ANNEXE
L'annexe du règlement (CE) no 440/2008 est modifiée comme suit:
|
(1) |
Dans la partie A, le chapitre suivant est ajouté: «A.25 CONSTANTE DE DISSOCIATION DANS L'EAU (MÉTHODE VOLUMÉTRIQUE — MÉTHODE SPECTROPHOTOMÉTRIQUE — MÉTHODE CONDUCTIMÉTRIQUE) INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 112 (1981) de l'OCDE pour les essais de produits chimiques. Conditions préalables
Informations générales
Conditions particulières
Documents de référence La présente méthode d'essai est basée sur les méthodes indiquées dans les références énumérées dans la rubrique “Bibliographie” et sur le document guide préliminaire pour la notification avant fabrication (EPA, 18 août 1978. METHODE — INTRODUCTION, OBJET, PORTEE, PERTINENCE, APPLICATION ET LIMITES DE D'ESSAI La dissociation d'une substance dans l'eau est un paramètre important pour évaluer l'impact de la substance sur l'environnement. Ce paramètre fixe la forme de la substance qui, à son tour détermine son comportement et son transport. Il peut affecter l'adsorption de la substance chimique sur les sols et les sédiments ainsi que l'absorption à l'intérieur des cellules biologiques. Définitions et unités La dissociation est le partage réversible en deux ou plusieurs espèces chimiques qui peuvent être ionisées. Le processus est généralement indiqué par l'équilibre: RX ⇌ R ++ X – et la constante d'équilibre gouvernant la réaction est:
Par exemple, dans le cas particulier où R est de l'hydrogène (la substance est un acide), la constante est:
ou
Substances de référence Quand on étudie une nouvelle substance, il n'est pas nécessaire d'utiliser à chaque fois les substances de référence ci-dessous. Elles sont surtout fournies pour pouvoir effectuer de temps en temps l'étalonnage de la méthode et pour permettre de comparer les résultats obtenus avec une autre méthode.
Il serait utile de disposer d'une substance qui possède plusieurs pK, comme cela est indiqué dans “Principe de la méthode”, ci-dessous. Une telle substance pourrait être:
Principe de la méthode Le processus chimique décrit ici ne varie en général que peu en fonction de la température, dans le domaine des températures rencontrées dans l'environnement. La détermination de la constante de dissociation implique la mesure des concentrations des formes dissociées et non- dissociées de la substance chimique étudiée. En connaissant la stœchiométrie de la réaction de dissociation indiquée dans la rubrique «Définitions et unités», ci-dessus, on peut déterminer la constante correspondante. Dans le cas particulier de cette méthode d'essai, la substance se comporte comme un acide ou une base, et il est plus commode d'effectuer la détermination en mesurant les concentrations relatives des formes ionisées et non-ionisées de la substance, ainsi que le pH de la solution. La relation qui existe entre ces différents termes est donnée dans l'équation du pKa dans la rubrique «Définitions et unités», ci-dessus. Certaines substances possèdent plus d'une constante de dissociation et on peut alors écrire des équations similaires. Quelques-unes des méthodes décrites ici sont également applicables aux dissociations non-acide/base. Critères de qualité Reproductibilité La mesure de la constante de dissociation doit être répétée au moins trois fois, les valeurs doivent se situer dans un intervalle de ± 0,1 unités de log. MODE OPERATOIRE Il existe deux façons de déterminer le pKa. L'une implique le dosage volumétrique d'une quantité connue de substance par un acide ou une base de référence selon le cas; l'autre consiste à déterminer la concentration relative des formes ionisées et non-ionisées, ainsi que leur variation en fonction du pH. Préparations Les méthodes basées sur ces deux principes peuvent être classées en méthodes volumétriques, méthodes spectrophotométriques et méthodes conductimétriques. Solutions d'essai Pour les méthodes volumétrique et conductimétrique, la substance chimique doit être dissoute dans l'eau distillée. Pour la méthode spectrophotométrique et les autres méthodes on emploie des solutions tampons. La concentration de la substance à tester ne doit pas dépasser 0,01 M ou la moitié de la concentration de saturation; pour faire les solutions on doit employer la substance sous la forme la plus pure qu'on puisse trouver. Si la substance n'est que faiblement soluble, elle peut être dissoute dans une petite quantité de solvant miscible à l'eau avant d'être diluée pour atteindre les concentrations indiquées ci-dessus. Si on a utilisé un co-solvant pour améliorer la solubilité, on doit vérifier l'absence d'émulsions dans les solutions, à l'aide d'un faisceau Tyndall. Quand des solutions tampons sont utilisées, la concentration du tampon ne doit pas excéder 0,05 M. Conditions expérimentales Température La température doit être contrôlée à ± 1 °C près au moins. L'expérience doit, de préférence, être réalisée à 20 °C. Si on pense que les résultats varient de façon significative avec la température, on doit répéter la détermination à deux autres températures, au moins. Dans ce cas, les intervalles de température doivent être de 10 °C, et la température doit être maintenue constante à ± 0,1 °C près. Analyses La méthode à employer sera déterminée par la nature de la substance à étudier. Elle doit être suffisamment sensible pour permettre la détermination des différentes espèces présentes à chaque concentration des solutions étudiées. Exécution de l'essai Méthode volumétrique La solution d'essai est dosée par titration avec une solution de référence d'acide ou de base, selon le cas; on mesure le pH après chaque addition du produit titrant. On doit faire au moins 10 additions avant le point d'équivalence. Si l'équilibre est atteint assez rapidement, on peut utiliser un potentiomètre enregistreur. Pour cette méthode, il est nécessaire de connaître de façon précise, à la fois la quantité totale de substance et sa concentration. On doit prendre soin d'éliminer le dioxyde de carbone. Les détails du mode opératoire, des précautions à prendre, et des calculs sont donnés dans les essais normalisés, par exemple dans les références (1), (2), (3) et (4). Méthode spectrophotométrique On doit trouver une longueur d'onde où les formes ionisées et non-ionisées de la substance ont des coefficients d'extinction suffisamment différents. On enregistre le spectre d'absorption UV- VIS de solutions de concentration constante, dans des conditions de pH où la substance est pratiquement non-ionisée, puis complètement ionisée, et enfin, à plusieurs pH intermédiaires. Ceci peut être réalisé, soit en ajoutant de l'acide (ou de la base) concentré à un volume relativement important d'une solution de la substance dans un tampon à plusieurs composants, initialement à pH élevé (faible) (réf 5), soit en ajoutant des volumes égaux d'une solution-mère de la substance, par exemple dans l'eau ou le méthanol, à des volumes constants de diverses solutions tampons, couvrant le domaine du pH désiré. A partir des valeurs de pH et d'absorbance à la longueur d'onde choisie, on calcule un nombre suffisant de valeurs du pKa en utilisant les données obtenues pour au moins 5 pH différents pour lesquels le taux d'ionisation de la substance se situe entre 10 % et 90 %. On trouvera d'autres détails expérimentaux et la méthode de calcul dans la référence 1. Méthode conductimétrique En utilisant une cuve, dont la constante est connue et petite, on mesure la conductivité d'une solution approximativement 0,1 M du composé dans l'eau. Sont également mesurées les conductivités d'un certain nombre de solutions obtenues par des dilutions précises de cette solution-mère. À chaque fois on diminue la concentration de moitié, et l'ensemble doit couvrir au moins un ordre de grandeur de concentration. On trouve la conductivité limite pour une dilution infinie, en effectuant une expérience similaire avec le sel de Na et en extrapolant. On peut alors calculer le degré de dissociation à partir de la conductivité de chaque solution, en utilisant l'équation d'Onsager, et à partir de là, en employant la loi de dilution d'Ostwald, la constante de dissociation peut être calculée par la formule: K = α2C/(1 – α) où C est la concentration en moles par litre et α est la fraction dissociée. On doit prendre soin d'éliminer le CO2. On trouvera d'autres détails expérimentaux et la méthode de calcul dans les textes de référence et dans les références 1, 6 et 7. RÉSULTATS ET RAPPORT Calcul des résultats Méthode volumétrique Le pKa est calculé pour 10 points mesurés sur la courbe de dosage. On calcule la moyenne et la déviation standard de ces pKa. On doit inclure une courbe du pH en fonction du volume de base ou d'acide de référence ajouté, ainsi qu'une présentation sous forme de tableau. Méthode spectrophotométrique Pour chaque spectre on fait entrer dans un tableau l'absorbance et le pH. A partir des points correspondant aux données sur les spectres intermédiaires, on calcule au moins cinq valeurs pour le pKa, ainsi que la moyenne et la déviation standard de ces résultats. Méthode conductimétrique La conductivité équivalente Λ est calculée pour chaque concentration acide et pour chaque concentration d'un mélange d'un équivalent d'acide plus 0,98 équivalent d'hydroxyde de sodium exempt de carbonate. L'acide est en excès afin d'éviter un excès en OH– dû à l'hydrolyse. Sur un graphique, on porte 1/Λ en fonction de √C, et on peut trouver le Λo du sel en extrapolant pour la concentration zéro. Le Λo de l'acide peut être calculé en utilisant les valeurs fournies par la bibliographie pour H+ et Na+. Le pKa peut être calculé à partir des formules α = Λi /Λo et Ka = α2C/(1 – α) pour chaque concentration. On peut obtenir de meilleures valeurs pour Ka en effectuant des corrections pour la mobilité et l'activité. On doit calculer les moyennes et les déviations standard des valeurs des pKa. Rapport Toutes les données brutes et les valeurs calculées pour le pKa doivent être fournies, ainsi que la méthode de calcul (de préférence sous la forme d'un tableau, comme cela est suggéré dans la référence 1) et les paramètres statistiques décrits ci-dessus. Pour les méthodes volumétriques, on doit donner les détails de la normalisation des produits titrants. Pour la méthode spectrophotométrique, tous les spectres doivent être fournis. Pour la méthode conductimétrique, on doit reporter les détails de la détermination de la constante de la cuve. On doit donner des informations sur la technique utilisée, les méthodes analytiques et la nature de chacun des tampons employés. Il convient de noter la ou les température(s) expérimentales. BIBLIOGRAPHIE
|


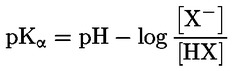
|
(2) |
Dans la partie B, le chapitre B.5 est remplacé par le texte suivant: «B.5 EFFET IRRITANT/CORROSIF AIGU SUR LES YEUX INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 405 (2012) de l'OCDE pour les essais de produits chimiques. Les lignes directrices de l'OCDE pour les essais de produits chimiques sont régulièrement réexaminées pour vérifier qu'elles intègrent les meilleures données scientifiques disponibles. Les précédents réexamens de cette méthode d'essai ont mis l'accent sur les possibilités d'éviter l'utilisation inutile des animaux de laboratoire afin de répondre aux préoccupations relatives au bien-être des animaux, moyennant l'évaluation préalable de toutes les informations existantes se rapportant aux produits chimiques d'essai. La ligne directrice 405 (adoptée en 1981 et mise à jour en 1987, 2002 et 2012) recommande d'analyser les résultats déjà disponibles en se fondant sur le poids de la preuve (1) avant d'envisager l'essai in vivo de l'effet irritant/corrosif aigu sur les yeux décrit dans cette ligne directrice. Il est conseillé de combler le manque de données par des essais séquentiels (2) (3). La stratégie d'essai inclut la réalisation d'essais in vitro validés et acceptés et est exposée dans un supplément à la présente méthode d'essai. Aux fins du règlement (CE) no 1907/2006 concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH) (2), une stratégie d'essai intégrée figure également dans le guide pertinent de l'ECHA (21). Les essais sur les animaux ne seront réalisés que s'ils s'avèrent nécessaires après examen des méthodes alternatives disponibles et mise en œuvre de celles jugées appropriées. À l'heure où la présente méthode d'essai est mise à jour, il existe encore des cas où le recours à cette méthode d'essai demeure indispensable ou est exigé par certains cadres réglementaires. La mise à jour la plus récente concerne essentiellement l'utilisation d'analgésiques et d'anesthésiques sans changer le concept de base et la structure de la ligne directrice. L'ICCVAM (3) et un groupe international d'experts scientifiques indépendants ont examiné l'utilité et les limites d'un recours en routine à des anesthésiques topiques, des analgésiques systémiques et des effets mesurés éthiquement acceptables lors d'essais in vivo d'irritation oculaire (12). Ils ont conclu que l'utilisation d'anesthésiques topiques et d'analgésiques systémiques permettait d'éviter la majeure partie, voire la totalité, de la douleur et de la détresse des animaux sans modifier le résultat de l'essai, et recommandé que ces substances soient systématiquement utilisées. La présente méthode d'essai tient compte des conclusions de cet examen. Il convient donc que les anesthésiques topiques, analgésiques systémiques et effets mesurés éthiquement acceptables soient utilisés en routine dans le cadre des essais in vivo de l'effet irritant/corrosif aigu sur l'œil. Toute exception à cet égard devra être justifiée. Les raffinements décrits dans cette méthode allègeront considérablement ou éviteront la douleur et la détresse chez les animaux dans la plupart des cadres expérimentaux qui exigent encore un essai de sécurité oculaire in vivo. Une gestion préventive et équilibrée de la douleur comprend: (i) un prétraitement en routine avec un anesthésique topique (p. ex. proparacaïne ou tétracaïne) et un analgésique systémique (p. ex. buprénorphine), (ii) un programme de traitement post-exposition en routine avec un analgésique systémique (p. ex. buprénorphine et méloxicam), (iii) un programme d'observation, de suivi et de consignation des signes cliniques de douleur et/ou de détresse chez les animaux, et (iv) un programme d'observation, de suivi et de consignation de la nature, de la gravité et de la progression de toutes les lésions oculaires. D'autres détails sont fournis dans les procédures mises à jour décrites ci-dessous. Après l'exposition au produit chimique d'essai, aucun anesthésique ou analgésique topique supplémentaire ne sera administré, de manière à éviter toute interférence avec l'essai. Les analgésiques dotés de propriétés anti-inflammatoires (comme le méloxicam) ne feront pas l'objet d'une application locale, et les doses systémiques employées ne devront pas perturber les effets sur l'œil. Les définitions sont données dans l'appendice de la présente méthode d'essai. REMARQUES PRÉLIMINAIRES Dans le souci de concilier la fiabilité des résultats scientifiques et le bien-être animal, on ne procédera pas aux essais in vivo avant d'avoir évalué, par une analyse du poids de la preuve, toutes les données relatives au caractère potentiellement corrosif ou irritant du produit chimique pour les yeux. Ces données comprennent les résultats d'études existantes menées sur des humains et/ou des animaux de laboratoire, des données établissant l'effet corrosif ou irritant pour l'œil d'une ou plusieurs substances structurellement proches de la substance d'essai ou de mélanges de ces substances, des données démontrant la forte acidité ou alcalinité de la substance (4) (5), et des résultats d'essais in vitro ou ex vivo de corrosion cutanée et de corrosion/irritation oculaires validés et acceptés (6) (13) (14) (15) (16) (17). Les études peuvent avoir été conduites avant, ou à la suite de, l'analyse du poids de la preuve. Pour certains produits chimiques, une telle analyse peut faire valoir la nécessité de mener des études in vivo du pouvoir corrosif/irritant pour l'œil du produit chimique. Le cas échéant, avant d'envisager un essai oculaire in vivo, il est préférable d'effectuer d'abord un essai in vitro et/ou in vivo de la corrosion cutanée induite par le produit chimique, et d'évaluer ces résultats conformément à la stratégie d'essai séquentielle de la méthode d'essai B.4 (7) ou à la stratégie d'essai intégrée décrite dans le guide de l'ECHA (21). Une stratégie d'essai de type séquentiel, comportant des essais in vitro ou ex vivo de corrosion/irritation de l'œil validés, est décrite dans un supplément à la présente méthode d'essai et, aux fins de REACH, dans le guide de l'ECHA (21). Il est recommandé d'appliquer une telle stratégie d'essai avant de procéder à des essais in vivo. Pour les produits chimiques nouveaux, une stratégie d'essai par étapes est préconisée pour obtenir des données scientifiques fiables sur l'effet corrosif ou irritant du produit chimique. En ce qui concerne les produits chimiques existants pour lesquels les données sur l'effet corrosif ou irritant sur la peau et les yeux sont insuffisantes, il est possible de suppléer ces lacunes en appliquant cette stratégie. Le choix d'une autre stratégie d'essai ou la décision de ne pas procéder par étapes fait l'objet de justification. PRINCIPE DE L'ESSAI IN VIVO Après l'administration d'un analgésique systémique et l'induction d'une anesthésie topique adaptée, le produit chimique d'essai est appliqué en une seule dose sur un des yeux de l'animal d'expérience, l'œil non traité servant de témoin. On évalue le score de l'irritation ou de la corrosion oculaires en cotant la gravité des lésions affectant la conjonctive, la cornée et l'iris, à intervalles déterminés. Les autres réactions de l'œil et les troubles systémiques sont également décrits de manière à fournir une évaluation complète des effets. La durée de l'étude doit être suffisante pour permettre d'évaluer la réversibilité des effets. Les animaux qui présentent des signes de douleur et/ou de détresse aiguës à tout stade de l'essai, ou des lésions correspondant aux effets mesurés éthiquement acceptables décrits dans la présente méthode d'essai (voir paragraphe 26) seront euthanasiés, et ces symptômes seront à prendre en compte dans l'évaluation du produit chimique d'essai. Les critères régissant la décision d'euthanasier les animaux moribonds et pris de fortes douleurs sont exposés dans un document d'orientation de l'OCDE (8). PRÉPARATION DE L'ESSAI IN VIVO Choix des espèces On choisira de préférence des lapins albinos et de jeunes adultes sains seront utilisés. L'utilisation d'une autre espèce ou souche fait l'objet de justification. Préparation des animaux Les deux yeux de chaque animal susceptible de participer à l'essai sont examinés dans les 24 heures précédant le début de l'essai. Les animaux qui présentent des signes d'irritation oculaire, des défauts oculaires ou une lésion de la cornée seront écartés. Conditions d'hébergement et d'alimentation Les animaux sont placés dans des cages individuelles. La température du local expérimental est réglée à 20 °C (± 3 °C) pour les lapins. S'il convient que l'humidité relative atteigne au moins 30 % sans excéder de préférence 70 %, en dehors des heures de nettoyage du local, on s'efforcera de maintenir le taux d'humidité autour de 50 à 60 %. L'éclairage est artificiel, avec une séquence alternant 12 heures de lumière et 12 heures d'obscurité. On évitera les éclairages trop intenses. Les lapins seront nourris avec un mélange classique pour animaux de laboratoire et boiront de l'eau potable à volonté. MODE OPÉRATOIRE Utilisation d'anesthésiques topiques et d'analgésiques systémiques Les procédures suivantes sont recommandées pour réduire ou éviter la douleur et la détresse dans le cadre des essais de sécurité pour l'œil. On pourra faire appel à des procédures de remplacement dont il est avéré qu'elles sont aussi efficaces ou plus efficaces pour réduire ou éviter la douleur et la détresse des animaux.
Application du produit chimique d'essai L'expérimentateur introduit le produit chimique d'essai dans le cul-de-sac conjonctival d'un des deux yeux de chaque animal, après avoir délicatement écarté la paupière inférieure du globe oculaire. Il ramène ensuite délicatement les deux paupières l'une contre l'autre et les maintient dans cette position pendant environ une seconde afin d'éviter toute perte de substance. L'autre œil, qui ne subit pas de traitement, sert de témoin. Irrigation Il convient de ne pas laver les yeux des animaux traités pendant au moins 24 heures après l'instillation du produit chimique d'essai, à moins que celui-ci soit à l'état solide (voir paragraphe 18) ou déclenche immédiatement des effets corrosifs ou irritants. Au besoin, un lavage pourra être effectué à l'issue de ce délai. L'utilisation d'un groupe d'animal satellite pour étudier l'influence du lavage n'est pas indiquée, à moins qu'elle ne se justifie d'un point de vue scientifique. Le cas échéant, on utilisera deux lapins. Les conditions du lavage sont décrites minutieusement: par exemple le moment du lavage, la composition et la température de la solution ophtalmique, la durée, le volume et la vitesse d'application. Niveau de dose (1) Essais sur des liquides Pour les liquides, on utilise une dose de 0,1 ml. Il convient d'éviter d'instiller le produit chimique directement dans l'œil avec un vaporisateur à pression; il est préférable d'expulser d'abord le produit chimique dans une fiole, d'en prélever 0,1 ml et de l'instiller dans l'œil. (2) Essais sur des solides Dans le cas des solides, pâtes ou substances particulaires, la quantité utilisée a un volume de 0,1 ml ou un poids ne dépassant pas 100 mg. Le produit chimique d'essai sera broyé finement. Il convient de mesurer le volume de la substance solide après l'avoir légèrement tassée, par exemple en tapotant le récipient de mesure. Si le produit chimique d'essai solide n'a pas encore été évacué de l'œil de l'animal par des mécanismes physiologiques au premier moment d'observation, à savoir une heure après le traitement, l'œil peut être rincé à l'aide d'une solution saline ou à l'eau distillée. (3) Essais sur des aérosols Il est recommandé de prélever une dose du contenu de tous les vaporisateurs à pression et aérosols avant de l'instiller dans l'œil. La seule exception concerne les produits chimiques conditionnés en bombes aérosol sous pression, qui sont impossibles à recueillir préalablement à l'instillation car ils se vaporisent. Dans ce cas, l'expérimentateur maintient l'œil de l'animal ouvert et administre le produit chimique à tester en un seul jet d'environ une seconde, émis à 10 cm et directement en face de l'œil. Cette distance peut être modulée en fonction de la pression du jet et de sa composition. Il convient de veiller à ce que la pression du jet n'endommage pas l'œil. Dans certains cas, il pourra être nécessaire d'évaluer l'ampleur des dégâts «mécaniques» risquant d'être causés à l'œil par la force du jet. La dose d'aérosol peut être estimée grâce à une simulation de l'application menée comme suit: le produit chimique est projeté sur du papier pour pesée à travers une ouverture de la taille d'un œil de lapin placée directement devant le papier. L'augmentation du poids du papier donne une idée approximative de la quantité administrée à l'œil de lapin. Pour un produit volatile, la dose peut être estimée à partir du poids du récipient (dans lequel elle est contenue) avant et après utilisation. Essai initial (essai in vivo de l'effet irritant/corrosif sur les yeux mené sur un seul animal) Il est fortement recommandé de commencer par pratiquer l'essai in vivo sur un seul animal (voir le supplément à la présente méthode d'essai: Stratégie d'essai séquentielle pour les essais d'irritation et de corrosion oculaires). Les observations qui en découlent doivent permettre de déterminer la gravité et la réversibilité des lésions avant de mettre en œuvre un essai de confirmation avec un animal supplémentaire. Si les résultats de cet essai, mené selon la procédure décrite, indiquent que le produit chimique est corrosif ou fortement irritant pour l'œil, il n'y a pas lieu de mener d'autres essais d'irritation oculaire. Essai de confirmation (essai d'irritation oculaire in vivo conduit sur des animaux supplémentaires) Si l'essai initial ne révèle aucun effet corrosif ou fortement irritant, il convient de confirmer la réaction irritante ou négative sur un ou deux animaux supplémentaires. Si l'essai initial produit un effet irritant, il est recommandé de conduire l'essai de confirmation sur un mode séquentiel en n'utilisant qu'un seul animal à la fois, plutôt que d'exposer les deux animaux simultanément. Si le deuxième animal manifeste des signes de corrosion ou de forte irritation, l'essai s'arrête là. Si les résultats de l'essai sur le deuxième animal suffisent à déterminer la catégorie de danger de la substance, l'essai s'arrête là. Période d'observation La durée de la période d'observation doit être suffisante pour permettre d'évaluer entièrement l'ampleur et la réversibilité des effets observés. Il convient cependant de mettre un terme à l'essai dès qu'un animal manifeste des signes de détresse ou de douleur aiguë (8). Pour déterminer la réversibilité des effets, les animaux sont normalement observés durant 21 jours après l'exposition au produit chimique d'essai. Si la réversibilité est constatée avant ce délai, l'expérience prend fin à ce moment-là. Observations cliniques et cotation de la gravité des réactions oculaires Les yeux font l'objet d'un examen complet pour repérer d'éventuelles lésions oculaires une heure après l'APCE, puis cette procédure est répétée au moins une fois par jour. Les trois premiers jours qui suivent l'exposition, on examinera les animaux plusieurs fois par jour afin de pouvoir mettre un terme à l'essai en temps opportun, s'il y a lieu. Les animaux sont soumis à des examens de routine tout au long de l'essai pour rechercher des signes cliniques de douleur et/ou de détresse (p. ex. coups de patte ou frottements de l'œil répétés, cillement excessif, larmoiement excessif) (9) (10) (11), au moins deux fois par jour à six heures d'intervalle minimum, ou plus fréquemment si besoin est. Ces examens sont nécessaires pour (i) évaluer correctement les signes de douleur et de détresse des animaux afin d'établir la nécessité d'augmenter le dosage des analgésiques en conséquence et (ii) déterminer si les effets mesurés éthiquement acceptables sont atteints, de manière à décider en connaissance de cause d'euthanasier ou non les animaux et veiller à ce que les décisions d'euthanasier soient prises en temps voulu. Pour faciliter la détection et la mesure des lésions oculaires et déterminer si les effets observés éthiquement acceptables motivant l'euthanasie ont été atteints, on utilisera une coloration à la fluorescéine en routine ainsi qu'un biomicroscope (lampe à fente), si nécessaire (p. ex. pour évaluer la profondeur de la lésion en cas d'ulcération cornéenne). Des photographies numériques des lésions observées pourront être collectées à titre de référence et pour garder une trace permanente attestant l'étendue de la lésion oculaire. Les animaux ne seront maintenus dans l'essai que le temps nécessaire pour obtenir un résultat définitif. Ceux qui manifestent des signes de douleur ou de détresse aiguë sont euthanasiés immédiatement, et ces symptômes sont à prendre en compte dans l'évaluation du produit chimique d'essai. Il convient également d'euthanasier les animaux qui présentent les lésions oculaires suivantes à la suite de l'instillation (voir le tableau 1 pour une description des scores des lésions): une perforation de la cornée ou une ulcération profonde de la cornée associée à un staphylome; la présence de sang dans la chambre antérieure de l'œil; une opacité cornéenne de niveau 4; l'absence de réflexe photomoteur (réaction iridienne de niveau 2) durant 72 heures; l'ulcération de la membrane conjonctivale; une nécrose des conjonctives ou de la membrane nictitante; ou un décollement du tissu nécrosé. Et ce, parce que ces lésions sont généralement irréversibles. Il est par ailleurs recommandé de considérer les lésions oculaires suivantes comme des effets mesurés éthiquement acceptables, dont la survenue motive l'arrêt d'un essai avant la période d'observation de 21 jours prévue. On estime que ces lésions en annoncent d'autres symptomatiques d'un effet corrosif ou fortement irritant, ou qui ne seront que partiellement réversibles dans le délai d'observation de 21 jours: lésions très profondes (p. ex. une ulcération cornéenne qui dépasse les couches superficielles du stroma), destruction du limbe > 50 % (traduite par un blanchiment du tissu conjonctival) et infection oculaire aiguë (écoulement purulent). Une vascularisation de la surface cornéenne (c'est-à-dire un pannus) associée à une surface colorée à la fluorescéine qui ne diminue pas au fil des examens quotidiens et/ou à l'absence de réépithélialisation 5 jours après l'application du produit chimique d'essai peuvent aussi constituer un faisceau de critères utiles pour justifier la décision clinique de mettre un terme à l'essai prématurément. Cependant, chacun de ces résultats pris isolément ne suffit pas à motiver l'arrêt prématuré de l'essai. Dès que des effets graves sur l'œil sont observés, il convient de faire appel à un vétérinaire traitant ou spécialisé dans les animaux de laboratoire, ou à du personnel formé à identifier les lésions cliniques, pour mener un examen clinique permettant de juger si l'association de ces réactions implique un arrêt prématuré de l'essai. On attribuera un score aux réactions oculaires (des conjonctives, de la cornée et de l'iris) 1, 24, 48 et 72 heures après l'application du produit chimique d'essai, et les résultats seront consignés (tableau 1). Les animaux qui ne présentent pas de lésions oculaires peuvent être écartés, mais seulement à partir du quatrième jour suivant l'instillation. Les animaux dont les lésions ne sont pas sévères demeurent en observation jusqu'à la disparition de ces lésions, ou pendant 21 jours, après quoi l'essai prend fin. Ces observations sont effectuées et consignées au moins à 1, 24, 48, 72 heures, puis 7, 14 et 21 jours après l'APCE afin de déterminer l'état des lésions ainsi que leur réversibilité ou leur irréversibilité. Il est parfois nécessaire de rapprocher les examens pour déterminer s'il convient d'euthanasier un animal pour des raisons éthiques ou de l'écarter de l'essai du fait de résultats négatifs. Le niveau des lésions oculaires (tableau 1) est consigné à chaque examen. Toutes les autres lésions oculaires (p. ex. pannus, coloration, modifications de la chambre antérieure) et les troubles systémiques sont également signalés. Pour examiner les réactions, on peut s'aider d'une loupe binoculaire, d'une lampe à fente portative, d'un biomicroscope ou d'un autre appareil approprié. Après l'enregistrement des observations effectuées à la vingt-quatrième heure, l'examen des yeux peut se poursuivre à la fluorescéine. L'attribution de score aux réactions oculaires est inévitablement subjective. L'harmonisation de l'attribution de scores aux réactions oculaires et l'appui aux laboratoires d'essai ainsi qu'au personnel chargé d'effectuer et d'interpréter les observations passent par une formation adéquate des expérimentateurs au système de score utilisé. RÉSULTATS ET RAPPORTS Évaluation des résultats Les scores d'irritation oculaire doivent être évalués en considération de la nature et de la gravité des lésions ainsi que de leur caractère réversible ou non. Les scores individuels ne sont pas un critère absolu de propriétés irritantes d'un produit chimique, car d'autres effets du produit chimique sont également évalués. En revanche, les scores individuels ont une valeur de référence et ne sont significatifs que lorsqu'ils sont étayés par une description et une évaluation complètes de toutes les observations. Rapport d'essai Le rapport d'essai comporte les informations suivantes:
Interprétation des résultats L'extrapolation à l'humain des résultats d'études d'irritation oculaire menées sur des animaux de laboratoire n'est valable que dans une certaine mesure. Dans bien des cas, le lapin albinos est plus sensible que l'espèce humaine aux substances irritantes ou corrosives pour l'œil. Lors de l'interprétation des résultats, il faut savoir reconnaître une irritation consécutive à une infection secondaire, laquelle ne devra pas être prise en compte. BIBLIOGRAPHIE
Tableau 1 Cotation des lésions oculaires
Appendice DÉFINITIONS Réserve acide/alcaline : pour les préparations acides, quantité (g) d'hydroxyde de sodium/100 g de préparation nécessaire pour obtenir un pH déterminé. Pour les préparations basiques, il s'agit de la quantité (g) d'hydroxyde de sodium qui équivaut à la masse (g) d'acide sulfurique/100 g de préparation nécessaire pour obtenir un pH déterminé (Young et al. 1988). Produit chimique : une substance ou un mélange. Substance non irritante : substance qui n'est pas classée comme irritant oculaire au sens des catégories I, II ou III de l'EPA, des catégories 1, 2, 2A, ou 2B du SGH ou de la catégorie 1 ou 2 de l'UE (17) (18) (19). Produit chimique corrosif pour l'œil : (a) produit chimique provoquant des lésions irréversibles des tissus oculaires; (b) produit chimique classé comme irritant oculaire au sens de la catégorie 1 du SGH, de la catégorie 1 de l'EPA ou de la catégorie 1 de l'UE (17) (18) (19). Produit chimique irritant pour l'œil : (a) produit chimique provoquant une modification réversible de l'œil; (b) produit chimique classé comme irritant oculaire au sens des catégories II ou III de l'EPA, des cartégories 2, 2A ou 2B du SGH ou de la catégorie 2 de l'UE(17) (18) (19). Produit chimique fortement irritant pour l'œil : (a) produit chimique provoquant des lésions tissulaires de l'œil qui ne sont pas réversibles dans les 21 jours suivant l'application ou entraînent une dégradation sévère de la vision; (b) produit chimique classé comme irritant oculaire au sens de la catégorie 1 du SGH, de la catégorie 1 de l'EPA ou de la catégorie I de l'UE (17) (18) (19). Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Stratégie à plusieurs niveaux : stratégie d'essai séquentielle consistant à examiner toutes les informations existantes sur un produit chimique d'essai dans un ordre déterminé, en ayant recours à chaque étape à un processus d'analyse du poids de la preuve pour déterminer si les informations disponibles sont suffisantes pour décider d'une classification dans une catégorie de danger, avant de passer à l'étape suivante. Si le potentiel d'irritation d'un produit chimique d'essai peut être déterminé sur la base des informations existantes, aucun essai supplémentaire n'est nécessaire. Dans le cas contraire, une procédure expérimentale progressive de type séquentiel sur des animaux est alors lancée jusqu'à ce qu'une classification sans équivoque puisse être effectuée. Poids de la preuve : procédé consistant à prendre en compte les forces et les faiblesses de divers éléments d'information pour aboutir à une conclusion concernant les dangers potentiels d'un produit chimique et étayer cette conclusion. SUPPLÉMENT À LA MÉTHODE D'ESSAI B.5 (4) DÉMARCHE EXPERIMENTALE SEQUENTIELLE POUR LES ESSAIS D'IRRITATION ET DE CORROSION OCULAIRES Généralités Pour concilier la fiabilité des résultats scientifiques et le bien-être animal, il importe d'éviter l'utilisation abusive d'animaux d'expérience et de réduire au minimum toute procédure expérimentale susceptible de déclencher des effets graves chez les animaux. Avant d'envisager un essai in vivo, on évaluera toutes les informations relatives à l'éventuel effet corrosif/irritant d'un produit chimique sur les yeux. Il se peut que les données disponibles suffisent à classer le produit chimique d'essai quant à son pouvoir irritant ou corrosif pour les yeux, sans qu'il soit nécessaire d'effectuer des essais sur animaux. Ainsi, le recours à l'analyse du poids de la preuve et l'adoption d'une stratégie d'essai séquentielle limiteront au maximum la nécessité de pratiquer des essais in vivo, surtout si le produit chimique risque d'engendrer des réactions violentes. Il est recommandé d'évaluer les informations existantes concernant le pouvoir irritant ou corrosif des produits chimiques pour les yeux en se fondant sur le poids de la preuve, afin d'établir la nécessité de réaliser des études supplémentaires, autres que des études oculaires in vivo, pour contribuer à caractériser ce pouvoir. Si cette nécessité se confirme, une démarche expérimentale séquentielle est préconisée pour produire les données expérimentales pertinentes. S'agissant des substances qui n'ont pas encore fait l'objet d'essais, il convient d'obtenir l'ensemble des données permettant d'évaluer le pouvoir corrosif ou irritant de la substance par une démarche séquentielle. La stratégie d'essai initiale décrite dans le présent supplément a été conçue lors d'un atelier de l'OCDE (1). Elle a ensuite été confirmée et étendue dans le Système harmonisé de classification intégrée des risques pour la santé humaine et l'environnement liés aux substances chimiques, adoptée par la vingt-huitième Réunion conjointe du Comité sur les produits chimiques et du Groupe de travail sur les produits chimiques en novembre 1998 (2), et mise à jour par un groupe d'experts de l'OCDE en 2011. Si cette stratégie d'essai séquentielle ne fait pas partie intégrante de la méthode d'essai B.5, elle constitue cependant la procédure recommandée pour déterminer l'effet irritant ou corrosif sur les yeux. Cette procédure représente à la fois la meilleure pratique et une référence éthique pour les essais in vivo dans ce domaine. La méthode d'essai décrit le mode opératoire de l'essai in vivo et récapitule les facteurs qui devraient être examinés avant d'entamer l'essai. La stratégie d'essai séquentielle indique comment évaluer les données existantes relatives aux propriétés irritantes ou corrosives des produit chimique pour l'œil en se fondant sur le poids de la preuve, et présente une stratégie à plusieurs niveaux permettant d'obtenir des données pertinentes sur les produits chimiques qui réclament d'autres études ou qui n'ont pas encore été étudiés. Cette démarche prescrit également de commencer par conduire des essais in vitro ou ex vivo validés et acceptés, puis, dans certaines circonstances, d'effectuer les études d'irritation/corrosion cutanées exposées dans la méthode d'essai B.4 (3) (4). Description de la stratégie d'essai par étapes Avant d'entreprendre les essais inscrits dans la démarche expérimentale séquentielle (graphique), il convient d'évaluer toutes les informations disponibles afin d'établir la nécessité d'effectuer des essais oculaires in vivo. Bien que l'évaluation de certains paramètres particuliers puisse livrer des informations capitales (p. ex. un pH extrême), la totalité des informations existantes est prise en considération. Toutes les données pertinentes sur les effets du produit chimique en question, et de ses analogues structurels, seront examinées en vue d'une prise de décision fondée sur le poids de la preuve, décision qui fait l'objet de justification. Il convient d'accorder le plus de poids aux données humaines et animales existantes, puis aux résultats des essais in vitro ou ex vivo sur le produit chimique. Les études in vivo sur des substances corrosives devront être évitées autant que faire se peut. Les facteurs pris en compte dans la stratégie séquentielle sont repris ci-après:
PROCÉDURE D'ESSAI ET D'ÉVALUATION DE L'IRRITATION ET DE LA CORROSION OCULAIRES
BIBLIOGRAPHIE
|
|
(3) |
Dans la partie B, le chapitre B.10 est remplacé par le texte suivant: «B.10 Essai d'aberration chromosomique in vitro chez les mammifères INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 473 (2016) de l'OCDE pour les essais de produits chimiques. Elle s'inscrit dans une série de méthodes d'essai sur la toxicologie génétique. Par ailleurs, un document de l'OCDE qui fournit des informations succintes sur les essais de génotoxicité et donne une vue d'ensemble des modifications récemment apportées à ces lignes directrices a été élaboré (1). L'essai d'aberration chromosomique in vitro est destiné à détecter les produits chimiques qui provoquent des aberrations chromosomiques structurales dans des cellules de mammifère cultivées (2) (3) (4). Les aberrations structurales peuvent être de deux types: chromosomiques ou chromatidiques. Il est possible que des cas de polyploïdie (y compris d'endoreduplication) surviennent dans les essais d'aberration chromosomique in vitro. Si les aneugènes peuvent induire une polyploïdie, la polyploïdie seule n'est pas le signe d'un potentiel aneugène et peut simplement indiquer une perturbation du cycle cellulaire ou une cytotoxicité (5). Cet essai n'est pas conçu pour mesurer l'aneuploïdie. Pour détecter l'aneuploïdie, il est recommandé de réaliser un test du micronoyau in vitro (6). L'essai d'aberration chromosomique in vitro peut être pratiqué sur des cultures de lignées cellulaires établies ou des cultures de cellules primaires d'origine humaine ou de rongeurs. Les cellules employées sont choisies en fonction de leur potentiel de croissance en culture, de la stabilité de leur caryotype (notamment leur nombre de chromosomes) et de la fréquence spontanée des aberrations chromosomiques (7). Les données disponibles à l'heure actuelle ne permettent pas d'émettre des recommandations fermes mais tendent à montrer qu'il importe de tenir compte, lors de l'évaluation des dangers chimiques, du statut du p53, de la stabilité génétique (caryotype), de la capacité de réparation de l'ADN et de l'origine (rongeurs ou humains) des cellules retenues pour l'essai. Les utilisateurs de la présente méthode d'essai sont donc invités à prendre en considération l'influence de ces caractéristiques cellulaires, et d'autres caractéristiques, sur les performances d'une lignée cellulaire quant à la détection de l'induction d'aberrations chromosomiques, sachant que les connaissances évoluent dans ce domaine. Les définitions utilisées sont données à l'appendice 1. REMARQUES PRÉLIMINAIRES ET LIMITES À moins que les cellules utilisées ne soient dotées d'un métabolisme compatible avec les produits chimiques testés, les essais conduits in vitro requièrent généralement une source exogène d'activation métabolique. Or, les systèmes d'activation métabolique exogène sont incapables de reproduire parfaitement les conditions in vivo. On prendra soin d'éviter les conditions susceptibles de conduire à de faux résultats positifs, c'est-à-dire à une lésion chromosomique qui ne soit pas causée par une interaction directe entre le produit chimique d'essai et les chromosomes; ces conditions peuvent être une modification du pH ou de l'osmolalité (8) (9) (10), une interaction avec les composants du milieu (11) (12) ou une cytotoxicité excessive (13) (14) (15) (16). Cet essai permet de détecter les aberrations chromosomiques pouvant résulter d'évènements clastogènes. L'analyse de l'induction d'une aberration chromosomique doit être effectuée sur des cellules en métaphase. Il est donc indispensable que les cellules des cultures traitées et des cultures témoins atteignent le stade de la mitose. Pour les nanomatériaux manufacturés, il peut s'avérer nécessaire d'apporter certaines adaptations spécifiques à cette méthode d'essai, mais ces adaptations ne sont pas décrites dans le présent document. Avant d'utiliser la méthode d'essai sur un mélange pour générer des données avec pour objectif recherché l'application réglementaire, on examinera si, et si oui, pourquoi, elle peut fournir des résultats adéquats à cette fin. De telles considérations ne sont pas nécessaires quand les exigences réglementaires stipulent que le mélange doit être testé. PRINCIPE DE L'ESSAI Des cultures cellulaires d'origine humaine ou provenant d'autres mammifères sont exposées au produit chimique d'essai, en présence et en l'absence d'une source exogène d'activation métabolique, à moins que les cellules utilisées ne soient dotées de capacités métaboliques idoines (voir paragraphe 13). Après avoir été exposées au produit chimique d'essai, les cultures de cellules sont, à intervalles préétablis, traitées par un produit chimique bloquant la métaphase (par exemple la colchicine ou le colcemide), récoltées et teintées. Les cellules en métaphase sont soumises à un examen microscopique permettant de déceler les aberrations chromatidiques et chromosomiques. DESCRIPTION DE LA MÉTHODE Préparations Cellules Diverses lignées cellulaires (par exemple, ovaire de hamster chinois (CHO), poumon de hamster chinois V79, poumon de hamster chinois (CHL)/IU, TK6) ou cultures de cellules primaires peuvent être utilisées, y compris des lymphocytes du sang périphérique humain ou d'autres mammifères (7). Le choix des lignées cellulaires utilisées doit être justifié scientifiquement. En cas d'utilisation de cellules primaires, pour des raisons relatives au bien-être des animaux, il conviendra d'envisager, lorsque cela est possible, le recours à des cellules primaires d'origine humaine, prélevées dans le respect des principes éthiques et de la réglementation en la matière. Les lymphocytes du sang périphérique humain utilisés sont issus de sujets jeunes (âgés de 18 à 35 ans environ), non fumeurs, ne souffrant d'aucune maladie connue et n'ayant pas été exposés récemment à des niveaux d'agents génotoxiques (produits chimiques, rayonnements ionisants, par exemple) susceptibles d'augmenter l'incidence de fond des aberrations chromosomiques, ceci afin de garantir que cette incidence soit faible et homogène. L'incidence de fond des aberrations chromosomiques augmente avec l'âge, et cette tendance est plus marquée chez la femme que chez l'homme (17) (18). Si des cellules issues de plusieurs donneurs sont mises en commun, le nombre des donneurs est précisé. Il est nécessaire de démontrer que les cellules se sont divisées entre le moment où elles ont été traitées avec le produit chimique d'essai et leur prélèvement. Les cultures cellulaires sont maintenues dans une phase de croissance exponentielle (lignées cellulaires) ou encouragées à se diviser (cultures primaires de lymphocytes) en vue de l'exposition des cellules à différents stades du cycle cellulaire, étant donné que la sensibilité des stades cellulaires aux produits chimiques d'essai peut ne pas être connue. En général, les cellules primaires dont la division doit être stimulée par des agents mitogènes ne sont plus synchronisées lors de leur exposition au produit chimique d'essai (les lymphocytes humains après une stimulation mitogène de 48 heures, par exemple). L'utilisation de cellules synchronisées pendant le traitement n'est pas recommandée mais peut être acceptable si elle est justifiée. Milieu et conditions de culture Il convient d'utiliser un milieu de croissance et des conditions d'incubation (récipients de culture, atmosphère humidifiée à 5 % de CO2 si nécessaire, température d'incubation de 37°C) appropriées pour les cultures. La stabilité du caryotype et l'absence de contamination par des mycoplasmes sont vérifiées régulièrement dans les lignées cellulaires (7) (19), et les cellules sont écartées si une contamination ou une modification du caryotype est constatée. La durée normale du cycle cellulaire des lignées ou des cultures primaires utilisées dans le laboratoire d'essai doit être établie et doit correspondre aux caractéristiques cellulaires publiées (20). Préparation des cultures Lignées cellulaires: les cellules sont multipliées à partir de cultures mères, placées dans un milieu de culture à une densité telle que les cellules en suspension ou en monocouche poursuivront leur croissance de manière exponentielle jusqu'au moment de la récolte (il convient par exemple d'éviter que les cellules qui se multiplient en monocouche arrivent à confluence). Lymphocytes: un sang total traité avec un anticoagulant (héparine, par exemple) ou des lymphocytes isolés sont mis en culture (pendant 48 heures pour les lymphocytes humains, par exemple) en présence d'un mitogène [phytohémagglutinine (PHA) pour les lymphocytes humains, par exemple] afin d'induire une division cellulaire avant l'exposition au produit chimique d'essai. Activation métabolique Le recours à un système d'activation métabolique exogène est nécessaire en cas d'utilisation de cellules dotées d'une capacité métabolique endogène inadéquate. Le système le plus couramment utilisé, recommandé par défaut, sauf justification contraire, est une fraction post-mitochondriale enrichie en cofacteur (S9), préparée à partir de foies de rongeurs (généralement des rats) traités avec des inducteurs enzymatiques comme l'Aroclor 1254 (21) (22) (23) ou un mélange de phénobarbital et ß-naphthoflavone (24) (25) (26) (27) (28) (29). L'utilisation de ce mélange n'est pas contraire à la Convention de Stockholm sur les polluants organiques persistants (30) et s'est révélée aussi efficace que celle de l'Aroclor 1254 pour l'induction d'oxydases à fonction mixte (24) (25) (26) (28). La fraction S9 est généralement utilisée à une concentration comprise entre 1 et 2 % (v/v) mais peut être portée à 10 % v/v dans le milieu d'essai final. Pendant le traitement, on évitera d'utiliser des produits entraînant une réduction de l'indice mitotique, en particulier des complexants du calcium (31). Le choix du type et de la concentration du système d'activation métabolique exogène ou de l'inducteur métabolique utilisé pourra dépendre de la classe des produits chimiques à tester. Préparation du produit chimique d'essai Les produits chimiques solides à tester sont dissous dans un solvant approprié puis, le cas échéant, dilués avant application (voir paragraphe 23). Les produits chimiques liquides peuvent être ajoutés directement ou après dilution au système d'essai. Les produits gazeux ou volatils nécessitent une modification appropriée des protocoles standards, par exemple l'utilisation de récipients de culture hermétiquement clos (32) (33) (34). Il convient de préparer les produits chimiques d'essai juste avant le traitement, à moins que les données concernant la stabilité ne démontrent qu'ils peuvent être stockés. Conditions de l'essai Solvants Le solvant doit être choisi de manière à optimiser la solubilité des produits chimiques d'essai, sans engendrer d'effets néfastes sur la conduite de l'essai, c'est-à-dire sans modifier la croissance cellulaire, nuire à l'intégrité du produit chimique d'essai, réagir avec les récipients de culture ou détériorer le système d'activation métabolique. On recommande d'envisager d'abord l'utilisation d'un solvant (ou milieu de culture) aqueux chaque fois que c'est possible. L'eau et le diméthylsulfoxyde sont des exemples de solvants couramment utilisés. En règle générale, les solvants organiques ne doivent pas dépasser 1 % (v/v) et les solvants aqueux (salin ou eau) 10 % (v/v) dans le milieu de traitement final. L'emploi d'un solvant/véhicule inhabituel (éthanol ou acétone par exemple) doit être justifié par des données faisant état de sa compatibilité avec le produit chimique d'essai et le système d'essai, ainsi que de son absence de génotoxicité aux concentrations utilisées. En l'absence de telles données, il est important d'inclure dans l'essai des témoins non traités (voir appendice 1) afin de démontrer que le solvant choisi n'entraîne aucun effet délétère ou clastogène. Mesure de la prolifération et de la cytotoxicité cellulaires et choix des concentrations d'essai Lors de la détermination de la plus forte concentration de produit chimique d'essai à tester, on évitera les concentrations susceptibles de produire de fausses réponses positives, notamment celles qui engendrent une cytotoxicité excessive (voir paragraphe 22), une précipitation dans le milieu de culture (voir paragraphe 23), ou une modification marquée du pH ou de l'osmolalité (voir paragraphe 5). Si le produit chimique d'essai provoque une modification marquée du pH du milieu au moment de son ajout, il est possible d'ajuster le pH par tamponnage du milieu de traitement final de manière à éviter les faux résultats positifs et à maintenir des conditions de culture appropriées. Des mesures de la prolifération cellulaire sont effectuées pour s'assurer qu'un nombre suffisant de cellules traitées a atteint la mitose pendant l'essai et que les applications sont réalisées à des niveaux de cytotoxicité appropriés (voir paragraphes 18 et 22). La cytotoxicité doit être mesurée lors de l'expérience principale avec et sans système d'activation métabolique, au moyen d'un indicateur pertinent de mort et de croissance cellulaires. Un essai préliminaire visant à évaluer la cytotoxicité peut s'avérer utile pour mieux cerner les concentrations à utiliser dans l'essai principal, mais il n'est pas obligatoire. S'il est réalisé, il ne doit pas remplacer la mesure de cytotoxicité effectuée dans le cadre de l'expérience principale. La mesure du doublement relatif de la population (Relative Population Doubling, RPD) et celle de l'augmentation relative du nombre de cellules (Relative increase in cell count, RICC) sont des méthodes adaptées pour évaluer la cytotoxicité dans les essais de cytogénétique (13) (15) (35) (36) (55) (voir formules à l'appendice 2). En cas de traitement de longue durée et lorsque les prélèvements sont effectués au-delà de 1.5 fois la durée normale du cycle cellulaire après le début du traitement (soit plus de 3 cycles cellulaires au total), il se peut que le RPD sous-estime la cytotoxicité (37). Dans ces circonstances, la RICC pourrait constituer une meilleure mesure, mais l'évaluation de la cytotoxicité à l'aide du RPD après une durée équivalente à 1.5 fois la durée normale du cycle cellulaire fournit néanmoins une estimation précieuse. Si l'indice mitotique (MI) mesure les effets cytotoxiques/cytostatiques sur les lymphocytes dans les cultures primaires, il est aussi influencé par le temps écoulé entre le début de l'exposition et le moment de la mesure, par le mitogène employé et par une éventuelle interruption du cycle. Toutefois, le MI constitue une mesure acceptable, car d'autres mesures de toxicité peuvent s'avérer laborieuses et peu pratiques, et ne pas forcément s'appliquer à la population cible de lymphocytes se développant en réponse à une stimulation par PHA. Les paramètres de cytotoxicité recommandés sont la RICC et le RPD pour les lignées cellulaires, et le MI pour la culture primaire des lymphocytes; néanmoins, d'autres indicateurs (tels que l'intégrité cellulaire, l'apoptose, la nécrose et le cycle cellulaire) peuvent fournir d'autres informations utiles. Il convient d'évaluer au moins trois concentrations d'essai (sans compter les témoins de solvant et les témoins positifs) remplissant les critères d'acceptabilité (cytotoxicité appropriée, nombre de cellules, etc.). Quel que soit le type de cellules (lignées cellulaires ou cultures primaires de lymphocytes), chacune des cultures réalisées en un seul ou plusieurs exemplaires peut être utilisée à chaque concentration d'essai. Alors que l'utilisation de cultures en double exemplaires est recommendé, l'utilisation de cultures en exemplaire unique est aussi acceptée à condition que le nombre total de cellules analysées par concentration reste identique qu'il s'agisse de cultures uniques ou de répliques. L'utilisation de cultures uniques est pertinent en particulier lorsqu'on évalue plus de 3 concentrations (voir paragraphe 31). Les résultats obtenus pour chacune des répliques (cultures réalisées en plusieurs exemplaires) à une concentration donnée peuvent être regroupés pour l'analyse des données (38). Pour les produits chimiques dont la cytotoxicité est faible ou nulle, des niveaux de concentrations espacés d'un facteur de 2 à 3 environ conviendront généralement. En cas de cytotoxicité, les concentrations d'essai retenues doivent couvrir une plage englobant la concentration produisant une cytotoxicité telle que décrite au paragraphe 22 et les concentrations pour lesquelles une cytotoxicité modérée, faible ou nulle est observée. De nombreux produits chimiques d'essai présentent des courbes concentration-réponse à forte pente et, pour obtenir des données dans le cadre d'une cytotoxicité faible et modérée ou étudier la relation dose-réponse en détail, il faudra donc utiliser des concentrations plus rapprochées et/ou plus de trois concentrations (cultures uniques ou répliques), notamment dans les cas où il est nécessaire de répéter l'expérience (voir paragraphe 47). Si la concentration maximale est basée sur la cytotoxicité, la concentration la plus forte doit viser une cytotoxicité de 55 ± 5 % selon les paramètres de cytotoxicité recommandés (à savoir une réduction de la RICC et du RPD pour les lignées cellulaires et une réduction du MI pour les cultures primaires de lymphocytes à 45 ± 5 % du témoin négatif concomitant). Les résultats positifs présents uniquement dans la tranche la plus haute de la plage de cytotoxicité 55 ± 5 % doivent être interprétés avec prudence (13). Pour les produits chimiques d'essai peu solubles qui ne sont pas cytotoxiques à des concentrations inférieures à la concentration insoluble la plus faible, la plus forte concentration analysée doit produire une turbidité ou un précipité visible à l'œil nu ou à l'aide d'un microscope inversé à la fin du traitement avec le produit chimique d'essai. Même si une cytotoxicité intervient au-delà de la concentration insoluble la plus faible, il est recommandé de tester une seule concentration produisant une turbidité ou un précipité visible, car de fausses réponses pourraient découler de ce précipité. À la concentration produisant un précipité, il convient de s'assurer que ce dernier n'interfère pas avec la conduite de l'essai (coloration ou analyse, par exemple). Il peut être utile de déterminer la solubilité dans le milieu de culture préalablement à l'essai. Si aucun précipité ou aucune cytotoxicité limitante ne sont observés, la concentration d'essai maximale doit correspondre à la plus basse parmi 10 mM, 2 mg/ml ou 2 μl/ml (39) (40) (41). Lorsque la composition du produit chimique d'essai n'est pas définie, par exemple dans le cas d'une substance de composition inconnue ou variable, de produits de réaction complexes ou de matériels biologiques (UVCB) (42), de produits extraits de l'environnement etc., il peut être nécessaire d'augmenter la la concentration maximale (5 mg/ml par exemple), en absence de cytotoxicity suffisante, afin d'accroître la concentration de chacun des composants. Il convient toutefois de noter que ces exigences peuvent être différentes pour les produits pharmaceutiques à usage humain (43). Témoins Des témoins négatifs concomitants (voir paragraphe 15), constitués uniquement du solvant dans le milieu de traitement et testés de la même façon que les cultures traitées, doivent être inclus pour chaque moment de récolte. Des témoins positifs concomitants sont nécessaires pour démontrer la capacité du laboratoire à identifier les clastogènes dans les conditions du protocole d'essai utilisé, ainsi que l'efficacité du système d'activation métabolique exogène, le cas échéant. Le tableau 1 ci-dessous présente des exemples de témoins positifs. D'autres produits chimiques peuvent être utilisés comme témoins positifs, si cela est justifié. Étant donné que les essais in vitro de génotoxicité sur cellules de mammifères sont suffisamment normalisés, l'utilisation de témoins positifs peut être limitée à un clastogène nécessitant une activation métabolique. À condition qu'elle soit réalisée simultanément à l'essai non activé et sur la même durée de traitement, cette réponse de témoin positif unique démontrera à la fois l'activité du système d'activation métabolique et la réactivité du système d'essai. Un traitement de longue durée (sans S9) doit toutefois disposer de son propre témoin positif, étant donné que la durée du traitement sera différente de celle de l'essai ayant recours à une activation métabolique. Chaque témoin positif doit être utilisé à une ou plusieurs concentrations devant normalement donner lieu à une augmentation reproductible et détectable par rapport à la valeur de fond afin de démontrer la sensibilité du système d'essai (c'est-à-dire que les effets sont nets mais que l'identité des lames codées n'est pas évidente pour l'examinateur) et la réponse ne doit pas être compromise par une cytotoxicité supérieure aux limites fixées dans la présente méthode d'essai. Tableau 1 Produits chimiques de référence recommandés pour la vérification des compétences du laboratoire et pour la sélection des témoins positifs
MODE OPÉRATOIRE Traitement avec le produit chimique d'essai Les cellules en prolifération sont traitées avec le produit chimique d'essai en présence et en l'absence d'un système d'activation métabolique. Délai de récolte des cultures Pour une évaluation complète, laquelle serait nécessaire pour conclure à un résultat négatif, les trois conditions expérimentales suivantes doivent être respectées pour un traitement de courte durée avec et sans activation métabolique, et pour un traitement de longue durée sans activation métabolique (voir paragraphes 43, 44 et 45):
Si l'une de ces conditions expérimentales entraîne une réponse positive, il n'est pas forcément nécessaire d'étudier les autres régimes de traitement. Préparation des chromosomes Les cultures de cellules sont traitées au colcemide ou à la colchicine pendant une période de une à trois heures avant la récolte. Chaque culture est récoltée et traitée séparément en vue de la préparation des chromosomes. La préparation des chromosomes passe par un traitement hypotonique des cellules, leur fixation et leur coloration. Il est possible que les cultures monocouches présentent des cellules mitotiques (reconnaissables à leur forme ronde et se détachant de la surface) à l'issue du traitement de 3 à 6 heures. Ces cellules se détachant facilement de la culture, elles risquent d'être emportées lors de l'élimination du milieu contenant le produit chimique d'essai. Si l'on constate une augmentation substantielle du nombre de cellules mitotiques par rapport aux témoins, ce qui indiquerait l'arrêt probable de la mitose, les cellules doivent être récoltées par centrifugation puis réintroduites dans les cultures afin d'éviter de perdre les cellules en phase de mitose, et présentant un risque d'aberration chromosomique, au moment de la récolte. Analyse Toutes les lames, y compris celles des témoins positifs et négatifs, doivent être codées individuellement avant l'analyse au microscope pour déceler les aberrations chromosomiques. Le procédé de fixation provoquant souvent la perte de chromosomes pour une certaine fraction des cellules en métaphase, les cellules examinées doivent donc contenir un nombre de centromères égal au nombre modal ± 2. Il y a lieu d'examiner au moins 300 cellules en métaphase bien étalées par concentration et par témoin pour conclure qu'un produit chimique d'essai est clairement négatif (voir paragraphe 45). Les 300 cellules doivent être réparties équitablement entre les répliques, lorsque les cultures ont été réalisées en plusieurs exemplaires. Lorsque des cultures en un seul exemplaire sont utilisées pour chaque concentration (voir paragraphe 21), il y a lieu d'examiner au moins 300 cellules en métaphase bien étalées par culture. L'examen de 300 cellules présente l'avantage d'augmenter la puissance statistique de l'essai. En outre, il est rare d'observer des valeurs nulles (de l'ordre de 5 % seulement) (44). Le nombre de métaphases examinées peut être réduit lorsqu'un grand nombre de cellules présentant des aberrations chromosomiques est observé et s'il a été conclu que le produit chimique d'essai est clairement positif. Les cellules présentant une/des aberration(s) chromosomique(s) structurale(s), lacunes comprises et non comprises, doivent être examinées. Les cassures et les lacunes sont définies à l'appendice 1 conformément à (45) (46). Il convient de consigner séparément les aberrations chromatidiques et chromosomiques et de les classer par sous-catégories (cassures, échanges). Les procédures en cours dans le laboratoire doivent assurer que l'analyse des aberrations chromosomiques est réalisée par des examinateurs qualifiés sous le contrôle de pairs si nécessaire. Bien que l'essai soit destiné à détecter les aberrations chromosomiques structurales, il est important de rapporter la fréquence des cas de polyploïdie et d'endoreduplication lorsqu'ils se présentent. (Voir paragraphe 2.) Compétence du laboratoire Afin d'établir qu'il possède une expérience suffisante pour mener à bien l'essai avant de l'utiliser en routine, le laboratoire doit avoir réalisé une série d'expériences avec des produits chimiques positifs de référence agissant selon des mécanismes variés, et avec plusieurs témoins négatifs (faisant appel à différents solvants/véhicules). Ces réponses de témoins positifs et négatifs doivent être cohérentes par rapport à la littérature. Cette exigence ne s'applique pas aux laboratoires possédant déjà une expérience, c'est-à-dire qui disposent une base de données historiques telle que définie au paragraphe 37. Une sélection de produits chimiques utilisés comme témoins positifs (voir tableau 1, paragraphe 26) doit être testée dans le cadre de traitements de courte et de longue durée en l'absence d'activation métabolique, ainsi que dans le cadre d'un traitement de courte durée en présence d'une activation métabolique, l'objectif étant de démontrer que le laboratoire possède la compétence nécessaire pour détecter les produits chimiques clastogènes et pour déterminer l'efficacité du système d'activation métabolique. Il conviendra de définir une plage de concentrations des produits chimiques sélectionnés qui permette d'obtenir des augmentations reproductibles et liées à la concentration par rapport aux valeurs de fond, afin de démontrer la sensibilité et la plage dynamique du système d'essai. Données des témoins historiques Le laboratoire doit établir:
Lors de l'acquisition initiale de données en vue d'établir une distribution des témoins négatifs historiques, les données des témoins négatifs concomitants doivent être cohérentes avec les données publiées, lorsqu'elles existent. Puis, à mesure que de nouvelles données expérimentales viennent étoffer la plage de distribution des témoins, les données des témoins négatifs concomitants doivent idéalement se situer dans les limites de contrôle 95 % de cette distribution (44) (47). La base de données historiques du laboratoire relatives aux témoins négatifs doit à l'origine être constituée à partir d'au moins 10 expériences, sachant qu'il serait préférable qu'elle en compte au moins 20, réalisées dans des conditions expérimentales similaires. Les laboratoires doivent avoir recours à des méthodes de contrôle de la qualité telles que des graphiques statistiques (cartes C ou cartes X-barre, par exemple (48)), afin de déterminer la variabilité de leurs données de témoins positifs et négatifs et de démontrer leur maîtrise de la méthodologie (44). On trouve dans la littérature (47) d'autres recommandations sur la façon de constituer et d'utiliser ces données historiques (critères d'inclusion et d'exclusion des données dans la base et critères d'acceptabilité pour une expérimentation donnée). Toute modification du protocole expérimental doit être étudiée en termes de cohérence avec les bases de données des témoins historiques existantes du laboratoire. Toute incohérence majeure doit conduire à l'établissement d'une nouvelle base de données des témoins historiques. Les données des témoins négatifs désignent l'incidence des cellules présentant des aberrations chromosomiques issues d'une seule culture ou de l'ensemble des cultures réalisées en plusieurs exemplaires, comme décrit au paragraphe 21. Les témoins négatifs concomitants se situent idéalement dans les limites de contrôle à 95 % de la distribution des données historiques des témoins négatifs contenues dans la base de données du laboratoire (44) (47). Lorsque les données des témoins négatifs concomitants se situent en dehors des limites de contrôle à 95 %, leur inclusion dans la distribution des témoins historiques peut être acceptable, à condition que ces données ne soient pas exagérément extrêmes et qu'il soit prouvé que le système d'essai est «sous contrôle» (voir paragraphe 37) et qu'il n'y a pas eu de défaillance technique ou humaine. RÉSULTATS ET RAPPORT Présentation des résultats Le pourcentage de cellules présentant une/des aberration(s) structurale(s) doit être évalué. Les aberrations chromatidiques et chromosomiques classées par sous-catégorie (cassure, échange) doivent être consignées séparément, avec leur nombre et leur fréquence pour les cultures traitées et les cultures témoins. Les lacunes sont enregistrées et rapportées séparément, mais elles ne sont pas incluses dans la fréquence totale des aberrations. Le pourcentage de cellules polyploïdes et/ou endoredupliquées est rapporté le cas échéant. Il convient aussi d'effectuer des mesures parallèles de cytotoxicité et de les consigner pour toutes les cultures traitées et les témoins négatifs et positifs dans la ou les expériences principales sur les aberrations. Les données doivent être indiquées individuellement pour chaque culture. En outre, toutes les données doivent être résumées sous forme de tableaux. Critères d'acceptabilité L'acceptation de l'essai repose sur les critères suivants:
Évaluation et interprétation des résultats À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement positif si, dans les conditions expérimentales étudiées (voir paragraphe 28):
Lorsque tous ces critères sont remplis, le produit chimique d'essai est considéré comme capable d'induire des aberrations chromosomiques dans les cellules de mammifères en culture dans ce système d'essai. Des recommandations concernant les méthodes statistiques les plus appropriées sont également disponibles dans la littérature (49) (50) (51). À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement négatif si, dans toutes les conditions expérimentales étudiées (voir paragraphe 28):
Le produit chimique d'essai est alors considéré comme incapable d'induire des aberrations chromosomiques dans les cellules de mammifères en culture dans ce système d'essai. Il n'est pas nécessaire de vérifier une réponse clairement positive ou négative. Lorsque la réponse n'est ni clairement négative ni clairement positive, tel que décrit ci-dessus, ou afin d'établir la signification biologique d'un résultat, les données doivent être soumises à un jugement d'experts et/ou des investigations plus poussées. Il peut être utile d'examiner des cellules supplémentaires (le cas échéant) ou de répéter l'expérience, éventuellement dans des conditions expérimentales modifiées [espacement des concentrations, autres conditions d'activation métabolique (concentration de S9 ou origine de S9), par exemple]. Dans de rares cas, même après des études complémentaires, l'ensemble de données ne permettra pas de conclure à un résultat positif ou négatif. Dans ce cas, la réponse au produit chimique d'essai sera considérée comme équivoque. Une augmentation du nombre de cellules polyploïdes peut signifier que le produit chimique d'essai est capable d'inhiber les processus mitotiques et d'induire des aberrations chromosomiques numériques (52). Une augmentation du nombre de cellules présentant des chromosomes endoredupliqués peut indiquer que le produit chimique d'essai est capable d'inhiber la progression du cycle cellulaire (53) (54) (voir paragraphe 2). La fréquence des cellules polyploïdes et des cellules présentant des chromosomes endoredupliqués doit donc être consignée séparément. Rapport d'essai Le rapport d'essai doit comporter les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Aberration de type chromatidique : lésion chromosomique structurale se traduisant par une cassure d'une seule chromatide ou par une cassure et une réunion entre chromatides. Aberration de type chromosomique : lésion chromosomique structurale se traduisant par une cassure, ou par une cassure et une réunion, des deux chromatides sur un même site. Aberration numérique : modification du nombre de chromosomes par rapport au nombre normal caractéristique des cellules employées. Aberration structurale : modification de la structure des chromosomes, détectable par un examen au microscope des cellules au stade de la métaphase et apparaissant sous la forme de délétions, fragmentations et modifications intrachromosomiques ou interchromosomiques. Aneuploïdie : tout écart par rapport au nombre diploïde (ou haploïde) normal de chromosomes, d'un seul ou de plusieurs chromosomes, mais non d'un ou de plusieurs jeux de chromosomes (polyploïdie). Apoptose : mort cellulaire programmée caractérisée par une succession d'étapes menant à la désintégration des cellules en particules membranaires qui sont ensuite éliminées par phagocytose ou par excrétion. Augmentation relative du nombre de cellules (Relative increase in cell count, RICC) : augmentation du nombre de cellules dans les cultures exposées à un produit chimique par rapport aux cultures non traitées. Ratio exprimé en pourcentage. Cassure de chromatide : interruption de la continuité d'une chromatide, manifestée par un défaut d'alignement clair de l'une des chromatides. Clastogène : produit chimique induisant des aberrations chromosomiques structurales dans des populations cellulaires ou des organismes eucaryotes. Concentrations : désigne les concentrations finales du produit chimique d'essai dans le milieu de culture. Cytotoxicité : pour les essais visés par la présente méthode d'essai et utilisant des lignées cellulaires, la cytotoxicité correspond à une baisse du doublement relatif de la population (RPD) ou de l'augmentation relative du nombre de cellules (RICC) des cellules traitées par rapport au témoin négatif (voir paragraphe 17 et appendice 2). Pour les essais visés par la présente méthode d'essai et utilisant des cultures primaires de lymphocytes, la cytotoxicité correspond à une baisse de l'indice mitotique (MI) des cellules exposées par rapport au témoin négatif (voir paragraphe 18 et appendice 2). Doublement relatif de la population (Relative Population Doubling, RPD) : augmentation du nombre de doublements de population dans les cultures exposées à un produit chimique par rapport aux cultures non traitées. Ratio exprimé en pourcentage. Endoreduplication : processus selon lequel le noyau, après une période S de réplication de l'ADN, n'entre pas en mitose mais recommence une nouvelle période S. Il en résulte des chromosomes comptant 4, 8, 16,… chromatides. Fraction S9 de foie : surnageant d'homogénat de foie centrifugé à 9 000 g (extrait de foie cru). Génotoxique : terme générique qualifiant tous les types de lésions de l'ADN ou des chromosomes, tels que les cassures, délétions, adduits, liaisons et modifications des nucléotides, réarrangements, mutations génétiques, aberrations chromosomiques et aneuploïdies. Tous les types d'effets génotoxiques n'entraînent pas nécessairement de mutations ou de lésions chromosomiques stables. Indice mitotique (Mitotic Index, MI): nombre de cellules en métaphase divisé par le nombre total de cellules dans une population; une indication de la vitesse de prolifération. Lacune chromatidique : région non colorée (lésion achromatique) d'une seule chromatide pour laquelle on observe un défaut d'alignement minime de la chromatide. Mélange S9 : mélange de fraction S9 de foie et de cofacteurs nécessaires à l'activité des enzymes métaboliques. Mitose : division du noyau cellulaire, généralement décomposée en prophase, prométaphase, métaphase, anaphase et télophase. Mutagène : qui produit une modification héréditaire portant sur une ou plusieurs séquences de paires de bases d'ADN génique, ou sur la structure de chromosomes (aberrations chromosomiques). Polyploïdie : aberrations chromosomiques numériques dans des cellules ou des organismes, impliquant un ou plusieurs jeux de chromosomes et non un ou plusieurs chromosomes isolés (aneuploïdie). Prolifération cellulaire : augmentation du nombre de cellules résultant de la division cellulaire mitotique. Statut p53 : la protéine p53 intervient dans la régulation du cycle cellulaire, l'apoptose et la réparation de l'ADN. Les cellules déficientes en protéine p53 fonctionnelle, incapables d'arrêter le cycle cellulaire ou d'éliminer les cellules lésées par le biais de l'apoptose ou d'autres mécanismes (induction de la réparation de l'ADN, par exemple) liés aux fonctions de p53 en réponse à des lésions de l'ADN, devraient théoriquement être davantage sujettes aux mutations génétiques ou aux aberrations chromosomiques. Produit chimique : une substance ou un mélange. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Témoin de solvant : terme générique désignant les cultures témoins recevant uniquement le solvant utilisé pour dissoudre le produit chimique d'essai. Témoins non traités : cultures ne recevant aucun traitement (ni produit chimique d'essai ni solvant) mais préparées parallèlement et de la même façon que les cultures exposées au produit chimique d'essai. Appendice 2 FORMULES POUR L'ÉVALUATION DE LA CYTOTOXICITÉ Indice mitotique (MI):
L'augmentation relative du nombre de cellules (RICC) et le doublement relatif de la population (RPD) sont des mesures recommandées, qui tiennent compte de la proportion de cellules ayant effectué une division cellulaire.
où: Doublement de population = [log (nombre de cellules post-application ÷ nombre de cellules initial)] ÷ log 2 Par exemple, une RICC ou un RPD de 53 % indique une cytotoxicité/cytostase de 47 % et une cytotoxicité/cytostase de 55 % mesurée par le MI signifie que le MI réel représente 45 % du témoin. Quoi qu'il en soit, il convient de mesurer le nombre de cellules avant traitement, qui doit être identique dans les cultures traitées et les cultures témoins négatives. Le RCC (à savoir le ratio nombre de cellules dans les cultures traitées / nombre de cellules dans les cultures témoins) était utilisé auparavant comme paramètre de cytotoxicité, mais n'est plus recommandé car il peut induire une sous-estimation de la cytotoxicité. Dans les cultures témoins négatives, le doublement de la population doit être compatible avec l'obligation de prélever des cellules à l'issue d'une période équivalente à environ 1.5 fois la durée normale du cycle cellulaire après le début du traitement, et l'indice mitotique doit être assez élevé pour obtenir un nombre suffisant de cellules en mitose et calculer de façon fiable une réduction de 50 %. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

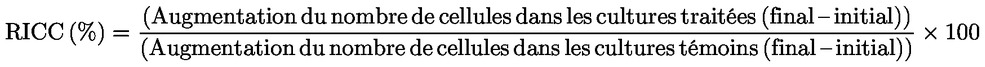
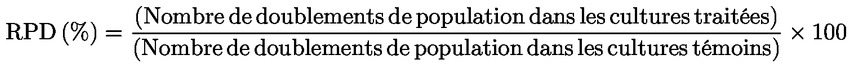
|
(4) |
Dans la partie B, le chapitre B.11 est remplacé par le texte suivant: «B.11 Essai d'aberration chromosomique sur moelle osseuse de mammifères INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 475 (2016) de l'OCDE pour les essais de produits chimiques. Elle s'inscrit dans une série de méthodes d'essai sur la toxicologie génétique. Par ailleurs, un document de l'OCDE qui fournit des informations succintes sur les essais de génotoxicité et donne une vue d'ensemble des modifications récemment apportées à ces lignes directrices a été élaboré (1). L'essai d'aberration chromosomique in vivo sur moelle osseuse de mammifères se prête particulièrement bien à l'évaluation de la génotoxicité car, malgré des variations entre les espèces, les facteurs du métabolisme in vivo, la pharmacocinétique et les processus de réparation de l'ADN sont actifs et contribuent aux réponses. L'essai in vivo est également considéré comme utile pour explorer plus avant un effet génotoxique détecté dans un système in vitro. L'essai d'aberration chromosomique in vivo est destiné à identifier les produits chimiques d'essai qui provoquent des aberrations chromosomiques structurales dans des cellules de moelle osseuse d'animaux, généralement des rongeurs (2) (3) (4) (5). Les aberrations structurales peuvent être de deux types: chromosomiques ou chromatidiques. Si la majorité des aberrations induites par des produits chimiques génotoxiques sont de type chromatidique, des aberrations chromosomiques se produisent aussi. Les lésions chromosomiques et les événements connexes sont à l'origine de beaucoup de maladies génétiques humaines et on a de bonnes raisons de penser que, lorsque ces lésions et événements connexes frappent des oncogènes et des gènes suppresseurs de tumeurs, ils jouent un rôle dans le cancer chez l'homme et dans les systèmes expérimentaux. Il est possible que des cas de polyploïdie (y compris d'endoreduplication) surviennent dans les essais d'aberration chromosomique in vivo. Or, une augmentation de la polyploïdie n'est n'indique pas en soi le signe d'un potentiel aneugène et peut simplement indiquer une perturbation du cycle cellulaire ou une cytotoxicité. Cet essai n'est pas conçu pour mesurer l'aneuploïdie. Pour détecter l'aneuploïdie, il est recommandé de réaliser un test du micronoyau in vivo sur érythrocytes de mammifère (chapitre B.12 de la présente annexe) ou un test du micronoyau in vitro sur cellules de mammifères (chapitre B.49 de la présente annexe). Les définitions utilisées sont données à l'appendice 1. REMARQUES PRÉLIMINAIRES Les rongeurs sont couramment utilisés dans cet essai, mais dans certains cas, d'autres espèces peuvent convenir si cela est justifié sur le plan scientifique. Cet essai se pratique sur la moelle osseuse, qui est un tissu très vascularisé formé d'une population de cellules au cycle rapide et faciles à prélever et à traiter. L'utilisation d'une espèce autre que le rat ou la souris doit être scientifiquement justifiée dans le rapport. Si des animaux autres que des rongeurs sont utilisés, il est recommandé que la mesure des aberrations chromosomiques sur la moelle osseuse soit intégrée à un autre essai de toxicité pertinent. Cet essai n'est pas pertinent s'il est prouvé que les substances chimiques d'essai, ou leurs métabolites, n'atteindront pas le tissu cible. Avant d'utiliser la méthode d'essai sur un mélange pour générer des données avec pour objectif recherché l'application réglementaire, on examinera si, et si oui, pourquoi, elle peut fournir des résultats adéquats à cette fin. De telles considérations ne sont pas nécessaires quand les exigences réglementaires stipulent que le mélange doit être testé. PRINCIPE DE LA MÉTHODE D'ESSAI Les animaux sont exposés au produit chimique d'essai par une voie d'exposition idoine et sont euthanasiés au moment approprié après le traitement. Avant l'euthanasie, les animaux sont traités avec un inhibiteur du fuseau (par exemple la colchicine ou le colcemide). Ensuite, les préparations chromosomiques faites à partir de cellules de la moelle osseuse sont colorées et les cellules en métaphase sont examinées pour mettre en évidence les aberrations chromosomiques. VÉRIFICATION DES COMPÉTENCES DU LABORATOIRE Épreuves de compétence Afin d'établir qu'il possède une expérience suffisante pour mener à bien l'essai avant de l'utiliser en routine, le laboratoire doit démontrer sa capacité à reproduire les résultats attendus d'après les données publiées ((6), par exemple) concernant les fréquences d'aberrations chromosomiques, avec un minimum de deux substances chimiques témoins positifs (y compris des réponses faibles induites par des doses faibles de témoins positifs) tels que ceux énumérés au tableau 1 et avec des témoins de véhicule/solvant compatibles (voir paragraphe 22). Les doses utilisées dans le cadre de ces expériences doivent produire des augmentations reproductibles qui sont fonction de la dose administrée, et démontrer la sensibilité et la plage dynamique du système d'essai sur le tissu en question (moelle osseuse); la méthode d'analyse retenue doit être celle qui sera employée par le laboratoire. Cette exigence ne s'applique pas aux laboratoires possédant déjà une expérience, c'est-à-dire qui disposent d'une base de données historiques telle que définie aux paragraphes 10 à 14. Données des témoins historiques Dans le cadre de la vérification des compétences, le laboratoire devra établir:
Lors de l'acquisition initiale de données en vue d'établir une distribution des témoins négatifs historiques, les données des témoins négatifs concomitants doivent être cohérentes avec les données publiées, lorsqu'elles existent. Puis, à mesure que de nouvelles données expérimentales viennent étoffer la plage de distribution des témoins historiques, les données des témoins négatifs concomitants doivent idéalement se situer dans les limites de contrôle à 95 % de cette distribution. La base des données historiques du laboratoire relatives aux témoins négatifs doit être statistiquement robuste pour permettre au laboratoire d'évaluer la distribution de ses données des témoins négatifs. La littérature suggère qu'un minimum de 10 expériences peut-être nécessaire, sachant qu'il serait préférable d'en compter au moins 20, réalisées dans des conditions expérimentales similaires. Le laboratoire doit avoir recours à des méthodes de contrôle de la qualité telles que des graphiques statistiques (cartes C ou cartes X-barre, par exemple (7)), afin de déterminer la variabilité de ses données et de démontrer sa maîtrise de la méthodologie. On trouve dans la littérature (8) d'autres recommandations sur la façon de constituer et d'utiliser ces données historiques (critères d'inclusion et d'exclusion des données dans la base et critères d'acceptabilité pour une expérimentation donnée). Si, au cours des expériences visant à vérifier sa compétence (comme décrit au paragraphe 9), le laboratoire n'a pas réalisé un nombre suffisant d'expériences pour établir une distribution des témoins négatifs statistiquement robuste (voir paragraphe 11), on peut accepter que la distribution soit construite au cours des premiers tests de routine. Cette approche devra suivre les recommandations établies dans la littérature (8) et les résultats des témoins négatifs obtenus lors de ces expériences devront être cohérents avec les données publiées des témoins négatifs. Toute modification du protocole expérimental doit être étudiée à la lumière de ses répercussions sur la cohérence des nouvelles données avec celles de la base de données des témoins historiques. Seules des incohérences majeures justifient l'établissement d'une nouvelle base de données des témoins historiques, après confirmation de la différence de distribution des données par des experts (voir paragraphe 11). Lors de la constitution de cette nouvelle base de données, le laboratoire n'a pas forcément besoin d'une base de données de témoins négatifs complète pour autoriser la conduite d'un essai, à condition qu'il puisse apporter la preuve que les valeurs des témoins négatifs concomitants sont cohérentes avec la précédente base de données ou avec les données publiées correspondantes. Les données des témoins négatifs désignent l'incidence des aberrations chromosomiques structurales (lacunes non comprises) chez chaque animal. Les témoins négatifs concomitants se situent idéalement dans les limites de contrôle à 95 % de la distribution des données historiques des témoins négatifs contenues dans la base de données du laboratoire. Lorsque les données des témoins négatifs concomitants se situent en dehors des limites de contrôle à 95 %, leur inclusion dans la distribution des témoins historiques peut être acceptable, à condition que ces données ne soient pas exagérément extrêmes, et qu'il soit prouvé que le système d'essai est «sous contrôle» (voir paragraphe 11) et qu'il n'y a pas eu de défaillance technique ou humaine. DESCRIPTION DE LA MÉTHODE Préparations Choix des espèces animales Il convient d'employer de jeunes animaux adultes sains issus de souches de laboratoire courantes. Les rats sont habituellement utilisés, mais les souris peuvent également convenir. Toute autre espèce appropriée de mammifère peut être employée à condition qu'une justification scientifique de ce choix soit donnée dans le rapport. Conditions d'encagement et d'alimentation des animaux Pour les rongeurs, la température de l'animalerie doit être maintenue à 22°C (± 3°C). L'humidité relative, qui est idéalement de 50 à 60 %, doit atteindre au moins 40 % et de préférence ne pas dépasser 70 %, sauf durant le nettoyage du local. L'éclairage est artificiel, la séquence d'éclairage étant de 12 heures de clarté et 12 heures d'obscurité. Le régime alimentaire des animaux est le régime classique de laboratoire avec eau potable à volonté. Le choix des aliments peut être influencé par la nécessité d'assurer une bonne incorporation du produit chimique dans la nourriture si l'administration se fait par cette voie. Les rongeurs sont mis en cage par petits groupes d'individus de même sexe (cinq au maximum par cage) recevant le même traitement, si aucun comportement agressif n'est à craindre, de préférence dans des cages à fond plein dotées d'un enrichissement environnemental approprié. Les animaux peuvent être encagés individuellement si cela est justifié sur le plan scientifique. Préparation des animaux On choisit habituellement de jeunes animaux adultes sains (les rongeurs sont idéalement âgés de 6 à 10 semaines au début du traitement, bien que des animaux un peu plus âgés soient également acceptables), qui sont répartis de manière aléatoire entre les groupes témoins et les groupes de traitement. Chaque animal est identifié individuellement selon une méthode sans cruauté, la moins invasive possible (par exemple, baguage, étiquetage, pose d'une puce électronique ou identification biométrique, en évitant l'entaillage des oreilles ou la phalangectomie) et gardés dans leurs cages pendant au moins cinq jours afin qu'ils s'acclimatent aux conditions du laboratoire. Les cages doivent être placées de manière à réduire au minimum l'influence éventuelle de leur disposition sur les résultats. Il convient d'éviter toute contamination croisée entre le témoin positif et le produit chimique d'essai. Au début de l'étude, la variation pondérale des animaux doit être minimale et ne pas dépasser ± 20 pour cent du poids moyen de chaque sexe. Préparation des doses Lorsque les produits chimiques testés sont solides, ils sont dissous ou mis en suspension dans des solvants ou des véhicules appropriés, ou incorporés aux aliments ou à l'eau de boisson avant d'être administrés aux animaux. Les produits chimiques liquides peuvent être administrés directement ou dilués avant d'être administrés. En cas d'exposition par inhalation, les produits chimiques d'essai peuvent être administrés sous forme de gaz, de vapeur ou d'aérosol solide ou liquide, en fonction de leurs propriétés physico-chimiques. On utilisera des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage et définissent les conditions de stockage appropriées. Solvant/véhicule: Le solvant/véhicule ne doit pas produire d'effets toxiques aux doses utilisées ni interagir avec les produits chimiques d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données de référence faisant état de leur compatibilité. Il est recommandé d'envisager en premier lieu l'utilisation d'un solvant/véhicule aqueux chaque fois que c'est possible. Parmi les exemples de solvants/véhicules compatibles couramment utilisés figurent notamment l'eau, le sérum physiologique, les solutions de méthylcellulose, les solutions de carboxyméthylcellulose sodique, l'huile d'olive et l'huile de maïs. En l'absence de données historiques ou publiées montrant que le véhicule/solvant inhabituel sélectionné n'induit aucune aberration structurale ou effet délétère, une étude initiale devra être réalisée afin d'établir l'acceptabilité du témoin de solvant/véhicule. Témoins Témoins positifs Chaque essai doit normalement inclure un groupe d'animaux traités avec une substance chimique utilisée comme témoin positif. Cette étape peut être évitée une fois que le laboratoire d'essai a apporté la preuve de ses compétences pour la conduite de l'essai et établi la plage des témoins positifs historiques. Lorsqu'aucun groupe témoin concomitant n'est utilisé simultanément, des témoins d'examen (lames fixes et non colorées) devront être inclus dans chaque expérience. Pour ce faire, on pourra intégrer à l'étape d'examen des échantillons de référence idoines obtenus et stockés lors d'un essai séparé sur témoin positif mené périodiquement (par exemple, tous les 6 à 18 mois) dans le laboratoire dans lequel l'essai est conduit, notamment au cours de l'épreuve de compétence et par la suite sur une base régulière, si nécessaire. Les susbtances utilisés comme témoins positifs doivent produire, de façon fiable, un accroissement détectable de la fréquence des cellules présentant des aberrations chromosomiques par rapport au niveau spontané. Les doses des témoins positifs doivent être choisies de telle sorte que les effets soient nets mais que l'identité des lames codées ne soit pas évidente pour l'examinateur. Il est possible d'administrer le témoin positif par une voie différente de celle du produit chimique d'essai, de suivre un autre programme de traitement et de n'effectuer qu'un seul prélèvement d'échantillon. De plus, l'emploi de témoins positifs appartenant à la même classe chimique peut être envisagé, s'il y a lieu. Des exemples de substances chimiques utilisées comme témoins positifs figurent au tableau 1 ci-dessous. Tableau 1 Exemples de substances chimiques utilisées comme témoins positifs
Témoins négatifs Pour chaque moment d'échantillonnage, il faut inclure un groupe d'animaux témoins négatifs, qui seront manipulés de la même façon que les groupes traités, mais ne recevront pas le produit chimique d'essai. Si un solvant/véhicule est utilisé pour administrer le produit chimique d'essai, ce solvant/véhicule doit être administré au groupe témoin. Toutefois, si des résultats obtenus antérieurement par le laboratoire d'essai démontrent que la variabilité interindividuelle et la fréquence des cellules comportant des aberrations chromosomiques provenant de témoins négatifs sont stables pour chaque moment d'échantillonnage, un seul prélèvement peut alors suffire pour les témoins négatifs. Dans ce cas, il doit intervenir au moment du premier prélèvement effectué dans le cadre de l'étude. MODE OPÉRATOIRE Nombre et sexe des animaux En règle générale, la réponse des micronoyaux est similaire chez les animaux mâles et femelles (9), et il est à prévoir qu'il en sera de même pour les aberrations chromosomiques structurales; la plupart des études pourrait donc être réalisée sur l'un ou l'autre des deux sexes. Des données démontrant des différences significatives entre les mâles et les femelles (par exemple sur le plan de la toxicité systémique, du métabolisme, de la biodisponibilité, de toxicité sur la moelle osseuse, etc comprenant également des données provenant par exemple d'études de détermination des doses) encouragent l'utilisation des deux sexes. Il peut donc être plus approprié dans ce cas de mener une étude sur les deux sexes, par exemple dans le cadre d'une étude de toxicité à doses répétées. Il pourrait être judicieux de recourir à un plan factoriel en cas d'utilisation des deux sexes. Des précisions sur cette méthode d'analyse des données sont fournies à l'appendice 2. La taille des groupes au début de l'étude doit permettre de disposer dans chaque groupe d'au moins cinq animaux analysables du même sexe, ou de chaque sexe si les deux sont utilisés. Si l'exposition humaine est spécifique pour un sexe, par exemple dans le cas de certains produits pharmaceutiques, l'essai sera pratiqué sur des animaux du sexe approprié. À titre d'information concernant le nombre maximum d'animaux généralement requis, une étude sur moelle osseuse comptant deux moments d'échantillonnage et impliquant trois groupes de traitement et un groupe de témoins négatifs concomitants, plus un groupe de témoins positifs (chaque groupe étant composé de cinq animaux du même sexe) nécessitera 45 animaux. Niveaux de dose Si l'on procède à une étude préliminaire de détermination des doses à administrer parce qu'on ne dispose pas de données fiables pour orienter le choix des doses, cette étude préliminaire doit être effectuée dans le même laboratoire, en utilisant une espèce, une souche, un sexe et un régime de traitement identiques à ceux de l'étude principale (10). Elle devra avoir pour objectif de déterminer la dose maximale tolérée (DMT), définie comme la dose la plus élevée qui sera tolérée sans faire apparaître de toxicité limitante, dans le cadre de la durée de l'étude (par exemple, induisant une baisse du poids corporel ou une cytotoxicité du système hématopoïétique), mais ne provoquant pas la mort ou des signes de douleur, de souffrance ou de détresse imposant de sacrifier les animaux (11). La dose la plus élevée peut aussi être définie comme celle qui donne lieu à certains symptômes de toxicité pour la moelle osseuse. Les substances chimiques dont les propriétés toxicocinétiques sont saturées ou qui induisent un processus de détoxification pouvant se traduire par une baisse de l'exposition après une administration à long terme peuvent faire exception aux critères d'établissement des doses et doivent être évaluées au cas par cas. Pour permettre d'obtenir des informations sur la relation dose-réponse, une étude complète doit comporter un groupe témoin négatif et au moins trois niveaux de doses, espacés en général d'un facteur de 2, mais pas de plus de 4. Si le produit chimique d'essai ne provoque aucune toxicité dans le cadre d'une étude de détermination des doses, ou d'après les données disponibles, la dose la plus élevée pour une administration unique doit être de 2 000 mg/kg de poids corporel. Néanmoins, si le produit chimique d'essai provoque une toxicité, la dose administrée la plus élevée devra correspondre à la DMT et les niveaux de dose employés devront de préférence s'étendre de la dose maximale à une dose induisant peu ou pas de toxicité. Lorsqu'une toxicité sur le tissu cible (moelle osseuse) est observée à tous les niveaux de doses administrés, il est conseillé de procéder à des études complémentaires à des doses non toxiques. Les études visant à caractériser davantage les informations sur la relation quantitative dose-réponse peuvent nécessiter un ou plusieurs groupes de traitement supplémentaires. Enfin, ces limites peuvent varier pour certains types de produits chimiques d'essai (par exemple les produits pharmaceutiques à usage humain) faisant l'objet d'exigences spécifiques. Essai limite Si les essais préliminaires de détermination des doses ou les données existantes concernant des souches de rongeurs apparentées indiquent qu'un régime de traitement égal ou supérieur à la dose limite (décrite ci-dessous) n'engendre pas d'effets toxiques observables (notamment aucune dépression de la fonction médullaire osseuse ni autre cytotoxicité pour le tissu cible), et si la génotoxicité n'est pas escomptée d'après des études de génotoxicité in vitro ou d'après les données relatives aux substances structurellement apparentées, une étude complète utilisant trois niveaux de doses peut ne pas être considérée comme nécessaire, à condition qu'il ait été démontré que le/les produit(s) chimiques d'essai atteignent le tissu cible (moelle osseuse). Dans ce cas, un seul niveau de dose, égal à la dose limite, peut s'avérer suffisant. Pour une période d'administration de plus de 14 jours, la dose limite est de 1 000 mg/kg de poids corporel/jour. Pour des périodes d'administration de 14 jours ou moins, la dose limite est de 2 000 mg/kg de poids corporel/jour. Administration des doses Lors de la conception d'un essai, il convient de tenir compte de la voie d'exposition humaine anticipée. Par conséquent, les voies d'administration telles que l'alimentation, l'eau de boisson, l'inhalation, l'implantation ainsi que les voies topique, sous-cutanée, intraveineuse, orale (par gavage) et intra-trachéale sont autant de choix valables, sous réserve qu'ils soient justifiés. Dans tous les cas, la voie retenue doit permettre une exposition adéquate du/des tissu(s) cible(s). L'injection intrapéritonéale n'est en général pas recommandée, car elle ne constitue pas une voie d'exposition humaine envisagée, et ne sera utilisée qu'en cas de justification scientifique spécifique. Si le produit chimique d'essai est mélangé à l'alimentation ou à l'eau de boisson, surtout dans le cas d'un dosage unique, il convient de s'assurer que le délai entre l'absorption de nourriture et d'eau et le prélèvement est suffisant pour permettre une détection des effets (voir paragraphes 33 et 34). Le volume maximal de liquide administrable en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne doit normalement pas excéder 1 ml/100 g de poids corporel, sauf pour les solutions aqueuses, où un maximum de 2 ml/100 g est acceptable. L'utilisation de volumes plus importants doit être justifiée. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, afin de garantir l'administration d'un volume constant pour un poids corporel donné, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration. Programme de traitement En règle générale, les produits chimiques d'essai sont administrés en une seule fois, mais si le volume est important, l'administration peut être fractionnée (à raison de deux traitements ou plus le même jour à intervalles de 2 à 3 heures maximum). En pareil cas, ou lors d'une administration du produit chimique d'essai par inhalation, le moment d'échantillonnage sera fixé en fonction du moment d'administration de la dernière fraction, ou de la fin de l'exposition. On ne dispose guère de données sur la pertinence d'un protocole de doses répétées pour cet essai. Cependant, dans les cas où il est conseillé d'intégrer cet essai à un essai de toxicité à doses répétées, il faut prendre soin d'éviter la perte de cellules mitotiques présentant des lésions chromosomiques, un phénomène susceptible de se produire à des doses toxiques. Une telle intégration est acceptable lorsque la dose la plus élevée est supérieure ou égale à la dose limite (voir paragraphe 29) et que cette dose limite est administrée à un groupe de traitement pendant toute la durée du traitement. Il convient de considérer le test du micronoyau (méthode d'essai B.12) comme le test in vivo de première intention pour la détection des aberrations chromosomiques lorsqu'une intégration à d'autres études est souhaitée. Les échantillons de moelle osseuse doivent être prélevés à deux moments différents à la suite de chaque traitement. Pour les rongeurs, le premier intervalle de prélèvement se situe à 1.5 fois la durée normale du cycle cellulaire (généralement de 12 à 18 heures après le traitement). Étant donné que le temps requis par l'absorption et la métabolisation des produits chimiques d'essai ainsi que leur effet sur la cinétique du cycle cellulaire peuvent influer sur le moment optimal pour la détection des aberrations chromosomiques, on recommande d'effectuer un autre prélèvement 24 heures après le premier. Au premier moment d'échantillonnage, tous les groupes de traitement doivent être traités et des échantillons collectés pour analyse; cependant, lors du/des prélèvement(s) ultérieur(s), seule la dose la plus élevée doit être administrée. Si l'on applique des programmes de traitement qui couvrent plus d'une journée sur justification scientifique, l'échantillonnage devrait normalement intervenir à l'issue d'une période équivalent à environ 1.5 fois la durée normale du cycle cellulaire après la fin du traitement. Après le traitement et avant le prélèvement des échantillons, les animaux reçoivent une dose appropriée d'un inhibiteur du fuseau (par exemple le Colcemide ou la colchicine) par injection intrapéritonéale et les prélèvements sont ensuite effectués dans un délai approprié, qui est de 3 à 5 heures environ pour la souris et de 2 à 5 heures pour le rat. Les cellules sont prélevées dans la moelle osseuse, dilatées, fixées et colorées, puis analysées en vue de détecter des aberrations chromosomiques (12). Observations Les animaux d'essai font l'objet d'un examen clinique général. Les signes cliniques doivent être consignés au moins une fois par jour, de préférence aux mêmes heures, en prenant en considération la période où les effets anticipés devraient être les plus marqués après l'administration. Au moins deux fois par jour pendant la durée du traitement, l'ensemble des animaux fait l'objet d'un constat de morbidité et de mortalité. Tous les animaux doivent être pesés au début de l'étude, au moins une fois par semaine au cours des études à doses répétées, puis lors de l'euthanasie. Pour les études dont la durée est égale ou supérieure à une semaine, la consommation de nourriture doit également être mesurée au moins une fois par semaine. Si le produit chimique testé est administré dans l'eau de boisson, la consommation d'eau est mesurée à chaque changement d'eau et au moins une fois par semaine. Les animaux montrant des signes non létaux de toxicité excessive sont euthanasiés avant la fin de l'essai (11). Exposition du tissu cible Lorsque cela se justifie et qu'il n'existe pas d'autres données sur l'exposition (voir paragraphe 44), il convient de réaliser un prélèvement sanguin au(x) moment(s) idoine(s) pour vérifier la concentration des produits chimiques d'essai dans le plasma afin de démontrer que la moelle osseuse a bien été exposée. Préparation de la moelle osseuse et des chromosomes Les cellules de moelle osseuse sont obtenues à partir des fémurs ou des tibias des animaux immédiatement après l'euthanasie, exposées à une solution hypotonique et fixées. Les cellules en métaphase sont ensuite étalées sur des lames et colorées selon des méthodes éprouvées (voir (3) (12)). Analyse Toutes les lames, y compris celles des témoins positifs et négatifs, doivent être codées individuellement avant l'analyse et randomisées afin que l'analyste ignore à quel traitement elles correspondent. L'indice mitotique sert de mesure de la cytotoxicité et doit être déterminé dans un minimum de 1 000 cellules pour tous les animaux traités (témoins positifs compris) et les animaux non traités ou témoins de véhicule/solvant. Un minimum de 200 métaphases par animal doit être analysé pour détecter les aberrations chromosomiques, lacunes comprises et non comprises (6). Cependant, si la base de données des témoins négatifs historiques indique une fréquence de fond moyenne des aberrations chromosomiques structurelles < 1 % dans le laboratoire, il convient d'envisager l'examen de cellules supplémentaires. Les aberrations de type chromatidique et chromosomique doivent être consignées séparément et classées par sous-types (cassures, échanges). Les procédures en cours dans le laboratoire doivent assurer que l'analyse des aberrations chromosomiques est réalisée par des examinateurs qualifiés, sous le contrôle de pairs si nécessaire. Étant donné que les procédures de préparation des lames provoquent souvent la rupture d'une certaine fraction des cellules en métaphase, entraînant une perte de chromosomes, toutes les cellules examinées doivent contenir un nombre de centromères égal ou supérieur à 2n ± 2, n étant le nombre haploïde de chromosomes pour cette espèce. RÉSULTATS ET RAPPORT Traitement des résultats Les résultats individuels pour chaque animal doivent être présentés sous forme de tableaux. L'indice mitotique, le nombre de cellules en métaphase examinées, le nombre d'aberrations par cellule en métaphase ainsi que le pourcentage de cellules présentant une/des aberration(s) chromosomique (s) structurale(s) doivent être indiqués pour chaque animal. Les différents types d'aberrations chromosomiques structurales doivent être consignés avec leur nombre et leur fréquence pour les groupes de traitement et témoins. Les lacunes, ainsi que les cellules polyploïdes et présentant des chromosomes endoredupliqués sont notées séparément. La fréquence des lacunes est rapportée mais n'est généralement pas incluse dans l'analyse de la fréquence totale des aberrations. S'il n'y a pas de différence de réponse entre sexes, les données des deux sexes peuvent être combinées dans l'analyse statistique. Les données relatives à la toxicité pour l'animal et aux signes cliniques doivent elles aussi figurer dans le rapport. Critères d'acceptabilité Les critères suivants déterminent l'acceptabilité de l'essai:
Évaluation et interprétation des résultats À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement positif si:
Si seule la dose la plus élevée est étudiée à un moment d'échantillonnage particulier, un produit chimique d'essai est considéré comme clairement positif si on constate une augmentation statistiquement significative par rapport aux témoins négatifs concomitants et que les résultats se situent à l'extérieur de la plage de distribution des données des témoins négatifs historiques (par exemple, limites de contrôle à 95 % d'une loi de Poisson). Des recommandations concernant les méthodes statistiques les plus appropriées sont également disponibles dans la littérature (13). Une analyse de la relation dose-réponse doit porter sur un minimum de trois groupes de traitement. Les méthodes statistiques doivent utiliser l'animal comme unité expérimentale. Des résultats positifs obtenus dans un essai d'aberration chromosomique indiquent qu'un produit chimique d'essai induit des aberrations chromosomiques dans la moelle osseuse de l'espèce testée. À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement négatif si, dans toutes les conditions expérimentales étudiées:
Des recommandations concernant les méthodes statistiques les plus appropriées sont disponibles dans la littérature (13). L'exposition de la moelle osseuse à une substance chimique d'essai peut être démontrée par une baisse de l'indice mitotique ou par la mesure de la concentration de la/des substance(s) chimique(s) d'essai dans le plasma ou le sang. Dans le cas d'une administration par voie intraveineuse, des preuves de l'exposition ne sont pas requises. Pour démontrer l'exposition de la moelle osseuse, on peut également avoir recours aux données ADME, obtenues dans le cadre d'une étude indépendante employant la même voie d'administration et la même espèce. Des résultats négatifs signifient que, dans les conditions de l'essai, le produit chimique d'essai n'induit pas d'aberrations chromosomiques structurales dans la moelle osseuse de l'espèce testée. Il n'est pas nécessaire de vérifier une réponse positive claire ou négative claire. Lorsque la réponse n'est ni clairement négative ni clairement positive, et afin d'établir la signification biologique d'un résultat (par exemple, une augmentation faible ou marginale), les données doivent être soumises à un jugement d'experts et/ou des investigations plus poussées sur les expériences déjà réalisées. Dans certains cas, il peut être utile d'examiner des cellules supplémentaires ou de répéter l'expérience en modifiant les conditions expérimentales. Dans de rares cas, même après des investigations complémentaires, les données ne permettront pas de conclure que le produit chimique d'essai provoque des résultats clairement positifs ou négatifs. Les résultats de l'étude seront alors considérés comme équivoques. La fréquence de métaphases polyploïdes et endoredupliquées par rapport au nombre total de métaphases doit être consignée séparément. Une augmentation du nombre de cellules polyploïdes/endoredupliquées peut signifier que la substance d'essai est capable d'inhiber les processus mitotiques ou la progression du cycle cellulaire (voir paragraphe 3). Rapport d'essai Le rapport d'essai doit contenir les informations suivantes: Résumé
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Aneuploïdie : tout écart par rapport au nombre diploïde (ou haploïde) normal de chromosomes, d'un seul ou de plusieurs chromosomes, mais non de multiples d'un ou de plusieurs jeux de chromosomes (cf. polyploïdie). Centromère : régions d'un chromosome auxquelles les fibrilles du fuseau sont associées pendant la division de la cellule et qui permettent le mouvement ordonné des chromosomes-filles vers les pôles des cellules-filles. Substance chimique : une substance ou un mélange. Aberration de type chromatidique : lésion chromosomique structurale se traduisant par une cassure d'une seule chromatide ou par une cassure et une réunion entre chromatides. Aberration de type chromosomique : lésion chromosomique structurale se traduisant par une cassure, ou par une cassure et une réunion, des deux chromatides sur le même site. Endoreduplication : processus par lequel le noyau, après une période S de réplication de l'ADN, n'entre pas en mitose mais recommence une nouvelle période S. Il en résulte des chromosomes comptant 4, 8, 16,… chromatides. Lacune : lésion achromatique inférieure à la largeur d'une chromatide, marquée par un défaut d'alignement minime des chromatides. Indice mitotique : ratio entre le nombre de cellules en mitose et le nombre total de cellules dans une population, donnant une mesure de la vitesse de prolifération de cette population de cellules. Aberration numérique : modification du nombre de chromosomes par rapport au nombre normal caractéristique des animaux employés (aneuploïdie). Polyploïdie : aberration chromosomique numérique se traduisant par une modification du nombre de jeux de chromosomes, et non par une modification numérique touchant une partie du jeu de chromosomes (cf. aneuploïdie). Aberration chromosomique structurale : modification de la structure des chromosomes, détectable par un examen au microscope des cellules au stade de la métaphase et apparaissant sous la forme de délétions, fragmentations et modifications intrachromosomiques ou interchromosomiques. Substance chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Appendice 2 PLAN FACTORIEL UTILISÉ POUR IDENTIFIER LES DIFFÉRENCES ENTRE SEXES DANS L'ESSAI D'ABERRATION CHROMOSOMIQUE IN VIVO Plan factoriel et analyse factorielle Selon cette démarche, un minimum de 5 mâles et de 5 femelles sont exposés à chaque concentration d'essai, ce qui conduit à utiliser un minimum de 40 animaux (20 mâles et 20 femelles, auxquels s'ajoutent les témoins positifs nécessaires). La démarche décrite ici, qui correspond à l'une des formes simples du plan factoriel, équivaut à une analyse de variance à deux facteurs, dans laquelle le sexe et la concentration sont les facteurs principaux. Les données peuvent être analysées à l'aide de nombreux progiciels statistiques standard tels que SPSS, SAS, STATA ou Genstat, ou en utilisant le logiciel R. À partir de l'ensemble de données, on détermine la variabilité entre les sexes, la variabilité entre les concentrations et la variabilité liée à l'interaction entre sexe et concentrations. Chacun de ces termes est comparé à une estimation de la variabilité entre les animaux répartis au sein des groupes d'animaux de même sexe exposés à la même concentration. On trouvera plus de précisions sur cette méthode dans les manuels de statistiques classiques (voir les références) et dans les fichiers d'aide fournis avec les logiciels statistiques. On examine ensuite le terme d'interaction sexe x concentration dans un tableau ANOVA (5). En l'absence de terme d'interaction significatif, la combinaison des valeurs inter-sexes ou inter-niveaux de concentration permet de réaliser des tests statistiques valides entre les niveaux, en se basant sur le terme de variabilité intra-groupe combinée fourni par l'ANOVA. L'analyse se poursuit par la partition de la variabilité estimée entre concentrations, de façon à obtenir des contrastes, ce qui permet d'établir les contrastes linéaires et quadratiques des réponses pour l'ensemble des niveaux de concentration. Lorsqu'il y a une interaction significative sexe x concentration, ce terme peut à son tour être partitionné en contrastes d'interaction linéaire x sexe et quadratique x sexe. Ces termes permettent de vérifier si les réponses aux concentrations sont parallèles pour les deux sexes ou si elles diffèrent selon le sexe. L'estimation de la variabilité intra-groupe combinée peut servir à tester l'écart entre les moyennes en les comparant deux à deux. Ces comparaisons peuvent se faire entre les moyennes pour les deux sexes et entre les moyennes pour les différents niveaux de concentration (comparaisons avec les témoins négatifs, par exemple). En cas d'interaction significative, des comparaisons peuvent être faites entre les moyennes des différentes concentrations pour un même sexe, ou entre les moyennes des deux sexes à la même concentration. Références De nombreux manuels de statistiques traitent de la théorie, de la conception, de la méthodologie, de l'analyse et de l'interprétation des plans factoriels, depuis les analyses les plus simples, à deux facteurs, jusqu'aux formes complexes utilisées dans la conception de l'expérimentation. La liste ci-dessous n'est pas exhaustive. Certains ouvrages comportent des exemples d'application de ce type de démarches, accompagnés parfois d'un code permettant l'exécution des analyses sous différents logiciels.
|
|
(5) |
Dans la partie B, le chapitre B.12 est remplacé par le texte suivant: «B.12 Test du micronoyau sur érythrocytes de mammifères INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 474 (2016) de l'OCDE. Elle s'inscrit dans une série de méthodes d'essai sur la toxicologie génétique. Par ailleurs, un document de l'OCDE qui fournit des informations succintes sur les essais de génotoxicité et donne une vue d'ensemble des modifications récemment apportées à ces lignes directrices a été élaboré (1). Ce test du micronoyau pratiqué in vivo chez les mammifères se prête particulièrement bien à l'évaluation de la génotoxicité, car, malgré des variations entre les espèces, les facteurs du métabolisme in vivo, la pharmacocinétique et les processus de réparation de l'ADN sont actifs et contribuent aux réponses. Le test est également considéré comme utile pour explorer plus avant un effet génotoxique détecté dans un système in vitro. Le test du micronoyau in vivo est pratiqué chez des mammifères en vue de détecter des lésions des chromosomes ou de l'appareil mitotique d'érythroblastes résultant de l'action d'une substance d'essai. Il évalue la formation de micronoyaux dans des érythrocytes prélevés dans la moelle osseuse ou dans les cellules du sang périphérique d'animaux, généralement des rongeurs. Le test du micronoyau a pour but d'identifier les produits chimiques qui engendrent des lésions cytogénétiques induisant la formation de micronoyaux contenant des fragments de chromosomes retardataires ou des chromosomes entiers. Lorsqu'un érythroblaste de moelle osseuse se transforme en érythrocyte immature (parfois désigné sous le terme d'érythrocyte polychromatique, ou de réticulocyte), le noyau principal est expulsé et les éventuels micronoyaux qui se sont formés peuvent subsister dans le cytoplasme. La visualisation ou la détection des micronoyaux dans ces cellules est facilitée par l'absence de noyau principal. Un accroissement de la fréquence des érythrocytes immatures micronucléés chez les animaux traités est le signe d'une lésion chromosomique structurale ou numérique induite. Les érythrocytes micronucléés nouvellement formés sont identifiés et dénombrés, une fois colorés, par examen visuel au microscope ou par analyse automatique. L'utilisation d'une plateforme d'analyse automatique facilite considérablement le dénombrement d'un nombre suffisant d'érythrocytes immatures dans le sang périphérique ou la moelle osseuse d'animaux adultes. Ce type de plateforme constitue une alternative acceptable à l'évaluation manuelle (2). D'après des études comparatives, de telles méthodes, couplées à l'utilisation de normes d'étalonnage idoines, offrent une reproductibilité et une sensibilité inter-laboratoires et intra-laboratoire supérieures aux performances de l'examen manuel au microscope (3) (4). Parmi les systèmes automatisés pouvant mesurer la fréquence des érythrocytes micronucléés, on peut citer les cytomètres en flux (5), les plateformes d'analyse d'images (6) (7) et les cytomètres à balayage laser (8). Bien que cela ne fasse habituellement pas partie de l'essai, les fragments de chromosomes peuvent être distingués des chromosomes entiers d'après un certain nombre de critères. Ceux-ci comprennent la détection de la présence ou de l'absence de kinétochore ou d'ADN centromérique, tous deux étant caractéristiques d'un chromosome intact. L'absence de kinétochore ou d'ADN centromérique indique que le micronoyau ne contient que des fragments de chromosomes, tandis que la présence de l'un de ces éléments signe une perte chromosomique. Les définitions des termes employés figurent à l'appendice 1 REMARQUES PRÉLIMINAIRES La moelle osseuse de jeunes rongeurs adultes est le tissu cible des lésions génétiques dans le cadre de cet essai, les érythrocytes étant produits dans ce tissu. Le comptage des érythrocytes immatures micronucléés dans le sang périphérique peut aussi être effectué chez d'autres espèces de mammifères qui ont démontré une sensibilité suffisante pour permettre la détection de produits chimiques provoquant des aberrations chromosomiques structurales ou numériques dans ces cellules (par induction de micronoyaux dans les érythrocytes immatures), et à condition que ce choix soit justifié sur le plan scientifique. La fréquence des érythrocytes immatures micronucléés est le principal effet mesuré. Il est également possible de mesurer la fréquence des érythrocytes matures micronucléés dans le sang périphérique chez les espèces ne présentant pas une forte sélection splénique contre les cellules micronucléées et lorsque les animaux sont traités en continu pendant une période supérieure à la durée de vie d'un érythrocyte chez l'espèce concernée (par exemple, quatre semaines au moins chez la souris). Cet essai n'est pas pertinent s'il est prouvé que le/les produit(s) chimique(s) d'essai, ou leur(s) métabolite(s), n'atteindront pas le tissu cible. Avant d'utiliser la méthode d'essai pour générer des données avec pour objectif recherché l'application réglementaire, on examinera si, et si oui, pourquoi, elle peut fournir des résultats adéquats à cette fin. De telles considérations ne sont pas nécessaires quand les exigences réglementaires stipulent que le mélange doit être testé. PRINCIPE DE LA MÉTHODE D'ESSAI Les animaux sont exposés au produit chimique d'essai par une voie d'exposition idoine. Si l'on utilise la moelle osseuse, les animaux sont euthanasiés au(x) moment(s) approprié(s) après le traitement; la moelle est extraite, préparée sur des lames et colorée (9) (10) (11) (12) (13) (14) (15). Lorsqu'on utilise le sang périphérique, ce dernier est prélevé au(x) moment(s) approprié(s) après le traitement; les préparations sont ensuite réalisées et colorées (12) (16) (17) (18). Lors d'une administration aigüe du traitement, il importe d'effectuer le prélèvement de moelle osseuse ou de sang périphérique à un moment où l'induction d'érythrocytes immatures micronucléés par le traitement peut être détectée. Dans le cas d'un prélèvement de sang périphérique, un laps de temps suffisant doit s'être écoulé pour que ces éléments apparaissent dans la circulation sanguine. Les préparations sont analysées en vue de la détection de micronoyaux, par visualisation directe à l'aide d'un microscope, par analyse d'image, par cytométrie en flux ou par cytométrie à balayage laser. VÉRIFICATION DES COMPÉTENCES DU LABORATOIRE Épreuves de compétence Afin d'établir qu'il possède une expérience suffisante pour mener à bien l'essai avant de l'utiliser en routine, le laboratoire doit avoir démontré sa capacité à reproduire les résultats attendus d'après les données publiées (17) (19) (20) (21) (22) concernant les fréquences de micronoyaux avec un minimum de deux produits chimiques témoins positifs (y compris des réponses faibles induites par des doses faibles de témoins positifs) tels que ceux énumérés au tableau 1, et avec des témoins de véhicule/solvant compatibles (voir paragraphe 26). Les doses utilisées dans le cadre de ces expériences doivent produire des augmentations reproductibles qui sont fonction de la dose administrée, et démontrer la sensibilité et la plage dynamique du système d'essai sur le tissu en question (moelle osseuse ou sang périphérique); la méthode d'analyse retenue doit être celle qui sera employée par le laboratoire. Cette exigence ne s'applique pas aux laboratoires possédant déjà une expérience, c'est-à-dire qui disposent d'une base de données historiques telle que définie aux paragraphes 14 à 18. Données des témoins historiques Dans le cadre de la vérification des compétences, le laboratoire devra établir:
Lors de l'acquisition initiale de données en vue d'établir une distribution des témoins négatifs historiques, les données des témoins négatifs concomitants doivent être cohérentes avec les données publiées, lorsqu'elles existent. Puis, à mesure que de nouvelles données expérimentales viennent étoffer la plage de distribution des témoins historiques, les données des témoins négatifs concomitants doivent idéalement se situer dans les limites de contrôle à 95 % de cette distribution. La base des données historiques du laboratoire relatives aux témoins négatifs doit être statistiquement robuste pour permettre au laboratoire d'évaluer la distribution de ses données des témoins négatifs. La littérature suggère qu'un minimum de 10 expériences peut-être nécessaire, sachant qu'il serait préférable d'en compter au moins 20, réalisées dans des conditions expérimentales similaires. Les laboratoires doivent avoir recours à des méthodes de contrôle de la qualité telles que des graphiques statistiques (cartes C ou cartes X-barre, par exemple (23)), afin de déterminer la variabilité de leurs données et de démontrer leur maîtrise de la méthodologie. On trouve dans la littérature (24) d'autres recommandations sur la façon de constituer et d'utiliser ces données historiques (critères d'inclusion et d'exclusion des données dans la base et critères d'acceptabilité pour une expérimentation donnée). Si, au cours des expériences visant à vérifier sa compétence (comme décrit au paragraphe 13), le laboratoire n'a pas réalisé un nombre suffisant d'expériences pour établir une distribution des témoins négatifs statistiquement robuste (voir paragraphe 15), on peut accepter que la distribution soit construite au cours des premiers tests de routine. Cette approche devra suivre les recommandations établies dans la littérature (24) et les résultats des témoins négatifs obtenus lors de ces expériences devront être cohérents avec les données publiées des témoins négatifs. Toute modification du protocole expérimental doit être étudiée à la lumière de ses répercussions sur la cohérence des nouvelles données avec celles de la base de données des témoins historiques. Seules des incohérences majeures justifient l'établissement d'une nouvelle base de données des témoins historiques, après confirmation de la différence de distribution des données par des experts (voir paragraphe 15). Lors de la constitution de cette nouvelle base de données, le laboratoire n'a pas forcément besoin d'une base de données de témoins négatifs complète pour autoriser la conduite d'un essai, à condition qu'il puisse apporter la preuve que les valeurs des témoins négatifs concomitants sont cohérentes avec la précédente base de données ou avec les données publiées correspondantes. Les données des témoins négatifs désignent l'incidence des érythrocytes immatures micronucléés chez chaque animal. Les témoins négatifs concomitants se situent idéalement dans les limites de contrôle à 95 % de la distribution des données historiques des témoins négatifs contenues dans la base de données du laboratoire. Lorsque les données des témoins négatifs concomitants se situent en dehors des limites de contrôle à 95 %, leur inclusion dans la distribution des témoins historiques peut être acceptable, à condition que ces données ne soient pas exagérément extrêmes et qu'il soit prouvé que le système d'essai est «sous contrôle» (voir paragraphe 15) et qu'il n'y a pas eu de défaillance technique ou humaine. DESCRIPTION DE LA MÉTHODE D'ESSAI Préparations Sélection des espèces animales Il convient d'employer de jeunes animaux adultes sains issus de souches courantes de laboratoire. On pourra choisir des souris, des rats, ou toute autre espèce de mammifère appropriée. Si l'examen porte sur le sang périphérique, il doit être établi que l'élimination par la rate des cellules micronucléées ne compromet pas la détection des micronoyaux induits chez l'espèce sélectionnée. Cela a été clairement démontré pour le sang périphérique chez la souris et le rat (2). L'utilisation d'une espèce autre que le rat ou la souris doit être scientifiquement justifiée dans le rapport. Si des animaux autres que des rongeurs sont utilisés, il est recommandé que la mesure des micronoyaux induits soit intégrée à un autre essai de toxicité pertinent. Conditions d'encagement et d'alimentation des animaux Pour les rongeurs, la température de l'animalerie doit être maintenue à 22°C (± 3°C). L'humidité relative, qui est idéalement de 50 à 60 %, doit atteindre au moins 40 % et de préférence ne pas dépasser 70 %, sauf durant le nettoyage du local. L'éclairage est artificiel, la séquence d'éclairage étant de 12 heures de clarté et 12 heures d'obscurité. Le régime alimentaire des animaux est le régime classique de laboratoire avec eau potable à volonté. Le choix des aliments peut être influencé par la nécessité d'assurer une bonne incorporation du produit chimique dans la nourriture si l'administration se fait par cette voie. Les rongeurs sont mis en cage par petits groupes d'individus de même sexe (cinq au maximum par cage) recevant le même traitement, si aucun comportement agressif n'est à craindre, de préférence dans des cages à fond plein dotées d'un enrichissement environnemental approprié. Les animaux peuvent être encagés individuellement si cela est justifié du point de vue scientifique. Préparation des animaux On choisit habituellement de jeunes animaux adultes sains (les rongeurs sont idéalement âgés de 6 à 10 semaines au début du traitement, bien que des animaux un peu plus âgés soient également acceptables), qui sont répartis de manière aléatoire entre les groupes témoins et les groupes de traitement. Chaque animal est identifié individuellement selon une méthode sans cruauté, la moins invasive possible (par exemple, baguage, étiquetage, pose d'une puce électronique ou identification biométrique, en évitant l'entaillage des oreilles ou la phalangectomie) et gardés dans leurs cages pendant au moins cinq jours afin qu'ils s'acclimatent aux conditions du laboratoire. Les cages doivent être placées de manière à réduire au minimum l'influence éventuelle de leur disposition sur les résultats. Il convient d'éviter toute contamination croisée entre le témoin positif et le produit chimique d'essai. Au début de l'étude, la variation pondérale des animaux doit être minimale et ne pas dépasser ± 20 % du poids moyen de chaque sexe. Préparation des doses Lorsque les produits chimiques testés sont solides, ils sont dissous ou mis en suspension dans des solvants ou des véhicules appropriés, ou incorporés aux aliments ou à l'eau de boisson avant d'être administrés aux animaux. Les produits chimiques liquides peuvent être administrés directement ou dilués avant d'être administrés. En cas d'exposition par inhalation, les produits chimiques d'essai peuvent être administrés sous forme de gaz, de vapeur ou d'aérosol solide ou liquide, en fonction de leurs propriétés physico-chimiques. On utilisera des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage et définissent les conditions de stockage appropriées. Conditions de l'essai Solvant/véhicule Le solvant/véhicule ne doit pas produire d'effets toxiques aux doses utilisées, ni pouvoir réagir avec les produits chimiques d'essai. Le recours à des solvants/véhicules inhabituels doit être justifié par des données de référence faisant état de leur compatibilité. Il est recommandé d'envisager en premier lieu l'utilisation d'un solvant/véhicule aqueux chaque fois que c'est possible. Parmi les exemples de solvants/véhicules compatibles couramment utilisés figurent notamment l'eau, le sérum physiologique, les solutions de méthylcellulose, les solutions de carboxyméthylcellulose sodique, l'huile d'olive et l'huile de maïs. En l'absence de données historiques ou publiées significatives montrant qu'aucun micronoyau ou effet délétère n'est induit par un solvant/véhicule inhabituel sélectionné, une étude initiale devra être réalisée afin d'établir l'acceptabilité du témoin pour le solvant/véhicule. Témoins Témoins positifs Chaque essai doit normalement inclure un groupe d'animaux traités avec un produit chimique utilisé comme témoin positif. Cette étape peut être évitée une fois que le laboratoire d'essai a apporté la preuve de ses compétences pour la conduite de l'essai et établi la plage des témoins positifs historiques. Lorsqu'aucun groupe témoin positif n'est utilisé simultanément, des témoins d'examen (lames fixées et non colorées ou échantillons de suspension cellulaire, selon les besoins de la méthode d'examen) devront être inclus dans chaque expérience. Pour ce faire, on pourra intégrer à l'étape d'examen des échantillons de référence idoines obtenus et stockés lors d'un essai séparé sur témoin positif mené périodiquement (par exemple, tous les 6 à 18 mois), notamment au cours de l'épreuve de compétence et par la suite sur une base régulière, si nécessaire. Les produits chimiques utilisés comme témoins positifs doivent produire, de façon fiable, un accroissement détectable de la fréquence des micronoyaux par rapport au niveau spontané. Si l'examen est effectué manuellement au microscope, les doses des témoins positifs doivent être choisies de telle sorte que les effets soient nets mais que l'identité des lames codées ne soit pas évidente pour l'examinateur. Il est possible d'administrer le témoin positif par une voie différente de celle du produit chimique d'essai, de suivre un autre programme de traitement et de n'effectuer qu'un seul prélèvement d'échantillon. De plus, l'emploi de témoins positifs appartenant à la même classe chimique peut être envisagé, s'il y a lieu. Des exemples de produits chimiques utilisés comme témoins positifs figurent au tableau 1 ci-dessous. Tableau 1 Exemples de produits chimiques utilisés comme témoins positifs Produit chimique et noCAS Méthanesulfonate d'éthyle [noCAS 62-50-0] Méthanesulfonate de méthyle [noCAS 66-27-3] Ethylnitrosourée [noCAS 759-73-9] Mitomycine C [noCAS 50-07-7] Cyclophosphamide (monohydratée) [noCAS 50-18-0 (noCAS 6055-19-2)] Triéthylènemélamine [noCAS 51-18-3] Colchicine [noCAS 64-86-8] ou vinblastine [noCAS 865-21-4] — comme aneugènes Témoins négatifs Pour chaque moment d'échantillonnage, il faut inclure un groupe d'animaux témoins négatifs, qui seront manipulés de la même façon que les groupes traités, mais ne recevront pas le produit chimique d'essai. Si un solvant/véhicule est utilisé pour administrer le produit chimique d'essai, ce solvant/véhicule doit être administré au groupe témoin. Toutefois, si des résultats obtenus antérieurement par le laboratoire d'essai démontrent que la variabilité interindividuelle et la fréquence des cellules comportant des micronoyaux provenant de témoins négatifs sont stables pour chaque moment d'échantillonnage, un seul échantillonnage peut alors suffire pour les témoins négatifs. Dans ce cas, il doit intervenir au moment du premier prélèvement effectué dans le cadre de l'étude. Si le sang périphérique est utilisé, un échantillon prélevé avant le traitement peut aussi tenir lieu de témoin négatif pour les études de courte durée, lorsque les résultats sont conformes aux valeurs de la base de données des témoins historiques du laboratoire. Il a été démontré chez le rat que le prélèvement de faibles volumes (par exemple, inférieurs à 100 μl/jour) avant le traitement avait un impact minime sur la fréquence de fond des micronoyaux (25). MODE OPÉRATOIRE Nombre et sexe des animaux En règle générale, la réponse des micronoyaux est similaire chez les animaux mâles et femelles (26), et la plupart des études pourrait donc être réalisée sur l'un ou l'autre des deux sexes. Des données démontrant des différences significatives entre les mâles et les femelles (par exemple sur le plan de la toxicité systémique, du métabolisme, de la biodisponibilité de la toxicité sur la moelle osseuse, etc. comprenant également des données provenant par exemple d'études de détermination des doses) encouragent l'utilisation des deux sexes. Il peut donc être plus approprié dans ce cas de mener une étude sur les deux sexes, par exemple dans le cadre d'une étude de toxicité à doses répétées. Il pourrait être judicieux de recourir à un plan factoriel en cas d'utilisation des deux sexes. Des précisions sur cette méthode d'analyse des données sont fournies à l'appendice 2. La taille des groupes au début de l'étude doit permettre de disposer dans chaque groupe d'au moins cinq animaux analysables du même sexe ou de chaque sexe si les deux sont utilisés. Si l'exposition humaine est spécifique pour un sexe, par exemple dans le cas de certains produits pharmaceutiques, l'essai sera pratiqué sur des animaux du sexe approprié. À titre d'information concernant le nombre maximum d'animaux généralement requis, une étude sur moelle osseuse conduite selon les paramètres établis au paragraphe 37, impliquant trois groupes de traitement et des groupes de témoins négatifs et de témoins positifs concomitants (chaque groupe étant composé de cinq animaux du même sexe) nécessitera entre 25 et 35 animaux. Niveaux de dose Si l'on procède à une étude préliminaire de détermination des doses à administrer parce qu'on ne dispose pas de données fiables pour orienter le choix des doses, cette étude préliminaire doit être effectuée dans le même laboratoire, en utilisant une espèce, une souche, un sexe et un régime de traitement identiques à ceux de l'étude principale (27). Elle devra avoir pour objectif de déterminer la dose maximale tolérée (DMT), définie comme la dose la plus élevée qui sera tolérée sans faire apparaître de toxicité limitante, dans le cadre de la durée de l'étude (par exemple, induisant une baisse du poids corporel ou une cytotoxicité du système hématopoïétique, mais ne provoquant pas la mort ou des signes de douleur, de souffrance ou de détresse imposant de sacrifier les animaux (28)). La dose la plus élevée peut aussi être définie comme la dose qui produit une toxicité dans la moelle osseuse (par exemple une diminution de plus de 50 % de la proportion d'érythrocytes immatures par rapport au nombre total d'érythrocytes dans la moelle osseuse ou le sang périphérique, sans que cette proportion devienne inférieure à moins 20 % de la valeur témoin). Toutefois, l'analyse des cellules positives au marqueur CD71 dans la circulation sanguine périphérique (par cytométrie en flux) révèle que cette fraction d'érythrocytes immatures très jeunes répond plus rapidement aux substances toxiques que la cohorte plus importante d'érythrocytes immatures positifs à l'ARN. Par conséquent, la toxicité peut apparaître supérieure dans le cadre d'expériences prévoyant une exposition aigüe suivie de l'examen des érythrocytes immatures positifs au marqueur CD71 par rapport aux dispositifs d'essai qui recensent les érythrocytes immatures en fonction de leur contenu ARN. Pour cette raison, lorsque l'expérience prévoit cinq jours de traitement ou moins, la dose la plus élevée du produit chimique d'essai entrainant une toxicité peut se définir comme la dose causant une réduction statistiquement significative de la proportion d'érythrocytes positifs au marqueur CD71 par rapport au total des érythrocytes, sans descendre en dessous de 5 % de la valeur témoin (29). Les produits chimiques d'essai dont les propriétés toxicocinétiques sont saturées ou qui induisent un processus de détoxification pouvant se traduire par une baisse de l'exposition après une administration à long terme peuvent faire exception aux critères d'établissement des doses et doivent être évalués au cas par cas. Pour obtenir des informations sur la relation dose-réponse, une étude complète doit comporter un groupe témoin négatif et au moins trois niveaux de doses, séparés en règle générale par un facteur de 2, et de 4 au maximum. Si le produit chimique d'essai ne provoque aucune toxicité dans le cadre d'une étude de détermination des doses à administrer, ou d'après les données disponibles, la dose la plus élevée par période d'administration de 14 jours ou plus doit être de 1 000 mg/kg de poids corporel/jour ou, pour des périodes d'administration de moins de 14 jours, de 2 000 mg/kg de poids corporel/jour. En revanche, si le produit chimique d'essai provoque une toxicité, la dose administrée la plus élevée devra correspondre à la DMT et les niveaux de dose employés devront de préférence s'étendre de la dose maximale à une dose induisant peu ou pas de toxicité. Lorsqu'une toxicité sur le tissu cible (moelle osseuse) est observée à tous les niveaux de doses administrés, il est conseillé de procéder à des études complémentaires à des doses non toxiques. Les études visant à caractériser davantage les informations sur la relation quantitative dose-réponse peuvent nécessiter un ou plusieurs groupes de traitement supplémentaires. Enfin, ces limites peuvent varier pour certains types de produits chimiques (par exemple les produits pharmaceutiques à usage humain) faisant l'objet d'exigences spécifiques. Essai limite Si les essais préliminaires de détermination des doses ou les données existantes concernant des souches de rongeurs apparentées indiquent qu'un régime de traitement égal ou supérieur à la dose limite (décrite ci-dessous) n'engendre pas d'effets toxiques observables (notamment aucune dépression de la fonction médullaire osseuse ni autre cytotoxicité pour le tissu cible), et si la génotoxicité n'est pas escomptée d'après des études de génotoxicité in vitro ou d'après les données relatives aux produits chimiques structurellement apparentés, une étude complète utilisant trois niveaux de doses peut ne pas être considérée comme nécessaire, à condition qu'il ait été démontré que les produits chimiques d'essai atteignent le tissu cible (moelle osseuse). Dans ce cas, un seul niveau de dose, égal à la dose limite, peut s'avérer suffisant. Pour une période d'administration égale ou supérieure à 14 jours, la dose limite est de 1 000 mg/kg de poids corporel/jour. Pour des périodes d'administration de moins de 14 jours, la dose limite est de 2 000 mg/kg de poids corporel/jour. Administration des doses Lors de la conception d'un essai, il convient de tenir compte de la voie d'exposition humaine anticipée. Par conséquent, les voies d'administration telles que l'alimentation, l'eau de boisson, l'inhalation, l'implantation ainsi que les voies topique, sous-cutanée, intraveineuse, orale (par gavage) et intra-trachéale sont autant de choix valables, sous réserve qu'ils soient justifiés. Dans tous les cas, la voie retenue doit permettre une exposition adéquate du/des tissu(s) cible(s). L'injection intrapéritonéale n'est en général pas recommandée, car elle ne constitue pas une voie d'exposition humaine envisagée, et ne sera utilisée qu'en cas de justification scientifique spécifique. Si le produit chimique d'essai est mélangé à l'alimentation ou à l'eau de boisson, surtout dans le cas d'un dosage unique, il convient de s'assurer que le délai entre l'absorption de nourriture et d'eau et le prélèvement est suffisant pour permettre une détection des effets (voir paragraphe 37). Le volume maximal de liquide administrable en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne doit normalement pas excéder 1 ml/100 g de poids corporel, sauf pour les solutions aqueuses, où un maximum de 2 ml/100 g est acceptable. L'utilisation de volumes plus importants doit être justifiée. Il convient de réduire au minimum la variation du volume administré en ajustant la concentration à tous les niveaux de doses, afin de garantir l'administration d'un volume constant pour un poids corporel donné, sauf pour les produits irritants ou corrosifs dont les effets sont généralement exacerbés lorsqu'on augmente la concentration. Programme de traitement On réalisera de préférence deux traitements ou plus, administrés à des intervalles de 24 heures, notamment lorsque l'essai est intégré à d'autres études de toxicité. Alternativement, on pourra administrer des traitements simples, si cela s'avère scientifiquement justifié (par exemple dans le cas de produits chimiques d'essai connus pour bloquer le cycle cellulaire). L'administration du produit chimique d'essai peut aussi être fractionnée, à raison de deux traitements le même jour à intervalle de deux à trois heures maximum, afin de faciliter l'administration d'un grand volume. En pareil cas, ou lors d'une administration du produit chimique d'essai par inhalation, le moment d'échantillonnage sera fixé en fonction du moment d'administration de la dernière fraction, ou de la fin de l'exposition. L'essai peut être réalisé chez la souris ou le rat de trois manières différentes:
D'autres régimes de traitement ou de prélèvement peuvent être envisagés le cas échéant et sous réserve qu'ils soient scientifiquement justifiés, ainsi que pour faciliter l'intégration avec d'autres essais de toxicité. Observations Les animaux d'essai font l'objet d'un examen clinique général. Les signes cliniques doivent être consignés au moins une fois par jour, de préférence aux mêmes heures, en prenant en considération la période où les effets anticipés devraient être les plus marqués après l'administration. Au moins deux fois par jour pendant la durée du traitement, l'ensemble des animaux fait l'objet d'un constat de morbidité et de mortalité. Tous les animaux doivent être pesés au début de l'étude, au moins une fois par semaine au cours des études à doses répétées, puis lors de l'euthanasie. Pour les études dont la durée est égale ou supérieure à une semaine, la consommation de nourriture doit également être mesurée au moins une fois par semaine. Si le produit chimique testé est administré dans l'eau de boisson, la consommation d'eau est mesurée à chaque changement d'eau et au moins une fois par semaine. Les animaux montrant des signes non létaux de toxicité excessive sont euthanasiés avant la fin de l'essai (28). Dans certaines circonstances, la température corporelle des animaux peut être surveillée, une hyper- ou une hypothermie causée par le traitement pouvant fausser les résultats (32) (33) (34). Exposition du tissu cible Lorsque cela se justifie et qu'il n'existe pas d'autres données sur l'exposition (voir paragraphe 48), il convient de réaliser un prélèvement sanguin au(x) moment(s) idoine(s) pour vérifier la concentration des produits chimiques d'essai dans le plasma afin de démontrer que la moelle osseuse a bien été exposée. Préparation de la moelle osseuse et du sang Les cellules de moelle osseuse sont généralement obtenues à partir des fémurs ou tibias des animaux, immédiatement après l'euthanasie. Le plus souvent, les cellules sont extraites, préparées et colorées suivant des méthodes bien établies. De faibles volumes de sang périphérique peuvent être prélevés, conformément aux normes en vigueur en matière de bien-être animal, soit selon une méthode permettant la survie de l'animal d'essai (par exemple prélèvement dans la veine caudale ou un autre vaisseau sanguin approprié), soit par ponction cardiaque ou prélèvement sur un vaisseau sanguin principal, après l'euthanasie de l'animal. Pour les érythrocytes provenant de la moelle osseuse ou du sang périphérique, suivant la méthode d'analyse, on peut effectuer immédiatement une coloration supravitale des cellules sanguines (16) (17) (18) ou des frottis qui sont ensuite colorés pour analyse microscopique, ou fixés puis colorés de manière idoine pour analyse par cytométrie en flux. L'emploi d'un colorant spécifique de l'ADN [par exemple, l'acridine orange (35) ou l'Hoechst 33258 avec la pyronine-Y (36)] peut éliminer une partie des artefacts associés à l'utilisation d'un colorant non spécifique de l'ADN. Cet avantage n'exclut pas l'utilisation de colorants classiques (par exemple le Giemsa pour l'analyse microscopique). D'autres méthodes, faisant par exemple appel à des colonnes de cellulose pour retirer les cellules nucléées (37) (38), peuvent être utilisées à condition que leur compatibilité avec la préparation d'échantillon dans le laboratoire ait été démontrée. Lorsque ces méthodes sont applicables, les anticorps anti-kinétochore (39), la méthode FISH associée à des sondes ADN pancentromériques (40) ou encore le marquage in situ par amorçage à l'aide d'amorces pancentromériques, avec un colorant de contraste de l'ADN approprié (41), peuvent être utilisés pour identifier le contenu (chromosomes/fragments chromosomiques) des micronoyaux afin de déterminer si le mécanisme d'induction des micronoyaux est le résultat d'une activité clastogène et/ou aneugène. D'autres méthodes de discrimination des effets clastogènes et aneugènes peuvent être utilisées, à condition que leur efficacité ait été prouvée. Analyse (manuelle et automatique) Toutes les lames et échantillons, y compris ceux des témoins positifs et négatifs, doivent être codés individuellement avant tout type d'analyse, et doivent être randomisés afin que l'analyste ne puisse pas connaître le traitement utilisé. Ce codage est inutile si l'on fait appel à un système d'analyse automatique, auquel cas l'analyse ne s'appuie pas sur un examen visuel et ne peut être affectée par un biais de l'opérateur. Pour chaque animal, on détermine la proportion d'érythrocytes immatures dans le nombre total d'érythrocytes (immatures + matures) en comptant au moins 500 érythrocytes dans le cas de la moelle osseuse et 2 000 érythrocytes dans le cas du sang périphérique (42). L'incidence des érythrocytes immatures micronucléés est déterminée sur la base de l'examen d'au moins 4 000 érythrocytes immatures par animal (43). Si la base de données des témoins négatifs historiques indique une fréquence de fond moyenne des érythrocytes immature micronucléés < 0,1 % dans le laboratoire, il convient d'envisager l'examen de cellules supplémentaires. À l'examen des lames, le pourcentage d'érythrocytes immatures par rapport au nombre total d'érythrocytes chez les animaux traités ne doit pas être inférieur à 20 % de cette valeur pour les témoins de véhicule/solvant lors de l'analyse microscopique, et pas inférieur à environ 5 % de cette valeur pour les témoins de véhicule/solvant lors de l'analyse par cytométrie des érythrocytes immatures positifs au marqueur CD71 (voir paragraphe 31) (29). Par exemple, pour un essai sur moelle osseuse avec examen microscopique, si la proportion témoin d'érythrocytes immatures dans la moelle osseuse est de 50 %, la limite supérieure de toxicité équivaudra à 10 % d'érythrocytes immatures. Étant donné que la rate de rat immobilise et détruit les érythrocytes micronucléés, pour maintenir la sensibilité de l'essai lors de l'analyse du sang périphérique chez le rat, il est préférable de circonscrire l'analyse aux érythrocytes immatures micronucléés les plus jeunes. En cas d'utilisation de méthodes d'analyse automatique, ces érythrocytes les plus immatures peuvent être repérés grâce à leur contenu élevé en ARN, ou au niveau élevé de récepteurs de la transferrine (positifs au marqueur CD71) exprimés à leur surface (31). Toutefois, une comparaison directe des différentes méthodes de coloration a révélé que plusieurs méthodes permettaient d'obtenir des résultats satisfaisants, y compris la méthode classique de coloration à l'acridine orange (3) (4). RÉSULTATS ET RAPPORT Traitement des résultats Les résultats individuels pour chaque animal doivent être présentés sous forme de tableaux. Le nombre d'érythrocytes immatures examinés, le nombre d'érythrocytes immatures micronucléés et la proportion d'érythrocytes immatures doivent être présentés séparément pour chaque animal analysé. Si des souris sont traitées en continu pendant au moins quatre semaines, il convient de fournir également les données relatives au nombre et à la proportion d'érythrocytes matures micronucléés, si elles ont été recueillies. Les données relatives à la toxicité pour l'animal et aux signes cliniques doivent elles-aussi être consignées. Critères d'acceptabilité Les critères suivants déterminent l'acceptabilité de l'essai:
Évaluation et interprétation des résultats À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement positif si:
Si seule la dose la plus élevée est examinée à un moment d'échantillonnage particulier, un produit chimique d'essai est considéré comme clairement positif si on constate une augmentation statistiquement significative par rapport aux témoins négatifs concomitants et que les résultats se situent à l'extérieur de la plage de distribution des données des témoins négatifs historiques (par exemple, limites de contrôle à 95 % d'une loi de Poisson). Des recommandations concernant les méthodes statistiques les plus appropriées sont disponibles dans la littérature (44) (45) (46) (47). Une analyse de la relation dose-réponse doit porter sur un minimum de trois groupes de traitement. Les méthodes statistiques doivent utiliser l'animal comme unité expérimentale. Des résultats positifs obtenus dans un test du micronoyau indiquent qu'un produit chimique d'essai induit la formation de micronoyaux, qui résultent de lésions des chromosomes ou de l'appareil mitotique d'érythroblastes chez l'espèce utilisée dans l'essai. Si l'on cherche à détecter des centromères dans les micronoyaux, la mise en évidence de tels micronoyaux pourvus de centromères (par détection d'ADN centromérique ou de kinétochore, signes d'une perte chromosomique) révèle le caractère aneugène du produit chimique d'essai. À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement négatif si, dans toutes les conditions expérimentales étudiées:
Des recommandations concernant les méthodes statistiques les plus appropriées sont disponibles dans la littérature (44) (45) (46) (47). L'exposition de la moelle osseuse à un produit chimique d'essai peut être démontrée par une baisse du ratio entre érythrocytes immatures et matures, ou par la mesure de la concentration du produit chimique dans le plasma ou le sang. Dans le cas d'une administration par intraveineuse, la preuve de l'exposition n'est pas nécessaire. Pour démontrer l'exposition de la moelle osseuse, on peut également avoir recours aux données ADME, obtenues dans le cadre d'une étude indépendante employant la même voie d'administration et la même espèce. Des résultats négatifs signifient que, dans les conditions de l'essai, le produit chimique d'essai n'induit pas de micronoyau dans les érythrocytes immatures chez l'espèce testée. Il n'est pas nécessaire de vérifier une réponse clairement positive ou clairement négative. Lorsque la réponse n'est ni clairement négative ni clairement positive, et afin d'établir la signification biologique d'un résultat (par exemple, une augmentation faible ou marginale), les données doivent être soumises à un jugement d'experts et/ou des investigations plus poussées sur les expériences déjà réalisées. Dans certains cas, il peut être utile d'examiner des cellules supplémentaires ou de répéter l'expérience en modifiant les conditions expérimentales. Dans de rares cas, même après des investigations complémentaires, les données ne permettront pas de conclure que le produit chimique d'essai provoque des résultats clairement positifs ou négatifs. Les résultats de l'étude seront alors considérés comme équivoques. Rapport d'essai Le rapport d'essai doit contenir les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Centromère : régions d'un chromosome auxquelles les fibrilles du fuseau sont associées pendant la division de la cellule et qui permettent le mouvement ordonné des chromosomes-filles vers les pôles des cellules-filles. Produit chimique : une substance ou un mélange. Érythroblaste : Stade précoce du développement d'un érythrocyte, précédant immédiatement la formation de l'érythrocyte immature, et lors duquel la cellule contient encore un noyau. Kinétochore : Structure protéique se formant au niveau du centromère des cellules eucaryotes, qui relie le chromosome à des polymères microtubulaires provenant du fuseau mitotique au cours de la mitose et de la méiose, et qui agit pendant la division cellulaire pour séparer les chromatides sœurs. Micronoyau : Petit noyau, présent en plus du noyau principal des cellules et séparé de celui-ci, produit pendant la télophase de la mitose (méiose) par des chromosomes ou des fragments de chromosomes retardataires. Érythrocyte normochromatique ou mature : Érythrocyte parvenu à maturité complète ayant perdu l'ARN résiduel demeurant après l'énucléation et/ou ayant perdu d'autres marqueurs cellulaires à vie courte qui disparaissent généralement après l'énucléation qui suit la division finale de l'érythroblaste. Érythrocyte polychromatique ou immature : Érythrocyte nouvellement formé, à un stade intermédiaire de son développement, réagissant aux composants bleus et rouges des colorants classiques du sang tels que la coloration Giemsa-Wright, en raison de la présence d'ARN résiduel. De telles cellules nouvellement formées sont pratiquement identiques aux réticulocytes, observables au moyen d'une coloration vitale sous l'action de laquelle l'ARN résiduel s'agglutine pour former un réticulum. D'autres méthodes, telles que la coloration monochromatique de l'ARN à l'aide de colorants fluorescents, ou le repérage des marqueurs de surface à vie courte tels que le CD71 au moyen d'anticorps fluorescents, sont aujourd'hui utilisées pour détecter les cellules sanguines nouvellement formées. Les érythrocytes polychromatiques, les réticulocytes et les érythrocytes positifs aux marqueurs CD71 sont tous des érythrocytes immatures, correspondant à différents stades d'évolution. Réticulocyte : Érythrocyte nouvellement formé coloré au moyen d'un colorant vital sous l'action duquel l'ARN cellulaire résiduel s'agglutine pour former un réticulum caractéristique. Réticulocytes et érythrocytes polychromatiques sont des érythrocytes à un stade de développement très proche l'un de l'autre. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Appendice 2 PLAN FACTORIEL UTILISÉ POUR IDENTIFIER LES DIFFÉRENCES ENTRE SEXES DANS LE TEST DU MICRONOYAU IN VIVO Plan factoriel et analyse factorielle Selon cette démarche, un minimum de 5 mâles et de 5 femelles sont exposés à chaque concentration d'essai, ce qui conduit à utiliser un minimum de 40 animaux (20 mâles et 20 femelles, auxquels s'ajoutent les témoins positifs nécessaires). La démarche décrite ici, qui correspond à l'une des formes simples du plan factoriel, équivaut à une analyse de variance à deux facteurs, dans laquelle le sexe et la concentration sont les facteurs principaux. Les données peuvent être analysées à l'aide de nombreux progiciels statistiques standard tels que SPSS, SAS, STATA ou Genstat, ou en utilisant le logiciel R. À partir de l'ensemble de données, on détermine la variabilité entre les sexes, la variabilité entre les concentrations et la variabilité liée à l'interaction entre sexe et concentrations. Chacun de ces termes est comparé à une estimation de la variabilité entre les animaux répartis au sein des groupes d'animaux de même sexe exposés à la même concentration. On trouvera plus de précisions sur cette méthode dans les manuels de statistiques classiques (voir les références) et dans les fichiers d'aide fournis avec les logiciels statistiques. On examine ensuite le terme d'interaction sexe x concentration dans un tableau ANOVA (6). En l'absence de terme d'interaction significatif, la combinaison des valeurs inter-sexes ou inter-niveaux de concentration permet de réaliser des tests statistiques valides entre les niveaux, en se basant sur le terme de variabilité intra-groupe combinée fourni par l'ANOVA. L'analyse se poursuit par la partition de la variabilité estimée entre concentrations, de façon à obtenir des contrastes, ce qui permet d'établir les contrastes linéaires et quadratiques des réponses pour l'ensemble des niveaux de concentration. Lorsqu'il y a une interaction significative sexe x concentration, ce terme peut à son tour être partitionné en contrastes d'interaction linéaire x sexe et quadratique x sexe. Ces termes permettent de vérifier si les réponses aux concentrations sont parallèles pour les deux sexes ou si elles diffèrent selon le sexe. L'estimation de la variabilité intra-groupe combinée peut servir à tester l'écart entre les moyennes en les comparant deux à deux. Ces comparaisons peuvent se faire entre les moyennes pour les deux sexes et entre les moyennes pour les différents niveaux de concentration (comparaisons avec les témoins négatifs, par exemple). En cas d'interaction significative, des comparaisons peuvent être faites entre les moyennes des différentes concentrations pour un même sexe, ou entre les moyennes des deux sexes à la même concentration. Références De nombreux manuels de statistiques traitent de la théorie, de la conception, de la méthodologie, de l'analyse et de l'interprétation des plans factoriels, depuis les analyses les plus simples, à deux facteurs, jusqu'aux formes complexes utilisées dans la conception de l'expérimentation. La liste ci-dessous n'est pas exhaustive. Certains ouvrages comportent des exemples d'application de ce type de démarches, accompagnés parfois d'un code permettant l'exécution des analyses sous différents logiciels.
|
|
(6) |
Dans la partie B, le chapitre B.15. est supprimé. |
|
(7) |
Dans la partie B, le chapitre B.16. est supprimé. |
|
(8) |
Dans la partie B, le chapitre B.18. est supprimé. |
|
(9) |
Dans la partie B, le chapitre B.19. est supprimé. |
|
(10) |
Dans la partie B, le chapitre B.20. est supprimé. |
|
(11) |
Dans la partie B, le chapitre B.24. est supprimé. |
|
(12) |
Dans la partie B, le chapitre B.47. est remplacé par le texte suivant: «B.47 Méthode d'essai d'opacité et de perméabilité de la cornée bovine pour l'identification des produits chimiques i) provoquant des lésions oculaires graves ou ii) ne relevant d'aucune classification pour irritation oculaire ou lésion oculaire grave INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 437 (2013) de l'OCDE pour les essais de produits chimiques. La méthode d'essai d'opacité et de perméabilité de la cornée bovine (OPCB) a été évaluée par le Comité de coordination inter-agences pour la validation des méthodes alternatives (ICCVAM) des États-Unis, en collaboration avec le Centre européen pour la validation de méthodes alternatives (ECVAM) et le Centre japonais pour la validation de méthodes alternatives (JaCVAM), en 2006 et 2010 (1)(2). La première évaluation visait à déterminer l'utilité de la méthode OPCB pour identifier les produits chimiques (substances et mélanges) provoquant des lésions oculaires graves (1). La seconde évaluation a consisté à déterminer l'utilité de la méthode OPCB pour identifier les produits chimiques (substances et mélanges) non classés comme irritants pour l'œil ou provoquant de graves lésions oculaires (2). La base de données de validation OPCB contient au total 113 substances et 100 mélanges (2)(3). À l'issue de ces évaluations et de leur examen par les pairs, il a été conclu que cette méthode d'essai peut identifier correctement les produits chimiques (substances et mélanges) provoquant des lésions oculaires graves (catégorie 1) ou ne relevant d'aucune classification pour irritation oculaire ou lésion oculaire grave, selon la définition du Système général harmonisé de classification et d'étiquetage des produits chimiques (SGH) des Nations Unies (ONU) (4) et du règlement (CE) no 1272/2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges (CLP) (7), et son utilité scientifique a donc été validée dans les deux cas. Une lésion oculaire grave est une lésion des tissus oculaires ou une dégradation sévère de la vue, provoquée par l'application d'un produit chimique testé sur la face antérieure de l'œil, et qui n'est pas totalement réversible dans les 21 jours suivant l'application. Les produits chimiques testés provoquant des lésions oculaires graves sont classées dans la catégorie 1 du SGH de l'ONU. Les produits chimiques non classés comme irritants pour les yeux ou provoquant de graves lésions oculaires sont définis comme ne répondant pas aux critères de classification des catégories 1 ou 2 (2A ou 2B) du SGH de l'ONU, autrement dit, ils sont considérés comme ne relevant d'aucune catégorie du SGH de l'ONU. La présente méthode d'essai indique les applications recommandées et les limites de la méthode OPCB au regard de ces évaluations. Les principales différences entre la version originale de 2009 et la mise à jour de 2013 de la ligne directrice de l'OCDE concernent notamment: l'application de la méthode OPCB pour identifier les produits chimiques ne relevant d'aucune classification au titre du SGH de l'ONU (paragraphes 2 et 7 de la version originale); des éclaircissements sur l'applicabilité de la méthode OPCB pour tester les alcools, les cétones et les solides (paragraphes 6 et 7) ainsi que les substances et les mélanges (paragraphe 8); des éclaircissements sur la façon de tester les substances tensioactives et les mélanges contenant des tensioactifs (paragraphe 28); des mises à jour et des éclaircissements relatifs aux témoins positifs (paragraphes 39 et 40); une mise à jour des critères de décision concernant la méthode OPCB (paragraphe 47); une mise à jour des critères d'acceptation de l'étude (paragraphe 48); une mise à jour concernant les éléments du rapport d'essai (paragraphe 49); une mise à jour de l'appendice 1 sur les définitions; l'ajout de l'appendice 2 pour la valeur prédictive de la méthode d'essai OPCB dans différents système de classification; une mise à jour de l'appendice 3 sur la liste des substances d'épreuve; et une mise à jour de l'appendice 4 sur le porte-cornée pour la méthode OPCB (paragraphe 1) et sur l'opacitomètre (paragraphes 2 et 3). Il est généralement admis que dans notre horizon de prévision, aucun essai unique d'irritation oculaire in vitro ne pourra remplacer le test oculaire in vivo de Draize pour établir le degré d'irritation oculaire provoqué par les différentes classes de produits chimiques. Cependant, la combinaison de plusieurs méthodes de substitution dans le cadre d'une stratégie d'essais (à plusieurs niveaux) pourrait remplacer le test oculaire de Draize (5). L'approche «top-down» (5) est indiquée lorsque, d'après les informations existantes, on s'attend à ce qu'un produit chimique soit fortement irritant, alors que l'approche «bottom-up» (5) est conçue pour être appliquée quand, au vu des informations existantes, un produit chimique devrait a priori ne causer aucune irritation oculaire suffisante pour nécessiter une classification. La méthode OPCB est une méthode d'essai in vitro pouvant être utilisée, dans certaines circonstances et compte tenu de ses limites particulières, pour classer et étiqueter les produits chimiques en fonction de leur caractère dangereux pour l'œil. Alors qu'elle n'est pas jugée valable pour remplacer purement et simplement la méthode d'essai in vivo sur les yeux de lapin, la méthode OPCB est recommandée comme première étape d'une stratégie d'essai telle que l'approche «top-down» suggérée par Scott et al. (5) pour identifier les produits chimiques provoquant des lésions oculaires graves, à savoir les produits relevant de la catégorie 1 du SGH de l'ONU, sans expérience supplémentaire (4). La méthode OPCB est aussi recommandée pour identifier les produits chimiques ne nécessitant aucune classification pour irritation oculaire ou lésion oculaire grave, ainsi que le définit le SGH de l'ONU («sans catégorie») (4) dans le cadre d'une stratégie d'essai telle que l'approche «bottom-up» (5). Cependant, un produit chimique qui, avec la méthode OPCB, semble ne causer aucune lésion oculaire grave ou ne relever d'aucune classification pour irritation oculaire/lésion oculaire grave nécessiterait d'être soumis à des essais complémentaires (in vitro et/ou in vivo) pour établir une classification définitive. L'objet de la présente méthode d'essai est de décrire les procédures utilisées pour évaluer le danger potentiel d'un produit chimique testé pour l'œil, mesuré par la propension du produit chimique à provoquer une opacité et une perméabilité accrue sur une cornée bovine isolée. Les effets toxiques pour la cornée sont mesurés par: (i) la diminution de la capacité de transmission de la lumière (opacité); et (ii) l'augmentation du passage de la fluorescéine sodique (perméabilité). Les mesures de l'opacité et de la perméabilité de la cornée suite à son exposition au produit chimique testé sont ensuite combinées pour établir le score d'irritation in vitro (SIIV), utilisé pour classer le produit chimique testé en fonction de son pouvoir irritant. Les définitions des termes utilisés sont données à l'appendice 1. REMARQUES PRÉLIMINAIRES ET LIMITES La présente méthode d'essai est fondée sur le protocole expérimental de la méthode OPCB de l'ICCVAM (6)(7), élaboré à l'origine à partir des informations provenant du protocole de l'Institute for in vitro Sciences (IIVS) et sur le protocole INVITTOX no 124 (8). Ce dernier protocole a été utilisé lors de l'étude de pré-validation conduite en 1997-1998 sous l'égide de la Communauté européenne. Ces protocoles étaient tous deux fondés sur la méthode d'essai OPCB décrite pour la première fois par Gautheron et al. (9). La méthode OPCB peut être utilisée pour identifier les produits chimiques provoquant des lésions oculaires graves, selon la définition du SGH de l'ONU, à savoir les produits chimiques relevant de la catégorie 1 du SGH de l'ONU (4). Appliquée dans ce cadre, la méthode d'essai OPCB présente une précision globale de 79 % (150/191), un taux de faux positifs de 25 % (32/126) et un taux de faux négatifs de 14 % (9/65), en comparaison avec les données de la méthode d'essai sur œil de lapin in vivo, classés selon le système de classification du SGH de l'ONU (3) (voir appendice 2, tableau 1). Si l'on exclut de la base de données les produits chimiques testés relevant de certaines classes chimiques (alcools, cétones) ou physiques (matières solides), la précision de la méthode OPCB au regard du système de classification du SGH de l'ONU est de 85 % (111/131), le taux de faux positifs de 20 % (16/81), et le taux de faux négatifs de 8 % (4/50). Les limites potentielles de la méthode OPCB pour identifier les produits chimiques provoquant des lésions oculaires graves (catégorie 1 du SGH de l'ONU) concernent le taux élevé de faux positifs pour les alcools et les cétones, et le taux élevé de faux négatifs pour les substances solides, relevés dans la base de données de validation (1)(2)(3). Toutefois, tous les alcools et cétones n'étant pas surévalués par la méthode OPCB et certains étant correctement identifiés comme relevant de la catégorie 1 du SGH de l'ONU, on ne considère pas que ces deux groupes fonctionnels organiques sortent du champ d'application de la méthode d'essai. Il appartient à l'utilisateur appliquant cette méthode d'essai de décider si une surévaluation possible d'un alcool ou cétone est acceptable ou s'il est nécessaire de réaliser des tests complémentaires fondés sur une analyse du poids de la preuve. Concernant les taux de faux négatifs pour les solides, il convient de relever que les solides peuvent entraîner des conditions d'exposition variables et extrêmes lors du test d'irritation oculaire in vivo de Draize, ce qui peut fausser la prédiction de leur véritable potentiel d'irritation (10). Il convient également de noter qu'aucun des faux négatifs identifiés dans la base de données de validation de l'ICCVAM (2)(3), lors de l'identification de produits chimiques provoquant des lésions oculaires graves (catégorie 1 du SGH de l'ONU), n'a produit un SIIV ≤ 3, soit le critère utilisé pour identifier un produit chimique testé ne relevant d'aucun catégorie du SGH de l'ONU. De plus, les faux négatifs obtenus avec la méthode OPCB dans ce contexte ne sont pas préoccupants car tous les produits chimiques testés qui produisent un SIIV > 3 et ≤ 55 feraient ensuite l'objet d'autres essais in vitro dûment validés, ou en dernier recours d'essais chez le lapin, en fonction des exigences de la réglementation, selon une démarche expérimentale séquentielle fondée sur l'analyse du poids de la preuve. Certains produits chimiques solides étant correctement identifiés avec la méthode OPCB comme relevant de la catégorie 1 du SGH de l'ONU, on ne considère pas non plus que cet état physique sorte du champ d'application de la méthode d'essai. Les chercheurs peuvent envisager d'utiliser cette méthode d'essai pour tous les types de produits chimiques, à condition d'admettre qu'un SIIV > 55 indique un effet provoquant des lésions oculaires graves et de classer le produit chimique testé dans la catégorie 1 du SGH de l'ONU sans mener d'essai complémentaire. Comme mentionné précédemment, il convient toutefois d'interpréter avec prudence les résultats positifs obtenus avec des alcools ou des cétones en raison du risque de surestimation. La méthode OPCB peut aussi être utilisée pour identifier les produits chimiques qui ne nécessitent aucune classification pour irritation oculaire ou lésion oculaire grave suivant le système de classification du SGH de l'ONU (4). Appliquée dans ce cadre, la méthode d'essai OPCB présente une précision d'ensemble de 69 % (135/196), un taux de faux positifs de 69 % (61/89) et un taux de faux négatifs de 0 % (0/107), en comparaison avec les données de la méthode d'essai in vivo sur les yeux de lapin classées selon le système de classification du SGH de l'ONU (3) (voir appendice II, tableau 2). Le taux de faux positifs obtenu (produits chimiques ne relevant d'aucune catégorie du SGH de l'ONU in vivo produisant un SIIV > 3, voir paragraphe 47) est considérablement élevé, mais pas préoccupant dans ce contexte car tous les produits chimiques testés qui produisent un SIIV > 3 et ≤ 55 feraient ensuite l'objet d'autres essais in vitro dûment validés, ou en dernier recours d'essais chez le lapin, en fonction des exigences de la réglementation, selon une démarche expérimentale séquentielle fondée sur l'analyse du poids de la preuve. La méthode OPCB ne montre aucune limite spécifique pour tester les alcools, les cétones et les solides quand l'objectif est d'identifier les produits chimiques qui ne nécessitent aucune classification pour irritation oculaire ou lésion oculaire grave («sans catégorie» du SGH de l'ONU) (3). Les chercheurs peuvent envisager d'utiliser cette méthode d'essai pour tous les types de produits chimiques, à condition d'admettre qu'un résultat négatif (SIIV ≤ 3) indique qu'aucune classification n'est requise («sans catégorie» du SGH de l'ONU). La méthode OPCB ne parvenant à identifier correctement que 31 % des produits chimiques ne nécessitant aucune classification pour irritation oculaire ou lésion oculaire grave, cette méthode d'essai ne doit pas être privilégiée pour lancer une approche «bottom-up» (5), si d'autres méthodes in vitro validées et acceptées avec une sensibilité élevée similaire mais une spécificité supérieure sont possibles. La base de données de validation OPCB contient au total 113 substances et 100 mélanges (2)(3). La méthode OPCB semble donc pouvoir s'appliquer aux essais des substances et des mélanges. La méthode OPCB n'est pas recommandée pour identifier les produits chimiques testés qui devraient être classés comme irritants pour l'œil (catégorie 2 ou catégorie 2A du SGH de l'ONU) ou les produits chimiques testés qui devraient être classés comme légèrement irritants pour l'œil (catégorie 2B du SGH de l'ONU) en raison du nombre considérable de produits chimiques de la catégorie 1 du SGH de l'ONU sous-classés dans les catégories 2, 2A ou 2B, du SGH de l'ONU et de produits chimiques ne relevant d'aucune catégorie du SGH de l'ONU surclassés dans les catégories 2, 2A ou 2B du SGH de l'ONU (2)(3). Aussi pourrait-il être nécessaire de mener des essais complémentaires avec une autre méthode adaptée. Tous les modes opératoires sur l'œil de bovin doivent respecter les réglementations applicables aux installations d'essai et les protocoles de manipulation de matériaux issus de l'homme et de l'animal, comprenant notamment, sans s'y limiter, les tissus et les fluides tissulaires. L'usage des précautions universelles de laboratoire est recommandé (11). Bien que la méthode OPCB ne prenne pas en considération les lésions conjonctivales et iridiennes, elle prend en compte les lésions cornéennes, qui sont le facteur principal dans la classification selon les effets in vivo, dans le cadre d'application du SGH de l'ONU. La réversibilité des lésions cornéennes ne peut pas, par définition, être évaluée avec la méthode OPCB. Il a été proposé, sur la base d'études menées sur l'œil de lapin, qu'une évaluation de la profondeur initiale de la lésion cornéenne puisse être utilisée pour distinguer les effets réversibles des effets irréversibles (12). Cependant, des connaissances scientifiques approfondies seront nécessaires pour comprendre comment les effets irréversibles qui ne sont pas liés à une lésion initiale importante peuvent survenir. Enfin, la méthode OPCB ne permet pas d'évaluer la toxicité systémique potentielle associée à l'exposition oculaire. La présente méthode d'essai sera mise à jour régulièrement, à mesure que de nouvelles informations et données seront examinées. Ainsi, l'histopathologie peut s'avérer utile lorsqu'une caractérisation plus complète de la lésion cornéenne est nécessaire. Comme indiqué dans le Document d'orientation no. 160 (13), les utilisateurs sont encouragés à conserver les cornées et à préparer des spécimens histopathologiques en vue d'établir une base de données et de définir des critères de décision susceptibles d'améliorer la précision de cette méthode d'essai. Pour tout laboratoire mettant en œuvre la méthode d'essai pour la première fois, la liste des produits chimiques d'épreuve de compétence est fournie à l'appendice 3. Un laboratoire peut utiliser ces produits chimiques afin de démontrer sa compétence technique dans la conduite de l'essai OPCB avant de soumettre des données résultant de l'application de la méthode OPCB à des fins réglementaires de classification des dangers. PRINCIPE DE L'ESSAI La méthode d'essai OPCB est un modèle organotypique qui permet de maintenir in vitro les fonctions physiologiques et biochimiques normales de la cornée bovine pendant une courte période. Selon cette méthode, les dommages causés par le produit chimique testé sont évalués par des mesures quantitatives des modifications de l'opacité et de la perméabilité de la cornée réalisées, respectivement, à l'aide d'un opacitomètre et d'un spectrophotomètre visible. Ces deux mesures entrent dans le calcul de l'IVIS, valeur utilisée pour assigner une catégorie de classification de danger d'irritation in vitro permettant d'estimer le potentiel d'irritation oculaire in vivo d'un produit chimique testé (voir «Critères de décision» au paragraphe 48). La méthode d'essai OPCB utilise des cornées isolées provenant d'yeux de bovins récemment abattus. L'opacité cornéenne est mesurée par la quantité de lumière traversant la cornée. La perméabilité est mesurée par la quantité de fluorescéine sodique traversant toute l'épaisseur de la cornée, et détectée dans le milieu de la chambre postérieure. Les produits chimiques testés sont appliqués sur la surface épithéliale de la cornée en étant ajoutés dans la chambre antérieure du porte-cornée. L'appendice 4 présente une description et un schéma de porte-cornée utilisé dans la méthode OPCB. Ces appareils sont disponibles dans le commerce auprès de différents fabricants, ou peuvent être construits. Provenance et âge des yeux de bovins et sélection des espèces animales Les bovins envoyés à l'abattoir sont généralement destinés à la consommation humaine ou à d'autres usages commerciaux. Seules les cornées de bêtes en bonne santé, considérées comme propres à intégrer la chaîne alimentaire humaine, sont prélevées pour la méthode OPCB. Le poids des bovins variant considérablement en fonction de la race, de l'âge et du sexe, il n'existe aucune recommandation spécifique en la matière au moment de l'abattage. L'utilisation d'yeux provenant d'animaux d'âges différents peut entraîner des variations de dimension des cornées. Les cornées dont le diamètre horizontal est supérieur à 30,5 mm et l'épaisseur cornéenne centrale (ECC) supérieure ou égale à 1 100 μm ont généralement été prélevées sur des bovins de plus de huit ans, tandis que les cornées ayant un diamètre horizontal inférieur à 28,5 mm et une ECC inférieure à 900 μm proviennent plutôt de bêtes de moins de cinq ans (14). En conséquence, les yeux des bovins âgés de plus de 60 mois ne sont généralement pas utilisés. En général, les yeux des bovins de moins de 12 mois ne sont pas non plus utilisés, car leur développement n'est pas terminé et l'épaisseur ainsi que le diamètre cornéen sont nettement inférieurs à ceux des yeux provenant de bovins adultes. Toutefois, les cornées issues d'animaux jeunes (entre 6 et 12 mois) sont acceptables car elles présentent certains avantages, tels qu'une plus grande disponibilité, une fourchette d'âge plus restreinte, et des risques moins importants d'exposition potentielle à l'encéphalopathie spongiforme bovine (15) pour le personnel. Une évaluation plus poussée des effets de la taille ou de l'épaisseur cornéenne sur la sensibilité aux produits chimiques corrosifs et irritants serait d'une grande utilité, aussi les utilisateurs sont-ils encouragés à consigner l'âge et/ou le poids estimés des animaux ayant fourni les cornées utilisées dans l'étude. Collecte et transport des yeux jusqu'au laboratoire Les yeux sont prélevés par les employés des abattoirs. Afin de limiter le plus possible tout endommagement, mécanique ou autre, des yeux, ceux-ci doivent être énucléés dès que possible après la mort de l'animal et immédiatement refroidies après énucléation et pendant le transport. Pour éviter d'exposer les yeux à des produits chimiques potentiellement irritants, les employés de l'abattoir ne doivent pas utiliser de détergent lors du rinçage de la tête de l'animal. Les yeux doivent être totalement immergés dans une solution froide saline équilibrée de Hank (HBSS), dans un récipient de taille adaptée, et transportés jusqu'au laboratoire en ayant soin de limiter le plus possible tout endommagement et/ou contamination bactérienne. Les yeux étant prélevés au cours de l'abattage, ils risquent d'entrer en contact avec du sang ou d'autres matières biologiques, notamment des bactéries ou autres microorganismes. Il importe donc de réduire au minimum le risque de contamination (par exemple en plaçant le récipient contenant les yeux sur de la glace mouillée, pendant la collecte et le transport, ou en ajoutant des antibiotiques à la HBSS utilisée pour le transport [100 UI/ml de pénicilline et 100 μg/ml de streptomycine, par exemple]). Il faut limiter au maximum l'intervalle de temps entre le prélèvement des yeux et l'utilisation des cornées dans le cadre de l'essai OPCB (l'utilisation doit avoir lieu le jour même du prélèvement) et démontrer qu'il ne compromet pas les résultats de l'essai. Ces résultats se fondent sur les critères de sélection des yeux, ainsi que sur les réponses des témoins positifs et négatifs. Tous les yeux utilisés au cours de l'essai doivent provenir d'un lot d'yeux collectés le même jour. Critères de sélection des yeux utilisés pour la méthode OPCB Une fois parvenus au laboratoire, les yeux sont examinés avec soin pour déceler d'éventuels défauts, tels qu'une opacité avancée, des éraflures ou une néovascularisation. Seules les cornées d'yeux exempts de tout défaut seront utilisées. La qualité de chaque cornée est également évaluée à des stades ultérieurs de l'essai. Les cornées présentant une opacité supérieure à sept unités d'opacité ou l'équivalent pour l'opacitomètre et le porte-cornée utilisés après une période initiale d'équilibration d'une heure doivent être écartées (NB: l'opacitomètre doit être étalonné en fonction des normes d'opacité utilisées pour définir les unités d'opacité, voir appendice 4). Chaque groupe de traitement (produit chimique testé et témoins négatifs et positifs) est composé de trois yeux au minimum. L'essai OPCB nécessite trois cornées servant de témoins négatifs. Toutes les cornées étant prélevées sur le globe oculaire entier puis placées dans les chambres du porte-cornée, les manipulations peuvent entraîner d'éventuels artefacts sur les valeurs de l'opacité et de la perméabilité cornéennes de chaque élément (y compris le témoin négatif). Par ailleurs, les valeurs d'opacité et de perméabilité mesurées sur les cornées témoins négatives servent à corriger les valeurs d'opacité et de perméabilité cornéennes des spécimens et des témoins traités positifs lors du calcul du score d'irritation in vitro. MODE OPERATOIRE Préparation des yeux Des cornées exemptes de tout défaut sont découpées en conservant un rebord de 2 à 3 mm de sclère destiné à faciliter les manipulations ultérieures, et en prenant soin de ne pas endommager l'épithélium et l'endothélium cornéens. Les cornées isolées sont placées dans des porte-cornées spécialement conçus à cet effet, qui se composent de deux compartiments, antérieur et postérieur, respectivement en contact avec les faces épithéliale et endothéliale de la cornée. Il convient de remplir jusqu'à débordement ces deux chambres d'un milieu essentiel minimum de Eagle (MEME) préalablement chauffé et ne contenant pas de phénol rouge (en commençant par la chambre postérieure), en évitant toute formation de bulles d'air. L'appareil est ensuite équilibré à une température de 32 ± 1°C pendant une heure au moins, afin que les cornées s'équilibrent avec le milieu et atteignent une activité métabolique aussi proche de la normale que possible (la température de la surface cornéenne in vivo est d'environ 32°C). Après la période d'équilibration, un nouveau MEME préalablement chauffé et ne contenant pas de phénol rouge est ajouté dans les deux chambres, et une première mesure d'opacité est effectuée pour chaque cornée. Les cornées présentant une altération macroscopique des tissus (éraflures, pigmentation ou néovascularisation, par exemple) ou une opacité supérieure à 7 unités d'opacité, ou l'équivalent pour l'opacitomètre et le porte-cornée utilisé, sont écartées. Au moins trois cornées sont sélectionnées pour servir de témoins négatifs (ou témoins de solvant). Les cornées restantes sont ensuite réparties entre le groupe de traitement et celui des témoins positifs. La capacité calorifique de l'eau étant supérieure à celle de l'air, l'eau procure des conditions de température plus stables pour l'incubation. Aussi est-il recommandé de plonger le porte-cornée dans un bain d'eau pour maintenir son contenu à 32 ± 1°C. Toutefois, il est également possible d'utiliser des incubateurs à air, à condition d'assurer la stabilité de la température (en chauffant au préalable les porte-cornées et les milieux, par exemple). Application du produit chimique testé Deux protocoles d'application différents sont utilisés, l'un pour les liquides et les tensioactifs (solides ou liquides), et l'autre pour les solides non tensioactifs. Les liquides sont testés non dilués. Les semi-solides, les crèmes et les cires sont typiquement testés comme les liquides. Les tensioactifs sont testés à une concentration de 10 % en masse volumique dans une solution de chlorure de sodium 0.9 %, d'eau distillée ou d'un autre solvant dont l'absence d'effets indésirables sur le système d'essai a été démontrée. Il conviendra de justifier de façon idoine l'utilisation de tout autre taux de concentration. Les mélanges contenant des tensioactifs peuvent être testés sans être dilués ou dilués à une concentration adéquate en fonction de la situation d'exposition in vivo. Il conviendra de justifier de façon idoine la concentration utilisée. Les cornées sont ensuite exposées aux liquides ou tensioactifs pendant 10 minutes. L'utilisation d'une autre durée d'exposition devra être scientifiquement justifiée. Il convient de se rapporter à l'appendice 1 pour la définition d'un tensioactif et d'un mélange contenant des tensioactifs. Les solides non tensioactifs sont généralement testés sous forme de solutions ou de suspensions à une concentration poids/volume de 20 % dans une solution de chlorure de sodium 0,9 %, d'eau distillée ou d'un autre solvant dont l'absence d'effets indésirables sur le système d'essai a été démontrée. Dans certaines circonstances et uniquement si cela est justifié scientifiquement, les solides peuvent aussi être testés purs par application directe sur la surface cornéenne, selon la méthode en chambre ouverte (voir paragraphe 32). Les cornées sont exposées aux solides pendant quatre heures, mais comme pour les liquides et les tensioactifs, des durées d'exposition différentes sont possibles moyennant une justification scientifique appropriée. Différentes méthodes d'application peuvent être utilisées, en fonction de la nature physique et des caractéristiques chimiques des produits chimiques testés (solides, liquides, liquides visqueux ou non visqueux, par exemple). Il est très important que le produit chimique testé recouvre convenablement la surface épithéliale puis soit correctement retirée durant les étapes de rinçage. Il conviendra généralement d'utiliser la méthode en chambre fermée pour les produits chimiques testés liquides non visqueux ou légèrement visqueux, tandis que la méthode en chambre ouverte sera privilégiée pour les produits chimiques testés semi-visqueux et visqueux et pour les solides purs. Dans la méthode en chambre fermée, une quantité de produit chimique testé suffisante pour recouvrir la face épithéliale de la cornée (750 μl) est introduite dans la chambre antérieure par les trous de dosage situés sur la face supérieure de celle-ci. Les trous sont ensuite occultés à l'aide de capuchons durant la durée d'exposition. Il importe de s'assurer que chaque cornée est exposée à un produit chimique testé pendant la durée qui convient. Dans la méthode en chambre ouverte, la goupille de fermeture et la fenêtre en verre de la chambre antérieure sont retirés avant l'application. Le produit chimique testé ou témoin (750 μl, ou une quantité suffisante pour recouvrir entièrement la cornée) est directement appliqué sur la surface épithéliale cornéenne à l'aide d'une micropipette. Si le produit chimique testé est difficile à pipeter, il est possible d'utiliser une pipette à déplacement positif qui permet un dosage plus précis. L'embout de la pipette à déplacement positif est inséré dans l'embout distributeur de la seringue de manière à ce que le produit chimique testé puisse être chargé dans la pipette sous pression. Il convient ensuite de pousser le piston de la seringue tout en tirant simultanément celui de la pipette. Si des bulles d'air se forment dans l'embout de la pipette, il faut alors retirer le produit chimique testé (en l'expulsant) et répéter le processus jusqu'à ce qu'aucune bulle d'air n'apparaisse au moment du remplissage de la pipette. Il est possible d'utiliser si nécessaire une seringue normale (sans aiguille), ce qui permet de mesurer un volume précis de produit chimique testé et de l'appliquer plus facilement sur la surface épithéliale de la cornée. Après le dosage, la fenêtre en verre est replacée sur l'ouverture de la chambre antérieure afin de recréer un système fermé. Incubation post-exposition Après la période d'exposition, le produit chimique testé, ou le produit chimique témoin négatif ou positif, est retiré de la chambre antérieure et l'épithélium est rincé au moins trois fois (ou jusqu'à ce qu'aucune trace du produit chimique testé ne soit plus visible) au moyen d'un MEME contenant du rouge de phénol. Le rinçage est effectué à l'aide d'un milieu contenant du rouge de phénol, dont le changement de couleur permet de déterminer l'efficacité du rinçage des produits chimiques d'essai acides ou alcalins. Il convient de laver à nouveau les cornées si, après trois rinçages, le rouge de phénol est encore altéré (jaune ou violet) ou si le produit chimique testé est toujours visible. Une fois le milieu débarrassé du produit chimique testé, les cornées subissent un dernier rinçage avec un MEME sans rouge de phénol, ce qui permet de s'assurer que la chambre antérieure est débarrassée de tout rouge de phénol avant la mesure d'opacité. La chambre antérieure est ensuite à nouveau remplie d'un MEME neuf sans rouge de phénol. Pour les liquides et les tensioactifs, les cornées sont incubées après rinçage pendant deux heures supplémentaires à 32 ± 1°C. Il peut être utile, dans certaines circonstances (déterminées au cas par cas), de prévoir une durée d'incubation post-exposition plus longue. Les cornées sur lesquelles des solides ont été appliqués sont soigneusement rincées après une période d'exposition de quatre heures, et ne requièrent pas d'incubation supplémentaire. Au terme de la période d'incubation post-exposition pour les liquides et les tensioactifs, et au terme de la période d'exposition de quatre heures pour les solides non tensioactifs, le degré d'opacité et de perméabilité de chaque cornée est enregistré. En outre, chaque cornée est observée visuellement et des commentaires pertinents sont consignés (exfoliation des tissus, résidus de produit chimique testé ou opacité non uniforme, par exemple). Ces observations peuvent s'avérer importantes car elles peuvent être reflétées par des variations de lecture de l'opacitomètre. Produits chimiques témoins Des témoins négatifs ou des témoins de solvant/véhicule ainsi que des témoins positifs doivent être inclus en parallèle dans chaque expérience. Lorsqu'une substance liquide non diluée est soumise à l'essai, il convient d'inclure dans la méthode d'essai OPCB une substance témoin négatif concurrente (solution de chlorure de sodium 0.9 % ou eau distillée, par exemple) afin de détecter des changements non intrinsèques au système d'essai et de fournir un point de référence pour les critères d'évaluation de l'essai. Cette méthode permet également de garantir que les conditions de l'essai n'entraînent aucune réaction d'irritation indésirable. Lorsqu'un liquide dilué, un tensioactif ou un solide est soumis à l'essai, il convient d'inclure dans la méthode d'essai OPCB un groupe témoin de solvant/véhicule concurrent afin de détecter des changements non intrinsèques au système d'essai et de fournir un point de référence pour les critères d'efficacité de l'essai. On utilisera uniquement des solvants/véhicules dont l'absence d'effets indésirables sur le système d'essai a été démontrée. Un produit chimique reconnu pour générer une réponse positive est inclus dans chaque expérience afin de vérifier l'intégrité du dispositif d'essai et de son bon fonctionnement. Cependant, afin de pouvoir évaluer la variabilité des réponses du témoin positif dans le temps, l'ampleur des effets irritants ne devra pas être excessive. Pour les produits chimiques liquides, les témoins positifs peuvent être composés de 100 % d'éthanol ou de 100 % de diméthylformamide. Pour les produits chimiques solides, les témoins positifs peuvent être composés d'imidazole à 20 % (en masse volumique) dans une solution de chlorure de sodium 0,9 %, par exemple. Des produits chimiques étalons peuvent s'avérer utiles pour évaluer le potentiel d'irritation oculaire de produits chimiques inconnus relevant de classes spécifiques de substances chimiques ou de produits, ou encore pour évaluer le potentiel d'irritation relatif d'un irritant oculaire dans une gamme spécifique de réactions d'irritation. Effets mesurés L'opacité est déterminée par la quantité de lumière traversant la cornée. L'opacité de la cornée est mesurée de manière quantitative à l'aide d'un opacitomètre, qui donne des valeurs d'opacité situées sur une échelle continue. La perméabilité est déterminée par la quantité de fluorescéine sodique pénétrant la totalité des couches cellulaires de la cornée (de l'épithélium sur la surface externe de la cornée à l'endothélium sur sa surface interne). Un volume de 1 ml de solution de fluorescéine sodique (respectivement 4 ou 5 mg/ml lors d'essais portant sur des liquides et des tensioactifs ou sur des solides non tensioactifs) est introduit dans la chambre antérieure du porte-cornée, en contact avec la face épithéliale de la cornée, tandis que la chambre postérieure, en contact avec la face endothéliale, est remplie d'un nouveau MEME. Le porte-cornée est ensuite incubé en position horizontale pendant 90 ± 5 min à une température de 32 ± 1°C. La quantité de fluorescéine sodique passant dans la chambre postérieure est mesurée quantitativement par spectrophotométrie UV-visible. Les mesures spectrophotométriques évaluées à 490 nm sont enregistrées en tant que valeurs de densité optique (DO490) ou d'absorbance, mesurées sur une échelle continue. La perméabilité de la fluorescéine est déterminée à l'aide des valeurs de DO490 données par un spectrophotomètre visible utilisant un trajet optique standard de 1 cm. Il est également possible d'utiliser un lecteur de plaques de microtitration 96 puits, à condition: (i) de pouvoir définir l'intervalle linéaire du lecteur de plaque servant à déterminer les valeurs de la DO490 par la fluorescéine; et (ii) d'utiliser le bon volume d'échantillons de fluorescéine sur la plaque 96 puits afin d'obtenir des valeurs de la DO490 équivalentes au trajet optique standard de 1 cm (ce qui peut nécessiter de remplir entièrement les puits [généralement 360 μl]). RÉSULTATS ET RAPPORT Évaluation des données Une fois que les valeurs d'opacité et de perméabilité moyenne (DO490) ont été corrigées de l'opacité de fond et des valeurs DO490 de perméabilité des témoins négatifs, l'opacité moyenne et les valeurs DO490 de perméabilité pour chaque groupe de traitement doivent être combinées dans une formule dérivée empiriquement pour calculer comme suit le score in vitro (IVIS) de chaque groupe de traitement: IVIS = valeur moyenne d'opacité + (15 × valeur DO490 moyenne de perméabilité) D'après Sina et al. (16), cette formule a été dérivée au cours d'études internes et inter-laboratoires. Les données générées pour une série de 36 composés dans le cadre d'une étude inter-laboratoires ont été soumises à une analyse à variables multiples afin de déterminer l'équation du meilleur ajustement entre les données in vivo et in vitro. Des scientifiques travaillant dans deux entreprises distinctes ont effectué cette analyse et ont dérivé des équations pratiquement identiques. Les valeurs d'opacité et de perméabilité doivent également être évaluées de manière indépendante afin de déterminer si un produit chimique testé a provoqué une corrosion ou une forte irritation pour un seul des deux paramètres (voir «Critères de décision»). Critères de décision Les valeurs critiques déterminante pour l'identification des produits chimiques induisant des lésions oculaires graves pour l'œil (catégorie 1 de SGH de l'O.N.U.) par rapport aux produits chimiques ne nécessitant pas de classifications pour l'irritation oculaire ou pour les dommages lésions oculaires graves (ne relevant d'aucune catégorie selon le SGH de l'O.N.U.) sont fournies ci-dessous:
Critères d'acceptation de l'étude Un essai est considéré comme acceptable si le témoin positif présente un IVIS compris dans un intervalle de deux écarts-types autour de la moyenne historique actuelle, qui doit être mise à jour au moins tous les trois mois, ou à chaque fois qu'un essai acceptable est mené dans des laboratoires conduisant peu fréquemment (c'est-à-dire moins d'une fois par mois) ce type d'étude. Les réponses du témoin négatif ou du témoin de solvant/véhicule doivent donner des valeurs d'opacité et de perméabilité situées en-deçà des limites maximales établies pour les valeurs d'opacité et de perméabilité de fond des cornées bovines traitées avec les témoins négatifs ou les témoins de solvant/véhicule correspondants. Une seule expérience composée d'au moins trois cornées doit être suffisante pour tester un produit chimique quand la classification résultante est sans équivoque. Cependant, dans le cas de résultats équivoques dans la première expérience, une seconde expérience devrait être considérer (mais pas nécessairement requise), ainsi qu'une troisième tentative dans le cas de résultats IVIS moyens discordants entre les deux premières expériences. Dans ce contexte, un résultat en première tentative est considéré comme équivoque si les prédictions résultant des trois cornées ne sont pas concordantes, comme suit:
Rapport d'essai Le rapport d'essai doit contenir les informations suivantes, dès lors qu'elles s'appliquent à l'étude:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Approche «bottom-up» : approche par étape utilisée pour un produit chimique pour lequel on anticipe qu'il ne relève d'aucune classification pour l'irritation oculaire ou les lésions oculaires graves. Cette approche commence par la détermination des produits chimiques ne relevant d'aucune classification (résultat négatif) par rapport aux autres produits chimiques (résultats positifs). Approche «top-down» : approche par étape utilisée dans le cas d'un produit chimique pour lequel on estime qu'il provoque des lésions oculaires graves; cette approche commence par la détermination des produits chimiques provoquant des lésions oculaires graves (résultat positif) par rapport aux autres produits chimiques (résultat négatif). Catégorie 1 du SGH : Voir «Lésions oculairse graves» Catégorie 2 du SGH : voir «Irritation oculaire» Cornée : Partie transparente située à l'avant du globe oculaire, recouvrant l'iris et la pupille et laissant pénétrer la lumière vers l'intérieur de l'œil. Danger : Propriété inhérente d'un agent ou situation susceptible de provoquer des effets indésirables lorsqu'un organisme, un système ou une (sous-) population est exposé(e) à cet agent. Effets irréversibles sur l'œil : voir «Lésions oculaires graves». Effets réversibles sur l'œil : voir «Irritation oculaire». Essai à plusieurs niveaux : Démarche expérimentale séquentielle consistant à examiner toutes les informations existantes sur un produit chimique testé dans un ordre déterminé, en ayant recours à chaque étape à un processus d'analyse du poids de la preuve pour déterminer si les informations disponibles sont suffisantes pour décider d'une classification des dangers, avant de passer à l'étape suivante. Si le potentiel d'irritation d'un produit chimique testé peut être déterminé sur la base des informations existantes, aucun essai supplémentaire n'est nécessaire. Si le potentiel d'irritation d'un produit chimique testé ne peut pas être déterminé sur la base des informations existantes, une procédure expérimentale progressive de type séquentiel sur des animaux est alors lancée jusqu'à ce qu'une classification sans équivoque puisse être effectuée. Fiabilité : Indique dans quelle mesure la mise en œuvre d'une méthode d'essai peut être reproduite dans un même laboratoire ou par plusieurs laboratoires au cours du temps, en utilisant le même mode opératoire. Pour l'évaluer, on calcule la reproductibilité intra-laboratoire et inter-laboratoires et la répétabilité intra-laboratoire. Irritation oculaire : Production de changements dans l'œil suite à l'application d'un produit chimique sur la surface antérieure de l'œil et qui sont totalement réversibles dans les 21 jours suivant l'application (2). L'expression est équivalente à «Effets réversibles sur l'œil» et avec «Catégorie 2 du SGH» (4). Lésions oculaires graves : Lésions des tissus oculaires ou dégradation sévère de la vue, provoquées par l'application d'un produit chimique testé sur la surface antérieure de l'œil et qui ne sont pas totalement réversibles dans les 21 jours suivant l'application. L'expression est équivalente à «Effets irreversibles sur l'œil» et avec «Catégorie 1 du SGH» (4). Mélange : un mélange ou une solution composée d'au moins deux substances qui ne réagissent pas entre elles (4). Mélange contenant des tensioactifs : Dans le contexte de la présente méthode d'essai, il s'agit d'un mélange contentant un ou plusieurs tensioactifs(s) à une concentration finale supérieure à 5 %. Méthode d'essai validée : Méthode d'essai ayant fait l'objet d'études de validation visant à déterminer sa pertinence (notamment sa précision) et sa fiabilité à des fins spécifiques. Il importe de noter que les performances d'une méthode d'essai validée peuvent être insuffisantes en termes de précision et de fiabilité pour qu'elle soit jugée acceptable pour les besoins envisagés. Non classé : produit chimique ne relevant d'aucune classification pour l'irritation oculaire (catégorie 2, 2A ou 2B du SGH de l'ONU) ou pour les lésions occulaires graves (catégorie 1 du SGH de l'ONU). L'expression est équivalente à «Sans catégorie SGH». Opacité cornéenne : Mesure de l'étendue du caractère opaque de la cornée suite à son exposition à un produit chimique testé. Une augmentation de l'opacité cornéenne indique un endommagement de la cornée. L'opacité peut être évaluée de manière subjective, par exemple lors du test de Draize réalisé sur les yeux de lapin, ou de manière objective à l'aide d'un instrument de mesure tel qu'un «opacitomètre». Opacitomètre : Instrument servant à mesurer l'«opacité cornéenne» en évaluant la quantité de lumière transmise à travers la cornée. En général, cet instrument est composé de deux compartiments, chacun disposant de sa propre source lumineuse et d'une cellule photoélectrique. L'un des compartiments est utilisé pour la cornée traitée tandis que l'autre sert à étalonner l'instrument et à le mettre à zéro. La lumière provenant d'une lampe halogène est dirigée à travers un compartiment témoin (chambre vide, sans vitre ni liquide) vers une cellule photoélectrique, et comparée à la lumière envoyée à travers le compartiment expérimental, qui abrite la chambre contenant la cornée, jusqu'à une cellule photoélectrique. La lumière transmise par les deux cellules photoélectriques est ensuite comparée, et une valeur chiffrée d'opacité s'affiche sur un écran numérique. Perméabilité cornéenne : Mesure quantitative de l'endommagement de l'épithélium cornéen via la détermination de la quantité de fluorescéine sodique traversant toutes les couches cellulaires de la cornée. Poids de la preuve : Prise en compte des atouts et des faiblesses de divers éléments d'information en vue d'aboutir à une conclusion concernant le danger potentiel d'un produit chimique testé et d'étayer cette conclusion. Précision : Degré de conformité entre les résultats de la méthode d'essai et les valeurs de référence acceptées. Elle constitue une mesure de performance de la méthode d'essai et l'un des aspects de sa «pertinence». Ce terme est souvent utilisé indifféremment à la place du terme «concordance» pour qualifier la proportion de résultats corrects d'une méthode d'essai. Produit chimique : une substance ou un mélange. Produit chimique étalon : Produit chimique utilisé en tant que référence, en comparaison avec un produit chimique d'essai. Un produit chimique étalon doit présenter les propriétés suivantes: (i) provenir d'une ou de plusieurs sources constantes et fiables; (ii) présenter une similitude structurale et fonctionnelle avec la classe des produits chimiques soumis à l'essai; (iii) posséder des caractéristiques physiques/chimiques connues; (iv) être accompagnée de données confirmant ses effets connus; et (v) avoir une activité connue dans la fourchette des réponses désirées. Produit chimique testé : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Sans catégorie SGH : produit chimique qui ne relève d'aucune classification dans la catégorie 1 ou 2 (2A ou 2B) du SGH. Expression équivalente à «Non classé» Score d'irritation in vitro (IVIS) : Formule dérivée empiriquement et utilisée dans les essais OPCB, dans laquelle les valeurs d'opacité moyenne et de perméabilité moyenne pour chaque groupe de traitement sont combinées pour obtenir le score in vitro unique de chaque groupe de traitement. IVIS = valeur d'opacité moyenne + (15 x valeur moyenne de perméabilité). SGH (Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations unies) : Système proposant la classification des produits chimiques (substances et mélanges) conformément à des types et des niveaux normalisés de dangers physiques, sanitaires et environnementaux, ainsi que la communication des éléments d'information correspondants, notamment par des pictogrammes, mentions d'avertissement, mentions de danger, conseils de prudence et fiches de données de sécurité, afin de diffuser des informations sur leurs effets indésirables dans l'objectif de protéger les personnes (en particulier les employeurs, employés, transporteurs, consommateurs et personnels des services d'urgence) et l'environnement (4). Substance : Élément chimique et ses composés, présents à l'état naturel ou obtenus grâce à un procédé de production. Ce terme inclut tout additif nécessaire pour préserver la stabilité du produit ainsi que toute impureté produite par le procédé utilisé, mais exclut tout solvant pouvant être extrait sans affecter la stabilité ni modifier la composition de la substance (4). Tensioactif : Aussi appelé agent de surface; substance telle qu'un détergent, qui peut réduire la tension superficielle d'un liquide et donc lui permettre de mousser ou pénétrer des solides; aussi appelant agent mouillant. Taux de faux négatifs : Proportion de produits chimiques positifs faussement identifiés comme négatifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Taux de faux positifs : Proportion de produits chimiques négatifs faussement identifiés comme positifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Témoin de solvant/véhicule : Échantillon non traité contenant tous les composants d'un système d'essai, y compris le solvant ou véhicule utilisé pour tester les échantillons témoins traités ou non avec la substance d'essai afin de déterminer une réponse de référence pour les échantillons traités avec la substance d'essai dissoute dans le même solvant ou véhicule. Testé avec un témoin négatif concurrent, cet échantillon indique également si le solvant ou véhicule interagit avec le système d'essai. Témoin négatif : Réplique non traitée contenant tous les composants d'un système d'essai. Cet échantillon subit les mêmes procédures que les échantillons traités avec la substance d'essai et les autres échantillons témoins afin de déterminer si le solvant interagit avec le système d'essai. Témoin positif : Réplique contenant tous les composants d'un système d'essai, et traité avec une substance induisant notoirement une réponse positive. Afin de pouvoir évaluer la variabilité des réponses du témoin positif dans le temps, la sévérité de la réponse ne devra pas être excessive. Appendice 2 VALEUR PRÉDICTIVE DE LA MÉTHODE D'ESSAI OPCB Tableau 1 Valeur prédictive de la méthode OPCB pour identifier les produits chimiques provoquant des lésions oculaires graves [Cat. 1 ou Pas cat. 1 (Cat. 2 + «sans catégorie») du SGH de l'ONU / CLP de l'UE; Cat. I ou Pas cat. I (Cat. II + Cat. III + Cat. IV) de l'EPA des États-Unis]
Tableau 2 Valeur prédictive de la méthode OPCB pour identifier les produits chimiques ne relevant d'aucune classification pour irritation oculaire ou lésion oculaire grave («non-irritants») [«sans catégorie» ou Pas «sans catégorie» (Cat. 1 + Cat. 2) du SGH de l'ONU / CLP de l'UE; Cat. IV ou Pas cat. IV (Cat. I + Cat. II + Cat. III) de l'EPA des États-Unis]
Appendice 3 PRODUITS CHIMIQUES A UTILISER POUR L'ÉPREUVE DE COMPÉTENCE RELATIVE A LA MÉTHODE D'ESSAI OPCB Avant d'utiliser en routine la présente méthode d'essai, les laboratoires doivent démontrer leurs compétences techniques en établissant correctement la classification des dangers pour l'œil présentés par les 13 produits chimiques préconisés dans le tableau 1. Ces produits chimiques ont été sélectionnés de façon à représenter la gamme des réactions selon le danger qu'ils représentent pour l'œil, sur la base des résultats de l'essai in vivo sur œil de lapin (LD 405) (17) et du SGH des Nations Unies (c'est-à-dire les catégories 1, 2A, 2B ou Non classé) (4). Les autres critères de sélection sont la disponibilité des produits chimiques dans le commerce, l'existence de données de référence in vivo de bonne qualité, et l'existence de données in vitro de bonne qualité obtenues grâce à la méthode OPCB. Les données de référence figurent dans le Récapitulatif simplifié (3) et dans les Background Review Documents de l'ICCVAM pour la méthode d'essai d'opacité et de perméabilité de la cornée bovine (2)(18). Tableau 1 Produits chimiques recommandés pour démontrer les compétences techniques relatives à la méthode d'essai OPCB
Appendice 4 LE PORTE-CORNÉE POUR LA MÉTHODE OPCB Les porte-cornées pour OPCB sont fabriqués à partir d'un matériau inerte (polypropylène par exemple). Ils se composent de deux parties (une chambre antérieure et une chambre postérieure) et possèdent deux chambres internes cylindriques identiques. Chaque chambre, d'une contenance de 5 ml, est fermée par une vitre à travers laquelle les mesures d'opacité sont enregistrées. Chacune des chambres internes mesure 1,7 cm de diamètre et 2,2 cm de profondeur (11). Un joint torique situé au niveau de la chambre postérieure empêche toute fuite. La cornée est placée face endothéliale dessous, sur le joint torique de la chambre postérieure, et la chambre antérieure est placée du côté épithélial de la cornée. Les chambres sont maintenues en place par trois vis en acier inoxydable situées sur les bordures extérieures du porte-cornée. Chaque chambre comporte à son extrémité une vitre amovible permettant d'accéder facilement à la cornée. Un joint torique placé entre la vitre et la chambre empêche toute fuite. L'introduction et le vidage du milieu et des produits chimiques d'essai se font par deux trous situés sur la face supérieure de chaque chambre. Pendant le traitement et les périodes d'incubation, ils sont fermés par des capuchons en caoutchouc. La transmission de la lumière à travers les porte-cornées peut varier dans la mesure où les effets de l'usure ou l'accumulation de certains résidus chimiques sur les trous des chambres internes ou sur les vitres peut modifier la répartition ou la réflectance de la lumière. En conséquence, on peut observer une augmentation ou une diminution de la lumière de référence transmise (et une variation inversement proportionnelle des mesures d'opacité de référence) à travers les porte-cornées, ces changements pouvant être marqués dès les premières mesures d'opacité de la cornée dans les chambres individuelles (autrement dit, les valeurs initiales d'opacité de la cornée dans des porte-cornées individuels spécifiques peuvent facilement varier de plus de 2 ou 3 unités d'opacité par rapport aux valeurs de référence attendues). Chaque laboratoire doit envisager d'élaborer un programme pour évaluer les variations de transmission de la lumière à travers les porte-cornées, selon la nature des produits chimiques testés et la fréquence d'utilisation des chambres. Pour établir des valeurs de référence, on peut contrôler les porte-cornées avant l'utilisation en routine en mesurant les valeurs d'opacité (ou de transmission de la lumière) de référence des chambres remplies de tout le milieu, sans cornées. Les porte-cornées peuvent ensuite être contrôlés périodiquement pour vérifier si la transmission de la lumière varie durant les périodes d'utilisation. Chaque laboratoire peut établir la fréquence de contrôle des porte-cornées, selon les produits chimiques testés, la fréquence d'utilisation et les variations observées par rapport aux valeurs d'opacité cornéenne de référence. Si l'on observe des variations notables de la lumière transmise à travers les porte-cornées, il convient d'appliquer les procédures appropriées de nettoyage et/ou de polissage de la surface interne des porte-cornées voire de la remplacer. Porte-cornée: vue éclatée 
Appendice 5 L'OPACITOMÈTRE L'opacitomètre est un appareil permettant de mesurer la transmission de la lumière. Par exemple, dans le cas de l'équipement OP-KIT d'Electro Design (Riom, France) utilisé pour valider la méthode OPCB, la lumière d'une lampe halogène est dirigée à travers un compartiment témoin (chambre vide, sans vitre ni liquide) vers une cellule photoélectrique, et comparée à la lumière envoyée à travers le compartiment expérimental, qui abrite la chambre contenant la cornée, jusqu'à une cellule photoélectrique. La lumière transmise par les deux cellules photoélectriques est ensuite comparée, et une valeur chiffrée d'opacité s'affiche sur un écran numérique. Les unités d'opacité sont alors définies. D'autres types d'opacitomètres présentant une configuration différente (par exemple, ne nécessitant pas de mesurer en parallèle le compartiment témoin et le compartiment expérimental) peuvent être utilisés s'il est prouvé qu'ils donnent des résultats similaires à ceux de l'équipement validé. L'opacitomètre fournit une réponse linéaire par le biais d'un éventail de mesures d'opacité couvrant les seuils utilisés pour les différentes classifications décrites par le modèle de prévision (c'est-à-dire, jusqu'aux seuils déterminant le caractère corrosif/fortement irritant). Afin de garantir des mesures linéaires et précises jusqu'à 75-80 unités d'opacité, il est nécessaire d'étalonner l'opacitomètre à l'aide d'une série de calibreurs. Les calibreurs sont placés à l'intérieur de la chambre d'étalonnage (une chambre de porte-cornée destinée à contenir les calibreurs) et une mesure des calibreurs est réalisée sur l'opacitomètre. La chambre d'étalonnage est conçue pour maintenir les calibreurs approximativement à la même distance de la source lumineuse et de la cellule photoélectrique que celle où se trouveront les cornées lors des mesures d'opacité. Les valeurs de référence et le point déterminé initialement dépendent du type d'équipement utilisé. Il convient de s'assurer de la linéarité des mesures d'opacité en appliquant les procédures appropriées (en fonction de l'instrument). Par exemple, pour l'équipement OP-KIT d'Electro Design (Riom, France), l'opacitomètre est tout d'abord étalonné à 0 unité d'opacité en utilisant la chambre d'étalonnage sans calibreur. Trois calibreurs différents sont ensuite placés l'un après l'autre à l'intérieur de la chambre d'étalonnage, et l'opacité est mesurée. Les calibreurs 1, 2 et 3 présentent des mesures d'opacité égales à leur valeur de consigne, soit respectivement 75, 150 et 225 unités d'opacité, ± 5 %. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
(13) |
Dans la partie B, le chapitre B.48 est remplacé par le texte suivant: «B.48 Méthode d'essai sur œil de poulet isolé pour l'identification des produits chimiques i) provoquant des lésions oculaires graves et ii) ne relevant d'aucune classification pour irritation oculaire ou lésion oculaire grave INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 438 (2013) de l'OCDE pour les essais de produits chimiques. La méthode d'essai sur œil de poulet isolé (OPI) a été évaluée par le Comité de coordination inter-agences pour la validation des méthodes alternatives (ICCVAM) des États-Unis, en collaboration avec le Centre européen pour la validation de méthodes alternatives (ECVAM) et le Centre japonais pour la validation de méthodes alternatives (JaCVAM), en 2006 et 2010 (1)(2)(3). La première évaluation a permis de valider scientifiquement l'utilité de la méthode OPI pour le dépistage des produits chimiques (substances et mélanges) provoquant des lésions oculaires graves (catégorie 1) selon la définition du Système général harmonisé de classification et d'étiquetage des produits chimiques (SGH) des Nations Unies (ONU) (1)(2)(4) et du règlement (CE) no 1272/2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges (CLP) (12). La seconde évaluation a consisté à déterminer l'utilité de la méthode OPI pour le dépistage des produits chimiques non classés comme irritants pour l'œil ou provoquant de graves lésions oculaires selon la définition du SGH de l'ONU (3) (4). Les résultats de l'étude de validation et les recommandations du panel d'examen ont confirmé la recommandation initiale selon laquelle il convient d'utiliser la méthode OPI pour la classification des produits chimiques pouvant provoquer de lésions oculaires graves (catégorie 1 du SGH de l'ONU), car la base de données disponible était restée inchangée depuis la première validation de l'ICCVAM. À ce stade, il n'a pas été recommandé d'étendre le champ d'application de la méthode OPI pour inclure d'autres catégories. Le jeu de données in vitro et in vivo utilisé lors de l'étude de validation a été réévalué dans le but de déterminer l'utilité de la méthode OPI pour identifier les produits chimiques ne nécessitant aucune classification pour irritation oculaire ou lésion oculaire grave (5). D'après les conclusions de cette réévaluation, la méthode OPI peut aussi être utilisée pour identifier les produits chimiques qui ne relèvent d'aucune classification pour irritation oculaire ou lésion oculaire grave selon la définition du SGH de l'ONU (4) (5). La présente méthode d'essai indique les applications recommandées et les limites de la méthode OPI au regard de ces évaluations. Les principales différences entre la version originale de 2009 et la mise à jour de 2013 de la ligne directrice de l'OCDE pour les essais de produits chimiques concernent notamment: l'application de la méthode OPI pour identifier les produits chimiques ne relevant d'aucune classification au titre du SGH de l'ONU, une mise à jour concernant les éléments du rapport d'essai, une mise à jour de l'appendice 1 sur les définitions, et une mise à jour de l'appendice 2 sur les substances d'épreuve. Il est généralement admis que dans notre horizon de prévision, aucun essai unique d'irritation oculaire in vitro ne pourra remplacer le test oculaire in vivo de Draize pour établir le degré d'irritation provoqué par les différentes classes de produits chimiques. Cependant, la combinaison de plusieurs méthodes de substitution dans le cadre d'une stratégie d'essais (à plusieurs niveaux) pourrait remplacer le test oculaire de Draize (6). L'approche «top-down» (5) est indiquée lorsque, d'après les informations existantes, on s'attend à ce qu'un produit chimique soit fortement irritant, alors que l'approche «bottom-up» (7) est conçue pour être appliquée quand, au vu des informations existantes, un produit chimique devrait a priori ne causer aucune irritation oculaire suffisante nécessitant une classification. La méthode OPI est une méthode d'essai in vitro pouvant être utilisée, dans certaines circonstances et compte tenu de ses limites particulières décrites aux paragraphes 8 à 10, pour classer et étiqueter les produits chimiques en fonction de leur caractère dangereux pour l'œil. Alors qu'elle n'est pas jugée valable pour remplacer purement et simplement la méthode d'essai in vivo sur les yeux de lapin, la méthode OPI est recommandée comme première étape d'une stratégie d'essai telle que l'approche «top-down» suggérée par Scott et al. (7) pour identifier les produits chimiques provoquant des lésions oculaires graves, à savoir les produits relevant de la catégorie 1 du SGH de l'ONU, sans expérience supplémentaire (4). La méthode OPI est aussi recommandée pour identifier les produits chimiques ne nécessitant aucune classification pour irritation oculaire ou lésion oculaire grave, ainsi que le définit le SGH de l'ONU («sans catégorie») (4), et peut donc être utilisée comme première étape dans le cadre d'une stratégie d'essai telle que l'approche «bottom-up» (7). Cependant, un produit chimique qui, avec la méthode OPI, semble ne causer aucune lésion oculaire grave ou ne relever d'aucune classification pour irritation oculaire/lésion oculaire grave nécessiterait d'être soumis à des essais complémentaires (in vitro et/ou in vivo) pour établir une classification définitive. En outre, les autorités réglementaires compétentes devraient être consultées avant de mettre en œuvre l'essai OPI dans une approche «bottom-up» dans le cadre d'autres systèmes de classification que le SGH de l'ONU. L'objectif de la présente méthode d'essai est de décrire les procédures utilisées pour évaluer le danger potentiel d'un produit chimique pour l'œil, mesuré par la propension du produit chimique à induire ou non une toxicité dans un œil de poulet énucléé. Les effets toxiques pour la cornée sont mesurés par (i) une estimation qualitative de l'opacité, (ii) une estimation qualitative des dommages subis par l'épithélium, fondée sur l'application de fluorescéine sur l'œil (rétention de fluorescéine), (iii) une mesure quantitative de l'augmentation de l'épaisseur (gonflement), et (iv) une évaluation qualitative des dommages morphologiques macroscopiques infligés en surface. L'opacité cornéenne, le gonflement et l'estimation des dommages après exposition à un produit chimique d'essai sont évalués individuellement, puis combinés pour établir une classification d'effet irritant pour les yeux. Les définitions des termes utilisés sont données à l'appendice 1. REMARQUES PRÉLIMINAIRES ET LIMITES La présente méthode d'essai est fondée sur le protocole suggéré dans le document d'orientation 160 de l'OCDE (8), élaboré à la suite de l'étude de validation internationale de l'ICCVAM (1)(3)(9), enrichie de contributions apportées par le Centre européen pour la validation des méthodes d'essais alternatives (ECVAM), le Centre japonais de validation des méthodes d'essais alternatives (JaCVAM), et le Quality of Life Department of Toxicology and Applied Pharmacology du TNO (Pays-Bas). Le mode opératoire s'appuie sur des informations dérivées de protocoles publiés, et de celui actuellement employé par le TNO (10) (11) (12) (13) (14). Les essais réalisés dans le cadre de l'étude de validation sous-tendant cette méthode d'essai ont porté sur un large éventail de produits chimiques; la base de données empiriques de cette étude totalisait 152 produits chimiques dont 72 substances et 80 mélanges (5). La présente méthode d'essai peut être utilisée pour tester des solides, des liquides, des émulsions et des gels. Les liquides peuvent être aqueux ou non; les solides peuvent être solubles ou insolubles dans l'eau. Les gaz et les aérosols n'ont pas encore fait l'objet d'une étude de validation. La méthode OPI peut être utilisée pour identifier les produits chimiques provoquant des lésions oculaires graves, à savoir les produits chimiques relevant de la catégorie 1 du SGH de l'ONU (4). Appliquée dans ce but, cette méthode présente pour limites un taux élevé de faux positifs pour les alcools et un taux élevé de faux négatifs pour les matières solides et les tensioactifs (1)(3)(9). Toutefois, le taux de faux négatifs (produits relevant de la catégorie 1 du SGH de l'ONU mais identifiés comme n'en relevant pas) n'est pas préoccupant dans ce contexte car tous les produits chimiques d'essai qui donnent des résultats négatifs feraient ensuite l'objet d'un ou plusieurs autres essais in vitro dûment validés, ou en dernier recours d'essais chez le lapin, en fonction des exigences de la réglementation, selon une démarche expérimentale séquentielle fondée sur l'analyse du poids de la preuve. Il convient de relever que les solides peuvent entraîner des conditions d'exposition variables et extrêmes lors du test d'irritation de l'œil in vivo de Draize, ce qui peut fausser la prédiction de leur véritable potentiel d'irritation (15). Les investigateurs peuvent envisager d'utiliser cette méthode d'essai pour tous les types de produits chimiques, à condition d'admettre qu'un résultat positif indique un effet provoquant des lésions oculaires graves, autrement dit de classer le produit chimique d'essai dans la catégorie 1 du SGH de l'ONU sans mener d'essai complémentaire. Toutefois, il convient d'interpréter avec précaution les résultats positifs obtenus avec des alcools, compte tenu du risque de surestimation des résultats positifs. Utilisée pour identifier les produits chimiques provoquant des lésions oculaires graves (catégorie 1 du SGH de l'ONU), la méthode OPI présente une précision d'ensemble de 86 % (120/140), un taux de faux positifs de 6 % (7/113) et un taux de faux négatifs de 48 % (13/27), en comparaison avec les données de la méthode d'essai in vivo sur les yeux de lapin classées selon le système de classification du SGH de l'ONU (4)(5). La méthode OPI peut aussi être utilisée pour identifier les produits chimiques qui ne nécessitent aucune classification pour irritation oculaire ou lésion oculaire grave suivant le système de classification du SGH de l'ONU (4). Les autorités réglementaires compétentes devraient être consultées avant de mettre en œuvre l'essai OPI dans une approche «bottom-up» dans le cadre d'autres systèmes de classification que le SGH de l'ONU. Cette méthode d'essai peut être utilisée pour tous les types de produits chimiques, un résultat négatif pouvant être accepté pour ne pas de classer un produit chimique pour irritation oculaire et lésion oculaire grave. Cependant, au regard d'un résultat de la base de données de validation, il est possible que les peintures antisalissures contenant des solvants organiques soient sous-évaluées (5). Utilisée pour identifier les produits chimiques qui ne nécessitent pas de classification pour irritation oculaire ou lésion oculaire grave, la méthode OPI présente une précision d'ensemble de 82 % (125/152), un taux de faux positifs de 33 % (26/79) et un taux de faux négatifs de 1 % (1/73), en comparaison avec les données de la méthode d'essai in vivo sur œil de lapin classées selon le système de classification du SGH de l'ONU (4)(5). Lorsque les produits chimiques d'essai de certaines catégories chimiques (à savoir, les peintures antisalissures contenant des solvants organiques) sont exclus de la base de données, la précision de l'OPI est de 83 % (123/149), le taux de faux positifs de 33 % (26/78), et le taux de faux négatifs de 0 % (0/71) au regard du système de classification du SGH de l'ONU (4)(5). La méthode OPI n'est pas recommandée pour identifier les produits chimiques d'essai qui devraient être classés comme irritants pour l'œil (catégorie 2 ou catégorie 2A du SGH de l'ONU) ou les produits chimiques d'essai qui devraient être classés comme légèrement irritants pour l'œil (catégorie 2B du SGH de l'ONU) en raison du nombre considérable de produits chimiques de la catégorie 1 du SGH de l'ONU sous-classés dans les catégories 2, 2A ou 2B, du SGH de l'ONU et de produits chimiques ne relevant d'aucune catégorie du SGH de l'ONU surclassés dans les catégories 2, 2A ou 2B du SGH de l'ONU. Aussi pourrait-il être nécessaire de mener des essais complémentaires avec une autre méthode adaptée. Tous les modes opératoires sur l'œil de poulet doivent respecter les réglementations applicables aux installations d'essai et les protocoles de manipulation de matériaux issus de l'homme et de l'animal, comprenant notamment, sans s'y limiter, les tissus et les fluides tissulaires. L'usage des précautions universelles de laboratoire est recommandé (16). Si la méthode OPI ne prend pas en considération les lésions conjonctivales et de l'iris évaluées par la méthode d'essai d'irritation oculaire chez le lapin, elle porte sur les effets observés sur la cornée qui sont les principaux critères de classification in vivo au regard du SGH de l'ONU. En outre, et bien qu'il soit impossible d'étudier la réversibilité des lésions cornéennes per se par la méthode d'essai OPI, on a proposé d'utiliser une estimation de la profondeur initiale d'une telle lésion afin de distinguer certains types d'effets irréversibles, en s'appuyant sur les études sur l'œil de lapin (17). En particulier, il apparaît nécessaire d'approfondir les connaissances scientifiques disponibles pour comprendre comment apparaissent les effets irréversibles non liés à une grave lésion initiale. Enfin, la méthode d'essai OPI ne permet pas l'évaluation du potentiel de toxicité systémique associé à une exposition oculaire. La présente méthode d'essai sera périodiquement actualisée après examen de nouvelles informations et de nouvelles données. Par exemple, l'histopathologie peut s'avérer utile lorsqu'une caractérisation plus détaillée des lésions cornéennes est nécessaire. Pour évaluer cet éventuel développement, les utilisateurs sont invités à conserver les yeux et à préparer des échantillons pour l'histopathologie en vue d'établir une base de données et des critères de décision susceptibles d'améliorer la précision de cette méthode d'essai. L'OCDE a élaboré un document d'orientation sur l'utilisation des méthodes d'essai de toxicité oculaire in vitro, qui intègre des procédures détaillées sur la collecte des échantillons d'histopathologie et indique où soumettre les échantillons et/ou les résultats histopathologiques (8). Tout laboratoire conduisant cet essai pour la première fois utilise les produits chimiques d'épreuve de compétence mentionnés à l'appendice 2. Il peut utiliser ces produits chimiques pour démontrer sa compétence technique en matière d'application de la méthode d'essai OPI préalablement à la soumission des données obtenues avec cet essai à des fins de classification réglementaire des dangers. PRINCIPE DE L'ESSAI La méthode d'essai OPI est un modèle organotypique qui permet le maintien à court terme d'yeux de poulet in vitro. Selon cette méthode, les dommages provoqués par le produit chimique d'essai sont évalués par détermination du gonflement, de l'opacité et de la rétention de fluorescéine de la cornée. L'observation des deux derniers paramètres est qualitative, alors que l'analyse du gonflement de la cornée est quantitative. Chaque mesure est reportée sous forme d'un score quantitatif utilisé pour calculer un indice d'irritation général, ou affectée à une catégorie qualitative utilisée pour appliquer une classification des produits chimiques dangereux pour l'œil in vitro, à savoir soit la catégorie 1 du SGH de l'ONU soit la catégorie Non classé du SGH de l'ONU. L'un ou l'autre de ces résultats peut ensuite être utilisé pour prédire si un produit chimique d'essai peut causer des lésions oculaires graves in vivo ou si elle ne nécessite aucune classification relative aux dangers des produits chimiques pour l'œil (se référer aux critères de décision). Cependant, on ne peut classer aucun produit chimique qui, avec la méthode OPI, ne semble causer aucune lésion oculaire grave ou ne relever d'aucune classification (voir le paragraphe 11). Source et âge des yeux de poulet Habituellement, on utilise dans cet essai des yeux prélevés sur des poulets provenant de l'abattoir où ils sont tués pour la consommation humaine, et les animaux de laboratoire sont donc inutiles. On utilise uniquement des yeux d'animaux sains dont l'entrée dans la chaîne alimentaire humaine est autorisée. Aucune étude contrôlée visant à évaluer l'âge optimal des poulets n'a encore été menée, mais l'âge et le poids des poulets habituellement utilisés dans cette méthode d'essai sont ceux des petits poulets couramment traités dans un abattoir de volailles (à savoir, environ 7 semaines et 1,5 — 2,5 kg). Collecte et transport des yeux au laboratoire Les têtes sont coupées immédiatement après sédation des poulets, généralement par choc électrique, et incision des cous pour la saignée. Il conviendra d'identifier une source locale de poulets proche du laboratoire, afin que le transfert des têtes entre l'abattoir et le laboratoire soit suffisamment rapide pour limiter autant que possible la détérioration et/ou la contamination bactérienne. Il faut réduire au minimum l'intervalle qui sépare la collecte des têtes de poulets et le placement des yeux dans la chambre de surfusion suivant l'énucléation (généralement deux heures) pour assurer le respect des critères d'acceptation de l'essai. Tous les yeux utilisés dans l'essai proviennent d'un groupe d'yeux collectés le même jour. Les yeux étant disséqués au laboratoire, les têtes sont transportées intactes depuis l'abattoir, à température ambiante (généralement entre 18 °C et 25 °C), dans des boîtes en plastique humidifiées par des tissus mouillés d'une solution saline isotonique. Critères de sélection et nombre d'yeux utilisés dans la méthode OPI Les yeux dont la coloration initiale par la fluorescéine ou le score d'opacité de la cornée sont élevés après énucléation (> 0,5 dans les deux cas) sont rejetés. Chaque groupe de traitement et chaque groupe témoin positif parallèle comprend au moins trois yeux. Le groupe témoin négatif ou le témoin de solvant (si on utilise un solvant qui n'est pas une solution saline) comprend au moins un œil. Dans le cas des matières solides se révélant non classées selon le SGH de l'ONU, il est recommandé de procéder à un second essai sur trois yeux afin de confirmer ou d'infirmer le premier résultat négatif obtenu. MODE OPÉRATOIRE Préparation des yeux Les paupières sont soigneusement excisées en veillant à ne pas endommager la cornée. L'intégrité de celle-ci est évaluée au moyen d'une goutte de fluorescéine sodique à 2 % (p/p) appliquée sur la surface cornéenne pendant quelques secondes, puis rincée par une solution saline isotonique. Les yeux traités à la fluorescéine sont examinés au biomicroscope pour vérifier si la cornée est ou non endommagée (ce qui se traduit par une rétention de fluorescéine et une opacité cornéenne ≤ 0,5). L'œil non endommagé est ensuite énucléé, en veillant à ne pas endommager la cornée. On tire le globe oculaire hors de l'orbite en maintenant fermement la membrane nictitante à l'aide de pinces chirurgicales, et les muscles de l'œil sont coupés avec des ciseaux courbes à pointes émoussées. Il est important d'éviter de léser la cornée par une pression excessive (artéfacts de compression). Lorsque l'œil est retiré de l'orbite, une portion visible du nerf optique reste attachée. Une fois retiré de l'orbite, l'œil est placé sur un tampon absorbant et la membrane nictitante et le reste des tissus conjonctifs sont découpés. L'œil énucléé est monté dans un clamp en acier inoxydable, en plaçant la cornée verticalement. Le clamp est ensuite transféré dans une chambre de l'appareil de surfusion (18). Dans l'appareil de surfusion, les clamps sont placés de telle sorte que la totalité de la cornée soit alimentée par le goutte-à-goutte de solution saline isotonique (3-4 gouttes par minute ou 0,1 à 0,15 ml/min). La température dans les chambres de l'appareil de surfusion est régulée à 32 ± 1,5 °C. L'appendice 3 propose un schéma d'un appareil de surfusion normal et des clamps pour œil, disponibles dans le commerce ou à construire. L'appareil peut être modifié en fonction des besoins d'un laboratoire particulier (par exemple, pour contenir un nombre d'yeux différent). Une fois placés dans l'appareil de surfusion, les yeux sont à nouveau examinés au biomicroscope afin de s'assurer qu'ils n'ont pas été endommagés pendant le protocole de dissection. L'épaisseur de la cornée est mesurée à ce moment au sommet de la cornée au moyen du dispositif de mesure dont est muni le biomicroscope. Il faut remplacer les yeux (i) dont la rétention de fluorescéine est > 0,5, (ii) dont l'opacité cornéenne est > 0,5; ou (iii) qui présentent tout autre signe de lésion. Les yeux qui satisfont tous ces critères, mais dont l'épaisseur cornéenne s'écarte de plus de 10 % de la valeur moyenne calculée pour l'ensemble des yeux sont également rejetés. La mesure de l'épaisseur cornéenne peut varier en fonction du réglage de la largeur de la fente du biomicroscope, et les examinateurs tiennent compte de cette contrainte en ajustant cette largeur à 0,095 mm. Une fois tous les yeux examinés et admis dans l'étude, ils sont incubés pendant environ 45 à 60 minutes pour s'équilibrer dans le système d'essai avant l'administration du produit chimique d'essai. Cette période est suivie des mesures au temps de référence zéro de l'épaisseur et de l'opacité cornéenne, qui serviront de mesures initiales (temps 0). Le score de fluorescéine à la dissection est la mesure initiale pour cet effet. Application du produit chimique d'essai Immédiatement après les mesures de référence au temps 0, l'œil (dans son support) est retiré de l'appareil de surfusion, placé en position horizontale et le produit chimique d'essai est appliquée sur la cornée. Les produits chimiques liquides sont généralement testés sans dilution, mais ils peuvent être dilués si besoin est (par exemple, dans une étape du modèle de l'étude). Il est préférable d'utiliser une solution physiologique pour diluer les produits chimiques d'essai. Il est toutefois possible d'utiliser d'autres solvants en conditions contrôlées, sous réserve de démontrer le bien-fondé de leur utilisation au lieu de la solution physiologique saline. Les produits chimiques liquides sont appliqués sur la cornée de façon à couvrir uniformément la totalité de la surface; le volume habituel est de 0,03 ml. Dans la mesure du possible, les produits chimiques solides sont broyés pour obtenir la plus grande finesse au moyen d'un mortier et d'un pilon, ou d'un outil de broyage comparable. La poudre est appliquée sur la cornée de façon à couvrir uniformément la totalité de la surface; la quantité habituelle est de 0,03 mg. Le produit chimique d'essai (liquide ou solide) est appliqué pendant 10 secondes, puis éliminée par rinçage de l'œil avec une solution saline isotonique (environ 20 ml) à température ambiante. L'œil (dans son support) est ensuite remis dans l'appareil de surfusion dans la position verticale originelle. En cas de besoin, il est possible d'effectuer un rinçage supplémentaire après l'application de 10 secondes ainsi qu'aux points de mesure ultérieurs (par exemple, si l'on découvre des résidus du produit chimique d'essai sur la cornée). En général, la quantité de solution saline utilisée en plus pour le rinçage n'est pas importante, contrairement en revanche à l'adhérence observée du produit chimique d'essai sur la cornée. Produits chimiques témoins Il convient d'inclure des témoins négatifs ou de solvant/véhicule et des témoins positifs concomitants dans chaque expérience. Dans le cas d'essais de liquides purs ou de solides, on utilise une solution saline physiologique comme témoin négatif parallèle dans la méthode d'essai OPI pour détecter des changements non spécifiques dans le système d'essai, et pour vérifier que les conditions de l'essai ne provoquent pas de réponse irritante indésirable. Lors des essais de liquides dilués, il convient d'inclure un groupe témoin de solvant/véhicule concomitants dans la méthode d'essai pour détecter des changements non spécifiques dans le système d'essai, et pour vérifier que les conditions de l'essai ne provoquent pas de réponse irritante indésirable. Comme le mentionne le paragraphe 31, il ne faut utiliser que des solvants ou des véhicules dont l'absence d'effet néfaste sur le système d'essai a été démontrée. Chaque expérience comprend un produit chimique irritant connu en tant que témoin positif parallèle, afin de vérifier l'induction d'une réponse appropriée. Dans cette méthode d'essai, l'essai OPI a pour vocation d'identifier des produits chimiques ayant des effets irritants graves ou corrosifs; le témoin positif est donc un produit chimique de référence qui induit une réponse forte dans la méthode d'essai. Toutefois, afin d'assurer que la variabilité de la réponse du témoin positif au cours du temps peut être évaluée, l'ampleur de la réponse forte ne doit pas être excessive. Les données in vitro obtenues pour le témoin positif doivent être suffisantes pour permettre le calcul d'une gamme de réponse acceptable défini statistiquement. S'il n'existe pas de données antérieures adéquates issues de la méthode d'essai OPI pour un témoin positif donné, il peut être nécessaire de mener des études susceptibles de fournir ces informations. L'acide acétique 10 % ou le chlorure de benzalconium 5 % sont des exemples de témoins positifs pour des produits chimiques d'essai liquides, tandis que l'hydroxyde de sodium ou l'imidazole peuvent jouer ce rôle pour les produits chimiques d'essai solides. Les produits chimiques étalons sont utiles pour évaluer le potentiel d'irritation oculaire de produits chimiques inconnus d'une catégorie chimique ou d'une catégorie de produits spécifique, ou pour évaluer le potentiel d'irritation relatif d'une substance irritante pour les yeux dans un intervalle spécifique de réponses irritantes. Effets mesurés Les cornées traitées sont évaluées avant le traitement et 30, 75, 120, 180, et 240 minutes (± 5 minutes) après le rinçage suivant le traitement. Ces points de mesure sont en nombre suffisant sur la période de traitement de 4 heures et les intervalles entre les mesures permettent d'effectuer les observations requises sur tous les yeux. Les effets mesurés sont l'opacité cornéenne, le gonflement, la rétention de fluorescéine et les effets morphologiques (par exemple, piqûres ou relâchement de l'épithélium). Tous les effets mesurés, à l'exception de la rétention de fluorescéine (qui est déterminée uniquement avant le traitement et 30 minutes après l'exposition au produit chimique d'essai) sont déterminés à chaque point temporel indiqué ci-dessus. Il est conseillé de prendre des photographies pour illustrer l'opacité cornéenne, la rétention de fluorescéine, les effets morphologiques et, le cas échéant, les observations histopathologiques. Après l'examen final à quatre heures, il est recommandé de conserver les yeux dans un fixateur approprié (par exemple, formol tamponné à pH neutre) en vue d'un éventuel examen histopathologique (voir le paragraphe 14 et la référence (8) pour des explications détaillées). Le gonflement cornéen est déterminé par des mesures d'épaisseur de la cornée réalisées au moyen d'un pachymètre optique monté sur un biomicroscope. Il est exprimé en pourcentage et calculé à partir de mesures de l'épaisseur de la cornée selon la formule suivante:
Le pourcentage moyen de gonflement de la cornée pour tous les yeux est calculé à chaque point temporel observé. Un score de catégorie général est attribué à chaque produit chimique d'essai, dérivé du score moyen de gonflement de la cornée le plus élevé observé sur tous les points temporels (paragraphe 51). Le score d'opacité cornéenne est calculé sur la surface de cornée qui est la plus fortement opacifiée comme le montre le tableau 1. La valeur moyenne d'opacité cornéenne pour tous les yeux est calculée pour chaque point temporel observé. Un score de catégorie général est attribué à chaque produit chimique d'essai, dérivé du score moyen d'opacité cornéenne le plus élevé observé sur tous les points temporels (paragraphe 51). Tableau 1 Scores d'opacité cornéenne
La rétention de fluorescéine est évaluée uniquement au point temporel d'observation de 30 minutes comme le montre le tableau 2. La valeur moyenne de rétention de fluorescéine est ensuite calculée pour tous les yeux observés au point d'observation de 30 minutes et utilisée pour déterminer le score de catégorie général de chaque produit chimique d'essai (paragraphe 51). Tableau 2 Scores de rétention de fluorescéine
Les effets morphologiques comprennent la «piqûre» des cellules épithéliales cornéennes, le «relâchement» de l'épithélium, la «rugosité» de la surface cornéenne et l'«adhésion» du produit chimique d'essai sur la cornée. La gravité de ces effets est variable et ils peuvent être observés simultanément. Leur classification est subjective et dépend de l'interprétation de l'observateur. RÉSULTATS ET RAPPORT Évaluation des données Les résultats d'opacité cornéenne, de gonflement et de rétention de fluorescéine sont évalués séparément et aboutissent à la détermination d'une catégorie OPI par effet mesuré. Ces catégories sont ensuite combinées en une classification d'effet irritant pour chaque produit chimique d'essai. Critères de décision Après évaluation de tous les effets, on peut assigner le produit chimique à une catégorie selon un barème prédéterminé. L'interprétation du gonflement de la cornée (tableau 3), de l'opacité (tableau 4) et de la rétention de fluorescéine (tableau 5) au moyen de 4 catégories d'OPI, suit les échelles indiquées ci-après. Il est important de relever que les scores de gonflement cornéen donnés au tableau 3 ne sont applicables que si l'épaisseur est mesurée au moyen d'un biomicroscope (Haag-Streit BP900, par exemple) par un dispositif de mesure de profondeur no 1 avec un réglage de largeur de fente à 9 Tableau 3 Critères de classification OPI pour le gonflement de la cornée
Tableau 4 Critères de classification OPI pour l'opacité
Tableau 5 Critères de classification OPI pour la rétention moyenne de fluorescéine
La classification in vitro d'un produit chimique d'essai est déterminée par la catégorie d'effet irritant qui correspond à la combinaison des catégories obtenues pour le gonflement cornéen, l'opacité cornéenne et la rétention de fluorescéine, ainsi que décrit au tableau 6. Tableau 6 Classification générale des effets in vitro
Critères d'acceptation de l'étude Un essai est considéré comme acceptable lorsque les témoins négatifs et de véhicule/solvant parallèles et les témoins positifs concurrents sont identifiés, respectivement, comme ne relevant pas de la classification du SGH et comme relevant de la catégorie 1 du SGH de l'ONU. Rapport d'essai Le rapport d'essai contient les renseignements suivants, dès lors qu'ils s'appliquent à l'étude:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Approche «bottom-up» : approche en plusieurs étapes appliquée à un produit chimique présumé ne pas nécessiter de classification pour irritation oculaire ou lésion oculaire grave, et dont la première étape consiste à distinguer les produits chimiques ne relevant d'aucune classification (résultat négatif) des autres produits chimiques (résultat positif). Approche «top-down» : approche en plusieurs étapes appliquée à un produit chimique présumé provoquer des lésions oculaires graves, et dont la première étape consiste à distinguer les produits chimiques provoquant des lésions oculaires graves (résultat positif) des autres produits chimiques (résultat négatif). Biomicroscope (lampe à fente) : instrument utilisé pour examiner directement l'œil sous le grossissement d'un microscope binoculaire en créant une image stéréoscopique droite. Dans la méthode d'essai OPI, cet instrument est utilisé pour visualiser les structures antérieures de l'œil de poulet et également pour mesurer objectivement l'épaisseur de la cornée au moyen d'un dispositif adjoint de mesure de la profondeur. Catégorie 1 du SGH : voir «Lésion oculaire grave» et/ou «Effets irréversibles sur l'œil». Catégorie 2 du SGH : voir «Irritation oculaire» et/ou «Effets réversibles sur l'œil». Cornée : partie transparente à l'avant du globe oculaire qui couvre l'iris et la pupille et laisse passer la lumière vers l'intérieur. Danger : propriété inhérente d'un agent ou d'une situation susceptible de provoquer des effets néfastes lorsqu'un organisme, un système ou une (sous-)population est exposé(e) à cet agent. Effets irréversibles sur l'œil : voir «Lésion oculaire grave» et «Catégorie 1 du SGH». Effets réversibles sur l'œil : voir «Irritation oculaire» et «Catégorie 2 du SGH». Fiabilité : mesure dans laquelle la mise en œuvre d'une méthode d'essai peut être reproduite au fil du temps par un même laboratoire ou par plusieurs laboratoires en utilisant le même protocole. Elle est évaluée en calculant la reproductibilité intra-laboratoire et inter-laboratoires ainsi que la répétabilité intra-laboratoire. Gonflement cornéen : mesure objective dans l'essai OPI de l'ampleur de la distension de la cornée après exposition à un produit chimique d'essai. Elle est exprimée en pourcentage et calculée au moyen des mesures initiales (avant administration de la dose) d'épaisseur cornéenne, et des mesures d'épaisseur relevées à intervalles réguliers après exposition au produit chimique d'essai dans l'essai OPI. Le degré de gonflement cornéen est un indicateur d'endommagement de la cornée. Irritation oculaire : modification de l'œil provoquée par l'application d'un produit chimique d'essai sur la face antérieure de l'œil, et qui est totalement réversible dans les 21 jours suivant l'application. Interchangeable avec «Effets réversibles sur l'œil» et avec «Catégorie 2 du SGH de l'ONU» (4). Lésion oculaire grave : lésion des tissus oculaires ou dégradation sévère de la vue, provoquée par l'application d'un produit chimique d'essai sur la face antérieure de l'œil, et qui n'est pas totalement réversible dans les 21 jours suivant l'application. Interchangeable avec «Effets irréversibles sur l'œil» et avec «Catégorie 1 du SGH» (4). Mélange : mélange ou solution composée de deux substances ou plus, qui ne réagissent pas entre elles (4) Méthode d'essai validée : méthode d'essai ayant fait l'objet d'études de validation visant à déterminer sa pertinence (notamment sa précision) et sa fiabilité à des fins spécifiques. Il importe de noter que les performances d'une méthode d'essai validée peuvent être insuffisantes en termes de précision et de fiabilité pour qu'elle soit jugée acceptable pour les besoins envisagés. Non classé : produit chimique qui n'est pas classé comme irritant oculaire (catégorie 2 du SGH de l'ONU) ou comme pouvant provoquer des lésions oculaires graves (catégorie 1 du SGH de l'ONU). Interchangeable avec «Sans catégorie dans le SGH de l'ONU». Opacité cornéenne : mesure de l'étendue de l'opacité de la cornée après exposition à un produit chimique d'essai. L'augmentation de l'opacité cornéenne est un indicateur d'endommagement de la cornée. Poids de la preuve : prise en compte des atouts et des faiblesses de divers éléments d'information en vue d'aboutir à une conclusion concernant le danger potentiel d'un produit chimique et d'étayer cette conclusion. Précision : degré de conformité entre les résultats de la méthode d'essai et les valeurs de référence acceptées. Elle constitue une mesure de performance de la méthode d'essai et l'un de ses aspects de pertinence. Le terme est souvent utilisé indifféremment à la place du terme «concordance» pour qualifier la proportion de résultats corrects d'une méthode d'essai. Produit chimique : une substance ou un mélange Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Produit chimique étalon : produit chimique utilisé comme référence dans une comparaison avec un produit chimique d'essai. Un produit chimique étalon présente les propriétés suivantes: (i) source(s) régulière(s) et fiable(s); (ii) similitude structurale et fonctionnelle avec la catégorie des produits chimiques à tester; (iii) caractéristiques physiques et chimiques connues; (iv) données confirmant les effets connus; et (v) puissance connue dans l'intervalle de la réponse désirée. Rétention de fluorescéine : mesure subjective dans l'essai OPI de la quantité de fluorescéine sodique qui est retenue par les cellules épithéliales de la cornée après exposition à un produit chimique d'essai. Le degré de rétention de fluorescéine est un indicateur d'endommagement de la cornée. Sans catégorie dans le SGH de l'ONU : substances qui ne répondent pas aux critères de classification des catégories 1 ou 2 (2A ou 2B) du SGH de l'ONU. Interchangeable avec «Non classé». SGH [Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations Unies (ONU)] : système proposant la classification des produits chimiques (substances et mélanges) conformément à des types et des niveaux normalisés de dangers physiques, sanitaires et environnementaux, ainsi que la communication des éléments d'information correspondants, notamment par des pictogrammes, mentions d'avertissement, mentions de danger, conseils de prudence et fiches de données de sécurité, afin de diffuser des informations sur leurs effets indésirables dans le but de protéger les personnes (en particulier les employeurs, employés, transporteurs, consommateurs et personnels des services d'urgence) et l'environnement (4). Stratégie d'essais à plusieurs niveaux : démarche expérimentale séquentielle consistant à examiner toutes les informations existantes sur un produit chimique d'essai dans un ordre déterminé, en ayant recours à chaque étape à un processus d'analyse du poids de la preuve afin de déterminer si les informations disponibles sont suffisantes pour décider d'une classification dans une catégorie de danger, avant de passer à l'étape suivante. Si le potentiel d'irritation d'un produit chimique d'essai peut être déterminé sur la base des informations existantes, aucun essai supplémentaire n'est nécessaire. Dans le cas contraire, une procédure expérimentale progressive de type séquentiel sur des animaux est alors lancée jusqu'à ce qu'une classification sans équivoque puisse être effectuée. Substance : un élément chimique et ses composés à l'état naturel ou obtenus par un processus de fabrication, y compris tout additif nécessaire pour préserver la stabilité du produit et toute impureté résultant du processus mis en œuvre, mais à l'exclusion de tout solvant pouvant être séparé de la substance sans en affecter la stabilité ni modifier la composition (4). Taux de faux négatifs : proportion de produits chimiques positifs faussement identifiés comme négatifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Taux de faux positifs : proportion des produits chimiques négatifs faussement identifiés comme positifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Témoin de solvant/véhicule : échantillon non traité contenant tous les composants d'un système d'essai, y compris le solvant ou véhicule utilisé pour tester les échantillons témoins traités ou non avec le produit chimique d'essai afin de déterminer une réponse de référence pour les échantillons traités avec le produit chimique d'essai dissoute dans le même solvant ou véhicule. Testé avec un témoin négatif concurrent, cet échantillon indique également si le solvant ou véhicule interagit avec le système d'essai. Témoin négatif : réplicat non traité contenant tous les composants d'un système d'essai. Cet échantillon subit les mêmes procédures que les échantillons traités avec le produit chimique d'essai et les autres échantillons témoins afin de déterminer si le solvant interagit avec le système d'essai. Témoin positif : réplicat contenant tous les composants d'un système d'essai et traité avec un produit chimique induisant notoirement une réponse positive. Pour permettre d'évaluer la variabilité de la réponse du témoin positif dans le temps, la gravité de la réaction ne doit pas être excessive. Tensioactif : aussi appelé agent tensioactif, il s'agit d'une substance, par exemple un détergent, capable de réduire la tension superficielle d'un liquide et de favoriser ainsi sa capacité à mousser ou à pénétrer les solides; il est aussi connu comme agent mouillant. Appendice 2 PRODUITS CHIMIQUES POUR L'ÉPREUVE DE COMPÉTENCE RELATIVE A LA MÉTHODE D'ESSAI OPI Avant d'utiliser en routine une méthode d'essai conforme à la présente méthode d'essai, les laboratoires démontrent leurs compétences techniques en déterminant correctement la catégorie de danger pour l'œil des 13 produits chimiques recommandés dans le tableau 1. Ces produits chimiques ont été sélectionnés de façon à représenter la gamme des réactions selon le danger qu'ils représentent pour l'œil, sur la base des résultats de l'essai in vivo sur œil de lapin (LD 405) et du SGH de l'ONU (c'est-à-dire les catégories 1, 2A, 2B ou «Sans catégorie») (4) (6). Les autres critères de sélection sont la disponibilité des substances dans le commerce, l'existence de données de référence in vivo de bonne qualité, et l'existence de données de bonne qualité issues de la méthode in vitro OPI constituent d'autres critères de sélection. Les données de référence sont disponibles dans le Récapitulatif simplifié (5) et dans les documents de l'ICCVAM (Background Review Documents) pour la méthode OPI (9). Tableau 1 Produits chimiques recommandés pour démontrer les compétences techniques relatives à la méthode OPI
Appendice 3 SCHÉMA D'UN APPAREIL DE SURFUSION NORMAL ET DES CLAMPS POUR OEIL (Voir Burton et al. (18) pour plus de détails génériques sur l'appareil de surfusion et les clamps pour oeil) 
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

 , soit 0,095 mm. Les utilisateurs tiennent compte du fait que les mesures d'épaisseur de cornée par un biomicroscope varient en fonction du réglage de la largeur de fente.
, soit 0,095 mm. Les utilisateurs tiennent compte du fait que les mesures d'épaisseur de cornée par un biomicroscope varient en fonction du réglage de la largeur de fente.|
(14) |
Dans la partie B, le chapitre B.49 est remplacé par le texte suivant: «B.49 Test du micronoyau in vitro sur cellules de mammifères INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 487 (2016) de l'OCDE pour les essais de produits chimiques. Elle s'inscrit dans une série de méthodes d'essai de toxicologie génétique. Par ailleurs, un document de l'OCDE qui fournit des informations succintes sur les essais de génotoxicité et donne une vue d'ensemble des modifications récemment apportées à ces lignes directrices a été élaboré (1). Le test du micronoyau in vitro (MNvit) est un essai de génotoxicité consistant à mettre en évidence d'éventuels micronoyaux (MN) dans le cytoplasme de cellules en interphase. Les micronoyaux peuvent avoir pour origine des fragments de chromosomes acentriques (c'est-à-dire dépourvus de centromère) ou des chromosomes entiers incapables de migrer vers les pôles de la cellule au cours de l'anaphase. Par conséquent, l'essai MNvit est une méthode in vitro qui, grâce à sa capacité de mise en évidence d'agents aneugènes et clastogènes, constitue un outil de base complet pour l'étude in vitro du potentiel de lésions chromosomiques (2) (3) dans des cellules ayant effectué une mitose pendant ou après l'exposition au produit chimique d'essai (voir paragraphe 13 pour des informations plus détaillées). Contrairement aux aberrations chromosomiques observées dans les cellules en métaphase, qui ne sont pas forcément transmises, les micronoyaux sont des lésions transmises aux cellules-filles. Dans les deux cas, les changements peuvent être incompatibles avec la survie des cellules. La présente méthode d'essai autorise l'utilisation de protocoles incluant ou non la cytochalasine B (cytoB), un inhibiteur de polymérisation de l'actine. L'ajout de cytoB avant la mitose donne lieu à des cellules binucléées, ce qui permet l'identification et l'analyse des micronoyaux uniquement dans les cellules ayant effectué une mitose (4) (5). Cette méthode d'essai autorise également l'utilisation de protocoles ne mettant pas en jeu le blocage de la cytokinèse, à condition de pouvoir démontrer que la population cellulaire analysée a bien effectué une mitose. Outre qu'il permet d'identifier des produits chimiques induisant la formation de micronoyaux, le test MNvit, associé au marquage immunochimique des kinétochores ou à l'hybridation avec sondes centromériques/télomériques [hybridation fluorescente in situ (FISH)], peut apporter des informations supplémentaires sur les mécanismes à l'origine des lésions chromosomiques et de la formation de micronoyaux (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Ces procédures de marquage et d'hybridation peuvent être suivies lorsqu'une augmentation de la formation de micronoyaux est constatée et que l'investigateur souhaite déterminer si cette augmentation est le résultat d'événements clastogènes et/ou aneugènes. Dans la mesure où les micronoyaux présents dans les cellules en interphase peuvent être évalués de façon relativement objective, le personnel du laboratoire a seulement besoin de déterminer le nombre de cellules binucléées en cas d'utilisation de la cytoB ainsi que l'incidence des cellules micronucléées dans tous les cas. L'examen des lames peut donc être relativement rapide, et l'analyse peut être automatisée. Chaque traitement permettant l'examen non plus de centaines mais de milliers de cellules, la puissance de l'essai s'en trouve accrue. Enfin, étant donné que les micronoyaux peuvent être issus de chromosomes retardataires, l'essai peut permettre de détecter la présence d'agents inducteurs d'aneuploïdie, difficiles à étudier lors de tests d'aberration chromosomique classiques, tels que celui décrit dans le chapitre B.10 de la présente annexe (18). Toutefois, l'essai MNvit décrit dans la présente méthode d'essai ne permet pas de distinguer les produits chimiques modifiant le nombre de chromosomes et/ou la ploïdie de ceux induisant la clastogénicité sans avoir recours à des techniques spécifiques, telles que la méthode FISH citée au paragraphe 4. L'essai MNvit est fiable et peut s'appliquer à plusieurs types de cellules, en présence ou en l'absence de cytoB. La validité de l'essai MNvit est attestée par de nombreuses données, et ce avec l'utilisation de différentes lignées cellulaires (cultures de lignées cellulaires ou de cellules primaires) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Ces données proviennent notamment des études internationales de validation coordonnées par la Société française de toxicologie génétique (SFTG) (19) (20) (21) (22) (23) et des rapports de l'International Workshop on Genotoxicity Testing (5) (17). Les données disponibles ont en outre fait l'objet d'une nouvelle évaluation lors d'une étude de validation rétrospective fondée sur le poids de la preuve, réalisée par le Centre européen pour la validation de méthodes alternatives (ECVAM) de la Commission européenne (CE), et la méthode d'essai a été déclarée scientifiquement valable par le comité consultatif scientifique de l'ECVAM (ESAC) (37) (38) (39). Le test de micronoyaux in vitro sur cellules de mammifères peut être pratiqué sur des cultures de lignées cellulaires ou des cultures de cellules primaires d'origine humaine ou de rongeurs. Dans la mesure où la fréquence des micronoyaux généralement constatée pour un type de cellule aura une influence sur la sensibilité de l'essai, il est recommandé d'utiliser des types de cellules pour lesquelles la fréquence habituelle de formation de micronoyaux est stable et définie. Les cellules employées sont choisies en fonction de leur capacité à bien se développer en culture, de la stabilité de leur caryotype (notamment leur nombre de chromosomes) et de la fréquence spontanée de micronoyaux (40). Les données disponibles à l'heure actuelle ne permettent pas d'émettre des recommandations fermes mais tendent à montrer qu'il importe de tenir compte, lors de l'évaluation des risques chimiques, du statut p53, de la stabilité génétique (caryotype), de la capacité de réparation de l'ADN et de l'origine (rongeurs ou humains) des cellules retenues pour l'essai. Les utilisateurs de la méthode d'essai sont donc invités à prendre en considération l'influence de ces caractéristiques cellulaires, et d'autres caractéristiques, sur les performances d'une lignée cellulaire quant à la détection de l'induction de micronoyaux, sachant que les connaissances évoluent dans ce domaine. Les définitions utilisées sont données à l'appendice 1. REMARQUES PRÉLIMINAIRES ET LIMITES À moins que les cellules utilisées ne soient dotées d'un métabolisme compatible avec les produits chimiques testés, les essais conduits in vitro requièrent généralement une source exogène d'activation métabolique. Or, les systèmes d'activation métabolique exogène sont incapables de reproduire parfaitement les conditions in vivo. On prendra soin d'éviter les conditions susceptibles de conduire à de faux résultats positifs ne reflétant pas la génotoxicité des produits chimiques d'essai. Ces conditions peuvent être une modification du pH (41) (42) (43) ou de l'osmolalité, une interaction avec le milieu de culture cellulaire (44) (45) ou une cytotoxicité excessive (voir paragraphe 29). Afin que l'analyse de l'induction de micronoyaux soit valable, il est essentiel que la mitose se soit produite aussi bien dans les cultures traitées et que dans les cultures non traitées. Pour que son intérêt soit maximal, l'examen des micronoyaux est effectué dans les cellules ayant achevé une mitose durant ou après l'application du produit chimique d'essai. Pour les nanomatériaux manufacturés, il est nécessaire d'apporter certaines adaptations spécifiques à la présente méthode d'essai, mais ces adaptations ne sont pas décrites dans le présent document. Avant d'utiliser la méthode d'essai sur un mélange pour générer des données avec pour objectif recherché l'application réglementaire, on considérera si, et si oui pourquoi, elle peut fournir des résultats adéquats dans cet objectif. De telles considérations ne sont pas nécessaires quand les exigences réglementaires stipulent que le mélange doit être testé. PRINCIPE DE L'ESSAI Des cultures cellulaires d'origine humaine ou provenant d'autres mammifères sont exposées au produit chimique d'essai, en présence et en l'absence d'une source exogène d'activation métabolique, à moins que les cellules utilisées ne soient dotées de capacités métaboliques idoines (voir paragraphe 19). Durant ou après l'exposition au produit chimique d'essai, les cellules sont mises en culture pendant une période suffisante pour que la lésion chromosomique ou d'autres effets sur le cycle cellulaire / la division cellulaire conduisent à la formation de micronoyaux dans les cellules en interphase. Le produit chimique d'essai est normalement présent au cours de la mitose pour qu'il y ait induction d'une aneuploïdie. Les cellules récoltées en interphase sont teintées et soumises à analyse en vue de la détection de micronoyaux. En principe, l'examen des micronoyaux ne devrait être réalisé que dans les cellules ayant effectué une mitose durant l'exposition au produit chimique d'essai, ou pendant la période qui suit le traitement, si l'essai la prévoit. Dans les cultures ayant été exposées à un agent de blocage de la cytokinèse, il suffit de procéder à l'examen des cellules binucléées. En l'absence d'agent de blocage de la cytokinèse, il importe de démontrer que les cellules analysées ont selon toute vraisemblance subi une mitose, d'après l'augmentation de la population cellulaire, pendant ou après l'exposition au produit chimique d'essai. Pour tous les protocoles, il convient de démontrer qu'une prolifération cellulaire a eu lieu tant dans les cultures témoins que dans les cultures traitées. Le degré de cytotoxicité ou de cytostase induit par le produit chimique d'essai est évalué pour toutes les cultures dans lesquelles est réalisé l'examen des micronoyaux. DESCRIPTION DE LA MÉTHODE Cellules Il est possible d'utiliser des cultures primaires de lymphocytes du sang périphérique humain ou d'autres mammifères (7) (20) (46) (47) et plusieurs lignées cellulaires issues de rongeurs telles que CHO, V79, CHL/IU, ainsi que les cellules L5178Y ou des lignées cellulaires humaines telles que les TK6 (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (voir paragraphe 6). D'autres lignées cellulaires telles que HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2 (52) (53), A549 et les cellules embryonnaires primaires de hamster doré (54) ont été utilisées pour des tests du micronoyau, mais elles n'ont pas encore été validées complètement. Par conséquent, le recours à ces types de lignées cellulaires doit être justifié via la démonstration de leur performance pour l'essai, ainsi qu'il est indiqué dans la section sur les critères d'acceptabilité. La CytoB est réputée pouvoir influer sur la croissance des cellules L5178Y et n'est donc pas recommandée avec cette lignée cellulaire (23). En cas d'utilisation de cellules primaires, pour des raisons relatives au bien-être des animaux, il conviendra d'envisager, lorsque cela est possible, le recours à des cellules d'origine humaine, prélevées dans le respect des principes éthiques et de la réglementation en la matière. Les lymphocytes du sang périphérique humain utilisés sont issus de sujets jeunes (âgés de 18 à 35 ans environ), non fumeurs, ne souffrant d'aucune maladie connue et n'ayant pas été exposés récemment à des niveaux d'agents génotoxiques (produits chimiques, rayonnements ionisants, par exemple) susceptibles d'augmenter l'incidence de fond des cellules micronucléées, ceci afin de garantir que cette incidence soit faible et homogène. L'incidence des cellules micronucléées augmente avec l'âge, et cette tendance est plus marquée chez la femme que chez l'homme (55). Si des cellules issues de plusieurs donneurs sont mises en commun, le nombre des donneurs est précisé. Il est nécessaire de démontrer que les cellules se sont divisées entre le moment où elles ont été traitées avec le produit chimique d'essai et leur prélèvement. Les cultures cellulaires sont maintenues dans une phase de croissance exponentielle (lignées cellulaires) ou encouragées à se diviser (cultures primaires de lymphocytes) en vue de l'exposition des cellules à différents stades du cycle cellulaire, étant donné que la sensibilité des phases cellulaires aux produits chimiques d'essai peut ne pas être connue. En général, les cellules primaires dont la division doit être stimulée par des agents mitogènes ne sont plus synchronisées lors de leur exposition au produit chimique d'essai (les lymphocytes humains après une stimulation mitogène de 48 heures, par exemple). L'utilisation de cellules synchronisées pendant le traitement avec le produit chimique d'essai n'est pas recommandée, mais peut être acceptable si elle est justifiée. Milieu et conditions de culture Il convient d'utiliser un milieu de croissance et des conditions d'incubation (récipients de culture, atmosphère humidifiée à 5 % de CO2 si nécessaire, température de 37°C) appropriées pour les cultures. La stabilité du caryotype (mode du nombre de chromosomes) et l'absence de contamination par des mycoplasmes sont vérifiées régulièrement dans les lignées cellulaires, et les cellules sont écartées si une contamination ou une modification du caryotype est constatée. La durée normale du cycle cellulaire des lignées ou des cultures primaires utilisées dans le laboratoire d'essai doit être établie et doit correspondre aux caractéristiques cellulaires publiées. Préparation des cultures Lignées cellulaires: les cellules sont multipliées à partir de cultures mères, placées dans un milieu de culture à une densité telle que les cellules en suspension ou en monocouche poursuivront leur croissance de manière exponentielle jusqu'au moment de la récolte (il convient par exemple d'éviter que les cellules qui se multiplient en monocouche arrivent à confluence). Lymphocytes: un sang total traité avec un anticoagulant (héparine, par exemple) ou des lymphocytes isolés sont mis en culture (pendant 48 heures pour les lymphocytes humains, par exemple) en présence d'un mitogène [phytohémagglutinine (PHA) pour les lymphocytes humains, par exemple] afin d'induire une division cellulaire avant l'exposition au produit chimique d'essai et à la cytoB. Activation métabolique Le recours à un système d'activation métabolique exogène est nécessaire en cas d'utilisation de cellules dotées d'une capacité métabolique endogène inadéquate. Le système le plus couramment utilisé, recommandé par défaut, à moins qu'un autre système soit justifié, est une fraction post-mitochondriale enrichie en cofacteur (S9), préparée à partir de foies de rongeurs (généralement des rats) traités avec des inducteurs enzymatiques comme l'Aroclor 1254 (56) (57) ou un mélange de phénobarbital et de ß-naphthoflavone (58) (59) (60). L'utilisation de ce mélange n'est pas contraire à la Convention de Stockholm sur les polluants organiques persistants (61) et s'est révélée aussi efficace que celle de l'Aroclor 1254 pour l'induction d'oxydases à fonction mixte (58) (59) (60). La fraction S9 est généralement utilisée à une concentration comprise entre 1 et 2 % v/v mais peut être portée à 10 % v/v dans le milieu d'essai final. Pendant le traitement, on évitera d'utiliser des produits entraînant une réduction de l'indice mitotique, en particulier des complexants du calcium (62). Le choix du type et de la concentration du système d'activation métabolique exogène ou de l'inducteur métabolique utilisé pourra dépendre de la classe des produits chimiques à tester. Préparation du produit chimique d'essai Les produits chimiques solides à tester sont préparés dans un solvant approprié puis, le cas échéant, dilués avant application. Les produits chimiques liquides peuvent être ajoutés directement ou après dilution au système d'essai. Les produits chimiques gazeux ou volatils nécessitent une modification appropriée des protocoles standards, par exemple l'utilisation de récipients hermétiquement clos (63) (64) (65). Il convient de préparer les produits chimiques d'essai juste avant le traitement, à moins que les données concernant la stabilité ne démontrent qu'ils peuvent être stockés. Conditions de l'essai Solvants Le solvant doit être choisi de manière à optimiser la solubilité des produits chimiques d'essai, sans engendrer d'effets néfastes sur la conduite de l'essai, et notamment sans modifier la croissance cellulaire, nuire à l'intégrité du produit chimique d'essai, réagir avec les récipients de culture ou détériorer le système d'activation métabolique. On recommande d'envisager d'abord l'utilisation d'un solvant (ou milieu de culture) aqueux chaque fois que c'est possible. L'eau et le diméthylsulfoxyde (DMSO) sont des solvants couramment utilisés. En règle générale, les solvants organiques ne doivent pas dépasser 1 % v/v. Si la cytoB est dissoute dans du DMSO, la quantité totale de solvant organique utilisée à la fois pour le produit chimique d'essai et pour la cytoB ne doit pas dépasser 1 % (v/v); sinon, on aura recours à des témoins non traités afin de vérifier que le pourcentage de solvant organique n'a pas d'effet néfaste. Les solvants aqueux (solution saline ou eau) ne doivent pas dépasser 10 % (v/v) dans le milieu de traitement final. L'emploi d'un solvant inhabituel (éthanol ou acétone, par exemple) doit être justifié par des données faisant état de sa compatibilité avec le produit chimique d'essai et le système d'essai, ainsi que de son absence de génotoxicité aux concentrations utilisées. En l'absence de telles données, il est important d'inclure dans l'essai des témoins non traités (voir appendice 1) ainsi que des témoins de solvant afin de démontrer que le solvant choisi n'entraîne pas d'effets délétères ou chromosomiques (aneuploïdie ou clastogénicité, par exemple). Utilisation de la cytoB comme agent de blocage de la cytokinèse Il est essentiel, pour garantir l'efficacité du test MNvit, que les cellules sur lesquelles est réalisé l'examen aient bien effectué une mitose durant le traitement ou pendant la période d'incubation qui suit le traitement, le cas échéant. L'examen des micronoyaux doit donc être limité aux cellules ayant effectué une mitose pendant ou après le traitement. La cytoB est l'agent le plus couramment utilisé pour bloquer la cytokinèse, car elle inhibe l'assemblage de l'actine, empêchant la séparation des cellules-filles après la mitose, d'où la formation de cellules binucléées (6) (66) (67). L'effet du produit chimique d'essai sur la cinétique de prolifération cellulaire peut être mesuré de façon concomitante, en cas de recours à la cytoB. L'usage de cytoB comme agent de blocage de la cytokinèse est indiqué lors de l'utilisation de lymphocytes humains, car la durée du cycle cellulaire est variable selon les donneurs et parce que tous les lymphocytes ne réagissent pas à la stimulation par la PHA. La cytoB n'est pas obligatoire pour les autres types de cellules s'il peut être établi que ces cellules ont subi une division tel que décrit au paragraphe 27. En outre, la cytoB n'est généralement pas utilisée lorsque la formation de micronoyaux dans les échantillons est évaluée par le biais de la cytométrie en flux. Pour chaque type de cellule, le laboratoire détermine la concentration de cytoB permettant d'obtenir la fréquence optimale de cellules binucléées dans les cultures témoins de solvant, et démontre qu'elle produit suffisamment de cellules binucléées pour que l'on puisse procéder à une analyse. La concentration appropriée de cytoB se situe habituellement entre 3 et 6 μg/ml (19). Mesure de la prolifération et de la cytotoxicité cellulaires et choix des concentrations d'essai Lors de la détermination de la plus forte concentration du produit chimique d'essai, on évitera les concentrations susceptibles de produire de fausses réponses positives, notamment celles qui engendrent une cytotoxicité excessive (voir paragraphe 29), une précipitation dans le milieu de culture (voir paragraphe 30) ou une modification marquée du pH ou de l'osmolalité (voir paragraphe 9). Si le produit chimique d'essai provoque une modification marquée du pH du milieu au moment de son ajout, il est possible d'ajuster le pH par tamponnage du milieu de traitement final de manière à éviter les faux résultats positifs et à maintenir des conditions de culture appropriées. Des mesures de la prolifération cellulaire sont effectuées pour s'assurer qu'un nombre suffisant de cellules traitées a effectué une mitose pendant l'essai et que les applications sont réalisées à des niveaux de cytotoxicité appropriés (voir paragraphe 29). La cytotoxicité doit être mesurée lors de l'expérience principale avec et sans activation métabolique, au moyen d'un indicateur pertinent de mort et de croissance cellulaires (voir paragraphes 26 et 27). Un essai préliminaire visant à évaluer la cytotoxicité peut s'avérer utile pour mieux cerner les concentrations à utiliser dans l'essai principal, mais il n'est pas obligatoire. S'il est réalisé, il ne doit pas remplacer la mesure de cytotoxicité effectuée dans le cadre de l'expérience principale. L'application de cytoB sur les cultures et la mesure des fréquences relatives de cellules mononucléées, binucléées et multinucléées dans chaque culture offrent un moyen précis de quantifier l'effet d'un traitement sur la prolifération cellulaire et l'activité cytotoxique ou cytostatique (6) et de s'assurer que seules les cellules ayant effectué une mitose pendant ou après l'application sont observées au microscope. Il est conseillé de mesurer l'indice de prolifération des cellules dont la division cytoplasmique a été bloquée (cytokinesis-block proliferation index, CBPI) (6) (27) (68) ou l'indice de réplication (Replication Index, RI) à partir d'au moins 500 cellules par culture (voir les formules à l'appendice 2) pour estimer l'activité cytotoxique et cytostatique d'un traitement en comparant les valeurs obtenues pour les cultures traitées et les cultures témoins. L'évaluation d'autres indicateurs de cytotoxicité (intégrité cellulaire, apoptose, nécrose, numération des cellules en métaphase, cycle cellulaire, etc.) peut fournir des informations utiles mais ne doit pas remplacer le CBPI ou le RI. Dans les études sans cytoB, il est nécessaire de démontrer que les cellules mises en culture se sont divisées, de sorte qu'une proportion importante des cellules examinées aient effectué une division cellulaire durant ou après l'exposition au produit chimique d'essai, faute de quoi on risque de produire de fausses réponses négatives. La mesure du doublement relatif de la population (Relative Population Doubling, RPD) ou de l'augmentation relative du nombre de cellules (Relative increase in cell count, RICC) est recommandée pour évaluer l'activité cytotoxique et cytostatique d'un traitement (17) (68) (69) (70) (71) (voir formules à l'appendice 2). Lorsque les prélèvements s'étalent sur une longue durée (par exemple, lorsque la durée du traitement équivaut à 1,5 à 2 fois la durée normale du cycle cellulaire et que la récolte a lieu à l'issue d'une durée équivalente après le traitement, ce qui engendre des moments d'échantillonnage allant au-delà de 3 à 4 fois la durée normale du cycle cellulaire au total (voir paragraphes 38 et 39), il se peut que le RPD sous-estime la cytotoxicité (71). Dans ces circonstances, la RICC pourrait constituer une meilleure mesure, mais l'évaluation de la cytotoxicité après une durée équivalente à 1,5 à 2 fois la durée normale du cycle cellulaire fournit néanmoins une estimation précieuse. L'évaluation d'autres marqueurs de cytotoxicité ou de cytostase (intégrité cellulaire, apoptose, nécrose, numération des cellules en métaphase, indice de prolifération, cycle cellulaire, ponts nucléoplasmiques ou bourgeons nucléaires, etc.) peut fournir d'autres informations utiles mais ne doit pas remplacer le RPD ni la RICC. Il convient d'évaluer au moins trois concentrations d'essai (sans compter les témoins positifs et les témoins de solvant) remplissant les critères d'acceptabilité (cytotoxicité appropriée, nombre de cellules, etc.). Quel que soit le type de cellules (lignées cellulaires ou cultures primaires de lymphocytes), chacune des cultures réalisées en un seul ou plusieurs exemplaires peut être utilisée pour chaque concentration d'essai. Alors que l'utilisation de cultures en double exemplaires est recommandée, l'utilisation de cultures en exemplaire unique est aussi acceptée à condition que le nombre total de cellules analysées par concentration reste identique qu'il s'agisse de cultures uniques ou de répliques. L'utilisation de cultures uniques est pertinent en particulier lorsque quand on évalue plus de 3 concentrations (voir paragraphes 44-45). Les résultats obtenus pour chacune des répliques (cultures réalisées en plusieurs exemplaires) à une concentration donnée peuvent être regroupés pour l'analyse des données. Pour les produits chimiques dont la cytotoxicité est faible ou nulle, des niveaux de concentration séparés par un facteur de 2 à 3 environ conviendront généralement. En cas de cytotoxicité, les concentrations d'essai retenues doivent couvrir une plage englobant la concentration produisant une cytotoxicité telle que décrite au paragraphe 29 et les concentrations pour lesquelles une cytotoxicité modérée, faible ou nulle est observée. De nombreux produits chimiques d'essai présentent des courbes de concentration-réponse à forte pente et, pour obtenir des données dans le cadre d'une cytotoxicité faible et modérée ou étudier la relation dose-réponse en détail, il faudra donc utiliser des concentrations plus rapprochées et/ou plus de trois concentrations (cultures uniques ou répliques), notamment dans les cas où il est nécessaire de répéter l'expérience (voir paragraphe 60). Si la concentration maximale est basée sur la cytotoxicité, la concentration la plus forte doit viser une cytotoxicité de 55 ± 5 % selon les paramètres de cytotoxicité recommandés (à savoir une réduction de la RICC et du RPD pour les lignées cellulaires sans cytoB et une réduction du CBPI et de l'indice de réplication quand la cytoB est utilisée à 45 ± 5 % du témoin négatif concomitant) (72). Les résultats positifs présents uniquement dans la tranche la plus haute de la plage de cytotoxicité 55 ± 5 % doivent être interprétés avec prudence (71). Pour les produits chimiques d'essai peu solubles qui ne sont pas cytotoxiques à des concentrations inférieures à la concentration insoluble la plus faible, la plus forte concentration analysée doit produire une turbidité ou un précipité visible à l'œil nu ou à l'aide d'un microscope inversé à la fin du traitement avec le produit chimique d'essai. Même si une cytotoxicité intervient au-delà de la concentration insoluble la plus faible, il est recommandé de tester une seule concentration produisant une turbidité ou un précipité visible, car de fausses réponses pourraient découler de ce précipité. A la concentration produisant un précipité, il convient de s'assurer que ce dernier n'interfère pas avec la conduite de l'essai (coloration ou analyse, par exemple). Il peut être utile de déterminer la solubilité dans le milieu de culture préalablement à l'essai. Si aucun précipité ou aucune cytotoxicité limitante ne sont observés, la concentration d'essai maximale doit correspondre à la plus basse parmi 10 mM, 2 mg/ml ou 2 μl/ml (73) (74) (75). Lorsque la composition du produit chimique d'essai n'est pas définie, par exemple dans le cas d'une substance de composition inconnue ou variable, de produits de réaction complexes ou de matériels biologiques (UVCB) (76), de produits extraits de l'environnement, etc., il peut être nécessaire d'augmenter la concentration maximale (5 mg/ml par exemple), en l'absence de cytotoxicité suffisante, afin d'accroître la concentration de chacun des composants. Il convient toutefois de noter que ces exigences peuvent être différentes pour les produits pharmaceutiques à usage humain (93). Témoins Des témoins négatifs concomitants (voir paragraphe 21), constitués uniquement du solvant dans le milieu de traitement et testés de la même façon que les cultures traitées, doivent être inclus à tous les moments de récolte. Des témoins positifs concomitants sont nécessaires pour démontrer la capacité du laboratoire à identifier les clastogènes et les aneugènes dans les conditions du protocole d'essai utilisé, ainsi que l'efficacité du système d'activation métabolique exogène, le cas échéant. Des exemples de témoins positifs figurent dans le tableau 1 ci-dessous. D'autres produits chimiques peuvent être utilisés comme témoins positifs, si cela est justifié. À l'heure actuelle, aucun aneugène connu ne nécessite une activation métabolique pour présenter une activité génotoxique (17). Étant donné que les essais in vitro de génotoxicité sur cellules de mammifères sont suffisamment normalisés en ce qui concerne les traitements parallèles de courte durée effectués avec et sans activation métabolique pendant la même durée de traitement, l'utilisation de témoins positifs peut être limitée à un clastogène nécessitant une activation métabolique. Dans ce cas, une seule réponse clastogène dans un témoin positif démontrera à la fois l'activité du système d'activation métabolique et la réactivité du système d'essai. Toutefois, un traitement de longue durée (sans S9) doit disposer de son propre témoin positif, étant donné que la durée du traitement sera différente de celle de l'essai ayant recours à une activation métabolique. Si un clastogène est sélectionné comme témoin positif unique pour le traitement de courte durée avec et sans activation métabolique, un aneugène devra être sélectionné pour le traitement de longue durée sans activation métabolique. Les témoins positifs tant pour la clastogénicité que pour l'aneugénicité sont utilisés pour des cellules métaboliquement compétentes ne nécessitant pas de préparation S9. Chaque témoin positif doit être utilisé à une ou plusieurs concentrations devant normalement donner lieu à une augmentation reproductible et détectable par rapport à la valeur de fond afin de démontrer la sensibilité du système d'essai (c'est-à-dire que les effets sont nets mais que l'identité des lames codées n'est pas évidente pour l'examinateur), et la réponse ne doit pas être compromise par une cytotoxicité supérieure aux limites fixées dans la présente méthode d'essai. Tableau 1 Produits chimiques de référence recommandés pour la vérification des compétences du laboratoire et pour la sélection des témoins positifs
MODE OPÉRATOIRE Déroulement du traitement Afin d'augmenter au maximum la probabilité de détecter une action aneugène ou clastogène à un stade précis du cycle cellulaire, il importe qu'un nombre suffisant de cellules représentant la totalité des stades de leur cycle cellulaire soit traité avec le produit chimique d'essai. Toutes les applications débutent et s'achèvent pendant la phase de croissance exponentielle des cellules, et les cellules poursuivent leur croissance jusqu'au moment de l'échantillonnage. Le déroulement du traitement pour les lignées cellulaires et les cultures de cellules primaires peut donc différer quelque peu de celui appliqué pour les lymphocytes qui nécessitent une stimulation mitogène pour débuter leur cycle cellulaire (17). En ce qui concerne les lymphocytes, l'approche la plus efficace consiste à commencer le traitement avec le produit chimique d'essai 44 à 48 heures après la stimulation par la PHA, au moment où les cellules se divisent de manière asynchrone (6). Les données publiées (19) indiquent que la plupart des aneugènes et clastogènes sont détectés à l'issue d'une application de courte durée, comprise entre 3 et 6 heures, en présence et en l'absence de préparation S9, suivie du retrait du produit chimique d'essai et d'une période de croissance équivalente à 1.5 à 2.0 fois la durée normale du cycle cellulaire (7). Toutefois, pour une évaluation complète, laquelle serait nécessaire pour conclure à un résultat négatif, les trois conditions expérimentales suivantes doivent être respectées pour un traitement de courte durée avec et sans activation métabolique et pour un traitement de longue durée sans activation métabolique (voir paragraphes 56, 57 et 58):
Si l'une de ces conditions expérimentales entraîne une réponse positive, il n'est pas forcément nécessaire d'étudier les autres régimes de traitement. S'il est connu ou supposé que le produit chimique d'essai affecte la durée du cycle cellulaire (par exemple si l'essai porte sur des analogues de nucléosides), notamment en ce qui concerne les cellules compétentes en p53 (35) (36) (77), les périodes d'échantillonnage et de récupération peuvent être prolongées d'une durée équivalente à 1,5 à 2,0 fois la durée normale du cycle cellulaire (soit au total 3,0 — 4,0 cycles cellulaires après le début des traitements de courte durée et de longue durée). Ces différents choix sont à faire en fonction des craintes d'interaction entre le produit chimique d'essai et la cytoB. En cas de recours à des périodes d'échantillonnage étendues (soit une durée de mise en culture équivalente au total à 3,0 — 4,0 cycles cellulaires), il faut veiller à ce que les cellules se divisent toujours activement. En ce qui concerne les lymphocytes, par exemple, la croissance exponentielle peut diminuer à partir de 96 heures après la stimulation et les cultures cellulaires monocouches peuvent devenir confluentes. Les déroulements proposés pour le traitement des cellules sont résumés dans le tableau 2. Ces déroulements de traitement généraux peuvent être modifiés (et doivent être justifiés) en fonction de la stabilité et de la réactivité du produit chimique d'essai, ou des caractéristiques de croissance des cellules utilisées. Tableau 2 Traitement des cellules et temps de récolte pour le test MNvit
Il est possible que les cultures monocouches présentent des cellules mitotiques (reconnaissables à leur forme ronde et se détachant de la surface) à l'issue du traitement de 3 à 6 heures. Ces cellules se détachant facilement de la culture, elles risquent d'être emportées lors de l'élimination du milieu contenant le produit chimique d'essai. Si l'on constate une augmentation substantielle du nombre de cellules mitotiques par rapport aux témoins, ce qui indiquerait l'arrêt probable de la mitose, les cellules doivent être récoltées par centrifugation puis réintroduites dans les cultures, afin d'éviter de perdre les cellules en phase de mitose, et présentant un risque de micronoyaux / d'aberration chromosomique, au moment de la récolte. Récolte des cellules et préparation des lames Chaque culture doit faire l'objet d'une récolte et d'un traitement séparés. La préparation des cellules peut comporter un traitement hypotonique, mais cette étape n'est pas indispensable si les cellules sont correctement étalées d'une autre façon. Différentes techniques peuvent être employées pour la préparation des lames, à condition que les préparations cellulaires obtenues pour l'examen soient de très bonne qualité. On retiendra les cellules dotées d'une membrane intacte et d'un cytoplasme intact pour permettre la détection d'éventuels micronoyaux et (dans la méthode du blocage de la cytokinèse) le repérage fiable des cellules binucléées. Les lames peuvent être colorées par différentes méthodes, au Giemsa ou à l'aide de colorants fluorescents spécifiques de l'ADN, par exemple. Le recours à des 'utilisation de colorants fluorescents appropriés [par exemple, l'acridine orange (78) ou l'Hoechst 33258 avec la pyronine-Y (79)] permet d'éliminer une partie des artefacts associés avec l'utilisation d'un colorant non spécifique de l'ADN. Les anticorps anti-kinétochore, la méthode FISH associée à des sondes ADN pancentromériques ou encore le marquage in situ par amorçage à l'aide d'amorces pancentromériques, avec un colorant de contraste de l'ADN approprié, peuvent être utilisés pour identifier le contenu des micronoyaux (les chromosomes entiers seront colorés tandis que les fragments de chromosomes acentriques ne le seront pas) si les informations d'ordre mécanistique concernant leur formation présentent de l'intérêt (16) (17). D'autres méthodes de discrimination des effets clastogènes et aneugènes peuvent être utilisées, à condition que leur efficacité ait été prouvée et validée. Ainsi, pour certaines lignées cellulaires, la mesure des noyaux sub-2N en tant qu'événements hypodiploïdes à l'aide de techniques telles que l'analyse d'images, la cytométrie à balayage laser ou la cytométrie en flux pourrait également fournir des informations utiles (80) (81) (82). Les observations morphologiques des noyaux pourraient aussi donner des indications sur une aneuploïdie éventuelle. De plus, un essai d'aberrations chromosomiques dans les cellules en métaphase, portant de préférence sur le même type de cellules et suivant un protocole d'une sensibilité comparable, pourrait également se révéler utile pour déterminer si les micronoyaux sont dus à une cassure chromosomique (sachant qu'une perte chromosomique ne serait pas détectée au cours de l'essai d'aberration chromosomique). Analyse Toutes les lames, y compris celles des témoins de solvant et des témoins non traités (s'il y en a) et positifs, doivent être codées indépendamment avant l'analyse au microscope des fréquences de micronoyaux. Il convient d'employer des techniques appropriées pour contrôler les biais ou dérives en cas d'utilisation d'un système d'analyse automatique tel que la cytométrie en flux, la cytométrie à balayage laser ou l'analyse d'images. Quelle que soit la plate-forme automatisée utilisée pour examiner les micronoyaux, le CBPI, le RI, le RPD et la RICC doivent être évalués simultanément. Dans les cultures traitées à la cytoB, la fréquence des micronoyaux est analysée dans un minimum de 2 000 cellules binucléées par concentration et par témoin (83), réparties équitablement entre les répliques, lorsque les cultures ont été réalisées en plusieurs exemplaires ou répliques. Lorsque des cultures en un seul exemplaire sont utilisées pour chaque concentration (voir paragraphe 28), il y a lieu d'examiner au moins 2 000 cellules binucléées par culture (83). Si le nombre de cellules binucléées disponibles pour l'examen de chaque concentration est largement inférieur à 1 000 par culture (pour les cultures dupliquées) ou à 2 000 (pour une culture en un seul exemplaire), et si aucune augmentation significative du nombre de micronoyaux n'est détectée, l'essai devra être renouvelé sur un nombre plus important de cellules, ou à des concentrations moins cytotoxiques, suivant la situation. Il convient également de veiller à ne pas examiner les cellules binucléées ayant une forme irrégulière ou dont les deux noyaux présentent une différence de taille trop importante. En outre, les cellules binucléées ne doivent pas être confondues avec des cellules multinucléées mal étalées. Les cellules possédant plus de deux noyaux principaux ne sont pas analysées, car la fréquence de référence des micronoyaux est susceptible d'être plus élevée dans ce type de cellules (84). L'examen des cellules mononucléées peut être envisagé s'il s'avère que le produit chimique d'essai interfère avec l'action de la cytoB. Dans ce cas, il peut se révéler utile de répéter l'essai sans cytoB. L'examen des cellules mononuclées outre celui des cellules binucléées pourrait fournir des informations utiles (85) (86), mais n'est pas obligatoire. En ce qui concerne les lignées cellulaires, lorsque l'essai est réalisé sans cytoB, l'examen des micronoyaux doit porter sur un minimum de 2 000 cellules par concentration d'essai (83) et par témoin, réparties équitablement entre les répliques, lorsque les cultures ont été réalisées en plusieurs exemplaires. Lorsque des cultures en un seul exemplaire sont utilisées pour chaque concentration (voir paragraphe 28), il y a lieu d'examiner au moins 2 000 cellules binucléées par culture. Si le nombre de cellules binucléées disponibles pour l'examen de chaque concentration est largement inférieur à 1 000 par culture (pour les cultures dupliquées) ou à 2 000 (pour une culture en un seul exemplaire), et si aucune augmentation significative du nombre de micronoyaux n'est détectée, l'essai devra être renouvelé sur un nombre plus important de cellules, ou à des concentrations moins cytotoxiques, suivant la situation. Lorsque l'essai est réalisé en présence de cytoB, il convient de déterminer un CBPI ou un indice de réplication (RI) afin d'évaluer la prolifération cellulaire (voir appendice 2), en utilisant 500 cellules au moins par culture. Lorsque les traitements sont effectués en l'absence de cytoB, il est essentiel de faire la preuve que les cellules en culture se sont divisées (voir paragraphes 24 à 28). Compétence du laboratoire Afin d'acquérir une expérience suffisante de l'essai avant de l'utiliser en routine, le laboratoire doit avoir réalisé une série d'expériences avec des produits chimiques positifs de référence agissant selon des mécanismes variés (au moins un avec activation métabolique et un sans activation métabolique, et un agissant selon un mécanisme aneugène, et sélectionné parmi les produits chimiques énumérés au tableau 1) et avec plusieurs témoins négatifs (y compris des cultures non traitées et divers solvants/véhicules). Ces réponses de témoins positifs et négatifs doivent être cohérentes par rapport à la littérature. Cette exigence ne s'applique pas aux laboratoires possédant déjà une expérience, c'est-à-dire qui disposent d'une base de données historiques telle que définie aux paragraphes 49 à 52. Une sélection de produits chimiques utilisées comme témoins positifs (voir tableau 1) doit être testée dans le cadre de traitements de courte et de longue durée en l'absence d'activation métabolique, ainsi que dans le cadre d'un traitement de courte durée en présence d'une activation métabolique, l'objectif étant de démontrer que le laboratoire possède la compétence nécessaire pour détecter les produits chimiques clastogènes et aneugènes, déterminer l'efficacité du système d'activation métabolique et démontrer la pertinence des procédures d'examen (analyse visuelle au microscope, cytométrie en flux, cytométrie à balayage laser ou analyse d'images). Il conviendra de définir une plage de concentrations des produits chimiques sélectionnés qui permette d'obtenir des augmentations reproductibles et liées à la concentration par rapport aux valeurs de fond, afin de démontrer la sensibilité et la plage dynamique du système d'essai. Données historiques Le laboratoire doit établir:
Lors de l'acquisition initiale de données en vue d'établir une distribution des témoins négatifs historiques, les données des témoins négatifs concomitants doivent être cohérentes avec les données publiées, lorsqu'elles existent. Puis, à mesure que de nouvelles données expérimentales viennent étoffer la plage de distribution des témoins, les données des témoins négatifs concomitants doivent idéalement se situer dans les limites de contrôle à 95 % de cette distribution (87) (88). La base de données historiques du laboratoire relatives aux témoins négatifs doit à l'origine être constituée à partir d'au moins 10 expériences, sachant qu'il serait préférable qu'elle en compte au moins 20, réalisées dans des conditions expérimentales similaires. Les laboratoires doivent utiliser des méthodes de contrôle de la qualité telles que des graphiques statistiques (cartes C ou cartes X-barre, par exemple (88)) afin de déterminer la variabilité de leurs données de témoins positifs et négatifs et de démontrer leur maîtrise de la méthodologie (83). On trouve dans la littérature (87) d'autres recommandations sur la façon de constituer et d'utiliser les données historiques (critères d'inclusion et d'exclusion des données dans la base et critères d'acceptabilité pour une expérimentation donnée). Toute modification du protocole expérimental doit être étudiée en termes de cohérence des données avec les bases de données historiques existantes du laboratoire. Toute incohérence majeure doit conduire à l'établissement d'une nouvelle base de données des témoins historiques. Les données des témoins négatifs désignent l'incidence des cellules micronucléées issues d'une seule culture ou de l'ensemble des cultures réalisées en plusieurs exemplaires, comme décrit au paragraphe 28. Les témoins négatifs concomittants se situent idéalement dans les limites de contrôle à 95 % de la distribution des données historiques des témoins négatifs contenues dans la base de données du laboratoire (87) (88). Lorsque les données des témoins négatifs concomitants se situent en dehors des limites de contrôle à 95 %, leur inclusion dans la distribution des témoins historiques peut être acceptable à condition que ces données ne soient pas exagérément extrêmes et qu'il soit prouvé que le système d'essai est «sous contrôle» (voir paragraphe 50) et qu'il n'y a pas eu de défaillance technique ou d'erreur humaine. RÉSULTATS ET RAPPORT Présentation des résultats Lorsque la technique du blocage de la cytokinèse est employée, seules les fréquences des cellules binucléées présentant des micronoyaux (indépendamment du nombre de micronoyaux par cellule) sont utilisées pour évaluer l'induction de micronoyaux. L'analyse des cellules présentant un ou plusieurs micronoyaux peut être rapportée séparément; elle peut apporter des informations utiles, mais n'est pas obligatoire. Il convient d'effectuer des mesures parallèles de cytotoxicité et/ou de cytostase pour toutes les cultures traitées et les témoins négatifs et positifs (16). On calculera également le CBPI ou l'indice de réplication (RI) pour toutes les cultures traitées et témoins, afin de mesurer le retard du cycle cellulaire en cas de recours à la méthode de blocage de la cytokinèse. En l'absence de cytoB, il convient de mesurer le doublement relatif de la population (RPD) ou l'augmentation relative du nombre de cellules (RICC) (voir appendice 2). Les données doivent être indiquées individuellement pour chaque culture. En outre, toutes les données doivent être résumées sous forme de tableaux. Critères d'acceptabilité L'acceptation de l'essai repose sur les critères suivants:
Évaluation et interprétation des résultats À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement positif si, dans les conditions expérimentales étudiées (voir paragraphes 36-39):
Le produit chimique d'essai est alors considéré comme incapable d'induire des cassures chromosomiques et/ou un gain ou une perte chromosomique dans ce système d'essai. Des recommandations concernant les méthodes statistiques les plus appropriées sont également disponibles dans la littérature (90) (91) (92). À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement négatif si, dans toutes les conditions expérimentales étudiées (voir paragraphes 36-39)
Le produit chimique d'essai est alors considéré comme incapable d'induire des cassures chromosomiques et/ou un gain ou une perte chromosomique dans ce système d'essai. Des recommandations concernant les méthodes statistiques les plus appropriées sont également disponibles dans la littérature (90) (91) (92). Il n'est pas nécessaire de vérifier une réponse clairement positive ou négative. Lorsque la réponse n'est ni clairement négative ni clairement positive, tel que décrit ci-dessus, ou en vue d'établir la signification biologique d'un résultat, les données doivent être soumises à un jugement d'expert et/ou des investigations plus poussées. Il peut être utile d'examiner des cellules supplémentaires (le cas échéant) ou de répéter l'expérience, éventuellement dans des conditions expérimentales modifiées [espacement des concentrations, autres conditions d'activation métabolique (concentration de S9 ou origine de S9), par exemple]. Dans de rares cas, même après des études complémentaires, l'ensemble de données ne permettra pas de conclure à un résultat positif ou négatif, et le résultat sera déclaré équivoque. Lorsque des produits chimiques d'essai induisent la formation de micronoyaux lors de l'essai MNvit, celle-ci est due soit à une cassure chromosomique, soit à une perte chromosomique, soit aux deux évènements combinés. Il est possible de procéder à d'autres analyses à l'aide d'anticorps anti-kinétochores, de sondes centromériques in situ ou d'autres méthodes afin de déterminer si le mécanisme d'induction de micronoyaux est le résultat d'une activité clastogène et/ou aneugène. Rapport d'essai Le rapport d'essai doit comporter les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Aneugène : produit chimique ou processus qui, via des interactions avec les composants de la cellule mitotique ou méiotique lors de la division cellulaire, provoque la formation de cellules ou d'organismes aneuploïdes. Aneuploïdie : tout écart par rapport au nombre diploïde (ou haploïde) normal de chromosomes, d'un seul ou de plusieurs chromosomes, mais non d'un ou de plusieurs jeux de chromosomes (polyploïdie). Apoptose : mort cellulaire programmée caractérisée par une succession d'étapes menant à la désintégration des cellules en particules membranaires qui sont ensuite éliminées par phagocytose ou par excrétion. Augmentation relative du nombre de cellules (Relative increase in cell count, RICC) : méthode de mesure de la cytotoxicité lorsque la cytoB n'est pas utilisée (voir formule à l'annexe 2). Cellules en interphase : cellules qui ne sont pas en phase de mitose. Centromère : région de l'ADN d'un chromosome où les deux chromatides sont reliées entre elles et sur laquelle les deux kinétochores sont fixés côte à côte. Clastogène : produit chimique ou événement induisant des aberrations chromosomiques structurales dans des populations cellulaires ou des organismes eucaryotes. Concentrations : désigne les concentrations finales du produit chimique d'essai dans le milieu de culture. Cytokinèse : processus de division cellulaire survenant immédiatement après la mitose pour former deux cellules-filles, contenant chacune un noyau unique. Cytostase : inhibition de la croissance cellulaire (voir formule à l'appendice 2). Cytotoxicité : pour les essais visés par la présente méthode d'essai effectués en présence de cytochalasine B, la cytotoxicité correspond à une baisse de l'indice de prolifération des cellules dont la division cytoplasmique a été bloquée (CBPI) ou de l'indice de réplication (RI) des cellules traitées par rapport au témoin négatif (voir paragraphe 26 et appendice 2). Pour les essais visés par la présente méthode d'essai effectués en l'absence de cytochalasine B, la cytotoxicité correspond à une baisse du doublement relatif de la population (RPD) ou de l'augmentation relative du nombre de cellules (RICC) des cellules traitées par rapport au témoin négatif (voir paragraphe 27 et appendice 2). Doublement relatif de la population (Relative Population Doubling, RPD) : méthode de mesure de la cytotoxicité lorsque la cytoB n'est pas utilisée (voir formule à l'appendice 2). Fraction S9 de foie : surnageant d'homogénat de foie centrifugé à 9 000g (extrait de foie cru). Génotoxique : terme générique qualifiant tous les types de lésions de l'ADN ou des chromosomes, tels que les cassures, délétions, adduits, liaisons et modifications de nucléotides, réarrangements, mutations génétiques, aberrations chromosomiques et aneuploïdies. Tous les types d'effets génotoxiques n'entraînent pas nécessairement de mutations ou de lésions chromosomiques stables. Indice de prolifération (Proliferation Index, PI) : méthode de mesure de la cytotoxicité lorsque la cytoB n'est pas utilisée (voir formule à l'appendice 2). Indice de prolifération des cellules dont la division cytoplasmique a été bloquée (Cytokinesis-Block Proliferation Index, CBPI) : proportion de cellules issues de la deuxième division dans la population traitée par rapport au témoin non traité (voir formule à l'appendice 2). Indice de réplication (Replication Index, RI) : rapport entre le nombre de cycles de division cellulaire achevés dans une culture traitée et ce même nombre dans le témoin non traité, au cours de la période d'exposition et la récupération (voir formule à l'appendice 2). Indice mitotique : nombre de cellules en métaphase divisé par le nombre total de cellules dans une population; une indication de la vitesse de prolifération cellulaire dans cette population. Kinétochore : assemblage de protéines situé au niveau du centromère d'un chromosome, auquel sont associées les fibres fusoriales lors de la division cellulaire, permettant le mouvement ordonné des chromosomes-fils vers les pôles des cellules-filles. Mélange S9 : mélange de fraction S9 de foie et de cofacteurs nécessaires à l'activité des enzymes métaboliques. Micronoyau : petit noyau, venant s'ajouter au noyau principal de la cellule et séparé de celui-ci, formé lors de la télophase de la mitose ou de la méiose à partir de chromosomes entiers ou de fragments de chromosomes retardataires. Mitose : division du noyau cellulaire, généralement décomposée en prophase, prométaphase, métaphase, anaphase et télophase. Mutagène : qui produit une modification héréditaire portant sur une ou plusieurs séquences de paires de bases d'ADN génique, ou sur la structure de chromosomes (aberrations chromosomiques). Non-disjonction : non-séparation d'une paire de chromatides, qui ne parviennent donc pas à migrer correctement lors de la division cellulaire, ce qui entraîne la présence d'un nombre anormal de chromosomes dans les cellules-filles. Polyploïdie : aberrations chromosomiques numériques dans des cellules ou des organismes, impliquant un ou plusieurs jeux de chromosomes et non un ou plusieurs chromosomes isolés (aneuploïdie). Produit chimique : une substance ou un mélange. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Prolifération cellulaire : augmentation du nombre de cellules résultant de la division cellulaire mitotique. Statut p53 : la protéine p53 intervient dans la régulation du cycle cellulaire, l'apoptose et la réparation de l'ADN. Les cellules déficientes en protéine fonctionnelle p53, incapables d'arrêter le cycle cellulaire ou d'éliminer les cellules lésées par le biais de l'apoptose ou d'autres mécanismes (induction de la réparation de l'ADN, par exemple) liés aux fonctions de p53 en réponse à des lésions de l'ADN, devraient théoriquement être davantage sujettes aux mutations génétiques ou aux aberrations chromosomiques. Témoin de solvant : terme générique désignant les cultures témoins recevant uniquement le solvant utilisé pour dissoudre le produit chimique d'essai. Témoin non traité : cultures ne recevant aucun traitement (ni produit chimique d'essai ni solvant) mais préparées parallèlement et de la même façon que les cultures exposées au produit chimique d'essai. Appendice 2 FORMULES POUR L'ÉVALUATION DE LA CYTOTOXICITÉ En cas d'utilisation de la cytoB , l'évaluation de la cytotoxicité se fonde sur l'indice de prolifération des cellules dont la division cytoplasmique a été bloquée (CBPI) ou sur l'indice de réplication (RI) (17) (69). Le CBPI indique le nombre moyen de noyaux par cellule, et peut servir au calcul de la prolifération cellulaire. Le RI indique le nombre relatif de cycles cellulaires par cellule effectués au cours de la période d'exposition à la cytoB dans les cultures traitées par rapport aux cultures témoins, et peut servir au calcul du pourcentage de cytostase: % cytostase = 100 – 100{(CBPIT – 1) ÷ (CBPIC – 1)} Avec:
Où:
Ainsi, un CBPI égal à 1 (toutes les cellules sont mononucléées) équivaut à une cytostase de 100 %. Cytostase = 100-RI
Ainsi, un RI de 53 % signifie que, par rapport au nombre de cellules ayant effectué une division cellulaire pour former des cellules binucléées et multinucléées dans la culture témoin, 53 % seulement de ces cellules ont effectué une division dans la culture traitée, soit une cytostase de 47 %. En l'absence d'utilisation de cytoB , l'évaluation de la cytotoxicité sur la base de l'augmentation relative du nombre de cellules (RICC) ou du doublement relatif de la population (RPD) est recommandée (69), ces deux mesures tenant compte de la proportion de cellules ayant effectué une division cellulaire.
Où: Doublement de population = [log (nombre de cellules post-application ÷ nombre de cellules initial)] ÷ log 2 Par exemple, une RICC ou un RPD de 53 % indique une cytotoxicité/cytostase de 47 %. En utilisant l'indice de prolifération (PI), on peut évaluer la cytotoxicité via le comptage du nombre de clones composés de 1 cellule (cl1), 2 cellules (cl2), 3 à 4 cellules (cl4) et 5 à 8 cellules (cl8).
Le PI est également un paramètre fiable et utile de cytotoxicité pour les lignées cellulaires cultivées in vitro en l'absence de cytoB (25) (26) (27) (28) et peut être considéré comme un autre paramètre utile. Quoi qu'il en soit, il convient de mesurer le nombre de cellules avant traitement, qui doit être identique dans les cultures traitées et les cultures témoins négatives. Le RCC (à savoir nombre de cellules dans les cultures traitées / nombre de cellules dans les cultures témoins) était utilisé auparavant comme paramètre de cytotoxicité mais n'est plus recommandé car il peut induire une sous-estimation de la cytotoxicité. En cas d'utilisation de systèmes d'analyse automatique tels que la cytométrie en flux, la cytométrie à balayage laser ou l'analyse d'images, le nombre de noyaux peut remplacer le nombre de cellules dans la formule. Dans les cultures témoins négatives, le doublement de la population ou l'indice de réplication doivent être compatibles avec l'obligation de prélever des cellules à l'issue d'une période équivalente à environ 1,5 à 2 fois la durée normale du cycle cellulaire après le début du traitement. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

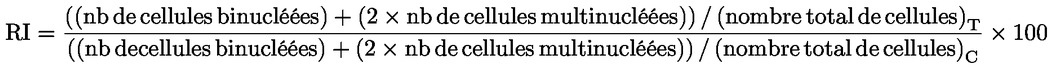
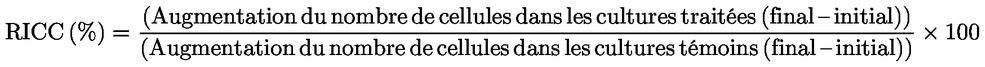

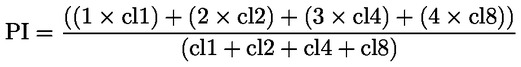
|
(15) |
Dans la partie B, les chapitres suivants sont ajoutés: «B.59 Sensibilisation cutanée in chemico: essai de réactivité peptidique direct (DPRA) INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 442C (2015) de l'OCDE pour les essais de produits chimiques. Un sensibilisant cutané est une substance qui provoque une réponse allergique suite à un contact avec la peau, selon la définition du Système général harmonisé de classification et d'étiquetage des produits chimiques (SGH) des Nations Unies (ONU) (1) et du règlement (CE) no 1272/2008 de l'Union européenne relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges (CLP) (18). La présente méthode d'essai décrit une procédure in chemico, à savoir l'essai de réactivité peptidique direct (Direct Peptide Reactivity Assay, DPRA), qui doit aider à distinguer les sensibilisants des non-sensibilisants cutanés, selon le SGH et le CLP. Les principales phases du processus biologique de sensibilisation cutanée font l'objet d'un consensus général. Les connaissances dont on dispose sur les mécanismes chimiques et biologiques associés à la sensibilisation cutanée sont résumées sous la forme d'une voie toxicologique impliquée dans les effets indésirables (AOP, Adverse Outcome Pathway) (2) allant de l'événement moléculaire initiateur jusqu'aux effets indésirables sur la santé que sont la dermatite de contact allergique chez l'homme ou l'hypersensibilité de contact chez les rongeurs, en passant par les étapes intermédiaires. Dans l'AOP relative à la sensibilisation cutanée, l'événement moléculaire initiateur est l'établissement d'une liaison covalente entre des substances chimiques électrophiles et les centres nucléophiles des protéines de la peau. Traditionnellement, l'évaluation de la sensibilisation cutanée a fait appel à l'expérimentation animale. Les méthodes classiques — test de maximisation chez le cobaye (GPMT) de Magnusson et Kligman et test de Buehler, également chez le cobaye [méthode d'essai B.6 (3)]– portent sur les phases d'induction et d'élicitation de la sensibilisation cutanée. Un essai sur la souris, l'essai de stimulation locale des ganglions lymphatiques (ELGL) [méthode d'essai B.42 (4)], et ses deux variantes n'utilisant pas d'isotopes radioactifs, l'ELGL: DA [méthode d'essai B.50 (5)] et l'ELGL: BrdU-ELISA [méthode d'essai B.51 (6)], tous trois portant sur la réponse à l'induction exclusivement, se sont également imposés, car ils présentent l'avantage, par rapport aux tests sur le cobaye, de préserver davantage le bien-être animal et de fournir une mesure objective de la phase d'induction de la sensibilisation cutanée. Plus récemment, des méthodes d'essai in chemico et in vitro de type mécanistique ont été considérées comme scientifiquement valides pour l'évaluation du danger de sensibilisation cutanée lié aux produits chimiques. Cependant des combinaisons de méthodes de substitution à l'expérimentation animale (méthodes in silico, in chemico, in vitro) seront nécessaires, dans le cadre des approches intégrées en matière d'essais et d'évaluation (IATA, Integrated Approaches to Testing and Assessment), pour qu'il soit possible de remplacer les essais sur l'animal actuellement pratiqués, compte tenu de la restriction de chacune de ces méthodes pour ce qui est de la couverture mécanistique de l'AOP (2) (7). Le DPRA est proposé pour l'étude de l'événement moléculaire initiateur dans l'AOP sensibilisation cutanée, nommément la réactivité protéique, par quantification de la réactivité des produits chimiques testés vis-à-vis de modèles peptidiques de synthèse contenant soit de la lysine, soit de la cystéine (8). Les taux de déplétion de la cystéine et de la lysine sont ensuite utilisés pour classer les substances dans l'une des quatre classes de réactivité, afin d'aider à distinguer les sensibilisants des non-sensibilisants cutanés (9). Le DPRA a fait l'objet d'une étude de validation du Laboratoire de référence de l'Union européenne pour les méthodes de substitution à l'expérimentation animale (EURL ECVAM), suivie d'un examen indépendant par des pairs sous la conduite du Comité scientifique consultatif (ESAC) de ce laboratoire, qui a considéré le DPRA comme scientifiquement valide (10), dans le cadre d'une approche intégrée de type IATA, pour aider à distinguer les sensibilisants des non-sensibilisants cutanés à des fins de classification et d'étiquetage des dangers. On trouvera dans la littérature des exemples d'utilisation de la méthode d'essai DPRA combinée à d'autres sources d'information (11) (12) (13) (14). Les définitions sont données à l'appendice 1. CONSIDÉRATIONS PRÉLIMINAIRES, APPLICABILITÉ ET LIMITES La corrélation entre la réactivité aux protéines et le pouvoir de sensibilisation cutanée est bien établie (15) (16) (17). Cependant, étant donné que la liaison aux protéines n'est qu'un des événements clés de l'AOP sensibilisation cutanée, même s'il s'agit de l'événement moléculaire initiateur, les données relatives à la réactivité aux protéines obtenues par des méthodes expérimentales et non expérimentales ne sont généralement pas suffisantes à elles seules pour conclure à l'absence de pouvoir de sensibilisation cutanée des produits chimiques. Il convient donc de considérer les données issues de l'application de la présente méthode d'essai dans le contexte d'une approche intégrée de type IATA, en combinant ces données à d'autres informations complémentaires provenant par exemple d'essais in vitro portant sur d'autres phases de l'AOP sensibilisation cutanée, ainsi que de méthodes non expérimentales telles que la prévision à partir de données croisées (read-across) relatives à des produits chimiques similaires. La présente méthode d'essai peut apporter une aide, en combinaison avec d'autres sources d'information complémentaires, à l'identification des sensibilisants cutanés (catégorie 1 du SGH/CLP) et des non-sensibilisants, dans le cadre d'une approche intégrée (IATA). Cette méthode d'essai ne peut pas être utilisée seule, ni pour le classement des sensibilisants cutanés dans les sous-catégories 1A et 1B du SGH/CLP, ni pour prédire la puissance de sensibilisation dans le cadre d'évaluations de sécurité. Cependant, en fonction du cadre réglementaire applicable, un résultat positif peut être considéré comme suffisant à lui seul pour classer un produit chimique dans la catégorie 1 du SGH de l'ONU/CLP. La transférabilité de la méthode d'essai DPRA à des laboratoires expérimentés dans le domaine de la chromatographie en phase liquide à haute performance (CLHP) a été démontrée. Le niveau de reproductibilité attendu des prédictions faites par la méthode d'essai sont de l'ordre de 85 % intra-laboratoire et 80 % inter-laboratoires (10). Dans l'ensemble, les résultats issus de l'étude de validation (18) et des études publiées (19) montrent que la précision de la méthode DPRA pour distinguer les sensibilisants (catégorie 1 du SGH/CLP) des non-sensibilisants est de 80 % (N=157), pour une sensibilité de 80 % (88/109) et une spécificité de 77 % (37/48), par comparaison avec les résultats obtenus par l'ELGL (LLNA en anglais). La méthode DPRA est susceptible de sous-prédire des produits chimiques ayant un potentiel de sensibilisation de la peau faible à modéré (c'est-à-dire sous-catégorie 1B du SGH/CLP) par rapport aux produits chimiques ayant un potentiel de sensibilisation de la peau élevé (c'est-à-dire sous-catégorie 1A) du SGH/CLP) (18) (19). Cependant, les valeurs relatives à la précision du DPRA utilisé seul n'ont qu'un caractère indicatif, car cette méthode doit être combinée à d'autres sources d'information dans le contexte d'une démarche IATA, et conformément aux dispositions énoncées au paragraphe 9 ci-dessus. Au demeurant, dans l'évaluation des méthodes d'étude de la sensibilisation cutanée sans expérimentation animale, il convient de tenir compte du fait que l'ELGL et les autres méthodes d'expérimentation animale ne reflètent peut-être pas parfaitement la situation chez l'espèce d'intérêt, à savoir l'être humain. Les données actuellement disponibles montrent que le DPRA est applicable à des produits chimiques couvrant divers groupes fonctionnels organiques, mécanismes de réaction, puissances de sensibilisation cutanée (telles qu'établies par des études in vivo) et propriétés physico-chimiques (8) (9) (10) (19). L'ensemble de ces données montre l'utilité du DPRA comme élément contribuant à l'identification des dangers de sensibilisation cutanée. Dans la présente méthode d'essai, le terme «produit chimique d'essai» désigne ce qui est testé, et ne fait pas référence à l'applicabilité de la méthode DPRA aux substances et/ou aux mélanges. La présente méthode d'essai n'est pas applicable à l'essai des composés métalliques, qui sont connus pour réagir avec les protéines par des mécanismes autres que la liaison covalente. Les produits chimiques d'essai doivent être solubles dans un solvant approprié à une concentration finale de 100 mM (voir paragraphe 18). Toutefois, les produits chimiques qui ne sont pas solubles à cette concentration peuvent être testés à des concentrations plus basses auxquelles ils sont solubles. Dans ce cas, un résultat positif pourra quand même être utilisé pour étayer l'identification du produit chimique d'essai comme sensibilisant cutané, mais aucune conclusion définitive quant à l'absence de réactivité ne devra être tirée d'un résultat négatif. On dispose actuellement d'informations limitées sur l'applicabilité du DPRA à des mélanges de composition connue (18) (19). On considère néanmoins que le DPRA est techniquement applicable aux essais de substances multi-constituants et de mélanges de composition connue (voir paragraphe 18). Toutefois, avant d'appliquer la présente méthode d'essai à un mélange pour obtenir des données à des fins réglementaires, il convient de vérifier si et, dans l'affirmative, pourquoi les résultats peuvent être acceptables dans le cadre réglementaire imposé. Cette vérification n'est pas nécessaire si l'essai du mélange répond à une exigence réglementaire. Le modèle prédictif actuel n'est pas applicable aux mélanges complexes de composition inconnue ni aux substances de composition inconnue ou variable, aux produits de réaction complexes ou aux matériels biologiques (substances UVCB), compte tenu des quotients molaires définis entre le produit chimique d'essai et le peptide. Pour ces produits, il sera nécessaire de mettre au point un nouveau modèle prédictif fondé sur une approche gravimétrique. Dans les cas où l'on peut apporter la preuve que la méthode d'essai ne peut s'appliquer à d'autres catégories spécifiques de produits chimiques, il convient de ne pas utiliser cette méthode pour tester les catégories en question. La présente méthode d'essai est une méthode in chemico, qui ne fait pas intervenir de système métabolique. Elle ne permet pas d'identifier les produits chimiques requérant une bioactivation enzymatique pour exercer leur pouvoir de sensibilisation cutanée (pro-haptènes). Dans certains cas, les produits chimiques qui deviennent sensibilisants après une transformation abiotique (pré-haptènes) apparaissent comme correctement identifiés par la méthode d'essai (18). A la lumière de ce qui précède, les résultats négatifs obtenus par la méthode d'essai devront être interprétés dans le contexte des limites indiquées et en lien avec d'autres sources d'information dans le cadre d'une démarche IATA. Les produits chimiques qui ne se lient pas de façon covalente au peptide mais qui facilitent son oxydation (p.ex. la dimerisation de la cystéine) peuvent amener à une surestimation potentielle de la déplétion en peptide, pouvant mener à un résultat faussement positif et/ou une assignation du produit chimique d'essai à une classe de réactivité supérieure (voir paragraphes 29 et 30). Comme indiqué, la méthode DPRA aide à distinguer les sensibilisants cutanés des non-sensibilisants. Cependant, utilisée dans le cadre d'une approche intégrée de type IATA, elle peut aussi contribuer à l'évaluation de la puissance de sensibilisation (11). Des travaux complémentaires, s'appuyant de préférence sur des données humaines, seront toutefois nécessaires pour déterminer de quelle façon les résultats du DPRA pourraient venir à l'appui de ce type d'évaluation PRINCIPE DE L'ESSAI Le DPRA est une méthode in chemico qui quantifie la concentration résiduelle de peptide contenant de la cystéine ou de la lysine après 24 heures d'incubation avec le produit chimique d'essai à 25 ± 2,5 °C. Les peptides de synthèse contiennent de la phénylalanine destinée à faciliter la détection. La concentration peptidique relative est mesurée par chromatographie en phase liquide à haute performance (CLHP) avec élution par gradient et détection UV à 220 nm. Les pourcentages de déplétion de la cystéine et de la lysine sont ensuite établis par calcul et utilisés dans un modèle prédictif (voir paragraphe 29) qui permet d'assigner le produit chimique d'essai à l'une des quatre classes de réactivité utilisées pour distinguer les sensibilisants des non-sensibilisants. Avant d'utiliser en routine la présente méthode d'essai, les laboratoires devront faire la preuve de leurs compétences techniques, en appliquant la méthode aux dix substances d'épreuve énumérées à l'appendice 2. MODE OPÉRATOIRE La présente méthode d'essai est basée sur le protocole DPRA DB-ALM no 154 (20), qui est celui utilisé pour l'étude de validation coordonnée par l'EURL ECVAM. Il est recommandé d'utiliser ce protocole lors de la mise en œuvre et de l'utilisation de la méthode au laboratoire. On trouvera dans ce qui suit une description des principaux éléments et modes opératoires du DPRA. Dans le cas où un autre système CLHP était utilisé, l'équivalence de ce système par rapport à celui validé dans le protocole DPRA DB-ALM devra être démontrée (p.ex. en testant les substances d'épreuve de compétence citées à l'appendice 2). Préparation des peptides contenant de la cystéine ou de la lysine Des solutions mères de cystéine (Ac-RFAACAA-COOH) et de lysine (Ac-RFAAKAA-COOH) contenant des peptides de synthèse de pureté supérieure à 85 %, et de préférence de l'ordre de 90-95 %, doivent être fraîchement préparées avant d'être incubées avec le produit chimique d'essai. La concentration finale du peptide à cystéine doit être de 0,667 mM dans un tampon phosphate de pH 7,5, la concentration finale du peptide à lysine de 0,667 mM dans un tampon acétate d'ammonium de pH 10,2. Les séquences d'analyse CLHP doivent être programmées avec pour objectif que le temps de l'analyse CLHP ne dépasse pas 30 heures. Dans le cas du système CLHP utilisé lors de l'étude de validation et décrit dans la présente méthode d'essai, jusqu'à 26 échantillons (comprenant le produit chimique d'essai, le témoin positif et un nombre approprié de solvants témoins, selon le nombre de solvants utilisés dans l'essai, chaque échantillon étant testé en triplicat) peuvent être analysés dans le même passage dans le système CLHP. Tous les réplicats analysés en un même passage doivent utiliser les mêmes solutions mères de peptide à cystéine et à lysine. Il est recommandé d'avoir la preuve de la solubilité des lots individuels de peptides avant leur utilisation. Préparation du produit chimique d'essai La solubilité du produit chimique d'essai dans un solvant approprié sera évaluée avant la conduite de l'essai, conformément au mode opératoire décrit dans le protocole DPRA DB-ALM pour la solubilisation (20). Un solvant approprié est un solvant qui dissout complètement le produit chimique d'essai. Dans le DPRA, le produit chimique d'essai est incubé en large excès avec les peptides à cystéine ou à lysine; une inspection visuelle de la solution formée est donc considérée comme suffisante pour vérifier que le produit chimique d'essai (et tous ses composants, dans le cas d'essais portant sur une substance multi-constituants ou un mélange) est bien dissous. Les solvants appropriés sont l'acétonitrile, l'eau, un mélange 1:1 eau:acétonitrile, l'isopropanol, l'acétone ou un mélange 1:1 acétone:acétonitrile. D'autres solvants peuvent être utilisés à condition qu'ils n'aient pas d'incidence sur la stabilité du peptide, d'après les essais réalisés sur des témoins de référence C (échantillons constitués du peptide seul dissous dans le solvant approprié; voir appendice 3). Une dernière option, si le produit chimique d'essai n'est soluble dans aucun de ces solvants, consiste à tenter de le solubiliser dans 300 μL de DMSO et de diluer la solution ainsi obtenue avec 2 700 μl d'acétonitrile; si le produit chimique d'essai n'est pas soluble dans ce mélange, on tentera de solubiliser la même quantité de produit chimique d'essai dans 1 500 μl de DMSO et de diluer la solution obtenue avec 1 500 μl d'acétonitrile. Le produit chimique d'essai doit être pré-pesé dans des flacons en verre, et dissous immédiatement avant l'essai dans un solvant approprié pour préparer une solution à 100 mM. Pour les mélanges et les substances multi-constituants de composition connue, un seul degré de pureté est établi d'après la somme des pourcentages relatifs de leurs constituants (à l'exception de l'eau) et une seule masse moléculaire apparente est établie à partir de la masse moléculaire de chacun des constituants du mélange (à l'exception de l'eau) et de leurs proportions respectives. La pureté et la masse moléculaire apparente obtenues sont alors utilisées pour calculer le poids de produit chimique d'essai nécessaire pour préparer une solution à 100 mM. Dans le cas des polymères pour lesquels il n'est pas possible d'établir une masse moléculaire prédominante, la masse moléculaire du monomère (ou la masse moléculaire apparente des divers monomères constituant le polymère) pourra être prise en compte pour préparer une solution à 100 mM. Cependant, lors de tests sur les mélanges, les substances multi-constituants ou les polymers de compositions connues, on devra envisager de tester aussi le produit chimique pur. Pour les liquides, le produit chimique pur devra être testé tel quel sans dilution préalable, en l'incubant dans des proportions de 1:10 et 1:50 (rapport molaire) avec les peptides lysine et cystéine, respectivement. Pour les solides, le produit chimique devra être dissous à sa concentration maximale de solubilité dans le même solvant utilisé pour la préparation de la solution à 10mM apparents. Le produit chimique devra alors être testé ainsi sans aucune autre dilution en l'incubant dans des proportions de 1:10 et 1: 50 avec les peptides lysine et cystéine, respectivement. Des résultats concordants (réactif ou non réactif) entre la solution à 100 mM apparents et le produit chimique pur devraient permettre de formuler une conclusion définitive. Préparation du témoin positif, des témoins de référence et des témoins de co-élution L'aldéhyde cinnamique (noCAS 104-55-2; pureté grade alimentaire ≥ 95 %) est utilisé comme témoin positif (TP) à une concentration de 100 mM dans l'acétonitrile. D'autres témoins positifs adaptés, donnant de préférence des valeurs de déplétion situées dans une fourchette moyenne, peuvent être utilisés si des données historiques sont disponibles pour dériver des critères d'acceptabilité comparables pour la séquence. En outre, des témoins de référence (échantillons contenant uniquement le peptide dissous dans le solvant approprié) doivent aussi être inclus dans la séquence d'analyse CLHP; ils permettent de s'assurer avant l'analyse que le système CLHP est adapté à l'usage prévu (témoins de référence A), de vérifier la stabilité des témoins de référence dans le temps (témoins de référence B) et de vérifier que le solvant utilisé pour dissoudre le produit chimique d'essai n'a pas d'incidence sur le pourcentage de déplétion des peptides (témoins de référence C) (voir appendice 3). Un témoin de référence approprié est utilisé pour chaque produit chimique afin de calculer le pourcentage de déplétion des peptides lié à cette substance (voir paragraphe 26). De plus, pour chaque produit chimique analysé, un témoin de co-élution constitué du seul produit chimique sera inclus dans la séquence afin de détecter une éventuelle co-élution du produit chimique d'essai avec le peptide à lysine ou à cystéine. Incubation du produit chimique d'essai avec les solutions de peptide à cystéine et à lysine Les solutions de peptide à cystéine et à lysine sont mises en incubation avec le produit chimique d'essai dans des flacons en verre pour auto-échantillonneur, dans des proportions de 1:10 et 1:50 respectivement. Si un précipité est observé dès l'ajout de la solution de produit chimique d'essai à la solution de peptide, du fait d'une faible hydrosolubilité du produit chimique d'essai, on ne peut savoir avec certitude quelle est la quantité de produit chimique d'essai restant dans la solution et susceptible de réagir avec le peptide. Dans ce cas, un résultat positif pourra être utilisé, mais un résultat négatif présentera une incertitude et devra être interprété avec prudence (voir également les provisions citées au paragraphe 11 dans le cadre d'essai avec des produits chimiques non solubles à une concentration finale de 100 mM). La solution de réaction doit être maintenue dans l'obscurité à 25 ± 2,5°C pendant 24 ± 2 heures avant l'analyse CLHP. Chaque produit chimique d'essai doit être analysé en triplicat pour les deux peptides. Une inspection visuelle des échantillons est nécessaire avant l'analyse CLHP. Si un précipité ou une séparation de phases sont observés, il est prudent de centrifuger les échantillons à vitesse modérée (100-400xg) afin d'entraîner le précipité au fond du flacon, un excès de précipité risquant de colmater les tubes ou les colonnes du chromatographe. Dans le cas où une précipitation ou une séparation de phases sont observées à la fin de la période d'incubation, la déplétion des peptides peut être sous-estimée et un résultat négatif ne permettra pas de conclure avec une certitude suffisante à l'absence de réactivité. Préparation de la courbe d'étalonnage CLHP Une courbe d'étalonnage doit être établie pour le peptide à cystéine et le peptide à lysine. Les étalons de peptides sont préparés dans une solution à 20 % ou 25 % acétonitrile:tampon, avec un tampon phosphate (pH 7,5) pour le peptide à cystéine et un tampon acétate d'ammonium (pH 10,2) pour le peptide à lysine. À partir d'étalons obtenus par dilution en série de la solution mère de peptide (0.667 mM), 6 solutions d'étalonnage sont préparées entre 0,34 et 0,0167 mM. Un blanc constitué du tampon de dilution doit être inclus dans la courbe d'étalonnage. Une courbe d'étalonnage adaptée aura un coefficient r2>0,99. Préparation et analyse CLHP Il convient de vérifier avant la conduite de l'analyse si le système est adapté à l'usage prévu. La déplétion de peptides est mesurée par CLHP couplée à un détecteur UV (détecteur à barrette de photodiodes ou détecteur à absorbance UV de longueur d'onde fixe égale à 220 nm). La colonne appropriée est installée dans le système CLHP. L'installation CLHP décrite dans le protocole validé utilise de préférence une colonne Zorbax SB-C-18 2,1 mm x 100 mm x 3,5 microns. Avec cette colonne CLHP à phase inverse, l'ensemble du système doit être équilibré à 30 °C avec 50 % de phase A (0.1 % (v/v) d'acide trifluoroacétique dans l'eau) et 50 % de phase B (0,085 % (v/v) d'acide trifluoroacétique dans l'acétonitrile) pendant 2 heures au moins avant la conduite de l'analyse. L'analyse CLHP doit être réalisée à un débit de 0,35 ml/min pour un gradient linéaire allant de 10 % à 25 % d'acétonitrile pendant 10 minutes, suivi d'une augmentation rapide jusqu'à 90 % d'acétonitrile pour éliminer les autres matériaux. Des volumes égaux de chaque étalon, échantillon et témoin doivent être injectés. La colonne doit être ré-équilibrée aux conditions initiales pendant 7 minutes entre les injections. Si une colonne CLHP à phase inverse d'un modèle différent est utilisée, il peut être nécessaire d'ajuster les paramètres ci-dessus pour assurer une élution et une intégration appropriées des peptides à cystéine et à lysine, en particulier le volume injecté, qui peut varier selon le système utilisé (habituellement entre 3 et 10 μl). Il est essentiel, si des paramètres CLHP différents sont utilisés, de démontrer qu'ils sont équivalents au paramétrage validé décrit ci-dessus (par des essais sur les substances d'épreuve de compétence de l'appendice 2, par exemple). L'absorbance est mesurée à 220 nm. Si un détecteur à barrette de photodiodes est utilisé, l'absorbance à 258 nm doit également être relevée. On notera que certains lots d'acétonitrile peuvent avoir une incidence négative sur la stabilité des peptides, qui doit être évaluée lorsqu'un nouveau lot d'acétonitrile est utilisé. Le rapport entre la surface du pic à 220 nm et la surface du pic à 258 nm peut servir d'indicateur de co-élution. Pour chaque échantillon, un rapport compris entre 90 et 100 % de la moyenne (19) des rapports des surfaces obtenue pour les échantillons témoins constitue un bon indicateur de l'absence de co-élution. Certains produits chimiques d'essai peuvent promouvoir l'oxydation du peptide à cystéine. Le pic du peptide à cystéine dimérisé peut être observé visuellement. S'il apparaît qu'une dimérisation s'est produite, il convient de le noter, car le pourcentage de déplétion du peptide peut être surestimé, amenant à une prédiction faussement positive et/ou une assignation à une classe de réactivité supérieure (voir paragraphes 29 et 30). L'analyse CLHP des peptides à cystéine et des peptides à lysine peut être réalisée le même jour, si l'on dispose de deux systèmes CLHP. Si les deux analyses ne sont pas réalisées le même jour, toutes les solutions de produit chimique d'essai doivent être chaque fois fraîchement préparées pour l'essai. L'analyse doit être programmée dans le temps de telle sorte que l'injection du premier échantillon commence 22 à 26 heures après le mélange du produit chimique d'essai avec la solution de peptide. Les séquences d'analyse CLHP doivent être programmées dans le souci de maintenir le temps d'analyse en-deçà de 30 heures. Dans le cas du système CLHP utilisé dans l'étude de validation et décrit dans la présente méthode d'essai, un maximum de 26 échantillons peuvent être analysés lors d'un même passage dans le système CLHP (voir aussi paragraphe 17). On trouvera à l'appendice 3 un exemple de séquence d'analyse CLHP. RÉSULTATS ET RAPPORT Évaluation des données On établit de manière photométrique à 220 nm pour chaque échantillon, la concentration de peptide à lysine ou à cystéine, en mesurant la surface des pics appropriés par intégration des pics (tout ce qui se situe sous la courbe) et en calculant la concentration peptidique d'après la courbe d'étalonnage linéaire établie à partir des étalons. On établit pour chaque échantillon le pourcentage de déplétion des peptides en mesurant la surface des pics et en la divisant par la surface moyenne des pics des témoins de référence C correspondants (voir appendice 3) selon la formule ci-après.
Critères d'acceptabilité Les critères suivants doivent être remplis pour qu'un essai soit considéré comme valide:
Si un seul ou plusieurs de ces critères ne sont pas remplis, l'essai doit être répété. Les critères suivants doivent être remplis pour que les résultats relatifs à un produit chimique d'essai soient considérés comme valides:
Modèle prédictif Le pourcentage moyen de déplétion de la cystéine et de la lysine est calculé pour chaque produit chimique d'essai. Une déplétion négative est considérée comme égale à 0 pour le calcul de la moyenne. Si l'on utilise le modèle prédictif cystéine 1:10/lysine 1:50 du tableau 1, c'est le seuil de 6,38 % de déplétion peptidique moyenne qui sera pris en compte pour l'identification des sensibilisants et des non-sensibilisants cutanés dans le cadre d'une approche IATA. L'application du modèle prédictif pour assigner un produit chimique d'essai à une classe de réactivité (réactivité faible, modérée ou forte) permettra peut-être d'obtenir des informations utiles pour évaluer la puissance de sensibilisation de ce produit chimique dans le cadre d'une approche de type IATA. Tableau 1 Modèle prédictif (20) cystéine 1:10/ lysine 1:50
Il peut arriver que le produit chimique d'essai (la substance, ou l'un ou plusieurs des composants d'une substance multi-constituants ou d'un mélange) donne lieu à une absorption significative à 220 nm et ait le même temps de rétention que le peptide (co-élution). La co-élution peut être résolues par un léger ajustement du système d'analyse CLHP afin de mieux séparer les temps de rétention entre le produit chimique d'essai et le peptide. Si un autre système d'analyse CLHP est employé dans l'objectif de résoudre cette problématique de co-élution, son équivalence au sytème CLHP validé dans la présente ligne directrice devra être démontrée (p.ex. au moyen des substances d'épreuve de compétence citées à l'appendice 2) Dans les cas de co-élution, le pic du peptide ne peut pas être intégré, et il n'est pas possible de calculer le pourcentage de déplétion des peptides. Si la co-élution pour ce produit chimique d'essai concerne à la fois les peptides à cystéine et à lysine, l'analyse est considérée comme «non concluante». Si la co-élution n'intervient que pour le peptide à lysine, le modèle prédictif du tableau 2 pour la cystéine 1:10 est applicable. Tableau 2 Modèle prédictif (22) cystéine 1:10
Il peut également y avoir des cas où les temps de rétention du produit chimique d'essai et de l'un des peptides ne se recouvrent pas complètement. Les pourcentages de déplétion peuvent alors être estimés et utilisés dans le modèle prédictif cystéine 1:10/lysine 1:50, sans qu'il soit possible de classer avec précision le produit chimique d'essai dans l'une des classes de réactivité. Pour un produit chimique d'essai, une seule analyse CLHP pour le peptide à cystéine et le peptide à lysine devrait suffire si le résultat est sans équivoque. Cependant, lorsque les données disponibles pour déterminer si un résultat limite est positif ou négatif sont proches du seuil, des tests complémentaires peuvent être nécessaires. Si le pourcentage moyen de déplétion se situe dans l'intervalle 3 % à 10 % pour la cystéine 1:10/lysine 1:50, ou si le pourcentage de déplétion de la cystéine se situe dans l'intervalle 9 % à 17 % pour la cystéine 1:10, une deuxième analyse doit être envisagée, voire une troisième en cas de résultats discordants entre les deux premières. Rapport d'essai Le rapport d'essai doit comporter les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DEFINITIONS Chemical : A substance or a mixture. Adéquation du système : Détermination des performances des instruments (sensibilité, par exemple) par l'analyse d'un étalon de référence préalablement à la mise à l'épreuve du lot analytique (22). AOP ( Adverse Outcome Pathway, voie toxicologique impliquée dans les effets indésirables) : séquence d'événements conduisant à la survenue d'un effet indésirable in vivo, à partir de la structure chimique d'un produit chimique cible ou d'un groupe de produits chimiques similaires et de l'événement initiateur au niveau moléculaire (2). Coefficient de variation : la mesure de la variabilité calculée pour un groupe de données issues de réplicats en divisant la déviation standard par la moyenne. Cette mesure peut être multipliée par 100 pour obtenir un pourcentage. Courbe d'étalonnage : Relation entre la réponse expérimentale et la concentration analytique (aussi appelée courbe étalon) d'une substance d'essai connue. Danger : Propriété inhérente d'un agent ou d'une situation susceptible de provoquer des effets indésirables lorsqu'un organisme, un système ou une (sous-) population est exposé(e) à cet agent. Événement moléculaire initiateur : Perturbation d'un système biologique induite par un produit chimique au niveau moléculaire, identifiée comme le point de départ de la voie impliquée dans un effet indésirable (AOP). Fiabilité : Indique dans quelle mesure la mise en œuvre d'une méthode d'essai peut être reproduite au cours du temps par un même laboratoire ou plusieurs laboratoires utilisant le même mode opératoire. Pour l'évaluer, on calcule la reproductibilité intra-laboratoire et inter-laboratoires et la répétabilité intra-laboratoire (21). IATA ( Integrated Approaches to Testing and Assessment , approches intégrées en matière d'essais et d'évaluation) : Approche structurée utilisée dans l'identification du danger (potentiel), la caractérisation du danger (puissance) et/ou dans l'évaluation'de la sécurité (potentiel de danger/puissance du danger et exposition) d'un produit chimique ou d'un groupe de produits chimiques, qui intègre de façon stratégique et pondérée toutes les données pertinentes dans le but d'informer une décision réglementaire sur le danger potentiel et/ou le risque et/ou le besoin d'effectuer d'autres tests ciblés. Mélange : Mélange ou solution constitué(e) de deux substances ou plus qui ne réagissent pas entre elles (1). Méthode d'essai validée : Méthode d'essai ayant fait l'objet d'études de validation visant à déterminer sa pertinence (notamment sa précision) et sa fiabilité à des fins spécifiques. Il importe de noter que les performances d'une méthode d'essai validée peuvent être insuffisantes en termes de précision et de fiabilité pour qu'elle puisse être jugée acceptable pour l'objectif envisagé (21). Pertinence : Décrit la relation entre l'essai et l'effet étudié, et rend compte de l'adéquation de l'essai et de son utilité à des fins spécifiques. La pertinence indique dans quelle mesure l'essai mesure ou prédit correctement l'effet biologique d'intérêt. Elle tient compte de la précision (concordance) de la méthode d'essai (21). Précision : Étroitesse de l'accord entre les résultats de la méthode d'essai et les valeurs de référence acceptées. Elle constitue une mesure de performance de la méthode d'essai et l'un des aspects de sa «pertinence». Ce terme est souvent utilisé au sens de «concordance», pour qualifier la proportion de résultats corrects d'une méthode d'essai (21). Produit chimique : une substance ou un mélange Produit chimique d'essai : Le terme «produit chimique d'essai» désigne ce qui est testé. Reproductibilité : Accord entre les résultats obtenus lors d'essais pratiqués sur le même produit chimique selon le même mode opératoire (voir Fiabilité) (21). Sensibilité : Proportion des produits chimiques positifs/actifs qui sont correctement classés par la méthode d'essai. Elle permet de mesurer la précision d'une méthode d'essai produisant des données catégorielles, et constitue un aspect important de l'évaluation de la pertinence d'une méthode d'essai (21). SGH (Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations Unies) : Système proposant la classification des produits chimiques (substances et mélanges) conformément à des types et des niveaux normalisés de dangers physiques, sanitaires et environnementaux, ainsi que la communication des éléments d'information correspondants, notamment par des pictogrammes, mentions d'avertissement, mentions de danger, conseils de prudence et fiches de données de sécurité, afin de diffuser des informations sur leurs effets indésirables dans l'objectif de protéger les personnes (en particulier les employeurs, employés, transporteurs, consommateurs et personnels des services d'urgence) et l'environnement (1). Spécificité : Proportion des produits chimiques négatifs/inactifs qui sont classés correctement par la méthode d'essai. Elle permet de mesurer la précision d'une méthode d'essai produisant des données catégorielles, et constitue un aspect important de l'évaluation de la pertinence d'une méthode d'essai (21). Substance : Élément chimique et ses composés, présents à l'état naturel ou obtenus grâce à un procédé de production. Ce terme inclut tout additif nécessaire pour préserver la stabilité du produit ainsi que toute impureté produite par le procédé utilisé, mais exclut tout solvant pouvant être extrait sans affecter la stabilité ni modifier la composition de la substance (1). Substance mono-constituant : Substance, définie par sa composition quantitative, dans laquelle un constituant principal est présent à une concentration d'au moins 80 % (P/P). Substance multi-constituants : Substance, définie par sa composition quantitative, dans laquelle plus d'un constituant principal est présent à une concentration comprise entre 10 % et 80 % (P/P). Une substance multi-constituants résulte d'un processus de fabrication. La différence entre un mélange et une substance multi-constituants est que le mélange est obtenu en associant deux substances ou plus sans réaction chimique. Une substance multi-constituants résulte d'une réaction chimique. Témoin de référence : Échantillon non traité contenant tous les composants d'un système d'essai, y compris le solvant ou le véhicule utilisé pour préparer les échantillons traités ou non avec le produit chimique d'essai, et servant à déterminer une réponse de référence pour les échantillons traités avec le produit chimique d'essai dissous dans le même solvant ou véhicule. Testé avec un témoin négatif concomitant, cet échantillon indique également si le solvant ou véhicule interagit avec le système d'essai. Témoin positif : Réplicat contenant tous les composants d'un système d'essai, traité avec une substance connue pour induire une réponse positive. Pour qu'il soit possible d'évaluer la variabilité dans le temps de la réponse du témoin positif, l'intensité maximale de celle-ci ne doit pas être excessive. UVCB : Substances de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques. Appendice 2 SUBSTANCES D'ÉPREUVE DE COMPÉTENCE Sensibilisation cutanée in chemico: essai de réactivité peptidique direct (DPRA) Avant d'utiliser en routine la méthode d'essai décrite dans la présente Ligne directrice, les laboratoires doivent démontrer leurs compétences techniques en obtenant les prédictions attendues par la méthode DPRA pour les 10 substances d'épreuve de compétence recommandées au tableau 1, et en obtenant des valeurs de déplétion de la cystéine et de la lysine se situant dans les domaines de référence respectifs de ces deux grandeurs pour au moins 8 des 10 substances d'épreuve de compétence. Ces substances ont été sélectionnées de façon à représenter la gamme des réponses possibles en ce qui concerne les dangers de sensibilisation cutanée. Les autres critères de sélection sont la disponibilité des substances dans le commerce, la qualité élevée des données de référence in vivo disponibles et des données in vitro obtenues par le DPRA, ainsi que l'utilisation de ces substances dans l'étude de validation coordonnée par l'EURL ECVAM pour démontrer que la méthode d'essai a été mise en œuvre avec succès dans les laboratoires participant à l'étude. Tableau 1 Substances d'épreuve recommandées pour démontrer les compétences techniques des laboratoires relatives à l'essai réactivité peptidique direct (DPRA)
Appendice 3 EXEMPLE DE SÉQUENCE D'ANALYSE
B.60 Sensibilisation cutanée in vitro: méthode d'essai ARE-Nrf2 luciférase INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 442D (2015) de l'OCDE. Un sensibilisant cutané est une substance qui provoque une réponse allergique suite à un contact avec la peau, selon la définition du Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations Unies (SGH) (1) et du règlement (CE) no 1272/2008 de l'Union europénne relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges (CLP) (27). La présente méthode d'essai décrit une procédure in vitro (essai ARE-Nrf2 luciférase) qui doit aider à distinguer les sensibilisants des non-sensibilisants cutanés, selon le SGH (1) et le CLP. Les principales phases du processus biologique de sensibilisation cutanée font l'objet d'un consensus général. Les connaissances dont on dispose sur les mécanismes chimiques et biologiques associés à la sensibilisation sont résumées sous la forme d'une voie toxicologique impliquée dans les effets indésirables (AOP, Adverse Outcome Pathway) (2) allant de l'événement moléculaire initiateur jusqu'aux effets indésirables sur la santé que sont la dermatite de contact allergique chez l'homme ou l'hypersensibilité de contact chez les rongeurs (2) (3), en passant par les étapes intermédiaires. L'événement moléculaire initiateur est l'établissement d'une liaison covalente entre des substances chimiques électrophiles et les centres nucléophiles des protéines de la peau. Le deuxième événement clé sur cet AOP se déroule dans les kératinocytes et comprend des réponses inflammatoires et des phénomènes d'expression génique, liés à des voies de signalisation cellulaire spécifiques telles que les voies dépendant de l'élément de réponse antioxydant/électrophile (ARE, Antioxidant Response Element). Le troisième événement clé est l'activation des cellules dendritiques, habituellement évaluée d'après l'expression de marqueurs de surface spécifiques de la cellule, les chimiokines et les cytokines. Le quatrième événement clé est la prolifération des lymphocytes T, évaluée indirectement par l'essai de stimulation locale des ganglions lymphatiques (ELGL) chez la souris (4). Traditionnellement, l'évaluation de la sensibilisation cutanée a fait appel à l'expérimentation animale. Les méthodes classiques — test de maximisation chez le cobaye (GPMT) de Magnusson et Kligman et test de Buehler, également chez le cobaye [méthode d'essai B.6 (5)] — portent sur les phases d'induction et d'élicitation de la sensibilisation cutanée. Un essai sur la souris, l'ELGL [méthode d'essai B.42 (4)], et ses deux variantes n'utilisant pas d'isotopes radioactifs, l'ELGL: DA (méthode d'essai B.50 (6)] et l'ELGL: BrdU-ELISA [méthode d'essai B.51 (7)], tous trois portant exclusivement sur la réponse à l'induction, se sont également imposés, car ils présentent l'avantage, par rapport aux tests sur le cobaye, de préserver davantage le bien-être animal et de fournir une mesure objective de la phase d'induction de la sensibilisation cutanée. Plus récemment, des méthodes d'essai in chemico et in vitro de type mécanistique ont été considérées comme scientifiquement valides pour l'évaluation du danger de sensibilisation cutanée lié aux produits chimiques. Cependant des combinaisons de méthodes de substitution à l'expérimentation animale (méthodes in silico, in chemico, in vitro) seront nécessaires, dans le cadre des approches intégrées en matière d'essais et d'évaluation (IATA, Integrated Approaches to Testing and Assessment), pour qu'il soit possible de remplacer complètement les essais sur l'animal actuellement pratiqués, compte tenu de la restriction de chacune de ces méthodes pour ce qui est de la couverture mécanistique de l'AOP (2) (3). La présente méthode d'essai (essai ARE-Nrf2 luciférase) est proposée pour l'étude de la deuxième étape décrite au paragraphe 2. Les sensibilisants cutanés ont été signalés comme provoquant l'induction de gènes qui sont régulés par l'élément de réponse antioxydant (ARE) (8) (9). Des substances électrophiles de petite taille telles que les sensibilisants cutanés peuvent agir sur la protéine détectrice Keap1 (Kelch-like ECH-associated protein 1), par exemple par une modification covalente de son résidu cystéine l'amenant à se dissocier du facteur de transcription Nrf2 (nuclear factor-erythroid 2-related factor 2). Le facteur Nrf2 dissocié peut alors activer les gènes dépendants de l'ARE, tels que ceux qui codent les enzymes détoxifiantes de phase II (8) (10) (11). A l'heure actuelle, le seul essai ARE-Nrf2 luciférase in vitro couvert par la présente méthode d'essai est l'essai KeratinoSensTM, qui a donné lieu à des études de validation (9) (12) (13) suivies d'un examen indépendant par des pairs, conduit par le Laboratoire de référence de l'Union européenne pour les méthodes de substitution à l'expérimentation animale (EURL ECVAM) (14). L'essai KeratinoSensTM a été considéré comme scientifiquement valide, dans le cadre d'une approche intégrée de type IATA, pour aider à distinguer les sensibilisants des non-sensibilisants cutanés à des fins de classification et d'étiquetage des dangers (14). Les laboratoires souhaitant appliquer cette méthode d'essai peuvent obtenir la lignée cellulaire recombinante utilisée dans l'essai KeratinoSensTM en établissant un accord de licence avec le développeur de la méthode (15). Les définitions sont données à l'appendice 1. CONSIDÉRATIONS PRÉLIMINAIRES, APPLICABILITÉ ET LIMITES L'activation de la voie Keap1-Nrf2-ARE ne concernant que la deuxième étape de l'AOP relative à la sensibilisation cutanée, les informations obtenues à l'aide de méthodes d'essai fondées sur l'activation de cette voie ne sont peut-être pas suffisantes à elles seules, pour établir le potentiel de sensibilisation cutanée de produits chimiques. Il convient donc de considérer les données issues de l'application de la présente méthode d'essai dans le contexte d'une approche intégrée de type IATA, en combinant ces données à d'autres informations complémentaires provenant par exemple d'essais in vitro portant sur d'autres étapes-clé de l'AOP sensibilisation cutanée, ainsi que de méthodes non expérimentales telles que la prévision à partir de données croisées (read-across) relatives à des produits chimiques similaires. On trouvera dans la littérature des exemples d'utilisation de la méthode d'essai ARE-Nrf2 luciférase combinée à d'autres sources d'information (13) (16) (17) (18) (19). La présente méthode d'essai peut apporter une aide à l'identification des sensibilisants cutanés (catégorie 1 du SGH/CLP) et des non-sensibilisants, dans le cadre d'une approche intégrée (IATA). Cette méthode d'essai ne peut pas être utilisée seule, ni pour le classement des sensibilisants cutanés dans les sous-catégories 1A et 1B du SGH/CLP optionnelles, ni pour prédire la puissance de sensibilisation dans le cadre d'évaluations de sécurité. Cependant, en fonction du cadre réglementaire applicable, un résultat positif peut être considéré comme suffisant à lui seul pour classer un produit chimique dans la catégorie 1 du SGH/CLP. D'après l'ensemble des données issues de l'étude de validation et des tests pratiqués en interne lors de l'examen indépendant de la méthode d'essai par des pairs, la méthode KeratinoSensTM est transférable à des laboratoires expérimentés dans le domaine des cultures cellulaires. Le niveau de reproductibilité attendu des prédictions est de l'ordre de 85 % intra- et inter-laboratoires (14). Par ailleurs, l'exactitude (77 % -155/201), la sensibilité (78 % — 71/91) et la spécificité (76 % — 84/110) des prédictions obtenues par l'essai KeratinoSensTM pour déterminer les sensibilisants (catégorie 1 du SGH/CLP) des non-sensibilisants, comparées aux résultats de l'ELGL, ont été calculées en prenant en compte toutes les données soumises à l'EURL ECVAM pour l'évaluation et l'examen par des pairs (14). Ces chiffres sont similaires à ceux récemment publiés à l'issue d'essais en interne portant sur quelque 145 substances chimiques (précision 77 %, sensibilité 79 %, spécificité 72 %) (13). L'essai KeratinoSensTM est susceptible de sous-prédire des produits chimiques ayant un potentiel de sensibilisation de la peau faible à modéré (c'est-à-dire sous-catégorie 1B du SGH/CLP) par rapport aux produits chimiques ayant un potentiel de sensibilisation de la peau élevé (c'est-à-dire sous-catégorie 1A) du SGH/CLP (13) (14). L'ensemble de ces données montre l'utilité de l'essai KeratinoSensTM comme élément contribuant à l'identification des dangers de sensibilisation cutanée. Cependant, les valeurs relatives à la précision de la méthode reposant uniquement sur l'essai KeratinoSensTM n'ont qu'un caractère indicatif, car cette méthode doit être combinée à d'autres sources d'information dans le contexte d'une démarche IATA, et conformément aux dispositions énoncées au paragraphe 9 ci-dessus. Au demeurant, dans l'évaluation des méthodes d'étude de la sensibilisation cutanée sans expérimentation animale, il convient de tenir compte du fait que l'ELGL et les autres méthodes d'expérimentation animale ne reflètent peut-être pas parfaitement la situation chez l'espèce d'intérêt, à savoir l'être humain. Dans la présente méthode d'essai, le terme «produit chimique d'essai» désigne ce qui est testé, et ne fait pas référence à l'applicabilité de la méthode d'essai ARE-Nrf2 luciférase aux substances et/ou aux mélanges. Les données actuellement disponibles montrent que l'essai KeratinoSensTM est applicable à des produits chimiques couvrant divers groupes fonctionnels organiques, mécanismes de réaction, puissances de sensibilisation cutanée (telles qu'établies par des études in vivo) et propriétés physico-chimiques (9) (12) (13) (14). Ce sont principalement des substances mono-constituant qui ont été testées, même si l'on dispose également d'un nombre limité de données relatives à l'essai de mélanges (20). La méthode est néanmoins techniquement applicable aux essais de substances multi-constituants et de mélanges. Toutefois, avant d'appliquer la présente méthode d'essai à un mélange pour obtenir des données à des fins réglementaires, il convient de vérifier si et, dans l'affirmative, pourquoi les résultats peuvent être acceptables dans le cadre réglementaire imposé. Cette vérification n'est pas nécessaire si l'essai du mélange répond à une exigence réglementaire. De plus, en cas d'essai portant sur des substances multi-constituants ou des mélanges, il convient de s'interroger sur l'interférence possible de constituants cytotoxiques avec les réponses observées. La méthode d'essai est applicable aux produits chimiques solubles ou formant une dispersion stable (colloïde ou suspension dans laquelle le produit chimique d'essai ne se dépose pas et ne se sépare pas du solvant en formant plusieurs phases) dans l'eau ou dans le DMSO (cela vaut pour tous les constituants du produit chimique d'essai si l'essai porte sur une substance multi-constituants ou un mélange). Les produits chimiques d'essai qui ne remplissent pas ces conditions à la concentration finale maximale requise de 2 000 μM (cf. paragraphe 22) peuvent néanmoins être testés à des concentrations plus faibles. Dans ce cas, les résultats remplissant les critères de positivité décrits au paragraphe 39 peuvent quand même être utilisés pour étayer l'identification du produit chimique d'essai comme sensibilisant cutané, tandis qu'un résultat négatif obtenu pour des concentrations < 1 000 μM sera considéré comme non concluant (voir le modèle prédictif au paragraphe 39). D'une manière générale, les substances ayant un LogP calculé inférieur ou égal à 5 ont été testés avec succès, alors que les substances extrêmement hydrophobes dont le LogP calculé est supérieur à 7 se situent en dehors des limites connues d'applicabilité de la méthode d'essai (14). Pour les substances ayant un LogP calculé compris entre 5 et 7, les informations disponibles sont limitées. Les résultats négatifs sont à interpréter avec prudence, car les substances réactives exclusivement vis-à-vis des résidus lysine peuvent être identifiées comme négatives par la méthode d'essai. De plus, en raison des capacités métaboliques limitées de la lignée cellulaire utilisée (21), ainsi que des conditions expérimentales, les pro-haptènes (produits chimiques nécessitant une activation enzymatique, par exemple via des enzymes P450) et les pré-haptènes (produits chimiques activés par auto-oxydation), en particulier ceux dont l'oxydation est lente, peuvent eux aussi donner des résultats négatifs. A l'inverse, les produits chimiques d'essai qui n'agissent pas comme des sensibilisants, mais sont néanmoins des facteurs de stress chimique, peuvent donner lieu à des faux positifs (14). De plus, lorsque les produits chimiques d'essai sont fortement cytotoxiques, ils ne peuvent pas toujours être évalués de façon fiable. Enfin, les produits chimiques d'essai qui interfèrent avec la luciférase peuvent créer un risque de confusion avec l'activité de la luciférase, dans les essais au niveau cellulaire, en provoquant soit une inhibition apparente soit une luminescence accrue (22). Ainsi, il a été signalé que des concentrations de phyto-œstrogènes supérieures à 1 μM interféraient avec les signaux de luminescence, dans d'autres essais avec gènes rapporteurs faisant intervenir la luciférase, du fait d'une suractivation du gène rapporteur de la luciférase (23). Il convient donc d'examiner attentivement l'expression de la luciférase obtenue à des concentrations élevées de phyto-œstrogènes ou de produits chimiques similaires suspectés d'induire une suractivation du gène rapporteur de la luciférase comparable à celle résultant des phyto-œstrogènes (23). Dans les cas où l'on peut apporter la preuve que la méthode d'essai ne peut s'appliquer à d'autres catégories spécifiques de produits chimiques d'essai, il convient de ne pas utiliser cette méthode pour tester les catégories en question. Outre qu'elle aide à distinguer les sensibilisants cutanés et des non-sensibilisants, la méthode KeratinoSensTM fournit sur la relation concentration-réponse des informations susceptibles de contribuer à l'évaluation de la puissance de sensibilisation, dans le cadre d'une approche intégrée IATA (19). Cependant, une étude complémentaire s'appuyant de préférence sur des données humaines fiables sera nécessaire pour déterminer de quelle façon les résultats de l'essai KeratinoSensTM peuvent contribuer à l'évaluation de la puissance (24) et à la sous-catégorisation des sensibilisants selon le SGH/CLP. PRINCIPE DE L'ESSAI La méthode d'essai ARE-Nrf2 luciférase utilise une lignée de cellules adhérentes immortalisées dérivées de kératinocytes humains HaCaT transfectés de façon stable avec un plasmide sélectionnable. Cette lignée cellulaire contient le gène de la luciférase sous le contrôle transcriptionnel d'un promoteur constitutif fusionné à un élément ARE d'un gène connu pour l'intensification de son expression sous l'effet de sensibilisants cutanés (25) (26). Le signal de la luciférase reflète l'activation par les sensibilisants de gènes endogènes dépendants du facteur Nrf2, et la dépendance du signal de la luciférase vis-à-vis de Nrf2 dans la lignée cellulaire recombinante a été démontrée (27). Cela permet la mesure quantitative (par détection de luminescence) de l'induction du gène de la luciférase, grâce à l'utilisation de substrats de luciférase produisant une luminescence satisfaisante, comme indicateur de l'activité du facteur de transcription Nrf2 dans les cellules après exposition à des substances électrophiles. Dans l'essai KeratinoSens™, les produits chimiques d'essai sont considérés comme positifs s'ils provoquent une induction statistiquement significative de l'activité de la luciférase dépassant un seuil donné (c.à.d. supérieure à 1,5 fois, soit 50 % d'augmentation, dans la méthode KeratinoSens™), au-dessous d'une concentration donnée n'affectant pas significativement la viabilité de la cellule (au-dessous de 1 000 μM, par exemple, et à une concentration pour laquelle la viabilité cellulaire est supérieure à 70 % dans la méthode KeratinoSens™ (9) (12)). On établit à cet effet le facteur maximal (Imax) par lequel est multipliée l'induction de l'activité de la luciférase par rapport au témoin (négatif) de solvant. De plus, les cellules étant exposées à une série de concentrations du produit chimique d'essai, la concentration nécessaire pour obtenir une induction statistiquement significative de l'activité de la luciférase au-delà du seuil (c.à.d. la valeur CE1,5 dans le cas de la méthode KeratinoSensTM) devra être interpolée à partir de la courbe dose-réponse (voir paragraphe 32 pour la méthode de calcul). Enfin, des mesures de cytotoxicité devront être réalisées en parallèle pour déterminer si l'induction de l'activité de la liuciférase survient à des concentrations sub-cytotoxiques. Avant d'utiliser en routine l'essai ARE-Nrf2 luciférase conformément à la présente méthode d'essai, les laboratoires devront faire la preuve de leurs compétences techniques, en appliquant la méthode aux dix substances énumérées à l'appendice 2. Des normes de performance (28) ont été établies pour faciliter la validation de méthodes d'essai ARE-Nrf2 luciférase in vitro nouvelles ou modifiées similaires à l'essai KeratinoSens™, et permettre de modifier rapidement la présente méthode d'essai afin de les y intégrer. L'acceptation mutuelle des données conformément à l'accord de l'OCDE ne sera garantie pour les méthodes d'essai validées conformément aux normes de performance que si ces méthodes d'essai ont été examinées et incorporées dans la ligne directrice correspondante par l'OCDE. MODE OPÉRATOIRE La seule méthode actuellement couverte par la présente méthode d'essai est l'essai scientifiquement validé KeratinoSens™ (9) (12) (13) (14). Les modes opératoires normalisés pour l'essai KeratinoSens™ sont disponibles et il convient de les appliquer lors de la mise en œuvre et de l'utilisation de cette méthode d'essai au laboratoire (15). Les laboratoires souhaitant appliquer cette méthode d'essai peuvent obtenir la lignée cellulaire recombinante utilisée dans l'essai KeratinoSens™ en établissant un accord de licence avec le développeur de la méthode. On trouvera dans les paragraphes qui suivent une description des principaux éléments et modes opératoires de la méthode d'essai ARE-Nrf2 luciférase. Préparation des cultures de kératinocytes Il convient d'utiliser une lignée de cellules transgéniques comportant une insertion stable du gène rapporteur de la luciférase sous le contrôle de l'élément ARE (lignée cellulaire KeratinoSens™). A réception, les cellules sont multipliées (2 à 4 repiquages, par exemple) afin de former un stock homogène conservé à l'état congelé. Les cellules provenant de ce stock d'origine peuvent être multipliées jusqu'à un nombre maximum de repiquages (25, dans la méthode KeratinoSens™) et peuvent ensuite être employées pour des essais de routine moyennant l'utilisation d'un milieu d'entretien approprié (DMEM additionné de sérum et de généticine, dans la méthode KeratinoSens™). Pour les essais, les cellules doivent présenter une confluence de 80-90 %, et il convient de veiller à ce qu'elles n'arrivent jamais à confluence complète. Une journée avant l'essai, les cellules sont récoltées et réparties dans des plaques 96 puits (10 000 cellules/puits dans la méthode KeratinoSens™). On veillera à éviter la sédimentation des cellules durant l'ensemencement, afin d'assurer une distribution homogène du nombre de cellules entre les puits. Si ce n'est pas le cas, cette étape peut donner lieu à une forte variabilité entre puits. Pour chaque répétition, trois réplicats sont utilisés pour mesurer l'activité de la luciférase, et un réplicat parallèle est utilisé pour le test de viabilité cellulaire. Préparation du produit chimique d'essai et des substances témoins Le produit chimique d'essai et les substances témoins sont préparés le jour du test. Dans l'essai KeratinoSens™, les produits chimiques d'essai sont dissous dans le diméthyl sulfoxyde (DMSO) à la concentration finale souhaitée (200 mM, par exemple). Les solutions de DMSO peuvent être considérées comme auto-stérilisantes, si bien qu'il n'est pas nécessaire de procéder à une filtration stérile. Les produits chimiques d'essai qui ne sont pas solubles dans le DMSO sont dissous dans de l'eau stérile ou un milieu de culture, et les solutions sont stérilisées, par exemple par filtration. Pour un produit chimique d'essai n'ayant pas de masse moléculaire (MM) définie, une solution mère est préparée à une concentration par défaut (40 mg/ml ou 4 % (m/v) dans l'essai KeratinoSens™). Dans le cas où des solvants autres que le DMSO, l'eau ou le milieu de culture sont utilisés, une justification suffisamment scientifique devra être fournie. A partir des solutions mères du produit chimique d'essai dans le DMSO, des dilutions en série sont préparées avec du DMSO pour obtenir 12 concentrations de référence du produit chimique à tester (de 0,098 à 200 mM dans l'essai KeratinoSens™). Si le produit chimique d'essai n'est pas soluble dans le DMSO, les dilutions permettant d'obtenir les concentrations de référence sont réalisées avec de l'eau stérile ou un milieu de culture stérile. Indépendamment du solvant utilisé, les concentrations de référence sont ensuite diluées 25 fois dans un milieu de culture contenant du sérum, et utilisées finalement pour le traitement après une nouvelle dilution d'un facteur 4, si bien que les concentrations finales du produit chimique d'essai vont de 0,98 à 2 000 μM, dans l'essai KeratinoSens™. Il est possible d'utiliser d'autres concentrations si cela est justifié (dans le cas d'un produit cytotoxique ou peu soluble, par exemple). Le témoin (de solvant) négatif utilisé dans l'essai KeratinoSens™ est le DMSO (noCAS 67-68-5, pureté ≥99 %), pour lequel six puits sont préparés par plaque. Il est soumis à une dilution identique à celle décrite au paragraphe 22 pour les concentrations de référence, de telle sorte que la concentration finale du témoin (de solvant) négatif est de 1 %, connue pour ne pas affecter la viabilité cellulaire (soit la même concentration de DMSO que dans le produit chimique testé et dans le témoin positif). Si le produit chimique d'essai n'est pas soluble dans le DMSO et a été dilué dans l'eau, la concentration de DMSO dans tous les puits de la solution d'essai finale doit être ajustée à 1 %, comme pour les autres produits chimiques d'essai et substances témoins. Le témoin positif utilisé dans l'essai KeratinoSens™ est l'aldéhyde cinnamique (noCAS 14371-10-9, pureté ≥98 %), pour lequel une série de 5 concentrations de référence allant de 0,4 à 6,4 mM sont préparées dans le DMSO (à partir d'une solution mère à 6.4 mM) et diluées selon la procédure décrite au paragraphe 22 pour les concentrations de référence, si bien que la concentration finale du témoin positif se situe entre 4 et 64 μM. D'autres témoins positifs adaptés, donnant de préférence une CE1,5 située dans la fourchette des valeurs moyennes, peuvent être utilisés si des données historiques sont disponibles pour dériver des critères d'acceptation issus de procédures comparables. Application du produit chimique d'essai et des substances témoins Pour chaque produit chimique d'essai et substance témoin positive, une expérience est nécessaire pour dériver une prédiction (positive ou négative), comportant au minimum deux répétitions indépendantes portant chacune sur trois réplicats (soit n = 6). En cas de résultats discordants entre les deux répétitions indépendantes, une troisième répétition portant sur trois réplicats (soit n = 9) devra être effectuée. Chaque répétition indépendante est effectuée à un jour différent au moyen d'une solution-mère de produit chimique d'essai, et de cellules collectées de façon indépendante. Les cellules peuvent cependant être issues du même repiquage. Après ensemencement selon le mode opératoire décrit au paragraphe 20, les cellules sont mises en culture pendant 24 heures dans les plaques de microtitrage 96 puits. Le milieu est ensuite retiré et remplacé par du milieu de culture frais (150 μl de milieu de culture contenant du sérum mais dépourvu de généticine dans l'essai KeratinoSens™) additionné de 50 μl du produit chimique d'essai et des substances témoins dilués 25 fois. Au moins un puits par plaque doit être laissé vide (sans cellules ni traitement) afin de déterminer les valeurs de fond. Les plaques traitées sont ensuite mises en incubation pendant environ 48 heures à 37°C ± 1°C sous 5 % de CO2 dans l'essai KeratinoSens™. Il convient d'éviter l'évaporation des produits chimiques d'essai volatils, ainsi que les contaminations croisées entre puits par les produits chimiques d'essai, en couvrant par exemple les plaques d'une feuille protectrice avant l'incubation avec les produits chimiques d'essai. Mesure de l'activité de la luciférase Trois facteurs sont déterminants pour une lecture correcte de la luminescence:
Avant l'essai, un test sera réalisé selon le mode opératoire décrit à l'appendice 3 afin de vérifier que ces trois exigences sont remplies. Après 48 heures d'exposition au produit chimique d'essai et aux substances témoins dans l'essai KeratinoSens™, les cellules sont lavées avec une solution physiologique tamponnée au phosphate, et le tampon de lyse approprié pour la lecture de la luminescence est ajouté à chaque puits pour une durée de 20 minutes à température ambiante. Les plaques contenant le lysat cellulaire sont ensuite placées pour lecture dans le luminomètre qui est programmé, dans l'essai KeratinoSens™, pour: (i) ajouter dans chaque puits le substrat de luciférase (50 μl), (ii) attendre une seconde, puis (iii) intégrer l'activité de la luciférase pendant 2 secondes. Si des paramètres différents sont utilisés compte tenu, par exemple, du modèle de luminomètre utilisé, il convient de les justifier. De plus, un substrat rayonnant peut également être utilisé, dans la mesure ou le contrôle de la qualité de l'expérience remplit les critères décrits à l'Appendice 3. Évaluation de la cytotoxicité Pour le test de viabilité cellulaire KeratinoSens™, le milieu est remplacé, après les 48 heures d'exposition, par du milieu frais contenant du MTT (bromure de 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium, bromure de thiazolyl tetrazolium bleu; noCAS 298-93-1) et les cellules sont mises en incubation pendant 4 heures à 37°C sous 5 % de CO2. Le milieu contenant du MTT est alors retiré et les cellules sont lysées pendant la nuit (p.ex. par ajout de solution SDS à 10 % dans chaque puits). Après agitation, l'absorption est mesurée à 600 nm au photomètre. RÉSULTATS ET RAPPORT Évaluation des données Les paramètres suivants sont calculés dans l'essai KeratinoSens™:
Équation 1:
où
CE1,5 est calculée par interpolation linéaire d'après l'équation 2, et la valeur résultante de l' CE1.5 est la moyenne géométrique de chacune des répétitions. Équation 2:
où
L'équation 3 permet de calculer la viabilité: Équation 3:
où
CI50 et CI30 sont calculés par interpolation linéaire d'après l'équation 4, et les valeurs résultantes de CI50 et CI30 sont les moyennes géométriques de chacune des répétitions. Équation 4:
où
Pour chaque concentration donnant une augmentation de l'induction de l'activité de la luciférase > 1,5 fois, on calcule la signification statistique (par un test t de Student bilatéral, par exemple) en comparant la luminescence pour les trois échantillons réplicats avec la luminescence pour les puits contenant le témoin de solvant (négatif), afin de déterminer si l'induction de l'activité de la luciférase est statistiquement significative (p < 0,05). La concentration la plus faible pour laquelle l'augmentation de l'induction de l'activité de la luciférase est > 1,5 fois est la valeur déterminant CE1,5. On vérifie dans chaque cas si cette valeur est inférieure à CI30, et donc si la réduction de la viabilité cellulaire est bien inférieure à 30 % à la concentration déterminant CE1,5. Il est recommandé de vérifier visuellement les données à l'aide de graphiques. En l'absence de courbe dose-réponse clairement observable, ou si la courbe dose-réponse obtenue est biphasique (c'est-à-dire qu'elle croise deux fois le seuil de 1,5), il convient de répéter l'expérience pour vérifier si ce résultat est spécifique au produit chimique d'essai ou s'il s'agit d'un artefact expérimental. Dans le cas où la réponse biphasique est reproductible au cours d'un essai indépendant, c'est la valeur CE1,5 la plus basse (la concentration à laquelle la courbe croise le seuil de 1,5 pour la première fois) qui doit être consignée dans le rapport. Dans les rares cas où l'on observe une induction statistiquement non significative supérieure à 1,5 fois, suivie d'une induction statistiquement significative à une concentration plus élevée, les résultats de cette répétition ne sont considérés comme valides et positifs que si une induction statistiquement significative supérieure au seuil de 1,5 a été obtenue à une concentration non cytotoxique. Enfin, pour les produits chimiques d'essai générant une augmentation de l'induction égale ou supérieure à 1,5 fois pour la concentration testée la plus faible de 0,98 μM, la valeur de CE1,5 est fixée à < 0,98 sur la base d'un examen visuel de la courbe dose-réponse. Critères d'acceptabilité Les critères d'acceptabilité suivants doivent être remplis lors de la mise en œuvre de l'essai KeratinoSens™. Premièrement, l'induction de l'activité de la luciférase obtenue avec le témoin positif (aldéhyde cinnamique) doit être significativement supérieure au seuil de 1,5 (d'après un test T, par exemple) pour au moins l'une des concentrations testées (de 4 à 64 μM). Deuxièmement, la valeur de CE1,5 doit se situer dans un intervalle de deux écarts types autour de la moyenne historique du laboratoire (entre 7 μM et 30 μM, par exemple, d'après les données utilisées pour la validation), qui devra être actualisée régulièrement. De plus, l'induction moyenne des trois réplicats pour l'aldéhyde cinnamique à 64 μM doit se situer entre 2 et 8. Si ce dernier critère n'est pas rempli, la relation dose-réponse pour l'aldéhyde cinnamique devra être vérifiée avec soin, et les tests ne pourront être acceptés que s'il existe une relation dose-réponse claire, l'induction de l'activité de la luciférase augmentant avec la concentration pour le témoin positif. Enfin, le coefficient moyen de variation de la luminescence pour le témoin (de solvant) négatif DMSO doit être inférieur à 20 % pour chaque répétition qui comprend 6 puits testés en triplicats. Si la variabilité est supérieure, les résultats ne doivent pas être pris en compte. Interprétation des résultats et modèle prédictif Une prédiction KeratinoSens™ est considérée comme positive si les 4 conditions suivantes sont remplies lors de 2 répétitions sur 2 ou sur 3; dans le cas contraire, la prédiction KeratinoSens™ est considérée comme négative (figure 1):
Si, lors d'une répétition donnée, les trois premières conditions sont remplies mais qu'il n'y a pas de relation dose-réponse clairement observable pour l'induction de la luciférase, le résultat de cette répétition doit être considéré comme non concluant et des tests supplémentaires peuvent être requis (figure 1). En outre, un résultat négatif obtenu avec des concentrations < 1 000 μM (ou <200 μg/ml pour les produits chimiques dont le poids moléculaire n'est pas défini) sera également considéré comme non concluant (voir le paragraphe 11). Figure 1 Modèle prédictif utilisé dans l'essai KeratinoSens™. Une prédiction KeratinoSens™ doit être envisagée dans le cadre d'une démarche IATA et conformément aux dispositions des paragraphes 9 et 11.
Dans de rares cas, les produits chimiques d'essai qui induisent une activité de la luciférase à des niveaux très proches des concentrations cytotoxiques peuvent être positifs à des concentrations non cytotoxiques (c.à.d. EC1,5 déterminant la concentration en-dessous (<) de l'IC30), et dans d'autres répétitions, uniquement à des concentrations cytotoxiques (c.à.d. EC1,5 déterminant la concentration en-dessus (>) de l'IC30). Ces produits seront re-testés en procédant à une analyse dose-réponse plus serrée, avec un facteur de dilution plus bas (par exemple, 1,33 ou √2 (=1,41) fois entre puits), afin de déterminer si l'induction s'est produite à des niveaux cytotoxiques ou non (9). Rapport d'essai Le rapport d'essai devra comporter les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS AOP (Adverse Outcome Pathway) : séquence d'événements conduisant à la survenue d'un effet indésirable in vivo, à partir de la structure chimique d'un produit chimique cible ou d'un groupe de produits chimiques similaires et de l'événement initiateur au niveau moléculaire (2). ARE : L'élément de réponse antioxydant (aussi appelé EpRE, élément de réponse électrophile) est un élément de réponse présent dans la région promotrice amont de nombreux gènes cytoprotecteurs et gènes de phase II. Lorsqu'il est activé par le facteur Nfr2, il médie l'induction transcriptionnelle de ces gènes. Coefficient de variation : la mesure de la variabilité calculée pour un groupe de données issues de réplicats en divisant la déviation standard par la moyenne. Cette mesure peut être multipliée par 100 pour obtenir un pourcentage. Danger : Propriété inhérente d'un agent ou d'une situation susceptible de provoquer des effets indésirables lorsqu'un organisme, un système ou une (sous-) population est exposé(e) à cet agent. EC1,5 : Concentration, établie par interpolation, à laquelle l'induction de luciférase est multipliée par 1.5. Fiabilité : Indique dans quelle mesure la mise en œuvre d'une méthode d'essai peut être reproduite au cours du temps par un même laboratoire ou plusieurs laboratoires utilisant le même mode opératoire. Pour l'évaluer, on calcule la reproductibilité intra-laboratoire et inter-laboratoires et la répétabilité intra-laboratoire (29). IATA (Integrated Approach to Testing and Assessment) : Approche structurée utilisée dans l'identification du danger (potentiel), la caractérisation du danger (puissance) et/ou dans l'évaluation'de la sécurité (potentiel de danger/puissance du danger et exposition) d'un produit chimique ou d'un groupe de produits chimiques, qui intègre de façon stratégique et pondérée toutes les données pertinentes dans le but d'informer une décision réglementaire sur le danger potentiel et/ou le risque et/ou le besoin d'effectuer d'autres tests ciblés. IC30 : Concentration pour laquelle la viabilité cellulaire, établie par le test au MTT (bromure de 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium, bromure de thiazolyl tetrazolium bleu; noCAS 298-93-1), est réduite de 30 %. IC50 : Concentration pour laquelle la viabilité cellulaire, établie par le test au MTT, est réduite de 50 %. Imax : Facteur d'induction maximale de l'activité de la luciférase, à quelque concentration du produit chimique d'essai, comparé au témoin (négatif) de solvant. Keap1 : Kelch-like ECH-associated protein 1, est une protéine détectrice pouvant réguler l'activité du facteur Nrf2. En conditions non induites, la protéine détectrice Keap1 cible le facteur de transcription Nrf2 et provoque son ubiquitination et sa dégradation protéolytique dans le protéasome. En cas de modification covalente des résidus cystéine réactifs de Keap1 par des molécules de petite taille, Nrf2 peut se dissocier de Keap1 (8) (10) (11). Mélange : Mélange ou solution constitué(e) d'au moins deux substances qui ne réagissent pas entre elles (1). Méthode d'essai valide : Méthode d'essai dont la pertinence et la fiabilité sont jugées satisfaisantes pour des fins spécifiques, et qui repose sur des principes scientifiquement valables. Une méthode d'essai n'est jamais valide dans l'absolu, mais seulement par rapport à une fin particulière (29). Nrf2 : Nuclear factor (erythroid-derived 2)-like 2, Facteur de transcription intervenant dans la voie de réponse antioxydante. Lorsque Nrf2 n'est pas ubiquitiné, il se développe dans le cytoplasme et se transloque dans le noyau, où il se combine à l'ARE dans la région promotrice amont de nombreux gènes cytoprotecteurs, dont il initie la transcription (8) (10) (11). Pertinence : Décrit la relation entre l'essai et l'effet étudié, et rend compte de l'adéquation de l'essai et de son utilité à des fins spécifiques. La pertinence indique dans quelle mesure l'essai mesure ou prédit correctement l'effet biologique d'intérêt. Elle tient compte de la précision (concordance) d'une méthode d'essai (29). Précision : Étroitesse de l'accord entre les résultats de la méthode d'essai et les valeurs de référence acceptées. Elle constitue une mesure de performance de la méthode d'essai et l'un des aspects de sa «pertinence». Ce terme est souvent utilisé au sens de «concordance», pour qualifier la proportion de résultats corrects d'une méthode d'essai (29). Produit chimique : une substance ou un mélange. Produit chimique d'essai : Le terme «produit chimique d'essai» désigne ce qui est testé. Reproductibilité : Accord entre les résultats obtenus lors d'essais pratiqués sur le même produit chimique selon le même mode opératoire (voir Fiabilité) (29). Sensibilité : Proportion des produits chimiques positifs/actifs qui sont correctement classés par la méthode d'essai. Elle permet de mesurer la précision d'une méthode d'essai produisant des données catégorielles, et constitue un aspect important de l'évaluation de la pertinence d'une méthode d'essai (29). SGH (Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations unies) : Système proposant la classification des produits chimiques (substances et mélanges) conformément à des types et des niveaux normalisés de dangers physiques, sanitaires et environnementaux, ainsi que la communication des éléments d'information correspondants, notamment par des pictogrammes, mentions d'avertissement, mentions de danger, conseils de prudence et fiches de données de sécurité, afin de diffuser des informations sur leurs effets indésirables dans l'objectif de protéger les personnes (en particulier les employeurs, employés, transporteurs, consommateurs et personnels des services d'urgence) et l'environnement (1). Spécificité : Proportion des produits chimiques négatifs/inactifs qui sont correctement classés par la méthode d'essai. Elle permet de mesurer la précision d'une méthode d'essai produisant des données catégorielles, et constitue un aspect important de l'évaluation de la pertinence d'une méthode d'essai (29). Substance : Élément chimique et ses composés, présents à l'état naturel ou obtenus grâce à un procédé de production. Ce terme inclut tout additif nécessaire pour préserver la stabilité du produit ainsi que toute impureté produite par le procédé utilisé, mais exclut tout solvant pouvant être extrait sans affecter la stabilité ni modifier la composition de la substance (1). Substance mono-constituant : Substance, définie par sa composition quantitative, dans laquelle un constituant principal est présent à hauteur de 80 % minimum (m/m). Substance multi-constituants : Substance, définie par sa composition quantitative, dans laquelle plus d'un constituant principal est présent dans une concentration ≥ 10 % (m/m) et < 80 % (m/m). Une substance multi-constituants résulte d'un processus de fabrication. La différence entre un mélange et une substance multi-constituants est qu'un mélange est obtenu en associant deux substances ou plus sans qu'il se produise de réaction chimique. Une substance multi-constituants est le résultat d'une réaction chimique. Témoin de solvant/véhicule : Réplicat contenant tous les composants d'un système d'essai, excepté le produit chimique d'essai, mais comprenant le solvant utilisé. Il sert à déterminer une réponse de référence pour les échantillons traités avec le produit chimique d'essai dissous dans le même solvant ou véhicule. UVCB : Substances de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques. Appendice 2 SUBSTANCES D'ÉPREUVE DE COMPÉTENCE Sensibilisation cutanée in vitro: méthode d'essai ARE-Nrf2 luciférase Avant d'utiliser en routine la présente méthode d'essai, les laboratoires doivent démontrer leurs compétences techniques en obtenant les prédictions attendues par la méthode KeratinoSensTM pour les 10 substances d'épreuve de compétence recommandées au tableau 1, et en obtenant des valeurs de CE1,5 et CI50 se situant dans les domaines de référence respectifs de ces deux grandeurs pour au moins 8 des 10 substances d'épreuve de compétence. Ces substances ont été sélectionnées de façon à représenter la gamme des réponses possibles en ce qui concerne les dangers de sensibilisation cutanée. Les autres critères de sélection sont la disponibilité des substances dans le commerce, ainsi que la qualité élevée des données de référence in vivo disponibles et des données in vitro obtenues par l'essai KeratinoSensTM. Tableau 1 Substances recommandées pour démontrer les compétences techniques relatives à l'essai KeratinoSensTM
Appendice 3 CONTRÔLE QUALITÉ DES MESURES DE LUMINESCENCE Expérience préliminaire visant à garantir des mesures de luminescence optimales lors de l'essai KeratinoSensTM Les trois paramètres suivants sont essentiels pour garantir l'obtention de résultats fiables au luminomètre:
Avant de procéder à l'essai, il est recommandé de vérifier la qualité des mesures de luminescence en testant une plaque témoin organisée comme indiqué ci-après (analyse en triplicat). Organisation de la plaque pour l'essai préliminaire
DMAEG= diméthacrylate d'éthylène glycol (CAS No: 97-90-5), produit chimique provoquant une forte induction AC= aldéhyde cinnamique, témoin positif (CAS No: 104-55-2) L'analyse de contrôle qualité doit établir:
B.61 Méthode d'essai de diffusion de fluorescéine pour identifier les substances corrosives et fortement irritantes pour l'œil INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 460 (2012) de l'OCDE pour les essais de produits chimiques. La méthode d'essai de diffusion de fluorescéine (DF) est une méthode d'essai in vitro qui, dans certaines circonstances et assortie de restrictions spécifiques, peut être utilisée pour classer les produits chimiques (substances et mélanges) comme corrosifs et fortement irritants pour l'œil, au sens du Système général harmonisé de classification et d'étiquetage des produits chimiques (SGH) des Nations Unies (ONU) (catégorie 1), du règlement (CE) no 1272/2008 de l'Union européenne relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges (CLP) (31) (catégorie 1), et de l'Agence de protection de l'environnement des États-Unis (EPA) (catégorie I) (1) (2). Aux fins de la présente méthode d'essai, les substances fortement irritantes pour l'œil sont définies comme des produits chimiques provoquant des lésions des tissus oculaires après application sur l'œil, lésions qui ne sont pas réversibles dans les 21 jours suivant l'application ou qui entraînent une dégradation sévère de la vue, tandis que les substances corrosives pour l'œil occasionnent des lésions irréversibles des tissus oculaires. Ces substances sont classées dans la catégorie 1 du SGH (ONU), la catégorie 1 du CLP (UE) ou la catégorie I de l'EPA (États-Unis). Bien que la méthode d'essai de DF n'ait pas été validée comme méthode substitutive complète à l'essai oculaire in vivo sur le lapin, elle est recommandée comme élément d'une stratégie d'essais à plusieurs niveaux à des fins de classification et d'étiquetage réglementaires. Le recours à la DF est en effet recommandé en phase initiale d'une approche «top-down» (descendante) pour identifier les substances corrosives et fortement irritantes pour l'œil, en particulier pour certains types de produits chimiques (substances et mélanges hydrosolubles) (3) (4). Il est généralement admis que dans notre horizon de prévision, aucun essai unique d'irritation oculaire in vitro ne pourra remplacer le test in vivo [méthode d'essai B.5 (5)] pour établir le degré d'irritation provoqué par les différentes classes de produits chimiques. Cependant, la combinaison de plusieurs méthodes de substitution dans le cadre d'une stratégie d'essais (à plusieurs niveaux) pourrait remplacer le test in vivo (4). L'approche «top-down» (45) est indiquée lorsque, d'après les informations existantes, on s'attend à ce qu'un produit chimique soit fortement irritant. Suivant le modèle prédictif détaillé au paragraphe 35, la méthode d'essai de DF permet, sans essai complémentaire, de classer les produits chimiques comme corrosifs ou fortement irritants pour l'œil dans un domaine d'applicabilité limité (catégorie 1 du SGH; catégorie 1 du CLP; catégorie I de l'EPA). On suppose que la méthode s'applique également aux mélanges bien qu'aucun mélange n'ait été utilisé pour la validation. La méthode d'essai de DF peut donc servir à identifier les substances chimiques irritantes/corrosives pour l'œil en suivant la stratégie d'essais séquentiels de la méthode d'essai B.5 (6). Toutefois, si un produit chimique n'est pas identifié comme corrosif ou fortement irritant pour l'œil à l'issue de l'essai de DF, il convient de le soumettre à une ou plusieurs autres méthodes d'essai (in vitro et/ou in vivo) capables d'identifier précisément i) les substances qui sont des faux négatifs pour les essais de DF in vitro visant à repérer les effets corrosifs/fortement irritants sur l'œil (catégorie 1 du SGH; catégorie 1 du CLP; catégorie I de l'EPA); ii) les substances non classées en ce qui concerne la corrosion/l'irritation oculaire (pas de catégorie dans le SGH; pas de catégorie dans le CLP; catégorie IV de l'EPA); et/ou iii) les substances modérément à légèrement irritantes pour l'œil (catégories 2A et 2B du SGH; catégorie 2 du CLP; catégories II et III de l'EPA). L'objet de la présente méthode d'essai est de décrire les procédures utilisées pour évaluer les éventuels effets corrosifs ou fortement irritants pour l'œil d'un produit chimique au regard de sa capacité d'induire des lésions sur une monocouche épithéliale confluente imperméable. L'intégrité de la perméabilité transépithéliale constitue une fonction primordiale d'un épithélium tel que celui présent dans la conjonctive et la cornée. La perméabilité transépithéliale est contrôlée par diverses jonctions serrées. Une corrélation a été démontrée entre l'augmentation de la perméabilité de l'épithélium cornéen in vivo et la sévérité de l'inflammation et des lésions de la surface de l'œil observées dans le développement d'une irritation oculaire. La méthode d'essai de DF permet de mesurer les effets toxiques induits après un court temps d'exposition au produit chimique d'essai en déterminant l'augmentation de la perméabilité à la fluorescéine-sodium à travers une monocouche épithéliale de cellules rénales de chien Madin-Darby (MDCK) cultivées sur des inserts perméables. La quantité de fluorescéine qui se diffuse est proportionnelle aux lésions provoquées par le produit chimique sur les jonctions serrées, les desmosomes et les cellules membranaires, et permet ainsi d'estimer sa toxicité potentielle pour l'œil. L'appendice 1 contient un diagramme représentant les cellules MDCK cultivées sur la membrane d'un insert pour la méthode d'essai de DF. Les définitions utilisées sont données à l'appendice 2. REMARQUES PRÉLIMINAIRES ET LIMITES La méthode d'essai s'inspire du protocole INVITTOX no 71 (6) qui a fait l'objet d'une évaluation dans le cadre d'une étude de validation internationale par le Centre européen pour la validation de méthodes alternatives (ECVAM), en collaboration avec le Comité de coordination inter-agences pour la validation des méthodes alternatives (ICCVAM) des États-Unis et avec le Centre japonais pour la validation de méthodes alternatives (JaCVAM). La méthode d'essai de DF n'est pas recommandée pour identifier les produits chimiques qui devraient être classés comme légèrement à modérément irritants, ou ceux qui ne devraient pas être classés comme irritants pour l'œil (substances et mélanges) (qui relèvent des catégories 2A/2B ou d'aucune catégorie dans le SGH; de la catégorie 2 ou d'aucune catégorie dans le CLP; des catégories II, III et IV de l'EPA), comme l'a démontré l'étude de validation (3) (7). La méthode d'essai ne s'applique qu'aux produits chimiques (substances et mélanges) hydrosolubles. Elle permet généralement de prévoir avec précision l'effet potentiel fortement irritant pour l'œil de produits chimiques qui sont hydrosolubles et/ou dont l'effet toxique n'est pas modifié par la dilution (7). Dans les conditions expérimentales, un produit chimique sera considéré comme hydrosoluble s'il est soluble dans une solution saline équilibrée de Hanks (HBSS) stérile avec calcium (à 1,0 — 1,8 mM) et sans rouge phénol, à une concentration supérieure ou égale à 250 mg/ml (soit une dose au-dessus de la valeur seuil fixée à 100 mg/ml). Néanmoins, si la substance d'essai est soluble à une concentration inférieure à 100 mg/ml, mais induit déjà une DF de 20 % à cette concentration (donc si DF20 < 100 mg/ml), elle peut quand même être classée dans la catégorie 1 du SGH ou dans la catégorie I de l'EPA. Les acides et bases forts, les fixateurs cellulaires et les substances très volatiles sont hors des limites d'application mises en évidence pour cette méthode d'essai. Ces produits chimiques participent en effet à des mécanismes qui ne sont pas mesurés par la méthode de DF, par exemple coagulation extensive, saponification ou réactions chimiques spécifiques. Les résultats obtenus avec les produits chimiques d'essai colorés et visqueux révèlent d'autres limites de la capacité de prévision de cette méthode (7). En effet, ces deux types de substances sont difficiles à éliminer de la monocouche après le court temps d'exposition, et il est suggéré que la qualité de prévision de la méthode d'essai pourrait être améliorée en augmentant le nombre d'étapes de lavage mises en œuvre. Par ailleurs, les produits chimiques solides en suspension dans un liquide ont tendance à précipiter, si bien qu'il peut être difficile de déterminer la concentration finale à laquelle sont exposées les cellules. Si l'on exclut de la base de données les produits chimiques relevant de ces classes chimiques et physiques, la précision de la méthode de DF au regard des systèmes de classification de l'UE, de l'EPA et de l'ONU se trouve considérablement améliorée (7). Dans l'optique de la présente méthode d'essai (qui vise exclusivement à identifier les substances corrosives/fortement irritantes pour l'œil), les taux de faux négatifs (voir paragraphe 13) ne sont pas préoccupants car les produits chimiques concernés feront l'objet d'essais chez le lapin ou d'autres essais in vitro dûment validés, conformément aux exigences de la réglementation, selon une démarche expérimentale séquentielle fondée sur l'analyse du poids de la preuve (5) (voir également les paragraphes 3 et 4). La méthode d'essai de DF révèle d'autres limites à l'examen des taux de faux négatifs et faux positifs. Quand l'essai de DF est mis en œuvre en phase initiale d'une approche «top-down» pour identifier les substances et mélanges hydrosolubles corrosifs/fortement irritants pour l'œil (catégorie 1 du SGH; catégorie 1 du CLP; catégorie I de l'EPA), le taux de faux positifs est situé entre 7 % (7/103; SGH et CLP) et 9 % (9/99; EPA) et celui de faux négatifs entre 54 % (15/28; EPA) et 56 % (27/48; SGH et CLP), par rapport aux résultats obtenus in vivo. Les groupes de produits chimiques induisant des faux positifs et/ou des faux négatifs avec la méthode d'essai de DF ne sont pas définis dans le présent document. Certaines limites techniques sont propres à la culture de cellules MDCK. Les jonctions serrées qui bloquent le passage du colorant (fluorescéine-sodium) à travers la monocouche baissent en qualité avec l'augmentation du nombre de repiquages des cultures. Or, la formation incomplète des jonctions serrées débouche sur une hausse de la diffusion de fluorescéine dans le témoin non traité. Il importe donc de définir un niveau de diffusion maximal admissible dans les témoins non traités (voir paragraphe 38: diffusion de 0 %). Comme dans tous les essais in vitro, les cellules employées risquent de se transformer au fil du temps, c'est pourquoi il est crucial de préciser la fourchette du nombre de passages des essais. Le domaine d'applicabilité actuel pourrait être étendu dans certains cas, mais seulement après analyse d'un ensemble élargi de données sur les produits chimiques testés, données obtenues de préférence lors d'essais (4). La présente méthode d'essai sera mise à jour en conséquence, après l'examen de nouvelles informations et données. Tout laboratoire qui fait appel au présent essai pour la première fois doit recourir aux produits chimiques d'épreuve de compétence indiqués à l'appendice 3. Les laboratoires peuvent utiliser ces substances pour apporter la preuve de leur compétence technique au regard de la mise en œuvre de la méthode d'essai de DF avant de soumettre les résultats des essais de DF à des fins de classification réglementaire dans une catégorie de danger. PRINCIPE DE L'ESSAI La méthode d'essai de DF est un essai in vitro fondé sur des paramètres de cytotoxicité et de fonctionnement cellulaire, réalisé sur une monocouche confluente de cellules épithéliales tubulaires MDCK CB997 cultivées sur des inserts semi-perméables et qui modélisent l'état non proliférant de l'épithélium cornéen in vivo. La lignée cellulaire MDCK est bien établie et forme des jonctions serrées et des desmosomes similaires à ceux que l'on observe sur la face apicale des épithéliums conjonctif et cornéen. In vivo, les jonctions serrées et les desmosomes empêchent les solutés et les substances exogènes de pénétrer l'épithélium cornéen. La perte d'imperméabilité transépithéliale, qui résulte de lésions des jonctions serrées et des desmosomes, est l'un des effets précoces de l'irritation oculaire d'origine chimique. Le produit chimique d'essai est appliqué sur la couche confluente de cellules cultivées sur la face apicale de l'insert. Un temps d'exposition réduit, de 1 min, est généralement appliqué pour refléter la vitesse de clairance normalement associée aux expositions humaines. L'un des avantages d'une courte durée d'exposition est que les substances et mélanges aqueux peuvent être évalués purs, s'il est possible de les éliminer facilement à l'issue du temps d'exposition. Cette possibilité permet d'effectuer des comparaisons plus directes avec les effets chimiques chez l'être humain. Le produit chimique d'essai est ensuite éliminé et la fluorescéine-sodium, un colorant non toxique et très fluorescent, est ajoutée sur la face apicale de la monocouche pendant 30 minutes. Les lésions induites par le produit chimique d'essai sur les jonctions serrées sont déterminées par la quantité de fluorescéine qui diffuse à travers la couche cellulaire pendant une durée définie. La quantité de colorant (fluorescéine-sodium) qui a traversé la monocouche puis la membrane de l'insert et se retrouve dans un volume donné de solution du puits (où diffuse le colorant) est déterminée par mesure spectrofluorimétrique de la concentration de fluorescéine dans le puits. L'ampleur de la diffusion de fluorescéine (DF) est calculée par rapport aux intensités de fluorescence (IF) relevées pour deux témoins: un témoin à blanc et un témoin de la diffusion maximale. Le pourcentage de diffusion, et donc l'ampleur des lésions des jonctions serrées, est exprimé, par rapport à ces deux témoins, pour chacune des concentrations du produit chimique d'essai. On calcule alors la DF20 (c'est-à-dire la concentration provoquant une DF égale à 20 % de la valeur relevée pour la monocouche confluente non exposée et les inserts sans cellules). La valeur de la DF20 (mg/ml) est alors utilisée dans le modèle de prévision pour identifier les produits chimiques corrosifs et fortement irritants pour l'œil (voir paragraphe 35). La capacité de récupération constitue un aspect important du profil toxicologique d'un produit chimique d'essai, également évaluée dans l'essai d'irritation oculaire in vivo. Des analyses préliminaires indiquent que les données sur la récupération (jusqu'à 72 h après l'exposition chimique) sont susceptibles d'améliorer la capacité de prévision du protocole INVITTOX 71, mais d'autres évaluations s'imposent et nécessitent des données supplémentaires, obtenues de préférence lors d'essais complémentaires (6). La présente méthode d'essai sera mise à jour en conséquence, après l'examen de nouvelles informations et données. MODE OPÉRATOIRE Préparation de la monocouche cellulaire La monocouche de cellules MDCK CB997 est préparée avec des cellules sub-confluentes cultivées en fioles dans le milieu de Eagle modifié de Dulbecco/mélange nutritif F12 (concentré x 1 avec L-glutamine, HEPES à 15 mM, calcium à 1,0 — 1,8 mM, et 10 % de SVF/SBF désactivé à chaud). Il importe que tous les milieux et solutions employés au cours de l'essai de DF contiennent du calcium à une concentration située entre 1,0 mM (111 mg/l) et 1,8 mM (200 mg/l), pour garantir la formation et l'intégrité des jonctions serrées. La fourchette du nombre de repiquages de cellules doit faire l'objet d'un témoin pour assurer une formation régulière et reproductible des jonctions serrées. On privilégiera les cellules obtenues après 3 à 30 repiquages à partir de la décongélation, car les cultures situées dans cette plage présentent une fonctionnalité similaire, ce qui contribue à la reproductibilité des résultats de l'essai En amont de l'essai de DF, les cellules sont détachées de la fiole par trypsinisation et centrifugées, puis la quantité nécessaire de cellules est repiquée dans les inserts placés dans des plaques 24 puits (voir appendice 1). Pour ce repiquage, on utilisera des inserts de 12 mm de diamètre équipés d'une membrane en esters cellulosiques mixtes de 80 à 150 μm d'épaisseur et percée de pores de 0.45 μm. L'étude de validation a été réalisée avec des inserts Millicell-HA 12 mm. Les propriétés de l'insert et du type de membrane sont importantes dans la mesure où elles peuvent modifier la croissance cellulaire et la formation de liaisons chimiques. Certains types de produits chimiques peuvent en effet se lier à la membrane de l'insert Millicell-HA, ce qui peut jouer sur l'interprétation des résultats. Il convient d'utiliser les produits chimiques d'épreuve de compétence (voir appendice 3) pour démontrer l'équivalence d'autres membranes, le cas échéant. La formation de liaisons chimiques avec la membrane de l'insert est plus courante avec les composés cationiques, comme le chlorure de benzalkonium, qui sont attirés par la membrane chargée (7). Ces liaisons chimiques sont susceptibles de prolonger le temps d'exposition au produit chimique étudié, et d'entraîner ainsi une surestimation de sa toxicité potentielle; elles peuvent aussi limiter physiquement la diffusion de fluorescéine à travers l'insert, en liant le colorant au composé cationique lui-même lié à la membrane, ce qui donne lieu à une sous-estimation de la toxicité potentielle du produit chimique d'essai. On peut évaluer ce phénomène directement en exposant la membrane seule à la concentration maximale du produit chimique testé, puis en ajoutant le colorant fluorescéine-sodium à la concentration normale et pendant le temps d'exposition standard (pas de témoins des cellules). Si la fluorescéine se lie à la membrane de l'insert, celle-ci paraît jaunie une fois que le produit chimique d'essai a été éliminé par lavage. Il est donc essentiel de connaître les affinités de liaison du produit chimique d'essai afin de pouvoir interpréter son effet sur les cellules. Le repiquage des cellules sur les inserts doit permettre d'obtenir une monocouche confluente au moment de l'exposition chimique. On ajoute 1.6 × 105 cellules par insert (400 μl de suspension cellulaire avec une densité de 4 × 105 cellules/ml). Dans ces conditions, une monocouche confluente apparaît généralement après 96 heures de culture. Il convient d'examiner visuellement les inserts avant le repiquage, pour s'assurer que les lésions éventuellement consignées à l'occasion de la vérification visuelle décrite au paragraphe 30 découlent de la manipulation. Les cultures de cellules MDCK sont maintenues dans des incubateurs en atmosphère humidifiée, à 5 ± 1 % de CO2 et 37 ± 1 °C. Les cellules sont exemptes de toute contamination bactérienne, virale, mycoplasmique ou fongique. Application des produits chimiques d'essai et témoins Une nouvelle solution-mère du produit chimique à tester doit être préparée pour chaque série d'essai et utilisée dans les 30 minutes qui suivent sa préparation. Les produits chimiques d'essai sont préparés dans le HBSS avec calcium (à 1,0 — 1,8 mM) et sans rouge phénol, pour éviter les liaisons avec les protéines sériques. La solubilité du produit chimique à 250 mg/ml est évaluée avant l'essai. Si, à cette concentration, il forme une suspension ou une émulsion stable (c'est-à-dire qui reste uniforme et ne décante pas ou ne se sépare pas en plusieurs phases) pendant 30 minutes, on peut maintenir le choix du HBSS comme solvant. Néanmoins, si le produit chimique s'avère insoluble dans le HBSS à cette concentration, on envisagera de recourir à d'autres méthodes que l'essai de DF. La prudence est de mise concernant l'utilisation d'une huile minérale légère comme solvant, dans les cas où le produit chimique se révèle insoluble dans le HBSS, car les données accessibles sont insuffisantes pour conclure sur la performance de l'essai de DF dans ces conditions. Tous les produits chimiques à évaluer sont préparés à partir de la solution-mère, dans du HBSS stérile avec calcium (à une concentration de 1.0 — 1.8 mM) et sans rouge phénol, en cinq dilutions dont les concentrations massiques sont fixées: 1, 25, 100 et 250 mg/ml ainsi qu'une solution pure ou saturée. En cas d'évaluation d'un composé solide, on inclura une très forte concentration de 750 mg/ml. Cette concentration de la substance d'essai doit parfois être appliquée sur les cellules à l'aide d'une pipette à déplacement positif. Si des effets toxiques apparaissent entre 25 et 100 mg/ml, il convient d'analyser également les concentrations suivantes à deux reprises: 1, 25, 50, 75 et 100 mg/ml. La DF20 sera déduite de ces concentrations, à condition que les critères d'acceptation soient satisfaits. Avant d'appliquer les substances d'essai sur les monocouches de cellules confluentes, le milieu de culture cellulaire est retiré et les monocouches sont lavées deux fois avec du HBSS stérile avec calcium (à 1,0 — 1,8 mM) et sans rouge phénol amené à 37 °C. On aura préalablement effectué une vérification visuelle des filtres à la recherche d'éventuelles lésions déjà présentes et qui pourraient être faussement imputées à une incompatibilité potentielle avec le produit chimique d'essai. Pour chaque concentration du produit chimique d'essai et pour les témoins, on met en œuvre au moins trois réplicats par série d'essai. Après 1 min d'exposition à température ambiante, le produit chimique d'essai est soigneusement éliminé par pipetage, la monocouche est lavée deux fois avec du HBSS stérile amené à 37 °C avec calcium (à 1,0 — 1,8 mM) et sans rouge phénol, et la diffusion de fluorescéine est immédiatement quantifiée. Les témoins concurrents négatifs (TN) et positifs (TP) sont présents dans chaque série d'essai pour démontrer que l'intégrité de la monocouche (TN) et la sensibilité des cellules (TP) sont dans une fourchette admissible, définie d'après les résultats historiques. Le composé recommandé comme TP est le Brij 35 (noCAS 9002-92-0) à 100 mg/ml. Cette concentration devrait induire environ 30 % de diffusion de fluorescéine (plage admissible: 20 — 40 % de DF, donc de lésions de la couche cellulaire). Le composé recommandé comme TN est le HBSS avec calcium (à 1,0 — 1,8 mM) et sans rouge phénol (témoin à blanc non traité). Un témoin de la diffusion maximale est également inclus dans chaque série d'essai pour permettre de calculer les DF20. La diffusion maximale est déterminée à l'aide d'un insert témoin ne contenant pas de cellules. Détermination de la perméabilité à la fluorescéine Immédiatement après avoir retiré les produits chimiques d'essai et témoins, ajouter aux inserts (Millicell-HA, par exemple) 400 μl de solution de fluorescéine-sodium à 0,1 mg/ml (soit 0,01 % en m/v dans du HBSS avec calcium [à 1,0 — 1,8 mM] et sans rouge phénol). Les cultures sont maintenues à température ambiante pendant 30 minutes. À l'issue de l'incubation avec la fluorescéine, les inserts sont délicatement retirés de tous les puits. On effectue une vérification visuelle de chaque filtre, et toute lésion éventuellement causée par la manipulation est consignée. La quantité de fluorescéine diffusée à travers la monocouche et l'insert est mesurée dans la solution qui reste dans les puits après le retrait des inserts. Ces mesures sont effectuées avec un spectrofluorimètre à des longueurs d'onde d'excitation et d'émission de 485 nm et 530 nm, respectivement. Il convient de régler la sensibilité du spectrofluorimètre de façon à obtenir une différence de valeur aussi large que possible entre la DF maximale (insert sans cellules) et la DF minimale (insert contenant la monocouche confluente traitée avec le TN). En raison des différences entre les spectrofluorimètres employés, il est suggéré de fixer la sensibilité de manière à obtenir une intensité de fluorescence > 4 000 pour le témoin de la diffusion maximale de fluorescéine. La valeur correspondant à la DF maximale ne doit pas dépasser 9 999. L'intensité de fluorescence correspondant à la diffusion maximale doit rester comprise dans la fourchette de fonctionnement linéaire du spectrofluorimètre utilisé. Interprétation des résultats et modèle de prévision La quantité de DF est proportionnelle aux lésions induites par le produit chimique d'essai sur les jonctions serrées. Le pourcentage de DF pour chaque concentration du produit chimique évalué est calculé à partir des DF obtenues pour ce produit en fonction des DF correspondant au TN (relevées pour la monocouche confluente de cellules traitées avec le TN) et au témoin de la diffusion maximale (relevés de la quantité de DF à travers un insert dépourvu de cellules). Valeur moyenne de l'intensité de fluorescence de la diffusion maximale = x Valeur moyenne de l'intensité de fluorescence pour une diffusion de 0 % (TN) = y La diffusion de 100 % moyenne est calculée en soustrayant la diffusion de 0 % moyenne de la diffusion maximale moyenne, soit x – y = z On calcule le pourcentage de diffusion pour chaque dose fixée en soustrayant la diffusion de 0 % de la moyenne des intensités de fluorescence relevées pour les trois réplicats (m), puis en divisant la valeur obtenue par la diffusion de 100 %, soit %DF = [(m – y) / z] x 100 %, où:
On applique l'équation suivante pour calculer la concentration de produit chimique induisant une DF de 20 %: DFD = [(A – B) / (C – B)] × (MC – MB) + MB Où:
La valeur seuil de la DF20 permettant de prévoir que les produits chimiques sont corrosifs/fortement irritants pour l'œil est indiquée ci-dessous:
La méthode d'essai de DF est recommandée exclusivement pour l'identification des substances hydrosolubles corrosives ou fortement irritantes pour l'œil (catégorie 1 du SGH, catégorie 1 du CLP, catégorie I de l'EPA) (voir paragraphes 1 et 10). Pour qu'un produit chimique (substance ou mélange) hydrosoluble (3) (6) (7) soit classé comme provoquant des «lésions oculaires graves» (catégorie 1 du SGH et du CLP) ou comme «corrosif ou fortement irritant pour l'œil» (catégorie I de l'EPA), il faut que le produit chimique d'essai induise une DF20 ≤ 100 mg/ml. Acceptation des résultats La valeur moyenne de la diffusion maximale de fluorescéine (x) doit dépasser 4 000 (voir paragraphe 31), la moyenne de la diffusion de 0 % (y) doit être inférieure ou égale à 300, et la moyenne de la diffusion de 100 % (z) doit tomber entre 3 700 et 6 000. Un essai est considéré comme acceptable si le témoin positif provoque des lésions sur 20 à 40 % de la couche de cellules (déterminées en % de diffusion de fluorescéine). RÉSULTATS ET RAPPORTS Résultats Pour chaque série, les données des puits correspondant à chaque réplicat (par exemple les intensités de fluorescence et les pourcentages de DF calculés pour chaque produit chimique d'essai, y compris leur classification) sont présentées sous forme de tableau. Il convient en outre de rapporter les moyennes ± écart-type des mesures correspondant à chaque réplicat pour chaque série d'essais. Rapport d'essai Le rapport d'essai contient les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DIAGRAMME REPRÉSENTANT LES CELLULES MDCK CULTIVÉES SUR LA MEMBRANE D'UN INSERT POUR LA MÉTHODE D'ESSAI DE DF On cultive une couche confluente de cellules MDCK sur la membrane semi-perméable d'un insert. Les inserts sont placés dans les puits de plaques 24 puits. 
Graphique tiré de: Wilkinson, P.J. (2006) Development of an in vitro model to investigate repeat ocular exposure. Thèse de doctorat, Université de Nottingham, Royaume-Uni Appendice 2 DÉFINITIONS Catégorie I de l'EPA : produits chimiques induisant un effet corrosif (destruction irréversible des tissus oculaires) ou des complications cornéennes ou une irritation persistant plus de 21 jours (2). Catégorie 1 du SGH : lésion des tissus oculaires ou dégradation sévère de la vue, provoquée par l'application d'un produit chimique d'essai sur la face antérieure de l'œil, et qui n'est pas totalement réversible dans les 21 jours suivant l'application. CLP de l'UE [Règlement (CE) no 1272/2008 de la Commission européenne relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges] : vise la mise en œuvre dans l'Union européenne (UE) du SGH de l'ONU pour la classification des produits chimiques (substances et mélanges). Danger : propriété inhérente d'un agent ou d'une situation susceptible de provoquer des effets néfastes lorsqu'un organisme, un système ou une (sous-)population est exposé(e) à cet agent. DF20 : peut être estimée en déterminant la concentration à laquelle le produit chimique d'essai induit 20 % de diffusion de fluorescéine à travers la couche de cellules. Diffusion de fluorescéine : quantité de fluorescéine qui traverse la couche cellulaire, mesurée par spectrofluorimétrie. Essai substitutif : essai conçu pour remplacer un autre essai utilisé en routine et accepté, servant à l'identification des dangers et/ou l'évaluation des risques, et dont il a été démontré qu'il assure, par rapport à l'essai accepté, une protection équivalente ou accrue de la santé humaine ou animale ou de l'environnement, selon les cas, pour toutes les situations et substances d'essai possibles. Fiabilité : mesure dans laquelle la mise en œuvre d'une méthode d'essai peut être reproduite au fil du temps par un même laboratoire ou par plusieurs laboratoires en utilisant le même protocole. Elle est évaluée en calculant la reproductibilité intra-laboratoire et inter-laboratoires ainsi que la répétabilité intra-laboratoire. Lésion oculaire grave : lésion des tissus oculaires ou dégradation sévère de la vue, provoquée par l'application d'un produit chimique d'essai sur la face antérieure de l'œil, et qui n'est pas totalement réversible dans les 21 jours suivant l'application. Mélange : dans le contexte du SGH ONU, désigne un mélange ou une solution composé(e) de deux substances ou plus qui n'interagissent pas. Méthode d'essai validée : méthode d'essai ayant fait l'objet d'études de validation visant à déterminer sa pertinence (notamment sa précision) et sa fiabilité à des fins spécifiques. Il importe de noter que les performances d'une méthode d'essai validée peuvent être insuffisantes en termes de précision et de fiabilité pour qu'elle soit jugée acceptable pour les besoins envisagés (8). Non classé : produit chimique qui n'est pas classé comme irritant oculaire au sens des catégories 1, 2A, ou 2B du SGH (ONU); des catégories 1 ou 2 du CLP (UE); ou des catégories I, II et III de l'EPA (États-Unis). Pertinence : description de la relation entre l'essai et l'effet étudié, et détermination de son adéquation et de son utilité à des fins spécifiques. Elle définit le degré auquel l'essai mesure ou prédit correctement l'effet biologique d'intérêt. La pertinence tient compte de la précision (concordance) d'une méthode d'essai (8). Poids de la preuve : prise en compte des atouts et des faiblesses de divers éléments d'information en vue d'aboutir à une conclusion concernant le danger potentiel d'une substance chimique et d'étayer cette conclusion. Précision : degré de conformité entre les résultats de la méthode d'essai et les valeurs de référence acceptées. Elle constitue une mesure de performance de la méthode d'essai et l'un des aspects de sa pertinence. Ce terme et celui de «concordance» sont souvent utilisés indifféremment pour qualifier la proportion de résultats corrects d'une méthode d'essai. Produit chimique : une substance ou un mélange. Produit chimique corrosif pour l'œil : (a) produit chimique provoquant des lésions irréversibles des tissus oculaires. (b) produit chimique classé comme irritant oculaire dans la catégorie 1 du SGH (ONU), la catégorie 1 du CLP (UE) ou la catégorie I de l'EPA (États-Unis). Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Produit chimique d'épreuve de compétence : produit chimique de la liste des produits chimiques de référence qu'un laboratoire peut employer pour démontrer sa compétence à mettre en œuvre la méthode d'essai de référence validée, s'il ne l'a encore jamais utilisée. Produit chimique fortement irritant pour l'œil : (a) produit chimique provoquant des lésions des tissus oculaires suite à une application sur la face antérieure de l'œil, qui ne sont pas réversibles dans les 21 jours suivant l'application ou entraînent une dégradation sévère de la vue; (b) produit chimique classé comme irritant oculaire dans la catégorie 1 du SGH (ONU), la catégorie 1 du CLP (UE) ou la catégorie I de l'EPA (États-Unis). Produit chimique irritant pour l'œil : (a) produit chimique provoquant une modification réversible de l'œil suite à une application sur la face antérieure de l'œil; (b) produit chimique classé comme irritant oculaire dans les catégories 2A ou 2B du SGH (ONU), la catégorie 2 du CLP (UE) ou les catégories II ou III de l'EPA (États-Unis). Sensibilité : proportion des produits chimiques positifs/actifs correctement classés par l'essai. Il s'agit d'une mesure de la précision d'une méthode d'essai produisant des résultats catégoriels, et d'un élément important à prendre en considération pour évaluer la pertinence d'une méthode d'essai (8). SGH [Système général harmonisé de classification et d'étiquetage des produits chimiques des Nations Unies (ONU)] : système proposant la classification des produits chimiques (substances et mélanges) conformément à des types et des niveaux normalisés de dangers physiques, sanitaires et environnementaux, ainsi que la communication des éléments d'information correspondants, notamment par des pictogrammes, mentions d'avertissement, mentions de danger, conseils de prudence et fiches de données de sécurité, afin de diffuser des informations sur leurs effets indésirables dans le but de protéger les personnes (en particulier les employeurs, employés, transporteurs, consommateurs et personnels des services d'urgence) et l'environnement. Spécificité : proportion des produits chimiques négatifs/inactifs qui sont correctement classées par l'essai. Il s'agit d'une mesure de la précision d'une méthode d'essai produisant des résultats catégoriels, et d'un élément important à prendre en considération pour évaluer la pertinence d'une méthode d'essai. Stratégie d'essais à plusieurs niveaux : démarche expérimentale séquentielle consistant à examiner toutes les informations existantes sur un produit chimique d'essai dans un ordre déterminé, en ayant recours à chaque étape à un processus d'analyse du poids de la preuve pour déterminer si les informations disponibles sont suffisantes pour décider d'une classification dans une catégorie de danger, avant de passer à l'étape suivante. Si le potentiel d'irritation d'un produit chimique d'essai peut être déterminé sur la base des informations existantes, aucun essai supplémentaire n'est nécessaire. Dans le cas contraire, une procédure expérimentale progressive de type séquentiel sur des animaux est alors lancée jusqu'à ce qu'une classification sans équivoque puisse être effectuée. Substance : dans le contexte du SGH de l'ONU, désigne un élément chimique et ses composés à l'état naturel ou obtenus par un processus de fabrication, y compris tout additif nécessaire pour préserver la stabilité du produit et toute impureté résultant du processus mis en œuvre, mais à l'exclusion de tout solvant pouvant être séparé de la substance sans en affecter la stabilité ni modifier la composition. Taux de faux négatifs : proportion de tous les produits chimiques positifs faussement identifiés comme négatifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Taux de faux positifs : proportion de tous les produits chimiques négatifs faussement identifiés comme positifs par une méthode d'essai. Il s'agit d'un indicateur de performance de la méthode d'essai. Témoin de solvant/véhicule : échantillon non traité contenant tous les composants d'un système d'essai, y compris le solvant ou véhicule utilisé pour tester les échantillons témoins traités ou non avec la substance d'essai afin de déterminer une réponse de référence pour les échantillons traités avec la substance d'essai dissoute dans le même solvant ou véhicule. Testé avec un témoin négatif concurrent, cet échantillon indique également si le solvant ou véhicule interagit avec le système d'essai. Témoin négatif : réplicat non traité contenant tous les composants d'un système d'essai. Cet échantillon subit les mêmes procédures que les échantillons traités avec le produit chimique d'essai et les autres échantillons témoins afin de déterminer si le solvant interagit avec le système d'essai. Témoin positif : réplicat contenant tous les composants d'un système d'essai et traité avec un produit chimique induisant notoirement une réponse positive. Pour permettre d'évaluer la variabilité de la réponse du témoin positif dans le temps, l'ampleur de la réponse positive ne doit pas être excessive. Appendice 3 PRODUITS CHIMIQUES D'ÉPREUVE DE COMPÉTENCE POUR LA MÉTHODE D'ESSAI DE DF Avant d'utiliser en routine la présente méthode d'essai, les laboratoires démontrent leurs compétences techniques en déterminant correctement la catégorie de corrosivité oculaire des 8 produits chimiques recommandés dans le tableau 1. Ces produits chimiques ont été sélectionnés de façon à représenter la gamme des réactions locales d'irritation/de corrosion oculaire, sur la base des résultats de l'essai in vivo sur œil de lapin [ligne directrice 405, méthode d'essai B.5 (5)] (c'est-à-dire les catégories 1, 2A, 2B, ou Non classé du SGH de l'ONU). Toutefois, compte tenu de l'utilité validée de ces essais (pour identifier uniquement les substances corrosives/fortement irritantes pour l'œil), seuls deux résultats expérimentaux obtenus à des fins de classification (substance corrosive/fortement irritante ou substance non corrosive/non sévèrement irritante) permettent de démontrer les compétences des laboratoires. Les autres critères de sélection retenus ont trait à la disponibilité des produits chimiques dans le commerce, à la disponibilité de données de référence in vivo de bonne qualité, et à l'existence de données de bonne qualité obtenues par la méthode d'essai de DF. C'est pourquoi les produits chimiques d'épreuve de compétence choisis sont issus du «Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing» (8), qui a servi à la validation rétrospective de la méthode d'essai de DF. Tableau 1 Produits chimiques recommandés pour démontrer la compétence technique des laboratoires au regard de l'essai de DF
B.62 Test des comètes in vivo en conditions alcalines sur cellules de mammifères INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 489 (2016) de l'OCDE pour les essais de produits chimiques. Le test des comètes in vivo en conditions alcalines (technique d'électrophorèse de cellules isolées en gel d'agarose), appelé simplement ci-après “test des comètes”, est utilisé pour la détection des cassures de brins d'ADN dans des cellules ou noyaux isolés à partir de divers tissus d'animaux, habituellement des rongeurs, qui ont été exposés à un/des produit(s) potentiellement génotoxique(s). Le test des comètes a été examiné et des recommandations ont été publiées par divers groupes d'experts (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). La présente méthode d'essai s'inscrit dans une série de méthodes d'essai sur la toxicologie génétique. Par ailleurs, un document de l'OCDE qui fournit des informations succintes sur les essais de génotoxicité et donne une vue d'ensemble des modifications récemment apportées à ces lignes directrices a été élaboré (1). Le test des comètes a pour objet d'identifier les produits chimiques causant des dommages à l'ADN. En conditions alcalines (pH > 13), le test des comètes peut détecter les cassures simple- et double-brin résultant, par exemple, d'interactions directes avec l'ADN, de sites alcali-labiles ou de cassures transitoires liées à des mécanismes de réparation par excision. Ces cassures peuvent être réparées, leur effet étant alors non persistant, elles peuvent être létales pour la cellule, ou elles peuvent se fixer sous forme de mutation stable se traduisant par une altération viable permanente. Elles peuvent aussi entraîner des dommages chromosomiques du type de ceux qui sont associés à de nombreuses maladies humaines comme le cancer. Des études de validation formelle du test des comètes in vivo sur les rongeurs ont été coordonnées en 2006-2012 par le Centre japonais pour la validation des méthodes alternatives (JaCVAM), en collaboration avec le Centre européen pour la validation des méthodes alternatives (ECVAM), le Comité de coordination inter-agences pour la validation des méthodes alternatives (ICCVAM) et le Centre inter-agences pour l'évaluation des méthodes toxicologiques alternatives du NTP (NICEATM) (12). La présente méthode d'essai indique les applications recommandées et les limites du test des comètes; elle est fondée sur le protocole final (12) utilisé dans les essais de validation, et sur d'autres données pertinentes, publiées ou non (données privées des laboratoires). Les termes-clés sont définis à l'appendice 1. On notera qu'un grand nombre de supports différents peuvent être utilisés pour ce test (lames de microscope, spots de gel, plaques 96 puits, etc.). Pour des raisons pratiques, le terme de «lame» désigne dans ce qui suit l'ensemble de ces supports. REMARQUES PRÉLIMINAIRES Le test des comètes est une méthode utilisée pour mesurer les cassures de brins d'ADN dans les cellules eucaryotes. Des cellules/noyaux isolés inclus dans un gel d'agarose déposé sur une lame sont lysés sous l'action d'un détergent et de sels à forte concentration. Cette étape de lyse digère les membranes cellulaires et nucléaires et libère des boucles d'ADN enroulé, généralement appelées nucléoïdes ou fragments d'ADN. L'électrophorèse à pH élevé produit des structures ressemblant à des comètes qui, si l'on utilise les colorants fluorescents appropriés, peuvent être observées par microscopie à fluorescence; les fragments d'ADN migrent de la «tête» vers la «queue» de la comète, selon leur taille, et l'intensité de fluorescence de la queue par rapport à l'intensité totale (tête plus queue) reflète l'ampleur des cassures de l'ADN (13) (14) (15). Le test des comètes in vivo en conditions alcalines se prête particulièrement bien à l'évaluation du risque génotoxique, dans la mesure où les réponses au test sont dépendantes de l'ADME (absorption, distribution, métabolisme et excrétion) in vivo, ainsi que des processus de réparation de l'ADN. Ces paramètres peuvent varier selon les espèces, les tissus et les types de dommages subis par l'ADN. Pour satisfaire aux exigences en matière de bien-être animal, en particulier en ce qui concerne la réduction de l'utilisation d'animaux (règle des 3Rs — Reduction, Refinement, Replacement, à savoir réduction du nombre d'animaux, réduction des souffrances des animaux, remplacement de l'expérimentation animale par d'autres méthodes), ce test peut aussi être intégré à d'autres études toxicologiques, telles que les études de toxicité à doses répétées (10) (16) (17), ou le paramètre étudié peut être combiné à d'autres paramètres de génotoxicité, dans le cas par exemple du test des micronoyaux sur érythrocytes de mammifères in vivo (18) (19) (20). Le test des comètes est le plus souvent réalisé sur des rongeurs, bien qu'il ait été appliqué à d'autres espèces, mammifères ou non. L'utilisation d'espèces autres que des rongeurs devra être justifiée au cas par cas du point de vue scientifique et éthique, et il est fortement recommandé de ne réaliser le test des comètes sur des espèces autres que des rongeurs que dans le cadre d'une autre étude de toxicité, et non en tant qu'essai indépendant. La sélection de la voie d'exposition et du/des tissu(s) à étudier sera fondée sur l'ensemble des connaissances existantes/disponibles concernant les produits chimiques testés, par exemple la voie d'exposition humaine prévue/anticipée, le métabolisme et la distribution, le potentiel d'effets au site de contact, les alertes structurelles, d'autres données de génotoxicité ou de toxicité, ainsi que l'objectif de l'étude. Le potentiel génotoxique des produits chimiques testés peut ainsi être étudié, s'il y a lieu, sur le(s) tissu(s) cible(s) d'un effet cancérogène et/ou d'autres effets toxiques. Le test est également considéré comme utile pour explorer plus avant un effet génotoxique détecté dans un système in vitro. Il est opportun de réaliser un test des comètes in vivo sur un tissu donné si l'on peut raisonnablement s'attendre à ce que ce tissu soit exposé de façon adéquate. Les études de validation les plus complètes dont le test ait fait l'objet ont porté sur les tissus somatiques de rats mâles, dans le cadre d'études interlaboratoires internationales du JaCVAM (12) ou dans Rothfuss et al. 2010 (10). Le foie et l'estomac ont été utilisés dans les études de validation internationales du JaCVAM. Le foie, parce que c'est l'organe le plus actif dans le métabolisme des produits chimiques, et que c'est fréquemment un organe cible pour la cancérogénicité. L'estomac, parce que c'est généralement le premier site de contact avec les produits chimiques en cas d'exposition orale, bien que d'autres régions du tractus gastro-intestinal, comme le duodénum et le jéjunum, puissent aussi être envisagées comme sites de contact et soient peut-être plus pertinentes pour l'homme que l'estomac glandulaire des rongeurs. On veillera à ce que ces tissus ne soient pas exposés à des concentrations trop élevées du produit chimique testé (21). La technique est applicable en principe à tout tissu dont il est possible de dériver des suspensions de cellules/noyaux isolés analysables. Les données privées de nombreux laboratoires démontrent qu'elle peut être appliquée avec succès à un grand nombre de tissus différents, et de nombreuses publications montrent son applicabilité à des organes ou tissus autres que le foie et l'estomac, comme le jéjunum (22), les reins (23) (24), la peau (25) (26), ou des cellules provenant de la vessie (27) (28) ou de lavages pulmonaires ou bronchoalvéolaires (dans le cas d'études portant sur des substances inhalées) (29) (30); des tests ont également été réalisés sur plusieurs organes simultanément (31) (32). S'il peut être intéressant d'étudier les effets génotoxiques sur des cellules germinales, on notera que le test des comètes standard en conditions alcalines tel qu'il est décrit dans la présente méthode d'essai n'est pas considéré comme approprié pour mesurer les cassures de brins d'ADN dans des cellules germinales matures. Étant donné que, s'agissant des dommages à l'ADN, des niveaux de fond élevés et variables ont été rapportés dans une revue de la littérature portant sur l'utilisation du test des comètes dans les études de génotoxicité sur les cellules germinales (33), des modifications du protocole et des études améliorées de validation et de standardisation sont jugées nécessaires avant que le test des comètes sur cellules germinales matures (cellules spermatiques, par exemple) puisse être inclus dans la présente méthode d'essai. De plus, le régime d'exposition recommandé dans cette méthode d'essai n'est pas optimal et seuls des temps d'exposition ou d'échantillonnage plus longs permettraient une analyse valable des cassures de brins d'ADN dans le sperme mature. Les effets génotoxiques mesurés par le test des comètes sur des cellules testiculaires à différents stades de différenciation ont été décrits dans la littérature (34) (35). Il convient cependant de noter que les gonades contiennent un mélange de cellules somatiques et de cellules germinales. Pour cette raison, des résultats positifs sur l'ensemble des gonades (testicules) ne témoignent pas nécessairement de dommages touchant les cellules germinales; ils indiquent néanmoins que le/les produit(s) chimique(s) testé(s) et/ou ses/leurs métabolites ont atteint les gonades. Les pontages ne peuvent pas être détectés de façon fiable dans les conditions expérimentales standard du test des comètes. Dans certaines conditions expérimentales modifiées, des pontages ADN-ADN et ADN-protéine, ainsi que d'autres modifications des bases telles que des bases oxydées pourraient être détectées (23) (36) (37) (38) (39). Des études complémentaires seraient nécessaires pour caractériser de façon adéquate les modifications à apporter au protocole. La détection des agents responsables de pontages n'est donc pas l'objectif premier du test décrit ici. Celui-ci n'est pas adapté, même en cas de modifications, pour la détection des substances aneugènes. Dans l'état actuel des connaissances, le test des comètes in vivo présente une série d'autres limites (voir l'appendice 3). Il faut s'attendre à ce que la présente méthode d'essai soit révisée à l'avenir et modifiée s'il y a lieu à la lumière des acquis de l'expérience. Avant d'utiliser la méthode d'essai sur un mélange pour générer des données avec pour objectif recherché l'application réglementaire, on examinera si, et si oui pourquoi, elle peut fournir des résultats adéquats à cette fin. De telles considérations ne sont pas nécessaires quand les exigences réglementaires stipulent que le mélange doit être testé. PRINCIPE DE LA MÉTHODE Les animaux sont exposés au produit chimique d'essai par une voie d'exposition idoine. Une description précise de l'administration des doses et de l'échantillonnage est donnée aux paragraphes 36-40. Au(x) moment(s) retenu(s) pour l'échantillonnage, les tissus à étudier sont disséqués et des suspensions de cellules/noyaux isolés sont préparées (une perfusion in situ peut être pratiquée si cela est jugé utile, par exemple, pour le foie) et incluses dans un gel d'agarose pour être fixées sur des lames. Les cellules/noyaux sont traités avec un tampon de lyse pour éliminer la membrane cellulaire et/ou nucléaire, et exposés à une base forte (de pH ≥ 13, par exemple) pour permettre le déroulement de l'ADN et la libération des boucles et fragments d'ADN déroulés. L'ADN nucléaire dans l'agar est alors soumis à l'électrophorèse. Les molécules d'ADN normales, non fragmentées, restent dans la position où l'ADN nucléaire se trouvait dans l'agar, tandis que tous les fragments d'ADN et boucles d'ADN déroulées migrent vers l'anode. Après l'électrophorèse, l'ADN est visualisé au moyen d'un colorant fluorescent approprié. Les préparations doivent être analysées au microscope et au moyen de systèmes d'analyse d'image automatiques ou semi-automatiques. L'ampleur de la migration de l'ADN au cours de l'électrophorèse et la distance de migration reflètent la quantité et la taille des fragments d'ADN. Différents paramètres peuvent être évalués dans le test des comètes. La proportion d'ADN présent dans la queue, ou intensité de la queue ( % tail DNA, ou % tail intensity) a été recommandée pour évaluer les dommages à l'ADN (12) (40) (41) (42). Après analyse d'un nombre suffisant de noyaux, les résultats du test sont interprétés à l'aide de méthodes d'analyse appropriées. On notera que les modifications apportées à des aspects de la méthodologie tels que la préparation des échantillons, les conditions d'électrophorèse, les paramètres d'analyse visuelle (intensité du colorant, intensité lumineuse de l'ampoule du microscope, par exemple, ou mise en place de filtres sur le microscope et paramètres dynamiques de la caméra) et les conditions ambiantes (éclairage de fond, par exemple) peuvent affecter la migration de l'ADN (43) (44) (45) (46). VÉRIFICATION DES COMPETENCES DU LABORATOIRE Chaque laboratoire doit établir ses compétences pour la réalisation du test des comètes, en démontrant son aptitude à obtenir des suspensions de cellules ou de noyaux isolés d'une qualité suffisante pour chaque tissu cible de chaque espèce étudiée. La qualité des préparations sera évaluée avant tout d'après le pourcentage d'ADN présent dans la queue des comètes chez les animaux traités par le véhicule, pour lesquels les valeurs doivent se situer dans une plage basse reproductible. Les données actuelles suggèrent que le pourcentage moyen d'ADN de queue du groupe (d'après la moyenne des médianes — voir le paragraphe 57 pour ces termes) dans le foie de rat devrait, de préférence, ne pas dépasser 6 %, ce qui serait cohérent avec les valeurs des essais de validation du JaCVAM (12) et d'autres données publiées ou privées. On ne dispose pas de données suffisantes à l'heure actuelle pour formuler des recommandations sur les plages optimales ou acceptables concernant d'autres tissus, ce qui n'exclut pas de recourir à d'autres tissus si cela se justifie. Le rapport d'essai devra rendre compte de façon appropriée de la conduite du test des comètes sur ces tissus en faisant référence à la littérature publiée ou à des données privées. Premièrement, un faible pourcentage d'ADN de queue chez les témoins est souhaitable afin de disposer d'une plage dynamique suffisante pour détecter un effet positif. Deuxièmement, chaque laboratoire doit être en mesure de reproduire les réponses attendues pour des mutagènes directs et des promutagènes, pour différents modes d'action, selon les exemples proposés au tableau 1 (paragraphe 29). Des substances positives peuvent être sélectionnées, par exemple à partir des essais de validation du JaCVAM (12) ou d'autres données publiées (voir paragraphe 9), s'il y a lieu, moyennant une justification et en établissant la preuve de réponses clairement positives dans les tissus d'intérêt. La capacité à détecter les effets faibles de mutagènes connus tels que l'EMS à faibles doses doit également être démontrée, en établissant par exemple des relations dose-réponse avec des nombres de doses et des écarts entre doses appropriés. Les efforts doivent porter dans un premier temps sur l'établissement des compétences pour les tissus les plus couramment utilisés, le foie de rongeurs par exemple, pour lesquels il est possible d'effectuer une comparaison avec les données existantes et les résultats attendus (12). Les données provenant d'autres tissus (estomac/duodénum/jéjunum, sang, etc.) pourraient être collectées simultanément. Le laboratoire devra faire la preuve de sa compétence pour chaque tissu de chaque espèce qu'il prévoit d'étudier, et démontrer qu'une réponse positive acceptable peut être obtenue avec un mutagène connu (l'EMS, par exemple) sur ces tissus. Il convient de collecter les données sur les témoins négatifs/traités par le véhicule afin de démontrer la reproductibilité des réponses négatives et d'établir que les aspects techniques de l'essai ont été correctement maîtrisés, ou de suggérer la nécessité d'établir de nouvelles plages de données des témoins historiques (voir paragraphe 22). On notera que, si plusieurs tissus peuvent être collectés lors de la nécropsie et traités en vue de l'analyse des comètes, le laboratoire doit être compétent pour recueillir différents tissus sur un même animal, assurant ainsi qu'aucune lésion potentielle de l'ADN ne sera négligée et que l'analyse des comètes ne sera pas compromise. L'intervalle de temps entre le sacrifice des animaux et le prélèvement des tissus en vue de leur traitement peut être critique (voir paragraphe 44). Le bien-être animal doit être pris en compte lors du développement des compétences pour ce test, et des tissus provenant d'animaux utilisés pour d'autres tests peuvent être utilisés pour se former aux différents aspects du test. De plus, il peut ne pas être nécessaire de conduire une étude complète lors des étapes d'établissement d'une nouvelle méthode d'essai dans un laboratoire, et il est possible de réduire le nombre d'animaux ou le nombre de concentrations étudiées lors de l'acquisition des gestes nécessaires. Données des témoins historiques Dans le cadre de la vérification des compétences, le laboratoire mettra en place une base de données historiques établissant les plages et distributions des témoins positifs et négatifs pour les tissus et espèces étudiés. On trouve dans la littérature (47) des recommandations sur la façon de constituer et d'utiliser ces données historiques (critères d'inclusion et d'exclusion des données dans la base, et critères d'acceptabilité pour une expérimentation donnée). Différents tissus et différentes espèces, ainsi que différents véhicules et voies d'administration, peuvent donner des pourcentages d'ADN dans la queue différents pour les témoins négatifs. Il importe donc d'établir les plages des témoins négatifs pour chaque tissu et chaque espèce. Les laboratoires doivent avoir recours à des méthodes de contrôle de la qualité telles que des graphiques statistiques [cartes C ou cartes X-barre, par exemple (48)], afin de déterminer la variabilité de leurs données et de démontrer leur maîtrise de la méthodologie. Le choix des substances chimiques utilisées comme témoins positifs, des gammes de doses et des conditions expérimentales (conditions de l'électrophorèse, par exemple) peut devoir être optimisé pour la détection d'effets faibles (voir paragraphe 17). Toute modification du protocole expérimental doit être étudiée en termes de cohérence avec les bases de données des témoins historiques existantes du laboratoire. Toute incohérence majeure doit conduire à l'établissement d'une nouvelle base de données des témoins historiques. DESCRIPTION DE LA METHODE Préparations Choix des espèces animales On choisit habituellement des souches communément utilisées en laboratoire de jeunes rongeurs adultes sains (âgés de 6 à 10 semaines au début du traitement, bien que des animaux un peu plus âgés soient également acceptables). Le choix de l'espèce de rongeur doit être fondé sur (i) les espèces utilisées dans d'autres études de toxicité (pour qu'il soit possible de corréler les données et pour permettre des études intégrées), (ii) les espèces qui ont développé des tumeurs dans une étude de cancérogénicité (lors d'investigations portant sur le mécanisme de cancérogenèse), ou (iii) les espèces dont le métabolisme est le plus proche de celui de l'homme, si elles sont connues. Les rats sont couramment utilisés dans cet essai. Il est toutefois possible de recourir à d'autres espèces si cela est justifié d'un point de vue éthique et scientifique. Conditions d'encagement et d'alimentation des animaux Pour les rongeurs, la température de l'animalerie est idéalement de 22 °C (± 3 °C). L'humidité relative, qui est idéalement de 50 à 60 %, doit atteindre au moins 30 % et de préférence ne pas dépasser 70 %, sauf durant le nettoyage du local. L'éclairage est artificiel, la séquence d'éclairage étant de 12 heures de clarté et 12 heures d'obscurité. Le régime alimentaire des animaux est le régime classique de laboratoire avec eau potable à satiété. Le choix des aliments peut être influencé par la nécessité d'assurer une bonne incorporation du produit chimique dans la nourriture si l'administration se fait par cette voie. Les rongeurs sont mis en cage par petits groupes comprenant généralement un maximum de cinq animaux du même sexe si aucun comportement agressif n'est à craindre. Les animaux peuvent être encagés individuellement si cela est justifié du point de vue scientifique. Dans la mesure du possible, le fond des cages doit être plein, les fonds grillagés pouvant provoquer de graves blessures (49). Un enrichissement environnemental approprié doit être fourni. Preparation des animaux Les animaux sont répartis de manière aléatoire entre les groupes traités et les groupes témoins. Ils sont identifiés individuellement et gardés dans leurs cages pendant au moins cinq jours avant le commencement de l'étude, afin qu'ils s'acclimatent aux conditions du laboratoire. La méthode d'identification individuelle des animaux doit être la moins invasive possible. Les méthodes appropriées sont notamment le baguage, l'étiquetage, la pose d'une puce électronique et l'identification biométrique. L'utilisation d'agrafes au niveau de la patte ou de l'oreille n'est pas scientifiquement justifiée pour ces essais. Les cages doivent être placées de façon à réduire au minimum l'influence éventuelle de leur disposition sur les résultats. Au début de l'étude, la variation pondérale des animaux doit être minimale et ne pas excéder ± 20 %. Préparation des doses Lorsque les produits chimiques testés sont solides, ils sont dissous ou mis en suspension dans des véhicules appropriés, ou incorporés aux aliments ou à l'eau de boisson avant d'être administrés aux animaux. Les produits chimiques liquides peuvent être administrés directement ou dilués avant d'être administrés. En cas d'exposition par inhalation, les produits chimiques testés peuvent être administrés sous forme de gaz, de vapeur ou d'aérosol solide ou liquide, en fonction de leurs propriétés physico-chimiques (50) (51). On utilisera des préparations fraîches, sauf si l'on dispose de données qui démontrent la stabilité des préparations dans les conditions du stockage et définissent les conditions de stockage appropriées. Conditions de l'essai Véhicule Le véhicule ne doit pas produire d'effets toxiques aux volumes administrés, ni être suspecté de réagir avec les produits chimiques d'essai. Le recours à des véhicules inhabituels doit être justifié par des données de référence faisant état de leur compatibilité eu égard aux animaux d'essai, à la voie d'administration et à l'effet mesuré. Il est recommandé d'envisager en premier lieu l'utilisation d'un solvant/véhicule aqueux chaque fois que c'est possible. On notera que certains véhicules (en particulier les véhicules visqueux) peuvent induire une inflammation et augmenter le niveau de fond des cassures de brins d'ADN au site de contact, en particulier en cas d'administration répétée. Témoins Témoins positifs A l'heure actuelle, chaque essai doit normalement inclure un groupe d'au moins trois animaux analysables du même sexe, ou de chaque sexe si les deux sont utilisés (voir paragraphe 32), traités avec un produit chimique utilisé comme témoin positif. Il se peut qu'à l'avenir, le laboratoire puisse démontrer que ses compétences permettent de réduire le nombre de témoins positifs. Si plusieurs moments d'échantillonnage sont prévus (par exemple dans le cas d'un protocole comportant une seule administration), il n'y a lieu d'inclure des témoins positifs que pour l'un des moments d'échantillonnage, mais il importe de veiller à une répartition équilibrée (voir paragraphe 48). Il n'est pas nécessaire d'administrer les substances chimiques utilisées simultanément comme témoins positifs par la même voie que le produit chimique testé, mais il importe d'utiliser la même voie pour mesurer les effets au site de contact. Il doit être établi que les substances utilisées comme témoins positifs induisent des cassures de brins d'ADN dans tous les tissus d'intérêt pour le produit chimique testé, et l'EMS sera sans doute le témoin positif de choix dans la mesure où il a provoqué des cassures de brins d'ADN dans tous les tissus sur lesquels il a été étudié. Les doses des substances chimiques utilisées comme témoins positifs sont sélectionnées de manière à produire des effets modérés permettant une évaluation éclairée des performances et de la sensibilité du test; elles peuvent être fondées sur les courbes dose-réponse établies par le laboratoire dans le cadre de la démonstration de ses compétences. Le pourcentage d'ADN de queue chez les témoins positifs simultanés doit être en cohérence avec la plage préétablie par le laboratoire pour chaque tissu et moment d'échantillonnage pour l'espèce considérée (voir paragraphe 16). Des exemples de substances chimiques utilisées comme témoins positifs et de certains de leurs tissus cibles (chez les rongeurs) figurent au tableau 1. Des substances chimiques autres que celles du tableau 1 peuvent être sélectionnées si cela est scientifiquement justifié. Tableau 1 Exemples de substances utilisées comme témoins positifs et de certains de leurs tissus cibles Substances et no CAS Éthyl méthanesulfonate (no CAS 62-50-0), tout tissu Éthyl nitrosourée (no CAS 759-73-9), foie et estomac, duodénum ou jéjunum Méthyl méthanesulfonate (no CAS 66-27-3), foie, estomac, duodénum ou jéjunum, cellules obtenues par lavage pulmonaire et bronchoalvéolaire, reins, vessie, poumons, testicules et moelle osseuse/sang N-Méthyl-N'-nitro-N-nitrosoguanidine (no CAS 70-25-7), estomac, duodénum ou jéjunum 1,2-Diméthylhydrazine 2HCl (no CAS 306-37-6), foie et intestin N-méthyl-N-nitrosourée (no CAS 684-93-5), foie, moelle osseuse, sang, reins, estomac, jéjunum et cerveau. Témoins négatifs Un groupe d'animaux témoins négatifs auxquels est administré le véhicule seul, et qui sont traités par ailleurs de la même façon que les groupes exposés, doit être inclus dans chaque essai pour chaque moment d'échantillonnage et chaque tissu. Le pourcentage d'ADN de queue chez les animaux témoins négatifs doit se trouver dans la plage de référence préétablie par le laboratoire pour chaque tissu et chaque moment d'échantillonnage pour cette espèce (voir paragraphe 16). En l'absence de données historiques ou publiées sur les témoins montrant que le véhicule choisi, le nombre d'administrations ou la voie d'administration n'induisent aucun effet délétère ou génotoxique, des études préliminaires seront réalisées avant d'entreprendre l'étude complète, afin d'établir l'acceptabilité des témoins recevant le véhicule. MODE OPÉRATOIRE Nombre et sexe des animaux Bien qu'il existe peu de données sur des animaux femelles permettant des comparaisons entre sexes dans le cas du test des comètes, les réponses aux autres tests de génotoxicité in vivo sont généralement similaires chez les animaux mâles et femelles, et la plupart des études pourrait donc être réalisée sur l'un ou l'autre des deux sexes. Des données démontrant des différences significatives entre les mâles et les femelles (par exemple sur le plan de la toxicité systémique, du métabolisme, de la biodisponibilité, etc comprenant également des données provenant par exemple d'études de détermination des doses) encouragent l'utilisation des deux sexes. Il peut donc être plus approprié dans ce cas de mener une étude sur les deux sexes, par exemple dans le cadre d'une étude de toxicité à doses répétées. Il pourrait être judicieux de recourir à un plan factoriel en cas d'utilisation des deux sexes. Des précisions sur cette méthode d'analyse des données sont fournies à l'appendice 2. La taille des groupes au début de l'étude (et lors de l'établissement des compétences) doit permettre de disposer d'au moins cinq animaux analysables du même sexe ou de chaque sexe si les deux sont utilisés dans chaque groupe (et d'un nombre inférieur dans le groupe témoin positif concomitant — voir paragraphe 29). Si l'exposition humaine est spécifique pour un sexe, dans le cas par exemple de certains produits pharmaceutiques, l'essai sera pratiqué sur des animaux du sexe approprié. À titre d'information concernant le nombre maximum d'animaux généralement requis, une étude conduite selon les paramètres établis au paragraphe 33, impliquant trois groupes de traitement et des groupes concurrents de témoins négatifs et de témoins positifs (chaque groupe étant composé de cinq animaux du même sexe) nécessitera entre 25 et 35 animaux. PROGRAMME DE TRAITEMENT Les animaux doivent recevoir un traitement quotidien sur une période de 2 jours ou plus (c'est à dire deux traitements ou plus à intervalles de 24 heures environ), et les échantillons doivent être collectés une fois à 2-6 h (ou à Tmax) après le dernier traitement (12). Des échantillons provenant de programmes de traitement prolongés (dose quotidienne pendant 28 jours, par exemple) sont acceptables. Il a été démontré que le test des comètes pouvait être combiné avec succès au test des micronoyaux sur érythrocytes (10) (19). Cependant, il convient d'accorder une grande attention à la logistique nécessaire pour pratiquer l'échantillonnage des tissus en vue du test des comètes tout en respectant les exigences relatives à celui effectué en vue d'autres types d'évaluations toxicologiques. La collecte 24 h après la dernière dose, classique pour une étude de toxicité générale, n'est pas appropriée dans la plupart des cas (voir le paragraphe 40 sur le moment d'échantillonnage). L'utilisation d'autres programmes de traitement et d'échantillonnage devra être justifiée (voir appendice 3). On pourra par exemple recourir à un traitement unique avec échantillonnages multiples, mais une étude comportant une seule administration nécessitera plus d'animaux, plusieurs moments d'échantillonnage étant requis; cette solution peut toutefois être préférable dans certains cas, par exemple lorsque la substance testée induit une toxicité excessive en cas d'administrations répétées. Quelle que soit la façon dont l'essai est réalisé, elle est acceptable si le produit chimique testé donne une réponse positive ou, en cas de réponse négative, si une preuve directe ou indirecte de l'exposition du/des tissu(s) cible(s) ou de la toxicité pour ce(s) tissu(s) est apportée, ou si la dose limite est atteinte (voir paragraphe 36). Pour faciliter l'administration de volumes importants du produit chimique d'essai, celle-ci peut aussi être fractionnée, à raison de deux traitements le même jour à intervalle de deux à trois heures maximum. En pareil cas, le moment d'échantillonnage sera fixé en fonction du moment d'administration de la dernière fraction (voir paragraphe 40). Niveaux de dose Si l'on procède à une étude préliminaire de détermination des doses à administrer parce qu'on ne dispose pas de données fiables provenant d'autres études pertinentes pour orienter le choix des doses, l'étude préliminaire doit être effectuée dans le même laboratoire, en utilisant une espèce, une souche, un sexe et un régime de traitement identiques à ceux de l'étude principale, selon les principes en vigueur pour la conduite des études de détermination des doses. Cette étude devra avoir pour objectif de déterminer la dose maximale tolérée (DMT), définie comme la dose induisant de légers effets toxiques liés à la durée de l'étude (avec des signes cliniques clairs tels qu'un comportement ou des réactions anormales, une perte pondérale mineure ou un effet cytotoxique sur un tissu cible, par exemple), mais ne provoquant pas la mort ou des signes de douleur, de souffrance ou de détresse imposant de sacrifier les animaux. Pour un produit chimique non toxique administré sur une période de 14 jours ou plus, la dose (limite) maximale est de 1 000 mg/kg de poids corporel/jour. Pour des périodes d'administration inférieures à 14 jours, la dose (limite) maximale est de 2 000 mg/kg de poids corporel/jour. Pour certains types de produits chimiques d'essai (produits pharmaceutiques à usage humain, par exemple) faisant l'objet de réglementations spécifiques, ces limites peuvent être différentes. Les produits chimiques testés dont les propriétés toxicocinétiques sont saturées ou qui induisent un processus de détoxification pouvant se traduire par une baisse de l'exposition après une administration à long terme peuvent faire exception aux critères d'établissement des doses et doivent être évalués au cas par cas. Pour les versions aiguë et subaiguë du test des comètes, outre la dose maximale (DMT, dose maximale possible, exposition maximale ou dose limite), une série supplémentaire d'au moins deux doses décroissantes présentant des écarts adaptés (facteur inférieur à √10, de préférence) doit être sélectionnée pour chaque moment d'échantillonnage afin de mettre en évidence d'éventuelles relations dose-effet. Cependant, les niveaux de dose utilisés doivent aussi couvrir dans la mesure du possible un intervalle compris entre la dose maximale et une dose produisant peu ou pas d'effet toxique. Lorsqu'une toxicité pour un tissu cible est observée à tous les niveaux de dose administrés, il est conseillé de procéder à des études complémentaires à des doses non toxiques (voir paragraphes 54-55). Les études visant à établir plus précisément l'allure de la courbe dose-réponse peuvent nécessiter un ou plusieurs groupes de traitement supplémentaires. Administration des doses Lors de la conception d'un essai, il convient de tenir compte de la voie d'exposition humaine anticipée. Par conséquent, les voies d'administration telles que l'alimentation, l'eau de boisson, l'inhalation, l'implantation ainsi que les voies topique, sous-cutanée, intraveineuse, orale (par gavage) et intratrachéale sont autant de choix valables, sous réserve qu'ils soient justifiés. Dans tous les cas, la voie retenue doit permettre une exposition adéquate du/des tissu(s) cible(s). L'injection intrapéritonéale n'est généralement pas recommandée, car elle n'est pas représentative de l'exposition humaine, et ne sera utilisée qu'en cas de justification spécifique (par exemple pour certaines substances chimiques utilisées comme témoins positifs, à des fins d'investigation, ou pour des médicaments administrés par voie intrapéritonéale). Le volume maximal de liquide administrable en une fois par gavage ou par injection dépend de la taille de l'animal. Ce volume ne devrait pas dépasser 1 ml/100 g de poids corporel, sauf dans le cas des solutions aqueuses où l'on peut aller jusqu'à 2 ml/100 g de poids corporel. L'utilisation de volumes plus importants (si elle est autorisée par la législation relative au bien-être animal) doit être justifiée. Chaque fois que cela est possible, les différents niveaux de dose doivent être obtenus en ajustant la concentration de la formulation administrée pour assurer à tous les niveaux de dose un volume constant rapporté au poids corporel. Moment d'échantillonnage Le moment d'échantillonnage est une variable critique car il dépend de la période nécessaire pour que les substances chimiques atteignent leur concentration maximale dans le tissu cible et que des cassures de brins d'ADN soient induites, mais doit se situer avant que ces cassures ne disparaissent, ne soient réparées ou ne provoquent la mort de la cellule. La persistance de certaines des lésions conduisant aux cassures de brins d'ADN détectées par le test des comètes peut être très brève, du moins pour certains produits chimiques testés in vitro (52) (53). En conséquence, si des lésions transitoires de ce type sont suspectées, des mesures doivent être prises pour en limiter la disparition, en s'assurant que les tissus sont échantillonnés suffisamment tôt, éventuellement avant les délais par défaut indiqués ci-dessous. Le moment d'échantillonnage optimal peut dépendre du produit chimique ou de la voie d'administration, se traduisant en général par une exposition rapide du tissu lors d'une administration par voie intraveineuse ou par inhalation. Par conséquent, les moments d'échantillonnage seront déterminés en fonction des données cinétiques, quand elles sont disponibles (par exemple, temps (Tmax) auquel le pic de concentration (Cmax) dans le plasma ou le tissu est atteint, ou à l'état stationnaire en cas d'administrations multiples). En l'absence de données cinétiques, un compromis approprié pour la mesure de la génotoxicité est l'échantillonnage à 2-6 h du dernier traitement pour deux traitements ou plus, ou à 2 6 h et à 16-26 h d'une administration unique, mais il faut veiller à nécropsier tous les animaux au même moment après la dernière (ou l'unique) dose. Des informations sur la survenue d'effets toxiques dans les organes cibles (lorsqu'elles sont disponibles) peuvent également être utilisées pour sélectionner les moments d'échantillonnage appropriés. Observations Des observations cliniques générales relatives à la santé des animaux seront recueillies et consignées au moins une fois par jour, de préférence à la ou aux mêmes heures, en prenant en considération la période où les effets anticipés devraient être les plus marqués après l'administration (54). Au moins deux fois par jour, l'ensemble des animaux fait l'objet d'un constat de morbidité et de mortalité. Dans les études de longue durée, tous les animaux doivent être pesés au moins une fois par semaine et à la fin de l'essai. La consommation d'aliments doit être mesurée à chaque renouvellement des aliments et au moins une fois par semaine. Si le produit chimique testé est administré dans l'eau de boisson, la consommation d'eau est mesurée à chaque changement d'eau et au moins une fois par semaine. Les animaux montrant des signes non létaux de toxicité excessive sont euthanasiés avant la fin de l'essai, et ne sont généralement pas utilisés pour le test des comètes. Collecte des tissus L'induction de cassures des brins d'ADN (comètes) pouvant être étudiée sur pratiquement tous les tissus, la justification du choix du/des tissu(s) à prélever doit être clairement établie, compte tenu de la raison présidant à la conduite de l'étude et de l'ensemble des données disponibles sur l'ADME, la génotoxicité, la cancérogénicité ou d'autres effets toxiques des produits chimiques étudiés. La voie d'administration (fondée sur la/les voie(s) d'exposition humaine potentielle(s)), la distribution et l'absorption tissulaires prévues, le rôle du métabolisme et le mécanisme d'action possible des produits chimiques étudiés sont des facteurs importants à prendre en considération. Le foie est le tissu qui a été le plus fréquemment étudié et sur lequel les données sont les plus nombreuses. C'est pourquoi, en l'absence de données préexistantes et d'éléments incitant à choisir un tissu donné, le choix du foie est justifié par le fait que c'est le siège principal du métabolisme des xénobiotiques et qu'il est souvent fortement exposé tant aux produits chimiques initiaux qu'à leur(s) métabolite(s). Dans certains cas, l'examen d'un site de contact direct (l'estomac glandulaire ou le duodénum/jéjunum pour les produits administrés par voie orale, par exemple, ou les poumons pour les produits inhalés) peut être particulièrement indiqué. Des tissus complémentaires ou différents pourront être sélectionnés compte tenu des raisons justifiant la conduite du test, mais il peut être utile d'examiner différents tissus prélevés sur les mêmes animaux, à condition que le laboratoire ait fait la preuve de ses compétences pour ces tissus et de son aptitude à manipuler simultanément différents tissus. Préparation des prélèvements Pour les opérations décrites dans les paragraphes suivants (44-49), il importe que toutes les solutions ou suspensions stables soient utilisées avant leur date d'expiration, ou que des solutions ou suspensions fraîches soient préparées si nécessaire. De plus, dans ce qui suit, le temps passé à (i) prélever chaque tissu après la nécropsie, (ii) traiter chaque tissu pour obtenir des suspensions de cellules/noyaux, et (iii) traiter la suspension et préparer les lames, est considéré chaque fois comme une variable critique (voir définitions à l'appendice 1), et des durées acceptables pour chacune de ces étapes doivent avoir été fixées lors de l'établissement de la méthode et de la démonstration des compétences du laboratoire. Les animaux seront sacrifiés conformément à la législation en vigueur sur le bien-être animal et à la règle des 3R, au(x) moment(s) approprié(s) après la dernière administration du produit chimique d'essai. Le(s) tissu(s) sélectionné(s) est/sont prélevé(s), disséqué(s), une portion est collectée pour le test des comètes et, au même moment, un échantillon de la même partie du tissu est découpé et placé dans une solution de formaldéhyde ou un fixateur approprié pour une éventuelle analyse histopathologique (voir paragraphe 55) selon les méthodes standards (12). Le tissu destiné au test des comètes est placé dans du tampon de broyage, rincé soigneusement au tampon de broyage froid pour éliminer le sang résiduel, et conservé dans du tampon de broyage glacé jusqu'au traitement. Une perfusion in situ peut aussi être pratiquée pour le foie ou les reins, par exemple. Un grand nombre de méthodes d'isolement des cellules/noyaux ont été publiées. Elles incluent le broyage de tissus tels que ceux du foie ou des reins, le raclage de la surface des muqueuses dans le cas du tractus gastro-intestinal, l'homogénéisation et la digestion enzymatique. Les essais de validation du JaCVAM n'ont porté que sur des cellules isolées; pour l'établissement de la méthode et pour pouvoir faire référence aux essais de validation du JaCVAM lors de la démonstration des compétences, on préférera donc les cellules isolées. Cependant, il a été établi que les résultats du test ne diffèrent pas fondamentalement selon que l'on a recours à des cellules ou des noyaux isolés (8). De même, différentes méthodes d'isolement des cellules/noyaux (homogénéisation, broyage, digestion enzymatique et filtration sur tamis) ont donné des résultats comparables (55). On peut donc utiliser soit des cellules isolées soit des noyaux isolés. Le laboratoire évaluera et validera soigneusement les méthodes d'isolement des cellules/noyaux par type de tissu. Comme indiqué au paragraphe 40, la persistance de certaines des lésions conduisant à des cassures de brins d'ADN détectées par le test des comètes peut être très brève (40) (41). Quelle que soit la méthode employée pour préparer les suspensions de cellules/noyaux isolés, il importe donc que les tissus soient traités dès que possible après que les animaux ont été sacrifiés, et placés dans des conditions visant à réduire la disparition des lésions (maintien des tissus à basse température, par exemple). Il convient de conserver les suspensions cellulaires à très basse température jusqu'à ce qu'elles soient prêtes à être utilisées, afin de pouvoir mettre en évidence une variation inter-échantillons minimale et des réponses appropriées chez les témoins positifs et négatifs. PRÉPARATION DES LAMES La préparation des lames doit intervenir dès que possible après la préparation des cellules/noyaux isolés (dans l'heure qui suit, idéalement), mais la température et le temps écoulé entre le sacrifice des animaux et la préparation des lames doivent être rigoureusement contrôlés et validés dans les conditions du laboratoire. Le volume de suspension cellulaire ajouté à l'agarose à bas point de fusion (habituellement de 0,5 à 1,0 %) pour préparer les lames ne doit pas réduire le pourcentage d'agarose à bas point de fusion à moins de 0,45 %. La densité optimale de cellules sera déterminée par le système d'analyse d'image utilisé pour le comptage des comètes. Lyse Les conditions de lyse constituent également une variable critique pouvant interférer avec les cassures de brins résultant d'un type spécifique de modifications de l'ADN (alkylations et formation d'adduits aux bases de l'ADN). Il est donc recommandé de maintenir les conditions de lyse aussi constantes que possible pour toutes les lames au cours d'une même expérimentation. Une fois préparées, les lames sont immergées dans une solution de lyse réfrigérée pendant au moins une heure (ou toute une nuit) à une température de l'ordre de 2 à 8 °C, en lumière tamisée (lumière jaune, par exemple) ou en environnement étanche à la lumière, pour éviter l'exposition à la lumière blanche, qui peut contenir des composantes UV. Après cette période d'incubation, on rince les lames pour les débarrasser du détergent et des sels résiduels avant l'étape de déroulement alcalin. On utilisera pour ce faire de l'eau purifiée, un tampon neutralisant ou un tampon phosphate. Un tampon d'électrophorèse peut également être utilisé. L'objectif est de maintenir des conditions alcalines dans la chambre d'électrophorèse. Déroulement et électrophorèse Les lames sont placées de façon aléatoire sur le plateau d'une unité d'électrophorèse sur gels immergés contenant suffisamment de solution d'électrophorèse pour que la surface des lames soit entièrement recouverte (la profondeur d'immersion doit, elle aussi, être cohérente d'une épreuve à l'autre). Dans un autre type d'unité d'électrophorèse utilisé pour le test des comètes, c'est-à-dire avec refroidissement actif, circulation du liquide de refroidissement et alimentation haute tension, l'intensité du courant électrique sera d'autant plus forte que la hauteur de solution sera élevée, à tension constante. Les lames doivent être réparties de façon équilibrée dans la cuve d'électrophorèse afin d'atténuer les effets d'éventuelles tendances ou les effets de bord dans la cuve et de limiter les variations entre lots; il faut donc que pour chaque épreuve d'électrophorèse, on utilise le même nombre de lames provenant de chaque animal, et des échantillons des différents groupes de doses, des témoins négatifs et des témoins positifs. Les lames doivent rester au moins 20 minutes dans la cuve pour que l'ADN se déroule, puis sont soumises à l'électrophorèse dans des conditions contrôlées assurant une sensibilité et une plage dynamique maximales pour l'essai (c'est-à-dire garantissant des pourcentages d'ADN de queue acceptables, chez les témoins négatifs et positifs, pour assurer une sensibilité maximale). Le degré de migration de l'ADN est proportionnel à la durée de l'électrophorèse, ainsi qu'au potentiel (V/cm). D'après les essais du JaCVAM, celui-ci pourrait être de 0,7 V/cm pendant au moins 20 minutes. La durée de l'électrophorèse est considérée comme une variable critique et le temps d'électrophorèse doit être réglé de façon à optimiser la plage dynamique. Des durées plus longues (30 ou 40 minutes, par exemple, pour gagner en sensibilité) se traduisent habituellement par des réponses positives plus nettes pour des mutagènes connus. Cependant, un allongement de la durée de l'électrophorèse peut aussi se traduire par une migration excessive dans les échantillons témoins. Chaque expérience doit être réalisée à tension constante, et la variabilité des autres paramètres doit se situer dans une plage étroite et spécifiée, par exemple, dans les essais du JaCVAM, 0,7 V/cm avec une intensité initiale de 300 mA. La profondeur du tampon doit être ajustée pour réaliser les conditions requises, et maintenue tout au long de l'expérimentation. Il convient de consigner l'intensité du courant au début et à la fin de l'électrophorèse. Les conditions optimales doivent donc être déterminées au cours de la démonstration initiale de compétences par le laboratoire pour chaque tissu étudié. La température de la solution d'électrophorèse au cours du déroulement et de l'électrophorèse doit être maintenue à un niveau bas (2-10 °C, habituellement) (10). Il convient de consigner la température de la solution d'électrophorèse au début du déroulement, au début de l'électrophorèse et à la fin de l'électrophorèse. Une fois l'électrophorèse terminée, les lames doivent être immergées/rincées dans le tampon de neutralisation pendant au moins 5 minutes. Les gels peuvent être colorés et observés à l'état «frais» (pendant 1 à 2 jours, par exemple) ou être déshydratés et observés ultérieurement (dans un délai de une à deux semaines après la coloration, par exemple) (56). Cependant, ces conditions doivent être validées lors de la démonstration de compétences et les données historiques doivent être recueillies et conservées séparément pour chacune de ces options. Dans le second cas, les lames doivent être déshydratées par immersion dans l'éthanol absolu pendant au moins cinq minutes, séchées à l'air libre puis stockées, soit à température ambiante, soit dans un récipient conservé au réfrigérateur jusqu'à la lecture. Méthodes de mesure L'évaluation quantitative des comètes fait appel à un système d'analyse d'image automatique ou semi-automatique. Les lames sont colorées par un colorant fluorescent approprié — SYBR Gold, Green I, iodure de propidium ou bromure d'éthidium, par exemple — et observées à un grossissement approprié (200x, par exemple) au microscope à épifluorescence équipé de détecteurs appropriés ou d'une caméra numérique (CCD, par exemple). Les cellules peuvent être classées en trois catégories décrites dans l'atlas des images de comètes (57), à savoir mesurable, non mesurable, cellule fantôme (voir le paragraphe 55 pour plus de précisions). Seules les cellules mesurables (tête et queue clairement définies, pas d'interférence avec les cellules voisines) doivent être classées en fonction du pourcentage d'ADN de queue, pour éviter les artefacts. Il n'est pas nécessaire d'indiquer la fréquence des cellules non mesurables. La fréquence des cellules fantômes est évaluée par un examen visuel (l'absence de tête clairement définie ayant pour conséquence que ces cellules ne sont pas bien détectées par le système d'analyse d'image) portant sur au moins 150 cellules par échantillon (voir paragraphe 56 pour plus de précisions) et mentionnée à part. Toutes les lames, y compris celles des témoins négatifs et positifs, doivent être codées indépendamment et évaluées «en aveugle», afin que l'analyste ignore à quel traitement elles correspondent. Pour chaque échantillon (par tissu et par animal), au moins 150 cellules (en dehors des cellules fantômes — voir paragraphe 56) doivent être analysées. L'évaluation de 150 cellules par animal chez au moins 5 animaux par dose (mais moins de 5 dans le groupe témoin positif concomitant — voir paragraphe 29) assure une puissance statistique satisfaisante, selon l'analyse de Smith et al., 2008 (5). Si des lames sont utilisées, cela peut représenter l'évaluation de 2 ou 3 lames par échantillon pour des groupes de 5 animaux. Plusieurs zones de la lame doivent être observées à une densité garantissant l'absence de chevauchement des queues. L'évaluation en bord de lame est à éviter. Les cassures de brins d'ADN dans le test des comètes peuvent être mesurées selon divers paramètres indépendants, tels que le pourcentage d'ADN de queue, la longueur de la queue ou le moment de la queue. Ces trois mesures peuvent être effectuées si l'on utilise un logiciel d'analyse d'image approprié. Cependant, le pourcentage d'ADN de queue (également appelé intensité de la queue) est recommandé pour l'évaluation et l'interprétation des résultats (12) (40) (41) (42); il est déterminé par l'intensité des fragments d'ADN présents dans la queue, exprimée en pourcentage de l'intensité totale de la cellule (13). Lésion des tissus et cytotoxicité Des résultats positifs au test des comètes peuvent ne pas être dus exclusivement à la génotoxicité, la toxicité pour les tissus cibles pouvant aussi se traduire par une augmentation de la migration de l'ADN (12) (41). A l'inverse, une cytotoxicité faible ou modérée est souvent observée dans le cas des substances génotoxiques connues (12); le test des comètes ne permet donc pas à lui seul de distinguer une migration de l'ADN induite par la génotoxicité d'une migration induite par la cytotoxicité. Toutefois, lorsqu'une augmentation de la migration de l'ADN est observée, il est recommandé d'étudier au moins un indicateur de cytotoxicité, ce qui peut aider à interpréter les résultats. Une augmentation de la migration de l'ADN en présence d'éléments attestant clairement de la cytotoxicité d'un produit doit être interprétée avec prudence. De nombreux critères de cytotoxicité ont été proposés et parmi eux, les altérations histopathologiques sont considérées comme fournissant une bonne évaluation de la toxicité tissulaire. Des observations telles qu'une inflammation, une infiltration cellulaire ou des modifications apoptotiques ou nécrotiques ont été associées à une augmentation de la migration de l'ADN; cependant, comme l'ont montré les essais de validation du JaCVAM (12), il n'existe pas de liste définitive de modifications histopathologiques systématiquement associées à une migration accrue de l'ADN. Les modifications de certains paramètres biologiques (AST, ALT, par exemple) peuvent aussi fournir des informations utiles sur les lésions des tissus, et d'autres indicateurs, tels que l'activation des caspases, le marquage des cellules apoptotiques par la méthode TUNEL, la coloration à l'annexine V, etc., peuvent aussi être envisagés. Cependant, les données publiées sur l'utilisation de ces indicateurs dans des études in vivo sont limitées et ne sont peut-être pas toutes également fiables. Les cellules fantômes sont des cellules dont l'image au microscope consiste en une tête de petite taille ou inexistante et une grande queue diffuse, et qui sont considérées comme très endommagées, bien que l'étiologie de ce phénomène soit incertaine (voir appendice 3). Du fait de leur apparence, la mesure du pourcentage d'ADN dans la queue par analyse d'image n'est pas fiable, et il convient donc de les évaluer séparément. L'occurrence de cellules fantômes doit être consignée et signalée, et toute augmentation importante de leur nombre attribuée au produit chimique testé doit être investiguée et interprétée avec soin. La connaissance du mode d'action potentiel des produits chimiques d'essai peut constituer une aide à cet égard. RESULTATS ET RAPPORT Traitement des résultats L'animal étant l'unité expérimentale, il convient de présenter sous forme de tableau les données animales individuelles et la synthèse des résultats. Du fait de la nature hiérarchique des données, il est recommandé de déterminer pour chaque lame le pourcentage médian d'ADN de queue et de calculer pour chaque animal la moyenne des médianes (12). La moyenne d'un groupe est obtenue en faisant la moyenne des moyennes individuelles des animaux qui le composent. Toutes ces valeurs doivent figurer dans le rapport. D'autres démarches peuvent être appliquées (voir paragraphe 53) si cela est justifié d'un point de vue scientifique et statistique. Pour l'analyse statistique, diverses approches sont possibles (58) (59) (60) (61). Lors du choix des méthodes statistiques, on tiendra compte de la nécessité de transformer éventuellement les données (en logarithmes ou en racines carrées, par exemple) et/ou d'ajouter un petit nombre (0.001, par exemple) à toutes les valeurs (même non nulles) pour atténuer l'incidence des valeurs nulles, comme il est expliqué dans les références ci-dessus. On trouvera à l'appendice 2 des précisions sur l'analyse des interactions traitement/sexe lorsque des animaux des deux sexes sont utilisés, et sur l'analyse subséquente des données selon que des différences sont observées ou non. Des données sur la toxicité et les signes cliniques doivent également figurer dans le rapport. Critères d'acceptabilité L'acceptation de l'essai repose sur les critères suivants:
Évaluation et interpretation des résultats À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement positif si:
Lorsque tous ces critères sont remplis, le produit chimique d'essai est considéré comme capable d'induire des cassures de brins d'ADN dans les tissus étudiés dans ce système d'essai. Si un ou deux seulement de ces critères sont remplis, voir le paragraphe 62. À condition que tous les critères d'acceptabilité soient remplis, un produit chimique d'essai est considéré comme clairement négatif si:
Le produit chimique d'essai est alors considéré comme incapable d'induire des cassures de brins d'ADN dans les tissus étudiés dans ce système d'essai. Il n'est pas nécessaire de vérifier une réponse clairement positive ou négative. Lorsque la réponse n'est ni clairement négative ni clairement positive (c'est-à-dire que tous les critères des paragraphes 59 ou 60 ne sont pas remplis) et afin d'établir la signification biologique d'un résultat, les données doivent être soumises à un jugement d'expert et/ou des investigations plus poussées si cela est justifié d'un point de vue scientifique. Il peut être utile d'examiner des cellules supplémentaires (le cas échéant) ou de répéter l'expérience, éventuellement dans des conditions expérimentales optimisées (espacement des doses, autres voies d'administration, autres moments d'échantillonnage ou autres tissus, par exemple). Dans de rares cas, même après des études complémentaires, l'ensemble de données ne permettra pas de conclure à un résultat positif ou négatif, et le résultat sera déclaré équivoque. Pour évaluer la signification biologique d'un résultat positif ou équivoque, des informations sur la cytotoxicité pour le tissu cible sont nécessaires (voir paragraphes 54-55). Lorsque des résultats positifs ou équivoques sont observés uniquement en présence de signes clairs de cytotoxicité, on conclura à une étude équivoque pour ce qui est de la génotoxicité, à moins de disposer d'informations suffisantes pour permettre une conclusion définitive. En cas de résultats négatifs s'accompagnant de signes de toxicité à toutes les doses, il peut être judicieux de procéder à une étude complémentaire à des doses non toxiques. Rapport d'essai Le rapport d'essai doit contenir les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DEFINITIONS Comète : nom donné à la forme que prennent les nucléoïdes soumis à un champ électrophorétique, en raison de sa similitude avec celle d'une comète: la tête correspond au noyau, et la queue est constituée de l'ADN migrant hors du noyau sous l'action du champ électrique. Électrophorèse de cellules isolées en gel d'agarose en conditions alcalines : technique sensible pour la détection de lésions primaires de l'ADN au niveau de cellules/noyaux individualisés. Intensité de la queue ou pourcentage d'AND de queue : correspond à l'intensité de fluorescence de la queue de la comète rapportée à l'intensité totale (tête plus queue). Reflète l'ampleur des cassures de l'ADN, exprimée sous forme de pourcentage. Produit chimique : une substance ou un mélange. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. UVCB : Substances de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques. Variable/paramètre critique : désigne une variable d'un protocole expérimental dont une modification mineure peut avoir une forte incidence sur la conclusion de l'essai. Les variables critiques peuvent être spécifiques d'un tissu. Il importe de ne pas modifier les variables critiques, particulièrement en cours d'essai, sans tenir compte de l'incidence de ces modifications sur la réponse à l'essai, signalée par exemple par l'ampleur et la variabilité de la réponse des témoins positifs et négatifs. Le rapport d'essai doit préciser les modifications apportées aux variables critiques au cours de l'essai, ou par rapport au protocole standard du laboratoire, et justifier chacune de ces modifications. Appendice 2 PLAN FACTORIEL UTILISÉ POUR IDENTIFIER DES DIFFÉRENCES ENTRE SEXES DANS LE TEST DES COMÈTES IN VIVO Le plan factoriel et son analyse Selon cette démarche, un minimum de 5 mâles et de 5 femelles sont exposés à chaque concentration d'essai, ce qui conduit à utiliser un minimum de 40 animaux (20 mâles et 20 femelles, auxquels s'ajoutent les témoins positifs nécessaires) La démarche décrite ici, qui correspond à l'une des formes simples du plan factoriel, équivaut à une analyse de variance à deux facteurs, dans laquelle le sexe et la concentration sont les facteurs principaux. Les données peuvent être analysées à l'aide de nombreux progiciels statistiques standard tels que SPSS, SAS, STATA ou Genstat, ou en utilisant le logiciel R. À partir de l'ensemble de données, on détermine la variabilité entre les sexes, la variabilité entre les concentrations et la variabilité liée à l'interaction entre sexe et concentrations. Chacun de ces termes est comparé à une estimation de la variabilité entre les animaux répartis au sein des groupes d'animaux de même sexe exposés à la même concentration. On trouvera plus de précisions sur cette méthode dans les manuels de statistiques classiques (voir les références) et dans les fichiers d'aide fournis avec les logiciels statistiques. On examine ensuite le terme d'interaction sexe x concentration dans un tableau ANOVA (35). En l'absence de terme d'interaction significatif, la combinaison des valeurs inter-sexes ou inter-niveaux de concentration permet de réaliser des tests statistiques valides entre les niveaux, en se basant sur le terme de variabilité intra-groupe combinée fourni par l'ANOVA. L'analyse se poursuit par la partition de la variabilité estimée entre concentrations, de façon à obtenir des contrastes, ce qui permet d'établir les contrastes linéaires et quadratiques des réponses pour l'ensemble des niveaux de concentration. Lorsqu'il y a une interaction significative sexe x concentration, ce terme peut à son tour être partitionné en contrastes d'interaction linéaire x sexe et quadratique x sexe. Ces termes permettent de vérifier si les réponses aux concentrations sont parallèles pour les deux sexes ou si elles diffèrent selon le sexe. L'estimation de la variabilité intra-groupe combinée peut servir à tester l'écart entre les moyennes en les comparant deux à deux. Ces comparaisons peuvent se faire entre les moyennes pour les deux sexes et entre les moyennes pour les différents niveaux de concentration (comparaisons avec les témoins négatifs, par exemple). En cas d'interaction significative, des comparaisons peuvent être faites entre les moyennes des différentes concentrations pour un même sexe, ou entre les moyennes des deux sexes à la même concentration. Références De nombreux manuels de statistiques traitent de la théorie, de la conception, de la méthodologie, de l'analyse et de l'interprétation des plans factoriels, depuis les analyses les plus simples, à deux facteurs, jusqu'aux formes complexes utilisées dans la conception de l'expérimentation. La liste ci-dessous n'est pas exhaustive. Certains ouvrages comportent des exemples d'application de ce type de démarches, accompagnés parfois d'un code permettant l'exécution des analyses sous différents logiciels.
Appendice 3 LIMITES ACTUELLES DE L'ESSAI Le test des comètes in vivo présente plusieurs limites liées à l'état actuel des connaissances. On peut s'attendre à ce que leur nombre diminue à l'avenir, ou à ce qu'elles soient définies de façon plus précise, à mesure que l'expérience acquise dans l'application du test apporte des réponses aux questions de sécurité, dans un contexte réglementaire.
Références
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||






|
(16) |
Dans la partie C, le chapitre C.13 est remplacé par le texte suivant: «C.13 Bioaccumulation chez le poisson: exposition via le milieu aquatique et via la voie alimentaire INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 305 (2012) de l'OCDE pour les essais de produits chimiques. Cette révision de la méthode d'essai poursuit deux objectifs. Tout d'abord, il s'agit d'intégrer à la méthode un essai de bioaccumulation via la nourriture (36), à même de déterminer le potentiel de bioaccumulation des substances très faiblement solubles dans l'eau. En deuxième lieu, on souhaite élaborer une méthode d'essai qui, dans un souci du bien-être animal, utilise le cas échéant un nombre plus réduit de poissons, et qui soit d'un meilleur rapport rendement-coût. Depuis l'adoption de la méthode d'essai C.13 (1), de nombreuses substances ont été testées, et les laboratoires comme les autorités réglementaires ont accumulé une expérience considérable. Il en ressort la conviction que l'essai peut être simplifié si des critères spécifiques sont remplis (voir paragraphe 88), et qu'une approche à plusieurs niveaux est possible. L'expérience a aussi montré que les facteurs biologiques comme le développement et la teneur en lipides du poisson ont une forte incidence sur les résultats et doivent probablement être pris en compte. De plus, on admet aujourd'hui que tester des substances très faiblement solubles dans l'eau n'est peut-être pas faisable techniquement. En outre, pour les substances très peu hydrosolubles, l'exposition par l'eau peut présenter moins d'intérêt que l'exposition par la nourriture. D'où le développement d'une méthode d'essai dans laquelle le poisson est exposé via son régime alimentaire (voir paragraphes 7 à 14, puis 97 et suivants). La validation (essai circulaire) de l'essai avec exposition par voie alimentaire a été effectuée en 2010 (51). Les principaux changements apportés à la méthode d'essai sont les suivants:
Avant de procéder à un essai de bioaccumulation quel qu'il soit, il convient d'avoir sur la substance d'essai les informations suivantes:
Indépendamment de la méthode d'exposition ou du dispositif d'échantillonnage retenus, cette méthode d'essai décrit une procédure permettant de caractériser le potentiel de bioaccumulation de substances chez le poisson. Bien qu'il faille accorder une nette préférence aux régimes d'essai en dynamique, les régimes semi-statiques sont acceptables à condition de respecter les critères de validité (voir paragraphes 24 et 113). Dans la méthode avec exposition par voie alimentaire, le régime dynamique n'est pas nécessaire pour maintenir dans le milieu aquatique les concentrations de la substance testée, mais il aidera à maintenir les concentrations adéquates d'oxygène dissous, à assurer la propreté de l'eau et à supprimer l'influence notamment des produits d'excrétion. Quelle que soit la méthode retenue, cette méthode d'essai offre suffisamment de détails pour que l'on puisse réaliser l'essai, tout en offrant une marge de liberté suffisante pour adapter le schéma expérimental aux conditions particulières des laboratoires et à la variation des caractéristiques des substances d'essai. L'essai par exposition via le milieu aquatique est le plus adapté pour les produits chimiques organiques stables avec des valeurs de log K OE situées entre 1,5 et 6,0 (13), mais peut aussi s'appliquer aux substances très hydrophobes (dont le log K OE est supérieur à 6,0), si l'on peut démontrer l'existence d'une concentration stable et pleinement dissoute de la substance d'essai dans l'eau. Si on ne peut démontrer une telle concentration, l'étude par exposition via le milieu aquatique n'est pas adaptée, aussi faut-il recourir à la méthode par la nourriture (encore que l'interprétation et l'utilisation des résultats de cet essai par voie alimentaire puissent dépendre du cadre réglementaire). Des pré-estimations du facteur de bioconcentration (FBC, parfois noté KB) pour les produits chimiques organiques dont les valeurs de log K OE sont supérieures à environ 9,0 peuvent être obtenues à l'aide de l'équation de Bintein et al. (14). La pré-estimation du facteur de bioconcentration pour des substances très hydrophobes de ce type peut être supérieure au facteur de bioconcentration à l'état stationnaire (FBCES) attendu pour des expériences en laboratoire, surtout si on utilise pour cette pré-estimation un modèle linéaire simple. Parmi les paramètres caractérisant le potentiel de bioaccumulation figurent la constante cinétique d'absorption (k 1), les constantes de perte dont la constante d'élimination (k2 ), le facteur de bioconcentration à l'état stationnaire (FBCES), le facteur de bioconcentration cinétique (FBCk) et le facteur de bioamplification alimentaire (FBA) (38). Le marquage radioactif des substances d'essai peut faciliter l'analyse des échantillons d'eau, de nourriture et de poisson, et être utilisé pour décider s'il y a lieu ou non d'identifier et de quantifier les métabolites. Si la totalité des résidus radioactifs est mesurée seule (par exemple par solubilisation des tissus ou par combustion), le FBC ou le FBA est déterminé à partir de la totalité de la substance parente, des métabolites retenus ainsi que du carbone assimilé. Le FBC ou le FBA déduit de la totalité des résidus radioactifs ne peut donc être directement comparable au FBC ou au FBA obtenu par une analyse spécifique de la substance parente seule. Des procédures de séparation, telles que la CCM, la CLHP ou la CG (39) peuvent être utilisées avant les études comportant un marquage radioactif pour déterminer le FBC ou le FBA correspondant à la substance parente. Quand on utilise des procédures de séparation, il convient d'identifier et de quantifier la substance parente et les métabolites pertinents (40) (voir paragraphe 65) si le FBC ou le FBA est déduit de la concentration de la substance parente dans le poisson plutôt que de la totalité des résidus radioactifs. Il est également possible de combiner une étude du métabolisme dans le poisson ou une étude de distribution in vivo et une étude de bioaccumulation, grâce à l'analyse et à l'identification des résidus présents dans les tissus. La possibilité de métabolisme peut être prévue par des outils adaptés (par exemple la boîte à outils QSAR de l'OCDE (15) et les programmes QSAR déposés). Le choix entre un essai par exposition via le milieu aquatique et un essai par voie alimentaire, et celui du dispositif d'essai, s'appuient sur les facteurs évoqués au paragraphe 3, ainsi que sur la réglementation en vigueur. Par exemple, pour des substances à log K OE élevé, mais présentant une bonne solubilité dans l'eau compte tenu de la sensibilité des techniques d'analyse disponibles, un essai par exposition via le milieu aquatique est considéré en premier lieu. Il se peut cependant que l'information sur l'hydrosolubilité ne soit pas définitive pour ce type de produits chimiques hydrophobes, aussi convient-il, avant de décider de la méthode à utiliser (16), d'étudier la possibilité de préparer des concentrations dissoutes dans le milieu aquatique stables et mesurables (les émulsions stables n'étant pas permises) utilisables pour une étude par exposition via le milieu aquatique. Il n'est pas possible de donner des instructions exactes sur la méthode à employer en fonction de critères d'exclusion d'hydrosolubilité et de coefficient de partage octanol-eau, car d'autres facteurs (techniques d'analyse, dégradation, adsorption, etc.) peuvent avoir une influence marquée sur la validité de la méthode, pour les raisons indiquées plus haut. Néanmoins, à partir d'un log K OE supérieur à 5 et d'une solubilité dans l'eau inférieure à ~0,01 — 0,1 mg/l les essais par exposition via le milieu aquatique deviennent de plus en plus difficiles. D'autres facteurs susceptibles d'influencer le choix de l'essai sont examinés, notamment le potentiel de la substance à l'adsorption vers les récipients d'essai et les appareils, sa stabilité dans la solution aqueuse par rapport à sa stabilité dans la nourriture pour poisson (17) (18), etc. D'autres études en milieu aquatique peuvent contenir des renseignements sur ces aspects pratiques. De plus amples informations sur l'évaluation des aspects liés à la performance des études de bioaccumulation sont disponibles dans la littérature [par exemple (19)]. S'agissant des substances pour lesquelles la solubilité ou le maintien de la concentration aqueuse et l'analyse de cette concentration ne représentent pas une contrainte pour la réalisation d'un essai par exposition via le milieu aquatique, cette méthode sera préférée pour déterminer le potentiel de bioconcentration. Dans tous les cas, on devra vérifier que la ou les concentrations de l'exposition via le milieu aquatique à appliquer s'inscrivent dans les limites d'hydrosolubilité dans les milieux d'essai. Différentes méthodes peuvent servir à maintenir des concentrations stables de la substance d'essai dissoute. On peut notamment recourir à des solutions mères ou à des systèmes de dosage passif (par exemple à la méthode d'élution sur colonne), pourvu qu'on puisse démontrer que des concentrations stables sont maintenues et que les milieux d'essai respectent les préconisations du paragraphe 27. Pour les substances très hydrophobes (log K OE supérieur à 5 et solubilité inférieure à ~ 0,01-0,1 mg/l), les essais par exposition via le milieu aquatique peuvent s'avérer ardus. La difficulté peut venir de ce qu'on ne parvient pas à maintenir la concentration aqueuse à un niveau considéré comme suffisamment constant (par exemple du fait de la sorption vers le verre des récipients ou de l'absorption rapide par le poisson) ou de ce que les concentrations aqueuses à appliquer sont si faibles qu'elles sont du même ordre de grandeur que la limite analytique de quantification ou inférieures à elle (41). Pour ces substances très hydrophobes, il est recommandé de pratiquer l'essai par voie alimentaire, si celui-ci respecte la réglementation en vigueur et les obligations d'évaluation des risques. Pour les tensioactifs, il convient d'examiner la faisabilité de l'essai de bioconcentration en milieu aquatique, compte tenu des propriétés de la substance, mais l'étude par voie alimentaire est probablement plus adaptée. Les tensioactifs sont des agents de surface, qui diminuent la tension interfaciale entre deux liquides. Amphiphiles (c'est-à-dire présentant une partie hydrophile et une partie hydrophobe), ils s'accumulent aux interfaces, par exemple eau-air, eau-nourriture et sur les parois en verre, ce qui empêche de déterminer leur concentration aqueuse. L'essai par voie alimentaire peut contourner certaines difficultés d'exposition pour les mélanges complexes dont les composants présentent des limites d'hydrosolubilité différentes, dans la mesure où il est plus probable d'obtenir une exposition comparable de tous les composants du mélange par voie alimentaire que par exposition en milieu aquatique [voir (20)]. Notons que la méthode par voie alimentaire permet d'obtenir un facteur de bioamplification alimentaire (FBA) et non un facteur de bioconcentration (FBC) (42). Il existe des méthodes pour estimer un facteur de bioconcentration cinétique (FBCk) à partir des données obtenues dans l'étude par voie alimentaire (voir appendice 8), mais elles sont à utiliser avec précaution. En général, ces méthodes supposent une cinétique du premier ordre, et ne sont applicables qu'à certains groupes de composés. Il est peu probable qu'elles puissent s'appliquer aux tensioactifs (voir paragraphe 12). L'essai réduit avec exposition via le milieu aquatique comportant moins de temps d'échantillonnage afin de réduire le nombre d'animaux et/ou les ressources (voir paragraphes 83 et suivants) ne devra être choisi que pour les substances pour lesquelles on a des raisons de penser que l'absorption et l'élimination obéiront approximativement à la cinétique du premier ordre (c'est-à-dire en général pour les substances organiques non ionisées, voir paragraphe 88). C.13 — I: Essai de bioconcentration chez le poisson par exposition via le milieu aquatique PRINCIPE DE L'ESSAI L'essai se déroule en deux phases: l'exposition (absorption) et la post-exposition (élimination). Pendant la phase d'absorption, un groupe de poissons d'une même espèce est exposé à la substance d'essai à une ou plusieurs concentrations, en fonction des propriétés de la substance (voir paragraphe 49). Ces poissons sont ensuite transférés vers un milieu dépourvu de la substance d'essai, pour la phase d'élimination. La phase d'élimination est toujours nécessaire, sauf si l'absorption de la substance au cours de la phase d'absorption est négligeable. La concentration de la substance d'essai dans ou sur le poisson (ou dans un tissu spécifié de cet animal) est suivie au cours des deux phases de l'essai. Outre le groupe exposé, un groupe témoin de poissons est élevé dans des conditions identiques, sans être exposé à la substance d'essai, afin de comparer les éventuels effets nocifs observés dans l'essai de bioconcentration au comportement d'un groupe témoin analogue et d'obtenir la concentration naturelle de la substance d'essai (43). Dans l'essai par exposition via le milieu aquatique, la phase d'absorption dure généralement 28 jours. Cette durée peut au besoin être prolongée (voir paragraphe 18), ou raccourcie s'il est démontré que l'état stationnaire a été atteint plus tôt (voir définitions et unités à l'appendice 1). Il est possible de prédire la durée de la phase d'absorption et le temps nécessaire à l'instauration de l'état stationnaire à partir des équations données à l'appendice 5. La phase d'élimination, au cours de laquelle les poissons ne sont plus exposés à la substance d'essai, débute ensuite, les poissons étant transférés dans un récipient propre contenant un milieu identique, mais dépourvu de la substance d'essai. Il est préférable, dans la mesure du possible, de calculer le facteur de bioconcentration de deux manières, d'une part le facteur de bioconcentration à l'état stationnaire (FBCES; voir définition à l'appendice 1), à savoir le rapport de la concentration dans les poissons (C p) à la concentration dans l'eau (C e) et, d'autre part, le facteur de bioconcentration cinétique (FBCk; voir définitions et unités à l'appendice 1), à savoir le rapport de la constante cinétique d'absorption (k 1) à la constante cinétique d'élimination (k2 ) dans l'hypothèse d'une cinétique du premier ordre (44). Si après 28 jours l'état stationnaire n'est pas encore atteint, il convient soit de calculer le FBC par la méthode cinétique (voir paragraphe 38) soit de prolonger la phase d'absorption. Si le temps requis pour atteindre l'état stationnaire est trop long dans la pratique (voir paragraphes 37 et 38 et appendice 5), on préférera la méthode cinétique. Pour les substances très hydrophobes, on envisagera de pratiquer l'essai par voie alimentaire (45), si celui-ci respecte la réglementation en vigueur. La constante cinétique d'absorption, la constante cinétique d'élimination (pertes) — ou les constantes, si des systèmes plus complexes sont en jeu –, le facteur de bioconcentration (à l'état stationnaire et/ou cinétique) et, si possible, l'intervalle de confiance de chacun de ces paramètres, sont calculés à partir du modèle qui décrit le mieux les concentrations de la substance d'essai mesurées dans les poissons et dans l'eau (voir appendice 5). La prise de poids des poissons durant l'essai provoquera chez eux une baisse de la concentration de la substance d'essai (c'est ce qu'on appelle la dilution par la croissance). Le FBC cinétique sera donc sous-estimé s'il n'est pas corrigé en conséquence (voir paragraphes 72 et 73). Le FBC est déduit à partir de la concentration totale dans le poisson (autrement dit en fonction du poids frais total du poisson). On pourra toutefois utiliser des tissus ou des organes déterminés (par exemple les muscles, le foie) pour des raisons particulières, si le poisson est suffisamment grand ou s'il peut être divisé en fractions comestibles (filets) et non comestibles (viscères). Il existe une nette relation entre le potentiel de bioconcentration et l'hydrophobicité pour beaucoup de substances organiques, qui implique une autre relation entre la teneur en lipides du poisson d'essai et la valeur observée de la bioconcentration de ces substances. Dès lors, afin de réduire cette source de variabilité dans les résultats des essais portant sur des substances très lipophiles (c'est-à-dire dont le log K OE est inférieur à 3), il convient d'exprimer la bioconcentration de façon normalisée rapportée à un poisson présentant une teneur en lipides de 5 % (de son poids corporel total), outre la bioconcentration obtenue directement de l'essai. Cela est nécessaire pour pouvoir comparer les résultats obtenus pour différentes substances et/ou espèces d'essai. Une teneur en lipides de 5 % est généralement utilisée, car c'est la teneur moyenne en lipides des poissons le plus souvent utilisés dans le cadre de cette méthode d'essai (21) INFORMATIONS SUR LA SUBSTANCE D'ESSAI En dehors des propriétés de la substance d'essai énumérées dans l'introduction (paragraphe 3), il y a lieu de connaître sa toxicité pour l'espèce de poisson utilisée dans l'essai, de préférence la CL50 asymptotique (indépendante du temps) et/ou sa toxicité estimée lors d'essais à long terme sur des poissons [voir notamment les méthodes d'essai C.47 (22), C.15 (23), C.14(24)]. Il convient de disposer d'une méthode d'analyse appropriée, dont on connaît la fiabilité, la précision et la sensibilité, pour quantifier la substance dans les solutions d'essai et dans le matériel biologique, ainsi que d'instructions précises pour la préparation et le stockage des échantillons. Il convient que la limite analytique de quantification de la substance d'essai dans l'eau et dans les tissus des poissons soit aussi connue. Lorsqu'on utilise une substance marquée à la radioactivité, il convient qu'elle présente une pureté très élevée (de préférence supérieure à 98 %) et que le pourcentage de radioactivité des impuretés soit connu. VALIDITÉ DE L'ESSAI La validité de l'essai est subordonnée à la réalisation des conditions suivantes:
SUBSTANCES DE RÉFÉRENCE Il serait utile de disposer de substances de référence à faible métabolisme et potentiel de bioconcentration connu pour vérifier le mode opératoire, le cas échéant (par exemple quand un laboratoire n'a encore jamais réalisé l'essai ou quand les conditions expérimentales ont été modifiées). DESCRIPTION DE LA MÉTHODE Appareillage Il convient qu'aucune partie du montage ne comporte de matériaux susceptibles d'être dissous, lessivés ou sorbés et de nuire aux poissons. Des récipients classiques, rectangulaires ou cylindriques, composés d'un matériau chimiquement inerte et dotés d'une capacité adaptée au taux de charge (voir paragraphe 43), peuvent être utilisés. Il convient de minimiser l'emploi des tubes en plastique souple. On choisira des tubes en polytétrafluoroéthylène, en acier inoxydable ou en verre. L'expérience a montré qu'en présence de substances d'essai à coefficient d'adsorption élevé, telles que les pyréthrines synthétiques, il sera peut-être nécessaire d'utiliser du verre silanisé. Dans ces circonstances, l'appareillage devra être jeté après usage. Il est préférable d'exposer les systèmes d'essai aux concentrations voulues de la substance d'essai aussi longtemps que nécessaire pour démontrer le maintien de concentrations d'exposition stables avant l'introduction des organismes d'essai. Eau Pour l'essai, on utilise généralement une eau naturelle obtenue à partir d'une source non contaminée et de qualité constante. Néanmoins, l'eau reconstituée (eau déminéralisée dans laquelle des nutriments spécifiques ont été ajoutés en quantités connues) peut être plus adaptée pour garantir une qualité uniforme dans le temps. La qualité de l'eau de dilution, c'est-à-dire de l'eau mélangée avec la substance d'essai avant d'être introduite dans le récipient d'essai (voir paragraphe 30), doit permettre à l'espèce de poisson choisie de survivre pendant la durée de l'acclimatation et de l'essai, sans présenter d'anomalies sur le plan du comportement ou de l'apparence. Idéalement, il faudrait démontrer que l'espèce testée peut survivre, croître et se reproduire dans l'eau de dilution (par exemple par un élevage en laboratoire ou par un essai de toxicité sur la totalité du cycle de vie). L'eau de dilution est au moins caractérisée par le pH, la dureté, les solides totaux, le carbone organique total (COT (47)) et de préférence aussi l'ammonium, les nitrites et l'alcalinité, ainsi que la salinité dans le cas des espèces marines. Les paramètres qui commandent le bien-être optimal des poissons ne sont pas entièrement connus, mais l'appendice 2 fournit les concentrations maximales recommandées de plusieurs paramètres pour des espèces d'eau douce et d'eau de mer. Il convient que la qualité de l'eau de dilution reste constante tout au long de l'essai. Le pH est compris entre 6,0 et 8,5 au début de l'essai, mais sans varier au-delà de ± 0,5 unité de pH au cours d'un essai donné. On analysera des échantillons prélevés à divers intervalles, afin de veiller à ce que l'eau de dilution n'influence les résultats de l'essai (par exemple par complexation de la substance d'essai) ou ne perturbe le comportement des poissons, au moins au début et à la fin de l'essai. Il convient de déterminer les métaux lourds (par exemple Cu, Pb, Zn, Hg, Cd, Ni), les principaux anions et cations (par exemple Ca2+, Mg2+, Na+, K+, C1-, SO4 2-), les pesticides (par exemple le total des organophosphorés et le total des organochlorés), le carbone organique total et les solides en suspension, par exemple tous les trois mois si on sait que l'eau de dilution présente une qualité relativement constante. S'il a été prouvé que la qualité de l'eau de dilution est demeurée constante pendant au moins un an, les déterminations peuvent être espacées et avoir lieu, par exemple, tous les six mois). La teneur naturelle de l'eau de dilution en particules et en carbone organique total est aussi basse que possible pour éviter l'adsorption de la substance d'essai sur la matière organique, ce qui réduirait sa biodisponibilité et conduirait à sous-estimer le FBC. La valeur maximale acceptable s'élève à 5 mg/l pour les matières particulaires (matière sèche retenue par un filtre de 0,45 μm) et à 2 mg/l pour le carbone organique total (voir appendice 2). Au besoin, l'eau de dilution est filtrée avant utilisation. Il convient que la contribution des excreta des poissons testés et des résidus alimentaires à la teneur en carbone organique de l'eau d'essai soit aussi faible que possible (voir paragraphe 46). Solutions d'essai On préparera une solution mère de la substance d'essai à la concentration appropriée. Il vaut mieux préparer la solution mère par simple mélange ou agitation de la substance d'essai dans l'eau de dilution. Une autre option adaptée dans certains cas consiste à utiliser un système de dosage de désorption de la phase solide. Il est généralement déconseillé de recourir à des solvants et à des dispersants (agents solubilisants) [voir (25)]; néanmoins, l'utilisation de ces produits peut être acceptable pour obtenir une solution mère à la concentration appropriée, mais il convient de s'efforcer d'y recourir aussi peu que possible et de ne pas dépasser leur concentration micellaire critique (le cas échéant). Les solvants qui peuvent être utilisés sont l'acétone, l'éthanol, le méthanol, le diméthylformamide et le triéthylène glycol; les dispersants qui ont été utilisés sont le Tween 80, la méthylcellulose à 0,01 % et l'HCO-40. Il convient que la concentration de solvant dans le milieu d'essai final soit la même dans tous les traitements (indépendamment de la concentration de la substance) et n'excède pas les niveaux de toxicité définis pour le solvant dans les conditions d'essai. La concentration maximale est de 100 mg/l (ou 0,1 ml/l). Il est peu probable qu'une concentration de solvant de 100 mg/l modifie beaucoup la concentration maximale de substance d'essai dissoute pouvant être obtenue dans le milieu d'essai (25). Il convient que la contribution du solvant et celle de la substance d'essai à la teneur en carbone organique total de l'eau de l'essai soient connues. Tout au long de l'essai, la concentration du carbone organique total dans les récipients d'essai ne devra pas dépasser la concentration du carbone organique provenant de la substance d'essai et, le cas échéant, du solvant ou du solubilisant (48), de plus de 10 mg/l (± 20 %). La teneur en matières organiques peut avoir un effet important sur le volume de substance d'essai dissoute dans les essais dynamiques, en particulier pour les substances très lipophiles. La microextraction en phase solide (voir paragraphe 60) peut fournir des informations importantes sur le ratio entre composés liés et dissous libres, ces derniers étant considérés comme la fraction biodisponible. Il convient que la concentration de la substance d'essai soit inférieure à la limite de solubilité de cette substance dans le milieu d'essai malgré l'utilisation d'un solvant ou d'un solubilisant. Des précautions s'imposent lors de l'utilisation de solvants facilement biodégradables, ceux-ci pouvant poser des problèmes de prolifération bactérienne dans les essais dynamiques. S'il n'est pas possible de préparer une solution mère sans recourir à un agent solubilisant, il convient d'évaluer l'intérêt d'un essai par exposition en milieu aquatique par rapport à un essai par voie alimentaire. Les essais dynamiques demandent un système capable de fournir et de diluer continuellement une solution mère de la substance d'essai (par exemple une pompe doseuse, un dilueur proportionnel, un système de saturation) ou un système de dosage de désorption de la phase solide, afin d'obtenir la concentration voulue dans les enceintes d'essai. Le volume sera remplacé de préférence cinq fois par jour dans chaque chambre d'essai. Le régime dynamique est préférable, mais lorsqu'il ne sera pas possible de l'instaurer (par exemple lorsque cela porte préjudice aux organismes testés), une technique semi-statique peut être utilisée à condition de respecter les critères de validité (voir paragraphe 24). Les débits des solutions mères et de l'eau de dilution sont vérifiés 48 heures avant l'essai et au moins une fois par jour pendant l'essai. Au cours de cette vérification, il faudra déterminer le débit dans chaque enceinte d'essai et veiller à ce que sa variation n'excède pas 20 % au sein des enceintes et entre les enceintes. Sélection des espèces Les critères importants dans la sélection des espèces sont leur disponibilité immédiate, leur taille adéquate et le fait qu'elles supportent bien les conditions du laboratoire. D'autres critères orientent la sélection des espèces de poissons, comme leur importance récréative, commerciale et écologique ainsi que leur sensibilité comparable, les bons résultats qu'elles ont donnés précédemment, etc. Des espèces recommandées pour les essais sont énumérées à l'appendice 3. D'autres espèces peuvent être utilisées, mais le mode opératoire devra éventuellement être adapté pour que l'essai se déroule dans des conditions appropriées. Dans ce cas, il convient de justifier le choix de l'espèce et d'exposer la méthode expérimentale. En général, l'utilisation de petites espèces réduira le délai d'établissement du régime stationnaire, mais un plus grand nombre de poissons (prélèvements) peut être nécessaire pour analyser correctement la teneur en lipides et la concentration de la substance d'essai dans les poissons. En outre, des différences de fréquence respiratoire et de métabolisme entre les poissons jeunes et les poissons plus âgés peuvent empêcher les comparaisons entre essais et entre espèces. Pratiquer l'essai sur des poissons à un stade précoce de leur vie (juvéniles) en croissance rapide peut compliquer l'interprétation des données. Conditions de vie des poissons en laboratoire (pertinentes pour les expositions via le milieu aquatique et par voie alimentaire) Le stock de poissons est acclimaté dans l'eau pendant au moins deux semaines (voir paragraphe 28) à la température de l'essai et être suffisamment nourri pendant toute cette période (voir paragraphe 45). L'eau et le régime alimentaire sont du même type que ceux qu'on utilisera durant l'essai. Après une période d'adaptation de 48 heures, on enregistre la mortalité et on applique les critères suivants:
On s'assurera que les poissons utilisés dans les essais ne présentent pas de maladies ou d'anomalies observables. Tous les poissons malades seront éliminés. Il convient de ne pas traiter les poissons pour une maladie durant les deux semaines qui précèdent l'essai ou pendant l'essai DEROULEMENT DE L'ESSAI Essai préliminaire Il peut être utile de conduire un essai préliminaire pour optimiser les conditions expérimentales de l'essai définitif, par exemple du point de vue de la sélection de la (des) concentration(s) de la substance d'essai. L'essai préliminaire est conçu de façon à obtenir les informations requises. On peut ainsi examiner si un essai réduit peut suffire à obtenir un FBC ou si une étude complète est requise (voir les paragraphes 83 à 95 sur l'essai réduit). Conditions d'exposition Durée de la phase d'absorption La durée de la phase d'absorption peut être prédite expérimentalement (par exemple à partir d'une étude précédente ou d'une étude d'accumulation portant sur une substance de structure voisine) ou d'après certaines relations empiriques qui exploitent la connaissance que l'on a du coefficient de partage n-octanol/eau ou de l'hydrosolubilité de la substance d'essai (à condition que l'absorption soit régie par une cinétique du premier ordre, voir appendice 5). La phase d'absorption dure 28 jours, sauf s'il est démontré que l'état stationnaire a été atteint plus tôt (voir définitions et unités à l'appendice 1). La courbe de la concentration de la substance d'essai dans le poisson (C p) en fonction du temps atteint un état stationnaire lorsqu'elle devient parallèle à l'axe du temps et que les résultats de trois analyses successives de la C p, réalisées sur des échantillons prélevés à au moins deux jours d'intervalle, ne s'écartent pas de plus de 20 % l'un de l'autre, et qu'il n'y a pas d'augmentation nette de C p dans le temps entre la première et la dernière analyse successive. Les échantillons regroupés font l'objet d'au moins quatre analyses successives. Pour les substances d'essai qui sont absorbées lentement, il serait plus approprié de prendre des intervalles de sept jours. Si l'état stationnaire n'a pas été atteint en 28 jours, il convient soit de calculer le FBC par la méthode cinétique seule, qui ne dépend pas de l'établissement du régime stationnaire, soit de prolonger la phase d'absorption, en poursuivant les mesures, jusqu'à ce que l'état stationnaire soit atteint ou jusqu'à 60 jours, suivant ce qui est le plus rapide. En outre, il convient que la concentration de la substance d'essai dans le poisson à la fin de la période d'absorption soit suffisamment élevée pour permettre une estimation fiable de la constante k2 lors de la phase d'élimination. Si aucune absorption significative n'est constatée après 28 jours, l'essai peut être arrêté. Durée de la phase d'élimination Pour les substances régies par une cinétique du premier ordre, une période égale à la moitié de la phase d'absorption suffit généralement à entraîner une diminution appropriée (par exemple 95 %) de la charge corporelle de la substance d'essai (voir appendice 5 pour une explication de l'estimation). Si le temps requis pour obtenir une élimination de 95 % est trop long dans la pratique et qu'il excède, par exemple, le double de la durée normale de la phase d'absorption (c'est-à-dire plus de 56 jours), on peut appliquer une période plus courte (jusqu'à ce que la concentration de la substance d'essai soit inférieure à 10 % de la concentration à l'état stationnaire, par exemple). Néanmoins, une phase d'élimination plus longue peut être nécessaire pour les substances dont l'absorption et l'élimination suivent des lois plus complexes que celles qui sont représentées par le modèle à compartiment unique pour les poissons, qui obéit à une cinétique du premier ordre. Si on observe de tels phénomènes et/ou si on s'attend à les observer, il est recommandé de demander conseil à un biostatisticien et/ou à un pharmacocinéticien pour garantir un bon dispositif d'essai. Lorsque la phase d'élimination est prolongée, le nombre de poissons à prélever peut devenir un obstacle et les différences dans la croissance des poissons peuvent influencer les résultats. La durée de la phase dépendra également de la période durant laquelle la concentration de la substance d'essai dans les poissons reste supérieure à la limite analytique de quantification. Nombre de poissons testés Le nombre de poissons par concentration d'essai est choisi de manière à ce que les échantillons comportent au moins quatre poissons à chaque temps d'échantillonnage. Les poissons ne sont regroupés que si l'analyse d'un seul poisson n'est pas praticable. Si on souhaite une plus grande précision dans l'ajustement des courbes (et dans les paramètres dérivés) ou si des études de métabolisme sont requises (par exemple pour faire la distinction entre les métabolites et la substance mère quand on utilise des substances marquées à la radioactivité), il faut un plus grand nombre de poissons par temps d'échantillonnage. La teneur en lipides et la concentration de la substance d'essai sont déterminées sur le même matériel biologique. Si ce n'est pas faisable, des poissons supplémentaires peuvent être nécessaires (voir paragraphes 56 et 57). Si l'on utilise des poissons adultes (c'est-à-dire sexuellement matures), il convient qu'ils ne soient pas en période de frai pendant l'essai ou qu'ils n'aient pas récemment frayé. Il faut aussi préciser s'il s'agit de mâles ou de femelles, ou des deux. Si les deux sexes sont utilisés, il convient de démontrer qu'il n'y a pas de différence significative entre les deux sexes du point de vue de la croissance et de la teneur en lipides avant le début de l'exposition, en particulier si on prévoit que le regroupement de mâles et de femelles sera nécessaire pour obtenir des concentrations détectables de la substance et/ou la teneur en lipides. Dans tous les essais, il convient de choisir des poissons de poids voisin de telle sorte que le poids des plus petits ne soit pas inférieur aux deux tiers du poids des plus gros. Il convient qu'ils soient tous de la même année et proviennent de la même source. Le poids et l'âge d'un poisson pouvant avoir une incidence importante sur le FBC (12), il y a lieu de consigner ces détails avec précision. On recommande de peser un sous-échantillon du stock de poissons peu de temps avant l'essai, afin d'estimer le poids moyen (voir paragraphe 61). Taux de charge On appliquera des rapports élevés du volume d'eau au poids des poissons afin de minimiser la réduction de la concentration du composé d'essai dans l'eau provoquée par l'introduction des poissons au début de l'essai et d'éviter une diminution de la concentration de l'oxygène dissous. Il est important d'adapter le taux de charge à l'espèce de poisson. Quoi qu'il en soit, on recommande normalement un taux de charge de 0,1-1,0 g de poissons (poids frais) par litre d'eau et par jour. Des taux de charge plus élevés peuvent être pratiqués, s'il a été prouvé que la concentration requise de la substance d'essai peut être maintenue à ± 20 % près et que la concentration de l'oxygène dissous ne tombe pas en dessous de 60 % de la saturation (voir paragraphe 24). L'habitat naturel du poisson guide le choix du régime de charge. Les espèces benthiques, par exemple, peuvent avoir besoin d'un aquarium doté d'un plus grand fond pour le même volume d'eau que les espèces pélagiques. Alimentation Pendant les périodes d'acclimatation et d'essai, les poissons recevront une alimentation appropriée, dont la teneur en lipides et en protéines totales est connue, en quantité suffisante pour être en bonne santé et conserver leur poids (une certaine croissance est permise). Il convient de les nourrir quotidiennement pendant les périodes d'acclimatation et d'essai suivant un dosage qui est fonction de l'espèce utilisée, des conditions expérimentales et de la valeur calorique des aliments (par exemple pour la truite, un dosage approximatif de 1 à 2 % du poids corporel par jour). La ration alimentaire est définie de manière à éviter un développement rapide et une forte augmentation de la teneur en lipides. Pour maintenir une ration stable, le volume de nourriture est recalculé s'il y a lieu, par exemple une fois par semaine. Pour ce calcul, le poids des poissons de chaque chambre d'essai peut être estimé à partir du poids des derniers poissons prélevés dans cette chambre. Il ne faut pas peser les poissons qui restent dans la chambre. Dans les chambres d'essai, les aliments non consommés et les excréments seront siphonnés chaque jour, peu après l'alimentation (de 30 minutes à une heure après celle-ci). Il convient que les chambres restent aussi propres que possible tout au long de l'essai, pour maintenir la concentration de matières organiques aussi basse que possible (voir paragraphe 29), la présence de carbone organique pouvant limiter la biodisponibilité de la substance d'essai (12). Comme beaucoup d'aliments sont à base de farine de poissons, il convient de veiller à ce qu'ils n'influencent pas les résultats de l'essai ou n'induisent pas d'effets négatifs, par exemple en contenant des (traces de) pesticides, de métaux lourds et/ou de la substance d'essai elle-même. Lumière et température Une photopériode de 12 à 16 heures est recommandée et il convient que la température (± 2 °C) soit adaptée à l'espèce testée (voir appendice 3). Il convient de connaître le type et les caractéristiques de l'illumination. Il convient de faire attention à une éventuelle phototransformation de la substance d'essai dans les conditions d'irradiation appropriée afin d'éviter d'exposer les poissons à des produits de photodégradation. Dans certains cas, il suffit d'utiliser un filtre pour éliminer les radiations ultra-violet inférieures à 290 nm. Concentrations d'essai Cet essai a tout d'abord été conçu pour les substances organiques non polaires. Pour ce type de produit, l'exposition d'un poisson à une concentration unique devrait être suffisante, puisqu'on n'attend pas d'effet de concentration, bien que le cadre réglementaire en vigueur puisse exiger deux concentrations. Si on teste d'autres types de substances, ou s'il existe d'autres indications de dépendance éventuelle à la concentration, l'essai est réalisé avec deux concentrations ou plus. Si on ne teste qu'une concentration, il convient de justifier le choix de cette concentration (voir paragraphe 79). En outre, il convient que la concentration testée soit aussi basse qu'il est pratiquement et techniquement faisable (autrement dit, elle n'approche pas la limite de solubilité). Dans certains cas on peut s'attendre à ce que la bioconcentration d'une substance dépende de sa concentration dans l'eau (par exemple pour les métaux, pour lesquels l'absorption par les poissons peut être au moins en partie régulée). Dans des cas de ce type, il peut être nécessaire de tester au moins deux concentrations, et si possible plus (voir paragraphe 49) pertinentes d'un point de vue environnemental. De plus, pour les substances pour lesquelles les concentrations testées doivent, pour des raisons pratiques, se rapprocher de la limite de solubilité, il est recommandé de tester au moins deux concentrations, ce qui peut donner une idée de la fiabilité des concentrations d'exposition. Parmi les concentrations d'essai figurent la concentration réaliste sur le plan environnemental ainsi que celle pertinente pour l'objet spécifique de l'évaluation. Il convient que la ou les concentrations de la substance d'essai soient inférieures au niveau auquel elles produisent un effet chronique ou à 1 % de la CL50 aiguë asymptotique, s'inscrivent dans une fourchette pertinente du point de vue de l'environnement et soient supérieures d'au moins une puissance dix à la limite de quantification dans l'eau par la méthode d'analyse utilisée. La concentration d'essai la plus élevée admise peut être déterminée en divisant la CL50 aiguë (96 h) par un rapport aigu/chronique (par exemple des rapports adéquats pour certaines substances chimiques se situent autour de 3, mais quelques-uns sont au-dessus de 100). Si une seconde concentration est utilisée, il convient qu'elle diffère de la première d'un facteur dix. Si ce n'est pas possible en raison du critère de toxicité (qui plafonne la concentration d'essai) et du seuil de détection analytique, il convient d'envisager d'appliquer un facteur inférieur à 10 et d'utiliser une substance d'essai marquée à la radioactivité (de la pureté la plus élevée, de préférence supérieure à 98 %). Il convient de veiller à ce que la concentration de la substance d'essai ne dépasse pas sa solubilité dans le milieu d'essai. Témoins Un groupe témoin traité avec de l'eau de dilution ou, le cas échéant, (voir paragraphes 30 et 31), avec le solvant est testé parallèlement aux groupes traités avec la substance d'essai. Fréquence des mesures de la qualité de l'eau Pendant l'essai, l'oxygène dissous, le carbone organique total, le pH et la température sont mesurés dans tous les récipients d'essai et témoins. La dureté totale et la salinité (le cas échéant) sont mesurées dans le(s) groupe(s) témoin(s) et dans le récipient. Si au moins deux concentrations sont testées, il convient de mesurer ces paramètres à la concentration la plus élevée. L'oxygène dissous et la salinité (le cas échéant) sont mesurés au moins trois fois pendant la période d'absorption (au début, vers le milieu et à la fin) et au moins une fois par semaine pendant la période d'élimination. Le carbone organique total est mesuré au début de l'essai (24 h et 48 h avant le début de la phase d'absorption) avant l'introduction des poissons et au moins une fois par semaine pendant les phases d'absorption et d'élimination. La température devrait être mesurée et enregistrée quotidiennement, le pH au début et à la fin de chaque période et la dureté une fois au cours de l'essai. La température est de préférence mesurée en continu dans au moins un récipient. Prélèvement et analyse des poissons et de l'eau Programme de prélèvement des poissons et de l'eau On prélèvera de l'eau dans les chambres d'essai afin de déterminer la concentration de la substance d'essai avant l'introduction des poissons et pendant les phases d'absorption et d'élimination. L'eau est prélevée en même temps que les poissons et avant leur alimentation. Des prélèvements plus fréquents peuvent être utiles pour garantir la stabilité des concentrations après l'introduction des poissons. Il convient de déterminer les concentrations de la substance d'essai pendant la phase d'absorption pour vérifier si les critères de validité ont été respectés (paragraphe 24). Si l'analyse de l'eau prélevée au début de la phase d'élimination ne détecte pas de substance d'essai, cela peut justifier de ne plus rechercher la présence de cette substance dans l'eau d'essai et l'eau témoin pour le restant de la phase d'élimination. Les poissons seront prélevés au moins cinq fois pendant la phase d'absorption et au moins quatre fois pendant la phase d'élimination de la substance d'essai. Comme dans certains cas il sera difficile de calculer une estimation raisonnablement précise du FBC avec ce nombre d'échantillons (en particulier lorsque l'absorption et l'élimination n'obéiront pas à une simple cinétique du premier ordre), il peut se justifier de prélever des échantillons plus fréquemment au cours des deux périodes (voir appendice 4). La teneur en lipides et la concentration de la substance d'essai sont déterminées sur le même matériel biologique au moins au début et à la fin de la phase d'élimination. Si ce n'est pas praticable, au moins trois poissons devront être prélevés pour déterminer leur teneur en lipides à chacun des trois mêmes temps d'échantillonnage. Il faudra ajuster en conséquence le nombre de poissons par récipient au début de l'expérience (49). Sinon, si on ne détecte pas un volume important de substance d'essai dans les poissons témoins (c'est-à-dire les poissons du stock), les poissons témoins de l'essai peuvent n'être analysés que pour leur teneur en lipides, et l'analyse de la substance d'essai dans le ou les groupes d'essai (ainsi que la constante cinétique d'absorption, la constante cinétique d'élimination et le FBC associés) peut être corrigée en fonction de la teneur en lipides du groupe témoin au cours de l'essai (50). Les poissons morts ou malades ne sont pas analysés à la recherche d'une concentration de la substance d'essai ou de leur teneur en lipides. Un exemple de programme de prélèvement acceptable est donné à l'appendice 4. D'autres programmes peuvent être établis facilement d'après d'autres valeurs supposées du K OE qui permettent de calculer le temps d'exposition correspondant à une absorption de 95 % (se reporter à l'appendice 5 pour les calculs). Pendant la phase d'absorption, les prélèvements se poursuivent jusqu'à ce que l'état stationnaire soit atteint (voir définitions et unités à l'appendice 1) ou que la phase d'absorption s'achève (après 28 ou 60 jours, voir paragraphes 37 et 38). Avant d'entamer la phase d'élimination, il convient de transférer les poissons dans des récipients propres. Prélèvement et préparation des échantillons On prélèvera les échantillons d'eau à analyser par exemple en siphonnant à travers un tube inerte à partir d'un point central de l'enceinte d'essai. La centrifugation ou la filtration ne réussissent pas toujours à séparer les fractions biodisponible et non biodisponible de la substance d'essai. Si on recourt à une technique de séparation, il convient de la justifier ou de la valider dans le rapport d'essai, compte tenu des difficultés de biodisponibilité (25). Les prélèvements de substances très hydrophobes, en particulier (c'est-à-dire dont le log K OE est inférieur à 5) (12) (26), pour lesquels une adsorption vers le filtre ou vers les récipients de centrifugation peut survenir, ne sont pas soumis à ces traitements. En revanche, il convient de s'efforcer de maintenir la plus grande propreté dans les aquariums (voir paragraphe 46) et de suivre la teneur en carbone organique totale pendant les phases d'absorption et d'élimination (voir paragraphe 53). Pour éviter les problèmes potentiels dus à la réduction de la biodisponibilité, on peut réaliser, pour les substances faiblement solubles et très hydrophobes, des prélèvements par techniques de microextraction en phase solide. Il convient d'euthanasier instantanément les poissons prélevés, suivant la méthode la plus appropriée et la moins cruelle (pour des mesures sur des poissons entiers, il suffit de rincer ceux-ci à l'eau (voir paragraphe 28) et de les sécher en surface). Il convient ensuite de peser les poissons et de mesurer leur longueur totale (51). Pour chaque poisson, le poids et la longueur mesurés sont reliés à la concentration de la substance analysée (et s'il y a lieu à la teneur en lipides), par exemple en attribuant un identifiant unique à chaque poisson prélevé. Il est préférable d'analyser les poissons et l'eau immédiatement après les avoir prélevés pour éviter les dégradations ou d'autres pertes et de calculer des constantes d'absorption et d'élimination approximatives pendant le déroulement de l'essai. En procédant à une analyse immédiate, on évite aussi de retarder la détermination du moment où le plateau (état stationnaire) a été atteint. À défaut de pouvoir pratiquer une analyse immédiate, on stockera les échantillons selon une méthode appropriée. Avant de commencer l'étude, on se renseignera sur la méthode de stockage adaptée à la substance d'essai, par exemple la congélation, la conservation à 4 °C, l'extraction, etc. La durée de stockage est choisie de façon à ce que la substance chimique ne se dégrade pas pendant le stockage. Qualité de la méthode d'analyse Comme toute la procédure est essentiellement subordonnée à la fiabilité, la précision et la sensibilité de la méthode d'analyse appliquée à la substance d'essai, il convient de vérifier expérimentalement que la précision et la reproductibilité de l'analyse chimique ainsi que l'isolement de la substance d'essai à partir de l'eau et du poisson sont satisfaisants pour la méthode en question. Ces vérifications s'effectuent au cours des essais préliminaires. Il convient de s'assurer également que la substance d'essai n'est pas détectable dans l'eau de dilution utilisée. Il faudra, au besoin, corriger les valeurs de la concentration de la substance d'essai dans l'eau et dans les poissons en fonction des valeurs fournies par les témoins pour l'isolement de la substance d'essai et sa concentration naturelle. Les échantillons d'eau et de poisson sont manipulés avec soin tout au long de l'essai de manière à minimiser la contamination et les pertes (provoquées par l'adsorption sur l'instrument de prélèvement, par exemple). Analyse des échantillons de poissons Si on utilise du matériel marqué pendant l'essai, il est possible d'analyser la radioactivité totale (c'est-à-dire celle de la substance parente et des métabolites) ou de purifier les échantillons pour analyser la substance parente séparément. Si le FBC doit s'appuyer sur la substance parente, les principaux métabolites sont caractérisés, au minimum à la fin de la phase d'absorption (voir paragraphe 6). Les principaux métabolites sont ceux qui représentent au moins 10 % des résidus totaux présents dans les tissus des poissons, ceux qui en représentent au moins 5 % à deux temps d'échantillonnage consécutifs, ceux en hausse tout au long de la phase d'absorption, enfin, ceux dont on sait qu'ils posent des problèmes toxicologiques. Si le FBC pour l'ensemble du poisson, en termes de résidus radiomarqués totaux, est supérieur ou égal à 500, il peut être conseillé, et même fortement recommandé dans le cas de certaines substances chimiques comme les pesticides — d'identifier et de quantifier les principaux métabolites. La quantification de ces métabolites peut être exigée par certaines autorités réglementaires. Si les produits de dégradation représentant 10 % ou plus (> 10 %) des résidus radioactifs totaux dans les poissons sont identifiés et quantifiés, il est alors aussi recommandé d'identifier et de quantifier les produits de dégradation dans l'eau d'essai. Si ce n'est pas praticable, il y a lieu de l'expliquer dans le rapport. La concentration de la substance d'essai est habituellement déterminée pour chaque poisson pesé individuellement. Si cela est impossible, on peut regrouper les échantillons d'un même prélèvement, mais cette opération limite le traitement statistique applicable aux résultats, aussi y a-t-il lieu d'inclure un nombre suffisant de poissons dans l'essai pour que le regroupement, la procédure et la précision voulus soient possibles. Les références (27) et (28) peuvent servir d'introduction sur les procédures de regroupement adaptées. Il convient d'exprimer le FBC de façon normalisée rapportée à un poisson présentant une teneur en lipides de 5 % (par rapport à son poids frais), outre le FBC obtenu directement de l'essai (voir paragraphe 21), à moins de pouvoir démontrer que la substance d'essai ne s'accumule pas prioritairement dans les lipides. Il faut, si possible, déterminer la teneur en lipides des poissons à chaque prélèvement, de préférence à partir du même extrait que la substance d'essai, l'extrait devant souvent être débarrassé de ses lipides avant d'être analysé par chromatographie. Néanmoins, l'analyse des substances d'essai nécessite souvent des procédures d'extraction spécifiques, qui peuvent être en contradiction avec les lignes directrices concernant la détermination des lipides. Dans ce cas (jusqu'à ce que des méthodes instrumentales non destructives adaptées soient disponibles), il est recommandé d'employer une stratégie différente pour déterminer la teneur en lipides des poissons (voir paragraphe 56). Les lipides sont déterminés suivant les méthodes appropriées (20). On recommande (30) habituellement la technique d'extraction chloroforme/méthanol (29), mais la méthode de Smedes (31) peut également être utilisée. Elle présente la même efficacité d'extraction, une grande précision, utilise moins de solvants organiques toxiques et facilite la performance. D'autres méthodes dont la précision soutient la comparaison avec les méthodes recommandées peuvent être utilisées, à condition de justifier correctement leur choix. Il est important de donner des détails sur la méthode mise en œuvre. Mesure du développement des poissons Au début de l'essai, il convient de peser et de mesurer un à un cinq à dix poissons du stock. Il peut s'agir des mêmes poissons utilisés pour l'analyse lipidique (voir paragraphe 56). Il convient de mesurer le poids et la longueur des poissons issus des groupes d'essai et témoins utilisés pour chaque échantillonnage avant de procéder à leur analyse chimique ou lipidique. Ces mesures peuvent servir à estimer le poids et la longueur des poissons restant dans les récipients d'essai et témoins (voir paragraphe 45). RAPPORT D'ESSAI ET RÉSULTATS Traitement des résultats On tracera la courbe de l'absorption de la substance d'essai en portant sur un diagramme arithmétique sa concentration dans/sur le poisson (ou dans des tissus spécifiés) pendant la phase d'absorption en fonction du temps. Si la courbe atteint un plateau, autrement dit si elle devient approximativement asymptotique par rapport à l'axe du temps, on calcule le FBC à l'état stationnaire (FBCES) à l'aide de la formule suivante:
Le développement de C p peut être influencé par la croissance des poissons (voir paragraphes 72 et 73). La concentration moyenne d'exposition (Ce) est influencée par la variation dans le temps. On peut s'attendre à ce qu'une concentration moyenne pondérée par rapport au temps soit plus pertinente, même si la variation s'inscrit dans les limites de la fourchette de validité (voir paragraphe 24). Une moyenne pondérée par rapport au temps de la concentration dans l'eau peut être calculée en suivant les instructions données à l'appendice 5, section 1 Il y a lieu de déterminer le facteur de bioconcentration cinétique (FBCk) en calculant le ratio k 1/k 2, k 1 et k 2 étant les deux constantes cinétiques du premier ordre. Les constantes cinétiques k 1 et k 2 et le FBCk peuvent être déduits en évaluant simultanément la phase d'absorption et la phase d'élimination. Une autre solution consiste à déterminer k 1 et k 2 l'un après l'autre (voir l'appendice 5 qui décrit et compare ces méthodes). La constante d'élimination (k 2) peut devoir être corrigée de l'effet de dilution par la croissance (voir paragraphes 72 et 73). Si la courbe d'absorption et/ou d'élimination n'obéit manifestement pas à une cinétique du premier ordre, il convient d'employer des modèles plus complexes (voir la bibliographie de l'appendice 5) et de demander conseil à un biostatisticien et/ou à un pharmacocinéticien. Poids et longueur des poissons Le poids frais et la longueur totale de chaque poisson, à chaque temps d'échantillonnage, sont présentés séparément pour les groupes d'essai et les groupes témoins pendant les phases d'absorption et d'élimination (y compris les poissons du stock au début de la période d'absorption). Pour chaque poisson, le poids et la longueur sont reliés à la concentration chimique analysée, par exemple en attribuant un identifiant unique à chaque poisson prélevé. Le poids est la mesure du développement à privilégier afin de corriger le FBC cinétique de l'effet de dilution par la croissance. Le paragraphe 73 et l'appendice 5 présentent la méthode utilisée pour cette correction. Correction de la dilution par la croissance et normalisation des lipides Le développement des poissons pendant la phase d'élimination peut réduire les concentrations chimiques mesurées chez eux, ce qui augmente la constante cinétique d'élimination globale (k 2) par rapport à ce que provoqueraient les processus d'élimination seuls (par exemple respiration, métabolisme, égestion). Les facteurs de bioconcentration cinétique sont corrigés de la dilution par la croissance. Le FBCES est aussi influencé par la croissance, mais il n'existe aucune procédure de correction convenue en la matière. Dans les cas de croissance importante, le FBCk corrigé de la croissance (FBCkg) est aussi calculé car il peut s'avérer plus pertinent. La teneur en lipides des poissons d'essai (fortement associée à la bioaccumulation des substances hydrophobes) peut suffisamment varier dans la pratique pour qu'une normalisation rapportée à une teneur en lipides prédéfinie (5 % du poids frais) soit nécessaire pour obtenir des facteurs de bioconcentration cinétique et à l'état stationnaire qui aient un sens — sauf si l'on peut démontrer que la substance ne s'accumule pas prioritairement dans les lipides (par exemple certaines substances perfluorées peuvent se lier aux protéines). L'appendice 5 présente les équations et donne des exemples de calculs de ce type. Pour corriger un FBC cinétique de la dilution par la croissance, il convient de corriger la constante cinétique d'élimination de la croissance. Cette constante cinétique d'élimination corrigée (k 2g) s'obtient en soustrayant la constante cinétique de croissance (kg, déduite du poids mesuré) de la constante cinétique d'élimination globale (k 2). Le facteur de bioconcentration cinétique corrigé de la croissance se calcule alors en divisant la constante cinétique d'absorption (k 1) par la constante cinétique d'élimination corrigée de la croissance (k 2g) (voir appendice 5). Dans certains cas, cette méthode est inadaptée. Par exemple, pour des substances s'éliminant très lentement testées chez des poissons à croissance rapide, la k 2g obtenue peut être très faible, aussi toute erreur commise dans les deux constantes cinétiques servant à la calculer est-elle cruciale, et dans certains cas la kg estimée peut être supérieure à k 2. Une autre méthode contournant la nécessité d'une correction de la dilution par la croissance consiste à utiliser le poids de substance d'essai par poisson (entier) lors de l'élimination plutôt que le poids de substance par unité de masse du poisson (concentration). Ce calcul est facile à faire, car dans le cadre d'essais respectant cette méthode d'essai, on associe les concentrations tissulaires aux poids individuels des poissons. L'appendice 5 présente la procédure de calcul simple. Il convient de souligner que le recours à cette méthode alternative n'empêche pas qu'il faille consigner la valeur de k 2. Les facteurs de bioconcentration cinétique et à l'état stationnaire sont aussi être consignés, rapportés à une teneur en lipides par défaut de 5 % (du poids frais du poisson), sauf si l'on peut prouver que la substance d'essai ne s'accumule pas prioritairement dans les lipides. Les données relatives à la concentration dans le poisson, ou le FBC, sont normalisées en fonction du ratio entre ces 5 % et la teneur réelle moyenne en lipides d'un poisson (en pourcentage de son poids frais) (voir appendice 5). Si l'analyse chimique et l'analyse lipidique ont été menées sur le même poisson, il convient d'utiliser ces données lipidiques individuelles normalisées pour calculer un FBC normalisé par rapport aux lipides. Autre possibilité, si la croissance des poissons est similaire dans les groupes exposés et les groupes témoins, on peut n'utiliser que la teneur en lipides des poissons témoins pour la correction en fonction des lipides (voir paragraphe 56). L'appendice 5 décrit une méthode de calcul du FBC normalisé par rapport aux lipides. Interprétation des résultats Les résultats sont interprétés avec prudence lorsque les valeurs mesurées de la concentration des solutions d'essai avoisinent le seuil de détection de la méthode d'analyse. La croissance moyenne dans le groupe d'essai et celle dans le groupe témoin en principe ne diffèrent pas beaucoup afin d'exclure les effets toxiques. Les constantes cinétiques de croissance ou les courbes de croissance des deux groupes sont comparées au moyen d'une procédure adaptée (52). Des courbes d'absorption et d'élimination clairement définies attestent que les résultats concernant la bioconcentration sont de bonne qualité. S'agissant des constantes cinétiques, il convient que le test du χ2 constate un bon ajustement du modèle de bioaccumulation [à savoir un faible pourcentage d'erreur dans les mesures (32)] pour que les constantes puissent être considérées comme fiables (voir appendice 5). Si on utilise plus d'une concentration d'essai, il convient que la variation des constantes d'élimination et d'absorption entre les concentrations d'essai soit inférieure à 20 % (53). Sinon, la dépendance à la concentration peut être indiquée. L'observation de différences significatives entre les vitesses d'absorption ou d'élimination mesurées aux diverses concentrations d'essai est signalée et donne lieu à des explications possibles. En général, si l'étude a été bien conçue, le seuil de confiance du FBC à 95 % tourne autour de 20 % du FBC obtenu. Si on teste deux concentrations ou plus, les chiffres obtenus à toutes ces concentrations servent à étudier la cohérence des résultats et à mettre en évidence une éventuelle dépendance à la concentration. Si une seule concentration est testée afin de réduire le nombre d'animaux et/ou les ressources utilisés, il y a lieu de justifier ce choix. Le FBC calculé est incertain si le FBCk est nettement supérieur au FBCES, car cela peut indiquer que l'état stationnaire n'a pas été atteint ou que la dilution par la croissance et les processus de pertes n'ont pas été pris en compte. Dans les cas où le FBCES est très supérieur au FBCk, le calcul des constantes cinétiques d'absorption et d'élimination est vérifié. Une procédure de validation de l'ajustement différente peut améliorer l'estimation du FBCk (voir appendice 5). Rapport d'essai Outre les informations sur la substance d'essai énumérées au paragraphe 3, le rapport d'essai comporte les renseignements suivants:
C.13 — II: Essai réduit d'exposition des poissons via le milieu aquatique INTRODUCTION L'expérience accumulée dans la conduite et l'interprétation de l'essai complet, que ce soit dans les laboratoires ou au sein des organes de réglementation, montre que — à quelques exceptions près — la cinétique du premier ordre s'applique pour estimer les constantes cinétiques d'absorption et d'élimination. Aussi l'estimation de ces constantes peut-elle se faire avec un minimum de temps d'échantillonnage, et permettre d'obtenir le FBC cinétique. L'étude d'autres concepts pour analyser le FBC avait pour objectif premier la mise au point d'un petit essai à mettre en œuvre lors d'une étape intermédiaire afin de réfuter ou confirmer les estimations du FBC basées sur K oe et sur les relations structure-activité quantitatives (RSAQ) et se passer ainsi de l'essai complet pour de nombreuses substances chimiques. Il s'agissait aussi de réduire le coût de l'essai et le nombre d'animaux utilisés, en diminuant les prélèvements et les séquences analytiques. Tout en respectant le concept général de la précédente méthode d'essai pour permettre l'intégration des résultats de l'essai aux données existantes sur le FBC et pour améliorer ces résultats et l'interprétation des données, l'objectif était aussi de fournir des estimations du FBC suffisamment précises pour évaluer les risques et prendre les décisions pertinentes. De nombreuses considérations s'appliquent de la même façon que dans l'essai complet, par exemple les critères de validité (voir paragraphe 24) et l'arrêt d'un essai dans le cas où aucune absorption notable n'est observée à l'issue de la phase d'absorption (voir paragraphes 16 et 38). Les substances pouvant se prêter à ce concept d'essai réduit relèvent du domaine général pour lequel la présente méthode d'essai a été développée, à savoir les substances organiques non polaires (voir paragraphe 49). S'il apparaît que la substance à l'étude se comporte différemment (net écart par rapport à la cinétique du premier ordre, par exemple), il convient, pour respecter la réglementation, de réaliser un essai complet. En règle générale, l'essai réduit n'est pas réalisé sur une période plus courte que l'essai type concernant le FBC, mais comprend moins de prélèvements de poissons (voir appendice 6 pour l'explication). Néanmoins, la période d'élimination peut être raccourcie pour les substances chimiques à élimination rapide afin d'éviter que les concentrations dans le poisson ne tombent sous le seuil de détection/quantification avant la fin de l'essai. Un essai réduit d'exposition via le milieu aquatique avec une seule concentration, peut servir à déterminer s'il est nécessaire de mener un essai complet; et si les données obtenues pour calculer les constantes cinétiques et le FBC sont fiables (voir paragraphe 93), on peut renoncer à l'essai complet à condition que le FBC obtenu soit loin des valeurs réglementaires critiques. Dans certains cas, il peut être avantageux de pratiquer l'essai réduit avec plus d'une concentration d'essai, comme un essai préliminaire, pour déterminer si les estimations du FBC pour une substance chimique sont dépendantes de la concentration. Si les estimations du FBC tirées de l'essai réduit sont dépendantes de la concentration, un essai complet s'imposera. Si en revanche, à l'issue d'un essai réduit, les estimations du FBC sont indépendantes de la concentration mais que les résultats sont jugés définitifs, tout essai complet ultérieur pourra être réalisé avec une seule concentration, ce qui réduira le nombre d'animaux utilisés par rapport à un essai complet avec deux concentrations (ou plus). Les produits chimiques pouvant se prêter à l'essai réduit:
PROGRAMME DE PRÉLÈVEMENT POUR LES ESSAIS RÉALISÉS SUIVANT L'ESSAI RÉDUIT Prélèvement des poissons Le prélèvement des poissons est réduit à quatre temps d'échantillonnage:
Prélèvement de l'eau Lors de l'essai réduit, l'eau est prélevée comme dans l'essai complet (voir paragraphe 54) ou au moins cinq fois à intervalles réguliers pendant la phase d'absorption et une fois par semaine pendant la phase d'élimination. Modifications du concept En fonction des propriétés de la substance d'essai, de la validité des prédictions des RSAQ et de l'objet spécifique de l'étude, il peut être envisageable de modifier le concept de l'essai:
Calculs Les raisons de cette approche tiennent du fait que le facteur de bioconcentration dans un essai complet peut être déterminé comme un facteur de bioconcentration à l'état stationnaire (FBCES) en calculant le rapport de la concentration de la substance d'essai dans les tissus du poisson à la concentration de la substance d'essai dans l'eau, ou en calculant le facteur de bioconcentration cinétique (FBCk), à savoir le rapport de la constante cinétique d'absorption k 1 à la constante cinétique d'élimination k 2. Le FBCk est valide même si une concentration à l'état stationnaire d'une substance chimique n'est pas atteinte durant l'absorption, à condition qu'absorption et élimination soient régies pour l'essentiel par des processus cinétiques de premier ordre. Au minimum, deux points sont nécessaires pour estimer les constantes cinétiques d'absorption et d'élimination, l'un à la fin de la phase d'absorption (soit au début de la phase d'élimination) et l'autre à la fin de la phase d'élimination (ou une fois la phase d'élimination bien avancée). Le temps d'échantillonnage intermédiaire est recommandé pour contrôler les cinétiques d'absorption et d'élimination (57). (Pour les calculs, voir appendices 5 et 6.) Interprétation des résultats Pour évaluer la validité et la valeur informative de l'essai, il convient de vérifier que la période d'élimination est supérieure à une demi-vie. De même, on compare le FBCkm (FBC cinétique obtenu à partir d'un essai réduit) à la valeur du FBCES minimisé (qui correspond au FBCES calculé à la fin de la phase d'absorption, en supposant que l'état stationnaire a été atteint, ce qu'on ne peut que supposer, puisque le nombre de temps d'échantillonnage ne suffit pas à le prouver). Si FBCkm < FBCES minimisé, le FBCES minimisé sera la valeur à privilégier. Si le FBCkm est inférieur à 70 % du FBCES minimisé, les résultats ne sont pas valides, et un essai complet s'impose. Si l'essai réduit donne un FBCkm avoisinant une valeur réglementaire critique, il faudra mener un essai complet. Si le résultat est loin de toute valeur réglementaire critique (nettement au-dessus ou en-dessous), un essai complet n'est pas forcément nécessaire, ou l'on pourra réaliser un essai complet avec une seule concentration si le cadre réglementaire l'exige. S'il apparaît à l'issue d'un essai réduit impliquant une seule concentration qu'un essai complet est nécessaire, ce dernier pourra être mené avec une seconde concentration. Si les résultats correspondent, on pourra se passer d'un essai complet avec une concentration différente, puisque la bioconcentration de la substance n'est apparemment pas dépendante de la concentration. Si l'essai réduit a été mené avec deux concentrations et que les résultats ne montrent aucune dépendance de la concentration, on pourra réaliser l'essai complet avec une seule concentration (voir paragraphe 87). Rapport d'essai Le rapport de l'essai réduit inclut toutes les informations exigées pour l'essai complet (voir paragraphe 81), à l'exception de celles qu'il n'est pas possible d'obtenir (à savoir la courbe montrant la durée avant l'établissement de l'état stationnaire et le facteur de bioconcentration à l'état stationnaire; à la place de ce dernier, il faudra indiquer le FBCES minimisé). Il conviendra aussi de préciser les raisons pour lesquelles il a été décidé de mener un essai réduit, et d'indiquer le FBCkm obtenu. C.13 — III: Essai de bioaccumulation chez le poisson avec exposition via la voie alimentaire INTRODUCTION La méthode décrite dans cette section s'applique pour les substances ne se prêtant pas à un essai par exposition via le milieu aquatique (par exemple parce qu'il n'est pas possible de maintenir des concentrations stables et mesurables dans l'eau ou d'obtenir des charges corporelles adéquates en 60 jours d'exposition; voir les sections précédentes pour la méthode d'exposition en milieu aquatique). Il convient néanmoins de noter que cet essai donnera au final un facteur de bioamplification alimentaire (FBA) et non un facteur de bioconcentration (FBC) (58). En mai 2001, une nouvelle méthode pour les essais de bioaccumulation de substances organiques peu solubles dans l'eau a été présentée lors de la conférence de SETAC Europe, organisée à Madrid (36). Ce travail s'appuie sur différentes études de bioaccumulation rapportées dans la littérature, utilisant une méthode de dosage avec une alimentation enrichie [voir par exemple (37)]. Début 2004, un projet de protocole (38), destiné à mesurer le potentiel de bioaccumulation des substances organiques peu hydrosolubles pour lesquelles la méthode de bioconcentration par exposition via le milieu aquatique ne convenait pas, a été soumis, avec un document de référence (39), au groupe de travail PBT de l'UE. Parmi les justifications avancées pour appliquer cette méthode, il a été indiqué que l'exposition potentielle de l'environnement à ces substances peu solubles (log K oe > 5) est sans doute largement liée à la voie alimentaire [voir (40) (41) (42) (43) (44)]. Aussi les essais avec exposition par voie alimentaire sont-ils mentionnés dans certains règlements relatifs aux produits chimiques (59). Il convient toutefois de noter que la méthode décrite ici évite soigneusement toute exposition via le milieu aquatique. En conséquence, un FBA obtenu à partir de cette méthode d'essai n'est pas directement comparable à un FBA obtenu avec une étude sur le terrain (qui permet de combiner exposition via le milieu aquatique et par voie alimentaire). Cette section de la présente méthode d'essai s'appuie sur ce protocole (38) et présente une nouvelle méthode qui ne figurait pas dans la version précédente de la méthode d'essai C.13. Avec cet essai de substitution, il est possible d'étudier directement l'exposition par voie alimentaire dans des conditions de laboratoire bien définies. Avant de procéder à cette étude, il est indispensable de se reporter aux paragraphes 1 à 14 de la présente méthode d'essai afin de comprendre les circonstances dans lesquelles l'essai avec exposition par voie alimentaire est préféré à l'essai par exposition via le milieu aquatique. Ces paragraphes apportent aussi des informations sur les substances, et il convient d'en prendre connaissance avant de réaliser un essai. Le marquage radioactif des substances d'essai peut être envisagé pour les mêmes raisons qu'avec la méthode d'exposition via le milieu aquatique (voir paragraphes 6 et 65). La méthode d'exposition par voie alimentaire peut servir à analyser plusieurs substances lors d'un seul essai, à condition que certains critères soient remplis; ces critères sont détaillés au paragraphe 112. Par souci de simplicité, la méthode décrit ici un essai pratiqué avec une seule substance d'essai. L'essai avec exposition par voie alimentaire est similaire à l'essai par exposition via le milieu aquatique à de nombreux égards, à l'exception manifeste du mode d'exposition. Aussi la méthode présentée ici recoupe-t-elle en de nombreux points la méthode d'exposition en milieu aquatique décrite à la section précédente. Dans la mesure du possible, il est fait référence aux paragraphes de la section précédente concernés, mais pour des raisons de lisibilité et de compréhension certaines répétitions n'ont pu être évitées. PRINCIPE DE L'ESSAI On peut appliquer des conditions d'essai dynamiques ou semi-statiques (voir paragraphe 4); il est recommandé de préférer les essais dynamiques pour limiter l'exposition potentielle à la substance d'essai via le milieu aquatique suite à une désorption de l'alimentation enrichie ou des excréments. L'essai comprend deux phases: l'absorption (substance d'essai-aliments enrichis) et l'élimination (aliments «propres», non traités) (voir paragraphe 16). Durant la phase d'absorption, on donne, tous les jours, à un groupe de poissons des aliments du commerce spécifiquement pour poisson et dont la composition est connue, enrichis de la substance d'essai. Idéalement, les poissons consomment toute la nourriture proposée (voir paragraphe 141). Durant la phase d'élimination, on leur donne cette même nourriture du commerce, mais pure, non traitée. Avec la méthode d'exposition via le milieu aquatique, il est possible, si nécessaire, d'utiliser plus d'un groupe en variant la concentration de la substance d'essai, mais pour la majorité des substances organiques très hydrophobes un groupe d'essai suffit (voir paragraphes 49 et 107). Dans des conditions semi-statiques, le poisson est transféré dans un nouveau milieu et/ou une nouvelle chambre d'essai à la fin de la phase d'absorption (au cas où le milieu ou l'appareillage utilisé pendant la phase d'absorption aurait été contaminé par la substance d'essai par lixiviation). Les concentrations de la substance d'essai dans le poisson sont mesurées lors des deux phases de l'essai. En plus du groupe de poissons nourris avec une alimentation enrichie (le groupe d'essai), un groupe de poissons témoin est maintenu dans des conditions identiques et nourri de la même façon, mais son alimentation n'est pas enrichie avec la substance d'essai. Ce groupe témoin permet de quantifier la concentration de la substance d'essai dans les poissons non exposés et sert de point de comparaison lorsque l'on observe chez le ou les groupes d'essai des effets nocifs liés au traitement (60). Cela permet aussi de comparer les constantes cinétiques de croissance entre les groupes pour contrôler que la nourriture proposée a été consommée dans les mêmes quantités (il convient aussi de tenir compte des qualités organoleptiques potentiellement différentes de l'alimentation pour expliquer la différence entre les constantes cinétiques de croissance; voir paragraphe 138). Il est important que pendant les phases d'absorption et d'élimination, les groupes d'essai et témoin reçoivent une alimentation équivalente. Une phase d'absorption qui dure entre 7 et 14 jours est généralement suffisante, à en juger par l'expérience des équipes ayant mis au point ces méthodes d'essai (38) (39). Cette durée devrait permettre de minimiser le coût de l'essai tout en garantissant une exposition suffisante pour la plupart des substances. Toutefois, il peut être préférable dans certains cas de prolonger la phase d'absorption (voir paragraphe 127). Durant cette phase d'absorption, la concentration de la substance dans les poissons peut ne pas atteindre l'état stationnaire si bien que le traitement des données et les résultats issus de cette méthode s'appuient en général sur une analyse cinétique des résidus présents dans les tissus. (Note: on peut appliquer ici les équations servant à estimer la durée nécessaire à l'établissement de l'état stationnaire comme lors de l'essai par exposition en milieu aquatique — voir appendice 5). La phase d'élimination commence au moment où l'on nourrit les poissons avec une alimentation non enrichie; elle dure habituellement jusqu'à 28 jours ou, si cela prend moins de temps, jusqu'à ce que la substance d'essai ne soit plus quantifiable dans le poisson (entier). La phase d'élimination peut être raccourcie ou prolongée au-delà de 28 jours, selon les variations dans le temps des concentrations mesurées et de la taille des poissons. Cette méthode permet de déterminer la demi-vie spécifique à la substance (t1/2, d'après la constante cinétique d'élimination, k 2), le rendement d'assimilation (absorption par voie intestinale; α), le facteur de bioamplification alimentaire cinétique (FBAk), le facteur de bioamplification alimentaire cinétique corrigé de la croissance (FBAkg) et le facteur de bioamplification alimentaire cinétique corrigé de la teneur en lipides (61) (FBAkL) (et/ou le facteur de bioamplification alimentaire cinétique corrigé de la croissance et des lipides, FBAkgL) pour la substance d'essai dans le poisson. S'agissant de la méthode d'exposition en milieu aquatique, la prise de poids des poissons durant l'essai provoquera chez eux une dilution de la substance d'essai. Le FBA (cinétique) sera donc sous-estimé s'il n'est pas corrigé en conséquence (voir paragraphes 162 et 163). En outre, si on estime que l'état stationnaire a été atteint en phase d'absorption il est possible de calculer un FBA à l'état stationnaire indicatif. Plusieurs méthodes permettent d'estimer un facteur de bioconcentration cinétique (FBCk) à partir des données obtenues dans l'étude par voie alimentaire [par exemple (44) (45) (46) (47) (48)]. Les pour et les contre de ces approches sont analysés à l'appendice 8. L'essai a été conçu en premier lieu pour les substances organiques non polaires peu hydrosolubles qui sont régies chez les poissons, pour l'essentiel, par des cinétiques d'absorption et d'élimination du premier ordre. Quand la substance testée n'est pas régie par des cinétiques d'absorption et d'élimination du premier ordre, il convient d'employer des modèles plus complexes (voir la bibliographie de l'appendice 5) et de demander conseil à un biostatisticien et/ou pharmacocinéticien. Normalement, on détermine le FBA en utilisant l'analyse de la substance d'essai pour tout le poisson (poids frais). Si cela est pertinent au regard des objectifs de l'étude, il est possible de prélever des tissus déterminés (par exemple les muscles, le foie) si le poisson est divisé en fractions comestibles et non comestibles (voir paragraphe 21). Par ailleurs, le prélèvement et l'analyse distincte du tractus gastro-intestinal peuvent aider à déterminer la contribution aux concentrations dans l'ensemble du poisson à différents temps d'échantillonnage, à la fin de la phase d'absorption et vers le début de la phase d'élimination, ou dans le cadre d'une approche fondée sur le bilan massique. La teneur en lipides des poissons (entiers) prélevés est mesurée pour que les concentrations puissent être corrigées en fonction de la teneur en lipides dans l'alimentation et dans le poisson (voir paragraphes 56 et 57, et appendice 7). Chaque poisson prélevé est pesé, et ce poids est consigné (par exemple en attribuant un identifiant unique à chaque poisson prélevé) et relié à la concentration chimique analysée par individu, afin de calculer le développement du poisson en cours d'essai. Dans la mesure du possible, il convient aussi de mesurer la longueur totale du poisson (62). Les données sur le poids sont aussi nécessaires pour estimer le FBC à partir des données relatives à la phase d'élimination durant l'essai avec exposition par voie alimentaire. INFORMATIONS SUR LA SUBSTANCE D'ESSAI Il convient de disposer des informations sur la substance d'essai décrites aux paragraphes 3 et 22. Une méthode permettant d'analyser les concentrations de la substance d'essai dans l'eau n'est habituellement pas nécessaire; il convient que les méthodes à appliquer présentent une sensibilité appropriée pour mesurer les concentrations dans l'alimentation et dans les tissus du poisson. La méthode utilisée peut servir à tester plus d'une substance lors d'un même essai. Cependant, il convient que les substances d'essai soient compatibles entre elles: elles ne doivent ni interagir ni changer d'identité chimique une fois ajoutées à l'alimentation du poisson. L'objectif est que les résultats mesurés pour chaque substance lors d'essais groupés ne diffèrent pas ou à peine des résultats qui auraient été obtenus avec des essais individuels. Il convient qu'une analyse préliminaire établisse que chaque substance peut être isolée à partir d'un échantillon de poisson ou de nourriture enrichi de plusieurs substances, avec i) des isolements élevés (par exemple > 85 % à la valeur nominale) et ii) la sensibilité nécessaire au bon déroulement de l'essai. Il convient que la dose totale des substances testées simultanément soit inférieure à la concentration combinée susceptible d'entraîner des effets toxiques (voir paragraphe 51). De plus, il convient que le schéma expérimental prenne en compte les éventuels effets nocifs chez le poisson et les interactions possibles (effets métaboliques) associés à l'essai simultané de plusieurs substances. Il convient d'éviter de tester simultanément des substances ionisables. En termes d'exposition, la méthode se prête aussi à des mélanges complexes (voir paragraphe 13, bien que les limites de l'analyse seront les mêmes qu'avec une autre méthode)). VALIDITÉ DE L'ESSAI La validité de l'essai est subordonnée à la réalisation des conditions suivantes (voir paragraphe 24):
SUBSTANCES DE RÉFÉRENCE Si un laboratoire n'a pas procédé à cet essai auparavant ou s'il a procédé à des modifications substantielles (changement de souche ou de fournisseur de poissons, espèce de poisson différente, changement notable de la taille ou de l'alimentation des poissons, ou de la méthode d'enrichissement, etc.), il est conseillé d'éprouver les compétences techniques disponibles, en utilisant une substance de référence. La substance de référence est principalement utilisée pour établir si la technique d'enrichissement de l'alimentation permet de garantir une homogénéité et une biodisponibilité maximales des substances d'essai. Par exemple, la substance de référence utilisée pour les substances hydrophobes non polaires est l'hexachlorobenzène (HCB), mais en raison des propriétés dangereuses du HCB (63). il convient de considérer d'autres substances pour lesquelles les données d'absorption et de bioamplification sont fiables. Le cas échéant, les informations générales sur la substance de référence devront figurer dans le rapport d'essai, en particulier le nom, la pureté, le numéro du CAS, la structure, la toxicité (si disponible) comme pour les substances d'essai (voir paragraphes 3 et 22). DESCRIPTION DE LA MÉTHODE Appareillage Le matériel et l'appareillage sont utilisés comme décrit pour l'essai par exposition en milieu aquatique (voir paragraphe 26). Il convient d'utiliser un système de renouvellement dynamique ou statique qui fournisse un volume d'eau de dilution suffisant aux récipients d'essai. Il convient aussi de consigner les débits. Eau L'eau de l'essai est utilisée comme décrit pour l'essai par exposition en milieu aquatique (voir paragraphes 27 à 29). Il convient que le milieu d'essai présente les caractéristiques requises et que sa qualité reste constante pendant toute la durée de l'essai. La teneur naturelle en particules et en carbone organique total est aussi basse que possible (≤ 5 mg/l pour les matières particulaires; ≤ 2 mg/l pour le carbone organique total) avant le début de l'essai. Le carbone organique total est mesuré uniquement avant l'essai au moment de la caractérisation de l'eau de l'essai (voir paragraphe 53). Alimentation Il est recommandé d'utiliser une nourriture pour poisson disponible dans le commerce (granulés flottant et/ou coulant lentement), caractérisée en termes au moins de protéines et riche en matières grasses. Il convient que les granulés présentent une taille uniforme pour accroître l'efficacité de l'exposition par voie alimentaire; de cette façon, les poissons mangeront plus, puisqu'ils ne se contenteront pas de manger les plus gros morceaux, délaissant les plus petits. Il convient aussi que la taille des granulés soit appropriée à la taille des poissons au début de l'essai (diamètre avoisinant 0,6 à 0,85 mm pour les poissons d'une longueur totale comprise entre 3 et 7 cm, et 0,85 à 1,2 mm pour les poissons d'une longueur totale comprise en 6 et 12 cm). On peut ajuster la taille des granulés en fonction du développement des poissons au début de la phase d'élimination. L'appendice 7 donne un exemple d'une nourriture du commerce adaptée. L'alimentation utilisée pour mettre au point cette méthode totalisait en général une teneur en lipides comprise en 15 et 20 % (du poids frais). Il est possible que des aliments pour poisson présentant une teneur en lipides aussi élevée ne soient pas disponibles dans certaines régions. Le cas échéant, l'essai pourrait être réalisé avec une teneur en lipides plus faible et, si nécessaire, d'ajuster la ration alimentaire pour maintenir les poissons en bonne santé (en fonction de l'essai préliminaire). La teneur totale en lipides de l'alimentation du groupe d'essai et du groupe témoin est mesurée et consignée avant le début de l'essai et à la fin de la phase d'absorption. Le rapport d'essai précise les détails fournis par le fabricant des aliments pour poisson dans l'analyse des nutriments, de la teneur en eau, des fibres et de la teneur en cendres, et si possible des minéraux et des résidus de pesticides (polluants prioritaires standard par exemple). Au moment d'enrichir l'alimentation avec la substance d'essai, il convient de veiller autant que possible à l'homogénéité des aliments utilisés pendant l'essai. La concentration de la substance d'essai dans la nourriture du groupe testé est sélectionnée en fonction de la sensibilité de la technique d'analyse, de la toxicité de la substance d'essai (CSEO si connue) et des données physico-chimiques pertinentes. Le cas échéant, il est préférable d'incorporer la substance de référence suivant une concentration d'environ 10 % de celle de la substance d'essai (ou en tout cas aussi faible que possible), en fonction de la sensibilité de l'analyse (par exemple pour l'hexachlorobenzène, une concentration dans la nourriture de 1 à 100 μg/g est jugée acceptable; voir (47) pour plus d'informations sur les rendements d'assimilation du HCB). La substance d'essai peut être ajoutée à la nourriture pour poisson de différentes manières, selon ses caractéristiques physiques et sa solubilité (voir annexe 7 pour plus de détails sur les méthodes d'enrichissement):
Dans certains cas, par exemple avec des substances d'essai moins hydrophobes plus susceptibles de se désorber des aliments, il peut être nécessaire d'enduire les granulés d'une petite quantité d'huile de poisson/germes de maïs (voir paragraphe 142). Il convient alors de traiter la nourriture témoin de même et de mesurer la teneur en lipides de la nourriture finale ainsi préparée. Le cas échéant, les résultats concernant la substance de référence sont comparables aux données des analyses décrites dans la littérature et menées dans des conditions similaires avec une ration alimentaire comparable (voir paragraphe 45), et les paramètres spécifiques à la substance de référence correspondent aux critères pertinents énoncés au paragraphe 113 (3e, 4e et 5e points). Si une huile ou un solvant est utilisé comme véhicule pour la substance d'essai, on mélange une quantité équivalente de ce véhicule (en excluant la substance d'essai) à la nourriture témoin de façon à maintenir l'équivalence avec les aliments enrichis. Il est important que pendant les phases d'absorption et d'élimination, les groupes d'essai et témoin reçoivent une alimentation équivalente. La nourriture enrichie est stockée dans des conditions qui maintiennent la stabilité de la substance d'essai au sein du mélange (par réfrigération par exemple) et ces conditions sont consignées. Sélection des espèces de poissons Cet essai peut être réalisé avec les espèces de poissons indiquées pour l'exposition en milieu aquatique (voir paragraphe 32 et appendice 3). Avant la publication de la présente méthode d'essai, les études de bioaccumulation par la nourriture menées avec des substances organiques utilisaient habituellement la truite arc-en-ciel (Oncorhynchus mykiss), la carpe (Cyprinus carpio) et le tête-de-boule (Pimephales promelas). Il convient que les espèces testées aient un comportement alimentaire tel que la ration administrée est consommée rapidement pour limiter au maximum l'influence potentielle d'un facteur quel qu'il soit sur la concentration de la substance d'essai dans l'alimentation (par exemple, lixiviation dans l'eau et exposition possible en milieu aquatique). La longueur et le poids des poissons utilisés sont compris dans les limites recommandées (voir appendice 3). Les poissons ne doivent pas être trop petits, ce qui gênerait les analyses par individu. Tester des espèces à une période où elles se développent rapidement peut compliquer l'interprétation des données, des taux de croissance élevés pouvant influer sur le calcul du rendement d'assimilation (64). Conditions de vie des poissons en laboratoire Les critères relatifs à l'acclimatation, à la mortalité et à d'éventuelles maladies sont les mêmes pour la méthode d'exposition en milieu aquatique (voir paragraphes 33 à 35). DÉROULEMENT DE L'ESSAI Étude préliminaire et essai de détermination de l'ordre de grandeur Une analyse préliminaire est nécessaire pour démontrer la possibilité d'isoler la substance de la nourriture enrichie ou des tissus du poisson. Il n'est pas toujours nécessaire de réaliser un essai de détermination de l'ordre de grandeur pour décider de la concentration appropriée de la substance chimique. Afin de montrer qu'aucun effet nocif n'est observé, d'évaluer les qualités organoleptiques de l'alimentation enrichie, de déterminer la sensibilité de la méthode d'analyse des tissus du poisson et des aliments, et de définir la ration alimentaire et les temps d'échantillonnage appropriés durant la phase d'élimination, etc., on peut procéder à des essais d'alimentation préliminaires, mais ce n'est pas obligatoire. Une étude préliminaire peut être utile pour estimer le nombre de poissons nécessaires aux prélèvements durant la phase d'élimination. Elle peut permettre de réduire notablement le nombre de poissons utilisés, surtout pour les substances d'essai très sensibles au métabolisme. Conditions d'exposition Durée de la phase d'absorption Une phase d'absorption de 7 à 14 jours est habituellement suffisante; durant cette phase, un groupe de poissons reçoit la nourriture témoin et un autre groupe la nourriture testée. La ration alimentaire qui leur est administrée chaque jour dépend de l'espèce testée et des conditions de l'essai; elle représentera par exemple entre 1 et 2 % du poids du poisson (poids frais) dans le cas de la truite. Cette ration devra être définie de façon à éviter un développement rapide et une augmentation importante de la teneur en lipides. Si besoin, il est possible de prolonger la phase d'absorption en fonction des enseignements tirés d'études antérieures ou des informations connues sur l'absorption ou l'élimination de la substance d'essai (ou analogue) chez le poisson. L'essai proprement dit commence avec la première administration de la nourriture enrichie. Un jour d'essai commence avec l'administration des aliments et se termine peu avant la ration suivante (par exemple une heure avant). Ainsi, en phase d'absorption, le premier jour de l'essai commence avec la première administration de la nourriture enrichie et se termine juste avant la deuxième ration enrichie. En pratique, la phase d'absorption prend fin juste avant (par exemple une heure avant) la première administration de la nourriture non enrichie avec la substance d'essai, sachant que le poisson continue à digérer les aliments enrichis et à absorber la substance d'essai au cours des 24 heures intermédiaires. Il est important de s'assurer que la charge corporelle de la substance d'essai est suffisamment élevée (non-toxique) au regard de la méthode d'analyse appliquée pour pouvoir mesurer une baisse d'au moins une puissance dix durant la phase d'élimination. Dans certains cas, on peut prolonger la phase d'absorption (jusqu'à 28 jours) en réalisant des prélèvements supplémentaires afin d'avoir une idée des cinétiques d'absorption. Pendant l'absorption, la concentration dans le poisson peut ne pas atteindre l'état stationnaire. Pour estimer le temps nécessaire avant atteinte de l'état stationnaire, et avoir ainsi une indication de la durée probable nécessaire avant atteinte de concentrations appréciables dans le poisson, il est possible d'appliquer les équations indiquées pour l'essai par exposition en milieu aquatique (voir appendice 5). Il arrive que l'on sache à l'avance qu'une durée d'absorption de 7 à 14 jours de la substance chimique dans le poisson ne suffira pas pour que la concentration utilisée dans l'alimentation permette d'atteindre une concentration dans le poisson suffisamment élevée pour analyser une baisse d'au moins une puissance dix durant l'élimination, en raison soit d'une faible sensibilité de la méthode d'analyse soit d'un rendement d'assimilation trop bas. Le cas échéant, il peut être utile d'étendre la phase initiale d'alimentation à plus de 14 jours, ou, surtout s'agissant de substances très métabolisables, d'envisager une concentration dans l'alimentation supérieure. Il convient toutefois de veiller à maintenir la charge corporelle durant l'absorption en-dessous de la concentration sans effet observé (CSEO) chronique (estimée) dans les tissus du poisson (voir paragraphe 138). Durée de la phase d'élimination En règle générale, l'élimination dure jusqu'à 28 jours; elle commence dès que l'on donne aux poissons du groupe d'essai une nourriture pure, non traitée, après la phase d'absorption. L'élimination commence avec l'administration de la première ration non enrichie, et non juste après l'administration de la dernière ration enrichie, puisque le poisson continue à digérer les aliments et à absorber la substance d'essai au cours des 24 heures intermédiaires, comme indiqué au paragraphe 126. C'est pourquoi le premier prélèvement de la phase d'élimination est réalisé juste avant la deuxième ration non enrichie. Cette période d'élimination vise à capturer les substances d'une demi-vie potentielle de 14 jours, ce qui correspond aux caractéristiques des substances bioaccumulables (65), un essai d'une durée de 28 jours couvre donc deux demi-vies de ces substances. Avec les substances fortement bioaccumulables, il peut être utile de prolonger la phase d'élimination (si indiqué par l'essai préliminaire). Si une substance est éliminée très lentement au point qu'on ne peut pas atteindre une demi-vie exacte durant la phase d'élimination, les informations obtenues peuvent néanmoins suffire à indiquer un niveau élevé de bioaccumulation et permettre de réaliser les évaluations. À l'inverse, si une substance est éliminée très rapidement au point qu'on ne peut déduire ni aucune concentration fiable au temps 0 (concentration à la fin de l'absorption ou au début de l'élimination, C0,d) ni k 2, on procédera à une estimation basse de k 2 (voir appendice 7). Si les premières analyses des poissons prélevés (à 7 ou 14 jours par exemple) montrent que la substance chimique a été éliminée en-dessous des niveaux prévus avant la fin de la période complète de 28 jours, on peut annuler les prélèvements suivants et arrêter l'essai. Dans certains cas, on ne constate aucune absorption mesurable de la substance d'essai à la fin de la période d'absorption (ou avec le deuxième prélèvement de l'élimination). Si l'on peut démontrer que i) les critères de validité énoncés au paragraphe 113 sont satisfaits, et que ii) cette faible absorption n'est pas due à un défaut de l'essai (par exemple durée d'absorption insuffisante, technique d'enrichissement inadéquate entraînant une faible biodisponibilité, sensibilité insuffisante de la méthode d'analyse, non ingestion des aliments par les poissons, etc.), alors il est possible d'arrêter l'étude sans avoir à renouveler l'essai avec une durée d'absorption plus longue. Si au vu de l'étude préliminaire cela semble être le cas, il peut être conseillé, si possible, d'analyser les excréments pour étudier la substance d'essai non digérée suivant une approche fondée sur le bilan massique. Nombre de poissons testés Comme pour l'essai par exposition via le milieu aquatique, il convient de sélectionner des poissons de poids et de longueurs similaires, le plus petit d'entre eux ne devant pas peser moins de deux tiers du poids du poisson le plus gros (voir paragraphes 40 à 42). Le nombre total de poissons utilisés pour l'étude est établi en fonction du programme de prélèvement (au minimum un prélèvement à la fin de la phase d'absorption et entre quatre et six prélèvements pendant la phase d'élimination, selon la durée de chaque phase). Il convient également de prendre en compte la sensibilité de la technique d'analyse, la concentration susceptible d'être atteinte à l'issue de la phase d'absorption (selon les informations disponibles au préalable) et la durée de l'élimination (si les informations disponibles au préalable permettent de l'estimer). Il convient de prélever chaque fois entre cinq et dix poissons et d'en mesurer le développement (poids et longueur totale) avant l'analyse chimique ou lipidique. En raison de la variabilité inévitable de la taille, du développement et de la physiologie des poissons et de la quantité probablement variable de nourriture ingérée par chacun, il est nécessaire de prélever à chaque temps d'échantillonnage au moins cinq poissons du groupe d'essai et cinq poissons du groupe témoin pour établir de façon appropriée la concentration moyenne et sa variabilité. La variabilité des paramètres à considérer chez les poissons est susceptible d'accroître plus la variabilité générale non maîtrisée de l'essai que la variabilité inhérente aux méthodes d'analyse appliquées, ce qui justifie l'utilisation dans certains cas de jusqu'à dix poissons par prélèvement. Toutefois, si les concentrations de référence de la substance d'essai dans les poissons témoins ne sont pas mesurables au début de l'élimination, l'analyse chimique de deux à trois poissons témoins au dernier temps d'échantillonnage suffira uniquement si l'on continue à prélever les poissons restants du groupe témoin en fonction de leur poids et de leur longueur totale (de façon à prélever chaque fois le même nombre dans les groupes d'essai et témoin pour tenir compte de leur développement). Les poissons sont stockés, pesés individuellement (même s'il s'avère finalement nécessaire de combiner les résultats des prélèvements) et mesurés (longueur totale). Ainsi, un essai type prévoyant une phase d'élimination de 28 jours avec cinq prélèvements nécessitera au total entre 59 et 120 poissons dans le groupe d'essai et entre 50 et 110 poissons dans le groupe témoin, en supposant que la technique d'analyse de la substance permette d'analyser la teneur en lipides sur un même poisson. S'il n'est pas possible d'analyser la substance chimique et la teneur en lipides sur le même poisson et que l'utilisation d'un poisson témoin pour analyser la teneur en lipides n'est pas envisageable (voir paragraphe 56), on devra ajouter 15 poissons (trois du stock de poissons au début de l'essai, trois respectivement du groupe témoin et du groupe d'essai au début de l'élimination et trois respectivement du groupe témoin et du groupe d'essai à la fin de l'expérience). On trouvera un exemple de programme de prélèvement avec les nombres des poissons utilisés à l'appendice 4. Taux de charge Les ratios eau/poissons sont aussi élevés qu'avec la méthode d'exposition en milieu aquatique (voir paragraphes 43 et 44). Bien que les taux de charge poissons/eau n'influent pas sur les concentrations d'exposition dans cet essai, il est recommandé d'appliquer un taux de charge compris entre 0,1 et 1,0 g de poisson (poids frais) par litre d'eau et par jour pour maintenir des concentrations adéquates d'oxygène dissous et minimiser le stress de l'essai sur les organismes. Nourriture testée et alimentation Durant la période d'acclimatation, les poissons reçoivent une alimentation appropriée (voir paragraphe 117). Si l'essai est réalisé avec un système de renouvellement dynamique, il convient d'interrompre le renouvellement au moment de nourrir les poissons. Durant l'essai, il convient que l'alimentation du groupe testé soit conforme aux critères décrits précédemment (voir paragraphes 116 à 121). En plus des caractéristiques de la substance testée, de la sensibilité de la méthode d'analyse, de la concentration attendue dans l'alimentation en fonction des conditions environnementales et des niveaux de toxicité chronique/charge corporelle, il convient de tenir compte, pour définir la concentration cible de la substance, des qualités organoleptiques des aliments (qui ne repoussent pas les poissons). La concentration nominale de la substance d'essai est consignée dans le rapport d'essai. D'expérience, les concentrations comprises entre 1 et 1 000 μg/g fournissent une fourchette d'étude adéquate pour les substances qui ne présentent aucun mécanisme toxique spécifique. S'agissant des produits chimiques dont l'action obéit à un mécanisme non spécifique, les résidus présents dans les tissus n'excèdent pas 5 μmol/g de lipides, sinon ils risquent d'entraîner des effets chroniques (19) (48) (50) (66). Concernant les autres substances, il convient de s'assurer qu'aucun effet nocif ne résulte de l'exposition cumulée (voir paragraphe 127). Cela est d'autant plus vrai si plusieurs substances sont testées en même temps (voir paragraphe 112). La quantité adéquate de la substance d'essai peut être ajoutée à la nourriture pour poisson de trois façons (voir paragraphe 119 et appendice 7). Les méthodes et procédures d'enrichissement utilisées sont spécifiées dans le rapport d'essai. Les aliments non traités sont donnés aux poissons témoins; si une huile non enrichie ou un solvant est utilisé comme véhicule dans la nourriture enrichie durant la phase d'absorption, la nourriture non traitée devra contenir une quantité équivalente de cette huile ou être traitée avec ce solvant à l'état pur. Il est nécessaire d'analyser au moins trois échantillons de la concentration de la substance d'essai dans les aliments, traités et non traités, avant le début et à la fin de la phase d'absorption. Après exposition à la nourriture traitée (phase d'absorption), les poissons (des deux groupes) reçoivent de la nourriture non traitée (phase d'élimination). Les poissons reçoivent une ration déterminée (en fonction de l'espèce; d'environ 1 à 2 % du poids frais par jour dans le cas de la truite). Cette ration alimentaire est déterminée de façon à éviter un développement rapide des poissons et une hausse importante de la teneur en lipides. Il convient de spécifier dans le rapport les rations administrées tout au long de l'essai. La première ration s'appuie sur les poids mesurés dans le stock de poisson juste avant le début de l'essai. La quantité est ajustée en fonction du poids frais des poissons prélevés aux différents temps prévus, pour tenir compte de leur développement en cours d'essai. Les poids et les longueurs des poissons dans les récipients d'essai et témoins peuvent être estimés à partir des poids et des longueurs totales des poissons utilisés à chaque échantillonnage; il ne faut pas peser ou mesurer le poisson restant dans les récipients d'essai et témoins. Il est important de respecter la ration alimentaire fixée tout au long de l'expérience. Il convient d'observer l'alimentation des poissons pour s'assurer qu'ils consomment visiblement toute la nourriture présentée et donc que les taux d'ingestion utilisés dans les calculs sont justes. On considérera la pertinence de réaliser des essais d'alimentation préliminaires ou de prendre en compte des expériences antérieures pour déterminer une ration alimentaire qui garantisse que toute la nourriture journalière administrée en une fois est consommée. Quand les poissons laissent systématiquement une partie de cette nourriture, il peut être judicieux de répartir la dose en ajoutant une prise par jour d'essai (même quantité journalière mais divisée par deux et administrée en deux fois au lieu d'une, par exemple). Au besoin, la seconde prise interviendra à un moment précis et sera chronométrée de façon à ce que le maximum de temps possible s'écoule avant le prélèvement des poissons (par exemple, cette seconde prise interviendra pendant la première moitié d'une journée d'essai). Bien que les poissons consomment en général rapidement la nourriture administrée, il est important de s'assurer que la substance chimique reste adsorbée dans les aliments. On évitera donc que la substance d'essai ne se disperse dans l'eau, ce qui exposerait les poissons à des concentrations aqueuses de la substance d'essai en plus de la voie alimentaire. Pour cela, il convient de retirer les aliments non consommés (et les excréments) des récipients d'essai et témoins dans l'heure voire, de préférence, dans les 30 minutes suivant l'administration. De plus, il est possible d'utiliser un système par lequel l'eau est nettoyée en continu au moyen d'un filtre à charbon actif, qui adsorbe tout contaminant dissous. Les systèmes de renouvellement dynamiques peuvent aider à évacuer rapidement les particules alimentaires et les substances dissoutes (67). Parfois, modifier la technique d'enrichissement des aliments peut contribuer à atténuer le problème (voir paragraphe 119). Lumière et température Concernant la méthode d'exposition via le milieu aquatique (voir paragraphe 48), une photopériode de 12 à 16 heures est recommandée et la température (± 2 °C) est adaptée à l'espèce testée (voir appendice 3). Il convient de connaître le type et les caractéristiques de l'illumination et de les spécifier dans le rapport. Témoins Il convient d'utiliser un groupe témoin auquel on donne la même ration qu'au groupe d'essai mais sans ajouter la substance d'essai à leur nourriture. Si une huile ou un solvant a été utilisé comme véhicule pour enrichir les aliments du groupe d'essai, il faudra traiter la nourriture du groupe témoin exactement de la même façon mais sans ajouter la substance d'essai de façon à ce que l'alimentation des groupes d'essai et témoin soit équivalente (voir paragraphes 121 et 139). Fréquence des mesures de la qualité de l'eau Les conditions décrites pour l'essai par exposition en milieu aquatique s'appliquent ici aussi, à l'exception du fait que le carbone organique total est mesuré uniquement avant l'essai au moment de la caractérisation de l'eau de l'essai (voir paragraphe 53). Prélèvement et analyse des poissons et alimentation Analyse des échantillons alimentaires Au moins trois échantillons de la nourriture d'essai et témoin sont analysés pour déterminer la concentration de la substance d'essai et la teneur en lipides, au minimum avant le début et à la fin de la phase d'absorption. Les méthodes d'analyse et les procédures appliquées pour garantir l'homogénéité de l'alimentation sont spécifiées dans le rapport d'essai. Il convient d'analyser la substance d'essai dans les échantillons suivant la méthode définie et validée. Une étude préliminaire est réalisée pour établir la limite de quantification, le pourcentage d'isolement, les interférences et la variabilité de l'analyse pour la matrice d'échantillon prévue. Si l'on teste du matériel radiomarqué, on devra considérer les mêmes aspects qu'avec la méthode d'exposition en milieu aquatique, l'analyse des aliments remplaçant l'analyse de l'eau (voir paragraphe 65). Analyse des poissons À chaque temps de prélèvement, on prélève entre 5 et 10 individus dans chacun des groupes, témoin et d'essai (dans certains cas, le nombre de poissons témoins peut être réduit; voir paragraphe 134). Les prélèvements ont lieu, chaque jour de l'expérience, au même moment (en fonction du moment auquel les poissons sont nourris) et sont chronométrés pour réduire au maximum la probabilité que des aliments restent dans les intestins durant la phase d'absorption et au début de la phase d'élimination, et éviter ainsi de fausser les concentrations totales de la substance d'essai (il convient donc de prélever les poissons à la fin d'une journée d'essai, sachant qu'un jour d'essai commence avec l'administration des aliments et se termine juste avant la ration suivante, soit approximativement 24 heures plus tard). L'élimination commence avec la première ration de nourriture «non enrichie» (voir paragraphe 128). Le premier prélèvement de la phase d'élimination (réalisé juste avant la deuxième ration non enrichie) est important, puisqu'il sert à extrapoler la concentration au temps 0, un jour plus tôt, (C0,d, la concentration dans le poisson à la fin de l'absorption/au début de l'élimination). En option, il est aussi possible de prélever et d'analyser séparément le tractus gastro-intestinal du poisson à la fin de l'absorption et aux jours 1 et 3 de l'élimination. À chaque temps d'échantillonnage, il convient de prélever les poissons des groupes d'essai et témoin et de les traiter de la même façon qu'avec la méthode d'exposition en milieu aquatique (voir paragraphes 61 à 63). On mesure les concentrations de la substance d'essai dans le poisson (poids frais) au moins à la fin de la phase d'absorption et pendant la phase d'élimination dans les groupes témoin et d'essai. Durant la phase d'élimination, quatre à six temps d'échantillonnage sont recommandés (par exemple aux jours 1, 3, 7, 14 et 28). En option, il est aussi possible d'inclure un temps d'échantillonnage supplémentaire après 1 à 3 jours d'absorption pour estimer le rendement d'assimilation à partir de la phase linéaire de l'absorption, puisqu'encore très proche du début de la période d'exposition. La méthode décrite tolère deux écarts: i) si on prolonge la phase d'absorption pour étudier les cinétiques d'absorption, il faudra prévoir un temps d'échantillonnage supplémentaire durant la phase d'absorption et donc inclure des poissons supplémentaires (voir paragraphe 126); ii) si aucune absorption mesurable n'intervient à la fin de la phase d'absorption, on devra mettre fin à l'essai (voir paragraphe 131). Chaque poisson prélevé est pesé (et sa longueur totale mesurée) pour permettre de déterminer les constantes cinétiques de croissance. On peut aussi mesurer les concentrations de la substance dans des tissus spécifiques du poisson (fractions comestibles et non comestibles) à la fin de l'absorption et à des moments précis de l'élimination. Si l'on teste du matériel radiomarqué, il est nécessaire de considérer les mêmes aspects qu'avec la méthode d'exposition en milieu aquatique, l'analyse des aliments remplaçant l'analyse de l'eau (voir paragraphe 65). En cas d'utilisation périodique d'une substance de référence (voir paragraphe 25), il est préférable de mesurer les concentrations dans le groupe d'essai à la fin de l'absorption et à tous les temps de l'élimination spécifiés pour la substance d'essai (poisson entier); il convient d'analyser uniquement les concentrations dans le groupe témoin à la fin de l'absorption (poisson entier). Parfois (par exemple quand les techniques d'analyse de la substance d'essai et de la substance de référence sont incompatibles au point qu'il est nécessaire d'utiliser des poissons supplémentaires pour respecter le programme de prélèvement), on pourra appliquer une autre approche pour minimiser le nombre de poissons supplémentaires nécessaires. Les concentrations de la substance de référence sont mesurées pendant l'élimination uniquement aux jours 1, 3 et lors de deux autres temps d'échantillonnage, sélectionnés de manière à obtenir des estimations fiables de la concentration au temps 0 (C0,d) et de k 2 pour cette substance de référence. Si possible, la teneur en lipides de chaque poisson est déterminée à chaque prélèvement, ou au moins au début et à la fin de la phase d'absorption et à la fin de la phase d'élimination (voir paragraphes 56 et 67). Selon la méthode d'analyse retenue (voir paragraphe 67 et appendice 4), il est possible d'utiliser les mêmes poissons pour déterminer la teneur en lipides et la concentration de la substance d'essai. Cela permet de minimiser le nombre de poissons nécessaires. Néanmoins, quand ce n'est pas possible, il convient d'appliquer l'approche décrite pour la méthode d'exposition en milieu aquatique (voir paragraphe 56 les différentes options envisageables pour mesurer la teneur en lipides). La méthode appliquée pour quantifier la teneur en lipides est spécifiée dans le rapport. Qualité de la méthode d'analyse Il est nécessaire de mener des essais préliminaires pour s'assurer de la spécificité, de la précision et de la reproductibilité de la technique d'analyse spécifique à la substance, ainsi que de l'isolement de la substance d'essai à partir de l'alimentation et du poisson. Mesure du développement des poissons Au début de l'essai, il convient de peser et de mesurer un échantillon de poissons du stock. Ces poissons sont prélevés juste avant la première administration de l'alimentation enrichie (une heure avant par exemple) et assignés au jour d'essai 0. Il convient que le nombre de poissons prélevés soit égal ou supérieur à celui des poissons prélevés ultérieurement pendant l'essai. Il peut s'agir des mêmes poissons utilisés pour l'analyse lipidique réalisée avant le début de la phase d'absorption (voir paragraphe 153). À chaque temps d'échantillonnage, les poissons sont d'abord pesés puis mesurés. Pour chaque poisson, le poids (et la longueur) est relié à la concentration chimique analysée (et s'il y a lieu à la teneur en lipides), par exemple en attribuant un identifiant unique à chaque poisson prélevé. Ces mesures peuvent servir à estimer le poids (et la longueur) des poissons restant dans les récipients d'essai et témoins. Évaluation de l'essai Il convient de consigner chaque jour les observations relatives à la mortalité. On observera et enregistrera aussi tout effet nocif, par exemple un comportement anormal ou une pigmentation. Les poissons sont considérés morts en l'absence de mouvement respiratoire ou de réaction à un léger stimulus mécanique. Tout poisson mort ou visiblement moribond devra être retiré. RAPPORT D'ESSAI ET RÉSULTATS Traitement des résultats Les résultats des essais sont utilisés pour calculer la constante cinétique d'élimination (k 2) en fonction du poids frais total du poisson. La constante cinétique de croissance, kg , basée sur la progression moyenne du poids du poisson est calculée et utilisée pour produire la constante cinétique d'élimination corrigée de la croissance, k 2g, s'il y a lieu. Il convient en outre de consigner le rendement d'assimilation (a; absorption par voie intestinale), le facteur de bioamplification cinétique (FBAk) (si nécessaire corrigé de la croissance, FBAkg), sa valeur corrigée des lipides (FBAkL ou FBAkgL, si corrigé de l'effet de dilution par la croissance) et la ration alimentaire. Par ailleurs, s'il est possible d'estimer le temps nécessaire à l'instauration de l'état stationnaire durant la phase d'absorption (par exemple 95 % de l'état stationnaire ou t95 = 3,0/k 2), on pourra inclure une estimation du FBA à l'état stationnaire (FBAES) (voir paragraphes 105 et 106 et appendice 5) si la valeur t95 indique que l'état stationnaire semble atteint. Il convient d'appliquer la même correction lipidique au FBAES qu'au FBA cinétique (FBAk) pour obtenir une valeur corrigée de la teneur en lipides, FBAESL (il n'existe aucune procédure convenue pour corriger un FBA à l'état stationnaire de l'effet de la dilution par la croissance). Les formules et des exemples de calcul sont présentés à l'appendice 7. Plusieurs méthodes permettent d'estimer un facteur de bioconcentration cinétique (FBCk) à partir des données obtenues dans l'étude par voie alimentaire. Elles sont discutées à l'appendice 8. Poids et longueur des poissons Le poids frais et la longueur de chaque poisson, à chaque temps d'échantillonnage, sont présentés séparément pour les groupes d'essai et les groupes témoins pendant la phase d'absorption [poissons du stock au début de la période d'absorption; groupe témoin et groupe d'essai à la fin de la période d'absorption et, le cas échéant, au début de la période (par exemple jours 1 à 3 de l'absorption) et pendant la phase d'élimination (par exemple jours 1, 2, 4, 7, 14, 28, pour le groupe d'essai et le groupe témoin)]. Le poids est la mesure du développement à privilégier afin de corriger les valeurs de l'effet de dilution par la croissance. Les paragraphes 162 et 163 et l'appendice 5 présentent la ou les méthode(s) utilisée(s) pour cette correction. Concentration de la substance d'essai dans les poissons Les mesures des résidus de la substance d'essai réalisées sur chaque poisson (ou sur des échantillons regroupés si des mesures individuelles ne sont pas possibles), exprimées en concentration du poids frais, sont présentées séparément selon les temps d'échantillonnage pour les groupes d'essai et témoins. Si une analyse lipidique a été réalisée sur chacun des poissons prélevés, on calculera et consignera les concentrations individuelles corrigées de la teneur en lipides, exprimées en teneur en lipides du poids frais.
Vitesse d'élimination et facteur de bioamplification Pour calculer le facteur de bioamplification, il convient tout d'abord d'obtenir le rendement d'assimilation (absorption de la substance d'essai par voie intestinale, α). Pour cela, il convient d'appliquer l'équation A7.1 à l'appendice 7, qui nécessite de connaître la concentration dans le poisson obtenue au temps 0 de la phase d'élimination (C0,d), la constante cinétique d'élimination (globale) (k 2), la concentration dans les aliments (C alim), la constante cinétique d'ingestion (I) et la durée de la phase d'absorption (t). La pente et l'ordonnée à l'origine de la relation linéaire entre ln(concentration) et le temps d'élimination sont consignées comme la constante cinétique d'élimination globale (k 2 = pente) et la concentration au temps 0 (C0,d = eordonn), comme indiqué précédemment. Il convient de contrôler la plausibilité biologique des valeurs obtenues (par exemple, le rendement d'assimilation sous forme de fraction n'est pas supérieur à 1). (I) est calculée en divisant la masse des aliments par la masse du poisson nourri chaque jour (s'il est nourri à 2 % de son poids, (I) sera égal à 0.02). Néanmoins, il peut s'avérer nécessaire d'ajuster la ration alimentaire utilisée dans les calculs en fonction du développement des poissons (en utilisant la constante cinétique de croissance connue pour estimer le poids du poisson à chaque temps de prélèvement durant la phase d'absorption; voir appendice 7). Dans les cas où il n'est pas possible d'obtenir k 2 et C0,d parce que, par exemple, les concentrations ont chuté sous le seuil de détection au moment du deuxième prélèvement d'élimination, on peut procéder à une estimation basse de k 2 et borner FBAk supérieurement (voir appendice 7). Une fois le rendement d'assimilation (α) obtenu, on peut calculer le facteur de bioamplification en multipliant α par la constante cinétique d'ingestion (I) et en la divisant par la constante cinétique d'élimination (globale) (k 2). Le facteur de bioamplification corrigé de l'effet de dilution par la croissance est calculé de la même façon mais en utilisant la constante cinétique d'élimination corrigée de la croissance (k 2g; voir paragraphes 162 et 163). Il est aussi possible d'estimer le rendement d'assimilation si on a analysé les tissus des poissons prélevés lors de la phase initiale linéaire de l'absorption (voir paragraphe 151 et appendice 7). Cette valeur représente une estimation indépendante du rendement d'assimilation pour un organisme quasiment non exposé (à savoir le poisson prélevé au tout début de la phase d'absorption). Le rendement d'assimilation estimé à partir des données de l'élimination sert habituellement à obtenir le FBA. Correction de la teneur en lipides et correction de la dilution par la croissance La croissance des poissons pendant la phase d'élimination peut réduire les concentrations chimiques mesurées chez eux, ce qui augmente la constante cinétique d'élimination globale, k 2, par rapport à ce que provoqueraient les processus d'élimination seuls (par exemple métabolisme, égestion) (voir paragraphe 72). La teneur en lipides des poissons d'essai (fortement associée à la bioaccumulation des substances hydrophobes) et la teneur en lipides des aliments peuvent suffisamment varier dans la pratique pour que leur correction soit nécessaire pour obtenir des facteurs de bioamplification qui aient un sens. Le facteur de bioamplification est corrigé de l'effet de dilution par la croissance (comme le FBC cinétique avec la méthode d'exposition en milieu aquatique) et corrigé de la teneur en lipides des aliments en fonction de celle du poisson (le facteur de correction en fonction des lipides). Les appendices 5 et 7 présentent les équations respectives et donnent des exemples de calculs de ce type. Pour corriger de la dilution par la croissance, il convient de calculer la constante cinétique d'élimination corrigée de la croissance (k 2g) (voir appendice 5 pour les équations). Cette constante cinétique d'élimination corrigée (k 2g) est utilisée pour calculer le facteur de bioamplification corrigé de la croissance, comme indiqué au paragraphe 73. Dans certains cas, cette méthode n'est pas possible. Une autre méthode contournant la nécessité d'une correction de la dilution par la croissance consiste à utiliser le poids de la substance d'essai par poisson (entier) lors de l'élimination plutôt que le poids de la substance d'essai par unité de masse du poisson (concentration). Ce calcul est facile à faire, car dans le cadre d'essais respectant la présente méthode d'essai, on associe les concentrations tissulaires aux poids individuels des poissons. L'appendice 5 présente la procédure de calcul simple. Il convient de souligner que le recours à cette méthode alternative n'empêche pas qu'il faille estimer et consigner la valeur de k 2. Pour corriger la teneur en lipides des aliments et du poisson quand l'analyse lipidique n'a pas été réalisée sur tous les poissons prélevés, on déduit les fractions lipidiques moyennes (du poids frais) dans le poisson et dans les aliments (68). Le facteur de correction de la teneur en lipides (Lc) est alors calculé en divisant la fraction lipidique moyenne du poisson par la fraction lipidique moyenne des aliments. Le facteur de bioamplification, corrigé de la croissance ou pas, est divisé par le facteur de correction de la teneur en lipides pour calculer le facteur de bioamplification corrigé des lipides. Si l'analyse chimique et l'analyse lipidique ont été menées sur le même poisson au même temps d'échantillonnage, il convient d'utiliser ces données corrigées des lipides par poisson pour calculer directement un FBA corrigé des lipides [voir (37)]. La courbe de la concentration corrigée en fonction des lipides donne C0,d et k 2. On peut ensuite procéder à une analyse mathématique en utilisant les équations à l'appendice 7, mais le rendement d'assimilation (a) est calculé en utilisant la constante cinétique d'ingestion normalisée par rapport aux lipides (Ilipides) et la concentration dans l'alimentation en fonction des lipides (C alim-lipides). Les paramètres corrigés de la teneur en lipides sont alors utilisés de façon similaire pour calculer le FBA (pour calculer le FBAkgL corrigé de la teneur en lipides et de la croissance, on applique aussi la correction de la constante cinétique de croissance à la fraction lipidique et non au poids frais du poisson). Interprétation des résultats La croissance moyenne dans le groupe d'essai et celle dans le groupe témoin en principe ne diffèrent pas beaucoup afin d'exclure les effets toxiques. Les constantes cinétiques de croissance ou les courbes de croissance des deux groupes sont comparées au moyen d'une procédure adaptée (69). Rapport d'essai Une fois l'étude terminée, il convient de rédiger un rapport final présentant les informations sur la substance d'essai, l'espèce testée et les conditions expérimentales énumérées au paragraphe 81 (pour la méthode d'exposition en milieu aquatique). En outre, ce rapport devra aussi inclure les renseignements suivants:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS ET UNITÉS
BIBLIOGRAPHIE
Appendice 2 QUELQUES CARACTERISTIQUE CHIMIQUES D'UNE EAU DE DILUTION ADMISSIBLE
Appendice 3 ESPÈCES DE POISSONS RECOMMANDÉES POUR L'ESSAI
Plusieurs espèces estuariennes et marines ont été utilisées plus rarement, notamment:
Les poissons d'eau douce mentionnés dans le tableau qui précède sont faciles à élever et/ou à se procurer tout au long de l'année, tandis que la disponibilité des espèces marines ou estuariennes est en partie limitée à leurs pays d'origine. Afin d'être en bonne santé et d'ascendance connue, les animaux à tester peuvent être élevés et se reproduire dans des fermes aquacoles ou en laboratoire, où ils sont protégés des maladies et des parasites. Ces poissons se trouvent dans beaucoup de parties du monde. BIBLIOGRAPHIE
Appendice 4 PROGRAMMES DE PRÉLÈVEMENT POUR LES ESSAIS D'EXPOSITION EN MILIEU AQUATIQUE ET PAR VOIE ALIMENTAIRE 1. Exemple théorique d'un programme de prélèvement pour un essai de bioconcentration avec exposition exclusivement en milieu aquatique pratiqué sur une substance dont le log K oe = 4.
2. Exemple théorique d'un programme de prélèvement pour un essai de bioaccumulation de la substance par voie alimentaire avec des phases d'absorption et d'élimination de respectivement 10 et 42 jours.
Note concernant la durée des phases et les temps d'échantillonnage: la phase d'absorption commence avec l'administration de la première ration enrichie. Le premier jour de l'essai commence avec la première administration de nourriture et se termine juste avant la suivante, 24 heures plus tard. Le premier prélèvement (1 dans le tableau) devrait intervenir juste avant la première administration de nourriture (une heure avant par exemple). Idéalement, il faudrait chaque fois prélever les poissons juste avant la ration du jour suivant (soit environ 23 heures après la dernière ration). La phase d'absorption prend fin juste avant la première administration de la nourriture non enrichie, quand la phase d'élimination commence (il est probable que les poissons du groupe d'essai digèrent encore les aliments enrichis dans les 24 heures suivant la dernière administration de nourriture enrichie). Autrement dit, le dernier prélèvement de la phase d'absorption intervient juste avant la première ration non enrichie, et le premier prélèvement de la phase d'élimination intervient environ 23 heures après la première ration non enrichie. Appendice 5 CALCULS GÉNÉRAUX
1. INTRODUCTION Le modèle général de bioaccumulation en milieu aquatique chez le poisson peut être décrit en termes de processus d'absorption et de perte, en ignorant l'absorption par voie alimentaire. L'équation différentielle (dC p/dt) qui décrit la vitesse de modification de la concentration dans le poisson (mg.kg– 1.jour– 1) est donnée avec (1):
Où
S'agissant de substances bioaccumulables, on peut s'attendre à ce qu'une moyenne pondérée par rapport au temps soit la concentration d'exposition dans l'eau (C e) la plus pertinente au sein de la fourchette de fluctuations autorisée (voir paragraphe 24). Il est recommandé de calculer une moyenne pondérée par rapport au temps de la concentration dans l'eau en suivant les instructions données à l'appendice 6 de la méthode d'essai C.20 (2). Notons que le ln-transformation de la concentration dans l'eau est approprié quand on s'attend à un déclin exponentiel entre les périodes de renouvellement, par exemple dans des conditions d'essai semi-statiques. Avec un système dynamique, le ln-transformation des concentrations d'exposition n'est pas forcément nécessaire. Si on obtient une moyenne pondérée par rapport au temps de la concentration dans l'eau, il convient de la consigner et de l'utiliser dans les calculs suivants. Dans un essai FBC type réalisé chez des poissons, l'absorption et l'élimination peuvent être décrites en termes de deux processus cinétiques de premier ordre.
À l'état stationnaire, en supposant que le développement et le métabolisme sont négligeables (les valeurs pour kg et km ne peuvent pas être distinguées de zéro), la cinétique d'absorption est égale à la cinétique d'élimination, et donc en combinant les équations A5.2 et A5.3 on obtient la relation suivante:
Où
Le ratio de k 1/k 2 correspond au FBC cinétique (FBCk) et devrait être égal au FBC à l'état stationnaire (FBCES) obtenu à partir du rapport entre la concentration à l'état stationnaire dans le poisson et la concentration à l'état stationnaire dans l'eau, mais des écarts sont possibles si l'état stationnaire est incertain ou si des corrections de la croissance ont été appliquées au FBC cinétique. Néanmoins, k 1 et k 2 étant des constantes, il n'est pas nécessaire que l'état stationnaire soit atteint pour obtenir un FBCk. Fondée sur ces équations du premier ordre, cette appendice 5 présente les calculs généraux nécessaires pour les deux méthodes de bioaccumulation, avec exposition en milieu aquatique et exposition par voie alimentaire. Les sections 5, 6 et 8 sont uniquement pertinentes pour la méthode d'exposition en milieu aquatique, mais ont été incluses ici parce qu'elles relèvent de techniques «générales». Les méthodes en mode séquentiel (sections 4 et 5) et simultané (section 6) permettent de calculer les constantes d'absorption et d'élimination qui servent à obtenir les FBC cinétiques. Utilisée pour déterminer k 2 (section 4), la méthode séquentielle est importante pour l'exposition par voie alimentaire, puisqu'elle aide à calculer à la fois le rendement d'assimilation et le FBA. L'appendice 7 détaille les calculs spécifiques à la méthode d'exposition par voie alimentaire. 2. PREDICTION DE LA DUREE DE LA PHASE D'ABSORPTION Avant de commencer l'essai, on peut estimer k 2 et, par conséquent, un certain pourcentage du temps requis pour atteindre l'état stationnaire, à partir des relations empiriques entre k 2 et le coefficient de partage n-octanol/eau (K oe) ou k 1 et le FBC. Il convient toutefois de tenir compte du fait que les équations présentées dans cette section s'appliquent uniquement quand l'absorption et l'élimination sont régies par une cinétique du premier ordre. Si cela n'est manifestement pas le cas, il est conseillé de demander l'avis d'un biostatisticien et/ou d'un pharmacocinéticien, pour savoir si des prédictions de la durée de la phase d'absorption sont souhaitables. Plusieurs méthodes permettent d'estimer k 2 (jour– 1) Par exemple, on pourra utiliser en premier lieu les relations empiriques suivantes (87):
ou
P= poids moyen du poisson traité (en grammes de poids frais) à la fin de l'absorption/au début de l'élimination (88) Pour d'autres relations associées, voir (6). Il peut être avantageux d'employer des modèles plus complexes pour estimer k 2 si, par exemple, il est probable qu'un métabolisme notable intervienne (7) (8). Néanmoins, en raison de la complexité accrue du modèle, on veillera à accorder une attention particulière à l'interprétation des prédictions. Ainsi, la présence de groupes nitro pourrait indiquer un métabolisme rapide, mais ce n'est pas toujours le cas. Aussi l'utilisateur doit-il considérer les résultats de la méthode prédictive au regard de la structure chimique et de toute autre information pertinente (notamment des résultats des études préliminaires) pour programmer une étude. Le temps nécessaire pour atteindre un certain pourcentage de l'état stationnaire peut être déduit, en appliquant l'estimation de k 2, de l'équation cinétique générale qui décrit l'absorption et l'élimination (cinétique du premier ordre), en supposant que le développement et le métabolisme sont négligeables. En cas de développement substantiel durant l'étude, les estimations décrites ci-dessous ne seront pas fiables. Il est alors préférable d'utiliser la k 2g corrigée de la croissance (voir la section 7 de la présente appendice):
ou, si C e est constante:
Lorsqu'on se rapproche de l'état stationnaire (t → ∞), l'équation A5.10 peut être réduite (voir. (9) (10)) à:
ou
FBC × C e est donc une approximation de la concentration dans le poisson à l'état stationnaire (C p ES). [Note: la même approche peut être utilisée pour estimer un FBA à l'état stationnaire lors d'un essai avec exposition par voie alimentaire. Dans ce cas, le FBC est remplacé par le FBA, et la C e par la C alim, la concentration dans les aliments, dans les équations ci-dessus.] L'équation A5.10 peut être reformulée de la façon suivante:
or
En appliquant l'équation A5.14, le temps nécessaire à l'obtention d'un certain pourcentage de l'état stationnaire peut être prédit lorsque k 2 est estimée à l'avance à l'aide de l'équation A5.5 ou A5.6. À titre d'orientation, la durée statistiquement optimale de la phase d'absorption, pour la production de données statistiquement acceptables (FBCk), est la période requise pour que la courbe du logarithme de la concentration de la substance d'essai dans le poisson en fonction du temps atteigne au moins 50 % de l'état stationnaire (soit 0,69/k 2), mais pas plus de 95 % de l'état stationnaire (soit 3,0/k 2) (11). Si l'accumulation dépasse 95 % de l'état stationnaire, le calcul d'un FBCES devient faisable. Le temps nécessaire pour atteindre 80 % de l'état stationnaire est égal à (en utilisant l'équation A5.14):
ou
De même, le temps pour atteindre 95 % de l'état stationnaire est donné par:
À titre d'exemple, la durée de la phase d'absorption (soit le temps nécessaire à l'obtention d'un certain pourcentage de l'état stationnaire, par exemple t 80 ou t 95) d'une substance d'essai dont le log Koe = 4 atteindrait (en appliquant les équations A5.5, A5.16 et A5.17): logk 2 = 1,47 – 0,414 · 4 k 2 = 0,652 jour– 1
ou Sinon, la formule suivante:
peut être utilisée pour calculer le temps nécessaire pour que l'état stationnaire réel (t eES) soit atteint (12). Pour une substance d'essai dont le log Koe = 4 cela donne: teES = 6,54 · 10 – 3 · 104 + 55,31 = 121 heures 3. PREDICTION DE LA DUREE DE LA PHASE D'ELIMINATION Une prédiction du temps requis pour ramener la charge corporelle à un certain pourcentage de la concentration initiale peut aussi être obtenue à partir de l'équation générale qui décrit l'absorption et l'élimination (en supposant une cinétique du premier ordre, voir l'équation A5.9 (1) (13). En ce qui concerne la phase d'élimination, C e(or C alim pour l'essai avec exposition par voie alimentaire) est supposée être nulle. L'équation peut donc être réduite à:
ou
où Cp ,0 est la concentration au début de la période d'élimination. t 50 correspond au moment où l'élimination aura atteint 50 %:
ou
De même, l'élimination s'élèvera à 95 % à t95:
Si on adopte 80 % d'absorption pour la première phase (1,6/k 2) et une perte de 95 % pour la phase d'élimination (3,0/k 2), alors la phase d'élimination vaut approximativement le double de la phase d'absorption. Il convient de noter que les estimations reposent sur l'hypothèse suivant laquelle les processus d'absorption et d'élimination sont régis par une cinétique du premier ordre. Si ces processus n'obéissent manifestement pas à une cinétique du premier ordre, ces estimations ne sont pas valides. 4. METHODE SEQUENTIELLE: DETERMINATION DE LA CONSTANTE CINETIQUE D'ELIMINATION (DE PERTE) K 2 On a fait l'hypothèse que la plupart des données concernant la bioconcentration étaient «raisonnablement» bien décrites par un modèle simple à deux compartiments/deux paramètres, comme le montre la courbe rectiligne qui relie approximativement les points représentant la concentration dans le poisson (sur un graphique logarithmique), pendant la phase d'élimination.
Remarquons que les écarts à la droite peuvent résulter d'un processus d'élimination plus complexe que celui régi par une cinétique du premier ordre. La méthode graphique peut être mise à profit pour traiter les processus d'élimination qui s'écartent d'une cinétique du premier ordre. Pour calculer k 2 pour des temps de prélèvement multiples, il convient de réaliser une régression linéaire de ln(concentration) par rapport au temps. La pente de la droite de régression est une estimation de la constante cinétique d'élimination k 2 (89). À partir de l'ordonnée à l'origine, la concentration moyenne dans le poisson au début de la phase d'élimination (C0,d; qui est égale à la concentration moyenne dans le poisson à la fin de la phase d'absorption) peut être facilement calculée (y compris les marges d'erreur) (89):
Pour calculer k 2 quand seulement deux temps de prélèvement sont disponibles (comme dans le concept d'essai réduit), il convient de substituer les deux concentrations moyennes dans l'équation suivante
Où ln(C p1) et ln(C p2) sont les logarithmes naturels des concentrations aux temps t1 et t2, respectivement, et t2 et t1 correspondent aux temps auxquels les deux prélèvements ont été réalisés par rapport au début de l'élimination (90). 5. METHODE SEQUENTIELLE: DETERMINATION DE LA CONSTANTE CINETIQUE D'ABSORPTION K 1 (METHODE D'EXPOSITION EN MILIEU AQUATIQUE UNIQUEMENT) Pour trouver une valeur pour k 1 d'après un ensemble de valeurs séquentielles de la concentration en fonction du temps pour la phase d'absorption, il est nécessaire d'utiliser un programme informatique qui corresponde au modèle suivant:
Où k 2 est donnée par le calcul précédent, Cp(t) et Ce(t) sont les concentrations dans le poisson et dans l'eau, respectivement, au temps t. Pour calculer k 1 quand seulement deux temps de prélèvement sont disponibles (comme dans le concept d'essai réduit), il convient d'utiliser la formule suivante:
Où k 2 est donnée par le calcul précédent, C p est la concentration dans le poisson au début de la phase d'élimination, et C e est la concentration moyenne dans l'eau durant la phase d'absorption (91). Pour évaluer la justesse de l'ajustement, on peut procéder à une inspection visuelle des pentes k 1 et k 2 par rapport aux données mesurées aux temps de prélèvement portées sur le graphique. S'il apparaît que la méthode séquentielle fournit une mauvaise estimation pour k 1, il convient d'appliquer la méthode simultanée pour calculer k 1 et k 2 (voir la section 6 ci-après). Une nouvelle fois, pour évaluer la justesse de l'ajustement, il est nécessaire de comparer visuellement les pentes obtenues aux données mesurées portées sur le graphique. Si l'ajustement n'est toujours pas satisfaisant, cela peut signifier que la cinétique du premier ordre ne s'applique pas et qu'il convient d'utiliser des modèles plus complexes. 6. METHODE SIMULTANEE DE CALCUL DES CONSTANTES CINETIQUES D'ABSORPTION ET D'ELIMINATION (PERTE) (METHODE D'EXPOSITION EN MILIEU AQUATIQUE UNIQUEMENT) Il est possible d'utiliser des programmes informatiques afin de calculer des valeurs pour k 1 et k 2 d'après un ensemble de valeurs séquentielles de la concentration en fonction du temps et le modèle:
où
Cette approche fournit directement des erreurs types pour les estimations de k 1 et k 2. Si k 1/k 2 est substituée par le FBC (voir équation A5.4) dans les équations A5.25 et A5.26, il est possible d'estimer également l'erreur type et l'intervalle de confiance de 95 % du FBC. Cela est particulièrement utile pour comparer des estimations différentes résultant d'une évolution des données. La variable dépendante (concentration dans le poisson) peut être ajustée avec ou sans ln transformation, et l'incertitude quant au FBC obtenu peut être évaluée. En raison de la forte corrélation entre les deux paramètres k 1 et k 2, s'ils ont été estimés simultanément, on conseille éventuellement de calculer d'abord k 2 à partir des seuls résultats de l'élimination (voir précédemment); dans la plupart des cas, k 2 peut être estimé à partir de la courbe d'élimination avec une précision relativement élevée. Par la suite, k 1 peut être calculée à partir des données d'absorption avec une régression non-linéaire(1). Il est conseillé de transformer les données de la même façon en cas d'ajustement séquentiel. Pour évaluer la justesse de l'ajustement, on peut procéder à une inspection visuelle des pentes obtenues en portant sur un graphique les données mesurées aux temps de prélèvement. S'il apparaît que cette méthode fournit une mauvaise estimation pour k 1, il convient d'appliquer l'autre méthode pour calculer k 1 et k 2. Une nouvelle fois, pour évaluer la justesse de l'ajustement, il faudrait comparer visuellement le modèle ajusté aux données mesurées portées sur le graphique, et les estimations des paramètres pour k 1, k 2 et le FBC obtenu ainsi que leurs erreurs types et/ou les intervalles de confiance doivent être comparés selon différents types d'ajustement. Si l'ajustement n'est toujours pas satisfaisant, cela peut signifier que la cinétique du premier ordre ne s'applique pas et qu'il convient d'utiliser des modèles plus complexes. L'une des complications les plus fréquentes est le développement des poissons durant l'essai. 7. CORRECTION DE L'EFFET DE DILUTION PAR LA CROISSANCE POUR LE FBC CINETIQUE ET LE FBA Cette section décrit une méthode standard pour corriger les données en fonction du développement des poissons en cours d'essai (autrement dit en fonction de l'effet de dilution par la croissance) qui est uniquement valide quand la cinétique du premier ordre s'applique. Quand il apparaît que la cinétique du premier ordre ne s'applique pas, il est recommandé de demander conseil à un biostatisticien pour bien corriger les données de l'effet de dilution par la croissance; on peut aussi appliquer la méthode fondée sur la masse décrite ci-après. Dans certains cas, cette méthode de correction de l'effet de dilution par la croissance manque de précision ou ne fonctionne pas (par exemple, pour des substances s'éliminant très lentement testées chez des poissons à croissance rapide, la constante cinétique d'élimination corrigée de la croissance, k 2g, peut être très faible, aussi toute erreur commise dans les deux constantes cinétiques servant à la calculer est-elle cruciale, et dans certains cas la k g estimée peut être supérieure à k 2). Il est alors possible d'utiliser une autre méthode (approche massique), qui évite d'apporter toute correction et fonctionne aussi en l'absence de cinétique du premier ordre. Cette méthode est présentée à la fin de cette section. Méthode de correction de la croissance par soustraction de la constante cinétique de croissance Selon la méthode standard, les poids et les longueurs individuels sont convertis en logarithmes naturels, et ln(poids) ou ln(1/poids) sont tracés sur deux graphiques distincts, pour le groupe témoin et le groupe d'essai, en fonction du temps (jour). On procède de même avec les données obtenues séparément pour les phases d'absorption et d'élimination. En général, pour corriger de l'effet de dilution par la croissance, il est plus approprié d'utiliser les poids de l'ensemble de l'étude pour obtenir la constante cinétique de croissance (kg), mais des écarts notables sur un plan statistique entre les constantes cinétiques de croissance obtenues pour la phase d'absorption et la phase d'élimination peuvent indiquer qu'il convient d'employer la constante cinétique de la phase d'élimination. Les taux de croissance globaux observés lors des études avec exposition en milieu aquatique pour les groupes témoin et d'essai peuvent servir à contrôler les effets associés à tout traitement. Une corrélation des moindres carrés linéaires est calculée pour ln(poids des poissons) en fonction du temps (jour) (pour ln(1/poids) en fonction du temps) pour chaque groupe (groupes d'essai et témoin, données individuelles, moyennes non journalières) pour l'ensemble de l'étude, les phases d'absorption et d'élimination en appliquant les méthodes statistiques standard. Les écarts des pentes des droites sont calculés et utilisés pour évaluer l'importance statistique (p = 0,05) de la différence entre les pentes (constantes cinétiques de croissance) à partir du test t (ou d'une ANOVA si plus d'une concentration est testée). On préfère en général utiliser les données sur le poids pour les corrections de la croissance. Les longueurs, traitées de la même façon, peuvent servir à comparer les effets du traitement sur les groupes témoin et d'essai. Si on ne constate aucune différence statistiquement notable lors de l'analyse des poids mesurés, on peut regrouper les données des groupes témoin et d'essai et calculer une constante cinétique de croissance du poisson globale pour l'étude (kg) à savoir la pente globale de la corrélation linéaire. Si on observe des écarts statistiquement notables, on consignera séparément les constantes cinétiques de croissance pour chaque groupe de poissons, et/ou chaque phase de l'essai. La constante cinétique pour chaque groupe traité est alors utilisée pour les corrections de l'effet de dilution par la croissance pour ce groupe. Si on constate des différences statistiques entre les constantes cinétiques d'absorption et d'élimination, il convient d'utiliser les constantes cinétiques tirées de la phase d'élimination. La constante cinétique de croissance calculée (kg exprimée en fonction de jour– 1) peut être soustraite de la constante cinétique d'élimination globale (k 2) pour donner la constante cinétique d'élimination, k 2g.
On divise la constante cinétique d'absorption par la constante cinétique d'élimination corrigée de la croissance pour obtenir le FBC cinétique corrigé de l'effet de dilution par la croissance, représenté par FBCKg (ou FBAKg).
La constante cinétique de croissance obtenue pour un essai avec exposition par voie alimentaire est utilisée dans l'équation A7.5 pour calculer le FBAkg corrigé de la croissance (voir appendice 7). Méthode de correction de la croissance fondée sur la masse Il est possible d'utiliser une autre méthode que celle par soustraction de la constante cinétique de croissance, décrite précédemment, pour contourner la nécessité d'une correction de la dilution par la croissance. Le principe consiste à utiliser les données relatives à l'élimination en fonction de la masse par poisson entier et non en fonction de la concentration. Convertir les concentrations observées dans les tissus lors de la phase d'élimination (masse de la substance d'essai/unité de masse du poisson) en masse de substance d'essai/poisson: mettre en parallèle, sous forme de tableau, les concentrations et les poids de chaque poisson (par exemple en utilisant un tableur informatique) et multiplier chaque concentration par le poids total du poisson pour que cette mesure donne un ensemble de masse de la substance d'essai/poisson pour tous les prélèvements de la phase d'élimination. Tracer le logarithme naturel obtenu avec les données de la masse de la substance chimique en fonction du temps (phase d'élimination) comme on le ferait normalement. Pour la méthode d'exposition en milieu aquatique, calculer la constante cinétique d'absorption comme d'habitude (voir sections 4 et 6; noter que la valeur k 2«normale» est utilisée dans les équations d'ajustement de la courbe pour k 1) et déduire la constante cinétique d'élimination des données ci-dessus. La valeur obtenue pour la constante cinétique d'élimination étant indépendante de la croissance, puisqu'elle découle d'une base massique par poisson entier, il convient de la représenter par k 2g et non par k 2. 8. NORMALISATION DES LIPIDES A 5 % DE LA TENEUR EN LIPIDES (METHODE D'EXPOSITION EN MILIEU AQUATIQUE UNIQUEMENT) Les résultats du FBC (cinétique et à l'état stationnaire) des essais par exposition en milieu aquatique sont aussi consignés en fonction d'une teneur en lipides par défaut de 5 % du poids frais des poissons, sauf si l'on peut prouver que la substance d'essai ne s'accumule pas prioritairement dans les lipides (par exemple certaines substances perfluorées peuvent se lier aux protéines). Il convient de convertir les concentrations dans les poissons, ou le FBC, en une teneur en lipides de 5 % par rapport au poids frais. Si les mêmes poissons ont été utilisés pour mesurer les concentrations de la substance et les teneurs en lipides à tous les temps d'échantillonnage, il est nécessaire de corriger les concentrations mesurées individuellement en fonction de la teneur en lipides des poissons.
Où
Si une analyse lipidique n'a pas été menée sur tous les poissons prélevés, on utilisera une valeur lipidique moyenne pour normaliser le FBC. S'agissant du FBC à l'état stationnaire, il convient d'utiliser la valeur moyenne enregistrée à la fin de la phase d'absorption dans le groupe testé. Quant à la normalisation du FBC cinétique il est parfois justifié d'appliquer une méthode différente, par exemple si la teneur en lipides a sensiblement changé pendant la phase d'absorption ou d'élimination. Cependant, il est conseillé de prévoir une ration alimentaire qui réduise au maximum tout changement spectaculaire de la teneur en lipides.
Où
BIBLIOGRAPHIE
Appendice 6 ÉQUATIONS RELATIVES À L'ESSAI PAR EXPOSITION EN MILIEU AQUATIQUE: CONCEPT D'ESSAI RÉDUIT Les raisons de cette approche tiennent du fait que le facteur de bioconcentration dans un essai complet peut être déterminé comme un facteur de bioconcentration à l'état stationnaire (FBCES) en calculant le rapport de la concentration de la substance d'essai dans les tissus du poisson à la concentration de la substance d'essai dans l'eau, ou en calculant le facteur de bioconcentration cinétique (FBCk), à savoir le rapport de la constante cinétique d'absorption k 1 à la constante cinétique d'élimination k 2. Le FBCk est valide même si une concentration à l'état stationnaire d'une substance chimique n'est pas atteinte durant l'absorption, à condition qu'absorption et élimination soient régies pour l'essentiel par des processus cinétiques de premier ordre. Si on mesure la concentration de la substance chimique dans les tissus (C p1) à la fin de l'exposition (t1) et qu'on mesure cette concentration dans les tissus (C p2) à nouveau après un certain temps (t2), il est possible d'estimer la constante cinétique d'élimination (k 2) avec l'équation A5.22 de l'appendice 5. La constante cinétique d'absorption, k 1, peut ensuite être déterminée de manière algébrique avec l'équation A5.23 de l'appendice 5 (où C p est égale à C p1 et t est égal à t1) (1). Le facteur de bioconcentration cinétique pour le concept d'essai réduit (désigné par FBCkm pour le distinguer des facteurs de bioconcentration cinétiques déterminés avec d'autres méthodes) correspond ainsi à:
Il convient de corriger les concentrations ou les résultats de l'effet de dilution par la croissance et de les normaliser par rapport à des poissons présentant une teneur en lipides de 5 % (voir appendice 5). Le FBCES minimisé correspond au FBC calculé à la fin de la phase d'absorption, en supposant que l'état stationnaire a été atteint. On ne peut que le supposer, puisque le nombre de temps d'échantillonnage ne suffit pas à le prouver.
Où C p-minES = Concentration dans le poisson à un état supposé stationnaire à la fin de l'absorption (mg kg– 1 du poids frais). C e-minES = Concentration dans l'eau à un état supposé stationnaire à la fin de l'absorption (mg l– 1). BIBLIOGRAPHIE
Appendice 7 ÉQUATIONS RELATIVES À L'ESSAI AVEC EXPOSITION PAR VOIE ALIMENTAIRE
1. EXEMPLE DE COMPOSITION D'UNE NOURRITURE POUR POISSON DU COMMERCE ADAPTEE
2. EXEMPLES DE TECHNIQUES D'ENRICHISSEMENT DE L'ALIMENTATION Généralités La nourriture témoin est préparée exactement de la même façon que la nourriture enrichie, mais sans ajout de la substance d'essai. Pour connaître les concentrations dans les aliments traités, il convient d'extraire trois échantillons de la ration alimentaire selon une méthode d'extraction adaptée, puis de mesurer la radioactivité ou la concentration de la substance d'essai dans ces échantillons. La possibilité d'isoler la substance d'essai à analyser (>85 %) ainsi que la faible variation entre les échantillons (trois concentrations de la substance mesurées sur des échantillons prélevés au début de l'essai ne varient pas de plus de ± 15 % de la moyenne) sont démontrées. Au cours de l'essai avec exposition par voie alimentaire, il convient de collecter trois échantillons de nourriture au jour 0 et à la fin de la phase d'absorption pour déterminer la concentration de la substance d'essai dans les aliments Préparation de la nourriture pour poisson avec une substance d'essai liquide (pure) On fixe une concentration d'essai nominale cible dans la nourriture traitée, par exemple 500 μg de substance d'essai/g de nourriture. La quantité appropriée (en fonction de la masse molaire ou radioactivité spécifique) de la substance d'essai pure est ajoutée à une masse connue de nourriture pour poisson dans un bocal en verre ou un ballon évaporateur rotatif. Il convient que la masse de nourriture suffise pour toute la durée de la phase d'absorption (prendre en compte la nécessité d'augmenter chaque ration en raison du développement des poissons). La nourriture pour poisson et la substance d'essai sont mélangées lentement pendant la nuit (par exemple au moyen d'un mixeur Roto-Rack ou, en cas d'utilisation d'un ballon évaporateur rotatif, par rotation). La nourriture enrichie est stockée dans des conditions qui maintiennent la stabilité de la substance d'essai au sein du mélange (par réfrigération par exemple) jusqu'à son utilisation. Préparation de la nourriture pour poisson avec un véhicule (huile de poisson/germes de maïs Les substances d'essai solides sont pilées dans un mortier en une fine poudre. Les substances d'essai liquides peuvent être ajoutées directement à l'huile de poisson ou de germes de maïs. La substance d'essai est dissoute dans une quantité connue d'huile de poisson ou de germes de maïs (par exemple entre 5 à 15 ml). L'huile dosée est transférée dans un ballon d'évaporation rotatif de taille appropriée. La fiole utilisée pour préparer l'huile dosée est nettoyée avec deux petites aliquotes d'huile qu'on ajoute ensuite dans le ballon pour veiller à ce que toute la substance d'essai dissoute soit bien transférée. Afin de garantir la dissolution/dispersion complète dans l'huile (ou si plus d'une substance d'essai est utilisée durant l'essai), on ajoute un micro-agitateur et on bouche la fiole pour que le mélange puisse être agité rapidement pendant la nuit. On ajoute une quantité appropriée de nourriture pour poisson (habituellement sous forme de granulés) dans le ballon, et le contenu du ballon de verre est mélangé de façon homogène par rotation continue du ballon pendant au moins 30 minutes, mais de préférence pendant toute la nuit. La nourriture enrichie est ensuite stockée de manière adéquate (réfrigérée par exemple) pour assurer la stabilité de la substance d'essai dans la nourriture jusqu'à son utilisation. Préparation de la nourriture pour poisson avec un solvant organique Une quantité appropriée de la substance d'essai (en fonction de la masse molaire ou radioactivité spécifique) suffisante pour atteindre la dose cible est dissoute dans un solvant organique adéquat (par exemple cyclohexane ou acétone; de 10 à 40 ml, ou un volume plus important si nécessaire, selon la quantité de nourriture à enrichir). Une aliquote, voire la totalité (ajoutée en plusieurs fois), de cette solution est mélangée à la masse appropriée de nourriture pour poisson, de façon à atteindre la dose nominale requise pour l'essai. On peut mélanger la nourriture pour poisson et la substance d'essai dans un récipient en acier inoxydable et laisser la nourriture fraîchement dosée dans ce récipient dans une hotte de laboratoire pendant deux jours (en agitant de temps en temps) pour permettre à l'excès de solvant de s'évaporer; on peut aussi effectuer ce mélange dans un ballon évaporateur à rotation continue. Si besoin, l'excès de solvant peut être retiré au moyen d'un courant d'air ou d'azote. Il convient de veiller à ce que la substance d'essai ne cristallise pas lors du retrait du solvant. Il convient que la nourriture enrichie soit stockée dans des conditions (par réfrigération par exemple) qui maintiennent la stabilité de la substance d'essai au sein du mélange jusqu'à son utilisation. 3. CALCUL DU RENDEMENT D'ASSIMILATION ET DU FACTEUR DE BIOAMPLIFICATION Pour calculer le rendement d'assimilation, il convient tout d'abord d'estimer la constante cinétique d'élimination globale comme indiqué à la section 4 de l'appendice 5 (avec la méthode séquentielle, c'est-à-dire avec une régression linéaire standard) à partir des concentrations moyennes mesurées dans les échantillons prélevés lors de la phase d'élimination. La constante de la ration alimentaire, I, et la durée d'absorption, t, sont des paramètres connus de l'étude. C alim, la concentration mesurée moyenne de la substance d'essai dans la nourriture, est une variable mesurée lors de l'étude. C 0,d, la concentration de la substance d'essai dans le poisson à la fin de la phase d'absorption, est habituellement donnée par l'ordonnée à l'origine sur le tracé de ln(concentration) en fonction du jour d'élimination. Le rendement d'assimilation de la substance chimique (a, absorption de la substance d'essai par voie intestinale) est calculé comme suit:
Où: C 0,d = concentration dans le poisson obtenue au temps 0 de la phase d'élimination (mg kg– 1); k 2 = constante cinétique d'élimination (non corrigée de la croissance) globale (jour– 1), calculée avec les équations présentées à l'appendice 5, section 3; I = constante cinétique d'ingestion (g de nourriture g– 1 de poisson jour– 1); C alim = concentration dans la nourriture (mg kg-1 de nourriture); t= durée de la période d'alimentation (jour) Néanmoins, il peut s'avérer nécessaire d'ajuster la ration alimentaire, I, utilisée dans les calculs en fonction du développement des poissons pour donner un rendement d'assimilation, a, précis. Dans un essai où les poissons se développent de façon notable pendant la phase d'absorption (durant laquelle on ne corrige jamais la quantité de nourriture afin de maintenir la ration alimentaire fixée), la ration alimentaire réelle à mesure que la phase d'absorption progresse sera inférieure à celle fixée, donnant lieu à un rendement d'assimilation «réel» supérieur. (Cet aspect n'est pas important pour les calculs généraux du FBA, puisque les termes I s'annulent entre les équations A7.1 et A7.4). La ration alimentaire moyenne corrigée de l'effet de dilution par la croissance, I g, peut être déduite de plusieurs façons, mais une méthode simple et rigoureuse consiste à utiliser la constante cinétique de croissance (k g) connue pour estimer les poids des poissons testés à certains temps de la phase d'absorption, soit:
où P p(t)= poids moyen des poissons au jour t de la phase d'absorption P p,0 = poids moyen des poissons au début de l'expérience De cette manière (au moins), on peut estimer le poids moyen des poissons au dernier jour d'exposition (P p,fin-absorption). La ration alimentaire ayant été fixée par rapport à P p,0, la ration alimentaire réelle pour chaque jour d'absorption peut être calculée en utilisant ces deux valeurs relatives au poids. La ration alimentaire corrigée de la croissance, I g (g de nourriture g– 1 de poisson jour– 1), qu'il convient d'utiliser à la place de I en cas de développement rapide durant la phase d'absorption, peut ensuite être calculée avec:
Une fois le rendement d'assimilation obtenu, on peut calculer le FBA en le multipliant par la constante de la ration alimentaire I (ou I g, si cette dernière a servi à calculer α) et en divisant le produit par la constante cinétique d'élimination globale k 2:
Le facteur de bioamplification corrigé de la croissance est calculé de la même façon, en utilisant la constante cinétique d'élimination corrigée de la croissance (obtenue comme indiquée à la section 7 de l'appendice 5). Une nouvelle fois, si I g a servi à calculer α, il convient de l'utiliser à la place de I:
Où: α = rendement d'assimilation (absorption de la substance d'essai par voie intestinale); k 2 = constante cinétique d'élimination (non corrigée de la croissance) globale (jour– 1), calculée avec les équations présentées à l'appendice 5, section 3; k 2g = constante cinétique d'élimination corrigée de la croissance (jour– 1); I = constante cinétique d'ingestion (g de nourriture g– 1 de poisson jour– 1); La demi-vie corrigée de la croissance (t 1/2) est calculée comme suit.
Le rendement d'assimilation de la substance chimique comprise dans la nourriture peut aussi être estimé si les résidus présents dans les tissus sont déterminés durant la phase linéaire de la phase d'absorption (entre les jours 1 et 3). On peut alors déterminer le rendement d'assimilation de la substance (α) comme suit:
Où C p (t) = la concentration de la substance d'essai dans le poisson au temps t (mg kg– 1 de poids frais). 4. CORRECTION EN FONCTION DE LA TENEUR EN LIPIDES Si la teneur en lipides a aussi été mesurée dans les poissons soumis à l'analyse chimique à tous les temps de prélèvement, alors il convient de corriger les concentrations individuelles en fonction des lipides et de tracer ln(concentration, corrigé de la teneur en lipides) par rapport à l'élimination (jour) pour obtenir C 0,d et k 2. Le rendement d'assimilation (équation A7.1) peut ensuite être calculé en fonction des lipides en utilisant Calim (Calim est multipliée par la fraction moyenne de lipides dans la nourriture). Des calculs complémentaires avec les équations A7.4 et A7.5 donneront le FBA corrigé de la teneur en lipides (et de l'effet de dilution par la croissance) directement. Dans le cas contraire, les fractions lipidiques moyennes (poids frais) dans le poisson et dans les aliments sont déduites pour les deux groupes, témoin et traité (s'agissant de la nourriture et des poissons du groupe témoin, cette fraction est déduite à partir des mesures relevées au début et à la fin de l'exposition; pour le groupe traité, elle découle habituellement des mesures relevées uniquement à la fin de l'exposition). Dans certaines études, la teneur en lipides du poisson peut augmenter sensiblement; il est alors préférable d'appliquer une concentration moyenne des lipides dans le poisson testé calculée à l'aide des valeurs mesurées à la fin de l'exposition et à la fin de l'élimination. En général, les données du groupe d'essai servent uniquement à obtenir les deux fractions lipidiques. Le facteur de correction de la teneur en lipides (Lc ) est calculé ainsi:
Où L p et L alim sont les fractions lipidiques moyennes respectivement dans le poisson et dans la nourriture. Le facteur de correction de la teneur en lipides sert à calculer le facteur de bioamplification corrigé par rapport aux lipides (FBAL):
5. ÉVALUATION DES DIFFERENCES ENTRE LA CONCENTRATION MESUREE AU TEMPS 0 (C 0,m) ET LA CONCENTRATION DEDUITE AU TEMPS 0 (C 0,d) Il convient de comparer la concentration mesurée au temps 0 (C 0,m) et la concentration déduite au temps 0 (C 0,d). Si elles sont très similaires, le modèle du premier ordre utilisé pour obtenir les paramètres d'élimination semble approprié. Dans certaines études, on observera une différence notable entre la valeur déduite au temps 0, C 0,d, et la concentration moyenne mesurée au temps 0, C 0,m (voir le dernier point du paragraphe 159 de la présente méthode d'essai). Si C 0,d est nettement inférieur à C 0,m (C 0,d << C 0,m), cet écart peut indiquer la présence, dans les intestins, d'aliments enrichis non digérés. Pour le savoir, on peut analyser séparément des intestins excisés si des poissons (entiers) supplémentaires ont été prélevés et stockés à la fin de la phase d'absorption. Dans le cas contraire, s'il apparaît à l'issue d'un test du point aberrant statistiquement valide appliqué à la régression linéaire de la phase d'élimination que le premier temps d'échantillonnage de l'élimination est erronément élevé, il peut être pertinent de poursuivre la régression linéaire pour obtenir k 2 mais en omettant la concentration au premier temps de l'élimination. Si la régression linéaire semble nettement moins incertaine et que le processus d'élimination semble manifestement régi par une cinétique du premier ordre, il peut être approprié d'utiliser les valeurs C 0,d et k 2 obtenues avec le calcul du rendement d'assimilation. Il faudra alors le justifier dûment dans le rapport d'essai. Il est également possible que la phase d'élimination ne soit pas régie par une cinétique du premier ordre. Si cette hypothèse est probable (le logarithme naturel formé à partir des données obtenues semble suivre une courbe par rapport à la droite de la régression linéaire), il est peu probable que les calculs de k 2 et C 0,d soient valides, et il est recommandé de demander conseil à un biostatisticien. Si C 0,d est nettement supérieure à la valeur mesurée (C 0,d >> C 0,m) cela peut signifier: que la substance a été éliminée très rapidement (le temps d'échantillonnage se rapproche très tôt de la limite de quantification de la méthode analytique lors de la phase d'élimination, voir la section 6 ci-après); que le processus d'élimination n'est pas régi par une cinétique du premier ordre; que la régression linéaire pour obtenir k 2 et C 0,d est erronée; ou qu'un problème concernant les concentrations mesurées lors de l'étude est survenu à certains temps de prélèvement. Il est alors nécessaire d'examiner le tracé de la régression linéaire pour identifier les prélèvements sur ou près de la limite de quantification, les points aberrants et toute courbure manifeste (suggérant une élimination non régie par une cinétique du premier ordre), et de les indiquer clairement dans le rapport d'essai. Toute réévaluation ultérieure de la régression linéaire visant à améliorer les valeurs estimées devra être décrite et justifiée. Si on observe une déviation notable de la cinétique du premier ordre, il est peu probable que les calculs de k 2 et C 0,d soient valides, et il est recommandé de demander conseil à un biostatisticien. 6. ORIENTATIONS POUR LES SUBSTANCES D'ESSAI S'ELIMINANT TRES RAPIDEMENT Comme mentionné au paragraphe 129 de la méthode d'essai, certaines substances s'éliminent si vite qu'il n'est possible de déduire ni une concentration fiable au temps 0, C 0,d, ni k 2 parce que très tôt durant la phase d'élimination (dès le deuxième prélèvement de l'élimination) la substance n'est plus réellement mesurée (concentrations à la limite de quantification). Cette situation, observée lors de l'essai comparatif interlaboratoires avec du benzo[a]pyrène, a été consignée dans le rapport de validation de cette méthode d'essai. Dans ce cas de figure, il n'est pas possible de poursuivre la régression linéaire de manière fiable, car elle donnerait probablement une estimation exagérément élevée de C 0,d, d'où un rendement d'assimilation en apparence nettement supérieur à 1. On peut alors procéder à une estimation basse de k 2 et borner le FBA supérieurement. En utilisant ces points de la phase d'élimination où une concentration a été mesurée, première concentration non détectée incluse (concentration fixée à la limite de quantification), une régression linéaire (fondée sur les concentrations transformées en logarithme naturel par rapport au temps) donnera une estimation de k 2. Cela impliquera souvent uniquement deux points (par exemple les jours de prélèvement 1 et 2 de l'élimination) et k 2 pourra ensuite être estimée avec l'équation A5.22 présentée à l'appendice 5. Cette estimation de k 2 peut servir à estimer un rendement d'assimilation avec l'équation A7.1, en remplaçant dans cette équation la valeur C0,d par la concentration mesurée au temps 0 (C0,m) quand d'après les estimations C0,d est nettement supérieure à ce que cet essai aurait permis d'atteindre. Si C0,m n'était pas mesurable, il convient d'utiliser le seuil de détection dans les tissus du poisson. Si, dans certains cas, cela donne une valeur α > 1, alors un rendement d'assimilation de 1 est supposé être le «pire cas de figure». Le FBAk maximum peut alors être estimé avec l'équation A7.4 et devra être indiqué comme une valeur «nettement inférieure à» (<<). Par exemple, pour une étude menée avec une ration alimentaire de 3 % et une demi-vie d'élimination inférieure à 3 jours, et un «pire cas de figure» α de 1, le FBAk risque d'être inférieur à environ 0,13. Étant donné l'objet de cette estimation et le fait que les valeurs seront basses par nature, il n'est pas nécessaire de les corriger de l'effet de dilution par la croissance ou de la teneur en lipides dans le poisson ou la nourriture. Appendice 8 MÉTHODES POUR ESTIMER DES FBC PROVISOIRES À PARTIR DES DONNÉES COLLECTÉES DANS L'ÉTUDE AVEC EXPOSITION PAR VOIE ALIMENTAIRE La méthode d'exposition par voie alimentaire est présentée dans la présente méthode d'essai pour l'essai de bioaccumulation impliquant des substances impossibles à tester avec la méthode d'exposition via le milieu aquatique. La méthode d'exposition via le milieu aquatique donne un facteur de bioconcentration, alors que celle par voie alimentaire fournit directement des informations sur le potentiel de bioamplification de la nourriture. De nombreux plans relatifs à la sécurité des produits chimiques requièrent des informations sur la bioconcentration aquatique (par exemple pour le système d'évaluation des risques ou le Système général harmonisé de classification). Aussi est-il nécessaire d'utiliser les données obtenues avec une étude d'exposition par voie alimentaire pour estimer un facteur de bioconcentration qui soit comparable aux essais menés selon la méthode d'exposition via le milieu aquatique (93). Cette section examine différentes approches en ce sens, tout en reconnaissant les limites inhérentes à ces estimations. L'étude par voie alimentaire mesure l'élimination pour donner une constante cinétique d'élimination, k 2. Si une constante cinétique d'absorption peut être estimée avec les données disponibles quand le poisson a été exposé à la substance d'essai dans l'eau, on pourra estimer un FBC cinétique. L'estimation d'une constante cinétique d'absorption pour l'exposition via le milieu aquatique à une substance d'essai repose sur de nombreuses hypothèses, toutes contribuant à l'incertitude des estimations. De plus, cette méthode d'estimation d'un FBC suppose que la cinétique d'élimination globale (y compris les facteurs contributifs tels que la répartition dans le corps et les processus d'élimination individuels) est indépendante de la technique d'exposition utilisée pour produire une charge corporelle de la substance d'essai. On peut résumer les principales hypothèses inhérentes à cette méthode d'estimation comme suit: L'élimination suivant l'absorption par voie alimentaire procède de même que suivant l'exposition via le milieu aquatique pour une substance donnée L'absorption par exposition via le milieu aquatique est régie par une cinétique du premier ordre Selon la méthode utilisée pour estimer l'absorption:
La base de données utilisée pour développer la méthode d'estimation de l'absorption est représentative de la substance considérée Plusieurs publications dans la littérature ont déduit des équations faisant le rapprochement entre l'absorption, depuis le milieu aquatique, de la substance par les branchies et un coefficient de partage octanol-eau de la substance, le poids du poisson (1) (2) (3) (4), le volume et/ou la teneur en lipides, l'imprégnation/diffusion par les membranes (5) (6), le volume de ventilation du poisson (7) et par une approche fugacité/bilan massique (8) (9) (10). Ces méthodes, appliquées dans ce contexte, sont évaluées en détail par Crookes et Brooke (11). Une publication de Barber (12), qui s'est attaché à modéliser la bioaccumulation associée à l'absorption par voie alimentaire, s'avère aussi utile dans ce contexte, puisqu'elle inclut les contributions de modèles de cinétique d'absorption par les branchies. Une section dans le document de référence du protocole de 2004 (13) est aussi consacrée à cet aspect. Pour la plupart, ces modèles semblent dériver de bases de données limitées. S'agissant des modèles dont les bases de données sont présentées en détail, il semble que les types de substances utilisés présentent souvent une structure similaire ou relèvent de la même classe (en termes de fonctionnalité, par exemple les composés organochlorés). Cela ajoute à l'incertitude qu'il y a à utiliser un modèle dans le but de prédire une constante cinétique d'absorption pour un type de substance différent, sans parler des considérations spécifiques à l'essai comme l'espèce testée, les températures en présence, etc. Une synthèse des techniques disponibles (11) montre qu'aucune méthode n'est «plus juste» que les autres. Aussi convient-il de justifier clairement le choix du modèle utilisé. Quand plusieurs méthodes sont justifiées, il est prudent de présenter plusieurs estimations de k 1 (et donc du FBC) ou une fourchette de valeurs pour k 1 (et pour le FBC) selon les différentes méthodes d'estimation d'absorption possibles. Néanmoins, étant donné les différences entre les types de modèle et les bases de données utilisées pour les développer, il ne serait pas approprié de prendre une moyenne des estimations obtenues avec ces différentes méthodes. Certains chercheurs posent comme postulat que ces estimations du FBC exigent une correction en fonction de la biodisponibilité pour tenir compte de l'adsorption d'une substance chimique par rapport au carbone organique dissous (COD) dans un milieu aquatique, afin que l'estimation corresponde aux résultats des études d'exposition en milieu aquatique [par exemple (13) (14)]. Cette correction n'est toutefois pas forcément appropriée en raison des faibles niveaux de COD requis dans une étude avec exposition en milieu aquatique pour une estimation dans le «pire cas de figure» (à savoir le ratio entre la substance biodisponible et la substance mesurée dans la solution). Avec les substances très hydrophobes, l'absorption par les branchies peut être limitée par le taux de diffusion passive près des ouïes; dans ce cas, il est possible que la correction tienne plus compte de cet effet que de son objet initial. Il est conseillé de s'intéresser aux méthodes qui nécessitent des données facilement disponibles sur les substances testées dans l'étude avec exposition par voie alimentaire décrite ici (à savoir le log Koe , et le poids des poissons). D'autres méthodes nécessitant des informations plus complexes peuvent s'appliquer, mais impliqueront de procéder à des mesures supplémentaires durant l'essai ou de disposer d'informations détaillées sur la substance d'essai ou sur l'espèce testée difficiles à obtenir. En outre, le choix du modèle peut être influencé par le niveau de validation et le domaine d'applicabilité [voir (11) pour une synthèse et une comparaison des différentes méthodes]. Il convient de garder à l'esprit que l'estimation de k 1 obtenue, et le FBC estimé, n'est pas sûre et peut nécessiter d'appliquer le poids de la preuve au FBA obtenu et aux paramètres relatifs à la substance (taille moléculaire par exemple) pour avoir une vue d'ensemble du potentiel de bioaccumulation d'une substance. L'interprétation et l'utilisation de ces paramètres peuvent varier en fonction du cadre réglementaire. BIBLIOGRAPHIE
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


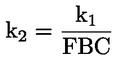



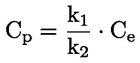

















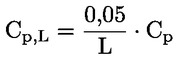

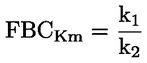


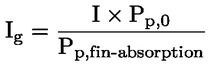
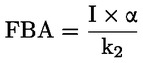
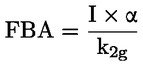




|
(17) |
Dans la partie C, le chapitre C.20 est remplacé par le texte suivant: «C.20 Daphnia magna, essai de reproduction INTRODUCTION La présente méthode d'essai est équivalente à la ligne directrice 211(2012) de l'OCDE pour les essais de produits chimiques. Les lignes directrices de l'OCDE pour les essais de produits chimiques sont régulièrement mises à jour pour intégrer les progrès de la science. La ligne directrice 211 a pour origine la ligne directrice 202 — Partie II: «Daphnia sp., essai de reproduction» (1984). Il était généralement admis que les résultats obtenus au cours d'essais pratiqués selon la ligne directrice 202 pouvaient être variables. De ce fait, des efforts considérables ont été déployés pour découvrir les raisons de cette variabilité, en vue de mettre au point une meilleure méthode d'essai. La ligne directrice 211 est le fruit des activités de recherche, des essais interlaboratoires et des études de validation réalisés en 1992 (1), 1994 (2) et 2008 (3). Les principales différences entre la version initiale (ligne directrice 202, 1984) et la deuxième version (ligne directrice 211, 1998) de la ligne directrice sont les suivantes:
Les définitions utilisées sont données à l'appendice 1. PRINCIPE DE L'ESSAI Le principal objectif de l'essai consiste à évaluer l'effet de produits chimiques sur le taux de reproduction de Daphnia magna. Pour ce faire, de jeunes femelles de Daphnia (les animaux parents), âgées de moins de 24 heures au début de l'essai, sont exposées au produit chimique d'essai ajouté à l'eau à différentes concentrations. L'essai dure 21 jours. À la fin de l'essai, le nombre total de descendants vivants produits est évalué. Le taux de reproduction des animaux parents peut s'exprimer autrement (p. ex. par le nombre de descendants vivants engendrés par animal et par jour, à partir du premier jour où des descendants ont été observés), mais ces résultats sont fournis en plus du nombre total de descendants vivants produits à la fin de l'essai. Du fait du mode opératoire particulier des essais semi-statiques par rapport aux autres méthodes d'essai sur la reproduction des invertébrés, il est également possible de compter le nombre de descendants vivants produits individuellement par chaque animal parent. Ainsi, contrairement aux autres méthodes d'essais sur la reproduction des invertébrés, on peut exclure de l'analyse les données correspondant aux descendants d'un animal parent décédé de manière accidentelle et/ou fortuite pendant l'exposition. En l'occurrence, si une mortalité parentale se produit dans les réplicats exposés à la substance d'essai, il convient d'analyser si cette mortalité suit une courbe concentration-response, par exemple s'il existe une régression significative de la réponse par rapport à la concentration testée du produit chimique d'essai, avec une pente positive (un test statistique tel que le test de tendance de Cochran-Armitage peut être utilisé à cet effet). Si la mortalité parentale ne suit pas de courbe concentration-réponse, alors les réplicats montrant une mortalité parentale sont exclus de l'analyse des résultats. Si la mortalité parentale suit une courbe de concentration-réponse, cette mortalité est assimilée à un effet du produit chimique testé et les réplicats ne sont pas exclus de l'analyse des résultats. Si le parent meurt au cours de l'essai de manière accidentelle ou fortuite non liée au produit chimique testé, ou se révèle être un parent de sexe masculin, alors le réplicat est exclu de l'analyse des résultats (voir paragraphe 51). L'effet toxique du produit chimique d'essai sur le taux de reproduction est mesuré par la CEx, les données étant ajustées à un modèle approprié au moyen d'une régression non linéaire en vue d'estimer la concentration qui entraînerait x % de réduction du taux de reproduction, respectivement, ou encore par celle de la CSEO/CMEO (4). Il est préférable que les concentrations d'essai encadrent la concentration efficace minimale (p. ex. CE10), de sorte que cette valeur soit déterminée par interpolation et non par extrapolation. Il convient aussi d'indiquer le taux de survie des animaux parents et le moment de la première portée. D'autres effets liés au produit chimique sur des paramètres tels que la croissance (p. ex. la longueur) et éventuellement le taux intrinsèque d'accroissement de la population peuvent aussi être examinés (voir paragraphe 44). INFORMATIONS SUR LE PRODUIT CHIMIQUE D'ESSAI Les résultats d'un essai de toxicité aiguë (voir chapitre C.2 de la présente annexe Daphnia sp., essai d'immobilisation immédiate) réalisé sur Daphnia magna peuvent être utiles pour sélectionner une gamme de concentrations d'essai adaptée aux essais de reproduction. Il convient que la solubilité dans l'eau et la pression de vapeur du produit chimique d'essai soient connues. De même, il convient de disposer d'une méthode d'analyse fiable pour doser le produit chimique dans les solutions d'essai dont le rendement de récupération et la limite de détermination sont connus. Des informations sur le produit chimique d'essai qui peuvent être utiles pour établir les conditions de l'essai comprennent la formule structurale, la pureté du produit chimique, la stabilité à la lumière, la stabilité dans les conditions de l'essai, le pKa, le coefficient de partage Poe et les résultats d'un essai de biodégradabilité immédiate [voir les chapitres C.4 (détermination de la biodégradabilité facile) et C.29 (Biodégradabilité facile — dégagement de CO2 dans des flacons hermétiquement clos) de la présente annexe. VALIDITÉ DE L'ESSAI Pour que l'essai soit valide, il convient que les témoins remplissent les critères de performance suivants:
Note: Le même critère de validité (20 %) peut s'appliquer pour la mortalité parentale aléatoire ou accidentelle chez les témoins ainsi que pour chacune des concentrations d'essai. DESCRIPTION DE LA MÉTHODE Appareillage Les récipients et autres équipements qui sont amenés à entrer en contact avec les solutions d'essai sont intégralement en verre, ou en un autre matériau chimiquement inerte. On utilisera en principe des béchers en verre. En outre, il sera nécessaire d'employer une partie ou la totalité du matériel suivant:
Organisme d'essai L'espèce à utiliser dans cet essai est Daphnia magna Straus (94). Le clone devrait de préférence avoir été identifié d'après son génotype. La recherche (1) a montré que la capacité reproductrice du clone A (originaire de l'IRCHA, en France) (5) répond de façon stable au critère de validité qui stipule une moyenne ≥ 60 descendants vivants par animal parent survivant lorsqu'il est élevé dans les conditions que décrit la présente méthode d'essai. D'autres clones sont toutefois acceptables à condition de prouver que la culture de Daphnia remplit les critères de validité pour l'essai. Au début de l'essai, les animaux sont âgés de moins de 24 heures et ne peuvent pas provenir d'une première génération de descendants. Ils sont issus d'un lot sain (c'est-à-dire qui ne présente pas de signes de stress, tels qu'une mortalité élevée, la présence de mâles et d'éphippies, un retard dans la production des premiers descendants, des animaux décolorés, etc.). Le lot d'animaux est maintenu dans des conditions de culture (lumière, température, milieu, alimentation et nombre d'animaux par unité de volume) semblables à celles qui seront appliquées au cours de l'essai. Si le milieu utilisé dans l'essai est différent de celui où sont normalement élevées les Daphnia, il convient de laisser aux Daphnia une période d'acclimatation, avant l'essai, qui dure habituellement trois semaines environ (c'est-à-dire une génération), afin d'éviter de stresser les animaux parents. Milieu d'essai Il est recommandé d'utiliser un milieu entièrement défini dans cet essai. On peut ainsi éviter d'employer des additifs (p. ex. des algues, des extraits de sol), qui sont difficiles à caractériser, et améliorer les possibilités de normalisation entre les laboratoires. Les milieux Elendt M4 (6) et M7 (voir appendice 2) se sont révélés pertinents à cette fin. D'autres milieux sont cependant acceptables [par exemple (7) et (8)], à condition que les Daphnia élevées dans ces milieux satisfassent aux critères de validité établis pour l'essai. Si le milieu utilisé contient des additifs non définis, ceux-ci seront spécifiés clairement et le rapport d'essai devra comporter des informations sur la composition, notamment la teneur en carbone, étant donné qu'elle peut contribuer au régime alimentaire fourni. On préconise de déterminer le carbone organique total (COT) et/ou la demande chimique en oxygène (DCO) de la solution mère de l'additif organique et d'estimer leur incidence sur le COT et la DCO du milieu d'essai. En outre, il est conseillé de maintenir les niveaux de COT dans le milieu (c'est-à-dire avant l'ajout des algues) inférieurs à 2 mg/l (9). Lorsque l'on teste des produits chimiques contenant des métaux, il est important de reconnaître que les propriétés du milieu d'essai (p. ex. la dureté, le pouvoir de chélation) peuvent influencer leur toxicité. C'est pourquoi il est souhaitable d'opérer dans un milieu entièrement défini. Néanmoins, dans l'état actuel des connaissances, les seuls milieux entièrement définis qui conviennent aux cultures à long terme de Daphnia magna sont Elendt M4 et M7. Ces deux milieux contiennent l'agent chélatant EDTA. Des travaux ont montré (2) que la «toxicité apparente» du cadmium est généralement moindre lorsque l'essai de reproduction est effectué dans les milieux M4 et M7, que dans des milieux ne contenant pas d'EDTA. M4 et M7 ne sont donc pas recommandés pour tester des produits chimiques contenant des métaux, de même que d'autres milieux contenant des agents chélatants connus. Il est souhaitable d'utiliser un autre milieu pour les produits chimiques renfermant des métaux, par exemple l'eau douce calcaire reconstituée d'ASTM (9), qui ne contient pas d'EDTA. La combinaison de l'eau douce calcaire reconstituée d'ASTM et de l'extrait d'algues (10) convient également aux cultures à long terme de Daphnia magna (2). La concentration de l'oxygène dissous est supérieure à 3 mg/L au début de l'essai et durant celui-ci. Le pH est compris entre 6 et 9 et normalement ne pas varier de plus de 1.5 unité au cours d'un même essai. Une dureté supérieure à 140 mg/l (en CaCO3) est recommandée. La capacité reproductrice des animaux dans les essais pratiqués à un niveau au moins égal à ce seuil s'est révélée conforme aux critères de validité (11) (12). Solutions d'essai Les solutions d'essai sont généralement amenées à la concentration voulue par dilution d'une solution mère. Idéalement, les solutions mères sont préparées sans solvants ni dispersants, dans la mesure du possible, en mélangeant ou en agitant le produit chimique d'essai dans le milieu d'essai par des moyens mécaniques comme l'agitation ou le traitement aux ultrasons, ou d'autres méthodes appropriées. Il est préférable d'exposer les systèmes d'essai aux concentrations du produit chimique d'essai qui seront employées dans l'étude, aussi longtemps que nécessaire pour démontrer que l'on peut maintenir des concentrations d'exposition stables avant d'introduire les organismes d'essai. Si le produit chimique d'essai est difficile à dissoudre dans l'eau, il convient de suivre les procédures décrites dans le document d'orientation de l'OCDE relatif à l'essai de substances difficiles (13). Le recours à des solvants ou à des dispersants devrait être évité, mais peut se révéler nécessaire dans certains cas pour obtenir une solution mère de concentration appropriée pour l'exposition. Outre les concentrations à tester, l'essai comprendra un témoin de l'eau de dilution et, si cela est impératif, un solvant témoin, avec le nombre de réplicats respectifs adéquat. Seuls les solvants ou les dispersants dont les effets sur la variable de réponse se sont avérés non significatifs ou minimes seront utilisés dans l'essai. Des exemples de solvants (p. ex. acétone, éthanol, méthanol, diméthylformamide et triéthylène glycol) et de dispersants (p. ex. Cremophor RH40, méthylcellulose 0,01 % et HCO-40) adéquats sont fournis dans le document (13). En cas d'utilisation d'un solvant ou d'un dispersant, sa concentration finale ne dépasse pas 0,1 ml/l (13) et il est identique dans tous les récipients d'essai, sauf pour le témoin de l'eau de dilution. Cependant, on mettra tout en œuvre pour limiter la teneur en solvant autant que faire se peut. MODE OPÉRATOIRE Conditions d'exposition Durée La durée de l'essai est de 21 jours. Charge Les animaux parents sont répartis individuellement dans des récipients d'essai contenant en général 50 à 100 ml de milieu chacun (avec Daphnia magna, on peut abaisser ce volume, notamment pour les daphnies de plus petite taille comme Ceriodaphnia dubia), à moins qu'un essai dynamique ne soit requis. Il est parfois nécessaire d'avoir recours à des volumes plus grands, afin de pouvoir appliquer la méthode d'analyse utilisée pour déterminer la concentration du produit chimique d'essai, bien qu'il soit aussi possible de regrouper les réplicats aux fins de l'analyse chimique. Si des volumes supérieurs à 100 ml sont employés, il faudra peut-être augmenter la ration distribuée aux Daphnia, afin que la nourriture disponible soit suffisante et que les critères de validité soient satisfaits. Animaux d'essai Pour les essais semi-statiques, on utilisera au moins 10 animaux répartis individuellement à chaque concentration d'essai et au moins 10 animaux répartis individuellement dans la série des témoins. Pour les essais dynamiques, il s'avère approprié d'utiliser 40 animaux répartis en quatre groupes de 10 à chaque concentration d'essai (1). Un plus petit nombre d'organismes d'essai peut être utilisé, mais on recommande d'employer au moins 20 animaux par concentration, répartis dans au moins deux récipients contenant le même nombre d'animaux (p. ex. quatre réplicats contenant cinq daphnies chacun). On notera qu'en ce qui concerne les essais où les animaux sont maintenus en groupes, il sera impossible d'exclure un descendant de l'analyse statistique en cas de mort fortuite/accidentelle d'un parent à partir du moment où la reproduction a commencé. Le cas échéant, il faudra donc exprimer le taux de reproduction par le nombre total de descendants vivants par parent présent au début de l'essai. Les traitements sont répartis de façon aléatoire entre les récipients d'essai, et toutes les manipulations ultérieures de ceux-ci se font de même. Faute de quoi, les résultats pourraient présenter un biais qui risquerait d'être interprété comme un effet de la concentration. Notamment, si les unités expérimentales sont manipulées par ordre de traitement ou de concentration, certains effets liés au temps, comme la fatigue du manipulateur ou d'autres erreurs, pourraient avoir un impact plus prononcé aux concentrations supérieures. En outre, si les résultats de l'essai sont susceptibles d'être affectés par une condition initiale ou liée à l'environnement, comme la situation dans le laboratoire, il faut envisager d'arrêter l'essai. Alimentation Dans les essais semi-statiques, on préconise d'administrer une ration quotidienne, ou au moins trihebdomadaire (ce qui correspond au renouvellement du milieu). Une dilution éventuelle des concentrations d'exposition due à l'administration de nourriture sera prise en compte et évitée autant que faire se peut, à l'aide de suspensions d'algues de concentration adéquate (voir paragraphe 29). Les écarts à ce régime (p. ex. dans les essais dynamiques) sont signalés. Au cours de l'essai, le régime alimentaire des animaux parents sera de préférence composé d'algues unicellulaires vivantes appartenant à une ou plusieurs espèces parmi les suivantes: Chlorella sp., Pseudokirchneriella subcapitata (anciennement Selenastrum capricornutum) et Desmodesmus subspicatus (anciennement Scenedesmus subspicatus). La nourriture est dispensée en fonction de la teneur en carbone organique (C) fournie à chaque animal parent. La recherche (14) a montré que pour Daphnia magna, des teneurs comprises entre 0,1 et 0,2 mg de C/Daphnia/jour dans la ration alimentaire suffisent pour produire le nombre de descendants vivants requis selon les critères de validité. On peut fournir des rations à teneur constante tout au long de l'essai ou utiliser des teneurs plus faibles au début, que l'on augmentera afin de tenir compte de la croissance des animaux parents. Dans ce cas, la ration devra toujours contenir entre 0,1 et 0,2 mg de C/Daphnia/jour, qui est la teneur recommandée. Si des paramètres de remplacement, comme le nombre de cellules d'algues ou l'absorbance de la lumière, sont utilisés pour doser la teneur en carbone requise dans la ration alimentaire (notamment pour des raisons pratiques, si la mesure de la teneur en carbone est trop longue), chaque laboratoire est tenu de produire son propre nomogramme reliant le paramètre de remplacement à la teneur en carbone de la culture d'algues (des conseils concernant l'établissement d'un nomogramme sont donnés à l'appendice 3). Les nomogrammes devraient être vérifiés au moins une fois par an, et plus souvent si les conditions de culture des algues ont changé. Il a été démontré que l'absorbance de la lumière donne une meilleure indication de la teneur en carbone que le nombre de cellules (15). Il convient d'administrer une suspension d'algues concentrée aux Daphnia afin de réduire au minimum le volume du milieu de culture des algues transféré dans les récipients d'essai. On peut concentrer les algues par centrifugation, puis remise en suspension dans le milieu de culture des Daphnia. Lumière 16 heures de lumière à une intensité ne dépassant pas 15-20 μE · m– 2· s– 1, mesurée au niveau de la surface de l'eau dans le récipient. En ce qui concerne les intruments de mesure de la lumière étalonnés en lux, la gamme de 1000 — 1500 lux pour la lumière blanche froide correspond étroitement à l'intensité lumineuse recommandée de 15-20 μE · m– 2 · s– 1. Température La température du milieu d'essai sera comprise entre 18 °C et 22 °C. Toutefois, dans un même essai, la température ne varie pas, si possible, quotidiennement de plus de 2 °C au sein de cet intervalle (p. ex. 18 — 20 °C, 19 — 21 °C ou 20 — 22 °C). Il peut être utile d'utiliser un récipient d'essai supplémentaire pour surveiller la température. Aération Les récipients d'essai ne sont pas aérés durant l'essai. Dispositif d'essai Essai préliminaire de détermination des concentrations Si nécessaire, un essai de détermination de l'ordre de grandeur sera mené avec, par exemple, cinq concentrations du produit chimique d'essai. On utilisera deux réplicats par concentration d'essai et par témoin. Des informations supplémentaires, provenant d'essais avec des produits chimiques similaires ou tirées d'études déjà publiées et portant sur la toxicité aiguë pour Daphnia et/ou d'autres organismes aquatiques peuvent également être utiles pour décider de la gamme de concentrations à utiliser dans l'essai de détermination de l'ordre de grandeur. L'essai de détermination de l'ordre de grandeur dure 21 jours ou suffisamment longtemps pour prévoir l'ampleur des effets de manière fiable. À l'issue de cet essai, on étudie la reproduction de Daphnia. Le nombre de parents et la production de descendants sont consignés. Essai définitif Normalement, on teste au moins cinq concentrations d'essai qui encadrent la concentration efficace (p. ex. CEx), et forment une série géométrique avec des concentrations successives séparées, de préférence, par un facteur inférieur ou égal à 3.2. Il convient d'utiliser un nombre approprié de réplicats pour chaque concentration d'essai (voir paragraphes 24-25). L'étude de moins de cinq concentrations fait l'objet d'une justification. Il convient de ne pas tester les produits chimiques au-dessus de leur limite de solubilité dans le milieu d'essai. Avant de réaliser l'expérience, il est souhaitable d'examiner l'efficacité statistique du mode opératoire et de faire appel à des méthodes statistiques appropriées (4). Lors de l'établissement de la gamme de concentrations, il convient de tenir compte des points suivants:
Si aucun effet n'est observé à la plus forte concentration utilisée lors de la détermination de l'ordre de grandeur (p. ex. 10 mg/l), ou s'il est très probable que le produit chimique d'essai présentera une toxicité faible voire nulle du fait de son innocuité pour d'autres organismes et/ou parce que ces derniers l'absorbent peu ou pas du tout, il est possible de mener l'essai de reproduction comme un essai limite, en testant un témoin et une concentration de la substance d'essai de 10 mg/l, par exemple. Il convient alors d'utiliser dix réplicats pour les groupes exposés et pour les témoins. Si l'essai limite requiert un système dynamique, moins de réplicats pourront convenir. Un essai limite permettra de démontrer l'absence d'effet statistiquement significatif à la concentration limite. Néanmoins, si des effets apparaissent à cette occasion, un essai complet sera normalement nécessaire. Témoins Il convient de tester une série de témoins du milieu d'essai et, le cas échéant, une série de témoins contenant le solvant ou le dispersant, parallèlement aux séries traitées avec le produit chimique d'essai. En cas de recours à un solvant ou à un dispersant, leur concentration est la même que dans les récipients qui contiennent le produit chimique d'essai. Il convient d'utiliser un nombre approprié de réplicats (voir paragraphes 23-24). Généralement, dans un essai correctement mené, le coefficient de variation autour du nombre moyen de descendants vivants par animal parent dans le ou les groupes témoins est ≤ 25 %, et cette information est communiquée pour les essais où les animaux sont maintenus dans des récipients individuels. Renouvellement du milieu d'essai La fréquence de renouvellement du milieu dépendra de la stabilité du produit chimique d'essai, mais devrait être au moins trihebdomadaire. Si les essais préliminaires de stabilité (voir paragraphe 7) montrent que la concentration du produit chimique d'essai n'est pas stable (c'est-à-dire qu'elle sort de la gamme de 80 — 120 % de la concentration nominale ou tombe en dessous de 80 % de la concentration initiale mesurée) durant la période de renouvellement la plus longue (3 jours), il faut envisager de renouveler le milieu plus fréquemment ou de pratiquer un essai dynamique. Lors du renouvellement du milieu dans les essais semi-statiques, on prépare une deuxième série de récipients d'essai en vue d'y transférer les animaux parents avec, par exemple, une pipette en verre d'un diamètre approprié. Il convient que le volume de milieu transféré avec les Daphnia soit le plus petit possible. Observations Les résultats des observations effectuées durant l'essai sont consignés dans des fiches de données (voir exemples aux appendicees 4 et 5). Si d'autres mesures s'imposent (voir paragraphe 44), des observations supplémentaires pourront être requises. Descendance Les descendants engendrés par chaque animal parent seront de préférence retirés et comptés quotidiennement, dès la première portée, pour qu'ils ne consomment pas la nourriture destinée aux parents. Aux fins de la présente méthode d'essai, on ne compte que les descendants vivants, mais la présence d'œufs avortés ou de descendants morts est signalée. Mortalité La mortalité des animaux parents est notée, de préférence quotidiennement, et au moins à chaque comptage des descendants. Autres paramètres Bien que la présente méthode d'essai soit avant tout destinée à évaluer les effets sur le taux de reproduction, il se peut que d'autres effets soient suffisamment chiffrés pour se prêter à une analyse statistique. On pourra ainsi rapporter le taux de reproduction par animal parent survivant, c'est-à-dire le nombre de descendants vivants produits pendant l'essai par parent survivant. Cette donnée pourra être comparée à la principale variable de réponse (taux de reproduction par animal parent présent au début de l'essai et qui n'est pas mort de manière fortuite ou accidentelle pendant l'expérience). Si une mortalité parentale se produit dans les réplicats exposés au produit chimique d'essai, il convient d'analyser si cette mortalité suit une courbe concentration-response, par exemple s'il existe une régression significative de la réponse par rapport à la concentration testée du produit chimique d'essai, avec une pente positive (un test statistique tel que le test de tendance de Cochran-Armitage peut être utilisé à cet effet). Si la mortalité parentale ne suit pas de courbe concentration-réponse, alors les réplicats montrant une mortalité parentale sont exclus de l'analyse des résultats. Si la mortalité parentale suit une courbe de concentration-réponse, cette mortalité est assimilée à un effet du produit chimique testé et les réplicats ne sont pas exclus de l'analyse des résultats. Voir détails au paragraphe 51. La mesure de la croissance est très intéressante, puisqu'elle fournit des indications sur les éventuels effets sublétaux, ce qui peut se révéler utile pour compléter les résultats sur la reproduction. On préconise de mesurer la longueur des animaux parents (la longueur du corps sans l'épine anale) à la fin de l'essai. D'autres paramètres peuvent être mesurés ou calculés: le moment de la première portée (et des portées suivantes), le nombre et la taille des portées par animal, le nombre de portées avortées, la présence de mâles parmi les néonates (OCDE, 2008) ou d'éphippies et, éventuellement, le taux intrinsèque d'accroissement de la population (voir définition à l'appendice 1 et procédures d'identification du sexe des néonates à l'appendice 7). Fréquence des dosages analytiques et des mesures La concentration de l'oxygène, la température, la dureté et le pH sont mesurés au moins une fois par semaine, avant et après le renouvellement des milieux, chez les témoins et dans les récipients qui renferment la concentration la plus élevée du produit chimique d'essai. Au cours de l'essai, les concentrations du produit chimique à tester sont déterminées à intervalles réguliers. Dans les essais semi-statiques, où l'on suppose que la concentration du produit chimique d'essai ne s'écartera pas de plus de 20 % de la concentration nominale (c'est-à-dire qu'elle restera comprise dans un intervalle de 80 à 120 %, voir paragraphes 6, 7 et 39), on recommande, au minimum, d'analyser les concentrations d'essai la plus élevée et la plus basse, dès leur préparation et au moment du renouvellement, dans les milieux qui viennent d'être renouvelés et dans ceux qui sont sur le point de l'être, une fois au cours de la première semaine de l'essai (les analyses seront pratiquées sur des échantillons prélevés dans la même solution, juste après la préparation de la solution et au moment du renouvellement). Ces déterminations sont ensuite répétées selon une fréquence au moins hebdomadaire. S'agissant des essais où la concentration du produit chimique d'essai n'est pas supposée demeurer dans un intervalle de ± 20 % de la concentration nominale, il est nécessaire d'analyser toutes les concentrations d'essai, juste après leur préparation et au moment du renouvellement. Cependant, pour les essais où la concentration initiale mesurée du produit chimique d'essai sort de l'intervalle de ± 20 % de la concentration nominale, mais pour lesquels on peut montrer de façon suffisamment convaincante que les concentrations initiales sont répétables et stables (c'est-à-dire comprises dans un intervalle de 80 à 120 % des concentrations initiales), les déterminations chimiques peuvent se limiter durant les deuxième et troisième semaines aux concentrations d'essai la plus élevée et la plus basse. En tout état de cause, la détermination de la concentration du produit chimique d'essai avant le renouvellement n'est nécessaire que sur un seul réplicat de chaque concentration d'essai. Si on pratique un essai dynamique, il convient de recourir à un régime de prélèvements identique à celui décrit pour les essais semi-statiques (mais l'analyse des solutions «anciennes» ne s'applique pas ici). Toutefois, il peut être souhaitable d'augmenter le nombre de prélèvements durant la première semaine (p. ex. trois séries de mesures) afin de vérifier la stabilité des concentrations d'essai. Dans ces types d'essai, le débit du diluant et du produit chimique d'essai sont contrôlés chaque jour. S'il s'avère que la concentration du produit chimique d'essai a pu être correctement maintenue tout au long de l'essai dans un intervalle de ± 20 % de la concentration initiale mesurée ou nominale, les résultats peuvent être déduits des valeurs initiales mesurées ou nominales. Si l'écart par rapport à la concentration initiale mesurée ou nominale est supérieur à ± 20 %, les résultats sont exprimés par rapport à la moyenne pondérée en fonction du temps (voir les conseils de calcul à l'appendice 6). RÉSULTATS ET RAPPORTS Traitement des résultats Le présent essai a pour objet d'établir l'effet du produit chimique d'essai sur le taux de reproduction. Le nombre total de descendants vivants par animal parent est calculé pour chaque récipient d'essai (c'est-à-dire pour chaque réplicat). En outre, la reproduction peut être évaluée en s'appuyant sur la production de descendants vivants par les organismes parents survivants. Cependant, la variable de réponse la plus pertinente du point de vue écologique est le nombre total de descendants vivants par animal parent qui n'est pas mort de manière accidentelle (95) ou fortuite (96) au cours de l'essai. Si un animal parent meurt, soit accidentellement, soit de manière fortuite, ou s'il se révèle être un mâle, le réplicat est exclu de l'analyse. L'analyse reposera alors sur un nombre réduit de réplicats. Si un animal parent meurt dans un récipient recevant le produit chimique d'essai, il convient d'examiner si la mortalité suit une fonction concentration-réponse, notamment s'il existe une régression importante de la réponse en fonction de la concentration de produit chimique d'essai avec une pente positive (on pourra appliquer un test statistique comme le test de tendance de Cochran — Armitage à cette fin). Si la mortalité ne correspond pas à une fonction concentration-réponse, les réplicats présentant une mortalité parentale sont exclus de l'analyse des résultats de l'essai. Si la mortalité suit une fonction concentration-réponse, la mortalité parentale est considérée comme un effet du produit chimique d'essai et les réplicats concernés ne sont pas exclus de l'analyse des résultats de l'essai. En résumé, si les effets sont exprimés par la CMEO et la CSEO ou par la CEx, il est recommandé de calculer les effets sur la reproduction selon les deux variables de réponse mentionnées précédemment, à savoir;
puis de choisir comme résultat final la valeur la plus faible des CSEO et CMEO ou CEx déterminées avec ces deux différentes variables. Avant de lancer l'analyse statistique, par exemple de type ANOVA ou la comparaison des groupes de traitement et des groupes témoins avec les tests de Student (test t), de Dunnett, de Williams, ou de Jonckheere — Terpstra (méthode descendante), il est conseillé de vérifier s'il est nécessaire de transformer les données pour satisfaire les exigences d'un test statistique particulier. Des méthodes non paramétriques peuvent être envisagées, comme les tests de Dunn ou de Mann — Whitney. Les intervalles de confiance à 95 % sont calculés pour les moyennes établies aux différentes concentrations. Le nombre de parents survivants dans les témoins non traités constitue un critère de validité crucial, et il convient qu'il soit consigné et signalé. Par ailleurs, on versera au rapport tout autre signe indiquant un effet nocif, par exemple un comportement anormal, ainsi que les résultats toxicologiques importants. CEx On calcule les CEx et leurs limites de confiance supérieures et inférieures en utilisant des méthodes statistiques adéquates (p. ex. fonction logistique ou de Weibull, méthode simplifiée de Spearman — Karber, ou simple interpolation). Pour calculer la CE10, la CE50 ou toute autre CEx, on soumet la série complète de données à une analyse de régression. CSEO/CMEO Dans le cas d'une analyse statistique visant à déterminer la CSEO ou la CMEO, il convient d'utiliser des méthodes statistiques adéquates (conformément au Document 54 de l'OCDE intitulé Current Approaches in the Statistical Analysis of Ecotoxicity Data: A Guidance to Application) (4). En général, les effets indésirables du produit chimique d'essai par rapport au témoin sont étudiés en procédant à une vérification de l'hypothèse unilatérale pour p ≤ 0,05. La distribution normale et l'homogénéité de la variance peuvent être vérifiées à l'aide d'un test statistique approprié, tel que le test de Shapiro — Wilk et le test de Levene, respectivement (p ≤ 0,05). Une analyse de la variance à un facteur (ANOVA), puis des tests multi-comparaisons peuvent être effectués. Des tests de comparaisons multiples (test de Dunnett, par exemple) ou des tests d'analyse de tendance descendante (tel que le test de William ou celui de Jonckheere — Terpstra, méthode descendante) peuvent permettre de calculer d'éventuelles différences significatives (p ≤ 0.05) entre les témoins et les diverses concentrations de produit chimique d'essai [on choisira le test recommandé conformément au Document d'orientation de l'OCDE 54 (4)]. On peut toutefois utiliser des méthodes non paramétriques (tels que le test U de Bonferroni conformément à Holm ou le test de tendance de Jonckheere — Terpstra) pour déterminer la CSEO et la CMEO. Essai limite Si un essai limite a été mis en œuvre (comparaison du témoin et d'un seul traitement) et que les conditions requises pour les procédures de tests paramétriques (normalité, homogénéité) sont remplies, on peut évaluer les réponses métriques par le test de Student (test t). Un test t de variance inégale (comme le test de Welch) ou bien un test non paramétrique tel que le test U de Mann — Whitney peuvent être employés lorsque ces conditions ne sont pas satisfaites. La détermination des différences significatives entre les témoins (témoin et solvant ou dispersant témoin), peut faire appel à l'analyse de plusieurs réplicats pour chaque témoin comme il est décrit pour l'essai limite. Lorsque les essais ne détectent aucune différence significative, il est possible de rassembler tous les réplicats des témoins et du solvant témoin. Dans le cas contraire, on compare tous les traitements avec le solvant témoin. Rapport d'essai Le rapport d'essai mentionne les informations suivantes:
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Les définitions suivantes sont utilisées dans le cadre de la présente méthode d'essai: Mortalité accidentelle : mortalité non liée au produit chimique testé, et causée par un incident (c'est-à-dire par une cause connue). Produit chimique : une substance ou un mélange. CEx : concentration du produit chimique d'essai dissous dans l'eau qui entraîne une diminution de x pour cent de la reproduction chez Daphnia, durant une période d'exposition définie. Mortalité fortuite : mortalité non liée au produit chimique testé, et de cause inconnue. Taux intrinsèque d'accroissement de population : mesure de l'accroissement de la population qui intègre la capacité reproductrice et la mortalité par tranche d'âge (1) (2) (3). Elle est nulle dans les populations à l'état stationnaire, positive dans les populations en croissance et négative dans les populations qui régressent. Cette dernière catégorie de population n'est évidemment pas durable et est vouée en fin de compte à l'extinction. Limite de détection : la plus basse concentration susceptible d'être détectée, mais non chiffrée. Limite de détermination : la plus basse concentration susceptible d'être mesurée quantitativement. Concentration minimale avec effet observé (CMEO) : concentration d'essai la plus basse à laquelle on a observé un effet statistiquement significatif du produit chimique sur la reproduction et la mortalité des animaux parents (à p < 0,05) par rapport au témoin, durant une période d'exposition définie. Cependant, il convient que toutes les concentrations d'essai supérieures à la CMEO produisent un effet nocif supérieur ou égal à celui observé à la CMEO. Si ces deux conditions ne peuvent être remplies, il convient de justifier de façon détaillée le choix de la CMEO (et donc de la CSEO). Mortalité : un animal est noté comme mort lorsqu'il est immobile, autrement dit lorsqu'il n'est pas capable de nager ou si aucun mouvement des appendices ou du postabdomen n'est observé dans les 15 secondes qui suivent l'agitation douce du récipient d'essai. (Si on utilise une autre définition, il convient que celle-ci soit stipulée avec sa référence). Concentration (maximale) sans effet observé (CSEO) : concentration d'essai immédiatement inférieure à la CMEO qui, comparée au témoin, n'a pas d'effet statistiquement significatif (à p < 0,05), durant une période d'exposition définie. Descendants : jeunes Daphnia engendrées par les animaux parents au cours de l'essai. Animaux parents : Daphnia femelles présentes au début de l'essai et dont la capacité reproductrice représente l'objet de cette étude. Taux de reproduction : nombre de descendants vivants issus d'animaux parents pendant la période d'essai. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. BIBLIOGRAPHIE
Appendice 2 PRÉPARATION DE MILIEUX ELENDT M7 ET M4 ENTIÈREMENT DÉFINIS Acclimatation aux milieux Elendt M7 et M4 Certains laboratoires ont éprouvé des difficultés à transférer directement les Daphnia dans les milieux M4 (1) et M7. Ils sont toutefois parvenus à un certain résultat en les acclimatant progressivement, c'est-à-dire en les transférant de leur milieu vers un milieu à 30 pour cent d'Elendt, puis à 60 pour cent d'Elendt et enfin à 100 pour cent d'Elendt. La période d'acclimatation peut prendre jusqu'à un mois. Préparation Élements traces Des solutions mères (I), contenant chacune un seul élément trace, sont tout d'abord préparées avec une eau d'une pureté adéquate, par exemple de l'eau désionisée, distillée ou obtenue par osmose inverse. A partir de ces différentes solutions mères (I), on prépare une deuxième solution mère unique (II) renfermant toutes les éléments traces (solution combinée), à savoir:
Milieux M4 et M7 Les milieux M4 et M7 sont préparés à partir de la solution mère II, des macronutriments et des vitamines, de la façon suivante:
La solution mère de vitamines combinées est congelée par petites parties aliquotes. Les vitamines sont ajoutées au milieu peu avant son utilisation.
Appendice 3 ANALYSE DE LA TENEUR EN CARBONE ORGANIQUE TOTAL (COT) ET ÉTABLISSEMENT D'UN NOMOGRAMME POUR LA TENEUR EN COT DANS LES ALIMENTS À BASE D'ALGUES Il est admis que la teneur en carbone des algues alimentaires n'est généralement pas mesurée directement, mais déduite de corrélations (par nomogramme) avec des paramètres de remplacement, comme le nombre de cellules d'algues ou l'absorbance de la lumière. Il faudrait mesurer le COT par oxydation à haute température plutôt que par les méthodes aux UV ou au persulfate. (Pour s'orienter voir: The Instrumental Determination of Total Organic Carbon, Total Oxygen Demand and Related Determinands 1979, HMSO 1980; 49 High Holborn, London WC1V 6HB). Pour établir le nomogramme, il y a lieu d'isoler les algues du milieu de croissance par une centrifugation suivie d'une remise en suspension dans de l'eau distillée. On mesure le paramètre de remplacement et la teneur en COT dans chaque échantillon, produit en triple exemplaire. Les témoins contenant uniquement de l'eau distillée devraient être analysés et la teneur en COT déduite de celle de l'échantillon contenant les algues. Les nomogrammes devraient être linéaires sur la gamme de teneurs en carbone utilisée. Des exemples sont présentés ci-dessous.

Chlorella vulgaris var. viridis (CCAP 211/12). Régression des mg/l de poids sec en fonction des mg de C/l. Données provenant de suspensions concentrées de cultures semi-continues de cellules, remises en suspension dans de l'eau distillée. Axe des x: mg de C/l d'algues alimentaires concentrées Axe des y: mg/l de poids sec d'algues alimentaires concentrées Coefficient de corrélation – 0,980 
Chlorella vulgaris var. viridis (CCAP 211/12). Régression du nombre de cellules en fonction des mg de C/1. Données provenant de suspensions concentrées de cultures semi-continues de cellules, remises en suspension dans de l'eau distillées. Axe des x: mg de C/1d'algues alimentaires concentrées Axe de y: nombre de cellules/1 d'algues alimentaires concentrées Coefficient de corrélation – 0,926 
Chlorella vulgaris var. viridis (CCAP 211/12). Régression de l'absorbance en fonction des mg de C/1 (longueur du trajet optique: 1 cm). Données provenant de suspensions concentrées de cultures semi-continues de cellules, remises en suspension dans de l'eau distillée. Axe des x: mg de C/1d'algues alimentaires concentrées Axe des y: Absorbance à 440 nm dans une dilution à 1/10 d'une solution d'algues alimentaires concentrées Coefficient de corrélation – 0,998 Appendice 4 EXEMPLE DE FICHE POUR CONSIGNER LE RENOUVELLEMENT DU MILIEU, LES DONNÉES DE SURVEILLANCE PHYSICO-CHIMIQUE, L'ALIMENTATION, LA REPRODUCTION DES DAPHNIES ET LA MORTALITÉ DES PARENTS
Appendice 5 EXEMPLE DE FICHE DE DONNÉES POUR CONSIGNER LES RÉSULTATS DE L'ANALYSE CHIMIQUE (a) Concentrations mesurées
(b) Concentrations mesurées en pourcentage de la concentration nominale
Appendice 6 CALCUL D'UNE MOYENNE PONDEREE SUR LE TEMPS Moyenne pondérée sur le [en fonction du] temps Etant donné que la concentration du produit chimique d'essai peut diminuer au cours de la période comprise entre les renouvellements du milieu, il est nécessaire de rechercher la concentration qui devrait être considérée comme représentative de la gamme de concentrations que subissent les Daphnia parents. Le choix devrait s'appuyer sur des considérations biologiques aussi bien que statistiques. Si on estime, par exemple, que la reproduction est surtout affectée par la concentration maximale, il convient d'utiliser la concentration maximale. Si, par contre, on juge que c'est l'effet cumulé ou à long terme du produit chimique toxique qui est prépondérant, il est plus pertinent de prendre une concentration moyenne. Dans ce cas, la concentration moyenne pondérée sur le temps convient, puisqu'elle tient compte de la variation de la concentration instantanée en fonction du temps. Figure 1 Exemple de moyenne pondérée sur le [en fonction] du temps 
La figure 1 illustre un essai (simplifié) qui dure sept jours et dans lequel le milieu est renouvelé aux jours 0, 2 et 4.
La moyenne pondérée sur le temps est calculée de façon que la superficie comprise en dessous de la moyenne pondérée en fonction du temps soit égale à la superficie située en dessous de la courbe de concentration. Le calcul correspondant à l'exemple figurant ci-dessus est illustré au tableau 1. Tableau 1 Calcul de la moyenne pondérée en fonction du temps
La superficie est celle comprise en dessous de la courbe exponentielle de chaque période de renouvellement. Elle se calcule par la formule suivante:
La moyenne pondérée sur le temps (moyenne PT) est la superficie totale divisée par le nombre total de jours. Bien entendu, dans le cas de l'essai de reproduction chez Daphnia, le tableau devrait être étendu à 21 jours. Il est clair que lorsque les observations ne sont faites qu'au début et à la fin de chaque période de renouvellement, il n'est pas possible d'affirmer avec certitude que la chute de concentration est effectivement exponentielle. Une courbe différente donnerait lieu à un calcul différent de la superficie. Cependant, une chute de concentration exponentielle est plausible et constitue probablement la meilleure courbe à utiliser en l'absence d'autres informations. Il convient toutefois d'être prudent si l'analyse chimique ne détecte aucun produit chimique à la fin de la période de renouvellement. Tant qu'il n'est pas possible d'estimer la vitesse de disparition du produit chimique dans la solution, il est impossible d'obtenir une superficie réaliste sous la courbe et, partant, une moyenne pondérée sur le temps qui soit raisonnable. Appendice 7 GUIDE POUR L'IDENTIFICATION DU SEXE DES NOUVEAUX-NÉS La production de mâles parmi les descendants peut avoir lieu sous certaines conditions environnementales changeantes, telles qu'un raccourcissement de la photopériode, des variations de températures, une diminution de la concentration en nourriture, et une densité de population accrue (Hobaek and Larson, 1990; Kleiven et al, 1992). La production de mâles est aussi connue comme une réponse à certains régulateurs de croissance d'insectes (Oda et al, 2005). Dans des conditions où les facteurs de stress chimiques induisent une diminution des descendants issus de femelles parthénogéniques, un nombre accru de mâles serait attendu (OECD, 2008). Sur la base des informations disponibles, il n'est pas possible de prédire, entre le sexe des descendants ou l'effet sur la reproduction, laquelle de ces mesures sera la plus sensible; cependant il existe des indications (OECD, 2008) tendant à montrer que l'induction de mâles dans la descendance pourrait être moins sensible que le nombre de descendants. Puisque l'objectif principal de la méthode d'essai est d'évaluer le nombre de descendants produits, l'apparation de mâle dans cette descendance est une observation optionnelle. Si cette mesure optionnelle est évaluée dans une étude, alors il convient d'appliquer un critère de validité supplémentaire aux témoins: les témoins ne contiennent pas plus de 5 % de descendants mâles. La manière la plus pratique et la plus aisée de différencier le sexe des daphnies est d'utiliser leurs caractéristiques phénotypiques, puisque les mâles et les femelles sont génétiquement identiques et leur sexe est déterminé par les conditions environnementales. Les mâles et les femelles diffèrent par leur longueur et par la morphologie de leurs premières antennes qui sont plus longues chez les mâles que chez les femelles (figure 1). Cette différence est reconnaissable dès la naissance, bien que d'autres caractéristiques sexuelles secondaires se développent à mesure que les animaux grandissent (voir figure 2 dans Olmstead et Leblanc, 2000). Afin d'observer le sexe morphologique, les descendants produits par chaque parent sont transférés au moyen d'une pipette et placés dans une boîte de pétri content le milieu d'essai. Le milieu d'essai est maintenu au minimum pour minimiser les mouvements des animaux. L'observation de la première antenne peut être effectuée sous un stéréomicroscope (x10-60). Figure. 1 mâle (à gauche) et femelle (à droite) de D. magna agés de 24 heures. Les mâles peuvent se distinguer des femelles par la longueur et la morphologie de leur première paire d'antennes, comme indiqué dans le cercle (Tatarazako et al., 2004). 
BIBLIOGRAPHIE Hobaek A and Larson P. 1990. Sex determination in Daphnia magna. Ecology 71: 2255-2268. Kleiven O.T., Larsson P., Hobaek A. 1992. Sexual reproduction in Daphnia magna requires three stimuli. Oikos 65, 197-206. Oda S., Tatarazako N, Watanabe H., Morita M., and Iguchi T. 2005. Production of male neonates in Daphnia magna (Cladocera, Crustacea) exposed to juvenile hormones and their analogs. Chemosphere 61:1168-1174. OCDE, 2008. Validation report for an enhancement of OECD TG 211 Daphnia magna reproduction test. Série sur les essais et l'évaluation, no 88. Organisation de coopération et de développement économiques, Paris. Olmstead, A.W., LeBlanc, G.A., 2000. Effects of endocrine-active chemicals on the development characteristics of Daphnia magna. Environmental Toxicology and Chemistry 19:2107-2113. Tatarazako, N., Oda, S., Abe, R., Morita M. and Iguchi T., 2004. Development of a screening method for endocrine disruptors in crustaceans using Daphnia magna (Cladocera, Crustacea). Environmental Science 17, 439-449. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
(18) |
Dans la partie C, au chapitre C.29, le paragraphe 66 est remplacé par le texte suivant:
|
|
(19) |
Dans la partie C, les chapitres suivants sont ajoutés: «C.47 Poisson, essai de toxicité aux premiers stades de la vie INTRODUCTION
PRINCIPE DE L'ESSAI
INFORMATIONS SUR LE PRODUIT CHIMIQUE D'ESSAI
VALIDITÉ DE L'ESSAI
DESCRIPTION DE LA MÉTHODE Enceintes d'essai
Choix des espèces
Soin des poissons géniteurs
Manipulation des œufs fécondés, des embryons et des larves
Eau
Solutions d'essai
MODE OPÉRATOIRE Conditions d'exposition Durée
Charge
Lumière et température
Alimentation
Concentrations d'essai
Témoins
Fréquence des dosages analytiques et des mesures
Observations 26. Stade du développement embryonnaire : il convient de vérifier aussi précisément que possible le stade embryonnaire au début de l'exposition au produit chimique testé. Ceci peut se faire en utilisant un échantillon représentatif d'œufs convenablement conservés et rendus translucides. 27. Éclosion et survie : il convient d'observer et dénombrer les éclosions et les survivants au moins une fois par jour. Si l'on observe la présence de champignos sur les œufs au début du stade de développement embryonnaire (le 1er ou le 2e jour de l'essai, par exemple), il faut compter et retirer les œufs en question. Les embryons, larves et juvéniles morts doivent être enlevés dès qu'ils sont repérés, car ils peuvent se décomposer rapidement et être mis brisés par les autres poissons. En retirant les individus morts, il convient d'être extrêmement attentif à ne pas endommager physiquement les œufs et les larves adjacents. Les signes létaux varient selon les espèces et les stades de développement. Par exemple:
28. Apparence anormale : il convient de noter le nombre de larves et de juvéniles présentant une anomalie corporelle, à des intervalles de temps adéquats qui dépendent de la durée de l'essai et de la nature de l'anomalie décrite. Notons que, chez les larves et les juvéniles, l'anormalité est un phénomène naturel pouvant toucher de l'ordre de plusieurs pour cent des individus dans le(s) témoin(s) de certaines espèces. En cas de malformations et donc d'apparence anormale telles que l'organisme est en état de souffrance considérable et irréversible, l'organisme peut être retiré de l'essai. Les animaux concernés doivent être euthanasiés et comptabilisés comme morts aux fins de l'analyse statistique ultérieure. Le développement embryonnaire normal a été décrit pour la plupart des espèces recommandées dans la présente méthode d'essai (9) (10) (11) (12). 29. Comportement anormal : des anomalies telles qu'une hyperventilation, une nage mal coordonnée et une immobilité ou un comportement alimentaire atypiques doivent être notées à des intervalles de temps adéquats, qui dépendent de la durée de l'expérience (une fois par jour pour les poissons d'eaux chaudes, par exemple). Ces effets, bien que difficiles à quantifier, peuvent, lorsqu'ils sont observés, contribuer à l'interprétation des résultats de mortalité. 30. Poids : à la fin de l'essai, tous les poissons ayant survécu dans chaque réplicat sont pesés (on note le nombre d'animaux présents dans le réplicat ainsi que le poids moyen par animal); il est préférable de noter le poids humide (poissons séchés par tamponnage), mais le poids sec peut également être consigné dans le rapport (13). 31. Longueur : à la fin de l'essai, les poissons sont mesurés. Il est recommandé de mesurer leur longueur totale, mais en cas de décomposition de la nageoire caudale ou d'érosion des nageoires, on peut utiliser la longueur standard. On utilisera la même méthode pour tous les poissons d'un essai donné. Les poissons peuvent être mesurés au moyen d'un pied à coulisse, d'un appareil photo numérique, d'un micromètre oculaire étalonné, etc. Les longueurs minimales types sont définies à l'appendice 2. RÉSULTATS ET RAPPORT Traitement des résultats
Rapport d'essai
BBLIOGRAPHIE
Appendice 1 DÉFINITIONS Longueur à la fourche (LF): longueur mesurée de la pointe du museau à l'extrémité des rayons centraux de la nageoire caudale, utilisée pour les poissons pour lesquels il est difficile de savoir où se termine la colonne vertébrale (www.fishbase.org). Longueur standard (LS) : longueur mesurée de la pointe du museau à l'extrémité postérieure de la dernière vertèbre ou à l'extrémité postérieure de la partie médio-latérale de la plaque hypurale. Autrement dit, cette mesure ne prend pas en compte la longueur de la nageoire caudale (www.fishbase.org). Longueur totale (LE) : longueur de l'extrémité du museau à l'extrémité du lobe le plus long de la nageoire caudale, généralement mesurée après avoir comprimé les lobes le long de la ligne médiane. La mesure se fait en ligne droite, sans suivre la courbe du corps (www.fishbase.org). Figure 1 Description des différentes longueurs utilisées Longueur standard Longueur à la fourche Longueur totale Produit chimique : une substance ou un mélange CEx (concentration efficace à x %): concentration qui engendre un effet de x % sur les organismes d'essai durant une période d'exposition déterminée, en comparaison avec un témoin. Par exemple, une CE50 est une concentration estimée produire un effet sur un paramètre évalué de l'essai dans 50 % d'une population exposée durant une période d'exposition déterminée. Concentration minimale avec effet observé (CMEO) : concentration la plus basse d'un produit chimique testé à laquelle on observe un effet statistiquement significatif (à p < 0,05) par rapport au témoin. Néanmoins, toutes les concentrations supérieures à la CMEO devraient avoir un effet néfaste supérieur ou égal à ceux observés à la CMEO. Lorsque ces deux conditions ne peuvent pas être remplies, il faut expliquer en détail la façon dont la CMEO (et donc la CSEO) a été choisie. Les appendices 5 et 6 donnent des indications à ce sujet. Concentration (maximale) sans effet observé (CSEO) : concentration d'essai immédiatement inférieure à la CMEO qui, comparée au témoin, n'a pas d'effet statistiquement significatif (à p < 0,05), durant une période d'exposition définie. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai UVCB : substances de composition inconnue ou variable, produits de réactions complexes ou matériels biologiques. IUPAC : Union internationale pour la chimie pure et appliquée. SMILES : Spécification d'écriture moléculaire linéaire simplifiée Appendice 2 CONDITIONS ET DURÉE DE L'ESSAI ET CRITÈRES DE SURVIE POUR LES ESPÈCES RECOMMANDÉES
Appendice 3 ORIENTATIONS EN MATIÈRE D'ALIMENTATION ET DE MANIPULATION DES POISSONS-GÉNITEURS ET DES POISSONS D'ESSAI ISSUS DES ESPÈCES RECOMMANDÉES
Appendice 4 QUELQUES CARACTERISTIQUES CHIMIQUES D'UNE EAU DE DILUTION ADMISSIBLE
Appendice 5 GUIDE D'ORIENTATION POUR L'ANALYSE STATISTIQUE DE LA DÉTERMINATION DE LA CSEO Généralités L'unité d'analyse est une série de réplicats. Ainsi, pour des mesures en continu telles que la taille, on calcule la moyenne ou la médiane des réplicats et les valeurs obtenues constituent les données destinées à l'analyse. La puissance des tests utilisés doit être démontrée, de préférence à partir une base de données historiques adéquate pour chaque laboratoire. L'ampleur de l'effet pouvant être détecté avec une puissance de 75-80 % doit être prévue pour chaque paramètre par le test statistique à utiliser. Les bases de données disponibles au moment de l'élaboration de la présente méthode d'essai établissent la puissance possible selon les procédures statistiques recommandées. Le laboratoire doit démontrer sa capacité à répondre à cette exigence de puissance, soit en procédant à sa propre analyse de puissance, soit en démontrant que le coefficient de variation (CV) pour chaque réponse ne dépasse pas le 90e percentile des CV utilisés pour développer la méthode d'essai. Le tableau 1 présente ces CV. Si l'on ne dispose que des moyennes et des médianes des réplicats, le CV intra-réplicats peut être ignoré. Tableau 1 CV au 90e percentile pour les espèces d'eau douce sélectionnées
Pour la quasi-totalité des tests statistiques utilisés pour évaluer les études toxicologiques en laboratoire, les comparaisons intéressantes concernent les groupes de traitement par rapport aux témoins. Ainsi, il n'y a pas lieu d'exiger un test F ANOVA statistiquement significatif avant d'utiliser le test de Dunnett ou de Williams, ou un test statistiquement significatif de Kruskal-Wallis avant d'utiliser le test de Jonckheere-Terpstra, de Mann-Whitney ou de Dunn (Hochberg et Tamhane 1987, Hsu 1996, Dunnett 1955, 1964, Williams 1971, 1972, 1975, 1977, Robertson et al. 1988, Jonckheere 1954, Dunn 1964). Le test de Dunnett permet des comparaisons multiples, et le fait d'utiliser le test F comme référence affecte négativement les taux de faux positifs et de faux négatifs. De même, le test de Williams et celui de Jonckheere-Terpstra utilisant un niveau de significativité de 0,05 à chaque étape conservent un taux global de faux positifs de 5 %, et ce taux ainsi que la puissance des tests sont affectés négativement par l'utilisation du test F ou du test de Kruskal-Wallis comme référence. Le test de Mann-Whitney et le test de Dunn doivent être corrigés aux fins de comparaison multiple et il est conseillé de leur appliquer une correction de Bonferroni-Holm. On trouvera dans OCDE (2006) un examen approfondi de la plupart des recommandations sur les tests paramétriques et la vérification des hypothèses sous-jacentes à ces tests, ainsi qu'une bibliographie détaillée. Traitement des témoins en cas d'utilisation d'un solvant Si l'on utilise un solvant, on doit prévoir un témoin-eau et un témoin-solvant. Les deux témoins doivent être comparés pour chaque réponse et combinés en vue de l'analyse statistique s'ils ne présentent aucune différence significative. Sinon, on doit utiliser le témoin-solvant pour le dosage de la CSEO ou l'estimation de la CEx et ne pas utiliser le témoin-eau. Voir la restriction applicable aux critères de validité (paragraphe 7). En ce qui concerne la longueur, le poids, la proportion d'œufs éclos, le taux de mortalité larvaire ou la proportion de larves anormales, et le premier ou le dernier jour de frai ou de remontée, un test T ou un test de Mann-Whitney doit être utilisé pour comparer les témoins au niveau de significativité de 0,05, en ignorant tous les groupes de traitement. Les résultats de ces tests doivent être notés dans le rapport. Observations morphologiques (longueur et poids) La longueur et le poids des poissons peuvent être distribués selon une loi normale ou log normale. Dans les deux cas, les valeurs moyennes par réplicat ont tendance à être normalement distribuées en vertu du théorème de la limite centrale et confirmées par les données provenant de plus d'une centaine d'études relatives aux stades de vie précoce de trois espèces d'eau douce. Par ailleurs, si les données ou les bases de données historiques font apparaître une distribution log normale en ce qui concerne les valeurs relatives à la taille des poissons, le logarithme des valeurs moyennes par réplicat peut être calculé, et les données pour l'analyse peuvent alors être les anti-logarithmes de ces logarithmes de moyennes par réplicat. L'évaluation doit porter sur la compatibilité des données avec une distribution normale et une homogénéité de la variance. A cet effet, on utilisera les résidus d'un modèle ANOVA où la concentration est la seule variable explicative. On peut avoir recours à la représentation sous forme de nuages de points et d'histogrammes ou bien de diagrammes à tiges et à feuilles. Sinon, on peut utiliser un test formel comme celui de Shapiro-Wilk ou d'Anderson-Darling. Il est possible d'évaluer la compatibilité des données avec l'homogénéité de la variance par un examen visuel du même nuage de points ou formellement grâce au test de Levene. L'évaluation de la normalité et de l'homogénéité de la variance est obligatoire uniquement pour les tests paramétriques (Williams, Dunnett, etc.). Une attention particulière doit être accordée aux éventuelles valeurs aberrantes et à leurs effets sur l'analyse. On peut avoir recours au test de Tukey et procéder à l'inspection visuelle des mêmes points de résidus décrits ci-dessus. Il convient de rappeler que les observations sont des réplicats entiers, de sorte que l'omission d'une aberration de l'analyse ne doit être effectuée qu'après un examen attentif. Les tests statistiques qui font appel aux caractéristiques du protocole expérimental et de l'attente biologique sont des tests de tendance régressifs comme le test de Williams ou celui de Jonckheere-Terpstra. Ces tests supposent une relation concentration-réponse monotone, et il faut vérifier que les données sont compatibles avec cette hypothèse. Cela peut être fait visuellement à partir d'un nuage de points des moyennes par réplicat par rapport à la concentration d'essai. Il sera utile de superposer ce nuage de points avec un diagramme linéaire reliant les concentrations moyennes pondérées par la taille d'échantillonnage dans le réplicat. Un écart important de ce diagramme linéaire par rapport à la monotonie indiquerait qu'il faut peut-être avoir recours à des tests non paramétriques. Par ailleurs, il est possible d'avoir recours aux tests formels. Un test formel simple consiste à calculer les contrastes linéaires et quadratiques des moyennes de concentrations. Si le contraste quadratique est significatif et que le contraste linéaire ne l'est pas, cela indique qu'il peut y avoir un problème avec la monotonie, qui devra alors être évaluée à partir des points. S'il peut y avoir un problème avec la normalité ou l'homogénéité de la variance, ces contrastes pourront être construits à partir de données hiérarchisées. Il est possible d'utiliser d'autres tests, tels que le test de Bartholomew (pour la monotonie), mais ces tests accroissent la complexité. Figure 2 Logigramme pour la CSEO — observations morphologiques (longueur et poids) 
À moins que les données ne soient pas conformes aux exigences des tests de Williams et de Jonckheere-Terpstra, on utilise ces tests pour déterminer la CSEO. On trouvera dans OCDE (2006) des informations détaillées sur ces procédures. Si les données sont non conformes aux exigences d'un test de tendance régressif, on peut utiliser le test de Dunnett ou celui de Tamhane-Dunnett (T3), qui permettent tous deux des comparaisons multiples. Ces tests supposent la normalité et, dans le cas du test de Dunnett, l'homogénéité de la variance. Si ces conditions ne sont pas remplies, on peut utiliser le test non paramétrique de Dunn. On trouvera dans OCDE (2006) des informations détaillées sur l'ensemble de ces tests. La figure 2 donne une vue d'ensemble permettant de choisir le test approprié. Éclosion des oeufs et survie des larves Les données à analyser sont les proportions d'œufs qui éclosent ou de larves qui survivent dans chaque réplicat. Ces proportions doivent être évaluées sous l'angle de la variance extra-binomiale, qui est fréquente mais pas universelle pour ces mesures. L'organigramme de la figure 3 donne des indications pour le choix du test approprié; le texte apporte des explications détaillées. Deux tests sont couramment utilisés: celui de la C(α) de Tarone (Tarone, 1979) et des tests du Chi2, chacun étant appliqué séparément à chaque concentration d'essai. En cas d'observation d'une variance extra-binomiale, même dans une seule concentration d'essai, les méthodes qui s'en accommodent doivent être utilisées. Formule 1 test de la C(α) de Tarone (Tarone, 1979)
̂ proportion moyenne pour une concentration donnée; m: nombre de réplicats; nj : nombre de sujets présents dans le réplicat j; xj : nombre de sujets répondants dans le réplicat j (non éclos ou morts, par exemple). Ce test est appliqué à chaque concentration séparément. Il peut être considéré comme un test du Chi2 corrigé, mais les simulations de faible puissance effectuées par Tarone ont montré qu'il était plus puissant qu'un test du Chi2. Figure 3 Logigramme pour la CSEO –éclosion des œufs et mortalité larvaire 
En l'absence de preuve significative de la variance extra-binomiale, on peut utiliser le test de Cochran-Armitage. Ce test ne tenant pas compte des réplicats, il est recommandé d'utiliser l'ajustement Rao-Scott du test de Cochran-Armitage (RSCA), qui prend en compte les répétitions, la taille des réplicats et la variance extra-binomiale, s'il existe des preuves de cette dernière. On peut également utiliser le test de Williams, le test de Jonckheere-Terpstra ou le test de Dunnett, tels que décrits pour les observations morphologiques. Ces tests s'appliquent qu'il y ait ou non variance extra-binomiale, mais ont une puissance un peu plus faible (Agresti 2002, Morgan 1992, Rao et Scott, 1992, 1999, Fung et al. 1994, 1996). Premier ou dernier jour d'éclosion ou de remontée La réponse est un nombre entier, indiquant le jour où l'observation indiquée est faite pour un réplicat donné. La plage de valeurs est généralement très limitée, et il y a souvent de fortes proportions de données liées; (par exemple, le premier jour d'éclosion est identique dans tous les témoins et éventuellement pour une ou deux faibles concentrations d'essai). Des tests paramétriques tels que le test de Williams et le test de Dunnett ne conviennent pas pour ce type de données. Sauf preuve sérieuse de non-monotonie, le test de Jonckheere-Terpstra est très puissant pour détecter les effets du produit chimique testé. Sinon, on peut utiliser le test de Dunn. Malformations larvaires La réponse est le nombre de larves plus ou moins atteintes de malformations. Cette réponse est souvent de faible incidence et pose certains des mêmes problèmes que le premier jour d'éclosion, avec une relation concentration-réponse parfois erratique. Si les données font apparaître une relation concentration-réponse globalement monotone, le test de Jonckheere-Terpstra est puissant pour détecter les effets. Sinon, on peut utiliser le test de Dunn. Bibliographie Agresti, A. (2002); Categorical Data Analysis, second edition, Wiley, Hoboken. Dunnett C. W. (1955); A multiple comparison procedure for comparing several treatments with a control, J. American Statistical Association 50, 1096-1121. Dunn O. J. (1964 ); Multiple Comparisons Using Rank Sums, Technometrics 6, 241-252. Dunnett C. W. (1964); New tables for multiple comparisons with a control, Biometrics 20, 482-491. Fung, K.Y., D. Krewski, J.N.K. Rao, A.J. Scott (1994); Tests for Trend in Developmental Toxicity Experiments with Correlated Binary Data, Risk Analysis 14, 639-648. Fung, K.Y, D. Krewski, R.T. Smythe (1996); A comparison of tests for trend with historical controls in carcinogen bioassay, Canadian Journal of Statistics 24, 431-454. Hochberg, Y. and A. C. Tamhane (1987); Multiple Comparison Procedures, Wiley, New York. Hsu, J.C. (1996); Multiple Comparisons: Theory and Methods; Chapman and Hall/CRC Press, Boca Raton. Jonckheere A. R. (1954); A distribution-free k-sample test against ordered alternatives, Biometrika 41, 133. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. OCDE (2006). Current approaches in the statistical analysis of ecotoxicity data: A guidance to application. Série sur les essais et l'évaluation no 54. Organisation de Coopération et de développement économiques, OCDE, Paris. Rao J.N.K. and Scott A.J. (1992) — A simple method for the analysis of clustered binary data, Biometrics 48, 577-585. Rao J.N.K. and Scott A.J. (1999) — A simple method for analyzing overdispersion in clustered Poisson data, Statistics in Medicine 18, 1373-1385. Robertson, T., Wright F.T. and Dykstra R.L. (1988); Order restricted statistical inference, Wiley. Tarone, R.E. (1979); Testing the goodness of fit of the Binomial distribution, Biometrika 66, 585-590. Williams D.A. (1971); A test for differences between treatment means when several dose levels are compared with a zero dose control, Biometrics 27, 103-117. Williams D.A. (1972); The comparison of several dose levels with a zero dose control, Biometrics 28, 519-531. Williams D. A. (1975); The Analysis of Binary Responses from Toxicological Experiments Involving Reproduction and Teratotlogy, Biometrics 31, 949-952. Williams D.A. (1977); Some inference procedures for monotonically ordered normal means, Biometrika 64, 9-14. Appendice 6 GUIDE D'ORIENTATION POUR L'ANALYSE STATISTIQUE DES ESTIMATIONS PAR RÉGRESSION Généralités Les observations utilisées pour ajuster un modèle sont les moyennes par réplicat (longueur et poids) ou les proportions d'œufs éclos et de larves mortes dans chaque réplicat (OCDE 2006). Il est généralement conseillé de procéder à une régression pondérée en utilisant pour ce faire la taille des échantillons réplicats. D'autres systèmes de pondération sont possibles (pondération en fonction de la réponse moyenne prédite ou bien combinaison de ce type de pondération et d'une pondération en fonction de la taille des échantillons réplicats, par exemple). En ce qui concerne les concentrations, la pondération en fonction de l'inverse de la variance dans l'échantillon à une concentration donnée n'est pas recommandée (Bunke et al. 1999, Seber et Wild 2003, Motulsky et Christopoulos 2004, Huet et al. 2003). Toute transformation des réponses avant l'analyse doit préserver l'indépendance des observations; la CEX et les limites de son intervalle de confiance doivent être exprimées dans les unités de mesure d'origine, plutôt que dans les unités transformées. Par exemple, une variation de 20 % du logarithme de la longueur n'équivaut pas à une variation de 20 % de la longueur (Lyles et al. 2008, Draper et Smith, 1999). Le logigramme de la figure 4 donne un aperçu des estimations de la CEX. Le texte y apporte des explications détaillées. Figure 4 Logigramme pour l'estimation de la CEx pour la longueur, le poids, la proportion d'œufs éclos ou la mortalité larvaire moyennes par réplicat; consulter le texte pour de plus amples informations. 
Considérations relatives aux éclosions et à la mortalité larvaire En ce qui concerne les éclosions et la mortalité larvaire, il est généralement préférable d'ajuster un modèle décroissant, à moins d'ajuster un modèle probit comme décrit ci-dessous. Il convient donc de modéliser la proportion d'œufs qui n'éclosent pas ou de larves qui meurent. En effet la CEx renvoie à une concentration à laquelle la variation est égale à x % de la réponse moyenne des témoins. Si 5 % des œufs témoins ne parviennent pas à éclore et que l'on modélise l'incapacité à éclore, la CE20 désigne une concentration à laquelle il se produit une variation égale à 20 % des 5 % d'œufs témoins non éclos, soit une variation de 0,2 * 0.05 = 0,01 autrement dit d'1 point de pourcentage qui se traduit par un taux d'œufs non éclos de 6 %. Une variation aussi faible ne peut pas être estimée de façon significative à partir des données disponibles et n'est pas biologiquement importante. En revanche, si l'on modélisait la proportion d'œufs qui éclosent, la proportion parmi les témoins serait de 95 % dans notre exemple, et une réduction de 20 % par rapport à la moyenne des réponses des témoins se traduirait par une réduction de 0,95 * 0,2 = 0,18, soit un taux d'éclosion passant de 95 % à 77 % (= 95 – 18); la concentration produisant cet effet peut être estimée et revêt sans doute un plus grand intérêt. Le problème ne se pose par avec les mesurages se rapportant à la morphologie, bien que les conséquences négatives sur la morphologie correspondent généralement à une diminution de la taille. Modèles concernant la morphologie (longueur ou poids) et le taux d'éclosion des œufs ou le taux de survie larvaire Hormis le modèle d'hormèse de Brain-Cousens, tous ces modèles sont décrits et préconisés dans OCDE (2006). Les modèles OCDE 2-5 sont également discutés pour des expériences d'écotoxicité dans Slob (2002). Il existe bien entendu beaucoup d'autres modèles qui pourraient être utiles. Bunke et al. (1999) énumère de nombreux modèles ne figurant pas ici, et les références à d'autres modèles sont abondantes. Les modèles énumérés ci-dessous sont jugés particulièrement adaptés aux expériences d'écotoxicité et sont largement utilisés. Avec 5 concentrations d'essai plus témoin
Avec minimum 6 concentrations d'essai plus témoin
Lorsqu'il existe une preuve visuelle de l'hormèse (peu probable en cas de d'éclosion des œufs ou de survie larvaire, mais parfois observée sur le plan morphologique)
Autres modèles pour les œufs non éclos et la mortalité larvaire
Qualité de l'ajustement d'un modèle unique
Comparaison des modèles
Qualité de l'estimation de la CEx L'intervalle de confiance (IC) de la CEx ne doit pas être trop large. Le jugement statistique est nécessaire pour décider de la largeur de l'intervalle de confiance et de l'utilité que peut encore avoir la CEx. Les simulations de modèles de régression ajustés aux données relatives à l'éclosion des œufs et aux données morphologiques montrent que 75 % environ des intervalles de confiance pour la CEx (x = 10, 20 ou 30) ne portent pas sur plus de deux concentrations d'essai. Cela donne une idée de ce qui est acceptable et réalisable. De nombreux auteurs affirment qu'il faut indiquer les intervalles de confiance pour tous les paramètres du modèle et que lorsque les intervalles de confiance des paramètres du modèle sont grands, cela signifie que les modèles sont inacceptables (Ott et Longnecker 2008, Alvord et Rossio 1993, Motulsky et Christopoulos 2004, Lyles et al. 2008, Seber et Wild 2003, Bunke et al. 1999, Environnement Canada, 2005). L'intervalle de confiance de la CEx (ou de tout autre paramètre du modèle) ne doit pas contenir zéro (Motulsky et Christopoulos 2004). Il s'agit de la régression équivalente à la différence significative minimale qui est souvent citée dans les méthodes de vérification des hypothèses (Wang et al. 2000, par exemple). Il correspond également à l'intervalle de confiance pour que les réponses moyennes à la CMEO ne contiennent par la moyenne des réponses des témoins. Les estimations des paramètres sont-elles scientifiquement plausibles ? Par exemple, si l'intervalle de confiance pour y0 est ± 20 %, aucune estimation de la CE10 n'est plausible. Si le modèle prédit un effet de 20 % à une concentration C et si l'effet maximal observé à la concentration C et aux concentrations en-deçà est de 10 %, alors la CE20 n'est pas plausible (Motulsky et Christopoulos 2004, Wang et al. 2000, Environnement Canada, 2005). La CEx ne doit pas être extrapolée en dehors de la plage de concentrations positives (Draper et Smith 1999, OCDE 2006). Par exemple, un guide général pourrait être que la CEx ne doit pas être inférieure de plus de 25 % environ à la concentration d'essai la plus faible ou supérieure supérieure de plus de 25 % à la concentration d'essai la plus élevée. BIBLIOGRAPHIE Alvord, W.G., Rossio, J.L. (1993); Determining confidence limits for drug potency in immunoassay, Journal of Immunological Methods 157, 155-163. Brain P. and Cousens R. (1989); An equation to describe dose responses where there is stimulation of growth at low doses. Weed res. 29: 93-96. Bunke, O., Droge, B. and Polzehl, J. (1999). Model selection, transformations and variance estimation in nonlinear regression. Statistics 33, 197-240. Collett, D. (2002); Modelling Binary Data, 2nde édition, Chapman and Hall, London. Collett, D. (2003); Modelling Survival Data in Medical Research, 2nde édition, Chapman and Hall, London. Draper, N.R. and Smith, H. (1999); Applied Regression Analysis, third edition. New York: John Wiley & Sons. Environnement Canada (2005), Document d'orientation sur les méthodes statistiques applicables aux essais d'écotoxicité, SPE1/RM/46 Huet, S., A. Bouvier, M.-A. Poursat, E. Jolivet (2003); Statistical Tools for Nonlinear Regression: A Practical Guide with S-PLUS and R Examples, Springer Series in Statistics, New York. Lyles, R. H., C. Poindexter, A. Evans, M. Brown, and C.R. Cooper (2008); Nonlinear Model-Based Estimates of IC50 for Studies Involving Continuous Therapeutic Dose-Response Data, Contemp Clin Trials. Novembre 2008; 29(6): 878–886. Morgan, B.J.T. (1992); Analysis of Quantal Response Data, Chapman and Hall, London. Motulsky, H., A. Christopoulos (2004); Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical Guide to Curve Fitting, Oxford University Press, USA. O'Hara Hines, R. J. and J. F. Lawless (1993); Modelling Overdispersion in Toxicological Mortality Data Grouped over Time, Biometrics Vol. 49, pp. 107-121 OCDE (2006), Current approaches in the statistical analysis of ecotoxicity data: A guidance to application, Série de l'OCDE sur les essais et évaluations, no 54, pp. 1-147, ENV/JM/MONO(2006)18, OCDE, Paris. Ott, R.L., M.T. Longnecker, An Introduction to Statistical Methods and Data Analysis, 6ème édition, 2008, Brooks-Cole, Belmont, CA Ratkowsky, D.A. (1993); Principles of nonlinear regression, Journal of Industrial Microbiology 12, 195-199. Seber, G.A.F., C.J. Wild, Nonlinear Regression, Wiley, 2003 Slob W. (2002); Dose-response modelling of continuous endpoints. Toxicol. Sci., 66, 298-312 Wang, Q., D.L. Denton, and R. Shukla (2000); Applications and Statistical Properties Of Minimum Significant Difference-Based Criterion Testing In a Toxicity Testing Program, Environmental Toxicology and Chemistry, Vol. 19, pp. 113–117, 2000. C.48 Essai à court terme de reproduction des poissons INTRODUCTION
REMARQUES PRÉMIMINAIRES ET LIMITES
PRINCIPE DE L'ESSAI
CRITÈRES DE VALIDITÉ DE L'ESSAI
DESCRIPTION DE LA MÉTHODE Appareillage
Eau
Solutions d'essai
Maintenance des poissons
Pré-expostion et sélection des poissons
CONCEPTION DE L'ESSAI
Sélection des concentrations d'essai
MODE OPÉRATOIRE Sélection et pesée des poissons d'essai
Conditions d'exposition Durée
Alimentation
Lumière et température
Fréquence des dosages analytiques et des mesures
Observations
Survie
Comportement et apparence
Fécondité
Euthanasie des poissons
Observation des caractères sexuels secondaires
Vitellogénine (VTG)
Évaluation de l'histopathologie gonadique
RÉSULTATS ET RAPPORT Évaluation de la réponse des biomarqueurs par l'analyse de la variance (ANOVA)
Rapport d'essai
ORIENTATIONS POUR L'INTERPRÉTATION ET L'ACCEPTATION DES RÉSULTATS DE L'ESSAI
BIBLIOGRAPHIE
Appendice 1 ABRÉVIATIONS & DÉFINITIONS Produit chimique : une substance ou un mélange CV : coefficient de variation ELISA (Enzyme-Linked Immunosorbent Assay) : procédé immunochimique d'absorption enzymatique Axe HHG : axe hypothalamo-hypophyso-gonadique Taux de charge : poids frais de poissons par unité de volume d'eau CMT : concentration maximale tolérée représentant environ 10 % de la CL50 Densité de peuplement : nombre de poissons par unité de volume d'eau Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'assai. VTG : la vitellogénine est une lipo-glyco-phospho-protéine précurseur des protéines du vitellus normalement produite par les femelles sexuellement actives de toutes les espèces ovipares Appendice 2 CONDITIONS EXPÉRIMENTALES DU PROTOCOLE DE DEPISTAGE DES SUBSTANCES AGISSANT SUR LE SYSTÈME ENDOCRINIEN DES POISSONS NON REPRODUCTEURS
Appendice 3 QUELQUES CARACTÉRISTIQUES CHIMIQUES D'UNE EAU DE DILUTION ACCEPTABLE
Appendice 4A SUBSTRAT DE FRAI POUR LES POISSONS-ZÈBRES Plateau de frai : plat à instruments en verre, mesurant par exemple 22 × 15 × 5,5 cm (L × l × h), recouvert d'une grille amovible en acier inoxydable (mailles de 2 mm de largeur). La base de cette grille se trouve plus bas que le bord du plat. 
Le substrat de frai est fixé sur la grille. Il forme une structure dans laquelle les poissons peuvent pénétrer. Des plantes d'aquarium artificielles en plastique vert, par exemple, peuvent convenir (NB: l'adsorption possible du produit chimique d'essai sur la matière plastique est prise en compte). Le plastique est lessivé dans un volume suffisant d'eau chaude et pendant un temps suffisant pour qu'aucun produit chimique ne risque de se retrouver dans la solution d'essai. Si des objets en verre sont utilisés, on veille à ce que les poissons ne soient ni blessés ni entravés lors de mouvements vigoureux. La distance entre le plateau et les parois du récipient est d'au moins 3 cm pour que le frai ne se fasse pas à l'extérieur du plateau. Les œufs déposés sur le plateau traversent la grille et peuvent être prélevés 45 à 60 minutes après le début de l'éclairage. Les œufs translucides n'adhèrent pas et peuvent facilement être comptés grâce une lumière transversale. Pour cinq femelles par récipient, le nombre d'œufs pondus peut être considéré comme faible s'il est inférieur ou égal à 20 par jour, moyen s'il est compris entre 20 et 100, et élevé s'il est supérieur à 100. Le plateau de frai est retiré, les œufs ramassés et le plateau de frai réintroduit dans le récipient d'essai, le plus tard possible dans la soirée ou bien très tôt le matin. La réintroduction du plateau se fait dans un délai maximum d'une heure car sinon le signal du substrat de frai peut induire un accouplement et un frai à un moment inhabituel. Si la situation nécessite une introduction ultérieure du plateau de frai, il convient d'attendre au moins neuf heures après le début de l'éclairage. À cette heure tardive de la journée, l'induction de frai ne se fait plus. Appendice 4B SUBSTRAT DE FRAI POUR LES TÊTES-DE-BOULE Deux ou trois plaques et plateaux de frai en plastique/céramique/verre ou en acier inoxydable combinés sont placés dans chaque enceinte d'essai (morceau de gouttière semi-circulaire grise de 80 mm de longueur posé sur un plateau à rebords de 130 mm de long, par exemple) (voir photo). Il est avéré que les plaques en PVC ou en céramique vieillies pouvaient convenablement servir de substrat de frai (Thorpe et al, 2007). Il est recommandé d'utiliser des plaques abrasées pour améliorer l'adhérence. Le plateau est aussi muni d'un écran de protection empêchant les poissons d'accéder aux œufs tombés, à moins que l'efficacité de l'adhérence des œufs ait été démontrée pour le substrat de frai utilisé. 
Le socle est conçu pour contenir tous les œufs qui n'adhèrent pas à la surface des plaques et tomberaient donc au fond de l'enceinte (ou les œufs déposés directement sur le socle en plastique plat). Tous les substrats de frai sont lessivés pendant au moins 12 heures dans l'eau de dilution avant leur utilisation. Thorpe KL, Benstead R, Hutchinson TH, Tyler CR, 2007. An optimised experimental test procedure for measuring chemical effects on reproduction in the fathead minnow, Pimephales promelas. Aquatic Toxicology, 81, 90–98. Appendice 5A ÉVALUATION DES CARACTÈRES SEXUELS SECONDAIRES DU TÊTE-DE-BOULE POUR LA DÉTECTION DE CERTAINS PERTURBATEURS ENDOCRINIENS Synthèse Chez les têtes-de-boule adultes, les caractéristiques physiques pouvant avoir de l'importance dans les essais de perturbateurs endocriniens sont les suivantes: couleur (claire/sombre), motifs de coloration (présence ou absence de bandes verticales), forme du corps (région de la tête et région pectorale, distension de l'abdomen) et caractères sexuels secondaires spécifiques de l'espèce (nombre et taille des tubercules nuptiaux, taille du bourrelet dorsal et ovipositeur). Les tubercules nuptiaux sont situés sur la tête (bourrelet dorsal) des têtes-de-boule adultes mâles reproducteurs, et sont généralement disposés bilatéralement et symétriquement (Jensen et al. 2001). Les femelles et les juvéniles mâles et femelles des témoins ne développent pas de tubercule (Jensen et al. 2001). Il est possible de dénombrer jusqu'à huit tubercules individuels autour des yeux et entre les narines des mâles. Les tubercules les plus importants en nombre et en taille forment deux lignes parallèles situées juste en dessous des narines et au-dessus de la bouche. De nombreux poissons possèdent des groupes de tubercules sous la mâchoire inférieure; tout près de la bouche on en trouve généralement une seule paire alors que le ventre peut comprendre jusqu'à quatre tubercules. Le nombre de tubercules dépasse rarement 30 (fourchette, 18-28; Jensen et al. 2001). Les tubercules les plus nombreux forment une seule et même structure de forme plutôt arrondie, dont la hauteur est à peu près égale au rayon. Chez la plupart des mâles reproducteurs, certains tubercules sont si étendus et protubérants qu'il est impossible de les distinguer les uns des autres. Certains types de perturbateurs endocriniens peuvent provoquer l'apparition anormale de caractères sexuels secondaires chez le sexe opposé; ainsi, les agonistes des récepteurs d'androgènes tels que la 17α-méthyltestostérone ou le 17β-trenbolone peuvent provoquer l'apparition de tubercules nuptiaux chez le tête-de-boule femelle (Smith, 1974; Ankley et al., 2001, 2003), tandis que les agonistes des récepteurs d'œstrogènes peuvent réduire le nombre ou la taille des tubercules nuptiaux chez les mâles (Miles-Richardson et al., 1999; Harries et al., 2000). On trouvera ci-après une description de la caractérisation des tubercules nuptiaux chez les têtes-de-boule, fondée sur le mode opératoire utilisé par le laboratoire de l'Agence pour la protection de l'environnement des États-Unis (USEPA) à Duluth, Minnesota. Les produits et/ou équipements spécifiques peuvent être remplacés par des matériaux comparables disponibles. L'utilisation d'une loupe à éclairage ou d'un microscope binoculaire de dissection à éclairage (3X) permet une observation optimale. On observera le poisson en position dorsale, partie antérieure vers l'avant (tête vers l'observateur).
Dénombrement et classement des tubercules Six zones spécifiques ont été identifiées pour l'évaluation de la présence et du développement de tubercules chez les têtes-de-boule adultes. Une matrice a été créée pour cartographier la localisation des tubercules et la quantité de tubercules présents (voir la fin du présent appendice). Le nombre de tubercules est consigné, et les tubercules peuvent être classés comme suit en fonction de leur taille: 0-absence, 1-présent, 2-agrandi et 3-protubérant pour chaque organisme (photo 1). Classe 0-absence de tout tubercule. Classe 1-tubercule présent, identifié comme tout tubercule dont un seul point est de hauteur presque égale à son rayon. Classe 2-tubercule agrandi, identifié par des tissus ressemblant à un astérisque, présentant généralement une large base radiale marquée de stries et de sillons partant du centre. La hauteur des tubercules est souvent plus irrégulière mais peut parfois être un peu arrondie. Classe 3-tubercule protubérant, de forme généralement plutôt large et arrondie et de structure moins bien définie. Ces tubercules s'agglomèrent parfois pour former une seule et même masse le long d'une zone ou de plusieurs zones (B, C et D, voir description ci-dessous). Leur couleur et leur forme sont similaires à celles de la classe 2, mais sont parfois assez indéterminées. Ce système de classement permet généralement d'obtenir un résultat global < 50 chez un mâle témoin normal possédant un nombre de tubercules compris entre 18 et 20 (Jensen et al. 2001). Photo 1 
Certains poissons peuvent présenter plus de tubercules que la matrice ne compte de cases pour une zone particulière. Dans ce cas, des chiffres supplémentaires peuvent être indiqués à l'intérieur, à droite ou à gauche de la case. La matrice n'a donc pas besoin d'afficher une symétrie. Une autre technique permettant de cartographier les tubercules allant par paires ou réunis verticalement le long du plan horizontal de la bouche pourrait consister à indiquer deux chiffres dans une seule case. Zones cartographiées:
RÉFÉRENCES
Appendice 5B ÉVALUATION DES CARACTÈRES SEXUELS SECONDAIRES DU MEDAKA POUR LA DÉTECTION DE CERTAINS PERTURBATEURS ENDOCRINIENS La mesure des tubercules papillaires (*10), qui sont les caractères sexuels secondaires du medaka (Oryzias latipes) est décrite ci-après.
Mesure
Fig.1. Schéma illustrant la différence de forme et de taille de la nageoire anale entre les sexes. A, mâle; B, femelle. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig.2. A, Tubercules papillaires situés sur les plaques conjointes de la nageoire anale. J.P: plaque conjointe; A.S.: espace axial;P: tubercule. B, Extrémité distale de la nageoire anale. Les actinotriches (Act.) sont situés à l'extrémité. Oka, T. B., 1931. On the processes on the fin rays of the male of Oryzias latipes and other sex characters of this fish. J. Fac. Sci., Tokyo Univ., IV, 2: 209-218. 
Fig.3. Photographie montrant le site de coupe lorsque la gonade est fixée dans une autre solution que du formol tamponné à 10 %. Dans ce cas, le corps restant sera coupé entre la région antérieure de la nageoire anale et l'anus à l'aide d'un rasoir (trait rouge). La tête sera placée dans la solution de fixation pour préserver la gonade, et la queue dans le formol tamponné à 10 %. 
Appendice 6 PROCÉDURES RECOMMANDÉES POUR LES PRÉLÈVEMENTS EFFECTUÉS À DES FINS DE DOSAGE DE LA VITELLOGÉNINE On prendra soin d'éviter la contamination croisée entre les échantillons de VTG des mâles et des femelles. Procédure 1A: tête-de-boule, prélèvement sanguin dans l'artère / la veine caudale Après anesthésie, le pédoncule caudal est partiellement sectionné avec une lame de scalpel, et un prélèvement sanguin est effectué dans l'artère / la veine caudale grâce à un tubule capillaire à microhématocrite héparinisé. Après le prélèvement sanguin, le plasma est rapidement séparé par centrifugation à température ambiante pendant 3 minutes à 15 000 g (ou bien à une température de 4°C pendant 10 minutes à 15 000 g). Le pourcentage d'hématocrite peut éventuellement être déterminé à l'issue de la centrifugation. Le plasma est ensuite retiré du tube à microhématocrite puis stocké dans un tube de centrifugeuse avec 0.13 unité d'aprotinine (un inhibiteur de protéase) à – 80°C jusqu'au dosage de la VTG. Selon la taille du tête-de-boule (qui dépend du sexe), les volumes de plasma pouvant être prélevés sont généralement de 5 à 60 μl par individu (Jensen et al. 2001). Procédure 1B: tête-de-boule, prélèvement sanguin par ponction cardiaque Il est également possible d'effectuer un prélèvement sanguin par ponction cardiaque effectuée à l'aide d'une seringue héparinisée (1 000 unités d'héparine par ml). Le sang est versé dans des tubes Eppendorf (maintenus dans de la glace) avant d'être centrifugé (5 minutes à 7 000 g à température ambiante). Le plasma est versé dans des tubes Eppendorf propres (dans des aliquotes si le volume de plasma le permet) puis rapidement congelé à – 80°C jusqu'à l'analyse (Panter et al., 1998). Procedure 2A: Medaka japonais, excision du foie Retrait des poissons d'essai de l'enceinte d'essai
Excision du foie
Après excision du foie, la carcasse est prête pour l'histologie gonadique et la mesure des caractères sexuels secondaires. Spécimens Stocker les spécimens hépatiques prélevés sur les poissons d'essai à une température ≤ – 70°C s'ils ne sont pas utilisés pour le pré-traitement juste après leur excision. Fig-1 Une incision est pratiquée avec des ciseaux juste avant les nageoires pectorales. 
Fig-2 La ligne médiane de l'abdomen est incisée avec des ciseaux jusqu'à un point situé environ 2 mm au-dessus de l'anus. 
Fig-3 Les parois abdominales sont écartées avec des pinces pour exposer le foie et les autres organes internes (elles peuvent aussi être épinglées latéralement). La flèche montre le foie. 
Fig-4 Le foie est disséqué et excisé avec des pinces. 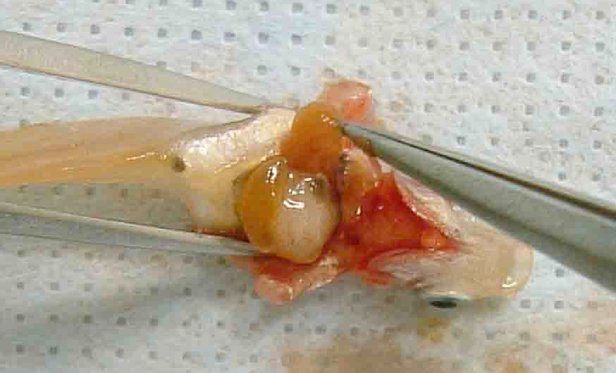
Fig-5 Les intestins sont délicatement retirés avec des pinces. 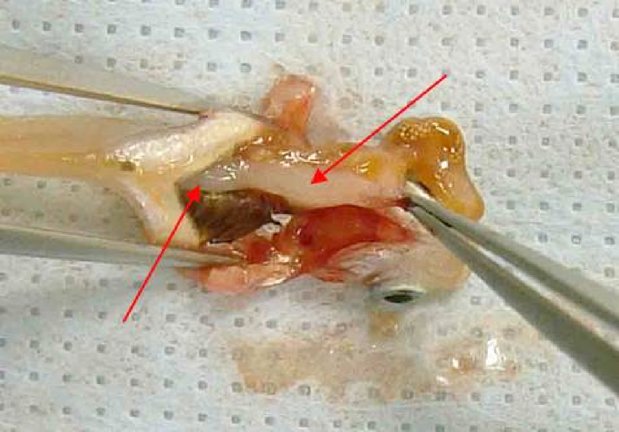
Fig-6 Les deux extrémités des intestins et les attaches du mésentère sont sectionnées avec des ciseaux. 
Fig-7 (femelle) La procédure est identique pour les femelles. 
Fig-8 Procédure achevée. 
Procédure 2 B: Medaka japonais (Oryzias latipes), pré-traitement du foie pour le dosage de la vitellogénine Retirer la bouteille contenant le tampon d'homogénat du kit ELISA et la refroidir avec de la glace pilée (température de la solution: ≤ 4 °C). Si un tampon d'homogénat provenant d'un kit ELISA EnBio est utilisé, laisser décongeler la solution à température ambiante, puis conserver la bouteille au frais avec de la glace pilée. Calculer le volume de tampon d'homogénat pour le foie à partir du poids de ce dernier (utiliser 50 μl de tampon d'homogénat par mg de foie). Exemple: Si le foie pèse 4,5 mg, le volume de tampon d'homogénat pour le foie sera de 225 μl. Dresser une liste des volumes de tampon d'homogénat pour tous les foies. Préparation du foie pour le pré-traitement
Déroulement du pré-traitement 1) Ajout du tampon d'homogénat Vérifier sur la liste le volume de tampon d'homogénat à utiliser pour un échantillon hépatique particulier et ajuster la micropipette (gamme de volumes: 100-1 000 μl) au volume approprié. Attacher un embout propre à la micropipette. Retirer le tampon d'homogénat du flacon de réactif et ajouter le tampon dans le microtube de 1,5 ml contenant le foie. Ajouter le tampon dans tous les microtubes de 1,5 ml contenant les échantillons hépatiques selon la procédure décrite ci-dessus. Il n'est pas nécessaire de remplacer l'embout de la micropipette par un embout neuf. Toutefois, si l'embout est contaminé ou suspect de contamination, il est changé. 2) Homogénéisation du foie
3) Centrifugation de l'homogénat hépatique en suspension
4) Collecte du surnageant
Stockage des spécimens Stocker les microtubes de 0,5 ml contenant le surnageant de l'homogénat hépatique à une température ≤ – 70 °C jusqu'à la conduite de l'essai ELISA. Procédure 3A: Poisson-zèbre, prélèvement sanguin dans l'artère / la veine caudale Immédiatement après anesthésie, le pédoncule caudal est sectionné transversalement, et un prélèvement sanguin est effectué dans l'artère / la veine caudale grâce à un tubule capillaire à microhématocrite héparinisé. Les volumes sanguins collectés varient entre 5 et 15 μl selon la taille du poisson. Un volume identique de tampon d'aprotinine [6 microgrammes/ml de solution tampon phosphate (PBS)] est ajouté dans le tube microcapillaire, et le plasma est séparé du sang par centrifugation (5 minutes à 600 g). Le plasma est versé dans des tubes à essais puis stocké à une température de – 20 °C jusqu'au dosage de la VTG ou d'autres protéines produisant des effets intéressants. Procédure 3B: Poisson-zèbre, prélèvement sanguin par ponction cardiaque Pour éviter la coagulation du sang et la dégradation de la protéine, les échantillons sont collectés sur une solution tampon phosphate (PBS) contenant de l'héparine (1 000 unités/ml) et de l'aprotinine, un inhibiteur de protéase (2 TIU/ml). Pour constituer le tampon, il est recommandé d'utiliser de l'héparine, du sel d'ammonium et de l'aprotinine lyophilisée. Pour le prélèvement sanguin, une seringue (1 ml) munie d'une fine aiguille fixe (Braun Omnikan-F, par exemple) est recommandée. La seringue est pré-remplie avec le tampon (approximativement 100 microlitres) afin d'éluer complètement les faibles volumes sanguins de chaque poisson. Les prélèvements sanguins sont effectués par ponction cardiaque. Tout d'abord, le poisson est anesthésié avec du MS-222 (100 mg/l). Le bon plan d'anesthésie est celui qui permet à l'utilisateur de distinguer le battement du cœur du poisson-zèbre. Pendant la ponction cardiaque, maintenir le piston de la seringue sous faible tension. Les volumes sanguins pouvant être collectés varient entre 20 et 40 microlitres. Après la ponction cardiaque, le mélange sang/tampon est placé dans le tube à essais. Le plasma est séparé du sang par centrifugation (20 minutes à 5 000 g) et est stocké à une température de – 80°C jusqu'à l'analyse. Procédure 3C: MON: Poisson-zèbre, homogénéisation de la tête et de la queue
Appendice 7 ÉCHANTILLONS SUPPLÉMENTÉS EN VITELLOGÉNINE ET ÉTALON DE RÉFÉRENCE INTER-ESSAIS Chaque jour où des essais d'induction de la VTG sont effectués, un échantillon supplémenté préparé à l'aide d'un étalon de référence inter-essais sera analysé. La VTG utilisée pour préparer l'étalon de référence inter-essais sera issue d'un lot différent de celui utilisé pour préparer les solutions-étalons pour l'essai en cours. Pour préparer l'échantillon supplémenté, on ajoutera une quantité connue d'étalon inter-essais à un échantillon de plasma de mâle témoin. L'échantillon sera supplémenté jusqu'à atteindre une concentration de VTG de 10 à 100 fois supérieure à la concentration de vitellogénine attendue chez les mâles témoins. L'échantillon de plasma ainsi supplémenté peut provenir d'un seul poisson ou de plusieurs poissons. Un sous-échantillon du plasma de mâle témoin non supplémenté sera analysé dans au moins deux puits dupliqués. L'échantillon supplémenté sera aussi analysé dans au moins deux puits dupliqués. La quantité moyenne de VTG dans les deux échantillons non supplémentés de plasma de mâle témoin sera ajoutée à la quantité calculée de VTG ajoutée pour fortifier les échantillons afin de déterminer la concentration attendue. Le rapport entre cette concentration attendue et la concentration mesurée sera noté avec les résultats de chaque série d'essais effectués le même jour. Appendice 8 ORDINOGRAMME D'ANALYSE STATISTIQUE 
C.49 Poisson, essai de toxicité aiguë au stade embryonnaire INTRODUCTION
PRINCIPE DE L'ESSAI
REMARQUES PRÉLIMINAIRES
VALIDITÉ DE L'ESSAI
DESCRIPTION DE LA MÉTHODE
Appareillage
Enceintes d'essai
Eau et conditions d'expérimentation
Solutions d'essai
Entretien des poissons géniteurs
Épreuve de compétence
Production des œufs
Différenciation des œufs
PROCÉDURE Conditions d'exposition
Concentrations d'essai
Témoins
Début de l'exposition et durée de l'essai
Répartition des œufs sur les plaques à 24 puits
Observations
Dosages analytiques
ESSAI LIMITE
RESULTATS ET RAPPORT Traitement des résultats
Rapport d'essai
BIBLIOGRAPHIE
Appendice 1 DEFINITIONS Effet apical : ayant des effets au niveau de la population. Blastula : formation cellulaire autour du pôle animal, qui recouvre une certaine partie du vitellus. Produit chimique : une substance ou un mélange Épibolie : prolifération massive de cellules principalement épidermiques lors de la phase de gastrulation de l'embryon et mouvement de ces cellules de la face dorsale vers la face ventrale, par lequel les couches de cellules entodermiques s'invaginent et le vitellus se retrouve à l'intérieur de l'embryon. Essai dynamique : essai avec un flux continu de solutions d'essai dans le système d'essai pendant toute la durée de l'exposition. Témoin à l'intérieur d'une plaque : témoin interne constitué de quatre puits remplis d'eau de dilution par plaque à 24 puits afin d'identifier la contamination potentielle des plaques par le fabricant ou par le chercheur au cours du protocole, et tout effet de la plaque pouvant influer sur le résultat de l'essai (gradient de température, par exemple). IUPAC : Union internationale pour la chimie pure et appliquée. Eau d'élevage : eau dans laquelle est pratiqué l'élevage des poissons adultes. Concentration létale médiane (LC50) : concentration d'un produit chimique d'essai qui devrait provoquer la mort de 50 % des animaux exposés au cours de l'essai. Essai semi-statique (essai avec renouvellement) : essai avec renouvellement régulier des solutions d'essai à l'issue de périodes définies (toutes les 24 h, par exemple). SMILES : Spécification d'écriture moléculaire linéaire simplifiée Somite : chez les vertébrés au stade de développement embryonnaire, les somites sont des masses mésodermiques situées de part de d'autre du tube neural, qui produiront ensuite le derme (dermatome), les muscles squelettiques (myotome) et les vertèbres (sclérotome). Essai statique : essai dans lequel les solutions d'essai restent inchangées pendant toute la durée de l'essai. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai UVCB : substances de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques. Appendice 2 ENTRETIEN, ELEVAGE ET CONDITIONS TYPES POUR LES ESSAIS DE TOXICITE AIGÜE SUR EMBRYONS DE POISSON-ZEBRES
Appendice 3 DEVELOPPEMENT NORMAL DU POISSON-ZEBRE A 26°C 
Appendice 4 Fig. 1 Plan des plaques 24 puits 
Fig. 2 Schéma du protocole des essais de toxicité chez le poisson-zèbre au stade embryonnaire (de gauche à droite): production d'œufs, collecte des œufs, pré-exposition immédiatement après la fécondation dans des cuves en verre, sélection des œufs fécondés au moyen d'un microscope inversé ou binoculaire et répartition des œufs fécondés sur des plaques à 24 puits préparées avec les concentrations d'essai/témoins respectifs, n = nombre d'œufs nécessaires par concentration d'essai/témoin (20 en l'occurrence), haf = heures après fécondation. 
Appendice 5 ATLAS DES PARAMETRES LETAUX POUR L'ESSAI DE TOXICITE CHEZ LE POISSON-ZEBRE AU STADE EMBRYONNAIRE Les paramètres apicaux ci-dessous donnent des indications sur la toxicité aiguë et, par conséquent, la mort des embryons: coagulation de l'embryon, non-détachement de la queue, absence de formation de somites et absence de détection de battements du cœur. Les micrographies ci-dessous ont été sélectionnées pour illustrer ces paramètres. Fig. 1 Coagulation de l'embryon: 
Par éclairage sur fond clair, on distingue diverses inclusions opaques sur les embryons de poisson-zèbre coagulés. Fig. 2 Absence de formation de somites 
Malgré un retard de développement de 10 h environ, l'embryon de poisson-zèbre de 24 h a des somites bien développées (→) en (a) tandis qu'il ne montre aucun signe de formation de somites (→) en (b). On observe la formation de somites bien distinctes (→) chez l'embryon de poisson-zèbre de 48 h en (c) malgré un œdème prononcé du sac vitellin (*) tandis qu'en (d) l'embryon de poisson-zèbre de 96 h ne montre aucun signe de formation de somites (→). On note également la déviation de la colonne vertébrale (scoliose) et l'œdème péricardique (*) chez l'embryon en (d). Fig. 3 Vue latérale du non-détachement du bourgeon caudal 
Fig. 4 L'absence de battements du cœur 
L'absence de battements du cœur est, par définition, difficile à illustrer au moyen d'une micrographie. La non-convulsion du cœur (double flèche) indique l'absence de battements du cœur. L'immobilité des cellules sanguines, par exemple dans l'aorte abdominale (→ dans l'encadré) n'est pas un indicateur d'absence de battements du cœur. On notera également l'absence de formation de somites dans cet embryon (*, apparence homogène plutôt que segmentaire des tissus musculaires). L'observation pour l'enregistrement d'une absence de battements du cœur doit durer au moins une minute, avec un grossissement supérieur ou égal à 80×. C.50 Essai de toxicité sur Myriophyllum spicatum dans un système sans sédiment INTRODUCTION
PRINCIPE DE L'ESSAI
INFORMATIONS SUR LE PRODUIT CHIMIQUE D'ESSAI
VALIDITÉ DE L'ESSAI
PRODUIT CHIMIQUE DE RÉFÉRENCE
DESCRIPTION DE LA MÉTHODE Appareillage
Organisme d'essai
Culture
Milieu expérimental
Solutions d'essai
Groupes traités et groupes témoins
Exposition
Conditions de l'essai
Durée
Mesures et déterminations analytiques
Fréquence des mesures et des déterminations analytiques
Essai limite
RÉSULTATS ET RAPPORT Variables de réponse
Taux de croissance spécifique moyen
Rendement
Temps de doublement
Courbes concentration-effet
Estimation de la CEx
Méthodes statistiques
Rapport
BIBLIOGRAPHIE
Appendice 1 DÉFINITIONS Biomasse : poids frais et/ou sec de la matière vivante présente dans une population. Dans cet essai, la biomasse comprend la tige principale, toutes les branches latérales et toutes les racines. Produit chimique : une substance ou un mélange. Chlorose : décoloration de l'organisme d'essai, en particulier des verticilles, passant du vert au jaune. CEx : concentration du produit chimique d'essai dissous dans le milieu expérimental qui entraîne une réduction de x % (par exemple 50 %) de la croissance de Myriophyllum spicatum pendant une période d'exposition définie (à mentionner explicitement si elle s'écarte de la durée normale ou totale de l'essai). Afin de distinguer les valeurs de la CE mesurées en fonction du taux de croissance de celles mesurées en fonction du rendement, on utilise les abréviations CEt s'agissant du taux de croissance, et CEr s'agissant du rendement, suivies par la mention de la variable de mesure utilisée, par exemple CEr (longueur de la tige principale). Croissance : augmentation de la variable de mesure, par exemple longueur de la tige principale, longueur totale des branches latérales, longueur totale des tiges, longueur totale des racines, poids frais, poids sec ou nombre de verticilles, pendant la période d'essai. Taux de croissance (taux de croissance spécifique moyen): accroissement logarithmique de la variable de mesure pendant la période d'exposition. Note: Les variables de réponse relatives au taux de croissance ne dépendent pas de la durée de l'essai tant que la croissance des organismes témoins non traités obéit à une loi exponentielle. Concentration minimale avec effet observé (CMEO) : concentration d'essai la plus faible à laquelle on observe que le produit chimique exerce un effet statistiquement significatif de réduction de la croissance (à p < 0,05) par comparaison avec le témoin, durant une période d'exposition donnée. Cependant, toutes les concentrations d'essai supérieures à la CMEO devraient avoir un effet néfaste supérieur ou égal à celui observé à la CMEO. Si ces deux conditions ne sont pas réunies, il convient de justifier de façon détaillée le choix de la CMEO (et donc de la CSEO). Variables de mesure : les variables de tout type qui sont mesurées pour exprimer le critère d'évaluation de l'effet au moyen d'une ou plusieurs variables de réponse. Dans la présente méthode d'essai, les variables de mesure sont la longueur de la tige principale, la longueur totale des branches latérales, la longueur totale des tiges, la longueur totale des racines, le poids frais, le poids sec et le nombre de verticilles. Monoculture : culture monospécifique. Nécrose : tissu mort (d'aspect blanc ou brun foncé) de l'organisme d'essai. Concentration sans effet observé (CSEO) : concentration d'essai immédiatement inférieure à la CMEO. Variable de réponse : variable permettant d'estimer la toxicité à partir de n'importe quelle variable mesurée décrivant la biomasse, selon différentes méthodes de calcul. Dans la présente méthode d'essai, le taux de croissance et le rendement représentent les variables de réponse dérivées des variables de mesure telles que la longueur de la tige principale, la longueur totale de tiges, le poids frais, le poids sec ou le nombre de verticilles. Essai semi-statique : essai dans lequel la solution d'essai est renouvelée périodiquement à intervalles définis durant l'essai. Essai statique : essai pendant lequel la solution d'essai n'est pas renouvelée. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Critère d'évaluation de l'effet : décrit le facteur général qui sera modifié, par rapport au témoin, par le produit chimique d'essai. Dans la présente méthode d'essai, le critère d'évaluation de l'effet est l'inhibition de la croissance, qui peut être exprimé par différentes variables de réponse dérivées d'une ou plusieurs variables de mesure. Milieu d'essai : milieu de croissance synthétique complet dans lequel les plantes mises à l'épreuve se développent pendant qu'elles sont exposées au produit chimique d'essai. Normalement, le produit chimique d'essai est dissous dans le milieu expérimental. UVCB : substance de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques Rendement : valeur de la variable de mesure choisie pour exprimer la biomasse à la fin de la période d'exposition moins la valeur de cette variable au début de la période d'exposition. Note: Lorsque la croissance des organismes non exposés obéit à une loi exponentielle, les variables de réponse fondées sur le rendement diminuent quand la durée du test augmente. Appendice 2 MILIEU D'ANDREWS MODIFIE POUR LES PRÉCULTURES ET LES CULTURES MÈRES Le milieu d'Andrews modifié nécessaire aux précultures et aux cultures mères est préparé à partir de cinq solutions mères nutritives élaborées séparément, auxquelles on ajoute 3 % de sucrose. Tableau 1 Composition de la solution nutritive d'Andrews (Norme ASTM E 1913-04)
Les solutions mères peuvent être conservées au réfrigérateur pendant six mois (entre 5 et 10 °C). Seule la solution mère no 5 a une durée de conservation inférieure (deux mois). Tableau 2 Production de la solution mère no 3.1 servant à préparer la solution mère no 3
Après obtention de la solution mère no 3.1 (Tableau 2), la répartir en aliquotes d'environ 11 ml qui seront congelées à une température inférieure ou égale à – 18 °C. Ces portions congelées se conservent cinq ans. Pour préparer la solution mère no 3, décongeler une solution mère no 3.1, en verser 10 ml dans une fiole volumétrique de 1 l et ajouter de l'eau ultra pure jusqu'à la jauge. Pour obtenir le milieu d'Andrews modifié, verser environ 2 500 ml d'eau ultra pure dans une fiole volumétrique de 5 l. Y ajouter 50 ml de chaque solution mère, remplir 90 % de la fiole volumétrique avec de l'eau ultra pure et amener le pH à 5,8. Ajouter ensuite 150 g de sucrose dissous (équivalant à 3 % dans 5 l), puis compléter jusqu'à la jauge avec de l'eau ultra pure. Enfin, verser la solution nutritive dans des fioles Schott de 1 l et autoclaver à 121 °C pendant 20 minutes. La solution nutritive ainsi obtenue peut être conservée stérile au réfrigérateur (entre 5 et 10 °C) pendant trois mois. Milieu d'Andrews modifié pour l'essai de toxicité dans un système sans sédiment Le milieu d'Andrews modifié concentré dix fois qui entre dans la composition des solutions d'essai est préparé à partir des cinq solutions mères nutritives déjà mentionnées dans les tableaux 1 et 2, après ajout de sucrose à hauteur de 30 %. Pour ce faire, verser environ 100 ml d'eau ultra pure dans une fiole volumétrique de 1 l. Ajouter 100 ml de chacune des solutions mères puis amener le pH à 5,8. Ensuite, dissoudre du sucrose pour une concentration de 30 % (soit 300 g dans 1 000 ml), puis compléter avec de l'eau ultra pure jusqu'à la jauge. Enfin, verser la solution nutritive dans des fioles Schott de 0,5 l et autoclaver à 121 °C pendant 20 minutes. La solution nutritive concentrée dix fois ainsi obtenue peut être conservée stérile au réfrigérateur (entre 5 et 10 °C) pendant trois mois. Appendice 3 ENTRETIEN D'UNE CULTURE MÈRE Le présent appendice 3 décrit la culture mère de Myriophyllum spicatum L (102), espèce de plante aquatique immergée dicotylédone de la famille des myriophylles. Entre juin et août, elle produit de discrètes fleurs roses et blanches qui pointent à la surface de l'eau. Les plantes sont enracinées dans le sol grâce à un système de rhizomes robustes. Elles poussent dans tout l'hémisphère nord dans des eaux stagnantes eutrophiques mais non polluées et plutôt calcifères qui contiennent un substrat boueux. Myriophyllum spicatum s'épanouit mieux en eau douce, mais se trouve aussi en eau saumâtre. La culture mère dans un système sans sédiment en conditions de laboratoire fait appel à des plantes stériles. On peut obtenir des plantes stériles auprès du laboratoire d'écotoxicologie de l'Umweltbundesamt allemand (Agence fédérale allemande pour l'environnement). Il est aussi possible d'obtenir des organismes d'essai à partir de végétaux non stériles, conformément à la norme ASTM E 1913-04. La méthode de culture de spécimens de Myriophyllum sibiricum prélevés dans la nature indiquée ci-dessous est tirée du guide ASTM: “Si l'on opte pour des plantes non stériles prélevées dans la nature, récolter des turions de M. sibiricum à l'automne. Placer ces turions dans un aquarium de 20 l contenant 5 cm de sédiment stérile recouvert de sable siliceux ou, par exemple, de Turface® et de 18 l d'eau de qualité réactif. Aérer l'aquarium, maintenir sa température à 15 oC ainsi que l'intensité lumineuse entre 200 et 300 μmol.m– 2.s– 1 pendant 16 heures par jour. La culture des plantes en aquarium peut fournir des végétaux de remplacement si les cultures stériles sont détruites par un dysfonctionnement mécanique dans l'enceinte de croissance, par une contamination ou d'autres causes encore. Les spécimens cultivés dans l'aquarium ne sont pas stériles, et les cultures stériles ne peuvent être conservées dans un système de culture en discontinu. Pour stériliser la culture, les plantes sont retirées de l'aquarium et rincées pendant une demi-heure environ dans un courant d'eau désionisée. Elles sont ensuite désinfectées en conditions aseptiques dans une hotte à flux laminaire pendant moins de 20 min (jusqu'à ce que la majorité des tissus végétaux aient blanchi et que seul l'apex en croissance reste vert) dans une solution d'hypochlorite de sodium à 3 % (m/v) contenant 0,01 % d'un tensioactif adapté. Agiter le désinfectant et le matériel végétal. Les segments comptant plusieurs nœuds sont transférés dans des tubes de culture stériles contenant 45 ml de milieu d'Andrews modifié stérilisé, ensuite clos avec leurs bouchons ordinaires. On ne place qu'un segment végétal par enceinte expérimentale. Pour s'assurer que les récipients de culture sont bien fermés, on les scelle avec du film de laboratoire. Une fois qu'une culture stérile a été préparée, il convient de transférer les segments végétaux comptant plusieurs nœuds dans de nouvelles enceintes expérimentales contenant du milieu nutritif liquide nouvellement préparé tous les dix à douze jours. Des cultures sur boîtes de gélose ont montré que les plantes doivent être stériles et le demeurer pendant huit semaines avant le lancement de l'essai.” Comme le milieu d'Andrews modifié contient du sucrose (qui stimule la croissance des champignons et des bactéries), l'ensemble du matériel, des solutions et des cultures seront manipulés en conditions stériles. Il convient de stériliser tous les liquides et le matériel expérimental avant de les utiliser. Cette stérilisation est effectuée par traitement à l'air chaud (210 oC) pendant 4 heures, ou en autoclave durant 20 minutes à 121 oC. D'autre part, tous les ballons, fioles, boîtes et autres équipements sont passés à la flamme sur une paillasse stérile avant d'être utilisés. Les cultures mères peuvent être conservées plus longtemps sous un éclairage et à une température réduits (50 μE.m– 2.s– 1, 20 ± 2 °C) sans qu'il faille les préparer à nouveau. Le milieu de croissance des myriophylles doit être à le même que celui utilisé pour l'essai, mais d'autres milieux riches en éléments nutritifs peuvent être utilisés pour les cultures mères. Les segments végétaux axéniques sont répartis dans plusieurs erlenmeyers de 500 ml et/ou dans des fioles Fernbach de 2 000 ml, chaque récipient contenant environ 450 ml (erlenmeyers) ou 1 000 ml (fioles Fernbach) de milieu d'Andrews modifié. Les récipients sont alors scellés avec des bouchons en cellulose pour garantir l'axénisation. D'autre part, il est impératif de minutieusement passer à la flamme l'équipement de la paillasse stérile juste avant de l'utiliser. En fonction de leur nombre et de leur taille, les plantes sont transférées dans une nouvelle solution nutritive toutes les trois semaines environ. Pour cette culture renouvelée, on peut utiliser les apex ou des segments de la partie médiane de la tige. Le nombre et la taille des plantes (ou segments de plante) transférées dépendent du nombre de plantes nécessaire. Par exemple, il est possible de transférer cinq segments de tige dans une fiole Fernbach et trois segments de tige dans un erlenmeyer, chaque segment ayant une longueur de 5 cm. Éliminer tout segment présentant des racines, des fleurs, des parties mortes ou qui se distinguent d'une manière ou d'une autre. Figure 1 Sectionnement des plantes pour la culture mère et la préculture après 3 semaines de culture. 
Les plantes sont cultivées dans des erlenmeyers de 500 ml et des fioles Fernbach de 2 000 ml dans un incubateur réfrigérant à 20 ± 2 °C avec un éclairage continu dont l'intensité est approximativement comprise entre 100 et 150 μE.m– 2.s– 1 ou 6 000-9 000 lux (émis par l'éclairage de l'enceinte avec une température de couleur correspondant à une «lumière blanche chaude»). Figure 2 Culture des plantes dans un incubateur réfrigérant éclairé. 
Il convient d'utiliser des récipients de culture en verre stériles et chimiquement propres (lavés à l'acide) et d'employer des techniques de manipulation aseptiques. En cas de contamination de la culture mère, notamment par des algues, des champignons et/ou des bactéries, il convient de préparer une nouvelle culture ou d'utiliser une culture mère provenant d'un autre laboratoire pour la remplacer. Appendice 4 ENTRETIEN D'UNE PRÉCULTURE ET PRÉPARATION DE L'ORGANISME POUR L'ESSAI Pour obtenir une préculture, couper les tiges de la culture mère en segments comptant deux verticilles chacun. Placer ces segments dans des fioles Fernbach remplies de milieu d'Andrews modifié contenant 3 % de sucrose. Chaque fiole peut recevoir jusqu'à 50 segments de tige. Cependant, on s'assurera que les segments sont vivants et ne présentent ni racines, ni branches latérales, ni bourgeons (voir graphique 1 de l'appendice 3). La préculture dure 14 à 21 jours en conditions stériles dans une enceinte environnementale alternant 16 heures de lumière et 8 heures d'obscurité. L'intensité lumineuse sera comprise entre 100 et 150 μE.m– 2.s– 1. La température des récipients d'essai est maintenue à 23 ± 2 °C. Étant donné que le milieu d'Andrews modifié contient du sucrose (qui stimule la croissance des algues, des champignons et des bactéries), les solutions du produit chimique d'essai sont préparées et la culture est effectuée en conditions stériles. L'ensemble des liquides et du matériel expérimental est stérilisé avant utilisation. Cette stérilisation se fait par traitement à l'air chaud (210 °C) pendant 4 heures, ou en autoclave durant 20 minutes à 121 °C. D'autre part, tous les ballons, fioles, boîtes et autres équipements sont passés à la flamme sur une paillasse stérile avant d'être utilisés. On retire les tiges des fioles de préculture en conditions axéniques, en choisissant un matériel végétal aussi homogène que possible. Chaque essai nécessite au moins 60 spécimens (essai portant sur huit concentrations du produit chimique d'essai). Aux fins de l'essai, prélever des branches latérales des végétaux précultivés, les raccourcir à 2,5 cm à partir de leur base (mesure effectuée à l'aide d'une règle) et les transférer dans un bécher contenant du milieu d'Andrews modifié stérile. Ces branches latérales fraîches peuvent servir à l'essai de toxicité sur Myriophyllum spicatum dans un système sans sédiment. Figure 2 Sectionnement des plantes précultivées pour l'essai de toxicité sur Myriophyllum spicatum dans un système sans sédiment. 
C.51 Essai de toxicité sur Myriophyllum spicatum dans un système eau-sédiment INTRODUCTION
PRINCIPE DE L'ESSAI
INFORMATIONS SUR LE PRODUIT CHIMIQUE D'ESSAI
VALIDITÉ DE L'ESSAI
PRODUIT CHIMIQUE DE RÉFÉRENCE
DESCRIPTION DE LA MÉTHODE Appareillage
Organisme d'essai
Culture de l'organisme d'essai
Sédiment
Milieu d'essai
Conception de l'essai
Concentrations du produit chimique d'essai et groupes témoins
Essai limite
Solutions d'essai
MODE OPÉRATOIRE
Phase d'implantation
Choix d'une série de plantes homogènes
Exposition via la phase aqueuse
Exposition via le sédiment
Maintien des niveaux d'eau pendant la durée de l'essai
Conditions expérimentales
Durée de l'essai
Mesures et déterminations analytiques
Fréquence des mesures et des déterminations analytiques
Mesures analytiques du produit chimique d'essai
ÉVALUATION DES DONNÉES
Variables de réponse
Taux de croissance spécifique moyen
Rendement
Courbes concentration-effet
Estimation de la CEx
Méthodes statistiques
RAPPORT
BIBLIOGRAPHIE
Appendice 1 COMPOSITION DU MILIEU DE SMART ET BARKO
Appendice 2 DÉFINITIONS Biomasse : poids frais et/ou sec de la matière vivante présente dans une population. Dans cet essai, la biomasse comprend la tige principale, toutes les branches latérales et toutes les racines. Produit chimique : une substance ou un mélange. Chlorose : décoloration de l'organisme d'essai, en particulier des verticilles, passant du vert au jaune. CEx : concentration du produit chimique d'essai dissous dans le milieu expérimental qui entraîne une réduction de x % (par exemple 50 %) de la croissance de Myriophyllum spicatum pendant une période d'exposition définie (à mentionner explicitement si elle s'écarte de la durée normale ou totale de l'essai). Afin de distinguer les valeurs de la CE mesurées en fonction du taux de croissance de celles mesurées en fonction du rendement, on utilise les abréviations CEt s'agissant du taux de croissance, et CEr s'agissant du rendement, suivies par la mention de la variable de mesure utilisée, par exemple CEr (longueur de la tige principale). Croissance : augmentation de la variable de mesure, par exemple longueur de la tige principale, longueur totale des branches latérales, longueur totale des tiges, longueur totale des racines, poids frais, poids sec ou nombre de verticilles, pendant la période d'essai. Taux de croissance (taux de croissance spécifique moyen): accroissement logarithmique de la variable de mesure pendant la période d'exposition. Note: Les variables de réponse relatives au taux de croissance ne dépendent pas de la durée de l'essai tant que la croissance des organismes témoins non traités obéit à une loi exponentielle. Concentration minimale avec effet observé (CMEO) : concentration d'essai la plus faible à laquelle on observe que le produit chimique exerce un effet statistiquement significatif de réduction de la croissance (à p < 0,05) par comparaison avec le témoin, durant une période d'exposition donnée. Cependant, toutes les concentrations d'essai supérieures à la CMEO devraient avoir un effet néfaste supérieur ou égal à celui observé à la CMEO. Si ces deux conditions ne sont pas réunies, il convient de justifier de façon détaillée le choix de la CMEO (et donc de la CSEO). Variables de mesure : les variables de tout type qui sont mesurées pour exprimer le critère d'évaluation de l'effet au moyen d'une ou plusieurs variables de réponse. Dans la présente méthode d'essai, les variables de mesure sont la longueur de la tige principale, la longueur totale des branches latérales, la longueur totale des tiges, la longueur totale des racines, le poids frais, le poids sec et le nombre de verticilles. Monoculture : culture monospécifique. Nécrose : tissu mort (d'aspect blanc ou brun foncé) de l'organisme d'essai. Concentration sans effet observé (CSEO) : concentration d'essai immédiatement inférieure à la CMEO. Variable de réponse : variable permettant d'estimer la toxicité à partir de n'importe quelle variable mesurée décrivant la biomasse, selon différentes méthodes de calcul. Dans la présente méthode d'essai, le taux de croissance et le rendement représentent les variables de réponse dérivées des variables de mesure telles que la longueur de la tige principale, la longueur totale de tiges, le poids frais, le poids sec ou le nombre de verticilles. Essai semi-statique : essai dans lequel la solution d'essai est renouvelée périodiquement à intervalles définis durant l'essai. Essai statique : essai pendant lequel la solution d'essai n'est pas renouvelée. Produit chimique d'essai : toute substance ou tout mélange soumis à un essai réalisé suivant la présente méthode d'essai. Critère d'évaluation de l'effet : décrit le facteur général qui sera modifié, par rapport au témoin, par le produit chimique d'essai. Dans la présente méthode d'essai, le critère d'évaluation de l'effet est l'inhibition de la croissance, qui peut être exprimé par différentes variables de réponse dérivées d'une ou plusieurs variables de mesure. Milieu expérimental : milieu de croissance synthétique complet dans lequel les plantes mises à l'épreuve se développent pendant qu'elles sont exposées au produit chimique d'essai. Normalement, le produit chimique d'essai est dissous dans le milieu expérimental. UVCB : substance de composition inconnue ou variable, produits de réaction complexes ou matériels biologiques Rendement : valeur de la variable de mesure choisie pour exprimer la biomasse à la fin de la période d'exposition moins la valeur de cette variable au début de la période d'exposition. Note: Lorsque la croissance des organismes non exposés obéit à une loi exponentielle, les variables de réponse fondées sur le rendement diminuent quand la durée du test augmente. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||








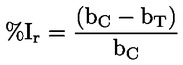
(1) Il n'existe pas de valeur pour 20 °C, mais on peut considérer que la variabilité des résultats des mesures est supérieure aux variations dues à la température
(2) Règlement (CE) no 1907/2006 du Parlement européen et du Conseil du 18 décembre 2006 concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH), instituant une agence européenne des produits chimiques, modifiant la directive 1999/45/CE et abrogeant le règlement (CEE) no 793/93 du Conseil et le règlement (CE) no 1488/94 de la Commission ainsi que la directive 76/769/CEE du Conseil et les directives 91/155/CEE, 93/67/CEE, 93/105/CE et 2000/21/CE de la Commission.
(3) US Interagency Coordinating Committee on the Validation of Alternative Methods (Comité de coordination interagences sur la validation des méthodes alternatives, aux Etats-Unis)
(*1) L'étendue de l'opacité cornéenne est précisée
(4) En ce qui concerne le recours à une stratégie d'essai intégrée pour l'irritation oculaire dans le cadre de REACH, voir également le guide des exigences d'information et évaluation de la sécurité chimique de l'ECHA, Chapitre R.7a:Informations spécifiques aux effets http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
(5) Les statisticiens qui suivent une démarche de modélisation telle que l'utilisation de modèles linéaires généralisés (MLG) peuvent conduire l'analyse d'une manière différente mais comparable; toutefois, ils n'obtiendront pas nécessairement le traditionnel tableau ANOVA, qui date des approches algorithmiques du calcul statistique développées à l'ère pré-informatique.
(6) Les statisticiens qui suivent une démarche de modélisation telle que l'utilisation de modèles linéaires généralisés (MLG) peuvent conduire l'analyse d'une manière différente mais comparable; toutefois, ils n'obtiendront pas nécessairement le traditionnel tableau ANOVA, qui date des approches algorithmiques du calcul statistique développées à l'ère pré-informatique
(7) Règlement (CE) no 1272/2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) no 1907/2006, JO L 353 du 31.12.2008, p.1.
(8) Des catégories chimiques ont été assignées à chaque produit chimique selon un schéma de classification standard, basé sur le système de classification des National Library of Medicine Medical Subject Headings (MeSH) (disponible à l'adresse: http//www.nlm.nih.gov/mesh).
(9) D'après les résultats issus de l'essai sur œil de lapin in vivo (LD OCDE 405) (17) et en utilisant le SGH de l'ONU (4).
(10) La classification dans les catégories 2A ou 2B dépend de l'interprétation des critères du SGH de l'ONU pour distinguer ces deux catégories, à savoir que l'on doit constater des effets chez 1 sur 3 ou 2 sur 3 animaux le septième jour pour classer le produit dans la catégorie 2A. L'étude in vivo est menée sur 3 animaux. Tous les effets sauf une rougeur conjonctivale chez un animal avaient disparu le septième jour ou avant. Le seul animal chez lequel la rougeur n'avait pas disparu le septième jour (score de 1) était pleinement rétabli le dixième jour.
(11) Les dimensions indiquées ici sont celles d'un porte-cornée utilisé pour des bovins âgés de 12 à 60 mois. Pour des animaux entre 6 et 12 mois, le porte-cornée est conçu de manière à posséder des chambres d'une contenance de 4 ml chacune et dont les dimensions intérieures sont de 1,5 cm de diamètre et 2,2 cm de profondeur. Pour tout porte-cornée nouvellement conçu, il est essentiel que le rapport entre la surface cornéenne exposée et le volume de la chambre postérieure soit le même que celui du porte-cornée classique. Cette condition est indispensable pour garantir que les valeurs de perméabilité sont correctement déterminées pour le calcul du SIIV par la formule donnée
(12) Règlement (CE) no 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) no 1907/2006, JO L 353 du 31.12.2008, p.1.
(*2) Score moyen le plus élevé observé sur tous les points temporels
(*3) Score moyen maximal observé sur tous les points temporels (à partir des scores d'opacité tels que définis au tableau 1).
(*4) À partir des scores d'opacité tels que définis au tableau 2.
(*5) Combinaisons moins probables.
(13) Les classes de produits chimiques ont été attribuées à chaque produit chimique d'essai à l'aide d'un système de classification normalisé, fondé sur le système de classification du Medical Subject Headings (MeSH) de la Bibliothèque nationale de médecine des États-Unis (disponible à l'adresse: http//www.nlm.nih.gov/mesh)
(14) D'après les résultats de l'essai in vivo sur œil de lapin (Ligne directrice de l'OCDE no 405) et en utilisant le SGH (ONU) (4)(6).
(15) D'après les résultats obtenus avec la méthode OPI comme décrit au tableau 6.
(16) Combinaison des scores OPI autres que ceux décrits au tableau 6 pour l'identification des produits ne relevant d'aucune catégorie du SGH ou relevant de la catégorie 1 du SGH (tableau 6).
(17) La classification dans les catégories 2A ou 2B dépend de l'interprétation des critères du SGH de l'ONU pour distinguer ces deux catégories, à savoir que l'on doit constater des effets chez 1 sur 3 ou 2 sur 3 animaux le septième jour pour classer le produit dans la catégorie 2A. L'étude in vivo est menée sur 3 animaux. Tous les effets sauf une rougeur conjonctivale chez un animal avaient disparu le septième jour ou avant. Le seul animal chez lequel la rougeur n'avait pas disparu le septième jour (score de 1) était pleinement rétabli le dixième jour.
(18) Règlement (CE) no 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) no 1907/2006.
(19) Le terme de «moyenne», dans le présent document, est entendu au sens de «moyenne arithmétique».
(20) Les chiffres correspondent à des valeurs seuils obtenues par traitement statistique, et ne se rapportent pas à la précision de la mesure.
(21) Une prédiction DPRA doit être envisagée dans le cadre d'une démarche de type IATA et conformément aux dispositions des paragraphes 9 et 12.
(22) Les chiffres correspondent à des valeurs seuils obtenues par traitement statistique, et ne se rapportent pas à la précision de la mesure.
(23) Une prédiction DPRA doit être envisagée dans le cadre d'une démarche de type IATA et conformément aux dispositions des paragraphes 9 et 12.
(24) La prédiction in vivo des dangers (et de la puissance sensibilisante) est fondée sur les données ELGL (19). La puissance in vivo est déterminée à partir de critères proposés par ECETOC (23).
(25) Une prédiction DPRA doit être envisagée dans le cadre d'une démarche de type IATA et conformément aux dispositions des paragraphes 9 et 11.
(26) Intervalles déterminés sur la base d'au moins 10 valeurs de déplétion établies par 6 laboratoires indépendants.
(27) Règlement (CE) no 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) no 1907/2006.
(28) La prédiction des dangers in vivo (et la puissance) est fondée sur les données ELGL (13). La puissance in vivo est déterminé au moyen des critères proposé par ECETOC (24).
(29) Une prédiction KeratinoSensTM doit être envisagée dans le cadre d'une démarche de type IATA et conformément aux dispositions des paragraphes 9 et 11 de la présente méthode d'essai.
(30) D'après les données historiques observées (12).
(31) Règlement (CE) no 1272/2008 du Parlement européen et du Conseil du 16 décembre 2008 relatif à la classification, à l'étiquetage et à l'emballage des substances et des mélanges, modifiant et abrogeant les directives 67/548/CEE et 1999/45/CE et modifiant le règlement (CE) no 1907/2006.
(32) Les classes de produits chimiques ont été attribuées à chaque produit chimique d'essai à l'aide d'un système de classification normalisé, fondé sur le système de classification du Medical Subject Headings (MeSH) de la Bibliothèque nationale de médecine des États-Unis (disponible sur http//www.nlm.nih.gov/mesh).
(33) D'après les résultats de l'essai in vivo sur œil de lapin (ligne directrice no 405 de l'OCDE, méthode d'essai B.5) et en utilisant le SGH (ONU) et le CLP de l'UE.
(34) D'après les résultats obtenus avec l'essai de DF [Protocole INVITTOX no 71(6)].
(35) Les statisticiens qui suivent une démarche de modélisation telle que l'utilisation de modèles linéaires généralisés (MLG) peuvent conduire l'analyse d'une manière différente mais comparable; toutefois, ils n'obtiendront pas nécessairement le traditionnel tableau ANOVA, qui remonte à des conceptions algorithmiques du calcul statistique développées à l'ère pré-informatique.
(36) Voir définitions et unités à l'appendice 1.
(37) Parfois noté POE, il est déterminé par agitation en flacon dans la méthode d'essai A.8 (4), par la méthode HPLC dans la méthode d'essai A.24 (5) et par brassage lent dans la méthode d'essai A.23 (6). La technique par colonne est parfois utilisée pour déterminer le log K OE. Un petit nombre d'études disponibles y recourent, essentiellement pour les biphényles chlorés et les dibenzodioxines (par exemple Li et Doucette, 1993) (3). Pour les substances susceptibles de s'ioniser, le K OE se réfère à la forme non ionisée.
(38) Voir définitions et unités à l'appendice 1.
(39) CCM: chromatographie en couche mince; CLHP: chromatographie en phase liquide à haute performance; CG: chromatographie en phase gazeuse.
(40) Dans certains cadres réglementaires, l'analyse des métabolites peut être obligatoire dans certaines conditions (voir paragraphe 65).
(41) En général, les concentrations mesurées dans l'eau durant la phase d'absorption sont supérieures d'au moins une puissance dix à la limite de quantification, de sorte que plus d'une demi-vie de la charge corporelle puisse être mesurée lors de la phase d'élimination de l'étude.
(42) Voir définitions et unités à l'appendice 1.
(43) Pour la plupart des substances d'essai, aucune concentration ne doit, dans l'idéal, être détectée dans l'eau témoin. Des concentrations ne doivent se retrouver que pour des matériaux d'origine naturelle (par exemple certains métaux) et des substances omniprésentes dans l'environnement.
(44) Si, de toute évidence, le système n'obéit pas à une cinétique du premier ordre, il convient de recourir à des modèles plus complexes (voir la bibliographie de l'appendice 5 et demander conseil à un biostatisticien).
(45) L'absorption peut être limitée par des concentrations d'exposition basses dues à la faible hydrosolubilité de la substance dans l'essai de bioconcentration; l'essai par voie alimentaire permettra, lui, d'atteindre des concentrations d'exposition beaucoup plus fortes.
(46) Pour les substances multi-composants comme les UVCB (substances de composition inconnue ou variable) et les mélanges, l'hydrosolubilité de chaque composant pertinent est examinée pour déterminer les concentrations d'exposition appropriées.
(47) Le COT comprend le carbone organique particulaire et le carbone organique dissous. En d'autres termes, COT = COP + COD.
(48) Bien que ce ne soit généralement pas recommandé, si un solvant ou un agent solubilisant est utilisé, le carbone organique en provenant est ajouté à celui issu de la substance d'essai pour évaluer la concentration en carbone organique dans les récipients d'essai.
(49) Si la teneur en lipides et la concentration de la substance d'essai ne sont pas mesurées sur le même poisson, les poissons utilisés ont au moins un poids similaire et (si approprié) être du même sexe.
(50) Cette solution alternative n'est valable que si les poissons de tous les groupes d'essai sont répartis en groupes de taille similaire, prélevés selon la même méthode et nourris de manière identique. Cela garantit que la croissance des poissons est la même dans tous les groupes d'essai, si la concentration testée est inférieure au seuil toxique. Si la croissance est similaire, la teneur en lipides devrait l'être aussi. Une différence de croissance dans le groupe témoin indiquerait un effet de la substance et invaliderait l'essai.
(51) Outre le poids, la longueur totale est enregistrée car l'augmentation de cette longueur au cours de l'essai est un bon indicateur de la survenue d'un éventuel effet négatif.
(52) On peut réaliser un test t sur les constantes cinétiques de croissance, pour tester si le développement entre les groupes témoins et d'essai varie, ou test F en cas d'analyse des écarts. Si besoin, on peut recourir à un test F ou à un essai fondé sur les rapports de probabilité pour faciliter le choix du modèle de croissance approprié [monographie OCDE no 54 (32)].
(53) Ces pourcentages supposent la fiabilité des méthodes d'analyse et une demi-vie inférieure à 14 jours. Si les méthodes d'analyse sont moins fiables ou si la demi-vie est (très) supérieure, les chiffres seront plus élevés.
IC: intervalle de confiance (quand estimation possible)
ET: écart-type (quand estimation possible)
(56) L'essai réduit peut en effet servir à démontrer un métabolisme rapide attendu comme tel.
(57) Si seulement deux points sont mesurés, on pourra estimer le seuil de confiance du FBCkm selon les méthodes «bootstrap». Lorsque des points intermédiaires sont également disponibles, le seuil de confiance du FBCkm peut être calculé comme dans l'essai complet.
(58) Voir définitions et unités à l'appendice 1.
(59) Aux fins du règlement (CE) no 1907/2006 concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH) (JO L 396 du 30.12.2006, p 1), cet aspect est abordé dans le «Guide des exigences d'information et évaluation de la sécurité chimique» qui lui est associé, chapitre R.7c, R.7.10.3.1, R.7.10.4.1, et figure R7.10-2.
(60) Pour la plupart des substances d'essai, on ne devrait idéalement rien détecter dans l'eau témoin. Les concentrations naturelles s'expliquent uniquement par la présence logique de matériaux (certains métaux par exemple) et substances omniprésents dans le milieu.
(61) Le FBA étant défini comme le rapport entre la concentration d'une substance dans un organisme et la concentration de la substance dans l'alimentation de cet organisme à l'état stationnaire, les lipides sont pris en compte dans la mesure où l'on corrige les valeurs obtenues en fonction des lipides présents dans l'organisme et dans l'alimentation, d'où le terme plus précis de «correction». Cette approche diffère de la «normalisation» par rapport à une teneur en lipides donnée dans l'organisme comme cela est fait pour l'essai de bioconcentration par exposition en milieu aquatique.
(62) La longueur totale est aussi être consignée durant l'essai, puisqu'elle constitue un bon indicateur des éventuels effets nocifs observés.
(63) Le HCB figure dans les listes des annexes A et C à la Convention de Stockholm, ainsi que dans les annexes I et III du règlement (CE) no 850/2004 concernant les polluants organiques persistants (JO L 158 du 30.4.2004, p. 7).
(64) En cas de développement rapide durant la phase d'absorption, la ration alimentaire réelle descendra en-dessous de celle définie au début de l'exposition.
(65) Dans le cadre d'une étude avec exposition via le milieu aquatique, une demi-vie de 14 jours correspondrait à un FBC d'environ 10 000 l/kg si on utilisait des poissons de 1 g avec une cinétique d'absorption avoisinant 500 l/kg/j [selon l'équation de Sijm et al. (46)].
(66) Les concentrations internes réelles ne pouvant être déterminées qu'à l'issue de l'essai, il est nécessaire d'estimer la concentration interne attendue (par exemple en s'appuyant sur le FBA attendu et sur la concentration dans la nourriture; voir à l'appendice 5 l'équation A5.8).
(67) La présence de la substance d'essai dans le milieu de l'essai à travers les excréments du poisson ou par lixiviation de la nourriture ne peut pas être totalement évitée. Aussi est-il possible par exemple de mesurer la concentration de la substance présente dans l'eau à la fin de la phase d'absorption, surtout si on utilise un système semi-statique, afin d'établir si une exposition en milieu aquatique est intervenue.
(68) Cette méthode est spécifique à l'étude fondée sur l'alimentation, et varie de la procédure appliquée pour l'exposition en milieu aquatique; aussi employons-nous le terme «correction» au lieu de «normalisation» afin d'éviter toute confusion — voir aussi la note de bas de page du paragraphe 106.
(69) On peut réaliser un test t sur les constantes cinétiques de croissance, pour tester si le développement entre les groupes témoins et d'essai varie, ou test F en cas d'analyse des écarts. Si besoin, on peut recourir à un test F ou à un essai fondé sur les rapports de probabilité pour faciliter le choix du modèle de croissance approprié [monographie OCDE no 54 (32)].
(70) Technique d'analyse des aliments s'attachant à la teneur en protéines, en lipides, en cellulose brute et en cendres. Ces informations sont habituellement disponibles auprès du fabricant.
IC: intervalle de confiance (quand estimation possible)
ET: écart-type (quand estimation possible)
(73) Meyer et al. (1)
(74) Il est préférable durant l'essai proprement dit de privilégier le poids pour mesurer les écarts des constantes cinétiques de croissance. Néanmoins il est reconnu que la longueur est une mesure plus pratique si le poisson doit être sélectionné à vue au début d'une expérience (au sein du stock de poissons).
(75) Cette fourchette de longueurs est indiquée dans la loi japonaise sur les substances chimiques: Testing Methods for New Chemical Substances etc., based on the Japan's Chemical Substances Control Law (CSCL).
(76) Les valeurs entre parenthèses correspondent au nombre d'échantillons (eau, poissons) à prélever lors d'un éventuel prélèvement supplémentaire.
(77) L'estimation avant l'essai du k 2 d'une substance dont le log K oe = 4 s'élève à 0.652 jours– 1. La durée totale de l'expérience est fixée à 3 × tES, soit 3 × 4,6 jours = 14 jours. L'estimation de tES est présentée à l'appendice 5.
(78) Prélever un échantillon d'eau après que l'équivalent du volume d'au moins trois enceintes ait été versé.
(79) Ces poissons sont prélevés dans le stock de poisson.
(80) Si une précision accrue ou des études de métabolisme sont requises, nécessitant plus de poissons, ces poissons devront être prélevés en particulier à la fin des phases d'absorption et de dépuration (voir paragraphe 40).
(81) Au moins trois poissons supplémentaires pourront être nécessaires pour analyser la teneur en lipides s'il n'est pas possible d'utiliser les poissons prélevés pour mesurer les concentrations de la substance au début de l'essai, à la fin de la phase d'absorption et à la fin de la phase d'élimination. Il convient de noter qu'il devrait être possible dans de nombreux cas d'utiliser seulement les trois poissons témoins (voir paragraphe 56).
(82) Trois échantillons de nourriture des groupes témoin et d'essai sont analysés pour mesurer les concentrations de la substance d'essai et la teneur en lipides.
(83) Les poissons sont prélevés du stock le plus tard possible avant le début de l'étude; au moins trois poissons du stock sont prélevés au début de l'essai pour mesurer la teneur en lipides.
(84) Le prélèvement (facultatif) au début de la phase d'absorption fournit les données nécessaires pour calculer l'assimilation de la substance d'essai ingérée par voie alimentaire, que l'on peut comparer au rendement d'assimilation calculé à partir des données obtenues lors de la phase d'élimination.
(85) On peut prélever cinq poissons supplémentaires pour l'analyse de tissus spécifiques.
(86) Au moins trois poissons supplémentaires pourront être nécessaires pour analyser la teneur en lipides s'il n'est pas possible d'utiliser les poissons prélevés pour mesurer les concentrations de la substance au début de l'essai, à la fin de la phase d'absorption et à la fin de la phase d'élimination. Il convient de noter qu'il devrait être possible dans de nombreux cas d'utiliser seulement les trois poissons témoins (voir paragraphes 56 et 153).
(87) Comme pour toute relation empirique, il convient de vérifier que la substance d'essai tombe dans le domaine d'applicabilité de la relation
(88) Le poids des poissons à la fin de la phase d'absorption peut être estimé en fonction des données d'une étude précédente ou des connaissances accumulées sur l'espèce d'essai, dont on sait qu'elle est susceptible de se développer à partir d'un poids au départ de l'essai habituel et sur une durée d'absorption habituelle (par exemple 28 jours).
(89) Dans la plupart des programmes qui permettent une régression linéaire, même les erreurs types et l'intervalle de confiance (IC) des estimations sont donnés, par exemple dans Microsoft Excel avec la commande Analyse des données.
(90) Contrairement à la méthode de régression linéaire, cette formule ne produira pas d'erreur type pour k 2.
(91) Contrairement à une procédure d'ajustement linéaire, cette méthode ne produit habituellement aucune erreur type ni aucun intervalle de confiance pour la k1 estimée.
(92) Dans certaines régions, il est possible qu'on ne puisse obtenir que de la nourriture pour poisson dont la teneur en lipides est très inférieure à ce plafond. Le cas échéant, il convient de réaliser un essai avec cette teneur en lipides plus faible, et d'ajuster la ration alimentaire pour maintenir les poissons en bonne santé. Il est préférable de ne pas augmenter artificiellement la teneur en lipides de l'alimentation en ajoutant trop d'huile.
(93) Dans la nature, l'ingestion est probablement le meilleur moyen d'exposer au maximum des poissons à des substances très hydrophobes dans un milieu aquatique; ainsi, un FBC estimé n'est pas strictement représentatif du potentiel de bioaccumulation d'une telle substance.
(94) On peut faire appel à d'autres espèces de Daphnia à condition qu'elles respectent les critères de validité pertinents (le critère relatif au taux de reproduction des témoins s'applique aux espèces de Daphnia). Si d'autres espèces de Daphnia sont employées, il y a lieu de les identifier clairement et de justifier leur utilisation.
(95) Mortalité accidentelle: mortalité sans lien avec le produit chimique d'essai et due à un événement accidentel (dont la cause est donc connue)
(96) Mortalité fortuite: mortalité sans lien avec le produit chimique d'essai, de cause inconnue
(*6) Indiquer quel récipient a été utilisé pour l'essai
(*7) Indiquer les portées avortées par les lettres “AB” dans la case appropriée
(*8) Indiquer la mortalité d'un quelconque animal parent par la lettre “M” dans la case appropriée
(97) La longueur totale moyenne minimale est obtenue à partir d'une sélection de données disponibles à l'heure actuelle.
(98) La souche particulière de truite arc-en-ciel étudiée peut nécessiter l'utilisation d'autres températures. Les poissons géniteurs doivent être maintenus à la même température que celle devant être utilisée pour les œufs. Après réception d'œufs du commerce, une adaptation de courte durée (1- 2 h, par exemple) à la température d'essai est nécessaire après leur arrivée.
(99) Obscurité pour les larves jusqu'à une semaine après l'éclosion sauf lors de leur inspection, puis éclairage tamisé pendant toute la durée de l'essai (photopériode de 12-16 heures)(4).
(100) Quelles que soient les conditions d'essai, les conditions d'éclairage doivent être constantes
(101) Quel que soit l'essai, elle ne doit pas varier de plus de ±20/00.
(*9) La nourriture doit être donnée à satiété. Le surplus et les excréta doivent être retirés quand cela s'avère nécessaire pour éviter l'accumulation de déchets.
|
AC |
artémies congelées; adultes du genre Artemia |
|
NA |
nauplies d'artémies; fraîchement écloses |
|
NA48 |
nauplies d'artémies; âgées de 48 h |
(1) les larves avec leur vitellus n'ont pas besoin de nourriture
(2) filtrés à partir de cultures mélangées
(3) granules provenant d'un processus de fermentation
(*10) Les tubercules papillaires sont normalement présents uniquement chez les mâles adultes, et se situent entre le deuxième et le septième ou le huitième rayon de nageoire en comptant à partir de l'extrémité postérieure de la nageoire anale (fig.1 et 2). Ils apparaissent rarement sur le premier rayon en comptant à partir de l'extrémité postérieure de la nageoire anale. Cette procédure opératoire standard (POS) permet de mesurer les tubercules présents sur le premier rayon de nageoire (le numéro du rayon est déterminé en comptant à partir de l'extrémité postérieure de la nageoire anale dans cette POS).
(*11) Tampon d'homogénéisaton:
|
— |
[50 mM de Tris-HCl de pH 7,4; 1 % de cocktail d'inhibiteurs de protéase (Sigma)]: 12 ml de Tris-HCl de pH 7,4 + 120 μl de cocktail d'inhibiteurs de protéase. |
|
— |
TRIS: TRIS-ULTRA PURE (ICN) de chez Bie & Berntsen (Danemark), par exemple. |
|
— |
Cocktail d'inhibiteurs de protéase: de chez Sigma (pour les tissus mammaliens). Numéro de produit P 8340. |
NOTE: Le tampon d'homogénéisation est utilisé le jour de sa fabrication. Placer sur de la glace pendant son utilisation.
(102) Carl von Linné (né le 23 mai 1707 à Råshult/Älmhult, décédé le 10 janvier 1778 à Uppsala).
(*12) eau déminéralisée (distillée ou désionisée)