymphoblasts Using Microtiter Plates. Anal. Biochem., 110 (1): 1-8.
|
|
(64)
|
Hayashi, M, Dearfield, K., Kasper, P., Lovell, D., Martus, H. J. and Thybaud, V. (2011). Compilation and Use of Genetic Toxicity Historical Control Data, Mutation Res., 723 (2): 87-90.
|
|
(65)
|
Ryan T.P. (2000). Statistical Methods for Quality Improvement. John Wiley and Sons, New York 2nd Edition.
|
|
(66)
|
OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 199.), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(67)
|
Fleiss J.L., Levin B. and Paik M.C. (2003). Statistical Methods for Rates and Proportions, Third Edition, New York: John Wiley & Sons.
|
1. függelék
FOGALOMMEGHATÁROZÁSOK
Aneugén
: olyan vegyi anyag vagy folyamat, amely a mitózissal vagy meiózissal történő sejtosztódási ciklus elemeivel kölcsönhatásba lépve a sejtek vagy organizmusok aneuploidiáját eredményezi.
Aneuploidia
: a normális diploid (vagy haploid) kromoszómaszámtól egyetlen kromoszómával vagy egynél több, szám feletti kromoszómával, de nem teljes kromoszómakészlettel (kromoszómakészletekkel) (poliploidia) való bármely eltérés.
Báziscserét okozó mutagének
: azok a vegyi anyagok, amelyek a bázispárok szubsztitúcióját okozzák a DNS-ben.
Vegyi anyag
: anyag vagy keverék.
Kolóniaképző képesség
: azon alacsony sűrűséggel leültetett sejtek százalékaránya, amelyek képesek megszámolható teleppé fejlődni.
Klasztogén
: bármely olyan vegyi anyag vagy folyamat, amely sejtek vagy szervezetek populációjában szerkezeti kromoszóma-rendellenességeket okoz.
Citotoxicitás
: az e vizsgálati módszerbe tartozó vizsgálatok esetében a citotoxicitás az egérlimfóma-vizsgálat esetében a relatív teljes növekedés (RTG), a TK6 vizsgálat esetében a relatív túlélés (RS) csökkenését jelenti.
Forward (előremutató) mutáció
: a mutáns alak szülőktől származó típusától eltérő olyan génmutáció, amely a kódolt protein enzimaktivitásának vagy funkciójának változását vagy elvesztését eredményezi.
Frameshiftet okozó mutagének
: azok a vegyi anyagok, amelyek egy vagy több bázispár addícióját vagy delícióját okozzák a DNS-ben.
Genotoxikus
: általános kifejezés, amely magába foglalja a DNS- vagy kromoszómakárosodás valamennyi típusát, köztük a DNS-töréseket, adduktok képződését, átrendeződéseket, mutációkat, kromoszómaaberrációkat és aneuploidiát. A genotoxikus hatások nem minden típusa eredményez mutációkat vagy stabil kromoszómakárosodást.
Mitotikus rekombináció
: a mitózis során a homológ kromatidok rekombinációja, ami kettős szálú DNS-töréseket vagy a heterozigótaság elvesztését okozhatja.
Mutagén
: örökletes elváltozást idéz elő a génekben lévő DNS-bázispár-szekvenciá(k)ban vagy a kromoszómák szerkezetében (kromoszómaaberrációk).
Mutációs gyakoriság
: a megfigyelt mutáns sejtek száma elosztva az életképes sejtek számával.
Fenotípusos expresszió ideje
: a kezelést követő időtartam, amely során a genetikai változás rögzül a genomban, és a korábban meglévő géntermékek olyan mértékben kiürülnek, hogy a fenotípus-tulajdonság megváltozik.
Relatív túlélés
: A relatív túlélést a kezelés okozta citotoxicitás mutatójaként használják a TK6 vizsgálatban. A relatív túlélés a közvetlenül a sejtkezelést követően leültetett sejtek relatív kolóniaképző képessége (CE) kiigazítva a kezelés során a sejtszámban bekövetkezett veszteséggel, és összevetve a negatív kontroll kolóniaképző képességével.
Relatív szuszpenziós növekedés (RSG)
: Az egérlimfóma-vizsgálatban a vizsgált sejtkultúra relatív teljes kétnapos szuszpenziós növekedése összevetve a negatív/oldószeres kontroll teljes kétnapos szuszpenziós növekedésével (Clive and Spector, 1975). A relatív szuszpenziós növekedésnek magában kell foglalnia a vizsgált kultúra által a kezelési időszakban a negatív/oldószeres kontrollhoz viszonyítva elért relatív növekedést.
Relatív teljes növekedés (RTG)
: Az egérlimfóma-vizsgálat esetében a relatív teljes növekedést a kezelés okozta citotoxicitás mutatójaként használják. A relatív teljes növekedés a vizsgált kultúrák relatív (a vivőanyagos kontrollhoz viszonyított) növekedését méri a vizsgálat kezelési, kétnapos kifejeződési és a mutánsok kiválasztására szolgáló kolóniaképzési szakaszában. Az egyes vizsgált kultúrák relatív teljes növekedését megszorozzák a vizsgált kultúra mutánsok kiválasztásának idején mért relatív kolóniaképző képességével, és a negatív/oldószeres kontroll kolóniaképző képességének arányában fejezik ki (Clive and Spector, 1975).
S9 májfrakciók
: 9 000 g centrifugálás utáni májhomogenátum felülúszója, azaz a nyers májextraktum.
S9 keverék
: az S9 májfrakció és a metabolikus enzimaktivitáshoz szükséges kofaktorok keveréke.
Szuszpenziós növekedés
: A sejtek számának növekedési faktora az egérlimfóma-vizsgálat kezelési és kifejeződési szakaszában. A szuszpenziós növekedés meghatározásához rövid idejű (3–4 órás) kezelésnél megszorozzák az 1. napon tapasztalt növekedési faktort a 2. napon észlelt növekedési faktorral. 24 órás kezelés alkalmazása esetén a szuszpenziós növekedés a 24 órás kezelés során mért növekedési faktor és az 1. és 2. expressziós napon mért növekedési faktorok szorzata.
Oldószeres kontroll
: a csak a vizsgálati vegyi anyag feloldására használatos oldószert tartalmazó kontrolltenyészetek meghatározására szolgáló általános kifejezés.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Nem kezelt kontrollok
: nem kezelt (azaz sem a vizsgálati vegyi anyaggal, sem oldószerrel nem kezelt), de a vizsgálati vegyi anyagot befogadó tenyészetekkel azonos módon feldolgozott tenyészetek.
2. függelék
KÉPLETEK
Citotoxicitás
Az egérlimfóma-vizsgálat mindkét (agarlemezes és mikrotiterlemezes) változatánál
A citotoxicitást a relatív teljes növekedés (RTG) alapján határozzák meg, amely magában foglalja a kétnapos expressziós időszak során elért relatív szuszpenziós növekedést (RSG) és a mutánsok kiválasztásának idején meghatározott relatív kolóniaképző képességet (RCE). Az RTG, az RSG és az RCE értékét százalékos arányszámként fejezik ki.
A relatív szuszpenziós növekedés (RSG) kiszámítása: Az 1. szuszpenziós növekedés (SG1) az 1. napon a 0. naphoz képest megfigyelt növekedési sebesség (1. napi sejtkoncentráció/0. napi sejtkoncentráció), a 2. szuszpenziós növekedés (SG2) pedig a 2. napon az 1. naphoz képest tapasztalt növekedési sebesség (2. napi sejtkoncentráció/1. napi sejtkoncentráció). Az RSG a kezelt kultúra teljes szuszpenziós növekedése (SG1 × SG2) a kezeletlen/oldószeres kontrollhoz viszonyítva. Azaz: RSG = [SG1(vizsgált) × SG2(vizsgált)] / [SG1(kontroll) × SG2(kontroll)] Az SG1 értékét a sejtkezelés kezdetén használt induló sejtkoncentráció alapján kell meghatározni. Ezzel megállapíthatók a vizsgált kultúrá(k)ban a sejtkezelés során a citotoxicitás terén bekövetkezett változások.
Az RCE a vizsgált kultúra relatív kolóniaképző képességének és a kezeletlen/oldószeres kontroll relatív kolóniaképző képességének aránya, amelyet a mutánsok kiválasztásának időpontjában határoznak meg.
Relatív teljes növekedés (RTG):
RTG=RSG × RCE
TK6
Relatív túlélés (RS):
A citotoxicitást a relatív túlélés alapján értékelik, ami a közvetlenül a kezelést követően leültetett sejtek kolóniaképző képessége (CE), kiigazítva a kezelés során a sejtszámban bekövetkezett veszteséggel, és összevetve a negatív kontrollok (túlélési arányuk 100 %-nak tekintendő) kolóniaképző képességével. A kezelés során végbement sejtpusztulás miatt szükséges kiigazítás az alábbi módon számítható ki:

A vizsgálati vegyi anyaggal kezelt tenyészetek relatív túlélése az alábbi módon határozható meg:

Mutációs gyakoriság az egérlimfóma- és a TK6 vizsgálat esetében
A mutációs gyakoriság (MF) a szelektív közegben növő mutáns telepek kolóniaképző képességének (CEM) és a nem szelektív közegben fejlődő telepek kolóniaképző képességének (CEV) aránya, amelyet a mutánsok kiválasztásának időpontjában határoznak meg. Azaz: MF=CEM/CEV. E két kolóniaképző készséget az agarlemezes és a mikrotiterlemezes kolóniaképző módszer esetén az alábbiakban leírtak szerint kell meghatározni.
Az egérlimfóma-vizsgálat agarlemezes változata: Az egérlimfóma-vizsgálat lágyagarlemezes változatában a szelekciós lemezen lévő telepek száma (CM) és a nem szelekciós, vagyis a kolóniaképző képesség (életképesség) meghatározására szolgáló lemezen lévő telepek száma (CV) a kolóniák közvetlen megszámlálásával állapítható meg. Amikor 600 sejtet ültetnek le a szelekciós lemezek (CM) és a nem szelekciós, vagyis a kolóniaképző képesség (életképesség) meghatározására szolgáló lemezek (CV) kolóniaképző képességének (CE) meghatározásához, és 3 × 106 sejtet használnak a mutánsok kiválasztására, akkor:
CEM = CM / (3 × 106) = (CM / 3) × 10-6
CEV = CV / 600
Mikrotiterlemezes egérlimfóma- és a TK6 vizsgálat:
Az egérlimfóma-vizsgálat mikrotiterlemezes változatában a CM és a CV értékét a mikrotiterlemezek teljes lyukszámának (TW) és a lyukankénti telepek feltételezhető számának (P) szorzataként határozzák meg.
CM = PM × TWM
CV = PV × TWV
A Poisson-elosztás nulla valószínűségi változójából kiindulva (Furth és munkatársai, 1981) a P-t a következő módon kapjuk meg:
P = - ln (EW / TW)
Ahol az EW az üres lyukak, a TW pedig az összes lyuk száma. Ebből következően:
CEM = CM / TM = (PM × TWM) / TM
CEV = CV / TV = (PV × TWV) / TV
Az egérlimfóma-vizsgálat mikrotiterlemezes változatában a kis és nagy telepek mutációs gyakoriságát azonos módon határozzák meg, a kis és nagy telepek üres lyukainak száma alapján.
A TK6 vizsgálatban a kis és nagy telepek mutációs gyakoriságát a gyorsan és a lassan megjelenő mutánsok száma alapján állapítják meg.
B.68. RÖVID EXPOZÍCIÓS IDEJŰ IN VITRO VIZSGÁLATI MÓDSZER i. A SÚLYOS SZEMKÁROSODÁST OKOZÓ VEGYI ANYAGOK ÉS ii. A SZEMIRRITÁCIÓ VAGY A SÚLYOS SZEMKÁROSODÁS TEKINTETÉBEN BESOROLÁST NEM IGÉNYLŐ VEGYI ANYAGOK AZONOSÍTÁSÁRA
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 491. vizsgálati iránymutatásában (2017) leírt módszerrel. A rövid expozíciós idejű (STE) vizsgálati módszer egy in vitro eljárás, amely bizonyos helyzetekben és meghatározott korlátok mellett alkalmazható a súlyos szemkárosodást okozó, valamint a súlyos szemkárosodás vagy szemirritáció tekintetében besorolást nem igénylő vegyi anyagok (anyagok és keverékek) veszélyességi osztályba sorolására és címkézésére, az Egyesült Nemzetek Szervezete (ENSZ) által kidolgozott, vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszerében (GHS) (1), valamint az Európai Unió (EU) anyagok és keverékek osztályozásáról, címkézéséről és csomagolásáról szóló 1272/2008/EK rendeletében (CLP-rendelet) (68) meghatározottak szerint.
|
|
2.
|
A vegyi anyagok szemet érintő veszélyességi potenciáljának értékelése évek óta elsősorban egy nyulakon végzett in vivo szemvizsgálattal (az OECD 405. vizsgálati iránymutatásával egyenértékű B.5. vizsgálati módszerrel (8)) történik. Általánosan elfogadott, hogy az előre látható jövőben egyetlen alternatívaként használható in vitro vizsgálat sem tudja majd teljes körűen helyettesíteni a nyulakon végzett szemvizsgálatot a különböző kémiai osztályoknál felmerülő súlyos szemkárosodási/szemirritációs válaszreakciók teljes skálájának előrejelzésében. Előfordulhat azonban, hogy egy-egy (többszintű) vizsgálati stratégián belüli alternatív vizsgálati módszerek stratégiai kombinációja teljes körűen helyettesíteni tudja a nyulakon végzett szemvizsgálatot (2). A felülről építkező megközelítést olyan vegyi anyagok vizsgálatához dolgozták ki, amelyek a rendelkezésre álló információk alapján várhatóan rendkívül irritáló hatásúak lehetnek, vagy súlyos szemkárosodást idézhetnek elő. Ezzel szemben az alulról építkező megközelítés olyan vegyi anyagok vizsgálatához alkalmazandó, amelyek a meglévő információk alapján várhatóan nem okoznak a besorolás szükségességéhez elegendő szemirritációt. Bár a rövid expozíciós idejű vizsgálati módszer nem helyettesíti teljes körűen a nyulakon végzett in vivo szemvizsgálatot, többszintű vizsgálati stratégia – például felülről vagy alulról építkező megközelítés – részeként, szabályozói követelmények szerinti osztályozásra és címkézésre alkalmazva azonban használható arra, hogy további vizsgálatok szükségessége nélkül azonosítsa i. a súlyos szemkárosodást okozó (vagyis az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) vegyi anyagokat és ii. a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő (azaz az ENSZ-GHS/CLP-rendelet „Kategória nélküli” csoportjába tartozó) vegyi anyagokat (kivéve a rendkívül illékony anyagokat és a felületaktív anyagoktól eltérő szilárd vegyi anyagokat) (1) (2). Azok a vegyi anyagok azonban, amelyek a rövid expozíciós idejű vizsgálati módszer alapján előreláthatólag nem okoznak súlyos szemkárosodást (ENSZ-GHS/CLP-rendelet szerinti 1. kategória), vagy az ENSZ-GHS/CLP-rendelet „Kategória nélküli” csoportjába tartoznak (nem váltanak ki sem szemirritációt, sem súlyos szemkárosodást), a végleges osztályba sorolásukhoz további vizsgálatot tesznek szükségessé. Ezenfelül a rövid expozíciós idejű vizsgálati módszernek az ENSZ-GHS/CLP-rendelettől eltérő osztályozási rendszeren belüli, alulról építkező megközelítés keretében történő alkalmazásáról egyeztetni kell a megfelelő szabályozó hatóságokkal. A legmegfelelőbb vizsgálati módszer kiválasztásához és e vizsgálati módszer alkalmazásához a súlyos szemkárosodási és bőrirritációs vizsgálatok és értékelések integrált megközelítéseiről szóló OECD-iránymutatás nyújt támpontokat (14).
|
|
3.
|
E vizsgálati módszer ismerteti azokat az eljárásokat, amelyek felhasználhatók a vizsgálati vegyi anyagok szemet érintő veszélyességi potenciáljának értékelésére az alapján, hogy képesek-e a rövid expozíciós idejű vizsgálati rendszerben citotoxicitást kiváltani. A vegyi anyagoknak a szaruhártya felhámsejtjeire gyakorolt citotoxikus hatása fontos hatásmechanizmus, ami a szaruhártya felhámjának károsodásához és szemirritációhoz vezet. A rövid expozíciós idejű vizsgálati módszerben a sejtek életképességét az alapján mérik, hogy az MTT (3–(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólium-bromid), más néven tiazolil kék tetrazólium-bromid vitális festéket élő sejtek enzimatikus úton kék formazán kristállyá alakítják, amelynek mennyiségét a sejtekből történő kivonás után meghatározzák (3). Az ily módon megállapított sejtéletképességet összevetik az oldószeres kontrollnál mért életképességgel (ez a relatív életképesség), majd ennek alapján megbecsülik a vizsgálati vegyi anyag szemet érintő veszélyességi potenciálját. Egy adott vizsgálati vegyi anyagot akkor sorolnak az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába, ha 5 %-os és 0,05 %-os koncentrációja egyaránt 70 %-os vagy az alatti (≤ 70 %) sejtéletképességet idéz elő. Ezzel szemben akkor jelzik előre, hogy valamely vizsgálati vegyi anyag az ENSZ-GHS/CLP-rendelet szerint „Kategória nélküli”, ha 5 %-os és 0,05 %-os koncentrációja egyaránt 70 %-ot meghaladó (> 70 %) sejtéletképességet eredményez.
|
|
4.
|
E vizsgálati módszer esetében a „vizsgálati vegyi anyag” kifejezés a vizsgálat tárgyát képező vegyi anyagra vonatkozik, és nem a rövid expozíciós idejű vizsgálati módszer anyagok és/vagy keverékek vizsgálatára való alkalmazhatóságához kapcsolódik. A fogalmak meghatározása a függelékben található.
|
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
5.
|
Ez a vizsgálati módszer a Kao Corporation által kidolgozott protokollon alapul (4), amelyen két különböző validálási vizsgálatot végeztek el: az egyiket a japán állatkísérletek alternatív módszereivel foglalkozó társaság validálásért felelős bizottsága (JSAAE) (5), a másikat pedig a japán alternatív módszerek validálásával foglalkozó központ (JaCVAM) (6) hajtotta végre. A validálási tanulmány jelentései és vizsgálati módszer felülvizsgálatára vonatkozó háttérdokumentumok alapján a NICEATM/ICCVAM szakértői vizsgálatot végzett (7).
|
|
6.
|
A súlyos szemkárosodást okozó (az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) (1) vegyi anyagok (anyagok és keverékek) azonosítására történő alkalmazásakor a rövid expozíciós idejű vizsgálati módszer 125 (anyagokat és keverékeket is tartalmazó) vegyi anyag vizsgálata során nyert adatainak általános pontossága 83 % (104/125), álpozitív aránya 1 % (1/86), álnegatív aránya pedig 51 % (20/39) a nyulakon végzett in vivo szemvizsgálat adataival való összevetéskor (7). Az álnegatív eredmények aránya ebben az összefüggésben nem kritikus jelentőségű, mivel minden 5 %-os koncentrációban legfeljebb 70 %-os, és 0,05 %-os koncentrációban 70 %-nál nagyobb sejtéletképességet kiváltó vizsgálati vegyi anyag vizsgálatára sor kerülne más, megfelelően validált in vitro vizsgálati módszerekkel, vagy – a szabályozási követelményektől függően, és a jelenleg ajánlott lépcsőzetes vizsgálati stratégiával és bizonyítékok súlyozásán alapuló megközelítéssel összhangban (1) (8) – utolsó lehetőségként nyulakon végzett in vivo szemvizsgálattal. Elsősorban egy összetevőből álló anyagok vizsgálatára került sor, bár a keverékek vizsgálatára vonatkozóan is rendelkezésre áll korlátozott mennyiségű információ. A vizsgálati módszer mindazonáltal a több összetevőből álló anyagok és keverékek vizsgálatára technikailag alkalmazható. E vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt azonban meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény. A rövid expozíciós idejű vizsgálati módszer nem mutat egyéb hiányosságokat az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó vizsgálati vegyi anyagok azonosítására történő alkalmazásakor. A vizsgálók mérlegelhetik e vizsgálati módszer vizsgálati vegyi anyagon való alkalmazását, amely esetben az 5 %-os és 0,05 %-os koncentrációban egyaránt legfeljebb 70 %-os sejtéletképességet további vizsgálat nélkül az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába sorolandó súlyos szemkárosodást okozó reakcióra utaló jelként kell elfogadni.
|
|
7.
|
A szemirritáció és a súlyos szemkárosodás tekintetében besorolást nem igénylő (az ENSZ-GHS/CLP-rendelet „Kategória nélküli” csoportjába tartozó) vegyi anyagok (anyagok és keverékek) azonosítására történő alkalmazásakor a rövid expozíciós idejű vizsgálati módszer 130 (anyagokat és keverékeket is tartalmazó) vegyi anyag vizsgálata során nyert adatainak általános pontossága 85 % (110/130), álnegatív aránya 12 % (9/73), álpozitív aránya pedig 19 % (11/57) a nyulakon végzett in vivo szemvizsgálat adataival való összevetéskor (7). Amikor a rendkívül illékony anyagok és a felületaktív anyagoktól eltérő szilárd vegyi anyagok nem szerepelnek az adatbázisban, a vizsgálati módszer általános pontossága 90 %-ra (92/102), álnegatív aránya 2 %-ra (1/54), álpozitív aránya pedig 19 %-ra (9/48) javul (7). Ebből következően a szemirritáció és a súlyos szemkárosodás tekintetében besorolást nem igénylő (az ENSZ-GHS/CLP-rendelet „Kategória nélküli” csoportjába tartozó) vizsgálati vegyi anyagok azonosítására történő alkalmazásakor a rövid expozíciós idejű vizsgálati módszer potenciális hiányossága az álnegatív eredmények magas aránya i. a 6 kPa-t meghaladó gőznyomású rendkívül illékony anyagok, valamint ii. a felületaktív anyagoktól eltérő szilárd vegyi anyagok (anyagok és keverékek) és a kizárólag felületaktív anyagokból álló keverékek körében. Ezeket a vegyi anyagokat kizárják a rövid expozíciós idejű vizsgálati módszer alkalmazási köréből (7).
|
|
8.
|
A 6. és a 7. pontban említett vegyi anyagokon túlmenően a rövid expozíciós idejű vizsgálati módszer adatbázisa magában foglalja 40 keverék belső vizsgálatokból származó adatait is, amelyek pontossága 88 % (35/40), álpozitív aránya 50 % (5/10), álnegatív aránya pedig 0 % (0/30) a Draize-féle in vivo szemvizsgálathoz képest, az ENSZ-GHS/CLP-rendelet szerinti osztályozási rendszerben besorolást nem igénylő keverékek előrejelzése terén (9). Ezért a rövid expozíciós idejű vizsgálati módszer alulról építkező megközelítés részeként alkalmazható az ENSZ-GHS/CLP-rendelet „Kategória nélküli” csoportjába tartozó keverékek azonosítására, kivételt képeznek ez alól – a szilárd anyagokra vonatkozó korlátozás kiterjesztésével – a csak felületaktív anyagokból álló keverékektől eltérő szilárd keverékek. Ezenfelül a 6 kPa-t meghaladó gőznyomású anyagokat tartalmazó keverékeket az esetleges alulkategorizálás elkerülése érdekében körültekintően kell értékelni, és előrejelzett besorolásukat eseti alapon kell megindokolni.
|
|
9.
|
Az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó, de a 2., 2A. vagy 2B. kategóriába alulosztályozott vegyi anyagok, valamint az ENSZ-GHS/CLP-rendelet szerinti „Kategória nélküli” csoportba tartozó, de a 2., 2A. vagy 2B. kategóriába felülosztályozott vegyi anyagok jelentős száma miatt a rövid expozíciós idejű vizsgálati módszer nem alkalmazható a vizsgálati vegyi anyagok ENSZ-GHS/CLP-rendelet szerinti 2. kategóriába tartozóként, illetve ENSZ-GHS/CLP-rendelet szerinti 2A. kategóriába (szemirritáló hatású) vagy 2B. kategóriába (enyhén szemirritáló hatású) tartozóként történő azonosítására (7). E célból egy másik megfelelő módszerrel végzett további vizsgálatra lehet szükség.
|
|
10.
|
A rövid expozíciós idejű vizsgálati módszer olyan vizsgálati vegyi anyagokhoz alkalmazható, amelyek oldhatók vagy legalább 5 perc alatt egyenletesen felszuszpendálhatók fiziológiás sóoldatban, 5 % dimetil-szulfoxidot (DMSO) tartalmazó sóoldatban vagy ásványolajban. A rövid expozíciós idejű vizsgálati módszer nem alkalmazható olyan vizsgálati vegyi anyagokhoz, amelyek nem oldhatók vagy egyenletesen nem szuszpendálhatók legalább 5 perc alatt fiziológiás sóoldatban, 5 % DMSO-t tartalmazó sóoldatban vagy ásványolajban. A rövid expozíciós idejű vizsgálati módszerben az ásványolaj használatát a rövid expozíciós idő teszi lehetővé. Ennek köszönhetően a rövid expozíciós idejű vizsgálati módszer alkalmas vízben oldhatatlan vizsgálati vegyi anyagok (pl. hosszú láncú zsíralkoholok vagy ketonok) szemet érintő veszélyességi potenciáljának előrejelzésére, amennyiben azok elegyednek legalább a három fent javasolt oldószer egyikében (4).
|
|
11.
|
E vizsgálati módszer esetében a „vizsgálati vegyi anyag” kifejezés a vizsgálat tárgyát képező vegyi anyagra vonatkozik (69) és nem a rövid expozíciós idejű vizsgálati módszer anyagok és/vagy keverékek vizsgálatára való alkalmazhatóságához kapcsolódik.
|
A VIZSGÁLAT ELVE
|
12.
|
A rövid expozíciós idejű vizsgálati módszer egy citotoxicitáson alapuló in vitro vizsgálat, amelyet 96 lyukú polikarbonát mikrolemezen tenyésztett, konfluens egyrétegű SIRC-sejteken (a Statens Serum Institut által előállított nyúl szaruhártya sejteken) hajtanak végre (4). A sejteket öt percen át kezelik a vizsgálati vegyi anyaggal, majd MTT-vizsgálattal mennyiségileg meghatározzák a citotoxicitást, amit a SIRC-sejtek relatív életképességeként fejeznek ki (4). A sejtéletképesség csökkenése alapján jelzik előre a szemkárosodáshoz vezető potenciális káros hatásokat.
|
|
13.
|
A vizsgálati jelentés szerint a nyúl szemébe csepegtetett oldatok 80 %-a három–négy percen belül kiürül a kötőhártyazsákon keresztül, míg az emberi szembe csepegtetett oldatok több mint 80 %-a egy–két percen belül kiürül (10). A rövid expozíciós idejű vizsgálati módszer igyekszik közelíteni egymáshoz ezeket az expozíciós időket, és a citotoxicitás mint végpont révén értékeli, hogy a vizsgálati vegyi anyaggal történő ötperces kezelésüket követően milyen mértékű károsodás keletkezett a SIRC-sejtekben.
|
A JÁRTASSÁG BIZONYÍTÁSA
|
14.
|
A vizsgálati módszerben ismertetett rövid expozíciós idejű eljárás rutinszerű alkalmazása előtt a laboratóriumoknak az 1. táblázatban ajánlott tizenegy anyag megfelelő besorolásával igazolniuk kell szakmai jártasságukat. Ezeket az anyagokat úgy választották ki, hogy a nyulakon végzett in vivo szemvizsgálat (405. vizsgálati iránymutatás) eredményei és az ENSZ-GHS/CLP-rendelet szerinti osztályozási rendszer (1) alapján a súlyos szemkárosodási vagy szemirritációs válaszreakciók teljes tartománya szempontjából reprezentatívak legyenek. A kiválasztás másik kritériuma az volt, hogy az anyagok kereskedelmi forgalomban beszerezhetők legyenek, azokra vonatkozóan magas minőségű in vivo referenciaadatok álljanak rendelkezésre, és a rövid expozíciós idejű vizsgálati módszerból származó, magas minőségű in vitro adatok álljanak rendelkezésre (3). Ha a jegyzékben szereplő valamely anyag nem áll rendelkezésre, illetve ha indokolható, megfelelő in vivo és in vitro referenciaadatokkal rendelkező más anyag is használható, amennyiben kiválasztása az itt ismertetett kritériumok szerint történik.
1. táblázat
A jártassági tesztanyagok jegyzéke
|
Anyag
|
CAS-szám
|
Kémiai osztály (70)
|
Halmazállapot
|
In vivo ENSZ-GHS/CLP kat. (71)
|
Rövid expozíciós idejű vizsgálat oldószere
|
Rövid expozíciós idejű vizsgálat ENSZ-GHS/CLP kat.
|
|
Benzalkónium-klorid (10 %, vizes oldat)
|
8001-54-5
|
Óniumvegyület
|
Folyadék
|
1. kategória
|
Sóoldat
|
1. kategória
|
|
Triton X-100 (100 %)
|
9002-93-1
|
Éter
|
Folyadék
|
1. kategória
|
Sóoldat
|
1. kategória
|
|
Acid Red 92
|
18472-87-2
|
Heterociklikus vegyület; Brómvegyület; Klórvegyület
|
Szilárd
|
1. kategória
|
Sóoldat
|
1. kategória
|
|
Nátrium-hidroxid
|
1310-73-2
|
Lúg; Szervetlen vegyi anyag
|
Szilárd
|
1. kategória (72)
|
Sóoldat
|
1. kategória
|
|
Butirolakton
|
96-48-0
|
Lakton; Heterociklikus vegyület
|
Folyadék
|
2A. kategória (CLP szerinti 2. kategória)
|
Sóoldat
|
Előrejelzés nem végezhető
|
|
1-oktanol
|
111-87-5
|
Alkohol
|
Folyadék
|
2A./B. (73) kategória (CLP szerinti 2. kategória)
|
Ásványolaj
|
Előrejelzés nem végezhető
|
|
Ciklopentanol
|
96-41-3
|
Alkohol; Szénhidrogén, ciklikus
|
Folyadék
|
2A./B. (74) kategória (CLP szerinti 2. kategória)
|
Sóoldat
|
Előrejelzés nem végezhető
|
|
2-etoxietil-acetát
|
111-15-9
|
Alkohol; Éter
|
Folyadék
|
Kategória nélküli
|
Sóoldat
|
Kategória nélküli
|
|
Dodekán
|
112-40-3
|
Szénhidrogén, aciklikus
|
Folyadék
|
Kategória nélküli
|
Ásványolaj
|
Kategória nélküli
|
|
Metil-izobutil-keton
|
108-10-1
|
Keton
|
Folyadék
|
Kategória nélküli
|
Ásványolaj
|
Kategória nélküli
|
|
1,1-Dimetil-guanidin-szulfát
|
598-65-2
|
Amidin; Kénvegyület
|
Szilárd
|
Kategória nélküli
|
Sóoldat
|
Kategória nélküli
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám.
|
|
ELJÁRÁS
Egyrétegű sejttenyészet elkészítése
|
15.
|
A rövid expozíciós idejű vizsgálati módszerhez nyúl szaruhártya eredetű sejtvonalat, SIRC-et kell használni. Ajánlatos a SIRC-sejteket egy megfelelő minősítéssel rendelkező sejtbankból, például az Amerikai Fajtakultúra Gyűjteményből (CCL60) beszerezni.
|
|
16.
|
A SIRC-sejteket 10 % magzati borjú szérummal (FBS), 2 mM L-glutaminnal, 50–100 egység/ml penicillinnel és 50–100 μg/ml sztreptomicinnel kiegészített Eagle-féle minimum esszenciális tápfolyadékot (MEM) tartalmazó sejttenyésztő edényben, valamint 37 °C-os, 5 %-os CO2-tartalmú és vízgőzzel telített környezetben kell tenyészteni. A sejttenyésztő edényben konfluenssé vált sejteket tripszin-etilén-diamin-tetraecetsavas oldattal fel kell szuszpendálni, sejtkaparó használatával vagy anélkül. A rutinszerű vizsgálat megkezdése előtt a sejteket (2–3 passzálással) felszaporítják a sejttenyésztő edényben, felolvasztás után azonban legfeljebb 25 passzálásig tartják fenn.
|
|
17.
|
Ezt követően a rövid expozíciós idejű vizsgálatban való felhasználásra kész sejteket megfelelő sűrűségben előkészítik, majd kiszélesztik 96 lyukú lemezekbe. A sejtek ajánlott kiszélesztési sűrűsége lyukanként 6,0 × 103 sejt, ha a sejtek a kiszélesztést követő negyedik napon kerülnek felhasználásra, illetve lyukanként 3,0 × 103 sejt, ha a sejteket a kiszélesztést követő ötödik napon használják fel, a tenyésztéshez használt térfogat 200 μl. A rövid expozíciós idejű vizsgálathoz használt, megfelelő sűrűségben kiszélesztett sejtek a vizsgálat idejére, vagyis az átoltást követő negyedik vagy ötödik napra 80 %-ot meghaladó konfluenciával rendelkeznek majd.
|
A vizsgálati vegyi anyagok és a kontrollanyagok alkalmazása
|
18.
|
A vizsgálati vegyi anyagok feloldásához vagy szuszpendálásához alkalmazott oldószer tekintetében a fiziológiás sóoldat legyen az első választás. Amennyiben a vizsgálati vegyi anyag rosszul oldódik, vagy legalább öt percig nem lehet egyenletesen szuszpendálni a sóoldatban, akkor az 5 % DMSO-t (CAS-száma: 67-68-5) tartalmazó sóoldat legyen a második választás. Azon vizsgálati vegyi anyagok esetében, amelyek sem sóoldatban, sem 5 % DMSO-t tartalmazó sóoldatban nem oldhatók vagy nem szuszpendálhatók egyenletesen legalább öt percig, az ásványolaj (CAS-száma: 8042-47-5) legyen a harmadik választás.
|
|
19.
|
A vizsgálati vegyi anyagot 5 tömegszázalékos koncentrációban feloldják vagy egyenletesen szuszpendálják a választott oldószerben, majd tízszeres oldatsorozatot létrehozva további hígítással elkészítik 0,5 %-os és 0,05 %-os koncentrációját. Minden vizsgálati vegyi anyagot tesztelni kell 5 %-os és 0,05 %-os koncentrációban is. A 96 lyukú lemezen fenntartott sejtkultúrákat szobahőmérsékleten öt percen át kezelik a lyukanként 200 μl mennyiségű vizsgálati vegyi anyag 5 %-os vagy 0,05 %-os koncentrációjú oldatával (vagy szuszpenziójával). A vizsgálati vegyi anyagok (egy vagy több összetevőből álló anyagok, illetve keverékek) nagyjából tisztának tekintendők, ha a vizsgálati módszerben meghatározottak szerint hígítják vagy szuszpendálják őket, függetlenül a valós tisztaságuktól.
|
|
20.
|
A 16. pontban ismertetett tápoldatot kell vivőanyagos kontrollként használni az összes ismétlés valamennyi lemezén. Emellett a sejteket az összes ismétlés minden lemezén vizsgálni kell oldószeres kontrollmintákban is. A 18. pontban felsorolt oldószerekről bizonyítást nyert, hogy nem befolyásolják károsan a SIRC-sejtek életképességét.
|
|
21.
|
A rövid expozíciós idejű vizsgálati módszerben az összes ismétlés valamennyi lemezén 0,01 % nátrium-lauril-szulfátot (SLS) tartalmazó sóoldatot kell használni pozitív kontrollként. A pozitív kontroll sejtéletképességének meghatározása érdekében minden ismétlés összes lemezének tartalmaznia kell egy sóoldatos oldószeres kontrollt is.
|
|
22.
|
Egy vakpróbára is szükség van az optikai sűrűség kompenzálásának meghatározásához, a vakpróbához használt lyukak csak foszfátpuffert tartalmazó sóoldatot tartalmaznak, nincs bennük se kalcium és magnézium (PBS-), se sejt.
|
|
23.
|
Minden egyes mintát (az 5 %-os és 0,05 %-os koncentrációjú vizsgálati vegyi anyagot, a tápoldatos kontrollt, az oldószeres kontrollt és a pozitív kontrollt) minden ismétlés során triplikátumban kell vizsgálni úgy, hogy a sejteket szobahőmérsékleten öt percig kezelik a megfelelő vizsgálati vagy kontrollanyag 200 μl-ével.
|
|
24.
|
A referenciaanyagok jól használhatók egy adott vegyianyag- vagy termékosztályba tartozó ismeretlen vegyi anyagok szemirritációs hatásának értékelésére vagy valamely szemirritációs anyag relatív irritatív hatásának az irritatív hatás egy adott tartományán belüli értékelésére.
|
A sejtek életképességének mérése
|
25.
|
A kezelést követően a sejteket kétszer átmossák 200 μl foszfátpuffert tartalmazó sóoldattal, majd hozzáadnak 200 μl MTT-oldatot (0,5 mg MTT/ml tápoldat). Két óra inkubációt (37 °C, 5 % CO2) követően az MTT-oldatot leszűrik, majd az MTT-formazán kivonásához 200 μl 0,04 N izopropanolos sósavval 60 percen át kezelik sötétben és szobahőmérsékleten, végül 570 nm-en mikrotiterlemez-olvasóval megmérik az MTT-formazán oldat abszorbanciáját. A vizsgálati vegyi anyagok csak abban az esetben befolyásolják az MTT-vizsgálat eredményét (színező vagy közvetlen MTT-redukáló hatásuk révén), ha a kezelést követő átmosás után jelentős mennyiségű vizsgálati vegyi anyag marad a vizsgálati rendszerben, ami előfordul a 3D-s rekonstruált emberi szaruhártya modell vagy a rekonstruált emberi felhámmodell szövetei esetén, azonban a rövid expozíciós idejű vizsgálati módszerben használt 2D-s sejtkultúrákra nem jellemző.
|
Az eredmények értelmezése és az előrejelzési modell
|
26.
|
Ezt követően az egyes vizsgálati vegyi anyagokra vonatkozó optikai sűrűség (OD) értéket kell felhasználni a 100 %-nak vett oldószeres kontrollhoz viszonyított sejtéletképesség kiszámításához. A százalékban kifejezett relatív sejtéletképességet úgy számolják ki, hogy a vizsgálati vegyi anyag OD-értékét elosztják az oldószeres kontroll OD-értékével, miután mindkét értékből kivonták a vakpróba OD-értékét.
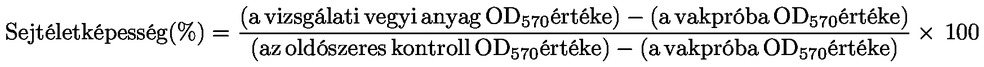
Hasonlóképpen határozzák meg az egyes oldószeres kontrollok százalékban kifejezett relatív sejtéletképességét is: minden oldószeres kontroll OD-értékét elosztják a tápoldatos kontroll OD-értékével, miután mindkét értékből kivonták a vakpróba OD-értékét.
|
|
27.
|
Három egymástól független ismétlést kell végezni, mindhármat triplikátum lyukkal (vagyis n=9). Az egyes független ismétléseken belül minden vizsgálati vegyi anyaghoz és oldószeres kontrollhoz tartozó három lyuk számtani átlaga alapján számítják ki a relatív sejtéletképesség számtani átlagát. A sejtéletképesség végső számtani átlagát a három független ismétlésből határozzák meg.
|
|
28.
|
A vizsgálati vegyi anyagok súlyos szemkárosodást okozóként (az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozóként), valamint a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő (az ENSZ-GHS/CLP-rendelet szerint kategória nélküli) vegyi anyagokként történő meghatározására szolgáló sejtéletképességi határértékek az alábbiakban szerepelnek.
|
2. táblázat
A rövid expozíciós idejű vizsgálati módszer előrejelzési modellje
|
Sejtéletképesség
|
ENSZ-GHS/CLP-rendelet szerinti besorolás
|
Alkalmazási kör
|
|
5 %-nál
|
0,05 %-nál
|
|
> 70 %
|
> 70 %
|
Kategória nélküli
|
Anyagok és keverékek, kivéve: i. a 6 kPa-t meghaladó gőznyomású rendkívül illékony anyagok (75), és ii. a felületaktív anyagoktól eltérő szilárd vegyi anyagok (anyagok és keverékek) és a kizárólag felületaktív anyagokból álló keverékek.
|
|
≤ 70 %
|
> 70 %
|
Előrejelzés nem végezhető
|
Nem alkalmazandó
|
|
≤ 70 %
|
≤ 70 %
|
1. kategória
|
Anyagok és keverékek (76)
|
Elfogadhatósági kritériumok
|
29.
|
A vizsgálat eredményei akkor ítélhetők elfogadhatónak, amikor az alábbi kritériumok mindegyike teljesül:
|
a)
|
A tápfolyadék (sejtekről leszívott tápoldat) kontroll optikai sűrűsége a vakpróba optikai sűrűségének kivonása után legalább 0,3.
|
|
b)
|
Az oldószeres kontroll tápoldatos kontollhoz viszonyított életképessége legalább 80 %. Amennyiben az egyes ismétlésekben több oldószeres kontrollt végeznek, minden kontrollnak 80 %-ot meghaladó sejtéletképességet kell eredményeznie ahhoz, hogy minősíteni lehessen az adott oldószerrel tesztelt vizsgálati vegyi anyagokat.
|
|
c)
|
A pozitív kontroll (0,01 % SLS) sejtéletképessége a dokumentált adatok átlaga körüli két szóráson belül van. A pozitív kontroll alsó és felső elfogadhatósági határértékét rendszeresen, vagyis háromhavonta, illetőleg a vizsgálatokat rendszertelenül, azaz havi egy alkalomnál ritkábban végző laboratóriumok esetében minden elfogadható vizsgálat után aktualizálni kell. Amennyiben valamely laboratórium nem végez elegendő számú kísérletet a pozitív kontrollok statisztikailag megbízható eloszlásának megállapítására, akkor elfogadható, hogy amíg az első rutinszerűen végzett belső vizsgálatok alapján meghatározzák az eloszlást, a módszer kidolgozói által megállapított alsó és felső elfogadhatósági határértékeket használják, amelyek laboratóriumuk dokumentált adatai szerint 21,1 % és 62,3 %.
|
|
d)
|
A három független ismétlés alapján meghatározott végső sejtéletképesség szórása sem a vizsgálati vegyi anyag 5 %-os, sem a 0,05 %-os koncentrációjánál nem érheti el a 15 %-ot.
Ha e kritériumok közül egy vagy több nem teljesül, az eredményeket érvénytelennek kell nyilvánítani, és másik három független ismétlést kell végezni.
|
|
ADATOK ÉS JELENTÉS
Adatok
|
30.
|
Minden ismétlés tekintetében az egyes lyukakra vonatkozó adatokat (pl. a sejtéletképességi értékeket), az összesített átlagot, a szórást és a besorolást bele kell foglalni a vizsgálati jelentésbe.
|
Vizsgálati jelentés
|
31.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálati vegyi anyag és kontrollként szolgáló anyagok
|
—
|
Egy összetevőből álló anyag: kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
—
|
több összetevőből álló anyagok, UVCB-k és keverékek: amennyiben lehetséges, az összetevők kémiai azonosságával (lásd fent), tisztaságával, mennyiségi előfordulásával és releváns fizikai-kémiai tulajdonságaival (lásd fent) jellemezve; amennyiben rendelkezésre állnak;
|
|
—
|
fizikai megjelenés, illékonyság, pH-érték, LogP-érték, molekulatömeg, kémiai osztály, valamint a vizsgálat elvégzése szempontjából releváns további fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
—
|
tisztaság, adott és a gyakorlatban megvalósítható esetben a szennyeződések kémiai azonosítója stb.;
|
|
—
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
—
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak.
|
|
|
|
Vizsgálati módszer körülményei és eljárásai
|
—
|
a megbízó, a vizsgálatot végző laboratórium és a vizsgálatvezető neve és címe;
|
|
—
|
az alkalmazott vizsgálati módszer leírása;
|
|
—
|
az alkalmazott sejtvonal, annak eredete, valamint a vizsgálathoz használt sejtek passzálásainak száma és konfluenciájának mértéke;
|
|
—
|
az alkalmazott vizsgálati eljárás részletei;
|
|
—
|
az alkalmazott ismétlések és replikátumok száma;
|
|
—
|
a vizsgálati vegyi anyag alkalmazott koncentrációi (ha eltér az ajánlottaktól);
|
|
—
|
az oldószer kiválasztásának indoklása minden egyes vizsgálati vegyi anyag esetében;
|
|
—
|
a vizsgálati vegyi anyagnak való expozíció időtartama (ha eltér az ajánlottól);
|
|
—
|
a vizsgálati eljárás bármilyen változtatásának leírása;
|
|
—
|
az alkalmazott értékelési és döntési kritériumok leírása;
|
|
—
|
hivatkozás a pozitív történeti kontrollok átlagértékére és szórásaira;
|
|
—
|
- a vizsgálati módszer végrehajtásában való laboratóriumi jártasság bizonyítása (például jártassági tesztanyagok vizsgálatával) vagy a vizsgálati módszer idővel reprodukálható végrehajtásának bizonyítása.
|
|
|
|
Eredmények
|
—
|
A replikátum lyukankénti OD-értékek, az egyes független ismétlések OD-értékeinek számtani átlaga, az egyes független ismétlések sejtéletképességi aránya és a három ismétlés alapján meghatározott átlagos sejtéletképességi arány és szórás táblázatba foglalása az összes vizsgálati vegyi anyagra és kontrollanyagra, valamint minden vizsgált koncentrációra vonatkozólag;
|
|
—
|
a tápoldatos, az oldószeres és a pozitív kontroll eredményei, amelyek igazolják a vizsgálat elfogadhatósági kritériumainak való megfelelést;
|
|
—
|
a megfigyelt egyéb hatások leírása;
|
|
—
|
az alkalmazott előrejelzési modellel/döntési kritériumokkal összhangban, az összesített eredmények alapján kialakított osztályozás.
|
|
|
SZAKIRODALOM
|
(1)
|
Egyesült Nemzetek Szervezete (ENSZ) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Elérhető a következő címen:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
|
(2)
|
Scott L, et al. (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In vitro 24, 1-9.
|
|
(3)
|
Mosmann T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to 7 Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(4)
|
Takahashi Y, et al. (2008). Development of the Short Time Exposure (STE) Test: an In vitro Eye Irritation Test Using SIRC Cells. Toxicol. In vitro 22, 760-770.
|
|
(5)
|
Sakaguchi H, et al. (2011). Validation Study of the Short Time Exposure (STE) Test to Assess the Eye Irritation Potential of Chemicals. Toxicol. In vitro 25, 796-809.
|
|
(6)
|
Kojima H, et al. (2013). Second-Phase Validation of Short Time Exposure Tests for Assessment of Eye Irritation Potency of Chemicals. Toxicol. In vitro 27, pp.1 855-1 869.
|
|
(7)
|
ICCVAM (2013). Short Time Exposure (STE) Test Method Summary Review Document, NIH. Elérhető a következő címen:[http://www.ntp.niehs.nih.gov/iccvam/docs/ocutox_docs/STE-SRD-NICEATM-508.pdf].
|
|
(8)
|
E melléklet B.5., Akut szemirritáció/szemkorrózió című fejezete.
|
|
(9)
|
Saito K, et al. (2015). Predictive Performance of the Short Time Exposure Test for Identifying Eye Irritation Potential of Chemical Mixtures.
|
|
(10)
|
Mikkelson TJ, Chrai SS and Robinson JR. (1973). Altered Bioavailability of Drugs in the Eye Due to Drug-Protein Interaction. J. Pharm. Sci.1 648-1 653.
|
|
(11)
|
ECETOC (1998). Eye Irritation Reference Chemicals Data Bank. Technical Report (No 48. (2)), Brussels, Belgium.
|
|
(12)
|
Gautheron P, et al. (1992). Bovine Corneal Opacity and Permeability Test: an In vitro Assay of Ocular Irritancy. Fundam Appl Toxicol. 18, 442-449.
|
|
(13)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34). Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(14)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 263). Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
Függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság
: a vizsgálati módszerrel kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálati módszer teljesítményének mutatója, valamint a relevanciájának egyik megítélési szempontja. E kifejezést gyakran használják az egyezés megfelelőjeként, amely egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát fejezi ki (13).
Referencia-vegyianyag
: a vizsgálati vegyi anyaggal való összevetéshez referenciaként használt anyag. A referencia-vegyianyagnak a következő tulajdonságokkal kell rendelkeznie: i. állandó és megbízható forrás(ok)ból kell származnia; ii. a vizsgálati vegyi anyagok osztályához hasonló szerkezettel és funkcionalitással kell rendelkeznie; iii. ismert fizikai-kémiai tulajdonságokkal kell rendelkeznie; iv. rendelkezésre kell állnia az ismert hatásait alátámasztó adatoknak; és v.. a kívánt hatástartományban ismert hatásúnak kell lennie.
Alulról építkező megközelítés
: a szemirritáció vagy a súlyos szemkárosodás tekintetében feltehetőleg besorolást nem igénylő vizsgálati vegyi anyag esetén alkalmazott lépcsőzetes megközelítés, amely a besorolást nem igénylő vegyi anyagok (negatív eredmény) más vegyi anyagokhoz (pozitív eredmény) viszonyított meghatározásával kezdődik.
Vegyi anyag
: anyag vagy keverék.
Szemirritáció
: a vizsgálati vegyi anyagnak a szem elülső felszínén történő alkalmazását követően megjelenő és az alkalmazástól számított 21 napon belül teljes mértékben visszafordítható szemelváltozás. Megfelel a „reverzibilis szemkárosodás” fogalmának és az ENSZ-GHS/CLP-rendelet szerinti 2. kategóriának.
Álnegatív arány
: a vizsgálati módszer alkalmazása során tévesen negatívnak minősített pozitív vegyi anyagok részaránya. A vizsgálati módszer teljesítőképességének egyik mutatója.
Álpozitív arány
: a vizsgálati módszer alkalmazása során tévesen pozitívnak minősített negatív vegyi anyagok részaránya. A vizsgálati módszer teljesítőképességének egyik mutatója.
Veszély
: valamely anyag vagy helyzet azon sajátossága, hogy káros hatásokat képes előidézni az élő szervezet, rendszer vagy (al)populáció anyagnak való expozíciójakor.
Tápoldatos kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó kezeletlen replikátum. Ezt a mintát a vizsgálati vegyi anyaggal kezelt mintákkal és más kontrollmintákkal együtt kell feldolgozni annak megállapításához, hogy az oldószer kölcsönhatásba lép-e a vizsgálati rendszerrel.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
MTT
: 3–(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólium-bromid; tiazolil kék tetrazólium-bromid.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő legalább 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
OD
: optikai sűrűség.
Pozitív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó, ismert pozitív hatást kiváltó anyaggal kezelt replikátum. Annak biztosítása érdekében, hogy a pozitív kontrollban jelentkező hatás időbeli változását fel lehessen mérni, a pozitív hatás nem lehet túlságosan erőteljes.
Relevancia
: a vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálati módszer pontosságát (az eredmények egyezését) figyelembe kell venni (10).
Megbízhatóság
: a vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. Megállapítása a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság, valamint a laboratóriumon belüli megismételhetőség kiszámításával történik (13).
Érzékenység
: az összes olyan pozitív/aktív vegyi anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (10).
Súlyos szemkárosodás
: a vizsgálati vegyi anyagnak a szem külső felületére való juttatását követően olyan szövetkárosodás kialakulása vagy a látás olyan súlyos fizikai romlása, amely a beadást követően 21 napon belül nem fordítható vissza teljes mértékben. Megfelel az „irreverzibilis szemkárosodás” fogalmának és az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriának.
Oldószeres/vivőanyagos kontroll
: a vizsgálati rendszer valamennyi alkotóelemét az oldószerrel vagy vivőanyaggal együtt tartalmazó nem kezelt minta, amelyet a vizsgálati vegyi anyaggal kezelt mintákkal és más kontrollmintákkal együtt kell feldolgozni az ugyanazon oldószerben feloldott vagy ugyanazon vivőanyaggal felvitt vizsgálati vegyi anyaggal kezelt minták kiindulási értékeinek megállapítása érdekében. Párhuzamos tápoldatos kontrollal elvégzett vizsgálata esetén ez a minta azt is bizonyítja, hogy az oldószer vagy vivőanyag kölcsönhatásba lép-e a vizsgálati rendszerrel.
Specificitás
: a vizsgálat elvégzésével helyesen besorolt összes negatív/inaktív vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (13).
Anyag
: olyan természetes állapotban előforduló vagy gyártási eljárásból származó kémiai elem és vegyületei, amely a stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott eljárásból származó szennyező anyagokat is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető.
Felületaktív anyag
: olyan vegyi anyag (például mosószer), amely csökkentheti a folyadékok felületi feszültségét, és így lehetővé teszi, hogy habozzanak vagy áthatoljanak szilárd anyagokon; nedvesítőszerként is ismert.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Többszintű vizsgálati stratégia
: olyan lépcsőzetes vizsgálati stratégia, amelynek keretében a vizsgálati vegyi anyagra vonatkozó valamennyi információt meghatározott sorrendben áttekintik, és az egyes szinteken az adatok bizonyító erejének elemzésével meghatározzák, hogy a következő szintre való továbblépés előtt a veszélyességi osztályozásra vonatkozó döntéshez elegendő információ áll-e rendelkezésre. Ha a vizsgálati vegyi anyag irritatív hatása a rendelkezésre álló információk alapján megállapítható, további vizsgálatra nincs szükség. Ha a vizsgálati vegyi anyag irritatív hatása a rendelkezésre álló információk alapján nem állapítható meg, több lépcsőben egymás után állatkísérleteket kell végezni mindaddig, amíg az egyértelmű besorolás meg nem állapítható.
Felülről építkező megközelítés
: a feltehetőleg súlyos szemkárosodást okozó vizsgálati vegyi anyag esetén alkalmazott lépcsőzetes megközelítés, amely a súlyos szemkárosodást okozó vegyi anyagok (pozitív eredmény) más vegyi anyagokhoz (negatív eredmény) viszonyított meghatározásával kezdődik.
A vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (ENSZ-GHS)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (1).
ENSZ-GHS/CLP-rendelet szerinti 1. kategória
: lásd: „Súlyos szemkárosodás”.
ENSZ-GHS/CLP-rendelet szerinti 2. kategória
: lásd: „Szemirritáció”.
ENSZ-GHS/CLP-rendelet szerint kategória nélküli
: az ENSZ-GHS/CLP-rendelet szerinti 1. és 2. kategóriába (illetve az ENSZ-GHS 2A. vagy 2B. kategóriába) nem besorolt vegyi anyagok.
UVCB
: ismeretlen vagy változó összetételű anyagok, valamint komplex reakciótermékek vagy biológiai anyagok.
B.69. AZ EMBERI SZARUHÁRTYÁHOZ HASONLÓ REKONSTRUÁLT FELHÁMON (RhCE) ALAPULÓ VIZSGÁLATI MÓDSZER A SZEMIRRITÁCIÓ VAGY A SÚLYOS SZEMKÁROSODÁS TEKINTETÉBEN BESOROLÁST NEM IGÉNYLŐ VEGYI ANYAGOK AZONOSÍTÁSÁRA
BEVEZETÉS
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 492. vizsgálati iránymutatásában (2017) leírt módszerrel. A súlyos szemkárosodás az Egyesült Nemzetek Szervezete (ENSZ) által kidolgozott, vegyi anyagok osztályozásának és címkézésének globálisan harmonizált rendszerében (GHS) (1), valamint az Európai Unió (EU) anyagok és keverékek osztályozásáról, címkézéséről és csomagolásáról szóló 1272/2008/EK rendeletében (CLP-rendelet) (77) meghatározottak szerint a vizsgálati vegyi anyagnak a szem külső felületére való juttatását követően olyan szövetkárosodás kialakulása vagy a látás olyan súlyos fizikai romlása, amely a beadást követően 21 napon belül nem fordítható vissza teljes mértékben. Szintén az ENSZ-GHS és a CLP-rendelet szerint a szemirritáció a vizsgálati vegyi anyagnak a szem elülső felszínén történő alkalmazását követően megjelenő és az alkalmazástól számított 21 napon belül teljes mértékben visszafordítható szemelváltozásokra utal. A súlyos szemkárosodást okozó vizsgálati vegyi anyagok az ENSZ-GHS és a CLP-rendelet szerinti 1. kategóriába, míg a szemirritációt kiváltó vizsgálati vegyi anyagok az ENSZ-GHS és a CLP-rendelet szerinti 2. kategóriába sorolandók. A szemirritáció vagy a súlyos szemkárosodás tekintetében be nem sorolt vizsgálati vegyi anyagok olyan vegyi anyagok, amelyek nem tesznek eleget az ENSZ-GHS és a CLP-rendelet szerinti 1. vagy 2. (2A. vagy 2B.) kategóriába való besorolás követelményeinek, vagyis azokra az ENSZ-GHS és a CLP-rendelet szerint kategória nélküliként történik hivatkozás.
|
|
2.
|
A súlyos szemkárosodás/szemirritáció vizsgálata jellemzően kísérleti állatokon történő vizsgálatot foglal magában (B.5. vizsgálati módszer (2)). A legmegfelelőbb vizsgálati módszer kiválasztásához és e vizsgálati módszer alkalmazásához a súlyos szemkárosodásra vonatkozó és bőrirritációs vizsgálatok és értékelések integrált megközelítéseiről szóló OECD-iránymutatás nyújt támpontokat (39).
|
|
3.
|
Ez a vizsgálati módszer egy olyan in vitro eljárást ismertet, amely lehetővé teszi a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást és címkézést nem igénylő vegyi anyagok (anyagok és keverékek) ENSZ-GHS és CLP-rendelet szerinti azonosítását. A módszerben az emberi szaruhártyahámhoz hasonló rekonstruált felhámot (RhCE) alkalmaznak, amely nagy hasonlósággal modellezi az emberi szaruhártya felhámjának szövettani, morfológiai, biokémiai és fiziológiai tulajdonságait. Négy másik in vitro vizsgálati módszert validáltak, nyilvánítottak tudományosan megalapozottnak és fogadtak el, a B.47. (3), B.48. (4), B.61. (5) és B.68. (6) vizsgálati módszert, amelyek mindegyike a súlyos szemkárosodással/szemirritációval mint az emberi egészség károsodásának egyik végpontjával foglalkozik.
|
|
4.
|
E vizsgálati módszer két validált, kereskedelmi forgalomban kapható RhCE-modellt alkalmazó vizsgálatot foglal magában. Az EpiOcular™ szemirritációs vizsgálat (EIT) és a SkinEthic™ emberi szaruhártya felhám (HCE) használatán alapuló szemirritációs vizsgálat (EIT) alkalmazásával elvégezték a szemirritáció/súlyos szemkárosodás felmérésének validálási tanulmányait (7) (8) (9) (10) (11) (12) (13). Mindkét vizsgálat kereskedelmi forgalomban beszerezhető RhCE-szövetkonstrukciót alkalmaz vizsgálati rendszerként, amelyre az alábbiakban validált referenciamódszerként – mint VRM1 és VRM2 – történik hivatkozás. E validálási tanulmányok és független szakértői vizsgálatuk (9) (12) alapján megállapítást nyert, hogy az EpiOcular™ EIT és a SkinEthic™ HCE EIT képes megfelelően azonosítani az ENSZ-GHS szerint szemirritáció vagy súlyos szemkárosodás tekintetében besorolást és címkézést nem igénylő vegyi anyagokat (anyagokat és keverékeket egyaránt), és a vizsgálatok e célra történő alkalmazását tudományosan megalapozottnak tekintik és ajánlják (13).
|
|
5.
|
Jelenleg általánosan elfogadott, hogy az előre látható jövőben egyetlen in vitro vizsgálati módszer sem tudja majd teljes körűen helyettesíteni a Draize-féle in vivo szemvizsgálatot (2) (14) a különböző kémiai osztályoknál felmerülő szemkárosodási/szemirritációs válaszreakciók teljes skálájának előrejelzésében. Előfordulhat azonban, hogy egy-egy (többszintű) vizsgálati stratégián, például az alulról/felülről építkező megközelítésen belüli több alternatív vizsgálati módszer stratégiai kombinációja teljes körűen helyettesíteni tudja a Draize-féle szemvizsgálatot (15). Az alulról építkező megközelítés (15) akkor alkalmazandó, amikor a meglévő információk alapján valamely vegyi anyag várhatóan nem okoz a besorolás szükségességéhez elegendő szemirritációt, míg a felülről építkező megközelítés (15) akkor alkalmazandó, amikor a meglévő információk alapján valamely vegyi anyag várhatóan súlyos szemkárosodást idéz elő. Az EpiOcular™ EIT és a SkinEthic™ HCE EIT vizsgálat ajánlott az ENSZ-GHS/CLP-rendelet szerint a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igényelő (kategória nélküli) vegyi anyagok további vizsgálatot nem igénylő azonosítására, olyan vizsgálati stratégia keretében, mint a Scott és munkatársai által javasolt alulról/felülről építkező megközelítés, például az alulról építkező megközelítés első lépéseként, vagy a felülről építkező megközelítés egyik utolsó lépéseként (15). Az EpiOcular™ EIT és a SkinEthic™ HCE EIT azonban nem alkalmas az ENSZ-GHS/CLP-rendelet szerinti 1. kategória (súlyos szemkárosodás) és az ENSZ-GHS/CLP-rendelet szerinti 2. kategória (szemirritáció) megkülönböztetésére. Ezt a különbségtételt a vizsgálati stratégiai egy másik szintjén kell megvalósítani (15). Azok a vizsgálati vegyi anyagok, amelyeket az EpiOcular™ EIT vagy a SkinEthic™ HCE EIT vizsgálattal szemirritáció/súlyos szemkárosodás tekintetében besorolást igénylőként azonosítottak, végleges kategorizálásuk (ENSZ-GHS/CLP-rendelet szerinti nem besorolt, 2. kategória vagy 1. kategória) érdekében további (in vitro és/vagy in vivo) vizsgálatokat tesznek szükségessé, például a B.47., B.48., B.61. vagy B.68. vizsgálati módszer alkalmazásával.
|
|
6.
|
E vizsgálati módszer ismerteti azt az eljárást, amely felhasználható a vizsgálati vegyi anyagok szemet érintő veszélyességi potenciáljának értékelésére az alapján, hogy az MTT-vizsgálat mérései szerint (16) képesek-e RhCE szövetkonstrukcióban citotoxicitást kiváltani (lásd a 21. pontot). A vizsgálati vegyi anyaggal való kezelését követően az RhCE szövet életképességét a negatív kontrollanyaggal kezelt szövetekkel való összevetésben határozzák meg (életképességi arány), amelyet azután felhasználnak a vizsgálati vegyi anyag szemet érintő veszélyességi potenciáljának előrejelzéséhez.
|
|
7.
|
Az EpiOcular™ EIT és a SkinEthic™ HCE EIT vizsgálathoz hasonló RhCE-alapú, új vagy módosított in vitro vizsgálatok validálásának megkönnyítéséhez rendelkezésre állnak az OECD 34. vizsgálati iránymutatásában (18) szereplő elvekkel összhangban kidolgozott teljesítményszabványok (17), amelyek lehetővé teszik az OECD 492. vizsgálati iránymutatásának időben történő módosítását az új vizsgálatok e vizsgálati iránymutatásba való beépítéséhez. Az adatok OECD-megállapodás szerinti kölcsönös elfogadása csak a teljesítményszabványoknak megfelelően validált vizsgálatok esetében lesz garantált, ha e vizsgálatokat az OECD felülvizsgálta és belefoglalta a megfelelő vizsgálati iránymutatásba.
|
FOGALOMMEGHATÁROZÁSOK
|
8.
|
A fogalommeghatározások az 1. függelékben találhatók.
|
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
9.
|
Ez a vizsgálati módszer kereskedelmi forgalomban kapható, háromdimenziós RhCE-szövetkonstrukció használatán alapul, amelyet (az EpiOcular™ OCL-200 esetében) primér humán epidermális keratinocita sejtekből vagy (a SkinEthic™ HCE/S esetében) immortalizált humán szaruhártya felhámsejtekből állítottak elő. Az EpiOcular™ OCL-200 és a SkinEthic™ HCE/S RhCE szövetkonstrukciók hasonlatosak az in vivo szaruhártya felhám háromdimenziós felépítéséhez, és e szövetkonstrukciókat a vizsgált faj sejtjeiből állították elő (19) (20). Emellett a vizsgálatok közvetlenül mérik a citotoxicitást, a vegyi anyag szaruhártyán történő áthatolása és az általa előidézett sejt- és szövetkárosodás alapján; ez a citotoxicitási reakció határozza azután meg az in vivo súlyos szemkárosodási/szemirritációs vizsgálat összesített eredményét. A sejtkárosodás számos hatásmechanizmuson keresztül valósulhat meg (lásd a 20. pontot), de a citotoxicitás fontos, ha nem elsődleges szerepet játszik egy adott vegyi anyag által kiváltott általános súlyos szemkárosodási/szemirritációs reakció mechanisztikus meghatározásában, ami a szövetkárosodás hátterében álló fizikai-kémiai folyamatoktól függetlenül in vivo többnyire a szaruhártya opálosodásában, szivárványhártya-gyulladásban, a kötőhártya vörösségében és/vagy kötőhártya-vizenyőben nyilvánul meg.
|
|
10.
|
A vizsgálati módszert alátámasztó validálási tanulmányban vegyi anyagok széles skáláját tesztelték, köztük számos, kémiai típus, kémiai osztály, molekulatömeg, LogP-érték, kémiai szerkezet stb. szempontjából eltérő anyagot. Az EpiOcular™ EIT validációs adatbázisa összesen 113 vegyi anyagot tartalmazott, amelyek – az OECD QSAR-eszköztár alapján végzett egyik elemzése szerint – 95 különböző szerves funkciós csoportba tartoznak (8). E vegyi anyagok többsége egykomponensű anyag, de a tanulmány kiterjedt néhány több összetevőből álló anyagra is (azon belül 3 homopolimerre, 5 kopolimerre és 10 kvázi polimerre). Halmazállapotuk és az ENSZ-GHS/CLP-rendelet szerinti kategóriák tekintetében a 113 vizsgálati vegyi anyag a következőképpen oszlik meg: 131. kategóriába tartozó folyadék, 151. kategóriába tartozó szilárd anyag, 6 2A. kategóriába tartozó folyadék, 102A kategóriába tartozó szilárd anyag, 7 2B. kategóriába tartozó folyadék, 7 2B. kategóriába tartozó szilárd anyag, 27 kategória nélküli folyadék és 28 kategória nélküli szilárd anyag (8). A SkinEthic™ HCE EIT validációs adatbázisa összesen 200 vegyi anyagot tartalmazott, amelyek 165 különféle szerves funkciós csoportba tartoznak (8) (10) (11). E vegyi anyagok többsége egy összetevőből álló anyag, de a tanulmány kiterjedt néhány több összetevőből álló anyagra is (azon belül 10 polimerre). Halmazállapotuk és az ENSZ-GHS/CLP-rendelet szerinti kategóriák tekintetében a 200 vizsgálati vegyi anyag a következőképpen oszlik meg: 271. kategóriába tartozó folyadék, 241. kategóriába tartozó szilárd anyag, 192A. kategóriába tartozó folyadék, 102A kategóriába tartozó szilárd anyag, 9 2B. kategóriába tartozó folyadék, 8 2B. kategóriába tartozó szilárd anyag, 50 kategória nélküli folyadék és 53 kategória nélküli szilárd anyag (10) (11).
|
|
11.
|
A vizsgálati módszer alkalmazható anyagokra és keverékekre, valamint szilárd anyagokra, folyadékokra, félszilárd anyagokra és viaszos anyagokra. A folyadékok lehetnek vizesek vagy nem vizesek; a szilárd anyagok lehetnek vízben oldódóak vagy oldhatatlanok. Ahol megoldható, a szilárd anyagokat alkalmazásuk előtt finom porrá kell őrölni; a mintát előzetesen nem szükséges más módon kezelni. Gázokat és aeroszolokat validálási tanulmány keretében nem vizsgáltak. Bár elképzelhető, hogy ezek is vizsgálhatók az RhCE-technikával, a jelenlegi vizsgálati módszer nem engedi meg gázok és aeroszolok vizsgálatát.
|
|
12.
|
Azok a vizsgálati vegyi anyagok, amelyek (jellegükből fakadóan vagy a kezelés után) ugyanabban a tartományban nyelik el a fényt, mint az MTT-formazán, és azok a vizsgálati vegyi anyagok, amelyek közvetlenül is képesek redukálni az MTT vitális festéket (MTT-formazánná), befolyásolhatják a szövet életképességének mérését, ennek korrigálása érdekében adaptált kontrollokra van szükség. Az esetlegesen szükséges adaptált kontrollok típusa függ a vizsgálati vegyi anyag által előidézett interferencia típusától és az MTT-formazán mennyiségének meghatározásához használt eljárástól (lásd a 36–42. pontot).
|
|
13.
|
Az elővalidálási (21) (22) és a teljes körű validálási (8) (10) (11) tanulmányok eredményei igazolták, hogy az EpiOcular™ EIT és a SkinEthic™ HCE EIT egyaránt alkalmazható a vizsgálatok elvégzésében tapasztalattal nem rendelkező laboratóriumokban, továbbá a laboratóriumon belül és a laboratóriumok között is reprodukálható. E tanulmányok alapján az EpiOcular™ EIT vizsgálat 113 vegyi anyag adataira vonatkozó előrejelzéseinek egyezése alapján meghatározott várható reprodukálhatósági szintje a laboratóriumokon belül 95 %, a laboratóriumok között pedig 93 %. A SkinEthic™ HCE EIT vizsgálat 120 vegyi anyag adataira vonatkozó előrejelzéseinek egyezése alapján meghatározott várható reprodukálhatósági szintje a laboratóriumokon belül 92 %, a laboratóriumok között pedig 95 %.
|
|
14.
|
Az EpiOcular™ EIT vizsgálat a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő vegyi anyagok ENSZ-GHS és CLP-rendelet osztályozási rendszere szerinti azonosítására alkalmazható. A validálási tanulmányban (8) gyűjtött adatok alapján az EpiOcular™ EIT általános pontossága 80 % (112 vegyi anyag alapján), érzékenysége 96 % (57 vegyi anyag alapján), álnegatív aránya 4 % (57 vegyi anyag alapján), specificitása 63 % (55 vegyi anyag alapján) és álpozitív aránya 37 % (55 vegyi anyag alapján) a referenciaként szolgáló nyulakon végzett in vivo szemvizsgálatból (B.5. vizsgálati módszer) származó (2) (14), az ENSZ-GHS és a CLP-rendelet osztályozási rendszere szerint azonosított adatokkal való összevetésben. Abban a tanulmányban, amelyben az EpiOcular™ EIT vizsgálattal 97 folyékony mezőgazdasági vegyszerkészítményt teszteltek, a vizsgálati módszer e keveréktípus tekintetében hasonló teljesítményt nyújtott, mint a validálási tanulmány során (23). A 97 készítmény eloszlása a következő: 211. kategóriájú, 192A. kategóriájú, 142B. kategóriájú és 43 kategória nélküli anyag, amelyek besorolása a referenciaként szolgáló nyulakon végzett in vivo szemvizsgálatból (B.5. vizsgálati módszer) származó adatok alapján az ENSZ-GHS osztályozási rendszere szerint történt (2) (14). A besorolás általános pontossága 82 % (97 készítmény alapján), érzékenysége 91 % (54 készítmény alapján), álnegatív aránya 9 % (54 készítmény alapján), specificitása 72 % (43 készítmény alapján) és álpozitív aránya 28 % (43 készítmény alapján) volt (23).
|
|
15.
|
A SkinEthic™ HCE EIT vizsgálat a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő vegyi anyagok ENSZ-GHS és CLP-rendelet osztályozási rendszere szerinti azonosítására alkalmazható. A validálási tanulmányban (10) (11) gyűjtött adatok alapján a SkinEthic™ HCE EIT általános pontossága 84 % (200 vegyi anyag alapján), érzékenysége 95 % (97 vegyi anyag alapján), álnegatív aránya 5 % (97 vegyi anyag alapján), specificitása 72 % (103 vegyi anyag alapján) és álpozitív aránya 28 % (103 vegyi anyag alapján) a referenciaként szolgáló nyulakon végzett in vivo szemvizsgálatból (B.5. vizsgálati módszer) származó (2) (14), az ENSZ-GHS és a CLP-rendelet osztályozási rendszere szerint azonosított adatokkal való összevetésben.
|
|
16.
|
A két RhCE vizsgálat anyagoknál vagy keverékeknél elért álnegatív aránya belül marad azon a 12 %-os általános valószínűségi arányon, amellyel a vegyi anyagokat az ENSZ-GHS és a CLP-rendelet szerinti 2. kategóriájába vagy az ENSZ-GHS és a CLP-rendelet kategória nélküli csoportjába sorolják a Draize-féle in vivo szemvizsgálatban, ismételt vizsgálatok esetén; ez a módszerre jellemző vizsgálaton belüli variabilitásnak tudható be (24). A két RhCE vizsgálati módszer anyagoknál vagy keverékeknél mért álpozitív aránya e vizsgálati módszerben nem kritikus jelentőségű, mivel a (44. pontban szereplő) megállapított határértékkel megegyező vagy az alatti szövet-életképességet kiváltó vizsgálati vegyi anyagokon további vizsgálatokat kell végezni más in vitro vizsgálati módszerekkel, vagy utolsó lehetőségként – a szabályozási követelményektől függően, az adatok bizonyító erejének mérlegelésén alapuló megközelítés keretébe tartozó lépcsőzetes vizsgálati stratégia segítségével – nyulakon végzett vizsgálatokkal. Ezek a vizsgálati módszerek valamennyi típusú vegyi anyagra alkalmazhatók, az alkalmazásuk során kapott negatív eredményt a szemirritáció és a súlyos szemkárosodás tekintetében nem besorolandóként kell elfogadni (az ENSZ-GHS és a CLP-rendelet kategória nélküli csoportja). Az EpiOcular™ EIT és a SkinEthic™ HCE EIT vizsgálat az ENSZ-GHS/CLP-rendelettől eltérő osztályozási rendszeren belüli alkalmazásáról egyeztetni kell a megfelelő szabályozó hatóságokkal.
|
|
17.
|
E vizsgálati módszer korlátai közé tartozik, hogy nem teszi lehetővé a szemirritáció/reverzibilis szemkárosodás (2. kategória) és a súlyos szemkárosodás/irreverzibilis szemkárosodás (1. kategória) ENSZ-GHS és CLP-rendelet szerinti megkülönböztetését, sem a szemirritáló hatás (opcionális 2A. kategória) és enyhén szemirritáló hatás (opcionális 2B. kategória) ENSZ-GHS szerinti megkülönböztetését (1). E célokból más in vitro vizsgálati módszerekkel végzett további vizsgálatra van szükség.
|
|
18.
|
E vizsgálati módszer esetében a „vizsgálati vegyi anyag” kifejezés a vizsgálat tárgyát képező vegyi anyagra vonatkozik (78) és nem az RhCE vizsgálati módszer anyagok és/vagy keverékek vizsgálatára való alkalmazhatóságához kapcsolódik.
|
A VIZSGÁLAT ELVE
|
19.
|
A vizsgálati vegyi anyagot legalább két háromdimenziós RhCE szövetkonstrukcióra viszik fel topikálisan, majd a kezelést és a kezelés utáni inkubációs időszakot követően meghatározzák a szövet-életképességet. Az RhCE szöveteket primér humán epidermális keratinocita sejtekből vagy immortalizált humán szaruhártya felhámsejtekből rekonstruálták, amelyeket több napon keresztül tenyésztettek, hogy egy olyan többrétegű, erősen differenciált laphámszövetet alkossanak, amely morfológiáját tekintve hasonló az emberi szaruhártya hámszövetéhez. Az EpiOcular™ RhCE szövetkonstrukció legalább 3 réteg élő sejtből és egy nem keratinizált felszínből tevődik össze, szaruhártyaszerű felépítése analóg az in vivo megtalálhatóval. A SkinEthic™ HCE RhCE szövetkonstrukció legalább 4 réteg élő sejtből áll, a normál emberi szaruhártya felhámhoz hasonlóan magában foglalva a bazális hengerhámsejteket, a közbenső szárnyas sejteket és a felszíni laphámsejteket (20) (26).
|
|
20.
|
A vegyi anyagok által kiváltott súlyos szemkárosodás/szemirritáció – amely in vivo főként a szaruhártya opálosodásában, szivárványhártya-gyulladásban, a kötőhártya vörösségében és/vagy kötőhártya-vizenyőben nyilvánul meg – egy kaszkád jellegű folyamat eredménye, amelynek első lépéseként a vegyi anyag a szaruhártyán és/vagy a kötőhártyán áthatolva sejtkárosodást idéz elő. Sejtkárosodás bekövetkezhet számos hatásmechanizmuson, többek között a következőkön keresztül: sejtmembrán lízis (pl. felületaktív anyagok, szerves oldószerek váltják ki); makromolekulák (különösen fehérjék) koagulációja (pl. felületaktív anyagok, szerves oldószerek, lúgok és savak váltják ki); lipidek szappanosodása (pl. lúgok váltják ki); és alkilációk, illetve a makromolekulákkal kialakított egyéb kovalens kapcsolatok (pl. fehérítők, peroxidok és alkiláló szerek váltják ki) (15) (27) (28). Kimutatták azonban, hogy a citotoxicitás a szövetkárosodás hátterében álló fizikai-kémiai folyamatoktól függetlenül fontos, ha nem elsődleges szerepet tölt be egy adott vegyi anyag által kiváltott általános súlyos szemkárosodási/szemirritációs reakció mechanisztikus meghatározásában (29) (30). Ezenkívül a vegyi anyagok súlyos szemkárosodási/szemirritációs potenciálját elsősorban a kezdeti sérülés mértéke határozza meg (31), amely korrelációt mutat a sejtpusztulás mértékével (29), valamint a kiváltott reakciók és a végső következmények nagyságrendjével (32). Ennek megfelelően a nagyon enyhén irritáló anyagok általában csak a szaruhártya felhámjának felszínén fejtenek ki hatást, az enyhén és mérsékelten irritáló anyagok többnyire a felhámot és a kötőszövet felszínét károsítják, míg a súlyosan irritáló anyagok a felhámban, a kötőszövet belső rétegeiben és esetenként a szaruhártya belhámjában tesznek kárt (30) (33). A vizsgálati vegyi anyaggal történő topikális kezelését követően az RhCE szövetkonstrukció életképességének – a súlyos szemkárosodás/szemirritáció tekintetében besorolást nem igénylő (az ENSZ-GHS/CLP-rendelet szerint nem besorolt) vegyi anyagok azonosítása céljából történő – mérése azon a feltevésen alapul, hogy a súlyos szemkárosodást vagy szemirritációt okozó összes vegyi anyag citotoxicitást idéz elő a szaruhártya felhámjában és/vagy a kötőhártyában.
|
|
21.
|
Az RhCE szövet életképességét hagyományosan az alapján mérik, hogy az MTT [3–(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólium-bromid; tiazolil kék tetrazólium-bromid; CAS-száma: 298-93-1)] vitális festéket a szövet élő sejtjei enzimatikus úton kék MTT-formazán kristállyá átalakítják, amelyet a szövetből történő kivonás után mennyiségileg meghatároznak (16). Azok a vegyi anyagok azonosíthatók az ENSZ-GHS/CLP-rendelet (nem besorolt) kategóriájának megfelelően besorolást és címkézést nem igénylő vegyi anyagokként, amelyek nem csökkentik a szövet-életképességet egy meghatározott határérték alá (vagyis az EpiOcular™ EIT és a SkinEthic™ HCE EITL (79) vizsgálatban 60 % alá, a SkinEthic™ HCE EITS (80) vizsgálatban pedig 50 % alá) (lásd a 44. pontot).
|
A JÁRTASSÁG BIZONYÍTÁSA
|
22.
|
Az RhCE vizsgálatok szabályozási célú rutinszerű alkalmazása előtt a laboratóriumoknak az 1. táblázatban felsorolt tizenöt jártassági tesztanyag besorolásának helyes előrejelzésével igazolniuk kell szakmai jártasságukat. Ezeket a vegyi anyagokat a vizsgálati referenciamódszereken végzett validálási tanulmányokban használt vegyi anyagok közül választották ki (8) (10) (11). A lehetőségekhez mérten olyan vegyi anyagok kerülnek kiválasztásra, amelyek: i. különféle halmazállapotúak; ii. a nyulakon végzett in vivo szemvizsgálat (B.5. vizsgálati módszer) (2) (14) magas minőségű eredményei, az ENSZ-GHS osztályozási rendszer (1., 2A., 2B. vagy nem besorolt kategória) (1) és a CLP-rendelet szerinti osztályozási rendszer (1., 2. vagy nem besorolt kategória) alapján lefedik a súlyos szemkárosodás/szemirritáció in vivo reakcióinak teljes tartományát; iii. lefedik az in vivo besorolást befolyásoló különböző hatásokat (24) (25); iv. a validálási tanulmányban használt kémiai osztályok tekintetében reprezentatívak (8) (10) (11); v. lefedik a megfelelő szerves funkcionális csoportok széles körét (8) (10) (11); vi. jól meghatározott kémiai szerkezettel rendelkeznek (8) (10) (11); vii. színesek és/vagy közvetlen MTT-redukáló hatásúak; viii. megismételhető eredményeket hoztak az RhCE vizsgálati módszereken végzett validálási tanulmányok során; ix. besorolásukat helyesen jelezték előre az RhCE vizsgálati módszerek validálási tanulmánya során; x. az RhCE vizsgálati módszerekből származó kiváló minőségű adatok alapján lefedik az in vitro válaszreakciók teljes tartományát (0–100 %-os életképesség); xi. kereskedelmi forgalomban kaphatók; és xii. nem kapcsolódnak hozzájuk kiemelkedően magas beszerzési és/vagy ártalmatlanítási költségek. Amennyiben valamely felsorolt vegyi anyag nem áll rendelkezésre, illetve más megalapozott okból kifolyólag nem alkalmazható, akkor a fenti kritériumoknak megfelelő – például a vizsgálati referenciamódszer validálása során alkalmazott vegyi anyagok közül származó – vegyi anyag is használható. Az ilyen eltéréseket azonban meg kell indokolni.
|
1. táblázat
A jártassági tesztanyagok jegyzéke
|
Kémiai név
|
CAS-szám
|
Szerves funkciós csoport (81)
|
Halmazállapot
|
VRM1 életképessége (%) (82)
|
VRM2 életképessége (%) (83)
|
VRM szerinti előrejelzés
|
MTT-redukciós hatás
|
Színinterferencia
|
|
In vivo 1. kategória (84)
|
|
|
Metil-tioglikolát
|
2365-48-2
|
Karbonsavészter; Tioalkohol
|
F
|
10,9 ± 6,4
|
5,5 ± 7,4
|
Előrejelzés nem végezhető
|
I (erősen)
|
N
|
|
Hidroxietil-akrilát
|
818-61-1
|
Akrilát; Alkohol
|
F
|
7,5 ± 4,7 (85)
|
1,6 ± 1,0
|
Előrejelzés nem végezhető
|
N
|
N
|
|
2,5-dimetil-2,5-hexándiol
|
110-03-2
|
Alkohol
|
Sz
|
2,3 ± 0,2
|
0,2 ± 0,1
|
Előrejelzés nem végezhető
|
N
|
N
|
|
Nátrium-oxalát
|
62-76-0
|
Oxokarboxilsav
|
Sz
|
29,0 ± 1,2
|
5,3 ± 4,1
|
Előrejelzés nem végezhető
|
N
|
N
|
|
In vivo 2A. kategória (84)
|
|
|
2,4,11,13-tetraaza-tetradekán-diimid-amid, N,N″-bisz(4-klórfenil)-3,12-diimino-, di-D-glükonát (20 %, vizes oldat) (86)
|
18472-51-0
|
Aromás szénhidrogén halogenid; Aril-halogenid; Dihidroxilcsoport; Guanidin
|
F
|
4,0 ± 1,1
|
1,3 ± 0,6
|
Előrejelzés nem végezhető
|
N
|
I (enyhén)
|
|
Nátrium-benzoát
|
532-32-1
|
Aril; Karbonsav
|
Sz
|
3,5 ± 2,6
|
0,6 ± 0,1
|
Előrejelzés nem végezhető
|
N
|
N
|
|
In vivo 2B. kategória (84)
|
|
|
Dietil-toluamid
|
134-62-3
|
Benzamid
|
F
|
15,6 ± 6,3
|
2,8 ± 0,9
|
Előrejelzés nem végezhető
|
N
|
N
|
|
2,2-dimetil-3-metilén-biciklo [2.2.1] heptán
|
79-92-5
|
Alkán, tercier szenekhez sorolva; Alkén; Bicikloheptán; áthidalt gyűrűs karbociklusok; Cikloalkán
|
Sz
|
4,7 ± 1,5
|
15,8 ± 1,1
|
Előrejelzés nem végezhető
|
N
|
N
|
|
In vivo nem besorolt (84)
|
|
|
1-etil-3-metilimidazolium-etil-szulfát
|
342573-75-5
|
Alkoxi; Ammóniumsó; Aril; Imidazol; Szulfát
|
F
|
79,9 ± 6,4
|
79,4 ± 6,2
|
Kat. nélküli
|
N
|
N
|
|
Dikaprilil-éter
|
629-82-3
|
Alkoxi; Éter
|
F
|
97,8 ± 4,3
|
95,2 ± 3,0
|
Kat. nélküli
|
N
|
N
|
|
Piperonil-butoxid
|
51-03-6
|
Alkoxi; Benzodioxol; benzil; Éter
|
F
|
104,2 ± 4,2
|
96,5 ± 3,5
|
Kat. nélküli
|
N
|
N
|
|
hidrogénezett ricinusolaj polietilén-glikolja (PEG-40)
|
61788-85-0
|
Acilál; Alkohol; Allil; Éter
|
Viszkózus
|
77,6 ± 5,4
|
89,1 ± 2,9
|
Kat. nélküli
|
N
|
N
|
|
1-(4-Klorofenil)-3-(3,4-diklorofenil)-karbamid
|
101-20-2
|
Aromás szénhidrogén halogenid; Aril-halogenid; Karbamidszármazékok
|
Sz
|
106,7 ± 5,3
|
101,9 ± 6,6
|
Kat. nélküli
|
N
|
N
|
|
2,2′-metilén-bisz(6-(2H-benzotriazol-2-il)-4-(1,1,3,3-tetrametil-butil)fenol)
|
103597-45-1
|
Alkán, kvaterner szenekhez sorolva; Fúziós aromás karbociklus; Fúziós telített heterociklusok; Kinoid vegyületek prekurzorai; Terc-butil
|
Sz
|
102,7 ± 13,4
|
97,7 ± 5,6
|
Kat. nélküli
|
N
|
N
|
|
Kálium-tetrafluorborát
|
14075-53-7
|
Szervetlen só
|
Sz
|
88,6 ± 3,3
|
92,9 ± 5,1
|
Kat. nélküli
|
N
|
N
|
|
Rövidítések:
CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám; ENSZ-GHS = a vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (1); VRM1 = az EpiOcular™ EIT validált referenciamódszer; VRM2 = a SkinEthic™ HCE EIT validált referenciamódszer; Színinterferencia = az MTT-formazán hagyományos abszorbancia (optikai sűrűség, OD) mérését zavaró színinterferencia.
|
|
23.
|
A jártassági vizsgálat részeként ajánlott, hogy a felhasználók az RhCE szövetkonstrukció gyártója által meghatározottak szerint, átvétel után ellenőrizzék a szövetek barrierjellemzőit (lásd a 25., 27. és 30. pontot). Ez különösen fontos, ha a szöveteket nagy távolságra/hosszú ideig szállították. Ha egy vizsgálatot már sikeresen igazoltak, valamint jártasságot szereztek az alkalmazásában és bizonyították azt, ilyen jellegű ellenőrzéseket nem szükséges rutinszerűen végezni. Ha azonban egy vizsgálatot rutinszerűen alkalmaznak, a barrierjellemzőket javasolt továbbra is rendszeres időközönként ellenőrizni.
|
ELJÁRÁS
|
24.
|
E vizsgálati módszerhez jelenleg a tudományosan megalapozottnak nyilvánított EpiOcular™ EIT és SkinEthic™ HCE EIT vizsgálat tartozik (9) (12) (13), amelyekre a szöveg validált referenciamódszerként is hivatkozik (az előbbi sorrend szerint VRM1 és VRM2). Az RhCE vizsgálati módszerekhez rendelkezésre állnak szabványműveleti eljárások, amelyeket alkalmazni kell e vizsgálati módszerek laboratóriumi végrehajtása és igénybevétele során (34) (35). A következő pontok és a 2. függelék ismertetik az RhCE vizsgálatok főbb összetevőit és eljárásait.
|
AZ RHCE VIZSGÁLATI MÓDSZER ÖSSZETEVŐI
Általános feltételek
|
25.
|
A megfelelő emberi eredetű sejtekből rekonstruált szaruhártyaszerű háromdimenziós felhámszövetet fokozatosan rétegzett, de nem elszarusodott sejtekből kell előállítani. Az RhCE szövetkonstrukciót úgy készítik, hogy porózus szintetikus membránt inzertálnak, amelyen keresztül a tápanyagok eljutnak a sejtekhez. A rekonstruált szaruhártyaszerű felhámban élő, nem keratinizált epitélsejtek több rétegének kell jelen lennie. Az RhCE szövetkonstrukció felhámjának felszíne közvetlenül érintkezzen a levegővel, hogy a szaruhártya felhámjának in vivo kezeléséhez hasonló módon közvetlenül kezelni lehessen topikálisan a vizsgálati vegyi anyaggal. Az RhCE szövetkonstrukciónak megfelelő ellenálló képességgel rendelkező funkcionális barriert kell képeznie a citotoxikus referencia-vegyianyagok, pl. a Triton X-100 vagy a nátrium-dodecil-szulfát (SDS) gyors áthatolása ellen. A barrierfunkciót bizonyítani kell, ennek értékeléséhez meghatározható az az expozíciós idő, amely alatt egy adott, állandó koncentrációjú referencia-vegyianyag (pl. 100 μl mennyiségű 0,3 térfogatszázalékos Triton X-100) hatására a szövetek életképessége 50 %-kal csökken (ET50), vagy az a referencia-vegyianyag koncentráció, amely meghatározott idejű expozíciót (pl. 30 perces kezelés 50 μl SDS-sel) követően 50 %-kal csökkenti a szövetek életképességét (IC50) (lásd a 30. pontot). Az RhCE szövetkonstrukció folyadékzáró tulajdonságainak meg kell akadályozniuk, hogy a vizsgálati vegyi anyag passzáljon az élő szövet szélei körül, amely a szaruhártya expozíciójának rossz modellezését eredményezné. Az RhCE szövetkonstrukció előállításához használt emberi eredetű sejteknek baktérium-, vírus-, mikoplazma- és gombafertőzéstől mentesnek kell lenniük. A forgalmazónak ellenőriznie kell, hogy a szövetkonstrukció steril, gomba- és baktériumfertőzéstől mentes.
|
Funkcionális feltételek
Életképesség
|
26.
|
A szövetek életképességének számszerű mérése MTT-vizsgálattal történik (16). Az RhCE szövetkonstrukció élő sejtjei az MTT vitális festéket kék MTT-formazán kicsapódássá redukálják, amelyet azután izopropanol (vagy hasonló oldószer) használatával kivonnak a szövetből. A kivont MTT-formazán mennyisége meghatározható hagyományos abszorbancia (optikai sűrűség, OD) méréssel vagy HPLC/UPLC spektrofotometriás eljárással (36). Az extraháló oldószer optikai sűrűségének (OD) eléggé alacsonynak, azaz 0,1 alattinak kell lennie. Az RhCE szövetkonstrukció felhasználóinak biztosítaniuk kell, hogy az alkalmazott RhCE szövetkonstrukció minden egyes tétele megfeleljen a negatív kontrollra vonatkozóan meghatározott kritériumoknak. A vizsgálati referenciamódszerek negatív kontrolljaira vonatkozó OD-értékek elfogadhatósági tartományát a 2. táblázat ismerteti. A HPLC/UPLC spektrofotometriás eljárás alkalmazása esetén a negatív kontroll elfogadhatósági feltételeként a 2. táblázatban szereplő negatív kontrollra vonatkozó OD-tartományok szolgálnak. A vizsgálati jelentésben dokumentálni kell, hogy a negatív kontrollanyaggal kezelt szövetek stabilak a tenyészetben (hasonló szövet-életképességi értékeket adnak) a vizsgálat során alkalmazott expozíciós időszak alatt. A szövet gyártójának a szövet készítés során hasonló eljárást kell lefolytatnia a szövettételek jóváhagyása előtti minőség-ellenőrzéskor, erre az esetre azonban a 2. táblázatban szereplőktől eltérő elfogadhatósági kritériumok alkalmazhatók. Az RhCE szövetkonstrukció előállítójának/forgalmazójának meg kell határoznia a negatív kontroll során nyert OD-értékek (a vizsgálati módszer minőség-ellenőrzési körülményei mellett érvényes) elfogadhatósági tartományát (alsó és felső határértékeket).
|
2. táblázat
A negatív kontroll OD-értékének elfogadhatósági tartománya (a vizsgálat felhasználói számára)
|
Vizsgálat
|
Alsó elfogadási határ
|
Felső elfogadási határ
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (mind a folyadékok, mind a szilárd anyagok protokolljánál)
|
> 0,8 (87)
|
< 2,5
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (mind a folyadékok, mind a szilárd anyagok protokolljánál)
|
> 1,0
|
≤ 2,5
|
Barrierfunkció
|
27.
|
Az RhCE szövetkonstrukciónak megfelelő vastagsággal és ellenálló képességgel kell rendelkeznie ahhoz, hogy például az ET50 alapján végzett becslések szerint (Triton X-100 esetében) vagy az IC50 alapján végzett becslések szerint (SDS esetén) (3. táblázat) meg tudja akadályozni a citotoxikus referencia-vegyianyagok gyors áthatolását. Az RhCE szövetkonstrukció előállítójának/forgalmazójának, a szövetek végfelhasználóhoz történő kiszállításakor bizonyítania kell az alkalmazott RhCE szövetkonstrukció minden egyes tételének barrierfunkcióját (lásd a 30. pontot).
|
Morfológia
|
28.
|
Az RhCE szövetkonstrukció szövettani vizsgálatának igazolnia kell az emberi szaruhártyáéhoz hasonló felhámfelépítést (legalább 3 élő epitélsejtekből álló réteg és egy nem keratinizált felszín). A validált referenciamódszerek tekintetében az előállítónak/forgalmazónak gondoskodnia kell a szövetkonstrukció megfelelő morfológiájáról, ezért ezt a vizsgálati módszer felhasználójának az egyes alkalmazott szövettételeknél nem szükséges ismételten igazolnia.
|
Reprodukálhatóság
|
29.
|
A vizsgálati módszer alkalmazása során végzett pozitív és negatív kontrollok eredményeinek igazolniuk kell a módszer időbeni megismételhetőségét.
|
Minőség-ellenőrzés
|
30.
|
Az RhCE szövetkonstrukció csak abban az esetben használható, ha előállítója/forgalmazója igazolja, hogy az alkalmazott RhCE szövetkonstrukció minden egyes tétele megfelel a gyártás utáni jóváhagyásra vonatkozóan meghatározott feltételeknek, amelyek közül az életképességre (26. pont) és a barrierfunkcióra (27. pont) vonatkozó kritériumok a leglényegesebbek. Az ET50 vagy IC50 érték által mért (lásd a 25. és a 26. pontot) barrierfunkció elfogadhatósági tartományát (alsó és felső határértékét) az RhCE szövetkonstrukció előállítójának/forgalmazójának kell meghatároznia. A 3. táblázat ismerteti az ET50 és az IC50 érték azon elfogadhatósági tartományát, amelyet (a validált referenciamódszerekben) az RhCE szövetkonstrukciók előállítója/forgalmazója a tételjóváhagyás előtti minőség-ellenőrzés során alkalmaz. A gyártás utáni jóváhagyásra vonatkozóan meghatározott feltételeknek való megfelelést igazoló adatokat az RhCE szövetkonstrukció előállítójának/forgalmazójának a vizsgálati módszer felhasználóinak rendelkezésére kell bocsátania, hogy ezeket az információkat fel tudják tüntetni a vizsgálati jelentésben. Kizárólag a gyártás utáni felszabadításra vonatkozóan meghatározott összes feltételnek megfelelő szövetekkel elért eredmények fogadhatók el a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást és címkézést nem igénylő vegyi anyagok ENSZ-GHS és CLP-rendelet szerinti besorolásának megbízható előrejelzéseként.
|
3. táblázat
Tételjóváhagyás előtti minőség-ellenőrzés kritériumai
|
Vizsgálat
|
Alsó elfogadási határ
|
Felső elfogadási határ
|
|
EpiOcular™ EIT (OCL-200) – VRM1 (100 μl 0,3 térfogatszázalékos Triton X-100)
|
ET50 = 12,2 perc
|
ET50 = 37,5 perc
|
|
SkinEthic™ HCE EIT (HCE/S) – VRM2 (30 perces kezelés 50 μl SDS-sel)
|
IC50 = 1 mg/ml
|
IC50 = 3,2 mg/ml
|
A vizsgálati vegyi anyag és a kontrollanyagok alkalmazása
|
31.
|
Minden vizsgálatmenetben valamennyi vizsgálati vegyi anyaghoz és kontrollanyaghoz legalább két szövetreplikátumot kell használni. Két különböző kezelési protokoll alkalmazható, az egyik folyékony, a másik szilárd vizsgálati vegyi anyagokhoz (34) (35). Mindkét módszer és protokoll esetében a vizsgálati vegyi anyaggal történő kezelés előtt a szövetkonstrukció felületét be kell nedvesíteni kalcium- és magnéziummentes Dulbecco-féle foszfátpuffert tartalmazó sóoldattal (Ca2+/Mg2+-mentes DPBS), megteremtve ezzel az emberi szemre jellemző nedves feltételeket. A szövetek kezelése a vizsgálati vegyi anyag(ok)nak és a kontrollanyagoknak való expozícióval veszi kezdetét. Mindkét validált referenciamódszer mindkét kezelési protokolljában a vizsgálati vegyi anyagot és a kontrollanyagot megfelelő mennyiségben kell alkalmazni ahhoz, hogy az egyenletesen fedje a felhám felszínét, ugyanakkor el kell kerülni a végtelen dózis alkalmazását (lásd a 32. és 33. pontot) (2. függelék).
|
|
32.
|
A validált referenciamódszerekben a 37 °C-on vagy alacsonyabb hőmérsékleten (szükség esetén finnpipettával) pipettázható vizsgálati vegyi anyagokat folyadékként kell kezelni, ellenkező esetben szilárd anyagokként kezelendők (lásd a 33. pontot). A validált referenciamódszerekben a folyékony vizsgálati vegyi anyagokat egyenletesen kell eloszlatni a szövet felszínén (ehhez legalább 60 μl/cm2 mennyiség szükséges) (lásd a 2. függeléket (33) (34)). A szövet megfelelő dózissal történő kezelése érdekében a lehetőségekhez mérten kerülni kell, hogy az inzertre (a szövet felszínére) felvitt vizsgálati vegyi anyag kis térfogata miatt kapilláris hatás (hajszálcsövesség) alakuljon ki. A folyékony vizsgálati vegyi anyaggal kezelt szöveteket a szokásos tenyésztési körülmények mellett (37 ±2 °C, 5 ±1 %-os CO2, legalább 95 %-os relatív páratartalom) 30 percig inkubálni kell. Az inkubációs idő lejártával a folyékony vizsgálati vegyi anyagot és a kontrollanyagokat óvatosan el kell távolítani a szövet felszínéről, ehhez szobahőmérsékleten Ca2+/Mg2+-mentes DPBS-sel alaposan át kell mosni a szövetet. Az átmosást követően a szövetet az alkalmazott validált referenciamódszertől függően változó, előre meghatározott időtartamig frissen készített, szobahőmérsékletű tápoldatba kell meríteni (ezáltal eltávolíthatók a szövetbe abszorbeált vizsgálati vegyi anyagok). Kizárólag a VMR1 esetén az MTT-vizsgálat végrehajtása előtt a szövetet frissen készített tápoldatban, szokásos tenyésztési körülmények között expozíció utáni inkubációnak kell alávetni (lásd a 2. függeléket (34) (35)).
|
|
33.
|
A validált referenciamódszerekben a 37 °C-os hőmérsékletig nem pipettázható vizsgálati vegyi anyagokat szilárd anyagokként kell kezelni. A vizsgálati vegyi anyagokat olyan mennyiségben kell alkalmazni, amely képes befedni a szövet teljes felületét, ehhez legalább 60 mg/cm2 mennyiségre van szükség (lásd a 2. függeléket). A szilárd anyagokat, amikor csak lehetséges finom por alakban kell vizsgálni. A szilárd vizsgálati vegyi anyaggal kezelt szöveteket egy előre meghatározott (az alkalmazott validált referenciamódszertől függő) időszakon keresztül a szokásos tenyésztési körülmények közepette inkubálják (lásd a 2. függeléket (34) (35)). Az expozíciós idő lejártával a szilárd vizsgálati vegyi anyagot és a kontrollanyagokat óvatosan el kell távolítani a szövet felszínéről, ehhez szobahőmérsékleten Ca2+/Mg2+-mentes DPBS-sel alaposan át kell mosni a szövetet. Az átmosást követően kerül sor az expozíció utáni merítésre, amikor a szövetet az alkalmazott validált referenciamódszertől függően változó, előre meghatározott időtartamig frissen készített, szobahőmérsékletű tápoldatba merítik (ezáltal eltávolíthatók a szövetbe abszorbeált vizsgálati vegyi anyagok), majd az MTT-vizsgálatot megelőzően frissen készített tápoldatban, szokásos tenyésztési körülmények között expozíció utáni inkubálásra kerül sor (lásd a 2. függeléket (34) (35)).
|
|
34.
|
Minden vizsgálatmenetben párhuzamos negatív és pozitív kontrollokkal bizonyítani kell, hogy a szövetek (negatív kontroll alapján meghatározott) életképessége és (pozitív kontroll alapján meghatározott) érzékenysége a dokumentált adatok alapján meghatározott elfogadhatósági tartományba esik. A párhuzamos negatív kontroll szolgáltatja emellett az alapvonalat (a 100 %-os szövet-életképességet) a vizsgálati vegyi anyaggal kezelt szövetek relatív életképességi arányának ((életképességvizsgált aránya) kiszámításához. A validált referenciamódszerekhez ajánlott pozitív kontrollanyag a tiszta metil-acetát (CAS-száma: 79-20-9, forgalmazza például a Sigma-Aldrich, katalógusszáma: 45997; folyadék). A VRM1, illetve a VRM2 vizsgálathoz alkalmazandó ajánlott negatív kontrollanyag az ultra nagytisztaságú víz, illetve a Ca2+/Mg2+-mentes DPBS. Ezeket a kontrollanyagokat használták a validált referenciamódszereken végzett validálási tanulmányokban, és ezekhez áll rendelkezésre a legtöbb ismert adat. A megfelelő alternatív pozitív vagy negatív kontrollanyagok alkalmazását tudományosan és kellő mértékben alá kell támasztani. A negatív és pozitív kontrollanyagokat ugyanazon protokoll(ok) alapján kell tesztelni, mint a vizsgálatmenetben részt vevő vizsgálati vegyi anyagokat (vagyis a folyadékokhoz és/vagy a szilárd anyagokhoz készült protokollal). A folyékony vizsgálati vegyi anyagok tesztelésével párhuzamosan végzett kontrollok (lásd a 32. pontot) vagy a szilárd vizsgálati vegyi anyagok tesztelésével párhuzamosan végzett kontrollok (lásd a 33. pontot) leírása szerint a kijuttatást követően és még az MTT-vizsgálatot megelőzően sor kerül az expozícióra, az átmosásra, az expozíciót követő merítésre és adott esetben az expozíciót követő inkubálásra (lásd a 35. pontot) (34) (35). Az azonos vizsgálatmenetben tesztelt azonos halmazállapotú (folyékony vagy szilárd) vizsgálati vegyi anyagok esetében elegendő egyetlen negatív és pozitív kontrollt vizsgálni.
|
A szövetek életképességének mérése
|
35.
|
Az MTT-vizsgálat egy szabványosított kvantitatív eljárás (16), amelyet e vizsgálati módszer során a szövetek életképességének méréséhez kell alkalmazni. A vizsgálat kompatibilis a háromdimenziós szövetkonstrukcióban történő használattal. Az MTT-vizsgálatot közvetlenül az expozíció utáni inkubációs időszakot követően hajtják végre. A validált referenciamódszerekben az RhCE szövetkonstrukciót a szokásos tenyésztési körülmények mellett 180 percre (±15 perc) 0,3 ml mennyiségű, 1 mg/ml koncentrációjú MTT-oldatba helyezik. Az MTT vitális festéket az RhCE szövetkonstrukció élő sejtjei kék MTT-formazán kicsapódássá redukálják. Ezt követően a kicsapódott kék MTT-formazán terméket megfelelő mennyiségű izopropanol (vagy hasonló oldószer) használatával kivonják a szövetből (34) (35). A folyékony vizsgálati vegyi anyaggal tesztelt szövetek esetében a szövetek tetejéről és aljáról is ki kell vonni az MTT-t, míg a szilárd vizsgálati vegyi anyagokkal és színes folyadékokkal tesztelt szöveteknél csak a szövet aljáról kell kivonni az MTT-t (ezáltal minimalizálni lehet az izopropanol extraháló oldat esetleges átszennyezését a szöveten maradt vizsgálati vegyi anyaggal). A nehezen lemosható folyékony vizsgálati vegyi anyaggal tesztelt szöveteknél szintén megengedhető, hogy a terméket csak a szövet aljáról vonják ki. A párhuzamosan vizsgált negatív és pozitív kontrollanyagokat a vizsgálati vegyi anyaggal azonos módon kell kezelni. A kivont MTT-formazán mennyisége meghatározható hagyományos abszorbancia (OD) méréssel egy sávszűrő alkalmazásával 570 nm hullámhosszon (maximum ±30 nm-es eltérés megengedett), vagy egy HPLC/UPLC spektrofotometriás eljárás útján (lásd a 42. pontot) (11) (36).
|
|
36.
|
A vizsgálati vegyi anyag optikai tulajdonságai vagy az MTT-re gyakorolt kémiai hatása folytán befolyásolhatja az MTT-formazán mérését, ami a szövet-életképesség helytelen megbecsüléséhez vezet. A vizsgálati vegyi anyagok befolyásolhatják az MTT-formazán mérését azáltal, hogy közvetlenül kék MTT-formazánná redukálják az MTT-t és/vagy azáltal, hogy színinterferenciát okoznak, ha a vizsgálati vegyi anyag jellegénél fogva vagy a kezelési eljárás következtében az MTT-formazánnal azonos OD-tartományban (vagyis kb. 570 nm-en) nyeli el a fényt. A vizsgálat előtt előzetes méréseket kell végezni, amelyek lehetővé teszik a potenciálisan közvetlen MTT-redukáló hatással rendelkező és/vagy színinterferenciát okozó vegyi anyagok azonosítását, és további kontrollokat kell alkalmazni e vizsgálati vegyi anyagok potenciális zavaró hatásának észleléséhez és korrekciójához (lásd a 37–41. pontot). Ez különösen fontos abban az esetben, amikor egy adott vizsgálati vegyi anyagot nem távolítottak el teljesen az RhCE szövetkonstrukcióból a mosás során, vagy amikor az anyag áthatol a szaruhártyaszerű felhámon, és így az MTT-vizsgálat elvégzése során jelen van az RhCE szövetkonstrukcióban. Azon vizsgálati vegyi anyagok esetében, amelyek (jellegüknél fogva vagy a kezelés következtében) az MTT-formazánnal azonos tartományban nyelik el a fényt, és amelyek túlságosan zavaró hatásúak, vagyis erőteljes adszorpciót mutatnak 570 ±30 nm-en, ezért az MTT-formazán hagyományos abszorbancia (OD) mérésével nem vizsgálhatók, az MTT-formazán HPLC/UPLC spektrofotometriás eljárással történő meghatározását lehet alkalmazni (lásd a 41. és 42. pontot) (11) (36). A validált referenciamódszerek szabványműveleti eljárásai részletes leírással szolgálnak arról, hogyan lehet észlelni és korrigálni a közvetlen MTT-redukáló hatást és a színezőanyagok okozta zavaró hatást (34) (35). A VRM1, illetve a VRM2 vizsgálathoz rendelkezésre álló, a III., illetve IV. függelékben közzétett szemléltető folyamatábrák iránymutatást nyújtanak ahhoz, hogyan lehet azonosítani és kezelni a közvetlen MTT-redukáló és/vagy színinterferenciát okozó vegyi anyagokat.
|
|
37.
|
Annak érdekében, hogy azonosítani lehessen azon vizsgálati vegyi anyagok potenciális zavaró hatását, amelyek (jellegüknél fogva vagy a kezelés következtében) az MTT-formazánnal azonos tartományban nyelik el a fényt, és dönteni lehessen a további kontrollok szükségességéről, a vizsgálati vegyi anyagot vízbe és/vagy izopropanolba helyezik, és megfelelő ideig szobahőmérsékleten inkubálják (lásd a 2. függeléket (34) (35)). Ha a VRM1 vizsgálatban (lásd a 3. függeléket) a vízbe és/vagy izopropanolba helyezett vizsgálati vegyi anyag 570 ± 20 nm-es tartományban kellő mennyiségű fényt nyeli el, vagy ha a VRM2 vizsgálatban (lásd a 4. függeléket) a vizsgálati vegyi anyag vízzel történő elegyítése során színes oldat keletkezik, a vizsgálati vegyi anyag feltehetően befolyásolja az MTT-formazán hagyományos abszorbancia (OD) mérését és további színezőanyag-kontrollokra van szükség, illetve alternatív megoldásként HPLC/UPLC spektrofotometriás eljárást lehet végezni, amelynek alkalmazásakor ezek a kontrollok nem szükségesek (lásd a 41. és 42. pontot, valamint a III. és IV. függeléket) (34) (35). Hagyományos abszorbancia (OD) mérés esetén az egyes zavaró hatású vizsgálati vegyi anyagokkal legalább két élő szövetreplikátumot kezelnek, amelyeken elvégzik a teljes vizsgálatot, de az MTT inkubációs fázis során MTT-mentes oldatba helyezik azokat, hogy létrehozzanak egy élő szöveteken végzett nem specifikus elszíneződés (NSCélő) kontrollt (34) (35). Az élő szövetekre jellemző biológiai változékonyságból kifolyólag az NSCélő kontrollt a színes vizsgálati vegyi anyag vizsgálatával párhuzamosan kell elvégezni, több vizsgálat esetén pedig (minden egyes vizsgálatmenetnél) minden vizsgálattal egyidejűleg független NSCélő kontrollt kell végezni. Ezután meghatározzák a szövetek valódi életképességét, ehhez a zavaró hatású vizsgálati vegyi anyaggal kezelt és MTT-oldatban inkubált élő szövetek életképességi arányából (Életképességvizsgált arány) levonják a zavaró hatású vizsgálati vegyi anyagnak kitett és MTT-mentes közegben inkubált élő szövetek nem specifikus elszíneződésének arányát, amelyet a korrigálandó vizsgálattal párhuzamosan vizsgáltak (NSCélő arány), tehát a szövetek valódi életképessége = [Életképességvizsgált arány] - [NSCélő arány].
|
|
38.
|
A közvetlen MTT-redukáló hatással rendelkező anyagok azonosítása érdekében minden vizsgálati vegyi anyagot friss MTT-oldatba kell helyezni. Az MTT-oldathoz megfelelő mennyiségű vizsgálati vegyi anyagot adagolnak, majd a keveréket mintegy 3 órán keresztül a szokásos tenyésztési körülmények közepette inkubálják (lásd a III. és a IV. függeléket) (34) (35). Amennyiben a vizsgálati vegyi anyagot tartalmazó MTT-keverék (illetve oldhatatlan vizsgálati vegyi anyag esetén szuszpenzió) színe kékre/lilára változik, a vizsgálati vegyi anyag feltételezhetően közvetlenül redukálja az MTT-t, ezért kiegészítő funkcionális ellenőrzést kell végezni elhalt RhCE szövetkonstrukciókon, független vizsgálat keretében, hagyományos abszorbancia (OD) mérés, illetve HPLC/UPLC spektrofotometriás eljárás útján. E kiegészítő funkcionális ellenőrzés során elhalt szöveteket használnak, amelyek reziduális metabolikus aktivitással rendelkeznek csupán, ugyanakkor az élő szövetekhez hasonló módon abszorbeálják és magukban tartják a vizsgálati vegyi anyagot. A VRM1 elhalt szöveteit úgy állítják elő, hogy a szöveteket alacsony hőmérsékletnek teszik ki („fagyasztással elölt szövet”). A VRM2 elhalt szöveteit pedig úgy állítják elő, hogy a szöveteket hosszabb ideig (legalább 24 ±1 órán keresztül) vízben inkubálják, majd alacsony hőmérsékleten tárolják („vízben elölt” szövet). Az egyes MTT-redukáló hatású vizsgálati vegyi anyagokkal legalább két elhalt szövetreplikátumot kezelnek, amelyeken elvégzik a teljes vizsgálatot azzal a céllal, hogy egy nem specifikus MTT-redukciós (NSMTT) kontrollt hozzanak létre (34) (35). A független vizsgálatok/vizsgálatmenetek számától függetlenül vizsgálati vegyi anyagonként elegendő egyetlen NSMTT-kontrollt végezni. Ezután meghatározzák a szövetek valódi életképességét, ehhez az MTT-redukáló anyaggal kezelt élő szövetekkel kapott életképességi arányból (Életképességvizsgált arány) levonják az ugyanazon MTT-redukáló anyaggal kezelt elhalt szövetekkel kapott nem specifikus MTT-redukció arányát, amelyet a korrigálandó vizsgálattal párhuzamosan végzett negatív kontrollhoz viszonyítva számítanak ki (NSMTT-arány), tehát a szövetek valódi életképessége = [Életképességvizsgált arány] - [NSMTT-arány].
|
|
39.
|
Azon hagyományos abszorbancia (OD) méréssel értékelt vizsgálati vegyi anyagok esetében, amelyek egyaránt okoznak színinterferenciát (lásd a 37. pontot) és közvetlen MTT-redukciót (lásd a 38. pontot), az előző pontokban ismertetett NSMTT és NSCélő kontrollok mellett szükség van egy harmadik fajta kontrollra is. Általában ez a helyzet áll elő a fényt 570 ±30 nm-en elnyelő, sötét színű (pl. kék, lila, fekete) vizsgálati vegyi anyagoknál, mivel saját színük akadályozza, hogy a 38. pontban leírtak szerint értékelni lehessen közvetlen MTT-redukáló hatásukat. Ilyen esetben alapértelmezetten el kell végezni mind az NSMTT, mind az NSCélő kontrollt. Előfordulhat, hogy azokat a vizsgálati vegyi anyagokat, amelyeken NSMTT és NSCélő kontrollt is végeztek, mind az elő, mind az elhalt szövetek abszorbeálják és magukban tartják. Ilyenkor az NSMTT kontroll nem csupán a vizsgálati vegyi anyag által előidézett közvetlen MTT-redukáló hatást korrigálja, hanem esetleg az anyag elhalt szövetek általi abszorpciójából és benntartásából eredő színinterferenciát is. Ez viszont a színinterferencia kétszeri korrekciójához vezethet, mivel az NSCélő kontroll már korrigálja a vizsgálati vegyi anyag élő szövetek általi abszorpciójából és benntartásából fakadó színinterferenciát. A színinterferencia esetleges kétszeri korrekciójának kiküszöbölése érdekében szükség van egy harmadik kontrollra, amely az elhalt szövetek esetében méri a nem specifikus elszíneződést (NSCelhalt) (lásd a III. és IV. függeléket) (34) (35). E kiegészítő kontroll során a vizsgálati vegyi anyaggal legalább két elhalt szövetreplikátumot kezelnek, amelyeken elvégzik a teljes vizsgálatot, de az MTT inkubációs fázis során MTT-mentes oldatba helyezik azokat. A független vizsgálatok/vizsgálatmenetek számától függetlenül vizsgálati vegyi anyagonként elegendő egyetlen NSCelhalt kontroll, azt azonban az NSMTT kontrollal egyidejűleg és ugyanazon szövettételen kell elvégezni. Ezután meghatározzák a szövetek valódi életképességét, ehhez a vizsgálati vegyi anyaggal kezelt élő szövetekkel kapott életképességi arányból (Életképességvizsgált arány) levonják az NSMTT-arányt és levonják az NSCélő arányt, majd hozzáadják a zavaró hatású vizsgálati vegyi anyaggal kezelt és MTT-mentes közegben inkubált elhalt szövetekkel kapott nem specifikus elszíneződési arányt, amelyet a korrigálandó vizsgálattal párhuzamosan végzett negatív kontrollhoz viszonyítva számítanak ki (NSCelhalt arány), tehát a szövetek valódi életképessége = [Életképességvizsgált arány] - [NSMTT-arány] - [NSCélő arány] + [NSCelhalt arány].
|
|
40.
|
Lényeges megjegyezni, hogy (hagyományos abszorbancia mérés esetén) a nem specifikus MTT-redukció és a nem specifikus színinterferencia a spektrofotométer linearitási tartománya fölé növelheti a szövetkivonat OD-értéket, és (HPLC/UPLC spektrofotometriás mérés esetén) a nem specifikus MTT-redukció a spektrofotométer linearitási tartománya fölé növelheti a szövetkivonat MTT-formazán csúcsterületét is. Ebből kifolyólag fontos, hogy minden laboratórium például MTT-formazán (CAS-száma: 57360-69-7, forgalmazza például a Sigma-Aldrich vállalat, Cat# M2003) segítségével az OD-értékre/csúcsterületre vonatkozólag meghatározza spektrofotométerének linearitási tartományát, mielőtt megkezdené a vizsgálati vegyi anyagok szabályozási célú vizsgálatát.
|
|
41.
|
A spektrofotométert használó hagyományos abszorbancia (OD) mérés akkor alkalmazható a közvetlen MTT-redukáló hatással bíró és színinterferenciát okozó vizsgálati vegyi anyagok értékelésére, amikor az MTT-formazán mérésekor tapasztalt zavaró hatás nem túl erőteljes (vagyis a szövetkivonatok vizsgálati vegyi anyaggal kapott, közvetlen MTT-redukció és/vagy színinterferencia tekintetében nem korrigált OD-értékei a spektrofotométer linearitási tartományán belül vannak). Mindazonáltal a negatív kontroll 60 %-ával (a VRM1, valamint a VRM2 folyadékokra vonatkozó protokollja esetén) vagy 50 %-ával (a VRM2 szilárd anyagokra vonatkozó protokollja esetén) egyező vagy annál nagyobb NSMTT-arányt és/vagy NSCélő arányt előidéző vizsgálati vegyi anyagok eredményei óvatosan értelmezendők, mivel ezeket a határértékeket használják a validált referenciamódszerekben a kategorizált és a nem kategorizált vegyi anyagok megkülönböztetésére (lásd a 44. pontot). Ha viszont a vizsgálati vegyi anyag túl erőteljesen zavarja az MTT-formazán mérését (vagyis a vizsgált szövetkivonatok nem korrigált OD-értékei kívül esnek a spektrofotométer linearitási tartományán), a standard abszorbancia (OD) nem mérhető. Az MTT-formazán hagyományos abszorbancia (OD) mérését túlzott mértékben befolyásoló színes vizsgálati vegyi anyagok, illetve vízzel vagy izopropanollal való érintkezés következtében elszíneződött vizsgálati vegyi anyagok mérése elvégezhető HPLC/UPLC spektrofotometriás eljárás útján (lásd a III. és IV. függeléket). A HPLC/UPLC rendszer ugyanis lehetővé teszi, hogy az MTT-formazánt mennyiségi meghatározása előtt elkülönítsék a vegyi anyagtól (36). Ezért HPLC/UPLC spektrofotometriás módszer használata esetén a vizsgálati vegyi anyagtól függetlenül soha nincs szükség NSCélő és NSCelhalt kontrollra. Az NSMTT-kontroll ugyanakkor alkalmazandó (a 38. pontban ismertetett módon), ha a vizsgálati vegyi anyag gyaníthatóan közvetlenül redukálja az MTT-t. Az NSMTT-kontroll akkor is alkalmazandó, ha a vizsgálati vegyi anyag (saját vagy vízzel való érintkezés következtében kialakult) színe miatt közvetlen MTT-redukáló hatását nem lehet a 38. pontban ismertetett módon felmérni. Amennyiben HPLC/UPLC spektrofotometriásan mérik az MTT-formazánt, a szövetek életképességi aránya a vizsgálati vegyi anyaggal kezelt élő szövetekkel kapott MTT-formazán csúcsterület és a párhuzamos negatív kontrollal kapott MTT-formazán csúcsterület aránya lesz. A közvetlen MTT-redukáló hatással rendelkező vizsgálati vegyi anyagok esetében a szövetek valódi életképességét a 38. pont utolsó mondatában leírt módon számítják ki: [Életképességvizsgált arány] mínusz [NSMTT-arány]. Végül megjegyzendő, hogy azok a közvetlen MTT-redukáló hatású, vagy színinterferenciát is okozó közvetlen MTT-redukáló vegyi anyagok, amelyek a kezelést követően visszamaradnak a szövetben és olyan erős MTT-redukáló hatással bírnak, hogy a vizsgált szövetkivonatok OD-értéke (hagyományos OD mérés esetén), illetve csúcsterülete (HPLC/UPLC spektrofotometriás mérés esetén) kívül esik a spektrofotométer linearitási tartományán, az RhCE vizsgálati módszerekkel nem értékelhetők, bár ilyen csak igen ritka esetben fordul elő.
|
|
42.
|
A HPLC/UPLC spektrofotometria bármely típusú vizsgálati vegyi anyagnál (színes, nem színes, MTT-redukáló vagy nem MTT redukáló hatású) alkalmazható az MTT-formazán mérésére (11) (36). A HPLC/UPLC spektrofotometriás rendszerek sokfélesége miatt a felhasználók nem tudnak azonos rendszerkörülményeket teremteni. Ezért a szövetkivonatok MTT-formazán-tartalmának meghatározása előtt igazolni kell a HPLC/UPLC spektrofotometriás rendszer minősítését azzal, hogy eleget tesznek az Egyesült Államok Élelmiszer- és Gyógyszerfelügyelete (FDA) által az iparág számára kiadott, bioanalitikus módszerek validálására vonatkozó iránymutatásban (36) (38) szereplő standard minősítési paramétereken alapuló elfogadhatósági kritériumoknak. E kulcsfontosságú paraméterekről és elfogadhatósági kritériumokról a 5. függelék nyújt tájékoztatást. Amint eleget tettek a 5. függelékben meghatározott elfogadhatósági kritériumoknak, a HPLC/UPLC spektrofotometriás rendszer minősítettnek tekintendő, és alkalmas arra, hogy az itt leírt vizsgálati módszerben bemutatott kísérleti feltételek mellett elvégezze az MTT-formazán mérését.
|
Elfogadhatósági kritériumok
|
43.
|
Minden olyan vizsgálatmenet esetében, amelynek során a minőség-ellenőrzési kritériumoknak megfelelő RhCE szövettételeket alkalmaznak (lásd a 30. pontot), a negatív kontrollanyaggal kezelt szöveteknek olyan OD-értéket kell mutatniuk, amely tükrözi a szöveteknek a szállítási és az átvételi lépéseket, valamint az összes protokollfolyamatot követő minőségét, és nem léphetik túl a dokumentált adatok alapján meghatározott, 2. táblázatban szereplő határértékeket (lásd a 26. pontot). Hasonlóképpen a pozitív kontrollanyaggal, vagyis a metil-acetáttal kezelt szövetek negatív kontrollhoz viszonyított átlagos életképességének 50 % alatt kell maradnia a VRM1 folyadékokra és szilárd anyagokra vonatkozó protokolljában, míg a VRM2 protokoll esetén legfeljebb 30 % (folyadékokra vonatkozó protokoll) vagy 20 % (szilárd anyagokra vonatkozó protokoll) lehet, mert ez igazolja, hogy a vizsgálati módszer feltételei mellett a szövetek képesek reagálni egy irritáló vizsgálati vegyi anyagra (34) (35). A vizsgálati vegyi anyagokkal és kontrollanyagokkal kezelt szövetreplikátumok variabilitásának az elfogadott határértékeken belül kell maradnia (vagyis két szövetreplikátum variabilitása közötti különbség nem haladhatja meg a 20 %-ot, illetve három szövetreplikátum közötti standard deviáció (SD) nem haladhatja meg a 18 %-ot). Amennyiben egy adott vizsgálatmenet negatív vagy pozitív kontrollja kívül esik az elfogadott tartományon, a vizsgálatmenetet érvénytelennek kell nyilvánítani, és meg kell ismételni. Amennyiben egy adott vizsgálati vegyi anyaggal kezelt szövetreplikátumok közötti variabilitás kívül esik az elfogadott tartományon, a vizsgálatot érvénytelennek kell nyilvánítani, és a vizsgálati vegyi anyagot újbóli vizsgálatnak kell alávetni.
|
Az eredmények értelmezése és az előrejelzési modell
|
44.
|
Az egyes vizsgálati vegyi anyagokkal kezelt szövetkivonat-replikátumokra vonatkozóan kapott OD-értékek/csúcsterületek alapján kell meghatározni a (100 %-nak vett) negatív kontrollra normalizált átlagos szövet-életképesség százalékos arányát (a szövetreplikátumok átlagát). A 4. táblázat ismerteti a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő (az ENSZ-GHS/CLP-rendelet szerinti kategória nélküli) vizsgálati vegyi anyagok azonosítására használt százalékos szövet-életképességi határértékeket. Az eredmények tehát az alábbiak szerint értelmezendők:
|
—
|
A vizsgálati vegyi anyagot nem szükséges az ENSZ-GHS/CLP-rendelet szerint osztályozni és címkézni (vagyis az anyag kategória nélküli), ha a szövet kezelést és expozíció utáni inkubációt követően megállapított átlagos életképességi aránya meghaladja (>) a szövet-életképesség meghatározott százalékos határértékét (lásd a 4. táblázatot). Ebben az esetben más vizsgálati módszerekkel végzett további vizsgálatra nincs szükség.
|
|
—
|
Amennyiben a szövet kezelést és expozíció utáni inkubációt követően megállapított átlagos életképességi aránya megegyezik a szövet-életképesség meghatározott százalékos határértékével vagy annál kisebb (≤) (lásd a 4. táblázatot), besorolása nem előrejelezhető. Ebben az esetben más vizsgálati módszerekkel végzett további vizsgálatra van szükség, mert az RhCE vizsgálati módszerek álpozitív eredményhez vezethetnek (lásd a 14. és 15. pontot), továbbá nem tudnak különbséget tenni az ENSZ-GHS/CLP-rendelet szerinti 1. és 2. kategória között (lásd a 17. pontot).
|
4. táblázat
Az ENSZ-GHS/CLP-rendelet osztályozási rendszere szerinti előrejelzési modellek
|
VRM szerinti
|
Kategória nélküli
|
Előrejelzés nem végezhető
|
|
VRM 1 – EpiOcular™ EIT (mindkét protokoll)
|
Átlagos szövet-életképesség > 60 %
|
Átlagos szövet-életképesség ≤ 60 %
|
|
VRM 2 – SkinEthic™ HCE EIT (folyadékokra vonatkozó protokoll)
|
Átlagos szövet-életképesség > 60 %
|
Átlagos szövet-életképesség ≤ 60 %
|
|
VRM2 – SkinEthic™ HCE EIT (szilárd anyagokra vonatkozó protokoll)
|
Átlagos szövet-életképesség > 50 %
|
Átlagos szövet-életképesség ≤ 50 %
|
|
|
45.
|
Egyértelmű eredmény esetén legalább két szövetreplikátum vizsgálatából álló egyszeri vizsgálatnak elegendőnek kell lennie az adott vizsgálati vegyi anyag esetében. Határértéken lévő eredmények esetében azonban, ha például az egyes szövetreplikátumok eredményei nem egyeznek és/vagy a szövetek átlagos életképességi aránya 60 ± 5 % (VRM1, valamint a VRM2 folyadékokra vonatkozó protokollja esetén) vagy 50 ± 5 % (a VRM2 szilárd anyagokra vonatkozó protokollja esetén), megfontolandó egy második vizsgálat lefolytatása, és ha az első és a második vizsgálat eredményei különbözőek, egy harmadiké is.
|
|
46.
|
A keverékek bizonyos típusainál, ahol megfelelő és indokolt, mérlegelni lehet más százalékos szövet-életképességi határértékek alkalmazását a besorolt és a nem besorolt vizsgálati vegyi anyagok közötti különbségtételhez, hogy az adott keveréktípusok tekintetében javítani lehessen a vizsgálati módszer általános teljesítményét (lásd a 14. pontot). A referencia-vegyianyagok hasznosnak bizonyulhatnak ismeretlen vizsgálati vegyi anyagok vagy termékosztályok súlyos szemkárosodási/szemirritációs potenciáljának értékelésére vagy valamely besorolt vegyi anyag szemre kifejtett relatív toxicitási hatásának a pozitív válaszreakciók egy adott tartományán belüli értékelésére.
|
ADATOK ÉS JELENTÉS
Adatok
|
47.
|
Mindegyik vizsgálati vegyi anyagra vonatkozóan táblázatos jelentést kell készíteni a vizsgálatmenetben tesztelt egyes szövetreplikátumok adatairól (például OD-értékek/MTT-formazán csúcsterületek, a vizsgálati vegyi anyagra és a kontrollanyagokra vonatkozólag meghatározott százalékos szövet-életképességi adatok és az RhCE vizsgálati módszer végső előrejelzése), amelyben szerepelnie kell a megismételt kísérletek adatainak is, ha voltak ilyenek. Emellett minden vizsgálati vegyi anyagra és kontrollanyagra vonatkozólag fel kell tüntetni a jelentésben az átlagos szövet-életképességi arányt és a két szövetreplikátum életképessége közötti különbséget (ha n=2 szövetreplikátum) vagy a standard deviációt (ha n≥ 3 szövetreplikátum). Ezenfelül bele kell foglalni a jelentésbe azt is, ha valamely vizsgálati vegyi anyagról megfigyelték, hogy közvetlen MTT-redukáló hatása és/vagy színinterferencia miatt befolyást gyakorol az MTT-formazán mérésére.
|
Vizsgálati jelentés
|
48.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
Vizsgálati vegyi anyag
|
|
Egy összetevőből álló anyag
|
—
|
kémiai azonosítása, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
—
|
fizikai megjelenés, illékonyság, pH-érték, LogP-érték, molekulatömeg, kémiai osztály, valamint a vizsgálat elvégzése szempontjából releváns további fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
—
|
tisztaság, adott és a gyakorlatban megvalósítható esetben a szennyeződések kémiai azonosítója stb.;
|
|
—
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
—
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak.
|
|
|
|
Több összetevőből álló anyagok, UVCB-k és keverékek
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítójával (lásd fent), tisztaságával, mennyiségi előfordulásával és releváns fizikai-kémiai tulajdonságaival (lásd fent) jellemezve; amennyiben rendelkezésre állnak;
|
|
—
|
fizikai megjelenés és a vizsgálat elvégzése szempontjából releváns további fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
—
|
tisztaság, adott és a gyakorlatban megvalósítható esetben a szennyeződések kémiai azonosítója stb.;
|
|
—
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
—
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak.
|
|
Pozitív és negatív kontrollanyagok
|
—
|
kémiai azonosítása, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
—
|
fizikai megjelenés, illékonyság, molekulatömeg, kémiai osztály, valamint a vizsgálat elvégzése szempontjából releváns további fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
—
|
tisztaság, adott és a gyakorlatban megvalósítható esetben a szennyeződések kémiai azonosítója stb.;
|
|
—
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
—
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
—
|
adott esetben az ultra nagytisztaságú víztől, illetve a Ca2+/Mg2+-mentes DPBS-től eltérő negatív kontrollanyag használatának indokolása;
|
|
—
|
adott esetben a tiszta metil-acetáttól eltérő pozitív kontrollanyag használatának indokolása;
|
|
—
|
a pozitív és negatív kontrollanyagok a vizsgálatmenet elfogadhatósági kritériumainak való megfelelőségét bizonyító korábbi eredményei.
|
A megbízó és a vizsgálatot végző laboratórium adatai
|
—
|
A megbízó, a vizsgálatot végző laboratórium és a vizsgálatvezető neve és címe.
|
|
—
|
Az alkalmazott RhCE szövetkonstrukció és protokoll (valamint használatuk indokolása, amennyiben szükséges)
|
A vizsgálati módszer körülményei
|
—
|
az alkalmazott RhCE szövetkonstrukció és gyártási szám;
|
|
—
|
az MTT-formazán mennyiségi meghatározására használt hullámhossz és (adott esetben) a sávszélesség, valamint a mérőeszköz (pl. spektrofotométer) linearitási tartománya;
|
|
—
|
az MTT-formazán számszerű mérésére alkalmazott módszer leírása;
|
|
—
|
az alkalmazott HPLC/UPLC-spektrofotometriás rendszer leírása, ha releváns;
|
|
—
|
az alkalmazott specifikus RhCE szövetkonstrukcióra vonatkozó teljes körű alátámasztó adatok, köztük a modell teljesítménye. Ennek többek között az alábbiakat kell magában foglalnia:
|
i)
|
a minőség-ellenőrzés során mért életképesség (forgalmazó által szolgáltatott adat);
|
|
ii)
|
a vizsgálati módszer feltételei mellett mért életképesség (felhasználó által szolgáltatott adat);
|
|
iii)
|
barrierfunkció minőség-ellenőrzése;
|
|
iv)
|
morfológia, ha rendelkezésre áll;
|
|
v)
|
reprodukálhatóság és előrejelző képesség;
|
|
vi)
|
az RhCE szövetkonstrukció egyéb minőség-ellenőrzési paraméterei, ha vannak;
|
|
|
—
|
hivatkozás az RhCE szövetkonstrukcióval kapott korábbi adatokra. Ennek többek között az alábbiakat kell magában foglalnia: a minőség-ellenőrzési adatok elfogadhatósága tekintettel a korábbi tételadatokra;
|
|
—
|
nyilatkozat arról, hogy a vizsgálatot végző intézmény a jártassági tesztanyagok vizsgálata révén bizonyította a vizsgálati módszer végrehajtásában való jártasságát annak rutinszerű használata előtt.
|
A vizsgálatmenet és a vizsgálat elfogadásának kritériumai
|
—
|
a pozitív és negatív kontrollok átlaga és elfogadhatósági tartománya a korábbi adatokon alapul;
|
|
—
|
a pozitív és negatív kontrollokban használt szövetreplikátumok közötti elfogadható mértékű variabilitás;
|
|
—
|
a vizsgálati vegyi anyaggal kezelt szövetreplikátumok közötti elfogadható mértékű variabilitás.
|
Vizsgálati eljárás
|
—
|
az alkalmazott vizsgálati eljárás részletei;
|
|
—
|
a vizsgálati vegyi anyag és a kontrollanyagok alkalmazott dózisa;
|
|
—
|
az expozíció, az expozíció utáni merítés és az expozíció utáni inkubáció időtartama és az alkalmazott hőmérséklet (adott esetben);
|
|
—
|
a vizsgálati eljárás bármilyen változtatásának leírása;
|
|
—
|
a közvetlen MTT-redukáló hatással rendelkező és/vagy színváltozást okozó vizsgálati vegyi anyagokhoz alkalmazott kontrollok feltüntetése, amennyiben releváns;
|
|
—
|
vizsgálati vegyi anyagonként és kontrollonként (pozitív kontroll, negatív kontroll, NSMTT, NSCélő és NSCelhalt, ha releváns) használt szövetreplikátumok száma.
|
Eredmények
|
—
|
az egyes vizsgálati vegyi anyagokra és kontrollanyagokra, minden vizsgálatmenetre (ideértve az esetlegesen elvégzett megismételt kísérleteket is) és replikátummérésre vonatkozó adatok, többek között az OD-érték vagy az MTT-formazán csúcsterület, a szövetek életképességi aránya, a szövetek átlagos életképességi aránya, a szövetreplikátumok közötti eltérések vagy standard deviáció és a végső előrejelzés táblázatba foglalása;
|
|
—
|
amennyiben alkalmazandó, a közvetlen MTT-redukáló hatású és/vagy színes vizsgálati vegyi anyagokhoz használt kontrollok eredményei, ideértve az OD-értéket vagy az MTT-formazán csúcsterületet, az NSMTT, az NSCélő és az NSCelhalt arányt, a szövetreplikátumok közötti különbséget vagy standard deviációt, a szövetek helyes végső életképességi arányát és a végső előrejelzést;
|
|
—
|
a vizsgálatmenet és a vizsgálat meghatározott elfogadhatósági kritériumai tekintetében a vizsgálati vegyi anyaggal/anyagokkal és a kontrollanyagokkal kapott eredmények;
|
|
—
|
az egyéb megfigyelt hatások leírása, például a szövetek valamely színes vizsgálati vegyi anyag okozta elszíneződése.
|
Az eredmények értékelése
Következtetések
|
SZAKIRODALOM
|
(1)
|
UN (2015). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York and Geneva: Egyesült Nemzetek. Elérhető a következő címen:http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(2)
|
E melléklet B.5., Akut szemirritáció/szemkorrózió című fejezete.
|
|
(3)
|
E melléklet B.47., Szarvasmarha-szaruhártya opacitásának és permeabilitásának mérésén alapuló vizsgálati módszer i. a súlyos szemkárosodást okozó vegyi anyagok és ii. a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő vegyi anyagok azonosítására című fejezete.
|
|
(4)
|
E melléklet B.48., Izolált csirkeszem vizsgálatán alapuló vizsgálati módszer i. a súlyos szemkárosodást okozó vegyi anyagok és ii. a besorolást nem igénylő vegyi anyagok azonosítására című fejezete.
|
|
(5)
|
E melléklet B.61., Fluoreszcein-szivárgáson alapuló vizsgálati módszer a szemkorróziót és a súlyos szemirritációt okozó anyagok azonosítására című fejezete.
|
|
(6)
|
E melléklet B.68., Rövid expozíciós idejű in vitro vizsgálati módszer i. a súlyos szemkárosodást okozó vegyi anyagok és ii. a szemirritáció vagy a súlyos szemkárosodás tekintetében besorolást nem igénylő vegyi anyagok azonosítására című fejezete.
|
|
(7)
|
Freeman, S.J., Alépée N., Barroso, J., Cole, T., Compagnoni, A., Rubingh, C., Eskes, C., Lammers, J., McNamee, P., Pfannenbecker, U., Zuang, V. (2010). Prospective Validation Study of Reconstructed Human Tissue Models for Eye Irritation Testing. ALTEX 27, Special Issue 2 010,261-266.
|
|
(8)
|
EC EURL ECVAM (2014). The EURL ECVAM - Cosmetics Europe prospective validation study of Reconstructed human Cornea-like Epithelium (RhCE)-based test methods for identifying chemicals not requiring classification and labelling for serious eye damage/eye irritation: Validation Study Report. EUR 28 125 EN; doi:10.2787/41680. Elérhető a következő címen:http://publications.jrc.ec.europa.eu/repository/handle/JRC100280.
|
|
(9)
|
EURL ECVAM Science Advisory Committee (2014). ESAC Opinion on the EURL ECVAM Eye Irritation Validation Study (EIVS) on EpiOcular™ EIT and SkinEthic™ HCE and a related Cosmetics Europe study on HPLC/UPLC-spectrophotometry as an alternative endpoint detection system for MTT-formazan. ESAC Opinion No 2014-03 of 17 November 2014; EUR 28 173 EN; doi: 10.2787/043697. Elérhető a következő címen:http://publications.jrc.ec.europa.eu/repository/handle/JRC103702.
|
|
(10)
|
Alépée, N., Leblanc, V., Adriaens, E., Grandidier, M.H., Lelièvre, D, Meloni, M., Nardelli, L., Roper, C.S, Santirocco, E., Toner, F., Van Rompay, A., Vinall, J., Cotovio, J. (2016). Multi-laboratory validation of SkinEthic HCE test method for testing serious eye damage/eye irritation using liquid chemicals. Toxicol. In vitro 31, 43-53.
|
|
(11)
|
Alépée, N., Adriaens, E., Grandidier, M.H., Meloni, M., Nardelli, L., Vinall, C.J., Toner, F., Roper, C.S, Van Rompay, A.R., Leblanc, V., Cotovio, J. (2016). Multi-laboratory evaluation of SkinEthic HCE test method for testing serious eye damage/eye irritation using solid chemicals and overall performance of the test method with regard to solid and liquid chemicals testing. Toxicol. In vitro 34, 55-70.
|
|
(12)
|
EURL ECVAM Science Advisory Committee (2016). ESAC Opinion on the SkinEthic™ Human Corneal Epithelium (HCE) Eye Irritation Test (EIT). ESAC Opinion No 2016-02 of 24 June 2016; EUR 28 175 EN; doi: 10.2787/390390. Elérhető a következő címen:http://publications.jrc.ec.europa.eu/repository/handle/JRC103704.
|
|
(13)
|
EC EURL ECVAM (2016). Recommendation on the Use of the Reconstructed human Cornea-like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Serious Eye Damage/Eye Irritation According to UN GHS. (Manuscript in Preparation).
|
|
(14)
|
Draize, J.H., Woodard, G., Calvery, H.O. (1944). Methods for the Study of Irritation and Toxicity of Substances Applied Topically to the Skin and Mucous Membranes. Journal of Pharmacol. and Exp. Therapeutics 82, 377-390.
|
|
(15)
|
Scott, L., Eskes, C., Hoffmann, S., Adriaens, E., Alépée, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., McNamee, P., Osborne, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielman, H., Stokes, W., Trouba, K., Van den Berghe, C., Van Goethem, F., Vassallo, M., Vinardell, P., Zuang, V. (2010). A Proposed Eye Irritation Testing Strategy to Reduce and Replace In vivo Studies Using Bottom-Up and Top-Down Approaches. Toxicol. In vitro 24, 1-9.
|
|
(16)
|
Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65, 55-63.
|
|
(17)
|
OECD (2016). Series on Testing and Assessment No 216: Performance Standards for the Assessment of Proposed Similar or Modified In vitro Reconstructed Human Cornea-Like Epithelium (RhCE) Test Methods for Identifying Chemicals not Requiring Classification and Labelling for Eye Irritation or Serious Eye Damage, Based on the Validated Reference Methods EpiOcular™ EIT and SkinEthic™ HCE EIT described in TG 492. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(18)
|
OECD (2005). Series on Testing and Assessment No 34: Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(19)
|
Kaluzhny, Y., Kandárová, H., Hayden, P., Kubilus, J., d’Argembeau-Thornton, L., Klausner, M. (2011). Development of the EpiOcular™ Eye Irritation Test for Hazard Identification and Labelling of Eye Irritating Chemicals in Response to the Requirements of the EU Cosmetics Directive and REACH Legislation. Altern. Lab. Anim. 39, 339-364.
|
|
(20)
|
Nguyen, D.H., Beuerman, R.W., De Wever, B., Rosdy, M. (2003). Three-dimensional construct of the human corneal epithelium for in vitro toxicology. In: Salem, H., Katz, S.A. (Eds), Alternative Toxicological Methods, CRC Press, pp. 147-159.
|
|
(21)
|
Pfannenbecker, U., Bessou-Touya, S., Faller, C., Harbell, J., Jacob, T., Raabe, H., Tailhardat, M., Alépée, N., De Smedt, A., De Wever, B., Jones, P., Kaluzhny, Y., Le Varlet, B., McNamee, P., Marrec-Fairley, M., Van Goethem, F. (2013). Cosmetics Europe multi-laboratory pre-validation of the EpiOcular™ reconstituted Human Tissue Test Method for the Prediction of Eye Irritation. Toxicol. In vitro 27, 619-626.
|
|
(22)
|
Alépée, N., Bessou-Touya, S., Cotovio, J., de Smedt, A., de Wever, B., Faller, C., Jones, P., Le Varlet, B., Marrec-Fairley, M., Pfannenbecker, U., Tailhardat, M., van Goethem, F., McNamee, P. (2013). Cosmetics Europe Multi-Laboratory Pre-Validation of the SkinEthic™ Reconstituted Human Corneal Epithelium Test Method for the Prediction of Eye Irritation. Toxicol. In vitro 27, 1 476-1 488.
|
|
(23)
|
Kolle, S.N., Moreno, M.C.R., Mayer, W., van Cott, A., van Ravenzwaay, B., Landsiedel, R. (2015). The EpiOcular™ Eye Irritation Test is the Method of Choice for In vitro Eye Irritation Testing of Agrochemical Formulations: Correlation Analysis of EpiOcular™ Eye Irritation Test and BCOP Test Data to UN GHS, US EPA and Brazil ANIVSA Classifications. Altern. Lab. Anim. 43, 1-18.
|
|
(24)
|
Adriaens, E., Barroso, J., Eskes, C., Hoffmann, S., McNamee, P., Alépée, N., Bessou-Touya, S., De Smedt, A., De Wever, B., Pfannenbecker, U., Tailhardat, M., Zuang, V. (2014). Retrospective Analysis of the Draize Test for Serious Eye Damage/Eye Irritation: Importance of Understanding the In vivo Endpoints Under UN GHS/EU CLP for the Development and Evaluation of In vitro Test Methods. Arch. Toxicol. 88, 701-723.
|
|
(25)
|
Barroso, J., Pfannenbecker, U., Adriaens, E., Alépée, N., Cluzel, M., De Smedt, A., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Templier, M., McNamee, P. (2017). Cosmetics Europe compilation of historical serious eye damage/eye irritation in vivo data analysed by drivers of classification to support the selection of chemicals for development and evaluation of alternative methods/strategies: the Draize eye test Reference Database (DRD). Arch. Toxicol. 91, 521-547.
|
|
(26)
|
Meloni, M., De Servi, B., Marasco, D., Del Prete, S. (2011). Molecular mechanism of ocular surface damage: Application to an in vitro dry eye model on human corneal epithelium. Molecular Vision 17, 113-126.
|
|
(27)
|
Hackett, R.B., McDonald, T.O. (1991). Eye Irritation. In Advances in Modern Toxicology: Dermatoxicology Marzulli F.N.and Maibach H.I. (Eds.), 4th Edition, pp. 749–815. Washington, DC, USA: Hemisphere Publishing Corporation.
|
|
(28)
|
Fox, D.A., Boyes, W.K. (2008). Toxic Responses of the Ocular and Visual System. In Cassaret and Doull’s Toxicology: The Basic Science of Poisons Klaassen C.D.(Ed.), 7th Edition, pp. 665–697. Withby, ON, Canada: McGraw-Hill Ryerson.
|
|
(29)
|
Jester, J.V., Li, H.F., Petroll, W.M., Parker, R.D., Cavanagh, H.D., Carr, G.J., Smith, B., Maurer, J.K. (1998). Area and Depth of Surfactant Induced Corneal Injury Correlates with Cell Death. Invest. Ophthalmol. Vis. Sci. 39, 922-936.
|
|
(30)
|
Maurer, J.K., Parker, R.D., Jester, J.V. (2002). Extent of Corneal Injury as the Mechanistic Basis for Ocular Irritation: Key Findings and Recommendations for the Development of Alternative Assays. Reg. Tox. Pharmacol. 36, 106-117.
|
|
(31)
|
Jester, J.V., Li, L., Molai, A., Maurer, J.K. (2001). Extent of Corneal Injury as a Mechanistic Basis for Alternative Eye Irritation Tests. Toxicol. In vitro 15, 115-130.
|
|
(32)
|
Jester, J.V., Petroll, W.M., Bean, J., Parker, R.D., Carr, G.J., Cavanagh, H.D., Maurer, J.K. (1998). Area and Depth of Surfactant-Induced Corneal Injury Predicts Extent of Subsequent Ocular Responses. Invest. Ophthalmol. Vis. Sci. 39, 2 610-2 625.
|
|
(33)
|
Jester, J.V. (2006). Extent of Corneal Injury as a Biomarker for Hazard Assessment and the Development of Alternative Models to the Draize Rabbit Eye Test. Cutan. Ocul. Toxicol. 25, 41-54.
|
|
(34)
|
EpiOcular™ EIT SOP, Version 8 (March 05, 2013). EpiOcular™ EIT for the Prediction of Acute Ocular Irritation of Chemicals. Elérhető a következő címen:[https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index].
|
|
(35)
|
SkinEthic™ HCE EIT SOP, Version 1. (July 20, 2015). SkinEthic™ HCE Eye Irritation Test (EITL for Liquids, EITS for Solids) for the Prediction of Acute Ocular Irritation of Chemicals. Elérhető a következő címen:https://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methodsAndProtocols/index.
|
|
(36)
|
Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M., McNamee, P. (2015). Use of HPLC/UPLC-Spectrophotometry for Detection of Formazan in In vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT-Reduction Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In vitro 29, 741-761.
|
|
(37)
|
Kaluzhny, Y., Kandárová, H., Handa, Y., DeLuca, J., Truong, T., Hunter, A., Kearney, P., d’Argembeau-Thornton, L., Klausner, M. (2015). EpiOcular™ Eye Irritation Test (EIT) for Hazard Identification and Labeling of Eye Irritating Chemicals: Protocol Optimization for Solid Materials and Extended Shipment Times. Altern. Lab Anim. 43, 101-127.
|
|
(38)
|
US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. 2001. május. Elérhető a következő címen:http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
|
|
(39)
|
OECD (2017). Guidance Document on an Integrated Approaches on Testing and Assessment for Serious Eye Damage and Eye irritation. Series on Testing and Assessment No 263. ENV Publications, Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
1. függelék
Fogalommeghatározások
Pontosság
: a vizsgálati módszerrel kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálati módszer teljesítőképességét mutatja, és „relevanciájának” egyik megítélési szempontja. E kifejezést gyakran használják az „egyezés” megfelelőjeként, amely egy adott vizsgálati módszer alkalmazásakor az azonos eredmények arányát fejezi ki (18).
Referencia-vegyianyag
: a vizsgálati vegyi anyaggal való összevetéshez referenciaként használt vegyi anyag. A referencia-vegyianyagnak a következő tulajdonságokkal kell rendelkeznie: i. állandó és megbízható forrás(ok)ból kell származnia, ami lehetővé teszi azonosítását és jellemzését; ii. a vizsgálati vegyi anyag(ok)éhoz hasonló szerkezeti, funkcionális és/vagy kémiai vagy termékosztályba kell tartoznia; iii. ismert fizikai-kémiai tulajdonságokkal kell rendelkeznie; iv. rendelkezésre kell állnia az ismert hatásait alátámasztó adatoknak; és v.. a kívánt hatástartományban ismert hatásúnak kell lennie.
Alulról építkező megközelítés
: a szemirritáció vagy a súlyos szemkárosodás tekintetében feltehetőleg besorolást és címkézést nem igénylő vizsgálati vegyi anyag esetén alkalmazott lépcsőzetes megközelítés, amely a besorolást és címkézést nem igénylő vegyi anyagok (negatív eredmény) más vegyi anyagokhoz (pozitív eredmény) viszonyított meghatározásával kezdődik.
Vegyi anyag
: anyag vagy keverék.
Egyezés
: Lásd a „pontosság” meghatározását.
Szaruhártya
: a szemgolyó elülső részén az íriszt és a pupillát fedő és a szem belsejébe a fényt bevezető, áttetsző rész.
CV
: variációs koefficiens.
Dev
: eltérés.
EIT
: Eye Irritation Test (szemirritációs vizsgálat).
EURL ECVAM
: Állatkísérletek Alternatíváinak Uniós Referencialaboratóriuma.
Szemirritáció
: a vizsgálati vegyi anyagnak a szem elülső felszínén történő alkalmazását követően megjelenő és az alkalmazástól számított 21 napon belül teljes mértékben visszafordítható szemelváltozások. Megfelel a „reverzibilis szemkárosodás” fogalmának és az ENSZ-GHS/CLP-rendelet szerinti 2. kategóriának.
ET50
: az az expozíciós idő, amely alatt egy adott, állandó koncentrációjú referencia-vegyianyag hatására a szövet életképessége 50 %-kal csökken.
Álnegatív arány
: a vizsgálati módszer alkalmazása során tévesen negatívnak minősített pozitív anyagok részaránya. A vizsgálati módszer teljesítőképességének egyik mutatója.
Álpozitív arány
: a vizsgálati módszer alkalmazása során tévesen pozitívnak minősített negatív anyagok részaránya. A vizsgálati módszer teljesítőképességének egyik mutatója.
Veszély
: valamely anyag vagy helyzet azon sajátossága, hogy káros hatásokat képes előidézni az élő szervezet, rendszer vagy (al)populáció anyagnak való expozíciójakor.
HCE
: SkinEthic™ Human Corneal Epithelium (emberi szaruhártya felhámszövet).
HPLC
: nagy teljesítményű folyadékkromatográfia.
IC50
: az a referencia-vegyianyag koncentráció, amely meghatározott idejű expozíciót (pl. 30 perces SDS-sel történő kezelést) követően 50 %-kal csökkenti a szövetek életképességét.
Végtelen dózis
: az RhCE szövetkonstrukcióra alkalmazott vizsgálati vegyi anyag azon mennyisége, amely meghaladja a felhám felszínének teljes és egyenletes befedéséhez szükséges mennyiséget.
Irreverzibilis szemkárosodás
: lásd: „Súlyos szemkárosodás”.
LLOQ
: mennyiségi meghatározás alsó határértéke.
LogP
: az oktanol/víz megoszlási koefficiens logaritmusa.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő legalább 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
MTT
: 3–(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazólium-bromid; tiazolil kék tetrazólium-bromid.
Negatív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó minta, amelyet egy olyan anyaggal kezelnek, amelyről ismert, hogy a vizsgálati rendszerben nem vált ki pozitív hatást. Ezt a mintát a vizsgálati vegyi anyaggal kezelt mintákkal és más kontrollmintákkal együtt feldolgozzák fel, és e minta alapján határozzák meg a 100 %-os szövet-életképességet.
Nem besorolt
: a szemirritáció tekintetében (az ENSZ-GHS/CLP-rendelet szerinti 2., és az ENSZ-GHS szerinti 2A. vagy 2B. kategóriába tartozóként) nem besorolt vagy a súlyos szemkárosodás tekintetében (az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozóként) nem besorolt vegyi anyagok. Megfelelnek az „ENSZ-GHS/CLP-rendelet szerinti kategória nélküli” vegyi anyagoknak.
NSCelhalt
: elhalt szöveteken megfigyelt, nem specifikus elszíneződés.
NSCélő
: élő szöveteken megfigyelt, nem specifikus elszíneződés.
NSMTT
: nem specifikus MTT-redukció.
OD
: optikai sűrűség.
Teljesítményszabványok
: hitelesített, tudományosan megalapozottnak nyilvánított referenciamódszeren alapuló szabványok, amelyek a javasolt, végrehajtás és funkcionalitás szempontjából hasonló vizsgálati módszer összehasonlíthatósági értékelésének alapjául szolgálnak. Ide tartoznak: i. a vizsgálati módszer alapvető összetevői; ii. a referencia-vegyianyagok minimális jegyzéke, amely a validált vizsgálati módszer által nyújtott teljesítmény elfogadhatóságának igazolására használt vegyi anyagokból került összeállításra; és iii. a validált vizsgálati módszer eredményei alapján meghatározott, hasonló megbízhatósági és pontossági szintek, amelyeket a javasolt vizsgálati módszernek teljesítenie kell a minimális jegyzék referencia-vegyianyagainak felhasználásával történő értékelése során (18).
Pozitív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó minta, amelyet egy olyan anyaggal kezelnek, amelyről ismert, hogy a vizsgálati rendszerben pozitív hatást vált ki. Ezt a mintát a vizsgálati vegyi anyaggal kezelt mintákkal és más kontrollmintákkal együtt dolgozzák fel. Annak biztosítása érdekében, hogy a pozitív kontrollban jelentkező hatás időbeli változását fel lehessen mérni, a pozitív hatás nem lehet túlságosan erőteljes.
Relevancia
: a vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálati módszer pontosságát (az eredmények egyezését) figyelembe kell venni (18).
Megbízhatóság
: a vizsgálati módszer laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. Megállapítása a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság, valamint a laboratóriumon belüli megismételhetőség kiszámításával történik (18).
Helyettesítő vizsgálat
: olyan vizsgálat, amelynek célja, hogy a veszélyek azonosítására és/vagy kockázatértékelésre rutinszerűen alkalmazott és elfogadott vizsgálatot helyettesítse, és amelyről megállapították, hogy az elfogadott vizsgálattal összehasonlítva, az összes lehetséges vizsgálati helyzetben és vegyi anyag esetében a humán vagy állati egészség egyenértékű vagy jobb védelmét biztosítja (18).
Reprodukálhatóság
: ugyanazon vizsgálati vegyi anyag ugyanazon vizsgálati protokollal végzett ismételt vizsgálata során kapott eredmények egyezése (lásd: „megbízhatóság”) (18).
Reverzibilis szemkárosodás
: lásd: „Szemirritáció”.
RhCE
: Reconstructed human Cornea-like Epithelium (az emberi szaruhártyahámhoz hasonló rekonstruált felhám).
Vizsgálatmenet
: egy vizsgálatmenet során egy vagy több vizsgálati vegyi anyagot vizsgálnak egy párhuzamos negatív kontrollal és egy pozitív kontrollal.
SD
: standard deviáció.
Érzékenység
: az összes olyan pozitív/aktív vizsgálati vegyi anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (18).
Súlyos szemkárosodás
: a vizsgálati anyagnak a szem külső felületére való juttatását követően olyan szövetkárosodás kialakulása vagy a látás olyan súlyos fizikai romlása, amely a beadást követően 21 napon belül nem fordítható vissza teljes mértékben. Megfelel az „irreverzibilis szemkárosodás” fogalmának és az „ENSZ-GHS/CLP-rendelet szerinti 1. kategóriának”.
Szabványműveleti eljárások
: formális, írásba foglalt eljárások, amelyek részletesen ismertetik bizonyos rutinjellegű és vizsgálatspecifikus laboratóriumi műveletek végrehajtási módját. Kidolgozásukat a helyes laboratóriumi gyakorlatok teszik szükségessé.
Specificitás
: a vizsgálat elvégzésével helyesen besorolt összes negatív/inaktív vizsgálati vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (18).
Anyag
: olyan természetes állapotban előforduló vagy gyártási eljárásból származó kémiai elem és vegyületei, amely a stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott eljárásból származó szennyező anyagokat is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető.
Vizsgálat
: egyetlen vizsgálati vegyi anyag egyidejű tesztelése legalább két szövetreplikátumon, a megfelelő szabványműveleti eljárásban meghatározott módon.
Szövet-életképesség
: egy sejtpopuláció rekonstruált szöveten belüli teljes aktivitását, például az életfontosságú MTT-festékanyag csökkentésének képességét mérő paraméter, amely a mért végponttól és az alkalmazott vizsgálat felépítésétől függően korrelál az élő sejtek teljes számával és/vagy az életképességükkel.
Felülről építkező megközelítés
: a feltehetőleg súlyos szemkárosodást okozó vegyi anyag esetén alkalmazott lépcsőzetes megközelítés, amely a súlyos szemkárosodást okozó vegyi anyagok (pozitív eredmény) más vegyi anyagokhoz (negatív eredmény) viszonyított meghatározásával kezdődik.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Többszintű vizsgálati stratégia
: lépcsőzetes vizsgálati stratégia, amely a vizsgálati módszereket egymást követően alkalmazza. A vizsgálati vegyi anyagra vonatkozó valamennyi információt áttekintik az egyes szinteken, és a bizonyítékok súlyozásával meghatározzák, hogy a stratégia következő szintjére való továbblépés előtt a veszélyességi osztályozásra vonatkozó döntéshez elegendő információ áll-e rendelkezésre. Ha a vizsgálati vegyi anyag veszélyességi potenciálja/hatáserőssége a rendelkezésre álló információk alapján az adott szinten megállapítható, további vizsgálatra nincs szükség (18).
ULOQ
: mennyiségi meghatározás felső határértéke.
A vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (ENSZ-GHS)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (1).
ENSZ-GHS és CLP-rendelet szerinti 1. kategória
: lásd: „Súlyos szemkárosodás”.
ENSZ-GHS és CLP-rendelet szerinti 2. kategória
: lásd: „Szemirritáció”.
ENSZ-GHS és CLP-rendelet szerint nem besorolt
: az ENSZ-GHS/CLP-rendelet szerinti 1. vagy 2. (illetve az ENSZ-GHS szerinti 2A. vagy 2B.) kategóriába való besorolás követelményeinek nem megfelelő vegyi anyagok. Megfelelnek a „Kategória nélküli” vegyi anyagoknak.
UPLC
: ultranagy teljesítményű folyadékkromatográfia.
UVCB
: ismeretlen vagy változó összetételű anyagok, valamint komplex reakciótermékek vagy biológiai anyagok.
Érvényes vizsgálati módszer
: olyan vizsgálati módszer, amely kellően relevánsnak és megbízhatónak minősül egy adott célra, és amely tudományosan megalapozott elveken alapul. Egy-egy vizsgálati módszer abszolút értelemben soha nem érvényes, kizárólag meghatározott céllal összefüggésben az (18).
Validált vizsgálati módszer
: olyan vizsgálati módszer, amelynek az adott céllal kapcsolatos relevanciája (pontosságát is ideértve) és megbízhatósága validálási vizsgálatok alapján megállapítást nyert. Fontos megjegyezni, hogy a validált vizsgálati módszer a javasolt felhasználási célra nem feltétlenül kínál megfelelő pontosságú és megbízhatóságú elfogadható eljárást (18).
VRM
: validált referenciamódszer.
VRM1
: az 1. validált referenciamódszer az EpiOcular™ EIT vizsgálatra utal.
VRM2
: a 2. validált referenciamódszer a SkinEthic™ HCE EIT vizsgálatra utal.
A bizonyítékok súlyozása
: a rendelkezésre álló különféle érvek és ellenérvek mérlegelésének folyamata az adott vizsgálati anyag veszélyességével kapcsolatban a megfelelő következtetés levonása és alátámasztása céljából.
2. függelék
A SZEMIRRITÁCIÓ VAGY A SÚLYOS SZEMKÁROSODÁS TEKINTETÉBEN BESOROLÁST ÉS CÍMKÉZÉST NEM IGÉNYLŐ VEGYI ANYAGOK AZONOSÍTÁSÁRA HITELESÍTETT RHCE VIZSGÁLATOK FŐBB ÖSSZETEVŐI
|
A vizsgálat összetevői
|
EpiOcular™ EIT
(VRM 1)
|
SkinEthic™ HCE EIT
(VRM 2)
|
|
Protokollok
|
Folyadékok
(15 percig pipettálható 37 ± 1 oC vagy alacsonyabb hőmérsékleten)
|
Szilárd anyagok
(nem pipettálható)
|
Folyadékok és viszkózus anyagok
(pipettálható)
|
Szilárd anyagok
(nem pipettálható)
|
|
A modell felülete
|
0,6 cm2
|
0,6 cm2
|
0,5 cm2
|
0,5 cm2
|
|
A szövetreplikátumok száma
|
Legalább 2
|
Legalább 2
|
Legalább 2
|
Legalább 2
|
|
Színinterferencia előzetes felmérése
|
50 μl + 1 ml H2O 60 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett (nem színes vizsgálati vegyi anyagok esetében), vagy 50 μl + 2 ml izopropanol 2–3 óráig szobahőmérsékleten keverve (színes vizsgálati vegyi anyagok esetében)
|
 ha a vizsgálati vegyi anyag OD-értéke 570 ±20 nm-en az izopropanol vagy a víz OD-értékének levonását követően nagyobb mint 0,08 (ami megközelítőleg a negatív kontroll OD-átlagának 5 %-a), élő szöveteken adaptált kontrollt kell végezni.
ha a vizsgálati vegyi anyag OD-értéke 570 ±20 nm-en az izopropanol vagy a víz OD-értékének levonását követően nagyobb mint 0,08 (ami megközelítőleg a negatív kontroll OD-átlagának 5 %-a), élő szöveteken adaptált kontrollt kell végezni.
|
|
50 mg + 1 ml H2O 60 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett (nem színes vizsgálati vegyi anyagok esetében)
és/vagy
50 mg + 2 ml izopropanol 2–3 óráig szobahőmérsékleten keverve (színes és nem színes vizsgálati vegyi anyagok esetében)
|
 ha a vizsgálati vegyi anyag OD-értéke 570 ±20 nm-en az izopropanol vagy a víz OD-értékének levonását követően nagyobb mint 0,08 (ami megközelítőleg a negatív kontroll OD-átlagának 5 %-a), élő adaptált kontrollt kell végezni.
ha a vizsgálati vegyi anyag OD-értéke 570 ±20 nm-en az izopropanol vagy a víz OD-értékének levonását követően nagyobb mint 0,08 (ami megközelítőleg a negatív kontroll OD-átlagának 5 %-a), élő adaptált kontrollt kell végezni.
|
|
10 μl + 90 μl H2O 30 ±2 percig szobahőmérsékleten (18–28 oC) keverve
|
 ha a vizsgálati vegyi anyag elszíneződik, élő szöveteken adaptált kontrollt kell végezni
ha a vizsgálati vegyi anyag elszíneződik, élő szöveteken adaptált kontrollt kell végezni
|
|
10 mg + 90 μl H2O 30 ±2 percig szobahőmérsékleten keverve
|
 ha a vizsgálati vegyi anyag elszíneződik, élő szöveteken adaptált kontrollt kell végezni
ha a vizsgálati vegyi anyag elszíneződik, élő szöveteken adaptált kontrollt kell végezni
|
|
|
Közvetlen MTT-redukáló hatás előzetes felmérése
|
50 μl + 1 ml MTT 1 mg/ml koncentrációjú oldata 180 ±15 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
 ha az oldat színe kékre/lilára vált, fagyasztással elölt adaptált kontrollt kell végezni
ha az oldat színe kékre/lilára vált, fagyasztással elölt adaptált kontrollt kell végezni
(negatív kontrollként MTT-oldatba helyezett 50 μl steril ionmentesített vizet használnak)
|
|
50 mg + 1 ml MTT 1 mg/ml koncentrációjú oldata 180 ±15 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
 ha az oldat színe kékre/lilára vált, fagyasztással elölt adaptált kontrollt kell végezni
ha az oldat színe kékre/lilára vált, fagyasztással elölt adaptált kontrollt kell végezni
(negatív kontrollként MTT-oldatba helyezett 50 μl steril ionmentesített vizet használnak)
|
|
30 μl + 300 μl MTT 1 mg/ml koncentrációjú oldata 180 ±15 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
 ha az oldat színe kékre/lilára vált, vízben elölt adaptált kontrollt kell végezni
ha az oldat színe kékre/lilára vált, vízben elölt adaptált kontrollt kell végezni
(negatív kontrollként MTT-oldatba helyezett 30 μl steril ionmentesített vizet használnak)
|
|
30 mg + 300 μl MTT 1 mg/ml koncentrációjú oldata 180 ±15 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
 ha az oldat színe kékre/lilára vált, vízben elölt adaptált kontrollt kell végezni
ha az oldat színe kékre/lilára vált, vízben elölt adaptált kontrollt kell végezni
(negatív kontrollként MTT-oldatba helyezett 30 μl steril ionmentesített vizet használnak)
|
|
|
Előkezelés
|
20 μl Ca2+/Mg2+-mentes DPBS
30 ± 2 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett, fénytől védve.
|
20 μl Ca2+/Mg2+ -mentes DPBS
30 ± 2 percig 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett, fénytől védve.
|
—
|
—
|
|
A kezelési dózisok és alkalmazásuk
|
50 μl (83,3 μl/cm2)
|
50 mg (83,3 mg/cm2) kalibrált eszközzel mérve (e.g. egy csapott kanál, amit 50 mg nátrium-kloridra kalibráltak).
|
10 μl Ca2+/Mg2+-mentes DPBS + 30 ± 2 μl (60 μl/cm2)
Viszkózus anyagokhoz nejlonháló használata ajánlott
|
30 μl Ca2+/Mg2+-mentes DPBS + 30 ± 2 mg (60 mg/cm2)
|
|
Expozíciós idő és hőmérséklet
|
30 perc (± 2 perc)
tápoldatban
37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
6 óra (± 0,25 óra)
tápoldatban
37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
30 perc (± 2 perc)
tápoldatban
37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
4 óra (± 0,1 óra)
tápoldatban
37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
|
Átmosás szobahőmérsékleten
|
3 alkalommal 100 ml Ca2+/Mg2+-mentes DPBS-ben
|
3 alkalommal 100 ml Ca2+/Mg2+-mentes DPBS-ben
|
20 ml Ca2+/Mg2+-mentes DPBS
|
25 ml Ca2+/Mg2+-mentes DPBS
|
|
Expozíció utáni merítés
|
12 perc (± 2 perc) szobahőmérsékleten tápoldatban
|
25 perc (± 2 perc) szobahőmérsékleten tápoldatban
|
30 perc (± 2 perc) 37 oC, 5 % CO2, 95 % RH tápoldatban
|
30 perc (± 2 perc) szobahőmérsékleten tápoldatban
|
|
Expozíció utáni inkubáció
|
120 perc (± 15 perc) tápoldatban 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
18 óra (± 0,25 óra) tápoldatban 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
Nincs
|
18 óra (± 0,5 óra) tápoldatban 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
|
Negatív kontroll
|
50 μl H2O
Párhuzamosan vizsgálva
|
50 μl H2O
Párhuzamosan vizsgálva
|
30 ± 2μl Ca2+/Mg2+-mentes DPBS
Párhuzamosan vizsgálva
|
30 ± 2μl Ca2+/Mg2+-mentes DPBS
Párhuzamosan vizsgálva
|
|
Pozitív kontroll
|
50 μl metil-acetát
Párhuzamosan vizsgálva
|
50 μl metil-acetát
Párhuzamosan vizsgálva
|
30 ± 2μl metil-acetát
Párhuzamosan vizsgálva
|
30 ± 2μl metil-acetát
Párhuzamosan vizsgálva
|
|
MTT-oldat
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
300 μl 1 mg/ml
|
|
MTT inkubációs idő és hőmérséklet
|
180 perc (± 15 perc) 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
180 perc (± 15 perc) 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
180 perc (± 15 perc) 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
180 perc (± 15 perc) 37 ±2 oC, 5 ±1 % CO2, ≥ 95 % RH mellett
|
|
Extraháló oldószer
|
2 ml izopropanol
(kinyerés az inzert tetejéről és aljáról a szövet átszúrásával)
|
2 ml izopropanol
(kinyerés az inzert aljáról a szövet átszúrásával)
|
1,5 ml izopropanol
(kinyerés az inzert tetejéről és aljáról)
|
1,5 ml izopropanol
(kinyerés az inzert aljáról)
|
|
Extrakciós időtartam és hőmérséklet
|
2–3 óra rázatással (~120 rpm) szobahőmérsékleten vagy éjszaka 4–10 oC-on
|
2–3 óra rázatással (~120 rpm) szobahőmérsékleten vagy éjszaka 4–10 oC-on
|
4 óra rázatással (~120 rpm) szobahőmérsékleten vagy rázatás nélkül legalább egész éjszaka 4–10 oC-on
|
Legalább 2 óra rázatással (~120 rpm) szobahőmérsékleten
|
|
OD leolvasás
|
570 nm (550–590 nm)
referenciaszűrő nélkül
|
570 nm (550–590 nm)
referenciaszűrő nélkül
|
570 nm (540–600 nm)
referenciaszűrő nélkül
|
570 nm (540–600 nm)
referenciaszűrő nélkül
|
|
Szövet minőség-ellenőrzése
|
Kezelés 100 μl 0,3 térfogatszázalékos Triton X-100-zal
12,2 perc ≤ ET50 ≤ 37,5 perc
|
Kezelés 100 μl 0,3 térfogatszázalékos Triton X-100-zal
12,2 perc ≤ ET50 ≤ 37,5 perc
|
30 perces kezelés nátrium-dodecil-szulfáttal (SDS) (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,5 mg/ml
|
30 perces kezelés nátrium-dodecil-szulfáttal (SDS) (50 μl)
1,0 mg/ml ≤ IC50 ≤ 3,2 mg/ml
|
|
Elfogadhatósági kritériumok
|
|
1.
|
A negatív kontrollal kezelt szövetreplikátumok átlagos OD-értékének 0,8-nál nagyobbnak és 2,5-nél kisebbnek kell lennie
|
|
2.
|
A pozitív kontrollal 30 percen át kezelt szövetreplikátumok átlagos, a negatív kontroll százalékában kifejezett életképessége nem érheti el az 50 %-ot
|
|
3.
|
A két szövetreplikátum életképessége közötti különbség nem érheti el a 20 %-ot.
|
|
|
1.
|
A negatív kontrollal kezelt szövetreplikátumok átlagos OD-értékének 0,8-nál nagyobbnak és 2,5-nél kisebbnek kell lennie
|
|
2.
|
A pozitív kontrollal 6 órán át kezelt szövetreplikátumok átlagos, a negatív kontroll százalékában kifejezett életképessége nem érheti el az 50 %-ot
|
|
3.
|
A két szövetreplikátum életképessége közötti különbség nem érheti el a 20 %-ot.
|
|
|
1.
|
A negatív kontrollal kezelt szövetreplikátumok átlagos OD-értékének 1,0-nál nagyobbnak, de legfeljebb 2,5-nek kell lennie
|
|
2.
|
A pozitív kontrollal 30 percen át kezelt szövetreplikátumok átlagos, a negatív kontroll százalékában kifejezett életképessége nem haladhatja meg a 30 %-ot
|
|
3.
|
A két szövetreplikátum életképessége közötti különbség nem érheti el a 20 %-ot.
|
|
|
1.
|
A negatív kontrollal kezelt szövetreplikátumok átlagos OD-értékének 1,0-nál nagyobbnak, de legfeljebb 2,5-nek kell lennie
|
|
2.
|
A pozitív kontrollal 4 órán át kezelt szövetreplikátumok átlagos, a negatív kontroll százalékában kifejezett életképessége legfeljebb 20 % lehet
|
|
3.
|
A két szövetreplikátum életképessége közötti különbség nem érheti el a 20 %-ot.
|
|
3. függelék
A VRM1 SZABVÁNYMŰVELETI ELJÁRÁSÁN ALAPULÓ SZEMLÉLTETŐ FOLYAMATÁBRA ARRÓL, HOGYAN LEHET AZONOSÍTANI ÉS KEZELNI A KÖZVETLEN MTT-REDUKÁLÓ ÉS/VAGY SZÍNINTERFERENCIÁT OKOZÓ VEGYI ANYAGOKAT
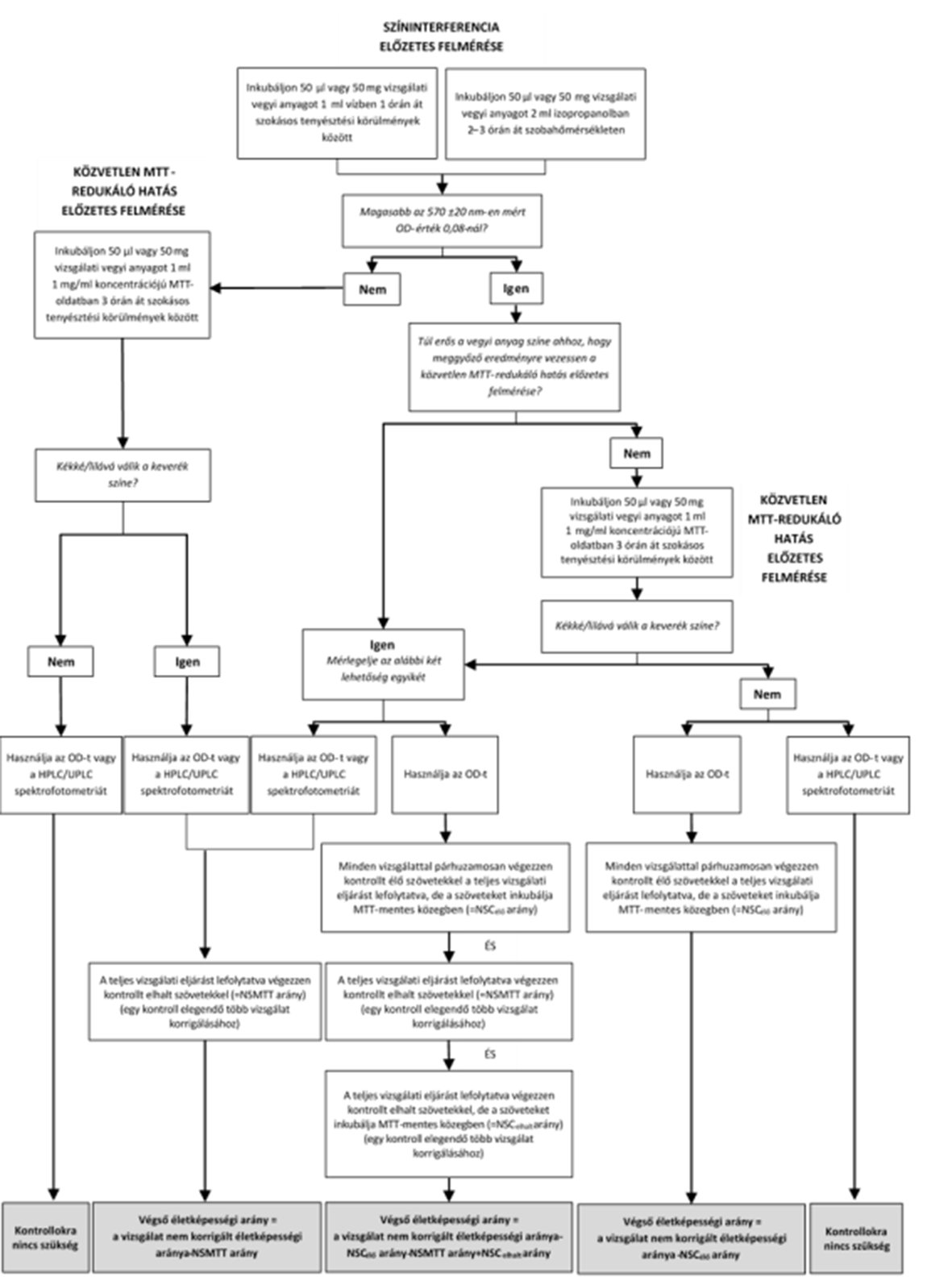
4. függelék
A VRM2 SZABVÁNYMŰVELETI ELJÁRÁSÁN ALAPULÓ SZEMLÉLTETŐ FOLYAMATÁBRA ARRÓL, HOGYAN LEHET AZONOSÍTANI ÉS KEZELNI A KÖZVETLEN MTT-REDUKÁLÓ ÉS/VAGY SZÍNINTERFERENCIÁT OKOZÓ VEGYI ANYAGOKAT

5. függelék
AZ RHCE SZÖVETKONSTRUKCIÓKBÓL KIVONT MTT-FORMAZÁN MÉRÉSÉRE HASZNÁLT HPLC/UPLC SPEKTROFOTOMETRIÁS RENDSZER MINŐSÍTÉSÉNEK FŐBB PARAMÉTEREI ÉS ELFOGADHATÓSÁGI KRITÉRIUMAI
|
Paraméter
|
Az FDA-iránymutatás szerinti protokoll (36) (38)
|
Elfogadhatósági kritériumok
|
|
Szelektivitás
|
Izopropanol elemzése, élő vakpróba (nem kezelt élő RhCE-szövetekből kivont izopropanol), elhalt vakpróba (nem kezelt elhalt RhCE-szövetekből kivont izopropanol), és egy festék (pl. metilénkék) elemzése
|
Területinterferencia ≤ TerületAMHÉ
(88) 20 %-a
|
|
Precizitás
|
Minőség-ellenőrzések (vagyis MTT-formazán 1,6 μg/ml, 16 μg/ml és 160 μg/ml mellett) izopropanolban (n=5)
|
Variációs koefficiens ≤ 15 % vagy ≤ az AMHÉ 20 %-a
|
|
Pontosság
|
Minőség-ellenőrzés izopropanolban (n=5)
|
Eltérési arány ≤ 15 % vagy ≤ az AMHÉ 20 %-a
|
|
Mátrixhatás
|
Minőség-ellenőrzés élő vakpróbán (n=5)
|
85 % ≤ Mátrixhatás % ≤ 115 %
|
|
Átvitel
|
Izopropanol elemzése egy FMHÉ (89) szabvány után
|
Területinterferencia ≤ TerületAMHÉ 20 %-a
|
|
Megismételhetőség (napon belüli)
|
3 független kalibrációs görbe (izopropanolba helyezett MTT-formazán 6 egymás utáni 1/3-os hígítása kezdve a FMHÉ-nél, vagyis 200 μg/ml-nél);
Minőség-ellenőrzés izopropanolban (n=5)
|
Kalibrációs görbék: Eltérési arány ≤ 15 % vagy ≤ az AMHÉ 20 %-a
Minőség-ellenőrzések: Eltérési arány ≤ 15 % és variációs koefficiens ≤ 15 %
|
|
Megismételhetőség (napok közötti)
|
1. nap: 1 kalibrációs görbe és minőség-ellenőrzések izopropanolban (n=3)
2. nap: 1 kalibrációs görbe és minőség-ellenőrzések izopropanolban (n=3)
3. nap: 1 kalibrációs görbe és minőség-ellenőrzések izopropanolban (n=3)
|
|
Az MTT-formazán rövid távú stabilitása az RhCE szövetkivonatban
|
Minőség-ellenőrzések élő vakpróbán (n=3) a készítés napján és 24 órás szobahőmérsékleten való tárolás után
|
Eltérési arány ≤ 15 %
|
|
Az MTT-formazán hosszú távú stabilitása az RhCE szövetkivonatban, ha szükséges
|
Minőség-ellenőrzések élő vakpróbán (n=3) a készítés napján és több napon keresztüli -20 °C-on való tárolás után
|
Eltérési arány ≤ 15 %
|
B.70. A VEGYI ANYAGOK ÖSZTROGÉNRECEPTOROKHOZ KÖTÉSI AFFINITÁSÁNAK HUMÁN REKOMBINÁNS ÖSZTROGÉNRECEPTORRAL (hrER) TÖRTÉNŐ ÉSZLELÉSÉRE SZOLGÁLÓ IN VITRO VIZSGÁLATOK
ÁLTALÁNOS BEVEZETÉS
Az OECD teljesítményalapú vizsgálati iránymutatása
|
1.
|
Ez a vizsgálati módszer egyenértékű az OECD 493. vizsgálati iránymutatásában (2015) leírt módszerrel. A 493. teljesítményalapú vizsgálati iránymutatás (PBTG) az ösztrogénreceptorokhoz kötési affinitást mutató anyagok észlelésére szolgáló humán rekombináns in vitro vizsgálatok (humán ösztrogénreceptor-kötési vizsgálatok) módszertanát ismerteti. Az iránymutatás az ösztrogénreceptort (vagyis az ERα-t) kötő anyagok azonosítását lehetővé tévő két, végrehajtás és funkcionalitás szempontjából hasonló vizsgálati módszert tartalmaz, és a veszély értékelésére szolgáló új és átdolgozott vizsgálati módszerek validálásáról és nemzetközi elfogadásról szóló OECD-irányelvben (1) megfogalmazott validálási elvekkel összhangban megkönnyíti az új – hasonló vagy módosított – vizsgálati módszerek kidolgozását. E teljesítményalapú vizsgálati iránymutatás alapjául szolgáló, teljes körűen validált (a 2. és a 3. függelékben ismertetett) vizsgálati referenciamódszerek a következők:
|
—
|
a teljes hosszúságú humán rekombináns ERα használatán alapuló Freyberger-Wilson-féle (FW) in vitro ösztrogénreceptor-kötési vizsgálat (2), valamint
|
|
—
|
a Chemical Evaluation and Research Institute (CERI) által kidolgozott in vitro ösztrogénreceptor-kötési vizsgálat, amely humán rekombináns fehérje ligandumkötő-doménjének használatán alapul (2).
|
A rendelkezésre álló teljesítményszabványok (3) megkönnyítik az ugyanezt a veszélyes végpontot célzó hasonló vizsgálati módszerek kidolgozását és validálását, és lehetővé teszik a 493. teljesítményalapú vizsgálati iránymutatás időben történő módosítását, hogy az új, hasonló vizsgálatokat be lehessen építeni az átdolgozott iránymutatásba. Hasonló vizsgálatok azonban csak azután foglalhatók bele az iránymutatásba, hogy az OECD felülvizsgálta és jóváhagyta a teljesítményszabványok teljesülését. A 493. vizsgálati iránymutatás alá tartozó vizsgálatok egységesen felhasználhatók az OECD-tagállamok ösztrogénreceptor-kötési vizsgálatok eredményeire vonatkozó követelményeinek kialakításához, ugyanakkor biztosítják az adatok kölcsönös elfogadásának előnyeit.
|
A vizsgálati módszerhez tartozó vizsgálatok háttere és elvei
|
2.
|
Az OECD 1998-ban kiemelt fontosságú tevékenységet indított az endokrin rendszert esetlegesen károsító vegyi anyagok kiszűrésére és vizsgálatára vonatkozó meglévő vizsgálati iránymutatások felülvizsgálata, valamint új vizsgálati iránymutatások kidolgozása érdekében. Az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére szolgáló fogalmi keretét 2012-ben átdolgozták. Az elvi keretrendszer eredeti és átdolgozott változatát mellékletként csatolták az endokrin rendszert károsító vegyi anyagok értékelésére vonatkozó szabványosított vizsgálati iránymutatásokról szóló iránymutatáshoz (4). Az elvi keretrendszer öt szintet határoz meg, amelyek mindegyike a biológiai komplexitás egy különálló szintjének felel meg. Az e vizsgálati módszer keretében ismertetett ösztrogénreceptor-kötési vizsgálatok a 2. szinten helyezkednek el, amely magában foglalja „a kiválasztott endokrin mechanizmus(ok)ról/útvonal(ak)ról adatokat szolgáltató in vitro vizsgálatokat”. Ez a vizsgálati módszer olyan in vitro receptorkötési vizsgálatokhoz használatos, amelyek célja a humán ösztrogénreceptor alfa (ERα) ligandumainak azonosítása.
|
|
3.
|
Egyértelműen bizonyították az in vitro ösztrogénreceptor-kötési vizsgálat biológiai funkciók terén betöltött jelentőségét. Az ösztrogénreceptor-kötési vizsgálatokat abból a célból hozták létre, hogy beazonosítsák az ösztrogén hormon szignalizációs útvonalát potenciálisan zavaró vegyi anyagokat, és az elmúlt két évtized során széles körben alkalmazták őket az ösztrogénreceptor szöveti eloszlásának jellemzésére, valamint az ösztrogénreceptor agonistáinak/antagonistáinak azonosítására. Ezek a vizsgálatok az ösztrogén szignalizációs útvonalának első lépését jelentő ligandum-receptor kölcsönhatással foglalkoznak, ami kulcsfontosságú szerepet tölt be minden gerinces élőlény szaporodási funkciójában.
|
|
4.
|
Az ösztrogének és az ösztrogénreceptorok kölcsönhatása befolyásolhatja az ösztrogénhatásra érzékeny gének transzkripcióját és okozhat nem-genomiális hatásokat, amelyek többek között a sejtosztódás, a normál magzati fejlődés és a szaporodási funkciók sejtszintű előidézéséhez vagy gátlásához vezethetnek (5) (6) (7). A normál ösztrogénrendszerek zavara károsan befolyásolhatja a normál egyedfejlődést (ontogenezist), a reprodukciós egészséget és a reproduktív rendszer integritását. A nem megfelelő ösztrogénreceptor szignál olyan hatásokat válthat ki, mint a hormonfüggő daganatos betegségek megemelkedett kockázata, a termékenység csökkenése, valamint a magzati növekedési és fejlődési rendellenességek (8).
|
|
5.
|
Az in vitro kötési vizsgálatok egy anyagnak a specifikus receptor ligandumkötő helyével történő közvetlen kölcsönhatásán alapulnak, ami a géntranszkripciót szabályozza. A humán rekombináns ösztrogénreceptor alfa (hrERα) kötési vizsgálat főként a radioaktívan jelölt ligandum (a [3H]-17β-ösztradiol) ösztrogénreceptorhoz történő kötését méri a vizsgálati vegyi anyag (vagyis a kompetítor) növekvő koncentrációjának jelenlétében. Az ösztrogénreceptorok iránt nagy affinitást mutató vizsgálati vegyi anyagok alacsonyabb koncentrációban versenyeznek a radioaktívan jelölt ligandummal azokhoz a vegyi anyagokhoz képest, amelyek kisebb affinitást mutatnak a receptor iránt. A vizsgálat két fő összetevőből áll: egy telítési kötési kísérletből, amelynek során meghatározzák a receptor-ligandum kölcsönhatás paramétereit és dokumentálják az ösztrogénreceptor-specifikusságot, majd egy kompetitív kötési kísérletből, amely jellemzi a vizsgálati vegyi anyag és a radioaktívan jelölt ligandum ER-kötőhelyekért folytatott vetélkedését.
|
|
6.
|
A CERI és a Freyberger–Wilson kötési vizsgálat validálási tanulmánya igazolta, hogy e vizsgálatok tervezett felhasználásuk tekintetében relevánsak és megbízhatóak (2).
|
|
7.
|
A vizsgálati módszerben használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A receptorkötési vizsgálatok alkalmazási területe és korlátai
|
8.
|
E vizsgálatokat szűrési és rangsorolási célra ajánlják, ugyanakkor a molekuláris kiváltó eseményre vonatkozólag is képesek adatokat szolgáltatni, amelyek felhasználhatók egy bizonyítékok súlyozásán (WoE) alapuló megközelítésben. A vizsgálatok tárgya az ösztrogénreceptor α ligandumkötő doménjén in vitro létrejövő kémiai kötés. Ezért az eredményekből nem lehet közvetlenül következtetni az intakt in vivo endokrin rendszerben megvalósuló összetett jelátvitelre és szabályozásra.
|
|
9.
|
A természetes ligandum, a 17β-ösztradiol megkötése az első lépése annak a molekuláris eseménysorozatnak, amely aktiválja a célgének transzkripcióját, majd végül egy fiziológiai változást idéz elő (9). Tehát az ösztrogénreceptor α ligandumkötő doménjéhez való kötést az ösztrogénreceptorok által kiváltott endokrin zavarok (ED) egyik fő mechanizmusaként tartják számon, bár más mechanizmusok is okozhatnak endokrin zavarokat, többek között i. az ösztrogénreceptor α ligandumkötő régióján kívüli részeivel létrejött kölcsönhatások, ii. az ösztrogén jelátvitel szempontjából releváns más receptorokkal, mint pl. az ösztrogénreceptor β-val, az ösztrogénreceptorokkal kapcsolt g-fehérjékkel, az endokrin rendszeren belüli más receptorokkal és enzimrendszerekkel létrejött kölcsönhatások, iii. hormonszintézis, iv. a hormonok metabolikus aktiválása és/vagy inaktiválása, v. hormonok elszállítása a célszövetekhez és vi. hormonok kiürülése a szervezetből. E hatásmechanizmusokkal a vizsgálati módszer alá tartozó vizsgálatok egyike sem foglalkozik.
|
|
10.
|
E vizsgálati módszer célja, hogy felmérje az anyagok humán ösztrogénreceptor α-hoz való kötődési képességét, és nem teszi lehetővé az ösztrogénreceptor α agonistái és antagonistái közötti különbségtételt. E vizsgálatok nem foglalkoznak továbbá az eseménysorozat későbbi történéseivel, például a géntranszkripcióval vagy a fiziológiai változásokkal. Mivel a validálás során csak különálló, egy komponensű anyagokat használtak, a vizsgálat keverékekre való alkalmazhatósága nem vetődött fel. Mindazonáltal a vizsgálatok elvileg alkalmazhatók több összetevőből álló anyagok és keverékek vizsgálatára. A vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért. Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény.
|
|
11.
|
A sejtmentes receptorrendszerek nem rendelkeznek saját metabolikus képességgel, metabolikus enzimrendszerekkel kombinált használatukat pedig nem validálták. Elképzelhető, hogy kidolgozható lenne egy metabolikus aktivitást magában foglaló vizsgálat, ez azonban további validálást tenne szükségessé.
|
|
12.
|
Azok a vegyi anyagok, amelyek denaturálhatják a fehérjét (vagyis a receptor fehérjét), például a felületaktív anyagok, vagy amelyek módosíthatják a vizsgálati puffer pH-értékét, nem vizsgálhatók, vagy csak olyan koncentrációkban vizsgálhatók, amelyek nem idéznek elő ilyen hatásokat. Ezen túlmenően egy adott vizsgálati vegyi anyag tesztelhető koncentrációtartományát a vizsgálati pufferben való oldhatósága is korlátozza.
|
|
13.
|
Az 1. táblázat tájékoztatási céllal ismerteti az e vizsgálati módszer által leírt mindkét teljes körűen validált vizsgálatban tesztelt 24 anyagra vonatkozó eredményeket. A szóban forgó anyagok közül 17-et ösztrogénreceptorhoz kötődő anyagként határoztak meg, 6-ot pedig nem kötődőnek minősítették a közzétett vizsgálati jelentésnek, többek között az in vitro ösztrogénreceptor transzkripciós aktivációs vizsgálatok és/vagy az uterotrofikus vizsgálat alapján (9) (10) (11) (12) (13) (14) (15). Az 1. táblázatban összefoglalt adatok vonatkozásában 10-4 M koncentrációig szinte 100 %-os egyezés mutatható ki a két vizsgálat között valamennyi anyag besorolása tekintetében, és minden anyagot helyesen osztályoztak ösztrogénreceptorhoz kötődő és nem kötődő anyagként. Erről az anyagcsoportról, valamint a validálási tanulmányok során az ösztrogénreceptor-kötési vizsgálatokban tesztelt egyéb anyagokról a humán ösztrogénreceptorhoz-kötési vizsgálat teljesítményszabványai nyújtanak bővebb tájékoztatást (3), lásd a 2. függeléket (1., 2., és 3. táblázat).
1. táblázat
A Freyberger–Wilson (FW) és a CERI humán ösztrogénreceptor-kötési vizsgálat során tesztelt anyagok ösztrogénreceptorhoz kötődő és nem kötődő anyagként való besorolása a várt válasszal való összevetésben
|
|
Az anyag neve
|
CAS-szám
|
Várt válasz
|
FW vizsgálat
|
CERI vizsgálat
|
MESH kémiai osztály
|
Termékosztály
|
|
|
Koncentráció- tartomány (M)
|
Besorolás
|
Koncentráció- tartomány (M)
|
Besorolás
|
|
1
|
17β-ösztradiol
|
50-28-2
|
Kötődő
|
1×10-11–1×10-6
|
Kötődő
|
1×10-11–1×10-6
|
Kötődő
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
2
|
Noretinodrel
|
68-23-5
|
Kötődő
|
3×10-9–30×10-4
|
Kötődő
|
3×10-9–30×10-4
|
Kötődő
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
3
|
Noretindron
|
68-22-4
|
Kötődő
|
3×10-9–30×10-4
|
Kötődő
|
3×10-9–30×10-4
|
Kötődő
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
4
|
Di-n-butil-ftalát
|
84-74-2
|
Nem kötődő (*5)
|
1×10-10–1×10-4
|
Nem kötődő (*6)
(1)
|
1×10-10–1×10-4
|
Nem kötődő (*6)
(1)
|
Szénhidrogén (ciklikus), észter
|
Lágyítószer, köztes vegyi anyag
|
|
5
|
Dietilstilbösztrol
|
56-53-1
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (ciklikus), fenol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
6
|
17α-etinilösztradiol
|
57-63-6
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
7
|
Mezo-hexestrol
|
84-16-2
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (ciklikus), fenol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
8
|
Geniszten
|
446-72-0
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (heterociklikus), flavonid
|
Természetes termék
|
|
9
|
Equol
|
531-95-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Fitoösztrogén metabolit
|
Természetes termék
|
|
10
|
Butil-parabén (nátrium-butil-4-hidroxi-benzoát)
|
94-26-8
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Parabén
|
Tartósítószer
|
|
11
|
Nonilfenol (keverék)
|
84852-15-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Alkil-fenol
|
Köztes vegyület
|
|
12
|
o,p’-DDT
|
789-02-6
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szerves klór
|
Rovarölő szer
|
|
13
|
Kortikoszteron
|
50-22-6
|
Nem kötődő (*5)
|
1×10-10–1×10-4
|
Nem kötődő
|
1×10-10–1×10-4
|
Nem kötődő
|
Szteroid
|
Természetes termék
|
|
14
|
Zearalenon
|
17924-92-4
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (heterociklikus), lakton
|
Természetes termék
|
|
15
|
Tamoxifén
|
10540-29-1
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (ciklikus)
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
16
|
5α-dihidrotesztoszteron
|
521-18-6
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szteroid, nem fenolos
|
Természetes termék
|
|
17
|
Biszfenol-A
|
80-05-7
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Fenol
|
Köztes vegyi anyag
|
|
18
|
4-n-heptilfenol
|
1987-50-4
|
Kötődő
|
1×10-10–1×10-3
|
Kétértelmű (5)
|
1×10-10–1×10-3
|
Kötődő
|
Alkil-fenol
|
Köztes
|
|
19
|
Kepone (klórdekon)
|
143-50-0
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Szénhidrogén (halogénezett)
|
Peszticid
|
|
20
|
Benz(a)antracén
|
56-55-3
|
Nem kötődő
|
1×10-10–1×10-3
|
Nem kötődő (6)
|
1×10-10–1×10-3
|
Nem kötődő (6)
|
Aromás szénhidrogén
|
Köztes
|
|
21
|
Enterolakton
|
78473-71-9
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
1×10-10–1×10-3
|
Kötődő
|
Fitoösztrogén
|
Természetes termék
|
|
22
|
Progeszteron
|
57-83-0
|
Nem kötődő (*5)
|
1×10-10–1×10-4
|
Nem kötődő
|
1×10-10–1×10-4
|
Nem kötődő
|
Szteroid
|
Természetes termék
|
|
23
|
Oktil-trietoxi-szilán
|
2943-75-1
|
Nem kötődő
|
1×10-10–1×10-3
|
Nem kötődő
|
1×10-10–1×10-3
|
Nem kötődő
|
Szilán
|
Felületmódosító
|
|
24
|
Atrazin
|
1912-24-9
|
Nem kötődő (*5)
|
1×10-10–1×10-4
|
Nem kötődő
|
1×10-10–1×10-4
|
Nem kötődő
|
Heterociklikus vegyület
|
Gyomirtó szer
|
|
A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI
A vizsgálat alapvető összetevői
|
14.
|
Ez a vizsgálati módszer olyan vizsgálatokra vonatkozik, amelyek ösztrogénreceptort és a receptorhoz kellően nagy affinitással kötődő ligandumot alkalmaznak, amely a vizsgálat során markerként/nyomjelzőként használható, és a vizsgálati vegyi anyagok növekvő koncentrációjával leszorítható. A kötési vizsgálatok az alábbi két fő összetevőből állnak: 1) telítési kötés és 2) kompetitív kötés. A telítési kötési vizsgálat során ellenőrzik a receptor preparátumok specifikusságát és aktivitását, a kompetitív kötési kísérlet során pedig értékelik a vizsgálati vegyi anyagok humán ösztrogénreceptorhoz való kötési képességét.
|
Kontrollok
|
15.
|
Meg kell határozni, hogy mi alapján javasolják a párhuzamosan vizsgált referenciaösztrogént és kontrollanyagokat. Az egyidejűleg elvégzett kontrollok (oldószeres (vivőanyagos), pozitív (ösztrogénreceptorhoz kötődő; erős és gyenge affinitással rendelkező) és negatív (nem kötődő) kontrollok) arra szolgálnak, hogy igazolják a vizsgálat tesztkörülmények közötti megfelelő működését, és megteremtsék az alapját az egyes kísérletek összevetésének; általában részét képezik a szóban forgó kísérlet elfogadhatósági kritériumainak (1). Minden vizsgálatmenetben ugyanazon a lemezen vizsgálni kell a referenciaösztrogén teljes koncentrációgörbéjét és a kontrollanyagokat (vagyis a gyengén kötődő és a nem kötődő anyagot). Az összes többi lemeznek az alábbiakat kell tartalmazniuk: 1) az E2 és a gyengén kötődő anyag magas koncentrációját (amely megközelítőleg teljesen leszorítja a radioaktívan jelölt ligandumot) és közepes koncentrációját (amely körülbelül az IC50 hatásának felel meg), három ismétlésben; 2) az oldószer és a nem specifikus kötődés vizsgálatát, három-három ismétlésben.
|
Standard minőség-ellenőrzési eljárások
|
16.
|
El kell végezni az egyes vizsgálatokhoz meghatározott standard minőség-ellenőrzési eljárásokat annak érdekében, hogy garantálni lehessen a receptorok aktivitását, a vegyi anyag megfelelő koncentrációértékeit, a tűréshatárok többszöri ismétlésen keresztüli stabilitását, valamint a várt ösztrogénreceptor-kötési válaszreakciók biztosításának hosszú távú képességét.
|
A laboratórium jártasságának igazolása
|
17.
|
Mielőtt ismeretlen vegyi anyagokat tesztelnének az e vizsgálati módszerhez kapcsolódó bármely vizsgálattal, a laboratóriumoknak igazolniuk kell az adott vizsgálat alkalmazásában szerzett jártasságukat azáltal, hogy végrehajtanak az ösztrogénreceptor preparátum specifikusságának és aktivitásának ellenőrzésére szolgáló telítési vizsgálatokat, valamint a referenciaösztrogént és a kontrollokat (gyengén kötődő és nem kötődő anyagokat) is tesztelő kompetitív kötési vizsgálatokat. A laboratóriumoknak adatbázist kell létrehozniuk a referenciaösztrogénnel és a kontrollanyagokkal különböző napokon végzett 3–5 független kísérletből származó eredményekből. Ezek a kísérletek teremtik meg az alapját annak, hogy a laboratóriumok a referenciaösztrogénre és a kontrollanyagokra vonatkozólag dokumentált adatokkal rendelkezzenek, amelyeket a jövőbeni vizsgálatmenetek során felhasználnak a vizsgálat elfogadhatóságának értékeléséhez.
|
|
18.
|
A vizsgálati rendszer reakcióképességét a 2. táblázatban felsorolt jártassági tesztanyagok vizsgálata révén is meg kell erősíteni. A jártassági tesztanyagok jegyzéke az ösztrogénreceptor-kötési vizsgálatok teljesítményszabványaiban (3) megadott referenciaanyagok egy csoportjából tevődik össze. Ezek az anyagok kereskedelmi forgalomban beszerezhetők, olyan kémiai osztályokat képviselnek, amelyeknél gyakori az ösztrogénreceptor-kötési aktivitás, a várt ösztrogénreceptor-kötési potenciál megfelelő (erőstől a gyengéig terjedő) tartományát képviselik, és vannak közöttük nem kötődő (azaz negatív) anyagok. Az egyes jártassági tesztanyagok vizsgált koncentrációértékeinek le kell fedniük a 2. táblázatban szereplő tartományt. Minden anyag esetében legalább három kísérletet kell végezni, és az eredményeknek összhangban kell lenniük a vegyi anyag várt aktivitásával. Az összes kísérletet független módon (vagyis a receptor, a vizsgálati vegyi anyagok és a reagens frissen készített oldataival) kell végrehajtani, minden egyes koncentrációértéket három ismétlésben vizsgálva. A jártasság az egyes jártassági tesztanyagok megfelelő (pozitív/negatív) osztályba sorolásával igazolható. A vizsgálatok elsajátítása során a jártassági vizsgálatot minden szakembernek el kell végeznie.
2. táblázat
A humán ösztrogénreceptor kompetitív kötési vizsgálatokban használt kontrollanyagok és jártassági tesztanyagok jegyzéke
(90)
|
Szám
|
Az anyag neve
|
CAS-szám (91)
|
Várt válasz (92)
(93)
|
Vizsgálati koncentrációtartomány (M)
|
MeSH kémiai osztály (94)
|
Termékosztály (95)
|
|
Kontrollanyagok (referenciaösztrogén, gyengén kötődő és nem kötődő anyag)
|
|
1
|
17-β-ösztradiol
|
50-28-2
|
Kötődő
|
1×10-11–1×10-6
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
2
|
Noretinodrel (vagy) Noretindron
|
68-23-5 (vagy) 68-22-4
|
Kötődő
|
3×10-9–30×10-6
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
3
|
Oktil-trietoxi-szilán
|
2943-75-1
|
Nem kötődő
|
1×10-10–1×10-3
|
Szilán
|
Felületmódosító
|
|
Jártassági tesztanyagok (95)
|
|
4
|
Dietilstilbösztrol
|
56-53-1
|
Kötődő
|
1×10-11–1×10-6
|
Szénhidrogén (ciklikus), fenol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
5
|
17α-etinilösztradiol
|
57-63-6
|
Kötődő
|
1×10-11–1×10-6
|
Szteroid
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
6
|
mezo-Hexestrol
|
84-16-2
|
Kötődő
|
1×10-11–1×10-6
|
Szénhidrogén (ciklikus), fenol
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
7
|
Tamoxifén
|
10540-29-1
|
Kötődő
|
1×10-11–1×10-6
|
Szénhidrogének (ciklikus)
|
Gyógyszer, állatgyógyászati hatóanyag
|
|
8
|
Geniszten
|
446-72-0
|
Kötődő
|
1×10-10–1×10-3
|
Heterociklikus vegyület, flavonid
|
Természetes termék
|
|
9
|
Biszfenol-A
|
80-05-7
|
Kötődő
|
1×10-10–1×10-3
|
Fenol
|
Köztes vegyi anyag
|
|
10
|
Zearalenon
|
17924-92-4
|
Kötődő
|
1×10-11–1×10-3
|
Heterociklikus vegyület, lakton
|
Természetes termék
|
|
11
|
Butil-parabén
|
94-26-8
|
Kötődő
|
1×10-11–1×10-3
|
Karbonsav, fenol
|
Tartósítószer
|
|
12
|
Atrazin
|
1912-24-9
|
Nem kötődő
|
1×10-11–1×10-6
|
Heterociklikus vegyület
|
Gyomirtó szer
|
|
13
|
Di-n-butil-ftalát (DBP) (96)
|
84-74-2
|
Nem kötődő (97)
|
1×10-10–1×10-4
|
Szénhidrogén (ciklikus), észter
|
Lágyítószer, köztes vegyi anyag
|
|
14
|
Kortikoszteron
|
50-22-6
|
Nem kötődő
|
1×10-11–1×10-4
|
Szteroid
|
Természetes termék
|
|
A vizsgálati vegyi anyagok oldhatósági vizsgálata és koncentrációtartományuk behatárolása
|
19.
|
Előzetes vizsgálatot kell végezni az egyes vizsgálati vegyi anyagok oldhatósági határértékének meghatározására, valamint a vizsgálat során alkalmazandó megfelelő koncentrációtartomány megállapítására. A vizsgálati vegyi anyagok oldhatósági határértékét először oldószerben kell meghatározni, majd a vizsgálati körülmények mellett meg kell erősíteni. A vizsgálat során tesztelt végső koncentráció nem haladhatja meg az 1 mM-t. A koncentrációtartomány-behatároló vizsgálatnak magában kell foglalnia egy oldószeres kontrollt és a legmagasabb elfogadható koncentrációtól (pl. 1 mM, illetve az oldhatósági határértéktől függően ennél alacsonyabb koncentrációtól) induló, nyolc koncentrációból álló logaritmikus hígítási sorozatot, az esetlegesen tapasztalt zavarosságot vagy kicsapódást fel kell jegyezni. A második és a harmadik kísérlet koncentrációértékeit úgy kell megválasztani, hogy jobban illeszkedjenek a koncentráció-válasz görbéhez.
|
A vizsgálatmenet elfogadhatósági kritériumai
|
20.
|
A vizsgálatmenetek elfogadására vagy elvetésére a minden kísérletben vizsgált referenciaösztrogén és kontrollanyagok eredményeinek értékelése alapján kerül sor. Először is, az 1. lemez tekintetében minden kísérlet referenciaként szolgáló kontrollanyagainak görbeillesztési paramétereiből (pl. az IC50 és a Hill-meredekség) létrehozott teljes koncentrációgörbéknek meg kell felelniük a teljesítménymutatóknak, amit a CERI és a Freyberger–Wilson vizsgálat protokolljában (2. és 3. függelék) feltüntetett eredmények, valamint a vizsgálatot végző laboratórium történeti kontrolladatai alapján értékelnek. Minden kísérletben helyesen kell besorolni valamennyi kontrollanyagot (a referenciaösztrogént, valamint a gyengén kötődő és a nem kötődő anyagot). Másodszor pedig fel kell mérni, hogy az összes többi lemezen végzett kontrollok eredményei egybevágóak-e az 1. lemez eredményeivel. A vizsgálati vegyi anyag megfelelő koncentrációtartományát kell vizsgálni ahhoz, hogy egyértelműen meg lehessen határozni a kompetitív kötési görbe telítési csúcsát. A vizsgálati vegyi anyag egyes koncentrációinak ismétlései közötti, illetve a három független vizsgálatmenet közötti eltéréseknek észszerűnek és tudományosan védhetőnek kell lenniük. A vizsgálat következetes végrehajtásának képességét bizonyítani kell azáltal, hogy a laboratóriumok adatbázist hoznak létre és tartanak fent a referenciaösztrogén és a kontrollanyagok adataiból. A laboratóriumon belüli reprodukálhatóság mérésére használhatók a referenciaösztrogén és a kontrollként szolgáló gyengén kötődő anyag több kísérletből származó görbeillesztési paraméterei átlagának standard deviációi (SD) vagy variációs koefficiensei (CV). Az egyes vizsgálatmenetek lemezein vizsgált kontrollanyagok és minden vizsgálati vegyi anyag eredményeinek értelmezése során szakmai megítélés alapján kell dönteni.
Ezenkívül érvényesülniük kell az elfogadhatósági kritériumokra vonatkozó alábbi elveknek is:
|
—
|
elegendő mennyiségű adattal kell rendelkezni ahhoz, hogy számszerűen értékelni lehessen az ösztrogénreceptor-kötést;
|
|
—
|
a vizsgált koncentrációknak a vizsgálati vegyi anyag oldhatósági tartományán belül kell maradniuk.
|
|
Az adatok elemzése
|
21.
|
A telítési és a kompetitív kötési adatok meghatározott adatelemzési eljárásának meg kell felelnie a receptor és a ligandum közötti kölcsönhatások jellemzésére vonatkozó főbb elveknek. A telítési kötési adatokat jellemzően egy nem-lineáris regressziós modellel elemzik, amellyel meghatározható mind a teljes, mind a nem specifikus kötődés. A Bmax és a Kd értékének meghatározásakor a ligandum csökkenése miatt szükség lehet korrekcióra (pl. Swillens, 1995 (19)). A kompetitív kötési vizsgálatok adatait általában transzformálják (pl. a specifikus kötődés arányát és a vizsgálati vegyi anyag koncentrációit (log M)). Az egyes vizsgálati vegyi anyagok log (IC50) értékének megbecsüléséhez egy megfelelő nem-lineáris görbeillesztő szoftvert kell használni, amely illeszteni tud a négyparaméteres Hill-egyenletre. A kezdeti elemzést követően meg kell határozni a görbeillesztési paramétereket, és vizuálisan ellenőrizni kell, hogy a kötési adatok mennyire illeszkednek a létrehozott kompetitív kötési görbére. Bizonyos esetekben a legjobban illeszkedő görbe meghatározásához további elemzésre lehet szükség (pl. a görbe tetejének és/vagy aljának szűkítésére, a 10 %-os szabály alkalmazására, lásd a 4. függeléket és a 2. referenciát (a III.A.2. részben)).
|
|
22.
|
Az elfogadhatósági kritériumok (20. pont) teljesítése jelzi a vizsgálati rendszer megfelelő működését, de nem garantálja egy adott vizsgálat adatainak pontosságát. Az adatok pontosságát az jelzi a legjobban, ha az első vizsgálat helyes eredményei megismételhetők.
|
Az adatok értelmezésének általános kritériumai
|
23.
|
Jelenleg nincs általánosan elfogadott módszer az ösztrogénreceptor-kötési adatok értelmezésére. Ugyanakkor a humán ösztrogénreceptor által közvetített aktivitás kvalitatív (pl. kötődő/nem kötődő) és/vagy kvantitatív (pl. log IC50, relatív kötődési affinitás (RBA) stb.) értékelésének empirikus adatokon és megalapozott tudományos véleményen kell nyugodnia.
|
Vizsgálati jelentés
|
24.
|
A vizsgálati jelentésnek a következőket kell tartalmaznia:
|
|
Vizsgálat:
|
—
|
az alkalmazott vizsgálat.
|
|
|
|
Kontroll/referencia/vizsgálati vegyi anyag
|
—
|
eredet, gyártási szám, felhasználási határidő, ha rendelkezésre áll;
|
|
—
|
a vizsgálati vegyi anyag stabilitása, ha ismert;
|
|
—
|
a vizsgálati vegyi anyag oldhatósága és stabilitása az oldószerben, ha ismert;
|
|
—
|
adott esetben a pH-érték, az ozmolalitás és a kicsapódás mérése abban a tápfolyadékban, amelyhez a vizsgálati vegyi anyagot hozzáadták.
|
|
|
|
Egy összetevőből álló anyag:
|
—
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok;
|
|
—
|
kémiai azonosítás, például IUPAC- vagy CAS-névvel, CAS-szám, SMILES- vagy InChI-kód, szerkezeti képlet alapján, tisztaság, adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb. alapján;
|
|
|
|
Több összetevőből álló anyag, UVCB-k és keverékek:
|
—
|
amennyiben lehetséges, az összetevők kémiai azonosítója (lásd fent), mennyiségi előfordulása és releváns fizikai-kémiai tulajdonságai által jellemezve.
|
|
|
|
Oldószer/vivőanyag:
|
—
|
jellemzés (az anyag természete, forgalmazója és gyártási száma);
|
|
—
|
az oldószer/vivőanyag kiválasztásának indokolása;
|
|
—
|
a vizsgálati vegyi anyag oldhatósága és stabilitása az oldószerben/vivőanyagban, ha ismert.
|
|
|
|
Receptorok:
|
—
|
a receptorok eredete (forgalmazó, katalógusszám, receptor faj, a forgalmazó által megadott aktív receptor koncentráció, a forgalmazó által kiadott tanúsítvány);
|
|
—
|
a receptorok jellemzése (többek között a telítési kötési vizsgálat eredményei): Kd, Bmax;
|
|
—
|
radioaktívan jelölt ligandum:
|
|
—
|
forgalmazó, katalógusszám, tétel, specifikus aktivitás.
|
|
|
|
Vizsgálati körülmények:
|
—
|
a vizsgálati körülmények között fennálló oldhatósági korlátok;
|
|
—
|
a kötési vizsgálat pufferének összetétele;
|
|
—
|
a receptor koncentrációja;
|
|
—
|
a nyomjelző (vagyis a radioaktívan jelölt ligandum) koncentrációja;
|
|
—
|
a vizsgálati vegyi anyag koncentrációi;
|
|
—
|
a vivőanyag aránya a végső vizsgálatban;
|
|
—
|
inkubációs hőmérséklet és idő;
|
|
—
|
a kötött és a szabad ligandumok elkülönítésére használt módszer;
|
|
—
|
pozitív és negatív kontrollanyagok/referenciaanyagok;
|
|
—
|
kritériumok, amelyek eldöntik, hogy a vizsgálatok pozitívnak, negatívnak vagy többféleképpen magyarázhatónak tekintendők-e.
|
|
|
|
Elfogadhatóság ellenőrzése:
|
—
|
a párhuzamos pozitív kontrollok/referenciaanyagok tényleges IC50 és Hill-meredekségi értéke.
|
|
|
|
Eredmények:
|
—
|
nyers adatok és a kötött/szabad ligandumokra vonatkozó adatok;
|
|
—
|
adott esetben denaturálást ellenőrző vizsgálat;
|
|
—
|
ha létezik, a legkisebb hatásos koncentráció (LEC);
|
|
—
|
adott esetben az RBA és/vagy az IC50 értéke;
|
|
—
|
amennyiben lehetséges, a koncentráció-válasz összefüggés;
|
|
—
|
statisztikai elemzések, ha végeztek ilyeneket, hiba- és megbízhatósági számításokkal (pl. SEM, SD, CV vagy 95 % CI), és annak leírása, hogy milyen módon kapták ezeket az értékeket.
|
|
|
|
Az eredmények értékelése:
|
—
|
a 10 %-os szabály alkalmazása.
|
|
Következtetések
|
SZAKIRODALOM
|
(1)
|
OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 34), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(2)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(3)
|
OECD (2015). Performance Standards for Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 222), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(4)
|
OECD (2012). Guidance Document on Standardized Test Guidelines for Evaluating Chemicals for Endocrine Disruption. Environmental, Health and Safety Publications, Series on Testing and Assessment (No 150), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(5)
|
Cavailles V. (2002). Estrogens and Receptors: an Evolving Concept, Climacteric, 5 Suppl 2: p.20-6.
|
|
(6)
|
Welboren W.J., et al. (2009). Genomic Actions of Estrogen Receptor Alpha: What are the Targets and How are they Regulated? Endocr. Relat. Cancer., 16(4): p. 1073-89.
|
|
(7)
|
Younes M. and Honma N. (2011). Estrogen Receptor Beta, Arch. Pathol. Lab. Med., 135(1): p. 63-6.
|
|
(8)
|
Diamanti-Kandarakis et al. (2009). Endocrine-Disrupting Chemicals: an Endocrine Society Sci. Statement, Endo Rev 30(4):293-342.
|
|
(9)
|
ICCVAM (2002). Background Review Document. Current Status of Test Methods for Detecting Endocrine Disruptors: In vitro Estrogen Receptor Binding Assays. (NIH Publication No 03-4504). National Institute of Environmental Health Sciences, Research Triangle Park, NC.
|
|
(10)
|
ICCVAM (2003). ICCVAM Evaluation of In vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(11)
|
ICCVAM (2006). ICCVAM Evaluation of In vitro Test Methods for Detecting Potential Endocrine Disruptors: Estrogen Receptor and Androgen Receptor Binding and Transcriptional Activation Assays.
|
|
(12)
|
Akahori Y. et al. (2008). Relationship Between the Results of In vitro Receptor Binding Assay to Human Estrogen Receptor Alpha and In vivo Uterotrophic Assay: Comparative Study with 65 Selected Chemicals, Toxicol. In vitro, 22(1): 225-231.
|
|
(13)
|
OECD (2007). Additional Data Supporting the Test Guideline on the Uterotrophic Bioassay in Rodents, Environment, Health and Safety Publications, Series on Testing and Assessment (No 67), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(14)
|
Takeyoshi, M. (2006). Draft Report of Pre-validation and Inter-laboratory Validation For Stably Transfected Transcriptional Activation (TA) Assay to Detect Estrogenic Activity - The Human Estrogen Receptor Alpha Mediated Reporter Gene Assay Using hER-HeLa-9903 Cell Line, Chemicals Evaluation and Research Institute (CERI): Japan. p. 1-188.
|
|
(15)
|
Yamasaki, K; Noda, S; Imatanaka, N; Yakabe, Y. (2004). Comparative Study of the Uterotrophic Potency of 14 Chemicals in a Uterotrophic Assay and their Receptor-Binding Affinity, Toxicol. Letters, 146: 111-120.
|
|
(16)
|
Kummer V; Maskova, J; Zraly, Z; Neca, J; Simeckova, P; Vondracek, J; Machala, M. (2008). Estrogenic Activity of Environmental Polycyclic Aromatic Hydrocarbons in Uterus of Immature Wistar Rats. Toxicol. Letters, 180: 213-221.
|
|
(17)
|
Gozgit, JM; Nestor, KM; Fasco, MJ; Pentecost, BT; Arcaro, KF. (2004). Differential Action of Polycyclic Aromatic Hydrocarbons on Endogenous Estrogen-Responsive Genes and on a Transfected Estrogen-Responsive Reporter in MCF-7 Cells. Toxicol. and Applied Pharmacol., 196: 58-67.
|
|
(18)
|
Santodonato, J. (1997). Review of the Estrogenic and Antiestrogenic Activity of Polycyclic Aromatic Hydrocarbons: Relationship to Carcinogenicity. Chemosphere, 34: 835-848.
|
|
(19)
|
Swillens S (1995). Interpretation of Binding Curves Obtained with High Receptor Concentrations: Practical Aid for Computer Analysis, Mol Pharmacol 47(6):1197-1203.
|
1. függelék
FOGALOMMEGHATÁROZÁSOK ÉS RÖVIDÍTÉSEK
10 %-os szabály
: annak lehetősége, hogy az elemzésből kizárják azokat az adatpontokat, amelyek esetében a [3H]17β-ösztradiol specifikus kötődési arányát mérő ismétlések átlaga legalább 10 %-kal meghaladja a valamely alacsonyabb koncentrációnál megfigyelt átlagértéket (lásd a 4. függeléket).
Elfogadhatósági kritériumok
: a kísérleti kontrollok és referenciaszabványok megfelelőségére vonatkozó minimális előírások. Ahhoz, hogy egy kísérlet érvényesnek minősüljön, meg kell felelni valamennyi elfogadhatósági kritériumnak.
Pontosság (egyezőség)
: a vizsgálati eredmények és az elfogadott referenciaértékek közötti egyezés eltérése. A vizsgálat megfelelőségének mutatója, és relevanciájának egyik szempontja. E kifejezést gyakran használják az „egyezőség” szinonimájaként, amely arra utal, hogy egy adott vizsgálat milyen arányban szolgáltat helyes eredményeket (1).
CF
: Az OECD endokrin rendszert károsító anyagok vizsgálatára és értékelésére szolgáló fogalmi kerete.
Vegyi anyag
: anyag vagy keverék.
CV
: variációs koefficiens.
E2
: 17β-ösztradiol
ED
: az endokrin rendszer károsítása.
hERα
: humán ösztrogénreceptor alfa.
ER
: ösztrogénreceptor.
Ösztrogénhatású aktivitás
: egy vegyi anyag azon képessége, hogy a 17β-ösztradiolhoz hasonlóan ösztrogénreceptorokhoz kötődjön. Ezzel a vizsgálati módszerrel a humán ösztrogénreceptor alfához való kötődés észlelhető.
IC
50
: egy gátló vizsgálati vegyi anyag maximális tényleges hatásának felét kiváltó koncentráció.
ICCVAM
: alternatív módszerek validálásával foglalkozó ügynökségközi koordinációs bizottság.
Laboratóriumok közötti reprodukálhatóság
: annak a mutatója, hogy különböző minősített laboratóriumok milyen mértékben képesek ugyanazon protokoll alkalmazásával és ugyanazon anyagok vizsgálatával hasonló kvalitatív és kvantitatív eredményeket elérni. A laboratóriumok közötti reprodukálhatóság az elővalidálási és validálási folyamat során kerül meghatározásra, és annak mértékét jelzi, hogy az adott vizsgálat mennyire sikeresen vihető át laboratóriumok között; más néven laboratóriumok közötti megismételhetőség (1).
Laboratóriumon belüli reprodukálhatóság
: annak meghatározása, hogy egy adott laboratóriumon belül dolgozó szakemberek milyen mértékben képesek egy meghatározott protokoll alapján különböző időpontokban sikeresen megismételni az eredményeket. Más néven „laboratóriumon belüli megismételhetőség” (1).
LEC
: a legkisebb hatásos koncentráció a vizsgálati vegyi anyag azon legalacsonyabb koncentrációja, amely választ vált ki (vagyis a vizsgálati vegyi anyag legkisebb koncentrációja, amelynél az indukciós tényező statisztikailag eltér a párhuzamos vivőanyagos kontrolltól).
Me-too vizsgálat
: olyan vizsgálatok köznapi elnevezése, amelyek strukturálisan és funkcionálisan hasonlóak egy validált és elfogadott, referenciaként szolgáló vizsgálati módszerhez. A hasonló vizsgálati módszer szinonimája.
PBTG
: teljesítményalapú vizsgálati iránymutatás.
Teljesítményszabványok
: validált vizsgálati módszeren alapuló szabványok, amelyek a javasolt, végrehajtás és funkcionalitás szempontjából hasonló vizsgálat összehasonlíthatósági értékelésének alapjául szolgálnak. Ide tartoznak: (1) a vizsgálat alapvető összetevői; (2) a referencia-vegyianyagok minimális jegyzéke, amely a validált vizsgálati módszer elfogadható teljesítményének igazolására használt vegyi anyagokból került összeállításra; és (3) a validált vizsgálati módszer eredményei alapján meghatározott, hasonló megbízhatósági és pontossági szintek, amelyeket a javasolt vizsgálatnak teljesítenie kell a minimális jegyzék referencia-vegyianyagainak felhasználásával történő értékelése során (1).
Jártassági tesztanyagok
: a teljesítményszabványokban megadott referenciaanyagokon belüli alcsoport, amelyet a laboratóriumok felhasználhatnak egy adott szabványosított vizsgálatban való szakmai jártasságuk igazolására. Ezeket az anyagokat általában az alapján választják ki, hogy összességükben lefedik a válaszreakciók lehetséges tartományát, kereskedelmi forgalomban kaphatók és rendelkezésre állnak rájuk vonatkozó minőségi referenciaadatok.
Jártasság
: annak igazolása, hogy a laboratórium képes megfelelően lefolytatni egy adott vizsgálatot, mielőtt ismeretlen anyagokat tesztelne.
Referenciaösztrogén
: 17ß-ösztradiol (E2, CAS-száma: 50-28-2).
Vizsgálati referenciamódszerek
: A 493. teljesítményalapú vizsgálati iránymutatás alapját képező vizsgálatok.
RBA
: relatív kötési affinitás. Valamely anyag relatív kötési affinitását az anyag log (IC50) értékének a 17β-ösztradiol log (IC50) értékéhez viszonyított arányaként határozzák meg.
Relevancia
: valamely vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálat pontosságát (az eredmények egyezését) figyelembe kell venni (1).
Megbízhatóság
: a vizsgálat laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. A laboratóriumon belüli és laboratóriumok közötti reprodukálhatóság kiszámításával állapítják meg.
SD
: standard deviáció.
Vizsgálati vegyi anyag
: bármely, e vizsgálati módszer alkalmazásával vizsgált anyag vagy keverék.
Validált vizsgálati módszer
: olyan vizsgálat, amelynek az adott céllal kapcsolatos relevanciája (pontosságát is ideértve) és megbízhatósága validálási vizsgálatok alapján megállapítást nyert. Fontos megjegyezni, hogy a validált vizsgálati módszer a javasolt felhasználási célra nem feltétlenül kínál megfelelő pontosságú és megbízhatóságú elfogadható eljárást (1).
Validálás
: az az eljárás, amelynek révén megállapítják egy bizonyos megközelítés, módszer, vizsgálat, eljárás vagy értékelés adott célra való megbízhatóságát és relevanciáját (1).
2. függelék
A FREYBERGER-WILSON-FÉLE TELJES HOSSZÚSÁGÚ REKOMBINÁNS ÖSZTROGÉNRECEPTOR Α (ERΑ) HASZNÁLATÁN ALAPULÓ IN VITRO TELÍTÉSI ÉS KOMPETITÍV KÖTÉSI VIZSGÁLATOK
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
1.
|
Ez az in vitro ösztrogénreceptor (ERα) telítési és kompetitív kötési vizsgálat teljes hosszúságú humán ERα (hrERα) receptort alkalmaz, amelyet bakulovírussal fertőzött rovarsejtekben termeltetnek és izolálnak. A Freyberger és Wilson által kidolgozott protokollt több laboratórium részvételével zajló, nemzetközi validálási tanulmánynak (2) vetették alá, amely bizonyította, hogy a vizsgálat tervezett felhasználása tekintetében releváns és megbízható.
|
|
2.
|
A vizsgálat egy szűrési eljárás, amelynek révén azonosíthatók a teljes hosszúságú humán ösztrogénreceptor α-hoz kötődni képes anyagok. A vizsgálatot annak meghatározására alkalmazzák, hogy egy adott vizsgálati vegyi anyag képes-e vetélkedni a 17β-ösztradiollal a humán ösztrogénreceptor α-hoz való kötődésért. A vizsgálat kvantitatív eredményei magukban foglalhatják az IC50 értéket (vagyis azt a koncentrációt, amelyre a vizsgálati vegyi anyagnak szüksége van ahhoz, hogy a [3H]-17β-ösztradiol felét leszorítsa a humán ösztrogénreceptor α-ról), valamint a vizsgálati vegyi anyag humán ösztrogénreceptor α-hoz való relatív – a 17β-ösztradiolhoz viszonyított – kötési affinitását. A vegyi anyagok szűrése céljából a vizsgálat elfogadható kvalitatív eredményei kiterjedhetnek a vizsgálati vegyi anyagok humán ösztrogénreceptor α-hoz kötődő, nem kötődő, illetve nem egyértelmű kategóriába sorolására, ezek meghatározására a kötési görbékre vonatkozó kritériumok alapján kerül sor.
|
|
3.
|
A vizsgálat radioaktív ligandumot használ, ezért a laboratóriumnak rendelkeznie kell radioaktív anyagok használatára vonatkozó engedéllyel. A radioaktív izotópokkal és veszélyes vegyi anyagokkal végzett valamennyi folyamat során be kell tartani a nemzeti jogszabályok által előírt rendelkezéseket és eljárásokat.
|
|
4.
|
E vizsgálat szabályozási célra való alkalmazása előtt el kell olvasni az „ÁLTALÁNOS BEVEZETÉS” és „A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részt. Az ebben a vizsgálati iránymutatásban használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLATI ELVEK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
5.
|
A humán ösztrogénreceptor α-kötési vizsgálat azt méri, hogy milyen mértékben képes egy radioaktívan jelölt ligandum (a [3H]17β-ösztradiol) az ösztrogénreceptorhoz kötődni egy adott vizsgálati vegyi anyag (a kompetítor) növekvő koncentrációjának jelenlétében. Az ösztrogénreceptorok iránt nagy affinitást mutató vizsgálati vegyi anyagok alacsonyabb koncentrációban versenyeznek a radioaktívan jelölt ligandummal azokhoz a vegyi anyagokhoz képest, amelyek kisebb affinitást mutatnak a receptor iránt.
|
|
6.
|
A vizsgálat két fő összetevőből áll: egy telítési kötési kísérletből, amelynek során meghatározzák a receptor és a ligandum közötti kölcsönhatás paramétereit, majd egy ezután végzett kompetitív kötési kísérletből, amelynek révén jellemzik a vizsgálati vegyi anyag és a radioaktívan jelölt ligandum ösztrogénreceptor-kötőhelyekért folytatott vetélkedését.
|
|
7.
|
A telítési kötési kísérlet célja, hogy a kompetitív kötési kísérlet előkészítéseként meghatározzák a receptorok egy adott tételének kötési affinitását és a receptorok számát. A telítési kötési kísérlet egyensúlyi állapotban méri egy fix koncentrációjú ösztrogénreceptor természetes ligandumához való kötési affinitását (amelynek mutatója a disszociációs állandó, a Kd) és a receptor aktív kötőhelyeinek koncentrációját (Bmax).
|
|
8.
|
A kompetitív kötési kísérletben azt mérik, hogy egy adott anyag milyen mértékű affinitással rendelkezik arra, hogy versenyezzen a [3H]17β-ösztradiollal az ösztrogénreceptorral formált kötésért. Az affinitást a vizsgálati vegyi anyag azon koncentrációjával számszerűsítik, amely egyensúlyi állapotban meggátolja a [3H]17β-ösztradiol specifikus kötődésének 50 %-át (ez az úgynevezett „50 %-os gátlást kiváltó koncentráció” vagy IC50). Az affinitás mérhető emellett a relatív kötési affinitással is (RBA, az ösztradiol ugyanazon vizsgálatmenetben, de külön mért IC50 értékéhez viszonyított affinitás). A kompetitív kötési kísérlet azt méri, hogy milyen mértékben kötődik egy fix koncentrációban alkalmazott [3H]17β-ösztradiol a vizsgálati vegyi anyag széles skálát (nyolc nagyságrendet) lefedő koncentrációja jelenlétében. Ezt követően – ahol lehetséges – az adatokat a Hill-egyenlet olyan formájára illesztik (Hill, 1910), amely meghatározza, hogy milyen mértékben szorítják le az egy kötőhelyet elfoglaló kompetitív kötődő anyagok a radioaktív ligandumot. A radioaktívan jelölt ösztradiol egyensúlyi állapotban történő leszorításának mértéke alapján jellemzik a vizsgálati vegyi anyagot kötődő, nem kötődő, vagy kétértelmű választ adó anyagként.
|
ELJÁRÁS
A humán ösztrogénreceptor α fehérje elfogadható teljesítményének igazolása
|
9.
|
A telítési és a kompetitív kötési vizsgálatok rutinszerű alkalmazása előtt a humán ösztrogénreceptor α minden új tételéről bizonyítani kell, hogy megfelelő teljesítményt nyújt abban a laboratóriumban, ahol használni fogják. A megfelelő teljesítmény egy két lépésből álló eljárással bizonyítható. Ez a két lépés a következő:
|
—
|
A humán ösztrogénreceptor α-specifikusságának és -telítettségének igazolása érdekében végezzünk el egy telítési [3H]-17β-ösztradiol kötési vizsgálatot. Az adatok nem-lineáris regressziós analízise (pl. BioSoft; McPherson, 1985; Motulsky, 1995), majd azt követő Scatchard-plot ábrázolása az egyes hrERα-tételekre vonatkozólag dokumentálja a [3H]-17β-ösztradiol humán ösztrogénreceptor α-hoz való kötési affinitását (Kd) és a receptorok számát (Bmax).
|
|
—
|
Végezzünk el egy kompetitív kötési vizsgálatot a kontrollanyagok (a referenciaösztrogén (17β-ösztradiol)), egy gyengén kötődő anyag (pl. noretinodrel vagy noretindron) és egy nem kötődő anyag (oktil-trietoxi-szilán, OTES) használatával. Valamennyi laboratóriumnak létre kell hoznia egy adatbázist annak dokumentálására, hogy a referenciaösztrogén és a gyengén kötődő anyag IC50 értékét és más releváns mutatóit következetesen határozta meg a különböző kísérletek és az eltérő hrERα-tételek vizsgálata során. A kontrollanyagok kompetitív kötési görbéjét alkotó paramétereknek a 95 %-os konfidenciaintervallumon belül kell lenniük (lásd az 1. táblázatot), amelynek értékeit a vizsgálat validálási tanulmányában részt vevő laboratóriumok adatai alapján határozták meg (2).
|
|
1. táblázat
A Freyberger–Wilson humán ösztrogénreceptor-kötési vizsgálatban használt referenciaösztrogén és gyengén kötődő anyag teljesítményére vonatkozó kritériumok
|
Anyag
|
Paraméter
|
Átlag (7)
|
Standard deviáció (n)
|
95 %-os konfidenciaintervallumok (8)
|
|
Alsó határ
|
Felső határ
|
|
17β-ösztradiol
|
Görbe teteje (%)
|
100,44
|
10,84 (67)
|
97,8
|
103,1
|
|
Görbe alja (%)
|
0,29
|
1,25 (67)
|
-0,01
|
0,60
|
|
Hill-meredekség
|
-1,06
|
0,20 (67)
|
-1,11
|
-1,02
|
|
LogIC50 (M)
|
-8,92 (9)
|
0,18 (67)
|
-8,97
|
-8,88
|
|
Noretinodrel
|
Görbe teteje (%)
|
99,42
|
8,90 (68)
|
97,27
|
101,60
|
|
Görbe alja (%)
|
2,02
|
3,42 (68)
|
1,19
|
2,84
|
|
Hill-meredekség
|
-1,01
|
0,38 (68)
|
-1,10
|
-0,92
|
|
Log IC50 (M)
|
-6,39
|
0,27 (68)
|
-6,46
|
-6,33
|
|
Noretindron (9)
|
Görbe teteje (%)
|
96,14
|
8,44 (27)
|
92,80
|
99,48
|
|
Görbe alja (%)
|
2,38
|
5,02 (27)
|
0,40
|
4,37
|
|
Hill-meredekség
|
-1,41
|
0,32 (27)
|
-1,53
|
-1,28
|
|
LogIC50 (M)
|
-5,73
|
0,27 (27)
|
-5,84
|
-5,62
|
A laboratórium jártasságának igazolása
|
10.
|
Lásd a vizsgálati módszer „A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részének 17. és 18. pontját, valamint 2. táblázatát. Minden (telítési és kompetitív kötési) vizsgálatnak három független (vagyis a receptor, a vegyi anyagok és a reagensek frissen készített oldataival végezett), különböző napokon lefolytatott vizsgálatmenetből kell állnia, az egyes vizsgálatmeneteknek pedig három ismétlést kell tartalmazniuk.
|
A receptor (hrERα) koncentrációjának meghatározása
|
11.
|
Az aktív receptor koncentrációja tételenként és tárolási feltételek szerint kissé változó. Ebből adódóan a forgalmazótól való átvételkor meg kell határozni az aktív receptor koncentrációját. Így megállapítható az aktív receptor vizsgálatmenet során alkalmazandó megfelelő koncentrációja.
|
|
12.
|
A kompetitív kötési vizsgálatnak megfelelő körülmények között (vagyis 1 nM [3H]-ösztradiollal) a receptort 0,25, 0,5, 0,75 és 1 nM névleges koncentrációban inkubálni kell 1 μM nem jelölt ösztradiol jelenlétében (nem specifikus kötődés) és annak hiányában (teljes kötődés). A specifikus kötődést, amely a teljes és a nem specifikus kötődés különbségeként határozható meg, ábrázolni kell a receptor névleges koncentrációja függvényében. A receptort abban a névleges koncentrációban kell használni a telítési és a kompetitív kötési kísérletek során, amelynél a specifikus kötődési értékei megegyeznek a hozzáadott radioaktívan jelölt ösztradiol kötési értékeinek 20 %-ával. A humán ösztrogénreceptor 0,5 nM-es végső koncentrációja többnyire eleget tesz ennek a feltételnek.
|
|
13.
|
Amennyiben a 20 %-os feltétel több próbálkozás után sem teljesül, ellenőrizni kell, hogy nincsenek-e potenciális hibák a kísérlet elrendezésében. A 20 %-os feltétel nem teljesülése jelezheti azt is, hogy a rekombináns tételben nagyon csekély az aktív receptorok száma, ilyen esetben fontolóra kell venni másik receptortétel alkalmazását.
|
Telítési vizsgálat
|
14.
|
A [3H]17β-ösztradiolt kell kiértékelni nyolc növekvő koncentrációban, három ismétlésben, az alábbi három kitétellel (lásd a 2. táblázatot):
|
—
|
Nem jelölt 17β-ösztradiol nélkül és ösztrogénreceptorral. Ez alapján határozható meg a teljes kötődés a csak [3H]17β-ösztradiolt tartalmazó lyukakban mért radioaktivitás alapján.
|
|
—
|
A jelölt 17β-ösztradiolhoz képest 1000-szeres koncentrációjú nem jelölt 17β-ösztradiol jelenlétében és ösztrogénreceptorral. Ez az állapot arra szolgál, hogy az aktív kötőhelyek telítődjenek nem jelölt 17β-ösztradiollal, így a lyukakban mért radioaktivitás alapján meghatározható a nem specifikus kötődés. A fennmaradó, receptorhoz kötődő radioaktív ösztrogének feltételezhetően nem specifikus helyre kötődőnek, mivel a nem jelölt ösztrogének olyan magas koncentrációban vannak jelen, hogy elfoglalják a receptor valamennyi szabad specifikus kötőhelyét.
|
|
—
|
Nem jelölt 17β-ösztradiol és ösztrogénreceptor nélkül (így meghatározható a teljes radioaktivitás).
|
|
A [3H]-17β-ösztradiol és a nem jelölt 17β-ösztradiol oldatainak elkészítése
|
15.
|
A [3H]-17β-ösztradiol oldatainak elkészítéséhez a [3H]-17β-ösztradiol 12 nM koncentrációjú törzsoldatához vizsgálati pufferrel beállítjuk a kezdeti, 0,12 nM-től 12 nM-ig terjedő koncentrációtartományt. Ezen oldatokból 40 μl-t adagolunk a 96 lyukú mikrotiterlemez megfelelő vizsgálati lyukaiba (160 μl végtérfogatban), így megkapjuk a vizsgálat végső, 0,03 és 3,0 nM közötti koncentrációértékeit. A vizsgálati puffer, a [3H]-17β-ösztradiol törzsoldatának és oldatainak elkészítését, valamint a koncentrációértékek meghatározását részletesen ismerteti a Freyberger–Wilson protokoll (2).
|
|
16.
|
A 17β-ösztradiol etanolos oldatait vizsgálati puffer hozzáadásával kell hígítani, elkészítve a nyolc növekvő elemből álló, 0,06 μM-től 6 μM-ig terjedő kezdeti koncentrációsorozatot. Ezekből az oldatokból 80 μl-t adagolunk a 96 lyukú mikrotiterlemez megfelelő vizsgálati lyukaiba (160 μl végtérfogatban), így megkapjuk a vizsgálat végső, 0,03 μM és 3,0 μM közötti koncentrációértékeit. A nem specifikus kötődést tesztelő lyukakban a nem jelölt 17β-ösztradiol 1000-szeres feleslegben kell legyen a jelölt [3H]-17β-ösztradiol végső koncentrációjához képest. A nem jelölt 17β-ösztradiol oldatainak elkészítését részletesen ismerteti a Freyberger–Wilson protokoll (2).
|
|
17.
|
A receptornak azt a névleges koncentrációját kell alkalmazni, amelyik 20 %-os (± 5 %) specifikus kötődést vált ki (lásd a 12–13. pontot). A humán ösztrogénreceptor α oldatot közvetlenül használat előtt kell elkészíteni.
|
|
18.
|
A 96 lyukú mikrotiterlemezeket a 2. táblázatban foglaltak szerint készítik el, az egyes koncentrációértékeket 3 ismétlésben vizsgálva. A [3H]-17β-ösztradiol, a nem jelölt 17β-ösztradiol, a puffer és a receptor lemezenkénti koncentrációértékeit és térfogatát a 2.2. függelék tartalmazza.
|
2. táblázat
Mikrotiterlemez elrendezése a telítési kötési vizsgálathoz
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3 H] E2 + ER
|
0,06 nM [3 H] E2 + ER
|
0,08 nM [3 H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Teljes kötődés (oldószer)
|
|
B
|
0,30 nM [3 H] E2 + ER
|
0,60 nM [3 H] E2 + ER
|
1,0 nM [3 H] E2 + ER
|
3,0 nM [3 H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3 H] E2 + ER + 0,03 μM E2
|
0,06 nM [3 H] E2 + ER + 0,06 μM E2
|
0,08 nM [3 H] E2 + ER + 0,08 μM E2
|
0,10 nM [ 3H] E2 + ER
+ 0,10 μM E2
|
Nem specifikus kötődés
|
|
E
|
0,30 nM [3 H] E2 + ER + 0,30 μM E2
|
0,60 nM [3 H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3 H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
|
|
|
|
|
|
H
|
|
|
|
|
|
[3H] E2:: [3H]-17β-ösztradiol
ER:: ösztrogénreceptor
E2:: nem jelölt 17β-ösztradiol
|
|
19.
|
A vizsgálati mikrotiterlemezeket 16–20 órán keresztül 2–8 °C-os hőmérsékleten inkubálni kell, a lemezeket az inkubáció ideje alatt rázógépre kell helyezni.
|
A humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiol mérése
|
20.
|
A humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolokat elkülönítjük a szabad [3H]-17β-ösztradioloktól, ehhez minden lyukhoz hozzáadunk 80 μl hideg DCC szuszpenziót, ezt követően a mikrotiterlemezeket 10 percen át rázatjuk, majd mintegy 2500 rpm sebességgel 10 percen keresztül centrifugáljuk. Annak érdekében, hogy e folyamat során minimálisra szorítsuk a kötött [3H]-17β-ösztradiolok humán ösztrogénreceptor α-ról történő disszociációját, nagyon fontos, hogy a pufferek és a vizsgálati lyukak hőmérsékletét 2 és 8 °C között tartsuk, és minden lépést gyorsan végezzünk el. A lemezek hatékony és gyors feldolgozásához szükség van egy mikrotiterlemez-rázógépre.
|
|
21.
|
50 μl felülúszóban az ösztrogénreceptor α-hoz kötött [3H]-17β-ösztradiolt átvisszük egy második mikrotiterlemezre, nagy figyelemmel végezve a transzfert, nehogy a DCC-hez hozzáérve átszennyezzük a lyukakat.
|
|
22.
|
Ezt követően mindegyik lyukhoz (A1–B12 és D1–E12) 200 μl szcintillációs folyadékot adunk, amely képes a radioaktív sugárzás mozgási energiáját fényenergiává alakítani. A G1–H12 lyukak (a teljes percenkénti bomlás (dpms) mutatói) a [3H]-17β-ösztradiol (40 μl) hígítási sorozatát tartalmazzák, amelyet közvetlenül a mérési lemez lyukaiba töltött szcintillációs folyadékba kell helyezni a 3. táblázat szerinti elosztásban, ezek a lyukak tehát csak 200 μl szcintillációs folyadékot és a [3H]-17β-ösztradiol megfelelő hígítását tartalmazzák. Ezek a mérőszámok azt jelzik, hogy mennyi dpms-ben kifejezett [3H]-17β-ösztradiolt adtunk hozzá a teljes kötődést és a nem specifikus kötődést vizsgáló lyukakhoz.
|
3. táblázat
Mikrotiterlemez elrendezése telítési kötési vizsgálathoz, radioaktivitás mérése
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
A
|
0,03 nM [3 H] E2 + ER
|
0,06 nM [3 H] E2 + ER
|
0,08 nM [3 H] E2 + ER
|
0,10 nM [3H] E2 + ER
|
Teljes kötődés (oldószer)
|
|
B
|
0,30 nM [3 H] E2 + ER
|
0,60 nM [3 H] E2 + ER
|
1,0 nM [3 H] E2 + ER
|
3,0 nM [3 H] E2 + ER
|
|
C
|
|
|
|
|
|
|
D
|
0,03 nM [3 H] E2 + ER + 0,03 μM E2
|
0,06 nM [3 H] E2 + ER + 0,06 μM E2
|
0,08 nM [3 H] E2 + ER + 0,08 μM E2
|
0,10 nM [ 3H] E2 + ER
+ 0,10 μM E2
|
Nem specifikus kötődés
|
|
E
|
0,30 nM [3 H] E2 + ER + 0,30 μM E2
|
0,60 nM [3 H] E2 + ER + 0,60 μM E2
|
1,0 nM [
3
H] E2 + ER
+ 1,0 μM E2
|
3,0 nM [3 H] E2 + ER + 3,0 μM E2
|
|
F
|
|
|
|
|
|
|
G
|
0,03 nM [ 3H] E2 (teljes dpms)
|
0,06 nM [3H] E2
|
0,08 nM [3H] E2
|
0,10 nM [3H] E2
|
Teljes dpms (*7)
|
|
H
|
0,30 nM [3 H] E2
|
0,60 nM [3 H] E2
|
1,0 nM [3 H] E2
|
3,0 nM [3 H] E2
|
|
[3H] E2:: [3H]-17β-ösztradiol
ER:: ösztrogénreceptor
E2:: nem jelölt 17β-ösztradiol
dpms:: percenkénti bomlás
|
|
23.
|
A mérés megkezdése előtt legalább 2 órát kell várni, a radioaktivitás mérési ideje pedig mintánként 40 perc legyen. A lyukankénti dpm értékének meghatározásához mikrotiterlemez-szcintillációs számlálót kell alkalmazni, és korrigálni kell az elnyelődött veszteség értékét. Amennyiben nem áll rendelkezésre mikrotiterlemezhez használható szcintillációs számláló, a minták hagyományos számlálóval is mérhetők. Ez esetben fontolóra lehet venni a számlálásra fordított idő csökkentését.
|
Kompetitív kötési vizsgálat
|
24.
|
A kompetitív kötési vizsgálat azt méri, hogy egy állandó koncentrációjú [3H]-17β-ösztradiol milyen mértékben képes kötődni az adott vizsgálati vegyi anyag növekvő koncentrációja mellett. Egy vizsgálatmeneten belül minden koncentrációt három párhuzamos ismétlésben kell vizsgálni. Emellett minden vizsgálati vegyi anyagot három nem egyidejűleg végzett vizsgálatmenetben kell tesztelni. A vizsgálatot egy vagy több 96 lyukú mikrotiterlemezen kell elvégezni.
|
Kontrollok
|
25.
|
A vizsgálat végrehajtása során minden egyes kísérletben párhuzamosan vizsgálni kell az oldószert és a kontrollanyagokat (a referenciaösztrogént, valamint a gyengén kötődő és a nem kötődő anyagot). Minden vizsgálatmenetben ugyanazon a lemezen vizsgálni kell a referenciaösztrogén teljes koncentrációgörbéjét és a kontrollanyagokat (vagyis a gyengén kötődő és a nem kötődő anyagot). Az összes többi lemeznek tartalmaznia kell i. az E2 és a gyengén kötődő anyag magas (maximális leszorítást eredményező) és közepes (körülbelül IC50 hatást kiváltó) koncentrációját, három ismétlésben; ii. az oldószeres kontrollt és a nem specifikus kötődés mérését, legalább három-három ismétlésben. A vizsgálati puffer, a kontrollanyagok, a [3H]-17β-ösztradiol, a humán ösztrogénreceptor α és a vizsgálati vegyi anyagok oldatainak elkészítési eljárását a 2. referencia ismerteti (K függelék, lásd a Freyberger–Wilson vizsgálati protokollt).
|
Oldószeres kontroll:
|
26.
|
Az oldószeres kontroll igazolja, hogy az oldószer nem lép kölcsönhatásra a vizsgálati rendszerrel, emellett méri a teljes kötődést (TB). Az ajánlott oldószer az etanol. Amennyiben a vizsgálati vegyi anyag legmagasabb koncentrációja nem oldható etanolban, akkor alternatívaként DMSO is használható. Az etanol, illetve DMSO használata esetén a DMSO koncentrációja a végső vizsgálati lyukakban 1,5 %, de nem haladhatja meg a 2 %-ot.
|
Puffer kontroll:
|
27.
|
A puffer kontroll (BC) nem tartalmazhatja sem az oldószert, sem a vizsgálati vegyi anyagot, csak a vizsgálat összes többi összetevőjét. A puffer kontroll eredményeit összevetik az oldószeres kontroll eredményeivel annak ellenőrzésére, hogy az alkalmazott oldószer nincs hatással a vizsgálati rendszerre.
|
Erősen kötődő anyag (referenciaösztrogén)
|
28.
|
A 17β-ösztradiol (CAS-száma: 50-28-2) az endogén ligandum, és magas affinitással kötődik az ösztrogénreceptor alfa altípusához. Minden humán ösztrogénreceptor α kompetitív kötési vizsgálathoz nem jelölt 17β-ösztradiollal el kell készíteni egy standard görbét, amely lehetővé teszi a variabilitás értékelését, ha a laboratóriumon belül egy későbbi időpontban ismét elvégzik a vizsgálatot. A nem jelölt 17β-ösztradiolból nyolc hígítást kell készíteni etanolban, és a vizsgálati lyukakba helyezni a következő koncentrációtartományban: 100 nM-tól 10 pM-ig (-7[logM] és -11[logM] között) és a következő elosztásban: (-7[logM], -8[logM], -8,5[logM], -9[logM], - 9,5[logM], -10[logM], -11[logM]). A nem jelölt 17β-ösztradiol legmagasabb koncentrációja (1 μM) egyben a nem specifikus kötődés indikátoraként is szolgál. Ezt a koncentrációt a 4. táblázatban az „NSB” rövidítés különbözteti meg, bár ez is részét képezi a standard görbének.
|
Gyengén kötődő anyag
|
29.
|
A vizsgálatba bele kell foglalni egy gyengén kötődő anyagot (a noretinodrel t (CAS-száma: 68-23-5) vagy a noretindront (CAS-száma: 68-22-4)), hogy igazolja az egyes kísérletek érzékenységét, és lehetővé tegye a variabilitás értékelését, ha a vizsgálatot egy későbbi időpontban megismétlik. A gyengén kötődő anyagból nyolc oldatot kell készíteni etanolban, és a vizsgálati lyukakba helyezni a következő koncentrációtartományban: 3 nM-tól 30 μM-ig (-8,5[logM] és -4,5[logM] között) és a következő elosztásban: -4,5[logM], -5[logM], -5,5[logM], -6[logM], -6,5[logM], -7[logM],-7,5[logM], -8,5[logM].
|
Nem kötődő anyag
|
30.
|
Negatív (nem kötődő) kontrollanyagként oktil-trietoxi-szilánt (OTES, CAS-száma: 2943-75-1) kell alkalmazni. Ez biztosítja, hogy a vizsgálat végrehajtása során detektálni lehessen, ha a vizsgálati vegyi anyagok nem kötődnek a humán ösztrogénreceptor α-hoz. A nem kötődő anyagból nyolc oldatot kell készíteni etanolban, és a vizsgálati lyukakba helyezni a következő koncentrációtartományban: 0,1 nM–1000 μM (-10[logM] és -3[logM] között), logaritmikus lépésközönként. Alternatív nem kötődő kontrollanyagként di-n-butil-ftalát (DBP) használható. Ez utóbbi anyagról kimutatták, hogy maximális oldhatósága -4[logM].
|
A humán ösztrogénreceptor α koncentrációja
|
31.
|
A receptort abban a mennyiségben kell alkalmazni, amelyik 1 nM radioaktív ligandum jelenlétében 20 %-os (± 5 %) specifikus kötődést vált ki (lásd a 2. függelék 12–13. pontját). A humán ösztrogénreceptor α oldatot közvetlenül használat előtt kell elkészíteni.
|
[3H]-17β-ösztradiol
|
32.
|
A [3H]-17β-ösztradiolt 1,0 nM koncentrációban kell a vizsgálati lyukakba helyezni.
|
Vizsgálati vegyi anyagok
|
33.
|
Első lépésként oldhatósági vizsgálatot kell végezni az egyes vizsgálati vegyi anyagok oldhatósági határértékének meghatározására, valamint a vizsgálati protokoll végrehajtása során alkalmazandó megfelelő koncentrációtartomány megállapítására. A vizsgálati vegyi anyagok oldhatósági határértékét először oldószerben kell meghatározni, majd a vizsgálati körülmények mellett meg kell erősíteni. A vizsgálat során tesztelt végső koncentráció nem haladhatja meg az 1 mM-t. A koncentrációtartomány-behatároló vizsgálatnak magában kell foglalnia egy oldószeres kontrollt és a legmagasabb elfogadható koncentrációtól (pl. 1 mM, illetve az oldhatósági határértéktől függően ennél alacsonyabb koncentrációtól) induló, 8 koncentrációból álló logaritmikus hígítási sorozatot, az esetlegesen tapasztalt zavarosságot vagy kicsapódást fel kell jegyezni (lásd még a 35. pontot). A vizsgálati vegyi anyagot a koncentrációtartomány behatárolására szolgáló előzetes vizsgálatban meghatározott, 8 log koncentrációintervallumot tartalmazó görbe alapján kell tesztelni. A második és a harmadik kísérlet koncentrációértékeit úgy kell megválasztani, hogy jobban illeszkedjenek a koncentráció-válasz görbéhez.
|
|
34.
|
A vizsgálati vegyi anyag oldatait megfelelő oldószerben kell elkészíteni (lásd a 2. függelék 26. pontját). Amennyiben a vizsgálati vegyi anyag legmagasabb koncentrációja sem etanolban, sem DMSO-ban nem oldható, és a hozzáadott oldószer mennyiségének növelése miatt a végső vizsgálathoz használt csőben az oldószer koncentrációja túllépné az elfogadható határértéket, a legmagasabb koncentráció az eggyel alacsonyabb koncentrációra csökkenthető. Ebben az esetben hozzá lehet adni egy további koncentrációértéket a koncentrációsorozat alacsonyabb értékeket tartalmazó végéhez. A sorozat többi koncentrációértékét változatlanul kell hagyni.
|
|
35.
|
A vizsgálati vegyi anyag mikrotiterlemezbe adagolt oldatait figyelemmel kell követni, mivel a vizsgálati vegyi anyag a vizsgálati lyukakba helyezést követően kicsapódhat. A kicsapódást tartalmazó vizsgálati lyukak adatait ki kell zárni a görbeillesztésből, és fel kell jegyezni az adatok kizárásának okát.
|
|
36.
|
Amennyiben a vizsgálati vegyi anyag log(IC50) értékére vonatkozólag léteznek más forrásokból származó korábbi adatok, érdemes lehet geometriailag elosztani az oldatokat (pl. hogy 0,5 log egységnyire helyezkedjenek el a log(IC50) várt értéke körül). A végső eredménynek a log(IC50) mindkét oldalán kellő távolságban eloszló koncentrációkat kell mutatnia, beleértve a görbe tetejét és alját, hogy a kötési görbét megfelelően lehessen jellemezni.
|
Vizsgálati lemez elrendezése
|
37.
|
A mikrotiterlemezeket fel kell címkézni, és kódokkal kell ellátni a hatszoros inkubálásnak alávetett oldószeres kontrollt, a referenciaösztrogén legmagasabb koncentrációját – amely egyben a nem specifikus kötődés (NSB) mutatója – és a puffer kontrollt, valamint a háromszor inkubált nem kötődő kontrollanyag (oktil-trietoxi-szilán) mind a nyolc koncentrációját, a referenciaösztrogén 7 alacsonyabb koncentrációját, a gyengén kötődő anyag nyolc dózist képviselő koncentrációit és minden egyes vizsgálati vegyi anyag (TC) 8 koncentrációját. Az alábbi 4. táblázat példát szolgáltat egy olyan lemezelrendezésre, amely biztosítja a referenciaösztrogén és a kontrollanyagok teljes koncentrációgörbéjét. A többi mikrotiterlemez a vizsgálati vegyi anyagok mérésére szolgál, emellett tartalmazniuk kell a lemezen belüli ellenőrzésre szolgáló kontrollanyagokat, vagyis 1) az E2 és a gyengén kötődő anyag magas (maximális leszorítást eredményező) és közepes (körülbelül IC50 hatást kiváltó) koncentrációját, három ismétlésben; 2) az oldószeres kontrollt és a nem specifikus kötődés mérését, hat-hat ismétlésben (lásd az 5. táblázatot). A 2.3. függelék példát szolgáltat arra, hogy milyen lemezelrendezést lehet alkalmazni a kompetitív vizsgálatban három ismeretlen vizsgálati vegyi anyag teszteléséhez. A 4. és az 5. táblázatban feltüntetett koncentrációértékek a vizsgálatban használt végső koncentrációkat jelzik. Az E2 maximális koncentrációja 1×10-7 M legyen, a gyengén kötődő anyag legmagasabb koncentrációja pedig az 1. lemezen használt legmagasabb koncentráció legyen. Az IC50 koncentrációját a laboratóriumnak kell meghatároznia, a kontrollanyagok adatait tartalmazó adatbázisa alapján. Ennek az értéknek közelítenie kell a validálási tanulmányok során nyert értékhez (lásd az 1. táblázatot).
|
4. táblázat
A kompetitív kötési vizsgálathoz használt mikrotiterlemez azon elrendezése, amely biztosítja a referenciaösztrogén és a kontrollanyagok teljes koncentrációgörbéjét (1. lemez).
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (csak oldószer)
|
TB (csak oldószer)
|
NSB
|
NSB
|
|
B
|
E2 (1×10-7)
|
E2 (1×10-8)
|
E2 (1×10-8,5)
|
E2 (1×10-9)
|
|
C
|
E2 (1×10-9,5)
|
E2 (1×10-10)
|
E2 (1×10-11)
|
Üres (*8)
|
|
D
|
NE (1×10-4,5)
|
NE (1×10-5)
|
NE (1×10-5,5)
|
NE (1×10-6)
|
|
E
|
NE (1×10-6,5)
|
NE (1×10-7)
|
NE (1×10-7,5)
|
NE (1×10-8,5)
|
|
F
|
OTES (1×10-3)
|
OTES (1×10-4)
|
OTES (1×10-5)
|
OTES (1×10-6)
|
|
G
|
OTES (1×10-7)
|
OTES (1×10-8)
|
OTES (1×10-9)
|
OTES (1×10-10)
|
|
H
|
Üres (radioaktívhoz) (*9)
|
Üres (radioaktívhoz) (*9)
|
Puffer kontroll
|
Puffer kontroll
|
|
Ebben a példában a gyengén kötődő anyag a noretinodrel (NE).
|
5. táblázat
A kompetitív kötési vizsgálathoz használt mikrotiterlemez azon elrendezése, amely biztosítja a vizsgálati vegyi anyagok és a lemezen belüli ellenőrzésre szolgáló kontrollanyagok teljes koncentrációgörbéjét
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
A
|
TB (csak oldószer)
|
TB (csak oldószer)
|
NSB
|
NSB
|
|
B
|
TC1 (1×10-3)
|
TC1 (1×10-4)
|
TC1 (1×10-5)
|
TC1 (1×10-6)
|
|
C
|
TC1 (1×10-7)
|
TC1 (1×10-8)
|
TC1 (1×10-9)
|
TC1 (1×10-10)
|
|
D
|
TC2 (1×10-3)
|
TC2 (1×10-4)
|
TC2 (1×10-5)
|
TC2 (1×10-6)
|
|
E
|
TC2 (1×10-7)
|
TC2 (1×10-8)
|
TC2 (1×10-9)
|
TC2 (1×10-10)
|
|
F
|
TC3 (1×10-3)
|
TC3 (1×10-4)
|
TC3 (1×10-5)
|
TC3 (1×10-6)
|
|
G
|
TC3 (1×10-7)
|
TC3 (1×10-8)
|
TC3 (1×10-9)
|
TC3 (1×10-10)
|
|
H
|
NE (IC50 )
|
NE (1×10-4,5)
|
E2 (IC50 )
|
E2 (1×10-7)
|
|
Ebben a példában a gyengén kötődő anyag a noretinodrel (NE).
|
A kompetitív kötési vizsgálat elvégzése
|
38.
|
Amint az a 6. táblázatból is látszik, a lyukakba 80 μl kontrollként szolgáló oldószert és puffert, referenciaösztrogént, gyengén kötődő anyagot, nem kötődő anyagot és vizsgálati pufferben előkészített vizsgálati vegyi anyagokat kell adagolni. Ezt követően minden egyes lyukhoz hozzá kell adni 4 nM koncentrációjú [3H]-17β-ösztradiolt 40 μl mennyiségben. Mintegy 10–15 perces, 2–8 °C hőmérsékleten végzett óvatos ráztatás után hozzá kell adni a 40 μl mennyiségű humán ösztrogénreceptor α oldatot. A vizsgálati mikrotiterlemezeket 16–20 órán keresztül 2–8 °C-os hőmérsékleten inkubálni kell, a lemezeket az inkubáció ideje alatt rázógépre kell helyezni.
|
6. táblázat
A humán ösztrogénreceptor kompetitív kötési vizsgálat mikrotiterlemezeire helyezett vizsgálati összetevők térfogata
|
Térfogat (μl)
|
Összetevő
|
|
80
|
Nem jelölt 17β-ösztradiol, noretinodrel , OTES, vizsgálati vegyi anyagok, oldószer vagy puffer
|
|
40
|
4 nM [3H]-17β-ösztradiol oldat
|
|
40
|
hrERα oldat, a meghatározott koncentrációban
|
|
160
|
Teljes térfogat az egyes vizsgálati lyukakban
|
|
39.
|
Ezt követően a telítési kötési vizsgálat 20–23. pontjában ismertetett módon minden lyukhoz hozzáadnak 80 μl hideg DCC szuszpenziót, hogy elkülönítsék a humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolt a szabad [3H]-17β-ösztradioltól, majd megmérik a humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolok mennyiségét.
|
|
40.
|
A H1 és H6 közötti lyukakban (amelyeket a 4. táblázat üres (radioaktívhoz) címen jelöl) mérik a 40 μl mennyiségű [3H] jelölt ösztradiol percenkénti bomlását. A 40 μl-nek megfelelő mennyiséget közvetlenül a H1–H6 lyukakban lévő szcintillációs folyadékhoz kell adni.
|
Elfogadhatósági kritériumok
Telítési kötési vizsgálat
|
41.
|
A specifikus kötődési görbének a [3H]-17β-ösztradiol fokozódó koncentrációban való alkalmazása miatt egy platóban kell végződnie, ami jelzi a humán ösztrogénreceptor α ligandummal való telítettségét.
|
|
42.
|
A [3H]-17β-ösztradiol 1 nM koncentrációjánál megfigyelt specifikus kötődésnek a minden vizsgálatmenetben mért átlagos teljes radioaktivitás 15–25 %-os elfogadhatósági tartományán belül kell maradnia. E tartománytól való esetenkénti kis mértékű elérés még elfogadható, de ha a vizsgálatmenetek eredménye következetesen kívül esik ezen a tartományon, illetve ha egy adott vizsgálatmenet eredménye jelentősen eltér tőle, akkor ki kell igazítani a fehérjekoncentrációt és meg kell ismételni a telítési vizsgálatot.
|
|
43.
|
Az adatoknak egy lineáris Scatchard-plotot kell alkotniuk.
|
|
44.
|
A nem specifikus kötődés nem lehet túlzott mértékű. A nem specifikus kötődés értéke jellemzően nem érheti el a teljes kötődés 35 %-át. Ugyanakkor a nem specifikus kötődés aránya esetenként meghaladhatja ezt a határértéket, ha a radioaktívan jelölt 17β-ösztradiol legalacsonyabb vizsgált koncentrációjánál nagyon alacsony percenkénti bomlást mérnek.
|
Kompetitív kötési vizsgálat
|
45.
|
A nem jelölt 17β-ösztradiol növekvő koncentrációban leszorítja a [3H]-17β-ösztradiolt a receptorról, és ez összhangban van az egy kötőhelyes kompetitív kötéssel.
|
|
46.
|
A referenciaösztrogén (vagyis a 17β-ösztradiol) IC50 és Kd értékének nagyjából egyenlőnek kell lennie a [3H]-17β-ösztradiol moláris koncentrációjával; a disszociációs állandót a telítési kötési vizsgálatban határozzák meg.
|
|
47.
|
A teljes specifikus kötődésnek rendre a 20 ± 5 %-os elfogadhatósági tartományon belül kell maradnia, ha a vizsgálatmenetekben az egyes lyukakhoz hozzáadott összes radioaktívan jelölt anyag átlagos mért koncentrációja 1 nM volt. E tartománytól való esetenkénti kis mértékű elérés még elfogadható, de ha a vizsgálatmenetek eredménye következetesen kívül esik ezen a tartományon, illetve ha egy adott vizsgálatmenet eredménye jelentősen eltér ettől, akkor ki kell igazítani a fehérjekoncentrációt.
|
|
48.
|
Az oldószer nem módosíthatja a vizsgálat érzékenységét vagy megismételhetőségét. Az oldószeres kontroll (Tb-ként jelölve) eredményeit összevetik a puffer kontroll eredményeivel annak ellenőrzésére, hogy az alkalmazott oldószer nincs hatással a vizsgálati rendszerre. Amennyiben a TB kontroll és a puffer kontroll hasonló eredményre vezet, akkor az oldószer nem befolyásolja a vizsgálatot.
|
|
49.
|
A nem kötődő anyag maximális 10-3 M (OTES) vagy 10-4 M (DBP) koncentrációnál legfeljebb 25 % [3H]-17β-ösztradiolt szoríthat le a humán ösztrogénreceptor α-ról.
|
|
50.
|
A Freyberger–Wilson humán ösztrogénreceptor-kötési vizsgálat validálási tanulmányából származó adatok alapján a referenciaösztrogénre és két gyengén kötődő anyagra (pl. noretinodrel , noretindron) vonatkozva teljesítménykritériumokat dolgoztak ki (2. referencia N. függeléke). A validálási tanulmányban részt vevő laboratóriumok 95 %-os konfidenciaintervallumot adtak meg az átlag (n) +/- SD értékéhez minden kontroll vizsgálatmenetben. 95 %-os konfidenciaintervallumot számoltak a görbeillesztési paraméterekre (vagyis a görbe tetejére, aljára, a Hill-meredekségre és a logIC50-re), a referenciaösztrogénre és a gyengén kötődő anyagokra, valamint a gyengén kötődő anyagok referenciaösztrogénhez viszonyított log10RBA értékére vonatkozólag, ami teljesítménykritériumként szolgál a pozitív kontrollanyagok számára. Az 1. táblázat ismerteti a görbeillesztési paraméterek várt tartományait, amelyek használhatók teljesítménykritériumként. A gyakorlatban az IC50 tartománya a receptor preparátum Kd értékétől és a ligandum koncentrációjától függően kissé változó lehet.
|
|
51.
|
A potenciális vizsgálati vegyi anyagok széles skálája, valamint potenciális affinitásuk és az eredmények (pl. teljes görbe, részleges görbe, nem illeszthető görbe) változatossága miatt a vizsgálati vegyi anyagok görbeillesztési paramétereire nézve nem dolgoztak ki teljesítménykritériumokat. Egy adott vizsgálati vegyi anyag egyes vizsgálatmeneteiből származó eredmények értékelése során azonban szakmai megítélés alapján kell dönteni. A vizsgálati vegyi anyag megfelelő koncentrációtartományát kell használni ahhoz, hogy egyértelműen meg lehessen határozni a kompetitív görbe (pl. 90–100 %-os kötődést jelző) platóját. A vizsgálati vegyi anyag különböző koncentrációin történő ismétlések eltérései közötti és a három független vizsgálatmenet közötti variabilitásnak észszerű mértéken belül kell maradnia és tudományosan védhetőnek kell lennie. Egy adott vizsgálati vegyi anyag minden vizsgálatmenetében elvégzett kontrollok eredményeinek meg kell közelíteniük a Freyberger–Wilson vizsgálat teljesítménymutatóit, és összhangban kell lenniük minden releváns laboratórium történeti kontrolladataival.
|
AZ ADATOK ELEMZÉSE
Telítési kötési vizsgálat
|
52.
|
Mérni kell mind a teljes, mind a nem specifikus kötődést. Ezen értékek alapján számolható ki a [3H]-17β-ösztradiol növekvő koncentrációin létrejött egyensúlyi állapotokban kiváltott specifikus kötődés úgy, hogy a teljes kötődésből kivonják a nem specifikus kötődést. A specifikus kötődés és a [3H]-17β-ösztradiol koncentrációi közötti függvényt ábrázoló grafikon a maximális specifikus kötődésre jellemző platóban végződik, amely azt az állapotot jelzi, amikor a humán ösztrogénreceptor α telítődött [3H]-17β-ösztradiollal. Emellett az adatok elemzése dokumentálja, hogy a [3H]-17β-ösztradiol egyetlen, nagy affinitású kötőhelyhez kapcsolódik. A nem specifikus, a teljes és a specifikus kötődést egy telítési kötési görbén kell ábrázolni. Az adatokat a továbbiakban nem-lineáris regressziós analízissel kell elemezni (pl. BioSoft; McPherson, 1985; Motulsky, 1995), végül pedig egy Scatchard-plot grafikonon ábrázolni.
|
|
53.
|
Az adatelemzés során kizárólag a teljes kötődési adatok alapján meg kell határozni a Bmax és a Kd értékét, feltételezve, hogy a nem specifikus kötődés lineáris, más módszer alkalmazását indokolni kell. A legjobban illeszkedő görbét pedig robusztus regresszióval kell meghatározni, az ettől való eltérést indokolni kell. A robusztus regresszió alkalmazásának módszerét meg kell adni. Abban az esetben, ha a Bmax és a Kd értékét a telítési kötési adatok alapján határozzák meg, mindig korrigálni kell a ligandum koncentrációjának csökkenését (például a Swillens, 1995 által leírt módszerrel).
|
Kompetitív kötési vizsgálat
|
54.
|
A kompetitív kötési görbe a [3H]-17β-ösztradiol specifikus kötődésének és a kompetítor (log10 egységekben feltüntetett) koncentrációjának összefüggését ábrázolja. A vizsgálati vegyi anyag azon koncentrációja, amely meggátolja a [3H]-17β-ösztradiol maximális specifikus kötődésének 50 %-át, az IC50 érték.
|
|
55.
|
A pozitív kontrollanyagok (pl. a referenciaösztrogén és a gyengén kötődő anyag) becsült log(IC50) értékének meghatározásához egy megfelelő nem-lineáris görbeillesztő szoftvert kell használni, amely illeszteni tudja egy négyparaméteres Hill-egyenlet görbéjét (pl. BioSoft; McPherson, 1985; Motulsky, 1995). A görbék illesztése során a görbe tetejét, alját, meredekségét jelző értékeket és a log(IC50) értékét általában szabadon ábrázolják. A legjobban illeszkedő görbét robusztus regresszióval kell meghatározni, az ettől való eltérést indokolni kell. A ligandum csökkenését nem kell korrigálni. A kezdeti analízist követően minden kötési görbét ellenőrizni kell, hogy megfelelően illeszkedik-e a modellre. A gyengén kötődő anyag relatív kötési affinitását a gyengén kötődő anyag log (IC50) értékének a 17β-ösztradiol log (IC50) értékéhez viszonyított arányaként kell meghatározni. A pozitív kontrollok és a nem kötődő kontroll eredményeit a vizsgálat e 2. függelék 45–50. pontjában ismertetett teljesítménymutatói alapján kell értékelni.
|
|
56.
|
Az összes vizsgálati vegyi anyag adatait lépésenként kell elemezni, biztosítva ezáltal az adatok megfelelő elemzését és a kompetitív kötési görbék helyes osztályozását. Javasolt vizsgálati anyagonként minden vizsgálat megkezdésekor egy szabványosított adatelemzést végezni, amely megegyezik a referenciaösztrogén és a gyengén kötődő anyag kontrolljainál alkalmazott analízissel (lásd a fenti 55. pontot). Az adatelemzés végeztével technikailag ellenőrizni kell a görbeillesztési paramétereket, valamint vizuálisan is ellenőrizni kell, hogy az adatok mennyire illeszkednek az egyes vizsgálatmenetek kompetitív kötési görbéjéhez. Amennyiben a technikai ellenőrzés során megfigyelhető a specifikusan kötődő [3H]-17β-ösztradiol arányának koncentrációfüggő csökkenése, a vegyi anyagok egyes koncentrációit tartalmazó ismétlések közötti csekély eltérés és a három vizsgálatmenet görbeillesztési paramétereinek következetessége jó indikátorai annak, hogy a vizsgálatot és az adatelemzést megfelelően hajtották végre.
|
Az adatok értelmezése
|
57.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag humán ösztrogénreceptor α-hoz kötődő anyagnak minősül, ha az adatokra illeszthető kötési görbe, és a válaszgörbe legalsó pontja az adattartomány 50 %-a alatt helyezkedik el (1. ábra).
|
|
58.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, valamely vizsgálati vegyi anyag akkor tekinthető humán ösztrogénreceptor α-hoz nem kötődő anyagnak, ha:
|
—
|
az adataira illeszthető kötési görbe, és az adatokra illesztett válaszgörbe legalsó pontja az adattartomány 75 %-a felett helyezkedik el, vagy
|
|
—
|
az adataira nem illeszthető kötési görbe, és az adatok koncentrációcsoportjai közül a legalsó simítatlan átlagos kötési arány 75 % felett helyezkedik el.
|
|
|
59.
|
A vizsgálati vegyi anyag válasza akkor minősül kétértelműnek, ha a fenti feltételek egyike sem teljesül (vagyis az adatokra illesztett válaszgörbe legalsó pontja 76 és 51 % között helyezkedik el).
|
7. táblázat
A vizsgálati vegyi anyagok kompetitív kötési görbén alapuló osztályba sorolásának kritériumai
|
Besorolás
|
Kritériumok
|
|
Kötődőa
|
Az adatokra illeszthető kötési görbe.
A válaszgörbe legalsó pontja az adattartomány 50 %-a alatt helyezkedik el.
|
|
Nem kötődőb
|
Ha az adatokra illeszthető kötési görbe,
az adattartományra illesztett válaszgörbe legalsó pontja 75 %-a felett helyezkedik el.
|
|
Kétértelműc
|
Ha az adatokra nem illeszthető kötési görbe,
az adatok koncentrációcsoportjai közül a legalsó simítatlan átlagos kötési arány 75 % felett helyezkedik el.
Bármely vizsgálatmenet,
amelynek eredménye sem a kötődő, sem a nem kötődő kategóriába nem sorolható (pl. az adatokra illesztett válaszgörbe legalsó pontja 76 és 51 % között helyezkedik el).
|
1. ábra
Példák a vizsgálati vegyi anyagok kompetitív kötési görbén alapuló osztályba sorolására.

|
60.
|
A vizsgálati vegyi anyag egy laboratóriumon belül elvégzett több vizsgálatmenetének eredményét összesítik úgy, hogy egy számértéket társítanak minden vizsgálatmenethez, majd meghatározzák a vizsgálatmenetek átlagát (lásd a 8. táblázatot). A laboratórium vizsgálatmeneteinek összesített eredményét azután összevetik az egyes vizsgálati vegyi anyagok várt besorolásával.
|
8. táblázat
Az egy laboratóriumon belül több vizsgálatmenetben vizsgálati vegyi anyagok osztályba sorolásának módszere
|
Számérték társítása az egyes vizsgálatmenetekhez:
|
|
|
Besorolás
|
Számérték
|
|
Kötődő
|
2
|
|
Kétértelmű
|
1
|
|
Nem kötődő
|
0
|
|
Besorolás a vizsgálatmenetek átlagos számértéke alapján:
|
|
|
Besorolás
|
Számérték
|
|
Kötődő
|
Átlag ≥ 1,5
|
|
Kétértelmű
|
0,5 ≤ Átlag < 1,5
|
|
Nem kötődő
|
Átlag < 0,5
|
VIZSGÁLATI JELENTÉS
|
60.
|
Lásd a vizsgálati módszer „A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részének 24. pontját.
|
2.1. függelék
SZAKKIFEJEZÉSEK LISTÁJA
[3H]E2
: radioaktív tríciummal jelölt 17β-ösztradiol.
DCC
: dextránbevonatú aktívszén.
E2
: nem jelölt 17β-ösztradiol (semleges).
Vizsgálati puffer
: 10 mM trisz, 10 mg/ml szarvasmarha szérum albumin, 2 mM DTT, 10 % glicerin, 0,2 mM leupeptin, pH-értéke 7,5.
hrERα
: humán rekombináns ösztrogénreceptor alfa (ligandumkötő domén).
Ismétlés
: egyike azon lyukaknak, amelyek azonos mintát tartalmaznak azonos koncentrációban, és vizsgálatuk egy adott vizsgálatmeneten belül, párhuzamosan zajlik. E protokoll keretén belül a vizsgálati vegyi anyagok minden koncentrációját három ismétlésben tesztelik; vagyis a vizsgálati vegyi anyagok minden koncentrációját egyidejűleg három ismétlésben vizsgálják.
Vizsgálatmenet
: párhuzamosan vizsgált mikrotiterlemez minden mintája, amely minden szükséges információt biztosít a vizsgálati vegyi anyagok humán ösztrogénreceptor α-hoz való kötődésének jellemzéséhez (nevezetesen a vizsgálati lyukba adagolt összes [3H]-17β-ösztradiol, a [3H]-17β-ösztradiol maximális kötődése a hrERα-hoz, nem specifikus kötődés és a vizsgálati vegyi anyagok különböző koncentrációi által előidézett teljes kötődés). Egy vizsgálatmenet állhat koncentrációnként egyetlen vizsgálati lyukból is (vagyis egy ismétlésből), mivel azonban ez a protokoll három ismétlésben való tesztelést ír elő, egy vizsgálatmenet koncentrációnként három vizsgálati lyukat foglal magában. Emellett a protokoll meghatározza azt is, hogy vegyi anyagonként három független (azaz nem egyidejűleg lefolytatott) vizsgálatmenetre van szükség.
2.2. függelék
HÁROM ISMÉTLÉSES TIPIKUS [3H]-17Β-ÖSZTRADIOL TELÍTÉSI VIZSGÁLAT
|
Három ismétléses tipikus [3H]-17β-ösztradiol telítési vizsgálat
|
|
Pozíció
|
Ismétlés
|
Lyuktípus kódja
|
Radioaktív E2 induló koncentrációja (nM)
|
Radioaktív E2 térfogata (μl)
|
Radioaktív E2 végső koncentrációja (nM)
|
Nem jelölt E2 induló koncentráció (μM )
|
Nem jelölt E2 térfogata (μl)
|
Nem jelölt E2 végső koncentrációja (μM)
|
Puffer térfogata (μl)
|
Receptor térfogata (μl)
|
A lyukak teljes térfogata
|
|
A1
|
1
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A2
|
2
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A3
|
3
|
H
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A4
|
1
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A5
|
2
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A6
|
3
|
H
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A7
|
1
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A8
|
2
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A9
|
3
|
H
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A10
|
1
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A11
|
2
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
A12
|
3
|
H
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B1
|
1
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B2
|
2
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B3
|
3
|
H
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B4
|
1
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B5
|
2
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B6
|
3
|
H
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B7
|
1
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B8
|
2
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B9
|
3
|
H
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B10
|
1
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B11
|
2
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
B12
|
3
|
H
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
80
|
40
|
160
|
|
D1
|
1
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D2
|
2
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D3
|
3
|
HC
|
0,12
|
40
|
0,03
|
0,06
|
80
|
0,03
|
—
|
40
|
160
|
|
D4
|
1
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D5
|
2
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D6
|
3
|
HC
|
0,24
|
40
|
0,06
|
0,12
|
80
|
0,06
|
—
|
40
|
160
|
|
D7
|
1
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D8
|
2
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D9
|
3
|
HC
|
0,32
|
40
|
0,08
|
0,16
|
80
|
0,08
|
—
|
40
|
160
|
|
D10
|
1
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D11
|
2
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
D12
|
3
|
HC
|
0,40
|
40
|
0,10
|
0,2
|
80
|
0,1
|
—
|
40
|
160
|
|
E1
|
1
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E2
|
2
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E3
|
3
|
HC
|
1,20
|
40
|
0,30
|
0,6
|
80
|
0,3
|
—
|
40
|
160
|
|
E4
|
1
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E5
|
2
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E6
|
3
|
HC
|
2,40
|
40
|
0,60
|
1,2
|
80
|
0,6
|
—
|
40
|
160
|
|
E7
|
1
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E8
|
2
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E9
|
3
|
HC
|
4,00
|
40
|
1,00
|
2
|
80
|
1
|
—
|
40
|
160
|
|
E10
|
1
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E11
|
2
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
E12
|
3
|
HC
|
12,00
|
40
|
3,00
|
6
|
80
|
3
|
—
|
40
|
160
|
|
G1
|
1
|
Radioaktív
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G2
|
2
|
Radioaktív
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G3
|
3
|
Radioaktív
|
0,12
|
40
|
0,03
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G4
|
1
|
Radioaktív
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G5
|
2
|
Radioaktív
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G6
|
3
|
Radioaktív
|
0,24
|
40
|
0,06
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G7
|
1
|
Radioaktív
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G8
|
2
|
Radioaktív
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G9
|
3
|
Radioaktív
|
0,32
|
40
|
0,08
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G10
|
1
|
Radioaktív
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G11
|
2
|
Radioaktív
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
G12
|
3
|
Radioaktív
|
0,40
|
40
|
0,10
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H1
|
1
|
Radioaktív
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H2
|
2
|
Radioaktív
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H3
|
3
|
Radioaktív
|
1,20
|
40
|
0,30
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H4
|
1
|
Radioaktív
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H5
|
2
|
Radioaktív
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H6
|
3
|
Radioaktív
|
2,40
|
40
|
0,60
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H7
|
1
|
Radioaktív
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H8
|
2
|
Radioaktív
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H9
|
3
|
Radioaktív
|
4,00
|
40
|
1,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H10
|
1
|
Radioaktív
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H11
|
2
|
Radioaktív
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
H12
|
3
|
Radioaktív
|
12,00
|
40
|
3,00
|
—
|
—
|
—
|
—
|
—
|
40
|
|
A „radioaktív” kifejezéssel jelölt lyukak az inkubálás alatt üresek. A 40 μl-t kizárólag a szcintillációs számlálás céljából adják hozzá a lyukakhoz.
|
2.3. függelék:
MINTÁK ELRENDEZÉSE A KOMPETITÍV KÖTÉSI VIZSGÁLATHOZ
|
Lemez
|
Pozíció
|
Ismétlés
|
Lyuk típusa
|
Lyuk kódja
|
Koncentráció kódja
|
Kompetítor induló koncentrációja (M)
|
hrER törzs (μl)
|
Puffer térfogata (μl)
|
Nyomjelző (radioaktív E2) térfogata (μL)
|
Hígítási lemezről adagolt térfogat (μL)
|
Végső térfogat (μl)
|
Kompetítor végső koncentrációja (M)
|
|
Sz
|
A1
|
1
|
teljes kötődés
|
TB
|
TB1
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A2
|
2
|
teljes kötődés
|
TB
|
TB2
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A3
|
3
|
teljes kötődés
|
TB
|
TB3
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A4
|
1
|
teljes kötődés
|
TB
|
TB4
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A5
|
2
|
teljes kötődés
|
TB
|
TB5
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A6
|
3
|
teljes kötődés
|
TB
|
TB6
|
—
|
40
|
|
40
|
80
|
160
|
—
|
|
Sz
|
A7
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
A8
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
A9
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
A10
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
A11
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
A12
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
B1
|
1
|
nem jelölt E2
|
Sz
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
B2
|
2
|
nem jelölt E2
|
Sz
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
B3
|
3
|
nem jelölt E2
|
Sz
|
S1
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
B4
|
1
|
nem jelölt E2
|
Sz
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
B5
|
2
|
nem jelölt E2
|
Sz
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
B6
|
3
|
nem jelölt E2
|
Sz
|
S2
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
B7
|
1
|
nem jelölt E2
|
Sz
|
S3
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
B8
|
2
|
nem jelölt E2
|
Sz
|
S3
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
B9
|
3
|
nem jelölt E2
|
Sz
|
S3
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
B10
|
1
|
nem jelölt E2
|
Sz
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
B11
|
2
|
nem jelölt E2
|
Sz
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
B12
|
3
|
nem jelölt E2
|
Sz
|
S4
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
C1
|
1
|
nem jelölt E2
|
Sz
|
S5
|
6,00 E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
Sz
|
C2
|
2
|
nem jelölt E2
|
Sz
|
S5
|
6,00 E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
Sz
|
C3
|
3
|
nem jelölt E2
|
Sz
|
S5
|
6,00 E-10
|
40
|
—
|
40
|
80
|
160
|
3,0 E-10
|
|
Sz
|
C4
|
1
|
nem jelölt E2
|
Sz
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1,0 E-10
|
|
Sz
|
C5
|
2
|
nem jelölt E2
|
Sz
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1,0 E-10
|
|
Sz
|
C6
|
3
|
nem jelölt E2
|
Sz
|
S6
|
2,00 E-10
|
40
|
—
|
40
|
80
|
160
|
1,0 E-10
|
|
Sz
|
C7
|
1
|
nem jelölt E2
|
Sz
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1,0 E-11
|
|
Sz
|
C8
|
2
|
nem jelölt E2
|
Sz
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1,0 E-11
|
|
Sz
|
C9
|
3
|
nem jelölt E2
|
Sz
|
S7
|
2,00 E-11
|
40
|
—
|
40
|
80
|
160
|
1,0 E-11
|
|
Sz
|
C10
|
1
|
üres
|
üres
|
B1
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Sz
|
C11
|
2
|
üres
|
üres
|
B2
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Sz
|
C12
|
3
|
üres
|
üres
|
B3
|
—
|
—
|
160
|
—
|
—
|
160
|
—
|
|
Sz
|
D1
|
1
|
noretinodrel
|
NE
|
WP1
|
6,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
D2
|
1
|
noretinodrel
|
NE
|
WP1
|
6,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
D3
|
1
|
noretinodrel
|
NE
|
WP1
|
6,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
D4
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1,0 E-05
|
|
Sz
|
D5
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1,0 E-05
|
|
Sz
|
D6
|
1
|
noretinodrel
|
NE
|
WP2
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
1,0 E-05
|
|
Sz
|
D7
|
1
|
noretinodrel
|
NE
|
WP3
|
6,00 E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
Sz
|
D8
|
1
|
noretinodrel
|
NE
|
WP3
|
6,00 E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
Sz
|
D9
|
1
|
noretinodrel
|
NE
|
WP3
|
6,00 E-06
|
40
|
—
|
40
|
80
|
160
|
3,0 E-06
|
|
Sz
|
D10
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
D11
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
D12
|
1
|
noretinodrel
|
NE
|
WP4
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
E1
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
E2
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
E3
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-07
|
40
|
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
E4
|
1
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
E5
|
2
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
E6
|
3
|
noretinodrel
|
NE
|
WP
|
2,00 E-07
|
40
|
|
40
|
80
|
160
|
1,0 E-07
|
|
Sz
|
E7
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
Sz
|
E8
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
Sz
|
E9
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-08
|
40
|
—
|
40
|
80
|
160
|
3,0 E-08
|
|
Sz
|
E10
|
1
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
E11
|
2
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
E12
|
3
|
noretinodrel
|
NE
|
WP
|
6,00 E-09
|
40
|
—
|
40
|
80
|
160
|
3,0 E-09
|
|
Sz
|
F1
|
1
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1,0 E-03
|
|
Sz
|
F2
|
2
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1,0 E-03
|
|
Sz
|
F3
|
3
|
OTES
|
N
|
OTES
|
2,00 E-03
|
40
|
—
|
40
|
80
|
160
|
1,0 E-03
|
|
Sz
|
F4
|
1
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1,0 E-04
|
|
Sz
|
F5
|
2
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1,0 E-04
|
|
Sz
|
F6
|
3
|
OTES
|
N
|
OTES
|
2,00 E-04
|
40
|
—
|
40
|
80
|
160
|
1,0 E-04
|
|
Sz
|
F7
|
1
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
F8
|
2
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
F9
|
3
|
OTES
|
N
|
OTES
|
2,00 E-05
|
40
|
—
|
40
|
80
|
160
|
3,0 E-05
|
|
Sz
|
F10
|
1
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
F11
|
2
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
F12
|
3
|
OTES
|
N
|
OTES
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
Sz
|
G1
|
1
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
G2
|
2
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
G3
|
3
|
OTES
|
N
|
OTES
|
2,00 E-07
|
40
|
—
|
40
|
80
|
160
|
3,0 E-07
|
|
Sz
|
G4
|
1
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
G5
|
2
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
G6
|
3
|
OTES
|
N
|
OTES
|
2,00 E-08
|
40
|
—
|
40
|
80
|
160
|
1,0 E-08
|
|
Sz
|
G7
|
1
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
G8
|
2
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
G9
|
3
|
OTES
|
N
|
OTES
|
2,00 E-09
|
40
|
—
|
40
|
80
|
160
|
1,0 E-09
|
|
Sz
|
G10
|
1
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1,0 E-10
|
|
Sz
|
G11
|
2
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1,0 E-10
|
|
Sz
|
G12
|
3
|
OTES
|
N
|
OTES
|
2,00 E-10
|
40
|
—
|
40
|
—
|
160
|
1,0 E-10
|
|
Sz
|
H1
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H2
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H3
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H4
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H5
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H6
|
1
|
radioaktív
|
H
|
H
|
—
|
—
|
—
|
40
|
—
|
40
|
—
|
|
Sz
|
H7
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Sz
|
H8
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Sz
|
H9
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Sz
|
H10
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Sz
|
H11
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
|
Sz
|
H12
|
1
|
puffer kontroll
|
BC
|
BC
|
—
|
40
|
80
|
40
|
—
|
160
|
—
|
A „radioaktív” kifejezéssel jelölt lyukak az inkubálás alatt üresek. A 40 μl-t kizárólag a szcintillációs számlálás céljából adják hozzá a lyukakhoz.
|
Minták elrendezése a kompetitív kötési vizsgálathoz
|
|
Lemez
|
Pozíció
|
Ismétlés
|
Lyuk típusa
|
Lyuk kódja
|
Koncentráció kódja
|
Kompetítor induló koncentrációja (M)
|
hrER törzs (μL)
|
Puffer térfogata (μL)
|
Nyomjelző (radioaktív E2) térfogata (μL)
|
Hígítási lemezről adagolt térfogat (μL)
|
Végső térfogat (μl)
|
Kompetítor végső koncentrációja (M)
|
|
P1
|
A1
|
1
|
teljes kötődés
|
TB
|
TBB1B1
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A2
|
2
|
teljes kötődés
|
TB
|
TB2
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A3
|
3
|
teljes kötődés
|
TB
|
TB3
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A4
|
1
|
teljes kötődés
|
TB
|
TB4
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A5
|
2
|
teljes kötődés
|
TB
|
TB5
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A6
|
3
|
teljes kötődés
|
TB
|
TB6
|
—
|
40
|
—
|
40
|
80
|
160
|
—
|
|
P1
|
A7
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
A8
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
A9
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
A10
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
A11
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
A12
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S0
|
2,00 E-06
|
40
|
—
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
B1
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
B2
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
B3
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
B4
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
B5
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
B6
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
B7
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
B8
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
B9
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
B10
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
B11
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
B12
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
C1
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
C2
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
C3
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
C4
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
C5
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
C6
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
C7
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
C8
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
C9
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
C10
|
1
|
1. vizsgálati vegyi anyag
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
C11
|
2
|
1. vizsgálati vegyi anyag
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
C12
|
3
|
1. vizsgálati vegyi anyag
|
TC1
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
D1
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
D2
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
D3
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
D4
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
D5
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
D6
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
D7
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
D8
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
D9
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
D10
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
D11
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
D12
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
E1
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
E2
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
E3
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
E4
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
E5
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
E6
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
6
|
—
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
E7
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
E8
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
E9
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
7
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
E10
|
1
|
2. vizsgálati vegyi anyag
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
E11
|
2
|
2. vizsgálati vegyi anyag
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
E12
|
3
|
2. vizsgálati vegyi anyag
|
TC2
|
8
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
F1
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
F2
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
F3
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
1
|
2,00 E-03
|
40
|
0
|
40
|
80
|
160
|
1,0 E-03
|
|
P1
|
F4
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
F5
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
F6
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
2
|
2,00 E-04
|
40
|
0
|
40
|
80
|
160
|
1,0 E-04
|
|
P1
|
F7
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
F8
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
F9
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
3
|
2,00 E-05
|
40
|
0
|
40
|
80
|
160
|
1,0 E-05
|
|
P1
|
F10
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
F11
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
F12
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
4
|
2,00 E-06
|
40
|
0
|
40
|
80
|
160
|
1,0 E-06
|
|
P1
|
G1
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
G2
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
G3
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
5
|
2,00 E-07
|
40
|
0
|
40
|
80
|
160
|
1,0 E-07
|
|
P1
|
G4
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
G5
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
G6
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
6
|
2,00 E-08
|
40
|
0
|
40
|
80
|
160
|
1,0 E-08
|
|
P1
|
G7
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
G8
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
G9
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
7
|
2,00 E-09
|
40
|
0
|
40
|
80
|
160
|
1,0 E-09
|
|
P1
|
G10
|
1
|
3. vizsgálati vegyi anyag
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
G11
|
2
|
3. vizsgálati vegyi anyag
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
G12
|
3
|
3. vizsgálati vegyi anyag
|
TC3
|
8
|
2,00 E-10
|
40
|
0
|
40
|
80
|
160
|
1,0 E-10
|
|
P1
|
H1
|
1
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H2
|
2
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H3
|
3
|
noretinodrel
|
NE
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H4
|
1
|
noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H5
|
2
|
noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H6
|
3
|
noretinodrel
|
NE
|
|
1,00 E-4,5
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H7
|
1
|
nem jelölt E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H8
|
2
|
nem jelölt E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H9
|
3
|
nem jelölt E2 S
|
|
|
IC50
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H10
|
1
|
nem jelölt E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H11
|
2
|
nem jelölt E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
|
P1
|
H12
|
3
|
nem jelölt E2 S
|
|
|
1,00 E-7
|
40
|
0
|
40
|
80
|
160
|
|
3. függelék
A CERI INTÉZET (CHEMICAL EVALUATION AND RESEARCH INSTITUTE) HUMÁN REKOMBINÁNS LIGANDUMKÖTŐ DOMÉNNEL RENDELKEZŐ ERΑ FEHÉRJE HASZNÁLATÁN ALAPULÓ IN VITRO ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLATA
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
1.
|
Ez az in vitro ösztrogénreceptor (ERα) telítési és kompetitív kötési vizsgálat a humán ösztrogénrecptor α (hrERα) ligandumkötő doménjét alkalmazza. A japán CERI Intézet által előállított fehérjekonstrukció egy glutation-S-transzferáz (GST) fúziós fehérje, amelyet E. coli-ban termeltetnek. A CERI protokollt több laboratórium részvételével zajló, nemzetközi validálási tanulmánynak (2) vetették alá, amely bizonyította, hogy a vizsgálat tervezett felhasználása tekintetében releváns és megbízható.
|
|
2.
|
A vizsgálat egy szűrési eljárás, amelynek révén azonosíthatók a humán ösztrogénreceptor α-hoz kötődni képes anyagok. A vizsgálatot annak meghatározására alkalmazzák, hogy egy adott vizsgálati vegyi anyag képes-e versenyezni a 17β-ösztradiollal a humán ösztrogénreceptor α ligandumkötő doménjéhez való kötődésért. A vizsgálat kvantitatív eredményei magukban foglalhatják az IC50 értéket (vagyis azt a koncentrációt, amelyre a vizsgálati vegyi anyagnak szüksége van ahhoz, hogy a [3H]-17β-ösztradiol felét leszorítsa a humán ösztrogénreceptor α-ról), valamint a vizsgálati vegyi anyag humán ösztrogénreceptor α-hoz való relatív – a 17β-ösztradiolhoz viszonyított – kötési affinitását. A vegyi anyagok szűrése céljából a vizsgálat elfogadható kvalitatív eredményei kiterjedhetnek a vizsgálati vegyi anyagok humán ösztrogénreceptor α-hoz kötődő, nem kötődő, illetve nem egyértelmű kategóriába sorolására, ezek meghatározására a kötési görbékre vonatkozó kritériumok alapján kerül sor.
|
|
3.
|
A vizsgálat radioaktív ligandumot használ, ezért a laboratóriumnak rendelkeznie kell radioaktív anyagok használatára vonatkozó engedéllyel. A radioaktív izotópokkal és veszélyes vegyi anyagokkal végzett valamennyi folyamat során be kell tartani a nemzeti jogszabályok által előírt rendelkezéseket és eljárásokat.
|
|
4.
|
E vizsgálat szabályozási célra való alkalmazása előtt el kell olvasni az „ÁLTALÁNOS BEVEZETÉS” és „A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részt. Az ebben a vizsgálati iránymutatásban használt fogalmak és rövidítések meghatározását az 1. függelék tartalmazza.
|
A VIZSGÁLATI ELVEK (LÁSD MÉG AZ ÁLTALÁNOS BEVEZETÉST)
|
5.
|
A humán ösztrogénreceptor α-kötési vizsgálat azt méri, hogy milyen mértékben képes egy radioaktívan jelölt ligandum (a [3H]17β-ösztradiol) az ösztrogénreceptorhoz kötődni egy adott vizsgálati vegyi anyag (a kompetítor) növekvő koncentrációjának jelenlétében. Az ösztrogénreceptorok iránt nagy affinitást mutató vizsgálati vegyi anyagok alacsonyabb koncentrációban versenyeznek a radioaktívan jelölt ligandummal azokhoz a vegyi anyagokhoz képest, amelyek kisebb affinitást mutatnak a receptor iránt.
|
|
6.
|
A vizsgálat két fő összetevőből áll: egy telítési kötési kísérletből, amelynek során meghatározzák a receptor és a ligandum közötti kölcsönhatás paramétereit, majd egy ezután végzett kompetitív kötési kísérletből, amelynek révén jellemzik a vizsgálati vegyi anyag és a radioaktívan jelölt ligandum ösztrogénreceptor-kötőhelyekért folytatott vetélkedését.
|
|
7.
|
A telítési kötési kísérlet célja, hogy a kompetitív kötési kísérlet előkészítéseként meghatározzák a receptorok egy adott tételének kötési affinitását és a receptorok számát. A telítési kötési kísérlet egyensúlyi állapotban méri egy fix koncentrációjú ösztrogénreceptor természetes ligandumához való kötési affinitását (amelynek mutatója a disszociációs állandó, a Kd) és a receptor aktív kötőhelyeinek koncentrációját (Bmax).
|
|
8.
|
A kompetitív kötési kísérletben azt mérik, hogy egy adott anyag milyen mértékű affinitással rendelkezik arra, hogy versenyezzen a [3H]17β-ösztradiollal az ösztrogénreceptorral formált kötésért. Az affinitást a vizsgálati vegyi anyag azon koncentrációjával számszerűsítik, amely egyensúlyi állapotban meggátolja a [3H]17β-ösztradiol specifikus kötődésének 50 %-át (ez az úgynevezett „50 %-os gátlást kiváltó koncentráció” vagy IC50). Az affinitás mérhető emellett a relatív kötési affinitással is (RBA, az ösztradiol ugyanazon vizsgálatmenetben, de külön mért IC50 értékéhez viszonyított affinitás). A kompetitív kötési kísérlet azt méri, hogy milyen mértékben kötődik egy fix koncentrációban alkalmazott [3H]17β-ösztradiol a vizsgálati vegyi anyag széles skálát (nyolc nagyságrendet) lefedő koncentrációja jelenlétében. Ezt követően – ahol lehetséges – az adatokat a Hill-egyenlet olyan formájára illesztik (Hill, 1910), amely meghatározza, hogy milyen mértékben szorítják le az egy kötőhelyet elfoglaló kompetitív kötődő anyagok a radioaktív ligandumot. A radioaktívan jelölt ösztradiol egyensúlyi állapotban történő leszorításának mértéke alapján jellemzik a vizsgálati vegyi anyagot kötődő, nem kötődő, vagy kétértelmű választ adó anyagként.
|
ELJÁRÁS
A humán ösztrogénreceptor α fehérje elfogadható teljesítményének igazolása
|
9.
|
A telítési és a kompetitív kötési vizsgálatok rutinszerű alkalmazása előtt a humán ösztrogénreceptor α minden új tételéről bizonyítani kell, hogy megfelelő teljesítményt nyújt abban a laboratóriumban, ahol használni fogják. A megfelelő teljesítmény egy két lépésből álló eljárással bizonyítható. Ez a két lépés a következő:
|
—
|
A humán ösztrogénreceptor α-specifikusságának és -telítettségének igazolása érdekében végezzünk el egy telítési[3H]-17β-ösztradiol kötési vizsgálatot. Az adatok nem-lineáris regressziós analízise (pl. BioSoft; McPherson, 1985; Motulsky, 1995), majd azt követő Scatchard-plot ábrázolása egy adott hrERα-tételre vonatkozólag dokumentálja a [3H]-17β-ösztradiol humán ösztrogénreceptor α-hoz való kötési affinitását (Kd) és a receptorok számát (Bmax).
|
|
—
|
Végezzünk el egy kompetitív kötési vizsgálatot a kontrollanyagok (a referenciaösztrogén (17β-ösztradiol), egy gyengén kötődő anyag (pl. noretinodrel vagy noretindron) és egy nem kötődő anyag (oktil-trietoxi-szilán, OTES)) használatával. Valamennyi laboratóriumnak létre kell hoznia egy adatbázist annak dokumentálására, hogy a referenciaösztrogén és a gyengén kötődő anyag IC50 értékét és releváns mutatóit következetesen határozta meg a különböző kísérletek és az eltérő humán ösztrogénreceptor α-tételek vizsgálata során. Emellett a kontrollanyagok kompetitív kötési görbéjét alkotó paramétereknek a 95 %-os konfidenciaintervallumon belül kell lenniük (lásd az 1. táblázatot), amelynek értékeit a vizsgálat validálási tanulmányában részt vevő laboratóriumok adatai alapján határozták meg (2).
|
1. táblázat
A CERI humán ösztrogénreceptor-kötési vizsgálatban használt referenciaösztrogén és gyengén kötődő anyag teljesítményére vonatkozó kritériumok
|
Anyag
|
Paraméter
|
Átlag (10)
|
Standard deviáció (n)
|
95 %-os konfidenciaintervallumok (11)
|
|
Alsó határ
|
Felső határ
|
|
17β-ösztradiol
|
Görbe teteje
|
104,74
|
13,12 (70)
|
101,6
|
107,9
|
|
Görbe alja
|
0,85
|
2,41 (70)
|
0,28
|
1,43
|
|
Hill-meredekség
|
–1,22
|
0,20 (70)
|
–1,27
|
–1,17
|
|
LogIC50
|
–8,93
|
0,23 (70)
|
–8,98
|
–8,87
|
|
Noretinodrel
|
Görbe teteje
|
101,31
|
10,55 (68)
|
98,76
|
103,90
|
|
Görbe alja
|
2,39
|
5,01 (68)
|
1,18
|
3,60
|
|
Hill–meredekség
|
–1,04
|
0,21 (68)
|
–1,09
|
–0,99
|
|
LogIC50
|
–6,19
|
0,40 (68)
|
–6,29
|
–6,10
|
|
Noretindron (12)
|
Görbe teteje
|
92,27
|
7,79 (23)
|
88,90
|
95,63
|
|
Görbe alja
|
16,52
|
10,59 (23)
|
11,94
|
21,10
|
|
Hill–meredekség
|
–1,18
|
0,32 (23)
|
–1,31
|
–1,04
|
|
LogIC50
|
–6,01
|
0,54 (23)
|
–6,25
|
–5,78
|
|
A laboratórium jártasságának igazolása
|
10.
|
Lásd a vizsgálati módszer „A humán ÖSZTROGÉNRECEPTOR–KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részének 17. és 18. pontját, valamint 2. táblázatát. Minden (telítési és kompetitív kötési) vizsgálatnak három független (vagyis a receptor, a vegyi anyagok és a reagensek frissen készített oldataival végezett), különböző napokon lefolytatott vizsgálatmenetből kell állnia, az egyes vizsgálatmeneteknek pedig három ismétlést kell tartalmazniuk.
|
A receptor (hrERα) koncentrációjának meghatározása
|
11.
|
Az aktív receptor koncentrációja tételenként és tárolási feltételek szerint kissé változó. Ebből adódóan a forgalmazótól való átvételkor meg kell határozni az aktív receptor koncentrációját. Így megállapítható az aktív receptor vizsgálatmenet során alkalmazandó megfelelő koncentrációja.
|
|
12.
|
A kompetitív kötési vizsgálatnak megfelelő körülmények között (vagyis 0,5 nM [3H]–ösztradiollal) a receptort 0,1, 0,2, 0,4 és 0,6 nM névleges koncentrációban inkubálni kell 1 μM nem jelölt ösztradiol jelenlétében (nem specifikus kötődés) és annak hiányában (teljes kötődés). A specifikus kötődést, amely a teljes és a nem specifikus kötődés különbségeként határozható meg, ábrázolni kell a receptor névleges koncentrációja függvényében. A receptort abban a névleges koncentrációban kell használni a telítési és a kompetitív kötési kísérletek során, amelynél a specifikus kötődési értékei megegyeznek a hozzáadott radioaktívan jelölt ösztradiol kötési értékeinek 40 %-ával. A humán ösztrogénreceptor 0,2 nM-es végső koncentrációja többnyire eleget tesz ennek a feltételnek.
|
|
13.
|
Amennyiben a 40 %-os feltétel több próbálkozás után sem teljesül, ellenőrizni kell, hogy nincsenek-e potenciális hibák a kísérlet elrendezésében. A 40 %-os feltétel nem teljesülése jelezheti azt is, hogy a rekombináns tételben nagyon csekély az aktív receptorok száma, ilyen esetben fontolóra kell venni másik receptortétel alkalmazását.
|
Telítési vizsgálat
|
14.
|
A [3H]17β-ösztradiolt kell kiértékelni nyolc növekvő koncentrációban, három ismétlésben, az alábbi három kitétellel (lásd a 2. táblázatot):
|
a.
|
Nem jelölt 17β-ösztradiol nélkül és ösztrogénreceptorral. Ez alapján határozható meg a teljes kötődés a csak [3H]17β-ösztradiolt tartalmazó lyukakban mért radioaktivitás alapján.
|
|
b.
|
A jelölt 17β-ösztradiolhoz képest 2000-szeres koncentrációjú nem jelölt 17β-ösztradiol jelenlétében és ösztrogénreceptorral. Ez az állapot arra szolgál, hogy az aktív kötőhelyek telítődjenek nem jelölt 17β-ösztradiollal, így a lyukakban mért radioaktivitás alapján meghatározható a nem specifikus kötődés. A fennmaradó, receptorhoz kötődő radioaktív ösztrogének feltételezhetően nem specifikus helyre kötődőnek, mivel a nem jelölt ösztrogének olyan magas koncentrációban vannak jelen, hogy elfoglalják a receptor valamennyi szabad specifikus kötőhelyét.
|
|
c.
|
Nem jelölt 17β-ösztradiol és ösztrogénreceptor nélkül (így meghatározható a teljes radioaktivitás).
|
A [3H]-17β-ösztradiol és a nem jelölt 17β-ösztradiol oldatainak és a humán ösztrogénreceptor α-nak az elkészítése
|
15.
|
A [3H]-17β-ösztradiolból 40 nM koncentrációjú oldatot kell készíteni 1 μM [3H]-17β-ösztradiol törzsoldatból DMSO-ban, úgy, hogy DMSO-t adunk hozzá (200 nM oldatot készítünk), majd szobahőmérsékleten a vizsgálati puffer hozzáadásával elkészítjük a 40 nM koncentrációjú oldatot. Ezzel a 40 nM koncentrációjú oldattal és a vizsgálati pufferrel szobahőmérsékleten el kell elkészíteni a [3H]-17β-ösztradiol 0,313 nM-től 40 nM-ig terjedő oldatsorozatát (a 2. táblázat 12. sávja alapján). A 0,0313 és 4,0 nM közötti végső vizsgálati koncentrációtartományt úgy kapjuk meg, hogy ezekből az oldatokból 10 μl mennyiséget teszünk egy 96 lyukú mikrotiterlemez megfelelő vizsgálati lyukába (lásd a 2. és 3. táblázatot). A vizsgálati puffer elkészítését, a [3H]-17β-ösztradiol eredeti törzsoldatának az ösztradiol specifikus aktivitásán alapuló kiszámítását, az oldatok elkészítését, valamint a koncentrációértékek meghatározását részletesen ismerteti a CERI protokoll (2).
|
|
16.
|
A nem jelölt 17β-ösztradiol oldatait a 17β-ösztradiol 1 nM koncentrációjú törzsoldatából kell elkészíteni vizsgálati puffer hozzáadásával, nyolc növekvő koncentrációból álló, 0,625 μM-től 80 μM-ig terjedő induló koncentrációsorozatot készítve. A 0,0625 és 8 μM közötti végső vizsgálati koncentrációkat úgy kapjuk meg, hogy ezekből az oldatokból 10 μl mennyiséget elhelyezünk egy 96 lyukú mikrotiterlemez nem specifikus kötődés mérésére szolgáló vizsgálati lyukaiba (lásd a 2. és 3. táblázatot). A nem jelölt 17β-ösztradiol oldatainak elkészítését részletesen ismerteti a CERI protokoll (2).
|
|
17.
|
A receptor azon koncentrációját kell alkalmazni, amelyik 40 %-os (± 10 %) specifikus kötődést mutat (lásd a 12–13. pontot). A humán ösztrogénreceptor α oldatot jéghideg vizsgálati pufferrel kell elkészíteni közvetlenül felhasználás előtt, vagyis azt követően, hogy előkészítettük a teljes kötődést és a nem specifikus kötődést mérő, valamint a csak a radioaktív ligandumot tartalmazó lyukakat.
|
|
18.
|
A 96 lyukú mikrotiterlemezeket a 2. táblázatban foglaltak szerint készítik el, a [3H]-17β-ösztradiol egyes koncentrációértékeit 3 ismétlésben vizsgálva. A [3H]-17β-ösztradiol, a nem jelölt 17β-ösztradiol, a puffer és a receptor térfogatát a 3. táblázat ismerteti.
2. táblázat
Mikrotiterlemez elrendezése a telítési kötési vizsgálathoz
|
|
1. (*10)
|
2. (*10)
|
3. (*10)
|
4. (*10)
|
5. (*10)
|
6. (*10)
|
7. (*10)
|
8. (*10)
|
9. (*10)
|
10
|
11. (*11)
|
12. (*11)
|
|
A TB méréséhez
|
A NSB méréséhez
|
Csak a radioaktív ligandum meghatározásához
|
|
Nem jelölt E2 oldatai a lemez 4–6. oszlopához
|
[3H]E2 oldatai a lemez 1–9. oszlopához
|
|
A
|
0,0313 nM [3H] E2+ ER
|
0,0313 nM [3H] E2+ 0,0625 μM E2+ ER
|
0,0313 nM
|
|
0,625 μM
|
0,313 nM
|
|
B
|
0,0625 nM [3H] E2+ ER
|
0,0625 nM [3H] E2+ 0,125 μM E2+ ER
|
0,0625 nM
|
|
1,25 μM
|
0,625 nM
|
|
C
|
0,125 nM [3H] E2+ ER
|
0,125 nM [3H] E2+ 0,25 μM E2+ ER
|
0,125 nM
|
|
2,5 μM
|
1,25 nM
|
|
D
|
0,250 nM [3H] E2+ ER
|
0,250 nM [3H] E2+ 0,5 μM E2+ ER
|
0,250 nM
|
|
5 μM
|
2,5 nM
|
|
E
|
0,50 nM [ H] E2+ ER
|
0,50 nM [3H] E2+ 1 μM E2+ ER
|
0,50 nM
|
|
10 μM
|
5 nM
|
|
F
|
1,00 nM [3H] E2+ ER
|
1,00 nM [3H] E2+ 2 μM E2+ ER
|
1,00 nM
|
|
20 μM
|
10 nM
|
|
G
|
2,00 nM [3H] E2+ ER
|
2,00 nM [3H] E2+ 4 μM E2+ ER
|
2,00 nM
|
|
40 μM
|
20 nM
|
|
H
|
4,00 nM [3H] E2+ ER
|
4,00 nM [3H] E2+ 8 μM E2+ ER
|
4,00 nM
|
|
80 μM
|
40 nM
|
|
TB
|
-
|
teljes kötődés
|
|
NSB
|
-
|
nem specifikus kötődés
|
|
[3H] E2
|
-
|
[3H]17β-ösztradiol
|
|
E2
|
-
|
nem jelölt 17β-ösztradiol
|
|
3. táblázat
A reagens térfogatértékei a telítési mikrotiterlemezhez
|
Sáv száma
|
1
|
2
|
3
|
4
|
5
|
6
|
7. (*12)
|
8. (*12)
|
9. (*12)
|
|
Előkészítési folyamat lépései
|
TB lyukak
|
NSB lyukak
|
Csak radioaktív ligandum
|
|
A fenti reagens lyukak összetevőinek térfogata és hozzáadási sorrendje
|
Puffer
|
60 μl
|
50 μl
|
90 μl
|
|
nem jelölt E2 a 2. táblázat 11. sávjából
|
—
|
10 μl
|
—
|
|
[3H]E2 a 2. táblázat 12. sávjából
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα
|
30 μl
|
30 μl
|
—
|
|
Reagens összes térfogata
|
100 μl
|
100 μl
|
100 μl
|
|
Inkubáció
|
EZT KÖVETŐEN REAGENS KÉT ÓRÁS INKUBÁLÁSA
|
Radioaktivitás mérése közvetlenül az elkészítés után. Nincs inkubáció.
|
|
Kezelés 0,4 % DCC-vel
|
Igen
|
Igen
|
Nem
|
|
A 0,4 %-os DCC térfogata
|
100 μl
|
100 μl
|
—
|
|
Szűrés
|
Igen
|
Igen
|
Nem
|
|
PERCENKÉNTI BOMLÁS MÉRÉSE
|
|
A szcintillációs folyadékhoz hozzáadott mérendő térfogat
|
100 μl (*13)
|
100 μl (*13)
|
50 μl
|
|
|
19.
|
A teljes kötődés és a nem specifikus kötődés meghatározásához használt vizsgálati mikrotiterlemezeket két órán keresztül kell inkubálni, szobahőmérsékleten (22 °C és 28 °C között).
|
|
A humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiol mérése
|
20.
|
A két órás inkubációt követően a humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolt el kell különíteni a szabad [3H]-17β-ösztradioltól, ehhez adjunk hozzá 100 μl 0,4 %-os jéghideg DCC szuszpenziót a lyukakhoz. Ezután a lemezeket 10 percre jégre kell helyezni, majd a reakciókeverék és a DCC szuszpenzió mikrotiterlemez szűrőn való átszűrésével el kell távolítani a DCC-t. A leszűrt anyagból 100 μl mennyiséget az LSC számláló berendezés csöveibe töltött szcintillációs folyadékba tesznek, és a folyadék szcintillációs számlálóval csövenként meghatározzák a (dpms-ben mért) percenkénti bomlást.
|
|
21.
|
Amennyiben nem áll rendelkezésre mikrolemez szűrő, a DCC eltávolítható centrifugálással is. A humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolt tartalmazó 50 μl felülúszót különös óvatossággal kell kivenni, nehogy a DCC-hez hozzáérve átszennyezzük a lyukakat, majd szcintillációs számlálást végzünk.
|
|
22.
|
A radioaktív ligandumot önmagában használják a vizsgálati lyukakba adagolt [3H]-17β-ösztradiol percenkénti bomlásának (dpm) mérésére. A radioaktivitás mértékét közvetlenül az elkészítés után kell számszerűsíteni. Ezeket a lyukakat nem szabad inkubálni és DCC szuszpenzióval kezelni, tartalmukat közvetlenül a szcintillációs folyadékhoz kell adagolni. Ezek a mérőszámok azt jelzik, hogy mennyi dpms-ben kifejezett [3H]-17β-ösztradiolt adtunk hozzá a teljes kötődést és a nem specifikus kötődést vizsgáló lyukakhoz.
|
Kompetitív kötési vizsgálat
|
23.
|
A kompetitív kötési vizsgálat azt méri, hogy egy állandó koncentrációjú [3H]-17β-ösztradiol milyen mértékben képes kötődni az adott vizsgálati vegyi anyag növekvő koncentrációja mellett. Egy vizsgálatmeneten belül minden koncentrációt három párhuzamos ismétlésben kell vizsgálni. Emellett minden vizsgálati vegyi anyagot három nem egyidejűleg végzett vizsgálatmenetben kell tesztelni. A vizsgálatot egy vagy több 96 lyukú mikrotiterlemezen kell elvégezni.
|
Kontrollok
|
24.
|
A vizsgálat végrehajtása során minden egyes kísérletben párhuzamosan vizsgálni kell az oldószert és a kontrollanyagokat (a referenciaösztrogént, valamint a gyengén kötődő és a nem kötődő anyagot). Minden vizsgálatmenetben ugyanazon a lemezen vizsgálni kell a referenciaösztrogén teljes koncentrációgörbéjét és a kontrollanyagokat (vagyis a gyengén kötődő és a nem kötődő anyagot). Az összes többi lemeznek tartalmaznia kell i. az E2 és a gyengén kötődő anyag magas (maximális leszorítást, vagyis a radioaktívan jelölt ligandum szinte teljes leszorítását eredményező) és közepes (körülbelül IC50 hatást kiváltó) koncentrációját, három ismétlésben; ii. az oldószeres kontrollt és a nem specifikus kötődés mérését, három-három ismétlésben. A vizsgálati puffer, a [3H]-17β-ösztradiol, a humán ösztrogénreceptor α és a vizsgálati vegyi anyag oldatainak elkészítési eljárását részletesen ismerteti a CERI protokoll (2).
|
Oldószeres kontroll:
|
25.
|
Az oldószeres kontroll igazolja, hogy az oldószer nem lép kölcsönhatásra a vizsgálati rendszerrel, emellett méri a teljes kötődést (TB). Az ajánlott oldószer a DMSO. Amennyiben a vizsgálati vegyi anyag legmagasabb koncentrációja nem oldható DMSO-ban, akkor alternatívaként etanol is használható. A végső vizsgálati lyukakba adagolt DMSO koncentrációja 2,05 % legyen, ez az érték legfeljebb 2,5 %-ra növelhető abban az esetben, ha a vizsgálati vegyi anyag az eredeti koncentrációban nem oldható. A DMSO 2,5 % fölötti koncentrációkban való használatát kerülni kell, mert az oldószer magasabb koncentrációi befolyásolhatják a vizsgálati eredményeket. A DMSO-ban nem, de etanolban oldódó vizsgálati vegyi anyagok esetében a vizsgálati eredmények befolyásolása nélkül az etanol legfeljebb 2 %-os koncentrációban alkalmazható.
|
Puffer kontroll:
|
26.
|
A puffer kontroll (BC) nem tartalmazhatja sem az oldószert, sem a vizsgálati vegyi anyagot, csak a vizsgálat összes többi összetevőjét. A puffer kontroll eredményeit összevetik az oldószeres kontroll eredményeivel annak ellenőrzésére, hogy az alkalmazott oldószer nincs hatással a vizsgálati rendszerre.
|
Erősen kötődő anyag (referenciaösztrogén)
|
27.
|
A 17β-ösztradiol (CAS-száma: 50-28-2) az endogén ligandum, és magas affinitással kötődik az ösztrogénreceptor alfa altípusához. Minden humán ösztrogénreceptor α kompetitív kötési vizsgálathoz nem jelölt 17β-ösztradiollal el kell készíteni egy standard görbét, amely lehetővé teszi a variabilitás értékelését, ha a laboratóriumon belül egy későbbi időpontban ismét elvégzik a vizsgálatot. A nem jelölt 17β-ösztradiolból nyolc oldatot kell készíteni DMSO-ban és vizsgálati pufferben, a standard görbe létrehozásához a végső koncentrációkat a következő elosztásban kell a vizsgálati lyukakba helyezni: 10–6, 10–7, 10–8, 10–8.5, 10–9, 10–9.5, 10–10, 10–11 M. A nem jelölt 17β–ösztradiol legmagasabb koncentrációja (1 μM) egyben a nem specifikus kötődést is jelzi. Ezt a koncentrációt a 4. táblázatban az „NSB” rövidítés különbözteti meg, bár ez is részét képezi a standard görbének.
|
Gyengén kötődő anyag
|
28.
|
A vizsgálatba bele kell foglalni egy gyengén kötődő anyagot (a noretinodrel t (CAS-száma: 68-23-5) vagy alternatívaként a noretindront (CAS-száma: 68-22-4)), hogy igazolja az egyes kísérletek érzékenységét, és lehetővé tegye a variabilitás értékelését, ha a vizsgálat egy későbbi időpontban megismétlik. A gyengén kötődő anyagból nyolc oldatot kell készíteni DMSO-ban és vizsgálati pufferben, az anyagot a vizsgálati lyukakba a következő végső koncentrációkban kell elhelyezni: 10–4,5, 10–5,5, 10–6, 10–6,5, 10–7, 10–7,5, 10–8 és 10–9 M.
|
Nem kötődő anyag
|
29.
|
Negatív (nem kötődő) kontrollanyagként oktil-trietoxi-szilánt (OTES, CAS-száma: 2943-75-1) kell alkalmazni. Biztosítja azt, hogy a vizsgálat végrehajtása során detektálni lehessen a humán ösztrogénreceptor α-hoz nem kötődő vizsgálati vegyi anyagokat. A nem kötődő anyagból nyolc oldatot kell készíteni DMSO-ban és vizsgálati pufferben, az anyagot a vizsgálati lyukakba a következő végső koncentrációkban kell elhelyezni: 10–3,10–4, 10–5, 10–6, 10–7, 10–8, 10–9, 10–10 M. Alternatív nem kötődő anyagként di-n-butil-ftalát (DBP, CAS-száma: 84-72-2) használható, ezt az anyagot azonban csak 10-4 M koncentrációig tesztelték. A DBP-ról kimutatták, hogy a vizsgálat során a maximális oldható koncentrációja a 10-4 M.
|
A humán ösztrogénreceptor α koncentrációja
|
30.
|
A receptort abban a mennyiségben kell alkalmazni, amelyik 40 %-os (± 10 %) specifikus kötődést vált ki (lásd a 3. függelék 12–13. pontját). A humán ösztrogénreceptor α oldatot közvetlenül használat előtt kell elkészíteni a funkcionális humán ösztrogénreceptor α jéghideg vizsgálati pufferben való hígításával.
|
[3H]-17β-ösztradiol
|
31.
|
A [3H]-17β-ösztradiolt 0,5 nM végső koncentrációban kell a vizsgálati lyukakba helyezni.
|
Vizsgálati vegyi anyagok
|
32.
|
Első lépésként oldhatósági vizsgálatot kell végezni az egyes vizsgálati vegyi anyagok oldhatósági határértékének meghatározására, valamint a vizsgálati protokoll végrehajtása során alkalmazandó megfelelő koncentrációtartomány megállapítására. A vizsgálati vegyi anyagok oldhatósági határértékét először oldószerben kell meghatározni, majd a vizsgálati körülmények mellett meg kell erősíteni. A vizsgálatban tesztelt végső koncentrációjuk nem haladhatja meg az 1 mM-t. A koncentrációtartomány-behatároló vizsgálat magában foglal egy oldószeres kontrollt és a legmagasabb elfogadható koncentrációtól (pl. 1 mM, illetve az oldhatósági határértéktől függően ennél alacsonyabb koncentrációtól) induló, legalább 8 koncentrációból álló logaritmikus hígítási sorozatot, az esetlegesen tapasztalt zavarosságot vagy kicsapódást fel kell jegyezni (lásd még a 3. függelék 35. pontját). Miután meghatározták a vizsgálat során alkalmazandó koncentrációtartományt, a vizsgálati vegyi anyagokat 8 logaritmikus koncentrációban kell tesztelni, a koncentrációtartomány behatárolására szolgáló előzetes vizsgálatban meghatározott eloszlás szerint. A második és a harmadik kísérlet koncentrációértékeit szükség esetén úgy kell kiigazítani, hogy egy jobb koncentráció-válasz görbét adjanak.
|
|
33.
|
A vizsgálati vegyi anyag oldatait megfelelő oldószerben kell elkészíteni (lásd a 3. függelék 25. pontját). Amennyiben a vizsgálati vegyi anyag legmagasabb koncentrációja sem DMSO-ban, sem etanolban nem oldható, és a hozzáadott oldószer mennyiségének növelése miatt a végső vizsgálathoz használt csőben az oldószer koncentrációja túllépné az elfogadható határértéket, a legmagasabb koncentráció az eggyel alacsonyabb koncentrációra csökkenthető. Ebben az esetben hozzá lehet adni egy további koncentrációértéket a koncentrációsorozat alacsonyabb értékeket tartalmazó végéhez. A sorozat többi koncentrációértékét változatlanul kell hagyni.
|
|
34.
|
A vizsgálati vegyi anyag mikrotiterlemezbe adagolt oldatait figyelemmel kell követni, mivel a vizsgálati vegyi anyag a vizsgálati lyukakba helyezést követően kicsapódhat. A kicsapódást tartalmazó vizsgálati lyukak adatait ki kell zárni a görbeillesztésből, és fel kell jegyezni az adatok kizárásának okát.
|
|
35.
|
Amennyiben valamely vizsgálati vegyi anyag log(IC50) értékére vonatkozólag léteznek más forrásokból származó korábbi adatok, érdemes úgy beállítani a hígítást, hogy geometriailag közelebb (pl. 0,5 log egységnyire) helyezkedjenek el a log(IC50) várt értéke körül. A végső eredményeknek a log(IC50) mindkét oldalán kellő távolságban eloszló koncentrációkat kell mutatniuk, beleértve a görbe tetejét és alját, hogy a kötési görbét megfelelően lehessen jellemezni.
|
Vizsgálati lemez elrendezése
|
36.
|
A mikrotiterlemezeken fel kell címkézni a hatszoros inkubálásnak alávetett oldószeres kontrollt, a referenciaösztrogén (E2) legmagasabb koncentrációját – amely egyben a nem specifikus kötődés (NSB) mutatója –, a puffer kontrollt, a háromszor inkubált nem kötődő kontrollanyag (oktil-trietoxi-szilán) mind a nyolc koncentrációját, a referenciaösztrogén (E2) hét alacsonyabb koncentrációját, a gyengén kötődő anyag (noretinodrel vagy noretindron) nyolc koncentrációját és minden egyes vizsgálati vegyi anyag (TC) nyolc koncentrációját. Az alábbi 4. táblázat példát szolgáltat egy olyan lemezelrendezésre, amely biztosítja a referenciaösztrogén és a kontrollanyagok teljes koncentrációgörbéjét. A többi mikrotiterlemez a vizsgálati vegyi anyag mérésére szolgál, emellett tartalmazniuk kell a lemezen belüli ellenőrzésre szolgáló kontrollanyagokat, vagyis i. az E2 és a gyengén kötődő anyag magas (maximális leszorítást eredményező) és közepes (körülbelül IC50 hatást kiváltó) koncentrációját, három ismétlésben; ii. az oldószeres kontroll (ami a teljes kötődés mutatója) és a nem specifikus kötődés mérését, hat-hat ismétlésben (lásd az 5. táblázatot). A 3.3. függelék példát szolgáltat arra, hogy milyen lemezelrendezést lehet alkalmazni a kompetitív vizsgálatban három ismeretlen vizsgálati vegyi anyag teszteléséhez. A lemezelrendezési ábrán, valamint a 4. és az 5. táblázatban feltüntetett koncentrációértékek az egyes vizsgálati lyukakba adagolt végső koncentrációkat jelzik. Az E2 maximális koncentrációja 1×10-7 M legyen, a gyengén kötődő anyag legmagasabb koncentrációja pedig az 1. lemezen használt legmagasabb koncentráció legyen. Az IC50 koncentrációját a laboratóriumnak kell meghatároznia, a kontrollanyagok adatait tartalmazó adatbázisa alapján. Ennek az értéknek közelítenie kell a validálási tanulmányok során nyert értékhez (lásd az 1. táblázatot).
|
4. táblázat
A kompetitív kötési vizsgálathoz használt mikrotiterlemez azon elrendezése (98)
(99), amely biztosítja a referenciaösztrogén és a kontrollanyagok teljes koncentrációgörbéjét (1. lemez)
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
Puffer kontroll és pozitív kontroll (E2)
|
Enyhén pozitív (Noretinodrel)
|
Negatív kontroll (OTES)
|
TB és NSB
|
|
A
|
Üres (*14)
|
1×10–9 M
|
1×10–10 M
|
TB (oldószeres kontroll) (2,05 % DMSO)
|
|
B
|
E2 (1×10–11 M)
|
1×10–8 M
|
1×10–9 M
|
|
C
|
E2 (1×10–10 M)
|
1×10–7,5 M
|
1×10–8 M
|
NSB (10–6 M E2)
|
|
D
|
E2 (1×10–9,5 M)
|
1×10–7 M
|
1×10–7 M
|
|
E
|
E2 (1×10–9 M)
|
1×10–6,5 M
|
1×10–6 M
|
Puffer kontroll
|
|
F
|
E2 (1×10–8,5 M)
|
1×10–6 M
|
1×10–5 M
|
|
G
|
E2 (1×10–8 M)
|
1×10–5,5 M
|
1×10–4 M
|
Üres (radioaktívhoz) (*15)
|
|
H
|
E2 (1×10–7 M)
|
1×10–4,5 M
|
1×10–3 M
|
5. táblázat
A kompetitív kötési vizsgálathoz használt mikrotiterlemez elrendezése, a vizsgálati vegyi anyagokhoz (TC) és a lemezen belüli ellenőrzésre szolgáló kontrollanyagokhoz használt további lemezekhez
|
|
1
|
2
|
3
|
4
|
5
|
6
|
7
|
8
|
9
|
10
|
11
|
12
|
|
|
1. vizsgálati vegyi anyag (TC-1)
|
2. vizsgálati vegyi anyag (TC-2)
|
3. vizsgálati vegyi anyag (TC-3)
|
Kontrollok
|
|
A
|
TC-1 (1×10–10 M)
|
TC-2 (1×10–10 M)
|
TC-3 (1×10–10 M)
|
E2 (1×10
-7
M)
|
|
B
|
TC-1 (1×10–9 M)
|
TC-2 (1×10–9 M)
|
TC-3 (1×10–9 M)
|
E2 (IC50)
|
|
C
|
TC-1 (1×10–8 M)
|
TC-2 (1×10–8 M)
|
TC-3 (1×10–8 M)
|
NE (1×10
–4,5
M)
|
|
D
|
TC-1 (1×10–7 M)
|
TC-2 (1×10–7 M)
|
TC-3 (1×10–7 M)
|
NE (IC50)
|
|
E
|
TC-1 (1×10–6 M)
|
TC-2 (1×10–6 M)
|
TC-3 (1×10–6 M)
|
NSB (10–6 M E2)
|
|
F
|
TC-1 (1×10–5 M)
|
TC-2 (1×10–5 M)
|
TC-3 (1×10–5 M)
|
|
G
|
TC-1 (1×10–4 M)
|
TC-2 (1×10–4 M)
|
TC-3 (1×10–4 M)
|
TB (Oldószeres kontroll)
|
|
H
|
TC-1 (1×10–3 M)
|
TC-2 (1×10–3 M)
|
TC-3 (1×10–3 M)
|
|
Ebben a példában a gyengén kötődő anyag a noretinodrel (NE).
|
A kompetitív kötési vizsgálat elvégzése
|
37.
|
A 6. táblázatban szereplő teljes kötődést vizsgáló és üres (radioaktívhoz) lyukak kivételével minden lyukba 50 μl vizsgálati puffert kell adagolni, amelyet össze kell keverni 10 μl oldószerrel vagy referenciaösztrogénnel (E2), gyengén kötődő anyaggal, nem kötődő anyaggal, vizsgálati vegyi anyagokkal, majd 10 μl 5 nM koncentrációjú [3H]-17β-ösztradiol oldattal. Ezt követően minden egyes lemezhez 30 μl jéghideg receptoroldatot kell adni, és a keveréket óvatosan össze kell keverni. Utolsó reagensként a humán ösztrogénreceptor α oldatot kell hozzáadni. A vizsgálati mikrotiterlemezeket két órán keresztül kell inkubálni, szobahőmérsékleten (22 °C és 28 °C között).
6. táblázat
A humán ösztrogénreceptor kompetitív kötési vizsgálat mikrotiterlemezeire helyezett vizsgálati összetevők térfogata
|
A sávok előkészítésének lépései
|
Nem TB lyukak
|
TB lyukak
|
Üres (radioaktívhoz)
|
|
A fenti reagens lyukak összetevőinek térfogata és hozzáadási sorrendje
|
Szobahőmérsékletű vizsgálati puffer
|
50 μl
|
60 μl
|
90 μl
|
|
Nem jelölt E2, gyengén kötődő anyag, nem kötődő anyag, oldószer és vizsgálati vegyi anyagok (*16)
|
10 μl
|
—
|
—
|
|
[3H]-17β-ösztradiol a végső, 0.5 nM (vagyis 5 nM) koncentráció biztosításához
|
10 μl
|
10 μl
|
10 μl
|
|
hrERα a meghatározott koncentrációban (lásd a 12–13. pontot)
|
30 μl
|
30 μl
|
—
|
|
Teljes térfogat az egyes vizsgálati lyukakban
|
100 μl
|
100 μl
|
100 μl
|
|
|
38.
|
Ezt követően a 3. függelék 21–23. pontjában a telítési kötési vizsgálatnál ismertetett módon minden lyukhoz hozzáadnak 100 μl jéghideg DCC szuszpenziót, hogy elkülönítsék a humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolt a szabad [3H]-17β-ösztradioltól, majd megmérik a humán ösztrogénreceptor α-hoz kötődött [3H]-17β-ösztradiolok mennyiségét.
|
|
39.
|
A G10–12 és H10–12 lyukakban (amelyeket a 4. táblázat üres (radioaktívhoz) címen jelöl) mérik a 10 μl mennyiségű [3H] jelölt ösztradiol percenkénti bomlását. A 10 μl-nek megfelelő mennyiséget közvetlenül a szcintillációs folyadékhoz kell adni.
|
Elfogadhatósági kritériumok
Telítési kötési vizsgálat
|
40.
|
A specifikus kötődési görbének a [3H]-17β-ösztradiol fokozódó koncentrációban való alkalmazása miatt egy platóban kell végződnie, ami jelzi a humán ösztrogénreceptor α ligandummal való telítettségét.
|
|
41.
|
A [3H]-17β-ösztradiol 0,5 nM koncentrációjánál megfigyelt specifikus kötődésnek a minden vizsgálatmenetben hozzáadott teljes radioaktivitás mért átlagának 30–50 %-os elfogadhatósági tartományán belül kell maradnia. E tartománytól való esetenkénti kis mértékű elérés még elfogadható, de ha a vizsgálatmenetek eredménye következetesen kívül esik ezen a tartományon, illetve ha egy adott vizsgálatmenet eredménye jelentősen eltér tőle, akkor ki kell igazítani a fehérjekoncentrációt és meg kell ismételni a telítési vizsgálatot.
|
|
42.
|
Az adatoknak egy lineáris Scatchard-plotot kell alkotniuk.
|
|
43.
|
A nem specifikus kötődés nem lehet túlzott mértékű. A nem specifikus kötődés értéke jellemzően nem érheti el a teljes kötődés 35 %-át. Ugyanakkor a nem specifikus kötődés aránya esetenként meghaladhatja ezt a határértéket, ha a radioaktívan jelölt 17β-ösztradiol legalacsonyabb vizsgált koncentrációjánál nagyon alacsony percenkénti bomlást mérnek.
|
Kompetitív kötési vizsgálat
|
44.
|
A nem jelölt 17β-ösztradiol növekvő koncentrációban leszorítja a [3H]-17β-ösztradiolt a receptorról, és ez összhangban van az egy kötőhelyes kompetitív kötéssel.
|
|
45.
|
A referenciaösztrogén (vagyis a 17β-ösztradiol) IC50 és Kd értékének nagyjából egyenlőnek kell lennie a [3H]-17β-ösztradiol moláris koncentrációjával; a disszociációs állandót a telítési kötési vizsgálatban határozzák meg.
|
|
46.
|
A teljes specifikus kötődésnek rendre a 40 ± 10 %-os elfogadhatósági tartományon belül kell maradnia, ha a vizsgálatmenetekben az egyes lyukakhoz hozzáadott összes radioaktívan jelölt anyag átlagos mért koncentrációja 0,5 nM volt. E tartománytól való esetenkénti kis mértékű elérés még elfogadható, de ha a vizsgálatmenetek eredménye következetesen kívül esik ezen a tartományon, illetve ha egy adott vizsgálatmenet eredménye jelentősen eltér ettől, akkor ki kell igazítani a fehérjekoncentrációt.
|
|
47.
|
Az oldószer nem módosíthatja a vizsgálat érzékenységét vagy megismételhetőségét. Az oldószeres kontroll (Tb-ként jelölve) eredményeit összevetik a puffer kontroll eredményeivel annak ellenőrzésére, hogy az alkalmazott oldószer nincs hatással a vizsgálati rendszerre. Amennyiben a TB kontroll és a puffer kontroll hasonló eredményre vezet, akkor az oldószer nem befolyásolja a vizsgálatot.
|
|
48.
|
A nem kötődő anyag maximális 10–3 M (OTES) vagy 10–4 M (DBP) koncentrációnál legfeljebb 25 % [3H]-17β-ösztradiolt szoríthat le a humán ösztrogénreceptor α-ról.
|
|
49.
|
A CERI humán ösztrogénreceptor-kötési vizsgálat validálási tanulmányából származó adatok alapján a referenciaösztrogénre és két gyengén kötődő anyagra (pl. noretinodrel , noretindron) vonatkozva teljesítménykritériumokat dolgoztak ki (2. referencia N. függeléke). A validálási tanulmányban részt vevő négy laboratórium 95 %-os konfidenciaintervallumot adott meg az átlag ± SD (n) értékéhez minden kontroll vizsgálatmenetben. 95 %-os konfidenciaintervallumot határoztak meg a görbeillesztési paraméterekre (vagyis a görbe tetejére, aljára, a Hill-meredekségre és a Log IC50-re), a referenciaösztrogénre és a gyengén kötődő anyagokra, valamint a gyengén kötődő anyagok referenciaösztrogénhez viszonyított Log10RBA értékére vonatkozólag. Az 1. táblázat ismerteti a görbeillesztési paraméterek várt tartományait, amelyek használhatók teljesítménykritériumként. A gyakorlatban az IC50 tartománya a receptor preparátum kísérleti úton meghatározott Kd értékétől és a ligandum vizsgálatban használt koncentrációjától függően kissé változó lehet.
|
|
50.
|
A potenciális vizsgálati vegyi anyagok széles skálája, valamint potenciális affinitásuk és az eredmények (pl. teljes görbe, részleges görbe, nem illeszthető görbe) változatossága miatt a vizsgálati vegyi anyagok görbeillesztési paramétereire nézve nem dolgoztak ki teljesítménykritériumokat. Egy adott vizsgálati vegyi anyag egyes vizsgálatmeneteiből származó eredmények értékelése során azonban szakmai megítélés alapján kell dönteni. A vizsgálati vegyi anyag megfelelő koncentrációtartományát kell használni ahhoz, hogy egyértelműen meg lehessen határozni a kompetitív görbe (pl. 90–100 %-os kötődést jelző) platóját. A vizsgálati vegyi anyag különböző koncentrációin történő ismétlések eltérései közötti és a három független vizsgálatmenet közötti variabilitásnak észszerű mértéken belül kell maradnia és tudományosan védhetőnek kell lennie. Egy adott vizsgálati vegyi anyag minden vizsgálatmenetében elvégzett kontrollok eredményeinek meg kell közelíteniük a CERI vizsgálat teljesítménymutatóit, és összhangban kell lenniük minden releváns laboratórium történeti kontrolladataival.
|
AZ ADATOK ELEMZÉSE
Telítési kötési vizsgálat
|
51.
|
Mérni kell mind a teljes, mind a nem specifikus kötődést. Ezen értékek alapján számolható ki a [3H]-17β-ösztradiol növekvő koncentrációin létrejött egyensúlyi állapotokban kiváltott specifikus kötődés úgy, hogy a teljes kötődésből kivonják a nem specifikus kötődést. A specifikus kötődés és a [3H]-17β-ösztradiol koncentrációi közötti függvényt ábrázoló grafikon a maximális specifikus kötődésre jellemző platóban végződik, amely azt az állapotot jelzi, amikor a humán ösztrogénreceptor α telítődött [3H]-17β-ösztradiollal. Emellett az adatok elemzése dokumentálja, hogy a [3H]-17β-ösztradiol egyetlen, nagy affinitású kötőhelyhez kapcsolódik. A nem specifikus, a teljes és a specifikus kötődést egy telítési kötési görbén kell ábrázolni. Az adatokat a továbbiakban nem-lineáris regressziós analízissel kell elemezni (pl. BioSoft; McPherson, 1985; Motulsky, 1995), végül pedig egy Scatchard-plot grafikonon ábrázolni.
|
|
52.
|
Az adatelemzés során kizárólag a teljes kötődési adatok alapján meg kell határozni a Bmax és a Kd értékét, feltételezve, hogy a nem specifikus kötődés lineáris, más módszer alkalmazását indokolni kell. A legjobban illeszkedő görbét pedig robusztus regresszióval kell meghatározni, az ettől való eltérést indokolni kell. A robusztus regresszió alkalmazásának módszerét meg kell adni. Abban az esetben, ha a Bmax és a Kd értékét a telítési kötési adatok alapján határozzák meg, mindig korrigálni kell a ligandum koncentrációjának csökkenését (például a Swillens, 1995 által leírt módszerrel).
|
Kompetitív kötési vizsgálat
|
53.
|
A kompetitív kötési görbe a [3H]-17β-ösztradiol specifikus kötődésének és a kompetítor (log10 egységekben feltüntetett) koncentrációjának összefüggését ábrázolja. A vizsgálati vegyi anyag azon koncentrációja, amely meggátolja a [3H]-17β-ösztradiol maximális specifikus kötődésének 50 %-át, az IC50 érték.
|
|
54.
|
A pozitív kontrollanyagok (pl. a referenciaösztrogén és a gyengén kötődő anyag) becsült log(IC50) értékének meghatározásához egy megfelelő nem-lineáris görbeillesztő szoftvert kell használni, amely illeszteni tudja egy négyparaméteres Hill-egyenlet görbéjét (pl. BioSoft; McPherson, 1985; Motulsky, 1995). A görbék illesztése során a görbe tetejét, alját, meredekségét jelző értékeket és a log(IC50) értékét általában szabadon ábrázolják. A legjobban illeszkedő görbét robusztus regresszióval kell meghatározni, az ettől való eltérést indokolni kell. A ligandum csökkenését nem kell korrigálni. A kezdeti analízist követően minden kötési görbét ellenőrizni kell, hogy megfelelően illeszkedik-e a modellre. A gyengén kötődő anyag relatív kötési affinitását a gyengén kötődő anyag log (IC50) értékének a 17β-ösztradiol log (IC50) értékéhez viszonyított arányaként kell meghatározni. A pozitív kontrollok és a nem kötődő kontroll eredményeit a vizsgálat e 3. függelék 44–49. pontjában ismertetett teljesítménymutatói alapján kell értékelni.
|
|
55.
|
Az összes vizsgálati vegyi anyag adatait lépésenként kell elemezni, biztosítva ezáltal az adatok megfelelő elemzését és a kompetitív kötési görbék helyes osztályozását. Javasolt vizsgálati anyagonként minden vizsgálat megkezdésekor egy szabványosított adatelemzést végezni, amely megegyezik a referenciaösztrogén és a gyengén kötődő anyag kontrolljainál alkalmazott analízissel (lásd e 3. függelék 54. pontját). Az adatelemzés végeztével technikailag ellenőrizni kell a görbeillesztési paramétereket, valamint vizuálisan is ellenőrizni kell, hogy az adatok mennyire illeszkednek az egyes vizsgálatmenetek kompetitív kötési görbéjéhez. Amennyiben a technikai ellenőrzés során megfigyelhető a specifikusan kötődő [3H]-17β-ösztradiol arányának koncentrációfüggő csökkenése, a vizsgálati vegyi anyagok egyes koncentrációit tartalmazó ismétlések közötti csekély eltérés és a három vizsgálatmenet görbeillesztési paramétereinek következetessége jó indikátorai annak, hogy a vizsgálatot és az adatelemzést megfelelően hajtották végre.
|
Az adatok értelmezése
|
56.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, a vizsgálati vegyi anyag humán ösztrogénreceptor α-hoz kötődő anyagnak minősül, ha az adatokra illeszthető kötési görbe, és a válaszgörbe legalsó pontja az adattartomány 50 %-a alatt helyezkedik el (1. ábra).
|
|
57.
|
Amennyiben valamennyi elfogadhatósági kritérium teljesül, valamely vizsgálati vegyi anyag akkor tekinthető humán ösztrogénreceptor α-hoz nem kötődő anyagnak, ha:
|
—
|
az adataira illeszthető kötési görbe, és az adatokra illesztett válaszgörbe legalsó pontja az adattartomány 75 %-a felett helyezkedik el, vagy
|
|
—
|
az adataira nem illeszthető kötési görbe, és az adatok koncentrációcsoportjai közül a legalsó simítatlan átlagos kötési arány 75 % felett helyezkedik el.
|
|
|
58.
|
A vizsgálati vegyi anyag válasza akkor minősül kétértelműnek, ha a fenti feltételek egyike sem teljesül (vagyis az adatokra illesztett válaszgörbe legalsó pontja 76 és 51 % között helyezkedik el).
|
7. táblázat
A vizsgálati vegyi anyagok kompetitív kötési görbén alapuló osztályba sorolásának kritériumai
|
Besorolás
|
Kritériumok
|
|
Kötődőa
|
Az adatokra illeszthető kötési görbe. A válaszgörbe legalsó pontja az adattartomány 50 %-a alatt helyezkedik el.
|
|
Nem kötődőb
|
Ha az adatokra illeszthető kötési görbe, az adattartományra illesztett válaszgörbe legalsó pontja 75 %-a felett helyezkedik el. Ha az adatokra nem illeszthető kötési görbe, az adatok koncentrációcsoportjai közül a legalsó simítatlan átlagos kötési arány 75 % felett helyezkedik el.
|
|
Kétértelműc
|
Bármely vizsgálatmenet, amelynek eredménye sem a kötődő, sem a nem kötődő kategóriába nem sorolható (pl. az adatokra illesztett válaszgörbe legalsó pontja 76 és 51 % között helyezkedik el).
|
1. ábra
Példák a vizsgálati vegyi anyagok kompetitív kötési görbén alapuló osztályba sorolására.

|
59.
|
A vizsgálati vegyi anyag egy laboratóriumon belül elvégzett több vizsgálatmenetének eredményét összesítik úgy, hogy egy számértéket társítanak minden vizsgálatmenethez, majd meghatározzák a vizsgálatmenetek átlagát (lásd a 8. táblázatot). A laboratórium vizsgálatmeneteinek összesített eredményét azután összevetik az egyes vizsgálati vegyi anyagok várt besorolásával.
|
8. táblázat
Az egy laboratóriumon belül több vizsgálatmenetben vizsgálati vegyi anyagok osztályba sorolásának módszere,
|
Számérték társítása az egyes vizsgálatmenetekhez:
|
|
Besorolás
|
Számérték
|
|
Kötődő
|
2
|
|
Kétértelmű
|
1
|
|
Nem kötődő
|
0
|
|
Besorolás a vizsgálatmenetek átlagos számértéke alapján:
|
|
Besorolás
|
Számérték
|
|
Kötődő
|
Átlag ≥ 1,5
|
|
Kétértelmű
|
0,5 ≤ Átlag < 1,5
|
|
Nem kötődő
|
Átlag < 0,5
|
VIZSGÁLATI JELENTÉS
|
60.
|
Lásd a vizsgálati módszer „A humán ÖSZTROGÉNRECEPTOR-KÖTÉSI VIZSGÁLAT ÖSSZETEVŐI” részének 24. pontját.
|
3.1. függelék
Szakkifejezések Listája
[3H]E2
: radioaktív tríciummal jelölt 17β-ösztradiol.
DCC
: dextránbevonatú aktívszén.
E2
: nem jelölt 17β-ösztradiol (semleges).
Vizsgálati puffer
: 10 mM trisz-HCl (pH-értéke 7,4), amely a következőket tartalmazza: 1 mM EDTA, 1 mM EGTA, 1 mM NaVO3, 10 % glicerin, 0,2 mM leupeptin, 1 mM ditiotreitol és 10 mg/ml szarvasmarha szérum albumin.
hrERα
: humán rekombináns ösztrogénreceptor alfa (ligandumkötő domén).
Ismétlés
: egyike azon lyukaknak, amelyek azonos mintát tartalmaznak azonos koncentrációban, és vizsgálatuk egy adott vizsgálatmeneten belül, párhuzamosan zajlik. E protokoll keretén belül a vizsgálati vegyi anyagok minden koncentrációját három ismétlésben tesztelik; vagyis a vizsgálati vegyi anyagok minden koncentrációját egyidejűleg három ismétlésben vizsgálják.
Vizsgálatmenet
: párhuzamosan vizsgált mikrotiterlemez minden mintája, amely minden szükséges információt biztosít a vizsgálati vegyi anyagok humán ösztrogénreceptor α-hoz való kötődésének jellemzéséhez (nevezetesen a vizsgálati lyukba adagolt összes [3H]-17β-ösztradiol, a [3H]-17β-ösztradiol maximális kötődése a hrERα-hoz, nem specifikus kötődés és a vizsgálati vegyi anyagok különböző koncentrációi által előidézett teljes kötődés). Egy vizsgálatmenet állhat koncentrációnként egyetlen vizsgálati lyukból is (vagyis egy ismétlésből), mivel azonban ez a protokoll három ismétlésben való tesztelést ír elő, egy vizsgálatmenet koncentrációnként három vizsgálati lyukat foglal magában. Emellett a protokoll meghatározza azt is, hogy vegyi anyagonként három független (azaz nem egyidejűleg lefolytatott) vizsgálatmenetre van szükség.
3.2. függelék
MINTÁK ELRENDEZÉSE A KOMPETITÍV KÖTÉSI VIZSGÁLATHOZ
|
Lemez
|
Pozíció
|
Ismétlés
|
Lyuk típusa
|
Lyuk kódja
|
Koncentráció kódja
|
Kompetítor induló koncentrációja (M)
|
hrER törzs (μl)
|
Puffer térfogata (μl)
|
Nyomjelző (radioaktív E2) térfogata (μL)
|
Hígítási lemezről adagolt térfogat (μL)
|
Végső térfogat (μl)
|
Kompetítor végső koncentrációja (M)
|
|
Sz
|
A1
|
1
|
Üres
|
BK
|
BK1
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Sz
|
A2
|
2
|
Üres
|
BK
|
BK2
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Sz
|
A3
|
3
|
Üres
|
BK
|
BK3
|
—
|
—
|
—
|
—
|
—
|
—
|
—
|
|
Sz
|
B1
|
1
|
nem jelölt E2
|
Sz
|
S1
|
1,00 E-10
|
30
|
50
|
10
|
10
|
100
|
1,0 E-11
|
|
Sz
|
B2
|
2
|
nem jelölt E2
|
Sz
|
S1
|
1,00 E-10
|
30
|
50
|
10
|
10
|
100
|
1,0 E-11
|
|
Sz
|
B3
|
3
|
nem jelölt E2
|
Sz
|
S1
|
1,00 E-10
|
30
|
50
|
10
|
10
|
100
|
1,0 E-11
|
|
Sz
|
C1
|
1
|
nem jelölt E2
|
Sz
|
S2
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
C2
|
2
|
nem jelölt E2
|
Sz
|
S2
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
C3
|
3
|
nem jelölt E2
|
Sz
|
S2
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
D1
|
1
|
nem jelölt E2
|
Sz
|
S3
|
3,16 E-9
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
Sz
|
D2
|
2
|
nem jelölt E2
|
Sz
|
S3
|
3,16 E-9
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
Sz
|
D3
|
3
|
nem jelölt E2
|
Sz
|
S3
|
3,16 E-9
|
30
|
50
|
10
|
10
|
100
|
3,2 E-10
|
|
Sz
|
E1
|
1
|
nem jelölt E2
|
Sz
|
S4
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
E2
|
2
|
nem jelölt E2
|
Sz
|
S4
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
E3
|
3
|
nem jelölt E2
|
Sz
|
S4
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
F1
|
1
|
nem jelölt E2
|
Sz
|
S5
|
3,16 E-8
|
30
|
50
|
10
|
10
|
100
|
3,2 E-9
|
|
Sz
|
F2
|
2
|
nem jelölt E2
|
Sz
|
S5
|
3,16 E-8
|
30
|
50
|
10
|
10
|
100
|
3,2 E-9
|
|
Sz
|
F3
|
3
|
nem jelölt E2
|
Sz
|
S5
|
3,16 E-8
|
30
|
50
|
10
|
10
|
100
|
3,2 E-9
|
|
Sz
|
G1
|
1
|
nem jelölt E2
|
Sz
|
S6
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
G2
|
2
|
nem jelölt E2
|
Sz
|
S6
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
G3
|
3
|
nem jelölt E2
|
Sz
|
S6
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
H1
|
1
|
nem jelölt E2
|
Sz
|
S7
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
H2
|
2
|
nem jelölt E2
|
Sz
|
S7
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
H3
|
3
|
nem jelölt E2
|
Sz
|
S7
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
A4
|
1
|
noretinodrel
|
NE
|
WP1
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
A5
|
2
|
noretinodrel
|
NE
|
WP1
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
A6
|
3
|
noretinodrel
|
NE
|
WP1
|
1,00 E-08
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
B4
|
1
|
noretinodrel
|
NE
|
WP2
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
B5
|
2
|
noretinodrel
|
NE
|
WP2
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
B6
|
3
|
noretinodrel
|
NE
|
WP2
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
C4
|
1
|
noretinodrel
|
NE
|
WP3
|
3,16 E-7
|
30
|
50
|
10
|
10
|
100
|
3,2 E-8
|
|
Sz
|
C5
|
2
|
noretinodrel
|
NE
|
WP3
|
3,16 E-7
|
30
|
50
|
10
|
10
|
100
|
3,2 E-8
|
|
Sz
|
C6
|
3
|
noretinodrel
|
NE
|
WP3
|
3,16 E-7
|
30
|
50
|
10
|
10
|
100
|
3,2 E-8
|
|
Sz
|
D4
|
1
|
noretinodrel
|
NE
|
WP4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
D5
|
2
|
noretinodrel
|
NE
|
WP4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
D6
|
3
|
noretinodrel
|
NE
|
WP4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
E4
|
1
|
noretinodrel
|
NE
|
WP5
|
3,16 E-6
|
30
|
50
|
10
|
10
|
100
|
3,2 E-7
|
|
Sz
|
E5
|
2
|
noretinodrel
|
NE
|
WP5
|
3,16 E-6
|
30
|
50
|
10
|
10
|
100
|
3,2 E-7
|
|
Sz
|
E6
|
3
|
noretinodrel
|
NE
|
WP5
|
3,16 E-6
|
30
|
50
|
10
|
10
|
100
|
3,2 E-7
|
|
Sz
|
F4
|
1
|
noretinodrel
|
NE
|
WP6
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
F5
|
2
|
noretinodrel
|
NE
|
WP6
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
F6
|
3
|
noretinodrel
|
NE
|
WP6
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
G4
|
1
|
noretinodrel
|
NE
|
WP7
|
3,16 E-5
|
30
|
50
|
10
|
10
|
100
|
3,2 E-6
|
|
Sz
|
G5
|
2
|
noretinodrel
|
NE
|
WP7
|
3,16 E-5
|
30
|
50
|
10
|
10
|
100
|
3,2 E-6
|
|
Sz
|
G6
|
3
|
noretinodrel
|
NE
|
WP7
|
3,16 E-5
|
30
|
50
|
10
|
10
|
100
|
3,2 E-6
|
|
Sz
|
H4
|
1
|
noretinodrel
|
NE
|
WP8
|
3,16 E-4
|
30
|
50
|
10
|
10
|
100
|
3,2 E-5
|
|
Sz
|
H5
|
2
|
noretinodrel
|
NE
|
WP8
|
3,16 E-4
|
30
|
50
|
10
|
10
|
100
|
3,2 E-5
|
|
Sz
|
H6
|
3
|
noretinodrel
|
NE
|
WP8
|
3,16 E-4
|
30
|
50
|
10
|
10
|
100
|
3,2 E-5
|
|
Sz
|
A7
|
1
|
OTES
|
N
|
OTES1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
A8
|
2
|
OTES
|
N
|
OTES1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
A9
|
3
|
OTES
|
N
|
OTES1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
Sz
|
B7
|
1
|
OTES
|
N
|
OTES2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
B8
|
2
|
OTES
|
N
|
OTES2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
B9
|
3
|
OTES
|
N
|
OTES2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
Sz
|
C7
|
1
|
OTES
|
N
|
OTES3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
C8
|
2
|
OTES
|
N
|
OTES3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
C9
|
3
|
OTES
|
N
|
OTES3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
Sz
|
D7
|
1
|
OTES
|
N
|
OTES4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
D8
|
2
|
OTES
|
N
|
OTES4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
D9
|
3
|
OTES
|
N
|
OTES4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
Sz
|
E7
|
1
|
OTES
|
N
|
OTES5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
E8
|
2
|
OTES
|
N
|
OTES5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
E9
|
3
|
OTES
|
N
|
OTES5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
F7
|
1
|
OTES
|
N
|
OTES6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
Sz
|
F8
|
2
|
OTES
|
N
|
OTES6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
Sz
|
F9
|
3
|
OTES
|
N
|
OTES6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
Sz
|
G7
|
1
|
OTES
|
N
|
OTES7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
Sz
|
G8
|
2
|
OTES
|
N
|
OTES7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
Sz
|
G9
|
3
|
OTES
|
N
|
OTES7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
Sz
|
H7
|
1
|
OTES
|
N
|
OTES8DBP7
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
Sz
|
H8
|
2
|
OTES
|
N
|
OTES88
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
Sz
|
H9
|
3
|
OTES
|
N
|
OTES8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
Sz
|
A10
|
1
|
teljes kötődés
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Sz
|
A11
|
2
|
teljes kötődés
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Sz
|
A12
|
3
|
teljes kötődés
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Sz
|
B10
|
4
|
teljes kötődés
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
-
|
|
Sz
|
B11
|
5
|
teljes kötődés
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Sz
|
B12
|
6
|
teljes kötődés
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
Sz
|
C10
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S1
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
C11
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S2
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
C12
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S3
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
D10
|
4
|
nem jelölt E2 (magas)
|
NSB
|
S4
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
D11
|
5
|
nem jelölt E2 (magas)
|
NSB
|
S5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
D12
|
6
|
nem jelölt E2 (magas)
|
NSB
|
S6
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
Sz
|
E10
|
1
|
Puffer kontroll
|
BC
|
BC1
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
E11
|
2
|
Puffer kontroll
|
BC
|
BC2
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
E12
|
3
|
Puffer kontroll
|
BC
|
BC3
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
F10
|
4
|
Puffer kontroll
|
BC
|
BC4
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
F11
|
5
|
Puffer kontroll
|
BC
|
BC5
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
F12
|
6
|
Puffer kontroll
|
BC
|
BC6
|
—
|
—
|
100
|
—
|
—
|
100
|
—
|
|
Sz
|
G10 (*17)
|
1
|
Üres (radioaktívhoz)
|
Radioaktív
|
H1
|
—
|
90
|
—
|
10
|
—
|
100
|
-
|
|
Sz
|
G11 (*17)
|
2
|
Üres (radioaktívhoz)
|
Radioaktív
|
H2
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Sz
|
G12 (*17)
|
3
|
Üres (radioaktívhoz)
|
Radioaktív
|
H3
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Sz
|
H10 (*17)
|
4
|
Üres (radioaktívhoz)
|
Radioaktív
|
H4
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Sz
|
H11 (*17)
|
5
|
Üres (radioaktívhoz)
|
Radioaktív
|
H5
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
Sz
|
H12
|
6
|
Üres (radioaktívhoz)
|
Radioaktív
|
H6
|
—
|
90
|
—
|
10
|
—
|
100
|
—
|
|
P1
|
A1
|
1
|
1. ismeretlen
|
U1
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A2
|
2
|
1. ismeretlen
|
U1
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A3
|
3
|
1. ismeretlen
|
U1
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
B1
|
1
|
1. ismeretlen
|
U1
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B2
|
2
|
1. ismeretlen
|
U1
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B3
|
3
|
1. ismeretlen
|
U1
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
C1
|
1
|
1. ismeretlen
|
U1
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C2
|
2
|
1. ismeretlen
|
U1
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C3
|
3
|
1. ismeretlen
|
U1
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
D1
|
1
|
1. ismeretlen
|
U1
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D2
|
2
|
1. ismeretlen
|
U1
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D3
|
3
|
1. ismeretlen
|
U1
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
E1
|
1
|
1. ismeretlen
|
U1
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E2
|
2
|
1. ismeretlen
|
U1
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E3
|
3
|
1. ismeretlen
|
U1
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F1
|
1
|
1. ismeretlen
|
U1
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F2
|
2
|
1. ismeretlen
|
U1
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F3
|
3
|
1. ismeretlen
|
U1
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
G1
|
1
|
1. ismeretlen
|
U1
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G2
|
2
|
1. ismeretlen
|
U1
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G3
|
3
|
1. ismeretlen
|
U1
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
H1
|
1
|
1. ismeretlen
|
U1
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H2
|
2
|
1. ismeretlen
|
U1
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H3
|
3
|
1. ismeretlen
|
U1
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
A4
|
1
|
2. ismeretlen
|
U2
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A5
|
2
|
2. ismeretlen
|
U2
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A6
|
3
|
2. ismeretlen
|
U2
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
B4
|
1
|
2. ismeretlen
|
U2
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B5
|
2
|
2. ismeretlen
|
U2
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B6
|
3
|
2. ismeretlen
|
U2
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
C4
|
1
|
2. ismeretlen
|
U2
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C5
|
2
|
2. ismeretlen
|
U2
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C6
|
3
|
2. ismeretlen
|
U2
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
D4
|
1
|
2. ismeretlen
|
U2
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D5
|
2
|
2. ismeretlen
|
U2
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D6
|
3
|
2. ismeretlen
|
U2
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
E4
|
1
|
2. ismeretlen
|
U2
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E5
|
2
|
2. ismeretlen
|
U2
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E6
|
3
|
2. ismeretlen
|
U2
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F4
|
1
|
2. ismeretlen
|
U2
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F5
|
2
|
2. ismeretlen
|
U2
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F6
|
3
|
2. ismeretlen
|
U2
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
G4
|
1
|
2. ismeretlen
|
U2
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G5
|
2
|
2. ismeretlen
|
U2
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G6
|
3
|
2. ismeretlen
|
U2
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
H4
|
1
|
2. ismeretlen
|
U2
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H5
|
2
|
2. ismeretlen
|
U2
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H6
|
3
|
2. ismeretlen
|
U2
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
A7
|
1
|
3. ismeretlen
|
U3
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A8
|
2
|
3. ismeretlen
|
U3
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
A9
|
3
|
3. ismeretlen
|
U3
|
1
|
1,00 E-9
|
30
|
50
|
10
|
10
|
100
|
1,0 E-10
|
|
P1
|
B7
|
1
|
3. ismeretlen
|
U3
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B8
|
2
|
3. ismeretlen
|
U3
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
B9
|
3
|
3. ismeretlen
|
U3
|
2
|
1,00 E-8
|
30
|
50
|
10
|
10
|
100
|
1,0 E-9
|
|
P1
|
C7
|
1
|
3. ismeretlen
|
U3
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C8
|
2
|
3. ismeretlen
|
U3
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
C9
|
3
|
3. ismeretlen
|
U3
|
3
|
1,00 E-7
|
30
|
50
|
10
|
10
|
100
|
1,0 E-8
|
|
P1
|
D7
|
1
|
3. ismeretlen
|
U3
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D8
|
2
|
3. ismeretlen
|
U3
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
D9
|
3
|
3. ismeretlen
|
U3
|
4
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,0 E-7
|
|
P1
|
E7
|
1
|
3. ismeretlen
|
U3
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E8
|
2
|
3. ismeretlen
|
U3
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E9
|
3
|
3. ismeretlen
|
U3
|
5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F7
|
1
|
3. ismeretlen
|
U3
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F8
|
2
|
3. ismeretlen
|
U3
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
F9
|
3
|
3. ismeretlen
|
U3
|
6
|
1,00 E-4
|
30
|
50
|
10
|
10
|
100
|
1,0 E-5
|
|
P1
|
G7
|
1
|
3. ismeretlen
|
U3
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G8
|
2
|
3. ismeretlen
|
U3
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
G9
|
3
|
3. ismeretlen
|
U3
|
7
|
1,00 E-3
|
30
|
50
|
10
|
10
|
100
|
1,0 E-4
|
|
P1
|
H7
|
1
|
3. ismeretlen
|
U3
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H8
|
2
|
3. ismeretlen
|
U3
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
H9
|
3
|
3. ismeretlen
|
U3
|
8
|
1,00 E-2
|
30
|
50
|
10
|
10
|
100
|
1,0 E-3
|
|
P1
|
A10
|
1
|
E2 kontroll (max)
|
Sz
|
E2max1
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,00 E-7
|
|
P1
|
A11
|
2
|
E2 kontroll (max)
|
Sz
|
E2max 2
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,00 E-7
|
|
P1
|
A12
|
3
|
E2 kontroll (max)
|
Sz
|
E2max 3
|
1,00 E-6
|
30
|
50
|
10
|
10
|
100
|
1,00 E-7
|
|
P1
|
B10
|
1
|
E2 kontroll (IC50)
|
Sz
|
E2IC501
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B11
|
2
|
E2 kontroll (IC50)
|
Sz
|
E2IC502
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
B12
|
3
|
E2 kontroll (IC50)
|
Sz
|
E2IC503
|
E2IC50x10
|
30
|
50
|
10
|
10
|
100
|
E2IC50
|
|
P1
|
C10
|
1
|
NE kontroll (max)
|
Sz
|
Nemax 1
|
1,00 E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 E-4,5
|
|
P1
|
C11
|
2
|
NE kontroll (max)
|
Sz
|
Nemax 2
|
1,00 E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 E-4,5
|
|
P1
|
C12
|
3
|
NE kontroll (max)
|
Sz
|
Ne max 3
|
1,00 E-3,5
|
30
|
50
|
10
|
10
|
100
|
1,00 E-4,5
|
|
P1
|
D10
|
1
|
NE kontroll (IC50)
|
Sz
|
NEIC501
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D11
|
2
|
NE kontroll (IC50)
|
Sz
|
NEIC502
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
D12
|
3
|
NE kontroll (IC50)
|
Sz
|
NEIC503
|
NEIC50 x10
|
30
|
50
|
10
|
10
|
100
|
NEIC50
|
|
P1
|
E10
|
1
|
nem jelölt E2 (magas)
|
NSB
|
S1
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E11
|
2
|
nem jelölt E2 (magas)
|
NSB
|
S2
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
E12
|
3
|
nem jelölt E2 (magas)
|
NSB
|
S3
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F10
|
4
|
nem jelölt E2 (magas)
|
NSB
|
S4
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F11
|
5
|
nem jelölt E2 (magas)
|
NSB
|
S5
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
F12
|
6
|
nem jelölt E2 (magas)
|
NSB
|
S6
|
1,00 E-5
|
30
|
50
|
10
|
10
|
100
|
1,0 E-6
|
|
P1
|
G10
|
1
|
teljes kötődés
|
TB
|
TB1
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G11
|
2
|
teljes kötődés
|
TB
|
TB2
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
G12
|
3
|
teljes kötődés
|
TB
|
TB3
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H10
|
4
|
teljes kötődés
|
TB
|
TB4
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H11
|
5
|
teljes kötődés
|
TB
|
TB5
|
—
|
30
|
60
|
10
|
—
|
100
|
—
|
|
P1
|
H12
|
6
|
teljes kötődés
|
TB
|
TB6
|
—
|
30
|
60
|
10
|
—
|
100
|
-
|
4. függelék
A HUMÁN ÖSZTROGÉNRECEPTOR KOMPETITÍV KÖTÉSI VIZSGÁLATBÓL NYERT ADATOK ELEMZÉSÉNEK SZEMPONTJAI
|
1.
|
A humán ösztrogénreceptor α kompetitív kötési vizsgálat azt méri, hogy milyen mértékben képes egy állandó koncentrációban alkalmazott [3H]-17β-ösztradiol kötődni egy adott vizsgálati vegyi anyag növekvő koncentrációjának jelenlétében. A kompetitív kötési görbe a [3H]-17β-ösztradiol specifikus kötődésének és a kompetítor (log10 egységekben feltüntetett) koncentrációjának összefüggését ábrázolja. A vizsgálati vegyi anyag azon koncentrációja, amely meggátolja a [3H]-17β-ösztradiol maximális specifikus kötődésének 50 %-át, az IC50.
|
A referenciaösztrogén és a gyengén kötődő anyag adatainak elemzése (1)
|
2.
|
A kontroll vizsgálatmenetekből nyert adatokat (vagyis a [3H]-17β-ösztradiol specifikus kötődésének arányát és a kontrollanyag logaritmikus koncentrációit) a további elemzés céljára transzformálják. A pozitív kontrollanyagok (pl. a referenciaösztrogén és a gyengén kötődő anyag) becsült log(IC50) értékének meghatározásához egy megfelelő nem-lineáris görbeillesztő szoftvert kell használni, amely meg tudja rajzolni a négyparaméteres Hill-egyenlet görbéjét (pl. BioSoft; GraphPad Prism) (2). A görbe illesztése során a görbe tetejét, alját, meredekségét jelző értékeket és a log(IC50) értékét általában szabadon ábrázolják. A legjobban illeszkedő görbét robusztus regresszióval kell meghatározni, az ettől való eltérést indokolni kell. A robusztus regresszió alkalmazásának módszerét meg kell adni. A Freyberger–Wilson és a CERI humán ösztrogénreceptor vizsgálata során nem kellett korrigálni a ligandum csökkenését, de ha szükséges, fontolóra vehető a korrekció alkalmazása. A kezdeti analízist követően minden kötési görbét ellenőrizni kell, hogy megfelelően illeszkedik-e a modellre. A gyengén kötődő anyag relatív kötési affinitását a gyengén kötődő anyag log (IC50) értékének a 17β-ösztradiol log (IC50) értékéhez viszonyított arányaként lehet meghatározni. A pozitív kontrollok és a nem kötődő kontroll eredményeit a vizsgálati módszerben meghatározott vizsgálati teljesítménymutatók és elfogadhatósági kritériumok szerint kell értékelni (20. pont, a 2. függelék (Freyberger–Wilson vizsgálat) 41–51. pontja és a 3. függelék (CERI vizsgálat) 41–51. pontja). A referenciaösztrogén és gyengén kötődő anyag 3 vizsgálatmenetére vonatkozólag az 1. ábra hoz példákat.
|
1. ábra
Példák a referenciaösztrogén és a gyengén kötődő anyag kompetitív kötési görbéire

A vizsgálati vegyi anyagok adatainak elemzése
|
3.
|
Az összes vizsgálati vegyi anyag adatait lépésenként kell elemezni, biztosítva ezáltal az adatok megfelelő elemzését és a kompetitív kötési görbék helyes osztályozását. Vizsgálati vegyi anyagonként minden vizsgálatmenet megkezdésekor egy szabványosított adatelemzést kell végezni, amely megegyezik a referenciaösztrogén és a gyengén kötődő anyag kontrolljainál alkalmazott analízissel. Az adatelemzés végeztével technikailag ellenőrizni kell a görbeillesztési paramétereket, valamint vizuálisan is ellenőrizni kell, hogy az adatok mennyire illeszkednek az egyes vizsgálatmenetek kompetitív kötési görbéjéhez. Amennyiben a technikai ellenőrzés során megfigyelhető a specifikusan kötődő [3H]-17β-ösztradiol arányának koncentrációfüggő csökkenése, a vegyi anyagok egyes koncentrációit tartalmazó ismétlések közötti csekély eltérés és a három vizsgálatmenet görbeillesztési paramétereinek következetessége jó indikátorai annak, hogy a vizsgálatot és az adatelemzést megfelelően hajtották végre. Egy adott vizsgálati vegyi anyag egyes vizsgálatmeneteiből származó eredmények értelmezése során szakmai megítélés alapján kell dönteni, és a vizsgálati vegyi anyagok kötődő vagy nem kötődő kategóriába sorolásához használt adatoknak tudományosan védhetőnek kell lenniük.
|
|
4.
|
Időnként előfordulhatnak olyan adatok, amelyekre fokozottabb figyelmet kell fordítani ahhoz, hogy a humán ösztrogénreceptor-kötési adatokat megfelelően lehessen elemezni és értelmezni. Az előző tanulmányok során előálltak olyan helyzetek, amikor a kompetitív receptor kötési adatok elemzését és értelmezését megnehezítette a legmagasabb koncentrációban vizsgálati vegyi anyagok specifikus kötődési arányának megugrása (2. ábra). Ez egy jól ismert probléma, ami kompetitív receptor kötődési vizsgálatok protokolljának használatakor fordul elő (3). Ezekben az esetekben alacsonyabb koncentrációértékeknél koncentrációfüggő választ figyeltek meg, de amint a vizsgálati vegyi anyag koncentrációja megközelíti az oldhatósági határértéket, a [3H]-17β-ösztradiol leszorítása már nem csökken. Ilyenkor a magasabb koncentrációk eredménye azt jelzi, hogy elérték a vizsgálat biológiai hatásának határát. Ez a jelenség gyakran kapcsolódik a vizsgálati vegyi anyag magas koncentrációknál megnyilvánuló oldhatatlanságához vagy kicsapódásához, ugyanakkor jelentheti azt is, hogy a vegyi anyag legmagasabb koncentrációinál a dextránbevonatú aktívszén az elkülönítési folyamat során már nem képes megkötni a szabad radioaktívan jelölt ligandumokat. Ha ezek az adatpontok is részt vesznek a kompetitív kötési adatok szigmoid görbére illesztésében, az esetenként az adott vizsgálati vegyi anyag receptorkötési potenciáljának helytelen kategorizálásához vezethet (2. ábra). Ennek elkerülése érdekében a Freyberger–Wilson és a CERI ösztrogénreceptor-kötési vizsgálatának protokolljai biztosítják annak lehetőségét, hogy az elemzésből kizárják azokat az adatpontokat, amelyek esetében az ismétlések [3H]17β-ösztradiol specifikus kötődési arányának átlagértéke legalább 10 %-kal meghaladja a valamely alacsonyabb koncentrációnál megfigyelt átlagértéket (ez az úgynevezett 10 %-os szabály). Ezt a szabályt egy görbére egyszer lehet alkalmazni, és csak akkor, ha legalább 6 koncentráció adatai megmaradnak, hogy a görbe alapján megfelelő kategóriába lehessen sorolni a vizsgálati vegyi anyagot.
|
2. ábra
Példák a 10 %-os szabályt alkalmazó és nem alkalmazó kompetitív kötési görbékre
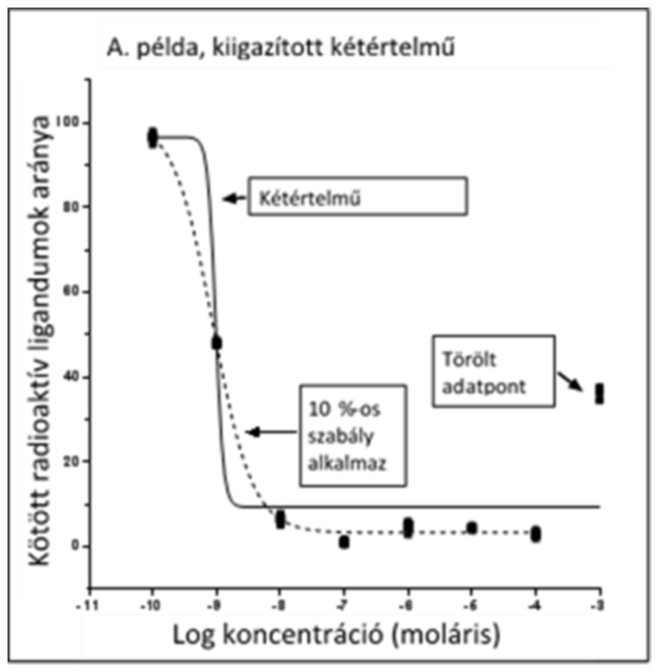
|
5.
|
A görbék korrigálására szolgáló 10 %-os szabály használatát alaposan mérlegelni kell, és csak azokban az esetekben szabad alkalmazni, amikor minden jel arra utal, hogy a receptorhoz kötődő anyagról van szó. A Freyberger–Wilson humán ösztrogénreceptor-kötési vizsgálat validálási tanulmányának keretében végzett kísérletek során megfigyelték, hogy a 10 %-os szabály alkalmazása esetenként nem szándékolt és előre nem látható következményekhez vezetett. Azok a vegyi anyagok, amelyek nem lépnek kölcsönhatásba a receptorral (vagyis a valódi nem kötődő anyagok) a radioaktív ligandumok közel 100 %-os kötésénél gyakran mutattak 10 %-nál nagyobb variabilitást a vizsgált koncentrációtartományban. Ha a legalacsonyabb értéket alacsony koncentráció idézi elő, a 10 %-os szabály alkalmazásakor előfordulhat, hogy az összes magasabb koncentráció adatait törölik az elemzésből, pedig e koncentrációk adatai hasznosak lennének a vegyi anyag nem kötődőként való azonosításához. A 3. ábra olyan helyzetekre hoz példát, ahol a 10 %-os szabály alkalmazása nem megfelelő.
|
3. ábra
Példák olyan kompetitív kötési adatokra, amelyek esetében a 10 %-os szabály alkalmazása nem megfelelő


Hivatkozások
|
(1)
|
OECD (2015). Integrated Summary Report: Validation of Two Binding Assays Using Human Recombinant Estrogen Receptor Alpha (hrERα), Health and Safety Publications, Series on Testing and Assessment (No 226), Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs.
|
|
(2)
|
Motulsky H. and Christopoulos A. (2003). The law of mass action, In Fitting Models to Biological Data Using Linear and Non-linear Regression. GraphPad Software Inc., San Diego, CA, pp 187-191. Www.graphpad.com/manuals/Prism4/RegressionBook.pdf
|
|
(3)
|
Laws SC, Yavanhxay S, Cooper RL, Eldridge JC. (2006). Nature of the Binding Interaction for 50 Structurally Diverse Chemicals with Rat Estrogen Receptors. Toxicological Sci. 94(1):46-56.
|
B.71. A BŐRSZENZIBILIZÁCIÓS KÁROS KIMENETI ÚT (AOP) EGYIK KULCSFONTOSSÁGÚ ESEMÉNYÉVEL, A DENDRITIKUS SEJTEK AKTIVÁCIÓJÁVAL FOGLALKOZÓ IN VITRO BŐRSZENZIBILIZÁCIÓS VIZSGÁLATOK
ÁLTALÁNOS BEVEZETÉS
A dendritikus sejtek aktivációján mint fő eseményen alapuló vizsgálati módszer
|
1.
|
A bőrszenzibilizáló anyagok a vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete (ENSZ) által globálisan harmonizált rendszere (ENSZ-GHS) (1), valamint az Európai Unió (EU) anyagok és keverékek osztályozásáról, címkézéséről és csomagolásáról szóló 1272/2008/EK rendelete (CLP-rendelet) (100) szerint olyan anyagok, amelyek a bőrrel való érintkezést követően allergiás válaszreakciót váltanak ki. A bőrszenzibilizációt előidéző főbb biológiai eseményeket illetően általános egyetértés van. A bőrszenzibilizációhoz kapcsolódó kémiai és biológiai mechanizmusokról már rendelkezésre álló ismereteket az OECD AOP-programjának keretében kidolgozott káros kimeneti út (Adverse Outcome Pathway, AOP) (2) foglalja össze a molekuláris kiváltó eseménytől a köztes eseményeken át a káros hatásig, amely az allergiás kontakt bőrgyulladás. A bőrszenzibilizációs káros kimeneti út esetében a molekuláris kiváltó esemény (vagyis az első kulcsfontosságú esemény) az elektrofil anyagok és a bőrben lévő fehérjék nukleofil centrumai között létrejövő kovalens kötés. E káros kimeneti úton a második kulcsfontosságú esemény a keratinsejtekben következik be, többek között gyulladást okozó válaszreakciók, valamint konkrét sejtjelző útvonalakhoz, például az antioxidáns/elektrofil válaszelemtől (ARE) függő útvonalakhoz kapcsolódó génexpresszió-változások formájában. A harmadik kulcsfontosságú esemény a dendritikus sejtek (DC) aktivációja, melynek értékelése jellemzően konkrét sejtfelületi markerek, kemokinek és citokinek expressziója alapján történik. A negyedik kulcsfontosságú esemény a T-sejtek aktivációja és proliferációja, melynek közvetett értékelése az egereken alkalmazott lokális nyirokcsomó-vizsgálati módszer (LLNA) (3) keretében történik.
|
|
2.
|
Ez a vizsgálati módszer egyenértékű az OECD 442E. vizsgálati iránymutatásában (2017) leírt módszerrel. A vizsgálati módszer a bőrszenzibilizációs káros kimeneti út egyik kulcsfontosságú eseményének, a dendritikus sejtek aktivációjának mechanizmusaival foglalkozó in vitro vizsgálatokat ismerteti (2). Olyan vizsgálatokat foglal magában, amelyekkel elősegíthető a bőrszenzibilizáló és a nem szenzibilizáló anyagok ENSZ-GHS és CLP-rendelet szerinti megkülönböztetése.
|
|
A vizsgálati módszer az alábbi vizsgálatokat ismerteti:
|
—
|
emberi sejtvonal aktiválási vizsgálat (h-CLAT);
|
|
—
|
U937 sejtvonal aktiválási vizsgálat (U-SENS™);
|
|
—
|
Interleukin-8 riportergén vizsgálat (IL-8 Luc vizsgálat).
|
|
|
|
3.
|
Az e vizsgálati módszer és az annak megfelelő OECD vizsgálati iránymutatás részét képező vizsgálatok adatgenerálási eljárásaik és a mért értékek tekintetében különbözhetnek egymástól, mindazonáltal megkülönböztetés nélkül használhatók arra, hogy eleget tegyenek a bőrszenzibilizációs káros kimeneti út egyik kulcsfontosságú eseményének, a dendritikus sejtek aktivációjának vizsgálati eredményeire vonatkozó nemzeti követelményeknek, ugyanakkor előnyükre válik az adatok kölcsönös elfogadása az OECD-megállapodás szerint.
|
A kulcsfontosságú eseményen alapuló vizsgálati módszer alá tartozó vizsgálatok háttere és elvei
|
4.
|
A bőrszenzibilizáció vizsgálata jellemzően vizsgált állatokon történő vizsgálatot foglal magában. A tengerimalacokon alkalmazott klasszikus módszerek, vagyis a Magnusson–Kligman-féle tengerimalac-maximizációs vizsgálat (GPMT) és a Bühler-féle vizsgálat (B.6. vizsgálati módszer) (4) a bőrszenzibilizáció indukciós és kiváltási fázisával egyaránt foglalkozik. Az egereken alkalmazott vizsgálatok – a lokális nyirokcsomó-vizsgálati módszer (LLNA, B.42. vizsgálati módszer) (3) és két nem radioaktív változata, az LLNA: DA (B.50. vizsgálati módszer) (5) és az LLNA: BrdU-ELISA (B.51. vizsgálati módszer) (6) – mindegyike csak az indukciós válaszreakciót vizsgálja, és szintén elfogadottá váltak, mivel állatjólét szempontjából előnyösebbek, mint a tengerimalacon végzett vizsgálatok, és objektív mérést tesznek lehetővé a bőrszenzibilizáció indukciós fázisában.
|
|
5.
|
Nemrégiben olyan mechanisztikus alapú in chemico és in vitro vizsgálati módszereket fogadtak el, amelyek a bőrszenzibilizációs káros kimeneti út első kulcsfontosságú eseményével (B.59. vizsgálati módszer; közvetlen peptidreaktivitási vizsgálat (7)) és második kulcsfontosságú eseményével (B.60. vizsgálati módszer; ARE-Nrf2 luciferáz alapú vizsgálati módszer (8)) foglalkoznak, és hozzájárulnak a vegyi anyagok bőrszenzibilizációs veszélyességi potenciáljának értékeléséhez.
|
|
6.
|
A vizsgálati módszerben ismertetett vizsgálatok mennyiségileg meghatározzák a monociták és a dendritikus sejtek szenzibilizáló anyagokkal való kezelést követően beinduló aktivációs folyamata által a sejtfelületi marker(ek) (pl. CD54, CD86) expressziójában előidézett változásokat, vagy a dendritikus sejtek aktivációja által egy citokin, az IL-8 expressziójában előidézett változásokat. A bőrszenzibilizáló anyagokról kimutatták, hogy a dendritikus sejtek aktivációjában részt vevő (2) sejtmembrán markerek (pl. CD40, CD54, CD80, CD83 és CD86), gyulladást okozó citokinek (pl. az interleukin-1 béta és a tumor nekrózis faktor alfa) és számos kemokin (pl. az IL-8 (CXCL8) és a CCL3) expresszióját (9) (10) (11) (12) idézik elő.
|
|
7.
|
Mindazonáltal, mivel a dendritikus sejtek aktivációja csupán egyike a bőrszenzibilizációs káros kimeneti út kulcsfontosságú eseményeinek (2) (13), a dendritikus sejtek aktivációjának markereit mérő vizsgálatokkal előállított információk önmagukban nem feltétlenül elegendőek ahhoz, hogy megállapítható legyen a vegyi anyagok bőrszenzibilizáló potenciálja van annak hiánya. Ezért az e vizsgálati módszerben leírt vizsgálatokkal kapott adatokat ajánlatos integrált vizsgálati és értékelési megközelítések keretében alkalmazni a bőrszenzibilizáló (ENSZ-GHS/CLP-rendelet szerinti 1. kategória) és a nem szenzibilizáló anyagok megkülönböztetésének támogatására olyan egyéb, releváns kiegészítő információkkal együtt, mint a bőrszenzibilizációs káros kimeneti út más kulcsfontosságú eseményeivel foglalkozó in vitro vizsgálatok eredményei, valamint a nem vizsgálatalapú módszerekkel, köztük a kémiai analógokból kereszthivatkozással kapott adatok (13). Közzétettek példákat az e vizsgálatokkal kapott adatok kidolgozott megközelítésekkel – vagyis az alkalmazott információforrások és az előrejelzéseket lehetővé tévő adatfeldolgozás tekintetében szabványosított megközelítésekkel – történő használatára (13), amelyek az integrált vizsgálati és értékelési megközelítések hasznos elemeként alkalmazhatók.
|
|
8.
|
Az e vizsgálati módszerben leírt vizsgálatok nem alkalmazhatók önmagukban sem a bőrszenzibilizáló anyagoknak az ENSZ-GHS/CLP-rendelet szerinti 1A. és 1B. alkategóriába sorolására azon hatóságok által, amelyek alkalmazzák e két opcionális alkategóriát, sem a hatáserősség előrejelzésére a biztonsági értékeléssel kapcsolatos döntésekhez. A szabályozási kerettől függően azonban az e módszerekkel kapott pozitív eredmény önmagában is felhasználható a vegyi anyagok ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába sorolására.
|
|
9.
|
E vizsgálati módszer esetében a „vizsgálati vegyi anyag” kifejezés a vizsgálat tárgyát képező vegyi anyagra vonatkozik (101), és nem a vizsgálatok egy összetevőből álló anyagok, több összetevőből álló anyagok és/vagy keverékek vizsgálatára való alkalmazhatóságához kapcsolódik. Jelenleg kevés információ áll rendelkezésre a vizsgálatok több összetevőből álló anyagokra és keverékekre való alkalmazhatóságával kapcsolatban (14) (15). Mindazonáltal a vizsgálatok technikailag alkalmazhatók több összetevőből álló anyagok és keverékek vizsgálatára. E vizsgálati módszer tervezett szabályozási célt szolgáló adatgenerálás érdekében, keveréken történő alkalmazása előtt azonban meg kell vizsgálni, hogy az megfelelő eredményeket biztosíthat-e erre a célra, és ha igen, miért (102). Ilyen megfontolások nem szükségesek, ha létezik a keverék vizsgálatára vonatkozó szabályozási követelmény. Ezenfelül a több összetevőből álló anyagok vagy keverékek vizsgálata során figyelembe kell venni a citotoxikus összetevőknek a megfigyelt válaszreakciókkal való lehetséges kölcsönhatását.
|
SZAKIRODALOM
|
(1)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Elérhető a következő címen:https://www.unece.org/trans/danger/publi/ghs/ghs_rev06/06files_e.html.
|
|
(2)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No. 168. Elérhető a következő címen:http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En.
|
|
(3)
|
E melléklet B.42., Helyi nyirokcsomó-vizsgálat című fejezete.
|
|
(4)
|
E melléklet B.6., Bőrszenzibilizáció című fejezete.
|
|
(5)
|
E melléklet B.50., Bőrszenzibilizáció: helyi nyirokcsomó-vizsgálat: DA című fejezete.
|
|
(6)
|
E melléklet B.51., Bőrszenzibilizáció: helyi nyirokcsomó-vizsgálat: BrdU-ELISA című fejezete.
|
|
(7)
|
E melléklet B.59., In chemico bőrszenzibilizáció: közvetlen peptid reaktivitás vizsgálat (DPRA) című fejezete.
|
|
(8)
|
E melléklet B.60., In Vitro bőrszenzibilizáció: ARE-Nrf2 luciferáz alapú vizsgálati módszer című fejezete.
|
|
(9)
|
Steinman RM. (1991). The dendritic cell system and its role in immunogenicity. Annu Rev Immunol 9:271-96.
|
|
(10)
|
Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, and Banchereau J. (1994). B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med 180:1841-7.
|
|
(11)
|
Aiba S, Terunuma A, Manome H, and Tagami H. (1997). Dendritic cells differently respond to haptens and irritants by their production of cytokines and expression of co-stimulatory molecules. Eur J Immunol 27:3031-8.
|
|
(12)
|
Aiba S, Manome H, Nakagawa S, Mollah ZU, Mizuashi M, Ohtani T, Yoshino Y, and Tagami. H. (2003). p38 mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and DNCB. J Invest Dermatol 120:390-8.
|
|
(13)
|
OECD (2016). Series on Testing & Assessment No 256: Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Annex 1 and Annex 2. ENV/JM/HA(2016)29. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen:https://community.oecd.org/community/iatass.
|
|
(14)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(15)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
1. függelék
IN VITRO BŐRSZENZIBILIZÁCIÓ: EMBERI SEJTVONAL AKTIVÁLÁSI VIZSGÁLAT (H-CLAT)
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
1.
|
A h-CLAT mennyiségileg meghatározza a THP-1 humán monocita leukémia sejtvonal monocitáinak és dendritikus sejtjeinek a szenzibilizáló anyagokkal való kezelést követően beinduló aktivációs folyamata által a sejtfelületi marker(ek) (CD86 és CD54) expressziójában előidézett változásokat (1) (2). Ezt követően a CD86 és a CD54 sejtfelületi marker mért expressziós szintjét a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetésének alátámasztására használják fel.
|
|
2.
|
A h-CLAT-ot az Állatkísérletek Alternatíváinak Uniós Referencialaboratóriuma (EURL ECVAM) által koordinált validálási vizsgálatban értékelték, az EURL ECVAM tudományos tanácsadó bizottsága (ESAC) pedig független szakértői értékelésnek vetette alá. A rendelkezésre álló bizonyítékok, valamint a szabályozó hatóságok és az érdekeltek álláspontja alapján az EURL ECVAM a h-CLAT-ot integrált vizsgálati és értékelési megközelítés keretében történő alkalmazásra ajánlotta (3) a veszélyességi osztályozás és címkézés céljából a bőrszenzibilizáló és nem szenzibilizáló anyagok megkülönböztetésének alátámasztására. A h-CLAT-ból származó adatok és más információk együttes használatára fellelhetők példák a szakirodalomban (4) (5) (6) (7) (8) (9) (10) (11).
|
|
3.
|
A h-CLAT-ról bebizonyosodott, hogy áthelyezhető a sejttenyésztési technikák és az áramlási citometriás elemzés terén tapasztalattal rendelkező laboratóriumokba. A vizsgálattól várható előrejelzések reprodukálhatóságának mértéke a laboratóriumokon belül és között mintegy 80 % (3) (12). A validálási vizsgálatban (13) és más közzétett tanulmányokban (14) kapott eredmények összességében azt jelzik, hogy a hCLAT a bőrszenzibilizáló (azaz az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) anyagokat az LLNA vizsgálati módszer eredményeihez képest 85 %-os (N=142) pontossággal különbözteti meg a nem szenzibilizáló anyagoktól, 93 % az érzékenysége (94/101), a specifikussága pedig 66 % (27/41) (az EURL ECVAM által végzett ismételt elemzés alapján (12), amely figyelembe vette az összes rendelkezésre álló adatot, ugyanakkor a 4. pontban leírtak szerint figyelmen kívül hagyta a 3,5-nél nagyobb Log Kow értékkel rendelkező vegyi anyagok negatív eredményét). A h-CLAT nagyobb valószínűséggel vezet hamis negatív előrejelzésekhez olyan vegyi anyagoknál, amelyeknek alacsonytól mérsékeltig terjedő a szenzibilizáló hatása (azaz az ENSZ-GHS/CLP-rendelet szerinti 1B. alkategóriába tartozó anyagoknál), mint a rendkívül bőrszenzibilizáló hatást mutató vegyi anyagoknál (azaz az ENSZ-GHS/CLP-rendelet szerinti 1A. alkategóriába tartozó anyagoknál) (4) (13) (15). Ezek az információk összességében azt jelzik, hogy a h-CLAT módszer érdemben hozzájárul a bőrszenzibilizáció veszélyeinek azonosításához. A h-CLAT mint önálló vizsgálat esetében itt megadott pontossági értékek azonban csak irányadóak, ugyanis a vizsgálatot az integrált vizsgálati és értékelési megközelítés keretében más információforrásokkal együtt és az Általános bevezetés 7. és 8. pontjának rendelkezéseivel összhangban kell figyelembe venni. Ezen túlmenően a bőrszenzibilizáció nem állatokon alkalmazott módszereinek értékelésekor nem szabad megfeledkezni arról, hogy az LLNA vizsgálat, valamint más állatkísérletek nem feltétlenül tükrözik teljes egészében az embernél fennálló helyzetet.
|
|
4.
|
Az aktuálisan rendelkezésre álló adatok alapján a h-CLAT módszerről bebizonyosodott, hogy különféle szerves funkciós csoportokat, reakciómechanizmusokat, (in vivo vizsgálatok során meghatározott) bőrszenzibilizáló hatásokat és fizikai-kémiai tulajdonságokat képviselő vizsgálati vegyi anyagokra alkalmazható (3) (14) (15). A h-CLAT módszer olyan vizsgálati vegyi anyagokra alkalmazandó, amelyek megfelelő oldószerben/vivőanyagban (lásd a 14. pontot) oldhatók, vagy azokkal stabil diszperziót alkotnak (azaz olyan kolloidot vagy szuszpenziót, amelyben a vizsgálati vegyi anyag nem ülepedik le, illetve nem válik szét az oldószertől/vivőanyagtól különböző fázisokra). A 3,5-nél magasabb log Kow értékkel rendelkező vizsgálati vegyi anyagok általában hamis negatív eredményre vezetnek (14). Ezért a 3,5-nél magasabb log Kow értékkel rendelkező vizsgálati vegyi anyagok negatív eredményét figyelmen kívül kell hagyni. Ugyanakkor a 3,5-nél magasabb log Kow értékkel rendelkező vizsgálati vegyi anyagok pozitív eredménye még felhasználható a vizsgálati vegyi anyag bőrszenzibilizáló anyagként való azonosításának alátámasztására. Ezen túlmenően az alkalmazott sejtvonal korlátozott metabolikus képessége (16) és a kísérleti körülmények miatt különösen a pro-hapténok (azaz például a P450 enzim közreműködésével enzimatikus aktivációt igénylő anyagok) és a lassú oxidációs rátájú pre-hapténok (azaz az oxidációval aktiválódó anyagok) is negatív eredményre vezethetnek a h-CLAT vizsgálatban (15). A h-CLAT vizsgálat alkalmas fluoreszcens vizsgálati vegyi anyagok értékelésére (17); mindazonáltal azok az erősen fluoreszcens vizsgálati vegyi anyagok, amelyek a fluoreszcein-izotiocianáttal (FITC), illetve a propídium-jodiddal (PI) azonos hullámhosszon bocsátják ki a fényt, zavarják az áramlási citometriás észlelést, ezért FITC-vel összekapcsolt ellenanyagok, illetve propídium-jodid használatával nem értékelhetők megfelelően. Ilyen esetben használhatók más fluorokrómmal jelzett ellenanyagok vagy más citotoxikus markerek, ha például az 1.2. függelékben szereplő jártassági tesztanyagok vizsgálata révén bizonyítást nyer, hogy a FITC-vel jelzett ellenanyagokhoz (lásd a 24. pontot) vagy a propídium-jodidhoz (lásd a 18. pontot) hasonló eredményt biztosítanak. A fentiek fényében: a negatív eredményeket a közölt korlátokra tekintettel és az integrált vizsgálati és értékelési megközelítés keretébe tartozó egyéb információforrásokkal együtt kell értelmezni. Olyan esetekben, amikor bizonyítékkal igazolható, hogy a h-CLAT módszer nem alkalmazható más, konkrét vegyianyag-kategóriákra, a módszert nem szabad e vegyianyag-kategóriákra alkalmazni.
|
|
5.
|
A fentieknek megfelelően a h-CLAT módszer elősegíti a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetését. Hozzájárulhat azonban a szenzibilizáló hatás erősségének értékeléséhez is (4) (5) (9), ha az integrált vizsgálati és értékelési megközelítéshez hasonló integrált megközelítések keretében alkalmazzák. Lehetőleg humán adatokon alapuló további munkára van azonban szükség annak meghatározásához, hogy a h-CLAT eredményei hogyan járulhatnak hozzá a hatáserősség vizsgálatához.
|
|
6.
|
A fogalommeghatározások az 1.1. függelékben találhatók.
|
A VIZSGÁLAT ELVE
|
7.
|
A h-CLAT módszer egy in vitro vizsgálat, amely mennyiségileg meghatározza a THP-1 humán monocita leukémia sejtvonal sejtjein kifejeződő sejtfelületi markerek (CD86 és CD54) expressziójában a vizsgálati vegyi anyaggal való 24 órás kezelés révén bekövetkező változásokat. Ezek a sejtfelszíni molekulák a monocita THP-1 aktiváció jellegzetes markerei, és képesek imitálni a dendritikus sejtek aktivációját, ami kulcsszerepet játszik a T-sejtek aktiválásában. A sejtfelületi markerek expressziójának változásait áramlási citometriával mérik, miután a sejteket megfestették fluorokrómmal jelzett ellenanyagokkal. Ezzel egyidejűleg citotoxicitási méréseket is végeznek annak felméréséhez, hogy a citotoxikus koncentrációknál alacsonyabb koncentráció mellett fokozódik-e a felületi markerek expressziója. Meghatározzák a felületi markerek oldószeres/vivőanyagos kontrollhoz viszonyított relatív fluoreszcencia-intenzitását, amelyet az előrejelzési modellben (lásd a 26. pontot) felhasználnak a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetésének alátámasztására.
|
A JÁRTASSÁG BIZONYÍTÁSA
|
8.
|
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak az 1.2. függelékben felsorolt tíz jártassági tesztanyag felhasználásával igazolniuk kell szakmai jártasságukat. Ezenfelül a vizsgálat felhasználóinak történeti adatbázist kell fenntartaniuk, amely tartalmazza a reaktivitási tesztekből (lásd a 11. pontot), valamint a pozitív és az oldószeres/vivőanyagos kontrollokból (lásd a 20–22.pontot) származó adatokat, ezekkel az adatokkal igazolva a vizsgálat laboratóriumon belüli hosszú távú reprodukálhatóságát.
|
ELJÁRÁS
|
9.
|
Ez a vizsgálat az állatkísérletek alternatív módszereire vonatkozó adatbázis-szolgáltatás (DB-ALM) h-CLAT-ot érintő 158. protokollján (18) alapul, amely az EURL ECVAM által koordinált validálási vizsgálathoz használt protokoll. A h-CLAT módszer laboratóriumi végrehajtása és alkalmazása során e protokoll alkalmazása ajánlott. Az alábbiakban a h-CLAT módszer főbb összetevőinek és eljárásainak leírása található; a módszer a következő két lépésből áll: dózisbehatároló vizsgálat és CD86/CD54 expresszió mérése.
|
Sejtek előkészítése
|
10.
|
A h-CLAT módszer végrehajtásához a THP-1 humán monocita leukémia sejtvonalat kell használni. Ajánlatos a (TIB-202™) sejteket egy megfelelő minősítéssel rendelkező sejtbankból, például az Amerikai Fajtakultúra Gyűjteményből beszerezni.
|
|
11.
|
A THP-1 sejteket 10 % magzati borjú szérummal (FBS), 0,05 mM 2-merkaptoetanollal, 100 egység/ml penicillinnel és 100 μg/ml sztreptomicinnel kiegészített RPMI-1640 tápoldatban, valamint 37oC-os, 5 %-os CO2-tartalmú és vízgőzzel telített környezetben kell tenyészteni. A tápfolyadékból a penicillin és a sztreptomicin elhagyható. Ez esetben azonban a felhasználóknak például az 1.2. függelékben felsorolt jártassági tesztanyagok tesztelésével meg kell bizonyosodniuk arról, hogy az antibiotikumok hiánya a tápfolyadékban nem befolyásolja az eredményt. A fertőzés kockázatának minimalizálása érdekében minden esetben a megfelelő sejttenyésztési gyakorlatok szerint kell eljárni, függetlenül attól, hogy van-e antibiotikum a sejttenyésztő tápfolyadékban. A THP-1 sejteket 2–3 naponta 0,1–0.2 × 106 sejt/ml sűrűségben rutinszerűen leoltják. A sejtek fenntartása során meg kell őrizni a 0,1–1,0 × 106 sejt/ml sejtsűrűséget. A vizsgálatban való felhasználásuk előtt a sejteket egy reaktivitási teszt során minősíteni kell. A sejtek reaktivitási tesztjét a felolvasztásuk után két héttel kell elvégezni, a pozitív kontrollanyagokkal, vagyis a 2,4-dinitro-klór-benzollal (DNCB) (CAS-száma: 97-00-7, legalább 99 %-os tisztaságban) és a nikkel-szulfáttal (NiSO4) (CAS-száma: 10101-97-0, legalább 99 %-os tisztaságban), valamint a negatív kontrollanyaggal, a tejsavval (LA) (CAS-száma: 50-21-5, legalább 85 % tisztaságban). Mind a DNCB-nak, mind a NiSO4-nak pozitív választ, a tejsavnak pedig negatív választ kell kiváltania a CD86 és a CD54 sejtfelületi markernél egyaránt. A vizsgálathoz kizárólag a reaktivitási tesztben megfelelt sejtek használhatók. A sejtek a felolvasztásukat követően két hónapig szaporíthatók. A sejteket legfeljebb 30 alkalommal szabad passzálni. A reaktivitási tesztet a 20–24. pontban leírt eljárások szerint kell végrehajtani.
|
|
12.
|
A vizsgálathoz a THP-1 sejteket vagy 0,1 × 106 sejt/ml vagy 0,2 × 106 sejt/ml sűrűségben átoltják, majd sejttenyésztő lombikban az előbbi esetben 72 óráig, az utóbbiban pedig 48 óráig előtenyésztik. Fontos, hogy közvetlenül az előtenyésztési időszak végét követően a sejttenyésztő lombikban minden (a fenti két előtenyésztési körülmény egyikét alkalmazó) kísérletben a sejtek sűrűsége nagyjából azonos legyen, mert közvetlenül az előtenyésztési időszak végét követően a sejttenyésztő lombikban fennálló sejtsűrűség befolyásolhatja az allergének által kiváltott CD86/CD54 expressziót (19). A vizsgálat napján a sejttenyésztő lombikból begyűjtött sejteket frissen készített tápfolyadékban 2 × 106 sejt/ml sűrűségben újraszuszpendálják. Ezt követően a sejteket elhelyezik egy 24 lyukú, lapos fenekű lemezben (500 μl, lyukanként 1 × 106 sejt) vagy egy 96 lyukú, lapos fenekű lemezben (80 μl, lyukanként 1,6 × 105 sejt).
|
Dózisbehatároló vizsgálat
|
13.
|
A dózisbehatároló vizsgálat során meghatározzák a CV75 értéket, azaz a vizsgálati vegyi anyag azon koncentrációját, amely az oldószeres/vivőanyagos kontrollhoz képest 75 %-os sejtéletképességet (CV) idéz elő. A CV75 értéket arra használják, hogy meghatározzák a vizsgálati vegyi anyagok koncentrációját a CD86/CD54 expresszió méréséhez (lásd a 20–24. pontot).
|
A vizsgálati vegyi anyagok és a kontrollanyagok előkészítése
|
14.
|
A vizsgálati vegyi anyagokat és a kontrollanyagokat a vizsgálat napján kell előkészíteni. A h-CLAT módszerben a vizsgálati vegyi anyagokat feloldják vagy stabilan diszpergálják (lásd még a 4. pontot) sóoldatban vagy tápoldatban, mint az első számú oldószer/vivőanyag választás, vagy dimetil-szulfoxidban (DMSO, legalább 99 %-os tisztaságban), mint második oldószer/vivőanyag választás, ha a vizsgálati vegyi anyag az előző két oldószerben/vivőanyagban nem oldható vagy nem diszpergálható stabilan, a végső koncentráció 100 mg/ml (sóoldat vagy tápoldat használata esetén) vagy 500 mg/ml (DMSO használata esetén). A fentiektől eltérő oldószerek/vivőanyagok megfelelő tudományos indokolással szintén használhatók. Figyelembe kell venni azt is, hogy a vizsgálati vegyi anyag mennyire stabil a végső oldószerben/vivőanyagban.
|
|
15.
|
A vizsgálati vegyi anyagok 100 mg/ml (sóoldat vagy tápoldat használata esetén) vagy 500 mg/ml (DMSO használata esetén) törzsoldatából kiindulva az alábbi hígítási lépéseket kell elvégezni:
|
—
|
Ha az oldószer/vivőanyag sóoldat vagy tápoldat: A megfelelő oldószer/vivőanyag használatával nyolc törzsoldatot (nyolc koncentrációt) készítenek, kétszeres oldatsorozatban. E törzsoldatokat ezután a tápfolyadékban 50-szeresükre tovább kell hígítani (munkaoldatok). Amennyiben a lemezre helyezett legnagyobb, 1 000 μg/ml-es végső koncentráció nem toxikus, a legmagasabb koncentrációértéket egy új citotoxicitási vizsgálatban újból meg kell határozni. A sóoldatban vagy tápoldatban feloldott vagy stabilan diszpergált vizsgálati vegyi anyagok lemezre helyezett végső koncentrációja nem haladhatja meg az 5 000 μg/ml értéket.
|
|
—
|
Ha az oldószer/vivőanyag DMSO: A megfelelő oldószer/vivőanyag használatával nyolc törzsoldatot (nyolc koncentrációt) készítenek, kétszeres oldatsorozatban. E törzsoldatokat ezután a tápfolyadékban 250-szeresükre tovább kell hígítani (munkaoldatok). A lemezre helyezett végső koncentráció még akkor sem haladhatja meg az 1 000 μg/ml értéket, ha ez a koncentráció nem toxikus.
|
Végül pedig felhasználják a munkaoldatokat a kezeléshez, ehhez a kétszeres továbbhígítás érdekében azonos mennyiségű munkaoldatot adnak hozzá a lemezen lévő THP-1 sejtszuszpenzióhoz (lásd a 17. pontot), a lemez végleges koncentrációértékei általában 7,81–1 000 μg/ml tartományban helyezkednek el.
|
|
16.
|
A h-CLAT módszerben alkalmazott oldószeres/vivőanyagos kontroll a tápfolyadék (a tápfolyadékban vagy sóoldatban feloldott vagy stabilan diszpergált vizsgálati vegyi anyagok esetében (lásd a 4. pontot)) vagy a DMSO (a DMSO-ban feloldott vagy stabilan diszpergált vizsgálati vegyi anyagok esetében), amelyet egyetlen végső, 0,2 %-os koncentrációban vizsgálnak a lemezen. Az oldószeres/vivőanyagos kontrollt a munkaoldatokhoz alkalmazott eljárás szerint kell hígítani (lásd a 15. pontot).
|
A vizsgálati vegyi anyagok és a kontrollanyagok alkalmazása
|
17.
|
A 15. és a 16. pontban leírt módon elkészített tápfolyadékot vagy munkaoldatokat 1: 1 térfogatszázalék arányban össze kell keverni a 24 lyukú vagy 96 lyukú lapos fenekű lemezen elkészített sejtszuszpenziókkal (lásd a 12. pontot). A kezelt lemezeket ezt követően 24 óráig (±0,5 óra) 37oC-on 5 % CO2 jelenlétében inkubálják. Ügyelni kell az illékony vizsgálati vegyi anyagok elpárolgásának elkerülésére, valamint a lyukak vizsgálati vegyi anyagokkal való keresztszennyeződésének elkerülésére, például a lemezek befedhetők fóliával a vizsgálati vegyi anyagokkal való inkubálás előtt (20).
|
Festés propídium-jodiddal
|
18.
|
A 24 órás (±fél óra) kezelést követően a sejteket mintacsövekbe helyezik át és centrifugálással kinyerik őket. A felülúszót eltávolítják, a maradék sejtet pedig 0,1 % borjú szérum albuminnal kiegészített 200 μl (96 lyukú lemez esetén) vagy 600 μl (24 lyukú lemez esetén) foszfátpuffert tartalmazó sóoldattal (festő puffer) újraszuszpendálják. 200 μl sejtszuszpenziót áthelyeznek egy 96 lyukú, gömbölyű fenekű lemezre (96 lyukú lemez használata esetén) vagy mikrocsőbe (24 lyukú lemez használata esetén), majd két alkalommal átmossák 200 μl (96 lyukú lemez használata esetén) vagy 600 μl (24 lyukú lemez használata esetén) festő pufferrel. Végül a sejteket (pl. 400 μl) festő pufferben újraszuszpendálják, és hozzáadnak (pl. 20 μl) propídium-jodidot (a propídium-jodid végső koncentrációja lehet például 0,625 μg/ml). Más citotoxicitási markerek, például 7-amino-aktinomicin D (7-AAD), tripankék vagy egyéb markerek is alkalmazhatók, amennyiben az alternatívaként használt festékről például az 1.2. függelékben felsorolt jártassági tesztanyagok tesztelésével kimutatták, hogy a propídium-jodidhoz hasonló eredményekre vezet.
|
Citotoxicitás mérése áramlási citometriával és a CV75 érték megbecslésével
|
19.
|
A propídium-jodid (PI) felvételét áramlási citometriával elemzik az FL-3 adatgyűjtő csatornán. Összesen 10 000 élő (PI negatív) sejt adatait gyűjtik össze. A sejtéletképességet az alábbi egyenlet alapján kalkulálja ki a citométer elemzőprogramja. Alacsony sejtéletképességnél az elhalt sejtekkel együtt legfeljebb 30 000 sejt adatait gyűjtik össze. Ennek alternatívája lehet az, amikor az adatokat az elemzés elindításától számítva egy percen át gyűjtik.

A CV75 értéket (lásd a 13. pontot), vagyis azt a koncentrációt, amelynél a THP-1 sejtek túlélési aránya 75 % (a toxicitás 25 %), logaritmikus-lineáris interpolációval számítják ki, az alábbi egyenlet alapján:
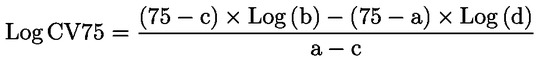
ahol:
a = a 75 % feletti sejtéletképesség minimumértéke
c = a 75 % alatti sejtéletképesség maximumértéke
b és d = az a és c sejtéletképességet előidéző koncentrációk

A CV75 érték más módon is meghatározható, amennyiben (pl. a jártassági tesztanyagok tesztelésével) igazolható, hogy az nem befolyásolja az eredményt.
|
CD86/CD54 expresszió mérése
A vizsgálati vegyi anyagok és a kontrollanyagok előkészítése
|
20.
|
A vizsgálati vegyi anyagokat feloldják vagy stabilan diszpergálják a megfelelő oldószerben/vivőanyagban (sóoldat, tápoldat vagy DMSO; lásd a 14. pontot). A vizsgálati vegyi anyagokat elsőként a dózisbehatároló vizsgálatban (lásd a 19. pontot) meghatározott CV75 érték 1,2-szerese 100-szorosának (sóoldat vagy tápoldat esetén) vagy 500-szorosának (DMSO esetén) megfelelő koncentrációra hígítják. Amennyiben a CV75 érték nem határozható meg (ha a dózisbehatároló vizsgálatban nem figyeltek meg kellő mértékű citotoxicitást), az induló koncentrációérték a vizsgálati vegyi anyag egyes oldószerekkel/vivőanyagokkal készített legmagasabb oldható vagy stabilan diszpergált koncentrációja legyen. A lemezre helyezett végső koncentráció azonban nem haladhatja meg az 5 000 μg/ml értéket (sóoldat vagy tápoldat esetén), illetve az 1 000 μg/ml értéket (DMSO esetén). Ezt követően a megfelelő oldószer/vivőanyag használatával 1,2-szeres oldatsorozatot készítenek (nyolc koncentrációval 100×1,2 × CV75 és 100×0,335 × CV75 között (sóoldat vagy tápoldat esetén), illetve 500×1,2 × CV75 és 500×0,335 × CV75 között (DMSO esetén)), létrehozva ezzel a h-CLAT módszerrel vizsgálandó törzsoldatokat (egy adagolási minta megtalálható a DB-ALM 158. protokolljában). A törzsoldatokat ezután a tápfolyadékban 50-szeresükre (sóoldat vagy tápfolyadék esetén), illetve 250-szeresükre (DMSO esetén) továbbhígítják (elkészítve a munkaoldatokat). Ezeket a munkaoldatokat végül felhasználják a kezeléshez, ehhez kétszeres hígítási tényező alkalmazásával elkészítik a lemezre helyezett végső oldatokat. Amennyiben az eredmények nem teljesítik a 29. és a 30. pontban szereplő sejtéletképességi kritériumokat, a CV75 értékének pontosabb meghatározása érdekében a dózisbehatároló vizsgálat megismételhető. A CD86/CD54 expresszió mérésére csak 24 lyukú lemezek használhatók.
|
|
21.
|
Az oldószeres/vivőanyagos kontrollt a 16. pontban leírtak szerint kell elkészíteni. A h-CLAT módszerben alkalmazott pozitív kontrollanyag a DNCB (lásd a 11. pontot), amelynek törzsoldatait DMSO-ban készítik el, a törzsoldatokat pedig a 20. pontban foglaltak szerint hígítják. A CD86/CD54 expresszió mérése során pozitív kontrollanyagként használt DNCB-t egyetlen végső (általában 4,0 μg/ml) koncentrációban vizsgálják a lemezen. Ahhoz, hogy 4,0 μg/ml koncentrációjú DNCB kerüljön a lemezre, DMSO-ban elkészítik a DNCB 2 mg/ml koncentrációjú törzsoldatát, majd a törzsoldatot tápfolyadékban a 250-szeresére hígítva elkészítik a 8 μg/ml koncentrációjú munkaoldatot. A pozitív kontrollanyag koncentrációjaként használható a DNCB CV75 értéke, amelyet minden vizsgálólaboratóriumban meghatároznak. Alkalmazhatók más, megfelelő pozitív kontrollok is, ha rendelkezésre állnak dokumentált adatok hasonló vizsgálatmenet-elfogadhatósági kritériumok származtatásához. A pozitív kontrollanyag egyetlen végső koncentrációban kerül a lemezre, amely nem haladhatja meg az 5 000 μg/ml értéket (sóoldat vagy tápoldat esetén), illetve az 1 000 μg/ml értéket (DMSO esetén). A vizsgálatmenet elfogadhatósági kritériumai megegyeznek a vizsgálati vegyi anyag elfogadhatósági kritériumaival (lásd a 29. pontot), az utolsó kritériumot leszámítva, mivel a pozitív kontrollanyagot egyetlen koncentrációban vizsgálják.
|
A vizsgálati vegyi anyagok és a kontrollanyagok alkalmazása
|
22.
|
Minden vizsgálati vegyi anyag és kontrollanyag esetében egy kísérletre van szükség az előrejelzéshez. Valamennyi kísérlet legalább két független CD86/CD54 expresszió mérési vizsgálatmenetből áll (lásd a 26–28. pontot). Mindegyik független vizsgálatmenetet más-más napon kell elvégezni, illetve a vizsgálatmenetek ugyanazon a napon is elvégezhetők, ha minden vizsgálatmenethez: a) a vizsgálati vegyi anyag és az ellenanyag oldatok egymástól független, frissen készített törzsoldatait és munkaoldatait használják és b) egymástól függetlenül begyűjtött sejteket (vagyis különböző sejttenyésztő lombikból gyűjtött sejteket) használnak; a sejtek azonban származhatnak ugyanabból a passzálásból. A vizsgálati vegyi anyagok és kontrollanyagok elkészített munkaoldatait (500 μl) 1: 1 arányban összekeverik 500 μl szuszpendált (1 x 106) sejttel, majd a sejteket a 20. és 21. pontban leírtak szerint 24 óráig (± fél óra) inkubálják. Minden vizsgálatmenetben elegendő a vizsgálati vegyi anyag és a kontrollanyag egyes koncentrációit egy replikátumban mérni, mivel az előrejelzést legalább két független vizsgálatmenet alapján határozzák meg.
|
Sejtek festése és adatok elemzés
|
23.
|
A 24 órás (± fél óra) kezelést követően a sejteket a 24 lyukú lemezről mintacsövekbe helyezik, centrifugálással begyűjtik, majd 1 ml festő pufferben kétszer átmossák (szükség esetén be lehet iktatni további mosási lépéseket). A mosást követően a sejtek növekedését meggátolják 600 μl blokkoló oldattal (festő puffer 0,01 tömegszázalékos globulinnal (II-es, III-as Cohn-frakció, humán eredetű; SIGMA, #G2388-10G vagy azzal egyenértékű)), majd a sejteket 4oC-on 15 percre inkubálják. A blokkolást követően a sejteket három egyenlő 180 μl mennyiségű részre osztva 96 lyukú gömbölyű fenekű lemezbe vagy mikrocsőbe helyezik.
|
|
24.
|
A centrifugálást követően a sejteket 4oC-on 30 percen át festik 50 μl FITC-vel jelzett anti-CD86, anti-CD54 vagy egér IgG1 (izotípus) ellenanyagokkal. A h-CLAT-ra vonatkozó 158. sz. DB-ALM protokollban (18) leírt ellenanyagokat kell alkalmazni festő pufferrel való 3: 25 térfogatszázalékos hígításban a CD86 esetében (BD-PharMingen, #555657; Fun-1 klón), illetve 3: 50 térfogatszázalékos hígításban a CD54 esetében (DAKO, #F7143; 6.5B5 klón) és az IgG1 esetében (DAKO, #X0927). Az ellenanyagok hígítási tényezőjét a vizsgálat kidolgozói határozták meg az alapján, hogy ezek biztosították a legjobb jel-zaj arányt. A vizsgálat kidolgozói által szerzett tapasztalatok arra utalnak, hogy az ellenanyagok fluoreszcencia-intenzitása a különböző tételeknél általában konzisztens. A legmegfelelőbb vizsgálati koncentrációértékek meghatározásához azonban a felhasználók fontolóra vehetik az ellenanyagok saját laboratóriumi körülményeik mellett történő titrálását. Más fluorokrómmal jelzett anti-CD86 és/vagy anti-CD54 ellenanyagok is alkalmazhatók, amennyiben például az 1.2. függelékben felsorolt jártassági tesztanyagok tesztelésével kimutatták, hogy a FITC-vel összekapcsolt ellenanyagokhoz hasonló eredményekre vezetnek. Megjegyzendő, hogy a h-CLAT-ra vonatkozó 158. sz. DB-ALM protokollban (18) leírt ellenanyag klón vagy forgalmazó lecserélése befolyással lehet az eredményekre. Miután legalább kétszer átmosták 150 μl festék pufferrel, a sejteket (pl. 400 μl) festék pufferben újraszuszpendálják, és hozzáadják a PI oldatot (pl. 20 μl mennyiségben, amellyel elérhető a végső 0,625 μg/ml koncentráció) vagy egy másik citotoxicitási marker oldatát (lásd a 18. pontot). A CD86 és a CD54 expressziójának mértékét és a sejtéletképességet áramlási citometriával elemzik.
|
ADATOK ÉS JELENTÉS
Az adatok kiértékelése
|
25.
|
A CD86 és a CD54 kifejeződését áramlási citometriával elemzik az FL-1 adatgyűjtő csatornán. A pozitív kontrollként szolgáló sejtek és a vegyi anyaggal kezelt sejtek CD86 és CD54 relatív fluoreszcencia-intenzitását (RFI) a fluoreszcencia-intenzitás mértani középértéke (MFI) alapján és az alábbi egyenlet szerint számítják ki:

Az izotípusos kontrollként szolgáló sejtek életképességét (amelyeket egér IgG1 (izotípus) ellenanyagokkal festettek meg) szintén a 19. pontban feltüntetett egyenlettel határozzák meg.
|
Előrejelzési modell
|
26.
|
A CD86/CD54 expressziós mérésnél minden vizsgálati vegyi anyagot legalább két független vizsgálatmenetben tesztelnek, amelyek alapján meghatározzák a (POZITÍV vagy NEGATÍV) előrejelzést. A h-CLAT előrejelzés akkor minősül POZITÍVNAK, ha két független vizsgálatmenetből kettőben vagy három független vizsgálatmenetből legalább kettőben az alábbi feltételek legalább egyike teljesül, minden egyéb esetben a h-CLAT előrejelzés negatívnak minősül (1. ábra):
|
—
|
a CD86 relatív fluoreszcencia-intenzitása bármely vizsgált koncentrációban legalább 150 % (legalább 50 %-os sejtéletképesség mellett);
|
|
—
|
a CD54 relatív fluoreszcencia-intenzitása bármely vizsgált koncentrációban legalább 200 % (legalább 50 %-os sejtéletképesség mellett).
|
|
|
27.
|
A fentiek alapján, ha az első két vizsgálatmenet pozitív eredménnyel zárult a CD86 és/vagy a CD54 esetében, akkor a h-CLAT vizsgálat előrejelzése POZITÍVNAK tekintendő, és harmadik vizsgálatmenetre nincs szükség. Hasonlóképpen, ha az első két vizsgálatmenet mindkét marker tekintetében negatív, akkor a h-CLAT előrejelzése NEGATÍVNAK minősül (figyelembe véve a 30. pontban foglaltakat), és harmadik vizsgálatmenetre nincs szükség. Ha azonban az első két vizsgálatmenet eredményei legalább az egyik marker (CD54 vagy CD86) esetében nem egyeznek, el kell végezni egy harmadik vizsgálatmenetet, és a végső előrejelzés a három független vizsgálatmenet többségi eredményén (a háromból kettőjén) fog alapulni. Ezzel kapcsolatban megjegyzendő, hogy ha két független vizsgálatmenetben az egyik csak a CD86 tekintetében pozitív (a továbbiakban P1) és a másik csak a CD54 esetében pozitív (a továbbiakban P2), harmadik vizsgálatmenetre van szükség. Amennyiben a harmadik vizsgálatmenet mindkét marker esetében negatív eredménnyel jár (a továbbiakban N), akkor a h-CLAT vizsgálat előrejelzése NEGATÍVNAK tekintendő. Másfelől viszont, ha a harmadik vizsgálatmenet bármelyik marker (P1 vagy P2) esetében vagy mindkét marker (a továbbiakban P12) esetében pozitív eredménnyel zárul, a h-CLAT vizsgálat előrejelzése POZITÍVNAK tekintendő.

1. ábra: A h-CLAT módszerben alkalmazott előrejelzési modell. A h-CLAT-tal végzett előrejelzést integrált értékelési és vizsgálati megközelítés keretében, valamint az Általános bevezetés 7. és 8. pontjának rendelkezéseivel összhangban kell figyelembe venni.
P1: csak a CD86 tekintetében pozitív vizsgálatmenet; P2: csak a CD54 tekintetében pozitív vizsgálatmenet; P12: a CD54 és a CD86 tekintetében egyaránt pozitív vizsgálatmenet; N: sem a CD54, sem a CD86 tekintetében nem pozitív vizsgálatmenet.
*A keretekben az első két vizsgálatmenet eredményeinek releváns kombinációi szerepelnek, az eredmények sorrendjétől függetlenül.
#A keretek a három vizsgálatmenetből származó eredmények releváns kombinációit jelenítik meg az első két vizsgálatmenet fenti keretben látható eredményei alapján, nem tükrözik viszont az eredmények sorrendjét.
|
|
28.
|
A h-CLAT vizsgálat által pozitívként előrejelzett vizsgálati vegyi anyagok esetében opcionálisan két hatáserősséget jelző koncentrációérték (EC) állapítható meg, a CD86 esetében az EC150 és a CD54 esetében az EC200, vagyis az a koncentráció, amelynél a vizsgálati vegyi anyagok relatív fluoreszcencia-intenzitása 150, illetve 200 százalék. Ezek az EC-értékek hozzájárulhatnak a szenzibilizáló hatás erősségének értékeléséhez (9), ha az integrált vizsgálati és értékelési megközelítéshez hasonló integrált megközelítések keretében alkalmazzák őket (4) (5) (6) (7) (8). Az alábbi egyenletekkel lehet kiszámítani értéküket:
EC 150 (CD86 esetén) = Bconc
+ [(150 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
EC 200 (CD54 esetén) = Bconc
+ [(200 - BRFI
)/ARFI - BRFI
) × (Aconc - Bconc
)]
ahol
az Akonc az a μg/ml-ben megadott legalacsonyabb koncentráció, amelynél a relatív fluoreszcencia-intenzitás nagyobb mint 150 (CD86) vagy 200 (CD54)
a Bkonc az a μg/ml-ben megadott legmagasabb koncentráció, amelynél a relatív fluoreszcencia-intenzitás kisebb mint 150 (CD86) vagy 200 (CD54)
az ARFI az a legalacsonyabb koncentrációjú relatív fluoreszcencia-intenzitás, ahol a relatív fluoreszcencia-intenzitás nagyobb mint 150 (CD86) vagy 200 (CD54)
a BRFI az a legmagasabb koncentrációjú relatív fluoreszcencia-intenzitás, ahol a relatív fluoreszcencia-intenzitás kisebb mint 150 (CD86) vagy 200 (CD54)
Az EC150 és az EC200 érték pontosabb meghatározása érdekében szükség lehet arra, hogy a CD86/CD54 expresszió mérését három független vizsgálatmenetben végezzék el. Ez esetben a végső EC150 és EC200 értéket a három független vizsgálatmenetben kiszámolt EC-értékek középértékeként határozzák meg. Amikor három független vizsgálatmenetből csupán kettőben teljesülnek a pozitív előrejelzés kritériumai (lásd a 26–27. pontot), a két kiszámított EC150 vagy EC200 érték közül a magasabbat fogadják el.
|
Elfogadhatósági kritériumok
|
29.
|
A h-CLAT módszer alkalmazásakor az alábbi elfogadhatósági kritériumoknak kell teljesülniük (22) (27).
|
—
|
A tápoldatos és az oldószeres/vivőanyagos kontroll sejtéletképességének meg kell haladnia a 90 %-ot.
|
|
—
|
Az oldószeres/vivőanyagos kontrollban sem a CD86, sem a CD54 relatív fluoreszcencia-intenzitása nem haladhatja meg a pozitív besoroláshoz szükséges kritériumot (CD86 esetén RFI ≥ 150 % és CD54 esetén RFI ≥ 200 %). Az oldószeres/vivőanyagos kontroll relatív fluoreszcencia-intenzitását a 25. pontban szereplő képlet alapján kell kiszámolni (a „vegyi anyag MFI értéke” helyett az „oldószer/vivőanyag MFI értékét”, az „oldószer/vivőanyag MFI értéke” helyett pedig a „tápoldatos kontroll MFI értékét” kell használni).
|
|
—
|
Mind a tápoldatos kontroll, mind az oldószeres/vivőanyagos kontroll során a CD86 és a CD54 esetében megállapított fluoreszcencia-intenzitás mértani középértékének izotípusos kontrollhoz viszonyított aránya meg kell, hogy haladja a 105 %-ot.
|
|
—
|
A pozitív kontroll (DNCB) során a CD86 és a CD54 relatív fluoreszcencia-intenzitásának teljesítenie kell a pozitív besoroláshoz szükséges kritériumot (CD86 esetén RFI ≥ 150 és CD54 esetén RFI ≥ 200), 50 %-nál nagyobb sejtéletképesség mellett.
|
|
—
|
A vizsgálati vegyi anyaggal elért sejtéletképességnek minden vizsgálatmenetben legalább négy vizsgált koncentrációban meg kell haladnia az 50 %-ot.
|
|
|
30.
|
A negatív eredmény csak akkor elfogadható, ha a vizsgálati vegyi anyag legmagasabb vizsgált koncentrációja (vagyis a 20. pontban ismertetett oldatsorozat szerint az 1,2 × CV75) 90 %-nál alacsonyabb sejtéletképességet eredményez. Amennyiben az 1,2 × CV75 koncentráció által kiváltott sejtéletképesség eléri vagy meghaladja a 90 %-ot, a negatív eredményt figyelmen kívül kell hagyni. Ilyen esetben ajánlatos a CV75 újbóli meghatározásával pontosítani a dózisválasztást. Amennyiben azonban a vizsgálati vegyi anyag maximális vizsgált koncentrációja 5 000 μg/ml sóoldatban (illetve tápoldatban vagy más oldószerben/vivőanyagban), 1 000 μg/ml DMSO-ban vagy a legmagasabb még oldható koncentráció, a negatív eredmény 90 % fölötti sejtéletképesség esetén is elfogadható.
|
Vizsgálati jegyzőkönyv
|
31.
|
A vizsgálati jegyzőkönyvnek a következőket kell tartalmaznia:
|
Vizsgálati vegyi anyag
|
Egy összetevőből álló anyag
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, Log Kow, vízoldékonyság, DMSO-ban való oldhatóság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Több összetevőből álló anyagok, UVCB-k és keverékek
|
amennyiben lehetséges, az összetevők kémiai azonosítójával (lásd fent), tisztaságával, mennyiségi előfordulásával és releváns fizikai-kémiai tulajdonságaival (lásd fent) jellemezve; amennyiben rendelkezésre állnak;
|
|
fizikai megjelenés, vízoldékonyság, DMSO-ban való oldhatóság és a további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
molekulatömeg vagy látszólagos molekulatömeg ismert összetételű keverékek/polimerek esetén, vagy a vizsgálat lebonyolítása szempontjából releváns egyéb információ;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
|
Kontrollok
|
Pozitív kontroll
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, log Kow, vízoldékonyság, DSMO-ban való oldhatóság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak és szükség szerint;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
adott esetben hivatkozás a pozitív történeti kontrollok eredményeire, amelyek a vizsgálatmenet elfogadhatósági kritériumainak való megfelelőséget bizonyítják.
|
|
|
|
Negatív és oldószeres/vivőanyagos kontroll
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
fizikai megjelenés, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben a vizsgálati iránymutatásban említett kontroll oldószerektől/vivőanyagoktól eltérő oldószerek/vivőanyagok használatosak, és amennyiben rendelkezésre állnak;
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Vizsgálati körülmények
|
a megbízó, a vizsgálatot végző laboratórium és a vizsgálatvezető neve és címe;
|
|
az alkalmazott vizsgálat leírása;
|
|
használt sejtvonal, annak tárolási feltételei és eredete (például az a létesítmény, ahonnan beszerezték);
|
|
az alkalmazott áramlási citometria rendszer (pl. modell) és annak műszerbeállításai, valamint az alkalmazott globulin, ellenanyagok és citotoxicitási markerek;
|
|
a vizsgálat végrehajtásában való, a jártassági tesztanyagok vizsgálatával megszerzett laboratóriumi jártasság bizonyítására alkalmazott eljárás és a vizsgálat idővel reprodukálható teljesítményének bizonyítására alkalmazott eljárás, pl. a dokumentált kontrolladatok és/vagy a dokumentált reaktivitási vizsgálatok adatai.
|
|
|
A vizsgálat elfogadhatósági kritériumai
|
az oldószeres/vivőanyagos kontroll során kapott sejtéletképesség, MFI és RFI az elfogadhatósági tartományokkal való összevetésben;
|
|
a pozitív kontroll során kapott sejtéletképesség és RFI az elfogadhatósági tartományokkal való összevetésben;
|
|
a vizsgálati vegyi anyag minden vizsgált koncentrációjában mért sejtéletképesség.
|
|
|
A vizsgálat menete
|
az alkalmazott vizsgálatmenetek száma;
|
|
a vizsgálati vegyi anyag koncentrációi, alkalmazása és a használt expozíciós idő (ha eltér az ajánlottól);
|
|
az alkalmazott értékelési és döntési kritériumok leírása;
|
|
a vizsgálati eljárás bármilyen változtatásának leírása.
|
|
|
Eredmények
|
az egyes vizsgálatmenetekben a vizsgálati vegyi anyaggal és a pozitív kontrollal kapott adatok táblázatba foglalása, feltüntetve a CV75 értéket (ha alkalmazandó), a fluoreszcencia-intenzitás mértani középértékét, a relatív fluoreszcencia-intenzitást, a sejtéletképességi értékeket, az EC150/EC200 értéket (ha alkalmazandó), valamint a vizsgálati vegyi anyag előrejelzési modell szerinti besorolását;
|
|
adott esetben bármely más releváns észrevétel leírása.
|
|
|
Az eredmények tárgyalása
|
a h-CLAT módszerrel kapott eredmények tárgyalása;
|
|
a vizsgálat eredményeinek integrált vizsgálati és értékelési megközelítés keretében történő tárgyalása, amennyiben rendelkezésre állnak más releváns információk.
|
|
|
SZAKIRODALOM
|
(1)
|
Ashikaga T, Yoshida Y, Hirota M, Yoneyama K, Itagaki H, Sakaguchi H, Miyazawa M, Ito Y, Suzuki H, Toyoda H. (2006). Development of an in vitro skin sensitization test using human cell lines: The human Cell Line Activation Test (h-CLAT) I. Optimization of the h-CLAT protocol. Toxicol. In Vitro 20, 767-773.
|
|
(2)
|
Miyazawa M, Ito Y, Yoshida Y, Sakaguchi H, Suzuki H. (2007). Phenotypic alterations and cytokine production in THP-1 cells in response to allergens. Toxicol. In Vitro 21, 428-437.
|
|
(3)
|
EC EURL-ECVAM (2013). Recommendation on the human Cell Line Activation Test (h-CLAT) for skin sensitisation testing. A dokumentum az alábbi helyről elérhető:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(4)
|
Takenouchi O, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Hirota M, Ashikaga T, Miyazawa M. (2015). Test battery with the human cell line activation test, direct peptide reactivity assay and DEREK based on a 139 chemical data set for predicting skin sensitizing potential and potency of chemicals. J Appl Toxicol. 35, 1318-1332.
|
|
(5)
|
Hirota M, Fukui S, Okamoto K, Kurotani S, Imai N, Fujishiro M, Kyotani D, Kato Y, Kasahara T, Fujita M, Toyoda A, Sekiya D, Watanabe S, Seto H, Takenouchi O, Ashikaga T, Miyazawa M. (2015). Evaluation of combinations of in vitro sensitization test descriptors for the artificial neural network-based risk assessment model of skin sensitization. J Appl Toxicol. 35, 1333-1347.
|
|
(6)
|
Bauch C, Kolle SN, Ramirez T, Fabian E, Mehling A, Teubner W, van Ravenzwaay B, Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potencials. Regul Toxicol Parmacol. 63, 489-504.
|
|
(7)
|
Van der Veen JW, Rorije E, Emter R, Natch A, van Loveren H, Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol. 69, 371-379.
|
|
(8)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, Natsch A, Emter R, Ashikaga T, Miyazawa M, Sakaguchi H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Parmacol. 71, 337-351.
|
|
(9)
|
Jaworska JS, Natsch A, Ryan C, Strickland J, Ashikaga T, Miyazawa M. (2015). Bayesian integrated testing strategy (ITS) for skin sensitization potency assessment: a decision support system for quantitative weight of evidence and adaptive testing strategy. Arch Toxicol. 89, 2355-2383.
|
|
(10)
|
Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, Matheson J, Jacobs A, Lowit A, Allen D, Casey W. (2016). Integrated decision strategies for skin sensitization hazard. J Appl Toxicol. DOI 10.1002/jat.3281.
|
|
(11)
|
Nukada Y, Ashikaga T, Miyazawa M, Hirota M, Sakaguchi H, Sasa H, Nishiyama N. (2012). Prediction of skin sensitization potency of chemicals by human Cell Line Activation Test (h-CLAT) and an attempt at classifying skin sensitization potency. Toxicol. In Vitro 26, 1150-60.
|
|
(12)
|
EC EURL ECVAM (2015). Re-analysis of the within and between laboratory reproducibility of the human Cell Line Activation Test (h-CLAT). A dokumentum az alábbi helyről elérhető:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-human-cell-line-activation-test-h-clat-for-skin-sensitisation-testing
|
|
(13)
|
EC EURL ECVAM (2012). human Cell Line Activation Test (h-CLAT) Validation Study Report A dokumentum az alábbi helyről elérhető:https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations
|
|
(14)
|
Takenouchi O, Miyazawa M, Saito K, Ashikaga T, Sakaguchi H. (2013). Predictive performance of the human Cell Line Activation Test (h-CLAT) for lipophilic with high octanol-water partition coefficients. J. Toxicol. Sci. 38, 599-609.
|
|
(15)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, NishiyamaN, Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Altern. Lab. Anim. 38, 275-284.
|
|
(16)
|
Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch Toxicol 87, 1683-1969.
|
|
(17)
|
Okamoto K, Kato Y, Kosaka N, Mizuno M, Inaba H, Sono S, Ashikaga T, Nakamura T, Okamoto Y, Sakaguchi H, Kishi M, Kuwahara H, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (6th report): A study for evaluating oxidative hair dye sensitization potential using h-CLAT. AATEX 15, 81-88.
|
|
(18)
|
DB-ALM (INVITTOX) (2014). Protocol 158: human Cell Line Activation Test (h-CLAT), 23pp. A dokumentum az alábbi helyről elérhető:http://ecvam-dbalm.jrc.ec.europa.eu/
|
|
(19)
|
Mizuno M, Yoshida M, Kodama T, Kosaka N, Okamato K, Sono S, Yamada T, Hasegawa S, Ashikaga T, Kuwahara H, Sakaguchi H, Sato J, Ota N, Okamoto Y, Ohno Y. (2008). Effects of pre-culture conditions on the human Cell Line Activation Test (h-CLAT) results; Results of the 4th Japanese inter-laboratory study. AATEX 13, 70-82.
|
|
(20)
|
Sono S, Mizuno M, Kosaka N, Okamoto K, Kato Y, Inaba H, , Nakamura T, Kishi M, Kuwahara H, Sakaguchi H, Okamoto Y, Ashikaga T, Ohno Y. (2010). The Japanese ring study of a human Cell Line Activation Test (h-CLAT) for predicting skin sensitization potential (7th report): Evaluation of volatile, poorly soluble fragrance materials. AATEX 15, 89-96.
|
|
(21)
|
OECD (2005). Guidance Document No 34 on The Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Series on Testing and Assessment. Organization for Economic Cooperation and Development, Paris, France, 2005, 96 pp.
|
|
(22)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment No 168. Elérhető a következő címen:http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2012)10/PART1&docLanguage=En
|
|
(23)
|
United Nations UN (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Fifth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Elérhető a következő címen:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
|
|
(24)
|
ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals (Technical Report No 87).
|
|
(25)
|
Ashikaga T, Sakaguchi H, Okamoto K, Mizuno M, Sato J, Yamada T, Yoshida M, Ota N, Hasegawa S, Kodama T, Okamoto Y, Kuwahara H, Kosaka N, Sono S, Ohno Y. (2008). Assessment of the human Cell Line Activation Test (h-CLAT) for Skin Sensitization; Results of the First Japanese Inter-laboratory Study. AATEX 13, 27-35.
|
1.1. függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság
: a vizsgálattal kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálat megfelelőségének mutatója, és relevanciájának egyik szempontja. A kifejezés gyakran használatos az „egyezés” szó megfelelőjeként, amely arra utal, hogy egy adott vizsgálat milyen arányban szolgáltat helyes eredményeket (21).
Káros kimeneti út (Adverse Outcome Pathway, AOP)
: valamely megcélzott vegyi anyag vagy hasonló vegyi anyagok csoportjának kémiai szerkezetéből kiinduló, a molekuláris kiváltó eseményen keresztül az érintett in vivo eredményig végbemenő eseménysorozat (22).
Vegyi anyag
: anyag vagy keverék.
CV75
: 75 %-os sejtéletképességet eredményező becsült koncentráció.
EC150
: a CD86 expressziója során 150 %-os relatív fluoreszcencia-intenzitást eredményező koncentrációk.
EC200
: a CD54 expressziója során 200 %-os relatív fluoreszcencia-intenzitást eredményező koncentrációk.
Áramlási citometria
: citometrikus technika, amelyben a folyadékáramban szuszpendált sejtek egyenként áthaladnak egy fókuszált gerjesztő fénynyalábon, amely a sejtekre és összetevőire jellemző minták szerint szóródik szét; a sejteket gyakran fluoreszcens markerekkel jelölik meg, amelyek az abszorbeált fényt eltérő frekvenciákon bocsátják ki.
Veszély
: valamely anyag vagy helyzet azon sajátossága, hogy káros hatásokat képes előidézni az élő szervezet, rendszer vagy (al)populáció anyagnak való expozíciójakor.
Integrált vizsgálati és értékelési megközelítés
: valamely vegyi anyag vagy vegyianyag-csoport veszélyeinek azonosításához (potenciál), jellemzéséhez (hatáserősség) és/vagy biztonsági értékeléséhez (potenciál, hatáserősség és expozíció) alkalmazott strukturált megközelítés, amely stratégiai szempontból beépít és mérlegel minden olyan releváns adatot, amely hozzájárul a lehetséges veszélyre és/vagy kockázatra és/vagy a további célirányos és ezért minimális vizsgálat szükségességére vonatkozó szabályozói döntéshozatalhoz.
Tápoldatos kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó kezeletlen replikátum. Ezt a mintát a vizsgálati vegyi anyaggal kezelt mintákkal és más kontrollmintákkal együtt kell feldolgozni annak megállapításához, hogy az oldószer/vivőanyag kölcsönhatásba lép-e a vizsgálati rendszerrel.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő legalább 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
Pozitív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó, ismert pozitív hatást kiváltó anyaggal kezelt replikátum. Annak biztosítása érdekében, hogy a pozitív kontrollban jelentkező hatás időbeli változását fel lehessen mérni, a pozitív hatás nem lehet túlságosan erőteljes.
Pre-hapténok
: abiotikus átalakulás révén szenzibilizálóvá váló vegyi anyagok.
Pro-hapténok
: olyan vegyi anyagok, amelyek enzimatikus aktivációt igényelnek bőrszenzibilizáló hatásuk kifejtéséhez.
Relatív fluoreszcencia-intenzitás (RFI)
: a vegyi anyaggal kezelt sejteknél mért fluoreszcencia-intenzitás mértani középértékének (MFI) az oldószerrel/vivőanyaggal kezelt sejteknél mért fluoreszcencia-intenzitás mértani középértékéhez viszonyított relatív értéke.
Relevancia
: a vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálat pontosságát (az eredmények egyezését) figyelembe kell venni (21).
Megbízhatóság
: a vizsgálat laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. Megállapítása a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság, valamint a laboratóriumon belüli megismételhetőség kiszámításával történik (21).
Vizsgálatmenet
: egy vizsgálatmenet során egy vagy több vizsgálati vegyi anyagot vizsgálnak egy párhuzamos oldószeres/vivőanyagos kontrollal és egy pozitív kontrollal.
Érzékenység
: az összes olyan pozitív/aktív vegyi anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálat pontosságának mutatója, valamint fontos szempont a vizsgálat relevanciájának megítélésében (21).
Festő puffer
: 0,1 % borjú szérum albuminnal kiegészített foszfátpuffert tartalmazó sóoldat.
Oldószeres/vivőanyagos kontroll
: valamely vizsgálati rendszernek a vizsgálati vegyi anyagon kívüli összes összetevőjét tartalmazó kezeletlen minta, amely azonban magában foglalja az alkalmazott oldószert/vivőanyagot. Az ugyanabban az oldószerben/vivőanyagban feloldott vagy stabilan diszpergált vizsgálati vegyi anyaggal kezelt minták esetében a kiindulási érték létrehozásához használatos. Párhuzamos tápoldatos kontrollal elvégzett vizsgálata esetén ez a minta azt is bizonyítja, hogy az oldószer/vivőanyag kölcsönhatásba lép-e a vizsgálati rendszerrel.
Specificitás
: a vizsgálat elvégzésével helyesen besorolt összes negatív/inaktív vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (21).
Anyag
: olyan természetes állapotban előforduló vagy gyártási eljárásból származó kémiai elem és vegyületei, amely a stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott eljárásból származó szennyező anyagokat is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető.
Vizsgálati vegyi anyag
: bármely, e módszer alkalmazásával vizsgált anyag vagy keverék.
A vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (ENSZ-GHS)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (23).
UVCB
: ismeretlen szerkezetű vagy változó összetételű, összetett reakcióban keletkezett vagy biológiai eredetű anyagok.
Érvényes vizsgálat
: olyan vizsgálat, amely kellően relevánsnak és megbízhatónak minősül egy adott célra, és amely tudományosan megalapozott elveken alapul. Egy-egy vizsgálat abszolút értelemben soha nem érvényes, kizárólag meghatározott céllal összefüggésben (21).
1.2. függelék
JÁRTASSÁGI TESZTANYAGOK
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak igazolniuk kell szakmai jártasságukat oly módon, hogy az 1. táblázatban javasolt 10 anyagra vonatkozóan a h-CLAT vizsgálattal várható előrejelzést helyesen kapják meg, továbbá a 10 jártassági tesztanyag közül legalább 8 esetében referenciatartományba eső CV75, EC150 és EC200 értékeket kapnak. A jártassági tesztanyagok kiválasztása oly módon történt, hogy az a bőrszenzibilizáció veszélye esetén bekövetkező különféle válaszreakciók tartományát illetően reprezentatív legyen. A kiválasztás többi kritériuma az volt, hogy a vegyi anyagok kereskedelmi forgalomban beszerezhetők legyenek, azokra vonatkozóan kiváló minőségű in vivo referenciaadatok álljanak rendelkezésre, és a h-CLAT módszer alapján kiváló minőségű in vitro adatok álljanak rendelkezésre. A h-CLAT módszerhez rendelkezésre állnak publikált referenciaadatok is (3) (14).
1. táblázat
A h-CLAT módszerben való szakmai jártasság bizonyításához ajánlott anyagok
|
Jártassági tesztanyagok
|
CAS-szám
|
Halmazállapot
|
In vivo előrejelzés (103)
|
CV75 referencia-tartomány μ g/ml-ben (104)
|
h-CLAT eredmények CD86 esetén (EC150 referencia-tartomány μg/ml-ben) (104)
|
h-CLAT eredmények CD54 esetén (EC200 referencia-tartomány μg/ml-ben) (104)
|
|
2,4-dinitro-klór-benzol
|
97-00-7
|
Szilárd
|
Szenzibilizáló (szélsőségesen)
|
2-12
|
Pozitív (0,5-10)
|
Pozitív (0,5-15)
|
|
4-Fenilén-diamin
|
106-50-3
|
Szilárd
|
Szenzibilizáló (erősen)
|
5-95
|
Pozitív (<40)
|
Negatív (>1,5) (105)
|
|
Nikkel-szulfát
|
10101-97-0
|
Szilárd
|
Szenzibilizáló (mérsékelten)
|
30-500
|
Pozitív (<100)
|
Pozitív (10–100)
|
|
2-merkapto-benzotiazol
|
149-30-4
|
Szilárd
|
Szenzibilizáló (mérsékelten)
|
30-400
|
Negatív (>10) (105)
|
Pozitív (10-140)
|
|
R(+)-limonén
|
5989-27-5
|
Folyadék
|
Szenzibilizáló (enyhén)
|
>20
|
Negatív (>5) (105)
|
Pozitív (<250)
|
|
Imidazolidinil-urea
|
39236-46-9
|
Szilárd
|
Szenzibilizáló (enyhén)
|
25–100
|
Pozitív (20-90)
|
Pozitív (20-75)
|
|
Izopropanol
|
67-63-0
|
Folyadék
|
Nem szenzibilizáló
|
>5 000
|
Negatív (>5 000 )
|
Negatív (>5 000 )
|
|
Glicerin
|
56-81-5
|
Folyadék
|
Nem szenzibilizáló
|
>5 000
|
Negatív (>5 000 )
|
Negatív (>5 000 )
|
|
Tejsav
|
50-21-5
|
Folyadék
|
Nem szenzibilizáló
|
1500-5 000
|
Negatív (>5 000 )
|
Negatív (>5 000 )
|
|
4-Aminobezoesav
|
150-13-0
|
Szilárd
|
Nem szenzibilizáló
|
> 1 000
|
Negatív (>1 000 )
|
Negatív (>1 000 )
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám.
|
2. függelék
IN VITRO BŐRSZENZIBILIZÁCIÓ: U937 SEJTVONAL AKTIVÁLÁSI VIZSGÁLAT (U-SENS™)
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
1.
|
Az U-SENS™ vizsgálat mennyiségileg meghatározza az U937 humán hisztiocitikus limfóma sejtvonal monocitáinak és dendritikus sejtjeinek a szenzibilizáló anyagokkal való kezelést követően beinduló aktivációs folyamata által a sejtfelületi marker (CD86) expressziójában előidézett változást (1). A CD86 sejtfelületi marker U937 sejtvonalban mért expressziós szintjét felhasználják a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetésének alátámasztására.
|
|
2.
|
Az U-SENS™ vizsgálatot egy, a L’Oréal által koordinált validálási vizsgálatban (2) értékelték, majd az állatkísérletek alternatív módszereinek uniós referencialaboratóriumának (EURL ECVAM) tudományos tanácsadó bizottsága (ESAC) elvégezte a vizsgálat független szakértői értékelését (3). A rendelkezésre álló bizonyítékok, valamint a szabályozó hatóságok és az érdekeltek álláspontja alapján az U-SENS™ vizsgálatot az EURL ECVAM (4) integrált vizsgálati és értékelési megközelítés keretében történő alkalmazásra ajánlotta, a bőrszenzibilizáló és nem szenzibilizáló anyagok veszélyességi osztályozás és címkézés céljából történő megkülönböztetésének alátámasztására. A bőrszenzibilizációs integrált vizsgálati és értékelési megközelítés keretében alkalmazott adatintegrálás és egyedi információforrások strukturált megközelítéseire vonatkozó jelentéstételről szóló iránymutatásában az OECD beszámol számos, különböző vizsgálati stratégiát és előrejelzési modellt ismertető esettanulmányról. A különféle kidolgozott megközelítések egyike az U-SENS™ vizsgálaton alapul (5). Az U-SENS™ alkalmazásából származó adatok és más információk, egyebek mellett a dokumentált adatok és az ismert hiteles humán adatok (6) együttes használatára a szakirodalom hoz példákat (4) (5) (7).
|
|
3.
|
Az U-SENS™ vizsgálatról bebizonyosodott, hogy áthelyezhető a sejttenyésztési technikák és az áramlási citometriás elemzés terén tapasztalattal rendelkező laboratóriumokba. A vizsgálattól várható előrejelzések reprodukálhatóságának mértéke laboratóriumon belül mintegy 90 %, laboratóriumok között pedig 84 % (8). A validálási vizsgálatban (8) és más közzétett tanulmányokban (1) kapott eredmények összességében azt jelzik, hogy az U-SENS™ a bőrszenzibilizáló (azaz az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) anyagokat az LLNA vizsgálati módszer eredményeihez képest 86 %-os (N=166) pontossággal különbözteti meg a nem szenzibilizáló anyagoktól, 91 % az érzékenysége (118/129), a specifikussága pedig 65 % (24/37). A humán vizsgálatokból származó eredményekkel összevetve a bőrszenzibilizáló (vagyis az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) anyagokat 77 %-os (N=101) pontossággal különbözteti meg a nem szenzibilizáló anyagoktól, 100 % az érzékenysége (58/58), míg a specifikussága 47 % (20/43). Az LLNA módszerrel összevetve az U-SENS™ nagyobb valószínűséggel ad hamis negatív előrejelzést olyan vegyi anyagoknál, amelyeknek alacsonytól mérsékeltig terjedő a szenzibilizáló hatása (azaz az ENSZ-GHS/CLP-rendelet szerinti 1B. alkategóriába tartozó anyagoknál), mint a rendkívül bőrszenzibilizáló hatást mutató vegyi anyagoknál (azaz az ENSZ-GHS/CLP-rendelet szerinti 1A. alkategóriába tartozó anyagoknál) (1) (8) (9). Ezek az információk összességében azt jelzik, hogy az U-SENS™ vizsgálat érdemben hozzájárul a bőrszenzibilizáció veszélyének azonosításához. Az U-SENS™ mint önálló vizsgálat esetében itt megadott pontossági értékek azonban csak irányadóak, ugyanis a vizsgálatot az integrált vizsgálati és értékelési megközelítés keretében más információforrásokkal együtt és az Általános bevezetés 7. és 8. pontjának rendelkezéseivel összhangban kell figyelembe venni. Ezen túlmenően a bőrszenzibilizáció nem állatokon alkalmazott módszereinek értékelésekor nem szabad megfeledkezni arról, hogy az LLNA vizsgálat, valamint más állatkísérletek nem feltétlenül tükrözik teljes egészében az embernél fennálló helyzetet.
|
|
4.
|
A jelenleg rendelkezésre álló adatok alapján az U-SENS™ vizsgálatról megállapították, hogy alkalmazható különféle szerves funkciós csoportokat, fizikai-kémiai tulajdonságokat, (in vivo vizsgálatok során meghatározott) bőrszenzibilizáló hatásokat és a bőrszenzibilizációval összefüggésbe hozható reakciómechanizmusok spektrumát (Michael-akceptor, Schiff-bázis képződés, aciltranszfer ágens, bimolekuláris nukleofil szubsztitúció [SN2] vagy aromás nukleofil szubsztitúció [SNAr]) képviselő vizsgálati vegyi anyagokra (ezen belül kozmetikai összetevőkre, pl. tartósítószerekre, felületaktív anyagokra, hatóanyagokra, festékekre) (1) (8) (9) (10). Az U-SENS™ vizsgálat olyan vizsgálati vegyi anyagokra alkalmazandó, amelyek megfelelő oldószerben/vivőanyagban (lásd a 13. pontot) oldhatók vagy azokkal stabil diszperziót alkotnak (azaz olyan kolloidot vagy szuszpenziót, amelyben a vizsgálati vegyi anyag nem ülepedik le, illetve nem válik szét az oldószertől/vivőanyagtól különböző fázisokra). Az U-SENS™ megfelelően előrejelezte azon vegyi anyagok besorolását is, amelyek az adatbázisban pre-hapténokként (oxidációval aktiválódó anyagokként) vagy pro-hapténokként (például a P450 enzim közreműködésével enzimatikus aktivációt igénylő anyagokként) tartottak nyilván (1) (10).A membránt károsító anyagok viszont álpozitív eredményre vezethetnek a CD86 expressziójának nem specifikus növelése miatt, mivel az in vivo referenciabesoroláshoz képest kapott 7 álpozitív eredményből 3 felületaktív anyagnál fordult elő (1). Bár a felületaktív anyagokkal elért pozitív eredményeket körültekintően kell mérlegelni, a felületaktív anyagok negatív eredménye használható a vizsgálati vegyi anyag nem bőrszenzibilizáló anyagként való azonosításának alátámasztására. Az U-SENS™ vizsgálat alkalmas fluoreszcens vizsgálati vegyi anyagok értékelésére (1), mindazonáltal azok az erősen fluoreszcens vizsgálati vegyi anyagok, amelyek a fluoreszcein-izotiocianáttal (FITC), illetve a propídium-jodiddal (PI) azonos hullámhosszon bocsátják ki a fényt, zavarják az áramlási citometriás észlelést, ezért FITC-vel összekapcsolt ellenanyagokkal (potenciális hamis negatív eredmény), illetve propídium-jodiddal (nem mérhető életképesség) nem értékelhetők megfelelően. Ilyen esetben használhatók más fluorokrómmal jelzett ellenanyagok vagy más citotoxicitási markerek, ha például a 2.2. függelékben szereplő jártassági tesztanyagok vizsgálata révén bizonyítást nyert, hogy a FITC-vel jelzett ellenanyagokhoz vagy a propídium-jodidhoz (lásd a 18. pontot) hasonló eredményt biztosítanak. A fentiek fényében: a felületaktív anyagokkal kapott pozitív eredményeket és az erősen fluoreszcens vizsgálati vegyi anyagokkal elért negatív eredményeket a közölt korlátokra tekintettel és az integrált vizsgálati és értékelési megközelítés keretébe tartozó egyéb információforrásokkal összefüggésben kell értelmezni. Olyan esetekben, amikor bizonyítékkal igazolható, hogy az U-SENS™ vizsgálat nem alkalmazható más, konkrét vegyianyag-kategóriákra, a vizsgálatot nem szabad e vegyianyag-kategóriákra alkalmazni.
|
|
5.
|
A fentieknek megfelelően az U-SENS™ vizsgálat elősegíti a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetését. Hozzájárulhat azonban a szenzibilizáló hatás erősségének értékeléséhez is, ha az integrált vizsgálati és értékelési megközelítéshez hasonló integrált megközelítések keretében alkalmazzák. Lehetőleg humán adatokon alapuló további munkára van azonban szükség annak meghatározásához, hogy az U-SENS™ eredményei hogyan járulhatnak hozzá a hatáserősség vizsgálatához.
|
|
6.
|
A fogalommeghatározások a 2.1. függelékben találhatók.
|
A Vizsgálat Elve
|
7.
|
Az U-SENS™ vizsgálat egy in vitro eljárás, amely a vizsgálati vegyi anyaggal való 45 ±3 órás kezelést követően mennyiségileg meghatározza a CD86 sejtfelületi markernek egy humán hisztiocitikus limfóma sejtvonal, az U937 sejtekben történő kifejeződésében előidézett változásokat. A CD86 sejtfelületi marker az U937 sejtek aktivációjának egyik jellegzetes markere. A CD86 egy ismert kostimuláló molekula, amely képes imitálni a monociták aktivációját, ami kulcsszerepet játszik a T-sejtek elsődleges aktiválásában. A CD86 sejtfelületi marker expressziójának változásait áramlási citometriával mérik, miután a sejteket megfestették, többnyire fluoreszcein-izotiocianáttal (FITC) jelzett ellenanyagokkal. Ezzel egyidejűleg citotoxicitás-méréseket is végeznek (például propídium-jodiddal) annak felméréséhez, hogy a citotoxikus koncentrációknál alacsonyabb koncentráció mellett fokozódik-e a CD86 felületi marker expressziója. Meghatározzák a CD86 felületi marker oldószeres/vivőanyagos kontrollhoz viszonyított stimulációs indexét (S.I.), amit felhasználnak az előrejelzési modellben (lásd a 19. pontot) a bőrszenzibilizáló és nem szenzibilizáló anyagok megkülönböztetésének alátámasztására.
|
A Jártasság Bizonyítása
|
8.
|
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak a 2.2. függelékben felsorolt tíz jártassági tesztanyag in vitro módszerekre vonatkozó bevált gyakorlatokkal összhangban történő felhasználásával igazolniuk kell szakmai jártasságukat (11). Ezenfelül a vizsgálat felhasználóinak történeti adatbázist kell fenntartaniuk, amely tartalmazza a reaktivitási tesztekből (lásd a 11. pontot), valamint a pozitív és az oldószeres/vivőanyagos kontrollokból (lásd a 15–16. pontot) származó adatokat, ezekkel az adatokkal igazolva a vizsgálat laboratóriumon belüli hosszú távú reprodukálhatóságát.
|
Eljárás
|
9.
|
Ez a vizsgálat az állatkísérletek alternatív módszereire vonatkozó adatbázis-szolgáltatás (DB-ALM) U-SENS™-t érintő 183. protokollján (12) alapul. Az U-SENS™ vizsgálat laboratóriumi végrehajtása és igénybevétele során alkalmazni kell a szabványműveleti eljárásokat. Az U-SENS™ vizsgálat automatizált rendszerrel is elvégezhető, ha például a 2.2. függelékben felsorolt jártassági tesztanyagok vizsgálatával kimutatták, hogy hasonló eredményekre vezet. Az alábbiakban az U-SENS™ főbb összetevőinek és eljárásainak leírása található.
|
Sejtek előkészítése
|
10.
|
Az U-SENS™ vizsgálat végrehajtásához az U937 humán hisztiocitikus limfóma sejtvonalat (13) kell használni. Ajánlatos a (CRL1593.2 klón) sejteket egy megfelelő minősítéssel rendelkező sejtbankból, például az Amerikai Fajtakultúra Gyűjteményből beszerezni.
|
|
11.
|
Az U937 sejteket 10 % magzati borjú szérummal (FCS), 2 mM L-glutaminnal, 100 egység/ml penicillinnel és 100 μg/ml sztreptomicinnel kiegészített RPMI-1 640 tápoldatban (teljes tápoldat), valamint 37 °C-os, 5 %-os CO2-tartalmú és vízgőzzel telített környezetben kell tenyészteni. Az U937 sejteket 2 vagy 3 naponta 1,5 vagy 3 × 105 sejt/ml sűrűségben rutinszerűen passzálják. A sejtsűrűség nem lépheti túl a 2 × 106 sejt/ml mennyiséget, a tripankék festékkizárásos teszttel mért sejtéletképességnek pedig legalább 90 %-osnak kell lennie (ez a felolvasztást követő első passzálásra nem vonatkozik). A vizsgálatban való felhasználásuk előtt a sejtek, a magzati borjú szérum és az ellenanyagok minden tételét egy reaktivitási teszt során minősíteni kell. A sejtek reaktivitási tesztjét a pozitív kontrollanyaggal, a pikrilszulfonsavval (2,4,6-trinitro-benzol-szulfonsav: TNBS) (CAS-száma: 2 508-19-2, legalább 99 %-os tisztaságban) és a negatív kontrollanyaggal, a tejsavval (LA) (CAS-száma: 50-21-5, legalább 85 %-os tisztaságban) kell végrehajtani, legalább egy héttel a felolvasztásukat követően. A reaktivitási tesztben mindkét kontrollanyag esetén hat végső koncentrációt kell tesztelni (TNBS: 1, 12,5, 25, 50, 75, 100 μg/ml és tejsav: 1, 10, 20, 50, 100, 200 μg/ml). A teljes tápoldatban feloldott TNBS-nak a CD86 pozitív és koncentrációfüggő válaszát kell előidéznie (pl. amikor egy pozitív eredményt, vagyis legalább 150-es CD86 S.I. értéket adó koncentrációt követő koncentrációérték a CD86 ennél magasabb S.I. értékét váltja ki), a teljes tápoldatban feloldott tejsavnak pedig a CD86 negatív válaszát kell előidéznie (lásd a 21. pontot). A vizsgálathoz kizárólag a reaktivitási tesztben 2 alkalommal megfelelt sejttételek használhatók. A sejtek a felolvasztásukat követően hét hétig szaporíthatók. A sejteket legfeljebb 21 alkalommal szabad passzálni. A reaktivitási tesztet a 18-22. pontban leírt eljárások szerint kell végrehajtani.
|
|
12.
|
A vizsgálathoz az U937 sejteket vagy 3 × 105 sejt/ml vagy 6 × 105 sejt/ml sűrűségben átoltják, majd sejttenyésztő lombikban az előbbi esetben 2 napig, az utóbbiban pedig 1 napig előtenyésztik. A fentiektől eltérő előtenyésztési körülmények is alkalmazhatók, ha megfelelő tudományos indokolással támasztják alá alkalmazásukat, és ha például a 2.2. függelékben felsorolt jártassági tesztanyagok vizsgálatával kimutatták, hogy hasonló eredményekhez vezetnek. A vizsgálat napján a sejttenyésztő lombikból begyűjtött sejteket frissen készített tápfolyadékban 5 × 105 sejt/ml sűrűségben újraszuszpendálják. Ezt követően a sejteket egy 96 lyukú, lapos fenekű lemezre helyezik 100 μl mennyiségben (lyukanként 0,5 × 105 sejt végső sejtsűrűségben).
|
A vizsgálati vegyi anyagok és a kontrollanyagok előkészítése
|
13.
|
A vizsgálat előtt felmérik az anyagok oldhatóságát. Ebből a célból a vizsgálati vegyi anyagokat 50 mg/ml koncentrációban feloldják vagy stabilan diszpergálják teljes tápoldatban, mint az első számú oldószer választás, vagy dimetil-szulfoxidban (DMSO, ≥ 99 %-os tisztaságban), mint második oldószer/vivőanyag választás, ha a vizsgálati vegyi anyag a teljes tápoldatos oldószerben/vivőanyagban nem oldható. A vizsgálati vegyi anyagot a vizsgálathoz 0,4 mg/ml végső koncentrációban oldják fel a teljes tápoldatban, amennyiben a vegyi anyag oldható ebben az oldószerben/vivőanyagban. Ha a vegyi anyag csak DMSO-ban oldható, akkor 50 mg/ml-es koncentrációban oldják fel. A fentiektől eltérő oldószerek/vivőanyagok megfelelő tudományos indokolással szintén használhatók. Figyelembe kell venni azt is, hogy a vizsgálati vegyi anyag mennyire stabil a végső oldószerben/vivőanyagban.
|
|
14.
|
A vizsgálati vegyi anyagokat és a kontrollanyagokat a vizsgálat napján kell előkészíteni. Mivel dózisbehatároló vizsgálatra nem kerül sor, az első vizsgálatmenetben 6 végső koncentrációt kell vizsgálni (1, 10, 20, 50, 100 és 200 μg/ml) a megfelelő oldószerben/vivőanyagban, amely vagy a teljes tápoldat, vagy 0,4 % DMSO-t tartalmazó tápoldat. A további vizsgálatmenetekben a megfelelő oldószerben/vivőanyagban legalább 4 munkaoldatot (vagyis legalább 4 koncentrációt) kell készíteni a vizsgálati vegyi anyagokból 0,4 mg/ml-től kezdve teljes tápoldat használata esetén, vagy 50 mg/ml-től kezdve DMSO használata esetén. A munkaoldatokat végül felhasználják a kezeléshez, ehhez a további kétszeres hígítás (12) érdekében a lemezbe helyezett munkaoldat térfogatával megegyező térfogatú U937 sejtszuszpenziót (lásd a fenti 11. pontot) adagolnak a munkaoldatokhoz. Az esetleges további vizsgálatmenetekben alkalmazott legalább 4 koncentrációt az előző vizsgálatmenetek eredményei alapján határozzák meg (8). A következő végső koncentrációértékek használhatók: 1, 2, 3, 4, 5, 7,5, 10, 12,5, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 120, 140, 160, 180 és 200 μg/ml. A maximális végső koncentráció a 200 μg/ml. Amennyiben a CD86 már 1 μg/ml koncentrációnál pozitív választ eredményez, akkor a 0,1 μg/ml koncentráció értékelésével megkeresik a vizsgálati vegyi anyag azon koncentrációját, amely nem indukál pozitív küszöbérték feletti CD86 kifejeződést. Ha a CD86 kifejeződés során pozitív koncentráció-válasz figyelhető meg, minden vizsgálatmenetben ki kell számolni az EC150 értéket (ez a vizsgálati vegyi anyag azon koncentrációja, amelynél a CD86 eléri a pozitív besoroláshoz szükséges 150 %-os küszöbértéket, lásd a 19. pontot). Amikor a vizsgálati vegyi anyag által előidézett pozitív CD86 válasz nem hozható összefüggésbe a koncentrációval, akkor előfordulhat, hogy az U-SENS™-re vonatkozó 183. sz. DB-ALM protokollban leírtak szerint (12) az EC150 érték a vizsgálat szempontjából nem releváns. Amikor csak lehetséges, minden vizsgálatmenetben meg kell határozni CV70 értéket (12) (ez a vizsgálati vegyi anyag azon koncentrációja, amelynél a citotoxicitás eléri a 70 %-os küszöbértéket, lásd a 19. pontot). Annak megállapításához, hogy a CD86 expresszió növekedése összefüggésben áll-e a koncentráció növekedésével, a használható koncentrációk közül kiválasztott koncentrációértékeknek egyenletesen kell eloszlaniuk az EC150 (vagy a legmagasabb, még negatív eredményhez vezető, nem citotoxikus koncentráció) és a CV70 (vagy a megengedett legmagasabb koncentráció, vagyis 200 μg/ml) között. Vizsgálatmenetenként legalább 4 koncentrációt kell vizsgálni, amelyek közül legalább kettőnek meg kell egyeznie az előző vizsgálatmenet(ek)ben alkalmazott koncentrációkkal, hogy összehasonlíthatók legyenek az eredmények.
|
|
15.
|
Az U-SENS™ vizsgálatban alkalmazott oldószeres/vivőanyagos kontroll a teljes tápoldat (a teljes tápoldatban feloldott vagy stabilan diszpergált vizsgálati vegyi anyagok esetében) (lásd a 4. pontot) vagy a 0,4 % DMSO-t tartalmazó teljes tápoldat (a DMSO-ban feloldott vagy stabilan diszpergált vizsgálati vegyi anyagok esetében).
|
|
16.
|
Az U-SENS™ vizsgálat során használt pozitív kontrollanyag a TNBS (lásd a 11. pontot), amelyet teljes tápoldatban készítenek el. A CD86 expresszió mérésénél pozitív kontrollanyagként használt TNBS-t egyetlen végső (50 μg/ml) koncentrációban vizsgálják a lemezen, amely 70 % feletti sejtéletképességet biztosít. Ahhoz, hogy 50 μg/ml koncentrációjú TNBS kerüljön a lemezre, teljes tápoldatban elkészítik a TNBS 1 M (azaz 293 mg/ml) koncentrációjú törzsoldatát, majd a törzsoldatot teljes tápoldattal a 2 930-szorosára hígítva elkészítik a 100 μg/ml koncentrációjú munkaoldatot. Negatív kontrollanyagként tejsavat (LA, CAS-száma: 50-21-5) kell alkalmazni, teljes tápoldatban 200 μg/ml koncentrációban feloldva (amelyet 0,4 mg/ml koncentrációjú törzsoldatból készítenek el). Minden vizsgálatmenet valamennyi lemezén a teljes tápoldatból álló kezeletlen kontrollt, az oldószeres/vivőanyagos kontrollt, a negatív és a pozitív kontrollt egyaránt triplikátumban kell elkészíteni (12). Alkalmazhatók más, megfelelő pozitív kontrollok is, ha rendelkezésre állnak dokumentált adatok hasonló vizsgálatmenet-elfogadhatósági kritériumok származtatásához. A vizsgálatmenet elfogadhatósági kritériumai megegyeznek a vizsgálati vegyi anyag elfogadhatósági kritériumaival (lásd a 12. pontot).
|
A vizsgálati vegyi anyagok és a kontrollanyagok alkalmazása
|
17.
|
A 14–16. pontban leírt módon elkészített oldószeres/vivőanyagos kontrollt vagy munkaoldatokat 1: 1 térfogatszázalék arányban összekeverik a 96 lyukú lapos fenekű lemezen elkészített sejtszuszpenziókkal (lásd a 12. pontot). A kezelt lemezeket ezt követően 45 óráig (±3 óra) 37 °C-on 5 % CO2 jelenlétében inkubálják. Inkubálás előtt a lemezeket félig áteresztő membránnal lezárják, hogy megelőzzék az illékony vizsgálati vegyi anyagok elpárolgását és a vizsgálati vegyi anyagokkal kezelt sejtek közötti keresztszennyeződés kialakulását (12).
|
Sejtfestés
|
18.
|
A 45 órás (±3 óra) inkubálást követően a sejteket V-aljú mikrotiterlemezekbe helyezik át és centrifugálással kinyerik őket. A vizsgálat kimenetelét zavaró oldhatósági problémát jelez, ha a 45 ±3 órás kezelés utáni inkubálást követően (még sejtfestés előtt) mikroszkóp alatt kristályok vagy cseppek figyelhetők meg. A felülúszót eltávolítják, a maradék sejteket pedig egyszer átmossák 100 μl jéghideg, 5 % magzati borjú szérummal kiegészített, foszfátpuffert tartalmazó sóoldattal (festő pufferrel). A centrifugálást követően a sejteket 100 μl festő pufferben újraszuszpendálják, majd 30 percen keresztül, 4 °C-on, fénytől védve megfestik 5 μl (pl. 0,25 μg) FITC-vel jelzett anti-CD86 vagy egér IgG1 (izotípus) ellenanyagokkal. Az U-SENS™-re vonatkozó 183. sz. DB-ALM protokollban (12) leírt ellenanyagokat kell használni (a CD86 esetében: BD-PharMingen #5556 57 Fun-1 klón vagy Caltag/Invitrogen #MHCD8601 BU63 klón; az IgG1 esetében: BD-PharMingen #5557 48 vagy Caltag/Invitrogen #GM4992). A vizsgálat kidolgozói által szerzett tapasztalatok arra utalnak, hogy az ellenanyagok fluoreszcencia-intenzitása a különböző tételeknél általában konzisztens. A vizsgálathoz használhatók a reaktivitási tesztben megfelelt egyéb ellenanyag klónok vagy forgalmazók is (lásd a 11. pontot). A legmegfelelőbb vizsgálati koncentráció meghatározásához azonban a felhasználók fontolóra vehetik az ellenanyagok saját laboratóriumi körülményeik mellett történő titrálását. Más detektálási rendszerek, többek között fluorokrómmal jelzett anti-CD86 ellenanyagok is alkalmazhatók, amennyiben például a 2.2. függelékben felsorolt jártassági tesztanyagok tesztelésével kimutatták, hogy a FITC-vel összekapcsolt ellenanyagokhoz hasonló eredményekre vezetnek. Miután 100 μl festő pufferrel kétszer, 100 μl jéghideg PBS-sel egyszer átmosták a sejteket, újraszuszpendálják őket jéghideg PBS-ben (pl. 125 μl-ben, ha a mintákat manuálisan, csövenként elemzik, vagy 50 μl-ben, ha automatikus mintavevő lemezt használnak), majd propídium-jodid oldatot adnak hozzájuk (3 μg/ml végső koncentrációban). Más citotoxicitási markerek – például 7-amino-aktinomicin D (7-AAD) vagy tripankék – is alkalmazhatók, amennyiben az alternatívaként használt festékről például a 2.2. függelékben felsorolt jártassági tesztanyagok vizsgálatával kimutatták, hogy a propídium-jodidhoz hasonló eredményekre vezet.
|
Áramlási citometria elemzés
|
19.
|
A CD86 expressziójának mértékét és a sejtéletképességet áramlási citometriával elemzik. A sejteket a méretük (FSC, előrefelé irányuló szórás) és granuláltságuk (SCC, oldalra irányuló szórás) alapján felhődiagramon (logaritmikus skálán) ábrázolják, amely lehetővé teszi az első kapun (R1) kiválasztott sejtpopuláció egyértelmű azonosítását és a sejttöredékek eltávolítását. Lyukanként összesen 10 000 célsejt adatait gyűjtik össze az R1 kapuzás során. Az azonos R1 kapunál mért sejteket FL3 vagy FL4/SSC arányt vizsgáló felhődiagramon jelenítik meg. Az élő sejteket egy második, R2 kapu elhelyezésével különítik el, amelynek segítségével kiválasztják a propídium-jodid negatív sejtpopulációt (FL3 vagy FL4 csatorna). A sejtéletképességet az alábbi egyenlet alapján kalkulálja ki a citométer elemzőprogramja. Alacsony sejtéletképességnél az elhalt sejtekkel együtt akár 20 000 sejt adatait is összegyűjthetik. Ennek alternatívája lehet az, amikor az adatokat az elemzés elindításától számítva egy percen át gyűjtik.

Ezt követően megmérik az R2 kapuval kiválasztott élő sejtek közötti FL1 pozitív sejtek arányát (R1-en belül). Az élő sejteken (R2) a CD86 sejtfelületi kifejeződését FL1/SSC arányt vizsgáló felhődiagrammal elemzik.
A teljes tápoldatot/IgG1-et tartalmazó lyukak esetében az elemző markert a fő sejtpopuláció közelébe helyezik, hogy a teljes tápoldat kontrollok IgG1 ellenanyagai a 0,6–0,9 %-os céltartományba essenek.
A színinterferenciát az FITC-vel jelzett IgG1 felhődiagram eltolódása jelzi (IgG1 FL1 S.I. mértani középértéke ≥ 150 %).
A kezeletlen vagy 0,4 %-os DMSO-val kezelt kontrollsejtek és a vegyi anyaggal kezelt sejtek CD86 expressziójának stimulációs indexét (S.I.) az alábbi egyenlettel számítják ki:.

A kezeletlen kontrollsejtek IgG1+ aránya: az elemző markerrel jelölt FL1 pozitív IgG1 sejtek aránya (elfogadhatósági tartomány: legalább 0,6 % és kisebb mint 1,5 %, lásd a 22. pontot) az élő kezeletlen sejtekhez képest.
A kontroll/kezelt sejtek IgG1+/CD86+ aránya: az FL1 pozitív IgG1/CD86 sejtek elemző marker áthelyezése nélkül mért aránya az élő kontroll/kezelt sejtekhez képest.
|
Adatok És Jelentés
Az adatok kiértékelése
|
20.
|
Az U-SENS™ vizsgálatban az alábbi paramétereket kell kiszámítani: CV70 érték, vagyis az a koncentráció, amely az U937 sejtek 70 %-os túlélését eredményezi (30 %-os citotoxicitás) és az EC150 érték, vagyis az a koncentráció, amelynél a vizsgálati vegyi anyag 150 %-os CD86 stimulációs indexet (S.I.) idéz elő.
A CV70 értéket logaritmikus-lineáris interpolációval számítják ki, az alábbi egyenlet alapján:
CV70 = C1 + [(V1 – 70) / (V1 – V2) * (C2 – C1)]
ahol:
V1 = a 70 % feletti sejtéletképesség minimumértéke
V2 = a 70 % alatti sejtéletképesség maximumértéke
C1 és C2 = a V1 és V2 sejtéletképességet előidéző koncentráció

A CV70 érték más módon is meghatározható, amennyiben (pl. a jártassági tesztanyagok tesztelésével) igazolható, hogy az nem befolyásolja az eredményt.
A EC150 értéket logaritmikus-lineáris interpolációval számítják ki, az alábbi egyenlet alapján:
EC150 = C1 + [(150 – S.I.1) / (S.I.2 – S.I.1) * (C2 – C1)]
ahol:
C1 az a μg/ml-ben megadott legmagasabb koncentráció, amelynél a CD86 stimulációs indexe kisebb mint 150 % (S.I. 1);
C2 az a μg/ml-ben megadott legalacsonyabb koncentráció, amelynél a CD86 stimulációs indexe legalább 150 % (S.I. 2).

Az EC150 és CV70 érték kiszámolása
|
—
|
minden vizsgálatmenet esetében: az egyes EC150 és CV70 értékek annak meghatározására szolgálnak, hogy a CD86 expressziójának növekedése összefüggésben áll-e a koncentráció növekedésével (lásd a 14. pontot),
|
|
—
|
az átlagos sejtéletképesség alapján meghatározzák az összesített CV70 értéket (12),
|
|
—
|
a CD86 értékek átlagos stimulációs indexe alapján meghatározzák az összesített EC150 értéket, amely szerint az U-SENS™ vizsgálatban előrejelzik a vizsgálati vegyi anyag POZITÍV besorolását (lásd a 21. pontot) (12).
|
|
Előrejelzési modell
|
21.
|
A CD86 expressziós mérésnél minden vizsgálati vegyi anyagot legalább négy koncentrációban és legalább két független (különböző napokon végzett) vizsgálatmenetben tesztelnek, amelyek alapján meghatározzák a (NEGATÍV vagy POZITÍV) előrejelzést.
|
—
|
Az U-SENS™ vizsgálat egyes vizsgálatmeneteinek eredménye akkor negatív (a továbbiakban N), ha a CD86 stimulációs indexe valamennyi nem citotoxikus koncentrációban (amikor a sejtéletképesség ≥ 70 %) kisebb mint 150 %, és ha nem állt elő a vizsgálat kimenetelét zavaró körülmény (citotoxicitási, oldhatósági interferencia: lásd a 18. pontot, vagy színinterferencia: lásd a 19. pontot, függetlenül az interferenciát előidéző nem citotoxikus koncentrációktól). Minden más esetben: a CD86 stimulációs indexe eléri vagy meghaladja a 150 %-ot és/vagy interferenciát figyeltek meg, akkor az U-SENS™ egyes vizsgálatmeneteinek eredménye pozitív (a továbbiakban P).
|
|
—
|
Az U-SENS™ vizsgálat előrejelzése akkor tekinthető NEGATÍVNAK, ha legalább két független vizsgálatmenet negatív (N) eredménnyel zárult (1. ábra). Ha az első két vizsgálatmenet mindegyike negatív (N) eredményre vezetett, az U-SENS™ vizsgálat előrejelzése NEGATÍVNAK minősül, és harmadik vizsgálatmenetre nincs szükség.
|
|
—
|
Az U-SENS™ vizsgálat előrejelzése akkor tekinthető POZITÍVNAK, ha legalább két független vizsgálatmenet pozitív (P) eredménnyel zárult (1. ábra). Ha az első két vizsgálatmenet mindegyike pozitív (P) eredményre vezetett, az U-SENS™ vizsgálat előrejelzése POZITÍVNAK minősül, és harmadik vizsgálatmenetre nincs szükség.
|
|
—
|
Mivel dózisbehatároló vizsgálatra nem kerül sor, ez alól kivételt képez az az eset, amikor az első vizsgálatmeneten a CD86 stimulációs indexe csak a legmagasabb nem citotoxikus koncentrációban éri el vagy haladja meg a 150 %-ot. Ekkor a vizsgálatmenetet NEM MEGGYŐZŐNEK (NC) kell tekinteni, és újabb vizsgálatmenetekben meg kell vizsgálni további, a legmagasabb nem citotoxikus koncentráció és a legalacsonyabb citotoxikus koncentráció közé eső koncentrációértékeket (lásd a 20. pontot). Amennyiben valamely vizsgálatmenetet nem meggyőzőnek értékelnek, legalább két további vizsgálatmenetet kell lefolytatni, illetve egy negyediket is, ha a 2. és a 3. vizsgálatmenet eredményei nem egyeznek (független vizsgálatban N és/vagy P) (1. ábra). A további vizsgálatmenetek abban az esetben is pozitívnak minősülnek, ha a CD86 expressziója csak egyetlen nem citotoxikus koncentrációban éri el vagy haladja meg a 150 %-ot, mivel a koncentrációértékeket az adott vizsgálati vegyi anyagra igazították. A végső előrejelzés a három vagy négy független vizsgálatmenet többségi eredményén alapul (vagyis 3-ból 2 vagy négyből 2 eredményén) (1. ábra).
|

1. ábra: Az U-SENS™ vizsgálatban alkalmazott előrejelzési modell. Az U-SENS™ vizsgálat előrejelzését integrált értékelési és vizsgálati megközelítés keretében, valamint a 4. pont és az Általános bevezetés 7., 8. és 9. pontjának rendelkezéseivel összhangban kell figyelembe venni.
N: olyan vizsgálatmenet, amelyben nem figyeltek meg sem pozitív CD86 expressziót, sem interferenciát;
P: olyan vizsgálatmenet, amelyben pozitív CD86 expressziót és/vagy interferenciá(ka)t figyeltek meg;
NC: nem meggyőző. Az első vizsgálatmenet akkor minősül nem meggyőzőnek, ha a CD86 csak a legmagasabb nem citotoxikus koncentrációban pozitív.
#: amennyiben csak az első vizsgálatmenetet nem meggyőzőnek (NC) nyilvánítják, automatikusan szükség van egy harmadik vizsgálatmenetre is, hogy 3 független vizsgálatmenetből legalább 2-ben pozitív (P) vagy negatív (N) többséget lehessen elérni.
$: a keretek a három vizsgálatmenetből származó eredmények releváns kombinációit jelenítik meg az első két vizsgálatmenet fenti keretben látható eredményei alapján.
°: a keretek a négy vizsgálatmenetből származó eredmények releváns kombinációit jelenítik meg az első három vizsgálatmenet fenti keretben látható eredményei alapján.
|
Elfogadhatósági kritériumok
|
22.
|
Az U-SENS™ vizsgálat alkalmazásakor az alábbi elfogadhatósági kritériumoknak kell teljesülniük (12).
|
—
|
A 45 ±3 órás expozíciós időtartam végére a triplikátumban vizsgált kezeletlen U937 sejtek átlagos életképességének meg kell haladnia a 90 %-ot, és a CD86 expressziója nem tolódhat el. A kezeletlen U937 sejtek CD86 bazális expressziójának a legalább 2 % és legfeljebb 25 % közötti tartományban kell lennie.
|
|
—
|
Ha DMSO-t használnak oldószerként, a DMSO vivőanyagos kontroll érvényességét úgy mérik fel, hogy meghatározzák a DMSO kezeletlen sejtekhez viszonyított stimulációs indexét, ugyanakkor a triplikátumban vizsgált sejtek átlagos életképességének meg kell haladnia a 90 %-ot. A DMSO vivőanyagos kontroll akkor tekinthető érvényesnek, ha a triplikátumban vizsgált CD86 stimulációs indexének átlaga nem éri el a kezeletlen U937 sejtek triplikátumban vizsgált CD86 S.I. átlagának 250 %-át.
|
|
—
|
A vizsgálatmenetek akkor minősülnek érvényesnek, ha a kezeletlen U937 sejtek három IgG1 értéke közül minimum kettő legalább 0,6 % és kisebb mint 1,5 %.
|
|
—
|
A párhuzamosan vizsgált negatív kontroll (tejsav) akkor tekinthető érvényesnek, ha a három replikátumból legalább kettő negatív (CD86 S.I. < 150 %) és nem citotoxikus (sejtéletképesség ≥ 70 %).
|
|
—
|
A pozitív kontroll (TNBS) akkor érvényes, ha a három replikátumból legalább kettő pozitív (CD86 S.I. ≥ 150 %) és nem citotoxikus (sejtéletképesség ≥ 70 %).
|
|
Vizsgálati jegyzőkönyv
|
23.
|
A vizsgálati jegyzőkönyvnek a következőket kell tartalmaznia:
|
Vizsgálati vegyi anyag
Egy összetevőből álló anyag
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, teljes tápoldatban való oldhatóság, DMSO-ban való oldhatóság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
Több összetevőből álló anyagok, UVCB-k és keverékek:
|
amennyiben lehetséges, az összetevők kémiai azonosítójával (lásd fent), tisztaságával, mennyiségi előfordulásával és releváns fizikai-kémiai tulajdonságaival (lásd fent) jellemezve; amennyiben rendelkezésre állnak;
|
|
fizikai megjelenés, teljes tápoldatban való oldhatóság, DMSO-ban való oldhatóság és a további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
molekulatömeg vagy látszólagos molekulatömeg ismert összetételű keverékek/polimerek esetén, vagy a vizsgálat lebonyolítása szempontjából releváns egyéb információ;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Kontrollok
Pozitív kontroll
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, DSMO-ban való oldhatóság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak és szükség szerint;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
adott esetben hivatkozás a pozitív történeti kontrollok eredményeire, amelyek a vizsgálatmenet elfogadhatósági kritériumainak való megfelelőséget bizonyítják.
|
Negatív és oldószeres/vivőanyagos kontroll
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
fizikai megjelenés, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben a vizsgálati iránymutatásban említett kontroll oldószerektől/vivőanyagoktól eltérő oldószerek/vivőanyagok használatosak, és amennyiben rendelkezésre állnak;
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Vizsgálati körülmények
|
a megbízó, a vizsgálatot végző laboratórium és a vizsgálatvezető neve és címe;
|
|
az alkalmazott vizsgálat leírása;
|
|
használt sejtvonal, annak tárolási feltételei és eredete (például az a létesítmény, ahonnan beszerezték);
|
|
az alkalmazott áramlási citometria rendszer (pl. modell) és annak műszerbeállításai, valamint az alkalmazott ellenanyagok és citotoxicitási markerek;
|
|
a vizsgálat végrehajtásában való, a jártassági tesztanyagok vizsgálatával megszerzett laboratóriumi jártasság bizonyítására alkalmazott eljárás és a vizsgálat idővel reprodukálható teljesítményének bizonyítására alkalmazott eljárás, pl. a dokumentált kontrolladatok és/vagy a dokumentált reaktivitási vizsgálatok adatai.
|
|
|
A vizsgálat elfogadhatósági kritériumai
|
az oldószeres/vivőanyagos kontroll során kapott sejtéletképesség és CD86 stimulációs index az elfogadhatósági tartományokkal való összevetésben;
|
|
a pozitív kontroll során kapott sejtéletképesség és stimulációs index az elfogadható tartományokkal való összevetésben;
|
|
a vizsgálati vegyi anyag minden vizsgált koncentrációjában mért sejtéletképesség.
|
|
|
A vizsgálat menete
|
az alkalmazott vizsgálatmenetek száma;
|
|
a vizsgálati vegyi anyag koncentrációi, alkalmazása és a használt expozíciós idő (ha eltér az ajánlottól);
|
|
az alkalmazott értékelési és döntési kritériumok leírása;
|
|
a vizsgálati eljárás bármilyen változtatásának leírása.
|
|
|
Eredmények
|
az egyes vizsgálatmenetekben a vizsgálati vegyi anyaggal és a pozitív kontrollal kapott eredmények táblázatba foglalása, feltüntetve a CV70 értéket (ha alkalmazandó), a stimulációs indexet, a sejtéletképességi értékeket, az EC150 értéket (ha alkalmazandó), valamint a vizsgálati vegyi anyag előrejelzési modell szerinti besorolását;
|
|
adott esetben bármely más releváns észrevétel leírása.
|
|
|
Az eredmények tárgyalása
|
az U-SENS™ vizsgálattal kapott eredmények tárgyalása;
|
|
a vizsgálat eredményeinek integrált vizsgálati és értékelési megközelítés keretében történő tárgyalása, amennyiben rendelkezésre állnak más releváns információk.
|
|
Következtetések
|
SZAKIRODALOM
|
(1)
|
Piroird, C., Ovigne, J.M., Rousset, F., Martinozzi-Teissier, S., Gomes, C., Cotovio, J., Alépée, N. (2015). The Myeloid U937 Skin Sensitization Test (U-SENS) addresses the activation of dendritic cell event in the adverse outcome pathway for skin sensitization. Toxicol. In Vitro 29, 901-916.
|
|
(2)
|
EURL ECVAM (2017). The U-SENS™ test method Validation Study Report. A dokumentum az alábbi helyről elérhető:http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations
|
|
(3)
|
EC EURL ECVAM (2016). ESAC Opinion No 2016-03 on the L'Oréal-coordinated study on the transferability and reliability of the U-SENS™ test method for skin sensitisation testing. EUR 28 178 EN; doi 10.2 787/8157 37. Elérhető a következő címen: [http://publications.jrc.ec.europa.eu/repository/handle/JRC103705].
|
|
(4)
|
EC EURL ECVAM (2017). EURL ECVAM Recommendation on the use of non-animal approaches for skin sensitisation testing. EUR 28 553 EN; doi 10.2 760/5889 55. Elérhető a következő címen:https://ec.europa.eu/jrc/en/publication/eur-scientific-and-technical-research-reports/eurl-ecvam-recommendation-use-non-animal-approaches-skin-sensitisation-testing.
|
|
(5)
|
Steiling, W. (2016). Safety Evaluation of Cosmetic Ingredients Regarding their Skin Sensitization Potential. doi:10.3390/cosmetics3020014.Cosmetics 3, 14.
|
|
(6)
|
OECD (2016). Guidance Document on The Reporting of Defined Approaches and Individual Information Sources to be Used Within Integrated Approaches to Testing and Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen: [http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(7)
|
Urbisch, D., Mehling, A., Guth, K., Ramirez, T., Honarvar, N., Kolle, S., Landsiedel, R., Jaworska, J., Kern, P.S., Gerberick, F., Natsch, A., Emter, R., Ashikaga, T., Miyazawa, M., Sakaguchi, H. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul. Toxicol. Pharmacol. 71, 337-351.
|
|
(8)
|
Alépée, N., Piroird, C., Aujoulat, M., Dreyfuss, S., Hoffmann, S., Hohenstein, A., Meloni, M., Nardelli, L., Gerbeix, C., Cotovio, J. (2015). Prospective multicentre study of the U-SENS test method for skin sensitization testing. Toxicol In Vitro 30, 373-382.
|
|
(9)
|
Reisinger, K., Hoffmann, S., Alépée, N., Ashikaga, T., Barroso, J., Elcombe, C., Gellatly, N., Galbiati, V., Gibbs, S., Groux, H., Hibatallah, J., Keller, D., Kern, P., Klaric, M., Kolle, S., Kuehnl, J., Lambrechts, N., Lindstedt, M., Millet, M., Martinozzi-Teissier, S., Natsch, A., Petersohn, D., Pike, I., Sakaguchi, H., Schepky, A., Tailhardat, M., Templier, M., van Vliet, E., Maxwell, G. (2014). Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 29, 259-270.
|
|
(10)
|
Fabian, E., Vogel, D., Blatz, V., Ramirez, T., Kolle, S., Eltze, T., van Ravenzwaay, B., Oesch, F., Landsiedel, R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1 683-1 696.
|
|
(11)
|
OECD. (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen:http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(12)
|
DB-ALM (2016). Protocol no 183: Myeloid U937 Skin Sensitization Test (U-SENS™), 33pp. A dokumentum az alábbi helyről elérhető: [http://ecvam-dbalm.jrc.ec.europa.eu/].
|
|
(13)
|
Sundström, C., Nilsson, K. (1976). Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer 17, 565-577.
|
|
(14)
|
OECD (2005). Series on Testing and Assessment No. 34: Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen:http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(15)
|
United Nations UN (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). ST/SG/AC.10/30/Rev.6, Sixth Revised Edition, New York & Geneva: United Nations Publications. Elérhető a következő címen:http://www.unece.org/fileadmin/DAM/trans/danger/publi/ghs/ghs_rev06/English/ST-SG-AC10-30-Rev6e.pdf.
|
|
(16)
|
OECD (2012). Series on Testing and Assessment No 168: The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen:http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(17)
|
ECETOC (2003). Technical Report No 87: Contact sensitization: Classification according to potency. European Centre for Ecotoxicology & Toxicology of Chemicals, Brussels. Elérhető a következő címen:https://ftp.cdc.gov/pub/Documents/OEL/06.%20Dotson/References/ECETOC_2003-TR87.pdf.
|
2.1. függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság
: a vizsgálattal kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálat megfelelőségének mutatója, és relevanciájának egyik szempontja. A kifejezés gyakran használatos az „egyezés” szó megfelelőjeként, amely arra utal, hogy egy adott vizsgálat milyen arányban szolgáltat helyes eredményeket (14).
Káros kimeneti út (Adverse Outcome Pathway, AOP)
: valamely megcélzott vegyi anyag vagy hasonló vegyi anyagok csoportjának kémiai szerkezetéből kiinduló, a molekuláris kiváltó eseményen keresztül az érintett in vivo eredményig végbemenő eseménysorozat (15).
CD86 koncentrációfüggő válasz
: koncentrációfüggésről (vagy koncentráció válaszról) akkor lehet szó, amikor egy pozitív választ (CD86 S.I. ≥ 150) kiváltó koncentrációt követő koncentráció növeli a CD86 stimulációs indexét.
Vegyi anyag
: anyag vagy keverék.
CV70
: 70 %-os sejtéletképességet eredményező becsült koncentráció.
Eltolódás
: eltolódásnak nevezik azt, ha i) a kezeletlen kontroll 3. replikátumának kiigazított CD86+ értéke nem éri el a kezeletlen kontroll 1. és 2. replikátumánál mért kiigazított CD86+ értékek átlagának 50 %-át; és ha ii) a negatív kontroll 3. replikátumának kiigazított CD86+ értéke nem éri el a negatív kontroll 1. és 2. replikátumánál mért kiigazított CD86+ értékek átlagának 50 %-át.
EC150
: a CD86 expressziója során 150 %-os stimulációs indexet eredményező becsült koncentrációk.
Áramlási citometria
: citometrikus technika, amelyben a folyadékáramban szuszpendált sejtek egyenként áthaladnak egy fókuszált gerjesztő fénynyalábon, amely a sejtekre és összetevőire jellemző minták szerint szóródik szét; a sejteket gyakran fluoreszcens markerekkel jelölik meg, amelyek az abszorbeált fényt eltérő frekvenciákon bocsátják ki.
Veszély
: valamely anyag vagy helyzet azon sajátossága, hogy káros hatásokat képes előidézni az élő szervezet, rendszer vagy (al)populáció anyagnak való expozíciójakor.
Integrált vizsgálati és értékelési megközelítés
: valamely vegyi anyag vagy vegyianyag-csoport veszélyeinek azonosításához (potenciál), jellemzéséhez (hatáserősség) és/vagy biztonsági értékeléséhez (potenciál, hatáserősség és expozíció) alkalmazott strukturált megközelítés, amely stratégiai szempontból beépít és mérlegel minden olyan releváns adatot, amely hozzájárul a lehetséges veszélyre és/vagy kockázatra és/vagy a további célirányos és ezért minimális vizsgálat szükségességére vonatkozó szabályozói döntéshozatalhoz.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő legalább 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
Pozitív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó, ismert pozitív hatást kiváltó anyaggal kezelt replikátum. Annak biztosítása érdekében, hogy a pozitív kontrollban jelentkező hatás időbeli változását fel lehessen mérni, a pozitív hatás nem lehet túlságosan erőteljes.
Pre-hapténok
: abiotikus átalakulás, például oxidáció révén szenzibilizálóvá váló vegyi anyagok.
Pro-hapténok
: olyan vegyi anyagok, amelyek enzimatikus aktivációt igényelnek bőrszenzibilizáló hatásuk kifejtéséhez.
Relevancia
: a vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálat pontosságát (az eredmények egyezését) figyelembe kell venni (14).
Megbízhatóság
: a vizsgálat laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. Megállapítása a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság, valamint a laboratóriumon belüli megismételhetőség kiszámításával történik (14).
Vizsgálatmenet
: egy vizsgálatmenet során egy vagy több vizsgálati vegyi anyagot vizsgálnak egy párhuzamos oldószeres/vivőanyagos kontrollal és egy pozitív kontrollal.
Érzékenység
: az összes olyan pozitív/aktív vegyi anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (14).
S.I.
: stimulációs index. A vegyi anyaggal kezelt sejteknél mért fluoreszcencia-intenzitás mértani középértékének az oldószerrel kezelt sejtekéhez viszonyított relatív értéke.
Oldószeres/vivőanyagos kontroll
: valamely vizsgálati rendszernek a vizsgálati vegyi anyagon kívüli összes összetevőjét tartalmazó kezeletlen minta, amely azonban magában foglalja az alkalmazott oldószert/vivőanyagot. Az ugyanabban az oldószerben/vivőanyagban feloldott vagy stabilan diszpergált vizsgálati vegyi anyaggal kezelt minták esetében a kiindulási érték létrehozásához használatos. Párhuzamos tápoldatos kontrollal elvégzett vizsgálata esetén ez a minta azt is bizonyítja, hogy az oldószer/vivőanyag kölcsönhatásba lép-e a vizsgálati rendszerrel.
Specificitás
: a vizsgálat elvégzésével helyesen besorolt összes negatív/inaktív vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (14).
Festő puffer
: 5 % magzati borjú szérummal kiegészített foszfátpuffert tartalmazó sóoldat.
Anyag
: olyan természetes állapotban előforduló vagy gyártási eljárásból származó kémiai elem és vegyületei, amely a stabilitásának megőrzéséhez szükséges adalékanyagot és az alkalmazott eljárásból származó szennyező anyagokat is tartalmazhat, de nem tartalmaz olyan oldószert, amely az anyag stabilitásának befolyásolása vagy összetételének megváltoztatása nélkül elkülöníthető.
Vizsgálati vegyi anyag
: bármely, e vizsgálat alkalmazásával vizsgált anyag vagy keverék.
A vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (ENSZ-GHS)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (16).
UVCB
: ismeretlen szerkezetű vagy változó összetételű, összetett reakcióban keletkezett vagy biológiai eredetű anyagok.
Érvényes vizsgálat
: olyan vizsgálat, amely kellően relevánsnak és megbízhatónak minősül egy adott célra, és amely tudományosan megalapozott elveken alapul. Egy-egy vizsgálat abszolút értelemben soha nem érvényes, kizárólag meghatározott céllal összefüggésben (14).
2.2. függelék
JÁRTASSÁGI TESZTANYAGOK
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak igazolniuk kell szakmai jártasságukat oly módon, hogy az 1. táblázatban javasolt 10 anyagra vonatkozóan az U-SENS™ vizsgálattal várható előrejelzést helyesen kapják meg, továbbá a 10 jártassági tesztanyag közül legalább 8 esetében referenciatartományba eső CV70 és EC150 értékeket kapnak. A jártassági tesztanyagok kiválasztása oly módon történt, hogy az a bőrszenzibilizáció veszélye esetén bekövetkező különféle válaszreakciók tartományát illetően reprezentatív legyen. A kiválasztás többi kritériuma az volt, hogy a vegyi anyagok kereskedelmi forgalomban beszerezhetők legyenek, azokra vonatkozóan kiváló minőségű in vivo referenciaadatok álljanak rendelkezésre, és az U-SENS™ vizsgálat alapján kiváló minőségű in vitro adatok álljanak rendelkezésre. Az U-SENS™ vizsgálathoz rendelkezésre állnak publikált referenciaadatok is (1) (8).
1. táblázat
Az U-SENS™ vizsgálatban való szakmai jártasság bizonyításához ajánlott anyagok
|
Jártassági tesztanyagok
|
CAS-szám
|
Halmazállapot
|
In vivo előrejelzés (106)
|
U-SENS Oldószer/vivőanyag
|
U-SENS CV70 referencia-tartomány μg/ml-ben (107)
|
U-SENS EC150 referencia-tartomány μg/ml-ben (107)
|
|
4-Fenilén-diamin
|
106-50-3
|
Szilárd
|
Szenzibilizáló (erősen)
|
Teljes tápoldat (108)
|
< 30
|
Pozitív (≤ 10)
|
|
Pikril-szulfonsav
|
2508-19-2
|
Folyadék
|
Szenzibilizáló (erősen)
|
Teljes tápoldat
|
> 50
|
Pozitív (≤ 50)
|
|
Dietil-maleát
|
141-05-9
|
Folyadék
|
Szenzibilizáló (mérsékelten)
|
DMSO
|
10–100
|
Pozitív (≤ 20)
|
|
Rezorcin
|
108-46-3
|
Szilárd
|
Szenzibilizáló (mérsékelten)
|
Teljes tápoldat
|
> 100
|
Pozitív (≤ 50)
|
|
Cinnamil-alkohol
|
104-54-1
|
Szilárd
|
Szenzibilizáló (enyhén)
|
DMSO
|
> 100
|
Pozitív (10–100)
|
|
4-Allil-anisol
|
140-67-0
|
Folyadék
|
Szenzibilizáló (enyhén)
|
DMSO
|
> 100
|
Pozitív (< 200)
|
|
Szaharin
|
81-07-2
|
Szilárd
|
Nem szenzibilizáló
|
DMSO
|
> 200
|
Negatív (> 200)
|
|
Glicerin
|
56-81-5
|
Folyadék
|
Nem szenzibilizáló
|
Teljes tápoldat
|
> 200
|
Negatív (> 200)
|
|
Tejsav
|
50-21-5
|
Folyadék
|
Nem szenzibilizáló
|
Teljes tápoldat
|
> 200
|
Negatív (> 200)
|
|
Szalicilsav
|
69-72-7
|
Szilárd
|
Nem szenzibilizáló
|
DMSO
|
> 200
|
Negatív (> 200)
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám.
|
3. függelék
IN VITRO BŐRSZENZIBILIZÁCIÓ: IL-8 LUC VIZSGÁLAT
ALAPVETŐ MEGFONTOLÁSOK ÉS KORLÁTOK
|
1.
|
A sejtfelületi markerek kifejeződését elemző vizsgálatokkal ellentétben az IL-8 Luc vizsgálat egy citokin, az IL-8 dendritikus sejtek aktivációja általi expressziójában előidézett változásokat számszerűsíti. A THP-1 sejtekből származó IL-8 riporter sejtvonalat (THP-G8, amelyet a THP-1 humán monocita akut leukémia sejtvonalból hoztak létre) szenzibilizáló anyagokkal kezelik, majd megmérik az IL-8 kifejeződését (1). Ezt követően a luciferáz kifejeződését felhasználják a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetésének alátámasztására.
|
|
2.
|
Az IL-8 Luc vizsgálatot értékelték egy, az Alternatív Módszerek Validálásával Foglalkozó Japán Központ (JaCVAM), a Gazdasági, Kereskedelmi és Ipari Minisztérium (METI) és a japán állatkísérletek alternatív módszereivel foglalkozó társaság (JSAAE) irányításával végzett validálási tanulmányban (2), amelyet később a JaCVAM és az Egészségügyi, Munkaügyi és Népjóléti Minisztérium (MHLW) égisze alatt, valamint az alternatív vizsgálati módszerekre vonatkozó nemzetközi együttműködési keret (ICATM) támogatásával független szakértői értékelésnek vetettek alá (3). A rendelkezésre álló bizonyítékok, valamint a szabályozó hatóságok és az érdekeltek álláspontja alapján az IL-8 Luc vizsgálat egy integrált vizsgálati és értékelési megközelítés részeként hasznosan alkalmazható a bőrszenzibilizáló és nem szenzibilizáló anyagok veszélyességi osztályozás és címkézés céljából történő megkülönböztetésére. Az IL-8 Luc vizsgálatból származó adatok és más információk együttes használatára fellelhetők példák a szakirodalomban (4) (5) (6).
|
|
3.
|
Az IL-8 Luc vizsgálatról bebizonyosodott, hogy áthelyezhető a sejttenyésztés és a luciferáz mérése területén tapasztalattal rendelkező laboratóriumokba. A laboratóriumon belüli reprodukálhatóság 87,7 %, a laboratóriumok közötti reprodukálhatóság pedig 87,5 % (2). A validálási tanulmányból (2) és más közzétett munkákból (1) (6) származó adatok azt jelzik, hogy az LLNA vizsgálati módszerhez képest az IL-8 Luc vizsgálat 143 vegyi anyagból 118-at minősített pozitívnak vagy negatívnak, és 25-öt nem meggyőzőnek, valamint hogy az IL-8 Luc vizsgálat a bőrszenzibilizáló (azaz az ENSZ-GHS/CLP-rendelet szerinti 1. kategóriába tartozó) anyagokat 86 %-os (101/118) pontossággal különbözteti meg a nem szenzibilizáló (vagyis az ENSZ-GHS/CLP-rendelet szerint nem besorolt) anyagoktól, 96 % az érzékenysége (92/96), a specifikussága pedig 41 % (9/22). A vizsgálat alábbiakban (az 5. pontban) ismertetett alkalmazási körén kívül eső anyagok kizárása esetén az IL-8 Luc vizsgálat 136 vegyi anyagból 113-at minősített pozitívnak vagy negatívnak, és 23 vegyi anyagot nem meggyőzőnek, így az IL-8 Luc vizsgálat pontossága 89 % (101/113), 96 % az érzékenysége (92/96), míg a specifikussága 53 % (9/17). Az Urbisch és munkatársai (7) által idézett humán adatok felhasználása alapján az IL-8 Luc vizsgálat 90 vegyi anyagból 76-ot minősített pozitívnak vagy negatívnak, és 14 vegyi anyagot nem meggyőzőnek, így pontossága 80 % (61/76), érzékenysége 93 % (54/58), míg a specifikussága 39 % (7/18). A vizsgálat alkalmazási körén kívül eső anyagok kizárása esetén az IL-8 Luc vizsgálat 84 vegyi anyagból 71-et minősített pozitívnak vagy negatívnak, és 13-at nem meggyőzőnek, így pontossága 86 % (61/71), érzékenysége 93 % (54/58), specifikussága pedig 54 % (7/13). Az IL-8 Luc vizsgálat nagyobb valószínűséggel vezet hamis negatív előrejelzésekhez olyan vegyi anyagoknál, amelyeknek alacsony/mérsékelt a bőrszenzibilizáló hatása (az ENSZ-GHS/CLP-rendelet szerinti 1B. alkategóriába tartozó anyagoknál), mint a rendkívül bőrszenzibilizáló hatást mutató (az ENSZ-GHS/CLP-rendelet szerinti 1A. alkategóriába tartozó) anyagoknál (6). Ezek az információk összességében azt jelzik, hogy az IL-8 Luc vizsgálat érdemben hozzájárul a bőrszenzibilizáció veszélyének azonosításához. Az IL-8 Luc vizsgálat mint önálló vizsgálat esetében megadott pontossági értékek csak irányadóak, ugyanis a vizsgálatot az integrált vizsgálati és értékelési megközelítés keretében más információforrásokkal együtt és az Általános bevezetés 7. és 8. pontjának rendelkezéseivel összhangban kell figyelembe venni. Ezen túlmenően a bőrszenzibilizáció nem állatokon alkalmazott vizsgálatainak értékelésekor nem szabad megfeledkezni arról, hogy az LLNA vizsgálat és más állatkísérletek nem feltétlenül tükrözik teljes egészében az embernél fennálló helyzetet.
|
|
4.
|
Az aktuálisan rendelkezésre álló adatok alapján az IL-8 Luc vizsgálatról bebizonyosodott, hogy különféle szerves funkciós csoportokat, reakciómechanizmusokat, (in vivo vizsgálatok során meghatározott) bőrszenzibilizáló hatásokat és fizikai-kémiai tulajdonságokat képviselő vizsgálati vegyi anyagokra alkalmazható (2) (6).
|
|
5.
|
Bár az IL-8 Luc vizsgálat során használt oldószer az X-VIVOTM 15, a vizsgálat megfelelően értékelte a 3,5-nél magasabb Log Kow értékkel és 100 μg/ml körüli vízoldékonysággal rendelkező vegyi anyagokat (az EPI SuiteTM számításai szerint), és a vízben nehezen oldódó szenzibilizáló anyagokat jobb hatékonysággal detektálja, mint az oldószerként dimetil-szulfoxidot (DMSO) alkalmazó IL-8 Luc vizsgálat (2). Azonban az oldószer 20 mg/ml koncentrációjában nem oldódó vizsgálati vegyi anyagok hamis negatív eredményhez vezethetnek, mivel nem oldhatók X-VIVOTM 15-ben. Ezért az ilyen vegyi anyagok negatív eredményeit figyelmen kívül kell hagyni. Az anhidridek esetében a validációs tanulmány magas hamisnegatív-arányt mutatott ki. Ezen túlmenően a sejtvonal korlátozott metabolikus képessége (8) és a kísérleti körülmények miatt a pro-hapténok (metabolikus aktiválást igénylő vegyi anyagok) és a pre-hapténok (a levegő oxidáló hatására aktiválódó anyagok) is negatív eredményre vezethetnek a vizsgálat során. Bár a feltételezhető pre-/pro-hapténok negatív eredményét elővigyázatosan kell kezelni, az IL-8 Luc vizsgálat az adatbázisában szereplő 11 pre-hapténból 11-et, a 6 pro-hapténból 6-ot és a 8 pre-/pro-hapténból 6-ot helyesen sorolt be (2). A három nem állatokon alkalmazott, pre- és pro-hapténokat észlelni képes vizsgálat (a DPRA, a KeratinoSens™ és a h-CLAT) közelmúltbeli átfogó felülvizsgálata alapján (9), és figyelembe véve, hogy az IL-8 Luc vizsgálatban használt THP-G8 sejtek a h-CLAT vizsgálatban alkalmazott THP-1 sejtekből származnak, az IL-8 Luc vizsgálat más vizsgálatokkal való együttes alkalmazása esetén hozzájárulhat ahhoz, hogy növekedjen a nem állatokon alkalmazott vizsgálatok érzékenysége a pre- és pro-hapténok detektálása terén. Az ez idáig vizsgált felületaktív anyagok típusuktól (pl. kationos, anionos vagy nem ionos) függetlenül (hamis) pozitív eredményekhez vezetnek. Végezetül a luciferázzal kölcsönhatásba lépő vegyi anyagok megzavarhatják a luciferáz aktivitását/mérését, látszólagos gátlást vagy megnövekedett lumineszcenciát okozva (10). Beszámoltak például arról, hogy az 1 μM-nél magasabb fitoösztrogén-koncentráció a luciferáz riportergén túlzott aktiválása miatt zavarja a lumineszcencia jeleit más, luciferáz alapú riportergén-vizsgálatokban. Következésképp a magas fitoösztrogén-koncentráció mellett vagy feltehetőleg a luciferáz riportergént a fitoösztrogénhez hasonló mértékben aktiváló vegyületek magas koncentrációja mellett kapott luciferáz-expressziót körültekintően meg kell vizsgálni (11). A fentiek fényében a felületaktív anyagok, az anhidridek és a luciferáz aktivitását zavaró vegyi anyagok kívül esnek a vizsgálat alkalmazási körén. Olyan esetekben, amikor bizonyítékkal igazolható, hogy az IL-8 Luc vizsgálat nem alkalmazható más, konkrét vegyianyag-kategóriákra, a vizsgálatot nem szabad e vegyianyag-kategóriákra alkalmazni.
|
|
6.
|
A fentieknek megfelelően az IL-8 Luc vizsgálat elősegíti a bőrszenzibilizáló és a nem szenzibilizáló anyagok megkülönböztetését. Szükség van további, lehetőleg humán adatokon alapuló kutatásra annak meghatározása érdekében, hogy az IL-8 Luc vizsgálat eredményei – más információforrásokkal együtt mérlegelve – hozzájárulhatnak-e a hatáserősség értékeléséhez.
|
|
7.
|
A fogalommeghatározások a 3.1. függelékben találhatók.
|
A VIZSGÁLAT ELVE
|
8.
|
Az IL-8 Luc vizsgálat THP-1 humán monocita leukémia sejtvonalat alkalmaz, amely az amerikai típustörzsanyag-gyűjteményből szerezhető be (Manassas, VA, USA). Ezt a sejtvonalat felhasználva a Tohokui Egyetem Orvostudományi Karának Bőrgyógyászati Tanszéke létrehozott egy THP-1 származék IL-8 riporter sejtvonalat, a THP-G8-at, amely IL-8 promóterrel szabályozott stabil narancssárga luciferáz (SLO) luciferáz gént és gliceraldehid-3-foszfát-dehidrogenáz (GAPDH) promóterrel szabályozott stabil vörös luciferáz (SLR) luciferáz gént hordoz (1). Ez lehetővé teszi a luciferázgén-indukció kvantitatív mérését, mégpedig az IL-8 és a GAPDH szenzibilizáló vegyi anyagokkal való kezelést követő, sejteken belüli aktivitásának mutatójaként szolgáló – jól bevált fénytermelő luciferáz szubsztrátok által kibocsátott – lumineszcencia észlelése révén.
|
|
9.
|
A két színt detektáló vizsgálati rendszer magában foglal egy narancssárga fényt kibocsátó luciferázt (SLO; λmax = 580 nm) (12), amely az IL-8 promóterrel szabályozott génkifejeződést jelzi, és egy vörös fényt kibocsátó luciferázt (SLR; λmax = 630 nm) (13), amely a belső szabályozó promóterrel, a GAPDH-val szabályozott génkifejeződést jelzi. A két luciferáz a d-luciferinnel reakcióba lépve eltérő színt bocsát ki; lumineszcenciájukat egyidejűleg, egylépéses reakció keretében mérik úgy, hogy optikai szűrő használatával elkülönítik a kibocsátást a vizsgálati keveréktől (14) (3.2. függelék).
|
|
10.
|
A THP-G8 sejteket 16 órán át kezelik a vizsgálati vegyi anyaggal, majd megmérik az IL-8 promóter aktivitását tükröző SLO luciferázaktivitást (SLO-LA) és a GAPDH promóter aktivitását tükröző SLR luciferázaktivitást (SLR-LA). A rövidítések értelmezésének megkönnyítése érdekében az SLO-LA a továbbiakban IL8LA, az SLR-LA pedig GAPLA néven szerepel. Az 1. táblázat ismerteti az IL-8 Luc vizsgálatban mért luciferázaktivitással kapcsolatos kifejezések leírását. A mért értékek alapján meghatározzák az IL8LA normalizált értékét (nIL8LA), amely az IL8LA és a GAPLA hányadosa; az nIL8LA indukcióját (Ind-IL8LA), amely a vizsgálati vegyi anyaggal kezelt THP-G8 sejtek négyszeresen mért nIL8LA értéke számtani átlagának a kezeletlen THP-G8 sejtek nIL8LA értékeinek számtani átlagához viszonyított aránya; és a GAPLA gátlását (Inh-GAPLA), amely a vizsgálati vegyi anyaggal kezelt THP-G8 sejtek négyszeresen mért GAPLA értéke számtani átlagának a kezeletlen THP-G8 sejtek GAPLA értékéinek számtani átlagához viszonyított aránya, és amely a citotoxicitás mutatója.
1. táblázat
Az IL-8 Luc vizsgálatban mért luciferázaktivitással kapcsolatos kifejezések leírása
|
Rövidítések
|
Fogalommeghatározás
|
|
GAPLA
|
A GAPDH promóter aktivitását tükröző SLR luciferázaktivitás
|
|
IL8LA
|
Az IL-8 promóter aktivitását tükröző SLO luciferázaktivitás
|
|
nIL8LA
|
IL8LA/GAPLA
|
|
Ind-IL8LA
|
A vegyi anyagokkal kezelt THP-G8 sejtek nIL8LA értéke/kezeletlen sejtek nIL8LA értéke
|
|
Inh-GAPLA
|
A vegyi anyagokkal kezelt THP-G8 sejtek GAPLA értéke/kezeletlen sejtek GAPLA értéke
|
|
CV05
|
A vegyi anyag azon legalacsonyabb koncentrációja, amely 0,05 alatti Inh-GAPLA értéket idéz elő.
|
|
|
11.
|
Az IL-8 Luc vizsgálathoz hasonló in vitro IL-8 luciferáz vizsgálatok validálásának megkönnyítéséhez rendelkezésre állnak teljesítményszabványok (15), amelyek lehetővé teszik az OECD 442E vizsgálati iránymutatásának időben történő módosítását az IL-8 luciferáz vizsgálatok e vizsgálati iránymutatásba való beépítéséhez. Az adatok OECD-megállapodás szerinti kölcsönös elfogadása csak a teljesítményszabványoknak megfelelően validált vizsgálatok esetében lesz garantált, ha e vizsgálatokat az OECD felülvizsgálta és belefoglalta 442E vizsgálati iránymutatásába (16).
|
A JÁRTASSÁG BIZONYÍTÁSA
|
12.
|
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak a 3.3. függelékben felsorolt tíz jártassági tesztanyag in vitro módszerekre vonatkozó bevált gyakorlatokkal összhangban történő vizsgálatával igazolniuk kell szakmai jártasságukat (17). Ezenfelül a vizsgálat felhasználóinak történeti adatbázist kell fenntartaniuk, amely tartalmazza a reaktivitási tesztekből (lásd a 15. pontot), valamint a pozitív és az oldószeres/vivőanyagos kontrollokból (lásd a 21–24. pontot) származó adatokat, ezekkel az adatokkal igazolva a vizsgálat laboratóriumon belüli hosszú távú reprodukálhatóságát.
|
ELJÁRÁS
|
13.
|
Az IL-8 Luc vizsgálathoz rendelkezésre állnak szabványműveleti eljárások, amelyeket alkalmazni kell e vizsgálat végrehajtása során (18). A vizsgálat elvégzésére vállalkozó laboratóriumok a rekombináns THP-G8 sejtvonalat az OECD sablonban szereplő feltételeknek megfelelően egy anyagátadási megállapodás aláírását követően a GPC Lab. Co. Ltd. vállalattól (Tottori, Japán) szerezhetik be. A következő pontok ismertetik a vizsgálat főbb összetevőit és eljárásait.
|
Sejtek előkészítése
|
14.
|
Az IL-8 Luc vizsgálathoz THP-G8 sejtvonalat kell használni, amely a GPC Lab. Co. Ltd. vállalattól (Tottori, Japán) szerezhető be (lásd a 8. és a 13. pontot). A sejteket a beérkezésüket követően (2–4 passzálással) fel kell szaporítani, és homogén állományként fagyasztva kell tárolni. Az ebből az állományból származó sejteket legfeljebb 12 passzálás erejéig vagy legfeljebb 6 héten át lehet szaporítani. A szaporításhoz használt közeg az RPMI-1640 tápfolyadék, kiegészítve 10 % magzati borjú szérummal (FBS), antibiotikumos/antimikotikus oldattal (100 egység/ml penicillin g, 100 μg/ml sztreptomicin és 0,25 μg/ml amfotericin B 0,85 %-os sóoldatban) (pl. GIBCO, kat.száma: 15240-062), 0,15 μg/ml puromicinnel (CAS-száma: 58-58-2) és 300 μg/ml G418-cal (CAS-száma: 108321-42-2).
|
|
15.
|
A vizsgálatban való felhasználásuk előtt a sejteket egy reaktivitási teszt során minősíteni kell. A reaktivitási tesztet a felolvasztás után 1–2 héttel vagy 2–4 passzálást követően kell elvégezni, a pozitív kontrollanyaggal, vagyis a 4-nitrobenzil-bromiddal (4-NBB) (CAS-száma: 100-11-8, legalább 99 %-os tisztaságban) és a negatív kontrollanyaggal, a tejsavval (LA) (CAS-száma: 50-21-5, legalább 85 % tisztaságban). A 4-NBB-nak pozitív választ (legalább 1,4-es Ind-IL8LA), a tejsavnak pedig negatív választ (1,4-nél kisebb Ind-IL8LA) kell kiváltania. A vizsgálathoz kizárólag a reaktivitási tesztben megfelelt sejtek használhatók. A tesztet a 22–24. pontban leírt eljárások szerint kell végrehajtani.
|
|
16.
|
A vizsgálathoz a THP-G8 sejteket 2–5 × 105 sejt/ml sűrűségben átoltják, majd sejttenyésztő lombikban 48–96 órán keresztül előtenyésztik. A vizsgálat napján a sejttenyésztő lombikból begyűjtött sejteket 10 % FBS-t tartalmazó, antibiotikum-mentes RPMI-1640 tápoldatban átmossák, majd 10 % FBS-t tartalmazó, antibiotikum-mentes RPMI-1640 tápoldatban 1 × 106 sejt/ml sűrűségben újraszuszpendálják. Ezt követően a sejteket 50 μl mennyiségben (lyukanként 5 × 104 sejt sűrűségben) elhelyezik egy 96 lyukú, lapos fenekű fekete lemezben (pl. Costar, kat.száma: 3603).
|
A vizsgálati vegyi anyag és a kontrollanyagok előkészítése
|
17.
|
A vizsgálati vegyi anyagot és a kontrollanyagokat a vizsgálat napján kell előkészíteni. Az IL-8 Luc vizsgálatban a vizsgálati vegyi anyagokat X-VIVOTM 15-ben, egy kereskedelmi forgalomban kapható, szérummentes tápoldatban (Lonza, 04-418Q) oldják fel, 20 mg/ml végső koncentrációban. Egy mikrocentrifuga csőbe töltött 20 mg vizsgálati vegyi anyaghoz (a vegyi anyag oldhatóságától függetlenül) hozzáadnak annyi X-VIVOTM 15 tápoldatot, hogy a keverék térfogata 1 ml legyen, majd a keveréket 30 percig, körülbelül 20 oC-os környezeti hőmérsékleten intenzív örvénykeverésnek vetik alá és rotorral legfeljebb 8 rpm sebességen rázatják. Amennyiben a szilárd vegyi anyagok ilyen módon nem oldódnak fel, a csövet ultrahanggal kezelik mindaddig, amíg a vegyi anyag teljesen feloldódik vagy stabilan diszpergálódik. Az X-VIVOTM 15 tápoldatban oldható vizsgálati vegyi anyagok esetében az oldatot X-VIVOTM 15-ben 5-szörös tényezővel hígítják, és a kapott keveréket a vizsgálati vegyi anyag X-VIVOTM 15 (4 mg/ml-os) törzsoldataként hasznosítják. Az X-VIVOTM 15 tápoldatban nem oldható vizsgálati vegyi anyagok esetében a keveréket ismét legalább 30 percig rázatják, majd 5 percen keresztül 15000 rpm (≈20000 g) sebességen centrifugálják; a létrejött felülúszót a vizsgálati vegyi anyag X-VIVOTM 15 törzsoldataként hasznosítják. Más oldószerek, például DMSO, víz vagy a tápfolyadék használatát tudományosan meg kell indokolni. A vegyi anyagok feloldásának részletes eljárását a 3.5. függelék ismerteti. A 18–23. pontban leírtak szerint elkészített X-VIVOTM 15 oldatokat 1: 1 térfogatszázalék arányban össze kell keverni a 96 lyukú, lapos fenekű fekete lemezen elkészített sejtszuszpenziókkal (lásd a 16. pontot).
|
|
18.
|
Az első vizsgálatmenet célja a citotoxikus koncentráció meghatározása és a vegyi anyagok bőrszenzibilizáló potenciáljának felmérése. A vizsgálati vegyi anyagok X-VIVOTM 15 törzsoldataiból X-VIVOTM 15 tápoldattal, kétszeres hígítási tényezővel oldatsorozatot készítenek (lásd a 3.5. függeléket) egy 96 lyukú vizsgálati lemezen (pl. Costar, kat.száma: EW-01729-03). Ezután lyukanként 50 μl mennyiségű hígított oldatot adagolnak a 96 lyukú, lapos fenekű fekete lemezen elkészített 50 μl mennyiségű sejtszuszpenziókhoz. Tehát az X-VIVO TM 15-ben oldható vizsgálati vegyi anyagok esetében a vizsgálati vegyi anyagok végső koncentrációértékei a 0,002–2 mg/ml-es tartományban helyezkednek el (3.5. függelék). A 20 mg/ml koncentrációjú X-VIVO TM 15-ben nem oldható vizsgálati vegyi anyagoknál csak a 2 és 210 közötti hígítási tényező határozható meg, a vizsgálati vegyi anyagok tényleges végső koncentrációértékei bizonytalanok, mivel attól függnek, hogy a vizsgálati vegyi anyagoknak mekkora a telítési koncentrációja az X-VIVO TM 15 törzsoldatban.
|
|
19.
|
A további vizsgálatmenetekben (vagyis a második, harmadik és negyedik ismétlésben) az X-VIVOTM 15 törzsoldat koncentrációja négyszerese annak a koncentrációértéknek, amely az első kísérlet során 05-ös sejtéletképességet eredményezett (CV05; az a legalacsonyabb koncentráció, amely 0,05 alatti Inh-GAPLA értéket idéz elő). Amennyiben az első vizsgálatmenetben alkalmazott legmagasabb koncentrációnál az Inh-GAPLA nem süllyed 0,05 alá, az X-VIVOTM 15 törzsoldatát az első vizsgálatmenet legmagasabb koncentrációjában készítik el. A CV05 értéket eredményező koncentráció kiszámításához elosztják az első vizsgálatmenet törzsoldatának koncentrációját a CV05 hígítási tényezővel (X) (CV05 hígítási tényező (X); az a hígítási tényező, amellyel a törzsoldat CV05 értéket eredményező koncentrációra hígítható) (lásd a 3.5. függeléket). A 20 mg/ml koncentrációjú X-VIVO-ban nem oldható vizsgálati anyagoknál a CV05 meghatározásához a törzsoldat koncentrációját megszorozzák 1/X-szel. A 2–4. vizsgálatmenethez egy második törzsoldatot készítenek, 4 × CV05 koncentrációban (3.5. függelék).
|
|
20.
|
Az X-VIVOTM 15 második törzsoldatából 1,5-szörös hígítási tényezővel készítenek oldatsorozatot, egy 96 lyukú vizsgálati lemezen. Ezután lyukanként 50 μl mennyiségű hígított oldatot adagolnak a 96 lyukú, lapos fenekű fekete lemez lyukaiba helyezett 50 μl mennyiségű sejtszuszpenziókhoz. Minden vizsgálati vegyi anyag valamennyi koncentrációját 4 lyukban vizsgálják. Ezt követően a mintákat lemezrázón összekeverik, és 16 órán át inkubálják 37 oC-on, 5 % CO2 mellett, majd az alább leírt módon megmérik a luciferázaktivitást.
|
|
21.
|
Az oldószeres kontroll a lyukanként 50 μl X-VIVOTM 15 és a lyukanként 50 μl, 10 % FBS-t tartalmazó RPMI-1640 tápoldatban lévő sejtszuszpenzió keveréke.
|
|
22.
|
Az ajánlott pozitív kontroll a 4-NBB. Egy 1,5 ml-es mikrocentrifuga csőben elhelyeznek 20 mg 4-NBB-t, majd hozzáadnak annyi X-VIVOTM 15-öt, hogy a keverék térfogata 1 ml legyen. A csövet legalább 30 percig intenzív örvénykeverésnek vetik alá és rotorral legfeljebb 8 rpm sebességen rázatják. Ezt követően a keveréket 5 percig 20000 g-n centrifugálják, majd a felülúszót 4-szeres tényezővel X-VIVOTM 15-tel hígítják, és 500 μl hígított felülúszót áthelyeznek a 96 lyukú vizsgálati lemez egyik lyukába. A hígított felülúszót 2-szeres és 4-szeres tényezővel tovább hígítják X-VIVOTM 15-tel, majd 50 μl mennyiségű oldatot adagolnak a 96 lyukú, lapos fenekű fekete lemez lyukaiba helyezett 50 μl mennyiségű THP-G8 sejtszuszpenziókhoz (3.6. függelék). A pozitív kontroll valamennyi koncentrációját 4 lyukban vizsgálják. A lemez tartalmát lemezrázón összekeverik, és CO2 inkubátorban 16 órán át inkubálják (37 oC-on, 5 % CO2 mellett), majd a 29. pontban leírt módon megmérik a luciferázaktivitást.
|
|
23.
|
Az ajánlott negatív kontrollanyag a tejsav. Egy 1,5 ml-es mikrocentrifuga csőben elhelyeznek 20 mg tejsavat, majd hozzáadnak annyi X-VIVOTM 15-öt, hogy a keverék térfogata 1 ml legyen (20 mg/ml). A 20 mg/ml koncentrációjú tejsavoldatot 5-szörös tényezővel X-VIVOTM 15-tel hígítják (4 mg/ml); majd a 4 mg/ml koncentrációjú tejsavoldatból 500 μl mennyiséget áthelyeznek a 96 lyukú vizsgálati lemez egyik lyukába. Ezt az oldatot 2-szeres tényezővel X-VIVOTM 15-tel hígítják, majd ismét 2-szeres tényezővel tovább hígítják, 2 mg/ml-es és 1 mg/ml-es oldatokat hozva létre. E 3 oldatból és a vivőanyagos kontrollból (X-VIVOTM 15) 50 μl mennyiséget adagolnak a 96 lyukú, lapos fenekű fekete lemez lyukaiba helyezett 50 μl mennyiségű THP-G8 sejtszuszpenziókhoz. A negatív kontroll valamennyi koncentrációját 4 lyukban vizsgálják. A lemez tartalmát lemezrázón összekeverik, és CO2 inkubátorban 16 órán át inkubálják (37 oC-on, 5 % CO2 mellett), majd a 29. pontban leírt módon megmérik a luciferázaktivitást.
|
|
24.
|
Alkalmazhatók más, megfelelő pozitív vagy negatív kontrollok is, ha rendelkezésre állnak dokumentált adatok hasonló vizsgálatmenet-elfogadhatósági kritériumok származtatásához.
|
|
25.
|
Ügyelni kell az illékony vizsgálati vegyi anyagok elpárolgásának elkerülésére, valamint a lyukak vizsgálati vegyi anyagokkal való keresztszennyeződésének elkerülésére, például a lemezek befedhetők fóliával a vizsgálati vegyi anyagokkal való inkubálás előtt.
|
|
26.
|
A vizsgálati vegyi anyagokat és az oldószeres kontrollt 2–4 vizsgálatmenetben kell vizsgálni ahhoz, hogy meg lehessen állapítani a besorolás pozitív vagy negatív előrejelzését (lásd a 2. táblázatot). Mindegyik vizsgálatmenetet más-más napon, a vizsgálati vegyi anyaggal frissen készített X-VIVOTM 15 törzsoldattal és egymástól függetlenül begyűjtött sejtekkel kell elvégezni. A sejtek származhatnak ugyanabból a passzálásból.
|
A luciferáz-aktivitás mérése
|
27.
|
A lumineszcenciát 96 lyukú mikrolemezhez való luminométerrel mérik, amelyet optikai szűrőkkel szerelnek fel, pl. Phelios (ATTO, Tokió, Japán), Tristan 941 (Berthold, Bad Wildbad, Németország) és az ARVO sorozat (PerkinElmer, Waltham, MA, USA). A reprodukálhatóság biztosítása érdekében a luminométert minden vizsgálat előtt kalibrálni kell (19). A kalibráláshoz rendelkezésre állnak rekombináns, narancssárga és vörös fényt kibocsátó luciferázok.
|
|
28.
|
A lemez valamennyi vegyi anyaggal kezelt vagy kezeletlen sejtszuszpenziót tartalmazó lyukába helyeznek 100 μl felmelegített Tripluc® luciferáz vizsgálati reagenst (Tripluc). A lemezt körülbelül 20 oC-os környezeti hőmérsékleten 10 percig rázatják. Ezután a lemezt a luciferáz-aktivitás méréséhez a luminométerbe helyezik. A biolumineszcenciát 3 másodpercig mérik optikai szűrő nélkül (F0) és optikai szűrővel (F1). Ha például a használt luminométer típusa miatt ettől eltérő beállításokat alkalmaznak, azt tudományosan indokolni kell.
|
|
29.
|
A mért értékek alapján meghatározzák az egyes koncentrációk paramétereit, például az IL8LA, a GAPLA, az nIL8LA, az Ind-IL8LA, az Inh-GAPLA értékét, az IL8LA átlagát ±SD, a GAPLA átlagát ±SD, az nIL8LA átlagát ±SD, az Ind-IL8LA átlagát ±SD, az Inh-GAPLA átlagát ±SD és az Ind-IL8LA 95 %-os konfidenciaintervallumát. Az ebben a pontban említett paraméterek meghatározását a 3.1. és a 3.4. függelék tartalmazza.
|
|
30.
|
A mérés előtt a több színt észlelő riporter vizsgálatokban a színek elkülönítését általában optikai szűrővel – például sharp-cut (long-pass vagy short-pass) szűrővel vagy sávszűrővel – felszerelt detektorokkal (luminométerrel és lemezolvasóval) végzik. Az egyes biolumineszcens jelzőszínek kiszűréséhez használt szűrők transzmissziós koefficiensét a vizsgálatot megelőzően a 3.2. függelékben foglaltak szerint kalibrálni kell.
|
ADATOK ÉS JELENTÉS
Az adatok kiértékelése
|
31.
|
A pozitív/negatív döntés kritériumai alapján minden vizsgálatmenetre érvényesek az alábbiak:
|
—
|
az IL-8 Luc vizsgálat előrejelzése akkor pozitív, ha az Ind-IL8LA legalább 1,4 és az Ind-IL8LA 95 %-os konfidenciaintervallumának alsó határértéke legalább 1,0;
|
|
—
|
az IL-8 Luc vizsgálat előrejelzése akkor negatív, ha az Ind-IL8LA kisebb mint 1,4 és/vagy az Ind-IL8LA 95 %-os konfidenciaintervallumának alsó határértéke kisebb mint 1,0.
|
|
Előrejelzési modell
|
32.
|
Azok a vizsgálati vegyi anyagok, amelyek az 1., 2., 3. és 4. vizsgálatmenetben két pozitív eredményre vezettek, pozitívnak minősülnek, míg azok, amelyek az 1., 2., 3. és 4. vizsgálatmenetben három negatív eredményre vezettek, feltételezett negatívnak tekintendők (2. táblázat). A feltételezett negatív vegyi anyagok közöl azok, amelyek oldódnak 20 mg/ml koncentrációjú X-VIVOTM 15-ben, negatívnak tekintendők, azok a vegyi anyagok pedig, amelyek nem oldódnak 20 mg/ml koncentrációjú X-VIVOTM 15-ben, nem minősíthetők (1. ábra).
2. táblázat
A pozitív és a feltételezett negatív anyagok azonosításának kritériumai
|
1. vizsgálatmenet
|
2. vizsgálatmenet
|
3. vizsgálatmenet
|
4. vizsgálatmenet
|
Végső előrejelzés
|
|
Pozitív
|
Pozitív
|
—
|
—
|
Pozitív
|
|
Negatív
|
Pozitív
|
—
|
Pozitív
|
|
Negatív
|
Pozitív
|
Pozitív
|
|
Negatív
|
Feltételezett negatív
|
|
Negatív
|
Pozitív
|
Pozitív
|
—
|
Pozitív
|
|
Negatív
|
Pozitív
|
Pozitív
|
|
Negatív
|
Feltételezett negatív
|
|
Negatív
|
Pozitív
|
Pozitív
|
Pozitív
|
|
Negatív
|
Feltételezett negatív
|
|
Negatív
|
—
|
Feltételezett negatív
|
|
1. ábra
A végső döntés előrejelzési modellje

Elfogadhatósági kritériumok
|
33.
|
Az IL-8 Luc vizsgálat alkalmazásakor az alábbi elfogadhatósági kritériumoknak kell teljesülniük.
|
—
|
Az Ind-IL8LA értékének minden vizsgálatmenetben a pozitív kontrollanyag, a 4-NBB legalább egyik koncentrációjában nagyobbnak kell lennie mint 5,0.
|
|
—
|
Az Ind-IL8LA értékének minden vizsgálatmenetben a negatív kontrollanyag, a tejsav minden koncentrációjában kisebbnek kell lennie mint 1,4.
|
|
—
|
Azon lemezek adatait, amelyeknél a sejteket és Tripluc reagenst tartalmazó, de vegyi anyag nélküli, kontrollként szolgáló lyukak GAPLA értéke nem éri el a csak a vizsgálati tápoldatot (vagyis lyukanként 50 μl, 10 % FBS-sel kiegészített RPMI-1640 és lyukanként 50 μl X-VIVOTM 15 keverékét) tartalmazó lyuk GAPLA értékének 5-szörösét, nem szabad figyelembe venni.
|
|
—
|
Szintén nem vehetők figyelembe azon lemezek adatai, amelyeknél a vizsgálati vagy kontrollként szolgáló vegyi anyagok összes koncentrációjánál az Inh-GAPLA kevesebb mint 0,05. Ez utóbbi esetben az első vizsgálatot meg kell ismételni, méghozzá úgy, hogy az ismétlésben alkalmazott legmagasabb végső koncentráció az előző vizsgálat legalacsonyabb végső koncentrációja legyen.
|
|
Vizsgálati jegyzőkönyv
|
34.
|
A vizsgálati jegyzőkönyvnek a következőket kell tartalmaznia:
|
Vizsgálati vegyi anyagok
Egy összetevőből álló anyag:
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, vízoldékonyság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
oldhatóság X-VIVOTM 15-ben. Az X-VIVOTM 15-ben oldhatatlan vegyi anyagok esetében: a centrifugálást követően megfigyeltek-e kicsapódást vagy flotációt;
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
nem X-VIVOTM 15 használata esetén az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
Több összetevőből álló anyagok, UVCB-k és keverékek:
|
amennyiben lehetséges, az összetevők kémiai azonosítójával (lásd fent), tisztaságával, mennyiségi előfordulásával és releváns fizikai-kémiai tulajdonságaival (lásd fent) jellemezve; amennyiben rendelkezésre állnak;
|
|
fizikai megjelenés, vízoldékonyság és a további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak;
|
|
molekulatömeg vagy látszólagos molekulatömeg ismert összetételű keverékek/polimerek esetén, vagy a vizsgálat lebonyolítása szempontjából releváns egyéb információ;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
oldhatóság X-VIVOTM 15-ben. Az X-VIVOTM 15-ben oldhatatlan vegyi anyagok esetében: a centrifugálást követően megfigyeltek-e kicsapódást vagy flotációt;
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak.
|
|
nem X-VIVOTM 15 használata esetén az oldószer/vivőanyag kiválasztásának indokolása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Kontrollok
Pozitív kontroll
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok), SMILES- vagy InChI-kód, szerkezeti képlet és/vagy más azonosítók alapján;
|
|
fizikai megjelenés, vízben való oldhatóság, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben rendelkezésre állnak és szükség szerint;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
vizsgálat előtti kezelés, ha történt ilyen (pl. melegítés, őrlés);
|
|
vizsgált koncentráció(k);
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
adott esetben hivatkozás a pozitív történeti kontrollok eredményeire, amelyek a vizsgálatmenet elfogadhatósági kritériumainak való megfelelőséget bizonyítják.
|
Negatív kontroll:
|
kémiai azonosítása, például IUPAC- vagy CAS-név (nevek), CAS-szám(ok) és/vagy más azonosítók alapján;
|
|
tisztaság, valamint adott esetben és amennyiben a gyakorlatban megvalósítható, a szennyeződések kémiai azonosítója stb.;
|
|
fizikai megjelenés, molekulatömeg, valamint további releváns fizikai-kémiai tulajdonságok, amennyiben a vizsgálati iránymutatásban említett negatív kontrollanyagoktól eltérő negatív kontrollanyagok használatosak, és amennyiben rendelkezésre állnak;
|
|
tárolási feltételek és stabilitás, amennyiben rendelkezésre állnak;
|
|
az oldószer kiválasztásának indoklása minden egyes vizsgálati vegyi anyag esetében.
|
|
|
Vizsgálati körülmények
|
a megbízó, a vizsgálatot végző laboratórium és a vizsgálatvezető neve és címe;
|
|
az alkalmazott vizsgálat leírása;
|
|
használt sejtvonal, tárolási feltételei és eredete (például az a létesítmény, ahonnan azt beszerezték);
|
|
a magzati borjú szérum gyártási száma és eredete, a forgalmazó neve, a 96 lyukú, lapos fenekű fekete lemez gyártási száma, valamint a Tripluc reagens gyártási száma;
|
|
a vizsgálathoz használt sejtek passzálásának száma és sűrűsége;
|
|
a vizsgálat előtt a leoltáshoz alkalmazott sejtszámlálási módszer és a homogén sejtszám-eloszlás biztosítása érdekében hozott intézkedések;
|
|
az alkalmazott luminométer (például modell), beleértve a műszer beállításait, a használt luciferáz szubsztrátot, valamint a megfelelő lumineszcencia-mérések szemléltetése a 3.2. függelékben ismertetett kontrollvizsgálat alapján;
|
|
a vizsgálat végrehajtásában való laboratóriumi jártasság bizonyítására alkalmazott eljárás (például jártassági tesztanyagok vizsgálatával) vagy a vizsgálat idővel reprodukálható végrehajtásának bizonyítására alkalmazott eljárás.
|
|
|
A vizsgálat menete
|
az alkalmazott replikátumok és vizsgálatmenetek száma;
|
|
a vizsgálati vegyi anyag koncentrációi, alkalmazási eljárása és az expozíciós idő (ha eltér az ajánlottól);
|
|
az alkalmazott értékelési és döntési kritériumok leírása;
|
|
az alkalmazott vizsgálat-elfogadhatósági kritériumok leírása;
|
|
a vizsgálati eljárás bármilyen változtatásának leírása.
|
|
|
Eredmények
|
az IL8LA és a GAPLA mért értékei;
|
|
az nIL8LA, az Ind-IL8LA és az Inh-GAPLA kiszámolt értékei;
|
|
az Ind-IL8LA 95 %-os konfidenciaintervalluma;
|
|
a luciferáz-aktivitás indukciójára és az életképességre vonatkozó dózis-válasz görbéket ábrázoló grafikon;
|
|
adott esetben bármely más releváns észrevétel leírása.
|
|
|
Az eredmények tárgyalása
|
az IL-8 Luc vizsgálattal kapott eredmények tárgyalása;
|
|
a vizsgálat eredményeinek integrált vizsgálati és értékelési megközelítés keretében történő tárgyalása, amennyiben rendelkezésre állnak más releváns információk.
|
|
Következtetés
|
SZAKIRODALOM
|
(1)
|
Takahashi T, Kimura Y, Saito R, Nakajima Y, Ohmiya Y, Yamasaki K, and Aiba S. (2011). An in vitro test to screen skin sensitizers using a stable THP-1-derived IL-8 reporter cell line, THP-G8. Toxicol Sci 124:359-69.
|
|
(2)
|
OECD (2017). Validation report for the international validation study on the IL-8 Luc assay as a test evaluating the skin sensitizing potential of chemicals conducted by the IL-8 Luc Assay. Series on Testing and Assessment No 267, ENV/JM/MONO(2017)19. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(3)
|
OECD (2017). Report of the Peer Review Panel for the IL-8 Luciferase (IL-8 Luc) Assay for in vitro skin sensitisation. Series on Testing and Assessment No 258, ENV/JM/MONO(2017)20. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(4)
|
OECD (2016) Guidance Document On The Reporting Of Defined Approaches And Individual Information Sources To Be Used Within Integrated Approaches To Testing And Assessment (IATA) For Skin Sensitisation, Series on Testing & Assessment No 256, ENV/JM/MONO(2016)29. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen: http://www.oecd.org/env/ehs/testing/series-testing-assessment-publications-number.htm.
|
|
(5)
|
van der Veen JW, Rorije E, Emter R, Natsch A, van Loveren H, and Ezendam J. (2014). Evaluating the performance of integrated approaches for hazard identification of skin sensitizing chemicals. Regul Toxicol Pharmacol 69:371-9.
|
|
(6)
|
Kimura Y, Fujimura C, Ito Y, Takahashi T, Nakajima Y, Ohmiya Y, and Aiba S. (2015). Optimization of the IL-8 Luc assay as an in vitro test for skin sensitization. Toxicol In Vitro 29:1816-30.
|
|
(7)
|
Urbisch D, Mehling A, Guth K, Ramirez T, Honarvar N, Kolle S, Landsiedel R, Jaworska J, Kern PS, Gerberick F, et al. (2015). Assessing skin sensitization hazard in mice and men using non-animal test methods. Regul Toxicol Pharmacol 71:337-51.
|
|
(8)
|
Ashikaga T, Sakaguchi H, Sono S, Kosaka N, Ishikawa M, Nukada Y, Miyazawa M, Ito Y, Nishiyama N, and Itagaki H. (2010). A comparative evaluation of in vitro skin sensitisation tests: the human cell-line activation test (h-CLAT) versus the local lymph node assay (LLNA). Alternatives to laboratory animals: ATLA 38:275-84.
|
|
(9)
|
Patlewicz G, Casati S, Basketter DA, Asturiol D, Roberts DW, Lepoittevin J-P, Worth A and Aschberger K (2016) Can currently available non-animal methods detect pre and pro haptens relevant for skin sensitisation? Regul Toxicol Pharmacol, 82:147-155.
|
|
(10)
|
Thorne N, Inglese J, and Auld DS. (2010). Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem Biol 17:646-57.
|
|
(11)
|
OECD (2016). Test No 455: Performance-Based Test Guideline for Stably Transfected Transactivation In Vitro Assays to Detect Estrogen Receptor Agonists and Antagonists, OECD Publishing, Paris. http://dx.doi.org/10.1787/9789264265295-en.
|
|
(12)
|
Viviani V, Uchida A, Suenaga N, Ryufuku M, and Ohmiya Y. (2001). Thr226 is a key residue for bioluminescence spectra determination in beetle luciferases. Biochem Biophys Res Commun 280:1286-91.
|
|
(13)
|
Viviani VR, Bechara EJ, and Ohmiya Y. (1999). Cloning, sequence analysis, and expression of active Phrixothrix railroad-worms luciferases: relationship between bioluminescence spectra and primary structures. Biochemistry 38:8271-9.
|
|
(14)
|
Nakajima Y, Kimura T, Sugata K, Enomoto T, Asakawa A, Kubota H, Ikeda M, and Ohmiya Y. (2005). Multicolor luciferase assay system: one-step monitoring of multiple gene expressions with a single substrate. Biotechniques 38:891-4.
|
|
(15)
|
OECD (2017). To be published - Performance Standards for the assessment of proposed similar or modified in vitro skin sensitisation IL-8 luc test methods. OECD Environment, Health and Safety Publications, Series on Testing and Assessment. OECD, Paris, France
|
|
(16)
|
OECD (2005). Guidance Document the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, OECD Series on Testing and Assessment No 34. OECD, Párizs, Franciaország.
|
|
(17)
|
OECD (2018). Draft Guidance document: Good In Vitro Method Practices (GIVIMP) for the Development and Implementation of In Vitro Methods for Regulatory Use in Human Safety Assessment. Gazdasági Együttműködési és Fejlesztési Szervezet, Párizs. Elérhető a következő címen: http://www.oecd.org/env/ehs/testing/OECD Final Draft GIVIMP.pdf.
|
|
(18)
|
JaCVAM (2016). IL-8 Luc assay protocol, elérhető a következő címen: http://www.jacvam.jp/en_effort/effort02.html.
|
|
(19)
|
Niwa K, Ichino Y, Kumata S, Nakajima Y, Hiraishi Y, Kato D, Viviani VR, and Ohmiya Y. (2010). Quantum yields and kinetics of the firefly bioluminescence reaction of beetle luciferases. Photochem Photobiol 86:1046-9.
|
|
(20)
|
OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins, Part 1: Scientific Evidence. OECD Environment, Health and Safety Publications, Series on Testing and Assessment No 168. OECD, Párizs, Franciaország.
|
|
(21)
|
United Nations (2015). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). Sixth revised edition. New York & Geneva: United Nations Publications. ISBN: 978-92-1-117006-1. Elérhető a következő címen: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
|
3.1. függelék
FOGALOMMEGHATÁROZÁSOK
Pontosság
: a vizsgálattal kapott eredmények és az elfogadott referenciaértékek közötti egyezés mértéke. A vizsgálat megfelelőségének mutatója, és relevanciájának egyik szempontja. A kifejezés gyakran használatos az „egyezés” szó megfelelőjeként, amely arra utal, hogy egy adott vizsgálat milyen arányban szolgáltat helyes eredményeket (16).
Káros kimeneti út (Adverse Outcome Pathway, AOP)
: valamely megcélzott vegyi anyag vagy hasonló vegyi anyagok csoportjának kémiai szerkezetéből kiinduló, a molekuláris kiváltó eseményen keresztül az érintett in vivo eredményig végbemenő eseménysorozat (20).
Vegyi anyag
: anyag vagy keverék.
CV05
: 05-ös életképesség, vagyis az a legalacsonyabb koncentráció, amelynél a vegyi anyag az Inh-GAPLA 0,05 alatti szintjét idézi elő.
FInSLO-LA
: az Ind-IL8LA értékre utaló rövidítés, amelyet az IL-8 Luc vizsgálatra vonatkozó validálási jelentésben és korábbi publikációkban használtak. Meghatározását lásd az Ind-IL8LA címszó alatt.
GAPLA
: a stabil vörös luciferáz (SLR) luciferáz-aktivitása (λmax = 630 nm), amelyet a GAPDH promóter szabályoz, és amely a sejtéletképességről és az elő sejtek számáról nyújt felvilágosítást.
Veszély
: valamely anyag vagy helyzet azon sajátossága, hogy káros hatásokat képes előidézni az élő szervezet, rendszer vagy (al)populáció anyagnak való expozíciójakor.
Integrált vizsgálati és értékelési megközelítés
: valamely vegyi anyag vagy vegyianyag-csoport veszélyeinek azonosításához (potenciál), jellemzéséhez (hatáserősség) és/vagy biztonsági értékeléséhez (potenciál, hatáserősség és expozíció) alkalmazott strukturált megközelítés, amely stratégiai szempontból beépít és mérlegel minden olyan releváns adatot, amely hozzájárul a lehetséges veszélyre és/vagy kockázatra és/vagy a további célirányos és ezért minimális vizsgálat szükségességére vonatkozó szabályozói döntéshozatalhoz.
II-SLR-LA
: az Inh-GAPLA értékre utaló rövidítés, amelyet az IL-8 Luc vizsgálatra vonatkozó validálási jelentésben és korábbi publikációkban használtak. Meghatározását lásd az Inh-GAPLA címszó alatt.
IL-8 (Interleukin-8)
: endoteliális sejtekből, fibroblasztokból, keratinsejtekből, makrofágokból és monocitákból származó citokin, amely a neutrofilek és a T-sejt limfociták kemotaxisát idézi elő.
IL8LA
: a stabil narancssárga luciferáz (SLO) luciferáz-aktivitása (λmax = 580 nm), amelyet az IL-8 promóter szabályoz.
Ind-IL8LA
: az nIL8LA indukciós faktora. Meghatározásához elosztják a vegyi anyaggal kezelt THP-G8 sejtek nIL8LA értékét a nem stimulált THP-G8 sejtek nIL8LA értékével, az Ind-IL8LA a vegyi anyagok által előidézett IL-8 promóter aktivitás mutatója.
Inh-GAPLA
: A GAPLA gátlása. Meghatározásához elosztják a vegyi anyaggal kezelt THP-G8 sejtek GAPLA értékét a nem kezelt THP-G8 sejtek GAPLA értékével, az Inh-GAPLA a vegyi anyagok által előidézett citotoxicitás mutatója.
Minimális indukciós küszöbérték (MIT)
: az a legalacsonyabb koncentráció, amelynél a vegyi anyag eleget tesz a pozitív besorolás kritériumainak.
Keverék
: két vagy több anyagot tartalmazó elegy vagy oldat.
Egy összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben az egyik fő összetevő legalább 80 tömegszázalékban van jelen.
Több összetevőből álló anyag
: olyan, a mennyiségi összetétele alapján meghatározott anyag, amelyben egynél több fő összetevő több mint 10 tömegszázalékban, de 80 tömegszázalékot nem meghaladó koncentrációban van jelen. A több összetevőből álló anyag gyártási folyamat eredménye. A keverék és a több összetevőből álló anyag között az a különbség, hogy a keverék két vagy több anyag összekeverésével, kémiai reakció nélkül jön létre. A több összetevőből álló anyag kémiai reakció eredménye.
nIL8LA
: az IL-8 promóter aktivitását tükröző SLO luciferáz-aktivitás (IL8LA) a GAPDH promóter aktivitását tükröző SLR luciferáz-aktivitással (GAPLA) normalizálva. A sejtéletképesség és a sejtszám figyelembevételét követő IL-8 luciferáz-aktivitás mutatója.
nSLO-LA
: az nIL8LA értékre utaló rövidítés, amelyet az IL-8 Luc vizsgálatra vonatkozó validálási jelentésben és korábbi publikációkban használtak. Meghatározását lásd az nIL8LA címszó alatt.
Pozitív kontroll
: a vizsgálati rendszer valamennyi alkotóelemét tartalmazó, ismert pozitív hatást kiváltó anyaggal kezelt replikátum. Annak biztosítása érdekében, hogy a pozitív kontrollban jelentkező hatás időbeli változását fel lehessen mérni, a pozitív hatás nem lehet túlságosan erőteljes.
Pre-hapténok
: abiotikus átalakulás révén szenzibilizálóvá váló vegyi anyagok.
Pro-hapténok
: olyan vegyi anyagok, amelyek enzimatikus aktivációt igényelnek bőrszenzibilizáló hatásuk kifejtéséhez.
Relevancia
: a vizsgálat és a vizsgált hatás kapcsolatát adja meg, valamint azt, hogy van-e a vizsgálatnak az adott cél szempontjából értelme és haszna. Azt tükrözi, hogy a vizsgálat mennyire pontosan méri vagy jelzi előre a vizsgált biológiai hatást. A relevancia meghatározása során a vizsgálat pontosságát (az eredmények egyezését) figyelembe kell venni (16).
Megbízhatóság
: a vizsgálat laboratóriumon belüli és laboratóriumok közötti időbeli reprodukálhatóságának mértéke ugyanazon protokoll alkalmazása mellett. Megállapítása a laboratóriumon belüli és a laboratóriumok közötti reprodukálhatóság, valamint a laboratóriumon belüli megismételhetőség kiszámításával történik (16).
Vizsgálatmenet
: egy vizsgálatmenet során egy vagy több vizsgálati vegyi anyagot vizsgálnak egy párhuzamos oldószeres/vivőanyagos kontrollal és egy pozitív kontrollal.
Érzékenység
: az összes olyan pozitív/aktív vegyi anyag aránya, amelyet a vizsgálat helyesen sorolt be. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (16).
SLO-LA
: az IL8LA értékre utaló rövidítés, amelyet az IL-8 Luc vizsgálatra vonatkozó validálási jelentésben és korábbi publikációkban használtak. Meghatározását lásd az IL8LA címszó alatt.
SLR-LA
: a GALPA értékre utaló rövidítés, amelyet az IL-8 Luc vizsgálatra vonatkozó validálási jelentésben és korábbi publikációkban használtak. Meghatározását lásd a GAPLA címszó alatt.
Oldószeres/vivőanyagos kontroll
: valamely vizsgálati rendszernek a vizsgálati vegyi anyagon kívüli összes összetevőjét tartalmazó kezeletlen minta, amely azonban magában foglalja az alkalmazott oldószert/vivőanyagot. Az ugyanabban az oldószerben/vivőanyagban feloldott vagy stabilan diszpergált vizsgálati vegyi anyaggal kezelt minták esetében a kiindulási érték létrehozásához használatos. Párhuzamos tápoldatos kontrollal elvégzett vizsgálata esetén ez a minta azt is bizonyítja, hogy az oldószer/vivőanyag kölcsönhatásba lép-e a vizsgálati rendszerrel.
Specificitás
: a vizsgálat elvégzésével helyesen besorolt összes negatív/inaktív vegyi anyag aránya. A kategorikus eredményt adó vizsgálati módszerek pontosságának mutatója, valamint fontos szempont a vizsgálati módszerek relevanciájának megítélésében (16).
Anyag
: természetes állapotban előforduló vagy termelőfolyamatból származó kémiai elemek és azok vegyületei, amelyek a termék stabilitásának megőrzéséhez szükséges adalékanyagokat és az alkalmazott folyamatból származó szennyeződéseket is tartalmazhatnak, de nem tartalmaznak olyan oldószereket, amelyek az anyag stabilitásának befolyásolása vagy összetételének megváltozása nélkül elkülöníthetők.
Felületaktív anyag
: olyan anyag (például mosószer), amely csökkentheti a folyadékok felületi feszültségét, és így lehetővé teszi, hogy habozzanak vagy áthatoljanak szilárd anyagokon; nedvesítőszerként is ismert. (TG437)
Vizsgálati vegyi anyag
: bármely, e módszer alkalmazásával vizsgált anyag vagy keverék.
THP-G8
: az IL-8 Luc vizsgálatban használt IL-8 riporter sejtvonal. A humán makrofágszerű THP-1 sejtvonalat transzfektálták az IL-8 promóterrel szabályozott stabil narancssárga luciferáz (SLO) luciferáz génnel és a GAPDH promóterrel szabályozott stabil vörös luciferáz (SLR) luciferáz génnel.
A vegyi anyagok osztályozásának és címkézésének az Egyesült Nemzetek Szervezete által globálisan harmonizált rendszere (ENSZ-GHS)
: a vegyi anyagoknak (anyagoknak és keverékeknek) a fizikai, egészségi és környezeti veszélyek szabványosított típusai és szintjei szerinti osztályokba sorolására és megfelelő kommunikációs elemekkel (például piktogramokkal, figyelmeztetésekkel, figyelmeztető mondatokkal, óvintézkedésekre vonatkozó mondatokkal és biztonsági adatlapokkal) történő jelölésére javaslatokat megfogalmazó rendszer, amelynek célja, hogy az emberek (köztük a munkáltatók, a munkavállalók, a fuvarozók, a fogyasztók és a sürgősségi segélyszolgálatok) és a környezet megóvása érdekében egységesítse a vegyi anyagok káros hatásaira vonatkozó információk továbbítását (21).
UVCB
: ismeretlen szerkezetű vagy változó összetételű, összetett reakcióban keletkezett vagy biológiai eredetű anyagok.
Érvényes vizsgálati módszer
: olyan vizsgálat, amely kellően relevánsnak és megbízhatónak minősül egy adott célra, és amely tudományosan megalapozott elveken alapul. Egy-egy vizsgálat abszolút értelemben soha nem érvényes, kizárólag meghatározott céllal összefüggésben.
3.2. függelék
A LUCIFERÁZ-AKTIVITÁS MÉRÉSÉNEK ÉS AZ SLO/SLR MÉRÉSÉHEZ HASZNÁLT OPTIKAI SZŰRŐ TRANSZMISSZIÓS KOEFFICIENSE MEGHATÁROZÁSÁNAK ELVE
Több riportert alkalmazó vizsgálati rendszer – Tripluc – használható, több színt detektálására képes rendszerrel működő mikrolemez típusú luminométerrel, amely felszerelhető optikai szűrővel (pl. Phelios AB-2350 (ATTO), ARVO (PerkinElmer), Tristar LB941 (Berthold)). A mérések során használt optikai szűrő 600–620 nm-es long vagy short pass szűrő, illetve 600–700 nm-es sávszűrő.
Két különböző színű luciferáz mérése egy optikai szűrővel
Az alábbi példában Phelios AB-2350 (ATTO) típusú szűrőt használtak. Ez a luminométer az SLO (λmax = 580 nm) és az SLR (λmax = 630 nm) lumineszcens jeleinek megkülönböztetésére egy 600 nm-es long pass szűrőt alkalmaz (R60 HOYA Co., 600 nm-es LP, 1. szűrő).
A 600 nm-es LP-szűrő transzmissziós koefficiensének meghatározásához tisztított SLO és SLR luciferáz enzim használatával megmérik i. az SLO és az SLR biolumineszcenciájának intenzitását szűrő nélkül (F0), ii. az SLO és az SLR biolumineszcenciájának intenzitását 600 nm-es LP-szűrőn (1. szűrőn) keresztül, valamint iii. kiszámolják a 600 nm-es LP-szűrő alábbi transzmissziós koefficienseit az SLO és az SLR méréséhez.
|
Transmission coefficients
|
Abbreviation
|
Definition
|
|
SLO
|
Filter 1 Transmission coefficients
|
=κOR60
|
The filter’s transmission coefficient for the SLO
|
|
SLR
|
Filter 1 Transmission coefficients
|
κRR60
|
The filter’s transmission coefficient for the SLR
|
Ha a vizsgálati minta SLO intenzitását O-val, SLR intenzitását R-rel jelöljük, akkor i) a fény (optikai) szűrő nélküli intenzitása (F0) és ii) a 600 nm-es LP-szűrőn (1. szűrőn) keresztül haladó fény intenzitása (F1) az alábbiak szerint határozható meg.
F0=O+R
F1=κOR60 x O + κRR60 x R
Ezeket a képleteket az alábbi módon is fel lehet állítani:

Ezt követően a kiszámított transzmissziós tényezők (κOR60 és κRR60), valamint a mért F0 és F1 alapján kiszámolható az O és az R értéke:

A transzmissziós tényező meghatározásához használat eszközök és módszerek
|
1.
|
Reagensek
Az egyes tisztított luciferáz enzimek:
|
Liofilizált tisztított SLO enzim
|
|
Liofilizált tisztított SLR enzim
|
|
(amelyeket a validálási tevékenységek céljára a GPC Lab. Co. Ltd. vállalattól (Tottori, Japán) szereztek be, a THP-G8 sejtvonallal együtt)
|
Vizsgálati reagens:
|
Tripluc® luciferáz vizsgálati reagens (beszerezhető például: TOYOBO, kat.száma: MRA-301)
|
Tápoldat: a luciferáz vizsgálathoz (30 ml, 2–8 °C-on tárolva)
|
|
Reagens
|
Konc.
|
Végső konc. tápoldatban
|
Szükséges mennyiség
|
|
RPMI-1640
|
—
|
—
|
27 ml
|
|
FBS
|
—
|
10 %
|
3 ml
|
|
2.
|
Az enzimoldat elkészítése
Csőbe helyezett liofilizált tisztított luciferáz enzimet oldjunk fel 10 tömegszázalékos glicerinnel kiegészített, 200 μl mennyiségű 10 ~ 100 mM Tris/HCl-dal vagy Hepes/HCl-dal (pH-értéke: 7,5 ~ 8,0), majd az enzimoldatot 10 μl mennyiségben adagoljuk ki 1,5 ml-es eldobható csövekbe, és tároljuk fagyasztóban, –80 °C-on. A fagyasztott enzimoldat akár 6 hónapig is felhasználható. Felhasználásukkor az enzimoldatot tartalmazó csövekhez adjunk hozzá 1 ml-t a luciferáz vizsgálat tápoldatából (10 % FBS-sel kiegészített RPMI-1640), így egy hígított enzimoldatot kapunk, amelyet az enzim deaktivációjának megelőzése érdekében jégen kell tartani.
|
|
3.
|
A biolumineszcencia mérése
Olvasszuk fel a Tripluc® luciferáz vizsgálati reagenst (Tripluc), és tartsuk szobahőmérsékletű vízfürdőben vagy szobahőmérsékletű levegőn. A fotomultiplikátor stabilizálódása érdekében a mérés előtt 30 perccel kapcsoljuk be a luminométert. Helyezzünk át 100 μl feloldott enzimoldatot egy fekete 96 lyukú (lapos fenekű) lemezre (az SLO referenciamintáját a B1, B2 és B3 lyukakba, az SLR referenciamintáját pedig a D1, D2 és D3 lyukakba). Ezt követően pipettával adagoljunk 100 μl felmelegített Tripluc reagenst a lemez feloldott enzimoldatot tartalmazó valamennyi lyukába. Lemezrázón rázassuk a lemezt szobahőmérsékleten (kb. 25 °C-on) 10 percig. Ha a lyukakban lévő oldatokban buborékok keletkeznének, távolítsuk el azokat. Helyezzük a lemezt a luciferáz-aktivitás méréséhez a luminométerbe. A biolumineszcenciát 3 másodpercig mérik optikai szűrő nélkül (F0) és optikai szűrővel (F1).
Az optikai szűrő transzmissziós koefficiensét az alábbi módon határozhatjuk meg:
Transzmissziós koefficiens (SLO (κOR60)) = (B1-nél mért F1+ B2-nél mért F1+ B3-nál mért F1) / (B1-nél mért F0 + B2-nél mért F0 + B3-nál mért F0)
Transzmissziós koefficiens (SLR (κOR60)) = (D1-nél mért F1+ D2-nél mért F1+ D3-nál mért F1) / (D1-nél mért F0 + D2-nél mért F0 + D3-nál mért F0)
Az ily módon meghatározott transzmissziós tényezők az adott luminométerrel végzett valamennyi méréshez felhasználhatók.
|
A berendezések minőség-ellenőrzése
Az IL-8 Luc vizsgálati protokollban leírt eljárásokat kell használni (18).
3.3. függelék
JÁRTASSÁGI TESZTANYAGOK
A B.71. vizsgálati módszer e függelékben ismertetett vizsgálatának rutinszerű alkalmazása előtt a laboratóriumoknak igazolniuk kell szakmai jártasságukat oly módon, hogy az 1. táblázatban javasolt 9 anyagra vonatkozóan az IL-8 Luc vizsgálattal várható előrejelzést kapják meg, továbbá a 9 jártassági tesztanyag közül legalább 8 esetében referenciatartományba eső értékeket kapnak (e jártassági tesztanyagokat úgy választották ki, hogy a bőrszenzibilizáció veszélye esetén bekövetkező válaszreakciók teljes tartományát képviseljék). A kiválasztás többi kritériuma az volt, hogy a vegyi anyagok kereskedelmi forgalomban beszerezhetők legyenek, azokra vonatkozóan kiváló minőségű in vivo referenciaadatok álljanak rendelkezésre, és az IL-8 Luc vizsgálat alapján kiváló minőségű in vitro adatok álljanak rendelkezésre. Az IL-8 Luc vizsgálathoz rendelkezésre állnak publikált referenciaadatok is (6) (1).
1. táblázat
Az IL-8 Luc vizsgálatban való szakmai jártasság bizonyításához ajánlott anyagok
|
Jártassági tesztanyagok
|
CAS-szám
|
Oldhatósági
|
állapot 20 mg/ml X-VIVO15-ben
|
In vivo előrejelzés (109)
|
IL-8 Luc előrejelzés (110)
|
Referenciatartomány μg/ml) (111)
|
|
|
CV05
(112)
|
IL-8 Luc MIT (113)
|
|
2,4-Dinitro-klór-benzol
|
97-00-7
|
Szilárd
|
Oldhatatlan
|
Szenzibilizáló (szélsőségesen)
|
Pozitív
|
2,3–3,9
|
0,5–2,3
|
|
Formaldehid
|
50-00-0
|
Folyadék
|
Oldható
|
Szenzibilizáló (erősen)
|
Pozitív
|
9–30
|
4–9
|
|
2-Merkaptobenzotiazol
|
149-30-4
|
Szilárd
|
Oldhatatlan
|
Szenzibilizáló (mérsékelten)
|
Pozitív
|
250–290
|
60–250
|
|
Etilén-diamin
|
107-15-3
|
Folyadék
|
Oldható
|
Szenzibilizáló (mérsékelten)
|
Pozitív
|
500–700
|
0,1–0,4
|
|
Etilén-glikol-dimetakrilát
|
97-90-5
|
Folyadék
|
Oldhatatlan
|
Szenzibilizáló (enyhén)
|
Pozitív
|
> 2000
|
0,04–0,1
|
|
4-Allil-anisol (esztragol)
|
140-67-0
|
Folyadék
|
Oldhatatlan
|
Szenzibilizáló (enyhén)
|
Pozitív
|
> 2000
|
0,01–0,07
|
|
Sztreptomicin-szulfát
|
3810-74-0
|
Szilárd
|
Oldható
|
Nem szenzibilizáló
|
Negatív
|
> 2000
|
> 2000
|
|
Glicerin
|
56-81-5
|
Folyadék
|
Oldható
|
Nem szenzibilizáló
|
Negatív
|
> 2000
|
> 2000
|
|
Izopropanol
|
67-63-0
|
Folyadék
|
Oldható
|
Nem szenzibilizáló
|
Negatív
|
> 2000
|
> 2000
|
|
Rövidítések: CAS-szám = Vegyianyag Nyilvántartási Szolgálat (CAS) nyilvántartási szám.
|
3.4. függelék
INDEXEK ÉS DÖNTÉSI KRITÉRIUMOK
nIL8LA (nSLO-LA)
Az IL8LA (SLO-LA) és a GAPLA (SLR-LA) tekintetében megmérik az i. koncentráció (i = 0–11) j. ismétlését (j = 1–4). A normalizált IL8LA, vagyis az nIL8LA (nSLO-LA), az alábbiak szerint határozható meg:
nIL8LAij = IL8LAij/GAPLAij
Ez a vizsgálat alapvető mutatója.
Ind-IL8LA (FInSLO-LA)
Az ismétlés átlagos nIL8LA (nSLO-LA) értékének az i. koncentrációban tapasztalt növekedési faktora összevetve a 0. koncentráció növekedési faktorával, ez a vizsgálat elsődleges mutatója. A két növekedési faktor aránya az alábbi egyenlettel írható le:

A vezető laboratórium javaslata szerint az 1,4-es érték a vizsgálati vegyi anyag pozitív besorolását eredményezi. Ez az érték a vezető laboratórium dokumentált adatainak vizsgálatán alapul. Az adatkezelő csoport ezt az értéket használta a validálási tanulmány valamennyi szakaszában. Az elsődleges eredmény, az Ind-IL8LA a két számtani átlag aránya, ahogyan az az egyenletből is látszik.
95 %-os konfidenciaintervallum (95 % CI)
Az Ind-IL8LA arányon alapuló 95 %-os konfidenciaintervallum (95 % CI) megbecsülésével kimutatható az elsődleges eredmény mérésének pontossága. Amennyiben a 95 %-os konfidenciaintervallum alsó határértéke legalább 1, az arra utal, hogy az i. koncentrációnál elért nIL8LA érték jelentősen nagyobb az oldószeres kontroll nIL8LA értékénél. A 95 %-os konfidenciaintervallum meghatározásának több módja is létezik. Ebben a vizsgálatban az úgynevezett Fieller-tételt alkalmaztuk. E tétel alapján a 95 %-os konfidenciaintervallumot az alábbi egyenlet segítségével határozhatjuk meg:

ahol: . a



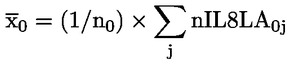



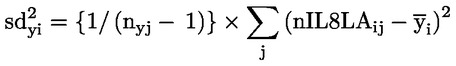
a centrális t eloszlás 97,5. percentilise ν szabadságfokkal, ahol

Inh-GAPLA (II-SLR-LA)
Az Inh-GAPLA az ismétlés i. koncentrációjában mért átlagos GAPLA (SLR-LA) érték és az oldószeres kontroll átlagos GAPLA (SLR-LA) értékének aránya, amely az alábbi egyenlettel írható le:

Mivel a GAPLA az nIL8LA nevezője, a szélsőségesen alacsony értékek az nIL8LA nagy variabilitását jelzik. Ezért előfordulhat, hogy a szélsőségesen alacsony (0,05-nál kisebb) Inh-GAPLA értékkel rendelkező Ind-IL8LA értékek csekély pontosságúak.
3.5. függelék
AZ IL-8 LUC VIZSGÁLATBAN A VEGYI ANYAGOK FELOLDÁSÁRA ALKALMAZOTT MÓDSZEREK SEMATIKUS ÁBRÁJA
|
a)
|
A 20 mg/ml koncentrációjú X-VIVOTM 15-ben oldódó vegyi anyagok esetében

|
|
b)
|
A 20 mg/ml koncentrációjú X-VIVOTM 15-ben nem oldódó vegyi anyagok esetében

|
3.6. függelék
AZ IL-8 LUC VIZSGÁLATBAN A POZITÍV KONTROLLANYAGKÉNT SZOLGÁLÓ 4-NBB FELOLDÁSÁRA ALKALMAZOTT MÓDSZER SEMATIKUS ÁBRÁJA
” |